

















































































































































































































































































































































































































































































































































































































































































































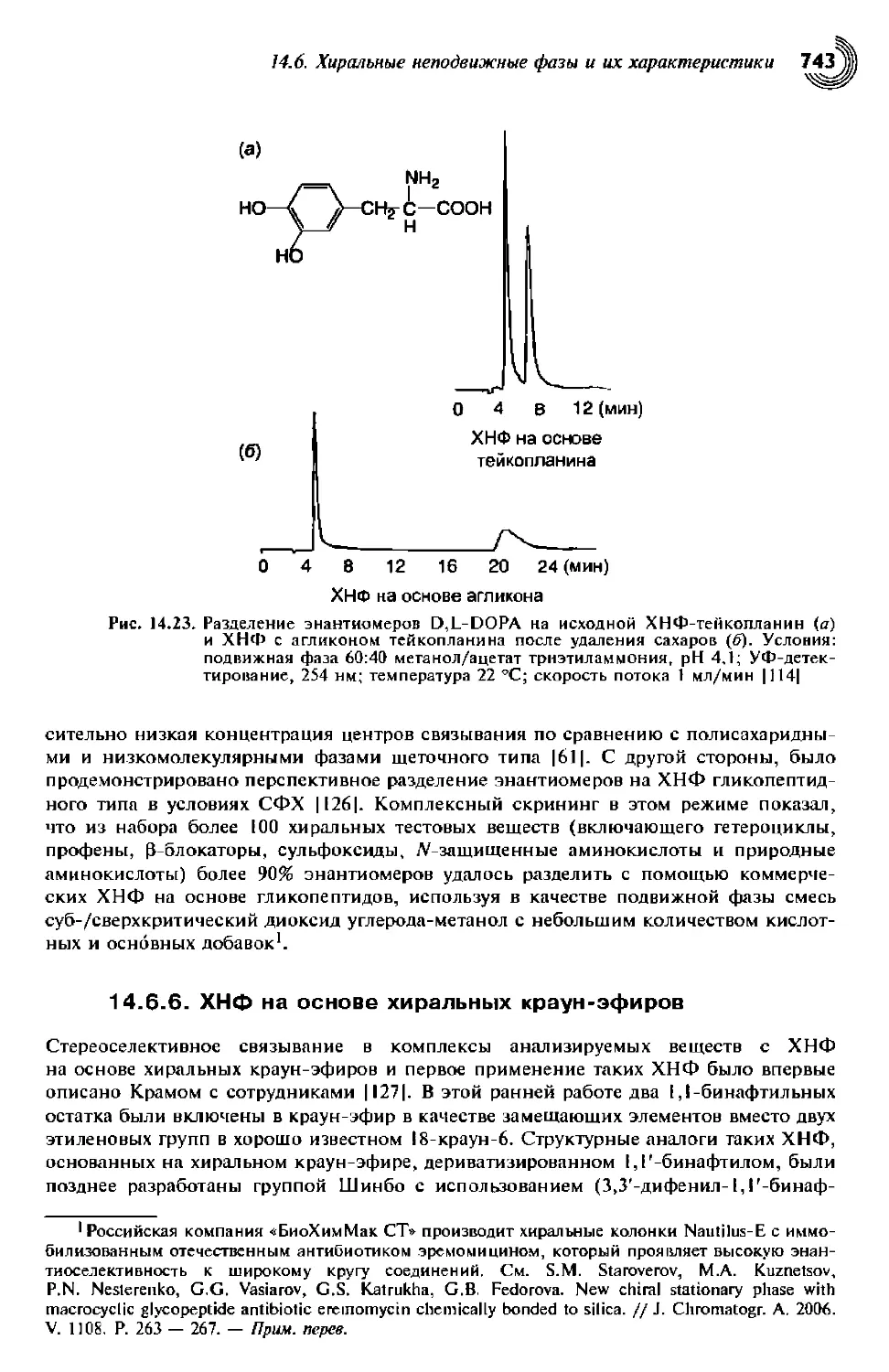



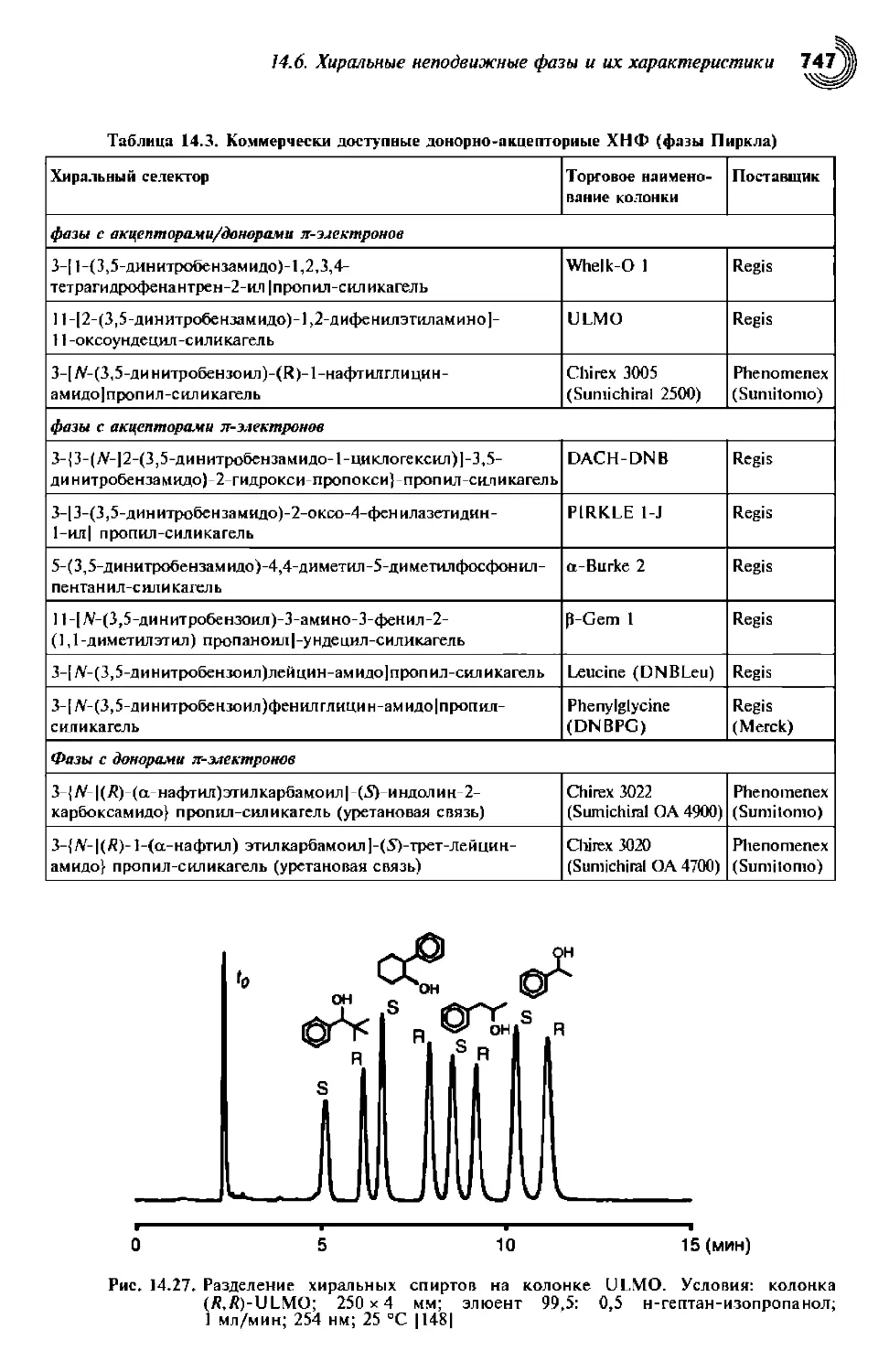








































































































































































































Author: Снайдер Л.Р. Ллойд Р. Джозеф Дж. Киркленд Джон У. Долан
Tags: другие физико-химические методы анализа (кроме оптических) физическая химия химическая физика химия жидкостная хромография хромография издательство техносфера
ISBN: 978-5-94836-600-5
Year: 2020




X И
ЛЛОЙД Р. СНАЙДЕР,
ДЖОЗЕФ ДЖ. КИРКЛЕНД,
ДЖОН У. ДОЛАН
Введение
в современную
жидкостную
хроматографию
Издание
3-е
|Яиц
~.ZZ
Л;^
ч***—*?
"
к.
<• Я Мм
ТЕХНОСФЕРА
р
и м и и
Ллойд Р. Снайдер, Джозеф Дж. Киркленд,
Джон У. Долан
Введение в современную
жидкостную хроматографию
Перевод с английского
к.т.н. М.Б. Бару
И.В. Важениной
д.ф.-м.н. Е.А. Козловского
к.х.н. И.А. Петухова
О.А. Петуховой
Под общей редакцией
к.т.н. М.Б. Бару
И.В. Важениной
д.х.н. СМ. Староверова
ТЕХНОСФЕРА
Москва
2020
УДК 543.544.5
ББК 24.58
С53
С53 Снайдер Ллойд Р., Киркленд Джозеф Дж., Долан Джон У.
Введение в современную жидкостную хроматографию
М.: ТЕХНОСФЕРА, 2020. - 960 с. + 17 с. цв. вкл.
ISBN 978-5-94836-600-5
Это третье издание книги «Введение в современную жидкостную
хроматографию» - на сегодняшний день одно из самых популярных в мире справочных
руководств по современной жидкостной хроматографии. Это и учебник,
и справочник, и даже энциклопедия по всем (или почти по всем) вопросам,
связанным с ВЭЖХ.
В книге освещено огромное количество вопросов, связанных с теорией
хроматографии, современным оборудованием ВЭЖХ, методами детектирования
и устройством детекторов, подробно рассмотрены теоретические и
практические аспекты выбора неподвижных и подвижных фаз. Особое внимание
уделено обращенно-фазовой, нормально-фазовой, гель-проникающей,
гидрофобной, гидрофильной и другим видам хроматографии. Отдельные главы
посвящены разделению синтетических и природных полимеров, препаративной
хроматографии, разделению энантиомеров, пробоподготовке, типовым
проблемам при работе с хроматографическим оборудованием и, что особенно
важно в современных условиях, валидации аналитических методов.
Книга предназначена для широкого круга специалистов, имеющих дело
с современной жидкостной хроматографией. Она будет полезна как тем, кто
только начинает знакомиться с жидкостной хроматографией, так и
специалистам уже имеющим опыт работы в этой области.
УДК 543.544.5
ББК 24.58
© 2010 by John Wiley & Sons, Inc. All rights reserved
Все права защищены. Авторизованный перевод
оригинального издания «Джон Вайли энд Сане Инк.»
© АО «РИЦ «ТЕХНОСФЕРА», перевод на
русский язык, оригинал-макет, оформление, 2020
INTRODUCTION ТО
MODERN LIQUID
CHROMATOGRAPHY
LLOYD R.SNYDER
I0SEPH |. KIRKLAND
I0HN W. D0LAN
ISBN 978-5-94836-600-5
ISBN 978-0^170-16754-0 (англ.)
СОДЕРЖАНИЕ
Предисловие редакторов перевода 29
Предисловие 30
Словарь символов и принятых сокращений 33
Глава 1. Введение 41
1.1. Предварительные сведения 42
1.1.1. Что такое ВЭЖХ? 42
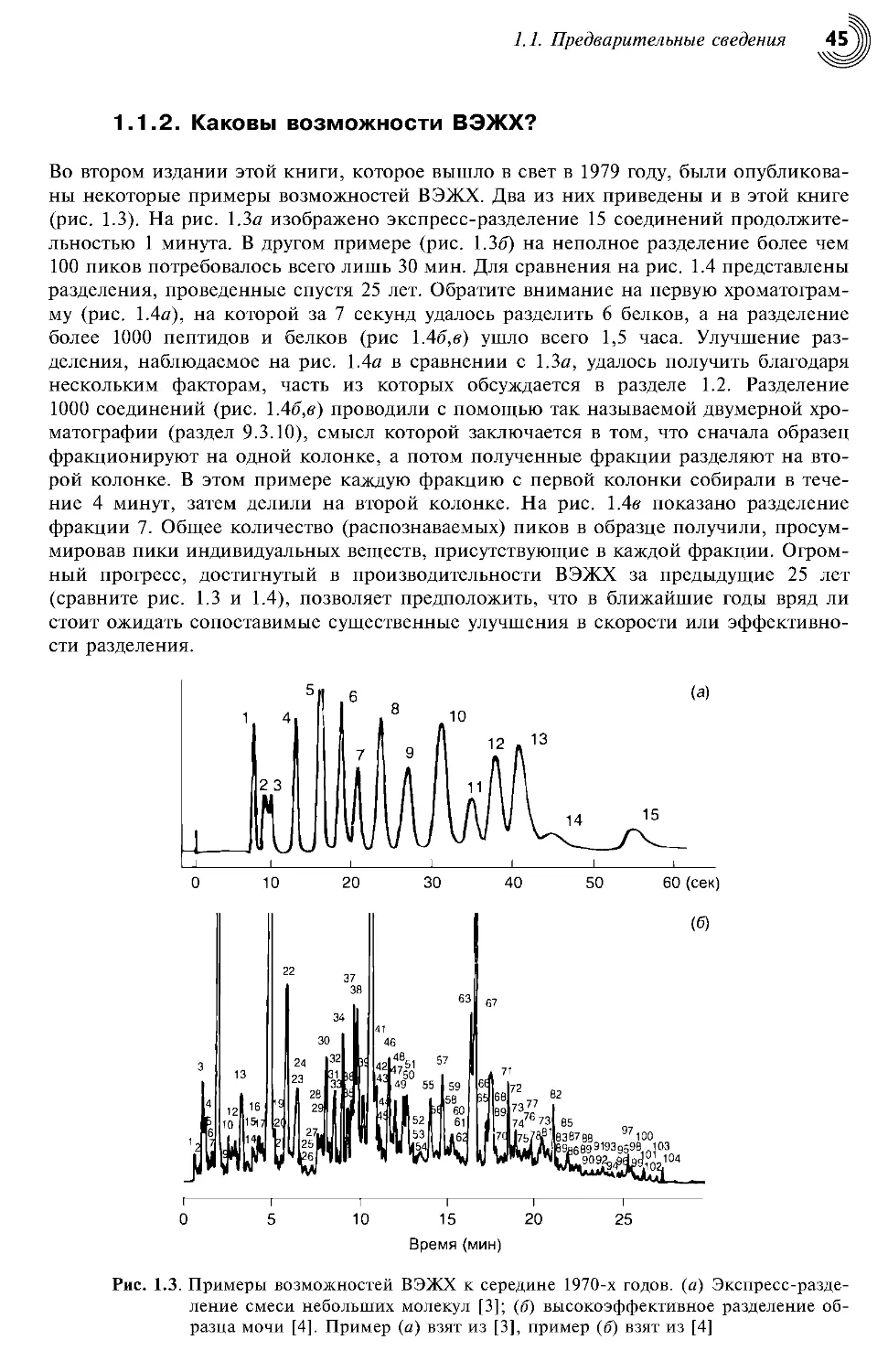
1.1.2. Каковы возможности ВЭЖХ? 45
1.2. Небольшой экскурс в историю ВЭЖХ 47
1.3. Некоторые альтернативы ВЭЖХ 50
1.3.1. Газовая хроматография (ГХ) 50
1.3.2. Тонкослойная хроматография (ТСХ) 50
1.3.3. Сверхкритическая флюидная хроматография (СФХ) 50
1.3.4. Капиллярный электрофорез (КЭ) 51
1.3.5. Противоточная хроматография (ПТХ) 51
1.3.6. Специальные виды ВЭЖХ 52
1.4. Другие источники информации о ВЭЖХ 52
1.4.1. Книги 52
1.4.2. Журналы 54
1.4.3. Обзоры 54
1.4.4. Краткие курсы 54
1.4.5. Интернет 54
Литература 54
Глава 2. Основные принципы и управление разделением 56
2.1. Введение 57
2.2. Хроматографический процесс 57
2.3. Удерживание 61
2.3.1. Фактор удерживания к и мертвое время колонки /0 61
2.3.2. Роль условий разделения и состава образца 65
2.3.2.1. Межмолекулярные взаимодействия 67
2.3.2.2. Температура 71
2.4. Ширина пика и число теоретических тарелок колонки N 71
2.4.1. Зависимость N от условий разделения 73
2.4.1.1. Процессы размывания пика, определяющие значения N 76
2.4.1.2. Несколько рекомендаций по выбору колонки 82
2.4.2. Форма пика 86
2.5. Разрешение и разработка метода 89
2.5.1. Оптимизация фактора удерживания к (член а в уравнении (2.24)) 93
2.5.2. Оптимизация селективности а (член б в уравнении (2.24)) 95
2.5.2.1. «Правильные» и «неправильные» образцы 96
2.5.3. Оптимизация числа теоретических тарелок N (член в в уравнении (2.24)). 97
2.5.3.1. Зависимость результатов разделения от параметров колонки. ... 97
2.5.3.2. Скоростная ВЭЖХ 99
2.5.4. Разработка метода 101
2.5.4.1. Оценка состава образца и целей разделения 101
2.5.4.2. Пробоподготовка 102
4 Содержание
2.5.4.3. Выбор вида хроматографии 102
2.5.4.4. Выбор детектора 102
2.5.4.5. Выбор хроматографических условий 102
2.5.4.6. Спрогнозировать, определить и решить возможные проблемы. . 103
2.5.4.7. Валидация метода и пригодность системы 104
2.6. Влияние количества пробы 105
2.6.1. Перегрузка по объему: влияние объема образца на разделение 105
2.6.2. Перегрузка по массе: влияние массы образца на разделение 107
2.6.3. Как избежать проблем, связанных со слишком большим количеством
образца 108
2.6.3.1. Если концентрация образца больше ожидаемой 109
2.6.3.2. Анализ следовых количеств вещества 109
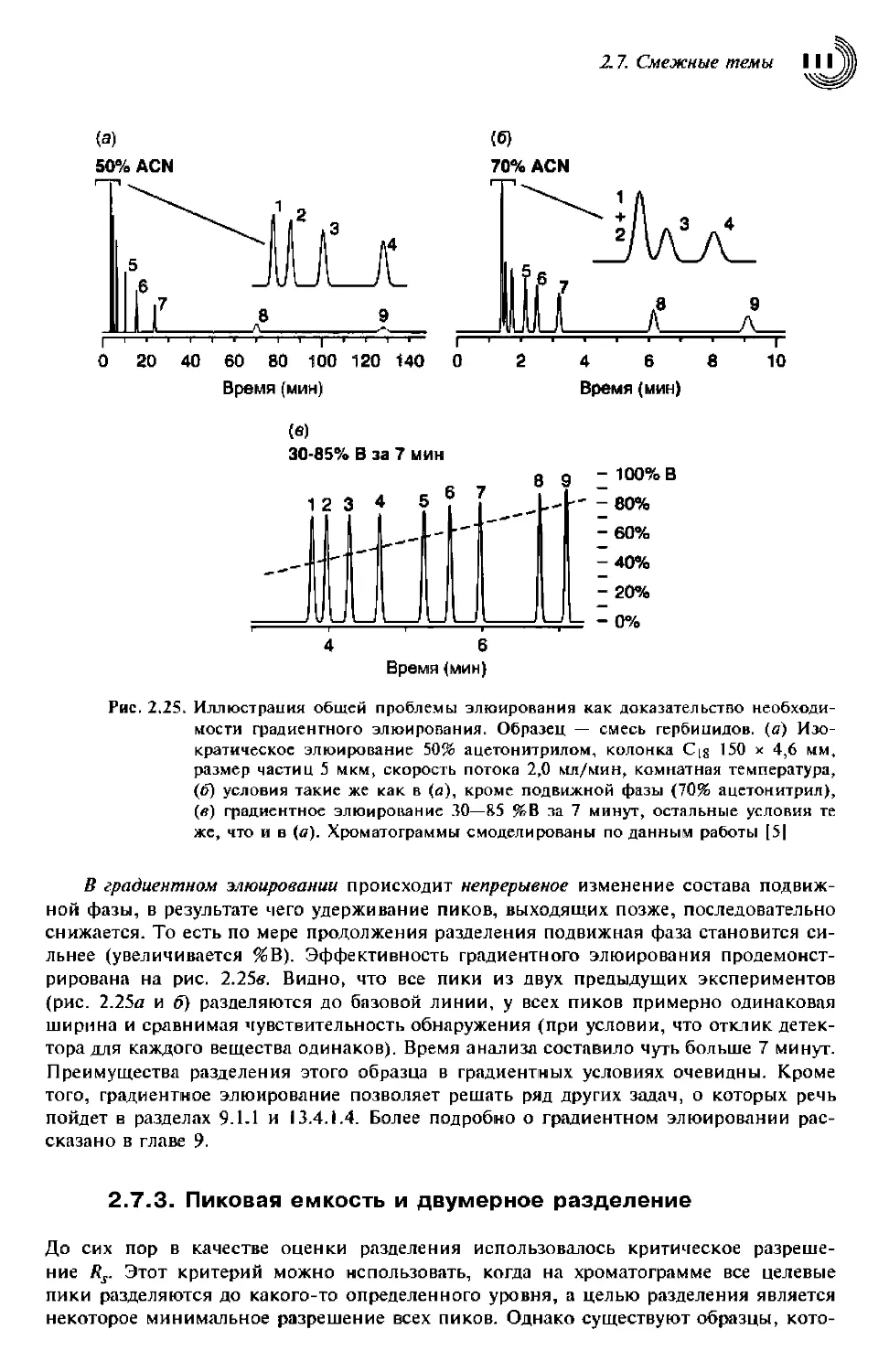
2.7. Смежные темы 110
2.7.1. Уравновешивание колонки ПО
2.7.2. Градиентное элюирование ПО
2.7.3. Пиковая емкость и двумерное разделение 111
2.7.4. Отслеживание пика 112
2.7.5. Вторичные равновесия 114
2.7.6. Переключение колонок 115
2.7.7. Предварительная оценка удерживания по структуре анализируемого
вещества 116
2.7.7.1. Модель сольватационных параметров 118
Литература 119
Глава 3. Оборудование 122
3.1. Введение 124
3.2. Резервуары и фильтрование элюентов 125
3.2.1. Устройство резервуара и его использование 126
3.2.2. Фильтрование подвижной фазы 127
3.3. Дегазация подвижной фазы 128
3.3.1. Требования к дегазации 128
3.3.2. Дегазирование гелием 130
3.3.3. Вакуумное дегазирование и онлайн-дегазирование 131
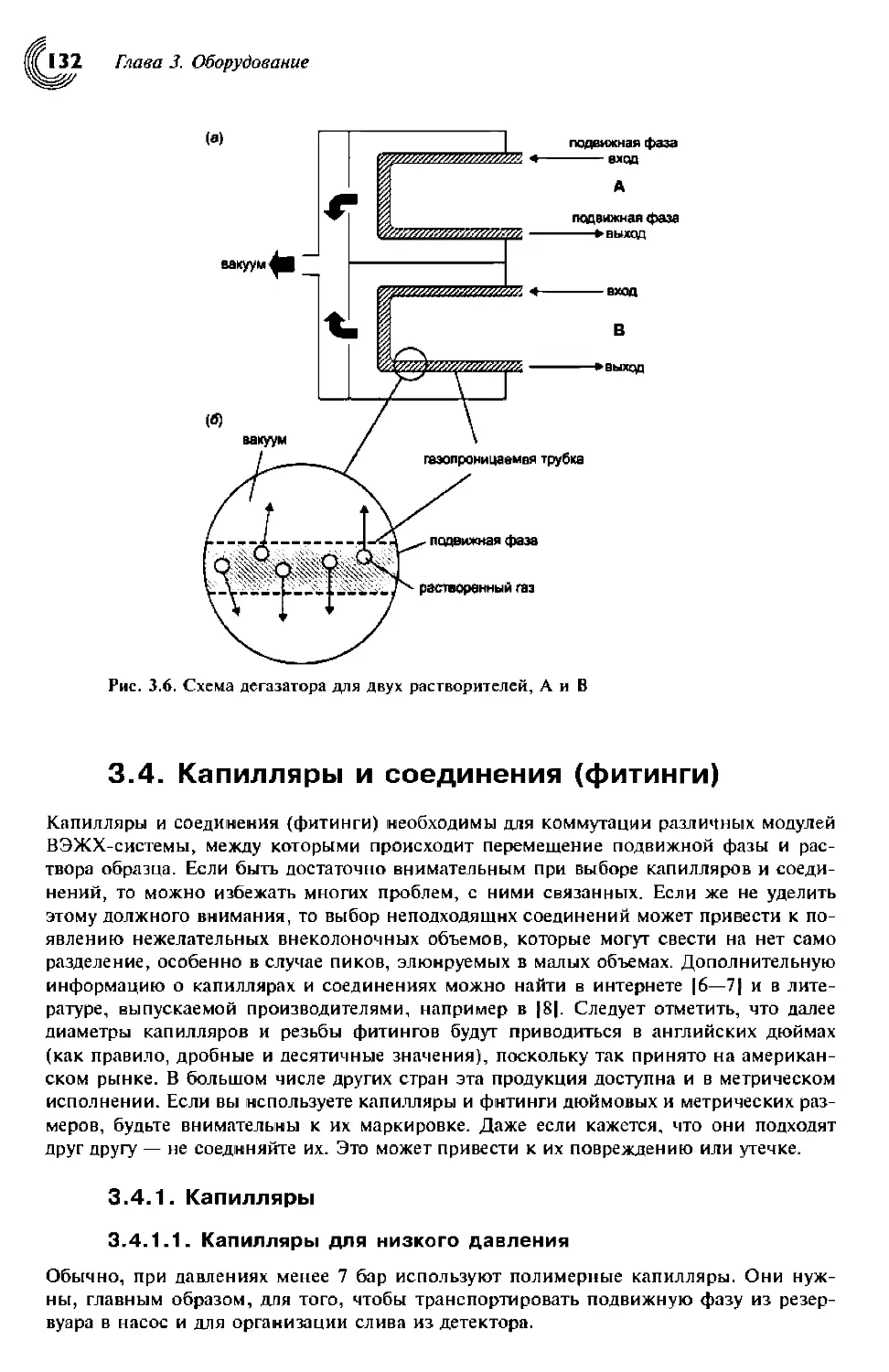
3.4. Капилляры и соединения (фитинги) 132
3.4.1. Капилляры 132
3.4.1.1. Капилляры для низкого давления 132
3.4.1.2. Капилляры для высокого давления 133
3.4.2. Фитинги 136
3.4.2.1. Фитинги для низкого давления 136
3.4.2.2. Фитинги для высокого давления 138
3.4.2.3. Специализированные фитинги 139
3.5. Насосные системы 141
3.5.1. Возвратно-поступательные плунжерные насосы 141
3.5.1.1. Двухплунжерные насосы 145
3.5.1.2. Накопительные плунжерные насосы 145
3.5.1.3. Активный клапан 146
3.5.2. Смешивание в потоке 146
3.5.2.1. Смешивание на стороне высокого давления 147
3.5.2.2. Смешивание на стороне низкого давления 148
3.5.2.3. Гибридные системы 148
3.5.3. Градиентные системы 149
6 Содержание
3.5.4. Специальные методы 150
3.5.4.1. Методы, использующие малые (микро- и нано-) скорости
потока 150
3.5.4.2. Методы, использующие высокие скорости потока
(препаративные разделения) 150
3.5.4.3. Методы, использующие высокое давление 151
3.6. Автосамплеры 151
3.6.1. Шестиходовой кран ввода образца 151
3.6.1.1. Ввод из заполненной петли 152
3.6.1.2. Ввод из частично заполненной петли 152
3.6.2. Конструкция автосамплера 154
3.6.2.1. Автосамплеры с заполнением петли засасыванием 155
3.6.2.2. Автосамплеры с заполнением петли нагнетанием 156
3.6.2.3. Автосамплер со встроенным петлевым дозатором 156
3.6.3. Эффекты, связанные с количеством образца 158
3.6.3.1. Вводимый объем 158
3.6.3.2. Растворитель образца 159
3.6.4. Другие области применения кранов 160
3.6.4.1. Переключение колонок 160
3.6.4.2. Коллекторы фракций 162
3.6.4.3. Направление в слив 162
3.7. Колоночные термостаты 163
3.7.1. Требования к температурному контролю 163
3.7.2. Устройство термостатов 164
3.7.2.1. Нагревательные термостаты 164
3.7.2.2. Воздушные термостаты 164
3.7.2.3. Термостаты с элементами Пельтье 164
3.8. Управляющие модули 165
3.8.1. Сопровождение эксперимента 166
3.8.2. Управление системой 167
3.8.3. Сбор данных 167
3.8.4. Обработка данных 168
3.8.5. Создание отчета 168
3.8.6. Регулирующие функции 169
3.9. Влияние внеколоночных объемов 169
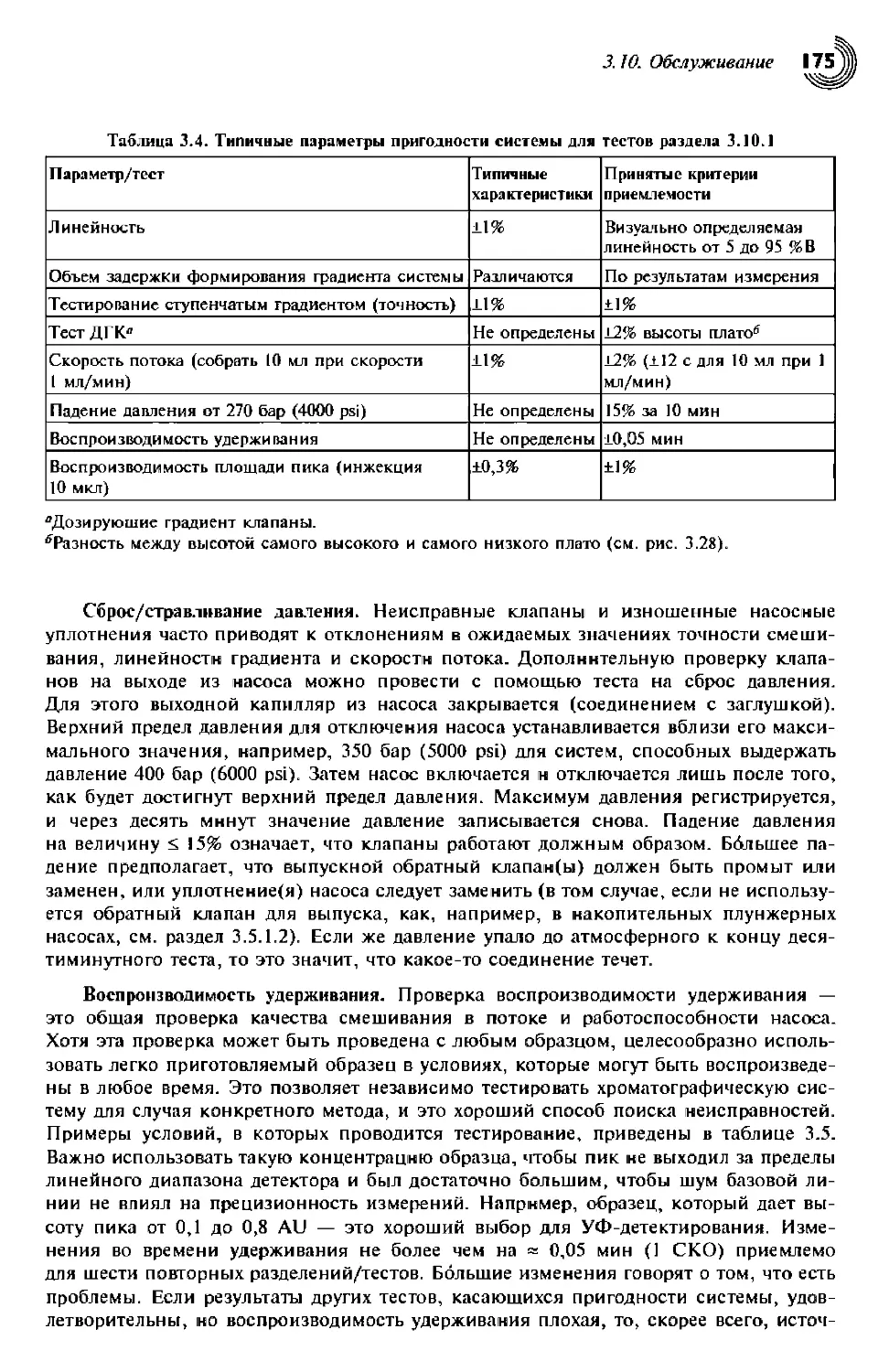
3.10. Обслуживание 170
3.10.1. Тест на работоспособность/пригодность системы 170
3.10.1.1. Установочная, операционная и эксплуатационная
квалификации 170
3.10.1.2. Градиентное тестирование 171
3.10.1.3. Дополнительная проверка системы 174
3.10.2. Профилактика 177
3.10.2.1. Периодическое обслуживание 177
3.10.2.2. Рекомендации для рутинных методов 180
3.10.3. Ремонтные работы 182
3.10.3.1. Персонал 182
3.10.3.2. Ведение учета 182
3.10.3.3. Специальные рекомендации по ремонту 183
Литература 184
Содержание 7
Глава 4. Детектирование 185
4.1. Введение 186
4.2. Характеристики детектора 187
4.2.1. Общие положения 187
4.2.2. Способы детектирования 189
4.2.2.1. Детектирование, основанное на измерении
общей характеристики вещества 189
4.2.2.2. Детектирование, основанное на измерении специфических
характеристик образца 190
4.2.2.3. Детектирование с помощью модификации подвижной фазы ... 190
4.2.2.4. Детектирование «независимыми» приборами 190
4.2.3. Сигнал, шум, дрейф и точность измерений 191
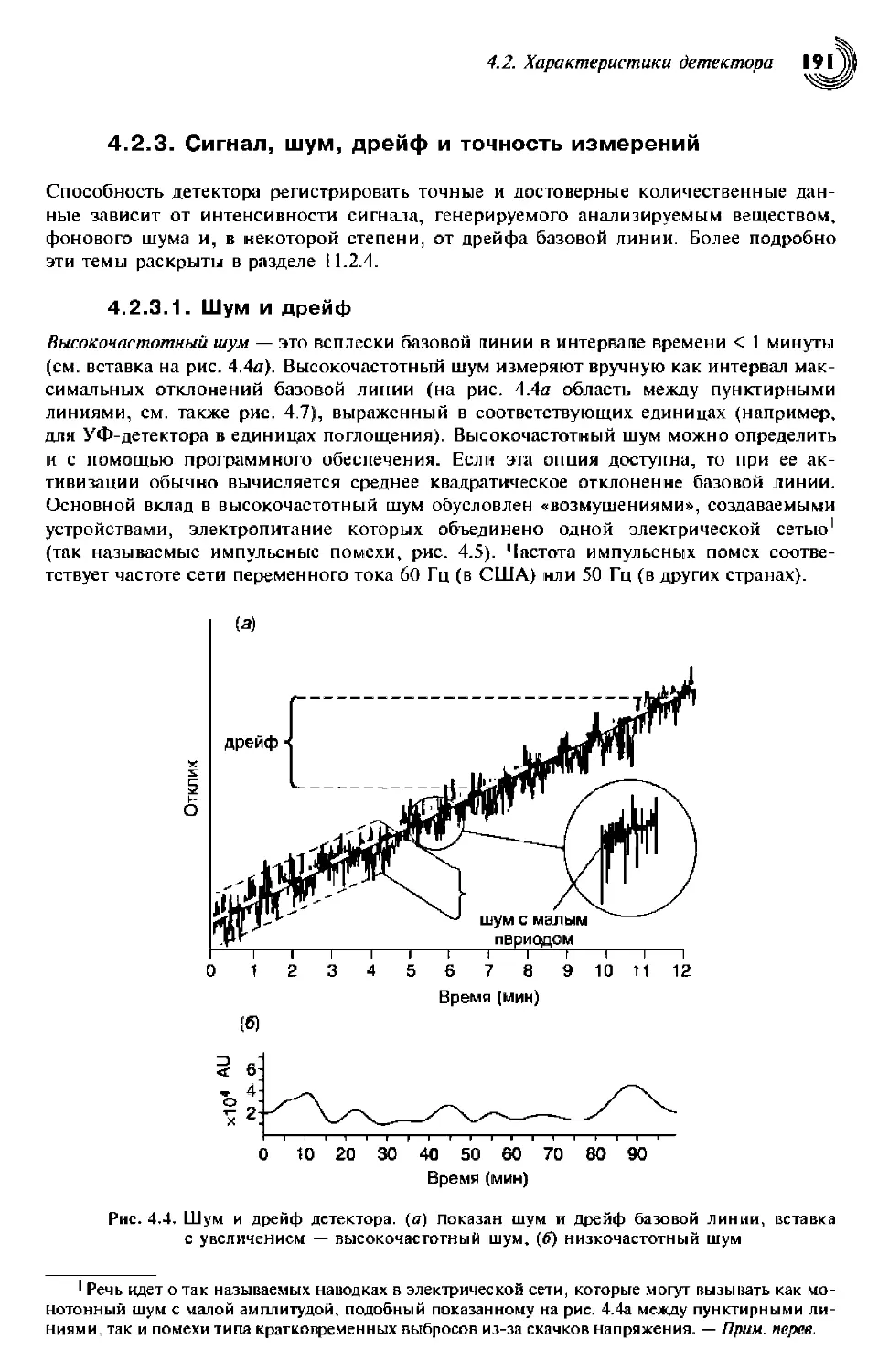
4.2.3.1. Шум и дрейф 191
4.2.3.2. Отношение сигнал/шум (S/N) 194
4.2.4. Пределы обнаружения 196
4.2.5. Линейность 198
4.3. Общее предисловие к различным типам детекторов 198
4.4. Детекторы, регистрирующие поглощение в УФ- и видимой областях
спектра света 198
4.4.1. Детекторы с фиксированной длиной волны 201
4.4.2. Детекторы с переменной длиной волны 202
4.4.3. Диодно-матричные детекторы 203
4.4.4. Общие характеристики УФ-детекторов 205
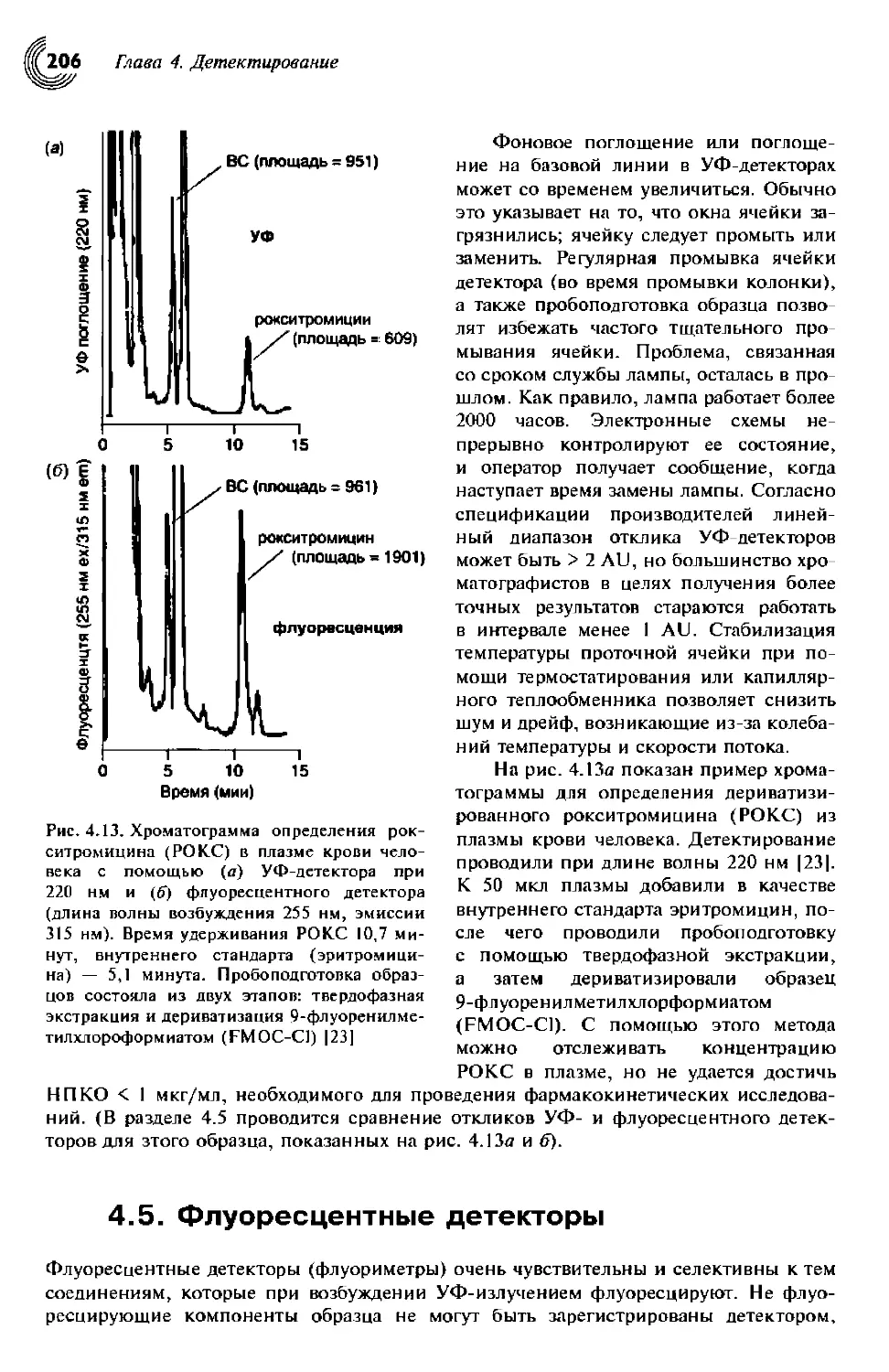
4.5. Флуоресцентные детекторы 206
4.6. Электрохимические (амперометрические) детекторы 209
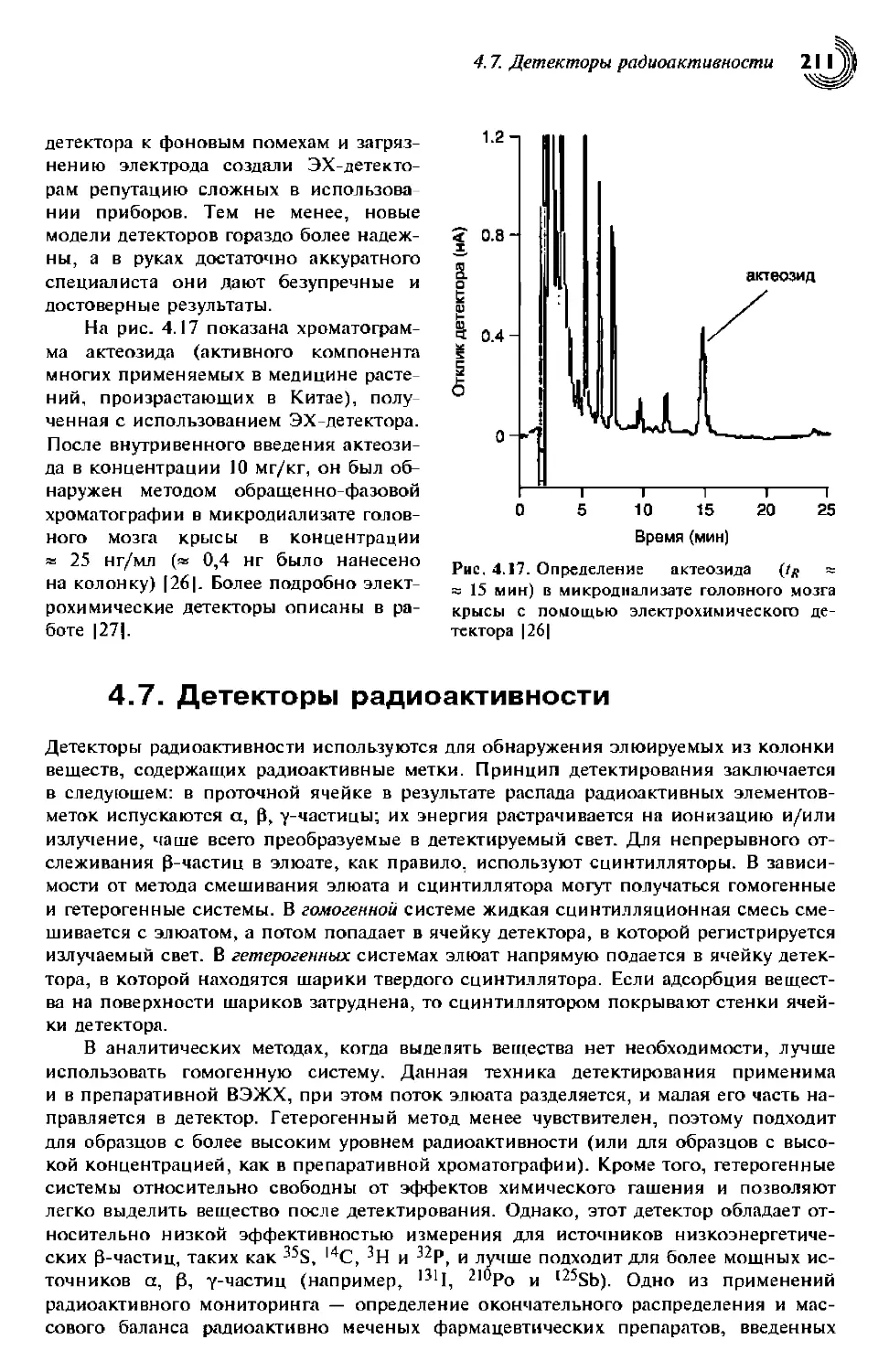
4.7. Детекторы радиоактивности 211
4.8. Кондуктометрические детекторы 212
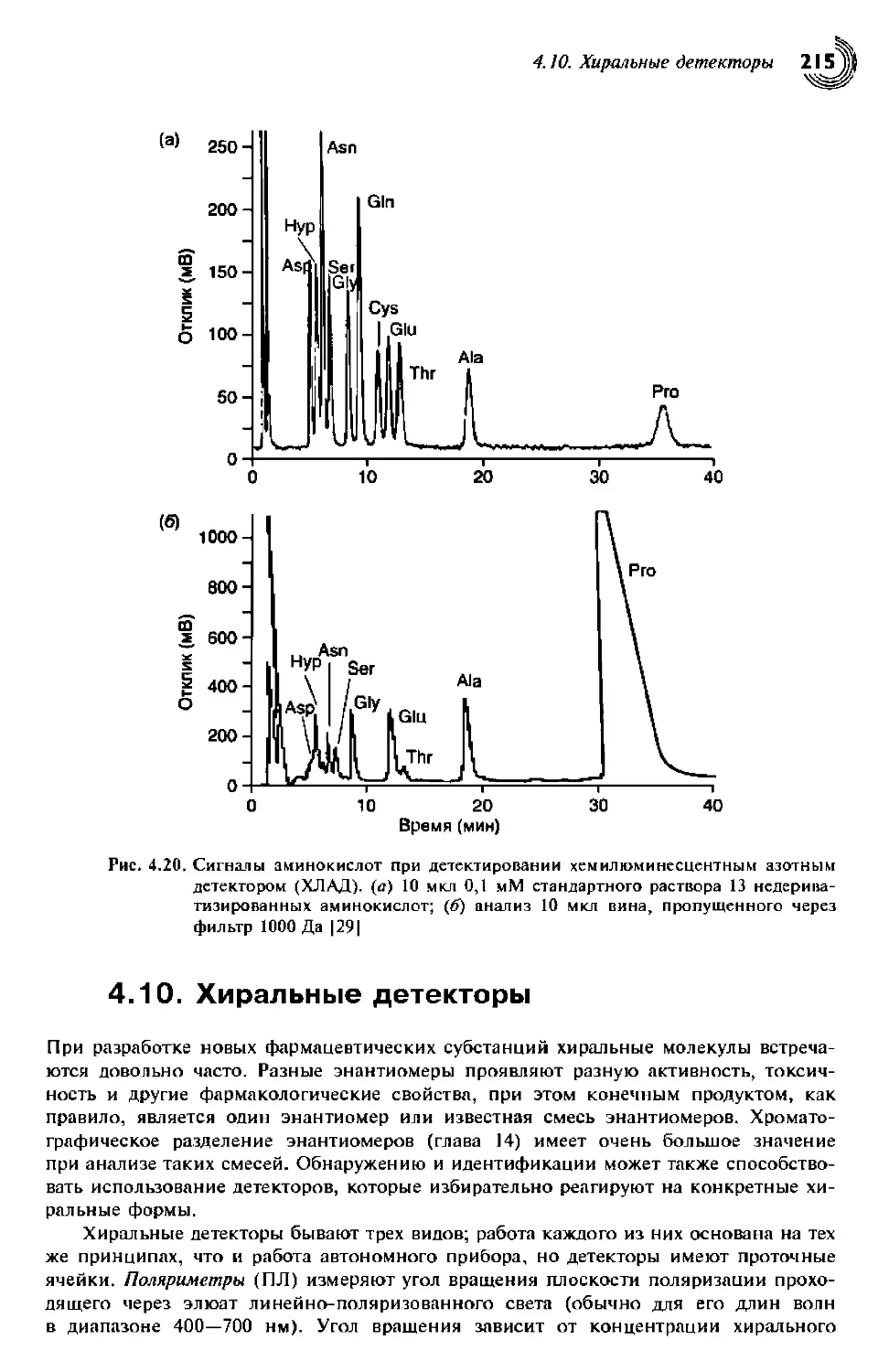
4.9. Хемилюминесцентный азотный детектор 213
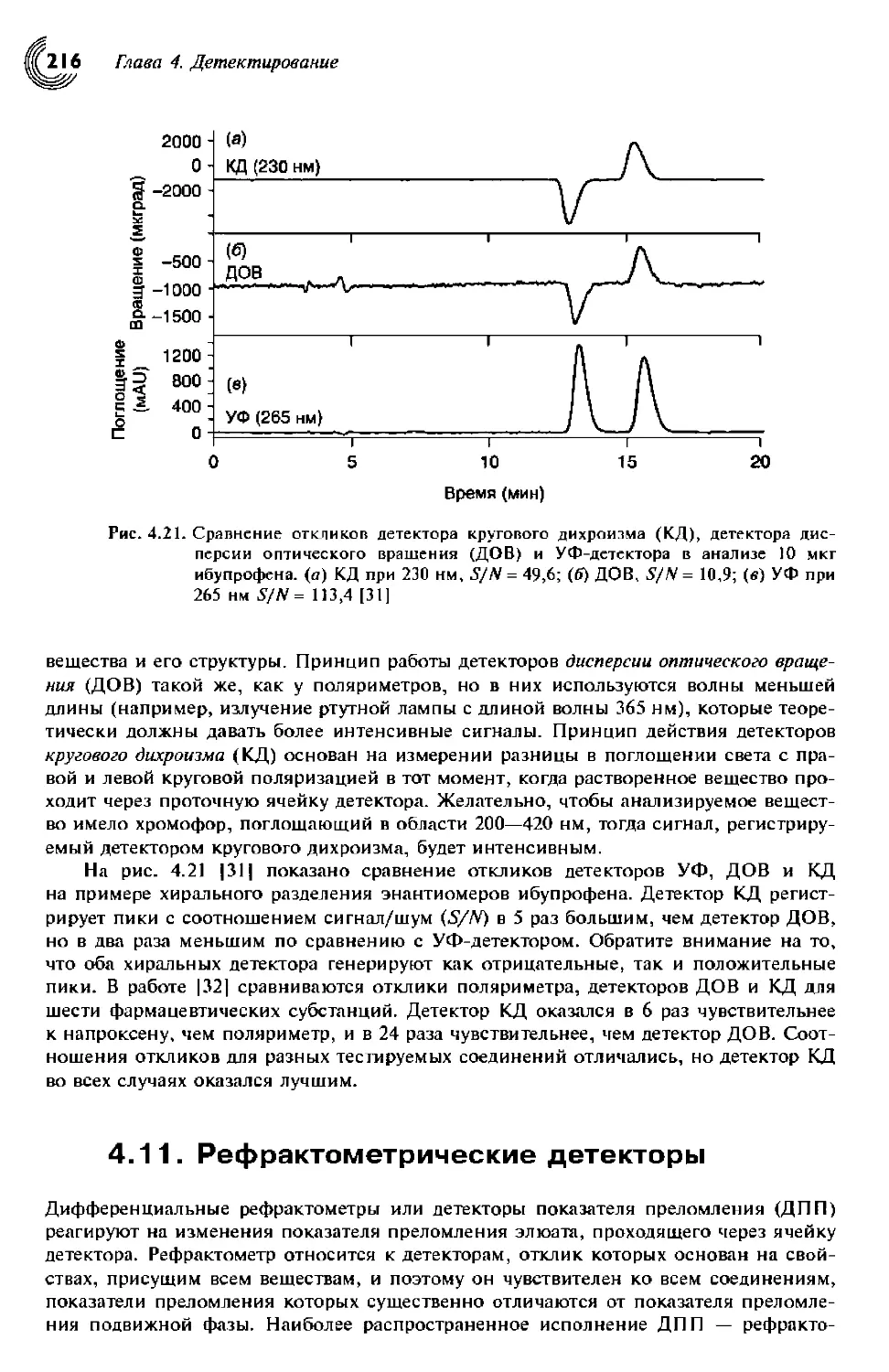
4.10. Хиральные детекторы 215
4.11. Рефрактометрические детекторы 216
4.12. Детекторы светорассеяния (ИДС) 219
4.12.1. Испарительный детектор светорассеяния 220
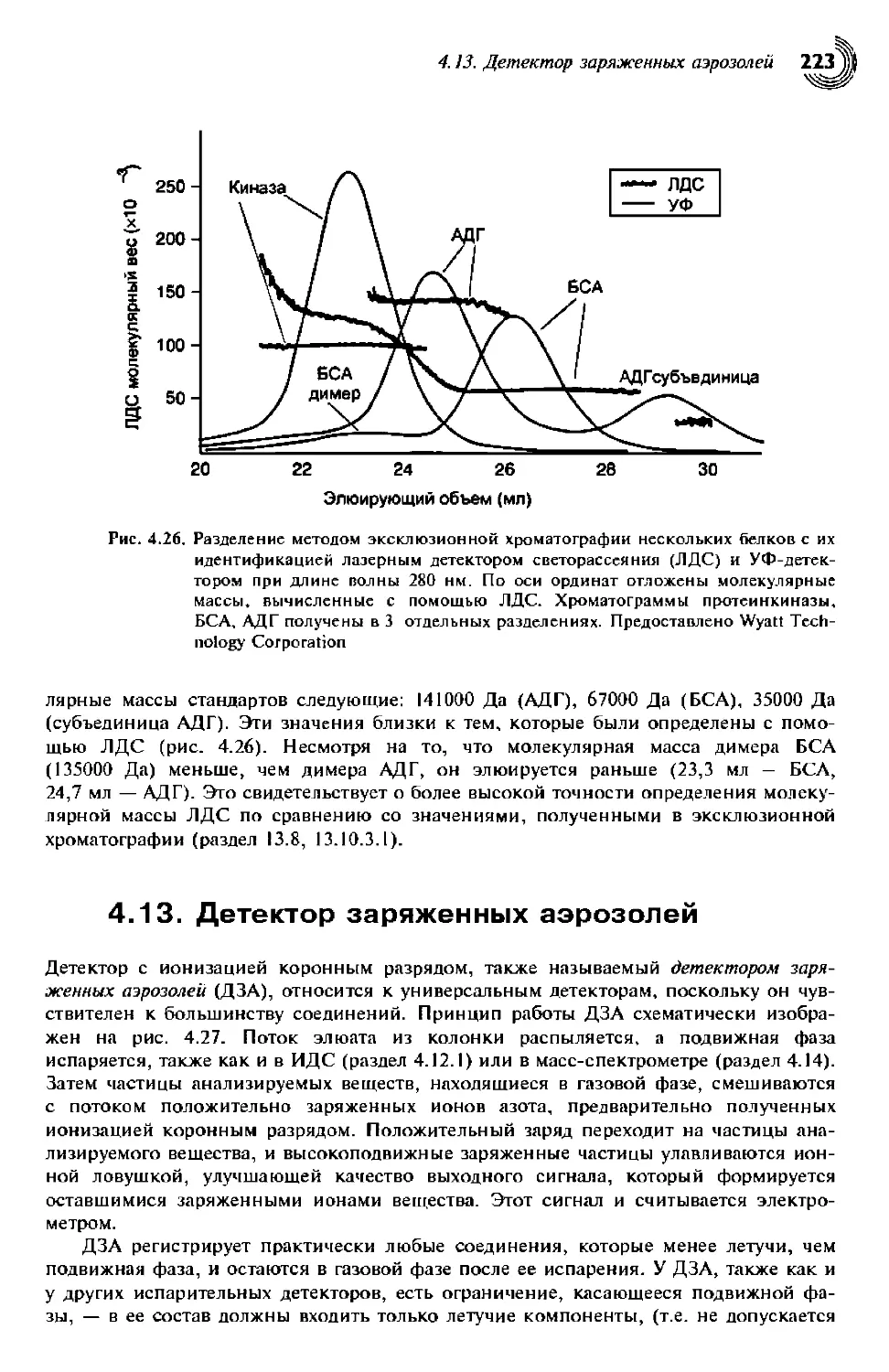
4.12.2. Детектор светорассеяния на центрах (ядрах) конденсации (ДСЦК) . . 222
4.12.3. Лазерные детекторы светорассеяния (ЛДС) 222
4.13. Детектор заряженных аэрозолей 223
4.14. Масс-спектрометрические детекторы (МС) 224
4.14.1. Интерфейсы МС-детекторов (способы ионизации образца) 225
4.14.1.1. Ионизация электрораспылением (ИЭ) 225
4.14.1.2. Химическая ионизация при атмосферном давлении (ХИАД) 226
4.14.1.3. Другие модификации интерфейса МС-детекторов 227
4.14.1.4. Соображения относительно скорости потока 227
4.14.2. Квадруполи и ионные ловушки 227
4.14.3. Другие виды МС-детекторов 229
4.15. Другие «независимые» детекторы 230
4.15.1. Инфракрасная спектроскопия с Фурье-преобразованием (ИКФП) . . 230
4.15.2. Ядерный магнитный резонанс (ЯМР) 231
4.16. Дериватизация образца и реакционные детекторы 233
Литература 235
Глава 5. Колонки 237
5.1. Введение 238
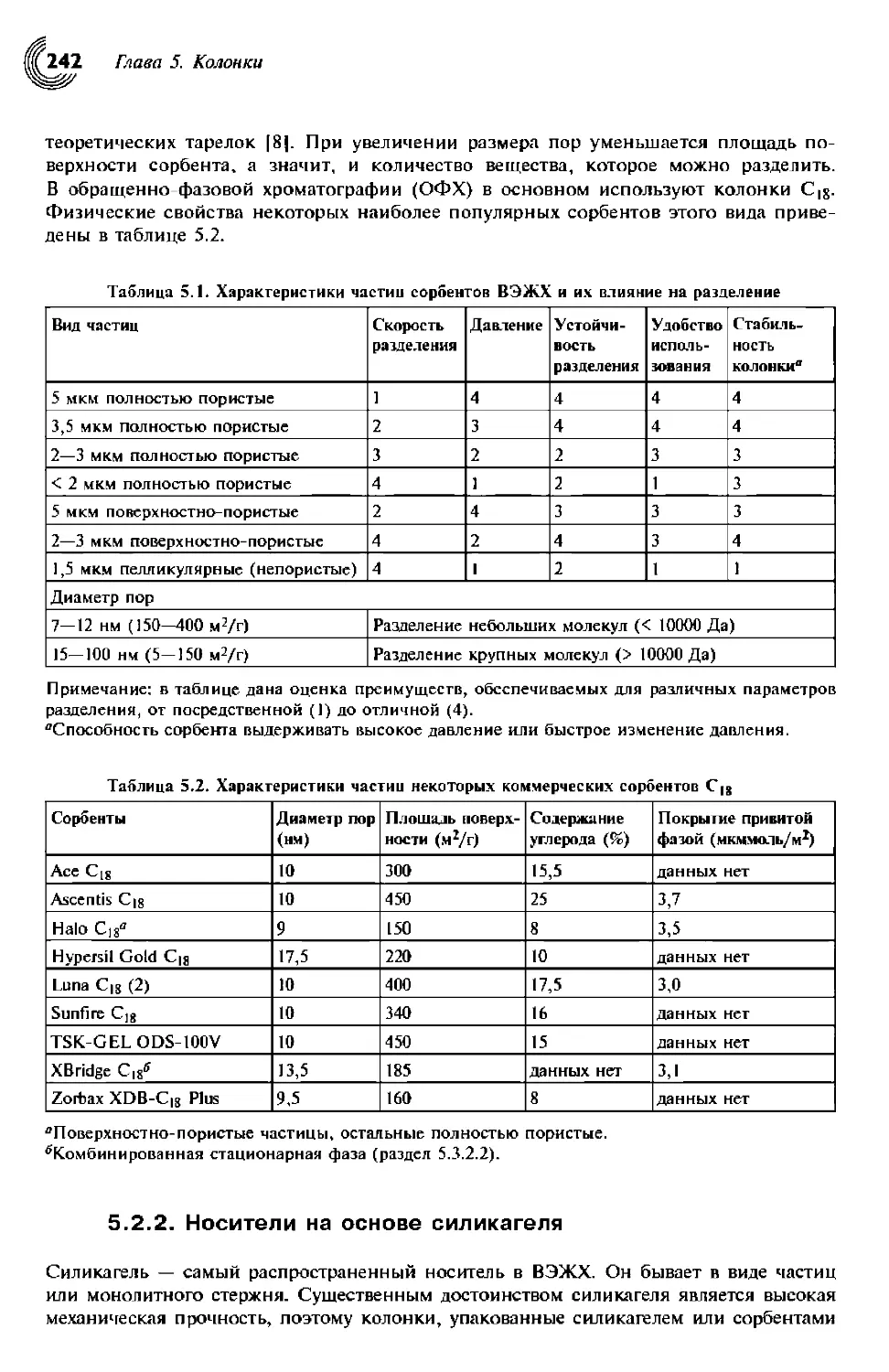
5.2. Носители 238
5.2.1. Характеристики частиц 239
8 Содержание
5.2.1.1. Типы частиц 239
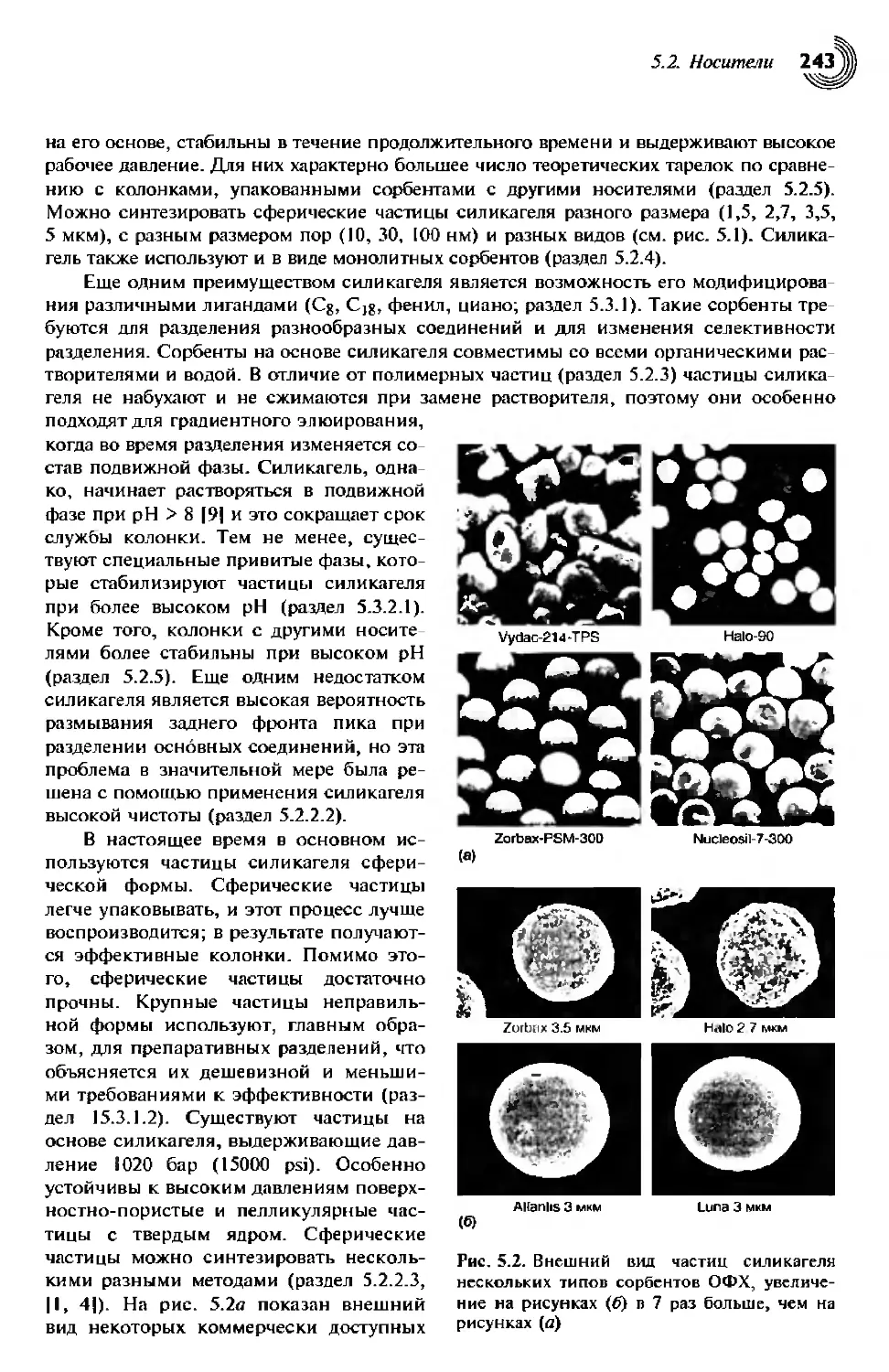
5.2.1.2. Размер частиц и диаметр пор 241
5.2.2. Носители на основе силикагеля 242
5.2.2.1. Эффективность колонки 244
5.2.2.2. Структура поверхности силикагеля 246
5.2.2.3. Подготовка частиц 248
5.2.3. Пористые полимеры 249
5.2.4. Монолитные сорбенты 250
5.2.4.1. Монолиты на основе силикагеля 251
5.2.4.2. Монолиты на полимерной основе 251
5.2.5. Другие неорганические частицы 252
5.2.5.1. Оксид циркония 253
5.2.5.2. Оксиды алюминия и титана 254
5.2.5.3. Графитированный углерод 254
5.3. Неподвижные фазы 255
5.3.1. «Привитые» неподвижные фазы 256
5.3.2. Другие органические неподвижные фазы 260
5.3.2.1. Нанесенные полимеры 260
5.3.2.2. Гибридные частицы 260
5.3.2.3. Колонки для подвижных фаз с высоким содержанием воды . . . 261
5.3.3. Колонки с различными функциональными группами сорбента 262
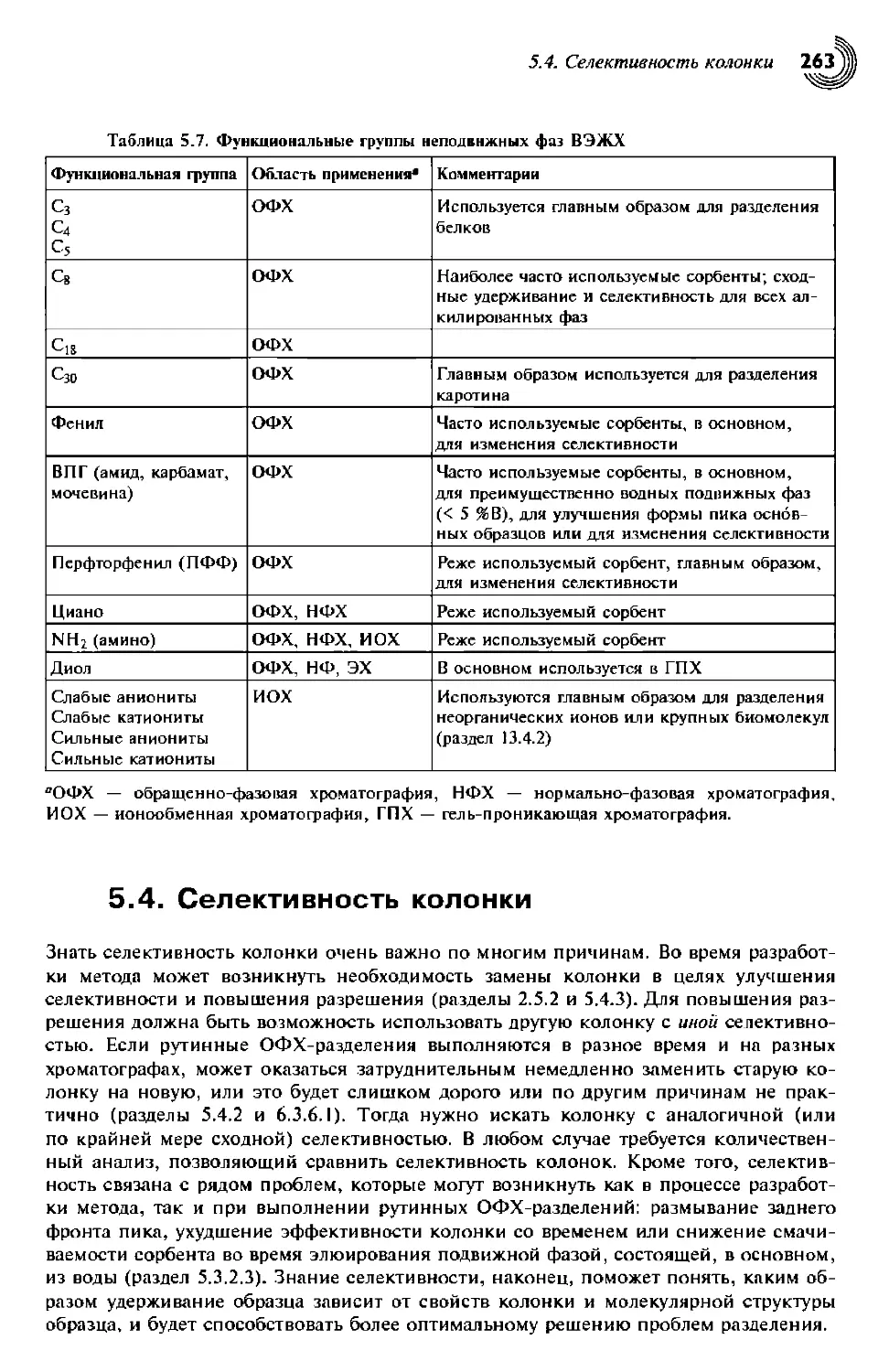
5.4. Селективность колонки 263
5.4.1. Основы селективности обращенно-фазовой колонки 264
5.4.1.1. Взаимодействия анализируемого вещества с сорбентом 266
5.4.1.2. Селективность по форме молекулы 269
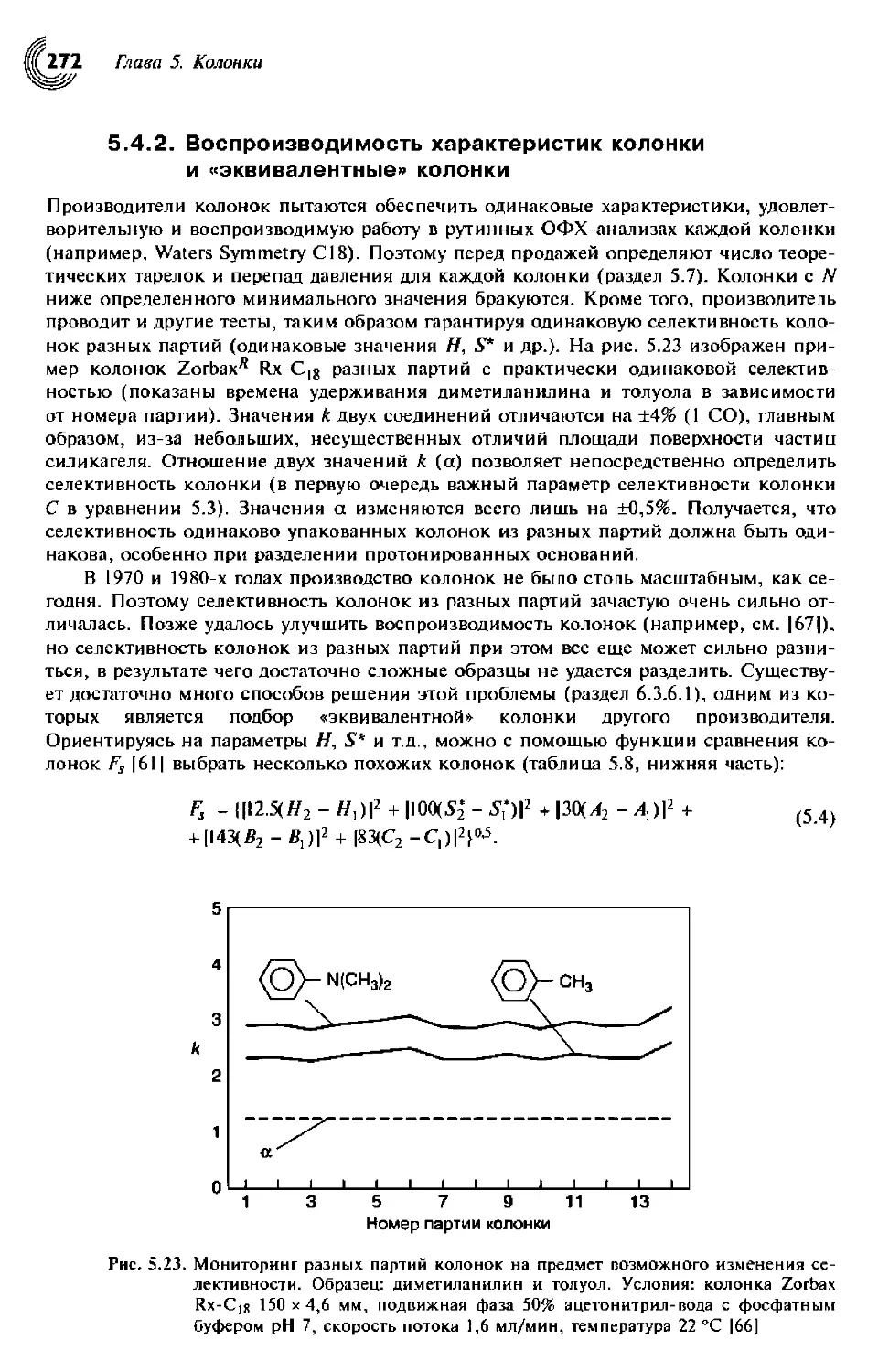
5.4.2. Воспроизводимость характеристик колонки и «эквивалентные»
колонки 272
5.4.3. Ортогональное разделение 273
5.4.4. В каких еще случаях нужно знать селективность колонки 273
5.4.4.1. Размывание заднего фронта пика 274
5.4.4.2. Несмачиваемость неподвижной фазы 274
5.4.4.3. Снижение эффективности сорбента 274
5.5. Типоразмеры колонок 275
5.5.1. Фитинги 275
5.5.2. Типоразмеры колонок 276
5.6. Методы упаковки колонок 277
5.6.1. Сухая упаковка 277
5.6.2. Суспензионная упаковка жестких частиц 277
5.6.2.1. Выбор растворителя для приготовления суспензии 278
5.6.2.2. Жесткие полимерные частицы 280
5.6.3. Упаковка мягких гелей 281
5.7. Спецификации колонок 281
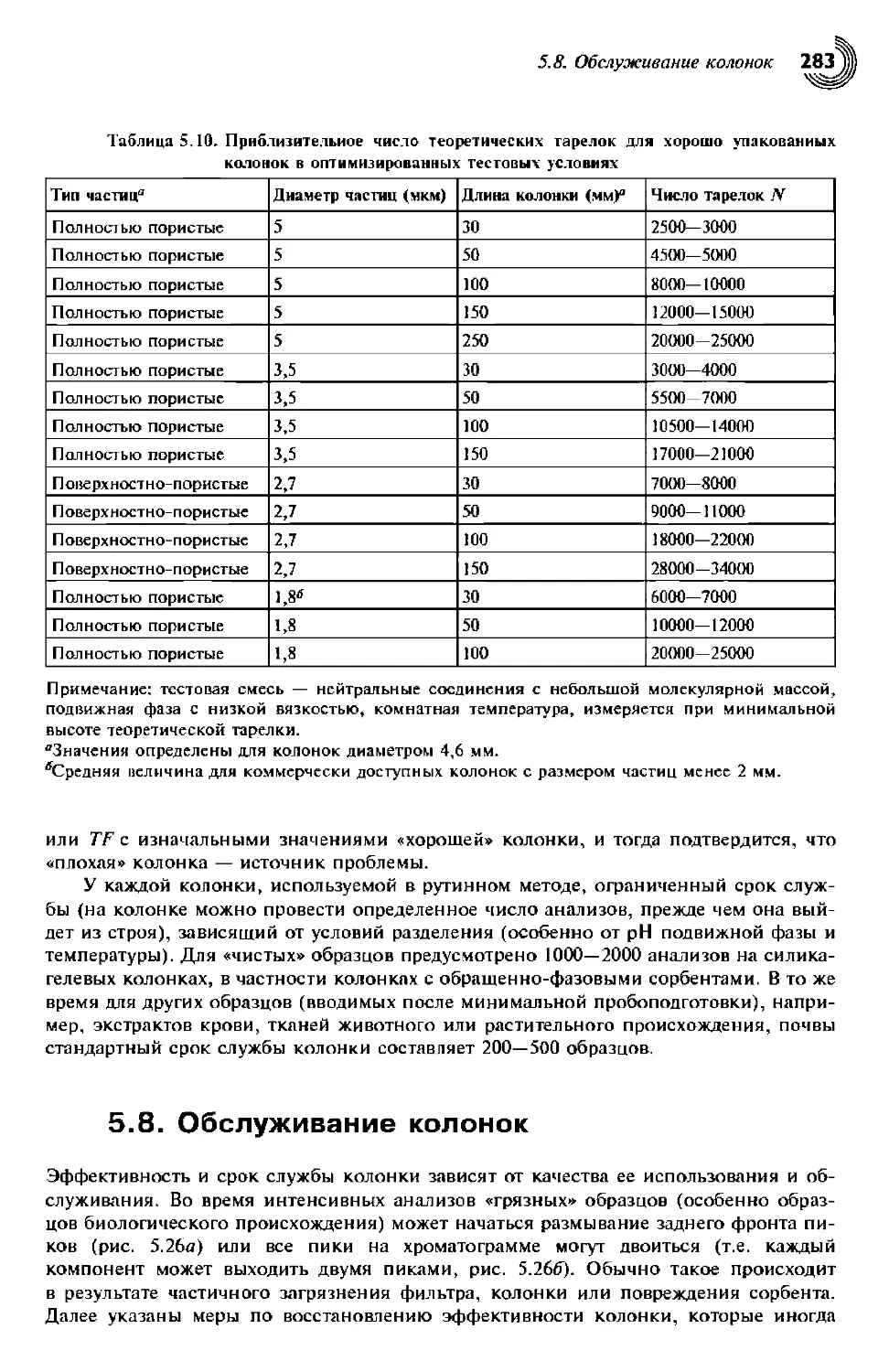
5.7.1. Стандарты производителя 281
5.7.2. Число теоретических тарелок колонки 282
5.8. Обслуживание колонок 283
Литература 287
Глава 6. Обращенно-фазовая хроматография нейтральных образцов 289
6.1. Введение 290
6.1.1. Краткая история обращенно-фазовой хроматографии 292
10 Содержание
6.2. Удерживание 292
6.2.1. Сила растворителя 294
6.2.2. Механизм удерживания в ОФХ 296
6.3. Селективность 300
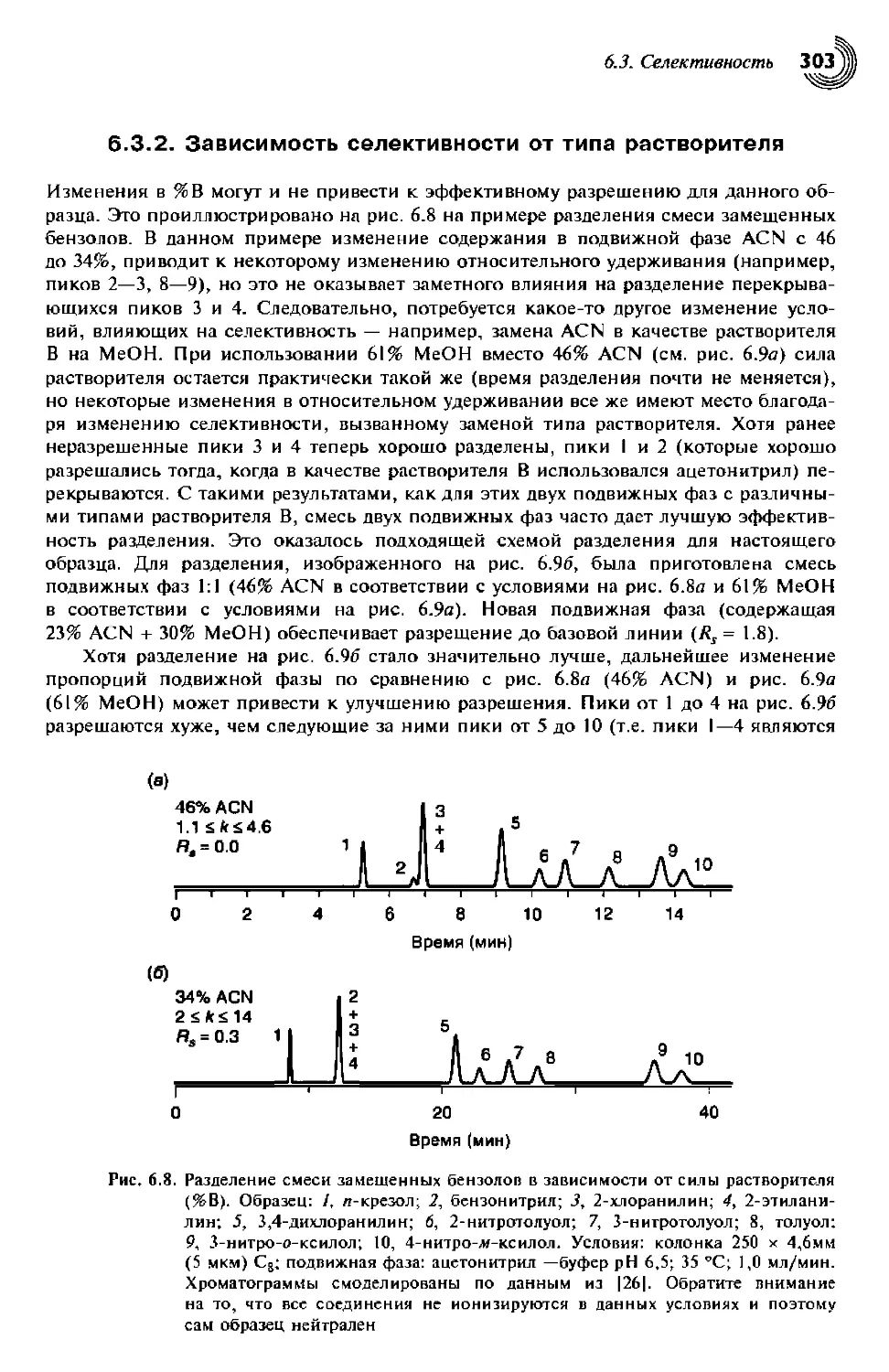
6.3.1. Зависимость селективности от силы растворителя 300
6.3.2. Зависимость селективности от типа растворителя 303
6.3.3. Зависимость селективности от температуры 306
6.3.3.1. Последующий анализ 309
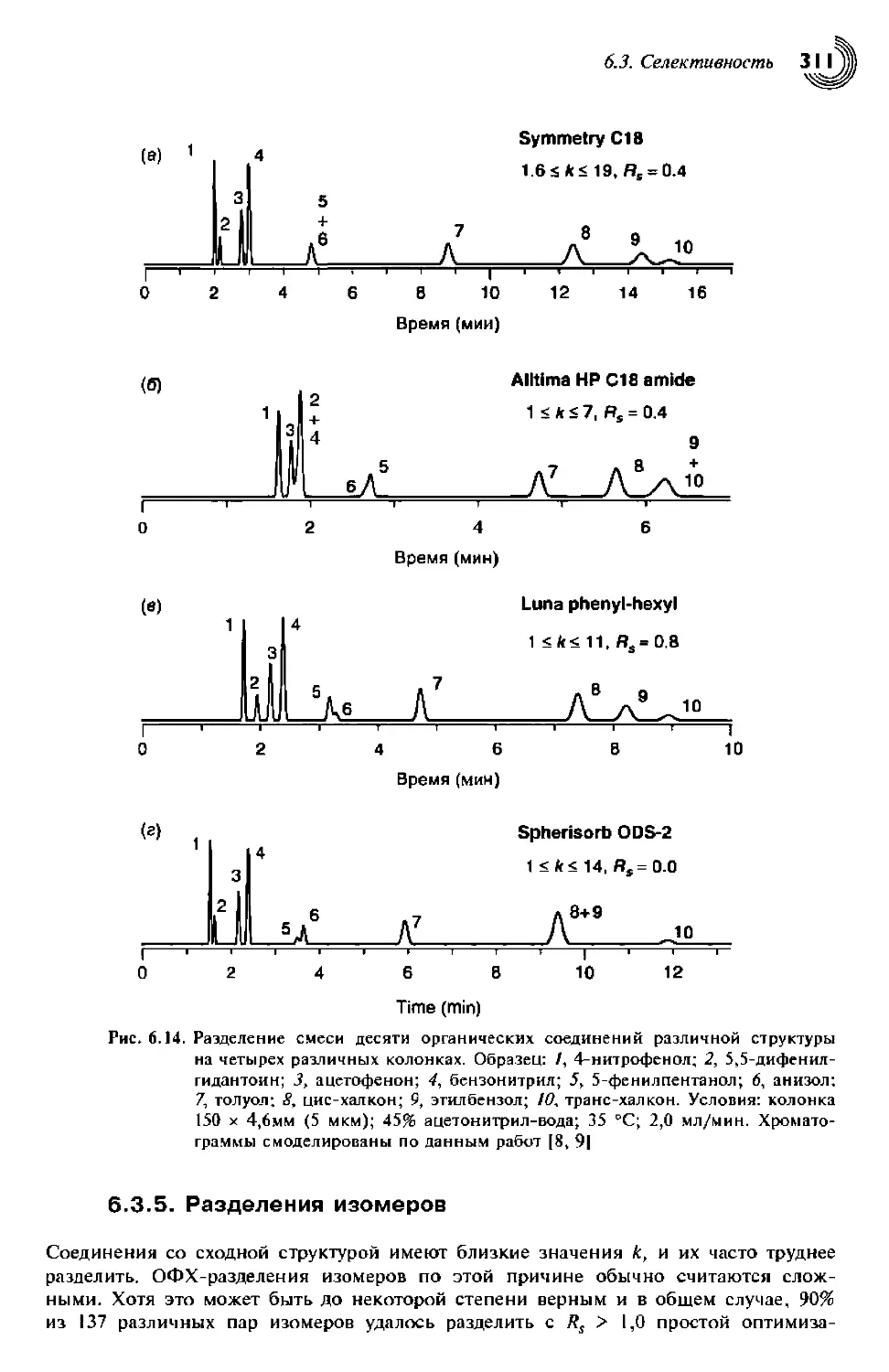
6.3.4. Селективность колонки 310
6.3.5. Разделения изомеров 311
6.3.5.1. Повышенная селективность при разделении изомеров 312
6.3.5.2. Зависимость селективности от формы молекулы 313
6.3.6. Другие соображения, касающиеся селективности 314
6.3.6.1. Эквивалентное разделение 314
6.3.6.2. Ортогональное разделение 318
6.4. Разработка метода и стратегия оптимизации селективности 319
6.4.1. Оптимизация нескольких параметров 321
6.4.1.1. Смеси различных органических растворителей 322
6.4.1.2. Одновременное изменение элюирующей силы
и типа растворителя 325
6.4.1.3. Одновременное изменение элюирующей силы и температуры
растворителя 325
6.4.1.4. Замена колонки с одновременным изменением
одного или более условий 328
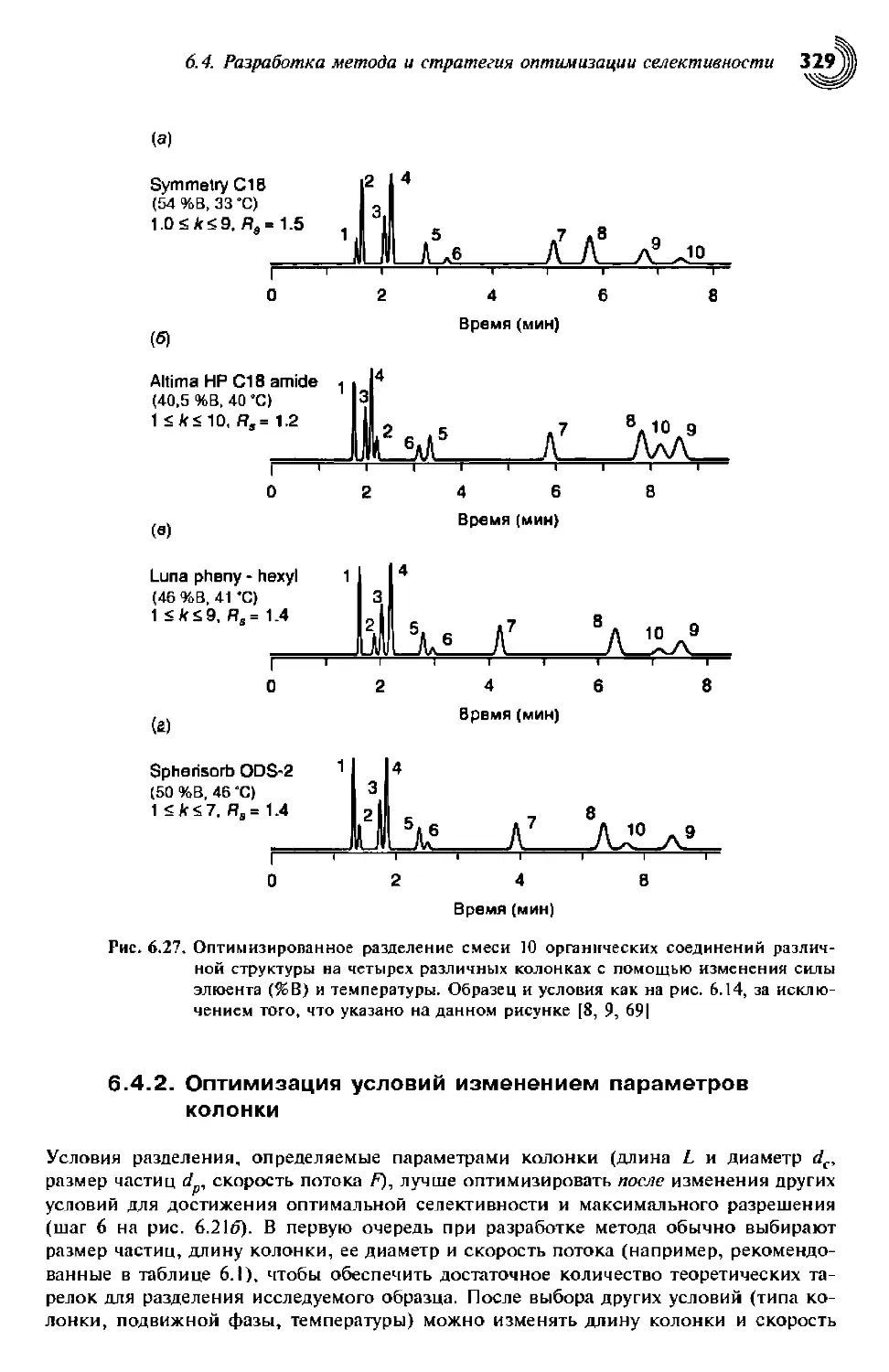
6.4.2. Оптимизация условий изменением параметров колонки 329
6.5. Неводная обращенно-фазовая хроматография (НОФХ) 331
6.6. Специфические проблемы 331
6.6.1. Слабое удерживание образцов с высокой полярностью 332
6.6.2. Уширение заднего фронта пика 333
Литература 333
Глава 7. Ионные образцы: обращенно-фазовая, ион-парная и ионообменная
хроматография 336
7.1. Введение 337
7.2. Кислотно-основные равновесия и удерживание в обращенно-фазовой
хроматографии 337
7.2.1. Выбор буферов 344
7.2.1.1. рКа и емкость буфера 344
7.2.1.2. Другие свойства буфера 347
7.2.1.3. Какие буферы лучше использовать 349
7.2.2. Как рКа зависит от структуры соединения 349
7.2.3. Влияние органических растворителей и температуры на рН подвижной
фазы и значения рКа образца 350
7.2.3.1. Влияние %В органического растворителя на эффективное
значение рКа вещества 351
7.2.3.2. Влияние температуры на значения рКа 352
7.3. Разделение ионных образцов методом обращенно-фазовой хроматографии
(ОФХ) 352
7.3.1. Как контролировать удерживание 352
7.3.2. Как контролировать селективность 353
11 Содержание
7.3.2.1. pH подвижной фазы 353
7.3.2.2. Сила растворителя (%В) и температура 355
7.3.2.3. Тип растворителя 356
7.3.2.4. Тип колонки 356
7.3.2.5. Другие условия, которые могут влиять на селективность 359
7.3.3. Разработка метода 359
7.3.3.1. Начальные условия (этап 1) 360
7.3.3.2. Оптимизация селективности (этап 3) 361
7.3.4. Нестандартные проблемы 361
7.3.4.1. Чувствительность к рН 361
7.3.4.2. Влияние силанольных групп 362
7.3.4.3. Слабое удерживание образца 364
7.3.4.4. Чувствительность к температуре 364
7.4. Ион-парная хроматография (ИПХ) 364
7.4.1. Основы удерживания 366
7.4.1.1. рН и образование ионной пары 367
7.4.1.2. Ион-парный реагент: тип и концентрация 367
7.4.1.3. Одновременное изменение рН и концентрации ион-парного
реагента (ИПР) 370
7.4.2. Разработка метода 371
7.4.2.1. Выбор начальных условий (этап 1) 372
7.4.2.2. Управление селективностью (этап 3) 375
7.4.2.3. Выводы 378
7.4.3. Нестандартные проблемы 378
7.4.3.1. Ложные пики 379
7.4.3.2. Медленное уравновешивание колонки 379
7.4.3.3. Искажение формы пиков 380
7.5. Ионообменная хроматография (ИОХ) 380
7.5.1. Основы удерживания 382
7.5.2. Роль противоиона 384
7.5.3. рН подвижной фазы 385
7.5.4. Колонки для ИОХ 385
7.5.5. Роль других условий 386
7.5.6. Разработка метода 386
7.5.7. Разделение углеводов 387
7.5.8. Разделения в смешанном режиме 387
Литература 389
Глава 8. Нормально-фазовая хроматография 391
8.1. Введение 392
8.2. Удерживание 393
8.2.1. Теория 396
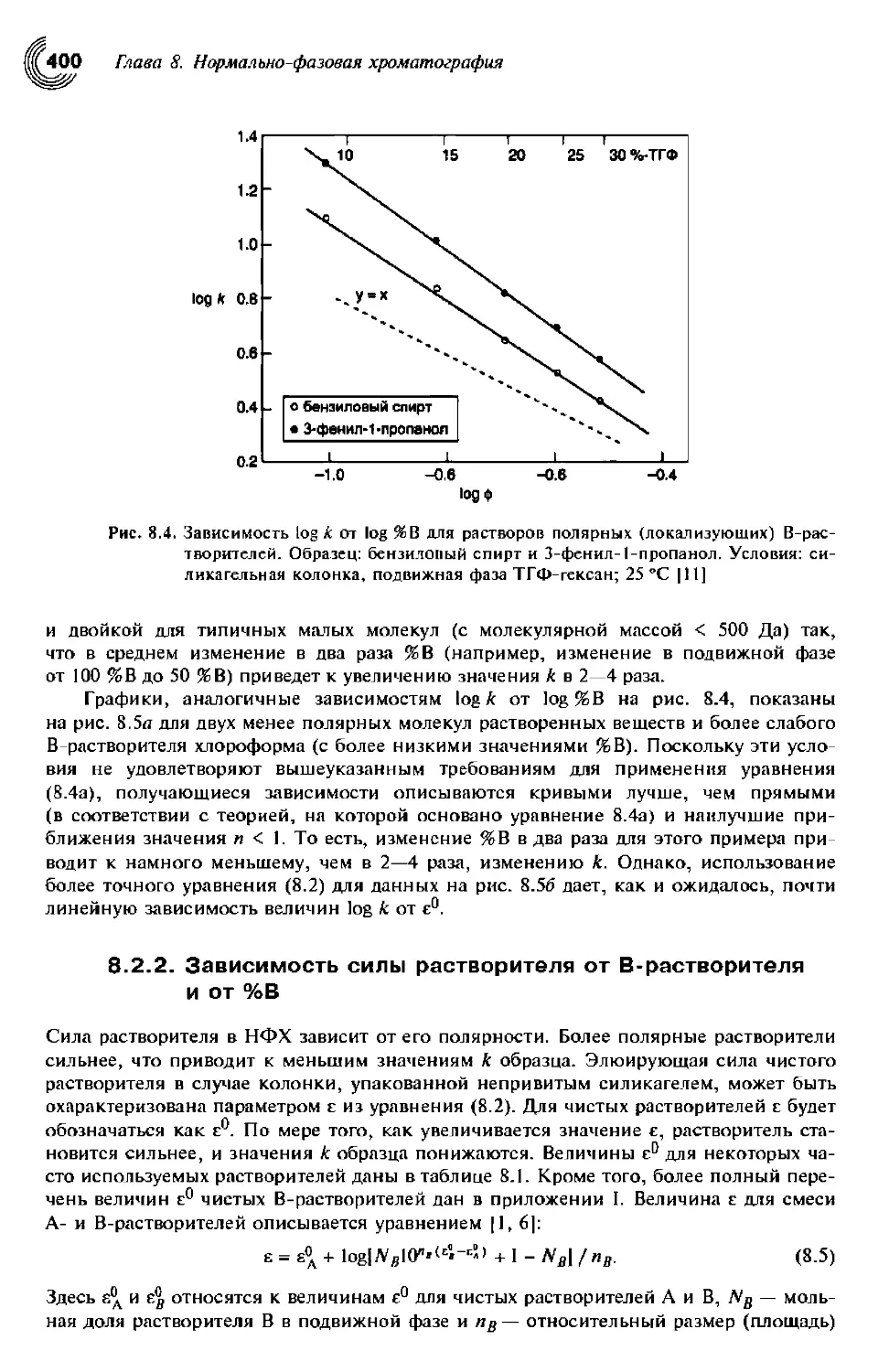
8.2.2. Зависимость силы растворителя от В-растворителя и от %В 400
8.2.3. Использование данных ТСХ для предсказания удерживания в НФХ. . . 403
8.3. Селективность 406
8.3.1. Зависимость селективности от элюирующей силы растворителя 406
8.3.2. Зависимость селективности от типа растворителя 406
8.3.3. Зависимость селективности от температуры 406
8.3.4. Селективность колонки 411
14 Содержание
8.3.5. Разделения изомеров 413
8.4. Рекомендации по разработке метода 416
8.4.1. Начальные условия для разработки НФХ-метода: выбор элюирующей
силы подвижной фазы и типа сорбента (шаги 1, 2; таблица 8.2) 418
8.4.2. Стратегии оптимизации селективности (шаг 3; таблица 8.2) 420
8.4.3. Пример разработки НФХ-метода 420
8.5. Трудности, возникающие при использовании НФХ 422
8.5.1. Плохая воспроизводимость разделения 422
8.5.2. Обеднение элюента растворителем и медленное уравновешивание
колонки 424
8.5.3. Уширение задних фронтов пиков 424
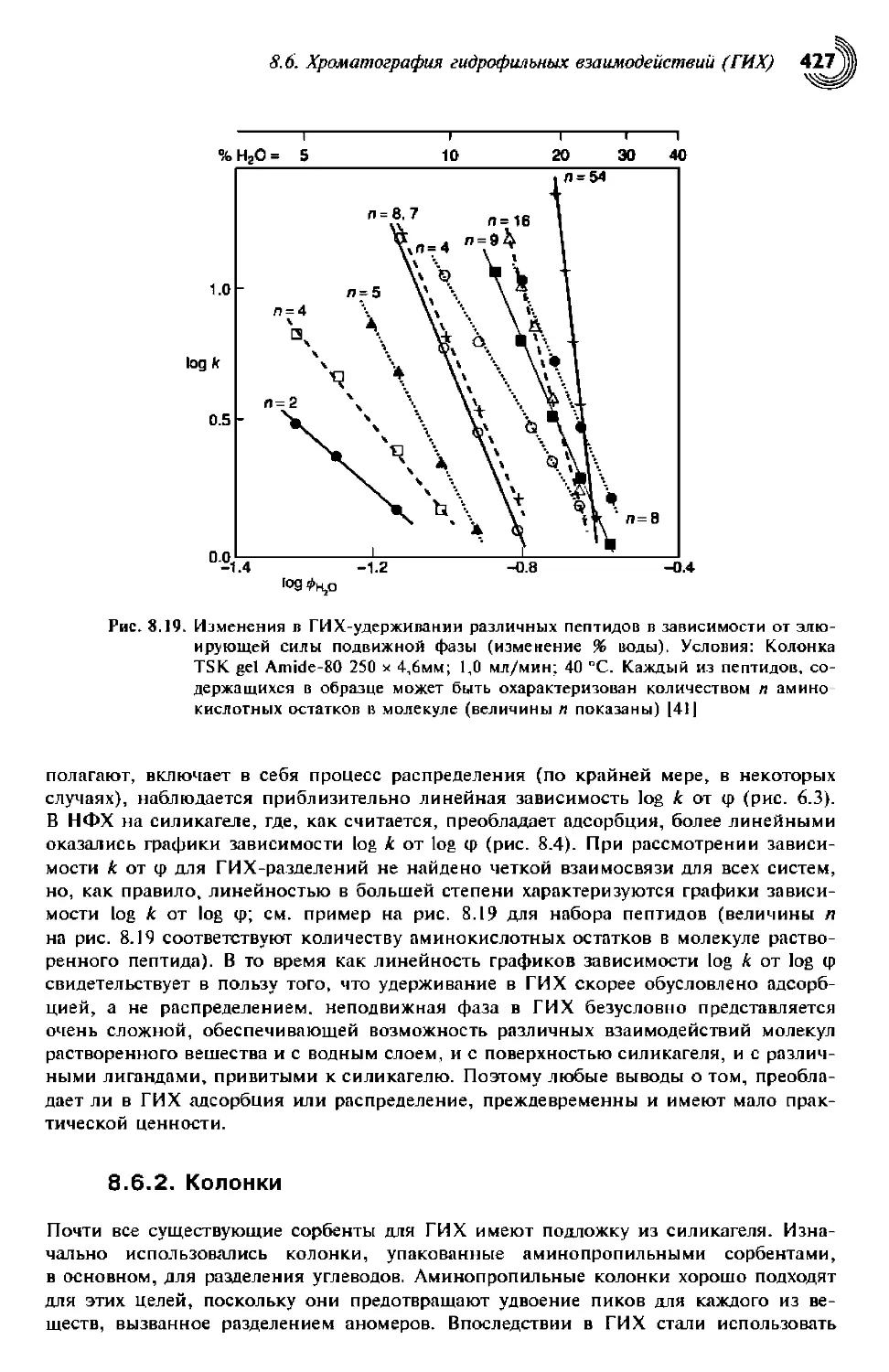
8.6. Хроматография гидрофильных взаимодействий (ГИХ) 425
8.6.1. Механизм удерживания 426
8.6.2. Колонки 427
8.6.3. Разработка метода ГИХ 429
8.6.4. Трудности, связанные с ГИХ 431
Литература 431
Глава 9. Градиентное элюирование 433
9.1. Введение 434
9.1.1. Причины для использования градиентного элюирования 435
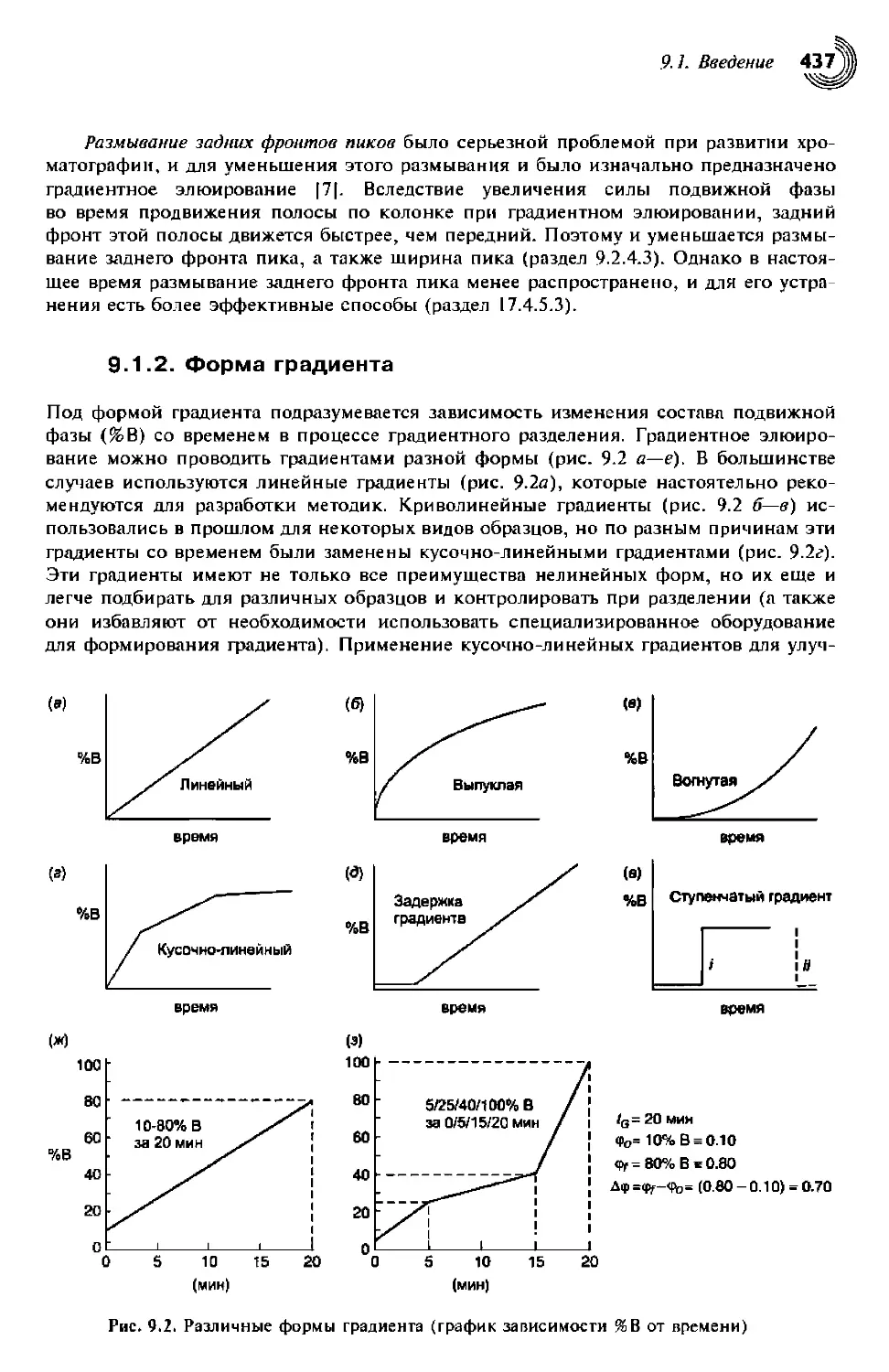
9.1.2. Форма градиента 437
9.1.3. Сходство изократического и градиентного элюирования 438
9.1.3.1. Модель линейного изменения элюирующей силы растворителя
(ЛИС) 439
9.1.3.2. Перемещение зон при градиентном элюировании 440
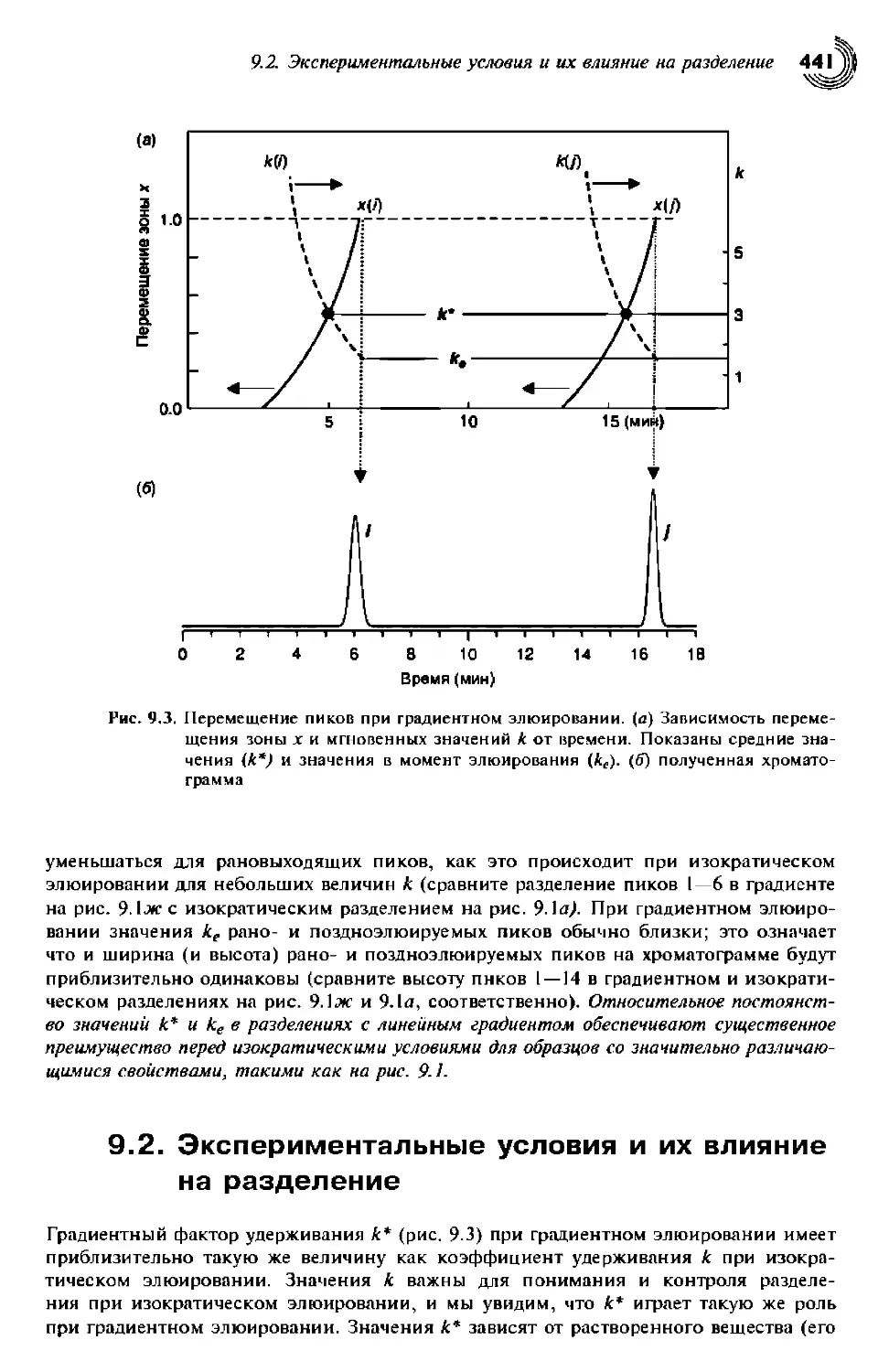
9.2. Экспериментальные условия и их влияние на разделение 441
9.2.1. Влияние изменения параметров колонки 444
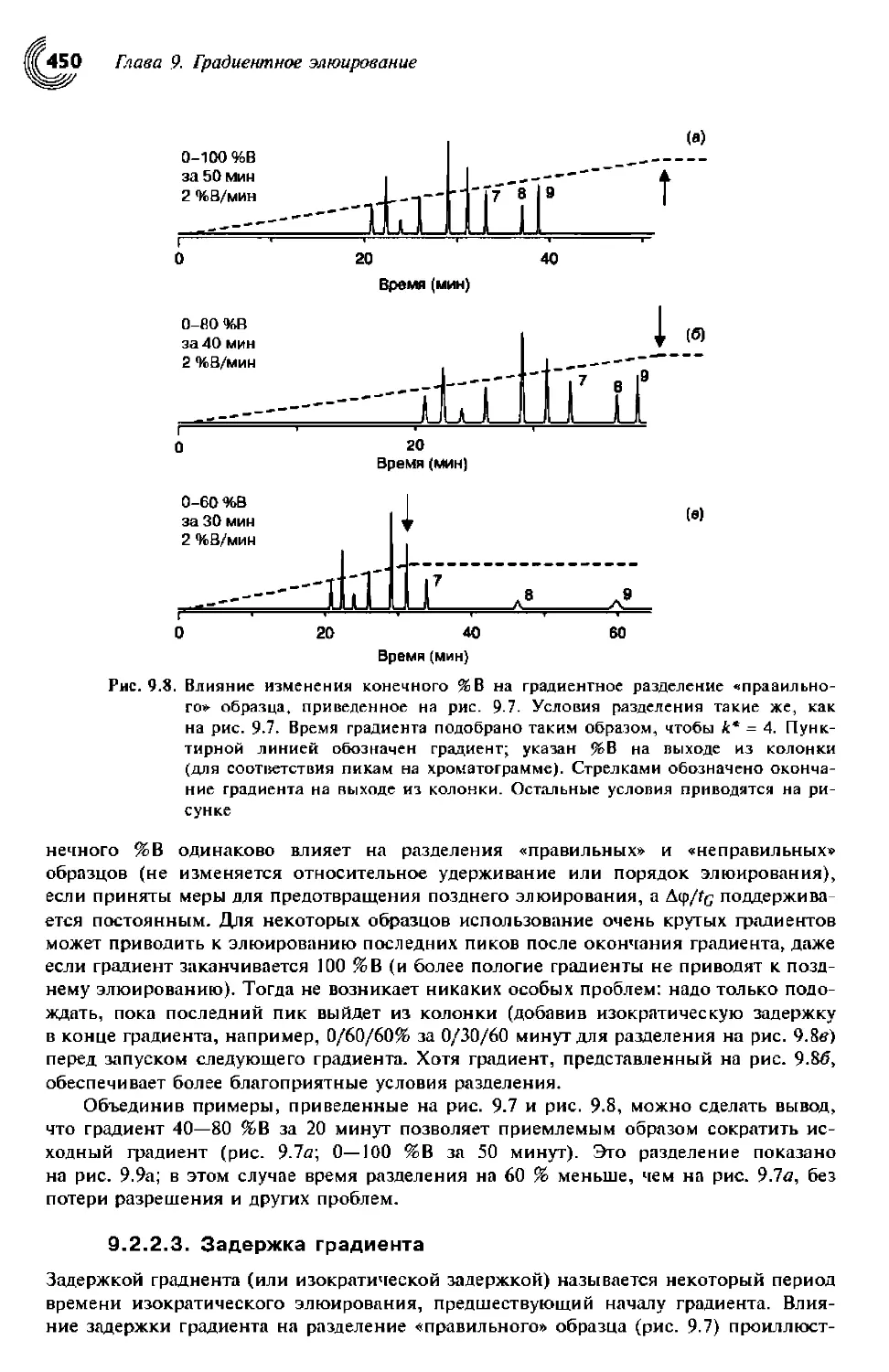
9.2.2. Влияние изменения градиента 447
9.2.2.1. Исходный %В 447
9.2.2.2. Конечный %В 449
9.2.2.3. Задержка градиента 450
9.2.2.4. Приборный объем задержки 452
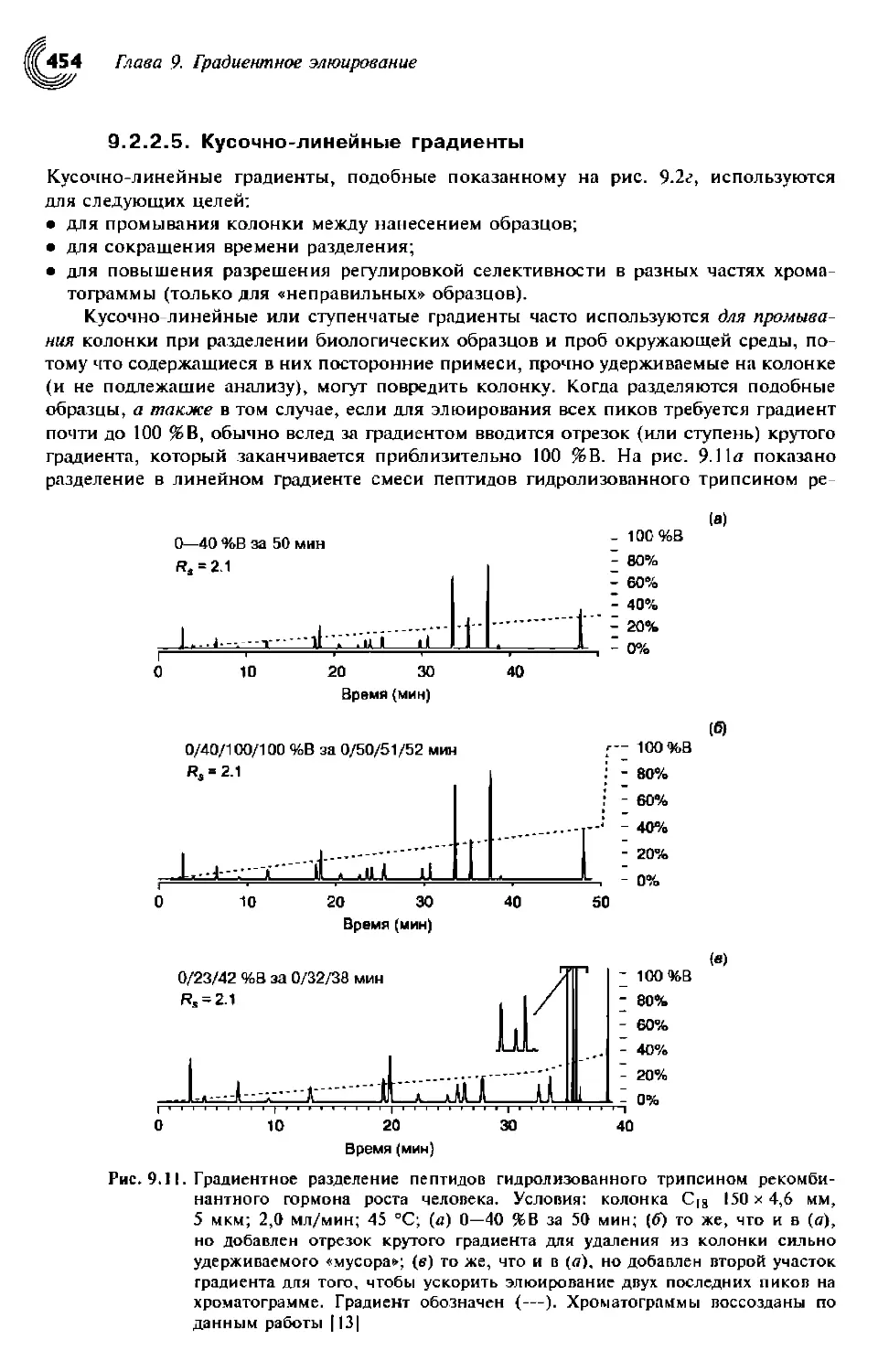
9.2.2.5. Кусочно-линейные градиенты 454
9.2.3. «Неправильные» образцы 456
9.2.4. Количественные соотношения 458
9.2.4.1. Время удерживания 459
9.2.4.2. Измерение величин S и kw 460
9.2.4.3. Ширина пика 461
9.2.4.4. Разрешение 462
9.3. Разработка метода 462
9.3.1. Первичное градиентное разделение 463
9.3.1.1. Выбор между градиентным и изократическим элюированиями . 465
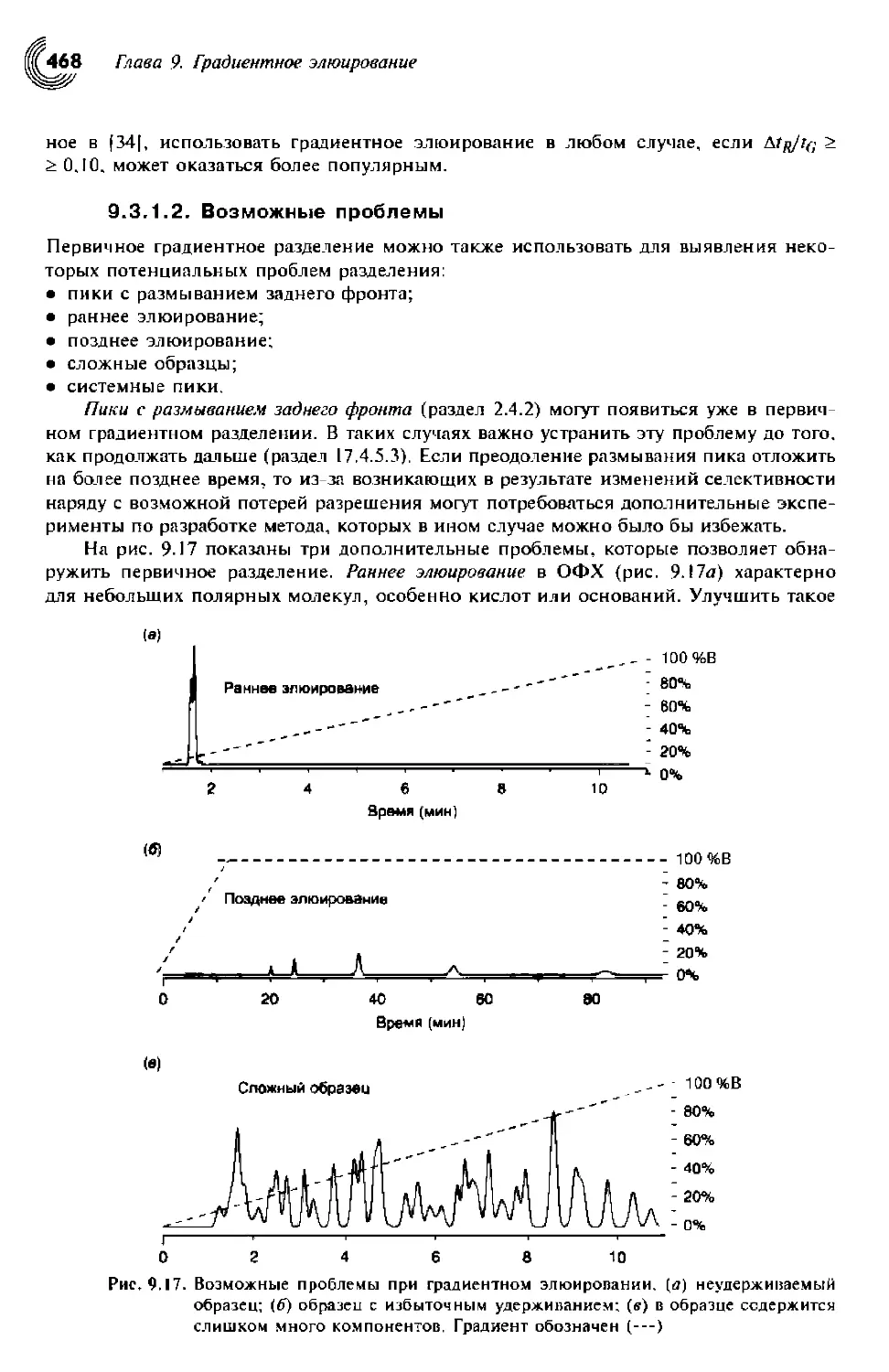
9.3.1.2. Возможные проблемы 468
9.3.2. Оптимизация к (этап 4 таблицы 9.2) 469
9.3.3. Оптимизация селективности градиента а (этап 5 таблицы 9.2) 470
9.3.4. Оптимизация диапазона градиента (этап 6 таблицы 9.2) 470
9.3.5. Ступенчатые (нелинейные) градиенты (продолжение этапа 6
таблицы 9.2) 472
16 Содержание
9.3.6. Оптимизация числа теоретических тарелок N* (этап 7 таблицы 9.2) . . . 473
9.3.7. Определение необходимого времени уравновешивания колонки
(этап 8 таблицы 9.2) 473
9.3.8. Воспроизводимость метода 476
9.3.8.1. Разработка метода 477
9.3.8.2. Рутинные анализы 477
9.3.9. Пиковая емкость и экспресс-разделения 478
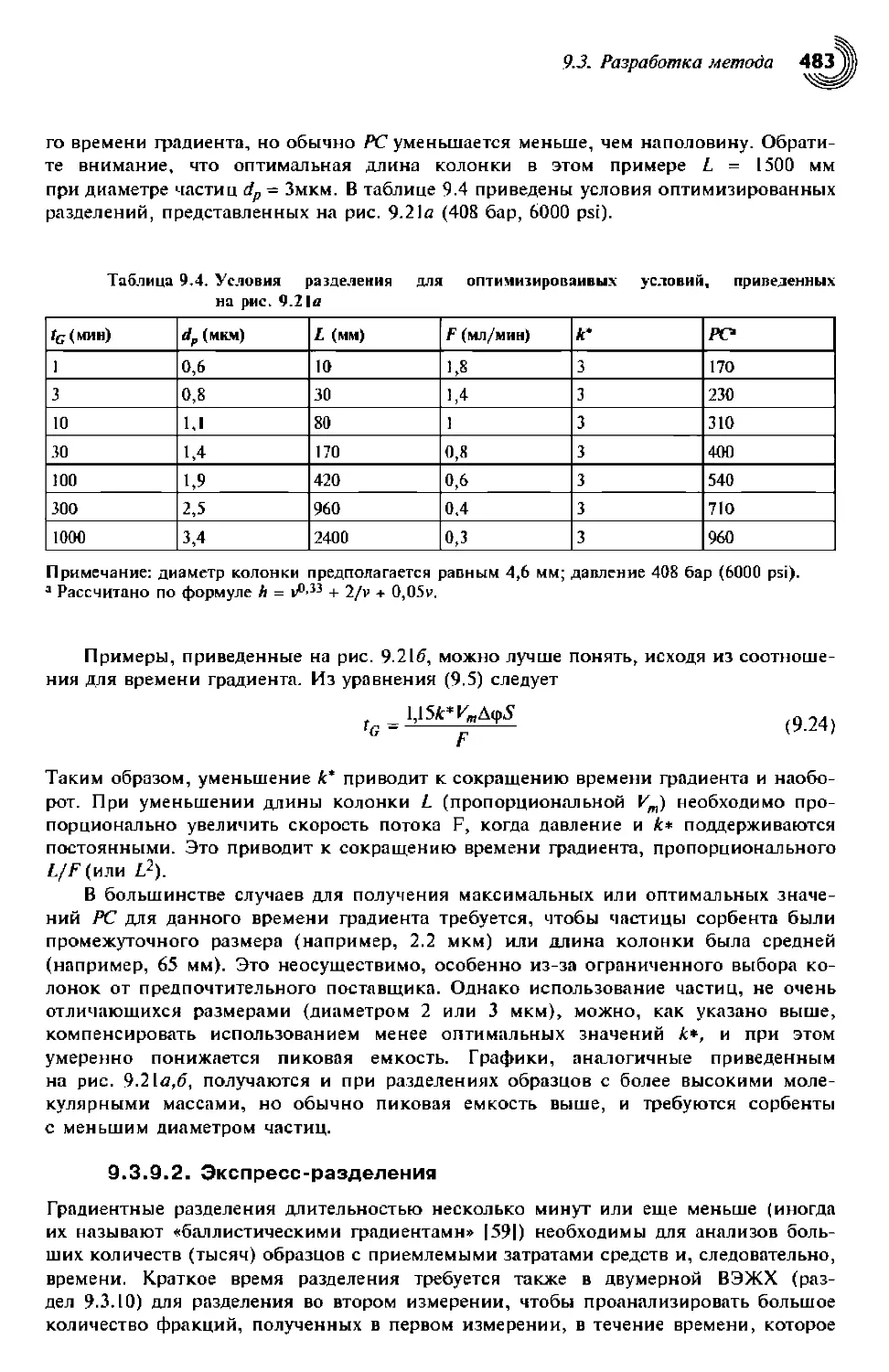
9.3.9.1. Оптимизированная пиковая емкость 480
9.3.9.2. Экспресс-разделения 483
9.3.10. Комплексная двумерная ВЭЖХ (в соавторстве
с Питером Шонмейкерсом) 485
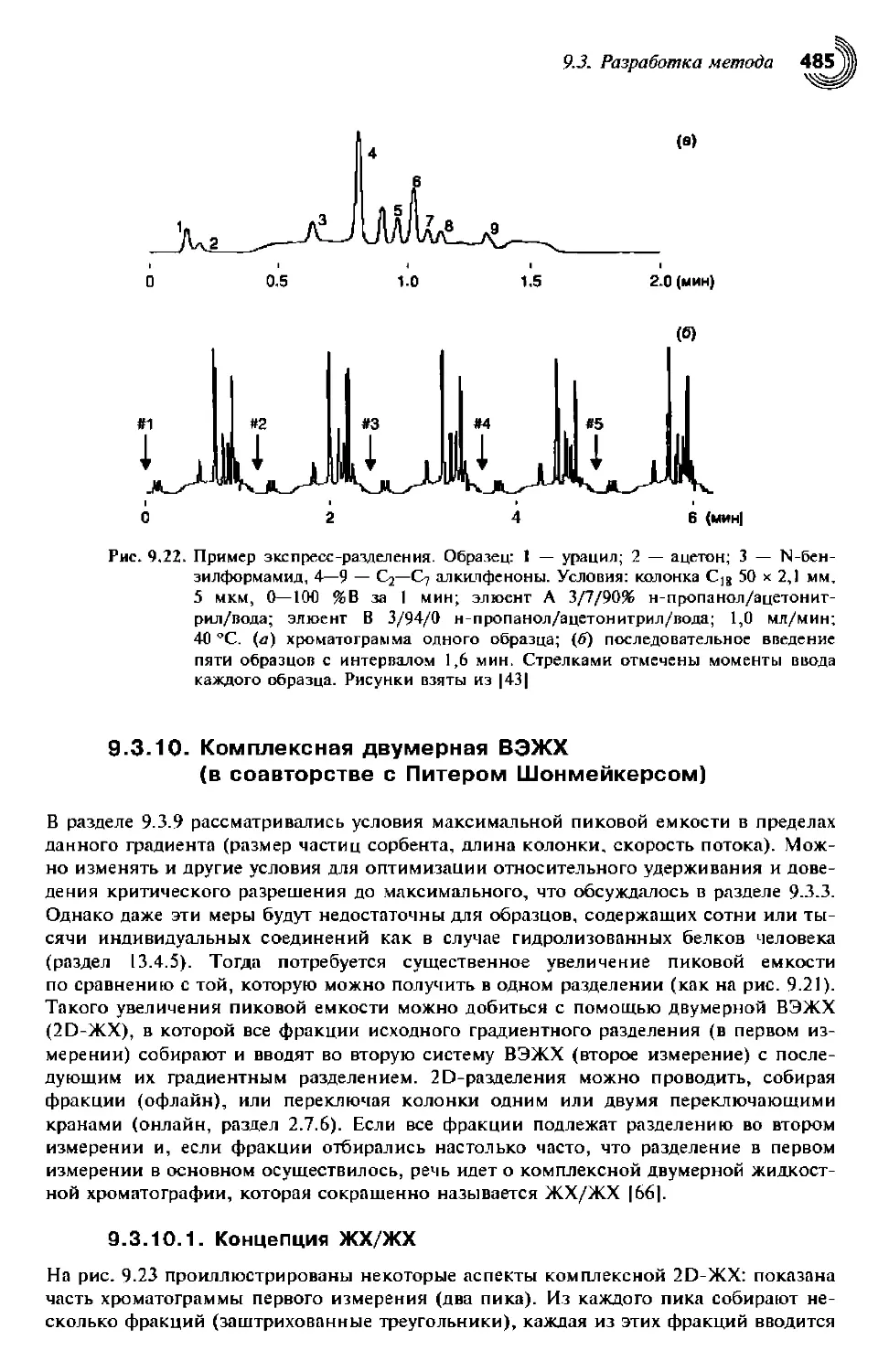
9.3.10.1. Концепция ЖХ/ЖХ 485
9.3.10.2. Пиковая емкость 488
9.3.10.3. Оборудование для ЖХ/ЖХ 488
9.3.10.4. Разработка метода для ЖХ/ЖХ 489
9.4. Разделение крупных молекул 491
9.5. Другие виды разделения 492
9.5.1. Теория 492
9.5.2. Нормально-фазовая хроматография 493
9.5.3. Гидрофильная хроматография (ГИХ) 493
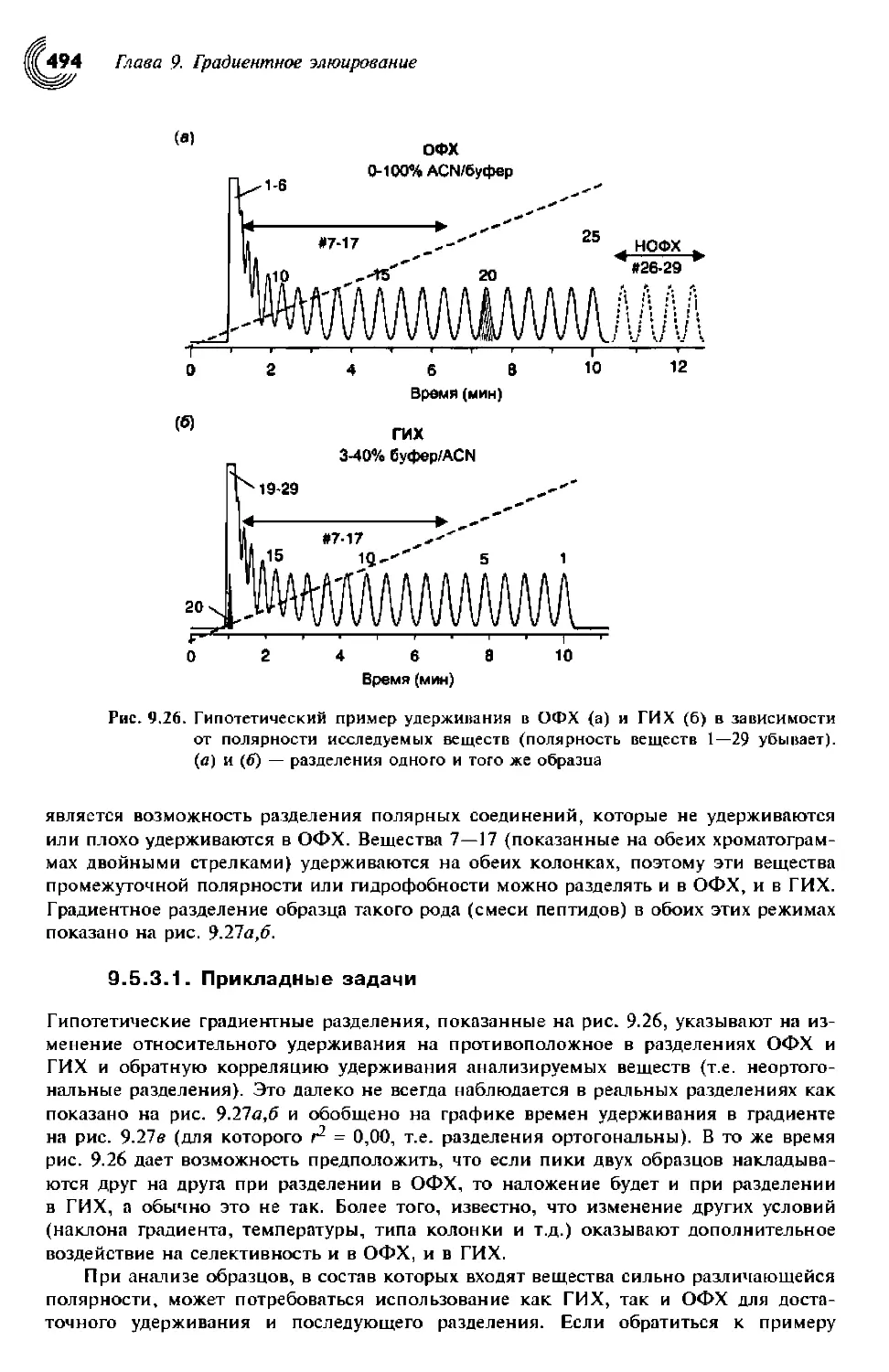
9.5.3.1. Прикладные задачи 494
9.5.3.2. Условия разделения 496
9.5.4. Ионообменная хроматография (ИОХ) 496
9.6. Проблемы 496
9.6.1. Обеднение растворителем 496
9.6.2. Системные пики 497
9.6.3. Дрейф базовой линии 497
Литература 497
Глава 10. Разработка метода с помощью программного обеспечения 500
10.1. Введение 501
10.1.1. Основы и история компьютерного моделирования 503
10.1.2. Когда использовать компьютерное моделирование 504
10.1.2.1. Преимущества 505
10.1.2.2. Недостатки 505
10.2. Программное обеспечение компьютерного моделирования 506
10.2.1. Работа с программой DryLab 507
10.2.2. Оптимизация градиента 509
10.2.3. Другие возможности 511
10.2.3.1. Прогнозирование изократического элюирования на основе
данных градиентного элюирования 511
10.2.3.2. Выбор определенного пика 512
10.2.3.3. Изменение других условий 512
10.2.3.4. Компьютерный подбор наилучшего кусочно-линейного
градиента 512
10.2.3.5. Уширение заднего фронта пика 514
10.2.3.6. Метод двух разделений для улучшения разрешения образца. 514
10.2.3.7. Примеры использования компьютерного моделирования
при разработке метода 514
10.2.4. Отслеживание пиков 515
18 Содержание
10.2.5. Источники коммерчески доступного программного обеспечения
для компьютерного моделирования хроматографических разделений. 516
10.3. Другое программное обеспечение для разработки метода 516
10.3.1. Удерживание анализируемого вещества и его молекулярная
структура 517
10.3.2. Величины рКа анализируемого вещества и его молекулярная
структура 517
10.3.3. Селективность ОФХ-колонки 517
10.3.4. Экспертные системы для разработки метода 518
10.4. Компьютерное моделирование и разработка метода 518
10.4.1. Пример 1: разделение смеси производных лизергиновой кислоты . . . 518
10.4.2. Пример 2: альтернативная стратегия разработки метода 520
10.4.3. Проверка устойчивости метода 522
10.4.4. Заключение 523
Литература 523
Глава 11. Качественный и количественный анализ 525
11.1. Введение 526
11.2. Измерение сигнала 526
11.2.1. Принцип работы интегратора 527
11.2.1.1. Сбор данных 527
11.2.1.2. Распознавание пика 530
11.2.1.3. Интегрирование неидеальных хроматограмм 530
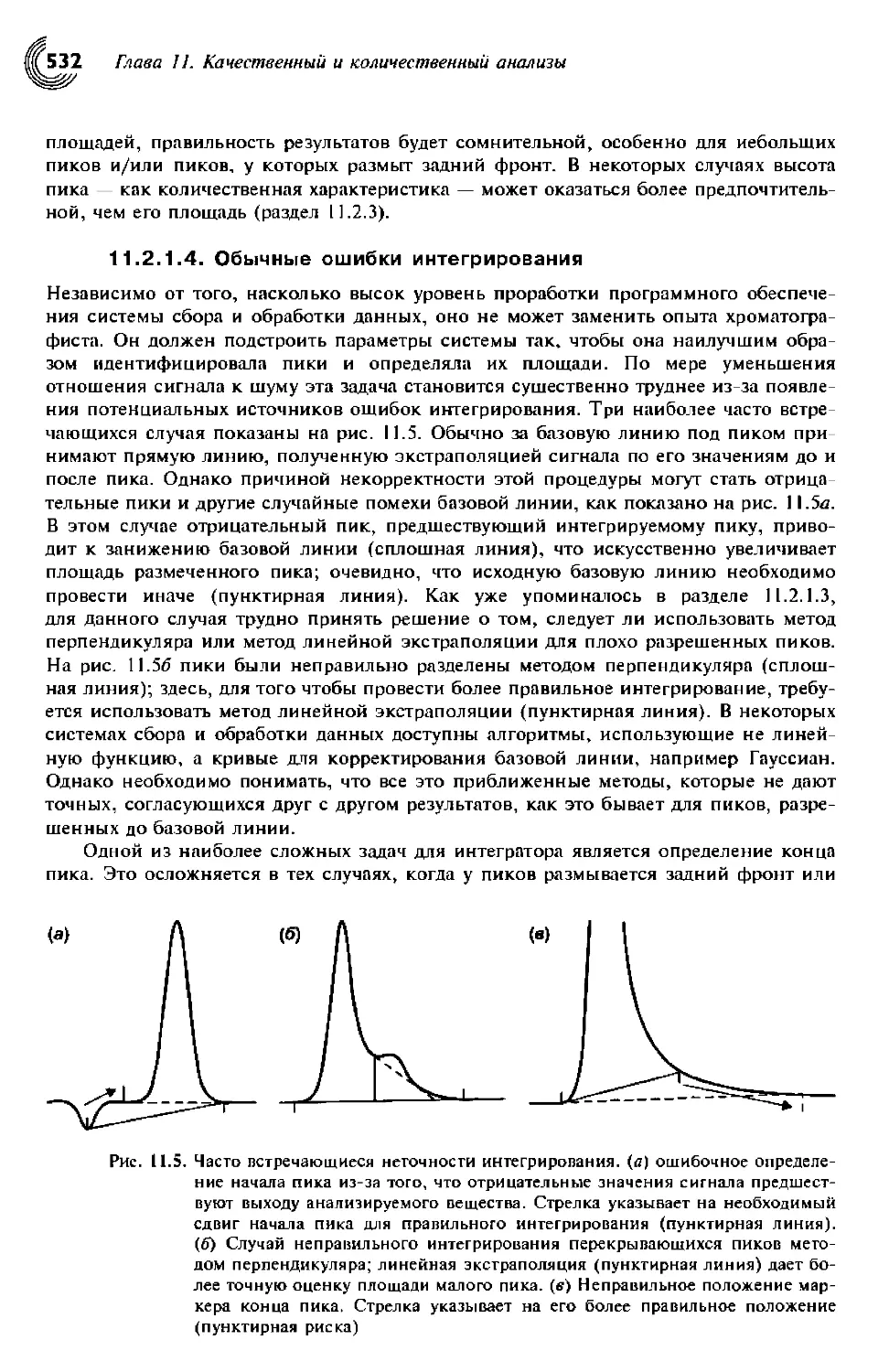
11.2.1.4. Обычные ошибки интегрирования 532
11.2.1.5. Дополнительные советы 533
11.2.2. Удерживание 534
11.2.3. Размер пика 535
11.2.4. Источники ошибок 535
11.2.4.1. Отбор проб и пробоподготовка 536
11.2.4.2. Хроматографическое разделение 536
11.2.4.3. Детектирование 536
11.2.4.4. Измерение пика 537
11.2.4.5. Калибровка 537
11.2.5. Пределы обнаружения и определения 539
11.2.5.1. Предел обнаружения (ПО) 540
11.2.5.2. Нижний предел количественного определения
(НПКО или ПКО) 542
11.2.5.3. Верхний предел 542
11.2.5.4. Образцы с концентрациями, лежащими вне пределов метода 543
11.3. Качественный анализ 544
11.3.1. Время удерживания 544
11.3.2. Качественный анализ в реальном времени 545
11.3.2.1. УФ-детекторы 546
11.3.2.2. Жидкостная хроматография с масс-спектрометрией
(ЖХ-МС) 546
11.3.2.3. ИК-спектроскопия с Фурье-преобразованием (ИКФП) .... 547
11.3.2.4. Жидкостная хроматография с детектором ядерного
магнитного резонанса (ЖХ-ЯМР) 547
11.3.2.5. Хемилюминесцентный азотный детектор (ХЛАД) 547
11.3.2.6. Лазерный детектор светорассеяния (ЛДС) 547
20 Содержание
11.3.2.7. Хиральные детекторы 547
11.3.2.8. Качественный анализ в автономном режиме (офлайн анализ). 548
11.4. Количественный анализ 548
11.4.1. Калибровка 548
11.4.1.1. Калибровка внешним стандартом 549
11.4.1.2. Калибровка внутренним стандартом 552
11.4.1.3. Нормирование площадей пиков 554
11.4.1.4. Метод стандартных добавок 555
11.4.1.5. Оценка калибровочных кривых 556
11.4.2. Анализ следовых количеств 558
11.5. Выводы 558
Литература 559
Глава 12. Валидация метода (совместно с Майклом Шварцем) 560
12.1. Введение 562
12.2. Термины и определения 565
12.2.1. Правильность 566
12.2.2. Прецизионность 568
12.2.2.1. Повторяемость 568
12.2.2.2. Внутрилабораторная (промежуточная) прецизионность .... 568
12.2.2.3. Воспроизводимость 569
12.2.2.4. Надежность 569
12.2.3. Специфичность 569
12.2.4. Предел обнаружения и предел количественного определения 569
12.2.5. Линейность и аналитическая область 571
12.2.6. Устойчивость 572
12.3. Пригодность системы 573
12.4. Документация 575
12.4.1. Валидационный протокол 575
12.4.2. Метод испытания 576
12.4.3. Валидационный отчет 577
12.5. Валидация для различных типов фармацевтических методов 578
12.5.1. Методы категории 1 578
12.5.2. Методы категории 2 578
12.5.3. Методы категории 3 579
12.5.4. Методы категории 4 580
12.6. Биоаналитические методы 580
12.6.1. Подготовка стандартного образца 581
12.6.2. Разработка и валидация биоаналитического метода 581
12.6.2.1. Избирательность 582
12.6.2.2. Правильность, прецизионность и степень извлечения 582
12.6.2.3. Калибровочные/стандартные кривые 583
12.6.2.4. Стабильность биоаналитического образца 584
12.6.3. Рутинное применение биоаналитического метода 585
12.6.4. Документирование биоаналитического метода 586
12.7. Передача (трансфер) методики анализа в принимающую
лабораторию (ТМА) 587
12.7.1. Опции передачи (трансфера) методики анализа 588
12.7.1.1. Сравнительное тестирование 588
12.7.1.2. Совместная валидация 589
12.7.1.3. Валидация и/или повторная валидация метода 589
12.7.1.4. Отказ от оформления передачи метода испытания 589
12.7.2. Основы ТМА 590
12.7.2.1. Предварительно утвержденный протокол плана
тестирования 590
12.7.2.2. Описание метода / процедуры испытаний 590
12.7.2.3. Описание и обоснование требований к тестам 592
12.7.2.4. Критерии приемлемости 592
12.7.2.5. Документирование результатов 593
12.7.3. Потенциальные ошибки при ТМА 593
12.7.3.1. Оценка оборудования 594
12.7.3.2. Колонки ВЭЖХ 594
12.7.3.3. Обучение оператора 594
12.8. Корректировка методики или модификация методики 595
12.8.1. Корректировка рН 597
12.8.2. Концентрация соли в буфере 597
12.8.3. Соотношение компонентов в подвижной фазе 597
12.8.4. Длина волны УФ/ВИД фотометрического детектора 598
12.8.5. Корректировка температуры 598
12.8.6. Изменение длины и диаметра колонки, размера частиц сорбента . . . 598
12.9. Контроль качества и обеспечение качества 599
12.9.1. Контроль качества 599
12.9.2. Обеспечение качества 599
12.10. Выводы 600
Литература 601
Глава 13. Разделение биополимеров и синтетических полимеров
(совместно с Тимоти Вером, Карлом Сканделлой и Петером Шонмейкерсом) 602
13.1. Биомакромолекулы 605
13.2. Молекулярная структура и конформация 605
13.2.1. Пептиды и белки (полипептиды) 606
13.2.1.1. Первичная структура 606
13.2.1.2. Вторичная структура 606
13.2.1.3. Третичная и четвертичная структуры 606
13.2.1.4. Посттрансляционные модификации 609
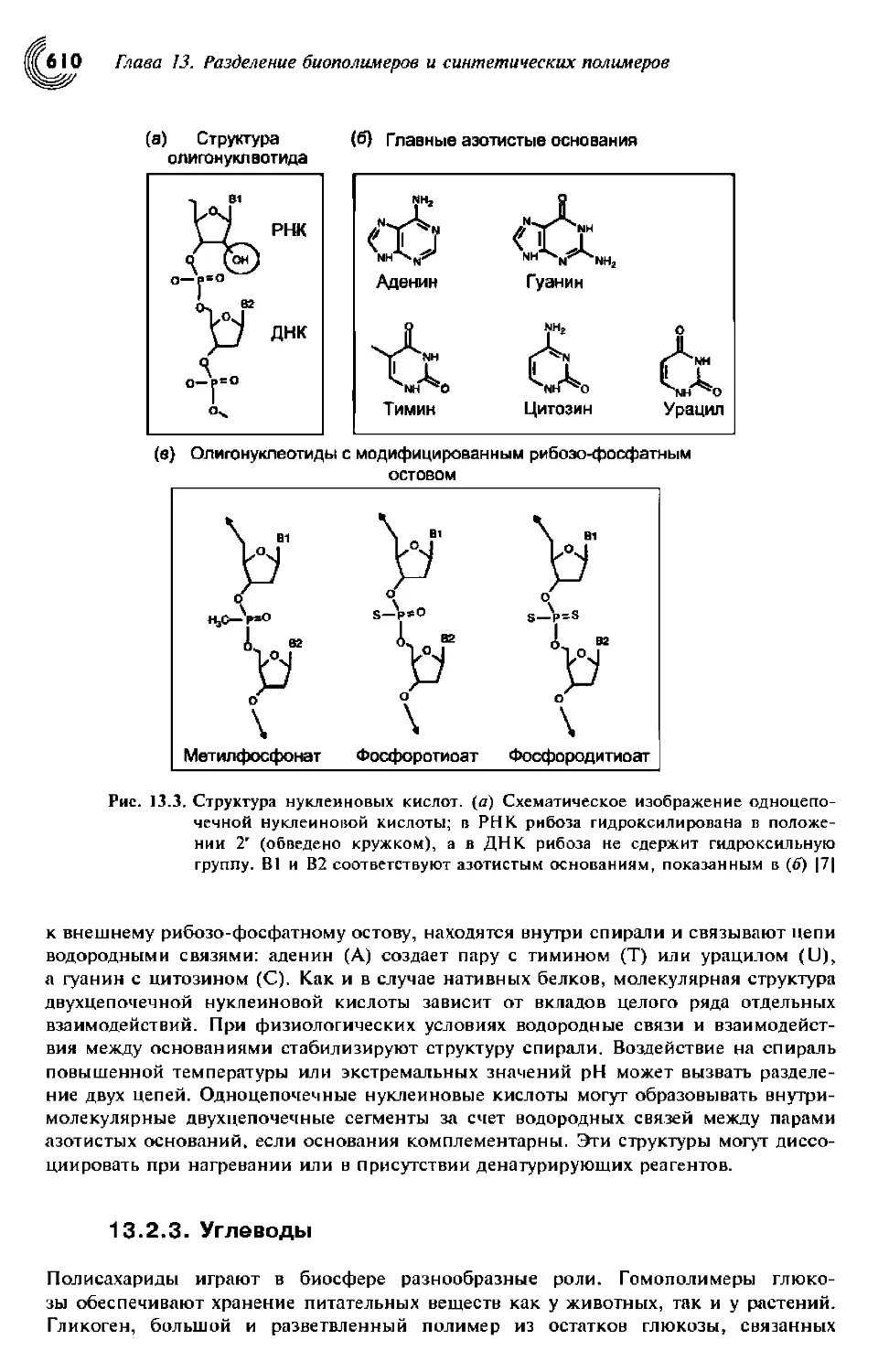
13.2.2. Нуклеиновые кислоты 609
13.2.2.1. Одноцепочечные нуклеиновые кислоты 609
13.2.2.2. Двухцепочечные нуклеиновые кислоты 609
13.2.3. Углеводы 610
13.2.4. Вирусы 612
13.3. Особенности ВЭЖХ биомолекул 614
13.3.1. Характеристики колонок 614
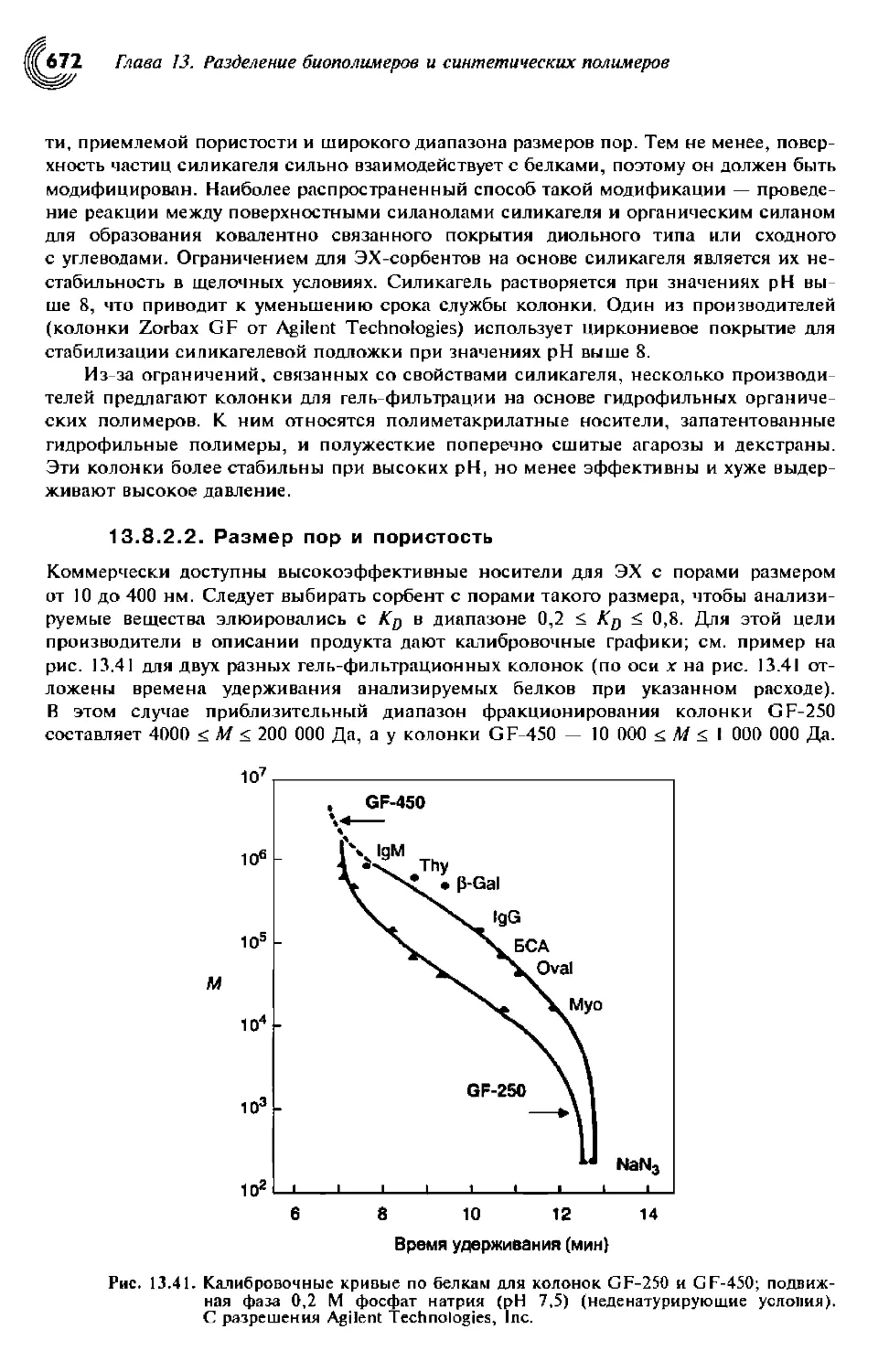
13.3.1.1. Размер пор 614
13.3.1.2. Размер частиц 615
13.3.1.3. Характеристики и стабильность носителя 617
13.3.1.4. Выход и биологическая активность 618
13.3.2. Влияние структуры белка на его поведение
при хроматографическом разделении 618
13.4. Разделение пептидов и белков 619
13.4.1. Обращенно-фазовая хроматография (ОФХ) 619
Содержание
13.4.1.1. Выбор колонки 620
13.4.1.2. Выбор подвижной фазы 621
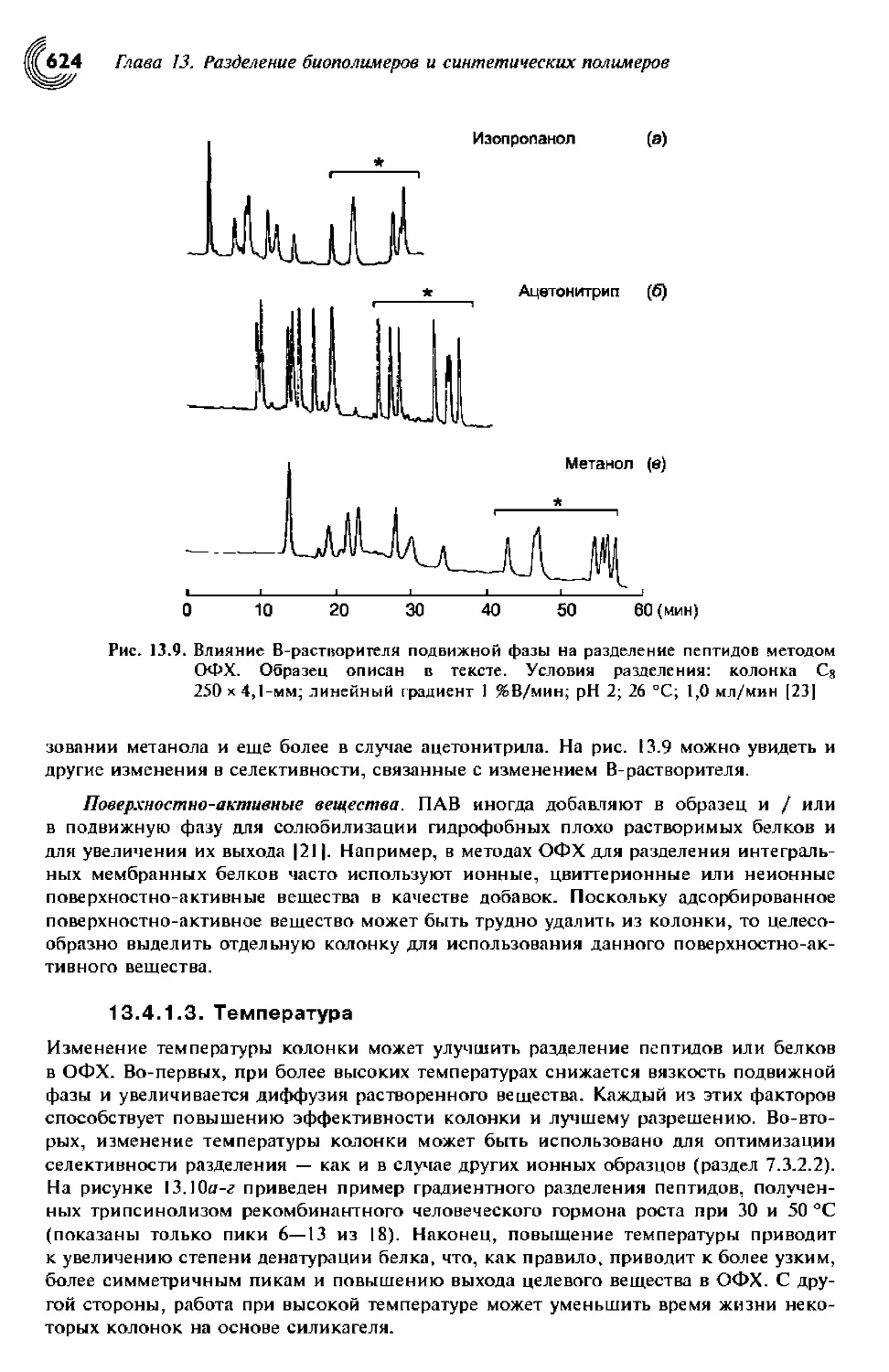
13.4.1.3. Температура 624
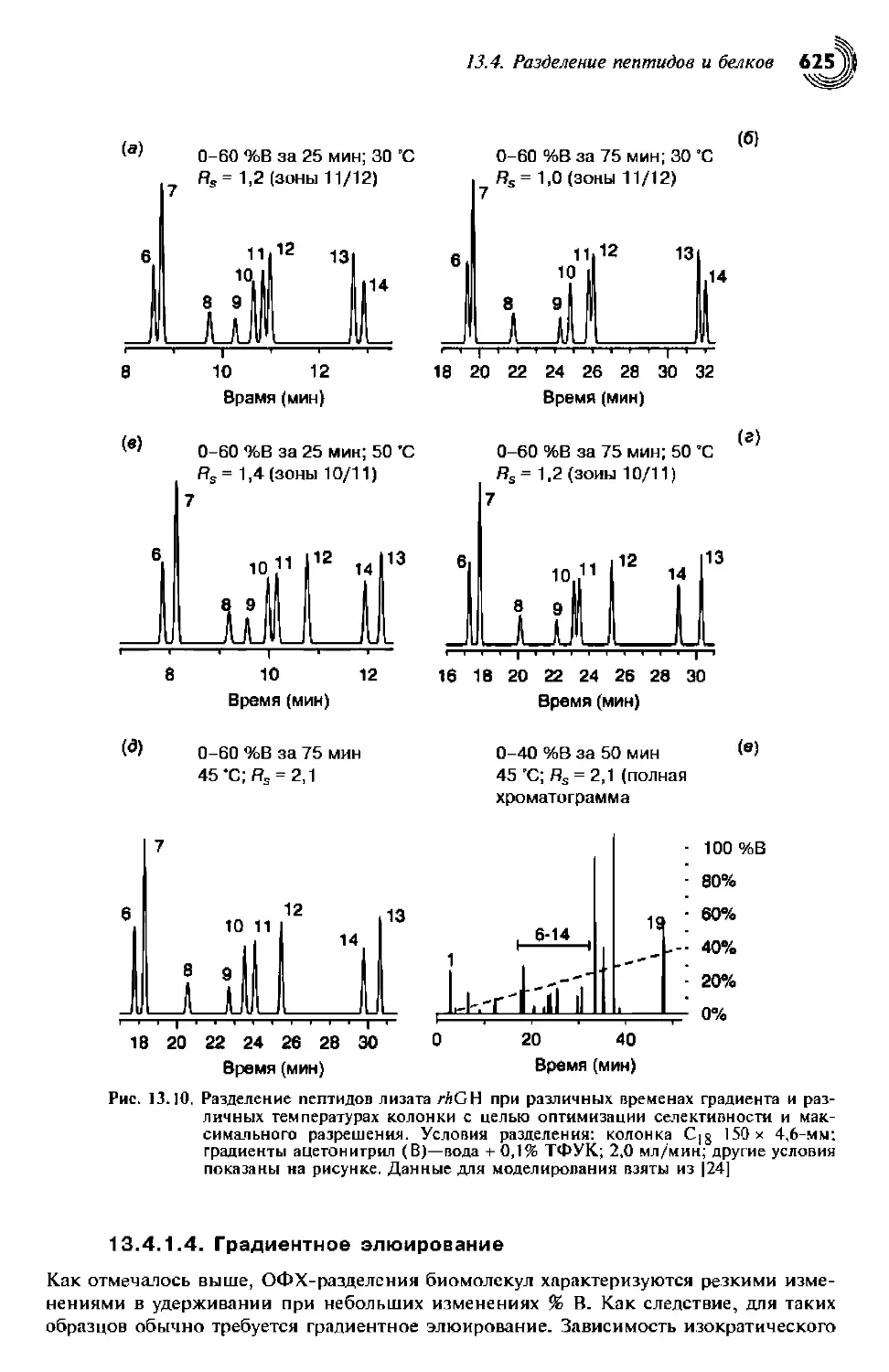
13.4.1.4. Градиентное элюирование 625
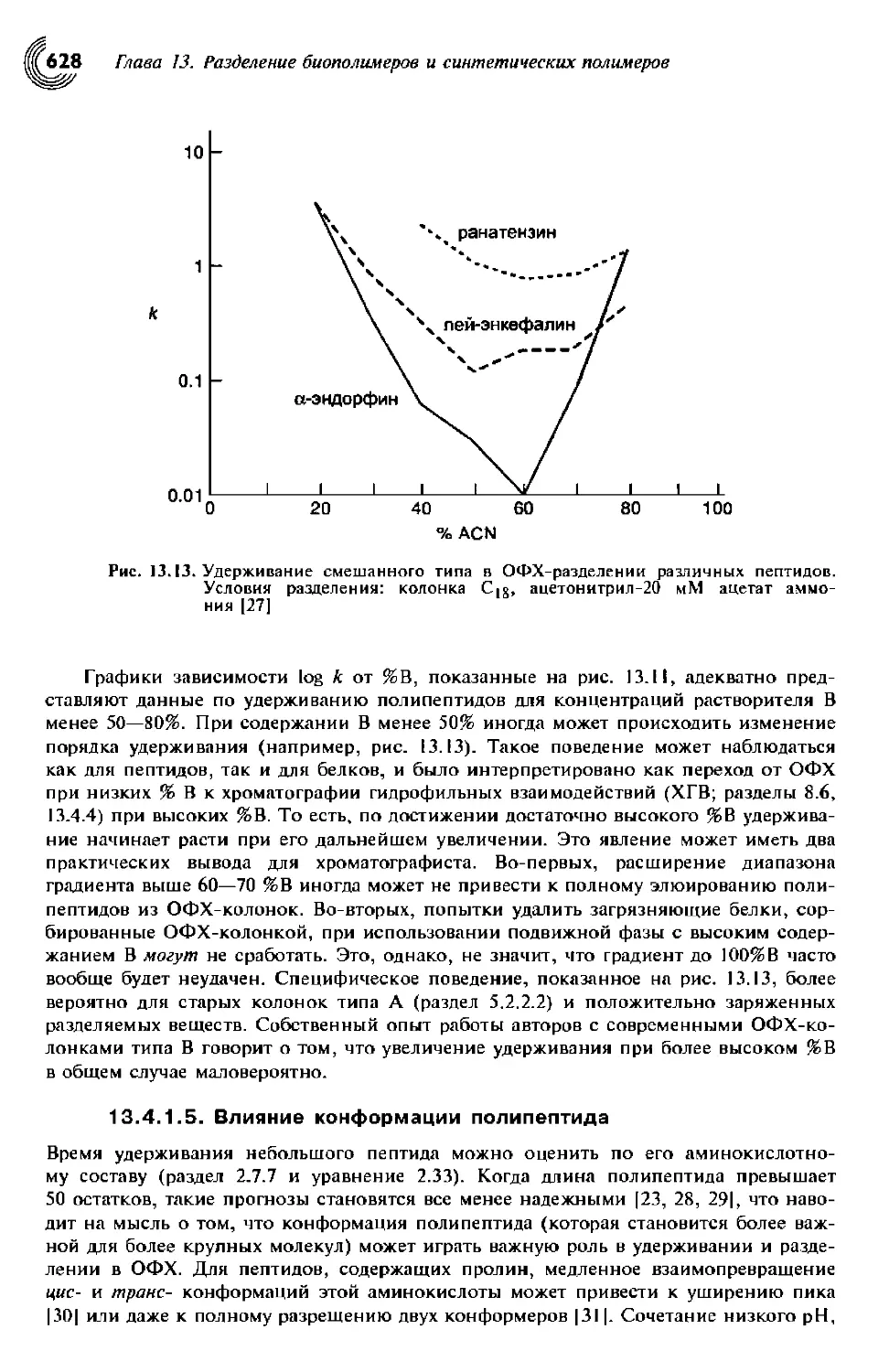
13.4.1.5. Влияние конформации полипептида 628
13.4.1.6. Капиллярные колонки и наноисточники ионизации 630
13.4.1.7. Разработка метода ОФХ 631
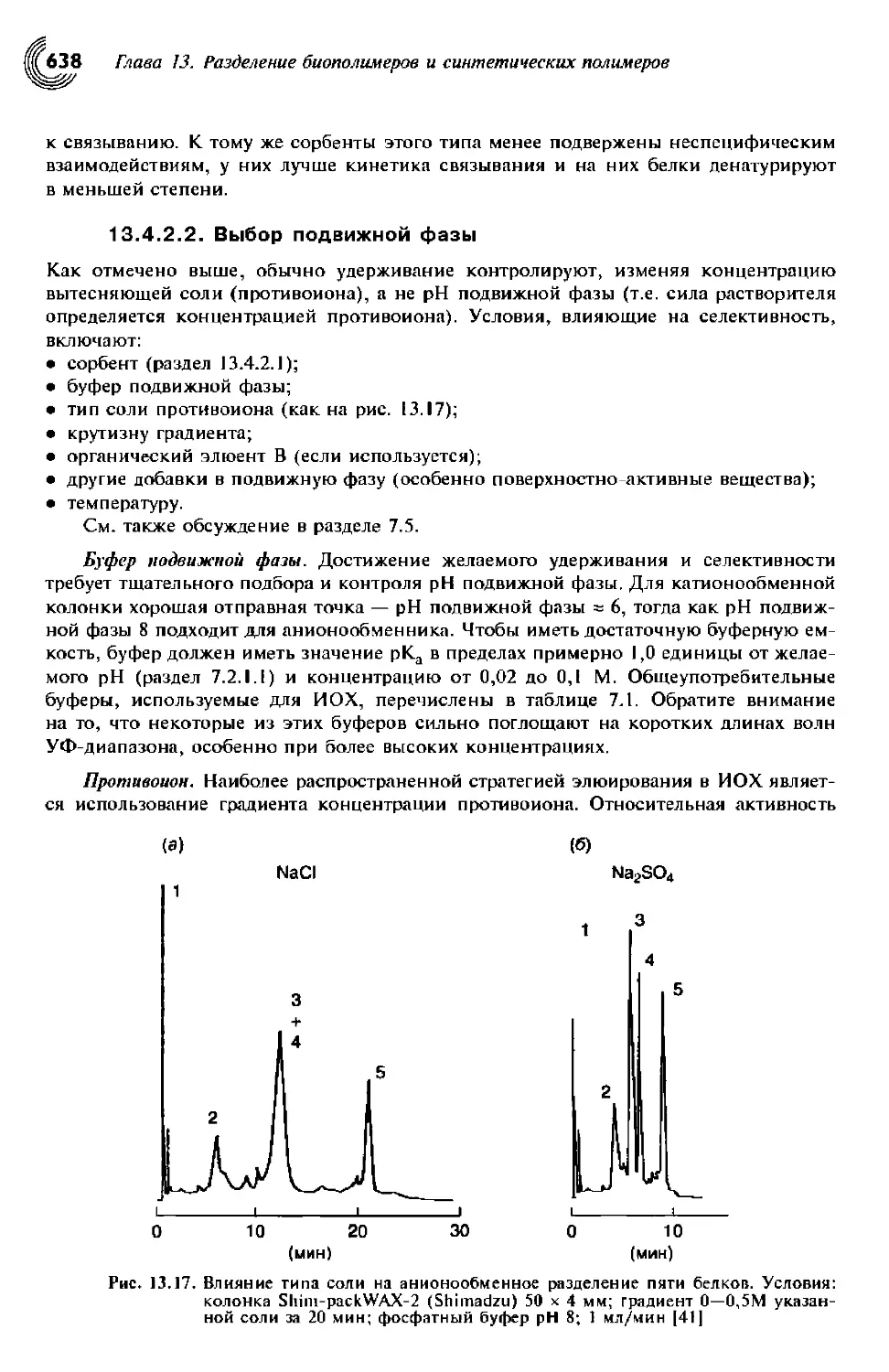
13.4.2. Ионообменная хроматография (ИОХ) и смежные методы 633
13.4.2.1. Выбор колонки 636
13.4.2.2. Выбор подвижной фазы 638
13.4.2.3. Хроматофокусирование 639
13.4.2.4. Хроматография на гидроксиапатите 640
13.4.2.5. Аффинная хроматография на сорбентах
с иммобилизованными металлами (АХИМ) 641
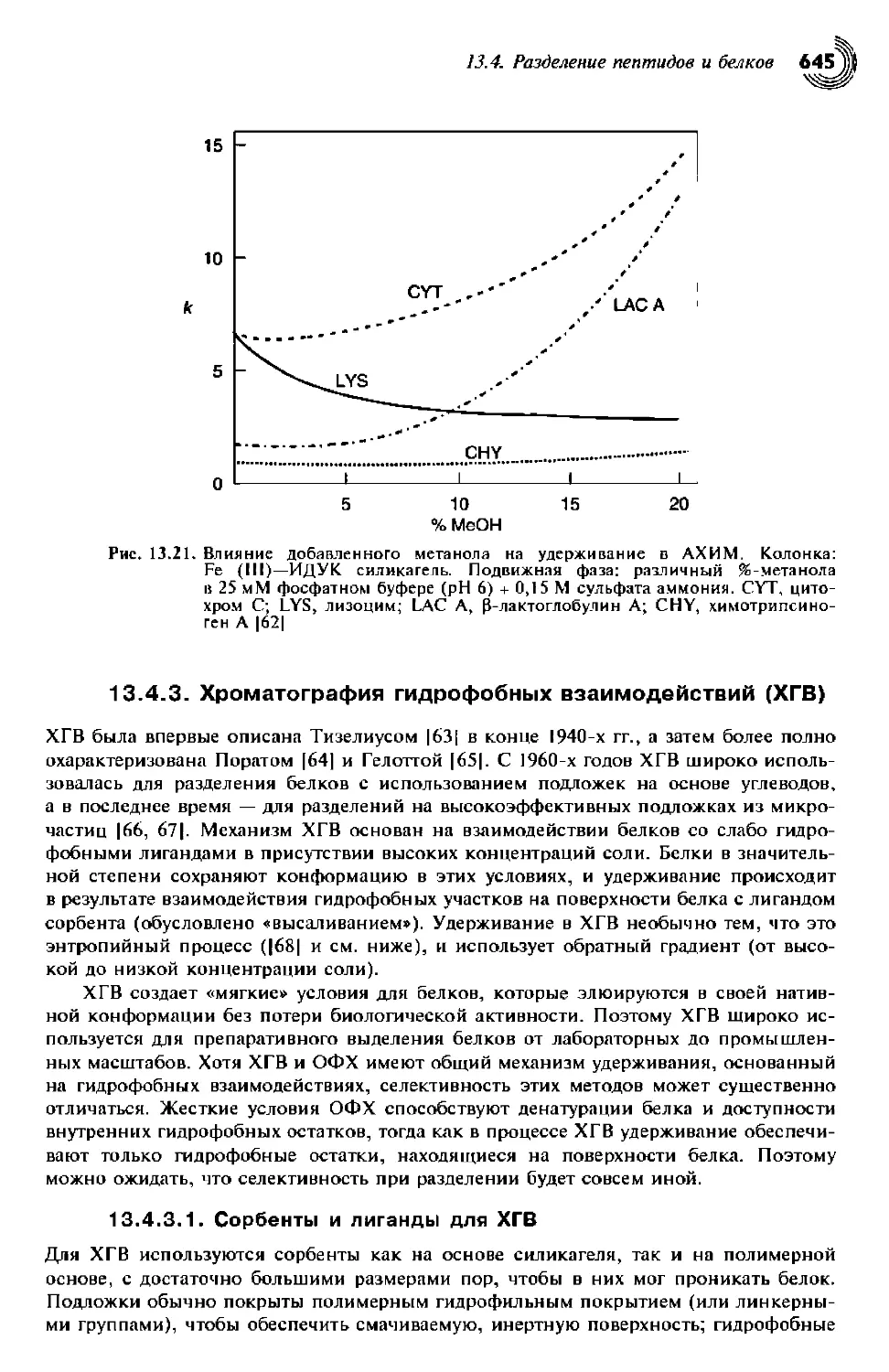
13.4.3. Хроматография гидрофобных взаимодействий (ХГВ) 645
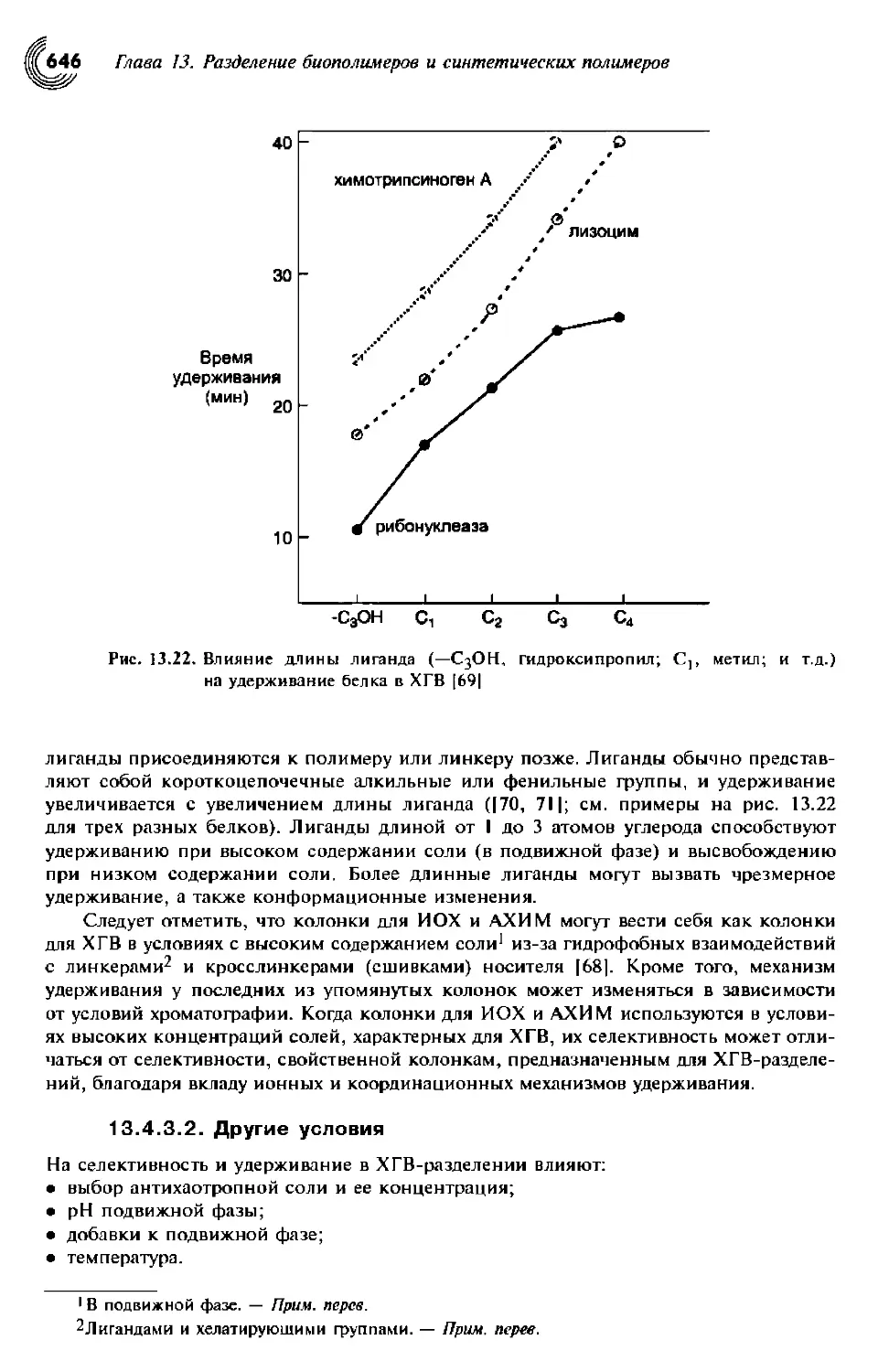
13.4.3.1. Сорбенты и лиганды для ХГВ 646
13.4.3.2. Другие условия 647
13.4.4. Хроматография гидрофильных взаимодействий (ГИХ) 648
13.4.4.1. Неподвижные фазы, используемые в ГИХ 650
13.4.4.2. Подвижные фазы для ГИХ 650
13.4.4.3. Применение ГИХ в разделении пептидов и белков 650
13.4.4.4. Хроматография гидрофильных взаимодействий
с электростатическим отталкиванием (ГИХЭО) 651
13.4.5. Многомерная жидкостная хроматография (МЖХ) в протеомике .... 652
13.4.5.1. Использование коллектора фракций 653
13.4.5.2. Непосредственно соединенные колонки для МЖХ 654
13.4.5.3. МЖХ с переключением колонок 654
Разделение нуклеиновых кислот 655
13.5.1. Анионообменная хроматография 655
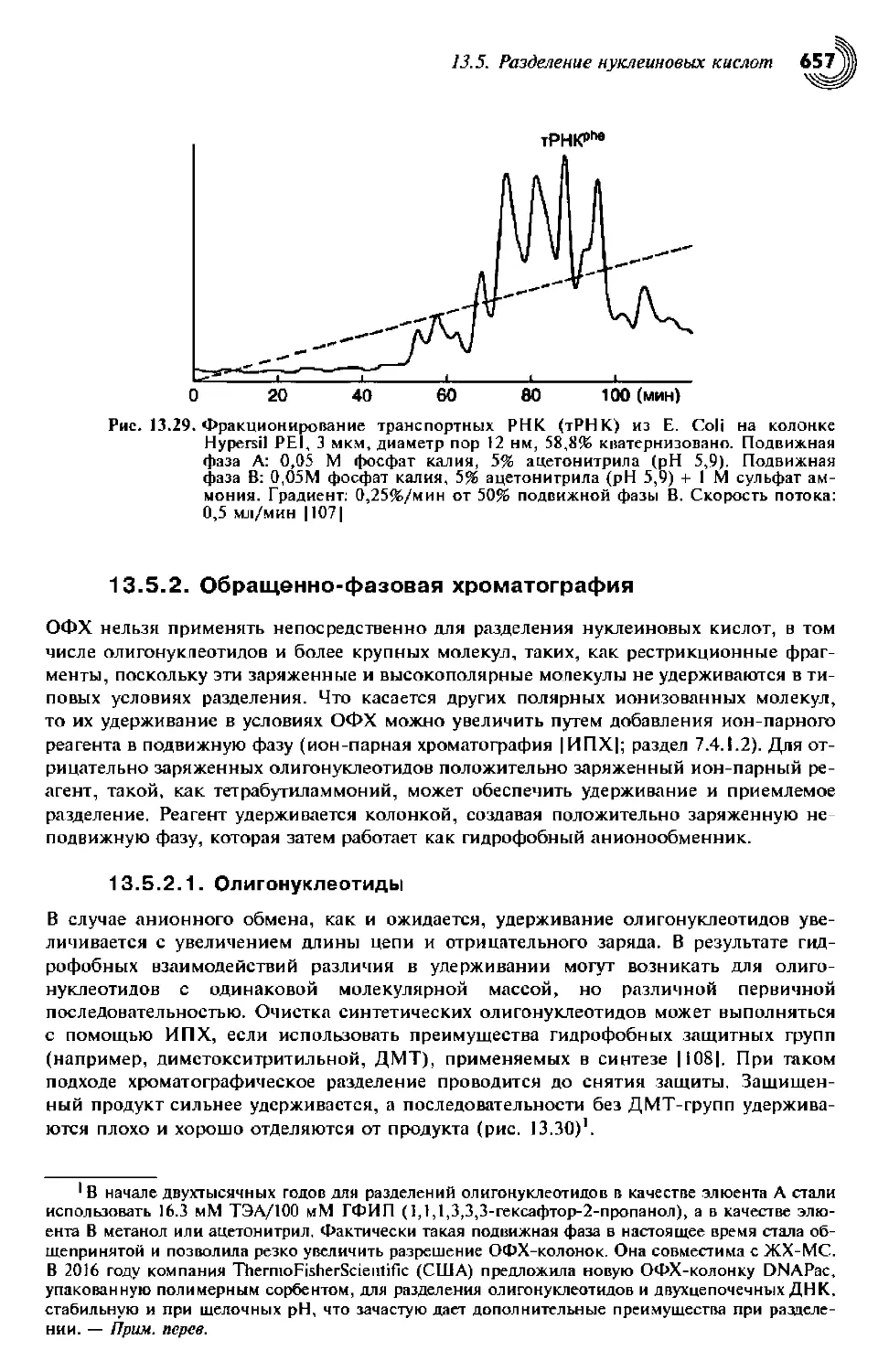
13.5.2. Обращенно-фазовая хроматография 657
13.5.2.1. Олигонуклеотиды 657
13.5.2.2. Фрагменты, полученные рестрикцией, и продукты ПЦР . . . 658
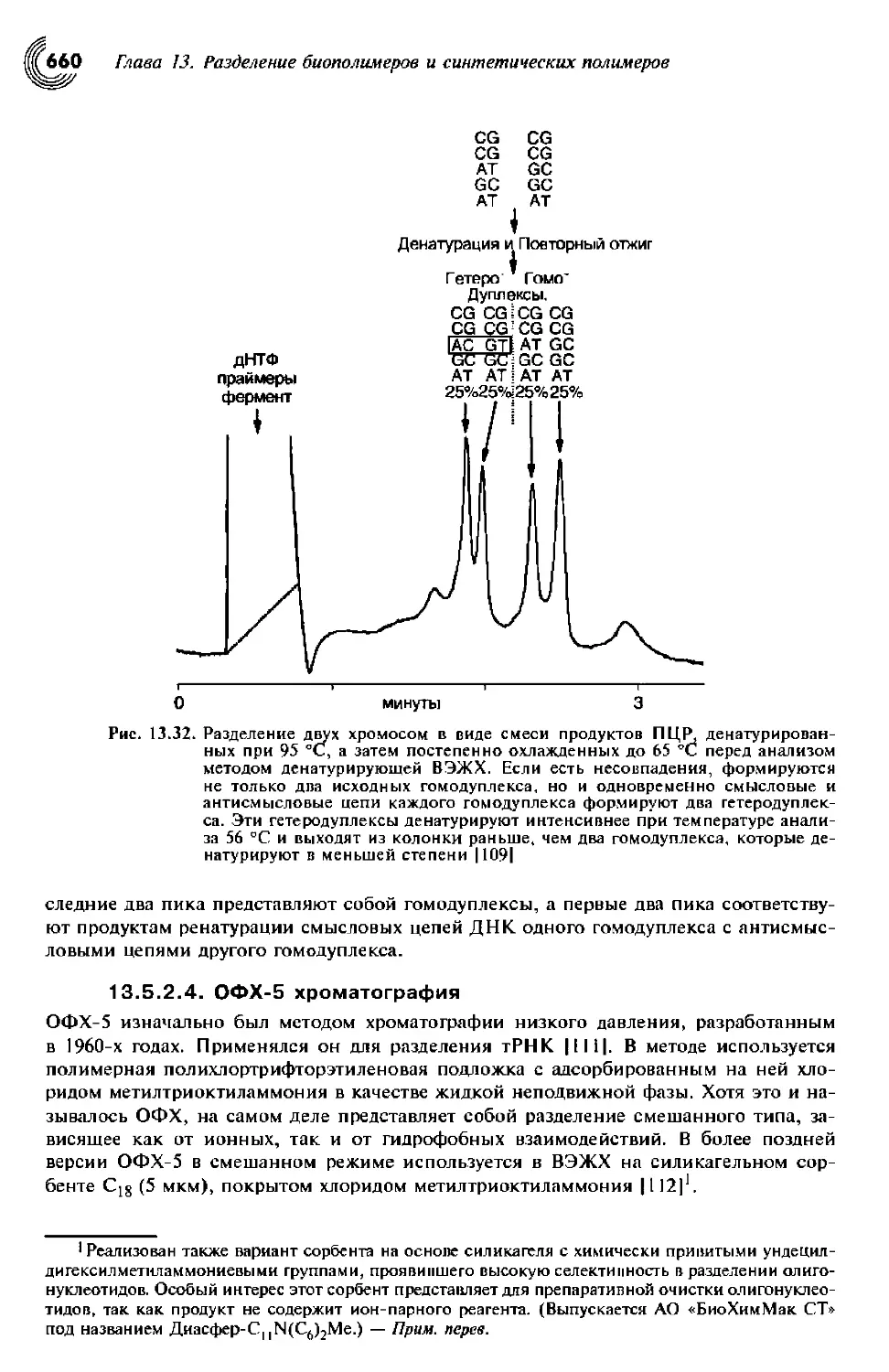
13.5.2.3. Денатурирующая ВЭЖХ 658
13.5.2.4. ОФХ-5 хроматография 660
13.5.3. Хроматография гидрофобных взаимодействий 661
Разделение углеводов 662
13.6.1. Хроматография гидрофильных взаимодействий 662
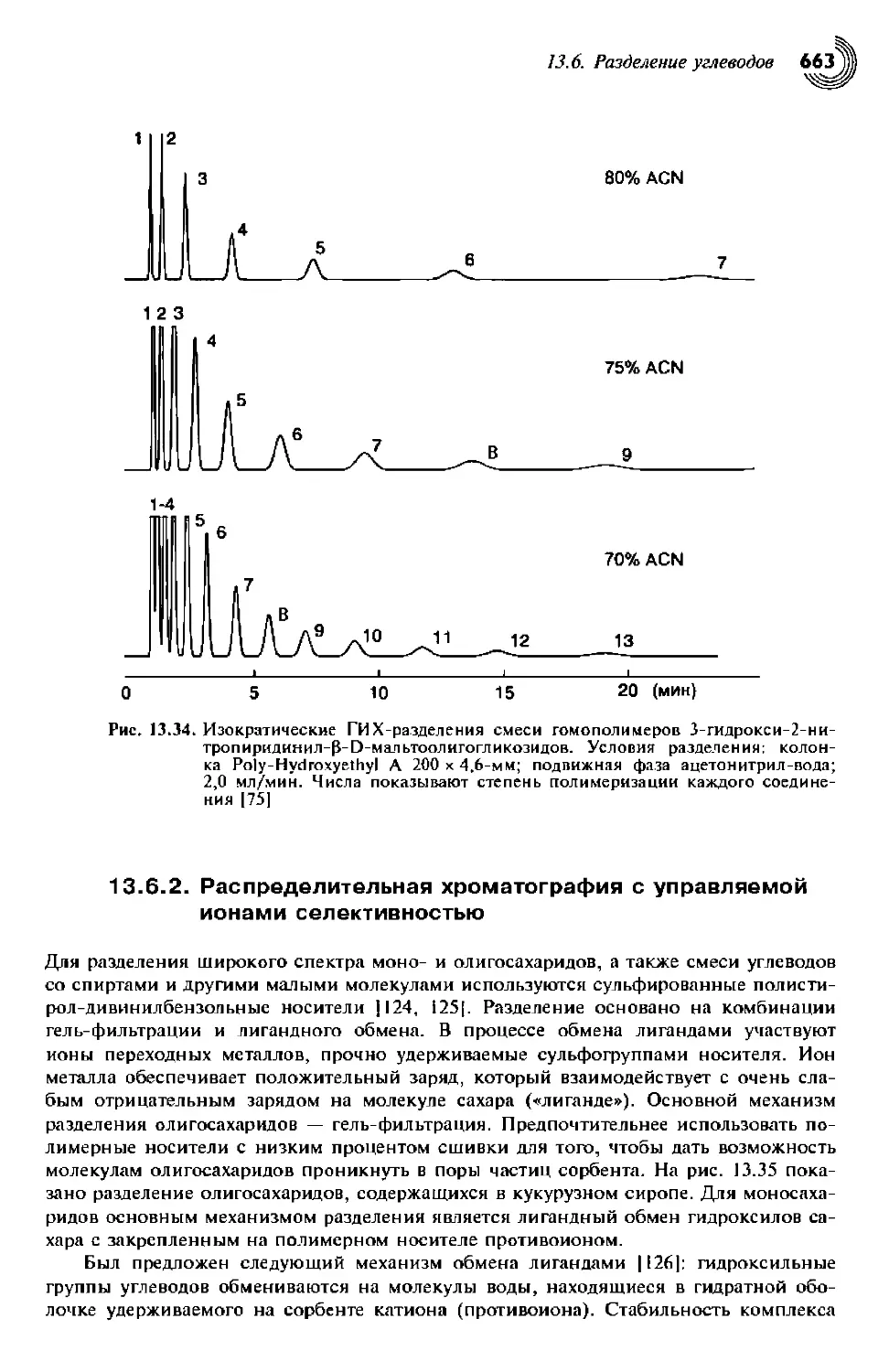
13.6.2. Распределительная хроматография с управляемой ионами
селективностью 663
13.6.3. Высокоэффективная анионообменная хроматография 665
Разделение вирусов 667
Эксклюзионная хроматография (ЭХ) 668
13.8.1. Процесс разделения в ЭХ 669
13.8.2. Колонки для ЭХ 671
13.8.2.1. Сорбенты 672
13.8.2.2. Размер пор и пористость 673
13.8.2.3. Диаметр частиц 673
13.8.2.4. Увеличение разрешения в ЭХ 674
13.8.3. Подвижные фазы для ЭХ 674
13.8.4. Соображения по эксплуатации 675
Hi
www.j-anatytics.ru
налитика
научно-технический журнал
ЖУРНАЛ «АНАЛИТИКА»
НАУЧНО-ТЕХНИЧЕСКИЙ ЖУРНАЛ О СОЗДАНИИ, ИЗУЧЕНИИ
И ПРИМЕНЕНИИ НОВЫХ ВЕЩЕСТВ И МАТЕРИАЛОВ - ОТ
ФУНДАМЕНТАЛЬНЫХ ИССЛЕДОВАНИЙ ДО ВНЕДРЕНИЯ
ПЕРЕДОВЫХ ПРОМЫШЛЕННЫХ ТЕХНОЛОГИЙ.
ОСНОВНАЯ ЦЕЛЬ ЖУРНАЛА - ФОРМИРОВАНИЕ ЕДИНОГО
ИНФОРМАЦИОННОГО ПРОСТРАНСТВА ДЛЯ
ВЗАИМОДЕЙСТВИЯ НАУКИ, БИЗНЕСА И ГОСУДАРСТВА В ЦЕЛЯХ СОЗДАНИЯ
И РАЗВИТИЯ ВЫСОК0ТЕХНОЛОГИЧНЫХИМП0РТОНЕЗАВИ-
СИМЫХ ОТЕЧЕСТВЕННЫХ ПРОИЗВОДСТВ И РЕШЕНИИ
ВОПРОСОВ МОДЕРНИЗАЦИИ ЭКОНОМИКИ РОССИИ.
ИЗДАТЕЛЬ - АО «РИЦ «ТЕХНОСФЕРА»
ЖУРНАЛ «АНАЛИТИКА» ВКЛЮЧЕН
s ВАК, s Российский индекс научного цитирования (РИНЦ)
Периодичность — 6 номеров в год
L E-mail: j-analytics@mail.ru
Тел.: +7(495)234-01-10
Факс: +7(495)956-33-46
13.8.4.1. Емкость колонки 675
13.8.4.2. Использование денатурирующих условий 675
13.8.4.3. Калибровка колонки 676
13.8.4.4. Использование неидеальных условий разделения 676
13.8.5. Преимущества и недостатки ЭХ 676
13.8.6. Применение ЭХ 677
13.8.6.1. Аналитические приложения 678
13.8.6.2. Препаративные приложения 679
13.9. Крупномасштабная очистка биомакромолекул 680
13.9.1. История вопроса 680
13.9.2. Очистка рекомбинантного инсулина человека в промышленном
масштабе 681
13.9.2.1. Цели очистки 681
13.9.2.2. Неподвижные фазы 682
13.9.2.3. Упаковка колонки 682
13.9.2.4. Стабильность продукта и колонки 682
13.9.2.5. Состав подвижной фазы 682
13.9.2.6. Разделение 682
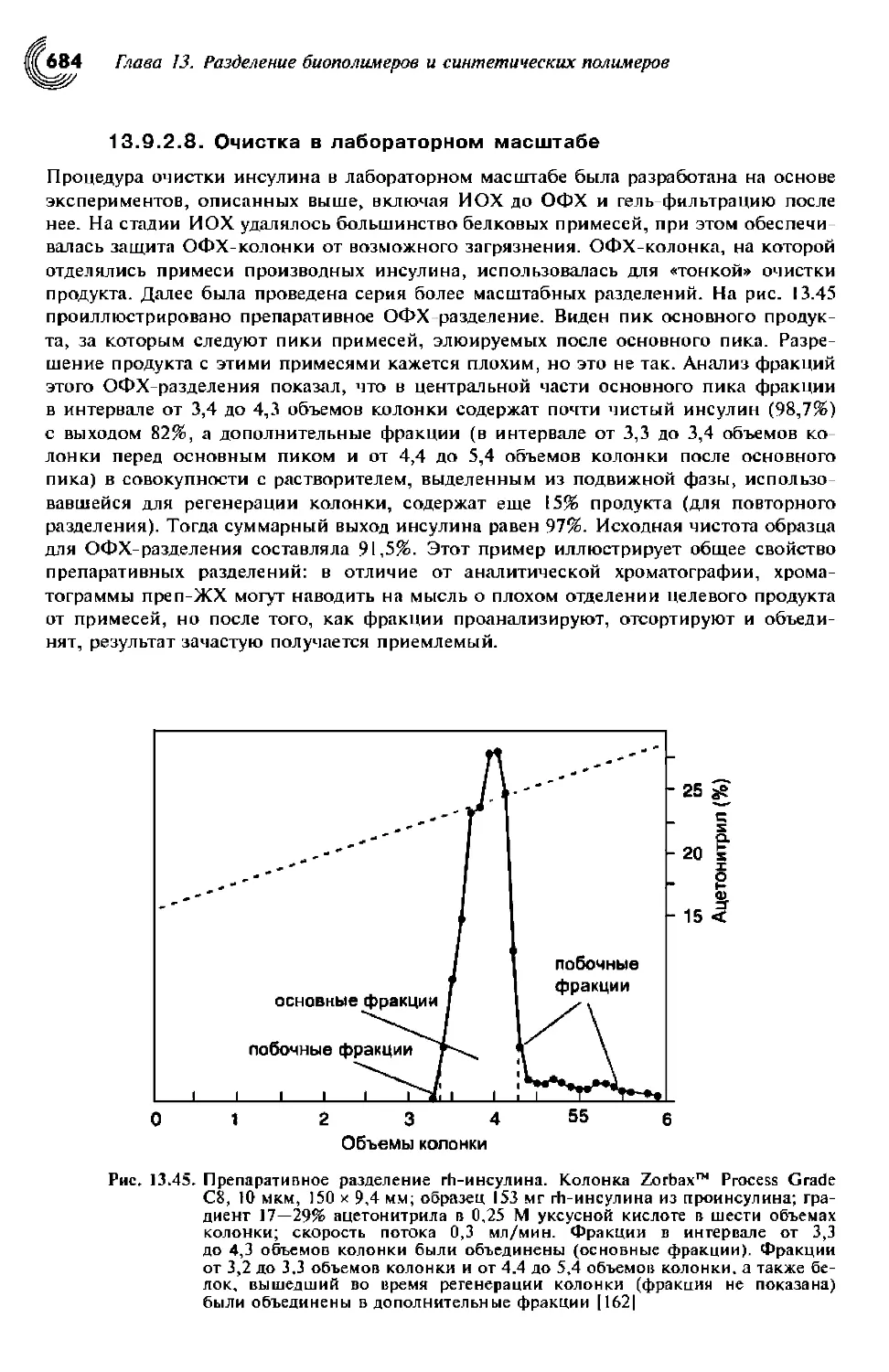
13.9.2.7. Регенерация колонки 684
13.9.2.8. Очистка в лабораторном масштабе 685
13.9.2.9. Масштабирование 685
13.9.2.10. Очистка в промышленных масштабах 686
13.9.3. Общие требования к препаративной очистке с использованием ЖХ . 687
13.10. Синтетические полимеры 688
13.10.1. Базовые понятия и принципы 688
13.10.2. Приемы анализа полимеров 691
13.10.3. Методы жидкостной хроматографии для анализа полимеров 692
13.10.3.1. Гель-проникающая хроматография (ГПХ) 692
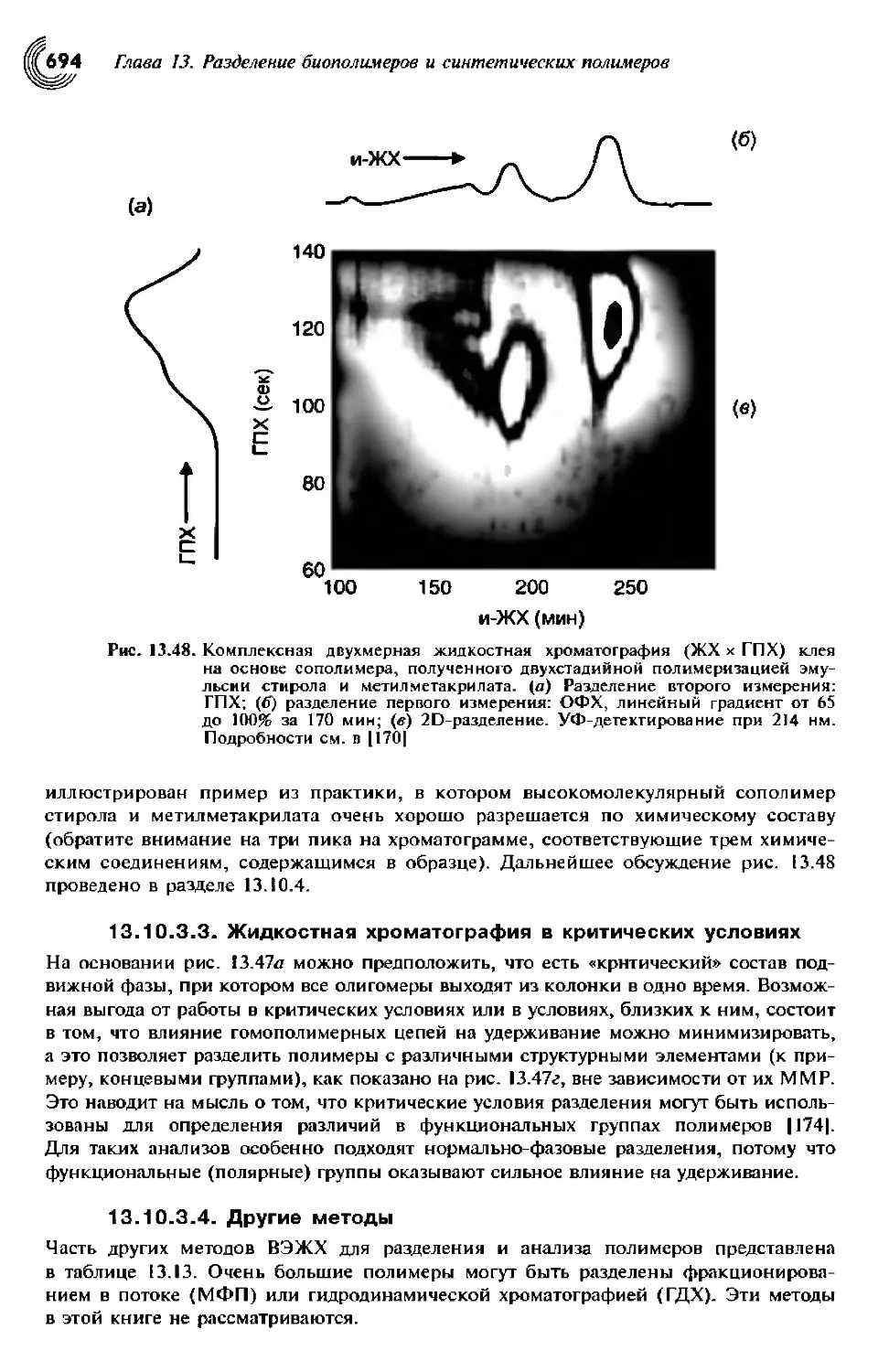
13.10.3.2. Интерактивная жидкостная хроматография 692
13.10.3.3. Жидкостная хроматография в критических условиях .... 695
13.10.3.4. Другие методы 695
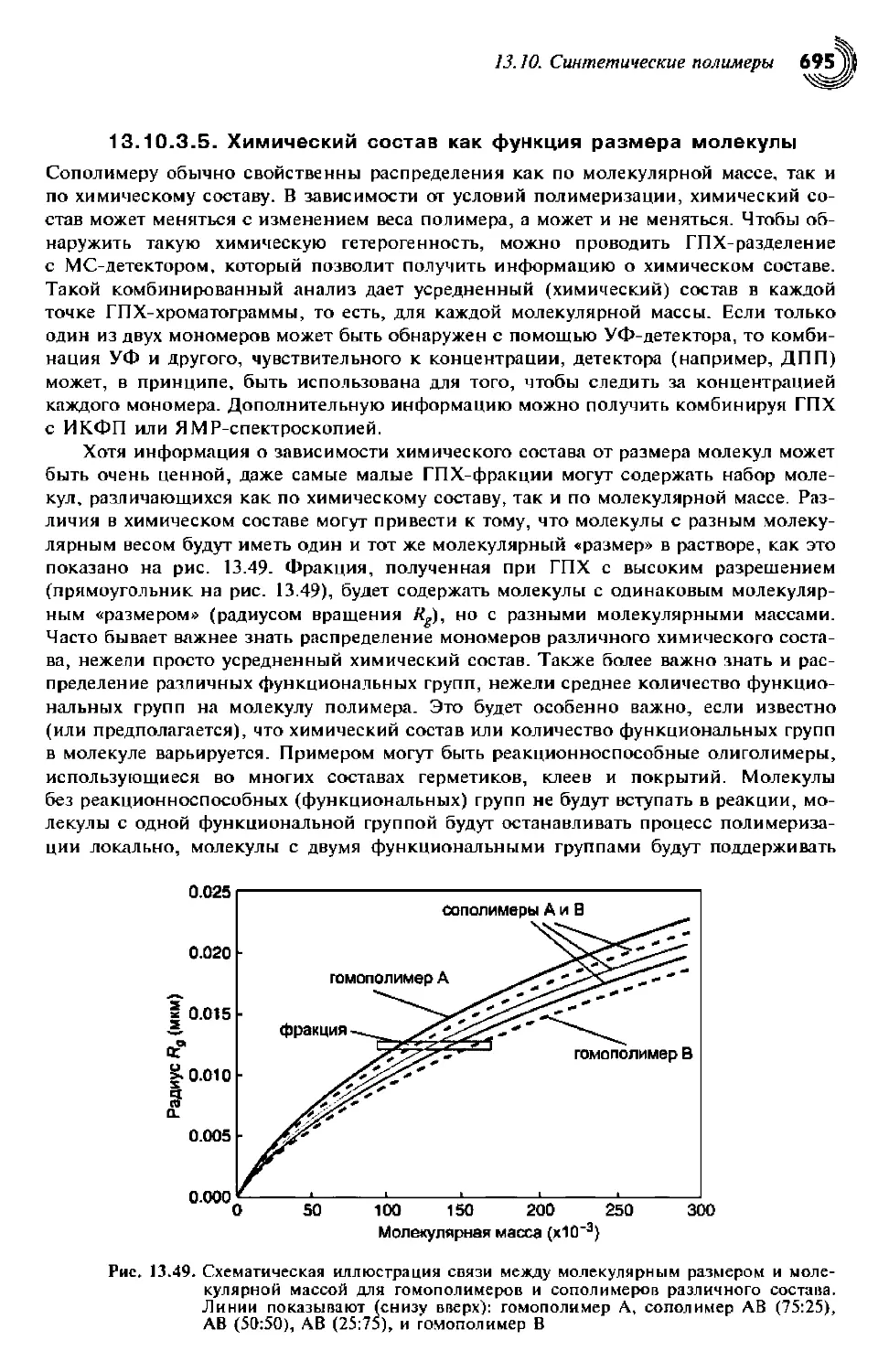
13.10.3.5. Химический состав как функция размера молекулы 696
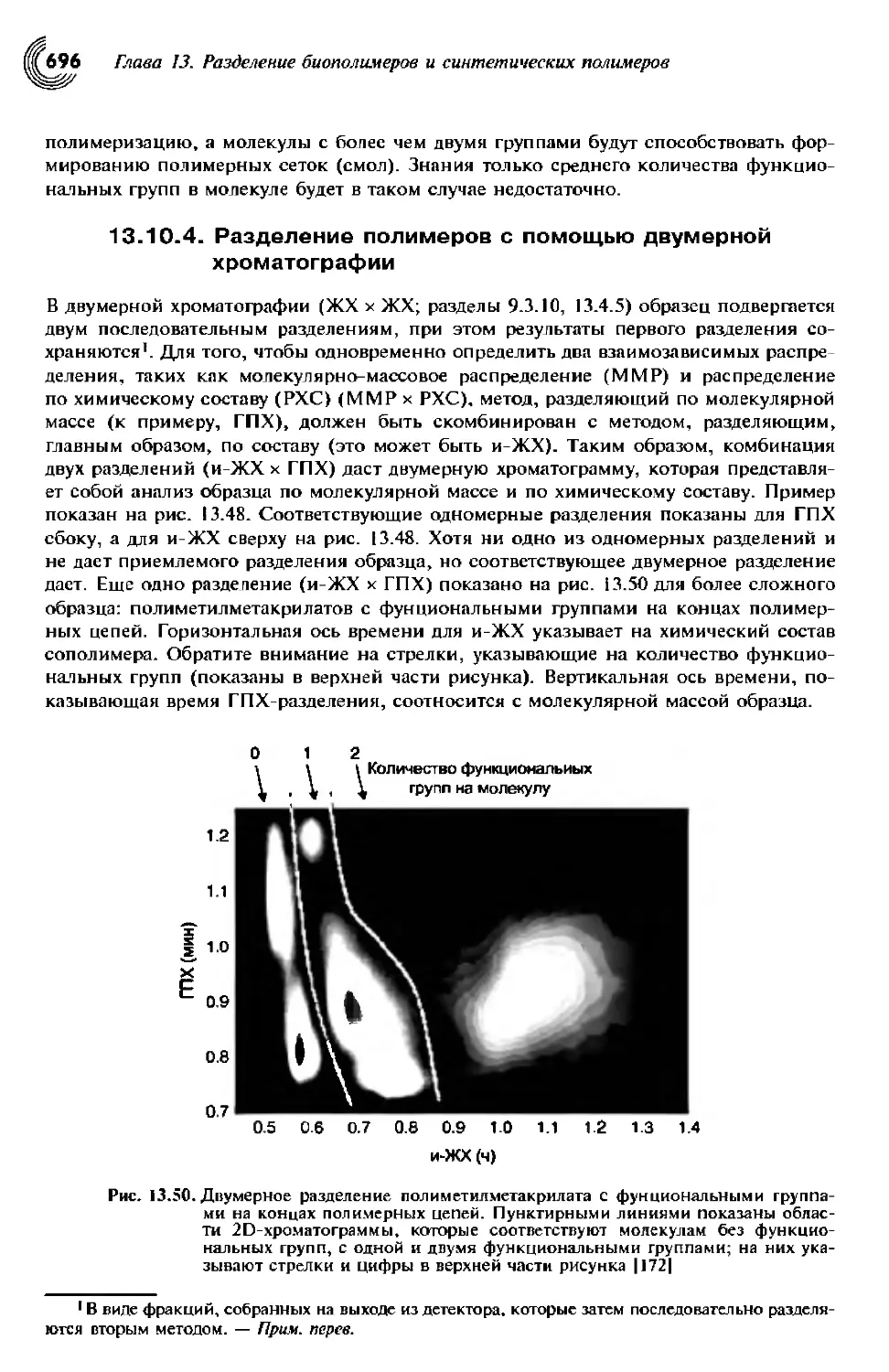
13.10.4. Разделение полимеров с помощью двумерной хроматографии .... 697
Литература 698
Глава 14. Разделение энантиомеров
(Михаель Лэммерхофер, Норберт М. Майер, Вольфганг Линднер) 703
14.1. Введение 704
14.2. Основы и определения 704
14.2.1. Изомерия и хиральность 705
14.2.2. Хиральное распознавание и разделение энантиомеров 707
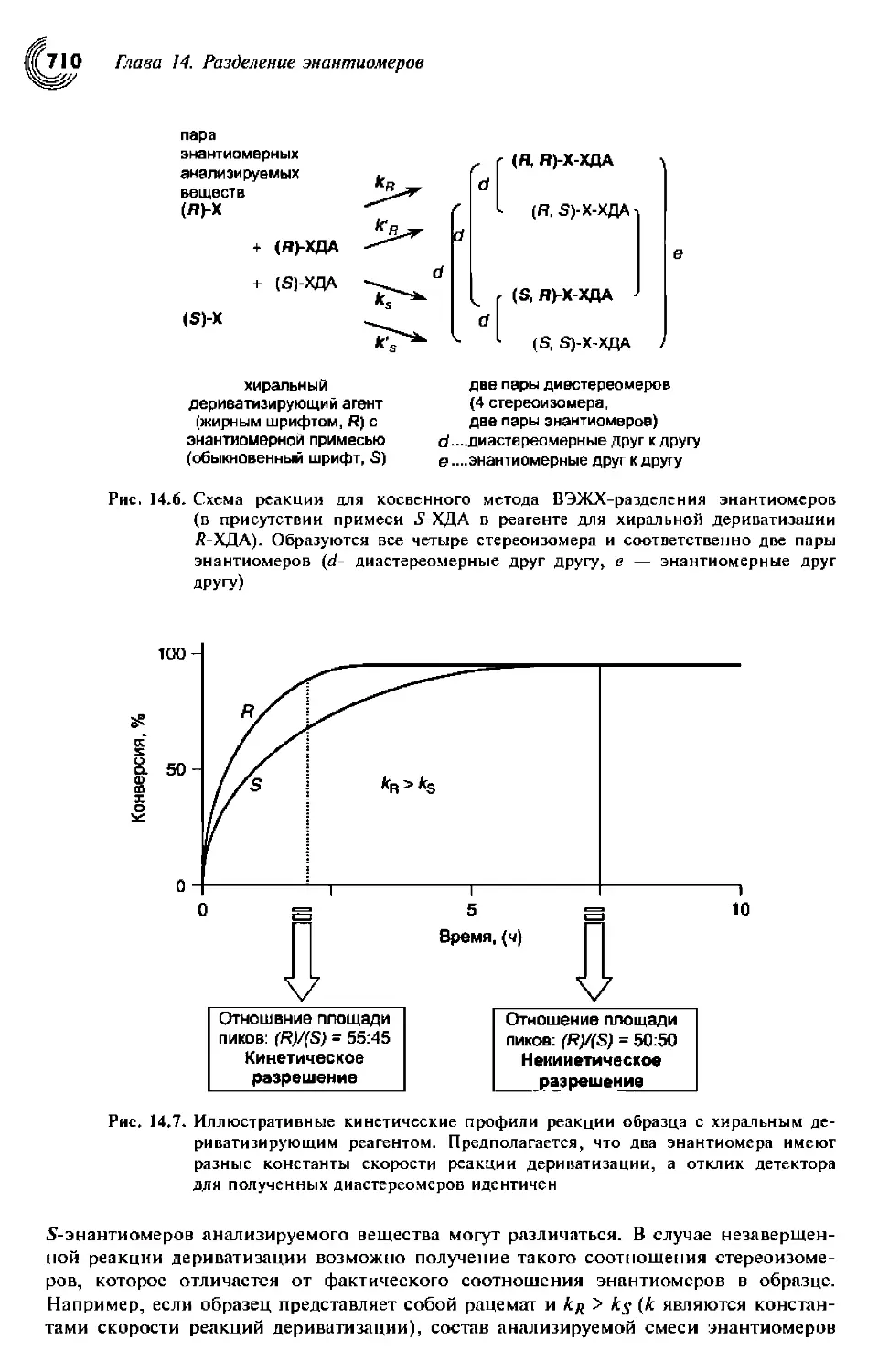
14.3. Косвенный метод 708
14.4. Прямой метод 712
14.4.1. Метод хиральных добавок к подвижной фазе (ХДПФ) или
аддитивный метод 712
14.4.2. Использование хиральной неподвижной фазы (ХНФ) 715
14.4.3. Принципы хирального распознавания 716
14.4.3.1. «Модель трехточечного присоединения» 716
14.4.3.2. Влияние подвижной фазы 718
14.5. Дисперсия и уширение задних фронтов пиков 719
Содержание 25
14.6. Хиральные неподвижные фазы и их характеристики 719
14.6.1. ХНФ на основе полисахаридов 720
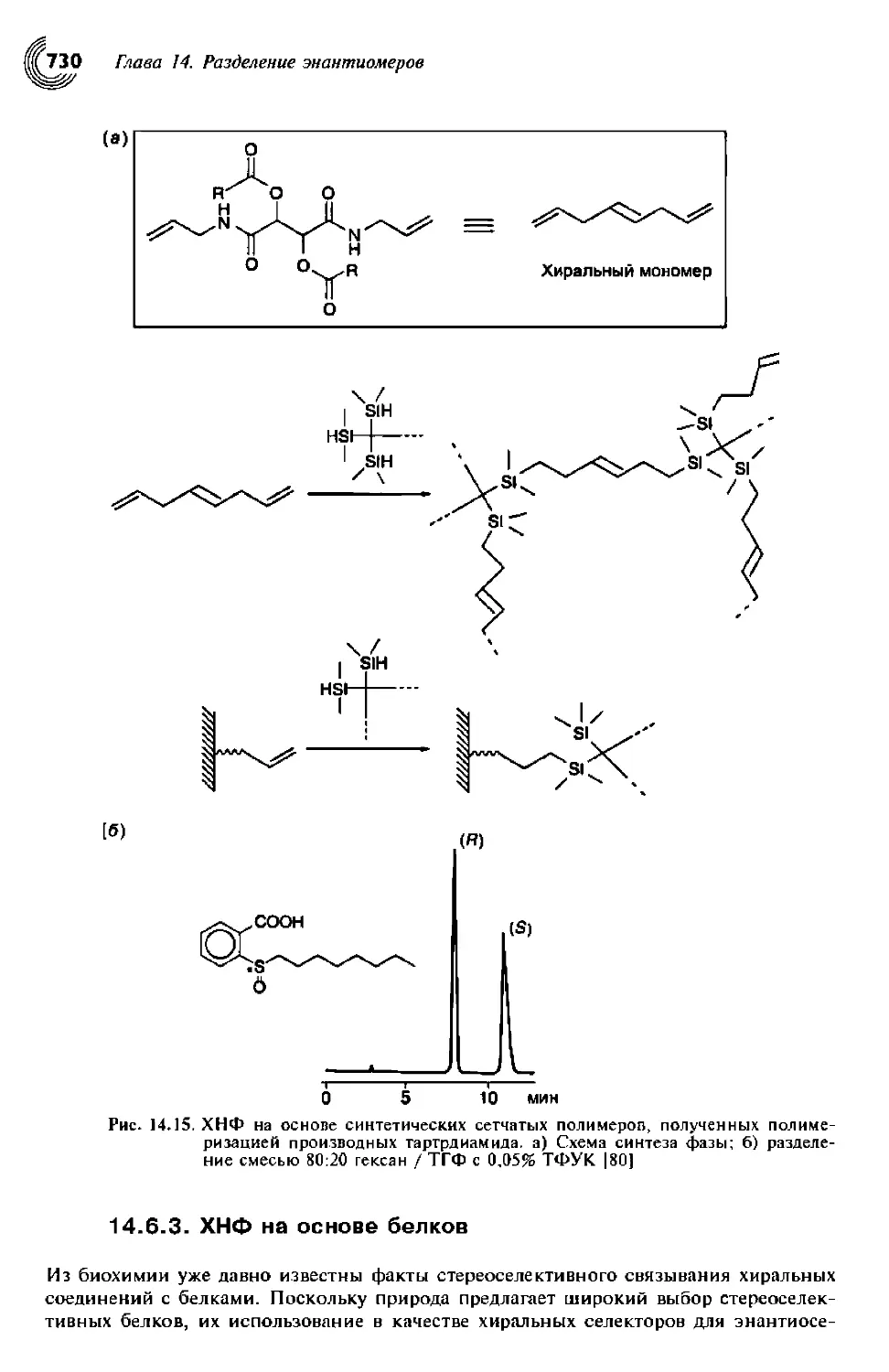
14.6.2. Синтетические полимерные ХНФ 727
14.6.3. ХНФ на основе белков 731
14.6.4. ХНФ на основе циклодекстрина 735
14.6.5. ХНФ на основе макроциклических антибиотиков 738
14.6.6. ХНФ на основе хиральных краун-эфиров 744
14.6.7. Донорно-акцепторные ХНФ 746
14.6.8. Хиральные ионообменники 749
14.6.9. Лигандообменные ХНФ. Хиральная лигандообменная
хроматография (ХЛОХ) 752
14.7. Термодинамические аспекты 754
14.7.1. Термодинамика ассоциации анализируемого вещества с селектором . 754
14.7.2. Термодинамика прямого хроматографического разделения
энантиомеров 755
14.7.3. Термодинамика гетерогенной хиральной поверхности 755
Литература 757
Глава 15. Препаративные разделения (совместно с Джеффом Коксом) 762
15.1. Введение 763
15.1.1. Перегрузка колонки и последствия этого 763
15.1.2. Масштаб разделения 764
15.1.2.1. Колонки большего диаметра 765
15.1.2.2. Оптимизированные условия для преп-ЖХ 765
15.1.2.3. Другие соображения 767
15.2. Оборудование для преп-ЖХ-разделения 767
15.2.1. Колонки 768
15.2.2. Введение образца 769
15.2.2.1. Инжекторы с петлей ввода 769
15.2.2.2. Насосный ввод 770
15.2.3. Детекторы 771
15.2.3.1. УФ-детекторы 771
15.2.3.2. Другие детекторы 771
15.2.4. Сбор фракций 772
15.2.5. Выделение продукта (удаление подвижной фазы) 773
15.3. Изократическое элюирование 775
15.3.1. Количество образца и разделение 775
15.3.1.1. Изотермы сорбции 775
15.3.1.2. Ширина пика при разделениях малых количеств образцов
в сравнении с шириной пика при разделениях больших
количеств образцов 777
15.3.2. Разделение по методу соприкасающихся пиков 777
15.3.2.1. Емкость насыщения колонки 779
15.3.2.2. Перегрузка по объему образца 780
15.3.2.3. Растворимость образца 781
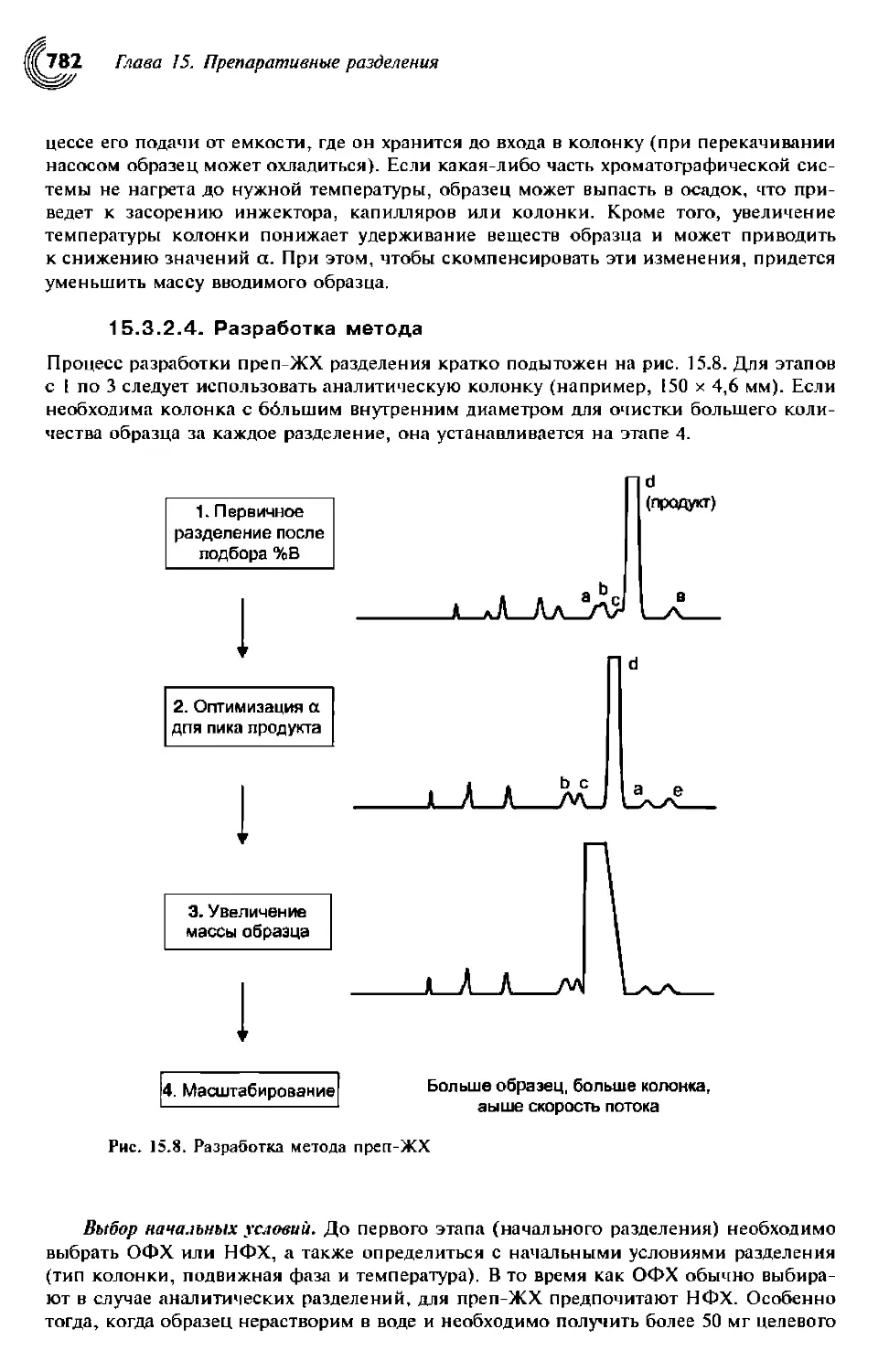
15.3.2.4. Разработка метода 783
15.3.2.5. Сбор фракций 785
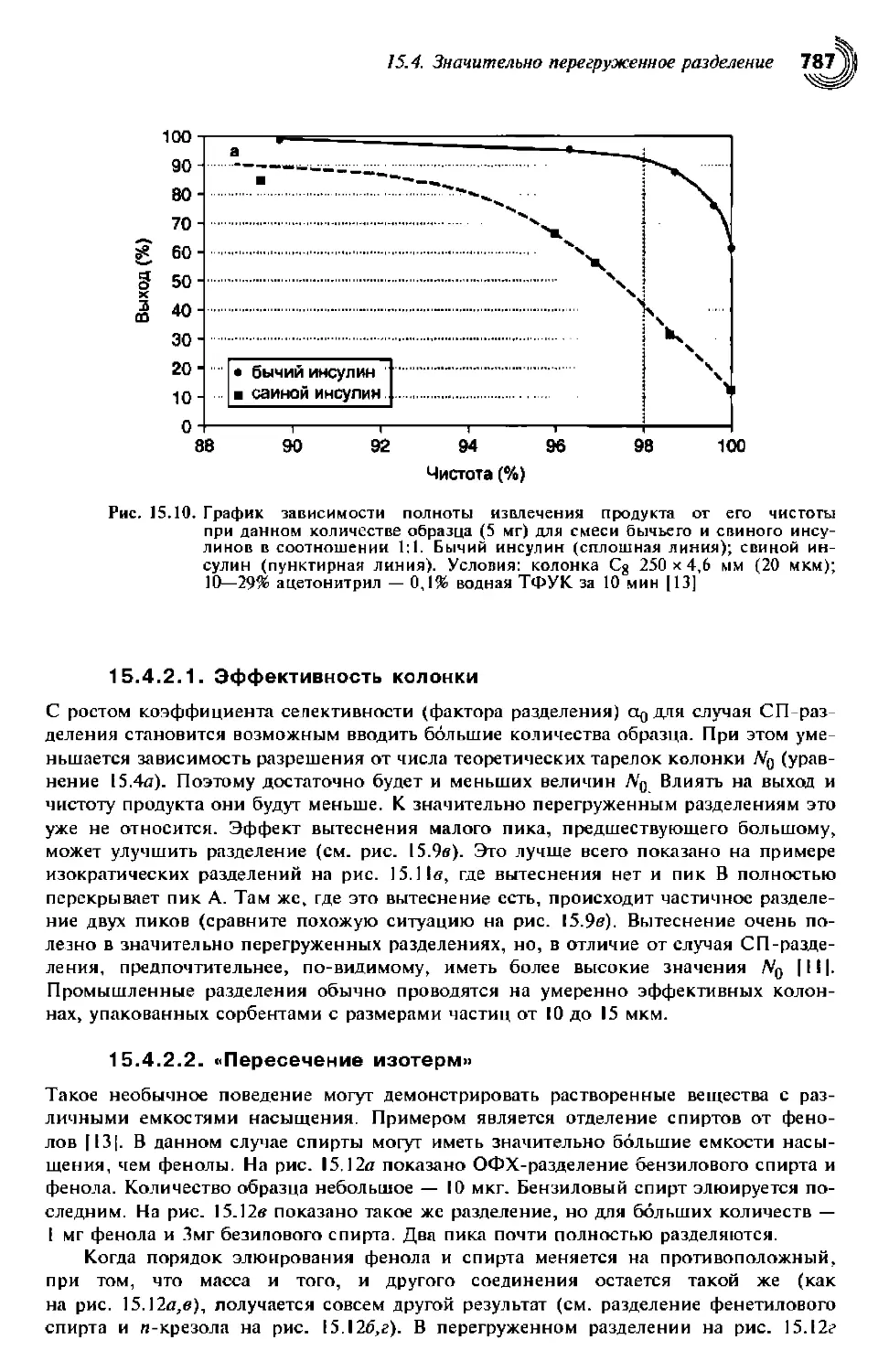
15.4. Значительно перегруженное разделение 786
15.4.1. Полнота извлечения или чистота 786
15.4.2. Разработка метода 786
26 Содержание
15.4.2.1. Эффективность колонки 788
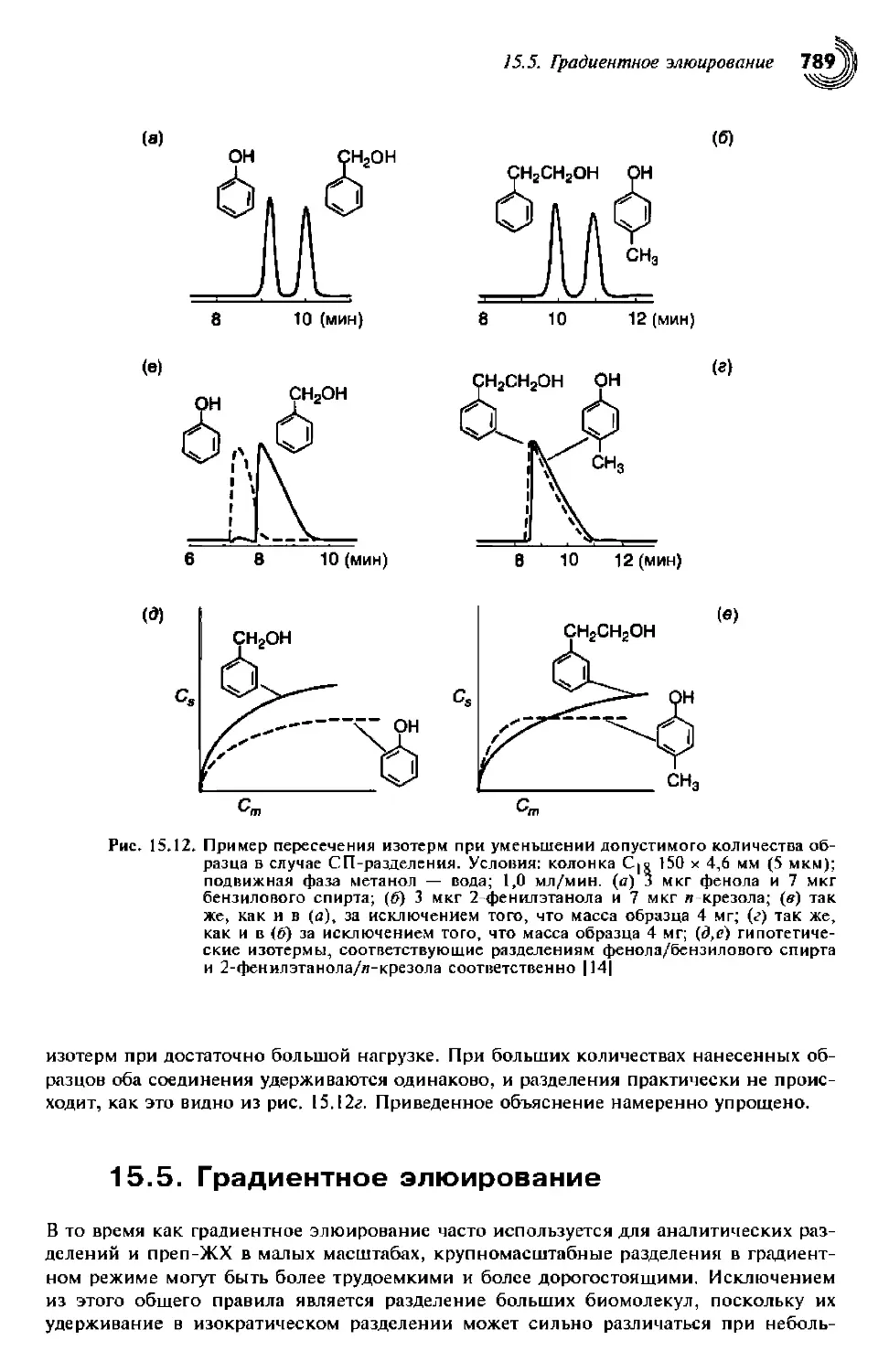
15.4.2.2. «Пересечение изотерм» 788
15.5. Градиентное элюирование 790
15.5.1. Сравнение изократической и градиентной преп-ЖХ 791
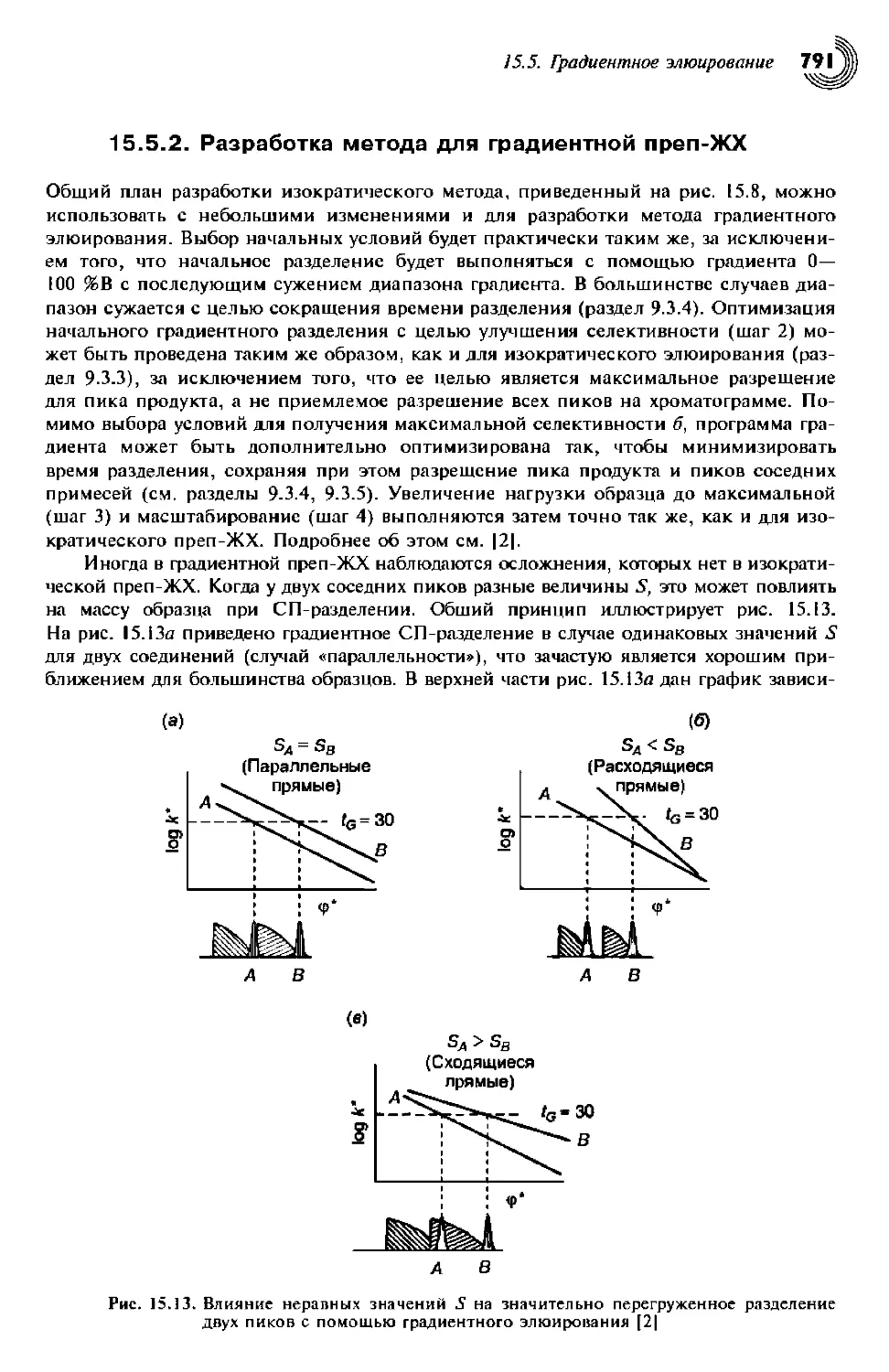
15.5.2. Разработка метода для градиентной преп-ЖХ 792
15.6. Промышленное разделение 795
Литература 796
Глава 16. Пробоподготовка (в соавторстве с Рональдом Мэджерсом) 797
16.1. Введение 799
16.2. Виды образцов 800
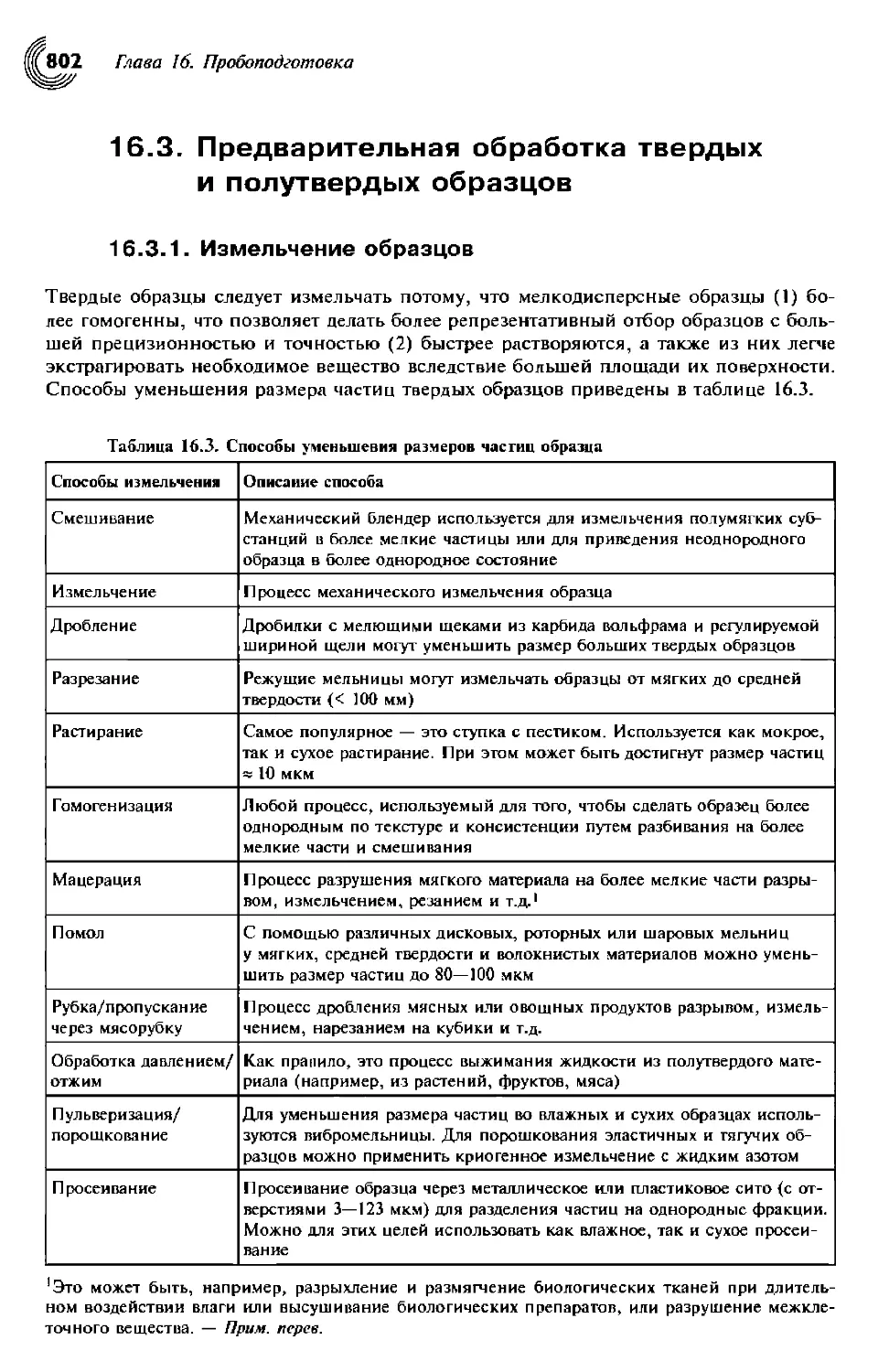
16.3. Предварительная обработка твердых и полутвердых образцов 803
16.3.1. Измельчение образцов 803
16.3.2. Высушивание образцов 804
16.3.3. Фильтрование 804
16.4. Подготовка жидких образцов 805
16.5. Экстракция в системе жидкость — жидкость 806
16.5.1. Теория 806
16.5.2. Практика 808
16.5.3. Трудности 810
16.5.3.1. Образование эмульсии 810
16.5.3.2. Адсорбция растворенного вещества 810
16.5.3.3. Связывание растворенного вещества 811
16.5.3.4. Взаимная растворимость фаз 811
16.5.4. Специальные подходы к ЖЖЭ 811
16.5.4.1. Микроэкстракция 811
16.5.4.2. Микроэкстракция методом одной капли (МОК) 811
16.5.4.3. ЖЖЭ на твердой фазе (ТФЖЖЭ) 812
16.5.4.4. Жидкостная экстракция на иммобилизованном носителе
(ИЖЭ) 812
16.6. Твердофазная экстракция (ТФЭ) 812
16.6.1. Сравнение ТФЭ и ВЭЖХ 813
16.6.2. Применение ТФЭ 814
16.6.2.1. Удаление примесей 815
16.6.2.2. Обогащение образца 815
16.6.2.3. Обессоливание 815
16.6.2.4. Другие применения 815
16.6.3. Приспособления для проведения ТФЭ 816
16.6.3.1. Картриджи 816
16.6.3.2. Диски 816
16.6.3.3. Другие приспособления для ТФЭ 817
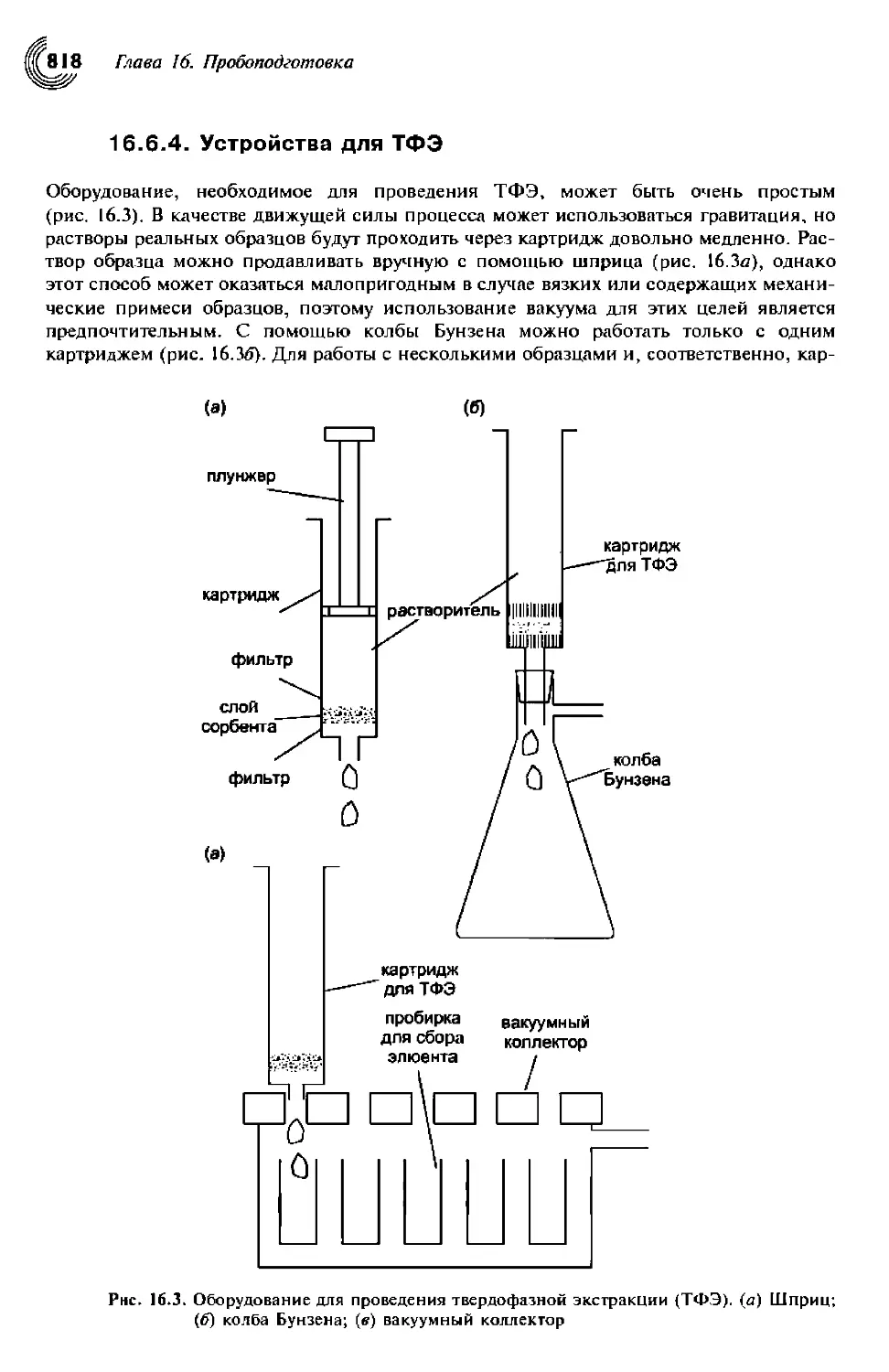
16.6.4. Устройство для ТФЭ 819
16.6.5. Разработка метода для ТФЭ 820
16.6.5.1. Этапы ТФЭ 821
16.6.5.2. Сорбенты для ТФЭ 823
16.6.6. Пример разработки ТФЭ-метода: выделение альбутерола из плазмы
крови человека 826
16.6.7. Специальные виды ТФЭ 827
16.6.7.1. Мультимодальная ТФЭ и ТФЭ со смешанными фазами. . . . 827
16.6.7.2. Адсорбенты с ограниченно доступной поверхностью (ОДП). 828
Содержание 27
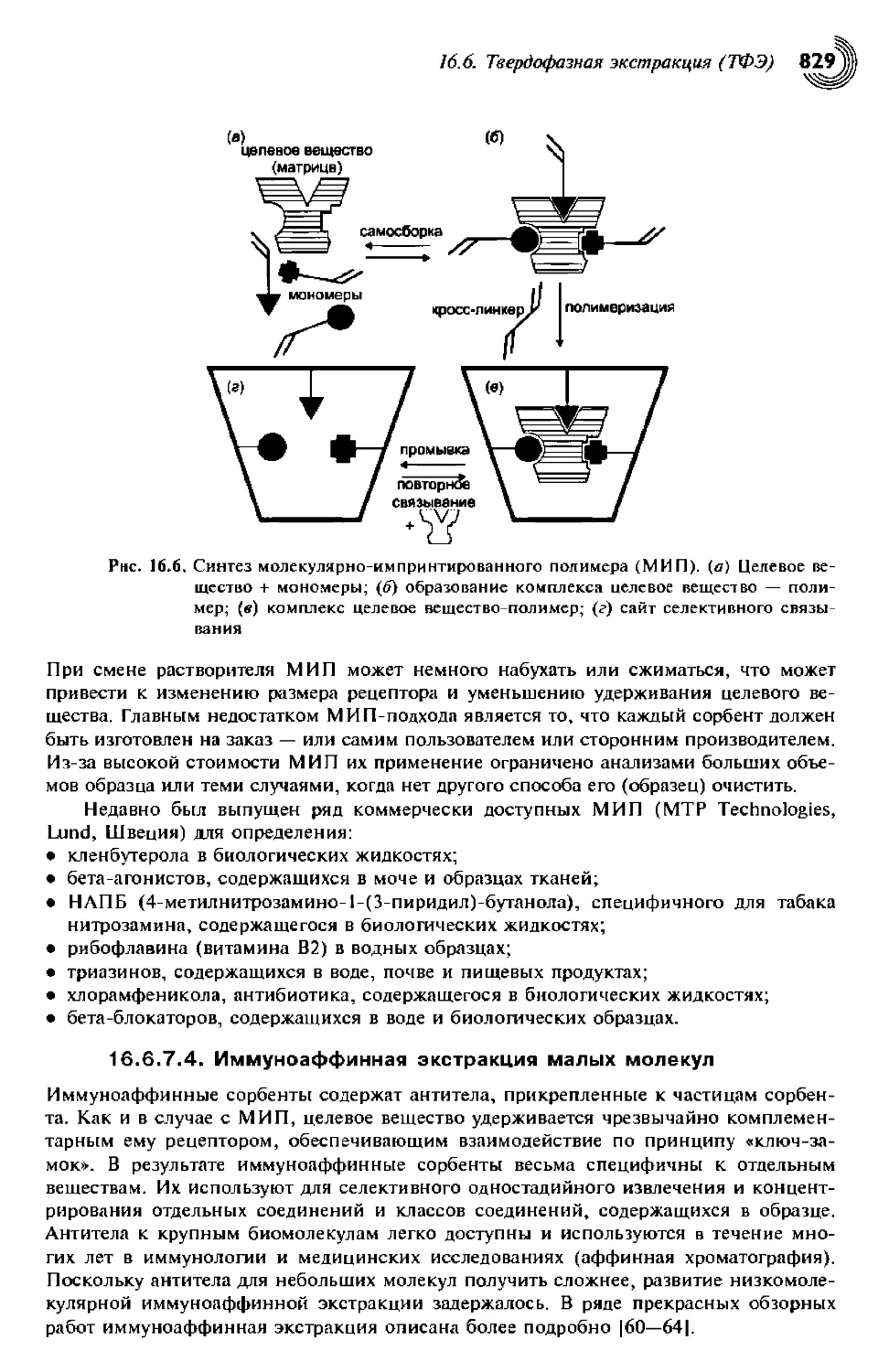
16.6.7.3. Молекулярно-импринтированные полимеры (МИП) 829
16.6.7.4. Иммуноаффинная экстракция малых молекул 830
16.6.7.5. QuEChERS и дисперсионная ТФЭ 831
16.6.7.6. Специализированные ТФЭ-картриджи для различных классов
соединений 832
16.7. Мембранные технологии в пробоподготовке 832
16.8. Методы пробоподготовки твердых образцов 833
16.8.1. Традиционные методы экстракции 834
16.8.2. Современные методы экстракции из твердых веществ 836
16.8.2.1. Экстракция в современном аппарате Сокслета 836
16.8.2.2. Сверхкритическая флюидная экстракция (СФЭ) 836
16.8.2.3. Жидкостная экстракция под давлением (ЖЭД) / ускоренная
экстракция растворителем (УСЭР) 837
16.8.2.4. Экстракция с микроволновым нагреванием (МЭ) 838
16.9. Переключение колонок 838
16.10. Пробоподготовка в биохроматографии 839
16.11. Пробоподготовка в ЖХ-МС 842
16.12. Дериватизация в ВЭЖХ 844
Литература 846
Глава 17. Неисправности и способы их устранения 850
17.1. Введение 852
17.2. Профилактика проблем 853
17.2.1. Тесты на пригодность (работоспособность) системы 853
17.2.1.1. Установочная квалификация (УК), операционная
квалификация (ОК) и эксплуатационная
квалификация (ЭК) 854
17.2.1.2. Тест на эффективность (рабочие характеристики) градиента. 854
17.2.1.3. Дополнительное тестирование системы 854
17.2.2. Профилактическое обслуживание 854
17.2.3. Тестирование пригодности системы 855
17.2.4. Хронологические записи 855
17.2.5. Полезные советы и методы 856
17.2.5.1. Удаление воздуха из насоса 856
17.2.5.2. Проверка гидравлического затвора растворителей 857
17.2.5.3. Предварительное смешивание для повышения
воспроизводимости удерживания в плавных градиентах .... 857
17.2.5.4. Очистка и обслуживание обратных клапанов 858
17.2.5.5. Обнаружение утечек 858
17.2.5.6. Устранение негерметичности соединений 859
17.2.5.7. Мытье химической посуды 859
17.2.5.8. Улучшение разделения с помощью трифторуксусной
кислоты 859
17.2.5.9. Получение высокоочищенной воды 860
17.2.5.10. Устранение остаточных примесей 860
17.3. Стратегии выявления проблем 862
17.3.1. Найти и исправить 862
17.3.2. Упрощать вместо усложнения 863
17.3.3. Менять за один раз один параметр 863
17.3.4. Устранение воспроизводимых сложностей 863
17.3.5. Замена частей 863
17.3.6. Вернуть на место 864
17.4. Общие симптомы ВЭЖХ-проблем 864
17.4.1. Утечки 865
17.4.1.1. Утечки на линии до насоса 865
17.4.1.2. Утечки в насосе 866
17.4.1.3. Утечки на стороне высокого давления 868
17.4.1.4. Утечки в автосамплере 868
17.4.1.5. Утечки в колонке 871
17.4.1.6. Утечки в детекторе 872
17.4.2. Аномальное давление 873
17.4.2.1. Слишком высокое давление 874
17.4.2.2. Слишком низкое давление 875
17.4.2.3. Нестабильность давления 877
17.4.3. Нестабильность времени удерживания 877
17.4.3.1. Проблемы, связанные со скоростью потока 878
17.4.3.2. Проблемы, связанные с размером колонки 878
17.4.3.3. Проблемы, связанные с подвижной фазой 878
17.4.3.4. Проблемы, связанные с неподвижной фазой 879
17.4.3.5. Проблемы, связанные с изменениями температуры 879
17.4.3.6. Симптомы проблем, связанных с удерживанием 880
17.4.4. Площадь пика 882
17.4.4.1. Слишком большая площадь пика 882
17.4.4.2. Слишком маленькая площадь пика 884
17.4.4.3. Изменяющаяся величина площади пика 884
17.4.5. Другие проблемы, связанные с хроматограммой 885
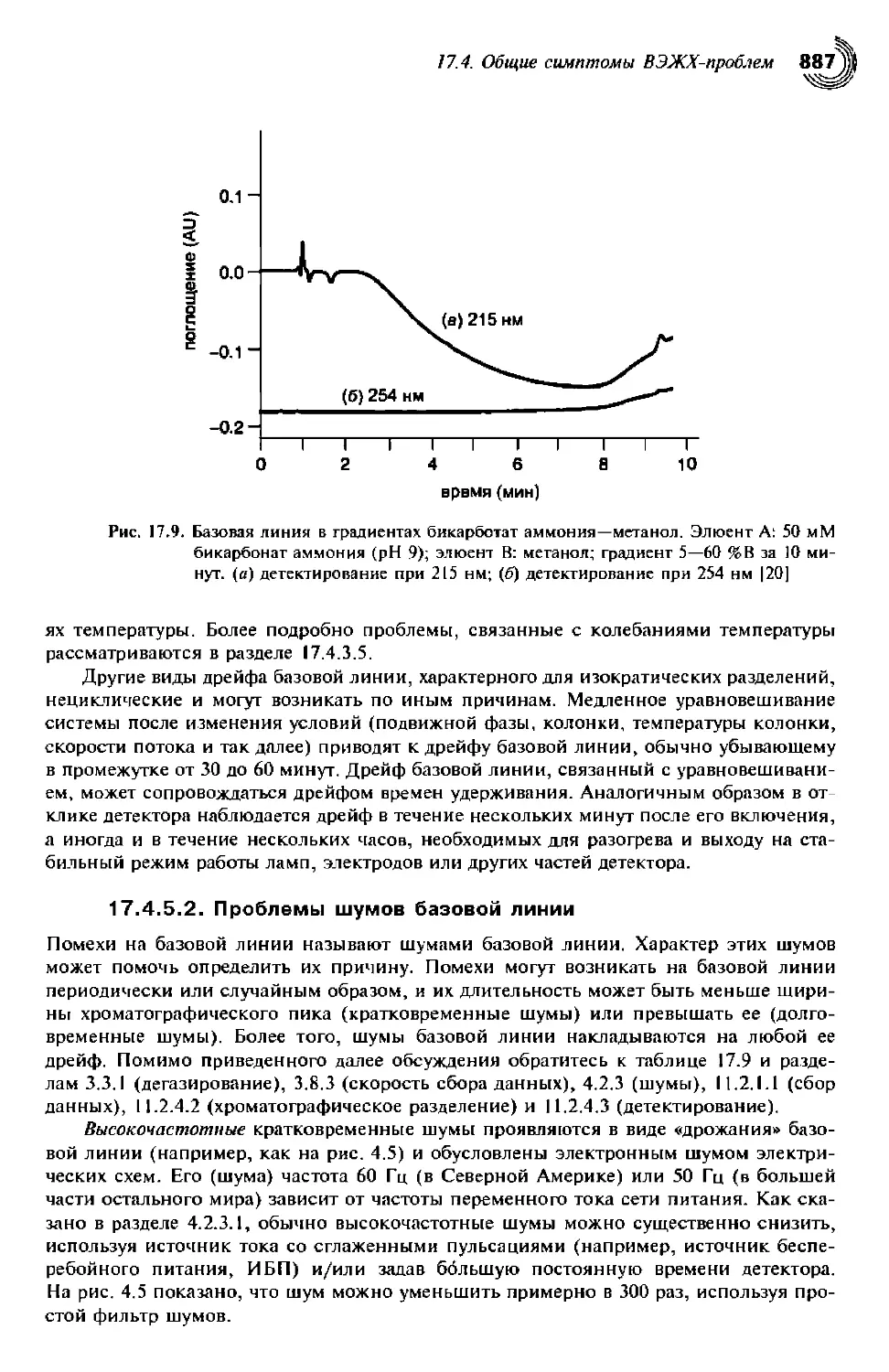
17.4.5.1. Проблемы, вызывающие дрейф базовой линии 885
17.4.5.2. Проблемы шумов базовой линии 888
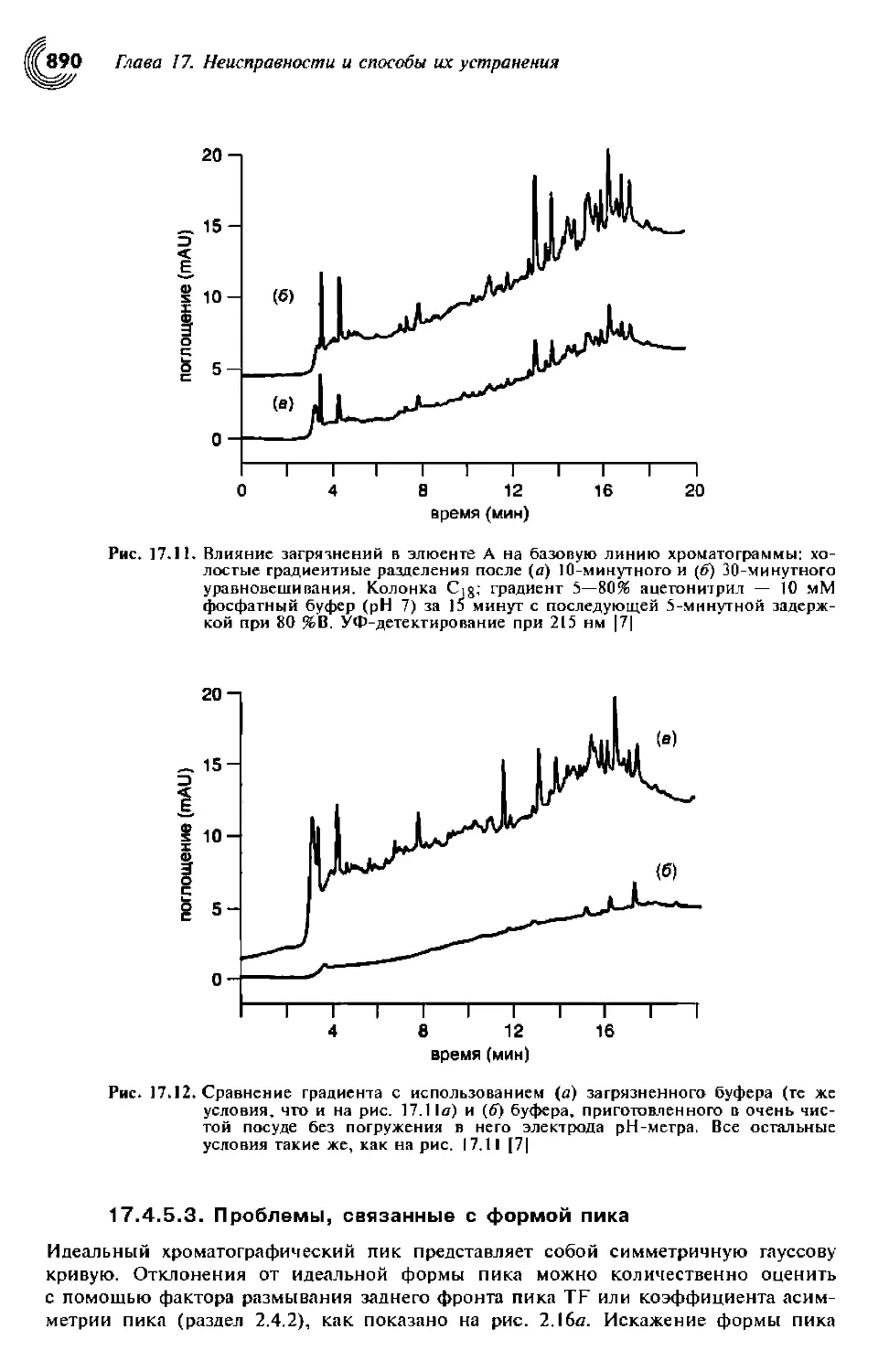
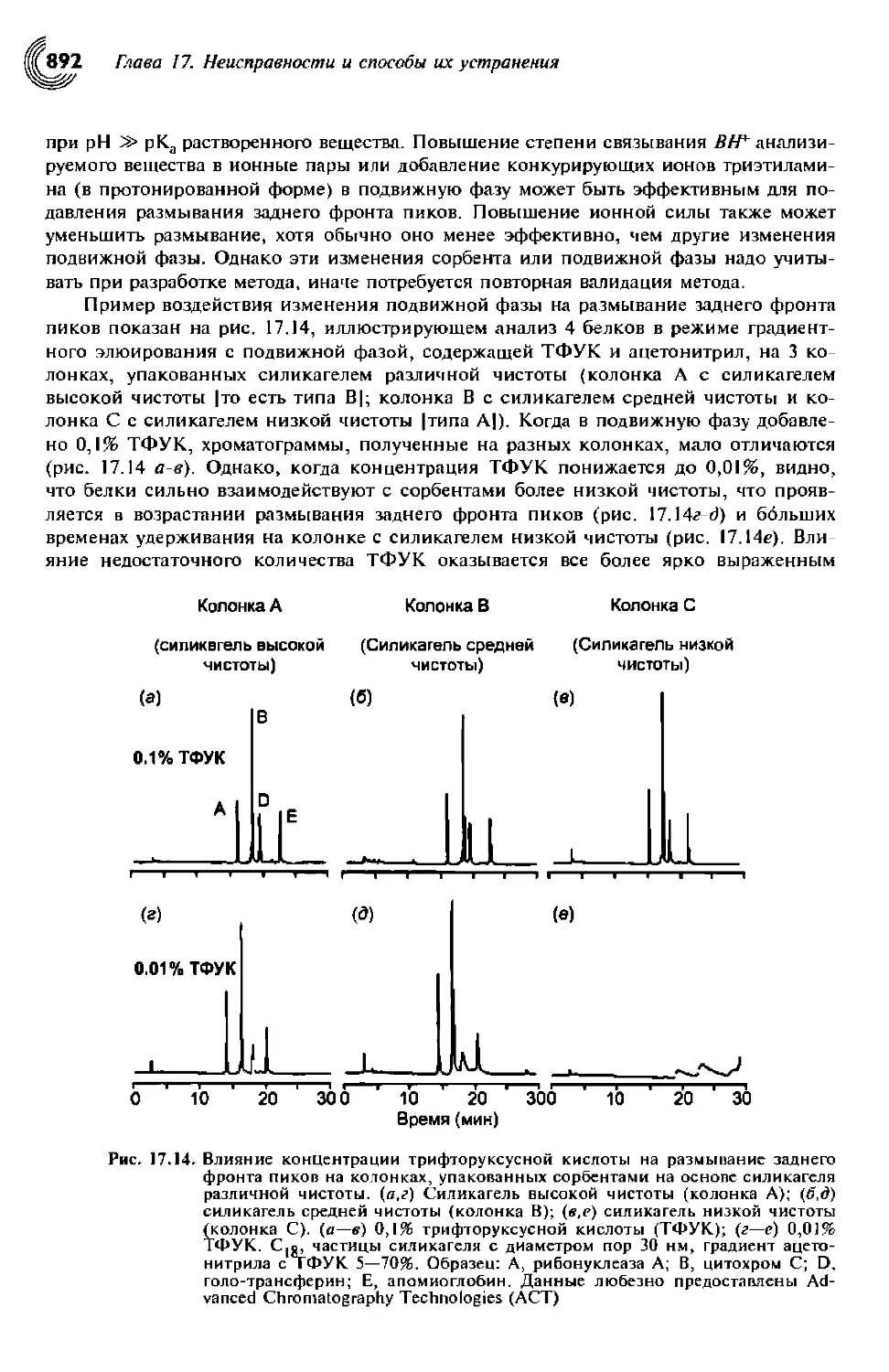
17.4.5.3. Проблемы, связанные с формой пика 891
17.4.6. Интерпретация тестов на пригодность (работоспособность) системы. 901
17.4.6.1. Интерпретация градиентного тестирования 901
17.4.6.2. Интерпретация дополнительных тестов системы 908
17.5. Таблицы по устранению неисправностей 909
Литература 920
Приложение I. Свойства растворителей, используемых в ВЭЖХ 922
1.1. Совместимость растворителя с детектором 922
1.1.1. Детектирование по поглощению в ультрафиолете 922
1.1.2. Детектирование по показателю преломления 925
1.1.3. Детектирование масс-спектрометром 925
1.2. Полярность и селективность растворителей 925
1.3. Безопасность при работе с растворителями 928
Литература 929
Приложение II. Приготовление буферных подвижных фаз 930
II. 1. Последовательность операций 930
II.2. Методики приготовления некоторых широко используемых буферов 931
Литература 932
Предметный указатель 932
ПРЕДИСЛОВИЕ РЕДАКТОРОВ ПЕРЕВОДА
Перед вами не просто книга, а титанический труд Ллойда Снайдера, Джека Кирк-
ленда и Джона Долана, посвященный ВЭЖХ. Это и учебник, и справочник, и даже
энциклопедия по всем (или почти по всем) вопросам, связанным с ВЭЖХ. На
сегодняшний день третье издание «Введения в современную жидкостную
хроматографию» — одно из самых популярных в мире справочных руководств на эту тему. Что
и говорить, перевод этой книги на русский язык задержался, но лучше поздно, чем
еще позднее.
В книге освещено огромное количество вопросов, связанных с теорией
хроматографии, современным оборудованием ВЭЖХ, методами детектирования и
устройством детекторов, подробно рассмотрены теоретические и практические аспекты
выбора неподвижных и подвижных фаз. Особое внимание уделено обращенно-фазовой,
нормально-фазовой, гель-проникающей, гидрофобной, гидрофильной и другим
видам хроматографии. Отдельные главы посвящены разделению синтетических и
природных полимеров, препаративной хроматографии, разделению энантиомеров, про-
боподготовке, типовым проблемам при работе с хроматографическим оборудованием
и, что особенно важно в современных условиях, валидации аналитических методов.
В.А. Жуковский говорил, что «переводчик в прозе — раб, а переводчик в
стихах — соперник». Коллектив переводчиков этой книги решил быть соперниками.
В тех случаях, когда оригинальный текст устарел, или необходимо было дать
дополнительные разъяснения, или у переводчиков имелось на этот счет другое мнение,
они писали примечания к переводу.
Перевод главы 13 сделан к.х.н. СВ. Шульгой-Морским, а главы 14 —
к.х.н. А.А. Новоженец. Коллектив переводчиков хотел бы поблагодарить
к.х.н. А.А. Колотвина за участие в редактировании главы 12, к.х.н. Л.Д. Аснина за
участие в редактировании главы 14 и К.Е. Маркачеву за помощь при оформлении
многочисленных рисунков и графиков.
к.т.н. М.Б. Бару
И.В. Важенина
д.х.н. СМ. Староверов
ПРЕДИСЛОВИЕ
В настоящее время высокоэффективная хроматография (ВЭЖХ) является основной
технологией химического анализа и его приложений. С ее помощью возможно
разделить, проанализировать и очистить практически любой образец. Второе издание этой
книги появилось в 1979 году и стало для десятков тысяч читателей справочником
по ВЭЖХ. Заметный успех второго издания (вкупе со значительными продажами
в первом десятилетии нынешнего века) может быть отнесен к ряду достоинств,
которые перешли и в эту книгу. Во-первых, все три издания выросли из кратких курсов,
прочитанных за последние сорок лет тремя авторами перед аудиторией, состоящей из
более чем десяти тысяч хроматографистов, работающих в промышленности,
исследовательских институтах и правительственных учреждениях. Преподавание позволяет
опробовать и оценить различные подходы к предмету изложения. Когда студенты
являются практикующими хроматографистами, особенно важен практический подход.
Во-вторых, во всех трех изданиях мы попытались объединить практические советы
(как?) с теоретической основой (почему?). И теория, и практика даны так, чтобы
читатель мог лучше понять и оценить данные в книге рекомендации. И, наконец, каждый
из трех авторов на протяжении большей части своей карьеры был активным
участником исследований в области ВЭЖХ и/или ее практического применения.
С момента подготовки второго издания в 1979 году существенно улучшились
колонки и хроматографическое оборудование. Кроме того, мы существенно
продвинулись в направлении (1) понимания ВЭЖХ-разделения, (2) нашей способности
решать проблемы, которые раньше было сложно решить, и (3) применения ВЭЖХ
для разделения новых типов образцов. В то время как во втором издании было
уделено равное внимание шести различным ВЭЖХ-методам, в третьем основное (но не
исключительное) внимание направлено на обращенно-фазовую хроматографию,
которая в настоящее время составляет около 80% всех ВЭЖХ-приложений. За
последние три десятилетия использование ВЭЖХ для работы с биологическими образцами,
для разделения энантиомеров и для очистки веществ возросло чрезвычайно и
сопровождалось более глубоким пониманием сути этих и других областей ее применения.
Продолжается совершенствование коммерчески доступных колонок. Кроме того,
разработано много новых видов колонок для конкретных приложений, для
ускорения процесса разделения и для того, чтобы повысить безотказность работы
ВЭЖХ-систем. До начала девяностых годов разработка ВЭЖХ-метода была
непредсказуемой. Зачастую требовалось несколько месяцев, чтобы добиться приемлемого
разделения образца. С того времени стало возможным существенное ускорение
разработки метода, особенно в случае использования подходящего программного
обеспечения. В то же самое время практика ВЭЖХ все больше стала проходить в
нормативно-правовом поле, что замедляет утверждение разработанного метода. Все эти
многочисленные и различные достижения в области ВЭЖХ и обусловили большую
часть изменений в настоящем издании.
Структура этой книги, хотя и похожа на структуру второго издания, существенно
переработана в свете современных исследований. В первой главе даются основы
ВЭЖХ с кратким изложением того, как использование этих знаний соотносится
с различными современными методами разделения. Кроме того, в первой главе
приведен обзор истории ВЭЖХ. В главе второй углубленно рассмотрены основы ВЭЖХ
и влияние различных экспериментальных условий на параметры разделения. Главы
третья и четвертая посвящены соответственно оборудованию и детектированию.
В 1979 году детекторы еще были «слабым звеном» в системе ВЭЖХ, но сегодня ши-
рокое использование диодно-матричных УФ-детекторов и масс-спектрометрического
детектирования, так же как и наличие ряда детекторов специального назначения,
решило эту проблему. В пятой главе описывается колонка: «сердце» всей хроматогра-
фической системы. В 1979 году у колонок существовало множество недостатков:
уширение заднего фронта пика (особенно для основных образцов), нестабильность
работы при повышенных температурах или ограничения по рН мобильной фазы,
а также нестабильность характеристик от партии к партии. Сегодня эти проблемы
встречаются гораздо реже. Мы теперь хорошо знаем, как эффективность
изменяется в зависимости от типа колонок, и это знание позволяет нам лучше их подбирать
для конкретных случаев. И, наконец, в значительной степени благодаря
усовершенствованию колонки стало возможным проведение сверхбыстрых разделений (в
которых время разделения составляет несколько минут или меньше) и более
качественное фракционирование смесей, содержащие сотни или даже тысячи компонентов.
Шестая глава, в которой рассматриваются разделения неионных образцов на
обращенных фазах, расширяет начатое во второй главе обсуждение важных
приложений ВЭЖХ. Подобное рассмотрение нормально-фазовой хроматографии (НФХ)
проводится в восьмой главе. При этом уделено специальное внимание хроматографии
гидрофильных влаимодействий (ГИХ). В седьмой главе обсуждается разделение ино-
низованных и ионизируемых образцов с помощью обращенно-фазовой, ион-парной
или ионообменной хроматографии. Разговор о градиентном элюировании образцов
низкомолекулярных веществ начинается в девятой главе для того, чтобы им
предварить обсуждение процесса разделения больших биомолекул в тринадцатой главе.
Рассмотрены и двумерные разделения, приобретающие все большее значение. В
десятой главе описано компьютерное моделирование хроматографических методов.
Другие важные общие темы освещены в главах 11 (количественный и качественный
анализ) и 12 (валидация метода).
Глава тринадцатая представляет собой введение в разделение
высокомолекулярных соединений, включая как биологические, так и синтетические полимеры.
Методы ВЭЖХ, использующиеся для этих целей, рассмотрены подробнее — обращен-
но-фазовая, ионообменная и эксклюзионная хроматография — а также связанные
с ними двумерные разделения. В четырнадцатой главе рассматривается особый
случай — разделение энантиомеров. Особый он постольку, поскольку для того, чтобы
разделить энантиомеры, требуется специально подбирать колонки и условия
для каждого образца. Этот подход не похож на те, что используются в большинстве
ВЭЖХ приложений, описанных в предыдущих главах.
Пятнадцатая глава посвящена препаративным разделениям (преп-ЖХ), в
которых на колонку вводят гораздо большие количества образца. С 1979 года в
препаративной хроматографии произошли большие изменения, мы теперь гораздо лучше
понимаем, как препаративные разделения зависят от изменения условий, что, в свою
очередь, дает возможность более эффективно и систематически разрабатывать метод
разделения. В шестнадцатой главе дается подробное введение в пробоподготовку,
важный раздел ВЭЖХ. Как и в случае с другими разделами ВЭЖХ, пробоподготовка
за последние тридцать лет претерпела существенные изменения, что позволило ее
превратить в рутинную процедуру, служащую дополнением к методу разделения.
Наконец, в семнадцатой главе рассматриваются возможные в ВЭЖХ проблемы и
неисправности. Несмотря на все наши достижения в усовершенствовании оборудования,
колонок, материалов и методов, гарантировать бесперебойную ВЭЖХ по-прежнему
невозможно. К счастью, мы стали более осведомленными, наши знания в большей
степени систематизированы и, как следствие, наши возможности предвидеть,
диагностировать и решать проблемы, связанные с ВЭЖХ, увеличились. Один из трех
авторов этой книги (Джон Долан) приложил большие усилия именно в этой области.
Разные читатели будут пользоваться этой книгой для решения разных задач. Тот,
у которого уже есть опыт, возможно, захочет выборочно изучить нужные ему темы
или станет искать ответы на конкретные вопросы. Таким читателям лучше начать
знакомство с книгой с изучения предметного указателя. Начинающие могли бы
сначала просмотреть главы с первой по седьмую, а затем с девятой по десятую: в них
наибольшее внимание уделено обращенно-фазовой ВЭЖХ. Эта последовательность
глав представляет собой в общих чертах ядро краткого курса ВЭЖХ, разработанного
авторами. После такого введения читатель может перейти к главам, которые
представляют для него особый интерес. Другие читатели, возможно, захотят начать
чтение с конкретных тем, перечисленных в оглавлении в начале книги, и в оглавлениях,
помещенных перед каждой главой. Книга так построена, что возможны разные
варианты ее прочтения.
Третье издание насыщено перекрестными ссылками с тем, чтобы позволить
читателю следовать за интересующей его темой или прояснить вопросы, возникающие
по ходу чтения. Поскольку большое количество перекрестных ссылок может
отвлекать, то зачастую рекомендуется их просто игнорировать (или откладывать, что
называется, на потом), чтобы не прыгать из одной главы в другую. Некоторые главы
содержат разделы, в которых детально рассматриваются те или иные вопросы. Это может
быть несколько в стороне от непосредственного интереса читателя, и потому такие
разделы выделены курсивом, чтобы читатель мог их пропустить. Кроме того, мы свели
определения всех символов, встречающихся в книге, в глоссарий и составили
всеобъемлющий предметный указатель. Наконец, следует обратить внимание на «наиболее
оптимальные методики» в предметном указателе, в которых объединены различные
рекомендации как для разработки метода, так и для рутинных методик.
Мы высоко ценим вклад, который внесли в создание настоящей книги восемь
наших коллег: Петер Шонмэйкерс (разделы 9.3.10, 13.10), Майк Шварц (глава 12),
Тим Вэр (разделы 13.1—13.8), Карл Сканделла (раздел 13.9), Вольфганг Линднер,
Михаель Лэммерхофер и Норберт Майер (глава 14), Джефф Кокс (глава 15) и Рон
Мэйджорс (глава 16). Они сотрудничают в следующих организациях:
Петер Шонмэйкерс University of Amsterdam
Майк Шварц Synomics Pharma
Тим Вэр BioRad Corp.
Карл Сканделла Consulting (4404 91st Avenue NE Bellevue, WA 98004)
Вольфганг Линднер, University of Vienna
Михаель Лэммерхофер
и Норберт Майер
Джефф Кокс Chiral Technologies
Рон Мэйджерс Agilent Technologies
Мы признательны рецензентам различных частей этой книги: Питеру Карру,
Тому Чэмберсу, Джеффу Коксу, Рою Экстену, Джону Фетцеру, Дику Хенри,
Владимиру Иоффе, Павлу Жандере, Питеру Джонсону, Тому Жупиль, Рону Мэйджорсу,
Дэну Марчанду, Дэвиду МакКэлли, Имре Мольнару, Тому Моурей, Уве Нойе, Рави
Равичандрану, Карен Руссо, Карлу Сканделле, Петеру Шонмэйкерсу и Лорен Рис-
лей. Тем не менее за все ошибки и недочеты в книге авторы берут ответственность
на себя.
Ллойд Р. Снайдер
Дж. Дж. (Джек) Киркленд
Джон В. Долан
Оринда. Калифорния
Уилмингтон, Делавэр
Амити, Орегон
СЛОВАРЬ СИМВОЛОВ И ПРИНЯТЫХ
СОКРАЩЕНИЙ
Этот раздел состоит из двух частей: основные символы и второстепенные символы.
Большинство обозначений, представляющих интерес, включены в список основных
символов. Уравнения, служащие определением данного символа, указаны рядом с
этим символом. Например, указание «уравнение (2.18)» отсылает к уравнению (2.18)
главы 2. Приведены единицы измерения для всех обозначений, используемых в
данной книге.
Основные символы и принятые сокращения
А(%А)
ACN
В(%В)
CV
Q> ci8
dc
d.
F
H
к
к*
L
М
МеОН
N
пс
Р
PC
рКа
«слабый» компонент бинарной подвижной фазы (А/В) и его объемный процент;
в обращенно-фазовой хроматографии растворитель А представляет собой воду или
водный буфер; также силикагель типа А (старый, более кислотный силикагель)
ацетонитрил
«сильный» компонент (и его объемный процент) в бинарной подвижной фазе
(А/В); в обращенно-фазовой хроматографии растворитель В представляет собой
органический растворитель, такой как ацетонитрил; также силикагель типа В
(современный, менее кислотный силикагель, раздел 5.2.2.2)
коэффициент вариации (эквивалентный относительному стандартному
отклонению, ОСО), раздел 4.2.3.2
обозначение обращенно-фазового лиганда, показывает длину алкильных цепочек,
привитых к поверхности сорбента
внутренний диаметр колонки (мм)
диаметр частиц сорбента (мкм)
объемная скорость подвижной фазы, скорость потока (мл/мин)
высота теоретической тарелки (равная L/N) раздел 2.4; см. также «менее часто
используемые символы»
фактор удерживания (то же самое, что и коэффициент емкости к');
равен (tit/to) - 1, раздел 2.3.1
градиентный фактор удерживания; уравнение (9.5), раздел 9.2
длина колонки (мм)
молекулярная масса (Да)
метанол
число теоретических тарелок, уравнение (2.9), раздел 2.4
«эквивалентная» пиковая емкость, обычно именуемая «условной» или «пиковой
емкостью образца» пс = PC (tz- <л)/{в^ раздел 9.3.9
перепад давления на колонке (psi); 1 бар или 1 атм = 14,7 psi; 1 МПа = 10 бар =
= 147 psi; также коэффициент распределения вещества между различными
растворителями (partition coefficient), раздел 6.2.2
пиковая емкость; уравнение (2.30), рис. 2.26а (изократика); уравнение (9.20),
рис. 9.20 (градиент)
логарифм константы ионизации кислоты или основания (-log^T„); уравнения (7.2),
(7.2а), раздел 7.2
Словарь символов и принятых сокращений
RF
Rs
s
Т
tD
TF
to
to
tR
uv
Vc
Vd
V
r m
Vr
W
^max
Щ
wx
a
Дф
£
£»
Ф
Ф*
V
Л
АХИМ
BAX
ВПМС
ГИХ
ГИХЭО
ГПХ
относительное перемещение зон (пятен) вещества в ТСХ; уравнение (8.6), рис. 8.8,
раздел 8.2.3
разрешение; уравнение (2.23), раздел 2.5
наклон прямой в координатах log£,cp (dlogfc/c^); уравнение (2.26), раздел 2.5.1
температура (°С)
время задержки (мин); эквивалентно Vp/F, рис. 3.26, раздел 3.10.1.2
фактор размывания заднего фронта пика (tailing factor), рис. 2.16а, раздел 2.4.2
время градиента (мин)
«мертвое» время колонки (мин); также время удерживания неудерживаемого
анализируемого вещества; эквивалентно Vm/F; уравнения (2.4а), (2.7), раздел 2.3.1
время удерживания (мин); уравнение (2.5), раздел 2.3.1
поглощение в ультрафиолетовой области спектра
объем колонки (мл)
приборный объем задержки, Кд = tpF, раздел 9.2.2.4
«мертвый» объем колонки; объем подвижной фазы внутри колонки (мл);
уравнение (2.7а), раздел 2.3.1
объем удерживания анализируемого вещества (мл); равен tgF
ширина пика при основании; рис. 2.10, раздел 2.4
максимальная масса образца, которую можно нанести на колонку без ее
перегрузки, раздел 2.6.3
емкость насыщения колонки (г), раздел 15.3.1.1
масса наносимого вещества (г), раздел 15.3.1.2
коэффициент селективности (фактор разделения); уравнение (2.24а), раздел 2.5
изменение ф в течение градиента; рис. 9.2, раздел 9.1.2
элюирующая сила подвижной фазы в НФХ; уравнения(8.2), (8.5); также
диэлектрическая постоянная
значение г (в НФХ) для чистого растворителя
объемная доля элюента В (равна 0,01 х%В)
значение ф в градиентной хроматографии, когда зона вещества достигает середины
колонки
приведенная скорость; уравнение (2.18а), раздел 2.4.1.1
вязкость подвижной фазы (сП)
аффинная хроматография на сорбентах с иммобилизованными металлами (immobi-
lized-metal affinity chromatography, IMAC) или металл-аффинная хроматография,
раздел 13.4.2.5
высокоэффективная анионообменная хроматография (high-performance anion-
exchange chromatography, HPAE), раздел 13.6.3
времяпролетный масс-спектрометр ( timeofflight (TOF) MS-detector), раздел 4.14.3
хроматография гидрофильных взаимодействий (hydrophilic interaction
chromatography, HILIC) раздел 8.6
хроматография гидрофильных взаимодействий с электростатическим
отталкиванием (electrostatic-repulsion hydrophilic-interaction chromatography, ERLIC),
раздел 13.4.4.4
гель-проникающая хроматография (gel-permiation chromatography, GPC),
раздел 13.10.3.1
Основные символы и принятые сокращения 35
ГФХ
ДПП
жх
жх-мс
ИАД
идс
и-ЖХ
ИКФП
иох
ИПР
ипх
КФГ
лдс
мжх
мс/мс-
детектор
НОФХ
нпд
нпко
НФХ
ок
осо
ОФХ
ПАВ
пл
пп
преп-ЖХ
ПС-ДВБ
СКО
С-П-раз-
деление
ТБА
типа А
типа В
гель-фильтрационная хроматография (gel-filtration chromatography) или эксклю-
зионная хроматография ЭХ, раздел 13.8
детектор показателя преломления или рефрактометр (refractive index, RI detector),
раздел 4.11
жидкостная хроматография
жидкостная хроматография с масс-спектрометрией (liquid chromatography—mass
spectrometry, LC-MS), раздел 4.14
импульсное амперометрическое детектирование (pulsed-amperometric detection,
PAD), раздел 13.6.3
испарительный детектор светорассеяния (evaporative light-scattering detector, ELSD),
раздел 4.12.1
интерактивная жидкостная хроматография (interactive LC), раздел 13.10.3.2
инфракрасная спектроскопия с Фурье-преобразованием (Fourier transform infrared
spectroscopy, FTIR) или соответствующий спектрометр; раздел 4.15.1
ионообменная хроматография (ion-exchange chromatography, IEC), раздел 7.5
ион-парный реагент
ион-парная хроматография (ion-pair chromatography, IPC), раздел 7.4
клапаны, формирующие градиент (для систем ВЭЖХ со смешиванием при низком
давлении), раздел 3.10.1.2
лазерный детектор светорассеяния (laser light-scattering detector, LLSD), раздел 4.12.3
многомерная жидкостная хроматография (multidimensional liquid chromatography,
MDLC), раздел 13.4.5, и 20-ЖХ для двумерного разделения, раздел 9.3.10
детектор с тандемной масс-спектрометрией (triple-quadrupole mass spectrometer),
раздел 4.12.2, или с другими сочетаниями нескольких МС-анализаторов
неводная обращенно-фазовая хроматография (nonaqueous RPC, NARP), раздел 6.5
нижний предел детектирования, раздел 4.2.4; иногда называется пределом
обнаружения, ПО, раздел 11.2.5
нижний предел количественного определения, раздел 4.2.4, или предел
количественного определения, ПКО, раздел 11.2.5
нормально-фазовая хроматография (normal-phase chromatography, NPC), глава 8
операционная квалификация (operational qualification) — тестирование новой
системы ВЭЖХ на соответствие техническим характеристикам, указанным
производителем, раздел 3.10.1.1
относительное стандартное отклонение или СО (relative standard deviation, RSD),
что эквивалентно относительному среднеквадратичному отклонению (СКО)
обращенно-фазовая хроматография (reversed-phase chromatography, RPC), глава 6
поверхностно-активные вещества
поляриметр (polarimeter, PL), раздел 4.10
показатель преломления (refractive index, RI)
препаративная жидкостная хроматография, глава 15
полистирол-дивинилбензол (PS-DVB)
относительное среднеквадратичное отклонение, эквивалентно ОСО
разделение соприкасающихся пиков (touching-peak separation), раздел 15.1.2
тетрабутиламин (ТВА)
старый, более кислотный силикагель, раздел 5.2.2.2
новый, менее кислотный силикагель, раздел 5.2.2.2
Словарь символов и принятых сокращений
ТФУК
ТФЭ
ТЭА
УВЭЖХ
УК
УФ
хгв
хлох
ХНФ
эк
эх
ЭХ-детек-
тор
трифторуксусная кислота (TFA)
твердофазная экстракция (solid-phase extraction, SPE), раздел 16.6
триэтиламин (TEA)
ультравысокоэффективная жидкостная хроматография (ultra-high-pressure liquid
chromatography, U-HPLC)
установочная квалификация (installation qualification) — тестирование новой
системы ВЭЖХ на соответствие установки системы условиям, указанным
производителем, раздел 3.10.1.1
ультрафиолетовый (область спектра, детектирование по поглощению в этой
области и т.д.)
хроматография гидрофобных взаимодействий (hydrophobic interaction
chromatography, HIC), раздел 13.4.3
хиральная лигандообменная хроматография (chiral ligand-exchange chromatography,
CLEC), раздел 14.6.9
хиральная неподвижная фаза, глава 14
эксплуатационная квалификация (performance qualification) — разработанная
пользователем совокупность тестов для проверки работы системы ВЭЖХ,
раздел 3.10.1.1
эксклюзионная хроматография (size-exclusion chromatography, SEC) или
гель-фильтрационная хроматография, ГФХ, раздел 13.8
электрохимический (амперометрический) детектор (electrochemical, EC (amperomet-
ric) detector), раздел 4.6
Менее часто используемые (или менее
общеупотребительные) символы и аббревиатуры
А
А
AAPS
АОАС
As
AU
Ь
В
с
с
cGMP
Cs
ю-гэ
Ю-ЖХ
поглощение
кислотность водородных связей сорбента колонки; уравнение (5.3), раздел 5.4.1.1
Американская ассоциация ученых-фармацевтов (American Society of Pharmaceutical
Scientists), глава 12
Ассоциация химиков-аналитиков, состоящих на государственной службе
(Association of Official Analytical Chemists), глава 12
коэффициент/фактор асимметрии пика (peak asymmetry factor), рис. 2.16а,
раздел 2.4.2
единицы абсорбции (детектирование по УФ); по-русски е.п. (единицы
поглощения), раздел 4.4
параметр крутизны градиента; уравнение (9.4), раздел 9.1.3.1
основность водородных связей сорбента колонки; уравнение (5.3), раздел 5.4.1.1
ионообменная емкость колонки, характеризующая электростатические
взаимодействия сорбента с анализируемым веществом; уравнение (5.3), раздел 5.4.1.1
концентрация анализируемого вещества в подвижной фазе
современная надлежащая производственная практика (current Good Manufacturing
Practice), глава 12
концентрация анализируемого вещества в стационарной фазе
двумерный гель-электрофорез (two-dimensional gel electrophoresis, 2D-GE),
раздел 13.4.5
двумерное разделение (two-dimensional separation, 2D-LC), раздел 9.3.10
Менее часто используемые (или менее общеупотребительные) символы
и аббревиатуры
37
Dm
ЕРА
Fopt
Fs
F/-C)
FDA
G
H
h
hp
ICH
ISO
ISPE
К
KD
ккв
kw
k0
mAU
m/z
N'
F
PhRMA
QuEChERS
R
R+
R-
«±
S'
SDS PAGE
коэффициент диффузии вещества (см2/с); уравнение (2.19), раздел 2.4.1.1
Агентство по охране окружающей среды, США (Enviromental Protection Agency),
глава 12
оптимальная объемная скорость подвижной фазы (мл/мин), раздел 2.4.1
функция сравнения колонок; уравнение (5.4), раздел 5.4.2
значение ^„для неионизированных образцов; уравнение (6.3), раздел 6.3.6.1
Управление по санитарному надзору за качеством пищевых продуктов и
медикаментов, США (US Food and Drug Administration), глава 12
коэффициент сжатия пика в градиенте; уравнение (9.15а), раздел 9.2.4.3
гидрофобность колонки; уравнение (5.3), раздел 5.4.1.1
приведенная высота теоретической тарелки; уравнение (2.18), раздел 2.4.1.1
высота пика
Международный совет по гармонизации технических требований к регистрации
фармацевтических продуктов, предназначенных для применения человеком, США
(Internationa 1 Council for Harmonization of Technical Requirements for
Pharmaceuticals in Human Use), глава 12
Международная организация по стандартизации (International Organization for
Standardization), глава 12
Международное общество фармацевтической инженерии (International Society for
Pharmaceutical Engineering), глава 12
коэффициент распределения анализируемого вещества между неподвижной и
подвижной фазами (называемый также постоянной Генри) в уравнении (15.2)
изотермы адсорбции Ленгмюра, равный (Cs/Cm), раздел 15.3.1.1
коэффициент распределения в ЭХ; рис. 13.39; также коэффициент распределения
в законе распределения Нернста, уравнение (16.1)
значение к этилбензола (для различных колонок при стандартных условиях);
уравнение (5.3), раздел 5.4.1.1
экстраполированные значения к веществ, когда подвижная фаза вода (или водный
буфер); уравнение (2.26), раздел 2.5
значение к вещества на старте градиентного элюирования, раздел 9.2.4.1
милли единицы абсорбции (детектирование по поглощению в УФ)
отношение массы молекулы к ее заряду
эффективное число теоретических тарелок при градиентном элюировании,
раздел 9.2.4.4
мера общей полярности растворителя, раздел 2.3.2.1, таблица 2.3
Американская ассоциация фармацевтических исследователей и производителей
(Pharmaceutical Research and Manufacturers of America), глава 12
технология экстракции для пробоподготовки образцов к анализу на содержание
пестицидов (Quick, Easy, Cheap, Effective, Rugged, and Safe), раздел 16.6.7.5
доля молекул анализируемого вещества в подвижной фазе, раздел 2.3
катионный реагент для ИПХ или катионная группа анионообменника, раздел 7.4
анионный реагент для ИПХ или анионная группа катионообменника, раздел 7.4
обозначает либо R+, либой-
характеристика стерических взаимодействий сорбента колонки с анализируемым
веществом; уравнение (5.3), раздел 5.4.1.1 (сопротивление неподвижной фазы
проникновению объемных молекул анализируемого вещества)
гель-электрофорез в полиакриламидном геле с додецилсульфатом натрия,
раздел 13.4.5
Словарь символов и принятых сокращений
S/N
Тк
TOF
и
ие
их
USP
Ув
Ум
К
ys
w0
w1/2
X
а*
а'
а
Р
Р'
AtR
г
Ze
Е/
£Г
Фс
Фо
Л'
к'
л*
о
о'
2
Ч*
отношение сигнал/шум, раздел 4.2.3.2
температура (в градусах Кельвина)
времяпролетный (масс-спектрометр) ( timeofflight MS-detector), раздел 4.14.3
линейная скорость подвижной фазы (мм/мин)
скорость подвижной фазы между частицами сорбента или интерстициальная
скорость подвижной фазы (мм/мин); ие > и, раздел 2.4.1.1
скорость полосы анализируемого вещества при перемещении по колонке
(мм/мин)
Фармакопея США (United States Pharmacopeia)
объем градиента (равен tg F) (мл)
объем смешивания оборудования при градиентном элюировании (мл),
раздел 9.2.2.4
объем пика (мл)
объем образца, наносимого на колонку; уравнение (2.29а), раздел 2.6.1; также
объем стационарной фазы в колонке (мл)
значение W в отсутствие внеколоночного размывания пика, раздел 2.4.1.1
ширина пика на полувысоте; рис. 2.10а, раздел 2.4
молярная доля
коэффициент селективности (фактор разделения) в градиентном элюировании,
раздел 9.2.4.4
кислотность водородных связей молекул анализируемого вещества и сорбента;
уравнение (5.3), раздел 5.4.1.1
кислотность водородных связей между подвижной фазой и анализируемым
веществом, раздел 2.3.2.1
основность водородных связей между подвижной фазой и анализируемым
веществом, раздел 2.3.2.1
основность водородных связей молекул анализируемого вещества и сорбента;
уравнение (5.3), раздел 5.4.1.1
разность времен удерживания компонентов анализируемого образца в градиенте;
рис. 9.15, раздел 9.3.1.1
диэлектрическая постоянная; также молярный коэффициент экстинкции
пористость сорбента (объем между частицами), раздел 2.4.1.1
пористость частиц сорбента (объем пор частиц), раздел 2.4.1.1
общая пористость колонки, раздел 2.4.1.1
конечное значение ср в градиентном разделении; уравнение (9.2а)
исходное значение ф в градиентном разделении; уравнение (9.2а)
гидрофобность молекул анализируемого вещества во взаимодействии с сорбентом;
уравнение (5.3), раздел 5.4.1.1
эффективный ионный заряд молекул анализируемого вещества во взаимодействии
с сорбентом; уравнение (5.3), раздел 5.4.1.1
полярность подвижной фазы, раздел 2.3.2.1
стандартное отклонение гауссовой кривой; уравнение (2.96), раздел 2.4
объемность молекул анализируемого вещества во взаимодействии с сорбентом;
уравнение (5.3), раздел 5.4.1.1
сумма значений а, (3 и ж* подвижной фазы, раздел 2.3.2.1
фазовое отношение, равно Vs/Vm, раздел 2.3.1
Менее часто используемые (или менее общеупотребительные) символы
и аббревиатуры
39
АФИ
ВС
ВПКО
вэ-тсх
гх
ГФМК
Да
ДЗА
ДМД
дмсо
ДОВ
дсн
дсцк
ДТМ
жжэ
жмэ
жэд
ИДУК
ижэ
ипс
кд
кк
кэ
кэх
ЛИУС
МБТЭ
мип
ММР
мок
мэ
активный фармацевтический ингредиент (фармацевтическая субстанция, ФС),
глава 12
водородная связь
верхний предел количественного определения, раздел 11.2.5
высокоэффективная ТСХ
газовая хроматография (gas chromatography, GC)
гептафтормасляная кислота (HFBA)
Дальтон (атомная единица массы или углеродная единица, внесистемная единица,
применяемая для оценки масс молекул, атомов, атомных ядер)
детектор заряженных аэрозолей (также называемый детектором ионизации
коронным разрядом; corona-discharge detector, CAD), раздел 4.13
диодно-матричный УФ-детектор (или детектор с фотодиодной матрицей;
diode-array detector, DAD), раздел 4.4.3
диметилсульфоксид (DMSO)
детектор дисперсии оптического вращения (optical rotary dispersion detector, ORD),
раздел 4.10
додецилсульфат натрия (SDS)
детектор светорассеяния на центрах (ядрах) конденсации (condensation nucleation
light-scattering detector, CNLSD), раздел 4.12.2
диспергирование твердой матрицы для экстракции из твердых образцов (matrix
solid-phase dispersion, MSPD); таблица 16.9, раздел 16.8
экстракция в системе жидкость — жидкость (liquid—liquid extraction, LLE),
раздел 16.5
жидкофазная микроэкстракция (liquid-phase microextraction, LPME), раздел 16.5.4.2
жидкостная экстакция под давлением (pressurized liquid extraction, PFE),
называемая также ускоренной экстракцией растворителем, УСЭР (accelerated solvent
extraction, ASE), раздел 16.8.2.3
иминодиуксусная кислота (IDA)
жидкостная экстракция на иммобилизованном носителе (immobilized liquid
extraction, ILE), раздел 16.5.4.4
изопропиловый спирт, изопропанол (IPA)
детектор кругового дихроизма (circular dichroism (CD) detector), раздел 4.10
контроль качества (quality control, QC), глава 12
капиллярный электрофорез (capillary electrophoresis, CE)
капиллярная электрохроматография (capillary electrochromatography, CEC)
Лабораторная информационно-управляющая система (Laboratory Information
Management System, LIMS), раздел 3.8.1
метил-трет-бутиловый эфир (МТВЕ)
молекулярно-импринтированные полимеры (molecular-imprinted polymer, MIP),
раздел 16.6.7.3
молекулярно-массовое распределение (molecular-weight distribution, MWD),
раздел 13.10.2
микроэкстракция методом одной капли (single-drop microextraction, SDME),
раздел 16.5.4.2
экстракция с микроволновым нагреванием (microwave-assisted extraction, МАЕ),
раздел 16.8.2.4
Словарь символов и принятых сокращений
нмп
НФ
одп
ОФ
ПАУ
пвх
пко
по
птм
ПТФЭ
птх
пээк
РХС
соп
СФХ
СФЭ
ТГФ
ТКМЭДА
ТМА
тмкц
тех
ТФЖЖЭ
ТФМЭ
ТЭАФ
УФЦ
ФКБ
ФС
ХДА
ХДПФ
ХЛАД
хс
цд
ЭАС
ЭДТА
наконечник микропипетки для микроэкстракции (micropipette tip, MPT),
раздел 16.6.3.3
нормально-фазовый (только для разделений на ХНФ), глава 14
адсорбенты с ограниченно доступной поверхностью (restricted access media, RAM),
раздел 16.6.7.2
обращенно-фазовый (только для разделений на ХНФ), глава 14
полициклические ароматические углеводороды (РАН)
поливинилхлорид (PVC)
предел количественного определения, раздел 11.2.5, называемый также нижним
пределом количественного определения, НПКО
предел обнаружения, раздел 11.2.5; также полярно-органический режим элюирова-
ния (только в разделениях на ХНФ), раздел 14.6.1
посттрансляционная модификация (post-translational modification), глава 13
политетрафторэтилен (PTFE)
противоточная хроматография (counter current chromatography, CCC), раздел 1.3.5
полиэфирэфиркетон (РЕЕК )
распределение по химическому составу (chemical-composition distribution, CCD),
раздел 13.10.2
стандартная операционная процедура, глава 12
сверхкритическая флюидная хроматография (supercritical fluid chromatography, SFC)
сверхкритическая флюидная экстракция (supercritical fluid extraction, SFE),
раздел 16.8.2.2
тетрагидрофуран (THF)
трис(карбоксиметил)этилендиамин (TED)
передача (трансфер) методики анализа (analytical method transfer, AMT), глава 12
триацетат микрокристаллической целлюлозы (хиральная неподвижная фаза),
раздел 14.6.1
тонкослойная хроматография (thin-layer chromatography, TLC)
экстракция в системе жидкость — жидкость на твердой фазе (solid-supported
liquid—liquid extraction, SLE), раздел 16.5.4.3
твердофазная микроэкстракция (solid-phase microextraction, SPME), раздел 16.6.3.3
триэтиламмония фосфат (ТЕАР)
ультрафильтрация (ultrafiltration), раздел 16.10
фенилборная кислота (РВА)
фармацевтическая субстанция (или активный фармацевтический ингредиент,
АФИ), глава 12
хиральный дериватизирующий агент (chiral derivatizing reagent, CDR), раздел 14.3
хиральные добавки к подвижной фазе (chiral mobile-phase-additive, CMPA),
раздел 14.4
хемилюминесцентный азотный детектор (chemiluminescent nitrogen detector,
CLND), раздел 4.9
хиральный селектор, хиральный комплексообразующий агент, раздел 14.4
циклодекстрин (CD), раздел 14.6.4
экстракция в автоматизированном аппарате Сокслета (automated Soxlet extraction,
ASE), раздел 16.8.2.1
1,2-этилендиамин-Аг,Лг,7У',7У'-тетрауксусная кислота (EDTA)
ГЛАВА I
ВВЕДЕНИЕ
1.1. Предварительные сведения 42
1.1.1. Что такое ВЭЖХ? 42
1.1.2. Каковы возможности ВЭЖХ? 45
1.2. Небольшой экскурс в историю ВЭЖХ 47
1.3. Некоторые альтернативы ВЭЖХ 50
1.3.1. Газовая хроматография (ГХ) 50
1.3.2. Тонкослойная хроматография (ТСХ) 50
1.3.3. Сверхкритическая флюидная хроматография (СФХ) 50
1.3.4. Капиллярный электрофорез (КЭ) 51
1.3.5. Противоточная хроматография (ПТХ) 51
1.3.6. Специальные виды ВЭЖХ 52
1.4. Другие источники информации о ВЭЖХ 52
1.4.1. Книги 52
1.4.2. Журналы 54
1.4.3. Обзоры 54
1.4.4. Краткие курсы 54
1.4.5. Интернет 54
Литература 54
Высокоэффективная жидкостная хроматография (ВЭЖХ) — один из нескольких хро-
матографических методов, позволяющий проводить разделение и анализ смесей
химических соединений (раздел 1.3). Среди других способов разделения ВЭЖХ
занимает особую нишу. Ее уникальность заключается в следующем:
• практически полная универсальность, только лишь небольшое количество
образцов не поддаются разделению методом ВЭЖХ;
• впечатляющая прецизионность измерений (±0,5%, а в большинстве случаев еще и
точнее);
• большой выбор оборудования, колонок и других сопутствующих компонентов,
позволяющих с помощью ВЭЖХ решить практически любую задачу;
• подавляющее большинство лабораторий, которые имеют дело с анализом
химических смесей, оснащены оборудованием для ВЭЖХ, это первое, что покупают в
лабораторию.
В настоящее время ВЭЖХ является наиболее используемым и широкоприменяе-
мым методом анализа. Масс-спектрометрия конкурирует с ВЭЖХ и во многом
дополняет ее. В сущности, оба этих метода уже применяют в комплексе (ЖХ-МС)
(раздел 4.14). В будущем значение такого сочетания будет расти.
В данной главе будут:
• изложены некоторые общие особенности ВЭЖХ
• обобщена история ВЭЖХ
• очень кратко рассмотрены альтернативы ВЭЖХ и их конкретные области
применения
• приведен список дополнительных источников информации о ВЭЖХ
1.1. Предварительные сведения
1.1.1. Что такое ВЭЖХ?
Жидкостная хроматография появилась в начале 1900-х (рис. \.\а-д) и в настоящее
время известна как «классическая колоночная хроматография». В стеклянный
цилиндр помещали мелко измельченный порошок, например, мел (рис. 1.1а), сверху
в колонку на слой порошка наносили образец (рис. 1.16) и наливали растворитель
(рис. Lie). Под действием гравитации растворитель проходил через слой сорбента
в колонке (рис. 1.1г), а компоненты образца (А, Б и В) вслед за растворителем
начинали перемещаться с разной скоростью. Таким образом происходило разделение.
Изначально процесс разделения исследовали на цветных образцах, поскольку их
перемещения по колонке можно было наблюдать визуально. Затем растворитель,
вытекающий из колонки, собирали порциями, упаривали и проводили
количественный анализ полученных компонентов образца или использовали в других целях
(рис. 1.13). В те далекие времена для разделения каждого образца требовалась
отдельная колонка, а сам процесс проводили вручную (а не в автоматическом режиме).
Поэтому разделение было трудоемким и отнимало много времени. Тем не менее,
даже на том этапе развития хроматография предоставляла уникальные возможности
по сравнению с другими методами анализа химических смесей.
Более простой вид жидкостной хроматографии, названный бумажной
хроматографией (рис. Lie), появился в 1940-х. Полоска бумаги заменила колонку,
изображенную на рис. 1.1 о. Образец наносится на нижний край бумажной полосы, которую
затем помещают в камеру, содержащую на дне растворитель. Благодаря капиллярным
силам растворитель перемещается вверх по полосе бумаги и происходит разделение,
аналогичное процессу, изображенному на рис. 1.1г, только в противоположном
направлении. Позже такой «открытый» вид хроматографии модифицировали и стали
вместо бумажной полоски, используемой в бумажной хроматографии, наносить
тонкий слой порошкообразного силикагеля на стеклянную подложку. Так появилась
тонкослойная хроматография (ТСХ)1. К преимуществам бумажной и тонкослойной
хроматографии относятся (1) удобство, (2) возможность одновременно разделять
несколько образцов на одной и той же полоске бумаги или пластине и (3) простота
обнаружения небольших количеств разделяемых веществ благодаря использованию
колориметрических реагентов уже после завершения разделения.
Вершиной развития современной жидкостной хроматографии стала ВЭЖХ
(рис. 1.1ж,з). Хроматографист начинает анализ с того, что помещает образцы в лоток
автоматического инжектора (автосамплера), из которого они попадают в колонку
(рис. 1.1ж). Насос непрерывно прокачивает элюент через колонку. Разделенные
компоненты образца покидают колонку и регистрируются детектором. Зависимость
интенсивности сигнала от времени представляет собой хроматограмму (рис. 1.1з),
которую можно сравнить с результатом разделения, изображенным на рис. Lid
(при условии идентичности состава образца А+Б+В и параметров разделения).
Компьютер полностью контролирует процесс разделения, от оператора требуется
только поместить образец в лоток автоматического инжектора. Кроме того, компьютер
сам для каждого образца может сформировать отчет об анализе образца. Помимо
полностью автоматизированного процесса, в ВЭЖХ используются насосы высокого
давления, что позволяет ускорить разделение, многоразовые и более эффективные
колонки, что повышает качество разделения, и надлежащий контроль всего процесса
'Тонкослойную хроматографию в 1938 году предложили Н.А. Измайлов и М.С. Шрайбер
См. Измайлов Н.А., Шрайбер М.С. Фармация, 1938, № 3. — С . 1—7. — Прим. перев.
1.1. Предварительные сведения
43
(а)
Классическая колоночная хроматография (а-д)
(д) (е)
S
S
3
га
а.
-е-
о
о
га
5
В
-| г-
_!_[__
Б
1
-
г
А
"Ь
5 10 15 20 25
Фракция
30
т.
Бумажная или тонкослойная
хроматография (е)
(з)
Емкость с
растворителем
]—HZI
Детектор
ВЭЖХ (ж-з)
(С
I
О
Время
Рис. 1.1. Этапы развития хроматографии
Глава 1. Введение
разделения, обеспечивающий более прецизионные и воспроизводимые результаты.
Более подробная информация об истории ВЭЖХ изложена в разделе 1.2.
С появлением ВЭЖХ в конце 1960-х начинается ее развитие (раздел 1.2).
На рис. 1.2а показано ежегодное количество публикаций, посвященных ВЭЖХ.
Первая статья появилась в 1966 [2]. Затем число статей каждый год росло
экспоненциально и достигло постоянного значения только в конце 1980-х. К 1990-м годам
удалось достичь понимания процесса разделения, стали доступны необходимые
оборудование и колонки. В настоящее время ВЭЖХ можно рассматривать как зрелый
метод анализа, которым теперь пользуются во всем мире. В то же время новые
специализированные приложения для ВЭЖХ продолжают появляться и после 1990 года.
Постоянное внимание уделяется оставшимся пробелам в нашем понимании сути
процесса, которое теперь вряд ли уже кардинально поменяется.
Хотя к 1990 году количество исследований, посвященных ВЭЖХ, перестало
расти, на аналогичную стабилизацию экономики ВЭЖХ ушло немного больше времени,
что подтверждает график (рис. 1.26) зависимости ежегодных расходов от времени для
всех продуктов ВЭЖХ (без учета инфляции). Ежегодные затраты на ВЭЖХ в
настоящее время превышают затраты на любую другую аналитическую технику.
is
S-
-|
го
ь;
С
vn
^
хг.
СТВО
Qj
J
"-,
О
Si
о
ш
X
*
Г1)
т
еннь
Ч
(Т
m
о
о
с
го
о
Q.
С
2
ш
fi
ID
О
00
О
О.
го
с;
с
о
сг
х:
ГО
I
о
S"
с;
с;
0 001
ш <п
0 0001
0 01 -
1960
1970
1980
1990
2000
2010
Рис. 1.2. Растущая актуальность исследований в области ВЭЖХ и прикладных работ,
начиная с 1966 г. (а) Количество публикаций, посвященных ВЭЖХ, в год [1];
(б) общий объем продаж ВЭЖХ оборудования и сопутствующих материалов
в год (приблизительные значения на основе различных источников)
1.1. Предварительные сведения
1.1.2. Каковы возможности ВЭЖХ?
Во втором издании этой книги, которое вышло в свет в 1979 году, были
опубликованы некоторые примеры возможностей ВЭЖХ. Два из них приведены и в этой книге
(рис. 1.3). На рис. 1.3а изображено экспресс-разделение 15 соединений
продолжительностью 1 минута. В другом примере (рис. 1.36) на неполное разделение более чем
100 пиков потребовалось всего лишь 30 мин. Для сравнения на рис. 1.4 представлены
разделения, проведенные спустя 25 лет. Обратите внимание на первую хроматограм-
му (рис. 1.4а), на которой за 7 секунд удалось разделить 6 белков, а на разделение
более 1000 пептидов и белков (рис \Лб,в) ушло всего 1,5 часа. Улучшение
разделения, наблюдаемое на рис. 1.4а в сравнении с 1.3а, удалось получить благодаря
нескольким факторам, часть из которых обсуждается в разделе 1.2. Разделение
1000 соединений (рис. \Лб,в) проводили с помощью так называемой двумерной
хроматографии (раздел 9.3.10), смысл которой заключается в том, что сначала образец
фракционируют на одной колонке, а потом полученные фракции разделяют на
второй колонке. В этом примере каждую фракцию с первой колонки собирали в
течение 4 минут, затем делили на второй колонке. На рис. 1.4в показано разделение
фракции 7. Общее количество (распознаваемых) пиков в образце получили,
просуммировав пики индивидуальных веществ, присутствующие в каждой фракции.
Огромный прогресс, достигнутый в производительности ВЭЖХ за предыдущие 25 лет
(сравните рис. 1.3 и 1.4), позволяет предположить, что в ближайшие годы вряд ли
стоит ожидать сопоставимые существенные улучшения в скорости или
эффективности разделения.
15
Время (мин)
Рис. 1.3. Примеры возможностей ВЭЖХ к середине 1970-х годов, (а)
Экспресс-разделение смеси небольших молекул [3]; (б) высокоэффективное разделение
образца мочи [4]. Пример (а) взят из [3], пример (б) взят из [4]
Глава 1. Введение
(a)
7 (сек)
О 10 20 30 40 50 60 70 80 90
(мин)
Рис. 1.4. Современные примеры возможностей ВЭЖХ. (а) Быстрое градиентное
разделение 6 белков на колонке 150 х 4,6 мм, пелликулярный сорбент с размером
частиц 1,5 мкм [5]; (б) первоначальное разделение пептидов и белков,
выделенных из клетки эмбрионального фибробласта человека с помощью
градиентной катионообменной хроматографии; (в) дальнейшее разделение
фракции 7, собранной в интервале 24—28 минут, на второй колонке с помощью
градиентной обращенно-фазовой хроматографии [6]. Рисунки взяты из
оригинальных статей [5, 6]
С 1979 года имели место и другие столь же значимые достижения в ВЭЖХ.
Начиная с 1980-х годов появляются колонки, пригодные для разделения белков и
других крупных биомолекул [7, 8], что открыло совершенно новую область применения
ВЭЖХ и способствовало значительному прогрессу в области биохимии. Благодаря
разработанным Пирклом и др. [9]1 хиральным колонкам для разделения энантиоме-
ров, такой же прогресс наблюдается в области фармацевтики и связанных с ней
науках о жизни.
Также растет интерес к ВЭЖХ очистке веществ в крупных масштабах благодаря
доступности соответствующего оборудования, глубокому пониманию того, как лучше
всего провести то или иное разделение, а также вследствие ужесточения требований
контрольных органов к чистоте фармацевтических препаратов.
1 Впервые разделение оптических изомеров в жидкостной хроматографии продемонстрировал
В.А. Даванков в 1971 году, предложив метод лигандообменной хроматографии. См. V.A. Davankov,
S.V. Rogozhin. Ligand chromatography as a novel method for the investigation of mixed complexes:
stereoselective effects in a-amino acid copper(II) complexes // J. Chromatogr. 1971. V. 60. P. 284—
312. — Прим. перев.
1.2. Небольшой экскурс в историю ВЭЖХ 47
1.2. Небольшой экскурс в историю ВЭЖХ
В разделе 1.1 описано развитие жидкостной хроматографии до появления ВЭЖХ.
Чтобы заполнить все пробелы в истории ВЭЖХ до 1965 года, можно обратиться
к обзорам, написанным «историком хроматографии» Лесли Эттером:
• предшественники хроматографии; разработки в период до 1900 [10, 11];
• создание хроматографии М.С. Цветом в начале 1900-х [12];
• повторное открытие хроматографии в начале 1930-х [13];
• создание А.Дж.П. Мартином распределительной и бумажной хроматографии в
начале 1940-х [14];
• разработка аминокислотного анализатора С. Муром и У.С. Штайном к концу
1950-х [15];
• разработка гель-проникающей хроматографии компанией Waters в начале
1960-х [16].
Карл Рунге, немецкий химик, занимавшийся красителями, родился в 1856 году.
Он первым сообщил о разделении неочищенных красителей методом, похожим на
бумажную хроматографию [10], однако ни он, ни другие исследователи не стали
развивать практические возможности этой работы. В конце 1890-х годов Дэвид Дэй из
Геологической Службы США разделял нефть методом, напоминающим классическую
колоночную хроматографию [11]. Однако целью этой работы была не разработка
методики разделения, а демонстрация того, что различное качество нефти из разных
месторождений обусловлено ее разделением при прохождении сквозь слои земли.
Исследования Дэвида Дэя, также как и труды Карла Рунге, не были продолжены. В начале
1900-х Михаил Цвет изобрел классическую колоночную хроматографию и
продемонстрировал разделение экстрактов различных растений [12]. Это событие, безусловно,
считается началом хроматографии, хотя в последующие два десятилетия эту работу
по достоинству так и не оценили. В начале 1930-х годов на работу Цвета вновь
обратили внимание [13], что привело к последующему бурному развитию хроматографии.
В 1943 году А.Дж.П. Мартин разработал бумажную хроматографию [14], наряду с этим
с конца 1930-х до середины 1950-х была введена в практику тонкослойная
хроматография [17]. Такое краткое извлечение из истории, охватывающее период до 1955 года,
неизбежно влечет за собой потерю огромного количества работ других исследователей,
которые также внесли свой вклад в развитие хроматографии.
В конце 1950-х [15] годов был создан аминокислотный анализатор — основной
предшественник ВЭЖХ. Смеси аминокислот на нем анализировали методом
ионообменной хроматографии в автоматическом режиме (раздел 7.5). За этим событием
последовала разработка Муром [18] гель-проникающей хроматографии (раздел 13.8),
а в начале 1960-х компания Waters [16] использовала ее на практике. Каждый из этих
методов был по своим принципам близок к тому, что позже стали называть ВЭЖХ, и
немногим отличался от схемы, изображенной на рис. 1.1ж,з. В каждом из этих
случаев через многоразовую наполненную мелкозернистым сорбентом колонку насос
под высоким давлением прокачивал растворитель, который после выхода из колонки
направлялся в детектор. Итогом разделения была хроматограмма (рис. 1.1з). Чего же
не хватало двум этим системам? Возможности разделять и анализировать разного
рода образцы. Аминокислотный анализатор ограничивался лишь анализом
аминокислотных смесей, а гель-проникающая хроматография служила исключительно
для определения распределения молекулярной массы синтетических полимеров.
Ни один из этих методов не был адаптирован для других образцов.
В начале 1960-х две группы, одна в США под руководством Чаба Хорвата,
другая — в Европе под руководством Йозефа Хубера, приступили к разработке
ВЭЖХ-системы универсального назначения. Оба ученых описали свои ранние рабо-
Глава 1. Введение
ты в личных воспоминаниях [19], а Эттре предоставил дополнительную информацию
о начале работы в лаборатории Хорвата [20]. Непосредственные работы этих групп,
а также связанные с ними работы других исследователей, проведенные несколькими
годами позже, описаны в статьях за период с 1966 по 1968 годы [2, 21—24]. Серийное
оборудование для ВЭЖХ появилось в конце 1960-х. Изначально монополистами
на рынке были компании Waters Associates и DuPont. Вскоре и другие фирмы смогли
предложить конкурентоспособные хроматографические системы. Началось
стремительное развитие ВЭЖХ (см. рис. 1.2а). В 1971 году появилась первая книга о ВЭЖХ
[25], а Американское Химическое Сообщество представило краткий курс лекций
(Современная жидкостная хроматография), авторами которого были Дж.Дж. Кирк-
ленд и Л.Р. Снайдер.
Прогресс в ВЭЖХ-разделениях показан на рис. 1.5д-е, на котором видно, что
продолжительность анализа за 50 лет, начиная с 1960 г., сократилась на несколько
порядков. На рис. 1.5ж видно, что сокращение времени разделения (О, —) связано
1960 (предшественники ВЭЖХ)
1970 (ВЭЖХ)
(б)
10 20 30 40 50
мин
(в)
1980
1990
(г)
2 4 6
мин
10 12
2 3
мин
(а)
2000
05
1 0
мин
1 5
02
2010
(е)
04 06
мин
1 0
Рис. 1.5. Хроматограммы, демонстрирующие как увеличилась эффективность ВЭЖХ
с 1960 по 2010 гг. Образец: смесь пяти гербицидов. Условия: подвижная фаза
50% метанол/вода, температура комнатная. Хроматограммы а-е созданы
с помощью компьютерного моделирования с использованием программы
DryLab® (Раздел 10.2) на основе данных [26], диаграммы ж а з содержат
дополнительную информацию об экспериментах а-е. Колонки диаметром
4,6 мм, заполнены сорбентом с одинаковой селективностью
1.2. Небольшой экскурс в историю ВЭЖХ 49
(ж)
690 бар
(10000 psi)
69 бар
(1000 psi)
6.9 бар
(100 psi)
0.7 бар
(10 psi)
0.07 бар
(1 psi)
(з)
100
10
1
0.1
1960 1970 1980 1990 2000 2010
Рис. 1.5. (Окончание)
с увеличением перепада давления в колонке (—) и уменьшением размера частиц
сорбента (•). Поэтому на ранних этапах развития ВЭЖХ этот метод иногда называли
«жидкостная хроматография высокого давления» или «высокоскоростная жидкостная
хроматография». На рис. 1.5з показаны соответствующие изменения длины колонки
(•) и скорости потока (О) для этих экспериментов (рис. \.5а-е).
Еще задолго до 1960-х годов были разработаны теоретические принципы
будущего развития ВЭЖХ. В 1941 г. Мартин сообщал [27]: «самые эффективные колонки
... должны быть заполнены частицами небольшого размера и работать при больших
перепадах давления». Так, в двух словах, можно сформулировать суть ВЭЖХ (как
показано на рис. 1.5ж). В начале 1950-х Мартин создал смежный метод — газовую
хроматографию [28]. Очень быстро этот метод стал популярен во всем мире [29], что
привело к ряду теоретических исследований, которые потом будут иметь отношение
к развитию ВЭЖХ. В начале 1960-х Гиддингз обобщил и продолжил эту работу уже
для определенных областей применения ВЭЖХ [30]. В будущем его работа оказалась
очень важна как для определения конструкции колонок, так и для выбора
подходящих экспериментальных условий.
Более подробно история ранних этапов развития ВЭЖХ описана в работах [19,
31—33]. Дополнительные исторические детали, касающиеся периода после 1980 года,
можно найти в собрании биографий некоторых разработчиков ВЭЖХ [34].
10
30 мкм „-'
О. 1
\ /
\ /
1 \
/ ^—
/ \
/
"" 4
100 мкм частицы
-
мкм
1.5 мкм
^#
3 мкм^.^
5 мкм ^~~
Давление (бар, psi)
Время разделения (мин)
1960 1970 1980 1990 2000 2010
Длина колонки \^
(CM) >«L
р_—ч> о--"'
/
/
/
/
// Скорость потока (мл/мин)
d
^-—о
1.3. Некоторые альтернативы ВЭЖХ
Газовая хроматография (ГХ) и тонкослойная хроматография (ТСХ) — два основных
метода, существовавшие еще до 1965 года, каждая из которых в определенных
ситуациях может быть использована вместо ВЭЖХ. Еще один метод, разработанный до
1965 года, называется противоточной хроматографией (ПТХ). В принципе, он может
составить конкуренцию ВЭЖХ в решении многих задач, но в скорости и
производительности с ВЭЖХ он не сравнится. После создания ВЭЖХ были разработаны
некоторые дополнительные, потенциально конкурентоспособные методы разделения:
сверхкритическая флюидная хроматография (СФХ) в 1970-х, капиллярный
электрофорез (КЭ) в 1980-х, капиллярная электрохроматография (КЭХ) в 1990-х.
1.3.1. Газовая хроматография (ГХ)
Методом ГХ [35] можно проанализировать только те образцы, которые будут
находиться в газовой фазе1 при температурах ниже 300 °С. То есть вещества, кипящие при
очень высоких температурах или вообще нелетучие, а это примерно 75% всех
известных соединений, невозможно разделить с помощью ГХ. С другой стороны, ГХ
отличается большей эффективностью (большим числом теоретических тарелок), а это
значит, что разделение проходит быстрее и/или лучше. Поэтому ГХ
предпочтительнее ВЭЖХ при анализе газов, большинства низкокипящих веществ и множества
образцов, кипящих при высоких температурах, но при этом термически стабильных
в условиях разделения. Кроме того, в арсенале ГХ имеются достаточно
чувствительные детекторы и/или специфические детекторы селективные к конкретным
элементам, благодаря чему можно существенно снизить пределы обнаружения.
1.3.2. Тонкослойная хроматография (ТСХ)
К преимуществу ТСХ [36] относится возможность одновременно на одной пластинке
разделять несколько проб, причем каждый компонент пробы в конце разделения
виден на пластинке (в ВЭЖХ сильно удерживаемые вещества могут вообще не элюиро-
ваться с колонки и отсутствовать на хроматограмме). При использовании
специального оборудования, способного под давлением направлять поток растворителя от одного
края пластины к другому, стала возможна так называемая высокоэффективная ТСХ
(ВЭ-ТСХ). Вне зависимости от того, как осуществляется ТСХ, по эффективности
(числу теоретических тарелок) разделения она, тем не менее, проигрывает ВЭЖХ, да и
количественный анализ в таких условиях менее удобен и точен. ТСХ к 2010 г в США
при количественных анализах использовали довольно редко, хотя для
полуколичественного анализа и определения примесей в образце этот метод очень удобен. ТСХ
широко используется для скрининга большого количества образцов без необходимости
существенной их очистки (например, контроль лекарственного препарата в плазме).
ВЭ-ТСХ более популярна в Европе, чем в США, но гораздо менее, чем ВЭЖХ.
1.3.3. Сверхкритическая флюидная хроматография (СФХ)
Оборудование и колонки для СФХ [37] похожи на использующиеся в ВЭЖХ.
Растворителем, по определению, является сверхкритическая жидкость, обычно это
углекислый газ при повышенном давлении и температуре. СФХ можно рассматривать, как
расширение возможностей ГХ вследствие способности сверхкритической жидкости
Будут находиться в значительной степени. — Прим. перев.
1.3. Некоторые альтернативы ВЭЖХ 5 I
растворять и разделять нелетучие в обычных условиях соединения. То есть, СФХ
занимает промежуточное положение между ГХ и ВЭЖХ и характеризуется большей
эффективностью разделения (больше N) по сравнению с ВЭЖХ, но меньшей в сравнении
с ГХ. Растворитель для СФХ имеет большее значение, чем в ГХ, но меньшее, чем
в ВЭЖХ. Чувствительность обнаружения в СФХ тоже находится между ВЭЖХ и ГХ.
СФХ в основном используют для анализа природных и синтетических полимерных
смесей, например, для разделения полифенолов [38]. Если в некоторых случаях ВЭЖХ
не позволяет разделить полимеры свыше некоторого максимума молекулярной массы,
то СФХ обычно значительно поднимает верхнюю границу предела молекулярной
массы. Кроме того, с помощью СФХ разделяют энантиомеры, у которых близки времена
удерживания, и поэтому требуется большая эффективность разделения (большее N).
1.3.4. Капиллярный электрофорез (КЭ)
КЭ [1, 39] не является одним из видов хроматографии, но в эффективности
разделения определенных классов соединений может составить конкуренцию ВЭЖХ.
Принцип разделения заключается в разной скорости миграции веществ внутри капилляра.
Под воздействием электрического поля молекулы разделяются в зависимости от
отношения массы к заряду (m/z). Соединения с небольшим соотношением m/z
мигрируют быстрее. Следовательно, с помощью КЭ можно разделять только заряженные
молекулы. По сравнению с ВЭЖХ у КЭ эффективность разделения выше (больше
N), особенно при разделении соединений с большой молекулярной массой. Однако
чувствительность обнаружения в случае КЭ обычно хуже, чем в ВЭЖХ. КЭ широко
применяется для анализа геномных образцов, основанного на фракционировании
ДНК-фрагментов. Кроме того, КЭ активно используют для разделения энантиоме-
ров, где его эффективность может быть выше ВЭЖХ по двум причинам: во-первых,
такие разделения, как правило, трудны, и для них требуется большее значение N, что
может обеспечить КЭ; во-вторых, ВЭЖХ разделения обычно проводят на хиральных
колонках. Прежде чем получить удовлетворительный результат, каждый конкретный
образец разделяют методом проб и ошибок на нескольких разных (и дорогостоящих)
колонках, в то время как для КЭ достаточно всего лишь небольшого количества
разных хиральных селекторов. В отличие от ВЭЖХ разделение происходит быстрее,
стоит дешевле, а метод является более универсальным. Дорогие хиральные комплек-
сообразующие реагенты нецелесообразно использовать в ВЭЖХ, поскольку скорости
потока в обычной ВЭЖХ много больше, чем в КЭ (мл/мин против мкл/мин). Кроме
того, существуют несколько вариантов КЭ, расширяющих использование этого
метода для других типов образцов. К примеру, с помощью мицеллярной
электрокинетической хроматографии можно разделять неионные соединения [40].
1.3.5. Противоточная хроматография (ПТХ)
ПТХ [41, 42] представляет собой более ранний вид жидкостно-жидкостной
распределительной хроматографии, впоследствии улучшенной различными способами.
ВЭЖХ с жидкой неподвижной фазой была вытеснена ВЭЖХ с привитыми фазами, и
потому ПТХ в качестве альтернативы ВЭЖХ стали использовать все реже. Часто
упоминаемая особенность ПТХ — отсутствие проблем, вызванных необратимой
сорбцией образца на обширной внутренней поверхности колонок в ВЭЖХ. Однако
используемые в настоящее время колонки для ВЭЖХ лишены этого недостатка. У ПТХ
есть некоторые преимущества при препаративном разделении энантиомеров [43].
В остальных случаях эта методика используется главным образом для выделения
лабильных природных соединений.
1.3.6. Специальные виды ВЭЖХ
В разделах 1.3.1—1.3.5 описаны пять методов разделения, существенно
отличающихся от ВЭЖХ. Еще четыре, о которых в данной книге речь не пойдет, можно отнести
к разновидностям ВЭЖХ. Тем не менее большая часть используемой в следующих
главах информации может быть применена и для них.
Капиллярная электрохроматография [44, 45] (КЭХ), в основном, подобна ВЭЖХ
за исключением одного момента: растворитель проходит через колонку не с
помощью нагнетающего его насоса, а посредством создания электрического потенциала
на концах колонки (эндоосмотический поток). Поскольку поток растворителя никак
не зависит от размера частиц сорбента (а эффективность колонки может быть выше
при малом размере частиц), то, в принципе, с помощью КЭХ можно получить
гораздо большие значения N, чем с помощью обычной ВЭЖХ. Кроме того, N также
увеличивается благодаря собственно эндоосмотическому потоку. Имея в виду
потенциально большие значения N в КЭХ по сравнению с ВЭЖХ, с 1995 года в практическую
реализацию этого метода инвестировались значительные средства, однако к моменту
выхода из печати этой книги оставались нерешенными важные технические
проблемы, не позволяющие КЭХ стать рутинной альтернативой ВЭЖХ.
ВЭЖХ на чипе [46] — недавно разработанная технология для удобного анализа
небольших количеств образца. Микроколонку (например, 43 х 0,06 мм), которая
является частью чипа, устанавливают между микронасосом и масс-спектрометром.
Принцип разделения такой же, как и в ВЭЖХ с обычными колонками и
оборудованием. К преимуществам можно отнести эффективность разделения и удобство
при разделении очень малых количеств образца.
Ионная хроматография [47, 48] — широко используемая методика анализа
смесей, содержащих неорганические анионы и катионы, например, С1~ и Na+
соответственно. Хотя принципы разделения такие же, как и в ионообменной ВЭЖХ
(раздел 7.5), для ионной хроматографии требуется особое оборудование. Эта методика,
главным образом, предназначена для анализа неорганических соединений.
Мицеллярная жидкостная хроматография — вариант обращенно-фазовой
хроматографии, только вместо водно-органических элюентов используют водный раствор
поверхностно активного вещества [49]. В настоящее время эта методика очень редко
применяется из-за низкой эффективности разделения.
1.4. Другие источники информации о ВЭЖХ
В дополнение к данной книге имеется огромное количество других доступных
источников информации. К ним относятся различные публикации (раздел 1.4.1-1.4.3),
краткие курсы (раздел 1.4.4) и интернет (раздел 1.4.5).
1.4.1. Книги
К настоящему времени изданы сотни книг, которые легко можно найти в интернете на
сайте Amazon.com и других сайтах. Книги по ВЭЖХ можно разделить на 2 группы:
1. специализированные книги, посвященные ВЭЖХ разделениям образцов
определенного типа (например, белков, углеводов, энантиомеров и др.) или
особенностям детектирования (например, масс-спектрометрии, химической дериватиза-
ции);
2. общие книги, такие как эта книга, охватывающие все аспекты ВЭЖХ.
1.4. Другие источники информации о ВЭЖХ 53
Далее в конце глав перечислены профильные книги по разным тематикам
ВЭЖХ. В таблице 1.1 приведен неполный список книг по общим темам ВЭЖХ,
опубликованных с 1995 года. Они могут оказаться полезным дополнением к
настоящей книге.
Таблица 1.1. Список наиболее интересных книг по общей ВЭЖХ, опубликованных
с 1995 года
Название
Автор(ы)
Дата печати
Издательство
Общая ВЭЖХ
Handbook of HPLC
High Performance Liquid Chromatography
High Performance Liquid Chromatography
HPLC, 2nd ed.
Modern HPLC for Practicing Scientists
Practical High- Performance Liquid
Chromatography, 4th ed.
E. Katz, R. Eksteen,
P. Schoenmakers,
and N. Miller, eds.
S. Lindsay
E. Prichard
M. С McMaster
M. W. Dong
V. R. Meyer
1998
2000
2003
2006
2006
2006
Dekker
Wiley
Royal Society of
Chemistry
Wiley-Interscience
Wiley-Interscience
Wiley-Interscience
Разработка методик
Practical HPLC Method Development,
2nd ed.
HPLC Made to Measure: A Practical
Handbook for Optimization
L. R. Snyder,
J. L. Glajch, and
J. J. Kirkland
S. Kromidas
1997
2006
Wiley-Interscience
Wiley
Устранение неисправностей
LC Troubleshooting
Troubleshooting HPLC Systems: A Bench
Manual
More Practical Problem Solving in HPLC
Pitfalls and Errors of HPLC in Pictures
2nd ed.
J. W. Dolan
P. C. Sadek
S. Kromidas
V. R. Meyer
С 1983 г.
по
настоящее время
1999
2005
2006
Ежемесячная колонка
в журнале LCGC
Magazine; предыдущие
статьи в свободном
доступе на сайте
www.chromatographyon-
line.com
Wiley
Wiley
Wiley
Препаративная ВЭЖХ
Practical Handbook of Preparative HPLC
D. A. Wellings
2006
Elsevier
ВЭЖХ-колонки
HPLC Columns: Theory, Technology and
Practice
U. D. Neue
1997
Wiley-VCH
ВЭЖХ-растворители
The HPLC Solvent Guide
P. C. Sadek
1996
Wiley
Градиентное элюирование
High-Performance Gradient Elution
L. R. Snyder and
J. W. Dolan
2007
Wiley
1.4.2. Журналы
Технические статьи по ВЭЖХ можно найти в большинстве химических и
биохимических журналов. Тем не менее, приведенный ниже список журналов будет особенно
ценен для тех читателей, которые хотят быть в курсе новых разработок в этой области.
• Analytical Chemistry, American Chemical Society
• Chromatographia, Springer
• Journal of Chromatographic Science, Preston
• Journal of Chromatography A, Elsevier
• Journal of Chromatography B, Elsevier
• Journal of Liquid Chromatography, Wiley
• Journal of Separation Science, Wiley
• LCGC, Advanstar (separate issues for North America and Europe)
1.4.3. Обзоры
Обзоры по ВЭЖХ можно найти не только в тех журналах, которые перечислены
выше, но и в других. Кроме того, есть ряд публикаций, частично посвященных
ВЭЖХ, изданных или в виде сборников обзорных статей
• Advances in Chromatography, Dekker
• High-Performance Liquid Chromatography. Advances and Perspectives, Academic Press
(публиковались только с 1980 г. по 1986 г.)
или как отдельные книги:
• Journal of Chromatography Library, Elsevier
1.4.4. Краткие курсы
В настоящее время существует огромное количество кратких курсов, которые
читаются как вживую, так и через интернет (раздел 1.4.5). Актуальный список кратких
курсов можно найти на последней странице журнала LCGC, либо в интернете по
запросу «ВЭЖХ тренинг».
1.4.5. Интернет
Динамичное развитие интернета гарантирует нам быстрое устаревание напечатанной
в книге информации. Нижеперечисленные сайты содержат ссылки на другие сайты
и, вполне очевидно, будут таким образом постоянно обновляться:
• http://www.lcresources.com
• http://matematicas.udea.edu.co/carlopez/index7.html
• http://lchromatography.com/hplciind/index.html
• thtp://tech.groups.yahoo.com/group/chrom-L/links
• http://userpages.umbc. edu/dfrey 1/Freylink
• http://www.infochembio.ethz.ch/links/en/analytchem_chromat.html
• http://www.chromatographyonline.com
Литература
1. R. L. Cunico, К. M. Gooding, and Т. Wehr, Basic HPLC and CE of Biomolecules, Bay Bioanalytical
Laboratory, Richmond, CA, 1998, p. 4.
2. С G. Horvath and S. R. Lipsky, Nature, 211 (1966) 748.
3. I. Halasz, R. Endele, and J. Asshauer, J. Chromatogr., 112 (1975) 37.
4. I. Molnar and С Horvath, J. Chromatogr. (Biomed App.), 143 (1977) 391.
5. T. Issaeva, A. Kourganov, and K. Unger, J. Chromatogr. A, 846 (1999) 13.
6. K. Wagner, T. Miliotis, G. Marko-Varga, R. Biscoff, and K. Unger, Anal. Chem., 74(2002) 809.
7. S. H. Chang, K. M. Gooding, and F. E. Regnier, /. Chromatogr., 125 (1976) 103.
8. W. W. Hancock, С A. Bishop, and M. T. W. Hearn, Science, 153 (1978) 1168.
9. W. H. Pirkle, D. W. House, and J. M. Finn, /. Chromatogr., 192 (1980) 143.
10. H. H. Bussemas and L. S. Ettre, LCGC, 22 (2004) 262.
11. L. S. Ettre, LCGC, 23 (2005) 1274.
12. L. S. Ettre, LCGC, 21 (2003) 458.
13. L. S. Ettre, LCGC, 25 (2007) 640.
14. L. S. Ettre, LCGC, 19 (2001) 506.
15. L. S. Ettre, LCGC, 243 (2006) 390.
16. L. S. Ettre, LCGC, 23 (2005) 752.
17. J. G. Kirchner, Thin-layer Chromatography, Wiley-Interscience, New York, 1978, pp. 5—8.
18. J. С Moore, /. Polymer Sci. Part A, 2 (1964) 835.
19. L. S. Ettre and A. Zlatkis, eds., 75 years of Chromatography—A Historical Dialog, Elsevier,
Amsterdam, 1979.
20. L. S. Ettre, LCGC, 23 (2005) 486.
21. J. F. K. Huber and J. A. R. Hulsman, /. Anal. Chim. Acta, 38 (1967) 305.
22. J. J. Kirkland, Anal. Chem., 40 (1968) 218.
23. L. R. Snyder, Anal. Chem., 39 (1967) 698, 705.
24. R. P. W. Scott, W. J. Blackburn, and T. J. Wilkens, J. Gas Chrommatogr., 5 (1967) 183.
25. J. J. Kirkland, ed., Modern Practice of Liquid Chromatography, Wiley-Interscience, New York,
1971.
26. T. Braumann, G. Weber, and L. H. Grimme, /. Chromatogr., 261 (1983) 329.
27. A. J. P. Martin and R. L. M. Synge, Biochem. J., 35 (1941) 1358.
28. A. T. James and A. J. P. Martin, Biochem. J., 50 (1952) 679.
29. L. S. Ettre, LCGC, 19 (2001) 120.
30. J. C. Giddings, Dynamics of Chromatography. Principles and Theory, Dekker, New York, 1965.
31. L. R. Snyder, J. Chem. Ed., 74 (1997) 37.
32. L. S. Ettre, LCGC Europe, 1 (2001) 314.
33. L. R. Snyder, Anal. Chem., 72 (2000) 412A.
34. C. W. Gehrke, ed., Chromatography—A Century of Discovery 1900—2000, Elsevier, Amsterdam,
2001.
35. R. L. Grab and E. F. Barry, Modern Practice of Gas Chromatography, 4th ed.,Wiley-Interscience,
NewYork, 2004.
36. B. Fried and J. Sherma, Thin-Layer Chromatography (Chromatographic Science, Vol.81), Dekker,
New York, 1999.
37. R. M. Smith and S. M. Hawthorne, eds., Supercritical Fluids in Chromatography and Extraction,
Elsevier, Amsterdam, 1997.
38. T. Bamba, E. Fukusaki, Y. Nakazawa, H. Sato, K. Ute, T. Kitayama, and A. Kobayashi, J.
Chromatogr. A, 995 (2003) 203.
39. K. D. Altria and D. Elder, /. Chromatogr. A, 1023 (2004) 1.
40. A. Berthod and C. Garcia-Alvarez-Coque, Micellar Liquid Chromatography, Dekker, New York,
2000.
41. Y. Ito and W. D. Conway, eds., High-Speed Countercurrent Chromatography, Wiley, New York,
1996.
42. J.-M. Menet and D. Thiebaut, eds., Countercurrent Chromatography, Dekker, NewYork, 1999.
43. E. Gavioli, N.M.Maier, C.Minguillon, and W. Lindner, Anal. Chem., 76 (2004) 5837.
44. K. D. Bartle and P. Meyers, Capillary Electrochromatography (Chromatography Monographs),2001.
45. F. Svec, ed., /. Chromatogr. A, 1044 (2004).
46. H. Yin and K. Killeen, J. Sep. Sci., 30 (2007) 1427.
47. J. S. Fritz and D. T. Gjerde, Ion Chromatography, 3rd ed., Wiley-VCH, Weinheim, 2000.
48. J. Weiss, Handbook of Ion Chromatography, 3rd ed., Wiley, 2005.
49. A. Berthod and M. C. Garcia-Alvarez-Coque, Micellar Liquid Chromatography, Dekker, New York,
2000.
ГЛАВА 2
ОСНОВНЫЕ ПРИНЦИПЫ И
УПРАВЛЕНИЕ РАЗДЕЛЕНИЕМ
2.1. Введение 57
2.2. Хроматографический процесс 57
2.3. Удерживание 61
2.3.1. Фактор удерживания к и мертвое время колонки /0 61
2.3.2. Роль условий разделения и состава образца 65
2.3.2.1. Межмолекулярные взаимодействия 67
2.3.2.2. Температура 71
2.4. Ширина пика и число теоретических тарелок колонки N 71
2.4.1. Зависимость N от условий разделения 73
2.4.1.1. Процессы размывания пика, определяющие значения N 76
2.4.1.2. Несколько рекомендаций по выбору колонки 82
2.4.2. Форма пика 86
2.5. Разрешение и разработка метода 89
2.5.1. Оптимизация фактора удерживания к (член а в уравнении (2.24)) 93
2.5.2. Оптимизация селективности а (член б в уравнении (2.24)) 95
2.5.2.1. «Правильные» и «неправильные» образцы 96
2.5.3. Оптимизация числа теоретических тарелок N (член в в уравнении (2.24)). 97
2.5.3.1. Зависимость результатов разделения от параметров колонки. ... 97
2.5.3.2. Скоростная ВЭЖХ 99
2.5.4. Разработка метода 101
2.5.4.1. Оценка состава образца и целей разделения 101
2.5.4.2. Пробоподготовка 102
2.5.4.3. Выбор вида хроматографии 102
2.5.4.4 Выбор детектора 102
2.5.4.5. Выбор хроматографических условий 102
2.5.4.6. Спрогнозировать, определить и решить возможные проблемы. . 103
2.5.4.7. Валидация метода и пригодность системы 104
2.6. Влияние количества пробы 105
2.6.1. Перегрузка по объему: влияние объема образца на разделение 105
2.6.2. Перегрузка по массе: влияние массы образца на разделение 107
2.6.3. Как избежать проблем, связанных со слишком большим количеством
образца 108
2.6.3.1.Если концентрация образца больше ожидаемой 109
2.6.3.2. Анализ следовых количеств вещества 109
2.7. Смежные темы 110
2.7.1. Уравновешивание колонки ПО
2.7.2. Градиентное элюирование ПО
2.7.3. Пиковая емкость и двумерное разделение 4
2.7.4. Отслеживание пика 112
2.7.5. Вторичные равновесия 114
2.7.6. Переключение колонок 115
2.7.7. Предварительная оценка удерживания по структуре анализируемого
вещества 116
2.7.7.1. Модель сольватационных параметров 118
Литература 119
2.1. Введение
Без понимания того, как разделение зависит от экспериментальных условий
(колонка, растворитель, температура, скорость потока и т.д.) оно успешным не будет.
В этой главе будут рассмотрены некоторые общие аспекты ВЭЖХ, которые помогут
разработать приемлемое разделение (разработка метода), выполнять рутинные
ВЭЖХ-анализы образцов и преодолевать трудности по мере их возникновения.
Как правило, описательный или качественный подход лучше всего позволяет понять
процесс разработки метода и использование ВЭЖХ в рутинных целях. Поэтому
читатель может пролистать или пропустить любые из последующих выводов формул,
по крайнее мере при ознакомлении с текстом. Важные уравнения, необходимые в
работе, помещены в рамочку, например, уравнение (2.5).
2.2. Хроматографический процесс
На рис. 2.1 схематически изображена ВЭЖХ-система. Сплошными стрелками
указано направление потока: из емкости с растворителем к детектору. Растворитель
обычно называют элюентом или подвижной фазой. В главе 3 более подробно описан
каждый узел ВЭЖХ-системы. После того, как образец из инжектора попадает в колонку,
происходит разделение. Разделенные компоненты образца покидают колонку (элюи-
руются или вымываются из нее) и с потоком подвижной фазы направляются в
детектор, в большинстве случаев ультрафиолетовый (УФ) или масс-спектрометриче-
ский (МС). В главе 4 подробно рассказано о каждом виде ВЭЖХ-детекторов.
Основные свойства или «режим» разделения определяются, главным образом,
выбором колонки (табл. 2.1). В настоящее время для анализа образцов преимущественно
используют обращенно-фазовую хроматографию (ОФХ), для которой характерно
использование неполярной неподвижной фазы (колонки) совместно с полярной
подвижной фазой (смесь воды и органического растворителя). В этой книге, если
не оговорено иное, в основном, рассматриваются разделения в режиме ОФХ. Другие
режимы ВЭЖХ можно найти в нижеследующих разделах этой книги (табл. 2.1).
В главах 2—8 мы будем считать состав растворителей на протяжении всего
разделения постоянным, что характерно для изократического элюирования. В случае
градиентного элюирования соотношение элюентов во время разделения сознательно
изменяют (раздел 2.7.2, глава 9).
Колонка представляет собой цилиндрическую трубку, наполненную чаще всего
небольшими (как правило, диаметром от 1,5 до 5 мкм) сферическими частицами
Образцы
i-»D
Колонка Детектор
Насос
Инжектор
Емкость с растворителем
Рис. 2.1. Схематическое изображение ВЭЖХ-системы.
Глава 2. Основные принципы и управление разделением
Таблица 2.1. Режимы ВЭЖХ-разделений
Хроматографи-
ческий режим
Обращенно-
фазовая
хроматография
(ОФХ)
Нормально-фазовая
хроматография (НФХ)
Неводная обра-
гценно-фазовая
хроматография
(НОФХ)
Хроматография
гидрофильных
взаимодействий (ГИХ)
Ионообменная
хроматография
(ИОХ)
Ион-парная
хроматография
(ИПХ)
Эксклюзионная
хроматография
(ЭХ)
Комментарии
Неполярная колонка (например, Qg), полярная подвижная
фаза — смесь воды и органического растворителя
(например, ацетонитрила). ОФХ очень популярна, особенно
для образцов, растворимых в воде
Полярная колонка (например, непривитый силикагель),
неполярная подвижная фаза — смесь неполярных и слабо
полярных растворителей (например, гексана и хлористого
метилена). НФХ подходит для анализа нерастворимых в воде
образцов, препаративной ВЭЖХ и разделения изомеров
Неполярная колонка (например, Qg), подвижная фаза —
смесь органических растворителей (например, ацетонитрила
и хлористого метилена). С помощью НОФХ разделяют
очень гидрофобные, нерастворимые в воде образцы
Полярная колонка (например, силикагель или силикагель
с привитой амидной фазой), подвижная фаза — смесь воды
и органического растворителя (например, ацетонитрила).
ГИХ используют для сильнополярных и поэтому слабоудер-
живаемых в ОФХ образцов
Колонка содержит заряженные группы, которые могут
взаимодействовать с противоположно заряженными ионами
образца, подвижная фаза — смесь водного раствора соли и
буфера. В режиме ИОХ разделяют ионизированные
молекулы, такие, как кислоты и основания.Особенно этот режим
подходит для разделения крупных биомолекул (например,
белков и нуклеиновых кислот)
Используют условия ОФХ, но в подвижную фазу
дополнительно добавляют ион-парный агент для взаимодействия
с противоположно заряженными ионами образца. С
помощью ИПХ разделяют кислоты или основания, слабо
удерживаемые в ОФХ
Используют нейтральную колонку с водной или
органической подвижной фазой. Принцип ЭХ — разделение веществ
по молекулярной массе, поэтому в этом режиме главным
образом разделяют крупные биомолекулы или
синтетические полимеры
Глава, в которой
можно найти
подробное описание
Глава 6,
раздел 7.3
Глава 8
Раздел 6.5
Раздел 8.6
Разделы 7.3,
13.4.2
Раздел 7.4
Раздел 13.8
(рис. 2.2а). Основой частицы в большинстве случаев является пористый силикагель.
На рис. 2.2 изображена пора в виде цилиндра определенного диаметра (обычно
около 10 нм для «небольших молекул» массой менее 1000 Да). Внутренняя поверхность
каждой поры покрыта неподвижной фазой, в этом примере группами Cjg, привитыми
к частице силикагеля. На рис. 2.2в можно видеть приближенное к реальности
изображение современных пористых частиц для ВЭЖХ. Частицы, как показано на
рисунке, формируются из небольших сферических субчастиц. Поры представляют собой
пространство между субчастицами. Поскольку практически вся поверхность частицы
находится внутри этих пор, то большинство молекул образца удерживаются внутри
частицы, а не на ее поверхности. На внутреннюю поверхность пор приходится более
99% всей площади поверхности частиц, поэтому влияние внешней поверхности
частицы на разделение в большинстве случаев ничтожно мало. Подвижная фаза,
2.2. Хроматографический процесс
(а) Колонка с изображением потока подвижной фазы
подвижная
фаза
вход выход
(б) Частица сорбента и окружающая подвижная фаза
подвижная фаза
частица
(в)
Пористая частица
(увеличенное
изображение)
хЮ
Рис. 2.2. ВЭЖХ-колонка. (а) Колонка упакована сферическими частицами; (б)
схематическое изображение частицы, демонстрирующее идеальную пору с
привитыми С[8-группами; (в) более реалистичное изображение сферической
пористой частицы (10-кратное увеличение)
перемещаясь внутри колонки, обволакивает каждую частицу, и благодаря диффузии
молекулы образца проникают в поры частиц. Обычно поток практически никогда не
течет сквозь частицу.
На рис. 2.3 изображено гипотетическое разделение образца, содержащего три
соединения (или три растворенных вещества). Вещество X обозначено •, Y— □, Z— а.
Молекулы элюента для упрощения не показаны, а молекулы растворителя, в
котором растворен образец, изображены в виде +. Нанесенный на колонку образец
(рис. 2.3а) под действием потока подвижной фазы перемещается по колонке
(рис. 23б-г) и, в итоге, растворенные вещества последовательно вымывается из нее
(рис. 2.3d). Детектор при этом регистрирует изменение сигнала в зависимости от
времени, такую запись разделения называют хроматограммой. Во время разделения
(рис. 2.3а-г) вещества X, Y, Z ведут себя по-разному: различаются скорости их
перемещения и распределение молекул этих веществ. К моменту, показаному на рис. 2.3г,
вещества X, Y, Z внутри колонки отделились друг от друга.
Различные скорости перемещения (разные средние скорости, с которыми
вещества X, Y, Z двигаются или перемещаются внутри колонки) являются основой хрома-
60
Глава 2. Основные принципы и управление разделением
(а) (б) (в) (е)
Рис. 2.3. ВЭЖХ-разделение. (а-г) Последовательное разделение внутри колонки (в
зависимости от времени); (д) конечная хроматограмма; (е) оценка значений к
по хроматограмме (д). Растворенные вещества X, Y, Z обозначены •, □, А,
соответственно, молекулы растворителя, в котором растворен образец,
показаны +
тографического разделения. Отсутствие разницы в скорости перемещения говорит
о невозможности разделения двух соединений. В этом примере быстрее всех
перемещаются молекулы X (•), а медленнее всех — Z (ж). Молекулы растворителя, в
котором растворен образец, как и молекулы подвижной фазы, вообще не удерживаются
на колонке, проскакивают сквозь нее быстрее всех и вымываются из колонки
раньше молекул образца. На рисунке молекулы растворителя образца показаны +
(рис. 2.36-й).
По мере перемещения вещества по колонке его молекулы все более существенно
распределяются по ее объему. Объем, который занимают молекулы вещества внутри
колонки, называют полосой или зоной. Ширина этого объема измеряется по
направлению потока и называется шириной полосы. На рисунке 2.3о-г ширина полосы для
вещества X (•) указана стрелкой и скобкой. Полоса, смытая с колонки и
зарегистрированная на хроматограмме (рис. 2.3d), называется пиком. Распознавание пика
2.3. Удерживание 61
проводят по времени его выхода из колонки (время удерживания ?д). Концентрация
растворенного вещества в образце пропорциональна размеру пика, определяемому
либо по его площади, либо по его высоте (раздел 11.2.3). При достаточно малом
количестве вещества, наносимом на колонку (от нескольких нанограмм до микрограмм,
что характерно для количественного ВЭЖХ-анализа), время удерживания не зависит
от концентрации образца (и размера получающегося пика). Далее в этой главе
зависимость разделения от экспериментальных условий будет изучена подробнее.
2.3. Удерживание
Время удерживания вещества tR — это промежуток времени, которое проходит с
момента ввода вещества в инжектор до появления вершины пика на хроматограмме.
Время удерживания веществ X, Y, Z соответственно 2, 3 и 5 минут (рис. 2.33). Время
удерживания пика растворителя, равное одной минуте, называют мертвым временем
?0 колонки (раздел 2.3.1) (иногда для обозначения мертвого времени колонки t0
заменяют tm). Степень перемещения или скорость их, с которой вещество X двигается
по колонке, определяется фракцией R молекул вещества X, находящихся в любой
момент времени в подвижной фазе. В среднем их равна R, умноженной на скорость
перемещения или скорость и молекул растворителя:
их = Ru. (2.1)
Например, если половина молекул X находится в подвижной фазе (R = 0,5),
а вторая половина адсорбировалась на неподвижной фазе, значит, в любой момент
времени только половина всех молекул перемещается с потоком и, следовательно,
средняя скорость вещества X будет равна половине скорости движения подвижной
фазы.
На рис. 2.4 показано, что фракция R молекул вещества X в подвижной
(движущейся) фазе определяется равновесным процессом:
X (подвижная фаза) <-> X (сорбент). (2.2)
Молекулы X в любой момент времени присутствуют в равном количестве в
подвижной и неподвижной фазах, в то время как молекулы вещества Z
преимущественно адсорбировались на неподвижной фазе. То есть вещество Z сильнее
удерживается по сравнению с веществом X, а значит, и перемещается медленнее (указано
в нижней части рис. 2.4 стрелками, длина которых отражает скорость перемещения).
На это равновесие и скорость перемещения вещества влияют структура молекулы,
химический состав элюента и сорбента (растворителя и колонки), а также
температура. Давление, изменяемое вдоль колонки, при разделении оказывает незначительное
влияние на удерживание вещества [1] (см. также раздел 2.5.3.1) и, как правило,
не учитывается при умеренных значениях (например, менее 345 бар/5000 psi).
2.3.1. Фактор удерживания к и мертвое время колонки t0
Для данных соединений фактор удерживания к (его до сих пор еще называют
коэффициентом емкости к') определяют по отношению количества вещества на
неподвижной фазе (s) к его количеству в подвижной фазе (т). Количество вещества
1 Надо все же понимать, что выходит из колонки не пик, а вещество, в то время как пик есть
лишь отражение этого процесса на хроматограмме. — Прим. перев.
Глава 2. Основные принципы и управление разделением
подвижная
фаза
частица
неподвижная
фаза
подвижная фаза
X
Z
Рис. 2.4. Равновесное распределение молекул растворителя и вещества между элюен-
том и сорбентом, и его влияние на скорость перемещения веществ. Значения
к для веществ X и Z равны 1 и 4 соответственно. Предполагается, что в
образце находится равное количество X и Z.
в каждой фазе равно его концентрации (Cs или С,
на объем фазы (Vs или Vm, соответственно), то есть:
^ Ч 'Ч ^S I ^1
соответственно), умноженной
к = -
V I V
Y s I у т
= КЧ,
(2.3)
где К= (Cs/Cm) — константа равновесия в уравнении (2.2), а Ч> = (Vs/Vm) — это
фазовое отношение, то есть соотношение объемов неподвижной и подвижной фаз внутри
колонки. Далее будет показано, что величина к — важная характеристика каждого
пика на хроматограмме. Значения к позволяют нам интерпретировать и улучшать
разделение. Молекулы анализируемого вещества должны присутствовать в обеих
фазах, то есть, если фракция молекул вещества, находящаяся в элюенте, будет R, то
фракция молекул вещества на сорбенте должна быть 1-R. Тогда из уравнения (2.3)
следует:
к = —- (2.3а)
R
R = ■
(2.36)
2.3. Удерживание
Время удерживания X (tg) можно вычислить, разделив расстояние на скорость
(скорость полосы), где расстояние — это длина колонки L, а скорость полосы — это их:
tR = А. (2.4)
их
Аналогичным образом определяют время удерживания пика растворителя
t0 = -. (2.4а)
и
где и — средняя скорость подвижной фазы. Если из уравнения (2.4а) выразить
величину L и подставить в уравнение (2.4), то получится:
tR = ^. (2.46)
их
Если из уравнения (2.1) выразим R = ujug и подставим в уравнение (2.36), то
получим:
(2.5)
tR =t0(l + к).
Кроме того, уравнение (2.5) можно выразить через объем удерживания VR = tRF,
где F — скорость подвижной фазы (мл/мин):
VR = Vn(l + к). (2.5а)
Здесь Vm — это мертвый объем колонки, равный t0F (далее величина Vm и уравнение
(2.5а) будут рассмотрены подробнее).
Уравнение (2.5) можно записать по-другому:
£ _ tR ~?0
(2.6)
По уравнению (2.6) можно вычислить значения к каждого пика на хроматограм-
ме. На практике к часто оценивают визуально по хроматограмме (на основе
уравнения 2.6), поскольку в рутинных анализах и при разработке разделения {разработка
метода) редко требуются точные значения к. Таким образом, к эквивалентен
разности времен удерживания (tg-tg), отнесенной к ?д или:
tR I 1. (2.6а)
to
По /д можно также примерно оценить к (рис. 2.3е, который соответствует
хроматограмме на рис. 2.3д). Так для соединения Xк равно одному отрезку to, считая от /д,
для Y— двум отрезкам /g, Z— четырем отрезкам.
Далее в разделе 2.4.1 описано почему к предпочтительно должно находиться
в интервале значений от 1 до 10. Поэтому очень важно уметь оценивать (или
вычислять) значения к различных пиков на хроматограмме, что, в свою очередь, требует
знания мертвого времени t0 колонки. Величину t0 обычно определяют визуально
по отклонению базовой линии в самом начале хроматограммы (рис. 2.5а-б). Иногда
первый всплеск базовой линии имеет характерную форму (рис. 2.5а), которая точно
указывает на пик неудерживаемого растворителя образца. Такой всплеск при /д
обычно происходит при смене показателя преломления (ПП) подвижной фазы (из-за
разницы ПП подвижной фазы и растворителя образца), что влияет на количество
света, проходящего через ячейку детектора. Пик при /д может отсутствовать, если
образец растворяют в элюенте (лучше всего так и делать).
(а)
Глава 2. Основные принципы и управление разделением
<б)
0 Т
(s)
0 t
to Г?
Рис. 2.5. Определение мертвого времени to колонки
(г)
тиомочевина
В остальных случаях, особенно в тех, когда речь идет об анализе реакционной
массы, проб из окружающей среды, растительных или животных экстрактов, в
начале хроматограммы (рис. 2.56) присутствует очень большой («неудерживаемый»
или «мусорный») пик. Здесь значение /д соответствует началу подъема пика. Иногда
на хроматограмме можно и не увидеть пика растворителя (рис. 2.5в), тогда его
определяют или вычисляют. Наиболее очевидный способ определения tg — анализ не-
удерживаемого (к = 0) легко детектируемого вещества, растворенного в воде или
подвижной фазе (рис. 2.5г). Если использовать УФ детектор с длиной волны менее
220 нм, то в качестве тестового образца можно взять тиомочевину. Для определения
?о также подходят урацил или концентрированные растворы солей, поглощающих в
УФ, например, нитрата натрия [2—4]. Наблюдаемое значение ?о для одной и той же
колонки может меняться в зависимости от состава подвижной фазы. Обычно
колебания ?о составляют менее 5% в диапазоне 20—80 %В (%В означает объемное
содержание в процентах органического компонента в подвижной фазе), а максимальные
отклонения достигают ±10-15% при крайних значениях %В (0, 100 %В) [5]. Чтобы
примерно оценить к (рис. 2.3d), достаточно предположить, что значение to,
измеренное при одном %В, будет таким же при всех значениях %В (при условии, что
меняется только %В).
С другой стороны, величину /q можно оценить, основываясь на размерах
колонки и величине скорости потока (для колонок, упакованных полностью пористыми
частицами):
to
5x10-
Ld}
(2.7)
Здесь L — длина колонки в мм, dc — внутренний диаметр колонки в мм, F —
скорость потока в мл/мин и /0 в минутах. Оказалось, что для нескольких сотен разных
ОФХ-колонок ?0, вычисленное по уравнению (2.7), совпадает с экспериментальными
значениями t0 со средней ошибкой ±10% (1 СКО) [6]. На практике такая точность
вполне достаточна. Мертвый объем колонки Vm связан с t0 следующей зависимостью:
Vm = t0F я 5 x\Q-ALd}. (2.7a)
где L и dc измеряются в мм. Мертвый объем колонки Vm — это общий объем
подвижной фазы внутри колонки, вне частиц сорбента и внутри их. Например, Vm =
= 2 мл, F = 0,5 мл/мин, тогда t0 = Vm/F = 2/0,5 = 4 мин. t0 можно расценивать как
время, требуемое для полной замены подвижной фазы, которой колонка была
изначально заполнена.
В общем случае для колонок с внутренним диаметром 4—5 мм удобно оценивать
Vm соотношением (объединив уравнения 2.7 и 2.7а):
Кт(мл) « 0,011. (2.76)
где длина колонки L в мм. Тогда зная Vm, можно получить значение t0: t0 = Vm/F.
Дальнейшее обсуждение измерения, точности и смысла мертвого времени и мертвого
объема колонки смотрите в работах [2—4]1.
2.3.2. Роль условий разделения и состава образца
Относительное влияние условий разделения на удерживание образца к обобщено
во второй колонке таблицы 2.2. Данные таблицы 2.2 применимы к любым режимам
разделения, но информация, изложенная ниже, касается обращенно-фазовой
хроматографии (ОФХ). Подвижная фаза, как правило, представляет собой смесь воды или
водного буфера (элюент А) и органического растворителя (элюент В), такого как
ацетонитрил или метанол. С увеличением объемной доли органического
растворителя (%В) снижается удерживание всех компонентов образца. Подвижная фаза,
при которой значения к уменьшаются, называется «сильной», то есть вода относится
к «слабым» элюентам, а органические растворители — к «сильным». При увеличении
доли элюента В на 10% значения к обычно снижаются в 2—3 раза. Влияние доли
растворителя В на удерживание образца показано на примере разделения смеси
из 5 гербицидов (рис. 2.6). Подвижная фаза, содержащая 80 %В, вызывает быстрое
элюирование компонентов образца (рис. 2.6а) с небольшими значениями к (0,3—
0,8), что приводит к плохому разделению. Если %В снизить (50 %В, рис. 2.66),
то разделение улучшится, но станет продолжительным (16 минут по сравнению
Таблица 2.2. Влияние условий разделения на удерживание (А), селективность (а) и число
теоретических тарелок (N)
Условия разделения
%В
Элюент В (ацетонитрил, метанол и т.д.)
Температура
Тип колонки (С[8, фенил, циано и т.д.)
рН подвижной фазы"
Концентрация буфера"
Концентрация ион-парного реагента"
Длина колонки
Размер частиц
Скорость потока
Давление
к
++
+
+
+
++
+
++
0
0
0
-
а
+
++
+
++
++
+
++
0
0
0
-
N
-
-
+
-
+
-
+
++
++
+
+е
Примечание: ++ сильное влияние, + умеренное влияние, - относительно небольшое влияние,
0 никакого влияния. Выделенные параметры (обозначенные ++) указывают на условия,
которые лучше использовать в первую очередь (рекомендуются) для контроля за к, а, N (например,
чтобы контролировать к или а, изменяют %В; длина колонки влияет на N).
"Для ионизированных соединений (кислот или оснований).
гБолее высокое давление позволит получить большие значения N при правильном выборе
остальных условий. Впрочем, давление само по себе мало влияет на N (см. раздел 2.4.1.1 и
2.5.3.1).
1 Приведенный в данном разделе поход предназначен лишь для оценки значения мертвого
времени, но не может быть использован для физико-химических измерений. — Прим. перев.
Глава 2. Основные принципы и управление разделением
80% метанол
(0.3<к<0.8)
0,2
50% метанол
(4<к<Щ
0.4
0.6 0.8
Время (мин)
~~Г~
1.0
1.2
и
2 3
_Л_
10'
Время (мин)
1.4
(б)
5
15
Рис. 2.6. Зависимость результатов разделения от %В в подвижной фазе (об. % метанола).
Образец состоит из смеси гербицидов: 1 — монолинурон, 2 — метобромурон,
3 — диурон, 4 — пропазин, 5 — хлороксурон. Условия: колонка Cig,
150 х 4,6 мм, 5 мкм, подвижная фаза метанол — вода, скорость потока
2,0 мл/мин, комнатная температура. Хроматограмма смоделирована по
данным работы [7]
с 1,5 минутами на рис. 2.6а). При этом из-за размывания высота пиков уменьшится.
Как правило, подбирая необходимый %В, можно регулировать к в желаемом
интервале. Кроме того, варьируя параметры, приведенные в таблице 2.2, можно
контролировать селективность разделения (б) и эффективность колонки (N). Более подробно
см. раздел 2.5.
ОФХ предполагает неполярную неподвижную фазу (например, С18) и полярный
водный элюент. Здесь работает принцип «подобное взаимодействует с подобным»:
то есть полярные соединения (1,3-пропандиол) будут преимущественно
взаимодействовать с полярной подвижной фазой (рис. 2.76), а значит, будут менее удерживаемы
(большие значения R и небольшие к), а неполярные вещества (н-гексан), напротив,
в основном будут взаимодействовать с неполярной стационарной фазой (рис. 2.7а),
что приведет к более сильному удерживанию (небольшие R и большие к). На
рисунке 2.7в изображена хроматограмма нескольких монозамещенных бензолов, имеющих
разную полярность или «гидрофобность» из-за наличия разных по своей природе
заместителей. Полярные (менее гидрофобные) группы, например, —NHCHO,
—СН2ОН или —ОН снижают удерживание по отношению к незамещенному бензолу
(заштрихованный пик). Менее полярные (более гидрофобные) группы, например,
хлор, метил, бром, йод и этил, наоборот, увеличивают удерживание.
Ионизированные кислоты и основания гораздо «полярнее», а значит, менее
удерживаемы по сравнению с их нейтральными формами. Изменение рН подвижной
фазы, которое приводит к повышению ионизации вещества, еще больше снижает
удерживание (раздел 7.2).
2.3. Удерживание
(а)
(б)
молекула образца.
НО-фН2-СН2-СН£нОН
C-ta Cie C-iig С18 С18 С1а С18
I I 1111 I
Неполярное (гидрофобное)
взаимодействие с неполярной
неподвижной фазой
Н
о—н
Полярное (образование водородной
связи) взаимодействие с полярной
подвижной фазой
(в)
-NH-CHO (бензилформамид)
-СНгОН (бензиновый спирт)
-ОН (фенол)
-СНО (бенэальдегид)
-СОСН3 (ацетофенон)
-CN (бензонитрил)
-N02 -СООСН3 (метилбензоат)
-ОСН3 (анизол)
бензол
■С1+ -СН3 (хлорбензол
+ толуол)
-СНгСН3
Время (мин)
Рис. 2.7. Полярность и удерживание образца. Показано взаимодействие неполярного
образца со стационарной фазой (о) и полярного образца с подвижной
фазой (б); (в) влияние различных заместителей монозамещенных бензолов на
удерживание. Условия: колонка Hypersil Сщ 150 х 4,6 мм, подвижная
фаза 50% ацетонитрил/вода, скорость потока 2 мл/мин, 25 °С, хроматограмма
смоделирована по данным работы [8]
2.3.2.1. Межмолекулярные взаимодействия
В этом разделе изложена информация, позволяющая более глубоко изучить вопрос
зависимости удерживания от состава образца, колонки и подвижной фазы. Кроме того,
здесь содержится больше материала, чем обычно требуется на практике. Поэтому
читатель может его пропустить и перейти к следующему разделу 2.3.2.2, а сюда
вернуться по мере необходимости.
Взаимодействие между ближайшими молекулами вещества и растворителя
происходит благодаря различным межмолекулярным силам (рис. 2.8). В принципе,
количественное понимание этих взаимодействий должно позволить оценить или даже
предсказать зависимость удерживания от структуры молекулы. Хотя в настоящее время1
сделать это практически невозможно (см. раздел 2.7.7), понимание этих взаимо-
На момент написания книги. — Прим. перев.
Глава 2. Основные принципы и управление разделением
действий в других ситуациях может оказаться полезным. Например, в выборе другой
колонки при попытке изменить разделение (раздел 5.4).
Дисперсионные взаимодействия (рис. 2.8а) возникают в результате случайных,
мгновенных смен положений электронов вокруг соседнего атома растворителя (S)
или вещества (X). Обычно в каждый определенный момент электроны асимметрично
окружают ядро S (рис. 2.8а), что приводит к тому, что электроны ближайшего атома
X (из-за кулоновского отталкивания) перемещаются так, как показано на рисунке.
В результате чего между молекулами S и X возникает мгновенный дипольный
момент, который вызывает электростатическое притяжение. Сила дисперсионных
взаимодействий зависит от поляризуемости двух соседних атомов. Чем больше размер
молекулы (количество атомов или молекулярная масса) и показатель преломления
[9], тем больше поляризуемость, поэтому дисперсионные взаимодействия сильнее
для ароматических соединений и для молекул, у которых заместителями являются
атомы с более высоким атомным весом (сульфид, хлорид, бромид и т.д.), при
условии, что молекулы имеют примерно одинаковый размер .
Дисперсионные взаимодействия существуют между каждыми двумя соседними
атомами, и это взаимодействие во многом объясняет физическое притяжение между
молекулами всех видов (особенно для менее полярных молекул). Из-за
неспецифической и универсальной природы дисперсионных взаимодействий они значительны
как в подвижной, так и в неподвижной фазе. Это приводит к тому, что
дисперсионные взаимодействия, как правило, мало влияют на селективность разделения при
смене подвижной фазы или колонки. Дисперсионные взаимодействия вносят вклад
в гидрофобные взаимодействия, которые так называются из-за того, что они обуслов-
(а) •
(б)
\
Дисперсионное
Д и п ол ь-ди л о л ь ное
(в)
СН30-Н - ■ ■ N(CH3)a -
О*
(г)
-J
с+_;>! х* )<-_+■>
>~\
Водородная связь
(д)
Перенос заряда (ге-гс)
Ионное
Рис. 2.8. Межмолекулярные взаимодействия, влияющие на удерживание образца и
селективность.
ливают притяжения менее полярных соединений к неполярному обращенно-фазо-
вому сорбенту (или вытеснение гидрофобных молекул из полярной водной фазы).
С возрастанием силы дисперсионного взаимодействия (для более крупных и менее
полярных образцов) происходит существенный рост удерживания.
Диполь-дипольные взаимодействия возникают между молекулами, имеющими
постоянные или наведенные диполи. На рис. 2.86 изображено взаимодействие
молекулы ацетонитрила (СНзС=1М) и нитроалкана (R—NO2). У функциональных групп
(—C=N и —NO2) этих двух молекул есть большой постоянный дипольный момент,
благодаря которому молекулы занимают такое положение, в котором
электростатическое взаимодействие максимально (положительно заряженный полюс одной
молекулы находится рядом с отрицательно заряженным полюсом другой). Сила диполь-
ного взаимодействия пропорциональна дипольному моменту каждой из двух
взаимодействующих групп, а не дипольному моменту целой молекулы (в которой
может быть много заместителей), так как дипольные взаимодействия возникают только
на очень близком расстоянии, например, между соседними атомами или групппами.
Водородные связи возникают в двух случаях: кислотный (или протон-донорный)
растворитель (метанол) взаимодействует с основным (протон-акцепторным)
веществом (7У,7У-диметиланилин), или кислотное вещество (фенол) взаимодействует
с основным растворителем (тетрагидрофуран, ТГФ) (рис. 2.8в). Сила
взаимодействия увеличивается с увеличением кислотности и основности
взаимодействующих групп (табл. 2.3).
Таблица 2.3. Параметры селективности растворителей
Растворитель
Уксусная кислота
Ацетонитрил
Алканы
Хлороформ
Диметилсульфоксид
Этанол
Этилацетат
Хлористый этилен
Метанол
Хлористый метилен
Метил-т-бутиловый эфир
Нитрометан
Пропанол (я- или изо-)
Тетрагидрофуран
Триэтиламин
Вода
Приведенная селективность"
Кислотность ВС
а/1
0,54
0,15
0,00
0,43
0,00
0,39
0,00
0,00
0,43
0,27
0,00
0,17
0,36
0,00
0,00
0,43
Основность ВС
Р/Е
0,15
0,25
0,00
0,00
0,43
0,36
0,45
0,00
0,29
0,0
=0,6
0,19
0,40
0,49
0,84
0,18
Полярность я*/Е
0,31
0,60
0,00
0,57
0,57
0,25
0,55
1,00
0,28
0,73
«0,4
0,64
0,24
0,51
0,16
0,45
р'б
6,0
5,8
0,1
4,1
7,2
4,3
4,4
3,5
5,1
3,1
«2,4
6,0
3,9
4,0
1,9
10,2
г"
6,2
37,5
1,9
4,8
4,7
24,6
6,0
10,4
32,7
8,9
«4
35,9
6,0
7,6
2,4
80
Примечание: дополнительная информация о растворителях приведена в Приложении I
(табл. 1.4).
"Значения из [11], Z — это сумма параметров а, (3 и л* каждого растворителя.
''Коэффициент полярности [12J.
"Диэлектрическая постоянная [13].
Глава 2. Основные принципы и управление разделением
Пример ионных (кулоновских) взаимодействий между положительно заряженным
ионом образца (Х+) и окружающими его поляризуемыми молекулами растворителя
показан на рис. 2.8г. Под действием положительно заряженного иона в молекулах
растворителя смещается заряд, что приводит к максимальному электростатическому
взаимодействию. Сила ионных взаимодействий увеличивается с увеличением
диэлектрической проницаемости е растворителя (табл. 2.3). Кроме того, ионные
взаимодействия могут возникать между заряженными ионами образца и любыми ионами в
подвижной или стационарной фазе. Более подробно этот вопрос обсуждается в разделе
7.4.1 (ион-парная хроматография) и разделе 7.5.1 (ионообменная хроматография).
Перенос заряда или л-л взаимодействия показаны на рисунке 2.$д на примере
1,3-динитробензола (обедненная электронами лжислота) и бензола (насыщенное
электронами л-основание). Взаимодействия такого рода возникают между любыми
ароматическими (или ненасыщенными) соединениями. Причем, чем сильнее
л-основания (такие как полициклические ароматические соединения, например,
нафталин и антрацен) и лжислоты (например, ароматические соединения,
замещенные электронакцепторными нитрогруппами), тем сильнее л-л взаимодействия.
Растворитель ацетонитрил, являющийся л-кислотой, тоже может вступать в л-л
взаимодействия с ароматическими образцами [10].
Полярные взаимодействия различных неионных алифатических растворителей,
применяемых в ВЭЖХ, можно представить в виде треугольника селективности
растворителей (рис. 2.9, [11]). Положение каждого растворителя в треугольнике
указывает на относительную кислотность его водородных связей a/Z, основность его
водородных связей |3/Z и полярность % /Z- Амины, например, относительно сильные
основания (судя по положению в верхней части треугольника, большое значение |3).
У нитроалканов, алифатических нитрилов, хлористого метилена есть группы с
большими дипольными моментами, поэтому они располагаются в нижней правой части
Н-В Основания
основные
растворителе
кислотные
растворители
ТГФ
дмсо
сложные
эф иры
нитрилы
• формам^ды - ™,'.
)_I-q * .' нмтросоединения
биполярные
растворители
персрторспирты
СНС1,',/
СН,С1~
Н-В Кислоты {«/!)
Биполярные (Jt*/Z)
Рис. 2.9. Треугольник селективности разного рода алифатических растворителей.
В таблице 2.3 приведены характеристики растворителей [11]
2.4. Ширина пика и число теоретических тарелок колонки N
71
треугольника. Перфторспирты — очень сильные доноры водорода (и одновременно
слабые акцепторы). Их, также как и карбоновые кислоты, можно найти в нижней
левой части треугольника (большие значения а). В таблице 2.3 приведены:
1. Относительные вклады дипольных взаимодействий, кислотности и основности
водородных связей в общую полярность растворителя;
2. Мера общей полярности растворителя (Р);
3. Диэлектрическая постоянная (е)
Чем больше е подвижной фазы, тем сильнее ионные взаимодействия в растворе
(рис. 2.8г), тем лучше ионные образцы растворяются в подвижной фазе и тем
меньше значения к для ионных образцов. Подробный обзор межмолекулярных
взаимодействий в хроматографии см. [14].
Далее, в главах 6 и 8, мы еще раз вернемся к треугольнику селективности
растворителей (рис. 2.9). В разделе 5.4 о селективности колонки аналогичным образом
объяснены взаимодействия между образцом и сорбентом.
2.3.2.2. Температура
Температура — важный параметр в ВЭЖХ. Она оказывает существенное влияние
на коэффициент удерживания. Удерживание большинства образцов в привычных
условиях разделения зависит от температуры в соответствии с уравнением Ван-Гоффа:
log k = А + —, (2.8)
Тк
где А и В не зависящие от температуры константы для данного вещества, Тк —
температура в градусах Кельвина (К). Обычно повышение температуры на один градус
снижает к на 1—2%, т.е. при увеличении температуры на 50 °С удерживание
уменьшается примерно в 2 раза. Повышение температуры зачастую негативно отражается
на разделении, хотя высота пиков растет (как при увеличении доли растворителя В
в элюенте, рис. 2.6). Следует отметить, что отклонения от уравнения (2.8) не так уж
и редки, в результате чего наблюдается криволинейный участок на графике
зависимости log к от 1/Jjf. Повышение температуры очень редко увеличивает коэффициент
удерживания. Исключения из уравнения (2.8) возможны, если при изменении
температуры:
1. происходит изменение ионизации образца [15, 16];
2. меняется конформация образца [17] или свойства стационарной фазы [18].
Кроме того, температура влияет на количество теоретических тарелок и на
перепад давления (раздел 2.4). Большинство термостатов хроматографических систем
имеют температурные ограничения: не более 80 °С (раздел 3.7.2). Период
эксплуатации колонки сокращается, если рабочая температура превышает 60 °С (раздел 5.8).
Более подробно о роли температуры в ВЭЖХ написано в разделе 2.5.3.1 и [19—20а].
2.4. Ширина пика и число теоретических
тарелок колонки N
Молекулы растворенного вещества (рис. 2.3), перемещаясь, распределяются по
колонке и занимают объем, превышающий исходный (или образуют более широкую
полосу). Вещество, вымываясь из колонки, на хроматограмме регистрируется в виде
пика, ширину которого можно вычислить различными способами. На рис. 2.10а
показана ширина W первого пика i на уровне базовой линии. К каждой стороне пика
проводят касательные (через точки перегиба), пересечение которых с базовой линией
Глава 2. Основные принципы и управление разделением
Ш
3.93 МИН
Ш
(а)
3.80 4.00 4.10 4.20
Время (мин)
4.30
80% В
(б)
70% В
0.3 0.8 1.3 (мин) 1.0 1.5 2.0 (мин)
60% В 50% В
2.6
3.1
3.6 (мин)
5.8
6,3
6.8 (мин)
Рис. 2.10. Ширина пика и ее измерение, (а) Определение ширины пика; (б)
зависимость ширины пика 3 рисунка 2.6 от %В. У всех пиков одинаковый
масштаб
образует отрезок W. Когда в этой книге упоминается ширина пика, предполагается,
что это значение W на уровне базовой линии. Относительная способность колонки
давать узкие пики называется эффективностью и определяется числом теоретических
тарелок N:
N
16^
(2.9)
Например, ширина W пика i (рис. 2.10а) равна (4,00 - 3,85) = 0,15 мин, время
удерживания tR= 3,93 мин, тогда N= 16 х (3,93/0,15)2 = 10980. Значения N могут
меняться в зависимости от образца, условий разделения и колонок (раздел 2.4.1). Чем
больше N, тем уже пики на хроматограмме и тем лучше разделение.
Ширину пика удобнее (и точнее) определять по ширине пика на полувысоте W\n
(показано для пика у на рисунке 2.10а). По данным систем сбора и обработки данных
ширина пика на полувысоте
определения N:
W\ji =0,588Ж. Величину W\n также используют для
(
N = 5.54
(2.9а)
2.4. Ширина пика и число теоретических тарелок колонки N
Пик идеальной формы имеет вид Гауссовой кривой (раздел 2.4.2), и поэтому ширину
пика иногда выражают как стандартное отклонение у Гауссовой кривой, то есть
W=4o. (2.96)
Тогда
N 1^-1 . (2.9в)
Уравнение (2.9в) можно представить в другом виде, например: N = 25(tR/iVia)2, где
W5a = 1,25 W так называемая ширина пика 5а.
Уравнение (2.9) можно записать в виде
W = 4N-°-5tR, (2.10)
или (заменив tR уравнением (2.5))
IV = 47V-°>5/0(l +k). (2.10a)
Поскольку для разных пиков на хроматограмме значения N практически постоянны,
то уравнение (2.10) говорит о том, что ширина пика W увеличивается
пропорционально времени удерживания. То есть на хроматограмме от ее начала и до ее конца
наблюдается непрерывное увеличение ширины пика (см. например, хроматограмму
на рис. 2.7в).
При изменении времени удерживания, которое меняется в зависимости от %В,
температуры или колонки, площадь пика, как правило, остается постоянной,
поэтому и произведение высоты пика hp на ширину пика W тоже должно остаться
постоянным. Таким образом
, (константа) (константа) .„ ,,.
h'"—ш—к—; • (2Л1)
W tR
Следовательно, с увеличением tR, высота пика падает. Пример зависимости
высоты пика от %В показан на рисунке 2.106 для пика 3 рисунка 2.6. Видно, что при
варьировании %В высота и ширина пика изменяются обратно пропорционально (что
подтверждает уравнение (2.11)).
2.4.1. Зависимость N от условий разделения
Начнем этот раздел с обобщения некоторых практических выводов относительно
зависимости числа теоретических тарелок N от колонки, образца и условий
эксперимента. Теория следующего раздела 2.4.1.1 именно на них и основывается. Число
тарелок также можно описать следующим уравнением
N = lJ[)L> <2Л2>
где Н = L/N — высота теоретической тарелки. Н показатель эффективности колонки
на единицу ее длины. Увеличение длины колонки (путем замены колонки длиной
150 мм на колонку длиной 250 мм) — очень удобный способ повысить N и улучшить
разделение (поскольку Н постоянна, отличается только длина колонок).
Рассмотрим следующий график в двойном логарифмическом масштабе
(рис. 2.11а), на котором показана зависимость N от скорости потока F и диаметра
частиц dp (2, 5 или 10 мкм), при этом другие параметры разделения остаются
постоянными. При увеличении скорости подвижной фазы F, начиная со значения
0,1 мл/мин, число теоретических тарелок N сначала увеличивается, а потом падает.
Так при данных условиях максимальное значение N (обозначено •) в зависимости
Глава 2. Основные принципы и управление разделением
10=
ю4
150 х 4.6-мм колонки
(а)
N
103
10й
-
: P{psi)
л"^*"^
Г""
-
"
*
;
~
-
-
"
= 2К 5К
,-—~~~ _ ^ "■* w
^.
— "^
15К
S
5ь ^
^ч^-—_Ч
■ч
X
5 ммГ^^
dp^ooTfitor-^
• максимум N
0 ш. раздел 2.4,1.2
, , ( ,
^Ч---^_ 2 мкм
Ч Ч ^\
^ЧА ^\
Ч v\ ч ^
^~~^ \ ч ч-~»
\\ ч ч \^
^^ ч ч ^\
^-^ч N ^ч
C<i \ ^
ч ^-ч_ ч
ч \"А
\ \ >
Р(МПа)= 14 34 100
i i ■'
0.1
1.0 10
F (мл/мин)
100х 4.6-мм, 3-мкм колонка
100
(б)
N
15,000
10,000
5,000
t ^»^ ..-■■•
у / \^ """ 80Х,
^ / ^-ч 200 Да
V зох\. -
/\ 200 Да ^~~~~~^__^^
i Ч ° "
Ч
ч
""--_ 30°С,
~--— 6,000 Да
• максимум N
i i i i i
0 12 3 4 5 6
F (мл/мин)
Рис. 2.11. Изменение числа теоретических тарелок N в зависимости от скорости
потока F, размера частиц dp и других условий. Подвижная фаза — 50% ацетонит-
рил в воде, (а) Условия: колонка 150 х 4,6 мм, 30 °С, молекулярная масса
образца 200 Да; (---) изобара зависимости N(F) при Р = 136 бар (2000 psi),
340 бар (5000 psi) и 1020 бар (15000 psi), соответственно, (б) Условия:
колонка 100 х 4,6 мм, размер частиц 5 мкм, остальные условия указаны на
рисунке. Все графике построены по уравнению (2.18а), где А = I, В = 2 и С =
= 0,05
от размера частиц dp наблюдается при значениях F = 0,2 - 1,0 мл/мин. Трехкратное
увеличение скорости потока относительно «оптимального» значения Fonm, при
котором N максимально, почти никак не сказывается на самом разделении (только лишь
снижает N примерно на 20%), но сокращает его продолжительность в три раза.
Поэтому на практике обычно используют скорость потока выше оптимального
значения. Не рекомендуется устанавливать скорость потока ниже Fonm, поскольку помимо
снижения числа тарелок увеличивается продолжительность разделения.
2.4. Ширина пика и число теоретических тарелок колонки N
Размер частиц влияет на число теоретических тарелок: чем меньше dp, тем
больше N (рис. 2.11а). При уменьшении размера частиц максимальное значение N
достигается при более высокой скорости потока, что позволяет сократить
продолжительность разделения (раздел 2.4.1.1). С маленькими частицами и высокими скоростями
потока перепад давления в колонке (будем его называть просто «давление» Р)
возрастает (штриховые линии на рисунке 2.11а при Р = 136 бар (2000 psi), 340 бар
(5000 psi) и 1020 бар (15000 psi)). Давление (в psi) в упакованной колонке можно
определить по уравнению
„ .. U5£2n
t0dp
(2.13)
или из уравнений (2.7) и (2.13)
2-^1, (2.13а)
где L— длина колонки в мм, г| — вязкость подвижной фазы в сП (значения т| в
зависимости от состава подвижной фазы и температуры приведены в Приложении I),
?0 в минутах, F — скорость потока в мл/мин, dc — внутренний диаметр колонки
в мм, d — диаметр частиц в мкм. В ВЭЖХ, помимо psi, иногда используют другие
единицы измерения давления: бар = атмосфера = 14,7 psi, мегапаскаль (МПа) =
= 10 бар = 147 psi.
На давление влияют также природа сорбента и качество упаковки колонки
(раздел 5.6). Капилляры от насоса до детектора, кран инжектора, ячейка детектора
вносят свой вклад в общее давление, определяемое на выходе из насоса. Однако
суммарный их вклад невелик (10—20%) по сравнению с давлением, вычисленным
по уравнению (2.13). Исключение составляют ВЭЖХ-системы, предназначенные
для работы при давлении > 400 бар, с размером частиц сорбента менее 3 мкм,
которые и в отсутствии колонки демонстрируют высокое сопротивление потоку
(например, > 70 бар). В зависимости от ВЭЖХ-оборудования или проницаемости колонки
отклонение от давления, определенного по уравнениям (2.13) и (2.13а), может
составлять ±20% и даже больше. Несмотря на способность большинства обычных
ВЭЖХ-систем работать в диапазоне 340—400 бар, рекомендуется ограничить перепад
давления до 200 бар (раздел 3.5). Со временем давление в колонке увеличивается,
поэтому давление на новой колонке, предназначенной для обычных анализов,
не должно превышать 150 бар. Впрочем последняя рекомендация устарела, так как
в настоящее время имеющиеся в продаже ВЭЖХ-системы для рутинных задач
вполне выдерживают 600-1000 бар и даже больше (раздел 3.5.4.3, [21]).
Теперь рассмотрим рисунок 2.116: разделение проводят на колонке 100 х 4,6 мм
с частицами 5 мкм, подвижная фаза содержит 50% ацетонитрила (обратите внимание
на двойной линейный масштаб и более узкий и типичный интервал значений F).
Графики зависимости N от F построены для трех различных условий: 1) 30 °С и образец
с молекулярной массой 200 Да, 2) 30 °С и образец с молекулярной массой 6000 Да
(крупный пептид), 3) 80 °С и образец с молекулярной массой 200 Да. Эти графики
для каждого из образцов выглядят сходным образом, за исключением того, что
максимум N или оптимальная Fonm сдвигается вправо (к большим значениям) при
увеличении температуры, и влево (к меньшим значениям) при увеличении размера молекулы.
Из этого можно сделать следующее заключение: разделение при повышенной
температуре лучше проводить при высокой скорости потока, что, в свою очередь, сокращает
продолжительность анализа (раздел 2.5.3.1). Похожие разделения образцов с большой
молекулярной массой при сравнимых значениях N, как правило, требуют уменьшения
скоростей потока, что удлинняет анализ. Зависимость N от геометрических размеров
колонки, молекулярной массы образца и других условий обобщена в таблице 2.4.
Глава 2. Основные принципы и управление разделением
Таблица 2.4. Влияние различных экспериментальных условий на Дг
Условия
Длина колонки L
Диаметр колонки dc
Размер частиц dp
Скорость потока
подвижной фазы F
Вязкость подвижной
фазы т|
Температура Т (К)
Молекулярная масса
образца М
Влияние на Fmm, величина F для
получения максимального Л"
Никакого
F ос (У2
1 опт ^- "с
Г опт ^ V"/>
Никакого
Fonm падает с увеличением т|
Fonm увеличивается с
увеличением Тб
F ос М-0.6
ronm J- lvi
Влияние увеличения указанных в графе
«условия» параметров на Nu Р"
N и Р увеличиваются пропорционально
Никакого, если F увеличивается
пропорционально d} (что рекомендуется)
N падает (рис. 2.12) и Р тоже
N падает (рис. 2.12), Р
пропорционально растет
N падает (уравн. 2.19), Р
пропорционально растет
N увеличивается (рис. 2.13<?), P
снижается
N падает (рис. 2.13в), никакого
влияния на Р
Примечание. См. обсуждение рисунков 2.12 и 2.13.
"Предполагается, что скорость потока равна или превышает оптимальное значение для
максимального N (Fmm), как это часто и делается.
гИз-за увеличения Ги уменьшения вязкости подвижной фазы (ур-ние 2.19).
2.4.1.1. Процессы размывания пика, определяющие значения N
Ширина W и время удерживания /д пика определяют число теоретических тарелок N
колонки (уравнение 2.9). Различные процессы, протекающие внутри и снаружи
колонки, влияют на объем, в котором выходит пик, или его ширину W, как это показано на
рис. 2.12. Обратите внимание на уширение полосы, происходящее в результате
каждого процесса (рис. 2Л2а-д). На это указывают скобки и стрелки около полосы образца.
Когда проба из инжектора попадает в систему, но еще не достигает колонки, она
занимает определенный, обычно небольшой, объем в капилляре (раздел 2.6.1). Это
показано на рисунке 2.12а, где каждая отдельная молекула образца обозначена •. Обычно
внеколоночное уширение полосы вещества можно не принимать во внимание (раздел
3.9), но эта возможность зависит от характеристик хроматографа и размеров колонки
(колонки небольшого объема, упакованные мелкозернистым сорбентом, больше всего
подвержены внеколоночному размыванию пика). Рассмотрим продольную диффузию
вещества в колонке (рис. 2.126). Этот процесс вызывает увеличение ширины пика со
временем, и он возникает вне зависимости от того, течет подвижная фаза или нет.
Время, затрачиваемое веществом при его прохождении через колонку, обратно
пропорционально скорости потока, поэтому вклад в уширение полосы от продольной
диффузии уменьшается при большей скорости потока.
Вихревая диффузия вносит свой вклад в уширение пика (рис. 2.12в). Молекулы
вещества по мере передвижения по колонке переносятся между частицами сорбента
потоком подвижной фазы в различных направлениях (указано стрелками).
Молекулы, попадающие в более медленно движущиеся потоки в суженных каналах между
частицами, отстают, а быстрые потоки в более широких каналах уносят молекулы
вперед. Такое распределение молекул почти не зависит от скорости потока, а
зависит от взаимного расположения и размера частиц сорбента. В плохо упакованной
колонке вихревая диффузия еще больше способствует размыванию полосы вещества.
Массообмен в подвижной фазе (рис. 2.12г) возникает вследстие неравномерной
скорости потока (в центре потока она выше, чем на переферии). С увеличением скорости
2.4. Ширина пика и число теоретических тарелок колонки N
77
(а)
(в)
о^о
О00
0W 0
Ввод образца
(внеколоночное размывание пика)
(в)
©Ж5
(в)
Вихревая диффузия
(независима от скорости потока)
частица
сорбента
пора
; i
Масооперенос s неподвижной фазе
(зависим от скорости потока)
Продольная диффузия
(зависима от времени)
W
Массообмен в подвижной фазе
(зависим от скорости потока)
(е)
Детектор
Поток через детектор +
соединительные трубки
(внеколоночное размывание пика)
Рис. 2.12. Иллюстрация различных процессов, влияющих на размывание пика в
процессе ВЭЖХ-разделения. Молекулы растворенного вещества до
перемещения обозначены О, после — •, > указывает направление движения
молекул вещества
элюента молекулы в центральной части потока перемещаются быстрее, что вызывает
еще большее размывание пика.
Последний процесс, который вносит свой вклад в размывание полосы вещества
внутри колонки, — массоперенос в стационарной фазе (рис. 2.12Й). Некоторые
молекулы образца попадают глубоко в поры частиц (благодаря диффузии) и застревают
там на какое-то время (например, молекула i на рис. 2.12Й). В это время другие моле-
Глава 2. Основные принципы и управление разделением
кулы (например, j) неглубоко проникают в поры, надолго там не задерживаются и
быстро вымываются из них. Молекулы, проводящие мало времени в частицах
сорбента, перемещаются быстрее внутри колонки, увеличивая тем самым размывание
полосы вещества. Чем выше скорость потока, тем больше размывание. В итоге пик
элюируется из колонки и проходит через детектор (рис. 2.12е), при этом происходит
дополнительное внеколоночное размывание, аналогичное размыванию на линии
в промежутке от инжектора до колонки (рис. 2.12а).
Каждый процесс вносит свой вклад в размывание пика (рис. 2.12) и влияет
на окончательную ширину пика W
W1 = T.W?, (2.14)
где Wj — вклад каждого (независимого) процесса i в окончательную ширину пика.
Можно разделить процессы, протекающие внутри колонки и вне ее. Пусть WEC —
суммарное внеколоночное размывание (рис. 2.12а и 2.Не), a W0 — суммарное внут-
риколоночное размывание, тогда
W2 = Wlc + W2. (2.14а)
В правильно собранной ВЭЖХ-системе внеколоночное размывание WEC должно
быть небольшим (раздел 3.9), поэтому далее это слагаемое не будет приниматься
во внимание при обсуждении. Величина WEC, в отличие от W0, не зависит от к
(уравнение 2.10а), поэтому вклад внеколоночного размывания в общую ширину пика
будет больше для раноэлюируемых соединений.
Оставшаяся часть этого раздела и раздел 2.4.1.2 могут оказаться очень полезными
для правильного понимания того, как величина N зависит от экспериментальных
условий. Кроме того, этот материал закладывает основы, позволяющие, с одной стороны,
понять как разработать методы экспресс-разделения (раздел 2.5.3.2), а с другой
стороны, правильно оптимизировать эффективность колонки и разделения. Информация,
изложенная ниже, не очень важна в повседневной работе, она может оказаться несколько
сложной и ее можно пропустить, перейдя сразу к разделу 2.4.2, а к разделам 2.4.1.1
и 2.4.1.2 вернуться позже. Тем не менее, материал, начинающийся с уравнения (2.17),
и особенно раздел 2.4.1.2 может оказаться полезным с практической точки зрения и
заслуживает внимания читателя. Развернутое обсуждение теории размывания пиков
также приводится в работах [22—25].
Величину Wg теперь будем считать эквивалентной ширине пика W
(уравнение 2.10), ее можно выразить через уравнение (2.14):
W1 = Wl + Wl + WlP + W2sp. (2.15)
Продольная Вихревая Массоперенос Массоперенос
диффузия диффузия в подвижной в неподвижной
фазе фазе
Объединив уравнения (2.10) и (2.12), получим:
W7
[тУ-
Н, (2.15а)
где значения Н = L/N для разных веществ в одних и тех же экспериментальных
условиях практически не зависят от времени удерживания tR для колонки длиной L.
Поэтому
W1 к (константа)Я. (2.156)
Если вместо W1 подставить величину Н из уравнения (2.156), то уравнение (2.15)
будет напрямую связано с N:
Н =HL + HE + HMP + HSP, (2.16)
где HL, НЕ, НМР и HSP коррелируют с соответствующими W в уравнении (2.15), т.е.,
например, параметр HL соответствует продольной диффузии и вносит свой вклад в Н
2.4. Ширина пика и число теоретических тарелок колонки N
и т.д. Если мы вернемся к обсуждению рисунка 2.12, отметив, что значения W2
пропорциональны значениям Н (уравнение 2.156), то исходя из теоретических
уравнений для каждого из четырех вкладов в величину W, можно получить следующее
выражение:
Н = А + B/F + CF. (2.16а)
Вихревая Продольный Массоперенос
диффузия массоперенос в подвижной
и неподвижной
фазах
где коэффициенты А, В и С — это константы для конкретных вещества, колонки
и экспериментальных условий. Если в уравнении (2.16а) вместо F подставить
линейную скорость подвижной фазы и, то получится уравнение Ван-Деемтера:
Н=А + - + Си, (2.166)
и
где параметры А, В и С обозначают различный набор констант, относящихся к
конкретному веществу, колонке и экспериментальным условиям.
Уравнения (2.16а и б) не совсем точны, поскольку допускают независимость всех
четырех слагаемых W друг от друга. Чего не скажешь о вихревой диффузии и массо-
переносе в подвижной фазе. Всякий раз, когда два потока, текущие между
частицами, встречаются, происходит смешивание и изменение профиля скоростей,
возникшего вследствие массопереноса в подвижной фазе (так называемое сопряжение).
Поэтому и вихревую диффузию, и массоперенос в подвижной фазе следует
рассматривать как единый процесс размывания полосы. Поскольку массоперенос в
подвижной фазе зависит от скорости потока (ocf1), а вихревая диффузия — нет (ос/0), то
уширение пика под действием этих двух процессов будет описываться степенной
зависимостью F с дробным показателем (F"). Эксперименты дают основание
предполагать, что объединённая величина Н для вихревой диффузии и массопереноса в
подвижной фазе зависит от скорости потока в степени 1/3. Тогда
Н = B/F + AFl'\ + CF
Продольная Вихревая диффузия + Массоперенос
диффузия массоперенос в неподвижной
в подвижной фазе фазе
(А, В, С — константы). И, наконец, обобщенную взаимосвязь уширения пика и
экспериментальных условий можно получить следующим образом: принять, что
параметры А, В и С в свою очередь в различной степени зависят от коэффициента
диффузии Dm и/или диаметра частиц d , поэтому уравнение (2.16) будет выглядеть так:
h = Лу0-33 + - + Cv
(2.17)
Это так называемое уравнение Нокса [25]. Для «усредненного» разделения
коэффициенты уравнения (2.17) можно принять равными А= \, В = 2, С = 0,05 (эти значения
очень приблизительны и в некоторой степени зависят от к, природы колонки и
качества ее упаковки). Теперь дадим определения приведенной высоты теоретической
тарелки h:
h=^- (2.18)
dp
и приведенной скорости v.
v = "^, (2.18а)
ие — интерстициальная скорость подвижной фазы (скорость движения элюента
между частицами сорбента), которая отличается от средней линейной скорости подвиж-
Глава 2. Основные принципы и управление разделением
ной фазы и; в уравнениях (2.18) и (2.18а) используются единицы измерения системы
СГС (см х г х с). Общая пористость колонки (доля свободного объема колонки)
обозначается еги включает объем пор частиц ег- и объем между частицами ге. При этом
ие равна (£т/се)и, где (ег/ее) » 1,6.
Коэффициент диффузии вещества Dm (см2/сек) зависит от его молекулярного
объема (Уд в мл), температуры (Т, в градусах Кельвина) и вязкости подвижной фазы
(г| в сП). Это уравнение Вилки-Чанга[26]:
г| КУ'6 с
где Afj — молекулярная масса растворителя, г[)£ — коэффициент ассоциации, равный
единице для большинства растворителей и больше единицы для растворителей,
способных формировать сильные водородные связи. Например, t|)£ воды равен 2,6, а
метанола — 1,9 (в условиях обычной ОФХ можно принять значение г[)5 к 2).
Уравнение (2.19) достаточно точно для веществ, молекулярные массы которых меньше
500 Да, и может оказаться полезным для приблизительной оценки величин Dm более
крупных молекул.
Уравнение Нокса (2.17) дает основу для оптимизации условий разделения,
то есть позволяет оценить, каким образом можно получить максимальное N за
минимальное время анализа. На результатах анализа этого уравнения также основаны
прогнозы, приведенные в таблице 2.4, и графики на рис. 2.11. Таким образом, можно
хотя бы приблизительно предположить, как будет меняться N при изменении тех или
иных экспериментальных условий. Для того, чтобы применить уравнение Нокса (2.17)
на практике, необходимо связать v с объемной скоростью потока. Это можно
сделать, выразив F через уравнения (2.4а) и (2.7):
F я 0.0005</|и, (2.20)
где F — объемная скорость потока в мл/мин, dc — диаметр колонки в мм, линейная
скорость подвижной фазы и в мм/мин. Преобразуем уравнение (2.18а) с учетом того,
что и в мм/мин, d в мкм, Dm в см2/с, а ие=1.6и:
2,7 х10-7мй?„
v = — '-. (2.20а)
Д.
Объединим уравнения (2.20) и (2.20а) и получим:
5,4 хЮ-4 Fd„
1850uf?vn,
dp
(2.206)
(2.20в)
На рис. 2.13а изображен график зависимости h(v) (—), построенный по
уравнению (2.17); при этом предполагается, что А = 1, В = 2, С = 0,05. Этот график не
зависит от экспериментальных условий, перечисленных в таблице 2.4, поэтому он
применим (приблизительно) ко всем условиям. Из рисунка 2.13а видно, что
минимальное значение h =honm я 2 соответствует значению v=vonm я 3.
(«оптимальные» А и v — это те значения, которые соответствуют максимальному N). Сплошная
кривая на рис. 2.136 повторяет сплошную кривую рис. 2.13а, за исключением того,
что по оси абсцисс отложена не приведенная скорость потока v, а объемная скорость
F (при определенных условиях: 50% ацетонитрил-вода, 30 °С, диаметр колонки
4,6 мм, молекулярная масса образца 200 Да). Изменение любых условий, перечне-
2.4. Ширина пика и число теоретических тарелок колонки N 81
(а)
6
h 4
2
|
\\ >/
1 \ v (оптимальные /
\ \ значения) / Av
ч\ \ У ■'
B/v\ ^~_—^ ^ - . Cv
\ -- ■'
ч -- ■
. *ъ
— — ~~~ ***■*. ■-■"'
1 ■! ,.l.,,itJ-lTfnl rJ — —t^.J—1—L I.LI.
0.1
1.0
10
100
(б)
h 4
N.
•*~^~^
30°C
6000' Да
i
\ 80°C
\ 200 Да
/
/
/
/
/
\ /
\ ■ ^
ЗОХ
200 Да
■-■' —*■
0,1
1
F (мл/мин)
10
Рис. 2.13. Уравнение Нокса. (а) Зависимость приведенной высоты теоретической
тарелки h от приведенной скорости v, где А = I, В = 2, С = 0,05. Сплошной
кривой изображена зависимость общей высоты теоретической тарелки А,
а пунктирными и прерывистыми линиями показаны зависимости трех
составляющих ее компонентов, (б) Влияние молекулярной массы образца
(6000 Да и 200 Да) и температуры (80 °С и 30 °С) на зависимость
приведенной высоты теоретической тарелки h от объемной скорости потока F. Все
графики построены по уравнению (2.17) с параметрами А = 1, В = 2,
С= 0,05
ленных в таблице 2.4, будет смещать график для данного образца (200 Да, 30 °С,
рис. 2.135) влево или вправо. Характер смещения будет зависеть от скорости потока,
соответствующей максимальному N (Fonm соответствует vonm я 3). В соответствии
с прогнозами таблицы 2.4 Fonm растет с увеличением температуры, что мы и видим
на рис. 2.136 (прерывистая кривая), а с увеличением молекулярной массы
происходит падение Fonm и график A(v) сдвигается влево (пунктирная кривая). Заменив
на оси ординат h на N (уравнения 2.12 и 2.18), мы из кривой на рис. 2.136 получим
график, изображенный на рис. 2.116.
Глава 2. Основные принципы и управление разделением
Как же зависит Fonm от условий, перечисленных в таблице 2.4? Любое изменение
условий разделения, которое влияет на размер частиц dp или коэффициент
диффузии Dm при неизменной линейной скорости и (или объемной скорости потока F),
влечет за собой изменение v. Чтобы компенсировать это влияние и сохранить
vonm ~ 3, при котором наблюдается минимальное h и максимальное N, надо
корректировать скорость потока F. Например, при повышении температуры коэффициент
диффузии Dm увеличивается пропорционально некоторой величине х
(уравнение 2.19), и, следовательно, v снижается в х раз (уравнение 2.18а). Значит, чтобы
компенсировать повышение температуры и удерживать постоянное значение v = v0„T,
надо скорость потока, соответствующую Fonm, также увеличить в х раз.
На рис. 2.13а изображены графики для каждого из трех компонентов
уравнения (2.17). Стрелкой указано значение v (v я 3, h x 2), соответствующее минимуму h
и максимуму N. Позже исходное уравнение Нокса было преобразовано, и были
получены новые соотношения, которые, как утверждается, дают более точные
результаты, пригодные для решения широкого спектра задач, и/или обеспечивают более
глубокое понимание основ эффективности колонки [27-32]. Кроме того, когда
учитывается диффузия в неподвижной фазе [35], параметры В и С будут зависеть от к
растворенного вещества [33, 34]. Следовательно, уравнение (2.17) полезно, главным
образом в практической полуколичественной работе. Его даже назвали «чисто
эмпирическим выражением» [36] (с чем авторы книги не согласны!). Тем не менее, ввиду
его простоты и удобства, а также фундаментальной теоретической базы это
уравнение по-прежнему рекомендуется использовать в повседневной работе.
2.4.1.2. Несколько рекомендаций по выбору колонки
Когда говорят о параметрах колонки, имеют в виду ее длину и диаметр, размер
частиц. При этом не забывают о скорости потока. Все вместе эти параметры
определяют число теоретических тарелок N, продолжительность эксперимента и перепад
давления на колонке Р. Чем больше нужно теоретических тарелок, тем, как правило,
продолжительнее разделение. Однако в случае, если колонка и система способны
выдерживать большой перепад давления, повышение давления решает проблему
выбора между числом теоретических тарелок и временем анализа: либо повышаем N
при неизменном времени анализа, либо сокращаем время анализа, не меняя N.
Выбор диаметра колонки зависит от задачи: в препаративных разделениях используют
больший диаметр (глава 15), ас колонками меньшего диаметра работают, если:
1. необходимо повысить чувствительность, но при этом количество образца
ограничено;
2. необходимо снизить объемную скорость потока в ЖХ-МС (раздел 4.14);
3. необходимо сократить потребление растворителей;
4. разделение проходит при очень высоком давлении (что позволяет свести к
минимуму проблемы, вызванные нагревом внутри колонки, раздел 3.5.4.3).
Диаметр колонки dc напрямую не оказывает какого-либо влияния на
соотношение между N, продолжительностью разделения и Р. Однако, чем уже колонка, тем
меньше объем пика и тем больше вклад внеколоночного размывания. Значит, чем
меньше диаметр колонки, тем больше возникает требований непосредственно к
самому хроматографу. Равно как и работа при высоком давлении (больше чем 400 бар)
требует наличия специального оборудования (раздел 3.3.5.4) и специальных колонок.
Когда принято решение относительно диаметра колонки, подобрать остальные
параметры колонки поможет теория, изложенная в разделе 2.4.1.1. Далее будем
считать, что в разделении участвуют небольшие молекулы массой менее 1000 Да. Как
разделять более крупные молекулы описано в разделе 13.3.1 (см. также материал
к рис. 2.116). Рассмотрим три варианта:
2.4. Ширина пика и число теоретических тарелок колонки N 83
A. в наличии есть колонки только одного типа (например, размером 150 х 4,6 мм,
упакованные частицами диаметром 5 мкм)
Б. в наличии есть колонки одного типа с одинаковым диаметром, но разной длины
B. широкий выбор колонок разной длины, упакованных частицами разного размера.
Рассмотрим вариант А. Можно менять только скорость потока (как
на рис. 2.11а), но существует максимально возможная скорость потока, которая
определяется максимальными возможностями хроматографической системы или
максимально возможным давлением для данной колонки (хотя часто желательно
работать при более низком давлении). То есть подбирают такое значение F, которому
будет соответствовать необходимое число теоретических тарелок N, но при этом не
будет превышено некое максимальное давление. Например, предположим, что для
колонки 150 х 4,6 мм (рис. 2.11а) требуется N более 10000, при этом давление не
должно превышать 136 бар. Видно, что скорость потока должна составлять примерно
2 мл/мин, что соответствует N = 10000 и Р < 136 бар.
Вариант Б: можно менять скорость потока и длину колонки. Максимальное N
за минимальное время будет тогда, когда комбинация F и L приведет к
максимальному допустимому давлению. Из уравнения (2.13а) видно, что это эквивалентно
тому, что произведение FL должно оставаться постоянным. На рис. 2.14а показана
получающаяся зависимость N от времени анализа (для значений ?д) для трех разных
размеров частиц 2, 5 и 10 мкм. Из данных, представленных на этом рисунке, можно
сделать следующий вывод: меньший размер частиц (2 мкм) позволяет сократить
время анализа (и, соответственно, меньше требуемое N). Таким образом, для tg = 1
мин наибольшее N будет у колонки, упакованной частицами 2 мкм, а для t$ =
= 1000 мин — у колонки с частицами 10 мкм. То есть, чем большее число
теоретических тарелок требуется, тем крупнее должны быть частицы в колонке. Если для
анализа достаточно небольшого числа теоретических тарелок, то лучше всего выбирать
колонку, упакованную частицами малого диаметра — это приведет к более
короткому времени анализа. Для анализов средней продолжительности (например, fy > 5
минут) соответственно подойдут колонки с частицами промежуточного размера
(5 мкм), которые, по сравнению с частицами меньшего размера, дают требуемое
число теоретических тарелок за более короткое время.
По данным рис. 2.14а построен кинетический график или график Поппе [37, 38]
(рис. 2.146). Это график зависимости времени, необходимого для образования одной
теоретической тарелки (to/N), от величины N. Видно, что величина «время на
теоретическую тарелку» растет с увеличением N. Кроме того, согласно этому графику,
если для разделения требуется небольшое число теоретических тарелок, то лучше
использовать сорбент с малыми частицами, и наоборот, если большое, то
предпочтителен сорбент с крупными частицами. Пунктирные линии, обозначенные «10 с»,
«100 с» и «1000 с», соответствуют условиям постоянного tg. Чтобы определить другие
значения ?о> проводят прямые, параллельные этим линиям. В настоящее время
графики Поппе широко используют для сравнения характеристик колонок, поскольку
они учитывают и эффективность и проницаемость колонок.
В варианте В допускается изменение длины колонки и размера частиц. Этот
вариант полезен, когда надо разработать либо очень быстрое разделение, либо
требуется очень большое число тарелок (экстремальные условия разделения). Последние
10 лет говорят о растущей потребности в очень быстрых ВЭЖХ-разделениях
продолжительностью минута и даже меньше (раздел 2.5.3.2). С другой стороны, сейчас
биохимики столкнулись с проблемой разделения образцов, содержащих тысячи
компонентов (раздел 13.4.5). В таких случаях важнее большее число N. Вне зависимости
от того, является ли целью сложного разделения минимальная продолжительность
или максимальное число теоретических тарелок, лучше подбирать условия, при
которых N в единицу времени будет максимальным (как на рис. 2.146). Можно пока-
84 Глава 2. Основные принципы и управление разделением
10
105
ю4
1.03
Р= 138 бар (2000 psi)
/ i
dp =
i
(а)
10 мкм.
2 мкм
0.1
1 10
'v (мин)
1000
10'
(j, = 10 сек'ч.
(„ = 100 сек '■■
10г
103
10"
N
105
10е
Рис. 2.14. Зависимость N от длины колонки. Для сохранения постоянного давления
изменяли скорость потока, (а) зависимость N от tg для частиц сорбента
разного размера; (б) «график Поппе» по данным (а). Вычисления проводились
по уравнению (2.17) при условии, что А = 1, В = 2, С = 0,05, вязкость —
0,6 сП (например, для смеси 60% ацетонитрил-вода, 35 °С), Dm = Ю-5 см2/с
(для образца с молекулярной массой 300 Да), перепад давления на колонке
136 бар (2000 psi)
зать, что выбор оптимальных длины колонки, размера частиц и скорости потока
всегда будет соответствовать минимальному значению h x 2 (или vs3). Поскольку
имеющиеся в продаже колонки ограничены в длине и размере частиц, можно только
подобрать приблизительные условия для v ~ 3 одновременно с требуемым значением
N за минимальное время анализа. К счастью, увеличение v в 2—3 раза слабо влияет
2.4. Ширина пика и число теоретических тарелок колонки N 85
на h или N, что существенно расширяет возможность выбора размера частиц или
колонки желаемого размера. См. также [38а].
График на рис. 2.15 построен по уравнению (2.17) при v = 3 и служит примером,
как можно подобрать условия, при которых достигается максимальное N для
определенного времени разделения при некотором заданном максимальном Р.
Предположим, максимальное давление составляет 136 бар (2000 psi), a k последнего пика
на хроматограмме равно 3 (для других значений к нужно подбирать время по
рис. 2.15 и уравнению (2.5)). В этих условиях, если требуется N = 5000,
продолжительность анализа может составить 15 секунд (на рисунке обозначено • и стрелкой),
а 300000 теоретических тарелок требуют 17-часового анализа (60000 с, на рисунке
обозначено о и стрелкой). В первом случае (N = 5000) необходимо использовать
частицы размером 1 мкм, во втором (N = 300000) — 9 мкм. То есть для быстрых
разделений лучше всего использовать колонки с малым размером частиц, если же требуется
105
Время
разделения
((сек)
ю4
103
100
1 мин ►
10
1
0.5 12 5 10 20
Давление (бар, psi x 0.001)
Рис. 2.15. Зависимость количества теоретических тарелок N от времени разделения /,
перепада давления на колонке Р и диаметра частиц сорбента dp.
Сплошными линиями (—) показаны различные значения N, штриховыми ( ) —
различный размер частиц, пунктирная линия (••■■) соответствует давлению
на колонке 136 бар (2000 psi). Все значения на рисунке указаны для
минимальной приведенной высоты теоретической тарелки h ~ 2 и к = 3. Вязкость
0,6 сП (например, для смеси 60% ацетонитрил-вода, 35 °С) и Dm =
= Ю-5 см2/с (относится к образцу с молекулярной массой 300 Да в
указанных условиях). По данным из [23]
86
Глава 2. Основные принципы и управление разделением
много теоретических тарелок, то подойдут и крупные частицы, но при этом
увеличится время анализа (см. предыдущее обсуждение рисунка 2.14). Увеличение
допустимого давления дает возможность либо сократить время анализа (при неизменном
N), либо увеличить N (при неизменном времени анализа). В этом можно убедиться,
сравнив графики на рис. 2.15 для Р = 1360 бар (20000 psi) и Р = 68 бар (1000 psi).
Повышение допустимого давления способствует использованию малых частиц для
более быстрого разделения или большего числа N. Очень большое количество
теоретических тарелок или очень быстрые разделения требуют экстремальных условий,
о чем свидетельствует рис. 2.15.
По разным причинам графики, изображенные на рис. 2.11 и 2.13—2.15, носят
приблизительный характер, и при варьировании условий, перечисленных в
таблице 2.4 (например, вязкости подвижной фазы, молекулярной массы компонентов
образца, температуры) они будут изменяться. Поэтому эти графики могут служить
прежде всего качественными ориентирами для выбора экспериментальных условий,
а не для количественных расчетов какого-то конкретного разделения.
Скорость потока и длину колонки, соответствующие графикам на рис. 2.15,
можно определить по уравнению (2.18), учитывая что Н = L/N и h = 2:
L = NH= Nhdp = 2Ndp. (2.21)
Здесь L и d выражено в сантиметрах. По уравнению (2.18а) для vs3
3D
и = ^"-, (2.21а)
dp
a F определяют по уравнению:
3V D
Р = m m, (2.216)
Ldp
где Vm — мертвый объем колонки (равный t0F, уравнение (2.7а)). Единицы
измерения в уравнении (2.216) следующие: мл/с(Р), мл (Vm), CM2/c(Dm), см (L), см (d).
2.4.2. Форма пика
До сих пор мы предполагали, что на окончательной хроматограмме у нас получатся
симметричные пики. В идеальных условиях пик имеет форму кривой Гаусса
у =(2n)-°'se-x1'2. (2.22)
Здесь х = (t - tR)/a, где t — это время, у = W/4, а у — это высота пика. Форма
настоящих пиков на хроматограмме, как правило, немного отличается от симметричной
кривой Гаусса, в большей или меньшей степени пикам свойственно размывание
заднего фронта (рис. 2.16а). Размывание заднего фронта пика можно охарактеризовать
двумя параметрами: коэффициентом асимметрии As (на рис. 2.16а показан слева) или
фактором размывания TF (tailing factor) (на рисунке справа). Величины As и TF
взаимосвязаны следующим образом:
As = 1 + 1,5(7У-1), (2.22а)
то есть значения As, как правило, немного больше TF (таблица 2.5).
На рис. 2.166 показано, как в зависимости от значений As и TF меняется форма
пика. У идеально симметричного пика As = TF = 1,0. Влияние размывания на
разделение двух соседних пиков продемонстрировано на рис. 2.16в, где изменяется только
размывание, охарактеризованное ТР. Два пика с А5 = ТР = 1,0 хорошо отделяются
друг от друга, но чем больше размывание, тем хуже разделение. Во многих случаях
2.4. Ширина пика и число теоретических тарелок колонки N 87
Коэффициент асе и метр и и
пика/1.
: В/А (10% величина
10% ВЫСОТЫ
пика
Фактор размывания
пика, TF
■- {А + В)/2А (5% величина)
О)
5% высоты
пика
Рис. 2.16. Определение размывания пика и его влияние на разделение, (а)
Определение коэффициента асимметрии и фактора размывания (As и TF), (б)
влияние As и TF на форму пика, (в) влияние размывания пика на разделение,
(г) размывание переднего фронта пика, (д) размывание из-за «перегрузки»
(сравните с «экспоненциальным» размыванием на рисунке (а))
Таблица 2.5. Соотношение коэффициента асимметрии и фактора размывания заднего
фронта пика
Коэффициент асимметрии пика (измеряется
на высоте равной 10% высоты пика)
1,0
1,3
1,6
1,9
2,2
2,5
Фактор размывания пика (измеряется на высоте
равной 5% высоты пика)
1,0
1,2
1,4
1,6
1,8
2,0
Глава 2. Основные принципы и управление разделением
размывание пика (TF < 1,2) мало и поэтому им можно пренебречь. Исключением
является ситуация, когда сразу же за большим пиком элюируется малый (сравните
рис. 2.17г и д). Гораздо реже встречается размывание переднего фронта пика (TF <
< 1,0, рис. 2.16г), которое оказывает аналогичное, потенциально отрицательное
влияние на разделение. Когда пик подвергается сильному размыванию TF > 2, это
сказывается не только на разделении, но и на количественном анализе, поэтому в
рутинных анализах рекомендуется придерживаться общего правила — показатель TF
всех пиков должен быть меньше 2. Количественная оценка по площади пика
(глава 11) требует интегрирования всего пика, что, в свою очередь, зависит от задания
правильных пределов интегрирования, а это осложняется для пиков с значительной
асимметрией.
Если на хроматограмме (неважно, во время рутинного анализа или в процессе
разработки метода) появились сильно размытые пики (TF> 2), то рекомендуется
незамедлительно решить эту проблему. Далее будем считать, что у нас симметричные пики,
если не оговорено иное. Можно выделить ряд причин, приводящих к неправильной
или размытой форме пика:
• «некачественная» колонка (загрязнен фильтр или в колонке между фильтром и
сорбентом образовалось пустое пространство, раздел 5.8);
• засорившаяся колонка (накопление на сорбенте сильно удерживаемых примесей,
раздел 5.8);
• перегрузка колонки (слишком много образца, раздел 2.6.2);
• образец растворен в слишком полярном растворителе (раздел 2.6.1);
• внеколоночное размывание пика (раздел 2.4.1, 3.9);
• силанольные взаимодействия (включая загрязнение сорбента следовыми
количествами металла, раздел 5.4.4.1, 7.3.4.2);
• неправильно выбран буфер или недостаточна его буферная емкость (раздел 7.2.1.1);
• прокачивание холодной подвижной фазы через нагретую колонку (неправильное
термостатирование, раздел 2.5.3.1).
См. также раздел 17.4.5.3. Прежде чем решать проблему размывания, надо
определить: это экспоненциальное размывание или размывание в результате перегрузки.
Экспоненциальное размывание характеризуется постепенным возращением пика
к базовой линии (рис. 2.16а). При перегрузке колонки пик имеет форму
прямоугольного треугольника (рис. 2.16<Э). В последнем случае (рис. 2.163) уменьшить
размывание можно, введя меньшее количество образца. Однако, при экспоненциальном
размывании этот способ не решает проблему, а может ее даже усугубить [39, 40].
Вообще размывание пиков желательно сокращать до значения TF < 1,5.
Были предприняты попытки математически описать форму размытого пика
разными способами [41, 42] и дать такое определение числа теоретических тарелок N,
которое бы включало негативное влияние размывания на разделение. Значения N
размытых пиков, вычисленные по уравнению (2.9) (и особенно по уравнению (2.9а)),
завышают эффективность колонки. Реальная эффективность, которую определяли
по фактическому разделению пиков на хроматограмме, говорит о меньшем числе
теоретических тарелок. Уравнение Дорси-Фоли широко используется для определения
скорректированного значения ^для пиков с размыванием заднего фронта [43]:
N - 41'7(W"V2. (2.226)
(В I А) + 1,25
Параметры А и В определяют как показано на рисунке 2.16а (на высоте, равной 10%
высоты пика от базовой линии), W0 j — это величина W, измеренная на той же
высоте.
2.5. Разрешение и разработка метода
Существует еще один способ определения скорректированного значения N для
асимметричного пика с помощью так называемого значения 5-а:
N = 25|-^-| . (2.22в)
Здесь W$c — ширина пика на высоте, равной 4,5% высоты его максимума. Такой
способ вычисления N похож на вычисление числа теоретических тарелок по ширине
пика на его полувысоте (уравнение 2.9а). Чем ближе к базовой линии определяют
ширину пика, тем сильнее влияет размывание пика на число теоретических тарелок,
т.е. И^/2 (незначительное влияние) < W < W5a (существенное влияние). Когда в
разделении присутствуют ассиметричные пики, уравнения (2.226) и (2.22в), также как и
другие способы [41], могут в лучшем случае дать лишь приблизительную оценку
эффективности колонки. При сравнении двух пиков на рисунках 2.16а и е можно
видеть, что асимметричные пики нельзя описать одной общей формулой (в отличие
от симметричных пиков, для которых есть уравнение (2.22)), и для описания пиков
при TF > 1,2 одного значения N недостаточно. Для характеризации таких пиков
рекомендуется использовать N, вычисленное по уравнению (2.9а), вкупе со
значениями As или TF. Во многих случаях уширение заднего фронта пика — один из
параметров, измеряемых при определении пригодности системы, чтобы отследить ухудшение
эффективности колонки. Для этих целей подойдет практически любой метод,
который позволит отслеживать изменения формы пика, происходящие со временем.
2.5. Разрешение и разработка метода
Разработка метода ВЭЖХ предполагает подбор хроматографических условий,
обеспечивающих приемлемое разделение данного образца. Более подробная информация
(но не такая современная, как в этой книге) дана в работе [44]. Разделение двух
пиков / и у (рис. 2.10а) обычно описывается их разрешением Rs, которое можно
определить по формуле (разность времен удерживания)/(средняя ширина пика) или:
R = 2[?Л(Л -?Л(/)]
W, + W,
(2.23)
Wt и Wj — ширина пиков i и у, соответственно, на уровне базовой линии. Чем
больше разность времен удерживания пиков и/или уже пики, тем лучше разделение
(больше разрешение). Для проведения точного количественного анализа разделений,
подобных представленным на рис. 2.17а, предпочтительно иметь разрешение пиков
до базовой линии, т.е. впадина между двумя пиками должна находиться на уровне
базовой линии. Два пика примерно одинакового размера будут иметь разрешение
до базовой линии (рис. 2.17а) при Rs > 1,5. В препаративной хроматографии
(раздел 15.3.2) разрешение до базовой линии позволяет полностью выделить каждый пик
со 100%-ной чистотой.
На рис. 2.17 изображены примеры различного разрешения (Rs = 1,0, 1,5, 2,0)
при разном соотношении размеров пиков (1:1, 10:1, 100:1, 1000:1). Когда разрешение
двух симметричных пиков (7У =1,0) больше 1,5, то видно, что перекрывание
достаточно мало вне зависимости от их относительных размеров. Небольшое размывание
заднего фронта пика (TF = 1,2, рис. 2.17с*) может приводить к значительной потере
разрешения, если следом за крупным пиком элюируется малый пик. В этом случае
нужно подбирать условия, чтоб разрешение было больше двух. С практической точки
зрения уменьшить размывание пика до TF я 1,0 очень сложно. Следовательно, если
90 Глава 2. Основные принципы и управление разделением
Я, =1.0
1.5
2.0 (а)
1:1
10:1
100:1
1000:1
(б)
(е)
(г)
1А
1000:1
ФР = 1.2
1:1000
ФР=1.2
(изменение порядка
элюирования пиков) у
(д)
J.
(в)
А1
Рис. 2.17. Зависимость разделения от разрешения при различных относительных
размерах пиков (1:1, 10:1 и т.д.), и размывании заднего фронта пика (д, е)
концентрация примеси в образце низкая относительно ближайшего основного пика,
то желательно, если вообще возможно, чтобы первым элюировался пик примеси, а
после него шел целевой пик. Это необходимо сделать для того, чтобы минимизировать
влияние размывания заднего фронта пика на разрешение (сравните рис. 2А7д и ё).
Изменить порядок элюирования пиков, т.е. сделать так, чтобы первым выходил
небольшой пик, а потом уже большой, иногда можно в процессе разработки метода
разделения, варьируя условия, влияющие на селективность (раздел 2.5.2).
Обычно при разработке ВЭЖХ-метода стоит задача отделить каждый
интересующий нас пик от соседних до базовой линии (Rs > 2) с коэффициентом запаса,
равным 1/3 (а не задача разделения до базовой линии пиков примерно одинакового
размера cS,s 1,5). Подбор условий для получения Rs > 2 проводится для того, чтобы
компенсировать незначительное размывание фронта пика, умеренное отличие пиков
по размеру и обычное медленное ухудшение эффективности колонки, которое также
приводит к размыванию и уширению пиков. В некоторых случаях может
потребоваться даже большее значение Rs. В разделах 2.5.1 и 2.5.3 даны рекомендации по
улучшению Rs.
Когда требуется разделить больше двух пиков, подбирают такие условия, в
которых разрешение наименее разделяющейся пары пиков было бы не менее двух. Такая
пара пиков называется критической, а их разрешение — критическим разрешением
2.5. Разрешение и разработка метода
разделения. Примером могут служить соседние пики 1 и 2 с разрешением Rs = 0,8
(рис. 2.18а) или пики с разрешением Rs = 3,9 (рис. 2.18г). Разрабатывая метод,
стараются, чтобы разрешение критической пары пиков было приемлемым. Это значит,
что разделение остальных пиков также будет удовлетворительным с Rs > 2. В этой
книге при упоминании разрешения Rs имеется в виду разрешение критической пары
пиков (если не оговорено иное).
При разработке метода удобно использовать другое, приближенное выражение
для разрешения, полученное из уравнений (2.5а) и (2.10а) (при условии равенства
ширины двух пиков):
*=li.
d+k)
(a-l)iV0
(2.24)
(а)
(б) (в)
Из уравнения видно, что разрешение зависит от удерживания к первого пика i
(член а), коэффициента селективности а (член б) и эффективности колонки или
80% метанол
(0.3 <к< 0.8)
0.2 0.4
0.6 0.8 1.0
Время (мин)
1.2
1.4
70% метанол
(0.8 £ /с й 2.3)
60% метанол
(1.8 < к < 6.6)
Время (мин)
г ь л- л4
(в)
.а:
2 4
Время (мин)
50% метанол
(4<k<19) t
J Ll
-Л_
Л-
(а)
10
Время (мин)
15
Рис. 2.18. Зависимость разделения от доли растворителя В в подвижной фазе.
Образец — смесь гербицидов: 1 — монолинурон, 2 — метобромурон, 3 — диурон,
4 — пропазин, 5 — хлороксурон. Условия: колонка Cig 150 х 4,6 мм, 5 мкм,
подвижная фаза метанол — вода, скорость потока 2,0 мл/мин, комнатная
температура. Хроматограмма смоделирована по данным работы [7]
Глава 2. Основные принципы и управление разделением
числа теоретических тарелок TV (член в). Коэффициент селективности а (его еще
называют селективностью разделения или относительным удерживанием) можно
вычислить по формуле:
а =
*/
(2.24а)
Значения kt и к: — это удерживание к соседних пиков i и у (рис. 2.10а). Для этого
разделения к = кх = 1,55, а = 1,12, N = 11000, следовательно, Rs = (1/4)[1,55/2,55] х
х (1,12 - 1)110000'5 = 1,9. На основании уравнения (2.24) можно сделать вывод:
разрешение можно улучшить изменяя к, а или N, однако наиболее эффективно улучшение
селективности разделения (параметр а).
Хотя уравнение (2.24) дает не такие точные результаты, как уравнение (2.23), все
же оно более полезно в процессе разработки метода. Все численные значения Rs
в этой книге всегда определяли по уравнению (2.23). Уравнение (2.24) необходимо,
главным образом, для понимания того, как разрешение зависит от
экспериментальных условий, а также в качестве инструкции по системной разработке метода.
Разрешение Rs можно также выразить следующим образом:
TV0-5. (2.25)
К,
1
4
Г к 1
La + *)J
Га -11
а
Производные формулы (2.24) и (2.25) получены при разной аппроксимации ширины
двух пиков. Вычисленные по обоим уравнениям значения Rs одинаково точны.
Единственное преимущество уравнения (2.24) — простота в использовании при
разработке метода.
Разработка изократического ВЭЖХ-метода разделения заключается в пошаговом
изменении («оптимизации») экспериментальных условий до тех пор, пока
разделение не станет приемлемым с критическим разрешением, желательно, больше двух.
В разделах 2.5.1—2.5.3 показано, что уравнение (2.24) служит удобным руководством
по разработке изократического метода. Каждый из членов а-в уравнения (2.24)
можно контролировать, изменяя определенные условия разделения (табл. 2.2). Обычно
сначала выбирают колонку с достаточным числом теоретических тарелок, способную
разделить образец указанной сложности. В большинстве случаев лучше начинать
с TV» 10000. Такой эффективностью обладают колонки длиной 150 мм, упакованные
частицами 5 мкм, либо колонки длиной 100 мм с 3 мкм частицами. Затем подбирают
долю растворителя В в подвижной фазе. Она должна быть такой, чтобы
коэффициент удерживания укладывался в соответствующий интервал (например, 1 < к < 10),
после чего оптимизируют а. На последнем этапе корректируют число теоретических
тарелок колонки, пытаясь найти компромисс между несовместимыми параметрами:
приемлемым разрешением и небольшим временем анализа.
Финальные условия разделения вряд ли можно назвать действительно
оптимальными, но они лучшие из возможных. Тем не менее в литературе часто встречается
термин «оптимизированный», который означает относительно улучшенные или
рекомендуемые условия, но не абсолютно идеальные. Кроме того, следует отметить
разницу между «локальной» и «глобальной» оптимизациями. Локальная
оптимизация — это улучшение одного или двух (редко больше) условий разделения в
ограниченном интервале значений каждого из этих условий. При этом все остальные
условия, обычно не оптимальные, такими же и остаются. Глобальная оптимизация
предполагает подбор всех условий, от которых зависит разделение. Когда говорят об
«оптимизированной» методике, в большинстве случаев имеют ввиду улучшенное
разделение или локальный оптимум. Будем и далее в этой книге придерживаться этой
формулировки: «оптимум» не есть синоним глобально лучшего разрешения.
2.5. Разрешение и разработка метода
2.5.1. Оптимизация фактора удерживания к
(член а в уравнении (2.24))
В изократических методах удерживание вещества обычно контролируют путем
изменения доли растворителя В в подвижной фазе (%В). На первом этапе подбирают
такое значение к, которое было бы не слишком большим, но и не слишком малым.
Зависимости относительных значений разрешения Rs (уравнение 2.24) и высоты пика
(уравнение 2.11) от коэффициента удерживания изображены на рис. 2.19, чтобы
показать, как они влияют на к (предполагается, что к не влияет на а). В нижней части
рис. 2.19 наглядно представлено как меняется разрешение и высота пиков при
изменении %В в интервале значений к от 1 до 10.
Как правило, стремятся к тому, чтобы удерживание всех пиков на хроматограмме
было < 10, т.к. это условие соответствует более высоким и узким пикам, что улучшает
их детектирование, а также приводит к уменьшению времени анализа, следовательно,
в день можно анализировать больше образцов. Значения & <ЗС 1 могут приводить к
плохому разрешению, особенно из-за возможного наложения зон анализируемых веществ
на зоны скапливающихся в области ?д компонентов матрицы (см. «мусорный» пик
на рис. 2.56). Следовательно, после завершения разработки разделения, в
окончательной методике времена удерживания всех пиков должны находиться в интервале 1 < к < 10.
Впрочем, по выбору хроматографиста этот интервал значений к можно несколько
расширить, например, от 0,5 до 20. С другой стороны, органы контроля могут
рекомендовать значение к не менее двух для всех целевых пиков на хроматограмме [46], чтобы
уменьшить влияние вспомогательных веществ или других неустановленных пиков
(«мусор»), элюируемых с колонки вблизи ?д. Если для детектирования используется
масс-спектрометр (ЖХ-МС), то из-за возможных эффектов подавления ионов
подбирают к > 3. Когда понятно, что все интересующие нас пики не помещаются в
некоторый максимальный интервал (т.е. 0,5 < к < 20), переходят к градиентному элюирова-
нию (раздел 2.7.2, глава 9).
&
(J?
о:
1.0
0.8
0.6
0.4
0.2
- относительное
разрешение Rs
ir [М1+Ч1 _—
V
Is-
/ V
/ *ч
/ ^--^
i i
относительная высота
пика hp, [1/(1 + А)]
! 1
10'
к=Л
* = 10
Рис. 2.19. Зависимость относительного разрешения Rs и относительной высота пика hp
от к или продолжительности анализа. Продолжительность анализа
пропорциональна величине (1+fe) для последнего пика. Значение а предполагается
постоянным
Глава 2. Основные принципы и управление разделением
На рис. 2.18 показано, как изменение доли органического растворителя в
подвижной фазе влияет на обращенно-фазовое разделение. При 80% метанола в воде
(80 %В) значения к слишком малы: 0,3 < к < 0,8 и, как следствие, разрешение низкое
(рис. 2.18а). Снижение доли растворителя В до 50% (рис. 2.18г, 4 < к < 19) приводит
к очень хорошему разрешению (Rs = 3,9), но время разделения слишком велико, и
последние пики стаовятся широкими и низкими, что понижает чувствительность
детектирования. К разумному компромиссу между разрешением (Rs= 2,6),
чувствительностью детектирования и продолжительностью анализа (6 минут), удается прийти,
установив промежуточное значение В (60%, рис. 2.18в). При этом и удерживание
укладывается в приемлемый диапазон 1,8 < к < 6,6. Подвижная фаза, содержащая
60—70 %В, возможно, будет даже лучше, так как достигается разрешение Rs > 2,0,
затрачивается еще меньше времени на анализ и ещё повышается чувствительность.
В ОФХ удерживание зависит от доли органического растворителя (как показано
на рис. 2.18) и эта зависимость описывается эмпирическим уравнением [47]
log к = log kw - S<p, (2.26)
где ф — объемная доля растворителя В (равная 0,01 х %В), kw — экстраполированное
значение к соединения X в подвижной фазе, состоящей из чистой воды, т.е. при ф =
= 0, a S — константа для соединения X, когда изменяется только ф. Вывод
уравнения (2.26) приведен в работе [48]. Для «малых» молекул с молекулярным весом
100—500 Да константа S я 4 [49], поэтому увеличение ф на 0,1, т.е. изменение
подвижной фазы на +10 %В, приведет к среднему уменьшению к всех пиков в образце
примерно в
10(0,1x4)
раз или в 2,5 раза. Поэтому уравнение (2.26) подразумевает
последовательный порядок действий, в ходе которых осуществляется поиск
удовлетворительного %В — такого, чтобы к находился в диапазоне от 1 до 10 (или в любом
другом желательном интервале).
В соответствии с этим порядком действий первое разделение выполняется с 80%
или 90 %В. Такое высокое содержание органического растворителя в подвижной
фазе гарантирует полное элюирование всего образца с колонки за короткое время.
Например, в первом эксперименте с 80 %В (рис. 2.18а) был получен интервал
удерживания 0,3 < к < 0,8. Применив правило 2,5-кратного увеличения к при снижении
на каждые 10 %В, можно оценить к для меньших долей растворителя В: для 70%
2,5 х 0,3 я 0,8 (к первого пика), 2,5 х 0,8 я 2,0 (к последнего пика), т.е. 0,8 < к < 2,0.
Аналогично рассчитываем для 60 %В и получаем 2,0 < к < 5. Этот интервал вполне
соответствует требуемому 1,0 < к < 10 (как и наблюдаемый 1,8 < к < 6,6). Поэтому
в качестве подвижной фазы во втором эксперименте по разработке метода лучше
использовать 60 %В (остальные условия разделения остаются неизменными).
Для образца, содержащего соединения, удерживающиеся слабее, чем
представленные на рис. 2.18, начальное элюирование 80 %В, приведет к тому, что все пики
на хроматограмме будут иметь к « 0 (т.е. все вещества в образце выйдут в одном
пике). Чтобы подобрать приемлемые значения к, необходимо существенно сократить
долю органического растворителя по крайней мере на 30%. Поэтому второй
эксперимент целесообразно проводить с (80 %В - 30 %В) = 50 %В. Затем, как в
приведенном выше примере (рис. 2.18), следует изменять %В до тех пор, пока 1 < к < 10.
Поскольку «правило 2,5» носит приблизительный характер, то между экспериментами
нежелательно изменять %В более чем на 30%, иначе при маленькой доле
органического растворителя можно упустить поздноэлюируемые пики. Существует и другой
способ подбора %В (обычно предпочтитают его): разрабатывая методику, первый
эксперимент проводят в градиентном режиме (раздел 9.3.1).
2.5. Разрешение и разработка метода
2.5.2. Оптимизация селективности а
(член б в уравнении (2.24))
Если и дальше требуется улучшение разделения, то следующим шагом является
регулирование относительного удерживания (расстояние между пиками на хроматограм-
ме, селективность или коэффициент селективности а) путем изменения любого
из первых семи параметров таблицы 2.2: %В, растворитель В (обычно метанол или
ацетонитрил), температура, тип колонки или — если в образце есть кислоты или
основания — рН, концентрация буфера, концентрация ион-парного агента. Заметим,
что любое изменение каждого из параметров таблицы 2.2 влияет на значение к,
поэтому необходимо одновременно корректировать %В, чтобы коэффициент
удерживания оставался в допустимом диапазоне (как правило, не более 0,5 < к < 20).
На рис. 2.20 показано изменение селективности при разделении образца,
содержащего 6 соединений. Первоначально %В был подобран так, чтобы коэффициент
удерживания находился в интервале 2 < к < 4 (рис. 2.20а). Для улучшения
селективности и разрешения будем далее изменять %В и температуру. В условиях 45 %В и
Т = 30 °С (рис. 2.20а) пики 1-3 и 5/6 плохо разрешены (Rs = 0,3). Снижение доли
органического растворителя в элюенте до 30% (рис. 2.206) положительно отразилось
на разделении всех 6 пиков с подходящим интервалом удерживания (4 < к < 12),
но разрешение пиков 3 и 4 стало предельно допустимым, в то время как в
первом эксперименте (рис. 2.20а) они разделились хорошо. Кроме того, при 30 %В
(рис. 2.206) пики 5 и 6 поменялись местами. По этим двум хроматограммам можно
сделать вывод: промежуточная доля растворителя В, вероятно, улучшит разрешение,
т.е. увеличится расстояние между пиками 3 и 4, а пики 1/2 и 5/6 не слишком сбли-
(а)
i_JU
45% В, 30°С
2 < к < 4,
R» = 0.3
5„ 6
Время (мин)
30% В, 30°С (б)
4<к< 12
R» =14
4 6 8 10
Время (мин)
(в)
Я
45% В, 45°С
1 < к < 34
Rs = 0.4
5
_J\1
Время (мин)
20% В, 47°С
Л<к<20
R„ = 4.0
(г)
[£Л1
0 2 4 6 8 1012141618
Время (мин)
Рис. 2.20. Влияние доли органического растворителя в подвижной фазе и/или
температуры на изократическое разделение шестикомпонентного образца.
Образец: 1 — 3-фенилпропанол, 2 — 1-нитропропан, 3 — оксазепам, 4 — и-хлор-
фенол, 5 — эвгенол, 6 — метилбензоат. Условия: колонка Сщ 150 х 4,6 мм,
5 мкм, подвижная фаза ацетонитрил — вода, скорость потока 2,0 мл/мин;
на рисунках указана температура и %В (изменения выделены жирным
шрифтом). Обратите внимание, на всех хроматограммах высоты пиков
нормализованы по отношению к самому высокому пику. Хроматограммы
смоделированы по работам [50, 51]
Глава 2. Основные принципы и управление разделением
зятся. На самом деле подвижная фаза с 33 %В и температура 30 °С значительно
улучшают разрешение (Rs = 2,1, хроматограмма не показана).
Если не менять состав подвижной фазы (оставить такой как на рисунке 2.20а),
но увеличить температуру с 30 до 45 °С (рис. 2.20в), относительное удерживание (или
селективность) изменятся, но в целом разделение останется неприемлемым (Rs =
= 0,4). Можно путем проб и ошибок найти подходящие %В и температуру и
определить максимальное разрешение. Оно будет составлять Rs = 4,0 при 20 %В и 47 °С
(рис. 2.20в), но значение к при этом достигнет 20. Целью повышения селективности
является либо увеличение разрешения или сокращение продолжительности анализа,
либо одновременное изменение обоих параметров. При выборе условий приемлемого
разделения (например, во время разработки метода) следует отдать предпочтение
изменению селективности, поскольку она одновременно влияет и на разрешение, и на
продолжительность анализа. Это в первую очередь справедливо для трудноразделимых
образцов с большим количеством компонентов (когда на хроматограмме
наблюдается очень много пиков) или с компонентами, чьи времена удерживания очень близки
(например, изомеры).
2.5.2.1. «Правильные» и «неправильные» образцы
Настоящий раздел поможет понять как и почему меняется разделение в зависимости
от %В, особенно в градиентном режиме. Тем не менее, этот материал не так важен
в повседневной работе или при разработке методов разделения в ОФХ. Поэтому его
можно пропустить, перейти к разделу 2.5.3, а сюда вернуться, если возникнет
необходимость.
В обращенно-фазовых разделениях образцы можно поделить на «правильные»
или «неправильные» [52]. При изменении только лишь %В хроматограмма
«правильных» образцов как аккордеон, либо сжимается, либо растягивается с минимальным
изменением относительного удерживания пиков на хроматограмме. Хороший
пример «правильного» образца (смесь гербицидов) дан на рис. 2.18, а на рис. 2.20
изображено разделение «неправильного» образца. Об этом говорит инверсия пиков 5 и 6
при изменении %В (сравните рис. 2.206 и 2.20а), а также небольшое изменение в
относительном удерживании пиков 1 и 4. Относительное удерживание (или
селективность) «неправильного» образца зависит от %В, «правильного» — нет. Пики
«неправильного» образца отслеживать сложнее из-за возможного изменения порядка
элюирования и ошибочного распознавания пиков (раздел 2.6.4).
Обычно компоненты «правильных» образцов имеют родственную структуру,
тогда как «неправильные» образцы, такие как на рисунке 2.20, являются смесями
соединений различной структуры. Тем не менее, очень сложно заранее сказать какой
образец «правилен», а какой «неправилен». Более или менее точный ответ дают
эксперименты с разной долей растворителя В в элюенте (рис. 2.18). Кроме того,
можно по уравнению (2.26) определить, к какой группе отнести образец. У
правильных образцов наблюдается сильная корреляция между значениями S и log kw
(например, графики зависимости этих величин смещены друг относительно друга и
практически параллельны), в то время как у неправильных образцов такая корреляция
не наблюдается (т.е. графики зависимости S от log kw пересекаются). Многие
образцы демонстрируют промежуточное поведение (среднее между тем, что изображено
на рис. 2.18 и 2.20), у них менее выражены изменения в относительном удерживании
при разном %В. Более подробно правильные и неправильные образцы еще раз
обсудим в разделе 6.3.1 (изократическое элюирование) и разделе 9.2.3 (градиентное элю-
ирование).
2.5. Разрешение и разработка метода
97
2.5.3. Оптимизация числа теоретических тарелок N
(член в в уравнении (2.24))
2.5.3.1. Зависимость результатов разделения от параметров
колонки
Правильно подобранная селективность, обеспечивающая максимальное разрешение
соединений и оптимальное расстояние между пиками (раздел 2.5.2), во многих
случаях обеспечивает удовлетворительное разделение. Тем не менее, можно
дополнительно улучшить разделение, подобрав нужные параметры колонки (длину колонки,
скорость потока, размер частиц), таким образом увеличив число теоретических
тарелок N (член в уравнения (2.24)). Заметим, что в изократическом режиме при
изменении параметров колонки относительное удерживание и расстояние между пиками
(величины к к а) остаются прежними. Т.е. оптимальное расстояние между пиками,
полученное путем подбора соответствующего значения а (член 6 уравнения (2.24)),
не будет нарушено.
Увеличение N влечет за собой повышение разрешения (уравнение 2.24), но
при этом обычно увеличивается продолжительность анализа. И наоборот,
уменьшение числа теоретических тарелок сокращает время разделения. Это может оказаться
полезным в том случае, если после оптимизации селективности Rs 2> 2 (см. ниже).
При равенстве других параметров число теоретических тарелок пропорционально
длине колонки (уравнение 2.12) и, как правило, увеличивается при уменьшении
скорости потока или размера частиц. Если к не меняется, то время разделения
пропорционально tQ (уравнение 2.5), а Гд пропорционально L/F (уравнение 2.7). Поэтому
продолжительность разделения увеличивается пропорционально увеличению длины
колонки или снижению скорости потока. Аналогичным образом, перепад давления
возрастает с увеличением длины колонки и скорости потока или с уменьшением
размера частиц сорбента (уравнение 2.13а). Следовательно, при изменении параметров
колонки в целях улучшения разделения необходимо сбалансировать время
разделения, разрешение и давление (раздел 2.4.1.2).
Рассмотрим пример улучшения разрешения с помощью изменения
параметров колонки (рис. 2.206, Rs = 1,4). Если селективность не оптимизирована (как
на рис. 2.20г) — такая оптимизация не всегда возможна для некоторых образцов —
всегда можно улучшить разрешение, увеличив длину колонки. На рис. 2.21а
представлен результат увеличения длины колонки с 150 мм (рис. 2.206) до 300 мм
(например, при последовательном соединении двух колонок длиной 150 мм). Теперь пики
разделяются до базовой линии с разрешением Л^ = 1,9. Хотя это разрешение немного
меньше рекомендованного в этой книге (Rs > 2,0), для большинства методик оно
может оказаться достаточным. Цена такого повышенного разрешения — увеличение
вдвое продолжительности разделения (до 20 мин) и давления (до 117 бар или
1700 psi). В этом примере допустимо повышение давления.
Желательно во время оптимизации 6 (раздел 2.5.2) стремиться к избыточному
разрешению, поскольку позже им можно пожертвовать в пользу сокращения
времени анализа (взяв более короткую колонку или повысив скорость потока). Для
примера возьмем рисунок 2.20г (Rs = 4,0) и покажем, как подбирая параметры колонки,
можно сократить время анализа. Если уменьшить длину колонки в 3 раза (с 150
до 50 мм), а скорость потока увеличить в 1,5 раза (до 3 мл/мин), то в результате
время анализа сокращается в 4,5 раза до 4 минут (рис. 2.216). В то же время разрешение
(Rs = 2,0) и давление (33 бар = 480 psi) остаются в допустимых пределах. Многие
образцы состоят из меньшего количества соединений, пики которых к тому же выходят
с большими временными интервалами, чем в этом примере, что позволит (вкупе
98
Глава 2. Основные принципы и управление разделением
<">
300 мм колонка, 5 мкм частицы,
2 мл/мин, Р= 117 бар (1700 psi)
Rs = 1.9, время разделения = 20 мин
.А.
ю
Время (мин)
20
(б)
JLJ
50 мм колонка, 5 мкм частицы
3.0 мл/мин, Р= 33 бар (480 psi)
Rs - 2.0, время разделения = 4 мин
/\
(•)
JLJ
0.2
Время (мин)
30 х 1.0 мм колонка, 1.5 мкм частицы
0.5 мл/мин, Р = 759 бар (11000 psi)
Rs= 2.0, время разделения = 0.7 мин
0.4
Время (мин)
0.6
0.8
Рис. 2.21. Изменение длины колонки или скорости потока с целью улучшения
разрешения или сокращения продолжительности разделения, (я) Разделение
в тех же условиях, как на рисунке 2.206, только на колонке в 2 раза длиннее
(300 мм); (6) разделение в тех же условиях, как на рис. 2.20г, только при
уменьшении длины колонки от 150 до 50 мм и увеличении скорость потока
от 2,0 до 3,0 мл/мин; («) разделение такое же как в случае (б), но
используется колонка 30 к 1,0 мм и скорость потока 0,5 мл/мин при повышенном
давлении. Все параметры колонок указаны на рисунке. Хроматограммы
смоделированы по данным работ [50, 51]
с выбранными подходящими параметрами колонки) потратить на их разделение
менее I минуты.
Ранее в большинстве лабораторий предпочитали использовать колонки,
упакованные сорбентом с частицами диаметром 5 мкм. Такие колонки менее
требовательны к оборудованию (раздел 3.9) и меньше засоряются механическими примесями
образца. Сейчас же растет популярность колонок, упакованных частицами диаметром
3 мкм и менее (см. следующий раздел 2.5.3.2). Если какой-либо параметр колонки
изменяют с целью повышения разрешения или сокращения продолжительности
разделения, то настоятельно рекомендуется использовать колонку с таким же типом
сорбента (например, Symmetry C;g). Это позволит избежать каких-либо изменений
селективности (раздел 5.4).
Больше советов по выбору параметров колонок можно найти в разделе 2.4.1.2.
2.5. Разрешение и разработка метода
99
2.5.3.2. Скоростная ВЭЖХ
Все большее внимание уделяется очень скоростным разделениям, например, с
продолжительностью анализа минута и менее для относительно простых образцов
(менее 10—15 соединений) или нескольких минут для более сложных. Допустим, у вас
есть подходящее оборудование и соответствующая колонка. Время, требуемое
для разделения, зависит от к последнего пика и от а пары пиков с самым низким
(«критическим») разрешением. После того, как подобраны «оптимальные» значения
к и а (оптимизация селективности), разрешение и время анализа зависят только
от N. Для скоростных разделений предпочтительны следующие условия: небольшой
размер частиц, короткая колонка и высокая скорость потока.
Сократить время анализа без потери эффективности (N), можно с помощью од
ного или нескольких способов:
• ультра-вые о кое давление (> 400 бар или 6000 psi);
• высокая температура;
• частицы особого строения.
Работа при высоком давлении. Несомненно высокое давление позволяет без
потери в количестве тарелок N (или разрешении) сократить время разделения (рис. 2.15,
раздел 2.4.1.1). Как и в случае разделений при более низком давлении, следует
использовать высокую скорость потока и (относительно) короткую колонку,
упакованную мелкозернистым сорбентом. Использование более высокого, чем на обычных
ВЭЖХ-системах давления (максимум 400 бар), относится к жидкостной
хроматографии сверхвысокого или ультравысокого давления (УВЭЖХ)1. Последняя при давлении
выше 400 бар позволяет либо улучшить разрешение, повысив jV, либо сократить
время анализа. Первую работу на эту тему опубликовал Роджерс |53|, более
подробно изучил Иоргенсон [54|. Позже появились коммерческие ВЭЖХ-системы, которые
позволяют работать при давлениях 1000 бар и выше.
Пример того, как с помощью высокого давления можно сократить анализ,
приведен на рис. 2.2\в, на котором изображена хроматограмма образца, который
анализировали ранее (рис. 2.20г). Эксперимент проводили в следующих условиях: колонка
30 х 1,0 мм, упакованная 1,5 мкм сорбентом, скорость потока 0,5 мл/мин. Рабочее
давление 750 бар. Изначально разделение занимало 17 минут, УВЭЖХ позволила его
сократить до 0,7 минут без потерь в разрешении (Rs = 2,0). Если сравнить этот
эксперимент с проведенным ранее (рис. 2.21я, 6), то преимущество высокого давления
становится очевидным. (Хотелось бы обратить внимание на то, что не все
имеющиеся в продаже УВЭЖХ-системы могут обеспечить скорость потока более 5 мл/мин и
потому, чтобы в быстрых разделениях можно было задать высокую линейную
скорость потока, используют колонки небольшого диаметра.) В работе |21] приведено
несколько похожих примеров, в которых благодаря переходу на УВЭЖХ, удалось
сократить продолжительность анализа в 2—6 раз.
Тем не менее, следует отметить, что некоторые используемые нами формулы
перестают работать, когда давление в колонке превышает 400 бар [55|. Вязкость
подвижной фазы увеличивается с давлением, поэтому давление больше уже не растет
пропорционально скорости потока. Время удерживания и селективность становятся
зависимыми от давления |1|, а значит, и от параметров колонки. При низком давле-
' Авторы книги предлагают аббревиатуру U-HPLC — ultra—high-pressure liquid chromatography,
что дословно переводится как жидкостная хроматография сверхвысокого давления или ЖХСВД,
но в настоящее время в России более распространена аббревиатура УВЭЖХ — ультра
высокоэффективная или сверхпроизводительная жидкостная хроматография. В дальнейшем мы будем
пользоваться именно ей. Прилагательное ультра высокоэффективная, впервые ввела в хроматогра-
фическую практику компания Waters. Она же запатентовала аббревиатуру UPLC. Все же остальные
должны писать UHPLC. — Прим. перев.
100
Глава 2. Основные принципы и управление разделением
нии это не так заметно. Наконец, подвижная фаза, перемешаясь по колонке,
нагревается пропорционально перепаду давления в колонке. Разница температур внутри
колонки негативно влияет на форму пика и число теоретических тарелок |56|, кроме
того, вызывает дальнейшее (нежелательное!) изменение А и а.
Несмотря на то, что у УВЭЖХ есть явные преимущества, для организации хро-
матографического процесса необходимо специальное оборудование (раздел 3.5.4.3) и
колонки (раздел 5.6.2). Помимо этого, для УВЭЖХ труднее разрабатывать метод |1,
55|, поскольку к, Dm, вязкость подвижной фазы и другие параметры, влияющие
на разделение, начинают в большей степени зависеть от давления, которое обычно
растет с увеличением срока эксплуатации колонки (при более низких давлениях эти
параметры практически не зависят от давления). Из-за этих и других причин
(безопасность, нормативные требования, стоимость, а также проблемы, связанные с рабо
той при очень высоком давлении) к моменту написания этой книги было неясно,
насколько УВЭЖХ сможет заменить ВЭЖХ в рутинных анализах.
Работа при высокой температуре. Обычно ВЭЖХ разделение проводят в интерва
ле от температуры окружающей среды до 50 °С. Какое-то определенное значение
внутри этого интервала выбирают, опираясь на оптимальную селективность (как
в примере на рис. 2.20г). Для сокращения времени анализа и улучшения разрешения
посредством увеличения числа теоретических тарелок в единицу времени при
заданном давлении, было предложено проводить разделение при более высокой
температуре (например, более 100 СС) |57—59|. Если мы вернемся к рис. 2.15, то вспомним,
что продолжительность анализа, число теоретических тарелок и перепад давления
на колонке взаимозависимы. С повышением температуры N еще больше увеличится,
при условии, что мы выбираем «оптимальные» параметры процесса (размер частиц,
длину колонки и скорость потока), при которых число тарелок в единицу времени
максимально. Это связано с тем, что повышение температуры снижает вязкость
подвижной фазы (приложение I) и увеличивает коэффициент диффузии вещества Dm.
Снижение вязкости подвижной фазы позволяет увеличить скорость потока без
изменения перепада давления на колонке (уравнение 2.13й). Это равнозначно разделению
при сверхвысоком давлении (УВЭЖХ). Увеличение Dm приводит к такому же
значению N при более высокой скорости потока (рис. 2.1 \6) и к более короткому времени
анализа. Следовательно, повышение температуры, в принципе, позволяет либо
сократить время разделения при неизменном /V, либо увеличить число теоретических
тарелок при той же продолжительности разделения.
Разделения при высоких температурах, наряду с вышеописанными
преимуществами, имеют ряд недостатков. Во-первых, раньше ВЭЖХ проводили при комнатной
температуре или близкой к этому значению, поскольку опасались разложения
образца во время разделения при повышенной температуре. Многие хроматографисты
стараются избегать этой проблемы, но вероятность того, что образец разрушится
в новых условиях, несомненно, очень мала |60—62|. Вторая проблема заключается
в возможном появлении радиальных градиентов температуры внутри колонки.
Без надлежащего термостатирования колонки и подвижной фазы пики приобретают
неправильную форму (раздел 3.7.1). Радиальные градиенты температуры могут стать
еще более серьезной проблемой с увеличением температуры колонки. Хотя и это
преодолимо: можно взять колонку с меньшим диаметром — в ней быстрее
происходит процесс уравновешивания температуры. Третий недостаток — нестабильность
сорбента при высоких температурах, особенно если рН подвижной фазы выходит
за пределы интервала 2—8. И, наконец, обычно при повышении температуры падает
селективность, хотя для выбранной пары пиков это может быть и не так.
«Идеальная» температура — это компромисс между максимальным N и максимальным а.
Частицы особого строения. Помимо широко используемых колонок, упакованных
сорбентом с полностью пористыми частицами, существуют и другие, упакованные
2.5. Разрешение и разработка метода
101
пелликулярными или поверхностно-пористыми частицами (раздел 5.2.1.1), и
монолитные колонки (раздел 5.2.4). Относительные достоинства каждого из этих типов
колонок приводятся в разделе 5.2. Крупные молекулы лучше всего разделять на пеллику-
лярных или поверхностно-пористых сорбентах поскольку влияние CV на высоту
теоретической тарелки (уравнение 2.17) становится меньше. Пелликулярные частицы
представляют собой твердые, непроницаемые для растворенных веществ, шарики,
покрытые тонким слоем пористого материала. Такие сорбенты легко перегрузить,
поэтому их использование ограничивается разделением очень малого количества образца.
Колонки с поверхностно-пористыми частицами, в отличие от пелликулярных,
покрыты более толстым слоем пористого материала, поэтому они выдерживают практически
такую же нагрузку, как и колонки с полностью пористыми частицами. У монолитных
колонок проницаемость гораздо выше по сравнению с колонками, заполненными
частицами, что позволяет проводить более быстрые разделение с более высокой
скоростью потока при прочих равных условиях. Относительные достоинства этих и других
традиционных колонок можно оценить с помощью графиков Поппе (рис. 2.146).
2.5.4. Разработка метода
В разделах 2.5.1—2.5.3 описано, как подобрать экспериментальные условия для
приемлемого разделения, включающего в себя разрешение пиков до базовой линии и
разумное время анализа. Однако, само по себе приемлемое разделение еще не все.
Чтобы разработать полноценную методику разделения необходимо |44|:
• оценить состав образца и цели разделения;
• провести пробоподготовку;
• выбрать вид хроматографии;
• выбрать детектор;
• подобрать условия разделения;
• спрогнозировать, определить и решить возможные проблемы;
• валидировать метод и определить критерии пригодности системы.
Возможно, какой-то из вышеперечисленных пунктов можно пропустить или
уделить ему минимум внимания. Для «сложных» образцов и/или сложных методик
анализа могут потребоваться не только эти действия, но и какие-то еще, подробно
изложенные в других главах этой книги. То есть в процессе разработки метода могут
возникнуть специфические трудности, требующие логического решения, которое
будет основано на предыдущем опыте, знаниях или на литературных источниках.
Некоторые примеры разработки метода, подобные этим, смотрите в работах |62, 631.
2.5.4.1. Оценка состава образца и целей разделения
Прежде чем приступить к разработке метода, рекомендуется изучить всю доступную
информацию о химическом составе образца. Если в пробе есть основания или
кислоты, то в подвижную фазу необходимо будет добавить буфер, чтобы
контролировать рН. Очень полезной, если она известна, может оказать величина рКа
образца (глава 7). Кроме того, молекулярная масса образца может повлиять на выбор
хроматографических условий (раздел 9.I.1, 13.4.1.4). Для проб, содержащих энантио-
меры, придется разрабатывать свою, особую методику (глава 14).
Цель разделения — это не только приемлемое разрешение компонентов за
минимальное время. Нужно еще учитывать имеющееся оборудование, на котором будет
реализована рутинная методика. Вполне возможно,что градиент нельзя будет реализовать
и поэтому придется разрабатывать изократическую методику. Определение следовых
количеств, например, при анализе посторонних примесей, может накладывать допол-
102
Глава 2. Основные принципы и управление разделением
нительные требования на стадию пробоподготовки и повлиять на выбор детектора.
Количественный анализ требует определенной минимальной точности (например,
+ 1—2% для основного вещества и +10—20% для следовых примесей). Впрочем, обычно
требуется разделить и определить лишь некоторые компоненты образца, например,
лекарственные препараты в крови или моче, пестициды в воде или почве.
2.5.4.2. Пробоподготовка
Прежде чем образец попадет в колонку, необходимо провести пробоподготовку,
чтобы устранить примеси, которые могут повредить колонку или мешать разделению
интересующих вас соединений. Процедура пробоподготовки (глава 16)
многостадийна, в ней используется множество различных сорбентов, и поэтому ее разработка
может быть сложнее, чем последующее ВЭЖХ-разделение. Если уже есть методика
пробоподготовки для аналогичного или похожего образца (разработанная в этой же
лаборатории или взятая из литературных источников), то ее имеет смысл
использовать в том виде, как она есть, или адаптировать к новому образцу и, тем самым,
облегчить себе задачу, избежав разработки полного цикла пробоподготовки. В
отношении ВЭЖХ-разделения такой подход не годится: вряд ли ВЭЖХ-метод удастся
применить без дополнительной оптимизации для нового образца (раздел 2.5.4.5).
2.5.4.3. Выбор вида хроматографии
Как правило, разрабатывать ВЭЖХ-метод начинают на обращенной фазе. При этом
в зависимости от природы образца могут быть предпочтительны другие виды
хроматографии. Часто это неочевидно до тех пор, пока не будут проведены первые
неудачные эксперименты на обращенной фазе. Если, например, в образце присутствуют
изомеры, которые сложно разделять на обращенной фазе, разумнее использовать
нормальную фазу с непривитым силикагелем. Кроме того, для препаративной
хроматографии предпочтительнее использовать нормальную фазу (глава 15).
2.5.4.4 Выбор детектора
Если компоненты образца имеют хромофорные группы, то в первую очередь, как
правило, выбирают УФ-детектор с переменной длиной волны. Структура молекул
компонентов образца может помочь с выбором наиболее подходящего детектора
(глава 4). Когда образец содержит неизвестные или не поглощающие в УФ-спектре
компоненты, имеет смысл использовать неспецифические детекторы — детектор
светорассеяния или рефрактометрический (используется реже).
Масс-спектрометрический детектор может быть использован как дополнение к УФ-детектору. Он
отличается универсальностью при обнаружении многих веществ и решении различных
задач. Ожидается, что в будущем роль ЖХ-МС существенно возрастет, и
масс-спектрометры станут рутинно использоваться практически во всех ВЭЖХ-методах.
2.5.4.5. Выбор хроматографических условий
О выборе условий ВЭЖХ-разделения говорилось ранее в разделах 2.5.1—2.5.3. Эта
тема будет упоминаться и в последующих главах, в которых разбираются отдельные
виды разделения или специфические образцы. Для большинства образцов
используется системный подход, основанный на методе проб и ошибок, который состоит
из трех последовательных этапов. В первую очередь подбирают нужную долю
растворителя В в элюенте (%В) до тех пор, пока не будет достигнуто время удерживания,
попадающее в правильный интервал, например, 1 < к < 10. На втором этапе
оптимизируют селективность а и разрешение. Для оптимизации селективности сначала
2.5. Разрешение и разработка метода
103
изменяют %В {например, на +10%) и температуру (например, 30—50 СС). Если
на хроматограмме остаются плохо разделенные или перекрывающиеся пики,
изменяют другие параметры разделения (описание этого можно найти в последующих
главах). На третьем этапе подбирают параметры колонки: ее длину, размер частиц
и/или скорость потока. Изменение этих параметров может приводить к умеренному
повышению числа теоретических тарелок и улучшению разрешения обычно за счет
увеличения времени разделения. Если после оптимизации селективности разрешение
становится больше необходимого (Я, 3> 2), то уменьшив длину колонки и/или
увеличив скорость потока можно существенно сократить время анализа. Во многих
случаях благодаря нескольким приведенным выше экспериментам можно разработать при
емлемыи метод разделения в течение одного-двух дней.
Бывают ситуации, когда необходимо разработать метод для разделения большого
количества образцов. Причем этот метод должен сочетать приемлемое разрешение и
минимально возможную продолжительность анализа. Тогда имеет смысл потратить
больше времени и провести «поисковые» эксперименты. Для этого
экспериментируют с разными сорбентами, с различными растворителями В, с рН подвижной фазы
или температурой. Если каждый эксперимент будет градиентным, то не надо
оптимизировать %В для каждого из исследуемых вариантов.
Существует еще один способ — поиск метода разделения этого или похожего
образца в литературе. Потом методом проб и ошибок подгоняют найденную методику
под имеющийся образец. Авторы книги не рекомендуют этот способ, поскольку в
литературе могут содержаться неточности, которые могут затянуть время разработки
окончательного приемлемого метода разделения. Системный подход с начальными
условиями, предложенными в этой книге, потребует меньше экспериментов и
приведет к лучшей окончательной методике. Тем не менее, помимо условий разделения
литературные данные могут оказаться полезными при выборе детектора и условий
детектирования (глава 4), а также на этапе пробоподготовки (глава 16)
специфического образца.
2.5.4.6. Спрогнозировать, определить и решить возможные
проблемы
Во время разработки метода и его дальнейшего использования при выполнении
рутинных ВЭЖХ-анализов могут возникнуть различные проблемы. Большинство
из них (неправильная форма пика, шум базовой линии и т.д.) можно сразу
обнаружить, и связаны они с недостатками используемых материалов, оборудования или
лабораторной техники. В главе 17 (о неполадках) рассмотрены вероятные причины
таких проблем и предложены пути их решения. Некоторые сложности можно
заблаговременно предупредить, проводя эксперименты, которые позволят свести к
минимуму вероятность их появления:
• слабое удерживание сильно полярных образцов;
• невыявленные пики;
• плохая воспроизводимость колонок от партии к партии;
• нестабильные условия разделения;
• изменения в оборудовании.
Слабое удерживание сильно полярных веществ — это общая проблема в ОФХ
(раздел 6.6.1, 7.3.4.3). Для неионизированных соединений, возможно, имеет смысл
проводить разделение с помощью НФХ (глава 8), в которой полярные вещества хорошо
удерживаются. Для ионизированных слабоудерживаемых соединений подойдет
ион-парная (раздел 7.4) или ионообменная хроматография (раздел 7.5).
Проблема невыявяенных пиков возможна в двух случаях: (1) низкая
чувствительность детектора, (2) неправильная хроматографическая методика для разделения двух
104
Глава 2. Основные принципы и управление разделением
соседних (перекрывающихся) пиков. Первая проблема обычно решается установкой
дополнительного неспецифического детектора (разделы 4.11—4.13), что целесообразно
при использовании УФ-детектирования для образцов, состав которых не до конца
известен на момент разработки методики. Перекрывающиеся пики очень легко не
заметить, особенно когда один пик значительно больше другого. Эта задача частично
решается с помощью масс-детектора (раздел 4.14), который способен с помощью
деконволюции извлечь информацию о перекрывающихся пиках. Другим способом
решения этой проблемы является разработка вслед за «первичной» методикой, в которой
все наблюдаемые в образце пики хорошо разрешаются, альтернативного ортогональ
ного разделения (раздел 6.3.6.2), селективность которого очень сильно отличается
от селективности первичной методики. Ортогональное разделение должно обеспечить
перемещение невыявленных пиков из одной части хроматограммы в другую, где
вероятность их обнаружения повысится. Скрытые пики могут появиться во время разра
ботки метода или позже при выполнении рутинного анализа, если пик
непредвиденного вещества из нового образца перекрывается с другим пиком на хроматограмме.
Плохая воспроизводимость колонок от партии к партии — в настоящее время ред
ко встречающаяся проблема. Вероятнее всего она проявится во время разделения
сложных образцов, когда на хроматограмме присутствует множество пиков с
предельным разрешением. Небольшое непреднамеренное изменение в селективности
колонки (раздел 5.4.2) может привести к снижению разрешения одного или нескольких
пиков. Чтобы избежать проблем, связанных с воспроизводимостью колонок от
партии к партии, придерживаются следующего правила: по окончании разработки ме
тода все его условия тестируют на колонках из других партий, чтобы удостовериться
в одинаковом разрешении. Обычно разделения на всех протестированных колонках
оказываются одинаковыми, а значит, в будущем можно не беспокоиться об их
воспроизводимости. Дополнительные способы компенсации возможных изменений
селективности колонки можно найти в разделах 5.4.2 и 6.3.6.1.
Нестабильные условия разделения приводят к ухудшению разрешения при
небольшом непреднамеренном изменении одного или нескольких условий разделения.
Например, в обычной лаборатории затруднительно избежать минимальных изменений
рН подвижной фазы, а они могут привести к значительным изменениям в
разрешении, если в образце есть ионизированные соединения (раздел 7.3.4). Для
подтверждения стабильности разработанного метода необходимо определить влияние
несущественных изменений условий на разрешение (раздел 12.2.6). Как правило, можно
изменить условия разделения таким образом, чтобы повысить стабильность метода.
Возможные изменения в оборудовании и их влияние на разделение также следует
изучить в процессе разработки метода. Самое важное требование — разработать
стандартные тесты, которые гарантируют удовлетворительную работу оборудования
(раздел 3.10.1 и 3.10.2). Время запаздывания градиента, которое варьируется от
системы к системе, необходимо учитывать при переносе метода, иначе это может
привести к сбою в работе метода из-за невоспроизводимости относительных времён
удерживания на разных системах. В разделе 9.3.8 можно найти разные способы решения
этой проблемы.
2.5.4.7. Валидация метода и пригодность системы
Разработав ВЭЖХ-метод, его тестируют на пригодность, как правило, согласно
рекомендациям надзорного органа (например, FDA1 и ICH2). Прецизионность и
точность метода может быть определена при повторных анализах подходящих образцов
1 FDA — Управление по санитарному надзору за качеством пищевых продуктов и
медикаментов (США). — Прим. перев.
2|СН — Международный совет по гармонизации (США). — Прим. перев.
2.6. Влияние количества пробы 105
согласно спецификациям по точности, разрешению, форме пика и другим
параметрам, относящимся к пригодности системы.
Методы, находящиеся под контролем надзорного органа, должны пройти
официальную валидацию с полным комплектом документов. Поскольку при неудачной
попытке валидации может потребоваться дополнительная документация
(отклонения, исследования и т.д.), целесообразно провести «предварительную валидацию».
Она подразумевает проверку некоторых параметров (например, прецизионности,
точности, линейности, возможности анализа всей серии образцов), которая покажет,
способен ли метод успешно пройти официальную валидацию. Опыт авторов показал,
что на предварительную валидацию стоит потратить дополнительное время. Это
существенно повысит вероятность прохождения валидации и сэкономит время и
деньги. Более подробно тема валидации и пригодности системы изложена в главе 12.
2.6. Влияние количества пробы
Пока объем и масса анализируемого образца достаточно малы, изменение массы или
объема отражается только на высоте или площади пиков, но не на удерживании,
ширине пиков или разрешении. При достаточно большом количестве (или объеме)
образца наступает перегрузка колонки: увеличивается ширина пиков, а разрешение
падает. Аналитическое ВЭЖХ-разделение обычно проводят на таком количестве
образца, которое никак не влияет на удерживание или разрешение. Если есть опасе-
ние.что масса или объем образца слишком велики, целесообразно повторить анализ,
уменьшив объем пробы в 2 раза. Если удерживание или разрешение не
изменились, значит, количество образца здесь ни при чем. Когда стоит задача провести
ВЭЖХ-очистку сырья, принято на колонку загружать гораздо большее количество
образца (нелинейное разделение), чтобы выход целевого продукта был максимален
(глава 15).
Обычно разделения проводят на колонках диаметром 4—5 мм и длиной 50—
250 мм, причем количество каждого компонента в образце должно быть не более
50 мкг, а объем образца i 25 мкл (при условии, что образец растворен в подвижной
фазе). Если диаметр колонки меньше, то количество и объем образца нужно
сократить пропорционально квадрату диаметра колонки. Однако, если проба содержит
ионизированные соединения, то разделение даже 1 мкг образца может привести
к перегрузке колонки (раздел 7.3.4.2 и 15.3.2.1). Понимание влияния объема и
количества образца на ВЭЖХ-разделение поможет:
ф избежать нежелательных изменений в разделении из-за слишком большого
количества образца;
• повысить чувствительность обнаружения микроколичеств, используя максимально
возможное количество образца;
• в препаративной ВЭЖХ получить максимально возможное количество
очищенного целевого продукта (глава 15).
Изменение в разрешении и/или удерживании, вызванное вводом слишком
большого количества или объема вводимого образца, называется перегрузкой по массе или
перегрузкой по объему соответственно.
2.6.1. Перегрузка по объему: влияние объема образца
на разделение
Если образец, растворенный в подвижной фазе, вводится в колонку в объеме Vs и
если объем пика на уровне базовой линии для образца очень малого объёма Vp0, то
106 Глава 2. Основные принципы и управление разделением
рассчитать объем пика образца большего объема V„ можно по следующей
формуле [64]:
Vp=Шк2+у1А ■ а27)
На рис. 2.22 можно увидеть как объем образца Vs влияет на размер и форму пика
при условии, что концентрация вещества в образце постоянна и нет перегрузки
по массе. В четырех экспериментах постепенно увеличивали объем пробы, в
результате чего сначала пик уширялся, а потом его вершина уплощалась и превращалась в
плато. Уравнение и рисунок предполагает, что вещество из инжектора переносится
на колонку в виде неискаженной, цилиндрической зоны. Однако, на практике зона
вещества размывается при смывании из инжектора, и ее объем 1^ увеличивается
примерно на 50%.
Объем пика вещества при малом объеме инжекции можно рассчитать по
уравнению Vpo= WF, а также по уравнениям (2.7) и (2.10а):
Vp0 ~ 0,002L4N-°Hl + к). (2.28)
Пока объем образца Vs < 0,4К,#, увеличение ширины пика и потеря в разреше
нии будут составлять не более 10% (уравнение 2.27), что вполне допустимо. (Если
допустимая потеря в разрешении 1%, то используйте Vs < 0,15f^n). Предположим,
что скорость потока оптимальна (Л = 2), тогда Н = 2dp, N = L/H = L/2dp. Если к
больше 1, то с учетом уравнения (2.28), можно определить допустимый объем
образца, который дает не более 10% потери в разрешении:
Vs< 0,14Z°'42<\ (2.29)
где Vs в мкл, L и dc в мм, d„ в мкм. В зависимости от размеров колонки и
упакованного в нее сорбента максимально допустимый объем образца будет меняться. Так
для колонок 150 х 4,6 мм с частицами 5 мкм Vs не более 80 мкл, для колонок
100 х 4,6 мм с частицами 3 мкм Vs не более 50 мкл, для колонок 30 х 4,6 мм с
частицами 3 мкм Vs не более 30 мкл, для колонок 30 х 2,1 мм с частицами 1,5 мкм У не
более 4 мкл. Если разрешение раноэлюируемых пиков не критично, можно вводить
и больший объем.
Образец может быть растворен в растворителе, отличном от подвижной фазы.
Если этот растворитель слабее подвижной фазы, то максимальный объем пробы может
быть увеличен без негативного влияния на ширину пика и разрешение. Если же
растворитель, наоборот, сильнее подвижной фазы, то это часто приводит к тому, что
на хроматограмме раноэлюируемые пики уширяются и/или искажаются |65—67|.
2/ \ \ 3
1
1 1
5 в
Время (мин)
Рис. 2.22. Влияние объема образца Vs на ширину и форму пика. Vs/Vc = 0,3 (пик 1),
3 (пик 2), 5 (пик 3), 15 (пик 4). Результат компьютерного моделирования
любезно предоставленный GeoffCox, Prochrom R&D
1
2.6. Влияние количества пробы
107
Вообще это распространенная ошибка и ее не следует допускать. Иногда, когда не
удается поменять растворитель, в котором приходится растворять образец, допускается
использование более сильного растворителя, но в этом случае на колонку наносят
очень маленький объем. Предварительное разбавление слабым компонентом
подвижной фазы (например, водой) в соотношении 1:1с последующей инжекцией двукратно
большего объема образца может также оказаться эффективным способом решения
проблемы сильного растворителя образца (особенно для тех образцов, растворитель
которых содержит компонент В в количестве более 50%). В градиентном элюировании
допускается растворение вещества в более сильном растворителе большого объема
(глава 9), поскольку в начальной точке градиента образец смешивается со слабой
подвижной фазой (малый %В). Если у вас есть сомнения, то хорошо бы сначала ввести
половину от нужного объема, а затем двукратно больший объем образца, понаблюдать
за разрешением и только после этого вносить соответствующие изменения.
Интересное исключение из приведенных выше выводов об использовании
сильного растворителя для образца было сделано в работах |68, 691 Для веществ, которые
при элюировании чистой водой или буфером имеют средние значения к. Для их
растворения можно использовать такие сильные растворители, как пропанол, тетрагид-
рофуран, изопропилацетат, 4-метил-2-пентанон, и это не влияет на форму и ширину
пика отрицательно до тех пор, пока вещества элюируются из колонки раньше
растворителя, в котором растворен образец. Об этом иногда полезно вспоминать
при работе с веществами недостаточно растворимыми в воде. Более подробно тема
влияния объема образца на разделение раскрыта в работах [70, 711.
2.6.2. Перегрузка по массе: влияние массы образца
на разделение
Даже небольшим объемом образца можно перегрузить колонку. Такое возможно
при высокой концентрации вещества в растворе. В результате пики на хроматограм-
ме уширяются и меняют форму. Причина перегрузки колонки по массе кроется
в ограниченной емкости сорбента к удерживанию молекул вещества. То есть
неподвижная фаза, соприкасающаяся с зоной вещества, насыщается образцом.
На рис. 2.23 показано, как размывается пик во время перегрузки колонки. Во всех
экспериментах использовали одинаковый объем образца и меняли только
концентрацию вещества в образце. Потом полученные хроматограммы наложили друг на
друга. В первых экспериментах (\а-в) анализировали небольшую массу вещества,
поэтому отсутствовало размывание и искажение пика, а высота была пропорциональна
массе вещества в образце. В последующих экспериментах (2, 3, 4) увеличили массу
вещества, и это привело к тому, что пики уширились и приобрели форму прямо-
т.
1
Рис. 2.23. Влияние массы образца на ширину и форму пика. Хроматограммы всех
четырех экспериментов наложены друг на друга. Пики 1 (а-в) соответствуют
образцам с малыми массами, и поэтому ширина этих пиков не зависит
от массы
108
Глава 2. Основные принципы и управление разделением
(а)
А
В'
•
ft
*
К
\
3 4
6 (мин)
6 (мин)
6 (мин)
2.5 мг Д
2.5 мгв
2.5 мг Л
10 мгв
2.5 мг А
25 мгв
Рис. 2.24. Влияние перегрузки по массе на разделение. Образец состоит из (3-гидро-
ксиэтилтеофиллина А и 7р-гидроксипропилтеофиллина В, вещества
анализировали раздельно и вместе. Массы веществ указаны на рисунке. Условия:
колонка 150 х 4,6 мм, 5 мкм, остальные (изократические) условия можно
найти в работе ]70|. Пики, обозначенные А'и В', получали при анализе
каждого компонента отдельно, обозначенные А и В — вместе [72|
угольных треугольников. Массу образца увеличивали в следующей
последовательности: 1а (самая маленькая масса) < 16 < 1в ■« 2 < 3 < 4 (самая большая масса).
До тех пор пока масса каждого отдельного компонента образца не превышает
определенного значения (для колонок диаметром 4,6 мм оно составляет не более
50 мкг, для колонок меньшего диаметра — еще меньше), на перемещение зоны
каждого вещества внутри колонки не влияют зоны других компонентов образца.
Следовательно, в общем случае, определяют перегрузку колонки и искажение формы
пиков массы индивидуальных компонентов образца, а не общая масса нагружаемого
образца. Это продемонстрировано на рис. 2.24. Масса образца составляет 2,5 мг
(рис. 2.24а) каждого из двух соединений (ксантинов) для (перегруженного)
ОФХ-разделения. Это примерно в 50 раз больше максимальной массы вещества
для неперегруженного разделения (поэтому у пиков размывается задний фронт).
Такое же количество вещества (2,5 мг) анализировали раздельно (сплошная линия А
и В ) и вместе (пунктирная линия А и В). Затем хроматограммы наложили друг на
друга (рис. 2.24а). Оказалось, что они практически не отличаются независимо от того —
вместе анализировали два соединения или каждое отдельно. То есть вещество,
перегружающее колонку, никак не влияет на разделение других компонентов в образце до
тех пор, пока пики этих компонентов разрешаются до базовой линии. Аналогичное
изменение происходит в следующем эксперименте (рис. 2.246), где массу вещества В
увеличили до 10 мг. Наблюдается небольшое снижение удерживания пика вещества А, но
оно вызвано слишком большим количеством В в образце. И только существенное
увеличение массы В влияет на отделение пика А (25 мг на рис. 2.24*). Из этого
эксперимента можно сделать следующий вывод: перегрузка колонки и размывание заднего
фронта пика происходит только при слишком большой массе отдельного вещества
в образце (например, более 50 мкг для колонок диаметром 4,6 мм).
2.6.3. Как избежать проблем, связанных со слишком
большим количеством образца
При анализе одним и тем же методом смесей, состоящей из разных соединений,
желательно, чтобы значения А, /V и Rs оставались постоянными. Так гораздо проще
определять количества вещества в образце и идентифицировать пики по временам
2.6. Влияние количества пробы
109
удерживания. Значения k, N и Rs, в свою очередь, будут постоянными, если
анализировать достаточно небольшие количества вещества. Проще говоря, следует избегать
перегрузки колонки, и тогда разделение не будет зависить от количества образца.
То есть существуют определенные ограничения по массе и по объему образца (это
описано выше). Обычно в ВЭЖХ-анализах объем пробы постоянен, так что
основной проблемой является слишком большая концентрация вешеств(а) в ней. Снова
повторим, что имеет значение wmax отдельного вещества в образце, а не общая масса
образца. Например, если содержание ни одного из компонентов не превышает 10%
от массы образца, то максимальная масса образца будет в 10 раз больше, чем у
образца, содержащего только одно вещество.
2.6.3.1. Если концентрация образца больше ожидаемой
Если концентрация вещества меняется от образца к образцу, то более концентриро
ванные образцы могут привести к перегрузке по массе, что влечет за собой
ухудшение разрешения и изменение во времени удерживания. Влияние концентрации или
массы вещества на разделение нужно учесть в финальной части разработки
ВЭЖХ-метода. Необходимо определить максимальную концентрацию вещества или
массу wmax, чтобы в будущем избежать перегрузки по массе, а также возможной
перегрузки детектора, которая приводит к нелинейному детектированию. Образцы,
концентрации которых превышают установленные значения, следует разбавить и
проанализировать снова.
2.6.3.2. Анализ следовых количеств вещества
Чтобы обнаружить следовые количества вещества в образце, желательно максимально
увеличить высоту пика, то есть необходимо повысить отношение сигнала к шуму.
Обычно количество определяемого вещества, присутствующего в пробе, настолько
мало, что оно не может перегрузить колонку, чего нельзя сказать об остальных
компонентах образца, которые могут не только привести к перегрузке, но и помешать
разделению нужных веществ, т.к. достаточно большое количество одного вещества влияет
на разделение второго, соседнего. Это проиллюстрировано на рис. 2.24. В первом
случае (рис. 2.24а) количество обоих соединений А и В небольшое, и они хорошо
разделяются. При увеличении содержания вещества В в 10 раз (рис. 2.24в), разделение и
количественное определение вещества А становится затруднительным. Если анализировать
отдельно 2,5 мг вещества А, то его время удерживания составит 3,6 мин., в совместном
анализе с избытком вещества В (25 мг) время удерживания сдвигается и становится
равным 3,1 мин. Полоса вещества А стала уже, разделение хуже.
Если в образце содержатся слишком большие количества примесей,
рекомендуется перед ВЭЖХ-анализом провести пробоподготовку (глава 16) для их удаления.
При анализе следовых количеств предпочтительно вводить максимально возможный
объем пробы. Если в наличии имеется достаточное количество образца, а целевой
пик хорошо отделяется от соседних, то с помощью введения больших объемов
образца можно увеличить высоту пика. Если же образец растворен в растворителе, элюи-
рующая сила которого слабее подвижной фазы, то можно ввести еще большие
объемы образца, и размер пика пропорционально увеличится без дополнительных
размываний. Этот метод особенно результативен в градиентном элюировании и
позволяет повысить предел обнаружения.
NO
Глава 2. Основные принципы и управление разделением
2.7. Смежные темы
Ниже приведены некоторые темы разной степени важности:
• уравновешивание колонки;
• градиентное элюирование;
• пиковая емкость и двумерное разделение:
• отслеживание пика;
• вторичные равновесия;
• переключение колонок;
• прогнозирование удерживания по структуре вещества.
Все эти темы обсуждаются в данном разделе, чтобы обеспечить более полное
понимание смысла последующих глав.
2.7.1. Уравновешивание колонки
Хроматографической ВЭЖХ-системе с момента общего включения и включения
насосов, качающих подвижную фазу, до перехода в состояние готовности к анализу
образцов может потребоваться от 30 до 60 минут. Это время уравновешивания можно
сократить, если хранить колонку заполненной подвижной фазой. Как правило,
желательно в конце каждого рабочего дня промывать систему и колонку сначала
водой, а потом 100% органическим растворителем (раздел 3.10.2.!). В течение рабочего
дня состав подвижной фазы может меняться из-за смены методики разделения
между повторяющимися градиентными разделениями или во время разработки метода.
После каждой смены подвижной фазы для уравновешивания колонки новым
составом элюента перед вводом образца потребуется 10 или более колоночных объемов
подвижной фазы. Новую колонку лучше уравновешивать еще большим объемом
подвижной фазы. При каждой смене элюента или колонки и до начала анализа или
разработки методики необходимо убедиться, что колонка уравновешена. Качество
уравновешивания определяют серией анализов одного и того же вещества. Если
на хроматограммах отсутствуют какие-либо изменения, колонку считают
уравновешенной. Более подробно об уравновешивании колонки рассказано в разделах 7.4.3.2
(ион-парная хроматография), 8.5.2 (нормально-фазовая хроматография), 9.3.7
(градиентное элюирование).
2.7.2. Градиентное элюирование
Изократическое элюирование — это самая простая форма жидкостной
хроматографии, которая характеризуется постоянным составом подвижной фазы и хорошо
подходит для разделения многих образцов. Однако, некоторые образцы не
представляется возможным разделить в изократических условиях, как это продемонстрировано
на примере ОФХ-разделения девяти компонентного образца смеси гербицидов
(рис. 2.25). При элюировании 50% ацетонитрилом последние пики на хроматограмме
довольно сильно уширяются и имеют слишком большое время удерживания
(рис. 2.25а). При этом продолжительность разделения составляет 140 минут,
а последние пики затруднительно детектировать, поскольку, например, высота
девятого пика составляет всего лишь 3% от высоты первого пика. Чтобы решить
проблему последних пиков, можно перейти на 70% ацетонитрил. Сразу же возникает
другая проблема — низкое разрешение 1-3 пиков. Этот пример говорит о
существовании общей проблемы элюирования: невозможности в рамках одного изократического
элюирования разделить образец, состоящий из компонентов с большим диапазоном
удерживания (пики с сильно отличающимися значениями к), за приемлемое время.
2.7. Смежные темы
(а)
50% ACN
(б)
70% ACN
г -\—г т 1—1—1—I—е—
i 1 'I ■
20 40
60 80 100 120 140
Время (мин)
4 6
Время (мин)
10
30-85% В за 7 м
12 34 I
__—
■
l
i
J
I
u
„.
Bpe»,
ИН
.-
J
1Я <
--■
I
мин
r 8
6
)
1 £
,
- 100% В
-' -80%
-60%
-40%
-20%
— - 0%
Рис. 2.25. Иллюстрация общей проблемы элюирования как доказательство
необходимости градиентного элюирования. Образец — смесь гербицидов, (а) Изо-
кратическое элюирование 50% ацетонитрилом, колонка C|g 150 х 4,6 мм,
размер частиц 5 мкм, скорость потока 2,0 мл/мин, комнатная температура,
(б) условия такие же как в (о), кроме подвижной фазы (70% ацетонитрил),
(в) градиентное элюирование 30—85 %В за 7 минут, остальные условия те
же, что и в (а). Хроматограммы смоделированы по данным работы [5|
В градиентном элюировании происходит непрерывное изменение состава
подвижной фазы, в результате чего удерживание пиков, выходящих позже, последовательно
снижается. То есть по мере продолжения разделения подвижная фаза становится
сильнее (увеличивается %В). Эффективность градиентного элюирования
продемонстрирована на рис. 2.25в. Видно, что все пики из двух предыдущих экспериментов
(рис. 2.25а и 6) разделяются до базовой линии, у всех пиков примерно одинаковая
ширина и сравнимая чувствительность обнаружения (при условии, что отклик
детектора для каждого вещества одинаков). Время анализа составило чуть больше 7 минут.
Преимущества разделения этого образца в градиентных условиях очевидны. Кроме
того, градиентное элюирование позволяет решать ряд других задач, о которых речь
пойдет в разделах 9.1.1 и 13.4.1.4. Более подробно о градиентном элюировании
рассказано в главе 9.
2.7.3. Пиковая емкость и двумерное разделение
До сих пор в качестве оценки разделения использовалось критическое
разрешение Rs. Этот критерий можно использовать, когда на хроматограмме все целевые
пики разделяются до какого-то определенного уровня, а целью разделения является
некоторое минимальное разрешение всех пиков. Однако существуют образцы, кото-
112
Глава 2. Основные принципы и управление разделением
рые содержат так много компонентов, что становится практически невозможным
добиться хорошего разрешения всех интересующих нас пиков. Поэтому требуется
какая-то иная оценка «степени разделения» в разных экспериментальных условиях.
Пиковая емкость разделения есть общее число пиков, которые умещаются на хро
матограмме при условии, что каждый пик отделяется от соседнего с Rs = 1.
На рис. 2.26а показано разделение с диапазоном удерживания 0 < к < 20 и N = 100.
Пиковая емкость для изократических условий определяется по уравнению [73|:
К = | + [—] ^[—) = 1 + О^ЛГ0-5 logfM, (2.30)
где to — это время удерживания последнего пика на хроматограмме. Для подобных
разделений с к последнего пика меньше 20 и N равным 20000, PC = 108. Если
исключить пики с к < 0,5, то есть 0,5 <, к <, 20, то величина пиковой емкости упадет до 93.
Если для пиков, находящихся в интервале 0,5 < к < 20, Rs должно быть равно двум,
то PC будет 47.
В сложных образцах, состоящих из множества компонентов, пиковая емкость
имеет большое значение. Редко удается разделить такие сложные смеси с
приемлемым разрешением всех пиков. В таких случаях пиковая емкость становится лучшей
оценкой всего разделения, чем Rs. Разделение сложных образцов обычно проводят
в градиентных условиях, в которых значение пиковой емкости более актуально
(раздел 9.3.9.1). Особое значение пиковая емкость играет в так называемом двумерном
(2D-)KX) разделении (раздел 9.3.10), в ходе которого фракции после первого
разделения направляют на второе разделение (рис. 1.46, в). На рисунке видно, что группу
перекрывающихся пиков после первого разделения (фракция 7) направляют на
второе разделение, в ходе которого пики распределяются по всей хроматограмме
(ортогональное разделение). При этом общая пиковая емкость двух разделений будет
произведением пиковых емкостей каждого разделения. В приведенном выше примере
пиковая емкость изократического разделения PC = 100, а для 2D-)KX разделения она
составит PC — 100 х 100 = 10000. То есть объединенные хроматограммы в двумерном
разделении обеспечивают гораздо больше «места» для пиков, что дает возможность
разделять сложные образцы, состоящие из сотен или тысяч соединений.
Пиковую емкость не стоит путать с количеством соединений, которые реально
будут разделены с Rs— I, поскольку на практике практически невозможно добиться
одинакового расстояния между всеми пиками на хроматограмме (как на рис. 2.26а) |73|.
На рисунке 2.266 показана требуемая пиковая емкость РСтре6 для разделения образца,
состоящего из и соединений, с Rs = 1 для всех пиков. Перед оптимизацией
селективности (раздел 2.5.2) можно предположить более или менее случайное расположение
пиков на хроматограмме. В таком случае для того, чтобы все пики образца, состоящего
из 10 соединений («случайная» кривая на рис. 2.266), разделились с Л, > 1, требуемая
пиковая емкость должна быть примерно 80. Однако, после оптимизации
селективности расстояние между критическими парами пиков увеличится, тогда требуемая
пиковая емкость снизится до 40 («оптимизированная» кривая на рис. 2.266). Более
подробно эта тема изложена в работе |74].
2.7.4. Отслеживание пика
Для интерпретации разделений, проведенных в ходе раразработки метода,
необходимо отслеживать пики или их соотносить. Пики на разных хроматограммах,
соответствующие тем или иным компонентам образца, должны быть пронумерованы или
обозначены каким-либо другим путем (как на рис. 2.20). Например, пик 1 в первом
анализе соответствует веществу А с известной или неизвестной химической структу-
2.7. Смежные темы I 13
I
(в)
4 6
Время (мин)
10
Требуемая пиковая емкость PC (PCntf) для
разделения образца, состоящего из л
соединений с Rs = 1.0
(б)
500
400
300
200
100
—
-
"случайная" /
кривая
_/
/ "оптимизированная"
У кривая
/*s^ идеальное распределение (PCwq - PC)
10
20
30
п
40
50
Рис. 2.26. Пиковая емкость, (я) Пример разделения с PC = 8, Л' = 100 и Rs — 1 для
каждого пика; (б) пиковая емкость РСтрее, требующаяся для разделения
образца, содержащего п компонентов с J,= 1 [74]; «идеальное распределение»
пиков в соответствии с уравнением (2.30)
рой. Необходимо установить, какой пик соответствует этому же веществу во втором
анализе. Для многих образцов это сделать нетрудно. Например, шесть пиков в
приведенном на рис. 2.20б,г примере можно соотнести по относительному удерживанию
и плошали, которые обычно при изменении условий разделения существенным
образом не меняются. Пики 3 и 4 на двух хроматограммах меняются местами, но
площади этих и других пиков достаточно разные, что позволяет однозначно их
отследить. В процессе ручного отслеживания пика руководствуются его площадью и
формой, а также учитывают тот факт, что случаи изменения порядка элюирования
(если оно и возникает) обычно редки, то есть пик одного и того же вещества на хро-
матограмме обычно появляется в одной и той же области хроматограммы.
Тем не менее, в некоторых случаях довольно сложно отслеживать пики,
особенно если некоторые из них накладываются друг на друга (рис. 2.20а,в). Несмотря
на то, что существуют некоторые способы отслеживания пиков при помощи УФ-де-
тектора |75—791, универсальной методики, которая подходила бы для всех образцов,
все же нет. Для того чтобы практически исключить проблему, связанную с
отслеживанием пиков, во время разработки метода широко используется
масс-спектрометрическое детектирование (ЖХ-МС), которое позволяет (1) распознать каждый из
перекрывающихся пиков и (2) определить (обычно единственную) молекулярную массу
каждого пика на хроматограмме [75|.
114
Глава 2. Основные принципы и управление разделением
2.7.5. Вторичные равновесия
Хроматографическое удерживание основывается на (первичном) равновесии между
молекулами вещества X, находящимися в подвижной и стационарной фазах (рис. 2.4
и уравнение (2.2)):
Х( подвижная фаза)<-»Х(стационарная фаза). (2.2)
Молекулы вещества могут находиться в дополнительных (вторичных)
равновесиях, в ходе которых происходит ионизация кислот и оснований, образование
ионных пар, комплексообразование или взаимное превращение изомеров. В результате
молекулы вещества во время перемещения по колонке могут находиться в двух
формах. Распространенный пример — разделение частично ионизированной карбоновой
кислоты, которая находится в состоянии равновесия между ионизированной и неио-
низированной формами:
R-COOH<->R-COO+H+. (2.31)
Относительные концентрации каждой формы определяются кислотностью
вещества (значением рКа) и рН подвижной фазы (раздел 7.2), поэтому некоторая часть
молекул /-""находится в растворе в ионизированной форме, а другая часть (l-F~) —
в нейтральной форме. Если значение к ионизированной кислоты равно к~, а Ар — это
к неионизированной кислоты, то на хроматограмме обе формы молекул будут
присутствовать в виде одного пика для обеих форм, удерживание которого равно:
к = F~k~ + (I - F-Щ. (2.32)
При изменении рН подвижной фазы, меняется ионизация молекул кислоты,
а вслед за ней содержание доли F~ и значение к (раздел 7.2).
Кислотно-основные равновесия для кислотных или основных веществ,
описываемые уравнением (2.31), можно считать достаточно быстрыми процессами, так что
равновесие устанавливается гораздо быстрее, чем происходит миграция молекул
внутри колонки. То есть при движении молекул вещества по колонке они много раз
переходят из одной формы в другую, и обратно, а удерживание этих молекул будет
иметь среднее значение, описываемое уравнением (2.32). Вторичные равновесия
существенно не влияют на ширину и форму пика, несмотря на частые утверждения
об обратном. МакКалли |80] как-то заметил: «распространенное утверждение о том,
что смешанный механизм удерживания неизбежно приводит к размыванию заднего
фронта (пика), безосновательно». С другой стороны, иногда происходит размывание
заднего фронта пиков кислот и оснований, что, главным образом, связано со
свойствами колонки (раздел 5.4.4.1) или недостаточной буферной емкостью подвижной
фазы (раздел 7.2.1.1).
Если равновесие между двумя формами достигается достаточно быстро, то
на хроматограмме наблюдается один пик. Примером может служить разделение
частично ионизированных кислот, у которых во время перемещения по колонке между
двумя формами R-COOH и R-COO" быстро наступает равновесие, и на
хроматограмме будет присутствовать только один пик. Если же равновесие устанавливается
медленно, форма пиков становится размытой, искаженной и/или появляются отдельные
пики. Рассмотрим пример взаимного превращения цис и транс изомеров
пептидов |81|. При высоких температурах взаимное превращение происходит быстро, и
на хроматограмме пептида наблюдается один острый пик несмотря на
изомеризацию. При низких температурах взаимное превращение протекает медленно, в
результате на хроматограмме наблюдаются два отдельных пика. При промежуточных
температурах пик будет один, но широкий и искаженной формы.
2.7. Смежные темы I 15
2.7.6. Переключение колонок
Метод, основанный на переключении колонок, подразумевает наличие двух
колонок, соединенных между собой посредством крана-переключателя {раздел 3.6.4.1).
Образец сначала попадает в первую колонку, после которой одна или несколько
фракций последовательно направляются для разделения во вторую колонку. Метод
переключения колонок может быть полезен в следующих случаях:
• пробоподготовка образца (раздел 3.6.4.1, 16.9);
• двумерная жидкостная хроматография (2D-XX) (раздел 9.3.10, 13.4.5, 13.10.4);
• увеличение частоты ввода анализируемых образцов1.
Метод переключения колонок полезен при пробоподготовке или 2D-)KX, когда
из сложной смеси необходимо выделить одно или несколько веществ, при этом
в первом разделении все целевые пики могут полностью перекрываться (а = 1,00).
Чтобы разделить вещества с очень близким удерживанием, во втором разделении
обычно изменяют селективность, что достигается использованием другой колонки и
подвижной фазы. Пример использования метода с переключением колонок
изображен на рис. \Л6,в.
Другое приложение метода переключения колонок в рутинных анализах
позволяет увеличить частоту ввода образцов после оптимизации условий разделения с
целью максимального сокращения времени анализа. Рассмотрим гипотетический
пример рутинного анализа пиков в или г (или их обоих, рис. 2.27а). Общее время
анализа составляет 52 минуты, что означает чуть более одного разделения в час.
Пробоподготовка могла бы позволить удалить сильноудерживаемые соединения д и
е, тогда продолжительность анализа сократится до 25 минут (и это будет уже 2,4
разделения в час). Если в день надо проанализировать большое количество образцов,
то благодаря методу переключения колонок, который в этом случае удобнее всего
назвать хроматографией разделений внахлест |82|2, можно значительно увеличить
частоту ввода анализируемых образцов.
Из рис. 2.27я видно, что пики в и г хорошо отделяются от других пиков
на хроматограмме. Их можно отделить от других примесей, содержащихся в
образце, на более короткой колонке с большей скоростью потока. При этом общее
время разделения составит менее 2 минут (рис. 2.276), и тогда в час можно обработать
более 30 образцов. Их вводят каждые 2 минуты на колонку 1, перенаправляя
фракцию с пиками в ч г переключением крана в колонку 2, используемую для
разделения, представленного на рисунке 2.27а. При таком переключении колонок
(рис. 2.27ff) отдельный насос должен подавать во вторую колонку подвижную фазу
со скоростью 0,5 мл/мин, для того чтобы обеспечить такое же разделение пиков в
1 Мы бы добавили сюда увеличение эффективности разделения (циркуляционная
хроматография); возможность концентрирования и выделения примеси в автоматическом режиме N-Rich и
возможность препаративной очистки компонента, окруженного с обеих сторон примесями в
непрерывном режиме MCSGP. Два последних подхода (N-Rich и MCSGP) требуют отдельного
разговора, более уместного при обсуждении препаративной хроматографии. Здесь мы только
добавим, что из названия метода MCSGP (Multicolumn Counlercurrent Solvent Gradient Purification),
которое дословно нужно было бы перевести, как «многоколоночная противоточная очистка в
градиенте растворителя», с нашей точки зрения следует исключить прилагательное «противоточная»,
поскольку в подавляющем большинстве случаев это двухколоночная хроматография, в которой
нет никакого противотока. Вещество и смешанные зоны вводят в обе колонки сверху вниз. Исходя
из сути процесса, его следовало бы называть «многоколоночной градиентной препаративной
хроматографией» (МГПХ). — Прим. перев.
^Оригинальное название метода boxcar (иногда его называют stacked injections или box-car
stacking) мы перевели как хроматографию разделений внахлест, ориентируясь не столько на
дословный перевод, сколько на смысл самого процесса, заключающийся в том, что на окончание одного
разделения накладывается начало следующего однотипного разделения. — Прим. перев.
116
Глава 2. Основные принципы и управление разделением
и г, как на рис. 2.27а (т.е. с приемлемым разрешением). Поскольку при разделении
образца на второй колонке зоны виг занимают небольшой ее объем, то
одновременно можно разделять несколько образцов (рис. 2.27г). Из рисунка видно, что
на колонке может одновременно находиться до 12 фракций (показано продольное
сечение второй колонки и разделение фракций 1, 6, 10 и 12, а другие фракции
не показаны).
Окончательный вид разделений на второй колонке показан на рис. 2.216. После
двадцатипятиминутной задержки со второй колонки элюируются пики »и гсо
скоростью 30 образцов в час. Хроматография разделений внахлест подразумевает
последовательное разделение разных образцов на второй колонке, причем они не должны
перекрывать друг друга в процессе их перемещения по колонке. Чтобы избежать
перекрывания, достаточно согласовать скорость введения образцов в первую колонку
со временем, затрачиваемым на элюирование целевых пиков (например, виг)
на второй колонке.
Разделения внахлест редко встречаются в литературе [S3]. В настоящее время
преимущества масс-спектрометрического детектирования привели к тому, что
методом разделений внахлест стали гораздо реже пользоваться. Только когда для
разделения требуются чрезвычайно большие значения jV (например, в препаративном разде
лении соединений, различающихся лишь по изотопному составу), хроматография
разделений внахлест обеспечивает гораздо более высокую производительность, чем
другие методы'.
2.7.7. Предварительная оценка удерживания, основанная
на структуре анализируемого вещества
Очевидно, что предсказание времен удерживания, исходя из экспериментальных
условий и молекулярной структуры соединений, было бы полезно для подбора
оптимальных условий разделения. К сожалению, ко времени публикации этой книги
невозможно было делать достаточно точные прогнозы на этот счет. А такое
предсказание удерживания может быть полезным для подтверждения идентичности
неизвестного пика на хроматограмме. Удерживание к соединения определяется его
структурой и условиями разделения. При определенных условиях разделения log A
можно приблизительно определить по формуле:
\ogk=A + YARU0), (2.33)
где А — log к исходной молекулы (например, бензола), ARMii) — увеличение log к
в результате введении группы (0 в исходную молекулу (например, введение нитро-
группы i в бензол с образованием нитробензола). Смит |84| определил значения
АЛмц) ряда типичных заместителей в различных условиях ОФХ, которые позволяют
оценить удерживание по структуре молекул образца (для очень небольшого
количества возможных образцов и условий разделения).
' На наш вгляд этот абзац дает несколько неверное представление о возможностях и
перспективах систем с переключении! потоков, Метод разделений внахлест позволяет заметно увеличить
производительность и сэкономить растворитель и в этом споем качестве всегда будет полезен
(разумеется, в изократическом режиме). Он никак не увеличиват числа теоретических тарелок N.
Это увеличение обеспечивается циркуляционной хроматографией, которая проводит разделение
на колонке условно неограниченной длины. Т.е. зона, содержащая смесь, после прохождения
колонки направляется с помощью крана на вторую колонку, а со второй снопа на первую. N
увеличивается кратно количеству переключений. Количество разделений ограничено размыванием
зоны, содержащей смесь. Метод предназначен для препаративного выделения компонентов плохо
отделяющихся друг от друга. — Прим. перев.
2.7. Смежные темы I 17
£ £
■1
Колонна-2
400 х 4.6 мм
3 мкм
0.5 мп/мин
«
Л.
20
40
Время (мин)
Колонка-1
50 х 4.6 мм
3 мкм
2.0 мп/мин
(б)
■^Д-^.
0.2
—1 1 1 1 1 1 1 1 1 г-
0.4 0.6 0.8 1.0 12 1.4 1.6 1.8
Время (мин)
Переключение колонок
колонка-1
(в)
I—»насос-1
^насос-
ф*й+0*сли
переключающий кран
слив
2—Н^
UZZZZZZZZ2ZZZZi-*t}\
колонка-2 детектор
пробоотборный
кран
12
Г
к
Перемещение внутри колонки-2
Л6
к
(г)
Л'д
вход
Последовательный анализ
5 10 15 20
выход
иши1шд1ш)Ш
20
30
40
50
60
70 (мин)
Рис. 2.27. Хроматография однотипных разделений гипотетического образца, (а)
Оптимизированное разделение образца с приемлемым разрешением (колонка 2),
(6) быстрое разделение образца на более короткой колонке с большей
скоростью потока (колонка 1), (в) схема разделения того же образца методом
хроматографии однотипных разделений, (г) перемещение выбранных
фракций образца (1, 6, 10 и 12) внутри второй колонки непосредственно перед
элюированием фракции образца 1, (д) начальная часть хроматограммы со
второй колонки
118
Глава 2. Основные принципы и управление разделением
В случае гомологических рядов уравнение (2.33) приобретает вид:
log к = А + пасту (2.34)
где п — количество метиленовых звеньев (-СН2) в молекуле, а^^2 — увеличение
log к при добавлении одной -СН2-группы в молекулу. В результате график
зависимости log А гомологических рядов от л, как правило, имеет линейный вид (учитывайте
исключение, описанное в разделе 6.2.2 и на рис. 6.5). Зависимости, похожие на
уравнение (2.34), можно применять к другим рядам соединений, у которых есть
некоторое количество п эквивалентных групп в молекуле (например, олигомеры
поливинилового спирта с повторяющимися СЬ^СН^О-группами, полистирол
с СН2(С,,Н5)СН2-группами и т.д.). Уравнения (2.33) и (2.34) называют уравнениями
Мартина в честь АДж.П. Мартина, который первым использовал эти зависимости.
Когда дело касается градиентного элюирования, уравнение (2.33) записывают
в виде:
tR « А + TAtm, (2.35)
где А — время удерживания исходной молекулы, ХД/ад — константа для данной
группы г, введенной в исходную молекулу. Уравнение (2.35) использовалось
для предсказания градиентного времени удерживание целого ряда молекул,
например, триацилглицеринов |85|, пептидов |86|, полисахаридов |87|. В каждом случае
эти предсказания работают только для определенных условий разделения.
Хотя уравнение (2.33) или (2.35) может иногда оказаться полезным для оценки
того, где должен находиться пик на хроматограмме, гораздо большее влияние, чем
количество заместителей и их тип, на удерживание особенно сложных полярных
молекул, как правило, присутствующих в ВЭЖХ-образцах, оказывают другие факторы.
С 1950-х годов многие хроматографисты исследовали влияние структуры образца
на удерживание в надежде однажды научиться без экспериментов предсказывать
удерживание (поиски хроматографического «Священного Грааля»). В целом
оказалось невозможным прогнозировать ВЭЖХ удерживание с точностью достаточной
для разработки метода (неудавшийся эксперимент описан в [88|). В 2007 году |89|
сообщалось об интересном исключении из, как правило, неудачных попыток
предсказать удерживание по молекулярной структуре образца. Тогда
масс-спектрометрическое детектирование в сочетании с предварительным прогнозом удерживания
позволили идентифицировать отдельные пептиды в белковом гидролизате.
2.7.7.1. Модель сольватационных параметров1
Хорошо известный и широко используемый подход связан с сольватационными
параметрами. Он применяется для объяснения зависимости удерживания в ОФХ от
образца, колонки и условий разделения (см. работы |90, 91] и особенно |14|).
Предполагают, что удерживание неионизированного образца определяется гидрофобными
взаимодействиями и водородными связями между образцом, подвижной фазой и
сорбентом. Уравнение модели сольватационных параметров имеет вид:
log к = С, + vKx + rR2 + s-n.!.{ + al.a.% + finp2. (2.36)
(/) (ji) (tfi) <fv) (v)
Коэффициент удерживания вещества к связан, во-первых, с постоянной С(,
зависящей от колонки и условий разделения, во-вторых, с величинами, зависящими от
разделяемого вещества (v,r^,a,b), в-третьих, с величинами, независимыми от разде-
1 В хроматографической литературе эта модель обычно называется моделью сольватационных
параметров Абрахама. — Прим. ред.
Литература I I9j)|
ляемого вещества (Vx, R2, щ , Xa| , ^Рг)- Слагаемые i—ш относятся к гидрофобным
взаимодействиям, а № и v определяются образованием водородных связей между
веществом и сорбентом или подвижной фазой. Значения параметров v,r,s,a,b
для большого количества различных веществ сведены в таблицу, в то время как
параметры Vx, Rj, л.", laf, ZP2 можно определить для колонки и данных условий,
используя соответствующие тестовые соединения.
Уравнение (2.36) дает представление о факторах, определяющих обращение фа
зовое разделение, однако большая погрешность (примерно +20%) в определении
значений к не позволяет использовать его при разработке метода. Кроме того,
уравнение (2.36) нельзя применять к ионизированным образцам и оно не учитывает ряд
дополнительных взаимодействий, влияющих на удерживание (см. дополнительно
раздел 5.4).
Литература
1. М. М. Fallas, U. D. Neue, M. R. Hadley, and D. V. McCalley, J. Chromatogr. A, 1209(2008) 195.
2. C. A. Rimmer, C. R. Simmons, and J. G. Dorsey, J. Chromatogr. A, 965 (2002) 219.
3. J. M. Bermudez-Saldana, L. Escuder-Gilabert, R. M. Villanueva-Camafias, M. J. Medina-
Hernandez, and S. Sagrado, J. Chromatogr. A, 1094 (2005) 24.
4. M. Shibukama, Y. Takazawa, and K. Saitoh, Anal. Chem., 79 (2007) 6279.
5. M. A. Quarry, R. L. Grab, and L. R. Snyder, J. Chromatogr., 285 (1984) 19.
6. L. R. Snyder, unreported data. For a single mobile phase (50% acetonitrile/buffer) andseveral hun-
dered different columns.
7. T. Braumann, G. Weber, and L. H. Grimme, J. Chromatogr., 261 (1983) 329.
8. L. С Tan, P. W. Carr, and M. H. Abraham, J. Chromatogr. A, 752 (1996) 1.
9. R. A. Keller, B. L. Karger, and L. R. Snyder, in Gas Chromatography. 1970, R. Starkand S. G.
Perry, eds., Institute of Petroleum, London, 1971, p. 125.
10. K. Croes, A. Steffens, D. H Marchand, and L. R. Snyder, J. Chromatogr. A, 1098 (2005) 123.
11. L. R. Snyder, P. W. Carr, and S. С Rutan, /. Chromatogr., 656 (1993) 537.
12. L. R. Snyder, J. Chromatogr. Sci., 16 (1978) 223.
13. J. A. Riddick and W. B. Bunger, Organic Solvents, Wiley-Interscieiice, New York, 1970.
14. M. Vitha and P. W. Carr, J. Chromatogr. A, 1126 (2006) 143.
15. S. M. С Buckenmaier, D. V. McCalley, and M. R. Euerby, J. Chromatogr. A, 1060 (2004) 117.
16. S. Heinisch, G. Puy, M.-P. Barrioulet, and J.-L. Rocca, J. Chromatogr., 1118 (2006) 234.
17. W. R. Melander, A. Nahum and Cs. Horvath, J. Chromatogr., 185 (1979) 129.
IS. D. Guillarme and S. Heinisch, /. Chromatogr. A, 1052 (2004) 39.
19. J. W. Dolan. J. Chromatogr. A, 965 (2002) 195.
20. G. Vanhoenacker and P. Sandra, J. Sep. Sci., 29 (2006) 1822.
20a. S. Heinisch and J. -L. Rocca, J. Chromatogr. A, 1216 (2009) 642.
21. S. A. C. Wren and P. Tchelitcheff, J. Chromatogr. A, 1119 (2006) 140.
22. J. C. Giddings, Dynamics of Chromatography, Part I, Principles and Theory, Dekker, New York,
1965.
23. G. Guiochon, in High-Performance Liquid Chromatography. Advances and Perspectives, Vol. 2,
C. Horvath, ed., Academic Press, New York, 1980, p. 1.
24. S. G. Weber and P. W. Carr, in High Performance Liquid Chromatography, P. R. Brown and
R. A. Hartwick, eds., Wiley-lnterscience, New York, 1989, p. 1.
25. J. H. Knox, in Advances in Chromatography, Vol. 38, P. R. Brown and E. Grushka.eds., Dekker,
New York, 1998, p. 1.
26. С R. Wilke and P. Chang. Am. Inst. Chem. Eng. J., 1 (1955) 264.
27. J. H. Knox, /. Chromatogr. A, 960 (2002) 7.
28. L. Kirkup, M. Foot, and M. Mulholland, J. Chromatogr. A, 1030 (2004) 25.
29. F. Gritti and G. Guiochon, Anal. Chem., 78 (2006) 5329.
30. F. Gritti and G. Guiochon, J. Chromatogr. A, 1169 (2007) 125.
31. J. G. Dorsey and P. W. Carr, eds., J. Chromatogr. A, 1126 (2006) 1-128.
120
Глава 2. Основные принципы и управление разделением
32. К. М. Usher, С. R. Simmons, and J. G. Dorsey, /. Chromalogr. A, 1200 (2008) 122.
33. R. W. Stout, J. J. Destefano, and L. R. Snyder, J. Chromalogr., 282 (1983) 263.
34. J. H. Knox and H. P. Scott, J. Chromalogr., 282 (1983) 297.
35. K. Miyabe, J. Chromatogr. A, 1167 (2007) 161.
36. G. Desmet, K. Broekhoven, J. De Smet, S. Deridder, G. V. Baron, and P. Gzi[,J. Chromatogr. A,
1188 (2008) 171.
37. H. Poppe, J. Chromatogr. A, 778 (1997) 3.
38. G. Desmet, D. Clicq, and P. Gzil, Anal. Chem., 77 (2005) 4058.
38a. P. W. Carr, X. Wang, and D. R. Stoll, Anal. Chem., 81 (2009) 5342.
39. D. V. McCalley, J. Chromatogr. A, 793 (1998) 31.
40. D. M. Marchand, L. R. Snyder, and J. W. Dolan, J. Chromatogr. A, 1191 (2008) 2.
41. J. J. Kirkland, W. W. Yau, H. J. Stoklosa, and С H. Dilks, Jr., J. Chromatogr. Sci., 15(1977) 303.
42. K. Lan and J. W. Jorgenson, J. Chromalogr. A, 915 (2001) 1.
43. J. P. Foley and J. G. Dorsey, Anal. Chem., 55 (1983) 730.
44. L. R. Snyder, J. J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, 2nd ed., Wi-
ley-lnterscience. New York, 1997.
45. L. R. Snyder and J. J. Kirkland, Introduction to Modem Liquid Chromatography, 2nded., Wiley-ln-
terscience, New York, 1979, pp. 34—36.
46. Reviewer Guidance. Validation of Chromatographic Methods, Center for Drug Evaluation and
Research, Food and Drug Administration (Nov. 1994).
47. K. Vaiko, L. R. Snyder and J. L. Glajch, J. Chromatogr., 656 (1993) 501.
48. L. R. Snyder, J. W. Dolan, and J. R. Gant, J. Chromatogr., 165 (1979) 3.
49. L. R. Snyder and J. W. Dolan, J. Chromatogr. A, 721 (1996) 3.
50. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, R. G. Wolcott, and P. W. Carr, J. Chro
matogr. A, 961 (2002) 171.
51. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, and P. W. Carr, J. Chrematogr.A, 961
(2002) 195.
52. L. R. Snyder and J. W. Dolan, High-Performance Gradient Elution, Wiley-lnterscience, New York.
2007, ch. 1.
53. B. A. Bidlingmeyer, R. P. Hooker, С H. Lochmuller. and L. B. Rogers, Sep. Sci., 4 (1969) 439.
54. J. E. MacNair, К. Е. Lewis, and J. W. Jorgenson, Anal. Chem., 69 (1997) 983.
55. F. Gritti and G. Guiochon, J. Chromatogr. A, 1187 (2008) 165.
56. R. G. Wolcott, J. W. Dolan, L. R. Snyder, S. R. Bakalyar, M. A. Arnold, and J. A. Nichols.
J. Chromatogr. A, 869 (2000) 211.
57. T. Greibrokk and T. Andersen, J. Chromatogr. A, 1000 (2003) 743.
58. X. Yang, L. Ma and P. W. Carr, J. Chromatogr. A, 1079 (2005) 213.
59. F. Lestremau, A. Cooper, R. Szucs, F. David, and P. Sandra, J. Chromatogr. A, 1109 (2006) 191.
60. F. D. Antia and С Horvath, J. Chromatogr., 435 (1988) I.
61. J. D. Thompson and P. W. Carr, Anal. Chem., 74 (2002) 1017.
62. K. P. Xiao, Y. Xiong, F. Z. Liu, and A. M. Rustum, J. Chromatogr. A, 1163 (2007) 145.
63. L. Deng, H. Nakano, and Y. Wwasaki, J. Chromatogr. A, 1165 (2007) 93.
64. J. C. Sternberg, in Adv. Chromatogr., 2 (1966) 205.
65. M. Tsimidou and R. Macrae, J. Chromatogr., 285 (1984) 178.
66. N. E. Hoffman, S.-L. Pan, and A. M. Rustum, J. Chromatogr., 465 (1989) 189.
67. N. E. Hoffman and A. Rahman, J. Chromatogr., 473 (1989) 260.
68. E. Loeser and P. Drumm, J. Sep. Sci., 29 (2006) 2847.
69. E. Loeser, S. Babiak, and P. Dnimm, J. Chromatogr. A, 1216 (2009) 3409.
70. H. Colin, M. Martin, and G. Guiochon, J. Chromatogr., 185 (1979) 79.
71. S. R. Bakalyar, С Phipps, B. Spruce, and K. Olsen, J. Chromatogr. A, 762 (1997) 167.
72. J. E. Eble, R. L. Grob, P. E. Antle, and L. R. Snyder, J. Chromatogr., 405 (1987) 51.
73. J. M. Davis and J. С Giddings, Anal. Chem. 55 (1983) 418.
74. J. W. Dolan, L. R. Snyder, N, M. Djordjevic, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 857 (1999) 1.
75. G. Xue, A. D. Bendick, R. Chen, and S. S. Sekulic, /. Chromatogr. A, 1050 (2004) 159.
76. J. L. Glajch, M. A. Quarry, J. F. Vasta, and L. R. Snyder, Anal. Chem., 58 (1986) 280.
77. H. J. Issaq and K. L. McNitt, J. Liq. Chromatogr., 5 (1982) 1771.
78. J. K. Strasters, H. A. H. Billiet, L. de Galan, and B. G. M. Vandeginste, J. Chromatogr., 499
(1990) 499.
Литература 121 ТЙ
79. Е. P. Lankmayr, W. Wegscheider, J. Daniel-Ivad, I. Kolossvary, G. Csonka, and M.Otto, J. Chro-
malgr., 485 (1989) 557.
80. D. V. McCalley, Adv. Chromatogr., 46 (2007) 305.
81. W. R. Melander, J. Jacobson and Cs. Horvath, J. Chromalogr., 234 (1982) 269.
82. L. R. Snyder, J. W. Dolan, and Sj. van der Wal, J. Chromatogr., 203 (1981) 3.
83. A. Nazareth, L. Jaramillo, R. W. Giese, B. L. Karger, and L. R. Snyder, J. Chromatogr., 309
(1984) 357.
84. R. M. Smith, J. Chromatogr. A, 656 (1993) 381.
85. J.-T. Lin, L. R. Snyder, and T. A. McKeon, J. Chromatogr. A, 808 (1998) 43.
86. C. T. Mant and R. S. Hodges, in HPLC of Proteins, Peptides and Polynucleotides, M. T. W. Hearn,
ed., VCH, New York, 1991, p. 277.
87. K. Yanagida, H. Ogawa, K. Omichi, and S. Hase, J. Chromatogr. A, 800 (1998) 187.
88. T. Baczek, R. Kaliszan, H. A. Claessens, and M. A. van Straten, LCGC Europe, 13 (2001) 304.
89. V. Spicer, A. Yamchuk, J. Cortens, S. Sousa, W. Ens, K. G. Standing, J. Q. Wilkens, and O. V.
Korkhin, Anal. Chem., 79 (2007) 8762.
90. P. С Sadek, P. W. Carr, R. M. Doherty, M. J. Kamlet, R. W. Taft, and M. H. АЬгапат.Лля/.
Chem., 57 (1985) 2971.
91. С F. Poole and S. K. Poole, J. Chromatogr. A, 965 (2002) 263.
ГЛАВА 3
ОБОРУДОВАНИЕ
3.1. Введение 124
3.2. Резервуары и фильтрование элюентов 125
3.2.1. Устройство резервуара и его использование 126
3.2.2. Фильтрование подвижной фазы 127
3.3. Дегазация подвижной фазы 128
3.3.1. Требования к дегазации 128
3.3.2. Дегазирование гелием 130
3.3.3. Вакуумное дегазирование и онлайн-дегазирование 131
3.4. Капилляры и соединения (фитинги) 132
3.4.1. Капилляры 132
3.4.1.1. Капилляры для низкого давления 132
3.4.1.2. Капилляры для высокого давления 133
3.4.2. Фитинги 136
3.4.2.1. Фитинги для низкого давления 136
3.4.2.2. Фитинги для высокого давления 138
3.4.2.3. Специализированные фитинги 139
3.5. Насосные системы 141
3.5.1. Возвратно поступательные плунжерные насосы 141
3.5.1.1. Двухплунжерные насосы 145
3.5.1.2. Накопительные плунжерные насосы 145
3.5.1.3. Активный клапан 146
3.5.2. Смешивание в потоке 146
3.5.2.1. Смешивание на стороне высокого давления 147
3.5.2.2. Смешивание на стороне низкого давления 148
3.5.2.3. Гибридные системы 148
3.5.3. Градиентные системы 149
3.5.4. Специальные методы 150
3.5.4.1. Методы, использующие малые {микро- и нано-) скорости
потока 150
3.5.4.2. Методы, использующие высокие скорости потока
(препаративные разделения) 150
3.5.4.3. Методы, использующие высокое давление 151
3.6. Автосамплеры 151
3.6.1. Шестиходовой кран ввода образца 151
3.6.1.1. Ввод из заполненной петли 152
3.6.1.2. Ввод из частично заполненной петли 152
3.6.2. Конструкция автосамплера 154
3.6.2.1. Автосамплеры с заполнением петли засасыванием 155
3.6.2.2. Автосамплеры с заполнением петли нагнетанием 156
3.6.2.3. Автосамплер со встроенным петлевым дозатором 156
3.6.3. Эффекты, связанные с количеством образца 158
3.6.3.1. Вводимый объем 158
3.6.3.2. Растворитель образца 159
3.6.4. Другие области применения кранов 160
3.6.4.1. Переключение колонок 160
3.6.4.2. Коллекторы фракций 162
3.6.4.3 Направление в слив 162
3.7. Колоночные термостаты 163
3.7.1. Требования к температурному контролю 163
3.7.2. Устройство термостатов 164
3.7.2.1. Нагревательные термостаты 164
3.7.2.2. Воздушные термостаты 164
3.7.2.3. Термостаты с элементами Пельтье 164
3.8. Управляющие модули 165
3.8.1. Сопровождение эксперимента 166
3.8.2. Управление системой 167
3.8.3. Сбор данных 167
3.8.4. Обработка данных 168
3.8.5. Создание отчета 168
3.8.6. Регулирующие функции 169
3.9. Влияние внеколоночных объемов 169
3.10. Обслуживание 170
3.10.1. Тест на работоспособность/пригодность системы 170
3.10.1.1. Установочная, операционная и эксплуатационная
квалификации 170
3.10.1.2. Градиентное тестирование 171
3.10.1.3. Дополнительная проверка системы 174
3.10.2. Профилактика 177
3.10.2.1. Периодическое обслуживание 177
3.10.2.2. Рекомендации для рутинных методов 180
3.10.3. Ремонтные работы 182
3.10.3.1. Персонал 182
3.10.3.2. Ведение учета 182
3.10.3.3. Специальные рекомендации по ремонту 183
Литература 184
124 Глава 3. Оборудование
3.1. Введение
Оборудование для современной ВЭЖХ разработано достаточно хорошо. За
некоторыми исключениями (например, устройства высокого давления, описанные в
разделе 3.5.4.3), принципиальные отличия в конструкции хроматографических систем
не часто встречаются. В то же время для повышения надежности оборудования
продолжают вноситься разнообразные изменения от небольших для одних моделей до
полного исчезновения с рынка других, однако прежние опасения относительно
быстрого морального устаревания оборудования ВЭЖХ для большинства приложений
неактуальны.
Те, кто только начинает использовать аналитическую ВЭЖХ, часто спрашивают:
какая хроматографическая система или какой производитель «лучше всех»? В
настоящее время различия между коммерчески доступными хроматографическими
системами минимальны, и можно с уверенностью сказать, что «плохих» хроматографов
на рынке нет. Это означает, что выбор того или иного хроматографа больше зависит
от услуг и технической поддержки, предоставляемых поставщиком оборудования,
а не от характеристик самого оборудования. Раньше пользователи зачастую
выбирали компоненты хроматографических систем от разных производителей и составляли
из них «идеальную» ВЭЖХ-систему. Сегодня так почти никто не поступает —
отчасти из-за того, что компоненты эти у разных производителей практически не
отличаются качеством, отчасти из-за того, что в хроматографической системе они
взаимосвязаны. Обычно компоненты, приобретенные у одного и того же производителя,
работают вместе лучше, чем модули разных производителей в одной системе. Так
например, насос, автосамплер и колоночный термостат обычно покупают у одного
и того же производителя в виде моноблока или в наборе.
Детектор можно купить и у другого производителя, особенно, если он
специальный — такой, как МС/МС (см. раздел 4.10). Поскольку большинство
производителей систем сбора и обработки данных часто закладывают в них возможности
управления оборудованием других производителей, то эту систему (и, соответственно,
блок управления) можно выбрать от одного производителя, а исполнительные
устройства от другого1. Однако, большинство лабораторий, принимая во внимание
обслуживание, обучение, ремонт и совместимость оборудования, старается
приобрести как можно больше компонентов хроматографических систем от одного
производителя. Особенно в тех случаях, когда в одном месте работают несколько систем.
Противоположный подход практикуют большие лаборатории, особенно те, которые
занимаются переносом методик в другие подразделения (раздел 12.7). В этих случаях
выбирают оборудование от нескольких производителей, чтобы оценивать
работоспособность метода на различных хроматографических системах. При передаче метода
такой подход позволяет выявить потенциальные проблемы, с тем, чтобы решить их
заранее.
Основные компоненты ВЭЖХ-системы показаны на рис. 3.1. Подвижная фаза
засасывается из резервуара насосом, который контролирует скорость потока и
создает давление, достаточное для того, чтобы прокачивать подвижную фазу через
колонку. Для ввода образца в колонку без остановки насоса, прокачивающего
подвижную фазу, используются инжектор или автосамплер. Разделение происходит внутри
1 И все же, при всей внешней привлекательности такого подхода, мы бы не советовали так
поступать, поскольку дьявол, как известно, кроется в мелочах. В данном случае это «мелочи»
программного обеспечения всех компонентов хроматографической системы. Стыковка всех модулей
системы может занять у вас довольно длительное время. Кроме того, даже если все будет работать,
то какие-то опции, доступные в том случае, если все модули куплены у одного производителя,
могут не быть активными. — Прим. персе.
3.2. Резервуары и фильтрование элюентов 125
Насос I Автосамплер
Резервуар
для подвижной фазы
Управляющий
модуль
Детектор
Рис. 3.1. Принципиальная схема ВЭЖХ-системы
колонки, которая обычно помещена в колоночный термостат. Детектор реагирует
на изменения концентрации анализируемого вещества во время разделения. Система
сбора данных отслеживает сигнал, выходящий из детектора, и предоставляет
результаты его обработки для графического и табличного вывода данных. Системный
контроллер (часто соединенный с системой обработки данных) управляет работой
различных модулей.
ВЭЖХ-система может содержать группу отдельных блоков (такие системы часто
называют «модульными») или же она может быть конструктивно объединена в один
блок. Такие системы называют «интегрированными». Из-за вечной нехватки места
на лабораторном столе модульные ВЭЖХ-системы сконструированы так, чтобы
занимать на нем минимум места. Это можно сказать и об интегрированных системах.
И еще одно дополнение. Хроматографические системы могут быть специально
сконструированы для низких расходов (микро) или высоких (препаратив) или для
хроматографии при высоких давлениях (разделы 3.5.4 и 15.2). Большинство аналитических
методов основаны на применении УФ-детекторов, но для других специальных
приложений доступны и другие детекторы (глава 4).
В этой главе обсуждаются различные составляющие ВЭЖХ-систем, за
исключением детекторов (глава 4) и модулей обработки данных (глава 11). Если это
специально не оговорено, то во всех случаях рассматривается коммерчески доступное
оборудование.
3.2. Резервуары и фильтрование элюентов
Резервуары для подвижной фазы (рис. 3.2) — хоть и простая, но важная часть
ВЭЖХ-системы. Для изократического элюирования, использующего подвижную
фазу, в которой заранее смешаны все компоненты, необходим всего один такой
резервуар.
В тех случаях изократического элюирования, когда компоненты подвижной
фазы смешиваются онлайн, или в случае градиентного разделения используют
несколько резервуаров. Поскольку подвижная фаза должна быть свободна от твердых
частиц, то ее перед заполнением резервуара нужно профильтровать.
126 Глава 3. Оборудование
(а) отверстие для
резервуар
Рис. 3,2. Резервуар для подвижной фазы
3.2.1. Устройство резервуара и его использование
Большинство резервуаров (рис. 3.2<з) сделано из стекла, хотя в некоторых случаях,
когда, например, определяют Ыа+-ионы с помощью ионообменной хроматографии,
стекло использовать нельзя. Чаще всего в качестве резервуаров используется
лабораторная посуда (например, колбы Эрленмейера), толстостенные стеклянные бутылки
или бутылки, в которых поставляются растворители. Некоторые производители хро-
матографических систем поставляют резервуары, специально сделанные для их
оборудования.
Одним из самых важных требований к резервуару, кроме инертности к
подвижной фазе, является его чистота. Стеклянные резервуары необходимо регулярно мыть
(например, еженедельно), используя стандартные методы мытья лабораторной
посуды. Необходимо чем-нибудь закрывать резервуар, чтобы предотвратить попадание
внутрь пыли и, вместе с тем, свести к минимуму испарение подвижной фазы.
При этом он не должен быть закрыт плотно, чтобы при откачивании подвижной
фазы не образовывался вакуум. Чаще всего резервуар закрывают резьбовой крышкой
с отверстием для капилляра входной линии подвижной фазы, диаметр которого
больше диаметра капилляра, или с двумя отверстиями (рис. 3.2я). Иногда горло
резервуара обжимают алюминиевой фольгой. Эти «способы» позволяют давлению
в резервуаре быстра уравновешиваться при выкачивании из него элюента.
Использования полимерных пленок (например, Paratilm®) для закрывания следует избегать,
поскольку некоторые подвижные фазы могут экстрагировать из них компоненты,
способствующие загрязнению системы'.
' Не экономьте «на пуговицах», раз уж вы раскошелились на покупку хроматографа. Вместо
того, чтобы позориться с алюминиевой фольгой и плоскодонными колбами, купите специальные
резервуары для элюентов, укомплектованные специальными крышками, в которых заранее
сделаны нужные отверстия для капилляров входной линии и предусмотрены уплотнения для того,
чтобы капилляры случайно не выскользнули из отверстия. Купите к этим крышкам специальные
фильтры, которые предотвращают попадание пыли и иных загрязнений из атмосферы, а также
способствуют быстрому уравновешиванию давления при выкачивании элюента. Коммерчески
доступными являются также и встроенные в линию краны, с помощью которых можно, в случае
необходимости, быстро перекрыть линию. — Прим. перев.
3.2. Резервуары и фильтрование элюентов I27jl
Капилляр, который идет из резервуара к насосу, оканчивается в резервуаре
входным фильтром (рис. 3.2а,б). Основная функция фильтра из пористого материала —
защищать хроматографическую систему от попадания механических частиц (пыли)
из резервуара с элюентом. Кроме того, он удерживает капилляр на дне резервуара.
Поскольку это не первое фильтрование элюента, то этот фильтр не должен создавать
большое сопротивление потоку. Чтобы растворитель свободно проходил через него,
лучше всего, если пористость фильтра будет > 10 мкм. Для того чтобы убедиться
в том, что это так, проделайте простой опыт. Отсоедините капилляр от входа в насос
или от клапанной коробки (в том случае, когда смешивание происходит на стороне
низкого давления). Если элюент не течет свободно — устройте сифон, т.е. засосите
в капилляр элюент из резервуара при помощи пипетки. Поток через такой сифон
должен быть > 10 х требуемой скорости потока. При этом резервуары с элюентом
должны быть расположены, как минимум, на полметра выше, чем точка измерения
скорости потока. Например, если обычная рабочая скорость потока от одного
до двух мл/мин, то самотеком элюент должен вытекать со скоростью не менее
20 мл/мин. Как правило, скорости потока в этих условиях больше 50 мл/мин. Если
жидкость при таком способе течет медленно, то необходимо заменить фильтр или
прочистить капилляр. Во время работы резервуар должен быть расположен выше,
чем входное отверстие насоса (например, > 50 см), чтобы обеспечить небольшое
избыточное давление элюента для более надежной работы насоса.
Множество входных фильтров различных конструкций из нержавеющей стали,
керамики, ПЭЭК и других инертных по отношению к подвижной фазе материалов
являются коммерчески доступными. Одна из часто встречающихся конструкций
входного фильтра изображена на рис. 3.26. В ней входной фильтр расположен снизу,
а не по боковой поверхности. Такое устройство позволяет выкачать элюент до
«последней капли» подвижной фазы1.
3.2.2. Фильтрование подвижной фазы
Работа различных узлов ВЭЖХ-системы может быть нарушена при попадании в них
твердых частиц. В перечень этих узлов входят: клапанная коробка, клапаны насосов,
капилляры и входной фильтр колонки. Поэтому очень важно, чтобы подвижная фаза
была свободной от твердых частиц. В том случае, если заранее отфильтрованные
растворители (для ВЭЖХ) недоступны, элюент перед помещением его в резервуар
нужно отфильтровать. Для лабораторий, которые работают согласно регуляторным
требованиям, должны быть написаны стандартные операционные процедуры (СОП).
В них должно быть указано, когда требуется дополнительное фильтрование
подвижной фазы, а когда нет.
Самый простой способ избежать попадания частиц в подвижную фазу —
использование предварительно отфильтрованных растворителей. Коммерчески доступные
растворители для ВЭЖХ перед упаковкой фильтруют через субмикронные фильтры
(обычно с диаметром пор 0,2 мкм). Воду для ВЭЖХ, подготовленную в лабораторных
условиях (например, очищенную на установке Milli-Q) на последней стадии очистки
' Кроме входных фильтров, способных выкачать весь элюент до «последней капли»,
коммерчески доступны специальные резервуары, в коническом дне которых есть небольшое углубление
для фильтров. Чаще всего используются цилиндрические входные фильтры из пористой
нержавеющей стали, в которых вся поверхность является рабочей — и боковая, и донная. Пористость
таких фильтров может довольно сильно различаться. Для систем УВЭЖХ 2 мкм, для обычных
ВЭЖХ-систем 10 мкм и для препаративных систем от 10 до 20 мкм. Необходимо отметить — чем
больше рабочая скорость потока, тем больше должна быть площадь и пористость фильтра для
беспрепятственного доступа элюента к насосу. — При», перев.
128 Глава 3. Оборудование
мемйранныи
фильтр
инертное
уплотнение
воронка для
фильтрования
_ подложка
*■ вакуум
пропускают через фильтр 0,2 мкм. Если
вы обычно используете растворители
класса «для ВЭЖХ», то, как правило,
перед использованием их не фильтруют.
Тем не менее, если вы не используете
специальные растворители для ВЭЖХ и,
к тому же, добавляете в подвижную фазу
твердые реагенты (например, в случае
фосфатного буфера), то целесообразно
фильтровать все подвижные фазы перед
использованием.
Подвижная фаза может быть легко
отфильтрована с помощью установки
для вакуумного фильтрования — такого,
который показан на рис. 3.3. Мембран
ный фильтр (обычно пористостью
0,5 мкм) укладывают на подложку,
которая тоже представляет собой фильтр
(только более грубый), в воронке
для фильтрования, установленной на
колбе Бунзена. Растворитель заливается
в воронку и фильтруется под вакуумом.
Производители фильтров снабжают по
требителей справочниками для выбора
подходящего фильтровального
материала в каждом конкретном случае. Например, тефлоновые фильтры гидрофобны, и
через них лучше всего фильтровать чистые органические растворители, такие как
метанол или ацетонитрил, но слишком неполярны, чтобы быстро профильтровать через
них воду. Уплотнение между колбой и воронкой делается или в виде инертной
(например, силиконовой) пробки или в виде двух пришлифованных стеклянных
конусов. Последнее лучше, поскольку нет опасности контакта элюента с пробкой.
колба Бунзена
Рис. 3.3. Установка для вакуумного
фильтрования подвижной фазы
3.3. Дегазация подвижной фазы
Присутствие пузырьков воздуха в подвижной фазе является общей проблемой
для функционирования ВЭЖХ-системы. Эти пузырьки могут привести к проблемам
в работе насосов и паразитным пикам на хроматограмме. В большинстве случаев
пузырьки можно удалить дегазацией подвижной фазы перед ее использованием.
3.3.1. Требования к дегазации
До тех пор, пока воздух растворен в подвижной фазе, проблемы с пузырьками
возникают редко. В принципе, подвижную фазу для изократического элюирования,
смешанную вручную, можно использовать и без дегазации, но раствор, насыщенный
воздухом, может выделять газ уже при небольшом разрежении, например, в том
случае, когда подвижная фаза проходит через встроенный в линию фильтр или когда
она входит в пространство относительно низкого давления в ячейке детектора.
По этой причине, а также для повышения общей эксплуатационной надежности
ВЭЖХ-системы, все растворители, используемые в обращенно-фазовой хроматогра-
3.3. Дегазация подвижной фазы 129
фни, настоятельно рекомендуется дегазировать. В случае нормально-фазовой ВЭЖХ
проблема выделения пузырьков газа при смешивании растворителей стоит менее
остро и потому дегазацию можно и не делать. Количество растворенного газа,
который должен быть удален из подвижной фазы, будет разным в зависимости от
конструкции ВЭЖХ-насосов. Некоторые насосы терпимы к растворенному газу, в то
время как другие требуют тщательной дегазации, без которой они надежно не работают.
Образование пузырей, как это видно изданных, представленных на рис. 3.4,
может приводить к серьезным сложностям в случае обращение фазовой хроматогра
фии. Предположим, например, что чистая вода и чистый этанол насыщены
кислородом, как это случается, когда элюенты находятся на воздухе. В том случае, когда
растворители смешаны, смесь содержит некоторое количество кислорода и элюента,
пропорциональное относительным концентрациям каждого растворителя {на рис. 3.4
показано пунктирной линией). Однако кислород менее растворим в смеси
растворителей (сплошная линия на рис. 3.4), и поэтому смесь теперь им перенасыщена. В
подобных случаях кислород или выделяется в виде пузырьков немедленно, или в месте
контакта с шероховатой поверхностью, например, с шероховатой поверхностью
встроенного в линию фильтра. Хотя на рис. 3.4 приведены данные для кислорода,
воды и этанола, тот же принцип верен и для воздуха, содержащего преимущественно
азот и кислород, в буферных водных растворах и других органических растворите
лей, таких, как ацетонитрил или метанол 111. Эти данные, кроме того,
свидетельствуют о том, что нет нужды удалять весь растворенный воздух из раствора —
достаточно, если количество растворенного воздуха в смеси будет ниже, чем на кривой
насыщения (сплошной) на рис. 3.4.
Для большинства работ дегазация важна, прежде всего, для улучшения работы
насоса. Тем не менее, в некоторых случаях, присутствие растворенного кислорода
может ухудшить работу детектора. В работе |2| описано, что УФ-детектирование
(раздел 4.3) на длинах волн до 185 нм возможно (подвижная фаза ацетонитрил/вода),
если элюент барботировали гелием для того, чтобы удалить кислород с оптического
пути в ячейке детектора. В этих условиях сигнал детектора вырос, а шум базовой
линии понизился. Даже при регистрации на более длинных волнах растворенный в
подвижной фазе кислород может повышать уровень базовой линии, как это показано
на рис. 3.5а. При 254 нм подвижная фаза, насыщенная воздухом, дает повышение
молярная доля этанола
Рис. 3.4. Растворимость кислорода в этаноле. ( ), концентрация кислорода после
смешивания насыщенной воздухом воды с этанолом (до выделения
избыточного кислорода); ( ) кривая насыщенной концентрации кислорода
в смеси воды и этанола [ 1 ]
130 Глава 3. Оборудование
0)
t
z
3
F
о
с
э
(а)
*
->
?
сч
*
(б)
1111 I ij.ijjjlI
-i—i—i—i—i—i—
0 8 0
t 1
He Воздух
T
0
t
He
-i—
16
Время (мин)
Рис. 3.5. Влияние барботирования гелием на отклик детектора при регистрации
нафталина, (а) УФ-детектирование при 254 нм, (б) детектирование
флюоресценции (возбуждение 250 нм, испускание 340 нм). (tHe) — начало
барботирования подвижной фазы гелием; (Твоздух) — начало барботирования
подвижной фазы воздухом. Данные из |1|
уровня фона (базовой линии) по сравнению с подвижной фазой, дегазированной
гелием. По-видимому, это происходит из-за того, что у подвижной фазы, насыщенной
воздухом, изменяется показатель преломления. В тех же условиях, но с
флуоресцентным детектором (раздел 4.5) около 75% интенсивности сигнала нафталина теряется
(рис. 3.56) в случае барботирования подвижной фазы воздухом, а не гелием |1].
Когда электрохимический детектор (раздел 4.6) работает в режиме восстановления,
растворенный кислород создает неприемлемый фоновый сигнал, и поэтому его
(кислород) необходимо удалить барботированием подвижной фазы гелием (раздел 3.3.2).
И, наконец, растворенный кислород может в процессе разделения вступать в
реакцию с некоторыми компонентами образца. Поэтому важно выбрать методику
дегазации, решающую как химические проблемы (например, отклик детектора), так и
физические (например, пузырьки в насосе), которые могут появиться из-за
растворенного газа в подвижной фазе.
При использовании предварительного дегазирования (в режиме офлайн) с
помощью гелия (из баллона) или вакуумной установки, элюент начнет снова
уравновешиваться с воздухом, как только дегазация прекратится. Для ВЭЖХ-систем, очень
чувствительных к растворенным газам в подвижной фазе, однократной дегазации
недостаточно. В таких случаях лучше всего непрерывное барботирование гелием
(Раздел 3.3.2) или постоянное онлайн вакуумное дегазирование (раздел 3.3.3.).
3.3.2. Дегазирование гелием
Дегазирование гелием является наиболее эффективным способом удаления
растворенного газа из подвижной фазы [3] (за исключением кипячение с обратным
холодильником и перегонку). Оно удаляет 80-90% растворенного воздуха. Обычно в
резервуар с элюентом помешают фильтр для диспергирования и подают через него под
3.3. Дегазация подвижной фазы 131Jj
небольшим давлением (например, 0,345 бар) гелий. При этих условиях необходим
лишь один объем гелия, чтобы дегазировать равный объем подвижной фазы |4|. Это
означает, что всего за несколько минут интенсивного барботирования гелием вы
п роде газируете подвижную фазу. Гелий сам по себе имеет такую низкую
растворимость в ВЭЖХ-растворителях, что пробарботированные им растворы практически
не содержат растворенного воздуха. Чрезмерное барботирование подвижной фазы
нежелательно, поскольку оно может изменить ее состав вследствие испарения
летучих компонентов. Тем не менее, интенсивное барботирование элюентов для обра-
щенно-фазовой хроматографии в течение нескольких минут не приведет к
проблемам. Ситуация с элюентами для нормально-фазовой хроматографии иная и, так как
они гораздо более летучи, гелий для их барботирования надо использовать
осторожно, если вообще это стоит делать. Впрочем, барботирование чистых растворителей
до того, как их смешали в потоке, к сложностям не приводит.
3.3.3. Вакуумное дегазирование и онлайн-дегазирование
Для большинства ВЭЖХ-систем использование вакуума для дегазации подвижной
фазы позволяет удалить достаточное количество растворенного газа, чтобы избежать
связанных с ним проблем. Вакуумная дегазация в течение 10—15 минут удаляет
60—70% растворенного газа |3|. В самом простом варианте частичное дегазирование
происходит при фильтровании растворителей, как это показано на рис. 3.3. Степень
этой дегазации может быть повышена до необходимой, если вместо воронки с
фильтром вставить в колбу с элюентом инертную пробку и в течение нескольких минут
вакуумировать содержимое колбы. Некоторые считают, что, если колбу во время ва-
куумирования поместить в ультразвуковую ванну, то это еще больше усилит
дегазацию.
В настоящий момент наиболее популярным способом является онлайн-дегазация.
Большинство производителей ВЭЖХ-систем включают онлайн-дегазаторы и как
стандартное, и как дополнительное оборудование в комплект приобретаемых
хроматографов. Принцип работы онлайн-дегазатора (для двух растворителей А и В) показан на
рис. 3.6. Коммерчески доступны дегазаторы для нескольких растворителей (от одного
до четырех). Их работа основана на селективной проницаемости для газов некоторых
полимерных трубок. Дегазатор монтируется перед насосом(-ами) в случае, если
смешивание происходит на стороне высокого давления (раздел 3.5.2.1), или перед
клапанами формирования градиента (клапанными коробками) в случае смешивания на
стороне низкого давления (раздел 3.5.2.2) или в случае гибридных систем (раздел 3.5.2.3).
Растворитель проходит через отрезок полимерной трубки, которая, в свою очередь,
помещена внутрь вакуумной камеры. Вакуум заставляет растворенные газы выходить
сквозь стенки трубки, а жидкая подвижная фаза остается внутри трубки. Детали
процесса можно посмотреть на рис. 3.66.
Если система сконструирована должным образом, то достаточно, чтобы объем
трубки для каждого растворителя был меньше миллилитра. При этом эффективность
дегазирования будет эквивалентной обычной дегазации для ВЭЖХ в статических
условиях |5|. Конечно, такой метод дегазации не столь эффективен, как
барботирование гелием, но удобство и дешевизна в сравнении с гелием сделало его
популярнее. Для тех случаев, когда концентрация растворенного кислорода критична
(например, при флуоресцентном и электрохимическом способах детектирования), или
там, где необходимо постоянно поддерживать минимальные концентрации
растворенных газов (например, при рефрактометрическом методе детектирования с
максимальной чувствительностью), непрерывное барботирование гелием с последующей
вакуумной дегазацией онлайн является оптимальным вариантом [5|.
132 Глава 3. Оборудование
подвижная фаза
вход
подвижная фаза
2 ► выход
вакуум
*выход
подвижная фаза
растворенный газ
Рис. 3.6. Схема дегазатора для двух растворителей, А и В
3.4. Капилляры и соединения (фитинги)
Капилляры и соединения (фитинги) необходимы для коммутации различных модулей
ВЭЖХ-системы, между которыми происходит перемещение подвижной фазы и
раствора образца. Если быть достаточно внимательным при выборе капилляров и
соединений, то можно избежать многих проблем, с ними связанных. Если же не уделить
этому должного внимания, то выбор неподходящих соединений может привести к
появлению нежелательных внеколоночных объемов, которые могут свести на нет само
разделение, особенно в случае пиков, элюируемых в малых объемах. Дополнительную
информацию о капиллярах и соединениях можно найти в интернете [6—7| и в
литературе, выпускаемой производителями, например в |8|. Следует отметить, что далее
диаметры капилляров и резьбы фитингов будут приводиться в английских дюймах
(как правило, дробные и десятичные значения), поскольку так принято на
американском рынке. В большом числе других стран эта продукция доступна и в метрическом
исполнении. Если вы используете капилляры и фитинги дюймовых и метрических
размеров, будьте внимательны к их маркировке. Даже если кажется, что они подходят
друг другу — не соединяйте их. Это может привести к их повреждению или утечке.
3.4.1. Капилляры
3.4.1.1. Капилляры для низкого давления
Обычно, при давлениях менее 7 бар используют полимерные капилляры. Они
нужны, главным образом, для того, чтобы транспортировать подвижную фазу из
резервуара в насос и для организации слива из детектора.
3.4. Капилляры и соединения (фитинги) 133 jl
На входе в насос обычно используют капилляр с наружным диаметром 1/86" и
внутренним 1/16" или меньше. Инертность фторуглеродных капилляров (например,
тех, которые сделаны из Teflon®) делает их наиболее пригодными для
транспортировки растворителей к насосу. Для этих целей могут быть пригодны и другие
полимеры (например, полипропилен и полиэтилен), но, на всякий случай, лучше всего
такие капилляры покупать у производителей оборудования для ВЭЖХ, чтобы быть
уверенным в достаточной чистоте этих полимеров для хроматографических целей.
Тефлоновые капилляры до некоторой степени проницаемы для газа и поэтому
воздух может диффундировать сквозь входной капилляр в подвижную фазу. Обычно
наличие воздуха не является проблемой, но в некоторых случаях (например,
при восстановительном электрохимическом детектировании) растворенный кислород
в подвижной фазе может привести к сложностям. Другой подходящий материал
для входных капилляров — ПЭЭК (полиэфирэфиркетон). Он непроницаем для
газов, но непрозрачен и потому увидеть в нем пузырьки воздуха невозможно. Кроме
того, капилляры из ПЭЭК имеют некоторые ограничения с точки зрения
химической стойкости (раздел 3.4.1.2).
От детектора в слив можно направлять капилляры, изготовленные из Тефлона,
полиэтилена, ПЭЭК и других относительно устойчивых материалов. Для удобства
к выходу детектора обычно присоединяют на слив капилляры с внешним
диаметром 1/16". Использование капилляров с внутренним диаметром 0,010" или меньше
создаст дополнительное противодавление, которое удержит воздух в растворенном
состоянии (не позволит образоваться пузырькам), пока он не выйдет из ячейки
детектора. Однако, такие капилляры надо применять с осторожностью, поскольку
при резком возрастании скорости потока может возникнуть слишком большое
избыточное давление в ячейке детектора. Как правило, более разумно для таких целей
использовать капилляры большего внутреннего диаметра (например, > 0,20") и рест-
риктор (дроссель) потока (коммерчески доступный и производимый многими
фирмами), чтобы поддерживать противодавление от 3,5 до 5,2 бар при любой скорости
потока.
Длина капилляров, рассчитанных на низкое давление, некритична, но в общем
случае эту длину лучше, насколько это возможно, минимизировать. Чрезмерная
длина капилляра может привести к увеличению времени промывки и к увеличению
времени, которое понадобится для уравновешивания. Большинство капилляров,
рассчитанных на низкое давление, можно разрезать лезвием бритвы. Для резки капилляров
ПЭЭК можно использовать резак, который поставляют производители капилляров.
Необходимо плоское лезвие, перпендикулярное оси разрезаемого капилляра.
3.4.1.2. Капилляры для высокого давления
В то время как капилляры низкого давления используются, в основном, для
транспортировки подвижной фазы из резервуара в насос или для организации слива
из детектора, капилляры высокого давления нужны в других частях хроматографиче-
ской системы. Обычные ВЭЖХ-системы сконструированы с учетом того, что
давление между насосом и детектором может достигать 414 бар (6000 psi), и капилляры его
должны выдерживать. Кроме того, капилляры, предназначенные для
транспортировки образца от автосамплера до колонки и от колонки к детектору, должны быть
достаточно инертны, чтобы образец не сорбировался и не разлагался по пути.
Внутренний диаметр и длина капилляров должны быть такими, чтобы не происходило
значительного уширения пиков (раздел 3.9). Для большинства случаев капилляры
из нержавеющей стали и ПЭЭК удовлетворяют этим требованиям.
Обычно для изготовления капилляров берут нержавеющую сталь марки AISI 316.
Она инертна по отношению почти ко всем растворителям и имеет очень высокий
134 Глава 3. Оборудование
предел прочности на разрыв. Стальные капилляры более жесткие, поэтому они
менее удобны, чем капилляры из ПЭЭК. Капилляры из ПЭЭК с внутренним
диаметром < 0.030" пригодны до я 483 бар (7000 psi), при более высоком давлении они
разрушаются. Для более высоких давлений требуются капилляры из нержавеющей
стали. Капилляры из ПЭЭК совместимы с большинством подвижных фаз,
используемых в ВЭЖХ, но набухают и становятся хрупкими при контакте с ТГФ, хлорсодер-
жащими растворителями или ДМСО (см. |6 или 7|, в которых есть полный список
совместимости ПЭЭК-капилляров с растворителями). Лучше всего в присутствии
таких растворителей не использовать капилляры из ПЭЭК. Если вы регулярно
заменяете соединительные капилляры, например, между автосамплером и колонкой, а
также колонкой и детектором, то использовать капилляры из ПЭЭК удобно, поскольку
они гибкие. Во всех случаях, когда подвижная фаза содержит компоненты,
вызывающие коррозию нержавеющей стали, использование капилляров из ПЭЭК поможет
минимизировать связанные с этим проблемы.
В большинстве ВЭЖХ-систем используются капилляры с наружным диаметром
1/16". Для некоторых случаев предпочитают использовать капилляры с наружным
диаметром 1/32", но их легче повредить, и они менее долговечны. Типовые
внутренние диаметры для капилляров с наружным диаметром 1/16" приведены в таблице 3.1.
Капилляры из нержавеющей стали производятся с внутренними диаметрами
от 0,005" до 0, 046", тогда как капилляры из ПЭЭК — от 0,0025" до 0,040".
Таблица 3.1. Внутренние диаметры типичных ВЭЖХ-капилляров (внешний диаметр 1/16" )
Внутренний диаметр (дюймы)
0,0025»
0,005
0,007
0,010
0,020
0,030
0,040
0,046»
Внутренний диаметр (ммУ
0,06
0,13
0,18
0,25
0,50
0,75
1,00
1,20
Объем (мкл/см)
0,03
0,13
0,25
0,51
2,03
4,56
8,11
10,72
"Номинальная величина (рассчитанная исходя из размеров в дюймах).
^Только для капилляров из ПЭЭК.
Только для капилляров из нержавеющей стали.
Капилляры с внутренними диаметрами 0,010" и 0,020" используются для подачи
растворителя от насоса к автосамплеру. В этом случае дополнительный объем
не принимается во внимание и, что немаловажно, капилляры с такими внутренними
диаметрами вряд ли забьются. В капилляре между автосамплером и колонкой
(по нему проходит раствор образца), а также в капилляре между колонкой и
детектором внутренний диаметр капилляра должен быть меньше (например, <, 0.007"), чтобы
избежать уширения пиков. Большие внутренние диаметры (£ 0.030") используются,
в основном, для изготовления петель ввода из-за относительно большого объема
на единицу длины капилляра (таблица 3.1).
Для того, чтобы минимизировать вклад капилляров в уширение пиков,
необходимо тщательно отнестись к выбору их длины и диаметра. Таблица 3.2
демонстрирует влияние различных комбинаций длины капилляра и его диаметра на
уширение пика для ряда колонок с определенными характеристиками. Несмотря на то,
3.4. Капилляры и соединения (фитинги)
135
что внутренний диаметр капилляра 0,007", он вполне достаточен для обычных
колонок с геометрическими размерами > 100 х 4,6 мм, упакованных частицами сорбента
> 3 мкм, колонки меньшего диаметра требуют капилляров с меньшим внутренним
диаметром. Для методик, в которых используются сорбенты с частицами менее
2 мкм и/или длинные отрезки капилляров (например, ЖХ-МС), необходимы
капилляры с внутренним диаметром 0,0025". Чем меньше внутренний диаметр капилляра,
тем скорее он забьется микрочастицами, которые появляются из подвижной фазы,
из образца, из уплотнений насоса и крана. Следовательно, при использовании
капилляров с внутренним диаметром < 0,005" нужно соблюдать специальные меры
для того, чтобы предотвратить их засорение. Необходимо помнить, что длина
капилляров в таблице 3.2 — это общая длина, включающая длину капилляра от автосамп
лера до колонки и от колонки до детектора. Иногда удобно использовать между
пробоотборником и колонкой короткий отрезок капилляра с внутренним диаметром
0,007" (чтобы свести к минимуму вероятность засорения) и отрезок капилляра
с внутренним диаметром < 0,005" между колонкой и детектором, чтобы
минимизировать уширение пиков. См. раздел 3.9 с дополнительным обсуждением внеколоночно-
го уширения пиков.
Таблица 3.2. Руководство по выбору длины капилляров
Характеристики колонки
Дмм)
150
150
100
100
100
100
50
50
50
50
50
50
dK (мм)
4,6
2,1
4,6
2,1
2,1
1,0
4,6
2,1
1,0
4,6
2,1
1,0
rf, (мкм)
5,0
5,0
3,0
3,0
1,8
1,8
3,0
3,0
3,0
1,8
1,8
1,8
N(h « 3)
10000
10000
11100
11100
18500
18500
5500
5500
5500
9200
9200
9200
Максимально допустимая длина (см), при
которой уширение полосы не превышает 5%"
0,0025"
1450
300
580
120
70
15
290
60
15
170
35
*
0,005"
90
20
35
*
*
*
20
•
•
10
*
*
0,007"
25
*
10
*
*
4
•
*
*
а
*
*
*< 10 см
"Условия: линейная скорость = 2,5 мм/сек внутри колонки (например, 2 мл/мин для колонки
с внутренним диаметром 4,6 мм), it = I
Резать капилляры из нержавеющей стали так же хорошо, как и в заводских
условиях, довольно трудно, поэтому лучше купить заранее нарезанные. Такие (заранее
нарезанные) капилляры хороши тем, что на заводе их заранее тщательно отмывают
и пассивируют, и потому их можно использовать без предварительной обработки.
Для тех случаев, когда невозможно купить предварительно нарезанные капилляры
из нержавеющей стали (рекомендуется марка AISI 316), покупают их оптом и
нарезают. Стальные капилляры легко можно нарезать специальным резаком, купленным
у поставщика капилляров. Нарезанные капилляры перед использованием
нужно промыть несколькими миллилитрами растворителя (присоедините один конец
136 Глава 3. Оборудование
капилляра к насосу и промойте капилляр, направив поток в слив), чтобы удалить из
него остатки масел или металлической пыли. ПЭЭК-капилляры легко режутся и в
лабораторных условиях, поэтому их покупают большими кусками. Для наилучшего
результата резаком для капилляров ПЭЭК наносится зарубка по периметру
капилляра, потом капилляр сгибается таким образом, чтобы он сломался в месте надреза.
При этом конец капилляра получается более правильной формы, чем при отрезании
резаком по всей толщине капилляра.
3.4.2. Фитинги
3.4.2.1. Фитинги для низкого давления
Такие фитинги используются в тех случаях, когда давление не превышает ж 7 бар.
На рис. 3.7 показаны две широко используемые конструкции фитингов. Для
фитингов с квадратной головкой «под ключ», которые используются для развальцованных
капилляров (например, Cheminert®, рис. 3.7а), требуется специальный инструмент,
чтобы развальцевать конец капилляра. Между фитингом и развальцованным концом
(а)
капилляр
(б)
фитинг
внутренний
конус
шайба фланец
(развальцованный
конец капилляра)
рифленый,
затягиваемый
пальцами винт
перевернутая
ферула
(в)
перевернутая
ферула
плоское дно
конец капилляра
резьбовая часть
гайки
соединение
Рис. 3.7. Фитинги для хроматографии низких давлений, (а) Фитинг Cheminert®,
использующий для уплотнения шайбу и развальцованный капилляр; (б) Фитинг
Fingertight*, использующий для уплотнения перевернутую ферулу; (в)
Неполное сечение соединения низкого давления для двух капилляров с помощью
соединения перевернутых ферул (б) (гайки не показаны), (а) С разрешения
VICI Valco Instruments Co. Inc.; (6) С разрешения Upchurch Scientific, Inc.,
подразделения IDEX Corporation в IDEX Health&Science Croup
3.4. Капилляры и соединения (фитинги) 1371)1
капилляра проложена шайба, чтобы предохранить капилляр от повреждений. Такие
фитинги требуют определенных навыков при работе с ними, но стоят недорого, и
потому пользуются популярностью у производителей хроматографического
оборудования, поскольку снижают их собственные затраты1. В альтернативной конструкции
использована ферула (коническое уплотнение), которая предохраняет окончание
капилляра от деформации (например, Fingertight®, рис. 3.76). Ферула повернута на 180°
по отношению к тому положению, в котором она используется при уплотнении
ВЭЖХ-соединений (так, как это показано на рис. 3.8я) — ее плоский конец прижат
к плоскости дна ответного соединения. Винт, уплотняющий все соединение, имеет
в себе внутренний конус и при закручивании прижимает ферулу к капилляру. Такой
тип соединений легко собирать (рифленый винт затягивают пальцами), и потому он
стал популярной альтернативой варианту с развальцованным на конце капилляром.
В промышленности используют стандартный винт 1/4", в котором на дюйм резьбы
приходится 28 витков. Таким образом, фитинги для хроматографии низкого
давления от разных производителей являются взаимозаменяемыми.
Соединение капилляров с помощью фитингов, предназначенных для низкого
давления, делается так, как это показано на рис. 3.7в. Концы двух капилляров соеди-
конец капилляра
(г)
рифленый, затягиваемый пальцами винт
Рис. 3.8. Фитинги, предназначенные для соединения капилляров при высоком
давлении, (а) Стандартный винт из нержавеющей стали и ферула на конце
капилляра; (б) корпус соединителя; (в) правильно собранное соединение; (г)
затягиваемый пальцами ПЭЭК-винт, ферула и ответная часть. С разрешения
Upchurch Scientific, Inc., подразделения IDEX Corporation в IDEX He-
alth&Science Group
' В настоящее время такие развальцованные капилляры почти никто не использует. Гораздо
проще использовать при уплотнении капилляров ферулы. Мало того, что не требуется никаких
специальных навыков и дополнительных инструментов, так еще и то давление, при котором
соединение сохранит свою герметичность, будет выше. — Прим. перев.
138 Глава 3. Оборудование
няются с помощью перевернутых на 180° ферул, которые устанавливаются встык
друг против друга, и таким образом образуется соединение (сравните это соединение
с аналогичным соединением для высокого давления на рис. 3.8 6,в). В других случаях
(например, в клапанных коробках хроматографических систем, раздел 3.5.2.2)
образуется сопрягаемое соединение, частичный разрез которого показан на рис. 3.7е.
Усилия пальцев достаточно, чтобы затянуть фитинги, предназначенные для
низкого давления, и поэтому их рекомендуют затягивать только руками, даже если
головка фитинга сделана под ключ. Если же для этих целей используют ключ или
плоскогубцы, то можно легко перетянуть соединение и сорвать резьбу. В тех случаях,
когда фитинг может открутиться (например, в клапанной коробке хроматографиче
ской системы низкого давления), некоторые производители предлагают для
дополнительной безопасности устанавливать контргайку. Помните, что в
хроматографических системах низкого давления открутившийся фитинг позволит воздуху попасть
в поток подвижной фазы. При этом наружу не вытечет ни капли жидкости.
Ослабленный фитинг — это совсем необязательно лужа подвижной фазы.
3.4.2.2. Фитинги для высокого давления
Все соединения капилляров в хроматографических системах высокого давления
включают в себя винт и ферулу, предохраняющую окончание капилляра (рис. 3.8а),
которые вставлены в резьбовое отверстие соединителя (рис. 3.86). Корпус фитинга
(является ли он составным, или обратным клапаном, или чем-то иным) имеет
резьбовую часть, конус для ферулы и цилиндрическую часть, в которую заходит
окончание капилляра. Для того, чтобы собрать фитинг, нужно последовательно надеть
на капилляр гайку и ферулу, вставить конец трубки в ответную часть фитинга
до упора, а потом затянуть гайку, чтобы уплотнить капилляр. Во время закручивания
гайки ферула обжимает капилляр и, таким образом, обеспечивает герметичность
и безопасность соединения. В правильно собранном фитинге высокого давления
не должно быть ни малейших зазоров между капилляром и корпусом фитинга
(рис. 3.8е). Такие соединения относятся к соединениям с нулевым мертвым объемом.
Большинство производителей выпускают гайки со стандартной резьбой UNF
10-32 и используют одинаковые ферулы для фитингов, обжимающих капилляры
1/16" (для метрических размеров используются другие стандарты). Это означает, что
фитинги разных производителей номинально взаимозаменяемы. На практике
фитинги разных производителей могут иметь разную глубину отверстия под капилляр,
и это означает (в случае стальных капилляров, где ферула плотно обжимается
на капилляре), что расстояние от ферулы до конца капилляра (рис. 3.9а) может быть
у разных производителей различным.
К примеру, отверстия для капилляров в инжекционных кранах Rheodyne
существенно глубже, чем в большинстве фитингов других производителей. Если конец
капилляра не доходит до конца отверстия, просверленного под него, это может
привести к проблемам (см. рис. 3.96). Например, если конец капилляра выпущен из ферулы
на слишком большое расстояние, то он достигнет дна отверстия раньше, чем ферула
конуса, в котором она должна уплотниться (левая сторона рис. 3.96). Это приведет
к утечкам. Обычно ферулу при закручивании гайки можно сдвинуть вниз по
капилляру и уплотнить. В этом случае и фитинг будет правильно собран и утечка
прекратится. Если же ферула сидит слишком близко к концу капилляра, то капилляр не
доходит до конца отверстия (правая сторона рис. 3.96). Это соединение будет
герметично, но при этом образуется небольшой мертвый объем между окончанием
капилляра и дном отверстия под него, что может привести к размыванию полосы
вещества. Чтобы избежать всех этих проблем, лучше всего покупать все фитинги
и капилляры из нержавеющей стали у одного и того же производителя.
3.4. Капилляры и соединения (фитинги)
Eia расстояние от конца
ферулы до конца
капилляра
(б)
утечки
мертвый объем
Рнс. 3.9. Влияние расстояния от конца ферулы до конца капилляра на качество
уплотнения фитингов, (а) Капилляр с показанным на нем расстоянием от конца
ферулы до конца капилляра; (б) сборка с капилляром, который слишком
сильно выпущен (слева), что приводит к утечкам, или слишком мало выпущен
(справа), что приводит к мертвому объему
Кроме нержавеющей стали фитинги для высокого давления делаются из ПЭЭК.
Ферулу, изготовленную из ПЭЭК (или из другого полимера) легко сдвинуть в
нужное положение1, в то время как вы просовываете капилляр в ответную часть
фитинга. Дополнительное удобство этого популярного конструктивного решения
заключается в том, что для затягивания ПЭЭК-ферулы можно использовать рифленый
ПЭЭК-винт (рис. 3.8г) и делать это пальцами вместо ключа. Такие затягиваемые
вручную ПЭЭК-фитинги используют в обычной ВЭЖХ, когда давление не
превышает 400 бар (6000 psi). Для более высоких давлений используют стальные соединения.
Если ПЭЭК-соединение течет, то хорошо бы выключить насос, ослабить соединение
(немного выкрутить винт, прижимающий ферулу) задвинуть капилляр в ответную
часть соединения на максимально возможную глубину, повторно затянуть винт и
снова включить насос. Это позволит избежать проскальзывания капилляра и
образования зазора в процессе уплотнения (см. рис. 3.95).
Винты, ферулы и корпуса фитингов могут быть выполнены из ПЭЭК или
нержавеющей стали. Часто вкручивают винты и ферулы из ПЭЭК в корпуса из
нержавеющей стали, но наоборот (установка стальных винтов в корпуса из ПЭЭК) делают
редко.
3.4.2.3. Специализированные фитинги
Две модификации стандартных фитингов для ВЭЖХ обеспечивают особые
преимущества:
• проточные (инлайн) фильтры;
• миксеры малого объема.
Проточный фильтр (рис. З.Юо) представляет собой модификацию стандартного
соединения, которое используется для соединения двух отрезков капилляров.
Для удаления механических примесей из потока используется мелкопористая филь-
1 В отличие от стальной ферулы, которая обжата на стальном капилляре на одном месте,
навсегда. — Прим. перев.
140 Глава 3. Оборудование
Рис. 3,10. Специализированные фитинги, (а) Проточный фильтр. Показан корпус
фильтра, винт и сменный фильтр, (б) ПЭЭК-миксер малого объема,
представляющий собой тройник. С разрешения Upchurch Scientific, Inc.,
подразделения 1DEX Corporation в IDEX Health&Science Group
трующая перегородка (обычно с порами 0,5 мкм). Хорошей практикой является
установка такого фильтра в каждой ВЭЖХ-системе после автосамплера. Хотя это и
незначительно увеличивает внеколоночный вклад в уширение пика, зато позволяет
удалить ненужные частицы от сальников насосов, уплотнений кранов, из подвижной
фазы и из образцов до того момента, как они достигнут входного фильтра колонки.
Некоторые насосы имеют встроенные в корпус выходного клапана мелкопористые
фильтры. Эти фильтры служат для улавливания твердых частиц из уплотнений
плунжеров насосов или других устройств, находящихся в гидравлической линии до
насоса. В отсутствие таких фильтров некоторые пользователи устанавливают
проточные фильтры между насосом и автосамплером, чтобы уловить частицы, которые
могут попасть в автосамплер и создать проблемы. Фильтр в колонке почти никогда
невозможно заменить без того, чтобы не испортить колонку, а проточный фильтр
сконструирован так, чтобы его было легко менять. Как только фильтр засоряется,
о чем сигнализирует повышение давления в системе, фильтрующую перегородку
заменяют, и ВЭЖХ-система очень скоро снова готова к работе. Проточные фильтры,
как из нержавеющей стали, так и из ПЭЭК с фильтрующими перегородками (часто
их называют фритами) разной пористости можно приобрести у самых разных
производителей.
Миксер малого объема может быть использован для того, чтобы приспособить
насосы, предназначенные для смешивания на линии высокого давления (раздел 3.5.2),
к задачам, которые выполняют насосы в ЖХ-МС-системах. Обычные миксеры
для систем высокого давления имеют объем I мл и больше, в то время как статиче-
3.5. Насосные системы
141
ские миксеры малого объема часто имеют объем менее 10 мкл. К примеру, миксер,
показанный на рис. З.Юб, изготовлен из ПЭЭК и имеет фильтрующую перегородку
с пористостью 10 мкм для улучшения перемешивания, а его рабочий объем всего
2,2 мкл. Доступны и другие конструкции миксеров с малым объемом.
3.5. Насосные системы
Почти все используемые сегодня в ВЭЖХ насосные системы являются вариациями
возвратно-поступательных плунжерных насосов. Насосы с гидравлическим
усилением используются для упаковки колонок и могут быть использованы в препаративных
системах (раздел 15.2). Шприцевые насосы используются для калибровки ЖХ-МС
систем, но не для подачи элюентов. Плунжерно-диафрагменные насосы когда-то
использовались для ВЭЖХ-систем, но теперь популярностью не пользуются.
Плунжерные возвратно-поступательные насосы с годами эволюционировали в надежные
устройства, способные проработать сотни часов без поломок и обслуживания.
Прецизионная работа насоса и его устройство для смешивания подвижной фазы — это
залог успеха при использовании ВЭЖХ для проведения анализов.
Большинство насосных ВЭЖХ-систем, продаваемых для проведения рутинных
анализов, предназначены для работы при давлении до 400 бар, но большая часть
хроматографистов эксплуатирует такие системы при давлении 150—200 бар.
ВЭЖХ-системы, предназначенные для быстрых анализов или для работы при
высоких давлениях, особенно с колонками, упакованными частицами сорбента меньше
2 мкм, имеют более высокие рабочие диапазоны давления 550—1000 бар или выше.
Следует отметить, что обычные системы, работающие при давлении до 400 бар, дают
возможность проводить быстрые анализы с колонками, упакованными мелкими
частицами без необходимости приобретать специализированное оборудование. Однако
когда обычные ВЭЖХ-системы используются при более высоких давлениях
(например, > 200 бар), нужно позаботиться о том, чтобы не было протечек. Например,
роторные уплотнения инжекторов должны быть затянуты с учетом того, что им
придется работать при повышенном давлении. С этой же целью необходимо подтянуть и
фитинги1. Кроме того, скорости механического износа уплотнений насоса,
уплотнений роторов инжекторов, и других движущихся частей обычно увеличиваются
при работе в условиях повышенного давления.
3.5.1. Возвратно-поступательные плунжерные насосы
Одноплунжерный насос, изображенный на рис. 3.11, является основой всех
насосных систем для ВЭЖХ. Основные части такого насоса:
• двигатель;
ф плунжер;
• уплотнение плунжера;
• клапаны.
Вращение кулачка, связанного с валом насоса и плунжером, приводит к
возвратно-поступательному движению плунжера в голове насоса. Плунжер обычно
изготавливают из сапфира, хотя в некоторых насосах он керамический. Уплотнение
плунжера, сделанное из полимерного материала, необходимо для предотвращения утечек
' Подтягивать надо с осторожностью, в соответствии с инструкцией, которую дает
производитель, к примеру, инжекторов. В противном случае можно или сорвать резьбу или деформировать
уплотнение. Все же лучше всего использовать оборудование, предназначенное для работы в
данных условиях и не подтягивать сверх того, что указано в инструкции. — Прим. перев.
142 Глава 3. Оборудование
из головы насоса. Входной и выходной клапаны управляют направлением потока
подвижной фазы. Во время заполнения (рис. 3.11а) плунжер выходит из головы насоса,
создавая тем самым разрежение внутри насосной камеры. При этом выпускной
клапан находится в закрытом положении, а впускной открывается, позволяя подвижной
фазе войти в камеру. Во время подачи (рис. 3.116) плунжер совершает обратное
движение — входит в камеру насоса. При этом впускной клапан закрывается, а
выпускной открывается, что позволяет подвижной фазе двигаться по направлению к ко
лонке.
Надежная работа клапанов — залог надежной работы насоса. Устройство обычно
используемых шариковых клапанов показано на рис. 3.12а. Клапан содержит
рубиновый шарик и сапфировое седло. Такое сочетание материалов дает надежную
герметизацию, при которой нет необходимости использовать дополнительные средства
помимо силы тяжести и разности давлений в камере насоса. В некоторых насосах
(а) Цикл заполнения
Рис. 3.11. Одноплунжерный возвратно-поступательный насос, (а) Заполнение, (б)
подача
(б) уплотнение
Рис. 3.12. Устройство клапана, (а) Шарик и седло; {б, «) активный клапан
используются керамические обратные клапаны. Иногда они снабжены маленькими
вспомогательными пружинками для более надежного закрытия.
Принцип действия шариковых клапанов достаточно прост. Когда давление
на входе в клапан (под шариком на рис. 3.12а) выше, чем давление на выходе, шарик
приподнимается над седлом, и растворитель проходит через клапан. В том случае,
когда давление выше на выходе (над шариком рис. 3.12а), шарик будет плотно
прижат к седлу, и жидкость через клапан не пройдет. Таким образом (рис. 3.11),
впускной и выпускной обратные клапаны позволяют насосу попеременно заполнять
камеру подвижной фазой из резервуара и направлять ее к колонке. Альтернативой
шариковому обратному клапану является активный обратный клапан, показанный
на рис. 3.126 и описанный в разделе 3.5.1.3.
У одноплунжерного насоса, работающего с постоянной скоростью от линейного
привода, половина времени уйдет на всасывание и половина на нагнетание раство-
144 Глава 3. Оборудование
(а)
п
л п л
Время
(б)
м
.^^/zziz^v
£.
Л.
(в)
z:
-v v?—
А
Рнс. 3.13. Устройство насоса для ВЭЖХ с соответствующими профилями потока и
давления, (а) одноплунжерный возвратно-поступательный насос; (6)
влияние ведущего кулачка; (в) двухплунжерный насос; (г) тандемная двухплун-
жерная конструкция (или конструкция с накопительной камерой)
ригеля. Профиль суммарного потока (и давления) показан на рис. 3.1 За. Такие
экстремальные пульсации потока нежелательны для ВЭЖХ.
Альтернативная одноплунжерная конструкция использует ведущий кулачок
специальной формы (например, эллиптический), сидящий на валу двигателя, или
шаговый двигатель с разной скоростью вращения, связанный с плунжером. С помощью
таких устройств скорость движения плунжера может изменяться внутри каждого
цикла работы насоса таким образом, чтобы больше половины времени уходило
на нагнетание, а всасывание происходило быстро (рис. 3.136). В некоторых насосах
этот процесс оптимизируется таким способом; контроллер насоса запоминает давле-
3.5. Насосные системы 145
ние, которые было при предыдущем ходе плунжера, и быстро начинает перемещать
плунжер вперед к началу следующего хода нагнетания до того момента, пока не
будет достигнуто предыдущее значение давления. После чего плунжер замедляется и
с постоянной скоростью перемещается до самого конца хода. Даже тогда, когда
плунжер перемещается с различной скоростью (его движение оптимизировано) и
пульсации постоянно демпфируются, одноплунжерный насос все равно не дает
стабильного потока и давления, необходимого для работы в режиме аналитической
ВЭЖХ. На рис. 3.1 За и г показаны два усовершенствования одноплунжерного меха
низма: двухплунжерный насос (раздел 3.5.1.1) и одноплунжерный насос,
совмещенный с накопительной насосной камерой (раздел 3.5.1.2).
В дополнение к узлам насоса, показанным на рис. 3.11, большинство насосов
имеют кран для экстренной промывки, расположенный на выходе из насоса,
который позволяет направить поток из насоса в слив при промывке насоса, смене рас
творителя и при удалении пузырька воздуха. Многие насосы имеют, кроме того,
фитинг на входе, позволяющий вручную промывать насос при помощи шприца.
Раньше для гашения пульсаций использовались механические демпферные
устройства (например, трубки Бурдона). Сегодня механическое гашение пульсаций
происходит благодаря специальным конструкциям насосов (например, двухплунжерные или
накопительные плунжерные насосы, см. разделы 3.5.1.1 и 3.5.1.2). Кроме того,
демпферные устройства создают нежелательный объем задержки в градиентном режиме
(раздел 3.5.3).
3.5.1.1. Двухплунжерные насосы
Одним из способов минимизации пульсаций одноплунжерного насоса является
использование двух насосных голов, работающих параллельно следующим образом:
в то время как одна из них заполняется элюентом, из другой он подается в выходной
капилляр. Это проиллюстрировано на рис. 3.13* — два плунжера, прикрепленных
к одному кулачку, работающих в противофазе. Несмотря на то, что большинство
конструкций двухкулачковые с приводом от одного двигателя, плунжеры
смонтированы параллельно, друг возле друга для удобства эксплуатации. При использовании
приводного кулачка со специально сформированным контуром, к которому
прикреплены два плунжера, поток на выходе из насоса может быть достаточно
равномерным, и для него требуется небольшое подавление пульсаций или вовсе можно без
него обойтись. Двухплунжерные насосы на сегодняшний день одни из двух видов
насосов, наиболее широко используемых в аналитической ВЭЖХ.
3.5.1.2. Накопительные плунжерные насосы
Альтернативой конструкции с двумя плунжерами является тандемная
конструкция с дополнительным плунжером-аккумулятором (накопителем), показанная
на рис. 3.13г. В этом случае один плунжер (питающий) работает с удвоенной
скоростью. Например, если необходима скорость потока 1 мл/мин, то верхний плунжер
(рис. 3.1 Зг) будет перекачивать элюент со скоростью I мл/мин. В тот момент, когда
верхний плунжер будет нагнетать элюент в линию, верхний (выходной) клапан будет
открыт, а промежуточный клапан (входной в камеру верхнего плунжера) будет закрыт.
В это время нижний плунжер будет заполнять свою камеру со скоростью 2 мл/мин и
входной клапан в эту камеру будет при этом открыт. Затем нижний плунжер будет
нагнетать растворитель со скоростью 2 мл/мин (заполняя камеру верхнего плунжера),
что приведет к закрытию входного обратного клапана и открытию промежуточного.
Половина этого потока в 2 мл/мин будет использована для заполнения камеры
верхнего плунжера, а половина будет непосредственно закачана в колонку.
146 Глава 3. Оборудование
Конструкция с плунжером-аккумулятором, по крайней мере теоретически, имеет
ряд преимуществ по сравнению с двухплунжерной. Растворитель непрерывным
потоком идет в колонку, и поэтому пульсации потока и давления должны быть
минимальны. Поскольку клапаны представляют собой самые сложные в обслуживании
части всей ВЭЖХ-системы, сокращение их количества с четырех (в двухплунжерной
конструкции) до трех (в конструкции с плунжером-аккумулятором) может
уменьшить эти сложности. Кроме того, поскольку растворитель всегда из верхней камеры
непрерывно течет по направлению к колонке, получается, что в самом верхнем
клапане нужды нет, и его можно исключить. Таким образом, остается всего два клапана.
Хотя простота конструкции с плунжером-аккумулятором представляется более
надежной, возникает целый ряд других сложностей (программное обеспечение,
материалы, усложнение сборки, механические допуски, и т.д.). Большинство
пользователей достигает сравнимой производительности при работе и с двухплунжерной
конструкцией, и с конструкцией, использующей плунжер-аккумулятор.
3.5.1.3. Активный клапан
И, наконец, еще одно усовершенствование может быть применено как к
двухплунжерной конструкции, так и к конструкции, использующей плунжер-аккумулятор.
Обратный клапан шарового типа (рис. 3.12я) легко может протекать даже в том
случае, если между седлом клапана и шариком попадет мельчайшая соринка.
Поверхности последних очень твердые (обычно они изготовлены из сапфира или рубина) и,
несмотря на давление насоса, которое придавливает шарик к седлу, клапан все равно
будет протекать, если в зазор между ними попала частица.
Активный клапан является альтернативной конструкцией, которая хорошо
работает в качестве входного клапана на стороне низкого давления в насосе. Как
показано на рис. 3.126, работа активного клапана зависит от полимерного уплотнения и
плунжера с механическим приводом. Плунжер и уплотнение обеспечивают
герметизацию. Во время фазы заполнения камеры плунжер поднимается над уплотнением,
и растворитель входит в насос. Во время фазы нагнетания плунжер прижимается
к уплотнению. Возросшая площадь поверхности по сравнению с шаровым клапаном
и мягкий материал уплотнения позволяет активному обратному клапану эффективно
уплотнять даже в присутствии мелких посторонних частиц. В конструкции активного
обратного шарового клапана (рис. 3.12в) для закрытия клапана используется сильная
пружина1. Плунжер, расположенный ниже шарика, приподнимает последний над
седлом для открытия клапана.
В двухплунжерной насосе с активными обратными клапанами (рис. 3.1 Зв)
достаточно двух выпускных шаровых клапанов. В конструкции с
плунжером-аккумулятором (рис. 3.13.?) необходим только один клапан шарового типа между верхней и
нижней рабочими камерами. Выпускной обратный клапан в данном случае не нужен.
В обоих случаях уменьшение количества шаровых клапанов приводит к повышению
надежности насоса.
3.5.2. Смешивание в потоке
Для работы в изократических режимах подвижную фазу можно смешать вручную.
После этого дополнительного перемешивания в ВЭЖХ-системе не потребуется.
С другой стороны, большая часть хроматографистов пользуется более удобным сме-
' Сильные пружины делают, как правило, из хромистых сталей, которые имеют обыкновение
ржаветь даже в воде, не говоря о средах с рН, отличающимся от нейтрального. Нержавеющие
стали, наоборот, недостаточно упруги, поскольку в них довольно большое содержание никеля. —
Прим. персе.
3.5. Насосные системы 147
шиванием в потоке и использует ВЭЖХ-систему для смешивания компонентов
подвижной фазы при работе в изократическом режиме точно так же, как и в
градиентном, где без смешивания в потоке не обойтись. Даже тогда, когда есть возможность
смешивания в потоке, предварительное смешивание часто дает более ровную
базовую линию. В некоторых случаях, например, когда используется рефрактометр
(раздел 4.11), смешивание в потоке и вовсе неприменимо, поскольку оно не дает
возможности получить приемлемую базовую линию при высокой чувствительности.
Смешивание в потоке можно осуществить тремя способами:
• смешивание на стороне высокого давления;
• смешивание на стороне низкого давления;
• гибридное смешивание.
3.5.2.1. Смешивание на стороне высокого давления
При использовании систем, где смешивание происходит на стороне высокого
давления (рис. 3.I4), каждый растворитель подается в миксер отдельным насосом.
Соотношение растворителей в подвижной фазе регулируется относительными
расходами насосов. Например, если требуется смесь 60/40 метанол/вода (60 %В)
со скоростью 1 мл/мин, то насос, доставляющий метанол, имеет расход 0,6 мл/мин,
а насос, доставляющий воду — 0,4 мл/мин. В процессе выполнения градиента, когда
%В изменяется в ходе разделения, скорость каждого насоса изменяется в
соответствии с программой градиента. Поскольку растворители смешивают под высоким
давлением, дегазация (раздел 3.3.1) не так важна, как при смешивании при низком
давлении. В этом случае проблемы, связанные с дегазацией, минимальны. Системы
для смешивания на стороне высокого давления обычно ограничены одновременным
использованием двух растворителей, поскольку для каждого растворителя требуется
отдельный насос. Некоторые ВЭЖХ-системы имеют краны выбора элюентов,
установленные на одном или на обоих насосах, что позволяет неодновременную подачу
двух или более растворителей одним и тем же насосом. Это может быть удобным при
разработке метода (например, ацетонитрил и метанол в разных разделениях) или
при автоматической промывке системы (например, промывка водой для удаления
буфера из системы). Системы смешивания на стороне высокого давления имеют
преимущество перед смешиванием на стороне низкого давления — стандартный
миксер может быть заменен микромиксером (раздел 3.4.2.3) для таких случаев, как
ЖХ-МС (раздел 4.14), требующих минимальных объемов задержки. Компоненты
гидравлической схемы, которые вносят вклад в объем задержки системы, на рис. 3.14
и 3.15 обведены пунктирной линией (см. также раздел 3.5.3).
блок
управл
□
—
6 НИИ
Л
А
s *
П..-——"
Г^
В
инжектор
объем задержки
Рис. 3.14. ВЭЖХ-система со смешиванием на стороне высокого давления. Объем
задержки локализован внутри пунктирной линии
148 Глава 3. Оборудование
инжектор
□
блок
управления
дозирующий
клапан миксер
объем
задержки
Рис. 3.15. ВЭЖХ система со смешиванием на стороне низкого давления. Объем
задержки локализован внутри пунктирной линии.
3.5.2.2. Смешивание на стороне низкого давления
В системах со смешиванием на стороне низкого давления (рис. 3.15) компоненты
подвижной фазы смешиваются до того, как они попадут в насос. Следовательно,
для таких систем нужен только один насос для доставки подвижной фазы к колонке.
Смешивание происходит в клапанной коробке, в которой одновременно можно
обычно смешивать до четырех элюентов. Насос работает с постоянным расходом, и
дозирующие клапаны для каждого растворителя открываются мгновенно на время
(обычно меньше одной секунды), пропорциональное их (растворителей)
концентрации в подвижной фазе. Таким образом, для того, чтобы подавать в колонку со
скоростью 1 мл/мин подвижную фазу состава 75/25 ацетонитрил/буфер, клапан на аце-
тонитриле будет открыт 75% времени, а буферный — 25% времени. Градиенты
формируются путем непрерывного изменения соотношений времен открытия
дозирующих клапанов. Поскольку растворители смешиваются на стороне низкого
давления при атмосферном давлении, при выделении газа могут образоваться пузырьки,
которые приводят к перебоям в работе насоса. Это означает, что дегазация
подвижной фазы нужна для всех систем, где смешивание происходит на стороне низкого
давления. Кроме того, при смешивании на стороне низкого давления особенно
важно убедиться, что входные фильтры в резервуарах и капилляры, по которым течет
подвижная фаза, не засорены и не создают избыточного сопротивления. Засорение
хотя бы одного из входных фильтров (такое чаще всего случается с фильтрами в
водной фазе) приводит к уменьшению доли этого компонента, подаваемого в
смеситель, и к ошибкам в дозировании растворителей. Чтобы эти сложности не
возникали, имеет смысл регулярно проверять фильтры на засоренность с помощью
сифонного теста (раздел 3.2.1). Делайте это регулярно. Например, ежемесячно1.
3.5.2.3. Гибридные системы
Хотя системы со смешиванием на стороне высокого или низкого давлений
популярны, но, по крайней мерю, две компании (Thermo и Varian) производят насосные
системы, которые используют смешанный принцип. В этих гибридных системах дозиру-
I Еще эффективнее регулярно отмывать входные фильтры в ультразвуковой бане с
растворителем от различных механических загрязнений, не дожидаясь, пока результаты сифонного теста
будут неудовлетворительными. — Прим. перев.
Рис. 3.16. Гибридная система для смешивания с дозирующими клапанами для растно-
рителей А, Б и В, смонтированная на голове насоса, (а) Сечение; (б) вид
спереди.
ющие клапаны смонтированы непосредственно у входов в голову насоса (рис. 3.16).
При этом активные клапаны (раздел 3.5.1.3) стоят на каждом входе с растворителем.
Растворители попадают в голову насоса раздельно в очень маленьких объемах и
смешиваются в ней под высоким давлением. Таким образом, минимизируются
проблемы с дегазацией (всевозможные потенциальные пузырьки, которые могут
возникнуть в результате смешивания, остаются в растворе под давлением), и нет
необходимости увеличивать объем миксера. При установке дозирующих клапанов
непосредственно на голове насоса исключается дополнительный объем задержки,
который всегда присутствует при смешивании на стороне низкого давления. В
сочетании с небольшим объемом поршня (24 мкл) в одном из исполнений такого
устройства (Accela от Thermo) получается объем задержки 65 мкл (раздел 3.5.3). Это делает
такую конструкцию очень привлекательной для градиентного элюирования при
работе с пиками, выходящими в малых объемах, например, в условиях быстрой ВЭЖХ
или ЖХ-МС.
3.5.3. Градиентные системы
Во время градиентного элюирования (глава 9) состав подвижной фазы должен
меняться по ходу градиента, и потому без смешивания в потоке (раздел 3.5.2) не
обойтись. Для случаев, где необходим градиент, широко используются как системы
со смешиванием на стороне высокого, так и низкого давлений. Объем задержки
системы, не имеющий значения для работы в изократическом режиме, представляет
собой серьезную проблему в градиентном. Разница в объеме задержки двух ВЭЖХ-сис-
тем может сильно повлиять на получаемые хроматограммы (раздел 9.2.2.4), и это
одна из основных причин трудностей переноса градиентных методов (раздел 9.3.8.2).
Объем задержки включает в себя объем системы от точки смешивания
растворителей до точки входа в колонку. На рис. 3.14 можно видеть, что основным вкладом
в объем задержки в системе со смешиванием на стороне высокого давления
являются смеситель, автосамплер (инжектор) и соединительный капилляр. Для системы
со смешиванием на стороне низкого давления (рис. 3.15) в объем задержки
дополнительно включаются объем соединительного капилляра от миксера к голове насоса
и объем головы насоса. Объем задержки должен быть измерен для каждой
градиентной ВЭЖХ-системы. В некоторых случаях систему можно модифицировать, чтобы
уменьшить объем задержки. Например, путем замены миксера в системах со
смешиванием на стороне высокого давления (раздел 3.4.2.3).
150
Глава 3. Оборудование
3.5.4. Специальные методы
Обычные насосные системы, способные создавать градиенты при расходах от 0,1
до 10 мл/мин и давлениях до 414 бар (6000 psi), позволяют выполнить большинство
задач ВЭЖХ. Существуют три области, в которых обычные ВЭЖХ-системы могут
не удовлетворять требованиям аналитической хроматографии:
• малые скорости потока;
• высокие скорости потока;
• высокие давления.
Эти области, особенно первые две, достаточно специализированы и описаны
в книгах, которые им посвящены, а мы здесь даем только краткий обзор.
3.5.4.1. Методы, использующие малые {микро- и нано-)
скорости потока
Разделения в протеомике и в других «омиках» часто имеют на порядок меньший (или
еще меньший) масштаб, чем обычная ВЭЖХ (обзор этих методов см. в [9—Ю|). Такие
разделения, если их грубо классифицировать по внутреннему диаметру колонки,
называются микро-ЖХ (при внутреннем диаметре 100—1000 мкм) и нано-ЖХ (при
внутреннем диаметре 75—300 мкм) без четкой границы между ними. Для подобных
разделений требуются скорости потока, которые значительно ниже нижнего предела
в 0,1 мл/мин, доступного в насосах, описанных ранее. Специально сконструированные
насосы способны давать потоки вплоть до 1 мкл/мин непосредственно или 50 нл/мин
с делителем потока при давлениях до 414 бар (6000 psi) (и выше для насосов,
сконструированных для использования при высоких давлениях, раздел 3.5.4.3). Само собой
разумеется, что капилляры, фитинги, автосамплеры и детекторы должны соответствовать
таким потокам. В противном случае эффекты, связанные с увеличением внеколоноч-
ного объема, сделают результаты неприемлемыми. В таких случаях обычно используют
специальные капиллярные ячейки для УФ-детекторов или непосредственно
направляют поток из колонки в МС или МС/МС детектор.
3.5.4.2. Методы, использующие высокие скорости потока
(препаративные разделения)
В препаративной ВЭЖХ используются колонки большего размера и требуются более
высокие скорости потока, чем те, которые могут обеспечить обычные насосы ВЭЖХ
(раздел 15.2). Для полупрепаративных разделений может оказаться достаточной
скорость 10 мл/мин, которая вполне доступна для большинства насосов ВЭЖХ.
Несколько производителей предлагают модификации обычных насосов ВЭЖХ с
увеличенной максимальной скоростью потока от = 10 мл/мин до скоростей в интервале от 20
до 50 мл/мин. Давление в этом случае обычно ниже, чем в аналитических
разделениях, так что нет оснований беспокоиться о превышении его пределов. В продаже
имеются насосные системы, обеспечивающие расход от 300 до 2000 мл/мин при
давлениях до 122 бар (1800 psi). Так как препаративные разделения часто проводятся
в изократическом режиме и нет необходимости строго контролировать скорость
потока, в некоторых случаях можно использовать насосы с гидропневматическим
усилением (насосы постоянного давления)1. При использовании высоких скоростей по-
' В этом случае давление на жидкость создается за счет давления газа. Газ из баллона давит
на поршень большого сечения, который передает давление на поршень меньшего сечения. В
результате давление увеличивается пропорционально отношению сечения площадей этих
поршней. — Прим. перев.
3.6. Автосамплеры 151
тока расходуются большие количества растворителей, поэтому необходимы системы
для их переработки и регенерации.
3.5.4.3. Методы, использующие высокое давление
Для коммерчески доступных колонок, упакованных частицами размером менее 2 мкм
(раздел 5.2.1.2), пределы давления обычной ВЭЖХ [обычно <414 бар (6000 psi)| могут
не позволить извлечь все преимущества использования частиц такого небольшого
размера (например, сверхбыстрые разделения, см. разделы 2.5.3.1, 9.3.9.2). Ряд
производителей предлагает ВЭЖХ-системы, способные работать при давлении больше 414 бар
(6000 psi). В методах, ориентированных на высокое давление, часто делается акцент на
высокую пропускную способность, поэтому время разделения можно понижать с
помощью высоких скоростей потока и используя более короткие колонки (обычно
длиной 50 или 100 мм). Для повышения чувствительности (увеличения высоты пика)
также используются колонки малого диаметра (например, с внутренним диаметром 1,0
или 2,1 мм). Эти параметры разделения уменьшают объемы пиков и часто требуют
модификации ВЭЖХ-насосов, фитингов и других компонентов системы для
минимизации внеколоночного объема. Большая часть оборудования, предназначенного для
использования при высоких давлениях, имеет ту же конструктивную основу, что и
обычное ВЭЖХ оборудование, но с дополнительными возможностями работы при
высоких даачениях и в малых объемах, которые могут ограничить диапазон некоторых
параметров системы. Например, у системы УВЭЖХ (Waters' Acuity) заявлена скорость
потока 2 мл/мин при давлении до 620 бар (9000 psi) и I мл/мин при более высоких
давлениях |до максимума в 1034 бар (15000 psi)|. Впрочем, при использовании колонок
малого диаметра (например, с внутренним диаметром 1—2,1 мм) эти скорости потока
вполне приемлемы.
3.6. Автосамплеры
Ввод образца в колонку представляет собой операцию, при которой его
определенное количество должно быть добавлено к потоку подвижной фазы, находящейся
под давлением. Для открытых колонок, в случаях, когда поток остановлен, или в
некоторых препаративных разделениях образец можно ввести и вручную. Для
автоматизированного, проводимого без присутствия оператора анализа, при работе с
сотнями образцов в день, ввод образца должен представлять собой точную, надежную и
автоматизированную процедуру. Для этих задач используют автосамплер. Ручной
ввод, популярный в прошлом, сегодня используется редко, за исключением тех
случаев, когда проводится обучение операторов или очень медленный анализ образцов
из окружающей среды. Представленное здесь описание процесса ввода образца и
оборудование, необходимое для этого, ориентировано на использование автосампле-
ров (пробоотборников). К ручному вводу применимы те же принципы.
3.6.1. Шестиходовой кран ввода образца
Кран ввода проб, первоначально сконструированный для ручного ввода, является
основным компонентом автосамплера. Хотя существуют и другие конструкции,
шестиходовой поворотный кран используется чаще всего. Корпус крана, показанного
на рис. 3.17, сделан из нержавеющей стали. У крана есть две позиции: загрузка
(заполнение петли) и ввод. На рисунках (о) и (6), соответствующих этим двум
позициям, показано расположение каналов, соединяющих вход образца (о), слив (с), петлю
ввода, насос (п) и колонку (к). В положении загрузка (рис. 3.17я) отверстия для вхо-
152 Глава 3. Оборудование
(a) (6)
Заполнение петли Ввод образца
Рис. 3,17. Шестиходовой кран ввода образца с заполняемой петлей, (а) Положение,
при котором заполняется петля (загрузка); (б) положение, при котором
вводится образец (ввод). Стрелки показывают направление потоков; (о) вход
образца в петлю; (с) — в слив; (п) — подвижная фаза из насоса; (к) — к
колонке
да образца и сливное соединены с противоположными отверстиями входа и выхода
из петли, а насос соединен с колонкой. Для ввода образца ротор поворачивается
в положение ввод (рис. 3.176), и содержимое петли вымывается из нее в колонку.
Одновременно с этим отверстие, через которое вводится образец, соединяется
со сливным каналом для промывки, если она необходима.
Полимерное уплотнение ротора служит для соединения канавками трех пар
отверстий (см. также рис. 17.3). Уплотнение ротора обычно изготавливают из
фторированных полимеров или ПЭЭК с добавлением дополнительных материалов
(наполнения) для повышения прочности. ПЭЭК, из которого делают уплотнения роторов,
имеет другой состав, чем тот, который используется для экструдирования
капилляров, и он стоек в ТГФ и хлорированных растворителях [111.
3.6.1.1. Ввод из заполненной петли
В режиме ввода из заполненной петли объем инжектируемого образца не может
превышать объем петли. Например, при использовании петли объемом 20 мкл, образец
вводится в петлю до тех пор, пока его избыток не покажется из сливного отверстия.
В тот момент, когда кран переключается в положение ввода, 20 мкл образца,
находящихся в петле, закачиваются насосом в колонку.
Ввод образца из заполненной петли может быть очень точным, если петля отка-
либрована и при заполнении переполняется. Менять петли неудобно и поэтому, если
вводимый объем надо регулярно изменять, метод заполненной петли, как правило,
не используется.
3.6.1.2. Ввод из частично заполненной петли
Альтернативой использованию крана с полностью заполненной петлей является
режим частичного заполнения петли, который часто называют вводом из частично
заполненной петли.
Последовательность действий такая же, как и в случае ввода с полностью
заполненной петлей, за исключением того факта, что объем петли больше, чем объем об-
3.6. Автосамплеры
(a)
(6)
Ввод образца из частично
заполненной петли
вход
образца
слив
Заполнение петли
Q образец
О подвижная фаза
Ввод образца
Рнс. 3.18. Работа шестиходового крана ввода образца в режиме ввода из частично
заполненной петли, (я) Положение, при котором заполняется петля; (6)
положение, при котором вводится образец. Стрелки показывают направление
потоков; о — вход образца в петлю; с — в слив; п — подвижная фаза из
насоса; к — к колонке
разца, измеренное количество которого должно быть введено в петлю. Например,
на шестиходовом кране может быть смонтирована петля объемом 100 мкл. В этом
случае 20 мкл образца нужно отмерить калиброванным шприцем и ввести в
положении загрузка в петлю (рис. 3.18а). (Как правило, оставшийся объем петли заполнен
подвижной фазой.) После этого ротор крана повернется в позицию ввод (рис. 3.186),
и содержимое петли насосом подается в колонку. Обращаем внимание на то, что
схема коммутации такова, что направление потока через петлю с образцом в
положениях загрузка и ввод противоположное. Это дает гарантию того, что размывание
образца при вводе будет минимальным. Чем больший путь проделает образец по петле
прежде чем достигнет колонки, тем больше будет нежелательное уширение пика.
Ввод из частично заполненной петли может быть точным и надежным, если
петлю заполнять аккуратно и, если используется менее половины объема петли. Одна
из возможных проблем, связанных с точностью ввода, отражена на рис. 3.19а |12|.
100
40 60
введенный объем, мкл
Рис. 3.19. Влияние ламинарного характера потока на точность ввода образца.
(а) Сравнение отклика детектора при вводе различных объемов образца
в петлю объемом 20 мкл; (6) профиль ламинарного потока образца. |12|
154 Глава 3. Оборудование
В данном случае вводились различные объемы образца через петлю объемом 20 мкл,
смонтированную на кране-инжекторе. При введении менее десяти и более сорока
микролитров в петлю, отклик детектора точно отражал введенный объем. Однако,
в области, которая находится между десятью и сорока микролитрами, отклик
детектора был меньше ожидаемого. Эта проблема связана с ламинарным характером
потока в капилляре до петли ввода образца и внутри нее. Когда жидкости проходят
по капиллярам, молекулы у стенок тормозятся из-за трения, и в результате
формируется характерный для ламинарного течения и похожий на пулю профиль потока.
Молекулы в центре потока движутся примерно в два раза быстрее, чем около стенок.
Следовательно, если 20 мкл образца вводится в петлю объемом 20 мкл (объем,
обозначенный на рис. 3.196 пунктирными линиями), то часть образца в начале «пули»
выйдет за пределы петли в слив, в то время как «хвосты» могут еще не войти в
петлю. В результате объем введенного образца получается меньше, чем предполагалось.
Это упрощенное описание процесса еще усложняется, если учесть промывку
обратным током и перемешивание, которое имеет место при изменении диаметра
капилляра или других изменениях в структуре потока. При ручном вводе для
максимальной точности лучше всего поддерживать объем вводимого образца постоянным, и
его объем должен быть < 50% или > 200% от объема петли.
3.6.2. Конструкция автосамплера
Автосамплеры в основном заменили ручные инжекторы — в первую очередь из-за
удобства использования. Кроме того, автосамплеры обеспечивают такую
воспроизводимость, которую невозможно обеспечить при ручном вводе. Многие из доступных
на сегодняшний день автосамплеров способны обеспечить < 0,5% СКО площади
пика для объемов пробы > 5 мкл. Точность измерения вводимого объема может
не соответствовать такому уровню из-за ошибок в калибровке инжекционного
шприца или петли. Частичное заполнение петли вследствие ламинарного характера
потока (раздел 3.6.1.2) тоже может привнести ошибки в точности вводимого объема.
Обычно, поскольку используются стандартные образцы и калибровочные растворы,
прецизионность ввода более важна, чем точность вводимого объема. Автосамплеры
лучше всего использовать для ввода постоянного объема образца внутри одной
методики, чтобы один и тот же одинаковый (хотя и не обязательно точно отмеренный)
объем образца вводился как в случае калибровочных стандартов, так и в случае
исследуемых образцов. Если следовать этой практике, то точность ввода менее важна,
а отличная прецизионность обеспечит удовлетворительные результаты анализа.
Перенос образца из предыдущего анализа подтверждается наличием небольшого
пика(ов) на холостой хроматограмме, которая получается при разделении без
введения образца, следующем за разделением с образцом. Перенос образца из
предыдущего разделения происходит оттого, что часть образца остается в системе после
завершения разделения. Это особенно неприятно тогда, когда анализ образца с низкой
концентрацией следует за анализом образца с высокой концентрацией
(раздел 17.2.5.10). Многие автосамплеры, чтобы избежать переноса образца,
запрограммированы на промывку между инжекциями иглы и мест, контактировавших с
образцом. Эти промывки варьируются от обычной промывки иглы в виале до активной
промывки. При использовании промывки состав промывочного растворителя
выбирается таким, чтобы он растворял образец и был совместим с подвижной фазой.
Образцы обычно помещают в отдельные виалы или в планшеты на 96 или 384
лунок. Чаще всего виалы изготавливают из стекла. Иногда их специально обрабатывают
для уменьшения потерь вещества за счет адсобрции. Стандартные виалы имеют объем
от одного до полутора миллилитров, а микровиалы — от ста до трехсот (и меньше)
3.6. Автосамплеры 155
микролитров; такой же объем можно получить со вставкой в стандартных виалах. Виа-
лы закрываются крышкой, снабженной септой, которая обычно делается из
силиконовой резины и/или фторопластовой пленки. Планшеты для образцов обычно
изготавливают из пластика с объемами лунок < I мл. Чаще всего к планшетам, чтобы их
прикрыть, прижимают септу или металлизированную полимерную пленку.
Забор образца, как правило, происходит через иглу, которая передвигается в
заданное положение относительно осей xyz к контейнеру с образцом. Некоторые
автосамплеры используют вращающийся лоток для того, чтобы доставить виалы с
образцами к игле, в то время как другие берут нужную виалу и перемещают ее к игле.
Время цикла — это время, необходимое автосамплеру для того, чтобы завершить
ввод образца, считая с момента получения исходного сигнала о старте операции.
Минимальное время цикла включает в себя время, необходимое для забора образца
и ввода его в колонку. Добавление промывочных операций может увеличить время
цикла. Это не столь важно до тех пор, пока время цикла работы автосамплера
не оказывает существенного влияния на производительность системы. Так от
минутного цикла автосамплера при тридцатиминутной длительности градиентного
разделения ничего не зависит, но эта же минута при продолжительности разделения
четыре минуты значительно уменьшит производительность системы. Время цикла,
составляющее < 5% от общего времени разделения, считается приемлемым. На
момент написания этой главы не многие автосамплеры имели время цикла < 15 сек,
сохраняя при этом приемлемый уровень переноса образца из предыдущего анализа
и воспроизводимость ввода. Один из способов уменьшить негативный вклад времени
цикла, необходимого автосамплеру для забора и ввода пробы, является предлагаемая
некоторыми системами опция «подготовленного ввода». В этом случае автосамплер
запрограммирован таким образом, чтобы производить промывку и забор образца
в то самое время, когда еще элюируется предыдущий образец. Как только разделение
закончится, образец можно будет тотчас же вводить. Это сократит эффективное
время цикла буквально до нескольких секунд.
Самыми распространенными являются три типа конструктивных решений авто-
самплеров:
• с заполнением петли засасыванием;
• с заполнением петли нагнетанием;
• со встроенным петлевым дозатором.
3.6.2.1. Автосамплеры с заполнением петли засасыванием
Конструкция автосамплера с заполнением петли засасыванием представлена
на рис. 3.20. Шприц имеет механический привод и связан с инжектором. Петля
нужного объема для ввода образца смонтирована на инжекторе. Иглу шприца соединяют
с отрезком капилляра, который, в свою очередь, соединен с инжектором. Игла
может быть перемещаема к виале с образцом или виала может быть перемещена к игле,
установленной стационарно. В положении загрузка (рис. 3.20я) игла прокалывает
септу виалы, и поршень шприца отводится, чтобы засосать образец через иглу и
соединительный капилляр до того момента, пока петля не заполнится полностью,
и лишнее количество образца не выйдет из петли. Затем ротор крана поворачивается
в положение ввод (рис. 3.206), и образец закачивается в колонку. Обратите внимание
на то, что в этой конструкции автосамплера уплотнения для иглы нет.
Использование автосамплера с заполнением петли под вакуумом приводит к
потере части образца из-за относительно большого внутреннего диаметра
соединительных капилляров, который необходим для предотвращения их засорения. Кроме того,
необходим избыток раствора образца для промывки петли (раздел 3.6.I.I). Такой
автосамплер лучше всего использовать, когда количество образца не ограничено, на-
156 Глава 3. Оборудование
/\ J~L J~U JV/\
Рис. 3.20. Конструкция автосамплера с заполнением засасыванием, (а) Перенос
образца из виалы в петлю (загрузка); (6) ввод, н — поток из насоса; к — поток
в колонку
пример, в случае мониторинга производственного процесса. Эта конструкция
проста, надежна и, поскольку в ней используется переполненная петля фиксированного
объема, может обеспечить хорошую точность и прецизионность.
3.6.2.2. Автосамплеры с заполнением петли нагнетанием
Автосамплер с заполнением петли нагнетанием представляет собой
автоматизированную версию ручного инжектора. В положении загрузка (рис. 3.21а) игла отбирает
образец из виалы в капилляр, соединенный со шприцем, имеющим механический
привод. Затем игла извлекается из виалы с образцом и вводится в порт инжекцион-
ного крана, снабженный уплотнением, способным выдержать невысокое давление
(рис. 3.215). Далее образец заполняет инжекционную петлю. После этого ротор
крана поворачивается в положение ввод (рис. 3.21в), и образец насосом закачивается
в колонку.
Автосамплер с заполнением петли нагнетанием может использоваться в режимах
работы с полностью или частично заполненной петлей (разделы 3.6.1.1 и 3.6.1.2).
В режиме работы с частично заполненной петлей воспроизводимость зависит от
воспроизводимости работы контроллера шприца. Поскольку можно ввести в колонку
практически весь образец, автосамплер такой конструкции не потеряет много
образца — возможно, 10 мкл останется в игле или в соединительных капиллярах. В авто-
самплере с заполнением петли нагнетанием для иглы используется уплотнение, рас-
читанное на низкое давление, что обычно гарантирует надежность в работе. Такие
автосамплеры очень популярны и используются для различных приложений.
3.6.2.3. Автосамплер со встроенным петлевым дозатором
В автосамплере со встроенным петлевым дозатором игла и петля являются частями
одного целого (рис. 3.22а). В положении загрузка, как показано на рис. 3.22а, игла
забирает образец из виалы. Затем игла перемещается и вводится в порт инжекцион-
ного крана, снабженный уплотнением, способным выдержать высокое давление.
Ротор крана при этом поворачивается в положение ввода образца (рис. 3.226).
3.6. Автосамплеры
JTJT Г1П
уплотнение низкого давления в
инжекторе
Рис. 3.21. Автосамплер с заполнением петли нагнетанием, (а) Забор образца из пиалы
в иглу и шприц; (б) заполнение петли; (в) ввод; н — поток из насоса; *: —
поток в колонку; с — в слив
(б)
ллдлл
уплотнение высокого давления
в инжекторе
Рис. 3.22. Автосамлер с петлевым дозатором, (о) Перенос образца из виалы в петлевой
дозатор; (6) ввод; н — поток из насоса; к — поток в колонку; с — в слив
Поскольку поток подвижной фазы в позиции ввод проходит через конец иглы,
автосамплер со встроенным петлевым дозатором вводит весь образец, забранный
из виалы, в которой он находился, и поэтому при такой технологии ввода потерь
образца нет. Это делает автосамплер подобной конструкции наиболее популярным
в тех случаях, когда количество образца очень мало. Такие автосамплеры обычно
158 Глава 3. Оборудование
используют петлевые дозаторы объемом 100 мкл, который годится для большинства
аналитических методик. Если необходимо ввести больший объем образца, то
придется заменить все устройство, состоящее из неразрывно связанных иглы и петли.
Такой тип петлевого дозатора гораздо дороже, чем обычная петля для ввода образца.
Кроме того, функционирование автосамплера с петлевым дозатором зависит от
расчитанного на высокое давление уплотения в месте ввода иглы в кран инжектора.
В некоторых исполнениях оно является слабым местом. Автосамплеры подобного
типа популярны из за малых потерь образца и, как правило, минимального переноса
образца из предыдущего анализа.
3.6.3. Эффекты, связанные с количеством образца
Количество вводимого образца может повлиять на вид хроматограммы. Это
отразится не только на высоте пиков или их площади, но также на форме и времени
удерживания. На уширение пиков может влиять объем вводимого образца Vs, и, как это
обсуждается ниже, сила растворителя, в котором растворен образец (по сравнению
с подвижной фазой). Эффекты, связанные с массой вводимого образца и
перегрузкой, обсуждаются в разделе 2.6.2.
3.6.3.1. Вводимый объем
Влияние объема вводимого образца на ширину пика обсуждалось в разделе 2.6.1.
Итог этому обсуждению подведен таблицей 3.3 и уравнением (2.27), где V„ — это
наблюдаемый объем пика, Кд — объем пика с учетом уширения полосы внутри
колонны, a Vs — объем вводимого образца в тех случаях, когда подвижная фаза
используется для растворения образца в изократических разделениях.
1/2
(2.27)
"> =
ш
у} +»%
Уравнение (2.27) можно использовать для определения максимально возможного
объема вводимого образца при заданном увеличении объема пика. Если 5% потеря
в разрешении допустима, то можно допустить 5% увеличение ширины пика.
Допустимые объемы ввода, перечисленные в таблице 3.3, рассчитаны для пятипроцентного
уширения пика, различных колонок и коэффициентов удерживания. (Обратите
внимание на то, что допустимые объемы ввода в таблице 3.3 меньше рассчитанных
по уравнению 2.27. Это происходит потому, что Vs увеличивается приблизительно
наполовину при вымывании образца из петли. См. раздел 2.6.1.) Очевидно, что
требуется вводить меньшие объемы образцов в колонки небольшого объема, колонки
с большим количеством теоретических тарелок и/или для разрешения раноэлюируе-
мых пиков. Во всех этих случаях пики элюируются в меньших объемах (становятся
более узкими). С другой стороны, колонки с внутренним диаметром 4,6 мм дают
довольно широкие пики, даже если они упакованы суб-2мкм частицами и длина их
(колонок) 50 мм. Например, колонка 50x4,6 мм, упакованная частицами сорбента
размером 1,8 мкм, имеет Кд к 30 мкл для А; = 0,5. Допустимый объем образца при
этом Vs « 5мкл. Большинство автосамплеров дают погрешность 5 0,5% СКО для
объема инжекций ;>5мкл.
Некоторые автосамплеры могут обеспечивать аналогичную прецизионность
при объеме ввода от 1 до 2 мкл или меньше. Для колонок с внутренним диаметром
< 2,1 мм, упакованных частицами размером <,3 мкм, автосамплер должен точно
вводить очень маленькие объемы. В изократических разделениях ширина пика всегда
растет при увеличении времени удерживания, и поэтому для сильно удерживаемых
3.6. Автосамплеры 159
пиков допустимы ббльшие объемы инжекции. Изменение методики с целью
увеличения времени удерживания является одним из подходов для минимизации влияния
вне колоночного уширения пика, редко используемым, поскольку суммарный
эффект уменьшения высоты пика и увеличения времени разделения обычно сводит
на нет эти усилия.
Таблица 3.3. Допустимые объемы инжекции, приводящие к уширению полосы, не
превышающему з%
Параметры колонки
L (мм)
150
150
100
100
100
100
50
50
50
50
50
50
dc (мм)
4.6
2.1
4.6
2.1
2.1
1.0
4.6
2.1
1.0
4.6
2.1
1.0
dp (мкм)
5.0
5.0
3.0
3.0
1.8
1.8
3.0
3.0
3.0
1.8
1.8
1.8
N (A J 3)
10,000
10,000
11,100
11,000
18,500
18,500
5,500
5,500
5,500
9,200
9,200
9,200
Объем пика (объем инжекции)* (мкл)
ft = 0,5
90* (10)"
20(2)
60(6)
12(2)
9(1)
2(<1)
40(4)
8(1)
2(<1)
30(4)
7(1)
2(<1)
ft-2
180(20)
40(4)
115(15)
25(15)
20(2)
5(<1)
80(10)
20(2)
5(<1)
65(7)
15(2)
3(<1)
ft-=20
1,270(130)
265(25)
800(90)
170(20)
130(15)
30(3)
565(60)
120(80)
25(3)
440(50)
90(10)
20(2)
"Уравнение (2.27), VJV^ = 0,16; отметим, что последняя (теоретическая) величина объема
ввода Vs разделена на 1.5, чтобы учесть размывание зоны образца по мере того, как он выходит
из петли.
*06ъем пика (мкл).
"Объем инжекции (мкл).
3.6.3.2. Растворитель образца
Как уже говорилось в разделе 2.6.1, в случае, если растворитель образца не
идентичен подвижной фазе, уравнение (2.27) не выполняется. Если растворитель образца
существенно слабее подвижной фазы, образец будет концентрироваться в начале
колонки. При изменении состава подвижной фазы на 10 %В к меняется в два с
половиной раза (раздел 6.2.1). Растворитель образца, который слабее подвижной фазы
на 10 %В, должен существенно замедлять пробу на входе в колонку. Если различия
в силе между растворителем образца и подвижной фазой будут еще больше, то
эффективность замедления еще возрастет. При необходимости работы с большими
объемами образца, разбавление водой может позволить вводить большие количества
образца (раздел 2.6.1). Использование для растворения образца более сильного
растворителя, чем подвижная фаза, будет отрицательно влиять на раноэлюируемые
пики больше, чем на удерживаемые сильнее (раздел 17.4.5.3). Кроме того, важно
приводить в соответствие растворители для стандартов и образцов.
Ввод образца в растворителе более сильном, чем подвижная фаза, будет
приводить к размыванию образца по длине колонки до тех пор, пока он полностью не
разбавится подвижной фазой. Как и при использовании разбавленных растворителей
160 Глава 3. Оборудование
образца, наблюдаемые эффекты зависят как от объема ввода, так и от разницы
между силой растворителя образца и подвижной фазы. Обычно при работе с колонками
150 х 4,6 мм вполне допустимы объемы инжекции < 10 мкл в сильном растворителе
(к примеру, в 100 %В). При работе с большими объемами инжекции и/или с
колонками меньшего объема целесообразно сопоставить наблюдаемое удерживание и
форму пика с результатами разделения, в котором образец растворен в небольшом
объеме подвижной фазы, чтобы убедиться в приемлемости полученных данных.
Для подвижных фаз, содержащих < 50% В, разбавление образца, растворенного
в 100 %В, водой в соотношении 1:1 часто позволяет вводить ббльшие объемы и
массы образца без негативных последствий.
3.6.4. Другие области применения кранов
Автоматические краны для ввода образца чаще всего используются в автосамплерах,
но, кроме того, они применяются и для других целей. Например:
• для переключения колонок;
• для сбора фракций в коллекторах фракций;
• для направления потока в слив.
3.6.4.1. Переключение колонок
Краны высокого давления для переключения колонок доступны в самых различных
исполнениях, а не только в виде шестиходового крана, представленного на рис. 3.17.
Их могут поставлять с электрическим приводом и с переключением, управляемым
от внешнего контроллера системы ВЭЖХ. Распространены две конфигурации
кранов: двухпозиционные, показанные на рис. 3.17, и многопозиционные,
позволяющие соединить вход с одним из множества выходов (см. ниже обсуждение рис. 3.25).
Здесь обсуждаются три способа применения кранов, а еще один описан в
разделе 2.7.6. На сайтах производителей
кранов (например, |6, 13|) можно узнать о том,
в каких еще областях они применяются.
Одним из распространенных способов
применения двухпозиционного крана
является концентрирование (обогащение)
образца, как это показано на рис. 3.23. Цель
состоит в том, чтобы сконцентрировать
разбавленный образец и затем ввести
концентрат в аналитическую колонку.
Примером может быть концентрирование
неполярного анализируемого вещества
из водного образца (например, метод
мониторинга окружающей среды). Во время
фазы концентрирования кран установлен
так, как это показано на рис. 3.23а, где
первый насос прокачивает разбавленный
образец через концентрирующую колонку,
в то время как предыдущий образец
разделяется на аналитической колонке, через
которую его прокачивает второй насос.
В этом примере образец, растворенный
в воде, можно пропустить через
упакованную C|g колонку, уравновешенную слабой
от насоса 1
(через
автосамплер).
в слив
концентрирующая
колонка
насос 1
Рис. 3.23. Переключение колонок для
концентрирования образца, (а)
Положение крана при вводе образца на
колонку, где происходит концентрирование;
(б) смыв образца противотоком с
концентрирующей колонки на аналитическую
3.6. Автосамплеры 161
подвижной фазой, чтобы сорбировались неполярные компоненты. После того, как
весь образец сконцентрируется на колонке, кран переключается (рис. 3.235), и
образец противотоком направляется на аналитическую колонку. Из-за перемены
направления потока, а также из-за резкого увеличения силы подвижной фазы, образец вы
мывается из концентрирующей колонки в аналитическую узкой полосой. Другие
области применения, в которых используется способ переключения колонок,
показанный на рис. 3.23, обсуждаются в разделе 16.9.
На рис. 3.24 показана конфигурация кранов, с помощью которой можно регене
рировать одну колонку, в то время как в другой происходит разделение образца с
последующим детектированием. Это может увеличить производительность системы и
оказаться полезным в градиентных разделениях. Таким образом также повышается
коэффициент использования дорогостоящего масс-спектрометрического детектора,
например, для анализа лекарственных веществ в образцах плазмы крови. В
конфигурации, показанной на рис. 3.24я, образец вводят и анализируют на колонке 2 обыч
ным способом с использованием градиентного элюирования. В это время колонка I
регенерируется подвижной фазой, прокачиваемой насосной системой 1. Как только
образец вымоется из колонки 2 в детектор, краны поворачивают, получая
конфигурацию, показанную на рис. 2.246, и новый образец будет инжектирован в колонку I,
в то время как колонка 2 будет регенерироваться. Такая схема работы увеличивает
производительность за счет исключения времени, которое обычно тратится на
ожидание момента уравновешивания колонки до стартовых условий после градиентного
разделения. Следует отметить, что существуют и другие способы минимизации
времени, необходимого для уравновешивания колонки после градиентного разделения
(раздел 9.3.7).
На рис. 3.25 показано, как использовать многоходовой кран для выбора одной из
трех колонок. При такой конфигурации три самостоятельные колонки могут быть
использованы в автоматическом режиме при разработке метода. Такую же
технологию можно применить и с набором из тридцати двух хиральных колонок,
смонтированных с помощью двух 32-ходовых кранах (см. раздел 14.6.1). При таком подходе,
от насоса 1
от насоса 2 +^
автосамплер
колонка 1
(б)
насос 1 *■
насос 2*
<*■ в спив
Регенерация
колонки 1
Регенерация
колонки 2
Рис. 3.24. Коммутация колонок для регенерации, (а) Образец вводится в колонку 2 и
направляется в детектор (МС), в то время как колонка I регенерируется
насосом 1; (б) образец разделяется на колонке 1, в то время как колонка 2
регенерируется. VI и V2 — шестипортовые краны
162 Глава 3. Оборудование
насоса
колонка 1
колонка 2
колонка 3
■+К
детектору
Рис. 3.25. Использование многоходовых автоматических кранов для выбора одной
колонки из нескольких. Как показано на рисунке, кран V1 направляет поток
от насоса к колонке 1 и далее через кран V2 к детектору. Переключение
кранов (—), которое обеспечивает выбор нужной колонки, осуществляется
с помощью программного обеспечения
когда колонки соединены как в многоствольной пушке Гатлинга, образец
последовательно вводится в каждую колонку в ходе запрограммированной серии инжекций.
После завершения процесса хроматограммы анализируют с целью выяснения, в
какой колонке разделение было наилучшим.
3.6.4.2. Коллекторы фракций
В препаративной хроматографии (раздел 15.2.4) требуется сбор фракций для: (I)
выделения пиков индивидуальных веществ по данным хроматограммы или (2) сбора
межпиковых фракций в том случае, если они перекрываются (как часть процесса
очистки какого-нибудь соединения). Коллектор фракций, обычно используемый
для этих целей, напоминает собой автосамплер, только работает наоборот. Поток
элюата, выходящий из колонки, распределяется по виалам с помощью механизма,
перемещающего выходной капилляр к нужной виале. В простейшем варианте
коллектор фракций работает по времени. В какое-то заранее заданное время после
инжекций начинается сбор фракций, и образец собирается в течение фиксированного
времени в каждую пробирку коллектора. Например, если интересующий вас пик
элюируется через семь минут, коллектор фракций можно запрограммировать на сбор
двадцатисекундных фракций, начиная с шестой минуты и до восьмой. Таким
способом будут собраны несколько частей пика. Другим распространенным режимом
работы коллектора фракций является электронная обратная связь с выходом детектора.
При этом фракции собирают только тогда, когда пик элюируется. Между детектором
и коллектором фракций может быть установлен дополнительный отрезок капилляра,
обеспечивающий временную задержку для того, чтобы не упустить нужные фракции
после того, как они выйдут из детектора.
3.6.4.3. Направление в слив
Область возможного использования многоходовых кранов в ВЭЖХ практически
безгранична. В этом разделе обсуждается два способа применения: направление потока
в слив с целью защиты ячейки детектора и рециркуляция подвижной фазы в случае
изо к рати чес ко го элюирования.
Одним из методов очистки образца для анализа лекарственных веществ в плазме
крови является преципитация белков плазмы (таблица 16.11). Хотя преципитация
плазмы проходит быстро и обходится недорого, часто при этом в образце остаются
значительные количества нелетучего белка, который может накапливаться в
интерфейсе МС-детектора. Один из способов избежать этого заключается в переключении
крана таким образом, чтобы направить часть потока, содержащую наибольшее коли-
3.7. Колоночные термостаты 163
чество белка, в слив. Обычно это та часть хроматограммы, которая находится ближе
к мертвому объему колонки. Если процесс управляется системным контроллером, то
программируемое автоматическое направление в слив будет минимизировать
загрязнение интерфейса нелетучими компонентами, содержащимися во вводимом образце.
Хотя затраты на подвижную фазу не являются основными в общей стоимости
анализа, это может быть важным, если принимается во внимание стоимость ее
утилизации. Чтобы сократить эти расходы, а также и по экологическим причинам,
некоторые пользователи пытаются уменьшить объем расходуемых растворителей. Один
из способов — повторное использование подвижной фазы. Самый простой — на
правлять подвижную фазу не в слив, а в установленный на мешалке резервуар с элю
ентом. Поскольку идущий в слив поток смешивается с оставшейся подвижной
фазой, примеси разбавляются и снова прокачиваются насосом через колонку до рав
новесного состояния, и поэтому не появляется никаких мешающих пиков. Со
временем, однако, концентрация загрязняющих примесей в резервуаре с элюентом будет
расти. Проще всего минимизировать этот нежелательный процесс, если отправлять
на рециркуляцию только ту часть подвижной фазы, которая не содержит пиков об
разца. Коммерчески доступны несколько устройств (например, SolventTrak
компании Axxiom Chromatography), которые включают переключающий кран и сенсор,
подключенный к выходу детектора. При обнаружении пика вещества кран
переключается, направляя поток в слив, а при отсутствии пиков кран направляет поток
в исходный резервуар с элюентом. Таким образом, на рециркуляцию идет только
«чистая» подвижная фаза. Количественную оценку процесса рециркуляции
подвижной фазы можно найти в работе |14|.
Альтернативным способом уменьшения расхода подвижной фазы является
уменьшение диаметра колонки. Расход растворителя пропорционален квадрату поперечного
сечения колонки, и поэтому замена колонки с внутренним диаметром 4,6 мм на
колонку с внутренним диаметром 2,1 мм уменьшит расход растворителя (4,6) /(2,1) я
= 5 раз (скорость потока должна быть одновременно уменьшена во столько же раз
для поддержания постоянной линейной скорости). При этом следует помнить о
возрастании влияния внеколоночного уширения пиков при использовании колонок
малого диаметра.
3.7. Колоночные термостаты
Давно известно, что температура колонки вносит важный вклад в удерживание
веществ и селективность процесса ВЭЖХ-разделения. Для дополнительной
информации см. раздел 6.3.3 и |15-17| (и ссылки в указанных публикациях).
3.7.1. Требования к температурному контролю
Согласно эмпирическому правилу для изократического обращенно-фазового
разделения повышение на один градус температуры колонки понижает величины к
приблизительно на 2%. Кроме того, температура может влиять на селективность
(разделы 6.3.3, 7.3.2.2, 8.3.3, |I5|), и поэтому важно контролировать температуру колонки
особенно в случаях разделений с предельным разрешением (например, Rs < 2). Если
входящая в колонку подвижная фаза предварительно не нагрета до температуры
колонки, это может привести к искажению формы пиков. Чтобы избежать этого
искажения, температура подвижной фазы при ее входе в колонку должна быть ±6 °С
от температуры колонки.
164 Глава 3. Оборудование
Влияние температуры на хроматографическое разделение указывает на то, что
в большинстве случаев необходимо контролировать температуру колонки. Несмотря
на то, что лаборатория в целом может иметь удовлетворительный климат-контроль,
в конкретном месте в ней1 возможны температурные колебания в несколько
градусов вследствие цикличности работы систем отопления, вентиляции и
кондиционирования. Для поддержания постоянной температуры настоятельно рекомендуется
использовать в каждой ВЭЖХ-системе колоночный термостат. Если этот термостат
не подогревает предварительно подвижную фазу (как правило, это требуется только
для методов, работающих при температуре > 40 °С), то можно смастерить простой
нагреватель из полуметра стального нержавеющего капилляра с внутренним
диаметром 0,005". Этот отрезок капилляра должен быть присоединен ко входу колонки
и смонтирован внутри термостата предпочтительно в контакте с нагревающей
поверхностью.
3.7.2. Устройство термостатов
Три самых популярных конструкции колоночных термостатов:
• нагревательные без принудительной циркуляции воздуха;
• воздушные;
• с элементами Пельтье.
Водонагревательные термостаты являются простым возможным решением,
но они неудобны, и в настоящее время используются редко. Если по какой-либо
причине колоночный термостат не используется, необходимо минимизировать
температурные колебания с помощью заключения колонки в термоизолирующий
материал (например, в пенопласт).
3.7.2.1. Нагревательные термостаты
В нагревательных термостатах происходит непосредственный контакт колонки с
источником тепла. Тепло из нагревателя кассетного типа поступает в рифленый
алюминиевый блок, который отдает его колонке, закрепленной внутри этого блока.
Колонка и нагревательный блок находятся в изолированном отсеке. В другом
исполнении вокруг колонки обернуто гибкое полотно нагревательной ленты.
Непосредственный контакт колонки с нагревательным элементом обеспечивает
эффективное нагревание |12|. Если необходим дополнительный предварительный нагрев
растворителя, то его обеспечивают так, как это описано выше.
3.7.2.2. Воздушные термостаты
Воздушный колоночный термостат сконструирован так же, как и термостат для
газового хроматографа. Он позволяет эффективно контролировать температуру колонки.
Воздух хуже проводит тепло, чем металл, и поэтому температурное уравновешивание
колонки может занять больше времени, чем в термостате с нагревателем кассетного
типа.
3.7.2.3. Термостаты с элементами Пельтье
Термостаты с элементами Пельтье широко используются в ВЭЖХ-системах. В
дополнение к нагреву такие термостаты могут поддерживать пониженную температуру,
не требуя для этого вспомогательных устройств. Однако большинство термостатов
с элементами Пельтье не очень эффективно проводят тепло, потому что колонка
'Там, где находится хроматографическая система и колонка. — Прим. перев.
3.8. Управляющие модули 165 JSJJ
редко находится в контакте с нагреваемой поверхностью. В большинстве
конструкций термостатов с элементами Пельтье имеется устройство для предварительного
нагрева, которое представляет собой отрезок капилляра, встроенного в алюминиевый
нагревательный блок термостата. Часто этот отрезок капилляра вносит самый
большой вклад в нагрев колонки 115|. Если такое устройство не используется в
термостате, то, скорее всего, заданная температура не будет соответствовать реальной
температуре колонки. Должным образом сконструированный термостат с элементами
Пельтье будет включать в себя нагревательный блок с рифленой поверхностью,
на которой закреплена колонка, и устройство для предварительного нагрева,
заключенное в изолированный отсек.
3.8. Управляющие модули
Во втором издании этой книги в общей сложности три параграфа были посвящены
описанию хроматографических систем сбора и обработки данных. Возможно,
ни один другой аспект ВЭЖХ так не обсуждался в предшествующие годы, как
обработка данных. Уже в 1979 году были коммерчески доступны несколько разных
специализированных интеграторов от разных производителей, но персонального
компьютера (ПК) с широкими возможностями программного обеспечения для ВЭЖХ еще
и на горизонте не было видно. ВЭЖХ-система состояла из отдельных модулей,
каждым из которых нужно было управлять с помощью ручных настроек. Со временем
устройства, с помощью которых осуществляется процесс настройки,
эволюционировали от реостатов к микропроцессорам, и в настоящее время каждый модуль хрома-
тографической системы содержит множество микропроцессоров. Разработка этих
микропроцессорных устройств позволила оперативно контролировать все
аппаратные функции ВЭЖХ-систем (скорость потока, длину волны детектора, управление
автосамплером и т.д.) с помощью специального системного контроллера. В то время
как системный контроллер управляет ВЭЖХ-системой, отдельная система сбора и
обработки данных обеспечивает сбор хроматографических данных, их обработку и
выведение на монитор. Сегодня управление системой и обработка данных слились
до такой степени, что одно устройство (с соответствующим программным
обеспечением) выполняет все эти функции и называется управляющим модулем. Мы будем
использовать этот термин, если не будет необходимости в разъяснении.
Нынешние хроматографические управляющие модули выполняют множество
функций, работая в режиме ВЭЖХ. Эти функции перечислены ниже:
• сопровождение эксперимента (раздел 3.8.1);
• управление системой (раздел 3.8.2);
• сбор данных (раздел 3.8.3);
• обработка данных (раздел 3.8.4);
• создание отчета (раздел 3.8.5);
• регулировочные функции (раздел 3.8.6).
В целом, чтобы реализовать все возможности управления системой ВЭЖХ,
управляющий модуль должен быть от того же производителя, что и оборудование.
Тем не менее, некоторые производители управляющих модулей предлагают их
для управления приборами, поставляемыми основными производителями ВЭЖХ-
оборудования. Сбор и обработка данных более универсальны, чем конкретное
оборудование, и поэтому многие лаборатории выбирают в качестве стандарта систему
сбора и обработки данных одного производителя и используют централизованную
систему сбора и обработки данных, получаемых от оборудования различных типов
и разных производителей.
166 Глава 3. Оборудование
3.8.1. Сопровождение эксперимента
При разработке метода систематический подход уменьшит объем работ и увеличит
вероятность достижения приемлемого разделения. В десятой главе обсуждается, как
использовать компьютерное моделирование при разработке метода. Программное
обеспечение для разработки метода доступно в виде самостоятельного программного
обеспечения, которое может использоваться в сочетании с любой ВЭЖХ-системой
(например, программа DryLab®), или же оно может быть специально разработано
для использования с одним типом оборудования.
В то же самое время, когда компьютер подключен к ВЭЖХ-системе, может
быть доступно и другое программное обеспечение, полезное для решения задач,
непосредственно не связанных с разделением. Экспертные программы,
вспомогательные файлы и другая информация может помочь пользователю быстро выявить и
решить проблемы. Электронные лабораторные журналы и базы данных могут
упростить ведение записей о проведенных разделениях и учет профилактических работ.
Некоторые программы позволяют устанавливать напоминания о том, что нужно
поменять уплотнения насоса, или о проведении других видов технического
обслуживания. Некоторые производители поставляют вместе с оборудованием аудио-,
видеофайлы, которые могут использоваться как руководство при обслуживании
оборудования.
Лабораторная информационно-управляющая система (ЛИУС)' — это еще одна
категория программного обеспечения, которая позволяет пользователям отслеживать
поток образцов и результаты работы с ними. Некоторые из возможностей такой
системы помогут соблюдать нормативные требования (раздел 3.8.6 и глава 12),
некоторые используются для координации работы, а некоторые могут быть полезны для
сопровождения финансовой деятельности лаборатории (например, инициировать
процесс выставления счетов, когда анализ образцов завершен). Рассмотрим, к
примеру, использование ЛИУС в биоаналитической сервисной лаборатории,
занимающейся анализом лекарственных веществ в плазме крови. Когда образцы поступают
в лабораторию, им присваиваются номера, и эта информация добавляется в базу
данных. Например, можно ввести в базу дату и время получения образца, его
состояние, номер, по которому его отслеживают, и идентификационный номер пациента.
Пробирка с образцом будет снабжена штрих-кодом (если он еще не указан), и будет
перенесена в морозильную камеру (с указанием местоположения холодильника, даты
и времени регистрации) для сохранения до тех пор, пока анализ не будет сделан.
Когда наступит время делать анализ образца, оператор извлекает из базы данных
аналитическую таблицу с данными образца, а сам образец извлекается из
морозильной камеры и перемещается в лабораторию подготовки образцов (дата и время
записываются). После подготовки образца штрих-код оригинального контейнера с
образцом согласуется со штрих-кодом на виале в автосамплере.
Дополнительные сведения могут быть добавлены в базу данных в процессе про-
боподготовки — например, таких, как номера партий различных реагентов. Любой
оставшийся необработанным образец будет возвращаться в холодильник (с
указанием даты, времени и места хранения). Создается таблица последовательности
действий, в которой соотносится положение в лотке автосамплера со штрих-кодом
образца. После окончания анализа данные обрабатываются для создания отчета
о концентрации образца. Они будут автоматически введены в таблицы данных
в шаблоне отчета. Аналитик проверяет данные и прежде, чем отправить отчет
клиенту (каждое замечание или отзыв будут записаны), передает его в подразделение,
ответственное за качество, для дальнейшего рассмотрения.
'Англ. Laboratory Information Management System (L(MS). — Прим. перев.
3.8. Управляющие модули 167 j)l)
Когда отчет клиенту готов к отправке, в бухгалтерию будет отправлен другой отчет
с тем, чтобы был выставлен счет клиенту за проанализированные образцы. В будущем
в любой момент можно будет создать персонализированные отчеты со специальными
целями, такими, как цепь поставок, выявление проблем {например, корреляция
партии реагентов с аномальными результатами), количество разделений образцов в месяц,
проводимых на одной хроматографической системе, и т.д. Функции ЛИУС часто
настраиваются с учетом нужд конкретной лаборатории или задачи, и, таким образом,
возможности системы оказываются практически безграничными. В некоторых случаях
граница между ЛИУС и системой сбора и обработки данных размывается.
Программное обеспечение ЛИУС может дублировать, заменять или использовать программное
обеспечение отдельной системы сбора и обработки данных. Для получения более
подробной информации обратитесь к одному из поставщиков ЛИУС. Поиск в Google
«LI MS vendors» дал список из более чем двухсот компаний, которые поставляют ЛИУС
и соответствующее программное или аппаратное обеспечение.
3.8.2. Управление системой
Одним из самых распространенных приложений компьютера, обслуживающего
процесс ВЭЖХ, является управление системой. Функция системного управления
обеспечивает единый центр управления всеми рабочими параметрами. Например,
скоростью потока, составом подвижной фазы, температурой колонки и длиной волны
детектора. Все параметры конкретного метода могут быть сохранены для удобства
поиска и корректировки при использовании последующих вариантов метода, а также
для архивирования или переноса на другую систему ВЭЖХ. Модификация метода и
разработка новых методов упрощается, поскольку существующий метод можно
использовать в качестве шаблона, и поэтому нужно менять только необходимые
параметры. Большая часть программного обеспечения контроллера содержит функции,
облегчающие создание постоянной электронной записи конкретных параметров
устройства, используемых для проведения каждого лабораторного разделения. Эта
информация может помочь при устранении неполадок, возникающих в системе, и
упростить нормативные проверки.
3.8.3. Сбор данных
ВЭЖХ-детекторы преобразуют пик образца в набор данных: х-(время) и у-(интен-
сивность), а иногда вводится дополнительная z-переменная (например, длина
волны). Эти данные генерируются в аналоговом или цифровом формате, а затем
отправляются в систему сбора и обработки данных для сбора и записи. Большая часть
систем сбора и обработки данных могут принимать как аналоговые, так и цифровые
сигналы, а большинство детекторов имеют как аналоговые, так и цифровые выходы.
Для систем, в которых детектор и система сбора и обработки данных изготовлена
одним и тем же производителем, их соединение (детектора и системы сбора и
обработки данных) может осуществляться простым оптоволоконным кабелем. Когда
детектор и система сбора и обработки данных от разных производителей, то приходится
делать специальное электронное соединение.
Помимо сигнала детектора система сбора и обработки данных может записывать и
другие параметры, такие как скорость потока, длину волны, давление в системе и
другие. В самом сложном исполнении система сбора и обработки данных может хранить
в памяти и воспроизводить точные условия, используемые для анализа конкретного
образца, включая серийные номера колонки и оборудования, номера партий
растворителей, составляющих подвижную фазу и все результаты хроматографических разделений.
168 Глава 3. Оборудование
Частота передачи данных (скорость, с которой они передаются) системе сбора и
обработки должна быть достаточно высокой, чтобы собранная информация точно
отображала наблюдаемые пики на хроматограмме. С другой стороны, если скорость
передачи данных слишком высока, в них будет содержаться слишком много шума.
Хорошей скоростью сбора считается к 20 точек на пик. Для узких пиков, которые
получаются при разделениях на коротких колонках или на колонках, упакованных
частицами маленького размера, система сбора и обработки данных должна
обеспечивать достаточно высокую скорость передачи данных для того, чтобы успеть собрать
по 20 точек на пик. Например, в таблице 3.3 колонка 50 х 4,6 мм, заполненная
сорбентом с размером частиц 1,8 мкм, дает пик объемом 30 мкл при к = 0,5. При работе
со скоростью потока 1 мл/мин это означает, что пик имеет ширину = 2 с, и тогда
требуемая скорость/частота передачи данных » 10 Гц. Однако, при работе в режиме
с более высокой пропускной способностью, скорость потока может быть гораздо
выше. При скорости потока 5 мл/мин пик будет иметь ширину всего 0,4 с, требуя
скорости передачи данных и 50 Гц для его надлежащего отображения. Большее
время удерживания и более низкие скорости потока будут приводить к бодее широким
пикам, и потому не требуется такая высокая частота передачи данных. Большая
часть систем сбора и обработки данных будет корректировать частоту сбора во время
разделения, таким образом, для каждого пика будет собрано приблизительно
одинаковое количество точек. Если есть сомнения, то всегда лучше собирать данные с более
высокой частотой, так как во время обработки можно их усреднить (сгруппировать),
чтобы упростить (уменьшить шум) во время их обработки. Обработка не в состоянии
создать дополнительные точки, если исходная частота передачи данных слишком
низкая. Дополнительную информацию см. в разделе 11.2.1.
3.8.4. Обработка данных
Когда данные хранятся в системе сбора и обработки, их можно обрабатывать в
любое время. Большинство аналитиков используют для визуального контроля качества
данных графическую хроматограмму. Поскольку лаборатории переходят на
безбумажные записи, хроматограмму просматривают только на мониторе компьютера.
Для качественного и количественного анализа (глава II) собранные данные
обрабатываются для создания упрощенного представления в виде таблицы с временами
удерживания и площадями пиков (или их высотой). Дополнительная обработка
может включать данные калибровочных/контрольных разделений для последующего
преобразования времен удерживания и площадей в концентрации (раздел 11.4.1).
Дальнейшая обработка данных может дать оператору дополнительную
информацию — такую, как данные по УФ, МС или МС/МС спектрам и другую информацию
от таких специальных детекторов, как детекторы светорассеяния.
3.8.5. Создание отчета
Большинство систем сбора и обработки данных связаны с использованием
персональных компьютеров и, таким образом, способны работать с любым программным
обеспечением, совместимым с персональным компьютером. Это упрощает перенос
данных из места, где происходит их обработка, в Excel® или другое программное
обеспечение для создания отчетов. Таким образом, таблицы, хроматограммы,
графики, данные статистики и другие обработанные результаты могут быть включены
в доступные официальные отчеты. Некоторые программные пакеты могут
переносить результаты в отчеты автоматически или полуавтоматически. Помимо удобства
3.9. Влияние внеколоночных объемов 169 Ш
и экономии времени, сам по себе этот процесс может уменьшить переписывание и
ошибки, связанные с работой оператора. Если программы прошли валидацию,
количество времени, затрачиваемого на проверку ошибок в отчетах, может быть сведено
до минимума.
3.8.6. Регулирующие функции
Поскольку работа все большего числа лабораторий регулируется нормативными
документами правительства или других регламентирующих органов, то полнота,
сохранность и неприкосновенность хроматографических данных будут все более
важны. Согласно конкретным требованиям к электронным записям, таким как 21 CFR'
раздел 11 |18|, требуется электронный аудит в том случае, если данные хранятся
в электронном виде, зачастую без бумажных копий. Системы сбора и обработки
данных, «совместимые с разделом 11» имеют встроенные функции, которые
освобождают оператора от необходимости вести записи. К примеру, такие системы требуют,
чтобы во время автоматической обработки данных любая ручная корректировка
базовых линий включала запись причины этой корректировки, имя оператора и
отметку о времени. Системы сбора и обработки данных, отслеживающие все системные
параметры для каждой инжекции, могут упростить процесс проверки достоверности
конкретного результата во время просмотра данных аудитором. Ожидается, что
по мере того, как в ВЭЖХ-лаборатории будут вводиться новые регулирующие ее
деятельность нормы, производители программного обеспечения продолжат поставлять
программы, которые помогают пользователям соблюдать эти нормы.
3.9. Влияние внеколоночных объемов
Наблюдаемый на хроматограмме объем пика V„ зависит от уширения полосы
вещества внутри колонки VpQ, а также от других системных факторов, включающих вклады
объемов инжекции Vs, капилляров, фитингов, используемых для коммутации
системы Vpf, ячейки детектора Vdel и постоянной времени Vds, задаваемой системой сбора
и обработки данных.
v, = <Vpo + Щ2 + vl + VL + vl )0-5 (3-D
Из совокупности этих объемов получается, согласно уравнению (3.1), общий
пиковый объем Vp и соответствующая ширина пика W. Уширение пика, обусловленное
факторами, не связанными с разделением (Vpo), возникает из-за внеколоночных
эффектов. Для данного метода внеколоночные эффекты постоянны, но вклад колонки
будет изменяться в зависимости от k растворенного вещества. При изократическом
разделении внеколоночное уширение пика будет более выраженным для раноэлюи-
руемых пиков с меньшими значениями V^. В условиях разделений, приводящих
к меньшим пиковым объемам V^ то есть при использовании коротких колонок
малого диаметра, упакованных мелкими частицами сорбента {см. таблицу 3.3),
внеколоночные эффекты приобретают большее значение. При использовании колонок
с размерами 150x4,6 мм, упакованных пятимикронными частицами, допускаются
относительно большие объемы ввода, диаметры капилляров и размеры ячеек
детекторов. Тем не менее, ВЭЖХ-системы, работа которых связана с использованием
колонок небольшого объема, упакованных частицами диаметром меньше двух микрон.
1CFR — Code of Federal Regulations, Свод федеральных постановлений. — Прим. персе.
170 Глава 3. Оборудование
должны быть сконструированы так, чтобы минимизировать внеколоночные
эффекты. В противном случае система просто не даст приемлемых результатов. Для
получения дополнительной информации о влиянии различных факторов на уширение
пиков см. Vp {раздел 2.4), Vs (разделы 2.6, 3.6.3), ^(разделы 3.4.1.2, 3.4.2.2), ^е,
(раздел 4.2.4) и VrfS (разделы 3.8.3, 11.2.3).
3.10. Обслуживание
Качество результатов анализов, полученных на ВЭЖХ-системе данным методом,
в значительной степени зависит от надежности ее работы и от соответствия ее харак
теристик указанным производителем значениям. Для надежной работы системы
необходимо решить три основных задачи:
• убедитесь, что ВЭЖХ система работает должным образом (раздел 3.10.I);
• устраните как можно больше неисправностей (раздел 3.10.2);
ф проведите квалифицированный и эффективный ремонт (раздел 3. Ю.З).
Эти задачи рассматриваются в настоящем разделе. Хотя и не бывает ВЭЖХ-сис-
тем без проблем, пользователь может активно их устранять или сводить к минимуму
с помощью некоторых описанных здесь методов.
3.10.1. Тест на работоспособность/пригодность системы
Количественные показатели работоспособности/пригодности системы ВЭЖХ
позволяют пользователю сравнить характеристики системы в разные промежутки времени.
Таким образом можно убедиться в том, как система работает, когда она новая,
и включается в первый раз, спрогнозировать трудности, которые могут появиться
в будущем, и показать, что ремонт был эффективным.
3.10.1.1. Установочная, операционная и эксплуатационная
квалификации
В фармацевтической промышленности одним из способов демонстрации того, что
новая ВЭЖХ-система работает должным образом, являются установочная — УК1,
операционная — ОК2 и эксплуатационная —ЭК3 квалификации. Они проводятся
до того, как система вводится в эксплуатацию. УК тестирование гарантирует, что
прибор устанавливается в соответствии с инструкцией изготовителя. УК чаще всего
делается представителем производителя, если установка включена в стоимость
системы. Тестирование будет описано в документации, прилагаемой к системе.
ОК свидетельствует о том, прибор соответствует всем спецификациям, которые
прилагаются к нему производителем, или некоторой их совокупности. Результаты
ОК могут быть также внесены в документацию. Кроме того, результаты ЭК,
описанные в разделах 3.10.1.2 и 3.10.1.3, могут быть использованы для сравнения
результатов испытаний с техническими характеристиками, указанными производителем
(обычно их можно найти в конце одной или нескольких пользовательских
инструкций). ОК и ЭК тестирование часто включает в себя тестирование колонок
(раздел 3.10.1.3) с тем, чтобы подтвердить, что вся хроматографическая система работает
хорошо как единое целое.
'Англ. Installation Qualification. — Прим. перев.
^Англ. Operational Qualification. — Прим. перев.
Зангл. Performance Qualification. — Прим. перев.
3.10. Обслуживание
ЭК тестирование обычно разрабатывается пользователем и может включать
различное количество тестов от многочисленных эксплуатационных тестов, которые
описаны ниже, до анализа нескольких пробных образцов, показывающего, что ожи
даемые результаты могут быть получены для конкретного метода ВЭЖХ. После того,
как все три теста (УК, ОК и ЭК) будут выполнены, система должна быть готова
для рутинной работы.
3.10.1.2. Градиентное тестирование
Важнейшей частью комплекса тестирования для оценки пригодности системы
являются эксперименты по определению линейности и точности, а также системной
задержки формирования градиента. Эти тесты относятся к работе хроматографической
системы во всех случаях, когда используется смешивание в потоке как для изократи-
ческих, так и для градиентных методов. Для проведения тестов колонку демонтируют
и на ее место вставляют капилляр малого внутреннего диаметра. Например,
инжектор с детектором можно соединить отрезком капилляра = 1м с внутренним
диаметром 0,005" (к 0,13мм). Такой длины капилляр создаст давление, достаточное
для обеспечения надежной работы клапанов насоса, у него незначительный мертвый
объем (= 12мкл), мало влияющий на дисперсию градиента в большинстве хроматог-
рафических систем. Следующим шагом является наполнение водой резервуара А.
В резервуар В помещаем 0,1% водный ацетон. (Альтернативой этим элюентам
является использование в качестве элюента А метанола и в качестве элюента В 0,1%
раствора ацетона в метаноле). Для обоих элюентов должен быть использован один и тот
же базовый растворитель. На УФ-детекторе следует установить длину волны 265 нм.
Линейность градиента. Сначала система программируется на запуск градиента
полного диапазона (0—100 %В). Рекомендуемое для этого время — двадцать минут.
Скорость потока должна быть такой, чтобы давление в системе было достаточно
высоким для обеспечения надежной работы клапанов насоса, обычно достаточно от I
до 3 мл/мин. Автосамплер/инжектор должен находиться в положении ввод, чтобы
подвижная фаза прокачивалась через петлю. Поскольку петля инжектора обычно
промывается потоком в течение всего процесса разделения (рис. 3.176), ее объем
вносит свой вклад в объем задержки. |Если хроматографическая система обычно
стартует в положении, когда поток не идет через петлю, этот тест следует проводить
с инжектором в положении загрузка
(рис. 3.17о), а не в положении ввод].
Далее выполняется тестовый градиент.
График изменения базовой линии
должен отображаться как S-образная кривая
(сплошная кривая на рис. 3.26). Этот
холостой градиент можно использовать
в качестве грубой проверки линейности
градиента, а также для измерения
объема задержки системы (см. ниже).
Отклонения от линейности
градиента проще всего проверить сравнением
с прямой, проходящей через середину
градиентного профиля (пунктирная
линия на рис. 3.26). Реальный градиент
(сплошная кривая) должен быть
плавным и не отклоняться от пунктирной Рис 3 2б профиль градиента иода - вода/
линии за исключением небольших «гра- ацетон.
172 Глава 3. Оборудование
диентных закруглений» в начале и в конце градиента. Если наблюдаются
отклонения, то следует уделить дополнительное внимание тестированию системы в условиях
ступенчатого градиента (см. ниже). Для систем со смешиванием на стороне низкого
давления проблемы с линейностью градиента помогает выявить и тестирование
дозирующих клапанов (см. ниже) |19|. Часто наблюдаемые отклонения от линейности
проявляются в виде четко выраженного сдвига или угла на профиле градиента
(например, рис. 17.23 и раздел 5.5.3.3 работы |20|). Целесообразно увеличить количество
контрольных ступенек при тестировании ступенчатым градиентом в тех областях,
где наблюдаются отклонения от линейности.
В большинстве систем ВЭЖХ можно запрограммировать скорость градиента
от 10%/мин и выше. Если систему предполагается использовать в условиях крутого
градиента, например, для высокопроизводительных методов, или для случаев с очень
разными скоростями потока, линейность градиента может быть проверена с
помощью холостого градиента в рабочих условиях. Например, вместо двадцатиминутного
градиента со скоростью потока 2 мл/мин, как на рис. 3.26, может быть
целесообразнее протестировать систему пятиминутным градиентом со скоростью потока
0,5 мл/мин, если эти условия более приближены к рабочим. Градиентное
закругление, которое не является серьезной проблемой для большинства градиентных
условий, имеет тенденцию увеличиваться для крутых градиентов (или малых градиентных
объемов IqF, где tG — время градиента и F — скорость потока; см. также
разделы 8.1.6.2 и 9.2.2 работы |20|). Обычно градиентное закругление можно снизить
уменьшением объема системной задержки формирования градиента (см.
обсуждение рис. 17.28) и увеличением градиентного объема tGF.
Определение объема задержки формирования градиента. Профиль градиента (такой
как на рис. 3.26) может быть использован для определения объема задержки
системы. Этот объем можно измерить двумя способами. Первый заключается в
продолжении пунктирной линии на рис 3.26 до тех пор, пока она не пересечет продолженную
базовую линию. Время между моментом этого пересечения и стартом градиента и
есть, как это показано на рис. 3.26, время задержки. Время задержки ?д можно
пересчитать в объем задержки Кд умножением на скорость потока F, Кд = tDF. Этот метод
определения объема задержки прост, но на результаты, полученные с его помощью,
оказывают влияние любые ошибки, которые могут возникнуть из-за неточности
при проведении пунктирной линии через профиль градиента. Кроме того, не всегда
удобно делать эти построения непосредственно на мониторе компьютера, пользуясь
полученным на выходе графиком.
Второй метод измерения объема задержки менее подвержен ошибкам и более
удобен для работы с ним на компьютерном мониторе. Графически он показан
на рис. 3.26. Определите отклик детектора в начале базовой линии (0 %В) и в конце
градиента (100%). Исходя из этих двух значений найдите точку, в которой отклик
детектора соответствует 50 %В, и отметьте время t^, необходимое для достижения
этой точки. Время задержки /д равно Т]д минус половину времени градиента
(например, 10 минут для 20-минутного градиента). Объем задержки равен гд/7.
Тестирование ступенчатым градиентом. В этом тесте используются те же
настройки системы и растворители А и В. Тестирование ступенчатым градиентом определяет
точность дозирования для выбранных смесей растворителей (или подвижных фаз).
Если эта же хроматографическая система используется и для проведения изократиче-
ских разделений, то такое тестирование будет проверкой на точность смешивания
растворителей в потоке. Системный контроллер запрограммирован, чтобы
осуществить пошаговую подачу серии смесей растворителей. При тестировании ступенчатым
градиентом удобнее использовать десятипроцентные ступеньки. Таким образом,
образуются смеси с содержанием 0, 10, 20... 80, 90, 100 %В. Каждая из этих смесей про-
3.10. Обслуживание 173 J)
/ 59.67
Г 54.04
/ 49-63
/"44.70
/" 39.95
~1 1 1 1 1 1 1 1 1 1
18 20 22 24 26 28 30 32 34 36
время (мин)
Рнс. 3.27. Результаты тестирования ступенчатым градиентом. Измеренный %В
показан под каждым шагом
ходит через систему в течение трех минут. Чаще всего проблемы возникают в районе
50 %В, поэтому к общему количеству ступенек здесь нужно добавить еще две — 45 и
55 %В. В общей сложности их должно получиться тринадцать. Остальные условия
такие же, как для теста на линейность градиента. Результаты для смесей в области
от 40 до 60 %В для хорошо работающей системы показаны на рис. 3.27.
Затем подсчитывают фактический %В для каждого шага (как на рис. 3.27),
измеряя его высоту от базовой линии (0 %В) и деля на расстояние между шагами 100 %В
и 0 %В. Процент В для каждого шага нужно сравнивать с процентом, который
запрограммирован. Например, 40 % В предустановленные, как указано на рис. 3.27,
фактически означают 39,95 %В. Обычно производители указывают точность +0,5—1%
во всем диапазоне смешивания. Например, шаг 55 %В на рис. 3.27 фактически
оказался шагом с 54,04 %В — т.е. едва попал в заданные пределы. Для методов со скоростями
градиента > 1%/мин, точности + 1% обычно хватает. При использовании более пологих
градиентов может потребоваться более узкий интервал отклонений. Точность
дозирования, кроме того, можно улучшить, смешивая растворители заранее. Например,
точность дозирования градиента 15—25 %В может быть десятикратно повышена от ±1%
до +0,1% заменой 100% водного растворителя в резервуаре А на заранее приготовлнен-
ную смесь 15/85 органика/водный раствор и заменой 100% органического
растворителя в резервуаре В на заранее приготовленную смесь 25/75 органика/водный раствор.
Кроме того, градиентную программу изменяют, и вместо 15—25 %В программируют
градиент 0—100 %В. Точность смешивания в потоке для изократического алюирования
может быть повышена аналогично с помощью таких же операций.
Тестирование дозирующих градиент клапанов (ДГК). Третий тест с аналогичной
подготовкой системы к работе, как уже описано выше, полезен для хроматографов,
в которых смешивание происходит при низком давлении, но не используется в
случае смешивания под высоким давлением. Этим тестом проверяется точность
дозирующих клапанов и связанного с ними управляющего программного обеспечения.
Рассмотрим, к примеру, четырехкомпонентную систему растворителей (А, В, С и D).
Входы линий А и В помещаются в резервуар, содержащий воду, а входы линий С
и D — в резервуар, содержащий 0,1% водного ацетона. Базовая линия есть результат
прокачивания через систему подвижной фазы состава 50/50 А и В. Работу системы
дозирующих градиент клапанов тестируют с помощью различных комбинаций
растворителей, которые получают смешиванием 90% А или В с 10% С или D.
Например, результаты испытаний, показанные на рис. 3.28а (приемлемый результат
тестирования), соответствуют последовательности смены подвижных фаз, указанных
о
174 Глава 3. Оборудование
m
15-п
10
Ь
0
90/10
50/50 А/В
/1
А/С
Г \
L
г
1
J
A/D
1 /"
\ \
! т
8 10 12
время (мин)
14 16 18
15-
14-
90/10 А/С
A/D
В/С
B/D
1±2%
П "ГТ ГГП"
Рнс. 3,28. Результаты проверки дозирующих градиент клапанов (ДГК). (а) Результаты
теста ДГК; (б) увеличенное изображение с указанием допустимой разницы
между различными плато ±2%. Базовая линия формируется подвижной
фазой состава 50:50 А:В. Остальные плато справа налево 90:10 А:С, A:D, B:C и
B:D. Растворители А = В = вода; С = D = 0,1% ацетон в воде
в подписи к рисунку. Измеряется высота каждого плато над базовой линией, которая
сформирована смесью 90/10. Для того, чтобы определить в процентах точность
дозирования клапанами различных рабочих смесей, разницу между высотой самого
высокого и низкого плато делят на среднюю высоту плато. Обычно допускаются
колебания < 2% от высоты плато, хотя у хорошо отлаженных систем такие изменения
соответствуют < 1% (рис. 3.286; для примера неудачного теста ДГК см. рис. 17.25).
3.10.1.3. Дополнительная проверка системы
На надежность хроматографической системы, кроме точности и линейности
градиента, влияют и другие факторы. Для обеспечения правильной работы системы,
тесты, перечисленные ниже, могут использоваться периодически (например, ежегодно
или раз в полгода). Полученные в этих тестах типичные значения рабочих
параметров приведены в таблице 3.4.
Тестирование расхода/скорости потока. Хотя небольшие изменения в скоростях
потока добычно приводят лишь к незначительным изменениям в разделении,
большие ошибки в F могут привести к серьезным последствиям. Отсюда следует,
что нужно периодически проверять точность расхода. Точность расхода «секунда в
секунду» без специального оборудования трудно измерить в процессе разделения
(да это и не нужно), но более длительное волюмометрическое тестирование провести
легко, если по времени собирать подвижную фазу со скоростью I мл/мин в изокра-
тическом режиме в мерную емкость объемом 10 мл. Для систем со смешиванием
на стороне высокого давления скорость потока может зависеть от сжимаемости
растворителя, поэтому лучше проверять для репрезентативной выборки растворителей.
Например, проверьте расход 100%А, используя воду, и 100 %В, используя ацетонит-
рил и метанол. Обычно производитель в спецификации указывает точность расхода
в пределах ±1%. Измеренная точность скорости потока для рутинных операций
должна находиться внутри этого интервала. В таблице 3.4 указан критерий
допустимости ±2%, поскольку совокупность ошибок измерения (волюмометрические ошибки,
связанные с использованием стеклянной посуды), а также ошибки установления
времени начала/остановки внесут вклад в общую ошибку измерения.
3.10. Обслуживание 175
Таблица 3.4. Типичные параметры пригодности системы для тестов раздела 3.10.1
Параметр/тест
Линейность
Объем задержки формирования градиента системы
Тестирование ступенчатым градиентом (точность)
Тест ДГК°
Скорость потока (собрать 10 мл при скорости
1 мл/мин)
Падение давления от 270 бар (4000 psi)
Воспроизводимость удерживания
Воспроизводимость площади пика (инжекция
10 мкл)
Типичные
характеристики
±1%
Различаются
±1%
Не определены
±1%
Не определены
Не определены
±0,3%
Принятые критерии
приемлемости
Визуально определяемая
линейность от 5 до 95 %В
По результатам измерения
±1%
±2% высоты плато6
±2% (±12 с для 10 мл при 1
мл/мин)
15% за 10 мин
±0,05 мин
±1%
"Дозирующие градиент клапаны.
^Разность между высотой самого высокого и самого низкого плато (см. рис. 3.28).
Сброс/стравливание давления. Неисправные клапаны и изношенные насосные
уплотнения часто приводят к отклонениям в ожидаемых значениях точности
смешивания, линейности градиента и скорости потока. Дополнительную проверку кчапа-
нов на выходе из насоса можно провести с помощью теста на сброс давления.
Для этого выходной капилляр из насоса закрывается (соединением с заглушкой).
Верхний предел давления для отключения насоса устанавливается вблизи его
максимального значения, например, 350 бар (5000 psi) для систем, способных выдержать
давление 400 бар (6000 psi). Затем насос включается и отключается лишь после того,
как будет достигнут верхний предел давления. Максимум давления регистрируется,
и через десять минут значение давление записывается снова. Падение давления
на величину <, 15% означает, что клапаны работают должным образом. Ббльшее
падение предполагает, что выпускной обратный клапан(ы) должен быть промыт или
заменен, или уплотнение(я) насоса следует заменить (в том случае, если не
используется обратный клапан для выпуска, как, например, в накопительных плунжерных
насосах, см. раздел 3.5.1.2). Если же давление упало до атмосферного к концу
десятиминутного теста, то это значит, что какое-то соединение течет.
Воспроизводимость удерживания. Проверка воспроизводимости удерживания —
это общая проверка качества смешивания в потоке и работоспособности насоса.
Хотя эта проверка может быть проведена с любым образцом, целесообразно
использовать легко приготовляемый образец в условиях, которые могут быть
воспроизведены в любое время. Это позволяет независимо тестировать хроматографическую
систему для случая конкретного метода, и это хороший способ поиска неисправностей.
Примеры условий, в которых проводится тестирование, приведены в таблице 3.5.
Важно использовать такую концентрацию образца, чтобы пик не выходил за пределы
линейного диапазона детектора и был достаточно большим, чтобы шум базовой
линии не влиял на прецизионность измерений. Например, образец, который дает
высоту пика от 0,1 до 0,8 AU — это хороший выбор для УФ-детектирования.
Изменения во времени удерживания не более чем на <= 0,05 мин (1 СКО) приемлемо
для шести повторных разделений/тестов. Большие изменения говорят о том, что есть
проблемы. Если результаты других тестов, касающихся пригодности системы,
удовлетворительны, но воспроизводимость удерживания плохая, то, скорее всего, источ-
176 Глава 3. Оборудование
ником проблемы являются утечки и воздушные пузырьки. Альтернативный тест,
особенно в тех случаях, когда система используется только для изократических
методов, заключается в том, чтобы протестировать систему на воспроизводимость
удерживания в изократических условиях, указанных в таблице 3.5 (80 %В).
Воспроизводимость площади пика. Хроматограммы, полученные при
тестировании на воспроизводимость удерживания, можно использовать и для определения
воспроизводимости площади пика, которая, прежде всего, характеризует
пригодность автосамплера. Колебания в площадях пиков, рассчитанные из шести
разделений, должны быть < 1% СКО. Большинство современных автосамплеров
обеспечивают < 0,5% СКО для объемов инжекции > 5 мкл при работе в стандартных условиях
(например, в условиях, указанных в таблице 3.5). Неудовлетворительная
воспроизводимость площади пика может быть связана с плохой работой автосамплера.
Воспроизводимость площади пика должна быть проверена как для изократического, так и
для градиентного режима. При этом в обоих случаях должны быть получены сходные
результаты.
Таблица 3.5. Условия тестов на воспроизводимость удерживания и площади пика
Параметр
Скорость потока
Колонка
Температура
Детектирование
Подвижная фаза А
Подвижная фаза В
Градиентный/изократический режимы
Время уравновешивания
Образец
Вводимый объем
Удерживание (типичное) градиентный/
изократический режимы
Величина
1,5 мл/мин
Си 150 х 4,6 мм, 5 мкм
35 "С
280 ни УФ
Вода
Метанол
5—95 %В в теч. 20 мин»/80 %ВЙ
10 мин
5мкг/мл 1-хлор-4-нитробензол'
Юмкл
14 мин"/3 мин* (при этом Уд = 2,2 млг)
"Условия тестирования в градиентном режиме.
^Условия тестирования в изократическом режиме; при необходимости подберите условия так,
чтобы 2 < к < 10.
*Можно использовать и другие неполярные ароматические соединения (например, толуол,
метил бензоат).
•\Ддя других значений объема задержки Кй: времена удерживания в условиях градиентного
разделения будут изменяться на разницу в объемах задержки, разделенную на скорость потока
(1,5 мл/мин).
Тестирование новой колонки. Быстрый способ проверки работоспособности
ВЭЖХ-системы — повторение, в точном соответствии с указаниями производителя,
процедуры тестирования новой колонки. Этот тест должен быть выполнен как часть
процедуры операционной квалификации (ОК) или эксплуатационной квалификации
(ЭК) (раздел 3.10.1.1), чтобы перед началом работы продемонстрировать — система
функционирует нормально. Новая колонка, кроме того, служит инструментом
в стратегии «найти и исправить» (раздел 17.3.1) при выявлении неисправностей в
системе. Просто установите новую колонку и повторите эксплуатационный тест произ-
3.10. Обслуживание 177 ))|
водителя. Условия тестирования должны быть аналогичны изократическим
условиям, описанным в таблице 3.5, и должны быть указаны в тестовом листе колонки,
включенном в прилагаемую к колонке документацию. Если результаты теста не
отклоняются более чем на = 10% от результатов испытаний производителя (время
удерживания, количество теоретических тарелок), то система работает хорошо —
скорость потока, температура колонки, состав подвижной фазы выбраны правильно
и внеколоночные эффекты минимальны. (В редких случаях можно получить
количество теоретических тарелок, равное приводимому производителем: системы
тестирования колонок у производителя оптимизированы именно для таких задач, а не
для рутинной лабораторной работы.)
3.10.2. Профилактика
Хорошим способом повышения надежности ВЭЖХ-системы является
прогнозирование и предотвращение неисправностей до их возникновения. Неисправности
в ВЭЖХ-системах могут быть результатом обычного износа компонентов или
особенностей режима их работы. Поэтому количество неисправностей можно
минимизировать, периодически проводя техническое обслуживание, чтобы изнашиваемые
части системы поддерживались в рабочем состоянии или ремонтировались до того,
как они придут в негодность. Кроме того, существует целый ряд простых
лабораторных методов, которыми можно пользоваться для минимизации отказов системы
по вине пользователя.
3.10.2.1. Периодическое обслуживание
Регулярная очистка и/или замена комплектующих помогут сделать систему ВЭЖХ
более надежной. Некоторые из задач, которые решают с помощью периодических
профилактических работ, перечислены в таблице 3.6. Указанная в таблице частота
различных профилактических мероприятий рекомендуется для систем, используемых
несколько раз в неделю. В других лабораториях, исходящих из собственного опыта,
интервалы между профилактическими работами могут отличаться от рекомендуемых.
Суть метода состоит в том, что каждый прибор/модуль должен регулярно
обслуживаться. Более подробную информацию об отдельных частях и модулях, упомянутых
ниже, см. в описаниях первой части этой главы.
Резервуар. Резервуар, содержащий водный элюент {компонент А), должен быть
вымыт и пополнен так, чтобы в нем не происходило роста микроорганизмов. Чтобы
избежать появления микроорганизмов, необходимо еженедельно менять элюент
и мыть резервуар. Впрочем, некоторые лаборатории растягивают этот срок с недели
до двух. Лучше всего никогда не доливать новую порцию в использовавшийся водный
элюент, а заливать свежий элюент в чистый резервуар. Резервуар с органическим
растворителем (компонент В) можно не менять дольше, поскольку вероятность роста
микроорганизмов в подвижных фазах, содержащих > 30% органики, существенно
меньше по сравнению с водными подвижными фазами. Если А- или В-растворители
категории «для ВЭЖХ», то фильтровать подвижную фазу необязательно. В противном
случае все подвижные фазы должны быть профильтрованы через мембранный фильтр
пористостью < 0.5мкм. Входной фильтр, если он загрязнится, может быть источником
повторного загрязнения содержимого резервуара. Фильтр стоит недорого, и потому
его, в качестве меры предосторожности, следует менять каждые полгода. Дегазация
подвижной фазы является одним из самых простых способов повышения
эксплуатационной надежности системы, поэтому все растворители, используемые для обращен-
но-фазовой ВЭЖХ, должны быть дегазированы (см. раздел 3.3).
178 Глава 3. Оборудование
Таблица 3.6. Рекомендованные интервалы обслуживания
Наименование
Резервуар
Капилляры и
соединения
Насос(ы)
Автосамплер
Колоночный
термостат
Детектор
Сливной контейнер
Система сбора и
обработки данных
ЖХ-система
ЖХ-метод
Действие
Замена водного буфера, мытье
резервуара
Замена органического буфера, мытье
резервуара
Фильтрование подвижной фазы
Замена входного фильтра
Дегазация подвижной фазы
Осмотр на предмет утечек
Обработка в ультразвуковой бане или
замена обратных клапанов
Замена уплотнения(й) плунжеров
Промывка для удаления буферов
Осмотр на предмет утечек
Замена промывочного растворителя и
мытье резервуара
Замена уплотнения ротора крана
Замена уплотнения иглы
Осмотр на предмет утечек
Контрольная калибровка
Осмотр на предмет утечек
Осмотр на предмет утечек
Дополнительные проверки детектора
Проверка заполнения и опорожнение
В зависимости от проводимых
экспериментов
Контрольные проверки
Проверка пригодности
Частота''
Еженедельно
Ежемесячно
Каждую новую порцию
Раз в полгода
Ежедневно
Еженедельно
По мере необходимости
Через каждые шесть месяцев
Ежедневно
Ежедневно
Еженедельно
См. рекомендации производителя
Через каждые шесть месяцев
Ежедневно
Ежегодно
Ежедневно
Еженедельно
См. инструкцию по эксплуатации
детектора
Ежедневно
Через каждые шесть месяцев
Ежедневно
"Может варьироваться в зависимости от частоты использования системы, ее пригодности в
течение определенного промежутка времени или от СОП.
Капилляры и соединения. Если капилляры и сопрягающие их соединения
правильно собраны, то они должны работать без ограничений и проблем. Тем не менее,
утечки время от времени происходят, и поэтому рекомендуется проверять все
капилляры и фитинги на предмет утечек раз в неделю. Единственным признаком утечки
может быть капля влаги или небольшое белое солевое пятно, оставшееся от
протекшего буфера. Если утечка обнаружена, то насос должен быть отключен и фитинг
затянут. Затем снова включают насос. В тех случаях, когда используют фитинги и/или
капилляры из ПЭЭК, концы капилляров должны быть повторно установлены в
своих посадочных местах до затягивания1. Иногда капилляр из ПЭЭК., если его
затягивать под давлением, может скользить внутри фитинга, поэтому насос следует
отключать во время этой операции.
1 Необходимо осмотреть конец капилляра и, если он деформирован, отрезать
деформированную часть. Если ферула деформирована, то заменить и ее. Подготовленный капилляр вдвигается
через ферулу в посадочное место до упора и только потом затягивается. — Прим. перев.
3.10. Обслуживание 179 J)
Насосы. Залипшие или протекающие/перепускающие обратные клапаны
являются наиболее часто встречающейся неполадкой у насосов в большинстве хромато-
графических систем. Эта неполадка обычно проявляется в виде колебаний давления
в системе. Неоднократно перепускавший/протекавший клапан можно восстановить
обработкой в ультразвуковой бане в метаноле в течение нескольких минут. Перед
обработкой необходимо проверить сам обратный клапан, чтобы убедиться в том,
что его составляющие не выпадут из корпуса и не перепутаются. Если клапан
требует повторной сборки, следует делать это в перчатках, на которых нет пыли, чтобы
избежать повторного загрязнения. (См. раздел 17.2.5.4 для дополнительной
информации по ультразвуковой очистке клапанов.) Уплотнения насосных плунжеров могут
прослужить год или больше, но не стоит ждать момента, когда они износятся
окончательно. Стоят они недорого, но когда они выйдут из строя, могут возникнуть и
другие проблемы — такие, как засоренные фильтры. По этой причине уплотнения
насоса разумнее заменять два раза в год, то есть до того, как они существенно
износятся1. Остатки буферов в насосе, когда он не используется, могут высыхать,
кристаллизоваться и оставлять абразивные отложения, которые увеличивают износ
уплотнений или приводят к неисправностям обратных клапанов. Лучше всего
промыть ВЭЖХ-систему от всех буферов перед отключением ее на время большее, чем
час или два. Если система должна быть готова к процессу разделения с момента
включения, то насос может быть запрограммирован на минимальный поток (напри
мер, 0,1 мл/мин), чтобы через него все время протекала подвижная фаза. Как и
в случае капилляров и фитингов, насос периодически может протекать, и поэтому
рекомендуется проверять насос на наличие утечек каждый раз при запуске.
Автосамплер. Так же, как необходимо заменять подвижную фазу и чистить
резервуары, чтобы избежать микробного загрязнения, нужно периодически заменять
растворитель для промывания автосамплера и мыть резервуар, в котором он находится.
Если для промывки вы используете водный элюент, то его следует менять каждую
неделю и при этом мыть резервуар. Если же вы используете органический
растворитель, то все это вы проделываете ежемесячно. Уплотнение ротора инжектора
рассчитано на длительный срок службы: сто тысяч циклов работы. Проверьте в руководстве
пользователя рекомендованный производителем интервал профилактического
обслуживания/замены этого уплотнения. Уплотнение иглы снашивается от постоянного
использования. Если в руководстве пользователя нет других рекомендаций, то
замена этого уплотнения раз в полгода поможет избежать ненужных проблем.
Автосамплер является таким компонентом ВЭЖХ-системы, в котором утечки вероятнее всего,
и потому разумно проверять его на наличие утечек каждый день.
Термостат. Термостат должен быть тем блоком хроматографической системы,
который работает без неисправностей. Калибровку термостата следует делать при ее
установке, а затем каждый год. Проточный датчик температуры можно использовать
для контроля температуры подвижной фазы, выходящей из колонки, для того, чтобы
сравнить температуру в термостате и в колонке. Каждый день в начале работы, или
начиная работать с новым методом, концевые соединения колонки необходимо
проверять на предмет утечек.
Детектор. ВЭЖХ-детекторы работают при давлениях существенно более низких,
чем колонки, и поэтому утечки в них происходят реже, чем в других местах
хроматографической системы. Однако имеет смысл детекторы на предмет утечек
проверять еженедельно. Частота других проверок работоспособности детекторов может
'Уплотнения лучше менять в соответствии с рекомендациями производителя вашего насоса.
Не через какой-то временной промежуток, а когда через насос будет прокачан установленный
производителем объем подвижной фазы. — Прим. перев.
180 Глава 3. Оборудование
очень сильно отличаться в зависимости от модели самого детектора. Для получения
дополнительной информации необходимо заглянуть в руководство пользователя.
Сливная емкость. Сливная емкость представляет собой компонент системы,
который легко упустить из виду. Каждый день проверяйте эту емкость — не
переполняется ли? Размещение сливной емкости в пластиковом поддоне поможет
предотвратить разлив, когда она рано или поздно, но непременно переполнится.
Тестирование ВЭЖХ-системы. Тестирование пригодности системы, описанное
в разделах 3.10.1.2 и 3.10.1.3, должно выполняться один или два раза в год. Проверка
раз в полгода часто устраняет проблемы, которые могут возникнуть, если
тестировать систему раз в год. Если же обосновать проверку системы два раза в год нельзя,
то проверяйте один раз.
Тестирование ВЭЖХ-метода. Перед тем, как произойдет анализ образцов,
необходимо провести проверку пригодности системы. Это, может быть, самый важный
тест, который можно выполнить для проверки ВЭЖХ-системы потому, что он
показывает, подходят ли система и метод для проведения требуемого анализа.
3.10.2.2. Рекомендации для рутинных методов
Для получения достоверных данных ВЭЖХ-система должна работать надежно и
воспроизводимо. В этом разделе перечислены и рассмотрены некоторые
дополнительные рекомендации и методы, которые помогут обеспечить высокое качество
результатов.
Качество реагентов. Градиентное элюирование, как правило, концентрирует
неполярные примеси, присутствующие в А- и В-растворителях, на входе в колонку
с последующим их вымыванием по мере элюирования. Эти примеси могут
проявиться в виде пиков как на хроматограмме холостого разделения, так и на хроматограм-
мах разделения образца (см. пример на рис. 17.12 и разделы 17.2.5.9 и 9.6.2). По этой
причине очень важно для градиентного элюирования использовать реактивы,
предназначенные для ВЭЖХ. Реагенты более низкого качества могут быть использованы
для разделений в условиях изократического элюирования, но даже самые
незначительные примеси могут создать сложности при градиентном элюировании. Для
достижения наилучших результатов следует использовать для всех работ только реактивы
для ВЭЖХ. Водные реагенты и буферные растворы следует часто заменять
(например, еженедельно), чтобы избежать микробного загрязнения. Особенно
неприятными могут быть примеси в воде.
Промывка системы. Так же, как и качество реагентов, для минимизации артефак-
тных пиков имеет значение и промывка всей системы. Следует соблюдать
рекомендации раздела 3.10.2.1 (резервуары и насосы). Разливы, утечки и другие
потенциальные источники загрязнения следует быстро удалять.
Дегазация. Несмотря на то, что некоторые ВЭЖХ-системы смогут работать и
без дегазации подвижной фазы, каждая система будет работать более надежно с
дегазированными растворителями (раздел 3.3). Захваченные пузырьки воздуха и
выделение газов из растворителей являются общими проблемами, которых в значительной
мере можно избежать, используя дегазацию. Рекомендуется прокачивать на
повышенном расходе (например, 5 мл/мин) насос(ы) и входные капилляры ежедневно,
открывая сливной кран(ы) на несколько минут для удаления пузырьков воздуха.
Каждому методу — своя колонка. Каждому аналитическому методу должна
соответствовать отдельная колонка. Не рекомендуется использовать одну и ту же колонку
для разных методов, поскольку пики, которые не мешают в одном методе (напри-
3.10. Обслуживание 181 ))|
мер, поздноэлюируемые), могут накладываться на другие в остальных методах.
Компоненты образца могут изменять селективность или даже привести к деградации
сорбента в одном методе, а в другом ничего этого не случится.
Уравновешивание. Перед каждым разделением колонка должна быть
уравновешена в той же степени, что и для других разделений данной серии. Полное
уравновешивание для градиентных методов разделения может понадобиться или не
понадобиться (раздел 9.3.7).
Предварительные разделения. Некоторые методы дают лучшие результаты, если
перед первым разделением с образцом провести несколько предварительных. Эти
разделения, при которых вводятся вместо образцов стандарты или имитации образ
цов, могут помочь насыщению медленно уравновешивающихся активных сайтов
на сорбенте и, таким образом, можно получить более воспроизводимые разделения.
Предварительные разделения чаще всего используют для работы с биологическими
образцами (раздел 13.3.1.4). Иногда такими предварительными разделениями служат
тесты на работоспособность хроматографической системы.
Не принимайте в расчет первое разделение. Поскольку некоторые методы требуют
предварительных разделений, в первом разделении колонка может быть
уравновешена иначе, чем в последующих. При количественном анализе лучше не принимать
в расчет первое разделение. Для этих задач второе и последующие разделения будут
более надежными, чем первое. Предварительный тест на пригодность системы
служит для этих же целей (см. следующий пункт).
Пригодность системы. Большинство методик, выполняемых под надзором
контролирующих органов (таких как FDA1, EPA2, OECD3, USP4 и др.), требуют
предварительных тестов на пригодность хроматографа. Такой тест служит подтверждением
того, что оборудование и аналитическая методика обеспечат получение достоверных
результатов. Поскольку требования к тестам на пригодность оборудования весьма
разнообразны, следует обратиться к нормативным рекомендациям для выбора
подходящего теста (раздел 12.3). В качестве частичной поверки хроматографа многие
используют, как в отдельности, так и в совокупности, тестирование следующих
параметров: воспроизводимость времени удерживания или площади пика, отклик
сигнала (чувствительность детектора), ширину или размывание пика, разрешение,
противодавление колонки.
Тестовыми образцами могут служить разбавленные чистые стандартные
вещества, либо модельные соединения из выбранного образца, или иные образцы,
отобранные для проверки работы хроматографа. Выбор тестового образца определяется,
прежде всего, возможностью проверить, можно ли с помощью данного метода
провести необходимые анализы. В любом случае — потребуется или нет тест на
пригодность хроматографа — настоятельно рекомендуется провести тестовый анализ до
начала текущих работ, хотя бы стандартного образца, чтобы убедиться в том, что время
удерживания и размер пика соответствуют ожидаемому.
Стандартные образцы и градуировочные растворы. Для количественного
определения вещества в пробе (глава 11) полученный при его анализе сигнал (отклик детекто-
1 FDA — Food and Drag Administration, Управление по санитарному надзору за качеством
пищевых продуктов и медикаментов (США). — Прим. перев.
2ЕРА — Environmental Protection Agency, Агентство по охране окружающей среды (США). —
Прим. перев.
^OECD — Organisation for Economic Co-operation and Development, Организация
экономического сотрудничества и развития (ОЭСР). — Прим. перев.
4(JSP — United States Pharmacopeia, Фармакопея США. — Прим. перев.
182 Глава 3. Оборудование
pa) сравнивают с сигналом стандартного образца известной концентрации. Диапазон
концентраций градуировочных растворов, число повторных анализов и их
последовательность может зависеть от конкретного метода. Можно использовать метод либо
внутреннего, либо внешнего стандарта (абсолютной градуировки). В любом случае,
прежде чем наносить на колонку неизвестный образец, нужно, по крайней мере хотя
бы один раз проанализировать стандартный образец, что позволит удостовериться
в надежности аналитической методики.
Таблица 3.7. Рекомендации по ремонт}'
Уровень подготовки оператора
Начинающий/оператор
Опытный
Обученный производителем
Стандартные профилактические работы
Замена подвижной фазы, входного проточного фильтра,
проточного фильтра, предколонки, колонки
Проверка клапанов, уплотнений насоса, замена КФГ*, ротора
инжектора, лампы детектора, капилляров и соединений
Ремонт КФГ, электроники, ремонт узлов отдельных модулей,
ячейки детектора
*КФГ — Клапаны формирования градиента
3.10.3. Ремонтные работы
Рано или поздно, вне зависимости от того, как и насколько регулярно проводятся
профилактические работы, неисправности могут быть у любой ВЭЖХ-системы.
Глава 17 посвящена устранению этих неисправностей. В этом разделе рассматриваются
некоторые из наиболее философских аспектов ремонта хроматографических систем.
3.10.3.1. Персонал
Некоторые лаборатории полностью полагаются на обслуживание, положенное по
договору с производителем оборудования, который включает в себя техническое
обслуживание и ремонт, тогда как другие лаборатории выполняют все эти задачи
самостоятельно. Большинство лабораторий, однако, стремится избежать этих двух
крайностей. Даже если в сервисный контракт включено 100% работ по
обслуживанию, имеет смысл некоторые работы выполнять персоналом лаборатории. Хотя бы
для экономии времени. Хорошо, когда имеется план, в котором расписано, какой
персонал должен проводить ремонт того или иного оборудования. Некоторые
рекомендации перечислены в таблице 3.7 и обсуждаются в разделе 3.10.3.3.
3.10.3.2. Ведение учета
Лабораториям, работающим под надзором контролирующих органов, может
потребоваться учет мероприятий по техническому обслуживанию и ремонту, но даже в тех
лабораториях, для которых такой учет необязателен, следует каким-то образом
регистрировать информацию об обслуживании оборудования. Один из простейших
способов учета заключается в подготовке папки, имеющей три раздела для учета и
описания различных видов работ по обслуживанию системы. Для каждой системы
ВЭЖХ заводится отдельная папка с материалами, которые вполне доступны и могут
быть использованы при обслуживании системы. Удобно включить в эту книгу учета
три следующих раздела.
3.10. Обслуживание 183 ))|
Конфигурация системы. Необходимо переписать все модули, из которых состоит
данная конфигурация системы, а также модель и серийный номер каждого модуля.
Все изменения состава системы должны быть отражены в записях. Это облегчит
общение с поставщиками системы и ее компонентов в случае, если возникнет
необходимость в ремонте, а также сможет подтвердить проверяющим органам, что именно
эта конфигурация системы была использована для анализа конкретного образца.
Отчеты о профилактических работах. Оформление записей о произведенных ре
монтах может быть упрощено, если создать шаблон с пробелами, в которые
вписывается кто, когда и почему его производил и как решались вопросы технического
обслуживания. В книге учета профилактических работ необходимо иметь несколько
пустых бланков, чтобы иметь возможность их заполнить во время проведения этих
работ. Раз в год после проведения ряда профилактических мероприятий эти записи
можно рассортировывать по категориям (например, уплотнения плунжеров насосов,
клапаны), и они смогут быть вспомогательной информацией для разработки про
граммы профилактических работ для данной хроматографической системы или всей
лаборатории.
Проверка системы. Третий раздел может содержать записи о периодических
тестах системы (разделы 3.10.1.2, 3.10.1.3). Имея легкий доступ к этим записям, можно
отслеживать тенденции от одной проверки к другой. С помощью этих записей
можно создавать программы профилактического обслуживания.
Кроме того, можно использовать и другие технологии учета записей. Хорошие
возможности для этого предоставляют компьютерные базы данных или электронные
записные книжки. Важно при этом, чтобы они были достаточно удобными и данные
в них могли бы быть легко введены во время профилактических работ или ремонта.
В противном случае важные записи будут утеряны.
3.10.3.3. Специальные рекомендации по ремонту
В таблице 3.7 приведены различные варианты ремонта, которые могут выполнять
пользователи, имеющие различный уровень подготовки.
Начинающий/Оператор. Персонал, только обучающийся работе на ВЭЖХ-систе-
ме, или работники, выполняющие рутинную работу по разделению образцов и
ничего более, могут иметь ограниченный набор навыков или полномочий для проведения
ремонта. Однако для замены подвижных фаз, фильтров, предколонок и колонок
требуется небольшая подготовка. Избежать ошибок при проведении таких операций
поможет двойная проверка или утверждение документа вторым лицом.
Опытный. По мере того как пользователь приобретает опыт, его навыки по
профилактическому обслуживанию системы и устранению неполадок будут развиваться.
Кто-то на этом пути продвинется дальше, чем другие, но большая часть сотрудников
смогут научиться менять обратные клапаны, уплотнения насосов, капилляры и
соединения. Более сложные работы — такие, как замена ламп в детекторах или
клапанных коробок могут быть закреплены за более подготовленными сотрудниками.
Обученный производителем. Некоторые виды работ должны выполнять
специально обученные люди. Это могут быть обычные сотрудники лаборатории с большим
опытом устранения неполадок и склонностью к такого рода работам, группа
обслуживания внутри компании или работающий по контракту техник
фирмы-производителя оборудования. Неполадки, связанные с электроникой, обычно требуют высокой
квалификации и/или оборудования для их устранения. Устранение другие
неполадок, связанных с разборкой модулей системы или ремонтом ее узлов, требующих
тонкой настройки (таких, как КФГ), тоже может потребовать специальной
подготовки.
184 Глава 3. Оборудование
Литература
1. S. R. Bakalyar, M. Р. Т. Bradley, and R. Honganen, J. Chromatogr., 158 (1978) 277.
2. S. van der Wal and L. R. Snyder, J. Chromatogr., 255 (1983) 463.
3. J. N. Brown, M. Hewins, J. H. M. van der Linden, and J. H. M Lynch, J. Chromatogr., 204 (1981)
115.
4. L. R. Snyder, J. Chromatogr. Sci., 21 (1983) 65.
5. J. Thompson, personal communication, 2008.
6. http://www.vici.com (Valco Instruments).
7. http://www.idex-hs.com/products/Brand.aspx7Brand]D=l (Upchurch Scientific).
8. J. W. Batts, IV, All About Fittings, Upchurch Scientific, Oak Harbor, WA, 2003.
9. Y. Ishihama, J. Chromatogr. A., 1067 (2005) 73.
10. J. Hernandez-Borges, Z. Aturki, A. Rocco, and S. Fanali, J. Sep. Sci., 30 (2007) 1589.
U.S. Stearns, personal communication, 2008.
\2.Technical Notes 5, Rheodyne, Dec. 1983.
13. http://www.idex-hs.com/products/Brand.aspx7BrandlD=8 (Rheodyne).
14. O. Abreu and G. D. Lawrence, Anal. Chem., 72 (2000) 1749.
15. R. G. Wolcott, J. W. Dolan, L. R. Snyder, S. R. Bakalyar, M. A. Arnold, and J. A. Nichols,
J. Chromatogr. A, 869 (2000) 211.
16. J. W. Dolan, J. Chromatogr. A, 965 (2002) 195.
17. P.-L. Zhu, L. R. Snyder, J. W. Dolan, N. M. Djordjeveic, D. W. Hill, L. R. Sander, and T. J. Wa
eghe, J. Chromatogr. A, 756 (1996) 21.
18.Guidance for Industry: Part II, Electronic Signatures—Scope and Application, http://www.fda.gov/
cder/guidance/index.htm (2003).
19. J. J. Gilroy and J. W. Dolan, LCGC, 22 (2004) 982.
20. L. R. Snyder and J. W. Dolan, High-Performance Gradient Eiution, Wiley, Hoboken, NJ, 2007.
ГЛАВА 4
ДЕТЕКТИРОВАНИЕ
4.1. Введение 186
4.2. Характеристики детектора 187
4.2.1. Общие положения 187
4.2.2. Способы детектирования 189
4.2.2.1. Детектирование, основанное на измерении
общей характеристики вещества 189
4.2.2.2. Детектирование, основанное на измерении специфических
характеристик образца 190
4.2.2.3. Детектирование с помощью модификации подвижной фазы ... 190
4.2.2.4. Детектирование «независимыми» приборами 190
4.2.3. Сигнал, шум, дрейф и точность измерений 191
4.2.3.1. Шум и дрейф 191
4.2.3.2. Отношение сигнал/шум (S/N) 194
4.2.4. Пределы обнаружения 196
4.2.5. Линейность 198
4.3. Общее предисловие к различным типам детекторов 198
4.4. Детекторы, регистрирующие поглощение в УФ и видимой областях
спектра света 198
4.4.1. Детекторы с фиксированной длиной волны 201
4.4.2. Детекторы с переменной длиной волны 202
4.4.3. Диодно-матричные детекторы 203
4.4.4. Общие характеристики УФ-детекторов 205
4.5. Флуоресцентные детекторы 206
4.6. Электрохимические (амперометрические) детекторы 209
4.7. Детекторы радиоактивности 211
4.8. Кондуктометрические детекторы 212
4.9. Хемилюминесцентный азотный детектор 213
4.10. Хиральные детекторы 215
4.11. Рефрактометрические детекторы 216
4.12. Детекторы светорассеяния (ИДС) 219
4.12.1. Испарительный детектор светорассеяния 220
4.12.2. Детектор светорассеяния на центрах (ядрах) конденсации (ДСЦК) . . . 222
4.12.3. Лазерные детекторы светорассеяния (ЛДС) 222
4.13. Детектор заряженных аэрозолей 223
4.14. Масс-спектрометрические детекторы (МС) 224
4.14.1. Интерфейсы МС-детекторов (способы ионизации образца) 225
4.14.1.1. Ионизация электрораспылением (ИЭ) 225
4.14.1.2. Химическая ионизация при атмосферном давлении (ХИАД) . . 226
4.14.1.3. Другие модификации интерфейса МС-детекторов 227
4.14.1.4. Соображения относительно скорости потока 227
4.14.2. Квадруполи и ионные ловушки 227
4.14.3. Другие виды МС-детекторов 229
4.15. Другие «независимые» детекторы 230
4.15.1. Инфракрасная спектроскопия с Фурье-преобразованием (ИКФП) . . . 230
4.15.2. Ядерный магнитный резонанс (ЯМР) 231
4.16. Дериватизация образца и реакционные детекторы 233
Литература 235
Глава 4. Детектирование
4.1. Введение
В первых классических жидкостных разделениях Цвету в качестве детектора служили
собственные глаза [1, 2|. Многие годы количественный и качественный анализы
выполняли следующим образом: сначала собирали фракции элюата {рис. 4.1а), а потом
их анализировали традиционными химическими методами анализа растворов,
гравиметрическим, оптическим и различными другими методами. График, на который
наносилась концентрация анализируемого вещества в каждой фракции в зависимости
от номера фракции (рис. 4.16) напоминает примитивную хроматограмму. Сбор
фракций и их последующий анализ отнимает много времени, как правило, ухудшает хро
матографическое разрешение и вообще неудобен. Анализ в режиме реального
времени отличается целым рядом преимуществ. Одними из первых применили
детектирование в режиме реального времени Циелиус (он использовал
рефрактометрический детектор |3|), а также Мартин и Рэндалл (детектирование
электропроводности |4|). В 1966 году Хорват и Липски |5| ввели в хроматографическую практику
УФ-детектор. С тех он остается самым популярным детектором ВЭЖХ.
В 1968 году Киркленд усовершенствовал УФ-детектор. Проточная ячейка и
ртутная лампа существенно повысили чувствительность прибора (интенсивность шума со
ставила 1 х 10 AU). В 70-х годах созданные на основе этого принципа детекторы с
одной длиной волны (254 нм) были самыми популярными до тех пор, пока их
не вытеснили детекторы с переменной длиной волны и диодно-матричные детекторы.
Хроматографический детектор представляет собой устройство, преобразующее
физические или химические свойства элюированного из колонки вещества в
электрический сигнал, который может быть соотнесен с концентрацией анализируемого
вещества. В то время, когда ГХ уже второй десяток лет эти свойства активно
использовала, ВЭЖХ только начинала их применять. Это привело к тому, что и детекторы
для ГХ развивались с опережением. В 1960-70-х годах было предпринято много
попыток адаптировать ГХ-детекторы для ВЭЖХ. Существовало несколько детекторов
для ГХ, основанных на принципах пламенной ионизации |7, 8|, пламенной
фотометрии |9| и электролитической проводимости |10|. Однако для того, чтобы
адаптировать ГХ-детектор для ВЭЖХ, нужно перевести подвижную фазу в парообразное
состояние. Отработать надежную в эксплуатации методику этого преобразования
не удавалось до разработки электроспрея в ЖХ-МС. В результате эксплуатация
ВЭЖХ-детекторов, созданных на основе ГХ-детекторов, была непростой. И только
(а)
о=
(б)
т
Ш
Щ
4
ЕЙ
S5 Щ
РФ
тг
ий
и
и
и
►и
анализ в
автономном j-
' режиме 3
tbd
ш
номер фракции
Рис. 4.1. (я) Открытая ЖХ-колонка, (б) график, на котором указаны значения
концентрации анализируемого вещества в каждой фракции, элюированной с
колонки (а)
4.2. Характеристики детектора 1871)1
после изобретения эффективного способа распыления детекторы, переводящие
жидкую фазу в газообразную (масс-спектрометрический детектор, испарительный
детектор светорассеяния, детектор заряженных аэрозолей, инфракрасный
спектрофотометр с Фурье-преобразованием), стали удобными в работе.
Несмотря на отсутствие универсального чувствительного детектора такого, как,
например, пламенно-ионизационный детектор (ПИД) в ГХ, в ВЭЖХ есть множество
различных детекторов, успешно использующихся в общих или специальных методах.
В этой главе описаны принципы работы большинства популярных ВЭЖХ-детекто-
ров, приводятся примеры их применения и проводится сравнение достоинств и не
достатков специализированных детекторов, где это целесообразно.
4.2. Характеристики детектора
Во втором издании этой книги |11| перечислены девять характеристик идеального
ВЭЖХ-детектора:
• высокая чувствительность и предсказуемый отклик;
• реакция на все анализируемые вещества или. напротив, предсказуемый отклик
на специфические свойства элюата;
• нечувствительность к перепадам температуры и изменению состава подвижной
фазы;
• независимость отклика от состава подвижной фазы;
• не должен вносить вклад во внеколоночное уширение пиков;
• должен быть надежным и удобным в эксплуатации;
• отклик должен расти линейно с увеличением количества анализируемого вещества;
• не должен разрушать анализируемые соединения;
• должен давать качественную информацию о детектируемом пике.
В настоящее время, конечно, не существует детектора, который бы сочетал все
эти характеристики. И в ближайшем будущем не предвидится. Однако у самых
распространенных ВЭЖХ-детекторов есть большинство этих качеств.
4.2.1. Общие положения
ВЭЖХ-детекторы устанавливают сразу же после колонки (рис. 4.2) и таким образом
сводят к минимуму размывание пика на выходе из колонки (раздел 3.9). Элюат из
колонки направляется в ячейку детектора, где и происходит детектирование. В
большинстве детекторов подвижная фаза остается в жидком состоянии в процессе
детектирования, а элюат из ячейки детектора направляется в слив, коллектор фракций или
другой детектор. Проточная ячейка УФ-спектрофотометра в ходе своего развития
претерпела изменения, и в настоящее время она выглядит так, как показано на рис. 4.3а.
Подвижная фаза проходит по Z-образному каналу, а УФ-излучение проникает через
кварцевое окно на одном из торцов ячейки. На другом ее торце с помощью
фоточувствительного диода отслеживается разница в интенсивностях опорного луча света и
луча, прошедшего через ячейку; затем с помощью системы сбора и обработки данных
эта разница преобразуется в электрический сигнал и далее в хроматограмму. У разных
детекторов свои особенности конструкции проточных ячеек. Об этом будет сказано
далее в разделах, посвященных определенным детекторам.
Оптические детекторы — такие, как ультрафиолетовые и особенно
рефрактометрические детекторы, обычно чувствительны к малым колебаниям температуры
подвижной фазы. Эти колебания приводят к изменениям показателя преломления,
а значит, меняется и количество проходящего через ячейку света. Для стабилизации
Глава 4. Детектирование
Рис. 4.2. Схема расположения основных узлов ВЭЖХ-системы: (а) емкость для
подвижной фазы, (б) насос, (в) автосамплер или инжектор, (г) колонка, (д)
детектор, (е) система сбора и обработки данных
(а)
крышка
корпус ячейки кварцевое стекло
* \ уплотнение'
световой
поток
световой
поток
ШШштттжщ
vg*-vt^ >->---- <*&- <»•- <■•>■.■■>»-■. -^
(б)
ВХОД
корпус ячейки
^ ^U
WWWWWWWWW^
щщттттттт^
■-'Г -"-У -У- -г- -V -^ -^ х--' -<-
поток
внутреннее
светоотражающее
покрытие
^У ту V2? ^У -У ЧУ flT '^Г- 'Ж "Sk
У^ '*2> Sii 2S* &■ S3* ■&■■ ^- i& Si
^2* ■?& S& t&- ■&' *4* ^- Ъ&* &■ 52
1&- ^ •&- Ъ?' ^ Si"* "*$* $ё^ ^$* Й5
■&& tt*r ?Sv ?£v у§£ з1^ <5> #v^ ffS^" <S
Рис. 4.3. Проточные ячейки УФ-детекторов. (с) Типовая конструкция Z-образной
проточной ячейки; (6") световой путь проточной ячейки с внутренней
отражающей поверхностью
4.2. Характеристики детектора 189))|
температуры ячейка в корпусе детектора устанавливается там, где нет сквозняков,
а иногда и в помещении с контролируемой температурой. Конструкция детектора
обычно включает капиллярный теплообменник для стабилизации температуры
поступающей подвижной фазы. Одно из распространенных исполнений такого тепло
обменника — капилляр, навитый вокруг корпуса ячейки детектора и залитый вместе
с ней теплопроводящим герметикой.
Детекторы, проводящие измерения в жидкости, могут быть восприимчивы к
оптическим или электрическим помехам, вызванным наличием пузырьков. Чтобы
предотвратить образование пузырьков в детекторе, обычно достаточно дегазировать
подвижную фазу (раздел 3.3). В качестве дополнительной меры предосторожности
помогает небольшое избыточное давление на выходном капилляре. Для этих целей
авторы книги рекомендуют приобрести у поставщиков ВЭЖХ-оборудования
подпружиненный обратный клапан. Если вы его используете, будьте осторожны: не
превышайте максимально допустимое для ячейки детектора давление. Например, при
максимально возможном давлении в ячейке 10 бар (150 psi), регулятор избыточного
давления должен давать сопротивление в 3,5—5,0 бар (50—75 psi). Некоторые
хроматографисты используют в качестве регулятора избыточного давления отрезок
капилляра (например, длиной 1 м с внутренним диаметром 0,005 дюйма). Однако
давление, создаваемое таким образом, пропорционально скорости потока. То есть
увеличение скорости потока непроизвольно может привести к повышению
избыточного давления, что может стать причиной утечек или вызвать повреждение ячейки.
4.2.2. Способы детектирования
Существует четыре способа ВЭЖХ-детектирования':
• измерение какой-либо общей характеристики вещества и определение ее
изменения в присутствии образца (дифференциальное измерение);
• измерение специфических характеристик образца;
• с помощью модификации подвижной фазы;
• «независимыми» приборами.
4.2.2.1. Детектирование, основанное на измерении
общей характеристики вещества
Такой детектор можно считать универсальным, поскольку он измеряет некоторое
свойство, которое является общим для всех соединений. Детектор измеряет разность
значений, характеризующих это свойство подвижной фазы, содержащей образец и
его не содержащей. Самым популярным детектором такого рода является
рефрактометрический (раздел 4.11). У подобных детекторов есть преимущество: они
детектируют все соединения. Универсальность таких детекторов, в то же время, является их
недостатком, поскольку все вещества, элюируемые из колонки, дают вклад в наблю-
' Предлагаемая авторами классификация способов детектирования представляется
избыточной, так как некоторые детекторы можно отнести к двум или трем категориям. Например, МС-де-
текторы анторы относят одновременно к детекторам, модифицирующим подвижную фазу, и
«независимым» приборам, записываемым через дефис после сокращения ЖХ (жидкостная
хроматография). Принцип же детектирования масс молекулярных ионов, используемый в МС-детекторе,
соответствует измерению специфических характеристик (свойств) анализируемого вещества.
Таким образом, для детектирования используется два основных подхода: измерение интегральных
(общих) характеристик подвижной фазы и анализируемого вещества (с последующим
вычитанием фона подвижной фазы) и измерение индивидуальных характеристик анализируемого
вещества. — Прим. персе.
190 Глава 4. Детектирование
даемый сигнал, и может потребоваться дополнительная хроматографическая
селективность, чтобы компенсировать отсутствие селективности при детектировании. Как
правило, универсальные детекторы менее чувствительны, поскольку их отклик
определяется по разности двух больших величин, измеренных соответственно для
растворителя и совокупности растворителя и анализируемого соединения.
4.2.2.2. Детектирование, основанное на измерении
специфических характеристик образца
Некоторые свойства образца уникальны или, по крайней мере, не присуши всем
анализируемым веществам, и детекторы реагируют на эту уникальность. Самым
популярным из таких детекторов является УФ-детектор. Он регистрирует вещества,
поглощающие УФ-излучение с определенной длиной волны. Различие между
детекторами, основанными на измерении свойств, присущих всем веществам, и
специализированными детекторами несколько расплывчато: например, при малой длине
волны (< 210 нм) все органические соединения в некоторой степени поглощают
ультрафиолетовое излучение, поэтому УФ-детектор становится менее
специализированным и более универсальным на малых длинах волн. Принцип действия других
широко используемых детекторов, регистрирующих вещества по их специфическим
свойствам, основывается на способности веществ флуоресцировать (флуоресцентный
детектор, раздел 4.5), проводить электричество (кондуктометрический детектор,
раздел 4.8), или вступать в реакции окисления/восстановления в специфических
условиях (электрохимический детектор, раздел 4.6).
4.2.2.3. Детектирование с помощью модификации подвижной
фазы
Детекторы этого типа модифицируют элюат таким образом, что меняются свойства
анализируемого вещества. Под модификациями элюата подразумеваются
специфические химические реакции с анализируемым веществом в жидкой фазе (реакционные
детекторы, раздел 4.16), в газовой фазе (например, детекторы заряженных аэрозолей,
раздел 4.13; масс-спектрометрические детекторы, раздел 4.14) или получение частиц
анализируемого вещества, суспендированных в газовой фазе (например,
испарительные детекторы светорассеяния, раздел 4.12.1).
4.2.2.4. Детектирование «независимыми» приборами
В некоторых случаях для детектирования к ВЭЖХ-системе присоединяются
«независимые» аналитические приборы1. Обычно полное название таких систем строится
по схеме: ЖХ-(аббревиатура метода детектирования). Самой популярной системой
является ЖХ-МС (раздел 4.14), в которой к жидкостному хроматографу присоединен
масс-спектрометр. К другим менее распространенным системам относятся ЖХ-ИК
или ЖХ-ИКФС (раздел 4.15.1), ЖХ-ЯМР (раздел 4.15.2). Можно предположить, что
детектор, записываемый через дефис, в какой-то момент перестанет считаться
самостоятельным прибором, не зависимым от системы ВЭЖХ. Как правило, когда
прибор, используемый для детектирования, приобретает большую популярность, и его
цена становится сопоставимой с ценой хроматографа, его начинают рассматривать
как один из узлов системы ВЭЖХ. Вполне можно предположить, что
масс-спектрометры, которые в настоящее время в 2—5 раз дороже ВЭЖХ-системы, со временем
станут еще одним видом детекторов для ВЭЖХ.
' Независимыми эти приборы, «записываемые через дефис», как они именуются в английском
оригинале, вряд ли можно считать. Для присоединения к системам ВЭЖХ для них были
разработаны специальные интерфейсы, не позволяющие или сильно осложняющие введение пробы, минуя
систему ВЭЖХ. То есть использовать их как автономные приборы затруднительно. — Прим. перев.
4.2. Характеристики детектора "О)}
4.2,3. Сигнал, шум, дрейф и точность измерений
Способность детектора регистрировать точные и достоверные количественные
данные зависит от интенсивности сигнала, генерируемого анализируемым веществом,
фонового шума и, в некоторой степени, от дрейфа базовой линии. Более подробно
эти темы раскрыты в разделе 11.2.4.
4.2.3.1. Шум и дрейф
Высокочастотный шум — это всплески базовой линии в интервале времени < 1 минуты
(см. вставка на рис. 4.4я). Высокочастотный шум измеряют вручную как интервал
максимальных отклонений базовой линии (на рис. 4.4д область между пунктирными
линиями, см. также рис. 4.7), выраженный в соответствующих единицах (например,
для УФ-детектора в единицах поглощения). Высокочастотный шум можно определить
и с помощью программного обеспечения. Если эта опция доступна, то при ее
активизации обычно вычисляется среднее квадратическое отклонение базовой линии.
Основной вклад в высокочастотный шум обусловлен «возмущениями», создаваемыми
устройствами, электропитание которых объединено одной электрической сетью'
(так называемые импульсные помехи, рис. 4.5). Частота импульсных помех
соответствует частоте сети переменного тока 60 Гц (в США) или 50 Гц (в других странах).
(а)
Время (мин)
(б)
3 с"
- 4-
0 10 20 30 40 50 60 70 80 90
Время (мин)
Рис. 4.4. Шуи и дрейф детектора, (а) показан шум и дрейф базовой линии, вставка
с увеличением — высокочастотный шум, (б) низкочастотный шум
1 Речь идет о так называемых наводках в электрической сети, которые могут вызывать как
монотонный шум с мапой амплитудой, подобный показанному на рис. 4.4а между пунктирными
линиями, так и помехи типа кратковременных выбросов из-за скачков напряжения. — Прим. персе.
192 Глава 4. Детектирование
Высокочастотный шум можно
снизить или вообще устранить с помощью
электрического фильтра шумов. У
большинства детекторов есть встроенный
фильтр шумов, который оператор может
настроить сам. С другой стороны, можно,
руководствуясь базовыми законами
физики, своими руками сделать
сглаживающий частотный фильтр, купив недорогие
детали у RadioShack1.
Пример эффективного использова
ния электрического фильтра приведен
на рис. 4.5, на котором видно, что шум
необработанного сигнала удалось
уменьшить примерно в 300 раз благодаря час
тотному фильтру с постоянной
времени равной одной секунде |12|. Если
постоянная времени слишком велика, будет
уменьшаться и сам сигнал. Как
правило, если постоянная времени меньше 10%
от ширины пика, это никак не
отражается на пике вещества на хроматограмме.
Например, при использовании колонки
150 х 4,6 мм {5 мкм) с N = 10000 при
скорости потока 1,5 мл/мин и к = 2 пики
будут шириной примерно 7 с,
следовательно, приемлемая постоянная времени
будет < 0,7 с.
Кроме того, высокочастотный шум
может быть обусловлен и частотой сбора
данных системой (раздел 11.2.1.1).
Обратимся к рис. 4.6: показаны графики базовой линии при частоте сбора данных 15 Гц
(точек в секунду) и для сравнения при частоте I Гц |12|. Шум должен уменьшаться
обратно пропорционально квадратному корню из отношения частот сбора данных.
Значит, при переходе от частоты сбора данных 15 Гц (рис. 4.6а) к частоте 1 Гц
(рис. 4.66) шум должен понизиться примерно в 4 раза, что и наблюдается на рис. 4.6.
Шум (рис. 4.6) упал с = 12 до » 3 мкВ, хотя, глядя на рисунок, кажется, что
подавление шумов намного больше. Это может быть объяснено тем, что высокочастотный
шум, присутствующий в первом случае (рис. 4.6а), практически устранен во втором
(рис. 4.66). Существует эмпирическое правило для задания частоты сбора данных:
на пик должно приходиться не менее 20 зарегистрированных точек. Вернемся к
ранее рассмотренному примеру с N= I0000 и к = 2 (ширина пика примерно 7 с):
оптимальная частота сбора данных будет равна < 7/20 с или >20/7 с-1 » 3 Гц. Запомните:
лучше при запуске анализа изначально установить более высокую частоту сбора данных;
при дальнейшей обработке ее молено будет уменьшить, однако не существует
корректных способов ее увеличения (раздел 11.2.1.1).
Для того, чтобы сгладить шумы базовой линии, можно варьировать как
постоянную времени детектора (фильтр шума), так и частоту сбора данных системой (или
скорость отбора данных при обработке), но механизм сглаживания в каждом из этих
'Американская компания, специализирующаяся на розничной продаже электроники. —
Прим. персе.
исходный
сигнал
О 5 10
Время (мин)
Рис. 4.5. Влияние частотного фильтра
на шум детектора. Высокочастотный
шум необработанного сигнала составляет
~ 16 mAU. Когда была задана
постоянной времени I с, шум снизился до уровня
< 1 mAU
4.2. Характеристики детектора
ш
2
!
(б)
1—i—m—i i i i—i i i i—гп—п—гт
4 15-i
| 10-
-5-
-10-
-15
"1—I—Г
тп—i i i i—i i i i—ги—r~i—i-г-1
1 2 3 4 S
Время (мин)
Рис. 4.6. Влияние скорости сбора данных на шум детектора. Базовая линия
зарегистрирована при частоте (с) 15 Гц и (б) 1 Гц [12)
случаев будет разным. Поэтому стоит рассмотреть оба метода (отдельно или
совместно), чтобы определить, как эффективнее подавить шум — каким-то одним методом
или их комбинацией.
Низкочастотный шум — это колебания базовой линии, период которых того же
порядка величины, что и ширина хроматографических пиков (рис. 4.45). Если
для шумов характерен больший период, то в борьбе с ними не поможет введение
электрического фильтра в схему прибора. Появление такого рода шума часто говорит
об элюировании хорошо удерживаемых веществ из предыдущих разделений. По мере
возрастания удерживания ширина пика увеличивается и, в конце концов, пик
размывается до такой степени, что начинает выглядеть как базовая линия, а,
следовательно, приводить к ее искажению. Чтобы устранить низкочастотный шум, обычно
достаточно промыть колонку сильным растворителем (например, 100% ацетонитрилом
или метанолом).
Дрейфе базовой линии представляет собой постепенное смещение базовой линии
в течение одного или нескольких разделений (на рис. 4.4а он показан жирным
пунктиром). Дрейф, как правило, характерен для градиентных разделений и возникает
из-за различия в отклике детектора на подвижную фазу в начале и в конце
разделения (раздел 17.4.5.1). Откликом детектора называется величина сигнала детектора
(например, величина УФ-поглошения), пропорциональная концентрации вещества.
Дрейф базовой линии при УФ-детектировании можно компенсировать, добавив
в подвижную фазу с меньшей абсорбцией неудерживаемое и поглощающее в УФ-об-
ласти вещество (стр. 178 в книге |13|). Колебания температуры подвижной фазы
также могут стать причиной дрейфа базовой линии, особенно это касается
рефрактометрических детекторов и других детекторов, чувствительных к изменениям
показателя преломления. Тщательный контроль температуры колонки, капилляров и
детектора, а также устранение сквозняков в помещении, где установлена ВЭЖХ-сис-
тема, позволит свести к минимуму дрейф базовой линии. Пока система сбора и
обработки данных может адекватно интегрировать пики, умеренный дрейф не влияет
на качество результатов.
1 Природа дрейфа базовой линии рассмотрена также в главе 11. — Прим. перев.
194 Глава 4. Детектирование
Высокочастотный и низкочастотный шумы накладываются на дрейф базовой
линии (рис. 4.4я). Чтобы сгладить шумы и дрейф, рекомендуется, во-первых,
установить оптимальные значения постоянной времени детектора и частоты сбора данных,
во-вторых, регулярно промывать колонку сильным растворителем, в-третьих,
минимизировать колебания температуры внутри и около ВЭЖХ-системы. Помните, что
небольшой дрейф базовой линии в градиентных разделениях — обычное явление,
поскольку отклик детектора для растворителей А и В различен, и это различие
нельзя устранить приведенными выше способами.
4.2.3.2. Отношение сигнал/шум (S/N)
Отношение сигнал/шум гораздо более важный параметр, чем сигнал или шум,
взятые в отдельности. Именно оно в большей степени влияет на точность оценок
параметров конкретного пика, полученного с помощью детектора. Шум, изображенный
на рис. 4.7, измеряют так, как описано в разделе 4.2.3.1 (для высокочастотного
шума). Высота сигнала1 — это расстояние от середины амплитуды шума базовой
линии до вершины пика (рис. 4.7). Вклад отношения сигнал/шум (S/N) в точность
метода определяют следующим образом:
S/N
где CV— коэффициент вариации (эквивалентный относительному стандартному
отклонению, ОСО, выраженному в процентах). Нижний предел детектирования (НПД)
обычно соответствует отношению S/N =3, при котором CV=: 16%, а нижний предел
количественного определения (НПКО) соответствует S/N = 10, т.е. CV' = 5% (см.
также раздел 11.2.4). Эти значения СУ определяют вклад S/N в общую погрешность
метода, поэтому предполагается, что общая точность метода будет меньше, чем вклад
S/N. До тех пор пока погрешность, связанная с S/N (или любым другим отдельным
источником ошибок), составляет менее половины требуемой погрешности метода,
влияние S/N на общую точность метода будет небольшим (<15%) |см.
обсуждение уравнения (И.2)|. Например, если в методе подразумевается погрешность
не более 2%, то вклад S/N <1% считается удовлетворительным. Это значит, что для
общей погрешности метода менее 2% требуется отношение S/N = 50:1 или больше.
Рис. 4.7. Измерение хроматографического сигнала (.S) и шума (ЛГ)
1 Это определение сформулироиано не совсем точно. Полезный сигнал — это разность
максимального значения наблюдаемого пика и среднего значения шума базовой линии вне области этого
пика. — Прим. перев.
4.2. Характеристики детектора 195
Повысить отношение сигнал/шум можно, увеличив интенсивность сигнала или
уменьшив шум, а также используя оба эти способа (таблица 4.1). Увеличить
интенсивность сигнала данного пика или образца можно, изменив установки детектора,
например, установить ту длину волны УФ-детектора, при которой образец имеет
максимальное поглощение, или использовать более чувствительный детектор такого
же или другого типа.
Таблица 4.1. Способы увеличения отношения сигнал/шум
Способы увеличения сигнала
Установить другую длину волны (или
другие параметры детектора)
Использовать более чувствительный
детектор
Провести дериватизацию образца
Вводить большее количество образца
Уменьшить ширину пика
Уменьшить к
Уменьшить объем колонки
Увеличить число теоретических тарелок
Способы понижения шума
Увеличить постоянную времени детектора
Группировать большее число точек для усреднения
их значений во время сбора данных
Тщательнее контролировать температуру
Использовать более чистые растворители и реагенты
Лучше проводить пробоподготовку
Устранить пульсации потока подвижной фазы
Использовать метод переключения колонок
Дериватизация (раздел 16.12) или другие способы модифицирования
анализируемого вещества могут повысить чувствительность выбранного детектора. Самый
распространенный способ увеличить интенсивность сигнала — анализировать большее
количество образца (либо брать больший объем образца, либо его более
концентрированный раствор, раздел 3.6.3). Однако увеличение количества образца, в конце
концов, приведет к перегрузке колонки или детектора, что ограничит увеличение
сигнала каким-то определенным уровнем. Любое уменьшение ширины пика будет
приводить к увеличению высоты пика (при этом его площадь предполагается
постоянной). С этой целью можно использовать один из вариантов: снизить значения к
(увеличив %Ъ, см. пример на рис. 2.106), использовать более узкую колонку или
более эффективный сорбент с меньшими частицами.
Снижение шума базовой линии любым способом — например, увеличением
постоянной времени детектора или уменьшением частоты сбора данных — может
повысить отношение S/N. При этом излишнее сглаживание может приводить к
уменьшению этого отношения. Тщательный контроль температуры колонки, детектора и
помещения также уменьшает шум, особенно у детекторов, чувствительных к
изменениям показателя преломления. Использование более чистых растворителей
(например, предназначенных для ВЭЖХ) и качественная пробоподготовка образца
позволяют избежать примесей, порождающих шум. Иногда градиентные методы переносят
с одной ВЭЖХ-системы на другую с целью уменьшения мертвого объема
(раздел 9.2.2.4) и времени задержки градиента. В большинстве хроматографов миксер
является основным источником мертвого объема, но уменьшение объема миксера
чревато увеличением шума базовой линии. В некоторых ВЭЖХ-системах для
сглаживания базовой линии и уменьшения шума имеются дополнительные миксеры,
которые можно установить. Это особенно полезно в изократических анализах,
проводимых при максимальной чувствительности детектора. Можно использовать метод
переключения колонок (раздел 16.9), то есть нужную фракцию вещества переносят
196 Глава 4. Детектирование
с одной колонки, используемой для пробоподготовки, на другую, аналитическую
колонку. Таким образом, ненужные примеси направляют в слив и тем самым снижают
шум базовой линии.
4.2.4. Пределы обнаружения
Хотя отношение сигнал/шум характеризует качество сигнала детектора, решающим
фактором, свидетельствующим о пригодности детектора для определенного метода
анализа, обычно является минимальная детектируемая масса или концентрация
вещества. Когда говорят о ВЭЖХ-детекторе, то зачастую термин чувствительность
используют в качестве синонима предела обнаружения. Хотя, если быть точными,
чувствительность — это угол наклона калибровочного графика, то есть изменение сигнала
на единицу изменения концентрации (или массы) вещества. Под пределом
обнаружения подразумевают минимальную концентрацию (или массу) вещества, которую
может зарегистрировать детектор. Отклик детектора зависит или от концентрации
образца в ячейке детектора (например, у УФ-детектора) или от массы образца в
детекторе (например, у МС-детектора).
Пределы обнаружения, более подробно рассмотренные в разделе 11.2.5,
означают следующее: нижний предел детектирования (НПД)1 — минимальный сигнал,
который можно отличить от шума, и при этом быть уверенным, что пик действительно
присутствует. Обычно НПД приравнивают к S/N= 3. Нижний предел количественного
определения НПКО (иногда называемый пределом количественного определения или
предел количественного обнаружения ПКО) — это наименьший сигнал, который можно
измерить в соответствии с требуемой точностью метода. Обычно НПКО
соответствует S/N > 10, но для высокоточных методов выбирают S/N > 50. Существует растущая
потребность во все более низких пределах обнаружения для анализа следовых
количеств вещества. Наряду с этим все большее распространение приобретают
анализы, в которых количество вводимого образца в колонку < 1 нг.
НПД и НПКО напрямую зависят от концентрации (или массы) вещества в ячейке
детектора. Так, чем больше световой путь ячейки УФ-детектора, тем интенсивнее
сигнал. При этом объем ячейки детектора должен быть минимален и совместим
с другими требованиями к детектору. Избыточный объем ячейки приводит к
дополнительному вне колоночному размыванию пиков (раздел 3.9). Это в первую очередь
касается колонок небольшого объема; колонок, упакованных мелкими частицами, и
пиков с к < 2. Например, при разделении вещества с к < 3 на колонке 50 х 4,6 мм,
упакованной 3-мкм частицами, наблюдалось значительное уширение пиков с
ячейкой УФ-детектора объемом 8 мкл по сравнению с ячейкой объемом 1 мкл [14].
Чтобы минимизировать размывание раноэлюируемых пиков, объем ячейки детектора
Ум должен составлять приблизительно десятую часть от объема интересующего нас
пика Vp(Vp = WF, где W — ширина пика на уровне базовой линии в минутах, F —
скорость потока, мл/мин) |15|:
Vdel<%\Vp. (4.2)
(Вклад других факторов в V дан в уравнении (3.1) в разделе 3.9).
В таблице 4.2 приведены примеры зависимости объема раноэлюируемых
пиков VpQ (к = 2) от размеров колонки (рассмотрены несколько популярных размеров
колонок). (В хорошо отлаженных хроматографических системах согласно
уравнениям (2.27) и (3.1) объем элюируемого пика Ур не должен быть существенно
большим, чем VpQ.) В таблице 4.3 перечислены объемы ячеек некоторых УФ-детекторов.
'В литературе также встречается эквивалентный термин «предел обнаружения», ПО. В этой
книге используются оба синонима. — Прим. перев.
4.2. Характеристики детектора 197
В УФ-детекторах (таблица 4.4)
интенсивность сигнала
пропорциональна длине светового пути,
поэтому, чем больше длина светового
пути, тем ниже предел
обнаружения. Однако, у ячеек диаметром
менее 1 мм может возникнуть
проблема потери сигнала,
вызванная рассеянием света в ячейке.
Поэтому у ячеек малого диаметра
(более подробно см. раздел 4.4)
должна быть особая конструкция
(например, с полным внутренним
отражением). Данные таблиц 4.2 и
4.3 говорят о том, что для анализов
на колонках длиной более 100 мм
и диаметром dc = 4,6 мм,
упакованных частицами 5 или 3 мкм,
оптимальной будет стандартная
УФ-ячейка размером 10x0,1 мм
(см. уравнение (4.2)). Любые
другие комбинации колонок с мень-
шими диаметрами или с
меньшими частицами сорбента потребуют
меньших объемов ячеек для того,
чтобы избежать ненужного вне-
колоночного размывания пиков
(обратите внимание на то, что
уравнение (3.1) является
приближенным, поэтому расчет по этому
уравнению, а, следовательно, и
выводы из таблиц 4.2 и 4.3 также
носят приблизительный характер).
Таблица 4.4. Вилы ВЭАХ-детекторов
Селективное
детектирование специфических свойств
образца (разделы 4.4—4.10)
Спектрофотометрический
Флуоресцентный
Электрохимический
Радиоакти вности
Кондуктометрически й
Хемилюминесцентный
азотный
Хиральный
Универсальное детектирование
свойств, присущих всем
веществам (разделы 4.11—4.13)
Рефракто метрический
Светорассеяния
Заряженных аэрозолей
■ Независимые»
приборы для детектирования
(разделы 4.14—4.15)
Масс-спектрометриче-
ский
Инфракрасный
Ядерный магнитный
резонанс
Реакционный
(раздел 4.16)
Реакционный
Таблица 4.2. Среднестатистические объемы пиков V
L (мм)
250
150
100
50
dc (мм)
4,6
4,6
2,1
4,6
2,1
1,0
4,6
2,1
2,1
1,0
dp (мкм)
5
5
3
3
3
3
3
2
3
3
2
2
*io (мк-1)*
212
164
127
26
104
22
5
4
73
15
12
3
"Предполагается, что к = 2; приведенная высота
теоретической тарелки h = 2,5 (см. уравнения 2.27
и 3.1).
Таблица 4.3. Объемы ячеек УФ-детекторов
Длина
светового пути (мм)
10
5
1
Внутренний
диаметр (мм)
1,0
0,5
0,25
1
0,5
1
0,5
Объем
(мкл)
8
2
0,5
4
1
0,8
0,2
198 Глава 4. Детектирование
4.2.5. Линейность
Чтобы проводить количественный ВЭЖХ-анализ (раздел 11.4), отклик детектора
должен соотноситься с количеством присутствующего исследуемого вещества. Если
построить график зависимости отклика на вещество у от его концентрации х, то
простейшее, удобное и наиболее надежное уравнение будет иметь виду = тх, где наклон
т — константа, определяемая как чувствительность. Такая зависимость между
откликом на вещество и его концентрацией называется линейной. Чтобы точнее
определять концентрации вещества в широком диапазоне, желателен широкий линейный
динамический диапазон детектора. То есть тот диапазон концентраций, внутри
которого отклик детектора пропорционален концентрации вещества, например, 10 для
УФ-детекторов. Тогда в одном анализе можно будет определять, как основные
вещества, так и следовые количества примесей. Например, в методе определения
стабильности субстанции должны быть указаны пики, площади которых составляют > 0,1%
от площади пика субстанции (эта площадь принимается за 100%). Для этого
линейный диапазон детектора должен составлять, по крайней мере, 100/0,1 = 103. У
некоторых детекторов (например, светорассеяния) узкий линейный диапазон в 1-2
порядка. Если с изменением концентрации (или массы) вещества отклик детектора
меняется предсказуемо, можно использовать нелинейную калибровочную кривую
(например, квадратичную), хотя это не очень удобно и не очень достоверно.
4.3. Общее предисловие к различным типам
детекторов
В оставшейся части этой главы (разделы 4.4-4.16) будут рассмотрены все виды
наиболее популярных на сегодняшний день ВЭЖХ-детекторов. В таблице 4.4 детекторы
сгруппированы по принципам, положенным в основу обнаружения веществ
(селективное измерение характеристик образца, универсальное измерение общих свойств
образца и подвижной фазы). В каждой группе детекторы расположены в порядке
убывания популярности. Обзор начинается с детекторов, измеряющих некоторые
специфические свойства образца, и заканчивается реакционными детекторами. В
заключение коротко упомянуты редко используемые детекторы. Внутри каждого
раздела сначала излагается материал о принципах работы детекторов, а в конце
приводятся примеры методов, в которых они используются. Там, где это целесообразно,
проводится сравнение детекторов.
Более подробное изложение материала о том или ином детекторе выходит
за рамки этой книги. Дополнительную информацию о ВЭЖХ-детекторах можно
найти в ссылках, приводимых в каждом разделе, и в работах [16, 17|.
4.4. Детекторы, регистрирующие поглощение
в УФ- и видимой областях спектра света
Наиболее широко используемыми детекторами в современной ВЭЖХ являются
фотометры, принцип действия которых основан на поглощении ультрафиолетового
(УФ) и/или видимого света (УФ/ВИД фотометр). Такие детекторы отличаются
высокой чувствительностью ко многим анализируемым веществам, но эти вещества
должны поглощать излучение в УФ- или в видимой областях электромагнитного спект-
4.4. Детекторы, регистрирующие поглощение в УФ- и видимой областях спектра 199
света 4^g
pa (например, в диапазоне 190—600 нм). Концентрация вещества в проточной ячейке
связана с долей света, прошедшего через ячейку, в соответствии с законом Бера:
l°e(yl = abc' <4-3>
где fg — интенсивность падающего света, / — интенсивность прошедшего через
раствор света, £ —коэффициент молярного поглощения (или коэффициент молярной
экстинкции), Ь — длина оптического пути1 ячейки в см, с — концентрация вещества
в моль/л. УФ-детекторы для ВЭЖХ обычно на выходе регистрируют поглощение,
которое прямо пропорционально концентрации вещества в проточной ячейке:
А = '«В [у] = ейс, (4.4)
где А — поглощение.
Правильно сконструированный УФ-детектор не очень чувствителен к
перепадам в скорости потока и температуры. В настоящее время в продаже имеются
УФ-фотометры, диапазон линейности которых больше 2 единиц оптической
плотности на всю шкалу (AUFS) при уровне шума I x 10~5AU2. При такой
эффективности можно детектировать вещества с относительно низкой поглощающей
способностью, а также количества порядка нанограмм веществ со средним поглощением
в УФ-области. Кроме того, широкий диапазон линейности УФ-детекторов (j= 105)
дает возможность измерения в одном анализе как следовых, так и основных
компонентов пробы.
Обычно в УФ-детекторах установлена проточная ячейка Z-образной формы
(рис. 4.3д) диаметром 1 мм и длиной 10 мм (объем такой ячейки примерно 8 мкл).
Такая ячейка подходит для колонок длиной 100 мм и больше и диаметром от 4,6 мм,
упакованных частицами i 3 мкм (раздел 4.2.4). Если же эксперименты проводят
на колонках меньшего объема и/или с частицами меньшего размера, а в детекторе
установлена ячейка объемом 8 мкл, то возможно нежелательное внеколоночное
размывание пиков. Уменьшение длины светового пути будет приводить к уменьшению
объема ячейки, но при этом нужно учитывать, что интенсивность сигнала
пропорциональна длине светового пути (уравнение 4.4). То есть при выборе размеров ячейки
придется идти на компромисс между чувствительностью определения и внеколоноч-
ным размыванием пиков. Если внутри ячейки меняется показатель преломления
(например, в процессе градиентного элюирования), то количество света, достигающего
фотоприемник, тоже меняется. Когда пучок света падает на стенку проточной
ячейки, отношение интенсивности отраженного света к интенсивности поглощенного
света зависит от отношения показателей преломления подвижной фазы и стенки
ячейки (и угла, под которым пучок света падает на стенку ячейки). Влияние
показателей преломления, а также несовершенство изготовления оптических конструкций
затрудняют эффективное использование ячейки диаметром менее 1 мм. Чтобы
уменьшить влияние этих факторов, разработали специальную технологию (рис. 4.36):
'Термин «длина оптического пути» некорректен с точки зрения физики. В оптике оптической
длиной пути называется произведение геометрической длины пути световой волны в данной среде
на абсолютный показатель преломления этой среды. Однако в хроматографии принято
использовать указанный термин, характеризуя параметры ячеек детекторов. — Прим. перев.
^AU — (absorbance unit) единица абсорбции; AUFS — (absorbance units per full scale)
(количество) единиц оптической плотности на всю шкалу. В русскоязычной литературе для характеристики
поглощения вещества наряду с сокращением AU используется е.п., единицы поглощения. — Прим.
перев.
Щй Глава 4. Детектирование
внутреннюю поверхность ячейки покрыли светоотражающим материалом. Теперь
пучок света, попадая на зеркальную поверхность проточной ячейки, отражается
и доходит до фотоприемника |18, 19]. Такая технология позволяет уменьшить
диаметр ячейки детектора, что приводит к уменьшению ее объема (например,
до 0,25 мм х [0 мм ~ 0,5 мкл) и уменьшению размывания пиков при анализе на
колонках очень малого объема или колонках, упакованных мелкими частицами. С
другой стороны, можно использовать более длинную и более узкую ячейку (т.е.
увеличить параметр Ь в уравнениях (4.3) и (4.4)); объем такой ячейки меньше, а
поглощение (чувствительность) будет больше (например, 0,25 мм х 50 мм ~ 2,5 мкл).
Вовсе не обязательно в УФ-детекторе устанавливать длину волны,
соответствующую максимуму поглощения анализируемого соединения. Рассмотрим
гипотетический пример выбора длины волны (рис. 4.8). На рис. 4.8а изображены спектры
поглощения двух соединений А" и У. Их максимумы поглощения в УФ находятся
соответственно при ж 250 нми» 270 нм. На длине волны 280 нм вещество У все еще
сильно поглощает, поэтому площадь пика Y на хроматограмме больше при
одинаковых массах введенных образцов (рис. 4.86). При 260 нм абсорбция веществ А" и К
приблизительно одинакова и, значит, площади пиков на хроматограмме тоже
примерно равны (рис. 4.8в). На еще меньшей длине волны (210 нм) поглощение обоих
веществ еще сильнее, и пики на хроматограмме становятся еще больше (рис. 4.8г).
Обратите внимание на появление нового пика Z, которого ранее на предыдущих хро
матограмма при других длинах волн не было. Такая тенденция, при которой со
снижением длины волны возрастает общая чувствительность, и стала одной из причин
широкого применения детектирования на длинах волн 220 нм и меньше
(практически универсальное детектирование). Соответствующая потеря селективности
детектора на низких длинах волн может быть недостатком для некоторых разделений,
в которых, возможно, нежелательно «видеть» определенные составляющие образца
(например, компоненты матрицы образца).
Спектрофотометрические детекторы регистрируют сигналы при любых длинах
волн УФ и видимого диапазона (например, 190—600 нм), что позволяет
анализировать обширный спектр соединений. Практически все ароматические соединения
хорошо поглощают на длинах волн менее 260 нм, соединения с одной или более
двойными связями (например, карбонилы, олефины) можно детектировать при < 215 нм,
а большинство алифатических соединений обладают значительной абсорбцией
при <205 нм. В рутинных анализах, когда детектирование проводят на длине волны
200 нм, используют подвижные фазы для ОФХ, состоящие из ацетонитрила и воды
или фосфатного буфера. Не следует брать метанол, если необходима длина волны
меньше « 210—220 нм (зависимость поглощения от концентрации метанола в элюен-
те дана в таблице 1.2 приложения I). Правильно подобранная подвижная фаза
позволяет работать практически в универсальном режиме в области 200—215 нм, в
которой большинство органических соединений имеют некоторую поглощающую
способность. Разница в абсорбции воды (или фосфатного буфера) и ацетонитрила
при > 200 нм или метанола при > 220 нм незначительная, поэтому УФ-детекторы
идеально подходят для градиентных экспериментов. Если же УФ-поглощение
растворителей, входящих в состав подвижной фазы, сильно различается (например,
вода и ТГФ при < 240 нм), то их не следует использовать в градиентном элюиро-
вании.
Существует три конфигурации УФ-детекторов. Детекторы с фиксированной
длиной волны (раздел 4.4.1) позволяют определить поглощение при одной определенной
длине волны, излучаемой лампой. В детекторах с переменной длиной волны
(раздел 4.4.2) и диодно-матричных детекторах (раздел 4.4.3) можно выбрать одну или
несколько длин волн из полного спектра света, излучаемого лампой.
4.4. Детекторы, регистрирующие поглощение в УФ- и видимой областях спектра 201
света
(б)
210 220 230 240 250 260 270 260
Длина волны (нм)
280 нм
0 1 2 3 4 5
Время (мин)
Рнс. 4.8. Селективность по длине волны в УФ-детекторах. (а) Спектр поглощения
двух гипотетических соединений X и Y. Хроматограммы при (6) 280 нм,
(в) 260 нм, (г) 210 нм
4.4.1. Детекторы с фиксированной длиной волны
На рис. 4.9 изображена общая схема УФ-детектора с постоянной длиной волны.
До появления детекторов с переменной длиной волны и диодно-матричных
детекторов именно эти приборы в основном использовались для регистрации УФ-погло-
шения. На сегодняшний день они утратили свою актуальность. В настоящее время
202 Глава 4. Детектирование
щ\ щ^
и н
фильтр I
пампа светоделитель проточные ячейки фотоэлемент
для анализируемого
образца и стандарта
Рнс. 4.9. Схема УФ-дстектора с фиксированной длиной волны. Пунктиром указан ход
луча света
детекторы с постоянной длиной волны привлекают своей дешевизной и простотой
конструкции и поэтому применяются главным образом при обучении студентов или
в организациях с ограниченным бюджетом.
УФ-излучение ртутной лампы низкого давления с длиной волны 254 нм
проходит через полосовой фильтр, светоделительную призму и попадает в проточную
ячейку. Свет, прошедший через проточную ячейку, попадает в фотоприемник
(обычно фотодиод) и преобразуется в электрический сигнал. Большинство детекторов
работают в режиме дифференциального поглощения, когда свет проходит также через
ячейку сравнения. Разность интенсивностей света, прошедшего через измерительную
ячейку и ячейку сравнения, преобразуется в поглощение в соответствии с
уравнением (4.4). Хотя у некоторых детекторов ячейку сравнения и можно заполнить
подвижной фазой, чаще всего в ней в качестве среды сравнения используют воздух, что
позволяет вносить поправку на изменения интенсивности света лампы-источника,
но не на изменения поглощения подвижной фазы.
Чаще всего длина волны таких УФ-детекторов — 254 нм, причем источником
света, как правило, служит ртутная лампа низкого давления. Несмотря на то, что нет
реальной причины пользоваться именно этой длиной волны, исторически
сложилось, что эта длина волны популярна и по сей день в методах, где указаны детекторы
с переменной длиной волны и диодно-матричные детекторы. Благодаря
использованию других ламп (например, цинковых), люминофоров, или других спектральных
линий ртутных ламп, детектор с постоянной длиной волны позволяет регистрировать
сигнал на другой длине волны, например, 214, 220, 280, 313, 334 и 365 нм.
4.4.2. Детекторы с переменной длиной волны
УФ-спектрофотометры (детекторы с переменной длиной волны и диодно-матричные
детекторы) позволяют выбрать любую длину волны в УФ- и видимом диапазонах.
Такие приборы универсальны и способны работать при максимуме поглощения
вещества или при той длине волны, которой соответствует максимальная
селективность. Кроме того, есть возможность изменить длину волны непосредственно
во время анализа.
В настоящее время наиболее широко используются детекторы с переменной
длиной волны (рис. 4.10). Свет, излучаемый лампой (обычно дейтериевой) в широком
диапазоне УФ, проходит сквозь щель и попадает на дифракционную решетку.
Дифракционная решетка расщепляет свет на составляющие его компоненты, имеющие
разные длины волн. Затем решетка поворачивается на определенный угол, и свет
с нужной длиной волны (или волн в узком диапазоне их длин), проходя через щель
ъ
4.4. Детекторы, регистрирующие поглощение в УФ и видимой областях спектра
света
фотоэлемент
проточная ячейка
диффракционная
решетка
Рис. 4.10. Схематическое изображение УФ-детектора с переменной длиной волны,
ячейка сравнения не показана. Пунктиром указан ход светового луча
и ячейку детектора, попадает на фотоприемник. Для дифференциальной регистрации
у таких детекторов обычно установлены измерительная ячейка и ячейка сравнения
(раздел 4.4). Чтобы проводить регистрацию пиков в видимой области, вместо дейте-
риевой лампы устанавливают вольфрамовую.
Детекторы с переменной длиной волны можно запрограммировать, чтобы во
время анализа изменялась длина волны. Так, один пик можно зарегистрировать
при 280 нм, а другой — при 220 нм. Несмотря на то, что большинство моделей
детекторов позволяет быстро менять длину волны, чтобы снять УФ-спектр пика,
полученные результаты интерпретировать сложно, и ценность их ограничена, поскольку
концентрация растворенного вещества изменяется во время спектрального сканирования.
4.4.3. Диодно-матричные детекторы
Схема диодно-матричного детектора (ДМД, также называемого детектором с
фотодиодной матрицей, ДФДМ) изображена на рис. 4.11. Длина оптического пути ячейки
у ДМД такая же, как и у детектора с переменной длиной волны, но, в отличие от
последнего, белый свет, излучаемый лампой, пройдя через проточную ячейку,
направляется на дифракционную решетку. Дифракционная решетка разлагает белый свет
в спектр, компоненты которого пространственно локализуются в зависимости от их
длин волн, и далее этот спектр попадает на массив фотодиодов, называемый фотоди-
однои матрицей (ФДМ). Количество фотодиодов зависит от фирмы-производителя и
модели детектора, но стандартными являются количества 512 и 1024. Сигналы
каждого фотодиода обрабатываются и формируют спектр растворенного вещества.
Поскольку спектры формируются одновременно (в отличие от регистрации на одной
длине волны в детекторах с переменной длиной волны), ДМД можно использовать
для идентификации пиков. Кроме того, используя ДМД, можно в течение одного
разделения или собирать данные на одной или нескольких длинах волн, или снимать
полный спектр одного или нескольких веществ. Разумеется, размер файла с
данными анализа, содержащего полные спектры, гораздо больше обычного, но
современные методы сжатия файлов и недорогие способы хранения данных позволяют обойти
эту сложность, что раньше было почти невозможно.
204 Глава 4. Детектирование
фотодиодная
матрица
^
проточная ячейка
диффракционная
решетка
Рис. 4.11. Схематичное изображение диодно-матричного УФ-детектора. Пунктирными
линиями обозначен ход светового луча
Если у двух пиков с близкими временами удерживания достаточно отличаются
спектры, то этим можно воспользоваться для разделения пиков. Как это удается,
можно понять, рассмотрев пример на рис. 4.12, на котором изображена часть хрома-
тограммы с двумя близко элюируемыми пиками А" и У (рис. 4.12а). Если
анализируемые вещества имеют спектры, такие как на рисунке 4.126, и их регистрируют на
длине волны, при которой оба вещества в значительной степени поглощают (260 нм), то
на хроматограмме эти два пика сольются в один (X + Y, рис. 4.12я). Если каждое
вещество проанализировать отдельно, то получим два разных пика X и Y (рис. 4.12а).
Даже когда пики полностью перекрываются друг с другом на длине волны 260 нм,
Х(260нм)
Х+ У (260 нм)
У(260 нм)
210 220 230 240 250 260 270 280
Длина волны (нм)
Рис. 4.12. Иллюстрация восстановления пиков спектральным методом, (а)
Гипотетические хроматограммы веществ X и Y, проанализированных раздельно и
совместно на длине волны 260 нм; (6) спектры веществ X и У
4.4. Детекторы, регистрирующие поглощение в УФ- и видимой областях спектра
света
205
их все-таки можно разделить, выбрав другую длину волны. Например, при 240 нм
хорошо поглощает только соединение X, а при 280 нм — только Y. Дополнительная
селективность детектора может, по крайней мере, отчасти компенсировать неполное
хроматографическое разделение. Так, например, ДМД позволяет одновременно
в процессе разделения собирать информацию при 240 и при 280 нм, а построенные
по этим данным хроматограммы либо при 240 нм, либо при 280 нм дадут
возможность количественно определить содержание пиков А" и Кдаже при их частичном на
ложении в условиях, иллюстрируемых на рис. 4.12.
Еще одно достаточно распространенное применение ДМД — определение
чистоты пика. Программное обеспечение, поставляемое с ДМД, позволяет рассчитать
отношение поглощения на разных длинах волн в каждой точке пика. Один и тот же
массив данных, собранных при 240 и 280 нм, можно использовать для определения
чистоты пика. Для этого рассчитывается отношение абсорбции при 240 и 280 нм
в каждой точке пика. Когда пик соответствует чистому X или У, отношение
абсорбции будет постоянным. Если пик соответствует смеси, как на рис. 4.12, то
отношение абсорбции будет > 1 в случае преобладания вещества X, и < 1, если основным
компонентом является К Когда отношение абсорбции непостоянно, это говорит
о том, что в образце присутствует смесь веществ, пики которых на хроматограмме
могут накладываться друг на друга и регистрироваться в виде одного пика при длине
волны 260 нм. Алгоритмы определения чистоты пика выверяют согласованность
спектральных отношений при значениях времени, соответствующих выходу пика, и
иногда с помощью этих алгоритмов можно обнаружить незначительные примеси
(например, менее 1%), элюируюшиеся на заднем фронте пика основного вещества.
Дополнительные примеры определения чистоты пиков с помощью ДМД можно
найти в работах [20, 211 или в инструкциях производителей детекторов (например, |22|).
4.4.4. Общие характеристики Уф-детекторов
Общие характеристики УФ-детекторов приведены в таблице 4.5. УФ-детекторы
идеально подходят для анализов в градиентном режиме. В качестве подвижной фазы
можно использовать большинство обычных, прозрачных в УФ-диапазоне растворителей
со степенью чистоты «для ВЭЖХ» (таблицы 1.2 и 1.3 в приложении I). УФ-детектор
очень полезен для обнаружения следовых количеств веществ, поглощающих
в УФ-диапазоне. При этом его в значительной мере изменяющийся для различных
веществ отклик может стать недостатком, особенно если интересующее соединение
не поглощает в УФ (или видимой) области спектра. УФ-детекторы надежны и просты
в обращении, они подходят прежде всего для малоопытных специалистов. Встроенные
процедуры тестирования, которые можно выполнять при запуске, позволяют выявлять
ряд возможных проблем, а также проводить автоматическую калибровку длин волн.
Таблица 4.5. Характеристики УФ-иетекторов
Может иметь высокую чувствительность (для веществ, поглощающих в УФ-области)
Широкий линейный диапазон (> 105)
Может иметь ячейку небольшого объема, минимизирующую внеколоночное размывание пиков
Относительно нечувствителен к колебаниям температуры и скорости потока
Чрезвычайно надежен
Прост в обращении
Не разрушает образец
Значительно изменяющийся отклик при анализе различных веществ
Подходит для градиентного элюирования
Возможен выбор длин волн
Как правило, есть встроенная самодиагностика и калибровка
206 Глава 4. Детектирование
(а)
3
о
Е
о
с
В
>
ВС (площадь = 951)
(б) Е
д)
S
X
8
см
0)
3
£
рокситромицин
(площадь =
609)
-г-
10
-1
15
HJ
- ВС (площадь = 961)
рокситромицин
/ (площадь = 1901)
флуоресценция
Фоновое поглощение или
поглощение на базовой линии в УФ-детекторах
может со временем увеличиться. Обычно
это указывает на то, что окна ячейки за
грязнились; ячейку следует промыть или
заменить. Регулярная промывка ячейки
детектора (во время промывки колонки),
а также пробоподготовка образца
позволят избежать частого тщательного
промывания ячейки. Проблема, связанная
со сроком службы лампы, осталась в
прошлом. Как правило, лампа работает более
2000 часов. Электронные схемы
непрерывно контролируют ее состояние,
и оператор получает сообщение, когда
наступает время замены лампы. Согласно
спецификации производителей
линейный диапазон отклика УФ-детекторов
может быть > 2 AU, но большинство
хроматографистов в целях получения более
точных результатов стараются работать
в интервале менее 1 AU. Стабилизация
температуры проточной ячейки при по
мощи термостатирования или
капиллярного теплообменника позволяет снизить
шум и дрейф, возникающие из-за
колебаний температуры и скорости потока.
На рис. 4.13а показан пример хрома-
тограммы для определения дериватизи-
рованного рокситромицина (РОКС) из
плазмы крови человека. Детектирование
проводили при длине волны 220 нм |23|.
К 50 мкл плазмы добавили в качестве
внутреннего стандарта эритромицин,
после чего проводили пробоп од готовку
с помощью твердофазной экстракции,
а затем дериватизировали образец
9-флуорен ил метил хлорформиатом
(FMOC-C1). С помощью этого метода
можно отслеживать концентрацию
РОКС в плазме, но не удается достичь
НПКО < I мкг/мл, необходимого для проведения фармакокинетических
исследований. (В разделе 4.5 проводится сравнение откликов УФ- и флуоресцентного
детекторов для этого образца, показанных на рис. 4.13а и 6).
4J
L
-i 1—
5 10
Время (мин)
~I
15
Рис, 4.13. Хроматограмма определения
рокситромицина (РОКС) в плазме крови
человека с помощью (о) УФ-детектора при
220 нм и (6) флуоресцентного детектора
(длина волны возбуждения 255 нм, эмиссии
315 нм). Время удерживания РОКС 10,7
минут, внутреннего стандарта
(эритромицина) — 5,1 минута. Пробоподготовка
образцов состояла из двух этапов: твердофазная
экстракция и дериватизация 9-флуоренилме-
тилхлороформиатом (FMOC-C1) |23]
4.5. Флуоресцентные детекторы
Флуоресцентные детекторы (флуориметры) очень чувствительны и селективны к тем
соединениям, которые при возбуждении УФ-излучением флуоресцируют. Не
флуоресцирующие компоненты образца не могут быть зарегистрированы детектором.
4.5. Флуоресцентные детекторы 207
и поэтому процедура пробоподготовки может быть упрощена. Например, простая
экстракция раствором, состоящим из буфера и ацетонитрила, позволяет с помощью
ВЭЖХ с флуоресцентным детектором зарегистрировать менее 30 пг рибофлавина
(обладающего естественной флуоресценцией) в пищевых продуктах |24|. Если
вещества не флуоресцируют, то их можно дериватизировать таким образом, чтобы они
флуоресцировали (см., например, [25|). Такой подход может быть полезен для
селективного обнаружения соединений, для которых нет других чувствительных и
селективных методов детектирования.
Флуоресцентный детектор схематически изображен на рис. 4.14. Обычно в этих
детекторах в качестве источника света используется УФ-лампа, излучающая свет
в широком диапазоне длин волн, например, дейтериевая, как в УФ-детекторах,
или ксеноновая импульсная лампа. Нужную длину волны возбуждения получают с
помощью фильтра или монохроматора. Свет с заданной длиной волны проходит
через проточную ячейку, облучая образец и вызывая его флуоресценцию. Светофи
льтром или монохроматором из потока излучаемого образцом света (эмиссии)
выделяют свет с необходимой длиной волны, который и направляют на фотоприем
ник, где световой поток измеряется и преобразуется в электрический сигнал,
используемый для обработки данных. Поскольку интенсивность флуоресцентной
эмиссии одинакова во всех направлениях, то регистрируют, как правило, свет,
испускаемый под прямым углом к падающему свету. Такая компоновка упрощает
оптическую схему детектора и снижает шум. обусловленный фоновым
излучением1. В недорогих флуориметрах установлены фильтры, позволяющие выбирать
длины волн возбуждения и эмиссии, в то время как в самых дорогих флуоресцентных
детекторах есть два монохроматора, которые обеспечивают широкий выбор длин
волн как света возбуждения, так и света эмиссии. Помните, что явление
флуоресценции не имеет 100% эффективности, поскольку часть энергии возбуждения
теряется. Это значит, что энергия эмиссионного света всегда должна быть ниже
энергии света, используемого для возбуждения молекул; следовательно, длина волны
излучаемого света, обратно пропорциональная его энергии, всегда больше длины
волны поглощенного света.
Для многих образцов флуоресцентные детекторы в 100 раз чувствительнее
УФ-детекторов. Кроме того, флуоресцентный детектор — один из самых
чувствительных ВЭЖХ-детекторов. В некоторых случаях чувствительность флуоресцентного
детектора может быть ненамного больше, чем у УФ-детектора, но этого может
оказаться достаточно для выполнения задачи. Сравним отклики флуоресцентного и
УФ-детектора на примере анализа рокситромицина (РОКС), проведенного в режиме
ввод образца
выход образца
\^ " j/^~~~ фотоячейка
Рис. 4.14. Схема флуоресцентного детектора. Пунктиром указан ход луча света
фильтр или
монохроматор
1 Регистрация эмиссии под прямым углом к направлению возбуждающего света дает
возможность отделить флуоресцетное излучение от возбуждающего света. — Прим. перев.
208 Глава 4. Детектирование
ОФХ (рис. 4.13) |23|. РОКС не обладает собственной флуоресценцией, поэтому и
образец, и внутренний стандарт дериватизируют 9-флуоренилметилхлорформиатом
(FMOC-C1). На рисунке видно, что отклик УФ-детектора (рис. 4.13а) и
флуоресцентного детектора (рис. 4.136) для внутреннего стандарта примерно одинаков. В то же
время отклик для РОКС в случае флуоресцентного детектора в три раза больше, чем
с УФ-детектором. Шум базовой линии обоих детекторов примерно одинаков.
Увеличение отклика флуоресцентного детектора оказалось достаточным для снижения
НПКО до концентрации менее 1 мкг/мл РОКС в плазме крови человека (такая
концентрация требуется для фармакокинетических исследований).
За счет своей высокой чувствительности флуоресцентные детекторы особенно
полезны при анализах следовых количеств, когда образца немного или его
концентрация очень мала. Как правило, линейный динамический диапазон флуоресцентного
детектора меньше, чем у УФ-детектора, но в большинстве случаев его вполне
достаточно для определения следовых количеств веществ. В то время как динамический
диапазон (диапазон, в котором изменение концентрации образца вызывает
изменения показаний детектора) флуоресцентного детектора может быть довольно большим
(например, 104), линейный динамический диапазон для определенных образцов
может быть ограничен относительно узким диапазоном концентраций (всего лишь
10-кратным). Для всех количественных анализов, в которых используется
флуоресцентный (или любой другой) детектор, необходимо проводить соответствующую
калибровку линейного диапазона (раздел 11.4.1).
По сравнению с другими методами детектирования флуоресцентный метод, как
правило, обеспечивает гораздо большую чувствительность и не создает больших
сложностей, связанных с нестабильностью работы прибора (например, показания
детектора не зависят от изменения температуры и скорости потока). Если
растворители или добавки в подвижную фазу не содержат флуоресцирующих веществ, то
флуоресцентный детектор можно использовать для градиентных разделений. Самым
большим недостатком флуоресцентного детектора является то, что не все вещества
обладают флуоресценцией. Чувствительность флуоресцентного детектирования, как
и других флуоресцентных методов, может быть снижена вследствие фоновой
флуоресценции подвижной фазы или матрицы образца, и из-за эффектов подавления.
Пример подавления флуоресценции показан на рис. 4.15 |25]. Когда подвижную
фазу барботируют гелием, сигнал стабилен, но, если в подвижной фазе присутствуют
пузырьки воздуха, интенсивность сигнала падает, так как кислород подавляет
флуоресценцию нафталина (длина волны возбуждения 250 нм, эмиссии — 340 нм). Бар-
к
S
цеш.
рее
.. Illllllll
Время (мин)
Не воздух Не
Рис. 4.15. Подавление флуоресценции нафталина растворенным в подвижной фазе
кислородом. Показана интенсивность флуоресценции при использовании
подвижной фазы, барботированной гелием (Не) или воздухом |25|
4.6. Электрохимические (амперометрические) детекторы
209
вотируя гелием подвижную фазу, из нее удаляют кислород, и сигнал становится
нормальным. Кроме того, из-за присутствия кислорода в подвижной фазе слегка
сдвигается базовая линия, но этот эффект незначителен.
В детекторах ФИЛ (флуоресценции, индуцированной лазером) источником
возбуждения является лазер. По сравнению с обычной дейтериевой или ксеноновой
лампами, он дает пучки света с большей плотностью энергии, что позволяет
увеличить чувствительность детектора, но ограничивает диапазон длин волн возбуждения
до 300-700 нм (диапазон флоуриметра с лампами 200—700 нм). Детекторы ФИЛ
не нашли широкого применения в ВЭЖХ; чаще всего они используются, когда
для ограничения дисперсии результатов анализа необходима ячейка малого диаметра
(например, с внутренним диаметром 100 мкм). Это требуется для микро-ЖХ,
капиллярной ЖХ, капиллярного электрофореза и т.д.
4.6. Электрохимические (амперометрические)
детекторы
Многие соединения под воздействием электрического потенциала могут окисляться
или восстанавливаться. Такие вещества можно обнаружить в очень малых
концентрациях с помощью селективных электрохимических (ЭХ) измерений. При таком
подходе ток между поляризуемым электродом и электродом сравнения измеряется
как функция приложенного напряжения. Поскольку обычно между электродами
приложено постоянное напряжение, в ходе протекающей в растворе реакции
меняется только сила тока, и поэтому электрохимические детекторы точнее называть ампе-
рометрическими. ЭХ-детекторы могут быть чувствительны к целому ряду соединений
(таблица 4.6). ЭХ-детекторы обычно используют для определения катехоламинов
и других нейромедиаторов. Многие из приведенных в таблице 4.6 соединений можно
обнаружить и с помощью УФ-детекторов, но некоторые из них (например,
алифатические меркаптаны, гидропероксиды) регистрируются только ЭХ-детектором.
УФ-детекторы либо вообще не чувствительны к таким соединениям, либо про-
'I долина 4.6. Некоторые виды соединений, регистрируемые ЭХ-детектором
Окисление
Фенольные полимеры
Оксимы
Меркаптанты
Пероксиды
Гидропероксиды
Ароматические амины, диамины
Пурины
Гетероциклические соединения"
Восстановление
Кетоны
Альдегиды
Оксимы
Сопряженные кислоты
Сопряженные сложные эфиры
Сопряженные нитрилы
Сопряженные ненасыщенные соединения
Активированные галогены
Ароматические галогены
Нитросоеди нения
Гетероциклические соединения
Примечание: ЭХ-детекторы обычно нечувствительны к соединениям определенного типа,
включающим простые эфиры, алифатические углеводороды, спирты и карбон оные кислоты.
"Детектируются в зависимости от структуры.
210 Глава 4. Детектирование
цесс их регистрации сложен, его чувствительность низкая, и длины волн должны
быть короткими.
ЭХ-детекторы можно использовать только с подвижной фазой, обладающей
электрической проводимостью. Это несущественное ограничение, поскольку в
настоящее время большинство разделений проводят на обращенной фазе, элюируя
подвижной фазой, содержащей воду или водный буфер. Точная настройка потенциала
детектора позволяет увеличить чувствительность к электроактивным соединениям.
ЭХ-детектор один из самых чувствительных ВЭЖХ-детекторов. Пределы
обнаружения допамина, к примеру, составляют 50 фемтограмм. При этом чтобы работать
в условиях высокой чувствительности, необходимо все делать чрезвычайно
аккуратно, в частности, для снижения фоновых помех использовать высокочистые
подвижные фазы. В некоторых методиках подвижную фазу перед тем, как она достигнет ав-
тосамплера, пропускают через ячейку с высоким потенциалом для того, чтобы
восстановить или окислить содержащиеся в ней примеси и таким образом снизить
фоновые помехи.
Обычно в электрохимических ячейках установлен стеклоуглеродный электрод,
который омывается элюатом (конфигурация ячейки показана на рис. 4.16). В другой,
не менее популярной конфигурации, элюат течет через ячейку с пористым
графитовым электродом. Коммерчески доступны разные исполнения ячеек с электродами,
например, есть ячейки с двумя электродами (рис. 4.16#). Высокая чувствительность
ввод
образца
прижимной
штуцер ^s
(б)
блок
вспомогател ьн ых
электродов
О
о
вывод электрод ввод
блок
рабочего
электрода
быстросъемный
механизм
Рис. 4.16. Схематическое изображение электрохимического детектора, (л) проточная
ячейка в сборе (вид сверху); (б) детали ячейки; (в) фрагмент ячейки с двумя
электродами. (С разрешения BioanalyticalSystems, Inc.)
детектора к фоновым помехам и
загрязнению электрода создали ЭХ-детекто-
рам репутацию сложных в
использовании приборов. Тем не менее, новые
модели детекторов гораздо более
надежны, а в руках достаточно аккуратного
специалиста они дают безупречные и
достоверные результаты.
На рис. 4.17 показана хроматограм-
ма актеозида (активного компонента
многих применяемых в медицине
растений, произрастающих в Китае),
полученная с использованием ЭХ-детектора.
После внутривенного введения
актеозида в концентрации 10 мг/кг, он был
обнаружен методом обращен но-фазовой
хроматографии в микродиализате
головного мозга крысы в концентрации
= 25 нг/мл (<= 0,4 нг было нанесено
на колонку) |26|. Более подробно
электрохимические детекторы описаны в
работе |27|.
4.7. Детекторы радиоактивности
Детекторы радиоактивности используются для обнаружения элюируемых из колонки
веществ, содержащих радиоактивные метки. Принцип детектирования заключается
в следующем: в проточной ячейке в результате распада радиоактивных элементов-
меток испускаются а, Р, у-частицы; их энергия растрачивается на ионизацию и/или
излучение, чаще всего преобразуемые в детектируемый свет. Для непрерывного
отслеживания р-частиц в элюате, как правило, используют сцинтилляторы. В
зависимости от метода смешивания алюата и сцинтиллятора могут получаться гомогенные
и гетерогенные системы. В гомогенной системе жидкая сцинтилляционная смесь
смешивается с элюатом, а потом попадает в ячейку детектора, в которой регистрируется
излучаемый свет. В гетерогенных системах элюат напрямую подается в ячейку
детектора, в которой находятся шарики твердого сцинтиллятора. Если адсорбция
вещества на поверхности шариков затруднена, то сцинтиллятором покрывают стенки
ячейки детектора.
В аналитических методах, когда выделять вещества нет необходимости, лучше
использовать гомогенную систему. Данная техника детектирования применима
и в препаративной ВЭЖХ, при этом поток элюата разделяется, и малая его часть
направляется в детектор. Гетерогенный метод менее чувствителен, поэтому подходит
для образцов с более высоким уровнем радиоактивности (или для образцов с
высокой концентрацией, как в препаративной хроматографии). Кроме того, гетерогенные
системы относительно свободны от эффектов химического гашения и позволяют
легко выделить вещество после детектирования. Однако, этот детектор обладает
относительно низкой эффективностью измерения для источников
низкоэнергетических (3-частиц, таких как 35S, '*C, 3Н и 32Р, и лучше подходит для более мощных
источников а, Р, у-частиц (например, '-"i, 2|0Ро и l25Sb). Одно из применений
радиоактивного мониторинга — определение окончательного распределения и
массового баланса радиоактивно меченых фармацевтических препаратов, введенных
4.7. Детекторы радиоактивности
1.2 -i
< 0.8-
L
0--'
актеозид
UJj
-г"
10
15
~Г"
20
25
Время (мин)
Рис. 4.17. Определение актеозида (/д =
=s 15 мин) в микродиализате головного мозга
крысы с помощью электрохимического
детектора |26|
212 Глава 4. Детектирование
экспериментальному животному. Такие исследования достаточно сложны, если
не невозможны, без помощи лекарств, меченных радиоактивными изотопами.
Радиохимические детекторы имеют широкий диапазон детектирования и
нечувствительны к смене растворителя, поэтому они подходят для градиентных режимов
элюирования. При этом, возможно, придется снизить чувствительность, чтобы
улучшить хроматографическое разрешение и ускорить анализ. Чувствительность
детектирования пропорциональна числу радиоактивных распадов, которое, в свою очередь,
пропорционально объему ячейки и обратно пропорционально скорости потока.
То есть чувствительность пропорциональна времени пребывания элюата в ячейке,
что позволяет большему числу атомов распадаться во время прохождения
анализируемого вещества через проточную ячейку. Большие объемы проточных ячеек
увеличивают внеколоночное размывание пика и могут снизить разрешение, в то время как
низкие скорости потока приводят к возрастанию времени разделения. Поскольку
работают обычно на пределе чувствительности то, как правило, для радиоактивного
детектирования предпочитают ячейки больших объемов.
На практике размывание заднего и переднего фронтов пика в радиометрической
проточной ячейке можно минимизировать, если использовать колонки больших
объемов (предполагается, что можно нанести достаточно большое количество образца,
чтобы компенсировать его разбавление). При использовании детектора
радиоактивности рекомендуется найти компромисс между хроматографическим разрешением и
чувствительностью детектора, соотношение этих параметров зависит от
аналитических требований.
4.8. Кондуктометрические детекторы
В кондуктометрических детекторах используют ячейки малого объема, в которых
определяют изменение проводимости элюата по мере прохождения его через ячейку.
Такие детекторы наиболее популярны в ионной и ионообменной хроматографии
в тех случаях, когда вещества не имеют УФ-хромофоров. Лучше всего
кондуктометрические детекторы использовать для обнаружения неорганических ионов
(например, лития, натрия, алюминия, калия) при анализе воды, содержимого
гальванических ванн, охлаждающих жидкостей электростанций и т.п. Кроме того, с помощью
такого детектора удобно определять ионы органических кислот, такие как ацетаты,
формиаты и цитраты.
Кондуктометрическому детектированию может помешать присутствие в
подвижной фазе ионов буфера. Буфер значительно повышает проводимость подвижной
фазы, которая в присутствии ионов образца увеличивается лишь незначительно.
Один из способов минимизировать эту проблему — использовать подходящий буфер
вместе с подавительной колонкой (ионообменником) для снижения фоновой
проводимости подвижной фазы. Например, если необходимо обнаружить один или более
анионов в образце, то в качестве буфера используют NajCO^-NaHCC^ и катионооб-
менную подавительную колонку (в ионной хроматографии ее называют анионным
подавителем) в Н+-форме, которая удаляет Na+ и другие катионы из подвижной фазы
и переводит карбонат-ион в слабую угольную кислоту. Таким образом снижается
проводимость подвижной фазы, и облегчается детектирование анионов в образце
или определение их небольших концентраций. На рис. 4.18 показано, что подави-
тельная колонка значительно улучшает отклик кондуктометрического детектора
для F-, С!" и SO|".
4.9. Хемилюминесцентный азотный детектор
(а)
(б)
Подвижная
фаза
Аналитическая
колонка'
подавление
анионитов
н,о
V
Образец
"(F-, СГ, S04z")
NaF, NaCI.
^- Na2S04
в Na2C03
н,о
HF, HCI, H2S04
в H2C03
Кондуктометрический
детектор
w
Без подавления ионов
. противоионы
СГ SO/
Время
(в)
Время
Рис. 4.18. Использование анионной подавителъной колонки для улучшения отклика
кондуктометрического детектора на анионы образца, (а) Схема процесса,
происходящего в хроматографической системе с кондуктометрическим
детектором; (6) хроматограмма, зарегистрированная кондуктометрическим
детектором без подаиительной колонки; (в) хроматограмма,
зарегистрированная кондуктометрическим детектором при использовании подавительной
колонки. С разрешения Dionex
4.9. Хемилюминесцентный азотный детектор
Одно из преимуществ газовой хроматографии перед ВЭЖХ состоит в том, что для
газовых хроматографов есть несколько детекторов, специально созданных для
идентификации конкретных элементов, например, азота, серы или фосфора. Это позволяет
селективно обнаруживать соединения, содержащие эти элементы. В 1970-х годах
было приложено немало усилий для создания подобных детекторов для ВЭЖХ, но
в большинстве случаев результаты не оправдали ожиданий. Одним из исключений
стал хемилюминесцентный азотный детектор (ХЛАД), о котором сообщалось еще
в 1975 году |28|. Несколько коммерческих разработок и доработок конструкции
привели к созданию современного ХЛАД.
Элюат, выходящий из ВЭЖХ-колонки, распыляется в токе кислорода и
газа-носителя (аргона или гелия) и подвергается пиролизу при 1050 "С. Соединения,
содержащие азот (кроме Nj), окисляются до оксида азота (NO), который затем
взаимодействует с озоном с образованием диоксида азота в возбужденном состоянии (NO^).
NO2 распадается, переходя в основное состояние, и испускает фотон, детектируемый
фотометром. Сигнал прямо пропорционален количеству азота в исходном образце,
поэтому для количественного определения содержания азота в неизвестных образцах
можно использовать калибровочные образцы с известным содержанием азота. Это
показано на рис. 4.19а |291, где видно, что отклик детектора при анализе 7 разных
214 Глава 4. Детектирование
Ю
1 2 3 4 5 6 7
-JV I
V_ К
^
(б) 1
^^ЧЛй^
Рнс. 4.19. Отклик хемилюминесцентного азотного детектора (ХЛАД) на разные типы
соединений, (я) Отклик, эквивалентный 50 нг азота, 1 — Л'.М-диметилани-
лина, 2 — нитробензола, 3 — нитрата миконазола, 4 — никотинамида, 5 —
4-ацетамшюфенола, 6 - глицина, 7 — кофеина. (б) Отклик 1 (стандарта,
эквивалентного 1 нг азота) по сравнению с откликами при введении 1 mi
растворителя: 2 — ацетона, 3 — этилацетата, 4 — гексана, 5 — изопропанола,
6 — метанола, 7 — воды |29|
соединений с равным содержанием азота (50 нг) практически одинаков в пределах
+ 10%. При этом надо соблюдать осторожность и использовать элюенты, не
содержащие азот, и поэтому ацетонитрил следует исключить из состава подвижной фазы.
Многие растворители совместимы с ХЛАД (рис. 4.195). На рисунке показаны
интенсивности откликов при введении I мг 6 разных растворителей, не содержащих азот,
и стандарта, эквивалентного 1 нг азота.
Один из производителей детекторов утверждает, что пределы обнаружения
эквивалентны 0,1 нг азота. Практический пример можно увидеть на рис. 4.20а |30|. 13 не-
дериватизированных аминокислот анализировали методом ион-парной
хроматографии и детектировали с помощью ХЛАД. Отклик на каждый атом азота находится
в пределах 6% ОСО, а пределы обнаружения аминокислот лежат в интервале
ж 0,3—0,5 мкг/мл. На рис. 4.206 изображена хроматограмма вина (10 мкл),
пропущенного через фильтр, задерживающий вещества с молекулярной массой более
1000 Да (обратите внимание на пик пролина, имеющий из-за перегрузки меньшее
время удерживания и более сильное размывание заднего фронта пика по сравнению
с хроматограммой на рис. 4.20я; более подробно о перегрузке читайте в разделе 2.6).
4.10. Хиральные детекторы
250-
Pro
л_
—I—
30
40
20
Время (мин)
Рис. 4.20. Сигналы аминокислот при детектировании хемилюминесцентным азотным
детектором (ХЛАД), (а) 10 мкл 0,1 мМ стандартного раствора 13 недерива-
тизированных аминокислот; (б) анализ 10 мкл вина, пропущенного через
фильтр 1000 Да |29|
4.10. Хиральные детекторы
При разработке новых фармацевтических субстанций хиральные молекулы
встречаются довольно часто. Разные энантиомеры проявляют разную активность,
токсичность и другие фармакологические свойства, при этом конечным продуктом, как
правило, является один энантиомер или известная смесь энантиомеров. Хромато-
графическое разделение энантиомеров (глава 14) имеет очень большое значение
при анализе таких смесей. Обнаружению и идентификации может также
способствовать использование детекторов, которые избирательно реагируют на конкретные
хиральные формы.
Хиральные детекторы бывают трех видов; работа каждого из них основана на тех
же принципах, что и работа автономного прибора, но детекторы имеют проточные
ячейки. Поляриметры (ПЛ) измеряют угол вращения плоскости поляризации
проходящего через элюат линейно-поляризованного света (обычно для его длин волн
в диапазоне 400—700 нм). Угол вращения зависит от концентрации хирального
216 Глава 4. Детектирование
2000-
0
-2000 -
ф
3
га
о..
ш
I?
§1
о
-500-
-1000
-1500
1200-
300 :
400
(а)
КД (230 нм)
(б)
ДОВ
-V—V-
(в)
УФ (265 нм)
1
10
Время (мин)
15
20
Рис. 4.21. Сравнение откликов детектора кругового дихроизма (КД), детектора
дисперсии оптического вращения (ДОВ) и УФ-детектора в анализе 10 мкг
ибупрофена. (о) КД при 230 нм, S/N = 49,6; (б) ДОВ, S/N = 10,9; (в) УФ при
265 нм S/N= 113,4 [31]
вещества и его структуры. Принцип работы детекторов дисперсии оптического
вращения (ДОВ) такой же, как у поляриметров, но в них используются волны меньшей
длины (например, излучение ртутной лампы с длиной волны 365 нм), которые
теоретически должны давать более интенсивные сигналы. Принцип действия детекторов
кругового дихроизма (КД) основан на измерении разницы в поглощении света с
правой и левой круговой поляризацией в тот момент, когда растворенное вещество
проходит через проточную ячейку детектора. Желательно, чтобы анализируемое
вещество имело хромофор, поглощающий в области 200—420 нм, тогда сигнал,
регистрируемый детектором кругового дихроизма, будет интенсивным.
На рис. 4.21 1311 показано сравнение откликов детекторов УФ, ДОВ и КД
на примере хирального разделения энантиомеров ибупрофена. Детектор КД
регистрирует пики с соотношением сигнал/шум (S/N) в 5 раз большим, чем детектор ДОВ,
но в два раза меньшим по сравнению с УФ-детектором. Обратите внимание на то,
что оба хиральных детектора генерируют как отрицательные, так и положительные
пики. В работе |32] сравниваются отклики поляриметра, детекторов ДОВ и КД для
шести фармацевтических субстанций. Детектор КД оказался в 6 раз чувствительнее
к напроксену, чем поляриметр, и в 24 раза чувствительнее, чем детектор ДОВ.
Соотношения откликов для разных тестируемых соединений отличались, но детектор КД
во всех случаях оказался лучшим.
4.11. Рефрактометрические детекторы
Дифференциальные рефрактометры или детекторы показателя преломления (ДПП)
реагируют на изменения показателя преломления элюата, проходящего через ячейку
детектора. Рефрактометр относится к детекторам, отклик которых основан на
свойствах, присущим всем веществам, и поэтому он чувствителен ко всем соединениям,
показатели преломления которых существенно отличаются от показателя
преломления подвижной фазы. Наиболее распространенное исполнение ДПП — рефракто-
щель
4.11. Рефрактометрические детекторы 217
ячейка сравнения
измерительная двойной выходной сигнал
ячейка фотодиод детектора
(б)
птп
тпп
П
сротод йодная
матрица
Рис. 4.22. Принципиальная схема рефрактометрического детектора, (д) Детектор
с двумя фотодиодами; {б) диодно-матричный детектор (лампа и проточная
ячейка не показаны). Пунктиром обозначен ход светового луча
метр оптического отклонения (рис. 4.22я). Свет от лампы (обычно вольфрамовой)
проходит через пару клинообразных проточных ячеек. Одна из них, ячейка
сравнения, обычно заполнена подвижной фазой и используется как непроточная,
другая — измерительная, и в нее направляется элюат из колонки. Свет, проходя
через ячейки детектора, по-разному преломляется в зависимости от состава раствора,
находящегося в ячейке сравнения или проходящего через измерительную ячейку
в данный момент времени. Пара фотодиодов позволяет определить изменение
положения пучка света, а, следовательно, изменение показателя преломления жидкости,
проходящей через проточную ячейку. Соответствующая электронная схема
преобразует это изменение в выходное напряжение. В обычном рефрактометрическом
детекторе установлены два фотодиода (рис. 4.22а). По мере изменения показателя
преломления элюата свет отклоняется так, что меняется его количество, достигающее
каждого фотодиода. Совсем недавно технологию фотодиодной матрицы применили
к ДПП, что позволило использовать большее число фотодиодов для детектирования
света (рис. 4.226). Утверждается, что такая конфигурация позволяет расширить
динамический диапазон ДПП и улучшить его чувствительность.
В таблице 4.7 перечислены характеристики рефрактометрических детекторов.
Поскольку ДПП регистрируют любые соединения, они являются отличными
универсальными детекторами, если при этом правильно выбрана подвижная фаза. Чтобы
чувствительность детектора была максимальной, показатель преломления подвижной
фазы должен как можно больше отличаться от показателя преломления
растворенного вещества (таблица 1.3 в приложении I). Тем не менее, даже в оптимальных
условиях чувствительность детектора будет средней. Несмотря на то, что эти детекторы
обычно не используют в анализах следовых количеств, все-таки в оптимальных
условиях можно количественно оценить пики с концентрацией 0,1%. У ДПП есть
жесткое ограничение — они не подходят для градиентных режимов элюирования, так как
довольно сложно сопоставлять показатели преломления растворов, если их потоки
одновременно проходят и через ячейку сравнения, и через измерительную ячейку
218 Глава 4. Детектирование
(исключение можно найти в обсуждении рис. 4.25<з, раздел 4.12.1). Даже в изократи-
ческом режиме незначительные изменения состава подвижной фазы, которые не
обнаруживаются при УФ детектировании, проявляются в случае ДПП в виде шума или
пульсаций базовой линии. Базовая линия будет более гладкой, если смешивать
компоненты подвижной фазы вручную, а не при помощи встроенного миксера.
Несмотря на ограниченную чувствительность и неприменимость в градиентном элюи-
ровании, дифференциальные рефрактометры широко используются особенно
в эксклюзионной хроматографии, где чувствительность не так критична.
Таблица 4.7. Характеристики рефрактометрических детекторов
Отличная универсальность; детектируют все соединения
Средняя чувствительность
Как правило, не подходят для анализа следовых количеств
Не подходят для градиентных разделений
Необходим эффективный теплообменник
Чувствительны к температурным колебаниям
Надежны, достаточно просты в эксплуатации
Не разрушают образец во время детектирования
Чувствительность рефрактометрического детектора к изменению температуры
также представляет собой серьезное ограничение. Конструкция современных
приборов была тщательно проработана, что позволило минимизировать температурные
колебания благодаря использованию термостатированных ячеек и эффективных теп
лообменников, которые уравновешивают температуру потока подвижной фазы
перед тем, как он попадёт в ячейку. Чтобы повысить эффективность
рефрактометрического детектора, необходимо, чтобы он работал все время, или термостатировать
прибор перед использованием по крайней мере два часа. Существует еще один
хороший способ: обеспечить теплоизоляцию капилляра, соединяющего колонку и
детектор, минимизируя, таким образом, температурные колебания. Рефрактометры
удобны и надежны, хотя не так просты и беспроблемны, как УФ-детекторы.
Дрейф базовой линии рефрактометрического детектора может появиться после
замены одной емкости с «чистым» растворителем или «аналогичной» подвижной фазой,
смешанной вручную, на другую. Дрейф базовой линии может быть ярко выраженным,
пока из системы не будет вымыт предыдущий элюент. Только после того, как один
элюент полностью вытеснит другой, дрейф уменьшится. Чтобы на протяжении всей
серии анализов сохранить гомогенность состава подвижной фазы, необходимо
приготовить достаточное ее количество и обеспечить ее непрерывное перемешивание в
емкости, в которой она хранится. Также для достижения стабильности базовой линии
рекомендуется избегать каких-либо изменений в составе подвижной фазы (возникающих
в результате дегазации, испарения, поглощения паров воды и др.).
В прошлом использовались рефрактометры Френеля или интерферометрические
детекторы, но в настоящее время самыми популярными являются рефрактометры
с оптическим отклонением. Многие годы рефрактометрические детекторы были
единственными приборами для «универсального» детектирования в ВЭЖХ. В настоящее
время во многих методиках детекторы светорассеяния (раздел 4.12) вытеснили
рефрактометрические детекторы. В коротковолновом диапазоне (< 210 нм) чувствительность
УФ-детекторов ко многим соединениям, отличающимся очень слабым поглощением
на длинных волнах, выше, чем у рефрактометрических (см. рис. 4.25, на которых
сравниваются отклики УФ, рефрактометрического и светорассеивающего детекторов).
4.12. Детекторы светорассеяния
барбитал (B.C.)
5 10
Время (мин)
Рис. 4.23. Отклик рефрактометрического детектора для треосульфана с концентрацией
560 мкг/мл через час после начала его внутривенного введения; барбитал
используется в качестве внутреннего стандарта |33|
Чувствительность рефрактометрических детекторов обычно исключает их
использование в рутинных анализах лекарственных образцов, но в некоторых случаях
такие детекторы оказались полезными при определении концентрации лекарств
в биологических образцах, например, когда в образце высокая концентрация
лекарственного препарата, и другие способы детектирования неэффективны. Например,
рефрактометрический детектор используют для определения концентраций
треосульфана (Ь-треитол-1,4-метансульфоната) в плазме крови детей (рис. 4.23) |33|. Треосу-
льфан является противоопухолевым препаратом, токсичным по отношению к
стволовым клеткам. Перед трансплантацией стволовых клеток его вводят внутривенно
для уничтожения собственных стволовых клеток. На рис. 4.23 показана хроматограм-
ма, где концентрация треосульфана 560 мкг/мл в плазме крови ребенка после
введения препарата. С учетом пробоподготовки такое количество треосульфана
эквивалентно объему анализируемой пробы в 83 мкл плазмы.
4.12. Детекторы светорассеяния
В последние годы усовершенствование детекторов светорассеяния привело к тому,
что во многих методиках они вытеснили рефрактометрические детекторы. Это было,
в частности, обусловлено способностью детектора светорассеяния эффективно
распылять элюат и испарять подвижную фазу. (Это же обстоятельство способствовало
широкому применению масс-спектрометрических детекторов). Наиболее
популярным является испарительный детектор светорассеяния (ИДС). Детектор
светорассеяния на центрах (ядрах) конденсации (ДСЦК) — это модификация ИДС с повышенной
эффективностью. В верхнем ценовом диапазоне находится лазерный детектор
светорассеяния (ЛДС), который по сравнению с ИДС и ДСЦК занял более
специализированную нишу. Сравнение некоторых свойств рефрактометрических детекторов и
детекторов светорассеяния представлено в таблице 4.8.
ШГ220 Глава 4. Детектирование
Таблица 4.8. Сравнение рефрактометра и детекторов светорассеяния
Характеристика
Универсальный отклик
Чувствительность
Совместимость с градиентным режимом
Необходимость легколетучей подвижной фазы
Чувствительность к изменениям температуры
Обеспечение качественными данными,
помогающими в определении структуры
дппа
+
-
-
Нет
+
-
идс»
+
0
+
да
+
-
ДСЦК"
+
+
+
да
-
-
ЛДС"
+
нп
+
да
+
+
Примечание. Относительное качество характеристик обозначено следующим образом: (+)
высокое, (0) среднее, (-) низкое; нп означает неприменимость данного критерия (детектор
предназначен для получения качественной информации для расчетов, к нему не применимо
понятие чувствительности).
"ДПП — детектор показателя преломления, ИДС — испарительный детектор светорассеяния,
ДСЦК — детектор светорассеяния на центрах конденсации, ЛДС — лазерный детектор
светорассеяния.
4.12.1. Испарительный детектор светорассеяния (ИДС)
Принцип действия испарительного детектора светорассеяния (ИДС) основывается
на испарении подвижной фазы с последующим измерением величины рассеяния
света мелкими частицами нелетучего образца (рис. 4.24). Элюат распыляется в
потоке газа-носителя (азота или воздуха) и направляется в нагреваемую испарительную
трубку, в которой подвижная фаза испаряется, и остаются нелетучие частицы
образца, взвешенные в потоке газа-носителя. Свет, рассеиваемый этими частицами,
детектируется фотоприемником, установленным под постоянным углом к пучку
падающего света. ИДС детектирует большинство соединений, анализируемых методом
из
колонки
распылитель
__ газ-
распылитель
нагревательная
трубка
ж\ У ~
ячейка
светорассеяния
лампа
фотоэлемент
Рис. 4.24. Схематическое изображение испарительного детектора светорассеяния (ИДС)
4.12. Детекторы светорассеяния
ВЭЖХ, но может быть менее чувствителен к образцам с большей летучестью. Отклик
детектора зависит от абсолютного количества вещества в образце, а не от его
спектральных свойств.
Как и рефрактометрический детектор, ИДС считается универсальным
детектором, потенциально способным обнаружить «любое» соединение. У ИДС есть
преимущество перед ДПП — не зависящий от растворителя отклик, поэтому его можно
использовать в градиентных анализах. Кроме того, ИДС не чувствителен к
температурным колебаниям или колебаниям потока подвижной фазы. В выборе подвижной
фазы для испарительного детектора светорассеяния существуют такие же
ограничения, как и для масс-спектрометрического детектора (раздел 4.14): подвижная фаза
должна быть летучей и не содержать нелетучих добавок (например, фосфатного
буфера). Достаточно один раз настроить ИДС (например, зафиксировав скорость
газа-носителя, температуру в испарительной трубке), и он будет стабильно и
удовлетворительно работать. Диапазон линейности детектора в некоторой степени
ограничен (10—100), однако при выборе подходящих концентраций калибровочных
растворов ИДС может оказаться полезным для количественного определения
концентраций образцов в широких пределах.
В общем случае ИДС в 10—100 раз чувствительнее ДПП, его пределы
детектирования 1 — 100 нг на колонку. Для некоторых образцов чувствительность может быть
ещё выше, как это наглядно показано на рис. 4.25й на примере разделения триглице-
10
15 20
Время (мин)
25
30
(б)
ИДС
дпгг
10
-г-
15
20
Рис. 4.25. Сравнение отклика ИДС. (а) ИДС в сравнении с ДПП и УФ-детектором
при длине волны 20S нм на примере разделения смеси триглицеридов.
Колонка Shimadzu Premier C]8, градиент ацетонитрил — дихлорметан, скорость
потока 1 мл/мин, температура термостата 30 °С (6) ИДС в сравнении
с ДПП в анализе полистирольных стандартов методом ГПХ при высокой
температуре (160 °С); 200 мкг образца разделяли на колонке PL-Gel
Mixed В. Молекулярная масса образцов: 1 — 2560000 Да, 2 - 320000 Да, 3 —
59500 Да, 4 — 10850 Да, 5 — 580 Да. (д) Любезно предоставлено Shimadzu
Corporation, (fi) с разрешения Varian Polymer Laboratories
222 Глава 4. Детектирование
ридов с детектированием ИДС, в то время как УФ-детектирование на длине волны
205 нм и детектирование рефрактометром не позволяют обнаружить целевые
вещества. Обратите внимание на то, что это разделение проводилось в градиентном режиме
с помощью неводной обращение фазовой хроматографии (НОФХ, раздел 6.5).
Водно-органические градиенты несовместимы с рефрактометрическим детектированием,
но поскольку показатели преломления ацетонитрила и дихлорметана достаточно
близки, можно использовать ДПП в таком градиенте. На рис. 4.256 приведена хро
матограмма, демонстрирующая превосходство ИДС над ДПП при разделении
образца, содержащего полистиролы, методом высокотемпературной (160°С) ГПХ.
4.12.2. Детектор светорассеяния на центрах (ядрах)
конденсации (ДСЦК)
Детектор светорассеяния на центрах конденсации (ДСЦК) является результатом
модернизации стандартного ИДС для повышения чувствительности и расширения
диапазона линейности. После испарения подвижной фазы частицы, находящиеся
в потоке газа-носителя, насыщаются парами растворителя. Частицы выступают в
качестве центров (ядер) конденсации, и растворитель конденсируется на них. При
этом размер частиц увеличивается, что облегчает детектирование за счет рассеяния
света [34|. В ранних работах в этой области использовали газообразный бутанол,
а в настоящее время в качестве конденсирующегося растворителя выступает вода.
Область применения ДСЦК и ИДС одинакова. В целом чувствительность ДСЦК
в 10—100 раз выше, чем у классического ИДС. Производители таких приборов f351
показали, что их детекторы могут зарегистрировать неорганические ионы (Li1-, Na1",
К+) при нагрузке 0,5 нг на колонку. Линейный диапазон детектирования для
сахарозы составляет три порядка, а динамический диапазон — пять порядков.
4.12.3. Лазерные детекторы светорассеяния (ЛДС)
Лазерные детекторы светорассеяния (ЛДС), называемые также многоугловыми свето-
рассеивающими детекторами (МУСД) — это ВЭЖХ-детекторы, измеряющие
рассеяние света в растворе, в отличие от ИДС и ДСЦК, которые детектируют рассеяние
света частицами в газовой фазе. Источником света в ЛДС является лазер, луч
которого направлен на проточную ячейку с подвижной фазой. Интенсивность
рассеянного света измеряется под несколькими углами (например, от 3 до 18 разных
углов). По этим данным с помощью известных математических соотношений1 можно
определить массу молекул анализируемого вещества в отсутствии эталонных
образцов. Эти детекторы особенно полезны в эксклюзионной хроматографии (глава 13)
для определения молекулярных масс синтетических полимеров и биологических
молекул в диапазоне 103—106 Да. На рис. 4.26 показаны наложенные друг на друга хро-
матограммы разделений фрагмента протеинкиназы и трех белковых стандартов (три-
мера алкогольдегидрогеназы (АДГ), бычьего сывороточного альбумина (БСА),
и мономера (АДГ), полученные с УФ-детектированием при 280 нм. Кроме того,
показаны молекулярные массы образцов, определенные с помощью ЛДС в 3
независимых разделениях; (молекулярные массы отложены по оси ординат). У
протеинкиназы теоретическая масса 53,5 кДа; масса, измеренная лазерным детектором
светорассеяния, составляет 108000 Да и, значит, это пик димера. Ожидаемые молеку-
1 В рамках физических моделей — например статического и динамического рассеяния света —
были получены уравнения, связывающие углы рассеяния света и его поляризацию со свойствами
рассеивающих центров, в частности, с их размерами. — Прим. перев.
4.13. Детектор заряженных аэрозолей 223
20 22 24 26 26 30
Элюирующий объем (мп)
Рис. 4.26. Разделение методом эксклюзионной хроматографии нескольких белков с их
идентификацией лазерным детектором светорассеяния (ЛДС) и УФ-детек-
тором при длине волны 280 нм. По оси ординат отложены молекулярные
массы, вычисленные с помощью ЛДС. Хроматограммы протеинкиназы,
БСА, АДГ получены в 3 отдельных разделениях. Предоставлено Wyatt
Technology Corporation
лярные массы стандартов следующие: 141000 Да (АДГ), 67000 Да (БСА), 35000 Да
(субъединица АДГ). Эти значения близки к тем, которые были определены с
помощью ЛДС (рис. 4.26). Несмотря на то, что молекулярная масса димера БСА
(135000 Да) меньше, чем димера АДГ, он элюируется раньше (23,3 мл — БСА,
24,7 мл — АДГ). Это свидетельствует о более высокой точности определения
молекулярной массы ЛДС по сравнению со значениями, полученными в эксклюзионной
хроматографии (раздел 13.8, 13.10.3.1).
4.13. Детектор заряженных аэрозолей
Детектор с ионизацией коронным разрядом, также называемый детектором
заряженных аэрозолей (ДЗА), относится к универсальным детекторам, поскольку он
чувствителен к большинству соединений. Принцип работы ДЗА схематически
изображен на рис. 4.27. Поток элюата из колонки распыляется, а подвижная фаза
испаряется, также как и в ИДС (раздел 4.12.1) или в масс-спектрометре (раздел 4.14).
Затем частицы анализируемых веществ, находящиеся в газовой фазе, смешиваются
с потоком положительно заряженных ионов азота, предварительно полученных
ионизацией коронным разрядом. Положительный заряд переходит на частицы
анализируемого вещества, и высокоподвижные заряженные частицы улавливаются
ионной ловушкой, улучшающей качество выходного сигнала, который формируется
оставшимися заряженными ионами вещества. Этот сигнал и считывается
электрометром.
ДЗА регистрирует практически любые соединения, которые менее летучи, чем
подвижная фаза, и остаются в газовой фазе после ее испарения. У ДЗА, также как и
у других испарительных детекторов, есть ограничение, касающееся подвижной
фазы, — в ее состав должны входить только летучие компоненты, (т.е. не допускается
224 Глава 4. Детектирование
распылитель
образец в
газообразном'
состоянии
перенос
заряда
электрометр
Рис. 4.27. Схематическое изображение
детектора заряженных аэрозолей
использование, например, фосфатных
буферов). Кроме того, частицы вещества
должны быть способны к ионизации
в детекторе. В анализах Сахаров и других
углеводов ДЗА является альтернативой
рефрактометрическому детектору и
детектору светорассеяния. Пределы обна
ружения олигосахаридов составляют 5 нг
на колонку (S/N = 3), а динамический
диапазон ДЗА превышает 104 [36|. На
рис. 4.28 показан пример применения
ДЗА при анализе примесей с
концентрацией 0,05% относительно активной
фармацевтической субстанции (АФС) |37|.
В этом случае 5 нг каждого из пяти
родственных сульфамидных лекарственных
соединений (рис. 4.28 пики 1—5)
четко детектируются в присутствии 10 мкг
сульфадиметоксина (пик 6).
_1
_Л
V4
-г-
10
Время (мин)
Рис. 4.28. Детектирование ДЗА введенной в колонку смеси, состоящей из 10 мкг
сульфадиметоксина (6) и родственных соединений: 1 — сульфагуанидин, 2 — су-
льфамеразин, 3 — сульфаметазин, 4 — сульфаметизол и 5 — сульфаметокса-
зол, по 5 нг каждого [39]
4.14. Масс-спектрометрические детекторы (МС)
Существуют независимые1 аналитические приборы (например, МС, ЯМР, ИКФП),
присоединяемые к ВЭЖХ-системам для детектирования. В настоящее время самым
популярным из них является масс-спектрометрический (МС) детектор (описание
других детекторов этого типа можно найти в разделе 4.15). МС-детектор стал стан-
1 Относительно независимости приборов см. примечание в разделе 4.2.2.4. — Прим. персе.
4.14. Масс-спектрометрические детекторы (МС) 225
дартным прибором в биоаналитических методах, то есть в анализах
фармацевтических соединений в биологических растворах (например, в плазме крови или моче).
Кроме того, М С детектор широко используется в научно-исследовательских
разработках, когда необходимо установить структуру неизвестного соединения или
подтвердить ее, хотя используемый для детектирования вариант и не имеет такого
разрешения по массе, как обычный автономный масс-спектрометр. МС-детекторы
выпускаются в двух популярных конфигурациях. Одностадийный детектор, который
иногда называют МСД (масс-селективныи детектор), используют для регистрации
одной группы ионов образца, зачастую протонированных молекулярных ионов
(М+Н). (При проведении одного анализа можно отследить более одного иона
образца, переключая детектор с одного значения m/z на другое и возвращаясь к первому,
или сканируя диапазон масс.) Система, в которой используется этот тип
детектирования, называется ЖХ-МС. В детекторе более сложной конструкции выделяются
группы первичных ионов (родительские ионы или ионы-предшественники), которые
фрагментируются в дополнительные ионы (дочерние или ионы-продукты), и один
или несколько из них отслеживаются. Такой процесс, иногда называемый
селективной регистрацией множественных реакции распада ионов (СРМР), повышает
селективность детектирования при переходе от иона предшественника к дочернему иону.
Этот переход используется в качестве «характерного признака» конкретного
вещества. Такие системы называют ЖХ-МС/МС; это сокращение и будет указываться
в тексте, если использовался именно этот тип детектирования. Ссылка на ЖХ-МС
будет означать одностадийную технологию или случай, когда не важно, которая
из технологий была использована. В разделе 8.1 работы [13] можно найти более
подробное описание ГИС-детектирования в применении к градиентному режиму.
Большая часть этого обсуждения в равной степени применима и к изократическому элю-
ированию. Дополнительная информация о ЖХ-МС и ЖХ-МС/МС изложена
в книгах, посвященных этой теме (например, |38—40|).
4.14.1. Интерфейсы МС-детекторов (способы ионизации
образца)
Разработка различных способов ионизации молекул анализируемого вещества
в МС-детекторах, пожалуй, стала самым важным фактором успешного
использования масс-спектрометрии в качестве метода детектирования в ВЭЖХ. МС-детекторы
работают с ионами и детектируют их в газовой фазе. Поэтому, чтобы
масс-спектрометр был полезен в ВЭЖХ в качестве детектора, подвижная фаза должна быть
испарена, а компоненты образца ионизированы. За это и ответственен интерфейс
МС-детектора. Объем, занимаемый подвижной фазой при переходе из жидкого
состояния в газообразное, увеличивается примерно в 1000 раз; в то же время давление
должно быть снижено с атмосферного (760 торр) до 10-5-10-6 торр в пределах 5—
10 см пути, по которому движется газ в интерфейсе. Давление снижается за счет
перекачивания большей части испаренной смеси образца и подвижной фазы в слив
(в самом устройстве ничего не концентрируется). Непосредственно в МС-детектор
попадает лишь небольшая часть образца. Существуют два наиболее популярных
метода ионизации, реализованных в интерфейсах МС-детекторов: ионизация
электрораспылением (ИЭ) и химическая ионизация при атмосферном давлении (ХИАД).
4.14.1.1. Ионизация электрораспылением (ИЭ)
Интерфейс ионизации электрораспылением или электроспреем изображен
на рис. 4.29. Благодаря потенциалу (например, 3—5 кВ), приложенному к
распылительной игле из нержавеющей стали («капилляру» на рис. 4.29), анализируемые
226 Глава 4. Детектирование
нагреватель N,
3-5 кВ
от ВЭЖХ-системы
Рис. 4.29. Схематическое изображение ионизации электрораспылением в ЖХ-МС-де-
текторе
вещества в подвижной фазе приобретают заряд. Подвижная фаза распыляется внутри
нагретого интерфейса, растворитель испаряется, и образуются ионы, находящиеся
в газообразном состоянии. ИЭ наиболее часто используется в биоаналитических
методиках, поскольку этот вид ионизации наиболее «щадящий», и с меньшей
вероятностью может вызывать нежелательную деградацию образца.
4.14.1.2. Химическая ионизация при атмосферном давлении
(ХИАД)
При таком способе ионизации в интерфейсе МС-детектора (рис. 4.30) сначала
происходит испарение подвижной фазы, а затем в газовой фазе молекулы образца
ионизируются коронным разрядом. Такой способ ионизации используют для
соединений, которые не очень хорошо ионизируются электрораспылением (зачастую
это более стабильные вещества с меньшей молекулярной массой, а также некото-
игла коронного разряда
от ВЭЖХ-системы
Рис. 4.30. Схематическое изображение химической ионизации при атмосферном
давлении (ХИАД)
4.14. Mace-спектрометрические детекторы (МС) ТП
рые неполярные соединения). В этом случае ионизация проводится в более
жестких условиях, в которых, по сравнению с ИЭ, повышается вероятность разрушения
веществ, особенно термолабильных. У ИЭ и ХИАД разные механизмы ионизации,
поэтому отклик детекторов и селективность могут очень значительно отличаться.
Оба эти интерфейса работают в двух режимах с противоположно заряженными
ионами, а, значит, ионы образца могут быть заряжены положительно или
отрицательно (что чаще всего достигается добавлением протона к молекуле образца или
его удалением).
4.14.1.3. Другие модификации интерфейса МС-детекторов
Помимо ИЭ и ХИАД существуют и другие модификации интерфейса. Например,
в ХИАД молекулы образца ионизируются коронным разрядом, а в детекторе с фо
тоионизацией при атмосферном давлении (ФИАД) — фотонами, генерируемыми
УФ-лампой. По крайней мере в одной из моделей интерфейсов поток элюата,
выходящий из колонки, делится на два. Это позволяет одновременно ионизировать
образец разными способами (ИЭ и ХИАД) и может быть полезно в том случае,
когда неизвестно, каким способом ионизируется образец, и необходимо найти
оптимальный.
4.14.1.4. Соображения относительно скорости потока
Чтобы свести к минимуму работу интерфейса, внутренний диаметр колонки должен
быть меньше того, который обычно используют в классических ВЭЖХ-анализах, что
позволит снизить скорость потока. Хотя МС-детекторы способны работать при
расходе ] мл/мин, все-таки они более надежны при меньших скоростях потока. Работая
с колонкой, внутренний диаметр которой 2,1 мм, можно установить расход
0,2—0,5 мл/мин, и при этом линейная скорость подвижной фазы и характер
разделения будут сравнимы с получаемыми на традиционных колонках диаметром 4,6 мм
(1,0—2,5 мл/мин). На коротких колонках длиной 30—50 мм, упакованных частицами
сорбента 3—5 мкм, можно осуществлять быстрые разделения обычных (простых)
смесей, встречающихся в биоаналитических методиках и содержащих образец,
внутренний стандарт и один или два метаболита. Для разделения более сложных
образцов потребуются более длинные колонки (100—150 мм), поскольку необходимо
большее число теоретических тарелок (при этом продолжительность анализа
увеличивается). Обычно в методиках по разделению белковых образцов используют
капиллярные колонки (например, диаметром менее 1 мм) и интерфейс с «наноспре-
ем*, рассчитанным на низкие скорости потока (микролитры/мин и меньше)
(раздел 13.4.1.6).
4.14.2. Квадруполи и ионные ловушки
В настоящее время преобладают две конструкции фильтров масс для одностадийных
ЖХ-МС: квадруполи (квадрупольные анализаторы) и ионные ловушки. Кроме того,
набирает популярность и время пролетная (ВПМС1) схема (раздел 4.14.3).
В квадруполях для выделения выбранных ионов образца используется набор
из 4 стержней и тщательно контролируемое электрическое поле. Ионы с выбранным
отношением массы к заряду (m/z) потом попадают во вторично-электронный
умножитель, обеспечивающий селективный отклик на целевое соединение. В ионных
ловушках выделение целевых ионов для последующего детектирования достигается
'По-английски времяпролетный масс-спектрометр называется timeofllight (TOF).
Аббревиатуру TOF в отечественной литературе часто используют без перевода. — Прим. перев.
228 Глава 4. Детектирование
(а)
интерфейс квадруполь 1
квадрупапь 2
(реакционная ячейка)
•U_q—n
квадруполь 3
электронный
умножитель
Л 1 |\
(б)
А* В*
С* D*
А* —
I слив
■V
А/
С*
В* D*
Аь* -
I слив
А *
А *
"с
Рис. 4.31. Трехквадрупольный масс-спектромегр. (а) Схематическое изображение,
(6) МС/МС эксперимент с переходом от иона-предшественника к иону-
продукту: А+ > А£
с помощью кольцевого электрода в сочетании с торцевыми электродами. Квадруполи
и ионные ловушки могут быть настроены на быструю смену мониторинга одной
массы на другую, сканируя, таким образом, весь спектр диапазона масс. Другой режим
работы позволяет детектору «одновременно» отслеживать элюируемые соединения,
такие как образец и внутренний стандарт, попеременно переключаясь с одного
значения m/z на другое во время элюирования пиков. Ниже приведено обоснование
того, что МС-детекторы с квадруполями предпочтительны для количественных
анализов, а ионные ловушки имеют преимущества при анализе структуры исследуемых
соединений.
Одностадийные МС-детекторы вышеупомянутого типа используются в
недорогих ЖХ-МС системах. Однако для более селективного детектирования
необходима дополнительная разрешающая способность в отношении структуры вещества.
Для этого служат трехквадрупольные или тандемные МС-детекторы (рис. 4.31а).
Ионы образца (на рис. 4.316 А+, В+, С+, D+) вводятся в первый квадруполь, в
котором отделяются ионы с заданным m/z (А+) и направляются во второй квадруполь
(ячейку соударений), заполненный инертным газом (азотом или аргоном). Здесь
ионы подвергаются фрагментации (A"1"—»-AJ, A^, AJ) и направляются в третий
квадруполь. В третьем квадруполе специфические фрагменты ионов
(например, А},) изолируются и направляются во вторично-электронный умножитель
для измерения. Переход от исходного иона (предшественника или родительского)
к фрагментированному (продукту или дочернему иону) дает уникальную
«характеристику» (А+ > А£, рис. 4.316) образца и существенно повышает селективность
МС-детектора с тремя квадруполями (МС/МС) по сравнению с МС-детектором
с одним квадруполем. (Обратите внимание, условное обозначение А"1" > A J
означает переход от иона-предшественника А+ к иону-продукту А£. Дальше мы будем
использовать обозначение «А+—*А^, А£, AJ», когда речь пойдет о самом процессе
фрагментации).
Ионные ловушки выполняют многостадийную фрагментацию и выделяют
нужный ион в одном и том же физическом пространстве (а не в разных частях
анализатора, как в случае цепи из трех квадруполей, рис. 4.31). Образовавшиеся
ионы направляются в ионную ловушку (для накопления ионов, рис. 4.32). Ионы
4.14. Масс спектрометрические детекторы (МС) 119
с выбранным отношением m/z
удерживаются, а остальные отправляются в слив
(выделение ионов I). Выделенные ионы
(А+ на рис. 4.32) далее подвергаются
фрагментации (A+^AJ, А£, А£), и
из всех фрагментов отбираются ионы
с нужным отношением m/z (A£,
выделение ионов 2). Ионы затем могут быть
направлены во вторично электронный
умножитель, с которого и снимается
электрический сигнал при
детектировании, или же процесс можно
продолжить (последующая фрагментация А£>
выделение продуктов и т.д.). Ионная
ловушка в состоянии проводить эту
операцию раз за разом, выделяя фрагменты
ионов и разрушая их с образованием
продуктов со все уменьшающимся
отношением m/z (соответственно на каждом
последующем этапе чувствительность
снижается). Такие эксперименты
полезны для идентификации структуры, но
в количественном анализе ионные
ловушки всегда уступали квадруполям
из-за эффектов пространственного
заряда (взаимодействия ионов внутри
детектора) и нестабильности
интенсивности выходного сигнала. В
основном в количественных анализах
пользуются квадруполями (одним или тремя),
а ионные ловушки предпочитают
использовать, когда необходима
структурная идентификация, например,
при выделении и идентификации
метаболитов.
накопление ионов
выделение иона 1
фрагментация 1
выделение иона 2
спив В* D*
V
дополнительные этапы
выделения/фрагментации ионов
или детектирование
Рис. 4.32. Масс-спектрометр с ионной
ловушкой. МС/МС эксперимент с переходом
от иона-предшественника к иону-продукту:
А+ > А£. Обратите внимание на то, что
ионы в квадратах представляют
превращения, происходящие на разных стадиях
в разное время в одном и том же
физическом пространстве
4.14.3. Другие виды МС-детекторов
Помимо ЖХ-МС детекторов с квадрупольными анализаторами и ионной ловушкой
существуют и другие конфигурации, самой популярной среди них является времяпро-
летныи МС-детектор (ВПМС) (рис. 4.33). В ВПМС сначала генерируются ионы,
затем они ускоряются в электрическом поле до определенной энергии и направляются
в пролетную трубу детектора. Скорость, с которой ионы перемешаются по пролетной
трубе, будет зависеть от энергии, полученной ионами при ускорении. Поэтому ионы
с меньшей массой будут перемещаться к детектору быстрее, а ионы с большей
массой — медленнее. Следовательно, время, необходимое для пролета через трубу,
зависит от массы (m/z) иона. Разрешение масс определяется длиной пролетной трубы.
Существуют пространственные ограничения длины пролетной трубы, поэтому
используют популярную конфигурацию электростатического зеркала («рефлектрона»)
для увеличения эффективной длины пролетной трубы, и следовательно, улучшения
разрешения. При достаточной длине пути ВПМС может обеспечить более высокое
230 Глава 4. Детектирование
электронный
интерфейс ппййАойяя тпи6кя умножитель
>
дрейфовая трубка
Рис. 4.33. Линейный времяпролетный масс-спектрометр (ВПМС)
разрешение, чем квадрупольные анализаторы, поэтому этот детектор используют в
экспериментах по определению структуры вещества. Однако следует отметить, что
ни один из МС-детекторов не имеет такой разрешающей способности, как у
традиционных специализированных масс-спектрометров.
Для МС-МС детектирования сейчас доступны различные сочетания анализаторов.
Ранее в разделе 4.14.2 уже упоминалось о том, что в трехквадрупольном
масс-спектрометре первый из квадруполей используется для выделения иона-предшественника,
второй — как ячейка соударений для его фрагментации, а в третьем выделяются фраг-
ментированные ионы-продукты. Две секции ВПМС можно присоединить к ячейке
соударений, и тогда такая конструкция будет выполнять те же функции посредством
ВПМС ВПМС. Кроме того, можно одновременно использовать несколько видов
анализаторов для выделения ионов. Например, в квадрупольном ВПМС1 на первом этапе
используется квадруполь, потом ячейка соударений, а на последнем этапе — ВПМС.
Каждая конструкция имеет определенные достоинства {и недостатки), поэтому их
производители с радостью объяснят, почему их прибор лучше, чем у остальных!
4.15. Другие «независимые» детекторы
Независимыми детекторами (раздел 4.2.2.4) называют аналитические приборы,
используемые вкупе с системой ВЭЖХ. Кажется, что практически любой автономный
аналитический прибор пытались когда-либо присоединить к ВЭЖХ, и поэтому
существует большое количество возможных комбинаций детекторов. Если каждый
детектор может дать об образце количественную и/или качественную информацию, то
из этого следует, что, присоединив к ВЭЖХ несколько детекторов, можно получить
еще больше информации об образце. Известно, по крайней мере, об одном случае,
когда к одной ВЭЖХ-системе присоединили четыре спектрометра. Такой сложный
прибор описан в работе |41|. Однако признание получила только ЖХ-МС, широко
используемая в настоящее время практически во всех областях ВЭЖХ. Также
коммерчески доступны два других детектора, инфракрасный и ЯМР, с интерфейсами
для ВЭЖХ. Эти детекторы используются в первую очередь для определения
структуры неизвестных соединений в образце, а не в рутинных количественных анализах.
4.15.1. Инфракрасная спектроскопия
с Фурье-преобразованием (ИКФП)
Инфракрасная спектроскопия с Фурье-преобразованием (ИКФП) — популярный
метод детектирования, позволяющий установить структуру химического соединения.
Присоединив его к ВЭЖХ-системе, можно получить ИК-данные о разделяемых
компонентах, т. е. можно проводить ИК-анализ смеси. Спектр шума подвижной фазы
может приводить к неоднозначностям при расшифровке ИК-спектра, поэтому про-
' По-английски такой масс-спектрометр называется Q-TOF. — Прим. перев.
^Относительно независимости приборов см. примечание в разделе 4.2.2.4. — Прим. перев.
4.15. Другие «независимые» детекторы
4000
3500
3000
2500
2000
1500
«00
Волновое число (см"1)
Рис. 4.34. ЖХ-ИКФП хроматограмма экстрактов Ecoli. (а) Общая хроматограмма;
по оси ординат отложены максимальные значения ИК-поглощения;
(6) ИКФП-спектр одного пика с временем удерживания 23 минуты (*) |41|
точные И К-детекторы имеют ограниченное применение. Альтернативой является
прибор, в котором подвижную фазу испаряют и образец помещают на окно
(например, из ZnSe), откуда он автоматически передается в И К-спектрометр для анализа.
В этом случае детектирование проходит не в непрерывном режиме, а дискретно.
(Чувствительность можно повысить, увеличив время регистрации каждого ИК спект
ра, но при этом анализ становится продолжительным). Тем не менее, данных
достаточно для выполнения анализов, таких как анализ экстрактов E.coli в режиме
ЖХ-ИКФП (рис. 4.34) |42|. Образец разделяли на колонке C]S в градиентном
режиме, элюируя его 0,1% муравьиной кислотой с ацетонитрилом. Общая хроматограмма
(рис. 4.34я) получена построением графика зависимости максимальных значений
ИК-поглощения в каждом спектре от времени. Спектр одного пика с временем
удерживания 23 минуты (*) приведен на рис. 4.345. По разным полосам колебаний
амидов при 1642, 1550 и 1268 см-1 (указаны стрелками) и другим особенностям спектра
было установлено, что этот пик соответствует белку. Более подробная информация
о ЖХ-ИКФП изложена в |43].
4.15.2. Ядерный магнитный резонанс (ЯМР)
Ядерный магнитный резонанс (ЯМР) можно использовать для детектирования
в ВЭЖХ в трех разных режимах |44|. Самый простой вариант: элюат из колонки
направляется в проточную ЯМР-ячейку, где происходит непрерывный сбор ЯМР-дан-
ных. Так как время для сбора данных ограничено, то чувствительность в этих
условиях низкая, хотя 'Н-ЯМР-спектры оказались полезными. Последние достижения
науки и техники позволяют снимать 2Б-ЯМР и 13С-ЯМР-спектры в режиме
непрерывного потока. Существует и альтернативный режим с остановкой потока. Фракции,
соответствующие целевым пикам на хроматограмме, отбираются в петлю и
направляются в ЯМР-детектор, в котором идет регистрация спектра, пока поток подвижной
фазы остановлен. В обоих режимах ЖХ-ЯМР (как с остановкой потока, так и
без нее) подвижная фаза не должна содержать протоны, и поэтому вместо воды
232 Глава 4. Детектирование
10 15 20 25 30 35 40
используют D20, (и дейтерированный аце-
тонитрил или CD3OD вместо метанола).
Кроме того, диаметр колонок должен
быть небольшим, чтобы уменьшить расход
растворителя. Третий альтернативный
режим — ЖХ-ТФЭ-ЯМР, смысл которого
заключается в накоплении элюата в ТФЭ-кар-
тридже. Картридж затем обрабатывают,
чтобы удалить всю воду. Затем образец элю
ируют небольшим количеством ацетонитри
ла в ячейку ЯМР-детектора для анализа.
Два последних способа могут выполняться
короткими циклами (длительностью,
например, 20 с) для снижения потери хро
I матографического разрешения. Обзор ЖХ
4HWw^^W*tf4*i*f*<tih)* ямр ™»в работе и5'-
На рис. 4.35 показана ЖХ-ЯМР хрома-
тограмма анализа органического соедине
ния в водном образце и ЯМР-спектр с оста
новкой потока |46|. Градиентное ион
парное разделение проводили на колонке
C|jj, с подвижной фазой, состоящей
из 0,01 М тетрабутиламмоний гидросульфата
в D^O (элюент А) и ацетонитрила (элю-
ент В). На целевых участках хроматограммы
в петлю отбирались образцы с интервалом
20 секунд. На УФ-хроматограмме (рис. 4.35а) выбрали участок в районе 16
минуты {*), и сняли 'Н-ЯМР-спектр соответствующего образца (рис. 4.355, показана
ароматическая область).
8.0
7.5
7.0
Ч./МЛН.
6.5
Рис. 4.3S. Анализ органического
соединения в водном образце в режиме
ЖХ-ЯМР с остановкой потока, (о)
УФ-хроматограмма образца на длине
волны 280 нм, (6) ароматическая область
'Н-ЯМР-спектра, на участке,
отмеченном звездочкой )46]
4.16. Дериватизация образца и реакционные
детекторы
Несмотря на то, что в настоящее время существует целый ряд ВЭЖХ-детекторов, все
же некоторые вещества либо невозможно детектировать, либо пределы их
детектирования недостаточно низки для их обнаружения на практике. В таких случаях один
из способов решения проблемы — превращение анализируемого вещества в
производное (или комплекс), чтобы обеспечить удовлетворительный отклик на обычных
детекторах (например, на УФ-детекторе или флуориметре). Провести реакцию дериватиза-
ции можно до того, как вещество попадет в колонку или уже на выходе из нее.
О новых реакциях дериватизации постоянно сообщается в литературе, к которой
рекомендуется обращаться, когда необходимо разработать способ детектирования
отдельных специфических соединений или классов соединений. Кроме того, есть книги,
посвященные дериватизации (например, |47|; см. также список литературы в конце
раздела 16.12), но самый практичный источник информации — набор методик,
который производители реакционных детекторов прилагают к приборам (например, |48|).
Предколоночная дериватизация может расширить пределы детектирования или
изменить хроматографические характеристики вещества (нечасто встречающийся
вариант), либо и то, и другое одновременно. Предколоночную дериватизацию, как
часть процесса пробоподготовки образца, можно провести вручную или в автомати-
4.16. Дериватизация образца и реакционные детекторы 233
11
3
I 1 1 1 1 1 1 1 1 1 1
О 2 4 6 8 10
Время (мин)
Рис. 4.36. Предколоночная дериватизация органических кислот для получения
флуоресцирующих производных (длина полны возбуждения 256 нм, эмиссии —
412 нм). 1 — 9-хлорметилантрацен (дериватизирующий реагент), 2 — моно-
фторацетат, 3 — муравьиная кислота, 4 — уксусная кислота, 5 — пропионо-
вая кислота [48]
ческом режиме. Конструкция некоторых автосамплеров предусматривает дериватиза-
цию образца непосредственно перед введением в колонку. Реакции дериватизации
могут быть быстрыми или медленными, но если их проводить порциями, то и
медленные реакции обеспечат приемлемую работоспособность. На рис. 4.36 изображен
пример хроматограммы анализа низкомолекулярных кислот с предколоночной дери
ватизацией, улучшившей отклик флуориметра |49|. В процессе пробоподготовки
проводилась реакция образца с 9-хлорметилантраценом в присутствии бромида тет-
рабутиламмония (катализатора) при 80 °С в течение 50 минут. Детектирование
флуоресценции (длина волны возбуждения 256 нм, эмиссии — 412 нм) давало линейный
отклик для концентраций в диапазоне от 1 до 250 нг/мл монофторацетата в
сыворотке, а предел детектирования составил 0,25 нг/мл сыворотки (при S/N = 3).
Постколоночная дериватизация подразумевает проведение реакции в то время,
когда образец перемешается от колонки к детектору в реакторе, являющимся одним
из составляющих реакционного детектора. Реакция может быть химической,
фотохимической или совмещать оба механизма. В таблице 4.9 приведены некоторые из
характеристик, которыми желательно обладать постколоночному реакционному
детектору и выбранным химическим реагентам. Обычно реактор для дериватизации
представляет собой дополнительный капилляр между колонкой и детектором
для смешения реагентов и обеспечения необходимой продолжительности реакции.
Дополнительный капилляр, однако, приводит к увеличению внеколоночного
размывания пиков, поэтому система должна быть разработана таким образом, чтобы найти
баланс между требованиями к реакции дериватизации и эффектами размывания
пиков. Существует способ, позволяющий снизить внеколоночное размывание пиков
во время реакции дериватизации — использование реакционной петли. Она
представляет собой гибкий капилляр малого диаметра, свернутый в несколько витков с
малым радиусом. Такая конструкция позволяет увеличить турбулентность потока и
снизить дисперсию. В общем случае, чем быстрее проходит реакция — тем меньше
требований к минимизации объема реактора. Кроме того, реакции, которые
проходят полностью, более воспроизводимы, чем те, которые протекают не до конца.
Химическую реакцию следует выбирать таким образом, чтобы, во-первых, реагенты,
участвующие в ней, были стабильны на протяжении выполнения всей серии
анализов, и во-вторых, чтобы реагенты и продукты были растворимы в подвижной фазе.
Конечно, реагенты не должны мешать детектированию продуктов, поэтому их
отклики должны быть минимальными. В настоящее время имеются в продаже наборы
реагентов для наиболее распространенных постколоночных реакций (например,
234 Глава 4. Детектирование
ортофталевый альдегид и нингидрин для аминокислотного анализа). Покупка такого
набора очень удобна, поскольку в нем стандартные концентрации реактивов,
прошедших контроль качества. Последнее может быть недоступно, если реагенты
подготавливаются в лаборатории.
Таблица 4.9.Требования к постколоночном} реакционном}' детектированию
Минимальная дисперсия
Реакции должны проходить до конца за короткое время
Вое п ро и з води мость
Стабильность реагентов
Растворимость реагентов и продуктов
Минимальный отклик детектора на реагенты
На рис. 4.37 изображена хроматограмма анализа с популярной постколоночной
дериватизацией аминокислот нингидрином при детектировании на длине волны
405 нм. 45 аминокислот и родственных соединений разделяли на ионообменной
колонке, после чего проводили реакцию дериватизации нингидрином при 130 °С в
реакторе объемом 500 мкл (продолжительность реакции дериватизации ~ 45 с). Все же
дериватизация образца и реакционные детекторы постепенно теряют популярность,
поскольку появляются новые, более эффективные ВЭЖХ-детекторы.
—[ 1 г-
20 40
—1 1 1 1 1—
60 80 100
120
Время (мин)
Рис. 4.37. УФ-хроматограмма при 405 нм 45 аминокислот и родственных соединений
после дериватизации нингидрином. С разрешения Pickering Laboratories, Inc.
Литература 235
Литература
1. G. Hesse and H. Weil, eds., Michael Tswett's First Paper on Chromatography, M. Woelm, Eschwe-
ge, 1954.
2. V. G. Berezkin, ed., Chromatographic Adsorption Analysis: Selected Works of M. S. Tswett, Ellis
Horwood, New York, 1990.
3. A. Tiselius and D. Claesson, Arkiv. Kemi Mineral. Geol., 15B (18) (1942) 1.
4. A. J. P. Martin and S. S. Randall, Biochem. J., 49 (1951) 293.
5. С G. Horvath and S. R. Lipsky, Nature, 211 (1966) 748.
6. J. J. Kirkland, Anal. Chem., 40 (1968) 391.
7. E. Haahti and T. Nikkari, Acta Chem. Scand., 17 (1963) 2565.
8. R. P. W. Scott and J. G. Lawrence, /. Chromatogr. Sci., II (1970) 65.
9. B. G. Julin, H. W. Vandenborn, and J. J. Kirkland, J. Chromatogr., 112 (1975) 443.
10. J. W. Dolan and J. N. Seiber, Anal. Chem., 49 (1977) 326.
11. L. R. Snyder and J. J. Kirkland, Introduction to Modern Liquid Chromatography, 1st ed., Wiley,
New York, 1979, p. 136.
12. J. W. Dolan. LCGC, 14 (1996) 378.
13. L. R. Snyder and J. W. Dolan, High-Performance Gradient Button, Wiley, New York, 2007.
14. J. J. Kirkland, W. W. Yau, H. J. Stoklosa, and С H. Dilks, Jr., J. Chromatogr. Sci., 15 (1977) 303.
15. J. C. Sternberg, in Advances in Chromatography, Vol. 2, J. C. Giddings and R. A. Keller, eds., Dek-
ker, New York, 1966, p. 205.
16. D. Parriott, A Practical Guide to HPLC Detection, Academic Press, San Diego, 1993.
17. R. P. W. Scott, in Handbook of HPLC (Chromatographic Science Series, Vol. 78), E. Katz, R. Eks-
teen, P. Schoenmakers, and N. Miller, eds., Dekker, New York, 1998, ch. 15.
18. D. T. Bach, US Patent 4,867,559 (Sept. 19, 1989).
19. M. H. Garrett, US Patent 6,542,231 (Apr. 1, 2003).
20. G. Cameron, P. E. Jackson, and M. V. Gorenstein, Chem. AusL, 60 (1993) 288.
21. A. G. Frentch, J. R. Torres-Laasio, K. De Braekeleer, D. L. Massart, J. L. Martinez-Vidal, and
M. M. Galera, J. Chromatogr. A, 855 (1999) 487.
22. Waters 996 Photodiode Detector: Peak Purity III: Interpretation of Peak Purity Plots Appl. Note
970983, Waters, 1996.
23. F. K. Glowka and M. Karazniewicz-Lada, J. Chromatogr. B, 852 (2007) 669.
24. P. Vinas, N. Balsalobre, C. Lopez-Erroz, and M. Hernandez-Cordoba, /. Agr. Food Chem., 52
(2004) 1789.
25. S. R. Bakalyar, M. P. T. Bradley, and R. Honganen, J. Chromatogr., 158 (1978) 277.
26. Y.-T. Wu, T.-R. Tsai, L.-C. Lin, and T.-H. Tsai, J. Chromatogr. B, 853 (2007) 281.
27. 1. N. Ackworth, Coulometric Electrode Array Detectors for H PLC (Progress in HPLC-HPCE, Vol. 6),
Brill Academic, Boston, 1997.
28. R. E. Parks and R. L. Marietta, US Patent 4018562 (1975).
29. Model 8060 Nitrogen Specific HPLC Detector, brochure A-8060-99-25M, Antek Instruments,
Houston, TX, 1999.
30. K. Petritis, С Elfakir, and M. Dreux, LCGC Europe, 14 (2001) 389.
31. Jasco Incorporated, LCGC Applications Notebook (June 2003) 26.
32. L. Kott, W. B. Holzheuer, M. M. Wong, and G. K. Webster, J. Pharm. Biomed. Anal., 43 (2007)
57.
33. F. Glowka, M. K. Lada. G. Grund. and J. Wachowiak, J. Chromatogr. B, 850 (2007) 569.
34. L. B. Allen, J. A. Koropchak, and B. Szostek, Anal. Chem., 67 (1995) 659.
35. Quant Technologies, QT-500 datasheet (2/ 2007).
36. D. Asa, Amer. Lab., 38(7) (2006) 16.
37. T. Gorecki, F. Lynen, R. Szucs, and P. Sandra, Anal. Chem., 78 (2006) 3186.
38. W. M. A. Niessen, Liquid Chromatography-Mass Spectrometry (Chromatographic Science Series, Vol.
97), 3rd. ed., Taylor and Francis, London, 2006.
39. M. С McMaster, LC-MS: A Practical User's Guide, Wiley, Hoboken, NJ, 2005.
40. Current Practice in Liquid Chromatography-Mass Spectrometry, W. M. A. Niessen and R. D. Voyks-
ner, eds., Elsevier, Amsterdam, 1998,
41. I. D. Wilson and U. A. Th. Brinkman, J. Chromatogr. A., 1000 (2003) 325.
ist
Глава 4. Детектирование
42. S. W. Huffman, К. Lukasiewicz, S. Geldart, S. Elliott, J. F. Sperry, and C. W. Brown, Anal.
Chem., 75 (2003) 4606.
43. G. W. Somsen, G. Gooijer, and U. A. Th. Brinkman, J. Chromatogr. A., 856 (1999) 213.
44. S. Down, Spectro. Europe, 16 (2004) 8.
45. K. Albert, J. Chromatogr. A, 856 (1999) 199.
46. A. J. Simpson, L.-H. Tseng, M. J. Simpson, M. Spraul, U. Braumann, W. L. Kingery, B. P. Kelle-
her, and M. H. B. Hayes, Analyst, 129 (2004) 1216.
47. K. Blau and J. M. Halket, eds., Handbook of Derivatives for Chromatography, 2nd ed., Wiley, New
York, 1993.
48. Application Manual: Amino Acids, Pickering Laboratories, Mountain View, CA, 2002.
49. Z. Xie, W. Shi, L. Liu, and Q. Deng, J. Chromatogr. B, 857 (2007) 53.
ГЛАВА 5
КОЛОНКИ
5.1. Введение 238
5.2. Носители 238
5.2.1. Характеристики частиц 239
5.2.1.1. Типы частиц 239
5.2.1.2. Размер частиц и диаметр пор 241
5.2.2. Носители на основе силикагеля 242
5.2.2.1. Эффективность колонки 244
5.2.2.2. Структура поверхности силикагеля 246
5.2.2.3. Подготовка частиц 248
5.2.3. Пористые полимеры 249
5.2.4. Монолитные сорбенты 250
5.2.4.1. Монолиты на основе силикагеля 251
5.2.4.2. Монолиты на полимерной основе 251
5.2.5. Другие неорганические частицы 252
5.2.5.1. Оксид циркония 253
5.2.5.2. Оксиды алюминия и титана 254
5.2.5.3. Графитированный углерод 254
5.3. Неподвижные фазы 255
5.3.1. «Привитые» неподвижные фазы 256
5.3.2. Другие органические неподвижные фазы 260
5.3.2.1. Нанесенные полимеры 260
5.3.2.2. Гибридные частицы 260
5.3.2.3. Колонки для подвижных фаз с высоким содержанием воды . . . 261
5.3.3. Колонки с различными функциональными группами сорбента 262
5.4. Селективность колонки 263
5.4.1. Основы селективности обрашенно-фазовой колонки 264
5.4.1.1. Взаимодействия анализируемого вещества с сорбентом 266
5.4.1.2. Селективность по форме молекулы 269
5.4.2. Воспроизводимость характеристик колонки и «эквивалентные»
колонки 272
5.4.3. Ортогональное разделение 273
5.4.4. В каких еще случаях нужно знать селективность колонки 273
5.4.4.1 Размывание заднего фронта пика 274
5.4.4.2. Несмачиваемость неподвижной фазы 274
5.4.4.3. Снижение эффективности сорбента 274
5.5. Типоразмеры колонок 275
5.5.1. Фитинги 275
5.5.2. Типоразмеры колонок 276
5.6. Методы упаковки колонок 277
5.6.1. Сухая упаковка 277
5.6.2. Суспензионная упаковка жестких частиц 277
5.6.2.1. Выбор растворителя для приготовления суспензии 278
5.6.2.2. Жесткие полимерные частицы 280
5.6.3. Упаковка мягких гелей 281
5.7. Спецификации колонок 281
5.7.1. Стандарты производителя 281
5.7.2. Число теоретических тарелок колонки 282
5.8. Обслуживание колонок 283
Литература 287
238 Глава 5. Колонки
5.1. Введение
Колонка — это «сердце» хроматографической системы. С середины 1960-х годов —
начала развития ВЭЖХ — она претерпела существенные изменения. Колонки были
усовершенствованы, что позволило увеличить скорость и эффективность разделения,
повысить стабильность и воспроизводимость результатов. Были разработаны новые
подвижные фазы для расширения возможностей анализа методом ВЭЖХ самых
разнообразных образцов, а также для оптимизации разделения образцов, раньше
считавшихся трудными для разделения. Теперь редко бывает, что не удается подобрать
колонку для конкретного разделения ВЭЖХ. Первые колонки были стеклянными,
а сейчас из-за высокого рабочего давления используются, в основном, стальные
колонки. Со временем колонки стали короче, а размер частиц меньше (рис. 1.5ле и з).
В настоящее время широко используются колонки длиной 30—250 мм с частицами
диаметром 1,5—5 мкм (более крупные частицы подходят для препаративной
хроматографии, глава 15).
В этой главе мы рассмотрим колонки ВЭЖХ, упакованные немодифицирован-
ными частицами (называемыми также носителями) или химически
модифицированными («привитыми») неподвижными фазами, а также типоразмеры колонок. Самым
распространенным сорбентом является пористый силикагель (колонка, упакованная
силикагелем, показана на рис. 2.2). Служить сорбентами или подложками могут
не только силикагель, но и другие твердые вещества. Главное преимущество таких
неподвижных фаз — стабильность при высокой температуре или экстремальных
значениях рН. Особый интерес в настоящее время вызывают колонки, упакованные
сорбентами с частицами очень малых размеров (диаметром менее 3 мкм),
предназначенные для экспресс-разделений (раздел 2.5.3.2).
Хотя очень немногие хроматографисты сами изготавливают сорбенты с
привитыми фазами, мы кратко опишем метод их получения. Возможно, эта информация
окажется полезной при выборе колонки или при устранении неполадок. Параметры
колонки, определяющие удерживание и селективность в ОФХ (раздел 5.4), также
помогут выбрать нужную колонку для выполнения конкретного разделения. В
разделе 5.6 описаны способы упаковки колонок — это дает представление о том, как
получаются «хорошие» колонки. Тестирование колонок рассмотрено в разделе 5.7.
Наконец, в разделах 5.7 и 5.8, соответственно, представлены спецификации колонок и
инструкции по работе с ними, что поможет при реализации надлежащей
лабораторной практики.
5.2. Носители
Сорбент состоит из жесткого носителя и привитой к ней фазы (на рис. 2.26
изображены частицы силикагеля с привитыми Cjg-группами). Иногда неподвижной фазой
служит сам носитель, например, непривитый силикагель часто используется в
нормально-фазовой хроматографии (глава 8). Монолитный силикагель, появившийся
сравнительно недавно, является альтернативным вариантом сорбента. Монолитами
называют сорбенты, представляющие собой единый стержень, пронизанный
сообщающимися между собой порами, в отличие от сорбентов, состоящих из отдельных
частиц. То есть, монолит можно рассматривать как одну большую частицу,
заполняющую весь объем колонки. В этом разделе будут рассмотрены носители, их
характеристики и свойства, влияющие на разделение. Кроме того, будут обсуждаться
(1) способы модификации носителей для получения сорбентов для различных целей
(в разделе 5.3) и (2) методы упаковки колонок (в разделе 5.6).
5.2. Носители 239
5.2.1. Характеристики частиц
Частицы различаются формой или типом, физическими размерами (обычно
указывается диаметр частиц), природой и размером пор внутри частиц, а также площадью
поверхности. Влияние размера частиц на разделение уже обсуждалось в разделе 2.4
(рис. 1.5). Размер пор и площадь поверхности обычно обратно пропорциональны друг
другу: с увеличением диаметра пор площадь поверхности уменьшается примерно
во столько же раз. Фазовое отношение Ф (раздел 2.3.1) приблизительно
пропорционально площади поверхности, а удерживание пропорционально Ф (уравнение 2.3).
Поэтому удерживание возрастает с уменьшением размера пор и с увеличением площади
поверхности сорбента. Максимальное количество образца, которое можно разделить
на колонке, тоже пропорционально площади поверхности сорбента (раздел 2.6.2).
Поэтому желательно использовать сорбент с большей площадью поверхности, а,
следовательно, с наименьшим возможным диаметром пор, но при этом поры должны быть
достаточно большими, чтобы молекулы разделяемых веществ могли свободно
проникать внутрь пор. Так, для соединений массой менее 500 Да предпочтителен диаметр
пор примерно 9 нм и больше. Для более крупных молекул требуются поры большего
размера. Например, белки обычно разделяют на сорбенте с порами диаметром 30 нм
(раздел 13.3.1.1). Внутренний объем колонки — это пространство между частицами. Он,
как правило, составляет 40% от всего объема колонки.
Не менее важны и физические свойства частиц, поэтому производители
контролируют их различными способами. Особенно важен размер частиц, поскольку
именно он определяет эффективность упакованной колонки. Несмотря на то, что
оптическая микроскопия и фракционирование (классификация) в газовой или жидкой
фазах позволяют получить эту информацию, существуют и другие удобные и
количественные методы определения размера частиц (например, счетчик Коултера).
Счетчик Коултера позволяет также определить распределение частиц по размерам.
Предпочтительно узкое распределение, так как в этих условиях выше число N
(раздел 5.2.2.1) и при сравнимой эффективности ниже давление в колонке.
Площадь поверхности частиц, средний диаметр пор и распределение пор по
размеру обычно определяют методом газовой адсорбции азота или аргона (метод Бру-
науэра — Эмметта — Теялера (БЭТ)). Частицы со средним диаметром пор более
30 нм, не так часто используемые, удобнее характеризовать методом ртутной поро-
метрии, который подходит и для пор диаметром менее 3 нм1. Не рекомендуется
применять ртутную интрузию для анализа хрупких частиц (с большим объемом пор) или
мягких полимерных частиц. Дополнительную информацию об измерении
физических свойств частиц сорбентов ВЭЖХ можно найти в работе [1|.
5.2.1.1. Типы частиц
На рис. 5.1 показана структура частиц нескольких типов, используемых в настоящее
время для сорбентов ВЭЖХ. Чаще всего применяются полностью пористые частицы
силикагеля (рис. 5.1в) в силу большой емкости, позволяющей разделять большее
количество образца, и доступности в различных вариантах (сорбенты с различной
селективностью, размером частиц и пор, а также широкий выбор готовых колонок
разного размера и т.д.). Наиболее популярны частицы диаметром 1,5—5 мкм.
В настоящее время такие частицы часто получают путем агрегации гораздо меньших
сферических частиц.
Пе/ыикулярные частицы (рис. 5.16) представляют собой твердые сферы, покрытые
очень тонким слоем неподвижной фазы. Сорбенты, состоящие из таких частиц
1 Метод газовой адсорбции наиболее эффективен при измерении пор в диапазоне от 0,5 до 100 нм,
в то время как метод ртутной порометрии — в диапазоне от 20 нм до 1000 мкм. — Прим. перев.
240 Глава 5. Колонки
(б)
Микро-
пелликулярная
частица
Пористая частица
Проходные поры
Пористая
оболочка
Дифузионная
Повернохностно-пористая
частица
<в)
Перфузионная частица
Рис. 5.1. Различные виды частиц стационарной фазы
на основе силикагеля или полимера (диаметром 1,5—2,5 мкм, а раньше гораздо
большего размера) обеспечивают при разделении макромолекул более высокое число
теоретических тарелок вследствие улучшения массопереноса в неподвижной фазе
(меньше коэффициент С, раздел 2.4.1.1, [2, 3|). Так как площадь поверхности этих
частиц мала, удерживание существенно уменьшается, и разделить можно гораздо
меньшее количество образца (при этом пропорционально падает чувствительность
детектирования). Пелликулярные частицы в гораздо большей степени подходят для
анализа основных компонентов образца, а не следовых количеств примесей.
Главным образом их используют при разделении крупных биомолекул (глава 13).
У поверхностно-пористых частиц (их также называют частицами с плавленым
ядром/сердечником, частицами с оболочкой или частицами с контролируемой
пористостью поверхности*) твердое ядро и пористая внешняя оболочка (рис. 5.1в). Они
обычно имеют диаметр от 2 до 5 мкм, а толщина внешней оболочки варьируется
1 По-английски все это называется /used-core"
porosity particles, соответственно. — Прим. персе.
particles, shell particles или controlled-surface-
5.2. Носители 241
от 0,25 до 0,5 мкм. По сравнению с пелликулярными частицами у поверхностно-
пористых частиц гораздо больше площадь поверхности (она составляет примерно
3/4 площади поверхности полностью пористых частиц), поэтому удерживание у них
больше, и можно вводить большее количество образца. Относительно тонкая
внешняя пористая оболочка позволяет сократить время разделения, особенно для
макромолекул [4, 5|. Кроме того, число теоретических тарелок колонок, упакованных
такими частицами, несколько больше, чем у колонок с полностью пористыми
частицами (раздел 5.2.2.1).
Перфузионные частицы (рис. 5.1г) имеют достаточно большие (400—800 нм) поры
(проходные поры), соединенные сеткой мелких (30—100 нм) пор. Сравните: диаметр
пор большинства других частиц (диффузионных пор) находится в интервале 8—30 нм.
Растворенные вещества переносятся не только под действием диффузии, но и сквозь
перфузионные частицы потоком подвижной фазы при высоких скоростях пото
ка |6, 7|. Утверждается, что такой дополнительный вклад в массоперенос в
неподвижной фазе снижает размывание зоны вещества, особенно для крупных молекул
при более высоких скоростях потока. Перфузионные частицы обычно достаточно
крупны (10 мкм), поэтому они предпочтительно используются и при более низких
давлениях. Такие частицы в наибольшей степени подходят для препаративных
разделений макромолекул (например, белков). Их очень редко используют для разделения
небольших молекул.
5.2.1.2. Размер частиц и диаметр пор
В период с 1980 до 2000 годов рутинные разделения предпочитали проводить на
колонках с частицами диаметром ~5 мкм. Частицы такого размера позволяют найти
компромисс между эффективностью, перепадом давления, удобством использования,
требованиями к оборудованию и сроком службы колонки. Однако в настоящее вре
мя становятся более популярными частицы диаметром 3 мкм и меньше, главным
образом потому, что они способствуют сокращению продолжительности анализа и
увеличению количества анализируемых проб в единицу времени, т.е.
производительности (разделы 2.5.3.2 и 9.3.9.2). Колонки, упакованные частицами малого размера,
дают узкие пики, и поэтому необходимо сводить к минимуму внеколоночное
размывание (разделы 2.4.1.1 и 3.9), особенно при использовании колонок с меньшим
диаметром. Колонки с частицами диаметром 2 мкм используются преимущественно
в системах ВЭЖХ, которые могут работать при более высоком давлении (например,
до I020 бар или 15000 psi, раздел 3.5.4.3).
В таблице 5.1 приведены некоторые физические характеристики частиц сорбентов
ВЭЖХ и дана шкала для иллюстрации влияния этих свойств на параметры разделения
от благоприятного («4») до неблагоприятного («1»). Для разделения молекул массой
менее 10000 Да чаще всего используют полностью пористые и поверхностно-пористые
частицы с диаметром пор 8—12 нм. Площадь поверхности сорбента, состоящего из
таких частиц, составляет примерно 125—400 м2/г. Это значит, что на колонке с
внутренним диаметром 4—5 мм можно разделять около 50 мкг образца. Поскольку есть
возможность обработать пики, соответствующие нескольким нанограммам вещества,
в одном разделении (или анализе) на такой колонке можно провести количественное
определение как целевого продукта, так и следовых количеств примесей.
Для крупных молекул (в основном биомолекул, например, белков) нужны
крупные поры, чтобы не создавать препятствий для диффузии в порах, снижающих
эффективность колонки (раздел 13.3.1.1). Молекулы массой более 10000 Да обычно
разделяют на колонках, упакованных сорбентом с размером пор = 30 нм. Если диаметр
пор по крайней мере в 4 раза больше гидродинамического диаметра молекул
вещества, то можно избежать ограничения диффузии и, следовательно, уменьшения числа
242 Глава 5. Колонки
теоретических тарелок |8|. При увеличении размера пор уменьшается площадь
поверхности сорбента, а значит, и количество вещества, которое можно разделить.
В обращенно фазовой хроматографии (ОФХ) в основном используют колонки C|g.
Физические свойства некоторых наиболее популярных сорбентов этого вида
приведены в таблице 5.2.
Таблица 5.1. Характеристики частиц сорбентов ВЭЖХ и их влияние на разделение
Вид частиц
5 мкм полностью пористые
3,5 мкм полностью пористые
2—3 мкм полностью пористые
< 2 мкм полностью пористые
5 мкм поверхностно-пористые
2—3 мкм поверхностно-пористые
] ,5 мкм пелликулярные (непористые)
Скорость
разделения
1
2
3
4
2
4
4
Давление
4
3
2
1
4
2
1
Устойчивость
разделения
4
4
2
2
3
4
2
Удобство
использования
4
4
3
1
3
3
1
Стабильность
колонки'
4
4
3
3
3
4
1
Диаметр пор
7-12 нм (150^100 м2/г)
15—100 нм (5-150 м2/г)
Разделение небольших молекул (< 10000 Да)
Разделение крупных молекул (> 10000 Да)
Примечание: в таблице дана оценка преимуществ, обеспечиваемых для различных параметров
разделения, от посредственной (1) до отличной (4).
"Способность сорбента выдерживать высокое давление или быстрое изменение давления.
Таблица 5.2. Характеристики част и и некоторых коммерческих сорбентов С|8
Сорбенты
Асе C[g
Ascentis C|8
Halo Cig"
Hypersil Gold C|g
Luna Си (2)
Sunfirc C]8
TSK-GEL ODS-100V
XBridge С,/
Zorbax XDB-C|8 Plus
Диаметр пор
(HM)
10
10
9
17,5
10
10
10
13,5
9,5
Площадь
поверхности (мгД)
300
450
150
220
400
340
450
185
160
Содержание
углерода (%)
15,5
25
S
10
17,5
16
15
данных нет
8
Покрытие привитой
фазой (мкммоль/м1)
данных нет
3,7
3,5
данных нет
3,0
данных нет
данных нет
3,1
данных нет
"Поверхностно-пористые частицы, остальные полностью пористые.
^Комбинированная стационарная фаза (раздел 5.3.2.2).
5.2.2. Носители на основе силикагеля
Силикагель — самый распространенный носитель в ВЭЖХ. Он бывает в виде частиц
или монолитного стержня. Существенным достоинством силикагеля является высокая
механическая прочность, поэтому колонки, упакованные силикагелем или сорбентами
5.2. Носители 243
на его основе, стабильны в течение продолжительного времени и выдерживают высокое
рабочее давление. Для них характерно большее число теоретических тарелок по
сравнению с колонками, упакованными сорбентами с другими носителями (раздел 5.2.5).
Можно синтезировать сферические частицы силикагеля разного размера (1,5, 2,7, 3,5,
5 мкм), с разным размером пор (10, 30, 100 нм) и разных видов (см. рис. 5.1). Силика-
гель также используют и в виде монолитных сорбентов (раздел 5.2.4).
Еще одним преимуществом силикагеля является возможность его
модифицирования различными лигандами (Cg, C|g, фенил, циано; раздел 5.3.1). Такие сорбенты
требуются для разделения разнообразных соединений и для изменения селективности
разделения. Сорбенты на основе силикагеля совместимы со всеми органическими
растворителями и водой. В отличие от полимерных частиц (раздел 5.2.3) частицы
силикагеля не набухают и не сжимаются при замене растворителя, поэтому они особенно
подходят для градиентного элюирования,
когда во время разделения изменяется
состав подвижной фазы. Силикагель,
однако, начинает растворяться в подвижной
фазе при рН > 8 [9| и это сокращает срок
службы колонки. Тем не менее,
существуют специальные привитые фазы,
которые стабилизируют частицы силикагеля
при более высоком рН (раздел 5.3.2.1).
Кроме того, колонки с другими
носителями более стабильны при высоком рН
(раздел 5.2.5). Еще одним недостатком
силикагеля является высокая вероятность
размывания заднего фронта пика при
разделении основных соединений, но эта
проблема в значительной мере была
решена с помощью применения силикагеля
высокой чистоты (раздел 5.2.2.2).
В настоящее время в основном
используются частицы силикагеля
сферической формы. Сферические частицы
легче упаковывать, и этот процесс лучше
воспроизводится; в результате
получаются эффективные колонки. Помимо
этого, сферические частицы достаточно
прочны. Крупные частицы неправиль
ной формы используют, главным
образом, для препаративных разделений, что
объясняется их дешевизной и
меньшими требованиями к эффективности (раз
дел 15.3.1.2). Существуют частицы на
основе силикагеля, выдерживающие
давление 1020 бар (15000 psi). Особенно
устойчивы к высоким давлениям
поверхностно-пористые и пелликулярные
частицы с твердым ядром. Сферические
частицы можно синтезировать
несколькими разными методами (раздел 5.2.2.3,
|1, 4]). На рис. 5.2я показан внешний
вид некоторых коммерчески доступных
Zorbax-PSM-ЗСЮ
Mudeosil-7-ЗОО
(в)
(б)
AtJantis 3 мкм
Рис. 5.2. Внешний вид частиц силикагеля
нескольких типов сорбентов ОФХ,
увеличение на рисунках (б) в 7 раз больше, чем на
рисунках (а)
244 Глава 5. Колонки
частиц силикагеля (рельеф их поверхности, распределение по форме и размеру),
а увеличенное изображение широко используемых частиц диаметром около 3 нм
приведено на рис. 5.26. На рисунках видны существенные отклонения формы частиц
от истинной сферической.
5.2.2.1. Эффективность колонки
Размер частиц в первую очередь определяет эффективность колонки, измеряемую
числом теоретических тарелок N. Это проиллюстрировано на рис. 5.3, на котором
показаны графики зависимости высоты теоретической тарелки Н (обратно пропор
циональной N) от линейной скорости подвижной фазы и (пропорциональной
скорости потока) для нескольких колонок с разным диаметром частиц. При уменьшении
диаметра пористой частицы d от 5 до 1,8 мкм снижается высота теоретической
тарелки Н, что соответствует возрастанию эффективности колонки на мм ее длины.
Приведенная высота тарелки h = H/d„ колонки, хорошо упакованной полностью
пористыми частицами, примерно равна 2, что до 2006 года считалось нижним
пределом, характеризующим качественную упаковку частицами этого типа (раздел 2.4.1).
Тип частиц и их распределение по размеру также может влиять на
эффективность колонки, влияя, по-видимому, на однородность упакованного слоя. Пример
показан на рис. 5.3 для колонки, упакованной поверхностно-пористыми частицами
размером 2,7 мкм. Эта колонка (Л = 1,5) более эффективна, чем колонка,
упакованная полностью пористыми частицами диаметром 1,8 мкм. Более высокая
эффективность колонки с частицами диаметром 2,7 мкм может быть обусловлена двумя
разными причинами. Во-первых, это чрезвычайно узкое распределения этих частиц
по размеру (+5—6%, I СО). Можно сравнить его с распределением частиц по размеру
+ 15—20% (I СО) для большинства коммерческих сорбентов с аналогичным размером
частиц [5| (пример на рис. 5.4). Согласно компьютерному моделированию |10|
упаковка частиц с более узким распределением по размеру должна приводить к
получению колонок с более высоким значением N и улучшенной проницаемостью
20
18
16
14
— 12
2
3. ю
8
6
4
2
0 2 4 6 8 10 12
Линейная скорость подвижной фазы (мм/сек)
Рис. 5.3. Зависимость эффективности колонки от размера и типа частиц. Образец —
нафталин. Условия: колонка Си 50 х 4,6 мм, подвижная фаза 60% ацетонит-
рил-вода, 23 "С. Материал любезно предоставлен Advanced Materials
Technology
5 мкм полностью
пористые частицы
3,5 мкм полностью
пористые частицы
1 .В мкм полностью
пористые частицы
2,7 мкм поверхностно-
пористые частицы
5.2. Носители 245
3000
2500
з
Й 2000
со
S 1500
3
т
с 1000
о
500
12 3 4 5 6 7
Диаметр частиц, мкм
Рис. 5.4. Более узкое распределение по размеру поверхностно-пористых частиц
(Halo™), которыми упакована колонка (см. график рис. 5.3), по сравнению
с распределением по размеру полностью пористых частиц. Материал
любезно предоставлен Advanced Materials Technology
(т.е. с меньшим перепадом давления при эквивалентности остальных параметров
разделения) [11, 12|. Кроме того, колонки с более узким распределением частиц
по размеру гораздо стабильнее |13|.
Второй возможной причиной исключительно высокой эффективности колонки,
упакованной поверхностно-пористыми частицами (рис. 5.3), является более высокая
плотность этих частиц (~1,4 г/см3), которая на 30—70% выше, чем у полностью
пористых частиц. Частицы с более высокой плотностью можно упаковать более
эффективно, как и частицы большего размера. На рис. 5.5 изображена микрофотография
поверхностно-пористой частицы в поперечном сечении, сделанная с помощью
трансмиссионного электронного микроскопа. Такая частица состоит из плотного
ядра, покрытого пористой оболочкой. На рис. 5.6 показана хроматограмма
экспресс-разделения образца, состоящего из смеси гербицидов, на колонке,
упакованной поверхностно-пористыми частицами размером 2,7 мкм.
Рис. 5.5. Поперечное сечение поверхностно-пористых частиц (Halo™) с порами 9 нм
(электронная микрофотография). Материал любезно предоставлен Advanced
Materials Technology
Распределение по размеру
частиц Halo
Среднее значение = 2.77 мкм
(± 6%, 1 СО)
Колонка, упакованная 3 мкм
сорбентом
Среднее значение = 3,78 мкм
.19%, 1 СО)
246 Глава 5. Колонки
Рис. 5.6. Экспресс-разделение на колонке, упакованной поверхностно-пористыми
частицами. Образец: 1 — ацетон, 2 — тебутиурон, 3 — тидиазурон, 4 — фторме-
турон, 5 — диурон, 6 — пропан ил, 7 — сидурон, 8 — линурон, 9 — дифтор
бензурон. Условия: колонка Halo Cjs 50 х 4,6 мм (2,7 мкм), подвижная фаза
45% ацетонитрил-вода, скорость потока 4,0 мл/мин, 60 "С, 231 бар (3400 psi).
Материал любезно предоставлен Advanced Materials Technology
5.2.2.2. Структура поверхности силикагеля
Химическая структура поверхности немодифицированного силикагеля различна и
зависит от методов его получения. Она существенным образом влияет на свойства
силикагеля 114—17|. Поверхностный слой силанольных групп (-SiOH),
концентрация которых составляет = 8 мкмолей/м2, является характерной чертой всех
полностью гидратированных силикагелей. Силанольные группы силикагеля
взаимодействуют с силанами с образованием привитой фазы (раздел 5.3.1). При нагревании
силикагеля до температуры выше 800 "С большинство силанольных групп
отщепляется, и сорбент становится непригодным для ВЭЖХ. На поверхности гидратирован-
ного силикагеля силанольные группы находятся в трех разных формах 114, 16—20|
(рис. 5.7а—в). Характеристики колонки очень сильно зависят от кислотности
силанольных групп. Свободные (не связанные водородными связями) силанольные
группы в большей степени проявляют кислотные свойства, что приводит к меньшим
значениям N и увеличению размывания заднего фронта пика основных соединений.
Еще большее влияние на кислотность силанольных групп и характеристики
колонки оказывает чистота силикагелевых носителей. Возможные примеси
определенных металлов (особенно А1|1Н| и Fe|lll|) могут увеличивать кислотность силикагеля,
притягивая электроны атома кислорода силанольной группы (рис. 5.7d), а также могут
непосредственно взаимодействовать с хелатируюшими соединениями (рис. 5.7г, 5.20ц).
В результате загрязнение поверхности силикагеля металлами может приводить к
размыванию заднего фронта пика и низкому выходу некоторых анализируемых веществ.
Современные хроматографические силикагели намного чище, о чем свидетельствуют
данные для одного из видов силикагеля, приведенные в таблице 5.3 (кроме того, есть
тесты для непосредственного измерения кислотности силикагеля и способности
образовывать комплексы, раздел 5.4.1 и |18, 21|). По чистоте и катионообменным
свойствам частицы силикагеля, полученный из них сорбент и упакованные им колонки
можно подразделить на типы А и В |15| (раздел 5.4.1). Силикагели типа А менее
чистые и имеют большую кислотность. До 1990 года они использовались только в ВЭЖХ
и до сих пор находят применение в некоторых областях, где требования к ним не столь
5.2. Носители 247
(а)
ОН
1
„..Si-
Свободная
силанольная
группа <г)
(б)
НО
—■ s'~"
ОН
Геминальные
сипанопы
М+
_1_
Металл на
поверхности
(б)
И
ОН -■■ ОН
1 1
..„Si Si-
Ассоциирован н ые
силанолы
VI
-М+ --Si—-
Внутренний металл
(активированная
силанольная группа)
Рис. 5.7. Силанольные группы разного типа на поверхности силикагеля
жестки, например, при пробоподготовке
(раздел 16.6) и в препаративной
хроматографии (глава 15). Степень ионизации сила-
нольных групп зависит от типа силикагеля
и рН подвижной фазы и определяется по
относительному удерживанию
неорганического катиона, например, Li+. На рис. 5.8
иллюстрируется ионизация или «катионооб-
менная емкость» силикагелей типа А, В и
«гибридного» силикагеля в колонках C]g
(раздел 5.3.2.2). Относительная кислотность
этих трех колонок Сц$ уменьшается в ряду:
тип А 3> тип В > гибридный.
Обычно основные, ионные или
ионизируемые соединения лучше разделяются
на колонках типа В. На рис. 5.9 проводится
сравнение разделения некоторых основных
Таблица 5.3. Репрезентативные результаты
анализа следовых количеств металлов
в Zorbax Rx-SIL метолом АЭС-ИСП/МС*
Элемент
№
Са
К
А1
Fe
Mg
Zn
Содержание (10~~')
10
2
< 3
1,5
3
4
1
'Атомная эмиссионная спектроскопия
с индуктивно связанной плазмой/масс -
спектрометрия. — Прим. персе.
-^
Э
т
i
л
и
О
?
Ф
ПС
ш
X
т
V3
8
X
S
1-
ь;
Тип А
i ■ ■ i
Тип В __, .-*
--»
_, •■** Гибридный _
10
РН
Рис. 5.8. Относительная кислотность (степень ионизации силанольных групп)
силикагелей разного типа, измеренная по удерживанию иона Li+ [22]
(((Г 248 Глава 5. Колонки
(б)
Силикагель тип В
10
4 L^Ju
iii i i
15
20
25 (мин)
Рис. 5.9. Сравнение разделений протежированных оснопных соединений на колонках
типа А и типа В. Образец представляет собой смесь четырех трициклических
антидепрессантоп. Условия: колонка Сц 150x4.6 мм. подписная фаза 30%
ацетонитрил-фосфатный буфер с рН 2,5 |19]
лекарственных препаратов на колонках C|g типа А и В. Видно, что на колонке с сили-
кагелем типа В пики эже и симметричная (рис. 5.96). Соображения, изложенные в
разделе 5.4.4.1, помогут подобрать колонку с меньшей кислотностью для сведения к
минимуму размывания заднего фронта пика. После 1990 года производят главным
образом сорбенты на основе силикагеля типа В.
5.2.2.3. Подготовка частиц
Существует несколько методов подготовки частиц для колонок ВЭЖХ. Полностью
пористые частицы силикагеля получают по золь-гель методике, включающей
эмульгирование золя силикагеля в несмешивающейся с ним неполярной жидкости с
образованием мелких капель и их переводом в сферические частицы гидрогеля
силикагеля; последующую сушку и отбор узкой фракции частиц с минимальным разбросом
по размеру. Контролируя рН, температуру и концентрацию золя силикагеля,
получают частицы с нужным распределением по размеру частиц и пор. Есть и другой
способ получения эмульсии — распыление с высушиванием золя силикагеля или
растворов тонкоизмельченного силикагеля, чтобы образовались сферические полностью
пористые частицы. Полученную пористую структуру модифицируют
гидротермальной обработкой в водных средах (или паром) при повышенных температуре и
давлении.
Существует другой метод получения полностью пористых частиц силикагеля:
конденсация или сшивка микрочастиц (рис. 5.10). Одна из методик заключается
в диспергировании золя силикагеля в полярной жидкости с последующим
добавлением полимеризующегося органического соединения (формальдегида с мочевиной
или меламина). При этом происходит расслоение золя силикагеля и полимера с
образованием сферических агрегатов одинакового размера. Полученную смесь сначала
нагревают для удаления органического полимера, а затем спекают при более
высокой температуре для стабилизации сети сообщающихся пор в частицах золя силика-
5.2. Носители 249 ,
° о °0 о
°о°о о ° о
оо°о °о°
о о0о-оо
микрочастицы силикагеля сферический агрегат
Рис. 5.10. Конденсация микрочастице образованием полностью пористых частиц
геля. В дальнейшем из небольших частиц золя силикагеля получают частицы с
меньшими порами, а из крупных — частицы с порами большего размера. Этот метод
конденсации также подходит для получения полностью пористых частиц циркония.
Так называемые гибридные (органические/неорганические) частицы получают
сополимеризацией органических силанов с тетраалкил-о-силикатами или другими
органическими силикатами. Структуру пор частиц, полученных этим методом,
можно модифицировать подходящей гидротермальной обработкой. После гидролиза
можно провести дальнейшую модификацию поверхности частиц взаимодействием
с другими силанами для прививки нужного лиганда.
Поверхностно пористые частицы силикагеля получают, покрывая сферическое
твердое ядро силикагеля частицами золя силикагеля. Главная идея этого метода —
поочередно покрывать частицы слоями отрицательно заряженного золя силикагеля и
противоположно заряженного органического полимера до тех пор, пока не будет по
лучена нужная толщина оболочки. Затем при нагревании удаляют промежуточные
органические слои, а частицы стабилизируют спеканием. Небольшие частицы золя
силикагеля образуют небольшие поры, а крупные — поры большего размера.
Иной подход используется для получения частиц из графитированного углерода
(раздел 5.2.5.3). Например, полимеризуюшееся органическое соединение вводят
внутрь пор частиц силикагеля и затем проводят его (органического соединения)
полимеризацию внутри пор. Полученные частицы силикагеля с полимером обрабатывают
плавиковой кислотой для растворения силикагеля и удаления его из сферических
пористых полимерных частиц. Затем полимерные частицы нагревают в отсутствие
кислорода до высокой температуры, и происходит графитизация. Более подробную
информацию о получении частиц для колонок ВЭЖХ можно найти в работе [11.
5.2.3. Пористые полимеры
Пористые полимерные частицы (главным образом, на основе поперечно-сшитого
полистирола) используют в обращенно-фазовой, ионообменной и эксклюзионной
хроматографиях. Другие полимеры, например, коммерчески доступные замещенные
метакрилаты и поливиниловые спирты применяются реже. Полимерные подложки
можно дериватизировать обращенно-фазовыми лигандами -C]g, и такими как -NH2 и
-C-N, чтобы получить сорбенты с разной селективностью или разными ионными
группами для ионообменной хроматографии. Однако, по сравнению с количеством
имеющихся в продаже сорбентов на основе силикагеля выбор коммерчески доступных
сорбентов с разными ОФ-лигандами, привитыми к полимерным частицам, в
настоящее время совсем невелик. Основным преимуществом полистирольных сорбентов
является стабильность при любом значении рН (0 < рН < 14). Колонки с сорбентами
на основе полимеров можно использовать в ОФХ-разделениях оенбвных соединений
при рН > 10 для подавления ионизации образца и предотвращения размывания
заднего фронта пика, которое иногда наблюдается при разделении на сорбентах на основе
250 Глава 5. Колонки
силикагеля (раздел 7.3.4.2). Однако на смену этим полимерным сорбентам пришли
новые ОФХ-сорбенты типа В, стабильные при высоком рН.
Полимерные частицы, модифицированные ионизирующимися
функциональными группами (-СООН, SO3H, -NH2, NR3+), используют, главным образом, в
ионообменных разделениях (раздел 7.5, 13.4.2). Для разделения небольших молекул
можно производить полимерные сорбенты с узкими порами, а для крупных молекул,
например, белков — с широкими порами. С полимерных колонок, стабильных
при высоком рН, можно элюировать сильно удерживаемые примеси образца
0,1М NaOH (раздел 13.4.2.1).
У сорбентов на пористой полимерной основе есть несколько недостатков.
Колонки, упакованные такими сорбентами, имеют более низкое значение N по
сравнению с колонками, упакованными частицами на основе силикагеля такого же
размера. Кроме того, может возникнуть проблема, связанная с разной степенью набухания
(или сжатия) сорбентов на основе полимерных частиц в различных органических
растворителях. Это приводит к потере эффективности колонки или повышению
в ней давления. Побочные явления, вызванные набуханием сорбента, гораздо
сильнее проявляются в градиентных разделениях, поскольку происходит постепенное
изменение доли органического растворителя (%В) в подвижной фазе. Впрочем, были
разработаны сорбенты на основе полимерных частиц с минимальным набуханием
для разделений в градиентном режиме.
5.2.4. Монолитные сорбенты
С 1990 года растет интерес к монолитным сорбентам. Монолитная колонка
формируется как прочный жесткий цилиндр в процессе полимеризации силикагеля или
полимера. На рис. 5.11 показаны электронные микрофотографии монолитных
сорбентов из силикагеля (рис. 5.11я) и полимера (рис. 5.116). Монолиты выпускаются
в виде цилиндров или дисков. Последний вариант используется, главным образом,
для синтеза пептидов или других приложений в биологии. В этой книге к
монолитным дискам мы больше не будем возвращаться. Далее речь пойдет о монолитных
колонках на основе силикагеля и полимера, будут рассмотрены их достоинства и
недостатки, а также некоторые важные области их применения. Об истории, разработке и
характеристиках монолитных сорбентов можно узнать из литературы |24—27].
(в) (в)
Рис. 5.11. Электронные микрофотографии монолитных сорбентов на основе (а)
силикагеля; (6") полимера. Материал из |23| с разрешения EDM Chemicals Inc.
(а) и Ф. Свек (6)
5.2. Носители 251
5.2.4.1. Монолиты на основе силикагеля
Монолиты изготавливаются в виде стержней из одного куска пористого силикагеля,
поэтому не требуется упаковывать колонку частицами по всей ее длине (раздел 5.6).
Коммерчески доступные монолиты не изготавливают внутри пустой колонки, а
формуют, сушат и потом покрывают полимерной оболочкой. Модификацию
поверхности полученного монолита (например, прививку лигандов C|g) проводят позже.
У монолитного сорбента пиры двух типов: крупные макропоры диаметром ~2 мкм и
небольшие мезопоры диаметром ~10 нм. Площадь поверхности (например, 300 м2/г)
в основном образована мезопорами, а элюент в колонке течет сквозь макропоры.
Объем подвижной фазы в обычной колонке составляет около 40% от ее полного
объема и фактически не зависит от размера частиц или распределения частиц по
размеру. Монолитом же можно упаковать колонку так, что объем подвижной фазы (объем
макропор) будет превышать 80%.
Диаметр и общий объем макропор монолита определяют проницаемость колонки
и давление, требуемое для создания нужной скорости потока. Поскольку макропоры
монолитной колонки довольно велики, то необходимое давление для протекания
подвижной фазы у нее будет гораздо меньше, чем у обычной колонки, упакованной
частицами «эквивалентного» диаметра (вычисленного из приведенного далее
уравнения 5.5). По сравнению с колонками, упакованными частицами, для монолитных
колонок можно устанавливать большую скорость потока без потери в эффективности.
Исследование разделения низкомолекулярных образцов позволяет предположить, что
эффективность коммерчески доступных монолитных колонок и колонок, упакованных
4—5 мкм частицами, сопоставима [28]. При этом возникает важный вопрос: как
сравнивать перепад давления и число теоретических тарелок монолитной и обычной
колонок. Результаты другого исследования указывают на то, что монолитные колонки еще
полезнее при необходимости разделения с высоким разрешением (например, когда
требуется N > 100000) [29|, но не все исследователи согласны с этим выводом |30|. В
обзоре [24| проводится более подробное сравнение монолитных и обычных колонок.
У монолитных колонок на основе силикагеля есть ряд недостатков:
ограниченный выбор размеров колонок, имеющихся в продаже, ограниченный выбор
стационарных фаз, наблюдаемая тенденция размывания заднего фронта пиков (например,
фактор размывания ~1,2) даже для нейтральных (неионизируемых) соединений |28|.
На момент написания данной книги монолитные колонки находили лишь
ограниченное применение в рутинных анализах, однако результаты исследований в то
время были многообещающими [31], особенно для капиллярных монолитных
колонок. Немногочисленные исследования говорят также о том, что монолиты на основе
силикагеля могут изготавливаться с приемлемой для рутинного анализа
воспроизводимостью |28, 32|. Значение этих колонок в будущем еще предстоит оценить.
5.2.4.2. Монолиты на полимерной основе
В настоящее время в продаже имеются монолиты на основе органических полимеров
для решения широкого спектра задач. Такие колонки получают путем сополимериза-
ции мономеров, например, стирол/дивинилбензола с моновинил/дивинил метакри-
латами. Сополимеризация других мономеров с различными свойствами позволяет
получать частицы с разными лигандами и разной селективностью в ОФХ.
Полимерные монолиты формируются непосредственно в цилиндре колонки и даже внутри
капиллярной колонки диаметром 0,5 мм и еще меньше. При изготовлении монолита
можно контролировать одновременно два параметра: природу полимеризуемого
материала и структуру пор. Монолиты с широкими порами подходят для разделения
крупных биомолекул (пептидов, белков, олигонуклеотидов и т.д.), как и для
капиллярных колонок, которые при их достаточной длине можно использовать для
разделения сложных образцов, требующих большого значения N |33|.
252 Глава 5. Колонки
5.2.5. Другие неорганические частицы
Несмотря на то, что хроматографисты обычно предпочитают использовать сорбенты
на основе силикагеля, определенное применение нашли и другие неорганические
носители. В этом разделе приводится обзор неорганических носителей и описание
областей их применения. Поверхность немодифицированных оксидов металлов
отличается по химическому строению от поверхности силикагеля, поэтому прививка
неподвижной фазы проводится другими методами: (I) поверхность покрывают
полимером или слоем углерода, (2) лиганды ковалентно связывают с поверхностью,
(3) в подвижную фазу вводят добавки с высокой реакционной способностью. Будут
описаны частицы из оксидов циркония, алюминия, титана и из графитированного
углерода. В таблице 5.4 перечислены несколько коммерчески доступных колонок
с сорбентами на основе оксидов циркония и алюминия. В таблице 5.5 обобщаются
некоторые достоинства и недостатки колонок с сорбентами на основе различных
неорганических соединений.
Таблица 5.4. Имеющиеся в продаже колонки с сорбентами на основе оксида циркония
или алюминия
Колонка"
ZirChrom-PBD
ZirChnom-PS
Aluspher RP-select В
Millpore PBD
Unisphere
GammaBond RP-1 (PBD)
Gam ma Bond RP-8
Подложка
Оксид циркония
Оксид циркония
Оксид алюминия
Оксид алюминия
Оксид алюминия
Оксид алюминия
Оксид алюминия
Диаметр
частиц (мкм)
3 или 5
3 или 5
5
5
10
5
5
% углерода
данных нет
данных нет
данных нет
7,2
5,1
данных нет
данных нет
Размер
пор (ни)
30
30
10
9
22
8
8
Плошадь
поверхности (м2/г)
30
30
170
110
37
данных нет
данных нет
"Часть данных из [35].
Таблица 5.S. Сравнение хроматографических характеристик частиц силикагеля и оксидов
металла
Свойства
Монодисперсность частиц
Структура пор
Плошадь поверхности/диаметр пор
Химические свойства поверхности
Механическая прочность
Химическая стабильность
Термическая стабильность
Эффективность колонки
Энергетическая однородность"
Силнкагель
3
3
4
3
4
2
2
3
3
Оксид
титана
2
данных нет
2
данных нет
данных нет
2
данных нет
данных нет
1
Оксид
алюминия
2
данных нет
2
2
данных нет
3
данных нет
2
1
Оксид
циркония
2
2
2
2
4
4
4
3
1
Примечание: в таблице приведены оценки преимуществ, обеспечиваемых для хроматографиче-
ского разделения, от низкой или нейтральной (1) до высокой или отличной (4).
"В отличие от силикагеля у других частиц есть активные адсорбционные центры. Это
обусловливает низкие значения N и неполное извлечения образца.
5.2. Носители 253
5.2.5.1. Оксид циркония
В общем случае ковалентное связывание лиганда с частицами оксида циркония
провести не удается, поскольку в условиях ОФХ эта связь, как правило, нестабильна.
Обычно необходимый сорбент на основе оксида циркония получают, покрывая
поверхность частиц органической фазой или проводя полимеризацию органической
фазы на поверхности. Полибутадиен или полистирол в качестве неподвижной фазы
обеспечивают селективность сорбента, сходную с селективностью алкилированного
силикагеля (по крайней мере, для неионизированных образцов). Такие полимерные
сорбенты на основе оксида циркония демонстрирует поразительную стабильность
как при низких, так и при высоких значениях рН (I < рН < 13), а также при очень
высоких температурах (< 160 °С) [34|. На рис. 5.12 показана хроматограмма
разделения на колонке с сорбентом на основе оксида циркония при рН 13 и Т = 140 °С.
У первых колонок, упакованных частицами оксида циркония с полимерным покры
тием, была довольно низкая эффективность, по-видимому, из-за недостаточного
массопереноса в неподвижной фазе. В то время как производители продолжают
совершенствовать эффективность колонок с сорбентами на основе оксида циркония,
до сих пор (по крайней мере, до издания этой книги) еще не найдена область
применения, в которой преимущества этих колонок перед колонками на основе
силикагеля оказались бы очевидными. Хотя это может отражать ограниченное использование
высоких температур (> 60 °С) в настоящее время.
Вне зависимости от метода изготовления сорбента на основе оксида циркония
при разделении ионизируемых образцов форма пиков обычно искажена.
Следовательно, чтобы пики имели правильную форму, а число теоретических тарелок было
приемлемым, требуются специальные добавки в подвижную фазу (например,
фосфаты или фториды) при разделении ионизируемых образцов на сорбентах на основе
оксида циркония |34, 35|. Причем при достаточно большой концентрации добавок
значения N и форма пиков основных соединений будут такими же, как и при
разделении на колонках с алкилированным силикагелем (но при рН < 10). Когда
выходила из печати эта книга, для разделений пептидов и белков на колонках с сорбентами
на основе оксида циркония были получены несколько худшие результаты, чем
для их разделений на колонках с алкилированным силикагелем.
| 1 1 1 1 1 1 т 1 1
0.2 0.4 0.6 0.8
Время (мин)
Рнс. 5.12. Разделение смеси фармацевтических препаратов при высокой температуре.
Образец: 1 — доксиламин, 2 — метаперилен, 3 — хлорофенирамин, 4 —
меклизин, 5 — трипролидин. Условия: колонка ZirChrom-PBD* (окись
циркония) 100 х 4,6 мм, подвижная фаза 20% ацетонитрил/вода с добавлением
гидроксида тетраметиламмония до рН 13, скорость потока 4,2 мл/мин,
температура 140 "С, давление 194 бар (2850 psi). С разрешения ZirChrom
Separations, Inc.
254 Глава 5. Колонки
Ионообменники на основе оксида циркония также можно найти в продаже. Слабые
анионообменники получают, покрывая частицы оксида циркония полиэтиленими-
ном с последующей сшивкой диглицидным эфиром 1,4-бутандиола. Такие колонки
стабильны в интервале рН 3—9, подходят для разделения органических кислот,
неорганических анионов и высокополярных соединений, например, Сахаров. Кроме того,
имеются колонки с термически и химически стабильными сильными анионообмен-
никами для разделений при температурах < 100 °С и рН I —13. Такие сорбенты
получают при сшивке полиэтиленимина на поверхности частиц оксида циркония
1,10-дийододеканом или подобными ему соединениями |34].
Частицы оксида циркония, покрытые углеродом, — сорбент с уникальной
селективностью, отличающейся от селективности других обращение фазовых сорбентов.
Его получают, пропуская сквозь пористые частицы оксида циркония пары органи
ческих веществ при пониженном давлении и температуре ~ 700 °С |34|. От сорбентов
с алкилированным силикагелем сорбент с оксидом циркония, покрытым углеродом,
отличается большей гидрофобностью, на нем лучше разделяются полярные и
неполярные геометрические изомеры, а, кроме того, он способен вступать в п-п
взаимодействия (раздел 5.4.1). Этот сорбент устойчив в интервале рН 0,3—14 при
температуре 40 °С, а при нейтральном рН он выдерживает температуру >200 °С. При низкой
температуре (например, 35 °С) колонкам, упакованным сорбентом на основе оксида
циркония, свойственна низкая эффективность и неправильная форма пиков, но этот
сорбент разрабатывается еще очень недолго по сравнению с сорбентами на основе
силикагеля, поэтому вполне вероятно, что в будущем он будет усовершенствован.
5.2.5.2. Оксиды алюминия и титана
В 1980-х годах появились статьи об обращенно-фазовых колонках на основе оксида
алюминия [361, позже на эту тему был написан обзор |37|. Химические свойства
поверхности оксида алюминия в большей степени сходны со свойствами поверхности
оксида циркония, чем силикагеля, и сорбенты на основе оксида алюминия также
стабильны при высоких рН. Так как ковалентные связи с оксидом алюминия
неустойчивы, вместо дериватизации силанами используются полимерные покрытия или
полимеризация на поверхности частиц. Для этого сорбента характерны ярко
выраженные адсорбционные свойства, поэтому их редко применяют в обращенно-фазо-
вой хроматографии. В современных ВЭЖХ-разделениях непривитый оксид
алюминия не используют, а вот в пробоподготовке он применяется (раздел 16.6.5.2).
В продаже также имеются колонки, упакованные сорбентами на основе оксида
титана. Главным образом, они используются в нормально-фазовой хроматографии.
По сравнению с традиционными сорбентами на основе силикагеля или оксида
циркония, сорбенты с оксидом титана не имеют каких-либо преимуществ, поэтому они
не получили широкого распространения. По большому счету силикагель
безоговорочно остается самой популярной подложкой, поскольку в нем сочетаются наиболее
важные для сорбента свойства (табл. 5.5).
5.2.5.3. Графитированный углерод
Пористый графитированный углерод (ПГУ) сильно отличается от других сорбентов
тем, что он состоит из полностью пористых сферических частиц углерода,
образованных из плоских слоев графита, имеющих гексагональную структуру 138, 39]. У атомов
углерода полностью насыщены все валентности, поэтому удерживание и
селективность ПГУ сильно отличаются от соответствующих параметров других сорбентов. ПГУ
пригоден для разделения как в обращенно-фазовом, так и в нормально-фазовом
режимах, он стабилен при I < рН < 13 и Т < 200 °С. Однако пониженная прочность частиц
ограничивает максимальное давление, допустимое для таких колонок.
5.3. Неподвижные фазы
к
I
10
Время (мин)
Рис. 5.13. Разделение на колонке с ПГУ гиппуровой кислоты и ее метилзамещенных
изомеров. Образец: 1 — 2-метилгиппуропая кислота, 2 — гиппуровая
кислота, 3 — 3-метилгиппуровая кислота, 4 — 4-метилгиппуровая кислота.
Условия: колонка НурегсагЬ 100 х 4,6 мм, подвижная фаза состоит из 30% ацето-
нитркла, 30% изопропанола, 40% 0,1 ТФУК, скорость потока 1,0 мл/мин,
температура 25 "С. С разрешения Thermo Scientific
Удерживание полярных соединений на ПГУ обусловлено сочетанием сильных
гидрофобных, электростатических и поляризационных взаимодействий |38|, поэтому
полярные вещества преимущественно удерживаются даже в условиях ОФХ. В
отличие от колонок с традиционной привитой фазой селективность колонок с графити-
рованным углеродом сложно предсказать, что затрудняет разработку метода. Кроме
того, эффективность колонки и форма полученных пиков могут быть несколько
хуже, чем при использовании традиционных ОФХ-колонок. Тем не менее, пористый
углерод дает возможность разделять стерео- и диастереоизомеры, а также
позиционные изомеры, которые либо плохо разделяются, либо вообще не разделяются
на традиционных сорбентах. На рис. 5.13 показано разделение гиппуровой кислоты
и ее метилзамещенных изомеров на колонке, упакованной ПГУ, при элюировании
подвижной фазой с низким рН.
5.3. Неподвижные фазы
Неподвижная фаза колонки определяет удерживание и селективность разделения
(разделы 2.3 и 5.4) и должна соответствовать определенным требованиям: например,
упакованная ею колонка должна обеспечивать приемлемые стабильность,
воспроизводимость, форму пика и эффективность N. В этом разделе мы рассмотрим методы
приготовления, природу и свойства различных неподвижных фаз. Их селективность
обсуждается отдельно, в следующем разделе 5.4. Большинство стационарных фаз
по своей природе органические и они либо ковалентно связаны с частицами
носителя, либо механически осаждаются на поверхность частиц, что встречается реже.
В некоторых случаях поверхность немодифицированной частицы является
стационарной фазой, например, немодифицированный силикагель используют в
нормально-фазовой хроматографии, включая хроматографию гидрофильных взаимодействий
(ГИХ). В дальнейших разделах предполагается, что неподвижная фаза наносится
на частицы силикагеля. Методики, используемые для других носителей, приведены
ранее в разделах 5.2.3—5.2.5.
256 Глава 5. Колонки
5.3.1. «Привитые» неподвижные фазы
Обращенно-фазовый сорбент обычно получают при ковалентном взаимодействии
(«связывании») органосилана с силанолами на поверхности частиц силикагеля. Так
образуется неподвижная фаза или лиганд R:
X3-Si-R + ^Si-OH -> =Si-0-Si(X2)-R + HX. (5.1)
{силан) (силанол) (конечная фаза)
Функциональная группа X — это обычно -CI или -OEt и/или -СНз (рис. 5.14).
Побочным продуктом (НХ) реакции является НО или этанол. Кроме того, в качестве
функциональной группы X используют и другие заместители, о чем будет сказано далее.
Некоторые привитые фазы получают в реакции замещения по одному положению
(рис. 5.14а). То есть одна молекула силана взаимодействует с одной силанольной
группой на поверхности силикагеля, например, молекула хлордиметил-октадецилсилана
(с лигандом R = C[g), в результате образуется мономерная неподвижная фаза,
сорбент C|g. Другие коммерческие доступные сорбенты получают при
взаимодействии трехзамещенных (или двузамещенных) силанов с силанольными группами
(рис. 5.145 и в, на рисунке показаны две связи силана с силанольными группами,
но возможны и три связи). При определенных условиях реакции может произойти
полимеризация неподвижной фазы (когда используют двузамещенные и трехзамещенные
силаны). Так получают полимерную неподвижную фазу или колонку (не путайте с
«сорбентами на основе полимерных частиц», раздел 5.2.3). Далее будет объяснено, что
свойства мономерных и полимерных сорбентов сильно отличаются во многих отношениях.
На рис. 5.15 показаны различные типы реакций между силанами и
силанольными группами для получения ВЭЖХ-сорбентов. «Вертикальная» фаза полимеризации
(рис. 5.15а) проходит при взаимодействии с дву- или трехзамещенными силанами
(как и на рис. 5.146 или в). В примере на рис. 5.15в видно, что силан,
прореагировавший с поверхностью силикагеля, затем реагирует с одной или несколькими
молекулами силана с образованием полимерной фазы (в водной среде, см. ниже). По срав-
(а)
-^Si-OH + CI-Si(CH3)2R ► -^ Si-0-Si(CH3)2-R + HCI
(б)
-^Si— ОН —Si—О
' + CI3Si-R ► v ^Si(CI)-R + 2 HCI
-^Si— OH ">Si— °
M
-^Si— OH —Si—О
' + <EtO)3S(-H ► x ^Si(OEt)-R + 2ЕЮН
— Si—OH —Si—О
00
-^Si-OH + (EtO)Si(CH3)2-R ► -^Si-0-Si(CH3)z-R + EtOH
Рис. 5.14. Синтез различных неподвижных фаз при взаимодействии силанов с силика-
гелем. (а, г) мономерные неподвижные фазы, (б, в) потенциально
полимерные неподвижные фазы
5.3. Неподвижные фазы 257
(«>
О—Si-ОН
'Si ОН
/ \
О О ОН
-+ 1 1---
Si Si Si Si
Полимерная фаза
Вертикальная полимеризация
(б)
,o-.Si.o..Si .о-.зио^зио-.зио-
I I I I I
о о о о о
--I Ь -+ 1 I--
Si Si Si Si Si
Полимерная фаза
Горизонтальная полимеризация
(в),
(г)
^-Si-£
I
-Si-£
I
-■г
о он о он о
-i 1 1 1- г—
Si Si Si Si Si
Мономерная фаза
(диметил-замещенная)
—<■ метил
^ ^7<^ изо-бутил
о он он о он
-Ч 1 (- + t—-
Si Si Si Si Si
Мономерная фаза
(пространственно защищенная)
Рис. 5.15. Другие виды привитых неподвижных фаз, образующиеся при иных условиях
реакции [40|
нению с мономерными фазами такие сорбенты более стабильны при высоких или
низких значениях рН, так как более плотное покрытие поверхности замедляет
гидролиз подвижной фазой силикагеля и связи лиганд-силикагель. Тем не менее,
удерживание и селективность такого рода сорбентов менее воспроизводимы из-за
неравномерной (неконтролируемой) полимеризации силанов.
При «горизонтальной» полимеризации с самосборкой силанов (Сз и C|g)
образуется общая структура, изображенная на рис. 5.156". Атомы кремния соседних силанов
соединены друг с другом через атомы кислорода (силоксановые мостики Si—О—Si),
а каждый силан связан с силикагелем другой силоксановой связью. Сообщалось,
что такой сорбент более стабилен при низком и высоком рН |40|, однако, на момент
публикации этой книги в продаже такого рода колонок не было.
Мономерная фаза, показанная на рис. 5.15в, наиболее широко используется
в ОФХ-колонках. Коммерчески доступны также сорбенты с несколькими разными
функциональными группами- лигандами (раздел 5.3.3, таблица 5.7). Обычно
используют силаны с боковыми метальными группами (рис. 5.15в). Получают эти
неподвижные фазы при взаимодействии силикагелевого носителя с диметилхлор- или
диметилэтоксисиланами (рис. 5.14а и г), причем одна молекула силана реагирует
с одной силанольной группой. Достоинством такой реакции в соотношении один
к одному является хорошо воспроизводимое образование привитой фазы с четко
определенным покрытием. Колонки с сорбентом, полученным по этой методике,
258 Глава 5. Колонки
демонстрируют самую высокую эффективность благодаря быстрой диффузии
анализируемого вещества сквозь слой менее разветвленной неподвижной фазы. И,
наоборот, для сорбентов с многофункциональными лигандами и высокой степенью
полимеризации может быть характерна более медленная диффузия, что приводит
к низким значениям /V колонок особенно при высоких скоростях потока.
Реакции, изображенные на рис. 5.\Лв и г, обычно проводят с алкоксисила-
нами, имеющими реакционноспособные группы R (лиганды) R= C3 NHj
и R= Сз О СН(ОН) СН2ОН. В результате соответственно получаются аминофазы
и диольные фазы. Некоторые неподвижные фазы (например, с реакционноспособ
ними гидроксильными группами и аминогруппами) нельзя получить с
использованием хлорсиланов из-за нежелательных побочных реакций лиганда. Поэтому такие
неподвижные фазы получают из алкоксисиланов (рис. 5. \4в и г). Реакции с ними
идут немного медленнее, чем с хлор- и диметиламиносиланом, при этом для
эквивалентного выхода требуется более высокая концентрация силана.
Пространственно защищенная привитая неподвижная фаза (рис. 5.15г) |41—43| —
это один из вариантов мономерной фазы (рис. 5.15»), у которой заместители силанов
не метильные группы, а изопропильные или изобутильные (рис. 5.15г). Изобутиль-
ная группа разветвлена, объемна и препятствует гидролизу силанольной связи
(сравните рис. 5.16д и б). Каждая Si О—Si связь защищена двумя объемными боковыми
группами (см. рис. 5.166). Пространственная защита важна в разделениях при низ
ком рН (но не высоком), так как низкий рН катализирует разрыв связи O-Si |4I|.
Коммерчески доступны пространственно защищенные неподвижные фазы с
разными лигандами (например, С8, С]8, фенил, циано), и каждая стабильна в кислотных
условиях. Так как ионные соединения рекомендуется разделять в кислотных
условиях (раздел 7.3.4.2), то для разделения биологических образцов (пептидов, белков)
лучше всего подходят пространственно защищенные сорбенты. Поскольку
заместители силана достаточно объемны, их концентрация на поверхности сорбента низка
-Si-<- ->-Si-
d он о он о
-4 + 1 1— -f-
Si Si Si Si Si
(6)
H30'
^f^4 ^?^
1
он он о он
-+—I—t-—f—
Si SI Si Si
(e)
M
+ (снз)3а-с1
>-Si-£ ^-Si-«r
О OH OH OH
-4 4 1 +
Si si si a
TMC = триметилсилил = <CH3)3Si-
>-Si-£ 'JMfV ^-Sl~*
I 7" I
о о он он
-4 4 1 +■---
Si Si Si Si
Рис. 5.16. Способы повышения стабильности сорбентов с алкилированными силикаге-
лями. (я, 6) Зашита связи O-Si в пространственно защищенной фазе
(только в кислотных условиях), (в, г) защита привитой фазы эндкепированием
5.3. Неподвижные фазы 259
и поэтому, по сравнению с фазами, образованными диметилсиланами, такие
стационарные фазы отличаются слабым удерживанием (см. данные по колонкам с
мономерными фазами в таблице 5.6). Сорбенты с бидентатными лигандами (рис. 5.146)
более стабильны при высоком рН [44|.
Таблица 5.6. Влияние длины и разветвленное™ углеводородной цепи лиганда на плотность
прививки к носителю
Лиганд, привитый к носителю
триметил
диметил-3-цианопропил
ди метил -л-бутил
ди метил-л-о ктил
диметил-л-октадецил
Плотность прививки
(икмоль/м2)
4,1
3,8
3,8
3,5
3,2
Прореагировавшие
силанолы (%)
51
48
48
44
40
Пространственно защищенные сорбенты:
триизопропил
диизопропил-3-цианопропил
ди иэоп роп ил -и-октил
ди изобутил -и -октадецил
2,2
2,1
2,0
1,9
28
26
25
25
До взаимодействия с силанами концентрация поверхностных силанолов
полностью гидроксилированного силикагеля составляет « 8 мкммоль/м2. Однако,
присоединенные силаны настолько объемны, что препятствуют реакции соседних
силанолов с силановым реагентом. Поэтому концентрация лигандов у полностью
прореагировавшего сорбента будет немного превышать значение 4 мкммоль/м2
(при этом остаются непрореагировавшими половина или больше исходных
силанолов). Как показано в таблице 5.6, с увеличением длины углеводородной цепи или
поперечного сечения заместителей силанов падает процент прореагировавших сила-
нольных групп (и концентрация привитых лигандов). Почти 50% силанольных групп
остаются непрореагировавшими при взаимодействии с силаном, имеющим самые
маленькие заместители (триметилсиланом). Несмотря на то, что непрореагировав-
шие силанолы оказываются недоступны для силанов, они все же могут реагировать
с молекулами анализируемых веществ в процессе хроматографии (раздел 5.4.1).
Полимерные неподвижные фазы нашли применение, хотя и в ограниченной
степени, благодаря более высокой стабильности и специфической селективности
(раздел 5.4.1.2). Если мономерную фазу (рис. 5. 14й) необходимо получать в безводной
среде, то для образования полимерных фаз требуется присутствие воды. Степень
полимеризации контролируется количеством воды, добавляемой при проведении
реакции [45|. Реакцию алкилтрихлорсилана с частицами силикагеля проводят в
присутствие воды |45|.
Чтобы минимизировать нежелательное взаимодействие с остаточными силаноль-
ными группами (раздел 7.3.4.2), обращенно-фазовые сорбенты обычно эндкепируют,
то есть обрабатывают привитую фазу силанами с необъемными заместителями
(например, триметилхлорсиланом или диметилхлорсиланом, рис. 5.16г). При этом
концентрация непрореагировавших силанолов становится меньше, а также снижается
(но не исключается полностью) их взаимодействие с удерживаемыми молекулами
анализируемого вещества (эндкепинг повышает процент прореагировавших силанолов
всего лишь на 20—30% |46], что соответствует небольшому снижению количества
260 Глава 5. Колонки
(а)
н
1
1
-Si-
1
1
о
--1-
Si
-О
н
1
1
-Si-
1
1
о
-+-
SI
-О-
н
1
1
-Si-
1
1
о
-+-
Si
-о
н
1
1
-Si
1
1
О
-г-
Si
Рис. 5.17. Силикагель типа С (о) немодифицированный и (б) с привитой фазой (би-
дентатным лигандом Cjg)
непрореагировавших силанолов). Небольшие лиганды (например, триметилсилильные
группы, присоединяемые при эндкепинге) в большей степени подвержены гидролизу
и отщеплению в кислой среде, что может привести к изменению удерживания и
селективности. С другой стороны, эндкепированные колонки более стабильны в
нейтральных и оснбвных условиях.
Существует еще один вид привитой фазы на основе силикагеля «типа С».
Полярные силанолы обычного силикагеля типа В сначала обрабатывают для получения
неполярной гидрированной поверхности (рис. 5.17а). Такой силикагель типа С можно
использовать без дальнейших изменений как в обращенно-фазовой, так и в
нормально-фазовой хроматографии. Силикагель типа С можно также модифицировать
(для ОФХ) алкильными группами (рис. 5.176). Утверждается, что у таких сорбентов
хорошая воспроизводимость и стабильность даже в 100%-ном водном элюенте в
интервале рН 1,5—10 |47|. Ко времени издания данной книги этот сорбент был
относительно новым, поэтому информация о его свойствах и практическом применении
была достаточно скудна, и тем не менее фирма MicroSolve Technology (Eatontown,
NJ) его продавала.
Определить содержание алкильных и ароматических лигандов в сорбенте (в том
числе и введенных при эндкепировании) можно после их удаления с частиц
силикагеля |48]. Сорбент сначала обрабатывают водным раствором плавиковой кислоты,
которая расщепляет связь лиганд-силикагель. Затем отщепленные в ходе реакции
лиганды можно охарактеризовать методами ГХ, ЯМР и/или масс-спектрометрии.
5.3.2. Другие органические неподвижные фазы
5.3.2.1. Нанесенные полимеры
Полимеры, такие как поперечно-сшитые полистирол и полибутадиен, используются
в качестве стационарной фазы, нанесенные на частицы на основе силикагеля и
оксидов металлов (раздел 5.2.5) [49|. Колонки с такими сорбентами достаточно редко
применяются из-за низкой эффективности, плохой воспроизводимости, а также
из-за небольшого выбора фаз с разными функциональными группами.
5.3.2.2. Гибридные частицы
В результате сополимеризации двух мономеров (например, тетраметоксисилана и
тетраэтоксисилана) образуются гибридные частицы с органической/неорганической
структурой (рис. 5.18). Впервые химический процесс для получения таких частиц
(б)
СН
-Si „ Si „ Si Si
i^o-i-o-i-o-i-
о о о о
-Л + +—4—
Si Si Si Si
5.3. Неподвижные фазы
OEt
I
,Si
ЕЮ
ч
OEt
ЕЮ
OEt
I OEt
OEt
EtO-
R
I
-Si'
был предложен Унгером и его коллегами
в 1977 году |50|, а затем разработан в
деталях фирмой Waters Corporation.
Гибридные частицы получают из силанов
с устойчивыми к гидролизу связями
Si-C. Эти связи формируют подложку,
улучшая ее рН стабильность
относительно других сорбентов на основе силика
геля. Сначала гибридные частицы
получали, смешивая тетраэтоксисилан
с метилтриэтоксисиланом в
соотношении 2:1 (сорбент ХТегга). Позже стали
синтезировать сорбенты из гибридных
частиц с улучшенной рН стабильностью
из тетраэтоксисилана и этил-бис-три-
этоксисилана (XBridge). Такие частицы
обладают отличной устойчивостью как
в кислотных, так и в основных элюентах,
а также механической прочностью, что
позволяет их использовать при давлении
до 1034 бар {15000 psi). Как и силика-
гель, гибридные частицы можно
прививать различными лигандами (С8, C,g
и т.д.) |51|. На гибридных сорбентах
лучше всего разделять неионизированные основные соединения, элюируя их подвижной
фазой с высоким рЫ. В таких условиях улучшается форма пиков и можно разделить
большую массу образца (раздел 15.3.2.1).
EtO-
/
ЕЮ
/
Si.
-о
О
/
-Si—О
\
О
/
.Si—О
л
Полимер Полиэтоксиолигосилоксан
Рис. 5.18. Синтез
органической/неорганической гибридной частицы. Материал любезно
предоставлен фирмой Waters Corporation
5.3.2.3. Колонки для подвижных фаз с высоким содержанием
воды
В ОФХ-разделениях соединений с высокой полярностью могут потребоваться
подвижные фазы с небольшим содержанием растворителя В, чтобы получить к > I,
хотя существуют и другие способы повысить удерживание таких образцов
(разделы 6.6.1 и 7.3.4.3, глава 8). Работая в режиме ОФХ с подвижной фазой, содержащей
небольшой процент растворителя В, можно столкнуться с некоторыми проблемами:
со временем снижается удерживание образца, понижается число теоретических
тарелок, а также при переходе от одной подвижной фазы к другой требуется больше
времени на уравновешивание |52, 53|. Это связано со снижением смачиваемости
стационарной фазы (что иногда неправильно называют ее коллапсом), обусловленным
вытеснением подвижной фазы из пор частиц [54, 55|. Тогда давление Р, требуемое
для принудительного заполнения пор диаметром dpore подвижной фазой, определяют
по формуле:
—4у cos в
(5.2)
где у — поверхностное натяжение подвижной фазы, 9 — угол контакта между
подвижной и стационарной фазами. Для неподвижной фазы С]8 и элюента воды
в > 90е. Это означает, что вода будет проникать внутрь пор сорбента только
принудительно, то есть под давлением. Если давление недостаточно для того, чтобы
подвижная фаза с большим содержанием воды проникла во все поры частиц, то и
молекулы разделяемого вещества будут вытесняться из пор. Чем гидрофобнее колонка
262 Глава 5. Колонки
и меньше размер пор частиц, тем большее требуется давление (для колонок С3о
нужно большее давление, для колонок С, — меньшее). Требуемое давление также
возрастает с уменьшением %В. Можно повысить смачиваемость и удерживание колонки
промыв ее метанолом или другим органическим растворителем |56].
Проблемы с низкой смачиваемостью сорбента возникают, главным образом,
при падении давления в колонке. Если колонка изначально была заполнена элю-
ентом с > 50 % В, то им будут заполнены все поры сорбента, а сама колонка будет
находится под обычным для ВЭЖХ давлением. То есть, если колонка долгое время
не использовалась (рекомендуется хранить колонки в 100% ацетонитриле), то
до начала серии ОФХ-разделений все поры сорбента обычно будут заполнены
растворителем. Если подвижная фаза заменяется другой с понижением доли
органического растворителя, а при этом давление в колонке поддерживается неизменным,
то вряд ли снизится смачиваемость сорбента. Тем не менее, если вам предстоит
провести разделения с элюентом, содержащим меньше 5% растворителя В,
рекомендуется подобрать колонку, менее подверженную снижению смачиваемости
(с более короткими лигандами, более полярными лигандами, с меньшей
концентрацией лигандов, см. раздел 5.4.4.2).
Для решения проблемы снижения смачиваемости, были разработаны особые
«водные обращение фазовые» сорбенты, позволяющие использовать элюент,
содержащий более 95% воды. Например, это сорбенты с фазами, содержащими полярные
группы или эндкепированные полярными группами (такие колонки обозначаются
«polar», «AQ», «hydrosphere», «aqua», «aquasil»), или с небольшой концентрацией ал
кильного лиганда. Кроме того, сорбенты с широкими порами или с короткими
лигандами менее подвержены изменениям смачиваемости, однако удерживание на
таких сорбентах хуже, поэтому для ОФХ-разделения образцов с высокой полярностью
они не столь полезны.
5.3.3. Колонки с различными функциональными группами
сорбента
Помимо отличий ОФХ-сорбентов, описанных в разделах 5.3.1 и 5.3.2, может
различаться и химический состав их лигандов. В таблице 5.7 перечислены, а на рис. 5.19
схематически показаны некоторые коммерчески доступные сорбенты с разными
функциональными группами (на рис. 5.19а—г не указана группа -S1ICH3I). У ОФХ-
сорбентов лигандом часто является алкильная группа, например, С5, Cs, C(8
(рис. 5.19а). Может быть лигандом и фенилпропильная или фенилгексильная группа,
тогда фаза называется фенильнои (рис. 5.196). Если лиганд -C3-ON (рис. 5.19в), это
циана фаза. Кроме того, алкильную группу можно заменить другой функциональной
группой X (рис. 5.19г), тогда получатся фазы иных типов. Их названия перечислены
в нижней части рис. 5.19. Так называемые фазы с встроенными полярными
группами (ВПГ) приобрели популярность в силу своей совместимости с подвижной фазой,
содержащей малое количество растворителя В, и специфической селективности
(раздел 5.4.1); наряду с этим силанольные группы этих фаз менее реакционнеспособны.
Колонки, упакованные такими неподвижными фазами, обычно дают пики основных
образцов правильной формы. В состав лигандов фаз с ВПГ входят амидная группа,
остатки карбамата или мочевины (все это основания, образующие водородные
связи), а также другие полярные функциональные группы. Некоторые сорбенты с ВПГ
могут оказаться не такими стабильными, как схожие по структуре алкильные или
арильные сорбенты. Природа лиганда определяет главным образом селективность
колонки, а это тема уже следующего раздела.
5.4. Селективность колонки 263
Таблица S.7. Функциональные группы неподвижных фаз В'ЭЖХ
Функциональная группа
Сз
С4
с5
с8
Cis
С30
Фенил
ВПГ (амид, карбамат,
мочевина)
Перфторфенил (ПФФ)
Циано
NHj (амино)
Диол
Слабые аниониты
Слабые катиониты
Сильные аниониты
Сильные катиониты
Область применения11
ОФХ
ОФХ
ОФХ
ОФХ
ОФХ
ОФХ
ОФХ
ОФХ, НФХ
ОФХ, НФХ, ИОХ
ОФХ, НФ, ЭХ
ИОХ
Комментарии
Используется главным образом для разделения
белков
Наиболее часто используемые сорбенты;
сходные удерживание и селективность для всех ал-
килированных фаз
Главным образом используется для разделения
каротина
Часто используемые сорбенты, в основном,
для изменения селективности
Часто используемые сорбенты, в основном,
для преимущественно водных подвижных фаз
(< 5 %В), для улучшения формы пика
основных образцов или для изменения селективности
Реже используемый сорбент, главным образом,
для изменения селективности
Реже используемый сорбент
Реже используемый сорбент
В основном используется в ГПХ
Используются главным образом для разделения
неорганических ионов или крупных биомолекул
(раздел 13.4.2)
"ОФХ — обращенно-фазовая хроматография, НФХ — нормально-фазовая хроматография,
ИОХ — ионообменная хроматография, ГПХ — гель-проникающая хроматография.
5.4. Селективность колонки
Знать селективность колонки очень важно по многим причинам. Во время
разработки метода может возникнуть необходимость замены колонки в целях улучшения
селективности и повышения разрешения (разделы 2.5.2 и 5.4.3). Для повышения
разрешения должна быть возможность использовать другую колонку с иной
селективностью. Если рутинные ОФХ-разделения выполняются в разное время и на разных
хроматографах, может оказаться затруднительным немедленно заменить старую
колонку на новую, или это будет слишком дорого или по другим причинам не
практично (разделы 5.4.2 и 6.3.6.1). Тогда нужно искать колонку с аналогичной (или
по крайней мере сходной) селективностью. В любом случае требуется
количественный анализ, позволяющий сравнить селективность колонок. Кроме того,
селективность связана с рядом проблем, которые могут возникнуть как в процессе
разработки метода, так и при выполнении рутинных ОФХ-разделений: размывание заднего
фронта пика, ухудшение эффективности колонки со временем или снижение
смачиваемости сорбента во время элюирования подвижной фазой, состоящей, в основном,
из воды (раздел 5.3.2.3). Знание селективности, наконец, поможет понять, каким
образом удерживание образца зависит от свойств колонки и молекулярной структуры
образца, и будет способствовать более оптимальному решению проблем разделения.
264 Глава 5. Колонки
(а)
Сорбенты с привитой алкильной группой
СНэ .
О ОН ТЭ
wAwUwUu
с, с3
(ТМС)
ОН О ОН ^0 ОН
он
C-ia
(ОДС)
J30
(б)
(в)
(в)
о он о
VUUwUuu
Фен ильная фаза
f
он о он
\\U\uU\ui
Циано фаза
S
он о он
"Другие" фазы
Тип колонки
Привитая полярная группа
Фтороэамещенные фазы
R-NH-(C=0>-0-
R-NH-(C=0)-NH-
R-(C=0)-NH-
F F
*CFgCFgCF3
Подтип
Карбамат
Мочевина
Амид
Пврфторфенил (ПФФ)
Фторалкил
Рис. 5.19. Классификация ОФХ-колонок в соответствии с типом лиганда (на рисунках
не показана связующая группа [-Si(CH3)2-])
В оставшейся части раздела 5.4 сначала обсуждаются основы селективности
колонки, которые могут быть связаны с различными взаимодействиями между
молекулами вещества и сорбентом. Все это приведет к количественным значениям тех
свойств колонки, которые определяют ее селективность. Ну, и напоследок приведен
материал о том, как знание свойств колонки, определяющих ее селективность,
может пригодиться при разработке метода и в рутинных разделениях.
5.4.1. Основы селективности обращенно-фазовой колонки
Удерживание вещества, как уже говорилось в разделе 2.3.2.1, определяется
различными взаимодействиями между анализируемым образцом, подвижной фазой и
стационарной фазой (колонкой). Относительная важность различных взаимодействий
5.4. Селективность колонки 265
между образцом и колонкой или селективности колонки зависит от состава сорбента
и молекулярной структуры образца. На рис. 5.20 изображены восемь видов
взаимодействий, влияющих на селективность:
(а) гидрофобное взаимодействие;
(б) пространственное исключение крупных молекул образца из неподвижной фазы
(здесь будем называть его истерическими взаимодействием»);
(в) водородные связи между акцепторной (основной) группой образца и донорной
(кислотной) группой внутри неподвижной фазы (обычно силанолом -SiOH);
(г) образование водородной связи между донорной (кислотной) группой образца и
акцепторной (основной) группой неподвижной фазы (на рисунке обозначена
буквой «X»);
(д) катионообменное или электростатическое взаимодействие между катионом
образца и ионизированным силанолом ( SiO~) внутри неподвижной фазы, а также от
талкивание ионизированной кислоты (например, R СОО~);
mwuwwo
Гидрофобное
взаимодействие
W
I
0=0
\
%
X
П ространствекное
взаимодействие
W
Водородная связь
(кислотное анализируемое
вещество)
\\\U\\U\\V\\\
Катионообменное
взаимодействие
' Й)
N
х i
wmmVwuu
Водородная связь
(основное анализируемое
(в) /ч/\
_^
С* х
чч\\Ч\\\\
Дипопь-дипольное
взаимодействие
(ж)
02N
it-я взаимодействие
(фенильная фаза)
(з)
ч
5°
#
о
л-л взаимодействие
(циано фаза)
(и)
«ЗНР
Комплексообразование с
хелатообразующей молекулой
анализируемого вещества
Рис. 5.20. Взаимодействия между анализируемым веществом и сорбентом,
определяющие ее селективность (на рисунках не показана связующая группа
|-И(СНз)Н)
266 Глава 5. Колонки
(е) диполь-дипольные взаимодействия между биполярной группой образца (нитрог-
руппой в данном случае) и биполярной группой сорбента (нитрильной группой
циано сорбента);
(ж, з) л-л взаимодействия ароматического образца с фенильной (рисунок ж, фени
льная фаза) или нитрильной (рисунок з, циано фаза) группами;
(и) образование комплекса между хелатообразующей молекулой образца и
примесями металла на поверхности частицы сорбента.
Очень важную роль для любого сорбента играют взаимодействия (ад). Диполь-
ные взаимодействия (е) характерны для циано сорбентов, л-л взаимодействия
(ж, э) — исключительно для фенильных и циано фаз |57). Ацетонитрил,
используемый в качестве растворителя В, подавляет как дипольные, так и л-л взаимодействия,
поэтому в таких разделениях эффект этих взаимодействий сводится к минимуму.
Комплексообразование с металлами примесей на поверхности сорбента (и)
наблюдается на не очень чистом силикагеле типа А, что вызывает уширение пиков и
размывание их заднего фронта (очень нежелательное явление). Для тестирования
сорбентов на склонность к комплексообразованию используют хелатообразующий реагент
а,а-бипиридил. Поскольку на фенильных и циано фазах не так часто работают,
а сорбенты на основе силикагеля типа А вообще не рекомендуют использовать,
то в этой главе обсудим взаимодействия (а—д) |57, 58|.
5.4.1.1. Взаимодействия анализируемого вещества с сорбентом
Представление о различных взаимодействиях между анализируемым веществом и
сорбентом, определяющих селективность последнего, сформировалось в общем виде
в 1980-х годах. Первые попытки охарактеризовать селективность сорбента хорошо
изложены в работе |591- Тем не менее, только после 2000 года появилась
возможность надежно охарактеризовать селективность ОФХ сорбента (и колонки) с точки
зрения этих взаимодействий. Это было достигнуто благодаря разработке и
применению модели гидрофобного вычитания [60—62|, признающей, что гидрофобные
взаимодействия являются, безусловно, самыми важными в удерживании на обращенной
фазе. Если бы учитывались только лишь гидрофобные взаимодействия, то график
зависимости значений log k для одной колонки от значений log к для другой выглядел
бы как прямая, на которую ложились бы без отклонений все данные. Как видно
на рис. 5.21, приблизительно это и наблюдается для двух колонок C|g (Inertsil ODS-3
и Stablebond C|g), хотя значения log к алифатических амидов {□) и протонированных
сильных оснований (О) лежат ниже прямой, наиболее оптимальным образом
аппроксимирующей эти данные. Отклонения значений log к алифатических амидов и
протонированных сильных оснований можно объяснить взаимодействием молекул
этих веществ с силанольными группами (силанольные взаимодействия имеют
большее значение для колонки Stablebond C|g). Эти и другие менее значительные
отклонения 6log к от графика (см. вставку с частью графика в увеличенном масштабе
на рис. 5.21) отражают вклады в удерживание негидрофобных взаимодействий
(рис. 5.205—д). Можно проанализировать значения Slog к для разных образцов и
разных сорбентов/колонок для того, чтобы раздельно оценить величину каждого
из пяти взаимодействий а—д на рис. 5.20. Значения к для сорбентов, в которых
значительно проявляются только а—д взаимодействия (рис. 5.20) (без учета фенильных
и циано фаз), могут зависеть от взаимосвязанных свойств анализируемого вещества
и сорбента следующим образом:
logf —I =r\'H -n'S* + Р'А + а.'В + к'С. (5.3)
\ki:B)
(/> (/О (ДО (/V) (у)
5.4. Селективность колонки
Здесь к и к£д — факторы удерживания анализируемого вещества и образца
сравнения этилбензола (ЕВ), соответственно. Члены i—v уравнения (5.3) соответствуют
взаимодействиям а—d, представленным на рис. 5.20. Коэффициенты г|\ о', р', а', к'
относятся к свойствам молекул анализируемого вещества: гидрофобности (rf),
«объемности» (а"), оснбвности водородных связей ф'), кислотности водородных
связей (а1), эффективному ионному заряду (к'). Основной практический интерес
вызывают соответствующие параметры сорбента (Н — гидрофобность, S* — стерические
взаимодействия или сопротивление неподвижной фазы проникновению объемных
молекул анализируемого вещества, А — кислотность водородных связей (ВС), В —
основность ВС, С — ионобменная емкость или электростатическое (кулоновское)
взаимодействие). Сорбенты с похожими значениями Н, S* и других перечисленных
параметров обладают схожей селективностью и удерживанием (обеспечивают выход
пиков на хроматограмме в одинаковой последовательности). Соответственно,
селективность и относительное удерживание будут отличаться у сорбентов с разными
значениями Н, S* и т.д. Модель гидрофобного вычитания и уравнение (5.3) лучше всего
обобщают современные представления об удерживании в ОФХ и селективности
колонки. |59|.
Далее соотнесем каждый из членов уравнения (5.3) с взаимодействиями,
показанными на рис. 5.20я—д. Гидрофобные взаимодействия (рис. 5.20а) показаны на при
мере взаимодействия 2 я октанона (СНзСОС6Н|з) с лигандами колонки С8.
Гидрофобность колонки Н увеличивается, если лиганды длиннее (сравните Cjj и C]g), их
концентрация на поверхности силикагеля выше (мкммоль/м2), диаметр пор меньше
(сравните поры 8 нм и 30 нм), и колонка эндкепирована. Чем выше Н, тем лучше
удерживаются гидрофобные молекулы, у которых значения г|' больше.
2.0
1.5
1.0
ю
о os
'й
с
ш
ё о.о
Ас
а
-0.5
-1.0
-1.5
1.0
у = 0.21 + 1.01 х
г2 = 0.995
D амиды
О протонированные
основания
jt>
/1.'
о
о
° о
~*^*
^jj%*
^J?**
i '4i »5*п
• о
♦ ;
Slog/с J
-0.5
0.0 0.5
logfe(StableBondC18)
1.0
1.5
Рис. 5.21. Сравнение удерживаний на двух разных колонках Cjg. Приведены данные
по 90 различным органическим соединениям. Условия: колонка
150x4,6 мм, подвижная фаза 50% ацетонитрил-вода, рН фосфатного
буфера 2,8, скорость потока 2,0 мл/мин, температура 35 °С |61)
268 Глава 5. Колонки
Пространственное исключение или «стерическое взаимодействие» (рис. 5.206)
показано на примере удерживания двух изомеров полициклических ароматических
углеводородов (ПАУ): неразветвленного, но длинного нафтацена и более «объемного»
трифенилена. Нафтацен лучше проникает между соседними лигандами, но, если
расстояние между ними увеличивается (т.е. концентрация лигандов на поверхности
становится меньше), трифенилену становится легче проникнуть в неподвижную фазу.
Параметр колонки S* оценивает «непроницаемость» неподвижной фазы для молекул
анализируемого вещества или трудности, возникающие при проникновении
объемных молекул между лигандами. Ббльшие значения S* говорят о большей
«непроницаемости» сорбента и относительно слабом удерживании объемных молекул. Чем
длиннее лиганд, чем выше его концентрация (т.е. лиганды располагаются ближе друг
к другу) и меньше диаметр пор, тем больше параметр S*. Объемность молекулы
анализируемого вещества определяется величиной а'. Пространственное исключение
в достаточной мере сложное явление, подробнее оно рассмотрено в разделе 5.4.1.2.
Водородные связи (ВС), возникающие между силанольными группами сорбента
и неионизированным основным соединением (например, пиридином), показаны
на рис. 5.20в. Кислотность водородных связей А сорбента обусловлена присутствием
силанолов на поверхности неподвижной фазы и поэтому она снижается после эндке-
пирования благодаря удалению некоторых силанолов и блокированию других
(см. рис. 5.\6г). Силанолы сорбентов типа А обычно имеют более высокую
кислотность по сравнению с сорбентами типа В и, следовательно, значения А первых выше,
чем вторых. Основность ВС анализируемого вещества определяется его значением
а'. Непротонированные амины и амиды имеют гораздо более сильные основные
свойства, их значения а' больше, в то время как нитрилы и нитросоединения менее
основные, следовательно, и а' для них меньше. Для большинства других полярных
соединений характерны промежуточная основность ВС и значения а'.
Образуются водородные связи и с неионизированными кислотами (например,
масляной кислотой, рис. 5.20«г). Донорная (основная) группа сорбента «Х> не
указывается, поскольку ее природа отличается у разных ОФХ-сорбентов. До сих пор нет
внятного объяснения, какие группы можно отнести к донорам «Х> в алкилирован-
ных сорбентах на основе силикагеля типа В. Исходя из обратно пропорциональной
зависимости значений А и Н и других аргументов, было выдвинуто предположение,
что этим группам «Х> соответствует вода, растворенная в стационарной фазе. Что
касается некоторых сорбентов типа А с большими значениями В, то группами «Х>,
по-видимому, являются примеси металлов. Кроме того, вполне вероятно, что
группам «X:» соответствуют встроенные в сорбент полярные группы (обычно
проявляющие основные свойства при образовании ВС). Чем больше В, тем сильнее
удерживаются карбоновые кислоты с большими значениями а'. Удерживание других
соединений с кислотными свойствами, например, спиртов и фенолов, в меньшей
степени зависит от В (поскольку значения а' у них меньше), хотя фенолы
удерживаются лучше на сорбенте с встроенными полярными группами (т.е. на таких
сорбентах фенолы играют роль сильных кислот).
Ионобменная емкость С колонки — это мера ионизации силикагеля и
доступности заряженных силанолов. Ионизация силанолов и/или доступность силанольных
групп (и значения С) растут с увеличением рН подвижной фазы. Кроме того, эти
показатели больше у неэндкепированных колонок и колонок типа А (по сравнению
с колонками типа В). В основном ббльшие значения С приводят к повышению
удерживания протежированных оснований, в то же время удерживание заряженных
кислот уменьшается в силу электростатического отталкивания. Величина к' вещества
примерно равна его молекулярному заряду (например, у полностью протониро-
ванных оснований +1, у полностью протонированных кислот -I). Главное отличие
колонок типа А и В определяется значением параметра С в кислотном диапазоне.
5.4. Селективность колонки 269
При рН 2,8 у колонок типа В С < 0,25, у колонок типа А С > 0,25. Считается, что
колонки с С < 0,00 в кислотных условиях имеют суммарный положительный заряд |63|.
Возможно, это обусловлено введением протонированных аминогрупп в процессе
производства некоторых колонок. Предполагается, что параметры Н, S*, А и В не
зависят от рН подвижной фазы.
Значения параметров селективности Н, S* и других были определены для более
400 разных колонок. Краткий список можно найти в работе |64|, за полным списком
можно обратиться к одному из авторов. Средние значения этих параметров для
некоторых типов ОФХ колонок приведены в верхней части таблицы 5.8. Для колонок
определенного типа значения Н, S* и других параметров также заметно отличаются
(см. некоторые колонки C|g типа В в нижней части таблицы 5.8). Следовательно,
не все колонки данного типа могут быть эквивалентны по селективности. Не только
параметры Н, S*, но и, например, среднее удерживание, которое определили по
значениям k этилбензола (последняя колонка таблицы 5.8), растет с увеличением
площади поверхности частиц сорбента.
5.4.1.2. Селективность по форме молекулы
В данном небольшом отступлении рассматриваются две разные аЪормы пространствен
ного исключения. Читатель может этот раздел пропустить и перейти к следующему
разделу 5.4.2.
В основе пространственного исключения при разделении лежат два явления:
пространственное взаимодействие (определяемое слагаемым и в уравнении 5.3) и
селективность по форме молекулы \65\. Различия между этими явлениями показаны
на примере разделения двух изомерных углеводородов на полимерном сорбенте
(рис. 5.22а) и мономерном сорбенте (рис. 5.226). Суть селективности по форме
молекулы объясняется на примере «узкой» (i) и «широкой» (/) молекул (рис. 5.22а)
для полимерного алкилсиликагелевого сорбента. «Широкая» молекула вытесняется
из неподвижной фазы, поскольку ее минимальное поперечное сечение (показано
двунаправленной стрелкой) больше расстояния между лигандами (молекула j не
может «протиснуться» между лигандами). В мономерной фазе (рис. 5.226) лиганды
расположены дальше друг от друга, поэтому и «широкие», и «узкие» молекулы могут
свободно проникать в стационарную фазу. Когда молекула проникает внутрь
сорбента, пространственное исключение иначе влияет на удерживание: теперь важную роль
играет гидродинамический диаметр молекулы, а не ее минимальное поперечное
сечение. Если гидродинамический диаметр молекулы сравним с расстоянием между
лигандами (как, например, для молекулы /), она становится ограниченной в своей
возможной ориентации внутри сорбента. При этом снижается ее удерживание таким
же образом, как и в эксклюзионной хроматографии (раздел I3.8.I). Отметим, что
селективность по форме молекулы и пространственное взаимодействие по-разному
влияют на зависимость удерживания от формы молекулы. То есть становится ясно,
что они вносят разные вклады в удерживание |61|. Природа селективности по форме
молекулы, предусматривает только два варианта — проникновение в неподвижную
фазу или невозможность проникновения и может привести к сравнительно большим
изменениям в относительном удерживании, в то же время пространственное
взаимодействие меньше влияет на ОФ-селективность.
На рис. 5.22в, г показано потенциальное преимущество селективности по форме
молекулы на примере разделения полициклических ароматических углеводородов
(ПАУ). Селективность по форме молекулы ярче всего проявляется на колонках,
упакованными полимерными фазами (рис. 5.22в), значит, и разделяются изомерные ПАУ
0-12 на них гораздо лучше, чем на колонках, упакованными мономерными фазами
(рис. 5.22г), на которых селективность по форме молекулы минимальна. Длинные
270 Глава 5. Колонки
Таблица 5.8. Характеристики селективности колонки, определенные на основании модели
гидрофобного вычитания (уравнение 5.3)
Тип колонки
H
S*
A
В
C(pH 2,8) C(pH 7,0)
*M
Разные типы колонок
Ci (тип В)
С3 (тип В)
С8 (тип В)
С|8(тип В)
С]8 (тип В, широкопористая)
С is (тип В, монолитная)
C|g (тип В, гибридная)
C|g (полярная, эндкепированная)
С|8 (тип А)
С3о (тип В)
Со встроенными полярными группами
Фенил (тип В)
Циано (тип В)
Перфторфенил (ПФФ)
Фторал кил
На основе окиси циркония
0,41
0,60
0,84
0,99
0,95
1,01
0,98
0,90
0,94
1,05
0,74
0,63
0,43
0,65
0,66
0,97
-0,08
0,12
0,00
0,01
0,01
0,02
0,01
-0,04
-0,05
-0,01
0,00
-0,12
-0,09
-0,11
-0,07
0,01
-0,08
0,08
-0,12
-0,01
-0,05
0,12
-0,14
-0,02
0,14
0,09
-0,22
-0,20
-0,49
-0,25
-0,11
-0,62
0,02
0,04
0,02
0,00
0,01
-0,02
-0,01
0,02
0,01
-0,02
0,12
0,02
0,00
0,01
0,03
0,00
0,04
-0,08
-0,03
0,00
0,22
0,11
0,13
-0,02
0,79
-0,08
-0,27
0,13
0,02
0,40
0,87
2,01
0,66
0,81
0,25
0,24
0,31
0,31
0,05
0,40
1,18
0,45
0,53
0,68
0,72
0,96
1,18
2,01
1,2
2,8
5,4
8,8
3,2
3,2
6,3
7,4
6,4
13,0
5,9
2,7
1,0
4,3
3,7
0,8
Разные колонки C|g типа В с узкими порами"
Halo-CIS*
Zoitax SlableBond CI8"
Zoitax Eclipse XDB-C18"
Kromasil 100-508^
ProntoSIL 120-5C18 SH<»
Inertsil ODS-3*
Alltima C18*
Nucleodur CI 8 Gravity3
Ace 5 CI
e'en romolith*
Luna СЩ2У
Gemini CIS UOA"
Discovery С18*
Hypurity CIS"
Hypersil Gold"
Symmetry CI 8°
Xterra MS CIS"
Sunfire C18°
TSKgel ODS-100Z"
1,11
1,00
1,08
1,05
1,03
0,99
0,99
1,06
1,00
1,00
1,00
0,97
0,98
0,98
0,88
1,05
0.98
1,03
1,03
0,05
-0.03
0,02
0,04
0,02
0,02
-0,01
0,04
0,03
0,03
0,02
-0,01
0,03
0,03
0,00
0,06
0,01
0,03
0,02
0,01
0,26
-0,06
-0,07
-0,11
-0,15
0,04
-0,10
-0,10
0,01
-0,12
0,03
-0,13
-0,09
-0,02
0,02
-0,14
0,04
-0,13
-0,05
0,00
-0,03
-0,02
-0,02
-0,02
0,01
-0,02
-0,01
-0,01
-0,01
0,01
0,00
0,00
0,04
-0.02
-0.01
-0,01
-0,03
0,06
0,14
0,05
0,04
0,11
-0,47
0,09
-0,08
0,14
0,10
-0,27
-0,09
0,18
0,19
0,16
-0,30
0,13
-0,19
-0,06
0,04
1,04
0,09
-0,06
0,40
-0,33
0,39
0,32
0,10
0,19
-0,17
0,19
0,15
0,17
0,48
0,12
0,05
-0,10
-0,16
6,1
7,6
9,1
12,5
8,7
10,9
11,5
11,0
7,9
3,1
9.6
8,0
4,8
5,6
3,9
9,8
6.3
9,9
11,6
"Все колонки с частицами 5 мкм |73|.
^Advanced Materials Technology; "Agilent; 'AkzoNobel; *BischofF; eCL Science; •>cGrace-AI1tech;
3Macherey Nagel; "ACT; *Merck; ■'Phenomenex; "Supelco; "Thermo/Hypersil; "Waters; "Tosoh Bios-
cience.
5.4. Селективность колонки
(а) Минимальное поперечное
сечение (ширина)
(6) Гидродинамический диаметр
О
О
О
Р
О—Si-OH У О—Si-OH
"Si он Si он
/ \ / \
Р ,,?,,,?,,,??,,
ЧЧЛЧЧ\\\ЧЧ\Л\\\\ЧЧ\ЧЧЧ\
селективность по форме молекулы
Полимерный сорбент с
близкорасположенными лигандами
(в)
н он о он он с>
пространственное взаимодействие
Мономерный сорбент с
широкорасположенными лигандами
&?
W
.л*Л—ID
селективность по форме
молекулы
Полимерный сорбент С18
45 fcS16
пространственное
взаимодействие
Мономерный сорбент С1в
5£*£7
WW
L
ю
20
30
10 15
(мин)
Рнс. S.22. Различные проявления пространственного исключения. Селективность по
форме молекулы (а) в сравнении с пространственным взаимодействием (б),
(в) Разделение смеси из 13 полициклических ароматических углеводородов
на полимерной фазе, (г) Разделение того же образца на мономерной фазе.
Примеры (в) и (г) взяты из работы [65]
и узкие молекулы (по сравнению с широкими и короткими) преимущественно
удерживаются, поскольку в удерживании таких молекул важную роль играет прежде всего
селективность по форме молекулы. Короткие и широкие молекулы с той же
молекулярной массой, наоборот, лучше удерживаются, когда превалируют пространственные
взаимодействия. Как правило, можно сказать, что на Сзо и полимерных колонках
большее значение имеет селективность по форме молекулы при разделении крупных
молекул с сильно разнящимся отношением длины к ширине. Большинство ОФХ-раз-
делений осуществляют на мономерных фазах (а не на Сзд), на которых главным
образом пространственное взаимодействие и значения S* определяют влияние
пространственного исключения на селективность колонки. Более подробно практическое
применение селективности по форме молекулы описано в разделе 6.3.5.2.
272 Глава 5. Колонки
5.4.2. Воспроизводимость характеристик колонки
и «эквивалентные» колонки
Производители колонок пытаются обеспечить одинаковые характеристики,
удовлетворительную и воспроизводимую работу в рутинных ОФХ-анализах каждой колонки
(например, Waters Symmetry CI8). Поэтому перед продажей определяют число
теоретических тарелок и перепад давления для каждой колонки (раздел 5.7). Колонки с N
ниже определенного минимального значения бракуются. Кроме того, производитель
проводит и другие тесты, таким образом гарантируя одинаковую селективность
колонок разных партий (одинаковые значения //, S* и др.). На рис. 5.23 изображен
пример колонок ZorbaxA Rx-C|g разных партий с практически одинаковой
селективностью (показаны времена удерживания диметиланилина и толуола в зависимости
от номера партии). Значения к двух соединений отличаются на ±4% (1 СО), главным
образом, из-за небольших, несущественных отличий площади поверхности частиц
силикагеля. Отношение двух значений к (а) позволяет непосредственно определить
селективность колонки (в первую очередь важный параметр селективности колонки
С в уравнении 5.3). Значения а изменяются всего лишь на ±0,5%. Получается, что
селективность одинаково упакованных колонок из разных партий должна быть
одинакова, особенно при разделении протонированных оснований.
В 1970 и 1980-х годах производство колонок не было столь масштабным, как
сегодня. Поэтому селективность колонок из разных партий зачастую очень сильно
отличалась. Позже удалось улучшить воспроизводимость колонок (например, см. |67|),
но селективность колонок из разных партий при этом все еще может сильно
разниться, в результате чего достаточно сложные образцы не удается разделить.
Существует достаточно много способов решения этой проблемы (раздел 6.3.6.1), одним из
которых является подбор «эквивалентной» колонки другого производителя.
Ориентируясь на параметры Н, S* и т.д., можно с помощью функции сравнения
колонок Fs |6I | выбрать несколько похожих колонок (таблица 5.8, нижняя часть):
Fs = |[12.5(//2 - Я,)|2 + |100(^ - S'W + POM2 - 4>12 + (5 4)
+ [143(в2-В1)12 + |83(С2-С|)р^.
0 1—1 I 1 I I I I I I 1 I I I 1_1
1 3 5 7 9 11 13
Номер партии колонки
Рис. 5.23. Мониторинг разных партий колонок на предмет возможного изменения
селективности. Образец: диметиланилин и толуол. Условия: колонка Zorbax
Rx-Cjg 150x4,6 мм, подвижная фаза 50% ацетонитрил-вода с фосфатным
буфером рН 7, скорость потока 1,6 мл/мин, температура 22 "С |66]
5.4. Селективность колонки 273
Величины Ht и //2 — это параметр Н колонок I и 2, то же самое касается и других
параметров в уравнении. Неравенство Fs < 3 указывает на взаимозаменяемость двух
колонок (селективность их приблизительно одинакова) в любом ОФХ разделении
(для несложных разделений допускаются большие знамения Fs). Способность с
помощью уравнения (5.4) определять колонки эквивалентной селективности была
продемонстрирована на примере двенадцати разных рутинных ОФХ-разделений,
которые были разработаны и использовались в некоторых лабораториях |68| (пример
приведен на рис. 6.19). Программу для сравнения селективности колонок с помощью
уравнения (5.4), а также параметры Н, S* и т.д. для более 400 колонок, можно найти
на сайте Фармакопеи США. Можно также связаться с одним из авторов для
получения доступа к базе данных значений Н, S* и т.д. для разных ОФХ-колонок.
5.4.3. Ортогональное разделение
Иногда необходимо найти две разные колонки со сходной селективностью, в других
же случаях нужна вторая колонка с отличающейся селективностью и, как правило,
иной подвижной фазой. Можно привести несколько причин необходимости
изменения селективности. Во первых, в процессе разработки метода может потребоваться
существенно изменить селективность для оптимального разрешения определенных
пар пиков на хроматограмме. Во-вторых, разрабатывая метод разделения для образца
неизвестного состава, можно столкнуться с проблемой перекрывания пиков, то есть
минорные пики могут скрываться в крупных пиках. В окончательном варианте
метода мелкие пики можно просто потерять. В-третьих, может возникнуть похожая
ситуация: образцы с дополнительными неучтенными пиками (которых не было раньше)
не разделяются должным образом в условиях рутинного метода. Облегчить решение
последних двух проблем может «ортогональное разделение», селективность которого
отличается от селективности исходного метода. То есть, если в рутинном методе два
пика накладываются друг на друга, то вполне вероятно, что в ортогональном
разделении они разделятся (и это можно будет увидеть). В работе |69| и на рис. 6.21
на примере дюжины различных методов показано, как с помощью уравнения (5.4)
найти колонки с разной селективностью для решения последних двух проблем.
В-четвертых, ортогональные колонки требуются для температурно регулируемой
оптимизации последовательно соединенных колонок |70|, т.е. для лучшего контроля
селективности разделения при разных температурах на двух колонках, расположенных
друг за другом. Ну и наконец, ортогональные колонки нужны для двумерных
разделений, в ходе которых образец разделяют последовательно на двух разных колонках
(раздел 9.3.10). Во всех этих методах две ОФХ-колонки с сильно отличающейся
селективностью соответствуют двум колонкам с большими значениями /^(уравнение 5.4).
В работе |71| предлагалось при выборе ортогональных колонок акцентировать
внимание на разделение нейтральных образцов (для которых параметр С не
учитывается при определении селективности колонки). Селективность колонки для
нейтральных образцов определяется следующим образом:
FA-Q = {Ц2.5(Я2 - Я,)|2 + |100<52 - *,*)|2 + тл2 - Л,)12 + Щ3(в2 - Я,)|2>ад. (5.4а)
Для максимальной ортогональности колонок рекомендуется одновременное
выполнение условий: Fs(-C) > 50 и Fs > 100 [711.
5.4.4. В каких еще случаях нужно знать селективность
колонки
Значения Н> S* и т.д. отражают природу и свойства неподвижной фазы, а также
связаны с некоторыми распространенными проблемами колонок:
274 Глава 5. Колонки
• размыванием заднего фронта пика;
• «несмачиваемостью» стационарной фазы;
• снижением эффективности колонки в рутинных анализах.
5.4.4.1 Размывание заднего фронта пика
Ранее в разделе 2.3.2 размывание заднего фронта пика рассматривалось с точки
зрения его влияния на разрешение. Размывание в ОФХ возникает прежде всего
при разделении протонированных оснбвных образцов |72| или во время работы на
алкилсиликагелевых колонках типа А |73|. Как правило, на колонках типа В (при
значениях С]2.8| < 0,25) эта проблема не возникает. В разделе 7.3.4.2 рассказано
о дополнительных способах снижения размывания заднего фронта пика основных
образцов. При этом не исключено, что и на колонках типа В пики могут
размываться; к этому склонны полностью ионизированные соединения всякий раз, когда
масса образца превышает I мкг на колонке диаметром 4,6 мм. Причиной является
взаимное отталкивание ионизированных молекул образца (с одинаковым зарядом),
сконцентрированных в стационарной фазе (раздел 15.3.2.1 и |72|). Пики неионизи-
рованных веществ не размываются до тех пор, пока не вводится в 50 раз большее
количество образца.
Не так часто можно наблюдать размывание заднего фронта пика карбоновых
кислот, элюируемых подвижной фазой с низким рН |73|. Чаще всего это может
случиться на колонках типа А, хотя и для более основных колонок типа В (у которых
параметр В имеет большие значения) оно также характерно. Неионизированные
кислоты в отличие от протонированных оснований размываются при нанесении
небольшого количества образца (< I мкг), но если масса образца находится в интервале от 1
до 50 мкг, то на хроматограмме пики будут симметричными. Более подробно эта
тема раскрыта в работе |73|.
5.4.4.2. Несмачиваемость неподвижной фазы
Несмачиваемость неподвижной фазы (раздел 5.3.2.3) характерна для гидрофобного
сорбента с узкими порами, т.е. для колонок с большими значениями Н. Среднее
значение Н для колонок С|8 типа В (самая популярная колонка) с узкими порами
(< 12 нм) составляет 0,99. Колонки с меньшими значениями Н менее подвержены
несмачиваемости. Колонки эндкепированные полярными группами (среднее
значение И = 0,90) были специально разработаны для минимизации несмачиваемости,
а в остальном с точки зрения селективности они аналогичны колонкам С]8 типа В,
поскольку параметр Н меньше влияет на селективность (об этом свидетельствуют
коэффициенты в уравнении 5.4).
5.4.4.3. Снижение эффективности сорбента
Неподвижная фаза колонки, используемой в рутинных анализах, постепенно теряет
свою эффективность. Происходит это в результате ее разрушения подвижной фазой
либо связей силан-силикагель (в кислотных условиях), либо самого силикагеля
(при высоких рН). Любой из этих процессов приводит к увеличению количества си-
ланольных групп (а значит, растут значения параметров, отвечающих за
селективность колонки А и С) тл к. снижению гидрофобности колонки (т.е. значений Н) |73|.
Поэтому со временем селективность колонки постепенно меняется. Когда
селективность (или число теоретических тарелок) падает до такой степени, что пики
накладываются друг на друга, колонку заменяют новой.
5.S. Типоразмеры колонок 275
5.5. Типоразмеры колонок
5.5.1. Фитинги
В настоящее время можно приобрести упакованные разными сорбентами колонки
из нержавеющей стали со стандартными фитингами (раздел 3.4.2) любых размеров.
Таким колонкам свойственны максимальная эффективность, воспроизводимость и
надежность. Кроме того, в продаже имеются недорогие предколонки (картриджи)
из нержавеющей стали. Упаковывают их различными сорбентами и используют,
главным образом, в рутинных несложных разделениях. Предколонки продаются
без каких-либо фитингов, поэтому для них нужно отдельно приобретать
многоразовые держатели.
Сегодня многие ВЭЖХ-системы рассчитаны на работу при высоком давлении
(= I5000 psi = 1000 бар = 100 МПа). Поэтому конструкция колонок должна,
во-первых, выдерживать такое давление, во-вторых, быть химически устойчивой к
подвижной фазе. Чтобы эти требования выполнялись, для соединения большинства колонок
используют капилляры из нержавеющей стали. В редких случаях бывает, что сталь
может подвергнуться воздействию элюентов или прореагировать с образцом и тогда
рекомендуется использовать титановые колонки или стальные с внутренней
поверхностью, покрытой стеклом. Для масс-спектрометрии подойдут капиллярные колонки
из плавленого кварца, а для препаративной хроматографии при давлении, не
превышающем = 40 бар (600 psi), — толстостенные стеклянные колонки. Если же рабочее
давление может достигать = 400 бар (6000 psi), лучше использовать толстостенную
колонку из полимерного материала ПЭЭК. Внутренние стенки любой колонки
должны быть гладкими с зеркальной полировкой. Состояние стенок колонок
существенно влияет на гомогенность упакованного сорбента, следовательно, на
эффективность колонки1.
Фитинги и соединители должны иметь такую конструкцию, которая
обеспечивает минимальный мертвый объем и не допускает значительного внеколоночного
размывания зоны пика (разделы 2.4.1.1, 3.9). Составляющие ВЭЖХ-системы (фитинги,
работающие под давлением, капилляры, соединители, ячейка детектора и т.д.)
должны быть собраны таким образом, чтобы минимизировать неомываемые углы или
застойные зоны, которые могут выполнять роль миксера и размывать пики. На входе
в колонку, где давление может превышать 400 бар (6000 psi), между металлическими
соединительными частями используют уплотнения. Некоторые уплотнения
выдерживают давление до 1000 бар (15000 psi). Если рабочее давление небольшое
(например, меньше 400 бар или 6000 psi), то подойдут уплотнения и фитинги из
полимерного материала (например, ПЭЭК). Более подробно эта тема обсуждалась
в разделе 3.4.
Для достижения наилучшего результата рекомендуется образец вводить в
колонку быстро, в минимальном объеме. Поэтому находящиеся с одной и с другой
сторон фильтры2 или сеточки, удерживающие сорбент в колонке, не должны
способствовать размыванию пика. Очень часто используют пористые фильтры
толщиной 0,2 мм из нержавеющей стали, титана или сплава на основе никеля. Тонкие
сеточки из нержавеющей стали обеспечивают наименьшее размывание пика,
но достаточно сложны в использовании, особенно когда речь идет о высоком
давлении на входе в колонку. Пористость фильтров или сеточек должна быть сущест-
'Чем хуже качество внутренней поверхности колонки — тем медленнее скорость потока у ее
стенок и тем больше искажение и размывание зоны вещества. — Прим. перев.
2 Иногда их называют фриттами, поскольку такие фильтры получают спеканием частиц
нержавеющей стали или титана соответствующего размера. — Прим. перев.
276 Глава 5. Колонки
венно меньше размера частиц сорбента. Особенно это касается сорбентов с
большим распределением частиц по размеру, у которых самые маленькие частицы
могут проникать сквозь фильтр или засорять его. Например, для сорбента с
частицами 5 мкм или 3 мкм с узким диапазоном распределения подойдут фильтры
пористостью 2 мкм. У большинства колонок с 3 мкм-сорбентом установлены
фильтры пористостью 0,5 мкм, у колонок с 2 мкм сорбентом — выходные фильтры
имеют размер пор 0,2 мкм. Поскольку 0,2 и 0,5 мкм фильтры очень быстро
забиваются, рекомендуется подвижную фазу и образцы тщательно фильтровать при
использовании колонок с частицами малых размеров.
В хроматографии практически всегда используют прямые колонки (а не изог
нутые или свернутые в спираль). Практика показала, что для разделения даже
очень чувствительных биологических образцов (пептидов или белков) редко
требуются стеклянные колонки. Несмотря на то, что в продаже имеются колонки из не
ржавеющей стали, внутренняя поверхность которых покрыта стеклом, или из проч
ного полимера (ПЭЭК) в алюминиевом держателе, они тоже довольно редко
используются.
Колонки с радиальной компрессией (стр. 67—69 |12|) представляют собой мягкий
полимерный цилиндр, неплотно заполненный сорбентом. Перед использованием
частицы сорбента под действием гидравлического давления в радиальном направлении
сжимаются и образуют компактный слой. Такие колонки пригодны как для
аналитических, так и для препаративных задач. К достоинствам можно отнести низкую
стоимость и отсутствие металлических деталей. В настоящее время ввиду трудности
температурного контроля и других неудобств, а также увеличения стоимости при сборке
серии колонок (для увеличения N), на практике их достаточно редко применяют.
Колонки с аксиальной компрессией сначала заполняют суспензией сорбента, а потом
с помощью поршня ее подпаковывают. Такие колонки служат исключительно
для крупномасштабных препаративных разделений |74, 75|.
5.5.2. Типоразмеры колонок
В настоящее время под любую задачу можно подобрать колонки нужного размера.
В таблице 5.9 представлены некоторые имеющиеся в продаже размеры колонок
с различными сорбентами. Для разработки рутинных методов обычно используют
колонки с внутренним диаметром 3—4,6 мм, упакованные сорбентом с размером
частиц 2,5—5 мкм и оснащенные фитингами, выдерживающими давление. Такие
колонки представляют собой разумный компромисс между удобством,
эффективностью и сроком службы. Колонки с внутренним диаметром 2,1 мм лучше всего
подходят для ЖХ-МС, поскольку скорость подвижной фазы соответствует
требованиям детектора (раздел 4.14). У колонок с внутренним диаметром < 2,1 мм число
теоретических тарелок меньше на 15—25%, во-первых, из-за внеколоночного
размывания пиков и, во-вторых, из-за сложности упаковки колонок с маленьким диаметром.
Автосамплеры могут оказаться серьезным источником внеколоночного размывания
пиков на колонках с небольшим внутренним диаметром, поскольку для них
требуются очень маленькие объемы пиков. Колонки с внутренним диаметром 1 мм и
капиллярные колонки диаметром 50 мкм иногда применяют в масс-спектрометрии,
но они, как правило, не подходят для рутинных анализов. Микроколонки очень
сложно эффективно упаковать. Для работы с ними нужно специальное
оборудование, которое позволяет минимизировать внеколоночное размывание.
5.6. Методы упаковки колонок 277
Таблица 5.9. Наиболее распространенные размеры колонок
Назначение колонки
Аналитическая
Предколонка (картридж)
Микрокапиллярная
Палупрепаратипная
Препаративная
Внутренний диаметр (мм)
1-4,6
3-4,6
1, 2,1
8-10
20-50
Длина (мм)
30-250
50-100
50-250
100-250
100-250
Размер частиц (мкм)
1,5-10
3-10
2-8
5-200
50-200
Примечание: в таблице приведены размеры колонок из нержавеющей стали; стеклянные,
покрытые изнутри стеклом, пластиковые и ПЭЭК колонки можно приобрести не всех
перечисленных в таблице размеров.
5.6. Методы упаковки колонок
Немногие читатели найдут какое-либо практическое применение в лаборатории
сведениям из этого раздела. Поэтому его можно пропустить. Сегодня у большинства
хроматографистов нет нужды упаковывать колонки, они покупают их уже готовыми.
Хроматографические колонки совершенствовались несколько десятилетий. У
большинства хроматографических лабораторий, занимающихся разработкой ВЭЖХ
методов и анализом образцов, нет возможности упаковывать колонки, да еще и делать
это так, чтобы новая колонка не уступала по эффективности, надежности и
воспроизводимости предыдущей. Не нужно воспринимать этот раздел как руководство
по упаковке ВЭЖХ-колонок. Мы всего лишь рассмотрим некоторые принципы
основных методов упаковки, чтобы читатель смог хотя бы оценить, насколько
хороши, упакованные этими способами, колонки. Если же кто-то пожелает
самостоятельно упаковать колонку, рекомендуем поподробнее изучить материал [76, 77|.
5.6.1. Сухая упаковка
Первые ВЭЖХ колонки упаковывали частицами, размер которых варьировал в
диапазоне 45—50 мкм. Такие колонки упаковывались вибрационным методом, который
ранее использовался для газовой хроматографии. После появления в конце 1960-х
годов плотных сферических частиц размером = 25 мкм с твердой сердцевиной (как
в случае пелликулярного, так и в случае поверхностно-пористого сорбентов), сухую
упаковку стали осуществлять «постукиванием» вдоль колонки. Такие колонки
оказались более эффективными и стабильными |77[. Этот прием до сих пор используется
для упаковки сорбентом с крупными частицами, особенно в препаративной
хроматографии (глава 15). С появлением частиц меньшего размера (< 10 мкм) ВЭЖХ
колонки, упакованные сухим методом, оказались неэффективными и нестабильными.
Требовались другие методы упаковки.
5.6.2. Суспензионная упаковка жестких частиц
На первых порах частицы для ВЭЖХ имели относительно большое распределение
по размеру, а в некоторых случаях были неправильной формы. Очевидно, что такие
частицы имеют тенденцию распределяться в зависимости от размера по поперечному
сечению колонки, в результате чего слой сорбента становился неэффективным и
нестабильным. Чтобы минимизировать эту проблему требовались иные методы
упаковки. Оказалось, что лучше всего упаковать колонку небольшими жесткими частицами
278 Глава 5. Колонки
можно, закачивая суспензию частиц в пустую колонку под давлением [26, 76, 77|.
Чтобы во время упаковки избежать распределения частиц по размеру, используют
разные подходы:
• жидкости со сбалансированной плотностью;
• жидкости с очень высокой вязкостью;
• несбалансированную суспензию с невысокой вязкостью.
Для приготовления суспензии со сбалансированной плотностью используют
жидкости, плотность которых сходна с плотностью частицы, заполненной жидкостью
(например, смесь тетрабромэтана с растворителем меньшей плотности). Можно
использовать жидкости с очень высокой вязкостью (например, глицерин). В обоих
случаях условия препятствуют оседанию частиц и распределению их по размеру
во время упаковки. Однако оба эти метода требуют много времени и, как правило,
не обеспечивают высокой эффективности при упаковке колонки.
Появление пористых микросфер силикагеля (< 5 мкм) с относительно узким
распределением частиц по размеру позволило использовать несбалансированную
суспензию с невысокой вязкостью. Поскольку распределение частиц по размеру во время
упаковки колонки не столь критично для очень мелких частиц, то первые два вида
суспензий оказались больше не нужны. Невязкие растворители (например, тетрагид-
рофуран, хлороформ, смеси этих растворителей с другими растворителями с низкой
вязкостью) позволяют быстро проводить упаковку колонки эффективно и стабильно.
5.6.2.1. Выбор растворителя для приготовления суспензии
Процесс упаковки колонки суспензией, приготовленной любым способом,
достаточно прост. Суспензию из растворителя и сорбента помещают в емкость,
присоединенную к насосу, который перекачивает суспензию в пустую колонку, снабженную
фильтром и фитингом (рис. 5.24). Во время перекачки частицы остаются в колонке
(благодаря фильтру или сеточке, которая установлена на выходе из колонки), а
растворитель вытекает из колонки. Когда колонка наполнится суспензией,
устанавливают входной фильтр и закручивают верхний фланец. Несмотря на простоту описания
процесса, для эффективной, стабильной и воспроизводимой упаковки необходимо
учитывать многие важные детали |26, 76|.
Емкость для
растворителя
д
Концевой фитинг
и фильтр
Слив
Рис. 5.24. Схема оборудования для суспензионной упаковки колонки |76|
5.6. Методы упаковки колонок 279
(а) Равномерно (б) Агрегированные частицы
распределяющиеся частицы
Рнс. 5.25. Сравнение равномерно распределяющихся и агрегированных частиц
сорбента (в разных растворителях)
Решающим критерием является отсутствие в суспензии конгломератов частиц
сорбента. То есть важную роль в приготовлении суспензии играет выбор жидкости,
препятствующей ассоциации частиц. Чтобы определить, как ведут себя частицы
сорбента в той или иной жидкости, обычно используют следующий простой способ.
Сорбент добавляют в жидкость, помещают в ультразвуковую баню, после чего
суспензию исследуют под обычным микроскопом. Если частицы свободно
перемещаются и не слипаются, как показано на рис. 5.25д, значит, жидкость подходит для сус-
пендирования. Если же в суспензии частицы образуют агрегаты (рис. 5.256), то эту
жидкость лучше не использовать. Но иногда даже при равномерном распределении
частиц в суспензии жидкость может оказаться неоптимальной для упаковки
определенного сорбента. Единственный способ проверить это — протестировать уже
упакованную колонку.
Для суспендирования растворитель с низкой вязкостью выбирают в зависимости
от природы сорбента. Чтобы предотвратить ассоциацию частиц сорбента в суспензии,
нужно чтобы взаимодействие между частицами и растворителем было сильнее, чем
между самими частицами. Например, частицы, модифицированные Cg или С]8,
(содержащие полярные непрореагировавшие силанольные группы) рекомендуется
упаковывать в слегка полярных растворителях — таких, как тетрагидрофуран,
метил -/п/>е/п-бутиловый эфир или в смесях ацетонитрил/хлороформ,
хлороформ/метанол, хлороформ/ацетон (не стоит использовать такие углеводороды, как гексан,
поскольку с ними упаковка неэффективна). Более полярные сорбенты (немодифициро-
ванный силикагель или силикагель с полярными лигандами, например, амино или
диол) лучше упаковывать в метаноле или в каком-либо другом полярном растворителе.
Пока неясно, влияет ли каким-то образом агрегация частиц на эффективную упаковку
капиллярных колонок. Этот процесс требует дальнейшего изучения. Тем не менее,
для упаковки традиционных колонок с внутренним диаметром более 1 мм
настоятельно рекомендуется готовить суспензию в жидкости, препятствующей агрегации частиц.
После того, как мы определились с выбором жидкости для суспензии,
рассмотрим другие аспекты суспензионной упаковки.
Размер частиц сорбента. Частицы диаметром более 3 мкм сравнительно нетрудно
упаковать так, чтобы получить стабильную и эффективную колонку. У упакованных
колонок минимальное приведенное число теоретических тарелок А должно быть
2—2,5, а фактор размывания < 1,2 (для небольших неполярных анализируемых
веществ). Для поверхностно-пористых частиц диаметром 2,7 мкм было получено
значение h <, 1,5 [5, 13|. Частицы диаметром менее 2 мкм упаковывать сложнее, колонки
могут оказаться менее эффективными.
Распределение частиц по размеру. Многие коммерчески доступные сорбенты
характеризуются относительно узким распределением частиц по размеру со стандарт-
280 Глава 5. Колонки
ным отклонением от среднего 15—20%. У поверхностно-пористых частиц диаметром
2,7 мкм стандартное отклонение всего лишь 5%, возможно, из-за этого колонки,
упакованные таким сорбентом, оказываются более эффективными {раздел 5.2.2.1).
Концентрация суспензии. Обычно самая оптимальная концентрация частиц в сус-
пензии составляет 7—15%. Точная концентрация для идеальной упаковки зависит
от типа частиц, растворителя, аппаратуры для упаковки и определяется эмпирически.
Конструкция устройства для суспензионной упаковки. Важную роль в упаковке
колонки может сыграть конструктивное исполнение емкости, из которой суспензия
попадает в колонку. Чтобы достичь лучших результатов рекомендуется, чтобы
внутренний диаметр выходного капилляра емкости и входного капилляра
колонки, по которым перекачивают суспензию, были одинаковыми (как показано
на рис. 5.24). Кроме того, для упаковки следует отдать предпочтение насосу с
постоянным давлением (например, насос Haskell), а не насосу с постоянным расходом.
Давление упаковки. Давление, требуемое для плотной упаковки, зависит от
размера частиц, длины и внутреннего диаметра колонки. Чтобы колонка была стабильна,
давление упаковки должно более, чем на 50%, превышать максимальное рабочее
давление колонки. При этом помещать суспензию в колонку необходимо максимально
быстро.
Обработка поверхности стенок колонки. Во время упаковки под давлением
стенки колонки слегка расширяются или «растягиваются», а частицы сорбента
сжимаются |76|. После того, как колонка упакована и давление сброшено, частицы
расправляются, и некоторые из них вынуждены перемещаться вдоль стенок колонки.
Следовательно, внутренние стенки колонки должны быть гладкими с зеркальной
полировкой, чтобы избежать истирания частиц и образования более мелких частиц, что
снижает эффективность колонки.
Прочность частиц. Процесс упаковки проходит под давлением, при этом
частицы сжимаются и упруго деформируются. Частично это сжатие будет компенсировать
внутренние напряжения, возникающие во время работы при более высоком
давлении или температуре. Если частицы недостаточно прочные, они будут в конечном
счете разрушаться или раздавливаться. В результате колонка будет терять
эффективность и работать при более высоком давлении. Некоторые колонки рассчитаны
на максимальное рабочее давление 600—1000 бар (9000—15000 psi). Частицы таких
колонок должны быть достаточно прочными и сохранять свою структуру во время
упаковки при более высоком давлении.
5.6.2.2. Жесткие полимерные частицы
Для упаковки колонок жесткими полимерными частицами («гелями»), такими как
сшитый полистирол, используют еще и суспензию сбалансированной плотности.
Общий алгоритм метода и оборудование то же самое, что и для упаковки жестких
частиц силикагеля. В отличие от жестких частиц плотные гели сначала нужно поместить
в жидкость, в которой они набухнут, а потом проводить упаковку. Поскольку
плотность органических гелей меньше плотности твердых тел, то и жидкости для
суспензии используются менее вязкие (например, смесь ацетон/тетрахлорэтилен). По
прочности жесткие полимерные частицы уступают жестким частицам силикагеля,
поэтому давление упаковки должно быть меньше. Полимерные частицы ионооб-
меннников («смолы») также упаковывают в водном растворе сбалансированной
плотности. При этом рекомендуется соблюдать следующую последовательность
действий: сначала готовят густую суспензию с набухшей ионообменной смолой в
солевом растворе (например, хлориде кальция), плотность которого соответствует
плотности смолы. Затем, также, как и в случае с жесткими частицами, суспензию
перекачивают под высоким давлением в пустую колонку. Давление упаковки зависит
5.7. Спецификации колонок 281
от прочности частиц смолы, которая, в свою очередь, обычно зависит от степени
сшивки смолы. Для самых прочных ионообменных смол может использоваться
давление приблизительно 350 бар (-5000 psi). Перед использованием колонку
необходимо тщательно промыть подвижной фазой, чтобы удалить из нее соль.
5.6.3. Упаковка мягких гелей
Частицы мягких гелей, которые используются в гель-фильтрации (исключение
по размеру, раздел 13.8), не упаковывают сухим методом или суспензионным
методом при высоком давлении. Мягкий гель сжимается и деформируется даже при
относительно небольшом давлении. Поэтому для такого сорбента обычно используют
гравитационный метод или суспензионно-седиментационный метод. Лучше всего
использовать метод упаковки, рекомендованный производителем.
5.7. Спецификации колонок
5.7.1. Стандарты производителя
Чтобы быть уверенным в воспроизводимой эффективности колонки, производитель
к каждой выпускаемой колонке (с присвоенным серийным номером) прилагает
стандарты или спецификацию. Правда, у производителей стандарты сильно разнятся.
Одни прилагают письменную спецификацию к каждой колонке, которая включает
показатели характеристик различных колонок и фактические данные
эффективности. Другие считают, что достаточно данных для одной или нескольких колонок
из одной производственной серии или партии, которые соответствуют всем
колонкам этой партии. Кроме того, характеристики колонки могут отличаться не только
у разных производителей, но и у разных партий одного производителя.
Важно различать специфические параметры, характерные для каждой колонки, и
параметры, зависящие от свойств сорбента (тест серии). К специфическим
параметрам колонки относятся число теоретических тарелок /V, коэффициент асимметрии
пика As (или фактор размывания) и давление колонки Р. К каждой колонке должна
прилагаться тестовая хроматограмма с указанием подробных условий теста, чтобы
хроматографист мог сам на своем приборе повторить его и подтвердить значения N,
As и Р. Однако, полученные пользователем значения N, As и Р могут отличаться
от заводских в силу различия хроматографов, на которых выполняются тесты.
К параметрам теста серии относятся физико-химические данные (например,
площадь поверхности, размер пор, объем пор, размер частиц, содержание углерода,
мкммоль/м2 лиганда) и/или тестовая хроматограмма серии. Физико-химические
параметры не являются первоочередной информацией для большинства
хроматографистов, но в будущем они могут оказаться полезными во время устранения неполадок,
поэтому их следует хранить. Рекомендуется в тестовую смесь для тестирования серии
включать вещества разной природы (например, кислоты, основания, нейтральные
соединения), удерживание которых укажет на нежелательные изменения в
селективности колонки от серии к серии (раздел 5.4.2). Данные по удерживанию этих
веществ (к и а) должны укладываться в узкий диапазон значений, который обычно
устанавливает производитель. Важно сохранить эти данные и хроматограммы
для того, чтобы ими можно было воспользоваться, когда возникнет проблема с
рутинными анализами.
Не все производители прилагают к каждой колонке полный пакет документов.
Иногда ддя колонок одной партии сорбента сообщаются только средние значения
282 Глава 5. Колонки
параметров, и эта информация не скомпонована в один документ: что-то описано
во вложенной в упаковку хроматограмме, что-то можно найти на сайте
производителя, в литературных данных компании или научных публикациях. Некоторые
производители дают гарантию на свои колонки на период их использования и тогда
хроматографист может быть уверен в определенном сроке службы колонки. Кроме того,
дополнительную полезную информацию можно найти в руководствах по
обслуживанию, в брошюрах и на сайтах, например, рекомендуемые рабочие условия (рН и
температура), а также перечень растворителей, не рекомендуемых для колонок
определенного типа.
5.7.2. Число теоретических тарелок колонки
Обычно число теоретических тарелок N определяют в условиях тестовых
экспериментов, близких к «идеальным» (подвижная фаза с низкой вязкостью, небольшие
нейтральные молекулы анализируемого вещества, практически оптимальная
скорость потока). По ряду причин (раздел 2.4.1) для других образцов и/или в других
рабочих условиях значения N часто отличаются от полученных при тестировании.
Приведенное ниже уравнение служит для оценки числа теоретических тарелок
колонок, хорошо упакованных полностью пористыми частицами в условиях,
оптимизированных для максимального /V:
/V,50^. (5.5)
В этом уравнении L — это длина колонки в мм, d„ — диаметр частиц в мкм. В
таблице 5.10 сведены значения числа теоретических тарелок (при тестировании
нейтральными соединениями с молекулярной массой ж 200 Да), типичные для хорошо
упакованных колонок разной длины с разным размером и типом частиц сорбента.
Предполагается, что диаметр всех упомянутых в таблице колонок составляет 4,6 мм.
У колонок с диаметром меньше 2 мм значения N могут быть меньше, возможно,
из-за сложности упаковки таких колонок, но главным образом, из-за внеколоночно-
го размывания пиков.
Условия, используемые для определения числа теоретических тарелок, обычно
устанавливает производитель. Поэтому хроматографист может сам протестировать
колонку, если он подозревает ухудшение эффективности колонки. Если полученное
значение N составляет менее 80% от заявленной производителем величины,
ВЭЖХ-система поверена, и колонку использовали надлежащим образом или не слишком
интенсивно, то ее (колонку) следует вернуть производителю. Вообще, редко бывает,
чтобы параметры колонки не совпадали со значениями, указанными в
спецификации производителя. Заниженное число теоретических тарелок и завышенный
коэффициент асимметрии пика, измеренные хроматографистом на новой колонке,
практически всегда являются результатом ненадлежащей работы хроматографической
системы с чрезмерным внеколоночным размыванием пика. И это размывание будет
тем больше, чем меньше диаметр колонки и/или ее длина, особенно если она
упакована частицами размером менее 5 мкм.
Желательно для рутинных ВЭЖХ-анализов, проводимых в течение длительного
периода времени, выделить одну или несколько колонок. Это может оказаться
полезным для отслеживания значений N и As (или фактора размывания) одного или
нескольких образцов. Спустя какое-то время эти значения, определенные изначально
для данного метода, можно сравнить со значениями, полученными на новой
колонке, соответствующей спецификации производителя. Если в рутинном методе
возникла проблема, причиной которой может быть колонка, надо сравнить параметры N, As
5.8. Обслуживание колонок 283
Таблица S. 10. Приблизительное число теоретических тарелок для хорошо упакованных
колонок в оптимизированных тестовых условиях
Тип частиц"
Полностью пористые
Полностью пористые
Полностью пористые
Полностью пористые
Полностью пористые
Полностью пористые
Полностью пористые
Полностью пористые
Полностью пористые
Поверхностно-пористые
П оверх ностн о-пористые
Поверхности о-пористые
Поверхностно-пористые
Полностью пористые
Полностью пористые
Полностью пористые
Диаметр частиц (мкм)
5
5
5
5
5
3,5
3,5
3,5
3,5
2,7
2,7
2,7
2,7
1,8s
1,8
1,8
Длина колонки (мм)0
30
50
100
150
250
30
50
100
150
30
50
100
150
30
50
100
Число тарелок ./V
2500-3000
4500—5000
8000-10000
12000—15000
20000-25000
3000-4000
5500-7000
10500-14000
17000-21000
7000—8000
9000-11000
18000-22000
28000-34000
6000—7000
10000-12000
20000-25000
Примечание: тестовая смесь — нейтральные соединения с небольшой молекулярной массой,
подвижная фаза с низкой вязкостью, комнатная температура, измеряется при минимальной
высоте теоретической тарелки.
"Значения определены для колонок диаметром 4,6 мм.
^Средняя величина для коммерчески доступных колонок с размером частиц менее 2 мм.
или TF с изначальными значениями «хорошей» колонки, и тогда подтвердится, что
«плохая» колонка — источник проблемы.
У каждой колонки, используемой в рутинном методе, ограниченный срок
службы (на колонке можно провести определенное число анализов, прежде чем она
выйдет из строя), зависящий от условий разделения (особенно от рН подвижной фазы и
температуры). Для «чистых» образцов предусмотрено 1000—2000 анализов на силика-
гелевых колонках, в частности колонках с обрашенно-фазовыми сорбентами. В то же
время для других образцов (вводимых после минимальной пробоподготовки),
например, экстрактов крови, тканей животного или растительного происхождения, почвы
стандартный срок службы колонки составляет 200—500 образцов.
5.8. Обслуживание колонок
Эффективность и срок службы колонки зависят от качества ее использования и
обслуживания. Во время интенсивных анализов «грязных» образцов (особенно
образцов биологического происхождения) может начаться размывание заднего фронта
пиков (рис. 5.26й) или все пики на хроматограмме могут двоиться (т.е. каждый
компонент может выходить двумя пиками, рис. 5.266). Обычно такое происходит
в результате частичного загрязнения фильтра, колонки или повреждения сорбента.
Далее указаны меры по восстановлению эффективности колонки, которые иногда
Глава 5. Колонки
(э)
размывание
Рис. 5.26. Примеры размывания заднего фронта пика (а) и раздвоения пиков (б)
оказываются эффективными. При этом время и ресурсы, затрачиваемые на восста
новление колонки, часто не оправдывают себя экономически. Обычно гораздо
целесообразнее просто поменять колонку на новую.
Засорившийся фильтр или загрязненную колонку можно иногда восстановить,
периодически промывая колонку сильным растворителем. Для ОФХ-колонок
наиболее эффективна промывка двадцатью колоночными объемами (то есть около 30 мл
для колонки 150 х 4,6 мм) смеси 96% дихлорметан — 4% метанола с 0,1% гидроксида
аммония. Для нормальной фазы лучше всего воспользоваться чистым метанолом или
изопропанолом. Если (хотя бы) ежедневно промывать ОФХ-колонки сильным
растворителем (метанолом или ацетонитрилом), то для изократических разделений
колонка сохранит эффективность в течение длительного времени, и срок ее службы
увеличится. В этом случае из колонки вымываются сильноудерживаемые
компоненты образца, которые могут накапливаться на входе в колонку. Чтобы примеси не
попадали в колонку, а вымылись из нее, гораздо надежнее промывать колонку в
обратном направлении на скорости 0,2—0,5 мл/мин. Однако, многие производители против
такого способа промывки. Объясняют они это тем, что поры у входного фильтра
крупнее, а промывка в обратном направлении спровоцирует вымывание через них
самого сорбента. Прежде чем это делать, изучите инструкцию по использованию и
обслуживанию конкретной колонки. При градиентном элюировании промывку
колонки выполняют сильным компонентом с крутым или ступенчатым градиентом
(раздел 9.2.2.5). Фильтр с пористостью 0,5 мкм, установленный в линии перед
входом в колонку, (раздел 3.4.2.3) может очень эффективно предотвращать накопление
загрязнений на входном фильтре колонки, поэтому его настоятельно рекомендуют
устанавливать.
Чтобы снизить возможное влияние «грязных» образцов на срок службы колонки,
необходимо проводить пробоподготовку образца (глава 16). Кроме того, для защиты
колонки используется предколонка. Ее рекомендуют устанавливать в рутинных
анализах. Предколонка представляет собой короткую колонку (длиной, например,
10—20 мм), упакованную преимущественно тем же сорбентом, что и основная
колонка, или сходным с ним (раздел 5.4.2). На предколонке оседают
сильноудерживаемые компоненты образца (или частицы), что предотвращает загрязнение самой
аналитической колонки. Предколонку нужно менять через одинаковые промежутки
времени, не дожидаясь полного ее насыщения примесями, которые затем могут
попасть в аналитическую колонку. Однако некоторые хроматографисты не очень любят
пользоваться предколонками, поскольку они доставляют неудобства и приводят
к дополнительным расходам. Для них проще почаще менять саму колонку.
Использование предколонок с высокоэффективными колонками (например, упакованными
частицами диаметром менее 2 мкм), имеющими малый объем, требует особого
мастерства, поскольку здесь большую роль играет внеколоночное размывание пиков.
5.8. Обслуживание колонок 285
У плохоупакованных колонок внезапный скачок давления (например, во время
ввода пробы) может привести к образованию пустот на входе в колонку, которые снижают
ее эффективность. К счастью, у известных производителей колонок такой проблемы
больше нет. Если для колонок на основе силикагеля проблемы, связанные с давлением,
достаточно редки, то колонки, упакованные другими сорбентами (например,
органическими гелями, графитированным углеродом), достаточно чувствительны и не могут
выдерживать внезапных перепадов скорости потока, давления или температуры.
Во время работы может происходить потеря привитой фазы, что существенно
снижает эффективность колонки и сокращает срок ее службы. Советуем изучить
рекомендации производителя, особенно в отношении рН подвижной фазы и
температуры. В кислотной среде (например, при рН < 2,5) может происходить гидролиз
связей Si О и потеря привитого силана (рис. 5.16а). Короткие лиганды (например, Сз,
-Сз-C^N) наименее устойчивы в кислотных условиях, а длинноцепочечные алкиль-
ные группы (Cijj, Cg) достаточно стабильны в интервале рН 2,5—7 и при температуре
<40 °С. Ранее уже отмечалось, что пространственно защищенная неподвижная фаза
(рис. 5.166) обеспечивает дополнительную стабильность в кислотных условиях.
При высоком рН подвижной фазы (например, рН > 8) происходит медленное
растворение силикагелевого сорбента, что приводит к снижению эффективности колон
ки. Сорбенты на основе гибридных частиц (раздел 5.3.2.2) и окиси циркония
(раздел 5.2.5.1) особенно устойчивы и не разрушаются при высоком рН подвижной
фазы. Другие особые сорбенты (например, привитые бидентантные силаны,
рис. 5.146 и в) также более стабильны при высоком рН |44|.
Разрушение сорбента на основе силикагеля в кислотной среде ускоряется с
повышением температуры. Поэтому следует соблюдать осторожность при повышении
температуры, как для увеличения эффективности колонки, так и селективности
разделения. Многие хроматографисты сообщают о положительных результатах работы
колонок C|g и Cg в интервале 40—60 °С. Пространственно защищенные сорбенты
могут выдерживать регулярную работу при температуре до 90 °С, а полимерная
неподвижная фаза на основе оксида циркония чувствует себя удовлетворительно по
крайне мере до 150 °С |34|.
В основных условиях сорбент на основе силикагеля разрушается. Происходит это
по причине его растворения, и процесс ускоряется с повышением температуры.
На рис. 5.27 приведен пример, из которого видно, что общее разрушение силикагеля
при 60 °С в 20 раз больше, чем при 40 °С. Следует избегать нагревания колонок с
сорбентами на основе силикагеля выше 40 °С в анализах с фосфатным или карбонатным
буферами при нейтральном или высоком рН. В этих условиях силикагелевая подложка
быстро растворяется |78|. С другой стороны, органические буферы (например, Трис,
HEPES, цитрат) при таком же значении рН, наоборот, повышают срок эксплуатации
колонки |79|. Предполагают, что основные частично гидрофобные органические
буферные соединения склонны взаимодействовать с непрореагировавшими силанольны-
ми группами сорбента, в результате чего образуется дополнительный барьер,
препятствующий растворению силикагелевой подложки [791- Хотя преимущество
органических буферов может вводить в заблуждение, поскольку действительное
значение рН подвижной фазы, состоящей из буфера и органического растворителя, в
некоторой степени ниже измеренного значения рН самого буфера (а рН фосфатного и
карбонатного буферов в присутствии органических растворителей немного больше,
чем в водной среде [80]). Более подробно этот материал изложен в разделе 7.2.3.
На эффективность и срок эксплуатации колонки также может влиять
неправильное хранение подвижной фазы. Водные буферы, особенно с рН к 7, хранящиеся
при комнатной температуре более одного дня, являются благоприятной средой для
роста бактерий. Образовавшиеся частицы, в свою очередь, засоряют колонки,
снижают N и повышают давление. Поэтому лучше готовить буферы ежедневно. С другой
Глава 5. Колонки
л500
с 400
300
200
%
100
обОХ
°40Х
/_
15
0 5 10
Объем подвижной фазы (л)
Рис. 5.27. Влияние повышения температуры на растворение силикагелепой подложки.
Колонка Zorbax Rx-C|g 15 х 0,46 см; непрерывное элюирование не
подвергавшимся переработке элюентом 20% ацетонитрил / 80% буфер 0,25М
фосфат натрия рН 7,0. Скорость потока 1,0 мл/мин. Данные из |78]. (Ось х —
объем подвижной фазы в литрах (л))
стороны, даже небольшие добавки органического растворителя (>20%) в подвижную
фазу или барботирование гелием для удаления кислорода подавляют рост бактерий
и продлевают срок годности буфера.
Если не планируется использовать колонку в течение длительного времени, ее
отсоединяют от хроматографической системы, предварительно законсервировав в апро-
тонном растворителе, например, 100% ацетонитриле. Периодически используемую
колонку (в течение нескольких дней или в ночное время) удобно (и это допускается)
оставлять в хроматографе заполненную подвижной фазой. Тем не менее, не стоит
хранить колонку в буферных растворах, особенно с высоким содержанием воды или
спирта. Перед консервацией колонку рекомендуется сначала промыть 5—10 колоночными
объемами рабочей водно-органической подвижной фазы, но без буфера, затем 5
колоночными объемами 100% органического растворителя. Именно такая
последовательность действий позволяет избежать выпадения компонентов буфера в осадок внутри
колонки. Не стоит долго промывать колонку чистой водой, это приводит к потере
смачиваемости сорбента, особенно более гидрофобного (C|g). Чтобы предотвратить
пересыхание колонки во время хранения, ее плотно завинчивают заглушками.
Особого ухода требуют колонки с частицами < 2 мкм. В таких колонках
установлены фильтры с очень узкими порами (например, 0,2 мкм), позволяющие удерживать
очень мелкие частицы сорбента. Следовательно, и образец, и подвижная фаза должны
пройти сквозь 0,2 мкм фильтры, не засоряя их и не снижая эффективность колонки.
Обычно мелкозернистые колонки используют в экспресс-анализах
продолжительностью менее I минуты. При этом получаются пики очень малой ширины, измеряемой
временем или объемом. К системам, на которых выполняют такие разделения,
предъявляют особые требования — у них должны быть минимизированы потенциальные
внеколоночные эффекты, приводящие к искусственному уширению пиков:
• капилляры, соединяющие инжектор, колонку и детектор, должны быть короткими
с минимальным объемом;
• детектор с микроячейкой минимального объема (например, I мкл);
• небольшие объемы анализируемых образцов (предпочтительно 1—2 мкл);
• быстрый отклик детектора (например, <,0,\ с);
• высокая скорость сбора данных (по крайней мере, 20 точек/с или 20 Гц).
Более подробно этот материал изложен в работах [81, 82|.
Литература 287
Литература
1. К. К. Unger, Porous Silica, Elsevier, Amsterdam, 1979, p. 294.
2. K. Kalghatgi and С Horvath, J. Chromatogr., 443 (1988) 343.
3. K. K. Unger and H. Giesche, Ger. Pat. DE-3543 143.2 (1985).
4. J. J. Kirkland, F. A. Truszkowski, С H. Dilks, Jr., and G. S. Engel, J. Chromatogr. А,Ш (2000) 3.
5. J. J. Kirkland, T. J. Langlois, and J. J. DeStefano, Amer. Lab., 39 (2007) 18.
6. N. B. Afeyan, N. F. Gordon, I. Mazsaroff, L. Varady, S. P. Fulton, Y. B. Yang, and F. E.Regnier,
J. Chromatogr.. 519 (1990) 1.
7. D. Whitney, M. McCoy, N. Gordon, and N. Afeyan, J. Chromatogr. A, 807 (1998) 165.
8. L. R. Snyder and M. A. Stadalius, in High-performance Liquid Chromatography: Advances and
Perspectives, Vol. 4, C. Horvath, ed.. Academic Press, San Diego, 1986.
9. R. K. Her, The Chemistry of Silica, Wiley, New York, 1979, p. 639.
10. J. Billen. P. Gzil, F. Lynen, P. Sandra, P Van der Meeren, and G. Desmet, Poster, HPLC2006,
San Francisco, June 2006.
11. K. K. Unger and E. Weber, A Guide to Practical HPLC, Git Verlag, Darmstadt, I999,p. 45.
12. U. D. Neue, HPLC Columns, Wiley-VCH, New York, 1997, p. 82.
13. J. J. DeStefano, T. J. Langlois and J. J. Kirkland, J. Chromatogr. Scl, 46 (2008) 1.
14. J. Kohler, D. B. Chase, R. D. Farlee, A. J. Vega, and J. J. Kirkland, J. Chromatogr.. 352 (1986)
275.
15. J. Kohler and J. J. Kirkland, J. Chromatogr., 385 (1987) 125.
16. J. Nawracki, Chromatographia, 31 (1991) 177.
17. J. Nawracki, Chromatographia, 31 (1991) 193.
18. H. Engelhardt, H. Low, and W. Gotzinger, J. Chromatogr., 544 (1991) 371.
19. J. J. Kirkland, B. E. Boyes, and J. J. DeStefano, Amer. Lab., 26 (1994) 36.
20. D. W. Sindorf and G. E. Maciel, J. Am. Chcm. Soc., 105 (1983) 1487.
21. P. G. Dietrich, K.-H. Lerche, J. Reusch, and R. Nitzsche, Chromatographia, 44 (1997) 362.
22. A. Mendez, E. Bosch, M. Roses, and U. D. Neue, J. Chromatogr. A, 986 (2003) 33.
23. M. Jacoby, Chemical and Engineering News, Dec. 11, 2006.
24. F. Svec and С G. Huber, Anal. Chem., 78 (2006) 2100.
25. G. Guiochon, J. Chromatogr. A, 1168 (2007) 101.
26. K. K. Unger, R. Skudas, and M. M. Schulte, J. Chromatogr. A, 1184 (2008) 393.
27. F. Svec, T. Tennikova, and Z. Deyl (eds.), Monolithic Materials: Preparation, Properties and
Applications, J. Chromatogr. Library, Vol 67, Elsevier, Amsterdam, 2004.
28. M. Kele and G. Guiochon, J. Chromatogr. A, 960 (2002) 19.
29. X. Wang, D. R. Stoll, P. W. Carr, and P. J. Schoenmakers, J. Chromatogr. A, 1125 (2006) 177.
30. S. Eeltink, G. Desmet, G. Vivy-Truyols, G. P. Rozing, P. J. Schoemnmakers, and W. Th.Kok,
J. Chromatogr. A, 1104 (2006) 256.
31. Т. Нага, H. Kobayashi, T. Ikegami, K. Nakanishi, and N. Tanaka, Anal. Chem., 78 (2006) 7632.
32. B. Bidlingmeyer, K. K. Unger, and N. von Doehren, J. Chromatogr. A. 832 (1999) 11.
33. L. Geiser, S. Eeltink, F. Svec, and J. M. J. Frcchet, J. Chromatogr. A, 1140 (2007) 140.
34. J. Nawracki, С Dunlap, J. Li, J. Zhao, С V. McNeff, A. McCormick, and P. W. Carr, J.
Chromatogr. A, 1028 (2004) 31.
35. J. Nawrocki, С Dunlap, A. McCormick, and P. W. Carr, J. Chromatogr. A, 1028 (2004) 1.
36. U. Bien-Vogelsang, A. Deege, H. Figge, J. Kohler, and G. Schomburg, Chromatographia, 19
(1984) 170.
37. J. J. Pesek and M. T. Matyska, J. Chromatogr. A, 952 (2001) 1.
38. P. Ross, LCGC, 18 (2000) 14.
39. J. H. Knox, B. Kaur. and G. R. Millward, J. Chromatogr., 352 (1986) 3.
40. M. J. Wirth and H. O. Fatunmbi, Anal. Chem., 65 (1993) 822.
41. J. J. Kirkland, J. L. Glajch, and R. D. Farlee, Anal. Chem., 61 (1988) 2.
42. J. L. Glajch and J. J. Kirkland, US Pat. 4,705,725, 1987
43. J. L. Glajch and J. J. Kirkland, US Pat. 4,847,159, 1989.
44. J. J. Kirkland, J. B. Adams, M. A. Van Straten, and H. A. Claessens, Anal. Chem., 70 (1998) 4344.
45. L. С Sanders and S. A. Wise. Anal. Chem., 56 (1984) 504.
46. N. S. Wilson, J. Gilroy, J. W. Dolan, and L. R. Snyder J. Chromatogr. A, 1026 (2004) 91.
47. L. Brown, B, Ciccone, J. J. Pesek, and M. T. Matyska, Amer. Lab., Dec. (2003) 23.
288 Глава 5. Колонки
48. К. Miyabe and N. Orita, Talanta, 36 (1989) 897.
49. M. R. Buchmeiser, J. Chromatogr. A, 918 (2001) 233.
50. K. K. Ungerand J. Schick-Kalb, US Pat. 4,017,528, 1977.
51. K. D. Wyndham, J. E. O'Gara, T. H. Walter, K. H. Glose, N. L. Lawrence, B. A. Alden,
G. S. Izzo, С J. Hudalla, and P. С Iraneta, Anal. Chem., 75 (2003) 6781.
52. R. E. Majors, LCGC, 20 (2002) 516.
53. B. A. Bidlingmeyer and A. D. Broske, J. Chromatogr. Set., 42 (2004) 100.
54. T. H. Walter, P. Iranetaimd, and M. Capparella, J. Chromatogr. A, 1075 (2005) 177.
55. R. Eksteen and J. Thoma, in: Chromatography, the State of the Art, Vol. 1, Akademia Kiado,
Budapest, 1985.
56. L. Zhang, L. Sun, J. I. Siepmann, and M. R. Shure, J. Chromatogr. A, 1079 (2005) 127.
57. K. Croes, A. Steffens, D. H. Marchand, and L. R. Snyder,/ Chromatogr. A, 1098 (2005)123.
58. M. R. Euerby, P. Pctersson, W. Campbell, and W. Roe, J. Chromatogr. A, 1154 (2007)138.
59. U. D. Neue, J. Sep. Sci., 30 (2007) 1611.
60. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, R. G. Wolcott, and P. W. Carr, J.
Chromatogr. A, 961 (2002) 171.
61. L. R. Snyder, J. W. Dolan, and P. W. Carr, J. Chromatogr. A, 1060 (2004) 77.
62. L. R. Snyder, J. W. Dolan, and P. W. Carr, Anal. Chem., 79 (2007) 3255.
63. D. H. Marchand and L. R. Snyder, J. Chromatogr. A, 1209 (2008) 104.
64. L. R. Snyder and J. W, Dolan, High Performance Gradient Elution, Wiley-lnterscience, Hoboken,
NJ, 2007, p. 420.
65. L. С Sander and S. A. Wise, J. Chromatogr. A, 656 (1993) 335.
66. J. J. Kirkland, Amer. Lab., 26 (1994) 28K.
67. U. D. Neue, E. Serowik, P. Iraneta, B. A. Alden, and T. H. Walter, J. Chromatogr. Л.849 (1999)
87.
68. L. R. Snyder, A. Maule, A. Heebsch, R. Cuellar, S. Paulson, J. Carrano, L. Wrisley, C.C. Chan,
N. Pearson, J. W. Dolan, and J. Gilroy, J. Chromatogr. A, 1057 (2004) 49.
69. J. Pellett, P. Lukulay, Y. Mao, W. Bowen, R. Reed, M. Ma, R. С Munger, J. W. Dolan,L.
Wrisley, K. Medwid, N. P. Toltl, С. С. Chan, M. Skibic, K. Biswas, K. A. Wells, andL. R. Snyder, J.
Chromatogr. A, 1101 (2006) 122.
70. Y. Mao and P. W. Carr, LCGC, 21 (2003) 150.
71. J. W. Dolan and L. R. Snyder, J. Chromatogr. A, 1216 (2009) 3467.
72. D. V. McCalley, Adv. Chromatogr., 46 (2008) 305.
73. D. M. Marchand, L. R. Snyder, and J. W. Dolan, J. Chromatogr. A, 1191 (2008) 2.
74. H. Colin. P. Hilaireau, and J. De Toumemire, LCGC, 8 (1990) 302.
75. F. Couillard, Chromatography Apparatus, US Patent 4,597,866, 1986-07-01.
76. J. J. Kirkland and J. J. DeStefano, J. Chromatogr. A, 1126 (2006) 50.
77. L,- R. Snyder and J. J. Kirkland, Introduction to Modern Liquid Chromatography, 2nd ed., Wiley,
New York, 1979, ch. 5.
78. H. A. Claessens, M. A. Van Straten, and J. J. Kirkland, J. Chromatogr. A, 728 (1996)259.
79. J. J. Kirkland, M. A. Van Straten, and H. A. Claessens, J. Chromatogr. A, 797 (1998)111.
80. G. W. Tindall and R. L. Perry, J. Chromatogr. A, 988 (2003) 309.
81. J. J. Kirkland, J. Chromatogr. Sci., 38 (2000) 535.
82. F. Gerber, M. Krummen, H. Potgeter, A. Roth, C. Siffrin, and C. Spoendlin, /. ChromatogrA,
1036 (2004) 127.
ГЛАВА 6
ОБРАЩЕН НО-Ф АЗОВ АЯ
ХРОМАТОГРАФИЯ
НЕЙТРАЛЬНЫХ ОБРАЗЦОВ
6.1. Введение 290
6.1.1. Краткая история обращенно-фазовой хроматографии 292
6.2. Удерживание 292
6.2.1. Сила растворителя 294
6.2.2. Механизм удерживания в ОФХ 296
6.3. Селективность 300
6.3.1. Зависимость селективности от силы растворителя 300
6.3.2. Зависимость селективности от типа растворителя 303
6.3.3. Зависимость селективности от температуры 306
6.3.3.1. Последующий анализ 309
6.3.4. Селективность колонки 310
6.3.5. Разделения изомеров 311
6.3.5.1. Повышенная селективность при разделении изомеров 312
6.3.5.2. Зависимость селективности от формы молекулы 313
6.3.6. Другие соображения, касающиеся селективности 314
6.3.6.1. Эквивалентное разделение 314
6.3.6.2. Ортогональное разделение 318
6.4. Разработка метода и стратегия оптимизации селективности 319
6.4.1. Оптимизация нескольких параметров 321
6.4.1.1. Смеси различных органических растворителей 322
6.4.1.2. Одновременное изменение элюирующей силы
и типа растворителя 325
6.4.1.3. Одновременное изменение элюирующей силы и температуры
растворителя 325
6.4.1.4. Замена колонки с одновременным изменением одного или
более условий 328
6.4.2. Оптимизация условий изменением параметров колонки 329
6.5. Неводная обрашенно-фазовая хроматография (НОФХ) 331
6.6. Специфические проблемы 331
6.6.1. Слабое удерживание образцов с высокой полярностью 332
6.6.2. Уширение заднего фронта пика 333
Литература 333
290 Глава 6. Обращение-фазовая хроматография нейтральных образцов
6.1. Введение
В этой главе описано разделение нейтральных образцов посредством обращенно фа
зовой хроматографии (ОФХ). Под нейтральным образцом мы понимаем тот,
который не содержит молекул, несущих положительный или отрицательный заряд
в результате кислотной или основной ионизации. Хотя нейтральный образец и
подразумевает отсутствие кислотных и основных растворенных веществ, это не всегда
так. В зависимости от рН подвижной фазы в образце могут присутствовать любые
кислоты или основания большей частью (например, 90%+) в нейтральной (неиони
зированной) форме. В этом случае их хроматографическое поведение будет
аналогичным поведению неионизируемых соединений. Разделение «ионных» образцов
(которые содержат одно или более ионизированных соединений) с помощью ОФХ
описано в главе 7.
Обычно к ОФХ прибегают в первую очередь, когда хотят разделить как нейтра
льные, так и ионные образцы на сорбенте, содержащем менее полярную привитую
фазу — такую как Cg и Сц{. В большинстве случаев подвижная фаза представляет
собой смесь воды и либо ацетонитрила (ACN), либо метанола (МеОН). Другие
органические растворители |например, изопропиловый спирт (ИПС), тетрагидрофуран
(ТГФ)| используются гораздо реже. Наилучшим для подвижной фазы в ОФХ будет
смешивающийся с водой относительно невязкий органический растворитель,
стабильный в условиях его использования, прозрачный на минимально возможной длине
волны при УФ-детектировании и легко доступный по умеренной цене. Обычно
используемые В-растворители могут быть ранжированы в соответствии с этими
требованиями следующим образом:
ACN (предпочтительно) > МеОН > И ПС 3> ТГФ (наименее используемый).
В некоторых странах существуют ограничения по широкому использованию ACN
из-за его токсичности, но так происходит не повсеместно. Для получения
дополнительной информации о свойствах растворителя и выборе растворителя В для
конкретного метода см. Приложение I. Образцы, содержащие кислоты или основания,
обычно требуют буферной подвижной фазы для поддержания постоянного рН
во время разделения (глава 7). Для сильно удерживаемых, очень гидрофобных
образцов может потребоваться безводная подвижная фаза |неводная обращенно-фазовая
хроматография (НОФХ), раздел 6.5|. Нормально-фазовая хроматография (НФХ,
глава 8) тоже может обеспечить приемлемые разделения крайне гидрофобных образцов,
поскольку гидрофобность вносит небольшой вклад в удерживание в этом виде
хроматографии. Предпочтительные условия для изократического разделения
нейтральных образцов с помощью ОФХ даны в таблице 6.1.
По сравнению с другими видами ВЭЖХ [НФХ, ионообменная хроматография
(ИОХ) и т.д.; таблица 2.11, разделения с помощью ОФХ гораздо более удобны,
устойчивы и универсальны. Кроме того, обращенно-фазовые колонки, как
правило, более эффективны и результаты, полученные с их помощью, более
воспроизводимы. Они доступнее в самых разных исполнениях — это и широкий выбор
геометрических размеров, и размеров частиц, и разные типы сорбентов (С,—Сзд,
фенил-, циано-фазы и т.д.; раздел 5.3.3). Растворители, используемые для ОФХ,
обычно менее огнеопасны или токсичны и более совместимы с УФ-детектировани-
ем на длинах волн менее 230 нм, которые используются для повышения
чувствительности обнаружения вещества (таблица 1.2 Приложения I). Дополнительным
преимуществом ОФХ является, как правило, быстрое уравновешивание колонки после
смены подвижной фазы или между разделениями при использовании градиентного
6.1. Введение 291
элюирования (раздел 9.3.7). Наконец, поскольку с конца семидесятых годов
прошлого века ОФХ является превалирующим видом ВЭЖХ, улучшилось понимание
практической стороны метода. Как правило, это означает более короткий путь
к более эффективным разделениям. Все вышеизложенные причины и
способствовали нынешней популярности ВЭЖХ.
Таблица 6.1. Предпочтительные условия для разделения нейтральных образцов с
помощью ОФХ
Условия
Колонка"
Подвижная фаза
Скорость потока
Температура
%В
Образец
к
Комментарии
Тип: Сц или С|я (тип Б)
Размеры: 100 х 4,6 мм
Размер частиц: 3 мкм
Диаметр пор: 8—12 нм
Ацетон итрил/вода
2.0 мл/мин*
30 или 35 °С«
Определяется в процессе разделения"
Объем < 25мкл
Масса £ 50 мкг
1 < к < 10
°В качестве альтернативы используйте колонку размером 150 х 4,6, упакованную частицами
размером 5 мкм; скорость потока, размеры колонки и размер частиц могут варьироваться в
зависимости от сложности предстоящего разделения и максимума допустимого давления
(раздел 2.4.1).
^Начальные значения, которые могут быть изменены в процессе разработки метода
(раздел 2.5); в большинстве случаев рекомендуется температура на 5—10 "С выше температуры
окружающей среды. Кроме того, следует запросить у производителей информацию о
максимально допустимой температуре, при которой может работать колонка.
"Зависит от образца; начинаем с 80 %В и далее корректируем так, как это описано в
разделе 2.5.1.
Множество органических соединений ограниченно растворимы в воде или
в водно-органических подвижных фазах, используемых для ОФХ, но на практике
это редко приводит к затруднениям, поскольку вводят в колонку очень малые
количества (нанограммы или несколько микрограмм) индивидуальных веществ.
Вследствие этого требуемая концентрация образца обычно несколько мкг/мл или
меньше. В тех случаях, когда образец очень плохо растворяется в воде или в
водно-органических смесях (супергидрофобные образцы), может быть целесообразным
применение нормально-фазовой хроматографии (НФХ) в неводных подвижных
фазах (раздел 8.4.1).
Некоторые образцы в режиме ОФХ разделяются хуже. Например, очень
полярные молекулы в условиях ОФХ могут слабо удерживаться (к <К 1) даже при
условии, что подвижная фаза будет чистой водой. К таким образцам может
потребоваться другой подход (раздел 6.6.1). Аналогично, для разделения энантиомеров
требуются условия с хиральной селективностью (глава 14). Хотя многие ахираль-
ные изомеры и могут быть разделены с помощью ОФХ (раздел 6.3.5), их лучше
разделять в режиме НФХ, используя колонки, упакованные непривитым силикаге-
лем (раздел 8.3.4.1). В конечном счете, НФХ часто лучший выбор для
препаративной ВЭЖХ (глава 15).
292 Глава 6. Обращенно фазовая хроматография нейтральных образцов
6.1.1. Краткая история обращенно-фазовой хроматографии
До изобретения ОФХ в I950 году А. Дж. П. Мартином |]| хроматографическое
разделение нейтральных образцов проводилось на полярной колонке (т.е. на полярной
стационарной фазе) с использованием менее полярной подвижной фазы. Такие
разделения теперь называют нормально-фазовой хроматографией (НФХ). Поскольку
в ОФХ применяется менее полярная неподвижная фаза и более полярная подвижная
фаза, то эти две фазы можно рассматривать как поменявшиеся местами из-за
изменения относительной полярности или «обращенные». Первые колонки для ОФХ
упаковывались частицами силикагеля, которые обрабатывались диметилдихлорсила-
ном для того, чтобы сделать поверхность частиц неполярной. Полученная
неполярная неподвижная фаза взаимодействовала с неполярным компонентом подвижной
фазы, что позволило эти частицы использовать для обращенно-фазовой
жидкость-жидкостной распределительной хроматографии |2|, когда вещество
распределяется между подвижной жидкой фазой и «неподвижной» жидкой фазой,
адсорбированной на силанизированной твердой фазе. Этот вид ВЭЖХ сегодня редко
используется.
Колонки с привитыми фазами явились следующим значительным шагом вперед
в направлении «высокопроизводительной» ОФХ. Поначалу сорбенты C)g получали
ковалентным связыванием полиоктадецилсилоксана с частицами силикагеля [3, 4].
Через несколько лет появились сорбенты, в которых к силикагельным частицам
присоединяли отдельные С|8-группы {а не полимер С\%) (раздел 5.3.1). С начала
семидесятых годов ОФХ-колонки последнего типа из-за их многочисленных преимуществ
использовались все чаще. В середине семидесятых был взрыв популярности ОФХ.
Несколько сотен разделений упоминаются в |5| за период с 1976 по 1979 гг. В то
время как в более ранних случаях применения ОФХ делался упор на разделение
супергидрофобных (липофильных) образцов, то в классической работе Хорвата |6|
продемонстрировано, что ОФХ также может использоваться для разделения
малополярных соединений. Особенно это касается водорастворимых образцов,
представляющих интерес для биохимии. Немногим позже ОФХ была дополнительно
приспособлена для разделения ионизируемых соединений (включая энантиомеры) путем
добавления ион-парных соединений к подвижной фазе |7| (ион-парная
хроматография, раздел 7.4). В конце семидесятых несколько групп исследователей сообщили
о первых ОФХ-разделениях больших пептидов и белков с использованием колонок,
упакованных широкопористыми сорбентами с короткими алифатическими цепями.
Сегодня ОФХ — наиболее употребительный вид ВЭЖХ, и в этом режиме проводится
большинство разделений в ВЭЖХ.
6.2. Удерживание
Удерживание в ОФХ уже кратко обсуждалось в разделе 2.2. Поскольку очень
полярные молекулы сильнее взаимодействуют с полярной подвижной фазой, эти
соединения удерживаются слабее и покидают колонку в первую очередь. Аналогично, менее
полярные соединения интенсивнее взаимодействуют с неполярной подвижной фазой
и выходят из колонки последними. Таким образом, молекулы сходных размеров
в ОФХ элюируются приблизительно в порядке уменьшения полярности. На рис. 6.1а
приведен пример, демонстрирующий, что более полярный бензонитрил (1)
появляется на хроматограмме первым, за ним следует менее полярный анизол (2) и,
наконец, выходит толуол (3). Более подробный пример, иллюстрирующий
зависимость удерживания в ОФХ от полярности растворенного вещества, представлен
6.2. Удерживание 293
О)
O-CsN(l)
<Э-ОСН3{2)
1
С18 колонка
40% ACN
30°С
0-СН3<3)
А
(б)
ю
12
С1В колонка
60%ACN
30°С
14 (мин)
10
12 14 (мин)
(в)
X
С18 колонка
40% ACN
70Х
10
12 14 (мин)
<«)
Циано колонка
40% ACN
ЗОХ
8
10
12
14 (мин)
Рис. 6.1. Зависимость удерживания в ОФХ от температуры и полярности растворенного
образца, подвижной фазы и сорбента. Образцы указаны на рисунке. Условия:
(я-e) колонка 150x4,0 мм, 5мкм Symmetry C|g и (г) колонка 150x4,6 мм,
5 мкм Zorbax StableBond cyano; 2 мл/мин; подвижная фаза ацетонитрил/вода,
состав которой (%В) и температура указаны на рисунке [жирным шрифтом
выделены значения, отличные от (я)|. Хроматотраммы смоделированы по
данным работ |8, 9|
на рис. 2.7е; см. также соответствующее обсуждение селективности ОФХ-колонок
в разделе 5.4.
Удерживание в ОФХ в значительной мере является результатом взаимодействий
между молекулой растворенного вещества и подвижной или неподвижной фазами
(раздел 2.3.2.1). Увеличение %В (объемный %В органического растворителя в
подвижной фазе) делает подвижную фазу менее полярной («сильнее») и увеличивает силу
взаимодействия между молекулами растворенного вещества и растворителя. В результате
при увеличении %В уменьшается удерживание молекул всех растворенных веществ.
Это иллюстрирует сравнение разделений, показанных на рис. 6.\б (в подвижной фазе
60 %В) и рис. 6.\а (40 %В). Повышение температуры ослабляет взаимодействие
молекул растворенного вещества как с подвижной фазой, так и с сорбентом и уменьшает
удерживание — сравните рис. 6. le (70 °С) и рис. 6.1а (30 °С). Наконец, уменьшение
гидрофобное™ колонки ослабляет взаимодействие между растворенным веществом и
сорбентом и уменьшает удерживание. Сравните рис. 6. \г (более полярная циано-ко-
лонка) и рис. 6.1а (менее полярная Cig-колонка).
Глава 6. Обращешю фазовая хроматография нейтральных образцов
6.2.1. Сила растворителя
Как уже было сказано в разделе 2.4.1, удерживание в ОФХ изменяется в зависимости
от %В в подвижной фазе как
(6.1)
log к - log кК - Sq>
где кК — экстраполированное значение к для О %В (вода в качестве подвижной
фазы), S — константа для данного растворенного вещества в том случае, если
изменяется только %В, и ф — объемная доля органического растворителя В в подвижной
фазе (ф s 0,01 %В). Величина %В, выбранная для окончательного разделения,
должна обеспечивать такие значения к для образца, которые находятся внутри
предпочтительного интервала (например, 1 < к S 10), и в то же время максимально
увеличивать селективность растворителя (раздел 6.3.1).
Подходящая величина %В может быть получена так, как это описано в
разделе 2.5.1: начинаем с 80 %В и затем понижаем %В ступенчато до тех пор, пока
не попадем в желательный интервал удерживания I < к < 10. На рис. 6.2
проиллюстрирован пример такого подхода. В первом разделении с 80 %В (рис. 6.2я) было
очень небольшое удерживание (к - 0,3), поэтому для следующего эксперимента
(рис. 6.26) использовали подвижную фазу с 50 %В. Диапазон, в котором находится
коэффициент удерживания образца, теперь приемлемый (1,9 < к < 4,2), но
разрешение недостаточно (Rs = 1,0). Дальнейшее уменьшение %В обычно (но не всегда)
увеличивает разрешение.
(а)
80% ACN 1-*
{А =0.3)
Яс = 0.0
I
(б)
50% ACN
(1.9<А<4.2)
0,-1.0
г 1 1 j 1 1
0.2 0.4 0.6 0.8
Время (мин)
Г
О
(в)
40% ACN
<4<*<М0)
Я,-1.4
г 1 1 1 1 1 1—
0 2 4 6
Время (мин)
Рис. 6.2. Разделение смеси четырех нитробензолов в зависимости от силы
растворителя (%В). Образец: 1, нитробензол; 2, 4-нитротолуол; 3, 3-нитротолуол;
4, 2-нитро-1,3-ксилол. Условия: колонка 100 х 4,6 мм (3 мкм) ZorbaxCs;
подвижная фаза содержит смеси ацетонитрил-вода (с изменяемым %В); 35 °С;
2 мл/мин. Хроматограммы смоделированы поданным из работы {10 J
6.2. Удерживание 295
Для образцов с молекулярной массой < 500 Да снижение на 10 %В приведет
к увеличению величин к в к 2,5 раза, что предполагает использование для
следующего эксперимента подвижной фазы с 40 %В (рис. 6.2е). Разрешение умеренно
возрастает (Rs = 1,4), но остается неудовлетворительным. Поскольку дальнейшее
уменьшение %В приведет к значениям к > 10, могут потребоваться и другие способы
увеличения разрешения: изменение селективности (раздел 6.3) или изменение
условий разделения, связанные с колонкой (раздел 2.5.3).
Следует отметить, что уравнение 6.1 не точное, а приближенное. Например,
на рис. 6.3 приведен график зависимости log А для типичного образца (4-нитротолу-
ол; соединение 2 на рис. 6.2) от %В при использовании в качестве растворителя В
ацетонитрила (ACN, □) и метанола (МеОН, •). В то время как уравнение (6.1)
предсказывает линейную зависимость, на практике результаты для ацетонитрила в
качестве растворителя В описываются кривой с небольшим отклонением от прямой.
Данные для метанола почти точно ложатся на прямую на рис. 6.3, что соответствует
предсказанию. Такое поведение характерно и для других образцов и
экспериментальных условий |П|. Графики с меньшим отклонением от линейности обычно
получаются для метанола в сравнении с ацетонитрилом или другими органическими
растворителями, взятыми в качестве растворителя В. Однако, в пределах обычно
используемого диапазона к (например, 1 < к < 10) уравнение (6.1) является достаточно
точным и для метанола и для ацетонитрила, взятых в качестве растворителя В.
J I I I I I L
0 20 40 60 80
%В
Рис. 6.3. Изменения log k в зависимости от %В. Образец: 4-нитротолуол. Условия:
колонка 250 х 4,6 мм (5 мкм) Zorbax C8; подвижная фаза водно-органическая;
35 "С; 2 мл/мин. График построен по данным из работы 1101
296 Глава 6. Обращение-фазовая хроматография нейтральных образцов
6.2.2. Механизм удерживания в ОФХ
Нижеследующее обсуждение дает углубленное понимание основ механизма удерживания
в ОФХ. Однако, в основном, это обсуждение представляет академический интерес,
не имея непосредственного отношения к практической ОФХ. Ориентированный на
получение практической пользы читатель может пропустить это обсуждение и перейти
к разделу 6.3.
Природа удерживания в ОФХ («механизм» удерживания) был предметом
большого числа исследований, обобщенных в нескольких обзорах и публикациях |5,
12—16]. Некоторые из вопросов, которые рассматривались в этих исследованиях,
включают:
• пространственное расположение молекулы растворенного вещества в
стационарной фазе (адсорбция или распределение?);
• зависимость удерживания молекул растворенного вещества от состава подвижной
фазы (к от ф);
• конформация образующих неподвижную фазу алкильных лигандов («развернутая»
или «сжатая»).
Ориентация растворенной молекулы внутри стационарной фазы может
осуществляться любым из способов, изображенных на рис. (\.4а-в для колонки, упакованной
сорбентом Cg. Сольвофобное взаимодействие (рис. 6.4д) предполагает, что
растворенная молекула ориентируется и связывается с лигандом неподвижной фазы (в данном
случае это Cg). Адсорбция (рис. 6.46) приводит к тому, что молекула растворенного
вещества не проникает в неподвижную фазу, а остается на границе между
неподвижной и подвижной фазами. Распределение (рис. 6.4ff) рассматривает неподвижную фазу
как подобие жидкой фазы, в которой растворяется молекула вещества. Обратите
внимание на то, что неподвижная фаза состоит из алкильных лигандов и молекул
органического растворителя. Последний избирательно экстрагируется Cg-группами
неподвижной фазы из подвижной фазы. Как при сольвофобных взаимодействиях,
так и при распределении молекула растворенного вещества находится внутри
неподвижной фазы.
При обсуждении механизма удерживания в ОФХ чаще всего ссылаются на
модель сольвофобного взаимодействия Хорвата |5|: (относительно) гидрофобные
молекулы вещества имеют склонность притягиваться к гидрофобным алкильным лиган-
дам. Такое поведение называется гидрофобным удерживанием. Вскоре после
введения ОФХ в практику ВЭЖХ было замечено, что удерживание в ОФХ
(величины к) коррелирует с коэффициентами распределения Р для распределения
растворенного вещества между октанолом и водой (рис. 6.4<?, |17|). Это говорит о том, что
распределительная модель лучше всего описывает механизм удерживания в ОФХ.
Однако, более поздние исследования показали, что корреляции между log P и log к,
подобные показанной на рис. 6Аг для аминокислот, менее выражены, когда образец
состоит из молекул с более разнообразным строением. Это делает последний
аргумент в пользу распределительного механизма менее убедительным.
В начале 1980-х годов |19| было сделано интересное наблюдение для
удерживания в ОФХ различных соединений гомологических рядов (СНз-|СН2|п_]-Х), где X
представляет собой функциональную группу, такую как -ОН или -СО2СН3. Было
показано, что нарушается непрерывность графика зависимости log А от количества
атомов углерода п для и, которое приблизительно равно числу атомов углерода п
у лиганда неподвижной фазы (например, л = 8 для CgsCH3-|CH2|7-). Такое
аномальное поведение иллюстрируется на рис. 6.5а гипотетическим графиком зависимости
log к от п для гомологического ряда и колонки Cg: наблюдается нарушение
непрерывности ожидаемого линейного графика (показанного пунктиром) при и равном 8
(показано стрелкой) для соединения (CH3-ICH2I7-X). Отсюда следовало, что вклад
6.2. Удерживание 297
(а)
(б)
молекула
анализируемого
образца
сопьвофобное
взаимодействие
адсорбция
(в)
сорбированная
подвижная фаза
распределение
Рис. 6,4, Различные механизмы удерживания молекул растворенного вещества в обра-
щенно-фазовой хроматографии, (я) Сольвофобное взаимодействие; (б)
адсорбция; (в) распределение; (г) сравнение ОФХ-удерживания (к) с
распределением в системе октанол-вода Р |17]; образец: восемь аминокислот;
колонка Cj; подвижная фаза: водный буфер (рН-6.7); 70 °С
в удерживание для цепочки, состоящей из CHj-rpynn в растворенном соединении,
становится немного меньше, когда длина молекулы растворенного вещества
превышает длину алкильного лиганда. Причина нарушения непрерывности графика
на рис. 6.5а проиллюстрирована на рис. 6.56, где л = 12 у растворенного вещества и
превышает значение л = 8 у лиганда сорбента (случай для л = и = 8 показан на
примере вещества 0- В примере, изображенном на рис. 6.56, когда п = 12, конец
молекулы, скорее всего, загибается назад к поверхности или внутрь неподвижной фазы,
а не выступает в подвижную фазу, как показано на рисунке.
Вероятно, становится слабее взаимодействие сорбента с теми молекулами
растворенного вещества, которые слишком длинны, чтобы полностью проникнуть
в него (или связаться с одним из лигандов). Соответствующим образом уменьшается
и удерживание Ch^-rpynn, которые «выступают» из неподвижной фазы. Вклад
в удерживание каждой СН2-группы может быть определен как ссснг= отношение
величин к для следующих один за другим гомологов, и этот вклад обычно принято
298 Глава 6. Обращение фазовая хроматография нейтральных образцов
Рис. 6.5. Зависимость удерживания от длины алкильной цепочки (для растворенного
вещества и сорбента), (а) График, иллюстрирующий зависимость log к
от числа СН2-групп п для гомологических рядов СН3—(CHj),,-!—X;
сорбент Cg; (б) график, иллюстрирующий «перекрывание» алкильных цепочек
в растворенном веществе и сорбенте; (в—д) экспериментальные зависимости
селективности асц от числа атомов углерода пс для указанных сорбентов
с лигандами различной длины. Усредненные данные для нескольких
гомологических рядов; 90% метанол-вода в качестве подвижной фазы; 25 °С.
Рисунки взяты из работы 1181
6.2. Удерживание 299
считать постоянным |см. обсуждение уравнения (2.34)|. Однако, для исключенных
СН2-групп величина аСц должна быть несколько меньше. Этот эффект
«исключения» экспериментально продемонстрирован на рис. 6.5е для колонки C|g графиком
зависимости асн2 от "• В области п = п = 18 наблюдается отчетливый перегиб
кривой (показанный вертикальной пунктирной линией). Похожие перегибы видны
на рис. 6.5г,д для колонок Cg и С6 соответственно, но для колонки, упакованной С|
(рис. 6.5d), никакого заметного проникновения растворенного вещества в сорбент не
наблюдается. Данные рис. 6.5в—д свидетельствуют о том, что небольшие молекулы
растворенного вещества могут проникать в неподвижную фазу С^, Cg или C|g, и
поэтому нельзя говорить о чисто адсорбционном механизме удерживания для других
небольших молекул (как на рис.6.46). Рис. 6.5 также служит подтверждением модели
сольвофобного взаимодействия, показанного на рис. 6.4а.
Рисунок 6.5 представляет собой иллюстративный материал к возможным
выводам относительно механизма удерживания в ОФХ. Что же касается адсорбции
и процессов распределения, то здесь были предложены более сложные объяснения
114, 15|. Следует отметить, что природа неподвижной фазы (и, вероятно, механизм
удерживания) меняется в зависимости от условий эксперимента. При увеличении
%В возрастающие количества растворителя В (например, ацетонитрила)
поглощаются неподвижной фазой. Также некоторые растворенные вещества могут
взаимодействовать с недериватизированными силанолами, присутствующими на поверхности
частиц сорбента (раздел 5.4.1). По этим и другим причинам конкретный механизм
удерживания, вероятно, будет зависеть от колонки, природы образца и
экспериментальных условий. Хорват предвосхитил эту ситуацию еще в работе |17|, отметив, что
«установление четкого различия между распределением и адсорбцией в ОФХ
неполярных |растворенных веществ| не имеет существенного термодинамического или
практического значения... и не имеет смысла заниматься им в дальнейшем». Авторам
этой книги трудно спорить с выводом Хорвата.
Рассуждения, касающиеся природы удерживания в ОФХ, тоже основывались
на том, что удерживание зависит от состава подвижной фазы (%В). Хотя
уравнение (6.1) чисто эмпирическое, были получены несколько теоретически
обоснованных уравнений для зависимости к от ф |11|. Полученные выражения для
зависимости к от ф в некоторых случаях чуть более достоверны, чем уравнение (6.1) |19|,
большей частью из-за дополнительных параметров подгонки. Однако уравнение (6.1)
в общем случае приемлемо для практического применения, и его гораздо удобнее
использовать. Основные допущения, связанные со всеми предыдущими
теоретическими выкладками для зависимости к от ф, сводят к нулю их использование для
интерпретации природы удерживания в ОФХ.
Утверждается, что конформация лигандов стационарной фазы играет
определенную роль в механизме удерживания в ОФХ. Использование преимущественно
водных подвижных фаз (ф = 0) может привести к значительному уменьшению
удерживания образца, что противоречит предсказанному уравнением (6.1). Когда впервые
обнаружили это уменьшение удерживания, то связали его с «фазовым коллапсом»,
при котором алкильные лиганды агрегируют и стремятся расположиться в
горизонтальной, а не в вертикальной плоскости над поверхностью частиц. Позже было
показано, что это изменение удерживания возникает не из-за фазового коллапса, а,
скорее, из-за исключения подвижной фазы и молекул образца из пор частиц сорбента
вследствие эффектов поверхностного натяжения («несмачиваемость», раздел 5.3.2.3 и
|2, 211). Более поздние исследования 122, 22а| показывают, что конформация лиган-
да не меняется в зависимости от состава подвижной фазы или от степени покрытия
частиц сорбента лигандами.
300 Глава 6. Обращение-фазовая хроматография нейтральных образцов
6.3. Селективность
Наиболее эффективный способ улучшить разрешение (или увеличить скорость) хро
матографического разделения — вызвать изменение относительного удерживания
(селективности). В ОФХ можно достичь разделения неионных образцов, изменяя
селективность посредством изменения силы растворителя (%В), температуры, типа
растворителя (например, метанол в качестве органического растворителя вместо аце
тонитрила) или сорбента (например, С]8 вместо циано-фазы). Относительная
эффективность изменения условий для изменения селективности возрастает в ряду:
температура (наименее эффективно) < %В < тип растворителя <
< тип сорбента (наиболее эффективно).
Однако, как это будет описано ниже, каждое из четырех вышеперечисленных
условий для изменения селективности может быть полезным для различных
образцов или целей разделения.
6.3.1. Зависимость селективности от силы растворителя
В примерах, представленных на рис. 6.I и 6.2, относительное удерживание не
изменяется при изменении силы растворителя (%В). По мере уменьшения %В (и роста к)
разрешение всех пиков улучшается, но их относительное расположение остается
практически неизменным. Образцы (такие как на рис. 6.I и 6.2) мы в разделе 2.5.2.1
назвали правильными. На рис. 6.6 показано разделение образца, в котором при
изменении %В относительное удерживание не остается неизменным. Первое разделение
с подвижной фазой, в которой 50 %В (рис. 6.6а), показывает полное перекрытие
пиков 2 и 3 (пик 3 заштрихован). Когда подвижную фазу меняют на 40 %В (рис. 6.66),
пик 3 перемещается по направлению к пику 4 и частично его перекрывает. Из этих
двух экспериментов видно, что промежуточное значение %В должно приводить
к улучшенному разделению, которое мы и наблюдаем на рис. 6.6* (45 %В). Образцы
(такие как этот), для которых относительное удерживание изменяется вместе с силой
растворителя, называются неправильными. Правильные образцы часто состоят
из структурно подобных молекул. Например, в разделении, показанном на рис. 6.2,
образец представляет собой смесь мононитроалкилбензолов, в то время как образец
на рис. 6.6 в этом смысле более разнообразен. Он представляет собой смесь алкил-
бензолов, содержащих 0, I или 2 нитрогруппы в качестве заместителей.
Изменения %В часто приводят к значительным изменениям относительного
удерживания. При этом максимальное разрешение достигается при промежуточных
значениях %В. Несмотря на этот очевидный факт, практики часто пренебрегают
зависимостью селективности от силы растворителя как полезным инструментом
для оптимизации относительного удерживания. Чтобы максимально использовать
зависимость селективности от силы растворителя, можно расширить допустимый
диапазон удерживания от 1 < к й 10 до 0,5 й к S 20. Когда меняют условия с целью
изменения селективности, важно следить за тем, чему соответствует каждый пик.
Нумерация каждого пика на хроматограмме может быть (как на рис. 6.6)
неочевидной. В таких случаях необходимо отслеживать пики (раздел 2.7.4).
Оставшаяся часть этого раздела, посвящена довольно подробному обсуждению
правильных и неправильных образцов. Читатель при желании может предпочесть бегло
просмотреть или пропустить это обсуждение и перейти к разделу 6.3.2.
Зависимость удерживания от %В для правильных образцов в противоположность
неправильным проиллюстрирована на рис. 6.7. На рис. 6.7а показаны графики зави-
6.3. Селективность 301
(а)
50% ACN
2bfc<,6
Я, = 0.0
(б)
40% ACN
4<*г<14
Я„=1.0
(в)
45% ACN
3<Л<6
Я, = 1.5
Время (мин)
11 2
-1 1 1 1 г-
2 4 6
Время (мин)
1 I 2
2 4
Время (мин)
Рис. 6.6. Разделение умеренно неправильного образца (смесь восьми нитроароматиче-
ских соединений) в зависимости от силы растворителя (%В). Образец: /,
нитробензол; 2, 2,6-динитробензол; 3, бензол (пик заштрихован); 4, 2-нитрото-
луол; 5, 3-нитротолуол; б, толуол; 7, 2-нитро-1,3-ксилол; £, 1,3-ксилол.
Условия: колонка 100 х 4,6 мм (3 мкм) Zorbax C8; подвижная фаза — смеси
ацетонитрил/вода; 35 "С; 2 мл/мин. Хроматограммы смоделированы по
данным из [10]
симости log k от %В для правильного образца — смеси из девяти гербицидов со
сходной молекулярной структурой (см. рис. 2.6, на котором показано разделение
нескольких из этих соединений в зависимости от %В). Наклон каждого графика
увеличивается для более удерживаемых растворенных веществ (в последовательности
1 < 2 < 3...). Для графиков зависимости log к от %В для правильных образцов это
приводит к «веерному» виду — без пересечения одного графика другим (без
изменений в относительном удерживании). Для правильных образцов величины а и
разрешения непрерывно растут по мере убывания %В. Кроме того, описать поведение
правильных образцов можно с помощью уравнения (6.1). Для правильных образцов
углы наклона S графиков на рис. 6.7а хорошо коррелируют с экстраполированными
значениями log к для случая, когда подвижная фаза — вода (log кК). На рис. 6.76
показан такой график для данных рис. 6.7а. Видно, что данные прекрасно коррелируют
(Й = 1,00). Соответствующие корреляции зависимостей log kw от S для правильных
образцов на рисунках 6.1<з, б и 6.2 составляют г2- - 0,99 для каждой.
На рис. 6.7в, г показаны результаты для неправильного образца, разделение
которого приведено на рис. 6.6; они получены таким же образом как результаты
для правильного образца (рис. 6.7 о,б). На рис. 6.7* графики зависимости log k от %В
302 Глава 6. Обращение фазовая хроматография нейтральных образцов
Рис. 6.7. Изменения log к в зависимости от %В для правильных и неправильных
образцов, (в) Правильный образец: смесь гербицидов |24], разделенных на
колонке, упакованной С|8 с использованием в качестве подвижной фазы смеси
метанол-вода (см. остальные условия на рис. 2.6); (б) график занисимо-
сти величин S от log kw для данных, показанных на (а); (в) неправильный
образец, разделение которого показано на рис. 6.6; условия такие же, как
на рис. 6.6; (г) график зависимости величин S от log кж для данных,
показанных на (в), (а,б) обработанные данные из |25|
часто пересекаются (пересечения обозначены •) в отличие от аналогичных графиков
для правильных образцов на рис. 6.7а. В результате в этом диапазоне %В несколько
пиков (например, пики 3—4) меняются местами. Аналогично, зависимость S
от log kw для неправильного образца на рис. 6.7г свидетельствует о несколько более
низкой корреляции (г2 = 0.87). Образцы, содержащие кислотные и/или основные
соединения (в отличие от примеров на рис. 6.7), имеют более выраженную тенденцию
проявлять свойства неправильных образцов. Впрочем, большинству образцов —
«ионных» или «нейтральных» — свойственна некоторая степень неправильности.
Поэтому зависимость селективности от силы растворителя может быть полезным
инструментом для улучшения разрешения таких образцов.
6.3. Селективность 303
6.3.2. Зависимость селективности от типа растворителя
Изменения в %В могут и не привести к эффективному разрешению для данного
образца. Это проиллюстрировано на рис. 6.8 на примере разделения смеси замещенных
бензолов. В данном примере изменение содержания в подвижной фазе ACN с 46
до 34%, приводит к некоторому изменению относительного удерживания (например,
пиков 2—3, 8—9), но это не оказывает заметного влияния на разделение
перекрывающихся пиков 3 и 4. Следовательно, потребуется какое-то другое изменение
условий, влияющих на селективность — например, замена ACN в качестве растворителя
В на МеОН. При использовании 61% МеОН вместо 46% ACN (см. рис. 6.9а) сила
растворителя остается практически такой же (время разделения почти не меняется),
но некоторые изменения в относительном удерживании все же имеют место
благодаря изменению селективности, вызванному заменой типа растворителя. Хотя ранее
неразрешенные пики 3 и 4 теперь хорошо разделены, пики 1 и 2 (которые хорошо
разрешались тогда, когда в качестве растворителя В использовался ацетонитрил)
перекрываются. С такими результатами, как для этих двух подвижных фаз с
различными типами растворителя В, смесь двух подвижных фаз часто дает лучшую
эффективность разделения. Это оказалось подходящей схемой разделения для настоящего
образца. Для разделения, изображенного на рис. 6.96, была приготовлена смесь
подвижных фаз 1:1 (46% ACN в соответствии с условиями на рис. 6.8а и 61% МеОН
в соответствии с условиями на рис. 6.9а). Новая подвижная фаза (содержащая
23% ACN + 30% МеОН) обеспечивает разрешение до базовой линии {Rs = 1.8).
Хотя разделение на рис. 6.96 стало значительно лучше, дальнейшее изменение
пропорций подвижной фазы по сравнению с рис. 6.8(7 (46% ACN) и рис. 6.9я
(61% МеОН) может привести к улучшению разрешения. Пики от 1 до 4 на рис. 6.96
разрешаются хуже, чем следующие за ними пики от 5 до 10 (т.е. пики I—4 являются
(в)
46% ACN I з
1.1Sfc<l4.6 I + ,5
я-°° 'I JF /1 ^ / л>
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1—
0 2 4 6 8 10 12 14
Время (мин)
(б)
34% ACN | 2
25*514 |+ ,
Я. = 0.Э II I3 1
| 1 1 1 !—
0 20 40
Время (мин)
Рис. 6.8. Разделение смеси замешенных бензолов в зависимости от силы растворителя
(%В). Образец: /, л-крезол; 2, бензонитрил; 3, 2-хлоранилин,' 4, 2-этилани-
лин; 5, 3,4-дихлоранилин; б, 2-нитротолуол; 7, 3-нитротолуол; 8, толуол;
9, 3-нитро-о-ксилол; 10, 4-нитро-л(-ксилол. Условия: колонка 250 х 4,6мм
(5 мкм) С8; подвижная фаза: ацетонитрил —буфер рН 6,5; 35 "С; 1,0 мл/мин.
Хроматограммы смоделированы по данным из |26|. Обратите внимание
на то, что все соединения не ионизируются в данных условиях и поэтому
сам образец нейтрален
Глава 6. Обращенно фазовая хроматография нейтральных образцов
(а)
61% МеОН
0.8<*<;3.0
Я, = 0.6
4 6
Время (мин)
(б)
30% МеОН + 23% ACN
1 sfcs4
Я, = 1.8
Г
1 I I
2 4
жЛф^
AjC
Б 8 10
Время (мин)
_г_—г^—г
12 14
Рис. 6.9. Зависимость селективности от типа растворителя. Разделение смеси
замещенных бензолов с использованием в качестве подвижной фазы метанола
или смеси метанол-ацетонитрил. Образец и условия такие же как на рис. 6.8,
за исключением тех, которые обозначены на рисунке. Хроматограммы
смоделированы поданным из [26]
«критическими»), и поэтому мы ограничим ими наше обсуждение. На рис. 6.10л в
повторены предыдущие хроматограммы, на которых изображены пики от первого
до четвертого с рис. 6.8 и 6.9 для демонстрации изменений относительных
пропорций пиков при изменении содержания ацетонитрила и метанола. По мере того, как
метанол заменяют на ацетонитрил (см. рис. 6.10д—в), видно, что пики 2 и 3
(заштрихованные) смещаются к передней части хроматограммы. На рис. 6.106 (при
использовании смеси подвижных фаз рис. 6.10а, в в соотношении 1:1) критическая
(наименее разрешенная) пара пиков — 2/3, и мы можем улучшить ее разрешение
дальнейшим смещением пика 2 от пика 3 к пику 1. Это может быть достигнуто
увеличением доли МеОН в окончательном варианте подвижной фазы (относительно
подвижной фазы на рис. 6.106). Как видно на рис. 6. Юг, смесь 57% подвижной фазы
рис. бЛОв (61%МеОН) и 43% подвижной фазы рис. 6.10а (46% ACN) элюирует пик 2
посередине между пиками 1и 3. Разрешение при этом максимальное (Rs = 2.0 против
Rs = 1,8 на рис. 6.106). Достижение окончательного оптимального разделения в
данном случае предполагает простую интерполяцию между двумя предыдущими
экспериментами (рис. 6.106,*), как и на рис. 6.6 при оптимизации %В. Подобную
оптимизацию селективности удобнее проводить с помощью компьютерного
моделирования (глава 10).
Несмотря на то, что улучшения разрешения и можно добиться с помощью
замены растворителя В (селективность, зависящая от типа растворителя), новая
подвижная фаза должна иметь сходную элюирующую силу, чтобы сохранить сопоставимые
с прежними величины к и времени разделения. В эксперименте на рис. 6.9л
использовался более высокий процент метанола (61% МеОН против 46% ACN) потому, что
метанол более слабый В-растворитель (обеспечивающий большее удерживание), чем
ацетонитрил. При изменении типа растворителя мы можем оценить необходимое
изменение в %В для нового В-элюента с помощью номограммы для растворителей,
приведенной на рис. 6.11. Здесь величины %В разных В-растворителей,
обеспечивающие одинаковую элюирующую силу, отмечены вертикальными рисками.
Напомним, что в предыдущем примере мы должны были заменить 46% ACN водно-метано-
льной подвижной фазой со сходной элюирующей силой. Из диаграммы на рис. 6.11
6.3. Селективность 305
<з)
46% ACN
1.1 sk S4.6
R. = 0.0
А
(б)
в
Время (мин)
50/50 смесь (а) и (в)
(30% МеОН + 23% ACN)
1 SKS4
Rs = 1.8
(в)
61% МвОН
0.8 £ к £ 3.0
Я =0.6
6
Время (мин)
л3
(г)
Время (мин)
57/43 смесь (в) и (а)
1 <fc<4
Rs = 2.0
Время (мин)
Рис. 6.10. Зависимость селективности от типа растворителя: точный подбор состава
элюента В. Образец и условия те же. что и на рис. 6.8 и 6.9 (только пики 1 —
4). плюс рисунок (г); (ff) 30% МеОН+23% ACN, и (г) 35% МеОН+20% ACN.
Хроматограммы смоделированы поданным из |26|
0 10
\ \~
20 30
40 50
0 20
н-ь
-\ 1 г
I I
I I
I I
30 40 50 60
-I) 1 1—«-U
60 70 80 90 100% ACN/HjO
Н—I 1—I 1
70
I
80
-+-
О 10 i 20
30
-+-
40
-+-
90
100% МеОН/НгО
50 60
701
80
-+-
90
Рис. 6.11. Номограмма элюирующей силы растворителей для ОФХ |28]. В качестве
примера две подвижные фазы равной элюирующей силы (46% ACN и
57%МеОН) отмечены •
можно видеть, что 46% ACN должен быть примерно эквивалентен по элюирующей
силе 57%МеОН (обе эти подвижные фазы отмечены • на рис. 6.11). Это близко к
составу подвижной фазы, фактически подобранному в эксперименте, результаты
которого изображены на рис. 6.9а (61%МеОН). Заметим, что время разделения
306 Глава 6. Обращенно фазовая хроматография нейтральных образцов
на рис. 6.9а (11 мин) несколько меньше, чем на рис. 6.8а (15 мин), чего и следовало
ожидать, исходя изданных на рис. 6.11: 61% МеОН сильнее, чем 57% МеОН,
рекомендованный на рис. 6.11.
Подводя итоги, можно сказать, что при замене типа растворителя с целью
изменения селективности и для улучшения разрешения, нужно действовать следующим
образом: если при первоначальном разделении использовался ACN и его необходимо
заменить МеОН, то с помощью диаграммы на рис. 6.11 можно оценить оптимальный
процент метанола для второго разделения. Затем изучают хроматограммы, в которых
использовались ацетонитрил и метанол, чтобы определить, может ли смесь ацето-
нитрила и метанола обеспечить более эффективное разделение, чем любой из этих
растворителей (как на рис. 6.96). Условия могут быть дополнительно
оптимизированы (как это показано на рис. 6.10 и будет обсуждаться позднее в разделе 6.4.1).
Если метанол был взят изначально в качестве В растворителя, используйте рис. 6.11
для расчета эквивалентного %ACN, чтобы при замене растворителя В элюирующая
сила подвижной фазы не изменилась.
Как описано в разделах 2.3.2.1 и 5.4.1, зависимость селективности от типа
растворителя возникает в результате взаимодействия между растворенным веществом,
В-элюентом и сорбентом. Селективное удерживание В-элюента в стационарной фазе
означает, что более сильное взаимодействие В-элюента и растворенного вещества
обычно приводит к увеличению удерживания (это, на первый взгляд, может
показаться противоречивым с точки зрения обсуждения того, что изображено на рис. 2.7а,б).
Однако, даже качественные предсказания влияния данного В-элюента на относите
льное удерживание различных классов растворенных веществ (например фенолов,
эфиров) делать, в лучшем случае, сложно. Дальнейшее обсуждение по этому вопросу
см. в [231.
6.3.3. Зависимость селективности от температуры
Зависимость удерживания от температуры обычно описывается формулой
]ogk= А+В/ТК, (6.2)
где А и В константы для данного соединения в том случае, когда изменяется только
абсолютная температура Т% (К). Поэтому графики зависимости log к от 1/7^ должны
быть прямыми, как показанные на рис. 6.12а графики для ряда полициклических
ароматических углеводородов (ПАУ). Увеличение температуры колонки на 1 °С
обычно приводит к уменьшению величин к на 1—2% для каждого пика на хромато-
грамме |29|. В случае разделения нейтральных образцов с помощью ОФХ влияние
температуры на селективность обычно незначительно. Однако, были описаны
исключения, как в случаях с ПАУ 130] и растительными пигментами |31|. На рис. 6.126
приведены дополнительные данные по трем ПАУ (пунктирные кривые А—С).
Соединения 1—6 представляют собой пленарные конденсированные молекулы, такие
как антрацен (пик 1), тогда как соединения А—С представляют собой трехмерные
(не пленарные) полиарилы, такие как 1,Г-бинафтил (пик А). Видно, что для пяти
соседних пар пиков при изменении температуры в интервале от 15 до 45 СС
наблюдаются инверсии времен удерживания (обозначенные •) на рис. 6.12#. (Обратите
внимание на шкалу °С в верхней части рисунка): В/5 при 42 °С, А/3 при 36 °С, С/6
при 31 °С, В/4 при 16 °С, С/5 при 6 °С. На этом примере видно, как значительно
изменяется селективность по мере изменения температуры. В результате был
определен ряд температур в интервале между 10 и 70 °С, при которых возможно
разрешение до базовой линии (Д5 > 1.5), например, при температуре 22 °С (отмеченной
вертикальной пунктирной линией на рис. 6.126).
6.3. Селективность 307
45°С 35°С 25°С 15°С
3.2 3.4
1000/7*
45°С 350С 25°С 15°С
3.2 3.4
1000/7V
Рис. 6.12. Зависимость удерживания полициклических ароматических углеводородов
от температуры, при которой проводится разделение. Условия: колонка
250x4,6 мм (5 мкм) сорбент Chromegabond-C18; 80% ацетонитрил-вода;
1мл/мин. (а) Образец (конденсированные ароматические углеводороды):
/, антрацен; 2, флуорантен; J, трифенилен; 4, хризен; 5, 3,4-бензофлуоран-
тен; б, 1,2,5,6-дибензантрацен. (б) Образец такой же, как и на (а) с
добавлением полиарилов: Л, 1,]'-бинафтил; В, 1,3,5-трифенилбензол; С, 9,10-дифе-
нилантрацен. Данные из J30]
Изменения разрешения при изменении температуры для данного образца лучше
всего видны на карте разрешения, изображенной на рис. 6.13я (график зависимости
критического разрешения от температуры). Более доступный для понимания и
подробный вариант карты разрешения дан на рис. 6.136. Так на рис. 6.136 обозначены
потенциально критические пары пиков (например, Б/4, В/6, А/3), которые
соответствуют различным сегментам ломаной линии на рис. 6.13а. Также стрелки
308 Глава 6. Обращенно фазовая хроматография нейтральных образцов
во 70
(б)
2.0
я.
-S
\\ B/4J
сЦ /
•
к А/3
!--Х---Яв=1.5
\ в'5/
I А/зД у
с/б-ууу w
i ? V 11
л А'4--
\алМ
i V 1
*^
0/4
1
0.0
(в) Г=11'С1
(э) Г =22'С
Л = 2.1
(И) 7= 46 "С
Я, -1.7
(е) Г = 61*С
Я5-1.7
о г 4 б в ю 12
Время {мин)
Рис. 6.13. Зависимость разделения полициклических ароматических углеводородов от
температуры. Образец и условия см. рис. 6.12. (о,б) карта разрешения; (в—лс) хро-
матограммы разделений при оптимальных температурах. Данные из [301
6.3. Селективность 309
на рис. 6.136 указывают на четыре значения температуры, при которых возможно
разделение до базовой линии (Rs > 1,5). Разделения, проведенные при указанных
четырех предпочтительных температурах, показаны на рис. 6.13е-л*с (обратите
внимание на измененный порядок удерживания в каждом из этих примеров). Можно
видеть, что три полиарила А-В (заштрихованные пики) удерживаются сильнее при
повышении температуры по сравнению с конденсированными ПАУ 1-6 (см. далее
обсуждение в разделе 6.3.3.1). Наилучшее разделение (Rs = 2.1) наблюдается при
температуре 22 °С. Изменения температуры иногда могут быть эффективны, если нужно
изменить селективность разделения нейтральных образцов. Гораздо более вероятны
изменения селективности при изменении температуры разделения частично
ионизированных кислот и оснований (раздел 7.3.2.2). Дальнейшее обсуждение влияния
температуры на селективность в ОФХ см. [32, 32а|.
6.3.3.1. Последующий анализ
Нижеследующее обсуждение дает более полную картину зависмости селективности
от температуры при разделении нейтральных образцов. Однако, читатель может, если
хочет, пропустить это обсуждение и продолжить с раздела 6.3.4.
Хотя уравнение (6.2) в целом удовлетворительно описывает зависимость
удерживания от температуры в ОФХ, иногда наблюдаются более сложные изменения к
при изменении температуры |7, 33—35|. Иногда к увеличивается с ростом
температуры, или графики зависимости log к от 1/Гмогут отклоняться от линейности. Эти
отклонения от уравнения (6.2) часто связаны с кислотной или основной природой об
разцов, а также с подвижными фазами, рН которых близок к рКа этих образцов
(см. раздел 7.2). В таких случаях изменение температуры может привести к
изменению относительной ионизации кислоты или основания (раздел 7.2.3) и к
существенному изменению удерживания растворенного вещества в дополнение к нормальному
воздействию температуры, описываемому уравнением (6.2), а иногда и
противодействуя ему.
Зависимость log к от температуры определяется величиной константы В в
уравнении (6.2), которая пропорциональна энтальпии удерживания АИ. Значения В
обычно растут при больших значениях к. В результате графики зависимости log к
от 1/Толя различных растворенных веществ (при одинаковых других условиях) часто
напоминают веерообразные графики на рис. 6.12а для нескольких
конденсированных ПАУ. Такое поведение можно сравнить с аналогичным примером на рис. 6.7я
для правильных образцов при изменении %В. В обоих случаях (изменение %В
или 7) изменение условий оказывает наибольшее влияние на к наиболее
удерживаемого образца (с наибольшими значениями к). Для большинства нейтральных
образцов изменения порядка удерживания при изменении температуры не ожидаются (как
показано на рис. 6.12а), но максимум разрешения тем не менее может наблюдаться
при промежуточной температуре. Однако изменение температуры, как правило,
менее результативно для улучшения разрешения нейтральных образцов (как и
изменение селективности за счет силы растворителя в случае «правильных» образцов).
Как показано на рис. 6.126", величины В имеют тенденцию к уменьшению
для более компактных молекул, таких как фенилнафталин (полиарил) по сравнению
с менее компактными, такими как антрацен (конденсированный ПАУ).
Аналогичные эффекты зависимости селективности от температуры наблюдались в газовой
хроматографии |36|. В этом случае величины В в уравнении (6.2) уменьшаются
в ряду и-алкины > w-алканы > разветвленные алканы > циклоалканы. То есть,
значения В в ГХ тоже уменьшаются для более короткой и более компактной молекулы.
Возможно, по аналогичным причинам.
310 Глава 6. Обращение фазовая хроматография нейтральных образцов
6.3.4. Селективность колонки
На ранних этапах развития ВЭЖХ сменой колонки часто пользовались как средством,
изменяющим селективность и улучшающим разрешение. Действительно,
селективность колонки — это мощный инструмент для изменения относительного
удерживания и улучшения разделения нейтральных образцов. Однако, использование одной
селективности колонки с целью систематического улучшения разделения имеет
серьезные ограничения по сравнению с изменениями %В, типа растворителя или
температуры. При изменении любого из последних параметров возможны непрерывные
изменения в селективности, и в ходе этих изменений часто можно найти промежуточные
условия, которые соответствуют максимальному разрешению. Как видно из примеров,
приведенных на рис. 6.13, это значительно увеличило вероятность приемлемого
разделения по сравнению с экспериментами, когда используются только одно или два
произвольно выбранных значения температуры. При смене колонки подбор
промежуточных условий разделения (между условиями, обеспечиваемыми каждой из колонок)
неудобен. В результате, смена только колонки с меньшей вероятностью приведет
к улучшению разделения (тем не менее, см. обсуждение в разделе 6.4.1.4). Кроме того,
смена колонки менее удобный вариант, чем изменения непрерывных переменных.
Примеры изменения селективности в тех случаях, когда меняют только колонку,
для нейтрального десятикомпонентного образца и четырех колонок с различной
селективностью показаны на рис. 6.14. Несмотря на значительные изменения в
селективности для этого образца при смене колонки, ни одна из колонок не дает
разрешения Rs > 0,8, если другие условия остаются неизменными (как в этом примере). Если
две разных колонки соединить вместе, то возникнут дополнительные возможности
для разделения, но этот способ неудобен и немногим лучше, чем использование
одной колонки (в примере на рис. 6.14 ни одна из комбинаций четырех колонок
не улучшила разделение, показанное на рис. 6.14е). Реальная польза от замены
колонки появляется тогда, когда замена последней сопровождается одновременным
изменением одного или нескольких условий разделения, например, %В, типа
растворителя и/или температуры для нейтральных образцов (раздел 6.4.1.4).
В настоящее время мы более полно понимаем основы селективности колонки
(сорбента, которым она упакована), чем других видов селективности (обусловленных
силой растворителя, типом растворителя, температурой и т.д.). Как обсуждалось
в разделе 5.4.1, селективность сорбента может быть количественно определена пятью
различными параметрами:
• гидрофобностью Н;
• стерическими препятствиями 5*;
• кислотностью водородных связей А;
• основностью водородных связей В;
• катионообменной емкостью С.
Для случая нейтральных образцов важны только первые четыре параметра
сорбента (Я, S', А, В). Разница в селективности двух сорбентов при разделении
нейтрального образца может быть описана функцией селективности F/-C), определяемой
уравнением (6.3) (см. далее раздел 6.3.1 и также раздел 5.4.3). Для колонок, на
которых проводились разделения рис. 6.14, величины F/-C) в сравнении со значением
для колонки Symmetry, составляют: 26 для колонки Alltima, 12 для колонки Luna и
16 для колонки Spherisorb. Если, как показано в этом примере, величина F/-C) > 5,
это может привести к существенным изменениям в относительном удерживании,
большие изменения относительного удерживания в результате смены колонки
наблюдаются для ионизированных образцов, поскольку для них параметр С гораздо
сильнее влияет на селективность.
6.3. Селективность 3 I I
(a) 1
I '
3
fl
4
5
+
7
л
1 1 1
Symmetry О 8
1.6£4c<19, Rs = 0.4
8 О
л 9Л jo
8 10
Время (мин)
12
14
16
Alltima HP C18 amide
1 <k<7, Rs = 0.A
9
Л7 Л'/ч"
Время (мин)
Luna phenyl-hexyl
1 <k< 11.Я„ = 0.8
1
10
4 6
Время (мин)
(г)
J\L
Spherisorb ODS-2
1 <k< 14. /?s = 0.0
8+9
К
10
~~Г~
10
12
Time (min)
Рис. 6.14. Разделение смеси десяти органических соединений различной структуры
на четырех различных колонках. Образец: /, 4-нитрофенол; 2, 5,5-днфенил-
гидантоин; 3, ацетофенон; 4, бензонитрил; 5, 5-фенилпентанол; 6, анизол:
7, толуол; 8, цис-халкон; 9, этилбензол; /ft, транс-халкон. Условия: колонка
150 х 4,6мм (5 мкм); 45% ацетонигрил-вода; 35 "С; 2,0 мл/мин. Хромато-
граммы смоделированы по данным работ [8, 9|
6.3.5. Разделения изомеров
Соединения со сходной структурой имеют близкие значения к, и их часто труднее
разделить. ОФХ-разделения изомеров по этой причине обычно считаются
сложными. Хотя это может быть до некоторой степени верным и в общем случае, 90%
из 137 различных пар изомеров удалось разделить с Я, > 1,0 простой оптимиза-
Глава 6. Обращенно фазовая хроматография нейтральных образцов
(а) 2а
Л »
40-С \?
J V1P
3.1 3.3 3.5
Время (мин)
(б)
28 *С
3.1 3.3
23д2а Л11Р
,/чл
3.5 3.7 З.Э
Время (мин)
Рис. 6.J5. Разделение трех изомеров гидрокситестостерона (2а, 2р\ lip) в зависимости
от температуры. Условия: колонка C|g 250 х 4,6 мм (5мкм); 40% ацетонит-
рил-вода; 2 мл/мин. Данные из |38|
цией %В (Л при этом изменялся четырехкратно) и температуры (Г изменялась на
25 °С) на колонках, упакованных алкилированным силикагелем, по данным обзора
|37|. Разделение трех изомеров гидрокситестостерона на колонке CiS в зависимости
от температуры показано на рис. 6.15. При 40 °С все три изомера элюируются вмес
те; при 28 °С изомеры разделяются с Rs = 0,8. Более низкие температуры обычно
способствуют лучшему разделению изомеров.
6.3.5.1. Повышенная селективность при разделении изомеров
Несмотря на то, что для некоторых изомеров разрешения до базовой линии можно
достичь с помощью ОФХ на колонках с алкилированным силикагелем,
использование циклодекстриновой неподвижной фазы может быть выигрышнее [39—411,
как показано на рис. 6.16. Повышенная селективность, которую обеспечивают цик-
лодекстриновые колонки, может быть обусловлена наличием ОН-групп в молекуле
циклодекстрина (способствующих образованию водородных связей между
растворенным веществом и сорбентом), большая, чем у гибких алкильных лигандов силикаге-
ля, жесткость трехмерной молекулы циклодекстрина может обеспечить более полное
стерическое соответствие одному изомеру по сравнению с другим. Однако, еще
более высокая селективность при разделении изомеров, как правило, возможна при
нормально-фазовой хроматографии на колонках с силикагелем (раздел 8.3.5).
СН-СООН
СНЭ
соЧ >-сн-соон
СНЭ
Q-~-Q
O-CO-Q-CH-COOH
ск
, J.
(1)
(2)
n
сн, О)
I
10
L.
-1
15 (мин)
Рис. 6.16. Разделение изомеров на колонке с циклодекстриновой неподвижной фазой.
Условия: колонка Cyclobond I 250 х 4,6 мм (5 мкм); 30% ацетонитрил —
буфер рН 4,5; 35 "С; 2,0 мл/мин. Данные из |40]
6.3. Селективность 3 13
Было установлено, что образование комплексов олефинов с ионами серебра
способствует улучшению ОФХ-разделения цис- и транс-олефинов. Добавление Ag+
к подвижной фазе приводит к его преимущественному взаимодействию с цис олефи
нами. Полученный комплекс Ag+ с олефинами более полярен и, следовательно,
предпочитает подвижную фазу (раздел 2.3.2), что приводит к уменьшению
удерживания по сравнению с транс-изомером. Соединения с различным содержанием цис-
изомеров могут быть отделены в зависимости от числа цис-двойных связей в
молекуле. Комплексообразование с ионами серебра также используется для разделения
ненасыщенных жирных кислот или их производных |42|. Кроме того, с его помощью
можно изменять селективность при разделении гетероциклических соединений азота
или серы |43|. В настоящее время комплексообразование с ионами серебра для
улучшения разделения цис- и транс-олефинов в большинстве случаев осуществляется
с помощью ионообменных колонок, насыщенных серебром |42|.
6.3.5.2. Зависимость селективности от формы молекулы
Эта специальная форма селективности обсуждалась в разделе 5.4.1.1. Несмотря на то,
что ей уделено значительное внимание в литературе, практическое применение ее
ограничено. Поэтому читатель может пропустить этот раздел и перейти к разделу 6.3.6.
Зависимость селективности от формы молекулы играет роль преимущественно
в удерживании планарных полициклических ароматических углеводородов (ПАУ) и
некоторых других соединений (каротинов, стероидов) на полимерных алкилсиликаге-
льных сорбентах [44—46|. На колонках, упакованными такими сорбентами, часто
можно наблюдать прекрасные разделения растворов изомеров. Два свойства
растворенного вещества способствуют увеличению удерживания изомеров ПАУ на
полимерном сорбенте: планарность молекулы (или уменьшение ее толщины) и большая
величина отношения ее молекулярной длины к ширине (L/B). Предполагается, что
привитая полимерная фаза доступна только в узких отверстиях или «щелях»
сорбента, в которые плоские и/или более узкие молекулы имеют больше шансов войти и
потому лучше удерживаются.
Эта «щелевая» модель удерживания на полимерных сорбентах
проиллюстрирована на рис. 6.17, где показано, как непланарная молекула ПАУ входит в узкую
«щель», образованную неподвижной полимерной фазой. Из-за большей, чем «щель»,
«толщины» этой непланарной молекулы она не сможет в нее войти полностью.
Для более плоской и менее «толстой» молекулы ПАУ проникновение в неподвижную
фазу легче, и удерживание, следовательно, будет больше. Установлено, что изомеры
ПАУ намного лучше разрешаются на сорбентах с привитой полимерной фазой
(которые имеют более высокую степень прививки алкильными лигандами), чем на
колонках, упакованных сорбентами с привитой мономерной фазой |45|. Это было
проиллюстрировано в главе 5 на примере разделения смеси двенадцати С^2 ПАУ-изомеров
lllll, i II I III
С18 привитаяфаза____
Рис. 6.17. «Щелевая» модель зависимости селективности от формы молекулы
314 Глава 6. Обращенно фазовая хроматография нейтральных образцов
на сорбенте с привитой полимерной фазой (рис. 5.23в) в сравнении с разделением
на сорбенте с привитой мономерной фазой (рис. 5.23г). Последние, однако, в других
отношениях обычно лучше, чем первые (раздел 5.2.3) и более широко используются.
Так как всего несколько классов соединений разделяются лучше на сорбентах с
привитой полимерной фазой, зависимость селективности от формы молекулы редко
используется при разработке метода в качестве средства улучшения разделения.
6.3.6. Другие соображения, касающиеся селективности
До сих пор наше обсуждение селективности разделения было сосредоточено на
стадии разработки метода — последовательное изменение условий для достижения
максимального разрешения и/или сокращения времени разделения. С одной стороны,
таким образом достигается требуемое разрешение, а с другой — у такого метода есть
и отрицательные стороны. После того, как разработан рутинный метод, он, скорее
всего, будет использоваться в течение длительного времени и в разных лабораториях.
Сорбенты, которыми упакованы колонки, в процессе рутинных анализов
деградируют, и потому их необходимо время от времени заменять. Как описано в
разделе 5.4.2, селективность сорбентов может незначительно отличаться от одной партии
колонок к другой. В этом случае замена на колонку того же типа того же
производителя может больше и не обеспечивать приемлемого разделения. Такая ситуация
более вероятна для разделений, в которых более десяти пиков, особенно тогда, когда
требуется предельное разрешение (Rs > 2). Когда новая колонка демонстрирует
другую селективность и не может обеспечить необходимое разрешение, необходимо
найти другую колонку, эквивалентную исходной, или изменить условия разделения
таким образом, чтобы минимизировать эффект изменения селективности
(раздел 6.3.6.1). Эквивалентная колонка может потребоваться и в том случае, если
исходную колонку перестали производить и ее невозможно купить, или она недоступна
там, где должно производиться разделение.
Другой проблемой, не связанной с воспроизводимостью характеристик колонок,
является вероятность того, что пик одного из компонентов образца может быть
перекрыт другим пиком и, таким образом, быть потерянным при анализе в режиме ОФХ.
Обнаружить такие скрытые пики может помочь использование ортогонального
разделения (раздел 6.3.6.2).
6.3.6.1. Эквивалентное разделение
Когда селективность колонки изменилась, можно использовать два способа
воспроизведения исходного разделения: (I) взять колонку из другого источника (например, из
другой партии) или (2) изменить условия разделения («настройка метода»). Какой из этих
способов применим, зависит от рекомендаций регуляторного органа (раздел 12.8) в том
случае, если ВЭЖХ-метод подпадает под государственное регулирование.
Чтобы изменить поставщика колонки, требуется охарактеризовать ее
альтернативный «эквивалент». Большинство колонок для ОФХ можно охарактеризовать
пятью параметрами, определяющими селективность (Н, S*, А, В и С), которые
обсуждались выше в разделе 5.4.1. Эквивалентные колонки будут иметь сходные значения
каждого из этих параметров с соответствующими значениями параметров исходной
колонки. Для двух колонок 1 и 2 и любого образца (ионизированного или
нейтрального) функция сравнения Fs определяется уравнением (5.4). Для нейтральных
образцов параметр С не нужен, потому уравнение (5.4) упрощается до [47]
только для нейтральных образцов:
FA-Q = {| 12,5{Я2 - Н,)|2 + | lOOtf? - S,*)|2 + \ЩЛг - Л,)|2 + [ 143(52 - S,)|2}4 <6.3)
6.3. Селективность 3 I 5
где индексы I и 2 относятся к значениям определенного параметра (Н, Уи т.д.)
для каждой из колонок. Для двух строго эквивалентных колонок Fs и Fs(-C) - 0.
Когда для двух колонок Fs или Fs(-C) < 3, весьма вероятно, что они обеспечат
эквивалентное разделение (т.е. критические величины Rs, которые будут отличаться
на < 0,5 единицы).
Пример использования уравнения (6.3) для градиентного метода и
ионизированного образца показан на рис. 6.18. Для ОФХ была разработана оригинальная
методика, в соответствии с которой провели отделение (показанное на рис. 6.18а)
лекарственного вещества от десяти примесей (всего 11 пиков, каждый из которых
маркирован *). На двух колонках, потенциально подходящих для замены
оригинальной колонки с Fs < 3 (по сравнению с колонкой рис. 6.18а), были проведены
разделения, показанные на рис. 6.186 и в. Очевидно, что эти три разделения (а—в) по
существу эквивалентны, и разрешение для каждого вещества имеет сходное значение.
Четвертая колонка (с Fs = 10) дала результат, показанный на рис. 6.18г; видно,
что два последних пика на этой хроматограмме перекрываются (см. по стрелке).
Следовательно, эта последняя колонка не эквивалентна исходной колонке, на
которой была получена хроматограмма рис. 6.18а, что и следовало ожидать, исходя
из большей величины ее F..
Luna C18(2)
(оригинальная колонка)
30 ^ 40
Prodigy ODS (3), F = 1
lAJvA*^^
-.—-т-
30 40 (мин)
Рис. 6.18. Пример использования величин Fs для выбора колонок со сходной
селективностью на замену в рутинном ВЭЖХ-анализе. Градиентные разделения,
в которых меняется только колонка а—г. Звездочками отмечены нужные
пики; величины Fs вычислены по уравнению (5.4) (ионизированный
[не нейтральный] образец) [481
Глава 6. Обращенно фазовая хроматография нейтральных образцов
Настройка метода для корректировки разницы в селективности колонок
используется в тех случаях, когда поиск эквивалентной колонки оказывается
нецелесообразным или невозможным. Вместо замены колонки изменяют условия разделения
для преодоления различий в селективности колонок. Пример такого подхода
показан на рис 6.19. Исходное разделение на колонке А с разрешением до базовой линии
(Rs - 1,7) показано на рис. 6.19а. Позже использовали новую колонку В (такую же,
но из другой партии) и получили разделение, показанное на рис. 6.196. Наблюдаемое
(а)
Колонка А
50% В, 35Х 1
(б)
Время(мин)
Колонка В
50% В, 35'С
Я = 1,2
I—
О
(в)
I
jLjjL
Время (мин)
Колонка В
50% В, 45">С
R„ = 0,5
I—
О
(в)
Колонка В
55% В, 35Х
Я* =2,1
1
■л 1 г-
2 4
Время (мин)
1
jt^X.
Время (мин)
Рис. 6.19. Пример корректировки метода для семикомпонентной смеси нейтральных
соединений. Образец: I, оксазепам; 2, флунитразепам; 3, нитробензол;
4, 4-нитротолуол; 5, бензофенон; б, цис-4-нитрохалкон; 7, нафталин.
Условия: колонка С]8 150 х 4,6 им (В отличается от А только меньшей на 10%
прививкой лигандрв); 2,0 мл/мин; подвижная фаза ацетонитрил-вода;
остальные условия указаны на рисунке. Разделения а-г смоделированы по
данным работ |8, 9]
6.3. Селективность 3 17 ))|
в этом случае разрешение неприемлемо из-за возросшего перекрывания пар
пиков 2/3 (Rs = 1,3) и 6/7 (Rs = 1,2), обусловленного изменением селективности
колонки. В первую очередь при проведении корректировки метода необходимо
определить, как разделение изменяется при изменении одного или нескольких условий
эксперимента. В случае нейтральных образцов возможные изменения условий
включают изменение %В и температуры (предпочтительно) или изменение соотношений
двух органических растворителей, составляющих элюент В (например, смесь ACN и
МеОН).
Результаты изменения температуры или %В для колонки В показаны
на рис. 6.19в,г. Мы видим, что при увеличении температуры (рис. 6.19в) уменьшается
разрешение пары пиков 2 и 3 (Rs уменьшается на 0,8 единиц). Однако это же
увеличение температуры оказывает противоположное действие на пару пиков 6 и 7 (Rs
увеличивается на 0,2 единицы). Следовательно, понижение температуры могло бы
восстановить разрешение до базовой линии пары пиков 2 и 3, но сделает еще хуже
разрешение пиков 6 и 7, которые и без того уже плохо разрешены. С другой
стороны, увеличение %В приводит к улучшению разрешения для каждой пары пиков
и приемлемому разрешению (Rs = 2,1).
Подход к разделению этого образца, проиллюстрированный на рис. 6.19,
достаточно прост, поскольку изменение только одного условия (%В) позволило
скорректировать селективность колонки В.
Зачастую все не так просто, и тогда может понадобиться одновременное
изменение двух (или более) параметров, чтобы добиться нужной селективности колонки.
Хотя выбор этих двух или более параметров может быть сделан методом проб и
ошибок, более эффективный подход был описан в работе |50|. Получив
необходимые данные по удерживанию в четырех эспериментальных разделениях, как
на рис. 6.19а—г, можно с помощью простых математических вычислений
предсказать условия для достижения максимального соответствия разделения на сменной
колонке (см. рис. 6.19й) исходному (см. рис. 6. 19й). О допустимых изменениях в
условиях разделений в процессе корректировки метода см. раздел 12.8.
Проблему восстановления эквивалентного исходному разделения (в том случае,
когда селективность колонок разных производственных партий отличается) лучше
всего не устранять, а предотвращать при разработке метода. Существуют три
варианта таких превентивных мер:
1) проверить используемые в окончательном варианте метода колонки из разных
партий на воспроизводимость;
2) удостовериться в идентичности одной или нескольких эквивалентных колонок
для замены;
3) выполнить корректировку метода для одной или более неэквивалентных колонок.
Вариант 1 нужно рассмотреть в качестве первого шага. Несколько разных партий
выбранной колонки можно оценить с точки зрения эквивалентности разделения.
Обычно выясняется, что колонки из всех проверенных партий обеспечивают
удовлетворительное разделение. Если это не так, то может потребоваться замена
исходной колонки на другую, более воспроизводимую.
Вариант 2 является полезным дополнением к первому варианту даже в том
случае, если проверка по варианту I подтверждает, что колонки из разных партий
эквивалентны (партии, выпушенные позднее, могут и не быть эквивалентными!).
Идентификация одной или нескольких эквивалентных колонок (см., например,
рис. 6.185,е) при разработке метода может обеспечить замену в том случае, если у
последующих партий исходной колонки будет отличная от исходной селективность и
они будут непригодны |48|.
Вариант 3 может использоваться всякий раз, когда вариант 2 невозможно
выполнить (ни одна альтернативная колонка не эквивалентна исходной). Если требуемые
318 Глава 6. Обращенно фазовая хроматография нейтральных образцов
изменения условий незначительны, можно использовать альтернативную колонку
в сочетании с корректировкой метода с получением равнозначного, не требующего
полной ревалидации метода (раздел 12.8). Другой вариант корректировки метода,
позволяющий не менять условий для подменной колонки, заключается в выборе таких
условий при его разработке, которые обеспечивают эквивалентное разделение как
для исходной, так и для одной или нескольких неэквивалентных колонок |51|.
6.3.6.2. Ортогональное разделение
Проблема потерянных или «скрытых» пиков может возникнуть при разработке
метода в том случае, когда два соединения остаются неразделенными несмотря на
изменение условий разделения. Если это упускается из виду, окончательный вариант
метода будет непригодным для использования из-за потерянных пиков. Даже если все
представляющие интерес соединения разделились в процессе разработки метода,
последующие образцы могут содержать дополнительные (неожидаемые) соединения,
которые могут быть пропущены, поскольку перекрываются другими пиками. Наибо
лее вероятно появление пропущенных пиков тогда, когда перекрываемый пик
(например, примеси) мал по сравнению с другими пиками в образце или в тех случаях,
когда изначально неизвестно возможное количество соединений в образце.
Проблема скрытых пиков может быть решена с помощью селективного
детектирования (в основном, масс-спектрометрии) и/или разработкой метода
ортогонального разделения. Последнее есть разделение с очень отличающейся селективностью,
с помощью которого можно разделить два пика, перекрывающихся в исходном
(основном) методе. Для разработки этого метода, в котором часто используется
масс-спектрометрическое детектирование (ЖХ-МС), было предложено несколько
вариантов ортогональных условий, предполагающих замену колонки, изменения
В-элюентов и/или рН подвижной фазы [52—53|. Два или более таких метода могут
использоваться для работы с данным образцом, чтобы свести к минимуму
вероятность перекрывания пиков двух растворенных веществ, пропущенных при
разработке методики. Специфичность ЖХ-МС в сочетании с очень разной селективностью
разделений делает маловероятным пропуск какого-либо компонента образца.
Когда разработка метода для данного (будем надеяться, репрезентативного)
образца завершена, всегда существует вероятность того, что в будущих образцах могут
содержаться соединения, не присутствовашие в исходном образце. Эти
дополнительные соединения могут появляться из-за изменений в производственном процессе,
загрязнения образца во время обработки или по другим причинам. Рутинные
методики ВЭЖХ-анализа обычно включают скорее УФ, чем МС-детектирование.
Следовательно, для лабораторий, где занимаются рутинным анализом, должна быть
доступна методика ортогонального разделения, основанная на УФ-детектировании.
Кроме того, требуется, чтобы ортогональный метод позволял полностью разделить
известные компоненты образца.
Общая методика разработки ортогонального разделения, соответствующая
вышеприведенным условиям для рутинного использования, была описана и опробована
в нескольких лабораториях |49|. Исходя из основного метода, используемого
для рутинных приложений, для ортогонального разделения выбирают колонку
с сильно отличающейся селективностью, ориентируясь на большую величину Fs(-C)
(уравнение 6.3) для ортогональных колонок по сравнению с оригинальными. Кроме
замены колонки меняется и элюент В — если для основного метода использовался
ацетонитрил, то для ортогонального используется метанол и наоборот. Поскольку на
данном этапе все компоненты образца, разрешаемые в рутинном разделении, могут
полностью не разделяться, теперь оптимизируются температура и %В, чтобы достичь
максимального разрешения (раздел 6.4.1.2). В конечном итоге, ортогональность раз-
6.4. Разработка метода и стратегия оптимизации селективности
319
Оригинальный (первичный) метод
L~ .« г
1 1 1 1 j i 1 1 1 1 i i 1 1 1 1 1 ! 1 1 1 1 T 1 1 Г
0 10 20 30 40 (мин)
(б)
3
. 2 "Ортогональный"
I метод
II II J»
1 ' ' ' I i | i ■ i i | i ■ i i | i i i i | i i ■ i | .
0 10 20 30 (мин)
Рис. 6.20. Сравнение оригинального и «ортогонального» разделений. Градиентные
разделения, в которых различаются колонки и органические растворители
(подвижная фаза имеет рН = 6,5 для а и 6). Звездочками отмечены
градиентные артефакты (пики не относящиеся к растворенным веществам).
Воспроизведено с разрешения [49]
работанното метода оценивается по зависимости log к для ортогонального метода
от log к для рутинного метода. Если и на этой стадии не была достигнута достаточная
ортогональность, можно и дальше пробовать изменять условия разделения
(использование другой колонки, изменение рН и т.д.). При работе с ионизированным
образцом (глава 7) следует подбирать большую величину Fs (уравнение 5.4), а не Fs(-C).
Окончательную общую методику, которая может быть распространена на
градиентное элюирование нейтральных и ионизированных образцов, можно дополнительно
улучшить (47, 54| для большей ортогональности двух разделений.
Пример вышеописанного ортогонального метода показан на рис. 6.20. Он был
разработан для исходного метода, основанного на градиентном элюировании.
На рис. 6.20я показано разделение образца рутинным методом. Основной компонент
3 и четыре примеси 1,2,4 и 5 разделяются до базовой линии. Когда рутинный метод
был только разработан, было установлено, что только эти пять компонентов
присутствуют в образцах, приготовленных в то время. Позднее, когда был разработан
и применен для новой партии образцов ортогональный метод, показанный на
рис. 6.205, была обнаружена примесь 6, пик которой при разделении рутинным
методом перекрывался основным компонентом 3 (и поэтому был упущен при
разделении на рис. 6.20а).
6.4. Разработка метода и стратегия
оптимизации селективности
Общий подход к разработке метода разделения описан в разделе 2.5.4 и подытожен
на рис. 6.21. Семь шагов разработки метода показаны на рис. 6.21д. Первый шаг
включает анализ информации об образце и предварительный выбор целей
окончательного разделения (требуемое разрешение, например, Rs > 2; желаемое время
разделения, например, < 10 мин и т.д.). Для некоторых образцов (больших биологических
или полимерных молекул, глава 13; энантиомеров, глава 14; неорганических ионов,
не обсуждаемых в данной книге) может быть показан специализированный подход.
\У 4
Глава 6. Обращенно фазовая хроматография нейтральных образцов
(в)
Разработка метода
(б)
Выбор условий разделения
1, Анализ состава образца и целей
разделения (Раздел 2.5.4.1)
"'
2. Профподготовка
(Глава 16)
■
'
3. Выбор вцда хроматографии
{обычно ОФХ)
1
'
4. Выбор детектора
(Глава 4)
•
'
5. Выбор условий разделения
(Рис. 6.216)
■
'
6. Прогнозирование, выявление
и решение возможных проблем
(Раздел 2.5.4.6)
'
'
7, Валидация метода и и проверка
пригодности хроматографической системы
(Глава12)
. Первоначальное
разделение
2. Трудности разделения?
(Разделы 2.4.2,9.3.1.1)
\
1а. Изменение %В до
получения 1 s k £ 10
(Раздел 6.2.1)
или
16. Проведение
градиентного разделения
(Раздел 9.3.1)
3. Пересмотр начальных
условий:
ОФХ (Глава 6, или Раздел 7.3)
ИПХ (Раздел 7.4)
ИОХ (Раздел 7.5)
НФХ (Глава В)
4. Соотнесение пиков с
кислотными, основными
и нейтральными
компонентами
(Раздел 7.2)
6. Оптимизация условии
колонки (N, Раздел 2.5.3)
S. Оптимизация
селективности (а)
Рис. 6.21. Общий подход к разработке метода разделения для использования в этой и
последующих главах
Он же показан в тех случаях, когда предполагается препаративное разделение
{глава 15). Второй шаг (предварительная обработка) требуется для образцов, которые
могут при введении испортить колонку или содержат мешающие вещества, пики
которых способны перекрывать интересующие вас пики. Третий шаг предполагает
первоначальный выбор вида хроматографии. В большинсте случаев выбирается
ОФХ, но решение может быть скорректировано после первых экспериментов, в
которых изменяют условия разделения (шаг 3 рис. 6.216). Выбор детектора (обычно это
УФ и/или МС детекторы) делается на четвертом шаге. Выбор условий разделения
(шаг 5) обычно считается основной частью разработки метода разделения. Он
детализирован на рис. 6.216 и обсуждается ниже. На шестом этапе рассматриваются
некоторые общие проблемы, которые могут возникнуть после того, как метод уже
разработан и используется для рутинных анализов. Седьмой шаг, представляющий
собой еще одну важную часть разработки, заключается в валидации метода и теста
системы на пригодность.
Подбор хроматографических условий дополнительно рассмотрен на рис. 6.216.
С учетом того, что известно об образце, выбираются исходные условия для
разделения (например, как в таблице 6.1), и проводится первоначальное разделение (шаг I
на рис. 6.216). Для первоначального изократического разделения %В меняют до
получения приемлемого удерживания 1 <к < 10 (шаг 1а). Как вариант может быть
проведено одно градиентное разделение (шаг 16; см. раздел 9.3.1). Первоначальное
разделение^) может выявить различные сложности (шаг 2): размывание заднего фронта
пиков, недостаточное удерживание образца (даже при использовании водной
6.4. Разработка метода и стратегия оптимизации селективности 321
подвижной фазы), чрезмерное его удерживание (даже в случае 100 %В) или слишком
большое количество пиков («сложный» образец). Какая бы проблема ни возникала
при первоначальном разделении(их), ее следует решить прежде, чем продолжать
работу (раздел 6.6). В некоторых случаях может потребоваться изменение условий
разделения или вида хроматографии (шаг 3). В этой ситуации вернитесь к шагу 1 и
начните заново.
Перед началом экспериментов по оптимизации селективности (шаг 5)
необходимо удостовериться, содержатся ли в образце кислотные, оснбвные и/или
нейтральные компоненты (шаг 4). Шаг 4 можно пропустить, если состав образца известен
до начала разработки метода. Для других образцов индивидуальные пики соединений
могут быть идентифицированы как кислоты, основания или нейтральные соедине
ния посредством изменения рН на 0,5 единиц (см. раздел 7.2 и примеры на рис. 7.2
и 7.3)'. Первостепенной важности выбор условий для оптимизации селективности
(шаг 5) будет разным для разных образцов и видов хроматографии. Эта тема
обсуждается ниже и в отдельных главах, посвященных рассмотрению образцов
определенного типа и/или видов хроматографии (главы 6—9, 13 и 14). Наконец, когда оптими
зация селективности завершена, условия разделения могут быть изменены (шаг 6)
либо с целью улучшения разрешения, либо с целью сокращения времени разделения.
Для образцов, которые легко разделяются, может потребоваться только выбор
значения %В, для чего достаточно нескольких экспериментов с изменением %В.
Если градиентное элюирование используется во время разработки первоначального
метода разделения, досточно провести лишь один эксперимент, чтобы выбрать %В
для 1 <к < 10 (раздел 9.3.1). Разделение многих образцов потребует дальнейшего
увеличения селективности (см. предыдущий раздел 6.3). Для некоторых образцов это
может означать, что придется одновременно изменять два или более параметров
разделения. Ниже будут описаны различные методики такой оптимизации нескольких
переменных.
6.4.1. Оптимизация нескольких параметров
Оптимизация нескольких параметров в каждом случае делается по схеме
эксперимента. На рис. 6.22 показан план экспериментов, требующихся для некоторых
комбинаций условий, которые влияют на селективность при разделении нейтральных
образцов. В каждом случае предполагается, что %В был изменен с целью определить
допустимый интервал %В (например 40—50 %В), в котором можно получить
приемлемое удерживание образца — такое, при котором 0,5<£<20для каждого пика. При
изменении %В с целью изменить селективность рекомендуется использовать более
широкий, чем обычно (1 <к < 10) диапазон к. В качестве иллюстрации сначала
рассмотрим рис. 6.22а. Эксперименты 1 и 2 выполняются первыми (например,
изменение только %В). Эти два эксперимента позволяют предположить, что успешное
разделение может произойти при некотором значении %В, для которого и 0,5<&<20,
и разрешение будет приемлемым. Если приемлемое разделение таким образом
получить невозможно, то затем проводятся эксперименты 3 и 4: повторяются
эксперименты 1 и 2 при более высокой температуре Т. Данные этих четырех экспериментов
можно интерполировать (или экстраполировать) для оценки величин Т и %В,
обеспечивающих оптимальную селективность и максимальное разрешение. После этого
проводится окончательный эксперимент в этих условиях для подтверждения
предсказанных условий разделения.
1 Времена удерживания нейтральных соединений не изменятся, а времена удерживания
кислотных и основных соединений уменьшатся или увеличатся. — Прим. перев.
322 Глава 6. Обращенно фазовая хроматография нейтральных образцов
(а)
низкий %В.
высокая 7
высокий %В,
высокая 7
(б)
низкий % ACN высокий % ACN
низкий %В,
низкая Т
высокий %В,
низкая Т
(в)
МеОН
низкий % МеОН высокий % МеОН
ACN
ТГФ
Рис. 6.22. Схемы экспериментов одновременной оптимизации различных условий
разделения для достижения оптимума селективности, (с) Сила растворителя
(%В) и температура (7); (б) сила растворителя и его тип (метанол или аце-
тонитрил); (в) тип растворителя (метанол, ацетонитрил и тетрагидрофуран).
Для (а)—(в) %-содержание воды варьируется, чтобы поддерживать
условие 0,5 < * < 20
Эксперименты, проведенные по такой схеме, из-за одновременного изменения
двух (или иногда более) различных условий может быть трудно интерпретировать
(особенно для образцов, содержащих большое количество компонентов, например,
более десяти). По этой причине такие схемы эксперимента часто сочетают с
компьютерным моделированием (глава 10). Кроме того, будет необходима надежная
система отслеживания пиков (раздел 2.7.4).
6.4.1.1. Смеси различных органических растворителей
Два органических растворителя {В-растворители) ранее были отмечены как наиболее
подходящие для ОФХ: ацетонитрил (ACN) и метанол (МеОН). Тетрагидрофуран
(ТГФ) используется меньше из-за его высокого порога прозрачности в УФ-диапазоне,
легкой окисляемости, более медленного уравновешивания колонок при смене
подвижной фазы (например, ТГФ/вода на ACN/вода) и несовместимости с ПЭЭК-
капиллярами (раздел 3.4.1.2). Однако, окисление ТГФ в процессе использования
может быть минимизировано добавлением воды |55|. Кроме того, многие образцы
не требуют детектирования в диапазоне ниже 230 нм, что делает ТГФ удобным в неко-
6.4. Разработка метода и стратегия оптимизации селективности 323
Н-В Кислоты (агн)
Биполярные (я")
Рис. 6.23. Предпочтительные растворители
для максимального изменения
селективности в зависимости от типа растворителя
торых случаях. В приложении 1 содер- Н-В Основания
■ (Рг)
жится дополнительная информация
о свойствах этих трех В-растворителей.
В оставшейся части раздела 6.4.1.1
описана методика, которая уже вышла из
широкого употребления, и потому читатель
может пропустить ее обсуждение.
Треугольник селективности
растворителей, показанный на рис. 2.9, испо
льзовался для сравнения зависимости
селективности от типа В-растворителя.
Как показано на рис. 6.23,
селективность, обусловленная различными
смесями ацетонитрила, метанола и ТГФ
с водой |56|, существенно различается.
Три этих В растворителя могут быть
использованы для оптимизации
селективности путем изменения содержания
каждого из них в подвижной фазе.
Начальные эксперименты с ацетонитрилом
в качестве В-растворителя определят такой %В для разделения образца, чтобы
0,5<А<20. Соответствующие значения %МеОН и %ТГФ (для получения сходной
элюирующей силы или значений к в нужном диапазоне) можно потом определить
с помощью номограммы на рис. 6.1]. Получившиеся три подвижные фазы,
содержащие по два растворителя (воду и В-растворитель), могут быть затем смешаны в
различных соотношениях, как это показано на рис. 6.22в. Пример использования схемы
эксперимента, приведенной на рис. 6.22в, для разделения 9-компонентной смеси
замещенных нафталинов показан на рис. 6.24. В этом примере первое
разделение проводится с элюентом 52% ACN/вода, второе с 63% МеОН/вода и третье
с 39%ТГФ/вода. Хотя селективность изменяется от первого к третьему разделениям
(см. рис. 6.24), в каждом из разделений есть два или более плохо разрешенных пика.
Следующим шагом является проведение экспериментов, в которых подвижную
фазу получают смешиванием равных частей подвижных фаз 1, 2 и 3. При этом
смешивание 1:1 (по объему) подвижных фаз I и 2 приводит к подвижной фазе 4:
26/32/42% ацетонитрила, метанола и воды соответственно. Аналогичным образом
получают подвижные фазы 5—7: подвижная фаза 5 представляет собой смесь 1:1
фаз I и 3 (26/20/54% ACN / ТГФ / вода); фаза 6, смесь 1: I подвижных фаз 2 и 3
(32/20/48% МеОН / ТГФ / вода); и фаза 7, смесь 1: 1: I подвижных фаз 1, 2 и 3
(17/21/13/49% ACN / МеОН / ТГФ / вода). Анализ последних четырех хроматограмм
на рис. 6.24 показывает, что разрешение до базовой линии достигается при
использовании подвижной фазы 5 (ACN/ТГФ). В большинстве случаев можно и дальше
улучшать селективность и разрешение, смешивая подвижные фазы от I до 7 и
получая промежуточные смеси. Такой подход может быть упрощен исключением
бесперспективных смесей. Например, из хроматограмм разделений I и 2 на рис. 6.24 видно,
что пики 6 и 7 перекрываются в каждом из них. Это говорит о том, что, скорее
всего, никакая смесь подвижных фаз 1 и 2 (к примеру, подвижная фаза 4) не улучшит
разделение пиков 6 и 7 (что и видно на хроматограмме разделения 4).
Схема эксперимента на рис. 6.22в и рис. 6.24 может быть дополнительно
улучшена при использовании компьютерного моделирования. С его помощью
прогнозируют, как будут зависеть результаты разделения от состава подвижной фазы |57|.
В другой работе предполагается расширить набор экспериментальных подвижных
фаз с 7 на рис. 6.22в до 15 [58| для того, чтобы более точно прогнозировать зави-
324 Глава 6. Обращенно фазовая хроматография нейтральных образцов
МеОН
©jut* ® iiii©
О S 10
О 5 10 15 (мин)
О 5 ЙГ~ ^5(мин)
О 5 10 15(мин)
5 10 15(мин)
■ш
* В |6
4 51 7 1 +
5 10 15(мин)
0
ACN
©
©
ТГФ
Рис. 6.24. Проведение семи экспериментов по изменению селективности в
зависимости от типа растворителя для разделения девяти замещенных нафталинов.
Заместители: /, l-NHCOCH3; 2, 2-SO?CH3; 3, 2-ОН; 4, 1-СОСН3; 5, l-N02;
6, 2-ОСНз; 7, -Н (нафталин); 8, 1-SCH3; 9, 1-С1. Условия: Колонка С%
150x4,6 мм; 40 "С; 2.0 мл/мин. Подвижные фазы (номера в кружках):
I, 52% ACN/вода; 2, 63% МеОН/вода; 3, 39% ТГФ/вода; 4, 1:1смесь 1 и 2;
5, 1:1 смесь 2 и 3; 6, 1:1 смесь 1 и 3; 7, 1:1:1 смесь 1, 2, и 3 |57|
симость времен удерживания от состава подвижной фазы (при использовании
компьютерного моделирования). Тем не менее, семи подвижных фаз, показанных
на рис. 6.22е, обычно достаточно для визуального выбора конечной оптимальной
подвижной фазы (без компьютера). Последовательно проводя эксперименты,
показанные на рис. 6.24 (1—>2, 2—»3, и т.д.), и анализируя результат каждого разделения
после его завершения, можно достичь приемлемого разделения без выполнения всех
семи экспериментов. К примеру, при таком подходе к образцу на рис. 6.24
потребуется лишь шесть экспериментов (разделения 1-6) или пять, если разделение 4 не
делается по причинам, описанным выше.
Читатель должен быть предупрежден, что при использовании этого (или
похожего) подхода к оптимизации с несколькими растворителями более сложные
подвижные фазы создают больше проблем, чем простые. По этой причине «приемлемое»
разделение с элюентом из трех растворителей (такое, как пятое разделение
на рис. 6.24) может быть предпочтительнее как более надежное по сравнению с
разделением, в котором используются все четыре растворителя для достижения немного
улучшенного результата.
6.4. Разработка метода и стратегия оптимизации селективности 325
6.4.1.2. Одновременное изменение элюирующей силы
и типа растворителя
Если для данного образца возможные недостатки ТГФ в качестве В-растворителя
неприемлемы, то альтернативой может быть изменение пропорций ацетонитрила,
метанола и воды в подвижной фазе |26]. Экспериментальная схема такого похода
показана на рис. 6.22ff. Начали с результатов разделений для двух подвижных фаз
с двумя различными значениями %ACN, которые обеспечивают попадание к в
нужный интервал 0,5 < ft < 20 (разделения I и 2), и с помощью номограммы на рис. 6.11
заменили их на подвижные фазы эквивалентной силы МеОН/вода (разделения 3 и
4). В конечном итоге выбираются подвижные фазы 5 и 6, получаемые смешиванием
подвижных фаз 1 и 3 (1:1) и 2 и 4 (1:1) соответственно. Использование данных,
полученных при проведении эксперимента в соответствии со схемой на рис. 6.225,
позволяет потом интерполяцией выбрать оптимальную подвижную фазу. Частный пример
такого подхода для оптимизации селективности продемонстрирован на рис. 6.8 и
6.10 (там нет данных для плохо разрешенных разделений 3 и 5 рис. 226).
Использование данных всех шести разделений, показанных на рис. 6.226, не изменило бы
конечных оптимальных условий, показанных на рис. 6.10г.
Более простая, но менее эффективная методика оптимизации %В и типа
растворителя заключается в постепенном изменении %В в подвижных фазах, которые
содержат сначала один, а затем другой В-растворитель (без смешивания различных
В-растворителей). На рис. 6.25 показан пример разделения шести стероидов. В
начальных двух экспериментах (рис. 6.26д,б) меняется %ACN. Поскольку пики 2 и 3
ни в одном из разделений не отделяются друг от друга, понятно, что используя
только ацетонитрил, эту проблему не решить.
В следующих двух экспериментах (рис. 6.25в,г) меняют %МеОН, выбрав с
помощью номограммы на рис. 6.11 те значения, которые обеспечат удерживание сходное
с показанным на рис. 6.25а,б. Теперь все пики могут быть разрешены почти до
базовой линии. Для критической пары пиков 4/5 на рис. 6.25г (35% В) Rs = 1,4.
Поскольку разрешение пары пиков 4/5 улучшается с увеличением %МеОН (в то время как
разрешение пары пиков 1/2 уменьшается), имеет смысл немного увеличить %МеОН.
Использование 37%МеОН позволило достичь разрешения Я, = 1,5 (до базовой
линии) вкупе с небольшим уменьшением времени разделения по сравнению
с 35%МеОН (разделение не показано).
Поскольку разделения, в котором пики едва разрешены, для окончательного
метода недостаточно (при том, что используется новая, не изношенная колонка),
то в качестве следующего В-растворителя можно рассмотреть ТГФ (рис. 6.25d,»r).
Разрешение еще возрастает, если использовать 15%ТГФ (рис. 6.25.Ж, Rs = 2.2), и этот
результат уже нельзя улучшить дальнейшим изменением %ТГФ без превышения к =
= 20. Процедуру оптимизации подбором типа растворителя и его силы,
представленную на рис. 6.25, можно прекратить на любом этапе, если будет достигнуто
приемлемое разрешение и время разделения. Таким образом, для некоторых образцов все
шесть экспериментов, показанных на рис. 6.25, не понадобятся, но они могут быть
недостаточны для других образцов.
6.4.1.3. Одновременное изменение элюирующей силы
и температуры растворителя
В таблице 2.2 показано, что изменение силы растворителя (%В) и/или температуры
(Т) менее эффективно, чем изменение типа растворителя. Однако, одновременной
оптимизации %В и температуры часто бывает достаточно для достижения разрешения
до базовой линии |60—65|. Кроме того, такая оптимизация удобнее и менее
трудоемка, чем альтернативные схемы одновременной оптимизации нескольких параметров.
Глава 6. Обращенно фазовая хроматография нейтральных образцов
(а)
35%ACN
1.5<fr<5
ft, = 0.0
А' л5 Л
<б>
25%ACN
Я. = 0.0
Время (мин)
Л' л5 Л6
(в)
45%МвОН
2<К<6
Я.= 1.1
(г)
35% МеОН
6<к<20
Яв=1.4
Г
(а)
25% ТГФ
1.4s*ri4
Я5=1.4
Время (мин)
20
i4 Л5
~i т 1 1 1 г"
Л'Л Л6
6
Время (мин)
10
Ж
А^
20
Время (мин)
Время (мин)
iQvL
20
Время (мин)
Рис. 6.2S. Разделение смеси шести стероидом с помощью изменения элюирующей силы
(%В) и типа растворителя. Образец: /, преднизолон; 2, гидрокортизон;
3, кортизон; 4, дексаметазон; 5, кортикостерон; 6, кортексолон. Условия:
Колонка С8 (5 мкм) 250 х 4,6 мм; различные подвижные фазы: смеси
органических растворителей и воды, как указано на рисунке; 35 "С; 2,0 мл/мин |59|
6.4. Разработка метода и стратегия оптимизации селективности 327
(а)
45 %В, 35 "С
2<к<13
Я, = 0.5
(б)
50 %В, 35 'С
1 <,к<,7
Rs=0.6
(г)
50 %В, 70 'С
1<fc<4
Я, = 0.2
Время (мин)
Ш
Л-
Время (мин)
-г
2
Время (мин)
Г
Время (мин)
Рнс. 6.26. Разделение смеси шести органических соединений различной структуры
с помощью изменения элюирующей силы (%В) и температуры. Образец:
/, метилбензоат; 2, бензофенон; 3, толуол; 4, нафталин; 5, фенотиазин;
6, 1,4-дихлорбензол. Условия: колонка C|g 125 х 3,0 мм; подвижная фаза:
смеси ацетонитрил/вода; 1 мл/мин |66|
Схема эксперимента для одновременного изменения %В и температуры показана
на рис. 6.22а, а пример применения этой схемы дан на рис. 6.26. На рис. 6.26а,б %В
изменяется при Т= 35 °С. Селективность изменяется значительно на фоне
изменения %В и при 41 %В Rs = 1,4. При этом 3<£<20 (разделение не показано). Затем
температуру повышают до 70 °С и получают результаты, продемонстрированные
на рис. 6.26»,г. При 45 %В и 70 °С получается приемлемое разделение (Rs = 1,8).
Дальнейшие эксперименты по изменению %В и Г не привели к увеличению
разрешения при условии сохранения удерживания в интервале 0,5 <к£20.
Применение одновременного изменения %В и Т приобретает все большую
популярность по следующим причинам: (1) необходимо только четыре начальных
эксперимента, если уже установлена величина %В для получения приемлемого интервала к;
(2) при смешивании А- и В-элюентов в потоке все четыре эксперимента можно
провести в автоматическом режиме без участия оператора (при условии, что контроль
температуры осуществляется системным контроллером); (3) нет никаких экспе-
328 Глава 6. Обращенно фазовая хроматография нейтральных образцов
риментальных сложностей, присущих другим способам оптимизации
селективности 126, 27]; (4) эта методика во многих случаях годится для достижения
необходимой селективности и разрешения образца; и (5) идентификация пиков легче, чем
в экспериментах по другим схемам. Более детально этот подход к оптимизации
селективности и разрешения описан в |63]. О модификациях метода см. |64|.
6.4.1.4. Замена колонки с одновременным изменением
одного или более условий
Как видно на рис. 6.14, использование четырех колонок разных видов {при прочих
одинаковых условиях) привело к максимальному разрешению Rs- 0.8. Такой результат
для этого конкретного образца не является чем-то необычным, поскольку замена
колонки не позволяет использовать «промежуточные» условия, как это сделано в случае
изменения других параметров в примерах, представленных на рис. 6.6 (%В), рис. 6.10
(смешанные В-элюенты) или рис. 6.13 и 6.14 (температура). Лучше совместить замену
колонки с дальнейшим изменением одного или более параметров, влияющих на
селективность. Методика, которую мы рекомендуем (и которая в настоящее время
используется во многих лабораториях), — это замена колонки в сочетании с одновременным
изменением %В и температуры (как это показано на рис. 6.22а). Применение этой
методики на четырех колонках, представленных на рис. 6.14, привело к получению
оптимизированных разделений, показанных на рис. 6.27а—г. В результате диапазон
разрешения для трех колонок вырос с 0,0 < Rs < 0,8 до 1,2 < Rs < 1,5. Если увеличить /V
колонки Symmetry Сщ (рис. 6.27а), взяв колонку большей длины (250 мм вместо
150 мм), и уменьшить скорость потока (от 2,0 до 1,0 мл/мин), можно достичь
разрешения Rs - 2,0 без возрастания давления (но при этом увеличивается время разделения
от 7,5 до 25 мин).
Во многих случаях просто оптимизация %В и температуры колонок с разной
селективностью (большие значения Fs) приведет к приемлемому разделению. Для
других более сложных образцов может потребоваться большее изменение
селективности. В таких случаях оптимизацию типа растворителя, подобную показанной
на рис. 6.226, можно сочетать с заменой колонки |68|.
В разделе 6.3.4 было отмечено, что неудобно постепенно изменять селективность
колонки, смешивая два различных сорбента в разных соотношениях в одной
колонке. Для этого были предложены два альтернативных подхода, ни один из которых
не нашел заметного практического применения. В одном из этих подходов
последовательно соединяли несколько разных колонок небольшой длины. Изменяя как тип
колонки, так и ее длину, можно дискретно изменять селективность (это так
называемая «фазово-оптимизированная жидкостная хроматография», ФОЖХ1). Определив
в экспериментах с отдельными колонками время удерживания образца для каждого
типа колонок, можно предсказать разрешение для различных сочетаний колонок
разной селективности и длины [71]. Другой подход использует две разных
последовательно соединенных колонки с независимым контролем температуры каждой
колонки (так назыавемый «температурно-регулируемый подход с тандемом колонок» 172|).
Поскольку время удерживания образца уменьшается при повышении температуры,
относительный вклад любой из колонок в суммарную селективность можно
увеличить, понизив температуру одной колонки относительно другой. Каждый из этих
двух подходов представляется сложноватым для рутинного применения.
'«phase-optimized liquid chromatography» или POPLC® от компании Bischoflf. — Прим. перев.
6.4. Разработка метода и стратегия оптимизации селективности 329
Symmetry C18
(54 %В, 33 'С)
1.0**5 9. flj-1.5 1
~\ г
6
(б)
Время {мин)
Altima HPC18amide 1
(40,5 %В, 40 *С)
1 </f<10, Я5= 1.2
сла!
(в)
п i i i
2 4 6
Время (мин)
л7 Л10.9
Л ЛлЛ
Luna pheny - hexyl
(46 %В, 41 *С)
1</fS9. Rs=1.4
I г"
Ajl_J\L
(a)
Spherisorb ODS-2
(50 %B, 46 *C)
1 <k<,7, Hs=1.4
т 1 r
4 6
Время {мин)
Г
Время (мин)
Рис. 6.27. Оптимизированное разделение смеси 10 органических соединений
различной структуры на четырех различных колонках с помощью изменения силы
элюента (%В) и температуры. Образец и условия как на рис. 6.14, за
исключением того, что указано на данном рисунке [8, 9, 69]
6.4.2. Оптимизация условий изменением параметров
колонки
Условия разделения, определяемые параметрами колонки (длина L и диаметр dc,
размер частиц d , скорость потока F), лучше оптимизировать после изменения других
условий для достижения оптимальной селективности и максимального разрешения
(шаг 6 на рис. 6.216). В первую очередь при разработке метода обычно выбирают
размер частиц, длину колонки, ее диаметр и скорость потока (например,
рекомендованные в таблице 6.1), чтобы обеспечить достаточное количество теоретических
тарелок для разделения исследуемого образца. После выбора других условий (типа
колонки, подвижной фазы, температуры) можно изменять длину колонки и скорость
Глава 6. Обращение-фазовая хроматография нейтральных образцов
потока либо для увеличения разрешения (за счет возрастания времени разделения),
либо для сокращения времени разделения (в тех случаях, когда начальное
разрешение 2> 2). На рис. 6.28 показаны примеры для каждого из этих случаев. Разделение
на рис. 6.28а имеет разрешение, близкое к минимальному допустимому (Rs = 1,1),
которое должно быть увеличено. Это увеличение эффективнее всего достигается
увеличением длины колонки, как это показанно на рис. 6.286, где длина колонки
увеличена со 150 до 250 мм. При этом почти достигается разрешение до базовой линии
за счет увеличения времени разрешения и давления на 2/3. Желательно еще больше
увеличить разрешение. Однако, предыдущий пример показывает, что увеличение
длины колонки приводит к увеличению времени разделения и давления, которые
фактически ограничивают возможный рост разрешения. Альтернативный вариант
(а)
L - 150 мм
2.0 мл/мин
Rs=1.1
Р=72бар
(1050 psi)
(б)
L = 250 мм
2.0 мл/мин
Rs=1.5
Р~ 121 бар
(1750 psi)
Li
г з
Время (мин)
Г
JL:
>л!_
^ А О
(в)
L = 250 мм
1.0 мл/мин
Rs=4.8
Р=68бар
(990 psi)
I
0
(з)
L- 100 мм
4.0 мл/мин
Rs=2.1
Р= 109 бар
(1560 psi)
4L
время (мин)
10
Время (мин)
20
Время (мин)
JL.
30
X
Рис. 6.28. Иллюстрация изменений условий колонки с целью улучшить разрешение
или уменьшить время разделения. Компоненты образца (в этих условиях
неионизированы; рН — 2.6): /, фталевая кислота; 2, 2-нитробензойная
кислота; 3, 2-фторбензойная кислота; 4, 3-нитробензойная кислота; 5, 2-хлор-
бензойная кислота; 6, 4-хлоранилин; 7, 3-фторбензойная кислота; £, 2,6-ди-
метилбензойная кислота; 9, 2-хлоранилин; 10, 3,4-дихлоранилин. Условия:
колонки диаметром 4, 6 мм С|а (5 мкм), длина L указана на рисунках;
подвижная фаза — 30% ACN-буфер в (я) и (ff); 40% ACN-буфер в (в) и (г),
40 "С в (а) и (б), 30 °С в (в) и (г); скорость потока указана на рисунках |73|
6.5. Неводная обращение фазовая хроматография (НОФХ) 331
увеличение разрешения — снижение скорости потока, но этого, как правило, делать
не стоит. Для примера, представленного на рис. 6.28я, понижение скорости потока
с 2,0 до 1,0 мл/мин приводит к незначительному росту разрешения (до Rs = 1,2)
в то время как время разделения удваивается до десяти минут {разделение не
показано). С другой стороны, можно уменьшить размер частиц сорбента и взять, например,
колонку 150 х4,6 мм с частицами диаметром 3 мкм. В этом случае время разделения
останется прежним, таким же как и на рис. 6.28а, но давление вырастет до 200 бар и
разрешение станет Rs = 1,6. В каждом из этих случаев разрешение, время разделения
и давление могут изменяться для достижения определенной цели. Разумного общего
компромисса можно достичь, если использовать колонку с 3-мкм сорбентом,
геометрическими размерами 250 х 4,6 мм и скорость потока 1,0 мл/мин. При этом Rs = 2,1,
давление 165 бар и время разделения 15 мин. См. также раздел 2.4.1.2.
На рис. 6.28в,г приведен пример уменьшения времени разделения при
избыточном разрешении. На рис. 6.28в время разделения составляет 30 мин и Rs = 4,8.
Уменьшая длину колонки и увеличивая скорость потока, время разделения можно
существенным образом сократить, поддерживая при этом Rs > 2,0 и давление < 138 бар.
На рис. 6.28г это сочетание изменений длины колонки и скорости потока приводит
к десятикратному уменьшению времени разделения с Rs = 2,1. Уменьшая длину
колонки и в таких же пропорциях увеличивая скорость потока, давление можно под
держивать постоянным (это не сделано в примерах на рис. 6.28е,г).
6.5. Неводная обращенно-фазовая
хроматография (НОФХ)
Разделение с помощью неводной обращенно-фазовой хроматографии (НОФХ)
зарезервировано за очень гидрофобными, сильно удерживаемыми образцами, не элюиру-
емыми 100% ACN в качестве подвижной фазы (липиды, синтетические полимеры
и т.д. 174—77|). Поэтому подвижная фаза для разделений в НОФХ будет содержать
смесь более полярного А-элюента и менее полярного В-элюента (органических
растворителей). Часто А-элюентом бывает ацетонитрил или метанол, в то время как
В-элюентом может быть ТГФ, хлористый метилен, хлороформ, метил-трети-бутило-
вый эфир (МТБЭ) или другие менее полярные органические растворители.
Удерживание образца контролируется изменением %В и/или полярности В-элюента,
которую можно приблизительно охарактеризовать величиной Р', приведенной
в таблице 1.4 Приложения 1.
На рис. 6.29 показан пример применения НОФХ для разделения различных ка-
ротиноидов (рис. 6.29д) в смеси стандартов (рис. 6.296) и в экстракте из томатов
(рис. 6.29е). Еще одной причиной использования НОФХ является нерастворимость
очень гидрофобных образцов в водных растворах. С практической точки зрения,
если уж вы выбрали для разделения НОФХ, то необходимо вымыть из ВЭЖХ-систе-
мы и колонки всю воду еще до перехода на неводную подвижную фазу. В общем
случае получаса промывки ацетонитрилом или метанолом будет достаточно.
6.6. Специфические проблемы
Одной из причин, по которой ОФХ более популярна, чем другие виды ВЭЖХ-разде-
лений, является то, что ее использование создает гораздо меньше сложностей.
В ОФХ две возможных проблемы требуют внимания: (1) слабое удерживание очень
полярных образцов и (2) уширение заднего фронта пика.
Глава 6. Обращенно фазовая хроматография нейтральных образцов
(«).
Р -каротин
и-каротин
ликопин
(б)
1
JH
—
2
С
(в)
10
15 мин
i
JJ
Ю
U
10 15 мин
Рис. 6.29. Неводное обращенно-фазовое (НОФ) разделение каротиноидоп. Условия:
колонка Си 250 х 4,6 мм; подвижная фаза 8% хлороформ — ацетонитрил:
2,0 мл/мин; комнатная температура |75|
6.6.1. Слабое удерживание образцов с высокой
полярностью
Этой проблеме было уделено внимание в разделе 6.I. Растворенные вещества,
характеризующиеся высокой полярностью, могут не удерживаться на колонке, имея к > I
даже в том случае, если в качестве подвижной фазы используется чистая вода (%В).
Такое чаще встречается в случае ионизированных веществ, которые намного слабее
удерживаются, чем их неионизированные аналоги (например, R-COO- и R-COOH).
Обычно удерживание ионизированных веществ в ОФХ может быть увеличено
изменением рН подвижной фазы (см. раздел 7.2, в котором описано как понизить
ионизацию растворенного вещества) или добавлением ион-парного реагента к подвижной
фазе (раздел 7.4).
При попытке разделения высокополярных неионизированных образцов с
помощью ОФХ на некоторых колонках наблюдается уменьшение времени удерживания
в том случае, если используется подвижная фаза с содержанием < 5 %В
(«несмачиваемость» сорбента). Некоторые сорбенты специально созданы для того, чтобы этих
сложностей избежать, но можно их свести к минимуму, следуя определенным
процедурам (раздел 5.3.2.3). Когда удерживание образца необходимо увеличить даже
Литература 333
при том, что в качестве подвижной фазы используется вода, выбор колонки может
обеспечить дополнительный контроль за удерживанием. Например, сорбенты с
развитой поверхностью (маленьким диаметром пор) обеспечивают, в общем случае,
более высокие значения к. Известно, что графитированные углеродные сорбенты
(раздел 5.2.5.3) преимущественно удерживают некоторые очень полярные неионизи-
рованные растворенные вещества. Впрочем, использование колонок, упакованных
этими сорбентами, ограничено их высокой стоимостью и ограниченной стабиль
н остью.
Когда образец слабо удерживается при использовании ОФХ, чаще всего
используют нормально-фазовую хроматографию, поскольку полярные раствореннные
вещества преимущественно удерживаются более полярной неподвижной фазой.
Хроматография гидрофильных взаимодействий (ГИХ; раздел 8.6), которая является
вариантом НФХ, особенно полезна в этом случае. Ее можно использовать с водными
подвижными фазами. Кроме того, у нее есть и другие преимущества, если ее
комбинировать с масс спектрометрическим детектированием (ЖХ-МС).
6.6.2. Уширение заднего фронта пика
Уширение заднего фронта пиков растворенных кислот или оснований
(разделы 5.4.4.1, 7.3.4.2) может возникать по целому ряду причин (раздел 17.4.5.3). Если
наблюдается заметное уширение заднего фронта пиков (например, с фактором
асимметрии As > 2), необходимо принять меры для устранения этого явления. Обычно
замена колонки или предколонки решает проблему, если уширение заднего фронта
пика наблюдается при выполнении рутинных анализов. Если же уширение заднего
фронта пика обнаружилось при разработке метода, важно еще до проведения
дальнейших экспериментов устранить это явление, изменяя условия разделения.
Дополнительную информацию об уширении заднего фронта пика см. в разделах 7.3.4.2,
7.4.3.3 и 17.4.5.3.
Литература
1. С A. Howard and A. J. P. Martin. Biochem. J., 46 (1950) 532.
2. L. R. Snyder and J. J. Kirkland, Introduction to Modem Liquid Chromatography, Wiley-lnterscien-
ce, New York, 1974, ch. 8.
3. J. J. Kirkland and J. J. DeStefano, J. Chromatogr. Sci., 8 (1970) 309.
4. J. A. Schmit, R. A. Henry, R. С Williams, and J. F. Dieckmann, J. Chromatogr. Sci., 9 (1971) 645.
5. W. R. Melander and C. Horvath, in High-Performance Liquid Chromatography. Advances and
Perspectives, Vol. 2, C. Horvath, ed., Academic Press, New York, 1980, pp. 113—319.
6. С Horvath, W. Melander, and I. Molnar, J. Chromatogr., 125 (1976) 129.
7. M. T. W. Hearn, in Ion-pair Chromatography, M. T. W. Hearn, ed., Dekkcr, New York, 1985.
8. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, R. G. Wolcott, and P. W. Carr, J.
Chromatogr. A, 961 (2002) 171.
9. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, and P. W. Carr, /. Chromatogr. A, 961
(2002) 195.
10. L. R. Snyder and M. A. Quarry, J. Liq. Chromatogr., 10 (1987) 1789.
U.K. Valko, L. R. Snyder, and J. L. Glajch, J. Chromatogr., 656 (1993) 501.
12. P. W. Carr, D. E. Martire, and L. R. Snyder, eds., Retention in Reversed-Phase HPLC (J.
Chromatogr., Vol. 656, 1993).
13. C. F. Poole, The Essence of Chromatography, Elsevier, Amsterdam, 2003.
14. L. С Tan and P. W. Carr, J. Chromatogr. A, 775 (1997) 1.
15. A. Ailaya and С Horvath, J. Chromatogr. A, 829 (1998) 1.
16. P. Nikitas, A. Pappa-Louisi, and P. Agrafiotou, J. Chromatogr. A, 1034 (2004) 41.
334 Глава 6. Обращенно фазовая хроматография нейтральных образцов
17. I. Molnar and Cs. Horvath, J. Chromatogr., 142 (1977) 623.
18. A. Tchapla, H. Colin, and G. Guiochon, Anal. Chem., 56 (1984) 62!.
19. J. Ко and J. С Ford, J. Chromatogr. A, 913 (2001) 3.
20. B. A. Bidlingmeyer and A. D. Broske, J. Chromatogr,. Sci. 42 (2004) 100.
21. T. H. Walter, P. Iraneta, and M. Capparella, J. Chromatogr. A, 1075 (2005) 177.
22. L. С Sander, K. A. Lippa, and S. A. Wise, Anal. Bioanal. Chem., 382 (2005) 646.
22a. J. L. Rafferty, J. I. Siepmann, and M. R. Schure, J. Chromatogr. A, 1204(2008) 11.
23. A. Klimek-Turek, T. H. Dzido, and H. Engelhardt, LCGC Europe, 21 (2008) 33.
24. T. Braumann, G. Weber, and L. H. Grimme, J. Chromatogr., 261 (1983) 329.
25. L. R. Snyder and J. W. Dolan, High-Performance Gradient Elation, Wiley, New York, 2007,
pp. 19-21.
26. L. R. Snyder, Today's Chemist at Work, 5 (1996) 29.
27. P. J. Schoenmakers, H. A. H. Billiel, and L. de Galan, J. Chromatogr., 185 (1979) 179.
28. P. J. Schoenmakers, H. A. H. Billiel, and L. de Galan, J. Chromatogr., 218 (1981) 259.
29. R. G. Wolcott, J. W. Dolan, and L. R. Snyder, J. Chromatogr. A, 869 (2000) 3.
30. J. Chmielowiec and H. Sawatzky, J. Chromatogr. Sci., 17 (9790) 245.
31. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, D. L. Saunders, L. Van Heukelem, and
T. J. Waeghe, J. Chromatogr. A, 803 (1998) 1.
32. J. W. Dolan, J. Chromatogr. A, 965 (2002) 195.
32a. S. Heinisch, and J. L. Rocca, J. Chromatogr. A, 1216 (2009) 642.
33. S. Heinisch, G. Puy, M.-P. Barrioulet, and J. L. Rocca, J. Chromatogr. A, 1118 (2006) 234.
34. J. W. Coym and J. G. Dorsey, / Chromatogr. A, 1035 (2004) 23.
35. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, D. L. Saunders, L. Van Heukelem, and
T. J. Waeghe, J. Chromatogr. A, 803 (1998) 1.
36. L. R. Snyder, J. Chromatogr., 179 (1979) 167.
37. L. R. Snyder and J. W. Dolan, J. Chromatogr. A, 892 (2000) 107.
38. P. L. Zhu, J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, J.-T. Lin, L. С Sander, and
L. Van Heukelem, J. Chromatogr. A, 756 (1996) 63.
39. D. W. Armstrong, W. Demond, A. Alak, W. L. Hinze, Т. Е. Riehl, and К. Н. Bui, Anal. Chem..
57 (1985) 234.
40. F. C. Marziani and W. R. Cisco, J. Chromatogr., 465 (1989) 422.
41. M. Paleologou, S. Li, and W. С Purdy, J. Chromatogr., Sci., 28 (1990) 311.
42. B. Nikolova-Damyanov, in HPLC of Acyl Lipids, J.-T. Lin and T. A. McKeon, eds., HNB
Publishing, New York, 2005. p. 221.
43. B. Voach and G. Schomburg, /. Chromatogr. 149 (1978) 417.
44. L. С Sander and S. A. Wise, J. Chromatogr. A 656 (1993) 335.
45. L. С Sander, M. Pursch, and S. A. Wise, Anal. Chem., 71 (1999) 4821.
46. L. С Sander, K. A. Lippa, and S. A. Wise. Anal. Bioanal. Chem., 382 (2005). 646.
47. J. W. Dolan and L. R. Snyder, J. Chromatogr. A, 1216 (2009) 3467.
48. J. W. Dolan, A. Maule, L. Wrisley, С. С. Chan, M. Angod, С Lunte, R. Krisko, J. Winston,
B. Homeierand, D. M. McCalley, and L. R. Snyder, J. Chromatogr. A, 1057 (2004) 59.
49. J. Pellett, P. Lukulay, Y. Mao, W. Bowen, R. Reed, M. Ma, R. С Munger, J. W. Dolan, L.
Wrisley, K. Medwid, N. P. Toltl, С. С. Chan, M. Skibic, K. Biswas, K. A. Wells, and L. R. Snyder,
J. Chromatogr. A 1101 (2006) 122.
50. J. W. Dolan, L. R. Snyder, T. H. Jupille, and N. S. Wilson, J. Chromatogr. A 960 (2002) 51.
51. J. W. Dolan, L. R. Snyder, and T. Blanc, J. Chromatogr. A, 897 (2000) 51.
52. G. Xue, A. D. Bendick, R. Chen, and S. S. Sekulic, J. Chromatogr. A, 1050 (2004) 159.
53. E. Van Gyseghem, M. Jimidar, R. Sneyers, D. Redlich, E. Verhoeven, D. L. Massart, and Y. Van-
der Heyden, /. Chromatogr. A, 1074 (2005) 117.
54. D. H. Marchand, L. R. Snyder, and J. W. Dolan, J. Chromatogr. A, 1191 (2008) 2.
55. J. Zhao and P. W. Carr, LCGC, 17 (1999) 346.
56. L. R. Snyder, J. Chromatogr. B, 689 (1997) 105.
57. J. L. Glajch, J. J. Kirkland, K. M. Squire, and J. M. Minor, /. Chromatogr., 199 (1980) 57.
58. A. C. J. H. Drouen, H. A. H. Billiet, P. J. Schoenmakers, and L. de Galan, Chromatographic, 10
(1982) 48.
59. L. R. Snyder, M. A. Quarry, and J. L. Glajch, Chromatographic, 24 (1987) 33.
60. P. L. Zhu, J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, J.-T. Lin, L. С Sander, and
L. Van Heukelem, J. Chromatogr. A, 756 (1996) 63.
Литература 335
61. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, D. L. Saunders, L. Van Heukelem, and
T. J. Waeghe, J. Chromatogr. A, 803 (1998) 1.
62. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 857 (1999) 1.
63. R. G. Wolcott, J. W. Dolan. and L. R. Snyder, J. Chromatogr. A, 869 (2000) 3.
64. A. Gonzalez, K. L. Foster, and G. Hanrahan, J. Chromatogr. A, 1167 (2007) 135.
65. J. W. Dolan, L. R. Snyder, T. Blanc, and L. Van Heukelem, J. Chromatogr. A, 897 (2000) 37.
66. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, D. L. Saunders, L. Van Heukelem, and
T. J. Waeghe, J. Chromatogr. A, 803 (1998) 1.
67. P. L. Zhu, L. R. Snyder, J. W. Dolan, N. M. Djordjevic, D. W. Hill, L. С Sander, and T. J.
Waeghe, J. Chromatogr. A, 756 (1996) 21.
68. J. J. DeStefano, J. A. Lewis, and L. R. Snyder, LCGC, 10 (1992) 130.
69. L. R. Snyder, J. W. Dolan, and P. W. Carr, J. Chromatogr. A, 1060 (2004) 77.
70. K. Valko, S. Espinosa, C. M. Du, E. Bosch, M. Roses, C. Bevan, and M. H. Abraham, J.
Chromatogr. A, 933 (2001) 73.
71. Sz. Nyiredy, A. Szucs, and L. Szepesy, J. Chromatogr. A, 1157 (2007) 122.
72. Y. Mao, and P. W. Carr, Anal. Chem., 73 (2001) 1821.
73. P. L. Zhu, J. W. Dolan, and L. R. Snyder, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 756 (1996) 51.
74. N. A. Parris, J. Chromatogr., 157 (1978) 161.
75. M. Zakaria, K. Simpson, P. R. Brown, and A. Krstulovic, J. Chromatogr., 176 (1979) 109.
76. N. E. Craft, S. A. Wise, and J. H. Soares, J. Chromatogr., 589 (1992) 171.
77. H. J. A. Philipsen, J. Chromatogr. A, 1037 (2004) 329.
ГЛАВА 7
ИОННЫЕ ОБРАЗЦЫ: ОБРАЩЕННО-
ФАЗОВАЯ, ИОН-ПАРНАЯ И
ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ
7.1. Введение 337
7.2. Кислотно-оснбвные равновесия и удерживание в обрашенно-фазовой
хроматографии 337
7.2.1. Выбор буферов 344
7.2.1.1. рКа и емкость буфера 344
7.2.1.2. Другие свойства буфера 347
7.2.1.3. Какие буферы лучше использовать 349
7.2.2. Как рКа зависит от структуры соединения 349
7.2.3. Влияние органических растворителей и температуры на рН
подвижной фазы и значения рКа образца 350
7.2.3.1. Влияние %В органического растворителя на эффективное
значение рКа вещества 351
7.2.3.2. Влияние температуры на значения рКа 352
7.3. Разделение ионных образцов методом обращенно-фазовой хроматографии (ОФХ) 352
7.3.1. Как контролировать удерживание 352
7.3.2. Как контролировать селективность 353
7.3.2.1. рН подвижной фазы 353
7.3.2.2. Сила растворителя (%В) и температура 355
7.3.2.3. Тип растворителя 356
7.3.2.4. Тип колонки 356
7.3.2.5. Другие условия, которые могут влиять на селективность 359
7.3.3. Разработка метода 359
7.3.3.1. Начальные условия (этап 1) 360
7.3.3.2. Оптимизация селективности (этап 3) 361
7.3.4. Нестандартные проблемы 361
7.3.4.1. Чувствительность к рН 361
7.3.4.2. Влияние силанольных групп 362
7.3.4.3. Слабое удерживание образца 364
7.3.4.4. Чувствительность к температуре 364
7.4. Ион-парная хроматография (ИПХ) 364
7.4.1. Основы удерживания 366
7.4.1.1. рН и образование ионной пары 367
7.4.1.2. Ион-парный реагент: тип и концентрация 367
7.4.1.3. Одновременное изменение рН и концентрации ион-парного
реагента (ИПР) 370
7.4.2. Разработка метода 371
7.4.2.1. Выбор начальных условий (этап 1) 372
7.4.2.2. Управление селективностью (этап 3) 375
7.4.2.3. Выводы 378
7.4.3. Нестандартные проблемы 378
7.4.3.1. Ложные пики 379
7.4.3.2. Медленное уравновешивание колонки 379
7.4.3.3. Искажение формы пиков 380
7.5. Ионообменная хроматография (ИОХ) 380
7.5.1. Основы удерживания 382
7.5.2. Роль противоиона 384
7.5.3. рН подвижной фазы 385
7.5.4. Колонки для ИОХ 385
7.5.5. Роль других условий 386
7.5.6. Разработка метода 386
7.5.7. Разделение углеводов 387
7.5.8. Разделения в смешанном режиме 387
Литература 389
7. Л Введение 337
7.1. Введение
В главе 6 описано, как методом обращен но фазовой хроматографии (ОФХ)
разделять нейтральные (неионизированные) молекулы. В этой главе рассматривается
разделение «ионных» образцов, в основном содержащих кислоты и/или основания
(иногда вместе с нейтральными соединениями). В состав таких образцов могут
входить и соединения, полностью ионизированные в интервале рН 2-12 (например, тет-
раалкиламмониевые соли, сульфокислоты). На начальном этапе развития ВЭЖХ
анализ ионных образцов зачастую вызывал определенные проблемы. Это было
вызвано отчасти отсутствием подходящих сорбентов, отчасти недостаточно глубоким
пониманием сути процесса разделения ионных образцов. Хотя в значительной
степени эти недостатки были преодолены, разделение ионных образцов по-прежнему
требует больших усилий, чем разделение нейтральных образцов. До 1980 года для
разделения кислот и оснований в большинстве случаев использовали ионообменную
хроматографию (ИОХ, раздел 7.5), но в настоящее время для разделения
«небольших» ионизируемых молекул (< 1000 Да) в основном предпочитают ОФХ (раздел 7.3)
и в меньшей степени ион-парную хроматографию (раздел 7.4). Тем не менее, ИОХ
широко применяется для разделения крупных биомолекул таких, как белки
(глава 13). Более подробно такие разделения рассмотрены в разделах 13.4.2, 13.5.1, 13.6.3.
7.2. Кислотно-оснбвные равновесия
и удерживание в обращенно-фазовой
хроматографии
Менее гидрофобные (более полярные) молекулы нейтральных образцов хуже
удерживаются на обращенной фазе (раздел 2.3.2.1, 6.2). Когда кислота (А) или
основание (В) подвергаются ионизации (т.е. переходят из незаряженной формы в
заряженную), соединение становится более полярным или гидрофильным. В результате
фактор удерживания на обращенной фазе к уменьшается в 10 и более раз:
ионизированная молекула
& А-+Н+ (7.1)
<=> ВН^ (7.1а)
гидрофильная (хуже
удерживается на ОФ)
подвижной фазы кислоты теряют протон и ионизируются,
протон при понижении рН и также становятся ионами.
О или основания (В) можно охарактеризовать константой
(кислоты) Ка = |А ||Н+1 (7.2)
|НА|
[RHH+1
Незаряженная молекула
(кислоты) НА
(основания) В -I- Н+
гидрофобная (лучше
удерживается на ОФ)
(основания) Ка = -—
|ВН+|
(7.2а)
При увеличении рН подвижной фазы кислоты теряют протон и ионизируются.
Основания акцептируют протон при понижении рН и также становятся ионами.
Ионизацию кислоты (НА) или основания (В) можно охарактеризовать константой
ионизации Ка:
(кислоты) Ка = |А ||Н+1 (7.2)
Здесь |НА] и [А~] — концентрации свободной и ионизированной кислоты НА, |В|
и |ВН+| — концентрации свободного и протонированного основания В. Величина
Ш 8 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^? хроматография
рКа ( =— logKa) кислоты или основания определяется по уравнению Гендерсона-Гас-
сел ьбаха:
(кислоты) рКа = рН - log] -Ь^—11 (7.3)
(основания) рКа = рН - log ' ' . (7.3а)
Например, значение рКа замещенного анилина (слабого основания) в воде находится
в пределах 4 < рКа < 6, а рКа алифатического амина (сильного основания) обычно
лежит в интервале 9—11. Указываемые в литературе значения рК.а кислот и
оснований обычно относятся к их водным (буферным) растворам при комнатной
температуре. Если подвижная фаза содержит органический растворитель или температура
раствора значительно отличается от комнатной, то значения рН и рКа могут
существенно измениться (раздел 7.2.3).
На примере разделения гипотетического образца, состоящего из карбоновой
кислоты НА (сплошная кривая, рис. 7.1а) и алифатического амина В (пунктирная
кривая, рис. 7.1я), показано, как удерживание зависит от рН и ионизации образца.
По вертикальной оси слева на рис. 7.1й отложена доля ионизированных молекул
каждого анализируемого вещества, а по горизонтальной оси — рН подвижной фазы.
Черными точками указаны значения рН, при которых половина молекул вещества
ионизирована (рН = рКа - 5,0 для кислоты НА и 9,0 для основания В). Значения к
(отложенные по вертикальной оси справа на рис. 7.1а) уменьшаются с увеличением
степени ионизации веществ, и их зависимость от рН и рКа описывается следующим
уравнением:
(кислоты, основания) к = к (I — FL) + klFL, (7-4)
где А0 — это к неионизированной молекулы (НА или В), к1 — значение к
полностью ионизированной молекулы (А~ или ВН+), F1 — доля ионизированных молекул
(0<1^< 1).
(кислоты) F* = (7.4а)
1+|Н+|/Ка
и
(основания) F* = . (7-46)
(l+(Ka/|H+l)}
Когда рН = рКа (или |Н+| = Ка), половина молекул вещества ионизирована (F± = 0,5).
На рис. 7.16 показаны хроматограммы разделения кислоты и основания при
разных рН подвижной фазы. По мере увеличения рН подвижной фазы от 4 до 10
кислота (заштрихованный пик) становится более ионизированной и менее удерживаемой,
в то время как степень ионизации основания убывает, а его удерживание возрастает
(при рН 7 к каждого из пиков равен 0,1). Из этого примера можно сделать
следующий вывод: изменение рН подвижной фазы — эффективный способ контроля
относительного удерживания (селективности) и качества разделения образцов,
содержащих кислоты и/или основания. Относительное удерживание двух кислот (или
оснований) может изменяться при варьировании рН, когда у них разные рКа, к
и/или kL (как обычно и бывает). Некоторые хроматографисты утверждают, что
при разделении частично ионизированных соединений (например, когда в растворе
одновременно присутствует значительное количество НА и А~) не избежать
искажения формы пиков. Однако, это утверждение ни экспериментально, ни теоретически
не подтверждено.
7.2. Кислотно основные равновесия и удерживание в обращенно-фазовой 339 ,
хроматографии
(а)
(б)
рН-4 В
рН-5 В
рН-6 В
рН-7
рН-8 НА
рН-Э НА
£
в
+
НА
Jll
НА
НА
рН-10 НА
Рис. 7.
1
6
8
10 (мин)
Гипотетические ОФХ-разделения кислоты (НА) и основания (В),
иллюстрирующие зависимость от рН. (я) Зависимость ионизации кислоты и
основания от рН подвижной фазы и влияние ионизации на к, (б) разделение
образца в зависимости от рН подвижной фазы; предполагается, что к0 кислоты
и основания равны, А* = 0 и /о = 1,0 мин
(Г340 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^ хроматография
Ч
На рис. 7.2 изображена зависимость времени удерживания /д гипотетического
слабого основания с рКа = 5,0 (например, замещенного анилина или пиридина)
от рН подвижной фазы. Когда рН изменяется в достаточно широком диапазоне,
график зависимости времени удерживания разделяемого вещества принимает
характерную S-образную форму. Как и рис. 7.1а, этот график отражает ионизацию образца.
В середине графика зависимости удерживания от рН (черная точка на рис. 7.2) рН
подвижной фазы равен рКа вещества. Обычно выбирают такое значение рН
подвижной фазы, которое позволяет контролировать селективность и разрешение. Когда рН
подвижной фазы близок к рКа наиболее сложного для разделения соединения или
соединений, изменение рН очень сильно повлияет на удерживание и разделение.
Если нужно изменить селективность и разрешение, то следует выбрать рН
подвижной фазы в области «II» (рис. 7.2, рН = рКа + 1,0). Впрочем, как будет сказано ниже,
значение рН подвижной фазы, обеспечивающее более существенное управление
селективностью (в пределах области II), может привести к худшей воспроизводимости
разделения. Это один из многих неизбежных компромиссов, с которыми приходится
сталкиваться во время разработки метода.
Когда кислота или основание наполовину ионизированы, изменение рН
подвижной фазы на 0,1 единицу приведет к изменению к примерно на 10%. Для
типичных условий разделения изменение к на 10% может привести к изменению
разрешения на +2,5 единицы, то есть разделение до базовой линии (Rs > 1,5) может
превратиться в полное перекрывание (Rs = 0). Таким образом, изменение рН
подвижной фазы на 0,1 единицу во время разделения наполовину ионизированного со
единения может вызвать полную потерю разрешения. Значит, для такого разделения
может возникнуть необходимость контролировать рН подвижной фазы с точностью
до 0,02 единиц. Для многих лабораторий это может оказаться затруднительным
(раздел 7.3.4.1). Чтобы избежать колебаний в удерживании, зависящих от рН, надо
подбирать такое его значение, которое будет отличаться от рКа всех компонентов пробы
по крайней мере на +1,5 единицы (области I и III на рис. 7.2). Поскольку у
большинства соединений значения рКа больше 4, разделения при низких рН подвижной
фазы (2 < рН < 3) с наибольшей вероятностью будут менее чувствительны к
незначительным колебаниям рН. Именно поэтому лучше начинать разработку метода с
низким рН подвижной фазы (что и рекомендуется в этой главе). В принципе для разра-
Рис. 7.2. Зависимость удерживания в ОФХ от рН. рКа основания предполагается
равным 5,0
7.2. Кислотно-основные равновесия и удерживание в обращенно-фазовои
хроматографии
ботки метода можно использовать и высокий рН (> 10), но для этого нужны особые
колонки, стабильные в оснбвных условиях (раздел 5.2.5, 5.3.2).
На рис. 7.3 приведен еще один пример зависимости от рН разделения
нескольких образцов разной кислотности и основности. На графике рис. 7.3а показана зави
симость времени удерживания от рН подвижной фазы пяти соединений: 1 —
салициловая кислота (относительно сильная карбоновая кислота), 2 — фенобарбитал
(слабая кислота), 3 — фенацетин (нейтральное соединение в приведенном интервале
рН), 4 — никотин (слабое основание), 5 — метиламфетамин (сильное основание).
На рис. 7.36—д изображены хроматограммы разделений этих веществ при разном рН
подвижной фазы. Например, обратите внимание на относительное (и абсолютное)
изменение удерживания сильного основания 5 (заштрихованный пик). С
увеличением рН ионизация метиламфетамина снижается, и он удерживается сильнее.
Пример, показанный на рис. 7.3, позволяет сделать некоторые важные выводы.
Во-первых, изменение значения рН при разделении образца, содержащего смесь
кислот, оснований и нейтральных соединений — эффективный способ управления
относительным удерживанием, а значит, и оптимизации разрешения. Разрешение
этого образца максимально (Rs = 7,2) при рН 8,3 и продолжительности анализа
28 минут (рис. 7.3е). С другой стороны, при рН 5 достигается разрешение до базовой
линии в кратчайшем разделении за II минут (рис. 7.3е). Впрочем, если не менять рН
подвижной фазы (рН 8,3), а вместо колонки длиной 300 мм взять колонку 50 мм и
увеличить скорость потока с 2,0 мл/мин до 5,0 мл/мин, то можно сократить
продолжительность разделения до 2 минуты (рис. 7.3яе). Разрешение при этом
останется > 2,0.
(а)
40
30
20-
I
е- 10
со
рН
5 сильное основание ( )
5.0'
•"8.3
1 сильная кислота ( )
4 слабое основание (—)
3 нейтральный (■--•)
2 слабая кислота (_.._)
Т
рн
Рис. 7.3. Влияние рН подвижной фазы на ОФХ-удерживание в зависимости от типа
образца. Образец: 1 —салициловая кислота, 2 — фенобарбитал, 3 —
фенацетин, 4 — никотин, 5 — метиламфетамин (заштрихованный пик). Условия
разделения (a—е): колонка C|g 300x4,0 мм, 10 мкм. Подвижная фаза 40%
метанол — фосфатный буфер, скорость потока 2,0 мл/мин, комнатная
температура. В эксперименте (ж) колонка длиной 50 мм, скорость потока
5,0 мл/мин. Данные из [1], хроматограммы (6—ж) смоделированы на
компьютере
1342 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
хроматография
рН-3
4 6
Время (мин)
т 1 I
8 10 12
рН-5
4 6 8
Время (мин)
т
10
рН-7
Время (мин)
рН-9
1
1
2
3 4
Л Л
0
рН-8.3 (300 м
_aJ
м колонка)
2
1 Л3
1 '
0
рн-8.:
3 (50 мм
колонка)
\2
т г
20
Время (мин)
л4
20
Время (мин)
5
1
40
5
Рис. 7.3. Продолжение
Время (мин)
(в)
(в)
(ж)
Во-вторых, такой образец содержит кислоты и основания с широким спектром
значений рКа (см. далее), следовательно, незначительное изменение рН во всем
диапазоне 3 < рН < 9 серьезно сказывается на относительном удерживании. Поэтому
для разделения такого образца необходимо либо тщательно контролировать рН
подвижной фазы (раздел 7.3.4.1), либо подобрать такие условия эксперимента, в ко-
7.2. Кислотно основные равновесия и удерживание в обращенно-фазовой 343 ^
хроматографии
торых разрешение будет намного больше 2. Например, разделение, показанное
на рис. 7.Зле (рН 8,3 и Rs - 2,0), можно сделать более надежным, используя колонку
длиной 10 см, а остальные параметры оставить прежними. Разрешение станет
равным 2,8, а продолжительность разделения — 4 минутам.
Ну и, наконец, по форме графика зависимости удерживания от рН можно
определить тип образца (кислота, основание или нейтральная молекула) и его примерное
значение рКа. Молекулы, лучше удерживаемые при повышении рН, являются
основаниями (соединения 4 и 5), а те, у которых удерживание уменьшается — кислотами
(соединения 1 и 2). Незначительное изменение удерживания говорит либо о том, что
вещество нейтрально (соединение 3) либо о том, что оно полностью ионизировано
в исследуемом диапазоне рН. Понятно, что приблизительные значения рКа
соединений 2 и 4 составляют соответственно 8 и 6,5. Сложно определить точные значения
рКа соединений I и 5, поскольку для этого нужен график зависимости удерживания
от рН в более широком интервале его значений, но вполне очевидно, что для 5 рКа >
> 9, а для 1 рКа <3.
Для амфотерных соединений, которые содержат и кислотные, и основные
группы, более сложная взаимосвязь между ОФХ-удерживанием и рН подвижной фазы.
Обратимся к рис. 7.4, на котором показана зависимость удерживания двух
аминокислот от рН. Молекулы каждой аминокислоты содержат кислотную группу -СООН
и основную группу -NH2. Вследствие этого минимальное удерживание наблюдается
при промежуточных значениях рН, поскольку в интервале 4 < рН < 8 карбоксильная
группа и аминогруппа ионизированы. Точнее, ионизация молекулы в этом
диапазоне рН максимальна, несмотря на то, что суммарный заряд молекулы равен нулю
(для различных ионизированных групп молекулы может быть предпочтительна более
полярная подвижная фаза, даже если эти группы имеют заряд разного знака).
25
20
15
10
5
RCHCOOH I RCHCOO" I
NH3+ Г NH3+ ~~И
\
\
\
- "ч \
b \ 1
< \ су'
1 1 1 1 1
RCHCOO"
1
NHj;
-
•
г
—оо—
70
60
50
40
30
20
10
10
12
14
РН
Рис. 7.4. Зависимость ОФХ-удержипания амфотерных соединений от рН подвижной
фазы. Образец: фенилаланин (•), лейцин (О). Условия: сорбент —
полистирол, подвижная фаза — 40 мМ фосфатный буфер |2|
344 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^1 хроматография
7.2.1. Выбор буферов
Всякий раз во время анализа кислот или оснований необходимо забуферить
подвижную фазу для поддерживания постоянного значения рН и воспроизводимости
удерживания. Показания рН-метра при измерении {и контроле) рН менее точны, если
в подвижной фазе содержится органический растворитель. Причина заключается
в том, что отклик электрода в водно-органических растворах подвержен дрейфу.
Поэтому рекомендуется сначала проводить измерения рН водного раствора (буфера А),
а потом уже добавлять в него органический растворитель для получения конечной
подвижной фазы. рН конечной подвижной фазы можно считать равным рН буфера А,
несмотря на то, что истинное значение рН несколько иное (раздел 7.2.3). Эта
неточность в определении рН конечной подвижной фазы не столь важна в рутинных
анализах методом ОФХ. Если буфер А готовится указанным образом, то следует
позаботиться о том, чтобы в других лабораториях смогли получить такие же значения рН
конечной подвижной фазы с точностью +0,04—0,05 единиц рН [3|. Указания о том,
что предпринять, если требуется более точное соблюдение рН подвижной фазы,
даны в разделе 7.3.4.1. Когда в данной книге упоминается рН подвижной фазы, обычно
имеется в виду рН буфера А. Инструкции по приготовлению буферных растворов
разных видов с различными значениями рН даны в приложении II.
При выборе буфера для разделения в режиме ОФХ следует учитывать следующие
его свойства:
• рКа и емкость буфера;
ф растворимость;
• поглощение в УФ-области (при использовании УФ-детектирования);
• летучесть (при использовании масс-спектрометра или испарительного детектора
светорассеяния);
• ион-парные свойства;
• стабильность и совместимость с оборудованием;
Обычно наиболее важны первые четыре свойства буфера.
7.2.1.1. рКа и емкость буфера
«Емкость буфера» или его способность поддерживать постоянный рН зависят от:
• значения рКа буфера
• концентрации буфера
• рН подвижной фазы
Уравнениями (7.2) или (7.2а) можно описать как ионизацию компонента образца,
показанную на рис. 7.1а, так и ионизацию буфера; т.е. ионизация буфера и
анализируемого вещества одинаково зависит от рН подвижной фазы и рКа. Буферная емкость
максимальна, когда концентрация двух форм буфера (например, НА и А-) одинакова,
т.е. когда рКа буфера равен рН подвижной фазы. Буферная емкость уменьшается
при увеличении разницы между значениями рН подвижной фазы и рК.а буфера.
Следовательно, в первую очередь требуется, чтобы значение рКа буфера было в интервале
±1,0 единиц рН относительно выбранного значения рН подвижной фазы (это
требование можно смягчить, расширив интервал до ±1,5 единиц рН при более высокой
концентрации буфера). Буферная емкость подвижной фазы пропорциональна
концентрации буфера, обычно имеющей величину от 5 до 25 мМ. Чтобы уменьшить до
минимума вероятность отличия рН образца от рН подвижной фазы во время ОФХ-разделе-
ния, в общем случае желательно растворять образец в подвижной фазе (или в буфере
с таким же значением рН). Это особенно важно в случае более низкой концентрации
буфера в подвижной фазе и/или больших объемов образца.
В таблице 7.1 перечислены буферы, которые можно использовать в ОФХ, а
также их характеристики, такие как рКа буфера и интервал значений рН подвижной
7.2. Кислотно-основные равновесия и удерживание в обращенно-фазовои 345
хроматографии ^§г
Таблица 7.1. Буферы, используемые в ВЭЖХ
Кислота буфера*
Трифторуксусная
кислота
Фосфорная кислота
Монофосфат
Дифосфат
Лимонная кислота
Моноцитрат
Дицитрат
Муравьиная кислота
Уксусная кислота
Угольная кислота
Монокароонат
Бис-трис-пропан-НО*
Бис-трис-п ропа н
Трис-HCl*
NH4C1
о-Борная кислота
Моно-о- борат
1-метилпиперидина
гидрохлорид
Триэтиламина
гидрохлорид
рК,
(25 "С)
>2
2,1
7,2
12,3
3,1
4,7
5,4
3,8
4,8
6,4
10,3
6,8
9,0
8,0
9,2
9,1
12,7
10,1
11,0
дПГ™НЬНЬ'Й
1,5-2,5
1,5-3,5
6,0-8,5
11,0-13,5
2,0-4,5
3,5-6,0
4,0-7,5
2,5-5,0
3,5-6,0
5,0-7,5
9,0-11,5
7,5-8,0
7,5-10,5
7,0-9,5
8,0-10,5
8,0-10,5
11,5-14,0
9,0-11,5
9,5-12,5
УФ предел
обнаружения6
210 нм (0,1%)
< 200 нм
(10 мМ)
< 200 нм
(10 мМ)
< 200 нм
(10 мМ)
230 нм (ЮмМ)
230 нм (ЮмМ)
230 нм (ЮмМ)
210 нм (ЮмМ)
210 нм (ЮмМ)
< 200 нм (ЮмМ)
< 200 нм (ЮмМ)
215 нм (ЮмМ)
225 нм (ЮмМ)
205 нм (ЮмМ)
200 нм (ЮмМ)
200 нм (ЮмМ)
200 нм (ЮмМ)
215 нм (ЮмМ)
< 200 нм (ЮмМ)
Комментарии
Ион-парный реагент,
летуч
Ограниченная
растворимость
Ограниченная
растворимость
Ограниченная
растворимость
Вызывает проблемы
с оборудованием"
Вызывает проблемы
с оборудованием"
Вызывает проблемы
с оборудованием"
Летуч
Летуч
Летуч'
Летуч
Может быть нестабилен'
Может быть нестабилен'
Может быть нестабилен'
Может быть нестабилен'
Может быть нестабилен'
"Буфер состоит из кислоты и ее нона, например, для фосфорной кислоты Н3РО4 и Н2ГО4.
^Водные растворы; поглощение < 0,5 AU при длинах волн выше предела обнаружения.
"Утверждается, что он повреждает нержавеющую сталь; у авторов этой книги возникали
проблемы с оборудованием, вызванные длительным использованием цитратного буфера.
'Использование такого буфера нецелесообразно, поскольку из емкости, в которой хранится
буфер, улетучивается С02.
<!1,3-Бис|трис(гидроксиметил)метиламино| пропан.
'Карбонат аммония нестабилен при рН 7,0 (улетучивается С02), но стабилен при рН 8,5 |11|;
буферы, содержащие амины, склонны к окислению, при этом значительно увеличивается
УФ-поглощение.
*Трис(гидроксиметил)аминометан.
фазы, в котором этот буфер эффективен. В разделениях с УФ-детектированием
при рН подвижной фазы < 8 широко используются фосфаты, трифторацетаты,
ацетаты и формиаты. Можно также рассмотреть возможность применения бикарбоната
аммония. Значения рКа аммиака (9,2) и бикарбоната (10,3) близки, поэтому интер-
346 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^ хроматография
вал буферной емкости бикарбоната аммония несколько расширяется (8,2 < рН <
< 11,3). Это летучий буфер и поэтому он совместим с ЖХ-МС. В то же время если
его рН < 8,5, может улетучиваться COj (например, при избыточной дегазации),
а в результате возможно непредусмотренное возрастание рН. Вследствие такой
нестабильности свежий бикарбонат аммония рекомендуется готовить каждый день.
В оставшейся части раздела 7.2.1.1 тема буферной емкости обсуждается более
подробно. Читатель может пропустить это обсуждение и при желании перейти к разде
лу 7.2.1.2, а при необходимости вернуться к данному разделу.
«Эффективную буферную емкость» подвижной фазы можно определить
как свойство, при возрастании которого уменьшаются проблемы, обусловленные
нестабильностью рН (недостаточной буферизацией). Эффективная буферная емкость
увеличивается при следующих условиях:
1) по мере уменьшения разницы между рКа буфера и рН подвижной фазы (при
изменении буфера или рН);
2) по мере увеличения разницы между рН подвижной фазы и рКа анализируемого
вещества (при значительном отличии этих значений анализируемое вещество либо
не ионизировано, либо полностью ионизировано; в этих случаях обеспечение
высокой буферной емкости менее важно);
3) при увеличении концентрации буфера;
4) при уменьшении объема наносимого образца;
5) при доведении рН образца до значения рН подвижной фазы.
Примерами недостаточно высокой буферной емкости являются хроматограммы
3,5-диметиланилина, полученные при различных рН подвижной фазы, в качестве
которой используется 25 мМ фосфат калия (рис. 7.5). Несмотря на значительную
концентрацию буфера наблюдается небольшое размывание заднего фронта пика,
когда подвижная фаза имеет рН = 3. При увеличении рН до 3,5 (рис. 7.56) и 4,0
(рис. 7.5в) пик все больше уширяется, и его форма становится все более искаженной
(из-за размывания переднего фронта пика). Это происходит в результате
уменьшения буферной емкости. Значения рКа разделяемого вещества и буфера равны
соответственно 3,8 и 2,1, поэтому, по мере возрастания рН от 3,0 единиц, в этом примере
различие между рКа буфера и рН подвижной фазы увеличивается (условие I в
приведенном выше перечне). В то же время различие между рН подвижной фазы и рКа
разделяемого вещества сокращается (условие 2 в приведенном выше перечне).
Оба эти обстоятельства приводят к уменьшению эффективной буферной емкости
при увеличении рН подвижной фазы от 3 до 4 единиц и к возрастающему
искажению формы пика разделяемого вещества. Форму пика в любом из примеров рис. 7.5
можно было бы улучшить, оптимизируя условия эксперимента, указанные в
пунктах 3—5, приведенных выше (увеличив концентрацию буфера и т.д.).
рН= 3.0 3.5 4.0
L
Рис. 7.5, Влияние недостаточной буферной емкости на форму пика. Условия:
образец — 3,5-диметиланилин, колонка 250x4,6 мм, сорбент — цианофаза,
5 мкм, подвижная фаза 25% метанол — буфер 50 мМ монофосфат калия,
35 "С, скорость потока I мл/мин |4|
7.2. Кислотно-основные равновесия и удерживание в обращенно-фазовои 347
хроматографии ^§g
7.2.1.2. Другие свойства буфера
Разрабатывая методику ОФХ-разделения ионного образца, следует также
рассмотреть остальные свойства буферов, перечисленные в конце раздела 7.2.1.
Растворимость буфера. Обычно органические буферы хорошо растворяются
во всех водно-органических подвижных фазах (0—100 %В), но многие
неорганические буферы характеризуются ограниченной растворимостью особенно в тех
подвижных фазах, которые преимущественно содержат органический растворитель
(высокий %В). Следовательно, при смешивании А- и В-элюентов существует опасность
выпадения буфера в осадок, что приведет к засорению колонки или капилляров хро
матографической системы. Если есть сомнения относительно того, не выпадет ли
буфер в осадок, сначала следует подтвердить полную его растворимость в конечной
подвижной фазе (во всем интервале предполагаемых значений рН). Особенно это
касается ВЭЖХ-систем, в которых происходит автоматическое смешивание элюен-
тов А и В. Для этого можно вручную смешивать в емкости элюенты А и В в разных
соотношениях и через 30 минут или около того наблюдать результат. Если в растворе
возникает помутнение или осадок при определенном составе подвижной фазы (%В),
то подвижную фазу такого состава (с данным %В и выше) следует исключить или
уменьшить концентрацию буфера (более подробно см. обсуждение в работах |5, 6|).
Необходимо уделять особое внимание растворимости буфера в градиентном элюиро
вании (раздел 9.3.1).
На растворимость буфера влияют несколько параметров разделения |6|. Одним
из таких параметров является противоион буфера. Для кислотных буферов, таких как
фосфатные, растворимость буфера растет в ряду
натриевая соль (наименее растворима) < калиевая соль <
< аммониевая соль (наиболее растворима)
Аналогичным образом растворимость буфера меняется с изменением
органического растворителя (В-элюента) при условии, что сравнение проведено для
одинаковых значений %В и рН:
тетрагидрофуран (наименее растворим) < ацетонитрил < метанол (наиболее растворим)
Кроме того, на растворимость буфера влияет его относительная ионизация.
Чем больше заряд иона буфера (например, НРО|" и Н2РО4), тем меньше
растворимость буфера в подвижной фазе с высоким содержанием органического
растворителя. Таким образом, выбор самого буфера, а также других условий разделения дает
широкие возможности контролировать растворимость буфера. Впрочем,
растворимость буфера в изократической ОФХ не такая уж и большая проблема, ведь
подвижная фаза для разделения ионных образцов обычно содержит менее 60%
органического растворителя. В меньшей степени это справедливо для градиентного
элюирования, поскольку буфер добавляют только в элюент А (в большинстве случаев
так поступают с неорганическими буферами), а значит, концентрация буфера в
подвижной фазе становится обратно пропорциональна доле органического
растворителя. Также это справедливо и для разработки изократического метода разделения.
Обычно проблемы с растворимостью характерны только для неорганических
буферов, особенно фосфатов. В одной из работ |6| сообщается, что при комнатной
температуре и рН 7 фосфат калия растворим в концентрации 10 мМ или в смеси 85%
МеОН-вода, или 75% ACN-вода, при этом его растворимость повышается при
уменьшении содержания органического растворителя и падает при более высоком %В.
При рН 3 ЮмМ фосфат калия растворим в присутствии 85% МеОН или ACN.
В фосфатный буфер с концентрацией от 1 до 2 мМ при наличии других
благоприятных условий для «эффективной буферной емкости» из перечня, приведенного в раз-
348 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
хроматография
деле 7.2.1.1, по-видимому, можно добавить до 90% метанола или ацетонитрила.
Но такие высокие значения %В очень редко требуются для ионизированных
образцов, поэтому проблема с растворимостью буферов не актуальна.
Буферы, удовлетворяющие требованиям детектора. Поглощение буфера пропор
ционально его концентрации и вносит свой вклад в поглощение водно-органической
смеси подвижной фазы. В таблице 7.1 приведены приблизительные оценки,
позволяющие установить, будет ли выбранный буфер существенно повышать УФ поглоще
ние подвижной фазы. Как буферы влияют на дрейф базовой линии можно увидеть
на рис. 17.7—17.10. Более подробная информация о поглощении буферов в
зависимости от длины волны приведена в таблице 1.2 приложения I.
Для масс-спектрометрического детектирования (ЖХ-МС) необходимы летучие
буферы. Обычно используют трифторуксусную (ТФУК), уксусную и муравьиную
кислоты и их соли аммония. Разделение оснбвных соединений проводят при низком
рН в летучих буферах: таких, как муравьиная или уксусная кислоты. При этом
желательно подбирать подвижную фазу с высокой ионной силой, то есть с высокой
концентрацией буфера при рН, обеспечивающем значительную степень его ионизации
(т.е. при рН подвижной фазы, близком к значению рКа буфера |7|). В противном
случае даже небольшое количество анализируемой пробы приведет к перегрузке
колонки и размыванию заднего фронта пика из-за электростатического отталкивания
удерживаемых протонированных оснований ВН+ (раздел 15.3.2.1). Например, если
нужен буфер с рН 3,5—4,0, то можно взять формиат аммония, в то время как
уксусная или муравьиная кислоты при более низком рН обеспечат меньшую
концентрацию ионов буфера. Чем меньше ионизация буфера, тем больше вероятность
перегрузки колонки или размывания заднего фронта пика. Чтобы выбрать оптимальный
буфер для ЖХ-МС, обратитесь к разделу 4.14.
Буфер как ион-парный реагент. До сих пор мы предполагали, что влияние буфера
на удерживание образца и разделение заключается только в обеспечении рН
подвижной фазы и ионизации образца. Помимо этого ионизированный буфер X может
взаимодействовать с ионизированным разделяемым веществом А~ или ВН+, образуя
ионную пару:
Ионизированное вещество Ионная пара
(кислоты) А"+Х+ <=> А"Х+ (7.5)
(основания) ВН+ + X" <=> ВН+Х" (7.5а)
гидрофильный образец Гидрофобный образец
(хуже удерживается в ОФХ) (лучше удерживается в ОФХ)
Обычно ионные пары образуют ионы органических буферов. В большинстве
случаев они более гидрофобны по сравнению с неорганическими ионами. Например,
в противоположность фосфату трифторацетат — сильный ион-парный агент
(раздел 13.4.1.2). Как правило, неорганические буферы образуют незначительное
количество ионных пар, хотя некоторые исследования позволяют предположить, что даже
фосфат может в некоторой степени образовывать ионные пары с протонированными
основаниями |8|. Более подробно образование ионных пар буферами рассмотрено
в разделе 7.4.2.1.
Стабильность буфера и совместимость его с оборудованием. Неорганические
буферы и карбоновые кислоты в основной массе отличаются стабильностью. Амины
подвержены окислению, вследствие чего их УФ-поглощение на длинах волн менее
250 нм возрастает. Например, чистый триэтиламин (ТЭА) не должен поглощать
на длинах волн более 200 нм. Тем не менее, в работе [9| сообщается, что для 1%
водного раствора ТЭА (90 мМ) на длине волны 220 нм наблюдалось поглощение
более 2 AU.
7.2. Кислотно-основные равновесия и удерживание в обращенно-фазовои 349
хроматографии ^§=5
Утверждается, что цитратные буферы вызывают коррозию нержавеющей стали,
хотя при эксплуатации ВЭЖХ-оборудования получены и другие результаты, которые
этому утверждению противоречат |10|. В одной из лабораторий время от времени
возникали проблемы с оборудованием, по-видимому, из-за использования цитратно
го буфера |11|, но причина неполадок не подтвердилась. Несмотря на то, что буфер
с концентрацией цитрата < 10 мМ можно использовать без опасений, и он
достаточно удобен для приготовления буферов с различным рН во всем интервале 2 < рН <
< 7, все же следует проявлять осторожность. В итоге реальные или воображаемые
проблемы, связанные с использованием цитрата: его относительно высокий предел
поглощения (230 нм, таблица 7.1) и доступность подходящих альтернативных
буферов (например, фосфатный и ацетатный) приводят к снижению популярности цит-
ратного буфера среди большинства хроматографистов.
7.2.1.3. Какие буферы лучше использовать
Обычно образцы, содержащие малые концентрации анализируемых веществ, плохо
поглощающих на больших длинах волн, регистрируют с помощью УФ-детекторов
в диапазоне 200—230 нм. Для таких задач лучше всего использовать фосфатные
буферы, которые позволяют задать рН подвижной фазы в интервалах, перечисленных
в таблице 7.1 (рН < 3,5, 6,0 < рН < 8,5 или рН > 11,0). Формиатный и ацетатный
буферы позволяют охватить интервал рН от 2,5 до 6,0, подходят для УФ-детектирования
на длине волны 210 нм и выше. Кроме того, они подходят и для ЖХ-МС, поскольку
обладают летучестью. Разделения оснований при низком рН с МС-детектированием
лучше проводить в растворах ацетата или формиата аммония. Эти буферы имеют
высокую ионную силу, что позволяет анализировать образцы с более высокими
концентрациями без размывания пиков |12|. Для подвижных фазе рН < 8,0 наиболее широко
используются фосфатный, ацетатный и формиатный буферы.
По мере разработки ОФХ-сорбентов, стабильных в основных условиях
(раздел 5.2.5), все чаще стали пользоваться подвижными фазами с высоким рН (рН > 8).
В качестве буферов с высоким значением рН иногда используют борат и гидроокись
аммония (аммиак), но нужно помнить о стабильности сорбента в основных условиях
(более подробно это обсуждается в разделе 5.8). В приложении II приведены
подробные методики приготовления некоторых широко используемых буферов с
требуемыми значениями рН.
7.2.2. Как рКа зависит от структуры соединения
Выбирая диапазон рН подвижной фазы для разработки метода (т.е. оптимизируя
рН), полезно знать хотя бы приблизительное значение рКа соединений, входящих
в состав образца. Эта информация позволит сузить диапазон рН подвижной фазы.
Например, чтобы с помощью рН контролировать удерживание и селективность,
следует использовать подвижную фазу с рН в пределах от рКа — I до рКа + 1. Если же
рН подвижной фазы находится вне этого интервала, метод становится более
устойчивым (робастным) и менее чувствительным к незначительным (непреднамеренным)
изменениям рН. У разных органических соединений значения рКа сильно разнятся,
при этом большое количество значений рКа уже определены экспериментально |13|,
а некоторые рКа можно оценить по молекулярной структуре вещества. Компания
Advanced Chemistry Development (Toronto, Canada) предоставляет в распоряжение
пользователей обширную базу данных экспериментальных и прогнозируемых
значений рКа (ACD/pKa DB: pKa prediction).
В процессе разработки метода ОФХ точные значения рКа не нужны, да и
во многих случаях может быть неизвестно химическое строение компонентов образ-
350 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^Ш^ хроматография
ца. Значения рКа определяют ионизируемые кислотные или оснбвные группы,
присоединенные к молекуле анализируемого вещества, например, -COOH,-NH2.
То есть, если у анализируемого вещества есть подобные функциональные группы, то
их рКа можно оценить по литературным данным для сходных соединений
(например, бензойная кислота является типичным представителем ароматических карбоно-
вых кислот). В таблице 7.2 приведены значения рКа в воде некоторых
распространенных кислотных или оснбвных заместителей, часто присутствующих в молекулах
образца. Можно также сделать предположения относительно значений рКа, исходя
из экспериментальных графиков зависимости удерживания от рН, как, например,
это было сделано для пиков 2 и 4 на рис. 7.3а.
Таблица 7.2. Приблизительные значения рК, кислотных и основных функциональных
групп (в полных растворах)
Группа
Сульфокислота, -SO^H
Аминокислота, -C(NH2)-COOH"
Карбоновая кислота, —СООН
Тиол, -SH
Пурин
Фенол, -ОН
Пиразин
Сульфоксид, -SO—
Тиазол
Амин, -NH2) -NR2
Пиридин
Имидазол
Пиперазин
рК,
Кислота
Алифатическая
1
3
5
10
Ароматическая
1
4
7
3
11
Основание
Алифатическое
10
1
2
2
10
10
Ароматическое
9
5
5
5
Примечание: соседние группы в молекуле влияют на рКа, вследствие чего его значения могут
меняться на 1—2 единицы или больше.
"Значения рКа аминокислот приведены на рис. 13.1.
7.2.3. Влияние органических растворителей и температуры
на рН подвижной фазы и значения рКа образца
Этот раздел не столь важен в повседневной работе, поэтому читатель может его
пропустить и перейти к разделу 7.3. Тем не менее, приведенные здесь выводы могут
оказаться полезными для правильной интерпретации взаимосвязи между удерживанием
вещества и рН подвижной фазы.
Приводимые в литературе значения рКа различных соединений (таблица 7.2)
обычно определяют в водных растворах при температурах, близких к комнатной.
Однако, достаточно часто ОФХ-разделения ионных образцов проводятся при более
высоких температурах в подвижной фазе, содержащей различные количества
органического растворителя. Добавление органического растворителя (%В) и температура
7.2. Кислотно-основные равновесия и удерживание в обращенно-фазовои 351
хроматографии ^§=
могут повлиять и на рН подвижной фазы, и на рКа образца. Хотя на практике, когда
речь идет о рутинных ОФХ-анализах, вовсе не обязательно знать, как истинное
значение рН подвижной фазы и рКа анализируемого вещества зависят от %В и темпера
туры. Важно только на протяжении всех экспериментов поддерживать эти параметры
постоянными, чтобы от анализа к анализу ионизация образца и его удерживание не
изменялись. Что же касается разработки метода, то желательно знать хотя бы
приблизительное значение рКа, чтобы можно было подобрать соответствующий рН
подвижной фазы (как сказано выше). В этой связи полезно определить «эффективное»
значение рКа анализируемого вещества, которое учитывает влияние %В и
температуры на рКа вещества и буфера (таблицы 7.1 и 7.2). Чтобы оценить, как степень
ионизации вещества и его удерживание зависят от рН, можно использовать эффективные
значения рКа вещества и рН буфера (не подвижной фазы, состоящей из буфера и
органического растворителя). Эффективное значение рКа эквивалентно значению,
которое можно определить из экспериментального графика зависимости удерживания
от рН подвижной фазы (т.е. рН буфера, как на рис. 7.2).
7.2.3.1. Влияние %В органического растворителя
на эффективное значение рКа вещества
рН подвижной фазы зависит от рКа буфера, а изменение %В органического
растворителя влияет и на рКа буфера, и на рКа анализируемого вещества |]4—16]. Если
буфер и вещество кислотной природы (например, буфер фосфатный, а вещество —
карбоновая кислота), то изменение доли органического растворителя одинаково
скажется на рКа буфера и рКа вещества — следовательно, изменения будут
скомпенсированы. В данном случае можно предположить, что эффективные значения рКа
вещества равны значениям, приведенным в таблице 7.2. То же самое справедливо и
в ситуации, когда анализируемое вещество и буфер — основания (т.е. эффективные
значения рКа вещества близки к значениям, измеренным в водном растворе
при комнатной температуре). Однако если буфер — кислота, а анализируемое
вещество — основание, или наоборот, кажущееся изменение рКа, зависящее от %В,
может быть существенным. Поэтому эффективные значения рКа вещества будут
отличаться от литературных данных, измеренных в водном растворе.
Обычно в ОФХ используются кислотные, а не основные буферы (например,
фосфат, ацетат, формиат). Когда анализируемое вещество является основанием,
а буфер — кислотой (и В-элюент — метанол), при увеличении доли
органического растворителя на 1% можно ожидать снижения наблюдаемых значений рКа
на 0,03 единицы (согласно экспериментальным данным |17, 18|). В качестве примера
рассмотрим разделение нескольких замещенных анилинов с подвижной фазой,
содержащей 25% метанола и фосфатный буфер |17|. Предположительно эффективные
значения рКа этих соединений будут на 25 х 0,03 = 0,75 единиц меньше
литературных данных. Приведенные в литературе значения рКа этих восьми разных
соединений составляют от 2,7 до 5,3, а экспериментальные рКа для этого образца (как и
на рис. 7.2) в среднем меньше на 0,7 + 0,1 единицы (I СО). В то же время, при
разделении шести замещенных бензойных кислот в ацетатном буфере с 40% метанола
|17|, эффективные значения рКа совпали с литературными данными (+0,1 единица,
1 СО). Добавление ацетонитрила оказывает такое же влияние на эффективные
значения рКа, как и добавление метанола.
Изменения эффективных значений рКа, зависящие от доли органического
растворителя, влияют на относительное удерживание таким же образом, как и
изменения рН подвижной фазы («эффективное» значение рН подвижной фазы). Это
позволяет предположить, что изменение доли органического растворителя оказывает
большее влияние на относительное удерживание и селективность разделения ионных
352 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^Ш^ хроматография
образцов по сравнению с нейтральными образцами. Подтверждением служит
градиентное алюирование, когда относительное удерживание и селективность изменяются
в зависимости от крутизны градиента [19, 20], что эквивалентно изменению %В
при изократическом элюировании (раздел 9.1.3).
В обсуждении, приведенном выше, рН подвижной фазы приравнивается к
значению, полученному при измерении рН водного раствора буфера. Именно так
рекомендуется определять рН подвижной фазы по ряду причин, приведенных в
разделе 7.2.1. Некоторые хроматографисты [14—16| предлагают определять рН подвижной
фазы уже после смешивания водного буфера с органическим растворителем. Это
менее удобно и результаты измерений хуже воспроизводятся. При этом требуется еще
вносить поправки для учета возможных изменений значений рКа анализируемого
вещества, зависящих от %В. То есть использование значений рН, измеренных в
водно-органической подвижной фазе, не решает проблему определения значений рКа
анализируемого вещества, изменяющихся в зависимости от %В. Еще раз
настоятельно рекомендуем определять рН водного буфера, а не конечной водно-органической по
движной фазы.
7.2.3.2. Влияние температуры на значения рКа
рКа как буфера, так и анализируемого вещества зависят от температуры |21—25|.
При нагревании уменьшаются значения эффективного рКа анализируемых протони-
рованных оснований |24|, и это может приводить к понижению степени ионизации
вещества и нетипичному повышению удерживания при более высоких температурах.
Поскольку рКа зависит от температуры, относительное удерживание ионных образцов
также может существенно изменяться под действием температуры — в большей
степени, чем для нейтральных образцов |19, 20|. То есть селективность, обусловленная
температурой, в целом будет выше у ионных образцов. Несмотря на то, что в
значения рКа, по-видимому, можно вносить поправки при изменении температуры
(как и при изменении %В, о чем шла речь выше), в настоящее время не существует
каких-либо простых способов это сделать. Так как температура редко изменяется в
широком диапазоне (как правило, это интервал от 30 до 50 СС), то на значение рКа
анализируемого вещества она обычно оказывает незначительное влияние, а значит, им
можно пренебречь и пользоваться расчетными значениями рКа при разработке метода.
7.3. Разделение ионных образцов методом
обращенно-фазовой хроматографии (ОФХ)
Для разделения смеси ионизирумых органических соединений одной из первых
следует рассматривать обращенно-фазовую хроматографию (ОФХ). Метод разделения
ионных образцов в ОФХ (раздел 7.3.3) разрабатывается таким же образом, каким и
для нейтральных образцов. Тем не менее, существуют некоторые важные отличия,
рассмотренные далее в этом разделе. Информация, приведенная в разделах 7.3.1 и
7.3.2, будет полезна не только для разработки методов разделения, но и для
устранения неисправностей при проведении рутинных анализов.
7.3.1. Как контролировать удерживание
Как и в случае нейтральных образцов (разделы 2.5.I, 6.2.1), сначала проводят
пробные эксперименты с подвижной фазой, содержащей различный %В, и определяют ее
оптимальный состав для получения нужного интервала удерживания (например,
7.3. Разделение ионных образцов методом обращение фазовой хроматографии (ОФХ) 353
1 < к < 10). Вместо этого можно (и предпочтительнее) первый пробный эксперимент
провести в градиентных условиях (раздел 9.3.1). Определив %В в подвижной фазе,
при котором наблюдается приемлемое удерживание образца, совершенствуют
селективность разделения, подбирая оптимальное относительное удерживание и
максимальное разрешение.
7.3.2. Как контролировать селективность
При разделении нейтральных молекул селективность можно варьировать, изменяя
долю органического растворителя (%В), температуру, сам растворитель В или
колонку. Эти параметры также оказывают влияние, зачастую даже большее, и на
разделение ионных образцов. Помимо этого, селективность при разделении ионных
образцов в значительной степени зависит от рН подвижной фазы и в меньшей степени
от типа и концентрации буфера. Раньше для подавления силанольной активности
сорбента в подвижную фазу добавляли алкиламины или четвертичные аммониевые
соединения, которые также могли изменять селективность. Впрочем, сегодня
предпочтение отдается колонкам типа В (раздел 5.2.2.2), поэтому необходимость
подавления взаимодействий с силанолами этими добавками отпала. В зависимости от
природы образца все перечисленные условия разделения могут в значительной степени
влиять на относительное удерживание и разрешение, как будет показано далее. В
работе |25] предложена расположить различные условия разделения в следующий ряд
в зависимости от степени их влияния на селективность ОФХ-разделения ионных
образцов:
рН (наиболее важен) > тип растворителя = тип колонки > %В > температура 3>
3> концентрация и тип буфера (наименее важны).
7.3.2.1. рН подвижной фазы
Изменение рН подвижной фазы может влиять на относительное удерживание
ионизируемых образцов, что очевидно из примера, изображенного на рис. 7.3. Ранее уже
отмечалось, что рН влияет на удерживание разделяемого вещества, только если рН
подвижной фазы отличается от рКа вещества приблизительно на +1,5 единицы рН
(рис. 7.2). Следовательно, когда надо с помощью рН изменять селективность
разделения, рН должен быть близок к рКа входящих в состав пробы компонентов. Карбо-
новые кислоты и амины — самые распространенные компоненты ионных образцов,
с которыми приходится сталкиваться на практике. Данные таблицы 7.2 говорят
о том, что при рН подвижной фазы 2—3 основания (рКа = 5—10) полностью
ионизированы, а кислоты (рКа = 5) присутствуют в нейтральной форме. Это утверждение
справедливо только отчасти, поскольку оно не учитывает влияние органического
растворителя (%В) и температуры на рКа, а также возможные изменения рКа при
наличии различных заместителей в молекуле образца. Поэтому, несмотря на то, что
селективность, обусловленная рН подвижной фазы, обычно снижается при низких
значениях рН, она все-таки может оставаться существенной в зависимости от
образца и %В.
Рассмотрим еще один пример изменения относительного удерживания в
зависимости от рН. На рис. 7.6 изображены хроматограммы разделения смеси замещенных
бензойных кислот (пики 1—4) и анилинов (пики 5—7). При повышении рН от 2,3
до 4,3 (рис. 7.6а—в) удерживание кислот I—4 снижается, а оснований
(заштрихованные пики 5—7) — увеличивается. При рН 4,3 и выше кислоты в основном полностью
ионизированы и удерживаются слабо, а основания практически не ионизированы,
поэтому удерживаются лучше. В результате при рН подвижной фазы выше четырех
354 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^? хроматография
(а)
1 -4 кислоты
5-7 основания
| Р 1 1 I 1 Г-
0 2 4 6
(б)
->—'—г
8 10
Время (мин)
JU
—i 1 1 1 1 г | г-
4 6 8 10
Время (мин)
рН-3.2
5<к-й 15
Я8 = 0.6
12 14
рН-3.7
2<к< 15
Я5=0.9
12
14
(в)
2I
I '
1
1—
3
,4
1
—1 1 i т 1
_5
Г 1 Г Г Г
рН
й °'5
7
А.
i i
0.5*/с<15
= 0.2
4 6 8 10
Время (мин)
12
14
(г)
6 8
Время (мин)
10
12
рН-3.4
45*515
Я* =2.7
Рис. 7.6. Влияние рН подвижной фазы на ОФХ-разделение смеси кислот и
оснований. Образец: 1 — 2-фторбензойная кислота, 2 — 3-хлорбензойная кислота,
3 — 3-нитробензойная кислота, 4 — 3-фторбензойная кислота, 5 — 3,5-диме-
тиланилин. 6 — 4-хлоранилнн, 7 — 3-хлоранилин. Условия: колонка Си
150 х 4,6 мм, 5 мкм, подвижная фаза 13% ацетонитрил — буфер (смесь цит-
ратного и фосфатного буферов), скорость потока 2,0 мл/мин, 35 "С. Пики
оснований заштрихованы [19]
кислоты и основания элюируются двумя группами пиков. При оптимальном рН
подвижной фазы 3,4 (рис. 7.6г) наблюдается приемлемое разделение. Тем не менее,
даже при таком относительно низком рН разделение (рис. 7.6г) остается до
некоторой степени чувствительным к незначительным колебаниям рН. Следовательно,
чтобы разделения были воспроизводимы, следует поддерживать рН буфера в пределах
+0,1 единицы от указанного значения. Присутствие частично ионизированных
кислот и оснований в образце при 3 < рН < 4 приводит как к изменениям
относительного удерживания с изменением рН, так и к низкой стабильности разделения,
показанного на рис. 7.6г.
7.3. Разделение ионных образцов методом обращение фазовой хроматографии (ОФХ)
7.3.2.2. Сила растворителя (%В) и температура
На рис. 7.7 показано, как относительное удерживание зависит от доли органического
растворителя и температуры. В экспериментах, изображенных на рис. 7.7 и 7.8,
используют один и тот же образец. Во всех экспериментах, хроматограммы которых
представлены на рис.7.7, использовалась такая же подвижная фаза, как на рис. 7.6,
с рН 3,2. Очевидно, что относительное удерживание сильно изменяется при
варьировании температуры или %В. Эти изменения могут быть обусловлены теми же
причинами, что и при разделении нейтральных соединений (например, как на рис. 6.26);
кроме того, происходят более важные в данном случае изменения «эффективного»
рН подвижной фазы из-за изменений %В и температуры (раздел 7.2.3).
Следовательно, доля органического растворителя и температура оказывают более существенное
влияние на относительное удерживание и разрешение ионных образцов, чем
нейтральных, как уже говорилось выше. На хроматограммах (рис. 7.7) видно, что с
увеличением %В или температуры относительное удерживание оснований (заштрихованные
пики 5—7) повышается по сравнению с удерживанием кислот (пики 1-4), в то время
как абсолютное удерживание всех соединений уменьшается. Это значит, что при
повышении %В или температуры значения рКа оснований образца уменьшаются, что
эквивалентно повышению рН подвижной фазы для этих оснований (в разделе 7.2.3
рассмотрен пример, когда образец имеет основные свойства, а буфер — кислотные).
При 19 %В и 49 °С наблюдается максимальное разрешение Rs = 3,3 (7.7d). Такие
оптимизированные условия разделения можно подобрать методом проб и ошибок,
но гораздо эффективнее воспользоваться программой компьютерного
моделирования (раздел 10.2.2).
13%В,35'С 1
5s*<;16
(а)
13 %В, 60 'С „ 3
5</fSl1 1
я-°°6 г и д \ : ыгг,
j—j—;—;—,ж-| "i i j—Г|—t—'—'—г I ' ' ' ' ^ ' ' ' ' Г~
О 2 4 в 8 10 12 1*
Время (мин)
(б)
О 2 4 6 8 10
Время (мин)
2 4
Время (мин)
28 %В, 60 'С
1 <к<А
Я8 = 1.0
(г)
3
2 4
Время (мин)
19 %В, 49'С
3£*<9
Я. = 3.3
Время (мин)
Рис. 7.7. Влияние силы подвижной фазы (%В) и температуры на разделение смеси
кислот и оснований. Образец такой же, как и на рис. 7.6, условия те же,
кроме рН буфера 3,2. Значения %В и температуры указаны на рисунке. Пики
основных соединений 5—7 заштрихованы |19]
356 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^1 хроматография
Во второй части этого раздела предлагается другое объяснение происходящих
изменений удерживания, показанных на рис. 7.7. Поскольку оно не так важно для понимания
природы влияния доли органического растворителя и температуры на удерживание, эту
часть можно пропустить и перейти к следующему разделу 7.3.2.3.
Влияние изменения %В, температуры или других условий на относительное
удерживание можно выразить следующим образом:
(кислоты, основания) к = А°(1 — F1) + kLFL, (7-4)
Так как к 3> к*-, это уравнение можно упростить
* = *°(1 - F1), (7.6)
где к — это значение к нейтральной (неионизированной) молекулы, F1 — доля
молекул разделяемого вещества, ионизированных при данном рН подвижной фазы.
Повышение %В или температуры приводит к уменьшению значений £° как
нейтральных, так и ионизированных веществ. Кроме того, условия, вызывающие
изменения «эффективного» рН подвижной фазы (а значит и значений /*), гораздо сильнее
влияют на разделение ионных образцов. Поэтому увеличение %В или температуры
(рис. 7.7) приводит к небольшому повышению рН подвижной фазы (что равноценно
снижению значений рКа этих соединений), и в результате удерживание оснований
5—7 возрастает.
7.3.2.3. Тип растворителя
Предполагается, что замена одного растворителя другим (например, метанола ацето-
нитрилом) оказывает сопоставимое влияние на относительное удерживание как
ионных, так и нейтральных образцов. Помимо этого, любое изменение «эффективных»
значений рКа при замене В-растворителя может дополнительно повлиять на
относительное удерживание ионных образцов, как и в случае изменения %В и температуры.
Благодаря этому дополнительному эффекту относительное удерживание ионных
образцов должно измениться сильнее, чем для нейтральных образцов, при замене
В-растворителя. И это действительно наблюдалось в исследовании |26|, в ходе
которого установлено изменение значения а на ±0,04 единицы для 45 нейтральных
образцов при замене 50% ACN на смесь 45% ACN + 5% МеОН. В этих условиях
для 22 ионных соединений значения а изменились на +0,09 единиц (вдвое больше,
чем для нейтральных образцов). В разделах 6.3.2 и 6.4.1 обсуждается влияние типа
растворителя на разделение неионных образцов.
7.3.2.4. Тип колонки
На рис. 6.14 был приведен пример разделения нейтрального образца на четырех
колонках с разными лигандами. На рис. 7.8 показаны аналогичные разделения ионных
образцов на трех тех же самых колонках (обратите внимание, что в «ионном»
образце могут также быть и нейтральные компоненты). Сравнение результатов этих
экспериментов (хроматограмм на рис. 6.14 и 7.8) показывает более значительное
изменение относительного удерживания ионных образцов (рис. 7.8). Этот ожидаемый
результат соответствует приводимому ниже обоснованию. Кроме того видно, что
в каждом из экспериментов (рис. 7.8) разрешение находится на пределе (Rs <l
для пары пиков с минимальным разрешением). Аналогичные результаты получены
при разделении нейтральных образцов на тех же самых колонках (рис. 6.14). То есть,
замена одной колонки на другую не обеспечивает существенного улучшения
разрешения в целом (как и разрешения «критических» пар пиков). Гораздо более
перспективной представляется замена колонки в совокупности с оптимизацией других
условий (например, %В и температуры), как показано на рис. 6.27, иллюстрирующем
7.3. Разделение ионных образцов методом обращение фазовой хроматографии (ОФХ)
Symmetry C18
2sfc<9,Rs = 0.3
К
(а)
Altima HP C18 amide
2<fcS7.Ra = 0.5
F. = 35
Время (мин)
4 7 л°
Время (мин)
(б)
Luna phenyi-hexyl
2<ft<8. Rs=0.4
Fs=33
'It
7
w
Время (мин)
Рнс. 7.8. Разделение ионного образца на разных колонках. Образец: 1 — 5-фенилпента-
нол, 2 — 4-л-гексиланилин, 3 — толуол, 4 — этклбензол, 5 — 4-н-бутилбен-
зойная кислота, 6 — транс-\апкон, 7 — мефенамовая кислота. Условия:
колонки 150x4,6 мм, 5 мкм, подвижная фаза 50% ацетонитрил — фосфатный
буфер рН 2,8, 35 °С, скорость потока 2,0 мл/мин. Приведены значения fs
относительно Symmetry C|g. Хроматограммы построены по данным из |27, 28]
разделение на тех же колонках нейтральных образцов. Различия в селективности
этих колонок можно описать функцией Fs (раздел 5.4.2): /^(Symrnetry/AItima) = 35,
/^(Symmetry/Luna) = 33. В каждом из этих случаев значения fs говорят о
существенных различиях в селективности, хотя можно было бы найти другие пары колонок,
у которых эти различия были бы еще больше.
Рассмотрим еще один пример влияния типа колонки на селективность (рис. 7.9).
В этом эксперименте проводится разделение смеси, состоящей из пяти полностью
протонированных сильных оснований (1—5), пяти частично ионизированных слабых
кислот (6—10) и одного нейтрального эталонного соединения (II, заштрихованный
пик). На рис. 7.9а показаны хроматограммы разделений смеси оснований и отдельно
смеси кислот на трех разных колонках (в каждой из смесей содержится нейтральное
соединение). На рис. 7.96 показаны соответствующие разделения всех 11 соединений
(кислот, оснований и нейтрального вещества). Относительное удерживание
полностью протонированных оснований (1—5) сильнее всего зависит от ионообменной
емкости С колонки (раздел 5.4.1). Чем больше С, тем лучше удерживаются прото-
нированные основания. При рН 2,8 ионообменная емкость С колонок составляет:
-0,47 (Intersil), -0,30 (Symmetry), 0,18 (Discovery). Как и предполагалось, в ряду
Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
хроматография
Сильные основания
Inertsil ODS-3
Время (мин)
Discovery C18
С- 0.16 "I ?
f
6.
}
7
я Discovery C18
I
Ш л°
Время (мин)
2 4
Время (мин)
10
SymmetryC18 основания 1-g
Время(мин)
Discovery C18
основания 1-5
Время(мин)
Рис. 7.9. ОФХ-разделение ионного образца на колонках разного типа. Образец:
(основания) 1 — пропранолол, 2 — пролинтан, 3 —димедрол, 4 — нортриптилин,
5 — амитриптилин, (кислоты) 6 — 2-фторбензойная кислота, 7 — 3-цианобен-
зойная кислота, 8 — 2-нитробензойная кислота, 9 — 3-нитробензойная
кислота, 10 — 2,6-диметилбензойная кислота, 11 — бензиловый спирт (нейтральное
соединение, заштрихованный пик). Условия: колонки 150 х 4,6 мм, 5 мкм.
подвижная фаза 30% ацетонитрил — фосфатный буфер рН 2,8, скорость потока
2,0 мл/мин, 35 "С. (с) Разделение кислот (образцы 6—10) или оснований
(образцы 1—5), в обоих случаях добавлено нейтральное соединение 11. (б)
Разделение всех 11 веществ. Хроматограммы построены по данным из |26, 29]
7.3. Разделение ионных образцов методом обращение фазовой хроматографии (ОФХ)
359
Inersil > Symmetry > Discovery относительное удерживание оснований I—5 возрастает
(обратите внимание на интервалы удерживания пиков 1—5 на рис. 7.96, показанные
стрелками в верхней части каждой из хроматограмм). Относительное удерживание
слабых кислот (пики 6—10) и нейтрального соединения на трех колонках сходно, так
как их удерживание не зависит от С.
7.3.2.5. Другие условия, которые могут влиять на селективность
К условиям, которые сейчас достаточно редко используются для контроля
селективности, относятся:
• тип буфера (например, фосфатный, ацетатный, аммониевый);
• концентрация буфера;
• аминомодификаторы.
Использование определенного типа буфера не является общепризнанным спосо
бом контроля селективности при разделении ионных образцов. Тем не менее, как
указано в разделе 7.2.3, тип буфера может влиять на «эффективные» значения рКа
разделяемого вещества, что эквивалентно изменению рН. Наиболее существенные
изменения относительного удерживания вызовет замена основного буфера
(например, содержащего ион аммония, триэтиламмин и т.д.) кислотным (фосфатным,
ацетатным) или обратная замена. Также тип буфера может повлиять на селективность,
если он способствует образованию ионных пар (раздел 7.2.1.2). Более гидрофобные
буферы, содержащие ионы трифторуксусной (ТФУК) и особенно гептафтормасляной
(ГФМК) кислот, могут образовывать ионные пары с протонированными
основаниями ВН+ и селективно увеличивать их удерживание |30|. Чем больше положительный
заряд протонированного основания — как, например, в пептидах, содержащих много
основных аминокислотных остатков (рис. 13.8, раздел 13.4.1.2), тем сильнее оно
удерживается на сорбенте. Буферы с ТФУК и ГФМК не вызывают каких-либо
проблем, которые часто встречаются в других ион-парных разделениях (раздел 7.4.3).
Концентрация буфера обычно вызывает незначительные изменения
относительного удерживания в разделениях при низком рН на современных алкилсиликагеле-
вых колонках типа В |26|. Однако, если эксперимент проводится при рН > 6 и/или
на старых колонках типа А (раздел 5.2.2.2), протонированные основания могут
удерживаться, взаимодействуя с ионизированными силанолами силикагеля.
Удерживание, обусловленное процессом ионного обмена, ослабевает с увеличением ионной
силы подвижной фазы (раздел 7.5.1), поэтому повышение концентрации буфера
будет способствовать уменьшению удерживания протонированных оснований [31].
Аминомодификаторы, например, ТЭА или соли тетрабутиламмония, раньше
добавляли в подвижную фазу в первую очередь для подавления нежелательных
взаимодействий с силанольными группами силикагеля (раздел 7.3.4.2). Реагируя с силано-
льными группами сорбента, аминомодификаторы подавляют ионообменные
взаимодействия между сорбентом и образцом, снижая таким образом удерживание
протонированных оснований. В настоящее время они редко используются,
поскольку, во-первых, у современных ОФХ-колонок типа В практически отсутствуют
нежелательные взаимодействия с силанольными группами. Во-вторых,
аминомодификаторы не всегда удобно использовать, так как в некоторых случаях приходится
длительное время уравновешивать колонку.
7.3.3. Разработка метода
Разработка метода ОФХ-разделения одинакова для ионных и нейтральных образцов.
На рис. 6.21а выделены семь этапов разработки метода, из которых существенно
отличается для ионных образцов только один этап (этап 3: выбор условий разделения).
360 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^ хроматография
Выбор условий разделения как для ионных образцов, так и для нейтральных
приведен на рис. 6.2Iff и включает следующие этапы:
1) подбор начальных условий;
2) подбор %В, при котором I < к < 10;
3) корректировка условий для достижения оптимальных селективности и
разрешения;
4) подбор размеров колонки для достижения наилучшего компромисса между
разрешением и продолжительностью анализа.
Разработка метода для ионных и нейтральных образцов отличается главным образом
на этапах 1 и 3. Разработку метода всегда следует начинать на новой (не
использовавшейся ранее) колонке, так как предыдущие образцы и условия разделения могут
повлиять на ее селективность, и тогда не удастся воспроизвести подобранные условия
на другой колонке.
7.3.3.1. Начальные условия (этап 1)
В таблице 7.3 предлагаются условия первоначального разделения смеси кислот и/или
оснований. Эти условия схожи с рекомендуемыми для первоначального разделения
нейтральных соединений (таблица 6.1). Главное отличие — необходимость буферной
подвижной фазы для ионизированных образцов. Обычно ионные образцы удержива-
ются слабее на ОФХ-сорбентах, поэтому в первом эксперименте, скорее всего, доля
органического растворителя должна быть ниже, чем для нейтральных соединений.
Тем не менее, начинать лучше всего с 80 %В, что позволит элюировать с колонки
все вещества — особенно те, которые в слабой подвижной фазе удерживаются
на сорбенте. Или же (и этот вариант наиболее предпочтителен), первый эксперимент
рекомендуется провести в градиенте, чтобы определить оптимальное значение %В
для изократического метода (раздел 9.3.1).
Таблица 7.3. Типичные условия разделения ионных образцов методом ОФХ
Условия
Колонка"
Подвижная фаза
Скорость потока
Температура
%В
Образец
к
Комментарии
Тип: Cj или С и (тип В)
Размеры колонки: 100 х 4,6 мм
Размер частиц: 3 мкм
Диаметр пор: 8—12 нм
80% ацетонитрил-буфер ЮмМ фосфат калия рН 2,55
2,0 мл/минв
30 или 35 °С«
Определяется методом проб и ошибок"
Объем <. 50 мкл
Масса < 10 мкг
1 <,к <10
"В качеств альтернативы можно взять колонку 150 х 4,6 мм с частицами 5 мкм. Важно:
разработку метода всегда проводят на новой (ранее не использовавшейся) колонке.
^Исходные значения будут изменяться в процессе разработки метода (раздел 2.5), также можно
изменять и исходное значение рН подвижкой фазы.
"Начните с 80 %В, а затем подберите нужное значение, как описано в разделе 2.5.1.
Выбор значения рН подвижной фазы и буфера для первоначального
эксперимента зависит, во-первых, от целей разделения и, во-вторых, от информации об
анализируемом образце, которая известна хроматографисту. Авторы книги рекомендуют
7.3. Разделение ионных образцов методом обращение фазовой хроматографии (ОФХ)
361
начинать с рН подвижной фазы 2,5—3,0, и фосфатного буфера, если используется
УФ-детектор, или с формиата аммония для ЖХ-МС. При таком рН подвижной фазы
маловероятны проблемы, связанные с размыванием заднего фронта пика
(раздел 7.3.4.2), нестабильностью колонки (раздел 5.3.1), недостаточной устойчивостью
(робастностью) метода (раздел 12.2.6). Образцы, содержащие сильные основания, все
чаще элюируют подвижной фазой с высоким рН (рН > 8) с целью минимизировать
ионизацию основных веществ во время разделения. Снижение ионизации образца
способствует возрастанию удерживания, благоприятствует получению симметричных
пиков и большей устойчивости ОФХ-метода. Однако, при рН > 8 требуются особые
колонки, чтобы предотвратить растворение частиц силикагеля, приводящее к разру
шению сорбента (разделы 5.2.5, 5.3). Основным из преимуществ подвижной фазы
с рН > 8 в анализах основных соединений является возможность разделять образцы
большей массы (раздел 15.3.2.1), при этом в количественных анализах повышается
чувствительность детектирования, а в препаративных разделениях — выход продукта.
Каким бы ни был рН подвижной фазы, следует обеспечить необходимую буферную
емкость (раздел 7.2.1.1).
7.3.3.2. Оптимизация селективности (этап 3)
Любое из условий разделения, перечисленных в разделе 7.3.2, можно изменять с
целью улучшения относительного удерживания и достижения максимального
разрешения. Более эффективным может оказаться одновременное изменение двух разных
параметров разделения. Такой способ, описанный в разделе 6.4.1 для нейтральных
образцов, может быть полезен и для ионных проб. Авторы книги рекомендуют
сначала изменять температуру и %В во всем диапазоне, в котором значение к
анализируемого вещества находится в интервале 0,5 < к < 20. Это иллюстрирует пример,
изображенный на рис. 7.7, и разъясняют аргументы, приведенные в разделе 6.4.1.3 и
в |32|. Если необходимы дальнейшие изменения селективности, то для
ионизируемых образцов можно одновременно изменять рН и силу растворителя |33|. При
одновременном изменении двух параметров разделения особенно полезна программа
для моделирования разделения (раздел 10.2), позволяющая определить условия
максимального разрешения.
Для контроля селективности можно одновременно оптимизировать более двух
параметров, например, популярной и эффективной стратегией является проведение
экспериментов на разных колонках (раздел 6.4.1.4) с изменением температуры и %В.
В работах [34, 35| сообщается о нескольких примерах, в которых одновременно
оптимизировали три разных параметра, каждый из которых можно было изменять
непрерывно (за исключением типа колонки). Однако, этот метод может потребовать
проведения огромного количества экспериментов. Например, чтобы одновременно
оптимизировать %В, температуру и рН для разделения одного образца, было
необходимо 32 эксперимента |34|.
7.3.4. Нестандартные проблемы
При ОФХ-разделении ионных образцов можно столкнуться с двумя проблемами,
которые в хроматографии нейтральных образцов не возникают.
7.3.4.1. Чувствительность к рН
Как отмечалось в разделе 7.2, при ОФХ-разделении ионизируемых образцов
относительное удерживание может быть достаточно чувствительно к небольшим
(непреднамеренным) изменениям рН подвижной фазы. Точность определения рН буфера
362 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^ хроматография
с помощью рН-метра в большинстве лабораторий обычно не превышает +0,05—
0,10 единиц. Для некоторых разделений могут оказаться неприемлемыми колебания
значений рН такой величины. Поэтому при разработке метода для ионных образцов
особое внимание следует уделять обеспечению устойчивости рН.
Есть несколько способов минимизации чувствительности к рН. Первый способ:
определяют чувствительность метода к изменению рН. Если рН подвижной фазы
нужно поддерживать с большой точностью (+0,1 единица и меньше), лучше его
контролировать, тщательно отмеряя компоненты буфера (по весу или по объему),
а не доводить рН буфера с помощью рН метра до нужного значения (несколько при
меров приведены в приложении II). Второй способ, не предполагающий точный
контроль рН, заключается в проведении экспериментов с подвижной фазой, рН
которой на 0,2 единицы больше и меньше требуемого значения. Эти хроматограммы
можно включить в метод, и оператор может по ним определять, есть ли
необходимость корректировать рН подвижной фазы (раздел 12.8).
Ну и наконец, лучший вариант для метода, слишком чувствительного к
изменениям рН, — заново провести оптимизацию условий разделения и разработать более
устойчивый метод. Иногда приходится подбирать такое значение рН, которое
больше, чем на 1 единицу отличается от рКа критических компонентов образца (чье
разрешение ухудшается от незначительных колебаний рН). Небольшие изменения
других условий разделения также могут способствовать повышению устойчивости
метода. Более подробно эта тема рассматривается в разделе 12.2.2.6 и в [36|.
7.3.4.2. Влияние силанольных групп
Между протонированными основными анализируемыми соединениями ВН+ и сила-
нольньши группами сорбента (в данном случае предполагается, что буфер содержит
ион калия) может происходить ионный обмен:
ВН+ + SiO"K+ <=> BH+SiO" + K+. (7.7)
Такое взаимодействие может приводить к возрастанию удерживания, размыванию
заднего фронта пика и потере воспроизводимости при замене одной колонки на
другую. Такого рода проблемы наиболее характерны для более старых колонок,
упакованных силикагелем типа А (раздел 5.2.2.2), поскольку он содержит примеси А1 ,
Fe2+ и ионов других тяжелых металлов. Такие примеси увеличивают кислотность
силанольных групп (что приводит к повышению концентрации SiO~-rpynn при любых
значениях рН подвижной фазы) и, вероятно, обусловливают недостаточную
воспроизводимость результатов при замене колонок. Новые колонки, заполненные
силикагелем типа В с более высокой степенью очистки, практически не содержат примеси
металлов, поэтому такого рода проблемы возникают реже.
Размывание заднего фронта пика может возникнуть при разделении основных
образцов даже на новой колонке типа В [37|. Причины размывания заднего фронта пика
на колонке типа В различны для разделений при высоком или низком рН подвижной
фазы. Если рН подвижной фазы < 5, то форма размытых пиков напоминает
закругленные прямоугольные треугольники (как на рис. 2.l5d). При рН > 6 чаще наблюдается
экспоненциальное размывание заднего фронта пика (как на рис. 2.15а). В настоящее
время полагают, что размывание в кислотных условиях происходит из-за отталкивания
одинаково заряженных удерживаемых ионизированных молекул (раздел 15.3.2.1, |38|).
Поэтому быстрее происходит перегрузка колонки основными образцами, чем
нейтральными, и размывание заднего фронта пика может стать заметным при нанесении
более 0,5 мкг оснбвного образца (для колонок диаметром 4—5 мм). Размывание
заднего фронта пика при низких рН подвижной фазы можно несколько уменьшить,
увеличив ионную силу подвижной фазы. Например, использование буферов при таком рН
7.3. Разделение ионных образцов методом обращение фазовой хроматографии (ОФХ) 363
подвижной фазы, которое благоприятствует ионизации буфера, приводит к
повышению ионной силы даже в тех случаях, когда молярность буфера не изменяется. Такой
подход для уменьшения размывания заднего фронта пика оснований рекомендуется
в ЖХ-МС с летучими формиатными буферами |39|.
Так до конца и не изучено, в чем причина размывания заднего фронта пика
оснбвных образцов на колонках типа В при рН 7 и выше; возможно, это
обусловлено медленной сорбцией и десорбцией молекул ВН+ |37|. На степень размывания
влияет природа растворителя В |40, 411:
ацетонитрил (пик размывается больше) > метанол >
> тетрагидрофуран (наименьшее размывание пика).
Обычно размывание заднего фронта пика становится меньше, если повысить
температуру колонки или увеличить %В, поскольку эти условия также
благоприятствуют снижению к. Вероятно, на колонках с небольшими значениями С (значения ка-
тионообменной емкости, раздел 5.4.1) размывание пиков уменьшается при рН
подвижной фазы менее 5. У «гибридных» частиц (раздел 5.3.2.2) силанольные группы
не ионизируются при рН подвижной фазы менее 8, и в этих условиях форма пиков
протонированных оснований правильная [16|. Хороший обзор по вопросам,
связанным с размыванием заднего фронта пиков оснбвных соединений дан в |16|.
На некоторых колонках (раздел 5.4.4.1) может происходить размывание заднего
фронта пиков недиссоциированных карбоновых кислот, нанесенных в небольшом
количестве, но с увеличением массы образца форма пиков улучшается. Причина
этого пока неизвестна. Тем не менее, можно определить, на каких колонках такая
проблема менее вероятна (раздел 5.4.4.1, |42|).
Раньше для снижения ионного обмена на колонках типа А (уравнение 7.7) и
предотвращения связанных с ним пагубных последствий |43, 44| применялись
различные меры:
• подавлялась ионизация силанолов (для этого использовали подвижную фазу
с низким рН);
• подавлялась ионизация оснбвных образцов (для этого использовали подвижную
фазу с высоким рН);
• подавлялся ионный обмен (использовалась подвижная фаза с высокой ионной
силой);
• блокировались ионизированные силанольные группы (в подвижную фазу
добавляли аминомодификаторы);
• использовали эндкепированные колонки.
ОФХ-сорбенты на основе силикагеля разрушаются гораздо быстрее при рН
подвижной фазы < 2,5 или > 8,0, что ограничивает использование экстремальных рН
для контроля ионизации силанольных групп или молекул образца. В настоящее
время есть колонки типа В, которые могут работать при экстремальных значениях рН
(см. раздел 5.3). Кроме того, минимизировать ионный обмен и связанное с ним
отрицательное воздействие силанольных групп можно, повышая концентрацию
буфера; тогда катионы буфера конкурируют с катионами анализируемого вещества в
равновесии, описываемым уравнением (7.7). Такие катионы буферов, как Ю", Li+, NHJ,
гораздо эффективнее, чем Na"1-, подавляют ионизацию силанольных групп и сводят
к минимуму размывание заднего фронта пика. В свое время часто добавляли в
подвижную фазу аминомодификаторы (например, ТЭА и диметилоктиламин) для
улучшения разделения оснбвных образцов. Сегодня, главным образом, используются
колонки типа В, и эти неудобные в использовании добавки оказались ненужными.
После эндкепирования (раздел 5.3.1) силанольные группы оказываются
экранированными от молекул анализируемого вещества, и форма пика обычно улучшается.
№364 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^? хроматография
7.3.4.3. Слабое удерживание образца
Как уже отмечалось в разделе 6.6.1 для нейтральных образцов, вещества с высокой
полярностью слабо удерживаются на ОФХ-сорбентах. Та же проблема еще более
характерна для ионных образцов, поскольку у ионизированных молекул полярность
еще выше. Однако, в этом случае существуют дополнительные способы повышения
удерживания образца. Причиной слабого удерживания ионного образца обычно
является его ионизация, при этом значения к могут понизиться более, чем в 10 раз.
Самый простой способ — изменить рН подвижной фазы так, чтобы снизить ионизацию
анализируемого вещества (кислоты или основания). Или можно использовать
ион-парную хроматографию (ИПХ, раздел 7.4), особенно для образцов, всегда
находящихся в ионизированной форме, например, четвертичных аммониевых
соединений. Для образцов с высокой полярностью также эффективна хроматография
гидрофильных взаимодействий (ГИХ) (раздел 8.6), и этот вариант обычно лучше ИПХ.
7.3.4.4. Чувствительность к температуре
Относительное удерживание ионизированных образцов, как правило, сильнее зависит
от температуры, чем в случае нейтральных соединений. Поэтому при разделении
ионных образцов важно тщательно термостатировать колонку (раздел 3.7). Не
рекомендуется проводить разделения любых образцов при комнатной температуре (без термоста-
тирования колонки), это создает проблемы, особенно в случае ионных образцов.
7.4. Ион-парная хроматография (ИПХ)
Ион-парную хроматографию (ИПХ) можно рассматривать как модификацию ОФХ
для разделения ионных образцов. Единственным отличием ИПХ от ОФХ является
добавление в подвижную фазу ион-парного реагента R+ или R", который потом
может прореагировать с ионизированной кислотой А" или основанием ВН+ в
равновесном процессе:
Ионизированное вещество Ионная пара
(кислоты) А" + R+ о A"R+ (7.8)
(основания) ВН+ + R" <=> BH+R" (7.8а)
гидрофильное вещество гидрофобная ионная пара
(слабо удерживается в ОФХ) (хорошо удерживается в ОФХ)
Следовательно, ИПХ влияет на относительное удерживание таким же образом,
как изменение рН подвижной фазы (раздел 7.2), но обеспечивает более широкие
возможности для управления удерживанием как кислот, так и оснований без
использования экстремальных значений рН подвижной фазы (например, рН < 2,5 или > 8).
К типичным ион-парным реагентам (ИПР) относятся алкилсульфонаты R-SOj(R~) и
тетраалкиламмониевые соли R4N+(R+), а также сильные (полностью ионизованные)
карбоновые кислоты: трифторуксусная кислота (ТФУК), гептафтормасляная кислота
(ГФМК) [R~|) и так называемые хаотропы (BRf, СЮ4, PF,j").
Оказалось, что в условиях высокоэффективной ИПХ, впервые примененной
в 1970-х годах, снижается размывание заднего фронта пиков основных соединений.
Это достоинство, а также возможность повышения удерживания до приемлемых
значений к для слабо удерживаемых ионизированных кислот и оснований стали
в то время главными причинами использования ИПХ. Кроме того, ИПХ создает
дополнительные возможности контролировать селективность при разделении ионных
образцов. В настоящее время преобладающее использование колонок типа В снизи-
7.4. Ионпарная хроматография (ИПХ) 365
ло важность преодоления размывания заднего фронта пика, и теперь мы лучше
понимаем, как эффективнее всего контролировать селективность в ОФХ.
Проблему слабого удерживания в ОФХ кислот и оснований, обладающих высокой гидро-
фильностью (особенно сильных оснований, которые остаются ионизированными
при рН < 8), можно решить другими способами, например:
1) используя подвижную фазу с высоким или низким рН, чтобы свести к минимуму
ионизацию анализируемого вещества и повысить его удерживание на колонках,
стабильных при экстремальных значениях рН;
2) с помощью хроматографии гидрофильных взаимодействий (ГИХ, раздел 8.6).
Поэтому из-за своей повышенной сложности и других проблем ИПХ на
сегодняшний день не столь популярна (раздел 7.4.3).
Разрабатывая метод для ВЭЖХ-разделения, авторы книги советуют начинать
с ОФХ, и только при необходимости добавлять ИПР. Для каких случаев можно было
бы порекомендовать ИПХ? В ИПХ есть две дополнительные переменные (тип и
концентрация ИПР), с помощью которых можно управлять селективностью. Если мы
знаем, что определенный пик соответствует кислоте, основанию или нейтральному
соединению, то влияние добавленного ИПР на удерживание анализируемого
вещества становится вполне предсказуемым. Далее мы в этом убедимся. Следовательно,
для оптимизации разделения кислот или оснований можно непрерывно изменять их
удерживание, в тех случаях, когда варьирование других параметров ОФХ-разделения
не дает приемлемого разрешения.
Кроме того, ИПХ позволяет сузить интервал времен удерживания образца,
поэтому образцы, для которых без ИПР потребуется разделение в градиентном режиме,
можно с ИПР разделить в изократическом. Рассмотрим, к примеру, запатентованный
образец (рис. 7.10), в состав которого входит лекарственное соединение X, несколько
консервантов и продукты разложения. На рис. 7.10а изображена хроматограмма
ОФХ-разделения этого образца в подвижной фазе, содержащей 30% метанола и буфер
(рН 3,5). Консерванты пропилпарабен (ПП) и метилпарабен (МП) — нейтральные
соединения и хорошо удерживаются на ОФХ-сорбенте, а лекарственное соединение X,
представляющее собой основание, и его продукты разложения Xj—Х3 удерживаются
слабо, поскольку находятся в протонированной форме ВН+. Два другие компонента
образца — кислоты: бензойная Б (еще один консервант) и гидроксибензойная ГБ
(продукт разложения парабена). На хроматограмме видно (рис. 7.10д), что у
компонентов пробы слишком широкий диапазон времен удерживания (0 < к < 30), что обычно
подразумевает необходимость градиентного разделения (раздел 9.1). Соединения
X—Хз являются сильными основаниями, и представляется нецелесообразным
повышать их изократическое удерживание (относительно остальных компонентов пробы)
за счет увеличения рН, поскольку потребовалась бы подвижная фаза с рН > 8 (и
колонка, стабильная при высоких значениях рН), а это привело бы к слабому
удерживанию кислотных соединений Б и ГБ (к <£ 1) и никак не повлияло на удерживание
нейтральных соединений МП и ПП. То есть, никакие изменения рН не помогли бы
сузить интервал времен удерживания образца так, чтобы значения к всех компонентов
пробы укладывались в приемлемый диапазон (например, 0,5 < к <. 20).
Для этого образца предпочтение было отдано изократическому разделению,
поэтому в качестве альтернативы градиентному режиму исследовали возможность
применения ИПХ. Добавление сульфонатного ИПР вполне предсказуемо приводит
к значительному повышению удерживания катионов образца (X—Х3), а также
умеренному снижению удерживания кислотных (Б и ГБ) и нейтральных компонентов
(МП и ПП). Обоснование этого приведено ниже в разделе 7.4.1.2. На рис. 7.106
показана хроматограмма разделения, проведенного с добавлением ИПР октансульфо-
ната (для избирательного повышения удерживания X—Хз) и увеличением доли
органического растворителя с 30% (рис. 7.10а) до 45% для снижения к нейтрального ПП.
366 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
*^^? хроматография
Хз „ (а)
+ без ион-парного реагента
i_Jb (0<*<Э0)
пл
20 40
Время (мин)
X с ион-парным реагентом (б)
(0.6<fc<9)
4 6
Время (мин)
Рис. 7,10. ОФХ-разделение запатентованной смеси кислот, сильных оснований и
нейтральных соединений. Образец: X — запатентованная лекарственная
субстанция, обладающая свойствами сильного основания; Х|, Х2, Хз —
продукты распада X, сильные основания; МП и ПП — метил- и пропилпарабено-
вые консерванты; Б — бензойная кислота; ГБ — гидроксибензойная
кислота (продукт разложения МП и ПП). Условия: (а) колонка 150x4,6 мм
5 мкм, подвижная фаза 30% метанол-ацетатный буфер рН 3,5; 30 "С,
скорость потока 2,0 мл/мин. (б) условия такие же, как в (а), кроме подвижной
фазы, которая теперь состоит из 45% метанола и буфера с 65 мМ октансуль-
фоната, скорость потока 1,5 мл/мин |46|
Таким образом удалось сузить диапазон времен удерживания компонентов образца
(0,6 <, к < 9) и разделить все компоненты образца до базовой линии за приемлемый
промежуток времени (11 минут).
В оставшейся части этого раздела кратко описаны основы ион-парного
разделения и обсуждается зависимость разделения от различных условий. Более подробно
эта тема раскрыта в [45] и главе 7 |46|.
7.4.1. Основы удерживания
В ИПХ существуют два возможных процесса или «механизма» удерживания. В
качестве примера рассмотрим образование ионной пары ионизированного кислотного
вещества А~ с тетраалкиламмониевым ИПР R+. Механизм образования ионной пары
между протонированным основным соединением В+ и алкилсульфонатным ИПР R~
такой же.
Одна из гипотез удерживания в ИПХ предполагает, что ионная пара
формируется в растворе в соответствии с уравнением (7.8). Образованная ионная пара
удерживается на сорбенте, т.е. равновесное распределение анализируемого вещества
между фазами, определяющее удерживание, описывается следующим уравнением
(аналогичным уравнению (2.2) в разделе 2.3):
A~R+ (подвижная фаза) <=> A~R+ (стационарная фаза). (7-9)
В соответствии с этой гипотезой удерживание определяется, во-первых, долей
ионизированных молекул А в подвижной фазе (которая зависит от рН подвижной фазы и
значения рКа анализируемого вещества), во-вторых, концентрацией ИПР и его
7.4. Ионпарная хроматография (ИПХ) 367
склонностью образовывать ионную пару {константой равновесия в уравнении (7.8)
или (7.8а)), и в-третьих, значением к ион-парного комплекса A~R+ (которое будет
больше у более гидрофобных ИПР).
Альтернативное представление об удерживании в ИПХ состоит в том, что ИПР
удерживается сорбентом, а потом в результате ионного обмена (раздел 7.5.1)
между R+X~ и, например, анионом кислоты А~ образуется ионная пара:
А~ (подвижная фаза) + R+X~ (неподвижная фаза) <=>
<=> A~R+ (неподвижная фаза) + Х~ (подвижная фаза) (7-9а)
То есть ИПР R+X~ сначала адсорбируется на неподвижной фазе, а затем анион
образца А~ замещает противоион Х~ на сорбенте. Любой из этих двух механизмов
(уравнения 7.9 и 7.9а) может превалировать в разделении, но роль которого из них
более важна определить непросто, да это и не столь существенно с практической
точки зрения. Показано [47], что эти два механизма удерживания фактически
равноценны, и оба они приводят к одинаковым результатам при прогнозировании
удерживания в зависимости от условий разделения. Следовательно, на практике можно
принять за основу любой из этих двух механизмов. Далее для упрощения изложения
материала будем иметь в виду ионообменный механизм (уравнение 7.9а), поскольку
по мнению авторов книги он проще для понимания и применения на практике.
7.4.1.1, рН и образование ионной пары
Рассмотрим подробнее, как удерживание в ИПХ зависит от рН подвижной фазы
на примере карбоновой кислоты RCOOH и ИПР катиона тетрабутиламмония ТБА+
(рис. 7.II). На рис. 1Л\а проиллюстрирован случай, когда в подвижную фазу не
добавляется ИПР (т.е. разделение проводится в ОФХ-режиме), и на Cg-сорбенте
(показанном в виде поверхности с присоединенными группами Cg) удерживается
преимущественно неионизированная кислота RCOOH. Справа на рис. 7.11а показан график
зависимости удерживания к от рН подвижной фазы. С увеличением рН удерживание
ослабевает, так как степень ионизации кислоты увеличивается (т.е. наблюдается
типичное для ОФХ изменение удерживания кислотного образца в зависимости от рН).
На рис. 7.1 Iff показано изменение удерживания того же вещества RCOOH,
но после добавления в подвижную фазу ТБА+ в концентрации, достаточной для
модификации всех групп Cg на поверхности сорбента; при этом блокируются
ОФХ-взаимодействия молекул образца (неионизированной кислоты RCOOH)
с Cg-группами сорбента. Теперь благодаря электростатическим взаимодействиям
с ТБА+ на сорбенте преимущественно удерживаются анионы кислоты RCOO"
(уравнение 7.9а). Зависимость к от рН подвижной фазы стала противоположной той,
которая изображена на рис. 7.1 \а (без образования ионных пар): с увеличением рН
удерживание возрастает (рис. 7.115) в результате повышения степени ионизации
кислоты благодаря образованию ионных пар. Если в пробе содержатся полностью про-
тонированные основания ВН^~ (например, сильные основания), то с увеличением
концентрации ИПР (ТБА+) их удерживание ослабеет. Происходит это из-за
отталкивания положительно заряженных ионов ВН+ положительно заряженными ионами
ТБА+ на поверхности сорбента. Кроме того, Cg-группы сорбента менее доступны,
поскольку они экранированы адсорбированными ионами ТБА+. Примеры для
иллюстрации этих обобщений даны в следующем разделе при обсуждении рис. 7.12.
7.4.1.2. Ион-парный реагент: тип и концентрация
Изменяя концентрацию ИПР в неподвижной фазе, можно непрерывно менять
природу удерживания от обращенно-фазового (рис. 7.11я) до ион-парного (рис. 7.115).
Это достигается изменением концентрации ИПР в подвижной фазе. Рассмотрим
368 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
*^^? хроматография
RCOO- + H*
RCOOH
А
М
/Cg Са ^в Cg Cg С
Cg Cg Cg Cg Cg Cg
- Cg Cg Cg Cg Cg Cj
Cg Cg Cg Cg Cg Cg
' Cg Cg Cg Cg Cg C.
Cg Cg Cg Cg Cg C|
]/////////////
RCOQ--t-H*-« ► RCOOH
(6)
it
' ТБА+ ТБА* ТБА
' ТБА* ТБА* ТБА*
' ТБА* ТБА+ ТБА*
' ТБА* ТБА* ТБА*
ТБА* ТБА* ТБА
ТБА* ТБА* ТБА*
У///////////У
Рис. 7.11. Описание зависимости удерживания кислоты RCOOH от рН подвижной
фазы, (я) ОФХ; (б) ИПХ с ионом тетрабугиламония (ТБА*) в качестве ИПР
в высокой концентрации
пример того, как концентрация ИПР влияет на удерживание вещества (рис. 7.12).
Предполагается, что разделение происходит на Cg-колонке, в качестве ИПР
используется фосфат тетрабутиламмония (ТБА+). Сначала обсудим, как устанавливается
равновесие ТБА* = R* при его адсорбции сорбентом (сплошная кривая ТБА* в
нижней части рис. 7.12а). На графике проиллюстрирована зависимость концентрации R+
в неподвижной фазе |R+|S от концентрации |R+]m в подвижной фазе; видно, что
с повышением [R+|m непрерывно растет |R+|S до насыщения сорбента. После того,
как произошло насыщение сорбента, при дальнейшем увеличении [R+|m
концентрация R+ на сорбенте больше не меняется. Вид этого графика типичен для адсорбции
анализируемого вещества или других молекул на ОФХ-колонке при увеличении
концентрация этого вещества в подвижной фазе (так называемая изотерма адсорбции
Ленгмюра, раздел 15.3.1.1, уравнение 15.1).
На рис. 7.12а изображены два графика: сплошная кривая для ТБА* и пунктирная
кривая для иона тетраэтиламмония (ТЭА+). Так как из этих двух ИПР ТБА* более
гидрофобный, он сильнее удерживается неподвижной фазой и насыщает ее при более
низких концентрациях R* в подвижной фазе. Количество образующихся ионных пар
определяется степенью насыщения неподвижной фазы, которая, как видно из
графика, зависит, во-первых, от концентрации ИПР в подвижной фазе и, во-вторых, от гид-
рофобности или удерживания ИПР. Гидрофобность и удерживание ИПР растут с
увеличением числа атомов углерода в молекуле ИПР (количества СН3- и СН2-групп
7.4. Ион парная хроматография (ИПХ) 369
(а) Адсорбция ИПР R* неподвижной фазой
ТБА* НгР04"
It
ТБА* НгР04-
Cg С8 С8 Св Cg Cg Cg
/у//?//////}///}////////
подвижная фаза
неподвижная фаза
,"" ТЭА+
[Rim
(б) Зависимость удерживания ионизрованной кислоты RCOO от
концентрации R+ в подвижной фазе [R+]m
RCOO"
+
НгР04*
+
ТБА+ НгР04+
Cg С8 С8 Св
ТБА+ RCOO"
Cg Са Св C8
Удерживание образца с
помощью ИПР
конкуренция
с Н2Р04-
Рис. 7.12. Адсорбция ИПР неподвижной фазой и влияние его количества на
удерживание образца
в молекуле ИПР). Поэтому ТБА+ (16 атомов углерода) более гидрофобен и сильнее
удерживается на сорбенте, чем ТЭА+ (8 атомов углерода). Кроме того, видно, что при
большей концентрации у в подвижной фазе менее гидрофобного ТЭА+ и меньшей
концентрации х более гидрофобного ТБА+ (точечные линии на рис. 7.12я)
неподвижная фаза адсорбирует одинаковое количество |R+|S, и при этом обеспечивается
одинаковое связывание анализируемого аниона А~ в ионные пары и, следовательно, его
одинаковое удерживание (см. далее рис. 7.15 и его обсуждение).
Теперь разберем, как меняется удерживание к образца по мере увеличения
концентрации ИПР (рис. 7.126). Предположим, что анализируемая кислота полностью
370 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^1 хроматография
ионизирована (RCOO-), и ее удерживание в режиме ОФХ практически нулевое
при некотором значении %В. При добавлении ИПР в подвижную фазу значение к
сначала возрастает в результате взаимодействия RCOO- с R+ в неподвижной фазе. После
насыщения сорбента ионами R+ удерживание RCOO- перестает возрастать. Однако,
дальнейшее увеличение концентрации R+ в подвижной фазе сопровождается
возрастанием концентрации его противоиона Х- (например, Н2РО4). Увеличение
концентрации Н2РО4 в подвижной фазе [Ь^РОд],,, приводит к конкуренции этих ионов с ионами
RCOO" во взаимодействии с R^~, обеспечивающем удерживание (уравнение 7.9а). В
результате к будет постепенно снижаться (рис. 7.126). То есть, рекомендуемые
концентрации ИПР в подвижной фазе не должны превышать значения, при которых проис
ходит насыщение колонки, иначе удерживание образца станет уменьшаться.
7.4.1.3. Одновременное изменение рН и концентрации
ион-парного реагента (ИПР)
Одновременное изменение рН подвижной фазы и концентрации ИПР позволяет
осуществлять весьма удобный контроль интервала удерживания и относительного
удерживания ионных образцов (как показано в примере на рис. 7.10). Это видно
также на графиках зависимости к модельного соединения от рН или концентрации
ИПР (рис. 7.116 и 7.126). Теперь будем считать, что подобные зависимости
существуют и для других образцов, но есть вероятность, что их к по-разному зависят от рН и
концентрации ИПР. На рис. 7.13 показан пример того, как разделение изменяется
при одновременном варьировании рН подвижной фазы и концентрации ИПР.
Образец состоит из нейтрального соединения (N, заштрихованный пик), слабо основного
соединения (В), кислотного соединения (НА|) и слабо кислотного соединения
(НА;)). Проведены три разделения (рис. 7.13а—в) при разном рН, но без добавления
ИПР в подвижную фазу. Точечными линиями показано, как меняется относительное
удерживание каждого пика. В четвертом эксперименте (рис. 7.13г) использовались
промежуточное значение рН подвижной фазы и добавка гексансульфоната (R-) в
качестве ИПР.
Рассмотрим сначала удерживание нейтрального соединения N (заштрихованный
пик). Как и ожидалось, его удерживание слабо зависит от изменений рН подвижной
фазы (рис. 7.13 а—в). При добавлении ИПР (R-) удерживание N ослабевает,
поскольку поверхность сорбента частично блокируется адсорбированными ионами R-.
Удерживание слабого основания В возрастает при рН > 5, а добавление ИПР
(рис. 7.13г) еще больше увеличивает его удерживание (стрелками указаны изменения
удерживания, происходящие в разделении на рис. 7.13г по сравнению с разделением
на рис. 7.136 при том же рН). Различные изменения в удерживании соединения В
вызваны понижением степени ионизации основания (ВН+—> В) при увеличении рН,
в то время как при добавлении ИПР ионы R- заряжают поверхность сорбента
отрицательно, способствуя притяжению положительно заряженных катионов ВН+. Ну и
наконец, удерживание кислотных соединений НА| и НА2 при увеличении рН
ослабевает и еще уменьшается при добавлении ИПР вследствие отталкивания анионов А"
от адсорбированных неподвижной фазой анионов R".
В итоге одновременное варьирование рН подвижной фазы и концентрации ИПР
приводит к следующим основным изменениям относительного удерживания
для каждого набора условий, представленных на рис. 7.13:
При низком рН без ИПР (а):
В < N < НА, - НА2.
При промежуточном рН без ИПР (б):
В < НА, < N < НА2.
7.4. Ион парная хроматография (ИПХ)
(в)
рН-7.5
ОФХ
(г)
N , нл РН"5-°
л nrtl ИПХ (R-)
Время (мин)
N
НА,
В
IAJ
НА,
Время (мин)
НА,
±л
|НАЭ
М Л^г
О 2
Время (мин)
Рис. 7.13. Пример разделения ионного образца с
изменением рН подвижной фазы и концентрации
ион-парного реагента. Образец: В — псевдоэфедрин,
N — гваяколат глицерина (нейтральное соединение,
на хроматограмме — заштрихованный пик), НА| —
бензоат натрия, HAj — метилпарабен (фенол).
Условия: колонка Cj 150 к 4,6 мм, 5 мкм, подвижная фаза
30% метанол — цитратный буфер (130 мМ гексан-
сульфоната добавлено в подвижную фазу при ИПХ-
разделении), 50 °С, скорость потока 3,0 мл/мин [51]
При высоком рН без ИПР (в):
НА, < N < В < НА2.
При промежуточном рН с ИПР (г):
НА, < N < НА2 < В.
Самое лучшее разделение этого образца достигается при рН 5,0 с добавлением ИПР
(рис. 7.1 Зг).
Резюмируя, можно сделать вывод, что подбор концентрации и типа ИПР
позволяет постепенно изменять степень образования ионных пар, предсказуемым образом
влияя на диапазон удерживания и относительное удерживание. При добавлении
в подвижную фазу анионного ИПР (например, алкилсульфоната), удерживание
основных соединений возрастает, а нейтральных, и особенно кислотных, —
снижается. Когда используется катионный ИПР (например, соль тетраалкиламмония),
удерживание ионизированных кислот увеличивается, а нейтральных и особенно
основных соединений — уменьшается. Чем больше концентрация ИПР в подвижной фазе,
тем существеннее изменения удерживания. Изменения значения рН подвижной
фазы, способствующие ионизации образца, повышают влияние ИПР на разделение.
7.4.2. Разработка метода
Принцип разработки метода разделения в ИПХ и ОФХ ионных образцов
(раздел 7.3.3) аналогичен. Для разработки метода все так же актуальны семь этапов,
перечисленные на рис. 6.2\а для нейтральных образцов. Только третий этап («выбор
условий разделения») отличается в случае ИПХ. При выборе условий разделения.
372 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^1 хроматография
как и в процессе разработки метода в ОФХ, рекомендуется придерживаться
следующей последовательности:
1) выбор начальных условий;
2) подбор %В, при котором I <к < 10 (с ИПР в подвижной фазе);
3) корректировка условий для достижения оптимальной селективности и
разрешения;
4) подбор колонок разной геометрии в поисках наилучшего компромисса между
разрешением и продолжительностью анализа.
Авторы этой книги рекомендуют сначала провести ОФХ-разделение без
добавления ИПР, в том числе исследовать влияние изменений рН подвижной фазы. Воз
можно, в результате удастся определить природу разделяемых веществ, отнеся
выходящие пики к кислотам, основаниям или нейтральным соединениям (как в примере
на рис. 7.3). Такой подход позволяет рассмотреть возможность ОФХ-разделения
без ИПР с подвижной фазой более простого состава и меньшей вероятностью столк
нуться с проблемами, сопряженными с ИПХ (раздел 7.4.3). Даже если с самого
начала разработки метода понятно, что без ИПХ не обойтись, первые эксперименты
нужно проводить так, как описано в разделе 7.3.3 (разработка метода ОФХ-разделе-
ния ионных образцов) без добавления ИПР в подвижную фазу. В этих
экспериментах можно будет подобрать приблизительный %В с приемлемым интервалом
удерживания (например, 1 < к < 10) или, по крайней мере, тот %В, при котором среднее
значение к находится в этом интервале. Как и в случае разработки обращен но-фазо-
вого метода разделения ионных образцов только первый и третий из перечисленных
этапов отличаются для ион-парной хроматографии от разработки метода разделения
нейтральных образцов.
7.4.2.1. Выбор начальных условий (этап 1)
Буферы и ИПР лучше растворяются в метаноле, поэтому желательно использовать
его в качестве растворителя В (хотя ТФУК и ГФМК хорошо растворяются и в ацето-
нитриле, но они являются слабыми ИПР). Если предварительные эксперименты
с ОФХ-разделениями (рекомендованные авторами книги) проводятся с ацетонитри-
лом и, если впоследствии возникают проблемы с растворимостью в этом
растворителе, то его лучше заменить метанолом (руководствуясь номограммой силы
растворителя на рис. 6. II). В большинстве случаев для образцов, содержащих основания,
в качестве ИПР выбирают алкилсульфонат, а для кислотных образцов используют
соль тетраалкиламмония. Если в образце содержатся и кислоты, и основания, любой
из этих ИПР может оказаться полезным, но не рекомендуется добавлять сразу два
реагента в подвижную фазу. Эти ИПР склонны образовывать между собой ионные
пары, при этом не оказывая какого-либо влияния на разделение. Использование
двух ИПР также усложнит разработку метода. Если необходимо селективно
увеличить удерживание оснований, то предпочтение следует отдать алкилсульфонату,
если же кислот — то соли тетраалкиламмония.
Для разделения смеси кислот и оснований лучше всего начать с низкого рН
подвижной фазы с добавлением алкилсульфоната. При низком рН подавляется
ионизация кислот, а ИПР способствует удерживанию оснований. Окончательный выбор
одного или другого ИПР можно будет сделать, исходя из результатов предварительных
ОФХ-разделений, в частности, зависимости удерживания от рН подвижной фазы.
И сульфонаты, и четвертичные аммониевые соли подходят для УФ-детектирования
на длине волны 210 нм и выше.
При обсуждении рис. 7.12я в разделе 7.4.1.2 указано на то, что эффективность
разделения будет одинаковой при использовании разных концентраций алкилсуль-
фонатов (или четвертичных аммониевых солей) разных видов, например, более низ-
7.4. Ион парная хроматография (ИПХ)
кой концентрации Cg-сульфоната или более высокой концентрации С^-сульфоната.
В общем случае это правильно |47, 48], но на практике возникает вопрос: какой
лучше использовать ИПР и в какой концентрации в первом ион парном разделении?
На рис. 7.14 представлена общая схема, позволяющая получить приблизительный
ответ на этот вопрос |49|: графики зависимости рекомендуемых начальных
концентраций разных сульфонатов (рис. 7.14а) или четвертичных аммониевых солей
(рис. 7.146) от доли метанола в подвижной фазе. Например, в подвижную фазу,
содержащую 40% метанола, планируется добавить алкилсульфонат. Судя по графику
на рис. 7.14я, подходящей начальной концентрацией ИПР будет 75 мМ октансуль-
фоната (Cg) или 15 мМ декансульфоната (Сю). В случае добавления тетраалкиламмо-
ниевых солей (рис. 7.146) для подвижной фазы с 40% метанола рекомендуется 70 мМ
соли ТБА (С4) или 20 мМ соли тетрапентиламмония (С5). Желательно выбрать
начальную концентрацию ИПР в интервале 5—100 мМ (на рис. 7.14а,б эти значения
лежат в неокрашенных областях).
Начальные концентрации ИПР, рекомендованные на рис. 7.14, будут
достаточны, чтобы покрыть около трети поверхности сорбента. Последующее увеличение
или уменьшение концентрации относительно начального значения позволяет
существенно изменять адсорбцию ИПР неподвижной фазой с предсказуемыми
изменениями относительного удерживания (раздел 7.4.1.2). Графики, изображенные
(а) Рекомендуемые концентрации различных алкилсупьфонатов
100
[R-L
ю
С^-
-
20 40 60 80 100
%МеОН
(б) Рекомендуемые концентрации различных тетраапкипаммониевых солей
100
[Rim
10
' (С,»4 :
| И<СЭ>4
20
40 60
%МеОн
80
100
Рис. 7.14. Рекомендуемые концентрации различных ион-парных реагентов в
зависимости от доли органического растворителя: (а) сульфонатные ИПР, (б)
четвертичные аммониевые ИПР |49|. Окрашенные области диаграмм
соответствуют условиям, не рекомендуемым для разделений
374 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
хроматография
на рис. 7.14, помогут определить концентрацию ИПР, если в качестве растворителя
В используется ацетонитрил, при этом эквивалентное значение %В для метанола
нужно увеличить приблизительно вдвое. Например, если подвижная фаза содержит
20% ацетонитрила, тогда выбирать ИПР и его концентрацию на графике (рис. 7.14)
нужно для 2 х 20 == 40% метанола j50|. To есть ацетонитрил более сильный
растворитель, чем метанол, поэтому более низкое содержание ацетонитрила эквивалентно
более высокому содержанию метанола.
Рассмотрим некоторые принципы, описанные выше, на примере разделения
смеси водорастворимых витаминов с разными ИПР, взятыми в разных концентраци
ях (рис. 7.15). Поскольку образец включает как кислотные (анионы), так и основные
компоненты (катионы), в качестве ион-парного реагента можно было бы
использовать либо сульфонат, либо четвертичную соль аммония. Если выбрать сульфонат,
то согласно схеме рис. 7.14 для подвижной фазы, состоящей из 15% метанола и
буфера (рН 3,2), подходят гексан-, гептан- или октансульфонат. На рисунке 7.15я—в
показаны хроматограммы разделений образца с разной концентрацией гексансуль-
фоната. Удерживание пиков 1—3 практически не изменяется, а значит, не зависит от
концентрации реагента, поэтому вещества, соответствующие этим пикам, можно
отнести к нейтральным соединениям. С увеличением концентрации ИПР удерживание
пиков 4 и 6 увеличивается, следовательно, это катионы (протонированные основа
ния или четвертичные аммониевые соединения). В противоположность этому
удерживание пиков 5 и 7 уменьшается при увеличении концентрации ИПР, и можно
(а)
(б)
(в)
гексансульфонат
2
3|4
5мМ
*А
т г г ^~^ 1 1
''■■-. 4 / 6
Время (мин)
\ l
\ '10мМ
\
5 7Ja6
4 ) е
Время (мин)
20 мМ'
4 6
Время (мин)
8 0
8 0
гвптансульфонат
1.3 мМ
г
11
в
2.5 мМ
^vLr£_
2 4 6
Время (мин)
5мМ
UL
«
т 1 т т 1
4 6 8
Время (мин)
(в)
(в)
4 6 8 10
Время (мин)
12
Рис. 7.IS. Ион-парное разделение водорастворимых витаминов с использованием
разных ИПР различной концентрации. Образец: 1 — аскорбиновая кислота.
2 —витамин ВЗ, 3 — никотинамид, 4 — витамин В6, 5 — фолиевая кислота,
6 — витамин В1, 7 — витамин В2. Условия: колонка Cs 83 х 4,6 мм, 3 мкм,
подвижная фаза 15% меганол-буфер (рН 3,2), 35 °С, скорость потока
2,0 мл/мин. Хроматограммы получены поданным работы [481
7.4. Ион парная хроматография (ИПХ) 375
предположить, что это анионы (ионизированные кислоты). Из этих первых
экспериментов следует, что концентрация гексансульфоната 6—7 мМ обеспечила бы
максимальное разрешение пиков 4—7 (пик 5 будет находиться посередине между пиками 4
и 6, при этом пик 7 не приблизится слишком близко к пику 6).
На рис. 7.15г—е показаны соответствующие разделения с гептансульфонатом;
в каждом из них концентрация ИП Р уменьшена в 4 раза по сравнению с
концентрацией гексансульфоната. Сравнивая хроматограммы а и ■?, 6 и д, в и е, можно сказать, что
они сходны, но не идентичны. То есть в принципе, можно получить одинаковое
разделение того же самого образца с более низкой концентрацией более гидрофобного
ИПР, но при этом селективность будет несколько иной. Поэтому не рекомендуется
произвольно заменять один ион-парный реагент другим в уже разработанной методике.
В ИПХ вместо обычных алкансульфонатов используют неорганические ион пар
ные реагенты (или «хаотропы»), например, ClOj, BF4", PF6" [52, 53]. Так как
неорганические реагенты удерживаются слабее, вполне вероятно, что механизм их
удерживания описывается уравнением (7.9) и соответствует образованию ионных пар
в подвижной фазе, а не уравнением (7.9а), предполагающим сорбцию ион-парного
реагента. Преимущество хаотропов заключается в том, что они больше подходят
для градиентного режима (меньше шум и дрейф базовой линии) и лучше
растворяются в подвижной фазе с высоким содержанием органического растворителя.
Относительная способность разных анионов (включая и буферы, и ИПР) образовывать
ионные пары увеличивается в ряду:
Н2Р04 < НСОО- < CH3SO, < CI- < NOj « CF3COO- < BF4 < СЮ4 < PF6".
В ИПХ используются только последние четыре аниона. Поскольку неорганические
ион-парные реагенты (хаотропы) слабо удерживаются на сорбенте, может быть,
процесс уравновешивания колонки при смене подвижной фазы проходит быстрее
(раздел 7.4.3.2). Под действием хаотропов удерживание протонированных оснований
при использовании ацетонитрила изменяется гораздо больше по сравнению с
метанолом или тетрагидрофураном |54|.
7.4.2.2. Управление селективностью (этап 3)
В ион-парной хроматографии регулировать селективность можно с помощью:
• РН;
• типа ИПР (сульфонат, четвертичная аммониевая соль, хаотроп);
• концентрации ИПР;
• доли органического растворителя (%В);
• типа органического растворителя (ацетонитрил, метанол и т.д.);
• температуры;
• типа сорбента;
• типа буфера и его концентрации.
Несмотря на то, что для управления селективностью в ИПХ есть множество
способов, обычно во время разработки метода требуется исследовать только некоторые
из них. Кроме того, влияние некоторых вышеперечисленных условий на
удерживание взаимосвязано. Так в некоторых случаях изменение рН подвижной фазы даст
такой же результат, как и изменение концентрации ИПР; например, повышение рН
или концентрации алкилсульфоната приведет к увеличению удерживания оснований
(или уменьшению удерживания кислот). Кроме того, мы уже убедились, что
основное воздействие изменения %В или температуры может быть обусловлено
связанными с этим изменениями «эффективного» рН подвижной фазы, а следовательно,
вызывать такие же изменения относительного удерживания, как и изменение рН.
Другие примеры такого рода приведены ниже.
#376
Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^? хроматография
рН подвижной фазы и тип или концентрация ион-парного реагента. Совместное
влияние этих условий на ион-парное разделение подробно рассматривалось выше
в разделе 7.4.1. Его лучше изучить в первую очередь в процессе разработки метода.
Удобнее изменять концентрацию ИПР (как на рис. 7.15а—в), а не его тип (сравните
рис. 7.15е и а).
Cu.ta органического растворителя. Когда в ОФХ-разделениях ионных образцов
варьируется %В (раздел 7.3.2.2, рис. 7.7), можно ожидать изменений как
абсолютного, так и относительного удерживаний. В некоторых случаях эти изменения могут
быть связаны с соответствующими изменениями «кажущихся» значений рН
подвижной фазы (или значений рКа анализируемых веществ, раздел 7.2.3).
Соответствующие изменения относительного удерживания, зависящие от %В, должны быть и
в ИПХ, но с дополнительными особенностями. Так, если увеличивается %В,
снижается адсорбция сорбентом и ИПР, и молекул образца. Следовательно, изменение %В
приведет к ожидаемым изменениям относительного удерживания пиков, на которые
сильно влияет образование ионной пары. Пример приведен на рис. 7.16, где
показано разделение того же образца, что и на рис. 7.10. Хроматограмма первого
разделения с 40% метанола и октансульфонатом в качестве ИПР представлена на рис. 7.16д.
Пики четырех протонированных оснований (X, Х|—Xj) заштрихованы, остальные
пики соответствуют кислотам или нейтральным соединениям. Когда повышается
доля органического растворителя до 45% МеОН (рис. 7.166) и до 50% МеОН
(рис. 7.16в), удерживание всех пиков снижается (под влиянием силы растворителя),
но у четырех оснований удерживание становится еще меньше относительно
остальных нейтральных и кислотных соединений (заштрихованные пики смещаются в сто-
40% МеОН (а)
*w\nn
( ! 1 ! г1 '1 Г 1 -1 1 Г
8 10 12 14 16 18
Время (мин)
45% МеОН (б)
ГБ
МП
i II А I А* А** а*з X
пп
| 1 1 1 Г "1 Г —] Г Г ~f"
0 2 4 6 8 10
Время (мин)
X
Имп
50% МеОН (в)
ГБ . /11 *Х1АХг _^Х3 А
0 2 4 6
Время (мин)
Рис. 7.16. Зависимость селективности ион-парного разделения от доли органического
растворителя. Образец и условия те же, что и в ранее представленном
разделении (рис. 7.105), кроме доли метанола, которая указана на хроматог-
раммах. Пики протонированных оснований X, Х|— Хз заштрихованы [451
7.4. Ион парная хроматография (ИПХ) 377
рону начала хроматограммы). Такое поведение оснований предсказуемо, так как
увеличение доли органического растворителя приводит к снижению удерживания ИПР
(R~) на сорбенте. Уменьшение концентрации R- на сорбенте приводит к тому, что
катионы X, Х|—Хз образуют меньше ионных пар, следовательно, их удерживание
становится еще слабее помимо общего снижения удерживания всех пиков при
увеличении %МеОН. Изменение %МеОН в этом примере демонстрирует исключительное
влияние изменения этого параметра на положение пика на хроматограмме и его
разрешение в ИПХ (обратите внимание на оптимизированное разделение
при 45% МеОН на рис. 7.166).
Тип растворителя. Замена одного органического растворителя другим приводит
к изменению селективности разделения в ИПХ как в ОФХ-разделениях нейтральных
образцов (раздел 6.3.2, рис. 6.9) и особенно ионных образцов (раздел 7.3.2.3).
Из-за дополнительного влияния растворителя В на адсорбцию ИПР сорбентом
изменение типа растворителя может повлечь еще большее изменение селективности, чем
в ОФХ. На рис. 7.17 изображен убедительный пример влияния замены растворите
ля В на селективность в ИПХ. Используя данные номограммы рис. 6.11 для
получения одинаковой силы растворителя, метанол (рис. 7. 17й) был заменен
эквивалентным количеством ацетонитрила (рис. 7.176). Совокупное изменение рН подвижной
фазы, концентрации ИПР и типа растворителя должно быть особенно эффективно
при разделении сложных ионных образцов |48, 50|.
Температура. Изменение температуры в ИПХ также должно существенно влиять
на относительное удерживание. Температура влияет на количество ИПР,
удерживаемого неподвижной фазой. Поэтому во время ион парного разделения особенно важно
контролировать температуру.
Тип сорбента и буфера. Ранее мы уже убедились, что вид сорбента оказывает
значительное влияние на селективность ОФХ-разделений ионных образцов.
Представляется вполне вероятным, что это справедливо и для разделений в ИПХ. Хотя
частичное покрытие поверхности сорбента ИПР может маскировать вклад собственно
сорбента в удерживание образца. Так как существует много других способов регули-
10% МеОН Л 1 <а>
1
2 4
Время (мин)
0 2 4 6
Время (мин)
Рис. 7,17. Зависимость селективности от типа растворителя в ион-парном разделении
катехоламинов. Образец: 1 — норадреналин, 2 — адреналин, 3 — октопа-
мин, 4 — 3,4-дигидроксифенилаланин, 5 — допамин, 6 — изопренол, 7 —
тирозин. Условия: колонка C|g 150x4,6 мм, 5 мкм, фосфатный буфер
(рН 2,5) с 2 мМ октансульфоната, 25 °С, скорость потока 1,0 мл/мин [50)
378 Глава 7. Ионные образцы: обращенно фазовая, ион парная и ионообменная
э^^ хроматография
ровать селективность в ИПХ, подбор для этой цели определенного вида сорбента
вряд ли стоит рассматривать в первую очередь, да это и не представляется особенно
перспективным. Таким же образом влияние вида и концентрации буфера
на ион-парное разделение может быть значительным, но гораздо более
существенные и предсказуемые изменения селективности можно получить при варьировании
рН, %В, температуры и/или типа или концентрации ИПР.
7.4.2.3. Выводы
Разрабатывая методику ион-парного разделения, рекомендуется придерживаться
следующей последовательности действий:
1) выбрать начальные условия для обращенно фазового разделения (раздел 7.3.3.1);
2) подобрать, как и в ОФХ-разделении, такое значение %В, чтобы определить
приемлемый интервал удерживания (например, 1 < к < 10);
3) изменять рН подвижной фазы, чтобы экспериментально определить, какие пики
на хроматограмме соответствуют кислотным, основным или нейтральным
соединениям (если анализируемые вещества не индентифицированы по стандартам);
4) на некотором этапе дальнейшей разработки обращенно-фазового метода
разделения рассмотреть целесообразность или необходимость применения ИПХ
(раздел 7.4);
5) если выбрана ИПХ, подобрать ИПР и его начальную концентрацию
(раздел 7.4.2.1).
Известный состав образца или предшествующие эксперименты с изменением рН
(этап 3) должны указать, какой ИПР лучше выбрать: соль четвертичного аммония
(если надо увеличить удерживание кислотных соединений) или алкилсульфонат
(для основных соединений или катионов). С другой стороны, для увеличения
удерживания основных соединений можно использовать хаотропы.
6) оптимизировать относительное удерживание (селективность).
Предполагается, что наиболее эффективно одновременно изменять %В и
температуру. Для дальнейшего изменения селективности можно варьировать рН
подвижной фазы и концентрацию ИПР (например, как на рис. 7.13, 7.15). Если требуется
дополнительная оптимизация селективности (что маловероятно), изменить другие
условия разделения, перечисленные в начале раздела 7.4.2.2;
7) чтобы дополнительно улучшить разрешение или оптимизировать
продолжительность анализа, подбирают параметры колонки.
7.4.3. Нестандартные проблемы
К разделениям ионных образцов методом ИПХ применимы некоторые из
требований, предъявляемых к ОФХ:
• необходимость в некоторых случаях точного контроля рН подвижной фазы
(например, +0,10 единиц или еще точнее);
• необходимость контроля температуры (в ИПХ важнее, чем в ОФХ).
Кроме того, в ИПХ есть некоторые проблемы, которые либо отсутствуют в ОФХ,
либо чем-то отличаются от проблем ОФХ:
• разделения в ИПХ сложнее проводить и интерпретировать;
• возможны ложные пики;
• медленное уравновешивание колонки после замены подвижной фазы;
• трудно объяснимое искажение формы пиков.
Ранее уже отмечалось, что разделения в ИПХ сложнее проводить, чем в ОФХ.
В ИПХ больше переменных, которые нужно учитывать при разработке метода и кон-
7.4. Ионпарная хроматография (ИПХ) 379
тролировать в рутинных анализах. Хотя сложность проведения ион-парных
разделений можно преодолеть, все-таки остается некоторая запутанность, делающая этот
метод не столь привлекательным. С другой стороны, размывание заднего фронта
пика протонированных основных соединений в ИПХ менее вероятно, чем в ОФХ
(слабее ионное отталкивание соседних молекул ВН+ на сорбенте). Кроме того, ИПХ
предоставляет больше возможностей для оптимизации относительного удерживания
ионных образцов (если возникает необходимость).
7.4.3.1. Ложные пики
Иногда во время холостого разделения (когда вместо образца в колонку вводится
подвижная фаза) в ИПХ наблюдаются как положительные, так и отрицательные пики.
Эти ложные пики могут препятствовать разработке метода и мешать в рутинных
анализах. Поэтому следует проводить холостые разделения и при разработке метода, и
при последующих рутинных анализах во избежание путаницы, вызванной ложными
пиками.
Появление ложных пиков обычно вызвано различиями состава подвижной фазы
и растворителя образца. Эту проблему могут усугубить недостаточная чистота ИПР,
буферов или других добавок к подвижной фазе. Общее правило в ИПХ — добиваться
как можно более точного соответствия состава подвижной фазы и растворителя
образца (вводя при необходимости в образец ИПР в нужной концентрации).
Рекомендуется также вводить меньший объем образца (например, < 25 мкл, если это
возможно). Если проблема ложных пиков сохраняется, то стоит попробовать ИПР из другой
партии или другого производителя. Общую информацию о ложных пиках можно
найти в разделе 17.4.5.2 или в 155, 56|.
7.4.3.2. Медленное уравновешивание колонки
При переходе к новой подвижной фазе необходимо для уравновешивания промыть
колонку достаточным ее объемом (раздел 2.7.1). В ИПХ и адсорбция ИПР сорбентом, и
его десорбция при определенных обстоятельствах могут происходить медленно, что
приводит к неполному уравновешиванию колонки новой подвижной фазой. Поэтому
важно подтверждать воспроизводимость удерживания образца при замене подвижной
фазы, если в состав старой или новой подвижной фазы входит ИПР. Для
полноценного уравновешивания колонки новой подвижной фазой может потребоваться несколько
часов (см. пример ниже). Уравновешивание может происходить особенно медленно,
если ИПР более гидрофобен (сравните, например, декансульфонат и октансульфонат),
или, когда происходит уравновешивание колонки типа А подвижной фазой,
содержащей четвертичные аммониевые соли |47|. Когда требуется заменить ИПР, необходимо
сначала вымыть старый ИПР из колонки специальным промывочным раствором
(см. ниже), а затем уравновешивать колонку подвижной фазой нового состава.
Для удаления из колонки анионных ИПР (например, алкилсульфонатов), ее
промывают 50—80% раствором метанола в воде. Четвертичные аммониевые соли
вымываются из колонок типа Ас помощью смеси 50% метанола с буфером (например, 100 мМ
фосфатом калия с рН 4—5). Фосфат калия необходим для подавления взаимодействий
между четвертичными аммониевыми группами и ионизированными силанольными
группами сорбента. В любом случае колонку сначала надо промыть 20 колоночными
объемами промывочного раствора, а потом уже проверять воспроизводимость
удерживания (т.е. полноту уравновешивания колонки) с новой подвижной фазой.
Первое уравновешивание колонки подвижной фазой с ИПР может оказаться
неожиданно медленным. Для разделения, показанного на рис. 7.106, сначала
предполагалось, что колонка уравновесилась после промывания 20—30 колоночными
объемами подвижной фазы |46|, так как при повторных вводах образца не наблюдалось
380 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
э^1 хроматография
существенного изменения времен удерживания. Однако, когда позже провели
несколько последовательных анализов в течение длительного периода времени,
оказалось, что медленное снижение удерживания оснбвных соединений X—Хз
происходило в течение II часов. Это указывает на очень медленное уравновешивание колонки.
Чтобы избежать 12-часового уравновешивания колонки перед каждой новой серией
анализов, пришлось оставлять колонку заполненной подвижной фазой с ИПР после
завершения каждой серии анализов. Такой прием позволяет гораздо быстрее
уравновешивать колонку перед началом серии анализов, и это та процедура, которую
рекомендуют авторы этой книги, когда в течение нескольких дней выполняются
одинаковые разделения. Хотя, если колонку хранить в такой подвижной фазе, срок ее
службы сокращается (раздел 5.8).
Медленное уравновешивание колонки подвижной фазой с более гидрофобными
ИПР может вызвать проблемы при использовании градиентного режима.
Удерживание может хуже воспроизводиться, базовая линия может быть нестабильной,
возможны и другие сложности. Поэтому ион-парные разделения не рекомендуется
проводить в градиентном режиме, особенно если в подвижную фазу добавлен более
гидрофобный реагент. Исключения можно сделать для слабого ИПР трифторуксус-
ной кислоты (ТФУК) и хаотропов ClOjJ.BEj" и PF<i~, поскольку все они в меньшей
степени замедляют уравновешивание колонки. Для приемлемого уравновешивания
колонки подвижной фазой, содержащей трифторуксусную кислоту или хаотропные
реагенты, обычно достаточно пропустить 10—20 колоночных объемов. По этой и
многим другим причинам популярность этих ион-парных реагентов растет.
Наконец, из-за медленного уравновешивания колонки подвижной фазой с ИПР
возможно, что не весь ИПР будет вымываться из колонки даже после активной
промывки. Поэтому авторы книги рекомендуют не использовать в ОФХ без ИПР колонки,
на которых ранее проводились ион-парные разделения (исключения составляют трифто-
руксусная кислота и хаотропы). В колонке могут остаться следовые количества ИПР,
что может привести к различиям в селективности, которая не будет
воспроизводиться после замены колонки на новую. Впрочем, замена одного ИПР на другой может и
не вызывать такие сложности.
7.4.3.3. Искажение формы пиков
В ИПХ обычно не происходит размывание заднего фронта пиков оснований,
поскольку разделения проводятся на колонках типа В, к тому же алкилсульфонаты
дополнительно минимизируют влияние силанольных групп (ИПР конкурирует с
ионизированными силанольными группами во взаимодействиях с протежированными
основаниями). Согласно некоторым исследованиям, размывание переднего фронта
пика в ИПХ можно устранить, повысив температуру колонки [57|. В другой работе
|45|, напротив, было показано, что при более низкой температуре пик приобретал
более правильную форму. В обоих этих исследованиях использовались колонки типа А.
Резонно предположить, что при разделении на колонках типа В в условиях ИПХ
пики будут иметь более правильную форму. Если в ИПХ наблюдаются пики
неправильной формы и/или низкое число теоретических тарелок, попробуйте изменить
температуру (повысить или понизить).
7.5. Ионообменная хроматография (ИОХ)
Ионообменная хроматография (ИОХ) — важный метод разделения, но в настоящее
время он имеет ограниченное применение:
• для разделения смесей неорганических ионов (ионная хроматография);
7.5. Ионообменная хроматография (ИОХ) 381
• для разделения биомолекул, включая аминокислоты, пептиды, белки и особенно
олигонуклеотиды (разделы 13.4.2, 13.5.1);
• для разделения углеводов (раздел 13.6.3);
• для разделения карбоновых кислот;
• для пробоподготовки (глава 16);
• в двумерных разделениях (разделы 9.3.10, 13.4.5).
Кроме того, побочные электростатические взаимодействия ионов могут вносить
свой вклад в разделения ионных образцов в условиях ОФХ (уравнение 7.7, раздел 5.4.1).
Когда в конце 1960-х годов ВЭЖХ стала общедоступным методом, ИОХ была
кандидатом, имеющим большие шансы на успех в качестве метода анализа любых
смесей, содержащих органические кислоты и основания. Наряду с жидкостной
распределительной и адсорбционной хроматографиями ИОХ использовалась для
проведения большинства разделений в ВЭЖХ. Однако, с тех пор для анализа небольших
молекул (с молекулярной массой < 1000 Да) стала в основном применяться ОФХ.
Снижение популярности ИОХ обусловлено несколькими причинами: (I) меньшим
количеством теоретических тарелок, чем в ОФХ; (2) лучшей осведомленностью
пользователей о разделении в режиме ОФХ; и (3) большей сложностью ИОХ-разделений
по сравнению с ОФХ разделениями. Кроме того, ВЭЖХ-оборудование в меньшей
степени приспособлено для условий ионообменной хроматографии (высокие
концентрации соли в подвижной фазе, некоторые соли, например, галогениды,
вызывают коррозию нержавеющей стали и т.д.).
Сначала мы дадим краткий обзор областей применения ИОХ, упомянутых выше,
а потом обсудим общие принципы ионообменных разделений.
Ионная хроматография стала основным направлением ИОХ с тех пор, как она
была разработана Смоллом в 1974 году. До этого смеси неорганических ионов
анализировали другими трудоемкими методами, требовавшими специального оборудования,
возможности которого были ограничены. Попыткам разделения неорганических
соединений с помощью ВЭЖХ препятствовало, главным образом, отсутствие достаточно
чувствительного детектора. В ионной хроматографии удалось решить эту проблему
с помощью кондуктометрического детектора с подавлением ионов. В этой книге тема
ионной хроматографии рассматриваться больше не будет, но читателю
предоставляются ссылки на литературу для более подробного изучения этого метода |59—611.
Примерно за 10 лет до внедрения ВЭЖХ биомолекулы разделяли с помощью
ИОХ. В 70-х годах появились ВЭЖХ-колонки, которые позволили методом
ионообменной хроматографии разделять аминокислоты, пептиды, белки, нуклеотиды,
олигонуклеотиды и нуклеиновые кислоты с высокой скоростью и большим
разрешением. Об этом важном применении ИОХ более подробно сказано в главе 13.
ВЭЖХ-разделение углеводов можно проводить либо с помощью хроматографии
гидрофильных взаимодействий (ГИХ, раздел 8.6), либо анионообменной
хроматографии (раздел 7.5.7). Ниже будет показано, что, как правило, предпочитают
использовать анионообменную хроматографию с амперометрическим детектором, особенно
тогда, когда требуется высокая чувствительность детектирования. Кроме того, в
разделе 13.6.3 описан анализ фрагментов углеводов, образующихся при расщеплении
гликозилированных белков.
Карбоновые кислоты часто разделяют на ионообменных колонках с помощью
ионной эксклюзии. В качестве подвижной фазы используют подкисленную воду
для подавления ионизации образца. Такие разделения основываются не на ионном
обмене, а на распределении, напоминающем процесс, лежащий в основе ОФХ.
Пример такого разделения приведен на рис. 7.18. Относительно гидрофильные образцы
такого рода слабо удерживаются на большинстве обращенно-фазовых колонок, а
более полярные ионообменные колонки обеспечивают большее их удерживание с
приемлемыми значениями к. Для анализа образцов, содержащих карбоновые кислоты,
7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
хроматография
"Г
8 10
Время (мин)
12
14
16
Рис. 7.18. Разделение смеси карбоновых кислоте помощью ион-экслюзионной
хроматографии. Образец: 1 — щавелевая, 2 — малеиновая, 3 — лимонная, 4 —
винная, 5 — глюконовая, 6 — яблочная, 7 — янтарная, 8 — молочная, 9 —
глутаровая, 10 — уксусная, 11 — левулиновая, 12 — пропионовая. Условия:
катионообменная колонка 300 х 7,8 мм, 9 мкм, подвижная фаза 0,006 N
серная кислота, 65 °С, скорость потока 0,8 мл/мин |57]
остается популярной ион-экслюзионная хроматография на ионообменных колонках,
как показывают примеры, приведенные в |62|. Более подробно влияние
экспериментальных условий ион-экслюзионной хроматографии на разделение карбоновых
кислот обсуждается в [63|.
Пробоподготовка (раздел 16.6.5.1) остается очень важной сферой приложения
ИОХ, хотя и является низкоэффективной предварительной процедурой,
предшествующей обращенно-фазовым и другим разделениям. Чтобы эффективно использовать
ИОХ при пробоподготовке, требуется понимание основ зависимости удерживания
от условий разделения. Об этом кратко рассказано в этом разделе.
Двумерные разделения, уже упоминавшиеся в разделе 2.7.3, обсуждаются также
в разделах 9.3.10 и 13.4.5. Этот метод предназначен для разделения очень сложных
образцов, содержащих слишком много компонентов, чтобы их можно было
разделить в процессе одного анализа ВЭЖХ. Принцип этого метода заключается в
следующем: сначала проводят частичное ВЭЖХ-разделение образца. Полученные в
результате разделения фракции сразу же направляются на вторую колонку
для выделения индивидуальных соединений с более высоким разрешением. Если
первым проводится разделение в режиме ИОХ с водной подвижной фазой, то
полученные фракции можно вводить непосредственно во вторую ОФХ-колонку.
7.5.1. Основы удерживания
Ионообменные разделения проводятся на колонках, заполненных сорбентами,
содержащими ионизированные или ионизируемые группы (раздел 7.5.4). Например,
катионообменная колонка, на которой благодаря электростатическим (ионным)
взаимодействиям удерживаются протонированные основания (ВН+), может содержать
сульфонатные группы -SO^, имеющие противоположный заряд. Аналогичным образом
анионообменный сорбент, удерживающий анионы кислот (А~), может содержать
четвертичные аммониевые группы такие, как -г<1(СНз)з- Удерживание в ИОХ
обусловлено конкуренцией между ионами образца и противоионами подвижной фазой во
взаимодействии с противоположно заряженными ионными группами сорбента.
Удерживание в ИОХ можно проиллюстрировать с помощью удерживания,
обусловленного обменом противоиона К+ на катион протонированного основания ВН+:
ВН+ +R"K+ oK+ + RBH+. (7.10)
Здесь R- — это анионная группа, присоединенная к сорбенту (например, -SOj"),
которая может связываться либо с ионом образца ВН'1", либо с противоионом
подвижной фазы К'1" благодаря кулоновскому притяжению. Уравнение (7.10) можно обоб-
7.5. Ионообменная хроматография (ИОХ) 383
щить как для кислотных, так и основных ионов образца, имеющих абсолютный
заряд \z\ = т (например, полностью ионизированная щавелевая кислота "СОО-СОО"
с z = -2, т = 2). Для образца, содержащего катион Х+/и и противоион Y+ удерживание
можно описать так:
X+ffl+m(R-Y+) «. X+mR^ + mY+. (7.II)
Здесь группа R~ — это анионная группа сорбента R- (например, —SOJ). Для образца,
содержащего анион Х~т, уравнение (7.11) приобретет вид:
Хт +m(R+Y) <=> XmR+ + тУ~. (7.12)
В ИОХ фактор удерживания к одновалентного противоиона Y+ или Y~
соответственно в катионном или анионном обмене можно определить из равновесия,
описываемого уравнением (7.11) или (7.12):
log к = а - ralog С, (7.13)
где С — молярная концентрация противоиона Y+ или Y" в подвижной фазе, а —
константа, равная log к при С = 1 М, т — абсолютная величина заряда z иона X образца.
Параметры ант — константы определенного соединения для данных условий:
колонки, соли, буфера, рН подвижной фазы и температуры. Уравнение (7.13)
иллюстрирует показанное на рис. 7.19 анионообменное разделение четырех полифосфатов,
имеющих зарядом z -3, -4, -6 и -8 (соответственно три-, тетра-, гекса- и октафос-
фат). Многочисленные примеры (приведенные, например, в |64, 65|) подтверждают
справедливость уравнения (7.13) в условиях изократической ИОХ.
В уравнениях (7.10)—(7.13) ионный обмен рассматривается как стехиометриче-
ский процесс, в котором одна молекула удерживаемого вещества замещает опреде-
2.0
1.5
- 1.0
0.5
-0.5 -0.4 з
togC
Рис. 7.19. Зависимость log* от концентрации противоиона (log С) в изократической
ИОХ. Образец — смесь четырех полифосфатов (указаны на рисунке).
Условия: анионообменная колонка TSKgel SAX, подвижная фаза — водный
раствор КС1 с EDTA рН 10,2, 30 °С |64]
384 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
хроматография
ленное количество противоионов, прочно удерживаемых благодаря
электростатическим взаимодействиям с отдельными зарядами на поверхности сорбента. Хотя
практические случаи эти уравнения вполне адекватно описывают, они все-таки
несколько упрощают реальный процесс ионного обмена. Фундаментальная природа
ионообменного взаимодействия и теория обусловленного этим процессом
удерживания подробно изложена в [66|.
7.5.2. Роль противоиона
В ИОХ обычно используется водная подвижная фаза, содержащая буфер для
контроля рН и соль (или противоион) для управления удерживанием образца (т.е. элюиру-
ющей силой подвижной фазы). Поскольку вещество обычно удерживается на
ионообменных сорбентах не только в результате ионных, но и ОФХ-взаимодействий,
добавление метанола или ацетонитрила в подвижную фазу дополнительно
увеличивает ее элюирующую силу. Тем не менее, удерживание контролируется главным
образом, изменением концентрации противоиона (С): с увеличением С удерживание
ионизированного вещества ослабевает (уравнение 7.13). У одновалентных
соединений с т = 1 увеличение С приводит к пропорциональному уменьшению значений к.
Если заряд разделяемого вещества т больше, то с увеличением С к будет
уменьшаться быстрее. Если же два вещества в образце имеют разные заряды т, то изменение С
приведет к изменению относительного удерживания (с возможной сменой порядка
элюирования веществ). Пример такой зависимости показан на рис. 7.20 для того же
образца, что и на рис. 7.19. По мере возрастания концентрации иона CI от 0,32
до 0,35 и 0,40 М снижается время удерживания каждого пика, но при этом
заштрихованный пик, соответствующий октафосфату, перемешается в начало хроматограм-
мы, т.е. меняется порядок элюирования (сравните рис. 7.20 и 7.19).
0.32М KCI
20
Время (мин)
(в)
0.35М
20
Время (мин)
(в)
0.40М
/V
0 10
Время (мин)
Рис. 7.20. Примеры влияния изменения концентрации противоиона (КС1) на
разделение образцов, охарактеризованных на рис. 7.19. Разделения смоделированы
по данным [64|. Предполагается, что используется колонка 150x4,6 мм,
5 мкм, скорость потока = 2,0 мл/мин, N = 1000
7.5. Ионообменная хроматография (ИОХ)
Различные противоионы подвижной фазы более или менее прочно
удерживаются сорбентом, поэтому замена одного противоиона на другой может быть также
полезна для увеличения или уменьшения элюирующей силы растворителя и в целом
изменения удерживания образца. Вообще противоионы с ббльшим зарядом гораздо
эффективнее понижают удерживание образца. Относительная способность иона
образовывать более прочные связи, подавлять удерживание образца и снижать
значение к увеличивается в указанной последовательности:
(анионный F~ (большие значения к веществ) < ОН" < CHjCOO" < С1~ < SCN" <
обмен) < Вг~ < NOJ < 1~ < оксалат2- < SO2,- < цитрат3- (меньшие значения к)
(катионный Li+(большие значения к веществ) <Н+< Na+< NHJ < К+< Rb+<
обмен) < Cs+ < Ag+ < Mg2+ < Zn2+ < Co2+ < Cu2+ < Cd2+ < Ni2+ < Ca2+ <
< Pb2+ <Ba2+ (меньшие значения к)
Замена одного противоиона другим (например, замена СГ ионом NO}") может
также повлиять на параметр а в уравнении (7.13), что, в свою очередь, возможно,
изменит селективность.
7.5.3. рН подвижной фазы
ИОХ обычно используют для разделения кислот или оснований. Так как на сорбенте
удерживаются только заряженные (ионизированные) молекулы благодаря ионному
обмену, значения к одновалентных соединений (т — I) пропорциональны их
ионизации. Например, при понижении рН подвижной фазы полностью ионизированные
кислоты становятся ионизированными наполовину, и тогда значение фактора
удерживания уменьшается вдвое. Аналогичным образом ионизация и удерживание
оснований будут уменьшаться при увеличении рН подвижной фазы. Такая зависимость
удерживания от рН противоположна характерной для ОФХ (рис. 7.11а), но сходна
с зависимостью этих параметров в ИПХ (рис. 7.116).
7.5.4. Колонки для ИОХ
В зависимости от типа ионогенной группы R1, образующей часть неподвижной
фазы, в ИОХ существует четыре основных вида сорбентов: сильные и слабые анио-
нообменники и сильные и слабые катионообменники1. Сильные ионообменники
содержат полностью ионизированные группы R1 во всем обычно представляющим
интерес диапазоне рН (2 < рН < 13). У сильных анионообменников самой
распространенной группой R+ является -N(CH5)3, у катионообменников — R~ это
-SOJ. У слабых ионообменников значения рКа групп R1 находятся в промежуточном
диапазоне (например, 4 < рКа < 10), поэтому ионизация этих групп (а значит, и
ионообменная емкость сорбента) может изменяться в зависимости от рН подвижной
фазы (см. рис. 13.16).
В ИОХ подбирают такое значение рН подвижной фазы, при котором
анализируемое вещество находится в ионизированном состоянии. Это значение может выходить
за рамки интервала 2 < рН < 8, который рекомендован для большинства силикагеле-
вых сорбентов. Поэтому в ИОХ в основном используют сорбенты на полимерной
основе такие, как полимеры метакрилатов или стирол-дивинилбензола.
1 В англоязычной литературе их обычно обозначают: SAX — strong anion exchanger (сильный
анионообменник) и WAX — weak anion exchanger (слабый анионообменник). Например,
ВЭЖХ-колонки ZORBAX SAX или ZEBRON ZB-WAX. SCX — strong cation exchanger (сильный ка-
тионообменник) и WCX — weak cation exchanger (слабый катионообменник). Например.
ВЭЖХ-колонки Spherisorb® SCX или ProPac™ WCX-10. — Прим. перев.
386 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
э^^ хроматография
У слабых анионообменников ионогенными обычно являются аминогруппы
(например, —NH2). Такие сорбенты начинают терять заряд и ионообменную емкость,
когда рН подвижной фазы существенно превышает рКа аминогруппы (5 < рКа < 10,
таблица 7.2). Поэтому слабые анионообменники в основном используются с
кислотными подвижными фазами (рН < 6), которые могут в значительной степени прото-
нировать аминогруппы. Слабые катионообменники в большинстве случаев содержат
карбоксильную группу ( СООН) с рКа ж 5, и ионизация этих сорбентов падает, ког
да рН подвижной фазы становится меньше 7. Слабые катионообменники в основном
используют с оснбвными подвижными фазами (рН > 8). Поскольку ионизацию и
ионообменную емкость слабых ионообменников можно уменьшить, используя
подвижную фазу с нужным рН, с них легче элюировать вещества, прочно
удерживаемые на сильных ионообменниках, с которых может быть затруднительно их
элюировать. Со слабого же ионообменника такие вещества можно элюировать, просто
изменив рН подвижной фазы. Кроме того, у слабых и сильных ионообменников
различная селективность, что также может послужить причиной использования слабых
ионообменников для разделения определенных образцов. Эта тема обсуждается
подробнее в разделе 13.4.2.1.
7.5.5. Роль других условий
Для изменения относительного удерживания и селективности в ИОХ в первую
очередь используют следующие условия:
• тип соли или буфера;
• добавление органического растворителя в подвижную фазу;
• температуру.
Было предложено несколько общих правил для обобщения влияния этих условий
на селективность в ИОХ небольших органических соединений. Однако, известно,
что каждое из них может изменить селективность.
7.5.6. Разработка метода
Обычно целью разделения в ИОХ является удерживание и разрешение смеси либо
анионов (ионизированных кислот), либо катионов (протонированных оснований).
Когда ИОХ используют в пробоподготовке, то подбирают условия селективного
связывания кислот или оснований для последующего ВЭЖХ-анализа. Если целью
является ВЭЖХ-разделение и анализ образца методом ионообменной хроматографии,
то можно разделить и проанализировать либо кислоты, либо основания, но не оба
вида образцов одновременно. Если разделить нужно кислоты, следует выбрать анио-
нообменник, для оснований — катионообменник. Обычно отдают предпочтение
сильным ионообменникам (по крайней мере, для первого эксперимента). Для
определения оптимальных условий разделения в ИОХ, как и при разработке всех методов
ВЭЖХ, эксперименты рекомендуется начинать проводить на новой, ранее не
использовавшейся колонке.
Общий подход для разработки ОФХ-метода (разделы 6.4, 7.3.3) подойдет и
для разработки метода ИОХ. Для первого эксперимента используется водная
подвижная фаза с добавлением подходящего буфера с концентрацией от I до 5 мМ
и какой-либо соли (например, NaCl). В последующих экспериментах методом проб
и ошибок (или с помощью градиентного элюирования) определяют оптимальную
концентрацию соли, при которой будет достигаться желательный интервал
удерживания (например, 1 < к <, 10). Потом можно будет подобрать оптимальную
селективность, как описано выше.
7.5. Ионообменная хроматография (ИОХ)
2
О 2 4 6 8 10 12
Время (мин)
Рис. 7.21. Разделение смеси стандартов углеводов методом анионообменной
хроматографии. Детектирование амперометрическим детектором. Образец: 1 —
л»ио-инозитол, 2 — D-сорбитол, 3 — лактитол, 4 — L-фруктоза, 5 — рамно-
за, 6 — D-галактоза, 7 — D-глюкозамин, 8 — D-глкжоза, 9 — D-манноза,
10 — D-фруктоза, 11 — D-рибоза. Условия: анионообменная колонка
300x4 мм, 5 мкм, подвижная фаза водный 5 мМ NaOH +1 мМ Ва(ОАс>2,
комнатная температура, скорость потока 1 мл/мин [68]
7.5.7. Разделение углеводов
Смеси углеводов можно разделять как методом хроматографии гидрофильных
взаимодействий (ГИХ, раздел 8.6), так и методом ИОХ. Углеводы имеют значения рКа
около 12 и это значит, что на их ионизацию будет влиять высокий рН подвижной
фазы. Это позволит разделить их на анионообменнике. В настоящее время отдают
предпочтение этому методу разделения углеводов, поскольку чувствительность ампе-
рометрического детектора выше (пределы детектирования менее I наномоля |67|),
а также есть возможность влиять на селективность при небольших изменениях рН
подвижной фазы. Пример такого разделения изображен на рис. 7.21 для образца,
состоящего из смеси 11 разных Сахаров. В других исследованиях было показано, что
существенную роль в порядке элюирования пиков и их разрешении играет
температура |69]. Дополнительные данные приведены в разделе 13.6.
7.5.8. Разделения в смешанном режиме
Сорбенты для смешанного режима разделения можно себе представить как
гидрофобные ионообменники в противоположность более гидрофильным сорбентам,
применяемым в традиционной ИОХ. У таких сорбентов сочетаются обращенно-фазовые
и ионообменные свойства. Изначально их стали использовать из-за специфической
селективности, отличающейся от селективности как обращенно-фазовых, так и
ионообменных сорбентов, по этой же причине продолжают использовать и до сих
пор. Пример показан на рис. 7.22, проводится сравнение ОФХ-разделения
(рис. 7.22а) и смешанного катионообменного разделения (рис. 7.226). В качестве
образца взята смесь небольших гидрофильных аминов (азотистый иприт). Очевидно,
что у пиков 1—6, элюируемых первыми, удерживание и разрешение лучше
на рис. 1.226, как и форма пиков 9 и 10. Еще один пример смешанного разделения
на катионообменной колонке PrimeSep SIELC приводится в |71]. В этом разделении
наблюдается «уникальное элюирование антоциана» из виноградного сока,
облегчающее идентификацию пиков и их количественное определение. В смешанных
разделениях удерживание можно контролировать, изменяя %В в подвижной фазе, ее рН и
концентрацию буфера или соли. Для изменения относительного удерживания также
подходят условия разделения, которые влияют на селективность в ОФХ или в ИОХ.
Смешанный режим разделения также решает проблему слабого удерживания
в ОФХ некоторых сильных оснований, поскольку их удерживание возрастает
благодаря взаимодействию с отрицательно заряженным сорбентом |72|. Дополнительным
№388 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
^^? хроматография
18 (мин)
Рис. 7.22. Сравнение обращенно-фазового разделения (а) и смешанного ион-парного
разделения (б) азотистого иприта. Образец: смесь небольших гидрофильных
аминов. Условия (б): колонка 150 х 2,1 мм PrimeSep 100, 5 мкм (S1ELC
Technologies, USA), подвижная фаза 40% ацетон итрил-буфер (0,1% ТФУК),
скорость потока 0,2 мл/мин [70|
преимуществом сорбентов для смешанного режима разделения является
возможность использовать большую нагрузку ионизированных образцов. Ну и наконец, та
кие сорбенты еще и уникальны в том отношении, что позволяют одновременно
разделять смеси анионов, катионов, цвиттер-ионов и нейтральных соединений |73|.
Незадолго до публикации этой книги сообщалось о катионообменниках для
смешанного режима разделения, которые особенно стабильны при низких рН и сохраняют
в этих условиях исключительную эффективность |74|. Сравнение хроматограмм
разделений, проведенных на колонках с этими сорбентами, обычных колонках C|g
и коммерчески доступных колонках для смешанного режима (гидрофобных
катионообменниках), представлено на рис. 7.23. Обратите внимание на более сильное
удерживание оснований на рис. 7.23«, несмотря на более высокую температуру разделе-
(б)
Свкопонка
-кислоты
и
/:
Покупная колонка
/ для смешанного режима
Экспериментальная колонка
для смешенного режима
8
10
12
14
16
Рис. 7,23. Разделение смеси кислот, оснований и трех аминокислот на разных
колонках. Условия: колонки 50x4,6 мм, 5 мкм, скорость потока 1,0 мл/мин.
(я) колонка Си, подвижная фаза 10% ацетонитрил-0,1% ТФУК, 40 °С,
(б) покупная колонка для смешанного режима (PrimeSep 200), 10% ацето-
нитрил-0,01% ТФУК, 40 "С, (в) экспериментальная колонка для
смешанного режима, 24% ацетонитрил-0,02% ТФУК, 65 °С |75|
Литература 389
ния и ббльшую долю органического растворителя в подвижной фазе (24 %В),
а также на улучшение разрешения оснований, которое, вероятно, есть результат
расширения диапазона времен удерживания. Сорбенты для смешанного режима
оказались эффективными и в твердофазной экстракции (ТФЭ), используемой при пробо
подготовке (раздел 16.6.7.1)
Литература
1. P. J. Twitchelt and А. С. Moffat, J. Chromatogr., HI (1975) 149.
2. E. P. Kroef and D. J. Pielrzyk, Anal. Chem., 50 (1978) 502.
3. L. R. Snyder, A. Maule, A. Heebsch, R. Cuellar, S. Paulson, J. Carrano, L. Wrisley, С. С Chan,
N. Pearson, J. W. Dolan, and J. Gilroy, J. Chromatogr. A, 1057 (2004) 49.
4. J. A. Lewis, J. W. Dolan, L. R. Snyder, and I. Molnar, J. Chromatogr., 592 (1992) 197.
5. U. D. Neue, A. Mendez, K. Van Tran, and D. M. Diehl, in HPLC Made to Measure, S. Kromidas,
ed., Wiley-VCH, Hoboken, NJ, 2006, pp. 71—87.
6. A. P. Schellinger and P. W. Carr, LCGC, 22 (2004) 544.
7. D. V. McCalley, Anal. Chem., 75 (2003) 3404.
8. F. Gritti and G. Guiochon, J. Chromatogr. A, 1033 (2004) 43—; 1132 (2006) 51.
9. J. B. Li, LCGC, 10 (1992) 856.
10. J. W. Dolan LCGC, 8 (1990) 212.
11. L. R. Snyder, unreported data.
12. D. V. McCalley, J. Chromatogr. A, 987 (2003) 17.
13. R. F. Doerge, ed., Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical
Chemistry, 8th ed., J. B. Lippincott, Philadepha, app. B.
14. M. Roses, /. Chromatogr. A, 1037 (2004) 283.
15. X. Subirats, E. Bosch, and M. Roses, J. Chromatogr. A, 1121 (2006) 170.
16. D. V. McCalley, Adv. Chromatogr., 46 (2008) 305.
17. J. A. Lewis, D. C. Lommen, W. D. Raddatz, J. W. Dolan, L. R. Snyder, and I. Molnar, J.
Chromatogr., 592 (1992) 183.
18. I. Canals, K. Vaiko, E. Bosch, and M. Roses, Anal. Chem., 73 (2001) 4937.
19. P. L. Zhu, J. W. Dolan, L. R. Snyder, D. W. Hill, L. Van Heukelem, and T. J. Waeghe, J.
Chromatogr. A, 756 (1996)51.
20. P. L. Zhu, J. W. Dolan. L. R. Snyder, N. M. Djordjevic, D. W. Hill, J.-T. Lin,L. С Sander, and
L. Van Heukelem, J. Chromatogr. A, 756 (1996) 63.
21. С. В. Castells, С. Rafols, M. Roses and E. Bosch, J. Chromatogr. A, 1002 (2003) 41.
22. С. В. Castells, L. G. Gagliardi, С Rafols, M. Roses, and E. Bosch, J. Chromatogr. A, 1042 (2004)
23.
23. L. G. Gagliardi, С. В. Castells, С. Rafols, M. Roses, and E. Bosch, J. Chromatogr. A, 1077 (2005)
159.
24. S. M. С Buckenmaier, D. V. McCalley, and M. R. Euerby, J. Chromatogr. A, 1060 (2004) 117.
25. J. Pellett, P. Lukulay, Y. Mao, W. Bowen, R. Reed, M. Ma, R. С Munger, J. W. Dolan, L.
Wrisley, K. Medwid, N. P. Toltl, С. С. Chan, M. Skibic, K. Biswas, K. Wells, and L. R. Snyder, J.
Chromatogr. A, 1101 (2006) 122.
26. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, and P. W. Carr, J. Chromatogr. A, 961
(2002) 195.
27. J. Gilroy, J. W, Dolan and L. R. Snyder, /. Chromatogr. A, 1000 (2003) 757.
28. J. J. Gilroy, J. W. Dolan, P. W. Carr, and L. R. Snyder J. Chromatogr. A, 1026 (2004) 77.
29. N. S. Wilson, M. D. Nelson, J. W. Dolan, L. R. Snyder, R. G. Wolcott, and P. W. Carr, J.
Chromatogr. A, 961 (2002) 171.
30. D. Guo, С. Т. Mant, and R. S. Hodges, J. Chromatogr., 386 (1987) 205.
31. J. L. Gtajch, J. C. Gluckman, J. G. Charikofsky, J. M. Minor, and J. J. Kirkland, J. Chromatogr.,
318(1985) 23.
32. R. G. Wolcott, J. W. Dolan, and L. R. Snyder, J. Chromatogr. A, 869 (2000) 3.
33. P. Haber, T. Baczek, R. Kaliszan, L. R. Snyder, J. W. Dolan, and С. Т. Wehr., J. Chromatogr.
Sci., 38 (2000) 386.
390 Глава 7. Ионные образцы: обращенно-фазовая, ион парная и ионообменная
s^1 хроматография
34. S. Pous-Torres, J. R. Torres-Lapasio, J. J. Baeza-Baeza, and M. C. Garcia-Alvarez-Coque,
J. Chromatogr. A, 1163 (2007) 49.
35. S. Pous-Torres, J. R. Torres-Lapasio, J. J. Baeza-Baeza, and M. C. Garcia-Alvarez-Coque,
J. Chromatogr. A, 1193 (2008) 117.
36. F. Vanbel, B. L. Tilquin, and P. J. Schoenmakers, J. Chromatogr. A 697 (1995) 3.
37. D. V. McCalley, LCGC, 17 (1999) 440.
38. S. M. С Buckenmaier, D. V. McCalley, and M. R. Euerby, Anal. Chem., 74 (2002) 4672.
39. D. V. McCalley, J. Chromatogr. A, 1075 (2005) 57.
40. D. V. McCalley, J. Chromatogr. A, 708 (1995) 185.
41. D. V. McCalley, J. Chromatogr. A, 738 (1996) 169.
42. D. M. Marchand, L. R. Snyder, and J. W. Dolan, J. Chromatogr. A, 1191 (2008) 2.
43. M. A. Stadalius, J. S. Berus, and L. R. Snyder, LCGC, 6 (1988) 494.
44. J. Nawrocki, /. Chromatogr. A, 779 (1997) 29.
45. M. T. W. Hearn, ed., Ion-pair Chromatography: Theory and Biological and Pharmaceutical
Applications, Dekker, New York, 1985.
46. L. R. Snyder, J. J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, 2nd ed., Wi-
ley-lnterscience. New York, 1997, ch. 7.
47. J. H. Knox and R. A. Hartwick, J. Chromatogr., 204 (1981) 3.
48. M. W. Dong, J. Lepore, and T. Tammoto, J. Chromatogr., 442 (1988) 81.
49. A. Bartha, G. Vigh, and Z. Varga-Puchony, J. Chromatogr., 499 (1990) 423.
50. A. Bartha, G. Vigh and J. StShlberg, J. Chromatogr., 485 (1989) 403.
51. A. P. Goldberg, E. Nowakowska, P. E. Antle, and L. R. Snyder, J. Chromatogr., 316 (1984) 241.
52. J. M. Roberts, A. R. Diaz, D. T. Fortin, J. M. Friedle, and Stanley D. Piper, Anal. Chem., 74
(2002) 4927.
53. J. Flieger, J. Chromatogr. A, 1113 (2006) 37.
54. J. Flieger, J. Chromatogr. A, 1175 (2007) 207.
55. J. W. Dolan and L. R. Snyder, Troubleshooting HPLC Systems, Humana Press, Clifton, NJ, 1989.
pp. 429—435.
56. S. Levin and E. Grushka, Anal. Chem., 58 (1986) 1602.
57. E. Rajakla, J. Chromatogr., 218 (198!) 695.
58. H. Small, T. S. Stevens, and W. С Bauman, Anal. Chem., 47 (1975) 1801.
59. P. R. Haddad and P. E. Jackson, Ion Chromatography, Elsevier, Amsterdam, 1990.
60. J. S. Fritz and D. T. Gjerde, Jon Chromatography, 3rd ed., Wiley-VCH, Weinheim, 2009.
61. J. Weiss, Handbook of Ion Chromatography, 3rd ed., Wiley, 2005.
62. M. More, M. I. H. Helaleh, Q. Xu, W. Hu, M. Ikedo, M.-Y. Ding, H. Taoda, and K.Tanaka, J.
Chrvmatogr. A, 1039 (2004) 129.
63. J. Qiu, J. Chromatogr. A, 859 (1999) 153.
64. Y. Baba and G. Kura, J. Chromatogr. A, 550 (1991) 5.
65. J. E. Madden and P. R. Haddad, J. Chromatogr. A, 850 (1999) 29.
66. J. St3hlberg, J. Chromatogr. A, 855 (1999) 3.
67. P. J. Andralojc, A. J. Keys, A. Adam, and M. A. J. Parry, J. Chromatogr. A, 814 (1998)105.
68. T. R. Cataldi, С Campa, M. Angelotti, and S. A. Bufo, J. Chromatogr. A, 855 (1999)539.
69. E. Landberg, A. Lundblad, and P. Pahlsson, J. Chromatogr. A, 814 (1998) 97.
70. H.-C. Chua, H.-S. Lee, and M.-T. Sng, J. Chromatogr. A, 1102 (2006) 214.
71. J. L. McCallum, R. Yang, J. C. Young, J. N. Strommer, and R. Tsao, J. Chromatogr. A,\ 148,
(2007) 38.
72. N.H. Davies, M. R. Euerby, and D. V. McCalley, J. Chromatogr. A, 1138 (2007) 65.
73. J. Li, S. Shao, M. S. Jaworsky, and P. T. Kurtulik, J. Chromatogr. A, 1185 (2008) 185.
74. H. Luo, L. Ma, Y. Zhang, and P. W. Carr, /. Chromatogr. A, 1182 (2008) 41.
75. H. Luo, L. Ma, С. Раек, and P. W. Carr, J. Chromatogr. A, 1202 (2008) 8.
ГЛАВА 8
НОРМАЛЬНО-ФАЗОВАЯ
ХРОМАТОГРАФИЯ
8.1. Введение 392
8.2. Удерживание 393
8.2.1. Теория 396
8.2.2. Зависимость силы растворителя от В-растворителя и от %В 400
8.2.3. Использование данных ТСХ для предсказания удерживания в НФХ. . . 403
8.3. Селективность 406
8.3.1. Зависимость селективности от элюирующей силы растворителя 406
8.3.2. Зависимость селективности от типа растворителя 406
8.3.3. Зависимость селективности от температуры 406
8.3.4. Селективность колонки 411
8.3.5. Разделения изомеров 413
8.4. Рекомендации по разработке метода 416
8.4.1. Начальные условия для разработки НФХ-метода: выбор элюирующей
силы подвижной фазы и типа сорбента (шаги I, 2; таблица 8.2) 418
8.4.2. Стратегии оптимизации селективности (шаг 3; таблица 8.2) 420
8.4.3. Пример разработки НФХ-метода 420
8.5. Трудности, возникающие при использовании НФХ 422
8.5.1. Плохая воспроизводимость разделения 422
8.5.2. Обеднение элюента растворителем и медленное уравновешивание
колонки 424
8.5.3. Уширение задних фронтов пиков 424
8.6. Хроматография гидрофильных взаимодействий (ГИХ) 425
8.6.1. Механизм удерживания 426
8.6.2. Колонки 427
8.6.3. Разработка метода ГИХ 429
8.6.4. Трудности, связанные с ГИХ 431
Литература 431
ь
Глава 8. Нормально фазовая хроматография
8.1. Введение
В начале девятисотых годов, когда хроматография только появилась как метод
(раздел 1.2), колонки упаковывались полярными неорганическими частицами — такими
как карбонат кальция или окись алюминия. Подвижная фаза, использовавшаяся
в этих экспериментах, была менее полярной (безводной) и представляла собой такой
растворитель как лигроин (насыщенная углеводородная фракция нефти). В течение
последующих шестидесяти лет эта методика продолжала оставаться наиболее
распространенным {«нормальным») способом проведения хроматографического процесса.
По этой причине использование полярной неподвижной фазы (вместе с менее
полярной подвижной фазой) сегодня называют нормально фазовой хроматографией
(НФХ). Для описания НФХ используют также понятие адсорбционной
хроматографии, исходя из признания того факта, что удерживаемые молекулы растворенных
веществ связываются с поверхностью частиц сорбента, которым упакована колонка,
или адсорбируются на ней (раздел 8.2).
После введения в практику в семидесятых годах высокоэффективной ОФХ НФХ
стала использоваться для ВЭЖХ-анализа все реже и реже. Отчасти это было
обусловлено тем, что ОФХ гораздо удобнее и имеет ряд преимуществ при разделении
многих представляющих интерес образцов биологического и/или медицинского
происхождения. Некоторые сложности, присущие НФХ (раздел 8.5), тоже сыграли свою
роль в уменьшении ее популярности по сравнению с ОФХ.
Сегодня НФХ используется, главным образом, для (1) аналитических разделений
с помощью тонкослойной хроматографии (ТСХ, раздел 1.3.2), (2) первичной очистки
необработанных препаратов (сырцов) (в препаративной хроматографии и пробопод-
готовке, главы 15, 16), (3) разделения высокополярных, слабо удерживаемых в ОФХ
образцов, или (4) разделения ахиральных изомеров (раздел 8.3.5). Кроме того, НФХ
может иногда оказаться полезной и для других образцов в силу их уникальных
свойств, например, для образцов, содержащих не представляющие интереса для
аналитика соединения с ярко выраженными неполярными свойствами. Эти соединения
будут сильно удерживаться в ОФХ, что потребует либо больших времен разделения,
либо пробоподготовки, либо использования градиентной элюции. При
использовании НФХ сильно неполярные соединения элюируются вблизи fy и их разделение
в изократическом режиме не создаст проблем (например, см. раздел 8.4.3). В любом
случае, часто лучше отложить использование НФХ до тех пор, пока не будет найден
и опробован ОФХ-вариант.
До 1970 года для НФХ использовались самые разнообразные сорбенты:
например, окись алюминия, окись магния, силикат магния (флорисил) и диатомит (целит,
кизельгур). Однако с появлением ВЭЖХ стали предпочитать синтетический
(непривитый) силикагель для колоночной хроматографии и ТСХ. Среди преимуществ сили-
кагеля для НФХ:
• более нейтральная и менее активная поверхность, которая с меньшей
вероятностью вступит в нежелательные реакции с образцом в ходе разделения;
• жесткие частицы контролируемого размера и пористости, выдерживающие
высокие давления, требуемые в ВЭЖХ;
• в целом большая площадь поверхности, позволяющая увеличить количество
вводимого образца и проводить разделение либо с повышенной чувствительностью
детектирования, либо с повышенным выходом, если это препаративная
хроматография;
• большая чистота и воспроизводимость, позволяющая проводить больше
разделений с идентичными результатами;
• разумная цена и доступность.
8.2. Удерживание 393
Хотя силикагель популярен и сегодня, со временем появились и другие варианты
сорбентов для НФХ. Три полярные привитые неподвижные фазы (раздел 5.3.3),
сходные по химическому строению с фазами, используемыми в ОФХ, были введены
в практику НФХ в течение семидесятых годов: (I) цианофазы, в которых к частицам
силикагеля привиты группы -(CH2)3-C=N, (2) диольные фазы с привитыми
группами -(СН2)3-0-СН2-СНОН-СН2ОН и (3) аминофазы с лигандами -(CH2)3-NH2.
Различные свойства этих привитых неподвижных фаз для НФХ обсуждаются в
разделе 8.3.4. При обсуждении различных аспектов использования в НФХ сорбентов
на основе силикагеля в разделе 8.5 приводится ряд аргументов в пользу применения
привитых неподвижных фаз вместо непривитого силикагеля.
В течение девяностых годов стали коммерчески доступными силикагели высокой
чистоты (тип В; раздел 5.2.2.2), и они постепенно вытеснили менее чистый
силикагель типа А, использовавшийся раньше для аналитических НФХ-разделений.
Некоторые преимущества силикагеля типа В для НФХ обсуждаются в разделе 8.5.
Последняя модификация НФХ — так называемая хроматография гидрофильных
взаимодействий (ГИХ; раздел 8.6), которую еще называют водной НФХ. Сорбенты
для ГИХ содержат либо (а) частицы силикагеля, к которым привиты полярные
гидрофильные группы, например, амидные, либо (б) просто частицы немодифициро-
ванного силикагеля. В отличие от безводных подвижных фаз, традиционно
используемых в НФХ, подвижная фаза для любой разновидности ГИХ представляет собой
смесь воды и органического растворителя. ГИХ несколько удобнее, чем ОФХ, и
при этом сводит к минимуму ряд сложностей, связанных с использованием силика
гельных колонок с неводными подвижными фазами (раздел 8.5).
В настоящей главе, если не указано специально, мы будем предполагать, что
используются непривитые силикагельные сорбенты типа В. Поверхность частицы
силикагеля покрыта силанольными группами =Si-OH (раздел 5.2.2.2), которые большей
частью и отвечают за ее хроматографические свойства. Эти силанольные группы
являются относительно сильными донорами протонов, могущими взаимодействовать
с молекулами растворенного вещества с образованием водородных связей между
донорными силанольными группами сорбента и акцепторными группами
растворенного вещества (любыми молекулами с доступными электронами или дипольным
моментом). Кроме того, поверхность силикагеля сильно притягивает маленькие
полярные молекулы, такие как вода, что приводит к некоторым сложностям, обсуждаемым
в разделе 8.5. Более подробную информацию о роли сорбента в НФХ-разделении
см. в разделе 8.3.4 (селективность колонок).
8.2. Удерживание
Поскольку сорбент в НФХ более полярен, чем подвижная фаза, то на нем (в
противоположность ОФХ) предпочтительнее будут удерживаться или адсорбироваться
более полярные растворенные вещества. Это проиллюстрировано на рис. 8.1й, на
котором показано разделение нескольких монозамещенных бензолов на силикагельной
колонке. Подвижная фаза — 20% хлороформ-гексан. Более полярный растворитель
хлороформ используется в качестве В-элюента, а менее полярный гексан в качестве
А-элюента. В данном случае менее полярные молекулы бензола (-Н) и хлорбензола
(-С1) выходят из колонки первыми, в то время как более полярные анилин (-NH2),
бензойная кислота (-СООН) и бензамид (-CONH2) покидают ее гораздо позже.
Такой характер удерживания контрастирует с удерживанием в ОФХ (рис. 2.7в), где оно
уменьшается с ростом полярности растворенного вещества. На рис. 8.15
сравнивается удерживание (log к) в НФХ и в ОФХ для нескольких монозамещенных бензолов.
Глава 8. Нормально фазовая хроматография
силикагельиая колонка
20% СНС13
(«)
-NO,
А
-со2снэ
-C-N
_А1
-сно„
-он-
-COCHj 4
Рнс. 8.
-NH,
6 в
Время(мин)
-СООН
I
ю
12
-CONH2
400
800
Время (мин)
1200
2.0
1.0
log к
(НФХ)
0.0 •
-1.0
• -СОСН3
№
-0.2 0.0 0.2 0.4
log к (ОФХ)
Пример зависимости удерживания в НФХ от полярности растворенного
вещества. Образец: монозамешенные бензолы (заместители указаны для
каждого пика; например, -Н — бензол, -CI — хлорбензол). Условия:
силикагельиая колонка 150x4,6 мм, 5мкм; подвижная фаза 20% хлороформ-гексан:
температура комнатная; 2,0 мл/мин. (а) Хроматограммы построены по
данным из |1|; (6) удерживание (а) сравнивается с ОФХ-удерживанием
на рис. 2.7в для бензолов, замещенных такой же функциональной группой
(подвижная фаза для ОФХ 50% ацетонитрил-вода)
Как и ожидалось, существует отрицательная корреляция удерживания в НФХ
по сравнению с ОФХ, что приблизительно соответствует изменению порядка
удерживания на противоположный. Несмотря на то, что наблюдаемая на рис. 8.15
корреляция достаточно велика (г2 = 0,76), значителен и разброс данных. Таким образом,
НФХ-разделение не может рассматриваться как полная противоположность таковому
в ОФХ.
Имейте в виду, что относительное удерживание как в НФХ, так и в ОФХ может
существенно изменяться в зависимости от колонки, подвижной фазы или
температуры. Все эти факторы влияют на разброс данных на графике, который мы видим
на рис. 8. Iff.
Помимо примерно обратного порядка удерживания образца в НФХ по сравнению
с ОФХ есть еще два дополнительных отличия в НФХ-удерживании, связанных с:
(1) числом п алкильных атомов углерода в молекуле растворенного вещества (его
углеродном числом Сп) и (2) изомерией образцов. Эти две основные характеристики НФХ
в сопоставлении с ОФХ проиллюстрированы разделением на рис. 8.2. На рис. 8.2я
8.2. Удерживание
ОФХ
(а)
С5
... „ , г-метил-4-н-
(4-Н-бутиланилин) 6утиланилин
НФХ
I 1 1 1 г 1
4 в в
Время (мин)
(б)
6
4Л
4
t
J№
6 в
Зремя (мин)
(в)
12 J 13 С»
I Л'
4 6 8
Бремя (мин)
(г)
(0)
С„
с,
С2
с,
с4
cs
среднее время удерживания (мин)
ОФХ (а)
1.2
1.5
1.9
2.6
4.5
6.5
НФХ (б-г)
7.5 (не показано)
6.6
5.4
4.6
8.7 (не показано)
5.1 (не показано)
диапазон к
ОФХ (я)
1.1
1.2
1.2
НФХ (б-з)
2.0
3.4
3.0
Рис. 8.2. Сравнение ОФХ-разделения (я) с НФХ-разделением (6—г) смеси алкиза-
мещенных анилинов. Условия: в (й) колонка Cg 150 х 4,6 мм, 5 мкм;
в (б— г) колонка цианофаза ]50 х 4,6 мм, 5 мкм; подвижная фаза в (а) 60%
метанол — буфер рН-7,0 в (б) 0,2% изопропанол — гексан; комнатная
температура и 2,0 мл/мин в (а) и (6). Образец (номера пиков): 1-3, 2-, 3- и 4-метил-
анилин; 4, 2,6-диметиланилин; 5, 2-этиланилин; 6, 2,5-диметиланилин;
7, 2,3-диметиланилин; 8, 2,4-диметиланилин; 9, 3-этиланилин; 10, 4-этилани-
лин; //, 3,4-диметиланилин; 12, 2,4,6-триметиланилин; 13, 2-изопропилани-
лин; 14, 4-изопропиланилин. Хроматограммы воссозданы по данным из |2]
показано разделение семнадцати алкилзамещенных анилинов на колонке Cg с
помощью 60% МеОН в качестве подвижной фазы. По мере роста величины Сп
удерживание в ОФХ растет (но не в НФХ). Видно, что изомеры с одинаковым углеродным
числом (например, С|_ содержащие о-, м-, я-метиланилин) сгруппированы, в то время как
вещества с разным числом атомов углерода (например, С\ и CJ) хорошо разделяются.
Как показано на рис. 8.2d, среднее время удерживания в ОФХ равномерно растет
с увеличением углеродного числа (в среднем, в 1,4 раза на каждый дополнительный
атом углерода в этом примере).
На рис. 8.25—г проиллюстрировано дальнейшее разделение с помощью НФХ
фракций С), Ci и Сз, полученных в ОФХ-разделен и и, показанном на рис. 8.2й.
В НФХ используется цианофаза в сочетании с подвижной фазой 0,2%
изопропанол — гексан. Видно, что по мере увеличения количества алкильных атомов углерода
396 Глава 8. Нормально фазовая хроматография
не происходит постоянного изменения времени удерживания в случае НФХ
(см. обощенные результаты на рис. 8.2d). Таким образом, с помошью НФХ можно
разделять вещества с разными функциональными группами {как на рис. 8.1а),
но различия в количестве атомов углерода в компонентах образца на удерживание
влияют намного слабее. По этой причине НФХ использовалась в прошлом для
разделения классов нефтепродуктов [3| и липидных образцов |4]. НФХ позволяет
фракционировать образцы нефтепродуктов на насыщенные углеводороды, олефины, бензолы
и различные полициклические ароматические углеводороды в зависимости от числа
двойных связей в молекуле. Однако различия в алкильных заместителях или
молекулярной массе растворенного вещества оказывают небольшое влияние на такое
разделение. Аналогичным образом можно разделить липидные образцы на моно-, ди- и
триглицериды (как и другие классы соединений).
Как видно из рис. 8.26 (фракция С, или метилзамещенные анилины), рис.8.2в
(фракция С2 или диметил- и этилзамещенные анилины) и рис. 8.2г (фракция Сз или
триметил-, метилэтил- и н пропил замещенные анилины), растворы изомеров
обычно лучше разделяются с помощью НФХ, чем в условиях ОФХ. Как правило,
изомеры в ОФХ (например, анилины фракции С]) имеют сходное удерживание
(с минимальным разрешением), в то время как для НФХ все наоборот (существеннее
различия величин к для разных изомеров и лучше разрешение). Большая
селективность НФХ в отношении изомеров по сравнению с ОФХ также подтвеждается диапа
зоном величин к для каждого ряда изомеров — то есть, отношением величин к
наиболее и наименее удерживаемых изомеров («диапазон величин к» на рис. 8.2d).
Интервал величин А: для ряда изомеров (фракции С|, С2 или С3) меняется от 1,1
до 1,2 в ОФХ, и во много раз больше в НФХ: от 2,0 до 3,4.
Когда два соединения — изомеры, их трудно разделить с помощью ОФХ
(раздел 6.3.5), НФХ, как правило, оказывается более эффективной. Так для
препаративных разделений (глава 15) ахиральных изомеров, где желательны максимальные
значения а, настоятельно рекомендуется НФХ на силикагельной колонке (о хиральных
изомерах см. главу 14). С обзором ОФХ-разделений изомеров можно ознакомиться в
разделе 6.3.5. Для дальнейшего обсуждения разделения изомеров с помощью НФХ
см. раздел 8.3.5.
Если у вас есть сомнения относительно природы удерживания образца при
разделении; доминирует ли нормально-фазовый или обращенно-фазовый механизм, то можно
сделать простои тест — изменить полярность подвижной фазы. Если удерживание
растет с увеличением полярности подвижной фазы, то имеет смысл использовать в
дальнейшей работе ОФХ. Если же оно уменьшается с увеличением полярности подвижной
фазы, то это задача для НФХ. Этот тест подходит вне зависимости от того,
является ли неподвижная фаза непривитым или привитым силикагелем, а подвижная фаза —
водной или неводной (обратите внимание на тот факт, что вода наиболее полярный
из растворителей).
8.2.1. Теория
Удерживание в НФХ удобнее всего описывать как процесс замещения, основанного
на том, что поверхность частиц силикагеля покрыта монослоем молекул
растворителя, адсорбированных из подвижной фазы |1, 5, б|. Следовательно, для удерживания
молекулы растворенного вещества в процессе НФХ одна или более предварительно
адсорбированных молекул растворителя должны быть вытеснены с поверхности
частицы силикагеля, чтобы освободить место для адсорбции молекулы растворенного
вещества. Замещение в НФХ проиллюстрировано на рис. 8.3я,б для относительно
неполярного растворенного вещества (хлорбензола) и более слабого, менее полярно-
8.2. Удерживание
го хлористого метилена, используемого в качестве подвижной фазы. Когда молекула
хлорбензола перемещается из подвижной фазы на рис. 8.3а в неподвижную фазу
на рис. 8.36, одна или более ранее адсорбированных молекул растворителя
хлористого метилена должна быть удалена с поверхности частицы сорбента и возвратиться
в подвижную фазу. В этом примере предполагается, что адсорбированная
молекула растворенного вещества лежит на поверхности частицы силикагеля и занимает
площадь, показанную на рис. 8.36 пунктирным прямоугольником, окружающим
молекулу удерживаемого хлорбензола. Как видно из рис. 8.3а, эта же площадь
первоначально была занята (приблизительно) двумя молекулами хлористого метилена.
Следовательно, устанавливающееся в результате удерживания равновесие можно описать
уравнением
Y(ra) + nM(s) <=> Y(s) + nM(m), (8.1)
.Подвижная
> фаза
/<3й££> <gH^£yJ""
Р ,н ,н
p.p.. P
77777777777777- J
у ci-
Нвпсяэ№кна|| ■''' «..•.|....Г;...|-..:Г;..!
► фаза ■{ ^ '^ ^
P..PP....
77777777777777-
Y{ni) + 2 Щя)
У8 + 2 M(m)
(г)
Подвижная
► фаза н
о о о
Poo
о о
н н
1 i
Негодвихдея
у фаза ч
О
I 777.
I
777777777777-
Y|m) + Mi
I»)
VW+M,
m
Рис. 8.З. Гипотетические примеры удерживания молекул растворенных веществ на
поверхности силикагеля: хлорбензола (а,б — нелокализованная адсорбция) и
фенола (е,г — локализованная адсорбция). Подвижная фаза: (а,б) — менее
полярный растворитель хлористый метилен (CH2CI2); (в,г) — более
полярный растворитель тетрагидрофуран (ТГФ)
398 Глава 8. Нормально фазовая хроматография
где п — это количество молекул растворителя М, замешенных удерживаемой
молекулой растворенного вещества Y (л — 2 в примере на рис. 8.За,б). Y(m) и Y(s) относятся
соответственно к молекуле растворенного вещества в подвижной и неподвижной
фазах, в то время как М(т) и M(s) относятся соответственно к молекуле растворителя
М в подвижной и неподвижной фазах. Таким образом, величина п в уравнении (8.1)
представляет собой отношение площадей молекул растворенного вещества к
площадям молекул подвижной фазы.
Случай, когда используется более полярный ТГФ в качестве подвижной фазы и
более полярное растворенное вещество — такое, как фенол, изображен на рис. 8.3в,г.
В отличие от более слабых и менее специфических взаимодействий, изображенных
на рис. %.Ъа,6, здесь взаимодействие растворителя и растворенных молекул с
поверхностными силанольными группами будет сильнее; это показано стрелками,
соединяющими две контактирующих группы. В результате поверхностная силанольная
группа взаимодействует в соотношении 1:1 с полярной группой в молекуле либо
растворенного вещества, либо подвижной фазы. Это называется локализованной
адсорбцией. В этих условиях адсорбированные молекулы могут принимать скорее
вертикальную, нежели плоскую горизонтальную конфигурацию, что и показано на
примере удерживания фенола на рис. 8.3г. В примере, проиллюстрирован на рис. $.3а,б,
вся молекула растворенного вещества (хлорбензола) притягивается к поверхности
силикагеля сильнее, чем молекулы менее полярного растворителя хлористого
метилена. На рис. 8.3в,г высокополярная -ОН группа растворенного фенола сильно
взаимодействует с силанольной -ОН группой на поверхности силикагеля, тогда как
менее полярная фенильная группа притягивается слабее, чем молекулы сильного и
очень полярного ТГФ. В результате фенильная группа не в состоянии конкурировать
с молекулами ТГФ за место на поверхности силикагеля и поэтому она смещается
в подвижную фазу, но остается связанной с поверхностью благодаря взаимодействию
силанольных групп с гидроксильными группами растворенного вещества. Для
растворенных веществ с некоторым числом п сильнее удерживаемых (очень полярных)
заместителей (например, -ОН, -NH2 групп) по-прежнему применимо уравнение
(8.1), причем н теперь будет относиться к числу полярных групп в молекуле
растворенного вещества, которые могут одновременно взаимодействовать с силанольными
группами силикагеля. Однако, для менее полярных молекул подвижной фазы,
изображенных на рис. &3а,6, я означает соотношение площадей, необходимых
соответственно для молекул растворенного вещества и для молекул подвижной фазы.
Конкуренция молекул растворенного вещества и растворителя за места на
поверхности силикагеля будет зависеть от взаимодействия каждой молекулы с подвижной
и неподвижной фазами. Поскольку в НФХ преобладают полярные взаимодействия,
полярные молекулы растворенного вещества или растворителя будут
взаимодействовать гораздо сильнее с более полярной поверхностью силикагеля, чем с менее
полярной подвижной фазой. В первом приближении можно пренебречь взаимодействиями
между молекулами растворенного вещества и растворителя и рассматривать только
взаимодействия молекул растворенного вещества и элюента со стационарной фазой.
Это позволяет получить простое уравнение для зависимости к от концентрации
элюента В в бинарной подвижной фазе А-В |1, 6|:
log к = log кд - А^ (8.2)
где кА — величина к для неполярного элюента А (для которого е равна нулю), Ач
означает площадь молекулы растворенного вещества, а сила растворителя
подвижной фазы е зависит от (I) силы чистых элюентов А и В zA и ев, соответственно; (2)
площади молекулы элюента В и (3) от концентрации элюента В (%В) в подвижной
фазе. Значение е можно вычислить, используя уравнение (8.5), приведенное в
следующем разделе 8.2.2.
8.2. Удерживание
В оставшейся части этого раздела проводится довольно подробное количественное
рассмотрение зависимости удерживания от природы растворенного вещества и
подвижной фазы. Читатель может, если захочет, перейти к разделу 8.2.2 (в котором да
ются практические выводы из нижеследующего обсуждения) и, при необходимости, вер
нуться к этому разделу.
Уравнение (8.2) применимо почти для всех растворителей и растворенных
веществ. В случае более полярных растворенных веществ и растворителей В, таких, как
на рис. 8.3в,г, их полярные группы могут связываться с отдельными силанольными
группами. Таким образом, в уравнении (8.1) теперь п равно числу полярных групп
в молекуле растворенного вещества (а не площади молекулы растворенного
вещества). Если %В и сила растворителя В достаточно велики, поверхность силикагеля
будет почти полностью покрыта молекулами растворителя В (до полного исключения
растворителя А). В этих условиях концентрация адсорбированного растворителя В
не будет сильно изменяться при изменении %В. Для этого случая можно получить
уравнение Сочевинского, преобразовав уравнение (8.1) [7,8]. Тогда константу
равновесия, описываемого уравнением 8.1, можно представить в виде
КЩ=ХУ^М^/ХУ^ХЫ/Ъ (8.3)
где Xysn X/tfm относятся к концентрациям (мольным долям) растворенного вещества
У в неподвижной (s) и подвижной (т) фазах, соответственно. Аналогично Ад^. и
Хцт — мольные доли В-растворителя М в неподвижной (s) и в подвижной (/я)
фазах. Коэффициент удерживания к может быть записан следующим образом
(8.3а)
где ф — фазовое отношение: отношение объемов неподвижной и подвижной фаз
во внутреннем объеме колонки. Если поверхность силикагеля почти полностью
покрыта В-растворителем, Хм к 1, тогда из уравнений (8.3) и (8.3а) получаем
log к = log kB — nlogXg (уравнение Сочевинского), (8.4)
где п— количество молекул В-растворителя, замещенных молекулами растворенного
вещества (примерно равное количеству полярных заместителей в молекуле
растворенного вещества); кв относится к величине к для чистого растворителя В в качестве
подвижной фазы (100 %В) и Хв — мольная доля растворителя В в подвижной фазе.
Уравнение (8.4) можно рассматривать как частный случай уравнения (8.2) всякий
раз, когда Хм^ и 1 (т.е. для больших концентраций растворителя В).
Уравнение 8.4 более удобно записать в приближенной форме:
log к ж log кв - wlog ф, (8.4а)
где ф — объемная доля полярного В-растворителя в подвижной фазе ( = 0,01 х %В).
На рис. S.4 показан пример использования уравнения (8.4а) для двух различных
(полярных) растворенных веществ и подвижной фазы состоящей из гексана (А) и ТГФ
(В), причем %В изменяется от 8 до 27%. В каждом случае наблюдается приблизительно
линейная зависимость log А от ф. Значения п из уравнения (8.4а) для двух графиков
на рис. 8.4 примерно равны 1,4, что несколько превышает ожидаемое значение 1,0
(поскольку в каждой молекуле растворенного вещества имеется всего лишь одна полярная
-ОН группа). Это — следствие приближенного характера уравнения (8.4а). Уравнения
(8.4) и (8.4а) верны для более полярных В-растворителей (г.д Э> 0,4) и более высоких
концентраций В-растворителя (> 10%) — для условий, обеспечивающих почти полное
покрытие поверхности силикагеля молекулами В-растворителя (вместо А-раствори-
теля). Обычно значения п в уравнении (8.4а) попадают в интервал между единицей
400 Глава 8. Нормально фазовая хроматография
1.4
1.2
1.0
log к 0.8
0.6
0.4
0.2
-1.0 -0.8 -0.6 -0.4
log Ф
Рис. 8.4. Зависимость log к от log %B для растворов полярных (локализующих) В-рас-
творителсй. Образец: бензиловый спирт и З-фенил-1-пропанол. Условия: си-
ликагельная колонка, подвижная фаза ТГФ-гексан; 25 "С ] 11 ]
и двойкой для типичных малых молекул (с молекулярной массой < 500 Да) так,
что в среднем изменение в два раза %В (например, изменение в подвижной фазе
от 100 %В до 50 %В) приведет к увеличению значения к в 2—4 раза.
Графики, аналогичные зависимостям log А: от log %В на рис. 8.4, показаны
на рис. 8.5я для двух менее полярных молекул растворенных веществ и более слабого
В-растворителя хлороформа (с более низкими значениями %В). Поскольку эти
условия не удовлетворяют вышеуказанным требованиям для применения уравнения
(8.4а), получающиеся зависимости описываются кривыми лучше, чем прямыми
(в соответствии с теорией, на которой основано уравнение 8.4а) и наилучшие
приближения значения л < 1. То есть, изменение %В в два раза для этого примера
приводит к намного меньшему, чем в 2—4 раза, изменению к. Однако, использование
более точного уравнения (8.2) для данных на рис. 8.56 дает, как и ожидалось, почти
линейную зависимость величин log к от е°.
8.2.2. Зависимость силы растворителя от В-растворителя
и от %В
Сила растворителя в НФХ зависит от его полярности. Более полярные растворители
сильнее, что приводит к меньшим значениям к образца. Элюирующая сила чистого
растворителя в случае колонки, упакованной непривитым силикагелем, может быть
охарактеризована параметром е из уравнения (8.2). Для чистых растворителей е будет
обозначаться как е . По мере того, как увеличивается значение с, растворитель
становится сильнее, и значения к образца понижаются. Величины е° для некоторых
часто используемых растворителей даны в таблице 8.1. Кроме того, более полный
перечень величин е° чистых В-растворителей дан в приложении I. Величина е для смеси
А- и В-растворителей описывается уравнением [1, 6]:
е = s°A + logl/V^O"'"^» + 1-^1 /пв. (8.5)
Здесь £аА и е^, относятся к величинам е° для чистых растворителей А и В, NB —
мольная доля растворителя В в подвижной фазе и пв— относительный размер (площадь)
о бензиловый спирт
• З-фенил-1-пропавол
8.2. Удерживание 401
w
2.0
1.5 -
logfc
<б)
2.5
2.0
%B = 0.5% 1%
2%
3% 4% 5%
1.5
logfc
1.0 -
0.5
0.0
"Т"
"Т"
"Т"
О нитробензол
• фенантрен
0.00
0.02
0.04
=0
0.06
0.08
Рис. 8.5. Зависимость log А от log %B для разбавленных растворов менее полярного
(нелокализующего) В-растворителя. Образец: нитробензол и фенантрен.
Условия: силикагельная колонка, подвижная фаза хлороформ-гексан; 25 "С.
(а) неудовлетворительные корреляции log А с log ф {уравнение 8.4); (6)
улучшение корреляции log к с с (уравнение 8.2) [II)
растворителя В (относительно величины rtg = 6 для бензола, взятого в качестве
В-растворителя). Уравнение (8.5) подразумевает полностью активированный
адсорбент (не дезактивированный водой), такой как силикагель.
Когда более слабый А-растворитель (например, гексан) смешивается с более
сильным В-растворителем (например, хлористым метиленом), получающаяся
подвижная фаза будет иметь промежуточную элюирующую силу и величину е,
определяемую уравнением (8.5). Рис. 8.6 представляет собой номограмму для НФХ на
колонке, упакованной силикагелем (аналогичную номограмме для ОФХ, показанной
402 Глава 8. Нормально фазовая хроматография
на рис. 6.11), и позволяет сравнить элюирующую силу различных смесей подвижных
фаз в терминах их величин е (см. величины в верхней части рис. 8.6, рассчитанные
по уравнению 8.5). Изменение е на 0,05 единиц влечет за собой изменение величин к
примерно в два раза. Например, подвижная фаза 50% хлористый метилен — гексан
имеет е = 0,24 (пунктирная вертикальная линия на рис. 8.6). Если необходимо
изменить значения к в два раза, то подвижная фаза 30% метиленхлорид-гексан (е = 0,19)
увеличит их примерно в два раза, в то время как подвижная фаза 95%метиленхло-
рид-гексан (е = 0,29) примерно в два раза их уменьшит.
Таблица 8.1. Элюирующая сила растворителя (с0), площадь молекулы (яд) и УФ-погло-
щение в зависимости от длины волны для растворителей, используемых
в НФХ
Растворители
Ацетонитрил"5
Этоксинонафгор-
бутан
Хлороформ"
Этилацетат*
Этиловый эфир*'-''
Гексан, гептан
Метанол"
Метилеихлорид*7
Метил-трет-бути-
ловый эфир1'
н-Пропанол"
Изопропанол1'
Тетрагидрофуранг<'
t0xc
0,52
0,01
0,26
0,48
0,43
0,00
0,70
0,30
0,48
0,60
0,60
0,53
"в
3,1
J
5,0
5,2
4,4
jT
3,7
4,1
4,1
4,4
4,4
5,0
УФ-поглощение (е.п.) при указанной длине волны (им)
200
0,03
210
0,2
220
0,00
230
0,00
240
0,00
250
0,00
260
0,00
Предполагается, что похож на этиловый эфир
> 1,0
> 1,0
> 1,0
0,35
> 1,0
> 1,0
> 1,0
> 1,0
> 1,0
> 1,0
> 1,0
> 1,0
> 1,0
0,20
0,53
> 1,0
0,70
0,65
0,44
> 1,0
> 1,0
> 1,0
0.46
0,07
0,23
> 1,0
0,54
0,35
0,20
0,60
> 1,0
> 1,0
0,27
0,03
0,10
> 1,0
0,45
0,15
0,11
0,40
> 1,0
> 1,0
0,18
0,02
0,04
0,09
0,28
0,07
0,05
0,21
0,25
> 1,0
0,10
0,01
0,02
0,00
0,10
0,03
0,03
0,18
0,08
> 1,0
0,05
0,00
0,01
0,00
0,05
0,01
0,02
0,09
Источник: [6,13].
"Не смешивается с гексаном.
^Неосновный локализующий.
"Нелокализующий.
^Основный локализующий.
''Легко окисляется и поэтому мало употребим на практике.
'Очень сильный (локализующий) растворитель, донор протонов. Классификация на
«основный» — «неосновный» не представляется адекватной.
■"Значения из работы |6|, полученные, как описано в работе |1|.
'Значения лвдля А-растворителей в уравнении (8.5) не требуются.
Подвижные фазы с одинаковыми значениями е на рис. 8.6 должны приводить
к сходным величинам к для конкретного образца. Например, предположим, что
подвижная фаза 50% хлористый метилен-гексан (с = 0,24) обеспечивает, как было
установлено, 1 < к < 10 для данного образца при разделении на силикагельной колонке.
По рис. 8.6 можно предсказать, что каждая из подвижных фаз 3,5% МТБЭ-гексан,
6% ТГФ-гексан или 4% этилацетат-гесан будет иметь близкую элюирующую силу и
поэтому должна обеспечивать диапазон удерживания 1 < к < 10 для одного и того же
образца. Таким образом, рис. 8.6 оказывается полезным при выборе другого В-рас-
8.2. Удерживание
—Г
0.1
-г-
0.2
-г-
0.3
I
0.4
I
0.5
I
0.6
I I I 1 1 1 I | IIIII
г 4 е ю го 50 «оо
т
%v СН2С12-гексан
m iiii i i 111
4 6 10 50 100
1—ГТН 1 1 I ! I I III
2 46 10 50 100
1 |i I I I I 1 I I I 111
%v МТБЗ-гексан
%v ТГФ-гаксан
2 4
%v Этилацвтат-гвксан
6 10 50 100
II llll 1 1 1 I I IIII %*Проланоп-
0,51 г 3 510 20
50 100
~l I llll %v МТБЭ-СН2С(,
10 20 50 100 ч
III I I I I I I I
10 50 100
III IIIII II
10 50 100
%v ТГФ-СНгС1г
%v Этилацетат-СНгС1г
I I I I MINI %vПропаноп-
10 20 50 100 CHjClj
Phc. 8.6. Номограмма элюентов для НФХ и силикагельных колонок |I2|
творителя для изменения селективности (раздел 8.3.2), при этом сохраняются сила
элюента и аналогичные величины к. Вследствие возможных больших изменений
селективности, зависящей от типа растворителя для НФХ (раздел 8.3.2), номограмма
для НФХ-растворителей на рис. 8.6 более приблизительна, чем номограмма
на рис. 6.11 для ОФХ.
На рис. 8.7 показан пример зависимости НФХ-разделения от %В для
произвольной смеси органических соединений на силикагельной колонке с этилацетатом (В)
и циклогексаном (А) в качестве подвижной фазы. Увеличение %В в два раза (40 %В на
рис. 8.76 относительно 20 %В на рис. 8.7я) приводит в этом примере к уменьшению
величин к в 2—3 раза. Кроме того, на рис. 8.7 видны изменения относительного
удерживания при изменении %В. Они будут описаны в следующем разделе 8.3.1 (зависимость
селективности от элюирующей силы растворителя). Вследствие этих изменений в
относительном удерживании при изменении %В наилучшее разрешение для данного
образца достигается в подвижной фазе промежуточного состава (рис. 8.7в).
8.2.3. Использование данных ТСХ для предсказания
удерживания в НФХ
Тонкослойная хроматография (ТСХ) может быть полезным дополнением, если вы
используете ВЭЖХ на силикагельной колонке. Соответствующие разделения с
помощью ТСХ и колоночной хроматографии (при том, что у вас один и тот же
образец, подвижная фаза, температура и особенно точно такой же силикагель в качестве
неподвижной фазы) должны привести к похожим значениям к для каждого
соединения в образце. Как показано на рис. 8.8, расположение отдельных полос (пятен)
на пластинке для ТСХ можно использовать для оценки величин к при
соответствующем разделении на колонке. Величина RF для растворенного вещества в ТСХ
определяется как отношение расстояния, на которое пятно переместилось от старта (того
Глава 8. Нормально фазовая хроматография
(а)
<в)
(в)
20% В, с -0.31
3£kS5,B„ = 0,1
Время (мин)
40% В.е = 0 36
1<fc£3.Rs = 1,3
1 2
Время (мин)
26% В, г = 0.33
2sks4,fis = 1,6
Время (мин)
Рис. 8.7. Зависимость селективности от элюирующей силы растворителя в НФХ.
Образец: I, 2-аминонафталин; 2, 2,6-диметилхинолин; 3, 2,4-диметилхинолин;
4, 4-нитрофенол; 5, хинолин; 6, изохинолин. Условия: силикагельная колонка
150 х 4,6мм, 5мкм; подвижная фаза — смесь этилацетат (В) — циклогексан
(А); комнатная температура; 2,0 мл/мин. Пики 1 и 4 затенены, чтобы
подчеркнуть изменение их относительного удерживания при изменении %В [14|
Яр
1.0 -
0.9 -
0.8 -
0.7 -
0.6 -
0.5 -
0.4 -
0.3 -
0.2 -
0.1 -
0.0 -
к
0.0
0.1
0.2
0.3
0.7
1.0
1.5
2.3
4.0
9.0
^» Г
^а№ В
^т Б
"
■*—
фронт
растворителя
пятно
образца
Рис. 8.8. Использование тонкослойной
хроматографии (ТСХ) для выбора
подвижной фазы, которая будет
использоваться в ВЭЖХ-разделении на сили-
кагельной колонке (гипотетический
образец)
места куда оно было нанесено), к расстоянию, которое было пройдено фронтом
растворителя в процессе разделения. Таким образом, если полоса (пятно) вещества
проходит половину того расстояния, которое проходит фронт элюента (например,
пятно Г на рис. 8.8), то оно будет иметь Яр = 0,5. Величины ftp веществ А—Г на рис. 8.8
соответственно равны 0,15, 0,25, 0,35 и 0,50.
8.2. Удерживание 405
Соответствующие значения к могут быть получены из уравнения
k = (\-Rp)/RF. (8.6)
В примере на рис. 8.8 значения Rp пятен изменяются от 0, 15 до 0,50, что
соответствует 1 < к < 5,7. Аналогичные значения к ожидаются для соответствующего
ВЭЖХ-разделения (при использовании такой же подвижной фазы в сочетании с си-
ликагельной колонкой). Следовательно, обеспечивается приемлемый диапазон
удерживания (1 < к < 10) для этого образца.
ТСХ иногда используют до разделения на колонке, что удобно как способ
заранее определить возможные проблемы и/или исследовать различные подвижные
фазы. Те компоненты (пятна) образца, которые не перемещаются {Rp к 0,0), будут
видны на ТСХ, тогда как растворенные вещества, попавшие в НФХ-колонку, в ней
и останутся и, следовательно, не будут обнаружены детектором и скорее всего
пропущены. Связанное с этим преимущество предварительного ТСХ-разделения
заключается в том, что образцы, которые могли бы при других обстоятельствах загрязнить
колонку и потому требуют предварительной обработки для колоночной
хроматографии (глава 16), как правило, можно непосредственно нанести на пластину ТСХ,
поскольку ТСХ-пластины одноразовые и их после использования можно выбросить.
Изменяя %В методом проб и ошибок (например, для смесей метиленхлорид-гексан)
получают тот %В, который может обеспечить 1 < к < 10, необходимые
для ВЭЖХ-разделения. Это можно проделать за короткое время с помощью ТСХ
при условии, что возможно изократическое элюирование.
Если цель 1 < к < 10 для всех пиков в режиме изократического элюирования,
величины Rp в соответствующем ТСХ-разделении должны быть в интервале между
0,1 и 0,5. Эксперименты с ТСХ могут показать, что образец невозможно разделить
в изократическом режиме на НФХ-колонке (нет значений %В, которые дают зоны
с 0,1 < Rp< 0,5). Изменения в относительном удерживании вследствие зависимости
селективности от элюируюшей силы растворителя (раздел 8.3.1) или его типа
(раздел 8.3.2), также можно исследовать с помощью ТСХ и часто это удобнее, чем с
помощью ВЭЖХ (см. пример в разделе 8.4.3).
Хотя использование ТСХ для целей, указанных выше, достаточно надежно,
следует отметить, что в некоторых случаях обеднение подвижных фаз одним из растворителей
(раздел 8.5.2) может приводить к неверным результатам. Когда в качестве подвижной
фазы в ТСХ используются разбавленные растворы (< 10% В) сильных В-растворителей
(е° > 0.4), В-растворитель может сильно удерживаться силикагелем, что приводит к его
удалению из подвижной фазы во время разделения. В результате концентрация В-рас-
творителя в подвижной фазе будет снижена, элюирующая сила подвижной фазы
уменьшится, и наблюдаемые значения Добудут слишком малы. Таким образом,
значения к, которые были приблизительно рассчитаны исходя из величин Rp, могут быть
завышены в тех случаях, когда происходит обеднение подвижной фазы сильным
растворителем. Эту проблему можно минимизировать, если сначала уравновесить
ТСХ-пластину подвижной фазой. Пластину следует поместить в камеру на 15 минут,
но так, чтобы подвижная фаза не касалась пластины и не началось перемещение
образца [9|. Тогда пары над подвижной фазой уравновесят пластину, и любое обеднение
жидкой подвижной фазы будет сведено к минимуму. После этого пластину опускают
так, чтобы она контактировала с жидкой подвижной фазой, и начинают разделение.
Особенно полезна ТСХ для мониторинга препаративных разделений. Так ряд
фракций, получающихся в результате разделения на колонке, легко
проанализировать одновременно, нанеся на достаточно большую пластину нескольких образцов.
Каждый образец наносится в своей отдельной полосе. Фракции, которые по данным
ТСХ содержат незагрязненный близкорасположенными примесями целевой продукт,
можно объединять для дальнейшей обработки.
406 Глава 8. Нормально фазовая хроматография
Для обнаружения в ТСХ зон компонентов образца, как на рис. 8.8, обычно
требуется добавить проявляющие реагенты. Различные способы детектирования зон
веществ в ТСХ рассмотрены в работе [10|.
8.3. Селективность
Изменения в порядке удерживания вследствие изменений условий разделения могут
быть довольно ярко выражены при фракционировании в режиме НФХ на силикагель-
ной колонке. Гораздо чаще, чем в ОФХ. Эта возможность сильнее влиять на
относительное удерживание в НФХ особенно полезна в препаративных разделениях, где
большие величины а очищаемых соединений позволяют работать с гораздо большими
количествами образца и снизить трудозатраты и стоимость очистки (раздел 15.3.2).
8.3.1. Зависимость селективности от элюирующей силы
растворителя
Пример зависимости селективности от элюирующей силы растворителя приведен
на рис. 8.7. Хотя относительное удерживание пиков 2, 3, 5 и 6 по мере изменения
%В сильно не изменяется, пики I и 4 (заштрихованные) смещаются к фронту хрома-
тограммы, когда %В растет. Важно, что каждая из молекул 2,3,5 и 6 имеет один и тот
же полярный заместитель (-N=). При этом пик 1 соответствует 2-аминонафталину
(-NH2), а пик 4 — 4-нитрофенолу (-N02, -ОН), т.е. молекулы соединений I и 4
замещены иными полярными группами, чем у соединений, выходящих в пиках 2,3,5 и
6. Изменение к при изменении %В для пика 4 больше, чем для других соединений
этого образца, и это можно объяснить наличием в молекуле двух полярных групп
(-N02, -ОН), а не одной как в других соединениях (-N= или —NH2). Таким образом,
п в уравнении (8.4а) должно равняться 2 для пика 4 (при п = 1 для остальных пиков),
и поэтому удерживание пика 4 должно измениться больше для данного изменения
%В. Несколько иное поведение пика I по сравнению с пиками 2,3,5 и 6 может быть
обусловлено отличием заместителя в соединении, соответствующем этому пику
(-NH2 группа), от заместителя в соединениях, соответствующих остальным пикам
(-N=rpynna).
Резюмируя, можно утверждать, что выраженные изменения относительного
удерживания, связанные с изменением %В (зависимость селективности от
элюирующей силы растворителя) более вероятны для образца, содержащего соединения с
различными полярными заместителями и особенно вероятны для соединений с
различным количеством полярных групп в молекуле и, следовательно, с различными
значениями п в уравнении (8.4а).
8.3.2. Зависимость селективности от типа растворителя
Перед тем, как заменить В-растворитель, следует подобрать %В таким образом,
чтобы достичь диапазона 1 < к £ 10 и в то же время получить максимальное разрешение
(как на рис. 8.7#). Если необходимы дальнейшие изменения селективности (а) и
разрешения, В-растворитель можно поменять, сохранив при этом силу растворителя
(приблизительно то же самое значение е по номограмме на рис. 8.6) с тем, чтобы
по-прежнему 1 < к < 10. Замена более полярного В-растворителя обычно является
наиболее эффективным средством для изменения относительного удерживания
в НФХ. Пример зависимости селективности от типа растворителя показан
на рис. 8.9. Это разделение двенадцати нафталинов с различными полярными заме-
8.3. Селективность
стителями. В каждом случае элюирующая сила подвижной фазы приблизительно
одинакова (е = 0,25, см. номограмму рис. 8.6 для подвижных фаз рис. 8.9а,б), так же
как и время разделения (7—Юмин), и поэтому зависимость селективности от элюи
рующей силы растворителя должна быть примерно одна и та же. В этих трех
разделениях наблюдаются многочисленные изменения относительного удерживания
вследствие зависимости селективности от типа растворителя:
(53%СН2С12) 1 <2<3<4<5<6<7 = 8<9< 10< II < 12,
(3,7%МТБЭ) 1<3<2<5<6<7<10<4<11<9<8<12,
<I,4%ACN) 1 <3<2<5<7<6=10<4= II <8<9< 12.
\
4»5
AUUb?_A!i
53% CH2CIZ
Rs = 0,5
11 12
(a)
Время (мин)
4J
1 I 10
3.7% МТБЭ
Bc = 0,8
Л
Луч!.
12
4 6
Время (мин)
1 , 10
—I г ■—i
в ю
1.4% ACN
RB = 0,3
.12
(2)
CHaCI2KACN
4 6
Время (мин)
(д)
СНгС1гкМТБЭ
{«)
(в)
10
(в)
ACN к МТБЭ
о
^ = 0.72 . *
•у'
• ^Г
>*4
log к
tog к
tog к
Рис. 8.9. Пример зависимости селективности от типа растворителя в НФХ. Образец:
/, 2-метоксинафталин; 2, 1-нитронафталин; 3, 1,2-диметосинафталин;
4, 1,5-динитронафталин; 5, 1-нафтальдегид; 6, метил-1-нафтоат; 7, 2-нафта-
льдегид; 8, 1-нафтилнитрил; 9, 1-гидроксинафталин; 10, 1-ацетилнафталин;
//, 2-ацетилнафталин; 12, 2-гидорксинафталин. Условия: силикагельная
колонка 150 х 4,6 мм, 5 мкм; состав подвижных фаз (в объемных %) указан
на рисунке (50% насыщение водой подвижных фаз), за исключением (в)
содержащей 6% метиленхлорида, добавленного чтобы получить смесь с аце-
тонитрилом (А-растворитель в данном случае гексан) 35 "С; 2 мл/мин.
(а—в) Разделения с указанными подвижными фазами; (г—е) корреляции
данных удерживания из (а—в) [15]
408 Глава 8. Нормально фазовая хроматография
Эксперименты показали [16|, что зависимость селективности от типа растворителя
в НФХ коррелирует главным образом с силой В-растворителя (в^). По мере
увеличения Eg В растворитель сильнее связывается с силанольной группой, что приводит
к локализованной адсорбции В-растворителя. Такая адсорбция растворителя показана
на рис. 8.3в стрелкой, которая соединяет силанольные группы на поверхности силика-
геля с молекулами В-растворителя (ТГФ). Такое локализованное взаимодействие
силанольной группы и растворителя можно противопоставить более слабому и более
диффузному взаимодействию силанолов с менее полярным (нелокализующим)
растворителем на рис. 8.3а. На хроматограммах, изображенных на рис. 8.9, В растворите
ли — хлористый метилен с ёд = 0,30 {рис. 8.9я), метил-трет-бутиловый эфир (МТБЭ)
с Eg = 0,48 (рис. 8.96) и ацетонитрил с sд = 0,52 (рис. 8.9в). МТБЭ и ацетонитрил
являются локализующими растворителями (как и подвижные фазы, содержащие эти два
растворителя), в то время как хлористый метилен таким растворителем не является.
По мере увеличения полярности растворенного вещества, оно тоже сильнее
связывается с одной или несколькими силанольными группами (или локализуется
на них), как это показано на примере фенола на рис. 8.3г. Поскольку В-растворитель
и растворенное вещество конкурируют друг с другом за место на поверхности сили-
кагеля, увеличение локализации В-растворителя приведет к относительно большему
уменьшению k для молекул растворенных веществ с более полярными (и,
следовательно, более локализованными) замещающими функциональными группами по
сравнению с менее полярными и менее локализованными группами. То есть,
локализованные молекулы растворенного вещества и В-растворителя конкурируют за одни и
те же адсорбционные центры на поверхности силикагеля. На нелокализующие
растворенные вещества меньше влияет локализация подвижной фазы на силанольных
группах, что приводит к изменениям в относительном удерживании (как на рис. 8.9).
Эти изменения определяются относительной локализацией молекул различных
растворенных веществ. См. |16, 17| для более подробного описания локализации
растворителя и растворенного вещества, а также ее влияния на относительное
удерживание и зависимость селективности от типа растворителя.
Локализующие В-растворители — такие как ацетонитрил и МТБЭ — могут
приводить к небольшим, но существенным различиям в селективности. Для девяти
различных локализующих растворителей [15—17| установлено, что четыре из них (ни-
трометан, ацетонитрил, ацетон и этилацетат) очень похожи по селективности.
Остальные растворители (диметилсульфоксид, триэтиламин, ТГФ, этиловый эфир и
пиридин) в этом отношении существенно различаются. Если сравнить положение
этих растворителей в треугольнике селективности растворителей на рис. 2.9, видно,
что последние пять попадают в группу, обозначенную как «основные растворители»,
в то время как первые четыре находятся в группе «биполярных растворителей» (или
неосновных). Поэтому представляется, что растворители могут быть
охарактеризованы с точки зрения селективности в НФХ как нелокализующие (например, метиленх-
лорид), основные локализующие (например, МТБЭ) и неосновные локализующие
(например, этилацетат или ацетонитрил). С помощью рис. 2.9 можно
классифицировать другие В-растворители как основные локализующие или неосновные
локализующие. Например, МТБЭ является обычно используемым (локализующим)
растворителем в НФХ. Как видно из рис. 2.9 эфиры, такие как МТБЭ, классифицируются
как основные растворители.
На рис. 8.9г-е проводится сравнение относительного удерживания,
обусловленного тремя В-растворителями, с которыми проводились разделения, показанные
на рис. 8.9а-в. Видно, что наибольшие различия в относительном удерживании (или
изменения селективности) происходят в случае нелокализующего В-растворителя
хлористого метилена, а не в случае неосновного локализующего ацетонитрила
(рис. 8.9г) или основного локализующего МТБЭ (рис. 8.9rf). To есть, корреляция на
8.3. Селективность 409 ,
этих графиках не столь велика (0,72 <г^ <0,80). Когда сравнивают удерживание,
обусловленное двумя локализующими В-растворителями (рис. 8.9е), относительное
удерживание хотя и сходно (г = 0,95), но все же достаточно различается, что
приводит к полезным различиям в селективности (как показано на рис. 8.9^ и рис. 8.9в).
Таким образом, в НФХ локализующие свойства растворителя в основном и
определяют селективность, зависящую от типа растворителя. Локализующие растворители
можно еще подразделить на основные, и неосновные. Обычно используемые в НФХ
растворители, собранные в таблице 8.1, охарактеризованы как нелокализующие,
основные локализующие, и неоснбвные локализующие. При исследовании
зависимости селективности от типа растворителя необходимо испытать В-растворитель
каждого типа (как на рис. 8.9). Спирты — очень сильные растворители и доноры
протонов. Их можно классифицировать как локализующие и оснбвные. Они должны
обеспечивать умеренное дальнейшее изменение селективности, особенно для
образцов, содержащих сильные акцепторы протонов.
После того, как проведены разделения с использованием нелокализую-
щих, основных локализующих и неосновных локализующих В-растворителей, как
на рис. 8.9, и подобран %В так, чтобы 1 < к < 10 для каждой подвижной фазы, две
или более из этих подвижных фаз можно смешать для достижения промежуточной
селективности и дальнейшего увеличения разрешения. На рис. 8.10 показано наи
лучшее разрешение, полученное для образца рис. 8.9, в результате смешивания
подвижных фаз разделений, показанных на рис. 8.9б,в (получается 0,03% ACN/0,1%
СН2С12/3,7% МТБЭ/96% гексан). Целенапра&гтенно подобрать наилучший состав
подвижной фазы можно таким же образом, как это сделано при оптимизации
зависящей от типа растворителя селективности для ОФХ (рис. 6.24). Дополнительная
информация приведена в разделе 8.4.2 и на рис. 8.15.
Наилучший выбор растворителей А и В для изменения селективности зависит
от нескольких факторов:
• тип В-растворителя: нелокализующий, основный локализующий или неосновный
локализующий;
• смешиваемость растворителей;
• граница пропускания для растворителя в УФ-диапазоне (для слабо поглощающих
в УФ образцов может потребоваться детектирование на более коротких длинах
волн).
Потенциально полезные для НФХ растворители и их свойства собраны в
таблице 8.1. Для подробного изучения зависимости селективности от типа растворителя
потребуются В-растворители трех типов: нелокализующий, основный локализующий
и неосновным локализующий. В качестве нелокализующего В-растворителя
хлористый метилен предпочтительнее хлороформа из-за его большей прозрачности в УФ
и меньшей токсичности. Однако, детектирование можно проводить при длинах
волн > 230 нм. Хлористый метилен смешивается со всеми растворителями,
приведенными в таблице 8.1.
2/98 смесь подвижных фаз
10 (в) и (б) (оптимум); Rs = 1.3
)иЛШСЛ1 9~Ла „12
2 5,
О 2 4 6 в 10
Время (мин)
Рис. 8.10. Оптимизированные селективность и разрешение для образца на рис. 8.9.
Условия такие же, как и на рис. 8.9, за исключением того, что подвижная
фаза представляет собой смесь подвижных фаз (б) и (в) в соотношении 98/2
410 Глава 8. Нормально фазовая хроматография
В таблице 8.1 указаны пять оснбвных локализующих растворителей. Диэтиловый
эфир и ТГФ легко окисляются кислородом воздуха, и по этой причине их
использовать не рекомендуется. Как нормальный пропанол, так и изопропанол обладают
достаточной элюирующей силой (еРв = 0,60), и это означает, что для подвижной фазы
с £° < 0,25 потребуется концентрация изопропанола <0.5% (как видно на рис. 8.6).
Использование очень низких концентраций В-растворителя может создать
сложности (раздел 8.5), и потому их следует по возможности избегать — особенно при
использовании колонок, упакованных непривитым силикагелем. При выборе оснбвно
го локализующего В-растворителя предлагается использовать МТБЭ для подвижных
фаз с е < 0,48, и нормальный пропанол или изопропанол для создания подвижных
фаз с е° > 0,48. Ни МТБЭ, ни пропанол сложностей при смешивании и
детектировании не создают.
Кандидатами на роль неоснбвных локализующих В-растворителей выступают
этилацетат и ацетонитрил. Этилацетат нехорош тем, что сильно поглощает в УФ
(детектировать в случае этилацетата можно лишь при длинах волн > 250 нм),
а ацетонитрил и гексан смешиваются не полностью. Добавление дополнительного
растворителя (например, метиленхлорида, как это показано на рис. 8.9в) позволяет
использовать гексан вместе с ацетонитрилом, но процесс нахождения нужного
дополнительного растворителя довольно утомителен. Непросто и рассчитать
величину е для смесей этих трех растворителей 118|, но в этом может помочь компьютерная
программа (LSChrom) |19, 20]. Можно не подбирать дополнительный растворитель,
а использовать этоксинонафторбутан в качестве растворителя А |2]| и ацетонитрил
в качестве растворителя В. Это позволит использовать УФ-детектирование на длинах
волн > 220 нм. Этоксинонафторбутан можно купить у компании ЗМ (он смешивается
с ацетонитрилом или метанолом, но стоит дорого). Считается, что с точки зрения
элюирующей силы растворителя (е° я 0,00) гексан и этоксинанофторбутан
взаимозаменяемы.
8.3.3. Зависимость селективности от температуры
Влияние температуры на селективность в НФХ недостаточно хорошо исследовано.
Один из нескольких описанных в литературе примеров приведен на рис. 8.11.
Разделение на рис. 8.1 \а,6 дано для 22 и 55 °С с использованием хлористого метилена
в качестве нелокализующей подвижной фазы. Видно, что селективность существенно
не зависит от температуры, и это может быть в общем случае справедливо для
менее полярных образцов и нелокализующих подвижных фаз. Для разделений
на рис. 8.11 в,г с локализующей подвижной фазой (2% ACN-гексан) изменение
температуры приводит к заметному изменению относительного удерживания
локализованных пиков анализируемых веществ 6 и 7. Такие изменения селективности в
зависимости от температуры, которые показаны на рис. 8.11е, могут быть следствием
относительного уменьшения удерживания локализующих растворителей при высоких
температурах. В этом случае аналогичные изменения в относительном удерживании
могут происходить и при замене локализующего В-растворителя на нелокализую-
щий. На практике изменение температуры как средство для оптимизации
селективности в НФХ ограничено относительно низкими температурами кипения
большинства наиболее употребляемых растворителей и применяется очень редко. С другой
стороны, существенные изменения селективности могут быть достигнуты сменой
В-растворителя, что сводит к минимуму необходимость применения таких
дополнительных средств, как изменение температуры.
8.3. Селективность 41 I
Подвижная фаза 100% CH2CI2
(а)
22 "С
4
55 "С
4
I 1 1 1 1 1 1 1 1 Г-
0 2 4 6 8
Время(мин)
(в)
22 "С
Подвижная фаза 2% ACN-гексан
70 "С
4 6 8 10 12
Время (мин)
(в)
I 1 1 1 1 Г"
0 2 4
Время (мин)
<е)
s | е | у
2 4 6
Время(мин)
Рис. 8.11. Влияние температуры на относительное удерживание в НФХ. Образец:
/, нитробензол; 2, метилбензоат; 3, бензальдегид; 4, ацетофенон; 5, альфа-
метилбензиловый спирт; б, бензиловый спирт; 7, З-фенил-1-пропанол.
Условия: Силикагельная колонка 250 х 4,6 мм, 5 мкм; 2 мл/мин; состав
подвижной фазы и температуру см. на рисунке. Данные из работы [22]
8.3.4. Селективность колонки
Поскольку силикагель в НФХ — наиболее широко используемый носитель и
поскольку перемена В-растворителя — эффективное средство изменения селективности,
то колонку меняют с этой целью нечасто. Более распространенная причина
использования сорбента с полярной привитой фазой связана с тем, что в отличие от
непривитого силикагеля он позволяет избежать проблем, описанных в разделе 8.5: плохой
воспроизводимости от разделения к разделению, долгого уравновешивания колонки
и сложности с использованием градиентного элюирования для образцов с широким
диапазоном удерживания. Это важно в первую очередь для методов анализа. В
нескольких работах |23—27| описано удерживание различных тестовых веществ на
полярных привитых НФХ-сорбентах трех типов (циано-, диол- и аминофазах).
По-видимому, селективность представляет собой сложную зависимость как от типа
сорбента, так и от выбора В-растворителя |27|. Это позволяет предположить, что
замена и сорбента, и В-растворителя методом проб и ошибок может привести к
значительным изменениям относительного удерживания.
На рис. 8.12 сравниваются разделения смеси ароматических соединений в гекса-
не в качестве подвижной фазы на (1) цианофазе, (2) диольной фазе и (3) аминофазе.
Приблизительное представление о разделении того же образца на силикагельной
колонке можно получить из рис. 8.12г, взятого из другой работы |1]. Сравнение этих
четырех разделений позволяет сделать вывод, что время разделения (как и
селективность колонки) изменяется в ряду
силикагель !3> аминофаза > диольная фаза > цианофаза.
Обратите внимание на логарифмическую шкалу времени на рис. 8.!2г для
разделения на силикагеле, верно отражающую тот факт, что интервал удерживания (и селек-
Глава 8. Нормально фазовая хроматография
Циано колонка
Время (мин
(б)
Диольная колонка
4 5
[LLA
Т // * "J "* I™
4 10 12
Время (мин)
Амино колонка
Сили катальная
колонка (оценочно)
t 'к а*
6
1 10 102 103 10* 10* 10е 10'
Время (мин)
Рис. 8.12. Сравнение удерживания и селективности различных НФХ-колонок.
Образец: /, хризен; 2, перилен; 3, 1-нитронафталин; 4, 1-цианокафталин;
5, 2-ацетонафталин; 6, нафталин-2,7-диметилкарбоксилат; 7, бензиловый
спирт. Условия: колонка 150 х 4,6 мм (тип колонки указан на рисунке);
подвижная фаза — гексан; 35 "С; 2,0 мл/мин. Хроматограммы (а-в)
реконструированы по данным из |26|; (г) по данным из [1] (обратите внимание на
экстремальное изменение интервала удерживания на силикагельной колонке г
по сравнению с колонками, упакованными полярными привитыми
сорбентами а—«)
тивности) для образца, разделяемого на силикагельной колонке, может быть гораздо
шире, чем для колонки, упакованной полярным привитым сорбентом.
Как и ожидалось, селективность разделения или относительное удерживание
изменяется умеренно от колонки к колонке на рис. 8.12. Молекулы растворенных
веществ, являющиеся донорами протонов, на аминофазе удерживаются сильнее, чем
другие молекулы, а на цианофазе слабее [26, 27|. Следует также отметить, что
данный образец можно разделить в изократическом режиме на любой полярной
привитой фазе (рис. 8.12а—в), тогда как разделение этого же образца на силикагельной
колонке (рис. 8.12г) потребовало бы градиентного элюирования (если оно вообще
возможно, см. раздел 8.5.2). Это наблюдение должно распространяться на любые
образцы: изократическое разделение скорее окажется возможным на полярном привитом
сорбенте, чем на непривитом сшшкагеле.
В конечном итоге, большие значения а (и возможность разрешения до базовой
линии) более вероятны при использовании силикагельной колонки. Кроме того, из-
8.3. Селективность 413 ))|
менения в селективности при смене В-растворителя (зависимость селективности
от типа растворителя) гораздо более выражены для силикагельных колонок по
сравнению с колонками, упакованными полярными привитыми сорбентами.
Следовательно, замена силикагеля на полярный привитый сорбент с последующим
изменением состава подвижной фазы (изменением %В и/или В-растворителя) вряд ли
улучшит разрешение пары пиков, которые не разделялись после замены
растворителя на силикагельной колонке.
8.3.5. Разделения изомеров
Мы уже отмечали, что изомеры в общем случае лучше разделяются с помощью
НФХ, нежели с использованием ОФХ. Аналогично, некоторые данные |15, 26| дают
основания предполагать, что разделение изомеров обычно проходит лучше на
силикагельных колонках по сравнению с полярными привитыми фазами. Тем не менее,
разделение на рис. 8.26—г с использованием колонки, упакованной цианофазой,
ясно показывает, что некоторые изомеры разделить можно. Селективность в
отношении изомеров в режиме НФХ на силикагельной колонке можно объяснить (как
минимум) тремя возможными свойствами молекул изомеров:
• стерическим препятствием, которое создает полярному заместителю соседний
неполярный;
• отрывом электрона или его оттягиванием от полярной группы вторым
заместителем молекулы растворенного вещества;
• относительным расположением различных полярных групп внутри молекулы и
планарностью самой молекулы.
На рис. 8.13 показан каждый из эффектов, обусловленных этими тремя
свойствами. На рис. %.\Ъа,б видно, что разделение двух изомеров метиланилина будет
затруднено из-за того, что о-метильная группа мешает взаимодействию полярной
NH2-rpynnbi с силанольной группой на поверхности (рис. 8.136). В том случае, когда
метильная группа находится в я-положении, стерических помех нет и этого не
происходит (рис. 8.13«). Стерическое препятствие, созданное о-метильной группой,
будет увеличиваться по мере приближения к поверхности силикагеля. Следовательно,
молекула, у которой есть стерически затрудненный полярный заместитель, должна
удерживаться хуже, чем та, у которой такого заместителя нет или он менее
затруднен. На рис. 8.25 представлен пример эксперимента, в котором демонстрируется
влияние стерического затруднения на селективность при разделении изомеров.
В этом НФХ-разделении я-метиланилин (пик 3) удерживается сильнее, чем о-мети-
ланилин (пик I).
На рис. 8.\Ъв,г метильный заместитель ни в м-, ни в о-положении не создает
для NH2-rpynnbi стерического препятствия. Однако, метильная группа в л-положении
эффективнее нагнетает электронную плотность по направлению к NH2-rpynne, что,
в свою очередь, приводит к повышению основности ее водородной связи и
удерживания. То есть, я-метильная группа имеет большую отрицательную величину
константы а Гаммета |28|, чем .м-метильная. В этом примере, следовательно, л-метильный
изомер должен удерживаться сильнее, чем лг-изомер. Это подтверждается рис. 8.26,
из которого видно, что и-метиланилин (пик 3) удерживается сильнее, чем ,и-метилани-
лин (пик 2). Аналогичные примеры влияния, оказываемого метильными
заместителями на удерживание, иллюстрируются примерами разделений на рис. 8.2в,г.
На рис. &.\3д,е относительное удерживание двух изомеров диметоксиэтилена
будет зависеть от того, какой из них сможет занять лучшую позицию у поверхности
частицы сорбента, чтобы каждая полярная метоксигруппа в молекуле могла
взаимодействовать с соседней силанольной группой на поверхности. Из этого примера вид-
Глава 8. Нормально фазовая хроматография
(а)
(б)
7
/кя\>.
,LJ„J„ J ft
7777777777777777Г/ /////////У////////
77777777777777777Т/ 77?
(в)
H-.N-
(г)
:
4
У J1 Р . . .
л
77777777777777777Т 77777777777777777Г
(а) (в) и
н чосн.
\
Hs. ^Н
W
/ \
t i ft t i
77777777777 7777777777Г ///////////
Рис. 8.13. Факторы, способствующие повышению селективности разделения изомеров
в НФХ на силикагеле. (а,б) Стерические затруднения; (в,г) электронодонор-
ные свойства; (д,е) относительным расположением полярных групп внутри
молекулы растворенного вещества; (ж) внутримолекулярная водородная
связь двух полярных групп
но, что цнс-1,2-диметоксиэтилен будет сильнее удерживаться. Правда, любое такое
предсказание следует рассматривать как предположительное. Силанольные группы
на поверхности силикагеля распределены случайным образом, а молекула
растворенного вещества может занимать на поверхности самые разные положения.
Следовательно, оснований для предсказания, у какого из двух пространственных изомеров
заместители ближе подойдут к соседним силанольным группам, как правило, довольно
мало. В некоторых случаях соседние полярные группы внутри растворенной
молекулы (как в цыс-1,2-дигидроксиэтилене) могут взаимодействовать друг с другом
(рис. S. 1 Зз*с). Это создает возможную конкуренцию межмолекулярным
взаимодействиям и уменьшает их, в то время как именно межмолекулярные взаимодействия
увеличивают удерживание. Эти и другие факторы, влияющие на разделение изомеров
на силикагеле (как на рис. 8.13о-е), подробно рассмотрены в главе 11 работы
MIES конечном итоге, молекулы, имеющие форму, близкую к планарной, легче
сопрягаются с планарной в первом приближении поверхностью силикагеля и, таким
образом, планарные изомеры лучше удерживаются. Пример разделения пяти
изомеров ретинола представлен на рис. 8.14. В ОФХ-разделении на рис. 8.14а эти изомеры
разделяются плохо, тогда как в НФХ-разделении на рис. 8.146 каждый пик хотя бы
частично, но разрешен. Конфигурация соединения, выходящего в пике 5 (все
/яряис-изомеры), ближе к планарной, и удерживание этого пика максимально. Чем
больше в молекулах растворенных веществ цис-связей, тем они менее планарны и
хуже удерживаются.
8.3. Селективность 415 ))|
(а) ОФХ (б) НФХ
15 20 15 20 (мин)
1 11,13-ди-«ис-рети нол
2 13-цис-ретинол
3 9,13-ди-«ис-ретинол
4 9-цис-ретинол
5 полностью транс-ретинол
Рис. 8,14. Сравнение селективности разделений изомеров в ОФХ(о) и НФХ(б).
Образец показан на рисунке (изомеры ретинола). Условия: (а) Колонка Си
200x4,4 мм; 80% метанол-вода; 1,0 мл/мин; 40 "С. (б) силикагельная
колонка 250x4,0 мм; 8% диоксан-гексан; 1,0 мл/мин. Рисунок получен по
данным из |29|
Теперь сравним вклады всех факторов, влияющих на разделение изомеров на си-
ликагеле и на их разделение на (1) полярных привитых НФХ-сорбентах или (2)
ОФХ-сорбентах. В случае полярных привитых НФХ-сорбентов стерические
препятствия (рис. 8.13а,б) будут не так важны, поскольку между поверхностью силикагеля и
молекулами растворенного вещества будут находиться привитые полярные циано-, ди-
ольные или аминогруппы, которые эти пространственные затруднения будут
уменьшать. Аналогичным образом взаимодействие полярных групп в молекуле
растворенного вещества с полярными группами неподвижной фазы будет происходить легче
в случае привитой полярной фазы (и на селективности разделения изомеров это
отразится в меньшей степени) потому, что циано-, диольные или аминогруппы не жестко
связаны с поверхностью силикагеля, а через гибкий -СЬ^-СЬ^-СЬ^-спейсер. В
конечном счете притяжение полярных групп молекулы растворенного вещества к полярной
неподвижной фазе слабеее для привитых полярных фаз, чем для силикагеля, что
в свою очередь снижает влияние каждого фактора на разделение, показанное
на рис. 8.13. Следовательно, по сравнению с разделениями на силикагеле, разделения
изомеров на привитых полярных фазах в общем случае будут менее эффективны.
В случае ОФХ-разделения взаимодействие полярных групп в молекуле
растворенного вещества с неподвижной фазой гораздо слабее, чем в НФХ-разделении. Это
уменьшает влияние всех факторов, отраженных на рис. 8.13 (как и в случае
сорбентов с привитыми полярными группами), и снижает селективность разделения
изомеров. Вместе с тем, возможны аналогичные взаимодействия с полярной подвижной
фазой. Последние, как правило, слабее, чем соответствующие взаимодействия
растворенного вещества и подвижной фазы с силанольными группами, и меньше
зависят от стерических эффектов. Однако, в ОФХ есть одно исключение из этого
правила — циклодекстриновые сорбенты (раздел 6.3.5). Разделение изомеров на этих
сорбентах происходит гораздо лучше, и это можно объяснить. Во-первых, молекула
цикл оде кс три на имеет полость, в которую может войти молекула растворенного
вещества (см. рис 14.17, 14.18). Некоторые молекулы могут помещаться в эту полость
лучше, чем другие. Во-вторых, молекула циклодекстрина имеет много заместителей
(гидроксильных групп), жестко закрепленных внутри молекулы. Эти —ОН группы
циклодекстрина могут играть такую же роль при разделении изомеров в ОФХ, как и
силанольные группы в НФХ (как на рис. 8ЛЗд,е).
А А А3/СН2°Н
полностью транс-ретинол
416 Глава 8. Нормально фазовая хроматография
8.4. Рекомендации по разработке метода
Первым шагом по разработке метода должен быть анализ целей разделения, включая
и анализ выбора НФХ в качестве инструмента разделения. Если не предвидятся
какие либо сложности при использовании ОФХ или они не возникали в пробных раз
делениях образца с помощью ОФХ, то, как правило, начиная разрабатывать метод,
лучше всего использовать ОФХ. Вот некоторые задачи, для которых изначально
стоит выбрать НФХ:
• очистка необработанных препаратов (сырцов);
• разделение изомеров;
• ортогональное разделение;
• образцы, содержащие гидрофобные загрязнения;
• образцы, содержащие очень полярные вещества (например, не удерживаемые
в ОФХ при использовании почти 100% водного элюента).
Очистку (препаративное разделение) растворимых в органических растворителях
образцов, как правило, лучше всего проводить с помощью НФХ на силикагельных
колонках. Органические растворители, используемые в НФХ, проще удалять из уже
разделенных фракций, чем высококипящую воду в том случае, если вы используете
ОФХ. Большие величины а растворенных веществ, которые нужно очистить,
как правило, возможны в НФХ и при использовании силикагеля. Это означает, что
за один раз можно разделить большее количество образца (при прочих равных
условиях). Наконец, удобно использовать ТСХ при предварительном анализе фракций
в препаративной НФХ.
Как отмечалось выше (раздел 8.3.5), изомеры обычно разделяются лучше в
условиях НФХ на силикагеле. Тем не менее, иногие изомеры легко разделяются с
помощью ОФХ, и поэтому может иметь смысл попробовать использовать для таких
образцов поначалу ОФХ, если предыдущие эксперименты с аналогичными образцами
не предполагают другое.
Непокализутащий СН,С1г
В-растворитель СНСЦ
Оснбвный локализующий Неосноеный локализующий
В-растворитель МТБЭ В-растворитель Этилацетат
ТГФ ACN
Рис. 8.15. Семь экспериментов для оптимизации селективности, зависящей в НФХ
от типа растворителя. А-растворитель: гексан, гептан или зтоксннонафтор-
бутан, или (для е° > 0,30) хлористый метилен. Для очень сильно
локализующих В-растворителей используют н-пропанол или изопропанол
8.4. Рекомендации по разработке метода 417 jffl
Ортогональные разделения (раздел 6.3.6.2) могут использоваться для того, чтобы
проверить, позволил ли предлагаемый метод разделить все соединения в образце.
В принципе, селективность ортогональной методики должна как можно сильнее
отличаться от селективности исходного метода. Раньше некоторые исследователи
предполагали, что разделения, основанные на различных принципах (например НФХ и
ОФХ), скорее дадут достаточно большую разницу в селективности — такую, при
которой любая пара пиков, перекрывающихся при использовании одного метода, будет
разделена ортогональным методом. В то время как это предполагает использование
НФХ для ортогональных разделений, можно также разработать два ортогональных
ОФХ-разделения (раздел 6.3.6.2), особенно для случая ионных образцов,
селективность которых больше зависит от условий разделения (раздел 7.3.2).
Одной из менее распространенных причин использования НФХ является нали
чие гидрофобных примесей в образце. Многие образцы содержат гидрофильные
добавки и примеси, которые хуже удерживаются в ОФХ, чем целевые вещества и,
соответственно, их пики. Эти не подлежащие анализу (нецелевые) вещества потом
появляются в виде «мусорных» пиков вблизи tfj (как в примере на рис. 2.56). Если
пики этих нецелевых веществ не перекрывают целевые пики, их можно и не удалять
из образца до ОФХ-разделения. Если же они гидрофобнее целевых пиков, они будут
в ОФХ элюироваться гораздо позже, что приведет к очень большому времени
разделения (как в примере в разделе 8.4.3 ниже). Для их удаления потребуется либо
предварительная обработка образца (глава 16), либо градиентное элюирование, которое
удалит эти примеси за приемлемое время. Любой из двух этих способов может не
подойти или быть неэффективным для некоторых образцов. В таких случаях НФХ раз
деление может стать наилучшим решением, поскольку гидрофобные нецелевые
вещества будут удерживаться слабее, чем целевые, и элюироваться из колонки
вблизи tfj.
Нередко очень полярные образцы плохо удерживаются в условиях ОФХ
(раздел 6.6.1), поэтому они, вероятно, будут хорошо удерживаться с помощью НФХ. В
таких случаях чаще всего начинают с ГИХ (раздел 8.6), особенно если образцы ионнные.
Разработка НФХ-метода проводится аналогично разработке ОФХ-метода.
Для НФХ применяются те же семь шагов, уже описанные в разделе 6.4:
1) определите цели разделения: разрешение, время разделения, пределы
детектирования и т.д.;
2) проведите пробоподготовку (глава 16);
3) выберите условия разделения;
4) проверьте воспроизводимость разделения на колонке, подберите альтернативные
колонки (раздел 6.3.6.1);
5) разработайте рутинное ортогональное разделение (раздел 6.3.6.2);
6) проведите валидацию метода (раздел 12.5);
7) разработайте тест пригодности системы (раздел 12.3).
Для случая препаративных разделений (глава 15) выбор условий имеет
первостепенное значение, а шагами I и с 4 по 7 при разработке метода чаще всего можно
пренебречь. Все НФХ-приложения от ОФХ-приложений отличаются выбором
условий разделения (шаг 3). Следующие четыре шага применяются для разработки
любого изократического ВЭЖХ-разделения (см. раздел 9.3.10 для градиентных методов):
1) выберите начальные условия;
2) подберите %В для получения приемлемого диапазона удерживания (например,
\<Lk< 10);
3) оптимизируйте селективность;
4) оптимизируйте длину колонки, скорость потока и (по возможности) размер частиц
сорбента, чтобы найти оптимальное соотношение между разрешением и временем
разделения.
418 Глава 8. Нормально фазовая хроматография
Шаги от 1 до 3 для НФХ обсуждаются ниже. В таблице 8.2 этому обсуждению
подводится итог. Шаг 4 — выбор конечных условий разделения — выполняется
так же, как и для ОФХ-разделения (раздел 2.4.3).
Таблица 8.2. Разработка метола лля проведения НФХ-разделения
Шаг
1. Начальные
условия
2. %В для
1 </t <10
3.
Оптимизация а
Подход
Выбор колонки
и А- и В-раство-
рителей из
таблицы 8.1
Изменение %
хлористого
метилена в гексане
Изменение
В-растворителя
Смешивание
В-растворителей
Комментарии
Силикагельные колонки, как правило, предпочтительнее.
Альтернатива — диол-модифнцированный силикагель. Гексан
и хлористый метилен — хороший первоначальный выбор
для А- и В-растворителей, соответственно
Можно использовать как ТСХ, так и ВЭЖХ. Начать со
100% CH2CI2. Изменение в два раза %В приведет к
изменению к примерно в три раза. Если нужен более сильный
В-растворитель, то попробуйте МТБЭ, затем изопропанол
Если можно использовать С/^СЬ/гексан в качестве
подвижной фазы, подберите по рис. 8.6 подвижные фазы МТБЭ/гек-
сан или этилацетат/гексан с такой же величиной С.
Если необходимы дальнейшие изменения селективности,
смешайте вышеупомянутые подвижные фазы так, как это
показано на рис. 8.15
"Если смеси хлористый метилен — гексан слишком слабы (к > 10), попробуйте другие
подвижные фазы, показанные на рис. 8.6.
8.4.1. Начальные условия для разработки НФХ-метода:
выбор элюирующей силы подвижной фазы и типа
сорбента (шаги 1, 2; таблица 8.2)
При разработке ОФХ-метода было рекомендовано начинать с колонки С]8 100x4,6 мм
(с частицами диаметром 3 мкм) и 80% ACN в качестве подвижной фазы
(таблица 6.1), а затем корректировать %ACN с тем, чтобы достичь I <, к 5 10. Для НФХ
приемлемой отправной точкой может быть колонка, упакованная непривитым сили-
кагелем или силикагелем с привитой полярной фазой. Силикагельные колонки
предпочтительнее для препаративных разделений и фракционирования изомеров, в то
время как на колонках, упакованных силикагелем с привитыми полярными фазами,
проще делать анализы. Кроме того, такие колонки имеют то преимущество, что
на них можно анализировать образцы с широким интервалом удерживания
компонентов, для которых может потребоваться градиентное элюирование при
использовании колонок с непривитым силикагелем.
Выбор исходной подвижной фазы включает в себя ряд соображений,
приведенных в разделе 8.3.2.1. Выбор элюирующей силы подвижной фазы (%В) удобно делать
на основе предварительных данных ТСХ-разделений на силикагельных пластинках
(раздел 8.2.3). В качестве альтернативы можно также использовать градиентное
элюирование на привитом полярном сорбенте (раздел 9.3.2), особенно если последний
будет использоваться для окончательного разделения (вместо силикагеля). В любом
случае, целью должно быть получение подвижной фазы такого состава, который
обеспечит 1 < к <. 10 в дальнейшем разделении (что эквивалентно 0,1 < Rp < 0,5
при ТСХ-разделении). Авторы рекомендуют проводить исходное ТСХ-раздедение
на силикагельных пластинках со 100% хлористым метиленом (В-растворителем)
в качестве подвижной фазы. Если значения RF слишком велики, то подвижная фаза
8.4. Рекомендации по разработке метода 4191)|
слишком сильная, и можно попробовать снизить процент хлористого метилена
(добавив гексан в качестве А-растворителя), используя в качестве руководства раздел
8.2.2. Если при использовании 100%-ого хлористого метилена величины Rf слишком
малы, значит для подвижной фазы нужно взять более сильный В растворитель.
В этом случае для последующих ТСХ-разделений лучше пробовать смеси МТБЭ
и гексана. Если требуется еще более сильный В-растворитель, попорбуйте смеси н-
или изопропанола с гексаном в качестве А-растворителя. В каждом из последних
случаев, номограмма для элюентов на рис. 8.6 может быть полезна при выборе
величин %В для проведения успешных разделений.
При корректировке %В с целью получить I < к < 10, уменьшение значений к в два
раза требует увеличения с на = 0,05 (уменьшение е на = 0,05 приведет к двукратному
увеличению к). Предположим, например, что ТСХ-разделение со 100% хлористым
метиленом в качестве подвижной фазы дает 0,5 < Rf < 0,8 для зоны образца, что
соответствует 0,2 <к < I (рис. 8.8 или уравнение 8.5). В этом случае нам нужно увеличить
к в 5—10 раз. Уменьшение е на 0,05, 0,10 или на 0,15 единиц должно увеличивать
значения к примерно в 2, 22 = 4 или в 23 = 8 раз, соответственно. Для этого примера
рекомендуется уменьшение е на 0,15. Обращались к рис. 8.6, мы видим, что
для 100% метиленхлорида е = 0,30. Это означает, что рекомендуемая подвижная фаза
будет иметь е° = 0,15 и согласно рис. 8.6 это будет подвижная фаза состава 18% мети
ленхлорид-гексан. Поскольку оценка зависимости к от %В в НФХ может быть менее
точной, обычно требуется дополнительная корректировка %В для достижения
желаемого диапазона удерживания 1 < к < 10, как и для того, чтобы воспользоваться
преимуществами влияния элюирующей силы растворителя на селективность.
По завершении пробных экспериментов с ТСХ на силикагельных пластинках
нам нужно выбрать колонку для НФХ (если ТСХ невозможно использовать для
рутинного анализа). Силикагельные колонки обеспечивают более высокие значения а
для препаративных разделений и разрешения изомеров, тогда как при работе с
колонками, упакованными привитыми полярными фазами, возникает меньше
трудностей, описанных в разделе 8.5. Кроме того, они более удобны при разделении
образцов, диапазон удерживания которых превышает 0,5 < к < 20 (по оценкам
предварительных ТСХ-разделений). Удерживание на привитых полярных сорбентах
намного слабее, чем на силикагеле (меньшие значения к). Если подвижная фаза,
подобранная как это описано ранее с помощью ТСХ на силикагельной пластинке,
имеет е < 0.15, удерживание на привитом полярном сорбенте может быть слишком
слабым, чтобы обеспечить I < к < 10 даже тогда, когда в качестве подвижной фазы
используется чистый гексан (е° = 0,00).В этом случае привитый полярный сорбент
лучше не брать.
После того, как по результатам предварительных ТСХ-экспериментов (для 0,1 <
< Я/г <. 0,5 или 1 < к <, 10) состав подвижной фазы выбран, эту подвижную фазу
можно использовать для разделений на колонке, упакованной силикагелем. Если
все же используется привитый полярный сорбент, то элюирующую силу
подвижной фазы следует уменьшить примерно на 0,15 единиц е. Например, если состав
рекомендуемой подвижной фазы должен был бы быть 40% метиленхлорид —
гексан (для силикагеля), соответствующая величина е° = 0,22. Для привитого
полярного сорбента величина е° на рис. 8.6 должна быть 0,22—0,15 и 0,07. Величина е° =
= 0,07 на рис. 8.6 соответствует подвижной фазе состава 5% метиленхлорид —
гексан, которую можно использовать для первоначального разделения на колонке
с привитым полярным сорбентом. Имейте в виду, что оценки элюирующей силы
подвижной фазы по рис. 8.6 для силикагеля (из расчета, что двукратное
изменение к достигается, если изменить е° на 0,05 единицы) являются
приблизительными, а распространение этого правила на колонки, упакованные привитой полярной
фазой, еще менее надежно.
420 Глава 8. Нормально фазовая хроматография
8.4.2. Стратегии оптимизации селективности
(шаг 3; таблица 8.2)
Нахождение подходящей величины %В для 1 <, к <. 10 обычно будет включать
эксперименты по ее изменению. В то же самое время можно исследовать зависимость
селективности от элюирующей силы растворителя (раздел 8.3.1). Если требуются
дальнейшие изменения селективности, необходимо исследовать ее зависимость
от природы растворителя (раздел 8.3.2), поскольку последний сильно влияет на
относительное удерживание и разрешение. Для исчерпывающего исследования
зависимости селективности от типа растворителя необходимы три растворителя: нелокали-
зующий, основный локализующий и неосновным локализующий (см. рис. 8.9). Две
из этих трех бинарных смесей растворителей могут быть в дальнейшем смешаны друг
с другом в различных соотношениях с целью получения промежуточной
селективности (как в примере на рис. 8.10). На рис. 8.15 представлена схема экспериментов
для реализации такого подхода со списком В-растворителей каждого типа.
Начальные эксперименты со смесью хлористый метилен — гексан могут быть
использованы для определения величины %В (и с), при которой можно достичь
приемлемого удерживания (1 S к й 10). Это разделение соответствует эксперименту I
на рис. 8.15. В том случае, если необходимо дополнительно повысить селективность и
разрешение, проводятся эксперименты 2 и 3, проиллюстрированные на рис. 8.15
(например, берутся смеси МТБЭ-гексан и этилацетат — гексан, имеющие такую же
величину е, как и в эксперименте 1, см. рис. 8.6). Рассмотрение трех последних хромато-
грамм покажет, может ли дальнейшее смешивание этих трех подвижных фаз
(эксперименты с 4 по 7) обеспечить какое-нибудь улучшение селективности и
разрешения. Эта процедура оптимизации селективности, зависящей от типа растворителя,
может быть достаточно полезной, особенно при использовании силикагельных
колонок. План на рис. 8.15 можно сравнить с аналогичной оптимизацией селективности
в ОФХ при использовании разных растворителей, которая проиллюстрирована на рис.
6.24 (см. подробности в соответствующем тексте). Когда смешиваются подвижные
фазы, использовавшиеся в экспериментах 1-3 по методике рис. 8.15, получающиеся
значения е могут и не оставаться одинаковыми, требуя изменений концентрации
А-растворителя. В литературе [30| сообщается о компьютерной программе для более
точного предсказания величин е в зависимости от состава подвижной фазы; программа
основана на методе, приведенном в |31]. Доступна ее демоверсия |19|.
8.4.3. Пример разработки НФХ-метода
Требовался метод разделения для образцов, содержащих полярный лекарственный
препарат паклитаксел в сочетании с более гидрофобным полимером (сополимером
себациновой и рицинолеиновой кислот) |32]. Структуры этих двух соединений
показаны внизу на рис. 8.16. Поскольку паклитаксел более полярен, чем полимер,
для ОФХ-анализа потребовалось бы либо предварительно отделить полимер от
лекарственного препарата (потому что в ОФХ полимер очень долго элюируется), либо
элюировать его в градиентном режиме. По этой причине попробовали НФХ-разделе-
ние с целью элюировать полимер перед препаратом (вблизи /а) и тем самым
исключить необходимость предварительной обработки образца. Первоначальные
эксперименты проводили на силикагельных ТСХ-пластинках. Используя чистый хлористый
метилен, для паклитаксела получили RF = 0.00, поэтому перешли к более
сильным В-растворителям ТГФ (S = 0,53) и метанолу (еР = 0,70) в смесях с хлористым
метиленом. Результаты, полученные на ТСХ с подвижными фазами, состоящими из
2—5% МеОН—СН2С12, показались многообещающими; эти результаты представлены
8.4. Рекомендации по разработке метода 411
(в)
Свежий образец
« Полимер ►
Паклитаксел
х\
<в)
I 1 И"
Деградировавший полимер
ИзС^оДс^о;
Рис. 8.16. НФХ-анализ паклитаксела в присутствии полимерной добавки. Условия:
силикагельная колонка 250x4,0 мм, 5 мкм; 1,5% МеОН—CH2CI2; 25 "С;
1 мл/мин. (а) Свежеприготовленный образец; (б) образец деградировавшего
полимера (хранился при рН 7,4 и 37 "С 60 дней); (с) деградировавший
образец полимера и паклитаксела |32|
в таблице 8.3 (ТГФ был бы менее желателен по соображениям, изложенным в
разделе 8.3.2.1). Подвижная фаза 1,5% МеОН—CHjC^ обеспечивает приемлемое
разделение, показанное на рис. 8.16а (УФ-детектирование при 240 нм). Отметим, что
в разделении на рис.8.16а для пика паклитаксела k = 3, в то время как ТСХ-разделе-
ние предполагало к = 6. Предсказания удерживания в НФХ, исходя из результатов
экспериментов с ТСХ, весьма приблизительны, но такие предсказания могут быть
полезны, как например, в настоящем примере.
422 Глава 8. Нормально-фазовая хроматография
Таблица 8.3. Экспериментальные ТСХ-разделения образцов смеси паклитаксела и
полимера
МеОН-СН2С12
<*МеОН
]
2
3
4
5
Я,,, п а клита ксела
0.04
0,22
0.29
0.35
0.46
Яр ноли мери
0,05
1,00
1,00
1,00
1,00
ТГФ -CH2CL2
%ТГФ
2
4
9
20
30
Rf паклитаксела
0,00
0,00
0,12
0,52
0,88
Af полимера
0.56
0,64
1.00
1,00
1.00
Источник: 1321.
Метод, проиллюстрированный на рис. 8.16а, предназначался также для работы
с термически деградировавшими образцами, хроматограммы которых приведены
на рис. 8.16 б,в. Видно, что термическое разложение полимера (рис. 8.166) не
приводит к образованию соединений, пики которых перекрывают пик паклитаксела и
таким образом негативно сказываются на этом анализе. Другие детали разработки
этого НФХ-метода см. в 1321.
8.5. Трудности, возникающие
при использовании НФХ
В процессе НФХ-разделения могут возникать сложности, связанные с упакованным
в колонку силикагелем:
• плохая воспроизводимость разделения (включая высокую чувствительность к
содержанию воды в подвижной фазе);
• обеднение подвижной фазы;
• медленное уравновешивание колонки при смене подвижной фазы;
• уширение задних фронтов пиков.
Первые три проблемы возникают вследствие очень сильных взаимодействий
небольших полярных молекул с силанольными группами на поверхности силикагеля.
Это, в свою очередь, может оказать существенное влияние на удерживание образца.
8.5.1. Плохая воспроизводимость разделения
Время удерживания образцов в НФХ может меняться день ото дня или даже в
течение одного дня в результате значительных перепадов влажности, что приводит к
изменению содержания воды в номинально «сухих» растворителях, составляющих
подвижную фазу. Этот эффект продемонстрирован на рис. 8.17я для элюирования
бензанилида с силикагельной колонки хлористым метиленом в качестве подвижной
фазы. В этом примере подвижная фаза была приготовлена смешиванием различных
объемов хлористого метилена, который был или сухим, или насыщенным водой.
Делалось это для того, чтобы достичь различных концентраций воды в получаемой
подвижной фазе (см. верхнюю часть рис. 8. \1а). Например, смешивание равных
объемов двух растворителей приведет к 50% насыщению водой конечной подвижной
фазы (0,08% воды). По мере увеличения содержания воды в подвижной фазе
удерживание растворенного вещества резко уменьшается (от к = 9 до к = 2) из-за
увеличения площади покрытия поверности силикагеля адсорбированной водой (вода очень
S.5. Трудности, возникающие при использовании НФХ 423
сильно взаимодействует с силикагелем). Из этого примера становится понятно, что
небольшие изменения влажности воздуха в помещении могут привести к
значительным изменениям процента насыщения элюента водой, а это, в свою очередь,
приведет к изменению удерживания образца. Эти изменения степени насыщения водой
подвижной фазы являются наиболее распространенной причиной нестабильного
удерживания в НФХ при использовании силикагельной колонки несмотря на то, что
колонка была должным образом уравновешена перед вводом образцов (раздел. 8.5.2).
Способом, который описан выше (смешиванием безводной подвижной фазы с во
донасыщенной) можно свести к минимуму изменения содержания воды в подвижной
фазе. Альтернативным и более удобным способом является добавление небольших
количеств очень полярного растворителя к подвижной фазе. Как видно на рис. 8.176,
добавление небольших количеств метанола приводит к уменьшению удерживания
образца — аналогично тому, как это происходит при добавлении воды, вызывающей
сопоставимую дезактивацию поверхности силикагеля. Скорее всего, добавление
метанола делает сорбент менее восприимчивым к изменениям количества воды.
Количество метанола, необходимое для дезактивации силикагеля, будет меняться в
зависимости от состава подвижной фазы и будет уменьшаться для более слабых подвижных фаз
(с меньшими значениями е). Ограниченная смешиваемость метанола и гексана
предполагает, что метанол в случае необходимости следует заменять пропанолом. Обратите
внимание, что по мере увеличения концентрации воды или метанола в подвижной
фазе удерживание образца уменьшается, и может потребоваться добавить гексан в
подвижную фазу для уменьшения е. Способность силикагеля разделять изомеры и другие
образцы будет снижена чрезмерной дезактивацией колонки.
Перемены в удерживании вследствие изменения количества воды в подвижной
фазе (как на рис. 8.17а) должны быть менее выраженными (1) для более полярных
подвижных фаз с большими значениями е или (2) для менее полярных привитых фаз
(поскольку вода не так прочно адсорбируется на поверхности таких сорбентов).
Следовательно, для полярных привитых фаз нестабильность удерживания не столь
характерна, как для силикагеля. Колебания времени удерживания на си.шкагельных
колонках могут и не создавать проблем, если контролируются образец, подвижная фаза
и относительное постоянство влажности в помещении и если растворители из своей
тары переносятся в резервуары для элюентов при минимальным контакте с
атмосферой. Поэтому имеет смысл подождать, пока колебания времен удерживания не
приведут к тому, что придется корректировать процент насыщения водой подвижной фазы
или добавлять такой полярный растворитель как метанол.
(а)
(б)
0.05 0.10 0.15
0 SO 100
%-нзсыщаемости водой
0.0S 0.10 0.15 0.20
%МеОН
Рис. 8.17. Влияние полярных дезактинаторов на удерживание в НФХ. Образец: бенза-
нилид. Условия: силикагельная колонка типа В; подвижная фаза —
хлористый метилен с изменяемой концентрацией воды (а) или метанола (6);
35 "С |37|
424 Глава 8. Нормально фазовая хроматография
Вторая возможная причина колебаний времен удерживания в НФХ может быть
следствием изменений состава подвижной фазы в результате барботирования
последней гелием. Происходящее при этом испарение подвижной фазы может привести
к преимущественной потере одного растворителя по сравнению с другим |33|, а
также к изменению в ней содержания воды. Такие изменения в составе подвижной
фазы можно уменьшить с помощью ее онлайн-дегазации (раздел 3.3.3).
8.5.2. Обеднение элюента растворителем и медленное
уравновешивание колонки
Когда разбавленные растворы полярного растворителя вступают в контакт с силика-
гельной колонкой или с ТСХ-пластинкой, полярный растворитель селективно
поглощается силикагелем. Это, как правило, приводит к обеднению подвижной фазы
полярным растворителем и к уменьшению наблюдаемых значений Л/г и к большим
величинам к. Обеднение подвижной фазы растворителем является, в основном,
проблемой при использовании ТСХ и градиентного элюирования, поскольку изократи-
ческому разделению обычно предшествует уравновешивание колонки растворителем
(раздел 2.7.1). Обеднение подвижной фазы понижает ее силу в ТСХ, поэтому
разделения с помощью ТСХ и колоночной хроматографии, как правило, неэквивалентны.
Следовательно, значения к, оцененные в ТСХ-разделениях, могут быть слишком
высокими, как в примере из раздела 8.4.3 (в этом примере они не обязательно связаны
с обеднением элюента). При градиентном элюировании на силикагельной колонке,
когда концентрация полярного В-растворителя растет в ходе разделения, обеднение
им элюента может привести к прерыванию градиента и к вызванному этим ухудшению
разделения. По этой причине часто избегают градиентного элюирования в НФХ на си-
ликагеле. Тем не менее, Майер показал |34|, что градиенты МТБЭ (е°= 0,48) в гексане
не приводят к подобным проблемам. То же самое справедливо и для других градиентов
на силикагельных колонках в том случае, если разница в значениях е для А- и В-элю-
ентов не более 0,5. См. также обсуждение градиентного элюиорвания в НФХ в |35|.
Для изократического НФХ-разделения обеднение элюента растворителем
обычно не представляет собой проблему, но может потребовать более длительного
предварительного уравновешивания колонки. Из-за обеднения элюента растворителем
при смене подвижных фаз уравновешивание колонки в НФХ происходит, как
правило, медленнее, чем в ОФХ. Для уравновешивания колонки требуется больший объем
подвижной фазы перед вводом образцов. Уравновешивание колонки может быть
особенно медленным в случае менее полярных подвижных фаз, если процент
насыщения водой новой подвижной фазы значительно отличается от того, что был в
предыдущей. Причина в том, что емкость колонки по воде (вес воды/вес неподвижной
фазы) намного больше, чем у подвижной фазы, и поэтому для того, чтобы
достаточное для уравновешивания неподвижной фазы количество воды было перенесено
подвижной фазой, необходимо прокачать через колонку ее большой объем. Привитые
полярные фазы в меньшей степени подвержены обеднению элюентов и медленному
уравновешиванию потому, что полярные растворители такими сорбентами меньше
удерживаются (по сравнению с силикагелем). Привитые полярные фазы, таким
образом, более пригодны для градиентного элюирования.
8.5.3. Уширение задних фронтов пиков
Уширение заднего форнта пика с давних пор было более серьезной проблемой
для НФХ, чем для ОФХ. Кроме того, количество теоретических тарелок N зачастую
меньше в НФХ, чем в ОФХ; возможно это обусловлено более медленной диффузией
8.6. Хроматография гидрофильных взаимодействии (ГИХ) 425
на поверхности силикагеля (см. диффузия на поверхности неподвижной фазы |36|).
Как и в случае с ОФХ (разделы 5.2.2.2 и 7.3.4.2), эффективность колонки в НФХ
значительно повышается при использовании силикагеля типа В вместо более старого
силикагеля типа А [37|. Пик, как правило, становится острее, и количество
теоретических тарелок больше. По этой причине настоятельно рекомендуется использовать
силикагель типа В для аналитических НФХ-разделений. Для препаративных
разделений применение более дорогого силикагеля типа В может быть неоправданным, тем
более, что эти разделения зачастую проводятся и при более низких величинах N
(раздел 15.4.1.1).
8.6. Хроматография гидрофильных
взаимодействий (ГИХ)
Хроматография гидрофильных взаимодействий (ГИХ) может рассматриваться как
нормально-фазовая хроматография с водно-органической подвижной фазой |38—40|.
По этой причине она иногда называется «водной нормально-фазовой
хроматографией». ГИХ-колонки более полярны чем ОФХ-колонки, а более полярная вода
служит более сильным В-растворителем в ГИХ, и увеличение % воды приводит к
уменьшению удерживания образца (в ОФХ все наоборот). На рис. 8.18 приведен пример
разделения смеси нейтральных олигосахаридов с помощью ГИХ с использованием
воды и ацетонитрила в качестве элюентов. По мере увеличения % воды удерживание
уменьшается (см. также похожий пример на рис. 8.19, где проиллюстрировано
ГИХ-разделение нескольких пептидов). Более полярные и/или ионизированные
растворенные вещества, как правило, при прочих равных условиях лучше удерживаются
в ГИХ (и снова в ОФХ все наоборот, но для НФХ обычная ситуация). В большинст-
(з)
40% H20/ACN
I ■ ' '
О ГО 20 30
16)
30% H20«CN
0 t0 20 30
1 (в)
20% HSCVACN
в 7
Г —— — г ■ ■ , 1
0 10 20 30
Время(мин)
Рис. 8.18. Разделение смеси дериватизированных олигосахаридов с помощью ГИХ
в подвижных фазах с различим % воды. Условия: колонка PolyHydroxy-
ethyl A 200 х 4,6, 5 мкм; подвижные фазы вода-ацетонитрил, содержание
воды указано на рисунке; 2 мл/мин. Значения л для каждого пика на
рисунке относятся к количеству мономеров в соответствующем олигосачариде.
Хроматограммы построены поданным из [381
I
-Жа^у_
llV*
426 Глава 8. Нормально-фазовая хроматография
ве ГИХ-разделений содержание воды в подвижной фазе изменяется от 3 до 40%. Как
правило, слабое удерживание (А: = 0) характерно для тех случаев, когда содержание
воды > 40%, хотя иногда, для некоторых образцов и сорбентов, удерживание может
начать расти по мере увеличения содержания воды выше 40% (т.е. начинает себя
вести как в ОФХ) |42|.
Преимущественное удерживание растворенных полярных веществ в ГИХ
означает, что многие образцы, плохо удерживаемые в ОФХ (к = 0). могут лучше разделяться
в условиях ГИХ. Кроме того, у ГИХ есть несколько других, потенциально выгодных
особенностей:
• хорошая форма пиков основных растворенных веществ;
• увеличивается чувствительность масс-спектрометрического анализа;
• возможность прямого ввода образцов, растворенных в основном в органическом
растворителе (что не годится для ОФХ, см. раздел 2.6.1);
• возможны более высокие скорости потока (или более низкие давления на
колонке) вследствие меньшей вязкости подвижной фазы (таблица 1.4. приложение I).
В то время как относительное удерживание для НФХ на силикагеле, как
правило, диаметрально противоположно удерживанию в ОФХ (рис. 8.16), удерживание
в ГИХ часто находится между двумя этими противоположностями. Последнее
наблюдение может свидетельствовать о том, что «полярность* растворенного вещества
представляет собой сложную зависимость от (I) структуры молекуры и (2) видов
взаимодействия образец-сорбент, которые существенным образом влияют на
удерживание и особенно на селективность. Многие (возможно большинство)
ГИХ-разделений проводятся в режиме градиентного элюирования. Тем не менее,
нижеследующее обсуждение изократического ГИХ-разделения одинаково приложимо и
для градиентного элюирования в ГИХ, которое более подробно рассмотрено далее в
разделе 9.5.3.
8.6.1. Механизм удерживания
Хотя «механизму удерживания» в ГИХ и уделено большое внимание в литературе, эта
тема представляет собой, главным образом, академический интерес. Поскольку
предмет обсуждения имеет очень ограниченное практическое применение, читатель может
этот раздел пропустить.
Считается, что удерживание в ГИХ включает в себя распределение молекул
растворенного вещества в слое воды, который образуется на поверхности упакованного
в колонку сорбента |38, 39, 42, 44|. Было показано |42|, что в условиях ГИХ для
упакованного в колонку силикагеля мертвый объем колонки Vm продолжает
уменьшаться по мере того, как увеличивается от 0 до 30% содержание воды в подвижной фазе.
Это объясняют тем, что на поверхности силикагеля нарастает слой воды. Экспери-
ментатьно установлено, что ГИХ-разделения требуют, чтобы в подвижной фазе
содержалось, как минимум, 2—3% воды, что подтверждает важность присутствия
при ГИХ-разделении этого слоя воды, который предположительно входит в состав
неподвижной фазы. Кроме того, возможно, в неподвижную фазу включается и
некоторое количество органического растворителя (ацетонитрила) из подвижной фазы,
в результате чего в удерживание могут вносить вклад взаимодействия растворенного
вещества как с поверхностью сорбента (силанольными группами), так и с любыми
его лигандами.
Можно разграничить адсорбцию на поверхности неподвижной фазы и
распределение в ней же, как в случае ОФХ (раздел 6.2.2). Некоторые исследователи считают,
что эти два процесса можно отличить путем сравнения зависимости величин k
от объемной доли (ф) В-растворителя (в данном случае воды). В ОФХ, которая, как
8.6. Хроматография гидрофильных взаимодействий (ГИХ) 427
1 1 1 1 1
%HjO=S 10 20 30 40
-1.4 -1.2 -0.8 -0.4
Рис. 8,19. Изменения в ГИХ-удерживании различных пептидов в зависимости от элю-
ирующей силы подвижной фазы (изменение % воды). Условия: Колонка
TSK gel Amide-80 250 х 4,6мм; 1,0 мл/мин; 40 °С. Каждый из пептидов,
содержащихся в образце может быть охарактеризован количеством я
аминокислотных остатков в молекуле (величины л показаны) [41]
полагают, включает в себя процесс распределения (по крайней мере, в некоторых
случаях), наблюдается приблизительно линейная зависимость log к от <р (рис. 6.3).
В НФХ на силикагеле, где, как считается, преобладает адсорбция, более линейными
оказались графики зависимости log к от log ф (рис. 8.4). При рассмотрении
зависимости к от ф для ГИХ-разделений не найдено четкой взаимосвязи для всех систем,
но, как правило, линейностью в большей степени характеризуются графики
зависимости log к от log ф; см. пример на рис. 8.19 для набора пептидов (величины п
на рис. 8.19 соответствуют количеству аминокислотных остатков в молекуле
растворенного пептида). В то время как линейность графиков зависимости log к от log ф
свидетельствует в пользу того, что удерживание в ГИХ скорее обусловлено
адсорбцией, а не распределением, неподвижная фаза в ГИХ безусловно представляется
очень сложной, обеспечивающей возможность различных взаимодействий молекул
растворенного вещества и с водным слоем, и с поверхностью силикагеля, и с
различными лигандами, привитыми к силикагелю. Поэтому любые выводы о том,
преобладает ли в ГИХ адсорбция или распределение, преждевременны и имеют мало
практической ценности.
8.6.2. Колонки
Почти все существующие сорбенты для ГИХ имеют подложку из силикагеля.
Изначально использовались колонки, упакованные аминопропильными сорбентами,
в основном, для разделения углеводов. Аминопропильные колонки хорошо подходят
для этих целей, поскольку они предотвращают удвоение пиков для каждого из
веществ, вызванное разделением аномеров. Впоследствии в ГИХ стали использовать
Глава 8. Нормально фазовая хроматография
YMC Sil
С-2.в = -0.15
15 (мин)
Рнс. 8.20. Уширение задних фронтов пиков пиримидинов на некоторых колонках
для ГИХ. Условия: силикагельные колонки (YMC SIL, Nucleosil silica и Zor-
bax SIL) 250 x 4,6 мм; подвижная фаза: 75% ацетонитрил/буфер (5 мМ
фосфорная кислота); 1,0 мл/мин; комнатная температура. Величины С-2,8 были
измерены для C]g ОФХ-колонок, полученных из того же источника, что и
колонки для ГИХ |48|
различные привитые сорбенты на основе силикагеля |39, 40, 45|. Классифицировать их
можно следующим образом: непривитый силикагель, полярные нейтральные
(например, цианопропильные) фазы, привитые диольные, амидные и полипептидные фазы,
положительно заряженная привитая аминофаза (анионообменник) и отрицательно за
ряженная (катионообменник), а также цвиттерионные фазы. В то время как различная
селективность колонок, упакованных этими сорбентами |46|, может оказаться
полезной при разработке метода разделения, непривитыи силикагель является наилучшим
выбором для множества ГИХ-разделений, в которых используется ЖХ-МС |39, 43, 47|
из-за того, что не разрушается структура привитой фазы и не происходит отшепления
привитых лигандов. Тем не менее, сообщалось о необратимом удерживании некоторых
соединений на силикагеле, и в этом случае обычно используют в качестве
альтернативы амидные фазы |40|. Подробный список колонок для ГИХ, обсуждение их свойств
и вебсайты их производителей приведены в работе [391.
В ранней работе [48| отмечено значительное уширение задних фронтов пиков
для ГИХ-разделения основных пиримидинов при низких значениях рН на
непривитом силикагеле (рис. 8.20). На рис. 8.20 видно, что уширение задних фронтов пиков
увеличивается для колонок с большими значениями параметра С-2,8 измеренными
для соответствующего привитого силикагеля Cjg (раздел 5.4.1.1). Значения С-2,8 >
> 0,25 для Cjs-колонок говорят о не очень чистом силикагеле типа А с более
выраженными кислотными свойствами (раздел 5.2.2.2), позволяя предположить, что такое
уширение заднего фронта пика, как на рис. 8.20, связано с использованием
силикагеля типа А для изготовления сорбентов. Поскольку большинство используемых
сегодня сорбентов изготовлено из силикагеля типа В, уширение задних фронтов
пиков, как на рис. 8.20б,е, должно быть менее вероятным в ГИХ-разделениях на
колонках, упакованных такими сорбентами. Необратимая адсорбция растворенного
вещества на колонках с силикагелем типа В также должна происходить реже.
8.6. Хроматография гидрофильных взаимодействий (ГИХ) 429
8.6.3. Разработка метода ГИХ
Ряд публикаций был посвящен обзору влияния различных экспериментальных
факторов на разделение в условиях ГИХ [38, 39, 44—46]. Разработка метода разделения
в условиях ГИХ молекул с небольшой молекулярной массой может быть проведена
аналогично разработке методов для разделения в условиях ОФХ или НФХ. Сначала
выбирают ГИХ колонку 100 — 150 х 4,6 мм (частицы 3 мкм). Как уже говорилось
ранее, часто предпочтителен непривитый «голый» силикагель в качестве сорбента.
При этом амидная фаза может быть хорошей альтернативой непривитому силикаге-
лю. Первоначальное разделение можно проводить, используя сильную подвижную
фазу — к примеру, буфер с 60% ацетонитрила (ацетонитрил является наиболее часто
используемым органическим растворителем для ГИХ). Затем % ацетонитрила можно
увеличивать с шагом в 10% до тех пор, пока не будет достигнуто желательный
интервал удерживания (например, \ < к < 10). См. аналогичную процедуру в разделе 2.4.1
для ОФХ, а также пример разделения в ГИХ на рис. 8.18. Еще более эффективным
может быть начальное градиентное разделение (раздел 9.3.1). В качестве буферов
для разделения ионизируемых соединений в ГИХ обычно используются муравьиная
кислота, ацетат или формиат аммония в концентрации от 2 до 20 мМ; рН
подвижной фазы при этом обычно находится в диапазоне от 3 до 7. Последние буферы
летучи (для использования в ЖХ-МС) и растворимы во всех смесях органики и воды.
Когда выбран % воды для получения приемлемого удерживания, можно
изменять условия для оптимизации селективности. Изменения относительного
удерживания могут возникать вследствие изменений %В, как можно предположить, исходя
из графиков на рис. 8.19. Таким образом, последовательность удерживания для пяти
последних пептидов на хроматограмме следующая:
л = 54 > 16 > 8 > 9 > 4 для 15% Н^О
и
п = 8 > 9 > 4 > 54 = 16 для 25% Н20.
Изменения селективности по мере изменения % воды наблюдались разными
исследователями [46, 44, 49, 50|. Несмотря на то, что такие изменения в относительном
удерживании часто незначительны, и поэтому от них мало пользы, их легко
обнаружить при корректировке % воды с целью достижения приемлемого удерживания
(например, 1 < к < 10). В нескольких работах были показаны изменения
селективности в ГИХ в зависимости от изменения температуры [44, 50|. Это говорит о том, что
может быть полезной одновременная оптимизация %В и температуры (как и в ОФХ,
раздел 7.3.2.2).
Влияние рН подвижной фазы на ионизацию растворенного вещества вполне
ожидаемо, как. это видно из примера на рис. 7.\а. С ростом рН ионизация кислот
увеличивается, а оснований — уменьшается. Поскольку удерживание в ГИХ растет
для более ионизированных соединений, удерживание кислот будет расти с ростом
рН, тогда как удерживание оснований будет уменьшаться. Когда рН подвижной
фазы находится в интервале ±1 единица от значения рКа, а величины рКа различны
(как часто и бывает в случае ионизируемых образцов), результатом изменения рН
должны быть большие изменения селективности. Кроме того, ожидается, что
произойдут большие изменения селективности при смене типа неподвижной фазы,
особенно с учетом широкого диапазона функциональных возможностей коммерчески
доступных неподвижных фаз (некоторые примеры приведены в работах |40, 511).
Для изменения селективности можно также заменить и органический растворитель А
(ацетонитрил), но этим пользуются нечасто. Другие растворители обычно сильнее
(рис. 8.21), что может привести к недостаточному удерживанию образца. Помимо
этого, ацетонитрил обычно способствует большим величинам N |44].
Глава 8. Нормально фазовая хроматография
Иэопропанол
Ацетонитрил
Ыл_
10
15
20
25 (мин)
Рис. 8.21. Влияние растворителя А на ГИХ-разделение. Образец: эпирубицин и его
аналоги. Условия: силикагельная колонка Kromasil KRIO0-5SIL 250 х
х 4,6 мм. Подвижная фаза 90% органический растворитель/буфер рН 2,9;
1,0 мл/мин; комнатная температура |52|
Кроме того, можно и просто изменить длину колонки и скорость потока для
увеличения разрешения или для сокращения времени разделения (раздел 2.5.3). Расход
будет ограничен допустимым давлением (раздел 2.4.1). Колонки меньшего диаметра
обычно используются для масс-спектрометрического детектирования (раздел 4.14).
Установлено, что эффективности колонок для ГИХ и ОФХ сравнимы, или колонки
для ГИХ могут иметь даже более высокую эффективность |40|. Хороший пример
этого представляют собой два разделения, показанные на рис. 8.22. Среднее число
теоретических тарелок jV для разделения на колонке длиной 150 мм, упакованной
частицами поверхностно пористого силикагеля диаметром 2,7 мкм, равно 36000
(рис. 8.22а), тогда как для колонки длиной 450 мм на рис. 8.225 N — 103000. Это
весьма показательно, учитывая небольшие времена разделений.
150мм колонка
97 бар (1400 psi)
(а)
5.0 (мин)
2
И
5
Г
450-мм колонка
290 бар (4200 psi)
»7 I
10
(б)
15 (мин)
Рис. 8.22. Высокоэффективное ГИХ-разделение. Образец: I, фенол; 2, нафталинсуль-
фокислота; 3, р-ксилолсульфокислота; 4, кофеин; 5, нортриптилин; 6, ди-
фенгидрамин; 7, бензиламик; <?, прокаинамид. Условия: колонка Halosilica
(Advanced Materials Technology, Wilmington, DE) 150x4,6 мм (а); три
колонки, как в (л), соединенные последовательно (6); 75% ацетонигрил/буфер
рН 3,0; 1,0 мл/мин; 30 "С [511
Литература 431
Обзор недавней литературы показывает, что описанные ГИХ-разделения часто
проводятся в градиентном режиме и с масс-спектрометрическим детектированием.
Замечания, сделанные выше для изократического элюирования, применимы и
для градиентного. В разделе 9.5.3 дана подробная информация о ГИХ-разделениях в
градиентном режиме.
8.6.4. Трудности, связанные с ГИХ
Представляется, что проблемы с формой пика (с размыванием как переднего, так и
заднего его фронта) несколько более присущи ГИХ, чем ОФХ |46, 50, 54]. Это может
отражать тот факт, что ГИХ стали пользоваться совсем недавно, и она не
использовалась так широко, как ОФХ, а также то, что использовали колонки, упакованные
силикагелем типа А — в особенности колонки, на которых работали до 1995 года
(например, см. рис. 8.20 и соответствующее обсуждение). Вероятно, произойдет
дальнейшее совершенствование колонок для ГИХ, а также станет более понятно,
как лучше справиться с проблемами уширения фронтов пика.
Когда вы сталкиваетесь с проблемами уширения фронтов пика, прежде всего
нужно попробовать увеличить концентрацию буфера в подвижной фазе. Для
получения приемлемой формы пика может потребоваться концентрация буфера до 100 мМ.
Образец следует растворять в подвижной фазе, но в некоторых случаях может
оказаться полезным увеличить процент органического растворителя в среде для
растворения образца. Если проблемы с формой пика остаются, то может потребоваться
замена колонки или изменение рН подвижной фазы. Подвижная фаза должна всегда
содержать воду, хорошо бы не менее 3—5%.
При использовании масс-спектрометрического детектирования у некоторых
колонок, упакованных привитыми фазами |41|, наблюдалось отщепление привитых ли-
гандов. Обычно эта проблема решается заменой колонки на иную с немодифициро-
ванным силикагелем или другой привитой фазой. Сообщалось о необратимой
сорбции некоторых образцов при использовании в ГИХ колонок с немодифициро-
ванным силикагелем. Если такая проблема возникает, то может потребоваться
переход на колонку с привитой фазой. Колонка может медленно уравновешиваться
подвижной фазой, в которой содержится другой буфер или изменена его
концентрация, но проблемы такого рода гораздо менее вероятны для ГИХ, чем для НФХ
на колонке, упакованной силикагелем.
Литература
1. L. R. Snyder, Principles of Adsorption Chromatography. The Separation of Nonionic Organic
Compounds, Ml Dekker, New York, 1968, chs. 8 and 10.
2. L.-AA. Truedsson and B. E. F. Smith, J. Chromatogr., 214 (1981) 291.
3. L. R. Snyder and D. L. Saunders, in Chromatography in Petroleum Analysis, К. Н. Altgelt and
Т. Н. Gouw, eds., Dekker, New York, 1979, ch. 10.
4. A. Kuksis, in Chromatography, E. Heftmann, ed., 5th ed., Elsevier, Amsterdam, 1993, p. Bl71.
5. L. R. Snyder and H. Poppe, J. Chromatogr., 184 (1980) 363.
6. L. R. Snyder, in High-Performance Liquid Chromatography: Advances and Perspectives, Vol. 3.
C. Horvath, ed., Academic Press, New York, 1983, p. 157. tab. 1
7. E. Soczewinski, Anal. Chem., 41 (1969) 179.
8. E. Soczewinski, J. Chromatogr. A, 965 (2002) 109.
9. L. R. Snyder, J. Chromatogr., 63 (1971) 15.
10. B. Fried and J. Sherma. Thin-Layer Chromatography (Chromatographic Science, Vol. 81), Ml
Dekker, New York, 1999.
432 Глава 8. Нормально фазовая хроматография
11. R. P. W. Scott and P. Kucera, J. Chromatogr., 112 (1975) 425.
12. V. R. Meyer and M. D. Palamareva, J. Chromatogr., 641 (1993) 391.
13. Solvent Guide, Burdick and Jackson Labs., Muskegon, Ml, 1980.
14. E. Soczewinski, T. Dzido, and W. Colkiewicz, J. Chromatogr., 131 (1977) 408.
15. J. L. Glajch, J. J. Kirkland, and L. R. Snyder, J. Chromatogr., 238 (1982) 269.
16. L. R. Snyder, J. Chromatogr., 63 (1971) 15.
17. L. R. Snyder, J. Planar Chromatogr., 21 (2008) 315.
18. L. R. Snyder, J. L. Glajch, and J. J. Kirkland, J. Chromatogr., 218 (1981) 299.
19. M. D. Palamareva and H. E. Palamareva, J. Chromatogr., All (1989) 235.
20. Ch. E. Palamareva and M. D. Palamareva, LSChrom, Ver. 2.1, Demo version, 1999,
http://www.lschrom.com.
21. M. Z. Kagan, J. Chromatogr. A, 918 (2001) 293.
22. L. R. Snyder and J. J. Kirkland, Introduction to Modem Liquid Chromatography, 2nd edn.,
Wiley- I nterscienee, New York, 1979, pp. 391—392.
23. L. R. Snyder and Т. С Schunk, Anal. Chem., 54 (1982) 1764.
24. E. L. Weiser, A. W. Salotto, S. M. Flach, and L. R. Snyder, J. Chromatogr., 303 (1984) 1.
25. W. T. Cooper and P. L. Smith, J. Chromatogr., 355 (1986) 57.
26. A. W. Salotto, E. L.Weiser, K. P. CafTey, R. L. Carty, S. C. Racine, and L. R. Snyder, J.
Chromatogr., 498 (1990) 55.
27. P. L. Smith and W. T. Cooper, J. Chromatogr., 410 (1987) 249.
28. L. P. Hammett, Physical Organic Chemistry, McGraw-Hill, New York, 1940, ch. 7.
29. B. Stancher and F. Zonta, J. Chromatogr., 234 (1982) 244.
30. M. D. Palamareva and H. E. Palamareva, J. Chromatogr., 477 (1989) 225.
31. J. L. Glajch and L. R. Snyder, J. Chromatogr., 214 (1981) 21.
32. B. Vaisman, A. Shikanov, and A. J. Domb, J. Chromatogr. A, 1064 (2005) 85.
33. L. R. Snyder, J. Chromatogr. Sci.. 21 (1983) 65.
34. V. R. Meyer, J. Chromatogr. A, 768 (1997) 315.
35. P. Jandera, J. Chromatogr. A, 965 (2002) 239.
36. R. W. Stout, J. J. DeStefano, and L. R. Snyder, J. Chromatogr., 282 (1983) 263.
37. J. J. Kirkland, С H. Dilks, and J. J. DeStefano, J. Chromatogr., 635 (1993) 19.
38. A. Alpert, J. Chromatogr., 499 (1990) 177.
39. P. Hemstrom and K. Irgum, J. Sep. Sci., 29 (2006) 1784.
40. T. Ikegami, K. Tomomatsu, J. Takubo, K. Horie, and N. Tanaka, J. Chromatogr. A, 1184 (2008)
474.
41. T. Yoshida, J .Chromatogr. A, 811 (1998) 61.
42. D. V. McCalley and U. D. Neue, J.Chromatogr. A, 1192 (2008) 225.
43. D. V. McCalley, J. Chromatogr. A, 1171 (2007) 46.
44. Z. Hao, B. Xiao, and N. Weng, J. Sep. Sci. 31 (2008) 1449.
45. B. Dejaegher, D. Mangelings, and Y. Vander Heyden, J. Sep. Sci., 31 (2008) 1438.
46. Y. Guo and S. Gaiki, J. Chromatogr. A, 1074 (2005) 71.
47. W. Naidong, J. Chromatogr. B, 19b (2003) 209.
48. B. A. Olsen, J. Chromatogr. A, 913 (2001) 113.
49. X. Wang, W. Li, and H. T. Rasmussen, J. Chromatogr. A, 1083 (2005) 58.
50. С Dell'Aversano, P. Hess, and M. A. Quilliam, J. Chromatogr. A, 1081 (2005) 190.
51. D. V. McCalley, /. Chromatogr. A, 1171 (2007) 46.
52. R. Li and J. Huang, J. Chromatogr. A, 1041 (2004) 163.
53. D. V. McCalley, J. Chromatogr. A, 1193 (2008) 85.
54. С Dell'Aversan, G. K. Eaglesham, and M. A. Quilliam, J. Chromatogr. A, 1028 (2004) 155.
ГЛАВА 9
ГРАДИЕНТНОЕ ЭЛЮИРОВАНИЕ
9.1. Введение 434
9.1.1. Причины для использования градиентного элюирования 435
9.1.2. Форма градиента 437
9.1.3. Сходство изократического и градиентного элюирования 438
9.1.3.1. Модель линейного изменения элюирующей силы растворителя (ЛИС) 439
9.1.3.2. Перемещение зон при градиентном элюировании 440
9.2. Экспериментальные условия и их влияние на разделение 441
9.2.1. Влияние изменения параметров колонки 444
9.2.2. Влияние изменения градиента 447
9.2.2.1. Исходный %В 447
9.2.2.2. Конечный %В 449
9.2.2.3. Задержка градиента 450
9.2.2.4. Приборный объем задержки 452
9.2.2.5. Кусочно-линейные градиенты 454
9.2.3. «Неправильные» образцы 456
9.2.4. Количественные соотношения 458
9.2.4.1. Время удерживания 459
9.2.4.2. Измерение величин S и kw 460
9.2.4.3. Ширина пика 461
9.2.4.4. Разрешение 462
9.3. Разработка метода 462
9.3.1. Первичное градиентное разделение 463
9.3.1.1. Выбор между градиентным и изократическим элюированиями 465
9.3.1.2. Возможные проблемы 468
9.3.2. Оптимизация к* (этап 4 таблицы 9.2) 469
9.3.3. Оптимизация селективности градиента (этап 5 таблицы 9.2) 470
9.3.4. Оптимизация диапазона градиента (этап 6 таблицы 9.2) 470
9.3.5. Ступенчатые (нелинейные) градиенты (продолжение этапа 6 таблицы 9.2) . . 472
9.3.6. Оптимизация числа теоретических тарелок N* (этап 7 таблицы 9.2) 473
9.3.7. Определения необходимого времени уравновешивания колонки
(этап 8 таблицы 9.2) 473
9.3.8. Воспроизводимость метода 476
9.3.8.1. Разработка метода 477
9.3.8.2. Рутинные анализы 477
9.3.9. Пиковая емкость и экспресс-разделения 478
9.3.9.1. Оптимизированная пиковая емкость 480
9.3.9.2. Экспресс-разделения 483
9.3.10. Комплексная двумерная ВЭЖХ (в соавторстве с Питером Шонмейкерсом) 485
9.3.10.1. Концепция ЖХ/ЖХ 485
9.3.10.2. Пиковая емкость 488
9.3.10.3. Оборудование для ЖХ/ЖХ 488
9.3.10.4. Разработка метода для ЖХ/ЖХ 489
9.4. Разделения крупных молекул 491
9.5. Другие виды разделения 492
9.5.1. Теория 492
9.5.2. Нормально-фазовая хроматография 493
9.5.3. Гидрофильная хроматография (ГИХ) 493
9.5.3.1. Прикладные задачи 494
9.5.3.2. Условия разделения 496
9.5.4. Ионообменная хроматография (ИОХ) 496
9.6. Проблемы 496
9.6.1. Обеднение элюента растворителем 496
9.6.2. Системные пики 497
9.6.3. Дрейф базовой линии 497
497
ffif 434 Глава 9. Градиентное элюирование
9.1. Введение
Понятие градиентного элюирования вводится в разделе 2.7.2 как средство для
анализа образцов, которые невозможно разделить в изократическом режиме. Обычно
градиентное элюирование используется для образцов, в которых интервал удерживания
пиков превосходит предел, разрешаемый в условиях изократического элюирования
(I < к < Ю). Как обсуждалось в разделе 2.5.1, этот интервал можно несколько
увеличить, например, до 0,5 < к < 20. Однако компоненты многих образцов имеют гораздо
более широкий интервал величин к, поэтому градиентное элюирование оказывается
чрезвычайно важным для их разделения. Пример образца, который невозможно
успешно разделить в режиме изократического элюирования, показан на рис. 9.1я.
В этом случае смесь 14 токсикологических стандартов наносится на колонку C|g.
(•>
г
0
7-8
1 9-10
1 Ji л
(б)
10%
В
—J
11
Г™
hi
12
20
(в)
25 %В
Изократическое
разделение, 50 %В
(0' к<50)
13
— —Т" — " Т" ■ - ■ "Г -—
40
Время (мим)
(a) (<Я
45 %В 52 %В
60
(
14
(в)
75 %В
10
J llf JUL JUT Ш"
12
2 4 6 8 246 4 6 468 468
Время (мин) Время (мин) Время (мин) бремя (мин) Время (мин)
(ж)
Градиентное разделение
5-90 %В за 13 мин (/с* = 3)
4-6
7-8. 9-11
- T2-f4
-
A^JJLlJ
-100 48
80%
-60%
-40%
-20%
-0%
6 8
Время (мин)
10
12
Рнс. 9.1. Пример общей проблемы элюирования. Образец: 14 токсикологических
стандартов. Условия: колонка C.|«; 250 х 4,6 мм. размер частиц 5 мкм: подвижная
фаза ацетонитрил (В) и фосфатный буфер рН 2,5 (А); 65 "С. 2 мл/мин.
(о) Изократическое разделение при 50 %В. (б)—(е) Изократические разделения
указанных фракций при 10%, 25%, 45%, 52% и 75 %В, соответственно (к ~ 3).
(ж) Градиентное разделение (пунктирной линией показан градиент). Хрома-
тограммы получены компьютерным моделированием данных, взятых в [1|
9.1. Введение 435
подвижной фазой является 50% ацетонитрил. Вследствие широкого интервала
удерживания этих образцов (величины к варьируются в пределах 0—50), рановыходящие
пики с 1 по 6 плохо разрешаются, а поздновыходящие пики 13 и 14 имеют
избыточное время удерживания. Эти последние пики также очень широкие и поэтому
невысокие и, следовательно, их измерение может быть неточным из-за плохого
соотношения сигнал/шум (раздел 4.2.3). Любое изменение %В в элюенте не смогло
бы обеспечить качественное разделение всех компонентов этого образца: большие
значение %В привели бы к уменьшению к, а, следовательно, к еще худшему
разрешению рановыходящих пиков. В то же время уменьшение %В привело бы к
увеличению к и времени анализа, а также еще больше усложнило бы измерение позд
новыходящих пиков. На рис. 9.1 я хорошо проиллюстрирована общая проблема
элюирования образцов с широким интервалом значений к, которая и является основ
ной причиной использования градиентного элюирования.
Если бы группы соседних пиков можно было бы элюировать раздельно (при
разных значениях %В), получились бы более оптимальные изократические разделения,
представленные на рис. 9Л6—е. Так при использовании подвижной фазы с 10 %В
для пиков I—3 (рис. 9.16) среднее значение к ~ 3, и они разделяются до базовой
линии. Аналогичным образом использование подвижной фазы с 25 %В позволяет
хорошо разрешить пики 4—6 тоже при среднем значении к ж 3 (рис. 9.1в). Также
для групп пиков 7—8, 9—II и 12—14 достигается то же значение к при
использовании подвижной фазы с 45, 62 и 75 %В, соответственно (рис. 9.1г—е). Таким образом,
каждую из этих фракций образца можно разделить изократически с приемлемым
разрешением (Rs > 2) за 6—8 мин, получив высокие узкие и хорошо детектируемые
пики. Единственным требованием является состав подвижной фазы,
обеспечивающий к « 3 для каждой группы пиков, но при этом имеющий различный %В.
Вследствие чего изократическое разделение всего образца с 1 < к < 10 невозможно.
Градиентное элюирование (рис. 9.1-ж) позволяет реализовать все преимущества,
показанные на рис. 9.16—е, в процессе одного разделения. В этом случае пики 1—3
в начале градиента проходят по колонке со средним значением Л к 3, в то время как
пики 4—14 находятся на входе колонки. По мере увеличения %В при градиентном
элюировании (пунктирная линия на рисунке показывает увеличения %В со
временем на выходе из колонки), постепенно вымываются и более удерживаемые пики
также со средним значением к я 3. Как будет показано в разделе 9.1.3.2, значения к
обычно одинаковы для всех пиков при градиентном элюировании, и их можно легко
оптимизировать, подбирая время градиента и скорость потока. В данной главе идет
речь о разделении в ОФХ, если не оговариваются иные условия. Однако те же общие
принципы и выводы применимы и для других видов ВЭЖХ (ионообменной,
нормально-фазовой и т.д.).
Более развернуто и подробно, чем в этой книге, градиентное элюирование
рассмотрено в |2|'.
9.1.1. Причины для использования
градиентного элюирования
Помимо того, что градиентное элюирование требуется для образцов, существенно
отличающихся полярностью, в ряде других случаев также удобнее или необходимо
использовать градиентное элюирование:
• для высокомолекулярных образцов;
' См. русский перевод книги Ллойд Р. Снайдер, Джон У. Долан. Высокопроизводительная
градиентная элюция: практическое применение модели линейного изменения элюирующей силы
растворителя. — Москва: ТЕХНОСФЕРА, 2015. — 584 с. — Прим. перев.
436 Глава 9. Градиентное элюирование
• для дженериков (например, комбинаторных библиотек);
• для разработки эффективных методик ВЭЖХ;
• для подготовки образцов;
• для предотвращения размывания задних фронтов пиков.
Образцы с большой молекулярной массой (высокомолекулярные), такие как пептиды,
белки и олигонуклеотиды, обычно плохо разделяются при изократическом элюиро-
вании, так как удерживание этих образцов очень чувствительно к малейшим
изменениям состава подвижной фазы (%В). Например, коэффициент удерживания к белка
с молекулярной массой 50000 Да может измениться втрое при изменении состава
подвижной фазы на 1 %В. Такое поведение может существенно осложнить получение
воспроизводимых результатов изократических разделений макромолекул в разных
лабораториях или даже в одной и той же лаборатории. Более того, изократическое
разделение смеси макромолекул обычно приводит к немедленному элюированию
некоторых компонентов образца (с к х 0 и без их разделения) и такому медленному
элюированию (с к » 100) других компонентов, что, кажется, они никогда не
пройдут через колонку. То есть интервал удерживания таких образцов часто чрезвычайно
велик (значения к компонентов образца различаются на несколько порядков
величины в условиях изократического разделения). В то же время при градиентном
разделении макромолекул, времена их удерживания не воспроизводятся редко, и образцы
разделяются быстро и эффективно (глава 13).
Разделение дженериков необходимо в тех случаях, когда ряд анализируемых образцов
содержит различные компоненты, например, А, Б и В в образце 1 и Г, Д и Е в образце 2
и так далее. Обычно каждый образец требуется разделить сразу за определенное время
(время анализа) без дополнительной отработки метода для каждого последующего
образца. Таким образом можно проанализировать сотни и тысячи похожих образцов,
имеющих различный состав, за минимальное время с минимальными затратами.
Автоматизированные разделения дженериков на обращенно-фазовых колонках
проводятся именно в режиме градиентного элюирования и используются для анализа
комбинаторных библиотек |3| и других образцов [4|. При разделении дженериков часто
используется масс-спектрометрический детектор [5|; это позволяет не только разделить,
но и идентифицировать компоненты образцов неизвестного состава. При этом не
требуется разделять до базовой линии пики, представляющие интерес.
Разработку эффективной методики ВЭЖХ разделения |6] предпочтительно
начинать с одного или нескольких экспериментальных градиентов (раздел 9.3.1). Один
экспериментальный градиент при разработке метода может заменить несколько
изократических разделений, проведенных методом проб и ошибок, для определения
оптимальной силы элюента (значения %В) для изократического разделения. Кроме
того, экспериментальный градиент позволяет сделать выбор между изократическими
или градиентными условиями разделения для данного образца.
Подготовка образца (пробоп од готовка, глава 16) требуется во многих случаях
из-за того, что некоторые образцы нельзя непосредственно наносить на колонку,
элюируемую изократически. Следует сначала удалить нерастворимые частицы,
сильно удерживаемые и мешающие примеси. В некоторых случаях градиентное
элюирование позволяет свести к минимуму подготовку образца или даже обойтись без нее.
Например, при градиентном элюировании вначале выходят мешающие (не
подлежащие анализу) примеси, которые обычно вымываются вблизи ^(сравните рис. 9.Ifl и
9. Ij*c), и их можно отделить от целевых пиков. Аналогичным образом сильно
удерживаемые примеси могут привести к неоправданному увеличению времени
разделения при изократическом элюировании, так как их необходимо вымыть из колонки
перед анализом следующего образца. Градиентное элюирование обычно позволяет
удалить эти поздноэлюирующиеся вещества в течение сравнительно недолгого
разделения.
9.1. Введение
Размывание задних фронтов пиков было серьезной проблемой при развитии
хроматографии, и для уменьшения этого размывания и было изначально предназначено
градиентное элюирование |7|. Вследствие увеличения силы подвижной фазы
во время продвижения полосы по колонке при градиентном элюировании, задний
фронт этой полосы движется быстрее, чем передний. Поэтому и уменьшается
размывание заднего фронта пика, а также ширина пика (раздел 9.2.4.3). Однако в
настоящее время размывание заднего фронта пика менее распространено, и для его
устранения есть более эффективные способы (раздел 17.4.5.3).
9.1.2. Форма градиента
Под формой градиента подразумевается зависимость изменения состава подвижной
фазы (%В) со временем в процессе градиентного разделения. Градиентное
элюирование можно проводить градиентами разной формы (рис. 9.2 а—е). В большинстве
случаев используются линейные градиенты (рис. 9.2я), которые настоятельно
рекомендуются для разработки методик. Криволинейные градиенты (рис. 9.2 6—в)
использовались в прошлом для некоторых видов образцов, но по разным причинам эти
градиенты со временем были заменены кусочно-линейными градиентами (рис. 9.2г).
Эти градиенты имеют не только все преимущества нелинейных форм, но их еще и
легче подбирать для различных образцов и контролировать при разделении (а также
они избавляют от необходимости использовать специализированное оборудование
для формирования градиента). Применение кусочно-линейных градиентов для улуч-
время
время
<е)
%в
время
Ступенчатый градиент
время
время
время
/Q= 20 мин
фь= io%e=o.io
<^=80%Ве0.80
Дф=Ф,-%. (0.80-0.10)-0.70
Рис. 9.2. Различные формы градиента (график зависимости %В от времени)
438 Глава 9. Градиентное элюирование
шения разделения для различных целей рассматривается в разделе 9.2.2.5. Задержка
градиента, или «изократическая задержка» (раздел 9.2.2.3) представлена на рис. 9.2d
Изократическую задержку можно также использовать в конце градиента.
Ступенчатые градиенты (рис. 9.2е), когда в процессе разделения производится мгновенное
изменение %В, представляют собой частный случай кусочно-линейных градиентов.
Они используются нечасто, за исключением случаев промывания колонки от позд-
новыходящих примесей в конце градиента. Это достигается резким увеличением %В
(i на рис. 9.2е). Ступенчатый градиент с резким уменьшением %В (п на рис. 9.2eJ
позволяет вернуть градиент к его начальному значению для следующего разделения.
В прошлом ступенчатые градиенты избегали использовать, опасаясь нестабильности
колонки. С современными колонками, хорошо упакованными сорбентами на основе
силикагеля, ступенчатые градиенты могут использоваться без опасения их повредить.
Линейный градиент можно описать (рис. 9.2ж) через исходный и конечный
состав подвижной фазы и время, в течение которого происходит изменение состава
элюента. Начальный и конечный состав подвижной фазы можно выразить в %В или
с помощью объемной доли ф растворителя В в подвижной фазе (эквивалентной
0,01 %В), то есть <ро и qy— объемные доли В в начале и в конце градиента,
соответственно. Изменение %В или ф за время анализа называют градиентным диапазоном и
обозначают Дер = qy — фо (или Д%В = конечный %В — начальный %В). Здесь
значения %В и ф взаимозаменяемы; то есть ф всегда соответствует 0,01 %В, а 100 %В
означает, что в состав подвижной фазы входит только органический растворитель
(ф = фу-= 1,00). По причинам, обсуждаемым в главе 5, емкости А и/или В могут
содержать смеси растворителей А и В, а не просто чистую воду и органический раствори
тель. Например, в емкости А будет находиться 5% ацетонитрила, а в емкости В —
95% ацетонитрила. В этом случае градиенту 0—100 %В будет соответствовать
изменение содержания ацетонитрила от 5 до 95%.
Под градиентной программой подразумевается описание изменения состава
подвижной фазы со временем. Линейные градиенты представляют собой простейший
случай, например, градиент от 10 до 80 % В за 20 минут (рис 9.2j*c), который также
можно описать как 10—80 %В за 0—20 минут (от 10 %В в нулевой точке времени
до 80 %В в конечной точке времени). Программы кусочно-линейного градиента, как
правило, выражают зависимость %В от времени для каждого линейного фрагмента,
например 5—25—40—100 %В за 0—5—15—20 минут (рис. [Аз).
9.1.3. Сходство изократического и градиентного
элюирования
Во время градиентного элюирования пик перемещается по колонке небольшими
«скачками», каждый из которых соответствует небольшому изменению состава
подвижной фазы (%В). То есть градиентное разделение можно рассматривать как
последовательность небольших изократических разделений. Можно рассчитать
параметры изократического и градиентного разделений таким образом, чтобы они имели
сходный результат. Разрешение, достигаемое для выбранных пиков при изократиче-
ском и градиентном элюированиях, будет приблизительно одинаковым, если средние
значения к при градиентном элюировании (при перемещении каждого пика по
колонке) сходны с величинами к при изократическом элюировании. Для этого также
необходимо, чтобы были одинаковы условия проведения элюирования (колонка,
температура, элюенты А и В и так далее). В этом случае изократическое и
градиентное разделения называются «соответствующими». Так примеры изократических
разделений, приведенные на рис. 9.\б—е можно сравнить с «соответствующим»
градиентным разделением рис. 9.1лс. Для каждого из изократических разделений
9.1. Введение
отдельных групп пиков на рис. 9.16—е к = 3, в то время как при градиентном
разделении на рис. 9.\ж эквивалентная величина к для каждого пика также ж 3. Этот
пример показывает, что разрешение пиков на рис. 9.1<э—е и рис. 9.1ж" приблизительно
одинаковое.
При изократическом разделении значения к можно изменять, варьируя силу
подвижной фазы (%В). При градиентном элюировании на средние значения к можно
влиять, подбирая условия хроматографии (как описано далее в разделе 9.2).
9.1.3.1. Модель линейного изменения элюирующей силы
растворителя (ЛИС)
В этом разделе даны основы количественной обработки градиентного элюирования. Од
нако соотношения, приведенные низке, редко применяются на практике (хотя и
необходимы для количественной обработки градиентного элюирования). Читатель может пе
рейти к разделу 9.1.3.2, а потом при необходимости вернуться к этому.
Удерживание в изократическом режиме ОФХ задается зависимостью от %В
(раздел 2.5.1):
log k = log кК - Sip. (9.1)
Для данного растворенного вещества значение кк— экстраполированная величина к
для ф = 0 (когда подвижная фаза вода или буфер), a S = 4 для небольших молекул
(< 500 Да). Линейный градиент можно описать следующим образом
%В = (%В)0 + W|(%B)/- (%B)0|. (9.2)
Здесь %В относится к составу подвижной фазы на входе в колонку, (%В)д —
значение %В в начале градиента (нулевое время), (%В)у— значение %В в конце градиента,
t — любой момент времени в течение градиента, а /с— время градиента. Уравнение
(9.2) можно переписать, используя ср, объемную долю В:
-](Ф/- Фо) = Ч>0 + f^V (9-2а)
'СJ \'G )
Ф = Ф0
где фо — значение <р в начале градиента, ц>г— значение <р в конце градиента, а Дф =
= (фг— Фо) — изменение <р в ходе градиента (градиентный диапазон); см. рис. 9.2ж\
Величина ф относится к значениям на входе в колонку, измеренным в различные
моменты градиента. Таким образом, состав подвижной фазы во время /0 (в начале
градиента) ф = фо в том случае, если нет задержки между входом в колонку и
градиентным миксером (раздел 9.2.2.3).
При объединении уравнений (9.1) и (9.2а) получаем
\ fG J
Для линейного градиента данного растворенного вещества и определенных
экспериментальных условий С| и Ci константы, так что log к линейно изменяется со
временем в ходе градиента (значение к в уравнении (9.3) относится к его значению,
измеренному на входе в колонку в любое данное время 1). Градиенты, описываемые
уравнением (9.3), называются градиентами линейного изменения элюирующей силы
(ЛИС). То есть, линейные градиенты в ОФХ практически являются градиентами ЛИС.
Уравнения для удерживания и ширины пика в градиентах ЛИС приводятся в
разделе 9.2.4. Разделения в режиме ЛИС гораздо легче понять и контролировать, чем гради-
440 Глава 9. Градиентное элюирование
енты других форм. Наконец, градиенты ЛИС обеспечивают более качественное
разделение большинства образцов, для которых требуется градиентная элюция.
Основное определение крутизны градиента b для данного растворенного вещества:
ь = ^mAtpS
или, поскольку /о= VmIF,
л = ГоДф5
to
Это определение крутизны градиента следует из уравнения (9-3), которое можно
записать следующим образом
,ogA = log*w-S%-(^Y^|
ИЛИ
log к = log kw - S<po - b j L1 (9.46)
где разность (log kw - Scpo) для данного градиента и растворенного вещества равна
log к в начале градиента (и следовательно, изменяется с изменением ф0, см.
уравнение (9.7) далее). Большая величина Ь соответствует более быстрому уменьшению к
со временем или более крутому градиенту. Выражения для времени удерживания и
ширины пиков при градиентном элюировании можно получить из приведенных
выше уравнений (см. раздел 9.2.4).
9.1.3.2. Перемещение зон при градиентном элюировании
Рассмотрим, как при градиентном элюировании перемешаются по колонке зоны
индивидуальных растворенных веществ (рис. 9.3). На рис. 9.3а сплошной кривой (дг|/|)
показано последовательное перемещение х зоны / по колонке в зависимости от
времени для первого из элюируемых веществ i (следует иметь в виду, что при у = I зона
находится на выходе колонки, а при у = 0 — на входе в колонку). Видно, что
передвижение по колонке со временем ускоряется, поэтому график зависимости х от /
представляет собой восходящую кривую. Также на рис. 9.3я пунктирной кривой к \i\
показано изменение мгновенного значения к для зоны i по мере ее перемещения
по колонке, к (/) соответствует значению к в момент времени t при изократическом
разделении в подвижной фазе, состав которой (%В) совпадает с составом подвижной
фазы, контактирующей с зоной в момент времени t в градиенте. Ширина пика и
разрешение при градиентном элюировании зависят от медианного значения к, то есть
мгновенного значения к в момент, когда зона прошла половину длины колонки. Это
медианное значение к называется градиентным фактором удерживания к*.
Ширина пика определяется значением к на выходе пика из колонки (ке равное к*/2).
На рис. 9.3fl показан также аналогичный график для второй зоны у (и
соответствующие значения х = х[/\ и к = k\j\). Хроматограмма, полученная в результате этого
разделения, показана на рис.9.36.
Рисунок 9.3я демонстрирует сходное поведение зон двух веществ /' и j при их
перемещении по колонке, за исключением задержки на старте зоны J вследствие ее
более сильного удерживания (большего значения kw). В частности, значения к* и ке
раньше и позже выходящих пиков веществ < и j приблизительно одинаковы. Это
позволяет предположить, что разрешение (и расстояние между пиками) не должно
(9.4)
(9.4а)
9.2. Экспериментальные условия и их влияние на разделение ААI
Т
8 10 12
Время (мин)
-г-
14
16
1В
Рис. 9.3. Перемещение пиков при градиентном элюировании. (й) Зависимость
перемещения зоны х и мгновенных значений к от времени. Показаны средние
значения (к*) и значения в момент элюирования (ке). (б) полученная хромато-
грамма
уменьшаться для рановыходяших пиков, как это происходит при изократическом
элюировании для небольших величин к (сравните разделение пиков I—6 в градиенте
на рис. 9Аж с изократическим разделением на рис. 9. \а). При градиентном
элюировании значения ке рано- и поздноэлюируемых пиков обычно близки; это означает
что и ширина (и высота) рано- и поздноэлюируемых пиков на хроматограмме будут
приблизительно одинаковы (сравните высоту пиков 1 — 14 в градиентном и
изократическом разделениях на рис. 9. \ж и 9.1я, соответственно). Относительное
постоянство значении к* и ке в разделениях с линейным градиентом обеспечивают существенное
преимущество перед изократическим и условиями для образцов со значительно
различающимися свойствами, такими как на рис. 9.!.
9.2. Экспериментальные условия и их влияние
на разделение
Градиентный фактор удерживания к* (рис. 9.3) при градиентном элюировании имеет
приблизительно такую же величину как коэффициент удерживания к при
изократическом элюировании. Значения к важны для понимания и контроля
разделения при изократическом элюировании, и мы увидим, что к* играет такую же роль
при градиентном элюировании. Значения к* зависят от растворенного вещества (его
442 Глава 9. Градиентное элюирование
значения S в уравнении (9.1)) и экспериментальных условий: времени градиента tG,
скорости потока F, размеров колонки и диапазона градиента Д<р [2|.
~^ШЩ. (9.5)
Здесь Vm — «мертвый» объем колонки (мл), который можно определить из
экспериментального значения t0 и скорости потока F (раздел 2.3.1; Vm - t0F). Значение S
для различных образцов, имеющих молекулярную массу от 100 до 500 Да, можно
принять равным 4. Это означает, что значения к* для различных веществ будут
приблизительно одинаковыми при разделении в линейном градиенте (при постоянных
значениях lG, F, Vm и Дф).
Теперь сравним изократические и градиентные разделения одного и того же
образца при сходных условиях (одинаковые элюенты А и В, колонка, скорость потока,
температура), сопоставляя значения к и к*. Изократические разделения, показанные
на рис. 9Аа-в, иллюстрируют влияние изменения %В (и к) для подвижной фазы 70,
55 и 40 %В. При градиентном элюировании близкие значения к* можно получить,
изменяя время градиента tG (уравнение (9.5) с S = 4), как показано на рис. 9.4г—е.
В этом случае к* = 1, 3 и 9 для lG= 3, 10 и 30 минут, соответственно. Изократическое
и градиентное разделения будут называться «соответствующими», если среднее
значение к в изократическом разделении равно значению к* в градиентном (для одного
и того же образца и одинаковых экспериментальных условий, отличающихся только
%В). В примере, показанном на рис. 9.4а и г, разделения «соответствующие», как и
разделения б и d, а также вне. «Соответствующие» разделения, такие как в
приведенных примерах, должны давать сходное разрешение и среднюю высоту пиков (хотя
в градиенте пики могут быть примерно вдвое выше).
И при изократическом, и при градиентном элюированиях А и А* возрастают
с увеличением времени разделения при прочих одинаковых условиях. При
изократическом элюировании разрешение возрастает с ростом значений к (уравнение 2.24),
как видно на рис. 9.4а—в (Rs = 0,4, 1,7 и 3,4). При градиентном элюировании
для близких значений к* в каждом из «соответствующих» разделений (рис. 9.4г—е)
наблюдается практически такое же разрешение (Rs= 0,4, 1,7 и 3,6). Наконец, ширина
пиков при изократическом элюировании увеличивается с ростом к (и уменьшением
%В), что приводит к уменьшению высоты пиков. Аналогичные изменения ширины
и высоты пиков наблюдаются при градиентном элюировании при изменении к*
на рис. 9.4г-е. Таким образом, изменения %В при изократическом элюировании или
времени градиента при градиентном элюировании приводят к сходным изменениям
времени разделения, разрешения и высоты пиков.
Образец, представленный на рис. 9.4, можно отнести к «правильным»
(раздел 2.5.2.1), так как относительное удерживание не изменяется при изменении к или
к* за счет варьирования %В или времени градиента, соответственно (при сохранении
других условий разделения постоянными). Следовательно, критическое разрешение
непрерывно возрастает на рис. 9Лг—е по мере увеличения времени градиента (и к*).
Ряд аналогичных экспериментов с «неправильным» образцом (раздел 2.5.2.1),
состоящим из смеси замещенных анилинов и бензойных кислот, показан на рис. 9.5.
Относительное удерживание пиков «неправильного» образца изменяется при
варьировании изократического %В или времени градиента. Как и на рис. 9.4, наблюдаются
такие же тенденции в изменении среднего разрешения, высоты пиков и
продолжительности разделения при увеличении времени градиента. Однако на рис. 9.5 помимо
этого изменяется относительное удерживание пиков образца при изменении времени
градиента (обратите внимание на изменение относительного времени
удерживания заштрихованного пика 3 и, в меньшей степени, пиков 7—10). Вследствие этих
9.2. Экспериментальные условия и их влияние на разделение
70 %В
12*52
Я„=0.4
'0
(в)
55 %В
2 SA 5 8
Я.» 1.7
Время(мин)
JULe_Jt
(б)
—i 1-
е
Время (мин)
40 %В
6 S к S 23
Яв = 3.4 .2
(в)
aJUL
4
5
-/V
10
Время (мин)
20
0-100 %В за 3 мин
Rs = 0.4
2.2 2.4 2.6 2.8 3.0 3.2 3.4
Воем я (мин)
0-100 %В за 10 мин
fc*=3
Я,-1.7
| 1 1 1 1 1 1 1 1 < | 1
6.0
Л*=9
| 1 I I I I 1 I I I | I I
7.0 8.0
Время (мин)
(е)
:3.6
Л.
L
10
12
14
Время (мин)
16
18
Рис. 9.4. Сравнение изократических (а—в) и градиентных (г—е) разделений с изменением %В
или времени градиента, соответственно. Образец: 1 — симазин; 2 — монолинурон; 3 — метобро-
мурон; 4 — диурон; 5 — пропазин. Условия: колонка С\% 150x4,6 мм, 5 мкм; подвижная фаза
метанол — вода (%В или условия градиента указаны на рисунках); скорость потока 2,0 мл/мин;
комнатная температура. Следует указать, что показаны реальные высоты пиков (без пересчета
к 100% высоты самого высокого пика). Хроматограмчы построены поданным работы |8|
Cr
Глава 9. Градиентное элюирование
градиент за 3 мин
ft* =1.5, Я, = 0.4
Время (мин)
(6)
градиент за 10 мин
ft* = 5, fis = 0.9
JiX
2 4
Время (мин)
градиент за 30 мин
ft*=15,Rs = 0.1
(«)
1 | 2
+
3
8 .9
4 5;7
б 8
Время(мин)
10
О!
12
Рис. 9.5. Разделения «неправильных» образцов в зависимости от времени градиента tc
Образец: смесь замещенных анилинов и бензойных кислот. Условия:
колонка С is 100 х 4,6 мм, 3 мкм; скорость потока 2,0 мл/мин; 42 °С; градиент 5 —
100% ацетонитрила в фосфатном буфере рН 2.6 за (а) 5 мин, (б) 15 мин.
(в) 30 мин. Пик 3 заштрихован для того, чтобы было заметнее изменение
относительного удерживания при варьировании времени градиента. Показаны
реальные высоты пиков (без пересчета к 100% высоты самого высокого
пика). Хроматограммы построены поданным работы |9]
изменений относительного удерживания в зависимости от ?с, максимальное
(«критическое») разрешение достигается при промежуточном значении времени
градиента 10 мин (рис. 9.56 Rs = 0,9), в то время как разрешение «правильного» образца
на рис. 9.4 продолжает расти при увеличении времени градиента (и к*). Для
«неправильных» образцов изменение к при изократическом разделении или к* при
разделении в градиенте приведет к таким же изменениям относительного удерживания.
Следовательно, максимальное разрешение образца может не соответствовать
максимальным возможным значениям к или к* для этих образцов.
9.2.1. Влияние изменения параметров колонки
Параметры колонки: ее длина и диаметр, скорость потока, размер частиц — влияют
на количество теоретических тарелок N (раздел 2.4.1) и длительность разделения.
Параметры колонки выбирают в начале разработки методики, потом иногда изменяют
после подбора других условий разделения или для оптимизации разрешения, или для
сокращения длительности разделения (раздел 2.5.3). При изократическом элюирова-
9.2. Экспериментальные условия и их влияние на разделение 445
нии изменение параметров колонки не влияет на значения к или относительного
удерживания. Разрешение, как и время разделения, обычно увеличивается при увеличении
длины колонки или при уменьшении скорости потока, в то время как высота пиков
уменьшается на более длинных колонках и при ббльших скоростях потока. Изменения
изократических разделений, вызванные только иной длиной колонки или скоростью
потока, показаны на рис. 9.6я—в для «правильного» образца, представленного
на рис. 9.4. На рис. 9.6о показано исходное разделение, а на рис. 9.66 и 9.6е приведены
результаты увеличения длины колонки или скорости потока, соответственно. Обратите
внимание на наблюдаемые в результате изменения длительности разделения,
разрешения и высоты пиков в этих изократических разделениях.
Подбирая экспериментальные условия при разработке методики изократическо
го разделения, желательно прежде всего изменять условия таким образом, чтобы
повлиять на к и а. для оптимизации селективности и разрешения. Если для
дальнейшего улучшения условий разделения требуется изменить параметры колонки,
предварительно подобранные значения к и а при изократическом разделении не из
менятся. При постоянных к и а также упрощается интерпретация последующих
экспериментов, так как могут измениться только N и длительность разделения. При
градиентном элюировании ситуация более сложная, так как значения к* изменяются
в зависимости от длины колонки и скорости потока (уравнение 9.5). Для того, чтобы
при градиентном элюировании остались неизменными к* и а, варьировать
параметры колонки следует таким образом, чтобы значение (t^F/L) оставалось постоянным
(уравнение 9.5; Vm пропорционален длине колонки, если не изменяется ее диаметр).
Для сохранения А* и а постоянными при изменении длины колонки L или скорости
потока F наиболее удобно изменять время градиента tg. При изменении длины
колонки кратно х во столько же раз надо изменить время градиента. А при изменении
скорости потока кратно х время градиента следует изменить пропорционально \/х.
Как в изократическом режиме, так и в градиенте изменение значений L и F
приводят к изменению времени разделения в том случае, если значения к*
поддерживаются постоянными с помощью изменения времени градиента.
Градиентные разделения, показанные на рис. 9.6г—е, иллюстрируют такое же
влияние изменения длины колонки и скорости подвижной фазы, как и в
изократических разделениях на рис. 9.6а—в, если значения к* поддерживаются постоянными
с помощью изменения времени градиента Iq. Для «соответствующих» разделений
на рис. 9.66,д увеличивается длина колонки от 100 до 300 мм (и время градиента
на рис. 9.6д увеличивается от 15 до 45 мин), при этом время разделения возрастает
втрое как и разрешение (Rs = 3 в изократике и Я,= 3,1 в градиенте). Высота пиков
уменьшается из-за их уширения. Таким же образом для «соответствующих» разделений
на рис. 9.6в,е при увеличении скорости подвижной фазы от 1,0 до 3,0 мл/мин
(и уменьшении времени градиента от 15 до 5 мин) втрое уменьшается время
разделения и разрешение (Rs - 1,2 в изократике и Rs = 1,2 в градиенте). Высота пиков
увеличивается, так как пики становятся более узкими. Примеры, приведенные
на рис. 9.6а—е, подтверждают одинаковое воздействие изменений параметров колонки
на изократическое и градиентное элюирование, если к и к* поддерживаются
постоянными. Характеристики разделений, приведенных на рис. 9.6, даны в таблице 9.1.
Когда при градиентном элюировании изменяются только размеры колонки или
скорость потока (т.е. время градиента остается неизменным), значения к* также
изменяются (уравнение 9.5, см. также уравнение 9.5в). Получающиеся в результате
градиентные разделения могут показаться неожиданными тем, кто ожидает сходства
с изократическими разделениями {рис. 9.6я—в). На рис. 9.6ж—з показано, что
происходит при таких же изменениях длины колонки и скорости подвижной фазы, как
на рис. 9.6 д,е, но при сохранении времени градиента равным 15 минут, так что к*
не остается постоянным. В этом случае уменьшается разрешение при увеличении
446 Глава 9. Градиентное элюирование
иэократическое
разделение
100 мм
1.0 мл/мин
Я,= 1.7
4 б
Воемя (мин^
иэократическое
разделение
300 мм
1.0 мп/мин
Я.= 3.0
10
(6)
X
А_
иэократическое
разделение
100 мм
3.0 мл/мин
Я,= 1.2
10 0
Время (мин)
j\ILj\ /i
30
(в)
г~
2.0
Время (мин)
3.0
прадиентиое разделение
(0-100% В за 15 мин)
100ММ р
1 О мл/мин
Я.= 1-7 >\ / \ 3
А
10
градиент за 45 мин
300 мм
1.0 мл/мин
Я. = 3.1
* л
Время (мин)
11
_л_
W)
JL
28
градиент за S мин
100 мм
3.0 мл/мин
Я =1.2
32
Время (мин)
И
—I—
3.0
3.2
3.4 3.6
Время(мин)
3.8
—I—
4.0
градиент за 15 мин
300-мм
1.0 мл/мин
Я, = 1.0
(ж)
13
14
Время (мин)
градиент за 15мин
100 мм
3.0 мп/мин
R, = 2.8
-УУ-
15
■*£ь.
16
.А.
в 7 в 9
Время (мин)
Рис. 9.6. Сравнение изократического и градиентного элюирования «правильного» образца
при изменении длины колонки или скорости потока. Образец и условия разделения даны на
рис. 9.4 (кроме различий в длине колонки или скорости потока, указанных на рисунках). Для
изократических разделений (а—в) 55 % В; 0 — 100 %В для градиентных разделений (г—з).
Показаны реальные высоты пиков (без пересчета к 100% высоты самого высокого пика). Хрома-
тограммы построены поданным работы |8|
9.2. Экспериментальные условия и их влияние на разделение 447
Таблица 9.1. Сравнение изменений разделения, вызванных изменением скорости
подвижной фазы F или длины колонки L при изократическом и градиентном элюи-
рованиях (примеры рис. 9.6)
Режим элюирования
Изократическое разделение"
Градиентное разделение
((с варьируется, к* постоянен)1'
Градиентное разделение
(>С постоянно, к* изменяется)"
Исходное разделение
я,
1,7
1,7
1,7
Средняя высота
пика'
(1,0)
(1,0)
(1,0)
Увеличение L втрое
*,
3,0
3,1
1,0
Средняя высота
пика'
0,6
0,6
1,0
Увеличение / втрое
Rs
1,2
1,2
2,8
Средняя высота
пика'
0,8
0,7
0,3
" Рис. 9.6а—е.
6 Рис. 9.вг—е.
* Рис. 9-бж1—з.
'Относительные величины (в пересчете к исходному разделению).
длины колонки (рис. 9.6ж, Rs~ 1,0), а при увеличении скорости потока оно
увеличивается (рис. 9.6з, Rs= 2,8). То есть изменение параметров колонки оказывает
противоположное воздействие на разрешение, чем в том случае, когда к* сохраняется
постоянным (рис. 9.6 д,е). По этой причине при изменении длины колонки или скорости
подвижной фазы при градиентном элюировании следует также изменять время
градиента, чтобы к* оставался постоянным, и — что еще более валено — оставались бы
прежними относительное удерживание и селективность.
В заключение укажем, что в «соответствующих» разделениях в изократическом и
градиентном режимах (т.е. в разделениях, имеющих сходные величины к и к*)
обычно наблюдаются приблизительно одинаковые разрешение и высота пиков.
Длительность разделения будет изменяться в одинаковой степени и при изократическом, и
при градиентном элюировании, если изменяются параметры колонки, но в то же
время к* (или к) поддерживается постоянным.
9.2.2. Влияние изменения градиента
Изменения градиента могут быть намеренными или ненамеренными, зависящими
от изменения оборудования. Эти изменения могут быть следующими:
• изменение %В в начале градиента (исходный %В, раздел 9.2.2.1);
• изменение %В в конце градиента (конечный %В, раздел 9.2.2.2);
• задержка градиента (раздел 9.2.2.3);
• замена оборудования (объем задержки оборудования, раздел 9.2.2.4);
• кусочно-линейные градиенты (раздел 9.2.2.5).
9.2.2.1. Исходный %В
Обычно исходный %В изменяют для того, чтобы сократить время разделения, удаляя
пустое пространство в начале градиентной хроматограммы, как показано на рис. 9.7.
Изменение исходного %В (а следовательно, и изменение градиентного интервала Дф)
без изменения времени градиента приводит к изменению величины к*
(уравнение 9.5), что может быть нежелательно. В этом разделе будет рассмотрено влияние
изменения исходного %В при сохранении к* постоянным (с помощью изменения
времени градиента ^-пропорционально Дф, сохраняя (Дф/Гс) и к* постоянными.
Следует иметь в виду, что если изменяется только %В при прочих неизменных условиях,
448 Глава 9. Градиентное элюирование
0-100 %В
за 50 мин
2 %В/мин
(а)
Т1,
10
—1—п—(—I—I—I—I—I—|—I—I—I—г-
20 30
Время (мин)
40
2-100 %В
за 40 мин
2 %В/мин
il!ii
(б)
10
' ' ' l
20
Время(мин)
Л
30
40-100 %В
за 30 мин
2 %В/мин
(а)
1—'—■—'—•-
0
60-100 %В
за 20 мин
2 %В/мин
1
10
Время (мин)
20
<*)
i 1 1 1 г-
-т т 1 г-
4 6
Время (мин)
10
Рис. 9.7. Влияние изменения исходного %В на градиентное разделение «правильного»
образца: смеси гербицидов. Условия: колонка C|g 150 х 4,6 мм, 5 мкм;
скорость потока 2,0 мл/мин; комнатная температура; подвижная фаза
метанол — вода. Время градиента подобрано таким образом, чтобы к* = 4.
Остальные условия указаны на рисунке
то возникающие изменения разделения будут обусловлены общим воздействием
изменения к* и начального %В.
Гораздо проще интерпретировать и оптимизировать разделение, если к*
поддерживается постоянным, а начальный %В (или другой параметр) варьируется (как
в предыдущем примере с изменением длины колонки или скорости подвижной фазы).
На рис. 9.7 проиллюстрировано влияние изменения начального %В на
разделение «правильного» образца, при этом в разделениях последовательно увеличивается
значение %В в начале градиента (что приводит к сокращению градиентного
интервала Дф). Одновременно сокращается время градиента г^для того, чтобы поддерживать
(Дф//д) и к* постоянными. При увеличении исходного %В от 0 до 20 % (рис. 9.76) Д<р
уменьшается на 20 %, поэтому требуется соответствующее сокращение времени
градиента на 20 % от 50 минут до 40 минут для сохранения к* постоянным
(уравнение 9.5). Разделение на рис. 9.76 остается практически таким же, как на рис. 9.7я
за исключением того, что пики элюируются на 10 минут раньше, и время разделения
9.2. Экспериментальные условия и их влияние на разделение 449
сокращается на 20 %. Когда начальный %В еще возрастает до 40 %В (рис. 9.7»),
наблюдается небольшое изменение для пиков I и 2: они становятся немного выше,
а их разрешение немного уменьшается (Rs= 2,7, в то время как Rs = 4,0 на рис. 9.7а).
Тем не менее разделение вполне приемлемое, а время разделения сократилось еще
на 10 мин. Наконец, на рис. 9.7г исходный %В увеличен до 60 %; это
сопровождается значительным возрастанием высоты пиков и заметным снижением разрешения
пиков 1 и 2 (Rs = 0,9). В этом случае наименьшее время разделения с приемлемым
разрешением достигается при 40 %В в начале градиента (рис. 9.7в).
Первые пики в градиенте 0—100 %В (рис. 9.7д) элюируются довольно поздно,
так как они прочно удерживаются на входе в колонку, и для них средние значения
к* и 3,7 в соответствии с уравнением (9.5). При увеличении начального %В до 20 %
(рис. 9.76) они все еще хорошо удерживаются, и по-прежнему к* » 3,7. Когда %В
в начале градиента еще увеличивается до 40 % (рис. 9.7е) и 60 % (рис. 9.7г), пики
выходят из колонки в еще более сильной подвижной фазе, и значения к* становятся
меньше (уравнение (9.5) строго выполняется только для пиков, которые прочно
удерживаются в начале градиента; для слабо удерживаемых пиков см.
уравнение (9.5в) в разделе 9.2.4.1). Это уменьшение значения к* для пиков, рановыходящих
при достаточном увеличении исходного %В, приводит к возрастанию их высоты и
обычно к ухудшению их разрешения.
Уменьшение значений к* рановыходящих пиков при достаточном увеличении
начального %В может также привести к изменению относительного удерживания
при разделении «неправильных» образцов. В результате этого в некоторых случаях
наблюдается увеличение разрешения пиков «неправильных» образцов при увеличении
начального %В 1101 несмотря на уменьшение к*. Градиентное разделение
«неправильных» образцов обсуждается далее в разделе 9.2.3.
9.2.2.2. Конечный %В
На рис. 98 проиллюстрировано влияние изменения конечного %В на разделение
«правильного» образца, приведенного на рис. 9.7я, с целью уменьшения времени
разделения. На рис. 9.8а показано разделение в градиенте 0—100 %В в течение 50 минут.
Последующее уменьшение конечного %В сопровождаются уменьшением времени
градиента для сохранения постоянными (Дф/?с) и к* (как и на рис. 9-7 при изменении
начального %В). Так при сокращении Дф на 20 %В до конечного 80 %В на рис. 9.86
время градиента также уменьшается на 20 % (от 50 до 40 минут). Изменений в разделении
нет, так как последний пик выходит из колонки до окончания градиента (обозначено
стрелкой на рисунке). Дальнейшее укорачивание градиента до 0—60 %В за 30 минут
(рис. 9Яе) приводит к тому, что пики 7—10 элюируются уже после окончания
градиента, то есть уже в изократическом режиме. В результате увеличивается ширина пиков
7—9 и их разрешение, как и время разделения вследствие более высоких значений к*
этих пиков. (Следует иметь в виду, что уравнение (9.5) справедливо только для пиков,
элюирующихся в течении градиента; для пиков, выходящих после окончания
градиента значения к* больше.) Можно сравнить рис. 9.8в с чрезмерно заниженным конечным
%В и рис. 9.7г с чрезмерно завышенным исходным %В; в каждом из этих случаев
разделение становится неудовлетворительным — или слишком низкое разрешение, или
слишком велико время разделения.
Так как последний пик выходит из колонки до завершения градиента (рис. 9.86),
уменьшение конечного %В не оказывает влияние на разделение (при условии что
Дф//с остается постоянным) — только сокращается время разделения. В большинстве
случаев рекомендуется завершать градиент после выхода последнего пика, но не
раньше. Элюирование пиков по окончании градиента приводит к неоправданно
длительному разделению и нежелательному уширению пиков (рис. 9.8е). Уменьшение ко-
450 Глава 9. Градиентное элюирование
0-100 %В
за 50 мин
2 %В/мин
1
" Г1Г
I 1 1-
(в)
20
40
бремя (мин)
0-80 %В
за 40 мин
2 %В/мин
i
(б)
iilDILJi
20
Время (мин)
0-60 %В
за 30 мин
2 %В/мин
i ■ '—
(в>
.8
.9
20
60
40
Время (мин)
Рис. 9.8, Влияние изменения конечного %В на градиентное разделение
«правильного» образца, приведенное на рис. 9.7. Условия разделения такие же, как
на рис. 9.7. Время градиента подобрано таким образом, чтобы к* = 4.
Пунктирной линией обозначен градиент; указан %В на выходе из колонки
(для соответствия пикам на хроматограмме). Стрелками обозначено
окончание градиента на выходе из колонки. Остальные условия приводятся на
рисунке
нечного %В одинаково влияет на разделения «правильных» и «неправильных»
образцов (не изменяется относительное удерживание или порядок элюирования),
если приняты меры для предотвращения позднего элюирования, а Д(рДс
поддерживается постоянным. Для некоторых образцов использование очень крутых градиентов
может приводить к элюированию последних пиков после окончания градиента, даже
если градиент заканчивается 100 %В (и более пологие градиенты не приводят к
позднему элюированию). Тогда не возникает никаких особых проблем: надо только
подождать, пока последний пик выйдет из колонки (добавив изократическую задержку
в конце градиента, например, 0/60/60% за 0/30/60 минут для разделения на рис. 9.8ff)
перед запуском следующего градиента. Хотя градиент, представленный на рис. 9.86,
обеспечивает более благоприятные условия разделения.
Объединив примеры, приведенные на рис. 9.7 и рис. 9.8, можно сделать вывод,
что градиент 40—80 % В за 20 минут позволяет приемлемым образом сократить
исходный градиент (рис. 9.7а; 0—100 %В за 50 минут). Это разделение показано
на рис. 9.9а; в этом случае время разделения на 60 % меньше, чем на рис. 9.7й, без
потери разрешения и других проблем.
9.2.2.3. Задержка градиента
Задержкой градиента (или изократической задержкой) называется некоторый период
времени изократического элюирования, предшествующий началу градиента.
Влияние задержки градиента на разделение «правильного» образца (рис. 9.7) проиллюст-
9.2. Экспериментальные условия и их влияние на разделение
рирован на рис. 9.9. На рис. 9.9а показана хроматограмма разделения в градиенте
40—80 %В без задержки градиента, при этом первый пик хроматограммы еще долго
удерживается в колонке после того, как градиент уже достиг выхода колонки
(«мертвое» время колонки /0 обозначено стрелкой). Когда используется пятиминутная
задержка градиента (рис. 9.96), время удерживания пиков возрастает на 2—5 мин,
но в остальном хроматограммы рис. 9.9а и 9.96 сходны (наблюдается также типичное
небольшое увеличение разрешения рановыходяших пиков на рис. 9.96).
Когда рано выходящие пики элюируются из колонки вскоре после запуска
градиента, его задержка может более существенным образом влиять на разделение,
особенно если рановыходящие пики плохо разрешаются. Этот случай показан
на рис. 9.9е (задержка градиента отсутствует) и рис. 9.9? (введена задержка
градиента) для того же образца, разделяемого в градиентах с иными исходными %В. В
разделении на рис. 9.9г пики I—3 элюируются из колонки во время задержки градиента
40-80 %В за 20 мин
2,0 %В/мин
i
U
idolZII'
8 8 12
Время (мин)
14 16
18
(а)
40/40/80 %В за 0/5/25 мин
2,0 % В/мин
bbiiilZA
Время (мин)
(б)
20
50-100 %В за 7,5 мин
6,7 %В/мин
(в)
4 6 в 10 12
Время(мин)
50/50/100 %В за 0/5/12,5 мин
6,7 %В/мин
I ' ' ■"
0 2 4
*ы
£
(г)
-, 1 1 1
6 8 10 12
Время (мин)
Рис. 9.9. Влияние задержки градиента на разделение образцов гербицидов,
представленное на рис. 9.4. Условия: колонка Сц 150x4,6 мм, 5 мкм; скорость
подвижной фазы 2,0 мл/мин; 30 °С; подвижная фаза метанол — вода. Время
градиента подобрано таким образом, чтобы к* = 4. Высота пиков не
нормализована к 100 %. Пунктиром показан градиент, а стрелками указано начало
градиента (измеренное на выходе из колонки). Остальные условия указаны
на рисунке
452 Глава 9. Градиентное элюирование
(стрелкой на рисунке показано время поступления градиента на выход колонки).
Эти два примера показывают, что пики 1 и 2 плохо разрешаются на хроматограмме
рис. 9.9e (Rs = 1,1), в то время как разрешение на рис. 9.9г гораздо лучше (Rs= 2,3).
Улучшение разрешения при введении задержки градиента можно отнести на счет
больших величин к* для этих пиков по сравнению с разделением на рис. 9.9в
(см. далее уравнение (9.5в)). У пиков 1—3 на рис. 9.9г, как и следовало ожидать,
увеличивается ширина, что характерно для изократического разделения, в то время
как пики, выходящие позднее в градиенте, имеют меньшую ширину, что типично
для градиентного разделения.
Когда пики элюируются вблизи окончания градиента, их времена удерживания
должны увеличиться на время задержки градиента без изменения относительного
удерживания. Например, у двух последних пиков на рис. 9.96,г времена удерживания
увеличивается на 5 мин по сравнения с временами удерживания на рис. 9.9а,в — это
в точности соответствует времени задержки градиента. Так ведут себя и
«правильные», и «неправильные» образцы.
Задержка градиента иногда используется для повышения разрешения рановыходя
ших пиков как на рис. 9.9г по сравнению с рис. 9.9tf. Хотя для разделений, которые
начинают с более высокого %В (например, рис. 9.9в) можно существенно улучшить
разрешение, просто понизив исходное значение %В градиента (сравните разделения
на рис. 9.7г и рис. 9.7в). С другой стороны, если начальный %В градиента близок
к нулю (и следовательно, существенное понижение %В невозможно), задержка
градиента может быть наиболее удобной альтернативой. Все же есть другие способы
увеличить к в этой ситуации (раздел 6.6.1). Следует иметь в виду, что относительное
удерживание «правильного» образца не изменяется при введении задержки градиента
(см. рис. 9.9). Однако относительное удерживание может изменяться у «неправильных»
образцов, так как задержка градиента может оказывать влияние на значения к* рано-
элюирующихся пиков (см. раздел 9.2.2.4 и сравните рис. 9.13е с рис. 9.13а).
9.2.2.4. Приборный объем задержки
Любое оборудование, используемое для градиентного элюирования, будет иметь
определенный удерживаемый объем (так называемый объем задержки Vq), равный объему
градиентного миксера плюс объем капилляров между миксером и входом в колонку
(рис. 3.13 и 3.14; раздел 3.5.3). Объемы задержки Vq могут варьироваться у разных
приборов от долей миллилитра у современного оборудования до нескольких миллилитров
у старых хроматографических систем. Наличие объема задержки эквивалентно
намеренному использованию задержки градиента, следовательно, о влиянии на разделение
изменяющегося времени задержки /д = Vq/F можно судить, исходя из данных рис. 9.9
для задержки градиента. Реальный градиент, поступающий на колонку, задерживается
на время Гд, в то время как градиент, выходящий из колонки, дополнительно отстает
из-за «мертвого» времени колонки /о (рис. 9.10). В разделе 3.10.1.2 описано, как можно
определить значения Кддля конкретной градиентной системы.
Когда градиентный метод переносится с одной системы ВЭЖХ на другую,
отличие объемов задержки Vq этих двух систем может привести к изменениям в
разделении. Часто метод ВЭЖХ разрабатывается на более современном оборудовании в
исследовательской лаборатории, а текущие анализы предстоит проводить на более
старом оборудовании производственной лаборатории. В результате объем задержки
оборудования будет больше при использовании разработанного метода для текущих
анализов по сравнению с исходным. Для «правильного» образца, такого как в
примерах на рис. 9.9, увеличение объема задержки приведет к возрастанию времени
удерживания всех пиков. При этом, возможно, уменьшится высота пиков и увеличится
разрешение рановыходящих пиков (как в примере на рис. 9.9г). Относительное удер-
9.2. Экспериментальные условия и их влияние на разделение
%В
реальный градиент на
входе в колонку(сдвинуто с
учетом (0)
реальный градиент на выходе из
колонки (сдвинуто с учетом (D + (0)
Рис. 9,10. Влияние объема задержки оборудования на градиент. ( )
запрограммированный градиент, выбранный пользователем; (—) реальный градиент
на входе в колонку с учетом объема задержки оборудования; (....) реальный
градиент на выходе из колонки, полагая, что время задержки оборудования
равно Гд
живание не изменится при различных значениях Vq. Однако если на оборудовании
с иным объемом задержки анализируются «неправильные» образцы, может
измениться относительное удерживание рановыходящих пиков, что может привести
к неприемлемому изменению их разрешения (сравните примеры на приведенных
далее рис. 9.13е и 9.13а). Эти и другие примеры, относящиеся к объему задержки
оборудования, обсуждаются в разделе 9.3.S.2.
Аналогичная ситуация может возникнуть, если изменяется объем колонки (и ее
«мертвый» объем Vm), так как влияние объема задержки на относительное
удерживание рановыходящих пиков определяется отношением Vg/Vm. При изменении объема
колонки может возникнуть необходимость подогнать объем задержки к объему
колонки для сохранения прежних значений относительного удерживания и разрешения
рановыходящих пиков. Например, если диаметр колонки dc уменьшен для
использования в хроматографической системе с масс-спектрометрическим детектором, объем
задержки в этой системе следует изменить пропорционально d}. (Обычно пользователь
имеет возможность уменьшить объем задержки в системе со смешиванием на стороне
высокого давления, но не в системе со смешиванием на стороне низкого давления).
Если диаметр колонки увеличивают для масштабирования препаративного разделения,
может потребоваться такое же увеличение объема задержки. Хотя более удобно
компенсировать это добавлением изократической задержки перед началом градиента.
Более подробно эти проблемы рассмотрены в |11, 12| и в разделе 3.5.3 |2|.
Когда выполняется тестовый градиент, как описано в разделе 3.10.1.2, обычно
наблюдается некоторое искажение в его начале и в конце (рис. 3.26). Градиентные
закругления обусловлены дисперсией растворителей А и В при попадании
подвижной фазы в градиентный миксер и на вход колонки. Градиентные закругления более
ярко выражены для градиентных систем со смешиванием на стороне низкого
давления. Степень закругления градиента возрастает при более значительных
величинах Vq ее можно количественно описать через объем смешивания оборудования V^
(Vm ж Vq). Градиентные закругления слабо влияют на разделение, если значение V^
не становится сравнимым с объемом градиента Vq = IqF. Более подробно влияние
объема смешивания на форму градиента и разделение рассмотрено в разделе 17.4.6.1
и на стр. 394—396 [2].
454 Глава 9. Градиентное элюирование
9.2.2.5. Кусочно-линейные градиенты
Кусочно-линейные градиенты, подобные показанному на рис. 9.2г, используются
для следующих целей:
• для промывания колонки между нанесением образцов;
• для сокращения времени разделения;
• для повышения разрешения регулировкой селективности в разных частях хрома
тограммы (только для «неправильных» образцов).
Кусочно-линейные или ступенчатые градиенты часто используются для
промывания колонки при разделении биологических образцов и проб окружающей среды,
потому что содержащиеся в них посторонние примеси, прочно удерживаемые на колонке
(и не подлежащие анализу), могут повредить колонку. Когда разделяются подобные
образцы, а также в том случае, если для элюирования всех пиков требуется градиент
почти до 100 %В, обычно вслед за градиентом вводится отрезок (или ступень) крутого
градиента, который заканчивается приблизительно 100 %В. На рис. 9.11а показано
разделение в линейном градиенте смеси пептидов гидролизованного трипсином ре
0—40 %В за 50 мин
Rs = 2.1
10
20
30
Время (мин)
0/40/100/100 %В за 0/50/51/52 мин
Я. = 2.1
1~
ли
10
20 30
Время (мин)
Т\
- 100 %В
- 80%
- 60%
" 40%
20%
0%
(а)
40
(б)
- 100 %В
- 80%
- 60%
- 40%
- 20%
- 0%
40
50
0/23/42 %В за 0/32/38 мин
Rs = 2.1
^A^^Jc.
Li
10
20
Время(мин)
30
- 100 %В
2 80%
- 60%
- 40%
- 20%
- 0%
40
Рис. 9.11. Градиентное разделение пептидов гидролизованного трипсином рекомби-
нантного гормона роста человека. Условия: колонка C|g 150x4,6 мм,
5 мкм; 2,0 мл/мин; 45 °С; (а) 0—40 %В за 50 мин; (6) то же, что и в (а),
но добавлен отрезок крутого градиента для удаления из колонки сильно
удерживаемого «мусора»; (в) то же, что и в (с), но добавлен второй участок
градиента для того, чтобы ускорить элюирование двух последних пиков на
хроматограмме. Градиент обозначен (—). Хроматограммы воссозданы по
данным работы [13|
9.2. Экспериментальные условия и их влияние на разделение
комбинантного гормона роста человека (rh-GH). 19 пептидов разделяются с
разрешением до базовой линии за 50 минут. На рис. 9.116 за разделением рис. 9.11а следует
ступень градиента 40 %В — 100 %В за 1 минуту для того, чтобы отмыть колонку от
любых примесей образца, не элюировавшихся в градиенте. За этим крутым градиентом
после разделения обычно следует изократическая задержка. Таким образом,
окончательный вариант градиента на рис. 9.1 16: 0/40/100/100 %В за 0/50/ 51/52 минут.
Сокращение времени разделения показано на рис. 9.1 \в для образца рис. 9. i la без за
ключительного промывочного градиента (который можно при необходимости
добавить). Поскольку последние пять пиков на хроматограмме разрешаются с Rs » 2,
для их разделения можно увеличить крутизну градиента, уменьшая их времена
удерживания и сохраняя Rs > 2 для всех пиков. Таким образом можно сократить время
разделения с 50 минут на рис. 9.1 la до 40 минут на рис. 9.1 \в.
Иногда можно повысить разрешение регулировкой селективности в разных частях
хроматограммы, используя кусочно-линейный градиент. Для различных критических
пар пиков подбирается оптимальная крутизна градиента (и значения к*). Пример
разделения смеси полициклических ароматических углеводородов показан на рис. 9.12;
пары пиков 3—4 и 14—15, отмеченные *, являются критическими. В то время как пара
пиков 3—4 лучше разделяется в пологом градиенте (большие к*), разделение пиков 14
и 15 улучшается в более крутом градиенте (меньшие значения к*). На рис. 9.12а
наклон линейного градиента был выбран для максимального критического разрешения
данного образца. Максимальное критическое разрешение соответствует одинаковому
разрешению этих двух пар пиков, так как изменение крутизны градиента увеличит
разрешение одной пары пиков, но уменьшит разрешение другой пары. Однако разреше
ние каждой пары пиков можно улучшить с помощью кусочно-линейного градиента,
показанного на рис. 9.126. В этом градиенте сочетается более пологий участок для
разрешения пиков 3 и 4 с более крутым участком для разрешения пиков 14 и 15.
Небольшое увеличение разрешения на рис. 9.126 (+ 0,3 единицы разрешения по сравнению
с рис. 9.12а) типично для кусочно-линейных градиентов. Кусочно-линейные градиен-
8 10 12
Время(мин)
40/50/100 %В
за 0/7,5/11,5 мин
«,= 1.7
—р т 1 т г
4 6 8
Время (мин)
И "'Ни
(б)
1
L
и
i
10
.,—100 %В
-'"' -80%
, * 160%
IT1 -40%
1 1
12
Рис. 9.12. Разделение смеси 16 полициклических ароматических углеводородов
(переработанный рис. 6.4 из [6]). Условия: колонка C]g 150x4,6 мм, 5 мкм;
2,0 мл/мин; 35 "С; (о) разделение в оптимизированном линейном
градиенте; (б) разделение в оптимизированном градиенте, состоящем из двух
участков. Градиент показан (—). Для более подробной информации см. [6]
456 Глава 9. Градиентное элюирование
ты редко позволяют увеличить разрешение более, чем на = 0,5 единиц. Время,
потраченное на отработку таких разделений, может оказаться неоправданным, если не
применять компьютерное моделирование (раздел 10.2.3.4).
Кусочно-линейные градиенты нечасто используются для улучшения разрешения
как на рис. 9.126, так как возможность повысить разрешение с помощью таких
градиентов без увеличения времени разделения обычно ограничена 114|. Для того,
чтобы использовать кусочно линейные градиенты для повышения критического разре
шения требуется, чтобы были, по крайней мере, две критические пары пиков, одна
из которых элюировалась бы рано, а другая поздно (как на рис. 9.12). В противном
случае частичное перемещение второй пары пиков под действием начального
сегмента градиента обеспечит лишь небольшое преимущество от использования второго
сегмента градиента или вообще сделает его бесполезным. Однако эти ограничения
на использование кусочно-линейных градиентов для повышения разрешения
образцов становятся менее важными в случае высокомолекулярных соединений, таких как
белки 115, 16|, поскольку поздновыходящие пики меньше перемещаются по колонке
в течение начального этапа градиента, который поэтому в меньшей степени
воздействует на разрешение поздновыходящих пиков. По этой причине более обосновано
использование кусочно линейных градиентов для повышения критического разре
шения при разделении смесей больших биологических молекул. Однако существуют
другие — обычно более эффективные — способы оптимизации разрешения с помо
шью изменения селективности и относительного удерживания (раздел 9.3.3). Также
разделения, в которых используются кусочно-линейные градиенты для повышения
разрешения, хуже воспроизводятся при переносе на другое оборудование.
Более подробно использование кусочно-линейных градиентов для повышения
разрешения анализируется в |17, 18|. Также есть сообщения о компьютерных
программах для автоматизированной разработки оптимизированных кусочно-линейных
градиентов |14, 19, 20|. Ступенчатое элюирование, включающее ступенчатые
градиенты, можно рассматривать как простой (в общем случае менее эффективный)
вариант кусочно-линейного градиента; теория таких разделений приводится в |21|.
9.2.3. «Неправильные» образцы
В этом разделе обсуждаются градиентные разделения, в которых изменяется
относительное удерживание «неправильных» образцов вследствие изменения некоторых
условии, влияющих на к* (время градиента, скорость потока и т.д.). Приведенные примеры
дополняют рассмотренные выше на рис. 9.4 и рис. 9.6—9.8 для «правшьных» образцов,
иллюстрируя изменения относительного удерживания для «неправильных» образцов в
зависимости от изменения условии, которые влияют на к*. Читатель при желании
может перейти к главе 9.3 и вернуться к этому разделу позже или в случае
необходимости. Однако этот разбор может способствовать интуитивному пониманию читателем
градиентного элюирования, а также оказаться полезным на практике.
к* может измениться вследствие изменения любых экспериментальных условий,
входящих в уравнение (9.5) (fg, Г, Ут или длина колонки L, Дер), а также при
изменении исходного %В, введения задержки градиента или изменения объема задержки
оборудования. Увеличение к* приведет в общем случае к возрастанию времени
удерживания, разрешения и ширины пика для всех образцов, как показано на рис. 9.4
и 9.5 в случае изменения времени градиента. В случае «неправильных» образцов
(рис. 9.5) изменение к* также приведет к изменению относительного удерживания,
которое может вызвать изменение разрешения некоторых пиков. Любое изменение
А* для данного «неправильного» образца приведет к аналогичным изменениям
относительного удерживания и разрешения вне зависимости от того, каковы изменения
к*. Это иллюстрируется далее в этом разделе для различных изменений градиента
9.2. Экспериментальные условия и их влияние на разделение 457
или параметров колонки на примере рис. 9.13 для выбранных пар пиков (2—3 и 8—
9) из «неправильного» образца, приведенного на рис. 9.5. Так как многие реальные
образцы попадают в категорию «неправильных», приведенное далее обсуждение,
по-видимому, отражает те изменения, которые большинство пользователей будут
наблюдать при изменении условий градиентного элюирования.
Исходное разделение пар пиков 2—3 и 8—9 «неправильного» образца рис. 9.5
показано на рис. 9.13а. Эти две пары пиков выбраны из тех соображений, что измене
ние к* влияет на их разрешение противоположным образом (из-за различий
значений S этих четырех веществ: £3 > $2! $8 < ^9< см- аналогичные примеры на рис. 6.7в).
5—100 %В за 5 мин
L - 50 мм
F = 2 0 мп/мин
*-=5
5—100 %В за 20 мин
L = 50 мм
F = 2.0 мл/мин
»г* = 20
5—100 %В за 5 мин
L = 100mm
F- 2.0 мл/мин
к-= 2.5
5—100 %В за 5 мин
L =50 мм
F~ 8.0 мл'мин
**=20
15/100 %В за 4,5 мин к'<Ь
L = 50 мм
F = 2.0 мл/мин
А*>5
5/5/100 %В за 0/5/10 мин
(. = 50 мм
F = 2.0 мп/мин
X
(в)
Рис. 9.13. Изменение расстояния между пиками при изменении условий градиента.
Образец состоит из пиков 2, 3, 8, 9 «неправильного» образца рис. 9.5.
Условия: 28 "С. На рис. 9.13s стрелками обозначено относительное смещение
пиков 2 и 9 вследствие увеличения времени градиента и к*. Градиент
обозначен (—)
458 Глава 9. Градиентное элюирование
Сначала рассмотрим увеличение времени градиента от 5 до 20 минут {рис. 9.136),
соответствующее изменению среднего к* от 5 до 20. В результате удерживание пика 2
увеличивается по сравнению с удерживанием пика 3, следовательно, увеличивается
разрешение этой пары пиков. В то же время удерживание пика 9 по сравнению
с удерживанием пика 8 уменьшается при увеличении времени градиента, и
разрешение этой пары пиков уменьшается. Аналогичные изменения относительного
удерживания и разрешения этих двух пар пиков следует ожидать при изменении любых
условий, приводящих к возрастанию к*. Относительное удерживание изменится
противоположным образом при уменьшении к*.
На рис. 9.l3ff длина колонки увеличена от 50 до 100 см, остальные условия
остаются такими же, как на рис. 9.13д. При этом значение к* вдвое уменьшается до к* =
= 2,5 (уравнение 9.5в, приведено далее). Как и следует ожидать при таком
уменьшении к* (по сравнению с разделением на рис. 9.1 За) относительное удерживание
на рис. 9.13в изменяется противоположным образом, чем на рис. 9.136: теперь пик 2
сближается с пиком 3, и разрешение падает, а пик 9 отодвигается от пика 8, и
разрешение увеличивается.
Влияние увеличения скорости потока (от 2,0 до 8,0 мл/мин) показано
на рис. 9.13г. Поскольку к* увеличивается от 5 до 20 (уравнение 9.3), следует ожидать
такое же изменение относительного удерживания, как при увеличении времени
градиента (рис. 9.135): снова пик 2 отдаляется от пика 3 с увеличением разрешения,
а пик 9 сближается с пиком 8 с уменьшением разрешения.
Когда увеличивается %В в начале градиента, возрастает tpg, а Л<рЛс
поддерживается постоянным (рис. 9.13d), значение к*, вычисленное по уравнению (9.5), остается
прежним. Однако реальные значения к* рановыходящих пиков уменьшаются
(уравнение 9.11) несмотря на постоянную величину Аф/fc1- Таким образом, уравнение (9.5)
оказывается неприменимым для рановыходящих пиков на хроматограмме, поэтому
пик 2 смещается к пику 3. На значение к* поздновыходящих пиков в меньшей степени
влияет увеличение исходного %В, поэтому относительное удерживание и разрешение
пиков 8 и 9 изменяются меньше (по сравнению с разделением рис. 9.13я).
Наконец, на рис. 9.13е вводится задержка градиента 5 минут (или возрастает
объем задержки оборудования гд) по сравнению с разделением на рис. 9.13а
(остальные условия не изменяются). Как и в предыдущем примере (рис. 9.13d) k*,
рассчитанный с использованием уравнения (9.5), не изменяется (к* = 5), но под влиянием
задержки градиента должно уменьшиться воздействие градиента на рановыходящие
пики на хроматограмме. Это в свою очередь обусловливает гораздо большие
значения к* этих рановыходящих пиков (уравнение 9.12). В результате наблюдается такое
же изменение сравнительного удерживания и разрешения, как на рис. 9.136 при
увеличении времени градиента, но в меньшей степени для поздновыходящих пиков 8 и
9 (на величины к* которых меньше влияют задержка градиента или изменение
исходного %В). Изменение объема задержки оборудования и соответствующего
времени задержки (из-за замены оборудования) таким же образом повлияет на разделение,
как введение градиентной задержки на рис. 9.13е.
Помимо изменений относительного удерживания на разрешение также влияют
изменения к* и N* (см. уравнение 9.15в далее). Вклад этих компонентов иногда
может искажать зависимость разрешения от относительного удерживания.
9.2.4. Количественные соотношения
Модель ЛИС позволяет получить ряд точных выражений для удерживания и ширины
пика; эти уравнения составляют основу компьютерного моделирования градиентного
элюирования (раздел 10.2). Помимо компьютерного моделирования и определения
зависимости к*от экспериментальных условий приведенные далее уравнения (9.5а)—
9.2. Экспериментальные условия и их влияние на разделение 459
(9.15) имеют ограниченное практическое применение. Поэтому можно при желании
перейти к уравнению (9.16), приведенному в конце этого раздела, и вернуться к
содержанию этого раздела в случае необходимости. Вывод уравнений этого раздела и их
приложения подробно рассмотрены в главе 9 |2|.
Предполагается, что все приведенные ниже уравнения выведены для линейных
градиентов в условиях обращенно-фазовой хроматографии. Значение к* можно
описать соотношением, соответствующим уравнению (9.1) для изократического элюиро
вания:
log к* = log кК - 5<р*, (9.5а)
где ф* относится к значению ф для подвижной фазы, контактирующей с зоной
вещества, находящегося в середине колонки. Значения кК и S одинаковы при изократиче-
ском и градиентном элюировании.
Vm можно вычислить, зная длину колонки L и внутренний диаметр колонки dc
(учитывая уравнение (2.7а), в котором предполагается общая пористость колонки
ег=0,65):
Vm « 5 х 1(Н Ld? (L и dc в мм). (9.56)
Для колонок обычного диаметра 4,6 мм Vm удобно аппроксимировать 0,01 длины
колонки в мм. Например, Vm в 1,5 для колонки размером 150 х 4,6 мм. Объединяя
уравнения 9.5 и 9.56, получаем
к*=™р£ (9.5в)
Ld?Aq>S
или при S к 4 для малых молекул
к* = —=@— (для веществ с молекулярной массой < 500 Да и S ж 4). (9.5в)
Z-rf/Дф
Таким образом, к* будет возрастать при увеличении tc и /""или уменьшении
длины колонки £, диаметра колонки dc или градиентного интервала А<р. Уравнения (9.4)
и (9.5) также показывают, что к* связан с параметром крутизны градиента Ь:
к* = W-. (9.6)
ь
То есть величина А* уменьшается в более крутых градиентах с возрастанием
значения Ь.
9.2.4.1. Время удерживания
Время удерживание вещества iR при градиентном элюировании рассчитывают
по-разному в зависимости от того (1) насколько велик объем задержки оборудования
{VD > 0) и (2) мала ли исходная величина к в начале градиента (А0). Значение fc0
описывается уравнением:
log k0 = log kw - 5ф0. (9.7)
Если ка велик и если Vq = 0, то
tR = ^]\og(2,3k0b+l) + t0, (9.8)
tR . №) log(2,3A06) + t0. (9.8a)
460 Глава 9. Градиентное элюирование
Если А0 велик и если Vq > О, то
f/t = f^]log(2,3A0b+ D + 'o + 'fl.
Здесь /д = Vq/F время задержки колонки.
Если ко мал и если Vq > О, то
r^^jlog 2,3*0й
+ ![ + !} + f„ + гд
(9-9)
(9.9а)
(9.10)
Уравнение (9.10) справедливо вне зависимости от величины Aq или Vq. В уравне
ниях (9.8)—(9.10) предполагается, что пики не элюируются до начала или после
окончания градиента. Эти уравнения рассматриваются также в |22|. Уравнение (9.9) часто
является хорошим приближением для градиентных разделений и широко цитируется
в литературе (хотя иногда используются иные обозначения, см. xxv—xxvi в [2|).
Значения градиентного коэффициента удерживания к* также могут изменяться
в зависимости от величины Ад и Vq. Для малых значений Ад и Vq = 0
(9.11)
1Д5* + {1/А0)
Для малых значений Ао и Кд > 0 (или любых значений ко и Vq)
2,ЗЫ(А„/2) -(Уо1Ут)\ + 1 2,36|<*о/2) -(W>o)l + Г
(9.12)
Таким образом, небольшая величина Ад приводит к уменьшению к* по
сравнению с ее значениями, вычисленными из уравнений (9.4) или (9-6). Аналогичным
образом, чем больше значения времени задержки 1q (или времени задержки
градиента Ijeiay), тем больше значения к* по сравнению со значениями, полученными
из уравнений (9.4) или (9.6).
9.2.4.2. Измерение величин 5 и kw
Значения S и kw можно получить из изократических значений к , зависящих от ф
в соответствии с уравнением (9.7), или из двух градиентных экспериментов,
отличающихся только временем градиента. Когда при градиентном алюировании
значения кй велики, удерживание в линейном градиенте ОФХ точно описывается
уравнением (9.9а). Для этого случая можно вычислить значения S и kw для любого
компонента образца, проведя два разделения, отличающихся только временем градиента.
Предположим, проведены эксперименты с временами градиентов fC! и t(% (tGl <
< ta)> а их отношение р = /ci/'C2- Величины Гд для данного образца в эксперименте 1
tg\, а в эксперименте 2 Igj, тогда значение Ь\ можно вычислить по формуле:
Аналогичным образом
log ко
fp log р*
-(/ю/Р)-(Го+Гц)(Р-1)/р
-log(2,36|).
(9.13)
(9.13a)
9.2. Экспериментальные условия и их влияние на разделение 461
Подставив ii в уравнение (9.4а) можно рассчитать значение S, a log kw тогда
рассчитывается как log kQ + Sq>Q.
9.2.4.3. Ширина пика
Ширина пика в градиентном элюировании определяется таким же образом, как и
в изократическом разделении (раздел 2.3), и может быть вычислена с
использованием любой из следующих эквивалентных формул:
IV = (4N*-°-5)GiJ [ +—)= (9.14)
\ 2,36
= (4Л,*-°'5)Сг011 + — ] = (9.14а)
= (4N*-°-5)GtB( I + /у. (9.146)
То есть W можно связать с крутизной градиента Ь, значением к* или значением к
в момент, когда пик элюируется с колонки (ке). Как отмечалось в разделе 9.1.3.2,
ке — к*/2. Коэффициент сжатия пика G описывает сужение пика при
градиентном элюировании благодаря более быстрому перемещению заднего фронта зоны
в подвижной фазе с большим %В по сравнению с фронтом пика в подвижной фазе
с меньшим %В [23, 24|. G можно выразить через крутизну градиента Ь |25|. Сначала
дадим определение величины р:
р=2ЛоЬ^22ь (9]5)
kfj + 1
для больших к0. Тогда G можно выразить через р
i0,5
W + P+\P2
\ (1 + Р)2
g=r \г''"2/3|)1 ■ <*,5»>
Значения С изменяются с увеличением крутизны градиента b следующим
образом: для 0,05 < b < 2 (соответствующего 0,4 < к* < 17) 0,6 < С < I; то есть большее
значение b или меньшее к* приводят к уменьшению G. Таким образом, значение G
изменяется от 0,6 при очень крутых градиентах до 1,0 при очень пологих. Более
удобное уравнения для С можно вывести, исходя из сходства уравнений для изокра-
тического и градиентного элюирования (уравнений 2.24 и 9Л5):
1 + к*
С « . (9.156)
\ + 2к*
Для значений к* > I уравнение (9.156) имеет точность в пределах нескольких
процентов.
Теория сжатия пиков при градиентном элюировании была вполне разработана
в 1981 году, но до 2006 не удавалось подтвердить это явление экспериментально
|24|. В настоящее время полагают, что эта неопределенность, касающаяся сжатия
пиков, в основном была обусловлена недостаточной точностью уравнения (9.1) и
использованием уравнения (9.14) вместо уравнения (9.146). Последняя формула
точнее, если графики зависимости к от ср не вполне линейны (из-за неточности
уравнения 9.1).
462 Глава 9. Градиентное элюирование
9.2.4.4. Разрешение
Для градиентного элюирования можно вывести уравнение, аналогичное уравнению
(2.24) для изократического |26]. В уравнение (2.23) можно подставить значения t^ и
tft, выраженные через уравнение (9.8а). Значения Wj и W, можно заменить шириной
одного пика ^(уравнение 9.14), а величину С можно аппроксимировать уравнением
(9.156). В результате получаем
RS= A
^jV*"2loga
(9.15в)
Используя аппроксимацию 2,3Iog(a) = (a — 1), получаем для малых значений а:
*. = (HIV^tW " О**0-5 ■ (9.16)
UJ[i+fc*J
Здесь а* величина фактора разделения а в тот момент, когда зона достигает
середины колонки (и к - к*), а N* величина N в момент, когда зона достигает середины
колонки. Значения /V* при градиентном элюировании такие же, как и значения N
при изократическом, когда к = к*. Уравнение 9.16 имеет в основном концептуальное
значение, описывая как разрешение зависит от к*, фактора разделения или
селективности и числа теоретических тарелок колонки. Это выражение будет полезно при
дальнейшем обсуждении разработки градиентной методики (раздел 9.3). Уравнение
(2.23), являющееся определением разрешения как при изократическом, так и при
градиентном элюировании, более точно, чем уравнение (9.16), и используется в
нашей книге для всех расчетов разрешения. Однако уравнение (2.23) не может служить
удобным ориентиром при разработке метода.
9.3. Разработка метода
Схема разработки метода для градиентного разделения (таблица 9-2, рис. 9.14) сходна
с таковой для изократического (раздел 6.4, рис. 6.21). Сначала следует рассмотреть
состав образца (этап I таблицы 9.2 и рис. 9.14) для того, чтобы определить
подходящие начальные условия. Потом следует определение цели разделения (этап 2),
например, разрешение до базовой линии (Rs> 2.0), наименьшее возможное время
разделения и условия, благоприятствующие (или не препятствующие) детектированию
и измерению отдельных пиков, представляющих интерес. Другие аспекты разработки
метода как для изократического, так и для градиентного разделений включают:
• возможную необходимость предварительной обработки образца (пробоподготовки)
перед вводом в колонку (глава 16);
• проверку воспроизводимости экспериментов (повторение разделений);
• проверку воспроизводимости работы колонки (пробовать разделение на двух или
более колонках из разных партий (раздел 9.3.8));
• проведение валидации метода и разработку теста для определения пригодности
системы после разработки метода (глава 12);
• разработку ортогонального метода, чтобы убедиться, что все пики,
представляющие интерес, были обнаружены при первичном анализе (раздел 6.3.6.2; |27|).
Указанные требования к разработке метода сходны с обсуждавшимися в
разделе 6.4 требованиями для изократических разделений, и эти последние не будут
обсуждаться в этой главе. Основным отличием разработки градиентного метода
и изократического являются эксперименты, используемые для окончательного
определения условий разделения (этапы 3—8 таблицы 9.2 и рис. 9.14).
9.3. Разработка метода 463
Таблица 9.2. План разработки рутинного градиентного разделения (сравните с рис. 9.14)
Этапы
1 .Соберите информацию об
образце
2. Определите цели разделения
3. Проведите первичное
разделение (эксперимент 1)
4. Оптимизируйте градиентный
фактор удерживания к*
5. Оптимизируйте селективность
разделения а*
5а. Если после пятого этапа
/?s « 2 или требуется очень
короткое время анализа, то
продолжайте изменять условия для
оптимизации расстояния между
пиками (для получения
максимального Rs или минимального
времени анализа)
6. Подберите диапазон и
профиль градиента
7. Подобрав лучшие условия
на этапах 5 или 6, найдите
оптимальный компромисс между
разрешением и временем анализа
8. Определите время,
необходимое для уравновешивания
колонки исходной подвижной фазой
между вводами проб
Комментарии
а. Молекулярная масса > 5000 Да? (см. главу 13)
б. Требуется придать подвижной фазе буферные свойства?
в. Необходима ли пробоподготовка?
Раздел 6.4
а. Условия табл. 9.3; время градиента 10 минут;
б. Возникли проблемы? (раздел 9.3.1.1, рис. 9.17)
в. Возможно ли проведение анализа в изократическом
режиме? (рис. 9.15)
Соблюдение условий табл. 9.3 должно привести к
приемлемому значению к* ~ 5
Увеличьте время градиента втрое (эксперимент 2, 30 минут);
повысьте температуру на 20 °С (эксперименты 3 и 4);
см. примеры на рис. 9.18
а. Замените ацетонитрил на метанол и повторите
эксперименты 1—4
б. Замените колонку и повторите эксперименты 1—4
в. Измените рН и повторите эксперименты 1—4
г. Рассмотрите вариант кусочно-линейного градиента
(раздел 9-3.5 — наименее перспективный вариант)
а. Выберите лучшее начальное и конечное значения %В
для минимального времени анализа с приемлемым
значением Rs
б. Добавьте ступень резко изменяющегося к 100 % В
градиента для «загрязненных» образцов (пример на рис. 9.116)
е. Добавьте ступень градиента для ускорения элюирования
сильно удерживаемых, далеко отстоящих друг от друга
пиков (рис. 9.11 в)
д. Добавьте изократическую задержку для улучшения
разделения пиков, элюирующихся на старте градиента (рис. 9.9?)
Поменяйте колонку (раздел 9.3.6)
Используя разработанную методику, проведите подряд
несколько одинаковых разделений, изменяя время
уравновешивания колонки между анализами; выберите минимальное
время уравновешивания, которое обеспечивает приемлемое
разделение (раздел 9.3.7)
9.3.1. Первичное градиентное разделение
В идеале для первичного разделения предпочтительнее полномасштабный градиент
(0—100 %В); если в образце содержатся кислоты или основания, то подвижная фаза
должна содержать буфер (раздел 7.2). Однако если градиент начинается с 0 %В, могут
возникнуть сложности с некоторыми колонками, связанные с не смачиваемостью
стационарной фазы в отсутствие органического компонента (раздел 5.3.2.3; см. также |28,
29|). Поэтому начинать градиент лучше с 5 %В или более в том случае, если нет
полной уверенности, что для колонки допустима водная подвижная фаза (0 %В) или что
464 Глава 9. Градиентное элюирование
Разработка градиентного (или изократического) разделения
1. Собрать информацию о
составе образца (Раздел 6.4)
2. Определить цели
разделения (Раздел 6.4)
3. Провести первичное
разделение (Раздел 9.3.1)
Градиентное
разделение?
4. Оптимизировать к*
(Раздел 9.3.2)
5. Оптимизировать о'
(Раздел 9.3.3)
в. Подобрать диапазон и
профиль градиента (Разделы
9.3.4, 9.3.5)
7. Оптимизировать N'
(Раздел 9.3.6)
в. Определить время для
уравновешивания колики
(Раздел 9.3.7)
Биологический
образец
(Глава 13)
Энантиомеры
(Глава 14)
Преп-ЖХ
(Глава 15)
Изократическое
разделение?
(Главы 6-8)
Рис. 9.14. План разработки градиентного метода разделения
давление на колонке можно непрерывно поддерживать после начального смачивания
элюентом, содержащим 100 %В. Наиболее вероятны проблемы со смачиванием
стационарных фаз с высокой плотностью привитых С)8 групп (большое значение Н, раздел
5.4.1), чем для колонок, упакованных сорбентами с меньшей плотностью привитых
Cis групп, содержащих внутренние полярные группы или эндкепированных
полярными группами (для большинства колонок несмачивания стационарной фазы можно
избежать, используя протокол, приведенный в разделе 5.3.2.3). Если буфер имеет
ограниченную растворимость при 100% ACN (в конце градиента 5— 100 %В), может быть,
потребуется снизить %В или концентрацию буфера. Однако если буфер добавляется
только к элюенту А (обычно так и делают), осаждение буфера не происходит.
9.3. Разработка метода 465
9.3.1.1. Выбор между градиентным и изократическим
элюированиями
Первое градиентное разделение имеет большое значение, так как оно помогает,
во-первых, выявить возможные трудности разделения, во-вторых, планировать
проведение дальнейших экспериментов. В таблице 9.3 представлены рекомендуемые
определенные начальные условия: от 5 до 100% ацетонитрила в буфере за 10 минут,
колонка С8 или С]8 100x4,6 мм, размер частиц 3 мкм, температура 30 или 35 "С,
скорость потока 2,0 мл/мин.
Эти условия должны обеспечить для разделения среднее значение к* = 5
(уравнение 9.5), что достаточно для приемлемого среднего разрешения, но ограничивает
перепад давления до = 170 бар (2500 psi). Также вполне приемлемы другие параметры
колонки и скорости потока (например, 150 х 4,6 мм, диаметр частиц 5 мкм, I —
2 мл/мин), если сохраняются приемлемые значения к* и давления. Для оценки
давления на колонке можно использовать уравнение (2.13а). При градиентном элюиро-
вании максимальное давление в процессе разделения определяется максимальной
вязкостью подвижной фазы (см. таблицу 1.5 в приложении 1). Можно изменять
время градиента, чтобы поддерживать к* я 5:
l,l5k*VmAtfS
или для к* = 5 и S = 4
'G—p —
Например, на колонке 100x4,6 мм с размером частиц 3 мкм (Vm ж 1,0 мл)
при использовании ацетонитрила в качестве элюента В при температуре 30 °С и
скорости потока 2,0 мл/мин должно быть давление ж 170 бар (2500 psi); для получения
к* ж 5 будет необходимо время градиента (23 х 1,5 х 0,95/2) = 11 мин. Можно
оптимизировать разделение по-другому, используя колонку меньшего диаметра при
скорости потока, уменьшенной пропорционально квадрату диаметра колонки.
Таблица 9.3. Предпочтительные условия для первичного эксперимента при разработке
градиентного метода (образец: малые молекулы 100—1000 Да)
Колонка
Подвижная фаза
Скорость потока
Температура
Градиент
Образец
к*
Тип
Размер
Размер частиц
Диаметр пор
В образце нет кислот или оснований
Образец содержит кислоты или
основания
Объем
Вес
Cg или С18 (тип В)
100 х 4,6 мм"
3 мкм
8—12 нм
Ацетонитрил/вода
Ацетонитрил/водный буфер
(рН 2,5-3,0)«
2,0 мл/мин
30 или 35 "С
5—100 %В за 10 мин
< 50 мкл
< 50 мкг
~5
"Можно использовать колонки другого размера с частицами иного диаметра как указано в
разделе 9.3.1.
*10—25 миллимолярный буфер, добавляется только в элюент А, подробно о составе буфера
и его концентрации см. раздел 7.2.1.
(9.17)
(9.17а)
466 Глава 9. Градиентное элюирование
Характерное первичное градиентное разделение показано на рис. 9.15 — это
разделение «неправильного» образца рис. 9.5, проведенное в условиях, приведенных
в табл. 9.3. Первый эксперимент можно использовать, чтобы ответить на два вопроса
|2, 30—33]: (1) требуется ли для этого образца градиентное элюирование и (2) если
возможно изократическое разделение, какую подвижную фазу следует испытать
в первую очередь, чтобы 1 < к < 10 для всех пиков? Перед тем, как делать какие-либо
выводы из этого первичного градиента (как это сделано в следующем абзаце), важно
убедиться, что колонка была должным образом уравновешена (см. обсуждение в
разделе 9.3.8.1).
В первичном разделении, показанном на рис. 9.15, времена удерживания первого
и последнего пиков (1-ого и 11-ого) равны 2,7 и 6,5 минут, соответственно. Второе
из этих времен удерживания определяет — возможно ли изократическое разделение.
Сначала вычислим разность времен удерживания (Д/д) пиков 1 и 11 (6,5—2,7). Дгд =
= 3,8 мин. Вычислим также, чему равно среднее время удерживания первого и
последнего пиков (/j?)avg = (6,5 + 2,7)/2 = 4,6 мин. Образцы с небольшими значениями Дгд
можно разделить в изократическом режиме так, чтобы 1 < к < 10. Для образцов с более
значительными Дгд может потребоваться градиентное элюирование. Существует
следующее эмпирическое правило, позволяющее решить, использовать изократический или
градиентный режим. Если &1r/1g ^ 0,25, используйте изократическое элюирование;
если Д'д/'с - 0.40 — градиентное. Для промежуточных значений отношения Atg/t^
оптимальным может оказаться либо изократическое, либо градиентное элюирование.
Для примера на рис. 9.15 Д/д/Гс = 3,8/10 = 0,38, поэтому вряд ли изократический режим
оптимален, более предпочтительным представляется градиентное элюирование.
Когда возможно изократическое разделение, рекомендуемый %В в подвижной
фазе можно приблизительно оценить, исходя из значения (//j)aV£ |2|- Для условий,
приведенных в табл. 9.3,
изократический %В = 9,5[(гд)0>? - tD] - 2. (9.18)
Здесь /д — время задержки градиентной системы, равное V^/F. Применим,
например, уравнение (9.18) к разделению на рис. 9.15, исключив пики 9—11(так, чтобы
для этого разделения больше подходил изократический режим). Объем задержки VD
и время задержки системы tg близки к нулю, а значения Гд первого и последнего
пиков 2,7 и 4,7, соответственно. Тогда А!ц = 2,0 мин; (?/j)av„ = 3,7 мин, а отношение
AlR/{(j= 2,0/10 = 0,2. Как сказано выше, при таком значении этого отношения
предпочтительнее изократическое элюирование.
Мв
1
5-100 %В
за 10 мин
1 L--"
1
10
11
„- - 100 %В
- 80%
- 60%
- 40%
: 20%
;0%
а
-г-
10
/R= (6.5-2.7) = 3.8 мин
<'*>„«,= (6-5+ 2.7)/2= 4.6
Дф = 0.01 (100-5) = 0.95
2 4 6
Время (мин)
Рис. 9.15. Использование стандартного градиентного разделения для определения
предпочтительности градиентного или изократического режима для анализа
образца. В данном примере представлен «неправильный^ образец рис. 9.4;
разделение проводилось в условиях, рекомендованных в таблице 9.3: 5 —
100% ацетонитрила за 10 минут, колонка C|g 100 х 4,6 мм (3 мкм),
2,0 мл/мин, 30 °С. Градиент показан (—)
9.3. Разработка метода
Для приведенного примера с (tft)aYS — 3,7 минут k/j = 0 оптимальный состав
подвижной фазы для изократического разделения 33 %В (уравнение 9.18). Это
разделение дано на рис. 9.16д (такие же колонка, температура и скорость потока) и 0,6 < к <
< 7. Интервал удерживания этого разделения находится за пределами целевого
интервала 1 < к < 10 (уравнение (9.18) — только приблизительная оценка!), но
удерживание можно скорректировать, немного уменьшив %В. Таким образом, значение к
разделения на рис. 9.16а следует увеличить примерно в полтора раза, чтобы к > I
для первого пика. В разделе 2.5.1 указано, что при уменьшении содержания В в
подвижной фазе на 10% log к увеличивается на 0,4 единицы («правило 2.5»), в то время
как желательно увеличить к на log 1,5 = 0,18 единиц. Это означает уменьшение %В
на (0,18/0,4) х 10% = 4,5% до величины 28,5 %В. Разделение в такой подвижной фазе
показано на рис. 9Л66, и в этом случае 1 < к < 10 соответствует целевому интервалу.
В нем 2 пары перекрывающихся пиков (2 + 3 и 5 + 6), которые, вероятно, можно
разделить, варьируя другие условия (см. в разделе 7.3.2 как можно повысить
селективность в изократическом режиме для такого ионного образца).
Приведенные выше рекомендации предполагают, что если образец можно
разделить в изократическом режиме с I < к < 10, то изократическое элюирование
предпочтительнее градиентного. Это предположение оспаривалось |34| на том основа
нии, что градиентное элюирование обычно протекает быстрее. Кроме того, оно
удовлетворительно во всех отношениях, если и для изократического, и для
градиентного разделений используются оптимизированные условия и, если время
уравновешивания колонки между последовательными градиентами сведено к минимуму
(раздел 9.3.7). На основании результатов разделения единственного образца |34|
рекомендуется градиентное элюирование в любом случае, если Дгд//(;- 0,10. Однако
в настоящее время во многих лабораториях предпочтение отдается изократическому
элюированию, несмотря на несколько более длительное время разделения,
поскольку по-прежнему считается, что градиентное в большей степени подвержено
осложнениям, чем изократическое, и его труднее воспроизводить в других лабораториях.
Должно быть, со временем это предубеждение против градиентного элюирования
уменьшится, а оборудование для создания градиента будет оптимизировано так,
чтобы можно было очень быстро уравновесить колонку. Тогда предложение, высказан-
33 %В
0.6S*S7
(в)
JU
X
Время (мин)
(б)
28,5 %В
1S*S10
yui
TV!
Время (мин)
Рис. 9.16. Изократическое разделение образца рис. 9.15 (только пики 1—8). (а)
Разделение при 33 %В, основанное на выводах из первичного градиентного
разделения рис. 9.15. Условия такие же, как на рис. 9.15, за исключением
изократического режима, (б) разделение при 28,5 %В; описание в тексте
468 Глава 9. Градиентное элюирование
ное в |34|, использовать градиентное элюирование в любом случае, если Af/j/ff; £
> 0.10. может оказаться более популярным.
9.3.1.2. Возможные проблемы
Первичное градиентное разделение можно также использовать для выявления
некоторых потенциальных проблем разделения:
• пики с размыванием заднего фронта;
• раннее элюирование;
• позднее элюирование;
• сложные образцы;
• системные пики.
Пики с размыванием заднего фронта (раздел 2.4.2) могут появиться уже в
первичном градиентном разделении. В таких случаях важно устранить эту проблему до того,
как продолжать дальше (раздел 17.4.5.3), Если преодоление размывания пика отложить
на более позднее время, то из-за возникающих в результате изменений селективности
наряду с возможной потерей разрешения могут потребоваться дополнительные
эксперименты по разработке метода, которых в ином случае можно было бы избежать.
На рис. 9.17 показаны три дополнительные проблемы, которые позволяет
обнаружить первичное разделение. Раннее элюирование в ОФХ (рис. 9. !7д) характерно
для небольших полярных молекул, особенно кислот или оснований. Улучшить такое
(в)
Раннее элюирование
в в
время (мин)
10
100 %В
80%
60%
40%
20%
0%
№
Позднее элюирование
20
40 80
Время (мин)
90
- 100 %В
: 80%
- 60%
"-_ 40%
- 20%
" 0%
0%
0 2 4 6 8 10
Рис. 9.17. Возможные проблемы при градиентном элюировании. (а) неудерживаемый
образец; (6) образец с избыточным удерживанием; (в) в образце содержится
слишком много компонентов, Градиент обозначен (---)
9.3. Разработка метода 469
разделение как на рис. 9.17а можно, понизив начальный %В градиента (если это
возможно) или введя изократическую задержку перед началом градиента как
на рис. 9.9г. Другие более эффективные способы преодоления раннего элюирования
можно узнать из раздела 6.6.1. В этом случае можно также попробовать использовать
нормально-фазовую хроматографию (глава 8), особенно ГИХ (раздел 8.6), которая
особенно удачно адаптирована для градиентного элюирования.
Позднее элюирование (рис. 9.176) или отсутствие пиков в градиенте означает, что
образец может быть слишком неполярным для разделения в условиях обычной ОФХ.
В таких случаях градиент ацетонитрил/буфер можно заменить градиентом менее
полярного растворителя, такого как тетрагидрофуран или (что лучше) метил-трет-бути-
ловый эфир. Любой из этих вариантов обладает большей элюирующей силой в ОФХ,
чем ацетонитрил (при этом следует проверить растворимость солей буфера в каждом
из двух предложенных градиентов, хотя часто буфер не требуется для очень
неполярных образцов). Кроме того, можно попробовать менее гидрофобную колонку (с
низким значением И, раздел 5.4.1) или нормально-фазовую хроматографию (глава 8).
Сложные образцы, содержащие более 15 компонентов, могут привести к такой
перегруженной пиками хроматограмме, как на рис. 9.17в. Маловероятно, что такие
образцы удастся разделить за один эксперимент так, чтобы все пики разрешались до
базовой линии. Если все компоненты образца представляют интерес, может возникнуть
необходимость разработать более мощную схему разделения. Двумерная (2D)
хроматография (разделы 9.3.10, 13.4.5) наиболее часто используется для сложных образцов:
фракции, полученные в исходном разделении, далее дополнительно разрешаются
в следующем «ортогональном». Если только некоторые компоненты образца
представляют интерес, лучше обратить внимание на пробоподготовку (глава 16), а потом
использовать обычное изократическое или градиентное разделение.
Еще одной проблемой, возникающей при градиентном элюировании, является
появление системных (артефактных) пиков, которые не соотносятся с компонентами
образца. Обычно системные пики образуются из-за загрязнений элюентов А или В,
используемых для формирования градиента, но иногда растворенный в образце
воздух может привести к возникновению «пика воздуха». Эту проблему можно
предвидеть, если каждый день перед началом работы запускать «холостой» градиент
(без ввода пробы). Холостой градиент используется также для проверки (и
корректировки) дрейфа базовой линии по ходу градиента (раздел 17.4.5.1). Подробно эта тема
обсуждается в разделе 7.4.3.1.
9.3.2. Оптимизация к" (этап 4 таблицы 9.2)
При дальнейшем совершенствовании разделения можно руководствоваться
уравнением (9.16), то есть оптимизировать к*, а* и УУ*. Этот подход к градиентному элюи-
рованию полностью совпадает с использованием уравнения (2.24) при разработке
метода изократического разделения и описан далее в разделах 9.3.3—9.3.6.
Начальные условия градиента, рекомендованные в таблице 9.3, обеспечивают
среднее значение к* « 5 для большинства низкомолекулярных образцов, имеющих
молекулярную массу менее 1000 Да (данные для более высокомолекулярных
образцов приведены в главе 13). Таким образом, в отличие от разработки изократического
метода первый же эксперимент с градиентным элюированием можно провести
в условиях, гарантирующих 1 < к' < 10. На рис. 9.15 показано исходное разделение
«неправильного» образца рис. 9.5 в этих условиях, оно повторяется на рис. 9.18а. Это
разделение с единственной парой перекрывающихся пиков 5 и 6 (отмечены
стрелкой) вполне перспективно для дальнейшего улучшения. На следующем этапе следует
изменить условия разделения таким образом, чтобы улучшить селективность
(расстояние между пиками) и разрешение.
470 Глава 9. Градиентное элюирование
9.3.3. Оптимизация селективности градиента а*
(этап 5 таблицы 9.2)
Значения а* можно изменить, варьируя любое из первых семи изократических
условий таблицы 2.2: силу растворителя {изменение tG эквив<гпентно изменению %В
при изократическом элюировании), растворитель В (например, замена ацетонитрила
метанолом), температуру, тип колонки, рН подвижной фазы, концентрацию
буфера или ион-парного агента. Каждая из этих семи переменных оказывает влияние
на относительное удерживание и селективность как при градиентной, так и при
изократической элюции. Возрастающее количество экспериментальных данных
свидетельствует о том, что в предварительных экспериментах по разработке метода
предпочтительным способом корректировки а* в первую очередь является
изменение времени градиента и температуры (сохраняя при этом 0.5 S к* <, 20). Поэтому
мы рекомендуем увеличение времени градиента в 2—3 раза во втором эксперименте
для разработки метода. По сравнению с исходным экспериментом, показанным
на рис. 9.18а, время градиента было увеличено от 10 до 30 мин, остальные
условия оставались без изменения. Получившееся разделение представлено на рис. 9.186,
к* я 15. В то время как относительное удерживание существенно изменилось,
разделение критической пары пиков 5 и 6 осталось прежним. Если трехкратное
изменение времени градиента существенно не влияет на разрешение перекрывающихся
пиков, вряд ли дальнейшие изменения времени градиента окажутся продуктивными
при сохранении остальных условий неизменными.
На следующем этапе изменяется температура. На рис. 9.18е,г
проиллюстрированы третье и четвертое разделения при разработке метода, воспроизводящие
разделения рис. 9.18я и б при температуре 50 "С. Поскольку пики 5 и 6 не разрешались
в первых двух разделениях, основной вопрос в том, можно ли разделить эти пики
при более высокой температуре. Значительное повышение разрешения 5 и 6 пиков
наблюдается на рис. 9.18e (Rs = 2,1), у пиков 6 и 7 теперь разрешение критическое
(Rs = 1,1). При увеличении времени градиента (рис. 918г) улучшается разрешение
пиков 6 и 7 (Rs= 1,9) и всего образца. Эти результаты позволяют предположить, что
дальнейшее увеличение времени градиента могло бы повысить общее разрешение,
но разрешение не возросло при увеличении tc для этого образца, так как при этом
стали перекрываться пики 2 и 3.
Разрешение, показанное на рис. 9.18г, можно было бы улучшить истинной
оптимизацией времени градиента и температуры с помощью компьютерного
моделирования (раздел 10.2.2), но на данном этапе будем считать условия на рис. 9.18г
приемлемыми.
9.3.4. Оптимизация диапазона градиента
(этап б таблицы 9.2)
Следующий этап разработки градиентного метода заключается в рассмотрении
возможности (1) сокращения диапазона градиента Дф (для сокращения времени
разделения) и (2) использования кусочно-линейного градиента (раздел 9.3.5) либо
для ускорения разделения, либо для улучшения разрешения. Оптимизированное в
первом приближении разделение рис. 9.18г воспроизведено на рис. 9.18d с
изображением градиента на выходе колонки (с задержкой на время tg). Время выхода из
колонки первого пика 1 3,2 мин, в это время в подвижной фазе 14 %В. Время выхода
последнего пика II 12,3 мин, в подвижной фазе в этот момент 42 %В.
Рекомендуется заканчивать градиент сразу после элюировании последнего пика.
В примере на рис. 9.18d время удерживания последнего пика 12,3 мин. Если сократить
9.3. Разработка метода 471
5— 100%В за 10 мин
30°С; к* - 5,
R, = 0.1
(а)
5—100 %В за 30 мин
30°С;**-15.
5—100%В за 10 мин
50"С;*с*«=5
R,= 1.1
50°С;к'=»15,
Rs=1.9
•о
u
Время (мин)
. vim. ГГ
6 в 10
Время (мин)
5-7
-1 1 1 1 1
12 14
1кА
.1°
la:
2 *
Время (мин)
2 8l9
LiJjlLf
10
6 в
Время (мин)
—Г~
10
г
(б)
(в)
(г)
6
4 5 | 7
10.
11
6 8
Время (мин)
l
10
12
100% В
80%
60%
40%
20%
0%
(б)
12
Рис. 9.18. Градиентное разделение «неправильного» образца рис. 9.15 в зависимости
от времени градиента и температуры (л —г). Условия: колонка Си 100 х
х 4.6 мм (3 мкм), 5—100% ацетонитрила —фосфатный буфер рН 2,6,
2,0 мл/мин. Время градиента и температура указаны на рисунке; (5)
подробно показан градиент (г)
472 Глава 9. Градиентное элюирование
градиент до этого времени, то конечное значение %В в градиенте следует
пропорционально уменьшить, чтобы оставить неизменным А* {чтобы сохранить оптимальное
расстояние между пиками как на рис. 9.l8d). To есть значение отношения fc/Дф в
уравнении (9.5) должно быть постоянным. В данном примере /(;/Дф = 30/0,95 = 31,6. Можно
также вычислить (р в момент элюирования пика из колонки <ре:
Ф^Фо+А^-Г°-Ч (9.19)
где ф0 значение ф на старте градиента, а /д время удерживания пика. Для последнего
пика на рис. 9.Ш фе-0,05 + 0,95(12,3 - 0,5 - 0,0)/30 = 0,42 (в этом примере ?0=0,5;
tD = 0,0). То есть новый (укороченный) градиент должен заканчиваться 42 %В. Тогда
новое значение Дф будет 42 - 5% = 37% или 0,37. Так как fg/Дф должно быть
постоянным (во избежание изменения относительного удерживания), новое значение
времени градиента fc = 31,6x0,37 = 11,7 мин (то есть градиент 5—42 %В за 11,7 минут
при остальных условиях, совпадающих с приведенными на рис. 9.18d). Новый гради
ент приведет к такой же хроматограмме, но закончится за 12,2 мин (fc + /0).
Рекомендуется несколько продлить градиент после выхода последнего пика
из-за градиентного закругления (раздел 3.10.1.2). Поэтому мы можем продлить
градиент до 13 мин, для чего потребуется увеличить конечный %В, чтобы к* остался
постоянным. Так как /с/Дф = 31,6 для «оптимизированного» разделения рис. 9.18d,
новое значение Дф — 13/31,6 - 0,41. Тогда конечный %В равен 41 + 5 = 46%; то есть
градиент от 5 до 46 % В за 13 минут.
В принципе, время градиента можно было бы еще сократить, увеличив
начальный %В (и уменьшив /g, чтобы к* остался постоянным). Однако в этом примере
разрешение уменьшалось при любом увеличении начального %В (из-за изменений
значений относительного удерживания в этом «неправильном» образце, как и в примере
на рис. 9.13d). Поэтому начальный %В оставили без изменения. Для других образцов
можно при увеличении исходного %В сократить время разделения без ухудшения
разрешения.
9.3.5. Ступенчатые (нелинейные)1 градиенты
(продолжение этапа б таблицы 9.2)
В предшествующем обсуждении градиентного элюирования предполагалось, что
используются линейные градиенты. Обзор различных причин возможного применения
ступенчатых градиентов вместо линейных дан в разделе 9.2.2.5: (1) для промывания
колонки между вводами образцов; (2) для сокращения времени разделения или
(3) для улучшения разделения оптимизацией селективности в разных частях хрома-
тограммы. Вследствие избыточного разрешения пиков, следующих за пиком 9 в
разделении рис. 9.18d, время разделения можно было бы сократить, увеличив крутизну
градиента после выхода пика 9. Аналогичный пример дан на рис. 9.1 \в. Однако
следует иметь в виду, что градиентные закругления могут различаться у разных
градиентных систем, и такие градиенты будут хуже воспроизводиться, а также необходимо
будет увеличивать конечный %В.
Ступенчатые градиенты обычно используются для промывания колонки, в то
время как сокращение времени разделения и его улучшение с помощью этих градиентов
возможны или желательны гораздо реже. Подробнее см. раздел 9.2.2.5.
Ступенчатый градиент есть частный случай кусочно-линейного. — Прим. персе.
9.3. Разработка метода 473
9.3.6. Оптимизация числа теоретических тарелок N'
(этап 7 таблицы 9.2)
На число теоретических тарелок /V=jV* влияют размеры колонки, размер частиц
сорбента и скорость потока (так называемые параметры колонки, раздел 2.5.3), а также
молекулярная масса образца (раздел 2.4.1.1). Размер частиц и диаметр колонки
обычно выбирают до разработки метода (например, в соответствии с рекомендациями
таблицы 9.3). Увеличение длины колонки обычно приводит к увеличению jV*,
разрешения и продолжительности разделения (как на рис. 9.6d по сравнению с рис. 9.6г).
Таким образом, время разделения можно сократить при уменьшении длины колонки
и/или при увеличении скорости потока (как на рис. 9.6е по сравнению с рис. 9.6е).
После оптимизации селективности а* (пятый этап, таблица 9.2), диапазона и
профиля градиента (шестой этап, таблица 9.2) может оказаться, что разрешение стало либо
слишком низким (меньше 2), либо гораздо больше необходимого значения (Rs 3> 2).
В любом случае изменение параметров колонки можно использовать для улучшения
разделения, но не следует забывать о возможных изменениях перепада давления
на колонке (уравнение 2.13).
В изократической хроматографии изменения длины колонки и скорости потока
не влияют на относительное удерживание или селективность, так как значения к и а
не зависят от изменения параметров колонки. Однако в градиентном режиме
изменение длины колонки L или скорости потока F влияет на значения А*
(уравнение 9.5в). Для «неправильных» образцов это может привести к изменению
селективности. Поскольку селективность оптимизируется до изменений параметров колонки
(пятый этап, таблица 9.2), то важно сохранить значения к* (и а*) при подборе
параметров колонки и числа тарелок N*. Это достигается поддерживанием отношения
tQpjL постоянным (уравнение 9.5в). Например, если длину колонки увеличить
вдвое, то продолжительность градиента также необходимо удвоить, чтобы величина
tc/L не изменилась; а если скорость потока увеличить вдвое, то продолжительность
градиента необходимо наполовину сократить, чтобы IqF оставалось постоянным.
Примеры этого подхода к оптимизации N* показаны на рис. 9.6г—е. Если значения
к* поддерживаются постоянными таким способом, изменение длины колонки или
скорости потока одинаково влияют на время разделения и разрешение как при изок-
ратическом, так и при градиентном элюировании. Небольшим исключением из этого
правила является разрешение рановыходящих пиков при больших величинах Vq вне
зависимости от того, что к* поддерживается постоянным (раздел 9.2.2.4 [11]) |11].
Когда параметры колонки изменяются в режиме кусочно-линейного градиента,
время каждого сегмента градиента tseg должно быть подобрано таким образом,
чтобы tseg F/L оставалось постоянным. Например, рассмотрим разделение, показанное
на рис. 9. Не, для которого использовался градиент 0/23/42 %В за 0/32/38 мин. Если
длину колонки удвоить, то длительность каждого сегмента tseg также потребуется
увеличить вдвое так, что новый градиент будет 0/23/42 %В за 0/64/76 мин.
9.3.7. Определения необходимого времени
уравновешивания колонки (этап 8 таблицы 9.2)
Разработанную методику в дальнейшем, как правило, применяют для рутинных
анализов образцов. Когда вы проводите эти анализы, то между концом предыдущего и
началом последующего градиентного разделения необходимо промывать колонку
достаточным количеством подвижной фазы, совпадающей по составу с подвижной
фазой в начале градиента (например, 5 %В в примере рис. 9.18). Такая стадия
уравновешивания колонки служит, во-первых, для внесения поправки на «мертвый» объем
474 Глава 9. Градиентное элюирование
системы (или на время задержки системы /D = VqJF), во-вторых, для корректировки
градиентного закругления (раздел 3.10.1.2) и, в-третьих, для постепенного
уравновешивания неподвижной фазы (удаления избыточного количества элюента В) при пе
реходе от более высокого %В, которым градиент обычно заканчивается, к более
низкому %В, с которого начинается следующий градиент.
На рис. 9.19 проиллюстрированы возможные последствия и корректировка
суммарного воздействия объема задержки, градиентного закругления и медленного
уравновешивания колонки, когда при рутинных анализах последовательно вводятся пробы
образцов. На рис. 9.19д сплошными линиями показаны несколько
запрограммированных градиентов, по горизонтальной оси отложено время. Стрелки указывают моменты
ввода образцов 1, 2 и так далее в начале каждого градиента. За этими градиентами
5—100 %В в течение 10 минут следуют этапы уравновешивания 5 %В в течение 1
минуты (время уравновешивания teq = 1мин). Таким образом, полный
запрограммированный градиент 5/100/5/5 %В за 0/10/10/11 минут. Если «мертвый» объем системы и
градиентные закругления пренебрежимо малы и, если колонка уравновешивается быстро,
реальный градиент должен быть таким как запрограммированный, и ввод образца
должен происходить через минуту после окончания предыдущего градиента. Это должно
привести к одинаковому разделению каждого образца.
На рис. 9.196 представлены некоторые дополнительные особенности реального
градиента, которые часто встречаются на практике: значительный объем задержки
оборудования, градиентные закругления и/или медленное уравновешивание
колонки. Пунктиром (—) на рис. 9.196 показаны реальные градиенты на входе в ко
лонку, наблюдается значительный объем задержки, при этом время задержки /д =
= 2 мин. Следовательно, градиент достигает входа колонки на 2 мин позже. Ввод
образца в момент запуска градиента (в обычном случае) по-прежнему приемлем
для первого образца, но последующие образцы вводятся на минуту раньше
окончания предыдущего градиента. Так как образцы 2, 3 и последующие вводятся в
подвижную фазу с гораздо более высоким %В (непосредственно перед окончанием
градиента), получающиеся значения к* первых пиков очень малы, и разрешение этих
пиков неприемлемо. Вследствие градиентного закругления и/или медленного
уравновешивания колонки %В снижается медленно от 100% до 5% в конце каждого
градиента. Для возвращения %В к исходному значению 5% требуется tx = 3 мин.
Заметим, что в этом примере смежные градиенты (значения %В) суммируются
(это не показано на рис. 9.196).
Неблагоприятное воздействие объема задержки и медленного возвращения
градиента к базовой линии можно устранить, введя время уравновешивания Хе„, равное
гд + fx, как показано на рис. 9.19е. При таком изменении градиента (5/100/5/5 %В
за 0/10/10/15 мин) каждый образец вводится в колонку в начале
запрограммированного градиента (и достигается исходное значение %В после окончания предыдущего
градиента). Теперь разрешение всех образцов становится одинаково приемлемым.
Требуемое значение f„ можно определить методом проб и ошибок, когда образец
последовательно вводится с одним значением te„, а потом несколько раз повторно
вводится с другим значением его значением. Предпочтительно наименьшее значение te„,
которое обеспечивает приемлемое разрешение при последовательном введении
образцов. Метод проб и ошибок имеет преимущество, так как во время
уравновешивания включается возможная задержка автосамплера.
При разработке методики колонку перед проведением каждого разделения
необходимо уравновешивать не менее чем 10 колоночными объемами (соответствующими
]0Vm/F или времени, равному Ю/q) подвижной фазы исходного состава (ср = щ).
Иначе любые изменения продолжительности уравновешивания между
последовательными разделениями (особенно в экспериментальных разработках) могут
приводить к различным равновесным состояниям колонки, что, в свою очередь, непред-
9.3. Разработка метода
Запрограммированный градиент на входе в колонку 5/100/5/5 %8 ,д\
за 0/10/10/11 мин
1 2 3
i i ■ ■ ■ i ■ ■ ■ i i ■ i ■ i i ■ i ■ i 1 ■ i i i i i i i ■ i
О S 10 15 20 25 30 (мин)
Реальный градиент на входе а колонку
Г 11 I3
(б)
запрограммированный,
градиент
реальный
градиент
i ■ i » i i ■ ■ i i i i ■ i i i 1 ■ i i i i I i ■ i
5 10 15 20 25 30 (мин)
Измененный градиент с откорректированным г„,. 5/100/5/5 %В
за 5/10/10/15 мин
эапрограммированны!
градиент
(в)
35 (мин)
Рнс. 9.19. Иллюстрация различных вкладов в неуравновешенное состояние колонки
при градиентном элюировании. (а) идеальный градиент; (б) более реальный
градиент; (в) добавление достаточного времени для уравновешивания
колонки между разделениями для градиента (б); (—) запрограммированный
градиент; (—) реальный градиент на входе колонки; 1, 2 и т.д. относится
к вводу образца 1, 2 и т.д.
сказуемо скажется на времени удерживания веществ и их разделении. В рутинных же
анализах (таких, как на рис. 9.19) время, необходимое для уравновешивания
колонки, можно уменьшить до teq с соответствующим сокращением времени разделения
(по сравнению с 10/г>) и возрастанием количества образцов, которые можно будет
проанализировать в течение рабочего дня. Более того, частичное уравновешивание
476 Глава 9. Градиентное элюирование
колонки (то есть неполное возвращение подвижной фазы к исходному %В) в
рутинных анализах вполне допустимо, как и сокращение времени уравновешивания
до значения < teq. Таким образом, если разрешение рановыходящих пиков не
ухудшается и, если время уравновешивания для каждого разделения одинаково, каждый
образец будет обрабатываться одинаково, и одинаковым будет каждое разделение,
несмотря на неполное уравновешивание колонки.
Для систем ВЭЖХ, не приспособленных для отсрочки ввода образца,
предпочтительное время частичного уравновешивания t eq для рутинных анализов определяется
выражением tQ<t е„ < /д 4- /Л, в котором / е„ следует определить методом проб и
ошибок (это должно быть минимальное допустимое значение t eq). Значения /Л обычно
сравнимы со значениями tp, поэтому можно считать, что teq = 2/д (далее следует
методом проб и ошибок определить?^). В системах, позволяющих ввести образец
в любой момент после запуска градиента, ввод можно запрограммировать на время
гд после запуска каждого градиента. Тогда соответствующее время уравновешивания
будет < tx. Однако методы с введением образца после начала градиента рассчитаны
только на градиентные системы с возможностью отсроченного ввода образца.
Независимо от того, какая продолжительность уравновешивания установлена
для процедуры рутинных анализов, следует удостовериться, что при повторных
разделениях получаются абсолютно одинаковые (приемлемые) хроматограммы, и
данные воспроизводятся (может быть, кроме первого эксперимента, результаты
которого можно не принимать во внимание). Время, необходимое для уравновешивания
колонки, будет меньше для систем с меньшим объемом задержки и градиентным
закруглением. Это время можно еще сократить, если система имеет опцию
отсроченного ввода образца. Для уменьшения градиентного закругления |43| и дальнейшего
сокращения teq можно изменить конфигурацию оборудования, хотя большинство
пользователей вряд ли захотят воспользоваться этой возможностью. (Может быть, в
будущем для усовершенствованного оборудования не потребуется в этом случае менять
его конфигурацию.) Особенности уравновешивания колонки при градиентном элюи-
ровании обсуждаются в работах |2, 44, 45|.
9.3.8. Воспроизводимость метода
Некоторые факторы, препятствующие воспроизводимости результатов или
обусловливающие низкую точность метода, одни и те же для изократических и градиентных
разделений (раздел 11.2). Другие причины отсутствия воспроизводимости
свойственны только градиентному элюированию или наиболее вероятны в этом режиме
разделения:
1) недостаточный контроль условий эксперимента в последовательных разделениях;
2) неисправность или низкое качество оборудования;
3) недостаточное уравновешивание колонки между градиентными разделениями;
4) разница объемов задержки при использовании разных хроматографов.
Перечисленные выше факторы, препятствующие воспроизводимости
разделений, могут проявиться как, собственно, при разработке градиентного метода
(следующий раздел 9.3.8.1), так и при его последующем рутинном использовании
(раздел 9.3.8.2). Кроме того, точность и надежность градиентного анализа (и
интерпретация результатов, полученных при разработке методики) могут искажаться
из-за дрейфа базовой линии и наличия ложных пиков, не имеющих отношения
к компонентам образца (раздел 17.4.5.2). Чтобы исключить такого рода проблемы,
настоятельно рекомендуется каждую серию экспериментов с градиентом начинать
с холостого градиента: градиента без ввода образца или лучше с вводом
растворителя, в котором будет растворен образец (раздел 3.10.1.2).
9.3. Разработка метода 477
9.3.8.1. Разработка метода
Рассмотрим в первую очередь необходимость нескольких повторных экспериментов
при разработке методики для контроля воспроизводимости полученных данных
в следующих друг за другом разделениях. Это особенно важно для экспериментов
по градиентному элюированию. Времена удерживания двух повторных опытов
не должны отличаться более чем на некоторую заданную величину, например,
+0,02 мин или +0,1% в зависимости от того, какая из этих величин больше.
Недостаточный контроль условии эксперимента и сбои оборудования (факторы 1 и 2)
становятся маловероятны при соблюдении правил надлежащей лабораторной
практики (GLP — Good Laboratory Practice). В данном обсуждении предположим, что все
экспериментальные условия контролируются в пределах, необходимых для
воспроизводимости разделения. Также предположим, что оборудование исправно, а эффективность
колонки соответствует сертификату производителя (раздел 3.10.1.2). Если отвлечься
от проблем, связанных с оператором и состоянием оборудования, главной целью
разработки является окончательный вариант методики, устойчивый к небольшим,
большей частью неизбежным изменениям условий градиента, например, температуры или
состава подвижной фазы (%В, рН и т.д.) со временем или при переходе от одной
градиентной системе к другой. Если методика оказалось ненадежной, следует приложить
усилия для выявления причин зависимости воспроизводимости результатов и
разрешения от экспериментальных условий (раздел 12.2.6).
Недостаточное уравновешивание колонки (фактор 3) является основной причиной
нестабильности времени удерживания при градиентном элюировании, поэтому
между каждым разделением или экспериментом необходима стадия уравновешивания
колонки (раздел 9.3.7). Вначале следует проверить воспроизводимость времени
удерживания. Для этого проводят два повторных анализа, между которыми колонка
уравновешивается в течение установленного минимального времени (например, 10/0),
а перед первым анализом в два раза дольше. Для некоторых образцов и/или условий
разделения может потребоваться более длительное уравновешивание > lOty- Впрочем,
и потом, в течение долгого времени, удерживание может продолжать очень медленно
изменяться |43|, но это изменение уже не будет влиять на разработку метода.
Для обеспечения воспроизводимости разделений при разработке методики разумно
сделать время уравновешивания больше минимального; потом его можно сократить
для уменьшения длительности разделения, когда разработка метода будет закончена.
Разные объемы задержки хроматографических систем (фактор 4) могут
существенно повлиять на результаты анализов (раздел 9.2.2.4). Поэтому во время разработки
методики настоятельно рекомендуется все эксперименты проводить на одном и том
же (или аналогичном) оборудовании, чтобы исключить разницу объемов задержки
во время экспериментов.
9.3.8.2. Рутинные анализы
Разрабатывая методику, необходимо учитывать возможные изменения в разделении
при ее переносе в другие лаборатории для рутинных анализов. Различия в
экспериментальных условиях, неисправности оборудования (описанные выше факторы !—2)
выявляются обычно при тестировании аппаратуры на соответствие заданным
требованиям (раздел 12.3.2.9).
Полноту уравновешивания колонки (фактор 3) в рутинных анализах регулируют
иначе, чем при разработке методики. Как правило, при разработке метода
приемлемое время уравновешивания колонки не менее Ю/q. Для рутинных анализов, когда
время уравновешивания колонки между разделениями фиксируется, желательно
сократить его до минимума, чтобы сократить время между вводами образцов
(раздел 9.3.7) как и общее время анализа.
478 Глава 9. Градиентное элюирование
Разные объемы задержки хроматографических градиентных систем (фактор 4)
являются распространенной причиной сбоя в работе метода при его переносе на другое
оборудование или повседневном использовании на разных хроматографах. У разных
градиентных систем иногда наблюдаются существенные различия в объеме задержки
Vq. Как правило, старое оборудование имеет больший объем задержки. Довольно
часто рутинные анализы по методикам, разработанным на одном хроматографе,
проводят на другом оборудовании. Если объемы задержки этих хроматографических
систем различны, то это приведет к неудовлетворительным результатам разделения
(раздел 9.2.2.4). Особенно это касается «неправильных» образцов. Если Vg второй
системы меньше, то это различие можно компенсировать, добавляя градиентную
задержку tieiay, что эквивалентно увеличению объема задержки. Продолжительность
градиентной задержки (в минутах) должна быть равна разности времен задержки ?д,
которые определяют делением объема задержки на скорость потока (/д = Vg/F).
Более сложная ситуация возникает в случае, когда у второго хроматографа
больший объем задержки (это более вероятно). Поэтому необходимо на стадии разработки
методики заранее учитывать максимально возможные объемы задержки, которые
могут быть присущи оборудованию других лабораторий. Первоначальный метод может
быть разработан с суммарной градиентной задержкой (tg + tdelay), фактически
увеличивающей время задержки до максимального значения, чтобы его можно было
адаптировать для оборудования в других лабораториях, использующих этот метод.
Значения tdelay можно корректировать, компенсируя разницу между объемами
задержки лабораторного и исходного оборудования, на котором разрабатывалась эта
методика (так что t0 + tdeJav остается постоянным для каждой из систем). Кроме того,
можно использовать задержку перед вводом пробы (если в системе есть такая
опция), чтобы эффективно уменьшить объем задержки второй системы. Более
подробно способы преодоления различий в объеме задержки оборудования обсуждаются
в |2| и [46|.
9.3.9. Пиковая емкость и экспресс-разделения
Пиковая емкость (PC — Peak Capacity) в изократическом разделении обсуждалась
в разделе 2.7.3. Определение пиковой емкости одинаково для изократического и
градиентного элюирования: PC равно максимальному количеству пиков, которые могут
разместиться в пределах данного хроматографического пространства (например,
градиентной хроматограммы) с разрешением Rs = 1 для всех соседних пиков (при этом
предполагается определенное время разделения). В градиентном разделении, когда
ширина каждого пика W приблизительно одинакова, пиковую емкость можно
описать через время градиента tc следующим образом:
PC = I + f-^-1 « X (9.20)
\W J W
На рис. 9.20 проиллюстрировано это определение пиковой емкости для
(гипотетического) разделения с временем градиента 10 мин. Для примера на рис. 9.20а
средняя ширина пика W= 0,2 мин. Поэтому PC для этого примера равна t(;/W= 10/0,2 =
= 50, как показано на рис. 9.206, где на хроматограмме 50 пиков шириной W =
- 0,2 мин с Rs - 1 для каждой пары соседних пиков. Понятие пиковой емкости
используется для оценки относительного показателя разделения при градиентном элю-
ировании вместо измерения числа теоретических тарелок колонки (что возможно,
но менее удобно, см. стр. 38 в |2|). В качестве меры эффективности разделения
значения PC особенно полезны для образцов, при разделении которых получается
большее количество пиков, чем можно разделить до базовой линии, т.е. «сложных»
9.3. Разработка метода 479
в
10 (мин)
(б)
Пиковая емкость PC = fG /W
10 20 30
.. 100%В
80%
- 60%
40%
20%
- 0%
10 (мин)
Рис. 9.20. Пиковая емкость в градиентной элюции. (а) гипотетическое разделение;
(б) иллюстрация пиковой емкости PC для разделения (а)
образцов, подобных показанным на рис. 9.17в или 9.20й (например, гидролизован-
ных пептидов, растительных экстрактов и т.д.).
«Пиковая емкость» — это гипотетическая (не всегда поддающаяся измерению)
величина, которая в общем случае дает завышенную оценку разделительной
способности реальной градиентной хроматограммы. Так в разделении на рис. 9.20а пики
появляются только между 2 и 9 минутами, и реально используется только часть
градиентной хроматограммы: (9 - 2)/!О = 70%. Если пики образца сосредоточены в
пределах части хроматограммы (как на рис. 9.20а,), а не распределены по всей хроматог-
рамме, эффективная пиковая емкость разделения меньше, чем значение PC, которое
определяется на рис. 9.206 для всего градиента. В реальных разделениях, таких как
на рис. 9.20а, разделительную способность лучше определять как число разрешенных
пиков между первым и последним пиками хроматограммы (не обязательно с начала
и до конца градиента). Последняя величина называется эквивалентной пиковой
емкостью пс = PC(lz - t^)/t(]\ nc называют также условной пиковой емкостью или пиковой
емкостью образца |47|. На практике эквивалентная пиковая емкость будет меньше
значения PC, заданного уравнением 9.20 (пс равно 35 для примера на рис. 9.20а).
Обратите внимание, что условия, в которых происходит увеличение PC до максимума
будут также способствовать возрастанию пс.
Так как одного градиентного разделения может оказаться недостаточно для
разрешения компонентов сложного образца, часто используются двумерные (2D)
разделения (разделы 9.10, 13.4.5, 13.10.4). В 20-разделении фракции с первой колонки
переносятся на вторую для дальнейшего разделения (как в примере на рис. \Аб,в).
Если разделения ортогональные (т.е. времена удерживания в них не коррелируют
между собой; раздел 6.3.6.2), пиковая емкость комбинации разделений равна произ-
480 Глава 9. Градиентное элюирование
ведению пиковых емкостей каждого разделения. Общая цель состоит в достижении
максимальной пиковой емкости при минимальном суммарном времени разделения
|47—58]. В этом разделе мы приближаемся к пониманию (и регулированию) пиковой
емкости, основываясь на соображениях, применявшихся для случая изократического
элюирования (раздел 2.4.1). Уместно заметить, что детектирование с использованием
масс-спектрометрии и поглощения на нескольких длинах волн добавляет
дополнительные измерения к одно- и двумерным разделениям |53|, и таким образом
увеличивает суммарную пиковую емкость.
9.3.9.1. Оптимизированная пиковая емкость
Основное внимание в данном разделе уделяется только двум практическим
приложениям: градиентным экспресс-разделениям (раздел 9.3.9.2) и двумерным ВЭЖХ-разделения-
ми (20-ЖХ) (раздел 9.3.10). Многое из дополнительного материала этого раздела не по
являлось в литературе с 2009 г. Если читателя не интересуют конкретно экспресс- или
2И-ЖХ-разделения, целесообразно было бы перейти к разделу 9.4.
Имеет смысл описать пиковую емкость через экспериментальные условия,
которые можно оптимизировать для получения максимальных значений PC и/или
минимального времени разделения. Для полного градиента (Дф = I) уравнение (9.20)
можно записать в виде:
PC = tz~*A. (9.21)
W
где 1А и ^относятся к времени удерживания пиков, которые элюируются,
соответственно, в начале и в конце градиента (например, пики I и 50 на рис. 9.206).
Значения W можно принять приблизительно равными для пиков А и Z, поэтому
уравнение (9.21) эквивалентно разрешению этих двух пиков (уравнение 2.23). Тогда
разрешение Rs в уравнении (9.15в) можно заменить PC, что дает:
PC = p^W'^loga*
1 +А*
(9.21а)
Если предположить, что значения 5 для пиков А и Z одинаковы (хотя S обычно
больше у пиков, выходящих позже на хроматограмме), то а* будет равно отношению
величин kw пиков Z и А (уравнение 9.5а) которое в свою очередь равно A<pS. Тогда
уравнение (9.21а) принимает вид:
РС = Ш- |ДфОТ*0'5
\ + к*
(0 (и) (ш)
(9.22)
Это выражение для пиковой емкости PC предполагает полномасштабный градиент
(Дф = 1), а величина S определяется молекулярной массой М образца
(раздел 13.4.1.4):
S * 0,25М0-5. (9.23)
Для образцов с М < 500 можно принять значение S « 4. Тогда член < в уравнении
(9.22) константа для образца с определенной молекулярной массой. Член и
зависит от длины колонки, размера частиц сорбента, скорости потока, температуры и
молекулярной массы образца (раздел 2.3.1). Наконец член Hi зависит от времени
градиента, скорости потока, длины колонки и S или молекулярной массы образца
(уравнение 9.5в). При подстановке в уравнение (9.22) всех перечисленных
экспериментальных переменных может получиться достаточно сложное выражение, которое
будет неудобно и использовать, и интерпретировать.
9.3. Разработка метода 481
Проще и удобнее использовать уравнение (9.22), если предположить наличие
условий, обеспечивающих максимальное значение PC для данного времени
разделения и максимальное допустимое давление в колонке Р. В разделе 2.4.1.1 доказано,
что оптимальным образом колонка используется (для некоторого требуемого
значения N в течение минимального времени) при минимальной приведенной высоте
теоретической тарелки А. Это значение соответствует величине приведенной скорости
потока подвижной фазы v = hdp/D„, as 3 (вариант В, раздел 2.4.1.2,
предусматривающий широкий выбор колонок разной длины с разным диаметром частиц). Тогда
оптимальные значения N (при изократическом элюировании) зависят (рис. 2.15)
от размера частиц dp, давления и времени разделения (для данных значений вязкости
подвижной фазы г| и коэффициента диффузии вещества Dm). Следовательно,
для каждого времени разделения и давления получаются конкретные значения
длины колонки, скорости потока и размера частиц, необходимые для оптимального
значения v = 3 (уравнения 2.13а и 2.216). При возрастании времени разделения должны
увеличиваться длина колонки и размеры частиц, а скорость потока должна
уменьшаться для того, чтобы давление поддерживалось постоянным (в то же время
необходимо сохранять v = 3, варьируя диаметр частиц). Можно показать (уравнение 9.22), что
значения PC пропорциональны к*-^/(\ + к*), откуда следует оптимальное
значение к* = 3.
На рис. 9.21а показана зависимость оптимальных величин PC сп эксперименталь
ных условий для низкомолекулярных образцов (обратите внимание на близкое
сходство этого графика для градиентного элюирования, с соответствующим графиком
в условиях изократического элюирования на рис. 2.15 для значений /V). Видно, что
в этих оптимизированных условиях пиковая емкость возрастает с ростом времени
градиента и давления, а требуемый диаметр частиц сорбента также увеличивается с
ростом времени градиента. Для очень быстрых разделений, особенно при повышенном
давлении, требуются недоступные в настоящее время сорбенты (rf„< 1,5 мкм)1.
Для таких случаев существуют различные (не столь эффективные) приемы
для получения максимальной пиковой емкости при очень коротких градиентах
(например, tQ < 1мин). Данные, приведенные на рис. 9.21а, также предполагают
обеспечение с помощью оборудования идеальных условий, трудно достижимых при
выполнении очень быстрых экспресс-разделений (раздел 9.3.9.2).
Когда оптимизация условий в соответствии с рис. 9.21д требует, чтобы диаметр
частиц был d„ < 1,5 мкм (то, что требуется для очень кратких градиентов), можно
использовать частицы большего размера с некоторой потерей пиковой емкости.
Также поскольку v оптимизирована (как и jV*), выбор других значений к* в диапазоне
1 < к* < 10 (т.е. при варьировании только времени градиента) приводит к S 10%
уменьшению оптимальной величины PC для данного времени разделения (значения t(;).
Это дает возможность варьировать время градиента (в пределах 10-кратного
интервала), поддерживая при этом близкие к оптимальным значения PC на протяжении
градиента. На рис.9.215 показаны неоптимизированные более короткие градиенты
на колонках различной длины, упакованных сорбентами с размером частиц 3 мкм;
скорость потока изменяется для поддерживания Р = 408 бар (6000 psi). Оптимальные
значения PC, взятые с рис. 9.21а для Р = 408 бар (6000 psi) (когда диаметр частиц и
длину колонки можно было изменять непрерывно) обозначены сплошной линией.
Каждая из остальных кривых на этом рисунке соответствует значениям к*, которые
изменяются от I до 10, при фиксированном диаметре частиц 3 мкм и постоянном
давлении Р = 408 бар (6000 psi). Видно, что при этих неоптимальных условиях
пиковая емкость несколько падает по сравнению с оптимальными значениями для данно-
1 Теперь уже доступны и сорбенты с частицами субмикронных размеров и колонки ими
упакованные. — Прим. ред.
482 Глава 9. Градиентное элюирование
1000
500
PC
200
100
1 мкм
15 мкм
J_
138 бар
I-- (2000psi)
414 бар
' {6000 psi)
1—1034 бар
(15П0И psi)
ю го
_ь
(в)
50 100 200 500 1000
toiMU»)
(в)
Р = 414 бар (6000 рв1) (о" ■ 3 мкм для отдельных колонок)
1000
500
PC
20 50
tg(UUH)
200 200 500 1000
Рис. 9.21. Зависимость оптимизированной пиковой емкости от диаметра частиц
сорбента, времени градиента и давления в колонке. Данные для
низкомолекулярного образца (S = 4, Dm = 10-5; 0—ТОО %В градиент буфер — ацетонитрил
(г| = 0,75); рассчитано по уравнению (2.17) сА=1,В = 2иС = 0,05. (а)
оптимизированные значения (v = 3, к' = 3) для Р = 136, 414 и 1034 бар (2000.
6000 и 15000 psi); (б) неоптимизированные значения для давления 408 бар
(6000 psi) и размера частиц 3 мкм (ft* варьируется для каждой длины
колонки)
9.3. Разработка метода 483
го времени градиента, но обычно PC уменьшается меньше, чем наполовину.
Обратите внимание, что оптимальная длина колонки в этом примере L = 1500 мм
при диаметре частиц dp — Змкм. В таблице 9.4 приведены условия оптимизированных
разделений, представленных на рис. 9.21<з {408 бар, 6000 psi).
Таблица 9.4. Условия разделения для оптимизированных условий, приведенных
на рис. 9.21а
tc (мин)
]
3
10
30
100
300
1000
dp (икм)
0,6
0,8
U
1,4
1,9
2,5
3,4
L (мм)
10
30
80
170
420
960
2400
F (мл/мин)
1,8
1,4
1
0,8
0,6
0,4
0,3
ft*
3
3
3
3
3
3
3
PC*
170
230
310
400
540
710
960
Примечание: диаметр колонки предполагается равным 4,6 мм; давление 408 бар (6000 psi).
л Рассчитано по формуле А = iA33 + 2/v + 0,05v.
Примеры, приведенные на рис. 9.215, можно лучше понять, исходя из
соотношения для времени градиента. Из уравнения (9.5) следует
1,15*-К,Аф5 <92А)
F
Таким образом, уменьшение к* приводит к сокращению времени градиента и
наоборот. При уменьшении длины колонки L (пропорциональной Vm) необходимо
пропорционально увеличить скорость потока F, когда давление и к* поддерживаются
постоянными. Это приводит к сокращению времени градиента, пропорционального
L/F (или L2).
В большинстве случаев для получения максимальных или оптимальных
значений PC для данного времени градиента требуется, чтобы частицы сорбента были
промежуточного размера (например, 2.2 мкм) или длина колонки была средней
(например, 65 мм). Это неосуществимо, особенно из-за ограниченного выбора
колонок от предпочтительного поставщика. Однако использование частиц, не очень
отличающихся размерами (диаметром 2 или 3 мкм), можно, как указано выше,
компенсировать использованием менее оптимальных значений к*, и при этом
умеренно понижается пиковая емкость. Графики, аналогичные приведенным
на рис. 9.2\а,б, получаются и при разделениях образцов с более высокими
молекулярными массами, но обычно пиковая емкость выше, и требуются сорбенты
с меньшим диаметром частиц.
9.3.9.2. Экспресс-разделения
Градиентные разделения длительностью несколько минут или еще меньше (иногда
их называют «баллистическими градиентами» |59|) необходимы для анализов
больших количеств (тысяч) образцов с приемлемыми затратами средств и, следовательно,
времени. Краткое время разделения требуется также в двумерной ВЭЖХ
(раздел 9.3.10) для разделения во втором измерении, чтобы проанализировать большое
количество фракций, полученных в первом измерении, в течение времени, которое
484 Глава 9. Градиентное элюирование
требуется для исходного разделения. Поскольку анализ занимает всего несколько
минут, то рабочие характеристики оборудования становятся решающим фактором.
Помимо уже обсуждавшихся требований к длине колонки, скорости потока и
размеру частиц (в связи с рис. 9.21), для выполнения экспресс-разделений требуются:
(1) очень малый объем задержки Vp, (2) ввод пробы, осуществляемый в течение од-
ной-двух секунд; (3) быстрый отклик детектора для пиков с W < 1 сек; (4)
возможность задавать очень крутой градиент {например изменение доли растворителя В
на 1—2% за сек.; рис. 17.28) и (5) быстрая (либо в режиме офлайн) обработка данных
(раздел 3.8.4). Обычно в экспресс-разделениях число теоретических тарелок N* и
пиковая емкость PC имеют меньшее значение, так как возможная величина PC
уменьшается для более коротких градиентов (рис. 9.21). Однако небольшие значения N* и
PC во многих случаях все-таки приемлемы:
• у образцов с небольшим количеством легко разделяемых компонентов;
• при полной оптимизации селективности разделения, особенно с использованием
двух или более факторов, влияющих на селективность (таблица 2.2);
• в случаях, когда допускаются небольшие значения Rs либо благодаря
селективности детектирования (например, в ЖХ-МС), либо если приемлема невысокая
точность результатов анализа;
• во втором разделении 2D ВЭЖХ.
Образцы с небольшим количеством легко разделяемых компонентов можно
анализировать при меньших значениях jV*. Также можно поступать и с образцами,
содержащими большое количество компонентов, когда селективность разделения эффективно
оптимизирована (с получением максимальных значений а), и с разделениями,
допускающими небольшие значения Rs. Второе разделение 2В-ВЭЖХ часто можно проводить
при небольшом значении N*, поскольку количество компонентов образца уже
существенно сократилось, а следовательно и величина а увеличилась ко второму
ортогональному разделению. Кроме того, для детектирования часто используется МС.
Помимо необходимого для экспресс-разделений оборудования минимальное
время разделения при требуемом значении PC определяется (теоретически)
выбором размера колонки и частиц сорбента, скорости потока и максимального
допустимого в системе давления. Таким образом, имеющиеся в распоряжении частицы
сорбента наименьшего размера и наибольшее допустимое давление в принципе
позволят добиться наиболее быстрого разделения при некотором требуемом
для образца разрешении (см. рис. 9.21а и обсуждение в |60]). Однако практические
ограничения для данной градиентной системы делают это утверждение не столь
категоричным (длину колонки можно ограничить до некоторой минимальной
величины, скорость потока также не может превышать определенных значений,
и внеколоночные эффекты нельзя преодолеть полностью). Наконец время
уравновешивания 1е„ между последовательными градиентами также следует максимально
сократить (раздел 9.3.7), а это целиком зависит от оборудования (его времени
задержки и закругления градиента).
В нескольких сообщениях |43, 59, 61—65| приведены примеры и подробные
описания экспериментов по проведению «быстрого» градиентного элюирования.
На рис. 9.22д показано разделение модельного образца за 1.6 минут, а на рис. 9.226
представлен результат последовательного ввода образца через 1,6 минут (стрелками
обозначены моменты ввода). Колонка, упакованная сорбентом с размером частиц
5 мкм, не очень подходит для быстрого разделения, но в данном случае быстрому
разделению способствуют большие величины а, по-видимому, достигнутые в
результате интенсивной оптимизации селективности. Скорость разделения можно также
увеличить, подняв температуру (раздел 2.4.1).
9.3. Разработка метода 485
6 (мин)
Рис. 9.22. Пример экспресс-разделения. Образец: 1 — урацил; 2 — ацетон; 3 — N-бен-
зилформамид, 4—9 — С2—С7 алкилфеноны. Условия: колонка С]8 50 х 2,1 мм,
5 мкм, 0—100 %В за 1 мин; элюснт А 3/7/90% н-пропанол/ацетонит-
рил/вода; элюент В 3/94/0 н-пропанол/ацетонитрил/вода; 1,0 мл/мин;
40 "С. (а) хроматограмма одного образца; (б) последовательное введение
пяти образцов с интервалом 1,6 мин. Стрелками отмечены моменты ввода
каждого образца. Рисунки взяты из |43|
9.3.10. Комплексная двумерная ВЭЖХ
(в соавторстве с Питером Шонмейкерсом)
В разделе 9.3.9 рассматривались условия максимальной пиковой емкости в пределах
данного градиента (размер частиц сорбента, длина колонки, скорость потока).
Можно изменять и другие условия для оптимизации относительного удерживания и
доведения критического разрешения до максимального, что обсуждалось в разделе 9.3.3.
Однако даже эти меры будут недостаточны для образцов, содержащих сотни или
тысячи индивидуальных соединений как в случае гидролизованных белков человека
(раздел 13.4.5). Тогда потребуется существенное увеличение пиковой емкости
по сравнению с той, которую можно получить в одном разделении (как на рис. 9.21).
Такого увеличения пиковой емкости можно добиться с помощью двумерной ВЭЖХ
(20-ЖХ), в которой все фракции исходного градиентного разделения (в первом
измерении) собирают и вводят во вторую систему ВЭЖХ (второе измерение) с
последующим их градиентным разделением. 20-разделения можно проводить, собирая
фракции (офлайн), или переключая колонки одним или двумя переключающими
кранами (онлайн, раздел 2.7.6). Если все фракции подлежат разделению во втором
измерении и, если фракции отбирались настолько часто, что разделение в первом
измерении в основном осуществилось, речь идет о комплексной двумерной
жидкостной хроматографии, которая сокращенно называется ЖХ/ЖХ |66].
9.3.10.1. Концепция ЖХ/ЖХ
На рис. 9.23 проиллюстрированы некоторые аспекты комплексной 20-ЖХ: показана
часть хроматограммы первого измерения (два пика). Из каждого пика собирают
несколько фракций (заштрихованные треугольники), каждая из этих фракций вводится
486 Глава 9. Градиентное элюирование
I I
Разделение первого измерения ->
Рис. 9.23. Гипотетический пример комплексного 20-разделения одного вещества.
Из каждого пика в разделении первого измерения отобраны несколько
фракций; каждая фракция представлена на нескольких хроматограммах
второго измерения. Пики / и у являются компонентами второго пика
разделения первого измерения
на колонку второго измерения. Получившиеся хроматограммы каждой фракции
приведены в верхней части рис. 9.23 (в этом примере второй пик от первого разделения
разрешается во втором как два пика / и у). Эти хроматограммы получены с
использованием одного детектора, установленного после колонки второго измерения. Наборы
хроматограмм (по одной на каждую фракцию разделения первого измерения)
сохраняются в компьютере, впоследствии эти данные можно представить различными
способами, например, трехмерными контурами или цветными графиками.
Пример трехмерного графика показан на рис. 9.24а, у каждого пика образца
указано время удерживания в разделениях первого и второго измерений. Высоте пика
соответствует поглощение, пропорциональное концентрации вещества. В этом случае
некоторые детали неясны (небольшие пики позади больших), но это можно прояснить,
вращая график на экране. Пример двумерного графика показан на рис. 9.24о,
представлен вид сверху (данные такие же, как на рис. 9.24а, но для другого образца).
Разная интенсивность окраски отражает разную величину пиков (более темные
изображения соответствуют более высоким пикам, как и на рис. 9.24а).
Если комплексный двумерный анализ проводится автоматически в режиме
реального времени, разделение второго измерения должно проходить гораздо быстрее,
чем первого. Например, если в первом разделении время градиента 250 минут, и
собраны 500 фракций (это не избыточное количество), то второе разделение может
занять не более 0,5 минуты. Если фракции собирают с помощью коллектора фракций,
их можно анализировать медленнее во втором измерении, но в этом случае
суммарное время анализа будет очень велико. Как видно на рис. 9.21а, при более быстром
разделении второго измерения существенно падает пиковая емкость; для второго
разделения предпочтительны более короткие колонки, частицы сорбента меньшего
размера и более высокие скорости потока.
Чтобы полностью использовать потенциальные возможности 2D-)KX (в
отношении пиковой емкости), любое разделение, проведенное в первом измерении, не
должно быть загублено при выполнении разделения второго измерения |67|. То есть,
не допускается повторное перемешивание фракций, разделенных в первом измере-
9.3. Разработка метода 487
(а)
mAU
"а*,
"**»«»*
25
(5)
0 5 10 15 20 25 30
Время удерживания в первом измерении (мин)
Рис. 9.24. Пример 20-разделения. (а) трехмерный график комплексной двумерной
хроматограммы экстракта мутантного маиса; (б) график комплексного
ЖХ/ЖХ разделения стандартной смеси метаболитов индола
нии (хотя можно предполагать, что каждая фракция с первой колонки повторно
перемешивается при ее перенесении на вторую колонку). Сначала был сделан вывод
|68|, что из каждого пика первого измерения надо отбирать по крайней мере 4
образца для второго разделения, чтобы избежать потери пиковой емкости. Однако это
может привести к очень большому количеству собранных фракций и, следовательно,
возрастанию объема работы. Последующий анализ влияния частоты отбора проб на
пиковую емкость говорит о том, что предпочтительнее отбирать по две фракции из
каждого пика первого измерения |69|, что тем не менее приводит к двукратной
потере пиковой емкости |57| из-за меньшего количества отобранных фракций.
488 Глава 9. Градиентное элюирование
Для хорошо спланированного разделения ЖХ/ЖХ (раздел 9.3.10.4) данный
компонент будет присутствовать в нескольких из собранных фракций и поэтому
появится на нескольких следующих друг за другом хроматограммах второго измерения
(см. рис. 9.23). Каждый компонент должен быть правильно идентифицирован на
соседних хроматограммах, и площади его пиков нужно просуммировать для
количественного анализа [711. Так как (1) пик разбивается на несколько фракций, (2) каждая
фракция разбавляется в разделении второго измерения и (3) базовые линии быстрых
градиентов часто негладкие и тогда детектирование может стать узким местом
ЖХ/ЖХ. Широко используются УФ-детекторы, но гораздо больше возможностей
по идентификации пиков и деконволюции сигнала предоставляет масс-спектромет-
рометрическое детектирование (раздел 4.14).
9.3.10.2. Пиковая емкость
Известно, что одномерная ЖХ высокого разрешения обеспечивает пиковую емкость
порядка сотен, в то время как ЖХ/ЖХ — порядка тысяч пиков. Рассмотрим, к
примеру, 200-минутное разделение первого измерения, за которым следует 100
разделений по 2 минуты во втором измерении (рис. 9.216). Предположим, что максимальное
давление на колонке 408 бар (6000 psi), тогда пиковая емкость PC для первого
измерения составит примерно 650. Аналогичным образом для второго измерения PC «
= 130; тогда PC == 650 х 130 = 85 000. Однако эффективная пиковая емкость будет
меньше по нескольким причинам:
• разделения в первом и втором измерениях не ортогональны;
ф используется только часть хроматограммы;
• недостаточное количество проб из разделения первого измерения.
Когда пиковая емкость 20-разделения рассчитывается как произведение
пиковых емкостей двух разделений, предполагается, что два разделения полностью ортого
налъны (времена удерживания в первом измерении не зависят от времен удерживания
во втором измерении). На практике этого никогда не происходит в ЖХ/ЖХ,
особенно в случае неионизируемых соединений, которые входят в состав образца [721- Так
как пики присутствуют только на части хроматограммы, пиковая емкость еще
понижается. Наконец, непрактично отбирать в первом измерении очень большое
количество образцов только для того, чтобы полностью избежать недостаточного
тестирования пиков первого измерения.
Перечисленные выше вклады в потери эффективной пиковой емкости трудно
контролировать, поэтому значения пс для ЖХ/ЖХ редко превышали к 2000 в то
время, когда была опубликована эта книга. Однако указанное значение можно
значительно увеличить, используя селективное детектирование (например,
масс-спектрометрическое). Основная цель заключается в том, чтобы выбрать два разделения,
которые были бы как можно более ортогональны. Обычно для этого требуются два
различных типа разделения (раздел 9.5), например, ионообменная хроматография
для первого измерения и обращенно-фазовая хроматография для второго измерения.
С другой стороны, в ЖХ/ЖХ можно использовать и два обращенно-фазовых
разделения, если для них можно подобрать достаточно ортогональные условия
(возможность таких условий обсуждалась в главах 5—7).
9.3.10.3. Оборудование для ЖХ/ЖХ
ЖХ/ЖХ с отбором фракций с помощью коллектора (офлайн) можно проводить
на обычном оборудовании, как описано в разделе 13.4.5. ЖХ/ЖХ с переключением
колонок несколько сложнее, но более удобна. В то время, когда одна фракция
анализируется на колонке второго измерения, вторая фракция разделения первого
измерения собирается и готовится к следующему вводу в колонку. В одной из распро-
9.3. Разработка метода
Рис. 9.25. Возможная конфигурация переключающего крана для ЖХ/ЖХ. На первой
стадии (а) в петлю 1 подается поток из колонки первого измерения, петля 2
соединяет насос 2 и колонку второго измерения. На второй стадии (б)
содержимое петли 1 вводится в систему второго измерения, в то время как
в петлю 2 поступает поток из колонки первого измерения
страненных систем используется двухходовый десятипортовый переключающий
кран, как показано на рис. 9.25.
На рис. 9.25а показано, что в петлю 1 подается поток из колонки 1. После
переключения крана содержимое петли 1 впрыскивается насосом 2 в колонку 2. Таким
образом получается хроматограмма второго измерения содержимого петли 1. В то же
время петля 2 заполняется следующей фракцией из колонки I. Когда кран снова
переключается (рис. 9.256), получается хроматограмма второго измерения содержимого
петли 2. Если объем петли существенно больше, чем объем фракции, в петлю
собирается вся фракция, и весь поступивший образец разделяется и анализируется.
Если \„ — время анализа второго измерения (его также называют временем цикла),
a ]F — скорость потока в разделении первого измерения, объем петли (V!oop) должен
отвечать следующему условию:
JF. (9.25)
V >2t
Система для ЖХ/ЖХ состоит из двух систем с насосами, но детектор требуется
только один. Детектор должен быть быстро срабатывающим (с малой постоянной
времени) и чувствительным, так как образец разводится при двух последовательных
разделениях.
9.3.10.4. Разработка метода для ЖХ/ЖХ
Условия для ЖХ/ЖХ подбираются иначе, чем для ID-разделений, в которых
основная цель — оптимизация селективности разделения для достижения некоего
минимального разрешения (например, Rs > 2) для всех пиков, представляющих интерес.
По разным причинам это нецелесообразно для ЖХ/ЖХ. При разработке полностью
автоматизированного 20-разделения преследуются следующие основные цели:
• ортогональные условия двух разделений;
• совместимость двух подвижных фаз;
490 Глава 9. Градиентное элюирование
• выбор условий для максимальной пиковой емкости в каждом разделении,
при этом время градиента второго разделения должно ограничиваться небольшой
долей времени градиента первого разделения.
Ортогональные условия. В ОФХ разделение основано на гидрофобное™ анализи
руемого вещества, в то время как в ИОХ — на его молекулярном заряде или
ионизации. Следовательно, эти два режима разделения можно рассматривать как
ортогональные, и по этой причине (а также по другим причинам, которые обсуждаются
далее) они часто используются вместе в 2Э-ЖХ. Обращенно фазовые разделения
также можно преобразовать в близкие к ортогональным, выбрав две ортогональные
колонки (раздел 5.4.3) с разными растворителями В, температурой и особенно рН
|73|. Возможны комбинации иных условий или режимов разделения, но они
привносят некоторые проблемы, обсуждаемые далее.
Совместимость подвижных фаз. Образец для разделения второго измерения рас
творен в подвижной фазе, используемой в разделении первого измерения. Двумя
важными характеристиками подвижных фаз разделений первого и второго
измерений являются смешиваемость и элюирующая сила. Введение значительного объема
растворителя, не смешивающегося с подвижной фазой, может привести к сильному
уширению пика и/или его искажению, если обращен но фазовая хроматография
сочетается с неводной нормально-фазовой. Если подвижная фаза фракций разделения
первого измерения имеет высокую элюирующую силу в разделении второго
измерения, (что обусловливает малое значение к) то возможно размывание или искажение
пика в разделении второго измерения (раздел 17.4.5.3). Использование ОФХ после
ИОХ позволяет в значительной степени избежать проблем, связанных со
смешиваемостью подвижных фаз, потому что водная подвижная фаза, применяемая в ИОХ,
во-первых, смешивается с подвижными фазами ОФХ и, во-вторых, является в этом
режиме очень слабой фазой.
При планировании системы ЖХ/ЖХ сначала надо выбрать приемлемые время
разделения первого измерения Iq и давление. На основании этого можно оценить PC
этого разделения и соответствующие задаче условия (размер частиц сорбента, длину
колонки, скорость потока; см. рис. 9.21а, таблицу 9.4 и обсуждение в разделе 9.3.9.1).
Получающаяся в результате пиковая емкость позволяет оценить среднюю ширину
пика ^(уравнение 9.20), которая чрезвычайно важна при отборе фракций пика
первого измерения (см. рис. 9.23). Рекомендуется |69| отбирать по две фракции каждого
пика (время отбора равно w/2). Более подробная информация о влиянии объема
фракции на пиковую емкость дана в |74|. Если известно время и давление в
разделении второго измерения, можно оптимизировать его условия таким же образом, как и
для разделения первого измерения. Возможная оптимизированная конфигурация
ЖХ/ЖХ представлена в таблице 9.5, она несколько сложнее чем широко
использовавшаяся в то время, когда издавалась эта книга.
Поскольку разделение первого измерения, приведенное в таблице 9.5,
проводится медленно, оптимальная длина колонки велика: к 950 мм или четыре колонки
по 250 мм. В противоположность этому колонка для разделения второго измерения
должна быть короткой (в данном примере 25 мм). Размер частиц сорбента для
первого измерения обычный (5 мкм), но очень мал (1,5 мкм) для второго измерения, так
как быстрым разделениям способствует малый размер частиц (рис. 9.21). Размывание
зоны во втором измерении можно снизить, минимизируя объем фракций с
первой колонки, чему способствует меньший диаметр первой колонки, по сравнению
со второй. Однако при таких условиях пик будет сильно разбавляться во втором
разделении (поскольку первая колонка малого диаметра будет иметь меньшую емкость
по образцу). Таким образом, выбирая диаметр колонки не нарушайте баланса между
шириной пика и чувствительностью детектирования. Дополнительные соображения
приводятся в |58|.
9.4. Разделения крупных молекул 491
Таблица 9.5. Типичные условия для разделения ЖХ/ЖХ
Параметр
Перепад давления (бар)
(psi)
Вязкость подвижной фазы (сП)
Коэффициент диффузии образца (см2/сек)
Время градиента
Диаметр колонки (мм)
Объем вводимой пробы (мкл)
Скорость потока (мкл/мин)
Размер частиц сорбента (мкм)
Длина колонки (мм)
к*
Пиковая емкость
Ширина пика (сек)/количество фракций
Коэффициент разбавления
* = 0
к = 3
Размывание зоны после ввода (%)
к = 0
А = 3
Первое измерение
408,2
6000
0,75
10-5
300 мин
1
5
20
5
950
4
700
26 сек/1400
1
3,5
21
1
Второе измерение
408,2
6000
0,75
10-5
30 сек
4,6
20
1500
1,5
25
3
100
1
1
264
33
9.4. Разделения крупных молекул
Градиентное разделение больших молекул (пептидов, белков, нуклеиновых кислот,
синтетических полимеров и т.д.) проводится практически также, как и разделение
малых молекул (100—1000 Да) |2]. Следовательно, отличия в градиентах или
параметрах колонки (tc, ipQ, qy, F, L и т.д.) влияют на разделения больших молекул таким же
образом, как описано в разделах 9.1—9.3 для малых молекул. Большие молекулы
действительно обладают некоторыми особенностями, играющими роль в их
градиентном разделении (разделы 13.3, 13.4.1.4). В этом разделе будет рассмотрена одна
из этих особенностей: возрастание величины S при увеличении молекулярной
массы М растворенного вещества; эти изменения приблизительно описываются
уравнением (9.23) (см. также примеры на рис. 13.11).
Основным последствием увеличения S с ростом М является его влияние на
значение к* (уравнение 9.4). Если условия градиента подбираются таким образом, чтобы
к* = 5 для низкомолекулярного образца (в соответствии с таблицей 9.3), то
высокомолекулярный образец в этих условиях будет иметь меньшее значение к*
(из-за ббльших значений S) и худшего разрешения. Чтобы получить такое же
значение к* = 5 для высокомолекулярного образца, надо увеличить время градиента
пропорционально отношению величин S этих образцов. Например, если для
высокомолекулярного образца М = 10 000 Да, S будет приблизительно 0,25 х (10 000)0,5 = 25
(уравнение 9.23). Поскольку для малых молекул 5«4, градиент для
высокомолекулярного образца должен быть в (25/4) = 6 раз дольше. Если время градиента для
низкомолекулярного образца 10 мин (таблица 9.3), то для высокомолекулярного образца
его следует увеличить примерно до одного часа, и в этом случае к*
высокомолекулярного образца также будет равен 5. Таким образом, градиентное разделение высо-
492 Глава 9. Градиентное элюирование
комолекулярных образцов требует больше времени или ббльших значений Xq при
прочих равных условиях. Разделение высокомолекулярных образцов (в большинстве
случаев в режиме градиентного элюирования) подробно рассмотрено в главе 13.
9.5. Другие виды разделения
Обсуждение градиентного элюирования в разделах 9.1—9.3 предполагало разделения
с помощью ОФХ. Существуют другие виды хроматографии, обзор которых приведен
в таблице 9.6. Градиентные разделения малых молекул с использованием
любой из методик, данных в таблице 9.6, происходят таким же образом, как и на
обрашенно-фазовых колонках, так что отличия градиентов или параметров колонок
аналогичным образом влияют на разделение, как описано в разделах 9.1—9.3
для ОФХ. Например, при увеличении времени градиента /с, или скорости потока F,
или при уменьшении объема колонки Vm, или диапазона градиента Д(р возрастает к
с предсказуемыми последствиями для среднего разрешения или ширины пика
{уравнение 9.4).
Таблица 9.6. Зависимость к от %Ъ для разных режимов разделения (уравнения 9.1
или 9.26)
Режим
разделения
Обраще и но -фа -
зовая
хроматография
Нормал ьно-фазо-
вая
хроматография (глава 8)
Ионообменная
хроматография
(ИОХ, глава 7)
Хроматография
гидрофобных
взаимодействий
(ХГВ, глава 13)
Гидрофильная
хроматография
(ГИХ, глава 8)
Зависимость
Аот%В
уравнение (9.1)
уравнение (9.24)
уравнение (9.24)
уравнение (9.1)
уравнение (9.24)
Определение к„ (уравнение
9.1) или кв (уравнение 9.26)
(к„) значение изократиче-
ского к для подвижной
фазы А (воды или буфера)
(kg) значение изократиче-
ского к для подвижной
фазы В (более полярный
растворитель)
(kg) значение изократиче-
ского к для подвижной
фазы с концентрацией
соли (<р) = 1,00 М
(кК) значение изократиче-
ского к для подвижной
фазы А (более высокая
концентрация соли)
(kg) значение изократиче-
ского к для органической
подвижной фазы А
Зависимость S или л от свойств
вещества
S возрастает с увеличением
размера молекулы вещества
(уравнение 9.21)
п увеличивается с
возрастанием числа полярных групп
в молекуле вещества
(раздел 8.2)
л = числу зарядов в молекуле
вещества (предполагается
одновалентный буфер)
5 = 0,14М °.37 [21
п увеличивается с
возрастанием числа полярных групп в
молекуле вещества (раздел 8.6)
9.5.1. Теория
Для каждого из видов разделения, приведенных в таблице 9.6, зависимость изокра-
тических значений к от %В описывается либо уравнением (9.1), либо выражением:
log к = log kB -n log <p. (9.26)
Определение log kg дано в таблице 9.6; п — константа для данных
экспериментальных условий и растворенного вещества (см. также таблицу 9.6), а ф относится
к объемной доли растворителя В (= 0,01 %В; в таблице 9.6 дано определение
растворителя В для разных видов разделения).
9.5. Другие виды разделения 493
Для разделений в линейном градиенте, описываемых уравнением (9.26),
градиентный коэффициент удерживании задается [2]:
к* = ^ . (9.27)
Здесь величина п различна для разных растворенных веществ и видов разделения
(объяснения в таблице 9.6); ер/ и Фо относятся к значениям <р = фд в начале (0) и
в конце (/) градиента соответственно.
Для низкомолекулярных образцов значение п обычно колеблется между 1 и 4,
значение л == 2 можно использовать как начальное приближение перед разработкой
метода. Таким образом, для нормально-фазового разделения на колонке 150 х 4,6 мм
при скорости потока 2 мл/мин и градиенте 0—100 %В за 10 минут значение к*
составит примерно (10 х 2)/(1,5 х 2 х log 10) = 6,7, то есть не слишком отличающееся
от значения к* <= 4 для близких условий в ОФХ. Для ИОХ разделения на колонке
150 х 4,6 мм при скорости потока 2 мл/мин и градиенте 10—100 ммоль за 10 минут
получим такое же значение к*, используя уравнение (9.26) (обратите внимание, что
концентрация соли ф в ИОХ равна сумме концентраций соли и буфера). Более
подробно теория градиентного элюирования в хроматографии различных видов
помимо ОФХ изложена в главе 13 |2| и в |75|.
9.5.2. Нормально-фазовая хроматография
В главе 8.5 приводится обзор некоторых недостатков изократической
нормально-фазовой хроматографии на колонках с непривитым силикагелем. За исключением
ГИХ (раздел 9.5.3), те же проблемы характерны и для градиентных разделений. Когда
подвижные фазы А и В существенно различаются по силе и полярности, серьезной
проблемой может оказаться обеднение элюента растворителем (раздел 8.5.2) |76| (за
последние 30 лет было очень мало сообщений о нормально-фазовых градиентных
разделениях). Использование колонок с полярными привитыми фазами (раздел 8.3.4) в основном
позволяет избежать обеднения растворителем при градиентном элюировании.
9.5.3. Гидрофильная хроматография (ГИХ)
В разделе 8.6 приведен обзор по изократической гидрофильной хроматографии.
Большинство сделанных выводов в равной степени применимы и к градиентному элюиро-
ванию. Использовать градиентное элюирование в ГИХ так же удобно, как в ОФХ,
и отсутствием проблем с ГИХ в этом режиме частично объясняется ее растущая
популярность. Применимость ГИХ к различного рода образцам иллюстрируют
гипотетические разделения рис. 9.26 а,б: на этом рисунке проводится сравнение градиентных
разделений в режимах обращенно-фазовой и гидрофильной хроматографии. Показано
разделение соединений (I—29) с понижающейся полярностью (возрастающей гидро-
фобностью), их удерживание на обращенной фазе увеличивается в этой
последовательности. Закрашенный пик 20 примерно соответствует толуолу и служит точкой отсчета
при сравнении этих разделений. Вещества 1—6 не удерживаются на обращенной фазе
(вследствие их более высокой полярности), в то время как вещества 19—29 не
удерживаются в ГИХ из-за их более значительной гидрофобное™. Соединения 26—29
настолько гидрофобны, что могут не вымываться с обращенной фазы данным
градиентом ацетонитрил/буфер, поэтому для их эффективного разделения необходимо
использовать неводную обращенно-фазовую хроматографию (НОФХ, раздел 6.5).
В соответствующем разделении в ГИХ на рис. 9.266 вещества 1—6 сильно
удерживаются и хорошо разделяются в отличие от ОФХ. Основным преимуществом ГИХ
494 Глава 9. Градиентное элюирование
(а)
ОФХ
0-100% ACN/буфер
о
(б)
6 В
Время (мин)
—Г"
10
12
ГИХ
3-40% буфер/ACN
4 в
Время (мин)
Рис. 9.2в. Гипотетический пример удерживания в ОФХ (а) и ГИХ (6) в зависимости
от полярности исследуемых веществ (полярность веществ 1—29 убывает),
(а) и (б) — разделения одного и того же образца
является возможность разделения полярных соединении, которые не удерживаются
или плохо удерживаются в ОФХ. Вещества 7—17 (показанные на обеих хроматограм-
мах двойными стрелками) удерживаются на обеих колонках, поэтому эти вещества
промежуточной полярности или гидрофобности можно разделять и в ОФХ, и в ГИХ.
Градиентное разделение образца такого рода (смеси пептидов) в обоих этих режимах
показано на рис. 9.27а,6.
9.5.3.1. Прикладные задачи
Гипотетические градиентные разделения, показанные на рис. 9.26, указывают на
изменение относительного удерживания на противоположное в разделениях ОФХ и
ГИХ и обратную корреляцию удерживания анализируемых веществ (т.е.
неортогональные разделения). Это далеко не всегда наблюдается в реальных разделениях как
показано на рис. 9.27а,б и обобщено на графике времен удерживания в градиенте
на рис. 9.27е (для которого г2 = 0,00, т.е. разделения ортогональны). В то же время
рис. 9.26 дает возможность предположить, что если пики двух образцов
накладываются друг на друга при разделении в ОФХ, то наложение будет и при разделении
в ГИХ, а обычно это не так. Более того, известно, что изменение других условий
(наклона градиента, температуры, типа колонки и т.д.) оказывают дополнительное
воздействие на селективность и в ОФХ, и в ГИХ.
При анализе образцов, в состав которых входят вещества сильно различающейся
полярности, может потребоваться использование как ГИХ, так и ОФХ для
достаточного удерживания и последующего разделения. Если обратиться к примеру
9.5. Другие виды разделения
J
(а)
0«Х
3. _ .7
uiili
гих
(6)
JP^Il^JjlLj^-
О 10 20
(в)
40
30
30
40
(ГИХ) 20
10
*чч •
•
•
•
1
.7
■
*ч%
чч
*ч
■
#.
у = 42 - Их
гй = 0.00
■
10
1Н(0ФХ)
15
20 (мин)
Рис. 9,27. Селективность при разделении смеси пептидов в ОФХ по сравнению
с ГИХ. Условия: (а) колонка Cig, 5—55% ацетонитрила /водный буфер
(рН 2) за 83 мин; (б) колонка ТСКгель Амид 80, 3—45% водный буфер
(рН 2)/ацетонитрил за 70 мин; (в) сравнение времен удерживания в
разделениях (б) и (а). Получено из данных [77]
на рис. 9.26, ОФХ можно использовать для анализа веществ 13—25, а ГИХ —
для остальных веществ 1 —12. Примером также может служить разделение в ГИХ
(рис. 9.28) неудерживаемой фракции обращенно-фазового градиентного разделения
гидролизованного глютена пшеницы.
^JWtfUiw
J !_
0 20 30 40 80 100 (мин)
Рис, 9.28. Разделение в ГИХ полярной неудерживаемой в ОФХ фракции
гидролизованного глютена пшеницы. Условия: Колонка ТСКгель Амид 80 250 х 1,5 мм;
10—40% буфера (рН 7,0)/ацетонитрил за 50 мин; 100 мкл/мин. Данные из |78|
496 Глава 9. Градиентное элюирование
9.5.3.2. Условия разделения
На колонках, упакованных силикагелем или привитой амидной фазой, часто
используются градиенты, начинающиеся с подвижной фазы 3—5% буфера (В)/ацетонитрил
(А) и заканчивающиеся 40—60 %В, время градиента от 20 до 60 минут. Остальные
условия сходны с используемыми в ОФХ (табл. 9.3). Градиентные разделения ГИХ
используются по тем же причинам, которые указаны в главе 13 как предпосылки
для применения изократической ГИХ (обычно для полярных образцов, которые
плохо удерживаются на обращенно-фазовых колонках и особенно в жидкостной
хроматографии в сочетании с масс-спектрометрией). Кроме того, для образцов,
удерживаемых и в ГИХ, и в ОФХ эти два режима разделения часто считают ортогональными,
что позволяет использовать их для 20-разделений. Однако фракции ОФХ
разделений первого измерения будут содержать сильный элюент для ГИХ разделения
второго измерения и наоборот (см. раздел 9.3.10.4).
9.5.4. Ионообменная хроматография (ИОХ)
Как отмечалось в разделе 7.5, в настоящее время ИОХ редко используется для
разделения низкомолекулярных соединений, за исключением углеводов и карбоновых
кислот. Ионная хроматография применяется для разделения неорганических ионов.
ИОХ в градиентном режиме широко используется для разделения биополимеров,
таких как полисахариды, белки, нуклеиновые кислоты и вирусы (глава 13).
9.6. Проблемы
В градиентном элюировании можно столкнуться с различными проблемами, редко
встречающимися или отсутствующими при изократическом разделении, среди них:
• обеднение элюента растворителем;
• системные пики;
• дрейф базовой линии.
9.6.1. Обеднение растворителя
Термин обеднение элюента растворителем в градиентном элюировании относится
к избирательному поглощению элюента растворителем В вследствие его большего
сродства к стационарной фазе. В большинстве случаев обеднение растворителя
не столь значительно в ОФХ, чтобы поставить под угрозу разделение. Его влияние
сводится к небольшому увеличению времени удерживания пиков, выходящих в
начале хроматограммы. Обеднение растворителем является серьезной проблемой в
нормально-фазовой хроматографии |79| при использовании колонок, упакованных
силикагелем, обладающим свойствами основания. Это может привести к полному
поглощению растворителя В в начале градиента с последующим внезапным
выбросом растворителя В в подвижную фазу. В результате все рановыходяшие пики могут
элюироваться вместе как один пик с потерей разрешения. Более подробно обеднение
растворителем в градиентном элюировании обсуждается в |2|.
Литература 497
9.6.2. Системные пики
Системными (ложными) пиками называют пики, не соответствующие компонентам
образца. Они могут быть вызваны загрязнением растворителей или буферов
подвижной фазы, а также другими примесями. Системные пики могут также возникать
при переносе образца из одного разделения в другое из-за адсорбции образца в авто-
самплере или на входном фильтре колонки. Общее обсуждение системных пиков
проведено в |80] и |81|. Меры по ликвидации системных пиков рассмотрены в
разделе 17.4.5.2.
9.6.3. Дрейф базовой линии
Это явление характерно для градиентного элюирования вследствие различного
отклика детектора на растворители А и В и может приводить к дрейфу базовой линии
на протяжении градиента. Дрейф базовой линии часто наблюдается в ОФХ с УФ-де-
тектированием, так как органические растворители {растворитель В) обычно имеют
большее поглощение, чем вода, особенно при малых длинах волн. Более подробно
дрейф базовой линии рассмотрен в разделе 17.4.5.1.
Литература
1. P. L. Zhu, L. R. Snyder, J. W. Dolan, N. M. Djordjevic, D. W. Hill, L. С Sander, and T. J. Wa-
eghe, J. Chromatogr. A, 756 (1996) 21.
2. L. R. Snyder and J. W. Dolan, High-Performance Gradient Elation, Wiley-lnterscience, New York,
2007.
3. F. Leroy, B. Presle, F. Verillon, and E. Verette, J. Chromatogr. Sci., 39 (2001) 487.
4. S. R. Needham and T. Wehr, LCGC Europe, 14 (2001) 244.
5. J. Ayrton, G. J. Dear, W. J. Leavens, D. N. Mallet, and R. S. Plumb, J. Chromatogr. B, 709 (1998)
243.
6. L. R. Snyder, J. J. Kirkland, and J. I_. Glajch, Practical HPLC Method Development, 2nd ed.,
Wiley-lnterscience, New York, 1997.
7. L. Hagdahl, R. J. P. Williams, and A. Tiselius, Arkiv. Kemi, 4 (1952) 193.
8. T. Braumann, G. Weber, and L. H. Grimme, J. Chromatogr., 261 (1983) 329.
9. P. L. Zhu, L. R. Snyder, J. W. Dolan, N. M. Djordjevic, D. W. Hill, L. С Sander, and T. J. Wa-
eghe, J. Chromatogr. A, 756 (1996) 21.
10. P. Jandera and M. Spacek, J. Chromatogr., 366 (1986) 107.
11. L. R. Snyder and J. W. Dolan, J. Chromatogr. A, 799 (1998) 21.
12. A. P. Schellinger and P. W. Carr, J. Chromatogr. A, 1077 (2005) 110.
13. W. Hancock, R. С Chloupek. J. J. Kirkland, and L. R. Snyder, J. Chromatogr. A, 686 (1994) 31.
14. T. Jupille, L. Snyder, and I. Molnar, LCGC Europe, 15 (2002) 596.
15. B. F. D. Ghrist, B. S. Cooperman, and L. R. Snyder, J. Chromatogr., 459 (1989) 1.
16. B. F. D. Ghrist and L. R. Snyder, J. Chromatogr., 459 (1989) 63.
17. B. F. D. Ghrist and L. R. Snyder, J. Chromatogr., 459 (1989) 25.
18. V. Concha-Herrera, G. Vivo-Truyots, J. R. Torres-Lapasio and M. C. Garcia-Alvarez-Coque,
J. Chromatogr. A, 1063 (2005) 79
19. S. V. Galushko and A. A. Kamenchuk, LCGC Intern., 8 (1995) 581.
20. H. Xiao, X. Liang, and P. Lu, J. Sep. Sci., 24 (2001) 186.
21. W. Markowski and W. Golkiewicz, Chromatographia, 25 (1988) 339.
22. P. Jandera, Adv. Chromatogr., 43 (2004) 1.
23. L. R. Snyder and D. L. Saunders, J. Chromatogr. Sci., 7 (1969) 195.
24. U. D. Neue, D. H. Marchand, and L. R. Snyder, J. Chromatogr. A, 1111 (2006) 32.
25. H. Poppe, J. Paanakker, and M. Bronkhorst, J. Chromatogr., 204 (1981) 77.
26. L. R. Snyder and J. W. Dolan, Adv. Chromatogr., 38 (1998) 115.
498 Глава 9. Градиентное элюирование
27. J. Pellett, P. Lukulay, Y. Мао, W. Bowen, R. Reed, M. Ma, R. С. Munger, J. W. Dolan, L. Wris-
ley, K. Medwid, N. P. Toltl, С. С Chan, M. Skibic, K. Biswas, K. A. Wells, and L. R. Snyder,
J. Chromatogr. A, 1101 (2006) 122.
28. Z. Li, S. C. Rutan and S. Dong, Anal. Chem., 68 (1996) 124.
29. R. Majors, LCGC, 20 (2002) 516.
30. L. R. Snyder and J. W. Dolan, J. Chromatogr. A, 721 (1996) 3.
31. P. J. Schoenmakers, H. A. H. Billiet, and L. De Galan, J. Chromatogr., 205 (1981) 13.
32. P. J. Schoenmakers, A. Bartha, and H. A. H. Billiet, J. Chromatogr., 550 (1991) 425.
33. P. Schoenmakers, in Handbook of HPLC, E. Katz, R. Eksteen, P. Schoenmakers, and N. Miller,
eds., Dekker, New York, 1998, pp.218-220 34.
34. A. P. Schellinger and P. W. Carr, J. Chromatogr. A, 1109 (2006) 253.
35. L. R. Snyder and J. W. Dolan, J. Chromatogr. A, 892 (2000) 107.
36. J. W. Dolan, L. R. Snyder, N, M. Djordjevic, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 857 (1999) 41.
37. P. L. Zhu, J. W. Dolan and L. R. Snyder, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 756 (1996) 51.
38. W. Hancock, R. С Chloupek, J. J. Kirkland, and L. R. Snyder, J. Chromatogr. A, 686 (1994) 31,
45.
39. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 857 (1999) 1.
40. J. W. Dolan, L. R. Snyder, T. Blanc, and L. Van Heukelem, J. Chromatogr. A, 897 (2000) 37.
41. J. W. Dolan, J. Chromatogr. A, 965 (2002) 195.
42. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, D. L. Saunders, L. Van Heukelem, and
T. J. Waeghe, J. Chromatogr., 803 (1998) 1.
43. A. P. Schellinger, D. R. Stall, and P. W. Carr, /. Chromatogr. A, 1064 (2005) 143.
44. A. P. Schellinger, D. R. Stoll, and P. W. Carr, J. Chromatogr. A. 1192 (2008) 41.
45. A. P. Schellinger, P. Adam, D. R. Stoll, and P. W. Carr, J. Chromatogr. A, 1192 (2008) 54.
46. J. Garcia, J. A. Martinez-Pontevedra, M. Lores, and R. Cela, J. Chromatogr. A, 1128 (2006) 17.
47. V. R. Meyer, /. Chromatogr. A, 1187 (2008) 138.
48. U. D. Neue and J. R. Mazzeo, J. Sep. Set., 24 (2001) 921.
49. U. D. Neue, J. Chromatogr. A, 1079 (2005) 153.
50. D. R. Stoll, X. Wang, and P. W. Carr, Anal. Chem., 78 (2006) 3406.
51. S. E. G. Porter, D. R. Stoll, S. С Rutan, P. W. Carr, and Cohen, Anal. Chem., 78 (2006) 5559.
52. X. Wang, W. E. Barber, and P. W. Carr/ Chromatogr. A, 1107 (2006) 139.
53. D. R. Stoll, X. Li, X. Wang, P. W. Carr, S. E. G. Porter, and S. С Rutan, J. Chromatogr. A, 1168
(2007) 3.
54. M. Gilar and U. D. Neue, J. Chromatogr. A, 1169 (2007) 139.
55. U. D. Neue,/. Chromatogr. A, 1184 (2008) 107.
56. D. R. Stoll, X. Wang, and P. W. Carr, Anal. Chem., 80 (2008) 268.
57. J. M. Davis, D. R. Stoll, and P. W. Carr, Anal. Chem., 80 (2008) 461, 8122.
58. X. Li, D. R. Stoll, and P. W. Carr, Anal. Chem., 81 (2009) 845.
59. L. A. Romanyshyn and P. R. Tiller, J. Chromatogr. A, 928 (2001) 41.
60. K. D. Patel, A. D. Jerkovich, J. С Link, and J. W. Jorgenson, Anal. Chem., 76 (2004) 5777.
61. H. Chen and С Horvath, J. Chromatogr. A, 705 (1995) 3.
62. S. K. Paliwal, M. de Fratos, and F. E. Regnier, in Methods in Enzymology, Vol. 270. W. S.
Hancock and B. L. Karger, eds., Academic Press, Orlando, 1996, p. 133.
63. J. J. Kirkland, F. A. Truszkowski, and R. D. Ricker, J. Chromatogr. A, 965 (2002) 25.
64. L. Xiong, R. Zhang, and F. E. Regnier, J. Chromatogr. A., 1030 (2004) 187.
65. P. Brown and E. Grushka, eds.. Design of Rapid Gradient Methods for the Analysis of Combinatorial
Chemistry Libraries and the Preparation of Pure Compounds, Dekker, New York, 2001.
66. P. J. Schoenmakers, P. Marriott, and J. Beans, LCGC Europe, 16 (2003) 335.
67. J. C. Giddings, Multidimensional Chromatography: Techniques and Applications; Dekker, New York,
1990.
68. R. E. Murphy, M. R. Schure, and J. P. Foley, Anal. Chem., 70 (1998) 1585.
69. K. Horie, H. Kimura, T. lkegama, A. Iwatsuka, N. Saad, O. Fiehn, and N. Tanaka, Anal. Chem.,
79 (2007) 3764.
70. D. R. Stoll, J. D. Cohen, and P. W. Carr, J. Chromatogr. A, 1122 (2006) 123.
Литература 499
71. S. Peters, G. Vivo-Truyols, P. J. Marriott, and P. J. Schoenmakers, J. Chromatogr. A, 1156 (2007)
14.
72. J. W. Dolan and L. R. Snyder, J. Chromatogr. ,4, 1216 (2009) 3467.
73. M. Gilar, P. Olivova, A. E. Daly, and J. C. Geblcr, Anal. Chem., 77 (2005) 6426.
74. K. Horie, H. Kimura, T. Ikegami, A. Iwatsuka, N. Saad, O. Fiehn, and N. Tanaka, Anal. Chem.,
79 (2007) 3764.
75. P. J. Schoenmakers, G. Vivo-Truyols, and W. M. C. Decrop, J .Chromatogr. A, 1120 (2006) 282.
76. P. Jandera, J. Chromatogr. A, 965 (2002) 239.
77. P. Jandera, J. Chromatogr. A, 1126 (2006) 195.
78. T. Yoshida, J. Chromatogr. A, 60 (2004) 265.
79. H. Schlichtherle-Cerny, M. Affolter, and C. Cerny, Anal. Chem., 75 (2003) 2349.
80. P. Jandera, J. Chromatogr. A, 965 (2002) 239.
81. S. Williams, У. Chromatogr. A, 1052 (2004) 1.
ГЛАВА 10
РАЗРАБОТКА МЕТОДА
С ПОМОЩЬЮ ПРОГРАММНОГО
ОБЕСПЕЧЕНИЯ
10.1. Введение 501
10.1.1. Основы и история компьютерного моделирования 503
10.1.2. Когда использовать компьютерное моделирование 504
10.1.2.1. Преимущества 505
10.1.2.2. Недостатки 505
10.2. Программное обеспечение компьютерного моделирования 506
10.2.1. Работа с программой DryLab 507
10.2.2. Оптимизация градиента 509
10.2.3. Другие возможности 511
10.2.3.1. Прогнозирование изократического элюирования на основе
пданных градиентного элюирования 511
10.2.3.2. Выбор определенного пика 512
10.2.3.3. Изменение других условий 512
10.2.3.4. Компьютерный подбор наилучшего кусочно-линейного
градиента 512
10.2.3.5. Уширение заднего фронта пика 514
10.2.3.6. Метод двух разделений для улучшения разрешения образца. 514
10.2.3.7. Примеры использования компьютерного моделирования
при разработке метода 514
10.2.4. Отслеживание пиков 515
10.2.5. Источники коммерчески доступного программного обеспечения
для компьютерного моделирования хроматографических разделений. 516
10.3. Другое программное обеспечение для разработки метода 516
10.3.1. Удерживание анализируемого вещества и его молекулярная
структура 517
10.3.2. Величины рКа анализируемого вещества и его молекулярная
структура 517
10.3.3. Селективность ОФХ-колонки 517
10.3.4. Экспертные системы для разработки метода 518
10.4. Компьютерное моделирование и разработка метода 518
10.4.1. Пример 1: разделение смеси производных лизергиновой кислоты . . . 518
10.4.2. Пример 2: альтернативная стратегия разработки метода 520
10.4.3. Проверка устойчивости метода 522
10.4.4. Заключение 523
Литература 523
JO. I. Введение 501
10.1. Введение
Разработка метода с помощью программного обеспечения — это общее определение,
которое может быть применено к использованию любого программного обеспече
ния, облетающего разработку метода. В этой главе мы остановимся на
коммерческом программном обеспечении компьютерного моделирования, которое может
предсказать результаты разделения в зависимости от одного или нескольких условий
эксперимента с помощью опытных данных, полученных в нескольких
предварительных разделениях. Поскольку это программное обеспечение позволяет предсказывать
оптимальные условия конечного метода разделения, мы можем уменьшить
количество экспериментов, необходимых для их поиска.
Простым примером для изократической ОФХ является предсказание
зависимости разделения от силы подвижной фазы (%В). Для компьютерного
моделирования необходимо два предварительных экспериментальных разделения. В примере
на рис. 10.1 эксперименты с использованием подвижной фазы с содержанием 10 и
20 %В (при неизменных остальных условиях) показаны на рис. 10. \а,б. Полученные
данные вводятся в компьютер: времена удерживания для каждого растворенного
вещества в каждом разделении, значения %В для двух разделений (10, 20 %В) и другие
экспериментальные условия (состав А- и В-растворителей, размеры колонки и
размер частиц сорбента, скорость потока, температура, см. раздел 10.2.1). После этого
можно с помощью компьютера построить модель разделения, т.е. предсказать, каким
оно будет при изменении %В в подвижной фазе, скорости потока, размеров колонки
и размера частиц сорбента.
Наиболее полезной информацией, которую можно получить с помощью
компьютерного моделирования, обычно является карта разрешения, показанная
на рис. 10. \в для смеси семи кислот и оснований. Карта разрешения представляет
собой график критического разрешения Rsm\n двух наименее разрешенных пиков в
зависимости от условия или условий, которые были изменены в предварительных
экспериментальных разделениях (дополнительная информация по картам
разрешения дана в разделе 6.3.3 и на рис. 6.135). В этом примере изменялся %В, и поэтому
на графике построена зависимость Rs от %В. На рис. 10.Is мы видим, что максимум
разрешения достигается при трех разных условиях: 9, 17 и 25 %В с наибольшим
разрешением в случае 17 %В. Программа позволяет построить модель хроматограммы
для любого значения %В, как и для «оптимальных» его значений (хроматограммы
при «оптимальных» значениях %В приведены на рис. 10.1г-е). Компьютер может
дать дополнительную информацию для каждого из этих (и других) разделений,
зависящих от %В. Особенно полезен в этом отношении диапазон величин к (указанный
для каждого из разделений на рисунках 10. \г-е). Видно, что разделение на рис. 10е
(25 %В, I < к < 6) лучше всех удовлетворяет обычно рекомендуемому условию 1 5 к i
< 10. Тем не менее, в некоторых лабораториях могут предпочесть разделение
на рис. 10.Id с элюентом, содержащим 17 %В (3 < к < 12), вследствие достижения
более высокого разрешения. Для данного образца, однако, диапазон удерживания
в любом из этих трех разделений может считаться приемлемым, если это согласуется
с целью разделения.
После того, как выбран приемлемый %В, появляется возможность сократить время
разделения за счет избыточного разрешения, изменив параметры колонки. Например,
разрешение в разделении с 25 %В (Rs = 2,4) можно было бы уменьшить без
отрицательных последствий, взяв более короткую колонку и/или увеличив скорость потока.
Компьютерное моделирование позволяет пользователю исследовать влияние изменения
размеров колонки, размера частиц сорбента или скорости потока. На рис. 10.\ж показана
одна из таких моделей процесса разделения (для 25 %В): уменьшение длины колонки
502
Глава 10. Разработка метода с помощью программного обеспечения
(а)
10 %В
п 1 i 1 г-
Время (мин)
Время (мин)
(г)
(б)
17 %В
3<к<12
1 I3
I 1 г (—i—i—i—i—i—i—i—1—i—г
0 20 0 2 4 6 8 10
Время (мин) Время (мин)
25 %в, 75-мм, 3 мкм колонка
Я, = 2.1
(ж)
I 1 1 1 1 1 т^? |~-
0 2 4 6 0 12 3
Время (мин)
Время (мин)
Рис. ЮЛ. Иллюстрация компьютерного моделирования разделения смеси кислот и
оснований. Образец (смесь кислот и оснований) и условия такие же, как и на
рис. 7.7. (а,б.) Предварительные экспериментальные разделения; (в) карта
разрешения, предсказанная на основе данных (а,б); (г—е) смоделированные хро-
матограммы для различных величин %В (соответствующих максимальному
разрешению); (ж) смоделированная хроматограмма для условий,
способствующих быстрому разделению (с приемлемым разрешением). Компьютерное
моделирование основано на данных, приведенных в работе |1|
наполовину (от 150 до 75мм), изменение размера частиц от 5 до 3 мкм при
сохранении неизменной скорости потока. Приемлемое разрешение (Rs = 2,1) и давление
(Р = 152 бар или 2200 psi) достигаются при времени разделения всего три минуты,
при этом сохраняется предпочтительный диапазон удерживания ] 2. к 2. 6.
Окончательный выбор «оптимизированного» разделения может зависеть от
соображений, не имеющих отношения к разрешению, времени разделения и диапазону
JO.]. Введение 503
удерживания. Он может зависеть, к примеру, от допустимого давления в колонке,
сокращения ширины пиков, необходимого для повышения чувствительности
детектирования, и относительной важности различных пиков на хроматограмме. По этой
причине программное обеспечение для компьютерного моделирования должно быть
гибким, чтобы позволить пользователю изучать последствия различных изменений
условий разделения, а не просто предлагать одно единственное «оптимизированное»
разделение.
10.1.1. Основы и история компьютерного моделирования
В компьютерном моделировании используют самые различные эмпирические и
теоретически выведенные формулы — например, уравнения (2.7), (2.I0), (2.18), (2.23),
(2.26) и уравнения (9.7)—(9.14а) — для прогнозирования времени удерживания и
ширины пика в изократическом и градиентном режимах элюирования. Часто
компьютерное моделирование начинается с уравнения, связывающего величины к и %В
(или ф = 0,01 %В). Для случая ОФХ это уравнение имеет вид:
log к = log kw - 5ф. (10.1)
Здесь kw — значение к для ф = 0 (например, для воды в качестве подвижной фазы),
a S — константа для данного растворенного вещества при прочих постоянных
условиях, кроме %В. Два реальных разделения, в которых изменяется либо %В (в случае
изократического элюирования), либо время градиента (в случае градиентного
элюирования, раздел 9.2.4.2), позволяют вычислить значения kw и S для каждого
компонента образца. Для НФХ аналогичная взаимосвязь между к и ф описывается
уравнением (8.4а). Как только значения kw и S получены, величины к и времени
удерживания tR могут быть предсказаны для любых значений %В (или для градиента
любой длительности), любых размеров колонки или скорости потока (уравнения 2.5
и 9.10).
Значения ширины пика Смогут быть рассчитаны различными способами: (1)
допущением, что колонка, на которой проведены предварительные разделения, имеет
некоторое количество теоретических тарелок (например, jV = 10000 для колонки
длиной 100 мм, упакованной сорбентом с частицами 3 мкм), (2) измерением ширины
пиков в предварительных разделениях или (3) расчетом ширины пиков, основанном
на значениях N из уравнения (2.17). С помощью различных уравнений |2]
компьютерное моделирование можно распространить на случай кусочно-линейных градиентов,
колонок различных размеров, изменений скорости потока и т.д. Корректность
компьютерного моделирования была подтверждена многочисленными
экспериментальными исследованиями, обобщенными в |2]. Времена удерживания, как правило,
достоверны с точностью до нескольких процентов и, что еще более важно, точность
предсказания величины разрешения Rs обычно укладывается в интервал ±10%. Этого,
в общем случае, достаточно для использования данных при разработке метода.
Впервые о применении компьютерного моделирования в ВЭЖХ сообщили
в 1978 году Лауб и Пернелл |3|. Они описали использование карты разрешения
для зависимости разделения в изократической ОФХ от температуры. Спустя
несколько лет Глайх, Киркленд и др. |4] опубликовали методику оптимизации типа
растворителя в изократических условиях, основанную на схеме экспериментов,
показанной на рис. 10.2г. В соответствии с этой схемой требуется семь
экспериментальных разделений с элюентами, содержащими ацетонитрил, метанол и ТГФ в
различных пропорциях (см. также раздел 6.4.1.1, рис. 6.24). Одновременно Деминг и сотр.
|5| представили похожую схему одновременной оптимизации рН подвижной фазы и
концентрации ион-парного реагента. Изократический метод Глайха и Киркленда
504
Глава 10. Разработка метода с помощью программного обеспечения
50'С
35 %В
(б)
50'С
1в = 10 мин
50'С
fg = 30 МИН
30 'С 30 "С
tG = 10 мин (с = 30 мин
рН2,5
35 %В
рН2,5
(<з = 10 мин
о
(а)
рН 3,0 рН 3,5
(G = 10 мин (G = 10 мин
рН 2,5 рН 3,0 рН 3,5
/G = 30 мин fG = 30 мин (G = 30 мин
(в)
30% ACN
40% МеОН
1:1:1
40% ACN
50% МеОН
МеОН
ТГФ
Рис. 10.2. Примеры различных системных подходов к оптимизации селективности
(схемы экспериментов); (а,в,д,ё) — эксперименты для изократических
разделений, (б,г) — для разделений в градиенте
был распространен на градиентное элюирование в 1983 |6|. Программа DryLabR6buia
внедрена в 1985 году и впоследствии доработана до наиболее универсального,
широко используемого и доступного в настоящее время программного продукта для
моделирования хроматографических разделений [7,8|. DryLabR описана в разделе 10.2 как
пример разнообразного возможного использования компьютерного моделирования.
Аналогичные программы для компьютерного моделирования разрабатывались и
после 1985. Об этих программах сообщалось в работах \Э—12|. Там же проводилось их
сравнение. Обзор этих программ дан в разделе 10.3.4.
10.1.2. Когда использовать компьютерное моделирование
Разработка метода может осуществляться с помощью или без помощи
компьютерного моделирования, и поэтому важно оценивать возможные «за и против» его
применения при разработке конкретного метода.
JO. I. Введение 505
10.1.2.1. Преимущества
Польза от компьютерного моделирования, вероятно, будет максимальной в том
случае, когда выполняются одно или несколько из следующих условий:
• разделения в режиме градиентного элюирования;
• многокомпонентные образцы, содержащие от 5 до 10 и более компонентов;
• когда требуются очень быстрые разделения (например, проводимые в течение
I минуты и менее);
• когда важна устойчивость (робастность) метода;
• когда имеет смысл немного улучшить разрешение или время разделения.
Способ изократического разделения образца, содержащего лишь несколько
компонентов, когда время разделения не является критическим, можно разработать
быстро и легко методом проб и ошибок. В таких случаях преимущества компьютерного
моделирования чаще всего минимальны, хотя стоит не забывать и о других его
преимуществах (возможность тонкой настройки разделения для сокращения его времени,
исследование устойчивости метода и т.д.). Градиентное элюирование, напротив,
включает исследование дополнительных параметров разделения, и это более удобно
делать с помощью компьютерного моделирования. Подобным же образом сложные
образцы с большим количеством близко элюируемых веществ (и соответственно их
пиков) часто требуют большого количества экспериментов для достижения
приемлемого разделения. Большая часть этих экспериментов можно заменить
компьютерным моделированием. Аналогичную задачу, также требующую ряда экспериментов,
могут представлять собой и очень быстрые разделения.
В тех случаях, когда важна надежность (робастность) метода, компьютерное
моделирование позволяет быстро изучить влияние на разделение изменений в
различных условиях без проведения дополнительных экспериментов. Небольшие улучшения
разделения с помощью изменений, вносимых методом проб и ошибок, легко
осуществить с помощью компьютерного моделирования. Влияющее на селективность
одновременное изменение двух условий разделения (таблица 2.2) — мощный инструмент,
который часто требуется для работы со сложными образцами или для проведения
других разделений, к которым предъявляются высокие требования. В таких случаях
компьютерное моделирование может быть использовано для определения
оптимальных условий с помощью минимального количества экспериментальных разделений
(рис. 10.2). Изменения относительного удерживания при изменении условий могут
затруднить идентификацию пиков и анализ результатов экспериментальных
разделений, особенно в тех случаях, когда образец многокомпонентный. При
компьютерном моделировании индивидуальные пики из разных разделений автоматически
сопоставляются (после обозначения пиков в начальных экспериментальных
разделениях). Интерпретация смоделированных разделений, таким образом, значительно
упрощается. Примеры для иллюстрации каждого из этих различных преимуществ
будут приведены в разделе 10.2.
10.1.2.2. Недостатки
Основными препятствиями для более широкого распространения компьютерного
моделирования являются высокая стоимость программного обеспечения и время,
необходимое хроматографисту для его освоения. Если разработками новых методов
занимаются редко, и образцы, как правило, легко разделяются, возможная польза
от компьютерного моделирования будет очень небольшой. Еще одним барьером для
использования компьютерного моделирования является убеждение в том, что не
нужно никакого моделирования при достаточном практическом опыте
хроматографиста. Хотя компьютерное моделирование редко играет важную роль при разработке
506
Глава 10. Разработка метода с помощью программного обеспечения
метода, оно зачастую может снизить стоимость и улучшить качество уже
разработанного метода даже при условии, что хроматографисты имеют достаточный опыт.
С другой стороны, компьютерное моделирование не подменяет собой опыт
хроматографиста. Последний по-прежнему важен, в том числе и для того, чтобы
пользоваться компьютерным моделированием.
Менее существенным возражением против применения компьютерного
моделирования является тот факт, что предсказанные хроматограммы получаются скорее
«идеальными», чем «реальными». Предполагается, что базовая линия не плывет,
пики симметричны (хотя компьютерное моделирование может учесть размывание
заднего фронта пика, см. раздел 10.2.3.5), артефакты базовой линии или
посторонние пики обычно не учитываются. Однако, такие артефакты часто ухудшают
качество разработанного метода и лучше всего их устранить перед началом компьютерного
моделирования. В отдельных случаях артефакты могут не влиять на анализ
результатов разделения или выбор окончательных условий для его проведения, и тогда
артефакты можно не принимать во внимание.
Наконец, любые ошибки при вводе данных (включая неправильное
соотнесение пиков разделяемым веществам) могут привести к серьезным ошибкам в
спрогнозированных разделениях. Однако количество ошибок при вводе данных можно
сократить с помощью автоматического переноса данных из устройства сбора и
обработки данных хроматографической системы в программу, осуществляющую
моделирование процесса разделения. Другие ошибки, которые могут возникнуть в
результате компьютерного моделирования, как правило, очевидны, и их легко
исправить. Например, в тех случаях, когда предсказанное разделение и то, что
получилось в результате эксперимента, который должен подтвердить это
предсказание, не согласуются.
10.2. Программное обеспечение
компьютерного моделирования
Компьютерное моделирование лучше всего использовать как составляющую общей
стратегии разработки метода (см. другие разделы книги и примеры в разделе 10.4).
Таким образом, на первом этапе моделирования нужно исследовать «наиболее
подходящий» диапазон удерживания (значений к для изократического элюирования или
к*дли градиентного) с помощью изменения %В в случае изократического
элюирования или длительности градиента в случае градиентного. В тоже время необходимо
обратить внимание на изменения селективности в зависимости от %В или t^.
Максимальное разрешение может соответствовать промежуточному значению %В (как
на рис. lO.le). Если же изменения в относительном удерживании недостаточны,
следует исследовать и другие факторы, влияющие на селективность. Различные схемы
экспериментов на рис. 10.2 обобщают некоторые из наиболее популярных подходов.
После того, как эксперименты, соответствующие одной из таких схем, проведены
(например, четыре экспериментальных разделения по схеме на рис. 10.2а), можно
приступать к моделированию эффектов одновременных изменений двух различных
условий разделения. В принципе, можно моделировать любые два условия
разделения, влияющие на селективность, как это показано на рис. 10.2. После проведения
экспериментов по любой из схем на рис. 10.2, компьютерное моделирование может
быть применено для выбора наилучших условий изократического или градиентного
элюирования (раздел 9.2.2). Кроме того, можно изменять параметры колонки
(геометрические размеры, размер частиц, скорость потока) для увеличения разрешения
или сокращения времени разделения.
10.2. Программное обеспечение компьютерного моделирования 507
10.2.1. Работа с программой DryLab
В этом разделе проиллюстрированы некоторые полезные функциональные
возможности компьютерного моделирования, которые представляют собой часть программы
DryLab. Многие из этих функциональных возможностей можно найти и в других
коммерчески доступных программах для компьютерного моделирования.
Прежде всего, выбирается схема эксперимента и метод разделения, которые
определяют количество экспериментальных калибровочных разделений,
требующихся для компьютерного моделирования (как на рис. 10.2). В этой главе мы
ограничимся обсуждением компьютерного моделирования для ОФХ. В качестве примера
выберем на рис. 10.25 экспериментальную схему и используем ее для изократического
разделения смеси из восьми кортикостероидов. В основе схемы лежат
предварительные градиентные разделения, требуется четыре экспериментальных разделения с
разными временами градиента ч7<20, 60 мин) и температурами Т (30, 60 °С) с целью
одновременной оптимизации %В и температуры Т. В компьютер вводятся следующие
данные эксперимента (см. рис. 10.За): время задержки системы (5,5 мл;
используемое только для предварительных экспериментов с помощью градиентного элюиро-
вания), размеры колонки (250 х 4,6 мм), размер частиц (5 мкм), скорость потока
(2,0 мл/мин), состав подвижной фазы (вода — растворитель А; ацетонитрил —
растворитель В), диапазон градиента (0—100 %В), времена удерживания и площади
пиков для каждого соединения в каждом из четырех разделений. Наконец, необходимо
соотнести данные по удерживания каждого пика с соответствующим компонентом
образца (раздел 10.2.4). Соотнесение пиков может быть выполнено вручную или
с помощью компьютера. Пики на рис. 10.3а соотнесены удовлетворительно (что
подтверждается значениями в колонке таблицы данных «area dev» (area deviation),
соответствующими измеренному % отклонения предсказанных площадей пиков от
наблюдаемых в реальности).
После того, как данные введены, можно выбрать окно для компьютерного
моделирования (рис. 10.36). Поскольку необходимы предсказания для изократического элюи-
рования, выбирается окно вывода %В и °С. Несколько вариантов вывода данных
показаны в таблицах над картой разрешения: карта разрешения, таблица с полученными
данными и т.д. В примере на рис. 10.35 выбрана карта разрешения. Значения Rs
показаны оттенками серого для различных сочетаний °С (отложенных по оси у) и %В
(отложенных по оси х). В этом случае максимум Rs = 1,34 достигается при Т = 30 °С
и %В = 38% (обратите внимание на перекрестие на рис. 10.36). Когда курсор
перемещается внутри карты разрешения в область значений %В и Т, при которых достигается
максимальное разрешение (или любой другой пары величин %В и 7), в нижней части
окна (нижней части рисунка 10.36) немедленно появляется хроматограмма для этих
условий. Выбор других условий (32,6 %В и 30 °С) при вводе в соответствующее поле
«status» («состояние») дает похожее разрешение {Rs = 1,30), но при этом на разделение
потребуется больше времени (22 мин по сравнению с 12 мин в примере на рис. 10.36).
Дальнейшие соображения не в пользу выбора Т= 30 °С и 32,6 %В следующие:
во-первых, диапазон удерживания не так хорош (4 < к < 16 в сравнении с 2 < к £ 8);
во-вторых, для разделения при 30 °С и 32,6 %В небольшие изменения %В могут приводить
к значительному уменьшению разрешения, что менее вероятно при Т = 30 "С и %В =
= 38%. Поэтому можно ожидать, что разделение в этих условиях будет более надежным
и устойчивым. Все перечисленные соображения сразу становятся очевидными, если
посмотреть на рис. 10.36.
Пользователь и далее может продолжать моделирование, например, изменить
параметры колонки, не проводя при этом дополнительных лабораторных
экспериментов. Обратите внимание, что на экране отображается значение давления 147 бар
(2131 psi), что позволяет избежать условий, при которых создается избыточное давле-
508
Глава 10. Разработка метода с помощью программного обеспечения
*!«.1<Ча||.
(a)
- ■■'■! М !=!'!-. --I !
**и
Illlll
Illlll
ЛЙ1- J gj
,— ГЛВ- —
■ни [3 fcfxtfe hf
L-5
sT :Г
L-?
Ml
■—par-.
э .';^., ^
S
ГЦ
ЯП
*
38 %B.30"C; 25 см, 3 мкм колонка; 1.0 мл/мин
R,= 2.1: P= 204 бар (2960 psi) (•)
JUL
10
Время (мин)
20
Рис. 10.3. Пример ввода данных градиентного элюирования (а) и результатов
компьютерного моделирования с помощью программы DryLab (6). Образец — смесь
восьми кортикостероидов [13|. Условия а (б): колонка Си 250x4,6 мм,
5 мкм; подвижная фаза ацетонитрил-вода 38 %В; 30 °С; 2,0 мл/мин:
(в) компьютерное моделирование для разделения образца в условиях (б)
за исключением изменений размера частиц сорбента (3 мкм) и скорости
потока (1,0 мл/мин)
ние в системе. Поскольку разрешение этого разделения может оказаться
неприемлемым (Rs = 1,3), можно его улучшить, либо увеличив длину колонки, либо используя
сорбент с частицами меньшего размера. На рис. Ю.Зв показан прогноз того, что
получится, если в колонке размерами 250 х 4,6 мм, упакованной пятимикронными
частицами, заменить сорбент на трехмикронный. Из-за увеличения давления вследствие
уменьшения размера частиц необходимо понизить скорость потока с 2 мл/мин
на рис. 10.35 до 1 мл/мин в разделении на рис. Ю.Зв. В результате получаются
приемлемые значения разрешения (Rs = 2,1) и давления 204 бар (2960 psi), хотя время
10.2. Программное обеспечение компьютерного моделирования 509
разделения удваивается, достигая 24 мин. После рассмотрения перечисленных выше
вариантов, разделение, в котором достигнуто наилучшее разрешение, могут все-таки
счесть неприемлемым из-за его слишком большой длительности. В этом случае
можно заменить колонку (или В-растворитель, которым был метанол, заменить ацето-
нитрилом) и всю процедуру повторить (еще четыре экспериментальных разделения,
в которых изменяют время градиента и температуру).
10.2.2. Оптимизация градиента
Четыре экспериментальных разделения на рис. 9.18 соответствуют схеме
эксперимента на рис. 10.25 (изменение температуры и времени градиента). В этом
случае, однако, будем стремиться получить оптимизированное градиентное разделение
(а не изократическое, как на рис. 10.3). Итоговая карта разрешения показана
на рис. 10.4а. Перекрестие (и стрелка) на этой карте разрешения указывают на
условия максимального разрешения (Rs = 2,1): градиент 5—100% за 35 мин при
температуре 47 °С. Полученная в результате хроматограмма показана на рис. 10.46.
Поскольку последнее вещество покидает колонку на 14 минуте (задолго до конца градиента),
градиент можно сократить до 5—43 %В за 14 минут (см. раздел 9.3.4). Ранее пробная
экспериментальная оптимизация времени градиента и температуры для этого
образца методом проб и ошибок привела к «оптимизированным» условиям 5—46 %В
за 14 мин при 50 °С с конечным разрешением Rs = 1,9. В этом примере (рис. 10.4)
компьютерное моделирование дает лишь небольшое улучшение разрешения по
сравнению с полученным вручную методом проб и ошибок. Тем не менее, компьютерное
моделирование подтверждает, что выбранные итоговые условия на самом деле луч-
Rs
И 2.2
1.9
1.6
1.3
1.1
0-8
0.5
| 0.3
0.0
50
45
С 40
35
30
V
V
И
4JL—*
^ «^^м
(а)
10 20 30 40 SO 60
Время градиента (с(мин)
70
80
5-100 %8 за
35 мин; 47 °С
R, = 2.1
в. I9
5т7
6 8 10
Время(мин)
12
|в)
,10 и
14
Рис. 10.4. Компьютерное моделирование с целью оптимизации времени градиента и
температуры разделения. Те же условия и «неправильный» образец — такой,
как на рис. 9.18 (калибровочные градиенты 5—100 %В). (а) Карта
разрешения; (б) оптимизированное разделение
510
Глава 10. Разработка метода с помощью программного обеспечения
шие, поскольку карты разрешения суммируют прогнозы для нескольких сотен
комбинаций условий (различных виртуальных экспериментов).
Дальнейшее изучение карты на рис. 10.4а показывает, что разрешение
уменьшается лишь незначительно при уменьшении времени градиента, особенно в том
случае, если параллельно происходит понижение температуры. Так для градиента
5—46 %В за 12 мин при 45 °С разрешение почти не изменяется (Rs - 2,0), а время
разделения сокращается на 2 мин. Компьютерное моделирование полезно для
точной настройки разделения и, таким образом, обеспечивает максимум разрешения
и/или минимизирует время разделения, проводя дальнейшее сканирование
изменений условий разделения методом проб и ошибок. Минимизация времени разделения
таким способом может быть важна в том случае, если метод планируется
использовать для сотен и тысяч образцов. Надежность метода может быть также проверена
путем небольших изменений в условиях эксперимента. В примере на рис. 10.4а
снижение температуры на два градуса приводит к серьезному ухудшению разрешения
(Rs = 1,3). Нередки колебания фактической температуры в интервале +1—2 °С в тех
случаях, когда разделение выполняется на различных ВЭЖХ-системах (раздел 3.7).
Чувствительность данного метода к случайным изменениям температуры можно
значительно снизить с помощью небольших изменений окончательных величин %В и Т.
При этом потери в разрешении будут минимальными. Например, выбор времени
градиента равным 13 минутам (5—46%) и температуры 48 °С дают разрешение Rs =
= 1,9, но изменение температуры в пределах ±2 °С не приводит к потере разрешения.
То есть, теперь метод устойчив по отношению к умеренным колебаниям
температуры. Такого рода компьютерное моделирование метода проб и ошибок может быть
выполнено за несколько секунд (новых экспериментов при этом не требуется).
За очень короткое время с помощью компьютерного моделирования можно
получить наглядное представление о большом количестве кусочно-линейных градиентов,
что позволяет легко разработать окончательный метод, который устроит пользователя.
На рис. 10.5 показано, как использовать окно программы DryLab для проектирования
форм градиентов. Отправной точкой является предыдущий пример (t(; = 13 мин и
48 °С). В процессе конструирования градиента каждую из трех введенных точек,
указанных стрелками на рис. 10.5, можно перетаскивать с помощью компьютерной мыши
к любым требуемым значениям времени и %В (тем самым изменяя программу
градиента). Одновременно с этим на экране будет отображаться, как на рис. 10.5,
получившаяся хроматограмма. Градиент, показанный на рис. 10.5, сокращает время
разделения до 9,5 мин, сохраняя при этом стабильное качество разделения при небольших
колебаниях температуры (см. соответствующее обсуждение в разделе 9.2.2.5).
%В
100
80
60
40
20
5/30/100 %В за 0/8.75/9.50 мин
Г=48*С:Я5=2.0
ОЬ^тт=
Рис. 10.5. Компьютерное моделирование метода проб и ошибок для подбора
кусочно-линейного градиента (с целью оптимизации разделения на рис. 10.460
10.2. Программное обеспечение компьютерного моделирования 511
10.2.3. Другие возможности
Программное обеспечение, с помощью которого производится моделирование,
может быть полезным хроматографисту рядом других опций, приведенных в
таблице I0.I (см. пункты 4—9).
Таблица 10.1. Оииин компьютерного моделирования
Опция
]. Моделирование хроматограм.ч для разного
%В в изократических или разных
градиентных условиях и температуры
2. Использование карт разрешения для
облегчения выбора оптимизированных условий
3. Выбор наилучших параметров колонки
4. Предсказание изократических условий
на основе градиентных разделений
5. Выбор определенного пика
6. Изменение других условий
7. Подбор на компьютере
кусочно-линейного градиента
8. Уширение заднего фронта пика
9. Методика двух разделений для улучшения
разрешения
Комментарии
Раздел 10.1
Раздел 10.1
Раздел 10.1
Моделирование изменений /с и 7* тоже
позволяет предсказать зависимость изократического
элюирования от %В и Г (разделы 10.2.1, 10.2.3.1)
Rs и Д^-карты рассчитываются только для пиков,
представляющих интерес. Пики, не
перекрывающиеся с пиком, представляющим интерес,
не рассматриваются (раздел 10.2.3.2)
Разделение можно моделировать для любых
факторов, влияющих на селективность (таблица 2.2).
При замене колонки для моделирования
необходимы реальные экспериментальные данные,
полученные на этой колонке (раздел 10.2.3.3)
Возможен и ручной, и автоматический подбор
(раздел 10.2.3.4)
Может моделироваться (раздел 10.2.3.5)
Различные градиенты для различных пиков
одного и того же образца (раздел 10.2.3.6)
10.2.3.1. Прогнозирование изократического элюирования
на основе данных градиентного элюирования
После того, как проведено компьютерное моделирование на основе двух градиентных
экспериментов, в которых менялось только время градиента, можно также
прогнозировать зависимость изократического разделения от %В. Так же, как после проведения
четырех экспериментов (см. рис. 10.25) можно предсказать зависимость
изократического разделения от %В и температуры, что проиллюстрировано на рис. 10.3. Эта
возможность предсказывать изократические разделения на основе градиентных
экспериментов позволяет оценить изократическое элюирование как возможную альтернативу
градиентному разделению. По мере того, как диапазон к для образца уменьшается,
изократическое элюирование становится все более предпочтительным. Прогнозы
относительно изократического разделения, сделанные на основе градиентных данных,
несколько менее надежны по сравнению с прогнозами, основанными на
предварительных изократических разделениях. Таким образом, если из данных
первоначального градиентного эксперимента можно сделать вывод, что изократическое элюирование
предпочтительнее (раздел 9.3.1), то лучше провести четыре дополнительные
изократические разделения (как на рис. 10.2а), а не градиентные как на рис. \0.26. Более
512
Глава 10. Разработка метода с помощью программного обеспечения
подробно прогнозирование изократического элюирования на основе градиентных
разделений обсуждается в |14|. Следует отметить, что хотя параметры изократического
разделения можно предсказать, основываясь на результатах градиентных разделений,
прогнозировать градиентное элюирование на основе изократических разделений
неудобно. Для облегчения адаптации метода к требованиям пользователя имеет смысл
начинать разработку метода с градиентных экспериментов.
10.2.3.2. Выбор определенного пика
Многие хроматограммы содержат пики, не представляющие непосредственного
интереса для хроматографиста. Например, анализ биологических образцов или проб
из окружающей среды может быть осложнен наличием многочисленных мешающих
пиков. Аналогичным образом в препаративном разделении (глава I5) мы часто
имеем дело с очисткой и выделением одного единственного вещества в образце. В том
случае, если интерес представляют только некоторые пики на хроматограмме, важно
добиться разрешения каждого из этих пиков со всеми остальными ненужными
пиками, но не разрешения перекрывающихся пиков примесей между собой, поскольку
в этом нет необходимости. В качестве иллюстрации процесса выбора определенного
пика рассмотрим пример на рис. 10.4 и предположим, что подлежат анализу только
пики 3,8 и 10 в присутствии оставшихся «ненужных» пиков. Таким образом,
потребуется окончательное разделение, в котором эти три пикя будут разрешены друг
с другом и отделены от всех других пиков, которые могут перекрыть пики 3,8 и 10
(это гораздо более простая задача, чем отделение всех 11 пиков друг от друга).
Используя компьютерное моделирование, мы можем обозначить пики 3,8 и 10
как пики, представляющие интерес. Когда потребуется карта разрешения (для пиков
3, 8 и 10 в этом примере), она будет содержать графики зависимости величин
критического разрешения Rs от температуры и времени градиента только для пиков 3,8
и 10. Это проиллюстрировано на рис. 10.6а, на нем отмечены наилучшие условия
для разделения (перекрестия и стрелки). Соответствующая хроматограмма показана
на рис. 10.66 (целевые пики пронумерованы). Как и ожидалось, возможное
критическое разрешение лишь трех из одиннадцати пиков гораздо выше (Rs = 4,9), чем при
разделении всех одиннадцати (Rs = 2,1 на рис. 10.4). Кроме того, на такое разделение
потребуется меньше времени (11 мин, а не 14). Полученное «избыточное»
разрешение можно «обменять» на сокращение времени разделения, изменив параметры
колонки. Например, уменьшение длины колонки наполовину и удвоение скорости
потока сокращает время разделения до трех минут, при том, что разрешение остается
Rs = 3,0 и давление в колонке не изменяется.
10.2.3.3. Изменение других условий
Другие условия, влияющие на селективность, можно моделировать на компьютере,
изменяя либо одно, либо два вида условий одновременно. В дополнение к вариантам
на рис. 10.2, можно моделировать любое сочетание %В или времени градиента,
температуры, смеси В-растворителей, рН подвижной фазы, буфера или концентрации
ион-парного реагента для ОФХ. Кроме того, можно моделировать изменение
большинства влияющих на селективность условий в нормально-фазовых и
ионообменных разделениях.
10.2.3.4. Компьютерный подбор наилучшего кусочно-линейного
градиента
Помимо промывки колонки, как это показано на рис. 9.I10, двухступенчатые
градиенты используются, в основном, для одной из трех общих задач. Во-первых,
разрешение группы выходящих близко друг к другу пиков в начале градиента часто может
10.2. Программное обеспечение компьютерного моделирования
С 40
я5
4.9
4.3
3.7
3.1
25
1.9
1.2
0.6
0.0
5-100 %В за 22 мии
49T;Ra = 4.9
(только пики 3. в, 10)
40 60 80
Время градиента Г0(мин)
100
(б)
±ик
I10
Li
4 6
Время (мин)
Т
10
Рис. 10.6. Оптимизация разделения отдельных пиков в «неправильном» образце
рис. 10.4. Использование компьютерного моделирования для выбора
оптимальных величин времени градиента и температуры для отделения пиков
3,8 и 10 от остальных пиков, (а) Карта разрешения; (6) наилучший вариант
разделения для градиента 5—100% за 22 мин при температуре 49 °С
быть улучшено изократической задержкой перед началом градиента (см., например,
рис. 9Эв,г). Во-вторых, разделение критических пар пиков в начале и в конце
разделения можно улучшить за счет использования ступенек/сегментов градиента
различной крутизны (как, например, на рис. 9.12а,б), если на этих пиках по-разному
отражаются изменения крутизны градиента. В-третьих, время разделения с избыточным
разрешением в конце часто можно сократить с помощью более крутого градиента,
поджимающего окончание хроматограммы (например, рис. 10.5). Компьютерное
моделирование может обеспечить тестирование большого количества двухступечатых
градиентов методом проб и ошибок (как на рис. 10.5), а затем выбор дающего
наилучшую селективность градиента, который, в свою очередь, обеспечит максимальное
разрешение и/или сократит время разделения. Однако, применение этого подхода
к нескольким образцам |15| приводит к выводу, что преимущество двухступенчатых
градиентов для дальнейшего повышения разрешения (как в примере на рис. 9.126)
чаще всего минимально, поскольку с помощью кусочно-линейного градиента редко
удается улучшить разрешение больше, чем на пол-единицы. Кусочно-линейные
градиенты для максимального увеличения разрешения можно разработать
автоматически с использованием специального программного обеспечения [15—18|.
По сравнению с одновременной оптимизацией времени градиента и
температуры разделения (что является предпочтительным подходом), использование
кусочно-линейного градиента только для увеличения разрешения, как представляется,
514
Глава 10. Разработка метода с помощью программного обеспечения
дает мало преимуществ. Небольшим исключением является использование
кусочно-линейных градиентов для разделения образцов, содержащих большие молекулы,
такие как белки, {раздел 9.2.2.5). Недостатком кусочно-линейных градиентов
является то, что они могут затруднить перенос метода вследствие градиентного
«закругления» (раздел 3.10.1.2; |19|).
10.2.3.5. Уширение заднего фронта пика
Уширение заднего фронта пиков следует устранять до того, как будут проведены
экспериментальные разделения в процессе разработки метода. Однако, для некоторых
образцов это может быть невозможно. Поскольку уширение заднего фронта пиков
может сильно влиять на разделение, при компьютерном моделировании необходимо
этот факт учитывать. Некоторые примеры уширения заднего фронта пиков показаны
на рис. 2.16 и 2.17. Все они были созданы с помощью компьютерного
моделирования программой DryLab. В то время как умеренное уширение заднего фронта пика
обычно оказывает небольшое влияние на разделение и разрешение, ситуация
радикально изменяется, если размеры пиков сильно различаются (сравните рис. 2Л1д и
рис. 2.17г). В этих случаях важно, чтобы программное обеспечение обеспечивало
адекватное моделирование уширения заднего фронта пика, наблюдаемого в
предварительных разделениях.
10.2.3.6. Метод двух разделений для улучшения разрешения
образца
Когда образец содержит 15 или более компонентов, достичь приемлемого
разрешения (например, Rs >2; см. раздел 2.3.4) в одном градиентном разделении может быть
трудно. Альтернативным, иногда успешным, подходом для работы с такими
образцами может быть использование различных градиентных процедур (или разделений)
для одного и того же образца [20] без замены колонки или элюентов А и В.
Например, для двух разделений можно использовать различные времена градиента tq и
различные температуры Т, позволяющие проводить разделения без какого-либо
дополнительного вмешательства оператора в процессе анализа ряда образцов. Все образцы
сначала анализируют в одних условиях, а затем проводят анализ, используя другие
условия (иные значения /g и Т). Цель таких экспериментов — приемлемое
разрешение для каждого представляющего интерес пика в одном или в другом из этих двух
разделений. Результаты этих разделений позволяют потом проанализировать все
пики в образце. С помощью компьютерного моделирования относительно несложно
выбрать наилучшие условия для каждого из двух разделений, чтобы максимально
увеличить разрешение для веществ, входящих в состав данного образца |20|.
Аналогичный подход, использующий два «дополняющих друг друга» разделения, был
описан для изократического разделения |21|.
10.2.3.7. Примеры использования компьютерного
моделирования при разработке метода
Примеров компьютерного моделирования в литературе описано много, их обзор
приведен в |22| и на стр.119 в \2\. Многие из этих работ иллюстрируют возможности
или точность компьютерного моделирования. В других публикациях сообщается
об использовании компьютерного моделирования для реальной разработки
рутинного метода. Пятнадцать примеров применения компьютерного моделирования при
разработке реальных методов сведены в таблице 10.2.
10.2. Программное обеспечение компьютерного моделирования 515
Таблица 10.2. Примеры компьютерного моделирования как составляющей разработки
метопов для различного практического применения
Функция при разработке
метода
Выбор определенного пика;
оптимизация времени
градиента [8|
Оптимальный
кусочно-линейный градиент |8|
Оптимальный
кусочно-линейный градиент |23|
Оптимальный кусочно линей
ный градиент |8, 18, 24|
Изменения 7" и fa с заменой
растворителя В |8, 251
Предсказания изократических
разделений на основе
градиентных данных [8|
Выбор оптимального
сочетания колонки и времени
градиента |26|
Выбор оптимального сочета
ния колонки и В-растворите-
ля |27|
Выбор оптимального
сочетания колонки, температуры и
времени градиента [28|
Изменения времени
градиента и соотношения ACN и
МеОН в В-растворителе |29]
Простое изменение времени
градиента или %В в
изократических разделениях |30|
Результаты (комментарии)
а. Определение единичной примеси в образце, содержащем
>15 компонентов
б. Препаративная очистка одного компонента
в. Определение трех компонентов в образце, содержащем
до 10 родственных соединений
Анализ 7-компонентного образна
С помощью многоступенчатых кусочно-линейных градиентов
удалось (в первый раз) разделить все 19 рибосомных белков
субчастицы 30S; аналогичным образом удалось разделить 33
из 34 рибосомных белков субчастицы 50S
Разделение 7-, 12-, и 19-компонентных образцов с помощью
кусочно-линейных градиентов
Если исходный растворитель В был выбран неудачно и цель
разделения не была достигнута, изменения Г и ^повторялись
с другими растворителями (ACN, МеОН или изопропанол)
Компьютерное моделирование изменений Г и Iq обеспечило
избыточное разрешение в изократическом режиме, далее были
изменены длина колонки и скорость потока для сокращения
времени разделения
Были протестированы 16 колонок с целью получить наилучшее
разделение 6- и 8-компонентных образцов
На трех колонках с использованием в качестве В растворителей
ацетонитрила и метанола было проведено по два градиентных
разделения (<с= 20 и 50 мин). Подобрав оптимальное сочетание
колонки и элюента, разделение совершенствовали далее,
используя экспериментальную схему Планкетта-Бэрманна
Время градиента и температура были оптимизированы
для 9 колонок, что позволило выбрать наилучшие условия для
различных целей разделения
Состав растворителя В изменялся в интервале 0—100%
ACN/MeOH (4 смеси растворителей). Каждая из этих смесей
использовалась в разделениях при двух временах градиента
для достижения оптимального разделения 8-компонентного
образца
Четыре примера для различных образцов
10.2.4. Отслеживание пиков
В процессе разработки метода при изменении условий, которые могут изменить
относительное удерживание, важно следить за тем, где какой пик элюируется на
каждой из хроматогромм. Способ отслеживания пиков обсуждался в разделе 2.7.4.
Отслеживание пиков еще важнее в случае компьютерного моделирования потому, что
ошибки в привязке пиков часто приводят к большим ошибкам в прогнозируемых
разделениях. Автоматическое отслеживание пиков, основанное на относительном
516
Глава 10. Разработка метода с помощью программного обеспечения
удерживании и подсчете площадей пиков, включено в программное обеспечение
DryLab. Поскольку эта программная опция предназначена для относительно простых
разделений (< 10 пиков, ограниченные изменения относительного удерживания,
не более чем десятикратные изменения площади пика), ее точность может быть
повышена при использовании дополнительного программного обеспечения (Peak
Match®; Molnar Institut, Берлин). Для образцов с большим количеством
компонентов, содержание которых отличается на несколько порядков, может потребоваться
масс-спектрометрическое детектирование. После того, как пики соотнесены с
определенными временами удерживания в исходных экспериментах, они отслеживаются
автоматически при последующем моделировании. Хотя точно отслеживать пики при
использовании компьютерного моделирования очень важно, ошибки в отслежива-
нии обнаружить нетрудно, если сравнить прогнозируемые и экспериментальные
разделения (в случае ошибки предсказанные времена удерживания для одного или
нескольких пиков не будут коррелировать с соответствующим экспериментом).
10.2.5. Источники коммерчески доступного программного
обеспечения для компьютерного моделирования
хроматографических разделений
Программное обеспечение для компьютерного моделирования продается рядом
компаний и групп. Например, ACD/LC Simulator® (Advanced Chemistry Development,
Toronto, Ontario, Canada); ChromSmart® (Agilent, Palo Alto); ChromSword® (Merck
KGaA, Darmstadt, Germany); DryLab® (Molnar Institut, Berlin); Turbo Method
Development (PerkinElmer, Shelton, CT); OsirisR |3I|; и Preopt-WR [32|. В большинстве своем
область применения этих программных пакетов ограничена ОФХ.
Кроме того, для прогнозирования разделений в ионной хроматографии доступно
программное обеспечение Virtual Column (Dionex, Sunnyvale, CA). Можно также
ознакомиться с несколькими обзорами и/или сравнительными описаниями
программного обеспечения по этой тематике [9—II, 33|. Подробный обзор некоторых
технических требований к компьютерному моделированию дан в |34]. (Новые
программные продукты появляются регулярно, в то время как старые прекращают
выпускать, и поэтому этот список ссылок неполон и скоро устареет).
10.3. Другое программное обеспечение
для разработки метода
Помимо описанного выше программного обеспечения для компьютерного
моделирования доступно и другое программное обеспечение для разработки метода
разделения. Некоторые возможности такого программного обеспечения включают в себя:
• предсказание удерживания анализируемых веществ, исходя из их молекулярной
структуры;
• предсказание величин рКа анализируемых веществ, исходя из их молекулярной
структуры;
• подбор обращенно-фазовых колонок со сходной или различной селективностью;
• экспертные системы для разработки метода.
В разделах 10.3.I —Ю.3.5 кратко рассмотрено программное обеспечение для этих
приложений, но и этим не исчерпываются возможности компьютерного
моделирования для разработки метода.
10.3. Другое программное обеспечение для разработки метода 517 )Jj
10.3.1. Удерживание анализируемого вещества
и его молекулярная структура
История попыток увязать хроматографическое удерживание молекул анализируемого
вещества с его молекулярной структурой довольно долгая. В принципе, если можно
было бы предсказать удерживание анализируемого вещества, опираясь на данные
о его молекулярной структуре и об экспериментальных условиях (подвижная и
неподвижная фазы, температура), не было бы нужды в проведении реальных разделений
в ходе разработки метода. Как уже обсуждалось в разделе 2.6.7, возможность
надежных предсказаний такого рода представляется делом отдаленного будущего. Тем
не менее, программное обеспечение для такого рода предсказаний уже предлагалось
неоднократно и, вероятно, будет предлагаться в будущем.
ChromSword и ACD/LC Simulator (см. раздел Ю.2.5) представляют собой
альтернативный вариант программного обеспечения, в которых одно или два
экспериментальных разделения заменяются предсказаниями удерживания, сделанными на основе
данных о молекулярной структуре анализируемого вещества. Независимая оценка этой
возможности |М| привела к выводу о том, что «предсказания, сделанные только
на основе данных о молекулярной структуре, не очень точны и вряд ли дадут полезную
информацию о разделении». Такое программное обеспечение было доступно для более
специализированных приложений (для гидролиза белка [35], метаболизма
лекарственных средств [36]), но его надежность находится под вопросом. Более поздние попытки
прогнозирования ОФХ-разделений несколько более перспективны |37|, но преследуют
другую цель: идентификацию пика в тех случаях, когда используется масс-спектромет-
рическое детектирование. Для этого случая необходима намного меньшая точность
прогноза, чем та, которая требуется для оптимизации разрешения.
10.3.2. Величины рКа анализируемого вещества
и его молекулярная структура
Величины рКа анализируемого вещества обуславливают зависимость удерживания
от рН подвижной фазы. Следовательно, знание величин рКа соединений,
присутствующих в образце, может оказаться полезным для планирования и интерпретации
экспериментов по разработке метода (раздел 7.2). Программное обеспечение для
поиска или оценки величин рКа (в воде при 25 °С) можно получить у следующих
разработчиков: Advanced Chemistry Development,Toronto, Ontario, Canada; Intertek ASG
Laboratory, Manchester, UK. Однако, как упоминалось в разделе 7.2.3, значения рКа
изменяются в зависимости от температуры и состава подвижной фазы.
Использование на практике величин рКа, рассчитанных с помощью программного обеспечения,
может иметь ограниченное применение поскольку, как правило, используются
водно-органические подвижные фазы, и температура может быть различной.
Эксперименты, в которых варьируется рН подвижной фазы, обычно дают для разработки
метода более надежные оценки значений рКа образца (см. раздел 7.2 и примеры
на рис. 7.3). В большинстве случаев приблизительные величины рКа различных
функциональных групп или классов соединений, приведенные в таблице 7.2,
должны быть удовлетворительны для разработки метода.
10.3.3. Селективность ОФХ-колонки
Селективность ОФХ-колонки может быть охарактеризована пятью параметрами
(раздел 5.4.1). Соответствующее программное обеспечение (ColumnMatch®, Molnar
Instilut, Berlin) обеспечивает возможность сравнения селективности различных коло-
518
Глава 10. Разработка метода с помощью программного обеспечения
нок, что, в свою очередь, позволяет выбирать колонки с похожей селективностью
на замену или колонки с сильно различающейся селективностью в тех случаях, когда
необходимо изменить относительное удерживание (раздел 5.4).
10.3.4. Экспертные системы для разработки метода
Подходы к разработке метода в полностью автоматическом режиме с помощью
программного обеспечения (экспертных систем) исследуются с середины восьмидесятых
годов |9, 38|. В идеальном случае информация о природе образца и целях разделения
вводится в компьютер, и экспертная система рекомендует условия первоначального
разделения. Образец вводится в колонку, система обрабатывает результаты первого
разделения и выбирает дополнительные эксперименты, используемые для
последующего моделирования — до тех пор, пока не будет получено успешное разделение.
Любые вопросы, возникающие в ходе разработки метода, будут решены с помощью
компьютера. Предыдущие попытки создания экспертных систем либо не стали
коммерчески успешными, либо ограничивались оптимизацией разделения после того,
как пользователь выбрал начальные условия |39|. Тем не менее, такой подход очень
привлекателен, и в будущем непременно будут внедрены усовершенствованные
программные продукты.
10.4. Компьютерное моделирование
и разработка метода
Компьютерное моделирование не предназначено для замены различных способов
разработки метода, описанных в предыдущих главах. Скорее, компьютерное
моделирование должно расширять возможности проведения подбора условий методом проб
и ошибок в процессе разработки. Зачастую выбор условий для приемлемого
разрешения при минимуме времени на разделение является основной целью при
разработке метода, и компьютерное моделирование может быть особенно эффективным
для ее достижения. Два примера подобного использования компьютерного
моделирования представлены в разделах 10.4.1 и 10.4.2.
10.4.1. Пример 1: разделение смеси производных
лизергиновой кислоты
Рассмотрим разработку ОФХ-метода для образца, содержащего двенадцать
производных диэтиламида лизергиновой кислоты (ЛСД). Сначала было проведено
градиентное разделение в условиях, рекомендованных в таблице 9.3 (рис. 10.7а).
Из рассмотрения этой исходной хроматограммы с десятиминутным градиентом
следует, что предпочтительным вариантом является градиентное элюирование
(раздел 9.3.1, A 1r/1(; = 0,33), хотя изократическое элюирование тоже возможно. Поэтому
следующим шагом является проведение тридцатиминутного градиента (при
неизменных прочих условиях). Это разделение показано на рис. 10.76. Разрешение (Л, = 0,6)
неприемлемо в обоих проведенных разделениях, но с помощью компьютерного
моделирования можно подобрать время градиента, при котором достигается максимум
разрешения. Карта разрешения на рис. 10.7е указывает два «оптимальных» времени
градиента: 19 мин (Rs = 0,9) и 80 мин (Rs = 1,1).Среднее значение к' = б при tc =
= 19 мин предпочтительнее, чем к' = 25 при 1q = 80 мин. В общем случае
предпочтительнее меньшее время градиента.
10.4. Компьютерное моделирование и разработка метода 519
5-100 %В за 10 мин
35 "С
Rs = 0.6 ,1
(в)
5-100 %В за 30 мин
35 'С 1
Rs = 0.6
Время(мин)
5-1,6
<б)
%!_jdt
Время (мин)
1.0 -
0.5 -,
40 SO 80
Время градиента (а(мин)
5-20 %В за 50 мин; 35 °С
300 мм колонка; 0.7 мл/мин
fia=1.7
100
(г)
I I ll I ll I
20
40
Время (мин)
Рис. 10.7. Иллюстрация к стратегии разработки метода, основанной на использовании
компьютерного моделирования для выбора окончательных условий
разделения. Образец: смесь 12 производных диэтиламида лизергиновой кислоты
(ЛСД). Условия: колонка Си; градиенты ацетонитрил/вода; другие условия
меняются, (а) колонка Си 100x4,6 мм, 3 мкм; 5—100 %В за 10 мин; 35 °С,
2,0 мл/мин; (б) то же, что и в (а) за исключением градиента: 5—100 %В
за 30 мин; (в) карта разрешения; (г) окончательное разделение в условиях,
указанных на рисунке. Смоделированные разделения основаны на данных |13|
Поскольку разрешение все еще далеко от удовлетворительного, лучшим
следующим шагом является изучение подходов к изменению селективности. Авторы
рекомендуют провести два дополнительных экспериментальных разделения, в которых
изменяется температура. Эти два разделения в совокупности с разделениями,
показанными на рис. \а.1а,6, приводят к схеме экспериментов на рис. 10.25 (времена
градиентов 10 и 30 мин, температуры 35 и 55 °С). К сожалению, такая одновременная
оптимизация времени градиента и температуры не помогла улучшить разрешение.
520
Глава 10. Разработка метода с помощью программного обеспечения
Так как на этом этапе разработки метода разрешение неудовлетворительно (Rs =
= 0,9), необходимо продолжить поиски путей улучшения разрешения, изменяя либо
селективность, либо параметры колонки. Изменять селективность можно заменой
В-растворителя (например, метанол вместо ацетонитрила), колонки (раздел 5.4.3)
или рН подвижной фазы (для образцов, содержащих кислотные или оснбвные
соединения). При использовании компьютерного моделирования для изменения
параметров колонки и времени градиента не требуется дополнительных экспериментов, и
поэтому эту возможность нужно использовать в первую очередь. Простейшим
изменением параметров колонки является увеличение ее длины, сопровождающееся
уменьшением скорости потока, необходимым для того, чтобы давление оставалось
прежним. Для данного образца колонка длиной 300 мм при скорости потока
0,7 мл/мин дает разрешение Rs = 1,7, но при этом время градиента возрастает
до 171 мин.
Следующим шагом может быть уменьшение диапазона градиента с тем, чтобы
сэкономить время. На рис. 10.1г показано полученное разделение в градиенте 5—20 %В
за 50 мин, в котором Rs = 1,7. В попытке достичь более удачного соотношения между
разрешением и временем разделения изменяли форму градиента, но эти изменения не
дали желаемых результатов. Учитывая сложность разделения, можно полученное
разрешение считать приемлемым. Однако, если пятидесятиминутная продолжительность
разделения неприемлема (из-за большого количества анализируемых образцов),
нужно, как было предложено выше, не оставлять попыток оптимизировать селективность.
Любое увеличение разрешения за счет роста селективности можно «обменять» на
более короткое время разделения (разделы 6.4.2, 9.3.6).
Хотя результат, полученный в примере на рис. 10.7, выглядит несколько
разочаровывающим, объем экспериментальных усилий, затраченный на исследования, был
минимален — всего четыре экспериментальных разделения. Немного времени
потребовалось и на компьютерное моделирование. В то же время было отсеяно несколько
неудачных вариантов, что позволило хроматографисту сосредоточиться на более
перспективных направлениях разработки.
10.4.2. Пример 2: альтернативная стратегия разработки
метода
В предыдущем примере акцентируется внимание на оптимизации времени
градиента и/или температуры с целью оптимизации селективности. Другие аналогичные
схемы разработки метода проиллюстрированы на рис. 10.2. Отличный от этого
подход заключается в изменении для каждого набора условий одного условия
за один раз (например, типа колонки) при оптимизации либо %В, либо времени
градиента. В работе |27| описан пример такого подхода для случая десятикомпо-
нентной смеси фармацевтических субстанций, содержащей три активных
ингредиента и семь примесей или продуктов разложения. Ни одно из растворенных
веществ не ионизируется, и поэтому буферные растворы в качестве элюентов
не понадобились. Этот образец потребовал градиентного элюирования. Подход
к разработке метода описан на рис. Ю.8й.
В первоначальных экспериментах оценивались возможности разделения на трех
различных колонках: NovaPak CIS, Luna C18, и Discovery RP Amide C16. За 20 и за
50 мин были проведены градиентные разделения от 10 до 90 %В с использованием
ацетонитрила или метанола в качестве В-растворителя (12 исходных предварительных
разделений). Результаты, полученные в каждой из шести пар экспериментов для
данной колонки и данного В-растворителя, использовались в качестве источника
данных для компьютерного моделирования (DryLab), чтобы с его помощью установить
10.4. Компьютерное моделирование и разработка метода
Подход к разработке метода [27]
Выбрать 3 различные
колонки
NovaPakC18
LunaCIS
Discovery RP Amide C16
10-90 %B за 20 и за 50 мин
В = МеОН или ACN (4
эксперимента для каждой
колонки)
Изменить содержание
растворителя В
25% MeOH/ACN
50% MeOH/ACN
75% MeOH/ACN
(а)
компьютерное
моделирование
Наиболее обещающие
результаты на колонке
Discovery RP Amide C16
Оптимальный вариант: содержание
растворителя В (50% MeOH/ACN) и
диапазон градиента (45-68 %В за 23 мин)
смоделированное разделение
Jd_l
(б)
0 5 10 15
экспериментальное разделение
L л
20 (мин)
jLaU.
(в)
10
15
20 (мин)
Рис. 10.8. Разделение десятикомпонентной смеси фармацевтических су&станций с
использованием компьютерного моделирования, (а) Последовательность
экспериментов; (б) смоделированное заключительное разделение (DryLab);
(в) экспериментальное заключительное разделение. Условия одинаковы для
(6) и (в): колонка Discovery RP Amide Об 250 х 4,6 мм; 45—68 %В за 23 мин
(В — 50% MeOH/ACN); 40 °С; 1,0 мл/мин [27|
оптимальное время градиента. Наиболее перспективные результаты были получены
на колонке Discovery RP Amide CI6 с ацетонитрилом в качестве В-растворителя.
Все же одна пара соединений при этом давала на хроматограмме перекрывающиеся
пики (Rs * 0). Однако сравнение разделений с разными В-растворителями на колонке
Discovery RP AmideC 16 давало основание предположить, что смесь ацетонитрила и
метанола в качестве В-растворителя позволит разделить все десять соединений образца.
Были протестированы смеси с 25, 50 и 75% MeOH/ACN в качестве В-растворителя.
Это привело к удовлетворительному разделению образца с 50%MeOH/ACN. В конце
концов, диапазон градиента был оптимизирован (45—68 %В за 23 мин), и
предсказанное разделение (рис. 10.86) было проведено, оно показано на рис. 10.8е.
Подобный подход к разработке метода описан в работе |40|, когда были оценены
двенадцать различных колонок. При этом изменялись температура и подвижные
фазы. Компьютерное моделирование использовалось для интерпретации результатов
и как ориентир при проведении дальнейших экспериментов. Однако такое большое
количество разделений при разработке метода редко бывает необходимым. Особенно
это касается случаев с небольшим количеством колонок с различной
селективностью, описанных в разделах 5.4.3, 6.3.6.2 и 7.3.2.4 (см. также |41|).
522 Глава 10. Разработка метода с помощью программного обеспечения
10.4.3. Проверка устойчивости метода
После выбора экспериментальных условий для разработанного метода, необходимо
проверить его устойчивость (робастность). Компьютерное моделирование может
использоваться для анализа влияния непредусмотренных изменений различных
условий, например:
• объема задержки системы Vq (только для градиентных методов);
• рН подвижной фазы;
• температуры;
• %В в подвижной фазе;
• скорости потока.
Эти пять параметров разделения указаны приблизительно в порядке убывания их
влияния на устойчивость метода.
Величины Vq варьируются от одной ВЭЖХ-системы к другой, что может привести
к значительным изменениям в случае градиентного разделения некоторых образцов
(раздел 9.2.2.4). С помощью компьютерного моделирования легко проанализировать
влияние изменения Vq на градиентное разделение. Для уже описанного метода
разделения производных ЛСД (рис. 10.7г) объем задержки системы составлял 1,1 мл.
Эффект от изменения Vq в интервале 0—5 мл (диапазон, который должен охватывать
большинство используемых сегодня систем) не превышает ±0,1 единицы Rs.
Следовательно, указанный метод устойчив к изменениям Кд. Компьютерное
моделирование особенно полезно при разработке градиентных методов, нечувствительных к
часто встречающимся изменениям Vjy от системы к системе (раздел 9.2.2.4).
Чрезмерная чувствительность к изменениям рН подвижной фазы является
распространенной причиной неустойчивости метода (см. раздел 7.3.4.1). С помощью
компьютерного моделирования, используемого для оптимизации рН подвижной
фазы, можно определять и устойчивость метода к небольшим изменениям рН.
Температуру ВЭЖХ-системы можно контролировать различными способами
(см. раздел 3.7), но возможны небольшие отклонения в ту или иную сторону от
выбранного значения. Компьютерное моделирование и в этом случае можно
использовать для определения влияния температурных изменений на разделение. Для метода
разделения производных ЛСД, описанного выше (рис. 10.7г), изменение
температуры в интервале ±2 °С приводит к ухудшению в разрешении не более, чем на 0,1
единицу Rs. Следовательно, этот метод устойчив к небольшим изменениям температуры,
которые происходят, в частности, при переходе от одной хроматографической
системы к другой.
В случае, если %В в подвижной фазе обеспечивается смешиванием в потоке, он
может изменяться на 1—2%. Для изократических методов, когда компьютерное
моделирование использовалось для выбора оптимизированного %В, влияние изменения
%В на +2% тоже можно предсказать с помощью моделирования. Сходный эффект
изменения %В при градиентном элюировании можно просчитать с помощью
компьютерного моделирования, вводя в программу значения %В, не входящие в диапазон
градиента (например, в случае градиента 5—100 %В, проверяются градиенты
4—100%, 6—100% и т.д.). В примере с ЛСД-производными колебания %В в
интервале ±2% приводят к ухудшению в разрешении < 0,1 единицу Rs.
Величина скорости потока обычно контролируется с точностью ±1—2% в
большинстве систем ВЭЖХ. Для ЛСД-метода, описанного выше, эффект изменения скорости
потока на ±2% можно точно так же, как и все остальное, просчитать с помощью
компьютерного моделирования: он приведет к ухудшению в разрешении < 0,1 единицу Rs.
Следует заметить, что использование компьютерного моделирования для
прогнозирования устойчивости (робастности) метода не отменяет необходимости под-
10.4. Компьютерное моделирование и разработка метода 523
тверждать эту устойчивость экспериментально (раздел 12.2.6). Тем не менее,
моделирование может использоваться при разработке метода, чтобы избежать проведения
разделения в нестабильных условиях, а также для выбора приемлемого диапазона
изменений каждого параметра при тестировании. Например, если моделирование
предполагает устойчивость рН только в пределах +0,2 единицы, экспериментальную
проверку робастности лучше проводить, изменяя рН именно на +0,2 единицы,
а не на 0,3, поскольку в этом случае результаты могут быть отрицательные.
10.4.4. Заключение
Какой бы подход ни применялся в процессе разработки метода, компьютерное
моделирование может заменить многие из экспериментальных шагов, необходимых
для оптимизации окончательного разделения, уменьшая затраченные усилия и
облегчая выбор оптимальных конечных условий. Компьютерное моделирование было
значительно усовершенствовано с момента его появления в 19S5 году, и теперь
позволяет получать более точные прогнозы разделения для более широкого диапазона
экспериментальных условий. Можно предполагать, что в будущем компьютерное
моделирование станет неотъемлемой частью системы ВЭЖХ, отвечающей за
автоматическую разработку метода. Однако для разработки метода в случае сложных
разделений лучше всего использовать компьютерное моделирование в сочетании с опытом
хроматографиста. Перефразируя слова Уинстона Черчилля, сказанные им во время
Второй мировой войны (об ученых, а не о компьютерах): компьютеры нужно
использовать, но лишь как средство.
Литература
1. P. L. Zhu, J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, J.-T. Lin, L. С Sander, and
L. Van Heukelem, J. Chromatogr. A, 756 (1996) 63.
2. L. R. Snyder and J. W. Dolan, High-Performance Gradient Elution, Wiley-lnterscience, Hoboken,
NJ, 2007.
3. R. J. Laub and J. H. Purnell, J. Chromatogr., 161 (1978) 49.
4. J. L. Glajch, J. J. Kirkland, K. M. Squire, and J. M. Minor, J. Chromatogr., 199 (1980) 57.
5. B. Sacliok, R. С Kong, and S. N. Deming, J. Chromatogr., 199 (1980) 317.
6. J. J. Kirkland and J. L. Glajch, J. Chromatogr., 255 (1983) 27.
7. I. Molnar, J. Chromatogr. A, 965 (2002) 175.
8. L. R. Snyder and L. Wrisley, in HPLC Made to Measure: A Practical Handbook for Optimization,
S. Kromidas, ed., Wiley-VCH, Weinheim, 2006, 56785.
9. J. L. Glajch and L. R. Snyder, eds., J. Chromatogr., 485 (1989)
10. P. J. Schoenmakers, J. W. Dolan, L. R. Snyder, A. Poile, and A. Drouen, LCGC, 9 (1991) 714.
11. T. Baczek, R. Kaliszan, H. A. Claessens, and M. A. van Straten, LCGC Europe, 14 (2001) 304.
12. S. Kromidas, ed., HPLC Made to Measure: A Practical Handbook for Optimization, Wiley-VCH,
Weinheim, 2006, pp. 565—623.
13. J. W. Dolan, L. R. Snyder, N, M. Djordjevic, D. W. Hill, D. L. Saunders, L. Van Heukelem, and
T. J. Waeghe, J. Chromatogr. A, 803 (1998) 1.
14. R. G. Wolcott, J. W. Dolan, and L. R. Snyder, J. Chromatogr. A, 869 (2000) 3.
15. T. Jupille, L. Snyder, and I. Molnar, LCGC Europe, 15 (2002) 596.
16. S. V. Galushko and A. A. Kamenchuk, LCGC Intern., 8 (1995) 581.
17. S. V. Galushko and A. A. Kamenchuk, and G. L. Pit, Amer. Lab., 27 (1995) 33G.
18. V. Concha-Herrera, G. Vivo-Trujols, J. R. Torres-Lapasio, and M. C. Garcia-Alvarez-Coque,
J. Chromatogr. A, 1063 (2005) 79.
19. D. D. Lisi, J. D. Stuart and L. R. Snyder, /. Chromatogr., 555 (1991) I.
20. J. W. Dolan, L. R. Snyder, N. M. Djordjevic, D. W. Hill, L. Van Heukelem, and T. J. Waeghe,
J. Chromatogr. A, 857 (1999) 21.
524
Глава 10. Разработка метода с помощью программного обеспечения
21. G. Vivo-Trayols, J. R. Torres-Lapasio, andM. С. Garcia-Alvarez-Coque, J. Chromatogr. A, 876
(2000) 17.
22. L. R. Snyder and J. W. Dolan, Adv. Chromatogr., 38 (1998) 115.
23. B. F. D. Ghrist, B. S. Cooperman, and L. R. Snyder, J. Chromatogr., 459 (1989) 43.
24. R. Bonfichi, J. Chromatogr. A, 678 (1994) 213.
25. I. Molnar, J. Chromatogr. A, 948 (2002) 51.
26. R. M. Krisko, K. McLaughlin, M. J. Koenigbauer, and С. Е. Lunte, J. Chromatogr. A, 1122 (2006)
186.
27. W. Li and H. T. Rasmussen, J. Chromatogr. A, 1016 (2003) 165.
28. L. Van Heukelem and С S. Thomas, J. Chromatogr. A, 910 (2001) 31.
29. M. R. Eurby, F. Scannapieco, H.-J. Rieger, and I. Molnar, J. Chromatogr. A, 1121 (2006) 219.
30. N. G. Mellisch, LCGC, 9 (1991) 845.
31. S. Heinisch, E. Lesellier, C. Podevin, J. L. Rocca, and A. Tschapla, Chromatographia, 44 (1997)
529.
32. R. Cela and M. Lores, Comput. Chem., 20 (1996) 175.
33. A. Tchapla, Analusis, 20(7) (1992) 71.
34. N. Lundell, J. Chromatogr., 639 (1993) 97.
35. C. T. Man! and R. S. Hodges, in High-Performance Liquid Chromatography of Peptides and
Proteins: Separation, Analysis and Confirmation, С. Т. Manl and R. S. Hodges, eds., CRC Press, Boca
Raton, 1991, p. 705.
36. K. Valko, G. Szabo, J. Rohricht, K. Jemnitz, and F. Darvas, J. Chromatogr., 485 (1989) 349.
37. V. Spicer, A. Yamchuk. J. Cortens, S. Sousa, W. Ens, K. G. Standing, J. A. Wilkins, and
O. V. Krokhin, Anal. Chem., 79 (2007) 8762.
38. T.-P. I, R. Smith, S. Guhan, K. Taksen, M. Vavra, D. Myers, and M. T. W. Hearn, J.
Chromatogr. A, 972 (2002) 27.
39. E. F. Hewitt, P. Lukulay, and S. Galushko, J. Chromatogr. A, 1107 (2006) 79.
40. M. PeiTer and H. Windt, Fresenius, J. Anal. Chem., 369 (2001) 36.
41. D. M. Marchand, L. R. Snyder, and J. W. Dolan, J. Chromatogr. A, 1191 (2008) 2.
ГЛАВА I I
КАЧЕСТВЕННЫЙ
И КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
11.1. Введение 526
11.2. Измерение сигнала 526
11.2.1. Принцип работы интегратора 527
11.2.1.1. Сбор данных 527
11.2.1.2. Распознавание пика 530
11.2.1.3. Интегрирование неидеальных хроматограмм 530
11.2.1.4. Обычные ошибки интегрирования 532
11.2.1.5. Дополнительные советы 533
11.2.2. Удерживание 534
11.2.3. Размер пика 535
11.2.4. Источники ошибок 535
11.2.4.1. Отбор проб и пробоподготовка 536
11.2.4.2. Хроматографическое разделение 536
11.2.4.3. Детектирование 536
11.2.4.4. Измерение пика 537
11.2.4.5. Калибровка 537
11.2.5. Пределы обнаружения и определения 539
11.2.5.1. Предел обнаружения (ПО) 540
11.2.5.2. Нижний предел количественного определения (НПКО или
ПКО) 542
11.2.5.3. Верхний Предел 542
11.2.5.4. Образцы с концентрациями, лежащими вне пределов метода. . 543
11.3. Качественный анализ 544
11.3.1. Время удерживания 544
11.3.2. Качественный анализ в реальном времени 545
11.3.2.1. УФ-детекторы 546
11.3.2.2. Жидкостная хроматография с масс-спектрометрией (ЖХ-МС). 546
11.3.2.3. ИК-спектроскопия с Фурье-преобразованием (ИКФП) 547
11.3.2.4. Жидкостная хроматография с детектором ядерного магнитного
резонанса (ЖХ-ЯМР) 547
11.3.2.5. Хемилюминесцентный азотный детектор (ХЛАД) 547
11.3.2.6. Лазерный детектор светорассеяния (ЛДС) 547
11.3.2.7. Хиральные детекторы 547
11.3.2.8. Качественный анализ в автономном режиме офлайн анализ). . 548
11.4. Количественный анализ 548
11.4.1. Калибровка 548
11.4.1.1. Калибровка внешним стандартом 549
11.4.1.2. Калибровка внутренним стандартом 552
11.4.1.3. Нормирование площадей пиков 554
11.4.1.4. Метод стандартных добавок 555
11.4.1.5. Оценка калибровочных кривых 556
11.4.2. Анализ следовых количеств 558
11.5 Выводы 558
Литература 559
526 Глава II. Качественный и количественный анализы
11.1. Введение
Система ВЭЖХ может давать как качественные, так и количественные данные.
Информация качественного характера позволяет идентифицировать исследуемые
вещества, тогда как количественные данные дают возможность определить, сколько
каждого исследуемого вещества содержится в образце. Три основных фактора влияют
на качество получаемых результатов. Во-первых, все компоненты системы ВЭЖХ
должны работать надежно, обеспечивая повторяемость измерений для одинаковых
образцов, то есть в рамках стандартных методик для таких образцов ВЭЖХ-данные
обязаны быть в достаточной мере близкими как друг к другу, так и к истинным
значениям. Во-вторых, управляющий модуль и соответствующее программное
обеспечение должны преобразовать выходной сигнал детектора ВЭЖХ в достоверную
качественную и количественную информацию. В-третьих, результаты стандартного
использования методик зависят от качества хроматограммы, то есть от разрешения,
формы пика, дрейфа базовой линии и т.д. В этой главе мы рассмотрим случай
идеальной работы хроматографа, когда нет никаких проблем с ВЭЖХ-оборудованием,
с системой сбора и обработки данных, а также с процессом хроматографии. За
исключением некоторых специфических примеров, диагностика неисправностей в
системе ВЭЖХ и способы их устранения сосредоточены в главе 17.
Раздел 11.2 посвящен описанию того, как система сбора данных измеряет сигнал
детектора. В том числе рассмотрены некоторые источники неточностей измерения
сигнала (раздел 11.2.4), а также устанавливаются методические ограничения для обнаружения
и количественного определения сигнала (раздел 11.2.5). Раздел 11.3 (качественный
анализ) охватывает некоторые из методов, используемых для идентификации
анализируемых веществ. Наконец, в разделе 11.4 (количественный анализ) рассмотрено, как надо
использовать полученные данные, чтобы ответить на вопрос «сколько»? Объем этой
главы ограничен, поэтому для получения более подробной информации по многим темам,
затронутым в этой главе, мы отсылаем читателя к литературе по аналитической химии и
математической статистике, а также к ссылкам, приведенным в этой главе. В частности,
ссылка |1| содержит больше информации об интегрировании хроматографических
пиков, чем большинству читателей может понадобиться за всю жизнь.
11.2. Измерение сигнала
ВЭЖХ-детектор (глава 4) — это датчик, который преобразует в электрический
сигнал величину концентрации (или массы) анализируемого вещества' на выходе из
колонки. Далее система сбора и обработки данных отображает этот сигнал в виде
графика зависимости интенсивности сигнала от времени (хроматограммы). Изначально
сигнал может быть представлен либо в аналоговом, либо в цифровом формате.
Для хранения и обработки сигнала используется только его оцифрованное
представление, поэтому аналоговые сигналы должны быть преобразованы в цифровой
формат. Затем программное обеспечение системы сбора и обработки данных или
какое-либо другое, умеющее работать с выходным форматом данных, преобразует
оцифрованный сигнал в хроматограмму, или таблицу, или какие-либо другие виды
отображения данных, полезные для хроматографиста. Ключевыми величинами в
любой точке хроматограммы являются время (его значения отложены по оси х) и
интенсивность сигнала (значения отложены по оси у), служащие для определения
времени удерживания (раздел 11.2.2), площади пика или его высоты (раздел 11.2.3).
'Детектор регистрирует определенный параметр, эквивалентный концентрации или массе
вещества. — Прим. персе.
11.2. Измерение сигнала 527
11.2.1. Принцип работы интегратора
К моменту выхода второго издания этой книги |2| результаты разделений в ВЭЖХ
регистрировались в основном с помощью ленточных самописцев, хотя уже тогда
для этой цели в некоторых лабораториях применяли специализированные
интеграторы. Практически все измерения и расчеты делались вручную с использованием
лишь линейки и микрокалькулятора. Появление в начале 1980-х годов
персонального компьютера (ПК), а также последующее развитие на его базе информационных
систем произвело революцию в сборе ВЭЖХ-данных. Сегодня для сбора и анализа
данных почти в каждой системе ВЭЖХ используется компьютер. Некоторые
пользователи называют интеграторами небольшие специализированные системы для сбора
хроматографических данных только с одной системы ВЭЖХ, выдающие очень
простые отчеты {например, таблицы с указанием времен удерживания и площадей
пиков). В противоположность этому, компьютерные системы сбора и обработки данных
обычно имеют дополнительные функции, включая управление блоками одной или
нескольких систем ВЭЖХ1 а также специальные возможности обработки всех
полученных данных. В рамках настоящего обсуждения мы будем считать синонимами
термины «интегратор» и «система сбора и обработки данных» (включая программное
обеспечение). В программном обеспечении системы сбора и обработки данных
используется специальный набор терминов (язык программирования), каждый из
которых описывает определенные параметры блоков системы ВЭЖХ или хроматогра-
фические характеристики. Некоторые из этих терминов используются в дальнейшем
обсуждении, они представлены на рис. 11.2. Словарь терминов несколько отличается
у разных производителей систем ВЭЖХ, однако для большинства систем одни и те
же функции являются общими. Примечание: Читатели, которые уже знают, как
работает интегратор, или же те читатели, которым это не интересно, могут
пропустить оставшуюся часть раздела 11.2.1.
11.2.1.1. Сбор данных
Система сбора данных регистрирует интенсивность сигнала как значения
(случайной) функции, зависящей от времени, в точках, которые отвечают высокой частоте
дискретизации абсциссы всей хроматограммы (как правило, 20—100 Гц |1|);
на рис. 11.1 это «сечения сигнала»2. Хроматографическая базовая линия редко
является истинно нулевой. Поэтому сначала определяются ее значения во всем диапазоне
изменения аргумента (времени), а затем эти величины вычитают из каждого
значения исходного сигнала. Поправленные таким способом данные далее
рассматриваются как хроматографический сигнал в каждой точке абсциссы хроматограммы (рис
11.1). Следствием высокой частоты дискретизации (сбора данных) является большой
объем файла данных; например, в течение 20-ти минутного анализа пробы при
частоте сбора данных 100 Гц регистрируется более 103 точек. Таким образом, объемы
файлов могут быть большими (в настоящее время, однако, хранение данных не
приводит к большим затратам). Для описания пика с точностью, близкой к
максимальной, нужно всего лишь 100 точек в области пика; большее число точек не улучшает
его описание |1|. Поэтому метод «группирования» исходных данных (рис. II.2а)
может уменьшить размер файла, сохранив информацию о пике. Например, при к = I и
'Эта часть программного обеспечения в данной книге называется управляющим модулем. —
Прим. персе.
2В теории случайных процессов сечением случайной функции называют случайную величину,
соответствующую фиксированному значению аргумента случайной функции. — Прим. перев.
528 Глава 11. Качественный и количественный анализы
сигнал детектора
базовая линия
хроматограммы
Рис. 11.1. Интегрирование пика по сечениям сигнала хроматограммы и вычитание
из него площади под базовой линией
(а)
сечения сигнала
сгруппированные
данные (единичное
значение)
конец пика
экстраполированная базовая линия
наклон
базовая линия
Рис. 11.2. Общие настройки интегратора, (я)
Группирование данных; (б) маркеры начала и конца пика; линейная
экстраполяция базовой линии в области пика по
значениям этих маркеров; (в) чувствительность определения
начала пика, зависящая от наклона хроматограммы
относительно базовой линии (сплошная линия)
jV = 10000 в колонке 150x4,6 мм с расходом I мл/мин пик будет иметь ширину 6а1,
соответствующую « II сек. Поэтому эффективная частота сбора данных оценивается
в = 9 Гц для 100 точек на пик, так что 10 Гц является достаточной частотой, при
которой полностью сохраняется информация об этом пике. (Обратите внимание на то,
что для устранения путаницы в этом разделе мы будем упоминать частоту сбора
данных для исходного непреобразованного сигнала и скорость сбора данных, имея
в виду частоту записи их в память ПК в конечном сжатом формате, в котором и
хранятся сгруппированные данные для дальнейшей обработки).
Гаким образом, каждые 10 соседних исходных точек могут быть объединены,
и эта операция понизит эффективную частоту сбора данных со 100 Гц до 10 Гц, что,
в свою очередь, позволит уменьшить размер файла данных. С помощью
программного обеспечения можно дополнительно сократить размер файла: (1) увеличивая число
'Правило «трех сигм»: вероятность того, что нормально распределенная случайная
величина (в соответствии с распределением Гаусса) отклонится от своего математического ожидания
более, чем на три среднеквадратических отклонения, не превышает = 0,0027. — Прим. персе.
11.2. Измерение сигнала
объединяемых точек на протяжении хроматограммы, поскольку ширина пиков
возрастает по мере увеличения удерживания; (2) используя другие более эффективные
методы сжатия данных.
Хотя выборка в 100 точек на пик позволяет полностью описать пик, для получе
ния его количественных характеристик достаточно 20 точек (сгруппированных)
данных. В этом случае помимо уменьшения размера файла данных, использование
сгруппированных данных снижает уровень шума базовой линии, так как величина
шума является функцией квадратного корня из числа объединенных точек1.
Снижение числа группируемых точек со 100 до 20 точек на пик уменьшает шум более, чем
в 2 раза; при этом практически нет потерь информации, содержащейся в данных.
На рисунке 11.3 показаны примеры хроматограмм, полученных при различных
способах группирования. На рис. 11.3а взято « 250 точек на пик, относительная высота
каждого пика принята за 1,00; после группирования, в результате которого осталось
* 25 точек на пик, высоты пиков стали равными 0,98 (рис. 11.36/ Следовательно,
если возрастает число группируемых точек исходного сигнала, то при
незначительной потере в оценке высоты пика величина шумов значительно уменьшается. С
другой стороны, сгруппировав слишком много точек, мы интерполируем пики
«слишком ступенчатой» функцией, использование которой приводит к заниженной оценке
высот пиков и завышенному значению величин впадин между ними (рис. П.Зг,
для ж 6 точек на пик его высота составляет 0,93 от начальной).
Сделаем следующее замечание. Необработанные данные, собранные при
наибольшей частоте сбора данных или данные, полученные группированием слишком
большого числа точек (в приведенном выше случае частота соответствует 100 Гц или
10 Гц), должны быть сохранены на протяжении всего анализа, поскольку именно
й/ V \м
Рис. 11.3. Влияние группирования данных на сигнал и шумы, (а) необработанный
сигнал; здесь взято ~ 250 точек на пик, видны значительные шумы;
(6) группирование по 10 точек из (о) дает относительную высоту пика 98%
от исходного; (в) для группирования по 20 точек из (в) получим
относительную высоту равную 97%; (г) для группирования по 40 точек из (а) —
относительная высота 93%
При группировании оценка величины сигнала равна уЛт1 = =!—±—, тогда как оценка сред-
N
неквадратического отклонения (разброса значений, определяющего вид хроматограммы) отвечает
aNm = "if———■ Здесь yt- и yNcw — соответственно значения сигнала на рис. П.За и б—г после
группировки; N— число группируемых интервалов. Также предполагается, что шумы в каждом
сечении сигнала взаимно некоррелированные случайные величины. — Прим. персе.
530 Глава II. Качественный и количественный анализы
они несут максимально полную информацию. Все последующие процедуры,
выполняемые с этими данными — например группирование до 20 точек на пик, —
проводятся без уничтожения необработанных данных.
Таким образом, исходные данные доступны для повторной обработки, если
допущены ошибки на каком-либо этапе работы с ними, а также для обработки данной
выборки в рамках другой методики. Объем выборки, полученной при высокой частоте
сбора данных, всегда может быть уменьшен путем группирования данных, но для выбор
ки, полученной при слишком низкой частоте сбора данных, невозможно создать большее
число сечений сигнала, чем уже содержится в ней. Для подстраховки более высокая
частота сбора данных используется при разработке метода, а также на первых этапах
его применения. Когда при регулярном использовании метод покажет свою
надежность, будут использованы более низкие скорости сбора данных, но такие, которые
адекватно описывают пик(и). Этот прием позволяет сэкономить память ПК при
хранении данных.
11.2.1.2. Распознавание пика
Одной из основных функций системы сбора и обработки данных является
распознавание пика на хроматограмме. Для этого текущее значение сигнала детектора
сравнивается с его значениями в последующих сечениях. Когда значение сигнала после
довательно растет в нескольких следующих друг за другом сечениях, — как правило,
в алгоритме распознавания берут от 5 до Ю интервалов, — программа распознает
пик, и текущее значение времени регистрируется как начало пика (рис. 11.26). Далее
аналогичная процедура применяется на заднем фронте пика, и когда в заданном
количестве сечений значение сигнала не становится меньше, программа выставляет
метку конца пика (рис 11.26). Заданная величина наклона кривой относительно
экстраполированной базовой линии, соответствующая чувствительности детектирования
пика (рис. 11.2в), указывает, насколько должен измениться наклон, чтобы программа
зарегистрировала начало или конец пика. Эта величина зависит от шума базовой
линии, от величины пика, от выбора пользователя или от других факторов. Система
сбора и обработки данных включает в себя алгоритмы обнаружения пика, если он
расположен на базовой линии, отклоняющейся от горизонтали, а также для случая
неполного разрешения пиков. Существуют алгоритмы и для многих других
неидеальных условий разделения пиков. После того, как установлены абсциссы (значения
времени) начала и конца пика, можно найти ширину пика. Если форма пика
подобна распределению Гаусса, то с высокой вероятностью можно считать, что ширина
пика при основании будет 6а, а на половине высоты пика («ширина на
полувысоте») — 2,35о'. Для оценки параметров пика на практике используются и другие
характеристики распределения Гаусса.
11.2.1.3. Интегрирование неидеальных хроматограмм
На хроматограмме легко идентифицировать и интегрировать пики, если они хорошо
разрешены, а базовая линия — прямая линия, параллельная оси абсцисс (рис. 11.4а).
Более сложны случаи, когда пики не разрешены полностью или наблюдаются шумы
или дрейф базовой линии. Тогда интегратор не может достаточно точно определить
моменты начала и конца каждого пика, а следовательно, сколько сечений сигнала
надлежит сделать для каждого пика. Спрямление базовой линии под пиком
(рис. 11.4б,в) является обычным приемом борьбы с ее дрейфом. Однако в рамках
этого метода могут возникнуть ошибки в оценках площади пика из-за
неопределенности, связанной с фактическим поведением базовой линии под пиком. Таким обра-
На самом деле, эта величина равна 2V2 In 2 а. — Прим. персе.
11.2. Измерение сигнала 531
О)
(в)
Рис. 11.4. Интегрирование пика, (о) Интегрирование двух хорошо разрешенных пиков
на базовой линии без шумов и дрейфа, (б) Занижение оценки площади пика
из-за линейной экстраполяции базовой линии, которая в действительности
вогнутая кривая, (в) Завышение оценки площади пика, если в
действительности базовая линия — выпуклая кривая, (г) Правильное применение
метода перпендикуляра для перекрывающихся пиков гауссовой формы; для
пиков, отличающихся по размеру, занижается плошадь меньшего пика.
(д) Метод перпендикуляра при оценке площади малого пика, выходящего
на заднем фронте большего пика, дает завышенную площадь малого пика,
(с) Использование метода линейной экстраполяции при интегрировании
меньшего пика, находящегося на заднем фронте большего пика |1|
зом, измеренная площадь пика будет либо меньше, если в действительности базовая
линия — вогнутая кривая (рис. 11.46), либо больше, если она — выпуклая кривая
(рис. 11.4в). Много усилий было затрачено на то, чтобы определить лучший метод
аппроксимации базовой линии (линейная, экспоненциальная зависимость и т.д.) —
оказалось, что линейная экстраполяция (рис. 11.1) является лучшим компромиссным
решением 11 [.
Наиболее распространенным методом разделения двух не полностью
разрешенных пиков является метод перпендикуляра, в котором границей между пиками
является перпендикуляр, опушенный на базовую линию из точки минимального
значения сигнала между пиками (рис. 11.4г)'. Для симметричных пиков с одинаковыми
характерными параметрами (шириной и высотой) этот метод дает правильные
оценки площадей под пиками, но если параметры симметричных пиков различны,
то площадь пика с меньшей высотой будет занижена, а с большей — завышена |3|.
Неопределенность в оценках площадей пиков возрастает, если пики имеют разные
параметры и если у одного из них или у обоих размыт задний фронт (рис. \\Ад).
В определенных случаях вполне оправдан переход от метода перпендикуляра
(рис. I l.4d) к методу линейной экстраполяции2 (рис 11.4с). Как правило, если высота
меньшего пика составляет < 10% от высоты большего, рекомендуется использовать
метод линейной экстраполяции; если же его высота > 10% — метод
перпендикуляра |1|. Тем не менее, независимо от используемого в таких случаях способа оценки
'Для интегратора эта точка отвечает концу первого пика и началу второго пика. — Прим. перев.
2Иногда его называют метод тангенты. — Прим. перев.
532 Глава II. Качественный и количественный анализы
площадей, правильность результатов будет сомнительной, особенно для небольших
пиков и/или пиков, у которых размыт задний фронт. В некоторых случаях высота
пика — как количественная характеристика — может оказаться более
предпочтительной, чем его площадь (раздел 11.2.3).
11.2.1.4. Обычные ошибки интегрирования
Независимо от того, насколько высок уровень проработки программного обеспече
ния системы сбора и обработки данных, оно не может заменить опыта
хроматографиста. Он должен подстроить параметры системы так, чтобы она наилучшим
образом идентифицировала пики и определяла их площади. По мере уменьшения
отношения сигнала к шуму эта задача становится существенно труднее из-за
появления потенциальных источников ошибок интегрирования. Три наиболее часто ветре
чающихся случая показаны на рис. М.5. Обычно за базовую линию под пиком при
нимают прямую линию, полученную экстраполяцией сигнала по его значениям до и
после пика. Однако причиной некорректности этой процедуры могут стать
отрицательные пики и другие случайные помехи базовой линии, как показано на рис. 11.5а.
В этом случае отрицательный пик, предшествующий интегрируемому пику,
приводит к занижению базовой линии (сплошная линия), что искусственно увеличивает
площадь размеченного пика; очевидно, что исходную базовую линию необходимо
провести иначе (пунктирная линия). Как уже упоминалось в разделе 11.2.1.3,
для данного случая трудно принять решение о том, следует ли использовать метод
перпендикуляра или метод линейной экстраполяции для плохо разрешенных пиков.
На рис. 11.5^ пики были неправильно разделены методом перпендикуляра
(сплошная линия); здесь, для того чтобы провести более правильное интегрирование,
требуется использовать метод линейной экстраполяции (пунктирная линия). В некоторых
системах сбора и обработки данных доступны алгоритмы, использующие не
линейную функцию, а кривые для корректирования базовой линии, например Гауссиан.
Однако необходимо понимать, что все это приближенные методы, которые не дают
точных, согласующихся друг с другом результатов, как это бывает для пиков,
разрешенных до базовой линии.
Одной из наиболее сложных задач для интегратора является определение конца
пика. Это осложняется в тех случаях, когда у пиков размывается задний фронт или
Рис. 11.5. Часто встречающиеся неточности интегрирования, (а) ошибочное
определение начала пика из-за того, что отрицательные значения сигнала
предшествуют выходу анализируемого вещества. Стрелка указывает на необходимый
сдвиг начала пика для правильного интегриролания (пунктирная линия).
(б) Случай неправильного интегрирования перекрывающихся пиков
методом перпендикуляра; линейная экстраполяция (пунктирная линия) дает
более точную оценку площади малого пика, (в) Неправильное положение
маркера конца пика. Стрелка указывает на его более правильное положение
(пунктирная риска)
J-
11.2. Измерение сигнала 533
когда наблюдаются дрейф и/или заметные шумы базовой линии. На рис. 11.5»
показан пример, когда метка конца пика была поставлена слишком рано, и,
следовательно, должна быть сдвинута на более позднее время (показано стрелкой на рис. 11.5в).
Пожалуй, это самая распространенная ошибка, встречающаяся при разметке пиков,
чьи размеры близки к пределу обнаружения или к нижнему пределу количественного
определения. Один из способов минимизировать вероятность такой ошибки — это
продолжать интегрирование до момента, когда пик (вещество) наверняка покинул
детектор. Это может быть достигнуто путем визуального определения момента, когда
значение сигнала стало близким к значению, наблюдаемому на базовой линии
(справа в конце пунктирной базовой линии на рис. М.5е). В этом случае в меню
программы обработки данных выбирается функция «force peak-end» («принудительно
назначить конец пика»). Заметим, что у разных фирм-изготовителей хроматографов эта
функция может иметь другие названия.
Теперь интегратор не будет искать конец пика, используя алгоритмы
программного обеспечения, а автоматически выставит принудительное время выхода пиков на
всей хроматограмме. Рекомендуется визуально проверять каждую хроматограмму,
чтобы убедиться в правильности интегрирования, а в случае ошибок внести необходимые
поправки. Некоторые пользователи считают, что перестраивать базовую линию после
того, как проведено интегрирование, — это жульничество. Но на самом деле система
сбора и обработки данных не может все время правильно интегрировать все пики. Ко
нечно, любые проводимые вручную изменения параметров базовой линии должны
выполняться последовательно и без подгонки к желаемому результату.
11.2.1.5. Дополнительные советы
Предшествующее обсуждение охватывает лишь часть факторов, которые
способствуют корректному интегрированию пиков хроматограммы. Программное
обеспечение современных систем сбора и обработки данных очень сложное и предназначено
для правильной идентификации пиков, корректировки базовых линий, определения
числа точек для группирования и так далее. Как правило, при апробации нового
метода наиболее целесообразно протестировать несколько стандартных образцов, позволяя
интегратору выбрать начальные параметры интегрирования. После того как получены
результаты анализа, предварительно установленные параметры можно
скорректировать для исправления систематических ошибок, например таких, как разделение двух
пиков с помощью метода перпендикуляра или метода линейной экстраполяции.
Другими словами, сначала надо позволить интегратору работать в обычном режиме, а
затем по мере необходимости вносить изменения в его настройки.
Для защиты данных фармацевтическим фирмам необходимо выполнять ряд
рекомендаций, называемых правилами электронной подписи (см. Свод федеральных
нормативных актов США1, раздел 21, часть II |5|). Одно из требований состоит
в том, что необходимо указывать любые изменения исходных данных (например,
настройка базовой линии или задание начала и конца пика), приводя имя оператора,
дату и время, а также причину внесения изменений. При этом оригинальные данные
должны быть сохранены для возможного последующего анализа. Большая часть
систем сбора и обработки данных удовлетворяют этим требованиям благодаря
встроенной в их математическое обеспечение функции «аудит». Если эта функция
активизирована, то при каждом изменении обработки она требует введения комментария,
к которому добавляется электронная подпись оператора и запись даты и времени.
Например, в случае корректировки, проведенной для хроматограммы на рис. 11.5е,
комментарий может быть таким: «неправильно размечен конец пика». Даже если
1 Code of Federal Regulations, CFR. — Прим. перев.
534 Глава II. Качественный и количественный анализы
такого сорта аудит не используется в вашей отрасли, активизация этой функции —
полезная практика, которая позволяет следить за изменениями, сделанными по ходу
обработки данных, если понадобится позднее повторно рассмотреть принятое
решение.
В последнем абзаце прекрасного справочника по интегрированию в
хроматографии [I, с. 1911 операторам сделано следующее предупреждение:
Если в интеграторах используются методы перпендикуляра и линейной экстраполя
ции, а базовые линии под пиками аппроксимируются прямыми, то такая обработка
данных применима только в контролируемых условиях, то есть в рамках разделения
с хорошим разрешением и минимальными помехами. Однако даже в этом случае работа
интегратора требует от оператора бдительности, умения оценивать параметры ин
тегрирования и при необходимости исправлять их. Сами по себе интеграторы не
способны улучшить процесс хроматографического разделения (выделено авторами), только
аналитики могут сделать это (усовершенствовав метод). В конце концов, это и есть
работа хроматографистов, за которую им платят.
11.2.2. Удерживание
Удерживание анализируемого вещества является основной характеристикой, которая
используется для его качественного определения {раздел 11.3.1). Удерживание чаще
всего измеряется как время удерживания 1ц, выраженное в минутах (например,
6,54 мин в десятичной системе счисления), но иногда используются секунды
(например, 36,4 с), если разделение протекает в секундном диапазоне. Изредка
удерживание измеряется в единицах объема (например, 4,35 мл); в последнее время это
встречается редко. Относительные времена удерживания используются во многих
методиках Американской фармакопеи1, а также в ряде других случаев. В этих случаях
удерживание — величина, указывающая на время выхода исследуемого пика
относительно времени выхода эталонного пика (например, отношение времен удерживания
двух пиков). Такой способ представления информации об удерживании в некоторой
степени компенсирует возникающие по какой-либо причине изменения
абсолютного времени удерживания, особенно когда анализ переносится с одной системы
ВЭЖХ на другую (конечно же, при переносе метода пик должен быть
корректно идентифицирован, а не только по относительному времени удерживания (см.
раздел 12.7)).
Время удерживания каждого анализируемого вещества измеряется от момента
ввода пробы до выхода вершины пика (рис. 2.3d). Время удерживания должно также
соответствовать моменту времени, которому отвечает наибольшее значение сечения
сигнала. Если неизменны все условия хроматографического разделения: скорость
потока, температура, состав подвижной фазы и т.д., то /д должно быть постоянным.
(Последнее справедливо, если количество образца невелико, раздел 2.6). Здесь таюке
предполагается, что ВЭЖХ-система работает должным образом, то есть значение
удерживания изменяется между вводами не более чем на 0,02—0,05 минуты в
течение дня.
Для расчета коэффициента удерживания к (раздел 2.3.1) надо также измерить
мертвое время колонки /q. В большинстве случаев достаточно определить /q как
время удерживания неудерживаемого пика растворителя в начале хроматограммы
(рис. 2.3d, 2.5я). Для чистых образцов изменение базовой линии при Го может не
детектироваться, в то время как некоторые детекторы (например, МС-детектор;
раздел 4.14) могут не регистрировать какие-либо изменения сигнала в точке Jq. В таких
случаях может потребоваться отдельное измерение Го (раздел 2.3.1).
1 United States Pharmacopeia, USP. — Прим. перев.
11.2. Измерение сигнала
11.2.3. Размер пика
Собираемые хроматографические данные
представляют собой значения
напряжения в зависимости от времени
(раздел II.2.l.l), при этом интенсивность
сигнала обычно приводится в единицах
мкВс1. Площадь пика соответствует сумме
сечений сигнала между точками начала и
конца пика, как показано на рис. 11.6а.
Высота пика соответствует
максимальному значению сечения сигнала, по
которому определяется время удерживания пика
(рис 11.66). В то время, когда
использовалось ручное интегрирование, часто
измеряли высоту пика, чтобы вносить
меньшую ошибку, так как требуется только
одно измерение (высота), в то время как
для определения площади пика требуется два измерения (высота и ширина). Однако
с современными интеграторами площадь пика стала наиболее популярным способом
для того, чтобы характеризовать размер пика. В некоторых случаях предполагается,
что высота пика дает лучшие результаты, например, при частично разрешенных
пиках, максимумы которых существенно не перекрываются. В таких случаях высота
пика будет по-прежнему соответствовать «чистому» пику, в то время как площадь
пика может быть поставлена под сомнение в результате неправильной разметки, как
показано на рис. 11.4г. Если у вас возникли сомнения, включите и площадь, и
высоту в отчет интегратора; затем сравните результаты в отношении правильности и
прецизионности (например, используйте указания, приведенные в разделах 12.2.1
и 12.2.2).
11.2.4. Источники ошибок
Рис. 11.6. (с) Измерение площади пиков
по сечениям сигнала, (б) Измерение высоты
пика при наибольшем значении сечения
сигнала
Правильность* результатов определяется как близость измеренного значения
параметра анализируемого вещества к его истинному значению. Правильность зависит
от (1) калибровки системы надежными стандартами и (2) разрешения соседних
пиков. Прецизионность^ результатов является мерой того, насколько близки к одной
величине значения, полученные при повторных измерениях, произведенных в разное
время, на различных измерительных приборах и/или с участием разных операторов.
Способность контролировать условия проведения измерений приборами для
выполнения конкретного метода является основой прецизионности метода. Желательно
получать точные результаты, которые были бы и правильными, и прецизионными.
Однако они могут быть правильными, но не прецизионными или прецизионными,
но неправильными. Прецизионными и правильными (точными) количественные
анализы будут только тогда, когда тщательно выполняются все этапы анализа,
от первоначального отбора проб до формирования окончательного отчета. Вот
некоторые из основных источников ошибок:
'Указанная единица соответствует плошали сгруппированного сигнала. — Прим. перев.
^Термины «правильность» и «прецизионность» соответствуют ГОСТ Р ИСО 5725-1-2002.
Термин «точность» соответствует совокупности правильности и прецизионности, так как «он
выражает суммарное отклонение результата от эталонного (опорного) значения, вызванное как
случайными, так и систематическими причинами». — Прим. перев.
536 Глава II. Качественный и количественный анализы
• отбор проб и пробоподготовка (раздел 11.2.4.1);
• хроматографическое разделение (раздел 11.2.4.2);
• детектирование (раздел 11.2.4.3);
• измерение пика (раздел 11.2.4.4);
• калибровка (раздел 11.2.4.5).
11.2.4.1. Отбор проб и пробоподготовка
Любой аналитический результат основан на предположении, что исследуемая проба
образца является репрезентативной для объекта, из которого она была отобрана. По
этому первая задача состоит в том, чтобы получить образец (воды, почвы, порошка,
плазмы крови, ткани и т.д.), который отвечает этим требованиям (раздел 16.1.1).
После того, как проба образца получена, ее надо транспортировать и хранить таким
образом, чтобы обеспечить сохранность образца до проведения анализа. Большинство
образцов требует пробоподготовки или очистки (глава 16) перед введением в систему
ВЭЖХ. Каждый этап обработки образца, в том числе все вспомогательные приборы
и реагенты, вносят свой вклад в общую погрешность метода. После того, как
подготовленный образец помещен в лоток автосамплера, все еще велика вероятность
погрешностей, связанных со стабильностью пробы, ее испарением и колебаниями
количества образца, вводимого для анализа. Некоторые из этих погрешностей можно
устранить, по крайней мере, частично за счет использования внутренних стандартов
(раздел 11.4.1.2); другие скорректировать невозможно.
11.2.4.2. Хроматографическое разделение
Несмотря на то, что подготовка образцов является наиболее вероятным источником
грубых ошибок и неточностей, несколько различных аспектов процесса хроматогра-
фического разделения способствуют нестабильности аналитических результатов.
Хроматографическое разделение имеет первостепенное значение. Как обсуждалось в
разделе 11.2.1, пики, которые разрешаются до базовой линии с малыми шумами,
не отклоняющейся от горизонтали, намного проще интегрировать. Такие пики дадут
более достоверные результаты. Очевидно, что известные пики должны быть
отделены друг от друга, но хороший метод должен также гарантировать, что потенциальные
примеси, введенные вместе с образцом (лекарственные препараты, метаболиты,
продукты распада, побочные продукты и т.д.) будут отделены от пиков, представляющих
интерес. Как было отмечено выше, размывание заднего фронта пика может привести
к недостоверным результатам при измерении его площади. В некоторых случаях
нелинейная адсорбция или другие взаимодействия с сорбентом, зависящие от
концентрации образца, ведут к росту разброса результатов. Часто это наблюдается при вводе
пробы с малым содержанием исследуемого вещества или с веществами из
биологических объектов. Когда отношение сигнал-шум (S/N) менее и 100, шум может
стать важным фактором, способствующим неточности измерений (раздел 4.2.3);
для больших пиков результаты, как правило, более прецизионные и правильные.
И, наконец, факторы, обусловленные оборудованием (старение колонки,
нестабильность скорости потока или температуры и т.д.) могут привести к неточности
измерения. Каждый из этих факторов может повлиять на общую прецизионность и
правильность метода.
11.2.4.3. Детектирование
ВЭЖХ-детекторы (глава 4) — это приборы, которые преобразуют в электрический
сигнал концентрацию или массу анализируемого вещества в подвижной фазе.
Некоторые детекторы, такие как рефрактометры (раздел 4.11), очень чувствительны
11.2. Измерение сигнала 537
к изменениям температуры или состава подвижной фазы; это может вызывать
значительный шум и недостоверность результатов при измерении. Наиболее
распространенные УФ-детекторы с переменной длины волны и диодно-матричные (раздел 4.4)
в значительно меньшей степени зависят от этих факторов, но грязная проточная
ячейка, пузырьки и старение лампы могут вносить вклад в неточность сигнала
детектора. Сигналы некоторых детекторов тоже можно поставить под сомнение из-за
подавления ионизации (МС-детекторы, раздел 4.14), тушения флуоресценции
(флуоресцентные детекторы, раздел 4.5) или иных факторов, характерных для других
методов детектирования. Все детекторы имеют ограниченный линейный диапазон,
вне пределов которого отклик на единицу массы анализируемого вещества
(чувствительность) изменяется. Если отклик детектора не проверять с помощью
калибровочной кривой, появятся ошибки при измерении. Наконец, любые погрешности в рабо
те электроники или программного обеспечения, которые приводят к ошибочному
преобразованию измеренных параметров, характеризующих количество
анализируемого вещества, в пропорциональный электрический сигнал, внесут свой вклад
в неточность метода; как правило, этот вклад незначителен и не влияет на качество
данных.
11.2.4.4. Измерение пика
Как обсуждалось в разделе II.2.1, возможны разные ошибки при преобразовании
выходного сигнала ВЭЖХ-детектора в значения времени удерживания
анализируемого вещества и плошади соответствующего ему пика. Определение начальной и
конечной точки пика, разделение частично разрешенных пиков методом
перпендикуляра или линейной экстраполяции, определение положения максимума пика,
степень группирования данных и оценка расположения базовой линии под пиком —
лишь некоторые из возможных источников ошибок. Методы с хорошо
разделенными пиками на ровной базовой линии с низким уровнем шума позволят свести к
минимуму количество ошибок в процессе интегрирования, и качество разделения
должно стать главной целью при разработке метода.
11.2.4.5. Калибровка
Ошибки, вызванные калибровкой, делятся на две основные категории: ошибки
при калибровке прибора и при калибровке метода. Калибровка прибора официально
проводится в серии тестов, называемых установочной, операционной и
эксплуатационной квалификациями (УК/ОК/ЭК), как описано в разделе 3.10.1.1. Это, а также
периодическое тестирование пригодности системы (раздел 17.2.1), может
гарантировать, что различные блоки системы (насос, автосамплер, детектор и т.д.) работают
приемлемым образом как по отдельности, так и при объединении в систему ВЭЖХ.
Правильно откалиброванный прибор должен вносить минимальный вклад в общую
погрешность метода, но всегда будут некоторые колебания скорости потока, состава
подвижной фазы, температуры и так далее.
Калибровка метода (раздел 11.4.1) относится к выбору стандартного образца,
построению калибровочной кривой (график зависимости интенсивности отклика
детектора от концентрации, также называемый стандартной кривой) и выбору
алгоритма аппроксимации эмпирической кривой. Каждый из этих этапов может внести свой
вклад в погрешность метода. Стандартные образцы должны быть известной чистоты;
если используется стандартный образец лаборатории (предприятия), он должен
проходить ту же процедуру пробоподготовки, что и анализируемые образцы, и хорошо
разрешаться на хроматограмме, чтобы можно было точно измерить площадь
соответствующего ему пика.
В Глава П. Качественный и количественный анализы
Ексе!
конц
(нг/мл)
1
2
5
10
20
50
100
сигнал
3.5
S.3
11.1
21.2
39.3
98.5
191.5
Регрессионная статистика
г
г*
Стандартная
ошибка
(для кривой)
Количество
измерений
Дисперсионный
анализ
(ANOVA)"
Ордината
пересечения(Ь)
Концентрация
Тпт7наклон, S")
0.9999
0.9999
0.8017
7
Коэффициенты
1.7774
/ТЛЮ42)
Стандартная
ошибка ординаты
{пересеченно, а)
(озвтг)
(3.3 к 0.39) П.90 = 0.67
ПО= 0.74 нг/мл
Рис. 11.7. Расчет предела обнаружения (ПО) по данным калибровочной кривой.
•От англ. ANalysis Of VAiance. ANOVA в настоящее время часто встречается
в русскоязычной специальной литературе. — Прим. персе.
Одним из измеряемых показателей метода является линейность, которая обычно
выражается через коэффициент корреляции г2' (хотя само по себе значение г2 не
полностью характеризует калибровку, как указано в разделе 11.4.1.5). «Линейность»
подтверждается, если график зависимости отклика детектора от концентрации
описывается уравнением у — тх + Ь и проходит достаточно близко к началу координат
(например, значение Ь, ординаты пересечения графиком оси у, < стандартной
ошибки), и линейная регрессия2 дает некоторое минимальное (допустимое) значение г2
(например, г2 ^0,98). В таких случаях может быть оправдана калибровочная кривая3
по одной точке. Калибровка по одной точке включает один калибровочный стандарт
(или среднее значение, полученное при многократном введении образца с
определенной концентрацией). Как правило, берется концентрация, близкая к
предположительной концентрации вещества в образце (например, 100% лекарственной
формы). Предполагается, что полученная зависимость описывается уравнением у = тх.
Калибровка по одной точке используется во многих фармакопейных методах.
Когда калибровочный график не проходит через начало координат или диапазон
возможных концентраций образцов охватывает несколько порядков, больше подойдет
многоточечная калибровка. Такой пример показан на рис. 11.7, в котором ожидаемые
1 Коэффициент корреляции характеризует близость экспериментальных значений и значений,
полученных при аппроксимации данных калибровки какой-либо функцией, например,
линейной. — Прим. персе.
^Термин «линейная регрессия» означает аппроксимацию линейной функцией. — Прим. перев.
3По утверждению авторов (раздел 11.4.1.1) калибровочный график принято называть кривой,
несмотря на то, что данные обычно аппроксимируются линейной функцией, как сказано
выше. — Прим. перев.
11.2. Измерение сигнала 539
концентрации образца охватывают два порядка (I —100 нг/мл), поэтому выбрана
многоточечная калибровка. Приведенные данные ложатся на прямую, что подтверждает
г = 0,9999, но ордината пересечения калибровочным графиком оси у (1,7774 нг/мл)
в 4 раза больше допустимой стандартной ошибки (0,3872), поэтому данные
калибровки более точно описывает уравнение у = тх + Ь, где ( Ь Ф 0): у - 1,9042х + 1,7774.
Двухточечная калибровка является разновидностью многоточечной калибровки,
ее использование обосновано и удобно, если калибровочный график линейный (на
пример, значение г* велико), но не проходит через начало координат, а диапазон
ожидаемых концентраций образцов узок (например, меньше, чем один порядок
величины). В таких случаях готовятся два калибровочных стандартных раствора с кон
центрациями, находящимися вблизи границы диапазона (например, 80 и 120%
для образцов, в которых ожидается содержание анализируемого вещества 100 +20%).
Любая экстраполяция за пределы концентрации стандартов, используемых при
построении любой многоточечной калибровочной кривой, увеличивает погрешность
измерений. В общем случае проще всего (и вполне корректно) использовать
линейную калибровку для методов ВЭЖХ, но в некоторых случаях может потребоваться
аппроксимация кривой квадратичной или иной функцией. Ошибка всегда
присутствует, когда обобщенная модель отклика (т.е. калибровочная кривая) используется
для количественной оценки конкретных образцов, так как с каждой точкой
калибровки связана своя ошибка, вносящая свой вклад в общую погрешность калибровки.
Поэтому важной частью валидации метода является проведение достаточного
количества экспериментов в контролируемых условиях с известными концентрациями
анализируемого вещества для того, чтобы определить правильность и
прецизионность во всем рабочем диапазоне метода (разделы 12.2.1, 12.2.2, 12.2.5).
11.2.5. Пределы обнаружения и определения
Данный метод ВЭЖХ должен давать результаты с приемлемой прецизионностью и
правильностью в пределах определенного диапазона концентраций анализируемого
вещества. Пределом обнаружения (ПО) является наименьшая концентрация, при
которой достоверно можно установить присутствие анализируемого вещества в
образце, но может оказаться невозможным точное определение его количественного
содержания. Нижним пределом количественного определения, называемым также
пределом количественного определения или количественным определением (НПКО или
ПКО), является самая низкая концентрация анализируемого вещества, которая
может быть представлена количественным значением с приемлемой прецизионностью
и правильностью. Верхним пределом количественного определения (ВПКО или просто
верхний предел) является наибольшая концентрация анализируемого вещества,
которая может быть представлена количественным значением с приемлемой
прецизионностью и правильностью. Независимо от того, какой способ используется для
определения этих предельных значений, настоятельно рекомендуется [6] проверить их,
введя образцы с предельными концентрациями. Эти три предела (ПО, НПКО,
ВПКО) более подробно рассмотрены ниже. Есть несколько различных способов
установить эти предельные значения для данного метода, и не все они дают
одинаковые результаты. Для методов, выполняемых под надзором регуляторного органа,
лучше всего использовать тесты пределов, предписанные этим регуляторным
органом. Если используется нерегламентированный метод, решение о том, как
тестировать указанные пределы, принимается в лаборатории.
Значения пределов для данного метода тесно связаны с использованием
конечных данных. Например, для биоаналитических методов (в частности, мониторинга
лекарственных средств в плазме крови), прецизионность и правильность должны
540 Глава II. Качественный и количественный анализы
быть не хуже, чем +15% ОСО при всех концентрациях выше НПКО, и +20% при
НПКО [6|. С другой стороны, предполагается, что способ высвобождения иди
стабильность лекарственного продукта обеспечиваете 1% ОСО при концентрации 100%
|7|. Отношение сигнал-шум (S/N), как показано на рис. 4.7, будет влиять на полную
погрешность метода. Простая оценка вклада S/N в полную погрешность метода
определяется как |8|
ОСО(%)=^_. (11.1)
S/N
Этот вклад в погрешность метода в сочетании с другими источниками погрешности
(отбор проб, пробоподготовка и т.д.) определяет полную погрешность метода1.
Погрешность метода jc, обусловленная каждым из источников, суммируется
с другими в виде значений дисперсии х .
Er=(Ef + El + ...+ EZ)V, (11.2)
где Е-] полная погрешность и Е\, Ej,---. , Е„ — вклады погрешностей каждого
источника 1, 2,..., п. Например, Е/ может быть погрешностью, обусловленной пробо
подготовкой, Ej погрешностью автосамплера, а Е-$ погрешностью из-за малого
значения отношения сигнала к шуму (уравнение 11.1). Как правило, если ОСО,
вызванное каким-либо из источников погрешности меньше, чем половина
суммарного значения ОСО, вклад этого источника в суммарное ОСО будет составлять
менее 15%. Суммарное ОСО будет определять источник самой большой погрешности
в уравнении (11.2), поэтому чтобы уменьшить полную погрешность, следует в
первую очередь свести к минимуму воздействие источника самый большой погрешности
(часто это S/N в условиях ПО или НПКО). Если один из источников приводит к
погрешности, превышающей допустимую полную погрешность, то ее желаемое
значение не будет достигнуто, пока этот источник погрешности не будет устранен.
11.2.5.1. Предел обнаружения (ПО)
Есть четыре способа оценки значения ПО:
• визуальная оценка;
• отношение сигнал/шум;
• стандартное отклонение сигнала и наклон;
• ОСО, выраженное в процентах.
Эти способы взаимосвязаны, поскольку являются разными подходами к
достижению одной и той же цели, но они могут давать несколько разные результаты.
Можно визуально оценить хроматограмму на наличие или отсутствие
анализируемого вещества, но такая оценка очень субъективна и зависит от ошибок аналитика.
Поэтому она не рекомендуется.
Для оценки ПО можно использовать отношение сигнал/шум. Обычно отношение
S/N = 3 (измеренное так, как показано на рис 4.7) принимается как определение
условий ПО. Если S/N измерить автоматически с помощью системы сбора и
обработки данных при многократном вводе пробы, среднее значение S/N = 3
соответствует ПО. Тем не менее, если и сигнал, и шум измеряются вручную, полученное
значение ПО может быть несколько субъективным из-за непреднамеренных ошибок
оператора. В любом случае необходимо несколько раз ввести пробу, чтобы
проверить, соответствует ли найденная концентрация анализируемого вещества ПО.
Из уравнения (11.1) концентрация, для которой S/N = 3, должна иметь ОСО и 17%.
1 Полная погрешность метода равна сумме систематической и случайной погрешности.
Случайную погрешность принято характеризовать относительным стандартным отклонением (ОСО).
т.е. усредненным разбросом экспериментальных значений. — Прим. перев.
11.2. Измерение сигнала 541
Приблизительно такое же значение ОСО должно получиться при многократных
вводах пробы с концентрацией вещества, близкой к ПО.
Для непосредственного определения ПО также используется метод стандартного
отклонения и наклона |9]:
ПО = ^£. (11.3)
S
где о — стандартное отклонение калибровочной кривой и S — ее наклон (например,
единичный отклик на концентрацию, выраженную в нг/мл). Пример пошагового
выполнения этой процедуры показан на рис. 11.7, где исходная электронная таблица
обработана программным пакетом Excel. Собранные для калибровочной кривой
данные вводятся в таблицу: значения концентрации анализируемого вещества и
соответствующие значения сигнала детектора (площадь или высота пика). В Excel
проводится линейный регрессионный анализ данных с получением таблицы регрессионной
статистики; а в уравнении (11.3) берется равным стандартной ошибке пересечения
линии регрессии с осью у (0,3872), a S устанавливается равным концентрационному
коэффициенту (1,9042). При подстановке в уравнение (11.3) значений, приведенных
на рис. 11.7, получаем ПО = (3,3 х 0,3872)/1,9042 = 0,67 нг/мл.
Рекомендации, данные в |9], позволяют использовать в качестве а в уравнении
(11.3) либо стандартную ошибку, вычисленную по всей кривой, либо стандартную
ошибку, определяемую ординатой пересечения калибровочной кривой с осью у.
Авторы данной книги не согласны с этой концепцией и рекомендуют использовать
ошибку, соответствующую ординате пересечения с осью.)> калибровочной кривой у =
= тх + Ь, где Ь * 0, даже если будет использоваться кривая, проведенная через начало
координат1. Стандартная ошибка кривой соответствует средней ошибке,
вычисленной по всем ее значениям (включая высокие и низкие концентрации). Для более
низких концентраций, которые в большей степени соответствуют условиям ПО,
стандартная ошибка уменьшается, а ОСО (%) увеличивается. Значения ПО
и НПК.О связаны в основном с ошибкой, вычисленной при низких концентрациях,
а не со средней ошибкой для всей кривой, то есть стандартная ошибка,
определяемая ординатой пересечения калибровочного графика с осью у, является лучшей
оценкой, чем стандартная ошибка, полученная для всей кривой.
Преимущество уравнения (11.3) по сравнению с критерием S/N = 3 заключается
в том, что не требуется совпадения полученной оценки ПО со значением
концентрации одного из калибровочных стандартов, использованных для построения
калибровочной кривой. Например, для регрессии, показанной на рис 11.7, были введены
стандарты с концентрациями 1 и 2 нг/мл, а для ПО было получено значение
0,67 нг/мл. Уравнение (11.3) является лишь оценкой ПО, поэтому важно
подтвердить, что установленная концентрация соответствует ПО. Для этого следует
проанализировать нескольких образцов с концентрацией 0,67 нг/мл, чтобы убедиться, что
пики действительно обнаруживаются системой сбора и обработки данных
(визуальный подход) и S/N и 3 или ОСО = 17% (уравнение 11.1).
Наконец, некоторые пользователи устанавливают ПО на основании конкретных
значений ОСО(%). Это значение ПО, основанное на относительном стандартном
отклонении, может быть определено как минимальная концентрация, ниже которой ОСО
превышает 17% (что соответствует S/N = 3 из уравнения 11.1). При использовании это-
1 Если в таблицу данных, приведенную на рис. 11.7, ввести точку (х,у) = (0;0), то при
регрессионном анализе программа вычислит ошибку и для этого значения у, которую авторы и рекомендуют
как о в уравнении (11.3). Однако при этом калибровочная кривая может как проходить, так не
проходить через начало координат. Существует и другая возможность: ввести значение b = 0 в качестве
ограничения — и тогда кривая точно пройдет через начало координат, но понятие ошибки ординаты
пересечения калибровочного графика с осью у теряет смысл. — Прим. перев.
542 Глава II. Качественный и количественный анализы
го подхода требуется рассмотреть зависимость величины ОСО от концентрации
анализируемого вещества, что, как правило, делается во время валидации метода.
Уверенность в том, что ПО был выбран правильно, можно получить только пу
тем многократного ввода пробы в концентрации, равной предполагаемому ПО.
Оценка ПО по отношению S/N или с помощью уравнения (11.3) может быть
подтверждена путем 5- или 6-кратного ввода пробы с оценочной концентрацией ПО,
а для дополнительного статистического подтверждения рассчитывается ОСО(%)
в этих условиях.
11.2.5.2. Нижний предел количественного определения
(НПКО или ПКО)
Значение НПКО можно определить с помощью трех процедур, которые
используются и для определения значений ПО:
• по отношению сигнал шум;
• по стандартному отклонению сигнала и наклону;
ф по относительному стандартному отклонению.
Если использовать подход, основанный на отношении сигнал-шум, то S/N = 10
обычно принимается в качестве НПКО, который можно проверить с помощью отно
шения ОСО < 5% (уравнение 11.1). Метод стандартного отклонения сигнала и
наклона, основанный на анализе калибровочной кривой, описан в разделе 11.2.5.1.
Для НПКО уравнение (11.3) принимает вид [9|:
НПКО = -^?. (11.4)
S
Используя данные рис. 11.7, из уравнения (11.4) можно вычислить НПКО =
= 2,0 нг/мл, при этом а равно стандартной ошибке определяемой ординатой
пересечения калибровочного графика с осью у (см. обсуждение в разделе 11.2.5.1).
Значение НПКО, основанное на относительном стандартном отклонении, непо
средственно связано с допустимой погрешностью результата, которая обычно
составляет 5% ОСО (для S/N ~ 10, уравнение (11.1)), но может быть любым другим
значением, которое соответствует требованиям испытаний. Для биоаналитических методов
НПКО определяется как наименьшая концентрация, для которой ОСО не
превышает 20% |6|. Для таких методов погрешность пробоподготовки образца может давать
значительный вклад в полную погрешность в дополнение к вкладу S/N при НПКО.
Таким образом, при многократном вводе калибровочных растворов резонно ожидать
S/N< 10 при НПКО (в соответствии с определением S/N для НПКО). Вклад S/N в
общую погрешность (уравнение 11.2) не должен превышать (50/10) = 5% (уравнение
11.1), и он вряд ли является основным источником погрешности. Тогда независимо от
того, какое значение ОСО считается допустимым, НПКО можно определить из
графика зависимости ОСО от концентрации анализируемого вещества.
Какой бы метод ни был выбран для определения НПКО (или ПО),
используемые данные должны быть в достаточной мере прецизионными и правильными.
НПКО и ПКО всегда должны быть проверены вводом образцов в соответствующих
концентрациях.
11.2.5.3. Верхний предел
В то время, как методы определения ПО и НПКО точно установлены нормативными
рекомендациями (например, |6, 9|), верхний предел в них определяется только как
верхний конец калибровочной кривой или как наибольшее измеряемое количество
анализируемого вещества в пределах требуемой прецизионности и правильности.
Для определения величины верхнего предела методик нет. Что касается аналитиче-
11.2. Измерение сигнала 543
ских применений ВЭЖХ, то для них, как правило, наиболее важны нижние пределы.
Верхний предел устанавливается самой высокой тестируемой концентрацией (самая
высокая концентрация калибровочной кривой) или точкой, в которой начинает про
являться нелинейность детектора или насыщение сорбента становится проблемой.
Анализ лекарственной субстанции (чистого вещества) или лекарственного
продукта (готовой лекарственной формы), в том числе сопутствующих примесей или
продуктов разложения, требует, чтобы метод хорошо работал на обоих пределах
количественного определения — верхнем и нижнем. В таких случаях указанные
пределы часто обозначаются как процент отклика детектора для активной
фармацевтической субстанции (АФС) при нормальной дозировке. Например, примеси в новых
лекарственных субстанциях должны быть на уровне 0,05%, а их количественное
определение должно быть проведено с точностью 0,1% по отношению к АФС [10|.
Для этого требуется, чтобы отклик в методе был линейным (или имел другую
заданную форму кривой) в диапазоне > 10 . Как правило, это не является проблемой
для таких детекторов, как УФ-спектрофотометры (раздел 4.4), но некоторые
детекторы, такие как испарительный детектор светорассеяния (ИДС, раздел 4.12.1), могут
иметь гораздо более ограниченный диапазон линейности, что исключает
возможность их применения для таких целей или требует изобретения оригинальных
методов, чтобы преодолеть недостатки детектора.
11.2.5.4. Образцы с концентрациями, лежащими вне пределов
метода
Метод валидации (раздел I2.5) предназначен для определения эффективности метода
в диапазоне рабочих концентраций, часто между ПО и верхним пределом. Когда
встречаются образцы, концентрации которых находятся вне пределов метода, может
потребоваться корректировка метода, если мы хотим получить адекватные значения.
Настоятельно не рекомендуется прибегать к экстраполяции калибровочной кривой,
если концентрация образца ниже или выше диапазона метода. Достаточно часто
наблюдается нелинейное поведение из-за адсорбции образца на нижнем конце кривой,
перегрузки детектора на ее верхнем конце или из-за других факторов, что приводит
к недостаточной достоверности экстраполированных данных. Поэтому
целесообразно выбрать один из вариантов, приведенных ниже.
Образцы, концентрация которых выше верхней границы калибровочной кривой,
часто можно разбавить до попадания в рабочий диапазон метода, и, таким образом,
получить достоверные результаты. Как правило, лучше всего разбавить образец
соответствующим раствором матрицы образца (включающим все составные части
образца, за исключением анализируемого вещества, например, плазму крови или
вспомогательные вещества, содержащиеся в фармацевтическом продукте) или раствором
для разбавления (т.е. растворителем для ввода пробы). Например, разбавьте в
известном отношении образец плазмы с превышающим предел содержанием
анализируемого вещества плазмой без образца до пробоподготовки или разбавьте препарат
пестицидов раствором для введения пробы перед вводом в систему ВЭЖХ. В методах,
где иногда могут встретиться образцы с превышающим предел содержанием
анализируемого вещества, например, в предварительных фармакокинетических
исследованиях, имеет смысл включить процедуру разведения в метод при его валидации.
Например, можно подготовить пробу с добавкой стандарта, чтобы продемонстрировать,
что 10-кратное разведение образца с превышающим предел содержанием
анализируемого вещества дает тот же результат, что и разбавленный в десять раз образец
известной концентрации. Затем в метод можно внести дополнение, допускающее
разведение любого образца вплоть до 10-кратного для попадания в утвержденный
для анализа диапазон концентраций.
544 Глава II. Качественный и количественный анализы
Для образцов, концентрация которых ниже, чем на стандартной кривой, может
потребоваться введение пробы большей массы. В некоторых случаях это может быть
достигнуто путем введения пробы большего объема. В других случаях решением
проблемы может стать концентрирование образца в процессе пробоподготовки или
меньшее его разбавление. В любом случае следует провести валидацию этой
процедуры, чтобы каждый был уверен в качестве полученных данных. Некоторые методы
могут быть разработаны для интервала концентраций анализируемого вещества в
образце между ПО и НПКО, т.е. для концентраций ниже предела количественного
определения. В этих случаях в отчете сообщается, что анализируемое вещество
присутствует, но в количестве недостаточном для определения его количественного
содержания. В любом случае вносимые изменения должны соответствовать
использованию конечных результатов.
11.3. Качественный анализ
Качественный анализ предполагает проведение измерений, которые способствуют
определению структуры анализируемого вещества. В общем случае хроматография
мало эффективна для качественного анализа, но отлаженная система ВЭЖХ в
сочетании с соответствующим детектором приспособлена для этого гораздо лучше.
Существуют три общих подхода для проведения качественного анализа с помощью
высокоэффективной жидкостной хроматографии:
• определение времени удерживания;
• качественный анализ в реальном времени (онлайн анализ);
• качественный анализ в автономном режиме (офлайн анализ);
Помните, что вне зависимости от используемого метода гораздо легче доказать,
что два пика не соответствуют одному и тому же соединению, чем пытаться доказать
обратное. То есть, соединения с похожей структурой обычно имеют сходные времена
удерживания и характеристики УФ-спектра, так что время удерживания плюс
УФ-спектры не могут быть достаточными для идентификации пика. Для
подтверждения идентичности пиков могут понадобиться масс-спектрометрия, ЯМР, ИКФП
или другие методы. Тем не менее идентифицировать пик, как правило, легче, если
можно показать, что его поведение и свойства совпадают со свойствами пика
конкретного соединения или стандартного образца в противоположность соединению,
у которого нет стандартов для сравнения.
11.3.1. Время удерживания
Наиболее распространенным методом качественного анализа с помощью ВЭЖХ
является сравнение времени удерживания tR анализируемого вещества и стандартного
образца (как говорилось в разделе 11.2.2, часто вместо абсолютного времени
удерживания используется относительное удерживание). Если все условия ВЭЖХ (состав
подвижной фазы, температура, расход и т.д.) сохраняются постоянными, время
удерживания должно быть постоянным. Конечно же, условия никогда не бывают
абсолютно одинаковы, и это приводит к малым колебаниям удерживания от ввода к
вводу (например, ±0,02—0,05 мин). Если введенное анализируемое вещество попадает
в диапазон удерживания стандарта, можно сделать вывод о том, что пики стандарта
и анализируемого вещества соответствуют одному и тому же соединению. Тем не
менее, время удерживания является характеристическим, но не специфическим
параметром: одинаковое время удерживания может быть у разных соединений.
11.3. Качественный анализ 545
Для того чтобы свести к минимуму возможность перепутать одно соединение
с другим с тем же временем удерживания, надо приложить усилия для разработки
такого метода, который бы гарантировал отвечающее требованиям разрешение
представляющего интерес анализируемого вещества и любых сходных с ним примесей.
Пример соседних пиков на рис. 2.17 позволяет оценить, какое для этой цели
требуется разрешение. Оно в значительной степени зависит от относительного размера
двух смежных пиков и уширения их задних фронтов. Кроме того, следует учитывать:
(1) вероятность того, что число теоретических тарелок колонки и степень
размывания заднего фронта пика могут изменяться со временем, (2) возможность изменений
относительного удерживания из-за непреднамеренных (обычно небольших)
изменений условий разделения и (3) возможное отличие наблюдаемого времени
удерживания от истинного времени удерживания при перекрывании двух пиков. На удержи
вание также может влиять матрица образца; например, время удерживания чистого
эталонного стандарта может слегка отличаться от гд того же соединения в плазме
крови. Это одна из причин, почему методы для определения лекарств в
биологических образцах следует калибровать, используя стандарты на основе матрицы |6|
(получаемые добавлением известного количества эталона в матрицу, не содержащую
анализируемое вещество). Поскольку несколько факторов могут создать
неопределенность при использовании времени удерживания для подтверждения идентичности
пика (в чем и состоит суть качественного анализа), лучше ограничить применение это
го метода условиями, в которых более вероятно присутствие анализируемого вещества
без других компонентов, пики которых могли бы перекрываться с пиком анализируемого
вещества.
Другой способ качественного анализа, связанный с определением удерживания,
заключается в совместном введении эталонного стандарта и исследуемого образца.
В этом случае вводится образец, затем контрольный стандарт добавляется к образцу,
и он вводится снова. Этот метод сходен с методом стандартных добавок
(раздел 11.4.1.4), используемым для количественного анализа. Если пики, наблюдаемые
после двух введений, имеют одинаковые времена удерживания, ширину и форму
(в пределах допустимых изменений), то это значит, что есть дополнительные
доказательства для подтверждения идентичности обоих соединений. С другой стороны,
если при совместном введении регистрируется более широкий пик, искаженный пик
или два пика, это убедительно доказывает, что эталонный стандарт и анализируемое
вещество не одно и то же.
И, наконец, использование расчетного времени удерживания, справочных
значений времени удерживания или времени удерживания родственных веществ никогда
не обеспечит достаточную точность для подтверждения идентичности соединений,
хотя такие оценки могут оказаться полезными для определенных целей (например,
сочетание оценки удерживания с данными масс-спектров для идентификации пика).
Использование только времени удерживания для идентификации анализируемого
вещества должно быть ограничено сравнением с удерживанием известного эталонного
стандарта в тех случаях, когда присутствие мешающих пиков маловероятно.
11.3.2. Качественный анализ в реальном времени
Расшифровку структуры неизвестных анализируемых веществ можно выполнить
с помощью ВЭЖХ-детекторов, но детекторы редко столь же эффективны, как
проведенное отдельно исследование образца. Несколько ВЭЖХ-детекторов (например,
УФ-, Я MP- или ИК-спектрометры) обеспечивают спектральную информацию,
характеризующую образец. Другие детекторы, такие как хемилюминесцентный
азотный детектор, лазерный детектор светорассеяния, МС- или хиральные детекторы,
546 Глава II. Качественный и количественный анализы
дают более конкретную и количественную информацию об анализируемом веществе,
например, позволяют определить содержание азота, приблизительную массу,
молекулярную массу или оптическое вращение, соответственно. Детекторы обычно
собирают информацию при прохождении образца через проточную ячейку детектора так,
что время, доступное для измерения при прохождении пика через ячейку,
соответствует ширине пика — часто это всего несколько секунд. Еще одно ограничение
состоит в том, что образец, как правило, очень разбавлен. Кроме того, возможна
остановка потока, что дает возможность увеличить время измерения.
Методы, работающие в реальном времени, дают меньше информации по
сравнению с теми же инструментальными методами, включающими измерения на
автономных приборах, когда время анализа не ограничено и концентрация анализируемого
вещества часто гораздо больше. По этой причине данные, полученные в реальном
времени, могут быть наиболее полезны для доказательства того, что определенный
пик не является конкретным соединением, а не для выявления химической
структуры вещества. Тем не менее, когда доступен эталонный стандарт, совпадение времен
удерживания стандарта и исследуемого вещества и данные одного детектора иногда
позволяют юридически доказать идентичность анализируемого вещества и стандарта.
Примером этого является использование ЖХ-МС (или ЖХ-МС/МС) в
судебно-медицинской экспертизе, контролирующей незаконный оборот наркотиков. И, нако
нец, во многих случаях предполагаемая идентичность пика (например, метаболита
препарата) в сочетании с характеристической информацией, полученной при
детектировании в реальном времени, может оказаться достаточной для предварительного
подтверждения структуры анализируемого вещества. Если объединить данные
нескольких детекторов ВЭЖХ (например, ИКФП, МС- и хемилюминесцентного азотного
детекторов), можно с большей уверенностью идентифицировать структуру
анализируемого вещества.
11.3.2.1. УФ-детекторы
Диодно-матричный УФ-детектор (раздел 4.4.3) и реже УФ-детектор с переменной
длиной волны (раздел 4.4.2) в режиме остановки потока могут сканировать УФ-спек-
тры хроматографических пиков, в то время как они проходят через проточную
ячейку детектора. УФ-спектры сами по себе независимо от того, получены они в онлайн
или офлайн режимах, редко содержат достаточно информации для определения
структуры анализируемого вещества. Эти спектры могут способствовать
подтверждению присутствия предполагаемого соединения в образце. Тем не менее, сходство
УФ-спектров родственных соединений, как правило, не позволяет сделать
окончательный вывод относительно структуры исследуемого образца. Например, УФ-спек-
тр может помочь подтвердить, какой из пиков соответствует активному ингредиенту
в образце растворенного лекарственного средства, но он не мог бы послужить
удовлетворительным доказательством присутствия наркотика в судебно-медицинской
экспертизе.
11.3.2.2. Жидкостная хроматография с масс-спектрометрией
(ЖХ-МС)
Масс-спектрометрический детектор (раздел 4.14), особенно в режиме МС-МС,
может дать достаточную спектральную информацию для подтверждения идентичности
пика. Квадрупольный ЖХ-МС в режиме МС-МС генерирует данные по переходам
ионов-предшественников в ионы-продукты, которые могут быть использованы
для выяснения структуры неизвестного вещества, особенно когда можно получить
несколько различных переходов для одного и того же вещества. ЖХ-МС с ионной
ловушкой позволяет получать дополнительную информацию о структуре в режиме
11.3. Качественный анализ 547
MS", когда ионы-продукты могут последовательно фрагментироваться во все более
мелкие ионы (раздел 4.14.2). Однако с каждой последующей фрагментацией образец
разбавляется, а качество данных снижается. Времяпролетный масс спектрометр тоже
дает информацию о молекулярной массе анализируемого вещества, что может
способствовать идентификации структуры. Так как разрешающая способность по массе
(например, m/z к 1) ЖХ-МС-детекторов значительно ниже, чем у автономных
масс-спектрометров, МС-детекторы нельзя использовать для структурного анализа
из-за неточности дробных значений масс.
11.3.2.3. ИК-спектроскопия с Фурье-преобразованием (ИКФП)
ИК-спектрометр с Фурье-преобразованием (раздел 4.15.1) наиболее часто
используется для измерений в собранных на выходе из колонки и испаренных аликвотах
элюата. Этот детектор может давать ценную информацию (см., например, рис. 4.34)
для определения или подтверждения химической структуры вещества, элюированно-
го при разделении.
11.3.2.4. Жидкостная хроматография с детектором ядерного
магнитного резонанса (ЖХ-ЯМР)
Детектор ядерного магнитного резонанса (раздел 4.15.2) в проточном режиме или
с остановленным потоком может дать ценную информацию о структуре вещества,
выходящего в пике (пример приведен на рис. 4.35). Для получения спектров 'Н Я MP
необходимо использовать дейтерированные растворители для подвижной фазы или
испарить подвижную фазу перед тем, как снимать спектр. Это обстоятельство может
ограничить область применения ЖХ-ЯМР.
11.3.2.5. Хемилюминесцентный азотный детектор (ХЛАД)
Хемилюминесцентный детектор (раздел 4.9) реагирует на азот, содержащийся в
анализируемом веществе. Поскольку сигнал детектора пропорционален молярной
концентрации азота в образце, детектор можно откалибровать по соединениям с
известным содержанием азота. Молярное содержание азота в анализируемом веществе
можно определить по отклику детектора, зная массу вещества во введенной пробе
(пример показан на рис. 4.20). Хотя эта информация недостаточна для определения
молекулярной структуры, она может помочь в структурном анализе.
11.3.2.6. Лазерный детектор светорассеяния (ЛДС)
Лазерный детектор светорассеяния (раздел 4.12.3) позволяет приблизительно
определить молекулярную массу высокомолекулярного анализируемого вещества без
эталонного стандарта. Точность определения достаточна для того, чтобы различить
мономерньсе и димерные формы анализируемого вещества (см. пример на рис. 4.26),
но намного ниже, чем точность определения молекулярного веса в ЖХ-МС.
11.3.2.7. Хиральные детекторы
Хиральные детекторы (раздел 4.I0) могут различать энантиомерные формы
анализируемого вещества (пример показан на рис. 4.21) и указать направление вращения.
Однако никакой другой информации о структуре анализируемого вещества они
не предоставляют.
548 Глава 11. Качественный и количественный анализы
11.3.2.8. Качественный анализ в автономном режиме
(офлайн анализ)
Если отбирать фракции на выходе ВЭЖХ-системы, работающей в
полупрепаративном или препаративном режиме (глава 15), можно собрать достаточное количество
чистого вещества, чтобы провести анализ для определения его структуры в
автономном режиме. Поскольку имеется большее количество образца, чем при
аналитическом разделении, и нет ограничения по времени как при анализе в проточном
режиме, офлайн анализ может обеспечить окончательную идентификацию структуры
вещества собранного пика. Можно провести традиционный анализ спектров И К
с Фурье-преобразованием, ЯМР и масс-спектров. Имея достаточное количество
образца, также можно применить аналитические химические реакции в растворе,
рентгеновскую кристаллографию или другие аналитические методы.
11.4. Количественный анализ
ВЭЖХ может дать некоторую информацию о химической структуре образца, но
предоставляемые ею возможности количественного анализа гораздо существеннее.
Помимо аналитических весов, рН-метра или градуированной пипетки, вероятно,
наиболее часто используемым инструментом для количественного определения
в аналитической лаборатории является ВЭЖХ. Чтобы получить надежные
результаты, необходимо выполнить пять требований. Во-первых, система ВЭЖХ и
связанный с ней метод должны работать с воспроизводимостью, обеспечивающей
требуемую прецизионность и правильность данных (разделы 11.2.4.2, II.2.4.3). Во-вторых,
система сбора и обработки данных (раздел II.2) должна прецизионно и правильно
преобразовать сигнал детектора в данные зависимости сигнала от времени.
В-третьих, система должна быть должным образом откалибрована (раздел I1.4.1), чтобы
обеспечить измерение неизвестных концентраций образцов по известному
количеству вещества в эталонных стандартах. Все данные должны быть обработаны таким
образом, чтобы гарантировать, что вся процедура выполняется на должном уровне
в соответствии с нормативными стандартами (см., например, раздел 12.5) или
другими требованиями конечного пользователя. И, наконец, условия разделения должны
быть такими, чтобы базовые линии были стабильны, и разрешение было
достаточным (Rs > 1,5). Последнее требование, однако, рассмотрено в других главах и не
будет повторяться здесь.
11.4.1. Калибровка
Калибровка представляет собой процесс, в котором определяется отклик детектора
на единицу концентрации (или массы) анализируемого вещества. Некоторые
детекторы реагируют на концентрацию анализируемого вещества (например, УФ-детек-
тор, раздел 4.4), в то время как другие реагируют на его массу (например,
испарительный детектор светорассеяния, раздел 4.12.1). В данном обсуждении мы будем
считать, что детектор чувствителен к концентрации. По большей части те же самые
процедуры применимы и для детекторов, чувствительных к массе. Есть два наиболее
распространенных метода калибровки — калибровка внешним стандартом и
внутренним стандартом. Нормирование площади часто используется для анализа чистоты
вещества и других приложений, в которых относительная концентрация более важна,
чем абсолютная или отсутствуют стандарты для калибровки. Метод стандартных
добавок является специализированным методом калибровки, особенно часто использу-
11.3. Качественный анализ 549
емым в случае, когда недоступен образец без анализируемого вещества (матрица),
а матрица образца может повлиять на время удерживания и/или площадь пика
анализируемого вещества. Если у образца такая матрица (например, плазма крови),
настоятельно рекомендуется (см., например, |6|) готовить калибровочные стандарты
в матрице (т.е. в образце должны быть все компоненты, которые там обычно
находятся, за исключением анализируемого вещества, вместо которого добавляется
стандарт). Это относится как к калибровке внешним стандартом, так и внутренним
(в методах нормирования площади и стандартных добавок уже присутствует
матрица). В тех случаях, когда матрица образца оказывает незначительное влияние
на удерживание или селективность, например, при анализе проб воды из
окружающей среды, или чистых веществ, или простых смесей, калибровка стандартами в
матрице образца может не потребоваться.
Для того чтобы метод давал точные результаты, калибровочная кривая должна
правильно отражать зависимость сигнала детектора от концентрации анализируемого
вещества. Одним из способов повышения точности калибровочной кривой является
оценка взвешенной кривой. Эта тема обсуждается в разделе 11.4.1.5.
11.4.1.1. Калибровка внешним стандартом
Готовится набор калибровочных стандартов с матрицей. Обычно для этого
взвешивают точное количество эталонного стандарта известной чистоты и его растворяют
в воде или буфере, получая основной (стоковый) раствор. Затем этот раствор
добавляют к матрице образца (например, к разбавителю образца, плазме крови, воде, поч
ве, или другой матрице в соответствии с типом образца), чтобы получить
концентрацию, соответствующую максимальной на калибровочной кривой. (В некоторых
лабораториях ее называют просто кривой или стандартной кривой несмотря на то,
что график чаще всего линейный). Далее готовят разбавления стандартов в матрице,
концентрации которых охватывают весь диапазон метода, включая минимальную
концентрацию на кривой. Иногда готовят и стандарт для предела обнаружения (ПО).
Принято включать в анализ также образец матрицы без анализируемого вещества,
чтобы продемонстрировать, что в ней нет примесей, выходящих в виде пиков
на хроматограмме.
Существует две наиболее популярные схемы разведения образцов для
построения калибровочной кривой: линейная и экспоненциальная (последнюю иногда
неправильно называют «логарифмической»). Например, если диапазон метода,
охватываемый стандартной кривой, составляет от 1 до 100 нг/мл, можно приготовить
стандарты с концентрациями 0, I, 20, 40, 60, 80, и 100 нг/мл, используя линейную
схему разведения, или 0, 1, 2, 5, 10, 20, 50, и 100 нг/мл, используя
экспоненциальную схему. Одновременно с калибровочными стандартами или во время подготовки
аналитических образцов рекомендуется приготовить стандарты контроля качества
(раздел 12.3), которые будут использоваться для проверки эффективности метода
при анализе партии образцов. Если для анализируемых образцов требуется пробо-
подготовка, калибровочные стандарты (и стандарты контроля качества) также
должны пройти все стадии пробоподготовки перед введением в колонку.
Для внешней калибровки в колонку вводится одинаковый объем калибровочного
стандарта с каждым из значений концентрации в последовательности от самой
низкой до самой высокой концентрации. При такой последовательности сводится к
минимуму вероятность любых остаточных примесей, связанных с неполным элюирова-
нием образца из колонки. После ввода стандарта с самой высокой концентрацией
можно ввести стандарт с нулевой концентрацией (содержащий только растворитель
или матрицу), чтобы проверить, не переносится ли часть образца из предыдущего
разделения (раздел 17.2.5.10), а также, чтобы избежать погрешности, обусловленной
550 Глава П. Качественный и количественный анализы
таким переносом в тех случаях, если следующий образец имеет низкую
концентрацию анализируемого вещества. Для построения калибровочной кривой лучше
вводить одинаковый объем растворов стандарта с различными концентрациями,
а не разные объемы раствора с одной и той же концентрацией. Большинство авто
самплеров дозирует вводимый объем с высокой прецизионностью, но правильность
(точность) вводимого объема может быть меньше (раздел 3.6.2).
Калибровочный график можно построить вручную с помощью электронных таб
лиц (например, Microsoft Excel) или программного обеспечения системы сбора и
обработки данных. Лучше использовать программное обеспечение, поскольку в
большинстве случаев его можно валидировать. Кроме того, упрощается передача
исходных данных для обработки. С Excel и аналогичными программами удобно
работать, но они не рассматриваются как валидированное программное обеспечение
(или как подлежащее валидации) в некоторых указаниях регуляторных органов,
таких как Управление по санитарному надзору за качеством пищевых продуктов и
медикаментов (США) или Международный совет по гармонизации (США), поэтому
необходимо будет сделать дополнительную проверку данных, чтобы убедиться, что
результаты не содержат ошибок. На рис. 11.8 представлен пример калибровочного
графика с внешним стандартом, построенного поданным таблицы 11.1. Наклон
калибровочного графика S' затем используется для расчета концентрации неизвестных
образцов:
Концентрация анализируемого вещества (нг/мл) =
площадь пика анализируемого вещества (П.5)
= J' '
На рис. 11.8 (график получен линейной регрессией и проведен через начало
координат, т.е. х = 0, у = 0) S' = 200,8 (единиц площади пика)/(нг/мл
анализируемого вещества). Таким образом, пик анализируемого вещества, имеющий площадь
862 единицы, будет иметь концентрацию (862/200,8) = 4,3 нг/мл. Уравнение (11.5)
предполагает, что график пересекает ось х при у = 0, хотя в действительности это
может быть не так (раздел 11.2.4.5). Проведем вместо этого линейную регрессию так,
чтобы график не проходил через точку (0,0); это обычный подход, используемый
в программном обеспечении для обработки данных. В этом случае данные,
приведенные в таблице 11.1, описываются уравнением регрессии у = 201дг-38 (график
не проходит через начало координат). Решая уравнение для х и подставляя у = 862,
получаем х = 4,5 нг/мл (с поправкой на незначительное отклонение графика
0 200 400 600 800 1000
Концентрация (нг/мл)
Рис. 11.8. Калибровочная кривая, построенная по данным калибровки внешним
стандартом, приведенным в таблице 11.1 (кривая проведена через начало
координат)
11.3. Качественный анализ 551
от нуля). Вычисленное значение представляет собой концентрацию анализируемого
вещества во введенном в колонку образце. До предоставления данных о
концентрации вещества в образце следует внести все необходимые поправки, учитывающие
погрешности взвешивания, разбавления или другой обработки образца.
Таблица 11.1. Данные калибровки для одного и того же анализируемого вещества и
одинаковых условий разделения, использованные на рис. 11.8—11.10
Сигнал
Концентрация (нг/мл)
0
]
2
5
10
20
50
100
200
500
1000
Внешний стандарт*
215
416
976
2056
4060
9921
20140
40163
99796
201123
Внутренний стандарт*
0,0408
0,0789
0,185
0,390
0,770
1,88
3,82
7,62
18,9
38,2
Метод стандартных добавок"
487
729
911
1435
2529
"Единицы площади (рис. 11.8).
^Отношение площади пика анализируемого вещества к площади пика внутреннего стандарта
(рис. 11.9).
"Площадь, полученная при калибровке методом стандартных добавок, концентрация добавки
приведена в первой колонке (рис. 11.10).
Поскольку метод внешнего стандарта предполагает, что площадь пика
анализируемого вещества точно отражает его концентрацию в исходном образце, такую
калибровку лучше использовать в методах, которые включают минимальную обработку
образца в промежутке между отбором пробы и вводом в хроматограф. Поэтому
для твердых или жидких образцов, которые взвешивают, растворяют, отмеривают
пипеткой, разбавляют и/или фильтруют, вполне подходит метод внешнего стандарта.
При анализе растворения в фармацевтике одну или несколько таблеток препарата
помешают в емкость для растворения известного объема, в определенные моменты
времени отбирают пробы, фильтруют их и вводят в хроматограф. Образец воды
определенного объема, отобранный из окружающей среды, встряхивают с
измеренным количеством растворителя, фильтруют и вводят в хроматограф. В обоих этих
случаях легко отследить концентрацию вводимого образца относительно исходного
необработанного образца, поэтому для этих образцов вполне подойдет калибровка
внешним стандартом.
Разновидностью метода внешней калибровки является калибровка по одной точке
(раздел 11.2.4.5). При использовании этой методики в процессе разработки и валида-
ции метода выполняются эксперименты, чтобы показать, что сигнал анализируемого
вещества пропорционален его концентрации в диапазоне метода. Затем вводят
единственный стандарт, и неизвестную концентрацию анализируемого вещества в
образце определяют по отношению площадей пиков анализируемого вещества в стандарте
и в образце (как в уравнении (11.5)). Обычно диапазон такого метода узкий, напри-
552 Глава II. Качественный и количественный анализы
мер, +20% от целевой дозы препарата в таблетке. Тест растворения лекарственных
средств разработан для немедленного высвобождения субстанции, и его можно
провести с калибровкой по одной точке при наличии данных, подтверждающих
правомерность этой методики 1111.
11.4.1.2. Калибровка внутренним стандартом
Всякий раз, когда есть стадия пробо под готовки (глава 16), сопряженная с возможной
потерей анализируемого образца, лучше использовать внутренний стандарт чем
внешний. Например, определение лекарственных средств в плазме крови часто
включает твердофазную или жидкофазную экстракцию с различной степенью
извлечения, выпаривание досуха и повторное растворение в заданном объеме подвижной
фазы для последующего ввода в хроматограф. На каждом из этих этапов исходное и
конечное количество анализируемого вещества в образце редко остается
одинаковыми, но внутренний стандарт помогает отслеживать эти изменения, что позволяет
получить прецизионные и правильные результаты.
Основным отличием внутреннего стандарта от внешнего является то, что
внутренний стандарт добавляют к образцам и калибровочным растворам перед
выполнением пробоподготовки. Расчеты концентрации анализируемого вещества основаны
на отношении площадей пиков анализируемого вещества и внутреннего стандарта.
Калибровочные растворы получают путем взвешивания, растворения стандарта и
последовательного разведения полученного раствора, так же как для метода внешнего
стандарта (раздел 11.4.1.1). Аликвоты калибровочных растворов стандарта
(например, 200 мкл плазмы со стандартом) смешивают затем с раствором внутреннего
стандарта (например, с 10 мкл), как и все другие образцы, затем стандарты и образцы
обрабатывают одинаковым образом. Внутренний стандарт растворяют в воде или
буфере в такой концентрации, чтобы небольшой объем этого раствора (например,
< 5% от объема образца) при вводе давал пик достаточного размера (например,
с S/N > 100) для его измерения с требуемой прецизионностью и правильностью. Как
и в случае внешней стандартизации, стандарты вводятся в последовательности от
меньшей концентрации к большей.
Вычисляется отношение площади пика анализируемого вещества к площади
пика внутреннего стандарта для каждого из калибровочных растворов, (например,
как в данных таблицы 11.1) и строится график зависимости этого отношения от
концентрации анализируемого вещества (рис. 11.9). Линейная регрессия по этим
данным дает у = 0,038\х - (0,0073 + 0,0172). Так как абсолютная величина ординаты
пересечения графика с осью у меньше стандартной ошибки (+0,0172), правомерно
провести кривую через начало координат (раздел 11.2.4.5); тогда у = 0,0381.x. Для
этого примера S = 0,0381 (единиц отношения площадей пиков)/(нг/мл анализируемого
вещества). Расчеты концентрации неизвестных образцов проводят таким же образом,
как и для калибровки внешним стандартом, за исключением того, что используется
отношение площади пика анализируемого вещества к площади пика внутреннего
стандарта вместо абсолютной величины площади анализируемого вещества:
отношение площадей пиков анализируемого
Концентрация анализируемого вещества и внутреннего стандарта ,..,
вещества (нг/мл) S
Таким образом, если площадь пика образца составляет 15345, а внутреннего
стандарта — 4725, то их отношение равно 3,25. Из уравнения (11.6) получаем
значение концентрации анализируемого вещества в образце: (3.25 единиц отношения
площадей пиков)/0,0381 = 85,2 нг/мл анализируемого вещества.
II.3. Качественный анализ 553
О 200 400 600 800 1000
Концентрация (нг/мл)
Рис. 11.9. Калибровочная кривая, построенная по данным калибровки внутренним
стандартом, приведенным в таблице 11.1
Для того чтобы внутренний стандарт надлежащим образом выполнял свои
функции, он должен обладать определенными свойствами. Некоторые из этих свойств
приведены в таблице 11.2. В образце не должно быть компонента с таким же
временем удерживания, как у внутреннего стандарта; в противном случае будет получен
неправильный (заниженный) результат анализа. Желательно, чтобы пик внутреннего
стандарта элюировался рядом с пиком анализируемого вещества, т. е. чтобы стандарт
и образец имели сходные хроматографические параметры. Лучше всего, если пик
внутреннего стандарта элюируется сразу после пика анализируемого вещества, так
как известно, что если внутренний стандарт имеет правильное время удерживания и
площадь пика, то все пики, вышедшие раньше него, также элюировались при
надлежащих хроматографических условиях. Пики, элюированные после внутреннего
стандарта, могут быть обусловлены пузырьками или ошибками оборудования, и
убедиться в этом не удастся при анализе пика внутреннего стандарта.
Таблица 11.2. Свойства внутреннего стандарта
!.
2.
3.
4.
5.
6.
7.
8.
Никогда не должен содержаться в образце
Имеет к, сходный с к анализируемого вещества, но предпочтительно элюирование после
анализируемого вещества
Проявляет такие же свойства, как анализируемое вещество, при пробоподготовке (рКа, ли-
пофильность — log Pa/W* и т.д.)
Имеет хорошее разрешение с анализируемым веществом (или стабильную метку
для МС-анализа)
Стабилен
Чистый или известной чистоты
Соответствующий требованиям отклик детектора (пик должен иметь S/N > 100)
По строению молекулы сходен с анализируемым веществом (желательно, но не
обязательно, если есть свойство 3)
*log Рв/ъ — это логарифм коэффициента распределения октанол-вода. — Прим. перев.
Одной из главных задач внутреннего стандарта является корректировка
непостоянства извлечения анализируемого вещества в процессе пробоподготовки. Поэтому
внутренний стандарт должен обладать химическими свойствами (коэффициентом
554 Глава II. Качественный и количественный анализы
экстракции, величиной рКа в частично ионизованном состоянии и т.д.), которые
подобны свойствам анализируемого вещества. Когда в одном образце присутствуют
несколько анализируемых веществ, желательно использовать более одного внутреннего
стандарта, но обычно это не существенно. Пик внутреннего стандарта должен
хорошо разрешаться с пиком анализируемого вещества, чтобы измерение пиков не
вызывало сомнений. Исключением является анализ ЖХ-МС, где меченое стабильными
изотопами соединение часто используется как внутренний стандарт. Для стандартов
с меткой не требуется хроматографическое разрешение, если разрешение по массе
МС-детектора позволяет четко различить анализируемое вещество и внутренний
стандарт (как это обычно и бывает). Стандарты, меченые изотопами, алюирующиеся
одновременно с анализируемым веществом (это характерно для стандартов с |3С),
не подвержены изменениям при разделении (например, образование пузырьков) или
при детектировании (например, непостоянство струи распыления в интерфейсе МС).
Таким образом, одновременно элюирующиеся стандарты более точно моделируют
анализируемое вещество, чем стандарты, имеющие иное время удерживания (так
часто бывает с дейтерированными стандартами).
Внутренний стандарт должен быть достаточно стабильным и не должен
изменяться в процессе пробоподготовки и хроматографического разделения. Хотя и нет не
обходимости, чтобы внутренний стандарт имел высокую чистоту, поскольку он
стабилен, но важно, чтобы он не содержал примесей, накладывающихся на сигнал
анализируемого вещества. Для него также требуется приемлемый отклик детектора.
Некоторые требования к внутреннему стандарту подразумевают, что его структура
должна быть подобна структуре анализируемого вещества. Нет, однако, никаких
оснований для этих требований, если внутренний стандарт имеет другие
необходимые свойства. Тем не менее вещество скорее всего лучше подойдет для внутреннего
стандарта, если оно структурно подобно анализируемому веществу, поэтому
большинство пользователей выбирают для стандарта структурные аналоги. Хорошими
кандидатами на роль внутреннего стандарта являются структурные аналоги
анализируемого вещества или родственные ему соединения, которые с высокой
вероятностью отсутствуют в образце. В некоторых случаях удобно «переключение» метода,
когда два родственных соединения используются как внутренние стандарты друг
для друга. Например, в методе I, соединение X используют в качестве внутреннего
стандарта для К, а в методе 2, соединение Y используется как внутренний стандарт
для анализируемого вещества X. Ограничением для этой методики, конечно же,
является то, что X и Y никогда не должны присутствовать в одном и том же образце.
Эта методика будет работать с двумя родственными лекарственными средствами,
которые никогда не добавляются в один препарат, но имеют сходные экстракционные
и хроматографические свойства.
11.4.1.3. Нормирование площадей пиков
Стандартной формой отчета системы сбора и обработки данных является
вычисление площади пика в процентах. Это достигается путем суммирования площадей всех
пиков на хроматограмме и предоставления информации о том, какой процент от
общей суммы составляет площадь каждого пика. Этот формат отчета удобен для
определения окончания химической реакции и приблизительной чистоты продукта,
а также для других применений. В отчете также проводится нормирование площадей
пиков, когда один пик считается основным, а площадь всех остальных пиков
приводится как процент площади основного пика. Нормирование площадей применяется
в методах, используемых для проверки стабильности лекарственных средств или
количественного анализа их примесей. В последнем случае любой пик, площадь
которого 2:0,1% площади пика активной фармацевтической субстанции (АФС), должен
11.3. Качественный анализ
быть включен в отчет и идентифицирован, в то время как пики, площади которых
> 0,05%, должны быть включены в отчет, но идентифицировать их не
обязательно |10|. Удобно приводить как нормированные значения площади, так и выражен
ные в процентах, поскольку тогда не требуется стандартов для каждого пика. Однако
оба метода основаны на предположении, что отклик детектора для неоткалиброван-
ных (например, неизвестных) пиков будет таким же, как для пиков, для которых
есть стандарты. Это предположение может быть или не быть верным в зависимости
от состава образца и выбора детектора. УФ-детектирование печально известно своей
чувствительностью, различающейся на порядок для различных веществ.
11.4.1.4. Метод стандартных добавок
Метод стандартных добавок может быть полезен, когда невозможно получить
раствор образца без анализируемого вещества (матрицу), а матрица образца может
повлиять на извлечение анализируемого вещества и/или сигнал детектора. Например,
при измерении уровня инсулина в плазме крови невозможно проанализировать
плазму без инсулина, поэтому можно использовать метод стандартных добавок.
В методе стандартных добавок может применяться одноточечная или
многоточечная калибровка. Для калибровки по одной точке образец разделяется на две
фракции. К одной из фракций добавляют раствор с известным содержанием стан
дарта и затем анализируют обе фракции. Калибровочный коэффициент получают
следующим образом
. _ площадьд - площадь^
концд
(11-7)
где площадЬд и площадь^ являются площадями пиков образцов со стандартной
добавкой и без нее, соответственно, а конц„ — концентрация стандарта, добавленного
к образцу. Используя данные таблицы 11.1, мы видим, что для конц = 2 нг/мл,
площадь,, = 911 и площадьбд = 487. S = (911 - 487)/(2 нг/мл) = 212 единиц площа-
ди/(нг/мл). Теперь мы можем определить концентрацию образца без стандартной
добавки (конц^д, площадь пика дана в строке с концентрацией стандарта 0 нг/мл
в таблице 11.1):
площадь^
конц6д = -,—'\ (11.8)
или кони6д = 487/212 = 2,3 нг/мл.
Для многоточечной калибровки методом стандартных добавок образец
разбивается на я + I фракцию, где я число стандартов, которые будут использоваться. Потом
в п образцов добавляется разное количество стандарта, и все образцы
анализируются. Строится калибровочная кривая по данным таблицы 11.1, как показано
на рис. 11.10. Обратите внимание, что линия регрессии продолжается влево до
пересечения с осью абсцисс (обозначено стрелкой на рис. 11.10). Величина отрезка,
отсекаемого на оси х, соответствует -конц6д. Линейная регрессия данных таблицы 11.1
дает у = 201дг + 496. Подставив у = 0, получаем х - -2,5, поэтому концбд = 2,5 нг/мл.
Результат (в пределах стандартного отклонения < I) совпадает с результатом,
полученным при использовании уравнений (11.7) и (11.8).
Следует подчеркнуть, что метод стандартных добавок не дает поправку на
отклонения базовой линии или другие помехи, обусловленные образцом. Эти проблемы
должны быть устранены перед добавлением стандарта. По сути дела, описанный
подход является упрощенной формой калибровки непосредственно в условиях
эксперимента и может быть очень полезным, когда невозможно применить более
традиционные методы калибровки внешним или внутренним стандартом. Как отмечалось
в разделе 11.3.1, метод стандартных добавок также может быть полезным при под-
556 Глава 11. Качественный и количественный анализы
Концентрация добавки (нг/мл)
Рис. 11.10. Использование метода стандартных добавок для определения
концентрации анализируемого вещества по величине отрезка, отсекаемого на оси
абсцисс (обозначено стрелкой); данные из таблицы ) 1.1
тверждении идентичности пиков, хотя в этом отношении он не более надежен, чем
сравнение времен удерживания.
11.4.1.5. Оценка калибровочных кривых
Графики калибровочных кривых для широкого диапазона концентраций, такие как
на рис. 11.8 и 11.9, трудно интерпретировать, когда используется линейный масштаб
для оси х, так как велика плотность точек, отвечающих низким концентрациям.
Другой способ анализа данных — построить график зависимости ошибки (%) от
логарифма концентрации {«график ошибки в процентах»). Ошибка (%) определяется
с использованием уравнения регрессии для расчета теоретических значений у
для каждой концентрации. Ошибка вычисляется следующим образом:
(экспериментальное значение — теоретическое значение)/теоретическое значение и выражается
в процентах.
Преимущество графика ошибки (%) показано на рис. 11.11 для данных из
гипотетического исследования для валидации метода. В хроматографическую систему
водились калибровочные растворы 10 концентраций: I, 2, 5, 10, 20, 50, 100, 200, 500
и 1000 нг/мл. График ошибки (%) относительно линейной шкалы концентраций
на рис 11.11л показывает, что относительная погрешность гораздо больше при более
низких концентрациях, но рисунок трудно интерпретировать из-за скученности
точек данных при низких концентрациях. График значений ошибки (%) в зависимости
от логарифма концентрации на рис ll.l Iff решает проблему скученности, и теперь
ошибку для каждой концентрации можно легко изучить. Данные распадаются на два
набора: первый с концентрациями выше 20 нг/мл и относительно постоянной
погрешностью = ±1% (I стандартное отклонение), и второй с остальными
концентрациями, при которых наблюдалась возрастающая относительная погрешность по мере
уменьшения концентрации (пунктирные линии на рис. 11.115). Такая зависимость
предполагает, что S/N вносит все больший вклад в относительную погрешность
при низких концентрациях (уравнение 11.1). Если не принимать во внимание
увеличение погрешности при низких концентрациях (что закономерно), данные рис. 11.11
имеют приемлемые статистические параметры регрессии (г2 = 0,9999; отрезок,
отсекаемый графиком на оси у, меньше стандартной ошибки). Поэтому правомерно
провести многоточечную калибровочную кривую через начало координат (у = тх;
разделы 11.4.1.1, 11.4.1.2).
График ошибки (%) может выявить проблемы с калибровочными
кривыми. Данные, аналогичные представленным на рис. 11.11а,б, показаны в примере
11.3. Качественный анализ 557
(а; 15
10
£ 5
1 о
-5
-10
-15
(6) 15
\- ■
г; :
i
i
i
•
•
1
200 400 600
Концентрация (нг/мп)
600
1000
10
I
-10
-15
\.
• \ч t ±1 осо
. ' >:-:
1
■
•
•
1
*
•
i
■
i
•
•
i
*
10 100
Концентрация (нг/мп)
1000
(в) 70
60
- 50
Г 40
30
20
10
»
•
\ Усредненная
ч величина
\ ■
ч *
• ч
*-^,
1
_f
'
10 100
Концентрация (нг/мп)
1000
Рис. 11.11. Использование графиков ошибки в процентах для изучения данных
калибровочной кривой, (с) График зависимости ошибки в процентах от
концентрации в линейном масштабе; (6) данные графика (а) в
полулогарифмическом масштабе (по оси абсцисс отложены значения логарифма
концентрации; отрезок, отсекаемый на оси у. меньше стандартной ошибки; кривая
проведена через начало координат); (в) зависимость ошибки в процентах
от логарифма концентрации (отрезок, отсекаемый на оси у, больше
стандартной ошибки; не следовало проводить кривую через начало координат)
558 Глава II. Качественный и количественный анализы
на рис. П.[Iff, в котором подчеркивается важность правильного выбора отрезка,
отсекаемого на оси ординат. Если выбрана многоточечная калибровка, и кривая
проведена через начало координат, получается график ошибки (%), приведенный
на рис. 11.1 [е. При более низких концентрациях наблюдается увеличение ошибки,
среднее значение которой достигает = 50% при I нг/мл. Для биоаналитического
метода, где максимальное допустимое отклонение при НПКО = ±20%, самая низкая
концентрация со средней погрешностью < 20% составляет в данном случае 5 нг/мл
(ошибка « 9%). Это ограничивает применение метода диапазоном концентраций от 5
до 1000 нг/мл. Для этого набора данных отрезок, отсекаемый на оси у, больше
стандартной ошибки, так что калибровочную кривую следует аппроксимировать
уравнением у = тх + b (с Ь * 0). Полученный график ошибки (%) (не показан) сходен
с графиком на рис. 11.116, средняя ошибка < 6% на всем протяжении кривой. С та
кой калибровочной кривой биоаналитический метод можно использовать в
диапазоне от 1 до 1000 нг/мл. Интересно отметить, что при обеих аппроксимациях кривой
(с Ь = 0 или Ь * 0) коэффициент корреляции г2 > 0,9999, так что значение г2 само
по себе не является достаточным для качественной многоточечной калибровки.
Малые значения г или г- указывают на проблемы с калибровочной кривой,
но обратное не всегда верно: большое л2 не гарантирует точную калибровочную
кривую. График зависимости ошибки в процентах от логарифма концентрации (график
ошибки (%)) полезен для визуального анализа данных. Мы рекомендуем
анализировать все данные калибровочной кривой, используя график ошибки (%) для выявления
потенциальных проблем с методом,
11.4.2. Анализ следовых количеств
ВЭЖХ используют для анализа образцов с различной концентрацией. Термин анализ
следовых количеств часто используется для описания образцов с малыми
концентрациями. Один из способов охарактеризовать анализ следовых количеств — это
определение образцов, для которых точность измерения зависит от концентрации, часто с
указанием точки перехода к анализу следовых количеств при S/N < ж 100 (раздел 4.2.3).
За исключением проблем, связанных с низкими концентрациями и малыми
сигналами, анализ следовых количеств мало чем отличается от анализа более
концентрированных образцов. В анализах такого рода предпочтительно измерение высоты пика, а не
площади. Мы рекомендуем измерять как высоту, так и площадь пика. Выбор
окончательного метода измерения зависит оттого, какой параметр после статистической
обработки даст лучшую прецизионность и правильность. Для получения дополнительной
информации см. разделы 2.6.3.2, 4.2.4 и 11.2.5. Для пояснений, связанных с
использованием конкретных детекторов, обратитесь к разделам 4.4—4.16.
11.5 Выводы
Использование ВЭЖХ для качественного или количественного анализа требует
работы хроматографической системы должным образом, и соответствующей настройки
системы сбора и обработки данных для правильного определения времени
удерживания и площади пиков или их высоты. Следует рассмотреть требуемое разрешение
целевых пиков, в том числе влияние относительного размера пика и его формы. Хотя
система ВЭЖХ не столь полезный инструмент для качественного анализа, как
некоторые специализированные приборы (например, ИКФП, ЯМР, или
масс-спектрометры высокого разрешения), с помощью соответствующего детектора (или детекто-
Литература 559
ров) она может обеспечить ценную информацию о структуре анализируемых
веществ, имеющую много областей применения. Жидкостная хроматография
предоставляет широкие возможности для количественного анализа. ВЭЖХ можно
использовать для анализа следовых количеств загрязняющих веществ в речной воде,
медицинских препаратов и их метаболитов в биологических системах или примесей
реактивов. Она также полезна для определения однородности содержимого
фармацевтических продуктов с высокой прецизионностью и правильностью.
Количественный анализ основан на выборе соответствующих стандартов и правильной
калибровки, чтобы качество результатов было высоким, и они могли пройти проверку
регуляторных органов.
Литература
1. N. Dyson, Chromatographic Integration Methods, 2nd ed., Royal Society of Chemistry, Letchworth,
UK, 1998.
2. L. R. Snyder and J. J. Kirkland, Introduction to Modem Liquid Chromatography, 2nd ed., Wiley,
New York, 1974.
3. V. R. Meyer, /. Chromatogr. Sci., 33 (1995) 26.
4. R. Q. Thompson, J. Chem. Ed., 62 (1985) 866.
5. Guidance for Industry: Part II, Electronic Records; Electronic Signatures—Scope and Application.
USFDA-CDER, Aug. 2003.
6. Guidance for Industry: Bioanalytical Method Validation, USFDA-CDER, May 2001,
http://www.fda.gov/cder/guidance/index.htm.
7. Reviewer Guidance: Validation of Chromatographic Methods, USFDA-CDER, Nov. 1994,
http://www.fda.gov/cder/guidance/index.htm.
8. L. R. Snyder, J. J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, 2nd ed., Wi-
ley-lnterscience. New York, 1997, p. 71.
9. Validation of Analytical Procedures: Text and Methodology Q2 (Rl), International Conference on
Harmonization, Nov. 2005, http://www.ich.org/LOB/media/MEDIA417.pdf.
10. Impurities in New Drug Substances Q3A (R2), International Conference on Harmonization, Oct.
2006, http://www.ich.org/LOB/media/MEDIA422.pdf.
11. Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug
Products: Chemical Substances Q6A, International Conference on Harmonization, Oct, 1999,
http://www.ich.org/LOB/media430.pdf.
12. J. C. Miller and J. N. Miller, Statistics for Analytical Chemistry, Halsted Press-Wiley, New York,
1984, sees. 4.9-4.10.
ГЛАВА 12
ВАЛИДАЦИЯ МЕТОДА
Совместно с Майклом Шварцем
12.1. Введение 562
12.2. Термины и определения 565
12.2.1. Правильность 566
12.2.2. Прецизионность 568
12.2.2.1 Повторяемость 568
12.2.2.2. Внутрилабораторная (промежуточная) прецизионность. . 568
12.2.2.3. Воспроизводимость 569
12.2.2.4. Надежность 569
12.2.3. Специфичность 569
12.2.4. Предел обнаружения и предел количественного определения . . . 569
12.2.5. Линейность и аналитическая область 571
12.2.6. Устойчивость 572
12.3. Пригодность системы 573
12.4. Документация 575
12.4.1. Валидационный протокол 575
12.4.2. Метод испытания 576
12.4.3. Валидационный отчет 577
12.5. Валидация для различных типов фармацевтических методов 578
12.5.1. Методы категории 1 578
12.5.2. Методы категории 2 578
12.5.3. Методы категории 3 579
12.5.4. Методы категории 4 580
12.6. Биоаналитические методы 580
12.6.1. Подготовка стандартного образца 581
12.6.2. Разработка и валидация биоаналитического метода 581
12.6.2.1. Избирательность 582
12.6.2.2. Правильность, прецизионность и степень извлечения. . . 582
12.6.2.3. Калибровочные/стандартные кривые 583
12.6.2.4. Стабильность биоаналитического образца 584
12.6.3. Рутинное применение биоаналитического метода 585
12.6.4. Документирование биоаналитического метода 586
12.7. Передача (трансфер) методики анализа в принимающую
лабораторию (ТМА) 587
12.7.1. Опции передачи (трансфера) методики анализа 588
12.7.1.1. Сравнительное тестирование 588
12.7.1.2. Совместная валидация 589
12.7.1.3. Валидация и/или повторная валидация метода 589
12.7.1.4. Отказ от оформления передачи метода испытания 589
Содержание 5 61 If)
12.7.2. Основы ТМА 590
12.7.2.1. Предварительно утвержденный протокол плана
тестирования 590
12.7.2.2. Описание метода / процедуры испытаний 590
12.7.2.3. Описание и обоснование требований к тестам 592
12.7.2.4. Критерии приемлемости 592
12.7.2.5. Документирование результатов 593
12.7.3. Потенциальные ошибки при ТМА 593
12.7.3.1. Оценка оборудования 594
12.7.3.2. Колонки ВЭЖХ 594
12.7.3.3. Обучение оператора 594
12.8. Корректировка методики или модификация методики 595
12.8.1. Корректировка рН 597
12.8.2. Концентрация соли в буфере 597
12.8.3. Соотношение компонентов в подвижной фазе 597
12.8.4. Длина волны УФ/ВИД фотометрического детектора 598
12.8.5. Корректировка температуры 598
12.8.6. Изменение длины и диаметра колонки, размера частиц сорбента 598
12.9. Контроль качества и обеспечение качества 599
12.9.1. Контроль качества 599
12.9.2. Обеспечение качества 599
12.10. Выводы 600
Литература 601
562 Глава 12. Валидация метода
12.1. Введение
Качество — это общепризнанный термин в области аналитической химии. Качество
определяет многие аспекты деятельности аналитической лаборатории; в данной
главе этот критерий рассматривается в приложении к разработке и применению
методик анализа, выполняемых с использованием высокоэффективной жидкостной
хроматографии (ВЭЖХ). Основное внимание будет уделено валидации методик
ВЭЖХ — процессу, который удостоверяет качество методики анализа и результатов
лабораторных исследований. Кроме того, в этой главе обсуждаются и другие
вопросы, связанные с качеством в лаборатории ВЭЖХ, особенно контроль качества и
обеспечение качества (раздел 12.9).
С помощью лабораторных исследований валидация метода устанавливает, что
представленные характеристики метода анализа соответствуют требованиям
предполагаемого аналитического применения. Валидация метода гарантирует надежность
метода при повседневном использовании; этот процесс иногда называют также
документированным подтверждением того, что этот метод делает то, что он должен
делать'. Контролируемые лаборатории обязаны проводить валидацию методик анализа
для того, чтобы соответствовать требованиям государственных или других регулятор-
ных органов. Четко определенный и документированный процесс валидации метода
должен не только удовлетворять установленным нормативным требованиям, но и
предоставлять доказательства того, что определенная система и методика анализа
пригодны для использования по предполагаемому назначению, а также могут быть
переданы в стороннюю лабораторию. В 1987 году Управление по санитарному
надзору за качеством пищевых продуктов и медикаментов США (FDA)2 впервые признало
спецификации, перечисленные в текущем издании Фармакопеи США (USP)3,
имеющими законную силу и соответствующими Федеральному акту по продуктам
питания, лекарствам и косметическим средствам |1—2|. Позже была опубликована новая
информация, которая содержит обновления предшествующих рекомендаций и
обеспечивает более полное согласование с руководящими принципами, изложенными
в трудах Международной конференции по гармонизации технических требований
к регистрации фармацевтических продуктов, предназначенных для применения
человеком |3—4|. Определения некоторых терминов в перечнях FDA, USP и
Международного совета по гармонизации технических требований к регистрации
фармацевтических продуктов, предназначенных для применения человеком (ICH)4 несколько
различаются, а также не все термины включены в документацию всех трех
организаций. Однако согласование на глобальном уровне обеспечило гораздо более глубокую
проработку терминов, чем это было доступно в прошлом. Это может быть полезным
для того, чтобы свести к минимуму любые различия между нормативными
требованиями в разных странах.
Методика ВЭЖХ может быть обозначена терминами «аналитическая методика»,
«аналитический метод», «метод испытания» или просто «метод». В данной главе
для любой методики обычно используется термин «метод испытания» или (реже)
' Использование валидированного метода анализа будет приводить к желаемому результату,
т.е. метод будет корректно работать от раза к разу. — Прим. перев.
-Здесь и в дальнейшем в этой главе эти и другие регуляторные органы США обозначаются
латинскими сокращениями, приведенными при первом упоминании в скобках, а также
включенными в словарь условных терминов и принятых сокращений. FDA — Food and Drug Administration —
Прим. перев.
^USP — United States Pharmacopeia — Прим. перев.
4|CH — International Council for Harmonization of Technical Requirements for Pharmaceuticals
for Human Use. — Прим. перев.
12. L Введение 563
«методикп анализа ВЭЖХ». Наибольшее число методик ВЭЖХ, используемых в
настоящее время во всем мире, применяется в фармацевтической промышленности.
По этой причине в данной главе будет описан подход к валидации ВЭЖХ-методик
анализа, применяющийся в фармацевтике. Другие регулируемые законами отрасли
промышленности также имеют четко определенные подходы к валидации методик
анализа. Например, лаборатории мониторинга окружающей среды находятся под
надзором Агентства по охране окружающей среды (ЕРА)1 |5|, в то время как
некоторые другие организации руководствуются указаниями Международной организации
по стандартизации (ISO)2 |6|.
Валидация и другие аспекты деятельности лабораторий регулируются FDA, USP,
ICH, ЕРА и связанными с ними организациями. Поскольку методики ВЭЖХ,
разработанные в промышленной аналитической лаборатории, часто используются в
производственном цикле, деятельность аналитической лаборатории может быть
ограничена производственным регламентом и соответствующими инструкциями. В этом
случае обычно руководствуются двумя стандартами: cGMP3 (современная
надлежащая производственная практика, см., например, |7—8|) и Всемирными стандартами
менеджмента ISO 9000 |9|, а также другими документами этой организации. Данные
аспекты регулирования лабораторной документации не обсуждаются в этой главе.
В нерегулируемых отраслях промышленности и научных лабораториях также
существует потребность в высококачественных методах испытаний, обеспечивающих
надежные данные. Предполагается, что в научных лабораториях используются
надежные и точные методы, хотя это и не всегда так. Поэтому валидация
методики анализа настоятельно рекомендуется, даже если это не требуется по
законодательству. Читателю следует принять во внимание представленную здесь информацию
для фармацевтической отрасли и использовать ее в качестве базовой инструкции
для применения в других областях.
Валидацию методики анализа можно рассматривать как всего лишь одну часть
общего процесса валидации, который охватывает, по меньшей мере, четыре
различных этапа: (1) валидацию программного обеспечения, (2) квалификацию
(аттестацию) или валидацию лабораторного оборудования (системы ВЭЖХ) (раздел 3.10.1),
(3) валидацию методики анализа и (4) проверку пригодности системы. Процесс
валидации начинается с проведения валидации программного обеспечения и системы
ВЭЖХ; затем разрабатывается метод испытания и проводится его валидация с
использованием квалифицированной системы ВЭЖХ. Наконец, применимость метода
испытания в данный момент может быть подтверждена с помощью теста на
пригодность системы. Каждый из этих четырех этапов имеет решающее значение для
подтверждения пригодности метода.
При проведении валидации метода важно руководствоваться двумя
нормативными документами: главой USP 1225 «Валидация фармакопейных методов испытаний»
|2|, а также руководством Международной конференции по гармонизации (ICH)
«Валидация аналитических методик: рекомендации и методология Q2 (R1)» |4|. Хотя
предметом настоящего обсуждения являются ВЭЖХ-методики, директивы USP и
ICH применимы к любой аналитической методике, методу или технологии,
используемым в контролируемой лаборатории. Следует отметить, что в USP опубликованы
официальные методы испытаний, часто называемые фармакопейными методами,
которые рассматриваются как уже валидированные. В USP приведены также
руководящие указания, которые следует применять при валидации методов испытаний,
неопубликованных в USP (предполагается, что опубликованные в USP методы разра-
' ЕРА — Enviromental Protection Agency — Прим. перев.
2ISO — International Organization for Standardization — Прим. перев.
3cGMP — current Good Manufacturing Practice — Прим. перев.
564 Глава 12. Валидация метода
ботаны в соответствии с теми же указаниями). В этой главе основное внимание
уделяется применению методических указаний USP (и других регуляторных органов)
при разработке методик ВЭЖХ независимыми лабораториями (т.е. не самими регу
ляторными органами).
Хотя методические указания USP являются единственным юридическим
документом, который признает FDA, в этой главе приводятся определения и методы
из руководств USP и ICH, так как директивы FDA, USP и ICH в значительной
степени согласованы между собой. Если методические указания не согласуются,
пользователь самостоятельно принимает решение об их надлежащем толковании. Часто
это является обязанностью отдела обеспечения качества (ООК) пользователя (раздел
12.9). Разобраться с выбором инструкций могут помочь и регулярно обновляемые
обзоры руководящих указаний (например, публикуемая FDA Форма 483 по
результатам пересмотра опубликованных рекомендаций). При дальнейшем обсуждении
публикуемые нормативные документы мы будем называть «методическими
рекомендациями». В дополнение к методическим рекомендациям тот или иной регуляторный
орган иногда публикует другую информацию, которая может быть полезной для
правильной интерпретации методических рекомендаций. Одним из таких изданий
является «Руководство для контролирующих инстанций» |10|, которое публикует FDA.
Этот документ предназначен для аудиторов FDA и содержит разъяснения
относительно того, что представляет собой адекватный метод испытания. Поэтому многие
пользователи стараются придерживаться рекомендаций, приведенных в этом
документе, чтобы получить гарантию, что используемый ими метод испытания будет одобрен
регупяторными органами. Помимо общей процедуры валидации методики анализа
обсудим также термины, определения и связанные с данной тематикой вопросы,
а также по возможности приведем примеры, чтобы показать, как эти общие
указания применяются к методу ВЭЖХ.
Основное различие между этой главой и другими главами книги обусловлено регу-
ляторным надзором за валидированием методик анализа в таких областях как
экология, фармацевтическая и некоторые другие отрасли промышленности. Правила,
относящиеся к валидации методик анализа, описаны в официальных документах, которые
появляются в разное время, написаны разными людьми (имеющими разные навыки
написания текстов), и выпускаются различными организациями (с различной
внутренней политикой). Этот бюрократический процесс неизбежно приводит к появлению
документов, которые могут быть неоднозначны, противоречивы и трудно
интерпретируемы. В этой главе мы пытаемся придать некоторое единообразие регуляторным
требованиям и указаниям, приведенным в различных нормативных положениях. Мы
также постараемся сделать обсуждение полезным с практической точки зрения для
обычного пользователя. Тем не менее, бюрократический язык нормативных
рекомендаций и требований невозможно полностью замаскировать. С другой стороны, вы
получите хорошую практику для работы с официальными документами.
Может показаться, что в протоколах валидации методик анализа используется
набор собственных терминов. Поэтому эта глава начинается с обсуждения важных
терминов и определений (раздел 12.2). В разделе 12.3 описано тестирование
пригодности хроматографической системы, гарантирующее, что используемый метод
испытания дает достоверные результаты. Без документального подтверждения проведения
валидации аналитической методики нет уверенности, что метод является надежным.
Некоторые аспекты оформления документации валидируемой методики анализа
описаны в разделе 12.4. Требования к валидации метода испытания
фармацевтической субстанции (чистого активного вещества) и лекарственного препарата
(лекарственной формы), изложенные в разделе 12.5, существенно отличаются от требований
к процедуре валидации биоаналитических методов определения лекарственных
веществ в биологических матрицах (раздел 12.6). После проведения валидации метода
12.2. Термины и определения 565
испытания его часто необходимо передать в другую лабораторию для повседневного
(рутинного) применения; некоторые принципы передачи (трансфера) методик
анализа (ТМА)1 обсуждаются в разделе 12.7. Довольно часто бывает, что переданные
методы испытаний не работают точно так, как в лаборатории, разработавшей эти
методы. Со временем большинство методов требуют определенной корректировки, чтобы
соответствовать требованиям пригодности хроматографической системы. Методы
можно скорректировать, чтобы удовлетворить требованиям пригодности
хроматографической системы, но если для этого необходимы более существенные изменения
(модификации), то эти методы испытаний должны пройти повторную валидацию (ре-
валидацию). Различие между корректировкой и модификацией метода
рассматривается в разделе 12.8. И, наконец, применение надежного метода испытания требует
эффективного контроля качества2, а, следовательно, необходимо разработать
программу обеспечения качества, как описано в разделе 12.9.
12.2. Термины и определения
Следующие валидационные характеристики могут быть исследованы в процессе ва-
лидации любой методики анализа:
• правильность (accuracy);
• прецизионность/надежность метода (precision/ruggedness);
• специфичность (specificity);
• предел детектирования (limit of detection);
• предел количественного определения (limit of quantitation);
• линейность (linearity);
• аналитическая область (range);
• устойчивость (robustness).
Хотя большинство этих терминов3 знакомы и ежедневно используются в любой
регулируемой лаборатории ВЭЖХ, иногда разные люди подразумевают под ними
разные понятия. Например, надежность, которая составляет часть любого хорошо
продуманного исследования прецизионности, часто путают с устойчивостью метода.
Приводимые далее в разделах I2.2.T —12.2.6 стандартные определения, применяемые
в фармацевтической промышленности, позволяют устранить любую путаницу в
терминологии. В этом контексте под лекарственным веществом подразумевается чистое
химическое вещество, также называемое активным фармацевтическим ингредиентом
(АФИ) или фармацевтической субстанцией (ФС ). Термин лекарственный препарат
относится к продукту, который продается потребителю; в лекарственном препарате
обычно содержатся одна или несколько фармацевтических субстанций, а также
вспомогательные вещества (химические вещества, не относящиеся к ФС: наполнители,
красители и т.д.).
Для определения содержания ФС и/или примесей, родственных соединений,
наполнителей и т.д. используются несколько типов методов испытаний. Основные
типы методов, обсуждаемые в этой главе — количественное определение,
определение примесей (а также родственных соединений), растворение и биоаналитические
методы. Метод испытания, называемый количественным определением,
подразумевает измерение концентрации активного вещества в лекарственном препарате или
' По-английски это называется АМТ — Analytical Method Transfer. — Прим. перев.
^получаемых аналитических данных. — Прим. перев.
^Термины по ГФ 14 РФ. — Прим. перев.
4 В соответствии с ГФ 14 РФ. — Прим. перев.
566 Глава 12. Валидация метода
фармацевтической субстанции. Метод определения однородности содержания
аналогичен методу количественного определения, но тест на однородность
содержания нацелен на определение отклонений концентрации активного вещества в об
разцах в пределах одной партии. При помощи метода испытания на примеси
измеряются концентрации компонентов с малым содержанием, присутствующих
в фармацевтической субстанции или лекарственном препарате (как правило,
ненамеренно внесенных), которые появляются в продукте из исходного сырья,
при производстве готовой продукции, или при разложении фармацевтической
субстанции во время хранения или обработки. Метод определения стабильности
используется для количественного определения присутствия примесей (продуктов
разложения), образующихся в результате принудительной деградации ФС.
Предполагается, что этот тест позволяет определить любые примеси, образующиеся в
процессе нормального или ускоренного испытания при определении срока годности
фармацевтической субстанции или лекарственного препарата. Любые продукты
разложения, установленные таким способом, могут быть впоследствии включены
в число определяемых примесей. В тесте на растворение измеряется концентрация
ФС в растворе; этот тест предназначен для имитации высвобождения
лекарственного вещества из формы лекарственного препарата в условиях его введения
(например, в моделируемых жидкостях желудка). В то время как перечисленные
методы испытаний предназначены для лекарственных препаратов или ФС, биоана
литическии метод (раздел 12.6) используется для определения концентрации
лекарственного вещества в биологической системе, чаще всего в плазме крови.
12.2.1. Правильность
Правильность является мерой точности аналитического метода или степени близости
значения, найденного в образце, к принятому опорному (истинному) значению.
Правильность измеряется во всей аналитической области метода как процентное
содержание анализируемого вещества, установленное при количественном
определении. Для фармацевтической субстанции правильность измерения определяется
сравнением результатов, полученных для испытуемого образца, со значением,
полученным для стандартного образца фармацевтической субстанции, или при сравнении
с результатами анализа, полученными другим хорошо охарактеризованным методом.
Для количественного определения лекарственного препарата правильность
оценивается с помощью анализа синтетических смесей с добавками известного количества
определяемого вещества. Для количественного определения примесей правильность
определяется при помощи анализа образцов (фармацевтической субстанции или
лекарственного препарата) с добавками известных количеств примесей (если примеси
не являются доступными, см. специфичность в разделе 12.2.3).
В таблице 12.1 проиллюстрировано типичное исследование правильности. Чтобы
документально подтвердить значение правильности, в нормативных указаниях
рекомендуется получить аналитические данные минимум девяти определений для как
минимум трех значений концентраций, охватывающих указанную аналитическую
область (т.е. три образца с различной концентрацией анализируются в трех повторах).
Полученные данные должны быть представлены как процент извлечения известного
или добавленного количества или как разность между средним полученным
значением и истинным значением с обозначением доверительного интервала (+I
стандартное отклонение, СО). В таблице 12.1 приведены значения извлечения относительно
100%, а средняя степень извлечения для л = 9 образцов составляет 98,62% с ОСО =
= 0,32%. В этом примере как правильность, так и прецизионность соответствуют
заданным критериям приемлемости 98—102% и <2% соответственно.
12.2. Термины и определения 567
Таблица 12.1. Определение правильности/степени извлечения и прецизионности методики
анализа
Концентрация образца
(мг/мл)
1.000
2.000
3.000
Среднее
Стандартное отклонение (СО)
Относительное стандартное
отклонение (ОСО)
Критерий приемлемости
результата
Оценка теста
Правильность/степень извлечения
Повтор 1
98,91%
99,08%
98,78%
98,62%
0,32%
0,32%
Правильность (среднее)
98-102%
выполнен
Повтор 2
98,79%
98,54%
98,68%
Прецизионность (ОСО)
<2,.0%
выполнен
Повтор 3
98,44%
98,39%
98,01%
Таблица 12.2. Определение повторяемости при повторных вводах одного и того же образца
Ввод образца
1
2
3
4
5
6
Среднее
Стандартное отклонение
Относительное стандартное отклонение (ОСО)
Критерий приемлемости (ОСО)
Оценка теста
Отклик
488,450
488,155
478,986
489,247
487,557
487,923
488,220
582.15
0,12%
5 2,0%
выполнен
Таблица 12.3. Измерение межллборяторной (промежуточной) прецизионности
Среднее
Стандартное отклонение
Относительное стандартное отклонение (ОСО) %
Расхождение средних значений %
Критерий приемлемости (ОСО)
Оценка теста
Количество
Первый аналитик
13,9 мг
0,05 мг
0,36
0,70
<2,0%
выполнен
Второй аналитик
14,0 мг
0,03 мг
0,21
568 Глава 12. Валидация метода
12.2.2. Прецизионность
Прецизионность аналитической методики определяется как степень близости друг
к другу отдельных результатов испытаний при повторных анализах однородного
образца. Для определения прецизионности обычно выполняют измерения трех
различных величин: повторяемости, внутрилабораторной (промежуточной) прецизионности
и воспроизводимости (межлабораторной прецизионности).
12.2.2.1 Повторяемость
Повторяемость — это способность методики анализа давать сопоставимые
результаты анализа в течение короткого промежутка времени в одинаковых условиях
(прецизионность в рамках одной серии анализов). Эту величину следует определять как
минимум в девяти повторах. Такая повторяемость должна охватывать определенную
аналитическую область методики анализа: т.е. необходимо провести анализ образцов
с тремя разными концентрациями в трех повторах для каждого значения
концентрации или проверить повторяемость как минимум в шести повторах для 100%
тестируемой или номинальной концентрации. Типичные результаты определения
повторяемости приведены в таблице 12.2, в которой обобщены результаты шести вводов
одного и того же образца. Полученное ОСО 0,12% вполне соответствует критерию
приемлемости <2%.
12.2.2.2. Внутрилабораторная (промежуточная) прецизионность
Под промежуточной прецизионностью подразумевается соответствие между
результатами анализов в условиях внутрилабораторных изменений, обусловленных
случайными событиями, которые обычно могут происходить при использовании методики
анализа в одной и той же лаборатории, например, в разное время разными
аналитиками на разном оборудовании. Для определения промежуточной прецизионности
следует спланировать эксперимент так, чтобы воздействие отдельных факторов (если
оно наблюдается) можно было контролировать. Типичные результаты определения
промежуточной прецизионности приведены в таблице 12.3. В этом исследовании
аналитики из двух разных1 лабораторий подготовили и проанализировали шесть
образцов препарата из одной партии и по одному образцу препарата из двух
дополнительных партий (подразумевается, что концентрация всех образцов была одинакова);
все данные, полученные обоими аналитиками, были объединены в таблице 12.3.
Аналитики независимо друг от друга подготовили свои собственные растворы
стандартных и испытуемых образцов, использовали для анализа колонки из разных
партий и разные системы ВЭЖХ. Оба аналитика успешно выполнили требования
по прецизионности < 2% ОСО, а разброс полученных ими средних значений
составил 0,7%. Это указывает на то, что нет никаких различий в полученных средних
значениях (f-критерий Стьюдента2 дает вероятность Р = 0,01).
'Авторы приводят не совсем корректный пример, т.к. аналитики были из разных лабораторий,
а промежуточная прецизионность предполагает рассмотрение вариабельности результатов в
пределах одной лаборатории. — Прим. персе.
2Статистическим критерием называется случайная величина, например, t, которая служит
для оценки статистической значимости модели, в частности, гипотезы об отсутствии различий
в результатах выборки. Буквой / обозначается случайная величина, подчиненная распределению
Стьюдента. Наблюдаемому значению критерия I соответствует определенная вероятность Р того,
что будет отвергнута указанная статистическая гипотеза. В исследованиях проверку гипотез
осуществляют при 1%-ном уровне значимости, т.е. при Р = 0,01. Практически уровень значимости
можно определить с помощью Excel. — Прим. перее.
12.2. Термины и определения 569
12.2.2.3. Воспроизводимость
В отчетные документы совместных исследований, проводимых в разных
лабораториях, следует включать значения стандартного отклонения, относительного
стандартного отклонения (или коэффициента вариации) и доверительного интервала.
В таблице I2.4 приведены некоторые результаты, типичные для исследования
воспроизводимости. Чтобы получить приведенные данные, аналитики из двух разных
лабораторий (но не те аналитики, которые участвовали в исследовании
промежуточной прецизионности), подготовили и проанализировали по шесть образцов из одной
партии продукта и по одному образцу из двух дополнительных партий
(подразумевается, что концентрация всех образцов была одинакова). Аналитики независимо друг
от друга подготовили свои собственные растворы стандартных и испытуемых
образцов, использовали колонки ВЭЖХ из разных партий и разные системы ВЭЖХ
для оценки содержания образца в растворах. Оба аналитика успешно выполнили
требование по прецизионности ОСО <2%, и расхождение полученных ими средних
значений составило 1,4%, что свидетельствует о том, что нет никакой разницы в
полученных средних значениях (/-критерий Стьюдента дает вероятность Р = 0,01).
Таблица 12.4. Измерение воспроизводимости
Среднее
Стандартное отклонение
Относительное стандартное отклонение (ОСО) %
Расхождение средних значений %
Критерий приемлемости (ОСО)
Оценка теста
Количество
Первая лаборатория
14,0 мг
0,07 мг
0,50
1,43
<2,0%
выполнен
Вторая лаборатория
13,8 мг
0,14 мг
1,01
12.2.2.4. Надежность
В предыдущих руководствах USP надежность определялась как степень
воспроизводимости результатов испытаний, полученных в результате анализа одних и тех же
образцов в различных условиях, таких как: различные лаборатории, аналитики,
приборы (системы ВЭЖХ), партии реактивов, температура при проведении анализа,
время, прошедшее с момента анализа, или, собственно, дата его проведения.
Надежность является мерой воспроизводимости результатов испытаний, ожидаемой в
общем случае при переходе от лаборатории к лаборатории и от аналитика к аналитику.
Однако термин надежность выходит из употребления, например, не используется
в руководстве ICH, но упоминается в тексте Q2 (R1) |4| при обсуждении внутри-
лабораторной прецизионности (раздел 12.2.2.2, внутрилабораторные изменения
из-за проведения анализов в разные дни разными аналитиками, на разном
оборудовании и т.д.) и воспроизводимости (раздел 12.2.2.3, межлабораторные изменения при
совместных исследованиях).
12.2.3. Специфичность
Специфичность определяется возможностью аналитической методики точно и
избирательно (специфично) измерять определяемое вещество в присутствии других
компонентов, которые предположительно могут присутствовать в испытуемом образце.
570 Глава 12. Валидация метода
Специфичность учитывает степень влияния погрешностей/помех, привносимых
другими активными соединениями, наполнителями, примесями, продуктами
разложения и т.д. Специфичность метода испытания гарантирует, что измеряемый отклик
определяемого соединения относится только к одному определяемому компоненту
(без перекрывания другими пиками). Специфичность для данного анализируемого
вещества обычно измеряется и документируется разрешением, числом теоретических
тарелок (эффективностью) и фактором размывания пика.
С целью точной идентификации определяемого вещества, специфичность
методики анализа должна быть наглядно продемонстрирована при помощи: (I) его
отделения от других соединений в пробе и/или (2) сравнения с известными
стандартными образцами.
Отделение от других соединений. В количественном определении и тестах
на определение примесей специфичность можно подтвердить с помощью
разрешения пиков двух близко элюируемых друг к другу соединений из тех, которые могут
присутствовать в образце. Этими соединениями, как правило, являются основной
компонент или фармацевтическая субстанция и элюируемая ближе всего примесь.
Если образцы примесей доступны, необходимо продемонстрировать, что
количественный анализ определяемого вещества не зависит от добавления примесей и/или
наполнителей. Если образцы примесей не доступны, проводятся испытания с
использованием второго хорошо изученного метода1. Для количественного анализа
сравниваются результаты, полученные при использовании обеих методик. Для тестов на
присутствие примесей сравниваются профили примесей. Критерии сравнения
результатов испытаний будут меняться в зависимости от конкретного метода испытания.
Они могут включать визуальное сравнение, а также сравнение времени удерживания,
площади (или высоты) пиков, их формы и т.д.
Сравнение с известными стандартами. В соответствии с рекомендациями USP 24 и
ICH в настоящее время для демонстрации специфичности в хроматографических
анализах используется тест чистоты пика, основанный на диодно-матричном
детектировании (ДМД) или масс-спектрометрии (МС). Современные технологии ДМД
(раздел 4.4.3) представляют собой мощный инструмент для оценки специфичности.
Детекторы ДМД могут снимать спектры во всем диапазоне длин волн для каждой
точки данных по периметру пика, а программное обеспечение позволяет сравнить каждый
спектр с другими спектрами для определения чистоты пика. Используя таким образом
детекторы ДМД, можно выделить мельчайшие спектральные и хроматографические
различия, которые нелегко определить при простом наложении спектров.
Тем не менее, в некоторых случаях детекторы ДМД могут быть ограничены
в применении при оценке чистоты пика из-за недостаточной чувствительности
в УФ-диапазоне, а также из-за шумов в системе ВЭЖХ и относительно невысоких
концентраций примесей. Кроме того, чем более схожи спектры и чем больше
отношение концентраций, тем труднее различить одновременно элюирующиеся
соединения. Масс-спектрометрическое (МС) детектирование (раздел 4.14) позволяет
преодолеть многие из упомянутых ограничений ДМД, и во многих лабораториях этот
способ детектирования становится предпочтительным при проведении валидации
методики анализа. МС может предоставить однозначную информацию о степени
чистоты пика, в том числе точную массу молекулярного иона и данные по структуре
и количеству анализируемого образца. Сочетание детекторов ДМД и МС в одной
системе ВЭЖХ позволяет получить взаимно дополняемую информацию и
гарантировать, что никакие помехи/перекрытия сигналов (пиков) не будут упущены из виду
во время проведения валидации методики анализа.
'Арбитражного метода. — Прим. перев.
12.2. Термины и определения 571
12.2.4. Предел обнаружения и предел количественного
определения
Предел обнаружения (ПО) определяется как самая низкая концентрация вещества
в испытуемом образце, которая может быть обнаружена/детектирована, но
количественная оценка которой не всегда возможна. Это тестирование предела, указывающее
на то, что содержание анализируемого вещества выше иди ниже определенного
значения. Предел количественного определения (ПКО) определяется как самая низкая
концентрация вещества в испытуемом образце, которую можно количественно
определить методом испытания с приемлемой прецизионностью и правильностью.
Определение ПКО представляет собой двухступенчатый процесс. Независимо
от метода, используемого для определения ПКО, во-первых, следует оценить
пределы величины на основе экспериментальных данных по отношению сигнал/шум или
наклону калибровочной кривой (разделы 4.2.4, 11.2.5). Во-вторых, полученное
значение необходимо подтвердить результатами анализов образцов с концентрацией,
равной ПКО. Более подробная информация приведена в разделе 11.2.5.
12.2.5. Линейность и аналитическая область
Линейность — способность метода испытания обеспечить результаты, которые
прямо пропорциональны концентрации определяемого вещества в заданной
аналитической области. Линейность в общем случае описывается как дисперсия наклона
линии регрессии (например, стандартная ошибка, полученная при регрессионном
анализе в Excel, как показано на рис. И.7). Аналитическая область представляет
собой интервал между максимальной и минимальной концентрациями анализируемого
вещества (включая максимальное и минимальное значения), в котором
использованная методика позволяет определять концентрацию анализируемого вещества с
приемлемой прецизионностью и правильностью при сохранении линейности, как было
продемонстрировано при проведении валидации методики анализа. Аналитическая
область обычно дается в тех же единицах концентрации, что и результаты,
полученные с использованием соответствующей методики анализа (например, нг/мл). В
руководствах указано, что для определения аналитической области и линейности
методики анализа необходимо использовать как минимум пять значений концентраций;
наряду с этим могут быть заданы некоторые минимальные аналитические области,
которые зависят от типа метода испытания |2,4|. В таблице 12.5 приведены
типичные минимальные аналитические области, указанные в руководствах |4|. Как
правило, в отчет должны быть включены уравнение и график калибровочной кривой с
коэффициентом корреляции (Й), как показано на рис. П.8 и П.9, на которых
приведены графики, построенные поданным таблицы II.I.
Таблица 12.5. Примеры рекомендованных минимальных аналитических областей
Тип метода
Количественный анализ
Примеси0
Однородность дозирования
Тест растворения
Рекомендованный минимальный диапазон
80—120% от номинальной концентрации определяемого вещества
От приводимой в отчетах концентрации каждой примеси до 120%
от концентрации, указанной в спецификации
70—130% тестируемой или номинальной концентрации
±20% от указанной аналитической области теста на растворение
"Для токсичных или более сильнодействующих примесей аналитическая область определения
должна включать концентрации, при которых эти примеси необходимо контролировать.
572 Глава 12. Валидация метода
12.2.6. Устойчивость
Устойчивость1 аналитической методики означает способность давать сравнимые и
приемлемые результаты при небольших, но преднамеренных изменениях указанных
в методике экспериментальных условий анализа.
Устойчивость дает представление о пригодности и надежности метода испытания
в условиях рутинного использования. При исследовании устойчивости условия
эксперимента (настройки оборудования) преднамеренно изменяются, чтобы определить
степень воздействия изменений на получаемые результаты анализов при
использовании данной методики анализа. Ключевым словом в данном определении
устойчивости является слово преднамеренно. Примеры изменений условий, вносимых в изокра-
тические и градиентные разделения ВЭЖХ, показаны в таблицах 12.6 и 12.7
соответственно.
Таблица 12.6. Типичные изменения лля проверки устойчивости в изократических
условиях разделения
Изменяемые условия
Концентрация органического (сильного)
растворителя
Концентрация буфера
рН буфера (если применимо)
Температура
Скорость потока
Длина волны детектирования
Объем ввода
Партия колонок
Пределы изменений
12-3%
±1-2%
±0,1—0,2 единицы рН
±3 °С
±0,1—0,2 мл/мин
±2—3 нм при ширине полосы 5нм
Зависит от объема ввода и типа инжектора
2—3 различные партии
Таблица 12.7. Типичные изменения для проверки устойчивости в градиентных условиях
разделения
Изменяемые условия
Исходное время задержки градиента
Наклон и длина
Конечное время задержки
Другие возможные изменения приведены
в таблице 12.6 (они зависят от конкретных
условий)
Пределы изменений
±10—20% времени задержки
Наклон определяется диапазоном градиента и
временем; установите время градиента ±10—20%
от исходного и дайте наклону измениться
Установите время, когда последнее из элюиро-
ванных веществ появится на хроматограмме
Изменения следует выбирать симметрично относительно значения параметра,
указанного в методе испытания (например, указанное изменение ±2% означает, что
изменять значение следует на +2% и -2% от значения параметра, указанного в мето-
'В руководствах по проведению валидации методик анализа, переведенных с английского
языка, вместо термина устойчивость зачастую используется калькированный термин робастность.
Однако в ГФ 14 РФ приведен именно термин устойчивость. — Прим. перев.
12.3. Пригодность системы 573
дике анализа), чтобы выбранный диапазон изменений немного превышал
изменения, которые могут произойти при использовании метода или его передаче в
стороннюю лабораторию. Например, если рН буфера доводится титрованием с рН-метром,
типичная лабораторная погрешность составляет +0,1 единицы рН. Для проверки
устойчивости метода испытания при изменении заданного рН буфера 2,5 можно
приготовить дополнительный буфер и испытать его при рН 2,3 и рН 2,7, чтобы
наглядно показать что данные изменения приемлемы и не приводят к значимым
изменениям анализа и, тем самым, гарантировать получение приемлемых результатов
анализа. Для определения допустимых отклонений настроек оборудования можно
использовать спецификации производителей. Диапазон изменений при проверке
устойчивости не следует выбирать настолько широким, чтобы тест преднамеренно
не получился. Необходимо выбирать диапазон изменений, близкий к изменениям,
обычно встречающимся в лабораторных условиях, и не доводить метод испытания
до сбоя, наглядно показать что данные изменения приемлемы и не приводят к
значимым изменениям анализа. При этом нет необходимости повторно валидировать
метод испытания (раздел 12.8).
Устойчивость следует тестировать при завершении разработки аналитической
методики, но обычно устойчивость исследуется одной из первых при валидации
методики анализа. Тем не менее, на протяжении всего процесса разработки метода следует
уделять внимание определению хроматографических условий наиболее
чувствительных к небольшим изменениям, чтобы эти условия проверить при испытании на
устойчивость. Исследования устойчивости также используются для тестирования
пригодности системы, чтобы убедиться, что вся система (как оборудование, так и метод
испытания) работают должным образом при внедрении и использовании метода
анализа. Кроме того, если результаты метода испытания или других измерений окажутся
чувствительными к изменениям некоторых условий эксперимента, эти условия
должны надлежащим образом контролироваться, а соответствующие предостережения
необходимо внести в сопроводительную документацию методики анализа.
Для измерения и документирования устойчивости надо контролировать
следующие характеристики:
• разрешение критической пары пиков Rs\
• число теоретических тарелок колонки N (или ширину пика при градиентном элю-
ировании);
ф время удерживания 1ц;
• фактор размывания пика TF;
• площадь (и/или высоту) пика и концентрацию.
В ходе исследования устойчивости следует вводить образец несколько раз
для уточнения оценки (например, по параметру ОСО (%)) влияния изменений
экспериментальных переменных. Во многих случаях контролируют несколько пиков, в
частности, когда в образце присутствует некоторая комбинация кислотных, нейтральных
или основных соединений. Может оказаться полезным включение в документацию
метода ряда хроматограмм с отклонениями, иллюстрирующих пределы устойчивости
(см. обсуждение рис. 1.5 в |11|). Такие примеры, а также корректирующие инструкции
могут быть полезны для устранения проблем при использовании методики анализа.
12.3. Пригодность системы
Проверка пригодности системы является неотъемлемой частью хроматографических
методик анализа |6|, хотя формально не требуется для валидации методики в
соответствии с указаниями USP. Тест на пригодность системы используется для провер-
574 Глава 12. Валидация метода
ки того, являются ли разрешающая способность и прецизионность системы
адекватными для выполнения анализа. Тесты на пригодность системы основаны
на комплексной оценке оборудования, электроники, аналитических операций и об
разцов как единой системы.
Тесты на пригодность системы предназначены для проверки системы на
соответствие рабочим характеристикам до или во время анализа проб. Измеряются такие
характеристики как число теоретических тарелок колонки, фактор размывания/
асимметрии пика, разрешение и прецизионность (повторяемость), и полученные
значения сравниваются со значениями, указанными в методе анализа в разделе
о пригодности системы. Параметры пригодности системы измеряются в процессе
анализа «образца для проверки пригодности системы», представляющего собой смесь
основных компонентов и предполагаемых продуктов разложения или примесей,
которая имитирует репрезентативный испытуемый образец. Тем не менее, можно
использовать образцы, дающие на хроматограмме только один пик (как, например,
при количественном определении лекарственного препарата, когда присутствует
только пик ФС) при условии, что в методе испытания указаны число теоретических
тарелок и фактор размывания/асимметрии пика. Проводится сравнение результатов
многократного введения образца для проверки пригодности системы, чтобы
определить, соблюдаются ли требования к прецизионности системы. Если иное не указано
в методе испытания, для вычисления относительного стандартного отклонения
используются данные пяти вводов образца, когда метод испытания требует ОСО < 2%;
данные шести вводов необходимы, если требуется ОСО > 2%.
В сообществе, деятельность которого контролируется регуляторными органами,
проверка пригодности системы должна предшествовать анализу любых образцов.
После ввода подвижной фазы, воды и/или раствора для разбавления образца и проведения
холостого разделения нужное количество раз вводят образец для проверки
пригодности системы, и результаты сравнивают с требованиями методики анализа. Если
требования пригодности системы выполняются, проводят последующие анализы. Если
требования пригодности системы не выполняются, необходимо провести диагностику
проблем, связанных с системой ВЭЖХ или методикой анализа, и устранить эти
проблемы (возможно, в рамках официального расследования причины несоответствия
спецификации [OOS]1). Система должна пройти тест на пригодность до
возобновления анализа образцов. Для того, чтобы обеспечить уверенность в том, что метод
тестирования пригодности работает должным образом, рекомендуется также через
определенные промежутки времени (между анализами испытуемых образцов каждой партии)
вводить дополнительные образцы для проверки пригодности системы (образцы
контроля качества или проверочные стандарты). В раздел «Пригодность системы»
методики анализа должно быть внесено максимально допустимое значение процента
расхождения результатов таких внеочередных анализов, чтобы убедиться, что система
по-прежнему работает надлежащим образом на протяжении анализа всех партий
образцов. В качестве альтернативы можно после завершения цикла анализов провести
тестирование со вторым набором образцов для проверки пригодности системы.
В 1994 году FDA опубликовало обзор рекомендаций по валидации хроматографи-
ческих методов, включающий в частности новейшие рекомендации по определению
пригодности системы |10|. Эти рекомендации приведены в таблице 12.8. Многие
практикующие специалисты считают, что методы тестирования, которые удовлетворяют
критериям, указанным в таблице 12.8, могут уменьшить риск критики при
аудиторской проверке контролирующими органами. Эти руководящие принципы служат
полезными примерами; однако фактические технические характеристики задаются
пользователем и могут значительно меняться в зависимости от методики анализа.
'Англ. «out-of-specification», 00S. — Прим. персе.
12.4. Документация 575
Таблица 12.8. Рекомендации FDA для оценки пригодности системы
Параметр
Фактор удерживания* k
Повторяемость ввода
Разрешение R,
Фактор размывания пика** TF
Число теоретических тарелок
колонки N
Рекомендации
* > 2
0С0 £ 1% для пг5
Rs>2
TF<7
N > 2000
Комментарии
Пик должен быть хорошо разрешен
относительно других пиков и
несдерживаемого пика, выходящего при to
Измеряется во время анализа образцов
Измеряется между целевым пиком и
ближайшим пиком, который потенци-
апьно может перекрывать целевой
Характеристики колонки не указаны
Источник: данные из 1101
*или коэффициент емкости. — Прим. перев.
"или фактор/коэффициент асимметрии As. — Прим. перев.
12.4. Документация
Валидационная документация включает: протокол, используемый для проведения
валидации; описание метода испытания; валидационный отчет. Эти документы
должны быть оформлены как контролируемые и должны быть частью системы качества
(раздел 12.9), которая обеспечивает соблюдение нормативных предписаний.
12.4.1. Валидационный протокол
В валидационном протоколе определены подлежащие выполнению требования к ва-
лидационным процедурам и критериям приемлемости. Там, где это возможно,
в протоколе следует приводить ссылки на стандартные операционные процедуры
(СОПы) для конкретных рабочих инструкций и аналитических методов. Протокол
должен быть подготовлен и утвержден до начала официальных работ по валидации.
Валидационный протокол, как правило, содержит следующие элементы:
• заголовок;
• назначение метода испытания, который подлежит валидации;
• описание испытуемых и стандартных образцов;
• краткое описание подлежащего валидации метода испытания, в том числе
оборудования, аналитической области методики анализа, а также описание испытуемых
и стандартных образцов; вместо этого можно сослаться на подробное описание
методики анализа или приложить ее к протоколу;
• валидационные характеристики, которые должны быть подтверждены;
• описание и обоснование критериев приемлемости для выбранных характеристик
валидации;
• подпись утверждающего протокол уполномоченного представителя отдела
контроля качества с указанием даты.
В названии протокола приводится краткое описание планируемой работы или
исследования, например, «Валидация методики анализа: количественное определение
ФС (X) в лекарственном продукте Y методом ВЭЖХ». Назначение должно определять
область и особенности применения метода испытания. В кратком описании должен
576 Глава 12. Валидация метода
быть надлежащим образом описан метод испытания (описание должно быть
достаточно подробным, чтобы его мог легко воспроизвести квалифицированный
специалист). Однако часто приводится ссылка на метод испытания, или он дается как
приложение к протоколу, чтобы сократить повторение. В протокол также включаются
подлежащие определению конкретные валидационные характеристики
(правильность, прецизионность и т.д.), потому что они зависят от типа аналитической
методики (раздел 12.5). Критерии приемлемости для валидации методики анализа
(например, допустимая ошибка или неточность) часто устанавливаются на заключительном
этапе разработки методики анализа (иногда этот этап называют «предвалидационны-
ми» экспериментами, см. раздел 12.4.2). Уполномоченный представитель отдела конт
роля качества проверяет и утверждает протокол, чтобы гарантировать соблюдение
надлежащих нормативных актов и соответствие предполагаемой работы своему
назначению (раздел 12.9).
Эксперименты, описанные в валидационном протоколе, могут быть
спланированы таким образом, чтобы измерить несколько валидационных характеристик
одновременно. Например, эксперименты для определения правильности и
прецизионности можно использовать как часть исследований линейности; ПО и ПКО можно
определить из данных для определения аналитической области и линейности,
а при тестировании стабильности растворов испытуемых образцов и стандартов
можно использовать те же растворы, с которыми проверяют правильность и
прецизионность. Выполнение экспериментов в такой последовательности позволяет наиболее
эффективно использовать время и материальные ресурсы.
12.4.2. Метод испытания
Метод испытания является официальным документом, в котором подробно описаны
все аналитические процедуры, подлежащие выполнению в каждодневном рутинном
анализе. Метод испытания представляет собой контролируемый документ, версии
которого подлежат контролю (т.е. требуется, чтобы любые изменения документа
были санкционированы и все версии документа были доступны для последующего
сравнения). Он утверждается на соответствующих уровнях (включая отдел контроля
качества, раздел 12.9) и изложен достаточно подробно, чтобы гарантировать
единственную возможную интерпретацию каждой инструкции и всего текста документа.
Типичный метод испытания включает следующие составляющие:
• заголовок описываемого метода;
• краткое описание метода или резюме;
• описание применимости и назначения, а также описание любых специальных мер
предосторожности (например, информацию, связанную с обеспечением
безопасности, хранения и выполнения необходимых процедур);
• перечень реагентов с указанием фирмы-производителя и категории чистоты;
• перечень оборудования, включающий систему ВЭЖХ и любое другое необходимое
оборудование (весы, центрифуги, рН-метры и т.д.);
• подробное описание условий эксплуатации системы ВЭЖХ, включая настройки
интегрирования;
• подробные инструкции по приготовлению всех растворов (подвижных фаз,
разбавителей), стандартов и испытуемых образцов;
• описание тестирования пригодности системы и критериев приемлемости;
• примеры хроматограмм, спектров или репрезентативных данных;
• подробное описание процедур, в том числе пример последовательности введения
образцов (порядок введения стандартов и испытуемых образцов в систему ВЭЖХ);
• формулы расчетов с описанием переменных;
12.4. Документация 577
• перечень зарегистрированных изменений;
• согласования.
После разработки методы испытаний часто подвергаются предварительной вали
дации, чтобы убедиться, что критерии приемлемости будут выполнены при
официальном проведении валидации. В ходе предварительной валидации, как правило,
тестируются линейность и правильность. Иногда вслед за этим проводится испытание
на устойчивость, если она уже не была проверена в ходе разработки методики
анализа. Процесс валидации обычно протекает более гладко и с меньшим риском неудачи,
если на стадии предварительной валидации подтверждается, что критерии
приемлемости удовлетворяют всем ключевым характеристикам валидации. Проект методики
анализа становится официальным методом испытания после полной валидации
методики по предполагаемому назначению.
12.4.3. Валидационный отчет
Валидационный отчет представляет собой сводку данных, полученных при
тестировании предложенного метода испытания согласно валидационному протоколу. Отчет
включает описание расчетов, репрезентативные хроматограммы, калибровочные
графики и другие результаты, полученные в процессе проведения валидации.
Приводятся также таблицы данных для каждого пункта протокола с заключением
«выполнено/не выполнено» для каждого из критериев приемлемости. Как правило,
валидационный отчет состоит из следующих разделов:
• титульный лист с указанием наименования валидационного отчета, авторов и
организации;
• страница с датой и подписью соответствующего персонала, который может
включать аналитиков, начальника группы, старшего менеджера, а также представителя
отдела контроля качества и/ или уполномоченного лица;
• подробный список установленных валидационных характеристик часто в виде
таблицы;
• введение или постановка задачи;
• краткое изложение метода испытания с указанием используемого оборудования и
методик приготовления растворов;
• результаты валидации, приведенные последовательно в отдельных подразделах
для каждой из изученных характеристик. Каждый подраздел должен включать
краткое изложение примененного пункта из валидационного протокола, среднее
значение полученной величины, ее стандартное отклонение, относительное
стандартное отклонение, критерии приемлемости и оценки (выполнен или не
выполнен);
• любые отклонения от валидационного протокола, запланированные или
наблюдавшиеся, а также их влияние (если это применимо) на результаты валидации;
• любые поправки к протоколу с пояснениями и согласованиями;
• вывод.
Правильно составленный валидационный протокол может служить шаблоном
при написании валидационного отчета. Например, в протоколе могут быть описаны
испытания и перечислены критерии приемлемости. При написании валидационного
отчета эта информация дополняется полученными результатами, ссылками на место
проведения испытаний и на место хранения подлинных исходных данных, а также
заключением о выполнении или невыполнении критериев приемлемости.
578 Глава 12. Валидация метода
12.5. Валидация для различных типов
фармацевтических методов
В USP официально признано, что не всегда необходимо оценивать каждую валида-
ционную характеристику для каждого метода испытания. Тип метода и его
назначение определяют, какие валидационные характеристики необходимо исследовать. Эти
данные обобщены в таблице 12.9 [12|. Как в USP, так и в документах ICH методы
испытаний подразделяются на четыре категории:
• количественное определение основных компонентов или активных веществ
(методы категории I);
• определение примесей или продуктов разложения (методы категории 2);
• определение характеристик готового лекарственного препарата (методы
категории 3);
• определение подлинности (методы категории 4).
В общем случае эти методы испытаний (и их разделение на категории)
применяются для анализа фармацевтических субстанций и лекарственных препаратов.
Подходы к валидации методов испытаний биоаналитических образцов рассматриваются
отдельно в разделе 12.6.
12.5.1. Методы категории 1
Методы категории 1 предназначены для анализа основных компонентов и включают
такие методы испытаний, как однородность содержания и количественное
определение активного вещества. Упомянутые количественные методы, как правило, не
подразумевают определение образцов с низкой концентрацией, поскольку
предназначены для определения ФС в лекарственном средстве. Из-за несложности разделения
(ФС должна разрешаться относительно всех примесей, а любые другие пики на хро-
матограмме нет необходимости разрешать друг относительно друга), наиболее важна
скорость1, а не разрешение. Для методов количественного определения категории I
оценка ПО и НПКО не нужна, так как измеряемый основной компонент или
активное вещество, как правило, присутствует в образце в высоких концентрациях.
Однако, так как требуется количественная информация, подлежат определению и все
остальные валидационные характеристики.
12.5.2. Методы категории 2
Методы категории 2 нацелены на анализ примесей или продуктов разложения (хотя
могут применяться и для других целей). Данные количественные определения, как
правило, подразумевают определение гораздо более низких концентраций
анализируемого вещества, чем в методах категории I, и методы категории 2 разделены на две
подкатегории: количественное определение и определение предела обнаружения
(ПО). Если требуется количественная информация, определение ПО не является
необходимым, но определение остальных валидационных характеристик является
обязательным. Методы, используемые для исследования стабильности (называемые
методами оценки стабильности), являются примером количественного определения
категории 2. Ситуация полностью меняется в случае тестирования предела
обнаружения. Поскольку количественное определение не требуется, достаточно измерить ПО
и подтвердить специфичность и устойчивость. Для теста категории 2 с определением
'проведения анализа. — Прим. перев.
12.5. Валидация для различных типов фармацевтических методов 579
ПО необходимо только показать, что определяемое соединение присутствует или
отсутствует в испытуемом образце (т.е. что его концентрация выше или ниже
определенного значения). Часто в эту категорию попадают методы испытаний,
предназначенные для вллидации процесса очистки1 и методы испытаний, используемые ЕРА
для контроля загрязнения окружающей среды. Хотя, как видно из таблицы 12.9, нет
необходимости измерять и ПО, и ПКО для любого из методов категории 2. Обычно
при вллидации определяют обе эти характеристики (больше в силу традиции, чем
по необходимости).
Таблица 12.9. Перечень подлежащих измерению вллидаиионных характеристик в
зависимости от применяемой категории метода
Валидаиионные
характеристики
Правильность
Прецизионность
Специфичность
ПО
ПКО
Линейность
Аналитическая область
Устойчивость
Категория 1:
Количестве иное
определение
Да
Да
Да
Нет
Нет
Да
Да
Да
Категория 2: примеси
Количественное
определение (ПКО)
Да
Да
Да
Нет
Да
Да
Да
Да
ПО
•
Нет
Да
Да
Нет
Нет
Нет
Нет
Категория: 3
Специфические тесты
*
Да
*
*
*
*.
•
Да
Категория: 4
Подлинность
Нет
Нет
Да
Нет
Нет
Нет
Нет
Нет
Данные из |12].
*Может потребоваться в зависимости от типа теста. Например, хотя тест на растворимость
попадает в категорию 3, поскольку этот тест количественный, проводятся измерения, типичные
для методов категории 1 (за некоторыми исключениями).
12.5.3. Методы категории 3
Характеристики, которые должны быть документированы для методов
испытаний, отнесенных к категории 3 по классификации USP (специфические тесты или
методы определения основных свойств продукта), зависят от природы теста.
Примером метода категории 3 является тест на растворимость. Так как это количественный
тест, оптимизированный для определения ФС в лекарственном препарате, для его
валидации оцениваются практически те же характеристики, что и в тесте категории 1
для лекарственного препарата, предназначенного для немедленного высвобождения.
Однако для лекарственного препарата с замедленным высвобождением может быть
необходимо подтвердить, что ни один активный ингредиент не высвобождается
из препарата до определенного момента времени. Тогда подлежат исследованию ва-
лидационные характеристики в большей мере сходные с теми, которые используются
в количественном тесте методов категории 2, включая тест ПКО. Поскольку цели
аналитических тестов категории 3 могут быть различными, подлежащие измерению
характеристики существенным образом зависят от конкретного метода испытания,
как указано в таблице 12.9.
1 Производственного оборудования или помещения. — Прим. перев.
580 Глава 12. Валидация метода
12.5.4. Методы категории 4
Тесты на подлинность, относящиеся к категории 4, носят качественный характер,
поэтому требуется только доказать специфичность. Определение подлинности может
быть выполнено, например, путем сравнения времени удерживания или спектра
исследуемого вещества с известным спектром или временем удержания стандартного
образца. С точки зрения хроматографического разделения требуется лишь, чтобы
не было наложения пиков примесей на пик определяемого соединения.
12.6. Биоаналитические методы
А" биоаналитическим1 относятся методы испытаний для анализа лекарственных
средств и их метаболитов в биологических испытуемых образцах. Обычно
испытуемыми образцами являются плазма крови или моча, также это могут быть ткани
животных. Иногда биоаналитические методы путают с анализом биомолекул, таких как
белки, пептиды и олигонуклеотиды — методы их разделения обсуждаются в главе 13.
Биоаналитические методы используются в клинической фармакологии,
токсикологии, при исследовании биодоступности и биоэквивалентности. Фармакокинетиче-
ские данные этих и других исследований требуются регуляторным органам, таким
как FDA, для подтверждения того, что данное лекарственное средство может быть
использовано для соответствующего применения. В сертифицированной
лаборатории биоаналитические методы для их использования по назначению должны быть
валидированы (как и любой другой аналитический метод), чтобы
продемонстрировать, что они являются надежными и воспроизводимыми.
При разработке и валидации методов испытаний готовой продукции, исходного
сырья, или фармацевтических субстанций (ФС) возникают специфические
трудности для каждой из этих категорий методов. Разработка биоаналитических методов
еще более осложняется из-за матрицы образца, следовых концентраций
лекарственного препарата и его метаболитов, а также (потенциально) из-за сложности
необходимого для анализа оборудования.
Чувствительность и селективность биоаналитических методов имеют решающее
значение для успешного проведения доклинических и клинических
фармакологических исследований. Как и в случае любого другого метода испытания, рабочие
характеристики биоаналитического метода должны быть подтверждены
задокументированными лабораторными данными, чтобы доказать надежность и воспроизводимость
метода для его использования по назначению. Для обсуждения этой темы
проводились совместные конференции представителей промышленности и регуляторных
органов (см., например, |13—15|). По результатам первых двух конференций в мае
2001 года FDA опубликовало руководство по валидации биоаналитических
методов |16]. В отличие от рабочих характеристик методов испытаний для лекарственной
субстанции или лекарственного препарата с четким перечнем конкретных
необходимых критериев (например, прецизионности и правильности), предписания для
биоаналитических методов приведены в виде рекомендаций. Это означает, что если
методы испытаний разработаны с учетом этих рекомендаций, то больше вероятность
того, что метод испытания будет утвержден регуляторным органом. Другими
словами, если не следовать рекомендациям руководства, необходимо подготовить
логичное и научно обоснованное заключение о применимости альтернативных рабочих
характеристик метода.
1 В русскоязычной специальной литературе эти методы анализа называют также
биофармацевтическими. — Прим. перее.
12.6. Биоашыитжеские методы 581
Сертифицированный метод проведения биоаналитических исследований обычно
предполагает использование системы ВЭЖХ с тандемным масс-спектрометром
(ЖХ-МС/МС, раздел 4.14). Чувствительность и селективность ЖХ-МС/МС
позволяет количественно определять анализируемое вещество с приемлемой
прецизионностью и правильностью при более низких концентрациях, чем большинство других
детекторов ВЭЖХ. Как правило, короткие колонки с малым размером частиц
сорбента (например, колонка 30—50 х 2,1 мм, упакованная частицами < 3 мкм)
используются для быстрых разделений, необходимых для большого числа образцов,
подлежащих анализу в ходе клинических исследований. В основном проводят
изократические или градиентные разделения длительностью < 5 мин. Разработка
методики пробоподготовки образца (глава 16), предназначенной для удаления избытка
белка и других потенциальных помех, может потребовать столько же усилий,
сколько и разработка метода ВЭЖХ. Обычно и пробоподготовку, и анализ проб
автоматизируют.
Разработку и использование биоаналитического метода испытания можно
разделить на три части:
• подготовка стандартного образца;
• разработка и валидация метода;
• применение валидированного метода испытания для рутинного анализа
лекарственных средств.
Каждый из этих процессов обсуждается в последующих разделах.
12.6.1. Подготовка стандартного образца
Стандартные образцы необходимы для количественного определения
анализируемого вещества в биологической матрице. Они используются как для построения
калибровочных (стандартных) графиков, так и для проверки применимости метода
(образцы контроля качества анализа). Существует три типа стандартных образцов:
(I) стандарты, чистота которых сертифицирована соответствующей уполномоченной
организацией (например, фармакопейные стандарты USP), (2) стандартные образцы,
полученные из другого коммерческого источника (например, от компании,
занимающейся продажей химических реактивов общего или специального назначения), и
(3) стандарты, синтезированные по заказу. По возможности стандарт должен быть
идентичен анализируемому веществу или, по меньшей мере, быть в определенной
химической форме (например, в форме свободной кислоты или основания, или
соли). В каждом случае чистота стандартов должна быть подтверждена
соответствующими документами — обычно это сертификат анализа. Сопроводительная
документация стандарта с указанием номера партии и срока годности, а также сертификаты
анализа, подтверждающие подлинность и чистоту, должны храниться вместе с
другими данными метода для проверки органами надзора. Соединения, используемые
в качестве внутренних стандартов (часто меченные изотопами лекарственные
средства) также должны иметь аналогичные сертификаты качества, подтверждающие их
чистоту.
12.6.2. Разработка и валидация биоаналитического метода
Основные биоаналитические характеристики, которые должны быть валидированы
для каждого определяемого вещества в матрице, включают правильность,
прецизионность, избирательность, аналитическую область, воспроизводимость и
стабильность. На практике для разработки метода испытания и его валидации исследуются
четыре группы параметров:
582 Глава 12. Валидация метода
• избирательность (селективность);
• правильность, прецизионность и степень извлечения;
• калибровочный/стандартный график;
• стабильность.
Данные исследований каждой группы параметров должны подтверждать
остальные характеристики.
12.6.2.1. Избирательность
Избирательность метода испытания показывает, что определяемое вещество может
быть точно определено, несмотря на потенциальные помехи, вызванные
присутствием других компонентов образца (в том числе компонентов его матрицы). Помимо
эндогенных компонентов матрицы (белков, липидов и т.д.) в составе образца могут
быть метаболиты лекарственного средства, продукты его разложения, сопутствующие
лекарственные вещества или другие вещества, которые также предстаачяют интерес.
В руководстве FDA рекомендуется проводить анализ холостых проб
соответствующей биологической матрицы, отобранных, по крайней мере, из шести различных
источников. Так, например, в плазму крови из каждого источника следует добавлять
известное количество определяемого вещества до достижения концентрации
нижнего предела количественного определения (ПКО или НПКО), чтобы показать, что
в этих условиях может быть достигнута приемлемая точность метода. Также следует
проанализировать холостые пробы экстрактов каждой матрицы, чтобы показать
отсутствие помех для определяемого вещества при разделении. В тех случаях, когда
матрица представляет собой редкий или трудно получаемый образец (например, если
это плазма крови экзотических организмов или ткани человека), требование о шести
образцах матрицы смягчается.
12.6.2.2. Правильность, прецизионность и степень извлечения
Правильность биоаналитического метода определяется как близость результатов
анализов к истинному значению, полученному при многократных анализах проб,
содержащих известные количества анализируемого вещества. Результаты анализа
представляются в виде отклонения среднего значения от истинного значения.
В руководстве FDA рекомендуется проводить минимум пять определений для
каждой концентрации и анализировать образцы с тремя концентрациями как минимум
из предполагаемой аналитической области (т.е необходимо проанализировать
минимум 15 отдельно подготовленных образцов). В соответствии с этими
рекомендациями среднее значение должно находиться в пределах ±15% от истинного значения, за
исключением собственно значения концентрации ПКО, при котором приемлемое
отклонение составляет +20%.
Прецизионность биоаналитического метода измеряется согласованностью
результатов испытаний, когда метод применяется многократно к нескольким выборкам
однородного образца. Согласно последним рекомендациям руководства 1СН
прецизионность может быть дополнительно разделена на повторяемость (в повторных
анализах испытуемых образцов в рамках одного цикла анализа или при анализе
образцов одной партии) и промежуточную прецизионность (в повторных анализах
испытуемых образцов в рамках разных циклов анализа, например в разные дни, или
в анализах испытуемых образцов разных партий) |4|. В рекомендациях FDA
рекомендуется проводить минимум пять определений для каждой концентрации и
анализировать образцы с тремя, как минимум, концентрациями из предполагаемой
аналитической области. Погрешность измерения для каждого значения концентрации
не должна превышать 15% ОСО, за исключением концентрации, соответствующей
ПКО, при которой погрешность не должна превышать 20% ОСО. Обычно для опре-
12.6. Биоаналитжеские методы 583
деления правильности и прецизионности используются одни и те же аналитические
данные.
Степень извлечения характеризует эффективность экстракции, и ее величина
определяется путем сравнения отклика от образца, извлеченного из матрицы,
со стандартным образцом (с соответствующими поправками на разведение
испытуемого образца и т.д.). Степень извлечения анализируемого вещества может быть
менее 100%, но извлечение должно быть количественным. То есть, оно должно быть
прецизионным и воспроизводимым. Эксперименты по извлечению должны
проводиться для трех концентраций (низкой, средней и высокой) с обязательным
сравнением результатов для экстрагированных и неэкстрагированных образцов (с поправкой
на разведение). Часто бывает непрактично анализировать неэкстрагированные об
разцы (например, ввод плазмы крови, не подвергнутой экстракции, приведет к
порче большинства колонок для ВЭЖХ), так что может потребоваться нестандартный
подход для измерения степени извлечения. Например, экстракт, полученный из
матрицы с добавкой определяемого соединения в системе жидкость-жидкость, можно
сравнить с экстрактом из водного раствора без матрицы. Можно также рассчитать
степень объемного извлечения после твердофазной экстракции, и сравнить отклик
детектора на экстрагированный образец с откликом на стандартный образец с
известной концентрацией.
12.6.2.3. Калибровочные/стандартные кривые
Калибровочная кривая (которую также называют стандартной кривой)
иллюстрирует взаимосвязь между откликом прибора и известной концентрацией
анализируемого вещества в пределах диапазона, заданного исходя из ожидаемых значений
концентрации. Следует использовать самую простую модель пропорциональной
зависимости (например, аппроксимация линейной функцией предпочтительнее
аппроксимации квадратичной функцией). Калибровка для биоаналитических
методов, как правило, более сложна, чем для анализов ФС, в которых калибровочные
графики обычно линейны и проходят через начало координат. Поэтому для
количественного определения ФС может быть достаточно калибровочного стандарта
с одной концентрацией. Для большинства биоаналитических методов
предпочтительно использование внутренних стандартов (раздел 11.4.1.2), поскольку
испытуемые образцы обычно проходят сложную процедуру пробоподготовки. По крайней
мере, четыре из шести ненулевых стандартов (67%) должны иметь концентрации
в пределах +15% от ожидаемой (+20% при ПКО). Калибровочную кривую следует
строить для каждого анализируемого вещества в соответствующем образце, и
образцы для калибровочной кривой нужно готовить, добавляя к матрице без
действующего вещества определенное количество анализируемого вещества. В руководстве
FDA указано, что калибровочная кривая должна быть построена по данным
анализа от шести до восьми образцов с ненулевой концентрацией, значения которой
охватывают ожидаемую аналитическую область, включающую ПКО. Кроме того,
следует подтвердить отсутствие помех со стороны матрицы, проанализировав
холостую пробу (прошедший пробоподготовку образец матрицы без добавки
анализируемого вещества и внутреннего стандарта) и нулевую пробу (прошедший
пробоподготовку образец матрицы без добавки анализируемого вещества, но с добавкой
внутреннего стандарта). Два условия должны быть выполнены для определения
ПКО: (I) отклик от анализируемого вещества при ПКО должен более, чем в 5 раз
превышать отклик от холостой пробы, и (2) пик анализируемого вещества должен
быть идентифицируемым, компактным и воспроизводимым с погрешностью < 20%
и правильностью в диапазоне 80—120%.
584 Глава 12. Валидация метода
12.6.2.4. Стабильность биоаналитического образца
Тесты на стабильность проводятся для подтверждения того, что анализируемое
вещество (и внутренний стандарт) не разрушаются при обычных лабораторных условиях,
а если разложение происходит, то об этом известно, как и о том, что разложение
можно предотвратить путем соответствующей обработки анализируемого образца. На
стабильность биоаналитического образца могут влиять различные факторы, в частности,
его химические свойства, условия хранения, а также матрица. Необходимо разработать
план исследований для оценки стабильности анализируемого вещества во время
отбора и обработки образцов, в условиях долговременного хранения (при предполагаемой
температуре) и краткосрочного хранения (при контролируемой комнатной
температуре во время пробоподготовки), а также при определенном количестве циклов
замораживания и оттаивания. Условия, используемые для любых исследований стабильности
образца, должны отражать фактические (реальные) условия, которые могут возникнуть
во время отбора, хранения и рутинных анализов образца (в том числе его рабочих и
исходных растворов). Исходные растворы образца известной концентрации должны
быть приготовлены в соответствующем растворителе. Следует удостовериться в
стабильности исходных растворов при комнатной температуре в течение, по крайней
мере, шести часов и в условиях хранения (например, в холодильнике). Кроме того,
поскольку в течение некоторого времени образцы обычно будут находиться на рабочем
столе или в автосамплере системы ВЭЖХ, также важно установить стабильность
обработанных образцов (например, лекарственного средства и внутреннего стандарта,
извлеченных из матрицы образца) в течение предполагаемого времени выполнения
анализа серии образцов. Рабочие стандарты следует приготовить из свежих исходных
растворов анализируемого вещества в матрице образца. Следует придерживаться
соответствующих стандартных операционных процедур (СОПов) при экспериментальных
исследованиях, а также при последующей статистической обработке данных.
В руководстве FDA рекомендуется минимальный протокол, включающий
определение стабильности при замораживании и оттаивании, а также краткосрочной и
долгосрочной стабильности при определенной температуре. Для исследования
стабильности при замораживании и последующем оттаивании следует подвергнуть трем
циклам замораживания-оттаивания три аликвоты образца с высокой и низкой
концентрациями, в которые добавлена матрица. Образцы следует выдержать 24 часа
при температуре хранения, а затем разморозить при комнатной температуре (без
нагревания). Когда образцы полностью оттают, их надо повторно заморозить на 12—
24 часа, а затем снова разморозить. Эту процедуру повторяют в третий раз, и после
завершения третьего цикла замораживания-оттаивания анализируют образцы.
Для исследования кратковременной температурной стабильности размораживают
по три аликвоты образца с низкой и высокой концентрациями и выдерживают их
при комнатной температуре столько времени, сколько предположительно они будут
находиться в помещении до их анализа (например, 4—6 часов).
Срок хранения для оценки долговременной стабильности должен охватывать
время между отбором первого образца и анализом последнего образца (часто 12 или более
месяцев). Отобранный объем пробы должен быть достаточным, по крайней мере,
для трех анализов через определенные промежутки времени. При каждом анализе
следует протестировать, как минимум, по три аликвоты образца с низкой и высокой
концентрациями, которые хранились в таких же условиях, как исследуемые образцы
(например, при -20 °С или -70 °С). При исследовании долговременной стабильности
концентрацию образцов, помещенных на хранение, следует определять с
использованием свежеприготовленных стандартных образцов. В отчете об исследовании
стабильности среднее значение полученных концентраций следует приводить по отношению
к среднему значению концентраций, полученных в первый день исследования.
12.6. Биоаналитические методы
585
12.6.3. Рутинное применение биоаналитического метода
После того, как биоаналитический метод был валидирован для рутинного
использования, образцы для контроля качества (КК) и определения пригодности системы
используются для контроля правильности и прецизионности, и для того, чтобы
определить, следует ли принять или отклонить результаты анализа для партии
исследуемых образцов. Образцы для контроля качества готовят отдельно и
анализируют с образцами, в которых содержание анализируемого вещества неизвестно.
Анализ образцов КК проводится с интервалами, зависящими от количества
испытуемых образцов в серии анализов. Как правило, используются два образца КК
(приготовленных из матрицы с добавкой анализируемого вещества), в трех
концентрациях (с концентрацией ниже или вблизи ПКО, с концентрацией,
соответствующей середине диапазона, и с высокой концентрацией). Рекомендуется, чтобы
минимальное количество образцов КК (в количестве кратном трем с низкой, средней
и высокой концентрациями) составляло не менее 5% от числа образцов с
неизвестной концентрацией или шести образцов КК, в зависимости от того, что больше.
Например, если надо проанализировать 40 образцов с неизвестной концентрацией,
40 х 5% = 2, таким образом, должны быть проанализированы 6 образцов КК (2
образца с низкой, 2 образца со средней, 2 образца с высокой концентрациями);
или на 200 образцов с неизвестной концентрацией, 200 х 5% = 10, следовательно,
должны быть проанализированы 12 образцов КК (по 4 образца КК для каждой
концентрации: низкой, средней и высокой). Как минимум, четыре из шести
результатов анализов для образцов КК должны быть в пределах ±15% от их
соответствующего номинального значения. Данные, отображающие типичные результаты,
полученные методом ЖХ-МС/МС при анализе образцов КК (с концентрациями
10, 35, 1000, 4400, и 5000 пг/мл плазмы), приведены в таблице 12.10. Как
упоминалось ранее, для приемлемой валидации метода как погрешность при каждой
концентрации (% ОСО), так и правильность (% отклонения) должны быть <, 15%
(< 20% для ПКО). В таблице 12.10 максимальные значения ОСО (<, 3,9%) и
отклонения (< 11,0%) для всех концентраций укладываются в пределы, указанные в
руководстве по валидации.
Таблица 12.10. Пример результатов анализа образцов КК биоаналитического метода
ЖХ-МС/МС
Измеренная концентрация образцов
для контроля качества, ОК (пг/мл)
OKI
11,8
11,1
11,4
10,4
10,8
10,9
10,9
10,8
10,3
11,4
ОК2
35,7
37,1
35,4
36,0
34,6
34,9
33,6
32,6
33,2
34,4
ОКЗ
1009,8
1036,0
1047,2
975,8
1047,8
986,5
971,8
960,4
956,7
977,8
ОК4
4670,3
4796,4
4684,9
4964,3
4628,6
4564,3
4491.9
4404,1
4539.5
4558,6
ОК5
5425,0
5334,5
5180,9
5241,6
5285,6
5049,0
5009.2
4883,7
5170,8
4802,7
Истинная
концентрация (пг/мл)
10,0
35,0
1000,0
4400,0
5000,0
п
10
10
10
10
10
Среднее
11,1
34,8
997,0
4630,0
5138,3
СО
0,402
1,37
35,45
160,5
199,4
% ОСО
3,6
3,9
3,6
3,5
3,9
%
отклонения
+11,0
-0,6
-0,3
+5,2
+2,8
586 Глава 12. Валидация метода
Определение пригодности системы и критериев приемлемости, анализ образцов
и указания для повторного анализа или изменения параметров интегрирования
данных — все должно выполняться в соответствии с утвержденными СОПами
{внутренними инструкциями). Основание для проведения повторных анализов или
изменения параметров интегрирования данных, а также форма отчета с результатами
анализа должны быть четко сформулированы в СОП. Вот некоторые из причин,
которые могут привести к необходимости повторного анализа проб: проблемы с
повторяемостью анализов, ошибки при пробоподготовке образцов, сбои оборудования или
плохое разделение испытуемых образцов. Кроме того, в последних версиях
руководств по биоаналитическим методам |15| указано, что определенное количество
образцов надлежит регулярно анализировать повторно, чтобы подтвердить пригодность
используемой методики анализа {такой тест иногда называют «проверкой
воспроизводимости метода с проверенными образцами»).
12.6.4. Документирование биоаналитического метода
Как обсуждалось ранее в разделе 12.4, четкое ведение записей и оформление СОПов
являются неотъемлемой частью любого валидированного метода испытания. После
того, как проведена валидация биоаналитического метода и его пригодность
подтверждена лабораторными исследованиями, соответствующая информация заносится
в отчет о валидации анализа. Данные, полученные в ходе разработки метода и
контроля качества, должны быть доступны для аудита и инспекции. Документация,
представляемая в FDA, должна включать следующую информацию: (1) краткую сводную
информацию, (2) отчеты по разработке и валидации методики анализа, (3) отчеты
о применении методики анализа для рутинного анализа испытуемых образцов
и {4) другую информацию (например, СОПы, список используемых сокращений
и ссылки).
Краткая сводная информация представляет собой табличный и пронумерованный
список всех отчетов, протоколов и кодов. В документации по разработке и валидации
метода должны быть приведены подробное описание экспериментальных методик и
проведенных исследований, доказательства чистоты и подлинности, особенности
валидации метода {результаты исследований для определения правильности,
прецизионности, степени извлечения и т.д.), а также любые отклонения от протокола
с обоснованиями. Документация, касающаяся применения метода испытания для
рутинного анализа образцов, как правило, весьма обширна. Описание должно включать:
• сводные таблицы, описывающие пробоподготовку и хранение образцов;
• подробные сводные таблицы аналитических разделений доклинических или
клинических образцов;
• данные калибровочной кривой;
• сводные данные анализа образцов КК, включая исходные данные, анализ данных
для прогнозирования дальнейшего применения метода испытания, а также
сводные данные статистической обработки полученных данных;
• примеры хроматограмм {испытуемых образцов, стандартных образцов, образцов
К К), до 20% от исследованных образцов;
• причины и обоснование отсутствия любых недостающих образцов или каких-либо
отклонений от описанных протоколов или СОПов;
• документирование любых повторных анализов или перерасчета данных с
изменением параметров интегрирования.
/2 7. Передача (трансфер) методики анализа в принимающую лабораторию (ТМА) 587
12.7. Передача (трансфер) методики анализа
в принимающую лабораторию (ТМА)
В сообществе, деятельность которого контролируется регуляторными органами,
лаборатория, разрабатывающая и валидирующая методику анализа, редко выполняет
все рутинные исследования испытуемых образцов. Вместо этого разработанные и ва-
лидированные (в лаборатории-разработчике или «передающей» лаборатории) методы
испытаний, как правило, переносят в стороннюю лабораторию («принимающую»
лабораторию) для дальнейшего применения. Однако, принимающая лаборатория
должна иметь возможность получить те же результаты в пределах погрешности
эксперимента, что и передающая лаборатория. Цель официального процесса передачи
метода испытаний заключается в том, чтобы обеспечить условия, при которых
персонал принимающей лаборатории был бы хорошо обучен, обладал достаточной
квалификацией для выполнения данного метода испытания и мог получить такие же
результаты (в пределах погрешности эксперимента), как и в передающей лаборатории.
Разработка и валидация устойчивых методов испытаний и строгое следование
хорошо проработанным СОПам — вот что обеспечит успешную передачу этого метода
в стороннюю лабораторию.
Процесс, обеспечивающий документальное подтверждение того, что методика
анализа работает в принимающей лаборатории так же хорошо, как в передающей
лаборатории, называется передачей (трансфером) методики анализа (ТМА)'. Тема
ТМА рассматривалась Американской ассоциацией ученых-фармацевтов (AAPS)2
в сотрудничестве с FDA и регуляторными органами Европейского союза,
Американской ассоциацией фармацевтических исследователей и производителей
(PhRMA)3 и Международным обществом фармацевтической инженерии (ISPE)4
|17—18|. В результате PhRMA был разработан документ «Приемлемая
аналитическая практика» (ПАП) |19|, который является приемлемым начальным
руководством по проведению ТМА.
В сущности, процесс ТМА обеспечивает подготовку принимающей лаборатории
к использованию передаваемого метода испытания. Для регуляторных органов
требуется документальное подтверждение того, что процесс переноса метода испытания
был успешно завершен. Только когда оба процесса (приобретение квалификации
персоналом лаборатории и оформление документации) завершены, принимающая
лаборатория может получить соответствующие cGMP «отчетные данные»,
основанные на результатах проведенных в этой лаборатории анализов. ТМА предназначен
именно для методов испытаний лекарственных средств и фармацевтических
субстанций, но те же принципы применимы к биоаналитическим методам (раздел 12.6).
Рассмотрим типичный пример, когда ТМА проводится между исследовательской
группой, которая разрабатывает метод испытания, и группой контроля качества,
ответственной за выпуск готового продукта. При передаче любой информации из
одной группы в другую (например, от фармацевтической компании в аналитическую
лабораторию, работающую по контракту), следует соблюдать правила ТМА. И у
передающей, и у принимающей лаборатории есть определенные обязанности в
процессе ТМА; они перечислены в таблице 12.11.
'От английского Analytical Method Transfer (AMT) — Прим. персе.
^American Association of Pharmaceutical Scientist — Прим. верее.
3 Pharmaceutical Research and Manufacturers of America — Прим. nepee.
''international Society for Pharmaceutical Engineering— Прим. иерее.
5 Acceptable Analytical Practice (AAP). — Прим. nepee.
588 Глава 12. Валидация метода
Таблица 12.11. Передача (трансфер) методики анализа: обязанности передающей и
принимающей лабораторий
Обязанности передаюшей лаборатории
Создание протокола передачи (трансфера)
Проведение обучения
Оказание помощи при анализе
Определение критериев приемлемости
Предоставление окончательного отчета
(вместе с принимающей лабораторией)
Принимающая лаборатория обеспечивает
Контрольно-измерительные приборы,
соответствующие требованиям
Персонал
Системы (оборудование)
Выполнение протокола
Предоставление окончательного отчета
(вместе с передающей лабораторией)
Перед началом ТМА следует провести несколько мероприятий, чтобы свести
к минимуму неожиданные проблемы при передаче метода. Если ранее принимающая
лаборатория не работала с передаваемым методом испытания, она должна иметь
возможность рассмотреть метод до передачи, а также провести его испытание, чтобы
выявить любые потенциальные проблемы, которые, возможно, потребуется решить
до завершения процесса передачи. Передающая лаборатория должна предоставить
принимающей лаборатории все результаты валидации, в том числе результаты
исследования устойчивости, а также документы об обучении персонала.
12.7.1. Опции передачи (трансфера) методики анализа
Основой успешного ТМА является правильно разработанная и валидированная
методика анализа. Поможет облегчить передачу методики и качественное исследование
ее устойчивости. Хорошо продуманный процесс ТМА требует исследования
достаточного количества образцов для подтверждения статистической оценки
пригодности методики анализа, поскольку один тест не является достаточным
доказательством того, что метод испытания будет корректно работать в дальнейшем. Однако
проведение официального ТМА не всегда необходимо. Для испытаний или
исследований методики не требуется ее официальной передачи. Основой для такой передачи
методики является выполнение теста пригодности системы. Во всех случаях
при определении требований ТМА следует руководствоваться обоснованной научной
оценкой конкретной ситуации.
Для ТМА можно использовать несколько различных подходов. К ним относятся:
• сравнительное тестирование;
• полная или частичная валидация методики анализа или ее повторная валидация;
• совместная валидация двумя лабораториями;
• невыполнение официальной передачи методики, которое иногда называют
отказом от передачи.
Выбор того или иного вариант» зависит от стадии разработки метода испытания
(ранней или поздней), на которой его планируют использовать; от типа метода
(например, фармакопейный или не фармакопейный, простой или сложный), а также
от опыта и квалификации персонала лаборатории.
12.7.1.1. Сравнительное тестирование
Наиболее распространенный вариант ТМА — сравнение данных анализа
испытуемых образцов в двух (или более) лабораториях. При этом две или более лаборатории
выполняют заранее утвержденный протокол, в котором указаны все критерии
12.7. Передача (трансфер) методики анализа в принимающую лабораторию (ТМА) 589
для определения того, достаточен ли уровень квалификации в принимающей
лаборатории для использования передаваемого метода испытания. Данные, полученные
при совместных испытаниях, сравниваются с набором заранее определенных
критериев приемлемости. Например, передающей и принимающей лабораториям могут
предоставить набор образцов и холостых проб без этикеток с известными
концентрациями; затем результаты, полученные в этих лабораториях, будут сравниваться с
истинными значениями, чтобы определить уровень квалификации принимающей
лаборатории. Сравнительные испытания также могут быть использованы в других
ситуациях после утверждения протокола, в частности в дополнительных
производственных подразделениях и/или в лабораториях, работающих по контракту. В общем
случае сравнительное тестирование наиболее часто используется для методик
анализа, находящихся на поздних стадиях разработки, и для передачи более сложных
методик анализа.
12.7.1.2. Совместная валидация
Обычно для проведения сравнительных испытаний (раздел 12.7.1.1) требуется, чтобы
метод испытания был валидирован. Тем не менее, существует еще один вариант
ТМА. Он заключается в том, чтобы привлечь принимающую лабораторию с самого
начала к проведению валидации метода испытания, подлежащего передаче. После
завершения исследований для совместной валидации персонал принимающей
лаборатории считается достаточно квалифицированным для выполнения анализов
испытуемых образцов с использованием данной методики. Для такой передачи метода
испытания принимающая лаборатория должна участвовать в определении
промежуточных валидационных характеристик и в планировании исследований. Если
включить в валидационный отчет данные всех лабораторий, участвовавших в
исследованиях, этот отчет может служить подтверждением ТМА; в этом случае от
принимающей лаборатории не требуется отдельных исследований для валидации метода
испытания.
12.7.1.3. Валидация и/или повторная валидация метода
Еще один вариант ТМА включает повторение принимающей лабораторией
некоторых или всех валидационных экспериментов лаборатории-разработчика. Как и в
случае совместной валидации (раздел 12.7.1.2) после выполнения части исследований
для валидации методики анализа квалификация персонала принимающей
лаборатории считается достаточной для выполнения рутинного тестирования испытуемых
образцов для выпуска производимой продукции. При этом сотрудники передающей
лаборатории и отдела контроля качества определяют, какое количество экспериментов
потребуется для удовлетворительного проведения ТМА.
12.7.1.4. Отказ от оформления передачи метода испытания
Отказ от оформления передачи (трансфера) метода испытания допустим, когда
не требуется официальный ТМА (например, для фармакопейных методов), и в
некоторых других ситуациях, когда есть основания не проводить официальный ТМА.
Отказ от передачи рассматривается в следующих случаях:
• принимающая лаборатория в настоящее время тестирует данный продукт другим
методом;
• метод испытания используется для лекарственной формы, сравнимой с новым
продуктом;
• метод испытания (или другой подобный метод) уже используется для другой цели;
• изменения, внесенные в новую методику анализа, не приводят к существенным
изменениям используемой методики анализа;
590 Глава 12. Валидация метода
• персонал передающей лаборатории сопровождает передачу методики испытания
из одной лаборатории в другую.
Когда заявляется отказ от передачи, принимающая лаборатория может
использовать методику испытания без получения каких-либо сравнительных данных. Тем
не менее, причины отказа должны быть документально обоснованы.
12.7.2. Основы ТМА
Для успешного проведения ТМА необходимы многие взаимосвязанные компоненты.
Как и в любом процессе валидации, важно документировать как процесс проведения
ТМА, так и его результаты. Все этапы от протокола ТМА до отчета о передаче
методики анализа должны быть отражены в документах с целью проверки их
соответствия поставленной задаче.
12.7.2.1. Предварительно утвержденный протокол плана
тестирования
Перед началом ТМА должен быть составлен протокол с описанием общего процесса
передачи методики анализа и критериев приемлемости. Этот документ, как правило,
оформляется в виде стандартной операционной процедуры (СОП), в которой
приведен подробный протокол ТМА или плана тестирования для конкретного продукта и
методики анализа. В этом документе следует четко определить сферу применения и
цели ТМА, все обязанности вовлеченных в процесс лабораторий, список всех
методик анализа, которые будут переданы (если ТМА включает более одной методики),
обоснование того, что какие-либо методики анализа не включены этот список (т.е.
отказ от передачи метода), а также критерии приемлемости. В документе также
следует указать, как отбирать материалы и образцы, которые будут использоваться
в ТМА. Протокол должен также содержать сертификаты анализов всех образцов
и стандартных образцов.
Следует использовать репрезентативные однородные образцы, которые одинаковы
для обеих лабораторий. Очень важно выбрать надлежащие образцы. Как правило,
выбирают образцы не из «официальных» партий готовой продукции (например,
продуктов, подлежащих аттестации по GMP) и не из «контрольной партии», чтобы не
потребовалась экспертиза несоответствия спецификации, если будет получен неожиданный
результат. Помните, что целью сравнительных испытаний методики анализа является
оценка ее рабочих характеристик, а не выявление изменений в образцах или матрице.
Следует также привести описание используемого оборудования и его настроек.
Лучше всего, если в обеих лабораториях будет использоваться одно и то же
оборудование (например, переданное передающей лабораторией в принимающую). Если это
не так, поскольку оборудование передается редко, передающая лаборатория должна
рассмотреть вопрос об использовании оборудования, сходного с имеющимся в
принимающей лаборатории (например, оборудование одного и того же производителя и той
же модели) для выявления потенциальных проблем до начала официального ТМА.
При исследовании внутрилабораторной прецизионности для валидации методики
анализа обычно принимаются во внимание и различия в используемом оборудовании.
12.7.2.2. Описание метода / процедуры испытаний
В документацию следует включать не только перечень действий для выполнения
метода испытания, но также данные валидации и любые специфические особенности
методики анализа. Любые меры предосторожности, которые следует принять
для обеспечения успешных результатов, должны быть включены в описании методи-
12.7. Передача (трансфер) методики анализа в принимающую лабораторию (ТМА) 591
Таблица 12.12. Планирование эксперимента и критерии приемлемости для передачи
аналитического метода
Тип
метода
Количественное
определение
Однородность
состава
Примеси,
продукты
разложе-
ния
Тест на
растворение
Количество
аналитиков
2
2
2
н/д
Ш
3 партии в 3-х
повторах
] партия
3 партии в 2-х
повторах6
6 единиц для
теста немедленного
высвобождения,
и 12 единиц
для замедленного
высвобождения
Критерии приемлемости
Требуется /-критерий
для сравнения 2 выборок
результатов с внутренним
разбросом значений < 2%
для доверительного
интервала 95%
Включает
непосредственное сравнение средних
значений < 3% и
разброса результатов (%ОСО)
как /-критерий для
сравнения 2 выборок
результатов с внутренним
разбросом значений <3%
для доверительного
интервала 95%
При высоком
содержании требуется /-критерий
для 2 выборок
результатов с внутренним
разбросом значений < 10%
для доверительного
интервала 95%; при низком
содержании критерии
основаны на абсолютной
разности средних
значений 125%
Требуется соответствие
спецификациям по
растворению в обеих
лабораториях, и два профиля
должны быть
сопоставимы или абсолютная
разность средних значений
должна быть ±5%
Примечания
Каждый аналитик должен
использовать различные
приборы и колонки, если
они имеются, и
независимо приготовить все
растворы. Должны быть
соблюдены все указанные
критерии пригодности системы
Если метод тестирования
однородности состава
эквивалентен методу
количественного определения
(например, одинаковые
концентрации стандартов
и образцов, условия ВЭЖХ
и критерии пригодности
системы), тогда не
требуется ТМА
Должны быть соблюдены
все применимые критерии
пригодности системы.
ПКО должен быть
подтвержден в принимающей
лаборатории, и должны
быть сопоставлены
профили примесей на хроматог-
раммах. Все образцы
должны быть схожими по
времени и условиям
хранения, однородности,
упаковке. Если образцы не
содержат примесей,
превышающих предельное
содержание, рекомендуется
использовать образцы
с добавками
Можно выполнить
статистическое сравнение
профилей или данных в конце
теста, полученных через
такой же интервал
времени, как и данные
количественного анализа
592 Глава 12. Валидация метода
Окончание табл. 12.12
Тип
метода
Определение
подлинности
Валидация
процесса
очистки
Количество
аналитиков
Партия* или
единица
дозирования
1 единица
2 образца с
добавкой, в одном
из которых
содержание
добавки больше при
веденного в
спецификации,
а в другом —
меньше
приведенного в
спецификации
Критерии приемлемости
Требуется подтверждение
хро мато графического
времени удерживания.
Возможна также
идентификация по спектрам и
химическим тестам, если
операторы достаточно
к вал ифицирован ы,
а
контрольно-измерительная аппаратура
позволяет обеспечить
эквивалентные результаты
Количество добавки
не должно отклоняться
от спецификации на
величину > 3-кратного
утвержденного
стандартного отклонения
методики анализа или 10%
от значения в
спецификации, в зависимости
от того, что больше
Примечания
По сути, тест предельного
содержания. Образцы
с низким и высоким
содержанием требуются
для подтверждения как
положительных, так и
отрицательных результатов.
Данные из 117].
л «Партия» или «серия» — это тестируемый материал, который был изготовлен одновременно
на одном и тем же оборудовании; образцы в партии считаются идентичными; образцы из
разных партий могут иметь (незначительные) различия; «Единица» — это единица дозирования
(например, таблетка, флакон, шприц или кусочек пластыря).
6 Нужно брать i партии в трех повторах, если примеси и содержание основного вещества
определяются в одном и том же испытании.
ки анализа. Метод испытания должен быть описан таким образом, чтобы обеспечить
однозначную интерпретацию текста (например, ввести обозначение об./об., если
проводилось измерение объема, а не взвешивание). В случае необходимости следует
приводить уравнения и четкие указания для проведения расчетов; примеры расчетов
часто помогают исключить неправильное толкование инструкций.
12.7.2.3. Описание и обоснование требований к тестам
Описание и обоснование требований к тестам должно включать конкретную
информацию, например, количество повторных анализов проб образца и партий образцов,
которые должны быть протестированы, а также обоснование того, как была выбрана
каждая характеристика. В этом разделе следует приводить описание всех параметров
пригодности системы, установленных для метода испытания.
12.7.2.4. Критерии приемлемости
Перед тем как произойдет передача метода, в документации оговаривается, как будут
оцениваться результаты. Так как обычно используется статистическая оценка, требу-
12.7. Передача (трансфер) методики анализа в принимающую лабораторию (ТМА) 593
ется, например, ясно указать количество партий исследуемых образцов и повторов
при тестировании. Обычно для оценки критериев приемлемости применяется
простая статистическая обработка, например, среднее значение и стандартное
отклонение при повторении испытаний в передающей и принимающей лаборатории. Также
широко применяются более сложные статистические методы, такие как ^-критерий'
или f-критерий Стьюдента. Надлежащее использование статистики может обеспечить
непредвзятое и объективное сравнение результатов с использованием заранее опре
деленной процедуры, приведенной в документации протокола трансфера. В поисках
более подробной информации рекомендуется обратиться к ссылкам по
статистической обработке (например, |20—22]). Поскольку технические характеристики
изменяются в зависимости от метода испытания, оборудования, образца и других
переменных, специфические критерии приемлемости в руководстве PhRMA не
перечислены [19|. Неполный обзор рекомендуемых в перечне ISPE |17] экспериментов и
критериев приемлемости представлен в таблице 12.12.
12.7.2.5. Документирование результатов
В отчете по ТМА обобщаются результаты выполнения ТМА. Отчет подтверждает, что
критерии приемлемости соблюдены, и что персонал принимающей лаборатории
полностью обучен и обладает достаточной квалификацией для проведения
испытаний в соответствии с переданной методикой анализа. В дополнение к кратким
сведениям обо всех выполненных экспериментах и полученных результатах в отчете
должны быть перечислены все приборы, используемые в переданной методике анализа.
Важно включить в отчет по ТМА любые наблюдения, сделанные при выполнении
передачи методики анализа. Эти наблюдения можно использовать для дальнейшей
оптимизации метода испытания, или они могут способствовать выявлению особых
проблем, которые могли не предвидеть сотрудники передающей лаборатории.
Конечно, иногда принимающая лаборатория может не соответствовать
критериям приемлемости протокола ТМА. При неудаче с переносом метода следует
прибегнуть к комплексу предусмотренных мер в поисках информации о том, как следует
поступать в данной ситуации. Необходимо инициировать расследование и составить
итоговый отчет, а затем принять меры для устранения нежелательной ситуации.
12.7.3. Потенциальные ошибки при ТМА
Многие из распространенных ошибок, возникающих при ТМА, можно
предотвратить с помощью небольшой предварительной подготовки. Еще раз необходимо
подчеркнуть, что исследования устойчивости, проведенные при окончании разработки
методики анализа или в начале ее валидации, играют решающую роль в успехе ТМА.
В ходе исследований устойчивости представляется возможность выявить многие
из критических параметров метода испытания и внести в методику анализа
информацию о мерах предосторожности. Исследование промежуточной (внутрилаборатор-
ной) прецизионности валидируемой методики анализа может способствовать
выявлению потенциальных проблем ТМА. Можно избежать многих распространенных
ошибок, предвидя, что в разных лабораториях могут различаться оборудование, опыт
и квалификация персонала, а также способы проведения тех или иных операций.
' /"-критерий или критерий Фишера — статистический критерий, используемый для
определения качества регрессионной модели в целом и по параметрам, например, для сравнения
дисперсий двух выборок. Этот критерий входит во многие пакеты статистической обработки, в частности
в Excel. — Прим. перев.
594 Глава 12. Валидация метода
12.7.3.1. Оценка оборудования
Различия в оборудовании, его конструкции и эффективности обусловливают многие
неблагоприятные последствия, с которыми сталкиваются в ходе ТМА. Время
рабочего цикла инжектора, точность установки длины волны детектора, точность
смешивания и подачи подвижной фазы и объем задержки системы в градиентном режиме
элюирования — вот всего лишь несколько различий, которые могут привести к
разным результатам анализа одного и того же испытуемого образца, анализируемого
одним и тем же методом испытания на двух разных системах ВЭЖХ |11, 23]. Особенно
большие проблемы может вызвать выполнение методик анализа, которые требуют,
чтобы система ВЭЖХ работала на пределе ее возможностей. Например, это методы
с высокой пропускной способностью, с высокой разрешающей способностью или
анализы следовых количеств определяемых веществ. Предвидение таких проблем
во время разработки методики анализа может помочь упростить процесс ТМА.
12.7.3.2. Колонки ВЭЖХ
Колонки в значительной степени обусловливают изменчивость результатов метода
испытания. Современные колонки, однако, в этом смысле вызывают гораздо меньше
беспокойства. В документации метода испытания должны быть указаны тип,
производитель и другие данные колонки, которую планируется использовать, а также
утвержденные альтернативные колонки (если таковые имеются). Использование
формулировки «похожая или эквивалентная» колонка может привести к проблемам,
особенно для неопытных сотрудников, и этого следует избегать. Для решения этой
проблемы в изданиях USP публикуются базы данных хроматографических тестов
для определения селективности колонок |24| (для дополнительного рассмотрения
эквивалентности колонок см. разделы 5.4.2, 6.3.6.1). С помощью нужной базы данных
пользователи могут быстро определить, какие колонки эквивалентны используемой
ими в настоящее время. Применение таких подходов, как этот, должно помочь
уменьшить проблемы с поиском колонки, действительно эквивалентной используемой,
в том случае если указанной колонки нет или она больше не обеспечивает требуемую
селективность.
Изменения температуры колонки также приводят к нестабильности данных;
во всех методах испытаний следует использовать термостат для колонки и
предварительно нагревать подвижную фазу (разделы 3.7.1, 6.3.2.1). Колебания времени
удерживания и изменения хроматографической селективности при изменении
температуры можно уменьшить, используя термостат колонки с контролируемой
температурой. Температурная калибровка колоночных термостатов и равномерность
нагрева в них может варьироваться для разных марок и моделей термостатов, так что
это тоже следует учитывать при составлении протокола ТМА.
12.7.3.3. Обучение оператора
Обучение может быть проведено в любое время, но лучше обучать новых
пользователей методу до официального ТМА. Несмотря на всю подготовительную работу,
ошибки все равно допускаются — это или простые оплошности, или ошибки
при выполнении методики, которые происходят из-за неясностей в протоколе
метода испытания. Методики анализа должны быть написаны так, чтобы можно было
однозначно понять, как выполнить метод анализа (и текст следует проверить). Кроме
того, описание должно быть достаточно подробным, чтобы ничто не было оставлено
на волю случая.
12.8. Корректировка методики или модификация методики 595
12.8. Корректировка методики
или модификация методики
«Официальные» или «валидированные» методики можно найти в нескольких
изданиях, таких как USP или «Официальные методы анализа Ассоциации
химиков-аналитиков, состоящих на государственной службе (АОАС)». Методы и в «Заявках на
регистрацию нового лекарственного средства» (NDA), и в «Заявках на сокращенную
процедуру регистрации препарата» (ANDA) также считаются стандартизированными,
официальными, валидированными методами. Чтобы впервые использовать
официальный метод «как есть», лаборатория должна выполнить верификацию (проверку),
чтобы продемонстрировать, что критерии пригодности как оборудования, так и
метода выполняются |25—27|. Однако, если не удается получить желаемые результаты,
может потребоваться корректировка или модификация (изменение) стандартного
метода.
Хотя методы испытаний в USP («фармакопейные методы») считаются
валидированными, корректировка методик USP была разрешена для того, чтобы выполнялись
требования пригодности системы. Такие инструкции могут быть включены в
фармакопейные статьи пользователя'. Однако, изменение методики, как правило, требует
некоторую дополнительную частичную валидацию. Поэтому возникает важный
вопрос, в какой момент корректировка становится изменением или модификацией?
Исторически сложилось так, что если корректировка методики испытания
проводится в пределах экспериментально установленной устойчивости, никакие дальнейшие
действия по валидации не требуются, при условии, что выполняются критерии
пригодности системы. Однако любая корректировка вне границ устойчивости приводит
к изменению методики испытания и может потребовать повторной валидации.
В 1998 году Фурман и его коллеги предложили способ классификации
допустимых корректировок [28|, но официальное руководство на эту тему появилось лишь
в 2005 году 127, 29—30|. Хотя руководство о допустимых корректировках метода было
недавно включено в главу 62I по хроматографии USP |l2j, Отдел обеспечения
соответствия нормативным требованиям (ORA) FDA располагает такого рода
руководством уже несколько лет |27|. В таблице 12.13 приведены разрешенные корректировки
различных параметров ВЭЖХ, взятые из обоих документов: USP и ORA.
Корректировки за пределами диапазонов, указанных в таблице 12.13, представляют собой
модификации или изменения, которые подлежат дополнительной валидации. Следует
провести серьезное научное обсуждение, прежде чем принять решение о том,
корректировать или модифицировать конкретный метод испытания. Например, если
исследования устойчивости показали, что условия методики анализа допускают
меньшие изменения переменной, чем перечисленные в таблице 12.13, или тестирование
устойчивости показало, что допускаются более существенные изменения, результаты
проверки устойчивости должны иметь приоритет (как указано в отчете по
валидации).
Хотя критерии, представленные в таблице 12.13, могут показаться довольно
простыми, многие из них не полностью учитывают последние достижения в области
технологии ВЭЖХ, особенно в отношении колонок с гораздо меньшими размерами
частиц (например, < 2 мкм), работающих при более высоких скоростях потока
(линейных скоростях) и более высоком давлении (например, более 400 атм (6000 psi)).
Кроме того, в руководствах отсутствует обсуждение
корректировки/модификации градиента. Например, кроме указанной необходимости корректировать состав
'например, производителя лекарственных средств. — Прим. перев.
^Office of Regulatory Affairs. — Прим. персе.
596 Глава 12. Валидация метода
элюента также должна быть рассмотрена компенсация объема задержки градиента
(раздел 9.2.2.4) для получения одинаковых результатов при переходе от колонки
одного размера к колонке другого размера. Помимо этого подразумевается
идентичность химических свойств сорбента, хотя в явном виде это не указано.
Таблица 12.13. Максимальный диапазон значений корректировки рабочих условий ВЭЖХ
Изменяемый параметр
рН
Концентрация соли в буфере
Концентрация минорных
компонентов в подвижной
фазе
Длина волны УФ-детектора
Длина колонки
Внутренний диаметр колонки
Скорость потока
Объем вводимой пробы
Размер частиц
Температура колонки
Максимальный диапазон значений"
±0,2 единицы
±10%
Рассматриваются только
компоненты с содержанием 50% и
меньше: ±30% или 2% по абсолютной
величине, в зависимости от того,
что больше; максимальное
изменение ±10% по абсолютной
величине; ни один из компонентов
не может быть уменьшен до нуля
Отклонений не должно быть
±70%
±25% USP, ±50% ORA
±50%
Можно уменьшать, пока
получаемые результаты согласуются с
допустимыми пределами
прецизионности и ПО
Допустимо уменьшение размера
до двукратного
±10% USP, ±20% ORA
Комментарии
Изменения рН только
в допустимых пределах
См. примеры и обсуждение
в разделе 12.8.3
Валидированная процедура
должна использоваться
для подтверждения того,
что ошибка в установке
длины волны не
превышает ±3 нм
Рекомендации USP
см. в |12]
Допустимо увеличить
объем до двукратного, если
это не приводит к
неблагоприятным последствиям
для хроматографического
разделения*
Данные из |7, 12, 26—30].
"См. примеры в разделе 12.8.
'Неблагоприятные последствия для базовой линии, формы пика, разрешающей способности,
линейности и времени удерживания.
Не следует настраивать систему ВЭЖХ для ее соответствия требованиям
пригодности системы, если требуется компенсировать износ колонки иди сбой в работе
системы. Для предотвращения «ползучести» технических характеристик корректировки
производятся только относительно исходных условий метода испытания и поэтому
не подлежат непрерывному изменению. Корректировки разрешаются только при
наличии соответствующих стандартных образцов для всех соединений, используемых
в тесте пригодности системы, и только если при использовании этих стандартов
показано, что корректировки улучшили качество хроматографического разделения
12.8. Корректировка методики или модификация методики 597
исследуемых соединений таким образом, чтобы соответствовать требованиям
пригодности системы. Пригодность метода испытания в новых условиях должна быть
проверена путем оценки соответствующих аналитических рабочих характеристик.
Поскольку многочисленные корректировки могут кумулятивно воздействовать на
работу системы, любые изменения должны быть тщательно продуманы до их
применения. И в заключении, еще одно предостережение: приведение в таблице 12.13
пределов допустимого изменения переменной еще не означает, что эту переменную можно
изменить безнаказанно: целесообразность каких-либо изменений в пределах,
указанных в таблице 12.13, определяется тестированием пригодности системы.
12.8.1. Корректировка рН
Как показано в таблице 12.13, значение рН буфера в подвижной фазе можно довести
с точностью до +0,2 единицы рН. Например, рН 2,5 подвижной фазы может быть
скорректирован в диапазоне от 2,3 < рН < 2,7. Однако при доведении рН следует
учитывать рКа подлежащих анализу соединений, так как при рН близком к рКа его
изменение всего на 0,1 единицы может привести к значительным (> 10%)
изменениям во времени удерживания |31|. Исследования показывают, что для многих
соединений допустимое изменение на +0,2 единицы имеет смысл только в случае, если
рабочее значение рН метода испытания существенно отличается от рКа
анализируемого соединения (например, рН < 4 для основных соединений, либо рН < 3 или
рН > 7 для кислотных соединений) |32|.
12.8.2. Концентрация соли в буфере
Концентрация солей, используемых при приготовлении водного буфера подвижной
фазы, может быть изменена в пределах +10%, при условии, что вызванное этим
изменение рН удовлетворяет требованиям, указанным в разделе 12.8,1. Например,
концентрацию буфера подвижной фазы, содержащего фосфатный буфер с
концентрацией 20 мМоль, можно корректировать в диапазоне от 18 до 22 мМоль. Влияние
буфера подробно обсуждается в разделе 7.2.1.
12.8.3. Соотношение компонентов в подвижной фазе
Корректирование соотношения компонентов подвижной фазы (% буфера или
органического растворителя) допускается в определенных пределах. Минорные
компоненты подвижной фазы (содержание которых <50%) можно изменять на ±30%
относительно их содержания. Тем не менее, изменение содержания какого-либо
компонента, не может превышать ±10% по абсолютной величине (т.е., по
отношению ко всей подвижной фазе). Например, для подвижной фазы с соотношением
воды и МеОН 50:50 изменение на 30% составляет 15% по абсолютной величине,
что превышает допустимый предел ±10% по абсолютной величине для любого
из компонентов. Поэтому допускается корректировка только в диапазоне от 60:40
до 40:60. Для подвижной фазы с соотношением воды и МеОН 95:5 30% от 5%
будет 1,5% по абсолютной величине, но поскольку допускается изменение вплоть
до ±2%, можно использовать подвижную фазу с соотношением воды и МеОН
от 93:7 до 97:3.
Корректировать можно только один минорный компонент в трехкомпонентной
смеси. Например, в подвижной фазе буфер — МеОН—ACN в соотношении 60:35:5
598 Глава 12. Валидация метода
при изменении на 30% содержание МеОН составит 10,5% по абсолютной величине,
что превышает допустимые изменения +10% по абсолютной величине, так что
содержание метанола должно оставаться в диапазоне 25—45%. Для ACN 30% от 5%
составляет 1,5%, но допускается +2%, поэтому содержание ACN может меняться
в диапазоне от 3 до 7%. В этой трехкомпонентной смеси можно изменять либо
концентрацию МеОН, либо концентрацию ACN, но не сразу обе. В каждом случае
следует вносить нужное количество компонента с самой высокой концентрацией, чтобы
получилось в общей сложности 100%. Дополнительные примеры изменения
содержания для двойных и тройных смесей описаны в главе 621 USP, где можно более
подробно ознакомиться с этой информацией 112|.
Хотя в таблице 12.13 допускается корректирование состава подвижной фазы,
следует подчеркнуть, что это может изменить хроматографическую селективность.
Поэтому следует действовать с осторожностью при корректировке состава
подвижной фазы существующих методик анализа.
12.8.4. Длина волны УФ/ВИД фотометрического детектора
Отклонения от длин волн, указанных в методе испытания, не допускаются. Для
подтверждения того, что ошибка установки длины волны детектора составляет <+3 нм,
следует использовать процедуру, указанную изготовителем детектора, или другую
валили рованную процедуру.
12.8.5. Корректировка температуры
Для температуры колонки ВЭЖХ допускаются изменения ±10% (или ±20%, в
зависимости от методического руководства). Следует отметить, что изменение температуры
может оказывать значительное влияние на селективность разделения и удерживание
разделяемых веществ (раздел 6.3.2.1).
12.8.6. Изменение длины и диаметра колонки, размера
частиц сорбента
В методических руководствах несколько не согласованы указания в отношении
критериев корректировки скорости потока, внутреннего диаметра и длины колонки,
а также размера частиц. Можно уменьшить скорость потока и внутренний диаметр
на величину, большую, чем приведенная в таблице 12.13, (допускается изменение
до 50% по ORA, до 25% по USP), и это не повлияет на удерживание (или
селективность), если линейная скорость подвижной фазы остается постоянной
(предполагается изократическое разделение). В последнем издании рекомендаций USP |12|
допускается еще большая корректировка при сохранении постоянной линейной
скорости.
Хотя диапазоны изменений длины колонки, ее внутреннего диаметра и размера
частиц указаны раздельно в таблице 12.13, эти переменные в действительности
нужно рассматривать вместе и правильно масштабировать в соответствии с известными
теоретическими принципами (как и постоянную линейную скорость). Вот тогда
результатом будет эквивалентное разделение, даже если изменения выходят за пределы
рекомендуемых критериев корректировки |33]. Если отношение длины колонки
к размеру частиц (L/dp) поддерживается постоянным, можно получить идентичное
разделение, выходя далеко за рамки рекомендуемых пределов, например, для 50-мм
12.9. Контроль качества и обеспечение качества 599
колонки с размером частиц 1,7 мкм и для 300-мм колонки с 10-мкм сорбентом
(L/d„ - 3 для обеих колонок) при сохранении условия, что увеличение скорости
потока обратно пропорционально размеру частиц. (Конечно требуется, чтобы хи
мический состав стационарных фаз колонок был одинаковым, внеколоночное
размывание пика было сведено к минимуму, а давление поддерживалось в допустимых
пределах). В таких случаях как этот, когда нормативные указания неполны,
целесообразно написать приложение к описанию метода испытания (или отдельную СОП)
с указанием допустимых корректировок методики анализа с соответствующими
доказательствами для подтверждения правомерности этих корректировок.
12.9. Контроль качества и обеспечение
качества
Термины «контроль качества» и «обеспечение качества» часто используются как
синонимы. Тем не менее, в правильно организованной и должным образом
управляемой системе эти два термина имеют разные и вполне определенные значения и
предполагают выполнение различных функций. Обеспечение качества (ОК) может
рассматриваться применительно к качеству процесса, в то время как контроль
качества (КК) относится к качеству продукта. Не имеет значения, как в конкретной
организации называют те или иные функции, но область ответственности для этих двух
видов деятельности должна быть четко определена. И обеспечение качества, и
контроль качества входят в компетенцию «отдела контроля качества», и они чрезвычайно
важны для получения высокоточных аналитических результатов, соответствующих
нормативным требованиям.
12.9.1. Контроль качества
Контроль качества является видом деятельности, которая определяет приемлемость
или неприемлемость продукта путем сравнения продукта с оригинальными
спецификациями, разработанными до изготовления продукта. В некоторых организациях
группа контроля качества отвечает за использование метода испытания для
проведения анализа продукта. Другие задачи, связанные с контролем качества, могут
включать обзоры руководящих указаний, калибровки, или дополнительные виды
измерительного тестирования (отбор проб и т.д.), и их приходится решать чаще, чем
выполнять работы, связанные с обеспечением качества. Контроль качества, как
правило, требует участия персонала, непосредственно связанного с исследованием,
разработкой или производством продукта. Например, при проверке лабораторного
журнала сотрудники группы контроля качества проверяют или контролируют качество
данных, производят поиск ошибок, допущенных при заполнении журнала,
проверяют расчеты и сличают подписи. Все эти действия помогают обеспечить получение
приемлемого продукта.
12.9.2. Обеспечение качества
Обеспечение качества определяется политикой руководства, аттестованными
методиками и рабочими инструкциями, а также государственными нормативными актами.
В начале валидационного процесса методики и инструкции по обеспечению качества
(предоставляемые отделом ОК) могут служить руководством для разработки или пе-
600 Глава 12. Валидация метода
ресмотра валидационных протоколов и других документов, касающихся валидации.
В ходе аналитического этапа в задачи обеспечения качества входит проверка
корректности используемой методики анализа или ее выполнения и подтверждение
соответствия качества полученных данных правилам и предписаниям компании и
государственных органов. Обеспечение качества можно рассматривать как деятельность,
направленную на определение задач контроля качества и последующую проверку их
должного выполнения. В отличие от проверок, осуществляемых группой контроля
качества (в которых внимание фокусируется на продукте), при обеспечении качества
основное внимание уделяется процессу тестирования продукта. Целесообразно
возложить обеспечение качества на менеджеров, администраторов корпоративного
уровня или сторонних аудиторов, которые будут анализировать эффективность
системы контроля качества, предоставления отчетности, хранения данных, обучения и
повышения квалификации персонала, выполняющего работу1.
12.10. Выводы
Процедура валидации методов испытаний постоянно совершенствуется и является
лишь частью общей деятельности по созданию законодательно контролируемой
среды. Процесс валидации начинается с квалификации системы ВЭЖХ (раздел 17.2.1.1)
перед ее вводом в эксплуатацию и продолжается еще долго после разработки
методики анализа, ее оптимизации и передачи в стороннюю лабораторию и все дальнейшее
время повседневного использования методики. Четко определенный и должным
образом документированный процесс валидации предоставляет регуляторным органам
доказательства того, что и система ВЭЖХ, и метод испытания пригодны для
использования по назначению. Он также гарантирует, что условия валидации и
спецификации метода отвечают утвержденным методическим рекомендациям.
Суть заключается в том, что все вовлеченные стороны должны быть уверены
в том, что метод ВЭЖХ дает результаты анализа, которые являются достаточно
точными, согласующимися между собой и воспроизводимыми. Официальная валидация
метода представляет собой просто набор тестов, используемых для выполнения этой
задачи. Независимо от того, требуется официальная валидация или нет, выполнение
серьезного научного исследования в рамках установленной системы контроля
качества будет способствовать тому, что полученный метод испытания, а также данные,
полученные при использовании этого метода, выдержат критику любого рецензента.
Основное внимание в данной главе уделено рассмотрению фармацевтических
методик анализа, но те же принципы применимы к любой методике ВЭЖХ и позволяют
гарантировать ее пригодность для использования по назначению.
1 В конце этой главы, как и во всех остальных главах этой книги, приведен список
рекомендуемой литературы. Особенность темы валидации в ВЭЖХ в том, что руководствоваться в своей
работе по валидации аналитических методик отечественному хроматографисту придется российскими,
а не американскими или европейскими нормативными документами. Прежде всего это общие
фармакопейные статьи ГФ XIV ОФС.1.1.0012.15 Валидация аналитических методик и
ОФС. 1.2.1.2.0001.15 Хроматография. См. также: Кейтлин И.М. Проблемы высокоэффективной
жидкостной хроматографии, валидации и трансфера аналитических методик в вопросах и
ответах. — Москва: ВИАЛЕК. — 2019. — 223с: ил.; Пятигорская Н.В., Ишмухаметов А.А., Беляев В.В.,
Аладышева Ж.И., Пятигорский A.M. Валидация в производстве лекарственных средств. — М.:
ОСЮ«ГРУППА РЕМЕДИУМ», 2019. — 328 с: ил.; И. Эрмер Дж. Миллер Валидация методик
в фармацевтическом анализе. — Москва: ВИАЛЕК. — 2013. — 495 с. — Прим. перее.
Литература 601
Литература
1. Guideline for Submitting Samples and Analytical Data for Methods Validation, US FDA-CDER
(February 1987), http://www.fda.gov/cder/guidance/ameth.htm.
2. United States Pharmacopeia No. 31-NF 26, (2008), ch. 1225.
3. Analytical Procedures and Methods Validation, USFDA-CDER (Aug. 2000), http://www.fda.gov/
cder/guidance/2396dft.htm.
4. Harmonized Tripartite Guideline, Validation of Analytical Procedures, Text and Methodology, Q2
(Rl), International Conference on Harmonization, (Nov. 2005), http://www.icli.org/LOB/media/
MEDIA417.pdf.
5. Guidance for Methods Development and Methods Validation for the Resource Conservation and
Recovery Act (RCRA) Program, US EPA, (1995), http://www.epa.gov/epawaste/hazard/testmethods/
pdfs/methdev.pdf.
6. ISO/1EC 17025, General Requirements for the Competence of Testing and Calibration Laboratories,
(2005), http://www.iso.org/iso/iso_catalogue/catalogiie_tc/catalogue_detail.htm?csnumber=39883.
7. Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding Of Drugs, 21
CFR Part 210, http://www.fda.gov/cder/dmpq/cgmpregs.htm.
8. Current Good Manufacturing Practice for Finished Pharmaceuticals, 21 CFR Part 211, http://www.
fda.gov/cder/dmpq/cgnipregs.htm.
9. International Organization for Standardization, http://www.iso.org/iso/home.htm.
10. Reviewer Guidance, Validation of Chromatographic Methods, USFDA (November 1994)http://www.
fda.gov/cder/guidance/cmc3.pdf.
11. L. R. Snyder, J. J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, 2nd ed., Wi-
ley-lnterscience. New York, 1997.
12. United States Pharmacopeia No. 31-NF 26, (2008), ch. 621.
13. V. P. Shah, K. K. Midha, and S. V. Dighe, Pharm. Res., 9 (1992) 588.
14. V. P. Shah, K. K. Midha, J. W. A. Findlay, H. M. Hill, J. D. Hulse, I. J. McGilvaray, G. McKay,
K. J. Miller, R. N. Patnaik, M. L. Powell, A. Tonelli, С. Т. Viswanathan, and A. Yacobi, Pharm.
Res., 17 (2000) 1551.
15. С. Т. Viswanathan, S. Bansal, B. Booth. A. J. DeStafano, M. J. Rose, J. Sailstad, V. P. Shah, J. P.
Skelly, P. G. Swann, and R. Weiner, AAPS J., 9(1), (2007) E30. See also: www.aapsj.org.
16. Guidance for Industry, Bioanalytical Method Validation, USFDA-CDER (May 2001),http://www.
fda.gov/cder/guidance/4252fnl.pdf.
17. ISPE Good Practice Guide: Technology Transfer, ISPE, Tampa, FL (Mar. 2003), http://www.ispe.
org/cs/ispe_good_practice_guides_section/ispe_good_practice_guides.
18. PhRMA Analytical Research and Development Workshop, Wilmington DE, 20 Sept. 2000.
19. S. Scypinski, D. Roberts, M. Oates, and J. Etse, Pharm. Tech., (Mar. 2004) 84.
20. J. C. Millerand J. N. Miller, Statistics for Analytical Chemistry, Ellis Horwood, Chichester, UK,
1986.
21. N1ST/SEMATECH e-Handbook of Statistical Methods, http://www.itl.nist.gov/div898/handbook.
22. P. C. Meierand R. E. Zund, Statistical Methods in Analytical Chemistry, Wiley, NewYork, 1993.
23. M. Swartz and R. Plumb, unpublished results.
24. H. Pappa and M. Marques, presentation at USP Annual Scientific Meeting, Denver, 28 September
2006. See also: http://www.usp.org/USPNF/columns.html.
25. M. E. Swartz and I. S. Krull, LCGC, 23 (2005) 1100.
26. Pharmacopeia! Forum, 31(2) (Mar.—Apr. 2005) 555.
27. FDA ORA Laboratory Procedure, ORA-LAB.5.4.5, USFDA (09/09/ 2005). See also: http://www.fda.
gov/ora/science ref/1m/vol2/section/5 04 05.pdf.
28. W. B. Furman, J. G. Dorsey, and L. R. Snyder, Pharm. Techno!., 22(6) (1998) 58.
29. Pharmacopeia! Forum, 31(3) (May-Jun. 2005) 825.
30. Pharmacopeia! Forum, 31(6) (Nov.—Dec. 2005) 1681.
31. M. E. Swartz and 1. S. Krull, LCGC, 23 (2005) 46.
32. M. E. Swartz, unpublished data on the analysis of tricyclicamines at pH-7.2.
33. M. E. Swartz and I. S. Krull, LCGC, 24 (2006) 770.
ГЛАВА 13
РАЗДЕЛЕНИЕ БИОПОЛИМЕРОВ И
СИНТЕТИЧЕСКИХ ПОЛИМЕРОВ
Совместно с Тимоти Вером, Карлом Сканделлой
и Петером Шонмейкерсом
13.1. Биомакромолекулы 605
13.2. Молекулярная структура и конформация 605
13.2.1. Пептиды и белки (полипептиды) 606
13.2.1.1. Первичная структура 606
13.2.1.2. Вторичная структура 606
13.2.1.3. Третичная и четвертичная структуры 606
13.2.1.4. Посттрансляционные модификации 609
13.2.2. Нуклеиновые кислоты 609
13.2.2.1. Одноцепочечные нуклеиновые кислоты 609
13.2.2.2. Двухцепочечные нуклеиновые кислоты 609
13.2.3. Углеводы 610
13.2.4. Вирусы 612
13.3. Особенности ВЭЖХ биомолекул 614
13.3.1. Характеристики колонок 614
13.3.1.1. Размер пор 614
13.3.1.2. Размер частиц 615
13.3.1.3. Характеристики и стабильность носителя 617
13.3.1.4. Выход и биологическая активность 618
13.3.2. Влияние структуры белка на его поведение
при хроматографическом разделении 618
13.4. Разделение пептидов и белков 619
13.4.1. Обращенно-фазовая хроматография (ОФХ) 619
13.4.1.1. Выбор колонки 620
13.4.1.2. Выбор подвижной фазы 621
13.4.1.3. Температура 624
13.4.1.4. Градиентное элюирование 625
13.4.1.5. Влияние конформации полипептида 628
13.4.1.6. Капиллярные колонки и наноисточники ионизации 630
13.4.1.7. Разработка метода ОФХ 631
13.4.2. Ионообменная хроматография (ИОХ) и смежные методы 633
13.4.2.1. Выбор колонки 636
13.4.2.2. Выбор подвижной фазы 638
13.4.2.3. Хроматофокусирование 639
13.4.2.4. Хроматография на гидроксиапатите 640
13.4.2.5. Аффинная хроматография на сорбентах
с иммобилизованными металлами (АХИМ) 641
13.4.3. Хроматография гидрофобных взаимодействий (ХГВ) 645
13.4.3.1. Сорбенты и лиганды для ХГВ 646
13.4.3.2. Другие условия 647
13.4.4. Хроматография гидрофильных взаимодействий (ГИХ) 648
13.4.4.1. Неподвижные фазы, используемые в ГИХ 650
13.4.4.2 Подвижные фазы для ГИХ 650
13.4.4.3. Применение ГИХ в разделении пептидов и белков 650
13.4.4.4. Хроматография гидрофильных взаимодействий
с электростатическим отталкиванием (ГИХЭО) 651
13.4.5. Многомерная жидкостная хроматография (МЖХ) в протеомике .... 652
13.4.5.1. Использование коллектора фракций 653
13.4.5.2. Непосредственно соединенные колонки для МЖХ 654
13.4.5.3. МЖХ с переключением колонок 654
13.5. Разделение нуклеиновых кислот 655
13.5.1. Анионообменная хроматография 655
13.5.2. Обрашенно-фазовая хроматография 657
13.5.2.1. Олигонуклеотиды 657
13.5.2.2. Фрагменты, полученные рестрикцией, и продукты ПЦР . . . 658
13.5.2.3. Денатурирующая ВЭЖХ 658
13.5.2.4. ОФХ-5 хроматография 660
13.5.3. Хроматография гидрофобных взаимодействий 661
13.6. Разделение углеводов 662
13.6.1. Хроматография гидрофильных взаимодействий 662
13.6.2. Распределительная хроматография с управляемой ионами
селективностью 663
13.6.3. Высокоэффективная анионообменная хроматография 665
13.7. Разделение вирусов 667
13.8. Эксклюзионная хроматография (ЭХ) 668
13.8.1. Процесс разделения в ЭХ 669
13.8.2. Колонки для ЭХ 671
13.8.2.1. Сорбенты 672
13.8.2.2. Размер пор и пористость 673
13.8.2.3. Диаметр частиц 673
13.8.2.4. Увеличение разрешения в ЭХ 674
13.8.3. Подвижные фазы для ЭХ 674
13.8.4. Соображения по эксплуатации 675
13.8.4.1. Емкость колонки 675
13.8.4.2. Использование денатурирующих условий 675
13.8.4.3. Калибровка колонки 676
13.8.4.4. Использование неидеальных условий разделения 676
13.8.5. Преимущества и недостатки ЭХ 676
13.8.6. Применение ЭХ 677
13.8.6.1. Аналитические приложения 678
13.8.6.2. Препаративные приложения 679
13.9. Крупномасштабная очистка биомакромолекул 680
13.9.1. История вопроса 680
604 Содержание
13.9.2. Очистка рекомбинантного инсулина человека в промышленном
масштабе 681
13.9.2.1. Цели очистки 681
13.9.2.2. Неподвижные фазы 682
13.9.2.3. Упаковка колонки 682
13.9.2.4. Стабильность продукта и колонки 682
13.9.2.5. Состав подвижной фазы 682
13.9.2.6. Разделение 682
13.9.2.7. Регенерация колонки 684
13.9.2.8. Очистка в лабораторном масштабе 685
13.9.2.9. Масштабирование 685
13.9.2.10. Очистка в промышленных масштабах 686
13.9.3. Общие требования к препаративной очистке с использованием ЖХ . 687
13.10. Синтетические полимеры 688
13.10.1. Базовые понятия и принципы 688
13.10.2. Приемы анализа полимеров 691
13.10.3. Методы жидкостной хроматографии для анализа полимеров 692
13.10.3.1. Гель-проникающая хроматография (ГПХ) 692
13.10.3.2. Интерактивная жидкостная хроматография 692
13.10.3.3. Жидкостная хроматография в критических условиях .... 695
13.10.3.4. Другие методы 695
13.10.3.5. Химический состав как функция размера молекулы 696
13.10.4. Разделение полимеров с помощью двумерной хроматографии .... 697
Литература 698
/3.1. Биомакромолекулы 605
13.1. Биомакромолекулы
С момента, когда была разработана жидкостная хроматография, она была важным
инструментом для выделения биомолекул и исследования их свойств. Однако
для того, чтобы ВЭЖХ можно было с успехом использовать для разделения
полипептидов, нуклеиновых кислот и углеводов, необходимо было разработать сорбенты
специально для этих молекул. В этой главе мы сконцентрируем внимание на ВЭЖХ
разделениях этих трех наиболее важных классов биомолекул с упором на
аналитические и полупрепаративные приложения. Можно предположить, что общие принци
пы ВЭЖХ-разделения «малых» молекул применимы к разделению биополимеров.
Однако размер и структура биополимеров обусловливают важные отличия, которые
будут рассмотрены в этой главе. В качестве введения в представленную главу,
читателю рекомендуется сначала вспомнить связанные с ней предыдущие главы, особенно
вторую о базовых принципах и контроле разделений, а также главу девятую о
градиентном элюировании.
Основными хроматографическими методами разделения биомолекул с помощью
систем низкого давления были ИОХ, ЭХ, ХГВ и аффинная, в частности металл
-аффинная хроматография на сорбентах с иммобилизованными металлами. В этой книге
будут рассмотрены их ВЭЖХ-версии. Аффинная хроматография детально
рассмотрена в работе [1|. Обращенно-фазовая хроматография была использована с большим
успехом для разделения и охарактеризования пептидов и служит одним из основных
инструментов для разработки и описания биофармацевтических средств, имеющих
белковую основу. Поэтому обращенно-фазовая хроматография пептидов и белков
будет главной темой этой главы. Для более общих рекомендаций относительно
препаративного разделения любых образцов см. главу 15.
13.2. Молекулярная структура и конформация
Макромолекулы, находящиеся в живых клетках, являются полимерами, состоящими
из субъединиц (мономеров) со сходными химическими свойствами, такими, как
аминокислоты, нуклеотиды и сахара. Аминокислотная последовательность белков и
нуклеотидная последовательность РНК и ДНК точно задается генетическим кодом.
Напротив, последовательности углеводов в боковых цепях гликопротеинов
определяются специфичностью биосинтетических ферментативных систем и доступностью
субстратов, и потому они отличаются большим разнообразием структуры и мест
присоединения к полипептидной цепи. Свойства собранного полимера зависят от
особенностей индивидуальных субъединиц (мономеров) и их положения в молекуле.
Эти два аспекта организации биополимеров (свойства субъединиц и трехмерная
структура) влияют как на их биологические функции, так и на поведение во время
хроматографического разделения. Хотя ранее считалось, что хроматография
биополимеров определяется иными принципами, нежели для малых молекул, в настоящее
время показано: хроматографическое поведение биополимеров такое же, как и у
малых молекул, но усложнено их размерами, конформацией и трехмерной
структурой |2, 3|. Поведение этих макромолекул, особенно белков, в растворах обусловлено
их структурой и стабильностью этой структуры, в частности, возможностью
образования агрегатов. Такое поведение накладывает ограничения на выбор хроматографи-
ческих условий.
606 Глава 13. Разделение биополимеров и синтетических полимеров
13.2.1. Пептиды и белки (полипептиды)
Базовыми субъединицами полипептидов являются аминокислоты, каждая из
которых состоит из карбоксильной группы, аминогруппы и боковой цепи (рис. 13.1).
Аминокислоты различаются боковыми цепями, которые могут быть
нейтральными и гидрофильными (серии и треонин), нейтральными и гидрофобными (лейцин и
фенилаланин), кислотными (аспарагиновая и глутаминовая кислоты) или оснбвны-
ми (лизин, аргинин, гистидин). При биосинтезе полипептидов карбоксильная группа
одной аминокислоты (или аминокислотного остатка) связывается с аминогруппой
следующей аминокислоты с потерей молекулы воды и образованием амидной
(пептидной) связи (—CONH—). Особый интерес представляет аминокислота цистеин,
у которой —SH группа боковой цепи может связываться с —SH группой другого
цистеина с формированием дисульфидной связи (—SS—). Также заслуживает
упоминания имидазольная группа гистидина, которая может формировать комплексы
с ионами металлов. Структуры 20 аминокислот, наиболее часто встречающихся
в белках, показаны на рис. 13.I вместе с их одно- и трехбуквенными кодами и
значениями рКа боковых цепей, способных к ионизации.
13.2.1.1. Первичная структура
Первичной структурой является последовательность аминокислот в молекуле
(рис. 13.2а). Пептиды состоят из 40 или менее аминокислот, их масса не превышает
приблизительно 5000 Да. Белки представляют собой более крупные полипептидные
цепи, которые содержат до нескольких сотен аминокислот с массами от 5000
до 250000 Да или более. Пептиды, содержащие менее 15 аминокислотных остатков,
существуют в растворе в виде статистических клубков1, и ведут себя при
хроматографии, в сущности, как и малые органические молекулы. Когда в пептиде более
15 остатков, молекулярные взаимодействия приводит к увеличению упорядоченности
структуры, как описано ниже.
13.2.1.2. Вторичная структура
Спонтанные взаимодействия внутри молекулы полипептида во время биосинтеза
приводят к образованию вторичных структур, которые определяют трехмерную
структуру конечной молекулы. Примеры вторичной структуры (рис. 13.25) включают
в себя а-спираль, которая стабилизируется водородными связями между
приблизительно каждым четвертым из аминокислотных остатков первичной
последовательности, и |3-складчатые структуры, которые образуются благодаря водородным связям
между соседними линейными сегментами первичной последовательности.
13.2.1.3. Третичная и четвертичная структуры
Конечной конформацией единичной полипептидной цепи является третичная
структура, которая может состоять из комбинаций а-спиралей, Р-структур, поворотов и
неструктурированных участков статистических клубков (рис. 13.2в). Комбинации
элементов вторичной структуры могут существовать как домены, фундаментальные
единицы третичной структуры. Каждый домен содержит индивидуальное гидрофобное
ядро, построенное из единиц вторичной структуры. Третичная структура
стабилизируется суммой большого числа слабых взаимодействий, включая водородные связи,
ионные связи и гидрофобные взаимодействия. Кроме того, третичная структура может
зависеть от дисульфидных связей между остатками цистеина, которые могут ковалентно
соединить удаленные сегменты первичной последовательности.
1 Не имеют в растворе определенной конформации. — Прим. перев.
13.2. Молекулярная структура и конформация 607
Базовые аминокислоты
Кислые
рК,=
3.9
ОН
НО NH,
Аспарагиноаая кислота (Asp, D)
О
НО
4.3
ОН
NH,
Глутэминоаая кислота (Glu, E)
Основные
^
РК.=
10.8
H,N-(
HN —
12.5
8.0 (
О
л 1
^^Y^oh
NH,
Лизин (Lys, К)
О
^^~^f"0H
NH2
Аргииин (Afg, R)
0
у
jT™
Гистидин (His. H)
Ароматически»
f*
V
но'
о
fY^r <*
Фенилаланин (Phe, F)
О
Триптофан (Tip, W)
О
л
I^N^Y^oh
X^J NH,
Тирозин (Туг. Y)
NH2
рК. = 8.8-10.8
Алифатические
Л
"ОН
NH,
Глицин (Gly, G)
О
"у^он
NH,
Алании (Ala, A)
О
ОН
NH,
Валин (Val. V)
О
"ОН
NH;
Лейцин (Leu, L)
О
ОН
NHj
Иэолейцин (Не. I)
И мин
\^NH
Пролин (Pro. Р)
Алифатические спирты
О
II
но^у^он
NH,
Серин (Ser. S)
НО О
1 II
^Y^oh
NH,
Треонин (The T)
Серосодержащие
HS^T-^OH
NH,
Цистеин (Cys. С)
О
ОН
N14,
Мвтионин (Mat, М)
Амиды
°WJLs°H
NH, NH,
Аспарагин (Asp, N)
О О
NHj
ОН
NH,
Глутамин (Gin, Q|
Рис. 13.1. Структуры аминокислот, обнаруживаемых в большинстве белков.
Аминокислоты подразделяются на несколько групп в соответствии с химическими
свойствами боковых цепей. Значения рКа ионогенных групп боковых цепей
показаны для кислотных и основных аминокислот |7]
Глава 13. Разделение биополимеров и синтетических полимеров
Уровни структуры белков
(а) Первичная структура (б) Вторичная структура
H2N- Asp-Glu- Phe- A rg- Asp-Se г
Gly-Tyr-Glu-Val-His-Gtn-Lys-Leu-COOH
(в) Третичная структура (г) Четвертичная структура
Рис. 13.2. Структуры полипептидов, (а) Линейное расположение аминокислот в
полипептиде определяет его первичную структуру, (б) Расположение
аминокислот в гомоолигомере из 14 остатков аланина, формирующих вторичную
структуру, представляющую собой а-спираль. Связи между атомами
показаны в виде стержневой модели, наложенная лента символизирует спираль,
(в) Ленточная схема скелета р-субъединицы гемоглобина, (г) Схема
фермента каталазы, состоящего из нескольких субъединиц |7. 8|
Четвертичная структура представляет собой ассоциацию двух или более
полностью свернутых белковых цепей с образованием комплекса (рис. 13.2г).
Четвертичная структура зависит от тех же взаимодействий что и третичная. Ассоциация
белковых субъединиц (и конформационные изменения внутри субъединиц) часто играют
функциональную роль в регуляции активности белка. Сходным образом агрегация
(ассоциация субъединиц) белка может быть изменена путем связывания с
субстратами и низкомолекулярными эффекторами.
Денатурация относится как к функциональному, так и к физическому
изменению состояния нативного (биологически активного) белка. Функционально
денатурация приводит к потере биологической активности. Физически денатурация
происходит при изменении или нарушении конформации белка, что приводит к потере
вторичной структуры и структур более высокого порядка. Денатурированные белки.
iff
13.2. Молекулярная структура и коиформацил 609
приобретающие конформацию статистических клубков, часто образуют агрегаты,
выпадающие в осадок из растворов. Причиной денатурации часто является среда,
в которой находится молекула белка (подвижная фаза, в которой белок растворен
при хроматографии, или хроматографический носитель, с которым он связан).
Денатурация с потерей вторичной, третичной и четвертичной структуры часто происходит
во время ОФ-ВЭЖХ, но менее вероятна при ИОХ, ХГВ или ЭХ.
13.2.1.4. Посттрансляционные модификации
Первичная структура белка, являющаяся прямым отражением нуклеотидной
последовательности в связанном с ним гене, в значительной степени определяет
конформацию. Однако многие белки модифицируются после трансляции (первоначального
синтеза белка) путем добавления одной или более групп, и эти посттрансляционные
модификации (ПТМ) не вытекают напрямую из последовательности гена. Одна и та
же нуклеотидная последовательность гена может направлять синтез белков с
разными ПТМ при экспрессии в разных клетках. Описано огромное разнообразие ПТМ,
но наиболее частыми из них являются добавление гликозидных групп к боковым
цепям остатков серина, треонина или аспарагина (гликозилирование) и фосфорили-
рование серина, треонина или тирозина. Некоторые ПТМ важны биологически,
потому что они участвуют в регуляции функции белка, в передаче сигнала и во
взаимодействиях рецепторов с лигандами, в то время как другие являются результатом
неправильного обращения с белком во время выделения и обработки. С точки
зрения хроматографического разделения, в присутствии ПТМ может измениться
взаимодействие белка с поверхностью неподвижной фазы и его удерживание.
13.2.2. Нуклеиновые кислоты
13.2.2.1. Одноцепочечные нуклеиновые кислоты
Одноцепочечные нуклеиновые кислоты состоят из линейной цепи нуклеозидов,
(рис. I3.3), где каждый нуклеозид является либо пуриновым (аденин или гуанин),
либо пиримидиновым основанием (цитозин или тимин для ДНК, цитозин или ура-
цил для РНК) (рис. 13.36), связанным с С-1 углеродом рибозы (РНК) или дезокси-
рибозы (ДНК) (рис. 13.За). Нуклеозидные остатки соединены фосфодиэфирными
связями между З'-гидроксилом одного нуклеозида и 5'-гидроксилом последующего
нуклеозида'. Олигонуклеотиды — это короткие (обычно одноцепочечные)
нуклеиновые кислоты, длиной от 13 до 25 оснований, хотя олигонуклеотидами иногда
называют и молекулы, содержащие до ста оснований. Олигонуклеотиды с
модифицированным рибозо-фосфатным остовом (рис. 13.Зв) — синтетические производные
(обычных нуклеиновых кислот) используемые в «антисмысловой»2 терапии, где
модифицированное соединение (олигонуклеотид) может образовывать комплекс с
патогеном и дезактивировать его матричную РНК, поскольку эти две молекулы
комплементарны (см. раздел 13.2.2.2).
13.2.2.2. Двухцепочечные нуклеиновые кислоты
Они состоят из двух комплементарных полинуклеотидных цепей, организованных
в спиральную структуру. Обе цепи оборачиваются вокруг общей оси и при этом
ориентированы в противоположных направлениях (рис. 13.4). Основания прикреплены
1 Сложные эфиры нуклеозидов и фосфорной кислоты называются нуклеотидами. — Прим. персе.
2«Антисмысловой» терапия называется потому, что мешает проявлению биологической
активности патогена, т.е. проявлению заложенного в нем биологического смысла. — Прим. перев.
Глава 13. Разделение биополимеров и синтетических полимеров
(а) Структура (б) Главные азотистые основания
опигонуклеотида
fix
Гуанин
Ч.Ао Ч„\
Цитозин Урацил
(в) Олигомукпеотиды с модифицированным рибоэо-фосфатным
остовом
V
с
\
Метилфосфонат
У
s-V*o
А *
\
Фосфоротиоат
V
\
Фосфородитиоат
Рис. 13.3. Структура нуклеиновых кислот, (а) Схематическое изображение одноцепо-
чечной нуклеиновой кислоты; в РНК рибоза гидроксилирована в
положении 2' (обведено кружком), а в ДНК рибоза не сдержит гидроксильную
группу. В1 и В2 соответствуют азотистым основаниям, показанным в (6) [7|
к внешнему рибозо-фосфатному остову, находятся внутри спирали и связывают цепи
водородными связями: аденин (А) создает пару с тимином (Т) или урацилом (U),
а гуанин с цитозином (С). Как и в случае нативных белков, молекулярная структура
двухцепочечной нуклеиновой кислоты зависит от вкладов целого ряда отдельных
взаимодействий. При физиологических условиях водородные связи и
взаимодействия между основаниями стабилизируют структуру спирали. Воздействие на спираль
повышенной температуры или экстремальных значений рН может вызвать
разделение двух цепей. Одноцепочечные нуклеиновые кислоты могут образовывать
внутримолекулярные двухцепочечные сегменты за счет водородных связей между парами
азотистых оснований, если основания комплементарны. Эти структуры могут
диссоциировать при нагревании или в присутствии денатурирующих реагентов.
13.2.3. Углеводы
Полисахариды играют в биосфере разнообразные роли. Гомополимеры
глюкозы обеспечивают хранение питательных веществ как у животных, так и у растений.
Гликоген, большой и разветвленный полимер из остатков глюкозы, связанных
NH,
Аденин
4nh"H>
Тимин
X = -0-Р02-0-
(фосфат)
13.2. Молекулярная структура и конформация 61 I
Основание
Углеводный X,
остов
Цепь-1
Рис. 13.4. Схематическое изображение спиральной структуры ДНК, пунктиром
показаны водородные связи между парами комплементарных оснований
сс-1,4-гликозидными связями главной цепи, служит основной формой хранения
глюкозы у животных. Крахмал, полимер, представляющий собой форму хранения сахара
в растениях, состоит из неразветвленных (амилаза) или разветвленных цепей (амило-
пектин) глюкозы с а-1,4-связями. И гликоген, и крахмал существуют в виде
спиральных структур. Целлюлоза, растительный структурный полисахарид, представляет
собой линейный полисахарид с р-1,4-гликозидными связями.
Олигосахариды играют важную функциональную роль в качестве компонентов
гликопротеинов, включающих интегральные мембранные белки и многие секретиру-
емые белки, такие как антитела и факторы свертывания крови. Олигосахариды
участвуют в распознавании иммунных клеток и передаче сигналов между клетками,
а также способствуют стабильности белков и поддержанию структуры клеток.
Олигосахариды присоединяются к белкам как непосредственно во время синтеза (котранс-
ляционная модификация белков), так и после него (посттрансляционная
модификация белков1); при этом они ковалентно связываются с боковыми цепями серина или
треонина (О-связанные олигосахариды) или с боковыми цепями аспарагина (ТМ-свя-
занные олигосахариды).
1 После синтеза большинство белков гликозилируется именно таким образом, причем сложные
структуры строятся непосредственно на молекуле белка из моно- и дисахаридов. — Прим. перев.
Глава 13. Разделение биополимеров и синтетических полимеров
ОН Н
P-L-Фукоза
(Fuc)
p-D-Галактоза
(Gal)
CHjOH
H/ir
\ OH
honT
i
H
f\
°\ OH
н /
_Ун
10
II
N-C-CHa
H
p-D-N-Ацетилгалактозамин p-D-N-Ацетилглюкозэмин
(GlcNAc)
Ha-C-^XR
H—С—OH
R - H—С—ОН
I
CH,OH
P-D-Манноэа
(Man)
ОН Н
Сиаловая кислота
(N-Ацетилнейраминат)
(Sia)
Рнс. 13.5. Сахарные остатки, обычно встречающиеся в гликопротеинах
Преобладающими сахарами в гликопротеинах (рис. 13.5) являются глюкоза,
галактоза, манноза, фукоза, N-ацетилгалактозамин (GalNAc) и N-ацетилглюкозамин
(GlcNAc). В О-связанных гликопротеинах углеводы присоединяются к белку через
остаток GalNAc, а в N-связанных олигосахаридах — через GlcNAc. В N-связанных
гликопротеинах {преобладающая форма у млекопитающих) олигосахарид состоит
из общего для всех олигосахаридов ядра из двух остатков GlcNAc и трех остатков
маннозы. Присоединенные к этому ядру дополнительные сахара образуют
разнообразное семейство олигосахаридных структур, включая олигосахариды с высоким
содержанием маннозы и комплексные олигосахариды, содержащие остатки GlcNAc,
галактозы, сиаловой кислоты и фукозы (рис. 13.6). Гликопротеины различаются
по степени гликозилирования (она может изменяться) и структуре олигосахаридов
в каждом из сайтов гликозилирования.
13.2.4. Вирусы
В ]990-х годах с появлением генной терапии возникла необходимость в рекомбинан-
тных (созданных в лаборатории) вирусах; хроматография оказалась
предпочтительным методом очистки больших количеств материала для доклинических и клини-
13.2. Молекулярная структура и конформация
Тип с высоким содержанием
манноэы
(б)
Сложный тип
Man
a1,2
Man
к1,2
i Man
! 1x1,3s
L__
Man
cc1,2
Man
а1,3\
Man
P1.4
GlcNAc
QIC
|31,4
W
\c
Man
a1,2|
Man
У'аЛА
Man |
a1,6 !
GlcNAc
Sia
a2.3
Gal
P1.4
GlcNac
01.2
Man
a1.3
01.4
Sia
o2,3
Gal
PI .4
GlcNac
01,2
Man
«1,6!
Man
(31.4
Fuc
a1,6
GlcNAc
I PL*
GlcNAc
Asn
»-~ч ■
Asn
Phc. 13.6. N-связанные олигосахариды (а) с высоким содержанием маннозы и (б) ком-
пдексные. Общее структурное ядро, состоящее из трех остатков маннозы
и двух остатков GlcNAc, показано пунктирной линией
ческих испытаний в генной терапии. Очистка вирусов также необходима
для производства некоторых вакцин. Традиционно очистка вирусов производилась
ультрацентрифугированием в градиенте плотности хлорида цезия |4|, но этот
процесс масштабируется с трудом, не обеспечивает постоянной степени очистки, выхода
продукта и может потребовать удаления CsCI |5|.
Рекомбинантные аденовирусы (rAd, общепринятое сейчас сокращение AAV)
являются распространенными векторами (носителями) для генной терапии. Они
послужили моделями для разработки хроматографических методов очистки и анализа
вирусов. Частица аденовируса содержит 85% белка и 13% ДНК, ее молекулярный
вес 167 х 10? Да. Она состоит из икосаэдрической белковой оболочки или капсида
(диаметром от 70 до 100 нм), окружающего белковое ядро, содержащее линейный
двухцепочечный ДНК-геном 1111. Кроме того, капсид содержит некоторые
дополнительные минорные полипептиды. Геном аденовируса состоит из линейной двухцепо-
чечной молекулы ДНК длинной от 35 до 36 килобаз. В том, что касается
хроматографических разделений, вирусы имеют одну особенность — их огромный
относительный размер ограничивает проникновение молекулы вируса в поры
сорбента.
614 Глава 13. Разделение биополимеров и синтетических полимеров
13.3. Особенности ВЭЖХ биомолекул
Размер и форма молекул биополимеров, а также необходимость сохранять их
биологическую активность при препаративной наработке требуют особого подхода при
выборе колонки, подвижных фаз и температуры — любой из этих параметров может
повлиять на сохранение биологической активности. Предпочтительные условия
варьируются для каждого хроматографического режима и типа образца, как описано
в разделах I3.4—13.7. Тем не менее, можно сделать некоторые общие замечания в
отношении характеристик колонок и их стабильности в зависимости от условий
разделения {раздел 13.3.1).
Общие принципы разработки методов для отдельных типов хроматографии
представлены в главах с 6 по 9, в то время как другие аспекты рассматриваются в главах
11 и 12. Все изложенное в значительной степени применимо для всех
фракционируемых молекул, больших и малых. В этой главе рассматриваются дополнительные
соображения, важные для ВЭЖХ биомолекул. Дополнительная информация приведена
в 19].
13.3.1. Характеристики колонок
Большой размер биомакромолекул требует особого внимания при выборе размера
пор и диаметра частиц сорбента. На окончательный выбор колонки, кроме того,
влияют стабильность и степень извлечения целевого вещества, возможная
потребность в условиях, не способствующих денатурации, и стабильность собственно
колонки.
13.3.1.1. Размер пор
Емкость колонки {раздел 15.3.2.1) и способность к удерживанию образца зависят
от количества неподвижной фазы, доступной для взаимодействия с образцом.
Количество неподвижной фазы, в свою очередь, пропорционально доступной площади
поверхности сорбента (раздел 5.2.1). Для небольших молекул (менее 1000 Да),
частицы носителя с диаметром пор от 8 до 12 нм обеспечивают свободный доступ
растворенных веществ внутрь пор, так что растворенное вещество может
взаимодействовать со всей площадью поверхности (обычно ж 250 м2/г для частиц с размером пор
10 нм) и свободно диффундировать в порах (не ставя под угрозу эффективность
колонки N). Напротив, большие биополимеры могут быть частично или полностью
исключены из пор такого размера, и будут взаимодействовать только с внешней
поверхностью частиц носителя (которая составляет < 1% от общей поверхности и
емкости колонки в порах). Для достижения адекватных емкости и эффективности
колонки, а также достаточного удерживания анализируемого вещества, следует
использовать частицы с порами такого размера, который бы позволил биомолекулам
легко войти в поры и выйти из них.
Соотношение между молекулярной массой М и размером молекул глобулярных
белков и неструктурированных белков в виде статистических клубков даны в
таблице 13.1. Чтобы избежать эффекта уширения хроматографических пиков из-за
ограниченной диффузии белков внутри пор, диаметр последних должен превышать диаметр
анализируемого вещества в 3 и более раз |2, 6]. Однако площадь поверхности
уменьшается приблизительно пропорционально увеличению размера пор (таблица 13.2),
так что оптимальный размер пор обеспечивает доступ белка к порам без чрезмерного
ущерба для площади поверхности и емкости колонки. Совместное влияние размера
молекул растворенного вещества и размера пор на удерживание растворенного веще-
13.3. Особенности ВЭЖХ биомолекул 615
ства на колонке показано на рис. 13.7. На рис. 13.7я максимальное удерживание
небольших пептидов ангиотензина I и II (примерно 1000 Да каждый) наблюдается
для сорбента с порами диаметром 10 нм. Для более мелких пор исключение обоих
пептидов (из пор) приводит к снижению удерживания; для ббльших пор площадь
поверхности уменьшается с соответствующим уменьшением емкости колонки и
удерживания образца. Для белков (рис. 13.76) значительное проникновение в поры
достигается только у носителей с размером пор примерно 30 нм, и все три белка
в значительной степени исключаются из частиц с меньшим диаметром пор. На
практике колонки с диаметром пор около 30 нм подходят для белков < 50 кДа, в то время
как колонки с диаметром пор от 100 до 400 нм предпочтительнее для больших
глобулярных и/или денатурированных белков. Помните, что носители с диаметром пор
> 100 нм имеют меньшую площадь поверхности и зачастую низкую механическую
прочность.
Следует помнить, что гидродинамический диаметр молекулы белка
увеличивается приблизительно в 2—3 раза при денатурации (таблица 13.1). Следовательно, если
белки должны быть разделены в денатурирующих условиях, требуется сорбент с
порами большего размера (особенно это важно для ЭХ).
Таблица 13.1. Сравнение диаметра молекулы белка и его молекулярной массы
Молекулярная масса, кДа
1
10
100
1000
Гидродинамический диаметр
Статистический клубок (нм)*
2.6
8.2
25.8
81.6
Глобулярный белок (нм)*
1.6
3.5
7.6
16.3
Источник: Данные из [9].
"Относится к разделениям в денатурирующих условиях (включая ОФХ).
*Как правило, это белок в нативном (неденатурированном) состоянии.
Таблица 13.2. Влияние диаметра пор на площадь поверхности
Диаметр пор (нм)
10
30
100
400
Площадь поверхности (м2/г)'
250
100
20
5-10
"Приблизительные значения, которые могут изменяться с объемом пор.
13.3.1.2. Размер частиц
В настоящее время колонки, упакованные полностью пористыми частицами
размером от 3,5 до 5 мкм, являются наилучшими для низкомолекулярных аналитических
разделений. Эти же колонки широко используются для фракционирования
биомолекул. Однако более медленная диффузия биополимеров по сравнению с диффузией
малых молекул приводит к уменьшению эффективности колонки и увеличению
ширины пика (раздел 2.4.1). Этому можно противодействовать, используя гораздо более
низкие скорости потока подвижной фазы, но при этом соответственно увеличивает-
Глава 13. Разделение биополимеров и синтетических полимеров
(в)
Пора(нм) = 30
(б)
15 10
200 400 600 800
покрытие лиганда (мкмоль/г)
Пора (нм) = 30
4| г—
миоглобин
15 10
0 200 400 600 800
покрытие пиганда (мкмоль/г)
Рис. 13.7. Влияние размеров пор и размеров анализируемых молекул на удерживание
в случае ОФХ на колонке C|g. (а) Малые пептиды ( = 1000 Да); 35% ацето-
нитрил/фосфатный буфер рН 2,3; (б) белки (> 10,000 Да); 49% ацетонит-
рил/ фосфатный буфер рН 2,3. Обратите внимание на диаметр пор
сорбента, указанный вверху каждого рисунка; «содержание привитых групп»
(отложенное по оси х) пропорционально площади поверхности J10J
си время разделения. Было предпринято несколько попыток повышения
эффективности колонок для разделения больших биомолекул. Как обсуждалось в разделе
2.4.1.1, влияние медленной диффузии на эффективность колонки всегда можно
уменьшить, используя более мелкие частицы. Все большее применение для разделения
больших молекул находят частицы носителя размером до 1,5 мкм.
Дальнейшее увеличение количества теоретических тарелок N в разделениях
больших биомолекул может быть достигнуто с помощью непористых («пелликулярных»)
частиц малого диаметра (раздел 5.2.1.1). Отсутствие пор в этих носителях исключает
медленную диффузию в порах, в то время площадь внешней поверхности, хотя и
довольно малая, может обеспечить достаточную емкость колонки для анализа
основных компонентов образца. Так называемые поверхностно-пористые частицы
(раздел 5.2.1.1) имеют непористое ядро, которое способствует ускорению движения
молекул в поры и из пор сорбента. При этом увеличивается количество
теоретических тарелок /V, но немного уменьшается емкость колонки. В так называемой перфу-
зионной хроматографии (раздел 5.2.1.1 |12]) используются носители, имеющие очень
большие сквозные поры, позволяющие потоку подвижной фазы проходить сквозь
13.3. Особенности ВЭЖХ биомолекул 617))|
частицу. В принципе, это также может свести к минимуму влияние медленной
диффузии внутрь частиц и из них, хотя эти сорбенты используются, в основном,
для препаративного разделения крупных биомолекул.
Наконец, еще одним вариантом является замена обычного носителя (состоящего
из отдельных частиц) на монолит (раздел 5.2.4). Монолитные колонки состоят из
непрерывного лабиринта сообщающихся между собой сквозных пор, обеспечивающих
прохождение подвижной фазы и растворенных веществ через колонку. В результате
монолиты могут работать при высоких скоростях потока и умеренных давлениях,
а эффективность колонки снижается в этом случае ненамного. Коммерчески
доступны монолиты на основе полимеров и силикагеля. Полимерные монолиты на основе
полиметакрилата и полистирола, сшитого дивинилбензолом, доступны в виде
колонок и в виде дисков для аналитической и препаративной хроматографии1. Эти
материалы также имеют бимодальную структуру пор с большими и малыми порами.
13.3.1.3. Характеристики и стабильность носителя
Пористый силикагель обладает рядом свойств, которые благоприятствуют его
использованию в качестве хроматографического носителя (раздел 5.2.2). К сожалению, он
обладает другими свойствами, ограничивающими его использование для разделения
биомолекул. Разделение пептидов с помощью ОФХ, как правило, проводится в
кислотных условиях (рН < 3), а ионообменные разделения часто проводятся в
нейтральных или щелочных условиях (рН > 7). У некоторых колонок на основе силикагеля
стабильность понижается за пределами интервала 2,5 < рН < 7,5 (разделы 5.3.1,
5.3.2.1). Разделение иногда проводят при повышенной температуре для того, чтобы
улучшить форму пиков или оптимизировать селективность, но стабильность
силикагеля с привитыми фазами при температуре выше 40 °С понижается, особенно
при экстремальных значениях рН. Однако использование подходящих колонок
и других условий может уменьшить неблагоприятное влияние рН и температуры
подвижной фазы на стабильность колонки.
Другая большая возможная проблема, связанная с использованием колонок
на основе силикагеля для разделения биомолекул, может возникнуть в результате
сильных взаимодействий между поверхностью диоксида кремния и разделяемым
веществом. Эти взаимодействия могут привести к уширению задних фронтов пиков и
потере образца из-за необратимой адсорбции. Проблемы такого рода более присущи
старым колонкам типа А, и поэтому настоятельно рекомендуются колонки,
упакованные высокоочищенным силикагелем типа В (раздел 5.2.2.2). Кроме того, для
минимизации нежелательных взаимодействий между образцом и колонкой эффективны
эндкепированные обращенно-фазовые колонки (раздел 5.3.1).
Были использованы различные способы для улучшения качества силикагеля
с привитыми фазами для хроматографии биополимеров. Наилучшими являются
современные обращенно-фазовые колонки на основе силикагеля. Их чаще всего
выбирают для большинства разделений пептидов. Из-за ограничений для сорбентов
на основе силикагеля в случае других видов хроматографии (например, ИОХ и ХГВ)
для этих целей используются, главным образом, колонки на основе полимерных
носителей (раздел 5.2.3). Такие полимеры, как сополимер стирола с дивинилбензолом
и полиметакрилат можно получать в виде пористых частиц, которые могут быть
использованы непосредственно для хроматографии (например, полистиролдивинил-
бензольные полимеры для ОФХ). С другой стороны, к полимерной основе сорбента
могут быть присоединены специфичные группы (например, ионогенные группы
для ИОХ; раздел 7.5.4). Эти носители стабильны в широком диапазоне рН (включая
'Такие колонки как ThermoMonolithSAX, очень хорошо подходят для анализа AAV вирусов,
например, для определения соотношения пустых вирусных частиц и частиц с ДН К. — Прим. персе.
618
Глава 13. Разделение биополимеров и синтетических полимеров
рН < 2 и рН > 10) и могут использоваться при более высоких температурах, которые
разрушили бы сорбент на основе диоксида кремния. Они также стабильны при
давлениях от 276 до 345 бар (от 4000 до 5000 psi). Основным преимуществом
полимерных материалов для препаративных и промышленных применений является возмож
ность очищать их сильными щелочами, чтобы удалить загрязняющие вещества,
например, такие как эндотоксины. Эндотоксины (обычно липополисахариды из
клеток продуцентов, используемых в производстве биофармацевтических препаратов)
вызывают воспалительные реакции, и их попадания в лекарственные препараты,
предназначенные для использования человеком, следует избегать.
13.3.1.4. Выход и биологическая активность
В тех случаях, когда ВЭЖХ используется для выделения целевого вещества и
изучения его свойств, выход целевого вещества должен быть высоким. Если
биологическая активность должна быть сохранена, биополимер должен еще и сохранить
нативную конформацию. Эти требования предполагают тщательный выбор хрома
тографического режима и условий разделения. Обращенно-фазовая хроматография
может вызвать денатурацию белков, и она, как правило, не используется для
извлечения крупных белков. Однако биологическая активность денатурированных пепти
дов или небольших белков, подученных в результате обращенно-фазовых
разделений, обычно может быть полностью восстановлена с использованием буферов
без органических растворителей (спиртов, ацетонитрила) в присутствии
определенных ионов в нужной концентрации. Использование других хроматографических
режимов с жесткими условиями разделения (экстремальные значения рН, повышенная
температура) также может поставить под угрозу извлечение нативных и
биологически активных молекул. Выход полипептида может уменьшиться вследствие
адсорбции на активных центрах сорбента или вследствие застревания в его порах. Потери
образца из-за адсорбции могут быть минимизированы предварительной обработкой
колонки суррогатным биополимером (например, бычьим сывороточным альбумином
для белковых образцов), чтобы дезактивировать колонку перед использованием.
Потери образца из-за денатурации белка в системе пор можно свести к минимуму,
используя носитель с порами большего размера или подобрав менее денатурирующие
условия. Разные виды ВЭЖХ можно расположить в следующем порядке, исходя из
того, в какой степени условия проведения разделений способствуют денатурации
белка: ОФХ 3> ХГВ я ИОХ > ЭХ. Более подробно об увеличении выхода
полипептидов см. [13|.
13.3.2. Влияние структуры белка на его поведение
при хроматографическом разделении
Удерживание белка в ВЭЖХ можно трактовать как взаимосвязь между структурой
белка и процессом разделения. В случае разделения малых молекул, большая часть
молекулы находится в контакте с неподвижной фазой. Для больших пептидов и
особенно белков, это может быть невозможным, потому что только поверхность этих
трехмерных молекул (поверхность контакта) может контактировать с неподвижной
фазой. Несколько обусловленных этим закономерностей для удерживания были
описаны в основополагающей публикации Ренье [14|, и они могут быть обобщены
в следующих постулатах:
• Слабые химические взаимодействия, определяющие конформацию белка и
поверхностное распознавание (электростатические, гидрофобные, водородные связи),
такие же, как и те, что участвуют в хроматографическом процессе.
13.4. Разделение пептидов и белков 619 ))|
• Невозможно, чтобы все аминокислоты в белке одновременно контактировали
с поверхностью неподвижной фазы (особенно у нативных молекул).
• Только аминокислотные остатки, расположенные на поверхности белка,
оказывают влияние на его поведение при хроматографическом разделении, и только часть
этих остатков (те, которые находятся в области контакта) участвуют во
взаимодействиях с неподвижной фазой.
• Неравномерное распределение аминокислотных остатков на поверхности белка
позволяет некоторым участкам поверхности определять хроматографическое
поведение; эти области превалирующего взаимодействия могут быть разными для
разных видов хроматографии.
• Структурные изменения, трансформирующие поверхность белка, могут изменить
его хроматографическое поведение, если они происходят в области контакта или
изменяют поверхность контактной зоны.
• Взаимодействие с неподвижной или подвижной фазами может изменить
вторичную, третичную и четвертичную структуры молекулы белка.
Мы еще вернемся к этим концепциям в нашем обсуждении различных хромато-
графических методов.
13.4. Разделение пептидов и белков
Выбор подходящего метода для хроматографии пептидов и белков диктуется целями
разделения. Если требуется высокий выход белка и сохранение его биологической
активности, предпочитают относительно мягкие хроматографические методы —
такие как ИОХ, ХГВ и гель-фильтрация. Если цель состоит в том, чтобы разделить
белки по размеру молекул, или провести разделение больших и малых молекул, то
метод гель-фильтрации является наилучшим выбором. Обращенно-фазовая
хроматография реже используется для очистки больших белков из-за склонности белков
к денатурации при проведении ОФХ-разделений. Это может ухудшить разделение
и поставить под угрозу как выход, так и биологическую активность. Однако для
очистки пептидов и небольших белков ОФХ оказалась удивительно эффективной. Она
является общепринятым предпочтительным методом для разделения пептидных
смесей. В области протеомики, когда требуется охарактеризовать весь белковый состав
клетки или ткани, необычайно сложные смеси пептидов должны быть разделены
до анализа МС/МС (раздел 4.I4); в этом случае часто используется двумерная (2-D)
ВЭЖХ на основе ИОХ с последующим разделением методом ОФХ (раздел I3.4.5).
13.4.1. Обращенно-фазовая хроматография (ОФХ)
Широкое использование в анализе и очистке пептидов и белков объясняют несколько
особенностей ОФХ. Высокая эффективность ОФХ-колонок, упакованных мелкими
частицами, обеспечивает повышенное разрешение и высокую пиковую емкость
(раздел 9.3.9.1) в случае разделения сложных смесей. Селективность ОФХ также
способствует разделению пептидов с очень близкими структурами. Растворители,
используемые в ОФХ, совместимы с УФ-детекторами, и летучесть этих растворителей позволяет
удалять их из собранных фракций. Важнее всего то, что водно-органические
подвижные фазы совместимы с ионизацией электроспреем, и поэтому ОФХ почти всегда
используют при работе с масс-спектрометрами (раздел 4.14.1.1). Для решения проблемы
конкретного разделения доступен большой выбор подвижных фаз и колонок, хотя
на практике для большинства образцов используются несколько общих методов.
620 Глава 13. Разделение биополимеров и синтетических полимеров
Важной особенностью поведения пептидов и особенно белков в ОФХ является
сильная зависимость удерживания разделяемого вещества от небольших изменений
в составе растворителя (раздел 13.4.1.4). Например, было показано, что белок массой
29 кДа меняет время удерживания на 20% при изменении концентрации
органического растворителя всего на ±0,1%. Обычно это делает изократическое разделение
даже простой полипептидной смеси нецелесообразным. Почти всегда требуется
градиентное элюирование.
13.4.1.1. Выбор колонки
Для обычно используемых условий разделения стабильность ОФХ-сорбентов
на основе силикагеля достаточна, и они зачастую предпочтительнее сорбентов на
полимерной основе, поскольку обеспечивают более высокую эффективность колонки.
Силикагельные сорбенты с малым диаметром пор (диаметр « 10 нм) подходят
для пептидов, но для разделения белков предпочтительнее сорбенты с большими
порами (диаметр пор >30 нм). После протеолитического расщепления пептиды могут
быть самых разных размеров (с массой 200—2500 Да после расщепления трипсином
или с большей массой в случае расщепления эндопротеазами LysC и AspN1),
поэтому для некоторых образцов могут понадобиться носители с порами 30 нм. Другие
соображения, принимаемые во внимание при выборе колонки, — лиганд неподвижной
фазы и плотность его прививки к носителю.
Данные по средней селективности сорбентов с различными лигандами сведены
в таблице 5.8 и обсуждаются в пояснениях к ней. Там же приведены величины гид
рофобности сорбентов — параметра Н. Самыми популярными сорбентами являются
сорбенты на основе силикагеля с привитыми к нему линейными алкильными
группами (С4, Cjj, C|g). При увеличении длины алкильной цепи растет удерживание
образцов на этих сорбентах, а также увеличивается стабильность самих сорбентов.
Триметильные (С|) сорбенты наименее гидрофобны (Н = 0,41). Их используют
для разделения белков, которые слишком сильно удерживаются на сорбентах с более
длинными лигандами (более гидрофобных сорбентах). Однако в условиях, обычно
используемых для разделения белков, С|-сорбенты легко гидролизуются и весьма
нестабильны. Как правило, для разделения пептидов в первую очередь берут
октальные (Cg, Н = 0,84) и октадецильные (Cjg, H = 0,99) сорбенты из-за их большей
стабильности и подходящих величин удерживания.
Концентрация подвижной фазы В (органической) при градиентном разделении
пептидов и белков редко превышает 60—80%. В этих условиях селективность бутиль-
ных, октальных и октадецильных сорбентов для белков и пептидов сопоставима [16|.
Селективность фенильных и цианофаз обычно отличается от селективности алкиль-
ных сорбентов с неразветвленной цепью, но циано-сорбенты в общем случае менее
стабильны, при низком или высоком рН. С^-сорбенты намного менее гидрофобны и
хуже удерживают; поэтому их предпочитают использовать для разделения
высокогидрофобных белков, таких как мембранные, и более гидрофобных (длинных)
полипептидов, образующихся при расщеплении белков бромцианом.
Привитые сорбенты на основе силикагеля (раздел 5.3.1) более эффективны, и их
чаще всего пробуют в первую очередь. Полимерные колонки более стабильны, но их
свойства могут отличаться от партии к партии из-за того, что реакция
полимеризации плохо воспроизводится. Кроме того, на стабильность сорбента влияет
концентрация лиганда (мкмоль/м ) и остаточные поверхностные силанольные группы.
1 Если после расщепления трипсином обычно получаются пептиды длиной от 3 до 25
аминокислотных остатков, то после расщепления LysC и AspN получаются пептиды длиной от 3 до 40—
50 аминокислотных остатков, причем длинноцепочечных пептидов получается больше. — Прим. ред.
13.4. Разделение пептидов и белков 621
13.4.1.2. Выбор подвижной фазы
В ОФХ подвижная фаза состоит из водного раствора буфера (элюент А) и
органического растворителя (элюент В). Для биохимического разделения «буфер» часто
представляет собой разбавленную кислоту, такую как фосфорная, трифторуксусная,
муравьиная или уксусная — с рН от 2 до 3,5. Удерживание образца в изократическом
режиме элюирования (значения к) можно контролировать, изменяя % В — как
и в случае низкомолекулярных образцов (раздел 2.3.2). Для градиентного разделения
увеличение крутизны градиента приводит к уменьшению значений к* и, как
правило, к ухудшению разделения (см. раздел [3.4.[.4 ниже). Состав подвижной фазы
может влиять на селективность разделения, совместимость с детектором, и в меньшей
степени на эффективность колонки.
Водный компонент. Гидрофобность (и, следовательно, удерживание) полипептида
в большой степени зависит от ионизации концевых карбоксильной группы и
аминогруппы (приблизительные значения рКа 2.4 и 9.8) и ионизируемых групп боковых
цепей остальных аминокислотных остатков (значения рКа на рис. [3.1). Таким
образом, рН подвижной фазы может сильно влиять на удерживание полипептида, и
потому необходимо контролировать рН подвижной фазы, добавляя буфер (раздел 7.2).
На практике разделение белков и пептидов чаще всего проводят при низком рН,
когда буфером может служить разбавленная кислота. В этих условиях подавляется
ионизация силанольных групп, и снижаются, таким образом, нежелательные
взаимодействия с протонированными разделяемыми веществами, что, в свою очередь,
приводит к уширению задних фронтов пиков этих веществ. Обычно используемые
органические кислоты (например, трифторуксусная кислота) также могут образовывать
ионную пару с протонированными молекулами разделяемых веществ, что приводит
к увеличению удерживания пептидов и улучшению формы пиков в случае белков.
Ионизация концевых и боковых карбоксильных групп подавляется при низком рН,
что еще больше увеличивает удерживание.
Наиболее широко для разделения пептидов и белков в ОФХ используется
трифторуксусная кислота (ТФУК) с концентрацией 0,05—0,1% (приблизительно 5—
10 мМ). Поскольку ТФУК более сильная кислота, чем, к примеру, муравьиная или
уксусная, ее требуется меньше для приготовления элюента и рН подвижной фазы
можно снизить (= 2). Кроме того, более низкие концентрации ТФУК уменьшают
фоновое поглощение в тех случаях, когда используется УФ-детектирование на коротких
длинах волн. Дополнительным преимуществом ТФУК является ее высокая летучесть,
что облетает удаление растворителя из собранных фракций образца в препаративных
разделениях. Однако, очищенные белки, как правило, удерживают значительные
количества ТФУК, которые могут быть удалены с помощью диализа или диафильтрации.
Более высокое поглощение органических кислот в УФ-диапазоне требует
детектирования на более высоких длинах волн, что увеличивает дрейф и шум базовой линии хро-
матограммы (все это отрицательно сказывается на чувствительности детектирования).
Для анализа малых количеств пептидов можно использовать вместо органической
кислоты фосфорную (которая прозрачна при 200 нм и выше), но в случае белков это
зачастую может приводить к размыванию задних фронтов пиков.
Использование ТФУК в качестве буфера при масс-спектрометрическом
детектировании (ЖХ-МС) может создать трудности при использовании ионизации
электрораспылением. При детектировании отрицательно заряженных ионов высокая
концентрация аниона ТФУК может подавлять ионизацию разделяемого вещества.
В режиме детектирования положительно заряженных ионов ТФУК образует такие
прочные ионные пары с пептидами, что выброс псевдомолекулярного иона пептида
в газовую фазу может подавляться. Эта проблема может быть частично решена с
помощью постколоночного добавления более слабой и менее летучей кислоты, такой
622 Глава 13. Разделение биополимеров и синтетических полимеров
как пропионовая |17|. Этот способ избежать подавления ионов пептида позволяет
использовать ТФУК с онлайн источником электрораспыления вкупе с квадруполь-
ными масс-спектрометрами. Более удобное решение этой проблемы состоит в
простой замене ТФУК уксусной или муравьиной кислотами.
В случае ОФХ-разделений при низких рН у пептидов иногда наблюдается уши-
рение задних фронтов пиков. Оно обычно связано с протонированием аминогрупп
в молекуле разделяемого вещества (раздел 7.3.4.2). Для современных сорбентов
из алкилированного силикагеля типа В уширение заднего фронта пика при низком
рН зависит, как правило, от количества введенного в колонку пептида. Оно
появляется при перегрузке колонки (из-за взаимного отталкивания положительно
заряженных ионов разделяемого вещества в неподвижной фазе; раздел 15.3.2.1). Перегрузка
колонки (и уширение задних фронтов пиков) обычно происходит в том случае, если
масса образца > 1 мкг пептида на колонку с внутренним диаметром 4,6 мм (или
при меньшем количестве для колонок с меньшим внутренним диаметром). Можно
вводить несколько большее количество пептида, увеличив ионную силу подвижной
фазы, например, путем использования полностью ионизированных кислот, таких
как ТФУК или фосфорная кислота, или сильно ионизированных буферов, таких как
ацетат или формиат аммония при более высоких рН |18, 19].
ТФУК способна к образованию ионных пар с протонированными основными
пептидами, как показано на рис. 13.8. Смесь синтетических пептидов, которые
содержат 0, 1, 2, 4 или 6 основных остатков, разделяли, используя элюенты с различ-
1
+
2
*~J \ 1
6
0,02%
ТФА
исх
0,05%
0,1%
ш
1 I I I
л)
0,2%
11
0,4%
0
л!
1 1
0,8%
1 1
18 20 22 24 20 22 24 26 (мин)
Рис. 13.8. Влияние концентрации ТФУК на удерживание основных пептидов.
Образец: синтетические пептиды с различным количеством основных
аминокислот в молекуле (количество основных групп указано для каждого пика).
Условия: колонка Си 250 х 4,6 мм; градиент ацетонитрил-вода с указанной
на рисунке концентрацией ТФУК; 26 °С; 1,0 мл/мин |20|
13.4. Разделение пептидов и белков 623
ными концентрациями ТФУК. При увеличении концентрации ТФУК увеличивается
удерживание оснбвного пептида из-за образования ионных пар (раздел 7.4.1), и этот
эффект сильнее для пептидов с ббльшим количеством оснбвных групп. В результате
изменяется относительное удерживание при изменении концентрации ТФУК.
Способность ТФУК изменять относительное удерживание в зависимости от заряда
пептида может быть полезна при разработке методов разделения.
Если нужно разделить белки, отличающиеся модификациями поверхности,
может быть целесообразным использование условий, не способствующих денатурации
|21|. Стандартные условия с низким рН, описанные выше, в этом случае не
подходят, и потребуются подвижные фазы, забуференные приблизительно до
нейтрального рН. Буферы на основе ацетата аммония, бикарбоната аммония и фосфата триэти
ламмония также могут оказаться более полезными при разделении вариантов
полипептидов с различными посттрансляционными модификациями, заменами
аминокислот или продуктами окисления и дезамидирования |21|.
Фосфат триэтиламмония в качестве буфера с ацетонитрилом в качестве В элю
ента изначально рекомендовался для ОФХ разделения пептидов |221 прежде всего
потому, что улучшал форму пиков и выход целевого продукта. Тем не менее, в
настоящее время, когда для разделения пептидов используются колонки типа В,
потребность в этом буфере отпала.
Органический компонент. Обычными В-растворителями для разделения пептидов
и белков методом ОФХ являются ацетонитрил, метанол, пропанол и изопропанол.
Самый популярный из них — ацетонитрил. Основные критерии выбора В-раствори-
теля: низкое поглощение в УФ-области и низкая вязкость. Лучше всего этим
критериям соответствует ацетонитрил. Кроме того, должны быть учтены селективность
растворителя, его стоимость, токсичность и чистота. Пептиды и белки могут быть
обнаружены по УФ-поглощению пептидной связи на длинах волн от 205 до 220 нм.
Ацетонитрил имеет хорошую оптическую прозрачность в этой области и позволяет
вести детектирование на длине волны 205 нм. Использование метанола или пропа-
нола требует детектирования на длинах волн > 210 нм во избежание чрезмерного
дрейфа базовой линии при градиентном элюировании. Интенсивность поглощения
полипептидов ослабевает на более длинных УФ-волнах. При этом уменьшается
чувствительность детектирования. Для пептидов и белков обычно используется УФ-де-
тектирование на длинах волн от 205 до 220 нм, поскольку оно «универсально»
для всех полипептидов. Остатки тирозина и триптофана имеют локальные
максимумы поглощения при 270 и 280 нм, соответственно, что позволяет селективно
детектировать пептиды и белки, содержащие эти остатки. Однако, поглощение этих
полипептидов при 270—280 нм намного ниже, чем при 205 нм.
Смеси ацетонитрил-вода обладают более низкой вязкостью, чем смеси спирт-вода,
что способствует большей скорости разделения с более высоким разрешением
(раздел 2.3.1). Поведение ацетонитрила, метанола и изопропанола в качестве В-раствори-
теля показано в сравнении на рис. 13.9 в градиентном разделении ряда синтетических
пептидов. Анализируемыми веществами в этом исследовании были октапептиды
одинаковой структуры, за исключением разных аминокислот во втором и третьем
положениях последовательности пептида |23|. Общее время анализа увеличивается в ряду
изопропанол < ацетонитрил < метанол, что говорит об обратном порядке увеличения
силы растворителя: метанол {самый слабый) < ацетонитрил < изопропанол (самый
сильный). Наиболее узкие и симметричные пики обычно наблюдаются при
использовании ацетонитрила в качестве В-растворителя. Селективность различна при
использовании этих трех В-элюентов. Это показано для группы из семи ликов, отмеченных
скобкой и звездочкой на рис. 13.9. При использовании изопропанола в качестве
В-растворителя, эти пептиды плохо разрешаются, но их разрешение улучшается при исполь-
624 Глава 13. Разделение биополимеров и синтетических полимеров
Иэопропанол (э)
Ацетонитрил (б)
Метанол (в)
60(мин)
Рис. 13.9. Влияние В-растиорителя подвижной фазы на разделение пептидов методом
ОФХ. Образец описан в тексте. Условия разделения: колонка Cs
250 х 4,1-мм; линейный градиент ] %В/мин; рН 2; 26 °С; 1,0 мл/мин [23]
зовании метанола и еще более в случае ацетонитрила. На рис. 13.9 можно увидеть и
другие изменения в селективности, связанные с изменением В-растворителя.
Поверхностно-активные вещества. ПАВ иногда добавляют в образец и / или
в подвижную фазу для солюбилизации гидрофобных плохо растворимых белков и
для увеличения их выхода |21|. Например, в методах ОФХ для разделения
интегральных мембранных белков часто используют ионные, цвиттерионные или неионные
поверхностно-активные вещества в качестве добавок. Поскольку адсорбированное
поверхностно-активное вещество может быть трудно удалить из колонки, то
целесообразно выделить отдельную колонку для использования данного
поверхностно-активного вещества.
13.4.1.3. Температура
Изменение температуры колонки может улучшить разделение пептидов или белков
в ОФХ. Во-первых, при более высоких температурах снижается вязкость подвижной
фазы и увеличивается диффузия растворенного вещества. Каждый из этих факторов
способствует повышению эффективности колонки и лучшему разрешению.
Во-вторых, изменение температуры колонки может быть использовано для оптимизации
селективности разделения — как и в случае других ионных образцов (раздел 7.3.2.2).
На рисунке 13.\0а-г приведен пример градиентного разделения пептидов,
полученных трипсинолизом рекомбинантного человеческого гормона роста при 30 и 50 °С
(показаны только пики 6—13 из 18). Наконец, повышение температуры приводит
к увеличению степени денатурации белка, что, как правило, приводит к более узким,
более симметричным пикам и повышению выхода целевого вещества в ОФХ. С
другой стороны, работа при высокой температуре может уменьшить время жизни
некоторых колонок на основе силикагеля.
13.4. Разделение пептидов и белков
<Ф
(а)
(в)
0-60 %В за 25 мин; 30 'С
Rs= 1,2 (зоны 11/12)
(6)
1 ' ' '—
10 12
Время (мин)
0-60 %В за 25 мин; 50 *С
Rs= 1,4 (зоны 10/11)
6
t 1
0-60 %В за 75 мин; 30
Rs = 1,0 (зоны 11/12)
11
1||
Ail
|' 1"'Г Т Т 1 1
12 13
■■■1— Т '^Г" у1!
С
I14
1
'
18 20 22 24 26 28 30 32
Время (мин)
0-60 %В за 75 мин; 50 °С
Rs= 1,2 (зоны 10/11)
(а)
I I | I I I I I I < I I |
8 10 12 16 18 20 22 24 26 28 30
Время (мин) Время (мин)
W 0-60 %В за 75 мин
45*C;RS = 2,1
0-40 %В за 50 мин <в>
45'С; Я3 = 2,1 (полная
хроматограмма
i—i—i—|—i—i-i—i—i—i—i—i—i—|—
18 20 22 24 26 28 30
Время (мин)
6-14
■k-rrii
iLi
1э
20 40
Время (мин)
100 %В
80%
60%
40%
20%
0%
Рис. 13.10. Разделение пептидов лизата /"ЮН при различных временах градиента и
различных температурах колонки с целью оптимизации селективности и
максимального разрешения. Условия разделения: колонка Cjg 150 х 4,6-мм:
градиенты ацетонитрил (В)—вода +- 0,1% ТФУК; 2,0 мл/мин; другие условия
показаны на рисунке. Данные для моделирования взяты из |24]
13.4.1.4. Градиентное элюирование
Как отмечалось выше, ОФХ-разделения биомолекул характеризуются резкими
изменениями в удерживании при небольших изменениях % В. Как следствие, для таких
образцов обычно требуется градиентное элюирование. Зависимость изократического
626 Глава 13. Разделение биополимеров и синтетических полимеров
удерживания к от состава подвижной фазы (% В) можно приближенно описать
уравнением:
log к - log kw - Sip, (13.1)
где ф (равно 0,01 х % В) — объемная доля органического растворителя, kw —
(экстраполированное) значение к для буфера (буфера А) в качестве подвижной фазы (ф = 0
или 0% В), a S представляет собой константу для конкретного растворенного вещества
и определенных экспериментальных условий помимо %В. Изменение зависимости
удерживания от %В для небольших молекул, пептидов и коротких белков показана
на рис. 13.11. Обратите внимание на то, что углы наклона этих графиков (значения 5)
увеличиваются с молекулярной массой растворенного вещества М. Связь между S и
молекулярной массой М может быть аппроксимирована уравнением:
S = 0,25 М0'5
(13.1а)
При градиентном элюировании удерживание связано с силой растворителя В
(раздел 9.2) соотношением, сходным с уравнением (13.1):
log к* = log kw - Лр*
(13.2)
где к* — среднее значение к во время градиентного элюирования, а <р* — среднее
значение ф (значение, когда анализируемое вещество дошло до половины колонки).
Значение к* зависит от градиентных условий
tr.F
к* =■
Vm&<f>S
(13.3)
где tG — время градиента, F — скорость потока, Дер — изменение ср в течение
градиента (например, Дф = 0,55 для градиента 5—60 %В), а Ут — мертвый объем
колонки (= IqF)- Уравнение (13.3) предсказывает, что приемлемые значения к* (1 <
< к* < 10) для молекул с большими значениями S (например, белков) могут быть
получены путем использования длительных градиентов с узким (если возможно)
log A
-0.5
М= 13000
бензол (М = 78)
Вещество
Бензол
Нонапептид
АКТГ (1-26)
Инсулин
Цитохром с
М
78
1400
4400
9000
13000
S
3
9
24
31
64
30 40
% ACN/буфар
Рис. 13.11. Зависимость изменений изократического удерживания к от изменении
во взаимосвязи с молекулярной массой вещества |25]
13.4. Разделение пептидов и белков
диапазоном градиента (небольшое значение Д<р, т.е. небольшая разница в
начальных и конечных значениях %В градиента). Обратите внимание на то, что
изменение условий градиента, влияющее на величину к*, может изменить селективность
как и изменение %В и к при изократическом элюировании. Незначительные
изменения такого рода в результате изменения времени градиента можно увидеть
на рис. 13.10. Аналогичным образом изменение скорости потока при сохранении
того же времени градиента может вызвать изменения к* и относительного
удерживания. Это бросается в глаза на рис. 13.12, на котором дана хроматограмма
разделения образца, состоящего из пептидов, полученных трипсинолизом. Часть каждой
хроматограммы на рис. 13.12а (отмеченная скобкой и стрелкой) приведена в
увеличенном масштабе на рис. ]ЪЛ26.
0.5 МЛ/МИН
0.5 МЛ/МИН
1.5 МЛ/мин
Рис. 13.12. Влияние скорости потока на селективность в градиентной ОФХ. Образец:
пептиды из лизированного трипсином миоглобина. Условия разделения:
колонка Со 80 х 6,2 мм (частицы 5 мкм); 10—70% ACN-буфер, градиент
за 60 мин; (а) полные хроматограммы; {б) области хроматограмм (а)
(показанные в (а) скобками и стрелкой) в увеличенном масштабе 126]
628 Глава 13. Разделение биополимеров и синтетических полимеров
0.01
40 60
% ACN
100
Рис. 13.13. Удерживание смешанного типа в ОФХ-разделении различных пептидов.
Условия разделения: колонка C|g, ацетонитрил-20 мМ ацетат
аммония [27]
Графики зависимости log k от %В, показанные на рис. 13.11, адекватно
представляют данные по удерживанию полипептидов для концентраций растворителя В
менее 50—80%. При содержании В менее 50% иногда может происходить изменение
порядка удерживания (например, рис. 13.13). Такое поведение может наблюдаться
как для пептидов, так и для белков, и было интерпретировано как переход от ОФХ
при низких % В к хроматографии гидрофильных взаимодействий (ХГВ; разделы 8.6,
13.4.4) при высоких %В. То есть, по достижении достаточно высокого %В
удерживание начинает расти при его дальнейшем увеличении. Это явление может иметь два
практических вывода для хроматографиста. Во-первых, расширение диапазона
градиента выше 60—70 %В иногда может не привести к полному элюированию
полипептидов из ОФХ-колонок. Во-вторых, попытки удалить загрязняющие белки,
сорбированные ОФХ-колонкой, при использовании подвижной фазы с высоким
содержанием В могут не сработать. Это, однако, не значит, что градиент до 100% В часто
вообще будет неудачен. Специфическое поведение, показанное на рис. 13.13, более
вероятно для старых колонок типа А (раздел 5.2.2.2) и положительно заряженных
разделяемых веществ. Собственный опыт работы авторов с современными ОФХ-ко-
лонками типа В говорит о том, что увеличение удерживания при более высоком %В
в общем случае маловероятно.
13.4.1.5. Влияние конформации полипептида
Время удерживания небольшого пептида можно оценить по его
аминокислотному составу (раздел 2.7.7 и уравнение 2.33). Когда длина полипептида превышает
50 остатков, такие прогнозы становятся все менее надежными [23, 28, 29|, что
наводит на мысль о том, что конформация полипептида (которая становится более
важной для более крупных молекул) может играть важную роль в удерживании и
разделении в ОФХ. Для пептидов, содержащих пролин, медленное взаимопревращение
цис- и транс- конформации этой аминокислоты может привести к уширению пика
1301 или даже к полному разрешению двух конформеров [3I|. Сочетание низкого рН,
13.4. Разделение пептидов и белков
наличия органических растворителей в подвижной фазе, и гидрофобных ОФХ-коло-
нок создает денатурирующую среду для поли пептидов, которая в еще большей
степени способствует денатурации при более высоких температурах.
Конформационные изменения при прохождении белка через колонку могут
оказать неблагоприятное влияние на его разделение. Ввод белка в гидрофобную ОФХ-
колонку приведет к полной потере четвертичной структуры, и частичной или полной
потере третичной структуры. Денатурация белка делает доступными гидрофобные
остатки, как правило, изолированные внутри нативного белка, что приводит к
увеличению удерживания последнего. Если частичная денатурация происходит до
начала миграции по колонке и если дальнейшая денатурация будет относительно
медленной, то разделение нативного и полностью денатурированного белка может привести
к появлению двух отдельных пиков. Если относительно медленная денатурация
происходит во время миграции, две формы (нативная и денатурированная) могут
перекрываться, что приводит к появлению широкого, деформированного пика.
Медленное взаимопревращение конформеров тоже может привести к увеличению ширины
пика без разделения его на два. Медленной денатурации сопутствуют низкий выход
(потери при хроматографии) и появление «пиков-призраков» (элюирование того же
пика в последующих разделениях).
Зависимость частичной денатурации от температуры во время ОФХ-разделения
1321 впервые была продемонстрирована для небольшого белка рибонуклеазы
(рис. 13.14). При низкой температуре, хроматограмма показывает острый, поздно-
элюирующийся пик с широким элюирующимся перед ним плечом. Спектральные
измерения показывают, что острый пик соответствует денатурированной молекуле
(белка). В виде широкого плеча, предшествующего пику, элюируется нативный
белок, образующийся при рефолдинге рибонуклеазы во время элюирования.
Хроматография при повышенных температурах способствует полной денатурации сразу после
ввода с превращением белка в один, более гидрофобный (и более удерживаемый
на колонке) пик.
37°С
20°С
15 (мин)
Рис. 13.14. Влияние температуры на форму пика рибонуклеазы. Условия: колонка
LiChrosplier СА 100 х 4,6-мм, 10 мкм; алюент А, 10 ммоль Н3РО4, рН 2,2;
алюент В, 55/45 Н^О/Ьпропанол + 10 ммоль Н3РО4; 5—85 %В за 30 мин;
скорость потока 1 мл/мин [32]
630 Глава 13. Разделение биополимеров и синтетических полимеров
Проблемы разделения, возникающие в ОФХ из-за конформационных эффектов,
могут быть сведены к минимуму при использовании условий, благоприятствующих
денатурации белков и пептидов — например, разделение при повышенных
температурах на более гидрофобной колонке в подвижной фазе с низким рН.
Предварительная обработка образцов в денатурирующих условиях с последующим разделением
при 60 °С были использованы для получения пиков приемлемой формы и для
повышения выхода разнообразных белков, различающихся по размеру и имеющих разные
изоэлектрические точки 133, 34|. Кроме того, на рисунке 6.21 приведен пример
из [3|, где выход и форму пика рекомбинантного белка изучали в интервале темпера
тур от 50 до 90 °С. При повышении температуры форма пика продолжала
улучшаться, но выход был максимальным при 70—80 °С. В методиках, где денатурация белка
нежелательна, рекомендуется выбрать более мягкий хроматографический режим,
такой как ИОХ или ХГВ.
13.4.1.6. Капиллярные колонки и наноисточники ионизации
ОФХ-разделение пептидов, полученных протеолизом белка, в сочетании с МС-де-
тектором, в котором источник ионизации представляет собой наноэлектроспрей,
является предпочтительным методом идентификации белков и описания структурных
характеристик в протеомных исследованиях. В большинстве случаев количество
образца ограничено, и требуется очень высокая чувствительность метода {для
достижения желаемых результатов). Для этих исследований используются капиллярные
ОФХ-колонки как для уменьшения объема пиков, так и для повышения
эффективности работы наноэлектроспрея. Капиллярные колонки с внутренним диаметром
от 50 до I50 мкм по сравнению с обычной колонкой диаметром 4,6 мм теоретически
могут улучшить чувствительность в 2000—10000 раз. Эти капиллярные колонки
работают при скоростях потока от 100 до 500 нл/мин, что значительно улучшает
эффективность десольватации и ионизации в наноэлектроспрее. Капиллярные колонки
должны использоваться вместе с системой доставки элюентов, у которой очень мал
мертвый объем. Такая система должна обеспечивать точность и стабильность
при этих низких скоростях потока, чтобы достичь воспроизводимых и приемлемых
во всех отношениях градиентов. Коммерчески доступны специально
сконструированные насосы для нано-ВЭЖХ, отвечающие этим требованиям пригодности
(раздел 3.5.4.1). В качестве альтернативы, можно настроить обычную систему ВЭЖХ
с обычным насосом, установив сплиттер для снижения скорости потока (мертвый
объем системы после сплиттера должен быть сведен к минимуму с помощью
капилляров и соединений)1.
Лизированные протеазами белки часто содержат соли, которые могут загрязнять
источник ионизации. Последние можно удалить, установив колонку, улавливающую
пептиды, и кран между инжектором и аналитической колонкой. После ввода образца
выход из колонки-ловушки переключается на контейнер для отходов, куда
смываются соли и другие низкомолекулярные загрязнители, а затем переключается на
аналитическую колонку, куда элюируются пептиды для последующего анализа на масс-
спектрометре. Альтернативным подходом является схема, в которой колонку-ловуш-
1 В настоящее время считается, что наносистемы не оправдали возложенных на них надежд
по анализу лекарственных субстанций на основе белков и пептидов и анализу протеом. Такие
системы, как правило, имеют низкую воспроизводимость и большие времена разделений. Учитывая
рост чувствительности обычных (не нано) ЖХ-МС систем, включающих в себя УВЭЖХ и
обеспечивающих высокую скорость, воспроизводимость разделений, а также гораздо более простых в
обслуживании, имеет смысл предпочесть их, а не наносистемы для работы в аналитической
лаборатории. Наносистемы все еще используются в исследовательских лабораториях академических
институтов и университетов (поскольку обладают высокой чувствительностью и селективностью),
где время разделения не является лимитирующей стадией. — Прим. перев.
13.4. Разделение пептидов и белков 631
ку соединяют непосредственно с аналитической колонкой через разветвитель |35|,
один из капилляров которого обеспечивает соединение аналитической колонки с на-
ноэлектроспреем, а на выходе из другого стоит кран, который может направлять
поток в слив (во время ввода образца или удаления соли) или перекрывается (во время
элюирования пептидов)1.
13.4.1.7. Разработка метода ОФХ
Рекомендуемые исходные условия для разработки метода разделения в ОФХ
для пептидов или белков перечислены в таблице [3.3. Если разделение с
использованием этих рекомендуемых начальных условий не решает поставленную задачу,
его можно улучшить с помощью стратегии, изложенной ниже. Вопросы формы
пика, переноса образца из предыдущего разделения и воспроизводимости следует
решить прежде, чем начинать оптимизацию разделения. Особенно это касается
разделения белков. Неприемлемая форма пика белка часто указывает на
присутствие конформеров или нескольких видов белка2 в исходных условиях. Для
улучшения формы пика могут потребоваться условия, преобразующие белок в одну
единственную конформацию — например, денатурирующие, которые достигаются
повышением температуры или использованием добавок к подвижной фазе, таких
как поверхностно-активные вещества. Кроме того, в таких условиях обычно не
происходит переноса образца из предыдущего разделения. Для подтверждения
воспроизводимости от разделения к разделению, линейности сигнала в зависимости
от концентрации и устранения переноса образца из предыдущего разделения
удобно использовать короткие градиенты.
После достижения приемлемой формы пика и воспроизводимости имеет смысл
заняться оптимизацией селективности. Это можно сделать, например, изменяя
температуру и время градиента (как это описано для малых молекул в разделе 9.3).
Кроме того, может оказаться, что быстрее и эффективнее выбирать оптимальные
условия разделения с помощью компьютерного моделирования (глава I0). В примере
на рис. 13.10 начальные четыре разделения а—г можно использовать для
прогнозирования наилучшего сочетания температуры и времени градиента для получения
оптимального разрешения (рис. 13.10d). После того, как достигается приемлемое
разделение пиков, диапазон градиента можно уменьшить, чтобы сократить общее время
разделения. Например, градиент может быть начат непосредственно перед первым
пиком и закончен сразу после выхода последнего пика (рис. 13.10е).
Если ни одна комбинация времени градиента и температуры не дает
приемлемого разрешения, следующим шагом может быть замена колонки или состава элюентов
А или В: например, увеличение концентрации ТФУК, изменение рН или замена
изопропанола на ацетонитрил в качестве В-элюента. После одного или нескольких
упомянутых изменений условий следует повторно изменить таким же образом
условия градиента и температуру для четырех разделений (аналогичных показанным
на рис. 13.10а—г), при этом надо изменить остальные условия (не касающиеся
параметров градиента и температуры).
Наконец, кусочно-линейные градиенты могут быть использованы для решения
конкретных проблем разделения. В случае сильно адсорбирующихся
загрязнений, которые должны быть удалены из колонки до следующего ввода образца,
для ее очистки на последнем этапе разделения может использоваться крутой
1 Проще говоря, имеются две колонки, между которыми встроен кран, переключающий поток
из первой колонки либо в слив, либо во вторую колонку, откуда поток идет в МС-детектор. Такая
схема может иметь смысл, когда имеются выходящие с первой колонки перед солями гидрофильные
пептиды и белки, которые необходимо проанализировать с помощью МС-детектора. — Прим. перев.
^Например, белка с различными пострансляционными модификациями. — Прим. перев.
632 Глава 13. Разделение биополимеров и синтетических полимеров
Таблица 13.3. Начальные условия для разработки методов ОФХ
Условия
Молекулярная масса образца, М
Обработка образца перед
анализом
Колонка"
Элюент А1
Элюент В1
Диапазон градиента
Температура
Скорость потока мл/мин
Время градиента, мин
к*
%В/мин
Принятое значение S
Величины для разных образцов
Пептиды
1 < М < 5 кДа
Нет
150 х 4,6 мм, Ci8
тип В (диаметр пор
8—12 нм), диаметр
частиц 3 мкм
0,1% ТФУК-вода
0,10% ТФУК—аце-
тонитрил
0-60 %В
30-35 °С
2,0
25
2
2,4
25
Белки
5 < М < 20 кДа
Добавить
мочевины до 8 М,
выдержать 30 мин
150 х4,6 мм, Cig
тип В (диаметр пор
12—30 нм),
диаметр частиц 3 мкм
0,1% ТФУК-вода
0,10%ТФУК-аце-
тонитрил
5—100 %В
30-35 "С*
1,0
50
1
1,2
40
М > 20 кДа
Добавить
мочевины до 8 М,
выдержать 30 мин
50 х 4,6 мм, С4
тип В (диаметр
пор > 30 нм),
диаметр частиц 3 мкм
0,1% ТФУК-вода
0,10% ТФУК-аце-
тонитрил
5—100 %В
30-35 'С6
0,5
50
1
1,2
70
"Колонки должны быть стабильны при низких значениях рН и температурах <60°С. Могут
быть использованы колонки другой длины или диаметра, или упакованные частицами другого
размера, при этом время градиента и скорость потока должны быть подобраны так, чтобы к*
оставался прежним и давление на колонке было приемлемым. Выбор длины лиганда (С8, С]8)
менее важен.
"Для некоторых белков, особенно для тех, у которых Л/ > 20 кДа, желательно проводить
разделения при более высоких температурах (например, 60—80 °С). В этих случаях необходимо
колонку перед работой в таких условиях проверить на стабильность при температуре > 50 "С
ирН< 2,5.
1 В последние годы рекомендуется использовать 1 % раствор ТФУК или иного модификатора
в ВЭЖХ-системах с градиентом на стороне низкого давления, оснащенных насосами, способными
создавать трехкомпонентные и четырехкомпонентные градиенты. Обычно этот раствор
помещается на линию В, линия А — вода, Б — ацетонитрил или иной органический растворитель. Такая
система приготовления подвижной фазы гораздо более удобна, чем традиционная, где ТФУК,
добавленная в воду, и органический растворитель испаряются быстрее, чем подвижная фаза в целом
и, как следствие, меняется изначальное содержание модификатора. В получающейся подвижной
фазе содержание ТФУК будет в десять раз меньше. По данным компании Waters подвижные фазы,
приготовленные традиционным способом, стабильны втечение 2—3 дней, а приготовленные с
помощью смешивания из трех или четырех компонентов могут быть стабильны две недели. Через две
недел и достаточ но заменить 1% раствор модификатора. Кроме того, поскольку чистые
растворители смешиваются в нужных соотношениях непосредственно в хроматографической системе в
процессе разделения, нет нужды выливать ранее приготовленные растворы уже ненужных
концентраций, что позволяет их (растворители) экономить. — Прим. перев.
13.4. Разделение пептидов и белков 633
градиент до 100 %В'. В случае сложных образцов с группами плохо разделяющихся
компонентов, в профиль градиента можно вставить пологий участок для улучшения
разделения. Эта стратегия годится далеко не для всех малых молекул. Вероятно, она
будет более успешной в случае пептидов, и особенно белков |36р.
13.4.2. Ионообменная хроматография (ИОХ) и смежные
методы
Ионообменная хроматография (ИОХ) может применяться для аналитического
разделения пептидов и белков, но чаше ее используют для выделения и очистки белков в
масштабах от лабораторных до промышленных |37|. Самыми важными преимуществами
ИОХ для выделения белков являются: (I) высокая вероятность сохранения
естественной конформации и биологической активности белков при разделении методом ИОХ,
(2) относительно высокая емкость сорбентов в ИОХ и (3) высокий процент выхода.
Реализации преимуществ (I) и (2) благоприятствует использование подвижных фаз
с умеренной ионной силой и значением рН, близком к физиологическому. Наиболее
важная особенность ИОХ для аналитических методик — это ее уникальная
селективность по сравнению с другими видами колоночной хроматографии. Три других хрома-
тографических метода (хроматофокусирование, хроматография на гидроксиапатите
и аффинная хроматография на сорбентах с иммобилизованными металлами;
разделы 13.4.2.3—13.4.2.5) родственны ИОХ в том смысле, что они также основаны на
ионном взаимодействии между сорбентом и образцом.
Ионный обмен основан на обратимом электростатическом взаимодействии
заряженных групп сорбента с противоположно заряженными группами полипептида
(раздел 7.4.1). Удерживание молекулы пептида или белка Р происходит в результате
вытеснения противоионов мобильной фазы X + ионами Р+г (или противоионов ЛГ
ионами Р~г).
(катионный обмен) Р+Цт) + z(R~)X+(s) «> (R-^P+Hs) + zX+im), (13.4)
(анионный обменам) + z(ir)X-(s) <т> (R+)ZP-Z(s) + zX~(m). (13.5)
Здесь Л~ или R+ относится к заряженной группе (лиганду) неподвижной фазы,
Z представляет собой заряд молекулы белка P+z или Р~г, а индексы (т) или (s)
указывают на расположение молекулы в подвижной или неподвижной фазах,
соответственно. В уравнениях (13.4) и (13.5) предполагается, что противоионы X* или Х~
одновалентны. В катионообменной хроматографии отрицательно заряженный лиганд
сорбента (Л~) связывается с катионными участками полипептида. В анионообменной
хроматографии, положительно заряженный лиганд (Л+) связывается с анионными
группами полипептида. Удерживание образца может быть изменено с помощью
изменения заряда разделяемого вещества или, в некоторых случаях, лиганда сорбента
' При этом необходимо помнить, что минимальным объемом для промывки колонки 100 %В
является ее полный объем. После этого колонку необходимо уравновесить как минимум,
тремя—пятью ее полными объемами элюента с исходным %В, в котором разделение начинается. —
Прим. персе.
^Хроматография биополимеров усложняется их возможной гетерогенностью и зачастую
высокой гидрофильностью. Можно ожидать появления множественных пиков на хроматограмме. В
таком случае можно использовать ступенчатый градиент или просто выбрать наиболее удобный пик
и использовать его для идентификации и определения количества биополимера. Необходимо,
однако, проверить, не изменится ли относительная плошадь этого пика в зависимости от партии
или производителя биополимера. В случае высокогидрофильных молекул, анализируемые
вещества могут помочь удержать колонки из пористого графита, если стандартные С8 или С]8ОФХ-ко-
лонки не справятся с задачей. — Прим. перев.
634 Глава 13. Разделение биополимеров и синтетических полимеров
(раздел 7.5.4) посредством изменения рН подвижной фазы. Более распространенной
стратегией элюирования является изменение концентрации Х+ или Х~ в подвижной
фазе, как описано в разделе 7.4.1, или использование градиентного элюирования, где
концентрация X* или Х~ увеличивается во время градиента (градиент соли). По
причинам, обсуждаемым ниже, кажущийся заряд белка +z в уравнениях (13.4) и (13.5)
может отличаться от его полного заряда.
На удерживание в процессе ИОХ сильно влияют заряженные концевые амино и
карбоксильные группы белка (а также заряженные группы боковых цепей аминокис
лот). Значения рКа этих групп лежат в интервале рН между 2 и 13 (таблица 13.4 и
рис. 13.1), поэтому удерживание будет сильно зависеть от рН подвижной фазы.
Обратите внимание на то, что локальное окружение заряженного аминокислотного
остатка белка (то есть, окружающая подвижная фаза и группы соседних аминокислот
внутри молекулы) может вызвать отклонение наблюдаемого кажущегося значения
рКа от его номинального значения для свободной аминокислоты.
Посттрансляционные модификации, сопряженные с введением дополнительных заряженных групп,
таких как сиаловая кислота, фосфатные и сульфатные группы, также могут
способствовать удерживанию, обусловленному ионными взаимодействиями.
Таблица 13.4. Значения рКа заряженных аминокислот
Остаток
Концевая аминогруппа
Аргинил
Гистидил
Лизил
Концевая карбоксильная группа
Аспаргил
Плута мил
рК, аминокислоты
8,8 - 10,8
12,5
6,0
10,8
1,8-2,6
3,9
4,3
рК, в белке
6,8 - 7,79
> 12
6,4-7,4
5,9-10,4
3,5-4,3
4,0-7,3
4,0-7,3
Источник: [37] с разрешения Validated Biosystems.
Полный заряд белка +z будет зависеть от рН подвижной фазы. Предполагается,
что при значении рН, для которого полный положительный и отрицательный заряды
равны (изоэлектрическая точка или pi), удерживание в ИОХ-разделении отсутствует.
При значениях рН ниже его pi белок будет иметь положительный заряд и должен
связываться с катионообменником. При значениях рН выше его pi, белок будет
обладать отрицательным зарядом и должен связываться с аниониообменником
(рис. 13.15я). Этой простой моделью можно руководствоваться при выборе колонки
и рН подвижной фазы, но на практике белок может связываться с сорбентом
аномальным образом вблизи своей изоэлектрической точки (рис. 13.156). Причина в том,
что заряд может быть неравномерно распределен по поверхности молекулы белка,
а сгруппирован в различных областях (областях контакта) (раздел 13.3.2). В
результате области с избыточным зарядом могут возникать в разных частях молекулы, и эти
области могут взаимодействовать с неподвижной фазой более или менее независимо
друг от друга. Аномальное поведение при связывании может проявляться в
связывании с сорбентом в изоэлектрической точке, связывании с анионобменником при рН
ниже р] белка или связывание с катионобменником при рН выше pi. Точно так же
белок может не связываться с анионообменником при рН выше своего pi или с
катионообменником при рН ниже своего pi. Например, Р-глюкозидаза (р! = 7,3)
13.4. Разделение пептидов и белков 635
(«)
Заряд белка
z, время
удерживания
р|
\ \
\ \
\ \
\ V
\\
КатионнообменникV
// '«
i /
i /
i /
1/
if
11 Анионнообменник
рН
(б)
<в>
20
(мин)
р-глюкоэидаэа
PI
химотрипсиноген
о катионнообменник
•анионнообменник
10 2
РН
Рис. 13.15. Влияние рН на удерживание белков ионобменниками. Идеальное поведение
(а); наблюдаемое поведение р-глюкозидазы (б) и химотрипсиногена (в) |38]
связывается при рН 7,3 с аниообменником, но не связывается с катиообменни-
ком до тех пор, пока рН подвижной фазы не станет на две единицы ниже ее pi
(рис. 13.156). Химотрипсиноген, имеющий pi 9, связывается при рН 9 как с
аниообменником, так и с катиообменником (рис. 13.15в).
Как правило, анионообменные разделения проводят при рН, превышающем pi
белка на I —1,5 единицы, а катионообменные разделения — при рН на 1 —1,5 единиц
ниже pi. Допустимые ионные условия разделения могут ограничиваться
растворимостью и стабильностью целевого белка (белков). Практически все схемы очистки
белков, используемые в биофармацевтической промышленности, содержат одну или
несколько стадий ИОХ.
Поскольку только ограниченное количество заряженных остатков на
поверхности белка может взаимодействовать с неподвижной фазой, небольшие различия в
характере и положениях этих заряженных остатков могут сильно влиять на
селективность в ионообменной хроматографии |14|. Кроме того, замены аминокислот внутри
молекулы белка могут изменить его конформацию и косвенно повлиять на
ионообменную селективность, изменив положения заряженных групп на поверхности
белка.
636 Глава 13. Разделение биополимеров и синтетических полимеров
13.4.2.1. Выбор колонки
Критерии выбора колонки включают в себя:
• размер частиц и диаметр пор;
• химическую структуру сорбента;
• тип лиганда;
• плотность лигандов.
Соображения, касающиеся размера частиц и диаметра, не отличаются от
приведенных для ОФХ сорбентов в разделах I3.3.1.I и I3.3.1.2.
Химическая структура сорбента. Первой неподвижной фазой для высокопроиз
водительной ИОХ был сорбент на основе диоксида кремния. Силикагель взяли
для этой цели, руководствуясь теми же соображениями, по которым он был выбран
для других режимов ВЭЖХ. Однако прежние сорбенты на основе диоксида кремния
были нестабильны в тех условиях, в которых предпочтительно проводить разделения
в ИОХ (физиологический рН, умеренная концентрация соли), и постепенно их заме
нили полимерными носителями на основе полистирола, поперечно сшитого диви
нилбензолом, или полиметакрилата. Хотя современные неподвижные фазы на
основе силикагеля и демонстрируют улучшенную стабильность при нейтральных и
щелочных рН, многие лаборатории продолжают использовать сорбенты на
полимерной основе. Для промышленной хроматографии предпочтительны сорбенты из
полужестких гелей, таких как сшитые декстран, агароза или полиакриламид, с крупными
частицами из-за их дешевизны и потому еще, что они могут выдерживать стадии
очистки с использованием высоких концентраций щелочи для удаления
эндотоксинов и других биологических загрязнений.
Тип и плотность прививки лигандов. В пределах соответствующих категорий кати-
онообменников и анионобменников, носители для ИОХ могут быть далее разделены
на «сильные» или «слабые» — в зависимости от рКа ионогенных лигандов
неподвижной фазы. Следовательно, заряд на сорбенте и его связывающая способность могут
изменяться в зависимости от рН подвижной фазы (рис. 13.16). Сильные ионообмен-
ники имеют значения рКа за пределами нормального рабочего диапазона рН
колонки и поэтому полностью ионизированы независимо от рН подвижной фазы. В
таблице 13.5 приведены некоторые примеры распространенных лигандов сорбентов
для ИОХ. Ионогенные группы сильных ионообменников включают —SOj для катио-
нобменников и —Ы(СНз)з для анионобменников. Слабые ионообменники имеют
значения рКа в рабочем диапазоне сорбента, поэтому их ионообменная емкость
зависит от рН подвижной фазы. Примеры ионных групп включают —N(C2H5)2H+
для слабого анионообменника и —СОО~ для слабого катионообменника. Можно
также упомянуть слабый анионообменник с высокой емкостью и стабильностью
в щелочных условиях, образованный плотным полимерным покрытием полиэтиле-
нимином подложки из диоксида кремния. Сильные ионообменники часто
предпочтительнее, так как их емкость не зависит от рН подвижной фазы, и их поведение
более предсказуемо. Связывающая способность ионообменников зависит от площади
поверхности носителя и плотности его заряда (мкмоль/м2). Стандартные емкости
(т.е. пригодные для максимального связывания образца сорбентом) для
ионообменных сорбентов на основе крупнопористых силикагеля или полимера варьируются
от 30 до I20 мг белка на миллилитр неподвижной фазы.
Линкерная группа, с помощью которой ионогенная группа присоединяется к
носителю, может внести свой вклад в хроматографические свойства сорбента.
Например, гидрофобные группы в линкере могут участвовать в гидрофобных (обращенно-
фазовых) взаимодействиях с разделяемым веществом. Такими взаимодействиями
можно объяснить различия в селективности колонок от разных поставщиков, ис-
13.4. Разделение пептидов и белков 637
(а)
Сильный АО
Сильный КО
Л
Рис. 13.16. Емкости ионообменных сорбентов, (а) Сильные ионообменники; (б)
слабые ионообменники |39|. КО — катионообменннк; АО — анионообменник
Таблица 13.5. Сильные и слабые ноногенные лиганаы
Анионообменники (АО)
Слабые
ДЕАЕ (диэтиламиноэтил)
-0-CH2-CH2-N+H(CH2CH3)2
ПЭИ (полиэтиленимин)
(-NHCH2CH2)„-N<CH2CH2-)„.
CH2CH2NH2
Сильные
Q {четвертичный амин) —СНОН-СН2-М+(СНз)3
Катионобменники (КО)
Слабые
КМ (карбоксиметил) — О—СН2—СОО-
С ильные
S (сульфонат) —СН2-СН2—СН2—S03
пользующих одни и те же функциональные ионогенные группы. Сорбенты Tentacle
для ИОХ имеют гибкий гидрофильный линкер (щупальце — tentacle), который
соединяет заряженную группу с носителем [40|. Эти сорбенты улучшают доступ белка
к заряженной группе носителя, таким образом увеличивая способность последнего
Глава 13. Разделение биополимеров и синтетических полимеров
к связыванию. К тому же сорбенты этого типа менее подвержены неспецифическим
взаимодействиям, у них лучше кинетика связывания и на них белки денатурируют
в меньшей степени.
13.4.2.2. Выбор подвижной фазы
Как отмечено выше, обычно удерживание контролируют, изменяя концентрацию
вытесняющей соли (противоиона), а не рН подвижной фазы (т.е. сила растворителя
определяется концентрацией противоиона). Условия, влияющие на селективность,
включают:
• сорбент (раздел 13.4.2.1);
• буфер подвижной фазы;
• тип соли противоиона (как на рис. 13.17);
• крутизну градиента;
ф органический элюент В {если используется);
• другие добавки в подвижную фазу (особенно поверхностно-активные вещества);
• температуру.
См. также обсуждение в разделе 7.5.
Буфер подвижной фазы. Достижение желаемого удерживания и селективности
требует тщательного подбора и контроля рН подвижной фазы. Для катионообменной
колонки хорошая отправная точка — рН подвижной фазы = 6, тогда как рН
подвижной фазы 8 подходит для анионообменника. Чтобы иметь достаточную буферную
емкость, буфер должен иметь значение рКа в пределах примерно 1,0 единицы от
желаемого рН (раздел 7.2.1.1) и концентрацию от 0,02 до 0,1 М. Общеупотребительные
буферы, используемые для ИОХ, перечислены в таблице 7.1. Обратите внимание
на то, что некоторые из этих буферов сильно поглощают на коротких длинах волн
УФ-диапазона, особенно при более высоких концентрациях.
Противоион. Наиболее распространенной стратегией элюирования в ИОХ
является использование градиента концентрации противоиона. Относительная активность
(мин)
Рис. 13.17. Влияние типа соли на анионообменное разделение пяти белков. Условия:
колонка Shim-packWAX-2 (Shimadzu) 50 х 4 мм; градиент О—0,5М
указанной соли за 20 мин; фосфатный буфер рН 8; 1 мл/мин [41]
13.4. Разделение пептидов и белков 639
разных противоионов соответствует их ранжированию в ряду Хофмайстера [37, 42|;
см. таблицу 13.6 или аналогичные последовательности в разделе 7.5.2. Тем не менее,
как для анионообменников, так и для катионообменников чаще всего используются
градиенты с увеличением концентрации NaCI. Помните о том, что хлорид-ион
вызывает коррозию нержавеющей стали при низком рН (< 5) и должен быть удален
из ВЭЖХ-системы после использования. В то же время были разработаны
специальные системы ВЭЖХ, позволяющие использовать хлориды в кислотных условиях.
Таблица 13.6. Ряды Хофмайстера для лнптропных и хаотропных ионов [36]
Увеличивающийся лиотропный эффект (эффект высаливания)
SCN- (наименьший) < СЮ4 < NQ3 < Вг- < С1~ < С00~ < SO2 < POj (наибольший)
Увеличивающийся хаотропный эффект
Ва2+ (наибольший) > Са2+ > Mg2+ > Li+ > Cs+ > Na+ > K+ > Rb+ > NHJ (наименьший)
Источник: [36|
Органические растворители и ПАВ. Органические растворители (например,
1 —10% метанола, пропанола или ацетонитрила) могут быть добавлены к подвижной
фазе для подавления гидрофобных взаимодействий с группами носителя или
линкера и для сокращения уширения пиков или их задних фронтов (в некоторых случаях
может потребоваться добавление 50% органического растворителя, как в примере
на рис. 11.15 из |43|). Для тех же целей могут быть использованы и неионные ПАВ.
Любая из этих добавок к подвижной фазе может, кроме того, способствовать
растворению высокогидрофобных разделяемых веществ, таких как мембранные белки.
Ионные ПАВ в ионообменной хроматографии использовать нельзя.
13.4.2.3. Хроматофокусирование
Хроматофокусирование — это специализированная форма ИОХ, в которой белки
элюируются с колонки градиентом рН [44—49|. Хроматофокусирование уникально
тем, что градиент рН образуется внутри колонки с помощью одной подвижной фазы,
которая представляет собой сложную смесь различных буферов. Хотя
хроматофокусирование может проводиться с катионо- или анионообменниками, коммерчески
доступные продукты ограничены анионобмениками |48|'. В начале разделения белки
связываются с анионообменником, предварительно уравновешенным буфером с высоким
рН для максимального удерживания образца. Затем в качестве подвижной фазы
используют буферную смесь с низким рН, которая, перемещаясь по колонке, постепенно
титрует заряд на ней так, что рН уменьшается вдоль колонки, от входа к выходу. Белки
мигрируют по колонке в ответ на изменение рН и элюируются в изоэлектрических
точках или вблизи них — при таких значениях рН они больше не могут оставаться
связанными с ионообменником. Элюирование происходит в порядке уменьшающихся
значений pi белков. Хроматофокусирование характеризуется очень высокой емкостью,
поэтому его рационально использовать для препаративного разделения. Благодаря
эффекту фокусирования метод может давать очень высокое разрешение, при котором
получаются узкие пики, выходящие в интервале от 0,04 до 0,05 единиц рН. Как и в
случае обычной ИОХ (рис. 13.15б,в) белок может элюироваться из хроматофокусирующей
колонки при рН, существенно отличающемся его pi.
Успешные и воспроизводимые разделения с помощью хроматофокусирования
зависят от использования элюентов, содержащих несколько видов буферов, значения
1 В настоящее время производство этих колонок и буферов прекращено. — Прим. перев.
640 Глава 13. Разделение биополимеров и синтетических полимеров
рКа которых охватывают диапазон градиента рН, и которые могут обеспечить
эффективную буферную емкость в этом диапазоне. Коммерчески доступные буферы
для хроматофокусирования состоят из смеси амфолитов {веществ, которые могут
действовать как кислота или основание). Можно также использовать комбинацию
биологических буферов, таких как буферные растворы Гуда |50|. Ионная сила
буфера должна поддерживаться низкой, чтобы минимизировать элюирование под
действием соли (вытеснение белков противоионами). Улучшение разрешения (или
повышение скорости разделения) белков, значения pi которых лежат в узком диапазоне
значений рН, может быть достигнуто путем сужения диапазона рН амфолита или
буферной смеси (аналогично уменьшению Дф в градиентном элюировании). Для
хроматофокусирования лучше брать сильные ионообменники, так как они полностью
ионизированы независимо от рН.
Одним из недостатков хроматофокусирования является пониженная раствори
мость белков в их изоэлектрической точке. Это ограничение усугубляется низкой
ионной силой элюирующего буфера. Растворимость белка можно повысить,
увеличив концентрацию соли, но при этом возрастет (ионная) сила подвижной фазы,
что поставит под угрозу качество разделения. Предпочтительной стратегией борьбы
с выпадением белка в осадок является добавление цвиттерионов в элюируюший
буфер. Добавки, такие как таурин, глицин и бетаин, способствуют растворимости
белка и могут использоваться в концентрациях до 2 М, не влияя на ионную силу
буфера. Добавление мочевины в концентрациях I—2 М также помогает солюбилизиро-
вать белки. Кроме того, могут быть использованы неионные и цвиттерионные ПАВ.
Не забывайте, впрочем, о том, что мочевина постепенно разлагается с образованием
карбаматов, которые могут (и будут!) ковалентно модифицировать белок.
Хроматофокусирование способно разделять изоформы белков, имеющие разные
заряды, например, белки с посттрансляционными модификациями, отличающиеся
количеством сиаловых кислот или фосфатных групп. Разделние изоформ может быть
проблемой, если целью является очистка белка. Целевой белок разделяется на
несколько соединений, выходящих отдельными пиками, при этом он разбавляется, и
увеличивается риск его совместного элюирования с примесями. С другой стороны, эта
особенность хроматофокусирования может быть преимуществом, если целью
является только лишь описание изоформ.
13.4.2.4. Хроматография на гидроксиапатите
Этот метод часто используется в промышленной хроматографии для очистки белка и
удаления загрязнений 137]. Гидроксиапатит (ГА) является кристаллическим
веществом Са]0(РО4)6(ОН)2, которое служит и подложкой, и неподвижной фазой |5l|. Его
многофункциональная поверхность состоит из положительно заряженных пар ионов
кальция (С-сайты) и кластеров из шести анионных атомов кислорода, связанных
с триплетами фосфат-ионов (Р-сайты). С- и Р-сайты и гидроксилы распределены
определенным образом на поверхности кристаллов |5]—53|, как показано
на рис. 13.18. Поначалу сорбенты из ГА были нестабильны, но современные
препараты ГА спекаются при высокой температуре с образованием пористого
керамического материала на основе гидроксиапатита (КГА), который стабилен в условиях
хроматографического разделения. Колонки, заполненные 5- или 10-микронными
частицами КГА коммерчески доступны как для аналитических, так и для
препаративных разделений.
Взаимодействие белка с КГА комплексное (рис. 13.18): электростатические
взаимодействия включают притяжение протонированных аминогрупп Р-сайтами и их
отталкивание С-сайтами (рис. 13.18а). Подобным же образом ионизированные
карбоксильные группы притягиваются С-сайтами и отталкиваются Р-сайтами (рис. 13.186).
13.4. Разделение пептидов и белков 641
С-сайты
Р-сайты
-ОН
-Са+»
г— Са+»
-ОН
-ро4-
f— ро4-
-ро4=
-он
«+н2,
(,tH2N-
•••■+H2N
...,+H2N-
J
о
i
(б)
КГА
Хотя сначала карбоксилы взаимодейст- (а) КГА
вуют с С-сайтами электростатически,
фактическое связывание вызывает фор
мирование гораздо более прочных
координационных комплексов между
С-сайтами и кластерами карбоксильных групп
белка |37|. Фосфатные группы белка
связываются с С-сайтами даже сильнее,
чем белковые карбоксильные группы.
Селективность КГА для основных
белков отличается от селективности
обычных катионобменников из-за
отталкивания аминов С сайтами. Связывание
слабоосновных белков может быть
усилено путем добавления ионов фосфата
в низкой концентрации, подавляющих
отталкивание аминов С-сайтами, но
не блокирующих их взаимодействие
с Р-сайтами |54|. Основные белки могут
быть элюированы градиентами хлорида
натрия или иона фосфата, может
потребоваться конечная концентрация соли
0,5 М. Связывание основных белков
увеличивается при более низком рН, но
КГА нестабилен ниже рН 5. Кислотные
белки невозможно элюировать хлоридом
натрия — даже при концентрациях
> 0,ЗМ. Их элюирование требует
использования фосфата, цитрата или
фторида. Эта особенность КГА позволяет
разделять основные белки, начиная
с градиента NaCI, с кислотными
белками, элюируемыми после этого в
градиенте фосфата.
КГА обычно обеспечивает отличные выходы, сохранение биологической
активности и используется для очистки белков в масштабах от лабораторных до
промышленных. Уникальная селективность КГА способствует разрешению таких
близкородственных соединений, как варианты белка, обусловленные посттрансляционными
модификациями, и гликоформы. КГА используется в биофармацевтической
промышленности для очистки антител и удаления загрязнений, таких как эндотоксины,
нуклеиновые кислоты и вирусы. Стабильность КГА по отношению к
концентрированным основаниям, органическим растворителям и хаотропам позволяют
применять жесткие способы очистки после использования.
Рис. П.18. Связывание с КГА основных (а)
и кислотных (б) белков. Двойными скобками
показано отталкивание, пунктирные линии
обозначают ионные связи, треугольником
обозначены координационные связи [37]
13.4.2.5. Аффинная хроматография на сорбентах
с иммобилизованными металлами (АХИМ)
Этот режим разделения, также известный как металл-аффинная' хроматография
(МАХ), основан на особом (характерном) взаимодействии белков с ионом
металла 155—57). Ион металла иммобилизуется хелатирующими группами, которые присо-
' В русскоязычной литературе часто употребляется термин металл-хелатная
хроматография. — Прим. перев.
642 Глава 13. Разделение биополимеров и синтетических полимеров
Исходная колонка
матрица
А хелатирующий
. агент
■£\ лиганд
+ Си**
Зар
7 < рН < в
+
Р
Шаг1.
ижвнный катион
металла
J
/\ХСи"
Шаг 2.
Связанный
белок (Р)
А 4
У. , 4<рН<5 О
|/чХси~+р —^г- i
;> Десорбция 4
' белка /
Си**- Р
+ ЭДТА
i
Шаг 4
Восстановленная
колонка
Я
|^sX
+ Си"ЭДТА
Рис. 13.19. Стадии аффинной хроматографии на сорбентах с иммобилизованными
металлами (АХИМ) [56|
единены к подложке через линкер; на рис. 13.19 показаны различные стадии работы
с таким носителем. Несколько боковых цепей аминокислот белков могут
образовывать координационные комплексы с металлами, поэтому АХИМ — это общий метод
разделения белков. Основное взаимодействие в АХИМ происходит с имидазольной
группой гистидина в непротонированной форме [58|. Сила связывания металлами
различных аминокислотных групп в молекуле белка уменьшается в следующем
порядке:
his > trp > tyr > phe > arg ~ met ~ gly.
Остатки цистеина могут связываться с металлами, но они бывают недоступны
на поверхности белка в восстановленном состоянии, так как легко окисляются в
присутствии ионов металлов 1591. Поэтому белкам, содержащим цистеин, может
потребоваться восстановительная среда (добавление 2-меркаптоэтанола или дитиотреи-
тола) для поддержания остатков цистеина в их активной {—SH) форме.
Ароматические остатки могут вносить косвенный вклад в удерживание, усиливая связывание
соседних гистидинов [59|.
Преимущественное связывание гистидина с сорбентами в процессе АХИМ
использовали, вводя методом генетической инженерии полигистидинные
последовательности в белки-мишени для упрощения очистки последних. После
преимущественного связывания, элюирования и получения целевого белка, полигистидиновая
последовательность может быть отщеплена с помощью карбоксипептидазы А |59|.
13.4. Разделение пептидов и белков
Фосфопротеины и фосфопептиды селективно связываются с АХИМ-сорбентами
с хелатированными Fe+3 и Ga+3. АХИМ стала ключевым инструментом в
идентификации продуктов фосфопротеон |60].
Высокая емкость колонок IMAC и высокий выход белка по массе и активности
делают этот метод полезным для препаративной и промышленной хроматографии.
Что касается очистки белка, то АХИМ выгодно отличается от аффинной
хроматографии с точки зрения силы связывания и емкости сорбента, она более стабильна
в широком диапазоне условий, выдерживает больше циклов использования,
проводится в относительно мягких условиях элюирования и не очень дорого стоит. В
промышленной хроматографии лучше всего использовать АХИМ в качестве первого
шага в схеме очистки, а последующие шаги, такие как ИОХ или ХГВ, позволяют
удалить любые металлы, вымытые из колонки в процессе АХИМ |37| (ионы
металлов могут катализировать окисление аминокислотных остатков белка).
Селективность в АХИМ может контролироваться выбором:
• хелатирующего лиганда (то есть сорбента);
• иммобилизованного иона металла;
• рН подвижной фазы и ее ионной силы;
• любыми добавками в подвижную фазу, используемыми для усиления связывания
или элюирования белков.
Хелатируюший лиганд. Комплекс хелатирующего лиганда с ионом металла
должен быть достаточно прочным и стабильным, но при этом также должны быть
доступны координационные центры этого иона для связывания белка. Металл должен
легко удаляться хелатирующим агентом, таким как ЭДТА, чтобы можно было
регенерировать сорбент и преобразовать его в другую форму с ионом другого металла.
Наиболее распространенные хелатирующие группы, используемые в АХИМ, имино-
диуксусная кислота (ИДУК) и трис(карбоксиметил)этилендиамин (ТКМЭДА),
которые соответственно образуют тридентатные и пентадентатные комплексы с ионами
металлов (рис. 13.20). Хотя пентадентатный ТКМЭДА обладает более сильным
сродством к металлу, тридентатный комплекс ИДУК-металл оставляет больше свободных
координационных центров у иона металла для связывания растворенного вещества,
поэтому сорбенты с комплексами ИДУК-металл могут иметь более высокое сродство
к белку [551. Стабильность комплексов ИДУК с ионами металлов на сорбенте с ага-
розной подложкой уменьшается в ряду |56|:
Си2+ > Ni2+ > Zn2+ > Со2+ > Fe2+ » Са2+.
Соответствующее сродство ТКМЭДА к металлам изменяется следующим образом:
Fe3+ > Cu2+ > Ni2+ > Zn2+ ~ Co2+ > Fe2+ > Ca2t
QH2p„CH2-CO
CH2-CO
иминодиуксусная кислота (ИДУК)
ОН,
трис(карбоксиметил)этилендиамин
(ТКМЭДА)
Рис. 13.20. Структура двух часто используемых лигандов для АХИМ [55]
644 Глава 13. Разделение биополимеров и синтетических полимеров
Ион металла. Селективность связывания белка в АХИМ зависит в первую
очередь от металла, находящегося в комплексе с хелатирующим лигандом.
Взаимодействия белка с хелатированным металлом включают кулоновские и координационные
связи, а при высоких концентрациях соли могут проявляться и гидрофобные взаимо
действия 155|. Самые популярные металлы для АХИМ — Cu2+, Ni2+ и Zn2+. Эта
популярность, вероятно, отражает сильное сродство этих металлов как к АХИМ-лиган-
дам, так и к белкам. Сродство Си к имидазолу в 15 раз больше, чем у Ni ,
у которого оно в 3 раза больше, чем у Zn2+ или Со2+ |57|. Оптимальные значения рН
и условия связывания специфичны для конкретных пар белок—ион металла и
должны быть определены экспериментально.
Связывание и элюирование белка. Помимо комплексообразования с ионами метал
лов АХИМ-сорбенты могут участвовать в ионном обмене, ионной эксклюзии и
в гидрофобных взаимодействиях. Начальная подвижная фаза для связывания белка и
финальная подвижная фаза для его элюирования могут быть разработаны таким
образом, чтобы минимизировать или использовать эти эффекты. Хелатируюшие
группы без иона металла функционируют как сайты катионного обмена и могут
взаимодействовать с катионными группами белков (при ионной силе < 0,1 М). Высокие
концентрации соли (> 0,5 М) подавляют ионообменные взаимодействия, но
способствуют гидрофобным взаимодействиям с хелатирующей группой или ее линкером.
Промежуточная ионная сила подвижной фазы (между 0,1 и 0,5 М) может подавлять
ионообменные взаимодействия и снижает риск агрегации белка, особенно для анти
тел |37|. Общий вывод заключается в том, что удерживание кислотных белков имеет
тенденцию увеличиваться с возрастанием концентрации соли, в то время как
удерживание основных белков в этих условиях сначала уменьшается, а затем
увеличивается |55|. Каждый из упомянутых параметров разделения может варьироваться с
целью оптимизации селективности.
Связывание и элюирование сильно зависит от рН подвижной фазы. Белки
связываются наиболее прочно при рН выше рКа имидазола гистидина (рКа ~ 7), и
связывание уменьшается, когда рН падает ниже этого значения (и гистидиновые группы
становятся более ионизированными). Поэтому общая стратегия заключается в
связывании белков при значениях рН от 7 до 8 с последующим резким или постепенным
изменением рН до значений между 4 и 5, чтобы перевести остатки гистидина
в ионизированную форму.
Альтернативной стратегией элюирования является использование вытесняющих
агентов, таких как имидазол, гистамин, гистидин, глицин или аммиак. Первые три
агента имеют эквивалентную алюирующую способность, и, как правило, она выше
элюирующей способности двух последних. Вытесняющие агенты с постепенно
увеличивающейся силой можно вводить последовательно для элюирования сначала
слабо удерживаемых белков, а затем сильнее связанных |61|. Белки также могут быть
разделены с помощью градиента концентрации одного вытесняющего агента.
Как и в других методах разделения, основанных на электростатических
взаимодействиях, АХИМ-лиганды взаимодействуют с группами на поверхности белка.
Поэтому условия, которые изменяют или нарушают конформацию белка, могут
изменить селективность в АХИМ. На рис. 13.21 показаны изменения в порядке
удерживания и элюирования, вызванные добавлением метанола в подвижную фазу
(возможно, отражающие изменения в конформации белка). АХИМ позволяет
разделить два белка, у которых количество остатков гистидина на поверхности молекулы
отличается всего на единицу.
13.4. Разделение пептидов и белков
15 -
10 -
5 -
CYT
/ LACA
CHY
10
% МеОН
15
20
Рис. 13.21. Влияние добавленного метанола на удерживание в АХИМ. Колонка:
Fe (III)—ИДУК силикагель. Подвижная фаза: различный %-метанола
и 25 мМ фосфатном буфере (рН 6) + 0,15 М сульфата аммония. CYT, цито-
хром С; LYS, лизоцим; LAC А, В-лактоглобулин A; CHY, химотрипсино-
ген А [62|
13.4.3. Хроматография гидрофобных взаимодействий (ХГВ)
ХГВ была впервые описана Тизелиусом |63| в конце 1940-х гг., а затем более полно
охарактеризована Поратом [64] и Гелоттой [65|. С 1960-х годов ХГВ широко
использовалась для разделения белков с использованием подложек на основе углеводов,
а в последнее время — для разделений на высокоэффективных подложках из
микрочастиц |66, 67|. Механизм ХГВ основан на взаимодействии белков со слабо
гидрофобными лигандами в присутствии высоких концентраций соли. Белки в
значительной степени сохраняют конформацию в этих условиях, и удерживание происходит
в результате взаимодействия гидрофобных участков на поверхности белка с лигандом
сорбента (обусловлено «высаливанием»). Удерживание в ХГВ необычно тем, что это
энтропийный процесс (|68| и см. ниже), и использует обратный градиент (от
высокой до низкой концентрации соли).
ХГВ создает «мягкие» условия для белков, которые элюируются в своей натив-
ной конформации без потери биологической активности. Поэтому ХГВ широко
используется для препаративного выделения белков от лабораторных до
промышленных масштабов. Хотя ХГВ и ОФХ имеют общий механизм удерживания, основанный
на гидрофобных взаимодействиях, селективность этих методов может существенно
отличаться. Жесткие условия ОФХ способствуют денатурации белка и доступности
внутренних гидрофобных остатков, тогда как в процессе ХГВ удерживание
обеспечивают только гидрофобные остатки, находящиеся на поверхности белка. Поэтому
можно ожидать, что селективность при разделении будет совсем иной.
13.4.3.1. Сорбенты и лиганды для ХГВ
Для ХГВ используются сорбенты как на основе силикагеля, так и на полимерной
основе, с достаточно большими размерами пор, чтобы в них мог проникать белок.
Подложки обычно покрыты полимерным гидрофильным покрытием (или линкерны-
ми группами), чтобы обеспечить смачиваемую, инертную поверхность; гидрофобные
646 Глава 13. Разделение биополимеров и синтетических полимеров
40 -
30-
Время
удерживания
(мин)
20-
-
-
-
-
/ .-'
/ / ^
/_
-С3ОН С,
с4
Рнс. 13.22. Влияние длины лиганда (—С^ОН, гидроксипропил; С], метил; и
на удерживание белка в ХГВ [69|
т.д.)
лиганды присоединяются к полимеру или линкеру позже. Лиганды обычно
представляют собой короткоцепочечные алкильные или фенильные группы, и удерживание
увеличивается с увеличением длины лиганда {[70, 711; см. примеры на рис. 13.22
для трех разных белков). Лиганды длиной от 1 до 3 атомов углерода способствуют
удерживанию при высоком содержании соли (в подвижной фазе) и высвобождению
при низком содержании соли. Более длинные лиганды могут вызвать чрезмерное
удерживание, а также конформационные изменения.
Следует отметить, что колонки для ИОХ и АХИМ могут вести себя как колонки
для ХГВ в условиях с высоким содержанием соли1 из-за гидрофобных взаимодействий
с линкерами2 и кросслинкерами (сшивками) носителя [68]. Кроме того, механизм
удерживания у последних из упомянутых колонок может изменяться в зависимости
от условий хроматографии. Когда колонки для ИОХ и АХИМ используются в
условиях высоких концентраций солей, характерных для ХГВ, их селективность может
отличаться от селективности, свойственной колонкам, предназначенным для ХГВ-разделе-
ний, благодаря вкладу ионных и координационных механизмов удерживания.
13.4.3.2. Другие условия
На селективность и удерживание в ХГВ-разделении влияют:
• выбор антихаотропной соли и ее концентрация;
• рН подвижной фазы;
• добавки к подвижной фазе;
• температура.
' В подвижной фазе. — Прим. перев.
^Лигандами и хелатируюшими группами. — Прим. перев.
13.4. Разделение пептидов и белков 647
<а>
05
log*
0.0
-0.5
(б)
■
Фазан
обыкновенный
рН =
j
/
/
/ /
/ t
/ * .
/ ' /
f t /
' s
8.0 7,0
J t
/ t
/ /
/ /
'" /
/
6.0
/
/
'
0.8
1.0 1.2 1.4 1.6
Концентрация соли (М)
1.0
0.5
tog*
Белок
куриных
яиц
рн= а.о 7,о б.о
о.е
1.0 1.2 1.4 1.6
Концентрация сопи (М)
1.в
Рис. 13.23. Влияние рН на удерживание в ХГВ двух видов лизоцима птиц (фазана
обыкновенного, белка куриных яиц) при разных молярных концентрациях
сульфата аммония |72|
Аитихаотропная соль. Основное внимание при разработке градиента в ХГВ
уделяется выбору соли и ее концентрации в начале и в конце градиента. Способность
солей способствовать удерживанию в ХГВ соответствует их эффективности в
высаливании белка, как показано в ряду Хофмайстера (таблица 13.6). При этом наиболее
широко используется дли ХГВ сульфат аммония'. Изменение вида соли, а также ее
концентрации2 могут дать возможность изменять как удерживание, так и
селективность, но при этом необходимо помнить о растворимости и чистоте соли.
Связывание белка достигается при начальной концентрации соли от 1,5 до 3,0 М. Для элюи-
рования используется обратный градиент до более низкой концентрации соли или
чистого буфера. В разделениях промышленных масштабов применять сульфат
аммония со щелочными подвижными фазами следует с осторожностью, так как может
высвобождаться свободный аммиак |37|. В таких случаях его можно с успехом
заменить сульфатом калия.
рН мобильной фазы. рН может повлиять на селективность, если кислотные или
основные аминокислоты расположены в зоне гидрофобного контакта. Это
иллюстрируется влиянием степени ионизацией гистидина на удерживание лизоцимов птиц
в ХГВ 1721 - Лизоцим фазана имеет гистидиновые остатки в области контакта, и
удерживание лизоцима меняется (рис. 13.23а) при варьировании рН в интервале,
включающем рКа имидазольной группы (рКа = 6). Удерживание куриного лизоцима,
который не имеет гистидинов в области контакта, слабо зависит от изменений рН
в том же диапазоне значений (рис. 13.236). Поэтому изменения в рН подвижной
фазы можно использовать для разделения этих двух видов белка.
Добавки в подвижную фазу. Удерживание в ХГВ основано на гидрофобном
взаимодействии, и поэтому на него влияют добавки поверхностно-активных веществ
в подвижную фазу (ПАВ могут связываться как с сорбентом, так и с белком, тем
самым снижая гидрофобность обоих). Таким образом, добавление неионных и цвит-
терионных ПАВ снижает удерживание белка [68, 73|. Включение ПАВ особенно
полезно при разделении высокогидрофобных образцов, таких как интегральные
мембранные белки, для солюбилизации которых могут понадобиться поверхностно-
активные вещества при подготовке образца к разделению. Добавление органического
' Вследствие его дешевизны, нетоксичности и доступности. — Прим. перее.
2В начальных и конечных условиях градиента. — Прим. перев.
648 Глава 13. Разделение биополимеров и синтетических полимеров
растворителя в подвижную фазу тоже уменьшит удерживание белка, но для ХГВ
создаст определенные трудности. Органические растворители могут вызывать кон-
формационпые изменения в белке, и их использование при высокой концентрации
соли увеличивает риск осаждения белка1.
Температура. Удерживание в ХГВ обусловлено энтропией и поэтому
увеличивается с температурой, в противоположность обычному влиянию температуры на
удерживание. Этот эффект усиливается из за конформационных изменений при более
высоких температурах, которые делают внутренние гидрофобные остатки
доступными для взаимодействия с колонкой. Конформационные эффекты можно определить
по уширению пиков при температурах, промежуточных между характерными для
естественных условий и вызывающими денатурацию белков |74|, например, между 10
и 35°Сна рис. 13.242.
13.4.4. Хроматография гидрофильных взаимодействий (ГИХ)
Высокогидрофильные и незаряженные водорастворимые молекулы представляют
проблему для хроматографиста, так как они плохо удерживаются в ОФХ и не
удерживаются в ИОХ. Решают эту проблему с помощью хроматографии гидрофильных
взаимодействий (ГИХ; раздел 8.6; [75, 76|). В ГИХ полярная неподвижная фаза
используется с водно-органической подвижной фазой. В отличие от ОФХ, водный
компонент подвижной фазы (например, вода или буфер) служит сильным
растворителем, а органический компонент (обычно ацетонитрил) здесь слабый растворитель;
то есть, удерживание увеличивается по мере возрастания процента органического
растворителя (рис. 13.25). Обратите внимание, что удерживание увеличивается
для более полярных молекул на рис. 13.25 (Arg |наиболее полярный| > p-Ser > Leu
|наименее полярный |) — снова не так, как в ОФХ. ГИХ можно рассматривать как
вариант нормально-фазовой хроматографии (НФХ; глава 8).
В дополнение к тому, что ГИХ помогает достичь достаточного удерживания и
разделения гидрофильных образцов, она имеет еще два других преимущества.
Во-первых, подвижные фазы с > 50% ацетонитрила являются менее вязкими, что
означает более низкие давления в системе и большее число теоретических тарелок.
Во-вторых, такие подвижные фазы с большим количеством органики прекрасно
испаряются при ионизации электроспреем в ЖХ-МС.
13.4.4.1. Неподвижные фазы, используемые в ГИХ
В ГИХ используются различные неподвижные фазы |76|, в том числе
^модифицированный |77|, аминопропильный |78|. амидный |79| и диольный |SO] силикагели,
сульфированный полистирол-дивинилбензол |81| и поли(2-гидроксиэтил аспарта-
мид) |75|. Использование немодифицированного силикагеля в качестве
неподвижной фазы устраняет проблему отщепления (смыва) лигандов в ЖХ-МС, которая мо-
'Также органические растворители при концентрации более 20% по объему могут вызвать
выпадение п осадок соли, что приведет к проблемам в хроматографической системе. — Прим. перев.
2Интересным приложением ХГВ для исследований стабильности антител и подбора состава
среды для хранения лекарственных средств на базе моноклональных антител является
количественное определение степени окисления белка. Обычно окисляется остаток метионина на участке
Fc моноклонального антитела, и окисленное антитело элюируется раньше нативного. Не всегда
удается подобрать условия для конкретного антитела, но если это получается, то простота и
скорость метода оправдывает затраты времени с лихвой. См. М. Haverick, S. Mengisen, M. Shameem,
A. Ambrogelly Separation of mAbs molecular variants by analytical hydrophobic interaction
chromatography HPLC: overview and applications. MAbs. 2014 Jul 1; 6(4): 852—858. — Прим. перев.
13.4. Разделение пептидов и белков 649
д_
50 "С
■ Г I 1 ■ L , . . ■ ■ | f | L . 1 1 I I I 1 I L_| | | I ,| I
A
40 °C
1 ' ' ' ' ' ' .....■■ ^ |_, i t t . , r
35 °C
....,,.,,..,
32 "C
'
L^
25 'C
r—T-i i—t-
\ I I I I I
10 °C
•'• ■ I I I I ■ ' - I . I I
О 5 10 15 20 25 30
Phc. 13.24. Влиянце температуры на элюирование в ХГВ а-лактальбумина с
дефицитом Са^+. Детали в тексте. Воспроизведено из |72] с разрешения авторов
Arg •
~-—~~~~^^ J
i i > t i i -,
/p-Ser
1
Leu
..-■■"
' <
20
40 60
%ACN
80
Рис. 13.25. Удерживание аминокислот в условиях хроматографии гидрофильных
взаимодействий (ГИХ). Условия разделения: катионообменная колонка (Poly-
Sulfoethyl А) в режиме ГИХ; подвижная фаза: 25-м М триэтиламмония
фосфат рН 5,0 с ацетонитрилом, содержание которого показано на рисунке |75|
650 Глава 13. Разделение биополимеров и синтетических полимеров
жет возникать при использовании привитых фаз. В то время как ионизация
силанолов при рН > 7 может привести к ионообменным взаимодействиям и
усложнить интерпретацию разделений, использование подвижной фазы с рН < 7 снимает
эту проблему. В качестве альтернативы можно использовать ГИХ-колонку с
привитой фазой: например, амидные колонки на основе силикагеля были успешно
использованы для разделения пептидов [79|. Диольные сорбенты на основе силикагеля
ближе всех к поведению недериватизированных силикагелей в ГИХ-разделениях,
и на них сведены к минимуму сложности, связанные с активными силанолами.
Поли(2-гидроксиэтил аспартамид) представляет собой неподвижную фазу,
специально разработанную для ГИХ |77|. Ее готовят путем введения этаноламина в покрыва
ющую сорбент оболочку полисукцинимида, ковалентно связанного с диоксидом
кремния. Полученное полиаспартамидное покрытие является производным аспара
гина — самой гидрофильной нейтральной аминокислоты. Сорбенты на основе
сополимера полистирола с дивинилбензолом с высокой степенью сульфирования могут
работать в режиме ГИХ с использованием ацетонитрильно-водных подвижных
фаз |81|. При этом полимерный носитель имеет перед другими преимущество — он
стабильнее. Однако эти сорбенты работают еще и как ионообменники. В то время,
когда писали эту книгу, чаще всего использовались для ГИХ колонки, упакованные
немодифицированным и амидным силикагелями.
13.4.4.2 Подвижные фазы для ГИХ
Типичными подвижными фазами для ГИХ являются смеси воды (или водного
буфера) и ацетонитрила с содержанием воды от 5 до 40% [75|. Обычно используются
буферы, представляющие собой соли аммония муравьиной или уксусной кислот,
растворимые в ацетонитриле. Эти соли также являются летучими и совместимы
с ЖХ-МС при ионизации электроспреем. Можно для этих целей рекомендовать
и трифторацетат триэтиламина. Хотя эта соль и является летучей, ТФУК, тем не
менее, печально известна тем, что подавляет ионизацию электроспреем |17|.
13.4.4.3. Применение ГИХ в разделении пептидов и белков
ГИХ успешно использовалась для разделения пептидов. Она обладает уникальной
селективностью в случае пептидов с гидрофильными посттрансляционными
модификациями. Гликопептиды с изменениями в структуре гликана были разделены на амидном
силикагеле [82, 83|, в то время как сорбенте поли(2-гидроксиэтиласпартамидной)
фазой был успешно применен для селективного выделения фосфопептидов |84|.
Взаимодополняемость ГИХ и ОФХ была продемонстрирована в исследовании поведения ам-
фипатических а-спиральных пептидов, обладающих гидрофобными и гидрофильными
участками |85|. Было показано, что замены в гидрофильной части молекулы
оказывают незначительное влияние на удерживание в ОФХ-разделении, но вызывают
изменение относительного удерживания в ГИХ и наоборот, замены в гидрофобной части
молекулы влияли на ОФХ-разделение, но не влияли на удерживание в ГИХ. Это
исследование также подтвердило, что в этих двух режимах разделения работают
различные области контакта.
ГИХ не нашла широкого применения при разделении белков из-за их
ограниченной растворимости в подвижных фазах с большими концентрациями
органических растворителей. В работах 186, 871 было зарегистрировано появление нескольких
пиков для отдельных белков в процессе ГИХ. Это объяснили денатурацией на
колонке в процессе разделения. Тем не менее, ГИХ была успешно использована
для разделения гистонов (семейство основных ДНК-связывающих белков с высоким
зарядом), включая варианты ацетилированных |88|, фосфорилированных |89| и
метилированных |90| гистонов.
13.4. Разделение пептидов и белков 651
13.4.4.4. Хроматография гидрофильных взаимодействий
с электростатическим отталкиванием (ГИХЭО)
Это вариант ГИХ, в котором используется ионообменный сорбент и элюент с
большим содержанием органики [9I|. С помощью ГИХЭО растворенные вещества могут
удерживаться с помощью гидрофильных взаимодействий (режим ГИХ), даже если
они имеют такой же заряд, как и неподвижная фаза. В результате можно
одновременно разделять смеси кислот и оснований, которые иным образом было бы трудно
разделить и в ГИХ, и в ИОХ. Механизм ГИХЭО можно рассматривать как
совокупность гидрофильных взаимодействий и процессов ионного обмена, на селективность
влияет и то, и другое. Важной особенностью этого сочетания является независимость
гидрофильных и электростатических взаимодействий, что позволяет управлять ими
независимым образом. Это показано на рис. 13.26 сравнением разделений в режимах
ГИХ и ГИХЭО смеси кислотных, нейтральных и основных пептидов. В процессе
ГИХ-разделения (рис. 13.26а) на нейтральном гидрофильном сорбенте то количество
ацетонитрила (% ACN), которое обеспечивает подходящее удерживание и разделение
основных пептидов, не может обеспечить приемлемое удерживание кислотных и
нейтральных пептидов. При разделении в режиме ГИХЭО на слабом анионообмен-
нике при низком рН (см. рис. 13.266) электростатическое отталкивание основных
кислый пептид
основный пептид
I I I I I I I I I [ 1
О 20 40 60 80 100
(мин)
Рис. 13.26. Разделение пептидных стандартов с помощью ГИХ (а) в сравнении
с ГИХЭО (б). Условия ГИХ-разделения: колонка PolyHydroxyethyl А (Ро-
lyLC, Columbia, Maryland); подвижная фаза 20 мМ Na-MeP04 (рН 2,0)
+63% ацетонитрила. Условия разделения в ГИХЭО: колонка PolyWAX LP:
подвижная фаза 20 мМ Na-MePO^ (рН 2,0) +70% ацетонитрила |91]
652 Глава 13. Разделение биополимеров и синтетических полимеров
пептидов уменьшает их удерживание. Это позволяет увеличить процент ACN так,
чтобы все пептиды были удержаны и разделены.
ГИХЭО была использована для обогащения и выделения фосфопептидов из смеси
белков клеток HeLa, полученных протеолизом трипсином |92|. В обычных условиях
разделения на слабых анионнообменниках отрицательного заряда одной фосфатной
группы недостаточно для противодействия электростатическому отталкиванию N-koh-
цевых аминогрупп и аминогрупп боковых цепей. Поэтому монофосфорилированные
пептиды элюируются вблизи мертвого объема колонки. Когда слабый анионообмен
ник работает в режиме ГИХЭО с более высоким процентом ACN, гидрофильное
взаимодействие усиливает взаимодействие фосфатов с сорбентом, обеспечивая
удерживание и разделение моно- и мультифосфорилированных пептидов. ГИХЭО также
применялась для обогащения и выделения сиалированных гликопептидов |93|.
13.4.5. Многомерная жидкостная хроматография (МЖХ)
в протеомике
В экспрессионной протеомике требуется охарактеризовать весь набор белков, экс-
прессируемых в клетке или ткани при определенных условиях с целью их
идентификации. Такие белки являются кандидатами в биомаркеры, используемые
при диагностике и мониторинге заболевания или в качестве мишеней для
терапевтического вмешательства. В протеомике «снизу-вверх» все содержащиеся в
клеточном или тканевом лизате белки расщепляются протеолитическим ферментом, а
полученные в результате пептиды разделяются с помощью хроматографических
методов и охарактеризовываются тандемной ЖХ-МС/МС. Трудность работы в
режиме «снизу-вверх» состоит в огромной сложности пептидной смеси. По оценкам
протеом' человека содержит от ста тысяч до двух миллионов белков, из которых от
десяти до двадцати тысяч белков экспрессируются в любой момент времени. При
протеолитическом расщеплении (например, трипсином) из каждого белка
получается приблизительно 20 пептидов, поэтому можно предположить, что в каждом
лизате содержится от 200 до 500 тысяч продуктов гидролиза. Уникальная особенность
эксперимента с протеомикой заключается в том, что почти каждый компонент
образца является целевым образцом и поэтому необходима экстраординарная
разрешающая способность. Разделение компонентов такой сложной смеси находится
за пределами возможностей какого-либо одного вида хроматографии.
Исследования в протеомике требуют сочетания двух или более видов хроматографии (МЖХ)
с МС-детектированием (которое добавляет дополнительное измерение к анализу).
См. раздел 9.3.10 с общим обсуждением МЖХ (или 20-ЖХ для случая двух
хроматографических режимов).
Ключом к достижению достаточной разрешающей способности в МЖХ является
последовательное использование ортогональных по селективности
хроматографических режимов. Это означает, что механизмы удерживания в этих разделениях
должны быть совершенно различны. Пример ортогональности — двумерный
гель-электрофорез (2D-T3 |94|), разделение в котором происходит по заряду (или
по изоэлектрической точке) в первом измерении (изоэлектрическая фокусировка) и
по размеру молекулы во втором измерении (SDS-PAGE)2. В этом случае
разрешающая способность или пиковая емкость (раздел 9.3.9.1) комбинированных разделений
' Протеомом называют совокупность белков, экспрессированных в данном типе клеток или
организме, в данный период времени при данных условиях. — Прим. персе.
2Гель-электрофорез в полиакриламидном геле с додецилсульфатом натрия (sodium dodecylsul-
fate-polyacrylamide gel electrophoresis). — Прим. перев.
13.4. Разделение пептидов и белков 653
является произведением пиковых емкостей каждого разделения. Таким образом,
если в каждом разделении можно выделить от 30 до 50 белков, то 2D-T3 разделение
сможет обеспечить разделение от тысячи до трех тысяч белков. Основным
ограничением в МЖХ является использование ОФХ для последнего разделения, поскольку
условия, в которых проводится ОФХ, совместимы с теми, которые необходимы для
подключения в режиме реального времени к системам с ионизацией электроспреем.
Выбор начального разделения (или разделений) будет зависеть от подхода к МЖХ.
ЭХ (эксклюзионная хроматография) и ИОХ совместимы с последующей ОФХ
потому, что подвижные фазы, в основном, будут водными и фракции, содержащие
образец, будут сильно удерживаться на ОФХ колонке (без чрезмерного уширения полосы
целевого вещества), что позволит вводить на колонку большие объемы образца. ЭХ,
однако, не самый лучший выбор из-за ее низкой пиковой емкости (раздел 13.8.5).
Более крупные полипептиды также имеют тенденцию элюироваться в ОФХ позже
(как и в ЭХ)1, поэтому эти два вида хроматографии не являются полностью
ортогональными (как это желательно в МЖХ).
Для первого измерения в МЖХ чаще всего выбирают ИОХ, потому что в этом
случае ортогональность двух измерений приблизится к 2D-T3. Предпочтительнее
всего катионный обмен при низких рН, поскольку все пептиды будут положительно
заряжены и удержатся на ионообменном сорбенте. Катионообменные сорбенты
обладают достаточной емкостью, чтобы с их помощью можно было обнаруживать и
извлекать пептиды как с высоким, так и с низким содержанием в образцах (поскольку
в ИОХ можно вводить большие объемы образцов). Было показано, что и ОФХ в ще
лочных условиях (рН 10) и ГИХ обеспечивают высокую степень ортогональной се
лективности в качестве разделений первого измерения |95|.
В протеомике для проведения МЖХ используются три подхода:
• дискретная МЖХ со сбором фракций;
• непосредственно соединенные колонки для МЖХ;
• МЖХ с переключением колонок.
13.4.5.1. Использование коллектора фракций
Самый простой и прямой подход к МЖХ — сбор фракций с колонки первого
измерения с последующим введением отдельных фракций на колонку второго
измерения |96]. Преимущество этого подхода состоит в том, что реагенты,
используемые для восстановления и алкилирования белков на стадии их расщепления
ферментами, элю пру юте я в слив без риска загрязнить ими источник ионизации.
Недостатком такого подхода является необходимость вмешательства оператора
для сбора фракций и повторного ввода их на колонку. Тем не менее, этот подход
может быть автоматизирован объединением разделений первого и второго
измерений в рамках одной многоканальной ВЭЖХ-системы, снабженной автоматическим
коллектором фракций и краном переключения колонок. Фракции собираются
в процессе разделения первого измерения, а затем кран выбора колонок
переключается, уже собранные фракции становятся образцами для разделения второго
измерения и последовательно анализируются на второй колонке. Несколько
производителей ВЭЖХ-оборудования предлагают автоматизированные 20-системы,
использующие ИОХ (или хроматофокусирование) для проведения разделения
первого измерения и ОФХ для второго измерения (Beckman, Waters, Michrom Bioreso-
urces, Microtech Scientific, США).
'Странно, в ЭХ крупные молекулы элюируются раньше. По-видимому, авторы имеют в виду
взаимодействие некоторых белков с сорбентами, что приводит к отклонению от механизма
«идеальной» ЭХ. См. раздел 13.8.3. — Прим. перев.
654 Глава 13. Разделение биополимеров и синтетических полимеров
13.4.5.2. Непосредственно соединенные колонки для МЖХ
Систему из непосредственно соединенных колонок для МЖХ группа Ятса положила
в основу многомерной технологии идентификации белка (МТИБ) [97, 98|. При
таком подходе одна капиллярная ВЭЖХ-колонка состоит из двух последовательных
сегментов двух ортогональных неподвижных фаз (первый сегмент — катионообмен-
ник, а второй — ОФХ сорбент) для разделения сложных пептидных смесей.
Проводится от 12 до 15 циклов алюирования, и пользователь получает ряд хроматограмм.
В типичном цикле элюирования часть образца вытесняется из катионообменного
сегмента водным буфером, после чего вытесненная фракция элюируется из ОФ-сег-
мента градиентом ацетонитрил — буфер в течение 90—120 мин. В последующих
циклах элюирования используются возрастающие концентрации ацетата аммония
(500 мМ в последнем цикле). Колонка соединена непосредственно с наноспреем
тандемного МС-детектора. Оценочный эксперимент с дрожжевыми лизатами
показал, что (в отличие от 2D ГЭ) этот подход не приводит к искажению данных при
небольшом содержании белков в образце, при обнаружении белков с большими или
малыми молекулярными массами, а также белков с ярко выраженными кислотными,
основными или гидрофобными свойствами [991.
13.4.5.3. МЖХ с переключением колонок
Многомерная жидкостная хроматография с переключением колонок может
обеспечить наибольшую гибкость и максимальную пиковую емкость, но все это дается
за счет усложнения оборудования и увеличения его стоимости. Переключение
колонок может также устранить проблему загрязнения примесями МС-источника
ионизации. Три примера с нарастающей сложностью (первый для пептидов, остальные
для белков) иллюстрируют различные возможные подходы к МЖХ с переключением
колонок. Первый пример 11001 представляет собой систему из сильного катионооб-
менника первого измерения и ОФХ для второго измерения. Образец в первом
измерении разделяется на две фракции: неудерживаемые пептиды и удерживаемая
фракция, элюируемая 0,5 М солью. Это простое бинарное фракционирование оптимально
для высокопроизводительных методик, но ограничено с точки зрения количества
разделенных пиков (только в 2 раза больше, чем при одном разделении).
Во втором примере (подробно см. 20-ЖХ; раздел 9.3.10) [101, 102| объединили
ЭХ в первом измерении и ОФХ во втором. Разделение в первом измерении
достигается с помощью набора из шести колонок, соединенных последовательно (общая
длина 1,8 м), с целью получения 90000 теоретических тарелок для повышения
пиковой емкости. Элюент, выходящий из колонок первого измерения, поочередно
отводится в одну из двух ОФХ-колонок, установленных параллельно. Пока пептиды
сорбируются на одной ОФХ-колонке, пептиды из предыдущей фракции разделяются
на другой ОФХ-колонке в режиме кратковременного градиента. Такое расположение
колонок вдвое сокращает необходимое общее время разделения. Основным
ограничением этого подхода является неполная ортогональность двух видов разделения
в ЭХ и ОФХ (а также ограниченное разрешение в ЭХ). Поэтому полученная пиковая
емкость составляла только около 30% ожидаемой (относительно небольшой). Эта
система средней сложности, требующая изократических и градиентных систем подачи
элюентов с переключающим краном (рис. 9.25).
Третий пример, разработанный для разделения белков методом МЖХ,
объединяет пробоподготовку с аналитическим разделением |103|. Проба наносится на
колонку для предварительного фракционирования, которая содержит носитель с
ограниченно доступной поверхностью (ОДП, раздел 16.6.7.2). Во время предварительного
фракционирования исключаются большие белки, а мелкие белки концентрируются и
разделяются по заряду. Разделение первого измерения аналитической системы МЖХ
13.5. Разделение нуклеиновых кислот 655
проводится на одной ионообменной колонке, работающей в градиентном режиме.
Фракции с этой колонки последовательно переносятся в две из четырех ОФХ-коло-
нок для разделения, в то время как две другие ОФХ колонки регенерируются.
Эта очень сложная МЖХ-система, в ней используются три градиентные системы
подачи элюентов, один изократический насос, автоматический инжектор и четыре де-
сятиходовых крана. См. также альтернативную методику в разделе 9.3.10.
13.5. Разделение нуклеиновых кислот
Нуклеиновые кислоты несут ионизированные фосфаты на сахарофосфатном остове,
а гидрофобные гетероциклические основания связаны с сахарами. Поэтому
анионный обмен, ОФХ и ХГВ являются кандидатами в методы для разделения
нуклеиновых кислот. Все три вида хроматографии были использованы в исследованиях
нуклеиновых кислот для аналитических и препаративных разделений.
13.5.1. Анионообменная хроматография
Анионообменная хроматография (на слабых или сильных анионообменниках)
наиболее часто используется для разделения олигонуклеотидов. Ионообменные
взаимодействия между фосфатными группами и колонкой возрастают с увеличением числа
фосфатных групп, и поэтому удерживание увеличивается с увеличением длины цепи
или молекулярной массы анализируемого вещества. Элюирование обычно
выполняется градиентом от низкой до высокой концентрации нейтральной соли, такой, как
хлорид натрия или фосфат натрия. В подвижную фазу для подавления
самоассоциации олигонуклеотидов может быть добавлен формамид1. Типичным применением
анионообменной хроматографии является определение чистоты синтетических
олигонуклеотидов, полученных твердофазным синтезом |1041. После отщепления
продукта от твердой фазы и удаления защитных групп, анионообменная хроматография
может отделить неполные последовательности от олигонуклеотидов полной
длины (рис. 13.27).
Продукт
О 10 20 (мин)
Рис. 13.27. Анализ неочищенной смеси, полученной при химическом синтезе олиго-
нуклеотида после деблокирования, на сильной анионообменной
колонке с использованием градиента от 1 мМ КНтРСЬ + 60% формамида
до 300 мМ КН2Р04 + 60% формамида |104]
1 Не надо только забывать о том, что он довольно токсичен. — Прим. перев.
656 Глава 13. Разделение биополимеров и синтетических полимеров
—г-
10
-г-
15
-г-
20
25 (мин)
Рис. 13.28. Разделение рестрикционных фрагментов на колонке TSK-DEAE-NPR
с непористым носителем с использованием градиента от 250 мМ
до 450 мМ NaCl в 20 мМ Трис-HCI (рН 9) 1106]
Обычная анионообменная хроматография применяется для разделения олиго-
нуклеотидов, содержащих до 30 оснований. Разделение нуклеиновых кислот
большей длины ограничено их исключением из пор носителя. В этом случае
предпочтительны носители с большими порами (диаметром S 30 нм) или непористые
носители. Фрагменты ДНК длиной от 50 до 1000 пар оснований, полученные
при расщеплении ферментами рестрикции (эндонуклеазами), разделяют на слабых
анионообменных фазах (рис. 13.28) на основе либо (1) силикагеля с порами 400 нм
|105], либо (2) непористых полимерных смол |107|'. Исключением из этого
правила является анионообменное разделение транспортных РНК (тРНК); эти
молекулы, состоящие из 75—90 нуклеотидов, имеют развитую вторичную структуру
и компактный размер, что позволяет им проникать в поры размером 12 нм
(рис. 13.29). Разделение, показанное на рис. 13.29, на удивление хорошее, если
учесть, что большинство тРНК приблизительно одинаковы по размеру и нет
оснований ожидать, что их можно разделить только на основе различий в зарядах.
Однако тРНК различаются первичной последовательностью и характером
метилирования, поэтому это разделение, вероятно, основано на эффектах смешанного
режима, возникающих в результате как гидрофобного, так и электростатического
взаимодействий |106, 107|2.
'С момента выхода последнего издания книги появился новый сорбент DNAPac,
производимый компанией ThermoFisherDionex. Он представляет собой мелкие (0,1 мкм) зерна непористого
полимера, посаженные на поверхность зерен большого диаметру (= 7 мкм), что создает сорбент
с высоким разрешением, высокой емкостью и с двумя типами взаимодействия — гидрофобным
и ионообменным. — Прим. перев.
2В настоящее время для анализа олигонуклеотидов методом ИОХ самыми популярными
являются колонки ThermoFisherScientific (США): DNAPac 100 и DNAPac 200. Первая считается
наиболее подходящей для разделения ДНК, а вторая — для РНК. — Прим. перев.
13.5. Разделение нуклеиновых кис/гот 657
тРНЮ"»
О 20 40 60 80 100 (мин)
Рис. 13.29. Фракционирование транспортных РНК (тРНК) из Е. СоМ на колонке
Hypersil PEI, 3 мкм, диаметр пор 12 нм, 58,8% кватернизовано. Подвижная
фаза А: 0,05 М фосфат калия, 5% ацетонитрила (рН 5,9). Подвижная
фаза В: 0.05М фосфат калия, 5% ацетонитрила (рН 5,9) + 1 М сульфат
аммония. Градиент: 0,25%/мин от 50% подвижной фазы В. Скорость потока:
0,5 мл/мин 11071
13.5.2. Обращенно-фазовая хроматография
ОФХ нельзя применять непосредственно для разделения нуклеиновых кислот, в том
числе олигонуклеотидов и более крупных молекул, таких, как рестрикцией ные
фрагменты, поскольку эти заряженные и высокополярные молекулы не удерживаются в
типовых условиях разделения. Что касается других полярных ионизованных молекул,
то их удерживание в условиях ОФХ можно увеличить путем добавления ион-парного
реагента в подвижную фазу (ион-парная хроматография |ИПХ]; раздел 7.4.1.2). Для
отрицательно заряженных олигонуклеотидов положительно заряженный ион-парный
реагент, такой, как тетрабутиламмоний, может обеспечить удерживание и приемлемое
разделение. Реагент удерживается колонкой, создавая положительно заряженную
неподвижную фазу, которая затем работает как гидрофобный анионообменник.
13.5.2.1. Олигонуклеотиды
В случае анионного обмена, как и ожидается, удерживание олигонуклеотидов
увеличивается с увеличением длины цепи и отрицательного заряда. В результате
гидрофобных взаимодействий различия в удерживании могут возникать для
олигонуклеотидов с одинаковой молекулярной массой, но различной первичной
последовательностью. Очистка синтетических олигонуклеотидов может выполняться
с помощью ИПХ, если использовать преимущества гидрофобных защитных групп
(например, диметокситритильной, ДМТ), применяемых в синтезе |Ю8|. При таком
подходе хроматографическое разделение проводится до снятия защиты.
Защищенный продукт сильнее удерживается, а последовательности без ДМТ-групп
удерживаются плохо и хорошо отделяются от продукта (рис. 13.30)1.
1 В начале двухтысячных годов для разделений олигонуклеотидов в качестве злюента А стали
использовать 16.3 мМ ТЭА/100 иМ ГФИП (1,1,1,3,3,3-гексафтор-2-пропанол), а в качестве элю-
ента В метанол или ацетонитрил. Фактически такая подвижная фаза в настоящее время стала
общепринятой и позволила резко увеличить разрешение ОФХ-колонок. Она совместима с ЖХ-МС.
В 2016 году компания ThermoFisherScientific (США) предложила новую ОФХ-колонку DNAPac,
упакованную полимерным сорбентом, для разделения олигонуклеотидов и двухцепочечных ДНК,
стабильную и при щелочных рН, что зачастую дает дополнительные преимущества при
разделении. — Прим. персе.
658 Глава 13. Разделение биополимеров и синтетических полимеров
Продукт, защищенный
п ДМТ
J
8 10 12 (мин)
Рис. 13.30. Градиентное разделение неочищенного 20-мерного олигонуклеотида с три-
тильной защитой с помощью ИПХ. Условия разделения: 0,1 М ацетат тет-
раэтиламмония (ТЭАА) как слабый растворитель и ацетонитрил как
сильный |107]
13.5.2.2. Фрагменты, полученные рестрикцией, и продукты ПЦР
Более крупные нуклеиновые кислоты, такие как двухцепочечные рестрикционные
фрагменты ДНК и продукты полимеразной цепной реакции (ПЦР), можно
разделить с помощью ИПХ либо на непористых частицах, либо на пористых полимерных
монолитах (рис. 13.31). Оба разделения на рис. 13.31 демонстрируют превосходную
эффективность на полистирол-дивинилбензольном сорбенте при ион-парном
разделении нуклеиновых кислот. В случае двухцепочечных молекул, элюирование
при умеренных температурах (< 50 °С) идет в порядке увеличения длины цепи. Из-за
расположения чрезвычайно гидрофильных групп фосфатов и Сахаров на наружной
поверхности спирали гидрофобный вклад оснований минимален 11091. При более
высоких температурах колонки (> 53 °С), происходит частичное или полное
разделение цепочек ДНК, и строгая зависимость удерживания от размера молекулы
нарушается под влиянием первичной последовательности олигоунклеотидов.
13.5.2.3. Денатурирующая ВЭЖХ
Денатурирующая ВЭЖХ (дВЭЖХ) представляет собой методику анализа вариаций
последовательностей ДНК у людей с целью выявления однонуклеотидных
полиморфизмов (ОНП)' и связанных с заболеваниями мутаций |Ю9, 110|. Эта методика
выявляет ОНП в полиморфных сайтах ПЦР-амплифицированных последовательностей
нормальной (эталонной) ДНК и целевой ДНК, в которой, как предполагается,
содержится модифицированный локус. Со-амплифицированные последовательности
нагревают до разделения отдельных цепей ДНК, затем повторно ренатурируют
постепенным охлаждением, и после этого анализируют, используя дВЭЖХ. Если сайт
ОНП в целевой ДНК совпадает с сайтом нормальной ДНК, наблюдаются только два
идентичных гомодуплекса. Если целевая ДНК содержит модифицированное
основание на сайте ОНП, образуются несовпадающие гетеродуплексы цепей нормальной и
целевой ДНК в дополнение к гомодуплексам.
В дВЭЖХ используется ИПХ при повышенной температуре для поддержания
амплифицированных последовательностей ДНК в частично или полностью денату-
1 В англоязычной литературе ОНП называется single nucleotide polymorphism (SNP). — Прим. перев.
13.5. Разделение нуклеиновых кислот 659
Колонка с
микрочастицами
10 (мин)
(б)
20 (мин)
Рис. 13.31. Сравнение разделений рестрикционных фрагментов двухцепочечной ДНК
с помощью (я) непористого сорбента PS-DVB-C18 (2 мкм) и (б)
капиллярной колонки с внутр. диам. 0,2 мм, заполненной монолитом P/S-DVB.
Подвижная фаза А: 100 мМ ТЭАА + ЫалЭДТА (рН 7,0). Подвижная фаза В:
100 мМ ТЭАА + ^фЭДТА (рН 7,0), 25% ацетонитрила. Температура
колонки: (я) 50 "С; (б) 49,7 "С. Большее время элюирования в случае
монолитной колонки является результатом задержки градиента при той
скорости потока, которая возможна в капиллярных колонах [109|
рированном состоянии. Этот метод может быть использован для
идентификации сайтов мутаций с помощью хроматографической «подписи» гомо- и гетеродуп-
лексов.
Несовпадающие области гетеродуплексов (содержащие непарные «пузырьки»)
имеют уменьшенную область контакта с поверхностью, покрытой адсорбированным
ион-парным агентом. В результате они хуже удерживаются, чем гомодуплексы,
и разрешаются друг с другом благодаря отличию последовательности оснований
в пузырьке. ИПХ проводится при температуре, поддерживающей стабильность
гетеродуплексов. Для смеси сегментов, являющихся гетерозиготными при замене одного
основания, характерна четырехкомпонентная хроматограмма (рис. 13.32), причем по-
Глава 13. Разделение биополимеров и синтетических полимеров
CG
CG
AT
GC
AT
СО
CG
GC
GC
AT
\
Денатурация и Повторный отжиг
Гетеро Гомо"
Дуплексы.
CG CG
CG CG
дНТФ
праймеры
фермент
CGCG
CGCG
IAC GTt AT GC
GCGC
О минуты 3
Рис. 13.32. Разделение двух хромосом в виде смеси продуктов ПЦР,
денатурированных при 95 "С, а затем постепенно охлажденных до 65 °С перед анализом
методом денатурирующей ВЭЖХ. Если есть несовпадения, формируются
не только два исходных гомодуплекса, но и одновременно смысловые и
антисмысловые цепи каждого гомодуплекса формируют два гетеродуплек-
са. Эти гетеродуплексы денатурируют интенсивнее при температуре
анализа 56 °С и выходят из колонки раньше, чем два гомодуплекса, которые
денатурируют в меньшей степени |109|
следние два пика представляют собой гомодуплексы, а первые два пика
соответствуют продуктам ренатурации смысловых цепей ДНК одного гомодуплекса с антисмыс-
ловыми цепями другого гомодуплекса.
13.5.2.4. ОФХ-5 хроматография
ОФХ-5 изначально был методом хроматографии низкого давления, разработанным
в 1960-х годах. Применялся он для разделения тРНК |1П|. В методе используется
полимерная полихлортрифторэтиленовая подложка с адсорбированным на ней
хлоридом метилтриоктиламмония в качестве жидкой неподвижной фазы. Хотя это и
называлось ОФХ, на самом деле представляет собой разделение смешанного типа,
зависящее как от ионных, так и от гидрофобных взаимодействий. В более поздней
версии ОФХ-5 в смешанном режиме используется в ВЭЖХ на силикагельном
сорбенте С18 (5 мкм), покрытом хлоридом метилтриоктиламмония I112]1.
1 Реализован также вариант сорбента на основе силикагеля с химически привитыми ундецил-
дигексилметиламмониевыми группами, проявившего высокую селективность в разделении олиго-
нуклеотидов. Особый интерес этот сорбент представляет для препаративной очистки олигонуклео-
тидов, так как продукт не содержит ион-парного реагента. (Выпускается АО «БиоХимМак СТ»
под названием Диасфер-С|^(С6)2Ме.) — Прим. перев.
13.6. Разделение углеводов 661
тРН1Са|-С-С
тРНК^'-С-С TpHKPhe-C-C
Л
10
40 (мин)
Рис. 13.33. Разрешение специфичных тРНК из дрожжей на аминопропильной фазе,
модифицированной капроновым ангидридом; элюирование в условиях
нисходящего градиента сульфата аммония. Перепечатано из [П4| с
разрешения авторов
13.5.3. Хроматография гидрофобных взаимодействий
ХГВ применялась для разделения тРНК с использованием слабо гидрофобных
сорбентов и нисходящих градиентов антихаотропных солей |Ю4]. Крупнопористые
подложки на основе силикагеля с привитыми С2- или С4-фазами |113|, а также с ами-
нопропильными фазами, дериватизированными введением алкильных цепей [114|,
использовались с градиентами сульфата аммония, как в примере на рис. 13.33.
Селективность может быть изменена добавлением небольших количеств 2-пропанола
к подвижной фазе. Транспортные РНК элюируются в порядке увеличения гидро-
фобности. В случае, если тРНК ацилирована по аминогруппе гидрофобной
аминокислотой, такой как валин, ацилированная форма тРНК элюируюется позже, чем
неацилированная. Разделение тРНК в режиме ХГВ также можно проводить на анио-
нообменных колонках |115, 116|. При этом подходе тРНК связываются с сорбентом
в условиях с высоким содержанием соли в подвижной фазе и элюируются обратным
градиентом до концентрации соли, которая является достаточно низкой для
освобождения тРНК, но все еще достаточно высокой, чтобы предотвратить
ионообменные взаимодействия.
13.6. Разделение углеводов
Хроматографический анализ нейтральных углеводов представляет собой серьезную
проблему, так как они плохо удерживаются в ОФХ и (при рН < 10) не имеют
ионных групп, что позволило бы использовать ион-парную или ионообменную
хроматографию. К тому же большинство углеводов не имеют хромофоров или флуорофо-
662 Глава 13. Разделение биополимеров и синтетических полимеров
ров, и поэтому обычно используются такие методы оптического обнаружения, как
детектирование показателя преломления или рассеяния света (с градиентным элюи-
рованием, однако, использовать ДПП нельзя).
Для разделения углеводов используются три вида хроматографии: НФХ или
ГИХ, распределительная хроматография с управляемой ионами селективностью и
анионообменная хроматография в области высоких значений рН. Пригодность
каждого из этих вариантов для конкретного применения зависит от требуемой
чувствительности, разрешения и доступного оборудования.
13.6.1. Хроматография гидрофильных взаимодействий
На ранних этапах развития ВЭЖХ колонки, упакованные аминофазами (аминопро-
пил, привитый на силикагель), с водой и ацетонитрилом в качестве подвижной фазы
широко использовалась для ГИХ-разделения углеводов. Основным недостатком ами-
нофаз является их низкая стабильность, в то время как важным преимуществом —
повышенная скорость мутаротации аномеров, что устраняет проблему двойных
пиков, образующихся при разделении аномеров (диастереомеров). Проблема короткого
времени жизни колонки была частично решена с помощью использования немоди-
фицированного силикагеля в сочетании с добавкой амина в подвижную фазу [117|.
Удобнее ГИХ-разделение углеводов проводить на других колонках: с немодифи-
цированным силикагелем, силикагелем с привитой амидной или диольной фазами,
гидрофильным силикагелем в полимерной оболочке, силикагелем с привитым цик-
лодекстрином и ионообменниками на основе полимеров |I1S|. Использование этих
сорбентов исключает реакции аминогрупп с восстанавливающими сахарами [119|;
они также стабильны при повышенных температурах. Хотя диольная фаза и
использовалась в режиме ГИХ для разделения углеводов, недостатком этих разделений
является уширение пика в результате частичного разделения аномеров. В подвижную
фазу можно добавить амин для ускорения мутаротации аномерных Сахаров |120|,
но эта же добавка ускоряет растворение силикагелевой подложки.
Для разделения углеводов могут использоваться сорбенты для ГИХ на основе
силикагеля, модифицированного полиаспартамидом, с подвижной фазой вода—ацето-
нитрил |75|. Обратите внимание на то, что в разделении олигогликозидов, показанном
на рис. 13.34, удерживание уменьшается при более низком % ацетонитрила (типичное
поведение в ГИХ). Как и в случае колонок, упакованных диольной фазой, добавление
амина к подвижной фазе может катализировать мутаротацию, чтобы предотвратить
уширение пика при разрешении аномеров. Неподвижные фазы с привитыми а- или
Р-циклодекстринами (циклодекстриновые колонки) используются с водно-ацето-
нитрильными подвижными фазами для разделения моно- и олигосахаридов |121|.
Удерживание растет при увеличении количества доступных гидроксильных групп в
молекуле. Эти наблюдения согласуются с механизмом ГИХ, который включает
взаимодействие анализируемого вещества с гид роке ильным и группами, расположенными
на внешней поверхности молекулы циклодекстрина, а не образование комплексов
включения в полости циклодекстрина (см. соответствующее обсуждение в разделе
14.6.4 и на рис. 14.18а). К преимуществам циклодекстриновых колонок относятся их
более высокая стабильность и приемлемая воспроизводимость удерживания.
Для разделения углеводов также использовались колонки, упакованные
сильными катионообменниками на полимерной основе |122|, и гидрофильные колонки
для гель-фильтрации [123| с обычными для ГИХ подвижными фазами (смесями аце-
тонитрил-вода). Механизм этих разделений нельзя отнести ни к ионному обмену,
ни к гель-фильтрации, о чем свидетельствует увеличение удерживания при
возрастании концентрации органического растворителя.
13.6. Разделение углеводов 663
J
1 2 3
/\_
80% ACN
75% ACN
10
11
12
70% ACN
13
10
15
20 (мин)
Рис. 13.34. Изократические ГИХ-разделения смеси гомополимеров З-гидрокси-2-ни-
тропиридинил-р-О-мальтоолигогликозидов. Условия разделения:
колонка Poly-Hydroxyethyl A 200 х 4,6-мм; подвижная фаза ацетонитрил-иода;
2,0 мл/мин. Числа показывают степень полимеризации каждого
соединения [75]
13.6.2. Распределительная хроматография с управляемой
ионами селективностью
Для разделения широкого спектра моно- и олигосахаридов, а также смеси углеводов
со спиртами и другими малыми молекулами используются сульфированные полисти-
рол-дивинилбензольные носители |I24, I25|. Разделение основано на комбинации
гель-фильтрации и лигандного обмена. В процессе обмена лигандами участвуют
ионы переходных металлов, прочно удерживаемые сульфогруппами носителя. Ион
металла обеспечивает положительный заряд, который взаимодействует с очень
слабым отрицательным зарядом на молекуле сахара («лиганде»). Основной механизм
разделения олигосахаридов — гель-фильтрация. Предпочтительнее использовать
полимерные носители с низким процентом сшивки для того, чтобы дать возможность
молекулам олигосахаридов проникнуть в поры частиц сорбента. На рис. 13.35
показано разделение олигосахаридов, содержащихся в кукурузном сиропе. Для
моносахаридов основным механизмом разделения является лигандный обмен гидроксилов
сахара с закрепленным на полимерном носителе противоионом.
Был предложен следующий механизм обмена лигандами |126]: гидроксильные
группы углеводов обмениваются на молекулы воды, находящиеся в гидратной
оболочке удерживаемого на сорбенте катиона (противоиона). Стабильность комплекса
664 Глава 13. Разделение биополимеров и синтетических полимеров
I I I
Ю 20 30 (мин)
Рис. 13.35. Анализ олигосахаридов кукурузного сиропа методом распределительной
хроматографии с управляемой ионами селективностью на сильном катио-
нообменнике с противоионами серебра (сшивка 4%). Образец: кукурузный
сироп (глюкоза, 1; 2—II, степень полимеризации 2—П). Условия
разделения: колонка Aminex НРХ 42-А 300x7,8 мм; подвижная фаза — вода;
85 °С; 0.4 мл/мин [124|
углевод — катион растет с увеличением доступности координационной сферы
комплекса, а удерживание углеводов увеличивается с ростом стабильности комплекса.
Селективность лигандного обмена зависит от природы противоиона и его размера,
а также от структуры и стереохимии углеводов. Разделения проводят в изократиче-
ском режиме с водой или разбавленными растворами серной кислоты (5—10 мМ)
в качестве подвижной фазы, обычно при повышенных температурах (40—85 °С).
Процент сшивки полимерной подложки и ионные формы, рекомендуемые для
конкретных методов, перечислены в таблице 13.7. Необходимо помнить, что это носители
с фиксированными противоионами. Перед упаковкой сорбент переводится
изготовителем в определенную ионную форму, и она сохраняется в таком виде в период
использования колонки. Пользователям не рекомендуется переводить сорбент из одной
формы в другую, поскольку носители могут сжиматься или набухать при изменении
ионной формы, что может привести к порче колонки. Сорбенты в натриевой форме
Таблица 13.7. Сильные катионообменннки для распределительной хроматографии
углеводов с управляемой ионами селективностью
Процент
сшивки
8
8
8
8
8
4
4
Ионная форма
(противонон)
кальция
свинца
водорода
натрия
калия
серебра
кальция
Приложения
Моносахариды и разделение по классам ди-, три- и тетрасахаридов
Пентозы и гексозы в продуктах древесины
Углеводы в растворах с жирными кислотами, спиртами и кетона.ми
Сахара в образцах с высоким содержанием солей
Моно-, ди- и трисахариды в кукурузном сиропе и сусле
Олигосахариды
Моно- и дисахариды в гидролизатах крахмала
13.6. Разделение углеводов 665
пригодны для образцов, содержащих высокие концентрации соли (например,
патоки); сорбенты в калиевой форме полезны для анализа кукурузного сиропа, колонки
с катионами серебра обеспечивают хорошую селективность при работе с олигосаха
ридами, а сорбенты с катионами кальция используются для анализа продуктов
гидролиза крахмала.
13.6.3. Высокоэффективная анионообменная
хроматография
Нейтральные углеводы не удерживаются на колонках в обычных условиях ИОХ,
но при рН > 12 они проявляют свойства слабых кислот, которые могут частично
ионизироваться (таблица 13.8). Их можно разделить в щелочных условиях с
помощью анионообменного сорбента на полимерной основе [127, 1281. Коммерчески
доступны колонки для высокоэффективной анионообменной хроматографии (ВАХ)
на основе полистирола/дивинилбензола (для разделения моносахаридов) или этил-
винилбензола/дивинилбензола (для разделения олигосахаридов). Оба сорбента
состоят из непористых частиц, покрытых тонким слоем сульфированных латексных
микрошариков (рис. 13.36). Сахарные спирты являются более слабыми кислотами,
чем невосстановленные сахара, и для их разделения с помощью ВАХ требуются
ионообменники большей емкости. Можно для этих целей использовать, к примеру,
макропористый винилбензол-хлорид/дивинилбензол с привитыми четвертичными
алкильными группами. Моносахариды (включая нейтральные и аминосахара) могут
быть разделены в изократическом режиме с использованием разбавленного
водного гидроксида натрия в качестве подвижной фазы. Разделение кислотных углеводов
(сиаловой кислоты, сиалированных и фосфорилированных олигосахаридов) может
быть достигнуто с помощью подвижной фазы, состоящей из гидроксида и ацетата
натрия, либо в изократическом режиме, либо в градиенте ацетата натрия. Олиго-
и полисахариды (включая олигосахариды с высоким содержанием маннозы,
гибридные и сложные) можно разделить, используя градиенты гидроксида натрия или
гидроксида натрия/ацетата натрия (рис. 13.37). См. также разделение углеводов
на рис. 7.21.
Таблица 13.8. Константы диссоинации часто встречающихся углеводов
Сахар
Фруктоза
Манноза
Ксилоза
Глюкоза
Галактоза
Дульцит
Сорбитол
а — метилгликозид
рК,
12,03
12,08
12,15
12,28
12,39
13,43
13,60
13,71
ВАХ обычно сочетается с импульсным амперометрическим детектированием
(ИАД). При высоком рН углеводы электрохимически окисляются на поверхности
золотого электрода при приложении положительного потенциала |129|. Измеренный ток
от этой реакции пропорционален концентрации углеводов. Пределы обнаружения
666 Глава 13. Разделение биополимеров и синтетических полимеров
о м+ >
поверхность с
микросферами
анионобменные
наносферы
Рис. 13.36. Частица пелликулярного анионообменного сорбента (Dionex). Частица
состоит из непористой сульфированной микросферы, поверхность которой
покрыта латексными наносферами с привитыми сильными анионообмен-
ными группами. Диаметр микросфер от 5,5 до 10 мкм в зависимости
от типа колонки. Диаметр наносфер от 43 до 275 нм, он также зависит
от типа колонки. Наносферы удерживаются на микросфере благодаря
электростатическим взаимодействиям между ионогенными группами |127]
__JLJ
1. дисиалированная,триантенная
2. дисиалированная,триантенная
3. трисиалированная, триантенная
4.трисиалированная, триантенная
5.тетрасиапированная,триантенная
6. тетрасиапированная, триантенная
7. трисиалированная, триантенная
6
10
20
30
40
50 (мин)
Рнс. 13.37. Разделение N-связанных олигосахаридных структур из бычьего фетуина
методом высокоэффективной анионообменной хроматографии с
использованием градиента 0,0—0,5М ацетата натрия в 100 мМ гидроксиде натрия.
С разрешения Dionex Corp. [128|
при этом находятся в низком пикомолевом диапазоне. В результате реакции
образуются продукты окисления, отравляющие поверхность электрода, поэтому напряжение
на электроде резко повышается в импульсном режиме для очистки его поверхности,
а затем резко понижается, чтобы восстановить оксид золота до металла.
13.7. Разделение вирусов 667
13.7. Разделение вирусов
Большие размеры частиц вирусов представляются определяющим фактором при их
хроматографическом разделении. Этот факт имеет несколько последствий.
Во-первых, от вирусов можно ожидать больших значений S (в ОФХ) или т (в ИОХ), таких,
что небольшие изменении в концентрации подвижной фазы будут оказывать
большое влияние на их изократическое удерживание |3|. Поэтому изократическое
разделение вирусов целесообразно проводить только в режиме ЭХ. В градиенте вирус
будет элюироваться в определенной точке градиента, в большинстве случаев с к* « 0.
Вторым следствием размера вируса является медленная диффузия. Например, Dm
для аденовируса равен 5 х Ю-8 см2/сек, то есть, он примерно в Ю раз меньше, чем
Dm большого белка. Медленная диффузия должна приводить к низким значениям N*
и широким пикам. Тем не менее, уширение пика из-за медленной диффузии
компенсируется малым значением к (в соответствии с уравнением (9.5) в
предположении очень большого значения 5), и наблюдаемая ширина пиков для вирусов
аналогична таковой для белков. Третьим следствием размера вируса является его (вируса)
большой гидродинамический объем, что ограничивает вход вирусов в систему пор
сорбента. По этой причине вирусы взаимодействуют только с небольшой долей
обшей площади поверхности сорбента, и емкость колонки для них в 20—50 раз ниже,
чем для белков.
Для очистки вирусов были использованы ИОХ, ХГВ и металл-аффинная
хроматография [130|. При очистке вируса на этапе выделения (из клеточного лизата) чаще
всего, однако, используется ионообменная хроматография, поскольку она дает
приемлемый выход и чистоту |3|. В случае аденовирусов изоэлектрическая точка
поверхности IP « 6, поэтому анионообменная хроматография для их разделения
предпочтительнее. Добавки, такие как сахароза, магний и глицерин, которые используются
для стабилизации вируса, совместимы с условиями анионного обмена.
Для разделения аденовирусов использовались как сильные, так и слабые анионо-
обменные сорбенты на основе сшитого полистирол-дивинилбензола, гидрофильных
полимеров, полиакриламидные или поперечно сшитые носители на основе агарозы
и декстрана |5|. Для разделения вирусов использовались и монолиты |3|, хотя эти
колонки не являются коммерчески доступными в размерах, которые требуются
для промышленной очистки. рН подвижной фазы должен быть примерно на две
единицы выше изоэлектрической точки вируса, чтобы избежать агрегации вирусов.
Поэтому обычно очистку аденовирусов проводят при рН между 7,5 и 9. Как правило,
используются градиенты хлорида натрия с концентрацией соли 0—0,3 М при вводе
образца и > 0,6 М при его элюировании. В исследовании зависимости
эффективности статического связывания от ионной силы было обнаружено, что емкость
колонки проходит через максимум вблизи значения 0,3 М. Рекомендуется вводить образец
на колонку при концентрации хлорида натрия 0,3 М или чуть выше, чтобы можно
было вымыть примесные белки из колонки на этапе ввода |3|. Повышения
температуры колонки следует избегать, чтобы минимизировать любую потерю вирусной
активности.
Преимущество ИОХ для очистки аденовируса заключается в ее способности
отличать агрегированные и разрушенные формы от интактного вируса [130|. Тем
не менее, отделение пустых капсидов от интактного вируса не всегда гарантировано
|5, 130|. Разрешение раннее элюирующегося аденовируса р53 и поздновыходящей
ДНК показано на рис. 13.38. Более подробная информация о разделениях вирусов
приводится в [3|.
Глава 13. Разделение биополимеров и синтетических полимеров
вирус
(ACN53)
30 мин
Рис. 13.38. Разделение аденовирусов методом анионообменной хроматографии.
Условия разделения: колонка Fractogel DEAE-650 М 50x6,6 мм; градиент
300—600 мМ NaCI (50 мМ Трис, рН-8,0, 2 мМ MgCI2 и 1% сахарозы)
за 10 мин 1130]
13.8. Эксклюзионная хроматография (ЭХ)
В ЭХ соединения разделяются в соответствии с размером их молекул в растворе в
результате исключения более крупных молекул из меньших по размеру пор сорбента,
которым упакована колонка |131|. В применении к синтетическим полимерам с
органическими растворителями в качестве подвижной фазы и полимерными
сорбентами (раздел 13.10.3.1), этот вид хроматографии называется гель-проникающей (ГПХ).
При разделении биополимеров, таких как белки, на гидрофильных сорбентах с
водными буферами в качестве подвижных фаз этот метод называют эксклюзионной
хроматографией (ЭХ) или гель-фильтрацией. Гель-фильтрация может использоваться
для препаративного выделения биологически активных веществ (зачастую совместно
с другими видами хроматографии в многостадийном процессе очистки) или в
качестве аналитического инструмента для получения информации о размере или форме
молекулы в растворе, степени агрегации исследуемого вещества или кинетики
связывания биополимера с различными лигандами.
Исторически для гель-фильтрации использовались мягкие гели, такие как декст-
ран, агароза или полиакриламид |132—134|. Эти носители подвержены сжатию
при увеличении скорости потока, и поэтому они выдерживают только поток
подвижной фазы, протекающий через колонку под действием гравитации или подаваемый
насосом низкого давления. Мягкие гели могут быть стабилизированы путем
поперечной сшивки, и в этом случае их можно использовать при более высоких
скоростях потока и при давлении в десятки атмосфер. Аналитическая гель-фильтрация
наиболее часто проводится с жесткими носителями: силикагелем, частицы которого
13.8. Эксклюзионная хроматография (ЭХ)
модифицированы гидрофильным покрытием, или поперечно-сшитыми
органическими полимерами. Эти сорбенты механически стабильны при давлениях в несколько
сотен атмосфер или выше, и колонки, ими упакованные, могут использоваться
в ВЭЖХ- системах.
13.8.1. Процесс разделения в ЭХ
ЭХ является самым простым видом хроматографии, в котором удерживание зависит
только от относительного проникновения молекул разделяемого вещества в поры
неподвижной фазы и выхода из них (т.е. происходит «фильтрация» молекул).
Молекулы разделяются в зависимости от их размера в растворе (у полимеров, имеющих
одинаковое химическое строение и пространственную структуру, этот размер
коррелирует с молекулярной массой). В отличие от других режимов хроматографии, таких
как ОФХ или ИОХ, в которых растворенные вещества удерживаются благодаря
взаимодействиям с неподвижной фазой, в ЭХ (в идеальных условиях) разделяемые
вещества не взаимодействуют с неподвижной фазой. Молекулы, слишком большие,
чтобы проникнуть в любую из пор, элюируются в объеме подвижной фазы, равном
объему между частицами неподвижной фазы в колонке (Vq). Молекулы, достаточно
малые, чтобы свободно проникать во все поры, элюируются в объеме, равном сумме
объема, не занятого частицами неподвижной фазы, и объема всех пор (V/).
Молекулы промежуточных размеров входят в некоторые из пор в зависимости от их размера
или формы и выходят из колонки между Vq и Vq + V) Полный объем подвижной
фазы или мертвый объем Vm колонки может быть выражен как сумма объема между
частицами и объема пор:
Vm = V0 + Vi. (13.6)
Степень проникновения растворенного вещества в систему пор определяется
коэффициентом распределения KD вещества, связанным с его объемом алюирования VR
уравнением:
KD=^°. (13.7)
' i
Уравнения {13.6) и (13.7) могут быть объединены:
*л = ^о + W 0 3-8)
Из приведенного выше уравнения видно, что если молекулы слишком велики, чтобы
войти в поры, все они будут иметь KD= 0 и будут элюироваться вместе в объеме V0.
Точно так же все молекулы достаточно малые и свободно проникающие во все поры
будут иметь KD = 1 и выходить все вместе из колонки при Vm. Молекулы
промежуточного размера будут иметь значения К0 от нуля до единицы и будут разделены
в соответствии с размером. Молекулы, имеющие больший размер, будут выходить
из колонки раньше молекул меньшего размера.
Соотношение между размером молекул и объемом удерживания может быть
использовано для оценки молекулярной массы М анализируемого вещества.
Калибровочный график зависимости log M от объема удерживания (или А"д) будет иметь
приблизительно линейный сегмент между Vq и V/, как на рис. 13.39л (часть кривой,
показанная сплошной линией). Диапазон молекулярных масс образцов,
соответствующий этому линейному сегменту, называют диапазоном разделения или
фракционирования. Если график построен с использованием стандартных белков, формы которых
сходны с формой анализируемого белка, время удерживания белка (или объем
удерживания, как на рис. 13.396), будет соответствовать определенной молекулярной
массе на графике, что позволит оценить молекулярную массу анализируемого белка.
670 Глава 13. Разделение биополимеров и синтетических полимеров
\ Полное
\проникновение
l
-нм <
-»■
Объем ►
,_ KD = 0.0 0.4 0.В 1.0
UUUL
i ■ ■ * i
о v0 vm
Объем ►
Рис. 13.39. Гипотетическая калибровочная кривая в ЭХ (и) и хроматограмма (б) |135|
Зависимость между log M и Ад почти линейная для значений Ад между 0,2 и 0,8, но
на концах графика наблюдаются криволинейные участки (как на калибровочной
кривой ЭХ на рис. 13.39а). Теоретически гель-фильтрационные колонки с порами
одинакового размера обеспечат разделение веществ, молекулярная масса которых
отличается на 1,5 порядка. На практике, однако, этот диапазон разделяемых
молекулярных масс будет охватывать примерно два порядка или чуть больше, так как
размер пор в конкретном материале носителя будет иметь некоторый разброс.
Хотя ЭХ часто используется для оценки молекулярной массы белка, следует
понимать, что удерживание на самом деле определяется гидродинамическим диаметром
молекулы растворенного вещества. Этот диаметр лишь косвенно связан с
молекулярной массой. Гидродинамический диаметр молекулы белка (или другой молекулы)
связан с его радиусом, вычисленным по формуле Стокса1, который может
изменяться в зависимости от степени гидратации вещества и формы его молекулы. Два белка
с одинаковыми молекулярными массами, но имеющие молекулы разной формы
(например, белки со сферическими молекулами по сравнению с белками, имеющими
форму яйца или цилиндра, нативные белки по сравнению с денатурированными)
будут иметь разные гидродинамические диаметры и, следовательно, существенно
отличающиеся объемы удерживания (рис. 13.40). Для получения точных оценок молеку-
1 Гидродинамический радиус рассчитывается, исходя из предположения о сферической форме
молекулы, по величине ее коэффициента диффузии в жидкости по формулам Стокса и
Эйнштейна. — Прим. перев.
Полное
исключение
log M
13.8. Эксклюзионная хроматография (ЭХ) 671
Гидродинамический диаметр молекулы определяется
как сфера с диаметром равным длине молекулы
Сфера Цилиндр
ММ = 10000 Да ММ = 10000 Да
Рис. 13.40. Сопоставление «размера» и формы молекулы 1135|
лярной массы с использованием гель-фильтрации необходимо, чтобы молекулы
белков, используемые для построения калибровочного графика, и молекулы
анализируемых белков имели схожие формы. Альтернативный подход заключается в
проведении калибровки и анализа в денатурирующих условиях, так чтобы и
калибровочные белки и анализируемые принимали конформацию статистического клубка, и их
время удерживания лучше коррелировало бы с молекулярной массой.
13.8.2. Колонки для ЭХ
Сорбенты колонок, используемых для ЭХ, должны быть как можно более
инертными, чтобы свести к минимуму любые взаимодействия с анализируемым веществом
(эти взаимодействия могут свести на нет взаимосвязь между молекулярной массой
растворенного вещества и удерживанием в ЭХ). Для гель-фильтрации это
достигается использованием гидрофильных носителей, взаимодействующих с водной
подвижной фазой сильнее, чем с растворенными белками. Объем пор в сорбенте должен
быть как можно больше, а сам носитель должен быть механически устойчивым
при требуемых скорости потока и давлении. Для разделения белков разной
молекулярной массы могут понадобиться сорбенты с различными размерами пор. Меньшие
поры используются для небольших молекул, большие поры — для больших молекул.
Если образец содержит белки с существенно различающейся молекулярной массой,
то две колонки с порами разного размера могут быть соединены последовательно
(порядок не важен). Чтобы диапазон фракционирования был максимальным,
подключенные колонки должны иметь сорбенты с порами, размер которых отличается
примерно в 10 раз. Кроме того, обе колонки должны быть сопоставимы с точки
зрения эффективности или значений Н (раздел 2.4.1).
13.8.2.1. Сорбенты
Для колонок ЭХ используются два типа сорбентов: гидрофильные привитые силикаге-
ли и гидрофильные органические полимеры. Силикагель является наиболее широко
используемым материалом для ВЭЖХ-сорбентов из-за его механической стабильное-
672 Глава 13. Разделение биополимеров и синтетических полимеров
ти, приемлемой пористости и широкого диапазона размеров пор. Тем не менее,
поверхность частиц силикагеля сильно взаимодействует с белками, поэтому он должен быть
модифицирован. Наиболее распространенный способ такой модификации — проведе
ние реакции между поверхностными силанолами силикагеля и органическим силаном
для образования ковалентно связанного покрытия диольного типа или сходного
с углеводами. Ограничением для ЭХ-сорбентов на основе силикагеля является их
нестабильность в щелочных условиях. Силикагель растворяется при значениях рН вы
ше 8, что приводит к уменьшению срока службы колонки. Один из производителей
(колонки Zorbax GF от Agilent Technologies) использует циркониевое покрытие для
стабилизации силикагелевой подложки при значениях рН выше 8.
Из-за ограничений, связанных со свойствами силикагеля, несколько
производителей предлагают колонки для гель-фильтрации на основе гидрофильных
органических полимеров. К ним относятся полиметакрилатные носители, запатентованные
гидрофильные полимеры, и полужесткие поперечно сшитые агарозы и декстраны.
Эти колонки более стабильны при высоких рН, но менее эффективны и хуже
выдерживают высокое давление.
13.8.2.2. Размер пор и пористость
Коммерчески доступны высокоэффективные носители для ЭХ с порами размером
от 10 до 400 нм. Следует выбирать сорбент с порами такого размера, чтобы
анализируемые вещества элюировались с К0 в диапазоне 0,2 < Кр < 0,8. Для этой цели
производители в описании продукта дают калибровочные графики; см. пример на
рис. 13.41 для двух разных гель-фильтрационных колонок (по оси х на рис. 13.41
отложены времена удерживания анализируемых белков при указанном расходе).
В этом случае приблизительный диапазон фракционирования колонки GF-250
составляет 4000 < М < 200 000 Да, а у колонки GF-450 10 000 < А/ < I 000 000 Да.
107
10е
105
м
ю4
103
ю2
6 в 10 12 14
Время удерживания (мин)
Рис. 13.41. Калибровочные кривые по белкам для колонок GF-250 и GF-450;
подвижная фаза 0,2 М фосфат натрия (рН 7,5) (неденатурирующие условия).
С разрешения Agilent Technologies, Inc.
-
-
-
"
GF-450
* j
*
l\ IqM
A *<L Thy
^ ^s? . p-Gal
>* ^Ччч,gG
>v \БСА
^V \Oval
^S* \^Myo
GF-250 \ I
~ 11
W NaN3
1 1 1 1 1 ■ 1 1
13.8. Эксклюзионная хроматография (ЭХ) 673
Соединив две колонки последовательно, мы получим гораздо более широкий
диапазон фракционирования: 4000 < М < 1 000 000 Да.
Колонки с узким распределением пор по размеру будут характеризоваться более
высоким разрешением в небольшом диапазоне молекулярных масс анализируемого
вещества, например, 1000 < М < 30 000 (для колонок одинаковых размеров). То есть
такие колонки будут иметь пологий калибровочный график и уменьшенный
диапазон фракционирования. Колонки с широким распределением пор по размерам будут
характеризоваться более широким диапазоном фракционирования и калибровочным
графиком с большим углом наклона. Разрешение образца или возможность
разделения двух анализируемых белков с разной молекулярной массой увеличивается
при уменьшении угла наклона графика зависимости log M от времени или объема
удерживания. Таким образом, выбор конкретной колонки для данного образца
представляет собой компромисс между разрешением и диапазоном разделения.
Пористость ЭХ колонки можно охарактеризовать ее фазовым отношением
(Vj/Vg), исходя из графика на рис. 13.39а. При прочих равных условиях разрешение
увеличивается при большем фазовом отношении. Мягкие сорбенты для ЭХ имеют
высокую пористость с фазовыми отношениями от 1,5 до 2,4 1136], в то время как
высокоэффективные ЭХ сорбенты имеют более скромные фазовые отношения от 0,5
до 1,5 |137|. Тем не менее, этот недостаток (более низкие фазовые отношения)
высокопроизводительных ЭХ-колонок более чем компенсируются их высокой
эффективностью и меньшей продолжительностью анализа. По мере увеличения диаметра пор
и их суммарного объема механическая прочность носителя уменьшается.
13.8.2.3. Диаметр частиц
Как и в других видах хроматографии, сопряженных с задержкой анализируемых
молекул на неподвижной фазе при их интерактивном взаимодействии (например,
в ИОХ или в ОФХ), уменьшение диаметра частиц в ЭХ повышает эффективность
колонки. Имеются сорбенты с диаметром частиц от 10 до 12 мкм для более грубых
разделений, таких, как препаративные, тогда как ЭХ-сорбенты с диаметром частиц
от 4 до 5 мкм могут быть использованы для методик, требующих более высокого
разрешения (например, для определения молекулярной массы или аналитического
разделения белковых смесей)1.
13.8.2.4. Увеличение разрешения в ЭХ
Разрешение в ЭХ контролируется двумя способами: (1) путем выбора колонки с
более пологим графиком зависимости log M от времени удерживания (при этом
существенно увеличивается селективность или значения а) и/или (2) путем увеличения
количества теоретических тарелок колонки N. Более высокие значения N могут быть
достигнуты путем использования сорбентов с более мелкими частицами, более
низких скоростей потока или при увеличении длины колонки.
Высокопроизводительные колонки для ЭХ с внутренним диаметром 8 мм обычно работают с расходом
1 мл/мин (или 0,5 мл/мин, если внутренний диаметр 4 мм). Поскольку
коэффициенты диффузии Dm белков и других биополимеров (или синтетических полимеров)
малы, N обычно значительно увеличивается при уменьшении расхода (раздел 2.3.1).
1 В настоящее время коммерчески доступны колонки для ЭХ белков, упакованные сорбентами
с диаметром частиц 1,7 мкм. Такие колонки могут работать в составе обычных ВЭЖХ-систем.
В системах УВЭЖХ эти колонки помимо увеличения разрешения, позволяют еще и сократить
время анализа до 3—5 минут (среднее время анализа на ЭХ-колонке, упакованной сорбентом с
частицами диаметром 5—10 мкм, составляет от 20 до 30 минут). — Прим. перев.
674 Глава 13. Разделение биополимеров и синтетических полимеров
13.8.3. Подвижные фазы для ЭХ
В отличие от интерактивных видов хроматографии, в которых подвижная фаза
выступает в роли активного участника процесса разделения, подвижная фаза в ЭХ
просто носитель, транспортирующий молекулы разделяемых веществ через колонку.
Подвижная фаза подбирается таким образом, чтобы анализируемое вещество оставалось
растворенным и сохраняло свою конформацию (например, нативную и не
денатурировало). Кроме того, подвижная фаза должна минимизировать взаимодействие
сорбента и анализируемого вещества и как можно больше продлить время жизни
колонки. Таким образом, подвижная фаза может содержать добавки, подавляющие
нежелательные взаимодействия образца с подложкой сорбента или связанной с ней
неподвижной фазой. Эти взаимодействия могут быть электростатическими по своей
природе, и в случае сорбентов на основе силикагеля остаточные ионизированные си-
ланолы на его поверхности часто создают отрицательный заряд. Для положительно
заряженных анализируемых молекул, таких как основные белки, ионизированные
силанолы могут приводить к увеличению удерживания вследствие катионного
обмена, и поэтому молекулы вещества будут элюироваться позже, чем это
предусматривает механизм «идеальной» ЭХ. Для отрицательно заряженных растворенных веществ,
таких как кислотные белки и нуклеиновые кислоты, исключение их ионов может
привести к более раннему элюированию, чем прогнозировалось. В крайних случаях
растворенные вещества могут элюироваться в объеме, превышающем V (при
ионном обмене), или в объеме меньше Vq (при исключении ионов). Второй причиной
неидеального поведения анализируемых веществ в ЭХ является их гидрофобное
взаимодействие с сорбентом, приводящее к увеличению удерживания.
Нежелательные взаимодействия образца с сорбентом часто можно свести к
минимуму, регулируя концентрацию соли: увеличение ионной силы уменьшает
электростатические взаимодействия, а понижение ионной силы уменьшает гидрофобные
взаимодействия. Таким образом, чтобы избежать неидеального поведения образца, требуется,
как правило, промежуточная ионная сила. Типичная подвижная фаза для
гель-фильтрации — 100 мМ фосфата калия + 100 мМ хлорида калия (рН 6,8). Гидрофобные
взаимодействия также можно уменьшить, добавив небольшое количество (например,
5—10%) органического растворителя, такого, как метанол, этанол или глицерин.
13.8.4. Соображения по эксплуатации
После выбора соответствующей колонки, подвижной фазы и скорости потока,
для успешного разделения в ЭХ могут потребоваться корректировки массы и
концентрации образца. Могут быть использованы добавки к подвижной фазе, такие как
ПАВ и соли, для поддержания растворимости образца или для подавления
нежелательных взаимодействий образца с сорбентом. И наоборот, такие взаимодействия
могут быть использованы для достижения желаемого разрешения (раздел 13.8.4.4).
13.8.4.1. Емкость колонки
Емкость колонок в ЭХ относительно невелика по сравнению с интерактивными
видами хроматографии вследствие того, что растворы образцов с высокой
молекулярной массой могут иметь высокую вязкость. Такие растворы могут привести к
нежелательному искажению пика и его уширению для образцов, имеющих слишком
высокую концентрацию или наносимых в большом объеме. Поэтому требуется либо
разбавить образец, либо уменьшить его объем. Простое эмпирическое правило
гласит, что объем образца, вводимого в колонку должен быть < 2% от объема колонки
для образца с молекулярной массой 10000 Да. Этот объем будет больше для образцов
13.8. Эксклюзионная хроматография (ЭХ) 675
с более низкой молекулярной массой и меньше для образцов с более высокой
молекулярной массой. Типичная аналитическая ЭХ-колонка размером 300 х 8,0 мм имеет
Vm = 10—II мл. Значит, максимальный объем вводимого образца должен быть
примерно 200 мкл. Поскольку количество образца в ЭХ ограничено, в основном,
объемом и вязкостью (которая увеличивается с ростом концентрации), предел массы
для образца белка, разделяемого на колонке 300 х 8,0 мм, будет приблизительно I —
2 мг. При больших массе или объеме наносимого на колонку образца разрешение
может ухудшиться. Количество и объем образца будут пропорциональны объему
колонки и будут расти или уменьшаться с увеличением или уменьшением полного
объема колонок.
13.8.4.2. Использование денатурирующих условий
Оценки молекулярного веса анализируемых нативных биополимеров при разделении
в условиях гель-фильтрации (как на рис. I3.4I) могут быть ошибочными из-за
различий в форме молекул. Такие различия менее вероятны для денатурированных
образцов, что предполагает использование денатурирующих условий для калибровочных
стандартов и анализируемых молекул, имеющих любую форму в нативной конфор-
мации. Добавление денатурирующих агентов, таких как 4—6 М гидрохлорида гуани-
дина, 4—6 М мочевины или 0,1 — ]% додецилсульфата натрия (ДСН), в подвижную
фазу можно использовать для перевода калибровочных стандартов и анализируемых
веществ в конформацию статистического клубка. Денатурирующие условия
уменьшают диапазон фракционирования гель фильтрационных колонок из-за увеличения
гидродинамических диаметров белков | !38|. Кроме того, ПАВ, такие как ДСН, могут
прочно связываться с сорбентом и плохо отмываться; поэтому желательно выделить
отдельную колонку для каждой методики такого рода (или использовать неудержива-
емые денатурирующие агенты, такие как мочевина или гуанидин). Высокие
концентрации хаотропных агентов, таких как мочевина и гидрохлорид гуанидина, в
подвижной фазе (для денатурирующих условий) могут вывести из строя уплотнения насоса
и инжектора. Никогда не следует оставлять систему ВЭЖХ после использования
непромытой.
13.8.4.3. Калибровка колонки
При установке новой гель-фильтрационной колонки следует определить значения Vg
и Vm с помощью соответствующих маркеров. Значение Vq можно измерить путем
введения большого биополимера, молекулярная масса которого превышает пределы
исключения сорбента. Часто используется, к примеру, высокомолекулярная ДНК
(например, ДНК тимуса теленка)1. Голубой декстран, используемый для измерения
Kq колонок, упакованных мягкими гелевыми сорбентами, может давать ошибочные
значения, если его использовать при калибровке некоторых
высокопроизводительных ЭХ-колонок из-за гидрофобного связывания с сорбентом, которое приводит
к увеличению удерживания. Значение Vm определяется с помощью очень
гидрофильных малых молекул, которые можно обнаружить с помощью УФ-детектора.
Популярными маркерами являются цианокобаламин (витамин BI2), глицилтирозин или
п-аминобензойная кислота |137|2.
Взаимодействия с сорбентом, способствующие отклонениям от механизма ЭХ,
можно оценить с помощью низкомолекулярных зондов [139, 140|, которые элюиру-
' На самом деле, чаще всего для калибровки используется не ДНК, а коммерчески доступные
наборы белков и пептидов. — Прим. перев.
2Можно также использовать кофеин или просто отфильтрованный кофе или 1% раствор
ацетона в воде — он быстро испаряется, но легко доступен. — Прим. перев.
676 Глава 13. Разделение биополимеров и синтетических полимеров
ются при Vm. Катионообменные взаимодействия определяются по завышению
объема удерживания аргинина или лизина, когда t% > Vm. Исключение ионов проявляется
в слишком раннем элюировании цитратов или глутаминовой кислоты {/д > Vm).
Гидрофобные взаимодействия могут быть обнаружены по завышению объема
удерживания фенилэтилового или бензилового спиртов 1ц > Vm. Поскольку белки могут
денатурировать при более высоких температурах, удержание в ЭХ может зависеть
и от температуры.
13.8.4.4. Использование неидеальных условий разделения
Хотя отклонения от идеальных условий разделения ЭХ могут помешать точным
оценкам молекулярной массы, взаимодействия, вызывающие эти отклонения, также
могут приводить к изменениям относительного удерживания разделяемых веществ и
способствовать улучшению их разрешения, особенно в препаративных разделениях.
Вышеупомянутые подходы для подавления взаимодействий образцов с сорбентом
(раздел 13.8.3: изменение концентрации соли, добавление органических
растворителей, изменение рН) дают представление о том, как при желании можно усилить те
или иные взаимодействия с целью повышения разрешения.
13.8.5. Преимущества и недостатки ЭХ
Эксклюзионная хроматография дает некоторые преимущества, которые делают ее
привлекательной как для препаративного, так и для аналитического разделения.
Во-первых, ЭХ-разделения происходят относительно быстро: при работе с
аналитической колонкой 300 к 8,0 мм и скорости потока I мл/мин все анализируемые
вещества должны элюироваться в течение 10 минут. Во-вторых, вследствие того, что
строение неподвижной фазы специально ориентировано на устранение взаимодействий
с образцом, ЭХ-колонки обычно дают отличные выходы и сохраняют биологическую
активность образцов. В-третьих, все разделения выполняются в изократических
условиях, что в общем случае удобно.
ЭХ имеет и некоторые недостатки. Во-первых, разрешающая способность у нее
довольно скромная по сравнению с интерактивной хроматографией. Максимальное
количество полностью разрешенных пиков при разделении обычно не превышает
5—10, в то время как градиентная ОФХ позволяет разрешить несколько сотен пиков.
Если гель-фильтрационная колонка имеет диапазон фракционирования от 10
до 500 кДа, это означает, что на ней можно разделить два белка, молекулярные
массы которых отличаются в два раза. Таким образом, аналитическая ЭХ полезна
только для образцов, которые содержат ограниченное количество компонентов с
существенно различающимися молекулярными массами. Вторым недостатком ЭХ является
ее низкая емкость по объему и массе образца. Вследствие этих двух ограничений, ЭХ
с большей вероятностью будет использоваться в качестве одного из последних шагов
в схеме очистки. Третий недостаток ЭХ — короткое время жизни сорбентов,
особенно сорбентов на основе силикагеля. При работе с водными буферами при
нейтральном рН срок службы ЭХ-колонки обычно короче, чем у ОФХ-колонки на основе
силикагеля, работающей в водно-органических подвижных фазах. Последнее
ограничение ЭХ - это точность оценок молекулярной массы, которые обычно весьма
приблизительны. Электрофорез в полиакриламидном геле в присутствии ДСН
и масс-спектрометрия дают более точные значения, но первый из этих методов
трудоемок, а второй дорог. Однако, при использовании ЭХ-колонки в комбинации
с лазерным детектором светорассеяния (и детектором, чувствительным к
концентрации), можно точно определить молекулярную массу |141|.
13.8. Эксклюзионная хроматография (ЭХ)
13.8.6. Применение ЭХ
ЭХ может быть использована для разделения молекул по размеру и определения их
свойств, а также для препаративного выделения очищенного вещества из снеси.
13.8.6.1. Аналитические приложения
Аналитические приложения ЭХ включают оценку молекулярной массы, мониторинг
или определение конформации и степени агрегации белка, а также исследование
взаимодействий между рецепторами и лигандами. Как обсуждалось выше,
удерживание биополимеров в ЭХ определяется размером и формой их молекул. Чтобы
получить точные оценки молекулярной массы, колонка должна быть откалибрована
по стандартам, молекулы которых имеют форму, сходную с формой молекул
анализируемого вещества, или же молекулы анализируемых веществ и стандартов должны
быть переведены в конформацию статистического клубка (денатурированную
форму) — и находиться в ней во время анализа. ЭХ также может быть использована для
определения конформации белка и степени его агрегации. Что касается
конформации, то на рис. 13.42 обобщены результаты нескольких экспериментов по
определению конформации лизоцима в присутствии различных концентраций гидрохлорида
гуанидина в подвижной фазе [142]. В отсутствии гуанидина (0,0 М) белок имеет на-
тивную конформацию (N), и его объем удерживания 15,5 мл. При концентрации
гуанидина 6,2 М полипептидные цепи белка полностью разворачиваются (if), он дена-
туририрует. В денатурированном состоянии молекула белка имеет больший размер,
и, следовательно, ее удерживание уменьшается (объем удерживания 14,5 мл). При
средней концентрации гуанидина молекулы белка существует одновременно в свер-
У
\
л
/"/
А
6,2 М
4,7 М
^
-ч 4,4 М
'ЛЧ 4,1 М
Ч\ 3,9 М
м ...
N~~~y V 0,0 М
1 1 1
10
15
20 (МЛ)
Рис. 13.42. Использование ЭХ для наблюдения за конформацией лизоцима при
повышении концентрации гидрохлорида гуанидина в подвижной фазе. U,
развернутые (денатурированные) молекулы; Л/, нативные молекулы J142J
678 Глава 13. Разделение биополимеров и синтетических полимеров
Олигомер
/\ Димер
6 8 10 12 14 (мин)
Рис. 13.43. ЭХ-разделение мономерных, димерных и олигомерных форм рекомбинант-
ного гормона роста человека. Перепечатано из [143] с разрешения авторов
нутой (нативной) и развернутой (денатурированной) конформации. Иллюстрацией
использования ЭХ для мониторинга агрегации белка (рис. 13.43) является
определение степени агрегации гормона роста человека по количеству этого белка в
мономерной, димерной и олигомерной формах [143|. Поскольку биологическая активность
белка зависит от степени его агрегации, измерения последней важны для
определения качества белковых лекарственных препаратов (как в этом примере)1.
ЭХ также позволяет проследить за взаимодействиями рецепторов с лигандами
(например, белков с лекарствами) с помощью зональной хроматографии, при
использовании метода Хуммеля—Дрейера и фронтального анализа |144|. В зональной
хроматографии смесь белка и лиганда вводится колонку. Белково-лигандный
комплекс элюируется первым и отделяется от свободного лиганда. Количественный
анализ соотношения этих двух продуктов позволяет рассчитать константу связывания
(аффинности). Зональную хроматографию можно использовать в тех случаях, когда
диссоциация белково-лигандного комплекса происходит медленнее, чем процесс
хроматографии. Метод Хуммеля—Дрейера |145| заключается в том, что подвижная
фаза содержит лиганд, а в колонку вводится небольшой объем белка. На профиле
элюирования мы сначала увидим пик, соответствующий комплексу белок-лиганд,
за которым будет следовать впадина, соответствующая подвижной фазе с
пониженным содержанием лиганда2. Преимуществом метода Хуммеля — Дрейера является
то, что белок постоянно находится в динамическом равновесии со свободным лиган-
дом, и то, что требуется небольшое количество белка. Метод применим к белко-
во-лигандным комплексам с быстрой кинетикой диссоциации-ассоциации. При
фронтальном анализе в колонку вводят большой объем белка и лиганда. На профиле
элюирования видны плато, соответствующие свободному белку, комплексу
белок-лиганд, находящемуся в равновесии с диссоциированными компонентами, и
свободному лиганду. Фронтальный анализ позволяет определить константы
связывания в условиях, когда концентрация молекул известна и постоянна, но в этом случае
требуются большие количества образцов.
1 Хотя ЭХ наиболее часто используется для определения процента белка в агрегированном
состоянии, необходимо понимать, что у этого метода есть ограничения. Основным ограничивающим
фактором является сорбция агрегатов на сорбенте. Причинами этого могут быть ионные и/или
гидрофобные взаимодействия. Такие потери части введенного образца приводят к значительному
дефициту количества агрегированных белков, и поэтому необходимо тщательно следить за
массовым балансом процесса. — Прим. персе.
2 Впади на образуется из-за связывания определенного количества лиганда белком. — Прим. пе-
рев.
Мономер
13.9. Крупномасштабная очистка биомакромолекул 679
13.8.6.2. Препаративные приложения
Скорость и мягкие условия элюирования делают ЭХ удобным методом для быстрого
выделения биополимеров с высоким выходом и сохранением биологической
активности. Хотя ее разрешающая способность умеренна по сравнению с другими видами
хроматографии, ЭХ полезна для очистки целевых продуктов от высоко- и
низкомолекулярных компонентов. Водные буферы, используемые в качестве подвижных фаз,
обычно совместимы с последующими этапами очистки, такими как ИОХ и ОФХ.
Инертный характер сорбентов для гель-фильтрации позволяет использовать
подвижные фазы с такими добавками, как ПАВ и органические модификаторы, которые
могут быть необходимы для увеличения растворимости гидрофобных молекул,
например, мембранных белков.
Соли, буферные агенты и другие малые молекулы при разделении будут элюи-
роваться все вместе в полном объеме колонки, хорошо отделенные от биополимеров,
таких, как белки и нуклеиновые кислоты. Время разделения обычно ограничивается
несколькими минутами, поэтому с помощью ЭХ можно быстро обессоливать
биологические образцы. Для перевода образца из одного буфера в другой, целевой буфер
используется в качестве подвижной фазы, и растворенные вещества элюируются в новом
буфере, отделенные от зоны, в которой находится исходный буфер образца. Таким
образом, в многостадийной схеме очистки ЭХ можно использовать не только для
фракционирования, но и в качестве связующего звена между несовместимыми
непосредственно друг с другом хроматографическими этапами как, например, ХГВ и ИОХ.
13.9. Крупномасштабная очистка
биомакромолекул
Общие принципы препаративной жидкостной хроматографии (преп-ЖХ)
обсуждаются в главе 15 и в равной степени применимы как для выделения, так и для
очистки крупных биомолекул. В этом разделе мы дадим краткую информацию об очистке
биомакромолекул с помощью преп-ЖХ, а также приведем типичные примеры.
13.9.1. История вопроса
Хроматографическая очистка пептидов и белков в лабораторном масштабе ведется
в течение последних пяти десятилетий. Основные виды хроматографии
первоначально были ограничены анионобменной и катионообменной хроматографией и
гель-фильтрацией. С тех пор и другие виды хроматографии стали использовать
для выделения и очистки биомолекул, включая очистку биомолекул в
промышленном масштабе. Другого метода, который мог бы конкурировать с преп-ЖХ в
разделении близкородственных белков и других биомолекул, на сегодняшний день нет.
Продажи биомолекул, очищенных методом препаративной ЖХ, сегодня составляют
десятки миллиардов долларов в год. Количество этих продуктов и их долларовая
стоимость продолжают расти. В настоящее время коммерчески доступен широкий
ассортимент сорбентов или «смол» разной пористости и с разными размерами частиц
для различных типов разделений. Автоматизированные системы и колонки
диаметром метр и более позволяют проводить препаративные разделения в любом
желаемом масштабе. Как обсуждается в главе [5, и аналитическая, и препаративная
хроматографии основаны на сходных принципах. В этом разделе мы акцентируем
внимание на некоторых аспектах преп-ЖХ, актуальных для крупномасштабных
разделений биомакромолекул.
680 Глава 13. Разделение биополимеров и синтетических полимеров
Несколько факторов способствовали недавнему быстрому развитию
крупномасштабной преп-ЖХ биомолекул. В настоящее время признано, что молекулы белка
могут модифицироваться в процессе обработки, и эти модифицированные молекулы
должны быть удалены из конечной фармацевтической субстанции |146|.
Высокопроизводительная преп-ЖХ — единственный практичный метод разделения этих
близкородственных видов белков в больших масштабах. Одним из первых примеров
таких разделений была очистка инсулина компаниями Eli Lilly (США) и Novo Nordisk
(Дания), использовавших крупномасштабную гель-фильтрацию и ИОХ |147|.
Последовавшее усовершенствование оборудования и сорбентов сделало этот подход более
универсальным, более привлекательным, и он нашел еще более широкое
применение [148]. Технология кипящего адсорбционного слоя позволила удалять твердые
частицы и растворимые примеси за одну стадию |149|. Кроме того, стали доступны и
другие режимы разделения |150, 1511. Наконец, появление технологии рекомбинант-
ных ДНК в конце 1970-х годов позволило производить большие количества белков
человека в клетках микроорганизмов или животных, что не только привело к
появлению продуктов с высокой добавочной стоимостью, но и к усложнению их хрома
тографической очистки.
Рекомбинантный инсулин человека (rh-инсулин; также называемый
человеческим инсулином, полученным из бактериальных клеток) был первым продуктом,
полученным по технологии рекомбинантных ДНК и одобренным FDA. Процесс
производства приводил к появлению примесей нескольких близкородственных
производных инсулина, которые могли быть отделены с помощью аналитической ОФХ,
но ни одним из методов преп-ЖХ, существовавших в то время [152, 153|. Позже был
разработан процесс ВЭЖХ-очистки инсулина в коммерческом масштабе (см.
раздел 13.9.2). В последующие годы препаративная очистка пептидов и
низкомолекулярных белков методом ОФХ стала наиболее популярной [152, 154]. Однако очистку
некоторых белков (например, моноклональных антител) нельзя провести с помощью
ОФХ, потому что они не переносят органических растворителей. Более удачными
оказались альтернативные хроматографические режимы, в которых используются
водные подвижные фазы (например, афинная и металл—аффинная хроматографии,
хроматография на гидроксиапатите, а также ХГВ |151|).
Очистка инсулина методом высокоэффективной ОФХ послужила примером
для получения многих других рекомбинантных продуктов, включая гормон роста
|155|, эритропоэтин |156], гирудин [157|, цитокины |158, 159|, инсулиноподобный
фактор роста I (ИФР-1) |160| и гранулоцитарный колониестимулируюший фактор
(Г-КСФ) |161|. Вскоре стало очевидным, что препаративная
высокопроизводительная ОФХ является мощным и универсальным методом очистки пептидов и
низкомолекулярных белков. Стоимость продуктов, очищенных методом
высокопроизводительной ОФХ, по оценкам, составляет около трети общего объема продаж
биотехнологической продукции [162].
13.9.2. Очистка рекомбинантного инсулина человека
в промышленном масштабе
Процесс очистки рекомбинантного инсулина человека представляет собой редкий
случай, когда подробности хроматографической очистки, а также история ее
разработки были опубликованы [162|, и поэтому полезно рассмотреть детали процесса
разделения, различные аспекты которого могут быть структурированы следующим
образом:
• цели очистки;
• неподвижная фаза;
13.9. Крупномасштабная очистка биомакромолекул 681
• упаковка колонки;
• стабильность продукта и сорбента/колонки;
• подвижная фаза;
• разделение;
• регенерация колонки;
• очистка в лабораторном масштабе;
• масштабирование;
• очистка в промышленных масштабах;
• анализ продукта.
13.9.2.1. Цели очистки
Целью ОФХ-очистки был продукт с чистотой > 97,5% и с выходами > 75%.
Первоначально рассматривались два разных процесса производства инсулина в
микроорганизмах: (I) процесс с получением двух цепей, в котором А- и В-цепи инсулина
получали в отдельных ферментациях (отдельных биореакторах) и затем объединялись, и
(2) процесс с получением одной цепи, в котором проинсулин получали в
ферментации, а затем превращали в инсулин обработкой ферментом протеазой. Для
обработки сырья, получающегося в обоих производственных процессах, был разработан
универсальный способ очистки, при условии, что инсулиноподобные примеси
не превышали 20%. В каждом случае были использованы ИОХ, ЭХ и
высокопроизводительная ОФХ. Разделение с помощью ОФХ мы обсудим далее. Требовалось,
чтобы подвижная фаза не влияла на стабильность инсулина и была совместима с
другими этапами процесса очистки. Допустимая цена этого этапа хроматографической
очистки за фунт инсулина была определена на основе ожидаемых продаж, которые
прогнозировались на уровне одной тонны в год.
13.9.2.2. Неподвижные фазы
Неподвижная фаза была выбрана после проверки сорбентов от пяти производителей.
С помощью упаковки в колонку размером 150x9,4 мм были проверены сорбенты
с привитыми алкильными цепями различной длины, различными диаметрами частиц
и размерами пор. Наилучшие результаты были получены с лигандами Cg или С)8
при диаметрах частиц < 12 мкм и размерах пор от 12 до 15 нм Эти носители
оказались менее хрупкими и легче упаковывались, чем частицы с порами > 30 нм. В
конечном итоге был выбран сорбент Zorbax™ Process Grade С8, исходя из того, что
всем упомянутым критериям он соответствовал, его можно было купить в
достаточном количестве, и производитель продемонстрировал воспроизводимость его свойств
от партии к партии.
13.9.2.3. Упаковка колонки
Для осуществления требуемого разделения в промышленном масштабе,
препаративные колонки должны были иметь то же количество теоретических тарелок /V, что и
аналитические. Колонки для разделения в лабораторном масштабе были упакованы
суспензией неподвижной фазы под высоким давлением'. Более крупные колонки,
содержащие от 5 до 50 кг сорбента, упаковывали с использованием аксиальной
компрессии под давлением 750 фунтов на квадратный дюйм (~ 52 бар). Для определения
Л' колонки проводилось тестирование низкомолекулярными стандартными
образцами. Полученные значения N составили от 30000 до 40000 ТТ/м для обычных
лабораторных колонок и от 45000 до 55000 ТТ/м для колонок большего размера,
упакованных с аксиальной компрессией. Последние сохраняли свою работоспособность
' Общепринятая технология упаковки аналитических колонок. — Прим. перев.
682 Глава 13. Разделение биополимеров и синтетических полимеров
в течение более длительного времени, благодаря сведению к минимуму пустот и
каналов, обычно возникающих при деградации и/или усадке неподвижной фазы.
13.9.2.4. Стабильность продукта и колонки
Очистку инсулина лучше всего проводить при рН подвижной фазы от 3,0 до 4,0, так
как растворимость инсулина уменьшается до минимума в его изоэлектрической
точке 5,4 1163]. В кислотных условиях инсулин дезамидируется с образованием А-21-мо-
нодезамидоинсулина |164|. Однако этой нежелательной реакции удалось избежать,
так как на разделение в ОФХ требовалось только несколько часов, и очищенный
инсулин можно было быстро извлечь из подвижной фазы кристаллизацией в виде соли
цинка |163|. При рН 7 или выше образуются нежелательные В-3-монодезамидные
производные. Колонка была стабильной в диапазоне рН от 2,0 до 8,0.
13.9.2.5. Состав подвижной фазы
Разделение инсулина и двух представляющих интерес примесей показано на рис. 13.44
в подвижных фазах, рН которых является либо кислотным {рис. 13.44а), либо
слабощелочным (рис. 13.446). Лучшее разрешение было получено в кислотных условиях, где
все примеси элюируются после инсулина (что очень желательно!); в слабощелочных
условиях примеси элюируются как до, так и после инсулина. Было опробовано
несколько кислот для приготовления подвижной фазы А: уксусная, муравьиная, пропио-
новая и фосфорная кислоты. Была выбрана 0,25 М уксусная кислота, так как было
обнаружено, что высококонцентрированный раствор инсулина (> 50 мг/мл) в уксусной
кислоте содержит его мономеры. Склонность инсулина к агрегации налагала
ограничения на состав подвижной фазы, потому что агрегация мешает разделению.
Было опробовано несколько В-растворителей, включая этанол, изопропанол,
ацетонитрил и ацетон. Этанол и изопропанол не обеспечили ни хорошего разрешения,
ни высокого выхода (50—60%). Кроме того, растворимость инсулина в изопропаноле
была низкой. С ацетоном выходы были еще ниже (< 50%) из-за осаждения инсулина.
Ацетонитрил обеспечил лучшее разделение, самые высокие выходы (75—85%) и не
мешал на стадии осаждения цинком. Ацетонитрил также был доступен в больших
количествах, и его можно было регенерировать перегонкой отработанной подвижной фазы.
По этим причинам он и был выбран в качестве В-растворителя.
13.9.2.6. Разделение
Было проведено сравнение изократического и градиентного элюирования инсулина.
С градиентным элюированием продукт может быть получен менее чем в одном объеме
колонки, в то время как при изократическом элюировании он выходит в двух и более
объемах. Градиентное элюирование было выбрано потому, что меньший объем
элюирования облегчает последующую обработку. Градиент от 15 до 30% ацетонитрила
обеспечивает удовлетворительную чистоту и выход в минимальном объеме подвижной фазы.
Разработать разделение со ступенчатым градиентом подвижной фазы с сопоставимой
производительностью не удалось. Емкость насыщения колонки (раздел 15.3.2.1) для
инсулина была примерно 85 мг инсулина/мл сорбента. Выход инсулина превысил 97%.
13.9.2.7. Регенерация колонки
Чтобы процесс был экономичным, колонка должна сохранять рабочие свойства в
течение многих разделений. Образцы клеточного происхождения обычно быстро
загрязняют хроматографические колонки из-за наличия в них (образцах) липидов,
нуклеиновых кислот, фрагментов клеточных стенок и клеточных мембран, сложных
углеводов и других клеточных компонентов. Загрязнение колонки может быть уме-
13.9. Крупномасштабная очистка биомакромолекул
i i i i i i i i
40 50 (мин)
Рис. 13.44. ОФХ-разделение rh-инсулина и его производных в (я) кислотной и
(б) основной подвижных фазах. Образец: 1, 7,5 мкг rh-инсулина и
по 1,3 мкг каждого производного инсулина; 2, А-21-дезамидоинсулин; 3,
)У-карбамоил-С1у-инсулин; 4, Л'-формил-СЛу-инсулин; 5, /V-карбамо-
ил-РЬе-инсулин; 6, димеры инсулина. Условия: колонка Zorbax™ Cg
250 х 3.5-мм; 35 °С; 1.0 мл/мин; градиенты: (а) подвижная фаза А
фосфатный буфер рН 2,1; подвижная фаза В 50% ацетон итрил/подвижная А;
(S) подвижная фаза А фосфатный буфер рН 7.3; подвижная фаза В 50%
ацетонитрил/подвижная фаза А |166]
ньшено при правильном планировании стадий, предшествующих ОФХ. Эти стадии
включают фильтрацию, осаждение и И ОХ. Было установлено, что после каждого
цикла выделения инсулина необходимо промыть колонку элюентом состава 60% аце-
тонитрил/буфер (50 мМ фосфат аммония; рН 7,4)'.
' В настоящее время основные производители инсулина используют обращенно-фазовые
сорбенты марок Kromasil и YMC, которые производятся большими единичными партиями (более
100 кг), отличаются воспроизводимостью и стабильностью, в том числе выдерживают регенерацию
щелочными растворами. — Прим. перев.
Глава 13. Разделение биополимеров и синтетических полимеров
13.9.2.8. Очистка в лабораторном масштабе
Процедура очистки инсулина в лабораторном масштабе была разработана на основе
экспериментов, описанных выше, включая ИОХ до ОФХ и гель-фильтрацию после
нее. На стадии ИОХ удалялось большинство белковых примесей, при этом
обеспечивалась защита ОФХ-колонки от возможного загрязнения. ОФХ-колонка, на которой
отделялись примеси производных инсулина, использовалась для «тонкой» очистки
продукта. Далее была проведена серия более масштабных разделений. На рис. 13.45
проиллюстрировано препаративное ОФХ-разделение. Виден пик основного
продукта, за которым следуют пики примесей, элюируемых после основного пика.
Разрешение продукта с этими примесями кажется плохим, но это не так. Анализ фракций
этого ОФХ разделения показал, что в центральной части основного пика фракции
в интервале от 3,4 до 4,3 объемов колонки содержат почти чистый инсулин (98,7%)
с выходом 82%, а дополнительные фракции (в интервале от 3,3 до 3,4 объемов
колонки перед основным пиком и от 4,4 до 5,4 объемов колонки после основного
пика) в совокупности с растворителем, выделенным из подвижной фазы,
использовавшейся для регенерации колонки, содержат еще 15% продукта (для повторного
разделения). Тогда суммарный выход инсулина равен 97%. Исходная чистота образца
для ОФХ-разделения составляла 91,5%. Этот пример иллюстрирует общее свойство
препаративных разделений: в отличие от аналитической хроматографии, хрома
тограммы преп-ЖХ могут наводить на мысль о плохом отделении целевого продукта
от примесей, но после того, как фракции проанализируют, отсортируют и
объединят, результат зачастую получается приемлемый.
основные фракции
побочные фракции
3 4
Объемы колонки
Рнс. 13.45. Препаративное разделение rh-инсулина. Колонка Zorbax™ Process Grade
CS, 10 мкм, 150 x 9,4 мм; образец 153 мг rh-инсулина из проинсулина;
градиент 17—29% ацетонитрила в 0,25 М уксусной кислоте в шести объемах
колонки; скорость потока 0,3 мл/мин. Фракции в интервале от 3,3
до 4,3 объемов колонки были объединены (основные фракции). Фракции
от 3,2 до 3,3 объемов колонки и от 4.4 до 5,4 объемов колонки, а также
белок, вышедший во время регенерации колонки (фракция не показана)
были объединены в дополнительные фракции |162|
13.9. Крупномасштабная очистка биомакромолекул 685
13.9.2.9. Масштабирование
Затем были проведены эксперименты по увеличению масштаба разделения с
использованием шести колонок, упакованных с аксиальной компрессией, с
последовательно возрастающим объемом сорбента от 10 мл до 80 л, как указано в таблицах 13.9 и
13.10. В каждом разделении вес инсулина, наносимого на колонку, был от 14 до 15 г
на 1 л объема колонки, скорость потока составляла 1,5 объема колонки/ч, а наклон
градиента был 2%/объем колонки. Незначительные изменения в наклоне
градиенте были необходимы для поддержания пригодности колонки, определяемой по
чистоте и выходу продукта. Скорости потока подвижной фазы были увеличены
пропорционально объему колонки при переходе от лабораторного до промышленного
масштаба. Чистота целевого продукта составила соответственно 98,5, 98,6 и 98,6% в
лабораторном, пилотном и промышленном масштабах, в то время как выход
продукта в основных фракциях составил 82, 79 и 83%. Может показаться примечательным,
что чистота, выход и объемы элюирования основных фракций оставались неизмен
ными от лабораторных до производственных масштабов при более чем 10
000-кратном увеличении объемов колонок, но так и должно быть при масштабировании
процесса, если оно проведено правильно (раздел 15.1.2.1).
Таблица 13.9. Размеры колонок, использованных в исследованиях масштабирования
очистки rh-инсулина
Размеры колонки, мм
150 х 9,4
300 х 22
500 х 50
450 х 150
500 х 300
500 х 450
Внутренний объем, л
0,01
0,12
0,59
8
35
80
Тип колонки
Неизменяемого объема
Неизменяемого объема
Неизменяемого объема
С аксиальной компрессией
С аксиальной компрессией
С аксиальной компрессией
Нагрузка колонки
инсулином,г
0,15
1J
8,9
120
525
1200
Источник: [162].
Таблица 13.10. Обобщенные данные по масштабированию процесса очистки rh-инсулина
Размер колонки
(объем)
150x9,4 мм (10 мл)
350 х 150 мм (6,2 л)
570 х 300 мм (40 л)
Масштаб
Лабораторный
Пилотный
Пилотный
Условия разделения
Скорость
потока, (ОК/ч)"
1,6
1,5
1,4
Градиент,
(%В/ОК/
2,2%
2,0%
2,1%
Нагрузка
колонки, (мг/мл)
13
t5
15
Продукт
Чистота
98,5%
98,6%
98,6%
Выход
82%
79%
83%
Источник: [162].
°ОК — объем пустой колонки.
^Изменение %В на один ОК. подвижной фазы; пропорционально крутизне градиента.
686 Глава 13. Разделение биополимеров и синтетических полимеров
13.9.2.10. Очистка в промышленных масштабах
Затем была проведена очистка rh-инсулина в промышленных масштабах для двухцепо-
чечного инсулина и проинсулина. Очистка двухцепочечного инсулина была более
сложной из-за того, что концентрация примесей, близких по структуре к
инсулину, была более чем на 20% больше. Условия разделения были следующие: колонка
48 х 30 см, масса загружаемого инсулина 500 г, градиент 17—30% ацетонитрила в
объеме 6 объемов колонки при скорости потока 1,4 объемов колонки/ч. (0,8 л/мин).
Чистота загружаемого инсулина была 80%, а чистота основных фракций после
разделения — 98,5%. Для инсулина, полученного из проинсулина, чистота исходного сырья
была выше (91%), что привело к получению очищенного продукта более высокой
чистоты (99,1%)- Соответственно, очистка образца весом I кг на колонке 48 х 45 см давала
сопоставимые результаты.
Фракции продукта после высокоэффективной ОФХ подвергали дополнительной
очистке ЭХ. Две партии rh-инсулина из каждого процесса затем сравнили с
инсулином, очищенным обычной хроматографией без использования
высокопроизводительной ОФХ. Результаты показали, что очистка с помощью ОФХ приводит к
повышению чистоты продукта, эквивалентной биологической активности и к приемлемо
низким уровням загрязнения эндотоксинами или белками клеток-продуцентов. В
целевом продукте не было обнаружено силоксанов (потенциальных продуктов распада
неподвижной фазы).
13.9.3. Общие требования к препаративной очистке
с использованием ЖХ
Необходимо определить целевые величины выхода и чистоты продукта, а также
методы определения этих величин (обычно это ОФХ). Требуется также оценить
потребность в продукте (например, тонн/год), определиться с планируемым временем
доставки и стоимостью очистки. Необходимо определить характеристики сырья
для очистки, а также определить ожидаемый диапазон содержания целевого
вещества в очищаемом сырье'. Полезно иметь стандарт целевого продукта, но можно и
получить его во время разработки процесса хроматографической очистки. Для
подтверждения идентичности и чистоты очищенного продукта могут быть полезны
масс-спектрометрия и другие физико-химические методы анализа. К процессу
хроматографической очистки могут быть предъявлены дополнительные требования,
если она предназначена для производства фармацевтических субстанций в
соответствии с действующей надлежащей производственной практикой (cGMP; [165,I66|) или
рекомендациями ISO 9000 |167].
Для продуктов белковой природы основным требованием является стабильность
целевого вещества во время разделения. Хотя, в принципе, белки могут быть
денатурированы в процессе ОФХ и ренатурированы впоследствии [168|, этот подход, как
правило, не одобряется для подобного рода разделений. Продукт должен не только
сохранить свою биологическую активность, но и не должно быть никаких обнаружи-
мых химических изменений, таких как окисление, дезамидирование или
расщепление пептидных связей. Методы масс-спектрометрии значительно упрощают задачу
обнаружения этих модификаций. Методы оценки стабильности белков при
различных условиях хорошо известны. Зачастую предпочтительнее оценить стабильность
1 В некоторых случаях необходимо определиться и с предельно допустимым содержанием
близкородственных примесей в сырье, поскольку отделение некоторых примесей может привести
к большой потере целевого вещества, а иногда и вовсе сделать очистку невозможной. — Прим. перев.
13. /0. Синтетические полимеры 687
белкового продукта до начала разработки методов хроматографической очистки,
чтобы не тратить время на исследования несовместимых с ним видов хроматографии
или подвижных фаз1.
13.10. Синтетические полимеры
Разделение синтетических полимеров обычно проводится для одной из двух целей:
(1) определение молекулярно-массового распределения образца (раздел 13.10.3.1),
и/или (2) определение различных типов или классов соединений в образце
(раздел 13.10.3.2). Методы, с помощью которых эти цели достигаются, принципиально
отличаются от других ВЭЖХ-разделений, описанных в этой книге. По этой и другим
причинам настоящий раздел представляет собой только введение в разделение
синтетических полимеров.
13.10.1. Базовые понятия и принципы
Синтетические полимеры — это созданные человеком макромолекулы; во всех
случаях они образуются из одного или нескольких разных мономеров, встречающихся
в молекуле много раз. Если полимеризуется один мономер, то в результате
получается гомополимер (рис. 13.46а). Короткоцепочечные полимеры такого вида называются
олигомерами. Например, р молекул этилена (мономера) реагируют друг с другом с
образованием полимера полиэтилена С2Н5—ICjH^—L_2—C^Hj. Если для создания
синтетического полимера используются два (или более) различных мономеров,
получается сополимер (рис. I3.46e). Гомополимеры могут отличаться по длине, как на
рис. 13.46a,ff,d. Длина молекулы выражается либо через степень полимеризации р
(где р — количество мономерных звеньев), либо через молекулярную массу
полимера. Наконец, гомополимеры могут отличаться по форме молекул или топологии.
Существуют линейные молекулы (рис. 13.46а-е), а также циклические (малые) олиго-
меры (рис. 13.46з) и сходные с ними по структуре полимеры. В зависимости
от процесса синтеза при производстве полимеров в линейной молекуле могут
случайно образоваться боковые цепи или они могут быть преднамеренно введены
(рис. 13.46 ж,к-м). Молекулы полимеров могут отличаться количеством боковых
цепей, их длиной и положением в структурах этих молекул. В случае сополимеров
весьма важной является последовательность мономеров. Если эта последовательность
1 К сожалению, авторы не осветили в этой главе такую тему, как хроматографическое
определение остаточных количеств реагентов, часто используемых при более или менее
крупномасштабных выделениях биополимеров. Для белковых или вирусных препаратов это белок А (или G),
малые количества белков клеток-продуцентов (так называемых host-cell proteins) и полимеры, такие
как Тритон Х-100, полиэтиленимин, Твин (полисорбат) 20 и 80, полоксамеры и основанные
на кремнеземах полимеры, предотвращающие вспенивание (Симетикон, например). Все
перечисленные выше соединения вызывают большой интерес контролирующих органов. Производители
обязаны показать, что вышеперечисленные соединения либо не присутствуют в лекарственных
формах и препаратах, либо имеются в меньших концентрациях, чем установлено правилами.
Некоторые методы для определения этих полимеров введены в USP, и этот источник должен быть
проверен в первую очередь. В общем случае, если полимер не имеет хромофоров и не может быть
дерикатизирован хромофором с более чем 95% выходом, необходимо использовать детектирование
ИДС или ДЗА. Популярной идеей является использование МС-детекторов для определения
концентрации остаточных молекул. Однако, высокая стоимость таких детекторов, а также
затруднения с переносом методов даже между одинаковыми детекторами, не говоря о переносе метода
с детектора одного производителя на другой, оставляют этот подход только большим компаниям
с достаточным уровнем финансирования лабораторий. — Прим. перев.
688 Глава 13. Разделение биополимеров и синтетических полимеров
О)
,<МКУО*Уа ,<К?ДЧУДЧ^ rgtfffgg)
"*^^^^^^т^^^ ^i^^^^F^^^F^^^ ^^^^^F^F^F^F
(б) (в) (г)
(в) Л\ (ж)
(") ^ W
(я) ^* (м)
Рнс. 13.46. Синтетические полимеры различной структуры, (а), (в) и (й), линейные
гомополимеры различной длины; (б), (в) и (г), линейные гомополимеры
с различными концевыми группами; (ж), полимеры с функциональными
группами, присоединенными вдоль цепи мономеров; (з), циклический го-
мополимер; (е), нерегулярный сополимер, (и), блок-сополимер; (л),
привитый сополимер; (к) и (м) раз нетленные гомополимеры с короткими и
длинными боковыми цепями, соответственно
определяется чисто статистическим процессом, образуются нерегулярные сополимеры.
В противоположность этим структурам блок-сополимеры (рис. 13.46м) — это
сочетание длинных последовательностей нескольких мономеров в молекуле сополимера.
Наконец, существуют привитые сополимеры (рис. 13.46л), в которых цепи,
образованные другим мономером, присоединены к скелету первичного полимера.
Особенности структуры полимера важны, поскольку они влияют на его свойства.
Более высокая молекулярная масса обычно приводит к большей прочности
полимера. Для полимеров, используемых в связующих и композиционных составах, таких
как клеи, герметики и покрытия (например, лакокрасочные) чрезвычайно важны
концевые и функциональные группы. Степень разветвленное™ полимеров обычно
влияет на их технологические свойства. Блок-сополимеры могут очень отличаться
по свойствам от нерегулярных сополимеров. Одно свойство структуры полимеров,
не отраженное на рис. 13.46, — это степень стереорегулярности цепи, обычно
называется изотактичностью. В атактическом полимере мономеры (заместители)
ориентированы случайным образом; в изотактическом (стереорегулярном) полимере все
мономеры (заместители) имеют одинаковую пространственную конфигурацию (или
разную, но чередующуюся с определенной периодичностью1, тогда полимер
называют синдиотактическим). Стереорегулярные полимеры обычно гораздо более склонны
к кристаллизации. В результате атактический полипропилен (обычно называемый
просто «полипропилен») представляет собой мягкий пластик, тогда как изотактиче-
ский полипропилен является твердым и прочным.
Поведение синтетического полимера в процессе хроматографии, как и его
свойства, обусловлено его молекулярной структурой. Это позволяет разделять полимеры
1 Мономеры (заместители) находятся по одну и другую стороны от плоскости цепи. — Прим. перев.
13.10. Синтетические полимеры 689
по молекулярной массе, химическому составу (содержанию функциональных групп),
степени стереорегулярности, степени разветвленности и так далее; разные методы
разделения указаны в таблице 13.11.
Таблица 13.11. Влияние молекулярной структуры синтетических полимеров на их хрома-
тографическое поведение
гпх
и-ЖХ
Хроматография в
градиенте температуры
Молекулярная
масса
•
о
■
Концевые
группы
а
О/в*
•
Химический
состав
о
•
о'
Стереоре-
гулярность
-
-
-
Разветв-
леннисть
о
в
-
Раздел
13.8
13.10.3.2
-
Примечания: • — сильное влияние; о — слабое влияние; незначительное влияние.
"Может наблюдаться значительное негативное влияние, если концевые или функциональные
группы сильно взаимодействуют с неподвижной фазой.
^Сильное влияние в изократической («критической») хроматографии полимеров. В
градиентной хроматографии это влияние обычно перекрывается влиянием молекулярной массы и,
в особенности, химического состава.
"Может наблюдаться значительное влияние, если в боковых цепях имеются различные или
дополнительные функциональные группы.
'Влияние может быть сильным, но этот метод обычно не используется для разделения
сополимеров.
Многие физические свойства синтетических полимеров важны для их поведения
в процессе хроматографии, но наиболее важна растворимость. Любой образец,
разделяемый методом жидкостной хроматографии, важно полностью растворить, так как
любые частицы или агломераты будут негативно влиять на разделение. Многие
полимеры с трудом растворяются полностью: к примеру, для растворения полиолефинов
(полиэтилена, полипропилена) нужна повышенная температура и растворитель с
высокой температурой кипения, такой, как трихлорбензол. Многие полярные полимеры
(полиэфиры, полиамиды, поликетоны) требуют специальных растворителей, таких,
как гексафторизопропанол. Также для растворения полимера может потребоваться
длительное время из-за сильных взаимодействий между его цепями (включая
физическое «переплетение» цепей друг с другом) и медленной диффузии больших молекул.
Другим важным моментом является детектирование синтетических полимеров
в процессе ВЭЖХ. Многие важные виды полимеров (полиолефины, полиакрилаты,
и т.п.) не имеют УФ-хромофоров. Поэтому в изократических разделениях
полимеров (особенно в ГПХ) обычно используется рефрактометр. Иногда могут дать
хорошие результаты детекторы, работающие на фиксированных длинах волн в
инфракрасной области спектра1. В градиентных разделениях широко используется
испарительный детектор светорассеяния (ИДС), но в этом случае количественный
анализ может быть осложнен2. Более перспективной альтернативой являются
1 Видимо, имеются в виду те полосы ИК-спектра молекулы, при которых поглощают ее
функциональные группы. — Прим. ред.
2При использовании ИДС площадь пиков анализируемого вещества не зависит линейно
от количества вещества, но эта проблема решается при обработке данных в Excel или подобных
программах, где легко найти квадратичную или кубическую функцию, хорошо описывающую
полученные калибровочные кривые. При этом по оси ординат откладывают концентрации
стандартов, а по оси абсцисс — площадь пика. — Прим. перев.
690 Глава 13. Разделение биополимеров и синтетических полимеров
детекторы заряженных аэрозолей (ДЗА)1. Хотя способы получения информации
о структуре (полярных) полимеров методами масс-спектрометрии были
значительно усовершенствованы с середины 1990-х годов, ЖХ-МС все еще редко
используется для этой цели. Некоторые распространенные синтетические полимеры,
типичные растворители, используемые в ГПХ, и наиболее часто используемые методы
детектирования приведены в таблице 13.12. Дополнительную информацию о
возможных детекторах для разделения полимеров можно найти в главе 3, главе 9
из книги [169| и в ссылках [170—172|.
Таблица 13.12. Наиболее распространенные синтетические полимеры, типичные
растворители, используемые в ГПХ, и часто используемые методы детектирования
Наиболее распространенные
синтетические полимеры
Полиэтиленоксид (полиэтиле-
ноксид, полиэтиленгликоль)
Полипропиленоксид (поли-
пропиленоксид, полипроли-
ленгликоль)
Полистирол
Поли (алкилакрилат) и поли
(алкилметакрилат)"
Алифатические полиэфиры,
алифатические полиамиды
Ароматические полиэфиры,
ароматические полиамиды
Полиэтен (полиэтилен), по-
липропен (полипропилен)
Типичные растворители
Вода
о
МеОН
•
ТГФ
•
■
ГФИП
•
о
ТХБ
•«
Методы детектирования
УФ
•
о
дпп
•
о
•
•
о
идс
Оя'г
.«,?
OV
Ов'г
ЛДС
•
•
•
О
•
Вискозиметрия
■
*
*
о
•
Сокращения: ГФИП — гексафторизопропанол; ТХБ — трихлорбензол; ДПП —детектор
показателя преломления; ИДС — испарительный детектор светорассеяния; ЛДС —лазерный
детектор светорассеяния.
Примечания: • указывает на применимость растворителя или детектора; о указывает на
ограниченную применимость растворителя или детектора,
'например, поли (метилакрилат) и поли (метилметакрилат).
Требует работы при высокой температуре (например, 150 °С).
"более реалистичный вариант для и-ЖХ, чем для ГПХ.
'детектор заряженных аэрозолей (ДЗА) может быть предпочтительнее.
13.10.2. Приемы анализа полимеров
Синтетические полимеры невозможно выделить как молекулы определенного вида.
Таким образом, они существенно отличаются от остальных образцов, анализируемых
с помощью ВЭЖХ. Синтетические полимеры представляют собой ряд молекул
разного размера со своим молекулярно-массовьш распределением (ММР)2. В то время
как для определения среднего значения молекулярной массы образца можно исполь-
'Они имеют более линейный отклик, нежели ИДС, и гораздо чувствительнее. Цдинственное,
что останавливает их широкое применение — это их высокая стоимость. — Прим. персе.
2 Молекулы синтетических полимеров неоднородны по молекулярной массе (в отличие от
биополимеров), а соотношение количеств молекул разной массы и есть ММР. — Прим. перев.
П. 10. Синтетические полимеры 691
зовать целый ряд методов, хроматографические методы, такие как ГПХ, позволяют
определить ММР, характеризующие среднечисловую и среднемассовую функции
распределения молекулярной массы |169|. Помимо ММР, сополимеры имеют
распределение и по химическому составу. Спектроскопические методы, такие как
инфракрасная спектроскопия с Фурье-преобразованием (ИК.ФП) и (особенно) ЯМР
могут дать детальную информацию о химическом составе анализируемого полимера,
а интерактивная ЖХ позволяет разделять различные типы химических молекул и
получать данные о распределении по химическому составу (РХС). Неполный обзор
ряда важных методов представлен в таблице 13.13. Видно, что ЯМР часто
упоминается в средней колонке таблицы (среди методов определения средних значений), тогда
как в правой колонке (характеризующей методы получения данных о
распределениях) преобладают хроматографические методы.
Таблица 13.13. Таблица методов для определения средней молекулярной структуры
н молекулярного распределения (структур) синтетических полимеров
Свойство полимеров
Молекулярная масса
Химический состав
Функциональность (концевые группы
и функциональные группы)
Регулярность цепей
Степень разветвленное™
Методы для определения
среднего значения
Осмометрия с
рассеиванием света
ЯМР, ИКФП, пиролиз,
ГХ-МС
Я М Р-титрова н ие
ЯМР
ЯМР"
Методы для определения
полного распределения
ГПХ, гидродинамическая
хроматография, седиментация,
ультрацентрифугирование
и-ЖХ (в основном в
градиентном режиме)
и-ЖХ (в основном в изокра-
тическом режиме)
Хроматография в
повышающемся градиенте температуры
Разделение по молекулярной
топологии
"Подходит для полимеров с относительно низкой молекулярной массой. Также ЯМР, в целом,
считается подходящим методом для определения коротких, но не длинных боковых цепей.
13.10.3. Методы жидкостной хроматографии для анализа
полимеров
13.10.3.1. Гель-проникающая хроматография (ГПХ)
Определение ГПХ дается в разделе I3.8. Этот вид хроматографии подробно
рассматривается в книге |169|. Она применяется для определения молекулярной массы или
молекулярно-массового распределения (ММР) синтетических полимеров; имеющая
такой же механизм эксклюзионная хроматография (ЭХ) или гель-фильтрация
применяется для исследования биополимеров. Между двумя этими приложениями
два основных отличия: если в случае синтетических полимеров ГПХ используется
для определения ММР |169], то в применении к биополимерам целью ЭХ является
оценка молекулярной массы образца1. Также отличаются и растворители,
используемые в качестве подвижной фазы. Для биополимеров обычно используются водные
подвижные фазы (ЭХ), а для синтетических полимеров — органические
растворители (ГПХ).
1 Молекулы биополимеров однородны по массе. — Прим. перев.
692 Глава 13. Разделение биополимеров и синтетических полимеров
В случае гомополимеров ГПХ может быть использована вместе с другими
методами характеризации полимеров, в частности с ИДС и вискозиметрией (для
сополимеров трудно точно интерпретировать полученные данные |170|). Статическое
светорассеяние может быть использовано для получения точной информации о сред-
немассовой молекулярной массе полимера в элюате ГПХ-колонки при условии, что
(1) мы знаем, как изменяется показатель преломления в зависимости от
концентрации полимера, и (2) детектор откалиброван. Кроме того, должна быть точно известна
концентрация полимера в элюате. Этого можно достичь использованием, к примеру,
ДПП в сочетании с ИДС.
13.10.3.2. Интерактивная жидкостная хроматография
В ГПХ условия выбираются таким образом, чтобы как можно полнее подавить
взаимодействия между анализируемым веществом и неподвижной фазой. В интерактивной
жидкостной хроматографии (и-ЖХ) эти взаимодействия используются для того чтобы
разделить молекулы по химическому составу или наличию тех или иных
функциональных групп. В то время как в и-ЖХ разделения полимеров во многом аналогичны
разделениям малых молекул методом ВЭЖХ, есть два важнейших отличия: (I) молекуляр-
но-массовое распределение полимеров (большое количество отдельных компонентов
в образце, отличающихся молекулярной массой), и (2) систематическое изменение
удерживания анализируемой молекулы при увеличении ее размера. Разделяемые
высокомолекулярные соединения, как правило, демонстрируют большее изменение к
для данного изменения %В, как показано в примерах на рис. 13.II для
нескольких пептидов и белков. Похожий пример для синтетических полимеров показан
на рис. 13.47а, который схематически иллюстрирует изменения удерживания при
изменении состава олигомеров и полимеров, отличающихся своими размерами или
степенью полимеризации р (т.е. количеством мономеров). Для олигомеров на рис. 13.47я
р равно 5—15; для полимеров р равно 25 и 100. У графиков для молекул большего
размера (с большими р) углы наклона увеличиваются. В конечном итоге получается, что
для больших полимеров остается очень узкий диапазон %В, в котором полимер может
быть смыт с колонки в изократическом режиме. По этой причине для анализа
полимеров методом и-ЖХ обычно используется градиентное элюирование.
Для разделений обычно используются линейные градиенты, которые
характеризуются (приблизительно) постоянными значениями градиентного фактора удерживания
к* (=к; см. раздел 9.1.3) для различных видов полимеров. В таких условиях время
выхода каждого пика в образце полимера будет соответствовать пересечению
графиков с горизонтальной линией, соответствующей данному значению к или к*
(см. рис. \ЪА1а). Для более высоких значений к* (соответствующих более длительным
градиентам; уравнение (9.5)) будут наблюдаться большие различия во времени
удерживания для соседних пиков (и соответственно, разрешение будет лучше) по сравнению
с разделениями, в которых используются кратковременные градиенты. И, наконец,
в достаточно быстром (кратковременном) градиенте (при достаточно малом значении
к*) все анализируемые молекулы покинут колонку в одно и то же время с одинаковым
временем удерживания, выйдя одним пиком. Такое поведение при разделении
низкомолекулярных полимеров в длительных и кратковременных градиентах показано
на рис. 13.475 и в. В длительном градиенте на рис. 13.476 удерживание индивидуальных
олигомеров отличается достаточно для того, чтобы происходило частичное разделение
образца. В кратковременном градиенте на рис. 13.47в все уже по-другому, и потому
наблюдается один пик, соответствующий выходу всех пиков в одно и то же время
(то есть, к ж I на рис. 13.47й).
Такое разделение как на рис. 13.47в или на рис. 13.47с при к - 1 иногда
называют псевдокритической хроматографией, чтобы отличить ее от (изократической)
13.10. Синтетические полимеры 693
100
Рнс. 13.47. Удерживание и разделение полимеров в интерактивной ЖХ (и-ЖХ). (а)
Иллюстрация поведения на колонке гомополимеров; (б) гипотетическая
иллюстрация элюирования низкомолекулярного гомополимера в
35-минутном градиенте; (в) такое же разделение как в (б), но время градиента
10 мин; (г) гипотетическая иллюстрация разделения смеси полимеров с
использованием и-ЖХ
хроматографии в критических условиях, описанной в разделе 13.10.3.3.
Псевдокритическая и-ЖХ (т.е. градиентное разделение) особенно полезна при разделении
полимеров в соответствии с химической структурой. Это проиллюстрировано
на рис. 13.47с" разделением двух полимеров разных видов. В этом примере
удерживание зависит от химической структуры, а не от молекулярного веса полимеров.
Такие псевдокритические условия могут быть достигнуты для (1) полимеров с
более высоким молекулярным весом и (2) при сокращении длительности градиентов.
В результате, и-ЖХ в сочетании с градиентными разделениями очень хорошо
подходят для определения распределения по химическому составу. На рис. 13.486 про-
694 Глава 13. Разделение биополимеров и синтетических полимеров
Рис. 13.48. Комплексная двухмерная жидкостная хроматография (ЖХ х ГПХ) клея
на основе сополимера, полученного днухстадийной полимеризацией
эмульсии стирола и метилметакрилата. (а) Разделение второго измерения:
ГПХ; (<Г) разделение первого измерения: ОФХ, линейный градиент от 65
до 100% за 170 мин; {в) 20-разделение. УФ-детектирование при 214 нм.
Подробности см. в [1701
иллюстрирован пример из практики, в котором высокомолекулярный сополимер
стирола и метилметакрилата очень хорошо разрешается по химическому составу
(обратите внимание на три пика на хроматограмме, соответствующие трем
химическим соединениям, содержащимся в образце). Дальнейшее обсуждение рис. 13.48
проведено в разделе 13.10.4.
13.10.3.3. Жидкостная хроматография в критических условиях
На основании рис. 13.47а можно предположить, что есть «критический» состав
подвижной фазы, при котором все олигомеры выходят из колонки в одно время.
Возможная выгода от работы в критических условиях или в условиях, близких к ним, состоит
в том, что влияние гомополимерных цепей на удерживание можно минимизировать,
а это позволяет разделить полимеры с различными структурными элементами (к
примеру, концевыми группами), как показано на рис. 13.47г, вне зависимости от их ММР.
Это наводит на мысль о том, что критические условия разделения могут быть
использованы для определения различий в функциональных группах полимеров |174|.
Для таких анализов особенно подходят нормально-фазовые разделения, потому что
функциональные (полярные) группы оказывают сильное влияние на удерживание.
13.10.3.4. Другие методы
Часть других методов ВЭЖХ для разделения и анализа полимеров представлена
в таблице 13.13. Очень большие полимеры могут быть разделены
фракционированием в потоке (МФП) или гидродинамической хроматографией (ГДХ). Эти методы
в этой книге не рассматриваются.
13. /0. Синтетические полимеры 695
13.10.3.5. Химический состав как функция размера молекулы
Сополимеру обычно свойственны распределения как по молекулярной массе, так и
по химическому составу. В зависимости от условий полимеризации, химический
состав может меняться с изменением веса полимера, а может и не меняться. Чтобы
обнаружить такую химическую гетерогенность, можно проводить ГПХ-разделение
с МС-детектором, который позволит получить информацию о химическом составе.
Такой комбинированный анализ дает усредненный (химический) состав в каждой
точке ГПХ-хроматограммы, то есть, для каждой молекулярной массы. Если только
один из двух мономеров может быть обнаружен с помощью УФ-детектора, то
комбинация УФ и другого, чувствительного к концентрации, детектора (например, ДПП)
может, в принципе, быть использована для того, чтобы следить за концентрацией
каждого мономера. Дополнительную информацию можно получить комбинируя ГПХ
с ИКФП или ЯМР-спектроскопией.
Хотя информация о зависимости химического состава от размера молекул может
быть очень ценной, даже самые малые ГПХ-фракции могут содержать набор
молекул, различающихся как по химическому составу, так и по молекулярной массе.
Различия в химическом составе могут привести к тому, что молекулы с разным
молекулярным весом будут иметь один и тот же молекулярный «размер» в растворе, как это
показано на рис. 13.49- Фракция, полученная при ГПХ с высоким разрешением
(прямоугольник на рис. 13.49), будет содержать молекулы с одинаковым
молекулярным «размером» (радиусом вращения Rg), но с разными молекулярными массами.
Часто бывает важнее знать распределение мономеров различного химического
состава, нежели просто усредненный химический состав. Также более важно знать и
распределение различных функциональных групп, нежели среднее количество
функциональных групп на молекулу полимера. Это будет особенно важно, если известно
(или предполагается), что химический состав или количество функциональных групп
в молекуле варьируется. Примером могут быть реакционноспособные олиголимеры,
использующиеся во многих составах герметиков, клеев и покрытий. Молекулы
без реакционноспособных (функциональных) групп не будут вступать в реакции,
молекулы с одной функциональной группой будут останавливать процесс
полимеризации локально, молекулы с двумя функциональными группами будут поддерживать
0.0251 1
Молекулярная масса (х10 )
Рнс. 13.49. Схематическая иллюстрация связи между молекулярным размером и
молекулярной массой для гомополимеров и сополимеров различного состава.
Линии показывают (снизу вверх): гомополимер А, сополимер АВ (75:25),
АВ (50:50), АВ (25:75), и гомополимер В
696 Глава 13. Разделение биополимеров и синтетических полимеров
полимеризацию, а молекулы с более чем двумя группами будут способствовать
формированию полимерных сеток (смол). Знания только среднего количества
функциональных групп в молекуле будет в таком случае недостаточно.
13.10.4. Разделение полимеров с помощью двумерной
хроматографии
В двумерной хроматографии (ЖХ х ЖХ; разделы 9.3.10, 13.4.5) образец подвергается
двум последовательным разделениям, при этом результаты первого разделения
сохраняются1. Для того, чтобы одновременно определить два взаимозависимых
распределения, таких как молекулярно-массовое распределение (ММР) и распределение
по химическому составу (РХС) (ММР х РХС), метод, разделяющий по молекулярной
массе (к примеру, ГПХ), должен быть скомбинирован с методом, разделяющим,
главным образом, по составу (это может быть и-ЖХ). Таким образом, комбинация
двух разделений (и-ЖХ х ГПХ) даст двумерную хроматограмму, которая
представляет собой анализ образца по молекулярной массе и по химическому составу. Пример
показан на рис. 13.48. Соответствующие одномерные разделения показаны для ГПХ
сбоку, а для и-ЖХ сверху на рис. 13.48. Хотя ни одно из одномерных разделений и
не дает приемлемого разделения образца, но соответствующее двумерное разделение
дает. Еще одно разделение (и-ЖХ х ГПХ) показано на рис. 13.50 для более сложного
образца: полиметилметакрилатов с фунциональными группами на концах
полимерных цепей. Горизонтальная ось времени для и-ЖХ указывает на химический состав
сополимера. Обратите внимание на стрелки, указывающие на количество
функциональных групп (показаны в верхней части рисунка). Вертикальная ось времени,
показывающая время ГПХ-разделения, соотносится с молекулярной массой образца.
0 1 2
\ \ \ Количество функциональных
V , ^ , ^ групп на молекулу
и-ЖХ (ч)
Рис. 13.50. Двумерное разделение полиметилметакрилага с фунциональными
группами на концах полимерных цепей. Пунктирными линиями показаны
области 20-хроматограммы, которые соответствуют молекулам без
функциональных групп, с одной и двумя функциональными группами; на них
указывают стрелки и цифры в верхней части рисунка |172|
1 В виде фракций, собранных на выходе из детектора, которые затем последовательно
разделяются вторым методом. — Прим. перев.
Литература 697
Двумерные хроматограммы, такие как на рис. 13.48 и рис. 13.50, могут дать
полезную качественную картину состава сополимера. Представляется возможность
детально сравнить различные образцы, и результаты такого сравнения могут быть
использованы для лучшего понимания свойств полимерного материала или
соответствующего процесса полимеризации |173|. К сожалению, гораздо труднее
получить из таких хроматограмм количественную информацию, так как возникают
некоторые сложности. Во первых, время удерживания в ГПХ зависит не только от
молекулярной массы полимера, но и от его химического состава и топологии
(например, степени разветвленное™). Во вторых, интенсивность отклика детектора
также зависит от этих свойств конкретного полимера.
Чтобы решить первую проблему (удерживание определяется не только
молекулярной массой), мы должны знать, как удерживание в ГПХ зависит от молекулярной
массы и химического состава анализируемого вещества. Эту зависимость можно
определить с помощью подходящих стандартов сополимеров. Вторую проблему
(изменение отклика детектора) решить сложнее. При анализе гомополимеров отклик
детектора может быть почти постоянным, то есть не зависеть от молекулярной массы
при УФ-детектировании. Однако многие полимеры не имеют хромофоров, что
обуславливает использование детектора показателя преломления. В таких случаях,
отклик детектора (обычно называемый увеличением коэффициента преломления или
А„/д€) зачастую непостоянен для отличающихся по длине олигомеров1.
Литература
1. С. Т. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, CA, 1996.
2. L. R. Snyder and M. A. Stadalius, High-Performance Liquid Chromatography: Advances and
Perspectives, Vol. 4, C. Horvath, ed. Academic Press, San Diego, 1986, p. 195.
3. L. R. Snyder and J. W. Dolan, High-Performance Gradient Elution, Wiley-lnterscience, Hoboken,
NJ, 2007.
4. J. O. Konz, R. C. Livingood, A. J. Belt, A. R. Goerke, M. E. Laska, and S. L. Sagar, Hum. Gene
Ther., 16 (2005) 1346.
5. E. I. Trilisky and A. M. Lenhofi, J. Chromatogr., 1142 (2007) 2.
6. M. A. Stadalius, B. F. D. Ghrist, and L. R. Snyder, J. Chromatogr., 387 (1987) 21.
7. J. S. Richardson, Adv. Protein Chem., 34 (1981) 167.
8. W. R. Melick-Adayan, V. V. Barynin, A. A. Vagin, V. V. Borisov, B. K. Vainshtein, B.K. Fita, M.
R. N.Munhy, and M. G. Rossman, J. Mol. Biol, 188 (1986) 63.
9. R. L. Cunico, K. M. Gooding, and T. Wehr, Basic HPLC and CE of Biomolecules, Bay Bioanalyti-
cal Laboratory, Richmond, CA, 1998.
10. M. T. W. Hearn and B. Grego, J. Chromatogr., 282, (1983) 541.
11. W. Doerfler, in Medical Microbiology, 4th ed., S. Baron (ed.), Univ. TX Medical Branch,
Galveston, 1996.
12. N. B. Afeyan, N. F. Gordon, I. Mazsaroff, L. Varady, S. P. Fulton, Y. B. Yang, and F. E. Regni-
er, J. Chromatogr, 519, (1990) 1.
13. F. B. Rudolph, D. P. Wiesenborn, J. Greenhut, and M. L. Harrison, in HPLC of Biological Mac-
romolecules, K. M. Gooding and F. E. Regnier, eds., Dekker, New York, 1990, p. 333.
14. F. E. Regnier, Science, 238 (1987).
15. M. A. Stadalius, H. S. Gold, and L. R. Snyder, J. Chromatogr., 296 (1984) 31.
16. M. Kawakatsu, H. Kotaniguchi, H. Freiser, and К. М. Gooding. J. Liq. Chromatogr., 18 (1995)
633.
17. A. Apfel, S. Fischer, G. Goldberg, P. C. Goodley, and F. E. Kuhlmann, J. Chromatogr. A, 712
(1995) 177.
1 Эту проблему можно решить, если применить детектор заряженных аэрозолей (ДЗА) или де-
риватизироватизировать полимер, т.е. химически модифицировать его с целью получить молекулы
с флуоресцентными или УФ-хромофорами. — Прим. перев.
698 Глава 13. Разделение биополимеров и синтетических полимеров
18. D. V. McCalley, LCGC, 23 (2005) 162.
19. D. V. McCalley, J. Chromatogr. A, 1075 (2005) 57.
20. D. Giro, С. Т. Mant, and R. S. Hodges, J. Chrvmatogr., 386 (1987) 205.
21. M. T. W. Hearn, in HPLC of Biological Macromolecules, 2nd ed., K. M. Gooding and F. E. Regni-
er, eds., Dekker, New York. 2002, pp. 195—312.
22. J. E. Rivier, J. Liq. Chromatogr., 1 (1978) 343.
23. D. Giro, С. Т. Mant, A. K. Taneja, J. M. R. Parker, and R. S. Hodges, J. Chromatogr., 359 (1986)
499.
24. W. Hancock, R. С Chloupek, J. J. Kirkland, and L. R. Snyder, J. Chromatogr. A, 686 (1994) 31.
25. S. Terabe, S. Nishi, and T. Ando, J. Chromatogr., 212 (1981) 295.
26. J. L. Glajch, M. A. Quarry, J. F. Vasta, and L. R. Snyder, Anal. Chem., 58 (1986) 280.
27. С. Т. Wehrand L. Correia, LC at Work, LC-121, Varian, Walnut Creek, CA 1980.
28. D. Guo, С. Т. Mant, A. K. Taneja, and R. S. Hodges, J. Chromatogr., 359 (1986) 519.
29. С. Т. Mant, Т. W. L. Burke, J. A. Black, and R. S. Hodges, J. Chromatogr., 458 (1988) 193.
30. M. T. W. Hearn and B. Grego, J. Chromatogr., 296 (1984) 61.
31. W. R. Melander, J. Jacobson, and С Horvath, J. Chromatogr., 234 (1982) 269.
32. S. Cohen, K. Benedek, Y. Tapuhi, J. С Ford, and B. L Karger, Anal. Biochem., 144 (1985) 275.
33. W. G. Burton, K. D. Nugent, T. K. Slattery, B. F. Johnson, and L. R. Snyder, J. Chromatogr., 443
(1988) 363.
34. K. D. Nugent, W. G. Burton, T. K. Slattery, B. F. Johnson, and L. R. Snyder, J. Chromatogr., 443
(1988) 381.
35. L. J. Licklider, С. С. Thoreen, J. Peng, and S. P. Gygi, Anal. Chem., 74 (2002) 3076.
36.B. F. D. Ghrist and L. R. Snyder, J. Chromatogr., 459 (1989) 43.
37. P. Gagnon, Purification Tools for Monoclonal Antibodies, Validated Biosystems, Tuscon, AZ, 1996.
38. W. Корасiewicz, M. A. Rounds, J. Fausnaugh, and F. E. Regnier, J. Chromatogr., 266 (1983) 3.
39. C. D. Scott in Modern Practice of Liquid Chromatography, J. J. Kirkland, ed., Wiley-lnterscience,
New York, 1971.
40. W. Muller, J. Chromatogr., 510 (1990) 133.
41. M. T. Ueda and Y. Ishida, J. Chromatogr., 386 (1987) 273.
42. R. Chicz and F. Regnier, Met. Enzymol., 182 (1990) 392.
43. L. R. Snyder. J. J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, 2nd ed.,
Wiley-lnterscience, New York, 1997, p. 515.
44. L. Sluyterman and O. Elcrmsa, J. Chromatogr., 150 (1978) 17.
45. L. Sluyterman and J. Wijdenes, J. Chromatogr., 150 (1978) 31.
46. L. Sluyterman and J. Wijdenes, J. Chromatogr., 206 (1981) 429.
47. L. Sluyterman and J. Wijdenes, J. Chromatogr., 206 (1981) 441.
48. Chromatofocusing with Polybuffer and РВЕ Handbook, ed. AB, Publication 18-1009-07, Amersham
Pharmacia Biotech, Uppsala, Sweden.
49. P. Gagnon, Quarterly Resource Guide to Downstream Processing, Validated Biosystems, Tuscon, AZ,
1999.
50. N. G. Good, G. D. Winget, W. Winter, T. N. Connally, S. Izawa, and R, M. M. Singh,
Biochemistry, 5 (1966) 467.
51. T. Kawasaki and S. Takahashi, Eur. J. Biochem., 152 (1985) 361.
52. T. Kawasaki, J. Chromatogr., 151 (1978) 95.
53. T. Kawasaki, J. Chromatogr., 157 (1978) 7.
54. M. J. GorbunofT, Anal. Biochem., 136 (1984) 425.
55. K. M. Gooding, Z. El Rassi, and С Horvath, in HPLC of Biologicial Macromolecules, 2nd ed.,
K. M. Gooding and F. E. Regnier, eds., Dekker, New York, 2002, pp. 247—280.
56. L. Kagedal, in Protein Purification, J. С Janson and L. Ryden, eds., VCH, New York, 1989.
pp. 227-251.
57. F. H. Arnold, Biotechnology, 151 (1991) 9.
58. J. Porath and B. Olin, Biochemistry, 22 (1983) 162.
59. E. Hochuli, W. Bannwarth, H. Dobeli, R. Gentz, and D. Stuber, Biotechnology. 6 (1988) 1321.
60. B. Bodenmiller, L. N. Mueller, M. Mueller, B. Domon, and R. Aebersold, Nature Methods, 4
(2007) 231.
61. J. Porath, J. Chromatogr., 443 (1988) 3.
62. Z. El Rassi and С Horvath, J. Chromatogr., 359 (1986) 241.
63. A. Tiselius, Ark. Kern. Min. Geo!., 26B (1948).
Литература 699
64. J. Porath, Biochem. Biophys. Acta, 39 (1960) 193.
65. B. Gelotte, J. Chromatogr., 3 (1960) 330.
66. Y. Kato, T. Kitamura, and T. Hashimoto, J. Chromatogr., 266 (1983) 49.
67. Y. Kato, T. Kitamura, and T. Hashimoto, J. Chromatogr., 292 (1984) 418.
68. R. E. Shansky, S.-L. Wu, A. Figueroa, and B. L. Karger, in HPLC of Biological Macromolecules,
K. M. Gooding and F. E. Regnier, eds., Dekker, New York, 1990, p. 95.
69. H. S. Frank and M. J. Evans, J. Chem. Phys., 13 (1945) 507.
70. S. Shaltiel, Z. Ег-el, Proc. Natl. Acad. Sci. USA, 52 (1973) 430.
71. D. L. Gooding, M. N. Schmuck, M. P. Nowlan, and К. М. Gooding, J. Chromatogr., 359 (1986)
331.
72. J. L. Fausnaugh and F. E. Regnier, J. Chromatogr., 359 (1986) 131.
73. D. B. Wetlaufer and M. R. Koenigbauer, J. Chromatogr., 359 (1986) 55.
74. S. L. Wu, K. Benedek, and B. L. Karger, J. Chromatogr., 359 (1986) 3.
75. A. J. Alpert, J. Chromatogr., 499 (1990) 177.
76. M. Lafosse, B. Herbreteau, M. Dreux, and L. Morinallorym, J. Chromatogr., 472 (1989) 209.
77. W. Naidong, J. Chromatogr. B, 796 (2003) 209.
78. B. A. Olsen, J. Chromatogr. A, 913 (2001) 113.
79. T. Yoshida, Anal. Chem., 68 (1997) 3038.
80. H. Tanaka, X. Zhou, and 0. Masayoshi, J. Chromatogr. A, 987 (2003) 119.
81. T. K. Chambers and J. S. Fritz, J. Chromatogr. A, 797 (1998) 139.
82. M. Wuhrer, C. A. M. Koeleman, A. M. Deelder, and С N. Hokke, Anal. Chem., 76 (2004) 833.
83. M. Wuhrer, C. A. M. Koeleman, C. H. Hokke, and A. M. Deelder, Anal. Chem., 11 (2005) 886.
84. A. J. Ytterberg, R. R. Ogorzalek-Loo, P. Boontheung, J. Wohlschlegel, and J. A. Loo, abstract
WP 523, 55th ASMS Conference on Mass Spectrometry and Allied Topics, Indianapolis, 2007.
85. С T.Mant, J. R. Litowski, and R. S. Hodges, J. Chromatogr. A, 816 (1998) 65.
86. C. A. Mizzen, A. J. Alpert, L. Levesque. T. P. A. Kruck, and D. R. McLachlan, J. Chromatogr. B,
144 (2000) 33.
87. A. Jungbauer, С Machold, and R. Harm, J. Chromatogr. A, 1079 (2005) 221.
88. H. Lindner, B. Sarg, С Meraner, and W. Helliger, J. Chromatogr. A, 743 (1996) 137.
89. H. Lindner, B. Sarg, C. Meraner, and W. Helliger, J. Chromatogr. A, 782 (1997) 55.
90. B. Sarg, W. Helliger, H. Talasz, E. Kooutzamani, and H. Lindner, J. Biol. Chem., 279 (2004)
53—58.
91. A. J. Alpert, Anal. Chem., 80 (2008) 62.
92. A. J. Alpert, G. Mitulovic, and M. Mechtler, poster P2412-W, 32nd Annual Symposium on High
Performance Liquid Phase Separations and Related Techniques, Baltimore, 2008.
93. U. Lewandrowski, K. Lohrig, R. P. Zahedi, D. Wolters, and A. Sickmann, Clin. Proteom., 4
(2008) 25.
94. P. H. O'Farrell, J. Biol. Chem., 250 (1975) 4007.
95. M. Gilar, P. Olivova, A. E. Daly, and J. С Gebler, Anal. Chem., 11 (2005) 6426.
96. S. P. Gygi, B. Rist, S. A. Gerber, F. Turecek, M. H. Gelb, and R. Aebersold, Nat. Biotechnol., 17
(1999) 994.
97. A. J. Link, J. Eng, D. M. Schieltz, E. Carmac, G. J. Mize, D. R. Morris, В. М. Garvik, and J. R.
Yates, Nat. Biotechnol.. 17 (1999) 676.
98. D. A. Wolter, M. P. Washburn, and J. R. Yates, Anal. Chem., 73 (2001) 5683.
99. M. P. Washburn, D. Wolters, and J. R. Yates, Nat. Biotechnol., 19 (2001) 5683.
100. M. T. Davis, J. Beierle, E. T. Bures, M. D. McGinley, J. Mort, J. H. Robinson, С S. Spahr, W.
Yu, R. Luethy, and S. D. Patterson, / Chromatogr. B, 752 (2001) 281.
101. G. J. Opiteck and J. W. Jorgenson, Anal. Chem., 69 (1997) 2283.
102. G. J. Opiteck, S. M. Ramirez, J. W. Jorgenson, and M. A. Moseley Anal. Biochem., 258 (1998)
349.
103. K. Wagner, T. Miliotis, G. Магко-Varga, R. BischoiT, and K. K. linger, Anal.Chem., 74 (2002)
809.
104. R. BischofTand L. W. McLaughlin, in HPLC of Biologicial Macromolecules, К. М. Gooding and
F. E. Regnier, eds., Dekker, New York, 1990, pp. 641—667.
105. R. Hecker, M. Colpan, and D. Riesner, J. Chromatogr., 326 (1985) 251.
106. S. Nakatani, T. Tsuda, Y. Yamasaki, M. Moriyama, H. Watanabe, and Y. Kato, Technical Report
78, TosoHaas, Tokyo, 1995.
107. R. R. Drager and F. E. Regnier, Anal Biochem., 145 (1985) 47.
700 Глава 13. Разделение биополимеров и синтетических полимеров
108. G. Zon, in Characterization of Proteins: New Methods in Peptide Mapping, W. S. Hancock, ed.,
CRC Press, Boca Raton, 1995, p. 301.
109. W. Xiao and P. J. Oefner, Human Mutation, 17 (2001) 439.
110. A. Premstaller and P. J. Oefner, in Methods in Molecular Biology,. 211, P.-Y. Kwok, ed., Humana
Press, Totowa, NJ, 2002, p. 15.
111. R. L. Pearson, J. F. Weiss, and A. D. Kelmers, Biochim. Biophys. Acta, 228 (1971) 770.
112. R. Bischoffand L. W. McLaughlin, Anal. Biochem., 151 (1985) 526.
113. J. D. Pearson, M. Mitchell, and F. E. Regnier, J. Liq. Chromatogr., 6 (1983) 1441.
114. R. BischofTand L. W. McLaughlin, J. Chromatogr., 296 (1984) 329.
115. Z. el Rassi and С Horv&h, J. Chromatogr., 326 (1985) 79.
116. Z. el Rassi and С Horvath, Chromatographia, 19 (1984) 9.
117. S. C. Cliurms, CRC Handbook of Chromatography: Carbohydrates, Vol. 2, CRCPress, Boca Raton,
1991.
118. S. С Churms, J. Chromatogr. A, 720 (1996) 75.
119. K. Koizumi, T. Utainura, Y. Kubota, and S. Hizukuri, J. Chromatogr., 409 (1987) 396.
120. С Brons and С Olieman, J. Chromatogr., 159 (1983) 79.
121. D. W. Armstrong and H. L. Jin, J. Chromatogr., 462 (1989) 219.
122. S. Honda and S. Suzuki, Anal. Biochem., 142 (1984).
123. A. S. Feste and I. Khan, J. Chromatogr., 607 (1992) 7.
124. Guide to Aminex®HPLC Columns, Bulletin 1928, Bio-Rad Laboratories.
125. T. Jupille, Amer. Lab., 13 (1981) 80.
126. R. W. Goulding, J. Chromatogr., 103 (1975) 229.
127. Analysis of Carbohydrates by High Performance Anion Exchange Chromatography with Pulsed Ampe-
rometric Detection (HРАЕ PAD), Dionex Technical Note 20 (2000).
128. Glycoprotein Oligosaccharide Analysis Using High-Performance Anion-Exchange Chromatography,
Dionex Technical Note 42 (1997).
129. Optimal Settings for Pulsed Amperometric Detection of Carbohydrates Using the Dionex ED40
Electrochemical Detector, Dionex Technical Note 21 (1998).
130. B. G. Huyghe, X. Liu, S. Sutjipto, B. J. Sugarman, M. T. Horn, H. M. Shepard, C. J. Scandella.
and P. Shabram, Hum. Gene Ther., 6 (1995) 1403.
131. W. W. Yau, J. J. Kirkland, and D. D. Bly, Modem Size-Exclusion Liquid Chromatography, Wi-
ley-lnterscience. New York, 1979.
132. J. Porath and P. Flodin, Nature (London), 183, (1959) 1657.
133. S. Hjerten and R. Mosbach, Anal. Biochem., 3, (1962) 109.
134. S. Hjerten, Arch. Biochem. Biophys., 99, (1962) 466.
135. E. L. Johnson and R. L. Stevenson, in Basic Liquid Chromatography, Varian, Walnut Creek, CA,
1978, p. 150.
136. L. Hagel and J. C. Janson, in Chromatography, 5th ed., E. Heftmann, ed., Elsevier, Amsterdam,
1992, A267.
137. K. M. Gooding and F. E. Regnier, in HPLC of Biological Macromolecules, 2nd ed., K.M Gooding
and F. E. Regnier, eds., Dekker, New York, 2002, p. 59.
138. B. F. D. Ghrist, M. A. Stadalius, and L. R. Snyder, J. Chromatogr., 387 (1987) I.
139. R. L. Cunico, K. M. Gooding, and T. Wehr, in Basic HPLC and CE of Biomolecules, Bay Bioana-
lytical Laboratory, Richmond, CA, 1999, p. 135.
140. E. Pfannkoch, К. С Lu, F. E. Regnier, and H. G. Barth, J. Chromatogr. Sci., 18, (1980) 430.
141. E. Folta-Stogniew and K. R. Williams, J. Biomol. Techniques, 10 (1999) 51.
142. V. N. Uversky. Biochem., 32 (1993) 13288.
143. L. Hagel, J. Chromatogr., 648, (1993) 19.
144. B. Sebille and N. Thuaud, In Handbook of HPLC for the Separation of Amino Acids, Peptides, and
Proteins, Vol. 2, W. S. Hancock, ed., CRC Press, Boca Raton, 1984, pp. 379-391.
145. J. P. Hummel and W. J. Dreyer, Biochim. Biophys. Acta, 63 (1962) 530.
146. J. Curling., Biopharm International, (Feb. 2007) 10.
147. G. Walsh., Appl. Microbiol Biotechnol., 67 (2005) 151.
148. L. Hagel, G. Jagschies, and G. K. Sofer, Handbook of Process Chromatography: Development,
Manufacturing, Validation and Economics, 2nd ed., Elsevier, Amsterdam, 2007.
149. H. Chase, Trends Biotechnol., 12 (1994) 296.
150. A. Jungbauer and E. Boschetti., J. Chromatogr. B, 662 (1994) 143.
151. A. Jungbauer, J. Chromatogr. A, 1065 (2005) 3.
Литература 701
152. P. Lu, С. D. Сагг, P. Chadwick, M. Li, and K. Harrison, BioPharm., (Sep. 2001) 19.
153. J. Rivier and R. McClintock, J. Chromatogr., 268 (1983) 112.
154. J. Rivier, R. McClintock, R. Galyean, and H. Anderson. J. Chromatogr., 288 (1983) 303.
155. E. I. Grimm and E. E. Logsdon, US Patent 4,612,367 (1986).
156. P. H. Lai and T. W. Strickland, US Patent 4,667,016 (1987).
157. R. Bischoff, D. Clesse, O. Whitechurch, P. Lepage, and С Roitsch, J. Chromatogr. A, Alb (1989)
245.
158. D. I. Urdal, D. Mochizuki, P. J. Conlon, C. J. March, M. L. Remerowski, J. Eisenman,
С Ramthun, and S. Gillis, J. Chromatogr. A, 296 (1984) 171.
159. S. Hershenson, Z. Snaked, and J. Thomson, US Patent 4,961,969 (1990).
160. C. V. Olsen, D. H. Reifsnyder, E. Canova-Davis, V. T. Ling, and S. E. Builder, J. Chromatogr. A
675 (1994) 101.
161. V. Price, D. Mochizuki, C. J. March, D. Cosman, M. C. Deeley, R. Klinke, W. Clevenger, S.
Gillis, P. Baker, and D. Urdal, Gene, 55 (1987) 28.
162. E. P. Kroeff, R. A. Owens, E. L. Campbell, R. D. Johnson, and H. I. Marks, J. Chromatogr., 461
(1989) 45.
163. J. Brange, The Physico-chemical and Pharmaceutical Aspects of Insulin, Springer, Berlin, 1987.
164. E. P. Kroeff and R. E. Chance, Proceedings of the FDA-USP Workshop on Drug and Reference
Standards for Insulins, Somatotrophins and Thyroid-Axis Hormones, United States Pharmacopeia
Convention, Rockville, MD, 1982, pp. 148—162.
165. Current Good Manufacturing Practice in Manufacturing, Processing, Pecking, or Holding Of Drugs,
21 CFR Part 210, http://www.fda.gov/cder/dmpq/cgmpregs.htm.
166. Current Good Manufacturing Practice for Finished Pharmaceuticals, 21 CFR Part 211,
http://www.fda.gov/cder/dmpq/cgmpregs.htm.
167. International Organization for Standardization, http://www.iso.org/iso/home.htm.
168. M. T. W. Hearn, Reversed-Phase High Performance Liquid Chromatography, Academic Press, New
York, 1984.
169. A. M Striegel, W. W Yau, J. J Kirkland, and D. D Bly, Modern Size-Exclusion Liquid
Chromatography, 2nd ed., Wiley-lnterscience, New York, 2009.
170. T. H. Mourey, Int. J. Polym. Anal. Charact., 9 (2004) 97.
171. A. M Striegel, Anal. Chem., 77 (2005) 104A.
172. W. F Reed, in Multiple Detection in Size-Exclusion Chromatography, A. M. Striegel, ed., ACS,
New York, 2005, ch. 2.
173. W. M. C. Decrop et al., submitted for publication.
174. X. Jiang, P. J. Schoenmakers, X. Lou, V. Lima, J. L. J. van Dongen, and J. Brokken-Zijp, J.
Chromatogr., 1055 (2004) 123.
175. X. Jiang, A. van der Horst, V. Lima, and P. J. Schoenmakers, / Chromatogr, 1076 (2005) 51.
ГЛАВА 14
РАЗДЕЛЕНИЕ ЭНАНТИОМЕРОВ
Михаель Лэммерхофер, Норберт М. Майер, Вольфганг Линднер
14.1. Введение 703
14.2. Основы и определения 703
14.2.1. Изомерия и хиральность 704
14.2.2. Хиральное распознавание и разделение энантиомеров 706
14.3. Косвенный метод 707
14.4. Прямой метод 711
14.4.1. Метод хиральных добавок к подвижной фазе (ХДПФ) или
аддитивный метод 711
14.4.2. Использование хиральной неподвижной фазы (ХНФ) 714
14.4.3. Принципы хирального распознавания 715
14.4.3.1. «Модель трехточечного присоединения» 715
14.4.3.2. Влияние подвижной фазы 717
14.5. Дисперсия и уширение задних фронтов пиков 718
14.6. Хиральные неподвижные фазы и их характеристики 718
14.6.1. ХНФ на основе полисахаридов 719
14.6.2. Синтетические полимерные ХНФ 726
14.6.3. ХНФ на основе белков 730
14.6.4. ХНФ на основе циклодекстрина 734
14.6.5. ХНФ на основе макроциклических антибиотиков 737
14.6.6. ХНФ на основе хиральных краун-эфиров 743
14.6.7. Донорно-акцепторные ХНФ 745
14.6.8. Хиральные ионообменники 748
14.6.9. Лигандообменные ХНФ. Хиральная лигандообменная
хроматография (ХЛОХ) 751
14.7. Термодинамические аспекты 753
14.7.1. Термодинамика ассоциации анализируемого вещества с селектором . 753
14.7.2. Термодинамика прямого хроматографического разделения
энантиомеров 754
14.7.3. Термодинамика гетерогенной хиральной поверхности 754
Литература 756
14.1. Введение 703
14.1. Введение
В предыдущих главах были описаны методы ВЭЖХ-разделения широкого
применения, которые можно использовать для многих различных типов образцов. Обычно
разработку метода можно начать с любыми из разнообразных хроматографических
колонок или условий разделения, с последующим систематическим улучшением
разделения путем изменения хроматографических условий, часто методом проб и
ошибок. Поэтому, как правило, существует множество способов успешного разделения
конкретного образца.
Однако, такой подход нельзя использовать для разделения энантиомеров,
имеющих одинаковое строение, но различную пространственную ориентацию, и
требующих в связи с этим высокоспециализированных методов и материалов. В
большинстве случаев выбор хроматографической колонки является определяющей стадией
исследования. Без выбора подходящей колонки последующие изменения других
хроматографических условий вряд ли приведут к успеху. Несмотря на сложности,
разделение энантиомеров является рутинным процессом, как в исследовательских, так и
в промышленных лабораториях, и имеет особое значение в фармацевтической
промышленности. Разработаны различные технологии и приемы, предоставляющие
богатый набор средств для разделения практически любого образца. Среди доступных
приемов — прямое разделение на хиральной неподвижной фазе (ХНФ).
Использование энантиоселективных или «хиральных» колонок стало преобладающей и наиболее
приемлемой, но не единственной методикой разделения энантиомеров. Однако
поиск наиболее подходящей комбинации ХНФ/подвижная фаза для конкретного
целевого вещества может быть достаточно сложной задачей. Из-за специфики
молекулярного распознавания в случае каждой комбинации ХНФ (хиралъного селектора) и
целевого вещества и чувствительности распознавания к незначительным
структурным изменениям как растворенного в подвижной фазе образца, так и хиральной
неподвижной фазы — надежно прогнозировать, исходя из молекулярной структуры
целевого вещества, какая колонка подойдет для работы с ним пока практически
невозможно.
Базы данных, такие как ChirBase (http://chirbase.ii-3mrs.fr), могут помочь в выборе
хроматографической колонки и начальной подвижной фазы на основе структуры
целевого вещества 11, 2|. Однако, такой подход может быть полезен только для
энантиомеров, уже включенных в базу данных или имеющих структуру, сходную с теми, которые
включены в базу данных, и проблематичен для совершенно новых структур. С другой
стороны, в крупных фармацевтических компаниях уже используются
автоматизированные процессы скрининга для эффективного решения этой проблемы |3, 4|. Таким
образом можно легко найти наиболее перспективную ХНФ для дальнейшего
исследования. Затем следует оптимизировать подвижную фазу. В общем и целом, прямое
разделение энантиомеров с помощью ВЭЖХ на ХНФ стало самой значимой технологией,
описание которой и является основной задачей настоящей главы.
14.2. Основы и определения
Методики неэнантиомерного разделения часто можно разработать, исходя только
из общего описания компонентов образца, например, учитывая, что анализируемые
вещества являются кислотными, щелочными или нейтральными соединениями.
В некоторых случаях, зная, к какому классу принадлежат соединения, содержащиеся
в образце (например, пептиды, белки, углеводы), уже можно предопределить ряд
подходящих условий анализа, включая тип колонки (но это не означает, что требует-
704 Глава 14. Разделение энантиомеров
ся совершенно определенная колонка). Следовательно, для разработки неэнантиосе-
лективного метода разделения нет необходимости иметь подробную информацию
о молекулярной структуре подлежащего анализу вещества. Это не относится к
разделению энантиомеров, где молекулярная структура разделяемого вещества и
связанные с ней физико-химические свойства играют решающую роль в разработке метода
анализа. По этой причине понимание основ поведения энантиомеров имеет важное
значение для их эффективного разделения.
14.2.1. Изомерия и хиральность
Изомеры — это вещества, молекулы которых обладают одинаковым атомным
составом и молекулярной массой, но не совпадают при наложении друг на друга |5, 6|.
Классификация изомерных структур приведена на рисунке I4.1 [6|. В отличие
от фактически идентичных «гомомеров», структурные изомеры могут определяться
различными свойствами. Например, 1-бутанол, 2-бутанол — структурные изомеры
с изомерией углеродного скелета. В противоположность им стереоизомеры имеют
одинаковые межатомные связи, но отличаются ориентацией атомов или групп
в трехмерном пространстве. Последние можно разделить на энантиомеры и диастере-
омеры (или «диастереоизомеры»). В то время как первые всегда являются хиральны-
ми, последние могут также включать ахиральные стереоизомеры, такие как цис/
транс-изомеры (например, цис/транс-1,2-дихлорэтилен).
Хиральность (от греческого хЕ^Р РУка)5 относится к геометрическому свойству
объекта не совмещаться в пространстве со своим зеркальным отражением подобно
тому, как правая рука не совмещается с левой. Такой объект не имеет элементов
симметрии второго рода — таких, как плоскость симметрии, центр инверсии или ось
вращения. Хиральность может быть обусловлена различными хиральными
элементами (рис. 14.2): центрами хиральности (асимметрическими атомами; рис. 14.2а),
хиральными осями (аксиальной хиральностью; рис. 14.26), хиральными плоскостями
(планарной хиральностью; рис. 14.2*), хиральными спиралями (спиральной хираль-
Молекулы с идентичным атомарным составом
Гомомеры
(идентичны)
Изомеры
>. (структурные
изомеры)
Диастереомеры
(SR SS)
Критерии различий:
Свойства Неэнантиомеры
Симметрия Структуры не
зеркальны
Энергия Различна
Энантиомеры
(R S)
(смесь 1:1= рацемат)
Энантиомеры
Нессамещаемые
зеркальные структуры
Идентична
Конституциональные
изомеры
14.2. Основы и определения 705
(а)
Центры хиральности
(»>
Пленарная хиральность
а а
у .„Hid : dn,„.^
Ь""'
,-х
X = С, SI. Ge, Sn. N\ Р\ As* X - N, S". P
четы рвхкоординироваиные трехкоординированные
Осевая хиральность
НООС
н \;оон
сн.
,jCOOH НООС,
QH^ '•2V
Спиральная хиральность
М(минус)
Рнс. 14.2. Структурные особенности, способствующие хиральности
Р (плюс)
ностью; рис. 14.2г) и топологическими хиральными элементами (топологической хи
ральностью). Наиболее известным примером является центр хиральности, который
обычно образует атом углерода с четырьмя различными заместителями, например,
атомы углерода 1 и 2 в паре эфедрин — псевдоэфедрин (рис. 14.3а).
Энантиомеры и диастереомеры можно различить по двум свойствам: симметрии
или свободной энергии молекул. Энантиомеры представляют собой молекулы,
являющиеся несовместимыми зеркальными отображениями друг друга и имеющие
одинаковую энергию (из-за идентичности межатомных расстояний, углов и вращений,
а также межатомных взаимодействий). Они неразличимы в ахиральной
(симметричной) среде и поэтому не могут быть разделены ахиральными хроматографическими
методами, такими как обычная обрашенно-фазовая хроматография (ОФХ).
Например, (\S, 2Я)-эфедрин и (1Д, 25)-эфедрин (рис. 14.За) представляют собой
энантиомеры. Более того, (I/J, 2Д)- и (15, 25)-псевдоэфедрины также энантиомерны друг
ДРУГУ (Рис- 14.35). Заметим, что энантиомерные друг другу молекулы всегда имеют
противоположные конфигурации в двух стереогенных центрах (рис. 14.3). Эквимо-
лярная смесь энантиомеров называется рацематом. Все другие смеси энантиомеров
с составом, отличающимся от 1:1, определяются как нерацемические смеси.
Энантиомеры снабжены стереохимическими дескрипторами, (например, L и £>,
или R и S, или + и -), по которым их различают (причем, Л и 5 наиболее
предпочтительны). В соответствии с правилами Кана — Ингольда — Прелога дескрипторы R и S
относятся к конфигурациям, в которых заместители в стереогенном центре
расположены по часовой стрелке и против часовой стрелки. При этом в соответствии с
правилами, самый младший с точки зрения стехиометрического старшинства заместитель
ориентирован в сторону от наблюдателя. Дескрипторы + и - относятся к свойствам
оптического вращения хиральной молекулы, то есть ее способности вращать плоскость
706 Глава 14. Разделение энантиомеров
с6н5
СеН
6nS
HI
HI
(б)
1
i2
снэ
(1S,2ff)-(+)
Q6H5
•OH
HOI
INHCHj ! H3CHNI
IH
■ H
1
эфедрин
трео
HOI
IH
INHCH3
CH3
(1Я.2ЯН-)
CH3
(1R.2SH-)
C«H
(a) эритро линейно поляризованного света вправо
или влево (правовращающая или левов-
ращающая конфигурации).
Дескрипторы L и D относятся к проекциям Фи
шера1 для аминокислот и Сахаров.
Проекции определяют относительные
конфигурации химических произвол
ных Д-(+)-глицеральдегида [6|.
Диастереомеры2 не являются
зеркальными отражениями друг друга.
Они характеризуются различными фи
зическими и химическими свойствами
и могут быть разделены с помощью
ахиральной хроматографии. В приме
ре на рис. 14.3 каждый стереоизомер
на рис. 14.3о является диастерео-
мером любого из стереоизомеров
на рис. 14.3<э, поскольку они отлича
ются только конфигурацией одного
стереогенного центра, а не обоих (как
у энантиомеров). Таким образом,
эфедрины и псевдоэфедрины
являются диастереомерами, поскольку они
не являются зеркальными
отражениями друг друга. Их также можно
назвать эпимерами, поскольку
инвертирован только один из нескольких
стереогенных центров. Это различие
между энантиомерами и
диастереомерами является фундаментальной основой для всех процессов хирального
разделения, включая все концепции разделения энантиомеров.
вп5
Hi
ЮН
H3CHNI
СНЭ
(1S.2SH+)
псеедо эфедрин
Рис. 14.3. Структуры эфедрина и
псевдоэфедрина
14.2.2. Хиральное распознавание и разделение
энантиомеров
Энантиомеры имеют одинаковые физико-химические свойства, поэтому их для
разделения необходимо превратить в (I) диастереомеры (косвенный метод) или (2) диа-
стереомерные комплексы {прямой метод) |7, 8|. В настоящее время косвенный
подход используют реже из-за некоторых проблем, обсуждаемых ниже |8|. Тем не менее,
косвенный метод используется для некоторых приложений. Его описание в
следующем разделе 14.3 будет охватывать вопросы, также имеющие отношение к прямому
методу, рассмотренному в разделе 14.4. При планировании энантиоселективного раз-
' D, L — номенклатура Фишера, где молекулы изображаются в виде проекции, и символ D
используется, если активная группа, например, аминогруппа а-аминокислот, расположена справа,
символ L — соответственно слева. — Прим. персе.
2Если в молекуле имеются два хиралъных центра, то, поскольку каждый центр может иметь
(R)- или (.^-конфигурацию, возможно существование четырех изомеров — RR, SSr RS и SR. Если
меняется конфигурация лишь одного асимметрического центра, то такие изомеры назынаютси
диастереомерами. Поскольку молекула имеет только одно зеркальное отображение, энантиомером
соединения (RR) может быть только изомер (SS). Аналогично другую пару энантиомеров образуют
изомеры (RS) и (SR). — Прим. перев.
14.3. Косвенный метод 707
деления прямым методом могут использоваться различные хроматографические
режимы, как и в случае ахиральной хроматографии. В этой связи мы выделим энанти-
оселективные разделения, обозначив их как обрашенно фазовые разделения (ОФ)
или нормально-фазовые разделения (НФ). Эти обозначения отличаются от ранее
использованных сокращений ОФХ и НФХ для ахиральных разделений.
14.3. Косвенный метод
Косвенный метод включает образование диастереомеров путем взаимодействия
анализируемого вещества (X) в конфигурации R или Sc энантиомерно чистым
соединением (далее с Л-конфигурацией), которое мы будем называть хиральным дериватизиру-
ющим агентом (ХДА):
(R)-X + (Ю-ХДА -> (Я, RyX-ХДА (14.1)
и
(S)-X + (Ю-ХДА -> (S, Ю-Х-ХДА (14.1а)
Реакции, приведенные выше, должны быть проведены до их полного завершения,
а диастереомерные продукты должны быть стабильными (химически и
конфигурационно). Полученные диастереомеры (Л, К)-Х-ХДА и {S,R)-X-XHA затем могут быть
разделены ахиральной хроматографией (обычно ОФХ).
Одним из преимуществ косвенного метода является использование
традиционных колонок ВЭЖХ, имеющих более высокое число теоретических тарелок по
сравнению с энантиоселективными («хиральными») колонками (см. раздел 14.4.) Более
высокая эффективность хроматографической колонки особенно важна для
определения примесей, содержащихся в количествах <0,1% в сложных смесях, как это и
требуется для фармацевтических продуктов. Еще более важна возможность
использовать ХДА для улучшения детектирования анализируемых веществ с помощью
флуоресцентных, электрохимических, масс-спектрометрических методов или по
поглощению в УФ-области. Хорошим примером являются флуоресцентные метки
для обнаружения аминокислот, трудно распознаваемых другими методами. Кроме
того, косвенный метод относительно экономичен в отличие от прямого метода, для
которого нужен набор разных (и в большинстве случаев дорогостоящих) энантиосе-
лективных колонок.
Было разработано большое количество ХДА, обеспечивающих приемлемую диа-
стереоселективность, и в некоторых случаях улучшающих детектирование |8|. Хира-
льные дериватизирующие реагенты должны быть как химически, так и стереохими-
чески устойчивы и коммерчески доступны в обеих энантиомерных формах
(например, R и S). Выбор R или S ХДА приводит к изменению порядка элюирования
двух энантиомеров. Они могут быть использованы для позиционирования минорного
пика перед основным пиком, когда один из энантиомеров находится в значительном
избытке (см. раздел 2.4.2, сравните рис. 2.17d,e в этом разделе). Некоторые широко
используемые ХДА приведены в таблице 14.1.
По-прежнему популярным косвенным методом для анализа энантиомерной
смеси аминокислот является использование реагента о-фтальдиальдегида (ОФА) с
хиральным тиолом, таким как N-ацетилцистеин (ХДА 4 в таблице 14.1). Схема реакции
показана на рис. 14.4.
Так как ОФА-дериватизация происходит быстро и поддается автоматизации,
нестабильность дериватизированного производного может быть преодолена
проведением реакции непосредственно перед вводом образца (избыток нефлуоресцирующего
исходного реагента не будет мешать обнаружению производного). Хроматограмма
Глава 14. Разделение энантиомеров
Таблица 14.1. Обычно используемые хиральные дериватизируюшие агенты (ХДА) и их
применение
ХДА
]. (R) или (5)-а-метокси-а-трифторметилфенил-
уксусная кислота и хлорангидрид этой кислоты
(реагент Мошера)
2. 0,(У-дибензоил-винной кислоты ангидрид
(ДВКА)
3. (К)- или (5)-1-(9-флуоренил) этилхлорформиат
(ФЭХФ)
4. орто-фтальдиалъдегид (ОФА) в комбинации
с хиральными тиолами, такими как (S)- или
(Л)-энантиомеры /V-ацетилцистеина, /V-t-Boc-UHC-
теина, iV-ацетилпеницилламина, 1-тио-р-глюкозы
5. 1-фтор-2,4-динитрофенил-5-(5)-аланинамид
(ДФАА) (реагент Мэрфи)
6. 2,3,4,б-тетра-0-ацетил-р-0-глюкопиранозил
изотиоцианат (ГИТЦ)
Класс анализируемого вещества
Спирты, амины
Первичные и вторичные
амины, спирты, амикоспирты
Первичные и вторичные
амины, аминокислоты
Первичные амины, первичные
аминокислоты
Первичные и вторичные
амины, аминокислоты, тиолы
Первичные и вторичные
амины, аминокислоты, тиолы
Литература
19]
|101
ПИ
|12, 13]
|14, 15]
|16|
HS.
но
сно
х:
я
соон
сил
анализируемое
вещество
СН3
VS-COOH
ССг£
часто используемые реагенты:
• ОФА / М-ацетилцистеин
• ОФА / Вос-цистеин
• ОФА / Изобутилцистеин
■ ОФА/ N-ацетил-О-пеницилламин
• ОФА/ 1-тио-р-глюкоза
■ ОФА/ 1-тио-Р-манноза
СООН
флуоресцентные производные
Рис. 14.4. Дериватизация ОФА с хиральным тиолом для стереоселектииного анализа
первичных аминов и аминокислот (косвенное разделение энантиомеров)
на рис. 14.5 демонстрирует анализ аминокислот в образце бацитрацина после его
гидролиза и окисления цистеина (Cys) до цистеиновой кислоты (Суа) ОФА-методом
|13|. Этот пример иллюстрирует возможность разделения нескольких пар
энантиомеров в течение одного анализа при косвенном подходе, что гораздо менее вероятно
при использовании прямого метода (если только не используется МС-детектиро-
вание, которое привносит дополнительную хемоселективность, и при этом
совместное элюирование веществ с различными МС-свойствами не оказывает влияния).
14.3. Косвенный метод
Asp
Суа
10
Gfu
^
20
30
L-His
L-Val
D-His
40
50
AN
60
l-lle
Л
D-Phe
L-Leu
D-Orn
L-Lys
£||D-Lys
70 (мин)
Рнс. 14.5. Разделение энантиомеров аминокислот, полученных при гидролизе бацит-
рацина А, с последующей дериватизацией ОФА с хиральным тиолом
(разделение косвенным методом с флуоресцентным детектированием; Суа = цис-
теиновая кислота) |13[
Тем не менее, несмотря на кажущуюся простоту, разработка косвенных методов
разделения энантиомеров далека от тривиальности |8|.
Важнейшим условием применимости косвенного метода является наличие у
анализируемого вещества функциональной группы, которую можно селективно дерива-
тизировать (такой как гидроксильная, аминогруппа, карбоксильная, карбонильная
или тиольная). Другим важным условием является химическая и (особенно) энанти-
омерная чистота дериватизирующего реагента, то есть избыток энантиомера должен
составлять > 99,9%. Если ХДА содержит значительное количество энантиомерной
примеси, то количественные результаты анализа будут ошибочными. Например,
примесь (5)-энантиомера в (Я) —ХДА при дериватизации, помимо основной диасте-
реомерной пары продуктов, приведет к образованию второй пары диастереомеров, и
тогда будут присутствовать все четыре стереоизомера (см. рис. 14.6). Отметим, что
примесь и ее диастереомерные продукты обозначены обычным (нежирным)
шрифтом. В ахиральной хроматографии (например, ОФХ) невозможно разделить парные
энантиомеры. Следовательно, стереоизомеры, образующиеся в результате энантио-
мерного загрязнения ХДА, будут совместно элюироваться с пиками
противоположных энантиомеров (то есть с энантиомерами противоположной конфигурации), и
будут наблюдаться только два пика. Понятно, что совместное элюирование такого рода
препятствует точному количественному анализу. Можно сделать поправку на
загрязнение ХДА, если известено количество энантиомерной примеси в нем, однако это
значительно усложняет как разработку методов, так и их валидацию.
Есть и дополнительные трудности при использовании косвенного метода.
Во-первых, для данного процесса дериватизации должна полностью сохраняться сте-
реохимическая целостность, не допускается рацемизация дериватизирующего
реагента, анализируемых веществ или диастереомерных продуктов [8]. Отсутствие
рацемизации можно легко проверить путем дериватизации и анализа образца известного
энантиомерного состава. Во-вторых, необходимо проверить, что дериватизация
прошла полностью, для того, чтобы избежать возможных проблем с кинетическим
разрешением (рис. 14.7) [8|. В частности, скорости реакции для дериватизации Я- и
710 Глава 14. Разделение энантиомеров
пара
энантиомерных
анализируемых
веществ
(Я)-Х
+ <Я)-ХДА
+ (S)-XflA
(S)-X
хиральныи
дериватизирующий агент
(жирным шрифтом, Я) с
энантиомерной примесью
(обыкновенный шрифт, S)
(Я, Я)-Х-ХДА
(Я, S)-X-XflA
(S, Я)-Х-ХДА
(S, SJ-X-ХДА
лае пары диастереомеров
(4 стереоизомера,
лае пары энантиомеров)
d.-..диастереомерные друг к другу
е ....энантиомерные друг к другу
Рис. 14.6. Схема реакции для косвенного метода ВЭЖХ-разделения энантиомеров
{в присутствии примеси J-ХДА в реагенте для хиральной дериватизации
Д-ХДА). Образуются все четыре стереоизомера и соответственно две пары
энантиомеров (d- диастереомерные друг другу, е — энантиомерные друг
другу)
100
зе
I
Отношение площади
пиков: (R)/(S) = 55:45
Кинетическое
разрешение
Отношение площади
пиков: (R)/(S) = 50:50
Некинетическое
разрешение
Рис. 14.7. Иллюстративные кинетические профили реакции образца с хиральным де-
риватизирующим реагентом. Предполагается, что два энантиомера имеют
разные константы скорости реакции дериватизации, а отклик детектора
для полученных диастереомеров идентичен
5-энантиомеров анализируемого вещества могут различаться. В случае
незавершенной реакции дериватизации возможно получение такого соотношения стереоизоме-
ров, которое отличается от фактического соотношения энантиомеров в образце.
Например, если образец представляет собой рацемат и кц > к$ (к являются
константами скорости реакций дериватизации), состав анализируемой смеси энантиомеров
14.4. Прямой метод 71 I
через 2 минуты (см. рис. 14.7) будет значительно отличаться от отношения 1:1,
которое ожидается для рацемата. Систематической погрешности из-за кинетического
разрешения можно легко избежать, доводя реакцию дериватизации до завершения.
Это обычно достигается увеличением температуры и времени реакции, а также с
помощью большого избытка соответствующего ХДА (ХДА обычно используют
в 10-кратном молярном избытке относительно энантиомеров).
Другим недостатком косвенного метода является то, что соотношения
энантиомеров не могут быть непосредственно рассчитаны из соотношений площадей пиков,
измеренных в конечном разделении (как это возможно в случае прямого метода), так
как отклики детектора на два диастереомерных продукта реакции могут значительно
отличаться |8[. Это справедливо для большинства аналитических методов, включая
детектирование в УФ, флуоресценцию и масс-спектрометрическое детектирование,
причем коэффициенты отклика отличаются обычно в 1,1—1,5 раза и, следовательно,
требуется коррекция этой разницы, для чего используется внешняя калибровка с
индивидуальными стандартами диастереомеров. Сложность в том, что такие стандарты
не всегда доступны.
Может показаться, что эти ограничения делают результаты, полученные
косвенным методом, сомнительными, что, безусловно, не так. Тем не менее, пользователи
должны тщательно исследовать все вышеперечисленные проблемы, а самое
главное — проконтролировать энантиомерную чистоту ХДА |17|. Нельзя полагаться
только на спецификации, предоставленные поставщиком. Кроме того,
многочисленные проблемы валидации методов, которые необходимо учитывать при разработке
косвенного анализа, делают этот подход трудоемким, требующим времени и более
подверженным систематическим ошибкам. Все же, некоторые энантиоселективные
разделения по-прежнему проводятся косвенным методом.
14.4. Прямой метод
Прямой метод разделения основан на обратимом образовании (переходных)
диастереомерных комплексов между двумя энантиомерами [(Л) — А" и (S) — X] и хираль-
ным комплексообразующим агентом, называемым хиралъным селектором (ХС). Как и
в случае косвенного метода, селектор должен быть энантиомерно чистым, но
для прямого метода это требование менее строго. В этом случае может
использоваться менее чистый ХС, если разница в устойчивости комплексов диастереомеров
достаточно велика:
(R)-X + (R)-XCo (R)-X-(R)-XC (14.2)
и
(S)-X+ (R)-XCo (S)-X-(R)-XC (I4.2a)
Прямой метод позволяет обойти трудоемкую дериватизацию ХДА. Существуют два
варианта экспериментального метода прямого разделения энантиомеров: метод хира-
льных добавок к подвижной фазе (ХДПФ) (или просто «аддитивный метод») и
использование хиральной неподвижной фазы (ХНФ).
14.4.1. Метод хиральных добавок к подвижной фазе
(ХДПФ) или аддитивный метод
В этом методе используется ахиральная неподвижная фаза (например, хроматогра-
фическая колонка, упакованная обращенно-фазовым или нормально-фазовым
сорбентом) с подвижной фазой, в которой содержится ХДПФ в соответствующей кон-
Глава 14. Разделение энантиомеров
(а) Удерживание энантиомера (R)-X
[(Я)-Х]т + [<Я)-ХС]т ^^ <[(Я)-Х]-[(Я)-ХС])т
Подвижная
фаза
^«.(«l-x
"а.1ЯухкЯ)ХС
Неподвижная
фаза
[(Я)ХЪ
[(Я)-ХС],
_Ь>
(■6) Удерживание энантиомера (Sf-X
Подвижная
фаза
Ли.(в\-х
([<Я)-Х]~[(Я)-ХС))Я
([(S)-X]-[(R)-XC])„
Неподвижная
фаза
[<S)-X]S
[(Я)-ХСЬ
([(S)-X]-[(ff)-XC|)s
Рис. 14.8. Равновесия, обуславливающие удеживание R- и 5-энантиомеров (метод
ХДПФ). Индексы т и s относятся к соответствующим компонентам в
подвижной и неподвижной фазах; К„ и Kj представляют собой константы
ассоциации и распределения, соответственно
центрации. ХДПФ (хиральный селектор) может присутствовать в подвижной фазе
и/или удерживаться неподвижной фазой в соответствии с коэффициентом
распределения К „,.. После ввода образца устанавливаются различные равновесия между
анализируемым веществом X и ХС (рис. 14.8). Следует обратить внимание на то, что
индексы т и s относятся к комплексам, находящимся соответственно в подвижной и
неподвижной фазах, и предполагается, что выбрана Л-форма селектора. Эти
равновесия включают:
• образование комплексов между хиральным селектором (R)-XC и энантиомерами
(fi)-X и (S)-X в подвижной фазе с константами ассоциации Кд^щ-х и Ka,(S)-Xi
• распределение (R)-Xv\ (S)-X между подвижной и неподвижной фазами с
константами распределения ^Ш-Х11 ^d.{S)-X''
• распределение комплексов анализируемых веществ Х-ХС между подвижной и
неподвижной фазами с константами распределения ^,(Я)-Х-(^-ХС и ^d,(S)-X-(R) -ХС-
Кроме того, не связанное в комплекс анализируемое вещество {R)-X может
непосредственно взаимодействовать с ХДПФ в неподвижной фазе (процессы i и j
на рис. 14.8). Следует учитывать такие же равновесия для S-энантиомера (S)-X.
Наблюдаемый фактор удерживания анализируемого вещества к представляет собой
средневзвешенную величину значений к для свободного и связанного в комплекс X.
Если ХДПФ очень сильно удерживается в колонке, она может насытить
неподвижную фазу, что приведет к образованию фазы, аналогичной динамически
модифицированной фазе ХНФ (обратите внимание на аналогичную ситуацию в случае с
удерживанием ион-парного реагента на неподвижной фазе, рассмотренную в разделе
7.4.1 и представленную на рис. 7.12).
Схема, изображенная на рис. 14.8, позволяет сделать следующий вывод: без
добавления хирального селектора (R)-XC невозможно разделение энантиомеров (R)-X
14.4. Прямой метод 7l3jl
и (S)-X, поскольку Kd,(R\-X = Kd,(S)-X- Если взаимодействие ХДПФ, присутствующей
в подвижной фазе, с анализируемым веществом является значительным и энантиосе-
лективным, то факторы удерживания к двух энантиомеров должны отличаться, что и
приведет к их хроматографическому разделению. Помимо ассоциации двух
анализируемых энантиомеров с ХДПФ (с различным удерживанием свободного и связанного
в комплекс анализируемого вещества) стереоселективность может также возникать
из-за различного удерживания ассоциатов диастереомеров, а также из-за
образования неодинаковых количеств адсорбатов (по ходу процессов i или у, рис. 14.8). Сте-
реоселективные вклады этих отдельных процессов могут либо усиливать, либо
ослаблять друг друга. По мере увеличения концентрации ХДПФ в подвижной фазе должно
улучшаться разделения двух энантиомеров. Однако при достаточно высоких
концентрациях ХДПФ возможно и ухудшение разделения. Причина заключается в том, что
достаточно высокая концентрация селектора может приводить к комплексообразова-
нию всего количества каждого из энантиомеров в подвижной фазе, в то время как
для различного удерживания энантиомеров предпочтительнее, чтобы один из них
был связан в комплекс в большей степени, чем другой.
Из описания рис. 14.8 должно быть ясно, что выбор ХДПФ и ее концентрации
в подвижной фазе являются решающими факторами энантиоселективности и
успешного разделения двух энантиомеров. Однако, определенную роль могут
играть и другие условия: например, рН подвижной фазы, ионная сила, различные
В-растворители (органические растворители в ОФХ) и температура. В качестве
ХДПФ для хирального разделения с помощью ВЭЖХ использовались а.-, (3- и
■у-циклодекстрины и их производные |18|, хинин и хинидин |19, 20|, (+)- и
(-)-Ю-камфорсульфоновая кислота [20|, /V-бензилоксикарбонил-защищенные ди-
и трипептиды [19|, хелатирующие агенты, такие как аминокислоты в сочетании
с ионами металлов (с применением метода хиральной лигандообменной
хроматографии) [21—23| и другие.
Использование ХДПФ представляется привлекательным из-за его практической
простоты и относительно невысокой стоимости колонок. Однако этот подход
страдает рядом недостатков, некоторые из которых подобны проблемам, возникающим
при добавлении ион-парного реагента к подвижной фазе (раздел 7.4.3):
• требуется строгий контроль температуры, необходимый из-за ее возможного
воздействия на различные равновесия, показанные на рис. 14.8, и, следовательно,
разделение менее надежно;
• наблюдаются системные пики, возникающие в результате различий в составе
растворителя образца и подвижной фазы (раздел 7.4.3.1);
• некоторые виды детектирования не совместимы с присутствием ХДПФ в
подвижной фазе (например, поглощающие в УФ-области добавки с УФ-детектированием,
подавляющие ионы добавки с МС-детектором);
• различны факторы отклика для двух энантиомеров, поскольку они могут
существовать частично в виде диастереомерных комплексов в подвижной фазе, проходя
через ячейку детектора (см. аналогичное обсуждение косвенного метода в
разделе 14.3);
• большой расход и ограниченная доступность реагентов ХДПФ для всех хиральных
соединений.
Из-за многочисленных присущих ему недостатков, сегодня ХДПФ-метод имеет
ограниченное практическое значение для ВЭЖХ-разделения энантиомеров. Однако
следует отметить, что этот метод прочно утвердился в качестве предпочтительного
метода для энантиомерных разделений капиллярным электрофорезом.
Глава 14. Разделение энантиомеров
14.4.2. Использование хиральной неподвижной фазы (ХНФ)
Использование хиральной неподвижной фазы (ХНФ), как правило, является самым
простым и удобным средством для хроматографического разделения энантиомеров,
подходящим как для аналитических, так и для препаративных целей. Хиральный
селектор в основном ковалентно связан с хроматографическим носителем или может
быть прочно физически адсорбирован (например, путем нанесения полимерного
селектора) на носителе (обычно на пористых частицах силикагеля). Подвижная фаза
является ахиральной, то есть не содержит никаких хиральных составляющих.
Во время перемещения образца по колонке энантиомеры удерживаются,
связываясь с селектором, находящимся на неподвижной фазе (аналогично процессу i/ j
на рис. I4.8). Таким образом, на неподвижной фазе образуются диастереомерные
комплексы (R)-X ... (R)-XC и (S)-X ... (R)-XC. Разделение двух энантиомеров
возможно, если значения к для них достаточно отличаются. Следовательно, для успешного
разделения требуется такая ХНФ, которая взаимодействует с одним энантиомером
сильнее, чем с другим. Значения к определяются устойчивостью полученных комплексов
или значениями констант равновесия Ktц и Kt $ на рис. 14.9, как описано в
следующем разделе 14.4.3.
ХНФ и упакованные ими колонки обеспечивают ряд исключительно важных
преимуществ по сравнению с косвенным и аддитивным методами. Например,
анализируемые вещества, не имеющие подходящих функциональных групп для дери-
ватизации, все-таки можно разделить методом ХНФ. Небольшое количество энан-
тиомерных примесей в селекторе может вызвать некоторые сложности только
в том случае, если они уменьшают коэффициент селективности а для двух
энантиомеров |24, 25|. Помимо этого, нет ни одного из осложнений, описанных выше
для косвенного метода с селектором, загрязненным примесями. Проблемы, связан-
Селектор
(ХС)
5-Энантиомер
(S)-X
Носитель
"идеальное соответствие
[(RKSk-KSbXI™ ^Д= (K*)-CS]-!(S)-X])S
Спейсер
Селектор
(ХС)
/?-Энаитиомер
<Я)-Х
НоситеяьЧ т (R)
.■»«**
„..''
неидеальное соответствие
\(R)-XC\S + [(«l-Xlr
K.s
([(RJ-XCl-KRJ-Xl),
Рис. 14.9. Модель трехточечного присоединения и связанные с ней взаимодействия
между R- или .У-энантиомерами и ХНФ
14.4. Прямой метод 7l5jl
ные с присутствием селектора в подвижной фазе на выходе из колонки, также
устраняются при использовании ХНФ. Таким образом, отсутствие селектора
позволяет избежать каких бы то ни было помех при детектировании. Детектор реагирует
одинаково на оба энантиомера, поэтому энантиомерный состав может быть непо
средственно вычислен из отношения площадей регистрируемых пиков R- и
5-энантиомеров без каких-либо поправок. Однако при использовании хирооптиче-
ских детекторов (раздел 4.10) для каждого из энантиомеров может быть
целенаправленно получен различный отклик, и это позволит соотнести пик с
определенным энантиомером как |+| и |-].
К недостаткам ХНФ-метода относятся в большинстве случаев относительно
высокая цена колонок, которые к тому же часто имеют меньший срок эксплуатации,
чем ОФХ колонки; их умеренная эффективность (раздел 14.5), а иногда и низкая хе-
моселективность для структурных аналогов (т.е. селективность для энантиомеров
часто выше, чем для структурных аналогов, которые также могут присутствовать в
образце, что приводит к перекрыванию пиков разделенных (Я) и (S) энантиомеров
пиками этих аналогов). Важно отметить, что выделение энантиомеров с
использованием ХНФ увеличило масштаб препаративного выделения энантиомеров от
лабораторного (от мг до г) до промышленного (от кг до тонны). Привлекательность
ХНФ-разделения заключается в малом времени разработки, простоте выделения
продукта упариванием летучих подвижных фаз, доступности высоких выходов обоих
химически и оптически чистых энантиомеров и в удобстве масштабирования
разделения (раздел 15.1.2.1).
14.4.3. Принципы хирального распознавания
14.4.3.1. «Модель трехточечного присоединения»
Ранние попытки логически обосновать хиральное распознавание на молекулярном
уровне привели к разработке геометрических моделей, таких как модель
трехточечного присоединения |26]. Эта модель все еще часто используется для визуализации и
формулирования требований к энантиоселективности при создании ХНФ. В своем
первоначальном виде эта модель постулирует, что для хирального распознавания
требуются, по крайней мере, три зависящие от конфигурации контактные точки
притяжения между хиральным рецептором и хиральным носителем. Однако часто
игнорируется четвертое существенное требование, а именно тот факт, что рецептор
доступен только с одной стороны, и поэтому к нему можно приблизиться только
с одной стороны. Упомянутая упрощенная модель вызывает споры с тех пор, как она
была впервые сформулирована. В настоящее время установлено, что не все три
взаимодействия должны быть притяжением (правило трех точек по Пирклу [27|).
Применение этого правила для хирального распознавания на ХНФ графически
проиллюстрировано на рис. 14.9.
Значительная путаница с моделью трехточечного взаимодействия возникла
из вопроса: «Что означает «взаимодействие»? |27|. Как отмечалось выше, понятие
«взаимодействие» включает силы притяжения и отталкивания, которые могут либо
стабилизировать, либо дестабилизировать образование комплекса анализируемого
вещества с ХНФ. Более того, в природе многие взаимодействия фактически
многоточечные, что сводит к минимуму необходимость дополнительных вспомогательных
взаимодействий. Например, водородные связи и взаимодействия зарядов,
сконцентрированных на концах диполей, рассматриваются как одноточечные взаимодействия
и поэтому считаются единичными взаимодействиями. В то же время взаимодействия
716 Глава 14. Разделение энантиомеров
между объединенными диполями и я-я-взаимодействия фактически являются
многоточечными, и каждое из них можно считать, по меньшей мере, двухточечным |27|.
Аналогичным образом, для молекул, в которых хиральные центры включены в
жесткие структурные элементы, такие как ароматический циклы, требуется меньшее
количество (или меньше точек) взаимодействий |28|. Две из четырех связей
асимметрических центров включены в жесткое кольцо, которое обеспечивает жесткость
молекулы и облегчает распознавание двух стереоизомеров. Общеизвестно, что одно
взаимодействие с жесткой плоскостью или ее поверхностью можно считать
эквивалентным как минимум двум центрам взаимодействия.
Теперь рассмотрим хиральную неподвижную фазу, которая имеет хиральный
селектор (ХС), связанный с поверхностью подходящей подложки через спейсер
(рис. 14.9). В этой идеализированной модели анализируемое вещество
взаимодействует исключительно с селектором, а не со спейсером или подложкой. Относительное
удерживание энантиомеров анализируемого вещества (R и S) определяется силой их
взаимодействия с селектором, показанным на рис. 14.9, что, в свою очередь, зависит
от трехмерной ориентации и пространственного расположения комплементарных
центров в связывающихся парах. В идеальном случае хиральный селектор и
соответствующий энантиомер анализируемого вещества будут полностью комплементарны
(идеальное соответствие). Этот энантиомер оказывается более крепко связанным
с подложкой \(S)-X на рис. 14.91, что характеризуется большей константой
связывания К,-s равновесной реакции. Поэтому он лучше удерживается и элюируется как
второй пик энантиомера. Для второго энантиомера (Я)-А' возникает значительное
пространственное несоответствие, по крайней мере, на одном из центров
взаимодействия. Следовательно, между (Л)-А" и ХС неидеальное соответствие, так что
соответствующая константа связывания Kt ц будет значительно меньше. Таким образом,
Я-энантиомер будет элюироваться раньше. Термодинамические аспекты ассоциации
анализируемого вещества и селектора, а также процесса адсорбции более подробно
рассматриваются в конце этой главы (см. раздел 14.7.).
Идеальное соответствие и эффективное связывание между анализируемым
веществом и селектором достигается:
• если они комплементарны по размеру и форме, когда анализируемое вещество
стерически соответствует центру связывания селектора, который часто
представляет собой предварительно сформированные карман или щель, как в случае цикло-
декстриновых селекторов (стерическое соответствие);
• если анализируемое вещество и селектор имеют комплементарные центры
взаимодействия (функциональные группы), нужным образом ориентированные в
пространстве, чтобы действовало притяжение, вызванное нековалентными
межмолекулярными взаимодействиями (функциональное соответствие, основанное на
комплементарных взаимодействующих группах). Эти взаимодействия приводят к
ассоциации между энантиомерами анализируемого вещества и селектора и, в основном,
являются электростатическими по своей природе. Они включают:
а ионные взаимодействия (электростатические взаимодействия между
положительно и отрицательно заряженными группами);
а водородные связи (между группами-донорами электронов и
группами-акцепторами с атомом водорода);
а. ион-дипольные, диполь-дипольные (обусловленные ориентационными силами)
взаимодействия, а также взаимодействия между диполями и индуцированными
диполями (обусловленные индукционными силами) и между индуцированными
диполями и мгновенными диполями (обусловленные дисперсионными силами);
а л-л-взаимодействия (электронно-насыщенных и электронно-ненасыщенных
ароматическиех групп, ориентированных параллельно или перпендикулярно);
а другие, такие как квадрупольные, я-катионные, я-анионные взаимодействия;
14.4. Прямой метод 7l7)Jj
• если гидрофобные области селектора и анализируемого вещества пространственно
совпадают, обеспечивая гидрофобное взаимодействие (гидрофобное соответствие);
• если гибкость анализируемого вещества и селектора позволяет оптимизировать
связывание в неподвижной фазе (динамическое соответствие). В частных случаях
индуцированное соответствие благодаря конформационному изменению молекул
анализируемого вещества и селектора при комплексообразовании может еще
больше повысить стабильность комплекса.
Интуитивно можно предположить, что хиральное распознавание улучшается
в случае более высокого сродства (аффинности) между анализируемым веществом и
селектором (то есть, большие константы связывания или низкие константы
диссоциации обычно увеличивают энантиоселективность). Эта концепция, в которой
утверждается, что стереоселективность лекарственных средств будет возрастать с их
эффективностью, давно известна в фармакологии как правило Пфайффера |29|.
Однако справедливость этого правила подвергается сомнению, и в настоящее время
считается, что по разным причинам оно имеет ограниченную применимость.
14.4.3.2. Влияние подвижной фазы
Хиральное распознавание обычно рассматривается как бимолекулярный процесс
взаимодействия анализируемого вещества с ХНФ, при этом влияние подвижной
фазы часто не учитывается. В действительности подвижная фаза может играть
значительную роль в энантиоселективности, определяя степень сольватации
интерактивных центров анализируемого вещества и селектора, а также доступность
центров для межмолекулярного контакта между анализируемым веществом и ХНФ.
Выбор растворителей, буферных солей и рН также являются основными
решающими факторами конформационных предпочтений и степени ионизации как
анализируемых веществ, так и ХНФ. Подвижная фаза также может подавлять
вредные неспецифические взаимодействия, ухудшающие энантиоселективность
(раздел I4.7.3). Следовательно, правильный выбор подвижной фазы представляет
собой важный шаг в разработке как непрямого, так и (особенно) прямого метода
разделения энантиомеров.
По причинам, приведенным выше, подвижная фаза должна рассматриваться
как важный фактор разделения энантиомеров на ХНФ, поскольку она определяет
среду взаимодействия, в которой происходит хиральное распознавание.
Растворители могут создавать помехи или стимулировать специфические взаимодействия
анализируемое вещество-селектор и, таким образом, влиять на энантиоселективное
распознавание. Растворители с высокой полярностью ослабляют силу
электростатических взаимодействий, а гидрофобные взаимодействия осуществляются только
в водных или водноорганических подвижных фазах. рН подвижной фазы особенно
важен в случае ионизируемых анализируемых веществ или ХНФ, так как ионные
взаимодействия требуют ионизированных объектов. Ионные взаимодействия могут
ослабевать при увеличении ионной силы подвижной фазы, как и в случае
удерживания в ионообменной хроматографии (раздел 7.5.2). Множество различных
факторов оказывают влияние на выбор подвижной фазы, но они одинаковы
для всех видов ЖХ: обращенно-фазовой, нормально-фазовой, ионообменной и т.д.
Дополнительная информация о выборе подвижной фазы для отдельных ХНФ
приводится в разделе I4.6; см. также соответствующие разделы в предыдущих главах
книги.
718 Глава 14. Разделение энантиомеров
14.5. Дисперсия и уширение задних фронтов
пиков
По сравнению с ОФХ-колонками эффективность энантиоселективных колонок
(см. раздел I4.6) часто довольно низкая. Число теоретических тарелок редко
превышает jV = 40 000/м для колонок с размером частиц 5 мкм, а задние фронты пиков
часто размываются в большей степени, чем в других видах ВЭЖХ. Можно
предположить, что эффективность энантиоселективных колонок зависит от тех же факторов,
что и для других ВЭЖХ-колонок (раздел 2.4.1), поэтому, вероятно, задействован
какой-то дополнительный фактор, а именно медленная кинетика
адсорбции-десорбции на хиральных центрах. Общепризнано, что процесс десорбции может быть
медленным из-за образования комплексов анализируемого вещества с селектором,
которые стабилизируются за счет одновременных множественных взаимодействий.
Медленную кинетику можно улучшить, используя более высокие температуры, но
при повышении температуры энантиоселективность часто уменьшается.
Следовательно, нужно признать, что более низкая эффективность многих ХНФ-колонок
представляет собой фундаментальную проблему, и очевидных способов ее решения нет.
14.6. Хиральные неподвижные фазы
и их характеристики
Успех в использовании энантиоселективных колонок зависит от выбора такой ХНФ,
на которой удается разделить целевые энантиомеры. Поэтому важно иметь сведения
об имеющихся в продаже хиральных фазах, а также об их основных характеристиках
и типичных условиях эксплуатации. Кроме того, может оказаться полезным
понимание механизма дискриминации энантиомеров и областей оптимального применения
конкретных ХНФ. Эти вопросы будут рассмотрены в настоящем разделе.
Был исследован широкий спектр хиральных молекул, как природного, так и
синтетического происхождения, с точки зрения перспективности их использования
для создания ХНФ. На сегодняшний день предлагается на коммерческой основе
несколько сотен ХНФ, среди которых наиболее часто используются 20—30 ХНФ.
Колонки, ими упакованные, позволяют разделять большинство энантиомеров, которые
с высокой вероятностью потребуется анализировать. Хиральные селекторы для этих
колонок относятся к следующим классам ХНФ:
• макромолекулярные селекторы полусинтетического происхождения
(полисахариды, раздел 14.6.1);
• макромолекулярные селекторы синтетического происхождения (поли(мет)акрила-
миды, политартрамиды, раздел 14.6.2);
• макромолекулярные селекторы природного происхождения (белки, раздел 14.6.3);
• макроциклические олигомерные селекторы или селекторы промежуточного
размера (циклодекстрины, макроциклические антибиотики, хиральные краун-эфиры,
раздел 14.6.4—14.6.6);
• синтетические нейтральные объекты с низкой молекулярной массой (фазы Пирк-
ла или ХНФ щеточного типа, раздел 14.6.7);
• синтетические низкомолекулярные иониты, обеспечивающие ионный обмен
(раздел 14.6.8);
• хелатирующие селекторы для хиральной лигандообменной хроматографии (раздел
14.6.9).
14.6. Хираяьные неподвижные фазы и их характеристики 719
В последующих разделах приведены наиболее важные характеристики наиболее
часто используемых коммерчески доступных ХНФ и энантиоселективных колонок.
Эти подробные сведения о том, как на различных ХНФ достигается энантиоселек-
тивность, могут быть полезны при первоначальном подборе перспективных ХНФ и
других условий хирального разделения. Далее следуют эксперименты методом проб и
ошибок для достижения приемлемого разделения. Конкретные рекомендации по
использованию определенных, более предпочтительных ХНФ и условий разделения выделены
курсивом.
14.6.1. ХНФ на основе полисахаридов
Полисахаридные селекторы традиционно используются в энантиоселективной ЖХ.
В 1973 году Хессе и Хагель предложили для энантиоселективной ЖХ триацетат
микрокристаллической целлюлозы (ТМКЦ) в качестве хроматографического материала
без опорной матрицы, обладающего свойствами полимерного селектора |30|. К
достоинствам ТМКЦ можно отнести широкий спектр энантиоселективности и
высокую адсорбционную емкость в препаративных разделениях, а к недостаткам -
низкую устойчивость при повышенном давлении, низкую скорость разделения и низкую
хроматографическую эффективность. Решение проблемы механической прочности
ТМКЦ было предложено Окамото и сотрудниками в I984 году. Производные
целлюлозы были нанесены в количестве примерно 20% по весу на поверхность
макропористых гранул силикагеля (с размером пор 100 или 400 нм) |31|. При этом значительно
улучшилась механическая прочность, повысились эффективность и скорость
разделения энантиомеров в условиях ВЭЖХ. Покрытые полисахаридами ХНФ считались
в течение нескольких десятилетий самыми передовыми.
В последующие годы Окамото с сотрудниками синтезировали большое
количество различных производных полисахаридов, главным образом сложных эфиров и кар-
баматов целлюлозы (состоящих из звеньев (1,4)-р-0-глюкозы) и амилозы (состоящих
из звеньев (1,4)-а-0-глкжозы). Эти производные были нанесены на
широкопористый силикагель (рис. 14.10). Коммерчески доступны три наиболее универсальных
полисахаридных ХНФ от Daicel Chemical Industries, Ltd. и Chiral Technologies:
• Chiralcel® OJ на основе трис(4-метилбензоата) целлюлозы;
• Chiralcel® OD на основе трис(3,5-диметилфенилкарбамата) целлюлозы;
• Chi ral pak® AD на основе трис(3,5-диметилфенилкарбамата) амилозы.
С момента истечения срока действия патентов на эти материалы стали доступны
дженерики, изготавливаемые несколькими поставщиками (например, Eka Chemicals,
Regis, Macherey-Nagel, Phenomenex) под разными торговыми названиями (например,
Kromasil®, CelluCoat™ и AmyCoat™ от компании Eka; Regis Cell фирмы Regis, Lux
Cellulose от компании Phenomenex). Однако общие характеристики этих дженериков
часто значительно отличаются от исходных продуктов с точки зрения способности
к хиральному распознаванию, удерживанию и эффективности колонок, поскольку
для их изготовления используются другие подложки и протоколы нанесения
покрытия (как и в случае неравнозначности качества всех ОФХ-колонок C|jj).
Daicel и Chiral Technologies (как и другие поставщики) предлагают эти ХНФ
для работы в нормально-фазовом режиме с алкано-спиртовыми элюентами (или
аналогичными элюентами) и в обращенно-фазовом режиме с водно-органическими
подвижными фазами (обычно смесями органических растворителей с водными
буферами). Предполагается, что у этих фаз селектор нанесен на носители различного типа.
В инструкции по эксплуатации рекомендуется использовать колонки с маркировкой
R в обращенно-фазовом режиме, в то время как для других колонок следует избегать
водных подвижных фаз. Какая бы ни была подвижная фаза, настоятельно рекомен-
Глава 14. Разделение энантиомеров
Полимерный каркас Остаток
о-
R_<o
Целлюлоза
CI
«<
сн.
Амилоза
Торговые названия Покрытые
a Chiracel OJ
b Chiracel OD
с Нет в продаже
d Chiralpak AD
Ограниченный
диапазон
растворителей
Название
Трис (4-
метилбенэоат)
целлюлозы
Трис (3,5-
диметилфенил-
карбамат)
целлюлозы
Трис (3,5-
дихлорфенил-
карбамат)
целлюлозы
Трис (3,5-
диметилфенил-
карбамат)
амилозы
Иммобилизованные
Нет в продаже
Chiralpak IS
Chiralpak 1С
Chiralpak IA
Полный
диапазон
растворителей
Торговое
название
а
Рис. 14.10. ХНФ на основе полисахаридов, полученные из производных целлюлозы и
амилозы, с соответствующими торговыми названиями колонок
дуется использовать колонку только в данном конкретном режиме и избегать
переключения между обращено-фазовыми и нормально-фазовыми условиями.
Маркировка Н означает сорбент для ВЭЖХ на основе частиц размером 5 мкм. Совсем
недавно для обеспечения более высокой эффективности колонки и большей
скорости разделения для высокопроизводительного хирального анализа стали производить
ХНФ на основе трехмикронных подложек (Chiralpak AD-3, Chiralcel OD-3' и
соответствующие обращенно-фазовые версии — Chiralpak AD-3R, Chiralcel OD-3R).
Однако из-за медленной кинетики процесса адсорбции-десорбции (раздел 14.5)
преимущество использования меньших частиц может быть менее значительным, чем
для ахирального разделения.
Уникальная универсальность хирального распознавания, свойственная полисаха-
ридным ХНФ, позволяет использовать их для очень широкого диапазона типов
образцов. Эта особенность обусловлена различными молекулярными и
надмолекулярными структурными особенностями этих полусинтетических макромолекул.
В настоящее время принципы хирального распознавания на полисахаридных ХНФ
все еще остаются неясными, несмотря на широкое использование и большое
значение этих ХНФ. В основном данные для анализа молекулярного распознавания
полисахаридами почерпнуты из хроматографических экспериментов, а количество
исследований, посвященных этой теме, ограничено. В частности, проведено изучение
методом ЯМР растворов олигомеров Сахаров 132—34| и твердых образцов [35|,
опубликованы результаты расчетных исследований |34—371, исследований методом инф-
14.6. Хиральные неподвижные фазы и их характеристики 721
ракрасной спектроскопии ослабленного полного отражения [351, изучения
термодинамики 136, 38, 391 и количественной зависимости свойств от структуры |39—42|.
Однако эти исследования не позволили получить конкретные рекомендации для
более эффективного использования полисахаридных ХНФ.
В работе 1331 показано, что глюкопиранозные цепи в производных целлюлозы
и амилозы образуют спирали, причем спиральное скручивание1 менее выражено
для производных целлюлозы по сравнению с производными амилозы (трис(3,5-ди-
метилфенилкарбамат) амилозы имеет структуру левой спирали 4/3). Хиральное
распознавание ХНФ полисахаридного типа может быть обусловлено тремя различными
функциональными особенностями: (I) молекулярной хиральностью из-за наличия
нескольких стереогенных центров у структурных единиц глюкопиранозы, (2) кон
формационной хиральностью из за спирального скручивания каркаса полимера и
(3) надмолекулярной хиральностью из-за группирования соседних полимерных
цепей, образующих хиральные полости. Эти особенности, еще более усиленные дери
ватизацией полисахарида, обеспечивают исключительные и универсальные
возможности избирательного стереораспознавания, свойственные современным селекторам
полисахаридного типа.
Из этой общей механически составленной картины ясно, что распознавание
энантиомеров ХНФ полисахаридного типа определяется не только типом, но и
специфической функциональностью соответствующего производного целлюлозы и
амилозы. Влияние ароматических заместителей на хиральное распознавание трис-фе-
нилкарбаматными производными целлюлозы подробно изучено Окамото |43|.
Хроматографические данные были получены для набора из 18 ХНФ, покрытых
различными моно- или дизамещенными фенилкарбаматными производными. Было
обнаружено, что индуктивные и стерические эффекты имеют решающее значение
для общей способности к хиральному распознаванию. Ортопроизводные,
независимо от природы заместителя, работали плохо. Пара-замещенные производные,
имеющие метил-, этил-, хлор- и трифторметильные группы, обеспечивали улучшение
распознавания энантиомеров. Наилучшие результаты были получены с 3,4- и 3,5-ди-
метилфенил- и дихлорфенилкарбаматными производными, которые впоследствии
были выбраны для изготовления коммерческих ХНФ. Однако для конкретных
анализируемых веществ ХНФ на основе каких-то других производных могут давать еще
более высокую энантиоселективность. По этой причине целесообразно включать в
процесс скрининга также и менее распространенные ХНФ, особенно для
препаративных разделений (см. подробные списки колонок Daicel и Chiral Technologies,
упакованных сорбентами, покрытыми производными полисахаридов).
Как отмечалось выше, надмолекулярная структура ХНФ полисахаридного типа
оказывает огромное влияние на их энантиоселективность. В ранних исследованиях
Хессе и Хагеля |30| микрокристаллической целлюлозы было продемонстрировано,
что этот полимер значительно измененял характеристики хирального распознавания
после растворения и повторного осаждения. Этот факт объяснялся с точки зрения
специфических энантиоселективных центров связывания (микрокристаллических
доменов). Аналогичным образом Франкотт и Чжан [44| обнаружили, что
надмолекулярная организация полисахаридных производных тоже оказывает большое влияние
на энантиоселективность покрытых этими производными ХНФ. Нанесение
покрытий трис-(4-метилбензоата) целлюлозы, растворенного в различных растворителях,
приводило к образованию фаз с совершенно различной энантиоселективностью, и
в некоторых случаях инвертировался порядок элюирования энантиомеров с этих
фаз [44|. Эксперименты с использованием рентгеновской дифракции подтверждают
1 Вероятно, имеется в виду, что расстояние между витками спирали в производных целлюлозы
больше, чем в производных амилозы. — Прим. перев.
722 Глава 14. Разделение энантиомеров
гипотезу о том, что различную энантиоселективность можно объяснить изменениями
надмолекулярной структуры, вызванными растворителем. Эти данные подчеркивают
тот факт, что селекторы полисахаридного типа очень чувствительны к внешним
факторам, таким как растворители, температура, а также добавки, которые могут
приводить к конформационным изменениям ХНФ, сопряженным с изменением процессов
связывания, эффектами памяти и т.д.
Сложность процессов хирального распознавания для ХНФ полисахаридного
типа затрудняет рациональный подход к разработке методов анализа. Современные
стратегии разработки методов хирального анализа основаны на подборе различных
полисахаридных ХНФ методом проб и ошибок в условиях многократного
варьирования подвижной фазы, часто с использованием полностью автоматизированной сие
темы переключения колонок и растворителей. В ряде исследований основное
внимание было уделено нахождению эффективных процедур скрининга для получения
максимальных шансов на успех [45-48|. Наиболее перспективными ХНФ в
нормально фазовом режиме являются
Chiralpak AD > Chiralcel OD > Chiralce] OJ.
Если проводится последовательный, а не параллельный скрининг, колонки следует
тестировать в указанной последовательности [45|. Интересно, что в обращенно-фазо-
вом режиме колонки Chiralce] OD-RH представляются, по-видимому, более
предпочтительными, чем Chiralpak AD-RH. Эти расширенные скрининговые исследования
также показывают, что полисахаридные ХНФ имеют чрезвычайно широкую сферу
применения. Например, Борман и сотр. опубликовали результаты комплексного скри-
нингового исследования для набора из более чем 100 рацематов различного
химического строения, проведенного с использованием методов и систем разделения ВЭЖХ,
СФХ (сверхкритической флюидной хроматографии) и капилярного
электрофореза |49|. Имея только три ХНФ полисахаридного типа (Chiralpak AD, Chiralcel OD и
Chiralcel OJ), авторы смогли разделить более 70% рацематов. Было также обнаружено,
что отдельные полисахаридные производные часто демонстрируют комплементарное
хиральное распознавание структуры анализируемого вещества, т.е. в разделениях
наблюдается различная энантиоселективность (а) и различный порядок элюирования
[50, 51 ]. Это особенно справедливо для трис-(3,5-диметилфенил)-производных
целлюлозы и амилозы (Chiralcel OD и Chiralpak AD), на которых для данного вещества часто
наблюдается противоположный порядок элюирования энантиомеров.
Полисахаридные ХНФ традиционно применялись для колонок,
использовавшихся в нормально-фазовом режиме, и предпочтительной подвижной фазой для них
является гексан или гептан с 2-пропанолом или этанолом в качестве сильных
растворителей (В-растворителей) |52]. Следует отметить, что при использовании Chiralpak
AD с 2-пропанолом селективность часто отличается от наблюдаемой с этанолом,
причем при замене одного из этих растворителей на другой возможно
инвертирование порядка элюирования |53|. Недавно независимое исследование с
использованием твердотельной ЯМР-спектроскопии подтвердило, что 2-пропанол более
эффективно вытесняет гексан из полимерного селектора, образуя более упорядоченные
«комплексы растворитель-ХНФ» |54|. При замене подвижной фазы необходимо
обеспечить достаточно длительное уравновешивание, чтобы полностью удалить
полярный растворитель. Для получения воспроизводимых результатов при переходе
от подвижной фазы без добавок к подвижной фазе с добавкой (например, 0,1%
трифторуксусной кислоты или диэтиламина) и наоборот, может потребоваться
длительное время для уравновешивания |55|.
В редких случаях использование определенных добавок может вызывать
временные или даже постоянные конформационные изменения полимерной структуры
14.6. Хиральные неподвижные фазы и их характеристики 723
производного полисахарида, усиливающие или ослабляющие его первоначальную
энантиоселективность. Было продемонстрировано устойчивое изменение
селективности ХНФ на основе амилозы после работы с подвижными фазами для
нормально-фазовой хроматографии, содержащими диэтиламин |56|. Однако было показано,
что после промывки 2-пропанолом колонка восстанавливает первоначальную
селективность. Обработка кислотой Chiralpak AD-H и Chiralcel OD-H также изменяла их
работоспособность, которая в значительной степени восстанавливалась при
промывке подвижными фазами, содержащими амины. Кроме того, изменения в энантиосе-
лективности могут возникать из-за конформационных изменений селекторов,
вызванных изменением температуры.
Хотя полисахаридные ХНФ используются, главным образом, в
нормально-фазовом режиме, они применимы, как было показано, и в других режимах. Помимо нор
мально-фазового режима и уже упомянутого обращенно фазового режима [57], эти
ХНФ можно использовать в полярно-органическом режиме (ПО). В ПО режиме испо
льзуются неводные подвижные фазы, состоящие из полярных органических
растворителей, таких как метанол или ацетонитрил, или смеси того и другого, с добавками
небольшого количества органических кислот (уксусной кислоты, муравьиной
кислоты) и оснований (триэтиламина, диэтиламина, аммиака) в качестве буферных
составляющих или конкурирующих агентов. Важным преимуществом ПО режима
по сравнению с нормально-фазовым разделением является лучшая совместимость
со все более популярным методом МС-детектирования с ионизацией электроспреем,
а также более высокая растворимость анализируемых веществ и полярных органиче
ских матриц образцов в полярной органической подвижной фазе.
Изменение энантиоселективности для одного и того же соединения часто
наблюдается при переходе от нормально-фазового режима к обращенно-фазовому [47|.
Как уже отмечалось, были разработаны специальные модификации Chiralpak
AD-RH, Chiralcel OD-RH и Chiralcel OJ-RH для анализов в обращенно-фазовом
режиме |57|. В этих сорбентах фазы нанесены на подложку, совместимую с ОФ-разде-
лениями. Для нейтральных анализируемых веществ можно использовать простые
водно-органические смеси (причем ацетонитрил предпочтительнее этанола, 2-пропа-
нола или метанола). В то время как основные или кислотные добавки мало влияют
на разделение нейтральных анализируемых веществ, при разделении ионизируемых
соединений, наоборот, требуются такие добавки для улучшения форм пиков.
Фосфорная кислота (рН 2) рекомендуется для кислотных анализируемых веществ. Энан-
тиомерное разделение основных веществ может быть улучшено добавлением хаот-
ропных противоионов, таких как перхлорат или гексафторфосфат. В качестве
альтернативы для эффективного подавления диссоциации основных анализируемых
веществ могут быть использованы основные буферные системы с рН 9
(предпочтителен боратный буфер относительно небольшой концентрации) [57|. В разделениях
с МС-детектированием необходимо использовать летучие буферные системы.
Для кислотных анализируемых веществ фосфат можно заменить муравьиной
кислотой с рН > 3, для основных борат может быть заменен бикарбонатом аммония
с рН 9. Тем не менее, следует иметь в виду, что длительное воздействие щелочной
среды с рН 9 неизбежно сократит срок службы колонок, как это происходит с
большинством неподвижных фаз на основе силикагеля.
Как упомянуто выше, полисахаридные ХНФ также полностью совместимы с
полярно-органическими подвижными фазами |58, 591- В достаточно подробном
исследовании, опубликованном Лайнемом и Стрингхэмом |59|, было показано, что из 80
тестируемых соединений около 30% можно разделить с использованием чистого
метанола, этанола или ацетонитрила в качестве подвижных фаз. При добавлении гекса-
на удерживание анализируемого вещества, как и ожидалось, возрастало, в то время
как влияние на энантиоселективность было незначительным. Авторы пришли к вы-
724 Глава 14. Разделение энантиомеров
воду, что процессы хирального распознавания, проходящие в полярно-органических
подвижных фазах, могут быть подобны процессам, характерным для
нормально-фазовых условий.
Наиболее серьезным ограничением для покрытых полисахаридами ХНФ является
их несовместимость с некоторыми видами растворителей, так называемыми
нестандартными растворителями, среди которых {помимо прочих) дихлорметан, хлороформ,
этилацетат, тетрагидрофуран, диоксан, толуол и ацетон (рис. 14.11). Под действием
указанных растворителей покрытые полисахаридами ХНФ набухают и/или происходит
растворение физически адсорбированного полимерного слоя с разрушением сорбента.
Таких растворителей следует строго избегать, даже в качестве компонентов матрицы
образца. Чтобы устранить это ограничение, несколько исследовательских групп
разработали полностью устойчивые к растворителям ХНФ с иммобилизованными на них
разными методами полисахаридами |61—63|. С 2005 года набор из трех ХНФ с
иммобилизованными полисахаридами стал коммерчески доступным от фирм Daicel и Chiral
Technologies:
ф Chiralpak® IA (иммобилизованная версия Chiralpak AD) |64|;
ф Chiralpak* IB (иммобилизованная версия Chiralce] OD) [60|;
ф Chiralpak^lC |65| с селектором трис-(3,5-дихлорфенилкарбаматом) целлюлозы,
который не производится в виде покрытия (см. рис. 14.10).
Было продемонстрировано, что иммобилизованные ХНФ полностью
совместимы с любыми органическими растворителями, включая нестандартные растворители,
которые нельзя использовать с ХНФ, покрытыми полисахаридами (рис. 14.11).
Кроме того, ХНФ с иммобилизованными полисахаридами нового поколения также хоро
шо работают в подвижных фазах, используемых в режимах ПО, ОФ и СФХ.
В то время как возможность применения дополнительных растворителей
предоставляет уникальные возможности и придает значительную гибкость разработке
методов, она обязательно предполагает более расширенный и тщательный скрининг,
сопряженный с дополнительными трудозатратами. Для решения этой проблемы в
результате систематических исследований была создана эффективная общая стратегия
Стандартные подвижные фазы
Нормально-фазовый режим:
Алкзны/спирты
Полярно-органический режим:
Ацетонитрил,
Спирты (Этанол, метанол)
Нестандартные подвижные
фазы
МТБЭ. Толуол, Хлороформ,
Дихлорметан, Этилацетат, ТГФ,
1,4-диоксан. Ацетон. ДМСО (сила
растворителей увеличивается в
таком порядке)
Рис. 14.11. Допустимые подвижные фазы для ХНФ на основе полисахаридов.
Различают стандартные растворители для использования со всеми ХНФ и
нестандартные растворители для использования только с иммобилизованными
ХНФ (МТБЭ, метил-трет-бутиловый зфир) |60|
Покрытые Иммобилизованные
ХНФ ХНФ
• •
Запрещены!
Они
необратимо
разрушат
покрытые
ХНФ
14.6. Хиральные неподвижные фазы и их характеристики 725
скрининга, основанная на трех коммерчески доступных иммобилизованных ХНФ и
ограниченном наборе подвижных фаз. Используя Chiralpak IA, IB и 1С и только пять
стартовых подвижных фаз для разработки метода (алкан-2-пропанол 80:20, алкан-этанол
80:20, МТБЭ-этанол 98:2, алкан-тетрагидрофуран 70:30 и алкан-дихлорметан-этанол
50:50:2), удалось разделить до базовой линии приблизительно 90% из 70 случайно
выбранных хиральных анализируемых веществ [66]. Интересно отметить, что эта
стратегия скрининга с нестандартными растворителями и ХНФ с иммобилизованными
полисахаридами обеспечивала иную энантиоселективность, которая не достигалась
на колонках, упакованных неподвижными фазами с полисахаридными покрытиями
и с подвижными фазами стандартного типа. В некоторых случаях разделение
значительно улучшилось |60|.
Полностью совместимые с растворителями, иммобилизованные ХНФ также обла
дают уникальными преимуществами для препаративных разделений. В частности,
низкая растворимость хиральных соединений в стандартных подвижных фазах
для нормально-фазовых разделений часто является самым опасным узким местом
в разработке препаративного разделения энантиомеров на полисахаридных ХНФ,
которые, как правило, имеют одну из самых высоких емкостей по загружаемому
образцу среди всех коммерчески доступных ХНФ [61]. Это особенно справедливо для поляр
ных анализируемых веществ, которые часто плохо растворяются в подвижных фазах
на основе алканов, применяемых в нормально фазовых разделениях. С
иммобилизованными ХНФ для растворения образцов могут быть использованы сильные растворители,
такие, как тетрагидрофуран, дихлорметан, хлороформ и даже диметилсульфоксид.
При этом можно не беспокоиться о стабильности колонок. Не являются проблемой и
остаточные нестандартные растворители в образце. Другой полезной особенностью
ХНФ с иммобилизованными полисахаридами является то, что они могут быть
регенерированы сильными нестандартными растворителями, такими как тетрагидрофуран,
диметилформамид и даже диметилсульфоксид, что позволяет эффективно удалять
прочно адсорбированные компоненты образца и улучшать воспроизводимость
последующих разделений.
Может возникнуть проблема, если метод, разработанный для ХНФ, покрытых
полисахаридами, переносится на иммобилизованные ХНФ. Сам процесс
иммобилизации, по-видимому, изменяет характеристики разделения энантиомера по
сравнению с разделением на ХНФ с покрытием [60, 67, 68|. Например,
энантиоселективность может быть потеряна при замене колонки Chiralcel OD (с полисахаридным
покрытием) на колонку Chiralpak IB (с иммобилизированным полисахаридом)
при использовании стандартной подвижной фазы на основе алканов (рис. 14.126-е).
При этом в обеих ХНФ используется одно и то же производное полисахарида. Тем
не менее, после повторной оптимизации состава подвижной фазы (включающей
использование соответствующих нестандартных растворителей) можно получить
значительно улучшенное разделение (сравните рис. 14А2а-б и в). Сопоставимые
результаты были получены с кислотными (рис. \4.)2а-в), основными (рис. 14.[2г-е) и
нейтральными соединениями. Эти примеры демонстрируют определенный уровень
взаимодополняемости (комплементарности) ХНФ, покрытых полисахаридами, и
ХНФ с иммобилизованными полисахаридами, которая дает преимущество при
разработке как аналитического, так и препаративного методов. Комбинированное
использование обоих типов ХНФ может обеспечить хроматографистов еще более
мощным инструментом для решения задач разделения все возрастающей сложности. Вне
всякого сомнения, внедрение технологии иммобилизации полисахаридов на ХНФ
стало важной вехой в развитии разделения энантиомеров.
Следует отметить, что полисахаридные ХНФ в настоящее время выбирают в
первую очередь в качестве хиральных фаз для разделения энантиомеров методом
СФХ [69—721.
726 Глава 14. Разделение энантиомеров
Chiralpak®IB
(иммобилизованный)
Chiralpak®IB
(иммобилизованный)
Chiralcel®OD-H
(покрытый)
W
гексан/СНС13/ЕЮН/ТФА
68/30/2/0.1 об/об
т
-гтр-
10
1
мин
гексан/ЕЮН/ТФА
90/10/0.1 об/об
У Ч
10
■*-|-г-
10
W
МТБЭ/ЕЮН/ЭДА
95/5/0,1 об/об
L
«I
гексан/ЕЮН/ЭДА
70/30/0,1 об/об
/ ч
W
LL
I " ' I ' ' > 1 I ■ ' I ■ I" "I I Г"|"
0 2 4 в 8 10 12 мин 0 2 4 6 8 10 12 14
I' " ч >' ч ' ■ ч " ' г' ■ ч ■ > < i'
0 2 4 в 8 10 12
Рис. 14.12. Влияние типа колонки (ХНФ с иммобилизованным полисахаридом или
полисахаридным покрытием) и подвижной фазы на энантиоселективность.
Разделение энантиомеров Л'-бензилоксикароонил-фенилаланина (о-в)
и лауданозина (г-е) на Chimlpak IB (с иммобилизованным полисахаридом)
и Chiralcel OD (с полисахаридным покрытием). Расход, 1 мл/мин,
температура, 25 "С, УФ-детектирование при 230 нм. Обратите внимание на то,
что подвижные фазы, используемые в (а) и (г), являются запрещенными
подвижными фазами для колонки Chiralcel OD с полисахаридным
покрытием (этилендиамин, МТБЭ) [60|
14.6.2. Синтетические полимерные ХНФ
В качестве потенциальных селекторов при попытке имитировать
энантиоселективность полусинтетических полисахаридов, описанных в разделе 14.6.1, был предложен
ряд хиральных синтетических полимеров. Как и полисахариды, хиральные
синтетические полимеры состоят из идентичных хиральных блоков. Тем не менее, с
помощью синтетических полимеров не достигается такое же эффективное энантиораспоз-
навание, как с помощью полисахаридных ХНФ. Это может быть результатом гораздо
менее упорядоченной структуры полимерных цепей, а также отсутствием
предварительно организованных полостей и щелей для включения анализируемого вещества.
Доказательство того, что только упорядоченная хиральная гиперструктура
является достаточной для эффективного хирального распознавания, было предоставлено
Окамото и др. |73|. ХНФ была изготовлена из поли-(трифенилметакрилата) с
одинаково направленными спиральными молекулами, который был получен анионной
полимеризацией ахиральных мономеров с использованием хирального катализатора.
14.6. Хиральные неподвижные фазы и их характеристики 727
Этим полимером затем покрывали макропористый силикагель. Позднее компании
Daicel и Chiral Technologies выпустили на рынок колонку, упакованную этим
сорбентом, под торговым названием Chiralpak OT+ (рис. 14.13д). На этой колонке
успешно разделили ряд хиральных соединений, обладающих несколькими
ароматическими функциональными группами, такими как 1,Г-бинафтил-2,2'-диол (а = 2,0
с метанолом в качестве подвижной фазы при 5 °С). Поскольку эта ХНФ
недостаточно химически устойчива, теперь она представляет скорее академический, чем
практический интерес.
В 1974 году Блашке разработал новые полимерные материалы в форме сшитых
поли(мет)акриламидных полимерных гранул без подложки |74], полученных
суспензионной полимеризацией акрил- или метакриламидов, причем хиральным
элементом является стереогенный центр в амидной боковой цепи. Так как эти полимеры
сильно набухали и обладали невысокой механической устойчивостью в разделениях
при высоким давлении, позднее были разработаны нанесенные на силикагель
композиционные материалы с более подходящими хроматографическими
свойствами |75|. ХНФ, полученная путем сополимеризации мономера этилового эфира iV-ак-
рилоил-(5^-фенилаланина с частицами силикагеля, модифицированного виниль-
ными группами, была выведена на рынок Merck KGaA (Дармштадт, Германия)
под торговым названием ChiraSpher (рис. 14.136). Эта ХНФ по-прежнему
используется не только для аналитических, но и для препаративных разделений |61]. В
частности, она оказалась полезной для разделения различных полярных
фармацевтических препаратов с донорно акцепторными функциональными группами в нор
мально-фазовом режиме (обычно применяли н-гексан или н гептан с полярными
(б)
(-)-спартеин или
другой хиральный
катализатор
Chiralpak OT(+) или ОТ(-)
(Daicel/Chiral Technologies)
ЕЮОС w
ChiraSpher
(Merck, Дармштадт, Германия)
—i—
10
—i г
15 20 (мин)
Рис. 14.13. ХНФ на основе хиральных полнметакрилатов. (а) ХНФ на основе поли(три-
тилметакрилата) со спиральной хиральностью, полученного изотактиче-
ской анионной полимеризацией в присутствии спартеина в качестве
катализатора (Chiralpak'^OT (+) или ОТ (-), Daicel и Chiral Technologies);
(б) ХНФ с лоли^-акрилоил-О^-фенилаланин-этиловым эфиром|
(ChiraSpher*' фирмы Merck KGaA, Дармштадт, Германия). Хроматографические
условия в (6): размер колонки 250x4,6 мм; подвижная фаза 80:20 н-гек-
сан/2-пропанол; скорость потока 1 мл/мин; температура, 25 °С. |77| (а)
и [78| (6)
728 Глава 14. Разделение энантиомеров
растворителями, такими как спирты, диоксан или ТГФ). Полный обзор по этой теме
был опубликован Кинкелем |76|.
Синтетические полимерные фазы обычно получают классическим методом
«прививки к» подложке, т. е. синтезом полимера из мономеров в растворе с
последующим закреплением его на носителе, предварительно модифицированном винильны-
ми группами для сополимеризации. Такие полимерные ХНФ часто создают
проблемы из за блокирования пор и неоднородного распределения полимера по по
верхности силикагеля. Это, в свою очередь, приводит к более низким значениям
числа теоретических тарелок для полимерных ХНФ по сравнению с фазами щеточ
ного типа (раздел 14.6.7).
Не так давно Гаспаррини с коллегами предложили альтернативный метод
для получения композитных ХНФ на основе силикагеля. Вместо «прививки к»
подложке готового полимера (рис. 14.14а) они прикрепляли хиральные фрагменты
селекторов к поверхности частиц силикагеля (рис. 14.146), после чего проводилась
полимеризация с ростом полимера от подложки (метод «прививки от») [79|. На первом
этапе радикальный инициатор («G») иммобилизуется на поверхности подложки.
После добавления мономеров и инициирования полимеризации путем нагревания,
полимерные цепи начинают расти на поверхности, образуя равномерный, хорошо упо-
рядоченный слой полимера. Побочные полимерные цепи, выросшие в растворе,
могут быть удалены на стадиях промывки. Эта методика была успешно выполнена
с использованием мономеров (транс- 1,2-диамино-1,2-дифенилэтана)-^У,Лг-диакрила-
мида и (транс-[,2-диаминоциклогексан)-Л,,/У-диакриламида (фирма ASTEC1
поставляла обе энантиомерные формы этих фаз под торговыми названиями Р CAP DP и
Р-САР). Синтез фазы со вторым мономером показан на рис. 14.14ff. ХНФ, подобные
изображенным на рис. 14.146, показали значительно более высокую эффективность
по сравнению с контрольными сорбентами, полученными обычной прививкой
готового полимера, как на рис. 14.14а. Широкий спектр хиральных соединений, таких
как бензодиазепины, карбоновые кислоты и сульфоксиды, сложные эфиры, амиды,
лактоны и аминокислоты с защищенной аминогруппой, можно разделить, главным
образом, в режимах НФ или ПО.
Новый класс ХНФ на основе полимеров сетчатого типа был предложен Аллен-
марком и другими |SO, 811. Получение этих ХНФ начинается с хиральных мономеров
0,0'-диароил-Л/,Л,,-диаллил-(Я,Д)-тартрдиамида, которые иммобилизуют на вини-
лизированном силикагеле путем сшивания мономеров с многофункциональными
гидросиланами, что приводит к получению сетчатого полимера, включающего
бифункциональный С2-симметричный хиральный селектор в виде тонкой пленки
на поверхности силикагеля (рис. 14.15а). Благодаря формированию полимерной
сетки и ее множественным сшивкам с винилинизированным силикагелем могут быть
получены высокостабильные ХНФ с прочно прикрепленной к подложке фазой (что
и является общим преимуществом таких ХНФ). Различные виды замещения гидро-
ксильных групп могут приводить к уникальной энантиоселективности. ХНФ, в
которых тартрамиды содержат О,0'-бис(3,5-диметилбензоильный) и 0,0'-бис|4-(трет-бу-
тил)бензоильный| заместители, поставляет фирма Eka Chemicals (Бохус, Швеция)
под торговыми названиями Kromasil CHI-DMB и CHI-TBB.2 Эти ХНФ хорошо
распознают в нормально-фазовых условиях энантиомеры различных используемых
в фармацевтике продуктов, включая кислотные, нейтральные и основные
соединения, имеющие донорные и акцепторные группы и центры я-л-взаимодействий (как
в разделении на рис. 14.156).
' В настоящий момент эти колонки продает SigmaAldrich. — Прим. перев.
2 В настоящее время эти фазы сняты с производства. — Прим. перев.
14.6. Хиральные неподвижные фазы и их характеристики
Прививка к
(а)
Прививка от
(б)
■■"I хиральныи
мономер
^>"-^/~->*
Рис. 14.14. Получение синтетических полимерных ХНФ (полиакрилатного типа).
(а) «прививка к» и (6) «прививка от»; (в) схема реакции синтеза поли(диа-
миноциклогексан-Л/,Л?-диакриламида) методом прививки (б) на
поверхности подложки ХНФ 1791
730 Глава 14. Разделение энантиомеров
(в)
ВТ О О
^^
н
° о. ,R
ы^^ =
о
Хирапьный мономер
(б)
соон
0 5 10 мин
Рис. 14.15. ХНФ на основе синтетических сетчатых полимероп, полученных
полимеризацией производных тартрдиамида. а) Схема синтеза фазы; б)
разделение смесью 80:20 гексан / ТГФ с 0,05% ТФУК |80]
14.6.3. ХНФ на основе белков
Из биохимии уже давно известны факты стереоселективного связывания хиральных
соединений с белками. Поскольку природа предлагает широкий выбор стереоселек-
тивных белков, их использование в качестве хиральных селекторов для энантиосе-
14.6. Хиральные неподвижные фазы и их характеристики
731
лективной ЖХ представляется очевидным. Исследователей Алленмарка, Херманссо-
на, Миуа, Хаджинаку и др. можно назвать пионерами в этой области. В литературе
описан широкий спектр ХНФ на основе белков |82—84|. Ряд таких белковых фаз
стал коммерчески доступным. В таблице 14.2 приведены наиболее важные белковые
фазы, а также некоторые их характеристики и торговые названия колонок. Среди
них кислый а,-гликопротеин или орозомукоид (АКТ) |85| и неочищенный овому-
коид (ОВМ) |86| (коммерчески доступные под названиями Chiral AGP и Ultron
ES-OVM, соответственно) демонстрируют самые широкие возможности разделения
энантиомеров, охватывающие большой набор нейтральных, кислотных и оснбвных
лекарственных препаратов. Для других белковых ХНФ диапазон применимых
анализируемых веществ значительно более узок, и поэтому их использование более
ограничено. Фаза с целлобиогидролазой I (ЦБГ1 I) [87] (коммерческое название колонки
Chiral CBH) является предпочтительной для оснбвных веществ (например, рблока-
торов), а фаза с человеческим сывороточным альбумином (ЧСА) |88] (коммерчески
доступная под торговым названием Chiral HSA) используется для разделения
кислотных анализируемых веществ. Помимо использования в анализе хиральных
соединений, на ХНФ на основе ЧСА обратили серьезное внимание те, кто исследует
связывание лекарственных препаратов с белком, поскольку характеристики связывания,
наблюдаемые в физиологических условиях в плазме крови, в основном сохраняются,
когда белок иммобилизуется на носителе из силикагеля. Это, к сожалению, не
относится к коммерческим колонкам Chiral AGP, у которых характеристики связывания
значительно изменяются после иммобилизации из-за особой методики поперечной
сшивки, используемой изготовителем для повышения стабильности колонок.
Таблица 14.2. Важные ХНФ на основе белков и торговые наименования колонок с
некоторыми характеристиками белковых селекторов [83]
Белок
Мол.
масса (КДа)
Углеводы (%)
Итоэлектри-
ческая точка
Торговое наименование колонки
(поставщик)
Сывороточный альбумин
ЧСА"
БСА°~
АКТ"
ОВМ^
ЦБГ"
Авидин
Пепсин
67
68
44
28
60-70
66
70-78
0
0
45
17-34
6
20,5
—
4,7
4,7
2,7
4,5
3,6
9,5-10
6,1—6,6
Chiral HSA (ChromTech)
Resolvosil BSA (Macherey Nagel)
Chiral AGP (ChromTech)
Ultron ES-OVM (Shinwa Chemical)
Chiral CBH (ChromTech)
Bioptic AV-1 (GL Sciences)
Ultron ES-Pepsin (Shinwa Chemical)
"Человеческий сывороточный альбумин.
*Бычий сывороточный альбумин.
вагкислый гликопротеин или орозомукоид.
'Неочищенный овомукоид.
<*Целлобиогидролаза.
Структурно орозомукоид состоит из одной пептидной цепи, содержащей
181 аминокислотный остаток и пять гетерополисахаридных фрагментов. Последние
включают 14 остатков сиаловой кислоты, которые придают орозомукоиду сильные
'Англ. Cellobiohydrolase (CBH). — Прим. nepes.
2 Англ. Human serum albumine (HSA). — Прим. персе.
732 Глава 14. Разделение энантиомеров
кислотные свойства (pi — 2,7) |89]. Орозомукоид, ковалентно прикрепленый
поперечной сшивкой к химически модифицированной поверхности силикагеля (Хер-
манссон |85|), допускает широкий диапазон изменений рН (3—7,5), высокие
концентрации органических растворителей (до 25%) и температуру до 70 °С. ХНФ,
основанные на неочищенном овомукоиде цыпленка (ОВМ) были впервые описаны
Миуа с соавторами; позже было установлено, что их способность хирального
распознавания обусловлена примесью (овогликопротеином, содержащимся в количестве
11% мас./мас. в неочищенном овомукоиде) 190, 911. Напротив, чистый овомукоид
цыпленка обладает незначительным потенциалом распознавания энантиомеров |92|.
ХНФ на основе овомукоида стабильна в диапазоне рН от 3 до 7,5 и допускает со
держание органических растворителей до 50%, температура разделения должна
быть < 40 °С.
Белковые фазы всегда используются с водными или водно-органическими
подвижными фазами (то есть в обращенно-фазовом режиме). Важными переменными
для контроля удерживания и энантиоселективности являются рН подвижной фазы,
тип и буферная емкость буфера, тип и содержание органических растворителей и
добавок, а также температура. Соответствующие условия должны выбираться
эмпирически и зависят от структуры анализируемого вещества. Для кислотных и основных
лекарственных средств удерживание будет варьироваться с изменением рН в
зависимости от ионизации анализируемого вещества и селектора. Изменение ионной силы
может быть важным инструментом для изменения удерживания и улучшения
селективности. Увеличение ионной силы может либо снижать удерживание путем
экранирования зарядов анализируемого вещества или ХНФ, либо увеличивать удерживание
путем высаливания вещества, тем самым усиливая его гидрофобные взаимодействия
с сорбентом. Удерживание нейтральных молекул также может корректироваться
изменением рН, которое в результате приводит к изменениям структуры/конформации
белка (и меняет характер связывания). Известно, например, что овомукоид может
подвергаться обратимому разворачиванию и сворачиванию в зависимости от рН [93|.
Добавление органических растворителей (особенно 1- или 2-пропанола или аце-
тонитрила) снижает удерживание путем ослабления гидрофобных взаимодействий.
Таким образом, энантиоселективность может либо уменьшаться, либо возрастать,
в зависимости от того, осуществляются ли эти гидрофобные взаимодействия
на энантиоселективных или неэнантиоселективных участках. В качестве примера
влияния замены органического растворителя рассмотрим разделение верапамила. Он
не разделялся на ХНФ АКГ-типа с подвижной фазой, содержащей 1-пропанол, но
полностью разделился при замене 1-пропанола ацетонитрилом |94|. Следует иметь
в виду, что добавление органических растворителей может вызвать обратимые
изменения вторичной структуры иммобилизованного белка, тем самым изменяя его
энантиоселективность. Слишком высокие концентрации органических растворителей
могут привести к необратимым изменениям и денатурации белка, и их следует
избегать, чтобы не повредить колонку. Как и с любой колонкой, следует строго
соблюдать инструкции производителя. Если используется смешивание растворителей
онлайн непосредственно перед поступлением в колонку, настоятельно рекомендуется
проследить за тем, чтобы ни один из резервуаров не содержал органический
растворитель с более высокой концентрацией, чем та, которая допускается по инструкции,
чтобы избежать случайного разрушения (дорогостоящего!) сорбента.
Для контроля удерживания и энантиоселективности используются также катион-
ные добавки (алкиламины или соли четвертичного аммония, такие как jVjJV-диокти-
ламин и тетрабутиламмонийбромид) и анионные добавки (гидрофобные карбоновые
кислоты, алкилсульфонаты), которые могут конкурировать с анализируемыми
веществами за центры как ионного, так и гидрофобного взаимодействий. Иногда
предлагают добавлять 50 мкмоль/л динатриевой соли 1,2-этилендиамин-Л, N, N', УУ-тетра-
14.6. Хиральные неподвижные фазы и их характеристики 733
уксусной кислоты (Трилон Б, ЭДТА-Nai) к подвижной фазе с целью экранирования
белковой фазы от загрязнения металлами, источником которых может стать
оборудование. Еще одной переменной величиной для точной настройки удерживания
и энантиоселективности является температура. Графики Вант Гоффа для колонок
с белковыми фазами, в отличие от графиков для фаз многих других колонок, часто
отклоняются от линейности (не выполняется уравнение (14.4)). Такое необычное
поведение при изменении температуры может быть также связано со структурными
изменениями белка. Например, температурная зависимость разделения энантиомеров
хинолин-замещенных карбоновых кислот на колонках Chiral AGP предполагает
такое изменение конформации белка между 30 и 35 °С [95|.
Из-за структурной сложности макромолекулярных белковых селекторов и их
частых конформационных изменений в зависимости от условий, о фундаментальном
процессе удерживания или о том, как это влияет на энантиоселективность, известно
мало. В начале 1980-х годов ХНФ на основе белка были особенно популярны
потому, что их можно было использовать в обращение фазовом режиме при анализе
водных растворов проб, что благоприятствовало применению в области биоаналитики.
Однако из-за некоторых серьезных ограничений их использование с тех пор
сократилось. Как отмечалось выше, существуют значительные ограничения в отношении
состава подвижной фазы. Неправильное использование или хранение, или
длительная эксплуатация в экстремальных условиях могут легко привести к химическим или
биохимическим изменениям хиральных селекторов белка. Это может отрицательно
повлиять на долговечность колонок или их энантиоселективность. Другим
ограничением является низкая или умеренная эффективность колонок, как показано
на примере разделения, приведенного на рис. 14.16 (N = 300—700). Даже при
больших значениях энантиоселективности (как на рис. 14.16) низкие значения числа
теоретических тарелок в сочетании с уширением задних фронтов пиков в некоторых
случаях могут сделать анализ энантиомерных примесей с содержанием 0,1%
невозможным. Это особенно важно, когда примесь является вторым пиком, как указано
15 МИН
Рнс. 14.16. Разделение энантиомеров пропранолола на белковой фазе (с целлобиогид-
ролазой 1, Chiral CBH I, ChromTech). Подвижная фаза: 0,01 М ацетатный
буфер, рН 5 [S3]
734 Глава 14. Разделение энантиомеров
в разделе 14.3. К сожалению, простой переход к ХНФ с противоположной
структурой и обратным порядком элюирования (для минимизации эффектов, связанных
с уширением задних фронтов пиков) невозможен для белковых колонок. Для
некоторых образцов при разделении на колонках AGP и OVM происходит
непредсказуемое изменение порядка элюирования. Сложность белковых селекторов затрудняет
понимание механизма хирального распознавания. Тем не менее, в этом направлении
было проведено несколько исследований |96—103|.
Использование белковых фаз значительно сокращается из-за их достаточно
ограниченной емкости |61|. Число специфических (энантиоселективных) центров
взаимодействия на поверхности (большой) белковой молекулы обычно невелико. Можно
было бы ожидать, что иммобилизация мелких фрагментов белка, включающих энан-
тиоселективные центры, приведет к возможности ввести больше образца на колонку,
но такие эксперименты до сих пор так и не привели к успеху |99, 100]. Следовательно,
белковые ХНФ для препаративных разделений большой ценности не представляют.
Резюмируя, можно сказать, что вклад белковых фаз в разделение энантиомеров
сегодня медленно снижается. Эти сорбенты все больше заменяются другими ХНФ,
например, сорбентами с полисахаридами и макроциклическими антибиотиками.
Хотя использование белковых колонок для контроля качества не рекомендуется, они
по-прежнему используются в некоторых биоаналитических исследованиях, не в
последнюю очередь из-за совместимости их подвижных фаз с интерфейсами
электроспреев (в отличие от элюентов, типичных для нормально-фазового режима
разделения) |104|.
14.6.4. ХНФ на основе циклодекстрина
ХНФ с привитыми а-, Р- или у-циклодекстринами (ЦД), представляющими
собой макроциклические структуры из 6, 7 и 8 остатков глюкозы, соответственно
(рис. 14.17), введены в употребление Армстронгом |105|. Остатки глюкозы (глюкопи-
ранозы) соединены а-1,4-связями. Макроциклические молекулы циклодекстринов
принимают форму усеченного конуса, а количество звеньев глюкозы определяет
размер полости (0,57, 0,78 и 0,95 нм в диаметре для а-, Р- и у-ЦД: соответственно),
в которую могут быть включены молекулы анализируемых веществ (предпочтительно
гидрофобных) или их заместители. Внутренняя поверхность ЦД-полости гидрофобна
из-за углеродного скелета молекул Сахаров. Верхний и нижний кольцеобразные края
молекулы ЦЦ гидрофильны благодаря присутствию первичных гидроксильных групп
(на нижнем более узком кольце) и вторичных гидроксильных групп (на верхнем
более широком кольце)1. ЦД обычно соединены с силикагелем через гидроксилы более
узкого кольца либо эфирной связью (колонки Cyclobond фирмы ASTEC2), либо кар-
баматной связью (колонки ChiraDex и ChiraDex Gamma, фирмы Merck, колонки -
Ultron ES-CD фирмы Shinwa). Помимо ХНФ с природными ЦД в качестве селектора,
также доступны несколько фаз с производными ЦД, например, Cyclobond I SP, RSP,
RN, SN или Ultron ES-PhCD) (рис. 14.17). Дериватизация может усилить хиральное
распознавание с помощью создания дополнительных центров взаимодействия или
путем изменения размера и доступности хиральной полости включения. Таким
образом, энантиоселективность ХНФ на основе дериватизированных ЦЦ может
значительно отличаться от энантиоселективности фаз с привитыми более простыми
(природными) ЦД.
1 Выше упоминалось о том, что молекула ЦЦ имеет форму усеченного конуса с нижним
отверстием меньшего диаметра и верхним отверстием большего диаметра. — Прим. перев.
2 В настоящий момент зги колонки продает SigmaAJdrich. — Прим. перев.
14.6. Хиральные неподвижные фазы и их характеристики 735
а-Циклодекстрин п = 0, m = 6
Р-Цикподвкстрин п = 1, m = 7
у-Циклодекстрин п = 2, m = 8
Коммерческие колонки:
Cyclobond I (природный р-ЦД)> /'{природный у-ЦД), и ///(природный пс-ЦД)
(от ASTEC) Cyclobond ISP или RSP((S)- или (Н5)-2-гидроксипропиловый
эфир-р-ЦД] (ASTEC) Cyclobond IRN или SN [(Я)-или (S)-1-(1-
нафтил)этилкарбамат-р-ЦД] (ASTEC) ChiraDex (природный р-ЦД) и ChiraDex
Gamma (природный у-ЦД) (от Merck) Ultron ES-CD (природный Р-ЦД) и Шюп
ES-PhCD (фенил карбамоилированный Р-ЦД) (от Shinwa)
Рнс. 14.17. Структура циклодекстринов и торговые наименования соответствующих
ХНФ
ХНФ на основе ЦД характеризуются существенным разнообразием условий элю-
ирования и механизмов хирального распознавания |18, 106|. Они могут работать
в полярно-органическом, обращенно- и нормально-фазовых режимах. Было
показано, что механизм молекулярного распознавания отличается при разных способах
элюирования. Общепризнано, что в обращенно-фазовом режиме при использовании
водных или водно-органических подвижных фаз (смесей ацетонитрила с буфером
или метанола с буфером) липофильные анализируемые вещества взаимодействуют
с ЦД-селекторами, образуя комплексы включения (рис. 14.186). Был предложен
двухстадийный (гидрофобный) механизм |107|: (1) проникновение гидрофобной
части молекулы анализируемого вещества в полость ЦД и (2) выделение из молекул
анализируемого вещества и ЦД связанных молекул растворителя (воды) с
последующей стабилизацией комплекса благодаря гидрофобному взаимодействию. Более того,
могут иметь место и положительно влиять на устойчивость комплекса гидрофильные
взаимодействия с гидроксильными группами на верхнем и нижнем кольце молекулы
ЦД (водородная связь, диполь-дипольные взаимодействия). Эти комбинированные
эффекты в сочетании с конформационными изменениями при комплексообразова-
нии (так называемая индуцированная подгонка) могут иногда приводить к
чрезвычайной стабильности комплексов.
Как и в ОФХ, концентрация органического растворителя в подвижной фазе
может использоваться для контроля удерживания (для 1 < к <10). Сила растворителя
для ХНФ на основе ЦД возрастает в той же последовательности, как и для ахираль-
ной ОФХ:
вода < метанол < этанол < пропанол — ацетонитрил < ТГФ.
736 Глава 14. Разделение энантиомеров
Степень включения анализируемого вещества обычно зависит от размера
полости ЦД. Согласно принципу соответствия по размеру для образования комплексов
включения, обычно более высокое сродство и бблыиую энантиоселективность
для пар хиралъных образцов обеспечивают ЦД, полость которых наиболее
соответствует по размеру гидрофобным фрагментам молекулы анализируемого вещества.
Замещенные фенильные, нафтильные и гетероароматические кольца могут легко
размещаться в полости РИД, в то время как для более крупных молекул, таких как
молекулы стероидов, больше подходят у-ЦД, а для меньших по размерам молекул —
а-ЦД. Эта взаимосвязь структуры и связывания может быть полезным начальным
ориентиром при выборе колонок.
В полярно-органическом режиме (например, при использовании смесей ацето-
нитрила и метанола) внутренняя полость ЦД блокируется молекулами растворителя,
поэтому образование комплексов включения с липофильными остатками становится
термодинамически невыгодным. Следовательно, анализируемые вещества с
гидрофильными группами связываются с полярной областью поверхности ЦД (верхним или
нижним кольцом, рис. 14.18д) |108|. Полярные гидроксилы окружены хиральной
средой, и энантиоселективность обусловлена различиями в силе этих полярных
взаимодействий (водородных связей, дипольных взаимодействий) для двух
энантиомеров.
Анализируемые вещества с более чем одной полярной функциональной группой, одна
из которых расположена в стереогенном центре или близко к нему, особенно хорошо
разделяются на циклодекстриновых сорбентах с полярно органическими растворителями.
Объемные группы, расположенные вблизи стереогенного центра, способствуют распо
знаванию энантиомеров. Поскольку многие хиральные лекарственные средства являются
полярными соединениями, этот режим элюирования может быть весьма полезен,
особенно в тех ситуациях, когда в обращенно- и нормально-фазовом режимах невозможно
разделить энантиомеры [109]. Полярно-органические подвижные фазы для ХНФ на
основе ЦД обычно состоят из О—15% метанола в ацетонитриле с добавкой 0,001—1,2%
уксусной кислоты и 0,001—1,2% триэтшюмина и были использованы, например, для энан-
тиомерных разделений fi-блокаторов [110].
В нормально-фазовом режиме (например, при элюировании смесями гексана
или гептана с полярными растворителями, такими как спирты) внутренняя полость
ЦД также заполняется молекулами растворителя, так что комплексы включения
не образуются. Вместо этого взаимодействие происходит на полярной поверхности
(а) Н [б)
Полярно-органический и Обращенно-фазовый режим
нормально-фазовый режим (комплексы включения)
Рис. 14.18. Механизмы молекулярного распознавания циклодекстриновыми
сорбентами в полярно-органических элюентах или элюентах, используемых в
нормально-фазовом режиме (а) и в обращенно-фазовом режиме (б) |108|
14.6. Хиральные неподвижные фазы и их характеристики 737
ЦД или ЦД-производного. Показано, что только дериватизированные
ароматическими соединениями ЦД, такими как (R) или (5)-1-{!-нафтил)этилкарбаматы, являются
эффективными ХНФ для разделения энантиомеров в нормально-фазовом режиме.
В этом случае полярные взаимодействия, такие как водородные связи и диполь-ди-
польные взаимодействия, поддерживаются л-л-взаимодействиями, которые,
по-видимому, приобретают решающее значение в этом режиме. Поскольку изменения
порядка элюирования наблюдались при изменении конфигурации стереогенного
центра заместителя в кольце на противоположный, можно сделать вывод, что
связывание энантиоселективного вещества в основном контролируется присоединенными
1-{1-нафтил)этилкарбаматными группами, а не полостью ЦД.
В последние годы значимость ХНФ на основе ЦД, по-видимому, снижается.
Они вытесняются более эффективными макроциклическими антибиотиками,
которые обычно обеспечивают более высокие уровни энантиоселективности. Кроме того,
их применение в препаративной хроматографии ограничено низкой емкостью. Одна
ко совместимость обращение фазового и полярно органического режимов с исполь-
зованием ионизации электроспреем для МС-анализа остается важным
преимуществом ХНФ с привитыми ЦД.
14.6.5. ХНФ на основе макроциклических антибиотиков
Вдохновленные возможностями стереоселективного включения, обеспечиваемого
макроциклическими циклодекстринами, Армстронг и коллеги исследовали свойства
других (более эффективных) макроциклических природных соединений, связанные
с образованием комплексов включения. Это исследование положило начало
разработке нового важного класса ХНФ на основе макроциклических антибиотиков.
Первой описанной ХНФ этого класса стал модифицированный ванкомицином силика-
гель, разработанный Армстронгом в I994 году [111|. Впоследствии было обнаружено,
что несколько структурных аналогов этого гликопептидного антибиотика являются
эффективными хиральными селекторами с дополнительной энантиоселективностью
(они обеспечивают энантиоселективность по отношению к разным группам веществ
или разный порядок элюирования для одной энантиомерной пары). Это привело
к созданию ХНФ на основе ванкомицина |111], тейкопланина [112|, ристоцетина А
11131 и агликона тейкопланина |114|, которые продает компания SigmaAldrich
под торговыми названиями Chirobiotic V, Chirobiotic T, Chirobiotic R и Chirobiotic
TAG1. В продаже имеются более старые варианты Chirobiotic V и Т (VI, Т1), а также
более новые (V2, Т2), полученные разными методами химического связывания
антибиотиков с силикагелем подложки. Эти ХНФ имеют широкий диапазон применения,
включающий разделение высокополярных соединений, таких как недериватизиро-
ванные аминокислоты. В настоящее время эти фазы являются одними из самых
эффективных ХНФ, они описаны в нескольких обзорах [115—118].
Структурно селекторы-антибиотики довольно сложны (рис. 14.19). Они имеют
одинаковое ядро: агликон, представляющий собой гептапептид с ароматическими
остатками, которые соединены друг с другом, образуя объемную фигуру в виде
корзины с неглубокими карманами для образования комплексов включения и
локализованными на поверхности углеводными остатками (рис. 14.20). Образование
комплексов включения часто обусловлено полярными взаимодействиями, особенно
для анализируемых веществ, содержащих карбоновые кислоты: связывание
карбоксильного конца (С-конца) тремя водородными связями стабилизируется другими
водородными связями, а также п-к и гидрофобными взаимодействиями (только
1 В настоящее время эти ХНФ изготовляет фирма Supelco. — Прим. перев.
Глава 14. Разделение энантиомеров
(а)
«"■
с Mb
Ристоцетин А
Рис. 14.19. Структуры гликопептидных антибиотиков: (с) ванкомицнн (1 сахарная группа,
3 полости включения А, В, С; молекулярная масса ж 1449 Да, рКа 2,9, 7,2, 8,6, 9,6, 10,4, 11,7;
pi 7,2 ); (б) тейкопланин (молекулярная масса ~ 1885 Да, 3 сахарных группы, R — остаток де-
кановой кислоты; 4 полости включения, А, В, С, D); (в) ристоцетин А (молекулярный вес
ж 2066 Да, 6 сахарных групп, 4 полости включения А, В, С, D)
14.6. Хиральные неподвижные фазы и их характеристики 739
в обращенно-фазовом режиме). Вместе
с энантиоселективностью, обусловленной
множеством стереогенных центров,
гетерогенная многофункциональность и
структурная специфичность гликопепти-
дов обеспечивают множество
потенциально стереоселективных вариантов
связывания, которые оказываются источ-
ником энантиоселективности для
различных фармацевтических препаратов.
Помимо их полезных структурных
особенностей, широкая применимость
ХНФ на основе макроциклических анти
биотиков частично является следствием
возможности их разнообразного
использования, которое включает режимы ОФ,
ПО и НФ (рис. 14.21) ЦП, 118, 119|.
Полярно-органический режим рекомендуется
в качестве первого шага, если
анализируемое вещество имеет более одной полярной
функциональной группы (что относится
ко многим хиральным препаратам), и, если
хотя бы одна из этих групп расположена
в стереогенном центре или рядом с ним.
Поскольку гидрофобные взаимодействия
не осуществляются в режиме ПО,
полярные взаимодействия, такие как ионное и
дипольное взаимодействия, а также
водородная связь, имеют первостепенное
значение для энантиоселективности. Типичная
начальная подвижная фаза состоит из
метанола с добавками 0,1% уксусной
кислоты и О,1% триэтиламина для ванкомици-
новых ХНФ (каждая из добавок должна
быть увеличена до 1% при использовании
тейкопланиновых ХНФ). Соотношение
уксусной кислоты и амина является ключевой
переменной для регулирования
энантиоселективности, тогда как общее количество
добавок при данном соотношении
регулирует, главным образом, удерживание. Если
Рис. 14.20. Объемная структура в виде
корзины и механизм молекулярного
распознавания ванкомицина.
Кристаллическая структура комплекса ванкомицина
с /Va,yVm-flHaueTHn-L-Lys-D-Ala-D-Ala,
установленная методом рентгеноструктурного
анализа, (о) Вид сбоку; (б) вид сверху.
Изображение структуры кристалла было
получено с помощью программного
обеспечения для молекулярного моделирования
SYBYL (Tripos, St. Louis, МО) по
относительным координатам, взятым из банка
данных Брукхейвена по белкам {http://www.
rcbs.org/pdb/)
анализируемое вещество не может элюиро-
ваться в ПО-режиме при содержании добавок примерно 1%, соединение слишком
полярное, и рекомендуется обращенно-фазовый режим. Типичными начальными условиями
для разделения в обращенно-фазовом режиме являются подвижные фазы, состоящие из
ТГФ и 20 мМ нитрата аммония с рН 5,5 (10:90, об./об.) для ванкомициновых ХНФ, и
метанола с 0,1% ацетататом триэтиламмония с рН 4.1 (20:80; об./об.) для
тейкопланиновых ХНФ. Более подробные процедуры разработки методов приведены в справочнике
Chirobiotic Handbook |119|.
В обращенно-фазовом режиме процесс образования комплексов включения
может осуществляться благодаря гидрофобным взаимодействиям или активно
поддерживаться ими, в то время как множественные водородные связи, а также вклады
740 Глава 14. Разделение энантиомеров
(а) ОФ
Бромацил
Девринол
Т
s-eH' Кумахлор
13
время (мин)
(б) ПО
Тербуталин
он
| и =1.30
1.30
(1=1.38
J
I
L
UL
МеОН/АсОН/
ТЭА
МеОН/
NH4AcO
МеОНЯФУК/
NH4OH
(в) НФ
? ?
15
время (мин)
30
Рис. 14.21. Разделения с помощью различных режимов элюирования колонки, упакованной
ХНФ с макроциклическим антибиотиком (ванкомицином). (а) Обращенно-фазовый (ОФ)
режим; (б) полярно-органический (ПО) режим; (в) нормально-фазовый (НФ) режим.
Экспериментальные условия: (<з) 10:90 ацетонитрил/1 % триэтиламмоний ацетатный буфер, рН 7;
(б) подвижные фазы (слева направо): 100:0,3:0,2 метанол/уксусная кислота/триэтиламин; 99:1
метанол/20 мМ ацетат аммония; 100:0,05:0,05 метанол/ТФУК/МН4ОН; (в) 50:50 гек-
сан/2-пропанол |111] и |119|
14.6. Хиральные неподвижные фазы и их характеристики
741
ионного и дипольного взаимодействий могут способствовать стереоселективному
позиционированию анализируемого вешества в связывающей полости макроцикличе-
ского селектора. Если анализируемые вещества не могут достаточно хорошо
удерживаться в ПО-режиме с добавками в количестве лишь 0,01% или если растворимость
образца слишком мала, альтернативой является нормально-фазовый режим.
Типичными начальными условиями являются смесь гексан-этанол (80:20, об./об.).
Некоторые из других обычно используемых условий элюирования указаны в подписи
к рис. 14.21. В целом использование различных режимов элюирования способствует
широкому применению ХНФ на основе макроциклических антибиотиков и обеспе
чивает значительную гибкость этих ХНФ при разработке методов разделения. Кроме
того, следует отметить, что эти ХНФ можно применять, переходя от одного режима
к другому, без необратимых изменений в работоспособности колонок.
Как показала статистическая оценка производителя колонок |120|, позже
подтвержденная работающими в промышленности исследователями |121|, наилучший
результат может быть достигнут в режиме с полярно-органическими растворителями
(~ 40%) или в обращение фазовом режиме (~ 40%), в то время как
нормально-фазовый режим, как оказалось, используется меньше (~ 5%). В этом контексте, однако,
следует отметить, что, как показал Ванг и другие |122|, многие из указанных в
публикациях «обращенно-фазовых» разделений с водно-органическими подвижными
фазами фактически проводились в режиме хроматографии гидрофильных
взаимодействий (ГИХ) (представляющей собой водную нормально-фазовую хроматографию,
раздел 8.6). Реальные обращен но-фазовые разделения могут быть проведены
на ХНФ Chirobiotic с подвижными фазами, содержащими менее 20% органического
растворителя, тогда как для подвижных фаз с более высоким содержанием
органических растворителей полярные взаимодействия могут быть усилены средой с низкой
диэлектрической постоянной, так что удерживание увеличивается с возрастанием
доли органического растворителя (что и предполагает режим ГИХ.)
Хотя различные макроциклические селекторы-антибиотики имеют близкое
структурное сходство, они обладают комплементарной энантиоселективностью
(рис. 14.22), что зачастую полезно для разработки метода. Когда с помощью
определенной ХНФ на основе гликопептидного антибиотика не удается достичь полного
разделения или вообще не удается ничего разделить, существует большая
вероятность того, что одна из других ХНФ на основе антибиотиков сможет обеспечить
полное разделение. Едва заметные различия в структуре ХНФ и характеристиках
связывания могут способствовать комплементарности разделения. Эти различия
приписывались отличающимся расстояниям между концами С-образного агликона,
которые становятся меньше в ряду ванкомицин, ристоцетин, тейкопланин, и его
спиральному скручиванию, которое увеличивается в том же ряду, а также влиянию
различных заместителей и сахарных групп [117|.
В целом широкое применение семейства ХНФ Chirobiotic включает разделения
хиральных кислот, оснований, а также амфотерных и нейтральных соединений,
причем Chirobiotic T и V эффективнее по сравнению с Chirobiotic R |121]. Chirobiotic V
особенно подходит для распознавания энантиомеров нейтральных анализируемых
веществ, амидов, сложных эфиров и аминов (включая аминоспирты и циклические амины).
На Chirobiotic Т можно разделять аминоспирты, недериватизированные и N-depuea-
тизированные аминокислоты и (ди)пептиды [116]; Chirobiotic R можно использовать
для разделения хиральных кислот, таких как гидроксикислоты, замещенные
алифатические кислоты и другие кислоты [117]. В связи с этим особого внимания заслуживает
способность тейкопланиновых ХНФ к разделению энантиомеров недериватизированных
природных и синтетических аминокислот [125]. С помощью этой фазы были получены
значения а. между 1,2 и 2,7 для природных аминокислот с разрешениями до базовой
Глава 14. Разделение энантиомеров
ХНФ на основе ХНФ на основе ХНФ на основе
ристоцетина
875 12.4
теикопланина
5.92
Время (мин)
7.88
\
Врем
10.5
L
я (мин)
Время (мин)
5.61
_
11.2
i
LL
Время (мин)
ванкомицина
4.70 4.91
\ /
14.8 -|
Время (мин)
3.73
\
JU
S.14
/
Время (мин)
Рис. 14.22. Комплементарностъ разделений на ХНФ с различными макроцикличес-
кими антибиотиками на примере разделений энантиомеров Z-Ala (вверху)
и 5-метил-5-фенилгидантоина (внизу). Экспериментальные условия: рис-
тоцетин А, 20:80 метанол/0,1% триэтиламмоний-ацетатный буфер,
рН 4,1; тейкопланин, 20:80 метанол/1% триэтиламмоний-ацегатный
буфер, рН 4,1; ванкомицин, 10:90 метанол/1% триэтиламмоний ацетатный
буфер, рН 4,1 [124|
линии (Rs > 1,5) для всех, кроме гистидина (Rs = 0,8). Разделение энантиомеров амино
кислот на ХНФ с тейкопланином лучше всего проводить с простыми подвижными
фазами этанол-вода (или метанол-вода), но для кислотных или основных аминокислот в
подвижную фазу следует добавлять 0,1% -ный триэтиламмонийныи буфер.
В работе |114| рассматривалась роль углеводных фрагментов в способности тей-
копланиновых ХНФ к хиральному распознаванию. Для хроматографи чес кого
сравнения с исходной ХНФ на основе теикопланина был подготовлен соответствующий
аналог тейкопланинового агликона (ТАГ), в котором все сахарные группы были
химически удалены, и модифицированный аналог был нанесен на силикагель. В то
время как полное удерживание на ХНФ-ТАГ было вполне сходно с удерживанием
на ХНФ с природным тейкопланином, профили распознавания энантиомеров
для большинства тестируемых соединений существенно изменились. Примечательно,
что остатки сахара значительно уменьшали энантиоселективность в отношении
аминокислот (рис. 14.23), что ясно указывает на то, что активные центры хирального
распознавания расположены в области агликона ХНФ. Для других анализируемых
веществ можно наблюдать противоположную тенденцию.
ХНФ типа Chirobiotic становятся все более популярными для энантиомерно-
го анализа лекарств и других ксенобиотиков в биологических жидкостях вследствие
(1) их совместимости с водными матрицами образцов и (2) их идеальной
совместимости с ионизацией электроспреем для МС-анализа, как в обращенно-фазовом, так
и в полярно-органическом режимах |104, 123|. Недостатки включают отсутствие
ХНФ, которые позволяют предсказуемым образом изменять порядок элюирования
в тех случаях, когда это необходимо для анализа следовых количеств энантиомера.
Несмотря на заявленную применимость этих фаз для препаративных разделений, их
нагрузочная емкость ограничена по той же причине, что и для белковых фаз: отно-
14.6. Хиральные неподвижные фазы и их характеристики 743
(а)
НО
NH,
—\ V-CHj-C—СООН
(б)
0 4 8 12 (мин)
ХНФ на основе
тейкопланина
0 4 8 12 16 20 24 (мин)
ХНФ на основе а гл и кона
Рис. 14.23. Разделение энантиомеров D,L-DOPA на исходной ХНФ-гейкопланин (л)
и ХНФ с агликоном тейкопланина после удаления Сахаров (б). Условия:
подвижная фаза 60:40 метанол/ацетат триэтиламмония, рН 4.1; УФ-детек-
тирование, 254 нм; температура 22 "С; скорость потока 1 мл/мин |П4|
сительно низкая концентрация центров связывания по сравнению с полисахаридны-
ми и низкомолекулярными фазами щеточного типа |61|. С другой стороны, было
продемонстрировано перспективное разделение энантиомеров на ХНФ гликопептид-
ного типа в условиях СФХ |126|. Комплексный скрининг в этом режиме показал,
что из набора более 100 хиральных тестовых веществ (включающего гетероциклы,
профены, р блокаторы, сульфоксиды, N защищенные аминокислоты и природные
аминокислоты) более 90% энантиомеров удалось разделить с помощью
коммерческих ХНФ на основе гликопептидов, используя в качестве подвижной фазы смесь
суб-/сверхкритический диоксид углерода-метанол с небольшим количеством
кислотных и основных добавок1.
14.6.6. ХНФ на основе хиральных краун-эфиров
Стереоселективное связывание в комплексы анализируемых веществ с ХНФ
на основе хиральных краун-эфиров и первое применение таких ХНФ было впервые
описано Крамом с сотрудниками |I27|. В этой ранней работе два 1,1-бинафтильных
остатка были включены в краун-эфир в качестве замещающих элементов вместо двух
этиленовых групп в хорошо известном 18-краун-6. Структурные аналоги таких ХНФ,
основанных на хиральном краун-эфире, дериватизированном 1,Г-бинафтилом, были
позднее разработаны группой Шинбо с использованием (3,3'-дифенил-1,Г-бинаф-
1 Российская компания «БиоХимМак СТ» производит хиральные колонки Nautilus-E с
иммобилизованным отечественным антибиотиком эремомицином, который проявляет высокую энан-
тиоселективность к широкому кругу соединений. См. S.M. Staroverov, M.A. Kuznetsov,
P.N. Nesterenko, G.G. Vasiarov, G.S. Katrukha, G.B, Fedorova. New chiral stationary phase with
macrocyclic glycopeptide antibiotic eremomycin chemically bonded to silica. // J. Chromatogr. A. 2006.
V. 1108. P. 263 — 267. — Прим. перев.
744 Глава 14. Разделение энантиомеров
(а)
(б)
Crownpak CH
си лика гель]— о—Si
ChiroSil
силикагель)—o-^i^^4^^! **o
'COOH
ChiroSil RCA
Рис. 14.24. Коммерчески доступные ХНФ на основе хиральных краун-эфиров (данные
из руководства для пользователей, приводимого на сайте поставщиков)
тил)- или (6,6'-диоктил-3,3'-дифенил-1,Г-бинафтил)-20-краун-6, динамически
нанесенных на октадецилсиликагель |128|. Полученные ХНФ коммерчески доступны —
например, под торговым названием Crownpak CR их производят компании Daicel и
Chiral Technologies (рис. 14.24я). Поскольку оба сорбента синтетического
происхождения, они доступны в двух формах с противоположным порядком элюирования:
Crownpak CR(+) и CR(-). Доступны их структурные аналоги на основе (18-кра-
ун-6)-2,3,11,12-тетракарбоновой кислоты, полученные включением в краун-эфир
винной кислоты, которая иммобилизована с помощью двух карбоксильных
функциональных групп на силикагель, модифицированный 3-аминопропилом [129|. Эти
ХНФ имеются в продаже под торговыми названиями ChiroSil RCA{+) и SCA(-)
(производители Regis, Мортон Гроув, США)
и Chiral Hyim-CR-1 (производитель
К-МАС, Корея) (рис. 14.246).
Применение таких хиральных ХНФ
на основе краун-эфиров по существу
ограничено первичными аминами, то есть
к потенциальным объектам анализа
принадлежат главным образом
аминокислоты, сложные эфиры аминокислот, ами-
носпирты и хиральные лекарственные
средства со свободной первичной амино-
функцией. Обычно для обеспечения
полного протонирования анализируемого
вещества требуются водные подвижные
фазы с рН от 1 до 3,5. Получающиеся
хиральные ионы аммония могут связываться
с макроииклическим краун-соединением
с образованием комплексов включения
за счет формирования трех водородных
связей между ионами аммония образца и
тремя атомами кислорода краун-эфира
(рис. 14.25). Энантиоселективность может
регулироваться стерическими факторами,
обусловленными заместителями в хираль-
соон
Рис. 14.25. Механизм молекулярного
распознавания ХНФ на основе хиральных
краун-эфиров. Схема взаимодействия
анализируемого вещества с селектором,
осуществляемого с помощью трех водородных
связей (данные взяты с веб-сайта Regis)
14.6. Хиральные неподвижные фазы и их характеристики 745
ных ионах аммония и остатками, присоединенными к хиральным фрагментам
18(20)-краун-6. Поддерживание сильнокислой среды также важно для подавления
взаимодействия с силанольными группами на поверхности силикагеля, которые
могут препятствовать энантиоселективности. Для этого можно использовать, например,
5 мМ раствор хлорной кислоты в воде или смеси метанол-вода (содержащей до 15%
метанола) в качестве подвижной фазы. Такие жесткие условия могут оказаться
вредными как для оборудования, так и для неподвижной фазы, что и стало причиной
ограниченной популярности этих ХНФ. Более поздние работы о ХНФ на основе
краун-эфиров можно найти в |130—133|.
14.6.7. Донорно-акцепторные ХНФ
(а)
OsN.
DNBPG
(б)
Первые ХНФ на силикагельной подложке с полностью синтетическими
селекторами были разработаны в конце 1970-х годов |134, 135|. Последующая работа Пиркла
и коллег привела в 1981 году к первым коммерческим ХНФ с производным
ДНБ-фенилглицина, ионно иммобилизованным на силикагеле. Позже этот
синтетический хиральный селектор был привит на силикагель через ковалентную амид-
ную связь. Этот хиральный сорбент по-прежнему коммерчески доступен, его
производят фирмы Regis, Machery Nagel и Merck под названием DNBPG (рис. 14.26а).
Такие донорно-акцепторные ХНФ (щеточного типа), основание на нейтральных
синтетических или полусинтетическиих хиральных селекторах с низкой
молекулярной массой, используются в
нормально-фазовом режиме. Они способны
создавать энантиоселективность с помощью
дополнительных неионных сил
притяжения [27|. Водородная связь, л-л
взаимодействия {между электронообогащенными и
электронодефицитными ароматическими
группами при их параллельной или
перпендикулярной взаимной ориентации) и
диполь-дипольные взаимодействия играют
важную роль в стабилизации комплекса се-
лектор-анализируемое вещество и в
распознавании энантиомеров.
Энантиоселективности часто способствуют стерические
взаимодействия объемных групп, которые
могут представлять собой эффективные
стерические барьеры для тесного контакта
между селектором и анализируемым
веществом в случае одного из энантиомеров.
Из-за относительно большого значения
водородных связей и других неионных
электростатических взаимодействий такие ХНФ
менее эффективны в полярных протонных
средах, а следовательно, и в
полярно-органических и водно-органических фазах,
используемых в обращенно-фазовой
хроматографии. Ввиду важного вклада группы
Пиркла в эту область исследований, такие
ХНФ донорно-акцепторного типа теперь
часто называют фазами Пиркла.
WHELK-0 1
(в)
0,N
ULMO
Рнс. 14.26. Структуры
норно-акцепторных
популярных до-
фаз Пиркла.
(в) DNBPG; (ff) WHELK-0 1; (в) ULMO
746 Глава 14. Разделение энантиомеров
Ряд важных ХНФ был разработан группой Пиркла в результате систематических
хроматографических [136| и спектроскопических исследований |137—139) явления хи-
рального распознавания, а также последовательного применения принципа обрати
мости хирального распознавания [140, 1411. Обратимость означает, что селектор и
анализируемое вещество взаимозаменяемы. Следовательно, один из энантиомеров
анализируемого вещества, которое хорошо разделяется на ХНФ с данным хиральным
селектором, позволит расщепить рацемат этого селектора, если его иммобилизовать
через группу, которая не участвует в процессе распознавания. Такие концепции и ме
тоды используются для направленной разработки новых передовых ХНФ [136, 142|.
Как отмечалось выше, в ХНФ донорно акцепторного типа наиболее важными
для образования связей с целью хирального распознавания являются тг-тг-стэкинг
взаимодействия между электронообогашенными и электронодефицитными
ароматическими группами. Первоначально разрабатывались либо ХНФ с я-кислотными
группами (с электронодефицитными ароматическими остатками, обычно с 3,5-ди-
нитробензоилом) для я-основных анализируемых веществ (с
электронообогашенными ароматическими группами), либо ХНФ с it основными остатками (например,
с остатком нафталина) для гс-кислотных анализируемых веществ. Вторые из
упомянутых ХНФ (например, ХНФ на основе производных N-2-нафтилаланин ундеци-
лового эфира) |143| оказались менее востребованы и поэтому исчезли из продажи.
Напротив, несколько ранее разработанных группой Пиркла п электронных акцеп
торных фаз по-прежнему доступны — их продает фирма Regis (например, DNBLeu,
DNBPG, р Gem 1, a Burke 2, PIRKLE 1-J, см. таблицу 14.3)
В конечном итоге оказалось, что наиболее широкое применение находят
те ХНФ, в селектор которых включены л-электронные донорные и акцепторные
группы. В соответствии с этой тенденцией была разработана фаза Whelk-O 1,
которая имеет предварительно организованные полости для размещения анализируемого
вещества и допускает одновременные л-я-взаимодействия между параллельно и
перпендикулярно ориентированными между собой ароматическими структурами
для облегчения хирального распознавания |144, 145| (рис. 14.266). Вдохновленные
результатами, полученными Пирклом, несколько других исследовательских групп
последовали этой концепции получения ХНФ с низкомолекулярными
синтетическими селекторами. Среди прочих Ои с сотрудниками разработали ХНФ амидного и
уретанового типа, которые теперь продаются упакованными в колонки Sumichiral OA
компанией Sumitomo (Япония), или в колонки Chirex компанией Phenomenex
(США). Доступен ряд структурных вариантов: тот, который называется Chirex 3005
(ХНФ амидного типа с донорами и акцепторами л-электронов) (см. таблицу 14.3),
по-видимому, популярнее всего, за ним следуют Chirex 3022 и в меньшей степени
востребована Chirex 3020 |ХНФ карбаматного типа с донорами л-электронов,
полученными при введении (5)-индолин-2-карбоновой кислоты и ^)-1-|а-нафтил] эти-
ламина, а также (5)-/прет-лейцина и ^)-1-|сс-нафтил| этиламина, соответственно
(таблица 14.3).
Группа Гаспаррини разработала другие важные ХНФ я-донорно-акцепторного
типа, в которых использовались С2-симметричные диаминовые соединения, такие как
бис-Л',ЛГ-(3,5-динитробензоил)-1,2-диаминоциклогексан |146, 147| (DNB-DACH*,
Regis) и УУ-3,5-динитробензоил-Л"-ундеканил-1,2-дифенил-1,2-диамин от Юрэй и
др. 1148| (ULMO®, Regis, рис. 14.26в). Последняя из перечисленных ХНФ позволяет,
например, хроматографически разделить недериватизированные арилкарбинолы, как
показано на рис. 14.27. Разработка ХНФ в лаборатории Пиркла, а также некоторые
стратегии химического проектирования и дизайна, приведшие к современным донор-
но-акцепторным фазам, всесторонне рассмотрены в обзоре Уэлча [142|. Позднее
Гаспаррини обобщил и провел обсуждение некоторых из новых разработок в этой
области [149|.
14.6. Хиральные неподвижные фазы и их характеристики
Таблица 14.3. Коммерчески доступные донорно-акцепторные ХНФ (фазы Пиркла)
Хиральный селектор
Торговое
наименование колонки
Поставщик
фазы с акцепторами/донорами л-электронов
3-[ 1-{3,5-динитрооензамидо)-1,2,3,4-
тетрагидрофенантрен-2-ш1|пропил-силикагель
11-[2-(3,5-динитро6ензамидо)-1,2-дифенилэтиламино]-
11 -оксоундецил-силикагель
3-[Л^(3,5-динитро6ензоил)-{Я)-1-нафтилглицин-
амидо]пропил-силикагель
Whelk-O 1
ULMO
Chirex 3005
(Sumichiral 2500)
Regis
Regis
Phenomenex
(Sumitomo)
фазы с акцепторами тг-злектронов
3-(3-{Л/-[2-{3,5-динитро6ензамидо-1-циклогексил)]-3,5-
динитробензамидо)-2-гидрокси-пропокси}-пропил-силикагелъ
3-13-(3,5-динитробензамидо)-2-оксо-4-фенилазетидин-
1-ил| пропил-силикагель
5-(3,5-динитробензамидо)-4,4-диметил-5-диметилфосфонил-
пентанил-силикагель
] l-[/V-(3,5-flHHMTpo6eH3omi)-3-aMHHO-3-<peHwi-2-
(1,1-ДИметилэтил) пропаноил|-ундецил-силикагель
3-[Л/-(3,5-динитробензо ил )лейцин-амидо]проп ил-сил икагель
3-[Л/-(3,5-динитрооензоил)фенилглицин-амидо|пропил-
силикагель
DACH-DNB
PIRKLE 1-J
a-Burke 2
p-Gem 1
Leucine (DNBLeu)
Phenylglycine
(DNBPG)
Regis
Regis
Regis
Regis
Regis
Regis
(Merck)
Фазы с донорами л-электронов
3-{Л/-|(Д)-(а-нафтил)этилкарбамоил|-(5)-индолин-2-
карбоксамидо} пропил-силикагель (уретановая связь)
3-{ЛГ-|(Л)- 1-(а-кафтил) этилкарбамоил]-(5)-трет-лейцин-
амидо} пропил-силикагель (уретановая связь)
Chirex 3022
(Sumichiral OA 4900)
Chirex 3020
(Sumichiral OA 4700)
Phenomenex
(Sumitomo)
Phenomenex
(Sumitomo)
10
15 (мин)
Рис. 14.27. Разделение хиральных спиртов на колонке ULMO. Условия: колонка
{Д,й)-иЬМО; 250x4 мм; элюент 99,5: 0,5 н-гептан-изопропанол;
I мл/мин; 254 нм; 25 °С |148|
748 Глава 14. Разделение энантиомеров
Поскольку донорно-акцепторные фазы почти всегда используются в нормально-
фазовом режиме, разработка метода проста. Обычно она начинается со смеси гексана
или гептана и 2 пропанола (1-10% полярного растворителя). Для оснбвных растворов
добавляют к подвижной фазе 0,1% оснбвного модификатора, такого как диэтиламин,
для кислотных растворов — 0,1% кислотной добавки, такой как ТФУК. После
первоначального разделения содержание полярного растворителя регулируется для
достижения приемлемого коэффициента удерживания (1 < к < 10). Если исходное разделение
не приводит к разрешению до базовой линии, 2-пропанол может быть заменен
другими полярными растворителями, таким как этанол, дихлорметан, диоксан,
метил -трет-бутиловый эфир или этилацетат. Если требуются обращенно-фазовые
условия, значения энантиоселективности обычно значительно уменьшаются, так как
полярные взаимодействия, определяющие удерживание и селективность, сводятся
на нет (или, по крайней мере, очень ослабевают) в таких сильных протонных раство
ригелях. Следует отметить, что фирма Regis предлагает скрининг фаз Пиркла.
Использование ХНФ типа фаз Пиркла дает ряд известных преимуществ. Так как
строительные блоки селекторов доступны в обеих энантиомерных формах, могут
быть разработаны ХНФ в обеих конфигурациях, что позволяет получить обратный
порядок элюирования для энантиомеров (раздел 14.3). Небольшая молекулярная
масса этих селекторов вкупе с ограниченными размерами молекул приводит к их
высокой концентрации на поверхности ХНФ. В результате емкость загрузки образцов
намного выше, чем для белковых фаз, ХНФ на основе макроциклических
антибиотиков и ХНФ на основе циклодекстрина |61|. Синтетические донорно акцепторные
фазы также оказались ценными инструментами для разделения энантиомеров
методом СФХ |150, 151|.
14.6.8. Хиральные ионообменники
В хиральных ионообменниках применяются ионизируемые селекторы для
использования ионных взаимодействий между противоположно заряженными селекторами и
анализируемыми веществами. Хотя несколько таких ХНФ имеют в качестве
селекторов большие молекулы (например, белковые и гликопептидные ХНФ), здесь мы
говорим о низкомолекулярных селекторах, подобных классическим ионообменникам,
но имеющим хиральную основу. Эти ХНФ можно также рассматривать как
подмножество фаз Пиркла, несущих ионизируемые функциональные группы. Таким
образом, для них уже не обязателен режим неионных взаимодействий, характерный
для ХНФ типа фаз Пиркла. Для энантиомерного разделения ионизируемых
хиральных соединений было разработано несколько хиральных ионообменников:
хиральные аниониты на основе производных цинхоновых алкалоидов' для хиральных
кислот |I52|, хиральные катиониты на основе хиральных аминосульфоновых кислот и
аминокарбоновых кислот для разделения хиральных оснований |153| и цвиттерион-
ные ионообменники для разделения как кислот и оснований, так и цвиттерионных
анализируемых веществ, таких как аминокислоты и пептиды |154|. Ко времени
выхода этой книги были коммерчески доступны только хиральные анионоообменники
с селекторами из карбамата цинхонана (под торговым названиями Chiralpak QN-AX
и Chiralpak QD-AX от Crural Technologies) (рис. 14.28я)2. Аббревиатура АХ относится
к их анионообменным характеристикам, тогда как QN и QD обозначают тип цинхо-
нового алкалоида, который используется в качестве основы селекторов —
хинина (QN) и хинидина (QD).
1 Цинхонин — простейший алкалоид такого типа; хинин, хинидин и хинуклидин — его
производные, поэтому их называют цинхоновыми алкалоидами. — Прим. перев.
^Сейчас доступны цвитгерионные фазы Chiralpak ZW1X. — Прим. перев.
14.6. Хиральные неподвижные фазы и их характеристики 749
<*>
(б)
J*
смликагель
СН,
л
соон
CHIRALPAK* QN-AX: (8S.9R)
CHIRALPAK* QD-AX: (8R.9S)
j_
(R)
i(S)
J
ajl
(R)
0 4 8 12 мин
CHIRALPAK® QN-AX
Хининовая ХНФ
0 4 8 12 мин
CHIRALPAK" QD-AX
Хинидиновая ХНФ
Рис. 14.28. коммерчески доступные хиральные акионообменники на основе иинхоновых
алкалоидов, (я) Структура: (б) иллюстрация изменения порядка элюирова-
ния путем замены ХНФ с хинином на ХНФ с псевдохинидином.
Экспериментальные условия: размер колонки 150x4 мм; подвижная фаза: 1%
уксусная кислота в метаноле; температура 25 "С; скорость потока 1 мл/мин:
УФ-детектирование при 230 нм [155|
Селекторы этих колонок обладают высокой энантиоселективностью благодаря
наличию пяти стереогенных центров. В то время как конфигурации в положениях
N], Cj, С-4 фиксируются как IS, ЗЛ, 4S, конфигурации у атомов углерода Cg и С9
противоположны в хинине (8S, 9Я) и хинидине (8Я, 9S), как и у полученных из них
ХНФ. Поведение этих ХНФ на основе производных цинхоновых алкалоидов в
условиях эксперимента часто находится под стереоконтролем стереогенного центра Сд.
Это придает им псевдоэнантиомерные свойства1, как результат противоположной
конфигурации двух алкалоидов в этом хиральном центре. Помимо этой
своеобразной особенности конфигурации природных алкалоидов, для хиральных
неподвижных фаз на основе карбамата цинхония характерна исключительная способность
распознавания энантиомеров, возникающая благодаря их нескольким свойствам:
наличию объемного хинуклидина, плоского хинолинового кольца и полуэластичной
карбаматной группы с объемным трет-бутиловым остатком. Эти функциональные
группы служат потенциальными центрами связывания, и они структурно
организованы для образования полужестких каркасов с заранее заданными хиральными
«карманами» для размещения молекул анализируемого вещества2.
' Суть в том, что хинин и хинидин — эпимеры по стереогенному центру С, и обеспечивают
обратный порядок злюирования при замене QN на QD, Это в данном случае и называется псевдоэ-
нантиомерными свойствами. — Прим. перев.
^Оригинальный метод иммобилизации хинина, включающий гидросилилирование двойной
связи и прямую прививку к силикагелю был предложен отечественными исследователями.
См. Nesterenko P.N., Kxotov V.V., Staroverov S.M. J. Chronwtogr. A. 1994, v. 667. p. 19. Сорбент Диа-
сорб-Хинин выпускался «БиоХимМак СТ». — Прим. перев.
750 Глава 14. Разделение энантиомеров
Об основных механизмах молекулярного распознавания у этих
полусинтетических ХНФ многое известно из различных хроматографических исследований |156—
158|, в частности с использованием Фурье- и ЯМР-спектроскопии |159—163|, из
термодинамических исследований 1163, 164|, из результатов молекулярного
моделирования |161, 1631 и рентгеноструктурных исследований |161 —163, 165]. Если в «гостевую
молекулу»1 включены комплементарные Н-донорно-акцепторные и ароматические
фрагменты, благоприятные межмолекулярные водородные связи и л-ге-взаимодейст-
вия могут приводить к образованию стабильных комплексов с исключительно
высокой энантиоселективностью. Целенаправленная оптимизация, основанная на данных
упомянутых выше исследований механизма распознавания, привела к созданию ряда
сильных ХНФ [166|, и те из них, которые коммерчески доступны, имеют широкое
применение.
Анионнообменные сорбенты на основе цинхоновых алкалоидов прекрасно раз
деляют с высоким разрешением хиральные карбоновые, фосфиновые, фосфорные и
сульфокислоты |166|. Предпочтительнее это делать в полярно-органических или
водно-органических растворителях (в обращенно фазовом режиме). Область
применения этих сорбентов охватывает разделения УУ-дериватизированных а-, р- и у-амино-
кислот (рис. 14.286), соответствующих аналогов фосфиновой кислоты, фосфо- и
сульфокислот, а также многих других применяемых в фармацевтической
промышленности хиральных кислот (например, арилкарбоновых, арилоксикарбоновых
кислот, гидроксикислот, пиретроидных кислот и некоторых недериватизированных
аминокислот).
Если использовать ХНФ на основе цинхоновых алкалоидов со слабокислыми по
движными фазами, азот хинуклидина протонируется и действует как
фиксированный заряд хирального анионообменника. Тогда кислотные анализируемые вещества
в основном удерживаются при анионном обмене, и удерживание может быть
объяснено с помощью модели стехиометрического замещения |166|. В этом случае
графики зависимости log А от log концентрации противоиона |Z| (т.е. аниона буфера)
линейны (раздел 7.5.1 и уравнение (7.13)). Как обсуждалось в главе 7, наклон графика
зависимости log А от log концентрации противоионов для данного сорбента
изменяется с изменением заряда анализируемого вещества и противоиона. Наклон графика
будет более крутым при большем заряде анализируемого вещества и менее крутым
при большем заряде противоиона. Изменение концентрации противоиона может
быть использовано для изменения удерживания. Зачастую это несущественно влияет
на энантиоселективность. Элюирующая сила (эффективность конкурирующего
аниона) увеличивается в ряду: ацетат < формиат < фосфат < цитрат. Была
протестирована серия противоионов (кислотных добавок) в режиме с полярно-органическими
растворителями, и эти тенденции подтвердились также и для подвижных фаз,
состоящих из неводных полярных растворителей |167|. Другие переменные оказывают
значительное влияние на энантиоселективность, что позволяет гибко варьировать
метод при его разработке: рН (в обращенно-фазовом режиме), кислотно-основное
соотношение (в режиме с полярно-органическими растворителями), а также тип и
содержание органического растворителя/ растворителей (в обращенно-фазовом
режиме и режиме с полярно-органическими растворителями).
Предпочтительные подвижные фазы состоят из метанола с добавкой 0,5—2%
ледяной уксусной кислоты, а также 0,1—0,5% ацетата аммония (в режиме с
полярно-органическими растворителями) или метанола с аммонийно-ацетатным буфером
(при общей концентрации буфера в подвижной фазе между 10 и 100 мМ, рН 5—6)
(в обращенно-фазовом режиме). Метанол можно заменить ацетонитрилом или
смесью метанол-ацетонитрил, которые до некоторой степени являются комплементар-
1 «Гостевая молекула» - это молекула, попадающая внутрь другой. — Прим. перев.
14.6. Хиральные неподвижные фазы и их характеристики
ными в отношении их возможностей распознавания энантиомеров {обеспечивая
различные энантиоселективность и порядок элюирования).
Как отмечено выше, хининовые и хинидиновые ХНФ представляют собой
фактически диастереомеры, ведущие себя как энантиомеры. Поэтому они
обычно (но не всегда) дают противоположные порядки элюирования, как показано на-
рис. 14.286. Такая комплементарность их хирального распознавания может
систематически использоваться в методиках для выявления энантиомерных примесей и
препаративного разделения энантиомеров, поскольку желательно, чтобы энантио-
мерная примесь элюировалась первой (раздел 14.5). ХНФ на основе карбаматов ци-
нина и хинидина также достаточно перспективны для препаративного разделения
энантиомеров в силу их большой нагрузочной емкости. Например, измерения
изотермы адсорбции для Fmoc-a-аллилглицина на ХНФ с привитым 0-9-трет-<Ьугпл-
карбамоилхинидином выявили адсорбционный механизм, близкий к гомогенному, с
массовой нагрузкой 20 мг/г ХНФ 1168|. Хотя в основном ХНФ цинхоно-карбаматно-
го типа применяли для разделения хиральных кислот, недавние исследования
показали, что они могут быть использованы и для нейтральных и основных соединений с
подвижными фазами, характерными как для обращен но фазовых |169|, так и для
нормально-фазовых условий |170|.
14.6.9. Лигандообменные ХНФ. Хиральная лигандообменная
хроматография (ХЛОХ)
Лигандообменные ХНФ позволили провести первое полное хроматографическое
разделение рацемата в конце 1960-х годов. Даванков иммобилизовал пролин на
носителе из полистирола и использовал эту энантиоселективную матрицу в сочетании с
подвижными фазами, содержащими ионы Cu(II), для энантиомерного разделения
аминокислот |171|. Основным принципом хиральной лигандообменной
хроматографии (ХЛОХ) является обратимая координация иммобилизованных селекторов и
анализируемых веществ в координационной сфере иона металла, благодаря которой
образуется смешанный тройной комплекс: ион металла / селектор / анализируемое
вещество (рис. 14.29) [ 172,173]. Энантиоселективность этих диастереомерных
комплексов определяется стерическими и функциональными свойствами анализируемых
веществ. В процессе хроматографического разделения координированные лиганды
(а)
(S.S)
(6)
<R.S)
Носитель из полистирола
Носитель из полистирола
Рис. 14.29. Механизм хиральной лигандообменной хроматографии (ХЛОХ). Тройные
диастереомерные комплексы иона меди Cu(ll), иммобилизованного лиган-
да 5-энантиомера пролина (X = Н) (или гидроксипролина X = ОН) с
анализируемыми веществами S- и Я-пролином, (а) и (Й), соответственно |173|
Глава 14. Разделение энантиомеров
обратимо замещаются другими лигандами из подвижной фазы, такими как аммиак и
вода. Важным аспектом этих разделений является то, что обмен лигандов в
координационной сфере катиона металла происходит быстро, в противном случае
эффективность колонки будет низкой.
Важнейшей предпосылкой для ХЛОХ является наличие хелатирующего иона
металла как в селекторе, так и в анализируемом веществе |172]. Подходящие структуры
имеют бидентатные или тридентатные лиганды с двумя или тремя
функциональными группами — донорами электронов, такими как аминогруппы, гидроксильные
и карбоксильные функциональные группы. Такие структуры обычно встречаются
в а-аминокислотах, аминоспиртах и сс-гидроксикислотах (рис. 14.30), типичных
соединениях, которые были разделены ХЛОХ. Cu(II) является предпочтительным хела-
тирующим ионом металла, но подходят также Zn(II) и Ni(II). Показано, что в
качестве селекторов для лигадообменных ХНФ лучшие результаты дают жесткие
циклические аминокислоты, такие как пролин и гидроксипролин, в сочетании
с Cu(II). Эти хелатирующие селекторы либо (1) ковалентно закреплены на
поверхности частиц силикагеля и органического полимера, соответственно, либо (2)
динамически нанесены на подложку с привитой обращенной фазой (обычно иммобилиза-
ция происходит за счет адсорбции, обусловленной гидрофобными взаимодействиями
с длинными алкильными цепями заместителей селекторов). При этом в подвижной
фазе допускается только небольшой процент органических веществ. Как
обсуждалось выше, из-за полярной природы анализируемых веществ (для разделения с
помощью ХЛОХ), подвижная фаза должна иметь водную основу. В подвижную фазу
обычно добавляют небольшие количества ионов металла, чтобы компенсировать
потерю металла из сорбента во время хроматографии, тем самым делая разделение
более стабильным.
Н3С
20
Время (мин)
40
Рис. 14.30. Разделение энантиомеров оксикислот в режиме хиральной лигандообмен-
ной хроматографией (ХЛОХ). Условия эксперимента: колонка
CH1RALPAK МА; подвижная фаза; 10% ACN/H20 + 2мМ CuS04 |8]
14.7. Термодинамические аспекты 753
Детектирование лишенных хромофоров аминокислот и гидроксикислот
возможно благодаря повышению их поглощения в УФ-области в присутствии Си**, в то
время как масс-спектрометрическое обнаружение присутствие ионов металлов в
подвижной фазе может затруднить. Экспериментальные условия, которые могут
варьироваться при разработке метода, включают рН подвижной фазы, тип и
концентрацию буферных солей, характер и содержание органического растворителя,
температуру и концентрацию ионов металлов в подвижной фазе.
Коммерчески доступны ряд ХНФ для ХЛОХ с ковалентно закрепленными на
подложке фазами и фазами, нанесенных в виде покрытий: Chiralpak MA+ (на основе N,
iV-диоктилА аланина, нанесенного на обращенную фазу C|g) от компании Chiral
Technologies, Nucleosil Chiral-1 (на основе А-гидроксипролина, химически связанного
с силикагелем), производитель Macherey-Nagel и Chirex 3126 (на основе N,S-auoK-
тил-пеницилламина, нанесенного на обращенную фазу С|8) фирмы Phenomenex. В
прошлом ХЛОХ была единственной методикой, которая позволяла проводить прямое
разделение энантиомеров аминокислот без дериватизации. Однако сегодня важность
хиральной лигандообменной хроматографии снижается в результате появления более
удобных вариантов анализа. Подробную информацию можно найти в обзоре [174|.
14.7. Термодинамические аспекты
В основе механизма удерживания анализируемого вещества и энантиоселективности,
конечно, лежит термодинамика процесса удерживания, как и при разделении ахира-
льных растворенных веществ, обсуждавшемся в предыдущих главах. Однако в случае
разделения энантиомеров имеются некоторые дополнительные термодинамические
соображения.
14.7.1. Термодинамика ассоциации анализируемого
вещества с селектором
Константа равновесия К/ {рис. I4.9) ассоциата анализируемое вещество-селектор
может быть описана через термодинамические параметры следующим образом
ДС?= -ДТТп К( = ДЯ? - TAS?. (14.3)
Здесь ДС?, Д#Р и AS? относятся к изменениям стандартной свободной энергии,
энтальпии и энтропии при образовании комплекса анализируемое вещество-селектор,
R — универсальная газовая постоянная, Т— абсолютная температура (в градусах К),
а нижний индекс i обозначает соответствующие соединения (т.е. здесь R- или
5-энантиомер); у — фазовое отношение (раздел 2.3.1). Дальнейшие преобразования
уравнения (14.3) дают два дополнительных уравнения
1аК,=-±Ж+&# (,4.4)
Т R R
и
ДДС^ = ДС£ - AG% = -R ■ Т ■ In -^ = -Я- Г • In a. (14.5)
Таким образом, графики зависимости In Kt от 1/Т должны быть прямыми с
наклоном, пропорциональным АН?. Аналогично, коэффициент селективности а для двух
энантиомеров R и S можно соотнести с изменением их стандартных свободных
энергий при ассоциации анализируемого вещества с селектором ДДС^5, а также с
соответствующими изменениями энтальпии AAH^S и энтропии AAS^S.
754 Глава 14. Разделение энантиомеров
14.7.2. Термодинамика прямого хроматографического
разделения энантиомеров
Если рассматривать только один вид взаимодействия между анализируемым
веществом и селектором — энантиоселективное, не принимая во внимание другие
механизмы адсорбции анализируемого вещества, то Kt в уравнениях (14.3)—(14.5) можно
связать с к и \) соотношением
к = К^. (14.6)
Значения АДСЙ $, ДД//5 s и ллуг; могут быть получены из значений а в
зависимости от Т, так как (обычно неизвестное) фазовое отношение у отсутствует в
уравнении (14.5), но не в уравнении (14.4)1. Графики зависимости In к от 1/Т обычно
имеют положительный наклон (к уменьшается с возрастанием Г), что подразумевает
отрицательную величину Afff или энтальпиино-контрояируемыи процесс
удерживания. Таким образом, нековалентные силы притяжения (большей частью
электростатической природы) между растворенным веществом и селектором приводят к
значениям Л^ 3> 1. Этим взаимодействиям, способствующим удерживанию, обычно
противодействуют энтропийные эффекты, поскольку комплекс анализируемого
вещества с селектором в большей степени упорядочен, чем анализируемое вещество
в подвижной фазе. То есть, Atfi > Д5° и ДЛЯ0 > ЛЛ,Ч°, что и наблюдается для
широкого спектра различных систем ХНФ-анализируемое вещество-подвижная фаза.
Обычно это приводит к уменьшению значений а при более высоких температурах.
Противоположное явление, увеличение энантиоселективности с ростом Т
(называемое энтропийно-контролируемым хиральным распознаванием), наблюдалось в
нескольких случаях при использовании полисахаридных ХНФ и ХНФ белкового типа.
Такое поведение было объяснено возможной (де)сольватацией центров связывания
|175| и/или конформационными изменениями в структуре селектора |176, 177].
Иногда наблюдались нелинейные графики зависимости In k от 1/Т |36|, Подобные
исключения, заключающиеся в линейном увеличение In А: с ростом 1/Т, наблюдаются
и в ахиральном разделении (раздел 2.3.2.2), возможно, по аналогичным причинам.
Необычное поведение другого типа, вызванное изменениями температуры,
наблюдалось при разделении хиральных дигидропиримидинонов на полисахаридных
ХНФ |178|. Графики зависимости \пк от 1/Т были получены в условиях: (I)
нагревания колонки от 10 до 50 °С и (2) ее охлаждения от 50 до 10 °С. Полученные графики
для колонки Chiralpak AD-H, сольватированной этанолом, не совмещались друг
с другом. То есть, в системе наблюдался значительный гистерезис, который не был
результатом конформационных изменений полисахаридного сорбента, а скорее
медленным уравновешиванием неподвижной фазы при изменении Т.
14.7.3. Термодинамика гетерогенной хиральной
поверхности
В приведенном выше обсуждении не упомянут тот факт, что энантиоселективное
удерживание не обязательно связано с одним центром удерживания |179|. Хотя это
наблюдение справедливо и для ахирального удерживания, существует важное
отличие в случае энантиомерного разделения. То есть, другие центры, скорее всего, будут
неэнантиоселективными, их называют центрами типа / в отличие от энантиоселек-
' Уравнение (14.4) можно переписать для kt с учетом уравнения (14.6)
1 АН ° AS ^
Ы К, = + + In 4\ — Прим. перев.
Т R R
14.7. Термодинамические аспекты
тивных центров типа И |179]. Центры типа I могут включать подложку (например,
силикагель), линкерные группы, спейсеры, лиганды эндкепированных силанольных
групп, покрывающих подложку, и даже фрагменты селектора, на которых нет энан-
тиоселективных центров связывания. Известно, что присутствие центров типа I
ухудшает энантиоселективность. Хотя аффинность связывания центрами типа I
обычно намного ниже, чем для центров типа II, концентрация центров типа I может
на несколько порядков превышать концентрацию центров типа II, особенно в случае
макромолекулярных селекторов, таких как белки (раздел 14.6.3). Поэтому вкладом
центров типа I в общее удерживание обычно пренебречь нельзя, а
экспериментальные данные по удерживанию представляют собой сумму неспецифических (ахираль
ных) и специфических (хиральных) вкладов в к:
kR = kl,R + kll,R (14-7>
и
ks = k/,S+k/,iS- (I4-7a)
Значения к в уравнениях (14.7) и (14.7а) даны для ввода небольшого количества
образца (неперегруженное разделение), а индексы I и II относятся соответственно
к центрам типа I и типа II. Индексы R и 51 относятся к значениям для R- и 5-энанти-
омеров, соответственно. Экспериментальный коэффициент энантиоселективности
определяется как а = кц (к$ (для кц > к$), или
к, v + кц с
(14.8)
Удерживание на центре I одинаково для обоих энантиомеров (т. е. оно неэнантиоселек-
тивное), поэтому kt R = kt s = к/ и
а = к,+к"*. (14.8а)
*/ + kl!,S
Если неспецифическое удерживание отсутствует, к/ = 0 и а = кцд/кцх-
Предположим, что Д-энантиомер лучше удерживается так, что кц ц /кц^ > 1. При к/ > 0
значение а в уравнении (14.8а) уменьшается с ростом к/ и приближается к 1 (когда
энантиоселективность отсутствует) при к/:» кц ц.
Очевидно, что максимальная селективность при разделении энантиомеров может
быть достигнута либо увеличением до возможного максимума селективности энанти-
оселективных центров типа II (кцц/кц <;), либо сведением к минимуму вклада
в удерживание неэнантиоселективных центров типа 1. Когда целью является
объяснение зависимости энантиоселективности селектора (т.е. активности центров
типа II) от природы анализируемого вещества, селектора и условий эксперимента,
подходящей для оценки количественной характеристикой является «истинная»
термодинамическая энантиоселективность (кцд/кцд), тогда как экспериментально
наблюдаемая энантиоселективность (соответствующая а в формуле (14.8а)) может
вводить в заблуждение |179, 180|.
Из предыдущего обсуждения ясно, что экспериментальные значения « лишь
косвенно связаны с различными взаимодействиями между анализируемым веществом и
селектором, так как эти значения а будут отражать как ахиральные, так и хиральные
взаимодействия анализируемого вещества с неподвижной фазой. Относительный
вклад хиральных и ахиральных центров в наблюдаемую энантиоселективность можно
определить, сопоставляя данные изотерм адсорбции для каждого энантиомера с
бинарной моделью Ленгмюра (для двух центров связывания) в широком диапазоне
концентраций анализируемого вещества. В этом случае методика дает значения к/ я, кцд,
756 Глава 14. Разделение энантиомеров
кц; и кц£ при небольшой концентрации анализируемого образца в подвижной фазе
(в линейной области изотермы Ленгмюра). Если данные для изотерм получены
при разных температурах, могут быть вычислены значения ДЯ/для каждого энантио-
мера на центрах обоих типов (I и II) [181, 182|. Можно также получить значения
АДС§ s, ДД//2 s и ДД-SJ! s и использовать их для определения основ энантиоселектив-
ности в данной системе. Используя эту методику измерений для построения изотерм
адсорбции при различных температурах, Гиошон и коллеги исследовали, например,
термодинамику связывания 2,2,2-трифтор-1-(9-антрил)-этанола (спирта Пиркла) |182|
и З-хлор-1-фенилпропанола |181] на силикагеле, модифицированном О-9-трет-бутил-
карбамоилхинидином, в условиях селективности центров связывания в неводных
растворителях (соотвествующих нормально-фазовой хроматографии).
Литература
1. С. Roussel, J. Pierrot-Sanders, I. Heitmann, and P. Piras, in G. Subramanian, Chiral Separation
Techniques, 2nd ed., Wiley-VCH, Weinheim, 2001, 95.
2. P. Piras and С Roussel, J. Pharm. Biomed. Anal., 46 (2008) 839.
3. T. Huybrechts, G. Torok, T. Vennekens, R. Sneyers, S. Vrielynck, and I. Somers, LCGC Europe,
20 (2007) 320.
4. H. A. Wetli and E. Francotte, J. Sep. Sci., 30 (2007) 1255.
5. V. A. Davankov, Pure Appl. Chem., 69 (1997) 1469.
6. B. Testa, Principles of Stereochemistry, Wiley-VCH, Weinheim, 1998.
7. M. Lammerhofer and W. Lindner, in Separation Methods in Drug Synthesis and Purification,
K. Valko, ed., Elsevier, Amsterdam, 2000, p. 337.
8. W. Lindner, in Stereoselective Synthesis. Series: Methods of Organic Chemistry', Vol. E2la, G. Helm-
chen, R. W. Hoffmann, J. Mulzer, E. Schaumann, eds., (Houben-Weyl), Thieme, Stuttgart, 1995,
p. 225.
9. R. J. Bopp and J. H. Kennedy, LCGC, 6 (1988) 514.
10. W. Lindner, С Leitner, and G. Uray, J. Chromatogr., 316 (1984) 605.
11. F. Lai, A. Mayer and T. Sheehan, J. Pharm. Biomed. Anal., 11 (1993) 117.
12. H. Bruckner, R. Winner, and H. Godel, J. Chromatogr., 476 (1989) 3.
13. H. Bruckner, T. Westhauser, and H. Godel, J. Chromatogr. A, 711 (1995) 201.
14. H. Bruckner and C. Gall, J. Chromatogr., 555 (1991) 81.
15. R. Bhushan and H. Bruckner, Amino Acids, 27 (2004) 31.
16. N. Nimura, H. Ogura, and T. Kinoshita, J. Chromatogr., 202 (1980) 75.
17. O. P. Kleidernigg, M. Lammerhofer, and W. Lindner, Enantiomer, 1 (1996) 387.
18. F. Bressolle, M. Audran, T.-N. Pham, and J.-J. Vallon, J. Chromatogr. B, 687 (1996) 303.
19. A. Karlsson and С Pettersson, Chiralily, 4 (1992) 323.
20. C. Pettersson and E. Heldin, in A Practical Approach to Chiral Separations by Liquid
Chromatography, G. Subramanian, ed., VCH, Weinheim, 1994, p. 279.
21. J. N. LePage, W. Lindner, G. Davies, D. E. Seitz, and B. L. Karger, Anal. Chem., 51 (1979) 433.
22. R. Marchelli, R. Corradini, T. Bertuzzi, G. Galaverna, A. Dossena, F. Gasparrini, B. Galli,
С Villani, and D. Misiti, Chirality, 8 (1996) 452.
23. S. Zeng, J. Zhong, L. Pan, and Y. Li, J. Chromatogr. B, 728 (1999) 151.
24. V. A. Davankov, Chromatograhia, 27 (1989) 475.
25. P. Levkin, N. M. Maier, W. Lindner, and V. Schurig, submitted (2008).
26. С. Е. Dalgliesh, J. Chem. Soc, 132 (1952) 3940.
27. W. H. Pirkle and Т. С Pochapsky, Chem. Rev., 89 (1989) 347.
28. V. A. Davankov, Chirality, 9 (1997) 99.
29. С. С. Pfeiffer, Science, 124 (1956) 29.
30. G. Hesse and R. Hagel, Justus Liebigs Annalen der Chemie, (1976) 996.
31. Y. Okanioto, M. Kawashima, and K. Hatada, J. Am. Chem. Soc, 106 (1984) 5357.
32. E. Yashima, С Yamamoto, and Y. Okamoto, J. Am. Chem. Soc, 118 (1996) 4036.
33. С Yamamoto, E. Yashima, and Y. Okamoto, J. Am. Chem. Soc, 124 (2002) 12583.
Литература 757
34. Y. К. Ye, S. Bai, S. Vyas, and M. J.Wirth, J. Phys. Chem. В., Ill (2007) 1189.
35. R. B. Kasat, N.-H. L. Wang, and E. I. Franses, Biomacromolecules, 8 (2007) 1676.
36. T. O'Brien, L. Crocker, R. Thompson, K. Thompson, P. H. Toma, D. A. Conlon, B. Feibush,
С Moeder, G. Bicker, and N. Grinberg, Anal. Chem., 69 (1997) 1999.
37. E. Yashima, M. Yamada, Y. Kaida, and Y. Okamoto, J. Chromatogr. A, 694 (1995) 347.
38. R. W. Stringham and J. A. Blackwell, Anal. Chem., 68 (1996) 2179.
39. T. D. Booth and I. W. Wainer, J. Chromatogr. A, 741 (1996) 205.
40. T. D. Booth and I. W. Wainer, J. Chromatogr. A, 737 (1996) 157.
41. T. D. Booth, W. J. Lough, M. Saeed, T. A. G. Noctor, and I. W. Wainer, Chirality, 9 (1997) 173.
42. T. D. Booth, K. Azzaoui, and I. W. Wainer, Anal. Chem., 69 (1997) 3879.
43. Y. Okamoto, M. Kawashima, and K. Hatada, J. Chromatogr., 363 (1986) 173.
44. E. Francotte and T. Zhang, J. Chromatogr. A, 718 (1995) 257.
45. C. Perrin, V. A. Vu, N. Matthijs, M. Maftouh, D. L. Massart, and Y. Vander Heyden, J.
Chromatogr. A, 947 (2002) 69.
46. C. Perrin, N. Matthijs, D. Mangelings, C. Granier-Loyaux, M. Maftouh, D. L. Massart, and Y.
Vander Heyden, J. Chromatogr. A, 966 (2002) 119.
47. N. Matthijs, С Perrin, M. Maftouh, D. L. Massart, and Y. V. Heyden, J. Chromatogr. A, 1041
(2004) 119.
48. N. Matthijs, M. Maftouh, and Y. Vander Heyden, J. Chromatogr. A, 1111 (2006) 48.
49. P. Borman, B. Boughtelower, K. Cattanach, K. Crane, K. Freebairn, G. Jonas, I. Mutton, A. Pa-
tel, M. Sanders, and D. Thompson, Chirality, 15 (2003) SI
50. Y. Okamoto and E. Yashima, Angew. Chem. Int. Ed., 37 (1998) 1021.
51. B. Chankvetadze, С Yamamoto, and Y. Okamoto, Chem. Lett., (2000) 1176.
52. E. Yashima, J. Chromatogr. A, 906 (2001) 105.
53. T. Wang, Y. W. Chen, and A. Vailaya, J. Chromatogr. A, 902 (2000) 345.
54. R. M. Wenslow, Jr. and T. Wang, Anal. Chem., 73 (2001) 4190.
55. S. Caccamese, S. Bianca, and G. T. Carter, Chirality, 19 (2007) 647.
56. R. W. Stringham, K. G. Lynam, and B. S. Lord, Chirality, 16 (2004) 493.
57. K. Tachibana and A. Ohnishi, J. Chromatogr. A, 906 (2001) 127.
58. B. Chankvetadze, C. Yamamoto, and Y. Okamoto, J. Chromatogr. A, 922 (2001) 127.
59. K. G. Lynam and R. W. Stringham, Chirality, 18 (2005) 1.
60. T. Zhang, D. Nguyen, P. Franco, T. Murakami, A. Ohnishi, and H. Kurosawa, Anal. Chim. Acta,
557 (2006) 221.
61. E. R. Francotte, J. Chromatogr. A, 906 (2001) 379.
62. P. Franco, A. Senso, L. Oliveros, and С Minguillorn, J. Chromatogr. A, 906 (2001) 155.
63. T. Ikai, C. Yamamoto, M. Kamigaito, and Y. Okamoto, J. Chromatogr. B, 875 (2008) 2.
64. T. Zhang, C. Kientzy, P. Franco, A. Ohnishi, Y. Kagamihara, and H. Kurosawa, J. Chromatogr. A,
1075 (2005) 65.
65. T. Zhang, D. Nguyen, P. Franco, Y. Isobe, T. Michishita, and T. Murakami, J. Pharm. Biomed.
Anal., 46 (2008) 882.
66. P. Franco and T. Zhang, J. Chromatogr. B, 875 (2008) 48.
67. A. Ghanem and H. Aboul-Enein, J. Liq. Chromatogr., 28 (2005) 2863.
68. A. Ghanem, H. Hoenen. and H. Y. Aboul-Enein, Talanta, 68 (2006) 602.
69. K. W. Phinney, Anal. Bioanal. Chem., 382 (2005) 639.
70. J. L. Bernal, L. Toribio, M. J. D. Nozal, E. M. Nieto, and M. I. Montequi, J. Biochem. Biophys.
Meth., 54 (2002) 245.
71. M. Maftouh, C. Granier-Loyaux, E. Chavana, J. Marini, A. Pradines, Y. Vander Heyden, and
С Picard, J. Chromatogr. A, 1088 (2005) 67.
72. R. W. Stringham, J. Chromatogr. A, 1070 (2005) 163.
73. Y. Okamoto, S. Honda, I. Okamoto, and H. Yuki, J. Am. Chem. Soc, 103 (1981) 6971.
74. G. Blaschke, Chem. Ber., 107 (1974) 237.
75. G. Blaschke, W. Broker, and W. Fraenkel, Angew. Chem. Int. Ed., 25 (1986) 830.
76. J. N. Kinkel, in A Practical Approach to Chiral Separations by Liquid Chromatography, G. Subrama-
nian, ed., VCH, Weinheim, 1994, p. 217.
77. T. Nakano, J. Chromatogr. A, 906 (2001) 205.
78. R. Cirilli, R. Costi, R. D. Santo, M. Artico, A. Roux, B. Gallinella, L. Zanitti, and F. L. Torre, J.
Chromatogr. A, 993 (2003) 17.
79. F. Gasparrini, D. Misiti, R. Rompietti, and C. Villani, J. Chromatogr. A, 1064 (2005) 25.
758 Глава 14. Разделение энантиомеров
80. S. G. Allenmark, S. Andersson, P. Moller, and D. Sanchez, Chirality, 7 (1995) 248.
81. S. Andersson, S. Allenmark, P. Moeller, B. Persson, and D. Sanchez, J. Chromatogr. A, 741
(1996) 23.
82. S. G. Allenmark and S. Andersson, J. Chromatogr. A, 666 (1994) 167.
83. M. С Millot, J. Chromatogr. B, 797 (2003) 131.
84. J. Haginaka, J. Chromatogr. B, 875 (2008) 12.
85. J. Hermansson, J. Chromatogr., 269 (1983) 71.
86. T. Miwa, M. Ichikawa, M. Tsuno, T. Hattori. T. Miyakawa, M. Kayano, and Y. Miyake, Chem.
Pharm. Bull., 35 (1987) 682.
87. P. Erlandsson, I. Marie, L. Hansson, R. Isaksson, C. Petterson, and G. Petterson, J. Am. Chem.
Soc., 112 (1990) 4573.
88. E. Domenici, C. Brett, P. Salvadori, G. Felix, 1. Cahagne, S.Motellier, and I. W. Wainer, Chro-
marographia, 29 (1990) 170.
89. W. J. M. Kremer and L. H. Janssen, Pharmacological Reviews, 40 (1988) 1.
90. J. Haginaka, C. Seyama, and T. Murashinia, J. Chromatogr. A, 704 (1995) 279.
91. J. Haginaka, С Seyama, and N. Kanasugi, Anal. Chem., 67 (1995) 2539.
92. J. Haginaka and H. Takehira, J. Chromatogr. A, 111 (1997) 241.
93. I. Kato, J. Schrode, W. J. Kohr, and M. Laskowski, Biochemsitry, 26 (1987) 193.
94. J. Hermansson, Trends Anal. Chem., 8 (1989) 251.
95. M. S. Waters, D. R. Sidter, A. J. Simon, C. R. Middaugh, R. Thompson, L. J. August, G. Bicker,
H. J. Perpall, and N. Grinberg, Chirality, 11 (1999) 224.
96. T. A. G. Noctor and 1. W. Wainer, Pharmaceut. Res., 9 (1992) 480.
97. H. Matsunaga and J. Haginaka, J. Chromatogr. A, 1106 (2006) 124.
98. F. Zsila, and Y. Iwao, Biochem. Biophys. Acta, 1770 (2007) 797.
99. S. Andersson, S. Allenmark, O. Erlandsson, and S. Nilsson, J. Chromatogr., 498 (1990) 8].
100. J. Haginaka and N. Kanasugi, J. Chromatogr. A, 694 (1995) 71.
101. K. Tomoko, S. Akimasa, and M. Katsumi, Pharmaceutical Research, 23 (2006) 1038.
102. I. Petitpas, A. B. Bhattacharya, S. Twine, M. East, and S. Curry, J. Biol. Chem., 276 (2001)
22804.
103. J. Stahlberg. H. Henriksson, C. Divne, R. Isaksson, G. Pettersson, G. Johansson, and T. A.
Jones, J. Mol. Biol., 305 (2001) 79.
104. J. Chen, W. A. Korfmacher, and Y. Hsieh, J. Chromatogr. B. 820 (2005) 1.
105. D. W. Armstrong and W. DeMond, J. Chrom. Set., 22 (1984) 411.
106. C. R. Mitchell and D. W. Armstrong, in Chiral Separations, G. Giibilz and M. G. Schmid, eds.,
Vol. 243, Humana Press, Totowa, NJ, 2004, p. 61.
107. M. V. Rekharsky and Y. Inoue, Chem. Rev., 98 (1998) 1875.
108. D. W. Armstrong. L. W. Chang, S. С Chang, X. Wang, H. Ibrahim, G. R. Reid, and T. Beesley,
J. Liq. Chromatogr., 20 (1997) 3279.
109. S. C. Chang, G. L. Reid, HI, S. Chen, C. D. Chang, and D. W. Armstrong, Trends Anal. Chem.,
12(1993) 144.
110. D. W. Armstrong, S. Chen, С Chang, and S. Chang, J. Liq. Chromatogr., 15 (1992) 545.
111. D. W. Armstrong, Y. Tang, S. Chen, Y. Zhou, C. Bagwill, and J.-R. Chen, Anal. Chem., 66
(1994) 1473.
112. D. W. Armstrong, Y. Liu, and К. Н. Ekborgott, Chirality, 7 (1995) 474.
113. K. Ekborg-Ott, Y. Liu, and D. W. Armstrong, Chirality, 10 (1998) 434.
114. A. Berthod, X. Chen, J. P. Kullman, D. W. Armstrong, F. Gasparrini, I. D'Acquarica, C. Villani,
and A. Carotti, Anal. Chem., 72 (2000) 1767.
115. T. J. Ward and A. B. Farris, 111., J. Chromatogr. A, 906 (2001) 73.
116. I. Ilisz, R. Berkecz, and A. Peter, J. Sep. Set., 29 (2006) 1305.
117. T. L. Xiao and D. W. Armstrong, in Chiral Separations, G. Gubitz and M. G. Schmid, eds., Vol.
243, Humana Press, Totowa, NJ, 2004, 113.
118. I. D'Acquarica, F. Gasparrini, D. Misiti, M. Pierini, C. Villani, Adv. Chromatogr., Vol. 46, CRC
Press, Boca Raton, 2008, 108.
119. ASTEC. Chirobiotic Handbook, ASTEC, Whippany, NJ, 1997
120. T. E. Beesley, J. T. Lee, and A. X. Wang, Chiral Separation Techniques, 2nd ed., G. Subramani-
an(ed.), Wiley-VCH, Weinheim, 2001, p. 25.
121. M. E. Andersson, D. Asian, A. Clarke, J. Roeraade, and G. Hagman, J. Chromatogr. A, 1005
(2003) 83.
Литература 759
122. С. Wang, С. Jiang, and D. W. Armstrong, J. Sep. Sci., 31 (2008) 1980.
123. H. Jiang, Y. Li, M. Pelzer, M. J. Cannon, С Randlett, H. Junga, X. Jiang, and Q. С Ji, J. Chro-
rnatogr. Л, 1192 (2008) 230.
124. K. H. Ekborg-Olt, J. P. Kullman, X. Wang, K. Cahm, L. He, and D. W. Armstrong, Chirality,
10 (1998)627.
125. A. Berthod, Y. Liu, С Bagwill, and D. W. Armstrong, J. Chromalogr. A, 731 (1996) 123.
126. Y. Liu, A. Berthod, С R. Mitchell, T. L. Xiao, B. Zhang, and D. W. Armstrong, J. Chroma-
togr. A, 978 (2002) 185.
127. L. R. Sousa, G. D. Y. Sogah, D. H. Hoffman, and D. J. Cram, J. Am. Chem. Soc, 100 (1978)
4569.
128. T. Shinbo, T. Yamaguchi, K. Nishimura, and M. Sugiura, J. Chromatogr., 405 (1987) 145.
129. M. H. Hyun, J. S. Jin, and W. Lee, J. Chromalogr. A, 822 (1998) 155.
130. M. H. Hyun, S. С Han, B. H. Lipshutz, Y.-J. Shin, and С J. Welch, J. Chromalogr. A, 910
(2001) 359.
131. R. J. Steffeck, Y. Zelechonok, and К. Н. Gahm, J. Chromalogr. A, 947 (2002) 301.
132. M. H. Hyun, J. Sep. Sci., 26 (2003) 242.
133. Y. Machida, H. Nishi, and K. Nakamura, Chirality, 11 (1999) 173.
134. W. Pirkle and D. House, J. Org. Chem., 44 (1979) 1957.
135. F. Mikes and G. Boshart, J. Chromalogr., 149 (1978) 455.
136. W. H. Pirkle, M. H. Hyun, A. Tsipouras, В. С Hamper, and B. Banks, J. Pharm. Biomed. Anal.,
2 (1984) 173.
137. W. H. Pirkle and Т. С Pochapsky, J. Am. Chem. Soc., 109 (1987) 5975.
138. W. H. Pirkle and S. R. Selness, J. Org. Chem., 60 (1995) 3252.
139. W. H. Pirkle, P. G. Murray, and S. R. Wilson, J. Org. Chem., 61 (1996) 4775.
140. W. H. Pirkle, D. W. House, and J. M. Finn, J. Chromalogr., 192 (1980) 143.
141. W. H. Pirkle and R. Dappen, J. Chromalogr., 404 (1987) 107.
142. С J. Welch, J. Chromalogr. A, 666 (1994) 3.
143. W. H. Pirkle, Т. С Pochapsky, G. S. Mahler, D. E. Corey, D. S. Reno, and D. M. Alessi, J. Org.
Chem., 51 (1986) 4991.
144. W. H. Pirkle and С J. Welch, Tetrahedron: Asymmetry, 5 (1994) 777.
145. M. E. Koscho, P. L. Spence, and W. H. Pirkle, Tetrahedron Asymmetry, 16 (2005) 3147.
146. F. Gasparrini, D. Misiti, and С Villani, Chirality, 4 (1992) 447.
147. F. Gasparrini, D. Misiti, С Villani, and F. La Torre, J. Chromatogr., 539 (1991) 25.
148. N. M. Maier and G. Uray. J. Chromatogr. A, 732 (1996) 215.
149. F. Gasparrini, D. Misiti, and C. Villani, J. Chromatogr. A, 906 (2001) 35.
150. P. Macaudiere, A. Tambute, M. Caude, R. Rosset, M. A. Alembik, and l. W. Wainer, J.
Chromatogr., 371 (1986) 177.
151. A. M. Blum, K. G. Lynam, and E. С Nicolas, Chirality, 6 (1994) 302.
152. M. Lammerhofer and W. Lindner, J. Chromatogr. A, 741 (1996) 33.
153. C. V. Hoffmann, M. Lammerhofer, and W. Lindner, J. Chromatogr. A, 1161 (2007) 242.
154. С V. Hoffmann, R. Pell, M. Lammerhofer, and W. Lindner, Anal. Chem., submitted (2008).
155. M. Lammerhofer, N. M. Maier, and W. Lindner, Nachrichten aus der Chemie, 50 (2002) 1037.
156. A. Mandl, L. Nicoletti, M. Lammerhofer, and W. Lindner, J. Chromatogr. A, 858 (1999) 1.
157. N. M. Maier, L. Nicoletti, M. Lammerhofer, and W. Lindner, Chirality, 11 (1999) 522.
158. M. Lammerhofer, P. Franco, and W. Lindner, J. Sep. Sci., 29 (2006) 1486.
159. J. Lesnik, M. Lammerhofer, and W. Lindner, Anal. Chim. Acta, 401 (1999) 3.
160. R. Wirz, T. Buergi, W. Lindner, and A. Baiker, Anal. Chem., lb (2004) 5319.
161. N. M. Maier, S. Schefzick, G. M, Lombardo, M. Feliz, K. Rissanen, W. Lindner, and К. В. Lip-
kowitz, J. Am. Chem. Soc, 124 (2002) 8611.
162. K. Akasaka, K. Gyimesi-Forras, M. Lammerhofer, T. Fujita, M. Watanabe, N. Harada, and W.
Lindner, Chirality, 17 (2005) 544.
163. W. Bicker, I. Chiorescu, V. B. Arion, M. Lammerhofer, W. Lindner, Tetrahedron: Asymmetry, 19
(2008) 97.
164. W. R. Oberleitner, N. M. Maier, and W. Lindner, J. Chromatogr. A, 960 (2002) 97.
165. C. Czerwenka, M. Lammerhofer, N. M. Maier, K. Rissanen, and W. Lindner, Anal. Chem., 74
(2002) 5658.
166. M. Lammerhofer and W. Lindner, Adv. Chromatogr., Vol. 46, CRC Press, Boca Raton, 2008, 1.
760 Глава 14. Разделение энантиомеров
167. К. Gyimesi-Forras, К. Akasaka, M. Lammerliofer, N. М. Maier, T. Fujita, M. Watanabe, N. На-
rada, and W. Lindner, Chirality, 17 (2005) SI34
168. R. Arnell, P. Forssen, T. Fornsledt, R. Sardella, M. Lammerliofer, and W. Lindner, J.
Chromatogr. A, 1216 (2009) 3480.
169. K. Gyimesi-Forras, J. Kokosi, G. Szasz, A. Gergely, and W. Lindner, J. Chromatogr. A, 1047
(2004) 59.
170. Gyimesi-Forras, N. M. Maier, J. Kokosi, A. Gergely, and W. Lindner, Chirality, 21 (2009) 199.
171. V. A. Davankov, Enantiomer, 5 (2000) 209.
172. V. A. Davankov, in Chiral Separations, G. Gubitz and M. G. Schmid, eds., Vol. 243, Humana
Press, Totowa, NJ, 2004, 207.
173. V. A. Davankov, J. Chromatogr. A, 666 (1994) 55.
174. G.Giibitz and M.G. Schmid, in Chiral Separation Techniques, 3rd ed.,G. Subramanian, ed.,
Wiley-VCH, Weinheim, Germany, 2007, p. 155.
175. W. H. Pirkle and P. G. Murray, J. High Resol. Chromatogr., 16 (1993) 285.
176. R. W. Stringham and J. A. Blackwell, Anal. Chem., 68 (1996) 2179.
177. O. Gyllenhaal and M. Stefansson, Chirality, 17 (2005).
178. F. Wang, D. Yeung, J. Han, D. Semin, J. S. McElvain, and J. Cheetham, J. Sep. Sci., 31 (2008)
604.
179. G. Gotmar, T. Fornstedl, and G. Guiochon, Chirality, 12 (2000) 558.
180. G. Gotmar, T. Fornstedl, and G. Guiochon, Anal. Cheat., 72 (2000) 3908.
181. L. Asnin, K. Kaczmarski, A. Felinger, F. Gritti, and G. Guiochon, J. Chromatogr. A, 1101 (2006)
158.
182. G. Gotmar, L. Asnin, and G. Guiochon, J. Chromatogr. A, 1059 (2004) 43.
ГЛАВА 15
ПРЕПАРАТИВНЫЕ РАЗДЕЛЕНИЯ
Совместно с Джеффом Коксом
15.1. Введение 762
15.1.1. Перегрузка колонки и последствия этого 762
15.1.2. Масштаб разделения 763
15.1.2.1. Колонки большего диаметра 764
15.1.2.2. Оптимизированные условия для преп-ЖХ 764
15.1.2.3. Другие соображения 766
15.2. Оборудование для преп-ЖХ разделения 766
15.2.1. Колонки 767
15.2.2. Введение образца 768
15.2.2.1. Инжекторы с петлей ввода 768
15.2.2.2. Насосный ввод 769
15.2.3. Детекторы 770
15.2.3.1. УФ-детекторы 770
15.2.3.2. Другие детекторы 770
15.2.4. Сбор фракций 771
15.2.5. Выделение продукта (удаление подвижной фазы) 772
15.3. Изократическое элюирование 774
15.3.1. Количество образца и разделение 774
15.3.1.1. Изотермы сорбции 774
15.3.1.2. Ширина пика при разделениях малых количеств образцов
в сравнении с шириной пика при разделениях больших
количеств образцов 776
15.3.2. Разделение по методу соприкасающихся пиков 776
15.3.2.1. Емкость насыщения колонки 778
15.3.2.2. Перегрузка по объему образца 779
15.3.2.3. Растворимость образца 780
15.3.2.4. Разработка метода 782
15.3.2.5. Сбор фракций 784
15.4. Значительно перегруженное разделение 785
15.4.1. Полнота извлечения или чистота 785
15.4.2. Разработка метода 785
15.4.2.1. Эффективность колонки 787
15.4.2.2. «Пересечение изотерм» 787
15.5. Градиентное элюирование 789
15.5.1. Сравнение изократической и градиентной преп-ЖХ 790
15.5.2. Разработка метода для градиентной преп-ЖХ 791
15.6. Промышленное разделение 794
Литература 793
I Глава 15. Препаративные разделения
15.1. Введение
Цель препаративной жидкостной хроматографии (преп-ЖХ) — выделение и сбор
одного или нескольких очищенных соединений из исходной смеси. Масштаб преп-ЖХ
может изменяться от нескольких микрограммов (в случае идентификации соединений)
до тонн и десятков тонн (в случае фармацевтического производства, как это описано
в разделе 13.9.2), но, как правило, это выделение от миллиграммов до граммов
вещества (этому отведено больше всего места в этой главе). Разделениям больших масштабов
будет уделено лишь небольшое внимание (раздел I5.6). Родственная сверхкритическая
флюидная хроматография |l] в настоящей главе рассматриваться не будет.
15.1.1. Перегрузка колонки и последствия этого
Преп-ЖХ предполагает перегрузку по массе, т.е. количество образца достаточно большое,
чтобы влиять на ширину пика и времена удерживания. При разработке аналитического
разделения внимание обращается на то, чтобы масса (или объем) образца не превышали
определенных пределов (раздел 2.6) и на то, чтобы времена удерживания и разрешение
не изменялись в зависимости от количества вводимой пробы. Поскольку масса
вводимого образца растет, то детектор, в конечном итоге, будет перегружен, времена
удерживания уменьшатся, пики уширятся и исказятся (сравните рис. I5.lu.ff).
(а)
(б)
Рис. 15.1. Гипотетические разделения, иллюстрирующие (а) аналитическое разделение
и (6) соответствующее разделение с перегрузкой по массе и
перекрывающимися пиками
/5.1. Введение 763
Часто перегрузка детектора или его нелинейность есть следствие перегрузки
колонки. Например, УФ-поглощение пика превышает одну или две единицы AU
(раздел 4.2.5). Нелинейность детектора зачастую можно обойти, используя проточную
ячейку с малой длиной оптического пути (препаративную ячейку) или изменяя
длину волны детектирования, чтобы целевое вещество (далее называемое продуктом)
поглощало менее интенсивно. Достаточно большое количество образца может, кроме
всего прочего, привести к изменениям в разделении, которые мы будем называть
перегрузкой колонки (как на рис. 15.16).
15.1.2. Масштаб разделения
Количество образца, которое необходимо разделить с помощью преп-ЖХ зависит
от того, как вы в дальнейшем предполагаете использовать очищенное вещество.
В таблице 15.1 для небольших молекул (молекулярная масса < 1000 Да) показаны
количества и цели, характерные для разработок в фармацевтической промышленности.
Обратите внимание на то, что перегрузка колонки определяется наибольшим
количеством целевого вещества (обычно это пик продукта), но не массой всего образца.
Для очистки загрязненного продукта, где исходная чистота часто > 80%, есть,
однако, небольшая разница между массой образца и массой продукта. Когда мы в этой
главе говорим о массе образца, то имеем в виду массу очищаемого продукта. Когда
необходимо выделить несколько мг (или меньше) очищенных веществ, масса
образца может расти до тех пор, пока расстояние между пиком продукта и ближайшим
соседним пиком не сократится до нуля (т.е. не станет соответствовать разрешению до
базовой линии). Результат этого сокращения характеризуется как разделение
соприкасающихся пиков (С-П, Т-Р — Touching Peak) или соприкасающихся зон. При таком
разделении вводят максимально возможное количество образца, при том, что
извлекается 100% продукта со 100% чистотой. Иллюстрация С-П-разделения показана
на рис. 15.16. На этом рисунке видно, что масса образца увеличивалась (по
сравнению с разделением на рис. 15.1с) до тех пор, пока пик основного вещества Б не
уширился так, что коснулся пика А. В этом примере масса вещества, которому
соответствует пик А, недостаточна для того, чтобы повлиять на время удерживания или
на ширину пика. Веществом, которому соответствует пик В, колонка несколько
перегружена, но недостаточно для того, чтобы на хроматограмме передний фронт
уширенного пика В коснулся пика Б. Следует отметить, что задние фронты обоих
перегруженных пиков элюируются почти там же, где элюировались максимумы пиков
веществ во время аналитического разделения (малое количество образца)
на рис. 15.1а (пунктирные линии на рис. 15.1). Фракцию, содержащую очищенное
целевое вещество (пик Б в примере 15.16), можно собирать после того, как она
покидает колонку, либо вручную, либо в автоматическом режиме (раздел. 15.2.4).
Продукт, свободный от растворителя/элюента может быть получен упариванием подвиж-
Таблица 15.1. Требования к очищаемым образцам
Цель
Предварительная идентификация инструментальными методами
Достоверная идентификация и подтверждение структуры
Использование аналитического стандарта (например, для
калибровки ВЭЖХ-акализа)
Токсикологический контроль
Первая фаза клинических испытаний
Требуемая масса продукта
~ 1 мг
1—100 мг
100 мг— 2 г
10-100 г
200 г — 2 кг
764 Глава 15. Препаративные разделения
ной фазы (раздел 15.2.5). Для выделения примерно до 10 мг очищенного целевого
вещества может потребоваться более одного разделения (ввода образца). Когда
необходимо намного более 10 мг очищенного продукта, то большое количество
разделений станет обременительным, особенно если их выполнять в ручном режиме. Для
образцов с большей массой существует два различных варианта разделения: (1)
использование колонки с большим диаметром (раздел 15.1.2.1) или (2) оптимизация
условий разделения для того, чтобы получить максимальное разрешение пика целевого
вещества и остальных пиков (раздел 15.1.2.2).
15.1.2.1. Колонки большего диаметра
Обычно при использовании колонки большего диаметра («полупрепаративной»), ее
длину оставляют прежней. Упаковывать эту колонку следует той же неподвижной
фазой. Скорость потока и объем образца увеличивают пропорционально площади
поперечного сечения колонки или d} (dc — внутренний диаметр колонки). Обратите
внимание на то, что оборудование должно быть в состоянии обеспечить такой
расход. Например, при замене обычной аналитической колонки (с внутренним
диаметром 4,6 мм) на колонку с внутренним диаметром 10 мм и скорость потока, и объем
образца должны быть увеличены в 102/4,62 = 4,73 раза. При этом условии результаты
разделения будут одинаковы для колонок с малым и большим диаметром
(одинаковые времена удерживания, ширины пиков, значения разрешения и давления в ко
лонках). Такая замена аналитической колонки на колонку большего диаметра будет
называться масштабированием. Обратите внимание на то, что длина колонки тоже
может быть изменена, и в этом случае количество образца должно быть
скорректировано пропорционально объему колонки. Более длинная колонка имеет большее
количество теоретических тарелок Л'дня аналитических разделений, но это
обстоятельство менее важно в условиях преп-ЖХ, поскольку /V лишь незначительно влияет
на препаративное разделение (раздел 15.3.1.2).
15.1.2.2. Оптимизированные условия для преп-ЖХ
Второй путь — это изменение селективности, обеспечивающее лучшее разделение
и очистку целевого вещества. Большинство аналитических разделений проводятся
таким образом, чтобы все нужные пики разделялись до базовой линии, как в
оптимизированном аналитическом разделении малого количества образца на рис. 15.2а.
В преп-ЖХ, где, как правило, только одно вещество должно быть выделено в
чистом виде, важно лишь разрешение пика продукта и пиков ближайших примесей.
Таким образом, разрешение для пика целевого вещества должно быть настолько
большим насколько это возможно. Это показано на рис. 15.26, где селективность
была оптимизирована именно для выделения вещества, которому соответствует
на хроматограмме пик 8. Для этого использовался тот же общий подход
(раздел 2.5.2), что и для разработки аналитического разделения, показанного
на рис. 15.2а (т.е., изменение условий разделения, улучшающих селективность).
Хотя пики некоторых примесей и перекрываются теперь на рис. 15.26 (пики 2-3 и
5-6), условия С-П-разделения для пика 8 позволяют вводить гораздо большее
количество образца (см. рис. 15.2в). Если такую же массу образца, как на рис. 15.2*,
ввести при условиях разделения, указанных на рис. 15.2а (см. рис. 15г), пик
целевого вещества не будет отделяться от пика примеси 7. Таким образом, если
используются условия, показанные на рис. 15.26, а не на рис. 15.2а, может быть
разделено гораздо большее количество образца (при выделении 100% чистого целевого
вещества, которому соответствует пик 8).
15. J. Введение 765
Оптимизированные
условия для
аналитического
разделения
в
(продукт)
(а)
6
Время (мин)
10
Оптимизированные
условия для
препаративного
разделения
1.
,2 4
ЗА Д-
5
+
6Л 7
в
Время (мин)
Условия для
перегруженного
препаративного
разделения
(б)
10
4 6
Время (мин)
10
Условия для
перегруженного
аналитического
разделения
1
(г)
1 ' 1 ■ Г"
4 6 8
Время (мин)
—i—
10
Рис. 1S.2. Сравнение аналитических и препаративных условий для нахождения
оптимальных условий разделения. Образец: смесь замешенных анилинов и
бензойных кислот. Условия: Cig-колонка, 150 х 4,6мм (5 мкм); элюенты ацето-
нитрил — буфер; скорость потока 2,0 мл/мин; остальные условия
приведены на рисунке, (л) Условия, оптимизированные для разделения и анализа
всех соединений в образце (небольшое количество образца); (б) условия,
оптимизированные для преп-ЖХ очистки вещества, которому соответствует
пик 8 на хроматограмме (небольшое количество образца); («) С-П-разде-
ление образца с помощью преп-ЖХ в условиях (б) (большое количество
образца); (г) ввод большого количества образца как в разделении (в), но
в условиях аналитического разделения (а). Перепечатано из [2] с
разрешения Wiley-lnterscience
766 Глава 15. Препаративные разделения
15.1.2.3. Другие соображения
Для больших количеств образца, встречающихся в гтреп-ЖХ, могут стать важными
другие вопросы, не связанные с размерами колонки или ее селективностью. Одним
из следствий хроматографического разделения является то, что компоненты
образца выходят из колонки сильно разбавленными и зачастую удаление растворителя
из собранных фракций элюента представляет собой серьезную проблему. Удалить
растворитель обычно легче в случае НФХ, чем при работе в режиме ОФХ потому,
что органические растворители упариваются легче, чем вода. В случае нескольких
миллиграммов продукта, растворенного в нескольких десятках миллилитров
водной подвижной фазы после ОФХ-разделения, растворитель можно легко удалить
с помощью роторного испарителя. Однако, удаление больших количеств водных
растворителей требует гораздо больших усилий и затрат. По этой причине многие
(но не все) препаративные разделения, как правило, выполняются скорее в
условиях НФХ, чем ОФХ.
Для образцов с массой больше 10 мг возможностей аналитического
оборудования для ВЭЖХ зачастую не хватает. Кроме того, аналитические ячейки детекторов
слишком чувствительны для преп-ЖХ. Для разделения этих больших количеств
может потребоваться специализированное оборудование, которое имеет в своем
составе высокопроизводительные насосы, автоматизированное устройство для ввода
образца, коллектор фракций и детектор, снабженный препаративной проточной
ячейкой.
15.2. Оборудование для преп-ЖХ разделения
Как уже отмечалось, многие микропрепаративные разделения могут быть проведены
на аналитических хроматографических системах (глава 3), для которых, возможно,
потребуются небольшие модификации для того, чтобы работать с возросшими
объемами образцов или понизить чувствительность детектора. При увеличении массы
образца более чем до нескольких мг удобнее увеличить размер колонки
(масштабировать разделение), чем использовать затратные по времени и трудоемкие
многократные вводы образца в аналитическую колонку. Это может потребовать
соответствующего изменения в оборудовании для системы, выделенной для преп-ЖХ,
поскольку необходимо обеспечивать более высокие скорости потока и обработку
образцов с большими массами. В таблице I5.2 сведены приблизительные
рекомендации по размерам колонок, типу оборудования и скоростям потоков для различных
масштабов работы. Для разделений в граммовых количествах, как правило, требуется
специальная преп-ЖХ система из-за того, что необходимы соответствующие
скорости потока. По существу, полу препаративное ВЭЖХ-оборудование аналогично
аналитическому, но при этом оснащено более производительным насосом и
устройством для сбора фракций. Малые и лабораторные системы для преп-ЖХ, как правило,
используются для выделения от десятков граммов до килограммов продукта. В таких
системах зачастую используют колонки с внутренним диаметром до II см. Колонки
большего диаметра, которые подходят для получения нескольких килограммов
целевого вещества, лучше всего использовать в составе препаративных лабораторных или
опытно-промышленных установок, взрывобезопасных и приспособленных для
работы с большими объемами растворителей. Такие системы с колонками большого
диаметра в данной главе не рассматриваются.
15.2. Оборудование для преп-ЖХ разделения 767
Таблица 15.2. Приблизительные размеры колонок и оборудование, используемое
для преп-ЖХ в лабораторных масштабах1
Необходимое
количество
продукта
< 1 мг
1 — 100 мг
100 мг - 5 г
5-100 г
200 г — 2 кг
Внутренний
диаметр
колонки (мм)
4,6
10
20—30
30—50
50—110
Оборудование
Аналитическое (~ 1 мл/мин)
Аналитическое (~ 5 мл/мин)
Полупрепаративное (20—50 мл/мин
Препаративное небольшого масштаба
(50—150 мл/мин)
Препаративное лабораторного масштаба
(100-600 мл/мин)
Коэффициент
масштабирования для скорости потока
и количества образца
1
4,7
19—42
42-120
120-570
15.2.1. Колонки
Лучше всего разработать преп-ЖХ на аналитической колонке, используя тот
сорбент, который будет в препаративной колонке большого диаметра. Использование
колонок малого диаметра в процессе разработки метода минимизирует ненужный
расход образца и подвижной фазы, а, кроме того, делает ненужной работу на более
дорогой хроматографической системе для преп-ЖХ. Колонки, используемые для
полупрепаративных и лабораторных преп-ЖХ разделений, очень похожи на колонки
для аналитических разделений (глава 5). Зачастую, при массе образца < I0 мг, можно
использовать аналитическую колонку, поскольку, в зависимости от разделяемой
смеси веществ, в такие колонки можно вводить до нескольких миллиграмм образца.
Колонка для преп-ЖХ должна быть упакована тем же самым сорбентом, которым была
упакована аналитическая колонка перед масштабированием. Это гарантирует
отсутствие изменений в относительном удерживании (селективности) между двумя
колонками, что очень важно для преп-ЖХ (раздел I5.3.2). Обратите внимание на то,
чтобы размер частиц сорбента был одинаковым как для аналитических, так и для
препаративных колонок2, чтобы полученная для аналитической колонки
эффективность и селективность были такими же и для большей колонки.
При переходе к колонкам большего диаметра (таблица 5.2) и, соответственно,
увеличенному расходу и большему объему образца, следует учитывать и другие
соображения. Убедитесь, что промежуток времени между выходом вещества из ячейки детектора
и входом в коллектор фракций достаточно мал для колонок и малого, и большого
диаметров. Фракции обычно собирают, ориентируясь на сигнал детектора. В том случае,
если объем капилляра между ячейкой детектора и входом в коллектор достаточно велик,
может произойти временной разрыв между детектированием пика и его сбором в
коллекторе фракций. Последнее, в свою очередь, может привести к ошибкам в определении
' Следует понимать, что предлагаемая здесь классификация масштабов довольно условна.
Для одних классов веществ {например, пептидов) уже 2 кг является полупромышленным и даже
промышленным масштабом производства, а для других (например, антибиотиков) 2 кг не более,
чем лабораторным. Это же касается и коэффициента масштабирования. Кроме того, и
внутренние диаметры аналитических колонок теперь далеко не ограничиваются диаметром 4,6 мм. —
Прим. перев.
2 Этот совет представляется, как минимум, спорным. Для работы с препаративными
колонками могут потребоваться куда большие скорости потока. При сохранении неизменным размера
частиц сорбента (например, 5 мкм в случае аналитической колонки с внутренним диаметром 4,6 мм)
давление в системе может так возрасти, что работать будет невозможно или очень трудно. Именно
поэтому размер частиц в препаративных колонках, как правило, больше, чем в аналитических
(чаще всего 10 мкм). — Прим. перев.
768 Глава 15. Препаративные разделения
момента начала и окончания работы коллектора фракций и, следовательно, либо к
потере целевого вещества, либо к загрязнению его близкой (по времени удерживания)
примесью. Помните о диаметре выходного капилляра из ячейки детектора, который может
быть значительно больше, чем у входяшего в ячейку капилляра. Проще всего рассчитать
внутренний объем капилляра и, сопоставив его с расходом, определить задержку по
времени. При более высоких расходах, используемых в преп-ЖХ, временная задержка
будет при прочих равных условиях пропорционально уменьшаться.
Если для преп-ЖХ используется то же самое аналитическое оборудование, то
более высокие скорости потока, используемые при работе с колонками большего
диаметра, будут приводить к большему падению давления в капилляре, соединяющем
ячейку детектора и коллектор фракций. Во многих ВЭЖХ-системах предусмотрен
капилляр с малым внутренним диаметром (0,005—0,010") для того, чтобы уменьшить
вне колоноч иное размывание пика (раздел 3.4). Если исходная аналитическая
ВЭЖХ-система будет работать при скоростях потока от 5 до 10 мл/мин с колонкой
диаметром 10 мм (вместо обычных 1—2 мл/мин, которые соответствуют колонке
с внутренним диаметром 4,6 мм), то капилляр малого внутреннего диаметра
приведет к большему перепаду давления и даже к возможному отключению системы1. Это
особенно актуально при работе с более вязкими, как в случае ОФХ, растворителями.
Особое внимание следует уделить капилляру, выходящему из детектора, так как
малый внутренний диаметр вкупе с высокими скоростями потока могут привести к
высокому противодавлению и к повреждению проточной ячейки детектора. Возможно,
потребуется замена исходного капилляра на капилляр с большим внутренним
диаметром (до 0,020" для колонок с внутренним диаметром 20 мм). В то же время,
не забывайте о том, что изменение диаметра этого капилляра повлечет за собой
изменение времени задержки между моментом детектирования пика и сбором фракции
(как это обсуждалось ранее). Производители ВЭЖХ систем тоже могут снабдить
пользователя рекомендациями по адаптации оборудования к преп-ЖХ разделениям.
15.2.2. Введение образца
При малых массах образца и использовании аналитической ВЭЖХ-системы,
образец, как правило, вводится одним из двух способов: (I) ввод через петлю ввода
инжектора, который является частью хроматографической системы или (2) ввод с
помощью отдельного насоса (насосный ввод). В специализированных преп-ЖХ
системах образец обычно вводится отдельным насосом, который отличается от
насосов для подвижной фазы2.
15.2.2.1. Инжекторы с петлей ввода
При использовании стандартных инжекторов, являющихся частью аналитической
ВЭЖХ-системы, максимальный объем петли инжектора может оказаться
недостаточным для преп-ЖХ. Штатная петля инжектора может быть заменена на петлю
большего объема. Такие большие петли поставляет целый ряд производителей хромато-
графического оборудования. Кроме того, петлю легко можно сделать самому
из отрезка нержавеющего капилляра. Зачастую для систем, оснащенных автосампле-
' В связи с выключением насоса/ов из-за превышения установленного верхнего предела
давления. Следует помнить, что при работе со скоростями потока от 5 до 10 мл/мин, очень быстро
изнашиваются уплотнения насосов. Кроме того, при высокой частоте возвратно-поступательных
движений плунжера температура в голове насоса может существенно повышаться и для случаев
НФХ-разделений или для термолабильных образцов это может быть серьезным препятствием. —
Прим. перев.
2Как правило, у него меньший расход. — Прим. перев.
15.2. Оборудование для преп-ЖХ разделения 769
рами, возможность вводить большие объемы образцов бывает предусмотрена
производителем. Важно, чтобы проба во время ввода сохраняла хотя бы приблизительно
постоянный объем и двигалась по соединительным капиллярам в виде цилиндра.
Любое разбавление образца подвижной фазой будет увеличивать его объем, что
может привести к неудачному разделению (раздел 15.3.2.2).
Следует проявлять осторожность при выборе петли большего объема, поскольку
зона образца размывается в открытом капилляре пропорционально внутреннему
диаметру капилляра, возведенному в шестую степень. Таким образом, при увеличении
диаметра капилляра, из которого сделана петля, образец размывается, и зона
уширяется. Использование для петли ввода капилляра большей длины (а не большего
диаметра) тоже увеличит размывание и уширение цилиндрической зоны образца, но,
как правило, в меньшей степени. Задний фронт этого цилиндра будет более размыт,
чем передний, потому что «хвост» цилиндра (но не его начало) должен пройти всю
длину петли, чтобы ввод был выполнен надлежащим образом (раздел 3.6.1.2).
Степень размывания заднего фронта образца может быть определена посредством
впрыскивания неудерживаемого и поглощающего в УФ вещества (например, с к = 0) и
последующего детектирования пика, покидающего колонку. Если входящая в
колонку цилиндрическая зона образца несильно размыта, то и детектируемый потом пик
будет симметричным.
Одним из способов устранения этой проблемы во время ввода образца является
частичное опорожнение петли ввода. Заполненная петля соединяется с колонкой
на время, достаточное для того, чтобы необходимое количество образца вошло в
колонку, но без того, чтобы задняя часть цилиндрической зоны входила тоже
(поскольку она будет разбавлена подвижной фазой). Для достижения этого результата
инжектор переключается из положения, при котором вводится образец, в положение, при
котором петля заполняется, до того момента, как петля полностью опорожнится. Это
гарантирует, что разбавленная часть вводимого образца не ухудшит разделение.
Однако, часть образца, оставшаяся в петле, может быть потеряна.
15.2.2.2. Насосный ввод
Этот способ более удобен и применим в случае больших вводимых объемов. Такая
техника требует систему с двумя насосами (например, градиентную) для изократи-
ческих преп-ЖХ разделений. Образец вводят в колонку с помощью одного насоса
с последующим элюированием вторым насосом, который перекачивает подвижную
фазу. Наилучшие результаты получены с помощью градиентной системы со
смешиванием на стороне высокого давления (раздел 3.5.2.1), поскольку мертвый объем
после градиентного миксера минимален. Один из двух насосов подает раствор образца,
а второй перекачивает (заранее смешанную) подвижную фазу. Насос,
осуществляющий ввод образца, сначала заполняется его раствором, после чего ввод
осуществляется простым переключением потока от насоса, подающего подвижную фазу, к
насосу, подающему образец, на время (зависящее от расхода), которое необходимо
для ввода требуемого объема пробы.
При насосном вводе в системах с градиентом на линии низкого давления
(раздел 3.5.2.2) один из входов линий для подачи элюентов в миксер используется для
закачивания образца. Образец вводится с помощью запрограммированного
ступенчатого градиента. Программа переключает линию подвижной фазы на линию образца
и обратно. Поскольку системы с градиентом на линии низкого давления имеют
больший мертвый объем, целесообразно измерять уширение пика в процессе ввода,
как это описано выше (раздел 15.2.2.1). Значительные потери образца возникают при
использовании систем с градиентом на линии низкого давления вследствие
заполнения образцом насоса и линий. Эти потери трудно предотвратить.
770 Глава 15. Препаративные разделения
Обычно в преп-ЖХ системах имеется отдельный от насоса(ов), подающего
подвижную фазу, насос для ввода образца. В тех случаях, когда объем образца
ограничен, а объем хроматографической системы велик, лучше всего использовать ручной
ввод или закачать образец через небольшой капилляр непосредственно в насос,
предназначенный для ввода. Ввод образца зачастую производится в режиме
остановленного потока. Насос, подающий подвижную фазу, останавливают, и включают
насос, подающий необходимый образец непосредственно в колонку. Такой прямой
ввод в колонку насосом исключает размывание зоны образца в отличие от систем,
где используется инжектор с большой петлей, в которую вводится образец.
15.2.3. Детекторы
Общее описание ВЭЖХ-детекторов дано в главе 4. В настоящем разделе внимание
будет акцентировано на детекторах, наиболее широко используемых в преп-ЖХ.
15.2.3.1. УФ-детекторы
В большинстве случаев один и тот же УФ-детектор может использоваться как
для аналитических так и для препаративных целей. Однако целесообразно снабдить
детектор проточной ячейкой с короткой длиной светового пути (= I мм), что
позволит работать на оптимальной длине волны без перегрузки детектора. Некоторые
ячейки продаются модифицированными — с переменной длиной светового пути, что
позволяет использовать их в различных разделениях. Несмотря на использование
проточных ячеек, предназначенных для преп-ЖХ, поглощение образца может все же
превышать 1 или 2 AU, и пики на хроматограмме могут выходить за пределы шкалы,
а кривая поглощения не возвращаться к базовой линии. Вследствие этого
мониторинг разделения станет затруднительным или вовсе невозможным. Чрезмерная
чувствительность при детектировании может быть понижена выбором подходящей
(неоптимальной, обычно сдвинутой в видимую область) длины волны. При этом
необходимо помнить, что на новой длине волны примеси могут поглощать гораздо
сильнее, чем целевое вещество, и это может привести к сложностям при сборе
фракций пика продукта. На рис. 15.3 показан пример, где в разделении с небольшим
количеством образца (рис. 15.3а, длина волны 280 нм) не видно никаких примесей
со значительным УФ-поглощением (видны только два основных пика). Однако когда
количество образца возрастает (рис. 15.36), сигнал детектора выходит за пределы
максимально возможной чувствительности детектора при длине волны 280 нм.
При попытке ограничить пик продукта пределами шкалы (детектирование при
большей длине волны — 375 нм), пики примесей (относительно целевого пика)
увеличиваются так, что пики основных компонентов уже невозможно четко
идентифицировать. Для такого образца может потребоваться другой детектор.
15.2.3.2. Другие детекторы
Рефрактометрическому (RI) детектору часто не хватает чувствительности для
мониторинга аналитических разделений, но он может быть весьма полезен в
препаративных разделениях именно благодаря своей низкой чувствительности. Поскольку
показатель преломления подвижной фазы может превышать показатель преломления
некоторых компонентов образца, возможны и отрицательные пики. Эту сложность
легко преодолеть, если фракции собираются вручную или если программное
обеспечение, используемое для сбора фракций, работает правильно в присутствии
отрицательных пиков. Для похожих компонентов (составляющих большинство образцов)
рефрактометрический детектор обеспечивает сходную чувствительность обнаружения
15.2. Оборудование для преп-ЖХ разделения
mAU
2000-
1500
1000-
500
О
детектирование
при 280 нм
(а)
10
Рис. 15.3. Трудности мониторинга преп-ЖХ разделения при изменении длины волны
для понижения чувствительности детектирования, (я) Аналитическая хро-
матограмма, 280 нм; (б) препаративная хроматограмма, 280 и 375 нм
и более ясное представление об относительной концентрации, чем УФ-детектор, что
в значительной степени устраняет проблему, проиллюстрированную на рис. 15.36.
В принципе, любой детектор может быть использован для мониторинга
препаративных разделений (глава 4). Размеры проточной ячейки детектора могут
препятствовать ее использованию в преп-ЖХ из-за высоких скоростей потока, обычно
используемых в преп-ЖХ. В таких случаях может использоваться делитель потока
(сплиттер) для обхода ячейки. На выход из колонки ставится тройник с малым
мертвым объемом и соединяется с детектором коротким капилляром с небольшим
внутренним диаметром. Поток через ячейку регулируется длиной и диаметром
капилляра, соединяющего другой конец тройника с коллектором фракций. Необходимо
следить за балансом скоростей потоков, чтобы быть уверенным — выход из
детектора синхронизирован с пиками, входящими в коллектор фракций. В идеале объемы
двух капилляров за сплиттером должны относиться друг к другу так же, как и
объемные скорости потока в них. Это должно гарантировать одновременное поступление
фракции/пика в ячейку детектора и коллектор фракций. Кроме того, делители
потоков должны использоваться в случае детекторов, разрушающих образец (детекторов
светорассеяния или МС-детекторов). МС-детекторы все чаще используют для
препаративных разделений небольших масштабов, главным образом, сложных смесей,
в которых вещества собирают на основе данных об их молекулярных массах.
15.2.4. Сбор фракций
Собирать фракции можно вручную, руководствуясь сигналом детектора для
определения времени начала и конца сбора той или иной фракции. Однако при повторных
разделениях оператор начинает уставать, и появляется опасность сбора не тех
фракций или слива целевого вещества в отходы. В случае нерегулярных,
мелкомасштабных преп-ЖХ разделений рекомендуется использовать коллектор фракций. Для не-
772 Глава 15. Препаративные разделения
которых препаративных систем коллектор фракций может стать неотъемлемой
частью. В дополнение к коллектору фракций можно приобрести устройство, которое
полезно запрограммировать на сбор фракций по сигналу детектора. Комбинируя
время сбора фракции, величину порогового сигнала и наклон базовой линии, можно
правильно собирать фракции целевого вещества. Такие коллекторы фракций могут
работать в автоматическом режиме непрерывно или же до тех пор, пока не будет
разделено необходимое количество образца.
Специализированные полупрепаративные и препаративные системы имеют
встроенные коллекторы фракций. Коллекторы фракций для микропрепаративной
ВЭЖХ могут использовать 96-ти луночные планшеты. Более крупные коллекторы
имеют фиксированный набор клапанов1, смонтированных на несущей конструкции
и позволяющих собирать отдельные фракции.
Хроматографические системы, использующие коллекторы фракций
фиксированного объема, такие как 96-ти луночные планшеты или ряд пробирок, обычно
собирают фракции по времени, чтобы быть уверенным в том, что объемы фракций
не превышены. Обязанность оператора после сбора фракций объединить те из них,
которые содержат целевое вещество. В том случае, если сбор происходит с помощью
переключающих кранов, фракции могут быть собраны в любой контейнер
подходящего размера. Необходимый объем контейнера рассчитывается, исходя из ширины
пика, скорости потока и количества разделений, которые должны быть проведены.
Важно выбрать коллектор фракций, отвечающий вероятным требованиям вашей
преп-ЖХ системы2. Если, как это часто бывает, собирается всего одно целевое вещество,
необходимо лишь несколько выходов для сбора фракций. При разделениях более
сложных образцов на большое количество фракций потребуется больше выходов для сбора
фракций. В последнем случае лучше всего взять коллектор фракций с большим
количеством пробирок (похожий на автосамплер), но при этом надо не забывать о том, что
резервуары, в которые собираются фракции, могут переполниться. К счастью, очень сложные
образцы, скорее всего, будут встречаться в микропрепаративных разделениях,
проводимых в исследовательских лабораториях, в то время как крупномасштабные разделения, в
которых, как правило, собирают одно или два целевых вещества, не требуют более пяти
или десяти выходов для сбора фракций. Поскольку часто обнаруживается, что небольшие
примеси могут элюироваться либо с передним фронтом, либо с задним фронтом пика, то
в преп-ЖХ общепринято, по крайней мере, при первых опытных разделениях собирать
узкие фракции переднего и заднего фронта пика, чтобы убедиться в том, что достигнута
необходимая чистота продукта. Таким образом, может потребоваться не менее трех
выходов коллектора фракций для каждого выделяемого вещества. Всегда лучше собрать
больше фракций выходящего пика и потом чистые фракции объединить, чем собрать одну
большую фракцию вещества с меньшей чистотой.
15.2.5. Выделение продукта (удаление подвижной фазы)
Способ выделения целевого вещества может повлиять на план разработки метода
преп-ЖХ. К примеру, на выбор между ОФХ и НФХ. Когда в результате ОФХ-разде-
ления получается несколько граммов вещества, растворенных в водной подвижной
фазе (возможно, содержащей нелетучий буфер), удаление подвижной фазы из про-
' Роль коллектора фракций может выполнять автоматический многоходовой кран. Например,
шести- или двенадцатиходовой. — Прим. перев.
2Как правило, в современных преп-ЖХ системах коллекторы фракций уже предусмотрены.
Поскольку управление системой (в том числе и коллектором фракций) осуществляется
специализированным программным обеспечением, то и коллектор фракций должен быть куплен у того же
производителя, а не выбран отдельно. — Прим. перев.
15.2. Оборудование для преп-ЖХ разделения 773
дукта уже не кажется второстепенной задачей. Основная трудность заключается
в удалении воды, поскольку она имеет большую температуру кипения и более
высокую теплоту испарения, чем обычные органические растворители. Это приводит
к длительным временам упаривания, что не только ограничивает количество
вещества, которое может быть выделено за данный промежуток времени, но и подвергает
опасности термического разложения продукт (как раз из-за необходимости более
длительного упаривания). По этим причинам многие хроматографисты обычно
выбирают неводную НФХ (раздел 8.4) для преп-ЖХ. В случае НФХ, проведенной
с растворителями, кипящими ниже 80 °С (см. таблицу 1.3 Приложения I) выделение
очищенного продукта относительно проще. Часто предпочитают НФХ из-за более
высоких значений фактора разделения а, которые соотносятся с более высокими
допустимыми массами образца (как в образце на рис. 15.2; см. также раздел 15.3.2). Это
особенно верно для близкородственных веществ, таких, как изомеры (раздел 8.3.5).
ОФХ не исключается из рассмотрения в преп ЖХ, поскольку существует неско
лько способов удаления подвижной фазы из продукта, которые зависят от его
(продукта) природы. Кроме того, любые необходимые буферы и добавки могут быть
выбраны из списка летучих соединений (раздел 7.2.1), чтобы в дальнейшем не было
необходимости удаления солей из продукта. Многие соединения имеют
относительно низкую растворимость в воде, особенно при тех значениях рН, когда они
не ионизированы. Например, сильнокислый рН для кислоты или щелочной рН
для основания (см. таблицы 7.2, 13.1 и 13.8 для веществ, величины рКа которых
зависят от молекулярной структуры). Соответствующая корректировка рН фракции
продукта с последующим упариванием раствора и удалением большей части
органического растворителя часто приводит к тому, что целевое вещество выпадает в
осадок и его можно отфильтровать. Альтернативным способом является непрерывная
экстракция целевой (частично водной) фракции органическим растворителем, не
смешивающимся с водой. Продукт таким образом переводят в органический
растворитель, который потом легко удаляют упариванием. При использовании обоих этих
способов любой (нелетучий) неорганический буфер остается в водной фазе.
В других случаях разбавление фракции водой с последующим пропусканием
раствора через картридж с сорбентом для твердофазной экстракции (ТФЭ)
(раздел 16.6) может удержать на нем продукт. Затем картридж можно промыть водой,
чтобы удалить нелетучий буфер или соль, и после того смыть целевое вещество
смешивающимся с водой растворителем, который легко упарить. Небольшие
количества воды, оставшиеся в растворе фракции после ТФЭ, могут быть удалены азе-
отропной перегонкой на роторном испарителе после того, как к раствору будет
добавлен подходящий растворитель, образующий азеотроп с водой. Хлороформ
особенно удобен для этой цели, поскольку любое количество оставшейся воды
будет визуально заметно в виде несмешивающихся с органическим растворителем
капель. Так как в отгоняемой смеси хлороформ/вода лишь 3% воды, возможно, что
процесс перегонки придется повторить один или два раза для того, чтобы удалить
воду полностью. Можно использовать для этой цели и другие растворители
(например, этанол, дихлорметан). При поиске подходящего растворителя на роль азе-
отропного реагента может оказаться полезной ссылка на таблицы физических
свойств смесей растворителей |3|.
Для менее устойчивых и очень хорошо растворимых в воде веществ может быть
предпочтительнее удаление воды лиофилизацией. Перед этим может потребоваться
предварительное удаление нелетучих буферов или добавок как, например, перед
ионообменной хроматографией (раздел 16.6.2.3). Поскольку лиофилизация проходит
при температуре < 0 °С, ее обычно используют для белков, чтобы предотвратить их
денатурацию или разложение в процессе сушки.
Глава 15. Препаративные разделения
15.3. Изократическое элюирование
С 1980 года достигнут значительный прогресс в нашем понимании и использовании
преп-ЖХ. В частности, группа Гиошона разработала для препаративной
хроматографии обширный математический аппарат, особенно для случаев крупномасштабных
разделений, в которых пики соединений не соприкасаются |4|. Для разделений
лабораторных масштабов (таблица I5.2) общие принципы преп-ЖХ более полезны, чем
применение специальных математических методов, которые потребуют
компьютерных расчетов для их реализации. В настоящей главе внимание будет уделено общим
принципам.
15.3.1. Количество образца и разделение
В настоящем разделе дается фундаментальное объяснение того, как перегрузка колонки
влияет на разделение. Впрочем, это имеет ограниченное непосредственное применение
на практике. По этой причине читатель может предпочесть перейти к разделу 15.3.2 и
вернуться к настоящему разделу в случае необходимости.
15.3.1.1. Изотермы сорбции
Изотерма сорбции описывает распределение растворенного вещества между
подвижной и неподвижной фазами при данной температуре в зависимости от концентрации
растворенного вещества в подвижной фазе. Большинство ВЭЖХ-разделений
описываются изотермой Ленгмюра, которая, в свою очередь, описывает распределение
молекул растворенного вещества между подвижной и неподвижной фазами:
г, аС,
+ Ь*Ст
(I5.1)
Здесь Cs — концентрация растворенного вещества в неподвижной фазе, Ст —
концентрация растворенного вещества в подвижной фазе, а а и Ь*— константы для данного
растворенного вещества, подвижной и неподвижной фаз и температуры. Эта модель
распределения образца в колонке предполагает, что удерживание растворенного
вещества происходит на плоской поверхности с известной максимальной емкостью —
полностью покрытой адсорбированным монослоем (разделы 8.2.1, 15.3.2.1; рис. 15.6я).
Для малых концентраций растворенного вещества Ст, Cs = aCm, и из
уравнения (15.1),
V IV
's I r m
= ЮГ, (15.2)
где Vs и Vm— соответственно объемы подвижной и неподвижной фаз внутри
колонки, К= (Cs/Cm) — коэффициент распределения растворенного вещества (также
известного как постоянная Генри), и Ч* = VJVm— фазовое отношение. Из уравнений 15.1
и 15.2 мы видим, что А" при малых значениях Ст равно а. Для больших значений Ст,
Cs = ajb* и соответствует заполненному монослою растворенного вещества.
Величина (a/b*)Vs эквивалентна максимальному насыщению колонки растворенным
веществом, которая определяется как емкость насыщения колонки ^(раздел 15.3.2.1).
Частичное заполнение (6) монослоя растворенным веществом эквивалентно Cs,
отнесенной к максимальной величине С, (равной а/Ь*) или
h*C
В= - т . (15.3)
\ + Ь*Ст
15.3. Изократическое элюирование
На рис. 15.4а показан график зависимости 9 от Ст для изотермы Ленгмюра
с а/Ь* = 25 и К = 13, а также соответствующие величины к. Значения к
уменьшаются по мере увеличения Cs. На рис. 15.46 представлен перестроенный
график рис. 15.4а с логарифмической шкалой для Ст. Последний график показывает,
что к становится константой (в данном случае это &д), когда Ст достаточно мала
(так называемая область линейных изотерм; для Ст > 0,0004 в этом примере А0 >
> к > 0,99*0).
15 20
С (мг/л)
(б)
■ о-асо—о-
область
линейности
изотермы
1.0
0.8
0.6
0.4
0.2
0.0
0.0001 0.001 0.01
Рис. 15.4. Изотерма сорбции для К = 25 и V = 0,2. (л) График, показывающий
влияние концентрации растворенного в подвижной фазе образца на
коэффициент емкости к и заполнение поверхностного монослоя 9; (б) тот же график
в полулогарифмическом масштабе, иллюстрирующий область линейности
изотермы
776 Глава 15. Препаративные разделения
15.3.1.2. Ширина пика при разделениях малых количеств
образцов в сравнении с шириной пика
при разделениях больших количеств образцов
Ширина W пика растворенного вещества может быть выражена как функция W
для малого количества образца (IV0) и дополнительного («термодинамического»)
уширения пика (IV,,,), связанного с увеличением массы образца |5, 6|:
W2=W* + W$,, (15.4)
где
И'о = f4rlro<1 + *o>2 <15-4а>
и
К=ЧЧ\—\ (15-46)
Nq — число теоретических тарелок для образца с малой массой, 1ц — мертвое время
колонки, &0 — фактор удерживания (коэффициент емкости) для образца с малой
массой, и^ — масса введенного образца и ws — емкость насыщения колонки
(раздел 15.3.2.1).
Согласно уравнению (15.4) влияние числа теоретических тарелок JVg на ширину
пика ослабевает с увеличением количества образца (W,^ становится больше Wq). Это
имеет важные следствия в случае преп-ЖХ. Например, при возрастании
селективности 6 в колонку могут быть введены большие количества образца для разделения
методом соприкасающихся пиков (СП-разделения). Требуемое число теоретических
тарелок становится меньше, а ширина пика будет меньше зависеть от факторов,
влияющих на N (длины колонки, размера частиц, скорости потока). Поскольку
большие значения Nq требуют более продолжительных времен разделения, можно
предположить, что в преп-ЖХ часто предпочтительнее более высокие скорости
потока и/или более короткие колонки. Это приводит к уменьшению Ng, но способствует
максимальной производительности очистки при минимальных потерях целевого
вещества и сохранении его высокой чистоты.
15.3.2. Разделение по методу соприкасающихся пиков
Принцип СП-разделения был сформулирован в разделе I5.I.1. Выбирают такое
количество вводимого образца, чтобы уширенный пик целевого вещества только
соприкасался с одним из двух окружающих его пиков (как на рис. \5.]5). Тогда это
позволит разделять максимальное количество образца и при этом выделять почти сто
процентов целевого вещества при почти стопроцентной его чистоте1. К
СП-разделению можно прийти методом проб и ошибок, руководствуясь нижеследующими
соображениями.
Если варьировать количество вводимого вещества, обозначенного на рис. 15.1
как пик Б, и потом наложить полученные хроматограммы друг на друга, то
получится серия вложенных друг в друга прямоугольных треугольников — похожих на те, что
'Это больше похоже на мечты хроматографиста, занимающегося препаративными
разделениями или даже на заклинания, чем на реальную ситуацию, но запретить мечтать нельзя никому.
Даже тем, кто занимается препаративными разделениями и хочет извлечь из исходной смеси
почти сто процентов целевого вещества с почти стопроцентной чистотой. — Прим. перее.
15.3. Изократическое элюирование 777
J I I 1 I L
6 7 8 9 10 11 (мин)
* №<пик4> ►
Рис. 15.5. Влияние массы образца на ширину пика для перегруженного разделения
(хроматограммы наложены друг на друга)
показаны на рис. 15.5. Здесь три перегруженных1 пика (1,2,4) целевого вещества
соответствуют трем относительным массам образцов 1,2 и 4. Время выхода хвостов
каждого из перегруженных пиков близко к времени удерживания пика,
соответствующего введению малой массы образца. Для различных «малых» количеств образца
не происходит изменений в ширине пика (линейная область изотермы; см. рис.
15.46). Ширины W перегруженных пиков увеличиваются примерно
пропорционально квадратному корню из массы образца (уравнение 15.46). В нижней части рис. 15.5
показана величина И^для наибольшей массы вводимого образца (пик 4), измеренная
от начала выхода пика на шестой минуте до выхода пика «малого» образца на
десятой минуте. Один из способов определения массы вводимого образца для
СП-разделения следующий: после первого ввода небольшого количества образца его масса
может быть произвольно увеличена (для второго разделения). Так, например, это
увеличение приведет к пику 1 на рис. 15.5. В этом случае W должна вырасти в два
раза, и фронт целевого пика переместится по направлению к соседнему пику
примеси (заштрихованному). Поскольку ширина пика увеличивается пропорционально
квадратному корню из массы образца, масса последнего должна возрасти
четырехкратно (40-5 = 2).
Масса вводимого образца для СП-разделения зависит от (1) величины
коэффициента разделения oq для малой массы вводимого образца, (2) природы образца,
(3) емкости насыщения колонки (раздел 15.3.2.1). Если молекулы целевого вещества
не ионизированы и предполагается, что колонка размером 150 х 4,6 мм упакована
сорбентом с порами 10 нм, то масса вводимого образца в случае СП-разделения
будет около трех миллиграммов для а$ = 1,5. Для колонок, отличающихся по длине
или по внутреннему диаметру, масса образца для СП-разделения пропорциональна
внутреннему объему колонки. Если ад равно 1,1 или 3,0, то соответствующие
количества образцов будут около 0,2 и 10 мг соответственно. Степень ионизации образ-
1 Строго говоря, пик перегрузить нельзя. Можно перегрузить только колонку образцом, в
котором содержится целевое вещество и вследствие этой перегрузки пик уширится, но для простоты
изложения авторы называют перегруженными сами пики. — Прим. перев.
778 Глава 15. Препаративные разделения
цов вместе с параметрами колонки определяет емкость насыщения колонки ws
(см. раздел 15.3.2.1). Тогда для СП-разделения масса вводимого образца wx может
быть приблизительно определена по формуле
*fj -1т II 1 w,. (15.5)
«)-
15.3.2.1. Емкость насыщения колонки
Определение емкости насыщения колонки ^(которую мы будем для простоты
называть емкостью колонки) является основным для любого обсуждения преп-ЖХ.
Величина ws соответствует максимально возможному поглощению молекул растворенного
вещества колонкой,' т.е, количеству растворенного вещества, заполняющему весь
монослой для адсорбции разделяемых молекул полностью (это соответствует
большой концентрации растворенного вещества в подвижной фазе); см. гипотетический
пример на рис. 15.6я для адсорбции бензола на типичной области поверхности непо
движной фазы. Емкость насыщения специфична для каждой конкретной колонки и
может несколько изменяться в зависимости от растворенного образца и
экспериментальных условий. Емкость колонки будет пропорциональна доступной площади по-
(а)
о>
<~> с>
° °QO
подвижная
фаза
OOOOOOQ
<о><о<о <о <о <о>,
неподвижная
фаза
(б)
з — з <£2>-МНз+
<£>NH3' <О^Н3* <1>™э
подвижная
фаза
C>NH3+ ,'<^>NH3+ !<£>МНз+ J
неподвижная
фаза
Рнс. 15.6. Иллюстрация зависимости емкости колонки от ионизации растворенного
вещества. Адсорбция бензола (я) и протонированного анилина (6)
' Поглощает, конечно, не колонка, а сорбент, в нее упакованный. — Прим. персе.
15.3. Изократичеекое элюирование 779
верхности сорбента, и для неионизированного растворенного вещества ws можно
аппроксимировать следующим образом
ws (мг) = 0,4 (площадь поверхности в м2). (15.6)
Такая зависимость наводит на мысль об использовании сорбентов с
максимально развитой площадью поверхности, однако необходимо помнить о том, что
сорбенты с более развитой поверхностью имеют поры меньшего диаметра. Если молекулы
растворенного вещества больше диаметра пор, они не могут войти в них, а
поскольку почти вся поверхность находится внутри пор, то емкость колонки будет снижена.
В то же время, для сорбентов с большими порами и площадь поверхности и емкость
колонки меньше. Таким образом, сорбенты с промежуточными размерами пор
обеспечивают наибольшие емкости колонок (например, диаметр пор = 8 нм для
растворенных веществ с молекулярной массой < 500 Да); см. соответствующее обсуждение
рис. 13.7 относительно зависимости удерживания больших молекул от диаметра пор
сорбента.
Кроме размера молекул важны и другие характеристики растворенного вещества.
Перпендикулярная ориентация небольших молекул растворенного образца к
поверхности сорбента в процессе адсорбции может привести к увеличению емкости
колонки по сравнению с молекулами, ориентирующимися горизонтально (сравните
рис. 8.36 с рис. 8.3а). Большие молекулы, такие, как белки, могут принимать
трехмерную структуру при удерживании, что приводит к значительно большей толщине
адсорбированного монослоя. В этом случае емкость насыщения может быть намного
больше предсказанной уравнением (15.6). Все это происходит до тех пор, пока
диаметр пор достаточно велик, чтобы молекулы белка могли сквозь них проходить.
Заряженные (ионизированные) молекулы растворенного вещества такого же типа будут
в процессе адсорбции отталкиваться друг от друга, и это электростатическое
отталкивание может снизить емкость колонки на два порядка. Это проиллюстрировано
на рис. 15.6, где адсорбция нейтрального растворенного вещества (бензола)
на рис. 15.6а совершенно не похожа на адсорбцию ионизированного вещества
(протонированного анилина) на рис. 15.66. Из-за гораздо меньшей емкости колонок
в случае разделения ионизированных молекул, преп-ЖХ предпочтительно проводят
в условиях, минимизирующих ионизацию образца. Однако из-за гораздо большего
удерживания неионизированной молекулы по сравнению с ее ионизированным
аналогом (например, непротонированного основания по сравнению с протонирован-
ным) для частично ионизированных растворенных веществ емкость колонки будет
гораздо больше, чем для полностью ионизированных, хотя и не такой большой, как
для нейтральных молекул.
15.3.2.2. Перегрузка по объему образца
Объем вводимого образца для СП-разделения зависит от того, какую массу продукта
требуется получить, и от концентрации целевого вещества в исходном растворе
образца (которая может быть ограничена растворимостью образца). Предварительно
было подсчитано |5|, что объем образца Vg будет мало влиять на преп-ЖХ
разделение до тех пор, пока он не превысит половины объема собираемого пика Vp, равного
ширине пика W, умноженной на скорость потока F. На рис. 15.7 показаны
смоделированные хроматограммы для объемов 0,1 мл (сплошная линия), I мл (пунктирная
линия) и 1,5 мл (штрихпунктирная линия) при сохранении постоянной массы
образца с помощью изменения его концентрации (36 г/л, 3,6 г/л и 2,4 г/л соответственно).
Объем образца в 1,5 мл привел к значительному перекрыванию пиков. Выделить
удалось лишь 85% чистого вещества А. Очевидно, что предпочтительнее
использовать более концентрированные образцы. СП-разделения (для случая с концентра-
Глава 15. Препаративные разделения
12 (мин)
Рис. 15.7. Хроматограммы, иллюстрирующие влияние перегрузки по объему на
разделение двух соседних пиков. Объемы образца: , 0,1 мл; 1,0 мл; ,
1,5 мл. Моделирование сделано для колонки 250 х 4,6 мм, скорость потока
1 мл/мин. Два компонента в соотношении 1:1, *(А) = 1,0, селективность
а= 1,5, N = 1000. Оба пика умеренно перегружены по массе
цией 2,4 г/л) можно достичь лишь уменьшая вводимый объем до величин менее I мл
и, следовательно, сокращая массу чистого вещества А, которое может быть выделено
в результате каждого разделения.
Чтобы избежать искажений пика и ухудшения разделения, а также других
проблем, лучше всего использовать одну и ту же смесь растворителей как для подвижной
фазы, так и для растворения образца. Однако для того, чтобы улучшить
растворимость образца, могут потребоваться и другие растворители (раздел 15.3.2.3). В
условиях, когда сила растворителя, в котором растворяется образец, и сила подвижной
фазы близки, но растворитель и подвижная фаза не одинаковы по составу, пик
может немного уширяться и искажаться.
15.3.2.3. Растворимость образца
Для СП-разделения требуется определенная масса вводимого образца. Последний
предпочтительнее вводить растворенным в подвижной фазе, составляющей по
объему менее половины объема пика WF (как обсуждалось выше). Растворители более
слабые, чем подвижная фаза, тоже подходят. Растворенные в них образцы могут
быть введены в больших объемах. Однако, растворимость образца часто уменьшается
при снижении %В, и потому масса образца, растворенного в большом объеме
слабого растворителя, может быть небольшой. Способы решения проблемы ограниченной
растворимости образцов включают в себя:
• замену подвижной фазы;
• замену растворителя образца;
• повышение температуры колонки.
Замена подвижной фазы. Выбирая метод преп-ЖХ (ОФХ или НФХ), нужно
принять во внимание, в каких элюентах лучше растворяется образец — в водных (ОФХ)
или в органических (НФХ). В каждом случае выбор В-растворителя может в
дальнейшем повлиять на растворимость образца. С другой стороны, выбор метода и
15.3. Изократичеекое элюирование 781
В-растворителя также влияет на селективность а и поэтому может статься, что
при выборе В-растворителя необходимо будет найти компромисс между
растворимостью образца в подвижной фазе и селективностью разделения.
Замена растворителя образца. Необязательно, чтобы растворитель образца был
таким же, как подвижная фаза, но, в общем случае, он не должен быть гораздо
сильнее. Так же как и в аналитических разделениях (разделы 2.6.1, 17.4.5.3),
использование более сильных растворителей образца в преп-ЖХ может приводить к уши-
рению и искажению пиков в тех случаях, когда растворитель образца сильнее
подвижной фазы. Обычно наилучшим выбором в качестве растворителя образца
будет тот растворитель, который по силе примерно равен подвижной фазе. Замена
В-растворителя для растворения образца может улучшить растворимость
последнего, особенно в случае НФХ, где такую замену можно провести без изменения силы
растворителя. Таким образом, одна и та же сила подвижной фазы е может быть
достигнута с помощью изменения %В разных В-растворителей (см. пример
на рис. 8.6) с учетом их полярности (или значения Eg). Поскольку растворимость
образца обычно выше в подвижной фазе с большим %В, это предполагает
использование в качестве растворителя образца такого, в котором %В будет
максимальным при условии сохранения той же силы подвижной фазы е. Например, если
подвижная фаза содержит 4% этилацетата в гексане, то следуя данным номограммы
на рис. 8.6, рациональнее использовать 50% хлористый метилен в гексане в
качестве растворителя образца, поскольку он, скорее всего, обеспечит более высокую
растворимость образца без ухудшения разделения.
Другая возможность при разделении кислотных и оснбвных соединений с
помощью ОФХ — изменение рН растворителя образца с целью увеличения его (образца)
растворимости. В этом случае лучше изменить рН подвижной фазы таким образом,
чтобы подавить ионизацию образца и увеличить емкость колонки (как это показано
на рис. 15.6), но не надо при этом забывать, что растворимости ионизированных
соединений часто выше, чем неионизированных. Это предполагает использование
такого рН растворителя образца, который обеспечит повышение ионизации и рас
творимости. Поскольку увеличение ионизации образца понижает время его удержи
вания, растворитель образца может быть слегка забуферирован таким образом, чтобы
при смешивании с подвижной фазой результирующий рН не сильно отличался от рН
подвижной фазы. Однако, эта процедура предполагает относительно небольшой
объем образца и более сильную забуференность подвижной фазы.
Независимо от изменении в растворителе образца следует учитывать возможность
выпадения образца в осадок (с засорением инжектора, капилляра или колонки) при
смешивании образца с подвижной фазой. В некоторых случаях выпадение в осадок может
происходить не мгновенно, позволяя образцу проникнуть в колонку до того, как оно
произойдет. По экспериментам, в которых аликвоты раствора образца смешиваются
с подвижной фазой и наблюдаются в течение нескольких минут, можно судить о
вероятности выпадения в осадок веществ, составляющих образец, во время разделения
(см. соответствующее обсуждение в разделе 7.2.1.2 по оценке растворимости буфера).
С другой стороны, вероятность осаждения образца при использовании для
растворения растворителя, отличного от присутствующего в подвижной фазе, может быть
уменьшена с помощью метода «разбавления в колонке» |7, 8|. Этот способ
предполагает введение образца с помощью отдельного насоса таким образом, что образец и
подвижная фаза смешиваются непосредственно перед входом в колонку. Таким
образом повышается вероятность того, что образец адсорбируется стационарной фазой
до того момента, как он выпадет в осадок.
Повышение температуры колонки. Повышение температуры обычно увеличивает
растворимость образца, но, кроме того, раствор образца необходимо нагревать в про-
782 Глава 15. Препаративные разделения
цессе его подачи от емкости, где он хранится до входа в колонку (при перекачивании
насосом образец может охладиться). Если какая-либо часть хроматографической
системы не нагрета до нужной температуры, образец может выпасть в осадок, что
приведет к засорению инжектора, капилляров или колонки. Кроме того, увеличение
температуры колонки понижает удерживание веществ образца и может приводить
к снижению значений а. При этом, чтобы скомпенсировать эти изменения, придется
уменьшить массу вводимого образца.
15.3.2.4. Разработка метода
Процесс разработки преп-ЖХ разделения кратко подытожен на рис. 15.8. Для этапов
с 1 по 3 следует использовать аналитическую колонку (например, 150 х 4,6 мм). Если
необходима колонка с бблыиим внутренним диаметром для очистки большего
количества образца за каждое разделение, она устанавливается на этапе 4.
1. Первичное
разделение после
подбора %В
jlJLJUJIa^
d
(прсщует)
2. Оптимизация а
для пика продукта
3. Увеличение
массы образца
Л—К—К.
JuJ\ Л лл
4. Масштабирование
Больше образец, больше колонка,
выше скорость потока
Рис. 15.8. Разработка метода преп-ЖХ
Выбор начальных условий. До первого этапа (начального разделения) необходимо
выбрать ОФХ или НФХ, а также определиться с начальными условиями разделения
(тип колонки, подвижная фаза и температура). В то время как ОФХ обычно
выбирают в случае аналитических разделений, для преп-ЖХ предпочитают НФХ. Особенно
тогда, когда образец нерастворим в воде и необходимо получить более 50 мг целевого
15.3. Изократичеекое элюирование 783
вещества1. Мы планируем обсудить НФХ ниже, а для выбора начальных условий
ОФХ см. главы 6 и 7.
Если требуется получить более 10 мг очищенного целевого вещества, убедитесь
заранее, что этот же сорбент доступен и в колонках больших геометрических
размеров для масштабирования (шаг 4). Затем выберите сильный (В) и слабый (А) элюен-
ты для подвижной фазы (см. главу 8 для НФХ; например, этилацетат и гексан) и
меняйте В% таким образом, чтобы для продукта и соединений, алюирующихся
позднее, I </с < 10 (если это возможно). Поскольку элюирующиеся перед продуктом
соединения необязательно разделять друг с другом, значения k для них могут быть
< 1. Аналогично, если некоторые примеси сильно удерживаются, их можно удалить
быстрее, промывая колонку более сильной подвижной фазой после элюирования
пика целевого вещества. После этого колонку необходимо уравновесить исходной
подвижной фазой перед вводом следующей порции образца (градиентное
элюирование является альтернативой для подобного рода образцов).
Шаг 1 на рис. 15.S. Затем, используя подход, аналогичный тому, что применялся
для разработки аналитического разделения, проводится первоначальное
препаративное разделение. Скажем, с использованием сильной подвижной фазы (например,
80 %В) для того, чтобы элюировать весь нужный образец из колонки в течение
приемлемого времени. Величина %В потом будет скорректирована методом проб и
ошибок для достижения 1 < k < 10 для пика целевого вещества, при этом
предпочтительны более низкие значения к для ускорения разделения и получения возможности
очистить большее количество продукта в час (см. разделы 2.5.1, 8.4). С другой
стороны, для этой цели может быть использована тонкослойная хроматография
(раздел 8.2.3) или первоначальное разделение может быть проведено с использованием
градиентного элюирования, которое, в свою очередь, позволит оценить желательный
%В для изократического разделения (раздел 9.3.1). Если у вас есть какие-либо
сомнения в принадлежности пика целевому веществу в этих первоначальных условиях,
их можно разрешить отдельной аналитической хроматограммой чистого продукта.
Шаг 2 на рис. 15.S. После подбора оптимального %В на первом шаге условия
разделения изменяются для достижения наилучшего возможного отделения пика продукта
от пиков близко выходящих примесей. Лучше всего, если пик продукта будет
находиться на одинаковом расстоянии от примесей, элюируемых до и после него. Предыдущие
главы содержат подробное обсуждение того, как селективность а может быть
оптимизирована в зависимости от природы образца и от того, какой вид хроматографии
используется — ОФХ или НФХ (см. в таблице 2.2 условия, влияющие на а)? Поскольку в
препаративных разделениях чрезвычайно важно обеспечить максимальную селективность
(уравнение 15.5), по-видимому, оправдано более интенсивное исследование на этапе 2,
чем при разработке аналитического метода. В отличие от аналитического разделения
в преп-ЖХ важно, если это возможно, избегать таких условий разделения, которые
приводят к превышающей 50% ионизации молекул целевого вещества (см. раздел 15.3.2.1
и обсуждение рис. 15.6). Большие изменения а (без ионизации продукта) могут быть
достигнуты, скорее всего, заменой В-растворителя или колонки.
Те же самые условия, используемые для этого оптимизированного разделения,
могут быть применены и для анализа фракций, собираемых в процессе преп-ЖХ
1 На самом деле псе обстоит куда сложнее, и выбор начальных условий зависит от многих
факторов, а не только от растворимости образца и количества целевого вещества, которое необходимо
получить. Например, если у вас есть хороший роторный испаритель, способный упаривать
большие количества поды при небольшой температуре, и нет условий для работы с большими
количествами пусть и легколетучих, но и легковоспламеняющихся, а часто и ядовитых растворителей,
то вы не колеблясь выберете ОФХ. — Прим. перев.
^Стратегия оптимизации а в НФХ рассмотрена в разделе 8.4.2. — Прим. перев.
784 Глава 15. Препаративные разделения
(но на аналитической колонке). Если разрешение пика продукта Л, 3> 2 (что
желательно для преп-ЖХ), анализ фракций можно делать и на более короткой колонке
с большей скоростью потока, чтобы его (анализ) ускорить.
Шаг 3 на рис. 15.8. Первоначальную оценку массы вводимого образца можно
провести с помощью (I) уравнения (15.5), (2) величины а, (3) емкости колонки ws
и (4) грубой оценки % содержания продукта в образце. При условии, что продукт,
растворенный в подвижной фазе, не ионизирован, емкость колонки 150 х 4,6 мм
с размерами пор сорбента 10 нм можно оценить как ws =s 150 мг (= 100 мг/г
сорбента). Отсюда получается, что масса образца, разделяемого в СП-разделении, равна
ws = (1/6) х (150) х (|% содержания продукта|/100) х (|а — 1|/а)2. Если продукт
частично или полностью ионизирован, допустимая масса образца может быть намного
ниже этой оценки. После разделения рассчитанного таким образом количества
образца, его массу можно методом проб и ошибок увеличить или уменьшить, чтобы
достичь СП-разделения. С другой стороны, использование полностью
автоматизированного оборудования позволяет провести ряд пробных разделений с разными
количествами образца. Результаты таких экспериментов позволят быстро
определить оптимальную массу образца. Как только таким способом будет достигнуто
нужное качество разделения, пик продукта должен быть собран и
проанализирован, чтобы подтвердить = 100% извлечение и чистоту1. На данном этапе имеет
смысл проверить, можно ли сохранить качество разделения при увеличении
расхода, тем самым сократив время разделения. Как правило, целью преп-ЖХ является
получение максимального количества очищенного продукта за минимальный
промежуток времени, что заставляет сокращать время разделения.
Шаг 4 на рис. 15.8. Последний шаг на рис. 15.8 (масштабирование) завершает
процесс разработки метода. С учетом доступности (1) колонок нужного внутреннего
диаметра и (2) оборудования, которое может обеспечить необходимый расход элюен-
та, нужный коэффициент масштабирования может быть рассчитан из результатов
шага 3 (см. раздел 15.1.2.1). Затем можно провести окончательное разделение на
колонке большого диаметра. Это позволит подтвердить результаты (% изатечения и
чистоты продукта), полученные на аналитической колонке. В принципе,
масштабирование должно приводить к такой же чистоте и степени извлечения продукта, какие
были получены в разделении малого масштаба.
15.3.2.5. Сбор фракций
Как правило, цель сбора фракций — вручную или с помощью автоматизированной
системы — состоит в том, чтобы получить максимальный выход достаточно чистого
продукта, прилагая как можно меньше усилий. Прежде всего необходимо пик
продукта разделить на несколько фракций, собрать их по мере выхода из детектора и
проанализировать на предмет содержания в них целевого вещества и его чистоты.
Результаты анализа пригодятся для определения времени, в течение которого нужно
собирать пик целевого вещества (точки начала и окончания сбора фракций) в
препаративном разделении(ях) с тем, чтобы достичь целей очистки (раздел 15.4.1). Перед
завершением разработки преп-ЖХ разделения можно провести контрольное разделе-
' Что касается стопроцентного извлечения и чистоты, то это, как уже было сказано выше,
мечта, которую в реальной жизни достичь трудно. Иногда к этому и не нужно даже стремиться,
поскольку стопроцентное извлечение и чистота могут потребовать таких затрат, которые сделают
ваш продукт непомерно дорогим. Все зависит от поставленной задачи. Стопроцентное извлечение
и чистота нужны далеко не всегда. Приходится искать золотую середину между затратами на
выделение (временными и материальными) и стоимостью продукта, при которой процесс выделения и
очистки еще будет рентабельным. — Прим. перев.
15.4. Значительно перегруженное разделение 785
ние для подтверждения того, что точки начала и конца сбора фракций выбраны
правильно. Для этого собирают несколько мелких фракций вокруг уже намеченных
точек начала и конца сбора. После того, как само разделение и точки начала и конца
сбора фракций определены, достаточно собирать лишь одну фракцию, в которой
содержится продукт. Однако, сбор дополнительных фракций может застраховать от
непредвиденных изменений в разделении.
15.4. Значительно перегруженное разделение
Подробное исследование сильной перегрузки колонок (например, с нанесением
больших количеств образцов, чем те, которые подходят для СП-разделений) выходит
за рамки настоящей книги, однако, полезно рассмотреть здесь некоторые аспекты
таких разделений. Такие сильно перегруженные разделения могут дать больший
выход очищенного продукта за единицу времени. При этом снизится расход подвижной
фазы, а, кроме того, можно будет обойтись колонками меньших размеров и
оборудованием меньшей производительности. Все это может дать большую пользу на
практике. Недостатком таких разделений является то, что для разработки метода
требуется больше усилий и в каждом разделении, как правило, требуется анализ нескольких
фракций с целевым веществом. Только так и можно получить достаточно чистый
продукт. Кроме того, может потребоваться повторная очистка (рехроматография)
недостаточно чистых фракций продукта. Интересующемуся читателю можно
порекомендовать несколько работ |4, 9, I9| для дальнейшего изучения.
15.4.1. Полнота извлечения или чистота
По мере увеличения количества наносимого образца до значительной перегрузки,
достоверность предсказания формы индивидуальных пиков уменьшается. Это
проиллюстрировано на рис. I5.9 для небольших количеств образцов (рис. 15.9о),
СП-разделений (рис. 15.96) и значительно перегруженных разделений (рис. 15.9е), где
относительные концентрации двух компонентов А и В меняются от 1:10 до 10:1.
На рисунке видно, что происходит с минорным пиком, который элюируется либо
до, либо после большего пика продукта. При значительной перегрузке (рис. 15.9е),
когда пик примеси предшествует пику продукта, он смещается, сжимается и, таким
образом, его высота увеличивается. Наряду с этим происходит некоторое перекрытие
пиков. Когда пик примеси выходит за пиком продукта, его «затягивает» внутрь пика
продукта (так называемый эффект «затягивания»). Степень влияния этих двух
эффектов предсказать трудно, и поэтому оптимальное количество образца должно
определяться экспериментально. Кроме того, эта оптимальная масса будет
изменяться в зависимости от относительных концентраций продукта и примесей.
15.4.2. Разработка метода
Выбор оптимального количества образца для значительно перегруженного преп-ЖХ
можно начать с оптимизированного СП-разделения (такого, как на рис. 15.26).
За этим разделением следуют другие, в которых последовательно увеличивается
масса образца. В каждом разделении (или для каждой массы образца) собирают и
анализируют ряд фракций непосредственно перед целевым пиком, во время выхода пика и
сразу за ним. Результаты анализов сводятся в электронную таблицу. Затем
объединяют фракции с требуемой степенью очистки, вычисляют полноту извлечения продук-
786 Глава 15. Препаративные разделения
1:10
10:1
(а)
0.005 г А
0.05 г В
2 3 4
(б)
0.5 г А
5гВ
(V
\ <
с-п
0.05 г А
0.005 г В
20 г А
2гВ
Рнс. 15.9. Зависимость результатов изократического разделения двухкомпонентного
образца ог его количества. Компьютерное моделирование на основе
изотермы Ленгмюра. Условия: колонка 250 х 50 мм (7 мкм); 210 мл/мин; N = 800;
it — 1 и 1,5, соответственно. Массы образцов указаны на рисунке |9|
та из образца (его выход) и строят зависимость полноты извлечения продукта из
образца от чистоты продукта как на рис. 15.10 для двух различных продуктов (бычий и
свиной инсулины). Аналогичные зависимости строятся для различных количеств
образцов, и анализ этих зависимостей позволяет выбрать наиболее подходящую массу.
Таким образом можно в первом приближении определить массу очищенного
продукта с некоторой заранее заданной чистотой (например, 98%). Для примеров,
приведенных на рис. 15.10, выход продуктов 98% чистоты составляет 92 и 42% для бычьего
и свиного инсулинов соответственно. Подобные графики для различных количеств
образцов помогут определить наилучшее приемлемое соотношение между массой
очищенного продукта и полнотой его извлечения из образца.
Разделения, результаты которых показаны на рис. 15.10, проводили в режиме
градиентного элюирования ОФХ, но такой же подход будет пригоден для НФХ или
изократического элюирования. Принцип оценки массы образца одинаков
независимо от того, используется изократическое или градиентное элюирование.
Оптимальные условия (помимо массы образца) для СП-разделения и для значительно
перегруженного разделения могут отличаться. В обоих случаях может потребоваться
довольно большое количество экспериментов, чтобы оптимизировать как количество
образца, так и условия разделения.
15.4. Значительно перегруженное разделение
50
40
30
20
10
• бычий инсулин
■ свиной инсулин
86
90
—i—
92
—\—
94
-1—
96
—i—
98
100
Чистота <%)
Рис. IS. 10. График зависимости полноты извлечения продукта от его чистоты
при данном количестве образца (5 мг) для смеси бычьего и свиного инсу-
линов в соотношении 1:1. Бычий инсулин (сплошная линия); свиной
инсулин (пунктирная линия). Условия: колонка Cg 250x4,6 мм (20 мкм);
10—29% ацетонитрил — 0,1% водная ТФУК. за 10 мин [13]
15.4.2.1. Эффективность колонки
С ростом коэффициента селективности (фактора разделения) ад для случая
СП-разделения становится возможным вводить большие количества образца. При этом
уменьшается зависимость разрешения от числа теоретических тарелок колонки /Vg
(уравнение 15.4а). Поэтому достаточно будет и меньших величин /Vq Влиять на выход и
чистоту продукта они будут меньше. К значительно перегруженным разделениям это
уже не относится. Эффект вытеснения малого пика, предшествующего большому,
может улучшить разделение (см. рис. 15.9е). Это лучше всего показано на примере
изократических разделений на рис. 15.1 Iff, где вытеснения нет и пик В полностью
перекрывает пик А. Там же, где это вытеснение есть, происходит частичное
разделение двух пиков (сравните похожую ситуацию на рис. 15.9в). Вытеснение очень
полезно в значительно перегруженных разделениях, но, в отличие от случая
СП-разделения, предпочтительнее, по-видимому, иметь более высокие значения iV0 [ll|.
Промышленные разделения обычно проводятся на умеренно эффективных
колоннах, упакованных сорбентами с размерами частиц от 10 до 15 мкм.
15.4.2.2. «Пересечение изотерм»
Такое необычное поведение могут демонстрировать растворенные вещества с
различными емкостями насыщения. Примером является отделение спиртов от
фенолов [13|. В данном случае спирты могут иметь значительно большие емкости
насыщения, чем фенолы. На рис. 15.12а показано ОФХ-разделение бензилового спирта и
фенола. Количество образца небольшое — 10 мкг. Бензиловый спирт элюируется
последним. На рис. 15.12в показано такое же разделение, но для больших количеств —
1 мг фенола и Змг безилового спирта. Два пика почти полностью разделяются.
Когда порядок элюирования фенола и спирта меняется на противоположный,
при том, что масса и того, и другого соединения остается такой же (как
на рис. 15.12д,е), получается совсем другой результат (см. разделение фенетилового
спирта и л-крезола на рис. 15.12б,г). В перегруженном разделении на рис. 15.12?
788 Глава 15. Препаративные разделения
Иэократическое элюирование
(а)
А,
А'
Я
В-
1
f\
\
2,5
2.5
5 6 (мин)
мг А
мгВ
6 (мин)
2,5 мг А
10 мгВ
2,5 мг А
25 мгВ
(г)
Градиентное элюирование
(<Э) (в)
10 11
2,5 мг А
2,5 мг В
13(мин> 10 11 12 13(мин) 10 11 12 13(мин)
2,5 мг А
ЮмгВ
2,5 мг А
25 мг В
Рис. 15.11. Аналогичные эффекты перегрузки колонки в соответствующих
разделениях: (а) изократическое и (б) градиентное элюирование. Разделение двух
ксантинов ((З-гидроксиэтилтеофиллин [А| и 7В-гидроксипропилтеофиллин
[В] с к (в изократическом режиме) равным к* (градиентному). Количества
образцов указаны на рисунке. Пики, обозначенные А' и В', относятся
к чистым образцам А и В, введенным отдельно; пики, обозначенные А и
В, относятся к разделениям смесей А и В |2]
пики практически полностью перекрываются в противоположность аналогичному
разделению на рис. 15. \2в.
Причина такого сильного различия в разделениях на рис. 15.12 довольно сложна,
но может быть проиллюстрирована «пересечением изотерм», как показано
на рис. I5.12d (фенол и бензиновый спирт) и на рис. 15.12^ (фенетиловый спирт и
л-крезол). При разделении фенола и бензилового спирта изотермы не пересекаются
(рис. 15.12d), т.к. фенол всегда удерживается сильнее, чем бензиловый спирт, и
поэтому эти два соединения хорошо разделяются. В случае разделения фенетилового спирта
и л-крезола (рис. 15.12е) большее удерживание п-крезола при нанесении небольшого
образца в совокупности с его меньшей емкостью насыщения приводит к пересечению
15.5. Градиентное элюирование
(а)
8 10 (мин)
(б)
12 (мин)
(в)
:нгснгон он
(г)
в 10 (мин)
8 10 12 {мин)
снгон
снгснгон
(в)
Рис. 15.12. Пример пересечения изотерм при уменьшении допустимого количества
образца в случае СП-разделения. Условия: колонка С|о 150 х 4,6 мм (5 мкм);
подвижная фаза метанол — вода; 1,0 мл/мин. (а) 3 мкг фенола и 7 мкг
бензилового спирта; (6) 3 мкг 2-фенилэтанола и 7 мкг и-крезола; (в) так
же, как и в {а), за исключением того, что масса образца 4 мг; (г) так же,
как и в (б) за исключением того, что масса образца 4 мг; (д,е)
гипотетические изотермы, соответствующие разделениям фенола/бензилопого спирта
и 2-фенилэтанола/л-крезола соответственно |14|
изотерм при достаточно большой нагрузке. При больших количествах нанесенных
образцов оба соединения удерживаются одинаково, и разделения практически не
происходит, как это видно из рис. 15.12г. Приведенное объяснение намеренно упрощено.
15.5. Градиентное элюирование
В то время как градиентное элюирование часто используется для аналитических
разделений и преп-ЖХ в малых масштабах, крупномасштабные разделения в
градиентном режиме могут быть более трудоемкими и более дорогостоящими. Исключением
из этого общего правила является разделение больших биомолекул, поскольку их
удерживание в изократическом разделении может сильно различаться при неболь-
790 Глава 15. Препаративные разделения
ших изменениях %В (раздел 13.4.1.4), что делает изократическое элюирование
неудобным или невозможным. Хроматографическая очистка в промышленном
масштабе с помощью градиентного элюирования обсуждается в разделе 13.9.2 на примере
биосинтетического человеческого инсулина. Разработка метода градиентного
разделения, как обсуждалось в главе 9, очень похожа на разработку метода для изократи-
ческого разделения. Таким образом, когда фактор удерживания к* для градиентного
элюирования такой же, как и к для изократического, и прочие условия тоже
одинаковы («соответствующее» разделение; раздел 9.1.3), отделение пика продукта
от примесей будет одинаковым как для градиентного, так и для изократического
элюирования. Точно так же и любое изменение условий, которое может улучшить
селективность в изократическом режиме, может использоваться таким же образом
для улучшения селективности при градиентном разделении. Следовательно,
практически все, что относится к изократической преп-ЖХ и изложено в разделе 15.3,
в равной степени относится и к градиентному элюированию. Это упростит наше
обсуждение градиентной преп-ЖХ в этом разделе. Для получения дополнительной
информации о градиентной преп-ЖХ см. [2|.
15.5.1. Сравнение изократической и градиентной преп-ЖХ
Рисунок 15.\\а-в уже был использован для сравнения влияния количества образца
на изократическое разделение при изменении только массы одного из соединений,
входящих в состав образца. Аналогичная серия разделений показана на рис. 15.11г-е
для градиентного разделения того же образца (соединений А и В) в тех же условиях
(за исключением того, что %В заменяет крутизна градиента). В каждом случае на
рисунке накладываются друг на друга хроматограммы, полученные при раздельном
нанесении каждого из соединений (А' и В'), и при разделении смеси (А + В); см.
соответствующее обсуждение рис. 2.24 в разделе 2.6.2. Хроматограммы изократических и
градиентных разделений с одинаковым количеством образца (например, рис. 15.1 \а
в сравнении с г, 6 в сравнении с д, в в сравнении с е) практически идентичны,
за исключением более скругленных, похожих на акулий плавник пиков для
значительно перегруженных градиентных разделений на рис. 15.\\г-е.
Это сходство изократических и градиентных разделений в сопоставимых
условиях уже обсуждалось в разделе 9.1.3. Для получения равноценных результатов в
«соответствующих» изократических и градиентных разделениях (как на рис. 15.11) фактор
удерживания для каждого пика в изократическом (к) и градиентном (к*) разделениях
должен быть приблизительно одинаковым, и все остальные условия разделения
(колонка, А- и В-растворители, скорость потока, температура) должны быть
одинаковыми. В градиентных разделениях, показанных на рис. 15.1 \г-е, условия были
подобраны таким образом, чтобы значения А:* для малых количеств образцов были равны
значениям А: в изократических разделениях на рис. 15.1 \а-в. Как уже обсуждалось
в разделе 9.2, величины А:* определяются условиями градиента:
Г-Ш£. (9.5)
VmAq>S
Здесь t(; — время градиента, F — скорость потока, Д<р — изменение <р = 0,01 х (%В)
за время градиента, S связано с изменением к для данного изменения <р или %В (оно
равно rf[log k\fd(p), и Vm— мертвый объем колонки (мл), который можно определить
из экспериментального значения t0 и скорости потока F (раздел 2.3.1; Vm = t0F).
Изменения в изократическом разделении при изменении %В могут быть
воспроизведены при градиентном элюировпнии с помощью изменения времени градиента tCi
(уравнение 9.5) таким образом, что новые значения к и к* совпадут.
/5. J. Градиентное элюирование 791
15.5.2. Разработка метода для градиентной преп-ЖХ
Общий план разработки изократического метода, приведенный на рис. [5.8, можно
использовать с небольшими изменениями и для разработки метода градиентного
элюирования. Выбор начальных условий будет практически таким же, за
исключением того, что начальное разделение будет выполняться с помощью градиента 0—
100 %В с последующим сужением диапазона градиента. В большинстве случаев
диапазон сужается с целью сокращения времени разделения (раздел 9.3.4). Оптимизация
начального градиентного разделения с целью улучшения селективности (шаг 2)
может быть проведена таким же образом, как и для изократического элюирования
(раздел 9.3.3), за исключением того, что ее целью является максимальное разрешение
для пика продукта, а не приемлемое разрешение всех пиков на хроматограмме.
Помимо выбора условий для получения максимальной селективности 6, программа
градиента может быть дополнительно оптимизирована так, чтобы минимизировать
время разделения, сохраняя при этом разрешение пика продукта и пиков соседних
примесей (см. разделы 9.3.4, 9.3.5). Увеличение нагрузки образца до максимальной
(шаг 3) и масштабирование (шаг 4) выполняются затем точно так же, как и для
изократического преп-ЖХ. Подробнее об этом см. |2|.
Иногда в градиентной преп-ЖХ наблюдаются осложнения, которых нет в изократи-
ческой преп-ЖХ. Когда у двух соседних пиков разные величины S, это может повлиять
на массу образца при СП-разделении. Общий принцип иллюстрирует рис. 15.13.
На рис. 15.13а приведено градиентное СП-разделение в случае одинаковых значений S
для двух соединений (случай «параллельности»), что зачастую является хорошим
приближением для большинства образцов. В верхней части рис. 15.13а дан график зависи-
(а)
SA - Sg
(Параллельные
прямые)
fe=30
А В
А В
<в)
SA>SB
(Сходящиеся
прямые)
Рис. IS. 13. Влияние неравных значений S на значительно перегруженное разделение
двух пиков с помощью градиентного элюирования [2|
794 Глава 15. Препаративные разделения
мости log к* от ер* для каждого пика, где ф* есть значение <р (= 0,01 х %В), когда пик1
находится в середине колонки (см. уравнение 9.5а). Величины ф* на рис. 15.13 определяют
время в процессе разделений, показанных в нижней части рис. 15.13а-е. Пунктирные
линии соединяют графики зависимости log А* - <р* для соединений А и В (малые
количества образца) с пиками на расположенной ниже хроматограмме. Значения к* и ср* для
каждого пика определяются условиями градиента. Предполагается, что для каждого
разделения на рис. 15.13 время градиента составляет 30 мин.
Теперь предположим, что было введено достаточно большое количество образца,
что позволило пику В занять все пространство между двумя пиками (в разделении
с малым количеством образца), т.е.получилось СП-разделение (см. широкие
заштрихованные пики на рис. 15.13а). Мы видим, что расстояние по вертикали между двумя
графиками зависимости log к* от ф* не изменяется (они параллельны) и равно log a
для каждого значения <р. Таким образом, в начале элюирования перегруженного
пика В (при меньшем значении (р*, соответствующем элюированию малого
количества образца А), а такой же (= ад), как и в конце элюирования — т.е. коэффициент
селективности не зависит от массы образца. (Обратите внимание, что уравнение
(15.5), которое связывает массу образца для СП-разделения с ад, предполагает
приблизительно равные величины S для двух соседних пиков.)
Рисунок 15.135 аналогичен рис. 15.13а (та же масса образца для СП-разделения,
что и на рис. 15.13я), за исключением того, что теперь графики зависимости ]ogk*
от ф* не параллельны, а расходятся при меньших значениях ф* (случай
«расходимости») — т.е. величина 5для соединения В больше, чем для соединения А. В случае более
значительной нагрузки колонки (при меньших значениях ф*) расхождение по вертика
ли между двумя графиками зависимости log Л* от ф* растет, что соответствует росту а
при увеличении массы образца. Большая величина а означает большую массу образца
при СП-разделении (уравнение 15.5) и поэтому того же самого количества введенного
образца (как на рис. 15.13а) будет уже недостаточно для того, чтобы пики
соприкасались. То есть, случай «расходимости» допускает большую массу вводимого образца
(при прочих одинаковых условиях) по сравнению со случаем равенства значений S в
уравнении (15.5), иллюстрируемым на рис. 15.13а.
На рис. 15.13* показана третья возможность: графики зависимости log Л* от ф*
сходящиеся при меньших значениях ф* (случай «сходимости»). Тогда S для
соединения В меньше, чем S для соединения А. Теперь а уменьшается с увеличением массы
образца, и введение такого же количества образца как на рис. 15.13а для
СП-разделения приводит к ускоренной перегрузке колонки с перекрытием двух пиков.
При подозрении на конвергентное поведение («сходимость») графиков (из-за
меньших, чем ожидалось, количеств образца для СП-разделения) необходимо задуматься
о дальнейших изменениях условий разделения с целью изменения порядка
элюирования двух пиков (продукта и ближайших к нему пиков примесей). Аналогичный
подход также может быть использован для минимизации проблемы пересечения
изотерм (раздел 15.4.2.2). Дальнейшее обсуждение последствий различия величин S
в градиентной преп-ЖХ см. в |2|.
15.6. Промышленное разделение
Промышленные разделения выходят далеко за рамки этой книги, но простые основы
теории и практики, изложенные здесь, актуальны по-прежнему. Разделения такого
рода, как правило, тщательно оптимизируются для того, чтобы обеспечить максималь-
' Никакого пика в середине колонки, конечно нет. Там вещество (его зона), а пик только
на хроматограмме. — Прим. персе.
Литература 795
но возможный выход продукта с требуемой чистотой за кратчайшее время. При таком
масштабе экономика процесса имеет первостепенное значение. Здесь целью является
такое сочетание чистоты продукта, степени его извлечения из образца и скорости
процесса, которое обеспечивает наименьшую стоимость в пересчете на килограмм
целевого вещества, включая стоимость удаления подвижной фазы из уже очищенного
продукта. Условия разделения обычно подбираются эмпирически, используя подход,
проиллюстрированный на рис. 15.10 для количеств образцов существенно больших,
чем те, которые нужны для СП-разделения. Например, см. раздел 13.9, в котором
описана промышленная очистка рекомбинантного человеческого инсулина.
Все более важным для разделений в промышленных масштабах становится
использование хроматографии с псевдодвижущимся слоем сорбента ПСС (SMB —
simulated moving bed). Это техника разделения двухкомпонентных смесей, основанная
на моделировании противоточной системы разделения с использованием нескольких
колонок и переключающих кранов. Отсылаем читателя к специализированным рабо
там по этой теме 114|. Хотя этот способ использовался в течение многих десятилетий
в нефтяной промышленности (Molex'-технология выделения л-ксилола) и пищевой
промышленности (Sorbex'-технология обогащения сиропов фруктозой), в
фармацевтической промышленности он стал использоваться лишь с 1990-ых годов.
Использование противоточного разделения в комбинации с непрерывным вводом образца и
постоянным отбором очищенного продукта позволяет более эффективно
использовать неподвижную фазу, чем в традиционном процессе, описанном в этой главе.
Таким образом, ПСС-технология уменьшает как размеры колонок, так и количество
используемого растворителя (и, следовательно, расходы). Для противоточного
разделения не требуется колонка с высокой эффективностью. Это позволяет использовать
очень короткие колонки-«блины». В частности, успешное разделение энантиомеров
проводят в ПСС условиях на колонках с внутренним диаметром 800 или 1000 мм и
высотой лишь 100 мм (упакованных, например, частицами 20 мкм).
Литература
1. М. S Villeneuve and L. A. Miller in Preparative Enantioseiective Chromatography, G. B. Cox, ed.,
Wiley- Blackwell, 2005.
2. L. R. Snyder and J. W. Dolan, High Performance Gradient Elution, Wiley, Hoboken, NJ, 2007, ch. 7.
3. Azeotropes: http://en.wikipedia.org/wiki/Azeotrope (data).
4. G. Guiochon, A. Felinger, D. G. Shirazi, and A. M. Katti, Fundamentals of Preparative
and Non-linear Chromatography, 2nd ed., Academic Press, Boston, 2006.
5. J. H. Knox and H. M. Pyper, J. Chromatogr., 363 (1986) 1.
6. L. R. Snyder, G. B. Cox, and P. E. Antle, Chromatographic, 24 (1987) 82.
7. U. D. Neue, C. B. Mazza, J. Y. Cavanaugh, Z. Lu, and Т. Е. Wheat, Chromatographia Suppl., 57
(2003) S-121.
8. U. D. Neue, T. E. Wheat, J. R. Mazzeo, С. В. Mazza, J. Y. Cavanaugh, F. Xia, and D. M. Diehl,
J. Chromatogr. A, 1030 (2004) 123.
9. L. R. Snyder, J. J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, 2nd ed., Wi-
ley-lnterscience, New York, 1997.
10. H. Schmidt-Traub, Preparative Chromatography of Fine Chemicals and Pharmaceutical Agents, Wi-
ley-VCH, New York, 2005.
11. G. Guiochon and S. Ghodbane, J. Phys. Chem., 92 (1988) 3682.
12. G. B. Cox and L. R. Snyder, J. Chromatogr., 590 (1992) 17.
13. G. B. Cox and L. R. Snyder, J. Chromatogr., 483 (1989) 95.
14. O. Dapremont in G. B. Cox, Preparative Enantioseiective Chromatography, Wiley-Blackwell, NewY-
ork, 2005.
'Торговые названия компании Honeywell UOP. — Прим. перев.
ГЛАВА 16
ПРОБОПОДГОТОВКА
в соавторстве с Рональдом Мэджерсом
16.1. Введение 798
16.2. Виды образцов 799
16.3. Предварительная обработка твердых и полутвердых образцов 802
16.3.1. Измельчение образцов 802
16.3.2. Высушивание образцов 803
16.3.3. Фильтрование 803
16.4. Подготовка жидких образцов 804
16.5. Экстракция в системе жидкость—жидкость 805
16.5.1. Теория 805
16.5.2. Практика 807
16.5.3. Трудности 809
16.5.3.1. Образование эмульсии 809
16.5.3.2. Адсорбция растворенного вещества 809
16.5.3.3. Связывание растворенного вещества 810
16.5.3.4. Взаимная растворимость фаз 810
16.5.4. Специальные подходы к ЖЖЭ 810
16.5.4.1. Микроэкстракция 810
16.5.4.2. Микроэкстракция методом одной капли (МОК) 810
16.5.4.3. ЖЖЭ на твердой фазе (ТФЖЖЭ) 811
16.5.4.4. Жидкостная экстракция на иммобилизованном
носителе (ИЖЭ) 811
16.6. Твердофазная экстракция (ТФЭ) 811
16.6.1. Сравнение ТФЭ и ВЭЖХ 812
16.6.2. Применение ТФЭ 813
16.6.2.1. Удаление примесей 814
16.6.2.2. Обогащение образца 814
16.6.2.3. Обессоливание 814
16.6.2.4. Другие применения 814
16.6.3. Приспособления для проведения ТФЭ 815
16.6.3.1. Картриджи 815
16.6.3.2. Диски 815
16.6.3.3. Другие приспособления для ТФЭ 816
16.6.4. Устройства для ТФЭ 818
16.6.5. Разработка метода для ТФЭ 819
16.6.5.1. Этапы ТФЭ 820
16.6.5.2. Сорбенты для ТФЭ 822
16.6.6. Пример разработки ТФЭ-метода: выделение альбутерола
из плазмы крови человека 825
Содержание 797
16.6.7. Специальные виды ТФЭ 826
16.6.7.1. Мультимодальная ТФЭ и ТФЭ со смешанными фазами . 826
16.6.7.2. Адсорбенты с ограниченно доступной
поверхностью (ОДП) 827
16.6.7.3. Молекулярно-импринтированные полимеры (МИП) . . . 828
16.6.7.4. Иммуноаффинная экстракция малых молекул 829
16.6.7.5. QuEChERS и дисперсионная ТФЭ 830
16.6.7.6. Специализированные ТФЭ-картриджи для различных
классов соединений 831
16.7. Мембранные технологии в пробоподготовке 831
16.8. Методы пробоп од готовки твердых образцов 832
16.8.1. Традиционные методы экстракции 833
16.8.2. Современные методы экстракции из твердых веществ 835
16.8.2.1. Экстракция в современном аппарате Сокслета 835
16.8.2.2. Сверхкритическая флюидная экстракция (СФЭ) 835
16.8.2.3. Жидкостная экстракция под давлением (ЖЭД)/
ускоренная экстракция растворителем (УСЭР) 836
16.8.2.4. Экстракция с микроволновым нагреванием (МЭ) 837
16.9. Переключение колонок 837
16.10. Пробоподготовка в биохроматографии 838
16.11. Пробоподготовка в ЖХ-МС 841
16.12. Дериватизация в ВЭЖХ 843
Литература
845
798 Глава 16. Профподготовка
16.1. Введение
Пробоподготовка является важной частью ВЭЖХ-анализа, дающая образцовый,
воспроизводимый, гомогенный раствор, пригодный для ввода в колонку. Цель
подготовки образца — снабдить хроматографиста аликвотой образца, которая (I)
относительно свободна от примесей, (2) не повредит колонку, (3) совместима
с предполагаемым методом ВЭЖХ-разделения и способами детектирования.
Растворитель образца должен смешиваться с подвижной фазой, не влияя на его (образца)
удерживание и не мешая детектированию. Кроме того, может возникнуть
необходимость сконцентрировать растворенные вещества и/или дериватизировать их для
улучшения детектирования или разделения.
Пробоподготовка начинается в момент сбора образца, продолжается до
введения образца в ВЭЖХ-колонку и включает в себя различные операции, сведенные
в таблице 16.1. Пункты от I до 4 таблицы I6.1, включающие сбор проб, их
транспортировку, хранение, предварительную обработку, лабораторный отбор проб и
последующее взвешивание/разбавление — все это является важными этапами пробоподго-
товки. Хотя эти четыре этапа ВЭЖХ-анализа могут иметь решающее влияние
на точность, прецизионность и удобство окончательного варианта метода, здесь
будет кратко обсужден только третий этап (предварительная обработка образца).
Обсуждение этапов 1, 2 и 4 см. 11—4]. В основном, эта глава будет посвящена этапам
с 5 по 8 таблицы 16.1, которые охватывают то, что обычно подразумевается под пред
варительной обработкой образца или пробоподготовкой.
Таблица 16.1 Этапы пробоподготовки
Номер этапа
1
2
3
4
5
6
7
8
Описание этапа
Отбор образцов
Хранение и консервация
образцов
Предварительная
обработка образцов
Взвешивание или
разбавление
Альтернативные методы
обработки образцов
Удаление механических
примесей
Экстракция образца
Дери ватизация
Комментарий
Получение репрезентативного образца с
использованием статистически достоверных методов
Используйте подходящие для этой цели инертные
плотно закрывающиеся контейнеры и условия
хранении. Стабилизируйте, если это необходимо, летучие,
нестабильные или химически активные образцы.
Биологические образцы могут нуждаться в замораживании
Диспергируйте или равномерно распределите образец
(с помощью сушки, просеивания, измельчения и т.д.)
с тем, чтобы получить более репрезентативную
выборку и улучшить растворение или экстракцию
Примите необходимые меры предосторожности в
случае работы с химически активными, нестабильными и
биологическими материалами. Используйте
калиброванную мерную стеклянную посуду для разбавления
Подумайте о замене растворителя, обессоливании,
упаривании или лиофильной сушке
Используйте фильтрование, центрифугирование и
твердофазную экстракцию
Методы обработки жидких образцов сведены в
таблице 16.2; твердых— в таблицах 16.8 и 16.9
Используется для улучшения обнаружения
растворенных вешеств или улучшения разделения.
Дополнительные этапы могут увеличить время обработки,
усложнить ее и привести к потерям образца (раздел 16.2)
16.2. Виды образцов 799
В то время как ВЭЖХ преимущественно автоматизированный процесс,
предварительная пробоподготовка часто делается вручную. В результате на
предварительную обработку образца может потребоваться 60% или больше от общего времени,
затраченного на рутинный анализ. Наконец, прецизионность и точность метода, как
правило, в значительной степени определяются процедурой предварительной пробо-
подготовки |5—6|, включая такие операции как взвешивание и разбавление. Исходя
из всех этих причин, разработка процедуры предварительной пробоподготовки
заслуживает тщательного планирования.
Эта процедура должна обеспечивать количественное извлечение растворенных
веществ, включать минимальное количество этапов и (если возможно) быть легко
автоматизируемой. Количественное (99+%) извлечение каждого растворенного вещества
повышает чувствительность и точность анализа, хотя это и не означает, что все
вещества, содержащиеся в исходном образце, должны быть включены в тот образец,
который будет введен в колонку. Например, в данном методе, включающем в себя ряд
шагов предварительной обработки образца, аликвоты промежуточных фракций можно
использовать для дальнейшей пробоподготовки или для ввода в колонку. Если степень
извлечения растворенного вещества меньше 100%, то она должна быть
воспроизводимой. Меньшее количество этапов предварительной обработки и автоматизация
сокращают общее время и необходимые усилия. При этом повышается точность анализа и
уменьшается возможность ошибок, которые может сделать аналитик.
Многие методы пробоподготовки автоматизированы, и оборудование для этого
можно купить. Подходы к автоматизации изменяются в диапазоне от использования
робота для выполнения ручных задач до специализированных устройств,
выполняющих определенную процедуру пробоподготовки. Часто необходимо анализировать
большое количество образцов и, хотя автоматизация дорога и требует детальной раз
работки методики, чрезмерные трудозатраты на ручную подготовку образцов
оправдывают ее применение. Решение об автоматизации процедуры пробоподготовки
часто основывается на анализе затрат или в некоторых случаях заботой о безопасности
оператора (например, для того, чтобы свести к минимуму воздействие токсичных
веществ или других возможных опасностей для здоровья). Рассмотрение полной
автоматизации пробоподготовки выходит за рамки этой главы. Читатель может быть
отослан к последним учебникам по этому предмету [7—8|.
16.2. Виды образцов
Основные виды образцов можно в общем случае классифицировать как
органические (включая биологические) или неорганические и дополнительно подразделить
на твердые, полутвердые вещества (включая кремы, гели, суспензии и коллоиды),
жидкости и газы. Почти для каждого вида образцов перед ВЭЖХ-анализом
потребуется предварительная обработка, даже если это только простое разбавление.
Газообразные образцы, как правило, анализируют с помощью газовой хроматографии, а не
ВЭЖХ. Для сбора и ввода газов используются такие методы, как отбор в емкости,
прямой отбор проб через петли и отбор проб из свободного пространства над
продуктом в таре, продувка и улавливание. Несмотря на это, летучие лабильные,
термолабильные растворенные вещества, склонные адсорбироваться на металлических
поверхностях из парообразного состояния, иногда лучше анализировать с помощью
ВЭЖХ. Улавливание требуется при анализе газообразных образцов с помощью
ВЭЖХ. Пробу газа либо (I) пропускают через сорбент и затем элюируют
растворителем, либо (2) барботируют через жидкость, улавливающую растворенное(ые)
вещество^). Примерами ВЭЖХ-анализа газового образца являются метод D5197-03 Амери-
800 Глава 16. Профподготовка
канского общества по испытанию материалов (American Society for Testing Materials,
ASTM) и метод ТО-11 для анализа летучих альдегидов и кетонов |9| Агентства по
охране окружающей среды (United States Environmental Protection Agency, EPA). Пробу
воздуха пропускают через поглотительную ловушку, заполненную 2,4-динитрофе-
н ил гидразином, который количественно превращает альдегиды и кетоны в 2,4-ди-
нитрофенилгидразоны. Гидразоны затем элюируют из ловушки ацетонитрилом и
разделяют с помощью ОФ-ВЭЖХ.
В таблице 16.2 представлен обзор типичных способов подготовки образцов,
используемых для жидкостей и суспензий. Оставшаяся часть этой главы будет посвящена
предварительной обработке образцов, представляющих наибольшие трудности:
полулетучих и нелетучих анализируемых веществ в различных жидких и твердых средах.
Пробоподготовка твердых образцов может быть более сложной, чем жидких.
В некоторых случаях образец легко растворяется и после этого готов для ввода в
колонку или для дальнейшей предварительной обработки. В других случаях матрица1
образца может быть нерастворимой в обычных растворителях, и подлежащие анализу
вещества необходимо экстрагировать из твердой матрицы. Бывают случаи, когда
анализируемые вещества нелегко отделить от нерастворимой матрицы из-за их
включения в матрицу или адсорбции на ней. Тогда могут потребоваться более жесткие
методы, такие как экстракция растворителем, например, в аппарате Сокслета,
жидкостная экстракция под давлением (ЖЭД) или обработка ультразвуком
(раздел 16.8.2). В таблице 16.8 перечислены некоторые традиционные методы извлечения
анализируемых веществ из твердых образцов, в то время как в таблице 16.9 описаны
некоторые недавно разработанные процедуры. Как только анализируемые вещества
количественно извлечены из твердого образца, полученная жидкая фракция может
быть как непосредственно введена в ВЭЖХ-систему/колонку, так и подвергнута
дальнейшей предварительной обработке.
Таблица 16.2. Типичные методы пробоподготовкн для жидких и суспендированных
образцов
Метод
пробоподготовкн
Твердофазная
экстракция
(ТФЭ)
Экстракция
жидкость —
жидкость (ЖЖЭ)2
Принцип действия
Процесс схож с ВЭЖХ. Образец
пропускают в жидкости через
колонку, заполненную твердой
фазой, которая выборочно
задерживает некоторые растворенные
вещества или примеси (раздел 16.6)
Образец распределяется между
двумя несмешивающимися
фазами. Затем растворенные
вещества без примесей извлекаются из
одной из двух фаз (раздел 16.5)
Комментарии
Доступно большое количество
разнообразных неподвижных фаз для
селективного удаления выбранных растворенных
примесей неорганического, органического
и биологического происхождения
Остерегайтесь образования эмульсий2.
Значения Ко могут быть оптимизированы
с помощью различных растворителей и
добавок. В случае малых величин Ко
можно использовать непрерывную
экстракцию или большие объемы экстрагента
1 Среда, в которой он распределен. — Прим перев.
2 Перевод ЖЖЭ в формат твердофазной экстракции позволяет устранить проблему
образования эмульсий и автоматизировать процесс (метод SLE — Solid-Supported Liquid—Liquid
Extraction). Проба, растворенная в воде, распределяется на поверхности частиц инертного
носителя в виде тонкой пленки, увеличивая поверхность контакта с экстрагентом — не смешивающимся
с водой органическим растворителем. При пропускании растворителя через патрон с носителем
эффективно извлекается целевой компонент без образования эмульсии. — Прим. перев.
[6.2. Виды образцов 801
Окончание табл. 16.2
Метод пробо-
подготовки
Разбавление
Испарение
Перегонка
Микродиализ
Лиофилизация
Фильтрование
Центрифугирование
Седиментация
Принцип действия
Образец разбавляется
растворителем, совместимым с
подвижной фазой для ВЭЖХ
Жидкость удаляется при мягком
нагревании и барботировании
воздухом или инертным газом
Образец нагревают до
температуры кипения растворителя, а
летучие вещества из паровой фазы
конденсируют и собирают
Между двумя водными жидкими
фазами помещается
полупроницаемая мембрана, и
растворенное вещество переходит из одной
жидкости в другую вследствие
разности концентраций
Водный раствор образца
замораживают и воду удаляют
испарением под вакуумом
Жидкость пропускается через
бумажный или мембранный
фильтр или ТФЭ-картридж/диск
для удаления взвешенных частиц
Образец помещают в коническую
центрифужную пробирку и
вращают с высокой скоростью (сила
тяжести в несколько раз
превышает обычную). Надосадочная
жидкость сливается
Образцу позволяют оседать,
не взбалтывая его. Скорость
седиментации зависит от радиуса
Стокса
Комментарии
Чтобы избежать чрезмерного уширения
или искажения пика, разбавляющий
растворитель должен смешиваться с
подвижной фазой для ВЭЖХ и предпочтительно
быть более слабым, чем она
Не испаряйте слишком быстро. Избегайте
потерь на стенках емкости. Не
перегревайте досуха. Лучше всего испарять
в токе инертного газа, например, азота
В основном, для образцов, которые легко
испаряются. Некоторые образцы могут
разлагаться, если их слишком сильно
нагреть. Для высококипящих соединений
используется вакуумная перегонка
Для концентрирования диализатов
необходимы методы обогащения, такие как
ТФЭ. Диализ с помощью мембран,
отсекающих вещества с определенной
молекулярной массой, может быть
использован для отделения белков от образцов
в режиме онлайн перед ВЭЖХ.
Аналогичным образом могут быть использованы
ультрафильтрация и обратный осмос
Хорош для нелетучих органических
соединений. Можно работать с большими
объемами образца. Возможны потери
летучих веществ. Удобно для извлечения
термолабильных растворенных веществ,
особенно биологической природы
Настоятельно рекомендуется для того,
чтобы предотвратить проблемы,
связанные с противодавлением, и продлить
срок службы колонки
Для простого удаления взвешенных
частиц ультрацентрифугирование обычно
не используется
Чрезвычайно медленный процесс.
Позволяет вручную отобрать частицы
различного размера, осевшие на разных уровнях
в зависимости от скорости их оседания
Жидкие образцы гораздо легче подготовить для ВЭЖХ, чем газообразные или
твердые. Значительная часть анализов ВЭЖХ основана на принципе «разбавь и
вводи», в соответствии с которым концентрация анализируемого вещества уменьшается
разбавлением, чтобы не перегружать колонку и детектор или чтобы сделать
растворитель образца более совместимым с подвижной фазой.
802 Глава 16. Профподготовка
16.3. Предварительная обработка твердых
и полутвердых образцов
16.3.1. Измельчение образцов
Твердые образцы следует измельчать потому, что мелкодисперсные образцы (1)
более гомогенны, что позволяет делать более репрезентативный отбор образцов с
большей прецизионностью и точностью (2) быстрее растворяются, а также из них легче
экстрагировать необходимое вещество вследствие большей площади их поверхности.
Способы уменьшения размера частиц твердых образцов приведены в таблице 16.3.
Таблица 16.3. Способы уменьшения размеров частиц образца
Способы измельчения
Смешивание
Измельчение
Дробление
Разрезание
Растирание
Гомоген изация
Мацерация
Помол
Рубка/пропускание
через мясорубку
Обработка давлением/
отжим
Пульверизация/
порошкование
Просеивание
Описание способа
Механический блендер используется для измельчения полумягких
субстанций в более мелкие частицы или для приведения неоднородного
образца в более однородное состояние
Процесс механического измельчения образца
Дробилки с мелющими щеками из карбида вольфрама и регулируемой
шириной щели могут уменьшить размер больших твердых образцов
Режущие мельницы могут измельчать образцы от мягких до средней
твердости (< 100 мм)
Самое популярное — это сгупка с пестиком. Используется как мокрое,
так и сухое растирание. При этом может быть достигнут размер частиц
~ 10 мкм
Любой процесс, используемый для того, чтобы сделать образец более
однородным по текстуре и консистенции путем разбивания на более
мелкие части и смеши вания
Процесс разрушения мягкого материала на более мелкие части
разрывом, измельчением, резанием и т.д.1
С помощью различных дисковых, роторных или шаровых мельниц
у мягких, средней твердости и волокнистых материалов можно
уменьшить размер частиц до 80—100 мкм
Процесс дробления мясных или овощных продуктов разрывом,
измельчением, нарезанием на кубики и т.д.
Как правило, это процесс выжимания жидкости из полутвердого
материала (например, из растений, фруктов, мяса)
Для уменьшения размера частиц во влажных и сухих образцах
используются вибромельницы. Для порошкования эластичных и тягучих
образцов можно применить криогенное измельчение с жидким азотом
Просеивание образца через металлическое или пластиковое сито (с
отверстиями 3—123 мкм) для разделения частиц на однородные фракции.
Можно для этих целей использовать как влажное, так и сухое
просеивание
'Это может быть, например, разрыхление и размягчение биологических тканей при
длительном воздействии влаги или высушивание биологических препаратов, или разрушение
межклеточного вещества. — Прим. перев.
16.3. Предварительная обработка твердых и полутвердых образцов 803
16.3.2. Высушивание образцов
Твердые образцы для анализа часто получают в сыром или увлажненном
состоянии. Для проведения достоверного анализа необходимо удалить воду или высушить
образец до постоянного веса. Неорганические образцы, такие, как почва,
необходимо нагреть до 100—110 °С, чтобы обеспечить удаление влаги. Гидрофобные
органические образцы редко требуют нагревания, поскольку они поглощают минимум
воды. Тем не менее, пары органики могут быть адсорбированы твердыми
органическими образцами, и нагревание может удалить эти загрязнения. Для
гигроскопичных или химически активных образцов (например, ангидридов кислот)
рекомендуется сушка в вакуумном эксикаторе. Образцы, которые могут окисляться
при нагревании в присутствии воздуха, должны быть высушены под вакуумом или
под азотом. Биологические образцы, как правило, не следует нагревать выше ста
градусов. Кроме того, чтобы предотвратить разложение образца, следует избегать
температур выше температуры окружающей среды. Образцы неустойчивых
биологических соединений (например, ферментов) чаще всего готовят в холодной
комнате при температуре < 4 °С, чтобы минимизировать разложение. С такими
образцами необходимо работать при этих низких температурах до начала ВЖЭХ-ана-
лиза. Сублимационная сушка (лиофилизация) часто применяется для сохранения
термолабильных образцов (особенно биологических). Она выполняется с помощью
быстрой заморозки образца с последующей возгонкой (сублимацией) замерзшей
воды в вакууме.
16.3.3. Фильтрование
Механические примеси должны быть удалены из жидких образцов перед вводом
в колонку из-за их неблагоприятного влияния на срок службы колонки, а также
из-за возможного повреждения капилляров, инжекторов и фильтров. Наиболее
распространенными методами удаления частиц из образца являются фильтрование,
центрифугирование и седиментация. Некоторые способы фильтрации описаны
в таблице 16.4. Чем меньше поры фильтра, тем чище фильтрат, но тем больше время
фильтрования. Фильтрование под вакуумом ускоряет процесс. Мембранные фильтры
в виде диска можно приобрести в комплекте вместе с фильтродержателями. Тем
не менее, большинство пользователей предпочитает одноразовые фильтры,
снабженные фитингами Luer. Образец помещают в шприц и фильтруют через мембрану под
небольшим давлением. В качестве фильтров коммерчески доступны разнообразные
мембранные материалы различной пористости и размеров, описанные в
сопроводительной документации производителей. Большие площади поперечного сечения
дисковых мембран обеспечивают их хорошую пропускную способность и сводят
к минимуму возможность засорения. Для большинства образцов, встречающихся
в ВЭЖХ, рекомендованы фильтры с номинальной пористостью от 0,25 до 2 мкм.
Величины пористости могут быть приблизительными. Кроме того, тип мембраны
может оказывать некоторое влияние на характеристики фильтрования. Наиболее
популярные размеры пор фильтров 0,25 и 0,45 мкм. Мембраны с порами 0,25 мкм
удаляют мельчайшие частицы (и крупные макромолекулы). Если образец содержит
коллоид или большое количество мелких частиц, то для его продавливания через
фильтр может потребоваться значительное давление. Иногда предварительный
крупнопористый фильтр (толстый фильтр большой емкости для улавливания более
крупных частиц) помещают поверх мембраны, чтобы предотвратить засорение мембраны
этими типами взвешенных частиц.
804 Глава 16. Профподготовка
Таблица 16.4. Фильтрование в ВЭЖХ
Тип фильтра
Бумажные
Мембранные
Специализированные
мембраны
ТФЭ-карт-
риджи
ТФЭ-диски
Материал, обычно
используемый для изготовления
Целлюлоза
Нейлон, ПТФЕ,
полипропилен, полиэфир, по-
лиэфирсульфон,
поликарбонат, поливинилпир-
ролидон
Ионообменные и
аффинные мембраны
На силикагельной и
полимерной основе
На ПТФЕ- и стекловоло-
конной основе
Рекомендации
по использованию
Удаление
крупных частиц
(< 40м км)
Удаление мелких
частиц (> 10 мкм)
Могут удалить как
отдельные
частицы, так и
примесные соединения
Могут удалить как
отдельные
частицы, так и
примесные соединения
Могут удалить как
отдельные
частицы, так и
примесные соединения
Комментарии
Остерегайтесь попадания волокон
бумаги в образец. Удостоверьтесь
в совместимости распюрителя и
фильтровальной бумаги
Дня загрязненных образцов перед
фильтрованием через мембрану
может понадобиться
предварительное фильтрование. Избегайте
несовместимости растворителей
Для загрязненных образцов перед
фильтрованием через мембрану
может понадобиться
предварительное фильтрование. Избегайте
несовместимости растворителей
Частицы привитого силикагельно-
го сорбента могут попадать
в фильтрат. Остерегайтесь
засорения
ПТФЕ-мембраны тонки.
Обращайтесь с ними осторожно.
Могут пропускать большие объемы
при большой скорости потока.
Остерегайтесь засорения
Важным фактором при выборе фильтра является совместимость растворителя
с мембраной. Если используется неподходящий растворитель, фильтр может
раствориться (или размягчиться) и загрязнить фильтрат. Производители мембранных
фильтров обычно предоставляют детальную информацию о совместимости их продукции
с растворителями. Более дорогие специализированные мембраны и ТФЭ-диски
и картриджи используются не только для удаления химических загрязнений, но
и для удаления механических примесей.
16.4. Подготовка жидких образцов
В таблице 16.2 дано введение в методы подготовки жидких образцов. Большинству
лабораторий нужны лишь некоторые из этих методик. К примеру, применение
перегонки ограничено летучими соединениями, хотя вакуумная перегонка высококипя-
ших соединений в пробах, взятых из окружающей среды, может расширить
применение этого метода |Ю]. Применение лиофилизации обычно ограничено очисткой и
обработкой биологических образцов. В настоящем обсуждении мы сделаем акцент
на двух методах, наиболее часто применяемых в большинстве ВЭЖХ-лабораториях:
экстракции в системе жидкость — жидкость (раздел 16.5) и экстракции в системе
жидкость — твердое тело или твердофазной экстракции (раздел I6.6).
/6.5. Экстракция в системе жидкость—жидкость
16.5. Экстракция в системе
жидкость—жидкость1
Экстракция в системе жидкость—жидкость (ЖЖЭ) полезна для отделения
анализируемых веществ образца от примесей посредством распределения образца между
двумя несмешивающимися жидкостями или фазами. Одна фаза в ЖЖЭ обычно бывает
водной, а вторая — органическим растворителем. Более гидрофильные соединения
охотнее уходят в полярную водную фазу, а более гидрофобные в органический
растворитель. Вещества, экстрагированные в органическую фазу, легко могут быть
выделены упариванием растворителя. Вещества, экстрагированные в водную фазу,
часто можно вводить непосредственно в ОФХ-колонку. Нижеследующее обсуждение
предполагает, что растворенное целевое вещество концентрируется
преимущественно в органической фазе. Когда вещество экстрагируют в водную фазу, можно
использовать аналогичные подходы.
На рис. I6.I приведены этапы разделения методом ЖЖЭ. Поскольку экстракция
является равновесным процессом с ограниченной эффективностью (только одна
теоретическая тарелка), значительные количества растворенного вещества могут
оставаться в обеих фазах — даже, когда К;) (или 1/Ад) 3> 1. Смещение равновесия за счет
изменения рН, образования ионных пар и комплексообразования может
использоваться для повышения величин Kg, увеличения степени извлечения растворенного
вещества и/или устранения примесей.
Желательно, чтобы органический растворитель, используемый для ЖЖЭ, имел
следующие свойства:
• низкую растворимость в воде {< 10%);
• летучесть, необходимую для того, чтобы можно было легко его удалить и
сконцентрировать образец после экстракции (таблица 1.3);
• совместимость с ВЭЖХ-детектором (например, низкое поглощение при малых
длинах волн в УФ-области) (таблица 1.3);
• полярность и способность образовывать водородные связи, способствующие
переходу компонентов образца в органическую фазу (раздел 2.3.2.1) (таблица 1.4);
• высокую чистоту для того, чтобы минимизировать загрязнение образца.
16.5.1. Теория
Закон распределения Нернста гласит, что любые вещества будут распределяться
между двумя несмешивающимися растворителями таким образом, что соотношение
концентраций между ними будет оставаться постоянным.
**.=£-. (I6-1)
где Кр — коэффициент распределения, С0 — концентрация растворенного вещества
в органической фазе и Cfl(, — концентрация растворенного вещества в водной фазе.
Более удобным выражением описывается количество Е экстрагированного
растворенного вещества, определяемое как
Е = С°^ = Ко* , (16.2)
CgVc + СщУщ I + KlW
где V0— объем органической фазы. Vaq — объем водной фазы и f- фазовое
отношение VJVaq.
' Liquid—liquid Extraction (LLE).
Глава 16. Профподготовка
Жидкий
образец
Выберите расворитель,
подходящий для
следующего шага
экстракции
Да
Отрегулируйте
условия
(например. рН)
Образец в
растворе?
Нет.
Поместите
образец е
делительную
воронку
Добавьте
несмешивающийся
растворитель и
сильно встряхните
Отделите фазы
друг от друга
Определите
количество
образца в
каждой фазе
Нет
Все вещества
экстрагировались
(повторите \^ количественно?.,
экстракцию)
Растворите в
подходящем
растворителе
Дайте фазам
расслоиться
Jfla
Упарьте до
необходимой
концентрации
Ввод в колонку
или дальнейшая
п робоподготовка
Рис. 16.1. Схема этапов экстракции а системе жидкость—жидкость (ЖЖЭ)
Многие ЖЖЭ-процедуры проводятся в делительных воронках и, как правило,
используются десятки или сотни миллилитров каждой фазы. Для одноступенчатых
экстракций А"д (или 1/А"д) должен быть большим (например, > 10) для
количественного извлечения растворенного вещества из одной из двух фаз, так как фазовое
отношение должно на практике поддерживаться в диапазоне значений 0.1 < Ч^ < 10
(см. уравнение 16.2). В большинстве операций ЖЖЭ с делительной воронкой
количественное извлечение (> 99%) требует двух или более экстракций. Для
последовательных многократных экстракций с объединением фаз, содержащих образец, после
каждой экстракции количество экстрагированного растворенного вещества Е равно
Е =1
\1+ Kirr)
(16.3)
/6.5. Экстракция в системе жидкость—жидкость 807
где л — количество экстракций. Например, если Kq — 5 для растворенного вещества
и объемы двух фаз равны (Ч1 = I), потребуются три экстракции для его извлечения
(> 99%). Для увеличения KD могут быть использованы несколько способов:
• можно заменить органический растворитель;
• Кр может быть увеличен подавлением ионизации целевого вещества, если оно
ионогенно или ионизируемо, и, таким образом, сделать его более растворимым
в органической фазе;
• целевое вещество может быть переведено в органическую фазу благодаря
образованию ионных пар при условии, что оно ионизировано, и в органическую фазу
добавлен ион-парный реагент;
• для уменьшения концентрации растворенного в водной фазе вещества может
использоваться высаливание при добавлении инертной нейтральной соли
{например, сульфата натрия) в водную фазу.
16.5.2. Практика
В таблице 16.5 приведены примеры типичных растворителей для экстракции, а
также некоторые растворители, не подходящие для этих целей (смешивающиеся с
водой). Помимо соображений смешиваемости, основным критерием является
полярность Р растворителя (таблица 2.3, 1.4) в сравнении с полярностью растворенного
образца. Кр достигает максимума, когда полярность растворителя для экстракции
соответствует полярности растворенного вещества. Например, экстракция полярного
растворенного вещества из водной фазы, в которой находится образец, лучше всего
пройдет, если использовать более полярный органический растворитель {с большей
величиной Р). Органический растворитель с оптимальной полярностью может быть
легко подобран смешиванием двух растворителей различной полярности (например,
гексана \Р = 0,1| и хлороформа \Р = 4,11) и контролем Ад в зависимости от состава
органической фазы |11|. Смесь растворителей, которая дает наибольшую величину
K.Q используется в дальнейшем для ЖЖЭ. Еще большее увеличение Ад может быть
достигнуто улучшением отделения растворенных веществ образца от примесей с
помощью изменения селективности органического растворителя (в дополнение к
полярности). Предполагается, что растворители, взятые из разных частей треугольника
селективности (рис. 2.9), обеспечат различия селективности; см. также обсуждение
в работе 112|.
При экстракции растворителем ионизируемые растворенные органические
вещества зачастую можно перевести в любую из фаз, подобрав соответствующие условия.
Рассмотрим, например, экстракцию органической кислоты из водного раствора.
Если водная фаза забуферена, по крайней мере, на 1,5 единицы рН выше
величины рКа, растворенная кислота будет ионизирована и предпочтительно перейдет
в водную фазу. Менее полярные примеси экстрагируются в органическую фазу. Если
рН водного раствора понизить до такой степени, что растворенное вещество не будет
ионизировано (<£ рКа), оно перейдет в органическую фазу, оставляя более полярные
примеси в водной фазе. Последовательные экстракции при высоком рН, следующие
за экстракциями при низких рН в состоянии отделить кислоту как от более, так и
от менее полярных примесей. Зависящие от рН равновесные состояния обсуждаются
более подробно в разделе 7.2. Обратите внимание, что принципы кислотно-основной
экстракции для ЖЖЭ в зависимости от рН такие же, как и для ОФХ.
Если Kq целевого вещества неблагоприятен для экстракции, могут потребоваться
дополнительные стадии экстракции для повышения выхода (уравнение 16.3). В
случае, когда целевое вещество растворимо в органическом растворителе, к водной фазе
для следующей экстракции добавляют свежую порцию органического растворителя.
808 Глава 16. Профподготовка
Таблица 16.5. Растворители для ЖЖЭ
Водные растворы
Чистая вода
Кислотный раствор
Основный раствор
Раствор с высокой
концентрацией соли
(эффект высаливания)
Комплексообразующие
агенты (ион-парные, хе-
латирующие, хиральные
и т.д.)
Комбинация из двух или
более растворителей
перечисленных выше
Не смешивающиеся с водой
органические растворители
Алифатические углеводороды (гексан,
изооктан, петролейный эфир и т.д.)
Днэтиловый эфир или другие эфиры
Метиленхлорид, хлороформ
Этилацетат и другие эфиры.
Алифатические кетоны (Qj и выше).
Алифатические спирты (Сь и выше)
Толуол, ксилолы (УФ-поглощение!).
Комбинация двух или более
растворителей, упомянутых выше
Смешивающиеся с водой
органические растворители
(не подходят для ЖЖЭ)
Спирты (низкомолекулярные)
Кетоны (низкомолекулярные)
Альдегиды
(низкомолекулярные)
Карбоновые кислоты
(низкомолекулярные)
Ацетон итрил
Диметилсульфоксид
Диоксан
Затем все экстракты объединяются. В общем случае для количественного извлечения
целевого вещества в данном объеме выбранного растворителя более эффективна
многократная, чем однократная экстракция. Реэкстракция может быть использована
для более полного удаления примесей. Рассмотрим, например, описанную выше
органическую кислоту. Если ее экстрагируют сначала при низком рН в органическую
фазу, полярные примеси (например, гидрофильные нейтральные соединения, прото-
нированные основания) остаются в водной фазе. Если для реэкстракции
используется свежая порция водного буфера с высоким рН, ионизированная органическая
кислота вновь переходит в водную фазу, оставляя менее полярные примеси в
органической фазе (последняя процедура аналогична последовательным экстракциям
при высоких рН, следующих за экстракциями при низких рН, описанным выше).
Таким образом, двухстадийная экстракция с изменением рН может помочь
в удалении как основных, так и нейтральных примесей, тогда как одностадийная
экстракция может устранить либо те, либо другие примеси, но не те и другие вместе.
Если Ад не намного больше I или необходимый объем образца велик, то, наверное,
нецелесообразно многократно экстрагировать для количественного извлечения
растворенного вещества, поскольку потребуется слишком много экстракций и общий
объем экстракта будет слишком велик (уравнение 16.3). Если экстракция проходит
медленно, то для установления равновесия может потребоваться длительное время.
В таких случаях может использоваться непрерывная экстракция жидкость-жидкость,
где свежий растворитель непрерывно рециркулирует сквозь растворенный в водной
фазе образец. Непрерывные экстракторы, использующие растворители тяжелее и
легче воды, описаны в работе |13]. Эти экстракторы могут работать продолжительное
время (12—24 ч), и с их помощью можно достигать количественного извлечения
(> 99%) даже в тех случаях, когда величины Ад не очень велики.
Противоточный экстрактор для ЖЖЭ может обеспечить тысячу или более
равновесных циклов (но за большее время и с большими трудозатратами). Это
позволяет извлекать растворенные вещества, имеющие Ад близкий к единице. Кроме того,
противоточное распределение обеспечивает лучшее отделение от примесей.
Небольшие экстракторы лабораторных масштабов коммерчески доступны. Более подробно
о них см. 114|.
16.5. Экстракция в системе жидкость—жидкость 809
В некоторых случаях ЖЖЭ может повысить концентрацию растворенного
вещества в экстракте относительно его концентрации в исходном образце. В соответствии
с уравнением (16.2) при выборе меньшего объема органического растворителя,
концентрацию растворенного вещества можно повысить, используя объемное
соотношение органической и водной фаз (при условии почти полной экстракции в органическую
фазу или при большом Kg). Например, дано 100 мл водного раствора образца, 10 мл
органического растворителя и очень большой Ад (например, А"д > 1000).
Концентрация растворенного вещества в органической фазе увеличится в десять раз.
Для больших отношений объемов водной и органической фаз даже небольшая
растворимость органического растворителя в водной фазе может значительно
уменьшить объем органического экстракта. Этих сложностей можно избежать,
предварительно насыщая водный растворитель органическим. Обратите внимание — когда
соотношение VJVaq невелико, работать (физически) с двумя фазами (включая
отделение органической фазы) становится трудно.
16.5.3. Трудности
Среди некоторых практических трудностей, связанных с ЖЖЭ:
• образование эмульсии;
• сильная адсорбция целевых веществ механическими примесями;
• связывание целевых веществ с высокомолекулярными соединениями (например,
взаимодействия белок-лекарство);
• взаимная растворимость двух фаз.
16.5.3.1. Образование эмульсии
Эмульсии представляют собой проблему, которая может возникать при работе
в определенных условиях с некоторыми образцами (например, имеющими жирную
матрицу). Извлечение растворенного вещества может стать менее полным, если
эмульсию не разбить так, чтобы получилась четкая граница между водной и
органической фазами. Зачастую эмульсии можно разбить с помощью:
• добавления соли к водной фазе;
• нагревания или охлаждения сосуда для экстракции;
• фильтрования через слой стекловаты;
• фильтрования через бумагу с разделением фаз;
• добавления небольшого количества другого органического растворителя;
• центрифугирования.
16.5.3.2. Адсорбция растворенного вещества
Если в образце присутствуют механические примеси, адсорбция растворенного
целевого вещества на этих частицах может привести к уменьшению полноты его
извлечения. В таких случаях промывка частиц после фильтрования более сильным
растворителем зачастую приводит к извлечению адсорбированного растворенного вещества.
Этот экстракт нужно объединить с экстрактом растворенного вещества, полученного
в результате ЖЖЭ. Определение «более сильный» растворитель может включать
изменение рН, увеличение ионной силы или использование более полярного
органического растворителя.
810 Глава 16. Пробоподготовка
16.5.3.3. Связывание растворенного вещества
Соединения, которые обычно количественно извлекаются с помощью ЖЖЭ, могут
при обработке образцов плазмы связываться с белками, что сильно понижает
степень их (соединений) извлечения. Связывание с белками создает особенные
трудности при определении лекарств и их метаболитов в физиологических жидкостях.
Методы разрушения связи анализируемых веществ с белками в образцах плазмы
включают:
• добавление детергента;
• добавление органического растворителя, хаотропных агентов или сильной кислоты;
• разбавление водой;
• замещение более сильным связывающим соединением.
16.5.3.4. Взаимная растворимость фаз
«Несмешивающиеся» растворители имеют небольшую, но конечную взаимную
растворимость, и растворенный в другой фазе растворитель может изменить
относительные объемы двух фаз. Поэтому рекомендуется насыщать одну фазу другой таким
образом, чтобы объем содержащей целевое вещество фазы мог быть точно измерен.
Это позволяет наиболее точно определять степень извлечения растворенного
вещества. Значения растворимости воды в растворителе (или растворителя в воде) можно
посмотреть в работе [15|.
16.5.4. Специальные подходы к ЖЖЭ
16.5.4.1. Микроэкстракция
Экстракции в такой модификации ЖЖЭ проводят при соотношениях органика/вода
от 0,001 до 0,01. Выход растворенного вещества может и уменьшиться по сравнению
с обычной ЖЖЭ, но его концентрация в органической фазе значительно
увеличивается, и расход растворителя сильно снижается. Такие экстракции удобно проводить
в мерной колбе. Органический растворитель для экстракции выбирают так, чтобы
его плотность была меньше плотности воды и, таким образом, его небольшой объем
будет скапливаться у горла колбы и его легко будет удалить. Для количественного
анализа нужно использовать внутренние стандарты и проводить экстракцию
калибровочных стандартов. Некоторые современные автосамплеры могут проводить
микроэкстракции в автоматическом режиме из небольших объемов водных образцов
во флаконах по 2 мл, при условии, что положение заборной иглы является
регулируемым [16|.
16.5.4.2. Микроэкстракция методом одной капли (МОК)1
Этот метод использует микрокаплю объемом I или 2 мкл несмешивающегося
органического растворителя, удерживаемого на конце иглы, для экстракции и
концентрирования веществ из водного раствора образца [17|. Растворенные вещества
диффундируют в каплю, что приводит к значительному увеличению их концентрации.
По достижении равновесия микрокаплю втягивают в иглу шприца и затем вводят
в колонку или разбавляют в микровиале для достижения совместимости с
подвижной фазой для ОФХ. Примером использования МОК является ОФХ-анализ гипери-
цинов в плазме крови и моче |18|. Иногда в качестве капли удобнее использовать
мембрану из полых волокон, заполненную небольшим объемом органического
растворителя. Такой метод называют жидкофазной микроэкстракцией (ЖМЭ).
'Single-Drop Micro Extraction (SDME).
16.6. Твердофазная экстракция (ТФЭ) 811
16.5.4.3. ЖЖЭ на твердой фазе (ТФЖЖЭ)1
ТФЖЖЭ представляет собой замену делительной воронки для ЖЖЭ небольшой
колонкой, содержащей инертную подложку, такую как кизельгур. Сначала водный
раствор образца наносится на колонку, чтобы покрыть им подложку. Затем через
колонку пропускают забуференный несмешивающийся с водной фазой растворитель
с тем, чтобы экстрагировать всевозможные растворенные гидрофобные вещества.
Образцы, для которых показана эффективность такого подхода, включают
разбавленную плазму, мочу и молоко. Растворитель проходит через колонку под действием
силы тяжести или под небольшим вакуумом. Поскольку образец не встряхивается
с растворителем для экстракции как в обычной ЖЖЭ, нет и возможности для
образования эмульсии. Упакованные колонки являются одноразовыми, и весь процесс
автоматизируем. Предупакованные несколькими сотнями миллиграмм твердой фазы
на лунку 96 луночные планшеты подходят для экстракции из водного раствора
образца объемом от 150 до 200 мкл. Примерами коммерчески доступных колонок
для ТФЖЖЭ являются Varian's Hydromax (PaloAlto, CA), Biotage's Isolute HM-N
(Charlottesville, VA), и Merck's Extrelut (Darmstadt, Germany).
16.5.4.4. Жидкостная экстракция на иммобилизованном
носителе (ИЖЭ)2
При ИЖЭ гидрофобные вещества из водного раствора образца экстрагируются в
полимерную пленку, содержащую фазу, аналогичную жидким фазам, используемым в
капиллярной ГХ. Полимерная пленка может быть нанесена на крышку виалы, на
внутренние стенки лунки 96-луночного планшета или на внутреннюю поверхность
наконечника микропипетки. Сначала образец помещают на поверхность пленки с тем,
чтобы произошла экстракция веществ из раствора, затем промывают пленку водой для
удаления полярных примесей и, наконец, промывают органическим растворителем
для извлечения целевых веществ. После этого образец может быть введен
непосредственно в хроматографическую систему или упарен и снова растворен в более
совместимом с подвижной фазой для ВЭЖХ растворителе. Устройства для ИЖЭ
поставляются ILE Inc. (Ferndale, CA) и другими поставщиками. ИЖЭ может быть
автоматизирована и в сравнении с ТФЭ выглядит более привлекательно [ 19|.
16.6. Твердофазная экстракция (ТФЭ)
ТФЭ является наиболее важным методом, используемым при пробоподготовке
для ВЭЖХ. ТФЭ можно использовать аналогично ЖЖЭ, но в то время как ЖЖЭ
является одностадийным процессом разделения, ТФЭ представляет собой хроматогра-
фический метод, напоминающий ВЭЖХ, и имеет ряд потенциальных преимуществ
по сравнению с ЖЖЭ:
• более полная экстракция растворенного вещества;
• более эффективное отделение примесей от растворенных целевых веществ;
• снижение расхода органического растворителя;
• легче собрать всю фракцию целевого вещества после отделения примесей;
• более удобные методики для работы в ручном режиме;
• удаление механических примесей;
• ее легче автоматизировать.
'Англ. Solid-Supported Liquid—Liquid Extraction (SLE). — Прим. перев.
2 Англ. Immobilized Liquid Extraction (ILE). — Прим. перев.
812 Глава 16. Профподготовка
Поскольку ТФЭ является более эффективным процессом разделения, чем ЖЖЭ,
легче получить более высокий выход целевого вещества. Методики ЖЖЭ,
требующие нескольких последовательных экстракций для того, чтобы выделить 99+%
целевого вещества, часто могут быть заменены одностадийными ТФЭ-методами. Кроме
того, ТФЭ позволяет достичь более полного отделения примесей от образца.
Методики ТФЭ на обращенной фазе наиболее популярны, поскольку для того, чтобы
выделить целевой компонент с высокой концентрацией, необходимы лишь небольшие
количества органического растворителя. Нет необходимости в разделении фаз (как
в ЖЖЭ) и, таким образом, в ТФЭ суммарную фракцию образца легко собрать,
избежав при этом ошибок, связанных с различными по объему или неточно
измеренными объемами экстракта. При ТФЭ не может образоваться эмульсия. Наконец, более
крупные нерастворенные частицы улавливаются картриджем ТФЭ и не попадают
во фракцию, содержащую образец.
Некоторые недостатки ТФЭ в сравнении с ЖЖЭ:
• возможные изменения свойств сорбентов от партии к партии в картриджах
для ТФЭ;
• необратимая адсорбция некоторых образцов на сорбенте в картриджах для ТФЭ;
• может потребоваться разработка более сложной методики (до четырех ступеней,
см. рис. 16.4).
Растворители, используемые в ЖЖЭ, как правило, чисты и хорошо
охарактеризованы, так что ЖЖЭ-разделение хорошо воспроизводимо. Хотя качество
использовавшихся ранее картриджей для ТФЭ изменялось от партии к партии, усилия,
предпринятые для улучшения их качества, привели к значительному улучшению
воспроизводимости процесса ТФЭ. Площадь поверхности устройства, в котором
проводят ЖЖЭ (например, делительная воронка) достаточно мала (и менее
эффективна) по сравнению с ТФЭ-картриджем (который заполнен сорбентом с высокораз
витой поверхностью), поэтому необратимая адсорбция образца (приводящая к более
низкому выходу) в случае ЖЖЭ менее вероятна, чем при ТФЭ.
16.6.1. Сравнение ТФЭ и ВЭЖХ
В самом простом варианте для ТФЭ используется маленькая пластиковая
одноразовая колонка или картридж, часто это цилиндрическая часть медицинского шприца,
заполненная 0,1 —1,0 г сорбента. Сорбент, как правило, представляет собой
обращенную фазу (например, силикагель C|g), которая по своей разрешающей
способности сходна с используемой в ОФХ. В следующем далее обсуждении предполагается,
что ТФЭ проводится с картриджами, упакованными обращенно-фазовыми
сорбентами (ОФ-ТФЭ), если не указано иное. Несмотря на то, что первыми стали
использовать привитые силикагельные фазы, в последние годы стали коммерчески доступны
полимерные сорбенты, приобретающие все большую популярность. В сравнении
с ТФЭ-сорбентами на основе силикагеля полимерные сорбенты имеют ряд
преимуществ: (1) более развитую поверхность (и, как следствие, бблыиую емкость), (2)
лучшую смачиваемость, (3) устойчивость к частичному осушению после
кондиционирования, которое не влияет на выход и воспроизводимость, (4) отсутствие силанолов
(меньше вероятность необратимой адсорбции сильнооснбвных соединений и (5)
широкий диапазон рН (большая гибкость при подборе условий).
В самом широко распространенном исполнении сорбент для ТФЭ
упаковывается в шприц между фильтрами, аналогично ВЭЖХ-колонке (рис. 16.2а). Размер
частиц сорбента (например, в среднем 40 мкм), как правило, больше, чем у сорбентов
ВЭЖХ (1,5—5 мкм). Поскольку в ТФЭ высота упакованного слоя меньше, частицы
крупнее и упакованы они хуже, картриджи для ТФЭ куда менее эффективны, чем
16.6. Твердофазная экстракция (ТФЭ) 813
(в)
реэевуар-
картридж
фильтр ,
слои
сорбента
(б)
держатель
__ префильтр
(опциональный)
диск
держатель
наконечник Пюэра
(в)
наконечник
пипетки
нанесенный
сорбент
Рнс. 16.2. Различные приспособления для проведения твердофазной экстракции
(ТФЭ). (а) Одноразовый картридж (в цилиндрической части шприца);
(6) диск; (в) наконечник микропипетки (НМП)
ВЭЖХ-колонки (jV < 100). По соображениям экономии в ТФЭ обычно используются
силикагели типа А с частицами неправильной формы (а не силикагели типа В
со сферическими частицами; раздел 5.2.2.2). В последнее время коммерчески
доступными стали силикагели со сферическими частицами для ТФЭ, но они не понизили
объем продаж наиболее популярных сорбентов. Полимерные сорбенты, которые
обычно имеют сферические частицы, дороже, чем сорбенты на основе силикагеля.
Тем не менее, в некоторых дисках для ТФЭ используются более дорогие сорбенты
со сферическими частицами диаметром около 7 мкм. В целом, принципы
разделения, выбора условий и разработка методик аналогичны как для ТФЭ, так и
для ВЭЖХ, за исключением того, что ТФЭ использует серию изократических шагов
для удерживания и элюирования образца. Одно из основных различий между ТФЭ
и ВЭЖХ состоит в том, что картридж для ТФЭ одноразовый, так как в нем могут
оставаться примеси, в то время как колонки для ВЭЖХ используются многократно.
16.6.2. Применение ТФЭ
ТФЭ используется для достижения шести основных целей пробоподготовки:
• удаление примесей и веществ, «убивающих колонку»;
• концентрирование или обогащение образца, содержащегося в следовых количествах;
814 Глава 16. Профподготовка
• обессоливание;
ф замена растворителя;
• дериватизация in situ;
• хранение и транспортировка образца.
16.6.2.1. Удаление примесей
Примеси, перекрывающие пики целевого вещества при ВЭЖХ-разделении,
усложняют разработку метода и могут отрицательно повлиять на результаты анализа. В
некоторых случаях, особенно при работе со сложными образцами (например,
натуральными продуктами и продуктами ферментативного расщепления белка), большое
количество примесей в исходном образце может сделать практически невозможным
их отделение от одного или двух пиков целевых веществ образца за одно ВЭЖХ-раз-
деление. ТФЭ может использоваться для уменьшения количества этих примесей или
для их устранения до проведения ВЭЖХ. Некоторые образцы содержат такие
компоненты, как гидрофобные вещества (например, жиры, масла, смазки), белки,
полимерные материалы или механические примеси, которые могут засорить или сделать
неэффективной ВЭЖХ-колонку. Эти «убивающие колонку» вещества зачастую могут
быть удалены с помощью ОФ ТФЭ.
16.6.2.2. Обогащение образца
ТФЭ можно использовать для увеличения концентрации компонентов, содержащих
ся в образце в следовых количествах. Если ТФЭ-картридж может быть выбран таким
образом, что для растворенного вещества 4» 1, то до того, как образец насытит
картридж и начнет с него элюироваться, можно нанести относительно большой его
объем (например, несколько мл). Увеличение концентрации целевого вещества
образца (обогащение следовых количеств) может быть достигнуто при условии, что
картридж промывается небольшим объемом сильного растворителя (к > 1). Примером
обогащения целевых веществ, находящихся в образце в следовых количествах, может
быть использование ТФЭ для концентрирования суб-наноколичеств полиядерных
ароматических углеводородов |20| или пестицидов |21| в пробах, взятых в
окружающей среде с помощью картриджа ОФ-ТФЭ. Сильный растворитель (например, аце-
тонитрил или метанол) элюирует эти целевые вещества в небольшом объеме, что
экономит время, необходимое для упаривания. Затем образец может быть
повторно растворен в растворителе, совместимом с растворителями, используемыми
для ВЭЖХ-разделения, и наоборот, элюированный с картриджа образец может быть
непосредственно разбавлен подходящим для ввода в колонку растворителем.
16.6.2.3. Обессоливание
ОФ-ТФЭ может использоваться для обессоливания образцов, особенно перед
ионообменной хроматографией (ИОХ), где желательно иметь образец в растворе с
небольшой ионной силой. Значения рН и % органики выбираются таким образом, чтобы
адсорбированный в картридже образец можно было отмыть от неорганических солей
водой. После чего образец (уже обессоленный) можно элюировать органическим
растворителем [22|.
16.6.2.4. Другие применения
Остальные области применения ТФЭ — замена растворителя, дериватизация in situ,
хранения и транспортировка образца — либо используются редко, либо менее
актуальны для ВЭЖХ. Детали см. в |21, 23, 24|.
16.6. Твердофазная экстракция (ТФЭ) 815
16.6.3. Приспособления для проведения ТФЭ
Применяется несколько конструкций (рис. I6.2):
• картридж;
• диск;
• наконечник пипетки;
ф 96-луночный планшет;
• покрытые волокна или перемешивающий элемент (магнит) с покрытием.
16.6.3.1. Картриджи
Наиболее популярное приспособление для проведения ТФЭ — картридж. Типичный
одноразовый картридж (в виде цилиндрической части шприца) изображен
на рис. 16.2й. Шприц, как правило, изготовлен из полипропилена медицинского
качества, снабжен наконечником Люер для того, чтобы можно было прикрепить иглу и
направить жидкие отходы в небольшой контейнер или виалу. Фильтры, между
которыми помещается сорбент в картридже, сделаны из фторопласта, полипропилена
или пористой нержавеющей стали с размером пор от 10 до 20 мкм и дают небольшое
сопротивление потоку. Картриджи для ТФЭ могут конструкционно отличаться, если
их необходимо встроить в автоматическую или роботизированную систему. Они
относительно недороги, и их выбрасывают после однократного использования, чтобы
избежать перекрестного загрязнения образцов.
Для работы с самыми разнообразными приложениями ТФЭ выпускают
картриджи с массой сорбента от 35 мг до 2 г, а также с объемом колонки/резервуара
(в который упакован сорбент) от 0,5 до 10 мл. Для больших количеств образцов
выпускают «мега» картриджи, содержащие до 10 г сорбента, упакованного в
резервуар объемом 60 мл. Картриджи с большим количеством сорбента следует
использовать для загрязненных образцов, способных перегрузить картридж малой
емкости. Правда, картриджи, содержащие 100 мг сорбента или менее, лучше
использовать для относительно чистых жидких образцов, в тех случаях, когда
емкость картриджа не играет большой роли, а также для небольших объемов
образцов. Вследствие более развитых площадей поверхности полимерных сорбентов для
ТФЭ, их требуется меньше, чем сорбентов на основе силикагеля (от 2 до 60 мг).
В большинстве случаев желательно собирать целевое вещество в минимально
возможном объеме растворителя. Из этого следует, что и картридж для ТФЭ должен,
в общем случае, быть как можно меньше.
16.6.3.2. Диски
Второе по популярности приспособление для проведения ТФЭ — диск (рис. 16.25).
Они сочетают в себе преимущества мембранной технологии (см. ниже) и ТФЭ.
По внешнему виду диски оченъ похожи на мембранные фильтры: они плоские, как
правило, < I мм толщиной и от 4 до 96 мм в диаметре. На самом деле строение
дисков отличается от мембранных фильтров. Диски могут быть куплены в любом из
следующих исполнений:
• полимерные или на основе силикагеля сорбенты, упакованные в гибкие или
растяжимые фторопластовые сетки;
• жесткие диски из стекловолокна с внедренными в них сорбентами;
• дериватизированные мембраны.
Диски с упакованным в них сорбентом. Доля сорбента в таких дисках обычно
составляет 60—90% от общего веса мембраны. Одни диски продаются отдельно от
держателя и во время работы должны быть установлены в многоразовом держателе.
816 Глава 16. Профподготовка
а другие уже закреплены в одноразовых держателях или картриджах с фитингами
Люер для удобного соединения со шприцами.
Диски и картриджи для ТФЭ отличаются, главным образом, соотношением
высоты и диаметра (L/d): у дисков L/d <1, а у картриджей L/d>\. Такая характеристика
диска, если сравнивать ее с аналогичной для ТФЭ-картриджа, позволяет работать
при более высоких скоростях потока и быстрее экстрагировать образец. «Грязная»
вода или сточные воды, содержащие механические примеси, могут закупорить как
пористые диски, так и картриджи. Таким образом, перед ТФЭ необходимо раствор
образца отфильтровать через предфильтр. Некоторые диски поставляются уже
со встроенными предфильтрами. Образование каналов, вызывающее неравномерный
поток через плохо упакованные картриджи, не создает проблем в случае дисков.
Однако из-за малой толщины диска (как правило, 0,5—2 мм), вещества с низкими
значениями k имеют меньшие объемы проскока, чем картриджи для ТФЭ.
Диски особенно полезны для экологических методик, таких как анализ следовых
количеств органических веществ в поверхностных водах. В таких случаях для
достижения необходимой чувствительности анализа зачастую необходим большой объем
образца. ЕРА (Американское агентство по защите окружающей среды, US
Environmental Protection Agency) одобрило ТФЭ-технологию в качестве альтернативы
методам ЖЖЭ |25|, которые требуют больших объемов растворителей при подготовке во
дных образцов для ВЭЖХ-анализов. Среди примеров утвержденных методов
методики для определения фенолов |26|, галоуксусных кислот в питьевой воде |27|,
а также пестицидов и полихлорированных бифенилов |28|.
Диски с внедренными сорбентами. Жесткие диски из стекловолокна с небольшой
массой от 1,5 до 30 мг суспендированного внутри стекловолокна1 сорбента
используются для предварительной обработки небольших количеств клинических образцов
(например, плазмы или сыворотки крови |29j). Уменьшенная масса и небольшой
объем этих дисков снижают расход растворителя (и любое, связанное с ним
загрязнение образца примесями растворителя). Преимуществом этого типа дисков
является отсутствие фильтров. Последние могут быть источником загрязнений.
Другие диски. Импрегнированные поливинилхлоридом (ПВХ) и модифицирован
ные мембраны используются в методиках ТФЭ очень редко и здесь не обсуждаются.
16.6.3.3. Другие приспособления для ТФЭ
Движение в сторону миниатюризации в аналитической химии привело к разработке
новых приспособлений для ТФЭ:
• наконечник микропипетки;
• 96-луночный планшет;
• покрытые волокна;
• экстракция при механическом перемешивании.
Конструкция наконечника микропипетки (НМП) (рис. 16.2в) позволяет работать
с субмикролитровыми количествами образцов — такими, как биологические
жидкости. ТФЭ была проведена с различными сорбентами, помещенными в наконечник
пипетки, или же сорбенты были включены в стенки наконечника или покрывали его
внутренние стенки. При работе с такими наконечниками жидкие образцы могут
быть отобраны и удалены из наконечника без проблем, связанных с перепадом
'Например, мембранные диски SUPELKO, США с пористой мембраной из стекловолокна,
содержащей химически модифицированный силикагель. Вместо стекловолокна может быть
использован, к примеру, фторопласт. Например, диски Empor для ТФЭ, выпускаемые Agilent, США,
представляют собой композиционные мембраны, в которых частицы полимеров суспендированы
в плотно переплетенные волокна фторопласта. — Прим. перев.
16.6. Твердофазная экстракция (ТФЭ) 817
давления или закупориванием. Образец поступает в наконечник, в котором он
взаимодействует с ТФЭ-сорбентом. Затем наконечник можно промыть для улучшения
очистки образца, а после этого элюировать сильным растворителем. Многие
популярные ТФЭ-методики были адаптированы для НМП, включая использование
различных видов хроматографии: обращенно-фазовой, ионообменной, гидрофобных и
гидрофильных взаимодействий, аффинной и металл-аффинной. В основном НМП
использовались для очистки, концентрирования и селективного выделения
{например, с помощью метал-аффинной хроматографии) белков и пептидов и являются
важным инструментом для МАЛДИ (матричная лазерная десорбционная ионизация)
и для других передовых методов МС (масс-спектрометрии) |30,31]. Одно из главных
преимуществ НМП состоит в том, что они могут быть использованы для работы
с микропипетками или для автоматизации работы с жидкими образцами.
96-яуночный ТФЭ-планшет также хорошо приспособлен для автоматизации и
обработки большого количества образцов малого объема в процессе ТФЭ. В этом
приспособлении 96 проточных ТФЭ-«колодцев» объемом от 0,5 до 2 мл, каждый из
которых содержит небольшое количество сорбента (< 100 мг). Сорбенты в «колодцах»
удерживаются небольшими фильтрами или включены в отдельные диски. Устройство
планшета аналогично батарее (8 х 12) миниатюрных ТФЭ-картриджей. С
планшетами можно работать с помощью роботизированных систем, чтобы полностью авто
матизировать процесс ТФЭ. Специализированные 96-канальные испарительные
станции могут в автоматическом режиме испарять элюируюший растворитель
из планшетов. После этого высушенные образцы можно вновь растворить в подходя
щем растворителе и ввести их в хроматографическую систему непосредственно
из планшета.
Покрытые волокна используются для твердофазной микроэкстракции (ТФМЭ).
В этой конструкции волокно из плавленого кварца покрыто полимерной
неподвижной фазой, такой как полидиметилсилоксан или полиакрилат 132, 33|. Волокно
погружается в анализируемый раствор, и растворенные в нем вещества диффундируют
внутрь неподвижной фазы и распределяются там в зависимости от их
коэффициентов распределения. Как только равновесие достигнуто, волокно удаляют из раствора
и вводят в канал ВЭЖХ-инжектора или в виалу автосамплера, из которых
анализируемые вещества вымывают сильным растворителем1.
Экстракция сорбентом, покрывающим поверхность перемешивающего элемента
магнитной мешалки |34| по сути своей похожа на использование покрытых волокон.
При этом значительно увеличивается площадь покрытой сорбентом поверхности,
что, в свою очередь, ведет к гораздо большей чувствительности метода к количествам
анализируемых веществ. Магнитный перемешивающий элемент помешается в
водный раствор и последний перемешивается, в то время как анализируемое вещество
и матрица разделяются. После уравновешивания перемешивающий элемент
извлекают, высушивают, чтобы удалить следы воды и переносят в специальное устройство,
где анализируемые вещества вводятся в колонку. Приспособления с покрытыми
волокнами и магнитные мешалки более популярны в газовой хроматографии, нежели
в ВЭЖХ, где термическая десорбция эффективнее при испарении сорбированных
веществ в газовую фазу, чем десорбция растворителя в жидкой фазе.
Для краткости понятие «типичный ТФЭ-картридж» будет использоваться
в оставшейся части раздела 16.6. Это же относится и к другим приспособлениям для
ТФЭ, упомянутым в разделах 16.6.3.2 и 16.6.3.3.
1 В качестве полого волокна может использоваться, к примеру, микрошприц, игла которого
покрыта сорбентом снаружи или изнутри. Шприц может иметь внутри иглы подвижный
относительно ее самой стержень, покрытый сорбентом. Подробнее о ТФМЭ и о классификации ее
вариантов см. обзор на русском языке В.Н. Зайцев, М.Ф. Зуй. Твердофазное микроэкстракционное
концентрирование // ЖАХ, 2014. — Т. 69. — № 8. — С. 1—14. — Прим. черев.
818 Глава 16. Профподготовка
16.6.4. Устройства для ТФЭ
Оборудование, необходимое для проведения ТФЭ, может быть очень простым
(рис. 16.3). В качестве движущей силы процесса может использоваться гравитация, но
растворы реальных образцов будут проходить через картридж довольно медленно.
Раствор образца можно продавливать вручную с помощью шприца (рис. 16.3а), однако
этот способ может оказаться малопригодным в случае вязких или содержащих
механические примеси образцов, поэтому использование вакуума для этих целей является
предпочтительным. С помощью колбы Бунзена можно работать только с одним
картриджем (рис. 16.36). Для работы с несколькими образцами и, соответственно, кар-
(•>
плунжер
картридж
фильтр
слой
сорбента"
(6)
растворитель
/Л Г
фильтр Q
о
(в)
картридж
' для ТФЭ
пробирка
для сбора
элюента
картридж
""для ТФЭ
колба
Бунзена
вакуумный
коллектор
/
о
Рис. 16.3. Оборудование для проведения твердофазной экстракции (ТФЭ). (а) Шприц;
(б) колба Бунзена; (в) вакуумный коллектор
16.6. Твердофазная экстракция (ТФЭ) 819
триджами рекомендуется использовать вакуумный коллектор1 (рис. 16.3в). Внутри
вакуумного коллектора расположена съемная стойка для пробирок, в которые
собирается элюент. В некоторые устройства вмонтированы клапаны стравливания давления,
клапаны-расходомеры и вакуумные манометры. Это позволяет лучше управлять
потоком растворителя. Кроме того, коммерчески доступны системы с повышенным
давлением, обеспечивающие индивидуальный контроль отдельных потоков в каждом
картридже. По мере усложнения таких устройств повышается и их цена. Для обеспечения
потока растворителя через картридж используют и центрифугирование, но реже.
Независимо от способа, используемого для создания потока через картридж или
другое приспособление для ТФЭ, скорость потока должна быть такой, чтобы
обеспечивать достаточное время контакта образца с сорбентом, упакованным в картридж.
При этом более высокие скорости снижают эффективность разделения (например,
количество теоретических тарелок N; раздел 2.4). Для типовых методик ТФЭ
рекомендуются скорости потока < 10 мл/мин |35| для картриджа и < 50 мл/мин для диска
диаметром 90 мм.
Когда количество образцов увеличивается до такой степени, что пробоподготов-
ка становится узким местом, становится выгодной автоматизация процесса.
Существует три основных подхода к автоматизации ТФЭ:
• специализированное оборудование для ТФЭ;
• модифицированные трехкоординатные системы для работы с жидкостями;
• автоматизированные рабочие станции.
Простейшее и недорогое специализированное устройство для ТФЭ способно
выполнить кондиционирование, загрузку, промывку и элюирование. Такие системы
могут использовать стандартные картриджи-шприцы, специальные картриджи,
предназначенные для установки в специализированное устройство, ТФЭ-диски или
96-луночные планшеты.
Модифицированные системы для работы с жидкостями используются, главным
образом, для разбавления, смешивания и добавления внутреннего стандарта.
Роботизированные системы являются наиболее универсальной аппаратурой
для выполнения операций, связанных с пробоподготовкой. Хотя робот и может быть
подключен к устройствам, каждое из которых выполняет отдельную стадию ТФЭ,
соединение робота со специализированной ТФЭ-станцией более экономично, и
времени на проведение работ потребуется меньше. Робот необходим для перемещения
контейнеров с образцами на рабочую станцию ТФЭ и с нее, а также с других
устройств для пробоподготовки (например, весов, миксеров, дилюторов, автосампле-
ров) и обратно. Коммерчески доступные роботизированные системы с различными
возможностями поставляются целым рядом производителей (например, Веск-
man-Coulter, Gilson, Hamilton, Tomtec).
16.6.5. Разработка метода для ТФЭ
В своем наиболее распространенном варианте применение ТФЭ обычно включает
четыре этапа (рис. 16.4):
1. Кондиционирование сорбента;
2. Нанесение образца;
3. Промывка сорбента (удаление примесей);
4. Извлечение целевого вещества.
Каждый из этих этапов может быть оптимизирован.
1 Вместо термина коллектор в русскоязычной литературе используется и калька с
английского — мэнифолд. — Примеч. персе.
820 Глава 16. Профподготовка
■ растворитель К
с о ^
п г
спив
образец
(примеси + целевое
вещество)
картридж
-"для ТФЭ"
Этап 1
Этап 2
растворитель П
' О ^
ЭтапЗ
целевое
''вещество
примеси
растворитель Э
С 0 г.
Этап 4
Рис. 16.4. Последовательность этапов в твердофазной экстракции (ТФЭ). (а)
кондиционирование сорбента; (б) нанесение образца; (в) промывка сорбента
{удаление примесей); (г) извлечение целевого вещества
16.6.5.1. Этапы ТФЭ
В этом обсуждении мы рассмотрим в первом приближении удерживание целевого
вещества на сорбенте при ОФ-ТФЭ. Для более подробного изучения других
неподвижных фаз и конкретных продуктов обратитесь к материалам производителей.
На этапе 1 (рис. 16.4д), выполняемом перед добавлением образца, сорбент «конди-
16.6. Твердофазная экстракция (ТФЭ) 821
ционируется» посредством пропускания через картридж растворителя К в количестве
нескольких свободных объемов сорбента. Обычно это метанол или ацетонитрил.
Стадия кондиционирования служит двум целям: (I) с ее помощью удаляются любые
примеси, которые могли накопиться в то время, как картридж находился в
лаборатории, или уже присутствовали в картридже, поставленном производителем, и (2) она
позволяет сольватировать сорбент. Сольватация необходима, так как упакованный
в картриджи обращенно-фазовый сорбент (особенно С8, С]? или фенильный)
пересыхает, и это часто приводит к значительному уменьшению удерживания образца.
Кроме того, различная степень высушивания сорбента приводит к
невоспроизводимым результатам при извлечении целевого вещества. С другой стороны,
упакованные полимерные сорбенты со сбалансированной гидрофобно-гидрофильной
поверхностью высыхают умеренно, и на их свойствах это не отражается.
После кондиционирования сорбента избыток кондиционирующего растворителя
должен удаляться потоком воздуха через картридж. Последний продувают до того
момента, пока растворитель не перестанет капать из нижней части картриджа
(этап la; не показан на рис. 16.4). Однако, продувка должна сразу после этого
прекратиться, поскольку можно пересушить сорбент, и это может неблагоприятно
сказаться на воспроизводимости анализа (особенно это касается ТФЭ-дисков;
полимерные сорбенты в этом отношении более устойчивы). Водой картридж промывают
непосредственно перед пропусканием через него водного раствора образца (этап 16;
не показан на рис. 16.4). Промежуток времени между промывкой водой и
нанесением образца не должен быть слишком большим (например, < 5 мин). Если сорбент
слишком долго находится в воде, сольватирующий растворитель может медленно
диффундировать в воду, тем самым ухудшая смачиваемость сорбента (раздел 5.4.4.2).
Этап 2 (рис. 16.46) в процедуре ТФЭ заключается в нанесении (введении)
образца. Образец, растворенный в слабом растворителе (воде или буфере, содержащем
< 10% органического компонента) вводится в картридж, сильно удерживающий
целевое вещество. Образец для ТФЭ может быть введен с помощью пипетки или
шприца, или закачан в картридж насосом. Последний способ более удобен в случае
больших объемов образца (> 50 мл), таких как пробы воды из окружающей среды.
Образцы и размеры картриджей должны соответствовать друг другу, чтобы избежать
перегрузки последних (см. емкость колонки; раздел 15.3.2.1). Помните, что емкость
картриджа должна быть достаточной для работы с образцами, их матрицами и
примесями. И те, и другие, и третьи могут сорбироваться на сорбенте в процессе ввода
в картридж. Раствор образца следует пропустить через картридж, не давая сорбенту
высохнуть. Скорость потока в ТФЭ не контролируется с такой точностью как
в ВЭЖХ, но ее можно регулировать, изменяя глубину вакуума или скорость подачи
раствора из шприца. Обычно хватает скорости потока от 2 до 4 мл/мин.
Этап 3 (рис. 16.4s) предусматривает удаление примесей с помощью промывки
картриджа растворителем П (промывочным) промежуточной силы. Сила и объем
П-растворителя должны быть тщательно подобраны, поскольку слишком большой
объем и/или чрезмерная сила растворителя могут привести к частичному элюирова-
нию целевого вещества. Лучше всего стадию промывки закончить непосредственно
перед тем, как целевое вещество начинает покидать картридж. В этом случае
примеси, которые удерживаются слабее, чем целевое вещество, вымываются из картриджа,
а потери целевого вещества не происходит. Часто для промывок в ОФ-ТФЭ
используют воду или буферы, но они не могут удалить максимальное количество примесей
из раствора, в котором содержится целевое вещество, собираемое на этапе 4
(рис. 16.4г). Небольшое измеренное количество органического растворителя может
быть добавлено к промывочному растворителю для того, чтобы удалить наиболее
гидрофобные примеси. При этом следует позаботиться о том, чтобы в то же самое
время не вымыть из картриджа целевое вещество. Из-за того, что результаты
822 Глава 16. Профподготовка
ТФЭ-разделения могут меняться от картридж» к картриджу, необходимо иметь
некоторый запас в силе и объеме промывочного растворителя, используемого для
удаления примесей. Основная цель — собрать 100% целевого вещества образца на
четвертом заключительном этапе. В противном случае степень извлечения этого вещества
будет низкой, и сам процесс каждый раз будет проходить по-разному.
Этап 4 (рис. 16.4г) предусматривает элюирование и сбор фракции целевого
вещества. Если чувствительность детектора является определяющим фактором, то задача
ТФЭ должна заключаться в сборе целевого вещества в минимально возможном
объеме. Этого можно достичь с помощью достаточно сильного элюирующего растворителя
Э так, что к х 0 для полосы вещества во время элюирования. Наоборот, использование
более слабого Э-растворителя, который все еще может элюировать целевое вещество
(например, к = I), минимизирует элюирование более сильно удерживаемых примесей,
которые преимущественно останутся на картридже (очень желательно, чтобы
ВЭЖХ-разделение было изократическим). Выпаривание досуха требуется часто,
поскольку Э-растворитель может быть слишком сильным для растворения образца,
вводимого в колонку. По этой причине выбирайте относительно летучий Э-растворитель.
В противном случае может потребоваться слишком много времени для упаривания.
Изменение рН промывочного или элюирующего растворителей может быть
эффективным способом уменьшения удерживания и/или высвобождения целевого вещества
(например, кислые вещества будут сильнее удерживаться при низких значениях рН
и слабее при высоких). Подбор условий процесса ТФЭ может быть усовершенствован
с помощью использования смешанных фаз (раздел 16.6.7.1). Например, сорбент,
имеющий свойства ионообменного и обращенно фазового, может быть использован
для очистки ионизируемых целевых веществ. Кроме того, пробоподготовка с помощью
ТФЭ, являющаяся «ортогональной» процессу разделения на хроматографической
колонке (т.е., имеющая отличающуюся селективность; раздел 6.3.6.2), вероятно, приведет
к меньшему перекрыванию пика на хроматограмме целевого вещества пиками
примесей. ТФЭ можно использовать для удерживания примесей и на этапе прохождения
образца через картридж. При этом целевые вещества через картридж проходят, не
удерживаясь. В этом случае сорбент подбирают таким образом, чтобы удержать примеси и
загрязнения, а не целевое вещество. Такая методика не предусматривает
концентрирования целевого вещества во фракции, собранной после ТФЭ. Невозможно в этом
случае и отделить целевое вещество от более слабо удерживаемых примесей. Поэтому этот
способ ТФЭ обычно дает более грязные фракции целевого вещества, в то время как
методика, иллюстрируемая на рис. 16.4, позволяет отделить его (целевое вещество) как
от слабо, так и от сильно удерживаемых компонентов образца. По этой причине
методика с удерживанием примесей на сорбенте используется гораздо реже и не будет
больше обсуждаться.
16.6.5.2. Сорбенты для ТФЭ
Поскольку ТФЭ представляет собой низкоэффективный вариант ВЭЖХ, многие
сорбенты, используемые в ВЭЖХ, могут применяться и в ТФЭ. В таблице 16.6
представлены наиболее популярные сорбенты для ТФЭ и типы веществ, для которых они
подходят. Привитые силикагели используются чаще всего, но коммерчески доступны
и другие неорганические и полимерные материалы. В дополнение к обычным
сорбентам, показанным в таблице 16.6, имеются специальные для выделения из мочи
наркотических средств |37—381, альдегидов и кетонов из воздуха |9|, катехоламинов
из плазмы крови [39|, и многие другие для широко используемых анализов. Флори-
сил (активированный силикат магния) и окись алюминия в настоящее время
для ВЭЖХ не используются, но могут быть полезны для ТФЭ. Опубликовано много
работ по методам выделения пестицидов с использованием флорисила |40|. Увеличи-
Таблица 16.6. ТФЭ-сорбеиты и условия их использования
Механизм
разделения
Нормальная фаза
(адсорбция)
Нормальная фаза
(полярная принитая
фаза)
Обращенная фаза
(неполярная
привитая фаза — сильно
гидрофобная)
Обращенная фаза
(неполярная
привитая фаза —
умеренно гидрофобная)
Обращенная фаза
(неполярная
привитая фаза — слабо
гидрофобная)
Обращенная фаза
(гидрофобно-гидрофильно
сбалансированная)
Типичные сорбенты
Силикагель, окись
алюминия, флорисил
Циано, амино, диол
Cia, Cg, сополимер
полистирола и дивинил-
бензола (ПС-ДВБ),
дивинилбензол (ДВБ)
(полимерные)
Циклогексил, фенил,
дифенил
Бутил, этил, метил
Полиамид, поли
[л-винилпирроли-
дон-дивинилбензол
(ДВБ)], метакри-
лат-ДВБ
Структура(ы)
-SiOH, АЮН,
Mg2Si03
-CN, -NH2
-СН(ОН)-СН(ОН)
С,8, С8, ПС-ДВБ, ДВБ
6 6 ак)
(-СН2-)3СНЭ,
—С2Н5, — СНз
Различные полимеры
Тип
растворенного вещества
От слабо
до умеренно
полярного
От умеренно
до сильно
полярного
Гидрофобный
(сильно
неполярный)
Умеренно
неполярный
От слабо
до умеренно
неполярного
Кислотный,
основный,
нейтральный
Растворитель, в котором
происходит нанесение
Небольшая величина е
(например, СНОуТек-
сан; рис. 8.6,
таблица 8.1)
Небольшая величина с
(например, гексан;
таблица 8.1)
Высокие /* (например,
вода, разбавленные
МеОН/Н20, ACN/HjO)
Высокие Р' (например,
вода, разбавленные
МеОН/Н20, ACN/HjO)
Высокие Р' (например,
вода)
Вода или буфер
Растворитель, которым злюируют
Большая величина е (например,
метанол, этанол; таблица 8.1,
рис. 8.6)
Большая величина с (например,
метанол, этанол; таблица 8.1)
Средние Р (например, метанол,
ацетонитрил)
Средние Р (например, метанол,
ацетонитрил)
Средние Р (например, метанол,
ацетонитрил)
Средние Р (например, метанол,
ацетонитрил)
Окончание табл. 16.6
Механизм
разделения
Анионообменник
(слабый)
Анионообменник
(сильный)
Катионообменник
(слабый)
Катионообменник
(сильный)
Типичные сорбенты
Первичный амин, 1,
2-диамин
Четвертичный амин
Карбоновая кислота
Ал к илсульфоновая
кислота
Арилсульфоновая
кислота
Структура(ы)
(_CH2-)3NH2,
(-CH2-)jNHCH2CH2NH2
(_CH2-);N- (CH3)j
(_СН2-)3СООН
(_CH2-)1S03H,
aS0,H
Тип
растворенного вещества
Ионный,
(ионизируемый),
кислотный
Ионный
(ионизируемый), кислый
Ионный
(ионизируемый),
основный
Ионный
(ионизируемый),
основный
Растворитель, в котором
происходит нанесение
Вода или буфер
(рН = рКа + 2)
Вода или буфер
(рН = рКа + 2)
Вода или буфер
(рН = рКа - 2)
Вода или буфер
(рН = рКа - 2)
Растворитель, которым злюируют
A. Буфер (рН = рКя - 2)
Б. рН, при котором сорбент или
растворенное вещество
нейтральны
B. Буфер с высокой ионной
силой
A. Буфер (рН = рК„ - 2)
Б, рН при котором растворенное
вещество нейтрально
B. Буфер с высокой ионной
силой
A. Буфер (рН = рКа + 2)
Б, рН при котором сорбент или
растворенное вещество
нейтральны
B. Буфер с высокой ионной
силой»
A. Буфер (рН = рКа + 2)
Б. рН при котором растворенное
вещество нейтрально
B. Буфер с высокой ионной
силой"
аДля ионного обмена существуют три возможных условия элюирования: А, буфер на две единицы выше (кислее) или ниже (основнее) рКа
целевого вещества; Б, рН, при котором или целевое вещество, или сорбент (слабые ионообменники) нейтральны; В, высокая ионная сила.
16.6. Твердофазная экстракция (ТФЭ) 825
вается использование графитированного углерода для ТФЭ, особенно для выделения
содержащих хлорофилл растительных экстрактов |411. Для получения инструкций
по использованию конкретных ТФЭ-сорбентов связывайтесь с производителями или
читайте одну из книг по этому вопросу |7, 42—46|.
Благодаря природе электростатических взаимодействий ионный обмен может
быть мощным и селективным средством для ТФЭ в случае ионных и ионизируемых
соединений. Катионообменные сорбенты удерживают протонированные основания и
другие катионы, в то время как анионообменные сорбенты удерживают
ионизированные кислоты и другие анионы. Ионообменные сорбенты бывают двух видов:
«сильные» и «слабые». Если необходимо, прежде всего, сильнее удержать целевое
вещество, как правило, предпочтительнее сильные ионообменники. Ионизация (и,
следовательно, удерживание) слабых ионообменников зависит от рН {рис. 13.16).
Выбор значения рН есть компромисс между поддержанием ионизированного
состояния неподвижной фазы и обеспечением ионизированного состояния целевого рас
творенного вещества. Таким образом, рН становится мощным регулятором как
для оптимизации удерживания, так и для элюирования целевого вещества со слабого
ионообменника.
Обычно качество и стоимость сорбентов для ТФЭ ниже, чем для ВЭЖХ, с этим
в частности связана проблема различного удерживания на сорбентах из разных пар
тий. Принимая во внимание то, что в ОФХ предпочитают колонки, упакованные
высокочистым силикагелем типа В (раздел 5.2.2.2), ОФ ТФЭ сорбенты в общем случае
более «кислотные» {тип А). Их силанольные взаимодействия, как правило, более
значительны и в большей степени подвержены изменениям от партии к партии. Од
нако, поскольку ТФЭ обычно используется как метод «on-off»1, небольшие различия
в удерживании менее важны, чем в ВЭЖХ.
16.6.6. Пример разработки ТФЭ-метода: выделение
альбутерола из плазмы крови человека
Выделение альбутерола (I) будет использовано в качестве иллюстрации типичного
применения ТФЭ |47|. Этот препарат широко применяют в качестве бронхолитика
при лечении астмы:
н он
^ iQCOH
\х^он
(I)
Альбутерол {молекулярный вес 239 Да) — полярное гидрофильное соединение
с двумя ионизируемыми функциональными группами: фенолом (рКа 9,4) и
вторичным амином (рКа 10,0). В водном растворе он существует, в основном, в
ионизированном состоянии при любом рН. По этой причине альбутерол плохо уходит
в органические растворители из водных растворов. Альбутерол имеет несколько
полярных и неполярных групп, которые могут удержать его в процессе ТФЭ
на сорбенте.
'Т.е. быстрый процесс адсорбции-десорбции. — Прим. перев.
826 Глава 16. Профподготовка
Можно ожидать, что любой из пяти типов неподвижных фаз (обращенная, кати-
онообменная, анионообменная, нормальная или аффинная) может удержать альбуте-
рол. Методом проб и ошибок на этих пяти типах неподвижных фаз было проведено
исследование с целью найти такой растворитель для промывки при ТФЭ, который
лучше всего удалял бы примеси, не смывая при этом целевое вещество. Был
исследован ряд из 17 различных ТФЭ-сорбентов вышеуказанных пяти типов на предмет
наилучшего выделения целевого вещества с 23-мя системами растворителей.
Некоторые растворители почти не смывали альбутерол из отдельных ТФЭ-картриджей, и
эти растворители были взяты на заметку с целью их возможного использования в
качестве промывочных растворителей на этапе 3 {рис. I6.4ff).
По окончании экспериментов четыре ТФЭ-картриджа (таблица 16.7) были
отобраны для дальнейшей работы. Эти сорбенты изначально казались
многообещающими. Экстракты имели низкий уровень эндогенной плазмы, приемлемый уровень
извлечения альбутерола и хорошую совместимость с условиями ВЭЖХ-разделения. Две
фазы (циано и силикагельная) оказались подходящими. Это показано на рис. 16.5.
При использовании этой методики альбутерол сильно удерживался в процессе
промывки (рис. 16.4в; этапы 4 и 5 на рис. 16.5); для элюирования потребовалось 0,5%
1 М NH4OAC в МеОН (рис. 16.4г; этап 6 на рис. 16.5).
Таблица 16.7. Результаты ТФЭ-выделения альбутерола из плазмы крови
Тип ТФЭ-
картриджа
Циано
Силикагель-
ный
Фенилборо-
натный
С|8
Элюент
19% 1 М NH4OAc +
90% МеОН
Такой же, как и
выше
0,1 М H2S04
Изопропанол
Полнота
выделения, %
89
94
90
92
Комментарии
Очищенный экстракт; малый объем;
приемлемые результаты
Очищенный экстракт; малый объем;
приемлемые результаты
Очищенный экстракт; малый объем, но элюиру-
ющий растворитель слишком кислый
для ВЭЖХ-разделения, неприемлемый результат
Недостаточно очищенный экстракт;
обогащение следовых количеств нестабильно;
неприемлемый результат
16.6.7. Специальные виды ТФЭ
16.6.7.1. Мультимодальная ТФЭ и ТФЭ со смешанными фазами
Большинство процедур ТФЭ включают использование разделений одного типа
(например, на обращенной фазе) и одно приспособление для его проведения
(например, картридж). Тем не менее, если интерес представляет более, чем один тип
целевого вещества или если для устранения примесей не хватает селективности,
мультимодальная ТФЭ может оказаться полезной. Мультимодальная ТФЭ
представляет собой заранее запланированное использование двух (или более)
последовательных типов разделений или двух картриджей (например, упакованных обращенной
фазой и ионообменником). Имеется два экспериментальных подхода к мультимода-
льной ТФЭ. В поэтапном подходе два (или более) ТФЭ-картриджа соединены
последовательно. Таким образом, для раздельного выделения кислот, сильных оснований
и нейтральных веществ можно последовательно соединить анионо- и катионообмен-
ный картриджи. При рН раствора образца и промывочного растворителя равном 7
кислоты и основания полностью ионизированы. В результате кислоты будут
удерживаться на анионообменном картридже, основания — на катионообменном, а ней-
16.6. Твердофазная экстракция (ТФЭ) 827
Циано ТФЭ-
картридж
Силикагельный
ТФЭ-картридж
1. Кондиционирование:
1 мл МеОН
2. Кондиционирование:
1 мл Н20
3. Нанесение:
1мл плазмы
4. Промывка:
1 мл Н20
5. Промывка:
1 мл ACN
тральные вещества будут проходить через
оба картриджа и таким образом отделятся
от кислот и оснований. Кислоты и
основания могут быть собраны отдельно
на выходе из каждого картриджа.
Другой подход к мультимодальной
ТФЭ — использование смешанных фаз.
В этом случае сорбент, упакованный
в один картридж, может иметь две (или
более) функциональные группы для
удерживания различных типов соединений
или для обеспечения специфической
селективности. Одним из широко
используемых приложений мультимодальной ТФЭ
является выделение наркотиков и других
лекарственных средств из биологических
жидкостей |48|. Еще одной версией
мультимодальной ТФЭ является
использование нескольких различных сорбентов,
упакованных слоями |49|, где два (или
более) различных слоя используются
для выделения различных типов молекул.
16.6.7.2. Адсорбенты
с ограниченно доступной
поверхностью (ОДП)1
ОДП — это особый класс ТФЭ-адсорбен-
тов, используемых для прямого введения
биологических жидкостей, таких, как
плазма крови, ее сыворотка или
собственно кровь в устройства, упакованные
такими неподвижными фазами. В
отличие от ТФЭ-картриджей, ОДП-сорбенты
фактически представляют собой
ВЭЖХ-сорбенты, и упакованные ими
колонки характеризуются высоким числом
теоретических тарелок и возможностью
повторного использования. Но
одновременно с разделением на них выполняется
и пробоподготовка. ОДП-сорбенты чаще
всего выбирают для анализа
низкомолекулярных лекарственных средств, их
примесей и метаболитов в биологических
жидкостях [50—52|. В литературе описаны различные модификации этих сорбентов:
(I) с обращенной фазой на внутренней поверхности пор, (2) экранированные2
гидрофобные фазы, (3) полупроницаемые поверхности, (4) двухзонные фазы и (5)
смешанные функциональные фазы. См. |50|, где описаны и сведены в таблицы
коммерчески доступные фазы.
6. Элюирование:
2 мл 99.5% МеОН
+ 0.5% NH4OAc
7. Сбор элюата и
упаривание досуха
8. Рекуперация:
МеОН
Рис. 16.5. Выделение альбутерола из
плазмы крови с помощью твердофазной
экстракции (ТФЭ)
'Англ. Restricted access media, RAM. Эти адсорбенты работают в смешанных адсорбцион-
но-эксклюзионных режимах. — Прим. перев.
2Гидрофильным внешним слоем. — Прим. перев.
828 Глава 16. Профподготовка
Бифункциональные пористые сорбенты применяются при упаковке большинства
используемых ОДП-колонок. Как правило, эти сорбенты применяются для анализа
лекарственных средств, содержащихся в крови потому, что белки, присутствующие
в этих образцах, накапливаются в ОФХ-колонке, и это приводит к тому, что она
не сможет работать уже после нескольких вводов образца. Сорбент состоит из частиц
с маленькими порами, внутренняя поверхность которых покрыта слоем лигандов Cg
или Си Наружную поверхность частиц покрывает не удерживающий белки
гидрофильный слой. Белки не могут проникнуть в поры из-за больших размеров молекул
и не удерживаются поверхностью частиц — следовательно, они проходят через
колонку с к х 0. Низкомолекулярные целевые вещества могут проникать в поры и
удерживаться так, чтобы элюироваться после белков (часто с помощью градиентного
элюирования). Поскольку белки не удерживаются на такого рода сорбентах, через
упакованные ими колонки можно пропустить значительное количество образцов.
Тем не менее, необходимо соблюдать осторожность при выборе рН подвижной фазы
и органического растворителя. В противном случае может произойти осаждение
белка в колонке и, следовательно, ее загрязнение.
В качестве альтернативного варианта, ОДП-колонка может быть соединена через
переключающий кран с обычной ОФХ-колонкой (двумерное разделение;
переключение колонок, раздел 16.9). Кран изначально стоит в положении, при котором проис
ходит элюирование ОДП-колонки в слив. При этом вводится образец. После того,
как белки элюируются с колонки, ее переключают на соединение с ОФХ-колонкой.
Далее целевые вещества элюируются с ОДП-колонки и поступают в ОФХ колонку
для дальнейшего разделения, которое обычно происходит с помощью градиентного
элюирования. ОФХ-колонка при таком режиме работы живет дольше, поскольку
белки крови никогда с ней не контактируют (см., например, |53|).
16.6.7.3. Молекулярно-импринтированные полимеры (МИП)1
МИП являются одними из наиболее селективных неподвижных фаз, используемых
в ТФЭ. Они предназначены для удерживания специфических целевых веществ.
МИП представляет собой стабильный полимер, у которого есть сайты
распознавания, адаптированные к трехмерной структуре и функциональным группам целевого
вещества (так же, как и в случае связывания антител). Наиболее распространенный
подход включает в себя нековалентный импринтинг. Схема синтеза МИП с
использованием этого подхода показана на рис. 16.6. Целевое вещество используется в
качестве матрицы. Оно химически связано с мономером (чаще всего с метакриловой
кислотой или метакрилатом; рис. [6.6а,6). После полимеризации связанное целевое
вещество отщепляется, и образуется селективный сайт связывания (рецептор;
рис. 16.6г). Селективные взаимодействия между целевым веществом и МИП
включают в себя водородные связи, ионные и/или гидрофобные взаимодействия. МИП
действует по принципу «ключ-замок». Селективный рецептор или полость на
поверхности полимерной частицы сорбента идеально подходят для целевого вещества,
которое использовалось для синтеза МИП. Эта концепция похожа на принцип
действия иммуноаффинных (ИА) ТФЭ-сорбентов (раздел 16.6.7.4). Однако, получение
антител для таких ИА-сорбентов может потребовать много времени. Вводная статья
|54| описывает основы МИП-технологии, а обзорные работы |55—58] и книга |59|
дают детальную информацию об использовании и потенциале МИП в ТФЭ.
Одна из основных проблем МИП-технологии — неполное отщепление матрицы
целевого вещества от МИП во время его получения. Это остаточное количество
целевого вещества мало-помалу вымывается, что приводит к дрейфу базовой линии и
влияет на качество анализа целевого вещества, особенно при низких его концентрациях.
'Англ. Molecular-Imprinted Polymers, MIPs. — Прим. персе.
16.6. Твердофазная экстракция (ТФЭ) 829
(я) №)
Рис. 16.б. Синтез молекулярно-импринтированного полимера (МИП). (а) Целевое
вещество + мономеры; {6) образование комплекса целевое вещество —
полимер; (в) комплекс целевое вещество-полимер; (г) сайт селективного
связывания
При смене растворителя МИП может немного набухать или сжиматься, что может
привести к изменению размера рецептора и уменьшению удерживания целевого
вещества. Главным недостатком МИП-подхода является то, что каждый сорбент должен
быть изготовлен на заказ — или самим пользователем или сторонним производителем.
Из-за высокой стоимости МИП их применение ограничено анализами больших
объемов образца или теми случаями, когда нет другого способа его (образец) очистить.
Недавно был выпущен ряд коммерчески доступных МИП (МТР Technologies,
Lund, Швеция) для определения:
• кленбутерола в биологических жидкостях;
• бета-агонистов, содержащихся в моче и образцах тканей;
• НАПБ (4-метилнитрозамино-1-<3-пиридил)-6утанола), специфичного для табака
нитрозамина, содержащегося в биологических жидкостях;
• рибофлавина (витамина В2) в водных образцах;
• триазинов, содержащихся в воде, почве и пищевых продуктах;
• хлорамфеникола, антибиотика, содержащегося в биологических жидкостях;
• бета-блокаторов, содержащихся в воде и биологических образцах.
16.6.7.4. Иммуноаффинная экстракция малых молекул
Иммуноаффинные сорбенты содержат антитела, прикрепленные к частицам
сорбента. Как и в случае с МИП, целевое вещество удерживается чрезвычайно
комплементарным ему рецептором, обеспечивающим взаимодействие по принципу
«ключ-замок». В результате иммуноаффинные сорбенты весьма специфичны к отдельным
веществам. Их используют для селективного одностадийного извлечения и
концентрирования отдельных соединений и классов соединений, содержащихся в образце.
Антитела к крупным биомолекулам легко доступны и используются в течение
многих лет в иммунологии и медицинских исследованиях (аффинная хроматография).
Поскольку антитела для небольших молекул получить сложнее, развитие
низкомолекулярной иммуноаффинной экстракции задержалось. В ряде прекрасных обзорных
работ иммуноаффинная экстракция описана более подробно |60—64|.
830 Глава 16. Профподготовка
Пока есть возможность получить антитела, количество иммуноаффинных
сорбентов может быть практически неограниченным. Однако, для их получения требуется
много времени и усилий, поэтому их использование ограничено по тем же причинам,
что и использование МИП (раздел 16.6.7.3). Тем не менее, несколько иммуноаффиных
сорбентов все же коммерчески доступны. Для различных фармацевтических, пищевых
и экологических методик доступны сорбенты, специфичные к конкретному классу
соединений [65|. В качестве примера использования иммуноаффинного сорбента
описана методика разделения группы из четырех афлатоксинов [66|.
16.6.7.5. QuEChERS и дисперсионная ТФЭ
QuEChERS
1. Отвесить 10 г
образца в пробирку
объемом 50 мл
2. Добавить
ЮмлАСМ
Взбалтывать 1 мин
3. Добавить 4 г
MgSQ4 + 1 г NaCI
Взбалтывать 1 мин
4. Добавить раствор
внутреннего
стандарта
Взбалтывать 30 сек;
отцентрифугировать
5. Отобрать
аликвоту. добавить
MgS04 + сорбент
(дисперсионная ТФЭ)
Взбалтывать 30 сек;
отцентрифугировать
6. Отрегулировать или
стабилизировать рН
(если образец
нестабилен)
7. Ввести и
проанализировать
Рис. 16.7. Метод QuEChERS
(Quick, Easy, Cheap, Effective,
Rugged, and Safe) — быстрая,
простая, дешевая, эффективная,
надежная и безопасная экстракция
пестицидов из образцов овощей,
содержащих большое количество воды
QuEChERS (Quick. Easy, Cheap. .Effective,
Hugged, and Safe) — быстрая, простая, дешевая,
эффективная, надежная и безопасная техника
экстракции, предназначенная для пробоподготовки
пестицидов из образцов с высоким содержанием
воды, таких как овощи |67|. В методике
используется простая стеклянная посуда и минимальное
количество органического растворителя, к
которому последовательно добавляется соль, затем
буфер и ТФЭ-сорбент (рис. 16.7). Сначала к
гомогенизированному образцу овоща добавляют
гидрофильный растворитель — такой как ацето-
нитрил или ацетон. Это позволяет
экстрагировать пестициды в органический растворитель
(шаг 2 на рис. 16.7). Последующее добавление
соли (шаг 3) приводит к удалению воды из
органического растворителя, которая попала туда
из образца, что способствует переходу
пестицидов в органический растворитель. Следующим
добавляется внутренний стандарт (шаг 4) и,
после встряхивания и центрифугирования,
аликвоту органической фазы подвергают дальнейшей
очистке, используя дисперсионную ТФЭ: к
экстракту присыпают небольшие количества
ТФЭ-сорбента (например, C|g, графитированный
углерод, аминофазу)1. Делается это для того,
чтобы удалить из экстракта примеси (шаг 5). После
очистки образца супернатант отбирают и
анализируют (шаг 7). Шаг 6 является дополнительным
в случае пестицидов, которые нестабильны при
промежуточных значениях рН.
Было установлено, что методика QuEChERS
особенно полезна для скрининга продуктов
питания на наличие нескольких пестицидов.
Существуют методики QuEChERS, утвержденные
Ассоциацией химиков-аналитиков, состоящих
на государственной службе (Association of Official
Analytical Chemists, AOAC, США). В настоящее
время доступен метод 2007.01 и утвержденный
' Обычно супернатант добавляют в пробирку с заранее насыпанным сорбентом. — Прим. перев.
}6.7. Мембранные технологии в пробоподготовке 831
DIN (Deutsches Institut tiir Normung, Немецким институтом по стандартизации)
европейский стандарт метода EN 15662. С помощью QuEChERS определяют
более пятисот пестицидов в различных фруктах и овощах |63,65,68]. Извлечение
целевого вещества (для концентраций 100 нг/г) обычно составляет от 70 до 110%
с дисперсией менее 10%. Методика была распространена на мясные и
рыбные продукты, а также на определение антибиотиков и других лекарственных
средств [69—701.
16.6.7.6. Специализированные ТФЭ-картриджи для различных
классов соединений
За годы существования ТФЭ были предложены сорбенты, специфичные к отдельным
соединениям или к различным классам соединений. В то время как МИП и иммуно-
аффинные сорбенты (разделы 16.6.7.3, 16.6.7.4) могут обеспечить высокую степень
специфичности или селективности, специализированные сорбенты со
специализированными функциональными группами могут избирательно взаимодействовать
с определенными классами соединений. В основных условиях иммобилизованная
фенилбороновая кислота (ФБК) селективно связывает соединения, содержащие ви
цинальные диолы (например, сахара и катехолы). Селективно удерживаются также
альфа-гидроксикислоты, ароматические о гидроксикислоты, амиды и соединения,
содержащие аминоспирты. Образуются ковалентные связи между сорбентом и
растворенным веществом, что позволяет вымыть различными растворителями примеси
из сорбента. После промывки ковалентные связи могут быть разорваны с помощью
промывки кислотным буфером/растворителем, гидролизующим ковалентные связи.
Широко используется метод выделения катехоламинов из биологических жидкостей
с применением ФБК-сорбентов |41|.
16.7. Мембранные технологии
в пробоподготовке
За исключением фильтрационных и ТФЭ-мембран, мембранные технологии не
нашли широкого применения в подготовке образцов для В.ЭЖХ. Благодаря размеру
своих микропор, микропористые полупроницаемые мембраны позволяют проводить
селективную фильтрацию. По сравнению с ТФЭ или ЖЖЭ разделения с помощью
мембран происходят медленнее и вероятность того, что концентрация целевого
вещества увеличится в результате этих процессов на порядки, куда меньше. Успешное
применение мембраны возможно либо при концентрировании (обогащении
следовых количеств), либо при изменении химического состояния целевого вещества
(например, при переходе от заряженного к незаряженному состоянию). Преимущество
методов мембранного разделения в случае анализов с использованием ОФ-ВЭЖХ
заключается в том, что как донорные, так и акцепторные растворы обычно
представляют собой воду или водные буферы. Целевые вещества могу проникать через
мембрану с помощью химических или электрохимических градиентов. Мембранные
разделения могут проводиться как в непроточной, так и в проточной системах,
причем последняя легче поддается автоматизации. Ультрафильтрация, обратный осмос,
диализ, микродиализ и электродиализ — примеры технологий, использующих
мембраны для концентрирования, очистки, и разделения целевых веществ. Для
получения более подробной информации см. [711.
832 Глава 16. Профподготовка
16.8. Методы пробоподготовки твердых
образцов
Образец должен быть в жидком состоянии перед ВЭЖХ анализом. Некоторые
нерастворимые твердые частицы содержат растворимые твердые вещества — такие,
как добавки в твердом полимере, жиры в пищевых продуктах и полиароматические
углеводороды в почве. Обработка образца растворителем позволяет экстрагировать
целевые вещества в растворитель. Растворитель отделяют от твердого остатка
декантированием, фильтрованием или центрифугированием, а фильтрат перед ВЭЖХ ана
лизом подвергают дальнейшей обработке, если это необходимо. В таблицах 16.8 и
16.9 обобщены некоторые методы, используемые для экстракции («вымывания»)
растворимых целевых веществ из твердой матрицы.
Таблица 16.8. Традиционные методы подготовки твердых образцов
Метод пробоподготовки
Экстракция твердое
вещество — жидкость
Экстракция в аппарате
Сокслета
Гомогенизация"
Обработка
ультразвуком"
Растворение"
Принцип метода
Образец и растворитель
помещают в закрытый
контейнер и перемешивают;
получившийся раствор фильтруют
и отделяют твердое вещество
(иногда этот метод
называется «встряхивание колбы»)
Образец помещен в гильзу
экстрактора; поток
растворителя из обратного
холодильника проходит через гильзу,
растворяет целевые вещества,
которые непрерывно
собираются в колбе с кипящим
растворителем (разделы 16.8.1,
16.8.2.1)
Образец и растворитель
гомогенизируют в блендере или
с помощью механического
гомогенизатора до тонко
измельченного состояния;
растворитель отделяется и с ним
работают дальше
Использование ультразвука
для интенсификации
перемешивания на поверхности
тонкоизмельченного твердого
материала; можно
использовать ультразвуковой зонд или
ультразвуковую панну
Образец растворяют и
используют непосредственно
в виде раствора после
химической обработки или без нее
Комментарии
Для улучшения растворимости
экстракцию иногда проводят кипящим
растворителем или с обратным
холодильником; для того, чтобы процесс
экстракции прошел быстрее, образец должен
быть тонко измельчен
Экстракция происходит во время
контакта с чистым растворителем; образец
должен быть стабилен в точке кипения
растворителя; процесс медленный, но
экстракция проходит до своего
завершения без участия оператора; степени
извлечений высокие; (используется как
стандарт, с которым сравниваются
другие методы экстракции из твердых
образцов)
Используется при работе с
растительными и животными тканями,
продуктами питания; может быть использован
органический или водный
растворитель; для удобства работы с образцами
добавляют сухой лед или кизельгур
Для увеличения скорости процесса
можно использовать подогрев;
безопасный, быстрый метод; лучше всего
подходит для грубых гранулированных
материалов
Некоторые образцы могут не
растворяться сразу (например, может
потребоваться ферментативное расщепление или
другая предварительная обработка);
после растворения может потребоваться
фил ьтрован ие
"В тексте не обсуждаются.
16.8. Методы пробоподготовки твердых образцов 833
Таблица 16.9. Современные методы экстракции из твердых образцов
Метод
предварительной обработки образца
Сверхкритическая
флюидная экстракция
(СФЭ)
Жидкостная
экстракция под давлением
(ЖЭД )/ускоре иная
экстракция
растворителем (УСЭР)
Экстракция в
автоматическом аппарате
Соке лета (ЭАС).
Сочетание
микроволнового нагревания и
экстракции (МЭ)
Диспергирование
твердофазной
матрицы (ДТМ)
Принцип метода
Образец помещается в проточную
ячейку и сверхкритический флюид
(например. СО2) пропускают сквозь
образец. После сброса давления
собирают экстрагированное целевое
вещество (раздел 16.8.2.2)
Образец и экстрагент помещают
в герметичный контейнер и нагревают
выше температуры кипения
растворителя, вызывая повышение давления
в контейнере. Экстрагированный
образец извлекают и переносят в виалу
для дальнейшей обработки
Сочетает промывку горячим
растворителем и экстракцию в аппарате Со-
кслета; гильзу с образцом сначала
погружают в кипящий растворитель, а
затем поднимают, чтобы продолжить
в режиме обычной
экстракции/промывки в аппарате Сокслета
(раздел 16.8.2.1)
Образец помещают в открытый или
закрытый контейнер и нагревают с
помощью микроволн (раздел 16.8.2.4)
Привитая фаза используется в
качестве абразива для разрушения структуры
образца (архитектуры его матрицы) и
как «связанный» растворитель для
полного разрушения образца во время
смешивания образцов
Комментарии
Изменяя плотность и внося
модификаторы в растворитель
можно влиять на полярность
СФЭ- экстра ге нта
Значительно увеличивается
скорость экстракции из твердых
частиц; экстракция может быть
автоматизирована; получается
разбавленный раствор
экстрагированного вещества, поэтому
требуется его концентрирование
Используется меньше
растворителя, чем в традиционной
экстракции в аппарате Сокслета;
время экстракции уменьшается
благодаря разделению процесса
на два этапа
Закрытый контейнер позволяет
использовать давление для
улучшения процесса экстракции
Твердый или вязкий образец
(и 0,5 г) вместе с 2 г ТФЭ-сор-
бента (например, С is)
гомогенизируется растиранием в
ступке или в другом подходящем
контейнере. Смесью заполняют
пустую колонку и целевые
вещества элюируют растворителем
16.8.1. Традиционные методы экстракции
Ни один метод экстракции нельзя использовать для всех образцов. В таблице 16.8
перечислены несколько традиционных методов предварительной обработки твердых
образцов. Экстракция в аппарате Сокслета используется уже более ста лет,
проверена временем и принята большинством ученых. Перечисленные классические методы
экстракции из твердых образцов утверждены контролирующими органами, такими
как Агенство по охране окружающей среды (ЕРА, США), Управление по
санитарному надзору за качеством пищевых продуктов и медикаментов (FDA, США) и
соответствующими инстанциями других стран. Однако в более старых методах часто
используются большие количества органических растворителей, поэтому в последнее
время в методиках пробоподготовки появилась тенденция сокращения масштабов
(миниатюризации). Экстракция в аппарате Сокслета как старейший вид
эффективной экстракции является признанным стандартом для сравнения с более новыми
методами экстракции из твердых образцов.
((((834 Глава 16. Профподготовка
Существует много видов экстракции растворителем. Метод «встряхивания
колбы», в котором к образцу добавляется растворитель, а потом суспензия
взбалтывается, хорошо работает, если целевое вещество хорошо растворяется в экстрагенте,
а образец достаточно пористый. Для увеличения скорости экстракции образец
следует тонко измельчать. Экстракцию может ускорить нагревание, в частности
нагревание образца в кипящем растворителе с обратным холодильником. Обработка
ультразвуком часто обеспечивает более эффективный контакт жидкости с твердыми
частицами, а также небольшое нагревание, что тоже способствует экстракции,
которая проходит быстрее и более полно. Обработка ультразвуком рекомендуется
для пробоподготовки многих твердых образцов из окружающей среды, в частности в
методе 3550 ЕРА (США) [72| для экстракции нелетучих и полулетучих органических
соединений из образцов почв, осадков и отходов. В этом методе рекомендуются
различные растворители для экстракции и разные условия обработки ультразвуком в
зависимости от типа экстрагируемых веществ и их содержания в образцах.
Экстракция в аппарате Сокслета является наиболее распространенным методом
экстракции из твердых веществ. В соответствии с этой методикой твердый образец
помещается в гильзу экстрактора (одноразовую пористую емкость из плотной
фильтровальной бумаги), и гильза устанавливается в аппарат Сокслета (рис. 16.8).
Растворитель для экстракции нагревается до кипения с обратным холодильником, потом
конденсируясь, стекает в гильзу и экстрагирует растворимые вещества, подлежащие
анализу. Конструкция аппарата такова, что экстракт сливается обратно в колбу с ки
Холодильник
Экстра кционная
гильза
Образец
Сифонная
трубка
Емкость с
растворителем
Рис. 16.8. Аппарат Сокслета для экстракции
16.8. Методы пробоподготовки твердых образцов 835
пищим растворителем каждый раз, когда камера с гильзой заполняется
растворителем до верхнего уровня сифона. Процесс повторяется до тех пор, пока все целевое
вещество не переносится из образца в колбу. Поскольку образец находится в
кипящем растворителе, он должен быть стабильным при повышенных температурах.
Для экстракции из твердого образца, находящегося в гильзе, используется только
очищенный (перегнанный) растворитель, поэтому эффективность экстракции
возрастает по сравнению с методом «встряхивания в колбе». Это является основным
преимуществом классической экстракции в аппарате Сокслета. Разработка метода
экстракции в данном случае заключается в том, чтобы подобрать летучий растворитель
(например, имеющий точку кипения < 100 °С), в котором хорошо растворяется
подлежащее анализу вещество, но плохо растворяется твердая матрица образца.
Обычно экстракция в аппарате Сокслета происходит медленно (12—24 часа или
дольше), но процесс проходит без участия оператора. Далее описано (раздел 16.8.2.1),
как можно ускорить экстракцию в 8—10 раз в современных экстракторах Сокслета.
В традиционном экстракторе используются сотни миллилитров очень чистого (и
дорогостоящего) растворителя, но имеются экстракторы и гильзы небольшого объема
для образцов в несколько миллиграммов. Необходимое количество целевого вещества
определяется его концентрацией, массой, нужной для получения репрезентативного
образца, и чувствительностью детектора хроматографической системы.
16.8.2. Современные методы экстракции из твердых
веществ
В таблице 16.9 перечислены несколько современных методов экстракции из твердых
образцов.
16.8.2.1. Экстракция в современном аппарате Сокслета
Классическая экстракция в аппарате Сокслета была усовершенствована с целью
сокращения времени экстракции в 8—10 раз [73]. В автоматическом аппарате гильза
с образцом опускается и поднимается для того, чтобы образец либо находился в
кипящем растворителе, либо экстрагировался обычным образом. Сначала образец
полностью погружается в кипящий растворитель, и тогда при более высокой
температуре кипящего растворителя экстракция целевого вещества ускоряется благодаря
увеличению его растворимости и возрастанию скорости диффузии. Затем для того,
чтобы вымыть оставшийся экстракт из образца, гильза поднимается над кипящим
растворителем, и продолжается экстракция в обычном для аппарата Сокслета
режиме. После завершения экстракции растворитель упаривается для концентрирования
целевого вещества. Экстракция в автоматическом аппарате Сокслета утверждена ЕРА
в качестве метода, пригодного для экстракции подлежащих анализу веществ из
твердых образцов почвы, осадков, отложений и отходов (метод 354! [74|).
Были предложены дополнительные усовершенствования описанной выше
методики для ускорения экстракции. При обработке твердых образцов из окружающей
среды время экстракции в аппарате Сокслета можно еще сократить, используя
направленный микроволновой подогрев растворителей, поглощающих микроволны
175, 76|. Если в качестве экстрагента применять воду, весь процесс наносит гораздо
меньше вреда окружающей среде [77].
16.8.2.2. Сверхкритическая флюидная экстракция (СФЭ)
Когда была разработана сверхкритическая флюидная экстракция (СФЭ) в начале
1990 годов, предполагалось, что она решит все проблемы с экстракций из твердых
образцов. СФЭ представлялась безопасным методом, не требующим растворителей
836 Глава 16. Профподготовка
и позволяющим без затруднений удалить диоксид углерода, используемый
для солюбилизации подлежащих анализу веществ. Более того, по прогнозам СФЭ
должна была обеспечить более полную и быструю экстракцию из самых
разнообразных твердых образцов, таких как почва, песок, осадки, зольная пыль, продукты
питания и полимеры. До сих пор публикуются сообщения о новых приложениях
СФЭ, но с 2000 года в продаже не появились новые приборы или дополнительные
устройства для существующих приборов. В то время как ТФЭ — важный метод
очистки промышленного масштаба, для аналитических целей (включая пробо-
подготовку образцов) она используется сравнительно редко. Можно привести
много причин того, почему ТФЭ все меньше используется для пробоподготовки,
но это выходит за рамки данной книги. Применение ТФЭ подробно освещено
в |78-82|.
16.8.2.3. Жидкостная экстракция под давлением
(ЖЭД)/ускоренная экстракция растворителем (УСЭР)
Изначально разработанный фирмой Dtonex (Sunnyvale, CA) метод был назван УСЭР,
впоследствии по инициативе ЕРА он получил обобщенное наименование ЖЭД.
В настоящее время ЖЭД рассматривается как современная альтернатива экстракции
по Сокслету — степень извлечения целевого вещества близка к получаемой в
аппарате Сокслета, но растворителя потребляется меньше, и процесс занимает от 10
до 20 минут. Возможная масса образца от 1 до 30 г, а ячейки для экстракции имеют
объем 1 —100 мл. В ЖЭД используются такие же растворители, как в классической
экстракции по Сокслету и при обработке ультразвуком, поэтому разработать метод
ЖЭД несложно. Тонко измельченный образец помещается в ячейку для экстракции,
установленную в термостате, и в нее насосом подается растворитель из одного или
более резервуаров. За один цикл можно провести экстракцию только из одного
образца, но имеется автосамплер, который позволяет обработать 24 образца в
автоматическом режиме.
Экстракцию можно проводить в статическом и/или динамическом режиме. Если
экстракция заканчивается в статическом режиме, экстракт переносится в виал
для сбора пробы током азота. В результате ЖЭД образец оказывается в большом
объеме растворителя, поэтому требуется дополнительная стадия для
концентрирования раствора целевого вещества (например, ОФХ, упаривание). Утвержденный ЕРА
метод 3545А [83|, который называется «Жидкостная экстракция под давлением»,
предусматривает экстракцию нерастворимых или слабо растворимых в воде
органических веществ из твердых образцов почвы, глины, осадков, отложений и отходов.
Этот метод может быть также применен для экстракции полулетучих органических
соединений, таких как пестициды, гербициды, полихлористые бифенилы и
диоксины. В ЖЭД, называемой также экстракцией растворителем под давлением (РЭД),
используются повышенные температуры (100—180 °С) и давления (100—135 атм,
1500—2000 psi).
В качестве примера ЖЭД можно привести экстракцию активных ингредиентов
фармацевтических препаратов, полученных из натуральных продуктов |84|,
при 100 °С и 100 атм (1500 psi). Капсулы с образцом открывают, перемолотый
образец в количестве 1—3 г помещают в ячейки для экстракции. Для каждого образца
используется всего « 15 мл растворителя ацетонитрила, экстракция проходит за 14 мин.
Анализ экстракта проводится в режиме ОФХ с внешней калибровкой. Результаты
(ОСО 3—4%) сравнимы с получаемыми при экстракции по Сокслету и при
обработке ультразвуком, но скорость экстракции выше, растворителя требуется меньше,
и процесс полностью автоматизирован.
16.9. Переключение колонок 837
16.8.2.4. Экстракция с микроволновым нагреванием (МЭ)
Экстракция подлежащих анализу органических веществ с микроволновым
нагреванием (иногда называемая также ускоряемой микроволновым излучением
экстракцией растворителем) |85|, представляет собой метод, альтернативный
традиционной экстракции жидкость—твердое тело, описанной выше. Нагревание
с помощью микроволнового излучения имеет преимущество, так как раствори-
тель-экстрагент разогревается изнутри, а не за счет конвекции. Температура в
контейнере для экстракции определяется растворителем, используемым для
экстракции, а не заданным значением температуры во внешнем термостате или
нагревателе. По мере возрастания температуры растворителя в закрытом контейне
ре повышается и давление, поэтому экстракция происходит быстрее, чем при
нагревании благодаря конвекции.
Вода имеет значительную диэлектрическую проницаемость и легко поглощает
энергию микроволн, но органические растворители, такие как углеводороды, не
поглощают энергию микроволн и поэтому не нагреваются. Если экстракция с
микроволновым нагреванием проводится органическим растворителем, не поглощающим
энергию микроволнового излучения, требуется дополнительное нагревание, для чего
в емкость для экстракции помещают объекты, поглощающее микроволны. Обычно
это стержни или порошок поглощающего микроволны вещества, например газовой
сажи, с покрытием из инертного полимера (например, ПТФЭ) во избежание
загрязнения образца. В микроволновой печи можно одновременно обрабатывать много
образцов, поэтому пропускная способность этого метода очень велика по сравнению
с некоторыми другими методами жидкостной экстракции из твердого тела.
Коммерчески доступные системы позволяют одновременно обработать до 24 контейнеров
с образцами.
Экстракция с микроволновым нагреванием — широко применяемый метод,
альтернативный экстракции по Сокслету, ЖЭД/УСЭР и другим методам экстракции.
Например, ЕРА утвержден метод 3546 |86|, представляющий собой методику МЭ
для экстракции нерастворимых или слабо растворимых в воде органических
соединений (пестицидов, гербицидов, полихлористых бифенилов и др.) из почвы, глины,
осадков, отложений и твердых отходов. Экстракция происходит при температурах от
100 до 115 °С и давлениях от 3 до 12 атм (50—175 psi) в закрытых контейнерах с
образцом и органическим растворителем (растворителями). Степень извлечения
целевых веществ эквивалентна достигаемой в экстракции по Сокслету (метод 3540 |87|),
но при этом требуется меньше растворителя и гораздо меньше времени.
16.9. Переключение колонок
Переключение колонок обсуждалось в разделах 2.7.6, 3.6.4.1, 9.3.10 и 13.4.5.3. Это
эффективный метод пробоподготовки (обсуждаемый в настоящем разделе), а также
он используется в случае двумерного разделения (раздел 9.3.10). Для подготовки
образца часть элюата из первой колонки (колонка I; например, обогатительная
колонка на рис. 3.23) вводится во вторую колонку (колонка 2; например, аналитическая
колонка на рис. 3.23) для дальнейшего разделения. Переключение колонок в ходе
пробоподготовки используется для:
• удаления веществ, способных вывести из строя вторую колонку;
• удаления поздноэлюируемых соединений до разделения на второй колонке;
• удаления примесей, способных перекрыть зоны целевых веществ при разделении
на второй колонке;
• обогащения следовых количеств.
838 Глава 16. Пробоподготовка
Достижение одной или нескольких из этих целей приводит, как правило, к
увеличению скорости пробоподготовки и анализа вследствие того, что на анализ
поступает более чистый образец. Нежелательные примеси могут быть направлены в слив
или вымыты из колонки обратным током элюента, чтобы предотвратить их
попадание в ВЭЖХ-колонку. Колонки переключают для того, чтобы отделить целевое
вещество от примесей. То есть, цель в данном случае такая же, как и в ТФЭ.
Хотя переключение колонок аналогично ВЭЖХ-анализу фракций после ТФЭ,
у этого метода существует ряд преимуществ:
• ТФЭ-картриджи используются только один раз и выбрасываются; первая колонка,
участвующая в переключении, используется повторно;
• первая колонка более эффективна (например, диаметр частиц ее сорбента 5 мкм),
если сравнить ее с ТФЭ-картриджем (например, диаметр частиц сорбента,
которым упакован картридж, 40 мкм);
• при переключении колонок потери образца меньше;
• возможны самые различные варианты переключения крана с целью вырезания
зоны примесей, вымывания их обратным током элюента, непосредственным
направлением их в слив и т.д.
Переключение колонок и пример обогащения следовых количеств образца
обсуждаются также в разделе 3.6.4.1 и показаны на рис. 3.23.
16.10. Пробоподготовка в биохроматографии
При разделении биомолекул пробоподготовка почти всегда включает один или
несколько методов предварительной обработки. Как и в случае ВЭЖХ других типов
образцов, универсального метода пробоподготовки для биологических образцов
не существует. Методы пробоподготовки, используемые в современной
биохроматографии, зачастую представляют собой те же методы, которые использовались в
классической биохимии — диализ, химическое осаждение, колоночная хроматография и
центрифугирование. В настоящее время растет интерес не только к применению
этих классических подходов, но и к новым технологиям пробоподготовки в областях
молекулярной биологии, биотехнологии и различных «омик» (например, протео-
мики, геномики, метаболомики). В этих областях образцы часто сложны, доступны
в небольших количествах и требуют особой осторожности при обращении.
Необходимость выделения биополимеров с сохранением структурной и функциональной
целостности требует быстрой и щадящей подготовки образцов.
Сложная природа большинства биологических образцов вынуждает делать пробо-
подготовку для достижения лучших результатов разделения и продления срока службы
ВЭЖХ-колонки. Фактически рекомендуемая методика(и) пробоподготовки будет
зависеть от природы образца (например, от молекулярной массы, наличия добавок,
эндогенных примесей, нерастворимых частиц или других нежелательных компонентов).
Хроматографические методы (включая аффинный) могут быть использованы
для очистки биологических образцов как дополнение к фильтрованию для удаления
микрочастиц. В таблице 16.10 приведен список методов подготовки образцов,
которые могут быть использованы в проточном режиме с использованием картриджа,
диска или колонки. Некоторые из этих методов могут быть реализованы в режиме
периодического процесса. В этом случае сорбент добавляют в раствор образца,
оставляют их контактировать, как правило, при перемешивании, а затем жидкость
удаляют фильтрованием, при этом целевое вещество(а) сорбируется на неподвижной
фазе или же остается в жидкой фазе. Хотя такой процесс и медленнее того, который
проходит на колонке, сорбцию в периодическом режиме осуществить легче.
16.10. Профподготовка в биохроматографии 839
Таблица 16.10. Методы пробоподготовки в биохроматографии
Задачи
Очистка
антител
Глубокая
очистка
буферов и
реагентов
Деионизация
Обессоливание и замена
буфера
Удаление
детергента
Концентрирование или
удаление
ионов
металлов
Удаление
механических
примесей
Наиболее часто
используемые методы
Аффинная
хроматография'
хроматография на гидро-
ксиапатите
Ионный обмен
Адсорбция
Смешанный
ионный обмен
Ионный обмен
Гель-фильтрация
Ионное
удерживание
Обращенная фаза
Ионный обмен
Адсорбция
Ионное
удерживание
Ионный обмен
Хелатообразующие
сорбенты
Адсорбция
Фильтрование
Удерживаемые
соединения
IgG и их
подклассы
Следы катионов
и анионов
Следы
органических веществ
Ионы
Белки
Катионы,
анионы
Большие
молекулы элюируют-
ся раньше солей
и малых молекул
Катионы,
анионы
Гидрофобные
целевые
вещества
Катионные и
анионные
детергенты
Неионные
детергенты
Анионные
детергенты
Катионы
Поливалентные
катионы
М еталлоор га ни -
ческие
комплексы
Нерастворимые
частицы
Типичные цеди применения
Концентрирование IgG из сыворотки
крови, асцитов и культуральных сред;
отделение флуоресцентно меченых антител
от непрореагиропавших флуоресцентных
меток
Удапение ионов, приводящих к уширению
зон или к высокому фону базовой линии
Нейтральный ПС-ДВБ, окись алюминия и
силикагель удалят полярные органические
соединения из буферов; вода может быть
удалена из органических растворителей
Деионизация углеводов перед ВЭЖХ;
отделение ионных примесей от белков;
приготовление реагентов; отделение анионов
от углеводов, декстранов и многоатомных
спиртов
Деионизация белков, содержащих
гидрофобные молекулы
Обессоливание аминокислот с целью
улучшения качества ТСХ и ВЭЖХ анализов
Обессоливание белков и нуклеиновых
кислот с массами > 6000 Да
Удаление солей и ионных детергентов
из белковых и аминокислотных образцов
Обессоливание растворов полипептидов
Очистка белков, реактивация ферментов
Удаление тритона Х-100 из растворов
белков
Удаление избытка ДСН" из растворов
белков
Удаление ионов металлов и солей из
водных сред
Удаление ионов меди, железа, тяжелых
металлов, кальция и магния
Образование комплексов металлов с
полярными или с гидрофобными комплексо-
образующими агентами
Предварительная обработка, чтобы
защитить фильтры и краны/клапаны системы
ВЭЖХ; фильтрование культуральных сред
840 Глава 16. Профподготовка
Окончание табл. 16.10
Задачи
Очистка
плазм ид и
образцов
Концентрирование
белков
Удаление
или
концентрирование
анионов и
катионов
Удаление
или
концентрирование
органических
вешеств
Наиболее часто
используемые методы
Гель-фильтрация
Адсорбция
Ионный обмен
Ионный обмен
Адсорбция
Ионный обмен
Адсорбция
Гель-фильтрация
(органическими
элюентами)
Удерживаемые
соединения
Низкомолекулярные примеси
Большие ДНК
Бромистый зти-
диум или
йодистый пропидиум
Вода
Белки
Гидрофобные
белки
Катионы,
анионы
Полярные
органические
вещества
Высокомолекулярные
соединения элюируются
раньше
низкомолекулярных
Типичные цели применения
Удаление невключенных радиоактивных
нуклеотидов из реакционных смесей
для введения меток
Удаление РНК, белка и других клеточных
компонентов
Удаление из плазмид использованных
красителей
Повышение чувствительности электрофо-
ретического и ВЭЖХ-анализов
Разделение белков и низкомолекулярных
соединений
С]8-ТФЭ для удаления гидрофобных
белков из гидрофильных белков
Удаление ионов из водных растворов;
концентрирование крупных белков;
удаление минеральных кислот
Удаление неионных детергентов и липи-
дов; отделение бромистого этидиума
от препаратов нуклеиновых кислот
Отделение растворимых органических
соединений с массами < 150 000 Да от
образцов со сложными матрицами
"ДСН — додецилсульфат натрия.
Проточный режим применяется более широко. Хотя в проточном режиме
возможно как следствие разбавление раствора образца, подход, в котором используется
проточная колонка, более пригоден для извлечения следовых количеств целевого
вещества. Производители поставляют удобные, заранее упакованные картриджи и
мембранные диски с меньшим сопротивлением потоку вследствие большой площади
их поперечного сечения. Иногда поставляются целые наборы, содержащие сорбенты,
реагенты и приспособления, необходимые для проведения процесса очистки.
Жидкости можно прокачивать через проточные устройства под давлением, под вакуумом
или с помощью центрифугирования. В целом ряде методов пробоподготовки,
упомянутых в таблице 16.10, практикуется удерживание ионных соединений посредством
ионного обмена или удерживания ионов. Другие методы используют гидрофобное
взаимодействие и адсорбцию для удерживания макромолекул, позволяя в это же
самое время ионным соединениям и более мелким молекулам пройти, не
задерживаясь.
Помимо хроматографических подходов, описанных в таблице 16.10, очистка
биологических образцов, при условии сохранения биологической активности, может
осуществляться и другими методами. Например, диализ — это проверенный време-
16. П. Профподготовка в ЖХ-МС 841
нем процесс, в котором для очистки и обессоливания биологических образцов
используются мембраны. Миниатюризированные наборы для диализа позволяют
эффективно диализовать небольшие объемы образцов. Электродиализ — более быстрый
метод для обессоливания или замены буфера. Это щадящая технология,
обеспечивающая прекрасное обессоливание без потери биологической активности белков.
При ультрафильтрации (УФЦ) в качестве движущей силы мембранной
фильтрации используется центрифугирование. Мембранные фильтры с отсечкой по
молекулярной массе в десятки КДа помещаются в центрифужный картридж. При
центрифугировании растворитель, соли и небольшие молекулы проходят сквозь мембрану,
в то время как макромолекулы, имеющие молекулярную массу больше порогового
значения, удерживаются и концентрируются над мембраной. Поскольку
подбираются мембраны с низкой неспецифической адсорбцией, УФЦ приводит к высокой
степени извлечения целевых веществ и небольшой потере биологической активности.
Многие из уже описанных методик пробоподготовки, таких как ЖЖЭ
(раздел 16.5), могут непосредственно использоваться для фракционирования
биологических образцов. Зачастую комбинация двух методик приводит к повышению общей
эффективности очистки. Например, использование хроматографических сорбентов
в сочетании с диализом может обеспечить прекрасное концентрирование белковых
растворов.
С появлением протеомики биологи стали искать новые мишени лекарственных
средств и биомаркеры заболеваний в организме человека в условиях их очень низких
концентраций в сыворотке/плазме. К сожалению, в этих жидкостях организма
преобладают представляющие меньший интерес белки, такие как человеческий
сывороточный альбумин (ЧСА) и иммуноглобулин (IgG), концентрация которых на
несколько порядков выше, чем концентрация белков, которые необходимо
идентифицировать и определить количественно. Классические методы удаления белков
с высоким содержанием могут, как правило, понизить концентрацию лишь одного
или нескольких белков. Новые аффинные сорбенты могут удалить белки с высоким
и средним содержанием (от 6 до 20 белков), при этом остаются тысячи белков с
низким содержанием, для удаления которых требуется дальнейшая пробоподготовка.
Аффинные сорбенты, содержащие антитела, специфичные для белков с высоким
содержанием, поставляют упакованными в проточные и спиновые колонки |65|. Белки
с высокими концентрациями удерживаются колонкой, в то время как белки с
низкими концентрациями проходят через колонку для сбора и последующего
концентрирования с помощью методик обогащения следовых количеств. Белки с высокой
концентрацией потом смываются с колонки при замене буфера и могут быть либо
выброшены, либо сохранены. В конце концов, колонку регенерируют, и она может
быть использована несколько сот раз. Для изучения следовых количеств
активирующих и подавляющих белков в биологических жидкостях используется сочетание
аффинного связывания белков с высоким и средним содержанием и многомерной
ЖХ-МС/МС |88|.
16.11. Пробоподготовка в ЖХ-МС
Для достижения максимальной эффективности ЖХ-МС И ЖХ-МС/МС без
пробоподготовки, как правило, не обойтись. Когда МС/МС только был введен в практику,
многие верили, что его специфичность исключит необходимость пробоподготовки.
По этой причине ВЭЖХ рассматривали как нечто большее, чем инструмент для
ввода образца. Однако, было наивно полагать, что один МС-анализ может решить все
проблемы разделения. Последующие годы показали, что и хроматографическое раз-
842 Глава 16. Пробоподготовка
деление до детектора и пробоподготовка перед вводом образца были необходимы
для того, чтобы избежать определенных сложностей — таких, как ранее неизвестные
«матричные эффекты» в ЖХ-МС и ЖХ-МС/МС.
Пробоподготовка может использоваться для минимизации проблем, связанных
с образцом и/или матрицей образца, таких как:
• спектральные помехи;
• сбой системы;
• образование аддукта;
• подавление ионного сигнала.
Ионы, которые образуются с тем же или почти с тем же соотношением m/z, что и
целевой ион, могут вызвать спектральные помехи. Существуют различные конструкции
МС-детекторов (раздел 4.14) с различной способностью различать ионы с одинаковым
m/z. МС-детекторы, используемые для рутинного ВЭЖХ-анализа, имеют достаточно
низкое разрешение в области малых масс, и поэтому может потребоваться
предварительная пробоподготовка для удаления совместно элюируемых соединений, имеющих
значения m/z, близкие к значениям целевого вещества (веществ).
Сбой системы может произойти, когда компоненты нелетучего образца или
подвижной фазы осаждаются в ЖХ-МС интерфейсе и ухудшают его работу.
Компоненты подвижной фазы, которые с наибольшей вероятностью осаждаются в источнике
ионизации при атмосферном давлении во время ионизации электроспреем,
представляют собой буферные соли и ион-парные реагенты, поэтому для того, чтобы
этого не было, используются летучие буферы и реагенты. Элементы матрицы образцов,
такие как белки, тоже могут осаждаться в ЖХ-МС-интерфейсе. Белки чаще всего
удаляют из образцов высаживанием, ЖЖЭ (раздел 16.5) или ТФЭ (раздел 16.6).
Дополнительные способы удаления белков приведены в таблице 16.11.
Образование аддукта между целевым и другим ионом изменяет значение m/z,
при котором целевой компонент появляется в спектре. Аддукты могут быть как
вредными, так и полезными, но в любом случае необходимо контролировать количество
образующегося аддукта. Источником ионов аддукта, таких как натрий, калий,
аммоний, могут быть как сам образец, реагенты или даже контейнер, в котором находится
образец. Кроме того, образование аддуктов можно использовать как способ для
улучшения сигналов макромолекул. Тем не менее, неконтролируемое образование
аддукта все же нежелательно и может потребовать специальной пробоподготовки для того,
чтобы его уменьшить или устранить. Методы удаления ионов перечислены в
таблице 16.10.
Подавление ионного сигнала происходит в тех случаях, когда есть примеси,
подавляющие ионный сигнал целевого вещества (или конкурирующие с ним). Подавление
ионного сигнала является наиболее критической помехой МС-детектированию,
поскольку оно может быть вызвано соединениями, не проявляющими себя в масс-
спектре. Фосфолипиды представляют собой класс компонентов матрицы, которые
могут особенно сильно подавлять ионный сигнал. Избирательно удалять эти
соединения можно с помощью сорбентов на основе диоксида титана [101|. В
биологических образцах естественное разнообразие концентраций эндогенных соединений
может приводить к различным уровням подавления ионного сигнала. Последнее,
в свою очередь, приводит к тому, что ответный сигнал целевых соединений
неприемлемо изменяется. Другой тип подавления ионного сигнала возникает при
образовании очень сильно связанных ионных пар, которые не разрушаются в ХИАД-интер-
фейсе. Было показано, что ион-парные агенты, такие как трифторуксусная кислота,
способствуют подавлению ионного сигнала |102|, и поэтому следует по возможности
избегать их использования в ЖХ-МС. ЖЖЭ (раздел 16.5) и ТФЭ (раздел 16.6) — два
метода, которые обычно используют для удаления подавляющих ионизацию
соединений из образца.
/6.12. Дериватизация в ВЭЖХ 843
Таблица 16.11. Методы удаления белков из биологических жидкостей
Метод
удаления белков
Осаждение
Адсорбенты
с
ограниченно-доступной
поверхностью
(ОДП)
Хроматография
турбулентных потоков
И ОХ
ЭХ
ОФХ
Сильное
снижение
концентрации белков
Принцип работы
Органический растворитель (например, ACN), раствор кислоты
(например, хлорной) или раствор соли (например, сульфата
натрия) добавляют при перемешивании к раствору плазмы крови.
Белок осаждается и образует глобулу при центрифугировании. Су-
пернатант может быть проанализирован с помощью ВЭЖХ
Раствор, содержащий белок, вводится в ОДП-колонку для
отделения белка от низкомолекулярных соединений. См. раздел 16.6.7.2
Через обращен но-фазовый привитый силикагель с крупными
(г= 50 мкм) частицами, упакованный в колонку малого диаметра
(внутренний диаметр 1 мм) идет поток с высокими линейными
скоростями (до S мл/мин). Хотя это и не настоящий турбулентный
поток, но эти высокие линейные скорости не позволяют медленно
диффундирующим белкам проникать в поры сорбента, и они
вымываются в слив. Более мелкие молекулы удерживаются в порах и
алюируются в аналитическую колонку или в МС-интерфейс
Белки удерживаются на ионообменной колонке при надлежащем
рН; незаряженные низкомолекулярные соединения не
удерживаются и могут элюироваться в аналитическую колонку
При выборе пор подходящего размера белки могут исключаться и
элюироваться из колонки первыми, в то время как
низкомолекулярные соединения элюируются позже
Использование Gj- или С4-фаз, привитых на широкопористом си-
ликагеле, позволяет обеспечить удерживание белков, а менее
удерживаемые полярные лекарственные соединения могут быть элкж-
рованы первыми
Плазма/сыворотка крови человека, содержащая в большом
количестве белки (до 20) очищается от них с помощью аффинных фаз,
содержащих антитела. См. раздел 16.6.7.4
Литература
89-90
50-54
91-92
93-94
95-96
97-98
99-100
16.12. Дериватизация в ВЭЖХ
Дериватизация подразумевает химическую реакцию между целевым веществом и
химическим реагентом с целью изменения химических и физических свойств целевого
вещества. У дериватизации в ВЭЖХ четыре основных задачи:
• улучшение детектируемости;
• изменение молекулярной структуры или полярности целевого вещества для
улучшения процесса хроматографии;
• изменение матрицы образца для улучшения разделения;
• стабилизация целевого вещества.
6 идеале реакция дериватизации должна быть быстрой, количественной и давать
минимум побочных продуктов. Избыток реагента не должен мешать анализу или
должен легко удаляться из реакционной смеси.
С ростом популярности ЖХ-МС и ЖХ-МС/МС, особенно в области анализа
биомолекул, во многих лабораториях предпочитают этот метод, обладающий высокой
чувствительностью и селективностью детектирования, вместо того, чтобы иметь дело
с относительно длительной и трудоемкой дериватизацией соединений. Зачастую
844 Глава 16. Профподготовка
при разработке метода к дериватизации прибегают в последнюю очередь. Введение
пред- и постколоночной дериватизации усложняет анализы, увеличивает их
продолжительность и служит источником дополнительных ошибок. Хотя эти процедуры и могут
быть автоматизированы, аналитик должен убедиться, что реакция дериватизации
прошла количественно (если это необходимое условие) и что в анализ не вносятся
дополнительные примеси. Несмотря на то, что у дериватизации есть недостатки, она все же
может потребоваться для решения конкретной проблемы разделения или детектирования.
Например, в тех случаях, когда МС-детектирование недоступно, зато доступны
реагенты, селективно реагирующие с конкретными функциональными группами и
образующие производные, которые легче детектировать в УФ или в условиях флуоресценции.
Некоторые из наиболее распространенных реакционноспособных функциональных
групп перечислены в таблице 16.12. Реагенты перечислены втаблице 16.13. На рис. 4.13,
4.36 и 4.37 показаны примеры дериватизации для повышения чувствительности
флуоресцентного обнаружения. Помимо повышения чувствительности детектирования
дериватизация применяется также для обеспечения возможности разделения энантио
меров, например, с помощью реагентов, перечисленных в таблице 14.1. Для получения
дополнительной информации об использовании дериватизации в ВЭЖХ см. разделы
4.16 и 14.3 или обратитесь к одной из книг, ей посвященных 1103—108|.
Таблица 16.12. Функциональные группы и реагенты для дериватизации
Функциональная группа
Карболовые кислоты
Жирные кислоты;
Фосфоновые кислоты
Спирты
Альдегиды
Кетоны
Амины, 1"
Амины, 1° и 2°
Аминокислоты (пептиды)
Изоцианаты
Фенолы
Тиолы
Реагенты для получения
производных, детектируемых вУФ ">*
ПН БДИМ
ДНБДИМ
ПБФБ
ДНБХ
Да6сил-С1
НИЦ-1
ПНБА
ДНБА
ДНБХ
СНФА
СДНФА
Дабсил-С1
НИЦ-1
Дабсил-С1
ПНБА
ДНБПА
ДНБХ
Дабсил-С1
НИЦ-1
Дабсил-О
Реагенты для получения
флуоресцирующих производных "■*
ВгМаК
ВгМмК
Дансил гидразин
Флуорескамин ОФА
НБД-С1
НБД F
Дансил-С1
Флуорескамин
ОФА
НБД-С1
НБД-F
НБД-С1
НБД-С1
НБД-F
Дансил-С1
НБД-С1
НБД-F
ОФА
"См. таблицу 16.13 с перечислением реагентов для дериватизации.
^Обычно ароматические производные, облегчающие УФ-детектирование при 254 нм.
•Обычно ароматические производные, облегчающие флуоресцентное детектирование.
Литература 845
Таблица 16.13. Реагенты для дериватизации
Реагенты для получения производных, детектируемых
в УФ
Дабсил-О
ДНБА
НИЦ-1
ПБФБ
ПНБА
ПНБДИМ
ДНБДИМ
ПНБПА
ДНБПА
СНФА
СДНФА
ДНБХ
4- ди метил ам и нобен зол -4 -сул ьфе н ил
3,5-Динитробензилоксиамина гидрохлорид
1-Нафтилизоцианат
л-бромфенацил-бромид
л-Нитробензилоксиамин гидрохлорид
л-Нитро-Л^ЛР-диизопропилизомочевина
3,5-Динитро-,/У,ЛР-диизопропилизомочевина
л-Нитробензил-Л'-и-пропиламин
гидрохлорид
3,5-Динитробензил-Л/-к-пропиламин
гидрохлорид
yV-Сукцинимидил-л-нитрофенилацетат
Л'-Сукцинимидил-3,5-динитрофенил ацетат
3,5-Динитробензил хлорид
Реагенты для получения
флуоресцирующих производных
НБД-С1
НБД F
Флуоре-
скамин
ОФА
Дансил-С1
ВгМмК
ВгМаК
7-хлор-4-нитробензо-2-
окса-1,3-диазол
7-фтор-4-нитро6ензо-2-
окса-1,3-диазол
4'-фенилспиро |2-6ензофу-
ран-3,2'-фуран|-1,3'-дион
о-Фталевый альдегид
5-Диметиламинонафта-
лин-1-сульфонил хлорид
4-Бромметил-7-метоксику-
марин
4-Бромметил-7-ацетокси-
кумарин
Реакционная способность по отношению к функциональным группам указана
в таблице 16.12.
Литература
1. P. Gy, Sampling for Analytical Purposes, Wiley, New York, 1998.
2. D. C. Harris, Quantitative Chemical Analysis, 7th ed., Freeman, New York, 2003, pp. 701—795.
3. S. Mitra, ed., Sample Preparation Techniques in Analytical Chemistry, Wiley-lnterscience, Hoboken,
NJ, 2003.
4. J. R. Dean, Methods for Environmental Trace Analysis, Wiley, New York, 2003, ch. 3.
5. L. R. Snyder and S. van der Wal, Anal. Chem., 53 (1981) 877.
6. V. Meyer, LCGC, 20 (2002) 106.
7. D. A. Wells, High Throughput Bioanalyiical Sample Preparation: Methods and Automation, Elsevier.
Amsterdam, 2003.
8. F. Settle, Ed., Handbook of Instrumental Techniques for Analytical Chemistry, Prentice-Hall, Upper
Saddle River, NJ, 1997.
9. C. M. Druzik, D. Grosjean, A. Van Neste, and S. Parmar, Int. J. Environ. Anal. Chem., 38 (1990)
495.
10. M. Hiatt, Anal. Chem., 67 (1995) 4044.
11. L. R. Snyder, Chemtech, 9 (1979) 750.
12. L. R. Snyder, Chemtech, 10 (1980) 188.
13. T. S. Ma and V. Horak, Microscale Manipulations in Chemistry, Wiley, New York, 1976.
14. N. B. Mandava and Y. Ito, eds., Countercurrent Chromatography: Theory and Practice, Dekker,
New York, 1988.
15. J. A. Riddick and W. B. Bunger, Organic Solvents, Wiley-lnterscience, New York, 1970.
16. R. E. Majors and K. D. Fogelman, Amer. Lab., 25(2) (1993) 40W.
17. M. A. Jeannot and F. F. Cantwell, Anal. Chem., 68 (1996) 2236.
846 Глава 16. Профподготовка
18. Е. М. Gioti, D. С. Skalkos, Y. С. Fiamegos, and С. D. Stalikas, J. Chromatogr. A, 1093 (2005) 1.
19. J. M. Stevens, M. Crawford, M. Halvorson, and R. Woleb, Pittsburgh Conference, paper 1550-4,
2008.
20. W. F. Lane and R. С Loehr, Environ. Set. Technol., 256(5) (1992) 983.
21. D. Barcelo, Analyst, 116 (1991) 681.
22. P. D. Mac Donald and E. S. P. Bouvier, eds., Solid Phase Extraction Applications Guide and
Bibliography, Waters, Milford, MA, 1995.
23. F. X. Zhou, J. M. Thome, and I. S. Kroll, Trends Anal. Chem., 11 (1992) 80.
24. D. R. Green and D. Le Pape, Anal. Chem., 59 (1987) 699.
25. A. Alfred-Stevens and J. W. Eichelberger, in Methods for Determination of Organic Compounds in
Drinking Water (Supplement I), Environmental Monitoring Systems Laboraotroy, Office of R&D,
US EPA, Cincinnati, OH, 1990, pp. 33-63.
26. JAf Empore Extraction Disks Method Summary: Phenols, Pub. 78-6900-3714-4 (113.05) Rl, 3M
Corp., St. Paul, MN, 1994.
27. 3M Empore Extraction Disks Method Summary, EPA Method 552.1: Haloacetic Acids and Dalapon in
Drinking Water, Pub. 78-6900-373-4 (113.05) Rl, 3M Corp., St. Paul, MN, 1994.
28. 3M Empore Extraction Disks Method Summary: Pesticides and Polychlorinated Biphenyls, Pub.
78-6900-3715-1 (113.05) Rl, 3M Corp., St. Paul, MN, 1994.
29. G. M. Hearne and D. O. Hall, Amer. Lab., 25(1) (1993) 28H.
30. R. Majors, LC/GC, 23 (2005) 358.
31. R. E. Majors and A. Shukla, LC/GC, 23 (2005) 646.
32. A. A. Boyd Boland and J. B. Pawliszyn, Anal. Chem., 68 (1996) 1521.
33. R. Shirey, Advances and Applications of Solid Phase Microextractios (SPME), paper 23602 at Analy-
tica, Munich, 1996, pp. 23—26.
34. E. Balrussen, P. Sandra, F. David, and C. A. Cramers, J. Microcol. Sep., 11 (1999), 737.
35. B. A. Bidlingmeyer, J. Liq. Chromatogr., 2 (1984) 578.
36. T. A. Dirksen, S. M. Price, and S. J. St. Mary, Amer. Lab., 25(18) (1993) 24.
37. G. E. Platoffand J. A. Gere, Forens. Sci. Rev., 3 (1991) 117.
38. X.-H. Chen, J.-P. Franke, K. Ensing, J. Wijsbeek, and R. A. De Zeeuw, J. Chromatogr. 613 (1993)
289.
39. V. Dixit and V. M. Dixit, J. Liq. Chromatogr., 14 (1991) 2779.
40. R.-C. Hsu, I. Biggs, and N. K. Saini, /. Agric. Food Chem., 39 (1991) 1658.
41. T. Tanaka, T. Hori, T. Asada, K. Oikawa, and K. Kawata, J. Chromatogr. A, 1175 (2007) 181.
42. N. J. K. Simpson, ed., Solid-Phase Extraction: Principles, Techniques, and Applications, Dekker,
New York, 2000.
43. E. M. Thurman and M. S.Mills, Solid-Phase Extraction: Principles and Practice, Wiley, New York,
1998.
44. J. S. Fritz, Analytical Solid-Phase Extraction, Wiley-VCH, New York, 1999.
45. N. Simpson and К. С van Home, Sorbent Extraction Technology Handbook, Varian, Harbor City,
CA, 1993.
46. M. J. Telepchak, Forensic and Clinical Applications of Solid Phase Extraction, Humana Press, Toto-
wa, NJ, 2004.
47. R. Bland, Proceedings of the 3rd Annual International Symposium on Sample Preparation and
Isolation using Bonded Silicas, Analytichem Internationa], Harbor City, CA, 1986, pp. 93—116.
48. E. J. Rook, M. J. X. Hillebrand, H. Rosing, J. M. van Ree, and J. H. Beijnen, J. Chromatogr. B,
824 (2005)213.
49. M. Raisglid and M. F. Burke, abstract 653, 48th Pittsburgh Conference on Analytical Chemistry
and Applied Spectroscopy, Atlanta, GA, 1997.
50. K.-S. Boos and A. Rudol'phi, LCGC, 15 (1997) 602.
51. K.-S. Boos and A. Rudolphi, LCGC, 15 (1997) 814.
52. N. M. Cassiano, V. V. Lima, R. V. Oliveira, A. C. de Pietra, and Q. B. Cass, Anal. Bioanal. Chem.,
384 (2006) 1462.
53. D. R. Doerge, M. L. Churchwell, and K. Berry Delclos, Rapid Commun. Mass Spectrometry, 14
(2000) 673.
54. K. Ensing, С Berggren, and R. E. Majors, LCGC, 19 (2001) 942.
55. S. G. Dmitrienko, V. V. Irkha, A. Yu. Kuznetsova, and Yu. A. Zolotov, J. Anal. Chem., 59 (2005)
808.
56. C. Baggiani, L. Anfossi, and C. Giovannoli, Current Pharma. Anal., 2 (2006) 219.
Литература 847
57. J. О. Mahony, К. Nolan, M. R. Smyth, and В. MizaikofT, Anal. Chim. Acta, 534 (2005) 31.
58. P. A. C. Cormack and A. Z. Elorza, J. Chromatogr. B, 804 (2004) 173.
59. S. Piletsky and A. Turner, Molecular Imprinting of Polymers, Landes Bioscience, Austin, TX, 2006.
60. M.-C. Hennion and V. Pichon, J. Chromatogr. A, 1000 (2003) 29.
61. V. Pichon, N. Delaunary-Bertoncini, and M.-C. Hennion, in Comprehensive Analytical Chemistry,
Vol. 37 J. Pawliszyn, ed., Elsevier, Amsterdam, 2002, p. 1081.
62. N. Delatmay, V. Pichon, and M.-C. Hennion, J. Chromatogr. B, 745 (2000) 15.
63. D. Stevenson, J. Chromatogr. B, 745 (2000) 39.
64. N. Zolotarjova, P. Mrozinski, and R. E. Majors, LCGC, 25 (2007) 118.
65. R. E. Majors, LCGC, 25 (2007) 16.
66. J. Stroka, E. Anklam, U. Jorissen, and J. Gilbert, JAOAC Int., 83 (2000) 320.
67. M. Anastassiades, S. J. Lehotay, D. Stajnbaher, and F. J. Schenck, JAOAC Int., 86 (2003) 412.
68. M. Anastassiades, http://www.quechers.com/docs/quechers recov.pdf (2004).
69. S. J. Lehotay, A. de Kok, M. Hiemstra, and P. Van Bodegraven, JAOAC Int., 88 (2005) 595—614.
70. С F. Fagerquist, A. R. Lightfield, and S. J. Lehotay, Anal. Chem., 77 (2005) 1473.
71. N. Jakubowska, Z. Polkowska, J. Namiesnik, and A. Przyjazny, Crit. Rev. Anal. Chem. 35 (2005)
217.
72. Method 3550C, Ultrasonic Extraction, US EPA, http://www.epa.gov/epawaste/hazard/ testmet-
hods/sw846/online/3 series, htm.
73. E. L. Randall, JAOAC Int., 57 (1974) 1165.
74. Method 3541, Automated Soxhlet Extraction, US EPA, http://www.epa.gov/ epawaste/hazard/test-
methods/sw846/online/3 series.htm.
75. J. L. Luque-Garcia and L. de Castro. J. Chromatogr A., 998 (2003) 21.
76. J. L. Luque-Garcia,M. J. Ramos,M. J. Martinez-Bueno, and M. D. Luque de Castro, Chromatog-
raphia, 62 (2005) 69.
77. J. L. Luque-Garcia and M. D. Luque de Castro., Anal. Chem., 73 (2001) 5903.
78. J. M Levy, LCGC, 17(6S) (1999) S14.
79. R. Smith, J. Chromatogr. A, 1000 (2003) 3.
80. L. T. Taylor, Techniques in Analytical Chemistry: Supercritical Fluid Extraction, Wiley, New York,
1996.
81. M. D. Luque De Castro, M. Valcarcel, and M. T. Tena, Analytical Supercritical Fluid Extraction,
Springer, New York, 1994.
82. E. D. Ramsey, ed., Analytical Supercritical Fluid Extraction Techniques, Kluwer Academic,
Dordrecht, 1998.
83. Method 3545A, Pressurized Fluid Extraction (PFE), US EPA, http://www.epa.gov/ epawaste/ha-
zard/testmethods/sw846/online/3 series.htm.
84. Accelerated Solvent Extraction (ASE) of Active Ingredients from Natural Products, Application note
335, Dionex Corp., Sunnyvale, CA.
85. G. LeBlanc, LCGC, 17(6S) (1999) S30.
86. Method 3546, Microwave Extraction, US EPA, http://www.epa.gov/epawaste/hazard/ testmet-
hods/sw846/online/3 series.htm.
87. Method 3540C, Soxhlet Extraction, US EPA, http://www.epa.gov/epawaste/hazard/ testmet-
hods/sw846/online/3 series.htm.
88. N. Tang and C. Miller, An Integrated Approach to Improve Sequence Coverage by Combining
LC-MALDI MS/MS and NanoLC-LC-MS/MS, Agilent Technologies, Santa Clara, CA, Publication
Number 5989-350OEN (2005).
89. S. R. Souverain and J.-L. Veuthey, J. Pharm. Biomed. Anal., 35 (2004) 913.
90. R. Herraez-Hernandez, P. Campins-Falcd, and A. Sevillano-Cabeza, Chromatographia, 33 (1992)
177.
91. L. Ynddal and S. H. Hansen, J. Chromatogr. A, 1020 (2003) 59.
92. J. Ayrton, G. J. Dear, W. J. Leavens, D. N. Mallett, and R. S. Plumb, Rapid Comm. Mass Spec,
II (1997) 1953.
93. K. N. Frayn and P. F. Maycock, Clin. Chem. 29 (1983) 1426.
94. Y.-J. Xue, J. B. Akinsanya, J. Liu, S. E. Unger, Rapid Comm. Mass Spec., 20 (2006) 2660.
95. J. Porath, J. Protein Chem., 16 (1997) 463.
96. F. Laborda, M. V. Vicente, J. M. Mir, and J. R. Castillo, Anal. Bioanal. Chem., 357 (1997) 1618.
97. С Shaw, Meth. Mol. Biol., 64 (1996) 101.
98. Y. Wang and J. Seneviratne, Curr. Proteomics, 5 (2008) 104.
848 Глава 16. Профподготовка
99. J. Martosella, N. Zolotarjova, H. Liu, G. Nicol, and В. Е. Boyes, Proleomics, 13 (2005) 3304.
100. N. Zolotarjova, J. Martosella, G. Nicol, J. Bailey, B. E. Boyes, and W. C. Barrett,
Electrophoresis, 25 (2004) 2402.
101. Y. Ikeguclii and H. Nakamura, Anal. Sci., 16 (2000) 541.
102. A. Apffel, S. Fishcher, G. Goldberg, P. C. Goodley, and F. E. Kuhlman, J. Chromatogr. A., 712
(1995) 177.
103. S. Ahuja, Selectivity and Detectability Optimization in HPLC, Wiley, New York, 1989.
104. B. King and G. S. Graham, Handbook of Derivatives for Chromatography, Heyden, Philadelphia,
1979.
105. H. Lingeman, Detection-Oriented Derealization Techniques in Liquid Chromatography, Dekker,
New York, 1990.
106. J. F. Lawrence and R. W. Frei, Chemical Derivatization in Liquid Chromatography, 3rd ed.,
Elsevier, Amsterdam, 1985.
107. T. Toyo'oka, Modern Derealization Methods for Separation Science, Wiley, West Sussex, UK,
1999.
108. K. Blau and J. M. Halket, eds., Handbook of Derivatives for Chromatography, 2nd ed, Wiley, West-
Sussex, UK, 1993.
ГЛАВА 17
НЕИСПРАВНОСТИ И
СПОСОБЫ ИХ УСТРАНЕНИЯ
17.1. Введение 851
17.2. Профилактика проблем 852
17.2.1. Тесты на пригодность (работоспособность) системы 852
17.2.1.1. Установочная квалификация (УК), операционная
квалификация (ОК) и эксплуатационная
квалификация (ЭК) 853
17.2.1.2. Тест на эффективность (рабочие характеристики) градиента. 853
17.2.1.3. Дополнительное тестирование системы 853
17.2.2. Профилактическое обслуживание 853
17.2.3. Тестирование пригодности системы 854
17.2.4. Хронологические записи 854
17.2.5. Полезные советы и методы 855
17.2.5.1. Удаление воздуха из насоса 855
17.2.5.2. Проверка гидравлического затвора растворителей 856
17.2.5.3. Предварительное смешивание для повышения
воспроизводимости удерживания в плавных градиентах .... 856
17.2.5.4. Очистка и обслуживание обратных клапанов 857
17.2.5.5. Обнаружение утечек 857
17.2.5.6. Устранение негерметичности соединений 858
17.2.5.7. Мытье химической посуды 858
17.2.5.8. Улучшение разделения с помощью трифторуксусной
кислоты 858
17.2.5.9. Получение высокоочищенной воды 859
17.2.5.10. Устранение остаточных примесей 859
17.3. Стратегии выявления проблем 861
17.3.1. Найти и исправить 861
17.3.2. Упрощать вместо усложнения 862
17.3.3. Менять за один раз один параметр 862
17.3.4. Устранение воспроизводимых сложностей 862
17.3.5. Замена частей 862
17.3.6. Вернуть на место 863
17.4. Общие симптомы ВЭЖХ-проблем 863
17.4.1. Утечки 864
17.4.1.1. Утечки на линии до насоса 864
17.4.1.2. Утечки в насосе 865
17.4.1.3. Утечки на стороне высокого давления 867
17.4.1.4. Утечки в автосамплере 867
17.4.1.5. Утечки в колонке 870
17.4.1.6. Утечки в детекторе 871
17.4.2. Аномальное давление 872
17.4.2.1. Слишком высокое давление 873
850 Содержание
17.4.2.2. Слишком низкое давление 874
17.4.2.3. Нестабильность давления 876
17.4.3. Нестабильность времени удерживания 876
17.4.3.1. Проблемы, связанные со скоростью потока 877
17.4.3.2. Проблемы, связанные с размером колонки 877
17.4.3.3. Проблемы, связанные с подвижной фазой 877
17.4.3.4. Проблемы, связанные с неподвижной фазой 878
17.4.3.5. Проблемы, связанные с изменениями температуры 878
17.4.3.6. Симптомы проблем, связанных с удерживанием 879
17.4.4. Площадь пика 881
17.4.4.1. Слишком большая площадь пика 881
17.4.4.2. Слишком маленькая плошадь пика 883
17.4.4.3. Изменяющаяся величина площади пика 883
17.4.5. Другие проблемы, связанные с хроматограммой 884
17.4.5.1. Проблемы, вызывающие дрейф базовой линии 884
17.4.5.2. Проблемы шумов базовой линии 887
17.4.5.3. Проблемы, связанные с формой пика 890
17.4.6. Интерпретация тестов на пригодность (работоспособность) системы. 900
17.4.6.1. Интерпретация градиентного тестирования 900
17.4.6.2. Интерпретация дополнительных тестов системы 907
17.5. Таблицы по устранению неисправностей 908
Литература 919
Неисправности и быстрые способы их устранения
Если ваша цель — быстро решить проблему, а не читать о том, как ищут и устраняют
неисправности, рекомендуем один из двух подходов. Просмотрите подробное оглавление
в начале книги и найдите раздел этой главы, касающийся вашей проблемы или же
переидите к разделу 17.5 в конце этой главы и используйте таблицы с 17.2 по 17.11, чтобы
по ним пройти весь процесс устранения неисправностей и получить перекрестную
ссылку на текст, в котором содержится дополнительная информация. Каждая глава в этой
книге содержит информацию об устранении неисправностей. Эту информацию легко
найти, обратившись к предметному указателю в конце книги.
17.1. Введение 851
17.1. Введение
К тому времени, когда было написано второе издание этой книги, устранение
неисправностей отнимало у оператора ВЭЖХ-системы большую часть рабочего времени.
Это проблемы, связанные с пульсациями насоса, скачками давления при
переключении ручных инжекторов, короткими сроками службы уплотнений насоса и ламп
детектора, а также общие проблемы, связанные с обслуживанием хроматографической
системы. Колонки могли не работать вследствие повреждений при транспортировке
или разрушения сорбента. Замена фильтра на входе колонки была настолько
распространенным явлением, что большинство производителей поставляли несколько
дополнительных фильтров с каждой колонкой. Представление о градиентном элюиро-
вании находилось в зачаточном состоянии и недопонимание его сути приводило
к неожиданным изменениям на хроматограмме при изменениях скорости потока или
других корректировках градиента. Вследствие этого перенос градиентных методов
был достаточно затруднительным.
Перенесемся на тридцать лет вперед и увидим, что ситуация кардинально
изменилась. ВЭЖХ аппаратура стала гораздо надежнее, а интервал планового
технического обслуживания с еженедельного или ежемесячного вырос до полугода и до года.
С появлением более чистых сорбентов типа В, более регулярных по размеру и форме
частиц и улучшенных процедур упаковки, проблемы, связанные с колонками, стали
малой частью тех проблем, которые преследовали хроматографистов тридцать лет
назад. Градиентное элюирование хорошо изучено и, при минимальной аккуратности,
оно может работать так же хорошо, как и изократические методы. Даже если его
используют не очень опытные хроматографисты.
Эта глава посвящена проблемам ВЭЖХ и способам их устранения. Принцип
устранения проблем — их предотвращение (раздел 17.2). Это и есть основная
стратегия надежной работы ВЭЖХ-системы. Проблемы легче всего вычленить с помощью
системного подхода (раздел 17.3) и выявить конкретные симптомы {раздел 17.4).
Устранению неисправностей и их предотвращению в значительной степени
помогает ясное понимание того как работает ВЭЖХ-система. Читателям рекомендуется
получить эту информацию из главы 3 (оборудование) и соответствующей части
(частей) главы 4 (детекторы). Раздел 3.10 (техническое обслуживание) следует, как
минимум, повторно просмотреть. Методики обслуживания тесно связаны с устранением
проблем, и на них в этой главе постоянно будут перекрестные ссылки. Конечно,
не забудьте заглянуть в разделы по устранению неисправностей и
профилактическому обслуживанию в инструкциях, относящихся к вашему ВЭЖХ-оборудованию.
Другим аспектом устранения проблем и профилактического обслуживания
является вопрос о том, кто будет производить необходимый ремонт и обслуживание. Эта
тема обсуждается в разделе 3.10.3.1. В таблице 3.7 перечислены работы,
рекомендуемые к проведению персоналом различного уровня. Окончательное решение — кто
имеет достаточную квалификацию для того, чтобы ремонтировать и обслуживать
хроматографическое оборудование, определяется сочетанием лабораторной
стратегии, нормативных требований, уровня обучения и квалификации персонала. В
любом случае, ясное понимание принципов работы хроматографической системы и
процесса устранения проблем пойдет на пользу сотрудникам всех уровней
компетентности и ответственности.
Наконец, важно понимать, что описание методов устранения неисправностей
в ВЭЖХ трудно объединить в одну главу, как это сделано в данной книге.
Существуют и другие источники, и вам предлагается изучить их для того, чтобы найти в них
дополнительные советы. На эту тему написано множество книг. Ссылки [1, 2|
хороши для того, чтобы начать изучать этот вопрос. Не надо забывать, что новые матери-
852 Глава 17. Неисправности и способы их устранения
алы появляются постоянно. На момент написания этой главы Google на запрос
«HPLC troubleshooting books» дал 13600 ссылок (но не 13600 книг). Другой
прекрасный источник регулярных советов на эту тему — колонка в ежемесячном журнале
LCGC под названием «LC Troubleshooting» одного из авторов этой книги |3|. В
дискуссионных онлайн группах можно получить совет по устранению проблем. Одной
из лучших групп является Chromatography Forum |4], на сайте которой обсуждается
устранение неисправностей и связанных с ними проблем. Следят за этим
обсуждением эксперты по всем аспектам ВЭЖХ. Кроме того, существуют экспертные онлайн
системы, такие, как HPLC Wizard |5|, которые могут помочь вам локализовать и
определить проблемы, возникающие при использовании ВЭЖХ.
Каждая лаборатория должна разработать свою собственную программу
профилактического обслуживания. Список возможных пунктов для включения в такую
программу можно найти в предметном указателе этой книги под заголовком
«Оптимальные подходы».
17.2. Профилактика проблем
Самая лучшая проблема в ВЭЖХ та, которая не возникает. Для того, чтобы такое
«невозникновение» стало общим правилом для системы ВЭЖХ, необходимо создать
структурированную программу профилактического обслуживания, описанную в
разделе 3.10.2. В этом процессе важны четыре составляющих. Во-первых, необходим
тест на пригодность системы (раздел 17.2.1), используемый для того, чтобы
установить — пригодна ли система для работы должным образом в идеальных условиях.
Во-вторых, необходимо проводить периодическое обслуживание (раздел 17.2.2) для
ремонта или замены деталей, имеющих ограниченный срок службы, вследствие
нормального износа. В-третьих, тест на пригодность системы (раздел 17.2.3) должен
проводиться перед каждой партией образцов, чтобы гарантировать, что метод и
оборудование работают на уровне, который обеспечит приемлемые данные.
Наконец, для каждой системы ВЭЖХ должны храниться записи о ремонте и техническом
обслуживании (раздел 17.2.4) для подтверждения технического обслуживания и для
определения характерных неисправностей.
17.2.1. Тесты на пригодность (работоспособность) системы
Тест на работоспособность системы описан в разделе З.ЮЛ. Краткий его обзор дан
здесь — за деталями обратитесь к разделу 3.10.I. Существует три типа тестов,
которые можно использовать для тестирования работоспособности системы:
установочная квалификация (УК), операционная квалификация (ОК) и эксплуатационная
квалификация (ЭК) (раздел I7.2.1.1). Все три группы тестов должны проводиться
сразу после покупки хроматографической системы. Градиентные тесты
(раздел 17.2.I.2) и дополнительные испытания (раздел 17.2.1.3) следует проводить
каждые 6—12 месяцев для того, чтобы обеспечить непрерывную надежную работу
системы ВЭЖХ.
На протяжении всей этой книги предполагается, что система ВЭЖХ работает
с колонками, упакованными обращенной фазой, и снабжена УФ-детектором.
Многие из описанных тестов лучше всего работают в условиях УФ-детектирования,
включая тесты, перечисленные в этом разделе, и тесты, на которые приведены
ссылки. Наверное, можно разработать и другие способы тестирования, с другими, не
использующими УФ, детекторами, чтобы достичь тех же целей, но некоторые тесты,
17.2. Профилактика проблем 853
такие как градиентные, будет очень трудно провести с определенного типа
детекторами (например, с ЖХ-МС или ЖХ-МС/МС). По этой причине мы рекомендуем
каждой лаборатории иметь хотя бы один УФ-детектор для облегчения тестирования
работоспособности и выявления неисправностей.
17.2.1.1. Установочная квалификация (УК),
операционная квалификация (ОК) и
эксплуатационная квалификация (ЭК)
Комбинация тестов, разработанная производителем (УК, ОК) и пользователем (ЭК)
показывает, что ВЭЖХ-система работает так, как было задумано производителем и
согласно прилагаемым спецификациям. Эти тесты выполняются сразу после
покупки системы, а ЭК-тест после этого периодически повторяется. Результаты этих
тестов играют важную роль в стратегии «разделяй и властвуй», направленной на
выявление ПРОБЛЕМЫ (раздел 17.3.1), что позволяет пользователю проверять текущую
пригодность системы по сравнению с ее начальной пригодностью, когда было
известно, что она работает должным образом. Это помогает ответить на вопрос «система
или метод?», который часто встает при возникновении проблем с ВЭЖХ-системой.
17.2.1.2. Тест на эффективность (рабочие характеристики)
градиента
Если ВЭЖХ-система может работать в градиентном режиме, следует произвести тест
на эффективность градиента (раздел 3.Ю.I.2) даже если система используется только
для изократических разделений со смешиванием подвижной фазы в потоке. Тест
на эффективность градиента проверяет линейность и точность приготовления
подвижной фазы, а также измеряет значение объема задержки градиента. Если ВЭЖХ-система
используется только для работы в изократическом режиме со смешиванием
подвижной фазы в потоке, следует провести альтернативный тест — ввести раствор стандарта
в изократических условиях. Затем заменяют резервуары с элюентами А и В и
программируют доставку той же подвижной фазы (например, 30 %А = вода + 70 %В = МеОН
становится 30 %В = вода и 70 %А = МеОН) и вводят снова раствор стандарта. Времена
удерживания в обоих случаях должны быть одинаковыми. Эти тесты гарантируют, что
запрограммированная в системном контроллере подвижная фаза нужного состава
доставляется в колонку. См. раздел 17.4.6.1, в котором описаны специальные случаи
неудачных тестов на эффективность градиента.
17.2.1.3. Дополнительное тестирование системы
Несколько дополнительных тестов завершат общую проверку пригодности
ВЭЖХ-системы (раздел 3.10.1.3). Проверка расхода определит, работает ли должным
образом насос, а сброс давления проверяет выпускные обратные клапаны и/или
состояние уплотнений насоса. Тест на воспроизводимость удерживания дважды
проверяет скорость потока и подтверждает, что подвижная фаза смешана в потоке
должным образом. Тест на воспроизводимость площади пика гарантирует, что автосам-
плер работает должным образом. В разделе 17.4.6.2 приведены конкретные примеры
неудачных дополнительных системных тестов.
17.2.2. Профилактическое обслуживание
Программа регулярного профилактического обслуживания позволит ВЭЖХ-системе
работать более надежно и по возможности избегать поломок. Список рекомендуемых
частей ВЭЖХ-системы для профилактического обслуживания содержится в табли-
854 Глава 17. Неисправности и способы их устранения
це 3.6 и обсуждается в разделе 3.10.2.1. Как правило, время, затрачиваемое на
профилактическое обслуживание, особенно то время, которое идет на регулярную
промывку системы и на своевременную, до выхода из строя, замену частей
с нормальным износом (например, уплотнений насоса), позволяет эффективно
работать и снижает общие эксплуатационные расходы. Лаборатории, в которых
дожидаются поломок, вместо того, чтобы проводить профилактическое обслуживание,
обычно тратят больше времени и денег на устранение неисправностей и ремонт, чем
те, кто регулярно обслуживает свои ВЭЖХ-системы.
17.2.3. Тестирование пригодности системы
Под пригодностью системы понимается серия вводов или инжекций, заранее
определенных для конкретного метода, которые делаются до разделений с настоящими
образцами. Делаются эти вводы для того, чтобы убедиться — система ВЭЖХ работает
должным образом и способна генерировать данные, соответствующие критериям
приемлемости для данного метода. Для выполнения этой задачи нужно, чтобы с помощью
образца, предназначенного для оценки пригодности системы, можно было адекватно
оценить эффективность метода. Обычно такой образец представляет собой смесь,
приготовленную из стандартов, смешанных в реальном или рассчитанном соотношении.
Эти тесты могут включать в себя подмножество теста эксплуатационной
квалификации (разделы I7.2.1, 3.10.1), а также других тестов — таких, как проверка времени
удерживания и/или воспроизводимости площади пика, демонстрирующие, что
ВЭЖХ-система работает в целом. Тестирование системы на пригодность должно
оценивать наиболее значимые параметры разделения (например, разрешение, точность,
воспроизводимость), чтобы гарантировать качество получаемых хроматографических
данных. Другими словами, тестирование системы на пригодность должно дать ответ на
вопрос: «Удовлетворительно ли работает ВЭЖХ-система для данного метода».
Независимо от того, требуются ли тесты на соответствие работы ВЭЖХ-системы правилам
контролирующего органа или местным эксплуатационным правилам, целесообразно
провести какой-нибудь тест до ввода образцов, чтобы минимизировать риск сбора
бессмысленных данных и не тратить понапрасну ценные образцы, время и ресурсы.
Хронологическая запись (раздел 17.2.4) тестов на пригодность системы может также быть
полезна для выявления источника проблемы. Для получения дополнительной
информации о проверке пригодности системы см. раздел 12.3.
17.2.4. Хронологические записи
Нельзя недооценивать ценность хронологических записей для выявления
последовательности отказов техники и при составлении программ профилактического
обслуживания. В разделе 3.I0.3.2 описан предлагаемый набор записей, включающий, как
минимум, три обязательных для каждой ВЭЖХ-системы элемента. Запись о
конфигурации системы будет включать в себя достаточную информацию для идентификации
каждого компонента системы (насос, автосамплер, детектор и т.д.), составляющего
конкретную ВЭЖХ-систему. Все это, в сочетании с записями о сериях (партиях)
образцов (например, обозначение образца, привязанное к номеру разделения) должно
позволить связывать каждое разделение с определенной конфигурацией
ВЭЖХ-системы. Записи о техническом обслуживании будут содержать письменный отчет
обо всех мероприятиях по обслуживанию системы. Результаты проверок системы,
таких как ОК и тестирование на эффективность градиента (раздел I7.2.1) тоже должны
быть включены в записи для каждой ВЭЖХ-системы.
Хронологические записи служат двум целям. Во-первых, для того, чтобы
предоставить контролирующему органу документацию, из которой следует, что необходи-
17.2. Профилактика проблем 855
мые процедуры для квалификации, использования и технического обслуживания
системы были своевременно проведены. Во-вторых, они необходимы для получения
данных, которые помогут разработать программу профилактического обслуживания.
По прошествии достаточного периода времени (например, года или двух) в
зависимости от интенсивности использования ВЭЖХ-системы, должно накопиться
достаточно данных для того, чтобы выявить ситуации, при которых происходят сбои
в работе оборудования. Если несколько хроматографических систем, имеющихся
в лаборатории, формально одинаковы (то есть, у них одинаковый производитель
и модель), то данные нескольких подобных систем могут быть объединены.
Например, для определения временного промежутка, через который следует менять
уплотнение насоса, можно объединить данные, полученные от всех насосов одинаковых
систем1. Обычно, лучше всего заменить уплотнения насоса до выхода их из строя,
чтобы частицы, образующиеся в результате износа уплотнения, не забивали
капилляры или фильтры, стоящие после насоса. Если замена уплотнения выполнялась
через 7, 6, 8, 12 и II месяцев во время пяти различных внеплановых ремонтов —
данные говорят о том, что было бы разумнее заблаговременно заменять уплотнения
каждые полгода, чтобы избежать последствий их выхода из строя. Аналогичная
процедура может быть использована для определения сроков проведения других замен
или ремонтных работ, которые должны быть добавлены к списку профилактических.
Как правило, если срок службы детали можно заранее рассчитать, замените ее
при 70—80% износе. Это будет разумным профилактическим мероприятием по
обслуживанию системы и принесет больше пользы при том, что снизится риск потери
данных при выходе этой части из строя.
17.2.5. Полезные советы и методы
Этот раздел содержит набор полезных советов и методов, которые можно
использовать для выявления и решения проблем, возникающих в ВЭЖХ, а также для
предотвращения их появления в будущем. Здесь будут освещаться некоторые наиболее
распространенные проблемные области при работе в режиме ВЭЖХ. Уделив этим
областям особое внимание, вы сможете уменьшить частоту возникновения
неисправностей, а также количество времени, потраченного для их устранения в тех случаях,
когда они возникают. В каждой главе даны перекрестные ссылки.
17.2.5.1. Удаление воздуха из насоса
Детали и конструкции, из которых состоит ВЭЖХ-насос и связанное с ним
оборудование, имеют множество небольших, часто угловых пространств, в которых могут
задерживаться пузырьки воздуха. Иногда постукивание по голове насоса деревянным
или пластиковым предметом (таким, как рукоятка отвертки) приводит к удалению
пузырьков. Иногда, если пузырьки не хотят удаляться и, несмотря на стук рукояткой
отвертки, дверь не открывают, помогают промывки полностью дегазированным
растворителем с низкой вязкостью и малым поверхностным натяжением, таким как
метанол. Такие промывки могут пузырьки растворить. Регулярное использование
дегазированных растворителей предотвратит накопление пузырьков в системе, поскольку
растворитель будет иметь дополнительную емкость для растворения газа, растворяя
пузырьки до того, как они станут проблемой. Любая ВЭЖХ-система будет работать
более надежно, если подвижная фаза дегазирована.
'Лучше, однако, придерживаться рекомендаций, которые дает в своих инструкциях к насосам
производитель. Обычно указывают не время, а объем прокачанного элюента, по достижении
которого необходимо менять уплотнения. — Прим. перев.
856 Глава 17. Неисправности и способы их устранения
17.2.5.2. Проверка гидравлического затвора растворителей
Все системы ВЭЖХ будут работать надежнее, если резервуары растворителей
приподнять относительно насоса так, чтобы слабое давление гидравлического затвора
способствовало доставке подвижной фазы к насосу. Чтобы обеспечить свободный
ток растворителя до миксера, необходимо периодически проверять входные
фильтры. Поскольку многие насосы имеют «асимметричный» цикл работы, в котором
времени на подачу растворителя уделено больше, чем на заполнение им камеры
насоса, скорость потока во время части цикла, отведенной на заполнение, намного
выше, чем общая средняя скорость потока. Следовательно, следует ожидать, что
резервуар сможет доставлять в несколько раз больше растворителя за счет действия
гидравлического затвора, чем будет необходимо насосу. Чтобы это проверить,
отсоедините линию входа растворителя в миксер (при смешивании на стороне низкого
давления) или во входной клапан насоса (при смешивании на стороне высокого
давления) и пустите растворитель самотеком по капиллярам. Десятикратное
превышение объема растворителя обеспечит приемлемое количество доставляемой жидкости.
Например, если система обычно работает со скоростью 1 мл/мин, то, по крайней
мере, 10 мл/мин растворителя должно проходить самотеком. В этом случае будет
поступать достаточное количество растворителя, так что насос никогда не окажется
без жидкости. Если поток будет ниже ожидаемого, следует проверить: не забиты ли
входные фильтры, нет ли перегиба входных капилляров, или разрежения в
резервуарах с подвижной фазой. Ограничение подачи растворителя в системах с
перемешиванием на стороне низкого давления также может приводить к неправильному
смешиванию компонентов подвижной фазы (раздел 17.4.6.1).
17.2.5.3. Предварительное смешивание для повышения
воспроизводимости удерживания в плавных градиентах
Сложные смеси часто требуют разделений с высоким разрешением. В таких
разделениях используются плавные градиенты. Кроме того, скорость нарастания градиента
менее, чем 1%/мин часто требуется для разделения высокомолекулярных
соединений, таких, как пептиды или белки, чтобы обеспечить приемлемое значение к* этих
соединений с высоким значением S (разделы 9.4, 13.4.1.4). В некоторых ВЭЖХ-сис-
темах нельзя сформировать пологий градиент с точностью, необходимой для
приемлемого разделения. Миксеры на линии низкого давления создают градиент
перемешиванием поочередно нагнетаемых растворителей А и В. Поскольку перемешивание
никогда не бывает полным, незначительные различия в составе (или элюирующей
силе) подвижной фазы остаются в момент достижения ею колонки. Это показано
на рис. 17.1 сплошной линией, наложенной на пунктирную линию, обозначающую
запрограммированный градиент. Таким образом происходит колебание значений %В
вокруг запрограммированных значений на всем протяжении разделения. Для такого
очень плавного градиента величина %В в точке 1 больше, чем в точке 2, хотя точка I
появляется в градиенте раньше. Для корректировки этой ситуации подвижные фазы
могут быть смешаны заранее.
Например, при использовании чистых растворителей в качестве элюентов А и В,
градиент 40—50 %В за 50 мин требует, чтобы система точно обеспечила градиент
с наклоном 0,2%/мин, для чего может не хватить точности некоторого оборудования.
Когда А и В предварительно смешаны до 40 %В (в резервуаре А) и 50 %В (в
резервуаре В), система может быть запрограммирована на подачу градиента подвижной
фазы от 0 до 100% в течение 50 минут или 2%/мин, что значительно легче выполнить
на большей части градиентного оборудования. Таким образом, предварительное
смешивание подвижной фазы (подобное вышеописанному) может преодолеть
ограничения, накладываемые хроматографическим оборудованием.
17.2. Профилактика проблем
95
is
65
Время
Рис. 17.1. Запрограммированный профиль плавного градиента ( ), действительно
наблюдаемый профиль (—). Элюирующая сила подвижной фазы в точке 1
больше, чем в точке 2, несмотря на то, что точка ] пояшшется в градиенте
раньше
Этот же метод предварительного смешивания может быть использован для
уменьшения шума базовой линии как в изократическом, так и в градиентном
разделении, в тех случаях, когда детектор работает с максимальной чувствительностью.
Например, в изократических методах с использованием рефрактометрического
детектора (раздел 4.11) базовая линия будет шуметь меньше, если подвижную фазу
предварительно смешать вместо того, чтобы использовать смешивание в потоке.
Базовые линии при градиенте обычно становятся лучше, если 5% В-растворителя
добавлено в резервуар А и 5 %А в резервуар В.
17.2.5.4. Очистка и обслуживание обратных клапанов
Плохо работающие клапаны насоса, вместо замены на новые, зачастую можно
восстановить обработкой ультразвуком в спирте. Следует отметить, что входные и
выходные клапаны редко взаимозаменяемы. Если они не имеют четкой маркировки, то
на корпусе клапана должны быть специальные отметки, указывающие положение и
направление потока. Некоторые клапаны могут разваливаться на части при
перевертывании и поэтому, прежде чем обрабатывать их ультразвуком, стоит это проверить,
иначе можно удивиться, обнаружив недостающие части в ультразвуковой бане после
процедуры очистки. Мы рекомендуем помешать каждый клапан в отдельный
стаканчик с достаточным количеством метанола или изопропанола, полностью
покрывающими клапаны и затем обрабатывать ультразвуком около 5-10 мин. Исходя из
нашего опыта, такая процедура в большинстве случаев устраняет перепускание клапанов,
удаляя, видимо, нежелательные загрязнения с клапанного шарика и/или седла
клапана. Если шарик выпал из клапана, все его части следует промыть в спирте, затем
аккуратно собрать с помощью пинцета. Следует избегать контакта внутренних частей
клапана с бумагой, тканью или пальцами, так как мельчайшие волоконные частицы
или отпечатки пальцев могут вызвать перепускание клапанов. В разделе 17.4.2.2
дополнительно обсуждаются возможные причины залипания клапанов и их
восстановление.
17.2.5.5. Обнаружение утечек
Утечка подвижной фазы может быть как легко обнаруживаемой, так и наоборот.
Капли, лужи или сигналы утечки облегчают обнаружение места утечек. При
использовании буферов белые кристаллические отложения без следов жидкости могут
указывать на медленное подтекание соединений. Малозаметную утечку несложно обна-
858 Глава 17. Неисправности и способы их устранения
ружить с помощью кусочка термобумаги (например, чеков, которые получают после
оплаты кредитной картой). Нарезают узкие заостренные полоски термобумаги
(примерно 1 х 5 см) и острым концом проверяют подозрительные соединения,
уплотнения или другие возможные источники протечки. Бумага начинает чернеть при
контакте с органическими растворителями. Такой способ поможет быстро установить
место утечки, если его трудно обнаружить другим способом.
17.2.5.6. Устранение негерметичности соединений
Если соединение подозревается в протечке, проще всего затянуть его на четверть
оборота и посмотреть, не прекратится ли утечка. Если это не поможет, то следует
разобрать соединение, промыть и вновь собрать либо заменить ферулы. Ферулы из
нержавеющей стали нельзя слишком сильно затягивать, так как они могут
расплющиться и их невозможно будет извлечь из соединения. Если соединения часто протекают,
лучше использовать ферулы из ПЭЭК (полиэфирэфиркетона), чем стальные,
поскольку первые достаточно деформируются, чтобы уплотнить любую шероховатую
поверхность. Если подтекают соединения и капилляры из ПЭЭК, то нужно остановить
поток, ослабить гайки, поправить капилляр в соединении и вновь затянуть гайки.
Иногда при затягивании гаек из ПЭЭК при включенном потоке подвижной фазы
конец капилляра может сместиться относительно ферулы, образуя полость. Это,
в свою очередь, может явиться причиной нежелательного уширения пиков (внеколо-
ночные эффекты, раздел 17.4.5.3).
17.2.5.7. Мытье химической посуды
Органические остатки в «чистой» химической посуде могут быть источником
ложных пиков или дрейфа базовой линии в холостом градиенте, поэтому очень важно
тщательно промывать используемые емкости. Предложен целый ряд способов мытья
стеклянной посуды |6|. Для того чтобы смыть моющие детергенты, достаточно 10 раз
ополоснуть посуду водопроводной водой и 10 раз — деионизированной водой |7|.
По другим источникам |8| лучше избегать моющих средств, предпочитая
ополаскивать водой стеклянную посуду (включая емкости, пипетки и градуированные
цилиндры), используемую только для подвижных фаз ВЭЖХ, и затем обмывать ее чистым
органическим растворителем [6|. Для того чтобы избежать случайного загрязнения
подвижной фазы из-за стеклянной посуды, используйте самую чистую стеклянную
посуду (из тех, что у вас имеется) и следите за тем, чтобы в процессе очистки не
возникало загрязнений на стеклянных поверхностях.
17.2.5.8. Улучшение разделения с помощью трифторуксусной
кислоты
Трифторуксусная кислота (ТФУК) широко используется при градиентных
разделениях в качестве добавки в подвижную фазу. Эта кислота хорошо смешивается и с
водой, и с органическими растворителями, создает низкий рН подвижной фазы (0,1%
ТФУК к рН 1,9), выступает как ион-парный реагент при разделении биомолекул
(раздел 13.4.1.2), применима при детектировании в УФ < 220 нм, обладает хорошей
летучестью, необходимой в масс-спектрометрии или для светорассеивающих
детекторов. ТФУК высокой степени чистоты, пригодной для ВЭЖХ, вполне доступна,
однако она быстро разлагается на воздухе. Для достижения наилучших результатов
приобретайте ТФУК для ВЭЖХ (или эквивалентной спектральной чистоты) в
ампулах по 1 мл и используйте всю ампулу за один раз. Имеются в продаже и большие
емкости ТФУК (например, по 25 мл), что намного дешевле в пересчете на
миллилитр, но, по опыту одного из авторов, невозможно предотвратить быстрое разложе-
17.2. Профилактика проблем 859
ние реактива после вскрытия бутыли даже при работе под инертным газом и
тщательном укупоривании бутыли после вскрытия. Однако водные растворы ТФУК
вполне устойчивы (по крайней мере в течение недели). При детектировании в УФ
< 220 нм иногда наблюдается некоторый дрейф базовой линии при использовании
подвижной фазы с ТФУК и ACN, поскольку поглощение ТФУК зависит от % ацето-
нитрила в подвижной фазе |9|. Чтобы минимизировать дрейф базовой линии,
добавьте одинаковое количество ТФУК и к А- и к В-растворителю и работайте
при 215 нм, поскольку в этой обрасти адсорбция смесей ТФУК/ACN достаточно
постоянна |9|. Альтернативой приготовлению ТФУК-содержащих элюентов в
лаборатории является покупка растворителей ВЭЖХ с заранее добавленной в них
производителями ТФУК (или другими добавками) (например, Burdick&Jackson, J.T. Baker)1.
17.2.5.9. Получение высокоочищенной воды
В примерах раздела 17.4.5.2 и сопровождающих раздел рисунках 17.11 и 17.12
обсуждается проблема загрязнения подвижной фазы примесями из воды или добавками.
Важно использовать реагенты самого высокого качества, чтобы избежать ненужных
фоновых пиков. Обычно это означает покупку реагентов для ВЭЖХ — всех солей,
буферов и органических растворителей. Большинство лабораторий приготовляет
собственную воду для ВЭЖХ с помощью системы для очистки, например, такой как
система Milli-Q (Millipore, США). В таких установках сочетаются механическая
фильтрация (<0,2 мкм), ионообменники и углеродные фильтры для удаления
органических примесей. Для уничтожения бактерий и окисления остатков органического
происхождения иногда применяют облучение ультрафиолетом |6].
Более глубокая очистка воды может потребоваться для максимального удаления
остаточных пиков, например, при анализе лекарственных образцов на стабильность,
когда необходимо рассчитывать площади пиков, составляющих > 0,05% от площади
основного пика. Воду можно очищать, пропуская ее через С^ ВЭЖХ-колонку, но
такой способ неудобен. Для систем со смешиванием на стороне высокого давления
возможен вариант установки защитной С18-предколонки между насосом А и смесителем
110—12|. Эта колонка «скруббер» будет улавливать органические соединения до того,
как они попадут в миксер, предотвращая их попадание в аналитическую колонку и
предупреждая, таким образом, образование фоновых пиков. Для окончательной
очистки воды были предложены другие устройства: С^-картридж для ТФЭ, через который
проходит вода перед использованием, или встроенный в поток фильтр с небольшим
противодавлением, установленный перед миксером (смешивание на стороне низкого
давления) или насосом А (смешивание на стороне высокого давления) |6, 13[.
17.2.5.10. Устранение остаточных примесей
«Остаточные примеси» есть не что иное, как пики, возникающие повторно в
последующих хроматограммах после однократного введения образца в первом разделении.
То есть остатки от образца, задержавшиеся после первого разделения, тем или иным
образом появляются в одном или более последующих холостых разделениях.
Существует три основных типа остаточных примесей:
• примеси, остающиеся во внутренних объемах системы;
• адсорбционные остаточные примеси;
• неполное элюирование.
Каждый из этих трех типов остаточных примесей рассматривается ниже, с
некоторыми рекомендациями относительно того, как их различить и устранить.
'ТФУК от разложения это вряд ли убережет, но заплатите вы за такие элюенгы втридорога. —
Прим. ред.
860 Глава 17. Неисправности и способы их устранения
Примеси, остающиеся во внутренних объемах системы, или объемные остаточные
примеси — это небольшие количества образца, задержавшиеся в системе ввода
образца, которые ненамеренно вводятся при следующем нанесении пробы. Отношения
площадей последующих пиков к площади предыдущего пика имеют постоянные
значение. Например, если при нанесении образца была получена площадь пика
в 100 000 единиц и наблюдался остаточный пик, составляющий 1% от исходного,
то холостой градиент, следующий за обычным разделением, будет давать пик с
площадью в 1% от площади первоначального пика (1000 единиц), причем с тем же
временем удерживания. Дополнительный холостой градиент будет давать пик, площадь
которого составляет 1% от площади пика в предыдущем холостом градиенте или
10 единиц. Такое постоянное постепенное разбавление в ряде последующих
холостых градиентов и характеризует объемные остаточные примеси. Из-за разбавления
остаточные примеси редко видны после двух-трех холостых разделений.
Если есть опасения, что без подобного рода примесей не обойдется, обратите
внимание на маленькие случайные объемы, в которых образец может застрять, а
потом вымываться. Возможно, это происходит из-за попадания вещества в зазоры
между плохо собранными фитингами, которые контактируют с подвижной фазой только
во время ввода образца. Иногда, если возникает избыточное давление в системе,
капилляры, которые удерживают ферулы, сделанные из ПЭЭК, могут немного
проскальзывать и создавать небольшие зазоры внутри фитинга. При этом фитинг
ослабляется, и конец капилляра смещается, как это описано в разделе 17.2.5.6.
Неудовлетворительная промывка автосамплера может также явиться источником таких пиков.
Система промывки автосамплера должна быть на должном уровне. Изношенный
ротор инжектора может внести свой вклад в появление примесей. Его замена должна
это устранить (см. раздел 17.4.1.4 с дополнительным обсуждением, связанным с авто
самплером). Когда входные фильтры в колонке засоряются, образец может
скапливаться в застойном канале, и его смыв происходит только во время очередного ввода.
Решает эту проблему промывка колонки в обратном направлении или замена
последней (см. расщепленные или искаженные пики в разделе 17.4.5.3).
Адсорбционные остаточные примеси иногда можно принять за объемные остаточ
ные примеси, но они не исчезают так быстро при последующих разделениях.
Например, первое холостое разделение, возможно, даст остаточную примесь в 1% от
первоначального пика, но во втором холостом разделении тоже может быть пик
с площадью 1% от первоначального пика или, может быть, немного меньше, чем 1%
а не 1% от однопроцентного пика. Источником таких примесей является адсорбция
образца на поверхностях внутри хроматографической системы или колонки.
Поверхности внутри хроматографической системы — это внутренняя полимерная
поверхность инжектора, петля автосамплера, внутренняя поверхность иглы инжектора. Все
эти поверхности являются распространенными источниками адсорбционных
остаточных примесей. Появление адсорбционных примесей, в основном, бывает
обусловлено вводом очень гидрофобного образца, растворенного в полярном
растворителе (например, в воде). Добавление нескольких процентов органического
растворителя в растворитель, в котором растворяют образец, может помочь избавиться от
адсорбционных примесей. Кроме того, может быть полезным увеличение силы и
объема растворителя, которым промывают автосамплер, или изменение природы
внутренних поверхностей (например, замена петли из нержавеющей стали на
петлю из ПЭЭК). Для более подробного обсуждения адсорбции образца в
инжекторе см. 114|.
Иногда перенос остаточных примесей из внутренних объемов системы и перенос
адсорбционных остаточных примесей могут протекать одновременно. В таких
случаях, в первом и, возможно, во втором холостом разделении наблюдалось бы
уменьшение части пика, поскольку «объемные» примеси исчезли бы, но адсорбционные
17.3. Стратегии выявления проблем 861
остались бы и в последующих разделениях. Когда все вышеупомянутые способы уже
опробованы, а остаточные примеси по-прежнему не исчезают, вам может
потребоваться изменение последовательности ввода проб таким образом, чтобы за вводом всех
образцов с высокой концентрацией следовало холостое разделение (то есть,
игнорируемое) или чтобы образцы с низкой концентрацией никогда не вводились
непосредственно после высококонцентрированных. Когда заранее неизвестны
приблизительные концентрации образцов, методика может быть написана так, чтобы
не учитывать результаты разделения, если за большим пиком идет маленький. Вслед
за этим разделением делается холостое, а вслед за ним снова вводится образец,
чтобы получить достоверный размер пика.
Неполное элюирование крайне маловероятно при градиентном разделении. Если
растворенное вещество не элюируется из колонки во время изократического
разделения, оно может элюироваться во время следующего разделения в виде неожиданно
широкого пика. См. раздел 17.4.4.1 для дальнейшего обсуждения поздноэлюируемых
пиков.
17.3. Стратегии выявления проблем
Способность выявлять проблемы — это навык, отточенный практикой и до некото
рой степени зависящий от личной склонности к такой деятельности. В этом разделе
мы опишем шесть практических правил, которые являются рекомендованными
составляющими стратегии выявления проблем. Опытные пользователи, вероятно,
включат хотя бы некоторые из этих правил в свой личный арсенал подходов к
выявлению проблем.
Не забывайте о безопасности во время работы с ВЭЖХ-системой. Необходимо
всегда надевать защитные очки. Несколько капель подвижной фазы, скорее всего,
вашей коже не повредят, но попав в глаза могут вызвать раздражение или более
серьезную травму. На первый взгляд может показаться, что высокое давление
в ВЭЖХ-системе опасно, но подвижная фаза протекает по системе медленно и
поэтому сломанный кусок капилляра или утечка могут создать некоторые
неприятности, но физической угрозы жизни это почти никогда не представляет. Тем не менее,
будьте осторожны и не пытайтесь остановить течь, зажав место течи большим
пальцем (и вообще пальцем) — давления в системе достаточно для того, чтобы
подвижная фаза проникла через кожу, что может привести к серьезному повреждению
тканей. Обычных лабораторных мер предосторожности, как правило, достаточно
при устранении неисправностей и для технического обслуживания ВЭЖХ-систем
(к примеру, защиты глаз, лабораторного халата и в отдельных случаях стойких к
растворителям перчаток).
17.3.1. Найти и исправить
Это важная часть методологии устранения проблем. Внесите изменения, которые
позволят вам устранить возможные проблемы. Чем больше источников таких проблем
устраняется при каждом изменении, тем лучше. Типичным примером является
тестирование новой колонки с целью определить, связаны ли сложности с вашим
аналитическим методом (включая колонку, на которой вы работали раньше) или с
оборудованием. Просто установите новую колонку и повторите эксплуатационную
квалификацию (раздел 3.10.1.3, таблица 3.5). Если вы получите те же самые
результаты что и производитель колонки (например, количество теоретических тарелок и
время удерживания, отличающееся на = 10%), вы можете быть уверены, что ваша
862 Глава 17. Неисправности и способы их устранения
ВЭЖХ-система работает удовлетворительно и, скорее всего, источником проблем
является метод (и/или колонка, на которой вы работали). ЭК колонки проверит
правильность выполнения изократического элюирования. Вы можете ЭК колонки и изок-
ратического элюирования дополнить тестированием линейности градиента или
тестированием ступенчатым градиентом (раздел 3.10.1.2).
17.3.2. Упрощать вместо усложнения
Чтобы сэкономить время, важно определить — какие тесты необходимо выполнять
в первую очередь. Например, если проблема состоит в том, что время удерживания
больше нормального и на хроматограмме присутствует более одного пика,
определите соотношение времен удерживания для каждого пика на исходной и текущей хро-
матограммах. Если это соотношение является постоянным для каждого пика, то,
вероятно, произошло снижение скорости потока. Это может быть выяснено простой и
быстрой проверкой скорости потока. Причина, однако, может быть в подвижной
фазе и стоит приготовить ее новую порцию, но проверку скорости потока имеет
смысл сделать в первую очередь, поскольку это быстрее и проще. Разумеется,
здравый смысл подскажет вам, что прежде всего следует сосредоточить свое внимание
на наиболее вероятных и часто встречающихся источниках трудностей, даже если их
не так легко обнаружить.
17.3.3. Менять за один раз один параметр
Этот принцип также называют правилом одного, и он соответствует научному подходу
к диагностике неисправности. Внесите изменение и оцените результат. Иногда
быстрее сделать несколько изменений одновременно, но это не приблизит к выявлению
истинного источника проблемы, знание о котором может помочь устранить
проблему и организовать профилактическое техническое обслуживание на будущее.
17.3.4. Устранение воспроизводимых сложностей
Так называемое правило двух. Убедитесь, что затруднение возникает, по крайней
мере, дважды. Хроматографические проблемы, которые невозможно воспроизвести,
трудно устранить и еще труднее узнать — исправлены ли они на самом деле.
Убедитесь, что проблема, которую вы пытаетесь решить, воспроизводима в достаточной
степени, чтобы вы могли быть уверены в ее исправлении. К примеру, если у вас есть
дополнительный пик или «всплеск» на хроматограмме, но вы не видите его при
повторном введении того же самого образца или при вводе других образцов, то как вы
узнаете, что устранили эту проблему, сделав некоторые изменения в системе?
17.3.5. Замена частей
Замена подозрительной части на исправную — будь то колонка, обратный клапан,
печатная плата, детектор или другая часть — является одним из самых простых
и действенных способов выявления проблемы. Эта стратегия — хороший аргумент
в пользу того, чтобы в лаборатории было несколько систем от одного производителя
и одной модели. В таком случае у вас будет больше возможностей для
эквивалентных замен. Всегда держите под рукой на замену побольше таких расходных
материалов, как фильтры, предколонки, колонки, капилляры и фитинги.
/7.4. Общие симптомы ВЭЖХ-проблем 863
17.3.6. Вернуть на место
Это правило идет рука об руку с правилом замены частей. Оно напоминает нам
о том, что, если мы заменили подозрительную часть системы на заведомо хорошую,
и это проблемы не устранило — верните на место замененную часть. Это позволяет
избежать накопления использованных частей подозрительного качества. Разумеется,
при этом нужно руководствоваться здравым смыслом — возвращать на место старое
уплотнение насоса не нужно, даже если замена уплотнения насоса проблему не
решила.
17.4. Общие симптомы ВЭЖХ-проблем
Один из наиболее эффективных способов вычленить и устранить ВЭЖХ-проблему —
всегда следить за симптомами распространенных неисправностей, перечисленными
ниже. Хорошее правило, помогающее выявить неисправности на ранних этапах
(до того, как начнутся реальные сложности) — тщательная ежедневная проверка
системы. Например, в начале дня, во время уравновешивания системы, проследите путь
потока через систему в поисках отклонений от нормы. Начните с резервуара
для элюента (достаточно ли элюента, нет ли видимых загрязнений), осмотрите
капилляр к насосу (нет ли утечек и постоянно ли давление), затем автосамплер (нет ли
лужиц растворителя, достаточно ли промывочного растворителя), колоночный
термостат (нет ли утечек), детектор (нет ли утечек) и сливную емкость (не
переполняется ли). Регулярно следуя этому правилу, вы можете обнаружить неисправности еще
до ввода образцов в систему. Кроме того, вы узнаете, как работает система, когда
она работает должным образом, чтобы понять, когда что-то работает не так. Вряд ли
вы станете записывать звуки, которые издает хроматограф во время работы, но вы
распознаете щелчки и гудение, которыми сопровождается нормальная работа
ВЭЖХ-системы. Это поможет вам, в случае необходимости, скорее понять, что
работает не так и расследовать это «не так».
Список часто встречающихся симптомов в этом разделе не является
исчерпывающим, но он должен охватывать наиболее распространенные проблемы ВЭЖХ.
Для получения дополнительной информации обратитесь к работам |1—5|, данным в
разделе 17.1. Текст настоящего раздела удобно использовать совместно с данными
таблиц 17.2—17.11, где каждый симптом связан с потенциальными источниками
неисправности и дана перекрестная ссылка на нижеследующее обсуждение. Обратите
внимание на то, что эти таблицы сгруппированы в разделе 17.5 в конце главы для того,
чтобы было удобнее ссылаться. Если же вы выходите за уровень своей
компетентности (см. раздел 3.10.3.1), не стесняйтесь обратиться за помощью к более опытному
сотруднику или вызвать сервисную службу. Первый раз, когда вы ремонтируете
механику, например, меняете уплотнения насоса, может быть полезным дополнить схе-
му(ы) замены, предлагаемую производителем, цифровыми фотографиями или
эскизом, чтобы вы могли произвести правильную сборку.
У неисправности может быть несколько симптомов. Например, плохо
закрученный фитинг может давать как утечку, так и пониженное давление в системе. Чтобы
не повторяться, каждый симптом в отдельности подробно обсуждаться не будет.
Наконец, описания симптомов и проблем, приведенные в этом разделе, основаны
на том предположении, что система и/или метод работали должным образом до того
момента, как симптом был обнаружен. По этой причине полезно рассмотреть, какие
изменения были внесены с тех пор, как система в последний раз работала
приемлемо — это может сузить область поисков возможных источников проблем.
864 Глава 17. Неисправности и способы их устранения
17.4.1. Утечки
Утечки подвижной фазы ямяются одним из самых распространенных источников
проблем в ВЭЖХ. Их локализовать и выявлять проще всего. Многие ВЭЖХ-системы
содержат датчики утечек, расположенные в местах наиболее вероятных утечек —
например, в термостате для колонок. Как правило, датчик содержит пару
электрических контактов, которые, при замыкании их небольшим количеством жидкости,
активируют сигнал тревоги, предупреждающий пользователя о наличии утечки. Эти
датчики расположены в нижней точке прибора/модуля таким образом, что любой
вытекший растворитель будет собираться в этой точке и включать датчик.
Некоторые утечки можно обнаружить при осмотре. Они и датчиком будут легко
обнаружены. Медленные утечки может быть не так просто обнаружить, поскольку
жидкость будет испаряться до появления капли (однако, можно увидеть выпавшие
кристаллы из буферных растворов в месте утечки). Для обнаружения таких
микроутечек может пригодиться самодельный детектор утечек (раздел 17.2.5.5).
Краткое описание причин утечек в ВЭЖХ-системе приведено в таблице 17.3 и
содержит перекрестные ссылки на последующие разделы.
17.4.1.1. Утечки на линии до насоса
Такие утечки находятся на стороне низкого давления системы ВЭЖХ (рис. 3.1) и чаще
всего происходят из-за плохо завинченного фитинга (раздел 3.4.2.1). Всякий раз, когда
свинчивают детали, изготовленные из двух разных материалов, происходит ослабление
такого соединения со временем из-за различий в коэффициентах теплового
расширения. Кроме того, ослабление ускоряется вибрациями, как это происходит в клапанной
коробке систем с формированием градиента на стороне низкого давления. Если такое
ослабление от вибраций происходит постоянно, можно сделать соединение более
надежным, приобретя фитинги низкого давления с контргайками, которые производят
несколько компаний. При нахождении ослабленного фитинга подтяните его
(например, на четверть или на пол-оборота). Если фитинг и после этого подтекает —
разберите и осмотрите его на предмет повреждений или загрязнений. В случае сомнений
замените его. Почти все фитинги низкого давления взаимозаменяемы (при условии, что
шаг резьбы у них совпадает, т.е. совпадает диаметр и количество ниток дюймовой или
метрической резьбы на единицу длины). Вследствие этого часто переходят на фитинги
другого производителя, если заводские выходят из строя.
В системах со смешиванием на стороне высокого давления (рис. 3.14)
единственные места, где есть фитинги низкого давления — это место, где входной капилляр
для подвижной фазы соединен с входным обратным клапаном, и место, где поток
разделяется при входе в насос в том случае, если он с двумя головами. В этом случае
источник утечки очевиден.
В системах со смешиванием на стороне низкого давления (рис. 3.15) в
дополнение к фитингам низкого давления, подходящим к голове насоса, имеются такие же
фитинги, установленные на клапанной коробке, используемой для смешивания
подвижной фазы в нужных пропорциях. Эти фитинги являются еще одним возможным
источником утечек. Дозирующие клапаны тоже могут протекать, хотя и случается это
редко. Если внизу под клапанной коробкой вы увидели жидкость — осторожно
подтяните винты, фиксирующие дозирующий(е) клапан(ы) на его месте. Винты могут
быть металлическими, а место, куда их ввинчивают — пластмассовым. Будьте
осторожны — болты легко перетянуть и сорвать резьбу в пластике. Если дозирующие
клапаны продолжают подтекать, лучше заменить всю клапанную коробку на новую,
поскольку детали для таких коробок тщательно подгоняют при сборке на заводе,
чтобы обеспечить эффективную эксплуатацию.
/7.4. Общие симптомы ВЭЖХ-проблем 865
Кроме того, возможно проникновение воздуха в капилляр на входе в насос.
Воздух может проникать даже через очень маленький зазор — такой, через который
жидкость не проникнет. Такое проникновение приведет к тому, что создаваемое
насосом давление будет ниже нормального (подробности см. в разделе 17.4.2.2).
17.4.1.2. Утечки в насосе
В насосе существует немало деталей и узлов, которые могут протекать. Утечки часто
сопровождаются падением или скачками давления (разделы 17.4.2.2 и 17.4.2.3).
Фитинги низкого давления обычно используются на входе в насос (раздел 17.4.1.1),
а фитинги высокого давления (раздел 17.4.1.3) на выходе из него. Для получения
более подробной информации обратитесь к этим разделам. Кроме того, источниками
утечек могут быть обратные клапаны, уплотнения насоса и вспомогательные узлы,
такие как устройства, демпфирующие пульсации насоса.
Внешние утечки подвижной фазы из клапанов есть следствие того, что клапан
или плохо закреплен/затянут, или перетянут, или его седло повреждено. Плохо
затянутый обратный клапан — наиболее распространенный источник утечек. Подтяните
с помощью гаечного ключа корпус обратного клапана (например, на 1/8—1/4
оборота), чтобы устранить эту проблему. Во время этой процедуры непременно
придерживайте насос, чтобы не повредить ни сам насос, ни капилляры. Если подтягивание
не устранит утечку, убедитесь, что клапан правильно ввинчен в голову насоса.
Выключите насос и снимите обратный клапан. Обратные клапаны должны свободно
поворачиваться пальцами после их ослабления1. Если для установки или снятия
обратного клапана требуется гаечный ключ, то это в некоторых случаях может
происходить от того, что резьба повреждена2. Проверьте резьбу на наличие повреждений и
замените поврежденный обратный клапан3. В некоторых случаях может быть
сорвана резьба внутри головы насоса, что потребует ее (головы) замены. После осмотра
корпуса обратного клапана осмотрите седло — то место, где нижняя часть клапана
соприкасается с головой насоса. Нижняя часть клапана может быть изготовлена из
жесткого полимерного материала и может треснуть. Если она треснула или
повреждена, необходимо ее заменить. В таком случае лучше всего почитать руководство по
обслуживанию конкретно вашего насоса для выяснения деталей его устройства.
Если уплотнения насоса не менять регулярно в соответствии с программой
профилактического обслуживания (раздел 17.2.2), то они будут самым
распространенным источником утечек. О назначении насосных уплотнений сказано в разделе 3.5.1.
Поскольку уплотнение насоса во время работы непрерывно истирается о
движущийся плунжер, оно является наиболее подверженной износу частью ВЭЖХ-системы.
По мере износа уплотнения оно становится все менее эффективным, что в итоге
приведет к утечкам и/или колебаниям давления. Если уплотнения насоса меняют
каждые 6—12 месяцев, то утечки будут редки. По этой причине мы настоятельно
рекомендуем периодически заменять уплотнения насоса в рамках профилактического
обслуживания. Помимо утечек, истирание насосного уплотнения приводит к
образованию мелких частиц, которые и сами по себе опасны, поскольку могут засорять ка-
1 Строго говоря, свободно поворачиваться должен картридж (в котором находится,
собственно, клапанная пара) внутри держателя, в который он вставлен. Именно держатель с нарезанной
на нем наружной резьбой и завинчивается в голову насоса. — Прим. перев.
2 Гаечный ключ для установки и снятия обратного клапана нужен всегда дня того, чтобы
окончательно подтянуть держатель клапана или отпустить в самом начале при откручивании. До или
после этих двух моментов держатель клапана должен завинчиваться/вывинчиваться пальцами.
Если же этого сделать невозможно, то, скорее всего, резьба сорвана или в нее попали
механические загрязнения. — Прим. перев.
3 Вовсе необязательно менять его весь. Достаточно заменить держатель картриджа клапана. —
Прим. перев.
866 Глава 17. Неисправности и способы их устранения
пилляры, фильтры и колонки. Как правило, если уплотнение протекает, под головой
насоса появляется жидкость, поскольку большая часть насосов имеет небольшое
дренажное отверстие в этом месте.
Перед заменой уплотнения насоса обратитесь к руководству по эксплуатации
насоса для получения конкретных рекомендаций. Общая схема замены уплотнения
следующая:
• Выключите насос и отсоедините входной и выходной капилляры у входного и
выходного клапанов.
• Удалите крепежные винты головы насоса. Обычно для этого требуется
шестигранный ключ. Последовательно понемногу ослабляйте каждый винт, пока голову
насоса нельзя будет снять (голова насоса не должна перекашиваться в процессе
разборки). Снимите голову так, чтобы не перекашивать ее и таким образом не
оказывать бокового давления на хрупкий плунжер. В некоторых случаях голова насоса и
плунжер конструктивно объединены в общий узел и потому извлекаются из
корпуса насоса как общее целое.
• Извлеките уплотнение насоса. Если насос поставляется с инструментом для
извлечения уплотнения — используйте его. В качестве альтернативы специальному
инструменту можно использовать латунный винт (аналогично тому, как используют
штопор). Если вы решите поддеть уплотнение — используйте пластмассовый
инструмент (не отвертку или другой металлический инструмент), чтобы не повредить
голову насоса. Не используйте для этих целей плунжер — он может сломаться.
Когда вы будете извлекать уплотнение — обратите внимание на то, как оно
установлено. Открытая сторона уплотнения (та, где видна браслетная пружина)
должна быть обращена к стороне высокого давления камеры насоса.
• Очистите плунжер. Как правило, для этого достаточно промыть его спиртом и
протереть одноразовой лабораторной салфеткой. Если же не удается полностью
очистить плунжер, то можно прибегнуть к его полировке небольшим количеством
зубной пасты для окончательной очистки (избегайте применения для этих целей
фторсодержащей зубной пасты в тех случаях, когда система используется для
ионной хроматографии). Зубная паста достаточно абразивна, чтобы удалить соли,
содержащиеся в буферах, или другие отложения, не царапая при этом сапфировый
плунжер.
• Осмотрите плунжер на предмет наличия царапин или дефектов. Проще всего это
сделать поднеся лазерную указку к концу плунжера — последний при этом будет
ярко светиться. Любые дефекты поверхности поршня представляют собой
царапины или присохшие частицы. Осмотрите конец плунжера, чтобы убедиться в том,
что он не имеет сколов, трешин и не шероховат. Если дальнейшая очистка
не устраняет дефекты поверхности, замените плунжер.
• Установите новое уплотнение. Обычно это можно сделать, вдавив его в
посадочное место в голове насоса кончиком пальца. Убедитесь в том, что вы установили
его в правильном направлении. Большинство уплотнений имеют фланец,
предотвращающий установку в неправильном направлении, но некоторые конструкции
уплотнений могут быть установлены и в обратном направлении.
• Смочите уплотнение и плунжер небольшим количеством спирта, чтобы уменьшить
трение между ними.
• Надвиньте корпус головы насоса на плунжер, стараясь не перекосить корпус и
не сломать плунжер.
• Последовательно начинайте понемногу закручивать крепежные винты головы,
стараясь не допускать перекоса, что может привести к поломке плунжера. Если в
инструкции по обслуживанию дана величина крутящего момента — затяните винты
в соответствии с этими величинами. В противном случае затяните винты
достаточно надежно.
/7.4. Общие симптомы ВЭЖХ-проблем 867
• Вновь присоедините все капилляры и прокачайте насос на максимальной
скорости потока (без присоединенной колонки, чтобы вымыть все посторонние
частицы). Теперь насос готов к использованию.
Утечка может произойти и в узлах, не относящихся непосредственно к голове
насоса — например, в демпфирующих устройствах, миксерах или датчиках давления.
Если утечка связана с фитингом, следуйте обычной процедуре устранения утечек
в фитингах высокого давления (раздел 17.4.1.3) — сначала подтяните фитинг и, если
это не устраняет утечку, очистите или замените его. Если источник утечки находится
в демпфирующем устройстве или миксере и причина утечки неочевидна или не
может быть легко устранена, возможно, придется этот узел заменить.
17.4.1.3. Утечки на стороне высокого давления
Чаще всего утечки в системах ВЭЖХ возникают в фитингах высокого давления.
Такие утечки обнаруживают благодаря сигналу датчика утечек или при образовании
лужицы. Менее очевидные признаки утечек — понижение или скачки давления
(разделы 17.4.2.2 и 17.4.2.3) или появление белого осадка кристаллов буфера в месте, где
гайка ввинчивается в фитинг. Иногда незначительные утечки удается обнаружить
с помощью полоски термобумаги (раздел 17.2.5.5). (Если вы не знаете о составных
частях и правильной сборке фитингов высокого давления, прочитайте раздел 3.4.2.2
и изучите рис. 3.8 и 3.9).
В большинстве случаев протекающий фитинг можно уплотнить, подтянув его
на четверть или на пол оборота. Обратите внимание, что, если фитинги из ПЭЭК,
следует выключить насос, ослабить гайку, плотно вставить капилляр в фитинг и
потом вновь затянуть гайку. В ином случае конец капилляра может сместиться в фи
тинге, образовав мертвый объем (см. рис. 3.9я). Если при повторном затягивании
утечка не устраняется, разберите соединение, промойте его и соберите вновь. Если
течет по-прежнему, замените ферулу. Можно использовать ферулы из ПЭЭК в
случае не совсем гладкой поверхности соединителя. Ферулы из ПЭЭК можно
использовать при обычных для систем ВЭЖХ давлениях (до 400 бар или 6000 psi). Следует
иметь в виду, что для систем с более высоким давлением для безопасных
соединений, гарантирующих отсутствие утечек, требуются фитинги из нержавеющей стали.
17.4.1.4. Утечки в автосамплере
В настоящее время почти все системы ВЭЖХ работают с автосамплерами. Но если
в вашей системе используется ручной инжектор, к нему применимы такие же
профилактические меры, потому что ручные инжекторы и краны ввода образцов в авто-
самплерах очень сходны. Конструкция и функционирование ручного инжектора и
автоматического инжектора в автосамплере обсуждаются в разделах 3.6.1 и 3.6.2,
соответственно. Утечки в фитингах низкого и высокого давления в автосамплере
следует устранять таким же образом, как утечки на линии до насоса (раздел 17.4.1.1)
или в других фитингах высокого давления (раздел 17.4.1.3). Остальные утечки можно
разделить на две категории: связанные с инжектором и не имеющие отношения
к нему.
Утечки, связанные с краном для ввода образца, возможны либо в одном из
фитингов или соединений, либо в результате протекания уплотнения ротора инжектора.
В автосамплерах с заполнением петли нагнетанием (раздел 3.6.2.2, рис. 3.21) и
в большинстве ручных инжекторов соединение иглы с краном для ввода образца
осуществляется через уплотнение, рассчитанное на низкое давление; оно показано
на схеме рис. 17.2. Гайка (г) и ферула (ф) используются для фиксирования
полимерной трубки (т) в корпусе крана (к). Вблизи ферулы трубка слегка обжимается (с),
герметизируя иглу. Эта трубка может износиться или деформироваться со временем
868 Глава 17. Неисправности и способы их устранения
Рис. 17.2. Схема уплотнения иглы, рассчитанного на низкое дапление. Корпус крана
(к), гайка (г), ферула (ф), полимерная трубка (т) и деформированная часть
трубки (претерпевшая усадку), образующая уплотнение вокруг иглы (у)
так, что жидкость будет вытекает сверху из трубки, когда образец вводится в петлю.
Слегка подтянув гайку, часто удается обжать трубку более плотно и ликвидировать
утечку. В других случаях может потребоваться замена трубки. Обратитесь к
инструкции руководства по обслуживанию автосамплера.
В автосамплере со встроенным петлевым дозатором (раздел 3.6.2.3, рис. 3.22)
требуется расчитанное на высокое давление уплотение в месте ввода иглы в кран
инжектора. Это уплотнение изготавливается из твердого пластика, такого как ПЭЭК,
или другого твердого материала, который может со временем износиться,
деформироваться или потрескаться. Иногда при затягивании гайки, фиксирующей
уплотнение высокого давления, можно устранить утечку, но обычно в случае утечки через
это уплотнение потребуется заменить его новым.
Повреждение уплотнения ротора инжектора может привести к утечкам в кране
инжектора. В стандартном шестиходовом кране инжектора используется
вращающееся уплотнение, ротор, который поворачивается относительно стационарного
статора, к которому подсоединяются капилляры. Обычно ротор изготовляется из твердого
полимера такого, как ПЭЭК1 или Веспел2 или другого фторсодержащего полимера.
В нем сделаны маленькие канавки, соединяющие каналы подвода жидкости,
находящиеся в статоре. Статор может быть частью корпуса крана из нержавеющей стали,
а может быть вставкой из керамики или другого материала, имеющего твердую
гладкую повехность. На схеме рис. 3.17 ротору соответствует часть поворотного крана,
находящаяся внутри окружности, а статору — часть, расположенная сразу же за ее
краями. В наиболее распространенных инжекционных кранах ротор представляет
собой плоский диск с тремя почковидными канавками на поверхности. Подобный
показан на схеме рис. 17.3л. Эти канавки совмещены с выходами капилляров,
присоединенных к статору, и показаны пунктирными окружностями на рис. 17.35. В одном
положении (например, загрузка) жидкость течет по образовавшемуся каналу,
соединяя позиции 1—2, 3—4 и 5—6, как видно на рис. 17.3ff. Когда ротор поворачивается
на 60° (например, в положение «ввод»), соединяются между собой другие позиции.
В данном примере 2—3, 4—5 и 6—1.
'Англ. РЕЕК — полиэфирэфиркетон. — Прим. перев.
2 Англ. Vespel* DuPont™ — полиимид. — Прим. перев.
17.4. Общие симптомы ВЭЖХ-проблем
Рнс. 17.3. Схема уплотнения ротора инжектора, (а) Вид почковидных канавок на
поверхности полимерного уплотнения; (б) то же, что и (о), показано, как
соединяются позиции (обозначенные цифрами внутри пунктирных
окружностей); (в) царапина между позициями 5 и 6, которая приводит к утечке
между ними, (г) Поперечное сечение почковидной канавки (нормальное
состояние); (д) изношенное уплотнение с заусенцами на краях (показаны
стрелкой)
Если небольшая твердая частица, такая как частица сорбента, которым
упакована колонка, или кусочек нержавеющей стали из плохо обрезанного капилляра
застревает в одной из канавок инжектора, он может поцарапать ротор. Таким образом
может образоваться канал между двумя канавками, как показано стрелкой
на рис. 17.3* между позициями 4 и 5. Это приводит к возникновению утечки между
позициями, когда жидкость из одной гидравлической части системы протекает в
другую. Это проявляется в вытекании жидкости из отверстия ввода или отверстия слива.
При этом нарушается точность дозирования образца из-за затекания жидкости в
петлю или ее вытекания из петли, а также может привести к задерживанию
«остаточных» примесей в системе (раздел 17.2.5.10).
Для устранения этой проблемы потребуется заменить уплотнение ротора (а
возможно и статор). Необходимо также тщательно промыть кран инжектора, чтобы
удалить все твердые частицы.
Чаще уплотнение ротора портится в результате износа за счет трения.
Уплотнения ротора расчитаны на использование в течение > 100000 циклов. Для многих
лабораторий это означает несколько лет работы до выхода из строя, поэтому рутинные
замены уплотнения ротора вряд ли целесообразны, за исключенем случая, когда
870 Глава 17. Неисправности и способы их устранения
в системе есть счетчик, автоматически регистрирующий количество циклов инжек-
ции и позволяющий оценить срок службы уплотнения.
В поперечном сечении канавки ротора, показанные на рис. 17.3о, имеют U-об-
разную форму (см. рис. 17.3г). По мере изнашивания поверхности при нормальной
работе ротор становится несколько тоньше, и часто на краях канавок образуются
небольшие заусенцы из материала ротора, показанные стрелкой на рис. 17.3d. Эти
заусенцы могут отломиться, образуя частицы, которые могут засорить капилляры или
фильтры. По мере изнашивания поверхности ротора уменьшается контактное
давление между ротором и статором, и со временем уплотнение начинает протекать.
Замена ротора решит эту проблему. Кран перед сборкой тщательно промойте.
Предел давления на уплотнение между ротором и статором крана связан с тем.
насколько плотно прижаты друг к другу их поверхности. Чем выше предельное
давление — тем плотнее прижаты поверхности ротора и статора, но при этом трение
между ними возрастает, как и усилие, прикладываемое для поворота крана.
Обычно инжекционные краны отрегулированы, чтобы выдерживать давление 400 бар
(6000 psi) при использовании в традиционной ВЭЖХ. В УВЭЖХ (> 400 бар,
раздел 3.5.4.3) поверхности ротора и статора ложны быть прижаты плотнее, поэтому
срок службы ротора должен стать меньше. Использование альтернативных
конструкций инжектора помогло бы решить эту поблему. После разборки инжекционного
крана для технического обслуживания, может потребоваться отрегулировать
давление на уплотнение между ротором и статором. Обратитесь к руководству по
обслуживанию за дополнительными инструкциями.
Основным исключением из обычно используемых поворотных инжекционных
кранов являются инжекторы, применяемые в некоторых моделях автосамлеров
фирмы Waters. В этих инжекторах обычно используются наборные/пакетные уплотнения
высокого давления в конструкции, включающей в себя золотниковые клапаны
вместо поворотных кранов. Некоторые из составляющих этого клапана могут
обслуживать пользователи, а некоторые требуют замены всей сборки или повторной сборки
всего узла. Обратитесь к руководству для пользователей за дополнительной
информацией по устранению проблем и ремонту.
Места и причины утечек в автосамплерах различаются у разных моделей или
часто характерны только для одной модели. Если затягивание фитинга или замена
соединения не устраняют утечку, обратитесь за дополнительной информацией к
руководству для пользователей.
17.4.1.5. Утечки в колонке
Утечки в колонке связаны с фитингами. Устранение утечек в местах соединения
капилляров описано в разделе 17.4.1.3. Если протекает соединение самой колонки,
можно попробовать устранить течь, затянув гайку на 1/4 оборота. Затягивание таких
гаек большего диаметра может потребовать большего усилия, чем затягивание
фитингов диаметром 1/16 дюймов, соединяющих капилляры. Если соединение все-таки
протекает, может быть лучше заменить колонку, потому что разборка концевого
фитинга колонки может привести к ее неустранимому повреждению. В прошлом
принято было удалять входной фитинг колонки, чтобы заменить фильтр, если
предполагали, что он засорился. Но при использовании современной технологии упаковки
колонок удаление концевого фитинга может привести к медленному вытеканию
упакованного сорбента и повреждению колонки. По этой причине удаление концевого
фитинга для обследования или починки более не рекомендуется.
Колонки типа картриджей состоят из одноразовой колонки и держателя,
подлежащего многоразовому использованию. Если подтекает концевой фитинг колон-
ки-катриджа, попробуйте подтянуть его для устранения проблемы. Если фитинг про-
/7.4. Общие симптомы ВЭЖХ-проблем
должает протекать, разберите держатель, промойте фитинг и вновь соберите
держатель. В некоторых случаях для устранения протечки может потребоваться
замена полимерного уплотнения между колонкой и держателем.
17.4.1.6. Утечки в детекторе
Давление на выходе колонки ниже на порядок или более давления на входе, так что
детекторы испытывают гораздо меньшее давление, чем составляющие, находящиеся
под высоким давлением. Так как проблемы утечек непосредственно связаны с
локальным давлением в системе, утечки в детекторах гораздо реже встречаются, чем
в других частях системы ВЭЖХ.
Фитинги на входе детектора обычно того же типа, что и в других частях системы,
находящихся под высоким давлением; утечки в соединениях этих капилляров
следует устранять так, как описано в разделе I7.4.1.3. В некоторых детекторах
используются капилляры диаметром 1/32 дюйма вместо обычных капилляров диаметром 1/16
дюйма, используемых в остальных компонентах системы. Эти капилляры меньшего
диаметра легко изгибаются и закручиваются, поэтому обращайтесь с ними
осторожно. Некоторые детекторы работают при достаточно низком давлении на входе, так
что можно использовать полимерные фитинги низкого давления; во многих
детекторах фитинги низкого давления установлены и на выходе. Устранение утечек в фи
тингах низкого давления описано в разделе 17.4.1.1.
УФ-детекторы (раздел 4.4) наиболее популярны в ВЭЖХ. Типовая ячейка детектора
показана на рис. 17.4. Обычно ячейка представляет собой корпус из нержавеющей стали
(длиной, например, 10 мм) с просверленным в нем отверстием (диаметром, например,
1 мм). Кварцевые окна на концах отверстия удерживаются кольцевыми О образными
уплотнениями. Капилляры на входе и выходе ячейки могут быть соединены
компрессионными фитингами, которые можно регулировать обычным образом (раздел 17.4.1.3)
в случае утечки. В других конструкциях капилляры припаяны или приварены к корпусу
проточной ячейки, и тогда потребуется фабричный ремонт или замена ячейки. На входе
ячейки обычно устанавливается тонкостенный капилляр малого диаметра (например,
с внешним диаметром 1/32 дюйма), который также функционирует как теплообменник,
стабилизирующий температуру в проточной ячейке. Будьте осторожны при затягивании
соединений, не перекручивайте и не изгибайте этот тонкий капилляр.
кварцевое стекло
крышка корпус ячейки
/ * \ уплотнение
/ | выход \
световой
Рис. 17.4. Схема типовой проточной ячейки УФ-детектора
872 Глава 17. Неисправности и способы их устранения
Так как пузырьки воздуха приводят к помехам в показаниях детектора, УФ-детек-
торы часто работают с противодавлением на выходе ячейки (раздел 4.2.1). Таким
образом создается достаточное давление (около 3,4—7 бар или 50—100 psi), чтобы
удерживать пузырьки в растворе, но не слишком большое для того, чтобы протекали
уплотнения, герметизирующие окна. Снабженное пружиной устройство для создания
противодавления хорошо это обеспечивает. Вместо него можно использовать
сливной капилляр малого диаметра (например, с внутренним диаметром <0,01 дюйм =
= < 0,25 мм), но если скорость потока увеличивается, то повышается и давление.
С ограничителем такого типа при высокой скорости потока давление может превысить
верхний предел для ячейки детектора (например, 10 бар или 150 psi), и может
образоваться утечка в уплотнениях окон. Ячейка детектора может продолжать подтекать, если
эти уплотнения деформировались. Некоторые ячейки могут обслуживать
пользователи, в то время как для починки других требуются услуги квалифицированного
специалиста или вообще ячейку приходится заменять. Следует уточнить детали относительно
модели используемого вами детектора в руководстве для пользователей.
В детекторах других типов конструкция ячейки иная, как и канал для подвижной
фазы. В любых проточных детекторах (флуоресцентных, электрохимических,
детекторах проводимости) могут возникнуть утечки в ячейке. Испарительные детекторы,
такие как испарительный детектор светорассеяния, детекторы, в которых
используется коронный разряд, или масс-спектрометрические детекторы могут иметь утечки
только в интерфейсе, так что обнаружить их несколько проще. Ознакомьтесь с соот
ветствующим разделом главы 4, в котором проведено общее обсуждение и
рассмотрены типовые конструкции конкретных детекторов. Эта информация и
дополнительные данные из руководства к детектору помогут вам обнаружить и устранить утечки
в конкретных детекторах.
17.4.2. Аномальное давление
Аномальное давление всегда является симптомом некой другой проблемы в системе
ВЭЖХ. Нормальное давление в различных системах ВЭЖХ и при различном их
использовании отличается, поэтому желательно регулярно записывать давление в
системе. Например, внесите значение давления в записи об анализе партии образцов
или о конкретном разделении как часть теста на пригодность системы; тогда вы
сможете сравнить вызывающие вопросы случаи повышения или понижения давления
в ретроспективе (раздел 17.2.4). Обычные системы ВЭЖХ могут работать при
давлении до 400 бар (6000 psi), но для большинства приложений рабочими являются
значения давления 100—200 бар (1500—3000 psi). В УВЭЖХ-системах рабочие давления
выше 400 бар (6000 psi): до 1000 бар (15000 psi) или выше. Однако обычно эти
системы работают при давлении 550—650 бар (8000—10000 psi).
В идеальном случае давление постоянно при изократическом разделении, но
небольшие флуктуации давления порядка 1—2% рабочего значения (то есть около I —
2 бар или 10—20 psi) считаются нормальными для многих приложений. Давление
также изменяется при изменении состава подвижной фазы, как показано в
таблице 17.1. Поэтому изменение давления при градиентном элюировании — нормальное
явление. Метанол имеет ббльшую вязкость, чем ацетонитрил, а смеси воды с
метанолом обладают гораздо ббльшей вязкостью, чем вода и метанол по отдельности.
С другой стороны, давление при работе со смесями воды с ацетонитрилом
понижается практически линейно при возрастании содержания ацетонитрила от 0% до 80%.
Более подробная информация о вязкости растворителей дана в таблице 1.3
приложения I.
17.4. Общие симптомы ВЭЖХ-проблем 873
Таблица 17.1. Примеры значений давления для различных смесей растворителей
% растворителя В"
0
20
40
60
80
100
Растворитель В
Ацетонитрнл
114 бар (1650 psi)6
127(1840)
117(1700)
96(1390)
70 (1010)
49 (710)
Метанол
114 бар (1650psi)s
174(2520)
225 (3265)
201 (2920)
148(2150)
75 (1080)
" растворитель А вода
''приблизительное (вычисленное по уравнению 2.13а) давление в колонке 250x4,6 мм,
диаметр частиц сорбента 5 мкм, 2 мл/мин, 35 "С.
Большая часть насосов ВЭЖХ имеет ограничение по давлению, при
превышении этого предельного значения насос выключается. Верхний предел давления
защищает систему от повреждения или утечек, если засорение вызывает избыточное
давление. Большинство пользователей задают верхний предел, намного превышающий
нормальное максимальное рабочее давление для всех методов. Например, если
нормальное для данного метода давление 130—200 бар (2000—3000 psi) верхний предел
может быть установлен в интервале 270—340 бар (4000—5000 psi). Некоторую защиту
это обеспечивает, но предел не такой низкий, чтобы насос слишком часто
отключался. Нижний предел давления обеспечивает отключение насоса, если давление
становится ниже заданного значения, например, при истощении подвижной фазы в ре
зервуаре. Опасность повреждения насоса при отсутствии растворителя не очень
велика, и он не будет качать воздух, поэтому колонка не пересохнет. Тем не менее,
рекомендуется задавать нижний предел по давлению примерно I—3 бар (20—50 psi)
так, чтобы насос прекращал работать, когда заканчивается растворитель. Проблемы,
связанные с давлением, обсуждаются далее и систематизированы в таблице 17.4.
17.4.2.1. Слишком высокое давление
Давление выше нормального является симптомом перекрывания потока элюента
(если настройки системы правильные). В наиболее общем случае давление
постепенно возрастает при анализе каждой последующей партии образцов с накоплением
загрязнений в проточном фильтре, предколонке или во входном фильтре колонки. Это
нормальная ситуация, если используется метод анализа образцов, которые могут
быть не полностью освобождены от мелкодисперсных частиц. Помните, что
давление может возрастать на величину до 60% по сравнению с исходным в течение
градиента (раздел I7.4.2, таблица 17.1) и устанавливайте верхний предел с учетом этих
нормальных колебаний давления. Важно также помнить, что давление связано с
размером частиц сорбента, диаметром и длиной колонки (уравнение 2.13а).
Фактически перепад давления на эквивалентных колонках может различаться (например,
на колонках Cjg 150 х 4,6 мм с диаметром частиц сорбента 5 мкм, если сорбенты от
разных производителей). Наконец, в системах ВЭЖХ, предназначенных для
работы при более высоком давлении (например, в УВЭЖХ), проходные отверстия
очень невелики, и внутренний диаметр капилляров меньше (например, отверстия
< 0,005 дюймов и внутренний диаметр капилляров < 0,12 мм). Это приводит к
давлению в системе без колонки > 68 бар (1000 psi), поэтому следует принимать во
внимание допуски на нормальное фоновое давление при поиске неисправностей.
874 Глава 17. Неисправности и способы их устранения
Иногда происходит внезапный скачок давления, который приводит к
срабатыванию триггера верхнего предела давления и выключению насоса. Это может
произойти при введении очень грязного образца, такого, как необработанная плазма крови,
если в образце присутствует достаточное количество микрочастиц, чтобы полностью
закупорить фильтр колонки или подводящий капилляр. Закупоривание линии может
также произойти при смешивании буфера и органического растворителя в потоке
в условиях низкой растворимости буфера и выпадения его компонентов в осадок.
Это случается при смешивании фосфатного буфера с ацетонитрилом в некоторых
системах ВЭЖХ.
Определить причину избыточно высокого давления достаточно просто. Следует
последовательно ослаблять все соединения от выхода колонки до насоса при
работающем насосе. {Если вы предполагаете, что засорилось какое-то определенное
соединение, такое, как проточный фильтр, начните с него и сэкономьте время). Если вы
ослабляете соединение, и давление резко падает, засорение произошло в следующем
соединении по направлению к выходу из системы. Помните, что нормальный
перепад давления на колонке > 68 бар (1000 psi), и он зависит от скорости потока.
Поэтому значительное падение давления при ослаблении соединения на входе колонки
вполне закономерно. В обычной системе ВЭЖХ, рассчитанной на работу
при 400 бар (6000 psi), после отсоединения колонки давление должно быть очень
низким (например, 7 бар (100 psi)). Как отмечалось ранее, в системах, рассчитанных
на работу при более высоких давлениях (раздел 3.5.4.3) и с сорбентами, диаметр
частиц которых менее 3 мкм, давление может достигать 68 бар (1000 psi) и выше даже
без присоединения колонки. Для получения контрольных значений нормального
давления в местах соединений может быть полезным использовать методику для
определения засоров при нормально работающей системе (при некоторой
определенной скорости потока).
Когда установлено место засора, нужно принять необходимые меры для его
ликвидации. Например, если засорен капилляр, его следует заменить. Если засорен
проточный фильтр, следует заменить в нем встроенный фильтр. Если засорена колонка,
может помочь ее подключение в обратном направлении (эта методика рассмотрена
при обсуждении расщепленных и искаженных пиков в разделе 17.4.5.3). В
противном случае может оказаться, что надо заменить колонку.
Перед тем, как вновь использовать систему, подумайте, что нужно предпринять,
чтобы проблема не возникла снова, то есть уменьшить вероятность ее появления
в следующий раз. Например, если проблема состоит в засорении фильтров
частицами примесей образца, можно ввести стадию фильтрования или центрифугирования
образца в процедуру пробоподготовки или использовать проточный фильтр
(раздел 3.4.2.3). Если вследствие осаждения буфера засорился капилляр насоса или
миксер, рассмотрите возможность уменьшения концентрации буфера или
предварительного смешивания буфера и органического растворителя.
17.4.2.2. Слишком низкое давление
Слишком низкое давление в системе говорит об утечках или проблемах с насосом
(если все настройки проведены правильно). Если давление нестабильно, скорее
всего, причина в неисправности насоса (это тема обсуждается также в разделе 17.4.2.3),
а если давление стабильное и низкое, более вероятна утечка. Так как проблемы
низкого давления и нестабильного давления тесно связаны, и иногда их трудно
разделить, обратитесь за дополнительной информацией к разделу 17.4.2.3.
Если насос отключился из-за того, что давление упало ниже установленного
предела, и причина не очевидна (очевидная причина — например, отсутствие в
резервуаре подвижной фазы), проведите повторный запуск системы и посмотрите, что
/7.4. Общие симптомы ВЭЖХ-проблем 875
будет. Иногда датчик давления, регистрирующий его нижний предел, слишком
чувствителен и выключает насос при мгновенном перепаде давления, например,
при прохождении пузырька воздуха через насос. Если это происходит регулярно,
может быть, лучше отключить датчик нижнего предела давления.
Утечки подвижной фазы также могут вызывать падение давления. Найдите
источник утечки и устраните его, используя указания, приведенные в разделе 17.4.1 и
таблице 17.3. Хотя чаще всего наблюдаются утечки подвижной фазы, возможно и
попадание воздуха в систему через недостаточно уплотненные фитинги низкого
давления (раздел 17.4.1.1). Если попытки найти утечку жидкости безуспешны, затяните
каждый из фитингов низкого давления, чтобы узнать, не устранит ли это проблему.
Если клапаны, формирующие градиент (КФГ), не уплотнены должным образом,
воздух может засасываться в систему через неиспользуемый капилляр подачи
растворителя.
Залипающий входной обратный клапан может препятствовать созданию в насосе
достаточного давления. Залипание обратного клапана особенно часто происходит
при использовании шариковых обратных клапанов (рис. 3.12а) с ацетонитрилом.
Когда в качестве растворителя используется ацетонитрил, механически обработанная
поверхность сапфирового седла клапана может катализировать полимеризацию
минорных примесей ацетонитрила (алифатических аминов) [15, 16]. Образующийся
полимер сглаживает контактную поверхность, так что рубиновый шарик прилипает
к седлу из-за увеличивающегося поверхностного натяжения. Обработка ультразвуком
в метаноле, по-видимому, устраняет эту проблему, по крайней мере, на время.
Предполагается также, что обработка ультразвуком в разбавленной азотной кислоте
может удалить этот полимерный налет |17|, но это предположение не было
подтверждено к моменту завершения этой книги. Насосы ВЭЖХ, в которых используется
активный клапан, не подвержены залипанию. К сожалению, нельзя заменить
активными обратными клапанами шариковые обратные клапаны, если они уже
установлены в насосе.
Насос, в который не поступает достаточное количество подвижной фазы,
не сможет создавать нужное давление. Проверьте, имеется ли достаточное
количество подвижной фазы для подачи в насос, проведя тест с сифоном, описанный в
разделе 3.2.1. Препятствиями для свободного течения подвижной фазы могут быть
засоренные фильтры на линии подачи, неисправные КФГ и пережатые или засоренные
капилляры подачи подвижной фазы.
Если подвижная фаза недостаточно дегазирована или насос недостаточно
промыт на максимальной скорости потока, в подвижной фазе остается воздух и у насоса
уменьшается расход, а давление падает. Обеспечьте эффективное дегазирование
подвижной фазы (раздел 3.3) и промойте насос на большой скорости 10—20 мл
дегазированной подвижной фазы в слив, открыв сливной вентиль (высокая скорость
потока иногда позволяет удалить пузырьки воздуха из насоса). Часто возникающие
проблемы с появлением пузырьков воздуха могут указывать на неисправность
модуля дегазирования. Если есть такие подозрения, попробуйте дегазировать другим
методом (раздел 3.3), чтобы исключить эту проблему.
Изношенное уплотнение насоса может приводить к утечкам (раздел 17.4.1.2),
но прежде, чем утечки будут заметны, износ уплотнений может помешать насосу
нагнетать нужное давление. Тщательно проверьте, не протекает ли уплотнение около
дренажного отверстия с нижней стороны головы насоса (между входным обратным
клапаном и корпусом насоса) и проверьте записи об обслуживания системы
(раздел 17.2.4), чтобы узнать, не пришло ли время заменить уплотнения. Замена
уплотнений (раздел 17.4.1.2) должна решить все проблемы. Когда вы вынете голову из
насоса, проверьте, не поврежден ли плунжер: сломанный или поцарапанный плунжер
также может быть причиной недостаточной эффективности насоса.
876 Глава 17. Неисправности и способы их устранения
17.4.2.3. Нестабильность давления
Как упоминалось выше, в системе ВЭЖХ давление изменяется в нормальном
режиме работы. Обычно эти колебания давления составляют 1—2% рабочего давления
(например, 1—2 бар или 10—20 psi), но колебания давления отличаются в разных
системах и режимах работы. Поэтому рекомендуется указывать характерные
флуктуации давления в отчетах для каждой партии образцов (раздел 17.2.4). Также следует
помнить об изменениях давления в каждом цикле градиентного элюирования.
Обзор признаков нестабильности давления и способов решения этой проблемы
дан в таблице 17.4. Наиболее распространенными причинами нестабильности
давления являются пузырьки воздуха в насосе, залипание в обратных клапанах,
изношенность уплотнений или поломка плунжера в насосе и недостаточная подача подвижной
фазы в насос. Определить и устранить эти неисправности можно таким же образом,
как проблемы со слишком низким давлением (раздел 17.4.2.2), поэтому
дополнительные инструкции не требуются. Основное различие между проблемами с низким
давлением и нестабильным давлением заключается в том, что нестабильность может быть
обусловлена дефектами лишь в одной из голов двухплунжерного насоса или одного
из двух насосов, используемых в системе. Таким образом, часть системы работает
нормально (тогда давление выше), а часть системы не доставляет нужное количество
подвижной фазы (давление ниже). Наиболее вероятных источников проблемы два:
пузырек воздуха в голове насоса или залипший обратный клапан. Для устранения
неисправности проще всего сначала промыть насос с большой скоростью и
посмотреть, не устранит ли это проблему. Если нет, обработайте клапан ультразвуком в
метаноле (раздел 17.4.2.2). Дополнительную информацию можно найти в таблице 17.4.
17.4.3. Нестабильность времени удерживания
Если обеспечено постоянство всех условий хроматографии, времена удерживания
анализируемых веществ должны оставаться постоянными для данной партии
(например, в течение рабочего дня или при использовании одной и той же партии
подвижной фазы). Если наблюдается изменение удерживания, это означает, что, по крайней
мере, один параметр изменился. Поскольку все параметры невозможно в точности
поддерживать постоянными, для каждого метода существуют допустимые
отклонения времени удерживания. Обычно эти значения лежат в интервале ±0,5% времени
удерживания или ±0,02—0,05 минут. Значение нормального отклонения можно
узнать из хронологических записей о проведенных анализах образцов. Когда
колебания времени удерживания превосходят допустимые пределы, следует принять меры
для поисков причины этих отклонений и способа их устранения.
Применяя подход «разделяй и властвуй» (раздел 17.3.1), следует рассчитать
коэффициент удерживания к (уравнение 2.6) и сравнить изменения к и времени
удерживания Гц с помощью таблицы I7.5. Таблица 17.5 укажет вам на один из следующих
разделов, содержащий дополнительную информацию о возможных изменениях
подвижной фазы, параметров колонки или ее температуры. Как более подробно
изложено далее, изменения подвижной фазы обычно происходят скачкообразно, когда
намеренно вносится какое-либо изменение, изменение состояния колонки
происходит в течение недель или месяцев, а изменения температуры имеют тенденцию
циклически изменяться в течение дня. Эти общие закономерности могут быть очень
полезны при определении источника проблемы.
Помочь разобраться в причинах изменений времени удерживания может также
характер наблюдаемых изменений гд: увеличивается оно, уменьшается или
колеблется. Если вы хотите использовать этот подход, обратитесь сначала к таблице 17.6 и
разделу 17.4.3.6. В приведенных ниже разделах (17.4.3.1—17.4.3.5) рассмотрен каждый
/7.4. Общие симптомы ВЭЖХ-проблем 877
из симптомов. На этом этапе можно рекомендовать тестирование системы
(разделы 3.10.1.3, 17.2.1) для того, чтобы определить, связана ли проблема с оборудованием
или методом. Наконец, многие причины нестабильности удерживания связаны друг
с другом, поэтому целесообразно прочитать все разделы с 17.4.3.1 до 17.4.3.5, чтобы
рассмотреть как можно больше версий происхождения отклонений, если проблему
не удается быстро решить.
17.4.3.1. Проблемы, связанные со скоростью потока
При изменении скорости потока изменится 1ц, но не к, потому что к не зависит
от скорости потока, в то время как г^ обратно пропорционально скорости потока
(за исключением условий, когда давление больше 340 бар (5000 psi) из-за некоторой
зависимости к от давления). Никогда не надо недооценивать возможность ошибки
оператора — полезно проверить, что задана нужная скорость потока. Если ошибка
в задании скорости исключена, единственной возможной причиной превышения
нормальной скорости потока является проблема с контроллером системы. Для
устранения этой проблемы нужен специалист.
Если скорость потока меньше заданной, времена удерживания возрастают. Это
может быть вызвано пузырьками воздуха в насосе, недостаточным количеством элю
ента для питания насоса, повреждением обратных клапанов или уплотнений насоса,
или утечками. В разделах 17.4.1 и 17.4.2.2 описаны меры, которые следует принять
для устранения этих проблем. Как сказано в разделе 17.4.2.3, часто нестабильность
давления и скорости потока обусловлена теми же причинами, что и занижение
давления и скорости потока.
17.4.3.2. Проблемы, связанные с размером колонки
Редко встречающейся, тем не менее, возможной причиной изменения гл, но не к,
является установка колонки нужного вида (например, С]8 Symmetry), но не того
размера. Безусловно, размер колонки не может измениться без участия оператора.
Очевидный способ решения проблемы состоит в том, чтобы сначала прочитать этикетку
на колонке, а уже потом устанавливать подходящую.
17.4.3.3. Проблемы, связанные с подвижной фазой
Изменение подвижной фазы (например, %В) может привести к изменению и гл, и к.
Если при приготовлении новой партии подвижной фазы допущена ошибка, любые
изменения удерживания будут сразу заметны вне зависимости от того,
использовалось ли предварительное смешивание или смешивание в потоке. Когда используется
смешивание в потоке, небольшие изменения состава подвижной фазы могут
возникнуть из-за проблем, связанных с системой КФГ. С другой стороны, небольшие
изменения состава подвижной фазы (и удерживания образца) могут постепенно
происходить с течением времени, хотя это случается гораздо реже. Например, может
испаряться летучий буфер, такой как карбонат аммония, изменяя рН подвижной
фазы. Если для дегазирования применяется непрерывное продувание гелием, может
избирательно испаряться органический компонент подвижной фазы.
Ошибки при приготовлении подвижной фазы являются вероятной причиной
сдвигов удерживания (с увеличением удерживания при уменьшении %В и наоборот).
«Правило 2,5» (раздел 2.5.1) указывает, что изменение %В на 10% вызывает
изменение коэффициента удерживания почти в 2,5 раза, а однопроцентная ошибка в %В
подвижной фазы может привести к изменению к на к 10%.
Ошибка в рН подвижной фазы может в гораздо большей степени повлиять
на удерживание кислотных или основных образцов (раздел 7.3.4.1), чем на удержива-
878 Глава 17. Неисправности и способы их устранения
ние нейтральных анализируемых веществ. Введение добавок в подвижную фазу,
таких как ион-парные реагенты (раздел 7.4.1.2), также может повлиять на
удерживание. При разработке метода следует проверить устойчивость разделения к
небольшим изменениям состава подвижной фазы (раздел 12.2.6). Результаты
тестирования устойчивости метода могут быть полезны при определении ошибки,
допущенной при приготовлении подвижной фазы. Однако с точки зрения практики
оптимальное решение — приготовить новую партию подвижной фазы, а потом
проверить, устранена ли проблема.
17.4.3.4. Проблемы, связанные с неподвижной фазой
При длительном использовании колонки обычно изменяются удерживание и
селективность, но /0 остается прежним; следовательно, изменятся и tg, и к. Сдвиги
удерживания, обусловленные изменением неподвижной фазы, редко являются
единственным наблюдаемым симптомом проблем. Обычно также уменьшается количество
теоретических тарелок /V, увеличивается размывание заднего фронта пиков и растет
давление на колонке. Отчеты об использовании метода (хронологические записи,
раздел 17.2.4) в сочетании с недавними данными о характеристиках колонки
(значения N, давления и т.д.) можно использовать, чтобы избежать выхода колонки
из строя во время анализа серии образцов. Как правило, время жизни колонки дол
жно соответствовать 500—2000 вводов образцов для большинства методов (и это
составляет менее 1% полной стоимости анализа). Некоторые методы могут привести к
деградации колонки за более короткое время, а некоторые позволят использовать ее
дольше. Следует ожидать сокращения времени жизни колонки, если она
используется за пределами интервала рН 2 < рН < 8 или при температурах > 50 °С. Обычно
срок службы колонки возрастает, если применяются проточные фильтры
(раздел 3.4.2.3) или предколонки. В любом случае колонка считается расходным
материалом, который со временем (желательно помедленнее) изнашивается.
Самым простым способом проверить проблемы, связанные с неподвижной
фазой, является замена колонки (замена частей, раздел 17.3.5). Если используется пред-
колонка, то в первую очередь следует заменить или удалить ее и посмотреть — не
решается ли таким образом проблема. Если колонка преждевременно неоднократно
выходит из строя, проверьте, соблюдается ли рекомендованный для нее режим
эксплуатации (обратитесь к инструкциям по эксплуатации колонки и уходу за ней). В
некоторых случаях может быть уместным найти и использовать эквивалентную более
стабильную колонку (раздел 5.4.2). Если повреждение колонки вызвано введением
грязных образцов, могут потребоваться дополнительные этапы пробоподотовки
(глава 16).
17.4.3.5. Проблемы, связанные с изменениями температуры
Изменение температуры колонки влияет на значения /r, и к. При увеличении
температуры колонки на I °С удерживание обычно уменьшается на 1—2%, поэтому, если
в методе не предусмотрен контроль температуры колонки, удерживание будет
изменяться в течение дня при изменении температуры в лаборатории. Изменения
температуры могут повлиять и на селективность (раздел 6.3.2), из-за этого также могут
наблюдаться сдвиги относительного удерживания. Во многих лабораториях
температура стабильна в течение дня, но с целью экономии энергии не
контролируется столь же тщательно ночью. Тем не менее, даже если температура в лаборатории
относительно постоянна (в соответствии с показаниями установленного на стене
термометра), возможны значительные локальные флуктуации температуры, особенно
если поток воздуха от обогревателя направлен на систему ВЭЖХ или сопредельное
пространство. По этой причине проблемы, связанные с температурой, имеют тен-
/7.4. Общие симптомы ВЭЖХ-проблем 879
денцию проявляться в виде циклических изменений удерживания в течение дня. Эти
проблемы с удерживанием можно преодолеть, используя колоночный термостат,
работающий в интервале температур, в котором он может обеспечить устойчивый
контроль температуры (раздел 3.7). Недостаточный контроль температуры колонки
может вызвать искажение формы пиков, как описано в разделе 17.4.5.3.
17.4.3.6. Симптомы проблем, связанных с удерживанием
В этом разделе обсуждаются признаки, свидетельствующие о проблемах с временем
удерживания. См. соответствующие пункты в таблице 17.6.
Резкие изменения удерживания обычно легко обнаружить. Если они происходят
при замене колонки, наиболее вероятной причиной является сама колонка.
Повторная установка прежней колонки должна это подтвердить. Различия в
характеристиках колонок гораздо реже наблюдаются в настоящее время при использовании
для упаковки силикагеля высокой чистоты типа В, но раньше это случалось сплошь
и рядом с колонками, упакованными силикагелем типа А более низкой чистоты.
Такие колонки могут использовать до сих пор в некоторых традиционных методиках.
Для выполнения этих методик может потребоваться подбор подвижной фазы к
каждой новой колонке для соблюдения пригодности системы. Альтернативой является
покупка нескольких колонок, упакованных сорбентом из одной партии. Другое
решение состоит в переработке метода для стабилизации результатов разделения, но
это может оказаться неэкономичным. Можно также заменить колонку
эквивалентной, но обеспечивающей большую воспроизводимость. Еще и учтите то, что вы,
возможно, просто по ошибке установили не ту колонку.
Если удерживание изменилось, когда начали работать с новой порцией
подвижной фазы, проще всего приготовить еще одну порцию и протестировать. Проверьте
соответствие рН подвижной фазы требуемому значению (раздел 7.2.1) и то, что рН
доводится до добавления органического растворителя.
Резкие изменения удерживания наблюдаются довольно часто, когда градиентный
метод переносится с одной системы ВЭЖХ на другую. Обычно это обусловлено
различием объемов задержки оборудования (раздел 9.2.2.4). Иногда эти различия можно
компенсировать изменением состава подвижной фазы, времени ввода образцов или
изменением конфигурации системы (раздел 9.3.8.2; также |18|, раздел 5.2.1).
Если удерживание резко изменяется в отсутствии перечисленных выше причин,
и нет очевидных изменений условий работы системы, наиболее вероятны проблемы
с оборудованием (например, несрабатывание обратного клапана), утечки
(таблица 17.3), пузырек воздуха в насосе (таблица 17.4) или изменение температуры
колонки (раздел 17.4.3.5).
Дрейф времен удерживания говорит о какой-то нестабильности в работе системы.
На стадии разработки метода зачастую времена удерживания колеблются при первых
нескольких вводах образца. Это может проявиться более явно при установке новой
колонки. Наиболее вероятной причиной дрейфа времен удерживания в ОФХ
является неполное уравновешивание колонки подвижной фазой. Неполное
уравновешивание может быть особенно резко выраженным в разделениях с ион-парным
реагентом, в которых для уравновешивания может понадобиться объем подвижной фазы,
равный 20 — 50 объемам колонки (раздел 7.4). Хотя для большей части изократиче-
ских методов времена удерживания должны стабилизироваться после первых двух —
трех вводов образца. При градиентном элюировании для стабилизации времен
удерживания может потребоваться увеличение времени уравновешивания между
градиентами, особенно если первые несколько пиков элюируются вблизи fy (раздел 9.3.7).
Менее распространенной причиной дрейфа времен удерживания является
наличие в колонке медленно уравновешивающихся активных сайтов, которые насыщают-
880 Глава 17. Неисправности и способы их устранения
ся лишь после нескольких вводов образца. В таком случае решить проблему могут
несколько предварительных инжекций для дезактивации колонки (разделы 3.10.2.2,
13.3.1.4). Введите подряд друг за другом несколько больших порций образца (обычно
не требуется проводить полное разделение после каждого ввода, достаточно
несколько раз ввести образец с интервалом между инжекциями 30 секунд), потом проведите
полное разделение указанным методом. Иногда для насыщения колонки достаточно
одного предварительного ввода образца, в то время как для других образцов
требуются предварительные инжекций перед каждым разделением.
Если дрейф времен удерживания непрерывно проявляется при анализе целой
партии образцов, это позволяет предположить, что при проведении разделений
данным методом что-то непрерывно изменяется, например, может быть нестабильна
подвижная фаза. При использовании летучего буфера (например, карбоната аммония)
в сочетании с барботированием гелием буфер может частично испаряться с
изменением рН подвижной фазы. Аналогичным образом может быть утрачена часть органи
ческого компонента подвижной фазы, но обычно это не заметно в течение дня.
Для некоторых методов может быть необходимо каждый день готовить новую
порцию подвижной фазы. Если используется барботирование гелием (раздел 3.3.2),
следует иметь в виду, что требуется всего один объем гелия для дегазирования
равного объема подвижной фазы (например, 1 л гелия для дегазирования 1 л подвижной
фазы), то есть нужно всего несколько минут интенсивно пропускать гелий. Если для
устойчивой работы насоса или детектора необходимо непрерывно пропускать гелий,
уменьшите ток гелия до минимума и не допускайте продолжительного интенсивного
барботирования. Если присутствие небольшого количества растворенного кислорода
не столь важно, в большинстве случаев более удобно и эффективно вакуумное
дегазирование онлайн, так как оно не приводит к испарению подвижной фазы.
Колебания времен удерживания некоторых или всех пиков в серии хроматограмм
свидетельствуют о том, что некоторые переменные не контролируются должным
образом. В одном из примеров наблюдалось изменение времен удерживания только
в середине градиентного разделения, что было связано с неправильным
дозированием подвижной фазы (см. |18] раздел 5.5.4.1). Перебои в работе обратного клапана
приводят к перебоям в подаче подвижной фазы, что и вызывает изменения
удерживания. Колебания температуры в лаборатории могут вызвать изменения времени
удерживания от разделения к разделению. Обычно причины и способы устранения
изменений времени удерживания и дрейфа удерживания сходны.
Уменьшение времен удерживания может быть вызвано несколькими причинами.
Если уменьшение времени удерживания коррелирует с увеличением массы
вводимого образца и пики на хроматограмме имеют форму треугольника (например,
рис. 17.15а), вероятна перегрузка колонки. Устранить проблему можно уменьшив
количество наносимого образца. Более подробная информация о перегрузке по массе
приводится при обсуждении искаженных пиков и пиков с размыванием заднего
фронта в разделе 17.4.5.3.
Когда у всех пиков на хроматограмме удерживание уменьшается, это может быть
связано с колонкой, подвижной фазой, температурой или со скоростью потока.
За дополнительной информацией обратитесь к таблице 17.5 и обсуждению в
разделах 17.4.3.1 — 17.4.3.5.
Если уменьшаются времена удерживания только некоторых пиков, можно
предположить, что в системе происходят некоторые неожиданные химические
изменения, например, изменяется степень ионизации кислотных или основных
анализируемых веществ. Проверьте рН подвижной фазы (до добавления органического
компонента). Обычно изменение %В влияет на все пики, элюируемые при
разделении (хотя не обязательно одинаковым образом). Если предполагается изменение %В,
приготовьте новую порцию подвижной фазы. Обратите внимания на то, что точ-
/7.4. Общие симптомы ВЭЖХ-проблем 881
ность смешивания в потоке подвижной фазы может отличаться у разных систем
ВЭЖХ. Старение колонки также может влиять на удерживание только некоторых
пиков; если установить новую колонку, станет понятно, была ли колонка причиной
изменения удерживания.
При разработке метода ОФХ иногда создает трудности неадекватное
удерживание полярных образцов. Если образец ионный, можно изменить рН подвижной
фазы, чтобы перевести его в неионизированную форму. Тогда образец станет менее
полярным и будет лучше удерживаться (раздел 7.3). Альтернативой является
использование ион-парного реагента для улучшения удерживания ионных образцов
(раздел 7.4). Если образец нейтральный, более полярная подвижная фаза (элюент
меньшей силы) должна увеличить удерживание. Однако если содержание органического
компонента < 5%, колонка на основе алкил-силикагеля может перестать смачиваться
(раздел 5.4.4.2), что приведет к уменьшению удерживания. Может быть полезно
в этом случае использовать колонку с привитыми полярными группами (колонку
типа «АО»). Если другие попытки удержать полярные соединения на обращенной
фазе оказываются безуспешными, для получения результата можно перейти к норма-
льнофазовой хроматографии (глава 8) или, что лучше, к гидрофильной
хроматографии (ГИХ, раздел 8.6). См. дополнительное обсуждение недостаточного удерживания
полярных соединений в разделе 6.6.1.
Слишком большие времена удерживания обычно обусловлены теми же причинами,
что и слишком малые. Обратитесь к таблице 17.5 и разделам 17.4.3.1 —17.4.3.5, а
также к обсуждению слишком малых времен удерживания.
17.4.4. Площадь пика
С современными системами обработки данных размер пика гораздо чаще
определяется по его площади, а не по его высоте (раздел П.2.3), поэтому при обсуждении мы
будем предполагать измерение площади пика. Однако методы устранения
неисправностей одинаковы при обоих способах определения размера пика. Если наряду с
изменением удерживания наблюдается изменение площади пика, сначала нужно
решить проблему удерживания, а потом разбираться с причинами изменения размера
пика.
Данные по площади пика очень хорошо воспроизводятся. Например, при
повторном вводе одного и того же хорошо удерживаемого образца (например, с к > 2)
с УФ-детектированием и отношением сигнал-шум S/N > 100 площадь пиков в
разделениях должна отличаться < 1% (раздел З.Ю. I.3). Однако для пиков меньшего размера
с меньшими временами удерживания и/или при использовании других детекторов
результаты воспроизводятся хуже. В приведенном ниже обсуждении проблем, связанных
с площадью пиков, они рассмотрены в такой последовательности: (!) пики, имеющие
площадь больше ожидаемой (раздел 17.4.4.1), включая пики в холостых градиентах и
пики, обусловленные адсорбционными остаточными примесями; (2) лики с площадью
меньше ожидаемой (раздел 17.4.4.2) и (3) пики, площади которых изменяются от
разделения к разделению (раздел 17.4.4.3). Обзор симптомов проблем и способов их
устранения дан в таблице 17.7. В этом разделе предполагается, что метод давал
правильные результаты при анализе предшествующих партий образцов.
17.4.4.1. Слишком большая площадь пика
Для пиков, площадь которых слишком велика, в первую очередь следует
установить — воспроизводится ли эта площадь и касается ли это одного образца или
растворенного соединения, или всех образцов. Чтобы ответить на эти вопросы, обыч-
fe
Глава 17. Неисправности и способы их устранения
но требуется повторно ввести один или несколько образцов и/или
проанализировать хроматограммы этой партии. Если величина площади не
воспроизводится при нескольких вводах одного и того же образца, си. раздел 17.4.4.3
(изменяющиеся знамения площади). Если размеры пиков изменяются в одном и том
же соотношении, проверьте, правильно ли выбран объем вводимого образца. Еще
одна из возможных причин — неправильная подготовка образца: проверьте,
правильно ли разведен образец и соблюдена ли нужная концентрация. Если площади
пиков на хроматограмме изменяются в разных соотношениях, настройки детектора
могут быть неверными. Проверьте длину волны (УФ-детектор, раздел 4.4),
настройки интерфейса (испарительные детекторы, разделы 4.12—4.14), постоянную
времени и так далее.
Пики, появляющиеся в холостом градиенте, в основном обусловлены двумя
причинами: поздним элюированием или адсорбционными остаточными примесями.
Пик, полностью не элюированный в одном разделении, может появиться в
следующем или в более позднем градиенте. Если в образце содержатся другие компоненты,
дополнительные пики будут гораздо шире, чем соседние. Это показано на рис. 17.5д:
широкий пик X (отмеченный стрелкой) появляется на хроматограмме примерно
при 2 минутах. На рис. 17.56 такое же разделение, как на рис. 17.5а, проведено в
течение более длительного времени, и видно, что пик X выходит, как и в первом
случае (= 2 мин), а потом на своем обычном месте (= 7 мин). Если нужно определить
размер пика X, время разделения следует увеличить как на рис. 17.56, чтобы пик
вышел в «свое время». Если этот пик не представляет интерес, возможно несколько
вариантов решения. Время разделения можно увеличить как на рис. 17.56; время
разделения можно подобрать таким образом, чтобы пик появился на следующей
хроматограмме в той области, где нет других пиков; можно использовать
ступенчатый градиент, чтобы вымыть пик из колонки или можно изменить предварительную
очистку образца, чтобы удалить соответствующий компонент из образца до ввода в
колонку. Адсорбционные остаточные примеси появляются, если небольшая часть
образца застаивается в автосамплере или адсорбируется на его поверхности, а потом
выходит в виде пика в холостом разделении. Проверяйте — не адсорбируются ли
остаточные примеси, как описано в раздел 17.2.5.10.
(а)
^JJUUL
4 6
Время (мин)
10
Рис. 17.S. Пример позднего элюирования. (а) Широкий пик (X) появляется на
хроматограмме «не в свое время»; (б) полная хроматограмма: при увеличении
времени разделения пик элюируется в нужное время (~ 7 мин)
/7.4. Общие симптомы ВЭЖХ-проблем 883
17.4.4.2. Слишком маленькая площадь пика
Площади пиков меньше ожидаемых могут быть обусловлены теми же
обстоятельствами, что и большие площади, и методику, описанную выше (в разделе I7.4.4.I),
можно применить для того, чтобы вычленить проблему и определить причины
уменьшения площади пика. Конечно, адсорбционные остаточные примеси и позднее
элюирование пиков с меньшей вероятностью могут способствовать появлению пиков
слишком малого размера. Другими не очень распространенными причинами
появления малых пиков могут быть слишком большая постоянная времени детектора
(раздел 4.2.3.1), заниженная скорость сбора данных (раздел 11.2.1.1), неправильно
выбранные пределы интегрирования или другие ошибки в интегрировании при расчете
площади пика (раздел 11.2.1.4).
17.4.4.3. Изменяющаяся величина площади пика
Если точность метода со временем ухудшается, то это может проявляться в том, что
площади пиков или их высоты могут отличаться все больше. Если нестабильны
также времена удерживания, рекомендуется в первую очередь скорректировать их
значения (раздел 17.4.3). Существует много возможных причин различий в значениях
площади пиков, некоторые из которых также обсуждаются в разделе 11.2.4.
Практически любой этап пробоподготовки и анализа может привести к изменению площади
пика. Некоторые из наиболее вероятных причин обсуждаются далее.
Во-первых, следует установить, согласуются ли между собой результаты,
полученные для одного и того же образца. Если при повторении вводов одного образца
значения площадей пиков изменяются одинаковым образом, все процессы, начиная
с ввода образца, происходят, как надо. Тогда изменения должны быть обусловлены
чем-то, что предшествовало помещению образца в емкость для отбора пробы.
Возможные проблемы этого типа включают ошибки при отборе пробы, погрешности
оборудования и ошибки при приготовлении образца. Отбор пробы — это процесс
отбора репрезентативной (в данном случае эквивалентной) порции образца
(раздел 16.3) — если сам образец неоднороден, отобранные порции могут быть
неэквивалентны одна другой. Если лабораторное оборудование для определения объема или
массы вещества неточно или не работает должным образом, могут быть допущены
ошибки при отборе пробы. Наиболее распространенный источник таких ошибок —
изношенная автоматическая пипетка, используемая дольше положенного времени.
Типичная процедура пробоподготовки (глава 16) включает много этапов, на каждом
из которых могут быть допущены небольшие погрешности, которые могут повлиять
на содержание анализируемого вещества в образце (например, фильтрование,
упаривание, разбавление). Следует пошагово изменять процедуру пробоподготовки или
дополнительные операции, чтобы выявить источник проблемы.
Если при повторных вводах одного образца значения площадей пика
изменяются по-разному, проблема, скорее всего, обусловлена процессами, протекающими
после инжекции. Наиболее вероятными ее источниками являются этапы, связанные
с работой автосамплера, насоса, детектора или с обработкой данных. Сначала
проверьте автосамплер, повторив тест на воспроизводимость из раздела 3.10.3, чтобы
сравнить его с результатами предшествующих тестов (раздел 17.2.4). Проведите
необходимый ремонт. Погрешности в работе насоса могут привести к изменению объемной
скорости подвижной фазы, что также может быть причиной изменения площади
пика. Проведите тестирование скорости потока, как описано в разделе 3.10.1.3.
Проблемы детектирования, такие как перегрузка детектора или неправильный выбор
длины волны, могут повлиять на один пик и не повлиять на другой. Если
предполагается перегрузка детектора (очень большие пики, например, > I AU для УФ-детек-
тора), разбавьте образец или введите меньший объем образца, чтобы определить —
884 Глава 17. Неисправности и способы их устранения
будут ли площади меньших пиков лучше согласовываться между собой. У
масс-спектрометрических детекторов с ионизацией электрораспылением (раздел 4.14.1.1)
из-за плохо работающего наконечника распылителя в детектор могут попадать
разные количества образца в процессе разделения. Случаются ошибки при
интегрировании пиков в процессе обработки данных, например, когда неправильно задается
начальная и конечная точки пика или слишком мала скорость сбора данных (раздел
11.2.1). Иногда изменяющиеся площади пиков наблюдаются, если замороженный
образец оттаял не полностью и/или недостаточно хорошо перемешан перед вводом.
Тогда между верхним и нижним краем емкости для пробы может возникнуть
градиент концентрации растворенного вещества. В этом случае при повторных вводах об
разца могут наблюдаться убывающие или возрастающие площади пиков (в
зависимости от природы растворенного вещества).
17.4.5. Другие проблемы, связанные с хроматограммой
Кроме отклонений от нормы, обсуждавшихся в предыдущих разделах, на хроматог-
раммах часто наблюдаются очевидные дефекты, которые также можно использовать
для определения проблемы в разделении. В этом разделе рассмотрены три таких
дефекта:
• дрейф базовой линии;
• шумы базовой линии;
• форма пика.
17.4.5.1. Проблемы, вызывающие дрейф базовой линии
Дрейфом базовой линии называется непрерывное отклонение базовой линии от
горизонтали вверх или вниз, продолжающееся от десятков минут до часов
(раздел 4.2.3.1). Иногда наклон базовой линии изменяется циклически или каким-то
иным образом. Обзор некоторых признаков и причин дрейфа дан в таблице 17.8.
Следует отметить, что некоторый дрейф базовой линии закономерен; например,
для одного УФ-детектора в соответствии со спецификацией дрейф <2х 1(Н AU/час
при длине волны 250 нм и постоянной комнатной температуре, а если в ячейке
детектора воздух, то дрейф составляет < 3 х 10^ AU/час при изменениях комнатной
температуры < 2 °С |19|.
Периодический дрейф проявляется в циклических изменениях отклонения
базовой линии от горизонтали, когда она поднимается, а потом опускается (или ее угол
наклона изменяется в обратном порядке) на протяжении одного или нескольких
разделений. Это обычно происходит в течение разделения при градиентном элюирова-
нии из-за различного отклика детектора на компоненты подвижной фазы А и В.
Примеры, иллюстрирующие отклонение базовой линии, приведены на рис. 17.6 |20|.
На рис. 17.6а представлена базовая линия разделения в градиенте 5—80%
вода/метанол при 215 нм, дрейф составляет « 0,9 AU (потому что поглощение метанола
на этой длине волны гораздо выше, чем поглощение воды; см. данные таблицы 1.2
в приложении I). Такой дрейф вполне нормален и может помешать только точному
интегрированию пиков на хроматограмме. Если дрейф неприемлем, есть три общих
подхода для преодоления этой проблемы. Один из них заключается в том, чтобы
добавить в элюент А реагент, поглощающий в ультрафиолете. На рис. 17.66 видно, что
при использовании вместо воды 10 мМ фосфатного буфера (с рН 2,8) дрейф
уменьшился почти в 30 раз по сравнению с базовой линией на рис. 17.6а. Влияние
изменения длины волны хорошо видно при сравнении рис. 17.6а (215 нм) с рис. 17.бе
(254 нм). Можно также выбрать органический растворитель с меньшим поглощени-
17.4. Общие симптомы ВЭЖХ-проблем 885
(в) 254 нм
1 1 1 1 1 1 1 1 1 Г
О 2 4 6 8 10
время (мин)
Рис. 17.6. Базовые линии, полученные в градиентах смесей вода—метанол и фосфат-
метанол 5—80 %В за 10 минут, (а) Градиент при 215 нм, шкала 1,0 AU; элю-
ент А вода; элюент В метанол; (б) то же самое, что (а), за исключением
элюента А: 10 мМ фосфат калия (рН 2,8) и шкалы 0,1 AU; (в) то же самое,
что (я), за исключением регистрации при 254 нм и шкалы 0,1 AU [20]
ем в ультрафиолете. В этом случае вместо метанола можно использовать ацетонитрил
(это не показано на рисунке); ацетонитрил дает пренебрежимо малый дрейф при
215 нм, и его можно с успехом использовать для градиентов при 200 нм и при
больших длинах волн. Безусловно замена компонентов подвижной фазы А и В
может изменить хроматографическую селективность, поэтому может потребоваться
дополнительная доводка метода (это применимо только при разработке метода).
Дрейф базовой линии в сторону отрицательных значений может создать более
серьезную проблему из-за того, что обычно система обработки данных прекращает
интегрировать, когда показания детектора становятся менее -0,1 AU (дрейф -10%).
Таким образом, если рассмотреть базовую линию на рис. 17.7я |20|, то можно
предположить, что она, вероятно, уйдет в область отрицательных значений с потерей
данных (приведенную на рисунке базовую линию удалось зарегистрировать, только
отключив функцию автоматического обнуления и вручную установив на старте
+ 1 AU). Как и на рис. \7.6а,б дрейф при 254 нм на рис 17.7 гораздо меньше.
Отрицательный дрейф на рис. 17.7д можно было бы преобразовать в более приемлемый
положительный дрейф, добавив поглощающий в ультрафиолете буфер к элюенту В
(рис. 17.8я |20|). Другой возможный способ устранения этой проблемы состоит в
настройке канала сбора данных на интервал 0,0 1,0 AU (если это позволяет система
сбора и обработки данных).
Однако в некоторых случаях добавки к элюентам (как на рис. 17.8а) не
помогают преодолеть значительный отрицательный дрейф. В примере, показанном
на рис. 17.9а, базовая линия в градиенте бикарбонат аммония—метанол при 215 нм
имеет впадину посередине. Настройка поглощения элюентов А и В не может решить
эту проблему. Хотя градиент этой подвижной фазы неприемлем для детектирования
при 215 нм (рис. 17.9о), детектирование при 254 нм (рис. 17.96) не создает никаких
проблем. Можно также использовать другой детектор; например, бикарбонатные
подвижные фазы широко используются с масс-спектрометрическими детекторами
без каких-либо трудностей с базовой линией. Другим возможным вариантом
является флуоресцентный детектор, который дает возможность получить гладкую базовую
линию при градиентном элюировании флюоресцирующих анализируемых веществ.
886 Глава /7. Неисправности и способы их устранения
<
S
о
с
время (мин)
Рис. 17.7. Базовая линия в градиентах ацетат аммония—метанол. Элюент А: 25 мМ
ацетат аммония (рН 4); элюент В: 80% метанол в воде; градиент 5—100 %В
за 40 минут, (а) детектирование при 215 нм; (б) детектирование при 254 нм
время (мин)
Рис. 17.8. Базовая линия в градиентах ацетат аммония—метанол: молярность буфера
такая же, как на рис. 17.7, но буфер добавлен к элюенту В. Элюент А: 25 мМ
ацетат аммония (рН 4) в 5% метаноле; элюент В: 25 мМ ацетат аммония
в 80% метаноле; градиент 5—100 %В за 40 минут, (л) детектирование
при 215 нм; {6) детектирование при 254 нм [20]
Еще одной серьезной причиной периодического дрейфа базовой линии является
изменение температуры колонки (и подвижной фазы). Изменение температуры
подвижной фазы приводит к изменению ее показателя преломления и передачи света
через ячейку УФ-детектора. Если колонка эксплуатируется без достаточного
контроля температуры (раздел 3.7.1), вероятен дрейф базовой линии по мере изменения
температуры в лаборатории. Зависимость дрейфа базовой линии от температуры
можно подтвердить тем, что времена удерживания также изменяются при колебани-
17.4. Общие симптомы ВЭЖХ-проблем
3
I
-0.1-
-0.2-
время (мин)
Рис. 17.9. Базовая линия в градиентах бикарботат аммония—метанол. Элюент А: 50 мМ
бикарбонат аммония (рН 9); элюент В: метанол; градиент 5—60 %В за 10
минут, (а) детектирование при 215 ни; (б) детектирование при 254 нм |20]
ях температуры. Более подробно проблемы, связанные с колебаниями температуры
рассматриваются в разделе 17.4.3.5.
Другие виды дрейфа базовой линии, характерного для изократических разделений,
нециклические и могут возникать по иным причинам. Медленное уравновешивание
системы после изменения условий (подвижной фазы, колонки, температуры колонки,
скорости потока и так далее) приводят к дрейфу базовой линии, обычно убывающему
в промежутке от 30 до 60 минут. Дрейф базовой линии, связанный с
уравновешиванием, может сопровождаться дрейфом времен удерживания. Аналогичным образом в
отклике детектора наблюдается дрейф в течение нескольких минут после его включения,
а иногда и в течение нескольких часов, необходимых для разогрева и выходу на
стабильный режим работы ламп, электродов или других частей детектора.
17.4.5.2. Проблемы шумов базовой линии
Помехи на базовой линии называют шумами базовой линии. Характер этих шумов
может помочь определить их причину. Помехи могут возникать на базовой линии
периодически или случайным образом, и их длительность может быть меньше
ширины хроматографического пика (кратковременные шумы) или превышать ее
(долговременные шумы). Более того, шумы базовой линии накладываются на любой ее
дрейф. Помимо приведенного далее обсуждения обратитесь к таблице 17.9 и
разделам 3.3.1 (дегазирование), 3.8.3 (скорость сбора данных), 4.2.3 (шумы), 11.2.1.1 (сбор
данных), 11.2.4.2 (хроматографическое разделение) и 11.2.4.3 (детектирование).
Высокочастотные кратковременные шумы проявляются в виде «дрожания»
базовой линии (например, как на рис. 4.5) и обусловлены электронным шумом
электрических схем. Его (шума) частота 60 Гц (в Северной Америке) или 50 Гц (в большей
части остального мира) зависит от частоты переменного тока сети питания. Как
сказано в разделе 4.2.3.1, обычно высокочастотные шумы можно существенно снизить,
используя источник тока со сглаженными пульсациями (например, источник
бесперебойного питания, ИБП) и/или задав большую постоянную времени детектора.
На рис. 4.5 показано, что шум можно уменьшить примерно в 300 раз, используя
простой фильтр шумов.
888 Глава 17. Неисправности и способы их устранения
Случайные и низкочастотные кратковременные шумы могут иметь несколько
разных источников. Недостаточное дегазирование приводит к попаданию пузырьков
воздуха в систему ВЭЖХ. Пузырьки, застрявшие в голове(ах) насоса, могут вызывать
помехи базовой линии, когда давление изменяется между тактами
возвратно-поступательного движения плунжера насоса. При этом шум базовой линии имеет
регулярную структуру. Как описано в разделе 17.4.2.3, пузырьки в голове насоса должны
приводить к флуктуациям давления. Пузырьки, проходящие через насос или
образующиеся за насосом при смешивании недостаточно дегазированных элюентов в
системах со смешиванием на стороне высокого давления, часто остаются в растворе
из-за давления в системе. Однако, когда растворенный воздух выходит из колонки,
давление сильно понижается, и пузырек может вновь образоваться. При
прохождения его через детектор на хроматограмме могут появляться случайные острые
всплески, особенно если используются оптические детекторы (например, УФ-детекторы,
раздел 4.4; флуоресцентные детекторы, раздел 4.5; рефрактометрические детекторы,
раздел 4.11). Детекторы, в которых подвижная фаза испаряется (например,
разделы 4.12—4.14), конечно, не чувствительны к проблемам, связанным с образованием
пузырьков в подвижной фазе. Если пузырек застревает в проточной ячейке
детектора, может наблюдаться значительное смещение базовой линии. Проблему пузырьков
в оптических детекторах можно решить, присоединив на выходе детектора клапан
обратного давления (раздел 4.2.1).
Электрические всплески проявляются как пузырьки. Для того, чтобы их
различить, выключите насос и следите за базовой линией. Если всплески продолжают
появляться, проблема связана с электроникой; если всплески прекращаются, и базовая
линия становится гладкой — виной всему пузырьки. Первая линия защиты от
пузырьков — оптимизированный метод дегазирования (раздел 3.3.1). Противодавление
(раздел 4.2.1) задержит пузырьки в растворе до момента, когда они выйдут из
детектора.
Выбор слишком высокой скорости сбора данных может привести к появлению
кратковременных шумов на базовой линии. Как описано в разделе 3.8.3, следует
задавать такую скорость сбора данных, чтобы на пик приходилось « 20 точек. Более
высокие скорости сбора данных увеличат шум базовой линии, но не обеспечат
особых преимуществ за счет количества собранных сигналов, так как ухудшится
отношение сигнал-шум (раздел 4.2.3). Меньшие скорости сбора данных могут понизить
шумы базовой линии, но при этом есть риск уменьшения самого сигнала, так что и
в этом случае отношение сигнал-шум может стать ниже.
Долговременные шумы проявляются в виде помех базовой линии, сравнимых
по длительности с нормальными пиками (или длящихся дольше). Обычно
долговременные шумы возникают из-за того, что в образце содержатся позвноэлюируемые
примеси (см. обсуждение рис. 17.5 в разделе 17.4.4.1). По мере того, как возрастают
времена удерживания растворенных веществ образца и его фоновых примесей, ширина
выходящего пика увеличивается, а его высота уменьшается. Поздноэлюируемые
пики от предыдущих разделений могут со временем накапливаться, приводя к
дрейфу базовой линии и помехам. Промывка сильным растворителем (например, 25 мл
метанола или ацетонитрила) часто удаляет сильно удерживаемые вещества из
колонки. Поэтому рекомендуется промывать колонку сильным растворителем после
окончания анализа партии образцов (имеются в виду разделения в изократическом
режиме). Для некоторых методов может потребоваться более частое промывание колонки.
Градиентные методы в меньшей степени подвержены влиянию поздноэлюируемых
примесей, так как они включают промывку сильным элюентом в конце каждого
разделения. Героические попытки удалить сильно удерживаемые примеси (например,
промывание кислотой, щелочью, хаоторопными реагентами или метиленхлоридом)
могут помочь, но могут и повредить сорбент. Лучше оптимизировать пробоподготов-
/7.4. Общие симптомы ВЭЖХ-проблем
э
<
i
о
40 50
Время (мин)
Рис. 17.10. Циклические шумы базовой линии, которые предположительно были
вызваны наводками от электронного воздушного фильтра
ку (глава 16), чтобы уменьшить содержание прочно удерживаемых примесей в
образце. Помните, что колонка — расходный материал. После анализа примерно 500
образцов стоимость колонки в пересчете на образец становится небольшой частью
общих затрат на анализы, поэтому лучше заменить колонку, чем пытаться ее отмыть
или изменить методику пробоподготовки.
Иногда долговременные шумы проявляются в виде регулярных колебаний
(флуктуации) базовой линии, как показано на рис. 17.10 (обратите внимание, что
шкала по оси у I mAU). Обычно циклические возмущения базовой линии вызваны
проблемами с насосом и сопровождаются колебаниями давления (раздел 17.4.2.3)
или утечками (раздел 17.4.1). В случае, показанном на рис. 17.10, отключение
системы ВЭЖХ от электропитания и устранение неисправностей в насосе и детекторе
не помогли решить проблему. А уменьшилась частота пульсаций базовой линии,
когда систему передвинули с того места, где она стояла, и на достаточном расстоянии
колебания базовой линии вообще исчезли. Хотя точно определить источник шумов
так и не удалось, предполагается, что это были наводки от электронного воздушного
фильтра |211.
Артефактные пики или пики«призраки» представляют собой особый вид
долговременных шумов. Пример такого пика показан на рис. 17.1 \а для градиента 5—80%
ацетонитрил-фосфатный буфер (рН 7) за 15 минуте задержкой при 80 %В. В
идеальном случае в этом холостом градиенте на базовой линии не должно быть никаких
пиков (дрейф базовой линии обусловлен различным поглощением элюентов А и В,
как говорилось ранее). Наиболее вероятной причиной появления пиков в холостом
градиенте является загрязнение элюента А, так как эти загрязнения обычно
концентрируются на входе колонки при уравновешивании между разделениями и потом
вымываются в градиенте. Простой способ подтвердить загрязнение элюентом А состоит
в том, чтобы увеличить время уравновешивания между разделениями (прокачивая
элюент А). Если загрязнение обусловлено элюентом А, все пики должны
увеличиться строго пропорционально возрастанию времени уравновешивания (то есть при
большем объеме элюента А возрастает количество сорбированных загрязнений). В
данном примере 10-минутное уравновешивание перед градиентом, показанным
на рис. 17.11а, было продлено до 30 минут, и градиент был повторен (рис. 17.116).
Очевидно, что пики увеличились примерно втрое, так что подтвердилось
загрязнение элюентом А. Дальнейшее изучение причин загрязнения выявило причастность
электрода рН-метра |7|. На рис. 17.12 проводится сравнение результатов холостых
разделений, проведенных с буферами, в один из которых при приготовлении
погружали электрод для доведения рН (рис. 17.12а), а в другой не погружали (рис.17.126).
Дополнительные примеры артефактных пиков в градиенте, возникающих вследствие
загрязнения воды и других реагентов, можно найти в |18|, разделе 5.5.4.
Артефактные пики в градиенте также превосходно рассмотрены в |6|.
890 Глава 17. Неисправности и способы их устранения
Рис. 17.11. Влияние загрязнений в элюенте А на базовую линию хроматограммы:
холостые градиентные разделения после (я) 10-минутного и (б) 30-минутного
уравновешивания. Колонка C]g; градиент 5—80% ацетонитрил — 10 мМ
фосфатный буфер (рН 7) за 15 минут с последующей 5-минутной
задержкой при 80 %В. УФ-детектирование при 215 им |7|
20-1
_. 15-
S 10-
I
0-
1 1 Г
8 12
время (мин)
Рис. 17.12. Сравнение градиента с использованием (а) загрязненного буфера (те же
условия, что и на рис. 17.11с) и (6) буфера, приготовленного в очень
чистой посуде без погружения в него электрода рН-метра. Все остальные
условия такие же, как на рис. 17.11 |7|
17.4.5.3. Проблемы, связанные с формой пика
Идеальный хроматографический пик представляет собой симметричную гауссову
кривую. Отклонения от идеальной формы пика можно количественно оценить
с помощью фактора размывания заднего фронта пика TF или коэффициента
асимметрии пика (раздел 2.4.2), как показано на рис. 2.16я. Искажение формы пика
/7.4. Общие симптомы ВЭЖХ-проблем 891
может сопровождаться или не сопровождаться аномальными временами
удерживания. Отклонения от симметричной формы пика можно классифицировать
следующим образом:
• пики с размыванием заднего фронта;
• пики с размываем переднего фронта;
• широкие пики;
• расщепленные или искаженные пики.
Далее обсуждаются эти отклонения формы пиков от идеальной, а в таблице 17.10
приведен обзор с детализацией их признаков, причин и методов устранения. Также
в разделе 2.4.2 подробно рассмотрены некоторые причины искажения формы пиков.
Размывание заднего фронта пика — наиболее распространенный тип
искажения пика. В спецификациях современных колонок часто дается допустимое значение
TF < 1,2, так что только небольшое размывание следует рассматривать как нормальное.
Поскольку в указаниях контролирующих органов |22| для фармацевтических методов
дается допустимое значение TF <2, предпочтительны пики с TF < 1,5. Размывание
заднего фронта пиков имеет тенденцию возрастать со временем вследствие изнашивания
колонки; когда наблюдаемый TF > 2, следует принять меры для понижения
размывания заднего фронта пика (например, заменить колонку). Если наблюдается
значительное размывание заднего фронта у всех пиков, обратитесь к приведенному далее
обсуждению расщепленных или искаженных пиков. Если размывание у рановыходящих
пиков больше, чем у пиков, выходящих позже, это может быть обусловлено внеколо-
ночным уширением пиков. Это иллюстрирует рис. 17.13, на котором фактор
размывания изменяется в пределах от TF = 2,5 для первого пика до TF = 1,2 для последнего
пика. Если предполагается внеколоночное уширение пиков, сократите внеколоночный
объем, например, используя более короткие капилляры меньшего диаметра и
обеспечивая должное соединение всех частей системы (разделы 2.4.1, 3.9).
В прошлом распространенной причиной размывания заднего фронта пиков было
сильное взаимодействие ионизированных основных компонентов образца ВН+
с ионизированными силанольными группами сорбента колонки SiO~ (раздел 7.3.4.2):
ВИ+ + SiO К+ <=> BfTSiO- + К+
Однако эта проблема встречается все реже при использовании колонок с менее
выраженными кислотными свойствами, упакованных силикагелем типа В
(раздел 5.2.2.2). Помимо использования сорбентов на основе силикагеля типа В
размывание фронта пиков этого вида можно понизить, изменив подвижную фазу. Ионизация
силанольных групп и, следовательно, размывание уменьшаются при понижении рН,
в то время как ионизация растворенного вещества и размывание уменьшаются
i i i i i i * i i
0 2 4 6 8
Время (мин)
Рис. 17.13. Размывание заднего фронта пиков из-за внеколоночных эффектов. У
первого пика TF = 2,5, а у последнего TF ~ 1,2
892 Глава 17. Неисправности и способы их устранения
при рН 3> рКа растворенного вещества. Повышение степени связывания ВН*
анализируемого вещества в ионные пары или добавление конкурирующих ионов триэтилами-
на (в протонированной форме) в подвижную фазу может быть эффективным для
подавления размывания заднего фронта пиков. Повышение ионной силы также может
уменьшить размывание, хотя обычно оно менее эффективно, чем другие изменения
подвижной фазы. Однако эти изменения сорбента или подвижной фазы надо
учитывать при разработке метода, иначе потребуется повторная валидация метода.
Пример воздействия изменения подвижной фазы на размывание заднего фронта
пиков показан на рис. 17.14, иллюстрирующем анализ 4 белков в режиме
градиентного элюирования с подвижной фазой, содержащей ТФУК и ацетонитрил, на 3
колонках, упакованных силикагелем различной чистоты (колонка А с силикагелем
высокой чистоты |то есть типа Bj; колонка В с силикагелем средней чистоты и
колонка С с силикагелем низкой чистоты (типа А|). Когда в подвижную фазу
добавлено 0,1% ТФУК, хроматограммы, полученные на разных колонках, мало отличаются
(рис. 17.14 а-в). Однако, когда концентрация ТФУК понижается до 0,01%, видно,
что белки сильно взаимодействуют с сорбентами более низкой чистоты, что
проявляется в возрастании размывания заднего фронта пиков (рис. 17.14г-<?) и больших
временах удерживания на колонке с силикагелем низкой чистоты (рис. 17.14е).
Влияние недостаточного количества ТФУК оказывается все более ярко выраженным
Колонка А
Колонка В
Колонка С
(силикзгель высокой (Силикагель средней (Силикагель низкой
чистоты) чистоты) чистоты)
(а)
0.1% ТФУК
<б)
(в)
jU i UL.
-1 1 ! I Г—I I I I
I I I I-
<г)
0.01% ТФУК
JJJl
10
(д)
20 30 0
(в)
л!__ _
10 20 300
Время (мин)
10
_-чЛ^
20
30
Рис. 17.14. Влияние концентрации трифторуксусной кислоты на размывание заднего
фронта пиков на колонках, упакованных сорбентами на основе силикагеля
различной чистоты. (а,г) Силикагель высокой чистоты (колонка А); (б,д)
силикагель средней чистоты (колонка В); (в,е) силикагель низкой чистоты
(колонка С), (а—в) 0,1% трифторуксусной кислоты (ТФУК); (г—е) 0,01%
ТФУК. С|о, частицы силикагеля с диаметром пор 30 нм, градиент ацето-
нитрила с ТФУК 5—70%. Образец: А, рибонуклеаза А; В, цитохром С; D,
голо-трансферин; Е, апомиоглобин. Данные любезно предоставлены
Advanced Chromatography Technologies (ACT)
/7.4. Общие симптомы ВЭЖХ-проблем
по мере понижения чистоты силикагеля от высокой (г) до средней (д) и низкой (е)
из-за возрастания ионизации силанольных групп. ТФУК используется как
ион-парный агент при разделении белков, поэтому преимущество более высокой
концентрации ТФУК может быть обусловлено возрастающей степенью связывания в ионные
пары, хотя сопутствующие понижение рН и возрастание ионной силы тоже могут
вносить свой вклад в улучшение формы пиков.
Введение слишком большой массы образца может привести к перегрузке колонки
по массе. Такая перегрузка может произойти для одного или более веществ хроматог-
раммы и тогда пики приобретают форму прямоугольного треугольника с
одновременным уменьшением времени удерживания по мере возрастания нагрузки на
колонку, как видно на рис. 17.15а (справа налево от 0,01 до 5 мкг |23|). Если
разведение образца или введение его меньшего объема приведет к возрастанию
времени удерживания и сокращению размывания заднего фронта пиков, то
предположение о перегрузке по массе подтвердится. Для полностью ионизированных образцов
перегрузка по массе наблюдается при введении примерно в 50 раз меньшей массы
образца, чем для других веществ (раздел 15.3.2.1). На колонках с внутренним
диаметром 4,6 мм массовая перегрузка и размывание заднего фронта пиков происходят
при введении > 1 мкг ионизированного анализируемого вещества, в отличие от ней
(б)
(г)
1_
Рис. 17.15. Примеры искажения формы пика, (а) Пик в виде прямоугольного
треугольника вследствие перегрузки колонки по массе; справа налево 0,01—5
мкг нортрипгилин; XTerra MS колонка C]g, 150x4,6 мм (3,6 мкм); 28%
ацетонитрил рН 2,7 (20 мМ муравьиная кислота); переработанные данные
из |23|. (б) Пик с плоской вершиной, характерный для перегрузки колонки
по объему или для перегрузки детектора, (в) Искажение формы пика
из-за введения слишком большого объема образца в слишком сильном
элюенте (30 мкл 100% ацетонитрила введено в подвижную фазу,
содержащую 18% ацетонитрила); (г) те же условия, что и в (в), за исключением
того, что образец растворили в подвижной фазе. Условия аля (в,г):
колонка Lichrosorb RP-18 250 х4 мм; подвижная фаза ацетонитрил-вода-уксус-
ная кислота (18:81:1) |29|
894 Глава 17. Неисправности и способы их устранения
Время (мин)
трального анализируемого вещества,
которого можно ввести примерно 50 мкг без
перегрузки колонки.
Размывание переднего фронта пика
происходит в ОФХ гораздо реже, чем
размывание заднего фронта. Как и в случае с
размыванием заднего фронта незначительное
размывание переднего фронта пика
допустимо; в спецификациях колонок многих
производителей допускается размывание
переднего фронта пика > 0,9. Обычно такое
размывание объясняют нарушением
образования ионных пар при повышении
температуры 124, 251, но эти сообщения касаются
колонок, упакованных силикагелем низкой
чистоты (типа А), а для колонок с более
чистым силикагелем типа В эта причина вряд
ли широко распространена. Обычно раз
мывание переднего фронта пика связыва
ют с образованием полости в колонке
(при проседании упакованного сорбента), и
результат может быть достаточно
серьезным, как показано на рис. 17.166 |26|.
В этом случае колонка Си
эксплуатировалась при рН 9, превышающем
рекомендованное рабочее значение рН. В процессе
анализа » 500 образцов форма пиков была
нормальной (рис. 17.16д), а потом внезапно
при каждом последующем вводе образца
стал размываться передний фронт пика (рис. 17.165). Изменения площади пика не
наблюдалось (данные, собранные для той части разделения, где выходит пик с
размытым передним фронтом, можно было использовать для количественной оценки).
Колонку не удалось регенерировать с помощью промывки и других мер по ее
восстановлению, поэтому колонку забраковали. Такое повреждение колонки типично для
использовавшегося метода и объясняется разрушением сорбента из-за растворения
силикагеля при высоком рН. Используя данный метод, более рационально заменять
колонку каждый раз, когда она выходит из строя, а не расходовать время и деньги на
переработку и повторную валидацию метода для более устойчивой колонки. Другие
потенциальные причины размывания переднего фронта пика включают
ограниченную растворимость анализируемого вещества в подвижной фазе, склонность его
молекул к агрегации и изменения их конформации.
Широкие пики могут быть предшественниками расщепленных или искаженных
пиков, поэтому к рассмотрению последних (далее в этом разделе) следует обратиться
в том случае, если предложенные ниже меры не помогут решить проблему. Широкие
пики имеют гораздо меньшее расчетное количество теоретических тарелок, чем
положено для новой колонки. Так как изготовители проводят исходное тестирование
колонки с «идеальными» образцами в «идеальных» условиях, полученное ими число
теоретических тарелок N может существенно превосходить значение для реальных
образцов и условий. Лучше всего измерить число теоретических тарелок (или
ширину пика при градиентном элюировании) на новой колонке с одним или несколькими
образцами для каждого метода. Это может послужить контрольным значением
для каждого метода. Приемлемые величины уширения пиков могут быть различны-
Рнс. 17.16. Размывание переднего фронта
пика вследствие разрушения сорбента
колонки, (а) Нормальная хроматограмма;
((f) размывание переднего фронта пика
после анализа ~ 500 образцов,
проводившегося при рН 9 на колонке C|g [26]
17.4. Общие симптомы ВЭЖХ-проблем 895
ми в разных прикладных задачах, и определение контрольного значения следует
включать в тест на пригодность системы. Как показывает практика, если значение /V
в рутинном тесте новой колонки меньше 75% величины, указанной производителем,
следует исследовать причину занижения значения N.
По мере старения колонки падает число теоретических тарелок, поэтому пики
становятся все шире в течение ее срока службы. Если уширение пиков наблюдается
после проведения 500 или более инжекций, вероятная причина — износ колонки.
Форма пиков может улучшиться после промывки колонки сильным элюентом
(например, 20—30 мл ацетонитрила или метанола), но часто целесообразнее заменить
колонку.
Если на хроматограмме один пик широкий среди остальных узких, скорее всего
этот единственный широкий пик — поздноэлюируюшийся пик из предыдущего
разделения. Можно подтвердить это предположение, увеличив время разделения
настолько, чтобы пик мог выйти в том разделении, в котором он был введен
(раздел 17.4.4.1, рис. 17.5).
Внеколоночные эффекты могут приводить к размыванию заднего фронта пика
с некоторым его уширением, как показано на рис. 17.13. Уменьшите внеколоночные
эффекты, соединяя колонку с автосамплером и детектором капиллярами меньшей
длины и меньшего диаметра.
Широкие пики также могут быть следствием проблем, связанных с обработкой
сигнала. Если используется постоянная времени детектора (фильтр шумов), то ее
значение не должно превышать 1/10 ширины самого узкого из анализируемых
пиков. Например, постоянная времени 1 секунда подходит для пика шириной 10
секунд (измеренной на базовой линии), а постоянная времени 5 секунд может
привести к избыточному уширению пика. Аналогичным образом, скорость сбора данных
системой обработки данных должна быть достаточно велика для того, чтобы каждый
пик описывался минимум 20 точками (раздел 11.2.1.1). Некоторые детекторы,
например, масс-спектрометрические, позволяют использовать сглаживание для улучшения
формы пиков. Однако избыточное сглаживание может привести к уширению пиков.
Слишком большой объем введенного образца может вызывать уширение пиков,
и они даже могут выглядеть как пики с плоской вершиной (см. рис. 17.156). Если
образец растворен в подвижной фазе, то его вводимый объем не должен превышать
= 15% объема первого из анализируемых пиков (разделы 2.6.1, 3.6.3, 15.3.2.2,
таблица 3.3). В этом случае ширина пика увеличится на 5%, что соответствует 5% потере
в разрешении. Если для растворения образца используются злюенты на и 10%
слабее, чем подвижная фаза, можно увеличить объем наносимого образца
(разделы 2.6.1, 3.6.3). Пики с плоской вершиной характерны также для перегрузки детектора.
При низких концентрациях отклик детектора будет расти в соответствии с
увеличением концентрации анализируемого вещества (на переднем фронте пика), но при
перегрузке детектора его отклик уже не возрастает при превышении определенной
концентрации, и получается пик с плоской вершиной.
Расщепление или искажение пиков может наблюдаться для одного, нескольких,
или всех пиков хроматограммы. На рис. 17.17 показана хроматограмма, на которой
все пики расщеплены. Расщепление всех пиков является классическим признаком
засорения входного фильтра или (реже) образования пустот в колонке, часто это
сопровождается повышением давления. Воздействие засоренного фильтра или полости
в колонке показано на рис. 17.18. На рис. 17.18а представлен вход в колонку с
находящимся на своем месте фильтром. Когда образец вводится в колонку, весь поток
образца (показанный стрелками) поступает в верхнюю часть колонки одновременно.
Таким образом, хроматографическое разделение начинается для всего переднего
фронта образца в одно и то же время. Когда фильтр частично засорен (рис. 17.185),
поток образца искажается таким образом, что некоторые порции образца попадают
896 Глава 17. Неисправности и способы их устранения
Рис. 17.17. Примеры одинакового искажения всех пиков на хроматограмме,
относимые в каждом случае за счет частичного засорения входного фильтра
колонки
Рис. 17.18. Искажение пика на входе колонки. Стрелками показано направление
потока компонентов образца, когда они поступают на колонку, (я)
Нормальное движение раствора образца через фильтр колонки во время ввода. (6")
Искажение потока образца вследствие частично засоренного входного
фильтра колонки, (в) Искажение потока образца из-за образования
полости п головной части колонки
в головную часть колонки поздно и это приводит к размыванию заднего фронта
пика. Полость в головной части колонки (рис. 17.18*) также может вызывать
искажение пика, но обычно размывается перелний фронт лика (как показано
на рис. 17.166). Поскольку эти искажения происходят еще до начала разделения, все
пики искажаются одинаково при их продвижении по колонке.
Промывка колонки в обратном направлении — лучший способ смыть
загрязняющие частицы с наружной стороны входного фильтра колонки и восстановить
правильную форму пиков. После установки колонки в обратном направлении пропустите
через нее 20—30 мл растворителя в слив (не в детектор), потом снова подсоедините
колонку, но в направлении, противоположном потоку (проблема может повториться,
если колонку установить в исходном направлении). Например, на хроматограмме,
показанной на рис. 17.19й, все пики раздвоены |27|. Введение контрольного
стандарта также дает раздвоенный пик (рис. 17.196), но установка колонки в обратном
направлении позволяет устранить проблему. На некоторых колонках не рекомендуется
17.4. Общие симптомы ВЭЖХ-проблем
(а)
_JL^aaa_
(б)
~~i г
6 8
Время (мин)
10
12
Рис. 17.19. Расщепление пиков, отнесенное за счет частично засоренного входного
фильтра колонки, (я) Расщепление, наблюдаемое для всех пиков
нормального образца; {б) расщепление пика контрольного стандарта [27|
постоянно работать, установив их в обратном направлении. Например, в некоторых
колонках, упакованных частицами размером 3 мкм, на выходе установлен фильтр
с порами 0,5 мкм, а на входе — фильтр с порами 2 мкм. Поры диаметром 2 мкм
недостаточно малы, чтобы удержать частицы диаметром 3 мкм в колонке, но недолгая
промывка колонки в обратном направлении обычно не приносит вреда, если потом
колонка вновь устанавливается в исходном направлении. Если вы сомневаетесь,
узнайте из инструкции по уходу за колонкой об ограничениях, касающихся
направления потока. В прошлом было принято заменять входной фильтр колонки, если
предполагали, что он засорился. Современные колонки можно полностью вывести
из строя, если снять концевой фитинг, поэтому такой способ более не
рекомендуется. Использование проточного фильтра с пористостью 0,5 мкм (раздел 3.4.2.3),
фильтрование или центрифугирование образца и/или более качественная очистка образца
могут предотвратить повторное засорение фильтров колонки.
Недостаточный контроль за температурой колонки может привести к искажению
пиков, особенно это касается поздноэлюируемых пиков. В примере, показанном
на рис. 17.20а, подвижная фаза подогревается до температуры колонки (56 СС), и
наблюдаются пики приемлемой формы. На рис. 17.206 температура подвижной фазы
на входе колонки была 39 °С (то есть на 17 °С ниже, чем температура колонки
из-за недостаточного предварительного подогревания подвижной фазы). В
результате пики шире, чем на рис. 17.20а, а поздновыходящие пики имеют явно искаженную
форму. Уширение и искажение пиков на рис. 17.206 вызваны радиальным
градиентом температуры вдоль продольной и поперечной осей колонки, показанным на ис.
17.21. На рис. 17.21а поступающая подвижная фаза и колонка находятся в
термическом равновесии, так что температура подвижной фазы одинакова во всем объеме
898 Глава 17. Неисправности и способы их устранения
(а)
О
Jul
(б»
и—i—i—i—i—1—
в 10 12
Время (мин)
—i—i—i—i
14 16
А
| 1 г-»-! 1 1 г"—I 1 1 1 1 1—
0 2 4 в в 10 12
Время (мин)
Рис. 17.20. Влияние несоответствия температуры подвижной фазы температуре
колонки, (я) Поступающий элюент 56 "С; термостат колонки 56 "С (соответствие
температур), (б) Поступающий элюент 39 °С; термостат колонки 56 °С
(несоответствие температур). Образец: А, урацил; В, нитроэтан; С, фта-
левая кислота; D, 3,5-диметиланилин; Е, 4-хлоранилин; F, мононитрил
терефталевой кислоты и G, 1-нитробутан. Колонка Zorbax SB-Cjg 150 х
х 4,6 мм (5 мкм); 90/10 50 мМ фосфат калия (рН 2,6)/ацетонитрил;
2 мл/мин |28|
(а)
56°С
й
56°С
56°С
56°С
(б)
39°С
ш
56°С
45°С
50°С
"'О
56°С
£м_
Рис. 17.21. Уширение зоны под влиянием температуры, (а) Идеальный случай,
термическое равновесие; (б) влияние подающейся на колонку подвижной фазы,
имеющей температуру ниже температуры колонки |28]
колонки, и зона образца (показанная точками) перемещается по колонке с
одинаковой скоростью. Когда в колонку вводится более холодная подвижная фаза
(рис. 17.216), подвижная фаза в центре колонки становится холоднее (и более
вязкой), чем у стенок, и тогда молекулы образца, близкие к центру, движутся
медленнее, чем находящиеся у стенок, поэтому зона образца уширяется и искажается.
Поскольку средняя температура колонки понижается при введении холодной
подвижной фазы, увеличиваются времена удерживания, и несколько изменяется
селективность (обратите внимание, что пики D и Е выходят на рис. \7.2\a и б в разной
последовательности). Решением проблемы, которую иллюстрирует рис. 17.20,
является более тщательный контроль температуры подвижной фазы, а именно предвари-
/7.4. Общие симптомы ВЭЖХ-проблем 899
тельное подогревание подвижной фазы (раздел 3.7.1). Когда температура
поступающей в колонку подвижной фазы находится в пределах +6 °С от температуры
колонки, искажение формы пиков вряд ли произойдет |28|.
Три причины искажения формы пиков связаны с вводом образца: искажение
вследствие слишком сильного растворителя образца, уширение из-за слишком
большого объема вводимого образца (см. обсуждение широких пиков) и размывание
заднего фронта пика из-за слишком большой массы вводимого образца (см. выше
обсуждение перегрузки по массе). Если образец растворен для инжекции в слишком
сильном растворителе, этот растворитель не разбавится достаточно быстро
подвижной фазой, поэтому часть образца может слишком быстро передвигаться по колонке
до того, как она полностью разбавится. Это может вызвать искажение пиков,
особенно рановыходящих пиков, как показано на рис. 17.15е. Если использовать
для растворения образца слишком сильный растворитель, время удерживания может
уменьшиться. На рис. 17.15е показана хроматограмма, полученная при введении
30 мкл образца, растворенного в ацетонитриле, в подвижную фазу, содержащую 18%
ацетонитрила. В этом случае растворитель образца слишком сильный для такого
объема пробы. Первый пик сильно искажен, а второй уширен. Кроме того, времена
удерживания меньше нормальных для этих пиков. Эту проблему можно устранить,
или (1) разбавив образец, чтобы его растворитель был эквивалентен по силе
подвижной фазе, или/и (2) уменьшив объем вводимой пробы до < 10 мкл (для колонки
150 х 4,6 мм; и меньший объем пробы для колонок меньшего размера). Когда
образец, разделение которого показано на рис. 17.15е, был разбавлен подвижной фазой,
при введении 30 мкл этого образца получились пики правильной формы с
нормальным временем удерживания (рис. 17.15г) [29|. Влияние растворителя образца
обсуждается более подробно в разделах 2.6 и 3.6.3.2.
Еше одной причиной искажения пиков являются разложение или химические пре
вращения анализируемого вещества при его прохождении через колонку, когда оно
нестабильно в условиях хроматографического разделения. Если разложение
происходит быстро на входе колонки, и возможно, катализируется металлом фильтра
колонки, то разложившийся образец разделяется без дальнейших изменений состава
образца. В результате исходный образец и продукты его разложения элюируются в виде
пиков правильной формы. Однако если скорость разложения низкая, образец может
постепенно распадаться, проходя по колонке, и тогда выходящие пики искажаются.
Это результат прохождения через колонку двух (или более) веществ с разной
молекулярной структурой, при этом соотношение их концентраций постоянно меняется
в процессе разделения (30, 31].
Пример быстрой и медленной реакции разложения представлен на рис. 17.22 |31,
32| — это градиентное разделение двух эпимеров типредана (его структура показана
на рисунке) в разных условиях. На рис. 17.22а показана хроматограмма, полученная
при введении чистого S-эпимера, и наблюдаются пики обоих эпимеров (то есть идет
превращение S-эпимера в R-эпимер). Так как пики острые и хорошо разрешенные,
реакция должна была в основном пройти до начала элюирования. При введении
чистого R-эпимера (рис. 17.226) реакция превращения в противоположный эпимер также
идет, но с меньшей скоростью. Оба разделения (рис. 17.22а и 17.226) проведены на
колонке Hypersil ODS. Когда колонку заменили на Resolve C|g, то вновь провели
разделение чистого S-эпимера (рис. 17.22е). В этом случае между пиками наблюдается
характерная седловина, свидетельствующая о том, что превращение образца происходит
медленнее — во время разделения, а не перед ним при нанесении образца.
Если предполагается, что происходит разложение образца на колонке, это
предположение можно подтвердить, изменив условия разделения (температуру, рН и
т.д.), чтобы ускорить или замедлить реакцию. Например, при возрастании или
понижении температуры колонки обычно ускоряется или замедляется превращение об-
900 Глава 17. Неисправности и способы их устранения
(а) (б) (в)
SCH3
но «YpvLf- s{0)ch2ch3
30 32 34 30 32 34 34 36
Время (мин)
Рис. 17.22. Разделение эпимеров типредана. Условия: (а,6) колонка Hypersil ODS
100 х 4,6 мм; 29/32/62% ацетонитрила/буфер рН 7,2 за 0/10/20 мин;
1,5 мл/мин; 26 °С. (е) колонка Resolve C^ 150 х 3,9 мм и сходные (но
не идентичные) условия градиента, (а) наносился S-эпимер; (б) наносился
R-эпимер; (в) наносился S-эгтимер [31, 32)
разца, предсказуемым образом влияя на форму пиков. Для образца, показанного
на рис. 17.22, установлено, что повышение температуры колонки ускоряет реакцию,
а повышение рН подвижной фазы ее замедляет.
17.4.6. Интерпретация тестов на пригодность
(работоспособность) системы
Тесты на работоспособность системы ВЭЖХ подробно описаны в разделе З.Ю.1,
а в разделе 17.2.1 дан их краткий обзор. Особое значение для определения проблем
с оборудованием имеет градиентное тестирование (раздел 3.10.1.2) и дополнительные
тесты системы (раздел 3.10.1.3). Мы рекомендуем, проводить эти тесты каждые 6—
12 месяцев на каждой системе ВЭЖХ, чтобы обеспечить оптимальную
эффективность оборудования. Обзор симптомов, проявляющихся при отрицательных
результатах тестов, и способов решения проблем дан в таблице 17.11. В нижеследующем
обсуждении использованы примеры изучения результатов тестов для того, чтобы
показать, как можно идентифицировать с их помощью некоторые проблемы.
17.4.6.1. Интерпретация градиентного тестирования
Градиентное тестирование, описанное в разделе З.Ю.1.2, включает следующие этапы:
• тест линейности градиента;
• определение объема задержки;
• тестирование ступенчатым градиентом;
• тестирование клапанов формирующих градиент (КФГ).
Обычно все эти тесты проводятся последовательно, и результаты одного теста
соотносят с результатами остальных. В других случаях результаты проведенного теста
указывают на то, что следует провести еще какой-то определенный тест. Примеры,
приведенные далее, иллюстрируют взаимосвязь между этими тестами, также, как и
взаимосвязь между их отрицательными результатами. Обсуждение построено в
форме последовательного разбора пяти случаев возникновения проблем, обнаруженных
по результатам градиентного тестирования. Каждый пример рассмотрен до
логического завершения, чтобы показать, как различные тесты соотносятся с реальными
проблемами.
/7.4. Общие симптомы ВЭЖХ-проблем 901
Случай I. Тест на линейность градиента описан в разделе 3.10.1.2. Этот тест
включает замену колонки на отрезок капилляра и выполнение градиента от [00%
воды до смеси воды с 0,1% ацетона при регистрации на длине волны 265 нм.
Типичный результат показан на рис. 3.26, видна задержка градиента в начале,
соответствующая объему задержки. За ней следует линейный переход к 100 %В с небольшими
закруглениями на концах градиента. Обычно достаточно посмотреть на график
для определения его линейности; график также можно распечатать и провести рядом
с ним прямую для сравнения, как показано на рис. 17.23 (пунктирная прямая).
В данном случае |33] график в целом линеен, но при 25%, 50% и 75 %В на нем
наблюдаются небольшие отклонения. Такой результат указывает на то, что процесс
дозирования подвижной фазы нарушается в каждой из этих трех точек кривой.
На следующем этапе проводится тестирование ступенчатого градиента. Он
также описан в разделе 3.10.1.2 и часть графика, полученного при успешном
тестировании, показана на рис. 3.27. Ступенчатый тест включает подачу серии смесей элюен
тов различного состава от 0 до 100 %В. Используются упомянутые выше элюенты
вода и ацетон. Рекомендуется использовать ступеньки с добавлением на каждой
по 10% В в интервале от 0 до 100 %В и еще две дополнительные ступеньки с 45 и
55 %В. В рассматриваемом примере полученный ряд ступенек (рис. 17.24<э)
удовлетворил критерию приемлемости: отклонение ±1 %В от целевых значений |33|. Тем
не менее, отклонения от линейности были очевидны в тесте на линейность
(рис. 17.23). Поэтому тестирование ступенчатым градиентом повторили в
окрестности результатов, вызывавших сомнения, и перепрограммировали систему на 1%
ступеньки. Результаты для области тестирования 45—55% показаны на рис. 17.245, где
все ступеньки выглядели нормально за исключением ступеньки 50—51 %В (показана
стрелкой), которая с очевидностью меньше остальных. Отклонения такого же типа
наблюдались при 25—26 %В и при 75—76 %В.
В этом случае в системе ВЭЖХ использовалось смешивание на стороне низкого
давления (раздел 3.5.2.2), поэтому следующим этапом в поиске проблемы стало
тестирование клапанов, аЬормирующих градиент (КФГ) (раздел 3.10.1.2). В основе этого
теста лежит попеременная подача воды и смеси ацетон-вода с использованием
различных комбинаций четырех линий подачи растворителя (линии А и В для подачи
воды и линии С и D для подачи смеси ацетон-вода); нормальный результат теста
показан на рис. 3.28. В рассматриваемом случае наблюдались результаты, показанные
на рис. 17.25 [33|. Максимальное приемлемое отклонение самого высокого плато
от самого низкого составляет 5%, в то время как на рис. 17.25 отклонение первого
т 1 1 1 1 1 1 1 1 1 1 1 г
0 2 4 6 в 10 12 14 16
Время (мин)
Рис. 17.23. График линейного градиента в системе с неисправными клапанами
формирующими градиент. Стрелками показаны отклонения от линейности.
Пунктирная прямая проведена под графиком для сравнения. Градиент 0 —
100 %В за 15 мин при I мл/мин; растворители: А — вода, В — 0,1% ацетон
в воде; детектирование при 265 нм |33|
902 Глава /7. Неисправности и способы их устранения
(б)
ступеньки 1%
(а)
ступеньки 10%
20
40
Время (мин)
Рис. 17.24. Результаты тестирования ступенчатым градиентом системы ВЭЖХ, график
линейного градиента которой дан на рис. 17.23. (а) Ступеньки 0, 10, 20, 30,
40, 45, 50, 55, 60, 70, 80, 90 и 100 %В; (5) верхняя кривая соответствует
интервалу 45—55 %В со ступеньками 1%. Стрелкой показана «низкая»
ступенька между 50 и 51 %В [33]
90/10
А/С
П
50/50 А/В,
A/D
ПВ/С В/О
п л
и
U 1_
"Т 1 Г-
Т
-1 1 1 1 1 1 г-
10
Время (мин)
20
Рис. 17.25. Тестирование клапанов формирующих градиент системы ВЭЖХ, график
линейного градиента которой дан на рис. 17.23. Базовую линию образует
отношение 50:50 для А:В. остальные плато — отношение 90:10 для А:С,
A:D, В:С и B:D слева направо. Растворители: А = В = вода; С = D = 0,1%
ацетон в воде 1331
плато от второго 12%. Так как на первых двух этапах растворитель подается по одной
и той же линии А, по-видимому, причина проблемы найдена. Сначала
предполагалось, что этой причиной является частично засоренный входной фильтр на линии
подачи растворителя или капилляр подачи растворителя между резервуаром и дози-
/7.4. Общие симптомы ВЭЖХ-проблем 903
рующим клапаном. Однако тест с сифоном (раздел 3.2.1) не показал, что входной
фильтр или соединительные капилляры существенно препятствуют течению. Были
приняты и другие меры, потенциально способствующие корректировке работы
системы: дегазирование подвижной фазы (раздел 3.3), обработка ультразвуком обратных
клапанов (раздел 17.2.5) и замена уплотнений в насосе (раздел 3.5.1). Были сделаны
попытки настроить в программном обеспечении алгоритм формирования градиента,
и, хотя были заметны улучшения, проблема оставалась. Наконец, заменили блок
клапанов формирующих градиент, и тогда при тестировании клапанов максимальное
отклонение составило 0,9% — вполне в пределах допустимых 5%.
В меньшей степени чреватый осложнениями, но более распространенный
вариант отрицательного результата теста линейности градиента представляет собой
график, состоящий из двух сегментов 0—50% и 50—100% с несколько различающимся
наклоном, полученный при программировании линейного градиента 0—100 %В.
Обычно так бывает при ошибках в программном обеспечении системного контрол
лера, и для некоторых систем ВЭЖХ его можно настроить. Обратитесь за специаль
ными рекомендациями к руководству по обслуживанию насоса или к изготовителю.
Случай 2. На рис. 17.26 показан еще более разительный пример того, как система
не прошла тестирование ступенчатым градиентом и тест на линейность градиента.
В этом случае оператору не удавалось получить воспроизводимые времена
удерживания [34]. При проведении тестирования ступенчатым градиентом (рис. 17.26с) и
теста на линейность градиента (рис. 17.266) были получены абсолютно неприемлемые
результаты. Предполагалось, что это вызвано пузырьком воздуха, застрявшим в
насосной системе, поскольку наблюдались нерегулярные колебания давления. После
тщательного промывания системы дегазированным растворителем с постукиванием
по каждому модулю системы ручкой отвертки (для удаления пузырьков воздуха,
прилипших к внутренним поверхностям) несколько пузырьков вышло в потоке
растворителя. При повторении тестов результат оказался удовлетворительным. (Смотрите
раздел 17.2.5.1, в котором указаны и другие способы удаления захваченного воздуха.)
Случай 3. В последнем примере тестирования ступенчатым градиентом с отрица
тельным результатом, сам метод работал хорошо с приемлемыми временами
удерживания, точностью, прецизионностью и разрешением. Однако так бывает не всегда:
при проведении этого теста были получены результаты, представленные на рис. 17.27
|34|. Видно, что между каждыми двумя большими ступеньками есть маленькая вторич-
М
I 1 1 1 1 1 1 1 I 1 1 1 1
о ю 20 зо о ю го
Время (мин) Время (мин)
Рис. 17.26. Неприемлемые результаты (а) тестирования ступенчатым градиентом и
(б) теста на линейность градиента |34|
904 Глава 17. Неисправности и способы их устранения
I 1 1 1 1 1 1—
О 10 20 30 40 50 60
Время (мин)
Рис. 17.27. Неприемлемые результаты тестирования градиентом с образованием
маленьких вторичных ступенек между основными ступенями [34]
ная ступенька (обратите внимание на отмеченную стрелкой маленькую ступеньку между
большими ступеньками при 10 и 20 %В). В процессе устранения вероятных причин ав-
тосамплер был заменен ручным инжектором (замена частей, раздел 17.3.5), при этом
проблема исчезла. Замена двух фильтров из нержавеющей стали в автосамплере решила
проблему. Не было ясно, почему засорение этих фильтров привело к появлению
вторичных ступенек на рис. 17.27. Ретроспективно можно предположить, что фильтры были
установлены в обводном канале, используемом в автосамплерах некоторых конструкций
для минимизации перепадов давления в колонке [35—37|. При такой конструкции часть
подвижной фазы проходит по обводному каналу, минуя кран инжектора, так что поток
не перекрывается во время поворота крана инжектора (для дополнительной
информации см. |38|, стр. 238). Один из авторов наблюдал сложности со временем удерживания
и шириной пиков, когда нарушалось течение по этому каналу; такое частичное
засорение фильтров тоже может привести к искажению градиента.
Случай 4. В некоторых случаях отрицательный результат теста линейности
градиента может свидетельствовать о неправильном обращении с оборудованием ВЭЖХ,
а не о неисправности этого оборудования. В разделе 3.10.1.2 было отмечено, что
в начале и в конце градиента есть закругления (рис. 3.26). Обычно эти закругления
незначительны и вряд ли могут повлиять на разделение. Однако, когда объем
градиента (= tcF) сравним с объемом задержки или меньше него, закругления становятся
более четко выраженными, так что могут вызвать искажение градиента. На рис. 17.28
показан пример с двумя различными градиентами |39|. Система со смешиванием
на стороне низкого давления (Кд » [ мл) использовалась с колонкой 100x2,1 мм
при скорости потока 0,2 мл/мин, и градиент достигал ячейки детектора через
ж 5 мин (см. профили ожидаемых градиентов на выходе колонки, обозначенные
пунктиром на рис. 17.28л,б). Градиент длительностью 9,9 минут UqF = 9,9 х 0,2 » 2 мл)
имел ожидаемый линейный профиль, показанный на рис. 17.27а сплошной линией,
заметны лишь небольшие искажения. За градиентом следовала минутная изократи-
ческая задержка и 0,1-минутная ступень градиента к исходным условиям. В этом
случае объем градиента (2 мл) значительно превышал объем задержки 1,0 мл.
/7.4. Общие симптомы ВЭЖХ-проблем
время (мин)
(б)
12
-1
15
время (мин)
Рис. 17.28. Искажение градиента из-за его слишком значительного закругления,
(а) Градиент 20/20/50/50/20 %В за 0,0/0,1/10/11/11,1 мин (небольшое
закругление); (б) 20/20/40/40/20 %В за 0,0/0,1/1,5/5/5,1 мин (чрезмерное
закругление). Сплошными линиями показан профиль градиента
вода/вода-ацетон; пунктирными линиями показан запрограммированный
градиент (сдвинут вверх для удобства восприятия) |39|
На рис. 17.286 показан крутой градиент длительностью 1,4 минуты с
последующей 3,5-минутной изократической задержкой и возвращением к исходным условиям
в течение 0,1 минуты. Объем градиента IqF'= 1,4 х 0,2 = 0,3 мл гораздо меньше
объема задержки. В этом случае можно предсказать существенное искажение градиента,
что и наблюдается на рис. 17.286. Хотя, может быть, и можно на каком-либо
хроматографе получить воспроизводимый градиент при условиях, указанных
на рис. 17.286", маловероятно, что такой метод с крутым градиентом удастся
перенести без проблем на другую систему ВЭЖХ. Дальнейшим подтверждением
существенного искажения градиента может служить фаза повторного уравновешивания
(медленное возвращение к исходному %В после градиента) на рис. 17.28а,6. Более
подробную информацию о влиянии закруглений градиента на разделение можно
найти в |181 стр. 393—396.
Случаи 5. Объем задержки градиента можно определить в эксперименте,
используемом для проверки линейности градиента (раздел 3.10.1.2 и рис. 3.26). Влияние
объема задержки на разделение обсуждается в разделе 9.2.2.4. Различия в объеме
задержки градиента у разных градиентных систем ВЭЖХ является одной из основных
причин того, что градиентные методы трудно переносить с одной системы на
другую. Как видно из рис. 17.29, обычно наблюдаются два эффекта в тех случаях, когда
метод используется на двух системах с разными объемами задержки. В данном
случае градиент 10—40 %В проводился 12 минут при скорости потока 1 мл/мин. Одна
система (рис. 17.29а) имела объем задержки 1 мл, а вторая (рис. 17.296) — 3 мл. Это
отличие объемов задержки нашло свое отражение в двух реальных градиентах,
показанных пунктирными линиями на рис. 17.29, с 1-минутной и 3-минутной отсрочкой
достижения градиентом колонки на рис. 17.29а и 6, соответственно. Для
анализируемых веществ, элюирующихся позже, значение k которых в начале градиента (Ао)
достаточно велико, различие объемов задержки в первую очередь выразилось в сдвиге
906 Глава 17. Неисправности и способы их устранения
(а)
VU
~~Г"
ю
—i—
12
—i—
14
(б)
-г-
10
Время (мин)
—г-
12
14
Рис. 17.29. Влияние изменения объема задержки градиента на удерживание и
селективность, (а) Система с объемом задержки 1 мл; (б) система с объемом
задержки 3 мл. Колонка 100 х 4,6 мм; градиент 10—40 %В за 12 минут
при скорости потока 1 мл/мин
времени удерживания пропорционально этому различию. Видно, что удерживание
пиков с 3 по 9 увеличилось на 2 минуты [(3 мл — 1 мл)/1 мл/мин = 2 мин|
на рис. 17.296". Однако может изменяться и относительное расстояние между пиками,
элюирующимися раньше, (как и а), особенно если это пики «неправильных»
образцов (раздел 2.5.2.2). Это иллюстрируют пики 1 и 2 (обратите внимание на ухудшение
разрешения этих пиков на рис. 17.295, несмотря на ожидаемое возрастание
разрешения рановыходяших пиков при увеличении времени задержки (раздел 9.2.2.3)).
Можно объяснить это так. На рис. 17.29а рановыходяшие пики 1 минуту перемещаются
по колонке в изократических условиях, и от 2 до 3 минут — в условиях градиента,
в то время как на рис. 17.296" они 3 минуты перемещаются в изократических
условиях и еще от 2 до 3 минут — в условиях градиента. Это изменение соотношения
длительности миграции в изократических и градиентных условиях приводит к
изменению эффективных значений к (к*) и расстояния между пиками. Для пиков,
выходящих позже, первоначальное движение пиков в изократических условиях не
имеет значения, поэтому наблюдается только сдвиг времен удерживания вследствие
отсрочки достижения градиентом входа колонки.
Есть несколько способов компенсировать различия объемов задержки у двух
систем ВЭЖХ. Для случая, иллюстрируемого рис. 17.29, есть три возможных решения,
если предположить, что метод, разработанный на системе рис. 17.29а, переносится
на систему рис. 17.296". Наиболее простым решением является программирование
отсрочки введения образца, если это позволяет оборудование. Сначала запускается
градиент в системе рис. 17.296", а введение образца программируется через 2 минуты
/7.4. Общие симптомы ВЭЖХ-проблем 907
после запуска градиента. Это способствовало бы сдвигу всей хроматограммы направо
относительно обозначенного пунктиром градиента, поэтому образец разделялся бы
точно в таком же градиенте, как и на рис. 17.29а. К сожалению, не у всех систем есть
опция отсрочки введения образца. Второй подход заключается в разработке метода
с максимальным объемом задержки. Для этого требуется заранее знать, каков объем
задержки системы рис. 17.295. Можно также объем задержки исходной системы
отрегулировать таким образом, чтобы он был равен наибольшему возможному объему
задержки любой системы, на которую будет переноситься метод. В данном случае
объем задержки системы рис. 17.29а будет отрегулирован при добавлении
2-минутной изократической задержки (2 мин при скорости потока 1 мл/мин = 2 мл) в начале
каждого разделения, так чтобы эффективный объем задержки составил 3 мл. При
перенесении метода эту дополнительную задержку следует исключить, тогда градиенты
будут одинаковыми для обеих систем. Если планируется использовать несколько
систем с разными объемами задержки, следует разрабатывать метод для максимального
объема задержки и потом подбирать изократическую задержку и истинный объем
задержки так, чтобы эффективный объем задержки был одинаков для всех систем.
Третий подход предполагает подбор исходного %В для системы 17.296. Он
подразумевает комбинацию запуска градиента с более высоким %В и изменение длительности
изократического и градиентного сегментов. Примеры использования этого подхода
приведены в |18|, в разделе 5.2.1.3. Еще один метод компенсации различий в объеме
задержки систем применяется, когда объем задержки исходной системы больше, чем
у второй, например, если данный метод был разработан на системе 17.296 и
перенесен на систему 17.29а. В этом случае перед градиентом рис. 17.29а можно добавить
2-минутную изократическую задержку, обеспечивая одинаковую отсрочку запуска
обоих градиентов. Этот подход подобен методу введения максимального объема
задержки, который обсуждался ранее. Более подробно подгонка градиента в системах
с разным объемом задержки рассмотрена в |18|, раздел 5.2.1.3.
17.4.6.2. Интерпретация дополнительных тестов системы
В разделе 3.10.I.3 описаны еще несколько дополнительных тестов, которые следует
регулярно проводить:
ф тестирование скорости потока;
ф тестирование сброса давления;
ф тестирование воспроизводимости удерживания;
ф тестирование воспроизводимости площади пика.
Если при тестировании скорости потока обнаружено отклонение от заданного
значения более ±2%, следует искать причину отклонения. Скорость потока ниже
заданной — достаточно распространенная проблема. Сначала проверьте, нет ли утечек
(раздел 17.4.1, таблица 17.3). Другими вероятными причинами снижения скорости
потока являются пузырьки воздуха в насосе, неисправные обратные клапаны и
изношенные уплотнения насоса. Иногда скорость потока органических растворителей и
воды отличаются на одном и том же насосе при одинаковых заданных значениях
вследствие большей сжимаемости органических растворителей. Пользователи
обычно оставляют без внимания сжимаемость растворителей, но многие насосы можно
настроить так, чтобы компенсировать сжимаемость растворителей (см. руководство
для оператора). Редко скорость потока превышает заданное значение (за
исключением неправильной настройки насоса в отношении сжимаемости). Сначала проверьте
правильность заданных значений скорости и повторите тестирование. Обратитесь
к инструкциям для оператора насоса за информацией по калибровке насоса, если
проблема не решается. Дополнительное обсуждение проблем, связанных со
скоростью потока, проведено в разделе 17.4.3.1, а работа насоса рассмотрена в разделе 3.5.
908 Глава 17. Неисправности и способы их устранения
Тестирование сброса давления представляет собой проверку способности насоса
держать давление в статических условиях. Если в этом тесте давление падает на величину
> 15% за 10 минут, следует установить причину и устранить ее. Так как в основе этого
теста лежит блокирование выходных капилляров насоса, он свидетельствует о
целостности ограничивающего давление узла насоса, расположенного ближе всего к
перекрытым выходным капиллярам. Сначала проверьте, нет ли утечек между насосом и
перекрытыми выходными капиллярами. Если в насосе на выходе установлены обратные
клапан(ы), они наиболее вероятная причина сбоя. Если в насосе нет обратных клапанов
на выходе, проблема может быть обусловлена уплотнением плунжера насоса, входным
обратным клапаном (двухппунжерные насосы, раздел 3.5.1.1) или промежуточным
обратным клапаном (накопительные плунжерные насосы, раздел 3.5.1.2). Замените любые
вызывающие сомнения уплотнения и обработайте ультразвуком (раздел 17.2.5.4) или
замените вызывающие сомнения обратные клапаны.
Воспроизводимость времени удерживания должна обычно гарантировать различие
времен удерживания не более чем на ±0,05 минуты (1 СКО), но в некоторых методах
воспроизводимость может быть хуже. Если вызывающие сомнение значения
точности времен удерживания относятся к методу, использовавшемуся раньше, сравните
их с полученными раньше результатами (раздел 17.2.4) и проведите вновь
тестирование воспроизводимости времен удерживания (раздел 3.10.1.3 и таблица 3.5). Если нет
утечек (раздел 17.4.1, таблица 17.3), наиболее вероятны проблемы, связанные с
работой насоса и/или с процессом смешивания в потоке подвижной фазы. Проведите
тестирование пригодности градиента (раздел 3.10.1.2) и вычлените проблему
(раздел 17.4.6.1).
Воспроизводимость площади пика зависит от области применения; приемлемые
значения колеблются в интервале от < 1—2% (при анализе состава образца)
до 15—20% (при анализе следовых примесей), поэтому лучше сравните результаты
теста с характеристиками метода, которые были определены раньше (раздел 17.2.4).
Когда стандартный тест на воспроизводимость площади пика (раздел 3.10.1.3) дает
значение более 0,5—1%, проверьте автосамплер. Убедитесь, что никакие отверстия, в
том числе отверстия игл, не засорены. Проверьте, не засорилась ли или не сломалась
ли игла для нанесения образца. Проверьте уплотнения и шприцы в системе отбора
пробы. Проверьте уплотнение иглы в инжекторе. Убедитесь, что не задано слишком
большое значение постоянной времени детектора. В поисках других причин можно
обратиться к разделу 3.6 или руководству для оператора автосамплера.
17.5. Таблицы по устранению неисправностей
Большая часть таблиц, поясняющих как устранять неисправности, сгруппирована
в этом разделе для удобства одновременного обращения к нескольким таблицам.
Для определения проблемы можно использовать разные подходы. Сначала
обратитесь к перечню разделов в начале этой главы и найдите тот, тема которого вас
интересует. Кроме того, можно использовать как указатель таблицу 17.2. В этой таблице
перечислены основные признаки неисправностей, с которыми вы, вероятно,
столкнетесь, перекрестные ссылки на другие таблицы этого раздела и на разделы этой
книги с полезными комментариями. Остальные таблицы содержат более
специализированную информацию, которая помогает вычленить определенные проблемы.
Большинство этих таблиц построено в виде плана действий. В левой колонке
приводится основной признак неисправности (например, местоположение утечки в
таблице 17.3). Изучая таблицу слева направо, в каждой следующей колонке вы
получаете все более подробную информацию для вычленения проблемы и определения ее
17.5. Таблицы по устранению неисправностей 909
возможных источников. В правой колонке обычно приводятся предлагаемые
решения и дополнительные перекрестные ссылки на тексты, посвященные данной
проблеме. Может оказаться, что для определения и решения проблемы достаточно одних
таблиц или вам могут понадобиться пояснительные тексты — это зависит от
особенностей проблемы и вашего опыта.
Таблица 17.2. Симптомы проблем в ВЭЖХ
Симптом проблемы
Утечки
Давление
Время удерживания
Площадь пика
Дрейф базовой линии
Шумы базовой линии
Форма пика
Отрицательный результат теста на пригодность системы
Дополнительная информация
Таблица 17.3, раздел 17.4.1
Таблица 17.4, раздел 17.4.2
Таблица 17.5, 17.6, раздел 17.4.3
Таблица 17.7, раздел 17.4.4
Таблица 17.8, раздел 17.4.5.1
Таблица 17.9, раздел 17.4.5.2
Таблица 17.10, разделы 17.4.5.3, 2.4.2
Таблица 17.11, раздел 17.4.6
Таблица 17.3. Утечки (раздел 17.4.1): симптомы и причины
Местоположение
Перед
насосом (17.4.1.1)
Насос
(17.4.1.2)
Фитинги
высокого
давления
(17.4.1.3)
Автосамплер
или ручной
инжектор
(17.4.1.4)
Уточненное
местоположение/симптом
Фитинги
Клапанная коробка
(смешивание на стороне
низкого давления)
Фитинги
Обратный клапан
Уплотнение насоса
Вспомогательный узел
Из нержавеющей стали
Из ПЭЭК
Фитинги
Возможная причина
Плохо затянутые фитинги
Плохо затянутый или
поврежденный клапан
Плохо затянутые или
поврежденные фитинги
Плохо затянутый клапан
Повреждение резьбы
Изношенное уплотнение
Ослабленные фитинги;
поврежденный узел
Плохо затянутые или
загрязненные
Плохо затянутые или
загрязненные
Плохо затянутые или
поврежденные фитинги
Решение проблемы
Подтянуть или заменить
Подтянуть клапан или
заменить клапанную
коробку
Подтянуть или заменить
Подтянуть
Заменить клапан; может
потребоваться новая
голова насоса
Заменить
Подтянуть или заменить
Подтянуть на 1/4 оборота,
промыть или заменить
Выключить насос,
переустановить капилляр, снова
затянуть; не использовать
при давлении > 400 бар
(6000 psi)
См. регулировка
фитингов, разделы 17.4.1.1,
17.4.1.3
910 Глава 17. Неисправности и способы их устранения
Окончание табл. 17.3
Местоположение
Автосамплер
или ручной
инжектор
(17.4.1.4)
Колонка
(17.4.1.5)
Детектор
(17.4.1.6)
Уточненное
местоположение/симптом
Уплотнение низкого
давления иглы инжектора
Уплотнение высокого
давления иглы инжектора
Перетекание жидкости
между портами
Внешние утечки не в
местах соединений
Проблемы в системе
уплотнений (только
для автосамплеров Waters)
Соединения капилляров
Концевые фитинги
Концевые фитинги пред-
колонки/картриджа
Соединительные фитинги
Утечки в ячейке
Детектор,
регистрирующий не поглощение в УФ
Возможная причина
Изношенное или
поврежденное уплотнение
Поврежденное или
изношенное уплотнение
Поврежденное
уплотнение ротора
Изношенное уплотнение
ротора
Изношенные или
поврежденные уплотнения
Плохо затянутые или
поврежденные
Плохо затянутые или
поврежденные
Плохо затянутые или
поврежденные фитинги,
поврежденное уплотнение
Плохо затянутые или
поврежденные
Плохо затянутые фитинги
Повреждение из-за
превышения давления
Нарушение герметичности
уплотнения окна
См. главу 4 и руководства
для пользователей
Решение проблемы
Подянуть или заменить
Подянуть или заменить
Промыть, заменить
уплотнение ротора; может
потребоваться замена
статора
Промыть, заменить
уплотнение ротора
Отремонтировать или
заменить
См. регулировка
фитингов, раздел 17.4.1.3
Подтянуть; может
потребоваться новая колонка
Подтянуть, заменить
уплотнение, может
потребоваться новая сборка
всего узла
См. регулировка
фитингов, разделы 17.4.1.1,
17.4.1.3
Подтянуть
Устранить причину
избыточного давления
Отрегулировать или
заменить
Таблица 17.4. Проблемы с давлением (раздел 17.4.2): признаки и причины
Симптом
Слишком
высокое
давление
(17.4.2.1)
Местоположение/признак
Вся система
Засоренные
капилляры
Возможная причина
Неподходящая подвижная фаза,
слишком низкая температура,
слишком высокая скорость
потока, неподходящая колонка,
неподходящий размер частиц
Выпадение осадка в буфере,
твердые частицы в образце
Решение проблемы
Скорректируйте условия
разделения
Заменить капилляры,
устранить источник засорения
/7.5. Таблицы по устранению неисправностей
Продолжение табл. 17.4
Симптом
Слишком
высокое
давление
(17.4.2.1)
Слишком
низкое
давление
(17.4.2.2)
Местоположение/признак
Засоренный
или частично
засоренный
фильтр
Превышение
предела
давления в градиенте
Вся система
Нижний предел
давления
Фитинги
высокого давления
Фитинги
низкого давления
Клапанная
коробка при
смешивании на
стороне
низкого давления
Входной
обратный клапан
В насос не
поступает
подвижная фаза
(отри цател ьн ы й
результат теста
с сифоном)
Пузырьки
воздуха в насосе
Уплотнение
насоса
Плунжер
насоса
Возможная причина
Инлайн фильтр
Предколонка
Аналитическая колонка
Естественное изменение
давления в ходе градиентного
разделения
Неподходящая подвижная фаза,
слишком высокая температура,
слишком низкая скорость
потока, неадекватная колонка
Недостаточное количество
подвижной фазы
Слишком чувствительный
датчик давления
Большая утечка
Плохо затянутые или
загрязненные фитинги
Плохо затянутые или
загрязненные фитинги
Поступление воздуха из
неиспользуемой линии подачи
растворителя, утечка в КФГ
Залипание клапана при
использовании подвижных фаз с аце-
тонитрилом
Засоренный входной фильтр
на линии всасывания
Неисправные КФГ
Деформированные или
засоренные капилляры
Недостаточное дегазирование
или промывание; неисправный
дегазатор
Изношенное уплотнение насоса
Сломанный или поцарапанный
плунжер
Решение проблемы
Заменить фильтр, рассмотреть
возможность дополнительной
очистки образца
Заменить предколонку
Установить колонку в
обратном направлении (если это
разрешено), заменить колонку,
рассмотреть возможность
использования инлайн фильтра
Отрегулировать верхний
предел давления для его
соответствия реальному давлению
Скорректировать условия
разделения
Заполнить резервуар
Откпючить датчик,
контролирующий нижний предел
давления
Найти и устранить
(раздел 17.3.1)
Подтянуть, промыть или
заменить
Подтянуть, промыть или
заменить
Промыть неиспользуемые
линии подачи органическим
растворителем; заменить
неисправные КФГ
Обработать ультразвуком
в метаноле; заменить
Заменить
Заменить
Промыть или заменить
Дегазировать подвижную фазу
и промыть насос с большой
скоростью; заменить дегазатор
Заменить
Заменить
912 Глава 17. Неисправности и способы их устранения
Окончание табл. 17.4
Симптом
Слишком
нестабильное давление
(17.4.2.3)
Местоположение/признак
Пузырьки
воздуха в насосе
Входной
обратный клапан
Фитинги высо
кого давления
Фитинги
низкого давления
Клапанная
коробка при
смешивании на
стороне
низкого давления
В насос не по
ступает
подвижная фаза
(отри цател ьн ы й
результат теста
с сифоном)
Уплотнение
насоса
Плунжер
насоса
Возможная причина
Недостаточное дегазирование
или промывка
Залипание клапана при
использовании подвижных фаз с аце-
тонитрилом
Плохо затянутые или
загрязненные фитинги
Плохо затянутые или
загрязненные фитинги
Поступление воздуха из
неиспользуемой линии подачи
растворителя, утечка в КФГ
Засоренный входной фильтр на
линии всасывания
Неисправные КФГ
Деформированные или забитые
капилляры
Изношенное уплотнение насоса
Сломанный или поцарапанный
плунжер
Решение проблемы
Дегазировать подвижную фазу
и промыть насос с большой
скоростью
Обработать ультразвуком
в метаноле; заменить
Подтянуть, промыть или
заменить
Подтянуть, промыть или
заменить
Промыть неиспользуемые
линии подачи органическим
растворителем; заменить
неисправные КФГ
Заменить
Заменить
Промыть или заменить
Заменить
Заменить
Таблица 17.S. Интерпретация изменений коэффициента удерживания к и времени
удерживания 1Я (раздел 17.4.3)
Изменения к
Нет
Нет
Есть
Есть
Есть
Изменения lR
Есть
Есть
Есть
Есть
Есть
Возможная причина
Скорость потока (заданные
значения, утечки, пузырьки воздуха,
проблемы с насосом)
Неподходящий объем колонки
Ошибка, связанная с подвижной
фазой (%В, рН, добавки)
Проблемы с неподвижной фазой
(старение, загрязненные образцы)
Проблемы с температурой
(недостаточный контроль)
Дополнительная информация
Раздел 17.4.3.1
Раздел 17.43.2
Раздел 17.43.3
Раздел 17.4.3.4
Раздел 17.4.3.5
17.5. Таблицы по устранению неисправностей 913
Таблица 17.6. Проблемы с временем удерживания (раздел 17.4.3.6): признаки и причины
Симптом
Резкое
изменение /R
Резкое
изменение /д
Дрейф
удерживания
Слишком
малое
удерживание
Местоположение/
признак
При замене
колонки
При изменении
подвижной фазы
При градиентном
элюировании
на другой системе
ВЭЖХ
Без видимых
изменений условий
Только при
введении нескольких
первых образцов
Во всей партии
образцов
Только при
введении больших масс
образцов для
некоторых или всех
пиков,
выходящие пики имеют
форму
прямоугольного
треугольника
Всех пиков
Некоторых пиков
Только пиков
полярных
анализируемых веществ
Возможная причина
Неподходящая
колонка; плохая
воспроизводимость от колонки
к колонке (особенно
для колонок с силика-
гелем типа А)
Ошибка в составе
подвижной фазы
Изменение объема
задержки
Проблемы с
оборудованием, пузырьками
воздуха или
температурой
Медленное
уравновешивание колонки;
«насыщение» колонки
образцом
Нестабильная
подвижная фаза; испарение
подвижной фазы;
избыточное барботирова-
ние гелием
Перегрузка по массе
Ошибка в составе
подвижной фазы;
изменение скорости потока
или температуры,
замена колонки
Проблемы с рН или
другие ошибки в
составе подвижной
фазы; старение
колонки
Ионные образцы
Решение проблемы
Использовать подходящую колонку;
использовать колонки из одной партии;
модифицировать метод для повышения
воспроизводимости; использовать
колонки, обеспечивающие более высокую
воспроизводимость (например, с силикаге-
лем типа В); раздел 2.5.4.6, таблица 17.5
Сделать новую порцию подвижной
фазы; таблица 17.5
Отрегулировать или компенсировать
объем задержки; раздел 9.3.8.2
Проверить утечки (таблица 17.3,
раздел 17.4.1), проверить давление
(таблица 17.4, раздел 17.4.2), проверить,
изменился ли к (таблица 17.5, раздел 17.4.3);
раздел 2.5.4.6
Нормальная ситуация для некоторых
колонок и образцов; увеличить время
уравновешивания; не использовать
данные нескольких первых инжекций
(раздел 3.10.2.2); разделы 2.7.1 (общая
теория), 7.4.3.2 (ион-парные реагенты), 8.5
(НФХ), 9.3.7 (градиентное элюирование)
Использовать стабильную подвижную
фазу; закрыть резервуар, чтобы
уменьшить испарение; сократить время барбо-
тирования гелием или заменить метод
дегазирования (раздел 3.3)
Уменьшить массу вводимого образца;
таблица 17.11, разделы 17.4.5.3, 2.6.2
См. таблицу 17.5
Сделать необходимые поправки;
раздел 7.3 (рН); заменить колонку
Изменить рН подвижной фазы
(раздел 7.3); использовать ион-парные
реагенты (раздел 7.4)
914 Глава 17. Неисправности и способы их устранения
Окончание табл. 17.6
Симптом
Слишком
малое
удерживание
Слишком
большое
удерживание
Местоположение/
признак
Только пиков
полярных
анализируемых веществ
Всех пиков
Некоторых пиков
Возможная причина
Нейтральные или
неионные образцы
Ошибка в составе
подвижной фазы;
изменение скорости потока
или температуры;
замена колонки; утечки
Проблемы с рН или
другие ошибки в
составе подвижной
фазы; старение
колонки
Решение проблемы
Пробовать колонки П П Г (типа AQ);
использовать нормальнофазовую
хроматографию (глава 8) или ГИХ (раздел 8.6)
См. таблицы 17.5, 17.3
Внести надлежащие поправки;
раздел 7.3 (рН); заменить колонку
Таблица 17.7. Проблемы с площадью пиков (раздел 17.4.4): признаки и причины
Симптом
Площадь
пиков
слишком
велика
(17.4.4.1)
Площадь
пиков
слишком
мала
(17.4.4.2)
Местоположение/
признак
Все пики больше
в одинаковом
соотношении
Площадь разных
пиков образца возросла
на разную величину
Пик(и) в холостом
разделении
Все пики меньше
в одинаковом
соотношении
Площадь разных
пиков образца
уменьшилась на разную
величину
Возможная причина
Слишком велик
объем вводимого образца
Ошибки в пробопод-
готовке
Неправильно
настроен детектор
Неправильно
настроен детектор
Позднее алюирование
пиков от предыдущей
инжекции
Остаточные примеси
Слишком мал объем
вводимого образца
Ошибки в пробопод-
готовке
Неправильные
настройки детектора
Неправильные
настройки детектора
Решение проблемы
Внести нужные изменения
Использовать правильную методику
Настроить
Настроить
Увеличить время разделения;
добавить промывку сильным элюентом;
измените пробоподготовку
Добавить этап промывки автосамп-
лера; заменить растворитель для
промывания; проверит соединения;
взять петлю из другого материала;
изменить порядок инжекции
Внести нужные изменения
Использовать правильную методику
Настроить
Настроить
17.5. Таблицы по устранению неисправностей 915
Окончание табл. 17.7
Симптом
Изменяющаяся
величина
площади пикоп
(17.4.4.3)
Местоположение/
признак
При повторных
вводах образца площадь
пиков не изменяется
При повторных
вводах образца площадь
пиков изменяется
Возможная причина
Проблемы,
предшествующие вводу
образца
Проблемы с
оборудованием, начиная с
инжектора и далее
Решение проблемы
Проверить методику отбора проб и
пробопод готовки
Проверить автосамплер, насос,
детектор и параметры
интегрирования
Таблица 17.8. Проблемы с дрейфом базовой линии (раздел 17.4.5.1): признаки н причины
Симптом
Периодический
дрейф
Другие виды
дрейфа
Местоположение/признак
1 цикл в течение
разделения
В течение нескольких
часов или 1 цикл в день
Чередуются участки
со стабильной базовой
линией и дрейфом
Возможная причина
Нормальная ситуация
для градиентного
алюирования
Цикл изменения
температуры в
лаборатории
Естественный процесс
уравновешивания;
прогрев детектора
Решение проблемы
Не принимать во внимание;
использовать для
регистрации ббльшую длину волны;
заменить растворитель;
добавить УФ-поглотитель1
Термостат; передвинуть
систему ВЭЖХ туда, где нет
сквозняков; отрегулировать
отопление, вентиляцию и
кондиционирование воздуха
в лаборатории
Подождать, пока
стабилизируется базовая линия
'Добавляют в элюент А небольшие количества УФ-поглощающих компонентов, не влияющих
на удерживание, например, фенол или бензойную к-ту. — Прим. перев.
Таблица 17.9. Проблемы шумов базовой линии (разлел 17.4.5.2): признаки и причины
Симптом
Кратковременные
шумы
Дол говременные
шумы
Местоположение/признак
Высокочастотные шумы
(50 или 60 Гц)
50—60 Гц < шумы <
< ширина пика
Циклические
Возможная причина
Помехи в цепи
питания; постоянная
величина тока в
течение лишь небольшой
части времени
детектирования; слишком
большая скорость
сбора данных
Недостаточное
дегазирование
подвижной фазы
Пузырьки воздуха
в насосе;
повреждение уплотнения
плунжера или самого
плунжера; залипание
или протекание
обратного клапана
Решение проблемы
Использовать ИБП
для стабилизации тока
питания; использовать
ббльшую постоянную
времени детектора или
сглаживающий фильтр;
использовать меньшую скорость
сбора данных
Использовать более
эффективный метод
дегазирования
Дегазировать подвижную
фазу; проверить и наладить
насос; промыть или
заменить обратный клапан
916 Глава 17. Неисправности и способы их устранения
Окончание табл. 17.9
Симптом
Долговременные
шумы
Местоположение/признак
Выглядит как хромато-
графический пик
Случайные отклонения
базовой линии
неправильной формы
Пики появляются в
холостых разделениях
(пики-«при зраки»,
рис. 17.11, 17.12)
Возможная причина
Поздноэлюируемое
анализируемое
вещество
Накопленные позд-
ноэлюируемые
неполярные примеси
образца
Загрязненная
подвижная фаза
Решение проблемы
Увеличить время
разделения; ввести в метод
промывку сильным элюентом;
оптимизировать очистку
образца
Внести в метод промывку
сильным элюентом;
заменить колонку;
оптимизировать очистку образца
Заменить реактивы для
подвижной фазы свежими
и/или реактивами более
высокой чистоты
Таблица 17.10. Проблемы с фермой пиков (раздел 17.4.5.3): симптомы и причины
Симптом
Размывание
заднего фронта
пика
Размывание
переднего
фронта
пика
Симптом
TF< 1,2
1,2 < TF<2
TF> 2
Пики в виде
прямоугольных
треугольников в
сочетании с меньшим
удерживанием
(рис. 17.15«)
TF > 0,9
Симптом/возможная причина
Нормальная ситуация
Если со временем возрастает:
старение или загрязнение
колонки; ошибка в составе
подвижной фазы
Если со временем не
изменяется, может быть нормой
Для всех пиков
У рановыходящих пиков
размывание больше, чем у позд-
новыходящих (рис. 17.13):
внеколоночные эффекты
Размываются больше пики
основных и ионных
соединений (рис. 5.9я): влияние сила-
нольных групп; загрязнения
следовыми количествами
металлов
Недостаточная буферная
емкость или концентрация
добавок в подвижной фазе
Массовая перегрузка колонки
Нормальная ситуация
Решение проблемы
Ничего не требуется
Промыть или заменить
колонку; приготовить новую порцию
подвижной фазы
Ничего не предпринимать или
искать решения для TF > 2
См. расщепленные или
искаженные пики
Уменьшить внеколоночный
объем (разделы 2.4.1, 3.9)
Довести рН; использовать
колонки с силикагелем более
высокой чистоты или менее
активные колонки (см.
разделы 5.4.4.1, 7.3.4.2)
Увеличить концентрацию
буфера или добавок; см.
раздел 7.2.1.1
Уменьшить вводимый объем
образца или его разбавить
для уменьшения массы
наносимого на колонку образца
(раздел 2.6.2)
Ничего не требуется
17.5. Таблицы по устранению неисправностей
Продолжение табл. 17.10
Симптом
Размывание
переднего
фронта
пика
Широкие
ПИКИ
Расщепленные
или
искаженные
пики
Симптом
TF < 0,9
/V > 75% значения,
полученного в
тесте производителя
Высокомолекулярные соединения
(белки, полимеры
и т.д.)
Постепенное уши-
рение после 500
инжекций
В присутствии
более узких пиков
(рис. 17.5)
В сочетании с
большими
временами удерживания
Более узкие пики
уширены
в большей
степени, чем пики
нормальной ширины
Ра но выходящие
пики широкие или
с плоской
вершиной (рис. 17.166)
Все пики
искажены одинаковым
образом
(рис. 17.17, 17.19)
Широкие или
искаженные пики,
особенно в конце
разделения
(рис. 17.206), в
сочетании с
изменением времени
удерживания
Симптом/возможная причина
При использовании
ион-парных реагентов, особенно
с сорбентами на основе сили-
кагеля типа А
Полость в колонке или
нарушение структуры упаковки
Можно считать нормальной
ситуацией
Некоторое уширение
нормально
Нормальное старение
колонки
Позднее элюирование
Слишком низкая температура
колонки
Слишком велика постоянная
времени детектора; слишком
мала скорость сбора данных
интегратором; чрезмерное
сглаживание (особенно
при масс-спектрометрическом
детектировании)
Слишком велик объем
наносимого образца
Частично засорен входной
фильтр колонки; полость
в колонке
Несовпадение температуры
подвижной фазы и колонки
Решение проблемы
Может помочь изменение
температуры на ±5—10 "С;
перейти на сорбенты типа В
Заменить колонку; работать
с колонкой при рН ниже
предельного
Не требуется никаких
действий; сравнить с результатами
нового теста колонки с
компонентами образца
Не требуется никаких действий
Промыть колонку сильным
растворителем, заменить
колонку
Увеличить время разделения;
отрегулировать
продолжительность разделения; использовать
промывку градиентом;
оптимизировать очистку образца
Повысить температуру
Задать постоянную времени
< 1/10 ширины пика; собирать
минимум 20 точек на каждый
пик; уменьшить сглаживание
Уменьшить объем наносимой
пробы, разбавить растворитель,
в котором вводится образец
(разделы 2.6.1, 3.6.3)
Промыть колонку в обратном
направлении; использовать
проточный фильтр или
улучшить очистку образца;
заменить колонку (раздел 5.8)
Оптимизировать контроль
температуры; подогревать
подвижную фазу перед поступлением
в колонку (раздел 3.7.1)
918 Глава 17. Неисправности и способы их устранения
Окончание табл. 17.10
Симптом
Расщепленные
или
искаженные
пики
Симптом
Искажение рано-
выходящих пиков
(рис. 17.1 бе),
обычно
сопровождается
уменьшением времени
удерживания
Симптом/возможная причина
Слишком велик объем
вводимого образца, образец
растворен в слишком сильном элю-
енте
Решение проблемы
Использовать более
разбавленный алюент для растворения
образца, уменьшить объем
вводимого образца (разделы 2.6.1,
3.6.2.2)
Таблица 17.11. Отрицательные результаты тестов пригодности системы (раздел 17.4.6):
признаки и причины
Тест с
отрицательным результатом
Линейность
градиента
Тестирование
ступенчатым
градиентом
Тестирование
КФГ
Различие объемов
задержки у
разных систем
Тестирование
расхода
Тест на
стравливание давления
Признаки
Ступеньки в градиенте
Линейный градиент
выглядит как
кусочно-линейный
Искажение градиента
(рис. 17.286)
Ступеньки
неодинаковой высоты и/или
имеют искаженную форму
Колебания высоты
плато КФГ > 5%
Изменения
удерживания и/или
селективности при переходе с
одной системы на другую
Скорость потока
отличается от заданной
на < ±2%
Давление падает
на > 15% за 10 минут
Признаки/возможная
причина
Засоренный фильтр
в резервуаре; дефект
дозирующего клапана
Ошибка программного
обеспечения
Объем смешивания
градиента слишком велик
по сравнению с
объемом градиента
Пузырьки воздуха,
неисправность обратного
клапана или утечка
Засоренный фильтр
в резервуаре;
ограничивающие поступление
растворителей
капилляры; неисправность КФГ
Нормальная ситуация,
если объемы задержки
систем различны
Пузырьки воздуха;
проблемы с обратным
клапаном или
уплотнением насоса; утечка;
неправильно задана
сжимаемость
Проблемы с обратным
клапаном или
уплотнением насоса; утечка
Решение проблемы
Проверить/заменить
фильтр; заменить
клапанную коробку
Настроить контрольные
параметры (см.
руководство по обслуживанию)
Использовать более
пологий градиент, уменьшить
объем смешивания
Дегазировать подвижную
фазу; промыть или
заменить обратный клапан;
устранить утечку;
засоренный фильтр в
резервуаре, неисправный
дозирующий клапан
Заменить фильтр;
промыть или заменить
капилляры; заменить КФГ
Скорректировать метод,
чтобы компенсировать
различие объемов
задержки
Дегазировать подвижную
фазу; промыть или
заменить обратный клапан;
заменить уплотнение
насоса; задать нужное
значение сжимаемости (или
не задавать)
Промыть или заменить
обратный клапан;
заменить уплотнение насоса;
устранить утечку
Литература 919
Окончание табл. 17.11
Тест с
отрицательным результатом
Тест на
воспроизводимость
времени удерживания
Тест на вопроиз-
водимость
площади пика
Признаки
> ±0,05 мин в
стандартном тесте; превышает
допустимое значение
для метода с
нормальными
характеристиками
Отклонение > 1%
в стандартном тесте;
превышает допустимое
значение для метода с
нормальными
характеристиками
Приз наки/'во зчожная
причина
Пузырьки воздуха;
утечки; проблемы с
обратным клапаном или
уплотнением насоса;
неисправность насоса
или погрешность сме
шивания
Проблема с автосампле-
ром
Решение проблемы
Дегазировать подвижную
фазу; устранить утечки;
промыть или заменить
обратный клапан;
провести тест на пригодность
градиента для выявления
возможных проблем
Промыть или заменить
иглу; заменить
уплотнения; см. руководство
для автосамплера
Литература
1. J. W. Dolan and L. R. Snyder, Troubleshooting LC Systems, Humana Press/Springer, Clifton, NJ,
1989.
2. V. R. Meyer, Pitfalls and Errors of HPLC in Pictures, Wiley-VCH, Weinheim, 2006.
3. J. W. Dolan, "LC Troubleshooting," in LCGC, a monthly column (1983-present).
4. http://www.chromforum.org.
5. http://www.lcresources.com/resources/TSWiz/.
6. S. J. Williams, J. Chramatagr. A, 1052 (2004) 1.
7. M. D. Nelson and J. W. Dolan, LCGC, 16 (1998) 992.
8. С. К. Cheung and R. Swaminathan, Clin. Chem., 33 (1987) 202.
9. C. T. Mant and R. S. Hodges, eds.. High-Performance Liquid Chromatography of Peptides and
Proteins: Separation, Analysis, and Conformation, CRC Press, Boca Raton, 1991.
10. J. W. Dolan, J. R. Kern, and T. Culley, LCGC, 14 (1996) 202.
11. D. W. Bristol, J. Chromatogr., 188 (1980) 193.
12. P.-L. Zhu, L. R. Snyder, and J. W. Dolan, J. Chromatogr. A, 718 (1995) 429.
13. J. W. Dolan, LCGC, 11 (1993) 640.
14. J. W. Dolan, LCGC, 9 (1991) 22.
15. M. L. Ledtje and D. Long, Jr., US Patent 5,002,662 (March 26, 1991).
16. J. W. Dolan, LCGC, 26 (2008) 532.
17. J. W. Dolan, personal communication.
18. L. R. Snyder and J. W. Dolan, High-Performance Gradient Elution, Wiley, Hoboken, NJ, 2007.
19. SPD-IOAvp UV-Vis Detector Instruction Manual, Shimadzu, Kyoto, 1997.
20. N. S. Wilson, R. Morrison, and J. W. Dolan, LCGC, 19 (2001) 590.
21. J. W. Dolan, LCGC, 23 (2005) 370.
22. Reviewer Guidance: Validation of Chromatographic Methods, USFDA-CDER, Nov. 1994,
http://www.fda.gov/cder/guidance/index.htm.
23. D. V. McCalley, Anal. Chem., 78 (2006) 2532.
24. M. T. W. Hearn, ed., Ion-pair Chromatography. Theory and Biological and Pharmaceutical
Applications, Dekker, New York, 1985.
25. E. Rajakyla', J. Chromatogr., 218 (1981) 695.
26. R. D. Morrison and J. W. Dolan, LCGC, 23 (2005) 566.
27. J. W. Dolan, LCGC, 25 (2008) 610.
28. R. G. Wolcott, J. W. Dolan, L. R. Snyder, S. R. Bakalyar, M. A. Arnold, and J. A. Nichols, J.
Chromatogr. A, 869 (2000) 211.
29. T.-L. Ng and S. Ng, J. Chromatogr., 389 (1985) 13.
30. W. R. Melander, H.-J. Lin, and С Horvath, J. Phys. Chem., 88 (1984) 4527.
920 Глава 17. Неисправности и способы их устранения
31. М. R. Euerby, С. М. Johnson, I. D. Rushin, and D. A. S. Sakunthala Tennekoon, J. Chromalogr.
/4,705(1995)229.
32. M. R. Euerby, C. M. Johnson, I. D. Rushin, and D. A. S. Sakunthala Tennekoon, J. Chromatogr.
A, 705 (1995) 219.
33. J. J. Gilroy and J. W. Dolan, LCGC, 22 (2004) 982.
34. T. Culley and J. W. Dolan, LCGC, 13 (1995) 940.
35. J. W. Dolan, LCGC, 13 (1995) 940.
36. U. D. Neue, personal communication (1995).
37. T. Eidenberger, personal communication (1995).
38. J. W. Dolan and L. R. Snyder, Troubleshooting LC Systems, Humana Press, Totowa, NJ, 1989.
39. G. Hendriks, J. P. Franke, and D. R. A. Uges, J. Chromatogr. A, 1089 (2005) 193.
ПРИЛОЖЕНИЕ I
СВОЙСТВА
РАСТВОРИТЕЛЕЙ,
ИСПОЛЬЗУЕМЫХ В ВЭЖХ
Растворители используются в ВЭЖХ для формирования подвижной фазы, для
растворения образцов и для проведения пробоподготовки. Растворители для подвижной
фазы особенно важны, так как их свойства должны соответствовать жестким
требованиям для надлежащего функционирования оборудования. Эти свойства влияют
также на выбор растворителя, используемого для растворения образца и для его
подготовки к вводу в хроматографическую систему. В таблице 1.1 перечислены
несколько свойств растворителей, которые могут иметь значение при выборе последних
для ВЭЖХ. Некоторые из этих свойств уже обсуждались в разделах этой книги
(ссылки приведены во второй колонке таблицы 1.1). В этом приложении даны
несколько таблиц, в которых перечислены значения одной или нескольких характеристик
растворителей (третья колонка таблицы 1.1). Краткие комментарии к каждому из
свойств растворителей приводятся в последней колонке таблицы 1.1. Они служат
введением для последующих разделов, в которых идет речь об отдельных
характеристиках растворителей.
1.1. Совместимость растворителя с детектором
1.1.1. Детектирование по поглощению в ультрафиолете
Желательно, чтобы поглощение подвижной фазы было А < 0,2 абсорбционных
единиц (АЕ) на длине волны, используемой для детектирования образца; более низкое
поглощение приводит к повышению точности регистрации сигнала и улучшению
результатов в режиме градиентного элюирования, но и более высокое поглощение
приемлемо для некоторых изократических разделений. В таблице 1.2 приводятся
значения поглощения растворителей, используемых в ОФХ (но не в нормально-фазовой
хроматографии) при разных длинах волн (200—260 нм). Очень редко может оказаться
целесообразным использовать детектирование на длинах волн менее 200 нм для
растворенных веществ с низким поглощением при больших длинах волн.
Так как вода не поглощает в ультрафиолетовом диапазоне, начиная с 200 нм,
поглощение водных подвижных фаз с растворителями равно поглощению чистых
растворителей, умноженному на объемную долю растворителя ф элюента В подвижной
фазы. Например, подвижная фаза, содержащая 25 %В, будет иметь следующие
значения поглощения А для различных растворителей при 215 нм: ацетонитрил —
0,00 АЕ; метанол — 0,09 АЕ; дегазированный метанол — 0,05 АЕ; тетрагидрофуран —
0,22 АЕ; изопропанол — 0,07 АЕ. Обратите внимание, что дегазирование метанола
понижает содержание кислорода в чистом растворителе, в результате чего
уменьшается его поглощение приблизительно на 1/3 в диапазоне 200—240 нм. Когда
подвижная фаза дегазируется, например, барботированием гелием, поглощение других
растворителей, используемых в качестве подвижной фазы В, также понижается
922 Приложение I. Свойства растворителей, используемых в ВЭЖХ
пропорционально количеству кислорода, обычно содержащегося в данном
растворителе. Кислород лучше растворяется в менее полярных растворителях, таких как тет-
рагидрофуран или изопропанол, поэтому значения поглощения этих растворителей,
указанные в таблице 1.2, могут превышать наблюдаемые на практике.
Таблица 1.1. Свойства растворителей, применяемых в ВЭЖХ
Свойство
Граница
пропускания
в УФ
Показатель
преломления
Полярность
Селе кти вность
Растворимость
образцов
Вязкость
Точка
кипения
Смешиваемость
Плотность
Стабильность
Безопасность
Ссылка на
раздел
4.4
4.11
2.3.2.1,
6.2.1, 8.2.1
6.3, 8.3.2
15.3.2.3
2.4.1
Таблица
значений
1.2
1.3
1.4
1.4
1.3, 1.5
1.3
1.6
1.7
Комментарий
Для детектирования по поглощению в УФ применение
растворителей зависит от длины волны, требующейся
для детектирования образца
Для детектирования по показателю преломления,в общем
случае предпочтительны низкие значения
Определяет силу растворителя для получения 1 < к < 10
Определяет различия в селективности, обусловленные
растворителем
Может быть важна при вводе больших количеств образца
в препаративной хроматографии или при анализе примесей
Определяет перепад давления на колонке; желательны
низкие величины вязкости
Влияет на работу насоса и безопасность; предпочтительны
высококипящие растворители
Важна для выбора органического растворителя дня ОФХ и
растворения образца
Требуется для (более точного) разведения подвижных фаз
по весу
Обычно не подлежит обсуждению
В общем случае важна, но обычно не имеет решающего
значения
Поскольку вода не должна поглощать свет при длинах волн > 200 нм,
предполагается, что она должным образом очищена. Характеристики воды со степенью
очистки для ВЭЖХ даны в методике D1I93 Американского Общества Тестирования
Материалов (ASTM), но для детектирования в коротковолновом УФ-диапазоне может
потребоваться вода с содержанием общего органического углерода1 порядка
50 млрд"'(ррЬ) и высоким удельным сопротивлением. В таблице 1.2 приведены
данные по УФ-поглощению некоторых широко используемых буферов, а в таблице 1.3
даны границы пропускания в УФ-диапазоне для нескольких растворителей.
Желательно, чтобы отклик детектора на подвижную фазу оставался одинаковым
в процессе градиентного элюирования. Для этого требуется, чтобы растворитель А
(вода) и растворитель В имели близкий по величине отклик. Когда детектирование
проводится на нижней границе УФ-диапазона, это может не выполняться, особенно
если растворитель В не ацетонитрил. Сходной проблемой является изменение
поглощения буфера или другой добавки к подвижной фазе по мере изменения %В.
Каждый из этих эффектов обсуждается в разделе 17.4.5.1.
'Англ. Total Organic Carbon. — Прим. персе.
/. I. Совместимость растворителя с детектором 923
Таблица 1.2. Поглощение в УФ-лиапазоне различных растворителей и буферов,
используемых в ОФХ, в зависимости от длины волны
Поглощение на волнах указанной длины (нм)
200
205
210
215
220
230
240
250
260
Растворители
Ацетонитрил
Метанол
Метанол (дегазированный)
Тетрагидрофуран
Изопропанол
0,06
1,0+
1,0+
1,0+
1,0+
0,02
1,0
0,76
1,0+
0,98
0,02
0,53
0,35
1,0+
0,46
0,01
0,35
0,21
0,85
0,29
0,00
0,23
0,15
0,70
0,21
0,00
0,10
0,06
0,49
0,11
0,00
0,04
0,02
0,30
0,05
0,00
0,02
0.00
0,17
0,03
0,00
0,01
0,00
0,09
0,02
Буферы
Ацетаты
Уксусная кислота, 1%
Соль аммония 10 нмоль
1,0+
1,0+
1,0+
0,94
1,0+
0,53
1,0+
0,29
1,0+
0,15
0,87
0,02
0,14
0,00
0,01
0,00
0,00
0,00
Карбонат
(NH4)HC03, 10 ммоль
0,41
0,10
0,01
0,00
0,00
0,00
0,00
0,00
0,00
Форчиат
Соль натрия, 10 ммоль
1,00
0,73
0,53
0,33
0,20
0,03
0,01
0,01
0,01
Фосфаты
Н3Р04, 1%
КН2Р04, 10 ммоль
К2НР04, 10 ммоль
(NH4)2HP04, 10 ммоль
Соль натрия, рН 6,8, 10 ммоль
0,00
0,03
0,53
0,37
0,20
0,00
0,00
0,16
0,13
0,08
0,00
0,00
0,05
0,03
0,02
0,00
0,00
0,01
0,00
0,01
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
0.00
0,00
0,00
0,00
0,00
0,00
0,00
0,00
Трифторуксусная кислота
0,1% в воде
0,1% в ацетонитриле
1,0+
0,29
0,78
0,33
0,54
0,37
0,34
0,38
0,20
0,37
0,06
0,25
0,02
0,12
0,00
0,04
0,00
0,01
Данные из 11, 2|.
Таблица 1.3. Различные свойства растворителей
Растворитель
Ацетон
Ацетонитрил
1-Бутанол
1 -Хлорбутан
Хлороформ
Циклогексан
Ди метил форм амид
Ди метисул ьфокс ид
1,4-диоксан
Граница
пропускания в УФ (нм)" [2]
330
190
215
220
245
200
268
268
215
Показатель
преломления [3]
1,359
1,344
1,399
1,402
1,446
1,424
1,430
1,478
1,422
Вязкость
(сП) 13]
0,36
0,38
2,98
0,45
0,57
1,00
0,92
2,24
1,37
Температура
кипения ('С) [3]
56
82
118
78
61
81
153
189
101
с(сили-
кагель)"
0,53
0,52
0,40
0,20
0,26
0,00
—
0,50
0,51
924 Приложение I. Свойства растворителей, используемых в ВЭЖХ
Окончание табл. 1.3
Растворитель
С^тилацетат
Гептан
Гексан
Изооктан
Метанол
Метил-третбутило-
вый эфир
Метилэтилкетон
Метиленхлорид
Изопропанол
н-Пропанол
Тетрагидрофуран
Толуол
Вода
Гранииа
пропускания в УФ (нм)" [2]
256
200
195
215
205
210
329
233
205
210
212
284
190
Показатель
преломления [3]
1,372
1,388
1,375
1,391
1,328
1,369
1,379
1,424
1,377
1,386
1,407
1,497
1,333
Вязкость
(сП) [3]
0,45
0,40
0,31
0,50
0,55
0,27
0,43
0,44
2,40
2,30
0,55
0,59
1,00
Температура
кипения {'С) [3]
77
98
69
99
65
55
80
40
82
97
66
111
100
£ (СИ.1И-
кагель)6
0,48
0,00
0,00
0,00
0,70
0,48
0,40
0,30
0,60
0,60
0,53
0,22
"Длина волны, на которой поглощение растворителя равно 1,0 АЕ в 10-мм кювете.
*Элюирующая сила растворителя [4].
1.1.2. Детектирование по показателю преломления
Для разделения в изократическом режиме выбор подвижной фазы обычно не
ограничен при детектировании по показателю преломления. Чувствительность
детектирования можно увеличить, выбрав подвижную фазу, показатель преломления
которой отличается в большей степени от показателей преломления компонентов
образца, чем их показатели преломления различаются между собой (таблица 1.3).
Детекторы, основанные на дифференциальном измерении (например, показателя
преломления) нельзя использовать в режиме градиентного элюирования из-за различия
в отклике детектора на компоненты подвижной фазы А и В, которое обычно велико.
1.1.3. Детектирование масс-спектрометром
В масс-спектрометре подвижная фаза испаряется, поэтому используется подвижная
фаза, содержащая воду, органический растворитель и летучие добавки (т.е. не
должно быть нелетучих буферов или солей). Так как подвижная фаза удаляется,
поглощение в УФ не имеет значения при детектировании масс-спектрометром, но в
растворителях не должно быть твердых частиц.
I.2. Полярность и селективность
растворителей
В таблице 1.4 перечислены свойства растворителей, которые влияют на элюирующую
силу, селективность и растворимость. Эти свойства «нормализованной
селективности» предполагают три вида взаимодействий растворенного вещества с растворите-
1.2. Полярность и селективность растворителей 925
леи: водородные связи с растворителем (Н-В) кислотного характера а2 /2, Н-В
основного характера Р2/2 и дипольные взаимодействия л*/2. Перечисленные
параметры составляют основу треугольника селективности растворителей (рис. 2.9).
Полярность растворителя Р является мерой суммарной полярности растворителя.
Растворимость образца имеет тенденцию коррелировать с величиной Р — «подобное
растворяется в подобном» — так, что образцы в большей степени растворимы в
растворителях, имеющих примерно такую же полярность, как у них. Менее полярные
вещества, такие как углеводороды, будут более растворимы в растворителях с
низкими величинами Р. Обратное будет справедливо для растворителей с высокими
величинами Р. Таким же образом диэлектрическая постоянная е связана со способно
стью растворителя растворять ионизированные соединения или буферы: высокие
значения е благоприятствуют повышенной растворимости ионизируемых веществ.
В таблице 1.5 приведены значения вязкости смесей метанол/вода и ацетон итр ил/вода
в зависимости от температуры. Эти данные полезны при оценке перепада давления
на колонке (уравнение 2.13). В таблице 1.6 даны значения плотности некоторых
широко используемых растворителей для того, чтобы более точно готовить подвижные
фазы для ОФХ, взвешивая каждый растворитель при смешивании.
Таблица 1.4. Свойства, определяющие селективность растворителей
Растворители
Уксусная кислота
Ацетон
Ацетонитрил
Бензол
Хлороформ
Диэтшювый эфир
Диметилсульфоксид
Этанол
Этилацетат
Этиленхлорид
Форма мид
Гликоль
Гексан
Изопропанол
Метанол
Метилацетат
Метиленхлорид
Метилэтилкетон
Метил-трет-бутиловый эфир
М,М-диметилформамид
Нитрометан
Нормализованная селективность'
Н-В
кислотность а$/Т.,
0,54
0,06
0,15
0,14
0,43
0,00
0,00
0,39
0,00
0,00
0,33
0,38
0,00
0,22
0,43
0,05
0,27
0,05
0,00
0,00
0,17
Н-В
основность pj/S
0,15
0,38
0,25
0,86
0,00
0,64
0,43
0,36
0,45
0,00
0,21
0,23
0,00
0,35
0,29
0,40
0,00
0,40
=0,6
0,44
0,19
Дипольные
взаимодействия л*/И
0,31
0,56
0,60
0,00
0,57
0,36
0,57
0,25
0,55
1,00
0,46
0,39
0,00
0,43
0,28
0,55
0,73
0,55
=Л,4
0,56
0,64
р.6
6,0
5,1
5,8
2,7
4,1
2,8
7,2
4,3
4,4
3,5
9,6
6,9
0,1
3,9
5,1
~5
3,1
4,7
=2,4
6,4
6,0
Е"
6.2
20,7
37,5
2,3
4,8
4,3
4,7
24,6
6,0
10,4
182
37,7
1,9
19,9
32,7
6,7
8,9
18,5
«4
36,7
35,9
926 Приложение I. Свойства растворителей, используемых в ВЭЖХ
Окончание табл. 1.4
Растворители
Пиридин
Сульфолан
Тетрагидрофуран
Толуол
Триэтиламин
Трифторэтанол
Вода
Нормализованная селективность"
0,42
0,00
0,00
0,17
0,00
0,68
0,43
0,58
0,17
0,49
0,83
0,84
0,00
0,18
0,00
0,83
0,51
0,00
0,16
0,32
0,45
Г*
5,3
4,0
2,4
1,9
10,2
F*
12,4
43,3
7,6
2,4
2,4
80
" Значения из [4|.
6 Индекс полярности; значения из |5|.
"Диэлектрическая постоянная; значения из |3].
Таблица 1.5. Вязкость подвижных фаз, используемых в ОФХ, в зависимости от состава
(%В) и температуры (Т)"
т
■с
15
20
25
30
35
40
45
50
55
60
%В
0
1,10
1,10
1,00
1,00
0,89
0,89
0,79
0,79
0,70
0,70
0,64
0,64
0,58
0,58
0,54
0,54
0,51
0,51
0,47
0.47
10
1,43
1,18
1,32
1,14
1,18
1,01
1,04
0,90
0,92
0,73
0,82
0,72
0,75
0,61
0,71
0,60
0,67
0,53
0,61
0.52
20
1,72
1,23
1,57
1,10
1,40
0,98
1,23
0,87
1,07
0,78
0,96
0,70
0,87
0,64
0,82
0,60
0,77
0,56
0,70
0.53
30
1,92
1,30
1,75
1,13
1,56
0,98
1,36
0,86
1,19
0,76
1,05
0,68
0,96
0,61
0,89
0,57
0,84
0,53
0,77
0.50
40
2,00
1,09
1,83
0,99
1,62
0,89
1,43
0,80
1,24
0,72
1,11
0,65
1,00
0,59
0,93
0,55
0,88
0,51
0,81
0.49
50
2,02
0,98
1,83
0,90
1,62
0,82
1,43
0,74
1,26
0,68
1,12
0,62
1,02
0,58
0,94
0,53
0,88
0,49
0,81
0.46
60
1,91
0,89
1,72
0,81
1,54
0,72
1,36
0,65
1,21
0,59
1,08
0,54
0,98
0,50
0,90
0,46
0,84
0,43
0,79
0.41
70
1,69
0,81
1,52
0,69
1,36
0,59
1,21
0,52
1,09
0,47
0,98
0,44
0,89
0,43
0,82
0,41
0,76
0,38
0,72
0.35
80
1,40
0,70
1,25
0,56
1,12
0,52
1,01
0,45
0,91
0,43
0,83
0,41
0,76
0,38
0,70
0,36
0,65
0,34
0,61
0.37
90
1,05
0,54
0,93
0,50
0,84
0,46
0,76
0,43
0,69
0,39
0,64
0,36
0,58
0,33
0,54
0,31
0,50
0,29
0,47
0.27
100
0,63
0,40
0,60
0,37
0,56
0,35
0,51
0,32
0,46
0,30
0,42
0,27
0,39
0,25
0,37
0,24
0,36
0,23
0,33
0.22
Данные из 16,7].
" Состав приводится в %В (ойьем/о&ьем), В — метанол (верхняя строка) или ацетонитрил
(нижняя строка). Например, пязкость воды, содержащей 30% метанола, при 30 "С равна 1,36.
В [8, 9| проводится сравнение вязкости при изменении состава, температуры, давления, а
также приведены данные по сжимаемости.
1.3. Безопасность при работе с растворителями 927
Таблица 1.6. Плотность растворителей, используемых для подвижных фаз в ОФХ [3]
Растворитель
Ацетонитрил
Метанол
2-пропанол
Тетрагидрофуран
Вода
Плотность при указанной температуре (г/мл)
20 "С
0,7822
0,7913
0,7855
0,8892
0,9982
22'С
0,7800
0,7894
0,7838
0,8874
0,9977
25'С
0,7766
0,7866
0,7813
0,8847
0,9970
Примечание: ошибка на 1 °С = 0,1%, ошибка по весу <£ 0,1%, ошибка относится
к объемному проценту.
1.3. Безопасность при работе
с растворителями
Растворители, обычно используемые в ВЭЖХ, часто огнеопасны и умеренно
токсичны. Поэтому большую часть этих растворителей следует хранить в надежном
металлическом шкафу. Воспламеняемость растворителей можно приблизительно оценить
по точке воспламенения, значения которой даны в таблице 1.7. Повседневный опыт
подсказывает, что метанол не очень легко воспламеняется и, следовательно,
растворители, имеющие точку воспламенения выше 12 °С, не должны при нормальных
условиях создавать пожароопасную ситуацию. Однако растворители с более низкой
температурой воспламенения опаснее, и с ними надо обращаться соответствующим
образом.
Токсичность растворителей определяется многими факторами, и с любыми
растворителями, кроме воды, следует работать под тягой. Очень грубой оценкой
непосредственного токсического действия является значение ЛДед (приводится в таблице 1.7),
это количество введенного в организм вещества, выраженное в миллиграммах на
килограмм веса тела, которое вызывает летальный исход у 50% пострадавших. Однако в
лаборатории растворители пьют' редко, в основном они действуют при попадании на
кожу или при вдыхании. Очевидно, что протирание спиртом (изопропанолом), у
которого ДДзд составляет 400, неопасно ни при контакте, ни при вдыхании. Перед тем как
пользоваться любым растворителем или реагентом, необходимо ознакомиться
с инструкциями по технике безопасности при работе с ним.
Таблица 1.7. Данные по воспламеняемости и токсичности различных растворителей
Растворитель
Ацетон
Ацетонитрил
Четыреххлористый углерод
Хлороформ
Этилацетат
Этиловый эфир
Точка воспламенения ('Су
-18
42
нет
нет
13
-45
ПДК* (млн-1)
1000
40
10
25
400
400
' Смотря какие. — Прим. персе.
928 Приложение I. Свойства растворителей, используемых в ВЭЖХ
Окончание табл. 1.7
Растворитель
Гептан
Гексан
Метанол
Метил-трет-бутиловый эфир
Метиленхлорид
н-Пропанол
Изопропанол
Тетрагидрофуран
Вода
Точка воспламенения VCf
-4
-26
12
-28
нет
27
12
-20
нет
ПДК* (илн"')
85
100
200
—
500
200
400
200
нет
"Данные из |2|.
6 Предельно допустимая концентрация паров растворителя на рабочем месте, утвержденная
постановлением правительства |2|.
Литература
I.J. В. Li, LCGC, 10 (1992)856.
2. High-Purity Solvent Guide, Burdick & Jackson Laboratories, Muskegon, Ml, 1980.
3. J. A. Riddick and W. B. Bunger, Organic Solvents, Wiley-lnterscience, New York, 1970.
4. L. R. Snyder, in High-performance Liquid Chromatography: Advances and Perspectives, Vol. 3,
С Horvath, ed., Academic Press, New York, 1983, p. 157.
5. K. Valko, L. R. Snyder and J. L. Glajch, J. Chromatogr., 656 (1993) 501.
6. L. R. Snyder, /. Chromatogr. Sci., 16 (1978) 223.
7. H. Colin, J. C. Diez-Masa, G. Guiochon, T. Czajkowska, and I. Miedziak, J. Chromatogr., 167
(1978)41.
8. M. A. Quarry, R. L. Grab, and L. R. Snyder, J. Chromatogr., 285 (1984) 1.
9. J. Billen, K. Broeckhoven, A Liekens, K. Choikhet, G. Rozing, and G. Desmet, J. Chromatogr. A,
1210 (2008) 30.
ПРИЛОЖЕНИЕ II
ПРИГОТОВЛЕНИЕ БУФЕРНЫХ
ПОДВИЖНЫХ ФАЗ
11.1. Последовательность операций
Для приготовления буферных подвижных фаз нужно выполнить следующие
операции:
1) смешать компоненты буфера с водой, чтобы получить водный буфер (фаза А);
2) проверить рН буфера или довести его до нужного значения с помощью рН-метра;
3) смешать определенный объем (например 200 мл) органического растворителя
(фаза В) с определенным количеством (например 800 мл) фазы А, полученной
на этапе 2, чтобы приготовить требуемую подвижную фазу (в этом примере буфер
с 20% органического растворителя);
4) проверить рН полученной подвижной фазы (при необходимости).
Так как измерение рН в подвижной фазе, содержащей органический
растворитель, дает ненадежный результат из-за дрейфа рН-метра, операция 4 позволяет
обнаружить только большие отклонения свойств раствора от заданных или сравнить
с раствором, содержащим столько же органического компонента. В большинстве
лабораторий операцию 4 не выполняют.
Обычно на этапе 1 готовят два водных буфера с разными рН (А1 и А2), а потом
смешивают эти два раствора в нужных пропорциях, чтобы получить подвижную фазу А
с нужным рН. Если рН доводится на этапе 2, титровать буфер до нужного рН по
показаниям рН-метра можно теми же исходными растворами. В большинстве лабораторий
точность измерений рН-метром обычно не превышает +0,05—0,10 единиц рН. Такие
отклонения рН могут вызвать существенные изменения разрешения некоторых образцов
(раздел 7.3.4.1). Когда в метод ВЭЖХ включен контроль рН, на этапе 2 только
приблизительно проверяется соответствие заданному рН. Если при приготовлении буфера
ингредиенты тщательно взвешиваются и растворяются в точно отмеренных объемах
дистиллированной дегазированной воды (без дальнейшего доведения рН), рН полученных
буферов можно контролировать с большой точностью (±0,02 единицы рН). Кроме того,
в продаже есть концентрированные буферы с точно измеренными значениями рН.
Обычно рН буферов доводят концентрированной кислотой. Например, раствор
А2 из таблицы II.I можно приготовить и оттитровать до нужного значения
концентрированной фосфорной кислотой. Таким образом получится буфер с нужным рН, но
ионная сила буфера будет выше, чем при смешивании эквимолярных растворов AI и
А2. Маловероятно, что использование подвижных фаз, приготовленных этими двумя
способами (с доведением рН концентрированной кислотой и смешиванием
эквимолярных растворов), приведет к различным результатам в ОФХ, но некоторые
разделения могут быть чувствительны к изменению ионной силы (например, в ИОХ).
Лучше всего в методической документации точно описать, как следует готовить
буфер, и выполнять эту инструкцию: важно следовать методике приготовления
подвижной фазы, это обеспечивает надежность результатов хроматографии.
Иногда для различных целей в подвижную фазу вводят кислотные или щелочные
добавки. Когда эти добавки не используются в качестве основного буфера, их
следует добавлять к общему количеству готового буфера, а потом доводить его рН
кислотой или щелочью.
930 Приложение I. Свойства растворителей, используемых в ВЭЖХ
Таблица 11.1. Приготовление фосфатных буферов с заданными низкими значениями рН
Заданные значения рН
2,0
2,2
2,4
2,6
2,8
3,0
3,2
Объем А1 (мл)"
565
455
345
250
175
ПО
55
Объем А2 (мл)9
435
545
655
750
825
890
945
"Раствор 0,1 М фосфорной кислоты; необходимо оттитровать фосфорную кислоту,
используемую для приготовления этого базового раствора, для подтверждения содержания фосфорной
кислоты в препарате.
6 Раствор 0,1 М однозамещенного фосфата натрия; растворите 13,8 г моногидрата №Н2Р04
в воде и доведите объем до I л в мерной колбе.
11.2. Методики приготовления некоторых
широко используемых буферов
рН буферного раствора практически остается постоянным при разбавлении
концентрированного буфера или при замещении одного ионизированного катиона
(например, Na+, К+) или аниона (СГ, Вг~ ) другим. В таблицах 11.1—II.3 описано
приготовление некоторых буферов, широко используемых в ОФХ (данные из |1|). Для этого
смешиваются два раствора, каждый из которых имеет одинаковую концентрацию
буферных компонентов. При смешивании указанных в таблицах количеств растворов
А1 и А2 получается буфер с требуемым значением рН. В таблицах 11.1—11.3
приведены соотношения для приготовления буферов с концентрацией 0,1 М и натрием в
качестве катиона. Из этих данных можно экстраполировать соотношения для других
концентраций буферов и/или других катионов (обычно предпочтителен К+). рН
более разбавленных или более концентрированных буферов или буферов с другими
катионами могут несколько отличаться от табличных значений. Точное значение рН
подвижной фазы обычно не имеет значения при разработке метода. Важно, чтобы
конечное значение рН воспроизводилось каждый раз при приготовлении новой
порции подвижной фазы (желательно с точностью ±0,02 единицы рН). Следует иметь
в виду, что растворы проявляют буферные свойства достаточно эффективно в
пределах ±1 единица рН от значения рКа ионизируемого ингредиента (раздел 7.2.1). Хотя
подвижную фазу можно использовать при температуре, отличающейся от
температуры окружающей среды, значения рН в таблицах II. 1—II.3 приведены для
температуры окружающей среды и их следует использовать для описания готовой подвижной
фазы.
Кроме таблиц II.1—II.3, в качестве руководства по приготовлению буферов
можно использовать онлайн калькуляторы буферов (следует искать HPLC buffer
calculator), в которых приведены расчеты для многих буферов. Например, один из таких
калькуляторов (The Buffer Wizprd, Zirchrom, Anoka, MN, www.zirchrom.com) позволяет
получать инструкции для приготовления нужных буферов. Следует ввести названия
кислоты, основания, требуемую концентрацию буфера и рН, и калькулятор выдает
инструкцию, содержащую, в частности, информацию о буферной емкости
полученного раствора, стабильности сорбента в нем и так далее.
/1.2. Методики приготовления некоторых широко используемых буферов 931
Таблица 11.2. Приготовление ацетатных буферов с заданными значениями рН
Заданные значения рН
3,6
3,8
4,0
4,2
4,4
4,6
4,8
5,0
5,2
5,4
5,6
Объем А1 (мл)"
926
880
820
736
610
510
400
296
210
176
96
Объем А2 (мл)"
74
120
180
264
390
490
600
704
790
824
904
" Раствор 0,1 М уксусной кислоты; 6,0 г (5,8 мл) ледяной уксусной кислоты доведите водой
до 1 л в мерной колбе.
6 Раствор 0,1 М ацетата натрия; растворите 8,2 г ацетата натрия (или 13,6 г тригидрата ацетата
натрия) в воде и доведите объем до 1 л в мерной колбе.
Таблица 11.3. Приготовление фосфатных буферов с заданными значениями рН в середине
диапазона рН
Заданные значения рН
5,6
5,8
6,0
6,2
6,4
6,6
6,8
7,0
7,2
7,4
7,6
7,8
8,0
Объем А1 (мл)"
948
920
877
815
735
685
510
390
280
190
130
85
53
Объем А2 (мл)*
52
80
123
185
265
315
490
610
720
810
870
915
947
" Раствор 0,1 М однозамещенного фосфата натрия; растворите 13,8 г моногидрата однозаме-
щенного фосфата натрия в воде и доведите объем до 1 л в мерной колбе.
6 Раствор 0,1 М двухзамешенного фосфата натрия; растворите 26,8 г Na2HP04 x 7Н20 в воде и
доведите объем до 1 л в мерной колбе.
Литература
1. G. Gomori, in Meth. Enzymology, S. P. Colowicxk and N. O. Kaplan, eds., Academic Press, New
York. 1955, p. 145.
предметный указатель
Автосамплеры 151 — 160; см. также
Инжекторы
воспроизводимость 176
конструкция 154—158
опция «подготовленного ввода» 155
перенос образца из предыдущего
нанесения 154
периодическое обслуживание 179
прецизионность и точность 154
проблемы 867-870, 883, 908
уплотнение иглы 155—156
Адсорбционная хроматография; см.
Нормально-фазовая хроматография
Азотный детектор; см. Детекторы, хеми-
люминесцентный азотный
Активный фармацевтический
ингредиент (АФИ) или фармацевтическая
субстанция (ФС) 565
Алкилированный силикагель, колонки
262—263; см. также Колонки
Алкилсульфонаты в качестве
ион-парных реагентов 372—374
Алкильные заместители в молекулах
образцов, разделение в ОФХ и НФХ 393—
396
Альбумин сывороточный человеческий;
си.Хиральные разделения, человеческий
сывороточный альбумин
Альбутерол, выделение 825—826
Амидные фазы, использование в ГИХ
428-429
Аминокислоты, значения рКа 607, 634
Аминомодификаторы, использование в
ОФХ 359
Амперометрические детекторы; см.
Детекторы, электрохимические
Анализ следовых количеств 109, 552—553
Анализируемые (разделяемые) вещества
59; см. также Образцы
амфотерные, в ОФХ 343
ионные 337—389
нейтральные 290—333
полярность и удерживание 66
Аналитический метод; см. Метод
испытания
Анионообменная хроматография; см.
также Ионообменная хроматография,
иох
вирусов 667—668
нуклеиновых кислот 655—657
углеводов 665—666
Антихаотропные соли 646—647
Аффинная хроматография на сорбентах
с иммобилизованными металлами
(АХИМ) или металл-аффинная
хроматография 641—645
а|-кислый гликопротеин или орозому-
коид; см. Хиральные неподвижные
фазы, на основе сц-кислого гликопротеина
Безопасность при работе с системой
ВЭЖХ 861, 927-928
Белки 606—609; см. также Полипептиды
агрегация, выявление при
гель-фильтрации 677—678
влияние структуры белка на его
поведение в процессе хроматографии 618—
619
закономерности удерживания белков
на неподвижной фазе (постулаты Ренье)
618-619
определение конформации с
помощью гель-фильтрации 677—678
поверхность контакта 618
Бидентатные силаны 285
Биоаналитические методы 225, 580—586;
см. также Биохроматография
валидация 581—582
документация 586
калибровочная кривая 583
правильность и прецизионность 582—
583
рекомендации для биоаналитических
методов 580—581
рутинное применение 585—586
стабильность образца 584
стандартные образцы 581
Биомакромолекулы; см. Биомолекулы
Биомолекулы, структура и конформация
605-612
Биохроматография 605—687
выход продукта и его биологическая
активность 61S
колонки 614—618
Бумажная хроматография 42, 47
Буферы 344-353
рКа 345
влияние на селективность 359
емкость 344—346
ион парные свойства 348
летучесть 348
несоответствие разделению 346, 892
осаждение 347—348
поглощение в УФ 348, 923
предпочтительные 349
приготовление 344, 929—931
растворимость 347—348
свойства 344—349
стабильность 348—349
эффективная буферная емкость 346
Валидация 104—105
биоаналитических методов 580—586
документация 575—577
методические рекомендации 562—565,
580-581
методов очистки 579
нормативные документы 562—564
обзор 562—565
предварительная 577
протокол 575—576
термины и определения 565—573
фармацевтических методик 578—580
Ванкомицин; см. Хиральные
неподвижные фазы, на основе ванкомицина
Взаимодействия межмолекулярные 67—71
водородные связи 68, 69, 265—268
гидрофобные 68—69, 265—267
диполь-дипольные 68, 69, 265—266
дисперсионные 68
кулоновские (электростатические)
или ионные 68, 70, 265—269
с переносом заряда или я-я 68, 70,
265-266
с молекулами сорбента 264—271
Вирусы 612-613, 667-668
Витамины водорастворимые, разделение
374-375
Вихревая диффузия 76—79
Предметный указатель 933
Влажность, влияние на НФХ 422—423
Внеколоночное уширение полосы
вещества 78, 169-170
Вода, анализ образца 231—232, 814, 816
Воспроизводимость
в НФХ 422-424
в рутинных анализах в градиентном
режиме 477—478
колонки 104
при градиентном элюировании 476
478
при разработке метода градиентного
разделения 477
ВПМС-детекторы; см. Детекторы, МС,
времяпролетные
Время задержки 452, 453; см. также
Объем задержки
Вторичная структура, полипептиды; см.
Полипептиды, вторичная структура
Вторичные равновесия 114
Высота теоретической тарелки 73—80
приведенная 79—80
ВЭЖХ 42-46
журналы 54
история 47—50
книги 52—53
краткие курсы сетевые 54
метод; см. Метод испытания
на чипе 52
оборудование 122—183
предшественники 47
режимы разделений 58
сравнение с ТФЭ 812-813
характеристики 42, 44
ВЭЖХ при высоких давлениях 151; см.
также Ультравысокоэффективная
жидкостная хроматография
ВЭЖХ при высоких скоростях потока
150-151
ВЭЖХ при малых скоростях потока 150
Вязкость
влияние на давление 75, 76
влияние на диффузию 79—80
растворителей; см. Растворители,
вязкость, величины
смесей ацетонитрил — вода в
зависимости от температуры 926
смесей метанол — вода в зависимости
от температуры 926
Предметный указатель
Газовая хроматография (ГХ) 50
Гауссова форма пика 73, 86, 530
Гель-проникающая хроматография (ГПХ)
668, 690—692; см. также Эксклюзионная
хроматография (ЭХ)
происхождение 47
Гель-фильтрация (гель-фильтрационная
хроматография, ГФХ) 668—669; см.
также Эксклюзионная хроматография (ЭХ)
колонки 671—673
отклонения от механизма
«идеальной» ЭХ 674, 676
подвижные фазы 674
применение 677—679
Гербициды, быстрое разделение 245—246
Гибридные частицы сорбента; си.
Частицы сорбента, гибридные
Гидродинамический диаметр молекул
615
в ЭХ 671
Гидрокс и апатит керамический 640—641
Гидрокситестостерона изомеры,
разделение 312
Гидрофобные взаимодействия; см.
Взаимодействия межмолекулярные,
гидрофобные
Гидрофобные примеси в образце 417
ГИХ; см. Хроматография гидрофильных
взаимодействий
Гликопротеины 611—612
Глобальная оптимизация разделения 92
Гомологи, удерживание 116—118
Гомополимеры 687
Гормон роста человеческий, разделение
454-455, 624-625
Градиентное элюирование 110—111,
434-497
«неправильных» образцов 442, 444
в биохимических разделениях в ОФХ
625-628
в ГИХ 493-496
в И ОХ 496
в НФХ 493
влияние температуры 470, 471
влияние экспериментальных условий
441-462
воспроизводимость 476—478
высокомолекулярных образцов 435—
436
диапазон градиента 438, 439, 470, 472
дрейф базовой линии 497, 884—887
задержка градиента 437—438, 450—452
искажение градиента 903—905
исходный %В 447-449
как пробоподготовка образца 436
когда использовать 434—437, 465—468
конечный %В 449-450
криволинейные градиенты 437
крутизна градиента 6 440
кусочно линейные градиенты 437
438, 454-456, 472
линейные градиенты как
оптимальный подход 437—438
макромолекул 491—492
миксер 149
обеднение элюента растворителем 496
объем задержки 149, 452^153; см.
также Объем задержки
проблемы при работе с разными
ВЭЖХ-системами 478, 905-907
объем и производительность
градиента 904-905
оптимальные подходы 445—447
оптимизация 469—473
оптимизация с помощью программы
Dry Lab 509 510
параметры колонки 444—447
первичное градиентное разделение
463-469
перемещение зон веществ 440—441
пиковая емкость 478—483
план разработки метода 463, 464
позднее элюирование 449—450, 469
полипептидов в ОФХ 625—628
правильных образцов 442—443
предварительное смешивание
подвижной фазы (оптимальные подходы)
856-857
применение 434—437, 452
проблемы 468-469, 904-905
проблемы переноса 452—453, 905—907
программирование 438
разделение дженериков 436
размывание заднего фронта пиков
437, 468
разработка метода 436, 462—491
разрешение 462
раннее элюирование 468—469
Предметный указатель 935
селективность 455—456, 463, 470
системные, или ложные, или арте-
фактные пики 469, 497; см. также
Устранение неисправностей, ложные
пики
сложных образцов 469
соответствующие разделения 442^147
сопоставление с изократическим элю-
ированием 434-435, 438-439, 465-468
ступенчатый градиент 437—438
теория 458—462
тестирование; см. Тесты на
пригодность (работоспособность) системы,
градиент
уравнения 458—462
уравновешивание колонки 473—476
условия градиента 447—458
фактор удерживания к* 440—441
форма градиента 437—438
экспресс-разделения 483—485
График Поппе 83—84
Графитированный углерод 254—255
получение частиц 249
Давление; еж. также Растворители,
вязкость, величины
внезапный скачок 285
и скорость потока 74, 75
тесты; см. Тесты на пригодность
(работоспособность) системы, сброс
(стравливание) давления
Двумерная ВЭЖХ (20-ЖХ) 112, 485-
491, 696—697; см. также Многомерная
жидкостная хроматография
оборудование 488—489
ортогональные условия 490
пиковая емкость 488
примеры 486—487
разработка метода 489—491
с переключением колонок 654—655
совместимость подвижных фаз 490
Дегазация 128—132
барботирование гелием 208
положительный эффект 855
Денатурирующая ВЭЖХ 658-660
Дериватизация 232—234; см. также
Образца пробоподготовка, дериватизация
Детектор(ы)
азотный; см. Хемилюминесцентный
азотный
выбор 102
детектирование с помощью
модификации подвижной фазы 190
для препаративных разделений 770—
771
дрейф базовой линии 191, 193—194,
530, 621
заряженных аэрозолей (ДЗА) (или
детектор ионизации коронным разрядом)
223-224
измерение общей характеристики
вещества (дифференциальные измерения)
189-190
инфракрасный спектрометр с
Фурье-преобразованием (ИКФП) 230—231
источники ошибок 536—537
кондуктометрические 212—213
линейность (детектора) 198
линейность (калибровочного
графика); см. Калибровка, линейность
МС (масс-спектрометрические) 224—
230
времяпролетные (ВПМС) 229—230
интерфейсы 225—227
ионные ловушки 227—229
ионы дочерние (или
ионы-продукты) 228-229
ионы родительские (или ионы-
предшественники) 228—229
квадрупольные анализаторы 227—
228
скорость потока 227
трехквадрупольные (тандемные) 228
«независимые» 190, 224—232
перегрузка 762—763
постоянная времени 192
пределы обнаружения 196—197; см.
также Калибровка, пределы
обнаружения
проблемы; см. Общие симптомы
ВЭЖХ-проблем
проточная ячейка 187-189, 199-200
профилактическое обслуживание 178,
179-180
радиоактивности 2! 1—212
реакционные 233—234
936 Предметный указатель
регистрирующие поглощение в
области видимого света; см. УФ-детекторы
регулятор избыточного давления 189
рефрактометрические или детекторы
показателя преломления 216—219
светорассеяния 219 223
испарительный (ИДС) 220—222
лазерные (ЛДС) 222-223
светорассеяния на центрах (ядрах)
конденсации 222
специфических характеристик
образца 190
теплообменники 189
тестирование чистоты пиков 570
УФ-детекторы (и поглощения в
области видимого света УФ/ВИД) 198-206
выбор длины волны 200—201
история 186
профилактическое обслуживание
206
селективность 200, 203—205
характеристики 205—206
флуоресцентные 206—209
тушение флуоресценции 208
характеристики 187—198
хемилюминесцентный азотный
(ХЛАД) 213-215
хиральные 215—216
дисперсии оптического вращения 216
кругового дихроизма 216
поляриметр 215—216
чувствительность (пределы
обнаружения) 196-197
шумы 191-193
электрохимические (амперометриче-
ские) 209-211
энантиомеров; см. Хиральные
ЯМР 231-232
Дженерики, разделение 436
Диапазон; см. Разработка метода,
диапазон градиента; Корректировка методики,
аналитическая область
Диастереомеры 706
Диольные фазы, использование в ГИХ
428
Дисперсии оптического вращения
детектор; см. Детекторы, хиральные
Диффузионные поры сорбента; см.
Поры частиц сорбента, диффузионные
Диэлектрическая постоянная, значения
для растворителей 925—926
Диэтиламида лизергиновой кислоты
производные, разделение 518—521
ДНК, хроматография 657—661
Документация; см. Валидация,
документация
Дрейф
базовой линии 191, 193, 530—531
при градиентном элюировании 497,
621, 623
Емкость насыщения колонки 778—779
Жидкостная экстракция под давлением
(ЖЭД) 836
Жирные кислоты, разделение изомеров
312
Журналы, статьи по ВЭЖХ 54
ЖХ-МС
детекторы 224—230
подавление ионного сигнала 842
Зависимость селективности от силы
растворителя; см. Селективность, влияние
силы элюента
Закон Бера 199
Замещение при удерживании в НФХ
396-400
Замещенные анилины, разделение 394—
396
Замещенные нафталины, разделение 323—
324
Изократическое элюирование 57
предсказание условий исходя из
градиентного разделения 463—468
Изомерия, хиральность 704—706
Изомеров разделение
в НФХ 394-396,413-415
в ОФХ 311-314
зависимость от температуры 312
Изомеры структурные 704
Изотактичность полимеров 688
Изотермы
адсорбции 774—775
«пересечение» 787—789
Инжекторы 151 — 158; см. также Авто-
самплеры
Предметный указатель 937
ввод из заполненной петли 152, 156
ввод из частично заполненной петли
152-154, 156
конструкция 151 — 152
ламинарный характер потока 153—154
точность 153—154
Инсулин человека рекомбинантный,
очистка в промышленном масштабе
680-686
Интегратор 165, 527
Интернет-ресурс 54
Инфракрасный спектрометр с Фурье-
преобразованием (ИКФП); см. Детекторы
Ионная хроматография 52, 380—381
детектирование 212
Ионные взаимодействия 68, 70—71; см.
также Взаимодействия
межмолекулярные, кулоновские (электростатические)
Ионные (ионизируемые) образцы
в ИОХ 380-389
в ИПХ 364-380
в ОФХ 352-359
проблемы при работе с ионизируемы
ми образцами 361—364
разработка метода 359—364
Ионизация
влияние %В 351—352
влияние температуры 352
образца 337—343
Ионообменная хроматография (ИОХ)
380—389, 633—645; см. также
Нуклеиновые кислоты, Углеводы
влияние рН 385
градиентное элюирование 496
колонки 385-386, 636-638
применение 380—382
противоионы 382—385
разработка метода 386
смешанный режим разделения 387—
389
удерживание 382—384, 386
Ион-парная хроматография (ИПХ) 364—
380
влияние буфера 378
влияние рН 366-367, 370-371
влияние температуры 377
ион-парный реагент 367—371
когда использовать (оптимальный
подход) 364—366
ложные пики 379
медленное уравновешивание
неподвижной фазы 379—380
преимущества 364—366
проблемы 378—380
размывание заднего фронта пиков 380
разработка метода 371—378
селективность 375—378
сила элюента 376—377
тип колонок 377—378
удерживание 367—371
хаотропы 375
Ион-парный реагент 364
буфер как ион-парный реагент 348
влияние концентрации 367—370
влияние на разделение 366—371
влияние на разделение типа реагента
367-370
Ионы-предшественники; см. Детекторы,
МС, ионы родительские
Ионы-продукты; см. Детекторы, МС,
ионы дочерние
ИОХ; см. Ионообменная хроматография
Испарительный детектор
светорассеяния; см. Детекторы, испарительные,
светорассеяния
Источники ошибок 535—539
при калибровке, см. Калибровка,
ошибки
Исходный %В, градиентное
элюирование; см. Градиентное элюирование,
исходный %В
Калибровка 537-544, 548-558
анализ следовых количеств 558
внешним стандартом 549—552
внутренним стандартом 552—554
вычисления по площади или по
высоте пика 558
графики ошибки (%) 556—558
двухточечная 539
калибровочные кривые 548—552, 556—
558
линейность 538
метод стандартных добавок 555—556
многоточечная 538—539
нормирование площадей пиков
(вычисление в процентах) 554—555
ошибки 537—539,
((((938 Предметный указатель
по одной точке 538
пределы обнаружения (ПО, НПКО,
ВПКО) 539-544, 571
образцы вне пределов метода 543—
544
и отношение сигнал/шум (S/N)
540-542
проблемы
с проведением через начало
координат 538
неправильное использование г2 556,
558
стандартная кривая; см.
Калибровочные кривые
стандарты 549
стандарты контроля качества 549
стандарты на основе матрицы образца
545
экстраполяция калибровочных
кривых 543
Кана — Ингольда — Прелога правила;
см. Хиральные неподвижные фазы,
Кана — Ингольда — Прелога правила
Капиллярная ЖХ 209
Капиллярная электрохроматография (КЭХ)
52
Капиллярный электрофорез 51
Капилляры 132—136
диаметры 134
длина 135
для высокого давления 133—136
для низкого давления 132—133
из нержавеющей стали 133—136
из ПЭЭК 133-136
периодическое обслуживание 178
Карбоновые кислоты, разделение в И ОХ
381-382
Каротиноиды, разделение 331—332
Карта разрешения; см. Разрешение,
карта
Катионный обмен 265
Качественный анализ 544—548; см.
также Пика идентификация
Квадрупольные масс-спектрометры; см.
Детекторы, МС
Квалификация; см. Тесты на
пригодность (работоспособность) системы
Кинетический график; см. График
Поппе
Кислород, проблемы при его
растворении в подвижной фазе 129
Кислотно-основное равновесие 337—343
Кислотность водородных связей,
значения для растворителей 925—926
Клапаны; см. также Насосы;
Устранение неисправностей, клапаны
активные 146
шариковые 142—143
очистка обратных клапанов (опта
мальный способ) 857
Книги о ВЭЖХ 52-53
Количественный анализ 548—558; см.
также Пика идентификация; Детекторы
Колонки 238-255
биохроматография 614—618
внутренний объем 239, 669
воспроизводимость характеристик
272-273
высокое давление 275
высота теоретической тарелки 73—82
гарантии 281—282
для ионообменной хроматографии
385-386
для ортогональных разделений 273
для полипептидов (ИОХ) 636—638
для ХГВ 645-646
емкость насыщения 774, 778—779
загрязненные 283—286
ионообменная емкость 268—269
каждому методу — своя колонка 180—
181
капиллярные (с малым диаметром)
276-277
картриджи (предколонки) 275, 284
качество трубки для колонки 275, 280
мертвое время 61, 63
мертвый объем 63
методы упаковки 277—281
обслуживание 284, 286
перегрузка 105, 762—763; см. также
Препаративные разделения
сильная 785-789
плохая воспроизводимость от партии
к партии 104
промывка в обратном направлении
284, 895-897
промывка сильным растворителем 284
с аксиальной компрессией 276
с радиальной компрессией 276
селективность 263—274, 376—377; см.
также Селективность, колонок
взаимодействия 264—266
для изомеров 312—314
для ионных образцов 356—359
для нейтральных образцов 310—311
для НФХ 411-413
отличия 272—273
параметры 270
сравнение 272
слабые и сильные ионообменники
636-638
снижение эффективности сорбента
274
соединители 275
сорбенты; см. Частицы сорбента;
Неподвижная фаза
спецификации 281—283
спецификация производителя 281—282
срок службы 283—286
стабильность285, 362—363, 617—618;
см. также Неподвижная фаза,
стабильность
стальные колонки с внутренней
поверхностью, покрытой стеклом 275
тесты для сравнения колонок из раз
ных партий 281
термостаты колоночные 163—165
периодическое обслуживание 179
требования к контролю температуры
163-164
типоразмеры 276—277; см. также
Колонок параметры
требования к температурному
контролю 163-164
упакованные частицами диаметром
менее 2 мкм 286
уравновешивание ПО
уравновешивание в градиентном
режиме 473—476
фильтры и сеточки (удерживающие
сорбент в колонке) 275—276
фитинги 275
хранение 286
число теоретических тарелок 282—283
эквивалентные 272—273, 314—318
эффективность 71—89, 244—246; см.
также Число теоретических тарелок
Предметный указатель 939
Колонок параметры 82-86, 97-98, 329-
331
при градиентном элюировании 444—
447, 473
Колонок переключение 115—116, 160—
162; см. также Многомерная
жидкостная хроматография; Двумерное
разделение (2Э-ЖХ)
выбор колонок для подключения
161 162
для подготовки образца 837—838
многомерная жидкостная
хроматография 652-653, 654-655
направление потока в слив 162—163
обогащение образца 160—161
параллельные колонки 161 — 162
повторное использование подвижной
фазы 163
регенерация колонок 161
сбор фракций 162
хроматография разделений внахлест
115-116
Колонок функции сравнения
Fs 272-273
Fs(-C) 314-315
Комплексообразование с ионом серебра
313
Кондуктометрические детекторы; см.
Детекторы, кондуктометрические
Конечный %В; см. Градиентное элюиро-
вание, конечный %В
Контроль качества (КК) 599—600
Корректировка методики 314—318, 565,
595—597; см. также Модификация
методики
аналитическая область 571
буфер 597
длина волны детектора 598
неэквивалентные колонки 317—318
оптимальный подход 596
подвижная фаза 597—598
размер колонки 598—599
размер частиц сорбента 598—599
рН 597
температура 598
Коррозия под действием цитратных
буферов 349
Косвенный метод в хиральных
разделениях 706, 707-711
Ш 940 Предметный указатель
Коэффициент асимметрии пика 86—89
Коэффициент диффузии 80, 82
Коэффициент емкости; см. Фактор
удерживания
Коэффициент полярности растворителя
(/") 69, 71, 922, 925-926
Коэффициент распределения А"д, в ЭХ
669-670
Кран ввода проб; см. Инжекторы
Краны высокого давления,
использование
для направления подвижной фазы в
слив 162—163
для переключения колонок 160—162
Краткие курсы по ВЭЖХ 54
Критическая пара пиков 90—91
Критическое разрешение 90—91
Кусочно-линейные градиенты; си.
Градиентное элюирование,
кусочно-линейные градиенты
Лазерные детекторы светорассеяния; см.
Детекторы, светорассеяния
Лекарственный препарат и
фармацевтическая субстанция, ФС (или активный
фармацевтический ингредиент, АФИ)
565
Лиганды; см. также Неподвижная
фаза
алкильные 262—263, 264
влияние длины привитой
углеводородной цепи 258—259
встроенные полярные группы 262—
263, 264
идентификация привитых лигандов
после отщепления плавиковой кислотой
260
типы 263, 264
фенильные 262—263, 264
циано- 262—263, 264
Линейность 571
Линейный динамический диапазон
детектора 198
ЛИУС; см. Системы сбора и обработки
данных, ЛИУС
Локализованная адсорбция 396—398,
408-410
Локальный оптимум 92
Макромолекулы, разделение 605, 614—
697
Макроциклические антибиотики,
компоненты хиральных неподвижных фаз
737-743
Мартин А. Дж. П. 47
Массообмен в подвижной фазе 76—79,
81-82
Масс-спектроскопические детекторы;
см. Детекторы, МС
Масштабирование разделения 763 766
Мертвое время 61, 63—65
Мертвый объем 63—65
Метод Брунауэра — Эмметта — Теллера
(БЭТ) 239
Метод испытания
биоаналитический 566, 580—586
документирование 575—577
количественное определение 565—567
однородность содержания 566
определение стабильности 566
синонимы термина 562—563
тест на примеси 566
тест на растворение 566
Метод ртутной порометрии 239
Метод рутинный; см. Метод испытания
Метод хиральных добавок к подвижной
фазе, хиральные разделения 711—713
Метода характеристики
специфичность 569—570
точность; см. Правильность
устойчивость 572—573
Методы (режимы) ВЭЖХ-разделений 58
Методы упаковки колонок 277—281
Методы (фармацевтические)
анализа однородности содержания
578
анализа примесей 578—579
анализа продуктов разложения 578—
579
биоаналитические; см.
Биоаналитические методы
валидации очистки 579
валидация; см. Валидация
категории I 578
категории 2 578—579
категории 3 579
категории 4 580
Предметный указатель 941
количественного определения 578—
579
количественного определения
активного вещества 578
определения основных свойств
продукта 579
тестирования на подлинность 580
тестирования на растворимость 579
тестирования предела обнаружения
578-579
Микро-ВЭЖХ 150, 209
Миксер малого объема 140—141
Минимизация продолжительности
разделения 329—331
Миоглобина трипсиновый лизат,
разделение 627
Мицеллярная жидкостная
хроматография 52
Многомерная жидкостная
хроматография (МЖХ) 652—655; см. также
Двумерная ВЭЖХ (2D->KX), с
переключением колонок
дискретная (с коллектором фракций)
653
полипептидов 652—655
с непосредственно соединенными
колонками 654
Многоугловые светорассеивающие
детекторы (МУСД); см. Детекторы,
светорассеяния, лазерные
Модель гидрофобного вычитания
266-269
Модель линейного изменения элюирую-
щей силы растворителя (ЛИС) 439—440
Модель сольватационных параметров
118-119
Модель трехточечного присоединения,
хиральные разделения 714—717
Модификация методики 595; см. также
Корректировка методики
Молекулярная масса синтетических
полимеров
средняя 691—694
молекулярно-массовое распределение
689-694
определение 668—670
Молекулярная структура как основа для
предсказания удерживания соединений
116, 118-119, 517
Монолитные сорбенты 238, 250—251
на основе силикагеля 251
преимущества и недостатки 251
Мономерные колонки; см. Неподвижная
фаза, мономерная
Монослой адсорбированный 396 401,
774-775
Моча, разделение 45
МС-детекторы; см. Детекторы, МС
Нано-ВЭЖХ 150
Наноисточники ионизации,
полипептиды после разделения в ОФХ 630—631
Насос(ы) 141-151
активный клапан 146
обратный клапан 142—144
периодическое обслуживание
(оптимальный подход) 178—179
сливной вентиль (кран для
промывки) 145
возвратно-поступательный
плунжерный 141—146
замена уплотнений 865—867
уплотнения, проблемы; см. Общие
симптомы ВЭЖХ-проблем, утечки
Нафталины замещенные, разделение
323-324
Начальные условия
в ГИХ 425-426
в ИПХ 372-375
в НФХ 418-419
в ОФХ 290-291, 360-361
в препаративных разделениях 782—784
при градиентном элюировании
463-468
при разделении полипептидов в ОФХ
632
Неводная обращенно-фазовая
хроматография (НОФХ) 221-222, 331-332
Неисправности и быстрые способы их
устранения 851-852, 908-919
Нелокализующие растворители 402, 408
410
Необходимость контроля температуры в
ИПХ 377
Неорганические носители (сорбентов)
252-255
Неподвижная (стационарная) фаза 255—
263
Ш 942 Предметный указатель
бидентатные силаны 285
горизонтальная и вертикальная
полимеризация 256—257
коллапс; см. Неподвижная фаза,
снижение смачиваемости
монолитные сорбенты 250
мономерная 256—259, 269—271
на основе оксида циркония 253—254
на основе полимеров 617—618
носители 238—255
площадь поверхности 239
полимерная 256, 259, 269—271
получение 256—262
различные функциональные группы
сорбентов в ОФХ 262-263
с алкильными лигандами 262—264
селективность; см. Колонки,
селективность
синтез; си. Неподвижная фаза, полу
чение
снижение смачиваемости 261—262,
274, 332-333
стабильность 257—260
стерически защищенная 258—259
со встроенными полярными группами
262-263
фенильная 263, 264
циано 263, 264
Нернста закон распределения 805—806
Неудерживаемый пик 63—64; см. также
Пик(и), неудерживаемые
Нижний предел детектирования; см.
ПО
Нижний предел количественного
определения или предел количественного
определения; см. ПК.О
Нитрозамещенные производные
бензола, разделение 294, 300—301
Номограмма; см. Растворители,
номограмма
Нормально-фазовая хроматография (НФХ)
392^31
воспроизводимость 422—424
градиентное элюирование 493
номограмма растворителей 402—403
обеднение элюента 424
применение 392—393
разделение изомеров 394—396, 413—
415
размывание заднего фронта пиков
424-425
разработка метода 416—422
исходные условия 418—419
пример (паклитаксел) 420—422
селективность 406—415
селективность колонки 411—413
селективность, зависимость от силы
растворителя 406
селективность, зависимость от темпе
ратуры 410 411
селективность, зависимость от типа
растворителя 406—410
сила растворителя 400—403
сравнение с ОФХ 393-396
трудности 422—425
удерживание 393—400
НОФХ; см. Неводная обращенно-фазо-
вая хроматография
НПКО, см. ПКО
Нуклеиновые кислоты 609—610, 655—661
двухцепочечные 609—610
ИОХ-разделение 655—657
нарушение последовательности нук-
леотидов 658—660
одноцепочечные 609
отделение неполных
последовательностей 655
ОФХ-разделение 657—661
ОФХ-5 (разделение смешанного
типа) 660-661
ХГВ-разделение 661
Обеспечение качества 599—600
Обессоливание, гель-фильтрация 679
Обзоры по ВЭЖХ 54
Оборудование 124—183; см. также
отдельные модули (Насосы, Детекторы и
т.д.)
для упаковки колонок 278, 280
изменения 103—104
Образцов
введение, препаративные разделения
768-770
концентрирование (обогащение) 160—
161
масса 105—109; см. также
Препаративные разделения
проблемы 108-109
влияние объема образца и
растворителя 158—160
разложение (химические
превращения) 899-900
растворимость 780—782
Образцов пробоподготовка 102, 798—845
адсорбенты с ограниченно доступной
поверхностью (ОДП) 827-828
аффинные сорбенты 841
в хроматографии биомолекул 838—
841
виды образцов 799—801
высушивание 803
дериватизация 232—233, 843—845
диализ 840-841
для ЖХ-МС 841-843
жидкие образцы 804—805
мембранные технологии 831
обзор 798-799
осаждение белков 842—843
предварительная обработка твердых и
полутвердых образцов 802—804
принцип «разбавь и вводи» 801
с помощью переключения колонок
837-838
твердофазная экстракция; см.
Твердофазная экстракция
твердые образцы 832—837
удаление белков из биологических
жидкостей 842—843
ультрафильтрация (УФ) 841
уменьшение размера частиц
(измельчение) 802
фильтрование 803—804
экстракция в системе жидкость —
жидкость (ЖЖЭ) 805-811
выбор растворителя 807—808
жидкостная экстракция на
иммобилизованном носителе (ИЖЭ) 811
микроэкстракция 810
микроэкстракция методом одной
капли (МОК) 810
проблемы 809-810
противоточное распределение 808
связывание растворенного
вещества 810
специальные подходы 810—811
схема ЖЖЭ 806
теория 805-807
Предметный указатель 943
экстракция в системе жидкость —
жидкость на твердой фазе (ТФЖЖЭ) 811
эмульсии 809
эффективность 805
электродиализ 841
Образцов твердых пробоподготовка
жидкостная экстракция под
давлением (ЖЭД) или ускоренная экстракция
растворителем (УСЭР) 836
метод «встряхивания колбы» 834
обработка ультразвуком 834
сверхкритическая флюидная
экстракция (СФЭ) 835-836
экстракция в аппарате Сокслета 833,
834-835
экстракция растворителем,
ускоряемая микроволновым излучением, или
экстракция с микроволновым
нагреванием (МЭ) 837
Образцы; см. также Анализируемые
(разделяемые) вещества
контроля качества анализа 574, 581,
585
«неправильные»; см. Обрашенно-фа-
зовая хроматография, «неправильные»
образцы
«правильные»; см. Обращен но фазо
вая хроматография, «правильные»
образцы
Обращенно-фазовая хроматография (ОФХ)
57, 290—333, 352—380; см. также
Сопутствующие аспекты разделений, например
Колонки
история 292
неионные образцы 290—333
«неправильные» образцы 96, 300—302
при градиентном элюировании 442—
444
«правильные» образцы 96, 300—302
при градиентном элюировании 442—
444
предпочтительные растворители 290
предпочтительные условия (изократи-
ка) 291
преимущества 290—291
процесс удерживания 296—299
разделение нуклеиновых кислот 657—
661
разделение полипептидов 619—633
Ш 944 Предметный указатель
Обслуживание оборудования 170; см.
также Профилактическое
обслуживание; Периодическое обслуживание
Обслуживание системы ВЭЖХ; см.
Периодическое обслуживание
Общая проблема элюирования 110—111
Общие симптомы ВЭЖХ-проблем 863—
919; см. также Устранение
неисправностей
адсорбционные остаточные примеси
882
артефактные (ложные) пики и пи-
ки-«призраки» 889—890
базовая линия в градиенте 884—885,
889-890
всплески на базовой линии 888
высокочастотные шумы 887
дрейф базовой линии 884—887
при изменении температуры 886—
887
засорение входного фильтра колонки
895-897
изменения соотношения площадей
пиков 882, 897-899
низкочастотные шумы 888
перегруженные пики 893—894
пики с плоской вершиной 893
поздноэлюируемые пики 882, 888—889
проблемы, связанные с давлением
872-876
проблемы, связанные с инжектором
867-868
проблемы, связанные с расчетом
площадей пиков 881—884
проблемы, связанные с удерживанием
876-881, 897-899
проблемы, связанные с формой
градиента 900-907
проблемы, связанные с изменением
температуры 878—879
проблемы с уплотнениями насоса
865-867
размывание заднего фронта пиков
891-893, 895
размывание переднего фронта пиков
894
расщепленные пики 895—900
регулярные колебания базовой линии
889
случайные шумы 888
утечки 864
в автосамплере 867—870
в детекторе 871—872
в колонке 870—871
в насосе 865—867
перед насосом 864—865
под давлением 867
при низком давлении 864—865
форма пиков 890 900
широкие пики 894 895, 899
шумы базовой линии 887—890
Объем вводимого образца 158—159
Объем задержки 104, 149, 452—453;
см. также Градиентное элюирование,
объем задержки
влияние на воспроизводимость 478
измерение 171—172
Овомукоид (ОВМ); см. Хиральные
неподвижные фазы, на основе овомукоида
Ограниченная диффузия 241—242
Окись алюминия, сорбент 252, 254
Олигомеры (синтетические) 687
Олигонуклеотиды, разделение 657—658;
см. также Нуклеиновые кислоты
Олигосахариды; см. Углеводы
Операционная квалификация (ОК); см.
Тесты на пригодность
(работоспособность) системы
Оптимальные подходы для
валидации метода (протоколы и
отчеты) 578
воспроизводимости градиентного
метода 476
выбора буфера 349
выбора колонок для разработки
метода 360
для разделения биомакромолекул 620—
628, 636-639, 643, 645-650, 669-676
выбора подвижной фазы 344—346
выбора реагентов 180
выявления проблем с ВЭЖХ-систе-
мой 861-863
графиков ошибки (%) 556—558
дегазации 180
замены запасных частей 855
замены модулей системы ВЭЖХ
862
интегрирования пиков 533—534
ион-парной хроматографии (когда
использовать) 364—366
использования калибровочных
стандартов в матрице 549
использования рефрактометрических
детекторов 2I8
использования фитингов из ПЭЭК
858
использования хиральных фаз 724—
725, 736, 739, 741-742
корректировки методики 596
линейных градиентов 437—438
мытья стеклянной химической
посуды S58
нахождения и исправления неполадок
861-862
обнаружения утечек 857—858
обслуживания резервуаров для элюен-
тов 177
определения пригодности системы
181
определения условий эксплуатации
колонки в градиентном режиме 445, 447
оценки устойчивости методики
анализа 572-573
очистки обратных клапанов 857
передачи (трансфера) методики
анализа 588
повышения контроля чистоты воды
859
подготовки образцов для ввода
181-182, 549-552
предварительного смешивания элю-
ентов 856-857
предварительных разделений 181
преодоления переноса образца из
предыдущего разделения 154
применения стандартов и градуиро-
вочных растворов 181 — 182
применения ТФУК 858-859
проверки гидравлического затвора
127, 856
промывки системы ВЭЖХ 180
профилактического обслуживания
системы ВЭЖХ 177
профилактического обслуживания
насосов 178-179
решения проблемы растворимости
буфера 347-348
Предметный указатель 945
тестирования пригодности
(работоспособности) системы 900
удаления воздуха из насоса 855
уравновешивания колонки 181
устранения неисправностей 909—919
устранения неисправностей: полезные
советы и методы 855—861
экстракции в системе жидкость —
жидкость 807-809
Оптимизация 92; см. также Разработка
метода; Удерживание; Селективность;
Число теоретических тарелок; Колонок
параметры; Оптимизация нескольких
условий разделения
Оптимизация нескольких условий
разделения, нейтральные образцы 321—329
Ортогональные разделения 103—104, 273,
318-319,417
Ортофтальдиальдегид (ОФА) 708
Основность водородных связей,
значения для растворителей 925—926
Отдел контроля качества 599
Относительное удерживание; см.
Селективность
рН; см. также Ионизация, образца;
Кислотно основное равновесие; По
движная фаза, оптимальный рН
в ионообменной хроматографии 385
в ион-парной хроматографии 366—371
рН-метр, использование 344, 361—362
чувствительность 361—362
рКа
аминокислот 606, 607
анализируемого вещества 337—343
буферов 344—346
определение 341—343
предсказание исходя из структуры
анализируемого вещества 517
углеводов 665
ПАУ; см. Полициклические
ароматические углеводороды
Пептиды; см. Полипептиды
Первичная структура, полипептиды; см.
Полипептиды, первичная структура
Перегрузка 105—108
колонки; см. Колонки, перегрузка
Перегрузка по массе 107-108, 762-763,
785, 786
Ш 946 Предметный указатель
Передача (трансфер) методики анализа в
принимающую лабораторию (ТМА) 587—
588
документ «Приемлемая аналитическая
практика» (ПАП) 587
документирование результатов 593
критерии приемлемости 593
обзор 591-592
опции 588-590
основы ТМА 590
отказ от оформления передачи метода
испытания 589—590
отчет 593
потенциальные ошибки при ТМА 593
при градиентном элюировании 478
протокол 590
Перенос образца из предыдущего
разделения («остаточные примеси») 859—861
«Пересечение изотерм» 787—789
Периодическое обслуживание системы
ВЭЖХ 177-180
Пестициды, подготовка образца 814, 816
Пик(и) 60—61; см. также Полоса (зона)
вещества
воспроизводимость площади; см.
Тесты на пригодность (работоспособность)
системы, воспроизводимость площади
пика
высота пика, зависимость от
удерживания 93
измерение площади 526—544
невыявленный 103—104
неудерживаемый (мусорный) пик 64
отслеживание 112—113,515—516
потерянный 273, 318
проблемы с формой; см. Общие
симптомы ВЭЖХ-проблем, расщепленные
пики, пики с плоской вершиной и т.д.
раздвоенный 283—284
размер 535
размывание фронта заднего 86—89,
283-285, 333, 362-363, 891-895, 899
в ион-парной хроматографии 380
в НФХ 424-425
вызванное использованием
несоответствующего буфера 344—346
при градиентном элюировании 468
при перегрузке 88, 105—108, 776—
777
причины 88, 274, 891-895, 899
прогнозирование 274
экспоненциальное 88
размывание фронта переднего 86—88,
894
скрытый 103—104
соотнесение пиков 112—113, 515—516
форма пика 86—89
соответствующая форме кривой
Гаусса 73
Пика идентификация 544 548; см. так
же Детекторы, специфических
характеристик образца
методом качественного анализа в
реальном времени 545—546
методом стандартных добавок 545
по времени удерживания 544—545
по УФ-спектру 546
при совместном введении со
стандартом 545
с анализом в автономном режиме
548
Пика ширина 71—73
в препаративном разделении 776
вклад детектора 196—197
зависимость от массы образца 776
Пиковая емкость 111 — 112, 478—483
в двумерной ВЭЖХ 488
оптимизированная 480—483
при разделении полипептидов 652—
653
Пики-призраки; см. Общие симптомы
ВЭЖХ-проблем, артефактные (ложные)
пики
jt-Ti взаимодействия; см. Взаимодействия
межмолекулярные, с переносом заряда
Пиримидины, разделение 428
Пиркла фазы; см. Хиральные
неподвижные фазы, донорно-акцепторные
ПКО 571; см. также Детекторы,
пределы обнаружения
ПО, пределы обнаружения; см.
Детекторы, пределы обнаружения; см. также
Калибровка, пределы обнаружения
Поверхностно-пористые частицы
сорбента; см. Частицы сорбента,
поверхностно-пористые
Подавление ионного сигнала, ЖХ-МС
842
Предметный указатель 947
Подвижная фаза 57; см. также
Растворители
Дегазация 128—132
оптимальный выбор 180, 344—346
оптимальный рН 338—443
повторное использование 163
предварительное смешивание 146—
147, 856-857
приведенная скорость 79—82
растворители для ОФХ 290
резервуары 125—127
селективность; см. Селективность,
влияние силы элюента, типа
растворителя
скорость (линейная) 63
фильтрование 127—128
Показатель преломления, значения для
растворителей 923—924
Полимерная (привитая) неподвижная
фаза 256-259, 269-271
Полимеры (синтетические); см.
Синтетические полимеры
Полипептидные фазы для ГИХ 428
Полипептиды 606—609
аффинная хроматография на
сорбентах с иммобилизованными металлами
(АХИМ) 641-645
влияние конформации на время
удерживания в ОФХ 628-630
влияние молекулярной массы на
удерживание 625—627
влияние температуры 624—625, 629—
630, 631
вторичная структура 606, 608
градиентное элюирование в ОФХ 625—
628
зависимость удерживания от рН в
ИОХ 634-635
колонки для ИОХ 636—638
колонки для ОФХ 619 620
многомерная жидкостная
хроматография (МЖХ) 652-655
первичная структура 606, 608
подвижные фазы в ИОХ 638—639
подвижные фазы в ОФХ 621—624
посттрансляционные модификации
609
разделение 619—655
разделение в ГИХ 648—650
разделение в ОФХ 619—633
разделение в ХГВ 645—648
разделение на гидроксиапатите 640—
641
разделение на капиллярных колонках
630-631
разделение с помощью
хроматографии гидрофильных взаимодействий с
электростатическим отталкиванием 651 —
652
разработка метода для ИОХ 633—639
разработка метода для ОФХ 631—633
третичная структура 606, 608
удерживание смешанного типа в ОФХ
628
хроматофокусирование 639—640
четвертичная структура 608
Полифосфаты, разделение в ИОХ 383
Полициклические ароматические
углеводороды (ПАУ)
зависимость удерживания от
температуры 306-309
зависимость селективности от формы
молекулы 313—314
разделение 269, 271
Полоса (зона) вещества 60; см. также
Пик(и)
перемещение по колонке 60
перемещение при градиентном элюи-
ровании 440—441
уширение 76—78
ширина 60
Поляриметры; см. Детекторы, хираль-
ные
Полярные привитые неподвижные фазы
392-393
Полярные эндкепированные колонки
270, 274
Пористые полимерные носители 249—
250
Поры частиц сорбента
диаметр 239
диффузионные 239—240
объем 669-670
проходные (перфузионных частиц
сорбента) 240—241
размер у сорбентов для разделения
биомолекул 614—615
размер у сорбентов для ЭХ 672—673
Ш 948 Предметный указатель
Посттрансляционные модификации
влияние буфера на удерживание в
ОФХ 623
полипептидов 609
Постулаты Ренье 618—619
«Правило одного» (изменять за один раз
только один параметр) 862
Правильность 535, 566—567
Пределы обнаружения; см. Калибровка,
пределы обнаружения
Предколонки (картриджи) 275, 277, 284
Препаративные разделения 150—151,
762—795; см. также Колонки,
перегрузка
введение образца 768—770
влияние температуры 781—782
градиентное элюирование 789—791,
794
детекторы 770—771
диаметр колонок 764
емкость насыщения колонки 774,
778-779
значительно перегруженное
разделение 785-789
изократическое элюирование 774—784
изотермы сорбции 774—775
исходные условия 782—783
колонки 767—768
масштабирование 763—766
оборудование 766—773
оптимизированные условия 764—765
перегрузка по массе 762—763, 786
перегрузка по объему 779—780
пересечение изотерм 787—789
подвижная фаза 780—781
полнота выделения или чистота 785
применение 762
промышленный масштаб 794—795
разделение соприкасающихся пиков
763-764, 776-785
разработка метода 782—784
растворимость образца 780—781
растворитель для растворения образца
780-781
сбор фракций 771-772, 784-785
сравнение изократического и
градиентного элюирования 790
удаление растворителя 766, 772—773
ширина пиков 776
Прецизионность 535, 567—569
в биоаналитических методах 582—583
в одной серии анализов
(повторяемость) 568
влияние отношения сигнал/шум
194-196
воспроизводимость (межлабораторная
промежуточная прецизионность) 569
надежность 569
промежуточная (внутрилабораторная)
567, 568
Приведенные параметры 79—80
Привитые стационарные фазы,
концентрация лигандов 259
Пригодность (работоспособность)
системы ВЭЖХ 104-105, 573-575, 854
Принцип обратимости хирального
распознавания 746
Проблемы 331—333; см. также
проблемы в конкретных случаях; Устранение
неисправностей
ГИХ431
градиентное элюирование 496—497
ионные образцы 361—364
НФХ 422-425
таблицы по устранению
неисправностей 909-919
Проверка гидравлического затвора 127,
856; см. также Устранение
неисправностей, тест с сифоном
Проверочные стандарты, стандартные
образцы 581; см. также Калибровка,
стандарты контроля качества
Программа DryLab 507—515; см. также
Программное обеспечение
компьютерного моделирования
кусочно-линейные градиенты 510,
512-514
метод двух разделений 514
оптимизация градиента 509—510
примеры разработки методов 518—
521
прогнозирование изократического
элюирования на основе данных градиентного
511-512
Программное обеспечение
компьютерного моделирования 501—516; ел*, также
Программа DryLab
история 503—504
Предметный указатель 949
источники коммерчески доступного
программного обеспечения для
компьютерного моделирования 516
карта разрешения 501—502
устойчивость (робастность) метода
505
опции компьютерного моделирования
511
отслеживание пиков 515—516
подбор селективности обращение фа
зовых колонок 517—518
предсказание удерживания исходя из
молекулярной структуры
анализируемого вещества 517
преимущества и недостатки 505—506
применение 504—506
примеры 518—521
проверка устойчивости (робастности)
метода 522—523
программа DryLab 507—515
программное обеспечение для поиска
и предсказания рКа анализируемого
вещества 517
разработка метода 518—521
условия эксперимента 505—506
экспертные системы для разработки
метода 518
Продольная диффузия 76—79
Продукты дезамидирования,
полипептиды в ОФХ 623
Продукты полимеразной цепной
реакции (ПЦР), разделение в ОФХ 658, 659
Промывка колонки в направлении,
противоположном потоку 284
Протеомика, многомерная жидкостная
хроматография 652—655
Противоион, ионообменная
хроматография 382-385
Противоточная хроматография (ПТХ)
51
Протокол; см. Валидация, протокол
Профилактическое обслуживание
177—180; см. также Обслуживание
оборудования, Ремонтные работы
Псевдокритическая хроматография 692—
694
Пфайффера правило; см. Хиральность,
определения, правило Пфайффера
Радиоактивности детекторы; см.
Детекторы, радиоактивности
Разделение
виды разделений (в ВЭЖХ) 58
выбор условий 102—103
макромолекул 605—697
ортогональные разделения 104, 318—
319
процесс 57—61
Разделение соединений, принадлежащих
к разным классам 396
Разделение соприкасающихся пиков; см.
Препаративные разделения, разделение
соприкасающихся пиков
Разделения оптических изомеров; см.
Хиральные разделения
Различные скорости перемещения
анализируемых веществ по колонке 59—60
Разработка метода 101-105, 319-331
ГИХ 429-431
градиентного элюирования 462—491
диапазон градиента 438, 470—472
ИОХ 386-387
ИПХ 372-378
исходного градиентного разделения
465-469
исходные условия 360—361
НФХ 416-422
оптимизация к* 469
параметры градиента 473
параметры колонки 97—98
предпочтительные условия для
градиентного элюирования 435—437
препаративного разделения 782—784
разделения ионных образцов 359—364
разделения нейтральных образцов
319-329
разделения пол и пептидов в режиме
ИОХ 633-639
разделения пол и пептидов в режиме
ОФХ 631-633
с использованием кусочно-линейных
градиентов 472
с помощью программного
обеспечения 501-523
селективность (ОФХ ионных
образцов) 361
селективность (НФХ) 420
((((950 Предметный указатель
селективность при градиентном элю-
ировании 470
стратегия 319—321
схема эксперимента для разработки
метода 321-324, 504, 506
Разрешение 89—92
в эксклюзионной хроматографии 673
и селективность; см. Селективность
и удерживание 93—94
карта 307-309
при градиентном элюировании 462
уравнения 89, 91
Разрушение; см. Неподвижная фаза,
стабильность; Образцов разложение
Распределительная хроматография с
управляемой ионами селективностью
663-665
Растворители; см. также Подвижная
фаза
безопасность 927—928
вязкость, величины 923—924, 926
локализующие (НФХ) 402, 408-409
нелокализующие (НФХ) 402, 408-409
неосновные (НФХ) 402, 408-410
номограмма для НФХ 401—403
номограмма для ОФХ 304—306
обеднение подвижных фаз (раствори
тел ем) 405
в НФХ 424
при градиентном элюировании 496
образца 159-160
основные в НФХ 402, 408-410
плотность, величины 927
поглощение в УФ 922—923
показатель преломления, величины
923-924
полярность, величины (F) 922, 924—
926
предпочтительные в ОФХ 290
свойства 922, 923-927
селективность, величины 922, 924—
926
сила 65-66, 294-295
в ион-парной хроматографии 373—
374
в НФХ 400-403
параметр к 400—403
прогнозирование 295
сильные 65—66
слабые 65—66
смеси (трех-, четырехкомпонентные)
322-324
совместимость с детектором 921—924
степень насыщения водой 422—423
температура кипения, величины 923—
924
треугольник селективности 70—71
фильтрование 125, 127—128
Реакционные детекторы; см. Детекторы,
реакционные
Резервуар 125—127
периодическое обслуживание 177
фильтр 127
Рекомбинантный инсулин человека; см.
Инсулин человека рекомбинантный
Ремонтные работы 182
ведение учета 182—183
описание конфигурации системы
ВЭЖХ 183
персонал 182
Ретинола изомеры, разделение 414—415
Рефрактометры; см. Детекторы,
рефрактометрические
Рибонуклеаза, конформационные
эффекты в ОФХ 629
Ристоцетин А; см. Хиральные
неподвижные фазы, на основе ристоцетина А
«Руководство для контролирующих
инстанций», валидация 564, 574
Сбор фракций 162, 770-773, 784-785
Сверхкритическая флюидная
хроматография 50—51
Селективность 92, 300-319
аминомодификаторы 359
в ионообменной хроматографии 386—
387
в ион-парной хроматографии 375—378
в НФХ 406-415
в ОФХ 300-319, 353-359
влияние концентрации буфера 359
влияние неподвижной фазы 328—329,
356-359, 377-378, 411-413
влияние силы элюента 355—356
в НФХ 406
и типа растворителя 325, 326
и температуры 325, 327—328
влияние температуры
Предметный указатель 951
в НФХ 410-411
в ОФХ 306-309
влияние типа буфера 359
влияние типа растворителя 303—306,
322-324, 356, 377
в Г ИХ 429 430
в НФХ 406-410
влияние рН 353—354
колонок 328-329, 356-359, 377-378,
411—413
оптимизация 95—96
по форме молекулы 269, 271, 313—314
при градиентном элюировании 470
при разделении изомеров 312—314
при разделении ионных образцов в
ОФХ 353-359
Сервисное обслуживание
(производителем) 182
Сертификат анализа 581
Сигнал, измерение 526—544
Сигнал/шум отношение 194—196
вклад в погрешность метода 540
и точность 194
улучшение 195
Силанольные группы 246—248
влияние на разделение 362—363
ионизация 246 247
кислотность 246—247, 268
эндкепирование 258, 259—260
Силикагель
загрязняющие ионы металлов и
измерение степени загрязнения 246—247
кислотность 246—247
колонки, для ГИХ 427—428
колонки, использование с
органическими буферами 286
носители 242—249
подготовка частиц
агрегаты золя силикагеля 248—249
распыление с высушиванием 248
преимущества в НФХ 392
растворение 285, 286
силанольные группы 246—248; см.
также Силанольные группы
следовые примеси 246—247
сравнение с оксидами металлов 252
строение поверхности 246—248
типа А 246-248, 268-269
типа В для 246-248, 268-269
типа В для НФХ 393
типа С 260
чистота 246—248
Сильный растворитель; см.
Растворитель, сильный
Симптомы и источники проблем в ВЭЖХ
909-919
Симптомы проблем в ВЭЖХ 908-909
Синтетические полимеры 687—697
анализ 690-691
двумерная хроматография (20-ЖХ)
696-697
зависимость разделения от
молекулярной структуры 688—690
интерактивная жидкостная
хроматография 692-694
критические условия 694
структура 687—690
химическое строение и молекулярная
масса 695—696
Системные, или ложные, или артефакт-
ные пики (пики-«призраки»)
градиентное элюирование 469, 497
ион-парная хроматография 379
Системный контроллер 165
Системы сбора и обработки данных
165 169
«группирование» данных 527—530
измерение сигнала 526—544
интегрирование 527—535
измерение удерживания 534
метод перпендикуляра 531—532
определение размера пика по его
площади и высоте 535, 558
ошибки 532—533; см. также
Детекторы, источники ошибок
распознавание пиков 530
спрямление базовой линии под
пиком 530—531
ЛИУС 166-167
обработка данных 168
программное обеспечение для
разработки метода 166—167
сбор данных 167-168, 527-530
«сечение сигнала» 528
создание отчета 168—169
управление системой 167
частота передачи данных при сборе
данных 167-168, 192-193, 527-530
((((952 Предметный указатель
Скоростная ВЭЖХ 99-101
Скорость потока (объемная, мл/мин)
и давление 75—76
оптимальная 73—74, 75
проверка; см. Тесты на пригодность
(работоспособность) системы, расход
(скорость потока)
Сливная емкость, периодическое
обслуживание 180
Сливной вентиль; см. Насосы, сливной
вентиль
Сложные образцы 469; см. также
Многомерная жидкостная хроматография
Смешивание; см. также Подвижная
фаза, предварительное смешивание
в гибридных системах 148—149
в потоке 146—149
на стороне высокого давления 147—
148
на стороне низкого давления 148
при градиентном элюировании 149
Снижение смачиваемости; см.
Неподвижная фаза, снижение смачиваемости
Соответствующие разделения
в ТСХ и НФХ 403-406
изократические и градиентные
438-439, 441-447
Сополимеры 687—688
нерегулярные 688
привитые 688
Спецификации колонок 281—283
Специфичность 569—570
отделение от других соединений 570
сравнение с известными стандартами
570
Стабильность; см. Неподвижная фаза,
стабильность
Стерическое взаимодействие 269, 271
Стерическое исключение 265—268, 270
Стерически защищенная неподвижная
фаза; см. Неподвижная фаза, стерически
защищенная
Суррогатные биополимеры 618
Тандемные МС-детекторы; см.
Детекторы, МС, трехквадрупольные
Твердофазная экстракция (ТФЭ)
811-831
96-луночный планшет 817
автоматизация 819
адсорбенты с ограниченно доступной
поверхностью (ОДП) 827-828
аппаратура
дериватизация in situ 814
диски 815-816
дисперсионная ТФЭ 830—831
замена растворителя 814
иммуноаффинная экстракция 829—
830
картриджи 815
кондиционирование сорбента 819—
821
молекулярно-импринтированные
полимеры (МИП) 828-829
мультимодальная ТФЭ 826—827
наконечник микропипетки для
экстракции (НМП) 816
нанесение образца 819—820, 821
обессоливание 814
обогащение 814
покрытые волокна для твердофазной
микроэкстракции (ТФМЭ) 817
преимущества и недостатки 811—812
применение 813—814
приспособления для ТФЭ 815—817
промывка картриджа 819—820, 821 —
822
разработка метода 819—826
роботизированные системы 819
сорбенты для ТФЭ 822-825
специализированные ТФЭ-картриджи
для различных классов соединений 831
сравнение с ВЭЖХ 812-813
ТФЭ со смешанными фазами 822,
827
удаление примесей 814
устройства для ТФЭ 818-819
экстракция QuEChERS (быстрая,
простая эффективная, надежная,
безопасная) 830-831
экстракция сорбентом, нанесенным
на поверхность перемешивающего
элемента магнитной мешалки (ЭММ) 817
элюирование 819—820, 822
этапы ТФЭ 819-822
Температура
адаптация метода 316—317
в ион-парной хроматографии 377
влияние на селективность в НФХ
410-411
высокая 100
и давление 75, 76
и удерживание 71, 364
последствия недостаточного контроля
897-899
проблемы 100
требования к контролю 163—164; см.
также Колонки, термостаты
колоночные
Температурно-регулируемый подход с
тандемом колонок 328
Тест новой колонки; см. Тесты на
пригодность (работоспособность) системы,
тест новой колонки
Тестирование ступенчатым градиентом;
си. Тесты на пригодность
(работоспособность) системы, градиентные тесты
Тесты на пригодность
(работоспособность) системы 170—177
воспроизводимость пощади пика 176
градиентные тесты 171 — 174:
клапанов, формирующих градиент
(КФГ) 173-174
линейности градиента 171 — 172
тестирование ступенчатым
градиентом 172—173
операционная квалификация (О К)
170-171
проблемы 900—908; см. также
Устранение неисправностей, тесты на
пригодность (работоспособность) системы,
воспроизводимость времени
удерживания
расход (скорость потока) 174
сброс (стравливание) давления 175
тест новой колонки 176—177
установочная квалификация (УК)
170-171
эксплуатационная квалификация (ЭК)
170-171
Термостаты; см. Колонки, термостаты
колоночные
Тетраалкил-о-силикаты 249
Типа А, силикагель; см. Силикагель,
типа А
Типа В, силикагель; см. Силикагель,
типа В
Предметный указатель 953
Типа С, силикагель; см. Силикагель,
типа С
Токсикологические стандарты,
разделение 434—435
Тонкослойная хроматография (ТСХ) 42,
403-406
значения Rp 403—404
Точка кипения, растворители 923—924
Точность; см. Правильность
Третичная структура, полипептиды; см.
Полипептиды, третичная структура
Треугольник селективности
растворителей для ОФХ 323
Трикарбоксиметилэтилендиамин
(ТКМЭДА), хелатирующий лиганд для
АХИМ 643
Трифторуксусная кислота (ТФУК)
влияние концентрации 892—893
образование ионных пар с полипеп
тидами 622—623
при разделении полипептидов в ОФХ
621-622
рекомендации по оптимальному
использованию 858—859
ТФЭ; см. Твердофазная экстракция
Углеводы 610 612, 661 666
анионообменная хроматография 665—
666
ГИХ 662-663
ионообменная хроматография 381,
387
распределительная хроматография с
управляемой ионами селективностью
663-665
значения рКа 665
Удерживание 61-71, 292-299
в ион-парной хроматографии 366—371
в НФХ 393-400
в ОФХ 65-67, 292-293
в ЭХ 669-671
влияние рН 337—343
воспроизводимость; см. Тесты на
пригодность (работоспособность)
системы ВЭЖХ, воспроизводимость
удерживания
зависимость от % В 94
зависимость от силы растворителя в
ОФХ 94
((((954 Предметный указатель
зависимость от температуры 71, 306—
307
зависимость от условий разделения
65-67
интервал, сужение 365
низкое 332—333, 364, 417
предварительная оценка по структуре
разделяемых веществ 116, 118, 517
проблемы; см. Общие симптомы
ВЭЖХ-проблем, проблемы с
удерживанием
рекомендуемый интервал 300
Удерживания
время, измерение 60—61, 63—65, 534;
см. также Система сбора и обработки
данных, интегрирование
процесс 296—299
уменьшение к при возрастании %В
на 10 94, 159
фактор к 61—65
в градиенте; см. Градиентное элюи-
рование, фактор удерживания к*
низкие величины 332—333
оптимизация 93—94
рекомендуемый интервал 93
Ультравысокоэффективная жидкостная
хроматография (УВЭЖХ) 99-100, 151
Уравнение Ван-Деемтера 79
Уравнение Вилки — Чанга 80
Уравнение Дорси — Фоли 88
Уравнение Нокса 79
Уравнение Сочевинского 399
Уравнение Хендерсона — Хассельбаха
338
Уравновешивание; см. также Колонки,
уравн овеш иван ие
в градиентном режиме 473—476
в ион-парной хроматографии 379—380
в НФХ 424
Установочная квалификация (УК); см.
Тесты на пригодность
(работоспособность) системы
Устойчивость (робастность) метода 572—
573
компьютерное моделирование 505,
522-523
недостаточная 104
Устранение неисправностей; см. также
Общие симптомы ВЭЖХ-проблем
адсорбция образца 860—861
аномалии давления 872—876
воздух (пузырьки), удаление из насоса
855
время удерживания, изменения 879—
881
выявление проблем 861—862
дрейф базовой линии 859, 884—887
замена частей (модулей) 862
засорение фильтров 856
исследование результатов тестов 900
908
источники ошибок в данных 535—539;
см. также Источники ошибок
ложные пики (пики-«призраки») 889—
890
мытье стеклянной химической
посуды 858
неисправности и быстрые способы их
устранения 850
обратные клапаны 857
обслуживание оборудования; of.
Обслуживание оборудования
определение приоритетности тестов
862
остаточные примеси образца из
предыдущего разделения 859—861
ошибки интегрирования 532—533
полезные советы и методы 855—861
получение высокоочишенной воды
859
предотвращение проблем 852—861
причина и следствие 908—919
проблемы с неподвижной фазой
878
проблемы с переносом градиента 905—
907
проблемы с подвижной фазой 877—
878
проблемы со скоростью потока 877
проблемы со смешиванием 856—857
проблемы, связанные с размером
колонки 877
симптомы проблем; см. Общие
симптомы ВЭЖХ-проблем
стратегия «найти и исправить»
861-862
таблицы по устранению
неисправностей 908-919
Предметный указатель 955
тест с сифоном {проверка
гидравлического затвора) 127, 856
тестирование новой колонки 861
тестирование пригодности системы
854
тесты на пригодность
(работоспособность) системы 852-853, 900-908
воспроизводимость времени
удерживания 908
линейность градиента 901—905
отрицательный результат
тестирования градиента 900—908
отрицательный результат
тестирования КФГ 901-903
отрицательный результат
тестирования ступенчатым градиентом 901—904
проблемы со сбросом давления 908
проблемы со скоростью потока 907
тест на рабочие характеристики
градиента 853
УК, ОК., ЭК 853
ТФУК 858
утечки в насосе и других модулях
ВЭЖХ-системы 865-872
утечки в фитингах, обнаружение и
устранение 857—858
фитинги, ПЭЭК 858
хронологические записи 854—855
шумы базовой линии 856—857,
887-890
эксплуатация ВЭЖХ-системы,
полезные советы 855—861
УФ-поглощение, растворители; см.
Растворители, поглощение в УФ
УФ-детекторы; см. Детекторы, УФ-де-
текторы
Фазовое отношение 62
и площадь поверхности сорбента 239
Фазово-оптимизированная жидкостная
хроматография 328
Фазы со встроенными полярными
группами (ВПГ) 262-263, 270
Фактор размывания пика (TF) 86—88
Фармакопейные методы испытаний
563-564, 595-597, 600
Фармацевтическая субстанция (ФС)
565-566
Фенильная фаза 262—263, 264, 270
Фильтрование; см. Объект
фильтрования, например Образцов, Подвижной
фазы и т.д.
Фильтры
входные фильтры 126, 127
проточные (инлайн) 139—140, 284
Фитинги 136—141; см. также Колонки,
фитинги
миксер малого объема 140—141
периодическое обслуживание 178
проточные (инлайн) фильтры 139 140
Фишера проекции; см. Хиральность,
определения, проекции Фишера
Флуоресценции детекторы; см.
Детекторы, флуоресцентные
Фосфатный буфер 344—349
Фосфопротеины (фосфопептиды),
разделение с помощью аффинной
хроматографии на сорбентах с иммобилизо
ванными металлами 643
Хаотропы 375
Хелатообразующие молекулы образца
265-266
Хемилюминесцентный азотный детектор
(ХЛАД); см. Детекторы,
хемилюминесцентный азотный
Хиральное распознавание 706—707; см.
также Хиральные разделения
бинарная модель Ленгмюра 755—756
необычное поведение, вызванное
изменениями температуры 754
неэнантиоселективные центры
754-756
термодинамика гетерогенной хираль-
ной поверхности 754—756
энантиоселективный центр 754—756
энтропийно контролируемый процесс
754
энтальпийно контролируемый
процесс 754
Хиральность, определения 704—706
гомомеры 704
диастереомеры 704, 706
динамическое соответствие 717
изомерия 704
классификация изомеров 704—706
комплементарность по размеру и
форме 716
H(f 956 Предметный указатель
константа распределения 712—713
конституциональные изомеры 704—
705
правило Пфайффера 717
проекции Фишера 706
спиральная хиральность 704—705
функциональное соответствие 716
эпимеры 706
Хиральные дериватизирующие агенты
(ХДА) 707-711
ортофтальдиальдегид (ОФА) 708
Хиральные детекторы; см. Детекторы,
хиральные
Хиральные колонки; см. Хиральные
неподвижные фазы (ХНФ)
Хиральные неподвижные фазы (ХНФ)
703, 714-715, 718-753
ChiraDex (ChiraDexGamma) 734
Chiralcel 719-726
Chiral Hyun-CR-1 744
Chiralpak 719-726, 727, 752
ChiraSpher 727
ChirBase 703
Chirobiotic 737, 739-742
ChiroSil 744
Crownpak 744
Crownpak CR 744
Cyclobond 734-735
Kromasil 719, 728
автоматизированные процедуры
скрининга 703
амилоза 720—721
ароматические заместители и хираль-
ное распознавание 721
влияние подвижной фазы 717
динамически модифицированная ХНФ
711-713
донорно-акцепторные ХНФ (фазы
Пиркла) 745-748
иммобилизованные полисахариды
724-726
ионообменники 748—751
Кана — Ингольда — Прелога правила
705
кинетика адсорбции-десорбции 718
константа ассоциации 712
лигандообменные ХНФ 751—753
микрокристаллический триацетат
целлюлозы 719
на основе аркислого гликопротеина
(АКТ) или орозомукоида 731—732
на основе ванкомицина 737—742
на основе овомукоида (ОВМ) 731
на основе ристоцетина А 737—738,
741-742
на основе тейкопланина 737—739,
741-743
на основе целлобиогидролазы I (ЦБ Г)
731-732
на основе целлюлозы и ее
производных 719-726
оптимальные подходы 724—725, 739,
741-742
полиакриламидные ХНФ, метод
«прививки от» 728, 729
преимущества и недостатки 714—715
прямой метод 706, 711—717
синтетические полимерные фазы
726-730
скрининг фаз для разработки метода
724-725
сшитые поли(мет)акриламидные
полимеры 727—728
фазы Пиркла (донорно-акцепторные
ХНФ) 745-748
ULMO 745-747
WHELK-Ol 745-747
ХНФ на основе белков 730—734
ХНФ на основе краун-эфиров 743—
745
ХНФ на основе макроциклических
антибиотиков 737—743
ХНФ на основе полимеров сетчатого
типа 726-730
ХНФ на основе полисахаридов 719—
726
нормально-фазовый режим 719—
720, 722-726
обрашенно-фазовый режим 719—
720, 722-726
полярно-органический режим 723—
726
ХНФ на основе циклодекстринов
734-737
ХНФ с карбаматом цинхонана 748—
751
ХНФ с производными динитробензо-
ила (ДНБ) 745-747
Предметный указатель 957
Хиральные разделения 703—756
71-тг-взаимодействия 716—717
асимметрические центры 705
ассоциация анализируемого вещества
с селектором 753
ахиральная среда 705
вращение плоскости поляризованного
света 705-706
гидрофобное соответствие 717
дисперсия пика и размывание заднего
фронта 718
жесткость молекулы 716
индуцированное соответствие 717
косвенный метод 706, 707—711
метод хиральных добавок к
подвижной фазе (аддитивный метод) 711—713
модель трехточечного присоединения
715-717
молекулярные взаимодействия 715—
717
нерацемические смеси 705
переходные диастереомерные
комплексы 711
пленарная хиральность 704—705
препаративное выделение энантиоме-
ров 715
принцип обратимости хирального
распознавания 746
принципы хирального распознавания
715-717
прямое ВЭЖХ-разделение энантио-
меров 703
прямой метод 706, 711—717
рацемат 705
сольватация интерактивных центров
анализируемого вещества 717
специфика молекулярного
распознавания 703
стереоизомеры 704
стереоселективность лекарственных
средств 717
стереохимические дескрипторы 705—
706
стерическое соответствие 716
термодинамические аспекты 753—
756
топологическая хиральность 705
хинидин 748-749, 751
хинин 748-749, 751
человеческий сывороточный
альбумин (ЧСА)731
энантиомеры как несовместимые
зеркальные отображения друг друга 704—
705
Хиральные селекторы 703, 711, 718; см.
также Хиральные неподвижные фазы
связывание с поверхностью сорбента
714, 715
соответствие анализируемому
веществу 714, 716-717
Хорват Ч. 47-48
Хранение колонок 286
Хроматограмма 42—43, 60
Хроматографические режимы, выбор 102
Хроматография
гель-фильтрационная; см.
Гель-фильтрационная хроматография (ГФХ)
гидрофильных взаимодействий (ГИХ);
см. Хроматография гидрофильных
взаимодействий
гидрофобных взаимодействий (ХГВ);
см. Хроматография гидрофобных
взаимодействий
ионообменная; см. Ионообменная
хроматография (ИОХ)
нормально фазовая; см. Нормально
фазовая хроматография (НФХ)
обращенно-фазовая; см. Обращен-
но-фазовая хроматография (ОФХ)
ОФХ-5 660-661
Противоточная; см. Противоточная
хроматография (ПТХ)
хиральная лигандообменная (ХЛОХ)
751-753
Хроматография гидрофильных
взаимодействий (ГИХ) 425-431
адсорбция и распределение 426—427
градиентное элюирование 493—496
колонки 427-428
оптимизация селективности подбором
растворителя 429—431
преимущества 426
проблемы 431
разработка метода 429—431
углеводов 662—663
удерживание 426—427
Хроматография гидрофобных
взаимодействий (ХГВ) 645-648
((((958 Предметный указатель
антихаотропные соли 647
влияние рН 647
влияние температуры 648
нуклеиновые кислоты, разделение 661
ПАВ 647-648
сорбенты 645—646
Хроматофокусирование, полипептиды
639-640
Хуберт Й. 47
Хуммеля — Дрейера метод 678
Цвет М. С. 47
Цвиттер-ионы; см. Анализируемые
(разделяемые) вещества, амфотерные
Целлобиогидролаза (ЦБГ); см. Хираль-
ные неподвижные фазы, на основе цел-
лобиогидролазы I
Цианофаза 270
в Г ИХ 428
Частицы сорбента; см. также
Неподвижная фаза
гибридные 249, 260-261
диаметр 239, 241, 242
для подвижных фаз с высоким
содержанием воды 261—262
пелликулярные 100—101, 239—240
перфузионные 240, 241
площадь поверхности 239
поверхностно-пористые (с оболочкой)
100-101, 240-241, 249
полимерные 249—250
полностью пористые 239, 240
получение 248—249
получение и конденсация золя сили-
кагеля 248—249
золь-гель-методика получения
частиц силикагеля 248
распыление с высушиванием золя
силикагеля 248
поры; см. Поры частиц сорбента
размер (диаметр) пор частиц сорбента
614-615
размер (диаметр) частиц сорбента
241-246
распределение частиц по размерам
239, 243-245
с контролируемой пористостью
поверхности; см. поверхностно-пористые
сферические и неправильной формы
243-244
типы 239-241
физико-химические данные 281
характеристики 239
электронные микрофотографии 243,
245
Четвертичные соли аммония как ион-
парные реагенты 372—375
Четвертичная структура, полипептиды;
см. Полипептиды, четвертичная
структура
Число теоретических тарелок 71—89; см.
также Колонки, эффективность; Пика
ширина
влияние экспериментальных условий
76
оптимизация 97—98
приблизительная оценка 283
хиральных колонок 707, 718
хорошо упакованной колонки 283
Чувствительность 196; см. также
Детекторы, чувствительность; рН,
чувствительность
низкая 103—104
Шумы 191 194; см. также Общие симп
томы ВЭЖХ-проблем
Шумы базовой линии, см. Шумы,
Устранение неисправностей, шумы
базовой линии
«Щелевая» модель зависимости
селективности от формы молекулы 313—
314
Эквивалентные колонки 272—273, 314—
318
Эквивалентные разделения; см.
Эквивалентные колонки, Корректировка
методики, неэквивалентные колонки
Эксклюзионная хроматография (ЭХ)
668—679; см. также Гель-фильтрация
калибровочный график 669—671
преимущества и ограничения 676
синтетических полимеров (гель-про-
никающая хроматография, ГПХ) 691 —
692
удерживание 669—671
Предметный указатель 959
Экспертные системы для разработки
метода 518
Эксплуатационная квалификация (ЭК);
см. Тесты на пригодность
(работоспособность) системы
Экспресс-разделения, градиентное элю-
ирование 483—485
Электрохимические детекторы; см.
Детекторы, электрохимические
Электростатическое отталкивание 264—
269
хроматография гидрофильных
взаимодействий с электростатическим
отталкиванием (ГИХЕО) 651-652
Элюент; см. Подвижная фаза
Энантиомеров разделение; см. Хираль-
ные разделения
Эндкепирование 259—260
Эпимеры; см. Хиральность,
определения, эпимеры
ЯМР-детекторы; см. Детекторы, ЯМР