/






























































































































Text
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ им. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ»
АНАЛИТИЧЕСКАЯ ХИМИЯ СЕЛЕНА и ТЕЛЛУРА
И. И. Наз аренко, А. Н. Ермаков
в
ИЗДАТЕЛЬСТВО«НАУКА»
Москва 1971
УДК 546.23-^-546.24:543
Серия «Аналитическая химия элементов'»
Главный редактор академик А. П. Виноградов
Редакционная коллегия:
И. П. Алимарин, А. И. Бусев, А. П. Виноградов, А. Н. Ермаков, Ю. А. Золотов, А. В. Карякин, П. Н. Палей, С. Б. Саввин,
И. В. Тананаев, М. П. Волынец (ученый секретарь)
Редактор тома «Аналитическая химия селена и теллура»
член-корреспондент АН СССР Ю. А. Золотов
Адрес редколлегии:
Москва, В-335, Воробьевское шоссе, 47а, Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
2-5-5
БЗ №33-1970 г. № 25
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернад*-ского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Эта серия —«Аналитическая химия элементов»— составит около пятидесяти томов. Потребность в подобного рода издании давно назрела. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий, и теперь стало возможным и необходимым его подытожить. Таким образом, возникло настоящее издание — серия «Аналитическая химия элементов», которое осуществляется впервые. Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы как физические, так и физико-химические и химические излагаются применительно для количественного определения данного элемента, начиная с анализа сырья, далее — типичных полупродуктов производства и, наконец, конечной продукции — металлов и сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей. Монографии содержат обширную библиографию, доведенную до последних лет; они рассчитаны на широкий круг химиков, в первую очередь химиков-
3
аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на химиков-преподавателей и студентов химических высших учебных заведений. К составлению монографий привлечены крупнейшие советские специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии аналитической химии элементов будут выходить самостоятельно по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию, урану, рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, бериллию, редкоземельным элементам и иттрию, никелю, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, селену и теллуру; готовятся к печати монографии по аналитической химии алюминия, нептуния.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
Памяти
нашего дорогого учителя Дмитрия Ивановича
Рябчикова посвящается эта книга
ПРЕДИСЛОВИЕ
В последнее время селен и теллур стали широко применяться в различных областях новой техники — автоматике, телемеханике, радиотехнике, полупроводниковой технике. Повышенный интерес к селену и теллуру способствовал расширению исследований в области неорганической и аналитической химии этих элементов. В советской и зарубежной литературе появилось большое число работ по аналитической химии селена и теллура. Подготавливая к изданию настоящий труд, авторы пытались представить на основании имеющихся литературных данных и своего личного опыта современное состояние аналитической химии этих элементов, обращая особое внимание на наиболее проверенные и чувствительные методы анализа.
Первые две главы посвящены общим вопросам, касающимся распространенности в природе селена и теллура, их применению, описанию основных физических свойств этих элементов и их химических соединений. В главе «Количественные методы определения» описаны фотометрические, флуориметрические, каталитические, электрохимические, спектральные, атомно-абсорбционные, пламенно-фотометрические, рентгенофлуоресцентные и нейтронно-активационные методы определения селена и теллура. Для анализа различных природных и промышленных объектов указаны специфические методы разложения и наиболее рациональные схемы проведения анализа материалов данного типа.
Авторы считают своим приятным долгом выразить глубокую благодарность рецензентам: академику И. В. Тананаеву, кандидату химических наук, старшему научному сотруднику Б. Н. Ку
5
ликовскому и большому специалисту в области аналитической химии селена и теллура И. А. Блюму. Авторы благодарят также кандидатов технических наук, старших научных сотрудников Ю. И. Беляева, Л. Г. Логинову и Е. И. Зайцева за ценные советы и замечания при написании разделов по физическим методам анализа, а также И. В. Кислову — за большую помощь при подготовке рукописи к печати.
И. И. Назаренко
А. Н. Ермаков
Глава 1
ОБЩИЕ СВЕДЕНИЯ О СЕЛЕНЕ И ТЕЛЛУРЕ
ПОЛОЖЕНИЕ в ПЕРИОДИ 4ЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВ
Селен и теллур принадлежат к VI группе периодической системы элементов Д. И. Менделеева и располагаются между серой —типичным металлоидом — и полонием — металлом. По своим химическим свойствам и условиям нахождения в природе селен во многом сходен с теллуром. Исследования последних лет, связанные в особенности с использованием полупроводников в ряде отраслей новой техники, выявили природу сходства селена и теллура и вместе с тем ряд существенных различий в их свойствах.
Распределение электронов в атомах элементов главной подгруппы VI группы дано в табл. 1.
Таблица 1
Распределение электронов по уровням у атомов 0,S,Se,Te,Po [203]
номер Главные уровни
к L м N о р
Ь я Подуровни
S Ф <5 Атом “1 2s 2р 3s Зр 3d 4s 4р id 4f 5s 5р 5d 5f 6s 6р
О S Se Те Ро 8 16 34 52 84 2 2 2 2 2 2 4 lift 1П1Ш1 2 6 2 6 2 6 2 6 2 4 ntnmui 2 6 10 2 6 10 2 6 10 2 4 им 2 6 10 2 6 1014 2 4 ini iitimi 2 6 10 2 4 1Ж1
7
Таким образом, все валентные электроны у атомов селена и теллура в невозбужденном состоянии размещаются на s- и р-элект-ронных уровнях.
С увеличением радиуса атома (для Se г = 1, 17 А, для Те г = = 1,37 А) уменьшается сродство к электрону, ослабевают неметаллические и усиливаются металлические свойства. Структура внешних электронных слоев атомов селена и теллура подобна структуре атома серы. Эти элементы обладают преимущественно металлоидными свойствами и имеют максимальную отрицательную валентность, равную двум. Селен и теллур — менее активные металлоиды, чем сера, а также стоящие с ними в одном горизонтальном ряду галоиды. Наличие во внешнем слое шести электронов определяет максимальную положительную валентность, равную шести. Однако наиболее устойчивы соединения, в которых селен и теллур положительно четырехвалентны.
Последовательные ионизационные потенциалы атомов селена и теллура приведены в табл. 2.
Таблица 2
Ионизационные потенциалы (в) атомов селена и теллура [267]
Элемент Ионизационные потенциалы отрыва электронов
1-го 2-го 3-го 4-го 5-го 6-го ,
Se 9,75 21,5 32,0 42,9 68,3 81,7
Те 9,01 18,6 31,0 38,0 60,0 72,0
Для селена и теллура известны также соединения, в которых они существуют в виде двухвалентных положительно заряженных ионов. Особенно интересны комплексные соединения двухвалентного теллура. Состояние положительно заряженного двухвалентного селена менее характерно.
Количество неспаренных электронов определяет число ковалентных связей, которое может образовать атом. У селена и теллура оно равно двум. Благодаря этим связям атомы могут соединяться в двухатомные или кольцевые молекулы. Это объясняет существование большого числа аллотропных модификаций.
ВАЛЕНТНЫЕ СОСТОЯНИЯ
Существование селена и теллура в форме двухвалентных отрицательно заряженных ионов обусловлено следующими реакциями:
Se° -J- 2е = Se®~,
Те0 + 2ё = Те®-.
8
Сродство атома селена и теллура к двум электронам составляет около 100 ккал!г-атом. Селен и теллур образуют селениды и теллуриды, аналогичные сульфидам. Отрицательно заряженные двухвалентные ионы селена и теллура — сильные восстановители. Окисляясь, они переходят в элементное состояние; при дальнейшем окислении образуются соединения четырехвалентных элементов. В то время как для соединений четырехвалентной серы восстановительные свойства характернее окислительных, для производных Se (IV) и Те (IV) справедливо обратное: они довольно легко восстанавливаются до элементных селена и теллура. Напротив, перевод четырехвалентных селена и теллура в шестивалентное состояние может быть осуществлен только действием сильных окислителей. Соединения шестивалентных селена и теллура являются сильными окислителями, особенно в кислой среде.
При частичном восстановлении четырехвалентного теллура в присутствии некоторых комплексообразующих веществ (например, тиомочевины) теллур переходит в двухвалентное состояние:
Те (IV) + 2е= Те (II).
Для двухвалентного теллура известны многочисленные комплексные соединения с тиомочевиной и ее производными, с тиосульфатом и некоторыми другими лигандами. Соединения двухвалентного селена, как уже говорилось, гораздо менее устойчивы.
Потенциалы окисления—восстановления приведены ниже [2031:
Кислые растворы
-0.408 +0,748 +1,158
H2Se----Se----SeO®"----SeO®";
—0,728 +0,5298 +1,028
H2Te----Те----TeO2-----HeTeOe крист.
Щелочные растворы
+0,928 +0,3008 +0,058
Se3-----Se----SeO®"----SeO®";
1,148 -0,578 „ +0,48
Те3"----Те----TeO|"----ТеОД.
ИЗОТОПНЫЙ СОСТАВ СЕЛЕНА И ТЕЛЛУРА
Методами масс-спектроскопии было доказано существование шести устойчивых изотопов у селена и восьми стабильных изотопов — у теллура [496]. Некоторые основные характеристики устойчивых и искусственно полученных изотопов для селена и теллура представлены в табл. 3 и 4.
Под действием протонов высоких энергий получены изотопы Те116 [332], а также Те127 из J1®7.
9
Таблица 3
Изотопы теллура [15]
Изотоп Распространенность в природе, % Период полураспада Изотоп Распространенность в природе, % Период полураспада
Те120 Те121 ре121т Те133 Те123 уе123т Те134 Те123 ye12sm Те126 0,089 2,46 0,89 4,74 7,03 18,72 17 дней 154 дня 104 дня 58 дней Те127 Те127т Те128 Те129 Те130 Те131 •ре131т Те133 Те133т 31,75 34,27 9,35 часа 105 дней 72 мин. 41 день 24,8 мин. 30 час. 2 мин. 53 мин.
• А. И. Алиев и др. «Яде анализа». М., Атомиздат, 196 риофизнческие ). Изотопы с константы для елена [15] нейтронного Tat активационного л и ц а 4
Изотоп Распространенность в природе, % Период полураспада Изотоп Распространенность в природе, % Период пол ураспада
Se73 Se74 Se75 Se76 Se77 Se”m Se78 Se79 0,87 9,02 7,58 23,52 7,1 часа 120,4 дня 17,5 сек. 6,5.104 лет Se7em Se89 Se81 Se8im Se82 Se83/n Se83 49,82 9,19 3,91 мин. 18,6 мин. 62 мин. 69 сек. 25 мин.
Приведенные в табл. 3 и 4 данные показывают, что у селена и теллура преимущественно преобладают более тяжелые изотопы.
СОДЕРЖАНИЕ В ЗЕМНОЙ КОРЕ. ГЛАВНЕЙШИЕ МИНЕРАЛЫ И РУДЫ
В 1798 г. Клапрот опубликовал данные о новом элементе, который он назвал теллуром (по латыни «теллус»—«земля»). Теллур был выделен из золотых руд и первоначально принимался за сурьму и висмут 142].
10
Таблица 5
Минералы селена и теллура [335]
Минералы селена <u S X Минералы теллура S X
08 CO £
Q,
название формула 8 название формула %
Селен самород- Se 100 Теллур само- Те 100
ный роднын
Селенотеллур TeSe 30 Селенотеллур TeSe 70
Селенистый H2Se 98 Теллуристый H2Se 97
водород водород
Парагуанахуа- BiaSeS 24 Вулканит CuTe 66
ТИТ
Лайтакаринт Bi4Se2S 16 Риккардит Cu7Ter, 60
Гуанахуатит BlgSeg 24 Вейссит Cu2_xTe 50
Клокманннт CuSe 56 Мелонит NiTe2 80
Берцелианит Cu2Se 40 Монтбрейит AuaTea 62
Умангит CusSe2 54 Калаверит AuTea 57
Науманнит AgaSe 27 Креннерит (Au,Ag)Tea 56
р-Науманнит AgaSe 27 Сильванит (Au,Ag)Te4 62
Агвиларит Ag4SeS 6 Мутманнит (Au.Ag)Te Пере-меи-
Кру кс ит (Cu,Tl,Ag)2Se 32 Петцит (Ag3Au)Te2 33
Эвкайрит CuAgSe 30 Гессит AgaTe 62
Эскеборнит Fe3CuSe4 52 Эмпрессит Ag5_xTe3 55
Тиррелит (Cu,Co,Ni)4Se4 55 Теллурэвисму- В^гТез 48
тит
Штиллеит ZnSe 54 Верлит 28
Кадмоселит CdSe 41 Хедлейит] Bi7Te3 20
Тиманнит HgSe 28 Тетрадимит Bi2Te2S 36
Клаусталит PbSe 27 Чикловаит B12TGS2 20
Фребольдит Трэгталит Хастит CoSe CoSe2 CoSea 57 72 72 Жозеит-А Жозеит-Б Грюнлингит Bi4TeS 12 20 Перемен-
Борнхардит Co3Se4 64 Оруэтит BieTeSi 6
Блокит NiSea 68 Колорадоит HgTe 39
Ферроселит FeSe2 73 Алтаит PbTe 38
Ашавалит FeSe 58 Фробергит FeTe2 82
Селенид палладия PdSe — Котульскит РсЦТе.В!)^ 44
Вейбуллит PbBi2(S,Se)4 13 Мончеит (Pt,Pd)(Te,Bi)2 33
Платинит PbBi2(S,Se)3 18 Майченерит (Pd0,75Pt0,25)TeBi 29-37
Виттит Pb5Bi6(S,Se)4 8 Нагиагит Pb5Au(TeSb)4S5-8 18
Джеромит AS(S,Se)2 7,5 Голдфилдит Cu6Sb2(S,Te)8 17
11
Таблица 5 (окончание)
Минералы селена Содержание, % Минералы теллура Содержание, %
название формула. название формула
Селенолит SeOa 71 Арсенотеллурит Т esAs^S? 40
Керстеиит PbSeO4-2H2O Перемен- Теллурит ТеО2 80
ное
Молибдоменит PbSeOs » Парателлурит ТеО2 80
Халькоменит CuSeOs-2H3O 49 Теллурат свии- РЬТеО4 Перемен-
ца ное
Селенит железа Данхемит РЬТеО3 »
Селенит ртути Монтанит РЬ2ТеО4(ОН)4 26
Альфельдит NiSeO3-2H2O Перемен- Тейиеит Cu(Te,S)O3-2H2O 48
ное
Кобальтоменит CoSeOs • nH2O Эммонсит Fe2(TeO3)s-2H3O 70
Маккейит Fe2(TeOa);i-rcH2O —
Блекеит Fes(TeO3)3 Пере-
ное
Магнолит Hg2TeO4 17
Селен был открыт в 1817 г. Берцеллиусом в шлаках свинцовых камер сернокислотного завода. Новый элемент по свойствам был очень похож на теллур. Этот элемент был назван в честь Луны — спутника Земли —«селене».
Селен и теллур относятся к группе рассеянных элементов. В большинстве случаев они не образуют самостоятельных месторождений, а входят как примеси в руды других металлов. Главным рудным элементом, с которым селен и теллур связаны близостью свойств, является сера.
Количественные соотношения S : Se : Те в земной коре, по Ферсману [403], близки 60000 : 100 : 1. По Гольдшмидту [123], соотношение S : Se в рудных месторождениях меняется от 400 : 1 до 250000 : 1 . В осадочных железных рудах эти отношения равны 100000 : 1 , в морской воде — 232 : 1
По последним данным, кларк селена равен 5-Ю~6 [92], кларк теллура — 1 • 10'7 [335, 336]. В водах морей и океанов содержание селена и теллура ниже кларковых — 3- 1Q7. Значительные примеси селена обнаружены в метеоритах: от 0,0001 до 0,23%. Теллур в них присутствует в виде следов.
Селен в земной коре находится как в рассеянной форме, фиксируясь в сульфидных минералах, так и в виде собственных минералов. В настоящее время известно около 40 собственных минералов, селена, относящихся к следующим классам: самородные элементы, селениды, окислы, селениты и селенаты. Наиболее распространенные собственные минералы — селениды (табл. 5).
12
В отличие от серы, образующей минералы более чем с 40 химическими элементами, селен соединяется с относительно небольшим числом элементов. Первичные природные соединения он образует преимущественно с элементами, характеризующимися высокими порядковыми номерами: Pb, Hg, Bi, Ag, Си, Со, Fe, Tl, Ni, Zn, Cd. Тот же примерно комплекс элементов характерен и для гипергенных минералов селена, в то время как у серы в гипергенных процессах характерна связь со щелочными и щелочноземельными элементами.
Помимо собственных минералов, селен входит в большое число минералов, преимущественно сульфидов (табл. 6).Для селена характерен изоморфизм с серой, а также теллуром и мышьяком, что обусловлено близостью химических и кристаллохимических свойств этих элементов.
Таблица 6
Содержание селена и теллура (в %) в некоторых минералах-носителях
Минерал Se Те
Галенит 0,1—20,0 0,0—0,37
Молибденит 0,0—0,1 0,04
Сера вулканическая 0,1-5,18 0,18
Халькопирит 0,0-0,1 0,05
Пирит 0,0—3,0 0,10—0,0002
Пирротин 0,0001-0,0059 0,0056
Сфалерит 0,0001—0,012 0,016
Висмутин 0,015-8,8 0,53
Антимонит 0,0—0,028 —
Урановая смолка 0,039 —
Арсенопирит 0,0001—0,0144 0,0—0,205
Киноварь 0,0—0,424 Не обнаружен
Блеклые руды Не обнаружен —
Магнетит 0,0—0,0003 Следы
Гидроокислы железа 0,0002—0,023 —
Сера 0,19 —
Касситерит — 1,42
Наличие или отсутствие серы в значительной степени определяет рассеяние или концентрирование селена в природных процессах. В магматических породах селен всегда изоморфно замещает серу в сульфидных минералах. Встречаются сульфиды с очень высоким содержанием селена. Значительные количества селена (до 5,18% и выше) наблюдаются в вулканической сере.
13
Отчетливо выявляется вторая после серы линия родства селена с медью, накопление которой в рудах сопровождается концентрированием селена. Представляет также интерес геохимическая ассоциация селен—уран—медь—кобальт.
При окислении сульфидов селен, обладая меньшим сродством к кислороду, или совсем не окисляется, образуя в сульфатных жилах селениды тяжелых металлов, или окисляется до селенитов, которые легко восстанавливаются до элементного селена. В окислительной среде селен частично переходит в растворимые в воде селенаты и селениты и переносится в такой форме грунтовыми водами. Селенаты сильно сорбируются окислами железа и могут концентрироваться в глинах и железистых гидроокисных остатках.
В осадочных месторождениях для селена характерен парагенезис с ураном и ванадием, а иногда и с серебром, молибденом, медью. Решающим фактором в процессах осаждения селена является восстановительная среда, которая в значительной мере создается наличием органического вещества. На породах, содержащих селен в повышенных концентрациях, развиваются почвы, обогащенные селеном. В зависимости от концентрации селена в почвообразу-ющих коренных породах, а также от окислительно-восстановительных условий почв и общих особенностей климата содержание селена в почвах меняется. А. П. Виноградов [92] выделил почвы нормальные (содержание Se составляло 1 • 10~в%) и почвы с высоким содержанием селена (ц-10“3%). В почвах под воздействием живых организмов и атмосферных агентов изменяются формы вхождения селена. В почвах с повышенным содержанием селен присутствует в виде селенитов и селенатов. Из почв селен усваивается многими растениями, в которых он замещает серу белков.
Теллур в земной коре находится как в рассеянной форме, фиксируясь в сульфидных минералах, так и в виде собственных минералов. Повышенные концентрации теллура характерны для золоторудных месторождений. В ряде случаев установлены корреляционные связи теллура с золотом.
В настоящее время известно более сорока минералов теллура, относящихся к следующим классам: самородные элементы, интерметаллические соединения, теллуриды, окислы, теллуриты и тел-лураты. Большинство минералов принадлежит к теллуридам (см. табл. 5). Вторичные минералы теллур образует с Bi, TI, Hg, Pt, Pd, Au, Ag, Sb, Cu, Ni, Fe. Щербина [455] установил следующий ряд возрастающего сродства этих металлов к теллуру:
Си—Pb—Ni— Bi—Hg—Ag—Au.
Содержание теллура в сульфидных минералах ниже, чем содержание селена, оно редко достигает десятых долей процента. Часто в сульфидах обнаруживают мельчайшие вкрапления теллуридов.
14
Таким образом, изоморфизм теллура и серы проявлен гораздо меньше, чем изоморфизм селена и серы.
В золоторудных месторождениях широко распространены минералы теллура, преимущественно теллуриды золота. При окислении сульфидов и теллуридов теллур окисляется до элементного, а также до Te(IV)H,Te(VI). Радиус Те (IV) (0,89 А) и Te(VI)(0,56A) более чем в два раза превосходит радиус S (IV) (0,32 А) и S (VI) (0,29 А). При окислении сульфидных минералов теллур почти полностью соосаждается с гидроокислами железа. В отличие от селена в осадочных породах теллур не дает повышенных концентраций, в почвах он практически отсутствует.
Наиболее крупными запасами селена и теллура являются месторождения медно-цинковых колчеданных руд, а также полиметаллических, в которых селен и теллур содержатся от десятитысячных до тысячных долей процента, а в некоторых месторождениях — до сотых процента. В медно-колчеданных рудах, как правило, преобладает селен. Отношение селена к теллуру в них колеблется от 3:1 до 2 : 1.
В полиметаллических рудах отношение Se к Те составляет от 1 : 1 до 1 : 2.
Перспективными являются селеноносные осадочные месторождения с ураном, ванадием и др. Заслуживают внимания богатые теллуром золоторудные месторождения и селен- и теллурсодержащие медно-молибденовые месторождения.
ПРОМЫШЛЕННЫЕ КОНЦЕНТРАТЫ
Медные концентраты, получающиеся при обогащении медноцинковых, а также полиметаллических, медно-молибденовых, мед-но-никелевых и других руд, содержат 10—25% общего количества селена и теллура в исходных рудах. Цинковые концентраты обычно бедны селеном и теллуром. Свинцовые концентраты содержат 0,003—0,01% Se и Те; в них содержится 20—25% селена и 30—35% теллура от общего количества их в исходных рудах.
В промышленности главным источником получения селена и теллура являются анодные шламы медьэлектролитных заводов, а также шламы сернокислотного и целлюлознобумажного производства. Анодные шламы получаются в процессе электролитического рафинирования черновой меди на медеплавильных заводах при переработке медных и медно-никелевых концентратов. Анодный шлам, содержащий селен и теллур, накапливается в виде тонкого порошка на дне электролитических ванн и периодически оттуда отгружается. В настоящее время анодные шламы — основное селеновое и теллуровое сырье, из которого в США производится более 90% селена [179]. Селен и теллур находятся в шламе в виде простых или сложных селенидов и теллуридов.
15
Примерный состав анодного шлама (в %) приведен ниже:
Медь......... 10—27 Сурьма...........0,2—3,6
Золото.........0,8—4,2 Мышьяк...........0,1—0,6
Серебро........ 30—50 Свинец................. 3—11,0
Селен.......... 3—14 Железо...........0,3—0,5
Теллур.........0,3—3,0 Никель...........0,2—0,9
Висмут.........0,1—1,5 Кремний........... 2—4
Селен и теллур извлекаются из анодного шлама попутно с серебром и золотом.
В ряде стран при переработке медных концентратов получают черновую медь, содержащую повышенные количества селена и теллура. Такая медь также служит сырьем для дальнейшего получения чистых селена и теллура.
Селен и теллур поступают в систему сернокислотного производства с газами от обжига серы или железного колчедана. При сжигании селенсодержащего пирита селен окисляется кислородом воздуха до SeO2, который восстанавливается сернистым газом до элементного селена. В процессе очистки сернистого газа от примесей селен осаждается вместе с пылью в отстойниках и холодильниках первых и вторых промывных башен, образуя селенсодержащие шламы. При дальнейшей очистке SeO2 на мокрых электрофильтрах селен осаждается в конденсаторах в количестве, которое меняется в зависимости от режима работы промывных отделений.
В шламах сернокислотного производства селен и теллур находятся почти целиком в элементном состоянии.
Примерный состав шлама (в %) приведен ниже:
Селен........0,9 —63,7 Свинец .... 4,82—30,4
Сера.............5—22 Железо..............8
Теллур.........0,2 —15 Вода..........0,2 —13,55
Мышьяк .... 0,3 —55,3
ПРИМЕНЕНИЕ СЕЛЕНА И ТЕЛЛУРА
Еще совсем недавно области применения селена и теллура ограничивались главным образом стекольной, резиновой и химической, а для теллура — радиохимической отраслями промышленности [352, 4711. В последние годы эти элементы нашли широкое применение в металлургии и в новой области техники — в полупроводниковой технике [203, 430, 431].
Селен, теллур и их соединения (селениды и теллуриды) в настоящее время широко применяются в полупроводниковой промышленности и электронике при изготовлении полупроводниковых
16
термоэлементов, фотоэлементов, датчиков Холла и других устройств для автоматики, радиоэлектроники, телемеханики. Селеновые электрические выпрямители, появившиеся в годы второй мировой войны, быстро вытеснили медные выпрямители. Увеличивающаяся потребность в полупроводниковых материалах заставляет сокращать потребление селена другими отраслями промышленности. Проводятся работы по использованию в качестве полупроводников различных соединений теллура: теллуридов кадмия, индия, галлия, сульфида теллура и др.
Фотоэлектрические свойства селена позволяют широко использовать его при изготовлении фотоэлементов, в измерительной аппаратуре, фототелеграфии, телевидении, сигнализации. Теллур применяется при изготовлении детекторов и термопар.
В металлургии селен и теллур находят применение как добавки к сплавам, улучшающие их качество. Так, добавка 0,30—0,35% селена к нержавеющей стали и сплавам меди улучшает их обрабатываемость, повышает устойчивость; небольшие присадки теллура (до 0,1—0,5%) резко усиливают антикоррозионные свойства сплавов меди, свинца, улучшают структуру и облегчают механическую обработку стали и чугуна. Добавки теллурида свинца (до 0,5% Те) к кислотоупорным материалам используются в сернокислотной промышленности, повышая вдвое срок службы аппаратуры.
В химической промышленности селен и теллур употребляются как катализаторы при органическом синтезе, в процессе переработки нефти. Высоко ценятся селеновые красители и антикоррозийные покрытия (в сочетании с кадмием, хромом, цинком), обладающие устойчивостью к теплу, свету, воздействию влаги и т. д. Селен является энергичным антиокислителем, что используется при изготовлении типографской краски, минеральных и растительных масел, смазочных материалов.
Некоторые соединения селена ядовиты и применяются в сельском хозяйстве для борьбы с вредителями. Известны соединения теллура, обладающие бактерицидными свойствами, используемыми в медицине.
Селен и теллур употребляются в качестве добавок к резине (0,1—2%) для повышения износоустойчивости, увеличения эластичности и теплостойкости. Примеси селена ускоряют процесс вулканизации.
Селен и теллур используются в стекольной промышленности для улучшения окраски стекла.
Обычной примесью в получаемом промышленностью селене является теллур, а в теллуре — селен. Кроме того, в них обычно присутствуют серебро, сера и иногда углерод. Возможны следы щелочи. Высокая степень чистоты препаратов селена и теллура является необходимым условием для использования их в полупроводниковой промышленности.
17
ТОКСИЧНОСТЬ СЕЛЕНА, ТЕЛЛУРА И ИХ СОЕДИНЕНИЙ
Селен и его соединения — очень ядовитые вещества, действующие на различные органы. Незначительные количества селена и его производных вызывают разрушение дыхательных путей, насморк и головную боль. При попадании на кожу соединения селена дают воспаление или сыпь. Селенистая кислота при попадании в организм вызывает рвоту, судороги, возможен паралич нервной системы. Галогениды селена вызывают удушье и расстройство сердечной деятельности.
Наиболее ядовитыми являются селенистый водород и теллуристый водород, двуокись селена, двуокись теллура, галогениды селена и соединения селена с тяжелыми элементами.
Первым признаком отравления селеном и теллуром является чесночный запах выдыхаемого воздуха. Известны случаи хронических отравлений соединениями селена и теллура. Отравления теллуром наблюдаются при концентрации в воздухе соединений теллура 0, 00001—0,0005 мг/л. Предельно допустимая концентрация теллура в воздухе 0,00001 мг/л. Предельно допустимая концентрация двуокиси селена 0,0003 мг/л.
При отравлении селеном или теллуром и их производными пострадавшему необходимо принять аскорбиновую кислоту, сделать внутривенное вливание раствора гипосульфита натрия или сыворотки с глюкозой.
Глава 2
ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА СЕЛЕНА И ТЕЛЛУРА И ИХ СОЕДИНЕНИЙ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Для элементного селена характерна способность изменять свое строение в зависимости от внешних условий. Это свойство является причиной существования большого числа аллотропных модификаций этого элемента. Основные аллотропные модификации селена можно свести к трем формам, обладающим различной внутренней структурой. Самой устойчивой из них является серый гексагональный селен. Две другие формы по отношению к этой метастабильны. Из них красный моноклинный селен имеет две кристаллические разновидности — Sea и Sep. Третья форма — аморфный селен — может быть порошкообразной или стекловидной.
При обычных температурах метастабильные формы селена в стабильную форму практически не переходят, но термодинамически устойчива только гексагональная модификация. На рис. 1 приведена диаграмма состояния селена по Саундерсу [1046]. Кристаллы гексагональной модификации селена состоят из бесконечных цепочек. Определение молекулярного веса селена указывает на формулу Se8.
Теллур представляет собой вещество серебристо-бел ого цвета с металлическим блеском. В твердом состоянии теллур, подобно селену, состоит из длинных спиральных цепочечных молекул. Теллур кристаллизуется в гексагональной решетке. Раньше
Рис. 1. Давление пара селена (р, мм рт. ст.) над различными его модификациями
19
считалось, что существует еще аморфная модификация. Однако методами рентгенографии было установлено, что «аморфный» теллур состоит из тонких кристалликов гексагонального теллура. Некоторые физические константы селена и теллура представлены в табл. 7.
Таблица 7
Основные физические свойства селена и теллура [203]
Свойства Селен Теллур
Порядковый номер 34 52
Атомная масса 78,96 127,60
Плотность, г[см3 4,79* 6,33
Температура плавления, °C 217 449,5—452
Температура кипения, °C 685,4 990,1
Атомный радиус, нм 0,117 0,137
Твердость, отн. ед. 2* 2,3
Электроотрицательность, отн. ед. (Li = 1) 2,4 2,1
Ионный радиус, нм
Э2’ 0,198 0,222
э4+ 0,069 0,089
эв+ 0,035 0,056
Скрытая теплота плавления, дж/г^кал/г) 6,91 (16,5) 133,9(32)
Теплота парообразования, дж)г 272,98 (65,2) 447,98(107)
Т еплоемкостьСтв, кдж/кг-град{кал1кг-град) 0,318(0,076) 0,201 (0,0481)
Теплоемкость Сж, кдж/кг-град (кал/кг
град) 0,494(0,118) 0,327(0,0783)
Удельная теплоемкость, дж!г-град 0,360(0,086)* 0,2022 (0,0483)
Теплопроводность, вт[м-град 0,293- 0,766 1,674
* Гексагональная модификация.
♦♦ Кристаллический.
Селен в темноте очень слабо проводит электрический ток. На свету электропроводность его возрастает примерно в 1000 раз,-в темноте снова понижается до первоначальной величины. На этом свойстве основано его применение в качестве фотосопротивлений. На электропроводность и фоточувствительность селена сильно влияют примеси других элементов, особенно сурьмы и теллура.
ПОЛУПРОВОДНИКОВЫЕ СВОЙСТВА [430 431]
Многие из селенидов и теллуридов являются полупроводниками. Полупроводниковые свойства обнаруживаются у тех селенидов и теллуридов, которые имеют смешанный характер сил связи, то есть занимают промежуточное положение между соединениями
20
с ионными и ковалентными типами связи. Среди теллуридов встречаются соединения, обладающие как электронной (n-типа), так и дырочной (р-типа) проводимостью, а в некоторых случаях — и смешанной проводимостью. Для теллуридов ширина запрещенной зоны колеблется в широких пределах — от 0,02 до 30 эв — и зависит от структуры кристаллов и их чистоты.
Почти во всех случаях ширина запрещенной зоны в пределах группы изменяется обратно пропорционально атомному весу металла и сумме атомных номеров. Наблюдается прямолинейная зависимость между температурой плавления металла и суммой атомных номеров.
ОСНОВНЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ
По химическим свойствам селен и теллур в общем похожи на серу. Из металлоидов они наиболее энергично взаимодействуют с фтором и хлором, ас кислородом соединяются лишь после предварительного нагревания. При этом теллур образует двуокись ТеО2; селен может образовывать несколько окислов: SeO, SeO2, Se2O3 и SeO3. Из них наиболее устойчив SeO2. С газообразным водородом частично реагирует при повышенных температурах только селен (до образования H2Se), теллур не реагирует. Со многими металлами селен и теллур дают при нагревании селениды и теллуриды. Теллур уже в обычных условиях медленно взаимодействует с водой по схеме:
Те + 2Н2О = ТеО2 + 2Н2.
При нагревании с водой реагирует и аморфный селен. Мелкораздробленные Se и Те растворяются в холодной концентрированной серной кислоте с образованием зеленой (Se) или красной (Те) жидкости, содержащей SeSO3 и TeSO3.
Разбавленные соляная и серная кислоты на селен не действуют. В смеси азотной и соляной кислот селен растворяется с образованием селенистой кислоты.
При окислении теллура раствором азотной кислоты в зависимости от условий могут быть получены либо двуокись теллура, либо основной нитрат теллура—4TeO2-N2O5-H2O.
В концентрированной HNO3 проходит реакция
2Те + 9HNO3 = Te2O3OHNO3 + 8NO2 + 4Н2О;
Te2OsOHNO3 = 2ТеО2 + HNO3.
В разбавленной HNO3 образуются селениты и теллуриты:
Те + 4НЫОз + Н2О = ЗН2ТеОз + 4NO;
Se + 4HNO3 + Н2О = ЗНгЗеОз + 4NO.
21
В концентрированных щелочах теллур и селен подвергаются реакции диспропорционирования:
нагревание
ЗТе + 6КОН ;------* К2ТеО3 + 2К2Те + ЗН2О.
охлаждение
При комнатной температуре и при 60—70° С селен и теллур в NaOH и КОН растворяются очень незначительно. Хлор и бром окисляют теллур и • селен:
Те + 2С12 = ТеСЦ.
При избытке хлора SeCl4 может также окисляться до шестивалентного состояния. Хлорноватой кислотой теллур окисляется до шестивалентного по схеме:
5Те + 6НС10з + 12Н2О = 5Н6ТеО6 + ЗС12.
При действии разбавленных кислот на селениды и теллуриды могут быть получены селеноводород H2Se и теллуроводород Н2Те. Водные растворы их показывают кислую реакцию:
X2H2Se = 1 •10-7 K1H2se = l-10’4; Х1НгТе = 2-10Л
где К — константы диссоциации кислот.
Под действием кислорода воздуха селениды и теллуриды окрашиваются в красноватый цвет вследствие образования аналогичных полисульфидам полителлуридов и полиселенидов.
H2Se и Н2Те медленно окисляются кислородом воздуха до двуокисей SeO2 и ТеО2.
Двуокись селена SeO2 — белое кристаллическое вещество, хорошо растворяется в воде с образованием селенистой кислоты H2SeO3 (Ki = 2-IO'3; К2 = 5• IO-9).
Двуокись теллура ТеО2 плохо растворима в воде. При растворении ТеО2 в щелочах образуются теллуриты — соли теллуристой кислоты. Теллуристая кислота в индивидуальном состоянии не получена, так как при выделении ее из раствора происходит частичное отщепление воды с образованием гидрата хТеО2-г/Н2О, который полностью обезвоживается уже при слабом нагревании. Ее кислотная функция (Ki = 3- 10-в) выражена несколько сильнее основной. При растворении ТеО2 в концентрированных кислотах происходит образование солей:
ТеО2 + 4HJ TeJ4 + 2Н2О.
Помимо галогенидов, были получены также основные сульфаты и нитраты. По растворимости селениты в общем сходны с соответствующими сульфитами, а из теллуритов хорошо растворимы в воде только производные наиболее активных одновалентных металлов (Na, К).
22
Селениты и теллуриты довольно легко восстанавливаются до элементных селена и теллура. Окисление селенитов и теллуритов до селенатов и теллуратов возможно только при взаимодействии с сильными окислителями.
Селеновая и теллуровая кислоты хорошо растворимы в воде. Селеновая кислота по силе приблизительно равна серной, тогда как теллуровая является слабой кислотой. Селенаты по свойствам похожи на сульфаты, в то время как теллураты сильно отличаются от последних. В воде растворимы только теллураты наиболее активных одновалентных металлов. Известны соли состава AgeTeOe, отвечающие ортотеллуровой кислоте НвТеОв.
Селеновая и теллуровая кислоты являются сильными окислителями; например, с НО взаимодействуют по схеме:
кислая среда
H2SeO4 + 2НС1 ,--------* HjSeOs + Cl2 + Н2О.
щелочная среда
SeO3 — белое кристаллическое вещество, хорошо растворимое в воде; ТеО3 — желтый кристаллический порошок, в воде почти нерастворим. Сильные щелочи взаимодействуют с ТеО3 до образования теллуратов.
Галоидные соединения селена и теллура могут быть получены путем взаимодействия элементов. Известны следующие галогениды: SeFe, SeF4, SeCl4, Se2Cl2, SeBr4, Se2Br2, TeF4, TeCl4, TeCl2, TeBr4, TeBr2, TeJ4, а также оксигалогениды — SeOCl2, SeOF2, SeOBr2, Te3O2F14.
Галогениды селена похожи по свойствам на соответствующие производные серы. Галогениды теллура резко отличаются от соответствующих производных серы. Для галогенидов характерно образование ацидокомплексных соединений с галоидоводородными кислотами и, особенно, некоторыми их солями. Обычно они отвечают общей формуле: Ме2ЭХв.
Более подробная характеристика свойств основных неорганических соединений селена и теллура дана в книге А. А. Кудрявцева «Химия и технология селена и теллура» [203], а также в монографии К- В. Бэгналла [506] «Химия селена, теллура и полония» и справочнике Р. С. Брестида [554]. Общие сведения о свойствах селена и теллура, запасах в рудах, технологии, областях применения приведены в книге А. М. Ланша «Селен и теллур» [865].
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
Если простым неорганическим соединениям селена и теллура посвящено несколько монографий, то многочисленные исследования комплексных соединений селена и теллура впервые систематизированы в небольшом обзоре [328]. В настоящей главе материал о комплексных соединениях теллура и селена значительно допол-
23
нен работами, появившимися в отечественной и зарубежной литературе за последние пять лет.
В некоторых соединениях селен и теллур резко различаются по свойствам — теллур проявляет себя как комплексообразователь в большой группе соединений, тогда как для селена известны лишь отдельные соединения этого типа. Разница в устойчивости комплексов теллура и селена, особенно ацидокомплексов, позволяет в ряде методов проводить количественное разделение этой близкой по свойствам пары элементов и селективное определение одного элемента в присутствии другого.
Образование тройных комплексов — соединений ацидокомплексов селена и теллура с органическими основаниями, например с основными красителями и аминами, успешно используется при разработке селективных методов определения теллура и отделения селена и теллура от других элементов. Экстракция аминами сложных ассоциатов, в состав которых входят ацидокомплексы, в настоящее время используется в технологических схемах очистки селена и теллура от примесей.
Особый интерес представляют соединения селена с ароматическими о-диаминами. Реакция селена с 3,3'-диаминобензидином была впервые предложена в 1946 г. Это одна из наиболее чувствительных и селективных реакций из всех известных в неорганическом анализе. Поэтому в последние годы появилось очень много работ по изучению различных аналогов 3,3'-диаминобензидина и исследованию механизма реакции их с селеном.
Было показано, что аналог 3,3'-диаминобензидина — 2,3-ди-иминонафталин — имеет несомненные преимущества: дает значительно большую чувствительность (0,002 мкг/мл), сохраняя в то же время высокую селективность по отношению к селену. Для селена разработаны и проверены на практике методы с 2,3-ди-аминонафталином, о-фенилендиамином, 3,3'-диаминобензидином для различных по составу материалов — руд, минералов, биологических образцов, полупроводниковых материалов.
Теллур не образует соединений с о-диаминами.
В поисках чувствительного и селективного реагента на теллур широко была обследована большая группа серусодержащих органических реагентов, а также изучены ацидокомплексы теллура с органическими основаниями.
Большое теоретическое значение имеют исследования комплексных соединений теллура с тиомочевиной и ее производными, поскольку эти соединения по своим свойствам (возможность существования нескольких модификаций и изомерные превращения одной модификации в другую) очень близки соединениям типичных металлов-комплексообразователей — никеля, кобальта, платиноидов.
Интересно, что этот класс соединений образует двухвалентный положительно заряженный ион теллура, что резко отличает -24
его от селена, восстанавливаемого тиомочевиной до элементного состояния. Существует возможность практического использования этой разницы в поведении селена и теллура.
Таким образом, различие в комплексообразующей способности этой пары элементов открывает широкие возможности перед аналитической химией, разрабатывающей специфичные методы разделения и чувствительные методы определения селена и теллура.
Галогеноводсродные кислоты селена и теллура и их соли
Для теллура известны галогеноводородные кислоты (НХ) состава: ТеС14-НС1-5Н2О, TeBr4-HBr-5H2O, TeJ4-HJ-8Н2О, полученные действием соответствующих кислот на ТеС14 [710,9251. Эти соединения образуются в виде игольчатых призм (желтых — для хлоридов, красных — для бромидов, черных — для иодидов). Они очень гигроскопичны и легко расплываются на воздухе, а при нагревании легко теряют молекулу НХ.
При добавлении CsF к раствору TeF« в HF было получено соединение CsTeF5 [1139]. Известны и другие пентафторотеллуриты NH4TeF5-H2O, KTeF5, Ba (TeF5)2-2H2O.
Описаны также оксифториды: TeF4-3TeO2-6H2O, TeF4-TeO2-•2Н2О, TeF4-TeO2-Н2О [925, 10041, для которых предполагается существование комплексного аниона [TeO2F2]2'.
Пентафторотеллуровая кислота HOTeF5 [647 , 648] была получена в виде стекловидных бесцветных кристаллов с т. пл. 40° С. В ИК-спектре наблюдалась сильная полоса ОН-группы при 3670 нм. В водном растворе HOTeF5 является сильной кислотой, быстро гидролизуется с образованием теллуровой кислоты и HF. С КС1 образуются соли KOTeF5.
При взаимодействии Те (ОН)6 с HF отмечена возможность образования ряда фторотеллуровых кислот с отношением F : Те = = 1, 2, 3, 4. Хроматографированием на бумаге эти кислоты были разделены. Синтезированы пиридиновая соль (C5H6N) [TeO(OH)F4l, а также соединение 3C5H5N (НО)4 TeFa-8HF; хроматограммы этой соли идентичны найденным для соединения с соотношением F : : Те = 2. В растворе соли титруется 8 молей HF [842].
Из теллурсодержащего раствора в HJ было выделено соединение с 8-оксихинолином —HTeJ5-20xin-10Н2О [697].
Эйнсли [5031 получил фторидный комплекс теллура с пиридином [C5H5NH] TeF5 и ряд галогеноводородных комплексов с мочевиной [CON2H5]TeX5, где X = Cl, Br, J [501].
Пентахлоротеллуриты были выделены также в форме соединения с мочевиной—(CON2H5) ТеС15.
Устойчивость аниона [ТеХ5]“ в соединениях с мочевиной возрастает от хлоридов к иодидам; хлориды легко превращаются под
25
действием соответствующей НХ в бромиды и иодиды. Фторидных мочевинных комплексов не удалось получить.
В настоящее время известны многочисленные гексагалогено-теллуриты — хлориды, бромиды и иодиды калия, рубидия и цезия [954, 11461, а также хлориды и бромиды аммония и таллия [630, 632, 10951. Возможность получения свободных гексагалогенокислот четырехвалентного Те пока окончательно не доказана. По утверждению Рипан и др. [1022], соединение Н2ТеС16-2Н2О можно получить при растворении свежеосажденной ТеО2 в HCl (1 : I) в виде хорошо образованных желтых кристаллов, которые при взаимодействии с НВт дают Н2ТеВг6. Добровольский [630, 632], Троицкий и Яковлева [478] пытались воспроизвести опыты Рипан [10221, однако гексагалогенокислот теллура им получить не удалось. Добровольский получил аммонийную и таллиевую соли состава (NH4)2 ТеС16, Т12ТеВг6.
По-видимому, устойчивость соединений с ионом [ТеХа]2- в значительной степени зависит от степени поляризации, вызываемой катионом. Сильно поляризующий ион водорода делает неустойчивым анион [ТеХа]2“. Этим объясняется неустойчивость кислых растворов, содержащих окрашенный анион [TeJ5]2- [244, 814, 815]. Вероятно, можно согласиться с Добровольским, который сомневается в возможности выделения в индивидуальном состоянии кислот Н2ТеС1а и Н2ТеВга. Очевидно, по этой же причине не были получены гексагалогенотеллуриты лития и натрия — катионов, также обладающих сильным поляризующим действием. С другой стороны, для больших, слабополяризующих катионов — К+ NH4, Tl+, Rb+ и Cs+ — получены вполне устойчивые гексахлоро-, бромо- и иодотеллуриты [954, 1095, 11461.
Из галогеноводородных кислот селена была выделена в чистом состоянии H2SeBra [729, 927]. Эта кислота растворяется в воде без разложения, но на воздухе разлагается с выделением брома и коллоидного селена. Аналогичная гексахлороселенистая кислота не получена. Соединения SeOa-2HCl и SeO2-2HBr идентичны гидратам SeOCl2-H2O и SeOBr2-H2O [9481.
Гексахлорселениты калия и аммония были получены в виде желтых кристаллических веществ, легко разлагающихся на воздухе [983]. Известны также гексабромоселениты (NH4)2 SeBra, K2SeBre и Rb2SeBra [734, 9541. Изучение кристаллической структуры соединений (NH4)2 SeBra (I), (NH4)2 Se (SO)2C14 (II) и (NH4)2 Те (SO)2 Cl4 (III) показало, что соединения II и III существуют в двух изомерных формах — ромбической и моноклинной [694].
В растворах, содержащих соединения селена с хлором и бромом [29, 30], существует несколько типов комплексов, причем в .определенных условиях могут образовываться оксигалогениды селена с отношением Se : Cl (Br) = 1:2.
26
Спектры поглощения растворов, содержащих SeO2, HF, НС! или НВг, указывают на образование соединений SeO3-4HX (X = = Cl, Вг) и SeO2-4,5-H2F2 [11581. С разбавлением растворов и повышением температуры устойчивость комплексов падает вследствие гидролиза.
Существование комплексных соединений было подтверждено методом электропроводности.
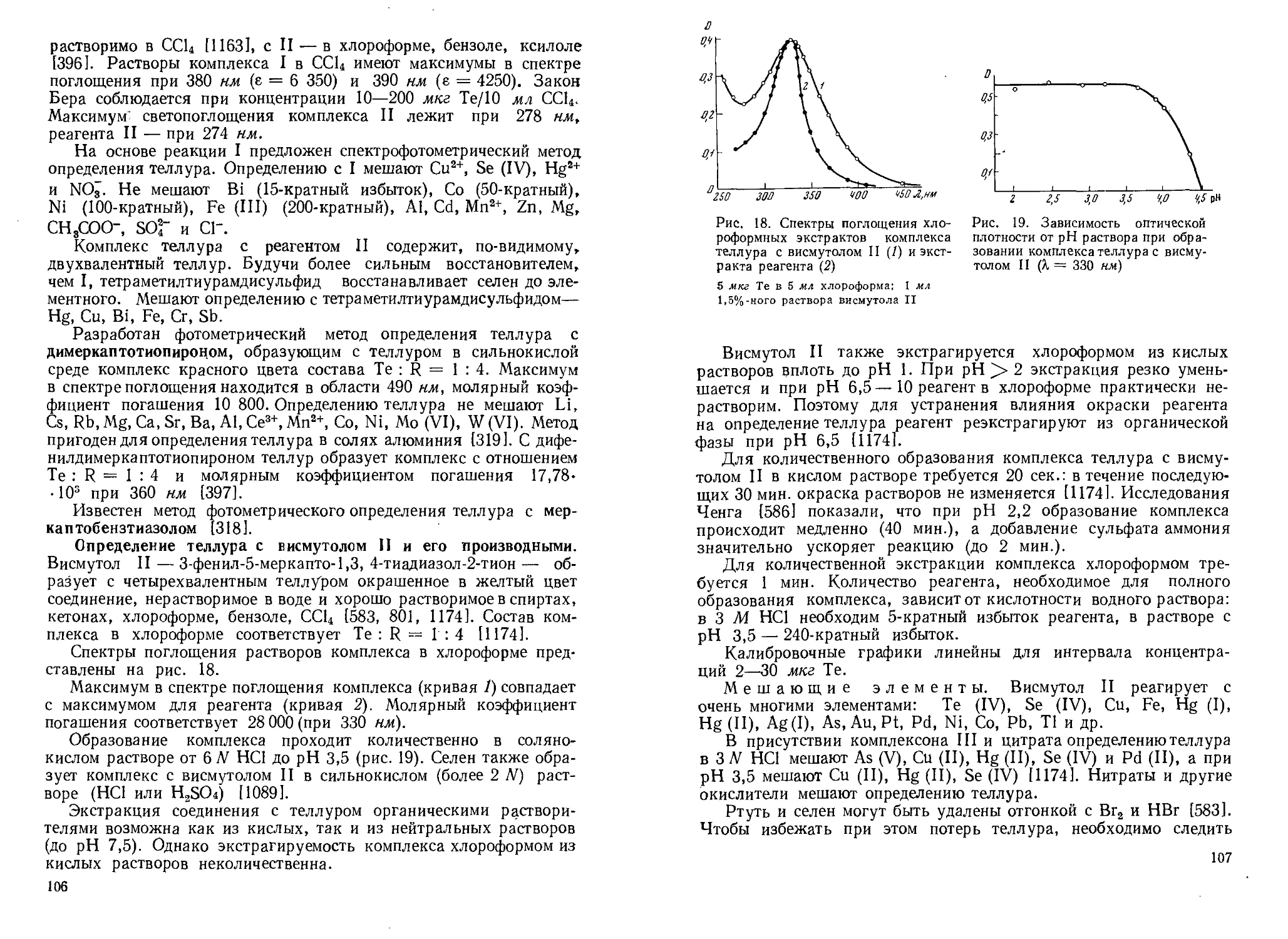
Спектрофотометрическое исследование солянокислых растворов, содержащих Те (IV), показало [745], что в таком растворе существует несколько ионов. При высоких концентрациях НС1 (9—12V) в спектре поглощения наблюдаются два максимума — при 269 и 376 нм. В 6N НС1 при 269 нм максимум исчезает, а при 376 нм заметно снижается. Поскольку галогенокомплексы теллура окрашены, растворы, содержащие эти соединения, в присутствии соляной кислоты, бромидов и иодидов используются в фотометрических методах определения теллура [244, 445, 559 , 672, 697 , 814, 8151.
Различие в свойствах соединений селена и теллура, существующих в солянокислом растворе, и возможность образования этими элементами комплексных ионов нескольких типов использованы в экстракционных и ионообменных методах разделения селена и теллура. Например, изучение сорбции селена и теллура ионитами в солянокислом растворе позволило Ермакову и сотр. [3261 найти условия количественного разделения теллура и селена на анионите ЭДЭ-10.
Соединения селена и теллура с органическими основаниями и аммиаком
При взаимодействии тетрахлорида или тетрабромида теллура, растворенных в безводном эфире, ледяной уксусной кислоте, бензоле, спирте или ацетоне, с ароматическими аминами образуются соединения (C6H8NH2)2-TeBr4, [(C6H3)2NH2]-TeBr4, (BrC6H4NH2)2-• TeBr4, (C10H7NH2)2-TeBr4, [С6Н4 (NHa)a]-TeBr4, [С7Н6 (NHa)ab • TeBr4, [NH2-C6H4-CeH4NH2]-TeBr4 и др. [393, 887, 888, 997—999]. Все эти соединения хорошо кристаллизуются, устойчивы на воздухе, но разлагаются водой; они нерастворимы в эфире, этаноле, хлороформе, бензоле, четыреххлористом углероде, но легко растворяются в кислотах. Некоторые производные, например соединения с толуидином и о-фенилендиамином, легко растворяются в. ацетоне. Тренев и сотр. [393] получили следующие соединения ТеС14 и SeCl4 с этилендиамином (Еп), пиридином (Ру), анилином (Ап), тиомочевиной (Thio) и аммиаком: SeCl4-4NH3, SeCl4-2Thio, SeCl4-2En, TeCl4-2NH3, TeCl4-6NH3, TeCl4-2En, TeCl4-2En-2NH3, TeCl4-2An, TeCl4-2An-2NH3, TeCl4-2Py, TeCl4-2Thio, TeCl4-6Thio. Была изучена термическая устойчивость этих соединений и показано, что связь селена и теллура с азотсодержащими лигандами
27
характеризуется довольно большой прочностью, особенно в случае этилендиамина.
ТеВг4 образует с диазинами (пирамидином, пиразином и др.) соединения состава (C2H4N2)-TeBr4. Аналогичные комплексные соединения типа 1 : 1 были получены для N4S4, N4S4H4 и диоксана [501 ]. Соединения с диазинами (C4H4N2)• ТеВг4 легко превращаются при нагревании в растворе НВг в кристаллические соли (C4H6N2)-TeBr6. Соответствующие хлориды и иодиды можно получить аналогичным образом. Исследования Эйнсли [501] показали, что устойчивость аниона ТеХв для диазиновых производных возрастает от хлорида к иодиду.
Для фторидов было получено белое кристаллическое вещество с пиридином (C5H5N)-TeF4, но не (C5H5N)2-TeF4, как можно ожидать по аналогии с хлоридами и бромидами. Пиридинтетрафтор-теллурит (C5H5N)-TeF4 не изменяется на воздухе, но легко разлагается водой с образованием теллуристой кислоты. В растворе HF образуется желтое кристаллическое соединение состава (C5H5NH)TeFB — пиридинпентафторотеллурит. Превратить это соединение в соответствующий хлорид, бромид или иодид с применением соответствующей галогеноводородной кислоты не удалось, так как во всех случаях образовывались соединения (CBHBNH)2-•ТеХ6.
Пентагалогенотеллуриты с общей формулой (CON2HB)-TeXB, где X = Cl, Br, J, были получены для мочевины [501].
Дипиридингексафторотеллурит (CBHBNH)2-TeF6 получен в растворе ТеО2 и пиридина в концентрированной HF. Это соединение при обработке HJ давало дипиридингексаиодотеллурит (CBHBNH)2-•TeJ6. Под действием НС1 и НВг дипиридингексафторотеллурит легко превращается в (CBHBNH)2-TeCl6 и (C5H5NH)2-TeBre. Дипиридингексаиодотеллурит образуется в результате обработки любого другого гексагалогенотеллурита иодистоводородной кислотой.
В настоящее время известны многочисленные гексагалогено-теллуриты с аминами (первичными, вторичными и третичными) [731—733, 868], N-ацетиламином [631], алкалоидами [872] и изучены их свойства. В случае ацетилпроизводных гексагалогенотел-луритов в некоторых случаях три и четыре молекулы N-ацетилами-на связываются с молекулой Н2ТеС1в [631].
В отличие от теллура, для которого комплексы с аминами (и некоторыми другими органическими основаниями) очень многочисленны, для селена известны лишь отдельные представители. Тренев [393] получил соединения SeCl4-4NH3, SeCl4>2Thio, SeCl4-2En. Известны также соединения аминов с оксигалогенидами селена: 2SeOCl2-3(CH3)2NH-HCl, SeO2-(CH3)2-NH-HCl, 2SeBr4-SeBr-• 3(CH3)2-NH-HBr, SeBr4-2(CH3)2*NH-HBr и некоторые другие [9701. Были получены аминопроизводные монобромида селена [997, 998] с анилином, о-толуидином, n-толуидином, о-анизидином, бензиламином, а-нафтиламином, бензидином и другими аминами 28
следующего состава:
для моноамина
для диамцна
‘CsHeNHa—» Se I
CsHeNH2 — Se
ВГ2
ГС6Н4—NH2—>Se‘ I I
C6H4— Nlh-^SeJ
Вгз
Эти соединения устойчивы на воздухе, нерастворимы в воде и органических растворителях, гидролизуются водой при кипячении и разрушаются разбавленными кислотами и щелочами.
Сведения о составе, строении и свойствах аммиакатов селена и теллура в литературе немногочисленны. Из старых работ известно, что при взаимодействии SeCl4 и ТеС14 с аммиаком возможно образование аммиакатов, амидов, имидов и нитридов различного состава [924, 1093, 1094]. При реакции с жидким аммиаком SeO2 дает азотистый селен (NSe)n [799, 800].
Тренев и сотрудники [392] исследовали ряд соединений, образующихся при действии безводного аммиака на ТеС14 и SeCh при повышенных давлении и температуре. Этим методом были получены SeCl4-4NH3, TeCl4-4NH3, TeCl4-6NH3, а также соединения смешанного типа: TeCl4-2En-2NH3, TeCl4-2An-2NH3.
Реакция диоксидифторида селена с аммиаком [646] дает смесь аммонийных солей циклического триселенамида (SeO2NNH4)3 и гомологическую серию кольчатых полиимидоамидов селенистой кислоты NH2SeO2N(NH4)nSeO2NH2.
Достал [634] получил и исследовал амидоселенаты серебра [Ag(SeO3NH)], а также щелочных и щелочноземельных металлов.
Соединения селена с 3,3'-дишинобензидином и некоторыми другими о-диаминами
В солянокислом растворе селенистая кислота реагирует с 3,3'-диаминобензидином и образует желтый кристаллический осадок, нерастворимый ни в кислой, ни в щелочной средах. В растворах, содержащих избыток селенистой кислоты, образуется дипиазоселе-нол с т. пл. 292—314° С [755, 756, 980]
При недостатке селена и избытке 3,3'-диаминобензидина может образоваться монопиазоселенол
— темно-красные кристаллы с т. пл. 202° С [980]. Теллур с ди-аминобензидином не реагирует.
29
Монопиазоселенол, перекристаллизованный из концентрированной НС1, имеет три формы, в зависимости от количества присоединенных ионов Н+: однопротонированную (Л), двупротонированную (В) и свободную непротонированную форму (С) [516]. Установлено, что спектры поглощения монопиазоселенола при больших значениях pH сдвигаются в сторону коротких волн. Были рассчитаны константы диссоциации для кислотной и основной форм монопиазоселенола при 22° С: Л1 = 3,0-10-1, М2 = 4,1 • 10~6 и = 3,3-• 10-14, К2 — 2,4-10-10 соответственно.
Коэффициенты молярного погашения растворов в толуоле равны: для Д-формы — 2,17-104 при 347 нм и 1,65- 10s при 410 нм; для S-формы— 1,57-Ю4 при 347 нм и 5,90-103 при 410 нм; для С-формы — 1,10-104 при 347 нм и 8,30-103 при 410 нм. Форма С плохо растворима в воде и хорошо экстрагируется органическими растворителями. При исследовании кинетики реакции селена с 3,3'-диаминобензидином при концентрациях H2SeO3 от 10~4 до 2-10~3 М и различном отношении Se : R было показано, что на первой стадии реакции образуется только монопиазоселенол, который затем может взаимодействовать с избытком селена и дает дипиазосе-ленол.
Последнее соединение имеет свойства слабого основания, плохо растворимо в воде и при pH 6 не извлекается в фазу органического растворителя; спектр поглощения раствора дипиазоселенола в толуоле имеет максимум при 356 нм.
На способности селена образовывать монопиазоселенол основан высокочувствительный и специфичный метод количественного определения малых (микрограммовых) содержаний селена.
Растворы в толуоле, содержащие монопиазоселенол, флуоресцируют в области 420 и 580 нм.
Исследования других ароматических о-диаминов показали, что о-фенилендиамин также реагирует с селеном [1119], причем чувствительность реакции в этом случае несколько выше, чем с 3,3'-диаминобензидином [4941.
Реакцию селенистой кислоты с о-фенилендиамином (I) изучал Барца [515] и показал, что реакция протекает по следующей схеме:
NH+ X\=N\
3 4- H2SeO3 ->1 I м >Se 4- ЗН2О 4- H-.
. NHo Is. >=!X1 /
Заместители X в бензольном ядре
3
h
снижают
основность аминогруппы и сдвигают максимум поглощения в видимую область спектра.
По данным рентгеноструктурного исследования, строение сое
30
динения селена с о-фенилендиамином можно представить формулой [8931:
Были изучены соединения с селеном производных о-фениленди-амина — 5-диметиламино-2,1,3-бензоселенодиазола [149, 622, 1049, 1050]
и 5-метилтио-2,1,3-бензоселенодиазола [1050]
Максимум в спектре поглощения растворов этих соединений расположен в видимой области спектра — при 500. и 572 нм соответственно.
В кислых растворах (pH 1—2) селенистая кислота реагирует и с пара-замещенными о-фенилендиамина — 4-метил-(П), 4-хлор-(Ш), 4-нитрофенилендиамином (IV) с образованием 5-метил-5-хлор-и 5-нитро-2,1,3-бензоселенодиазолов [1113]. Все эти соединения экстрагируются толуолом. Длины волн максимумов в спектрах поглощения экстрактов равны соответственно (нм): у соединения с о-фенилендиамином — 335, у производных — 337, 341, 350; е=17 750, 17 900, 18400,15460. Для о-фенилендиамина и его производных (спектрофотометрическим методом при 20°С и ионной силе, равной 1) определены константы диссоциации:
II .
III .
IV .
рк, 4,85 5,24 4,04 2,85
рК2
0,88
1,27 0,1
Все о-диамины могут быть использованы в качестве высокочувствительных и избирательных реагентов для экстракционноспектрофотометрического определения селена.
Реакция селенистой кислоты с 1,8-нафталиндиамином
nh2nh2
I I
дает устойчивое коричневое соединение, экстрагируемое
хлороформом при pH 4—6 [662, 1116].
31
Максимум в спектре поглощения растворов в хлороформе лежит при 372 нм. Соединение пригодно для количественного спектрофотометрического определения селена.
Были изучены свойства соединений селена с 1,2-диаминонафталином (V) и 2,3-диаминонафталином (VI) [886, 9811:
Соединения содержат пятичленное диазоловое кольцо. Поскольку здесь в реакцию с селеном вступают две аминогруппы из двух, то образуются или нейтральные, или очень слабоосновные селенодиазолы, в отличие от соединения селена с 3,3'-диаминобензидином, где реагируют две аминогруппы из четырех, и образуется относительно основный пиазоселенол, который может экстрагироваться только при высоких значениях pH растворов. Производные 2,3-диаминонафталина экстрагируются хорошо из кислых растворов такими растворителями, как циклогексан и декалин. Толуол меньше подходит для экстракции, так как в нем соединение менее устойчиво. При облучении растворов 4,5-бензопиазоселенола ультрафиолетовым светом растворы флуоресцируют с максимумом в спектре флуоресценции при 520 нм.
Выделенное в твердом виде соединение селена с 2,3-диаминонафталином — 4,5-бензопиазоселенол — представляет собой красное мелкокристаллическое вещество состава C^H^N^Se, устойчивое при нагревании до 290°С. В растворе (в присутствии НС1) вещество разлагается при температуре ниже 100°С. Максимум в спектре поглощения растворов лежит при 380 нм.
2,3-Диаминонафталин является наиболее чувствительным реагентом на селен из группы диаминов, позволяющим определять флуориметр ическим методом 0,002 мкг Se/мл.
Из азотсодержащих органических реагентов с селеном реагируют 1,1'-диантримид [8621 и 2,2'-диантримид [613, 8601.
О О
II II
uOj Ши II II
о о
Спектрофотометрическим методом в среде концентрированной серной кислоты было установлено существование двух комплексов 1,Г-диантримида с Se (IV). При малых концентрациях селена существует соединение с отношением Se: R = 2:1, а при больших 32
концентрациях — Se : R -= 1:1. Соединение состава 1 : 1 выделено в индивидуальном состоянии. ИК-спектры показали, что соединение содержит карбазольную связь. Методом изомолярных серий и модифицированным методом прямой линии определены константы устойчивости этого комплекса (5,6-108 и 6,1 • 108). Максимумы в спектре поглощения растворов находятся при 600 и 495 нм.
Комплексообразование между Se (IV) и 2,2'-диантримидом было изучено методами спектрофотометрии, ИК-спектроскопии и элементного химического анализа. Установлено существование двух комплексов: мономерного SeR и димерного Se2R2. В димере Два мономера соединены посредством водородных связей. При низкой концентрации селена и реагента доминирует димер. При увеличении концентрации компонентов водородная связь разрывается. Константа устойчивости мономера равна 7,7-103, а димера — 3,2-1012. Мономер был выделен в твердом виде.
2,2-Диантримид реагирует только с селеном и не взаимодействует с бором, германием и теллуром (с этими элементами реагирует 1,Г-диантримид). Попытка использования реакции селена с 2,2'-диантримидом для количественного спектрофотометрического определения селена показала, что метод малочувствителен; сильное поглощение реагента и необходимость проводить определение селена в среде концентрированной серной кислоты препятствуют практическому применению метода.
Соединения селена и теллура с тиомочевиной и ее производными
Соединение теллура с тиомочевиной было впервые получено в 1927 г. [658] в виде желтого кристаллического вещества с т. пл. 165°С, хорошо растворимого в спирте, эфире и других органических растворителях.
При взаимодействии ТеХ«, где X = С1 и Вг, с тиомочевиной в этанольном растворе образуются несколько соединений [5021 : (SCN2H4)2-TeCl4 (т. пл. 175°), (SCN2H4)• TeBr О4 (210°С), (SCN2H3),-• ТеС12, (SCN2H3)2-TeBr2 (215°С), (SCN2H5)2-TeCl6, (SCN2H5)2 • • TeBr6 (155°С), (SCN2H5)2.TeJ6, (SCN2H5)2-TeCl6-2SCN2H5Cl (155°С), (SCN2H5)2TeBr6-2SCN2H8Br (155°С).
Тренев и сотр. [393] получили соединения селена и теллура с тиомочевиной: SeCl4-2SCN2H4, TeCl4-2SCN2H4, TeCl4-6SCN2H4.
Вржештял [1131] выделил из водных растворов ряд тиомо-чевинных соединений теллура и показал, что все комплексы являются производными двухвалентного теллура, так как в кислом водном, растворе тиомочевина восстанавливает четырехвалентный теллур до двухвалентного. В растворах различных неорганических кислот двухвалентный теллур образует с тиомочевиной желтые кристаллические малорастворимые в воде следующие соединения: Te(Thio)4Cl2-2H2O (175°С), Te(Thio)2Cla (184°С), Те(ТЫо)2Вг2
2 Зак. № 1476
33
(две модификации с т. пл. 170 и 220°С), Te(Thio)4-(С1О4)2 (144°С), Te(Thio)4(N03)2-2H20 (117°С), Te(Thio)4S04-2H20 (141°С),
Те(ТЫо)4(НС204)2-2Н20 (170°С). Эти соединения хорошо растворимы в полярных растворителях и нерастворимы в неполярных.
Рябчиков, Назаренко (неопубликованные данные) получили соединения теллура с тиомочевиной в индивидуальном состоянии и определили его состав методом элементного анализа. Было определено число молей тиомочевины в комплексных соединениях и определена валентность теллура в этих комплексах. Комплекс двухвалентного теллура [Te(Thio)4]Cla-2H20 (рис. 2) был выделен из водного раствора, 0,5 N по НС1 и 3%-ного по тиомочевине, в виде желтых игольчатых кристаллов с т. пл. 175°С. Комплекс [Te(Thio)2]Cl2 (рис. 3) был получен в виде тонких ромбических пластинок из метанольного раствора, 3 N по НС1, при концентрации тиомочевины —0.05Л4. Валентность теллура в обоих комплексных соединениях равна двум.
Восстановление тиомочевиной четырехвалентного теллура до вухвалентного можно предггазагь уравнением:
Те (IV) + 4SCN2H4 + 2Х" ->j
- Ге (II) (SC4/^)2 Х2 4- [(Ч I.)2C-S —S -3 (N'l^]8*.
Тараян и Саркисян [377] определили константу нестойкости комплекса [Те(ТЮо)2Р+, которая оказалась равной 2-ЮЛ Исследование спектров поглощения растворов комплекса показало, что максимум в спектре поглощения находится при 428 нм, коэффициент молярного погашения при 428 нм равен 1 • 103, при 3ЭЭ —325 нм — 1,12-10’. Спектры поглощения в солянокислой, сернокислой и фосфорнокислой средах идентичны.
В 1959—1961 гг. была опубликована серия работ Фосса и сотр. [680—6861 по синтезу и изучению некоторых свойств комплексных соединений теллура с тиомочевиной и ее производными (этилентиомочевиной, пропилентиомочевиной, тетраметилтиомочевиной).
Для четырехвалентного теллура были получены шестикоординированные комплексы с тетраметилтиомочевиной (Tmthio): Te(Tmthio)2Cl4 (т. пл. 153—154°С), Те(Т nthio)2Br4 (176—177°С). При растворении в кислом (4 N НС1) водно-метанольном растворе эти вещества быстро превращались в соединения двухвалентного теллура.
Тетрахлор- и тетрабромсоединения теллура с тетраметилтиомочевиной — единственные представители соединений четырехвалентного теллура. Все остальные соединения с тиомочевиной являются производными двухвалентного теллура.
В водно-метанольном растворе, содержащем Те (IV) и НС1 (или НВг), тетраметилтиомочевина образует соединения Te(Tmthio)Cl2 (т. пл. 207°С) и Te(Tmthio)Br2 (226°С).
34
Рис. 2. Кристаллы комплексного соединения теллура с тиомочевиной [Те (Thio)4]Cl3 -2Н3О
Увеличение X 20 раз
Рис. 3. Кристаллы комплексного соединения теллура с тиомочевиной [Те (Thio)3]Cl3
Увеличение X 20 раз
С двумя молями тиомочевины (Thio), тетраметилтиомочевины (1'mthio), этилентиомочевины (Ethio) и пропилентиомочевины (Prthio) были получены четырехкоординированные комплексные соединения с общей формулой TeR2X2, где X = Cl, Br, J, SCN, a R = Thio, Tmthio, Ethio, Prthio. В частности, с тиомочевиной были получены соединения: Te(Thio)2Cl2 (т. пл. 220°С), Te(Thio)2Br2 (220°С), Te(Thio)2J2 и Te(Thio)2(SCN)2. Кристаллы этих соединений изоморфны и представлены zjuc-изомером. С этилентиомочевиной были выделены две модификации бромида: Te(Ethio)2Br2 — желто-коричневые ромбические призмы с т. пл. 186°С (/juc-изомер) и красно-желтые моноклинные призмы с т. пл. 192°С (трамс-изомер). Оба эти соединения нерастворимы в метиловом спирте и в ледяной уксусной кислоте, но хорошо растворяются в диметилформ-амиде.
Были получены также Te(Ethio)2J2 (т. пл. 198—200°С) и Te(Ethio)2(SCN)2 (т. пл. 142—144 °C).
С пропилентиомочевиной синтезированы хлорид, бромид, роданид и метилтиосульфонат, причем два первых соединения представлены цис-изомерами, а два последних — трамс-изомерами: Te(Prthio)2Cl2 (т. пл. 199°С), Te(Prthio)2Br2 (190°С), Te(Prthio)2(SCN)2 (214 °C) и Te(Prthio)2(S2O2CH3)2 (172 °C).
Для тетраметилтиомочевины получены траме-изомеры дихлоро-и дибромокомплексов: Te(Tmthio)2Cl2 (т. пл. 167°С) и Te(Tmthio)2Br2 (192°С) в виде красно-оранжевых кристаллов и две кристаллические модификации цис- и трамс-иодокомплексов: Te(Tmthio)2J2.
При образовании соединений типа Te(Thio)2X2 возможно замещение хлорид- и бромид-ионов метантиосульфонатом и бензолтиосульфонатом. Из соединений этого типа были выделены следующие: Te(Thio)2(S2O2CH3)2 (т. пл. 130— 131°С), Je(Ethio)2(S202CH3)2 (два изомера цис- и транс- с т. пл. 168°С), Te(Ethio)2(S2O2CeH6)2, Te(Tmthio)2(S2O2CH3)2, Te(Tmthio)2(S2O2CeH6)2.
Для комплексов типа Te(Thio)2X2 не было получено ни одного фторида.
Известны также соединения с тремя молекулами тиомочевины (а также этилентиомочевины и пропилентиомочевины): Te(Thio)3(HF2)2, Te(Ethio)3(ClO4)2, Te(Prthio)3(ClO4)2. Для перхлората пропилентиомочевины были выделены две модификации кристаллов желто-зеленого цвета: моноклинная и триклинная.
И, наконец, с четырьмя молекулами тиомочевины (и ее производных) были получены комплексы: Te(Thio)4Cl2-2H20, Te(Thio)4Cl2, Te(Thio)4Br2, Te(Thio)4(SCN)2, Te(Thio)4(NOs)2, Te(Thio)4(C104)2, Te(Thio)4(HF2)2, Te(Thio)4F2-2H2O, Te(Ethio)4Cl3-2H2O, Te(Ethio)4-• Br2-2H2O, Te(Ethio)4Cl2, Te(Ethio)4Br2, Te(Ethio)4(C104)2, Te(Ethio)4TeCle, Te(Prthio)4Cl2-2H20, Te(Prthio)4(ClO4)2. Кислые водные и этанольные растворы этих соединений, насыщенные тиомочевиной, устойчивы.
36
Дигидраты этилентиомочевины хорошо растворяются в кислом водном и метанольном растворах, а также в диметилформамиде с образованием оранжевых растворов, устойчивых при комнатной температуре. Кипящий метанол растворяет в 100 мл около 1,5 г веществ; при охлаждении таких растворов кристаллизуются безводные соли.
Следует заметить, что катионный комплекс [Te(Thio)J2+ образуют тиомочевина, этилентиомочевина и пропилентиомочевина, но не тетраметилтиомочевина. В последнем случае, вероятно, возникают пространственные затруднения. Однако оказалось возможным выделить соединения с двумя молекулами тиомочевины и двумя молекулами тетраметилтиомочевины: [Te(Thio)2(Tmthio)2]Cl2 и [Te(Thio)2(Tmthio)2]Br2.
Образование окрашенного комплекса [Те(ТЫо)2]2+ в растворах, насыщенных тиомочевиной, используется при количественном определении малых количеств теллура. Селен восстанавливается избытком тиомочевины до элемента и не мешает последующему определению.
При взаимодействии тиомочевинного комплекса четырехвалентного теллура с солью Рейнеке в кислой среде (НО, HNO3, НСЮ4, СН3СООН и др.) образуется кристаллический осадок розового или желтовато-розового цвета состава [Te(Thio)4][Cr(CNS)4(NH3)2]2. После растворения осадка в метилэтилкетоне возможно фотометрическое определение теллура [31, 65, 129].
Соединение теллура с тиомочевиной в присутствии роданида калия экстрагируется из солянокислого раствора (0,6—1,7 N НС1) трибутилфосфатом [749]. При избытке KSCN возможно определение очень малых количеств теллура.
Четырехвалентный теллур образует желтые комплексы с производными тиомочевины (VII) — симметричной дифенил- (VIII), фенил- (IX), дитолил- (X), этилентиомочевиной (XI) в среде 4—9N НС1 или HCIO4, причем светопоглощение комплексов увеличивается в ряду VII XI X IX <( VIII. Комплексы Te(IV) с дифенил-, фенил- и дитолилтиомочевиной экстрагируются хлороформом из 4,5—8N НС1 или НС1О4. Образование комплексов теллура с производными тиомочевины используется в экстракционно-фотометрическом методе определения теллура. Этот метод более прост и чувствителен, чем определение с тиомочевиной в водном растворе 11171].
В последнее время показана возможность образования комплекса селена с тиомочевиной [276, 277, 339]. Чем выше концентрация тиомочевины и НС1 + H2SO4, тем быстрее идет растворение элементного селена в избытке тиомочевины.
37
Соединения селена и теллура с тиосульфатом и его производными
В кислых растворах четырехвалентные селен и теллур восстанавливаются тиосульфатом натрия до двухвалентных и образуют желтые растворы, содержащие комплексные ионы [Se(S2O3)2]2- и [Te(S2O3)2]2-, которые по аналогии с пентатионатом [S(S2O3)2]2- названы селен- и теллурпентатионатами. Селен и теллур замещают в пентатионате средний атом серы, образуя нераз-ветвленную цепочку — S — S — Те — S — S—.
Реакция образования пентатионата протекает по схеме:
SeO2 -|- 4Na2S20s + 4НС1 = Na2 [Se (8203)2] + Na2S4Oc + 4NaCl -[- 2НгО.
Соединения селена и теллура с тиосульфатом в растворах легко разрушаются при действии щелочей с выделением элементных селена и теллура, но по отношению к кислотам эти растворы совершенно устойчивы. Разбавленные растворы соединений можно кипятить некоторое время в присутствии избытка тиосульфата, не опасаясь разложения, в воде же они медленно разрушаются.
Соли селен пентатионовой и теллур пентатионовой кислот впервые выделил Фосс [674, 675]. Он получил следующие соединения: Na2[Se(S2O3)2]-3H2O, Na2[Te(S2O8)2]-2H2O, K2[Se(S2O3)2b 1,5Н2О, K2lTe(S2O3)2], (NH1)2[Se(S2O3)2]-l,5H2O, (NH4)2[Te(S2O3)2],
Rb2[Se(S2O3)2] • 1,5H2O, Rb2[Te(S2O3)2]-1,5H2O, Cs2[Se(S2O3)2],
Cs2 [Te(S2Os)a] • 1,5H2O.
Соединения дают хорошо образованные кристаллы; гидраты обычно образовывали более правильные кристаллы. Все соединения окрашены в желтый или желто-зеленый цвет; гидраты, как правило, окрашены ярче, чем безводные соединения. При хранении веществ в вакууме над серной кислотой молекул воды из гидратов-не выделяется.
Соли, выделенные в чистом виде, вполне устойчивы и могут сохраняться несколько недель без разложения. Растворимость веществ в воде уменьшается в соответствии с рядом Na NH4
К > Rb > Cs, причем соли теллура несколько менее растворимы, чем соединения селена. Na- и NHa-соли обычно заметно растворимы в метаноле и немного — в 96%-ном этаноле, в то время как К-, Rb- и Cs-соли нерастворимы в спиртах.
Селенопентатионат и теллуропентатионат быстро и количественно реагируют с диэтилдитиокарбаматом, образуя соединения Se[S2CN(C2H5)2]2 и Te[S2CN(C2H3)2]2.
Исследование кристаллической структуры теллуропентатионата аммония (NH4)2[Te(S2O3)2] [685] показало, что ион [Te(S2O3)2]2-содержит неразветвленную цепочку —S — S — Те — S — S—, которая имеет транс-конфигурацию, а три кислородных атома расположены в вершинах тетраэдров. Водород связывает атомы кислорода и атомы азота ионов аммония.
38
Тиосульфатные комплексы двухвалентных серы, селена и теллура образуют изоморфные кристаллы.
Фосс синтезировал новый тип соединений — производные метан-тиосульфоната (676, 6881 Se(S2O2CH3)2 и Te(S2O2CH3)2. Эти соединения являются аналогами пентатионовых соединений, в которых гидроксильная группа кислоты замещена метилом. Были получены и другие производные тиосульфонатов селена и теллура — дибензолтиосульфонаты Te(S2O2C6H5)2 [677, 1008, 1009] и я-толуолтиосульфонаты [677]. Бензолсодержащие соединения более устойчивы, чем соединения с n-толуолом. Se(S2O2C6H5)2 довольно хорошо, a Te(S2O2C,H,)2 умеренно растворимы в теплом этил-ацетате, бензоле и хлороформе.
Двухвалентный теллур этих соединений в реакциях замещения с тиосульфатом и диэтилдитиокарбаматом легко освобождает тио-сульфонатную группу и дает теллурпентатионат и теллурдиэтил-дитиокарбамат.
Было получено еще одно соединение, образующее кристаллы, изоморфные с сульфонатами селена и теллура,— триселендисульфинат Se3(SO2R)2, в котором три атома серы замещены тремя атомами селена.
Фосс [6791 получил также селен- и диселендисульфинаты Se(SO2R)2 и Se2(SO2R)2.
Кристаллы Se(SO2C6H3)2 и Se(SO2C7H7)2 устойчивы на воздухе, на солнечном свету быстро выделяют селен, при нагревании разлагаются, нерастворимы в воде, умеренно растворимы в теплом эфире, этаноле и уксусной кислоте и легко растворимы в хлороформе, теплом бензоле и четыреххлористом углероде. Эфирные растворы 862(80267147)2 устойчивы при нагревании.
Тиосульфонаты теллура могут присоединять два моля тиомочевины и образовывать смешанные соединения: Te(Thio)2(S2O2CH3)2, Te(Ethio)2(S2O2CH3)2, Te(Ethio)2(S2O2CeH5)2, Te(Tmthio)2(S2O2CH3)2, Te(Tmthio)2(S2O2C6H3)2.
Все выделенные соединения представляют собой зеленовато-желтые кристаллы, только соединение Te(Tmthio)2(S2O2C6H5)3 красное. В твердом состоянии соединения вполне устойчивы, а в воде быстро разрушаются с выделением элементного теллура.
Способность селена и теллура образовывать тиосульфатные комплексы использована в ряде титриметрических методов определения этих элементов [697, 813].
Соединения селена и теллура с диэтилдитиокзрбамзтом и его производными
Теллур образует с диэтилдитиокарбаматом (DDTQ окрашенное нерастворимое в воде соединение Te(DDTC)4, которое легко пере-Ходит при экстракции в органический растворитель — хлороформ, четыреххлористый углерод и др. [537]. Селен образует с диэтил
39
дитиокарбаматом подобное соединение. Комплекс селена количественно переходит в органическую фазу при pH 4—6,2, а теллур экстрагируется в более широком интервале значений pH 4—8,8. Экстракционное разделение карбаматов селена и теллура обычно проводят в интервале pH 8,5—8,7 [536]. Присутствие в растворе комплексонов, а также цианидов, цитратов, тартратов, фосфатов и боратов не влияет на экстракцию селена и теллура.
Измерение оптической плостности растворов диэтилдитиокарбамата теллура в хлороформе проводят при 428 нм [538, 543, 751], молярный коэффициент погашения равен 3200. Водно-ацетоновые растворы, содержащие соединение теллура, имеют максимум при 340 нм [770].
Растворы, содержащие диэтилдитиокарбамат теллура, неустойчивы и быстро обесцвечиваются, особенно на свету. Поэтому фо-тометрирование растворов нужно проводить сразу же после развития окраски и работать в посуде из темного стекла.
Определение теллура с диэтилдитиокарбаматом применено к анализу железа и сталей [722], чистого селена [284], платиновых металлов [285]. Этот метод также использован при определении теллура в свинце и меди [891.
3,5-Дифенилпиразолин-1-дитиокарбамат натрия образует с теллуром соединение, окрашенное в желтый цвет, хорошо растворимое в хлороформе и четыреххлористом углероде и более устойчивое в растворах органических растворителей, чем диэтилдитиокарбамат теллура. Соединение теллура с 3,5-дифенилпиразолин-1-ди-тиокарбаматом образуется в интервале кислотности от 12 N НС1 до pH 9[64].
Спектр поглощения растворов в хлороформе имеет мак- симум при 415 нм, а кажущийся молярный коэффициент светопо-глощения при этой длине волны равен 20 400 [64].
Соединения селена и теллура с висмутолом II и другими азот- и серусодержащими органическими реагентами
Висмутол II образует с теллуром окрашенное соединение [583, 586, 794, 1170]:
~/==\__м_С—Я
Те |
N=C\
s-J4
Образование прочных окрашенных полимерных соединений с теллуром осуществляется через атомы серы с шестичленными гетероциклами. Так, например, для висмутола II комплексные сое
40
динения имеют следующую формулу:
S
/=\_N_C \
I >S Те/4
N=CZ /
Аналогичное соединение с висмутолом II дает селен [1089, 1172].
Соединение теллура образуется в солянокислом растворе при концентрации кислоты в пределах 10“4 — 644. Комплекс теллура (и селена) с висмутолом II —желтое вещество, плохо растворимое в воде, хорошо растворимое в спиртах,кетонах, хлороформе и ССН. Соединение устойчиво в водном растворе и в органических растворителях. Соединения селена и теллура с висмутолом II разрушаются при нагревании выше 50° С, они могут быть высушены в вакууме при комнатной температуре до постоянного веса.
Максимум поглощения растворов комплекса теллура с висмутолом II в хлороформе лежит при 335 нм и совпадает с максимумом поглощения реагента. Молярный коэффициент погашения 35 000.
В присутствии сильных окислителей соединение разрушается и реагент окисляется до дисульфида. Состав соединения теллура с висмутолом II был определен методом Жоба и отвечал соотношению теллур: висмутол, равному 1:4.
Комплекс селена с висмутолом II существует только в сильнокислом растворе (>2 А; НС1 или H2SO4). В этих условиях образуется устойчивое желтое соединение, хорошо экстрагируемое при этой кислотности СНС13, СС14. Максимум в спектре поглощения раствора соединения в хлороформе лежит при 330 нм, т. е. практически совпадает с максимумом для соединения теллура. С использованием висмутола II следы теллура определяют в рудах, пирите, теллуридах кадмия и ртути [584].
Димеркаптопирон (2,3-димеркапто-1,4-тиопиронкарбоновая кислота) дает с теллуром в сильнокислой [НС1 (1 : 1), H2SO4, Н.Л’0,1 среде соединение красного цвета с отношением Те : R = = 1 : 4 и максимумом поглощения при 490 нм (е = 10 880). На этой основе разработан фотометрический метод определения теллура в солях алюминия [319].
Меркаптобензтиазол (2-бензтиазолтион) образует с теллуром (IV) устойчивый желто-оранжевый комплекс состава Те : R = 1 : 4, количественно экстрагируемый СНС13, С6Н6, толуолом. Максимум в спектре поглощения раствора в СНС13 находится при 330 нм, е = 24 023 (при 360 нм) [318].
2-Меркаптобензимидазол дает с Se (IV) соединение желтого цвета. Из 3 N НС1 селен осаждается в виде желтого кристаллического осадка состава (C7H8N2S)2Se. Соединение не экстрагируется из 1—3N НС1 бензолом, толуолом, эфиром, плохо экстрагируется изо
41
амиловым и бутиловым спиртом, несколько лучше — дихлорэтаном, хлороформом, хорошо — смесью н. бутанола и хлороформа (1 : 5). Максимум светопоглощения водных растворов находится при 330 нм, е — 10500.
Методом изомолярных серий установлено, что селен и 2-меркап-тобензимидозол реагируют в отношении 1 : 4. На основании этой реакции разработан экстракционно-фотометрический метод определения селена в отсутствие теллура, поскольку теллур ведет себя аналогично [56, 72].
Диэтилдитиофосфорная кислота взаимодействует с селенистой кислотой с образованием Диэтилдитиофосфата двухвалентного селена:
C2H6-O S
/Р:\
С2Н5—(J S
Se
2
В результате реакции образуется устойчивое соединение в виде желтого кристаллического осадка, нерастворимого в воде, но растворимого в органических растворителях. Максимум поглощения растворов находится в области 315—320 нм- е = 1900 [75, 66].
Тиогликолевая кислота — SH—СН2 — СООН — образует с Se (IV) и Те (IV) в солянокислых растворах (1—5 V) растворимые желтые комплексные соединения,плохо растворимые в СНС13, СС14, С6Нв, хорошо экстрагируемые диэтиловым эфиром, н. бутанолом, амил-и этилацетатом и метилбутилкетоном [833]. Спектры поглощения имеют максимумы в области 260 нм, е = 1600 (Se), е = 330 (Те). Растворы устойчивы приблизительно 2 часа. Состав комплексов 1 : 3 (для селена), 1 : 4 (для теллура).
Производные тиогликолевой кислоты — N-меркаптоацетил-п-анизидин (XII) и N-меркаптоацетил-и-толуидин (XIII)
/V- NH-CO-CH2-SH NH-СО-СНг— SH
H.CO-11J у
НзС хи хш
осаждают селен из солянокислого раствора (0,05—I N HCI— для XII, 0,05—0,5 N НС1—для XIII). Состав соединений соответствует формуле: R2SeO. Лучшим экстрагентом для комплекса селена является смесь н. бутанола с СНС13 в отношении 1 : 5. Теллур ведет себя аналогично селену [56, 73].
Селенистая кислота реагирует с 1-фенилтиосемикарбазидом и дает окрашенное в желтый цвет соединение. Водные растворы неустойчивы и быстро разлагаются с выделением элементного селена. Соединение экстрагируется изоамиловым и изобутиловым спиртами; экстракты устойчивы во времени (12 час.) [74]. Наибольшая
42
оптическая плотность экстрактов наблюдается при кислотности 0,05—0,2М НС1, хотя экстракция идет полностью в более широком интервале кислотности — до 2N НС1.
Спектр светопоглощения экстрактов соединения селена с 1-фе-нилтиосемикарбазидом имеет максимум при 320 нм, е = 5200. Селенистая кислота взаимодействует с 1-фенилтиосемикарбазидом в молярном отношении 1 : 2. Теллуристая кислота реагирует подобным образом с образованием более слабоокрашенных растворов.
Так же селенистая кислота реагирует с 1,4-дифенилсемикарбази-дом С6Н5—NH — NH — С(О) — NH —СвН8. Продуктом реакции является вещество красно-бурого цвета, плохо растворимое в воде и в минеральных кислотах, хорошо — в органических растворителях. Хлороформные экстракты комплекса устойчивы несколько часов. Селенистая кислота реагирует с 1,4-дифенилсе-микарбазидом в солянокислом растворе (0,02—0,1 AQ. В спектре поглощения имеются два максимума — в области 200 и 325 нм, е = 44000 и 6940.
Теллур реагирует подобным образом, однако дает более слабо-окрашенные растворы и поэтому практически не мешает фотометрическому определению селена [360, 361].
Была изучена также реакция селена с дифенилкарбазидом и дифенилтиокарбазидом, в результате которой образуются комплексные соединения с отношением Se : R = 1 : 2 [361].
4,5-Диамино-6-тиопиримидин образует с селеном комплексное
nh2
I с / \ H3N—С N II I S=C CI \ Z N
соединение, окрашенное в желтый цвет, при pH 1,5—2,5 с максимумом в спектре поглощения растворов при 380 нм. В разбавленных растворах комплекс довольно устойчив, в более концентрированных соединение разлагается и выделяется элементный селен 15771.
Гетерополисоединения селена и теллура
Теллур образует гетерополикислоты, причем центральным ионом может быть и] четырехвалентный, и шестивалентный теллур. Известны отдельные гетерополисоединения и для селена.
Четырехосновная теллурито-12-вольфрамовая гетерополикислота H4[TeO4(H2WO4)12]-13H2O была синтезирована в виде желтого кристаллического порошка, хорошо растворимого в воде. Вод
43
ные растворы гетерополикислоты не разлагаются под действием концентрированной серной кислоты. В солянокислом растворе гетерополианион [TeO4(H2WO4)12]4- устойчив при концентрации НС1< 1,138 N [110].
Для ряда Те—W-гетерополикислот были выделены Cs-соли. В солянокислом растворе при разложении гетерополианионов образуются гексахлортеллуристая Н2ТеС1е и метавольфрамовая кислоты. В сернокислой среде преобладают Te-6W-и Te-14W-aHHOHbi. При повышении кислотности до 0,8 N вероятно существование гетерополикислот промежуточного состава от Te-6W до Te-12W. Были установлены также анионы Te-18W [111].
При восстановлении H4[TeO4(H2WO4)12]-13Н2О проволокой из углеродистой стали образуются растворы теллуристовольфра-мовой сини (в 0,04—1,1 N НС1), что может быть использовано для фотометрического определения теллура [109].
Гетерополианион [TeW6O24]6- образуется при добавлении азотной кислоты к раствору, содержащему Н6ТеО6 и Na2WO4 в молярном отношении 1 : 6. Наличие в растворе аниона [TeW6O2tr установлено потенциометрическим методом, полярографически и методом бумажной хроматографии. Комплекс образуется при pH 7—7,5 и устойчив до pH 5,3. В более кислых растворах он разлагается [1018].
Некоторые Те (У1)-молибдаты и Те(У1)-вольфраматы были выделены в виде солей [980, 1032]. Получено соединение(ЗМН4)20-•ТеО3-4МоО3-15Н2О— белое кристаллическое вещество, устойчивое при 20°С, растворяется в разбавленных кислотах и разлагается при нагревании с образованием МоО3 и ТеО3. Методом кондуктометрического титрования установлено [1001], что при добавлении соответствующих солей образуются: 3Ag2O-TeO3-4МоО3-4Н2О, ЗВаО-ТеО3-4МоО3-7Н2О, ЗРЬО-ТеО3-4МоО3-6Н2О, 3(UO3)-TeO3-4MoO3-11Н2О, Се2О3-ТеО3-4МоО3-12Н2О. Было получено соединение 3(NH4)2O-2TeO3-3P2O5-ЮН2О, а также Ва-, Си-, [Си (МН3)Д-соли и фосфаттеллуратмолибдат и его Ва-соль 11002].
Аммонийная соль теллурито-6-молибденовой кислоты — (NH4)2H6[TeMo6O24]-хН2О [438, 440] — была выделена в виде белого труднорастворимого мелкокристаллического порошка. Получена также свободная гетерополикислота с основностью, равной восьми (определено потенциометрически и термогравиметрически).
В присутствии Те (IV) в водном растворе кремнемолибденовой кислоты происходит замещение атома Si на Те и образуется более прочный бесцветный теллуристомолибдат состава Те : Мо =1:6-[221].
В присутствии Те (IV) в подкисленном растворе фосфорномо-либденовой кислоты существует окрашенная в желтый цвет тройная фосфорнотеллуристомолибденовая кислота состава Р : Те : : Mo = 1 : 1 : 11 [212].
44
Были синтезированы гетерополикислоты с центральным атомом Se [313]. Получены в свободном состоянии селеномолибдено-вая и селеновольфрамовая гетерополикислоты. Основность их была определена потенциометрическим титрованием раствором КОН.
Взаимодействием H2SeO3 и Н2МоО4 (1:4) в водном растворе NH3 при 110°С получен белый кристаллический порошок состава 3(NH4)2O-2SeO2-5MoO3-30HaO, нерастворимый в этаноле, эфире, бензоле, но растворимый в воде. Водные растворы при 20°С устойчивы, а при нагревании разлагаются до МоО3 и SeO2. Получены следующие соли:
3AgaO • 2SeOa • 5МоОз • 17НаО,
РЬО • 2SeOa • 5МоОз • ЗНаО,
FeaOs- 2SeOa- 5МоОз- 24НаО,
СеаОз 2SeOa • 5МоОз • 9НаО,
4 (NH3)a 2SeOa • 9МоОз 12НаО.
Состав комплексов подтвержден кондуктометрическим титрованием [1001].
Желтый селенистомолибденовый комплекс был выделен при взаимодействии Se(IV) с молибдатом [437, 4381.
Соединения теллура с оксикислотами
При взаимодействии дикарбоновых кислот [10001 с раствором ТеС14 в бензоле образуются соединения Те [(СН2)2(СОО)2]2 с янтарной кислотой, Те[(СН2)4(СОО)2]2 с адипиновой кислотой и Те[С6Н4(СОО)2]2 с фталевой кислотой.
Оксикислоты (салициловая, галловая, таннин, малоновая) образуют с теллуром соединения Те(ОНС6Н1СОО)4, Те[(ОН)3С6Н2СОО], Те(С14Н8О8)2, Те[СН2СН(ОН)(СОО)2]2. Все выделенные вещества растворимы в этаноле, ацетоне и этилацетоне, но нерастворимы в эфире, хлороформе и четыреххлористом углероде.
Глава 3
КАЧЕСТВЕННОЕ ОБНАРУЖЕНИЕ СЕЛЕНА И ТЕЛЛУРА
РЕАКЦИИ НА СЕЛЕН
Большинство методов обнаружения селена и теллура основано на восстановлении этих элементов до элементного состояния. Реакции обнаружения селена и теллура приведены в табл. 8 и 9.
Таблица 8
Обнаружение селена
Реактив Реакционноспособная форма Результат реакции Открываемый минимум, мкг/мл Мешающие элементы
Тио мочевина Se(IV) Розовое окрашивание или красный осадок 5 Те, NO2, Си, Hg, Bi, Au, Pt, Pd
Гидроксиламин солянокислый Se(IV) То же 5 Многие элементы; Те не мешает
Йодиды Se(IV) Красно-коричневый осадок 40 As (III), Ge (IV), Mo (VI); Те не мешает
Роданистоводород-ная кислота Se (IV) То же 2 As, Sb, Sn, Fe(II), MoO2~
Пиррол Se(IV) Пирроловая синь 0,5 Окислители; Se (VI), Те (IV), Те (VI)
Асимметричный ди-феиилгидразин Se(IV) Красное окрашивание 2 Окислители; Те не мешает
Метиленовый голубой и сульфид натрия Se° Обесцвечивание метиленового голубого 3 Окислители
Молибдат аммония Se(IV) Молибденово-селе-нистая синь 3 POf, sof
3,3'-Диаминобензидин Se(IV) Желтое окрашивание или красная флуоресценция 2 Окислители; Fe(III), Си (II)
2,3-Диаминонафта-лин Se(IV) То же 2 Окислители
46
Обнаружение теллура
Таблица 9
Реактив Реакционноспособная форма Результат реакции Открываемый минимум, мкг! мл Мешающие элементы
SnCh Те (IV) Черный ссадок или ксричневоеокраши-ваиие 10 Se, Au
SnCh в щелочной среде Те (IV) То же 30 Ag, Hg, Cu, Bi, Sb
CuSOa и персульфат натрия Те (IV) Желтее окрашивание 0,2 JO;, SeOf
Гипс фссфс ристая кислота Те (IV) и Те (VI) Коричневое скрашивание 10 Pt, Cu, Ag, Au, Se
Тисмочевина в присутствии если Рей-неке Те (IV) Розово-красный осадок 2 Восстановители
Антразо Полису лыф ид натрия Те (IV) Те» Синее окрашивание Коричневое скрашивание или черная муть 2,5 10 Sb, Sn, Hg, Pt, Bi Многие элементы
Обнаружение селенистой кислоты при помоши тиомочевины
Лучшими восстановителями для солей селенистой кислоты являются тиомочевина и солянокислый гидроксиламин. При действии на селениты тиомочевиной в зависимости от содержания селена выделяется красный осадок элементного селена или раствор окрашивается в розовый цвет [657]. Мешают нитриты и большие количества меди; Те, Hg и Bi дают желтые осадки. Возможно определение 5 мкг Se в присутствии 25 мкг Те. Мешают также Au, Pt и Pd. Открываемый минимум 5 мкг{мл при предельном разбавлении 1 : 200000. Чувствительность реакции можно увеличить в 5 раз при флотации элементного селена эфиром [661 ]. Осадок элементного селена флуоресцирует красно-голубым светом [655].
Выполнение реакции. На фильтровальную бумагу помещают немного порошка тиомочевины, которую смачивают каплей исследуемого раствора; при этом выделяется оранжево-красный осадок селена.
47
Обнаружение селена с гидроксиламином в присутствии других катионов
Для обнаружения селена в присутствии теллура применяется солянокислый гидроксиламин [3651. При открытии селена в сложных объектах (рудах, минералах) необходимо предварительное отделение мешающих катионов. Для этого используется обработка испытуемого раствора NaOH, при которой большинство мешающих катионов выпадает в осадок, в то время как селен (и теллур) остаются в растворе. В присутствии избытка щелочей адсорбции селена и теллура не наблюдается.
Выполнение реакции. К 3—5 мл испытуемого раствора добавляют в избытке 4 N раствор NaOH. Жидкость энергично взбалтывают, а затем осторожно кипятят. Полученный осадок отфильтровывают. К фильтрату добавляют конц. НС1 до сильнокислой реакции и немного (горошину) сухого солянокислого гидроксиламина. Жидкость кипятят в течение 2—3 мин. Если селена очень мало, выжидают несколько минут. Красный осадок или красная муть указывают на присутствие селена.
Выполнение реакции (в присутствии благородных металлов). К испытуемому раствору добавляют избыток 4 N раствора NaOH и несколько крупинок солянокислого гидроксиламина. После энергичного встряхивания раствор осторожно кипятят. Осадок отфильтровывают, к фильтрату добавляют конц. НС1 и дальше поступают так же, как в отсутствие благородных металлов.
Обнаружение селенистой кислоты при помощи иодидов
Селенистая кислота восстанавливается иодидами в кислом растворе; в результате образуются элементный селен и иод:
SeO' +- 4J -[- 6Н+ - Se° + ЗН2О + 2J2.
Окраска от иода исчезает при добавлении тиосульфата, а селен остается в виде красно-коричневого осадка. В этих же условиях теллуристая кислота реагирует с иодистоводородной кислотой с образованием красно-коричневого комплекса [TeJj2 . Этот комплекс разрушается и обесцвечивается при действии тиосульфата натрия. Селен, таким образом, может быть определен в присутствии значительных количеств теллура [926, 993]. Мышьяк и германий (IV) в отношении 100 : 1 подавляют реакцию. Мешает определению Mo (VI). Открываемый минимум 1 мкг Se/0,025 мл при предельном разбавлении 1 : 25 000.
Предложен метод последовательного обнаружения SeO|~, SeO^, TeOg“, ТеОГ селективным восстановлением этих анионов при помощи Na2SO3 или NHaJ [659].
Выполнение реакции. Каплю концентрированной иодистоводородной кислоты (или конц. KJ + конц. НО) помещают на фильтровальную бумажку. В центр пятна добавляют каплю исследуемого раствора. Появляется красно-коричневое окрашивание. Если селен отсутствует, пятно обесцвечивается каплей 5%-ного раствора тиосульфата натрия. Если Se есть, то красно-коричневое пятно остается.
48
Восстановление селенистой кислоты роданистоводородной кислотой HSCN позволяет обнаружить 2 мкг Se в 1 мл при разбавлении 1 : 200 000. Теллуриты уменьшают чувствительность реакции; мешают As3+, Sb3+, Sn2+, Fe2+, MoO4“ [885].
Обнаружение селенистой кислоты с пирролом
Селенистая кислота окисляет пиррол до «пирроловой сини» [5281. Эта реакция может служить для обнаружения селенистой кислоты, поскольку селеновая, теллуристая и теллуровая кислоты не реагируют с пирролом. Мешают выполнению реакции окислители: VO3, MoOg”, MnO], СгО4-, NO3, BrO3, JO3, JO7, [РО4- 12МоО3] 3-, Au3+, Hg2+, Sb(V).
Если реакция между селенистой кислотой и пирролом выполняется в концентрированном фосфорнокислом растворе, то чувствительность ее повышается в 25 раз при добавлении солей железа (III). В этих условиях образуется комплексный анион [Fe(PO4)2]3-, который связывает «пирроловую синь»; равновесие реакции пиррол + SeO2j2Se0 + пирроловая синь смещается вправо и таким образом увеличивается окислительный потенциал селенистой кислоты.
Выполнение реакции. Каплю 5%-ного раствора FeCl3 и 7 капель сиропообразной фосфорной кислоты добавляют к капле исследуемого раствора. Хорошо перемешивают. Затем добавляют каплю пиррола (1%-ный раствор в этаноле, не содержащем альдегидов) и снова перемешивают.
Зелено-голубое окрашивание указывает на присутствие селенистой кислоты. Чувствительность определения 0,5 мкг/Se, предельное разбавление 1 : 100 000.
Обнаружение селенистой кислоты с асимметричным дифенилгидразином
Селенистая кислота легко восстанавливается до элементного селена рядом органических соединений. Она окисляет асимметричный дифенилгидразин
NH2
до окрашенного в фиолетовый цвет хинонанилдифенилгидразона
Кислородные соединения теллура не реагируют с асимметричным дифенилгидразином. Эта реакция, таким образом, может слу
49
жить чувствительной и специфической реакцией на селен, поскольку элементный Se, селениды и селенаты могут быть легко в соответствующих условиях переведены в селениты [6641.
Мешают реакции окислители. Вольфраматы, молибдаты, соли Fe3+ и Сы2+ не мешают в присутствии оксалатов.
Выполнение реакции. Четыре капли асимметричного дифенилгидразина в ледяной уксусной кислоте смешивают с каплей 2 А НС! и каплей исследуемого раствора. В присутствии селенистой кислоты появляется красное окрашивание, которое постепенно переходит в коричнево-красно-фиолетовое. Если селена мало, то окрашивание появляется только через несколько минут. Чувствительность определения 0,05 мкг SeO2, предельное разбавление 1 : 1000000.
Теллуристая кислота обычно дает слабую реакцию, обусловленную содержанием примеси селена. По этой реакции можно определить 0,001% Se в ТеО2. После сильного прокаливания Те02 не дает этой реакции, по-видимому, из-за того, что следы SeO2 при прокаливании улетучиваются.
Выполнение реакции при анализе минералов, серы и теллура [€€4]. Анализируемый сульфидный минерал необходимо обработать смесью конц. НС1 и Н2О2 для переведения селена в селениты и селенаты и выпарить раствор досуха. Образец, кипятят в микротигле с 3—4 каплями 10%-ной Н2О2 и 4—5 каплями конц. НС1, операцию повторяют до полного растворения образца. Содержимое тигля затем выпаривают досуха для разложения следов Н2О2. Остаток обрабатывают 3—4 каплями конц. НО, добавляют несколько миллиграммов щавелевой кислоты и смесь выпаривают. Добавляют каплю разбавленной НО, 2 капли воды, 4 капли асимметричного дифенилгидразина. Если появится красное окрашивание (сразу или через несколько минут), то в пробе присутствует селен.
Ту же операцию (но без добавления щавелевой кислоты) можно использовать для определения малых количеств селена в сере или теллуре.
Проверка ТеО2 на содержание SeO2 показала, что практически все образцы содержат значительные количества селена. ТеО2 можно очистить длительным прокаливанием.
Для восстановления селенитов применяют также /-аскорбиновую кислоту. Эта реакция пригодна для обнаружения селена в присутствии теллура и других металлов. Реакцию проводят при pH 1. Нагревание способствует восстановлению селенитов [33, 1082].
Обнаружение селенистой кислоты метиленовым голубым [662]
Специфическая реакция на селен была предложена Файглем и Вестом в 1947 г. [666]. Она основана на каталитическом действии селена на восстановление метиленового голубого сульфидами щелочных металлов до бесцветного лейкосоединения. Другие сильные восстановители — гипофосфит натрия, фосфит натрия — не мешают реакции.
Селен может быть по этой реакции легко определен в сере при соотношениях Se : S = 1 : 48000, что соответствует 0,002% Se в сере. Предел определения Se 0,08 мкг при разбавлении 1 : 1000000.
50
Выполнение реакции. Каплю воды и каплю исследуемого щелочного раствора помещают на предметное стекло. Добавляют по капле 0,2 М раствора сульфида натрия. Затем добавляют по одной капле раствора метиленового голубого к «холостому» раствору и к исследуемому и наблюдают за изменением их окраски. Если содержится селен, то окраска в этом растворе исчезает раньше.
Для обнаружения селенитов и селенатов применяют фенилантраниловую кислоту [294], а также л1-(меркаптоацетамидо)фенол (994]. Описано также применение гидразина для открытия селена в органических веществах [661]. Селениты и селенаты в растворах минеральных кислот могут быть определены восстановлением до элементного Se раствором FeSO< (предел определения 10 мкг Se) 1635] или аскорбиновой кислоты (предел определения 1 мкг Se) 1879].
Качественное открытие селената в присутствии селенита основано на разнице окислительно-восстановительных потенциалов селенита и селената в сильнокислом бромидном растворе. Селенат реагирует с бромидом по схеме:
Se (VI) 4- 2Вг- -► 2Вг’ 4- Se (IV).
С соответствующим редоксиндикатором — п-этоксихризоиди-ном — оказалось возможным определение селената в присутствии элементного брома, а также сульфата и стократного избытка селенита. Предел обнаружения 0,2 мкг Se(VI) в 0,3 мл раствора [517, 518].
Обнаружение селенигоз на бумажных хроматограммах по образованию комплексной молибденово-селенисгоа кислоты
Описан метод обнаружения селенитов на бумажных хроматограммах, основанный на образовании комплексной молибденово-селенистой кислоты,восстанавливающейся при облучении ультрафиолетовым светом до соответствующей сини. Чувствительность метода 3—4 мкг Na2SeO3 в 1 мл раствора [1037].
Пробу наносят на полоску бумаги Ватман № 3 и в качестве подвижной фазы используют кислый (75 мл изопропанола, 25 мл воды, 5 г СН3СООН, 0,3 мл 25%-ного NH4OH) или щелочной растворитель (30 мл 2-метилпропанола-1,30 мл безводного этанола, 39 мл воды, 1 мл 25% -ного NH4OH).
Бумажную хроматограмму высушивают и опрыскивают смесью, состоящей из 1 г молибдата аммония, 85 мл воды, 10 мл 1 N НС1 и 5 мл 60%-ной НС1О4. После высушивания хроматограммы в струе горячего воздуха ее облучают несколько минут светом кварцевой лампы (при этом вся хроматограмма окрашивается в голубой цвет) и оставляют до следующего дня. Затем выдерживают над концентрированным раствором NH4OH, пары которого обесцвечивают хроматограмму за исключением пятен, содержащих SeO3“, РО®~ и SO^'-
При использовании кислого растворителя значения R; указанных анионов различаются между собой, в то время как в случае применения щелочного растворителя значения R^ для SeO3~ и ТеО2- совпадают.
51
Обнаружение селена с 3,3'-диаминобензидином [510]
Из всех известных методов обнаружения селена наиболее чувствительные и селективные методы основаны на образовании пиазо-селенолов с 3,3'-диаминобензидином [127, 514, 752, 755] и другими, ортодиаминами: 1,8-нафталиндиамином [753, 1040], 4-диметилами-но-1,2-фенилендиамином и 4-метилтио-1,2-фенилендиамином [1049],. с 2,3-диаминонафталином [886].
При больших содержаниях в слабокислых растворах селениты образуют коричнево-красные осадки пиазоселенолов. При меньших содержаниях селена наблюдается желтое окрашивание. Чувствительность реакции повышается с применением экстракции окрашенного комплекса селена органическими растворителями — толуолом, бензолом и др. При облучении ультрафиолетовым светом пиазоселенолы флуоресцируют красно-оранжевым светом.
Открываемый минимум для реакции селена с 4-диметиламино-1,2-фенилендиамином составляет 0,05 мкг Se при предельном разбавлении 1 : 4000000 [1049]. Реакция селена с 2,3-диаминонафта-лином еще более чувствительна [886]. Теллур не мешает определению селена. Мешают окислители и большие количества железа и меди.
Выполнение реакции. К 1 мл нейтрального или слабокислого анализируемого раствора прибавляют каплю 1 N НС1 и каплю 1%-ного раствора 3,3'-диаминобензидина. Через 5—6 мин. прибавляют 0,1 г CH3COONa -ЗН2О и 2—3 капли толуола. После встряхивания толуол в присутствии селена окрашивается в желтый цвет.
Для устранения мешающего влияния других элементов предложено отделять селен дистилляцией в виде SeBr4. К 0,1—0,2 мл анализируемого раствора прибавляют 0,01—0,2 г КВг, каплю бромной воды и 1 мл конц. H2SO4. Стакан покрывают часовым стеклом, на котором висит капля 5%-ного раствора NH2OH -НС! (для восстановления брома). Содержимое стакана нагревают до появления белого дыма H2SO4 и смывают висящую каплю в пробирку. После этого открывают селен, как описано выше. Чувствительность реакции 0,05 мкг Se.
Для обнаружения элементного селена Файгль [662] рекомендует использовать реакции, основанные на образовании селенида галлия, селенида серебра, селеносульфата.
РЕАКЦИИ НА ТЕЛЛУР.
Так же, как и при открытии селенистой кислоты, многие методы качественного обнаружения теллура основаны на реакциях окисления-восстановления.
Обнаружение теллура раствором щелочного станнита [973]
При действии на теллуриты раствором SnCl2 на холоду в зависимости от содержания теллура выделяется черный осадок или раствор окрашивается в коричневый цвет [199, 365, 558, 1125].
52
Открываемый минимум составляет 10 мкг Те/мл при предельном разбавлении 1 : 100000. Мешает определению теллура золото. Вместе с теллуром выделяется и красный осадок селена [1038]. Чаще всего проводят вначале открытие селена с гидроксиламином и после отделения селена теллур восстанавлиют раствором SnCl2.
Если осадок элементных селена и теллура обработать 3%-ным раствором Н2О2, то теллур растворяется, а селен остается в виде красного пятна [957].
При восстановлении теллуритов и теллуратов в щелочной среде SnCl2 селен не восстанавливается, а восстанавливается только теллур [993, 1124, 1125]:
TeOj- + 2SnO*- + Н2О = 2SnOj' + Те0 + 2О1Г;
ТеО4~ + 3SnO*’ + Н2О = 3SnO|“ + Те0 + 2О1Г
Теллур выделяется в виде черного хлопьевидного осадка. Серебро, ртуть, медь, висмут и сурьма также восстанавливаются (до-металлов). Поэтому следует проводить определение теллура после отделения других элементов обработкой NaOH, как это описано ниже.
Выполнение реакции. Каплю раствора SnCl2, каплю 25%-ного раствора NaOH и каплю щелочного исследуемого раствора смешивают на предметном стекле. Если появляется черный осадок или раствор темнеет, то значит в нем присутствует теллур. Если теллура очень мало, окраска может появиться только через 1—2 мин. Для проверки необходимо ставить холостой опыт.
Открываемый предел 0,6 мкг Те в 0,025 мл при разбавлении 1 : 41000.
В присутствии селена можно определить 0,8 мкг Те при 100-кратном избытке Se.
Выполнение реакции в рудах и минералах [961]. ~ 250 мг пробы кипятят в фарфоровом тигле с 3—4 каплями 30%-ной Н2О2 и 4—5 каплями конц. НС1. Эту операцию повторяют 2—3 раза до полного растворения пробы. Остаток выпаривают почти досуха, прибавляют 3—4 капли конц. НО, осторожно нагревают, разбавляют водой до 5—6 мл, подщелачивают 10—15 каплями 20%-ного раствора NaOH, добавляют 1 мл воды и отстаивают.
Отбирают весь прозрачный раствор над осадком гидроокисей, подкисляют насыщенным раствором Н2С2О4, упаривают до 4—6 капель и испытывают на присутствие ртути (по Си — J-бумажке). Если присутствует ртуть, то ее удаляют перед открытием теллура (выпариванием с концентрированной НО).
К порции анализируемого раствора, из которого удалена ртуть, прибавляют в тигле каплю 20% -ного раствора NaOH и каплю раствора SnCl2. Появление зеленовато-черного осадка указывает на присутствие теллура. Другую порцию-анализируемого раствора подкисляют на белом предметном стекле раствором конц. НО и прибавляют каплю SnCl2. В этих условиях селен и теллур образуют красный и черный осадки. При обработке черного осадка 3%-ной Н2О2 осадок теллура растворяется, выявляя красный осадок селена.
Обнаружение теллура с солями меди
Чувствительная реакция на теллур была предложена Файглем в 1936 г. [400, 665] на основании образования в сильно окислительной среде (в присутствии персульфата) красно-коричневого
53
комплекса теллурата с трехвалентной медью. Кроме таллуровой кислоты, такую реакцию дают еще только перйодаты, т. е. эта реакция исключительно избирательна на теллур. »
Применяя методику «а», 1 мкг Те можно определить в присутствии 500-кратного избытка селенатов. Селениты мешают реакции и должны быть окислены до селенатов (бромом) до проведения реакции на теллур. Применяя методику «б», 2,5 мкг Те можно определить в присутствии 20000-кратного избытка селенатов.
Выполнение реакции. «. Каплю раствора CuSO4 (1 : 50 000) и каплю раствора NaOH добавляют к капле щелочного испытуемого раствора и добавляют немного твердого Na- или К-персульфата. Смесь доводят осторожно до кипения. Желтое окрашивание указывает на присутствие теллуровой кислоты. Чувствительность 0,5 .икгТе при разбавлении 1 : 100000.
6. Каплю раствора, содержащего Си2^ и Мп2^, и каплю раствора гипобромита натрия добавляют к капле испытуемого раствора в микропробирке. Параллельно выполняют холостой опыт на воде.
Обе пробирки нагревают несколько минут на кипящей водяной бане. В присутствии теллуратов раствор образца становится бесцветным или слабо желтоватым, в то время как «холостой» раствор сохраняет цвет перманганата. Чувствительность 0,2 ккз Те. Предельное разбавление 1 : 250000.
Теллуриты и теллураты восстанавливаются до элементного теллура при выпаривании с гипофосфористой кислотой [6621:
ТеО2’ + Н2РО- = Те0 + PO3f 4-Н2О;
2ТеО2- + ЗН2РО" = 2Те° -f- ЗРО3" + 2Н2О + 2Н+.
Мешают определению Pt, Си, Ag, Au, Se, также восстанавливаемые до металлов, поэтому теллур определяют после отделения селена восстановлением SO2 или солянокислым гидроксиламином.
Как уже было упомянуто выше (стр. 51), селениты и селенаты количественно восстанавливаются до элементного селена при нагревании растворов с FeSCU. Теллуриты и теллураты в этих условиях не восстанавливаются. Однако в присутствии фосфорной кислоты, образующей с Fes+ прочный комплекс [Fe(PO4)2]3-, восстановительный потенциал Fe2+ повышается настолько, что становится возможным и восстановление теллуритов и теллуратов [662]. Таким образом, если теллур должен быть определен в присутствии селена, вначале кислый раствор обрабатывают раствором FeSOa и после полного осаждения и отделения селена добавляют каплю фосфорной кислоты к капле фильтрата. Появление черного осадка указывает на присутствие теллура. Чувствительность 0,5 мкг Те при предельном разбавлении 1 : 100000.
В солянокислом растворе С>8,5 N НС1) тиомочевина в присутствии соли Рейнеке образует с теллуром розово-красный осадок двойного комплексного соединения: [Te(SCN2H4)J [Cr(SCN)4(NH3)2]2. Открываемый минимум — 1,85 мкг Те при предельном разбавлении 1 : 432400 [32, 129].
54
Обнаружение теллура при помощи антразо [206]
Кузнецов [206] предложил новую качественную реакцию на теллур с реагентом антразо (антрахинон-1-азо-4-диметиланилин). Реакция протекает в солянокислом растворе через образование аниона ТеС1|~. В присутствии теллура образуется сине-фиолетовый осадок. Селен не мешает обнаружению теллура. Подобную реакцию с антразо дают Sb, Sn, Hg, Pt, Bi. Проведение капельной реакции на бумаге, импрегнированной реагентом антразо, позволяет определить 0,06 мкг Te(IV) при разбавлении 1 : 10000.
Выполнение реакции. На розовую реактивную бумагу, импрегнированную реагентом антразо, помещают маленькую каплю исследуемого раствора, слегка подсушивают и смачивают каплей НС1 (уд. вес 1,12—1,19). При наличии Те (IV) быстро образуется синее или сине-фиолетовое пятно с резко очерченными краями.
Яркие пятна, которые могут быть приняты за пятно от теллура, образуют Sb, Sn, Hg, Pt и Bi. Отделить теллур от этих элементов можно обычными приемами.
Исследуемый раствор, содержащий Те (IV), Sb, Sn и Bi, выпаривают досуха, сухой остаток смачивают разбавленной HNO3 (1 : 100) и нагревают до кипения. При этом теллур переходит в раствор, a Sb, Sn и Bi остаются в осадке.
Так как реакцию с антразо дает только четырехвалентный, но не шестивалентный теллур, окислители, переводящие Te(IV) в Te(VI), вызывают исчезновение синего пятна, образованного Te(IV). Цветные пятна от прочих элементов при действии окислителей не меняются. Это может быть использовано в качестве проверочной реакции на теллур. Для быстрейшего исследования пятна удобно поместить на него по капле раствора нитрита с обеих сторон бумаги.
Обнаружение теллура в минералах. Содержащие теллур минералы обычно легко растворяются в царской водке. Поэтому обнаружение теллура проводят без разложения минерала прижиманием к минералу реактивной бумажки с антразо, смоченной смесью HNO3 (уд. вес 1,27) и НО (уд. вес 1,19).
При наличии в исследуемом минерале теллура на розовой реактивной бумажке образуется синее пятно. Минералы, содержащие висмут и сурьму, дают сходную реакцию. Применяя проверочную реакцию с нитритом, можно отличить пятна, образованные теллуром, от пятен, обусловленных присутствием сурьмы и висмута.
Обнаружение элементного теллура может быть основано на образовании сульфотеллурата [471, 561]. Бесцветные растворы сульфида натрия или аммония растворяют элементную серу и элементный селен с образованием полисульфидов или селеносульфидов. Однако моносульфид натрия не действует на элементный теллур в отличие от полисульфида натрия, который растворяет элементный теллур с образованием сульфотеллурата:
Те0 + (NH4)a S4 е» (NI14)2 TeSf.
Если теллур находится в смеси со свободной серой, то и сульфид аммония также будет растворять теллур, поскольку при растворении серы в моносульфиде образуется полисульфид.1
55
Желтый или красно-коричневый раствор щелочного полисульфида или полиселеносульфида обесцвечивается при добавлении сульфита натрия в результате образования тиосульфата или селеносульфата:
(NH4)2 SSx 4- х Na2SO3 -♦ (NH4)2 S 4- x Na2S2O3;
(NH4)2 SSex 4- x Na2SO3 -» (NH4)2 S 4- x Na2SSeO3.
В противоположность этому желто-красный раствор сульфотел-лурата (NH4)2TeS4 не обесцвечивается, но при нагревании с сульфитом выпадает осадок элементного теллура:
(NH4)2 TeS4 4- 3Na2SO3 ->• Те» 4- (NH4)2 S 4- 3Na2S2O3.
Поэтому, чтобы определить в образце элементный теллур в смеси с селеном и серой, раствор образца нагревают сначала с полисульфидом аммония, отфильтровывают или центрифугируют осадок. Затем добавляют твердый сульфит натрия до получения прозрачного раствора и нагревают. Если присутствует теллур, появляется черный осадок или черно-серая муть.
Обнаружение S, Se и Те пиролизом с Hg(CN)2
Одновременное обнаружение S, Se и Те было предложено Файг-лем [663] и основано на пиролизе S, Se и Те с Hg(CN)2 с выделением HCN, который затем обнаруживают при помощи ацетатов меди и бензидина.
Выполнение реакции. Для открытия S, Se и Те пробу помещают в микропробирку, добавляют 20—30 мг Hg (CN)2, пробирку погружают в глицериновую баню, нагретую предварительно до 130° С, и накрывают кружком фильтровальной бумаги, увлажненной раствором ацетата меди с бензидином [0,286 г Си (СН3СОО)2 растворить в 100 мл воды и смешать с 66,7 мл насыщенного раствора бензидинацетата]. В присутствии S, Se и Те на бумаге появляется синяя окраска. Открываемый минимум 8—10 мкг.
Глава 4
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА
Широкое применение селена и теллура в различных областях современной техники способствовало быстрому развитию новых методов количественного определения этих элементов. Если еще до совсем недавнего времени, кроме классических гравиметрического и титриметрического методов определения, практически единственным фотометрическим методом было определение селена и теллура в форме золей, то в настоящее'время аналитическая химия селена и теллура располагает многочисленными селективными и чувствительными методами определения этих элементов, в том числе фотометрическими.
Наибольшее число публикаций относится к изучению новых органических реагентов на селен и теллур и исследованию условий проведения количественного определения этих элементов с помощью этих реагентов.
В результате обследования широкого класса ароматических ортодиаминов были предложены новые флуориметрические, фотометрические и гравиметрические методы определения селена, которые намного превосходят по чувствительности, селективности и точности все до сих пор известные методы определения этого элемента. Такие реагенты, как 3,3'-диаминобензидин, 2,3-диамино-нафталин, о-фенилендиамин, в настоящее время нашли широкое применение при анализе различных селенсодержащих объектов.
В результате изучения большого числа серусодержащих реагентов на теллур были предложены новые спектрофотометрические методы определения теллура с висмутолом II и его производными. Перспективными являются экстракционно-фотометрические методы, основанные на образовании ионных ассоциатов ацидокомплексов теллура с родаминовыми красителями. Однако по чувствительности и селективности все известные к настоящему времени методы с использованием реагентов на теллур намного уступают методам определения селена с применением о-диаминов.
Дальнейшее повышение чувствительности количественных методов определения селена и теллура связано с развитием новых
57
физических и физико-химических методов определения селена и теллура — атомно-абсорбционного, рентгеноспектрального и нейтронно-активационного. Метод атомно-абсорбционного определения теллура был предложен в 1962 г.; исследования последних лет позволяют рассматривать этот метод как один из наиболее селективных и чувствительных.
Несмотря на то, что для селена и теллура имеется немного сведений о возможности их количественного определения кинетическими методами, эти методы, несомненно, являются очень перспективными. Развитие этих методов для селена и теллура должно идти параллельно разработке новых, более селективных методов концентрирования.
Обзоры современного состояния методов определения селена и теллура были сделаны в свое время Бусевым и Симоновой [67], Тараян и Арустамян [376], Мурашовой и Сушковой [254], Троицкой [391], Боком [533].
ГРАВИМЕТРИЧЕСКИЕ МЕТОДЫ,
Широкое применение получили гравиметрические методы, основанные на выделении осадков элементных селена и теллура неорганическими и органическими восстановителями. Однако несмотря на большую распространенность этих методов, все они недостаточно избирательны: в присутствии посторонних элементов (Cu, Hg, Sn, Sb, Bi, Au, Pt, Pd) возможны значительные ошибки в результате загрязнения осадков. Органические восстановители имеют некоторые преимущества перед неорганическими, так как их применение позволяет получать более чистые осадки. Подобный обзор гравиметрических методов определения селена был сделан в свое время Гутбером 1730].
Среди других весовых методов в настоящее время наиболее надежными, по-видимому, являются, кроме выделения селена и теллура в элементном состоянии, весовой метод определения теллура в виде ТеОа и весовой метод определения селена, основанный на образовании пиазосел енола.
Электрогравиметрические методы не характерны для определения селена и теллура.
Для осаждения селена и теллура в элементном состоянии применяются такие неорганические восстановители, как сернистый газ, гидразин, гидроксиламин, гипофосфит натрия, хлористое олово и др.
Восстановление сернистой кислотой
При использовании сернистого газа образуются наиболее'чистые осадки. Из концентрированных солянокислых растворов (8,8 N) селен из селенитов и селенатов осаждается без примеси теллура
58
1870]. В 3,7—4,8 N соляной кислоте сернистый газ осаждает как селен, так и теллур [869].
Выполнение определения селена. Смесь окислов, в которой содержится не более 0,25 г селена или теллура, растворяют в 100 мл холодной конц. НС1. При непрерывном перемешивании, следя за тем, чтобы температура не поднималась выше 30° С, в раствор вводят 50 мл конц. НО, насыщенной сернистым газом при комнатной температуре, и оставляют раствор стоять до полного выделения красного селена.
Осадок отфильтровывают через взвешенный тигель Гуча, промывают холодной конц. НО, затем водой до исчезновения реакции на хлор, затем этанолом и эфиром. Сушат красный селен 3—4 часа при 30—40° С для удаления эфира, а затем — 2 часа при 120° С. Взвешивают в виде элементного селена.
Теллур количественно осаждается при концентрации соляной кислоты в пределах 0,5—6 N [542]. В присутствии меди осаждение теллура неполное. При температуре 100° С в 2—5 N НС1 теллур осаждается количественно. Медь соосаждается незначительно. Сильно соосаждается ртуть; анализ осадка указывает на образование соединения 2HgTe-HgCl2. Частично соосаждается олово. При осаждении теллура сернистым газом из 5 N НС1 при 100° С незначительно соосаждаются также Cd, Tl, Pb, Sb, Bi, Мо. Сильно захватываются осадком Ru, Os, Rh, Ir, количественно осаждаются Pt, Pd. Обычно при снижении кислотности соосаждение металлов увеличивается [539].
При осаждении теллура сернистым газом эффективным промотором является гидразин [967]. При этом осаждение теллура происходит почти мгновенно, если концентрация соляной кислоты в растворе равна 3 N. Метод применим к теллуритам и теллура-там [402].
Восстановление обычно проводят в фильтрате, полученном после выделения селена сернистым газом [597]. Однако, если растворы содержат очень мало теллура (микрограммовые количества), то возможно неполное осаждение [480].
Выполнение определения теллура восстановлением сернистой кислотой и гидразином. Раствор, содержащий примерно 0,2 г Te(VI) и Te(IV) в 50 мл 3 N НС1 (примерно 25%-ной по объему), нагревают до кипения и прибавляют сначала 15 мл насыщенного раствора сернистого ангидрида, затем 10 мл 15% -ного раствора солянокислого гидразина и,’наконец, еще 25 мл насыщенного раствора сернистого ангидрида.
Продолжают кипятить, пока осадок не приобретает форму, в которой он легко отфильтровывается и промывается. Для этого требуется не более 5 мин. Дают отстояться, осадок отфильтровывают через взвешенный тигель Гуча и немедленно промывают горячей водой до удаления хлоридов. Затем, во избежание окисления, возможно быстрей отмывают воду этанолом и сушат осадок при 105° С.
Восстановление селенистой кислоты сернистой представляет собой сложный процесс, идущий через нестойкие промежуточные соединения, различные при различных соотношениях реагирующих веществ [479, 707]. При повышенной температуре в качестве промежуточного продукта образуется летучий монохлорид селена.
59
Чтобы уменьшить вероятность образования монохлорида, осаждение селена необходимо проводить на холоду в присутствии большого избытка восстановителя.
Для раздельного определения четырех- и шестивалентного селена рекомендуется проводить осаждение при 100° С в закрытом сосуде; в 0,5—1 # НС1 селен осаждается из селенитов, а в 4 7VHC1 — из селенатов [540, 1154].
Скорость и полнота реакции восстановления селена сернистой кислотой растут с увеличением концентрации соляной кислоты. Ионы С1_ и Вг~ ускоряют восстановление, что объясняется образованием промежуточных галогенидных соединений селена, которые восстанавливаются с большей скоростью, чем селенистая кислота [29, 2401.
Для уменьшения потерь селена рекомендуется проводить осаждение элементного селена в сернокислом растворе (5,5 М по H2SO4) при 80—90° С [569]. В этих условиях количественно осаждается селен из селенитов. Аморфный осадок красного селена при кипячении растворов переходит в черную модификацию, которая склонна прочно удерживать воду, трудноудаляемую в процессе просушивания осадка при 100° С. Поэтому при осаждении селена из холодного солянокислого раствора осадок красной модификации селена промывают соляной кислотой, водой, затем этанолом и эфиром и сушат 3—4 часа при 30—40° С, а затем — 2 часа при 120° С [115]. Чтобы избежать окисления селена, осадок элементного селена рекомендуется сушить в атмосфере СО2, промывку осадка этанолом или эфиром можно в этом случае опустить [1070]. Есть указания на то, что лучше всего сушить осадок элементного селена в вакууме при комнатной температуре [1017].
Восстановление селенистой кислоты гидразином катализируется монохлориодом JC1. Если в отсутствие JC1 реакция за 2 часа протекает только на 30—50%, то в присутствии JC1 в солянокислом растворе (5%-ном) или в смеси 5%-ной НС1 и 10%-ной HNO3 при 40° С реакция заканчивается за 30 мин. [368].
Гидроксиламин количественно и селективно осаждает селен в присутствии теллура из горячей 17%-ной НС1 или в присутствии лимонной и винной кислот [870]. Сернокислый гидроксиламин неприменим.
Восстановление солями Fe2+, Sn2+, Cr2+, Cu+, V2+ и др.
Быстро и количественно селен и теллур восстанавливаются до элементного состояния хлоридом двухвалентного олова [401, 963]. Возможно проведение реакции в присутствии значительных количеств азотной кислоты. Однако при осаждении селена и теллура с помощью SnCl2 наблюдается соосаждение сопутствующих металлов и олова, поэтому эта реакция используется чаще для выделе
60
ния суммы элементных селена и теллура и реже — для окончательного весового определения.
Для гравиметрического определения селена и теллура в виде металлов применялись растворы солей двухвалентного хрома 155, 1115]. Быстрому выделению и лучшей коагуляции осадков способствует добавление NaCl [1115].
Растворы солей двухвалентного железа в присутствии этилен-диаминтетрауксусной кислоты, фосфорной кислоты(связывающих же-лезо(Ш)в прочные комплексы) количественно восстанавливаютSe(IV) и Те (IV) до элементных, что может быть использовано для весового определения [1003]. Предложен гравиметрический метод определения селена в виде элементного путем восстановления селенатов, селенитов и селеноорганических соединений раствором Си2С12 в среде 3—4 V НС1 [927]. Определению селена не мешают F“, С1_, БОГ, СОГ, РОГ, Р2ог, ВОГ, МоОГ, VO-3, СН3СОО-, С2ОГ-Cu2+, Zn, Cd, Pb, Mn2+, Co, Ni, Pd, Al, As (III), Bi, Sb (III), Cr3+, Ce3+, Ce4+, Th, Pt (IV), Sn (IV), Ti (IV), ПОГ и Cr (VI).
Выполнение определения селена с Cu2Cl2. К 10—30 мл анализируемого раствора добавляют конц. НС1 до концентрации 3—4 N и 0,1 N раствора Си2С12 в 2 А’ НО до окончания выделения красного осадка элементного селена. После отстаивания (15—30 мин.) осадок фильтруют через стеклянный фильтр № 4, промывают 2—3 раза конц. НС1, затем — водой до исчезновения реакции на СГ, далее — этанолом и диэтиловым эфиром, высушивают при 105 °C и взвешивают.
Для одновременного определения селена и теллура 20—30 мл раствора подкисляют до кислотности 7 N по НС! и проводят осаждение и определение селена, как описано выше. Фильтрат упаривают, разбавляют водой до 2 N НО, и элементный теллур осаждают раствором Си2С12, после чего заканчивают анализ, как описано при определении селена.
Для восстановления и весового определения селена и теллура можно также применять соли двухвалентного ванадия [938].
Восстановление гипофосфитом натрия, тиомочевиной и др.
Арстамян и Тараян [26] изучили восстановление селенистой и теллуристой кислот гипофосфитом натрия. Было установлено, что количественное восстановление селенистой кислоты до элементного селена происходит в 5—7 N НО при температуре 50—60° С в течение 30 мин., а при кипячении раствора — в течение 15— 20 мин. Теллуристая кислота восстанавливается до элементного состояния в 0,5—4,0 V НС1 при температуре выше 70° С. Дробное восстановление селенистой кислоты в присутствии теллуристой кислоты проходит в 6—7 N НС1 при нагревании до 50—60° С. В 6 V НС1 реакция восстановления теллуристой кислоты гипофосфитом натрия катализируется Си2+. На процесс восстановления селенистой кислоты медь не влияет. Предполагают [854], что фос-форноватистая кислота восстанавливает Си2+ до Си+, последняя
61
выделяет теллур в элементном состоянии. В условиях восстановления теллуристой кислоты гипофосфитом натрия количественна осаждаются также золото, мышьяк, селен.
Для выделения селена и теллура в элементном состоянии используются также органические восстановители. Тиомочевина в кислом растворе восстанавливает селен до элементного; теллур образует с тиомочевиной окрашенные комплексные соединения, восстанавливаясь тиомочевиной только до двухвалентного [199, 275, 952, 9931. Метод используется для весового определения селена в присутствии теллура.
При большом избытке тиомочевины в сильносолянокислом растворе селен восстанавливается неполностью. Тараянисотр. [275J предполагают, что в этом случае возможно образование комплексных соединений низших валентностей селена (двухвалентного селена).
Селениты и теллуриты в 3,5 N НС1 при 40° С восстанавливаются 1-амидино-2-тиомочевиной (гуанилтиомочевиной) до элементных Se и Те по следующему уравнению [11111:
H2SeOs + 2C2H6N4S = Se° + 2C2H4N4S + ЗН2О.
Гравиметрическому определению селена и теллура этим методом мешает серебро.
Z-Аскорбиновая кислота в кислой среде применяется для выделения и весового определения селена в присутствии теллура [33, 650, 673, 10231. Реакцию восстановления можно представить следующим образом:
2C0HsO0 + Me2SeO3 -* 2С6Н0О6 + Se° + 2МеОН + Ц2О.
Наилучшее выделение осадка селена проходит при pH 1 в случае нагревания до 80—90° С. Теллур в количестве менее 1 мг/мл не мешает определению.
Глюкоза восстанавливает селениты в слабощелочных или нейтральных средах до элементного состояния. Осадок промывают этанолом и сушат при 80—90° С [6681.
Молочный сахар восстанавливает селениты, селенаты, теллуриты и теллураты до элементного состояния в слабощелочной среде (при pH 10—11) при кипячении раствора.
Метод обеспечивает быстрое выделение и высокую точность определения. При определении теллура добавляют ферроцианид калия, который препятствует восстановлению меди и селена до элементных [278]. Чтобы предупредить осаждение Fe, As, Bi, Sn, Sb, реакцию проводят в присутствии винной кислоты.
Тиосемикарбазид восстанавливает селениты и теллуриты в солянокислом растворе до элементных селена и теллура. При гравиметрическом определении селена осадок отфильтровывают, промывают водой, этанолом и эфиром и сушат в вакуум-эксикаторе 25—
62
30 мин. [10241. При выделении элементного теллура одновременно образуется некоторое количество TeS и элементной серы. Последнюю переводят в раствор обработкой осадка смесью CSa и СаН5ОН (1 : 1). Результаты определения теллура этим методом бывают немного завышенными (в среднем на 1,63%) из-за образования TeS [1021]. Мешают определению теллура Au, Ag, Си, Bi.
Тетраэтилгиурамдисульфид (СаН5)а—N—C(S)—S—S—С (S)—N— —(С2Н5)а восстанавливает Se (IV) и Se (VI) до элементного состояния. Теллур не восстанавливается. На основе этой реакции возможно гравиметрическое определение селена [897, 928].
Диэтилдитиокарбамат натрия количественно восстанавливает четырехвалентный селен до элементного в 2,5—4МНС1 или HaSO4 при кипячении раствора в течение 15 мин. В условиях восстановления селена теллур образует комплексное соединение Те (DDTC)4, .которое при кипячении сильнокислого раствора довольно быстро разрушается и выделяется элементный Те [10121.
Меркаптобензимидазол восстанавливает селенит-ион до элементного селена при нагревании солянокислого раствора. Гравиметрическому определению селена мешают Ag, Pb, Hg, Au, Си, Bi, Те 15511.
Определение теллура в виде ТеО2
Имеется метод гравиметрического определения теллура в форме двуокиси ТеОа, «’выделяющейся при гидролизе теллуритов при pH 4—5 [270, 338, 582, 585, 9471 или при действии органических (оснований — уротропина [595, 746], пиридина [1120].
Гравиметрический метод определения теллура в виде двуокиси (был описан еще в 1909 г. [557], однако применялся редко. Причиной, вероятно, было то, что одновременно с осаждением НаТеО3 происходил гидролиз поливалентных ионов в кислой среде. Однако в присутствии комплексона в слабокислой среде (pH 4—5) возможно количественное и селективное выделение теллуристой кислоты НаТеО3, которая быстро и легко может быть отфильтрована и просушена до весовой формы ТеОа.
Преимуществом этого метода перед другими методами выделения элементного теллура является то, что с осадком выделяющейся теллуристой кислоты в присутствии комплексона III не соосажда-ются примеси. Существенным преимуществом является и то, что весовая форма ТеО2 имеет меньший фактор пересчета (0,7995). Выделение теллуристой кислоты возможно и из азотнокислого раствора, что также упрощает обычную схему анализа на теллур.
В условиях выделения теллуристой кислоты селенистая кислота растворима и, таким образом, может быть отделена от теллура. Определение растворимости теллуристой кислоты [582] показало, что при pH 4,5 и 25° С в 100 мл раствора содержится 0,1 мг ТеО2. .Растворимость в 10%-ном этанольном растворе еще меньше.
63
Для уменьшения потерь Те вследствие растворимости Н2ТеО3 рекомендуется проводить осаждение в ацетоновом растворе [585 L Теллуристая кислота начинает осаждаться при pH 0,7 и растворяется при pH 8. Минимальная растворимость наблюдается при pH 4,0—5,0. Выпадающий осадок теллуристой кислоты вначале очень объемистый и аморфный, но при кипячении раствора быстро переходит в кристаллическую, легкофильтруемую форму.
Растворимость теллуристой кислоты растет с ростом температуры (при 40° С она увеличивается до 0,2 мг ТеО2 в 100 мл раствора), поэтому необходимо хорошо охлаждать раствор перед фильтрованием. Хлориды, нитраты, перхлораты и ЭДТА не влияют на растворимость. Сульфаты несколько повышают растворимость, что обусловлено, вероятно, комплексообразованием.
Выполнение определения теллура в виде ТеО2. К азотнокислому раствору образца в объеме 50 мл добавляют раствор комплексона III в количестве, необходимом для полного связывания присутствующих примесей, нейтрализуют аммиаком до pH 4,0 (по универсальной индикаторной бумажке).
Раствор с белым аморфным осадком теллуристой кислоты доводят до кипения и выдерживают на горячей плитке до перехода аморфного осадка в кристаллический (5—10 мин.). После этого охлаждают и фильтруют теллуристую кислоту через взвешенный тигель Гуча. Промывают осадок тремя небольшими порциями холодной воды, затем — этанолом и эфиром и просушивают при 120°С. Фактор пересчета на теллур 0,7995.
Прочие методы
Определение селена и теллура возможно при выделении в виде сульфидов [1045, 1103, 1106], образующихся при разложении тиосолей щелочным сульфидом.
При гравиметрическом определении малых содержаний теллура в некоторых случаях применяют тяжелые органические катионы типа тетрафениларсония [535], тетрафенилфосфония или тет-рафенилстибония [1151]. Тетрафениларсоний осаждает из солянокислого раствора (4,5 N) соединение состава [(C6H6)4As]2TeCle. Осадок просушивают при 110° С и взвешивают. Фактор пересчета 0,1153. Определению мешают нитраты. Селен даже в больших количествах не мешает определению теллура.
При взаимодействии тиомочевинного комплекса теллура с солью Рейнеке образуется соединение [Te(SCN-2H<)2] [Cr (SCN)4 (NH3)2J. На основе этой реакции предложен гравиметрический метод определения теллура [128]. Возможно также определение теллуровой кислоты в виде комплекса [Cr(NH3)e]2 [HeTeOe]3 [33].
2,3-Диаминонафталин количественно осаждает миллиграммовые количества Se (IV) в виде красного пиазоселенола состава C10HeN2Se [886]. Теллур не взаимодействует с 2,3-диаминонафта-лином. В присутствии маскирующих агентов (комплексона III, фторида натрия, оксалата натрия) возможно весовое определение селена в присутствии Fe, Cu, Zn, Al; мешают определению Се (IV) 64
и Ti (IV). Осадок пиазоселенола тщательно промывают и сушат при 110° С; он устойчив до температуры 290° С. Метод дает стандартное отклонение 1,52%.
Электрогравиметрические методы
Предложен метод определения селена посредством внутреннего электролиза с использованием пары электродов Си—Pt [3901, основанный на совместном восстановлении (за счет близости потенциалов восстановления) H2SeO3 и Си2+ металлической медью до Cu2Se. Это соединение является удобной весовой формой, устойчиво при нагревании до 130° С и негигроскопично. Абсолютная ошибка определения 0,3%. Метод использован при определении селена в технической SeO2. Теллур мешает определению.
Селен выделялся электролитически совместно с медью на Pt-электроде из азотнокислого раствора [166].
Количественное электроосаждение теллура проводилось из раствора HF—H2SO4 [915]. Электроосаждение теллура на Pt-катоде возможно из сульфатного, тартратного и сукцинатного растворов. Одновременно с теллуром на катоде осаждаются селен и медь [889 , 972].
При исследовании возможности электролитического выделения микроколичеств теллура на медном или Си—Pt-катодах лучшие результаты были получены в случае электролиза из раствора 2М H2SO4, содержащего 0,4 М (NH4)2SO4; 0,144 Na2C20i и 0,0644 NaNO3. Мешают определению Bi, Se, Sb и Cu [855].
Наилучшей средой для электрогравиметрического определения селена в присутствии теллура является раствор 10%-ный по NaOH и 20%-ный по NaCl. После выделения теллура раствор нейтрализуют соляной кислотой до нейтральной реакции и проводят электролитическое выделение селена. Если концентрация NaOH менее 10%, происходит совместное осаждение селена и теллура [10].
Изучен процесс электролитического выделения селена и теллура на Pt-электроде из растворов различных электролитов (HNO3, H2SO4, НС1, NH4OH, KCN) при контролируемой величине потенциала [912].
ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
Для определений миллиграммовых содержаний селена и теллура при анализе концентратов, шламов, пылей, сплавов, сталей, полупроводниковых соединений и других материалов часто применяют титриметрические методы. Большой вклад в развитие титри-метрических методов определения селена и теллура внесли работы Мурашовой [247, 248], Князевой [190] и Сырокомского [362].
Большинство титриметрических методов основано на окислительно-восстановительных реакциях. В качестве восстановителей
3 Зак. № 1476
65
при титровании селена и теллура применяют тиосульфат натрия, иодид, соли двухвалентного железа, двухвалентного хрома или трехвалентного титана. Однако наиболее распространенными являются тиосульфатный и йодометрический методы титрования. В качестве окислителей для Se (IV) и Те (IV) используют перманганат калия и бихромат калия. Разработаны методы количественного восстановления селенитов и теллуритов до элементного состояния различными восстановителями с последующим титрованием элементных селена и теллура растворами окислителей.
Известны также титриметрические методы, основанные на образовании труднорастворимых соединений селена и теллура. Комплексонометрическое титрование использовано для косвенного определения теллура. Имеется несколько работ по алкалиметрическому титрованию теллуровой кислоты.
Методы окислительно-восстановительного
титрования
Окислительно-восстановительные реакции для селена и теллура разбиваются на следующие группы.
1. Восстановление четырехвалентных селена и теллура до элементного состояния.
2. Окисление элементных Se и Те до Se (IV) и Те (IV).
3. Окисление четырехвалентных селена и теллура до шестивалентных.
4. Восстановление шестивалентных Se и Те до четырехвалентных.
Селен и теллур значительно отличаются по величинам окисли-тел ьно-восстановительных потенциалов.
Se -±2iZa_£_ HaSeOs -^1’16--H2SeO4;
Те +^Те02 +1^н2ТеО4.
С повышением концентрации водородных ионов окислительно-восстановительный потенциал селена растет, а у теллура — почти не увеличивается.
Различие в величинах окислительно-восстановительных потенциалов объясняет то, что некоторые реагенты (например, солянокислый гидроксиламин) восстанавливают селен, но не теллур.
Окисление четырехвалентных элементов до шестивалентных основано на следующих реакциях:
SeO*~ - 2е + Н2О = SeOf + 2Н+;
SeO|- — 2е"+2ОН"= SeO*- + Н2О;
ТеО.^ - 2ё+ Н2О = ТеО|~ + 2Н+;
TeOg- - 2е + 2ОН~ = ТеО*~ + Н2О.
66
Поскольку теллур легче окисляется, чем селен, некоторые окислители, такие как бихромат калия (Ео = 1,36 в) или перйодат (Ео = 1,6 в) [2151, не действуя на селен (IV), окисляют теллур (IV) до шестивалентного. Это свойство используется для количественного определения теллура в присутствии селена. Большинство других титриметрических методов определения селена требует предварительного отделения теллура или внесения поправки на его содержание.
Сущность тиосульфатного метода определения селена заключается в восстановлении селенистой кислоты избытком титрованного раствора тиосульфата натрия в слабосолянокислой среде.
H2SeO3 + 4Na2S2O8 + 4HCl=Na2S4SeO6 + Na2%0, + 4NaCl + 3H2O;
E« (S.Oe / SX>3~)= + °>03 e‘
Возможно также титрование в среде бромистоводородной и серной кислот, но не в присутствии азотной и фосфорной кислот [599, 971]. Избыток Na2S2O3 оттитровывают раствором иода [248, 256, 919, 920].
Для уменьшения разложения избытка тиосульфата в кислой среде рекомендуется вести титрование при 0°С и избегать большого избытка тиосульфата [617]. Теллуристая кислота в солянокислом растворе не реагирует с избытком тиосульфата и малым количеством йодистого калия.
Тиосульфатный метод определения селена используется для установки титров стандартных растворов, для определения селена в селенитах и селенатах [236], а также в более сложных объектах — селеномышьяковых шламах и продуктах их переработки [239], в рудах и породах [235, 1079].
При определении 2:—100 мг селена тиосульфатным методом ошибка составляет 0,4% [843].
Если после определения селена тиосульфатным методом требуется дальше определять теллур Йодометрическим титрованием, то применяют не обычный раствор иода в иодистом калии, а уксуснокислый раствор, содержащий эквивалентное количество соли ртути (II). Последняя связывает образующиеся при титровании иодид-ионы в комплексный ион HgJT.
Метод в последнем варианте применим к любым природным и технологическим объектам: рудам, концентратам, пылям, возгонам, шлакам, огаркам [283]. Мешают определению большие количества меди и золота. В этом случае проводят предварительное выделение Se и Те.
Чувствительность метода при навеске 10 а —п-10-4%. Относительная ошибка определения +2—10%.
з*
67
Йодометрическое определение селена и теллура возможно в нескольких вариантах.
1. Se (IV) и Se (VI) восстанавливают KJ или иодистоводородной кислотой до элементного состояния, освободившийся иод титруют тиосульфатом:
SeOg- + 4J- + 6Н+ = Se + 2J2 4- ЗН2О.
2. Те (IV) образует с KJ иодотеллуритный комплекс ТеДГ. который быстро и количественно реагирует с тиосульфатом с образованием пентатионата теллура (II) [813]:
TeJjF + 4S2O2- -> 6J- + Те (S2O3)^ + S4O|’.
Комплекс теллура Те (82О3)Г легко гидролизуется по уравнению:
Те (S2O3)2- + 2Н2О--Те (ОН)2 + 2Н+ Ц- гз^О^";
2Те (ОН)2=Те + Н2ТеО3 + Н2О.
Для устранения гидролиза теллуропентатионата и его стабилизации необходим избыток тиосульфата, который определяют обратным титрованием раствором иода.
При изучении влияния различных факторов (объема титруемого раствора, кислотности среды, количества добавляемого KJ, присутствия Вг" или Fe3+) на йодометрическое титрование селенитов было установлено, что этот метод является более сложным по сравнению с прямым тиосульфатным методом [822, 1079].
При восстановлении миллиграммовых количеств Se (IV) иодидом выделившийся красный осадок селена затрудняет титрование в присутствии крахмала [201, 246]. Установление конечной точки для этого случая проводят амперометрическим или потенциометрическим титрованием [344]. Также возможно выделять осадок элементного селена в слой органического растворителя (CCh) или на разделе фаз [836]. На измерении скорости реакции селенита с иодидом в кислой среде основано хронометрическое определение микроколичеств селена (3—13 мкг) с ошибкой 1,3%. Этот метод применим для определения произведения растворимости некоторых селенитов (Со, Zn, Cd, Са, Sr, Мп, Pb) [1025].
Йодометрический метод определения селенитов и теллуритов был использован при анализе минерального сырья, концентратов, шлаков [217, 283, 314, 692, 920], сплавов [250, 346], полупроводниковых материалов [222], органических соединений [764] и сталей [244, 247].
Возможно также йодометрическое титрование Те (IV), первоначально выделенного в виде комплекса с хлоридом тетрафенилар-сония [(CeH5)4As]2TeCle [535]. Осадок растворяют в соляной кислоте и титруют раствором иода.
Восстановление селенитов и селенатов проводят также растворами солей двухвалентного железа (солью Мора). Необходимо
К
добавлять пирофосфорную кислоту, которая понижает окислительно-восстановительный потенциал системы Fe3+/Fe2+ от Ео = = + 0,71 в до Е = + 0,42 в и, таким образом, позволяет использовать эту реакцию для количественного определения селена. Теллуриты не мешают этой реакции [105, 362, 1052]. Известны также методы восстановления селенистой кислоты растворами солей двухвалентного хрома [883, 951] или треххлористого титана [1121, 1122].
Титрование раствором КМпО4. Метод, основанный на окислении Те (IV) избытком КМпО4 и дальнейшем титровании избытка раствором соли Fe2+, был предложен Шренком [1058]. Основной трудностью прямого титрования теллурита КМпО4 в кислой среде является выделение осадка МпО2. Коричневый цвет осадка затрудняет наблюдение конечной точки титрования. При добавлении к титруемому раствору NaF, который образует комплекс с трехвалентным марганцем, а также солей одновалентной ртути возможно прямое титрование Те (IV) перманганатом калия [788].
Реакция окисления селенитов избытком КМпО4 протекает очень медленно [828]. При нагревании растворов на водяной бане реакция значительно ускоряется, однако одновременно происходит термическое разложение КМпО4, что приводит к ошибкам. В присутствии Na2HPO4, который препятствует осаждению МпО2, селен может быть количественно окислен избытком КМпО4; избыток окислителя оттитровывают сульфатом Fe2+ [513, 1058] или арсенитом натрия [1123].
Критический обзор перманганатометрических методов определения селена и теллура был сделан Пацук [295].
Титрование селенитов и теллуритов КМпО4. Раствор, содержащий 25 мл 40%-ной H2SO4, разбавляют водой до 150 мл, прибавляют 12 г Na2HPO4 и добавляют избыток 0,1N КМпО4. Через 30 мин. раствор титруют солью Мора или прибавляют избыток раствора соли двухвалентного железа и проводят обратное титрование перманганатом калия. При этом 1 мл 0,1М КМпО4 соответствует 3,96 мг Se или 6,375 мг Те.
Ошибка определения селена составляет до 0,25 абс.% (в 99%-ном чистом селене).
Была изучена возможность определения H2SeO3 и SeO3“ методом термометрического титрования раствором КМпО4. Точку эквивалентности находят по излому кривой титрования в координатах «расход титранта — АТ1». Она соответствует молярному соотношению H2SeO3 : КМпО4 = 5 : 2. Определению не мешают Ni2+, Cd2+, Zn2+, ТеО4“, SeO4', NO3. Мешающее влияние Fe2+, As3+, Sb3+, TeO3-, SO3~, S2~ и S2O3” устраняют предварительным окислением раствором К2Сг2О7. Метод позволяет анализировать двойные и тройные смеси Se—Те2+, Se—Те4+, Se—Sb3+, H2SO4—Н2ТеО3— —H2SeO3; HNO3—H2TeO3—H2SeO3 и H2SeO4—H2TeO4—H2TeO3 [617].
69
Четырехвалентный селен может быть также точно определен окислением избытком КМпО4 в присутствии NaF в сернокислом растворе [787, 788].
В щелочном растворе (0,5—4,0 М NaOH) теллурит окисляют раствором манганата [547, 785, 791]. При этом Мп (VI) восстанавливается до Мп (IV). Титрование дает точные результаты в присутствии небольших количеств Н2ТеО4.
В кислом растворе восстановление КМпО4 четырехвалентным теллуром идет через образование Мп (III). Дальнейшее восстановление Мп (III) —> Мп (II) катализируется солями рутения [857]. Поэтому окончательная реакция может быть представлена следующим образом:
2Мп (VII) +5Те (IV) —> 2Мп (II) + 5Те (VI).
На основе реакций окисления перманганатом калия четырехвалентных селена и теллура до шестивалентных были разработаны титриметрические методы определения селена и теллура в селенитах и сульфоселенитах свинца [259], селенитах и селенидах [513], сплавах [384], рудах [379].
Определение теллура бихроматом калия. Четырехвалентный теллур в присутствии Se (IV) определяют окислением бихроматом калия; избыток последнего титруют солью Мора. Реакцию можно проводить в солянокислом и сернокислом растворах [698, 859, 1057] с ферроином в качестве внутреннего индикатора или с феррицианидом в качестве внешнего индикатора [873]. При точном соблюдении условий титрования метод дает хорошие результаты.
Выполнение определения. К раствору добавляют 15 мл НС1 (уд. вес 1,19), доводят объем до 200 мл и приливают избыток титрованного раствора бихромата калия. Через 30 мин. добавляют небольшой избыток FeSO4 и затем его оттитро-вывают К2СГ2О;. 1 мл 0,1V раствора К/ДО; соответствует 6,345 мг Те.
В качестве окислителей для теллуритов применяют также перйодаты [363] и солиСе (IV) [727, 1152]. Для повышения эффективности действия солей церия реакцию проводят в присутствии катализатора AgNO3 4- Мп (NO3)2 в сернокислом растворе при нагревании до 95° С или в присутствии солей Cr (III). Избыток окислителя оттитровывают солью Мора с ферроином в качестве индикатора.
Разработаны методы количественного восстановления селенитов, и теллуритов до элементного состояния различными восстановителями — аскорбиновой кислотой, солянокислым гидразином, глюкозой [1051], галактозой [342], молочным сахаром [278, 279], сернистой кислотой [295, 296], тиомочевиной [210] с последующим титрованием растворами окислителей КМпСЧили K2Cr2O7, Ce(SO«)2.
Методы, основанные на предварительном осаждении селена в виде элементного в кислых растворах и дальнейшем титровании раствором КМпО4, являются более точными, чем метод прямого окисления селенита, и позволяют определять микрограммовые концентрации селена.
70
Реакция окисления коллоидного селена бихроматом калия ускоряется в присутствии катализатора — монохлориода [600, 858].
Иодатометрическое определение малых количеств селена основано на способности элементного селена легко растворяться в водном растворе цианида калия с образованием KSeCN [600, 649, 1065]. Селеноцианид затем разрушают в сильносолянокислом растворе О 4V), и селен титруют раствором йодата или другими окислителями.
Если в образце присутствует сера, то образовавшийся SCN" титруется аналогично SeCN~ [144]. Иодатометрический метод, основанный на образовании селеноцианида, дает для 12,0—0,04 мг Se более точные результаты, чем при применении тиосульфатного и перманганатометрического титрования.
Известен вариант титриметрического определения SeO3~ и SeO^”, когда после восстановления селена до элементного состояния аскорбиновой кислотой и переведения селена и серы в комплексы SeC.V и SCN", избыток CN" связывают в Ni (CN)4-, а избыток Ni2+ оттитро-вывают комплексоном III [708].
Описан метод, использующий иодатометрическое титрование иода, образующегося при окислении селена до Se (IV), в 0,3—4jV НС1 растворами монохлориода:
Se + 4 JC1 + 3H2O=H2SeO8 + 2J2 + 4НС1.
Выделившийся иод титруют раствором KJO3 в присутствии хлороформа [484].
Восстановление селенатов сернокислым гидразином в 1—2 N HaSO4 с последующим титрованием избытка гидразина йодатом позволяет количественно определить селен с высокой точностью [859, 1098]. Избыток гидразина может быть оттитрован хлорамином Т в солянокислом растворе с монохлориодом в качестве катализатора [1083].
На основании образования селеноцианида возможно йодометрическое титрование [1061, 1064, 1066]:
SeCN- + 4Вг2 + 4Н2О BrCN + SeO2’ + 8Н+ + 7ВГ;
BrCN + 2J- J2 + Br" + CN".
Этот метод использовался при определении примеси селенита в селенате [1063].
Другие титриметрические методы
Титриметрические методы, основанные на образовании труднорастворимых соединений с AgNO, и Pb(NO3)a, применяют для определения теллуритов [737, 1135]. Аргентометрическое титрование проводят в присутствии индикатора бромфенолсинего до окра
71
шивания коллоидного осадка Ag2TeO3 в зеленый цвет [1135]. Относительная ошибка рпределения составляет 1 %.
Титрование раствором Pb(NO3)2 проводят в присутствии флуоресцеина или симметричного дифенилкарбазона в качестве индикаторов. В точке эквивалентности при титрованиии с флуоресцеином зелено-желтая флуоресцирующая окраска переходит в краснорозовую, при титровании дифенилкарбазоном — оранжево-красная в фиолетовую. Метод позволяет определять 10—500 мг ТеОз~ с ошибкой 1—2% [1136, 1137].
Для определения селена используют метод, основанный на образовании осадка PbSeO4 [220, 1075]; избыток РЬ2+ оттитровывают комплексоном III. Для полного перевода SeOl- в ЗеОД применяют окисление раствором КМпО4 [1075].
Теллуровую кислоту можно определить по образованию высокоустойчивого гексамолибдотеллурового комплекса в щелочной среде:
Н6ТеО6 + 6МоО2~ [ТеМобО24]6- + 6ОН~.
Селен, присутствующий в равных количествах, не мешает определению теллура [564].
При косвенном комплексонометрическом определении малых количеств Те (IV) в присутствии Se (IV) комплекс четырехвалентного теллура с диэтилдитиокарбаматом экстрагируют в СС14 при pH 8,7—8,8; после взаимодействия с аммиачным раствором меди и после реэкстракиии определяют медь комплексонометрическим титрованием [969, 1147].
В случае прямого алкалиметрического титрования теллуровой кислоты применяют фенолфталеин в этанольном растворе (в присутствии пропиленгликоля) в качестве индикатора [642, 985].
Теллуровая кислота образует с глицерином одноосновную глицеринтеллуровую кислоту, которая может быть оттитрована с фенолфталеином в качестве индикатора [1033].
ФОТОМЕТРИЧЕСКИЕ И ФЛУОРИМЕТРИЧЕСКИЕ МЕТОДЫ
Определение селена и теллура в форме золей
Классическими методами определения малых количеств селена и теллура являются методы, основанные на колориметрировании золей этих элементов. Методы определения по образованию золей отличаются друг от друга главным образом применяемыми восстановителями. Хлорид двухвалентного олова является удобным реагентом как для теллура, так и для селена, в то время как гипофос-фористая кислота особенно пригодна для определения теллура, а гидразин — для селена.
72
Колориметрический метод определения селена и теллура на основе реакции восстановления этих элементов до элементного состояния раствором SnCl2 в кислой среде предложил Земмель [164]. В дальнейшем Шахов [436] изучил устойчивость коллоидных растворов селена и теллура, выделенных действием раствора SnCl2. Золи, полученные при восстановлении раствором SnCl2, достаточно устойчивы в солянокислых растворах (— 5—6N). Эти золи содержат заметные количества соосажденных окислов олова, в то время как золи теллура при восстановлении гипофосфористой кислотой [811] и золи селена при восстановлении гидразином являются, по-видимому, золями чистых элементов.
При соблюдении соответствующих условий проведения реакций можно обеспечить достаточно хорошую воспроизводимость оптических характеристик золей.
Определение селена и теллура хлоридом двухвалентного олова
Восстановление селена и теллура до элементного состояния хлоридом двухвалентного олова проводится в буферной уксуснокислой среде по способу Волкова [96] или в солянокислой среде по способу Земмеля [164]. Наибольшей чувствительностью обладает фотометрический метод определения теллура в буферном уксуснокислом растворе. При проведении реакций в солянокислом растворе чувствительность фотометрического определения селена и теллура можно повысить добавками катионов — меди, висмута, сурьмы и др. [43, 45, 46, 82, 164, 230, 231, 337]. На основании этих наблюдений Блюм и Глазкова [45, 46] рекомендуют проводить определение селена и теллура по окраске коллоидных растворов, полученных с присадкой сульфата двухвалентной меди. Для повышения чувствительности определения селена некоторые авторы рекомендуют применять добавку трехвалентной сурьмы [82, 231].
Эффект от присадки меди и сурьмы при определении теллура в солянокислом растворе значительно слабее, чем в случае селена. Восстановление хлоридом двухвалентного олова производится при комнатной температуре.
В 2—3N солянокислом растворе получаемые золи сравнительно устойчивы в отношении аггломерации и могут использоваться без добавления защитного коллоида. При добавлении гуммиарабика или желатины оптическая плотность растворов несколько снижается, но растворы становятся более устойчивыми в течение нескольких часов.
Измерение оптической плотности коллоидных растворов селена и теллура проводят на фотоколориметрах или на спектрофотометрах (для селена — при 390 нм [845], для теллура — при 440 нм [492]).
73
Выполнение определения селена [82]. Анализируемый раствор, содержащий 0,05—0,3 мг Se н 0,05—0,6 мг Те, помещают в мерную колбу емкостью 50 мл. Добавляют 20 мл НС1 (1 ; 1), 3 капли HNO3 (уд. вес 1,14), воду до объема 35 мл, и растворы перемешивают.
Прибавляют 1,0мл 0,1%-ного раствора сульфата сурьмы (III), 4 мл 0,5%-ного раствора желатины и 6 капель 25%-ного раствора хлористого олова в 20%-ной (по объему) соляной кислоте. Растворы доводят до метки и перемешивают.
Через 40 мин. оптическую плотность растворов измеряют на фотоколориметре ФЭК-М с синим светофильтром в кюветах с толщиной слоя 2 см по отношению к нулевому раствору.
Выполнение определения селена и теллура в солянокислом растворе [96, 164, 608]. Анализируемый раствор объемом 25 мл в 3 N НС1 должен содержать 0,1—1 мг Те (IV) или 0,2—2 мг Se(IV). В присутствии в анализируемом растворе азотной кислоты концентрация соляной кислоты должна быть соответственно снижена.
Добавляют 2 мл 10%-ного раствора хлорида двухвалентного олова при перемешивании. После развития окраски добавляют 3 мл 4%-ного раствора гуммиарабика. Разбавляют до 50 мл и измеряют оптическую плотность со светло-синим светофильтром в кюветах с толщиной слоя 1 см.
Аналогично получают нулевой раствор и устанавливают по нему прибор на 100%-ное пропускание. Калибровочную кривую получают, исходя из стандартных растворов теллура или селена.
При высоких содержаниях золота (свыше 10 г/m) его отделяют гидрохиноном [96, 98].
Определение теллура гипофосфористой кислотой
Изучение свойств коллоидных растворов теллура, полученных при восстановлении теллурита гипофосфористой кислотой, было проведено Эвансом [652, 654] и Джонсоном [811, 812, 816]. Гидрозоли элементного теллура, полученные при восстановлении гипофосфористой кислотой, имеют две полосы поглощения — одну в видимой, а другую в ультрафиолетовой областях спектра (рис. 4). Полоса в видимой области спектра заметно сдвигается от красного к ультрафиолетовому участку спектра с уменьшением размера час-
тиц, и соответственно изменяется окраска золя от темно-синей (при размере частиц около 600 нм) к пурпурным и красноватым оттенкам (при размере частиц около 300 нм).
При уменьшении размеров частиц наблюдается увеличение коэффициента погашения. Положение максимума светопоглощения в ультрафиолетовой области не зависит от размера частиц, но коэффициент погашения изменяется.
Рис. 4. Спектры поглощения золей теллура 1 — синий: 2 — пурпурный; 3 — красный;
4— желто-оранжевый
74
В связи с изменением оптических свойств золей в зависимости от размеров частиц необходимо очень строго контролировать условия получения коллоидных растворов. Воспроизводимость измерений оптической плотности золей повышается при уменьшении размеров частиц; величина ошибки колеблется от 0,3 мкг/мл для синих золей до 0,1 мкг/мл — для красных золей. Свойства золей зависят и от концентрации гипофосфористой кислоты (табл. 10).
Таблица 10
Влияние концентрации гипофосфористой кислоты на окраску золя теллура
Концентрация НзРО2, Л1 Окраска золя ^тах, нм
0,06 Синяя 580
0,12 Пурпурная 500
0,25 Красная 400
0,4 Желто-оранжевая 350
Концентрация ионов водорода может изменяться от 0,01 до 0,3 N. При меньшей концентрации гидролиз уменьшает скорость реакции и полноту ее протекания. В более кислой среде ускоряется аггло-мерация частиц. Наибольшая точность достигается при кислотности в пределах 0,1—0,2 N.
Для стабилизации золей добавляют 0,3—0,5% гуммиарабика. Применение желатины при концентрации 0,01% возможно только при изменении оптической плотности в видимой области, так как она сильно поглощает в ультрафиолетовой области.
Скорость восстановления сильно зависит от температуры. Восстановление обычно проводят при комнатной температуре.
В видимой области светопоглощение золей подчиняется закону Бера при всех длинах волн. Максимум поглощения в видимой области несколько сдвигается для пурпурного золя в зависимости от концентрации теллура.
В ультрафиолетовой области закону Бера подчиняются синие золи в пределах длин волн 240—290 нм. Коэффициент погашения для красных золей при 290 нм равен 53, при 250 нм — 47.
Выполнение определения. В коническую колбу емкостью 125 мл помещают анализируемый раствор, содержащий 0,1—0,7 мг Te(IV) и 1—8 мг-эк.в соляной кислоты. Добавляют 3 мл 4%-ного раствора гуммиарабика и доводят объем водой до 35 мл. Нагревают до кипения. При перемешивании быстро добавляют 5 мл 3 М гипофосфористой кислоты. Поддерживают температуру смеси близкой к температуре кипения в течение 15 мин., затем охлаждают водой 15 мин.
Переводят золь в мерную колбу емкостью 50 мл, разбавляют до метки и изменяют лптичрскию плотность красного золя при 240—290 нм (можно проводить элыних длинах волн — до 420 нм).
п
Определение селена гидразином [46, 739, 874, 892]
Золи селена, полученные при восстановлении гидразином, поглощают свет в очень широком диапазоне длин волн. Максимум поглощения лежит при 250—260 нм (рис. 5).
Оптические характеристики таких золей селена мало зависят от концентрации восстановителя. Только при очень высокой концентрации гидразина или высокой концентрации щелочи получаются частицы золя иной формы, золь обладает сероватой окраской и имеет сравнительно небольшой максимум поглощения примерно при 360 нм. В связи с тем, что гидразин заметно поглощает при длинах волн менее 260 нм, желательно работать при низких его концентрациях и измерять оптическую плотность при 260 нм.
Восстановление четырехвалентного селена гидразином зависит от концентрации ионов водорода в растворе. Реакция протекает очень медленно при pH 11, а при pH 9 — за одну минуту. Максимальная однородность частиц наблюдается при pH 8—11.
В кислой среде золи селена быстро коагулируют, поэтому необходимо применять защитный коллоид. В слабощелочной среде золи сравнительно устойчивы даже в отсутствие защитного коллоида.
Количественное определение селена при восстановлении гидразином проводится при pH 8—9. Оптимальная концентрация гидразина 0,9—1,5 Л4 [7391; оптическую плотность измеряют при 260 нм. В муравьинокислом растворе в качестве защитного коллоида используют поливиниловый спирт; оптическую плотность измеряют при 315 нм [874, 875]. Величина е при 260 нм равна 47.
Теллуриты и теллураты не восстанавливаются гидразином, однако в присутствии теллура возрастает светопоглощение растворов коллоидного селена (по-видимому, вследствие образования соединения Se12Te) [876].
В качестве восстановителя для колориметрического определения селена можно применять аскорбиновую кислоту [150, 395], Z-аскорбиновую кислоту [619]. Для стабилизации золя используют желатину, гуммиарабик или солянокислый гидроксиламин.
Фотометрические и спектрофотометрические методы определения селена и теллура, основанные на образовании коллоидных рас-
Рис. 5. Спектр поглощения красного золя аморфного селена (восстановление 20 мкг/мл селена гидразином)
76
творов, до недавнего времени очень широко использовались при определении селена и теллура в сталях [875], рудах [46, 85, 98, 232], колчеданах и пиритах [837], сплавах [608], пылях [621], при определении малых количеств теллура в присутствии больших количеств висмута [714], при определении селена в водах [334].
Определение селена и теллура в сульфидной руде [85]. Навеску 3—5 или 10—15 г (в зависимости от содержания селена и теллура в исследуемом материале) помещают в стакан емкостью 300 или 500 мл, смачивают водой, прикрывают стакан стеклом и, поместив его в холодную воду, приливают небольшими порциями 60 мл (120 мл) HNO3 (уд. вес 1,4) и оставляют на ночь.
На другой день раствор нагревают и осторожно кипятят в течение 10 мин. (до прекращения обильного выделения окислов азота). Споласкивают стекло водой, и раствор упаривают на водяной бане до небольшого объема. Дав раствору охладиться, приливают осторожно небольшими порциями при перемешивании 20 мл (45 мл) НгЗО4 (уд. вес 1,84) и упаривают на плитке с умеренным нагреванием до выделения паров серного ангидрида.
Прикрывают стакан часовым стеклом и, не прекращая нагрева, приливают небольшими порциями раствор, содержащий 0,5 г нитрата аммония, и затем, сдвинув стекло, продолжают дымить в течение 5 мин. Споласкивают стекло и стенки стакана 10—20 мл воды и вновь упаривают до выделения паров серного ангидрида в течение 5 мин., неплотно прикрывая стакан в конце упаривания часовым стеклом и наблюдая за прекращением выделения влаги на внутренней поверхности стекла. Обработку водой и упаривание повторяют.
При анализе углефицированных материалов нитрат аммония прибавляют небольшими порциями до просветления раствора. Частицы угля, приставшие к стенкам стакана, счищают стеклянной палочкой, смывают водой, раствор вновь упаривают до паров серного ангидрида и продолжают добавлять нитрат аммония до полного сжигания углистого вещества. Затем производят денитрацию раствора, как указано выше.
После охлаждения разбавляют раствор водой до 100 мл (200 мл), прибавляют 10 мл (20 мл) НСГ (уд. вес 1,19), нагревают и кипятят в течение 2—3 мин. Дав осадку отстояться, раствор фильтруют через плотный фильтр в стакан емкостью 400 мл (800 мл) и промывают осадок несколько раз горячей водой.
Если исследуемый материал содержит золото свыше 10 г/т и при кипячении с азотной кислотой разлагается полностью, не оставляя темного остатка, то нерастворимый остаток отделяют еще из азотнокислого раствора. С этой целью после кипячения с азотной кислотой раствор упаривают до сокращения объема вдвое, прибавляют равный объем воды и нагревают до растворения азотнокислых солей. Раствор фильтруют через плотный фильтр, и остаток промывают несколько раз горячей водой.
Фильтрат упаривают на водяной бане до небольшого объема, прибавляют серную кислоту в соответствии с величиной навески и производят денитрацию раствора, как указано выше.
Сернокислый раствор разбавляют водой до 100 мл (200 мл), прибавляют 10 мл (20 мл) НС1 (уд. вес 1,19) и кипятят в течение 2—3 мин. Если при этом остается нерастворимый остаток, то его отфильтровывают и промывают 2—3 раза разбавленной соляной кислотой, собирая фильтрат в стакан емкостью 400 мл (800 мл).
Методы определения селена
Среди многочисленных известных к настоящему времени методов определения селена можно выделить следующие.
1. Фотометрические и флуориметрические методы, основанные на образовании пиазоселенолов — соединений селена с о-диами-
77
нами. Эти методы в настоящее время широко используются при анализе самых различных объектов. Среди о-диаминов наиболее чувствительным и селективным реагентом на селен является 2,3-ди-аминонафталин.
2. Методы, основанные на образовании комплексных соединений с серусодержащими органическими реагентами. Несмотря на высокую чувствительность фотометрических методов с такими реагентами, как дитизон, висмутол II и другие, они не находят такого широкого применения, как методы первой группы, ввиду недостаточной избирательности.
3. Методы, основанные на окислении органических соединений четырехвалентным селеном до диазониевых солей. Последние реагируют с ароматическими аминами, давая интенсивно окрашенные азосоединения. Эти методы высокочувствительны, но требуют предварительного отделения мешающих элементов, а также окислителей и восстановителей.
4. Методы, основанные на образовании комплексов с двухвалентным селеном (фенильных замещенных семикарбазида и тиокарбазида). Отличаются достаточно высокой избирательностью. Теллур не дает подобных реакций. Наиболее чувствительной является реакция с 1,4-дифенилтиосемикарбазидом.
Определение селена с ароматическими о-диаминами
Селен может реагировать с ароматическими о-диаминами. Эту реакцию впервые описал Гинсберг в 1889 г. для о-фенилендиамина [752]. Ароматические м- и n-диамины и алифатические диамины не дают реакции.
З, 3'-Диаминобензидин. Селенистая кислота образует с 3,3'-ди-аминобензидином в разбавленных растворах окрашенное в желтый цвет комплексное соединение — 3,4-диаминофенилпиазоселенол 1980].
Образующийся при этом монопиазоселенол содержит две свободные аминогруппы и является довольно сильным основанием. Это соединение не экстрагируется органическими растворителями из кислого раствора. Из нейтрального и щелочного растворов соединение экстрагируется толуолом, бензолом и другими растворителями [581].
Метод определения селена с 3,3'-диаминобензидином предложили Хост и Гиллис [755, 756]. Пиазоселенол образуется в водном растворе при pH 2—3. Экстракция органическими растворителями делает метод более чувствительным и селективным [580].
В водном растворе пиазоселенол имеет главную полосу поглощения при 340 нм и небольшой максимум при 420 нм (рис. 6)'. Положение и величина максимумов в спектре поглощения зависят от pH раствора. Если в нейтральном или щелочном растворе спектр 78
Рис. 6. Спектры поглощения (/, 2) и флуоресценции (3, 4) комплекса селена с 3,3'-диаминобензидином
1,3 — экстракт холостого опыта в толуоле; 2,4 — экстракт комплекса селена
Рис. 7. Спектры поглощения монопиа-зоселенола в водном растворе (12мкг/мл) при различной кислотности
Значения pH: 1 — 9; 2 — 4,1; 3 — 1; концентрация НС1, АГ: 4 —1; 5 —5
поглощения имеет два пика (около 340 и 420 нм), то в кислом растворе пик при 420 нм постепенно уменьшается, а пик при 340 нм увеличивается и сдвигается в сторону более длинных волн. В 1 N НС1 имеется только один максимум при 348 нм (рис. 7).
В толуоле сохраняется полоса поглощения при 340 нм (348,8 нм — [9351, 339 нм — [980]), а полоса поглощения при 420 нм (418 нм — [980]) значительно усиливается. Максимумы в спектре поглощения реагента (3,3'-диаминобензидина) лежат при 275,6 и 340 нм 1935].
Растворы пиазоселенола в толуоле флуоресцируют [980, 1133]. Спектр флуоресценции приведен на рис. 6. Флуориметрический вариант метода определения селена дает более высокую чувствительность, чем фотометрический метод.
Наибольшая чувствительность реакции достигается при возбуждении флуоресценции светом с длиной волны 436 нм. Пиазоселенол флуоресцирует при 580 нм, а 3,3'-диаминобензидин — при 420 нм.
С 3,3'-диаминобензидимом реагирует четырехвалентный селен и не реагирует шестивалентный. Для полной уверенности в переходе Se (VI) в Se (IV) необходимо нагревать селенсодержащий солянокислый раствор (4 N). Для быстрого переведения элементного селена в H2SeO3 применяют смесь 12N НС1 и 0,3%-ной Н2О2 (1 : 1) [797].
Влияние природы органического растворителя на экстракцию пиазоселенола. Из нейтрального и щелочного растворов соединение экстрагируется толуо
79
лом, бензолом, хлороформом, мезитиленом, изоамиловым спиртом, ксилолом, амилацетатом, не экстрагируется н. гексаном и эфирами. Лучшим растворителем пиазоселенола оказался толуол [891]; иногда применяют бензол [127].
При использовании хлороформа получаются неустойчивые результаты анализа, особенно в тех случаях, когда в образцах присутствует медь [891]. Наибольшая чувствительность экстракцион-но-флуориметрического метода была достигнута при применении мезитилена [639]. Константа распределения для пиазоселенола в системе толуол — вода равна 25,4 [744]. Таким образом, при соотношении объемов Уорр : Уводи ~ 1 •’ 2550% соединение переходит в органическую фазу, при соотношении 1 : 10 экстрагируется 74% пиазоселенола.
Зависимость полноты выделения селена в органическую фазу (50мл хлороформа) от концентрации селена в водном растворе приведена ниже:
Концентрация селена, мкг/№ мл Полнота выделения. %
360 36 3,6 98 92 85
Из органической фазы можно реэкстрагировать селен 7 N серной кислотой (из 36 мкг обратно реэкстрагировано 89% селена [533]).
Влияние pH на образование и экстракцию пиазоселенола. Оптимальными условиями для образования комплекса является интервал pH 2—3 [580, 639, 1128]. При повышении кислотности до 0,1 N реакция образования пиазоселенола замедляется. В растворах с pH 3 ускоряются побочные реакции окисления З.З'-диаминобензидина, возрастает значение холостого опыта (при фотометрическом определении селена). Поэтому большинство исследователей рекомендует устанавливать pH 2— 2,4. Есть также указания на возможность выполнения реакции
Рис. 8. Влияние pH на экстракцию пиазоселенола
80
при pH 1 [145]. Максимальная экстракция [580] наблюдается в интервале pH 5—10 (см. рис. 8). Для количественного определения селена проводят экстракцию обычно при pH 6,5—7.
Влияние времени и температуры. В обычных условиях при pH 2 для максимального образования пиазоселенола требуется от 30 до 80 мин., в сильнокислотном растворе (2 N НС1) — более 3 час. Большие количества сульфатов замедляют реакцию: для достижения максимального комплексообразования требуется 2,5—3 часа [616]. При стоянии растворов на солнечном свету оптическая плотность и флуоресценция растворов уменьшаются. Флуоресценция также понижается с увеличением температуры. Для очень точных измерений интенсивности флуоресценции пробы необходимо термостатировать.
Для устранения влияния температуры и света на определение селена некоторые авторы [797] предлагают нагревать растворы определенное время на кипящей водяной бане и защищать их от прямого солнечного света. При нагревании растворов в течение 45 мин. при 50° С пиазоселенол образуется количественно, а поправка холостого опыта при флуориметрическом определении селена значительно понижается.
Чувствительность метода. Метод позволяет определять 1,0 мкг Se в 10 мл толуола [173, 728, 744, 797, 891, 1043]. Ошибка метода при работе в интервале концентраций 6,0—60,0 мкг составляет -Д 4,2%. Чувствительность фотометрического метода не может быть повышена из-за сильного поглощения растворов реагента.
Чувствительность флуоресцентной реакции селена с 3,3'-ди-аминобензидином равна 0,02 мкг в 5 мл экстракта [581].
Молярные коэффициенты погашения пиазоселенола приведены ниже [580, 935]:
Водный раствор е 10~2 Экстракт в толуоле е10-’
при 347—349 нм . . . 287 при 340 нм . . . .200
340 нм . . . . 289 420 нм . . . . 199
420 нм . . . . 177 348,8 нм . . . . 170
Устойчивость окраски. На прямом солнечном свету окраска меняется, образуются коричневые продукты окисления 3,3'-диаминобензидина. Поэтому определение селена с 3,3'-диами-нобензидином рекомендуют проводить в затемненной комнате или при желтом свете.
Мешающие элементы. Сильные окислители мешают определению селена из-за образования окрашенных продуктов реакции с реагентом. Диаминобензидин окисляется при этом до фиолетового семихинона.
Восстановители мешают из-за осаждения элементного селена. Влияние небольших количеств олова, которое попадает в раствор
81
после экстракции селена дитиолом, можно исключить, вводя фторид.
В присутствии комплексона III определению селена не мешают Hg, As, МоО|~, Cr3+, №2+, Со, не слишком большие количества Fe, Си. Теллур не реагирует с 3, З'-диаминобензидином.
Для отделения селена от больших количеств железа, меди и других мешающих элементов используют методы соосаждения селена с элементным мышьяком [173 , 261, 891], соосаждения пиазэсе-ленола с органическим соосадителем, например фенолфталеином [209], экстракцию железа метилизобутилкетоном, дитиолом [891 к
Мешают определению селена любые количества азотной кислоты. Большие количества сульфатов также мешают определению, так как связывают 3,3'-диаминобензидин в труднорастворимый сульфат [616].
Мешают Ti (III), Ti (IV), Sn (II), Sn (IV), Zr, V (V), Au (III), PO4', WO4~, C2O4~ и большие количества Cu, Fe (II), Fe (III) [171].
Калибровочный график, построенный для серии растворов сульфатов с добавлением 5—20 мкг Se, в которых селен определялся после осаждения в виде элементного раствором NH2OH-HC1, проходит параллельно графику (но несколько ниже), построенному для растворов селена, без его предварительного выделения [413]. Содержание селена в чистых реактивах (NH4)2SO4 и Na2SO4 составляет 2-10~® и 1-10~6% соответственно [413].
Синтез хлоргидрата 3,3'-д иаминобензидина [755]. Получение диацетилбензидина. В круглодонную колбу, снабженную обратным холодильником, помещают 25 г бензидина и 80 мл ледяной уксусной кислоты. Раствор кипятят на песчаной бане в течение 2 час. Полученный диацетилбензидин отфильтровывают на воронке Бюхнера, прибавляют 100 мл этанола и кипятят 1—2 мин. После охлаждения ацетилированный бензидин снова отсасывают на воронке Бюхнера и сушат на воздухе.
Нитрование. В стакан емкостью 200 мл, помещенный в смесь льда и хлористого натрия, вливают 47,4 мл дымящей HNO3 (уд. вес 1,48) и при помешивании вносят небольшими порциями (по 1—2 г) 15 г диацетилбензидина, следя за тем, чтобы температура не поднялась выше 40° С.
После добавления последней порции диацетилбензидина реакционную смесь вливают в 2—3 л холодной воды. При этом выпадает нитропроизводное желтого цвета, которое отсасывают и промывают горячей водой до тех пор, пока промывные воды не будут бесцветными, и сушат на воздухе. Выход 24 г.
Гидролиз нитропроизводного. 24 г полученного продукта вносят в 150 мл этанола, добавляют раствор 10 г едкого кали в 50 мл воды и кипятят 5 мин. Нитропроизводное при этом растворяется, затем быстро выпадают темно-красные иглы 3,3'-динитробензидина. После охлаждения кристаллы отфильтровывают на воронке Бюхнера от красного раствора и сушат на воздухе.
Восстановление. Из расчета на 5 г динитробензидина готовят раствор 27,5 г кристаллического хлористого олова в 25 мл 10 V НС1. Раствор нагревают на водяной бане и к нему добавляют небольшими порциями динитробензидин. После прибавления последней порции нагревание продолжают в течение 2 час.
Выделение хлоргидрата. К восстановленному раствору добавляют 50 мл 10 У НС1 и оставляют на ночь. Кристаллический осадок отфильтровывают на воронке Бюхнера. Затем его растворяют в 200мл воды и пропускают через раствор сероводород для удаления олова.
82
Осадок отфильтровывают и отбрасывают. Фильтрат обрабатывают концентрированной НО с таким расчетом, чтобы концентрация ее в растворе была ЗУ-Хлоргидрат осаждается полностью в течение 30 мин. Затем его отсасывают, высушивают в вакууме и сохраняют в темном месте в банке с притертой пробкой.' Очистка реагента. Продажный реагент представляет собой обычно желтоватобелый легкий кристаллический порошок. Красноватый оттенок указывает на частичное разложение реагента.
Очистку реагента проводят следующим образом. 3,3'-Диаминобензидин растворяют в небольшом количестве, воды (~0,1 г в 10—20 .ил) и взбалтывают с гранулированным активированным углем в течение 3 мин. в цилиндре с притертой пробкой. Раствор быстро отфильтровывают и добавляют НС1 до получения 4 N раствора. Стакан оставляют на 1—1,5 часа в темном прохладном месте.
Выпавший осадок отфильтровывают под вакуумом, промывают несколько раз 4 N НС1 и высушивают под вакуумом в эксикаторе. Высушенный реагент хранят в темной склянке с притертой пробкой в холодильнике.
При работе с 3,3'-диаминобензидином необходимо иметь в виду концероген-ность реактива [1074].
Фотометрический метод определения селена 3,3'-диаминобен-зидином широко используется при анализе различных объектов £555], при определении селена в металлическом теллуре [1178], в свинце и меди [696,891], в циркониевых сплавах [571], серной кислоте [616, 320, 1006, 1108, 1109], соляной кислоте [50, 51], сульфидных рудах и минералах [53, 84, 149, 173, 261, 644, 821, 1043, 1185], сталях [144, 966, 984], пылях [1185], водах [1028], углефициро-ванных рудах [86], метеоритах [637].
Часто необходимо быстро и надежно определять селен при Se-токсикозах в крови и волосах [610], в различных биологических материалах [145, 744, 984], почвах [1088, 1182], воздухе [1182], органических веществах [823].
Флуориметрический метод применялся при определении малых содержаний селена в полупроводниковых материалах [94, 95], биологических объектах [1133].
Колориметрическое определение селена в рудах по реакции с 3,3'-диамино-бензидином [127]. К 0,25—0,3 г руды прибавляют 3—5 мл конц. HNO3 и через 2 часа нагревают до разложения. Раствор упаривают до небольшого объема, прибавляют 5 мл НСЮ4 (1 : 1) и выпаривают до появления белого дыма. К остатку добавляют 1 мл воды и повторяют упаривание.
Остаток разбавляют 15—20 мл воды, нагревают до кипения и фильтруют. Осадок промывают 5—6 раз 2%-ной НС1О4. Если содержание селена меньше 100 мкг, то к фильтрату прибавляют 100 мкг Те и нагревают. К нагретому раствору прибавляют 40%-ный раствор SnCl2 в 20%-ной НС1 до восстановления Fe(III) и еще 2 мл, затем вводят 1 г N2H4 -2НС1 и кипятят 2—3 мин.
На следующий день отфильтровывают осадок селена и теллура на тампоне из беззольной бумажной массы и промывают 10—12 раз 10%-ным раствором НС1, содержащим 0,5% N2H4-2HC1. Осадок вместе с тампоном помещают в колбу, где производилось осаждение селена, вводят 10 Мл конц. НС1, 3—5 капель конц. HNO3 и нагревают на кипящей водяной бане 30 мин.
После растворения селена добавляют 10—15 мл воды и раствор фильтруют. Аликвотную часть фильтрата, содержащую 5—30 мкг Se, нейтрализуют до pH 1, добавляют 3 мл 0,1 М раствора комплексона III, 50 мл воды, 2 мл 2,5 М муравьиной кислоты. Устанавливают pH 2,5 и добавляют 2 мл свежеприготовленного раствора 3,3'-диаминобензидина. Через час добавляют раствор NH4OH до pH 6—7 и экстрагируют образовавшийся пиазоселенол бензолом.
83
Органическую фазу фотометрируют в кювете с толщиной слоя 3 см на фотоэлектроколориметре ФЭК-Н-57 со светофильтром № 2.
При определении 0,0008—0,29% селена абсолютная ошибка составляет 0,0001—0,03%.
Из других о-диаминов были исследованы в качестве реагентов на селен также о-фенилендиамин [515] и его замещенные — 4-диме-тиламинофенилендиамин, 4-метилтиофенилендиамин [149, 622,
1049, 1050], а также 4-метил-, 4-хлор-, 4-нитрофенилендиамин [913, 957].
Все производные о-фенилендиамина являются более слабыми основаниями, комплексные соединения селена с которыми могут экстрагироваться толуолом (и другими органическими растворителями) из кислых растворов. Максимумы в спектрах поглощения растворов пиазоселенолов расположены в видимой области спектра.
Для аналитических целей чаще других используют о-фенилендиамин [450, 494, 852, 1114].
Экстракционно-фотометрический метод с о-фенилендиамином позволяет определять от 10 до 150 мкг Se. Комплекс экстрагируется толуолом и хлороформом и может бытв обратно извлечен из органической фазы конц. H2SOi [880]. о-Фенилендиамин образует пиазоселенол с селенистой кислотой при pH 1,5—2,5. Оптимальное значение pH для экстракции равно 2,5.
Комплекс селена с о-фенилендиамином дает максимум в спектре поглощения раствора в хлороформе при 335 нм (кривая 4, рис. 9) [460]. Водный раствор о-фенилендиамина имеет максимум в спектре поглощения около 480 нм и в спектре флуоресценции — около 570 нм. Комплекс селена с о-фенилендиамином в . водном растворе не показывает дополнительных максимумов в спектрах поглощения (только при 480 нм) и флуоресценции (только при 570 нм).
Рис. 9. Спектры поглощения (У, 3—6) и флуоресценции ^ раствора о-фенилендиамина и его комплекса с селеном
/, 2 — о-фенилендиамин в водном растворе, pH 2; 3 — холостой опыт в хлороформе; 4—100 мкг Se в хлороформе; 5, 6 — холостой и 100 мкг Se, реэкстрагированных серной кислотой
84
Минимально определяемое содержание селена составляет 0,5 мкг в 5 мл экстракта. Коэффициент распределения при использовании хлороформа равен 30, что при соотношении фаз (1:1) обеспечивает извлечение 96% селена. Молярный коэффициент погашения равен 17 800 [460].
При работе с о-фенилендиамином необходимо учитывать легкую окисляемость кислородом воздуха как растворов, так и сухого реагента.
Мешают определению селена Fe (III), Sn (IV), J“, V (V), Bi, Hg, Sb, причем Fe (III) и V (V) могут быть маскированы комплексоном III. Олово и хром снижают окраску комплекса селена с о-фенилендиамином; их рекомендуется отделять в виде оксихинолина-тов [913].
о-Фенилендиамин применялся при определении селена в технической серной кислоте [515], определении примесей селена и основных компонентов в сульфидах и селенидах кадмия и цинка [852], анализе сульфидных руд и минералов [216], определении селена в продуктах цветной металлургии [451].
1,8-Диаминонафталин реагирует с селеном, образуя устойчивое соединение, экстрагируемое хлороформом [662, 1116]. Максимум в спектре поглощения растворов в хлороформе лежит при 372 нм. Это соединение использовалось для фотометрического определения селена в металлической меди и медных рудах. Минимально определяемое количество селена равно 0,5 мкг [1117].
2,3-Диаминонафталин. Из всех известных в настоящее время органических реагентов на селен из серии о-диаминов наиболее селективным и вместе с тем наиболее чувствительным является 2,3-ди-аминонафталин.
Реакцию селенистой кислоты с 2,3-диаминонафталином (DAN) впервые исследовали Паркер и Херви [981]. В результате был предложен высокочувствительный флуориметрический метод определения селена, позволяющий определять 0,002 мкг Se/мл. В дальнейшем Лотт и Ку кор [886] провели исследование этой реакции и предложили использовать 2,3-диаминонафталин для весового, спектрофотометрического и флуориметрического определения селена. Эта реакция очень селективна.
Для больших содержаний селена используют весовой вариант метода, а для субмикрограммовых количеств — флуориметрический вариант. Таким образом, по реакции с 2,3-диаминонафталином возможно определение селена в очень широком диапазоне концентраций и в самых различных по составу образцах. Определение селена в органических веществах с 2,3-диаминонафталином описано в статье Уоткинсона [1134].
Селенистая кислота реагирует с 2,3-диаминонафталином, образуя селенодиазоловое пятичленное кольцо. Реакция протекает в кислом растворе и имеет ряд существенных преимуществ перед описанной выше реакцией селена с 3,3'-диаминобензидином.
85
Рис. 10. Влияние pH и времени образования пиазоселенола (с 2,3-диаминонафталином) на оптическую плотность раствора
Значения pH: 1 — 1,7; 2 — 2,2;
3 — 1,0; 4 — 0,5; 5 — 3, 0
1. Более высокий коэффициент молярного погашения и более интенсивная флуоресценция Se-содержащих растворов, позволяющая определять 0,002 мкг Se/мл.
2. Метод более селективен, поскольку комплекс экстрагируется из кислых растворов. Селенистая кислота реагирует только с единственной о-диаминогруппой 2,3-диаминонафталина, в то время как у 3,3'-диаминобензидина остается свободной вторая пара аминогрупп, так что образующийся в последнем случае пиазоселенол еще относительно основной и может экстрагироваться в органическую фазу только при достаточно высоких значениях pH, Это значительно усложняет ход анализа в присутствии металлов, гидроокиси которых осаждаются в нейтральном и щелочном растворах.
Влияние кислотности водного раствора [581, 980]. Скорость реакции уменьшается с ростом кислотности, и при концентрации НС10,1 N при комнатной температуре реакция проходит неполно даже через 2,5 часа. Влияние pH на оптическую плотность представлено на рис. 10 [581] и 11 [756]. Максимальная оптическая плотность наблюдается при pH 2 после 100-минутного стояния растворов. Окраска устойчива 4 часа.
Влияние температуры водного раствора. Нагревание ускоряет реакцию, но также увеличивает скорость
<Р
Рис. И. Влияние pH на флуоресценцию при образовании пиазоселенола (с 2,3-диаминонафталином)
86
окисления 2,3-диаминонафталина. Максимальная окраска развивается за 5 мин. на кипящей водяной бане [262, 264]. При 5.0° С в 0,1 МНС1 реакция проходит количественно в течение 20 мин. Влияние pH на флуоресценцию (F) растворов при нагревании до 50° С представлено в табл. 11 [756]. Время стояния растворов перед экстракцией 30 мин.
Таблица 11
Влияние pH на флуоресценцию растворов
Концентрация НС1, N pH F экстракта
0,01 2 60
0,1 1 55
1 0 39
Таким образом, флуоресценция циклогексанового экстракта меняется в интервале концентраций 0,1—0,01 N НС1. При увеличении кислотности до 1 N НС1 скорость реакции замедляется, реакция проходит неколичественно. Оптимальной кислотностью является 0,1 N НС1. При этой концентрации скорость реакции достаточно велика и в то же время кислотность достаточна, чтобы удержать в растворе большинство металлов.
Степень экстракции. При однократной экстракции 0,1 мкг Se в 5 мл циклогексана селен извлекается практически полностью. Кукор [609] указывает на то, что процент экстракции пиазоселенола циклогексаном равен 23,8, толуолом — 22,2. В толуоле растворы неустойчивы, постепенно выделяют белое аморфное вещество, которое, вероятно, является полимером. Спектры поглощения и флуоресценции растворов в толуоле расплывчаты и сдвинуты в сторону более коротких волн.
Наиболее удобные экстрагенты — циклогексан и декалин. Спектры флуоресценции и поглощение в этих растворителях идентичны.
Спектры флуоресценции и поглощения. Кривые светопоглощения для реагента и пиазоселенола в воде и толуоле показаны на рис. 12. Максимум оптической плотности пиазоселенола наблюдается при 380 нм [262, 264]. Молярный коэффициент погашения комплекса при 376,8 нм равен 23 800 [581]. Максимум светопоглощения реагента — при 303,3 нм. Спектр флуоресценции имеет максимум при 520 нм. Для возбуждения флуоресценции раствор облучают светом X = 390 нм [756], 366 нм [264].
Точность и воспроизводимость метода. Холостой опыт соответствует 0,002 мкг Se. Флуоресценция холостого раствора изменяется от присутствия следов примесей. Величина поправки холостого опыта может быть сведена к минимуму при
87
Рис. 12. Спектры поглощения (1) и флуоресценции (2) комплекса селена с 2,3-диаминонафталином
Кривые 3,4 — соответствующие холостые опыты
перекристаллизации реагента и экстракции декалином или цикло-гексаном раствора реагента в 0,1 N НС1. Определение лучше всего проводить в комнате с желтым светом.
Уоткинсон [1134] рассчитал стандартное отклонение, равное 3,6—3,1 отн.%.
Зависимость интенсивности флуоресценции циклогексанового экстракта от содержания в нем селена имеет линейный характер в пределах концентраций 0,002—0,04 мкг Se/мл.
Мешающие элементы. Влияние примесей на фотометрическое и флуориметрическое определение селена показано в табл. 12 [886]. При флуориметрическом определении селена не мешают Fe, Cu, Hg, Sb, As, Те, Bi при соотношениях Se : Me = = 1:10 000 (0,1 мкг Se и 1 мг Me), а V, Au, Pt — при соотношении 1 : 100 [264]. Для устранения влияния больших количеств железа и меди (до 10—20 мг) предложено применять комплексон Ш, фторид натрия и пирофосфат натрия [264, 282, 628].
Синтез 2,З-д и ами нон афтал ин а [264]. Исходными веществами для синтеза 2,3-диаминонафталина являются 2,3-нафталиндиол и 25% -ный водный раствор аммиака. Синтез проводят в стальном автоклаве, рассчитанном на давление порядка 100 атм с вкладышем из фторопласта-4; объем камеры вкладыша 180 см3. Заполнение камеры исходными продуктами должно составлять д/ю часть — 2,0 г 2,3-нафталиндиола и 20 мл раствора аммиака.
Автоклав помещают в вертикальную тигельную электропечь с автоматической регулировкой температуры и нагревают до 250—260° С. При этой температуре автоклав выдерживают 5—6 час. После окончания синтеза и полного охлаждения автоклава содержимое вымывают в стакан емкостью 200 мл минимальным количеством раствора NH4OH, осадок отфильтровывают через тигель с фильтрующим дном, промывают небольшим количеством раствора NH4OH и высушивают в эксикаторе с СаО.
Полученный 2,3-диаминонафталин представляет собой кристаллическое вещество желто-зеленого цвета с т. пл. 192°С. (По литературным данным, т. пл. равна 190е— 195° С.)
О метку полученного реагента проводят экстракционным методом: 2,3-ди-аминоиафталин растворяют в 0,1 IV НС1, и примеси экстрагируют циклогексаном (на 0,1 г 2,3-диаминонафталина 100 мл 0,1 N НС1 и 30 мл циклогексана). Полученный солянокислый очищенный раствор используют для определения селена.
88
Таблица 12
Влияние посторонних ионов на реакцию селена с 2,3-диаминонафталином [886]
Элемент D f J Элемент D F
Se (IV) 0,31 89 Sn (IV) 0,29 82
Al 0,31 82 Sr 0,31 89
Au 0,32 88 Те 0,37 89
Ba 0,31 89 Ti 0,30 82
Bi 0,31 89 U 0,30 87
Ca 0,30 89 Sb (III) 0,22 89
Ce 0,33 88 Zn 0,32 88
Cd 0,30 88 Br~ 0,34 88
Co 0,31 80 ЭДТА 0,26 88
Cr 0,31 93 CIOs" 0,32 98
Cu 0,33 88 CN“ 0,35 91
Fe (II) 0,33 80 CIO~4 0,34 87
Fe (III) 0,34 80 C2O4’-- 0,31 76
Hg (II) ' 0,28 88 Цитраты 0,32 97'
К 0,32 91 Fe- 0,34 89
Mg 0,30 89 po43- 0,32 90
Mn 0,32 93 SO?~ 0,30 80
Na 0,33 89 no2- 0,54 31
Ni 0,29 80 NO3- 0,36 88
Pb 0,31 82
Примечание. D — спектрофотометрический метод. 10 мкг Se -р маскирующий агент + 2 мл 0,01 М раствора мешающего иона в 50 мл. F — флуориметрический метод. 1 мкг Se + маскирующий агент + 1 мл 0,01 М раствора мешающего иона на 50 мл раствора.
Определение селена с 2,3-диаминонафталинЬм [262, 264]. 0,1 —1,0 г образца (в зависимости от ожидаемого содержания селена) разлагают смесью конц. HNO3 и конц. НС1О4 (2 : 1) при нагревании. Если после полного удаления азотной кислоты и появления паров хлорной кислоты навеска разложилась неполностью, добавляют еще некоторое количество (2—5 мл) конц. HNO3 и продолжают нагое-вание.
После полного разложения образца и появления паров хлорной кислоты раствор охлаждают, добавляют 2—5 мл дистиллированной воды и снова нагревают до появления паров хлорной кислоты. Операция повторного выпаривания раствора до паров хлорной кислоты обеспечивает полное удаление из раствора следов азотной кислоты. Следует избегать длительного выпаривания раствора с хлориой кислотой, поскольку при этом возможен переход четырехвалентного селена в нереакционноспособные соединения шестивалентного селена. Рекомендуется , проводить выпаривание до появления паров хлорной кислоты.
В стакан добавляют 20—30 мл дистиллированной воды и нагревают раствор с осадком до кипения. После охлаждения отфильтровывают осадок через фильтр с синей лентой, промывают разбавленной НСЮ4 (1 : 100), осадок с фильтром выбрасывают. Раствор переводят в мерную колбу емкостью 50—100 мл и разбавляют водой до метки.
89
К аликвотной порции раствора (1—20 мл — в зависимости от содержания селена) добавляют 0,1 N НС1 до общего объема раствора 20 мл и устанавливают значение pH раствора, равное 1, по универсальной индикаторной бумажке добавлением соответственно НС1 или NH4OH.
Добавляют 2 мл раствора комплексона III и 5 мл раствора 2,3-диаминонафта лииа. Колбочки с растворами нагревают 5 мин. на кипящей водяной бане. После охлаждения раствор из колбочки переводят в делительную воронку, добавляют 5 мл циклогексана и экстрагируют в течение 1 мин. Органическую фазу отфильтровывают через маленький фильтр в пробирку с притертой пробкой.
После окончания экстрагирования серии образцов (8—12 проб) экстракты переводят в специальные пробирки от прибора ФАС-2 и измеряют флуоресценцию растворов с первичным светофильтром 366 нм и вторичным № 04 (с границей скрещивания 530 нм).
Калибровочные графики строят для интервала содержаний селена 0,01— 0,2 мкг. Измеряют флуоресценцию растворов на ФАС-2 при чувствительности 10; для интервала концентраций селена 0,1—0,5 мкг—при чувствительности 5 или 2. В каждой партии определений проводят холостой опыт через весь ход анализа. Значение поправки холостого опыта учитывают при расчете концентраций селена.
Определение селена с серусодержащими органическими реагентами
Известны фотометрические методы определения селена с диэтилдитиокарбаматом [538, 5431, диэтилдитиофосфор ной кислотой [541], толуол-3,4-дитиолом [591], диацетилдитиолом [590], дитизоном [897], 2-меркаптобензимидазолом [72] и продуктами конденсации тиогликолевой кислоты с толуидином и анизидином [73]. Все эти реагенты взаимодействуют также с теллуром и многими другими элементами: Cu2+, Bi (III), Cd2+, Pd2+, Tl3+, Sn (II), Sn (IV), Fe (II), Fea+(III), Co2+, Ni2+, Zn2+.
Высокая чувствительность некоторых реакций селена (например, с дитизоном) позволяет применять их после отделения мешающих элементов.
Дитизон. Селен и теллур, как и многие другие элементы (Мп2+, Fe (II), Fe (III), Со, Ni, Си, Zn, Pd, Ag, Cd, In, Sn, Pt, Au, Hg, TI, Pb, Bi, TR), реагируют с дитизоном [174 , 459, 895 , 897, 1168]. Дитизон взаимодействует с селеном и теллуром в сернокислом (1— 13 N H2SO4) и солянокислом (1—8 N) растворах. Дитизонаты количественно экстрагируются четыреххлористым углеродом из 6—7 N НС1 и 11—12 N H3SO4 [460].
На рис. 13 представлены спектры поглощения экстрактов дити-зонатов селена и теллура в СС14. Положение максимумов в спектрах поглощения практически совпадает (405—420 нм), но интенсивность окраски комплекса селена немного выше, чем комплекса теллура. Молярный коэффициент погашения дитизоната селена в CCh равен 7,55-104. На прямом солнечном свету оптическая плотность экстрактов дитизоната селена быстро снижается, поэтому необходимо работать в затемненной комнате или при электрическом освещении. В этих условиях оптическая плотность сохраняется неизменной в течение 3—4 час.
90
Рис. 13. Спектры поглощения дитизонатов селена и теллура
1 — холостой опыт; 2 — 10 мкг Se;
3 — 100 мкг Те
Коэффициент распределения комплекса селена между органической и водной фазами равен 25, что при соотношении объемов фаз Кводн * Горг = 5 : 3 дает 94%-ное извлечение.
Калибровочные графики линейны для растворов, содержащих от 1 до 10—12 мкг Se. В случае спектрофотометрирования при 420 нм в кювете с толщиной слоя 3 см в 15 мл экстракта можно определить от 0,2 мкг Se. Коэффициент вариации для концентраций селена Ю-3—10-4 % на превышает 15%.
С дитизоном в кислом растворе реагируют многие элементы — Au, Си, Hg, Ag, Те; хромат, ванадат, Fe3+ окисляют дитизон. Мешают также Sn, Zn, Bi, W, Nb, Ta.
Для устранения влияния большинства мешающих элементов селен и теллур предварительно отделяют соосаждением с элементным мышьяком [459] или экстракцией дитизонатов при pH _> 2 [для отделения Си, Hg2+, Те (IV), Bi, Ag]. Селен (IV) при этом экстрагируется не более чем на 1,5% [897].
При осаждении с мышьяком в осадок могут переходить золото, ртуть, серебро и некоторое количество меди. Ртуть удаляют предварительно кратковременным нагреванием пробы с порошком металлического железа, серебро — в виде хлорида, золото — восстановлением до металла [85]. Для удаления меди экстракт дитизоната селена промывают раствором ферроцианида, при этом связываются в комплексы также свинец и цинк. Присутствие до 10 мкг Те не мешает определению селена. В случае содержания до 200 мкг Те последний следует определять в другой аликвотной части раствора и вычитать поглощение, соответствующее теллуру [459].
Выполнение определения [459]. Аликвотную часть раствора с содержанием 0,2—10 мкг Se переносят в делительную воронку, добавляют 1 \N H2SO4 до общего объема 25 мл, затем приливают 15 мл 0,002% -ного свежеприготовленного раствора дитизона и встряхивают 30 сек.
После расслоения фаз сливают органический слой в другую воронку, добавляют 20 мл 25%-ного раствора ферроцианида и встряхивают 2 мин. Еще раз сливают экстракт в делительную воронку, для удаления избытка дитизона вводят 30 мл раствора NH4OH (1 : 100) и встряхивают 10—20 сек. После разделения фаз органический слой колориметрируют в кюветах с толщиной слоя от 0,5 до 3 см.
Содержание селена рассчитывают по калибровочному графику, построенному по растворам, содержащим 0, 0,5; 1,0; 2,0; 5,0; 10,0 мкг Se в 25 мл UN H2SO4, с которыми проводят весь ход анализа, начиная с экстракции дитизоном; колориметрируют эталонные растворы одновременно с пробами. Экстракты проб и шкалы должны находиться в одинаковых условиях освещенности.
91
п
Рис. 14. Спектры поглощения растворов, содержащих соединение селена с меркаптобензтиазо-лом в 7,5 + 0,2 N HCI
1 — 4 мкг/мл Se + 4 мл 0,1%-ного раствора реагента; 2 — 2 мкг/мл Se 4- 4 мл 0,1%-ного раствора реагента; 3 — реагент
Теллур определяют с бутилродамином С в другой аликвотной порции раствора с фотометрическим [173] или флуориметрическим [456] окончанием анализа; при содержании в растворе 10—20 мкг Те рассчитывают оптическую плотность его ди-тизоната и вычитают из суммарного поглощения, измеренного при колориметрировании селена в экстрактах проб. Используя полученную разность, находят по калибровочному графику содержание селена.
2-Меркаптобензтиазол образует с Se (IV) в сильносолянокислом растворе устойчивое желтое комплексное соединение [519, 527], спектр поглощения которого представлен на рис. 14. В щелочной и нейтральной средах селен не реагирует с меркаптобензтиазолом, а постепенно восстанавливается до элементного. Максимальная окраска возникает от pH 6,5 до 10 М НС1. В сернокислом растворе окраска гораздо слабее, в азотнокислом растворе окисляется реагент.
Для получения максимальной окраски требуется не менее 14-кратного избытка реагента. При нагревании окрашенных растворов до 60° С селен выделяется в виде элементного. При обычной температуре окраска развивается очень быстро и не меняется в течение 6 час. При 370 нм закон Бера соблюдается в интервале содержания селена 1—7 мкг. Коэффициент молярного погашения при 370 нм равен 9,48-103. Окрашенный комплекс легко экстрагируется хлороформом и ССБ, но чувствительность реакции при этом понижается.
Мешают определению селена Bi, Au, Ag, Pt, Pd, Fe2+, Sn2+, SO3”, J-, допустим 10—12-кратный избыток Те. В присутствии цитрат- и тартрат-ионов не мешают As (III), Sb (III) и W (VI). Fe (III) и Си (II) могут быть отделены экстракцией купферонатов хлороформом [115, 691].
Большие количества Сг3+ и Ni2+ могут быть удалены экстракцией голубой надхромовой кислоты этилацетатом [556] и экстракцией диметилглиоксимата никеля хлороформом [741].
2-Меркаптобензимидазол был предложен Бусевым [56, 72] в качестве реагента для фотометрического определения селена в отсутствие теллура. Этот реагент образует с Se (IV) соединение желтого цвета. Максимум светопоглощения водных растворов находится при 330 нм, молярный коэффициент погашения составляет 92
10 500. Соединение не экстрагируется бензолом, толуолом, эфиром, экстрагируется смесью бутилового спирта и хлороформа (1 : 5).
Закон Бера соблюдается для экстрактов в интервале концентраций 1—10 мкг/мл Se при 360—380 нм и 1—40 мкг/мл Se при 420 нм.
Определению селена не мешают умеренные количества Hg, Ag, Au, Cd и Pb. Мешают Bi, Cu, Fe, BrO3 и JO3, Те. Теллур может быть отделен иодидным методом. Определение селена может быть проведено после предварительного выделения селена осаждением SO2 или гидразином.
Выполнение определения. К 0,1—0,5 г анализируемого вещества добавляют при нагревании на песчаной бане 5 мл HNO3 (1 : 1), 2—3 мл H2SO4 (1 : 1), 1 мл концентрированной НО и упаривают наполовину.
По охлаждении вводят 5 мл воды и 2 мл НО (1 : 1), нагревают до 60—70° С, прибавляют — 2 г мочевины и нагревают еще 15 мин. Затем охлаждают, разбавляют вдвое водой, фильтруют через неплотный фильтр и разбавляют до концентрации селена 100—200 мкг/мл. К 3 мл 1%-ного этанольного раствора 2-меркап-тобензимидазола в делительной воронке прибавляют 2 мл концентрированной НС1, 1—2 мл исследуемого раствора, хорошо встряхивают и разбавляют водой до 12 мл, после чего экстрагируют 4 мл смеси бутилового спирта с хлороформом (1 : 5). Через час экстракт фильтруют через сухой фильтр и спектрофотометри-руют при 420 нм в кювете с толщиной слоя 1 см относительно чистого экстрагента.
Тиогликолевая кислота образует с Se (IV) и Те (IV) в 1—5А1 солянокислых растворах растворимые желтые комплексные соединения, хорошо растворимые в диэтиловом эфире, н.бутаноле, амилацетате, этилацетате и бутилметилкетоне. На основе этой реакции предложен экстракционно-спектрофотометрический метод определения селена и теллура [833]. Максимумы в спектре поглощения экстрактов лежат при 260 нм, и молярный коэффициент погашения равен 1600 (для селена) и 330 (для теллура).
Закон Бера соблюдается для интервала концентраций 6—34 мкг/мл (для селена) и 5—36 мкг/мл (для теллура). Методом изомо-лярных серий установлено, что селен с тиогликолевой кислотой реагирует при соотношении R : Se = 3 : 1, а теллур — 4:1.
Мешают определению селена и теллура с тиогликолевой кислотой Au, Cr, Си, Fe, Ge, Hg, V, As, Bi, Mo, Sb.
Производные тиэгликолевой кислоты — N-меркаптоацетил-га-анизидин и N-меркаптоацетил-га-толуидин [56, 73] — взаимодействуют с селенистой кислотой и при больших концентрациях селена образуют желтые кристаллические осадки состава R2SeO. Эти соединения хорошо растворимы в смеси н.бутанола с СНС13 (1 : 5), максимумы в спектре поглощения экстрактов находятся в области 310—320 нм, е — 1200. Реакция проводится в 0,05—1А1 солянокислом растворе. Закон Бера соблюдается в интервале концентраций 8—70 мкг Se/мл. Этот экстракционно-фотометрический метод позволяет определять селен в различных материалах, не содержащих теллура.
93
Висмутол II [1089, 1170, 1172]. В сильнокислом растворе ( >= 2N НС1 или H2SO4) висмутол II образует с селеном устойчивое желтое соединение, хорошо экстрагируемое при этой кислотности хлороформом и четыреххлористым углеродом. Максимум в спектре поглощения экстракта совпадает с максимумом для соединения теллура (330 нм). Соотношение Se: висмутол II в комплексе равно 1 : 4 [960, 1172]. Мешают определению селена: Те, Си, Hg, Tl, As, Pd; Fe*1" не мешает в присутствии 1 г Na4P2O7.
При спектрофотометрическом определении селена с висмутолом II к раствору, содержащему не более 20 мкг Se в 3.V НС1, добавляют 0,1%-ный раствор висму-тола II и экстрагируют 10 мл хлороформа. Экстракт промывают буферным раствором с pH 7,5 (для удаления избытка реагента) и измеряют оптическую плотность экстракта при 330 нм. Закон Бера соблюдается для интервала концентраций селена 0,5—20 мкг в 10 мл хлороформа.
Методы, основанные на окислении органических соединений четырехвалентным селеном
Для определения селена было предложено применять сульфат или фосфат кодеина [922, 1184]. В концентрированном сернокислом растворе образуется окрашенное в голубой цвет соединение. По-видимому, здесь происходит не окисление кодеина четырехвалентным селеном, а каталитическое окисление кодеина кислородом воздуха в присутствии следов селена. Из-за плохой воспроизводимости результатов реакция эта не получила практического применения.
Подобное каталитическое действие селена на окисление метиленовой сини в присутствии сульфида натрия было использовано в методе определения селена [666, 719].
Следует упомянуть также реакции селена с морфином и другими алкалоидами [690, 922, 1054], с пирролом [528, 903, 1099], асимметричным дифенилгидразином [664, 726], 2,2'-диантримидом [861, 860, 864], о-дифениламинокарбоновой кислотой [294], фенилгид-радин-п-сульфоновой кислотой [834], о-фенанитролином [1107], фенилантраниловой кислотой [297].
Селенистая кислота окисляет арилгидразины (фенилгидразин, нафтилгидразин, n-нитрофенилгидразин и фенилгидразин-п-суль-фоновую кислоту) до диазониевых солей, которые могут реагировать с ароматическими аминами, давая интенсивно окрашенные азосоединения [322, 834, 996]. Реакция селенистой кислоты с ди-фенилгидразин-п-сульфоновой кислотой в кислой среде может быть представлена следующим образом:
HSO3-</~/NH-NHs + HSeO; + Н+ -» HSO3--<^2/N+sN + Se° + 3H2O, _ _ (1> HSO3-<Qn-' SN + <Q>NH2 -> HSO3-^~^-N=N-^2^NH2 -t- H+ (2)
94
Реакция (1) проходит наиболее полно в сильносолянокислом растворе (>4,5 N) за 35—60 мин. Реакция (2) выполняется при pH 1,8—2,2, и поглощение измеряется через 10 мин. при 520 нм. В этих условиях оптическая плотность не изменяется в течение 6 час. Спектры поглощения растворов имеют максимум в области 520 нм. При этой длине волны поглощение реагентов незначительно. Закон Бера соблюдается для интервала содержаний 2—40 мкг Se в 25 мл раствора. Чувствительность реакции 0,01 мкг Se/мл.
Мешают определению селена окислители Cr2O>, JO3~, СЮ3, МпОд, ВгО7, МоО7, VO3, Fe (III), восстановители Fe2+, Sn2+, S2“, S2O3 , SO>, J-, As (HI), Sb (III), аскорбиновая кислота. Мешают также Hg+, Ag+, Pt2+, Au (III), Cu2+, Co2+, Cr3+.
Se (VI) реагирует подобно Se (IV), но более слабо. Те (VI) не мешает даже в 100-кратном избытке; 50-кратный избыток Те (IV) необходимо учитывать.
Поскольку реакцию окисления дифенилгидразин-п-сульфоно-вой кислоты проводят в сильносолянокислом растворе, использование комплексообразующих веществ для устранения влияния Fe (Ш) и Сп2+ неэффективно. Поэтому рекомендуется проводить предварительное экстракционное отделение железа и меди в виде купферонатов хлороформом.
Выполнение определения селена с дифенилгидразин-п-сульфоновой кислотой. При анализе образцов, которые могут содержать большие количества железа и меди, переводят раствор образца (в объеме не более 5 мл) в 40-миллилитровую делительную воронку, добавляют НО до 0,1 N и 1 мл холодного свежеприготовленного 5%-ного раствора купферона и экстрагируют двумя порциями по 10 мл хлороформа. Добавляют НО до 1 /V, 1 мл раствора купферона и экстрагируют 3 раза порциями по 10 мл хлороформа.
Переводят водную фазу в стакан и нагревают до удаления хлороформа, охлаждают, доводят объем до 10 мл и берут аликвотную часть раствора для определения селена.
Раствор, содержащий 2—40 мкг Se (не более 2мл), помещают в стакан емкостью 50 мл. Добавляют 4 мл 12 N НС1 и 3 мл 0,5%-ной фенилгидразин-п-суль-фоновой кислоты. Оставляют на 40 мин. Добавляют 2 мл 0,5%-ного раствора 1-нафтиламина и 10 мл 4М раствора ацетата натрия. Доводят pH до 1,8—2,2, проверяют pH на потенциометре.
Переводят раствор в 25-миллилитровую мерную колбу, доводят до метки и вставляют на 10 мин. Измеряют поглощение при 520 нм по сравнению с холостым опытом.
Метод определения селена с фенилгидразин-п-сульфоновой кислотой использовался при определении микроколичеств селена в металлическом германии и двуокиси германия особой чистоты [322].
Экстракционно-фотометрический метод был разработан на основе применения 1,Г-дифенилгидразина (ДФГ) [253, 664 , 726]. Селенистая кислота в 1,0—1,5 N солянокислом растворе восстанавливается 1,Г-дифенилгидразииом с образованием красно-фиолетовых продуктов окисления органического реагента, которые хорошо экстрагируются хлороформом, изоамиловым спиртом, дихлор-
95
этаном; плохо — бутиловым спиртом; не экстрагируются бензолом, толуолом, ксилолом, трибутилфосфатом, СС14, эфирами.
Реакция протекает при соотношении H2SeO3 : ДФГ =1:2. Максимум в спектре поглощения находится при 550 нм. Молярный коэффициент погашения экстракта в хлороформе равен 36 800, в водном растворе — 25 700. Чувствительность метода 0,05 мкг!мл.
Реакция 1,1'-дифенилгидразина с теллуром проходит очень медленно. Мешают ионы-окислители: Fe (П1), V (V), W (VI).
4,5-Диамино-6-тиопиримидин взаимодействует с Se (IV) при pH 1,5—2,5 с образованием желтоокрашенных растворов [577]; при этом, по-видимому, происходит восстановление Se (IV) до элементного селена. Максимум в спектре поглощения находится около 380 нм, е = 19200. Закон Бера соблюдается до интервала концентраций 0,1—2,5 мкг/мл. Окраска развивается 30 мин. и затем медленно ослабевает из-за постепенного выделения элементного селена.
Определению мешают Fe3+, Fe2+, Cu2+, S2O3 , SO|~, BrO3, C1O3. He мешают С2О4_, C1O4, J'. На основании реакции селена с 4,5-ди-амино-6-тиопиримидином разработаны методы определения селена в селенидах и сульфидах кадмия.
Методы, основанные на образовании комплексных соединений низших валентностей селена
Дифенилкарбазид (I), 1,4-дифенилсеми карбазид (II), дифенилтиокарбазид (Ш), фенилтиосемикарбазид (IV) и дифенилтиосемикарбазид (V) в солянокислом растворе (4,0—4,5 N НС1 для I и II, pH 1—2 для III, IV, V) восстанавливают цетырехвалентный селен до двухвалентного и образуют с Se (II) окрашенные комплексные соединения с соотношением Se (II): реагент =1:2 [74, 252, 360, 361]. Теллур не мешает определению селена.
Комплексные соединения селена с серусодержащими реагентами III, IV, V экстрагируются органическими растворителями ССЦ, СНС13, С6Н6, С2Н4С12 и др. Эти соединения в водном растворе при стоянии неустойчивы, постепенно разлагаются с выделением элементного селена, в то время как органические экстракты их вполне устойчивы. Соединения селена с I и II неустойчивы как в водном растворе, так и в органических растворителях, постепенно разлагаются с выделением элементного селена.
Спектры поглощения хлороформного экстракта комплекса селена с 1,4-дифенилтиосемикарбазидом (рис. 15) показывают, что наибольшая разница между поглощением растворов селенового комплекса и холостого опыта, которая может быть использована при фотометрировании, наблюдается около 340 нм.
Молярные коэффициенты погашения комплексов в водном растворе равны: для I—5600 (при 486 нм), для II — 770 (при 500 нм), для экстракта в хлороформе комплекса с III — 39 700 (при 460 нм), 96
Рис. 15. Спектры поглощения растворов комплекса селена с 1,4-дифенилтиосемикарбазидом в хлороформе
1 — холостой опыт; 2 — 250 мкг Se
экстракта в смеси бутанола и хлороформа с IV — 5200 (при 380 нм), экстракта в хлороформе с V — 44 000 (при 390 нм) и 15 000 (при 400—420 нм) [4601.
Чувствительность реакции селена (в мкг/мл) составляет: для I и II — 3; для III — 0,02; для IV—1,0; для V — 0,04.
Реакция селена с 1,4-дифенилсемикарбазидом мешают Си, Аи, Pt, Pd, Hg, Fe, Ag. При добавлении к раствору комплексона III и винной кислоты можно маскировать влияние железа и меди. Мешают определению селена окислители Cu2+, Fe (III), Hga+, СгС>4 и др. Фенилтиосемикарбазид дает интенсивно окрашенные соединения с Cu2+, V (V), Fe (III).
На основе реакций селенистой кислоты с фенильными замещенными семикарбазида, тиокарбазида и тиосемикарбазида были разработаны фотометрические методы определения селена в присутствии теллура [74, 252, 360, 361].
Для определения селена в природных' и промышленных объектах рекомендуется использовать наиболее чувствительную реакцию с 1,4-дифенилтиосемикарбазидом [255, 460].
Окраска подчиняется закону Бера в широком интервале концентраций: 0,04 — 20,0 мкг/мл — при 320 нм и 0,4 — 80,0 мкг/мл при 413 нм. Экстракты устойчивы в течение нескольких дней.
Коэффициент распределения селена между водной фазой с pH 2 и хлороформом имеет величину порядка 10, что при соотношении объемов фаз 1 : 2 соответствует 96%-ному извлечению селена [91%-ное при соотношении фаз 1 : 1].
Выполнение определения селена с 1,4-дифенилтиосемикарбазидом [252]. Осадок суммы селена и теллура растворяют в 10 мл концентрированной НС1 и 5 каплях HNO3 (уд. вес 1,40). Раствор переводят в мерную колбу.
К аликвотной порции анализируемого раствора добавляют 0,1 — 0,2 г мочевины, каплю фенолфталеина и нейтрализуют концентрированным раствором NH4OH до появления розовой окраски. Избыток NH4OH нейтрализуют 1 У НС1 и устанавливают pH 1—2. Универсальным индикатором проверяют pH раствора. Затем добавляют 0,5 мл 10-2 М раствора 1,4-дифенилтиосемикарбазида, перемешивают и дают постоять 10—15 мин.
4 Зак. № 1476 97
Добавляют 4 мл хлороформа (или четыреххлористого углерода) и экстрагируют 1—2 мин. После разделения фаз нижний слой сливают в сухую пробирку. Оптическую плотность измеряют на спектрофотометре при 340 нм или ФЭК-56 со светофильтром №1 (Х= 365 нм). Параллельно проводят холостой опыт. В качестве раствора сравнения используют растворитель (хлороформ или СС14).
Построение калибровочной кривой проводят в аналогичных условиях. В зависимости от содержания селена в анализируемом материале применяют стандартные растворы селенистой кислоты с конц. 1 -Ю-6 или 1 -10-4 М.
Методы, основанные на образовании комплексов селена с трифенилметановыми красителями
Селен в ION H2SO4 и 3N КВг реагирует с красителем виктория голубая Б с образованием синего соединения, экстрагируемого смесью бензола и ацетона (10 : 1).
Максимум светопоглощения лежит в области 580—640 нм; кажущийся молярный коэффициент погашения 3500. Прямолинейная зависимость между концентрацией селена и оптической плотностью наблюдается в пределах 5 — 20 мкг/мл [170].
Четырехвалентный селен реагирует также с бриллиантовым зеленым и малахитовым зеленым с образованием комплексных соединений синего цвета с максимумом светопоглощения в области 620—630 нм. Реакцию проводят в солянокислом растворе в присутствии иодида.
Не мешают определению селена Ni2+, Со21_, Те (IV), Вг", СГ, NO; и SOt. Мешают Sb (III), S2~, Hg (II), Hgt Ag+, Fe (III), Bi (HI), Cu2+, Pb2+, Sn2+ [320].
Другие методы
Предложен люминесцентный метод определения четырехвалентного селена и теллура в стеклообразных замороженных растворах в 7,5 — 8 N НС1 и НВг при 77° К [40,457]. Полосы люминесценции селена в НС1 и НВг лежат в области 550 нм, полосы возбуждения — при 330 и 352 нм. Чувствительность метода (4 — 8)-10-8 г/мл в среде НС1 и 1,22-10“9 г/мл в среде НВг.
Этим методом определяют селен в присутствии Tl, Pb, Bi й Sb [40].
Возможно спектрофотометрическое определение селена по реакции образования кетоно-хлорокомплекса Se (IV) с циклогексаном в среде 7 N НС1; молярный коэффициент погашения составляет 5,6-103 при 345 нм [606].
Известен косвенный фотометрический метод определения селената в присутствии селенита, основанный на фотометрическом определении трибромида, образующегося при восстановлении селената бромистоводородной кислотой [531].
98
Методы определения теллура
Из методов определения теллура наиболее широко представлена группа фотометрических методов, основанных на образовании комплексных соединений с серусодержащими органическими реагентами. Реакции теллура с диэтилдитиокарбаматом, тиомочевиной и ее производными, висмутолом II были положены в основу методов анализа различных природных и промышленных объектов. Среди этой группы методов наиболее чувствительными являются методы с применением висмутола II и его производных.
Вторая большая группа методов основана на образовании ионных ассоциатов аци до комплексов теллура с органическими основаниями — производными пиразолона и родаминовыми красителями. Реакцию с теллуром обычно проводят в сильнокислом растворе — в условиях высокой прочности ацидокомплексов. Реакции этой группы успешно используются и в методах отделения теллура от других элементов.
Различия в свойствах ацидокомплексов теллура в зависимости от природы лиганда (хлорид, бромид, иодид, роданид), а также в свойствах их соединений с различными органическими основаниями открывают большие возможности при разработке селективных методов определения теллура, а также избирательных методов его отделения от других элементов.
Представляет несомненный интерес также применение гетеро-пол исоединений теллура в фотометрических методах определения. При анализе сталей, сплавов, концентратов часто применяют классический иодидный метод для определения теллура.
Следует, однако, отметить, что, несмотря на быстрое развитие и появление большого количества новых фотометрических и флуо-риметрических методов определения теллура, все они по чувствительности и селективности значительно уступают методам определения селена с 2,3-диаминонафталином и 3,3'-диаминобензиди-ном. Это заставляет искать новые пути повышения чувствительности и избирательности методов определения теллура.
Иодотеллуритный метод
В солянокислом растворе четырехвалентный теллур образует с иодидом желто-коричневый иодотеллуритный комплекс. Вначале образуется комплекс HTeJs, который при стоянии при достаточной концентрации НС1 и KJ переходит в более интенсивно окрашенный комплекс H2TeJe [559]. Спектр пропускания растворов иодотеллуритного комплекса приведен на рис. 16 [8141.
Изучение состава иодотеллуритного комплекса методом изо-молярных серий показало, что в 17V НС1 и 0,2 М KJ образуется комплекс H2TeJe, константа нестойкости которого равна 4,49-•10'8 [245].
4*
99
Рис. 16. Спектр пропускания растворов иодотеллуритного комплекса (1 мкг/мл) в смеси с 0,4Д4 KJ и 0,27V НС1
При избытке четырехвалентного теллура возможно образование других иодидных комплексов, которые менее интенсивно окрашены. Максимумы поглощения растворов иодотеллуритного комплекса лежат в области 285 и 325 — 345 нм. Молярные коэффициенты погашения для H2TeJ5 составляют 700 (при 285 «ж); для H2TeJe— 9,2-104 (при 285 нм) 1814], 325 (при 330 нм) [245] и 7,2-103 (при 413 нм).
Устойчивость комплекса H2TeJ6 зависит от концентрации в растворе соляной кислоты и иодида калия. При недостаточной кислотности раствора комплекс может гидролизоваться, в результате чего окраска исчезает. Оптимальными являются концентрация соляной кислоты 1N и концентрация иодида калия 0,2 М. Влияет также продолжительность стояния растворов. Для получения воспроизводимых результатов требуется 20-минутное стояние растворов перед измерением оптической плотности. Растворы иодотеллуритного комплекса подчиняются закону Бугера — Бера в интервале содержаний теллура 0,02—1,0 мг.
Определению теллура мешают окислители Fe (Ш), Cu2+, SeO|~ и другие, реагирующие в кислой среде с выделением свободного иода; мешает определению Bi (III).
Полоса поглощения трииодида находится в области 350 нм, а молярный коэффициент погашения равен 300. Поэтому измерение оптической плотности растворов иодотеллуритного комплекса проводят при длине волны 420 нм.
Для совместного определения селена и теллура иодидным методом следует вначале отделить селен в условиях, при которых комплексообразование для теллура еще не имеет места (в 0,5 — 1 N НС1 и 0,02 N KJ). В этих условиях восстановление селенистой кислоты количественно заканчивается за 1 час [370]. После отделения селена повышают концентрацию иодида до ОДУ KJ и определяют теллур иодотеллуритным методом.
Фотометрическое определение теллура с иодидом было применено при анализе сталей [244], сульфидных руд [370], медных и'свинцово-цинковых руд [846].
100
Выполнение определения теллура с иодидом. Осадок элементного теллура (не содержащий селена) растворяют в 10 мл конц. НС1 и в 5 — 6 каплях конц. HNO3 при нагревании. После полного растворения теллура раствор разбавляют дистиллированной водой до 30 — 40 мл, фильтруют в медную колбу емкостью 100 мл. Фильтрат в колбе охлаждают, добавляют 3 г мочевины, 10 мл 2 М KJ, разбавляют дистиллированной водой до метки и хорошо перемешивают.
После 20-минутного выдерживания в темном месте измеряют оптическую плотность раствора на ФЭК-М в кювете с I = 0,5 см с синим светофильтром (при 420 нм).
Для проведения холостого опыта поступают следующим образом-, в мерную колбу емкостью 100 мл добавляют 25 — 30 мл дистиллированной воды, 10 мл конц. НС1, 2 капли конц. HNO3, вводят 3 г мочевины и 10 мл 2 М КФ разбавляют дистиллированной водой до метки и перемешивают. Через 20 мин. измеряют оптическую плотность.
Для построения калибровочной кривой в мерные колбы емкостью 100 мл, содержащие 0,2—1 мг Те, добавляют по 25 мл дистиллированной воды, 10 мл конц. HCI, 10 мл 2 М К-1, вносят 3 г мочевины, хорошо перемешивают и ставят в темное место на 20 мин.
Взаимодействие теллура с серусодержащими органическими реагентами
Серусодержащие органические реагенты — тиомочевина, дитизон, висмутол II, диэтилдитиокарбамат и их производные — в настоящее время широко используются в фотометрических методах определения теллура. Методы определения теллура с серусодержащими органическими реагентами обладают высокой чувстви
тельностью.
Фотометрический метод определения теллура с тиомочевиной основан на образовании окрашенного комплекса [Те (Thio)al2+ в растворах, насыщенных тиомочевиной. Четырехвалентный теллур
в этих условиях восстанавливается до двухвалентного, и последний образует комплекс с константой нестойкости ас 2-1СГ3 [377]. Спектр поглощения комплекса представлен на рис. 17.
Максимум в спектре поглощения находится при 328 нм, молярный коэффициент погашения равен 1-Ю2 (при 428 нм), 1,12-103 (при 300 — 325 нм). Спектры поглощения в солянокислой, сернокислой и фосфорнокислой средах идентичны.
Илек и Вржештял [805 — 808] разработали фотометрический метод определения четырехвалентного теллура в присутствии шестивалентного. Эти авторы установили, что скорость реакции теллура
Рис. 17. Спектры поглощения комплекса Те(П1 с тиомочевиной в 2;V НС1
/ — Н2ТеО3; 2 — тиомочевина; 3 — комплексное соединение
101
с тиомочевиной зависит от pH раствора, концентрации тиомочевины и температуры. Максимальная окраска наблюдается в хлорнокислой среде (0,8 N НС1О4) в растворе, насыщенном по отношению к тиомочевине.
Нильш [962—9641 разработал метод определения теллура реакцией с тиомочевиной в азотнокислом (5— 10%-ная HNO3), фосфорнокислом (2— 10%-ная Н3РО4) и сернокислом (2 — 6%-ная H2SO4) растворах при концентрации тиомочевины 10—11%. Метод был применен к определению теллура в меди и селене.
Определение теллура в чугуне по реакции с тиомочевиной проводят в азотнокислой среде (6%-ная HNO3) [972]. Закон Бера соблюдается в интервале содержаний 0,05 — 0,5 мг Те в 50 мл. Оптимальная концентрация тиомочевины 8,5%. Окраска развивается в течение 2 мин. и не изменяется в течение 10 мин. Определение проводят при 350 нм в кювете с толщиной слоя 1 см.
Леонтович [8771 определял теллур в железных продуктах используя фосфорнокислую среду. Гладышев [120] применил тиомочевину для колориметрического определения теллура в пылях свинцового производства. Описан метод определения теллура в висмуте [549, 1041]. В присутствии вольфрама, галлия, молибдена, хрома, никеля, сурьмы и висмута теллур отделяют в элементном состоянии из солянокислого раствора при помощи хлористого олова.
Мешают определению теллура с тиомочевиной Pd, Hg (I) и Hg (II), Os (VIII), Rh (III), Pt (IV). He мешают Ag, Mg, Zn, Ba, Al, Sn (IV), Mn2+, Be, Cd, Се (IV), Pb.
Значительное повышение чувствительности реакции теллура с тиомочевиной (до 1 мкг 1&/мл) было достигнуто в солянокислом растворе (0,6—1,7 N НО) в присутствии избытка KSCN при экстракции комплекса трибутилфосфатом.
Определение теллура возможно также после переведения тио-мочевинного комплекса теллура в нерастворимое соединение с солью Рейнеке [Те (SCN2H4)4] [Сг (SCN)4 (NH2)2]2, извлекаемое метилэтилкетоном [65]. Измеряют оптическую плотность при 400 нм.
Четырехвалентный теллур в среде 4 — 9 N НС1 или НС1О4 образует желтые комплексные соединения с симметричной дифенилтиомочевиной (I), фенилтиомочевиной (II), дитолилтиомочевиной (III), этилентиомочевиной (IV) и тиомочевиной (V), причем свето-поглощение комплексов увеличивается в ряду V <( IV <( III <’ < II < I. Комплексы Те (IV) с I, II и III экстрагируются хлороформом. Светопоглощение экстрактов не зависит от кислотности водной фазы в пределах 4,5—8,0 N.
Закон Бера (при 380—410 нм) соблюдается при концентрации Те меньше 200 мкг/‘20 мл раствора [1171].
Выполнение определения теллура симметричной дифенилтиомочевиной [1171]. К раствору, содержащему 10 — 200 ^/caTe(IV) в объеме раствора не более 5 мл, прибавляют 5 мл конц. НС1 (или 6 мл 60%-ной НС1ОД разбавляют раствор, если 102
необходимо, до 10 мл, экстрагируют Te(IV) хлороформным раствором симм,-дифенилтиомочевины (1 г в 100 мл СНС13), экстракт центрифугируют или высуши вают над безводным Na2SO4 и фотометрируют в 1-сантиметровых кюветах при 380 — 410 нм, используя в качестве раствора сравнения холостую пробу.
Определению не мешают Cd, Со, Мп, Ni и Zn. Мешают Fe(III), Se(IV) и более чем 10-картные количества Bi.
Определение теллура с диэтилдитиокарбаматом и его производ-
S
ными. Диэтилдитиокарбамат натрия (GHs^N—образует с
SNa
теллуром окрашенное в желтый цвет комплексное соединение состава Те (DDTC)4 [537]. Соединение количественно образуется в 3 N H2SO4, 4 N НС1О4 и 3 N НС1 и хорошо растворяется в органических растворителях — бензоле, хлороформе, четыреххлористом углероде (538, 709, 722]. Под действием света комплекс разрушается до (DDTK)2Te и тетраэтилтиурамдисульфида [538].
Максимум в спектре поглощения комплексов в хлороформе лежит при 340 нм [770].
Закон Бера соблюдается для содержаний теллура до 50 мкг (при 340 нм) и до 100 мкг (при 400 нм).
Возможно спектрофотометрическое определение теллура в водно-ацетоновой среде (60% ацетона), е = 3200 (при 424 нм) [750]. Определение проводят в интервале концентраций!—20 мкг/мл. Определяемый минимум в последнем случае 0,04 мкг/мл.
Диэтилдитиокарбамат натрия образует окрашенные комплексы с большим числом катионов, поэтому для селективного определения теллура в присутствии мешающих элементов необходимо строгое соблюдение pH среды, применение маскирующих веществ и экстракция комплекса органическими растворителями. При экстракции комплекса СС14 из раствора с pH 8,5 — 8,7 возможно отделение теллура от селена, который также образует комплекс с DDTK, но при более высокой кислотности.
В присутствии комплексона III возможно определение теллура в присутствии Со, Hg (II), Ni, W(VI), Al, Ba, As (III), Ca, Cd, Mg, Mn2+, Mo (VI), Pb, Zn.
В присутствии комплексона III и цианида определению не мешают платиновые металлы [285]. Мешают Fe (Ш), Сн2+, Bi(III).
Предложен вариант определения теллура с DDTK, основанный на вытеснении теллура из комплекса (DDTK)dTe медью и последующем фотометрическом определении меди [545].
Фотометрический метод определения теллура с DDT К был применен к определению теллура в железе и стали [722], в индии и мышьяке [1251, в селене [284, 537, 11591, арсениде галлия [124], в материалах металлургического производства [355], при разделении Те (VI) и Te(IV) [770].
103
Изучена возможность фотометрического определения Bi, Со, Cu, Ni и Те с 7п-б«с-(2-октаэтил)дитиокарбаматом в цитратной среде. Чувствительность реакции 0,04 мгк/мл (при 420 нм) [546, 1175].
Выполнение определения теллура в свинце и меди. Свинец. 10,00 г анализируемого свинца помещают в коническую колбу емкостью 300 мл, добавляют 15 мл смеси НС1О4 и HNO3, накрывают и осторожно нагревают до растворения металла. Раствор упаривают до появления обильных паров хлорной кислоты для удаления азотной кислоты. Охлаждают, добавляют 50 мл воды, нагревают до слабого кипения и затем добавляют 50 мл НС1. Продолжают нагревание около 1 мин., время от времени перемешивая вращательным движением для предотвращения толчков, до коагуляции осадка. Охлаждают до 10—15° С в бане со льдом, фильтруют через неплотный фильтрат в коническую колбу емкостью 250 мл и промывают осадок 3 — 4 раза тонкой струей холодной НС1 (1 : 1). Осадок отбрасывают.
К фильтру добавляют 2 мл раствора мышьяка, а затем 10 мл 50%-ной гипо-фосфористой кислоты, перемешивают вращательным движением.
Охлаждают до 70—80° С, фильтруют через плотный бумажный фильтр и промывают колбу и фильтр 3—4 раза НС1 (1 : 1).
Переносят фильтр и осадок обратно в 250-миллилитровую колбу. Собирают все следы металла со стенок стеклянной воронки при помощи небольшого кусочка фильтровальной бумаги, которую помещают в ту же колбу.
Добавляют 10 мл HNO3 и 4 мл НС1О4, накрывают часовым стеклом и нагревают до разрушения бумаги. Когда окисление закончится, раствор упаривают до объема 2 мл, добавляют 2 мл H2SO4, упаривают до 1 мл и далее поступают, как при получении калибровочной кривой. Проводят контрольный опыт с реагентами.
Медь. Растворяют 5 г анализируемой меди в 25 мл смеси НС1О4 и HNO3 и упаривают до появления обильных паров до удаления азотной кислоты. Раствор охлаждают, добавляют 50 мл воды и нагревают для растворения солей. Добавляют 50 мл НО, 2 мл раствора мышьяка, 15 мл 50%-ной гипофосфористой кислоты и промывают осадок мышьяка несколько раз порциями по 10 мл НС1 (1 : 1) и далее поступают, как при определении теллура в свинце.
Построение калибровочной кривой дл/я теллура. Помещают 0; 0,5; 1,0 и 1,5 мл стандартного раствора теллура (100 мкг Те/ды) в конические колбы емкостью 250 мл. Добавляют около 0,25 мл НС1О4 и 2 мл H2SO4. Упаривают на пламени до объема 1 мл. Охлаждают, добавляют 50 мл воды и охлаждают до комнатной температуры. Затем добавляют 2 капли раствора л«-крезолового пурпурного и нейтрализуют раствором NH4OH до начала перехода красного цвета в желтый.
Добавляют 5 мл 5%-ного раствора KCN и затем NH4OH до появления пурпурного цвета индикатора. Раствор переводят в делительную воронку из темного стекла емкостью 125 мл.
Добавляют 2 мл раствора диэтилдитиокарбамата, перемешивают вращательным движением, добавляют 20,0 мл хлороформа, закрывают пробкой, немедленно встряхивают, приоткрывают пробку для уменьшения давления и затем энергично встряхивают 30 сек. Дают фазам разделиться и сливают большую часть нижней фазы в коническую колбу из темного стекла емкостью 50 мл, в которой находится около 3 г безводного Na2SO4. Перемешивают вращательным движением и быстро' измеряют оптическую плотность при 415 нм в кювете с толщиной слоя 5 см относительно хлороформа.
3,5-Дифенилпиразолин-1-дитиокарбамат натрия (пиразолиндитио-карбамат натрия) образует с теллуром комплексное соединение, более устойчивое, чем с DDTK [64]. Комплекс теллура экстрагируется хлороформом в широком интервале кислотности — от 12 N НС1 до pH 9. Максимум светопоглощения растворов находится, около 415 нм (е = 20400).
104
В присутствии тартратов при pH 8,0 возможно прямое определение микроколичеств теллура при соотношении Те : Se — = 1 : 100.
Добавляя маскирующую смесь (NaCN, комплексон III, тартрат натрия при pH 8,0—9,0), можно определить более 2 мкг Те в присутствии Se, As, Bi, Cd, Ni, Cu, Fe (III), Мп (II), Pd (II), Sb, Sn, Zn.
Мешают определению T1 (III) и Т1 (I), которые могут быть удалены экстракцией этим же реагентом при pH 14.
Метод был применен для определения теллура в селенистой кислоте, полупроводниковом селене, сере и фосфоре [14, 64].
Выполнение определения теллура. 0,5—1,0 г с ел енистой кислоты растворяют в минимальном количестве воды, добавляют маскирующую смесь (Юг NaCN, 1 г комплексона III, 5 г тартрата калия на 100 мл воды), доводят pH до 8,0—9,0 добавлением щелочи или кислоты, прибавляют 1—2 мл 1%-ного водного раствора 3,5-дифенилпиразолин-1-дитиокарбамата натрия и экстрагируют тремя порциями хлороформа —• 10; 10 и 5 мл натрия. Экстракт фильтруют через сухой бумажный фильтр в мерную колбу емкостью 25 мл, доводят до метки хлороформом, перемешивают и измеряют оптическую плотность на спектрофотометре СФ-4 при 415 нм относительно раствора, содержащего все компоненты, кроме теллура.
0,5—1 г металлического селена в виде порошка растворяют в HNO3 (уд. вес 1,42) при очень слабом нагревании на водяной бане, добавляя несколько капель НС1. Кислый раствор переносят в делительную воронку емкостью 500 мл, нейтрализуют раствором NaOH, затем добавляют маскирующую смесь и доводят pH до ~8,5, добавляют 1—2 мл 1%-ного раствора 3,5-дифенил-пиразолин-1-дитиокарбамата натрия, экстрагируют тремя порциями хлороформа — 10; 10 и 5 мл.
Экстракт сливают в делительную воронку емкостью 50 мл, в которой находится 10 мл маскирующей смеси, встряхивают 5 мин., и экстракт фильтруют в мерную колбу емкостью 25 мл, доводят до метки хлороформом, перемешивают и измеряют оптическую плотность, как указано выше.
Для построения градуировочной прямой навеску селенистой кислоты растворяют в воде, устанавливают pH 8,7—8,9 10%-ным раствором NaOH и приливают 2 мл 0,1%-ного водного раствора 3,5-дифенилпи-разолин-1-дитиокарбамата натрия. Выделяющуюся муть экстрагируют несколько раз порциями по 10 мл хлороформа, встряхивая 2—3 мин. и добавляя перед каждой экстракцией 0,5—1,0 мл реагента. Примеси экстрагируют до тех пор, пока прибавление реагента не перестанет вызывать помутнение раствора. Экстракты отбрасывают, а очищенный таким образом раствор селенистой кислоты фильтруют и стандартизуют обычным путем.
Для этого берут аликвотную часть раствора (—1 г Se), добавляют 10—15 мл маскирующей смеси, устанавливают рН~ 8,6 и прибавляют 0,5; 1,0; 1,5; 2,0; 2,5 мл стандартного раствора теллура (1 мкг Те/мл), 1—2 мл 1 %-кого раствора реагента и экстрагируют хлороформом порциями по 10; 10 и 5 мл, фильтруя экстракт в мерные колбы емкостью 25 мл. Доводят до метки хлороформом, перемешивают и измеряют оптическую плотность на спектрофотометре в кювете /= 5 см; при более высоком содержании теллура — в кювете I = 1 см.
Нулевой раствор как при построении градуировочной кривой, так и при -определении теллура в образцах содержит все компоненты, кроме теллура.
Тетраэтилтиурамдисульфид (или бис-диэтилтиокарбамил)ди-сульфид (C2H8)2N—C(S)—S—S—C(S)—N (C2H8)2 (I) и тетраметил-тиурамдисульфид (II) образуют с Те (IV) желтые не растворимые в солянокислом растворе осадки. Соединение теллура с I хорошо
105
растворимо в СС14 [11631, с II — в хлороформе, бензоле, ксилоле [396]. Растворы комплекса I в СС14 имеют максимумы в спектре поглощения при 380 нм (е = 6 350) и 390 нм (е = 4250). Закон Бера соблюдается при концентрации 10—200 мкг Те/10 мл СС14. Максимум' светопоглощения комплекса II лежит при 278 нм, реагента II — при 274 нм.
На основе реакции I предложен спектрофотометрический метод определения теллура. Определению с I мешают Cu2+, Se (IV), Hg2+ и NOg. Не мешают Bi (15-кратный избыток), Со (50-кратный), Ni (100-кратный), Fe (III) (200-кратный), Al, Cd, Mn2+, Zn, Mg, CHSCOO-, SOI" и Cl".
Комплекс теллура с реагентом II содержит, по-видимому, двухвалентный теллур. Будучи более сильным восстановителем, чем I, тетраметилтиурамдисульфид восстанавливает селен до элементного. Мешают определению с тетраметилтиурамдисульфидом— Hg, Си, Bi, Fe, Сг, Sb.
Разработан фотометрический метод определения теллура с димеркаптотиопироном, образующим с теллуром в сильнокислой среде комплекс красного цвета состава Те : R = 1:4. Максимум в спектре поглощения находится в области 490 нм, молярный коэффициент погашения 10 800. Определению теллура не мешают Li, Cs, Rb, Mg, Ca, Sr, Ba, Al, Ce3+, Mn2+, Co, Ni, Mo (VI), W (VI). Метод пригоден для определения теллура в солях алюминия [319]. С дифенилдимеркаптотиопироном теллур образует комплекс с отношением Те : R — 1 : 4 и молярным коэффициентом погашения 17,78-•103 при 360 нм [397].
Известен метод фотометрического определения теллура с мер-каптобензтиазолом [318].
Определение теллура с висмутолом II и его производными. Висмутол II — 3-фенил-5-меркапто-1,3, 4-тиадиазол-2-тион — образует с четырехвалентным теллуром окрашенное в желтый цвет соединение, нерастворимое вводе и хорошо растворимое в спиртах, кетонах, хлороформе, бензоле, СС14 [583, 801, 1174]. Состав комплекса в хлороформе соответствует Те : R = Г: 4 [1174].
Спектры поглощения растворов комплекса в хлороформе представлены на рис. 18.
Максимум в спектре поглощения комплекса (кривая /) совпадает с максимумом для реагента (кривая 2). Молярный коэффициент погашения соответствует 28 000 (при 330 нм).
Образование комплекса проходит количественно в солянокислом растворе от 6М НС1 до pH 3,5 (рис. 19). Селен также образует комплекс с висмутолом II в сильнокислом (более 2 N) растворе (НС1 или H3SO4) [1089].
Экстракция соединения с теллуром органическими растворителями возможна как из кислых, так и из нейтральных растворов (до pH 7,5). Однако экстрагируемость комплекса хлороформом из кислых растворов неколичественна.
106
Рис. 18. Спектры поглощения хлороформных экстрактов комплекса теллура с висмутолом II (/) и экстракта реагента (2)
5 мкг Те в 5 мл хлороформа; I мл 1,5%-ного раствора висмутола II
Рис. 19. Зависимость оптической плотности от pH раствора при образовании комплекса теллура с висмутолом 11 (Л = 330 нм)
Висмутол II также экстрагируется хлороформом из кислых растворов вплоть до pH 1. При pH > 2 экстракция резко уменьшается и при pH 6,5—10 реагент в хлороформе практически нерастворим. Поэтому для устранения влияния окраски реагента на определение теллура реагент реэкстрагируют из органической фазы при pH 6,5 [1174].
Для количественного образования комплекса теллура с висмутолом II в кислом растворе требуется 20 сек.: в течение последующих 30 мин. окраска растворов не изменяется [1174]. Исследования Ченга [586] показали, что при pH 2,2 образование комплекса происходит медленно (40 мин.), а добавление сульфата аммония значительно ускоряет реакцию (до 2 мин.).
Для количественной экстракции комплекса хлороформом требуется 1 мин. Количество реагента, необходимое для полного образования комплекса, зависит от кислотности водного раствора: в 3 М НС1 необходим 5-кратный избыток реагента, в растворе с pH 3,5 — 240-кратный избыток.
Калибровочные графики линейны для интервала концентраций 2—30 мкг Те.
Мешающие элементы. Висмутол II реагирует с очень многими элементами: Те (IV), Se (IV), Си, Fe, Hg (I), Hg (II), Ag(I), As, Au, Pt, Pd, Ni, Co, Pb, T1 и др.
В присутствии комплексона III и цитрата определению теллура в 3 А НС1 мешают As (V), Си (II), Hg (II), Se (IV) и Pd (II), а при pH 3,5 мешают Си (II), Hg (II), Se (IV) [1174]. Нитраты и другие окислители мешают определению теллура.
Ртуть и селен могут быть удалены отгонкой с Вг2 и НВг [583]. Чтобы избежать при этом потерь теллура, необходимо следить
107
за тем, чтобы выпаривание проводилось на водяной бане (ниже 100° С) в растворе, содержащем очень немного H2SOa. Для отделения ртути можно также использовать экстракцию дитизоном [584].
Серебро и таллий удаляются в виде нерастворимых хлоридов.
По данным Янковского [801 ], интенсивность окраски растворов комплекса несколько уменьшают некоторые анионы — F-, тартрат, цитрат, дитионат.
В последнем варианте использования висмутолового метода [9911 теллур отделяют от других элементов экстракцией 1,1,2,2-тетрахлорэтаном его комплекса с 4,4'-метилендиантипиреном.
Выполнение определения [991]. 5 мл анализируемого раствора выпаривают с H2SO4 (1 : 1) до появления белого дыма, остаток охлаждают, разбавляют НС1 (1 : 1) до ~25 мл и при помощи НС1 (1 : 1) переводят в делительную воронку.
Добавляют 2 мл 5%-ного раствора 4,4'-метилендиантипирена в СН3СООН (1 : 1), через 1 мин. приливают 10 мл тетрахлорэтана и встряхивают 90 сек.
Слой тетрахлорэтана отделяют и повторяют экстракцию еще с 10 мл тетрахлорэтана. Объединенный экстракт дважды промывают (порциями по 20 мл) солянокислым раствором реагента (25 мл 5%-ного уксуснокислого раствора 4,4'-метилендиантипирена смешивают с 400 ли 6 М НС1), и реэкстрагируют теллур водой (2 раза по 15 мл).
Реэкстракт упаривают до 20—25 мл, добавляют 2 мл буферного раствора с pH 3,5 (раствор содержит 1 г комплексона III и 20 г цитрата натрия в 100 мл воды, разбавлен НО до pH 3,5), устанавливают pH 3,0 — 3,5, приливают 1 мл I %-кого раствора висмутола II и через 20—30 сек. экстрагируют при помощи 22 — 24 мл хлороформа.
Экстракт промывают 25 мл буферного раствора с pH 7,5, органический слой фильтруют, разбавляют до 25 мл СНС13 и фотометрируют в 5-сантиметровых кюветах при 330 нм. Закон Бера выполняется в интервале 0,2 — 4 мкг Те1мл.
Одновременно проводят холостой опыт. При определении теллура в материалах на Cu-основе пробу растворяют в HNO3 (1 : 1) и далее поступают, как описано выше.
При содержании ~0,001% Те в пробе ошибка определения 2%.
Бусев и Симонова [68 — 70, 154] синтезировали производные висмутола II —5-мер капто-3(нафтил-2)-1,3,4-тиаДиазолтион-2 и 5-мер капто-3(2,3-диметоксибензил)-1,3,4-тиадиазолтион-2 и изучили свойства соединений этих реагентов с теллуром.
Теллур (IV) образует с производными висмутола II осадки желтого цвета, малорастворимые в воде и хорошо растворимые в циклогексане, бензоле, хлороформе. Соединения теллура образуются в широком интервале кислотности —от 18 N H2SO4 до pH 6 (для нафтильного производного) и от 6 N НС1 до pH 5,5 (для диметил-оксибензилпроизводного). Количественная экстракция комплексов проходит из растворов с рНО — 6. Реагенты также экстрагируются из кислых растворов до pH 3,5. Методом изомолярных серий, методом насыщения и элементным анализом показано, что Те (IV) взаимодействует с реагентами в молярном отношении 1 : 4.
Нафтильное производное висмутола II является более селективным реагентом на теллур — не реагирует с As (V) H-Hg(I). Хотя селен также реагирует с производными висмутола, комплексы 108
экстрагируются хлороформом при pH < 4,5 (рис. 20). Различие в значениях pH количественного извлечения в органическую фазу соединений селена и теллура позволяет определять теллур без разделения при pH 4,8 — 5,7 в присутствии винной кислоты [68, 69]. Мешают определению теллура 100-кратные количества TI (I), Hg (И), Sn (II), Ag (I).
Оптическая плотность хлороформных экстрактов комплекса с нафтилвисмутолом пропорциональна концентрации в интервале 1—50 мкг Те/мл.
Кривые светопоглощения имеют максимум при 330 нм (рис. 21). Истинный молярный коэффициент погашения, равный для комплекса с нафтилвисмутолом 29 550 (при 326 нм), для самого реагента составляет 9 250 (при 330 нм).
Нафтилвисмутол был применен для экстракционно-фотометрического определения теллура в Pb-концентрате и сере [68], в молибденовых гетерополисоединениях теллура [154], рудах [67], полупроводниковых материалах [70].
Выполнение определения теллура с нафтилвисмутолом [68]. Для построения калибровочного графика в ряд делительных воронок вводят по 10 мл буферного раствора с pH 4,8 — 5, 0,1 — 5 мл (с интервалом в 1 мл) стандартного раствора соли теллура (10 мкг/мл), 1 мл 0,1%-ного водного раствора реагента и 10 мл хлороформа. Окрашенный комплекс экстрагируют2 — Змин. Экстракт фильтруют в сухую кювету с толщиной слоя 0,5 см через сухой бумажный фильтр и измеряют оптическую плотность на фотоэлектроколориметре (светофильтр № 2) относительно раствора реагента.
Анализируемый раствор, содержащий 50—250 мкг теллура, разбавляют водой до объема 25 мл в мерной колбе. Для определения отбирают аликвотную часть 5,0 мл и определяют в ней теллур так, как описано при построении калибровочного графика.
Рис. 21. Спектры поглощения хлороформных экстрактов теллура с нафтилвисмутом (/), экстракта реагента (2), дисульфида
109
Синтез 5-меркапто-З (нафтил -2)-1,3,4 - тиадиазолтио-на-2 (нафтилвисмутола).
Получение fi-нафтилгидразина. 25 г тонкоизмельчениого 3-нафтиламииа растворяют в 50 мл НС1 (уд. вес 1,19) и затем добавляют еще 200 мл НС1. Стакан помещают в баию со льдом, и смесь охлаждают до 0° С. Медленно при постоянном перемешивании механической мешалкой по каплям вводят раствор 12 г NaNO3 в 25 мл воды.
После окончания диазотирования охлажденный раствор диазосоединения быстро фильтруют через воронку Бюхиера. Маточный раствор охлаждают до 0° С и при энергетичном помешивании вводят раствор 125 г SnCl2 в 100 мл HCI (уд. вес 1,19).
Смесь оставляют на 1 час, и затем отфильтровывают солянокислый р-иафтил-Гидразин через воронку Бюхнера, тщательно промывают на фильтре соляной кислотой и отжимают. Продукт перекристаллизовывают из горячей воды и сушат на воздухе.
Выход 14 г солянокислого fl-нафтилгидразина (49% от теоретического).
Получение калиевой соли 5-меркапто-3-(нафтил-2)-1,3,4-тиадиазолтиона-2.
Собирают прибор, состоящий из трехгорлой колбы, снабженной обратным холодильником, капельной воронкой и механической мешалкой (шлифы!). В колбу помещают 14 г солянокислого p-нафтилгидразина в 70 мл этанола. По каплям из капельной воронки при перемешивании приливают 21 г абсолютированного сероуглерода. Смесь нагревают на водяной бане, помещенной на электроплитку с закрытой спиралью, в течение получаса, затем вводят раствор 21 г КОН в 100 мл этанола. Реакционную смесь выдерживают на кипящей водяной баие в течение 8 — 9 час.
После окончания нагревания горячую смесь выливают в сухой стакан. По охлаждении из нее выделяются желтые кристаллы калиевой соли реагента. Продукт отсасывают от маточника на воронке Бюхиера, отжимают на фильтре и перекристаллизовывают из спирта. Выход сухой калиевой соли 15 г (58% от теорет.).
Калиевая соль реагента хорошо растворима в воде, устойчива на воздухе. При обработке водных растворов калиевой соли разбавленной соляной кислотой выделяется не растворимая в воде кислотная форма реагента, легко растворимая в хлороформе. Для соли, перекристаллизованной из бензола, температура плавления хорошо совпадает с литературными данными (157—158° С).
Определение теллура с производными пиразолона
Гексахлортеллуриты и гексабромотеллуриты взаимодействуют с антипирином, диантипирилметаном, диантипирилпропилметаном с образованием желто-оранжевых кристаллических осадков. Из растворов 1,5 — 6 (V НВг были выделены соединения следующего состава: с диантипирилметаном (С23Н24О2^)2-Н2ТеВг6, с дианти-пирилметилметаном (CsiHgoC^N^-H^TeBr 6, с диантипирилпропилметаном (С2бН30О2^)2-Н2ТеВг6 [57].
Из солянокислых растворов (5 — 7 N HCI) соединения экстрагируются дихлорэтаном [57, 58, 63].
Методом физико-химического анализа и элементным анализом выделенных продуктов реакции теллура с диантипирилметаном было установлено, что соотношение реагирующих компонентов Те (IV): диантипирилметан: Вг" =1:2:6. Растворимость комплекса в дихлорэтане равна 1,12 г/л [302].
ПО
Рис. 22. Спектры поглощения соединений теллура (в дихлорэтане) и селена (в хлороформе) с диантипирилпропилметаном
/, 2 — холостой опыт и 100 мкг Те при экстракции из HCI (1 : 1), 3,4 — холостой опыт и 50 мкг теллура при экстракции из 2./VH2SO4, насыщенной К В г; 5, 6 — холостой опыт и 100 мкг Se, при экстракции из НС1 (1:1)
D
1.5
Экстракты, содержащие хлоридные соединения теллура с производными антипирина, имеют два максимума в спектре поглощения — 325 и 380 нм, е = 790.
Хлоридные комплексы четырехвалентного селена не взаимодействуют с производными пиразолона во всем интервале кислотности и полностью остаются в водной фазе.
Бромидные комплексы теллура реагируют с производными антипирина в 1,5 — 6 44 НВг, растворы более интенсивно окрашены. Молярный коэффициент погашения при 330 нм равен 13 500, при 450 нм — 4200. Спектр поглощения бромидного комплекса теллура с диантипирилпропилметаном представлен на рис. 22.
Экстракты, содержащие бромидные соединения теллура с производными антипирина, имеют максимум в спектре поглощения при 450 нм. В растворах, содержащих бромистоводородную кислоту, селен также образует соединения с производными антипирина и частично экстрагируется дихлорэтаном.
Диатипирилпропилметан и другие производные антипирина успешно применялись при экстракционно-фотометрическом определении теллура в селене, свинце, висмуте, меди, конвекторной пыли, цинковом электролите [62]. Методы дают точные и хорошо воспроизводимые результаты.
Эти реагенты успешно используются для экстракционного отделения теллура от селена, золота, меди, свинца, висмута и других элементов и дают очень надежные результаты [58, 60, 63].
Синтез диантипирилпропилметана [62, 426]. Растворяют 5 г антипирина в небольшом количестве воды, прибавляют2мл НС1, 2 мл перегнанного масляного альдегида, нагревают 30 мин. на водяной бане. После охлаждения разбавляют водой до 200 мл и нейтрализуют аммиаком до появления слабого запаха. Маслянистая жидкость быстро кристаллизуется. Вещество отфильтровывают через воронку Бюхнера и промывают водой.
Выполнение определения [59]. 0,5 г металлического селена растворяют в 15-—20 мл конц. HNO3, добавляют 5 мл H2SO4 (I : 1) и выпаривают до появления паров SO3. После охлаждения осторожно приливают 5—10 мл воды и вновь упа
111
ривают до паров SO3, охлаждают и переводят в мерную колбу емкостью 50 мл соляной кислотой (1 : 1).
Отбирают в делительную воронку емкостью 100 мл аликвотную часть 5 или 10 мл (в зависимости от содержания теллура), прибавляют 1—2 мл 5%-ного раствора диантипирилпропилметана в уксусной кислоте (1 : 1), общий объем доводят до 20 мл соляной кислотой (1 : 1), приливают 5 мл дихлорэтана и встряхивают 1 мин.
После разделения фаз органический слой сливают в чистую делительную воронку емкостью 50 мл. К оставшейся водной фазе добавляют еще 5 мл дихлорэтана и снова встряхивают 1 мин.
Дихлорэтановые экстракты объединяют (водный слой отбрасывают), дважды промывают 10 мл НС1 (1 : 1), теллур из органического слоя реэкстрагируют 10 мл 0,02 N раствора комплексона III.
После расслаивания дихлорэтан сливают. К оставшейся водно й фазе добавляют 5 мл НВг и дают постоять 1 мни., затем прибавляют 0,5 мл аскорбиновой кислоты (1 г в 20 мл воды), 1 мл 1%-ного раствора диантипирилпропилметана и 5 мл дихлорэтана. Встряхивают 1 мин. После просветления органический слой фильтруют через сухой фильтр в кювету I = 1 см.
Оптическую плотность измеряют на приборе ФЭК-Н-57 со светофильтром № 3 относительно дихлорэтана.
Построение калибровочной кривой проводят, исходя из раствора, содержащего 0,01 мг!мл теллура. Отбирают от 10 до 100 мкг Те в делительные воронки емкостью 500 мл, добавляют воду до объема 100 мл, 5 мл перегнанной бромистоводородной кислоты и оставляют на 1—2 мин., затем добавляют 0,5 мл аскорбиновой кислоты (1 г в 20 мл воды), 1 мл 1%-ного раствора диантипирилпропилметана и 5 мл дихлорэтана. Встряхивают 1 мин.
После разделения фаз органический слой фильтруют через сухой фильтр в кювету на 1 см, измеряют оптическую плотность на приборе ФЭК-Н-57 со светофильтром № 3 относительно дихлорэтана. Окраска раствора устойчива длительное время.
Определение теллура с родаминовыми красителями
В последние время для определения теллура были применены методы, основанные на экстракции низкополярными растворителями ассоциантов галогенидных (хлоридных и бромидных) комплексов теллура с красителями группы родаминов. Щербов и Иванкова [457,458,4611 предложили экстрагировать хлортеллурит родамина С из 2—2,5 N НС1 смесью бензола с эфиром (3 : 1); определяемый минимум элемента 0,5—1 мкг Те.
В дальнейшем Иванкова и Блюм [173] провели системати- . ческое сравнение четырех красителей — родамина С, этилродамина С, бутилродамина С и родамина 6Ж как реагентов для флуоримет-рического и фотометрического определения теллура.
На рис. 23 представлены спектры поглощения и флуоресценции раствора бромтеллурита бутилродамина С в бензоле [460]. Коэффициент распределения ассоциата между смесью бензола с бутил-ацетатом (5 : 1) и водной фазой (—10 N H2SO4) составляет 8, между бензолом и водным раствором такого же состава — 4. При экстракции с помощью 6 мл смеси органических растворителей из 10 мл водной фазы извлечение ассоциата составляет 83%; с учетом этого
112
Рис. 23. Спектры поглощения (1) и флуоресценции (2) раствора бромтеллурита бутилродамина С в бензоле
условный молярный коэффициент погашения раствора ассоциата в экстракте при 565 нм соответствует 7,7-104.
Для выяснения возможности повышения чувствительности флуориметрического метода определения теллура было испытано влияние ряда органических растворителей на яркость свечения комплекса теллура с бутилродамином С [460]. Для этого 5 мкг Те экстрагировали при помощи 10 мл смеси бензола и бутилацетата; проводили также холостой опыт. По разделении фаз из экстрактов брали по 1 мл, разбавляли до 5 мл испытуемыми растворителями и измеряли яркость свечения на флуориметре ФО-1. Результаты этих определений представлены в табл. 13. В 3-й и 4-й графах этой таблицы показана измеренная сила фототока (мка), в 5-й и 6-й графах фототоки пересчитаны на доли от фототока «исходной» смеси растворителей, свечение которой принято за 1 (1/Ф)-В 7-й графе дано отношение яркости флуоресценции комплекса теллура к свечению холостого опыта (Ф/Х).
Таблица позволяет сделать следующие выводы. С увеличением диэлектрической проницаемости растворителя наблюдается некоторая тенденция к увеличению яркости свечения теллурового комплекса (графа 4), но у экстракта холостого опыта эта тенденция выражена сильнее (графа 3). Более отчетливо это видно в графах 5 и 6. Последняя графа показывает, что по величине отношения полезного сигнала к фону подавляющее большинство растворителей уступает смеси бензола с бутилацетатом, близок — ксилол, превосходит ее бензол.
Бутилродамин С был использован для определения теллура в рудах и полупроводниковых материалах [83, 93, 99, 173].
В условиях, рекомендуемых для проведения реакции четырехвалентного теллура с бутилродамином С (9,5 N H2SO4 и 0,18V КВг), в бензольный экстракт переходят и мешают определению окрашенные соединения бромидных комплексов индия, таллия (III), сурьмы (V), золота (III), ртути (I и II), железа (III), меди (I), олова (II и IV), серебра и висмута [93, 173]. Селен (IV) реакции с бутилродамином С не дает.
113
Таблица 13
Влияние природы органического растворителя на яркость флуоресценции комплекса теллура с бутилродамином С
Растворитель Диэлектрическая проницаемость /, мка 1/ф Ф/Х
холостой опыт комплекс теллура ХОЛОСТОЙ опыт комплекс теллура
1 2 3 4 5 6 7
Бензол бутилацетат 1,0 26 1 1 26
Нонан 1,97 0,3 6 0,3 0,23 20
Ксилол 2,27 0,7 20 0,7 0,77 30
Бензол 2,28 0,5 21 0,5 0,81 42
Дибутиловый эфир 3,06 0,7 12 0,7 0,46 17
Диэтиловый эфир 4,3 1,8 15,5 1,8 0,59 8
Хлороформ 4,8 2,2 40,0 2,2 1,54 18
Бутилацетат 4,77 2,2 25,0 2,2 0,96 11
Этилацетат 6,0 2,2 32 2,2 1,23 15
Бутиловый спирт 17,1 2,5 46 2,5 1,77 16
Ацетон 20,14 1,5 35 1,5 1,35 28
Этиловый спирт 24,3 2,7 42 2,7 1,61 16
Метиловый спирт 32,6 2,3 45 2,3 1,73 20-
Гликоль 38,7 2,5 54 2,5 2,08 22
Для отделения от сопутствующих элементов теллур соосаждают с мышьяком при восстановлении гипофосфитом в присутствии ионов меди в качестве катализатора. При соосаждении теллура с мышьяком осадком сорбируется медь. Если анализируемый материал содержит более 1% меди, осадок переосаждают. Из элементов, мешающих определению теллура при восстановлении гипофосфитом, в осадок переходят золото, ртуть и небольшие количества железа.
Золото восстанавливается до металла в процессе разложения пробы при упаривании раствора до паров SO3 и отделяется с нерастворимым остатком при фильтровании сернокислого раствора.
При высоком содержании ртути ее удаляют, обжигая смесь навески пробы с железом, восстановленным водородом, или разлагая навеску щелочным сплавлением. Ртуть в небольшом количестве (до 100 мкг) достаточно полно улетучивается в виде хлорида при обработке сернокислого раствора концентрированной соляной кислотой. Одновременно с ртутью удаляется и селен.
Мешающее действие железа (III) устраняют восстановлением его до железа (II) аскорбиновой кислотой. При этом теллур также начинает медленно восстанавливаться, поэтому экстракцию следует
114
проводить непосредственно после добавления аскорбиновой кислоты; неэкстрагируемая с бутилродамином С медь (II) частично восстанавливается до реакционноспособной меди (I), что вынуждает предварительно тщательно отделять теллур от следов меди. Органические вещества, сообщающие экстракту постороннюю окраску, следует разрушить при вскрытии пробы.
Синтез солей бутилродамина С [208]. 50 г продажной хлористоводородной соли родамина С растворяют в 200 мл воды при нагревании на кипящей водяной бане. Темно-розовый горячий раствор фильтруют и к фильтрату постепенно при перемешивании прибавляют раствор 25 г NaOH в 100 мл воды. Выпавшую в осадок натриевую соль родамина С отфильтровывают из горячего раствора на воронке Бюхнера и сушат на воздухе.
30 г вполне сухой натриевой соли родамина С помещают в круглодонную колбу, приливают 54 мл бромистого н.бутила, 200 мл бутилового спирта и нагревают с обратным холодильником на масляной бане в течение 10—12 час. (температура масляной бани 120° С). После охлаждения реакционную смесь переносят в фарфоровую чашку и удаляют избыток бромистого н.бутила и бутилового спирта выпариванием на водяной бане.
Получение хлористоводородной соли бутилродамина С. Продукт, оставшийся после удаления бромистого бутила и бутилового спирта, растворяют в горячей воде, и горячий раствор подкисляют конц. НС1 до кислой реакции на конго. Выпавшую при охлаждении хлористоводородную соль бутилродамина С отделяют от маточного раствора декантацией и сушат при 35—40° С.
Для определения теллура применяют хлористоводородную соль бутилродамина С. Однако продажный препарат — «бутилродамин С» — может быть азотнокислой солью, которая гораздо хуже растворима, чем хлористоводородная соль. Продажный препарат часто содержит, кроме бутилродамина С, также значительные количества непрореагировавшего родамина С.
Проверку пригодности бутилродамина С для определения теллура проводят следующим образом.
В пробирку № 1 вводят 0,1 мл 0,05% -ного водного раствора бутилродамина С и разбавляют до 10 мл 0,1 N НС1.
В пробирку № 2 вводят 0,1 мл 0,05% -ного водного раствора бутилродамина С, разбавляют до 10 мл 0,1 N НО, добавляют 4 мл толуола и встряхивают 1 мин.
После расслаивания окраска водного слоя во второй пробирке не должна отличаться от окраски в пробирке № 1.
Оптическая плотность водного раствора после экстракции при измерении на спектрофотометре СФ-4 при 560 нм в кювете с I = 1 см должна составлять 0,750.
Опыт, накопленный со времени первой публикации метода определения теллура с бутилродамином С (1961 г.), показал, что область его применения в анализе минерального сырья ограничивается сравнительно несложными по составу материалами или материалом с относительно высоким (более 0,002—0,003%) содержанием теллура. При анализе проб, содержащих миллиграммовые количества сурьмы или кислоторастворимого олова, более 100— 200 мг меди или более 50—100 мкг таллия, однократное и даже двукратное осаждение теллура на мышьяке не обеспечивает достаточно полного отделения его от перечисленных элементов.
При восстановлении 1 мг мышьяка в условиях, рекомендованных прописью (осаждение гипофосфитом из 5—6 N НС1), осадок сорбирует в среднем 0,5% содержащегося в растворе олова и
115
0,05—0,1% сурьмы. Адсорбционная способность осадка, есте-венно, возрастает с увеличением количества коллектора за счёт мышьяка, содержащегося в навеске пробы. Ртуть, платина и золото полностью или почти полностью соосаждаются с теллуром. Процессы, предусмотренные для отделения этих элементов (дымление сернокислых растворов для восстановления золота, отгонка ртути в виде хлорида), плохо контролируются и не всегда оказываются эффективными. Перечисленные помехи заставляли искать дополнительные способы отделения теллура от мешающих элементов.
Блюм и Чувилева [47] предложили проводить разделение совместно экстрагированных после растворения осадка от гипофосфита родаминатов теллура и мешающих элементов, используя различие их экстракционных свойств. Авторы нашли, что такое разделение эффективнее при применении в качестве реагента этилродамина С (этилового эфира родамина С), а не бутилродамина С, что хорошо согласуется с общим возрастанием реакционной способности родаминовых красителей с утяжелением заместителя карбоксильной группы [208]. Чувствительность фотометрического и флуориметрического определения теллура с обоими красителями приблизительно одинакова.
Ниже приводятся некоторые характеристики экстракции бром-теллурита этилродамина С бензолом х. На рис. 24—26 представлены кривые зависимости интенсивности флуоресценции экстракта (показатель, пропорциональный степени экстракции теллура) от состава водной фазы (концентрации серной кислоты, бром-иона и красителя). Максимальное извлечение теллура имеет место при [H2SOJ = 9—9,5 N-, [Вг] = 0,13—0,15 М; [Р] = 0,01%.
В этих условиях при однократной экстракции, если отношение объемов органической и водной фаз равно 0,6, в бензол переходит около 80% теллура. Температурный коэффициент экстракции в интервале 16—26° С невелик — менее 2% на 10° С.
Окраска (флуоресценция) экстракта до отделения его от водной фазы достаточно (до 1—1,5 час.) стабильна, но после отделения начинает убывать; поэтому целесообразно стабилизировать экстракт ацетоном. Условный молярный коэффициент погашения бензольно-ацетонового (2 : 1) раствора при 560 нм составляет (при отборе для стабилизации 10/12 объема бензольного экстракта) 6,5-101 (еИст ~ Юь). При содержании в исходном водном растворе 1 мкг теллура оптическая плотность стабилизированного ацетоном экстракта объемом 15 мл (при измерении на спектрофотометре в кювете сечением — 5 см2 и длиной 3 см) составляет 0,10, а холостого экстракта — не более 0,03 [поправка холостого опыта
1 Неопубликованные данные И. А. Блюма и Н. А. Бруштейн.
116
Рис. 24. Зависимость интенсивности флуоресценции экстрактов бромтеллурита этилродамина С от концентрации H3SO4 вводной фазе
1—2 мкг Те; 2 — холостой опыт [Р1 = 0,01%; [KBr]=0.1JV; ГНгО= 10 мл\ = 6 мл
Рис. 25. Зависимость интенсивности флуоресценции экстрактов от концентрации красителя в водной фазе
I — 2 мкг Те; 2 — холостой опыт; [H2SOj=9,5 JV; [КВг] = 0,1 N; VH2O =10 *л;^с,н, = 6 кл
Рис. 26. Зависимость интенсивности флуоресценции экстрактов от концентрации бромида в водной фазе
/ — 2 мкг Те; 2 — холостой опыт; [HsSOj = 9,5ЛГ; [Р] = 0,01%;
VH2O =10 мл- VC.H, = 6 мл
обусловлена главным образом наличием экстрагируемых в виде родаминатов примесей, например, железа (III), в реактивах].
В условиях, оптимальных для опредления теллура с этилрода-мином С, в различной степени экстрагируются те же элементы, что и при определении Те с бутил родамином С.
Для разделения совместно экстрагированных после растворения осадка от гипофосфита ассоциатов бромидных комплексов теллура и мешающих элементов с этилродамином С сначала встряхивают экстракт с сернокислым раствором, содержащим краситель, но не содержащим бром-ион, и промывают реэкстракт бензолом. При этом теллур и некоторые из сопутствующих ему элементов ведут себя по-разному. Сравнительно мало прочный анион ТеВга (а также бромиды железа, олова и меди) может существовать лишь в присутствии значительного избытка бром-ионов, поэтому теллур медь (I), олово (II) и (IV), железо (III) полностью экстрагируются, а при промывке реэкстракта бензолом теллур остается в водной фазе. В то же время ассоциаты обладающих высокой прочностью бромидных комплексов золота, ртути, платины, и таллия при реэкстракции остаются преимущественно в органической фазе; незначительные количества этих элементов, перешедшие в реэкстракт, удаляются из него при промывке бензолом.
Затем добавляют в водный раствор (реэкатракт) бромид и экстрагируют бромтеллурит этилродамина С. При этом основная масса реэкстрагированных вместе с теллуром элементов (— 90% олова, 99,9% меди и железа) остается в водной фазе.
Совокупность описанных операций достаточна для того, чтобы отделить (с погрешностью, эквивалентной не более 0,2 мкг Те) 1 мкг Те от 100 мкг Sn, Au и Pt, 10 мкг Т1 и Hg, 20 мг Fe и Си (табл. 14).
Таблица 14
Специфичность фотометрического определения теллура с этилродамином С и эффективность разделения теллура и сопутствующих элементов
Элемент 1 2 3 Преобладающий механизм разделения^ Элемент 1 2 3 I Преобладаю- щий механизм разделения *
Железо 0,7 >100 >150 1 Таллий 0,003 0,03 10 2
Медь 2 >100 >50 1 Платина 0,025 0,1—1,0** 4—40 2
Олово 0,04 0,50 12 1 Золото 0,005 0,1—1,0** 20—200 2
Сурьма 0,001 0,001 1 — Индий 0,001 0,001 1 —
Ртуть 0,001 0,025 25 2
• 1 — на основе различия коэффициентов экстракции в условиях экстрагирования этил-родамнната теллура; 2 — на основе различной прочности бромидных комплексов.
•* Результаты нестабильны.
118
Для удаления очень больших (Д> 10 мг) количеств сурьмы и ртути рекомендована отгонка летучих бромидов этих элементов при температуре дымления серной кислоты.
В графе 1 (табл. 14) показано количество сопутствующего элемента (мг), эквивалентного по оптической плотности экстракта 1 мкг Те при «прямом» определении; в графе 2 — такой же показатель 2
после разделения родаминатов; в графе 3 — отношение q---------«фак-
тор разделения» определяемого и сопутствующего элемента (валентное состояние сопутствующих элементов задается условиями растворения осадка от гипофосфита и условиями определения).
Отмечено, что даже при таком значительно усложненном по сравнению с методиками, описанными в [83, 173], ходе подготовки к определению помехи, обусловленные неполным отделением мешающих элементов, в некоторых случаях могут достигать (при навеске 1 г) величины, соответствующей 0,2 мкг теллура. Поэтому высокая чувствительность флуориметрического определения не может быть полностью реализована; целесообразно заканчивать анализ измерением оптической плотности экстракта, выдавая значащие результаты при величине плотности, эквивалентной не менее чем 1 мкг теллура.
Выполнение определения теллура с этилродамином С. Отбирают 10 мл или меньший объем (аликвота ие должна содержать более 12 мкг теллура) раствора в стакан емкостью 50 мл, добавляют 12 мл 14,8 N H2SO4 и нагревают до начала дымления. Если раствор темнеет, осторожно добавляют в дымящую H2SO4 несколько капель HNO3 или пергидроля до обесцвечивания раствора, затем добавляют немного воды и вновь доводят до дымления.
Охлаждают, обмывают стенки стакана небольшим количеством воды, приливают приблизительно 3 мл конц. НВг и упаривают на негорячей бане до начала дымления раствора *, обмывают стенки стакана водой и вновь доводят до дымления (следует прекратить нагревание раствора сразу после начала дымления, так как заметное уменьшение количества H2SO4 привело бы к отклонению условий последующего коицентрирования теллура от оптимальных).
Добавляют 3—4 мл воды, переводят раствор в мерный цилиндр и доводят объем водой до 12 мл, после чего переносят раствор (не обмывая цилиндр) в делительную воронку емкостью 50 мл. Приливают 4 мл раствора этилродамина С, перемешивают, приливают 12 мл бензола, 4 мл раствора КВг и экстрагируют 30 сек.
Переводят водный раствор в другую делительную воронку, приливают 12 мл бензола и вновь экстрагируют 30 сек. Сливают водную фазу и объединяют первый и второй экстракты в одной из делительных воронок (необходимо тщательно отделять бензольный экстракт от водной фазы, содержащей бромид, иначе реэкстракция теллура не будет достаточно полной). Приливают 12 мл 14,8 TV H2SO4, 4 мл этилродамииа С и, встряхивая 30 сек., реэкстрагируют теллур 1 2.
После расслоения фаз просматривают экстракт на белом фоне: если он окрашен, переводят водный раствор в другую делительную воронку и промывают его,
1 Если навеска, предназначенная для определения теллура, содержит более 10 мг ртути или сурьмы, операцию отгонки повторяют еще два раза.
2 Операцию концентрирования можно также проводить в пробирках с притертыми пробками, отбирая бензольный экстракт пипеткой с грушей. В этом случае объемы водной и органической фаз уменьшают вдвое, сохраняя концентрации реагентов.
119
встряхивая 30 сек. с 10—12 мл бензола и т. д., пока очередная порция бензола не окажется бесцветной; бензол отбрасывают. Эта операция имеет целью удаление из водной фазы микрограммовых количеств платины, ртути, золота. Если органический слой после реэкстракцин теллура окажется бесцветным, то ее опускают.
Переводят реэкстракт в пробирку с притертой пробкой, приливают 12 мл бензола, 4 мл раствора КВт и экстрагируют 30 сек. Через 10—15 мин. отбирают пипеткой с грушей в сухой мерный цилиндр 10 мл экстракта и переводят его всухую пробирку или небольшую колбу, содержащую 5 мл ацетона. Перемешивают, измеряют оптическую плотность при толщине слоя 3 см на спектрофотометре при X = 560 нм или на фотоколориметре с зеленым светофильтром.
Построение градуировочного графика: объемы стандартного раствора, содержащие 0; 2; 4; 6; 8; 10; 12 мкг теллура, помещают в делительные воронки, доводят объемы до 12 мл 14,8 N H2SO4, прибавляют этилродамин С, бензол и т. д. и проводят концентрирование и определение, как указано выше.
Если можно ожидать, что содержание золота и платины в навеске или ее части, используемой для определения теллура, превышает 1 мг, навеску вскрывают азотной кислотой без добавления соляной кислоты. В этом случае перед осаждением теллура гипофосфитом отфильтровывают нерастворимый остаток.
Определение теллура в виде гетерополисоединений
Предложен метод фотометрического опредэления теллура в виде теллуритовольфрамовой сини. Желтые растворы теллуритовольфра-мовой гетерополикислоты Н4 [ТеО4 (H2WO4)2]- 13Н2О восстанавливаются проволокой из углеродистой стали до теллуритовольфрамовой сини. В присутствии сильных восстановителей (SnCl2, гидразина, гидрохина) происходит выделение металлического теллура. Теллуритовольфрамовая гетерополикислота устойчива в интервале кислотности 0,04—1,17V НС1 [109].
На образовании теллуритомолибденовой гетерополикислоты основан косвенный фотометрический метод определения теллура [211]. В присутствии четырехвалентного теллура в водном растворе кремнемолибденовой кислоты при pH 2—3 происходит ослабление окраски этого раствора вследствие вытеснения кремния из кремне-молибдата теллуром с образованием более прочного бесцветного теллуритомолибдата со стехиометрическим соотношением Те : Мо = = 1:6. Комплекс образуется практически мгновенно. Присутствие селена и других элементов, дающих с молибдатом прочные комплексы, мешает определению теллура.
Известен фотометрический метод определения теллура, основанный на образовании окрашенной в желтый цвет тройной фосфорно-теллуристомолибденовой кислоты со стехиометрическим соотношением Р : Те : Мо =1:1:11. Метод пригоден для определения малых количеств теллура в присутствии селена [212].
Разработан фотометрический метод определения Те (IV) в полупроводниковых сплавах InTe, 1п2Те3 в виде теллуристомолибде-нованадиевого комплекса, образующегося в сернокислой среде (0,2— 0,3 N по H2SO4) при большом избытке Мо и V и отношении Мо : : V = 1 : 0,5 [312].
120
Определение теллура с производными триоксифлуорона
Шитарева и Назаренко [443—445] в качестве реагентов на теллур предложили производные 2,3,7-триокси-6-флуорона, замещенные в положении 9—9-пропил-2,3,7-триокси-6-флуорон (I) и 9-Р-окси-а-нафтил-2,3,7-триокси-6-флуорон (II). Оба реактива дают с Те (IV) окрашенные в розово-красный цвет соединения с соотношением Те : реактив =1:2, выделяющиеся в осадок (при больших количествах теллура), а в присутствии желатины образующие устойчивые золи. Оптимальные значения pH образования соединения с реагентом I равно 4—6, с II равно 4—5,5. Окраска развивается в течение 4 час. и далее остается практически постоянной.
Нагревание необратимо разрушает окраску. Молярный коэффициент погашения составляет для соединения теллура с I —22 000, с 11-^27 700.
Закон Бера при достаточном избытке реактива соблюдается при концентрациях 0,4—2,4 мкг Те /мл. Подобно теллуру, реагируют также Al, Fe, Se, Au (III), In, Ge, Sn (IV), Sb (III), Ti, Zr, W, Mo, U, Ta, Nb и V (V). Определению Те (IV) не мешают Те (VI), As (V), Sb (V), Se (IV) и Se (VI).
Введение в раствор комплексона III маскируют влияние А1, Fe, Au, Sc, Zn и других трехвалентных металлов.
Выполнение определения теллура с 9-пропил-2,3,7-триокси-6-флу<»роном. При определении теллура в присутствии Se, As, Au, Bi, Fe и Al к 3 мл анализируемого раствора (1 N по НО) прибавляют 2,5 мл 40%-ного раствора CH3COONa* 2 мл 2%-ного раствора комплексона III, 4 мл 1%-ного раствора желатины и 2,5 мл этанола, разбавляют водой до ~ 22 мл, вводят 2 мл 0,05%-ного раствора реагента, разбавляют водой до 25 мл, выдерживают 4 часа и спектрофотометри-руют при 530 нм.
Определение теллура косвенными фотометрическими методами
Для определения микрограммовых количеств теллура был разработан косвенный фотометрический метод, основанный на восстановлении комплекса Fe (III) с 1,10-фенантролином (феррин) до интенсивно окрашенного комплекса Fe2+ (ферроин) по реакции [651, 773, 802]: .
Те (IV) + 2Fe (С,2Н8М..)Д Те (VI) + 2Fe (C12H8N2)2+.
Скорость развития окраски зависит от кислотности раствора,, времени взаимодействия, концентрации реагента. В хлорнокислом растворе (0,05 N) при содержании теллура 1 мкг максимальное развитие окраски достигается через 10 мин. при концентрации ферроина ~ 5-10-6 М. Максимум светопоглощения ферроина находится в области 474—508 нм. Чувствительность метода по шкале Сендела
121.
0,007 мкг Те/мл [773] или 0,5 мкг в 1 г [651J. Ошибка определения составляет —0,2 мкг Те.
Мешают определению Mn2+, Cu2+, Се2+, Au, Pd, Rh, Ir, Sn,W, V. Селен не мешает.
Были описаны также методы определения теллура с акридином, а-нафтафлавоном и хинином [717]. Эти реагенты в присутствии иодида реагируют с теллуром так же, как с золотом и селеном (тушение флуоресценции этих реагентов выделенным элементным иодом). Методы мало избирательны.
каталитические методы
Изучение новых каталитических реакций позволяет разрабатывать высокочувствительные методы определения элементов. Однако по каталитическим методам определения селена и теллура имеется в настоящее время немного работ. Следует отметить метод определения теллура, основанный на каталитическом действии теллура на реакцию восстановления золота гипофосфористой кислотой. Этот метод позволяет определять 1 нг теллура.
Определение теллура
Для определения нанограммовых количеств теллура был предложен высокочувствительный каталитический метод [853], основанный на восстановительном осаждении элементного золота из раствора 6N НС1, содержащего хлорид золота, хлорид меди и гипо-фосфористую кислоту. Количество восстановленного золота пропорционально количеству присутствующего теллура. Этим методом можно определить 1 нг (1 • 10'9 г) Те в 50 мл раствора. Скорость восстановления хлорида золота в 6 N НС1 гипофосфорной кислотой зависит от концентрации Те, Au, Си, Н3РО2 и от температуры.
Чтобы избежать влияния примесей и различия в концентрации продажной гипофосфористой кислоты, необходимо использовать Н3РО2, синтезированную по методу Джонса [853].
Количество выделившегося при восстановлении золота определяют по родаминовому методу при измерении оптической плотности при 335 нм.
Мешают определению As, Se, Hg и Sb, бромиды и иодиды. Все эти элементы удаляются выпариванием образца с H2SO4 и НВг.
Следующие соединения не мешают, если они добавлены к 50 мл 6 А НС1, содержащей Си, Аи и Н3РО2: 100 мг NaNO3, 60 мг K2SO4 и Na2HPO4-7H2O; 50 мг FeNH4 (SO4)2-12Н2О; 30мг СгС13 и MgCl2-•6Н2О; 20 мг A12(SO4)3-12Н2О, ВаС12; Bi (NO3)3-3H2O, CdCl2-•г^аНгО, CaSO4-2H2O, СаС12-6Н2О, LiCl, MnSO4-H2O, NiSO4-6H2O,
122
NaF, Sr(NO3)2, K2TiO (C2O4)2 • 2H2O, VO2(NO3)2-6H2O, ZnSOv •7H2O, Na2B4O7-10H2O; 10 мг GeO2, Sc2O3, T12O3, La2O3, Ta2O5, Ga2O3, In2O3, SnCl2, AgNO3; 100 мкг Pt, Pd, Re, Be, W; 200 мкг Nb; 1000 мкг Mo; 200 мкг Pb.
Хотя предложенный метод в настоящем варианте и не дает желаемой точности (расхождение параллельных определений в полтора раза), высокая чувствительность этого метода позволяет определять очень малые содержания теллура в различных геологических материалах, образцах чистых минералов [853].
Выполнение определения. Навеску образца растворяют в смеси СС14 4-+ Вг2 с HNO3, окислы железа — сплавлением с пиросульфатом калия, силикаты — сплавлением с Na2CO3 и KNO3.
После первоначальной обработки образца добавляют 1 мл H2SO4 и 2 мл НВг; полученный раствор выпаривают до паров SO3, добавляют 2 мл НВг, н выпаривание повторяют три раза.
К холодному сернокислому раствору добавляют 50 мл 6 N НС1, содержащей 1 мг Au и 6 мг Си. Добавляют 1 мл 50% -ной Н3РО2, и раствор оставляют на 5 мин. при 15° С до фильтрования.
Количество теллура в образце рассчитывают по стандартной кривой, измеряя интенсивность окраски комплекса золота с родамином С.
Определение селена
Описан кинетический метод определения четырехвалентного селена [345, 1110], основанный на каталитическом участии селена в реакции восстановления 1,4,6,11-тетразонафтацена при помощи Н3РО2 в кислой среде с образованием 1,6-дигидропроизводного нафтацена синего цвета (Хтах при 570 и 600 нм). Для получения нафтацена (Хтах =478 нм) в условиях определения используется реакция конденсации 2,3-диаминофеназина (Хтах = 492 нм) с глиоксалем. Скорость реакции зависит от температуры, концентрации реагентов и pH раствора.
При оптимальных концентрациях соляной кислоты (0,1 Л4), 2,3-диаминофеназина (4-Ю'5 А4), глиоксальсульфата (10~4 М), гипофосфита (0,02 М) чувствительность определения селена составляет 0,02 мкг/мл.
Определению 0,1 мкг Se не мешают 100 мкг Мп, Са, Со, Al, Ni, Zn, 10 мкг Сг, As, Си, V5+, Fe. Теллур в количестве 1 мкг мешает определению. Мешают определению селена: 1 мг Ba, Fe(III) и W (VI), а также Bi3+, Sn2+ и J", изменяющие характер окраски смеси. Большую часть мешающих элементов рекомендуется отделять ионным обменом или экстракцией. Селен (VI) не мешает.
Выполнение определения [345]. В пробирке с пришлифованной пробкой смешивают анализируемый раствор (<0,15 мкг Se) с 0,10 мл 4%-ного раствора глиоксаля в 0,1 У НС1 и 1,0 мл 1,5 •! О-4 М раствора 2,3-диаминофеназина в в 0,1 А НО. Разбавляют до 4,5 мл 0,1А НС1, выдерживают смесь 10 мин. в термостате при 50° С, вводят 0,5 мл 50%-ной Н3РО2 и точно через 30 мин. после прибавления восстановителя погружают пробирку в ледяную баню для прекращения каталитической реакции.
123
Затем не позднее, чем через 30 мин., измеряют оптическую плотность раствора при 600 нм в кювете с I — 1 см и находят содержание селена по прямолинейному калибровочному графику.
Предложен метод определения следов селена, основанный на каталитическом участии его в реакции восстановления метиленового голубого сульфидом натрия в щелочной среде в присутствии ионов Fe (III) и комплексона III [1144]. Реакцию проводят при pH 10,8.
Определению 1 мкг селена мешают >• 10 мкг Си2+. В меньшей степени мешает присутствие Sb, Ag и Bi (> 25 мкг), не мешают 50-кратные количества Li+, Na+, К+, Ве2+, Mg2+, Са+, Sr2+, Ва2+, Zn2+, Cd2±, Hg2+,B4Ot, А13+, Се3+, НСОд, СОГ, SiOt, Zr (IV), Sn<IV>, Pb2+, NH4, NOg.PO3-, VO3, F_, СГ, Br и J~. Калибровочный график прямолинеен в интервале содержаний 0,1—1 мкг Se.
Обнаружено каталитическое действие селена (IV) на реакцию CIO3 и N2H4, что дает возможность определять малые количества селена (0,1—1 мкг в 5 мл раствора) со средней ошибкой 10% [548].
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Полярографическое определение теллура
Лингейн и Нидрач [884] исследовали полярографическое поведение четырехвалентного теллура в широком диапазоне pH в аммиачном и тартратном буферных растворах, а также в щелочном растворе и установили, что в аммиачном и виннокислом буферном растворах Те (IV) восстанавливается до элементного и дает хорошо выраженную волну, пригодную для его количественного определения.
Согласно данным кулонометрического анализа, суммарный диффузионный ток при pH 0,4—11 в буферных растворах обусловлен электродной реакцией
Н2ТеОз + 4Н+ + 4?^± Те» + ЗН2О.
В аммиачном буферном растворе с pH 9,4 в присутствии 0,003% желатины потенциал полуволны при восстановлении Те (IV) на капельном ртутном электроде равен —0,68 в (отн. н. к. э). При увеличении pH потенциал полуволны смещается в более отрицательную область [237, 398, 627].
На фоне 0,1 N NH4C1, NH4NO3 при pH 6,3 четырехвалентный теллур образует четкую диффузионную волну с Еуг = —0,71 в (отн. н. к. э.). Диффузионный ток значительно снижается при увеличении pH от 2 до 5, но при pH 6,3—9 достигает первоначального значения. Изменение концентрации фона от 0,025 до 9,5 М не влияет на высоту волны [627].
124
На фоне 1 V NHiCl и 0,5 N NHiOH при содержании в растворе 0,004—0,008% желатины возможно определение теллура в присутствии селена [8, 9, 288, 944]. На сульфатно-аммиачном фоне при pH 9 потенциал полуволны теллура равен —0,60 в; селен также не мешает определению теллура [23, 167, 347], возможно совместное определение этих элементов [194].
Влияние цинка, свинца, железа и других элементов в аммиачном буферном растворе в присутствии комплексона III изучалось Зелянской и др. [162]. Наличие в растворе РЬ2+ и катионов других металлов, образующих нерастворимые теллуриды, уменьшает диффузионный ток теллура. При определении теллура в сплавах свинца Загорский и др. [1183] проводили прямое определение теллура, которому не мешали большие количества свинца, а также Cu, Cd, As, Sb в количествах ~ 0,01%.
После выделения теллупа гидразином можно определить 0,05 мг Те в меди в присутствии мышьяка [572]. Отделить теллур можно также восстановлением SnCl2 и соосаждением с гидроокисью цинка [146].
Для отделения теллура от мешающих элементов эффективным оказался метод соосаждения теллура с элементной серой [182, 183].
В случае использования переменнотоковой полярографии на фоне буферного раствора NH4OH — NH4NOa линейная зависимость между концентрацией теллура и высотой его пика (Еп = —1,0 в отн. донной ртути) наблюдается при концентрации теллура в интервале 1 • 10'8—1-Ю-4 М (с капельным ртутным электродом) и 1,6-10'7— 4-10"® М (со стационарным ртутным электродом). Селен не мешает вплоть до соотношения Se : Те = 100 000 : : 1 [288].
На фоне тартратно-аммиачного буферного раствора можно определять до 2-10-5 М Те в присутствии сурьмы, свинца и железа [7,78]. В 1 М виннокислом растворе Е1/а = —1,0 в (отн. н. к. э.) 1291].
На полярограммах Те (IV) во всех электролитах (кроме NaOH) имеется большой максимум (рис. 27). Обычные поверхностноактивные вещества его не подавляют, из чего следует, что он не является полярографическим максимумом, вызванным движением поверхности. О причинах его возникновения были высказаны различные предположения [202, 882]. Аномальный максимум теллура подавляется в случае присутствия в растворе сульфита натрия [452].
Применение сульфатно-аммиачного фона позволяет проводить определение теллура (и селена) в медьсодержащих продуктах [78, 160, 161]. Описан метод определения теллура в селене [12]. В присутствии больших количеств меди теллур определяют в растворе цианида [76—78]. Потенциал полуволны Те (IV) на фоне 0,65 М KCN, 0,25 М NaOH, 0,2 М Na2SOa и 0,1—0,05%-ной желатины равен —1,20 в.
125
Рис. 27. Полярограммы Те (IV) в’растворах различной концентрации
1 — фон (1 М NH«OH + 1 М NH4C1 + 310—®% желатины); 2 — 0,2'10-•М Те; 3— 0,5-10-» М Те;
4 —1,010-» М Те;5 — 2,0-10~» МТе
В щелочной среде четырехвалентный теллур восстанавливается до теллурида. На фоне 2 М КОН и 0,05%-ной желатины теллур определяют в присутствии 10000-кратного избытка селена методом осциллографической полярографии [995].
В щелочных и аммиачных средах происходит необратимое восстановление теллура. Поэтому такие растворы не пригодны для высокочувствительного квадратно-волнового полярографического определения теллура. В отличие от полярографических волн» пульсполярографические пики в кислых растворах пропорциональны концентрации теллурита [181, 183]. Для квадратно-волновой полярографии наилучшим фоном является раствор КС1 с pH 1,5—2,5. Аналогичные полярограммы были получены на вектор-полярографе ЦЛА [34].
На катодной осциллополярограмме Те (IV) в растворах НС1 и H2SO4 имеются два пика — с Ад = —0,13 и Дщ —0,80 в, причем второй пик в 100 раз больше первого. Те (IV) восстанавливается до элементного, и последний адсорбируется на электроде. Количественное определение теллура возможно в интервале его концентраций 8-10-8—2-Ю-6 М [432].
Известен пульсполярографический метод определения теллура (на фоне 1 N НО) в полупроводниковых сплавах и тонких слоях, содержащих индий. Метод позволяет определять теллур до Ю'6 М без предварительного отделения индия [223]. При определении теллура в тонких пленках HgTe и CdTe методом осциллографической полярографии использована 0,01 N HNOa. Высота волны пика пропорциональна концентрации теллура в интервале концентраций 0,05—2 мкг/мл. Ртуть и кадмий при концентрациях до 30 мкг/мл не влияют на пик теллура [1149].
Описаны методы определения теллура в 1 М фосфорнокислом растворе в присутствии сурьмы и галлия [90].
На фоне 0,1 М (NH4)2SO4 + 0,08 М Na2P2O7 и 0,03%-ной желатины определению теллура не мешают 5-кратные количества меди.
126
50-кратные количества кадмия и 100-кратные количества цинка 11941.
Фосфорнокислые растворы успешно применяли при квадратноволновом определении микрограммовых количеств теллура после отделения серебра и меди соосаждением с Fe(OH)a, отделения селена отгонкой [7951. Потенциал восстановления теллура на фоне 1 М НаРО4 и 0,5 М HClO4(E1/2Te(iv) = —1,2 ^l/2Te(Vl) = —1,57 в) значительно отрицательнее потенциалов восстановления меди и свинца (—0,21 и —0,76 в соответственно), но близок к потенциалу восстановления цинка (Ео = —0,49 в). Поэтому возможно прямое определение теллура методом переменнотоковой полярографии в /Продуктах, содержащих медь и свинец 1287].
Описан метод определения следов теллура в латуни и чугуне на фоне 1,5 М Н3РО4 при помощи катодно-лучевого полярографа. Теллур предварительно отделяют от мешающих элементов соосаждением с Fe(OH)a. Высота катодного пика пропорциональна содержанию теллура в интервале концентраций 0,05—0,7 мкг/мл. Не мешают 20 мг Fe(III), мешают V и Сг [901].
На фоне 0,5 N HJ методом переменнотоковой полярографии можно определить 2-Ю”8 % Те в присутствии ряда элементов при следующих их соотношениях к теллуру:
Bi ... . 500 :1 Fe . . . . 6000 : 1 Au ... . 100:1 Cd ... . 100 : 1 Си ... . 1000 : 1
Mo . . . . 50:1
As ... . 1:1
Sn . . . . 100 : 1
Pd . . . . 100 :1
Pt ... . 5:1
Pb . . . . 500 :1
Se . . . . 100 : 1
Ag . . . . 100 :1
Sb ... . 1000 :1
Zn . . . . 1000 : 1
Мешают HC1 и HNOa [291, 293]. На фоне 0,25 N HJ и 0,07 N H2SO4 селен не мешает при соотношении Se : Те = 100000 : 1 [287]. При определении трифенилтеллурхлорида на полярограммах обнаруживаются две волны; реакции восстановления обратимы 1754].
В случае полярографического восстановления шестивалентного теллура на капающем ртутном электроде Те (VI) дает хорошо выраженную волну с Ех/2 = —1,21 в (на фоне 2—0,1 N NH4C1 — NH4OH при pH 8); ток прямо пропорционален концентрации теллура в пределах 5,7-10-3—5,7-10“eA4. Изменение pH от 6,2 до 9,2 сдвигает потенциал полуволны в более отрицательную область 19711.
На капающем ртутном электроде в универсальном буферном растворе pH 6,1—11,98 образуется одна волна, соответствующая восстановлению Те (VI) до элементного теллура, и потенциал смещается с увеличением pH от —1,24 до —1,58 в. На фоне 0,001—0,1V NaOH наблюдается также одна волна Е1/2 = —1,43 в, а на нейтральном фоне 0,1 N КС1, NaCl, NaClO4 и K2SO4 обнаружены две волны — Ех/2 = —1,15 и —1,5 в, что
127
объясняется наличием следующего равновесия в растворе [7921:
Н6ТеО6 + HsTeO- + Н+;
HsTeOe + 8е + 7Н+ -» Те2- + 6Н2О;
Н5ТеО; + бё + 8Н+ -» Те0 + 6Н2О.
На фоне 0,1 N (NH4)2 SO4 и 0,1 N Na2SO3 в присутствии желатины при pH > 8 теллурат-ион дает на полярограмме четкую волну,, пригодную для количественного определения теллура [194].
Теллурид-ион дает анодную волну при всех значениях pH. В аммиачном буферном растворе происходит окисление Те2- до элементного теллура. Реакция окисления обратима. В 1 У НС1 потенциал полуволны равен —0,72 в. В сильнощелочном растворе (среда NaOH) наблюдается дополнительная волна, связанная с окислением до иона теллурита [882—884].
Полярографическое определение селена
Полярографическое восстановление четырехвалентного селена в кислых растворах происходит в две стадии: сначала до элементного состояния, затем до селенида [348]. Восстановление селена до селенида сопровождается выделением водорода.
Исследование полярографического поведения четырехвалентного селена на фоне серной, соляной и хлорной кислот показало, что в кислой среде четырехвалентный селен дает две волны [588]. Потенциалы полуволны в вольтах (отн. н.к.э.) четырехвалентного селена при восстановлении на ртутном капельном электроде в различных электролитах представлены ниже:
Первая Вторая волна волна
0,1 М HNO3 . . .+0,083 -0,561 1 М HNO3 . . . +0,021 —0,411 1 М KNO3 . . . -0,185 —0,681
Первая Вторая волна волна
0,1 М НС1 . . .-0,011 -0,541 1 М НС1 ... —0,101 —0,511 0,1 М НС1О4 . . . +0,089 —0,541
В результате исследований Лингейна [882—884], изучавшего восстановление Se (IV) на капельном ртутном электроде в широком интервале значений pH, были получены систематические данные потенциалов полуволн и коэффициентов диффузии. Различные восстановленные состояния наблюдались методом кулонометрического анализа, основанного на электролизе с контролируемым потенциалом на ртутном катоде.
Кулонометрические исследования [292, 882—884] показали, что в сильнокислых растворах суммарный диффузионный ток обусловлен электродной реакцией:
HjSeOs + бё + 6Н+ -► H2Se + ЗН2О.
128
Рис. 28. Полярограммы Se (IV) в растворах с различным значением pH
1 — 1,63; 2 — 2,70; 3 — 3,60; 4 — 5,90; Г— 7,80; 6 — 8,90; 7 — 10,7
В сильнокислой среде и буферном растворе с pH > 8 восстановление селена происходит во всех случаях до Se2-, а в буферном растворе с pH 3—7 — до элементного селена.
В растворах с pH 4,85—5,90 четырехвалентный селен образует три волны [884]. При более высоких значениях pH (8—9) остается только одна волна, соответствующая восстановлению Se (IV) до элементного (рис. 28). Сложные волны, наблюдаемые для селена при промежуточных значениях pH, являются результатом сложения нескольких факторов, включающих восстановление до элементного селена и селенида и образование пленки элемента на поверхности капающего электрода. Состояние Se2+, на которое указывают в своей работе Шваер и Сухи [1069], не было подтверждено Лингейном.
Высокочувствительный метод определения селена в присутствии теллура при соотношении Те : Se = 50 : 1, а также в присутствии Fe (HI), Nb, Pb, Си при соотношении 1 : 1, Fe (II) при соотношении 40 000: 1 [292] основан на восстановлении четырехвалентного селена на фоне НС1, НВг, H2SO4, Н3РО4, винной и хлорной кислот с использованием стационарного ртутного электрода.
Метод переменнотоковой полярографии Se (IV) на фоне 1 N H2SO4 был применен для анализа полупроводниковой системы Си — Ge — Se [221, 224]. Разработан метод определения селена на фоне H2SO4 и НС1 на вектор-полярографе ЦЛА со стационарным электродом в виде ртутной капли. Метод позволяет определять 2,5-10“9 — 2-10“7Л4 Se с коэффициентом вариации 15% [290].
Для определения малых количеств селена в медной руде был применен переменнотоковый полярограф PZ-62. Селен и теллур определяли в разбавленном солянокислом растворе после выделения в элементном состоянии двухлористым оловом и солянокислым гидразином [847]. Известен метод осциллополярографического определения селена в селениде германия. В качестве фона для определения селена в присутствии германия служили 2N НС1 [419, 420], а также IV NH4C1 и 0,2 N НС1 [104].
5 Зак. № 1476
129
В аммиачном буферном растворе при pH 8—9,5 Шваер и Сухи [1069] наблюдали простую хорошо выраженную волну, соответствующую восстановлению Se (IV) до Se2~. При pH 8 диффузионный ток обусловлен электродной реакцией:
SeO2 + 6ё + ЗН2О Se2~ + 6ОН~.
Значение потенциала полуволны этой реакции соответствует значению £>/2 анодной волны Se2-, появляющейся вследствие следующей реакции [884]:
Н g + H2Se HgSe + ЗН+ + 2е.
Количественное полярографическое определение селена обычно проводят в аммиачном буферном растворе с pH 8 [21, 146, 237, 281, 347 , 404]. Лучшим электролитом для этой цели является раствор IV NH4C1, 0,1 — 1 V NH4OH, содержащий 0,001—-0,003% желатины. Потенциал полуволны селена несколько сдвигается с изменением значения pH: при pH 8£\/, = —1,44а, а при рН9Е/3 = = —1,54 в (отн. н.к.э).
Присутствие тяжелых металлов искажает волну селена, и перед полярографическим определением селена необходимо проводить отделение селена и теллура от мешающих элементов [161—163, 182, 350, 1023].
Арефьева и Позднякова [22] определяли селен в аммиачном буферном растворе с pH 8,0—8,2 в присутствии пирофосфата натрия без предварительного отделения от Fe, As, Те и Си. Потенциал полуволны для селена (отн. н.к.э.) был равен —1,20 в.
На фоне смеси состава 0,75 М NHiCl, 0,25 М NH4OH и 0,1 М Na2COa в присутствии 0,02% желатины можно раздельно определить селен и теллур [160].
В среде 0,1Л4 ацетата аммония в присутствии 0,005% желатины при pH 6,2 наблюдаются две волны с потенциалами полуволн —0,81 и —1,38 в. Дешмук [624] объясняет их восстановлением до Se2+ и элементного состояния.
На фоне подкисленного раствора тартрата калия (натрия) или цитрата лития при pH 1—5 в присутствии 0,01 % желатины четырехвалентный селен образует четкую волну, потенциал полуволны которой смещается от —0,55 до —0,60 в с изменением pH от 1 до 5 [534, 944].
Селенид-ион так же, как и S2", дает на капельном электроде анодную волну. В растворе 0,1 N NaOH можно определять селениды и сульфиды при совместном присутствии, так как потенциал селенида имеет на 0,2 в более отрицательное значение, чем потенциал полуволны сульфида. Электрохимическое поведение селеносульфид-иона SeS2~ на ртутном капельном катоде изучала Торопова и сотр.
[388].
При растворении элементного селена в растворе сульфита щелочного металла образуется селеносульфат-ион SeSOa~, который вос-130
станавливается на ртутном капельном электроде и образует хорошую полярографическую волну. Торопова [387] изучала механизм этой реакции и сделала вывод, что при восстановлении SeSO|~ образуется ион селенида:
SeSO|- + 2? -> Se2' + SO^.
Электродные процессы, соответствующие указанной полярографической волне, были изучены Зелионкайте и Яницким [158]. Каплан с сотр. [182], изучавшие механизм этой реакции, считают, что в результате разложения селеносульфата образуются сульфит и элементный селен, который адсорбируется на электроде и обратимо восстанавливается при потенциале пика селеносульфата до селенида. Метод позволяет определять до 10“5М селена в присутствии теллура, который в этих условиях не восстанавливается.
Пульсполярографическое определение селена в виде селеносульфата позволяет определять до 5-10 '8Л4 селена [182]. Для концентрирования микрограммовых количеств селена в последнем случае применяют серу в качестве коллектора, полярографирование проводят на фоне O,6VNH4C1 +0,6VNH4OH в присутствии сульфита. Полярографически определяли селен на сульфатно-аммиачном фоне при анализе медных концентратов [10901, шламов, серной кислоты [316], полупроводниковых материалов [386] и других объектов [78, 91, 227].
Полярографически четырехвалентный селен может быть определен по волнам пиазоселенола, который образуется при добавлении к раствору, содержащему Se (IV), 3,3'-диаминобензидина или о-фенилендиамина [593, 594]. Для маскирования примесей добавляют комплексон III. При определении микрограммовых количеств селена экстрагируют комплекс бензолом и экстракт используют для полярографического определения.
Фенилпиазоселенол на фоне 0,17V НС1О4 и 0,1 V NH4C1O4 при pH 2,5 дает две волны с потенциалами полуволн —0,17 и —0,64 в (отн. н.к.э.). Высота первой волны пропорциональна концентрации Se (IV) в интервале 0,5—20 М. Метод был применен при определении селена в сере, сульфидах, сульфатах.
Описан косвенный метод определения селена, в котором использовано количественное осаждение селена сульфатом гидразина в присутствии нитрата таллия. В осадке TISe полярографически определяют количество таллия и по нему рассчитывают соответствующее количество селена [737].
Амперометрическое титрование
Амперометрически определяют селен по току окисления избыточных иодид-ионов при потенциале +1,0 в. Реакция селенистой кислоты с иодидом обратима:
SeOg- + 4J- + 6Н+ Щ Se + 2J2 + ЗН2О.
5*
131
При достаточной концентрации Н+-ионов возможно прямое амперометрическое титрование избытка J" на вращающемся платиновом электроде [3431.
Взаимодействие селенистой кислоты с тиосульфатом по реакции H2SeO3 + 4S2O|" + 4Н+ -> Se (S2O3)f + S4Ot + 3H2O было использовано при амперометрическом микроопределении селена. Добавляли избыток тиосульфата, который оттитровывали раствором иода. Конечную точку титрования определяли по току восстановления иода на платиновом электроде [803].
Разработана методика амперометрического определения 0,1% Se в сере без предварительного выделения селена. Метод основан на восстановлении селенитов избытком тиомочевины и оттитровывании тиомочевины бихроматом при +1,0 в (отн. меркур-иодидного электрода) по току окисления тиомочевины. Относительная ошибка метода +5% [210].
Для амперометрического определения селена и теллура были исследованы в качестве титрантов 12 производных димеркаптотиопирона, дающих с селеном и теллуром нерастворимые в кислотах окрашенные осадки. Лучшие результаты получены с реактивом-2,6-димеркапто-3,5-диэтил-1,4-тиопирон. Титрование проводят с графитовым электродом по анодному току окисления реагента при +0,7<? (отн. н.к.э.).
Титрование селена и теллура (а также сурьмы) возможно на фоне растворов H2SO4 и НС1 в широких пределах их концентраций. Точка эквивалентности фиксируется при соотношении Se(Te) : R = = 1 : 3 (в растворе НС1) и Se(Te) : R = 1 : 4 (в растворе H2SO4). Селен и теллур образуют с реагентом более прочные соединения, чем сурьма, поэтому возможно последовательное титрование селена (или теллура) и сурьмы на фоне 3,5 N НС1. Последовательному титрованию селена (теллура) не мешает большой избыток Sn, Сг, Ni, W, Мо, Fe, Al и других элементов [24, 359].
Пиридилдимеркаптан использован при амперометрическом определении селена в теллуре,в полупроводниковых и теллурсодержащих материалах. При титровании применяют угольный индикаторный электрод. Определению не мешают Те, Hg, РЬ, Си; мешает золото. Относительная ошибка определения <( 2% [24].
Для определения селена и теллура в полупроводниковых сплавах без отделения сопутствующих элементов применимы гексил- и циклопентилдитиокарбаматы. В отличие от диэтилдитиокарбамата эти реагенты устойчивы в кислых средах и позволяют вести титрование в 3 N НС1 по анодному току реагента. Не мешают определению большие количества Zn, Al, Мп, Pb, Ni, 150-кратный избыток Cd, 100-кратный TI, Fe (II), 20-кратный Sb (V). Мешают определению медь и ртуть. При pH < 3,6 титруют сумму селена и теллура, а при pH 5,5 — 4,0 только теллур. Количество селена находят по разности [394].
132
Описан метод амперометрического титрования селена и теллура раствором этилксантогената калия на фоне НС1 по току восстановления Se (IV) и Те (IV) на капельном ртутном электроде. Селен титруют на фоне 1—6 N НС1 при —0,4 в (отн. н.к.э.), теллур титруют на фоне 4—5 N НС1 при —0,5 в. Состав осадков отвечает формулам (C2H5OCSS)4Se и (C2H5OCSS)4Te [626].
Для амперометрического титрования SeOa“ применен раствор РЬ (СНаСОО)2. На фоне KNOa (pH 3—6) кривая титрования имеет U-образнук), форму. Мешают определению ионы, осаждаемые свинцом (F", J", SO4~, ОН-, С2О4^, РО4", CrO2“, S2’), и ионы с более положительным потенциалом восстановления [Fe (III), Си (II), Sn (II), ТеОа~, ТеО'Д, Sb (III), Cd (II), Cr (III)] [1048].
Метод амперометрического титрования H2SeOa раствором NaOBr применен для определения Ag,Hg, РЬ, образующих в нейтральных и слабощелочных растворах нормальные селениты [625].
Разработан метод амперометрического титрования ТеО|- раствором КМпО4 на фоне 0,05—0,1V NaOH с ртутным электродом [790]. Этот метод1 после дополнительных исследований (использован платиновый вращающийся электрод,титрование проводят в 1ДА+ГаОН при +0,4 в) рекомендован для анализа промышленных объектов [344].
Амперометрическое установление эквивалентной точки использовано при кулонометрическом титровании H2SeO3 раствором Fe(CN)l~, образующимся при окислении Fe (CN)|+ на платиновом электроде. При титровании 3—36 мг H2SeOa в 100 мл ошибка составляет +1%. Мешают ТеОа~, S2‘, S2Oa~, SO'a' [902].
Кулонометрическое и потенциометрическое титрование
При титриметрическом определении теллура и селена отсчет объемов титранта можно заменить кулонометрическим титрованиём. В качестве восстановителей для Se (IV) и Те (IV) используют растворы Ti (III), Sn (II), J- [1, 3,421,422, 1036], электролитически генерируемые в процессе титрования.
На основании кривых зависимости «г — /(£)» установлены оптимальные условия генерации Sn (II) на платиновом электроде, обеспечивающие достижение максимальной эффективности тока в условиях определения Se (IV) и Те (IV) на фоне 6N НС1.
При потенциометрической индикации конечной точки титрования возможно совместное определение селена и теллура [5]. Для восстановления Те (IV) используют графитовый [4] и платиновый электроды [579]. Потенциометрический вариант индикации конечной точки позволяет определять Se (IV) и Те (IV), а также Те (IV) в присутствии Fe (III). Чувствительность метода для селена и теллура 1 • 10'b М [4].
133
Разработан метод кулонометрического определения четырехвалентного теллура при электролитическом восстановлении на платиновом и золотом электродах. На платиновом электроде в фосфатном буферном растворе можно определить Те (IV) в присутствии 50-кратного избытка Se (IV) с чувствительностью 0,7 мкг!мл Те [61.
Известен косвенный метод кулонометрического определения Se (IV) и Те (IV) окислением раствором КМпО4, избыток которого оттитровывают электрогенерированным Fe (II). Конечную точку титрования фиксируют потенциометрически или биамперометрически. Метод применен для определения селена и теллура в полупроводниковых сплавах, пылях и платиновых шламах [2].
Потенциометрическим титрованием можно определить Те (IV) и Se (IV) в щелочном растворе при помощи КМ.пО4. Избыток окислителя оттитровывают щавелевой и муравьиной кислотами [786, 7891. Предложен метод потенциометрического определения теллура в присутствии свинца и олова титрованием раствором КМпО4 с платиновым индикаторным электродом [602].
Косвенный потенциометрический метод описан для титрования селенита феррицианидом калия [689]. Потенциометрическое титрование НвТеО6 возможно в случае образования [ТеМовО34]6-. Если в растворе находится более чем 6-кратный избыток МоО4~, то при титровании раствором HNO3 со стеклянным электродом в точке эквивалентности наблюдается резкий скачок потенциала [1019].
СПЕКТРАЛЬНЫЕ МЕТОДЫ
Спектральный анализ широко применяется при исследовании элементного состава веществ. Однако вследствие особенностей строения и возбуждения атомных спектров селена и теллура, а также физико-химических свойств этих элементов и их соединений при спектральном анализе, выполняемом в обычных условиях, получение сведений о присутствии селена и теллура в анализируемом материале затруднительно. Оба элемента принадлежат к легколетучим, особенно селен и его соединения [203] (табл. 15).
Таблица 15
Температуры плавле! ия и кипения (°C) некоторых соединений селе! а и теллура
Соединение Т’пл гкнп Соединение Т'пл Т'кнп
Те (элемент) 452 1390 SeO2 340 317*
ТеО2 733 450* SeO3 118 —
Se (элемент) 217 680
* Темг.ература еозгоикн.
134
Относительно высокие потенциалы ионизации (Se — 9,75 эв, Те — 9,01 эв) и высокие потенциалы возбуждения линий (табл. 16) определяют малую чувствительность прямых спектральных методов определения селена и теллура. Длины волн наиболее чувствительных спектральных линий для селена и теллура приведены в табл. 16.
Таблица 16
Характеристика спектральных дуговых линий делена и теллура [88, 148, 511]
Длина волны спектральной линии, А Интенсивность линий Потенциал возбуж-дения, % Длина волны спектральной линии, А Интенсивность линий Потенциал возбуждения, %
в дуге в искре в дуге в искре
Селен. Теллур
2062,788 — 1000 6,33 * 2081,01 80 — 7,26
2039,85 — 800 6,33 * 2142,74 120 — 5,78
1961 — —- — 2147,19 30 — —
2074,793 — 100 5,98 2208,83 40 — 6,92
2164,160 — 100 5,97 2259,04 60 — 5,49
2413,517 — 100 6,32 2383,25 100 60 5,78*
2385,76 120 60 5,78 *
* Самые чувствительные линии.
Интенсивные дуговые линии селена Se 1961; Se 2039,85; Se 2062,78 находятся вне диапазона обычно употребляемых спектральных приборов — в дальней ультрафиолетовой области спектра. Дуговые линии селена в ближайшей и средней ультрафиолетовой областях спектра значительно менее интенсивны и появляются лишь при высоких содержаниях селена (целые проценты). С другой стороны, наиболее чувствительные линии теллура — Те 2383,25 и Те 2385,76 А— расположены в ближайшей ультрафиолетовой области спектра. ।
Положение чувствительных аналитических линий селена и теллура в разных областях ультрафиолета обусловливает различный методический подход к спектральному определению этих элементов. Так, для спектрального определения теллура можно применять обычные спектрографы — призменные и дифракционные типа ИСП-22 и ИСП-28. Можно получить спектры селена и теллура при условии однолинзового освещения щели.
Для работы в коротком ультрафиолете применяют вакуумные спектрографы типа ДФС-5М.
Трудная возбудимость селена и теллура требует специальных искровых источников возбуждения [7251. Генераторы ИГ-2 и ИГ-3
135
используются для получения искровых линий селена Se 3675 А, Se 5227,51 A., Se 5175,98 А [52, 143, 4661. Применение искрового генератора ИГ-2 позволило Смородиной и др. [340] достигнуть достаточно высокой чувствительности определения селена в теллуре (0,001%).
При возбуждении в искре была зарегистрирована чувствительность 2-КГ4 — 2-10"5 % Se по линии 2039,8 А [185]. При применении в качестве источника возбуждения газоразрядной трубки с полым катодом и спектрографа ИСП-51 можно определить свободный селен (по линии Se 4840,6 А) в селениде цинка с чувствительностью до 7,5-10"2% и коэффициентом вариации 15% [323].
При работе с дугой постоянного тока усиление интенсивности линий можно получить в результате снижения температуры анода. Возбуждение дугового разряда в атмосфере аргона значительно снижает температуру анода (при 6а до 965° С, при 12 а до 1150° С), в то время как в обычной воздушной атмосфере при 6 а температура анода 1250° С.
Понижение температуры анода позволило повысить чувстви-.ельность определения селена по линии Se 2039 А до 3-10-4%. Спектры селена регистрировались при этом на спектрографе PGS [504].
Более поздние исследования показывают, однако, что из графитового порошка в дуге постоянного тока (I = 5,2—5,5 а) селен испаряется как с поверхности, так и из глубины пробы, причем испарение в атмосфере воздуха происходит лучше, чем испарение в атмосфере аргона (интенсивность линии Se 2413,5 А уменьшается в атмосфере аргона) [976].
Для повышения чувствительности определения селена и теллура также используется метод вдувания пробы в разряд. Давлетшин и Айдаров [131] предложили вдувать порошки проб в разряд между горизонтально расположенными угольными электродами. Определение теллура в сере проводилось с использованием дуги переменного тока при 1 = 20 а. Спектры регистрировали на фотопластинках типа III на спектрографе ИСП-30. По линии Те 2385 А авторам удалось определить таким методом 5-10 5% Те.
Метод вдувания образца применяли также при определении селена [982]. По искровой линии Se 3637,5 А чувствительность определения селена в анодных осадках составляла 0,1%.
При использовании дуги переменного тока с подачей пробы просыпкой с воздушным дутьем удалось добиться гораздо более высокой чувствительности определения селена — 0,001% [233, 427]. Регистрация интенсивных линий селена — Se 1961 А и Se2150 А — проводилась на спектрографах ИСП-22 и ИСП-28. Использовали пластинки типа III, сенсибилизированные салицилатом натрия.
Значительное повышение чувствительности спектрального определения теллура происходит при добавлении к пробе угольного порошка. При этом увеличивается скорость поступления пробы в разряд и повышается его эффективная температура. По данным
136
Юделевич и др. [266, 468], чувствительность определения теллура в присутствии угольного порошка составляет 0,003%.
Усиление интенсивности линии селена Se 2039,85 А при добавлении СиО было отмечено в работах [592] и [466]. При определении селена в продуктах переработки шламов [466] применяли дугу переменного тока~(/ = 10а), чувствительность определения составляла 0,02%. Определяя селен в пирите, халькопирите и марказите, спектр воз бу жд а.ди в дуговом разряде постоянного тока (/ = 12 а).
Чувствительность определения селена по линии Se 2039,85 А составляла 0,0015% [592]. В качестве внутреннего стандарта использовали висмут. Этот элемент близок к селену по характеру поступления в дуговой разряд, потенциалу ионизации; кроме того, висмут имеет линии достаточной интенсивности вблизи линий селена: Se 2039,85 A, Bi 2041,96 A, Bi 2021,21 А. Спектры регистрировались на спектрографе ИСП-22 с двухлинзовой системой освещения щели на специальных пластинках, сенсибилизированных салицилатом натрия для коротковолновой области.
Для повышения чувствительности спектрального метода определения проводятся химические реакции на электроде, приводящие к получению более летучих соединений этих элементов.
Захария и сотр. [151, 152] предложили метод «сульфидизации», применяя элементную серу в качестве «носителя». Чувствительность определения селена и теллура в рудах и минералах при этом повысилась до 5'10"5% Те и 2 • 10-3% Se. Пробы смешивали с элементной серой и помещали в глубокий кратер угольного электрода. Спектры возбуждали в дуге переменного тока (220 в, 12 а) и фотографировали на спектрографе ИСП-28. Определение селена и теллура проводилось по аналитическим линиям Те 2385,76 A, Se 2039,85 А и Se 2062,79 А.
Фришберг и др. [265, 410] предложили проводить предварительное «иодирование» пробы. Образующийся при этом иодистый теллур — легколетучее соединение. Температура угольного электрода (600—700° С) обеспечивает максимальную скорость испарения йодистого теллура. При такой низкой температуре электрода другие элементы пробы не образуют летучих иодистых соединений. Это способствует избирательному испарению теллура из проб, поэтому результаты определения теллура в пробах руд мало зависят от их состава. В сульфидных рудах определяют до 3-10-4% Те.
Применяя предварительную отгонку при 1900° С и сжигание конденсата на угольном электроде в дуговом разряде переменного тока (/ = 6 а), можно определять теллур в пятиокиси ванадия с чувствительностью 10-4—10-5%. Спектры регистрируют на среднем кварцевом спектрографе [243].
Предварительная отгонка примесей (в том числе и теллура) в вакууме при 1300° С с последующей конденсацией их на охлаждаемом электроде использовалась при анализе высокочистого кремния 1165]. Концентрирование селена сублимацией повысило чувстви
137
тельность определения селена по дуговым линиям 2413 А до 0,01% [6671.
Явление фракционированной отгонки соединений теллура вместе с летучими соединениями Zn, Pb, Bi и Cd было использовано при определении теллура в продуктах цветной металлургии [54, 378]. Пробы и эталоны смешивали в отношении 1:5с буферной смесью, состоящей из 75 г ZnO, 20 г РЬО, 10 г CdO и 5 г Bi2Oa. Авторы работы отмечают, что сульфат свинца повышает чувствительность определения теллура. Спектры возбуждали в дуговом разряде переменного тока (10 а) и фотографировали на кварцевом спектрографе ИСП-28. Чувствительность определения составляла 0,002% Те. Замечено, что присутствие в пробе хлоридов железа и свинца значительно повышает интенсивность линии теллура Те 2385 А [265].
Наиболее эффективным методом повышения чувствительности спектральных методов определения селена и теллура является предварительное химическое обогащение. После осаждения теллура с Fe (ОН)а и при последующем спектральном определении его в концентрате удалось определить 0,1 мкг Те по линии Те 2385 А на спектрографе ИСП-28. Спектры возбуждали в дуге переменного тока при I = 10 а [54].
Высокая чувствительность определения теллура в сере (5-10“6%) была достигнута за счет предварительной химической обработки: пробу серы растворяли в азотной кислоте, образовавшуюся серную кислоту удаляли выпариванием на электрической плитке, а остаток прокаливали при 420° С, растворяли в 0,15 мл HNO3 (1 : 1) и пропитывали этим раствором угольный электрод [311]. Анализ проводили на спектрографе ИСП-28 по линии Те 2385 А. Спектр возбуждали в дуге переменного тока (/ = 8 а).
При анализе меди и ее соединений химическое обогащение было достигнуто предварительнымэлектролитическимвыделениемосновно-го компонента — меди на платиновом электроде [27]. После упаривания электролита остаток прокаливали при 550° С и помещали в кратер угольного электрода. Спектры возбуждали в дуге постоянного тока и фотографировали на спектрографе ИСП-28. Чувствительность определения теллура 10~3 — 10-6%.
При спектральном определении примесей селена и ряда других элементов в металлическом титане, тантале, ниобии и ванадии применяли предварительное концентрирование примесей экстракцией диэтилдитиокарбаматов хлороформом [247]. Полученные концентраты анализировали спектральным методом с использованием дуги постоянного тока (10 а, 220 в). Чувствительность метода составляла 5-10“5%.
Рокенбауэр и др. [1027] повысили чувствительность определения селена до 0,001%, используя для спектрального анализа остаток после химической обработки (пробы растворяли в HNOa и соли высушивали). Спектры возбуждали в дуге постоянного тока при 220 в и 18 а.
138
При спектральном определении микроэлементов (в том числе и селена) в минеральных лечебных водах использовали предварительное концентрирование микроэлементов на ионообменной смоле [565].
Спектральный метод применяли при определении теллура в селене 11167], сталях и сплавах [140, 415], чистых металлах [184], свинцовых концентрациях и пылях [89, 441, 464, 465, 467], теллуристом свинце [89] и в других объектах [810, 929, 1142].
ПЛАМЕННО-ФОТОМЕТРИЧЕСКИЕ, АТОМНО-АБСОРБЦИОННЫЕ И РЕНТГЕНОФЛУОРЕСЦЕНТНЫЕ МЕТОДЫ
Пламенная фотометрия
В литературе имеется несколько работ по пламенно-спектрофотометрическим методам определения селена и теллура. Для серы, селена, теллура и полония основными состояниями являются Зр2 — часть триплета 3/?23pi3p0.
Упрощенная картина электронных переходов для серы, селена и теллура [570], которые соответствуют спектральным резонансным линиям, представлена в табл. 17.
Таблица 17
Спектральные резонансные линии (нм)
Элемент Электронные переход
Зр2- 3°, Зр,~» 3s,0 Зр„- 3s?
S 180,7 182,0 182,6
Se 196,1 204,0 206,3
Те 214,3 238,6 238,3
Ро 245,0 417,0 300,3
Линии переходов Зр2 —> 3s ° и Зр2 —> 3s°2 не дают заметного поглощения и флуоресценции для всех этих элементов. Для селена линии 196,1 и 204,0 нм абсорбируют сильно, а линия 206,3 нм — слабо. Линия 196,1 нм основного состояния абсорбирует в 5 раз сильнее, чем для Зр-состояния, которое лежит ниже на 0,25 эв основного состояния. Линия Те 214,3 нм абсорбирует примерно в 10 раз сильнее, чем линии 238,6 и 238,3 нм. Уровни Зр0 и 3pt лежат на 0,6 эв ниже Зр2 основного состояния.
В пламенно-эмиссионном методе определения теллура [620] использовали кислородно-ацетиленовое пламя. В отличие от многих металлов, которые имеют слабые и плохие эмиссионные линии в пла-
139
ZSBfi Рис. 29. Интенсивность эмиссионных спектральных
У линий теллура (/), используемых для количествен-
17 кого анализа
мени, теллур дает эмиссионный спектр, пригодный для аналитического использо-вания. Наиболее интенсивной является Z2S9 двойная линия 238,3 и 238,6 нм, хотя бо-
/Г V лее УД°бны Для практики линии 214,3 и / 225,9 нм (рис. 29). Чувствительность опре-
I деления по линии 238,6 нм равна 2 мкг/мл
Те. Калибровочная кривая линейна до 4-1- 4 000 мкг/мл,.
Для отделения от других элементов при-2 „м меняли экстракцию комплекса теллура с
' диэтилдитиокарбаматом хлороформом из
водного раствора при pH 4 в присутствии комплексона III. Чтобы улучшить чувствительность эмиссии, СС14 удаляли выпариванием досуха и остаток растворяли в метилизобутилкетоне. Этот раствор вводили в пламя. Для определения теллура использовали пламенный спектрофотометр Бекмана.
В случае применения экстракции определению теллура не мешали Bi, Сг (III), Со, Си, РЬ, Мп, Ni. Железо мешало определению, так как линии железа 237,3; 238,2; 238,9; 239,6 нм накладываются на линии теллура. Максимальная чувствительность определения достигалась при введении органического аэрозоля (метилизобутил-кетон) в ацетилен-кислородное пламя.
Выполнение определения [620]. Образец растворяют в HNO3. Избыток кислоты удаляют выпариванием, разбавляют раствор до 100 мл и доводят pH раствора до 4 добавлением NH4OH. Добавляют комплексон III(5а на каждый грамм металла), переводят раствор в делительную воронку, добавляют 50 мл 0,06% -ного диэтилдитиокарбамата натрия в СС14 и встряхивают 2 мин.
Отделяют органическую фазу, промывают водную фазу 10 мл СО4 и прибавляют к экстракту. Выпаривают органическую фазу до 2—3 мл, не доводя до кипения, до маслянистой пасты и добавляют 10 мл метилизобутилкетона. Полученный раствор вводят прямо в пламя.
Калибровочную кривую строят в интервале содержаний 4—100 мкг Те.
Атомная абсорбция
Определение селена методом атомной абсорбции возможно по линиям 196,1, 204,0 и 206,3 нм. Линия 196,1 нм наиболее чувствительна в воздушно-водородном и воздушно-ацетиленовом пламени.
В первых работах по атомно-абсорбционному определению селена и теллура [488, 489, 10861 была достигнута чувствительность 5 мкг Se/мл и 0,5лпсг Те/ли по линиям соответственно 204,0 и 214,3 нм.
140
Чувствительность определения селена была повышена в дальнейшей до 1 мкг/мл по линии 196,1 нм с применением воздушно-ацетиленового и воздушно-пропанового пламени [835, 1013]. Источником излучения служила трубка с полым катодом из графита, пропитанным расплавленным в вакууме селеном, или высокочастотная лампа с парами селена. П]5и работе с высокочастотной лампой была отмечена зависимость величины отсчетов от режима разряда вследствие изменения самопоглощения света в источнике. Этот метод был применен для определения относительно больших концентраций селена (10-1— 10~3%) в пшенице и галените. Стандартное отклонение для 16,7 мкг/г составляло 0,75 лмсг/г, а для 0,11—0,17? Se - 0,004%.
Дальнейшее изучение условий атомно-абсорбционного определения селена показало, что абсорбция зависит от природы пламени, скорости поступления воздуха и ацетилена, характеристик лампы, силы тока и времени, ширины спектральной полосы [574]. В пламени смеси водород—воздух найдена следующая чувствительность определения селена: 1; 10; 69, 110 мкг/мл по линиям 196,1; 204,0; 206,3 и 207,5 нм, а в пламени ацетилен — воздух чувствительность в 1,6 раза выше. Газы пламени сильно поглощают излучение линии селена 196,1 нм. Посторонние элементы и анионы, присутствующие в 1000-кратных количествах, увеличивают поглощение света на 5—15%.
Определять селен в водных растворах можно при условии идентичности солевого состава и физических свойств анализируемых и стандартных растворов. Селен экстрагируют из водного раствора при pH 4—6,2 в виде диэтилдитиокарбаматного комплекса метилизо-бутилкетоном. Совместно экстрагирующиеся элементы не мешают определению. Введение селена в пламя в органическом растворителе повышает чувствительность в 1,5 и 2,4 раза для водород-воздушного и ацетилен-воздушного пламени.
При атомно-абсорбционном определении селена и теллура, выполненном на атомно-абсорбциометре Unicam SP 900 А, в качестве источников был использован свет безэлектродных разрядных высокочастотных ламп. В воздушно-пропановом и воздушно-ацетиленовом пламени [612] предел определения селена по линии 196,1 нм был равен 1 мкг/мл. Калибровочный график линеен в интервале концентраций 5—100 мкг/мл.
Для атомно-абсорбционного определения селена использовали спектрофотометр Бекмана, ламинарное пламя смеси ацетилена с воздухом и турбулентное — смеси водорода с воздухом. Применяли лампы с полым катодом, содержащим селен [1013]. Чувствительность определения селена по аналитической линии 196,1 нм составляла в этом случае 0,25 мкг/мл. Калибровочные графики в области концентраций 0—100 мкг Se/мл представляли собой кривые, изогнутые к оси абсцисс. Прямолинейная зависимость между оптической плотностью и концентрацией наблюдалась в области концентраций < 10 мкг Se/мл.
141
При атомно-абсорбционном определении теллура возможно использование следующих спектральных резонансных линий — 214,3; 225,9; 238,6 нм, причем линия 214,3 нм дает наибольшую чувствительность определения. Для очень больших концентраций теллура могут быть использованы две другие линии. Линию теллура 214,3 нм невозможно воспроизвести в воздушно-водородном пламени, но получается значительная адсорбция в кислород-ацетиленовом пламени при высокой скорости поступления кислорода (14—18 л) мин) и ацетилена (1,5—2,2 л/мин).
При анализе образцов металлургического производства, содержащих большие количества меди, нитратов и других мешающих элементов, проводили предварительное концентрирование теллура соосаждением с мышьяком или экстракцией [573]. Теллур экстрагировали метилизобутилкетоном в виде комплекса K2TeJe из раствора 5%-ной НО, содержащего KJ. Теллур также можно экстрагировать из почти нейтрального раствора (pH 8,5—8,8) в виде комплекса с диэтилдитиокарбаматом — Те (DDTC)4. Чувствительность определения теллура по линии 214,3 нм в этом случае составляла 0,23 мкг/мл', предел определения 0,076 мкг/мл.
Для повышения чувствительности определения теллура и устранения влияния других элементов была изучена экстракция комплексов теллура различными органическими растворителями [1162]. Наиболее полно теллур экстрагировался 2,4-пентандиолом в виде пирролидиндитиокарбамата при pH 4—6. Мешающее влияние Cu, Sb, Se и Hg устраняли, добавляя в раствор перед экстракцией комплексон III. Чувствительность определения теллура существенно зависела от качества ламп полого катода.
Для определения теллура в медных шламах и сере использовали атомно-адсорбционный спектрофотометр Перкин-Эльмер-303 и воздушно-ацетиленовое пламя [953]. Скорость воздуха 21 л/мин, скорость ацетилена 3,8 л/мин, скорость подачи раствора 5 мл/мин. Ток лампы полого катода 12 ма, ширина щели монохроматора 0,3 мм. Для линии 214,27 нм поглощение следовало закону Бера в интервале концентраций 0—25 мкг/мл со стандартным отклонением 2,3%. Чувствительность определения теллура в водных растворах 0,2мкг/мл. Большинство сопутствующих элементов, за исключением Zr1+ и Sn2+, а также HCI, HNO3 в интервале концентраций 0,5—0,6 N, H2SO4 и Н3РО4 в интервале 0,1—0,2 А1 не влияло на поглощение.
Определение 0,0005—0,030% теллура в железе и стали проводили после выделения элементного теллура раствором SnCl2 и экстракции комплекса с диэтилдитиокарбаматом амилацетатом. Полученный экстракт распыляли в пламя ацетилен — воздух и по линии 214,3 нм определяли до 0,3 мкг Те/мл. Такие элементы, как As, Bi, Cu, Se, в количестве 1 мг не мешали определению. Стандартное отклонение метода 3—5% [905].
Определение теллура в почвах и породах описано в работе Накагава [956].
142
Определение теллура в металлическом висмуте [1035] было основано на экстракции теллура метилизобутилкетоном и измерении оптической плотности ацетилен-воздушного пламени по линии 214,3 нм на атомно-абсорбционном спектрофотометре Перкин-Эльмер-303. Чувствительность определения 0,5 мкг/мл.
В полупроводниковых сплавах CuInTe2 теллур определяли по линии 214,3 нм на атомном абсорбциометре СФМ на базе ЗМР-З. Источником света служила трехэлементная лампа, содержащая Bi, Sb и Те, пламя — воздушно-пропановое, горелка удлиненная (8 см). В пламя вводили раствор образца, полученный после растворения сплава в кислотах.
Выполнение определения теллура в металлическом висмуте [1035], железе и стали [905]. Навеску металлического висмута промывают ацетоном, сушат и растворяют в 20 мл конц. HCI и 2 мл конц. HNO3. К сухому остатку добавляют 10 мл конц. HCI, 10 мл воды и нагревают до растворения. К полученным растворам добавляют 10 мл метилизобутилкетона и встряхивают 40 сек. Органическую фазу промывают 10 мл метилизобутилкетона и сушат безводным Na2SO4.
0,5—1 г железа или стали растворяют в H2SO4, карбиды окисляют добавлением HNOg и осаждают элементный селен добавлением 10 мл 10%-ного раствора SnCI2. Осадок теллура отфильтровывают, растворяют в смеси H2SOi + HNOa и экстрагируют теллур в виде диэтилдитиокарбамата 5 мл амилацетата.
Распыляют органическую фазу в пламя ацетилен — воздух длиной 10 см и полинии теллура 214,3 нм определяют концентрацию теллура на атомно-абсорбционном спектрофотометре.
Применение дейтериевого корректора фона позволяет определять теллур в сталях без отделения основы. В этом случае применяют ацетилено-воздушное пламя и прибор Перкин-Эльмер, модель 303 [522].
Рентгеновская флуоресценция
Рентгенофлуоресцентным методом селен определяют по линии селена Ка.. При анализе сложных по составу объектов образцы обычно предварительно химически обрабатывают и отделяют мешающие элементы. При определении селена в меди его выделяли соосаждением с элементным мышьяком. Для дальнейшего рентгенофлуоресцентного определения селена был применен спектрограф Филипс с W- и Mo-трубками и кристаллом-анализатором LiF. Внешним стандартом служила Си—Se-мишень. Изучение линии селена Ка регистрировали сцинтилляционным счетчиком. Чувствительность определения составляла 0,3 мкг Se в 5 г пробы, относительная ошибка ± 6% [894].
Рентгенофлуоресцентное определение микрограммовых количеств селена проводили после выделения селена в виде элементного с использованием в качестве восстановителя этанольного раствора 4,5-диамин-6-тиопирамидина. Для переведения элементного селена в гексагональную модификацию осадок нагревали в атмосфере гелия до 205—207° С. Дальше определяли селен на спектрографе ХРД-6 с кристаллом LiF и Мо-трубкой [576].
143
При совместном определении селена и мышьяка Мамаев и сотр. [229] применили двухканальный рентгенофлуоресцентный спектрограф, изготовленный на базе спектрометра КРУС. Взаимное влияние селена и теллура, а также других мешающих элементов (например, свинца) учитывали введением поправочных коэффициентов. Чувствительность определения была равна 0,1% Se, средняя квадратичная ошибка 6—7%.
При прямом рентгенофлуоресцентном анализе селенидов и теллуридов цинка окончательный расчет концентрации селена, теллура и цинка проводили с учетом взаимного влияния элементов [618].
Описан метод прямого определения микрограммовых количеств селена в присутствии Sb, Zn, As, Ge и Си на спектрофлуориметре с W-анодом (50 кв, 20 ма) и кристаллом LiF. Чувствительность определения 1 мкг Se, ошибка определения 10% при содержании элемента 5—10 мкг [1160].
Селен определяли в теллуре в интервале содержаний 0,05— 2,5% Se с коэффициентом вариации 1—6% [939]. Метод рентгенофлуоресцентного анализа использовали для определения селена и теллура в смеси газов [817], стекле [505], растительных материалах [743], различных геологических материалах [39], для определения теллура в различных сплавах [486, 563], следов селена — в меди [703], а также при изучении селенида германия и селенида серебра [941—943].
радиоактивдционные методы
Для определения малых содержаний селена и теллура в рудах, метеоритах, биологических веществах, животных тканях и в других объектах используется радиоактивационный метод анализа.
В табл. 18 описаны некоторые основные свойства стабильных и искусственных изотопов селена и теллура [15, 866].
Для определения селена чаще всего используется изотоп Se7s (Т>/а = = 127 дней). Этот изотоп легко определяется и идентифицируется по трем интенсивным у-пикам с энергиями 0,136; 0,267; 0,402 Мэв. Период полураспада этого изотопа позволяет проводить полное отделение всех других активных примесей. Вследствие малого процентного содержания Se74 в природном селене (0,87%) облучение необходимо проводить в достаточно интенсивном нейтронном потоке (1012—1013 п! см2-сек) и в течение длительного времени (7—14 дней), так как чувствительность нейтронно-активационного определения элементов определяется интенсивностью активирующего источника, поперечным сечением захвата нейтронов и периодом полураспада образующихся изотопов.
При определении селена в меди [126, 881] облучение образцов проводили в реакторе в потоке тепловых нейтронов 3* 1013 п!см2-сек в течение 10 и 100 час. Измерение у-спектров вели на сцинтилля-
144
Т аб'лица 18
Сечения активации тепловыми нейтронами для изотопов селена и теллура [15]
Распространенность стабильного изотопа в при-роде, % Сечения активации теплЛыми нейтронами, Сарны Образующийся изотоп Пэриод полураспада
Se71-- 0,87 26+6 Se75 120,4 дня
Se.76—9,02 7+3 Ss77m 17,5 сек.
Se8»—49,82 0,03+0,01 Sa,lm 62 мин.
0,5+0,1 Se81 18,6 мин.
Se82—9,19 0,05+0,025 • Ss43"1 69 сек.
0,004+0,002 Se8S 25 мин.
Те122—2,46 1,1+0,5 уе123т 104 дня
Те124—4,74 5+3 58 дне!
Те123—18,72 0,09+0,02 pe127m 105 дней.
0,8+0,2 Те127 9,35 часа
Те™—31,79 0,015+0,005 ye129m 41 день
0,13+0,03 Те129 72 мин.
Те13»—34,27 0,008 |ешм 30 час.
0,22+0,05 Те1»1 24,8 мин.
ционном т-спектрометре. Чувствительность метода 0,5 мкг Se в 0,1 г Си.
При определении селена в сере [434] была достигнута чувствительность 8»10"6% при облучении пробы в 1 г потоком нейтронов 8,7-1012 п!см2-сек.
В случае определения селена в фосфоре [475] навеску 1 г облучали в том же режиме 14 дней; была достигнута чувствительность 4-10~s% с относительной ошибкой определения + 10—20%.
Определение селена в платине [946] проводили из навески в 0,1 г с чувствительностью 2-10'5% при облучении пробы 12 дней в реакторе с потоком нейтронов 1012 п1см2-сек.
Чувствительность определения селена в сере повышенной чистоты составляла 3-10~s% с относительной ошибкой 10% при облучении в течение 48 час. в ядерном реакторе с потоком тепловых нейтронов 1,3-Ю1® п!см2’сек [122].
Изотоп Se7S был использован также при радиоактивационном определении селена в мышьяке и арсениде галлия [965], в породах, концентратах, шлаках и отходящих газах [562 , 669], в никотиновой кислоте [979].
При определении микроколичеств селена в биологических средах [917] по Se75 была достигнута чувствительность 0,02 мкг Se в
W
145
1 г образца. Использование специальной установки для счета совпадений, состоящей из двух одноканальных сцинтилляционных ^-спектрометров, настроенных на регистрацию y-пиков 0,130 и 0,270 М.эв, позволило достичь чувствительности 2-10~8 г Se при облучении образцов минералов и окиси теллура [748] в течение 18, час. в потоке тепловых нейтронов 1012 п1смг-сек.
Для радноактивационного определения селена по изотопу Se75' образец после облучения выдерживали — 2 недели. После этого протравливали на холоду HNO3 (1:1), промывали и растворяли в HNO3 в колбе с обратным холодильником. Селен отгоняли из растворов с НО и НВг либо при пропускании тока азота [552], либо при пропускании через раствор хлористого водорода [155, 434], а также при упаривании раствора с НВг [112, 475]. Для упрощения операций химической очистки, сокращения времени анализа и повышения выхода селена Конрад [603] применил метод сухой отгонки селена в специальной кварцевой трубке. Этот метод позволяет количественно отделить селен от других элементов. В дистилляте выделяли элементный селен солянокислым гидразином или солянокислым гидроксиламином из 8 N НО или осаждением при помощи SO2 из 6 jV НО при кипении раствора, переосаждали выделившийся осадок элементного селена, высушивали, взвешивали и определяли активность.
При определении селена и теллура в метеоритах [637, 10531 образец после нейтронной активации сплавляют с Na2O2, выделяют сумму селена и теллура осаждением при помощи SO2 из 6 N НО. Разделяют селен и теллур на колонке со смолой Дауэкс, селен элюируют 3 N НО, а теллур — 0,2—0,5 N НО. Из растворов селен и теллур осаждают при помощи SO2, осадки промывают, высушивают, взвешивают и определяют активность.
В настоящее время при радиоактивационном определении селена широко используются короткоживущие изотопы селена — Se77(Ti/2 = 17,4 сек.), Se81 (18,6 мин.) и Se81,n (57 мин.) [866]. Техника применения короткоживущих радиоизотопов в активационном анализе описана в [587].
Высокая чувствительность определения селена (0,017 мкг Se) была достигнута при использовании изотопа Se77,?!. Пробу облучали 35 сек. в потоке тепловых нейтронов 4,3-1012 п/см^сек, выдерживали 10 сек. и измеряли активность Se77"' по Y-пику 0,162 Мэе при помощи многоканального сцинтилляционного ^-спектрометра с тонким (6—13 лш) кристаллом NaJ с применением специального пластмассового фильтра для поглощения 3-излучения [671, 977].
Изотоп Se77 применяли также для определения селена в сере и ее соединениях (NH4SCN, FeSO4. (NH4)2SO4, (NH4)2SO4, H2SO4), в серной руде и аммиачной воде [973, 974]. Чувствительность определения 10~2—102 мкг из навески 0,1—0,5 г, коэффициент вариации 10%.
146
Сочетание методов быстрого химического отделения селена с Т-спектрометрией позволило использовать изотоп Se78”1 [899, 900] (Ti/2 = 3,9 мин.), но предельная чувствительность при этом составляла только 10 мкг.
Изотопы Se81 (Т1/г = 18 мин.) и Se8im(Ti/z = 57 мин.) применяли при определении селена в арсениде галлия [473], свинце [476], меди [126], чистом теллуре [1165], волосах [529].
При работе с изотопом Se81”1 пробы облучали в ядерном реакторе 20 час. в потоке тепловых нейтронов 0,87-1013 п/см2-сек [473, 476], чувствительность определения составляла 2,2-10"s% Se с ошибкой 10—20%. Селен отделяли от*других элементов и очищали методами отгонки с НВг и осаждением в дистилляте солянокислым гидроксиламином из кипящего раствора 6—8 N НО. Однако этот метод довольно длителен и не очень удобен при работе с короткоживущими изотопами селена.
Был предложен быстрый, эффективный и высокоизбирательный метод радиохимической очистки селена (особенно удобный для отделения от мышьяка и германия), основанный на извлечении Se(IV) из 4 Д'НВг эфиром, содержащим 1% фенола, позволяющий выделить >90% селена за 10 сек. Окислители и восстановители не мешают реакции [917].
При определении селена в волосах [529], а также для отделения As, Sb, Sn,J селен экстрагировали толуолом в виде пиазоселенола и реэкстрагировали 9 N НО, содержащей Н2О2 [614]. Чувствительность определения 0,2—0,05 мкг Se.
Высокая чувствительность (5-10”7%) была достигнута при использовании изотопа Se81 в случае определения селена в биологических материалах [552]. Образцы облучали в этом случае 18 мин. в реакторе в потоке нейтронов 1012 п!см?>сек. Селен определяли по p-активности Se81 на счетчике Гейгера. Радиохимическую чистоту препаратов проверяли по кривым распада образцов и стандартов.
Теллур определяли радиоактивационным методом по изотопу Те127 (Ti/2 = 9,3 часа). Более короткоживущие изотопы теллура (Те128, Те131), также образующиеся при захвате нейтронов, требуют очень быстрого проведения химического разделения. В зависимости от интенсивности потока тепловых нейтронов облучение проб в реакторе проводили в течение 1—3 суток.
При определении теллура в Ni-сплавах [945] и сере [434] была достигнута чувствительность определения теллура 5-Ю”7 %. Облучали 0,1 г образца в реакторе потоком тепловых нейтронов 1,2--1012 п!см2’сек в течение 3 суток.
При определении теллура в цинке с чувствительностью 1,5-10’6% {477] и мышьяке [474] образцы облучали 20 час. в реакторе с потоком нейтронов 8,7-1012 п1см2-сек. В случае анализа арсенида галлия чувствительность определения теллура составляла 3-10-4% 1218], п-Ю"6?^ [1010].
147
При сложном составе образцов после облучения проводили радиохимическое разделение элементов с выделением ТеО2 или элементного теллура. Разделяли селен и теллур ионообменным методом на колонке с Дауэкс [25, 1053]. После получения радиохимически чистых осадков теллура определяли активность Те127. На счетчике Гейгера 1 мкг Те127 дает примерно 105 3-импульсов в 1 мин.
Описан активационный метод определения теллура в латуни и белом чугуне по дочернему изотопу J131 (Ту, = 8,05 дня) [508, 5251. Образцы облучали в реакторе 5—10 мин., иод отделяли от примесей отгонкой и экстракцией и определяли у-активность J131 по у-пику 0,364 Мэв при помощи 400-канального сцинтилляционного у-спектрометра. Определению теллура этим методом мешает уран вследствие ядерной реакции U235 (n, y)J131. При определении теллура в присутствии урана последний предварительно (перед облучением) отделяют экстракцией эфиром [614].
Глава 5
МЕТОДЫ ОТДЕЛЕНИЯ СЕЛЕНА И ТЕЛЛУРА ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Экстракция стала одним из основных методов разделения элементов в химическом анализе. Чаще всего используют экстракцию внутрикомплексных соединений, ацидокомплексов. Широкому распространению экстракционных методов способствовали монографии Моррисона и Фрейзера [241], Стары [351], Даймонда и Така [133], Фомина [406], ряд сборников [388, 414, 462, 463] и статей [28, 168, 169, 207].
Экстракция тройных комплексов (металл — галогенид — органическое основание типа диантипирилметана) применялась для разделения селена и теллура и отделения их от Au, Си, Fe и других элементов.
Экстракция селена и теллура трибутилфосфатом позволяет количественно разделять такие трудноразделяемые другими методами комбинации элементов, как Se (IV) — Те (IV); Те (IV) — Те (VI); Те (IV) — Au (III) — Fe (III); Те (IV) — J"; Se (IV) — Те(IV) — SOf.
Из кислородсодержащих органических растворителей селен и теллур лучше всего экстрагируют кетоны. Экстракция метил-этилкетоном позволяет отделять селен от Си, Pb, Ag при анализе медь- и железосодержащих материалов. •
В настоящее время в промышленных масштабах для очистки селена и теллура от примесей успешно применяется экстракция высокомолекулярными аминами.
Экстракция трибутилфосфатом
20%-ный раствор трибутилфосфата (ТБФ) в керосине количественно экстрагирует Те (IV) из 4—10 N солянокислого раствора. С ростом концентрации НС1 от 1 до 5—6 N коэффициент распределения увеличивается от 1 до 200 [784]. При добавлении NH4SCN тел
149
лур начинает экстрагироваться уже из 0,4 N НС1, и максимальная экстракция (100%) наблюдается в 2 N НС1 в присутствии 20% NH4SCN [1118], Те (VI) остается в водной фазе [768]. При использовании 100%-ного трибутилфосфата количественное извлечение Те (IV) в органическую фазу происходит при более низкой кислотности — 2—10 N НО, тогда как Те (VI) полностью остается в водном растворе [767]. Селен не экстрагируется из 2—5 N НО, но извлекается в органическую фазу из 6—12 N НО на 95—97% [354, 772].
Те (IV) экстрагируется трибутилфосфатом из растворов бромистоводородной кислоты. Максимальная экстракция наблюдается из растворов 4,2 N НВг 20%-ным раствором триб ути лфосфата в толуоле. Коэффициент распределения равен 21,7 [428]. Из растворов 0,23 N НВг Те (IV) практически не экстрагируется трибутилфосфатом, селен переходит в органическую фазу на 80—95%.
В качестве разбавителей ТБФ применяли СС14 и СНС13; изучалось также влияние циклогексана, циклогексанона, метилэтилкето-на, бензола как разбавителей ТБФ [418].
Изучению механизма экстракции четырехвалентного теллура трибутилфосфатом посвящен ряд работ [42, 114, 10421.
Беляев и Птицын [41] изучили зависимость коэффициента распределения теллура между водной и органической фазами от концентраций экстрагента, Те (IV), Н+- и СК-ионов и установили, что экстрагирующейся частицей является тетрахлорид теллура, сольватированный тремя молекулами ТБФ. Механизм экстракции был представлен следующим образом:
ТеО2Н+ + ЗН+ + 4СГ + ЗТБФ Ц ТеСЦ • ЗТБФ • 2Н2О.
Исследование механизма экстракции Те(IV) трибутилфосфатом с привлечением методов ИК-спектроскопии, элементного анализа, а также изучение зависимости коэффициента распределения теллура от равновесной концентрации ТБФ в бензоле позволило сделать вывод о том, что из растворов 2—10 N НО теллур экстрагируется в виде трисольвата [Те-ЗТБФ- хН2О]С14, где х = 1—2 [113].
При низких концентрациях НО, но в присутствии H2SO4 образование комплекса идет за счет водородной связи, т. е. экстрагируется металлогалогенидная кислота [Н3О-ЗТБФ]-ТеС15.
В насыщенном по теллуру трибутилфосфате возможно существование и других типов сольватов. В ИК-спектре поглощения такого ТБФ было обнаружено дополнительное смещение максимума частоты колебания группы Р = О от 1280—1265 до 1190 см-1, в то время как образованию сольвата [Те-ЗТБФ-хН2О]С14 соответствовал сдвиг максимума от 1280—1265 до 1240—1225 см-1, а образованию [Н3О-ЗТБФ]-ТеС15 — смещение максимума до' 1208— 1213 см'1.
Логарифмическая зависимость коэффициента распределения тел
150
лура от равновесной концентрации ТБФ в бензоле также подтверж дает вывод о том, что из солянокислых растворов экстрагируется одно соединение с тремя молекулами ТБФ (тангенс угла наклона прямой близок 3). Из солянокислых растворов при высоких концентрациях ТБФ экстрагируется также одно соединение (тангенс угла наклона прямой также равен 3), а при низких концентрациях ТБФ угол наклона равен 2,4, т. е. в этих условиях в системе образуется новый комплекс с меньшим числом координированных молекул ТБФ. Дробное значение угла наклона указывает на существование в органической фазе двух комплексов с двумя и тремя молекулами ТБФ.
Наиболее полная экстракция теллура трибутилфосфатом происходит из солянокислых растворов с образованием сольвата [Те-ЗТБФ-хН2О]С14, где растворитель непосредственно связан с теллуром [114].
При экстракции Te(IV) из солянокислых растворов при помощи ТБФ в изооктане в некоторых случаях образуются две органические фазы [447]. Появление второй органической фазы зависит от концентрации НО и теллура в водном растворе. Отношение ТБФ : Те в обеих фазах равно 2, а отношение Те : С1 практически постоянно и в 4,5—6АНС1 равно 1 : 4. Состав теллурсодержащего сольвата в легкой и тяжелой органической фазах одинаков: ТеС14 (ТБФ)2. Тяжелая органическая фаза содержит повышенное количество воды.
Экстракция селена и теллура 30%-ным трибутилфосфатом в керосине из 6—12 N НО применялась для количественного отделения Те (IV) и Se (IV) от сульфатов [354, 772]. Для экстракционного разделения Se (IV) и Те (IV), а также Те (IV) и Те (VI) использовалась разница в отношении к ТБФ этих элементов в 4—5 N НО [738]; эффективность разделения Se (IV) и Те (IV) этим методом была подтверждена при помощи радиоактивных индикаторов Se75 и Те127 [772]. Количественное разделение Те (IV) и Mo(VI) было достигнуто экстракцией ТБФ в СС14 из 4 N НО [341].
100%-ный трибутилфосфат применялся для разделения Te(IV) и иодида (причем количественная экстракция иодида проходила из 0,2 N НО) [769], а также Те (IV) и Fe (III) [750].
Томпсон и Лэкин [1118] предложили экстракционный метод отделения Те (IV) от Au (III) и Fe (III). При использовании смеси трибутилфосфата и диэтилового эфира (3 : 7) в присутствии NH4SCN 5 г Fe полностью извлекались из 0,5 N НС1 за две экстракции. Теллур не экстрагировался из 0,5 N НО в отсутствие NH4SCN и количественно экстрагировался из 2 А НС1 в присутствии 20% NH4SCN. Золото экстрагировалось из разбавленного солянокислого раствора даже при низкой концентрации NH4SCN и, таким образом, было возможно отделение— 10 мг Au от 10 мкг Те. **
Из других сложных эфиров фосфорной кислоты для экстракции Те (IV) применяли ди-(2-этилгексил)фосфат [381, 830], а также диизоамилметилфосфат и триоктилфосфиноксид [380].
151
Экстракция кислородсодержащими растворителями и бензолом
Такие экстрагенты, как ди-н.бутиловый эфир, ди-н.амиловый эфир, 3-н.октиловый спирт, извлекают четырехвалентный селен только при высокой концентрации НО в водном растворе [196, 405]. Кислородсодержащие экстрагенты (спирты, кетоны, эфиры) извлекают Se (IV) в виде кислот состава [Н3О(Н2О)Х (орг)ж ]2SeCle.
Шестивалентный селен органическими растворителями экстрагируется гораздо хуже, чем четырехвалентный. Зависимость коэффициента распределения селена от концентрации НО при экстракции различными органическими растворителями приведена на рис. 30 [196].
Исследована экстракция Se (IV) бензолом и Те (IV) нитробензолом из растворов НВг и из смеси НВг с НО, H2SO4 и НС1О4. Показано, что теллур полностью извлекается из сернокислых растворов (> 17,5 N), содержащих 0,05 N НВг, и из хлорнокислых растворов, содержащих 0,05 N НВг [1111, 1114].
Изучение механизма экстракции четырехвалентного теллура из солянокислых растворов некоторыми спиртами, кетонами, простыми и сложными эфирами показало, что эти растворители экстрагируют Те (IV) в виде соединений, содержащих различные хлоридные комплексы теллура. Только простые эфиры экстрагируют теллур в виде одного соединения [Н3О+(Н2О)зс(эфир),/]2ТеС1в 1179].
На рис. 31—33 приведены значения коэффициентов распределения Те (IV) между растворами соляной кислоты и различными кислородсодержащими растворителями. Лучше всего экстрагируют теллур кетоны, плохо — простые эфиры. Для всех экстрагентов наблюдается увеличение коэффициента распределения с ростом концентрации НО в водной фазе. Как видно из рис. 33, коэффициенты распределения теллура в диапазоне его концентраций 5-10"11 — 2- 10~2 М не меняются, что указывает на отсутствие полимеризации теллура как в водной, так и в органической фазах.
В случае экстракции кетонами (см. рис. 32) увеличение коэффициента распределения при уменьшении концентрации теллура связано, по-видимому, с диссоциацией экстрагируемого соединения.
Экстракция селена и теллура сложными эфирами была изучена Тараян, Арстамян, Микаелян [373]. Максимальная экстракция Те (IV) (87%) достигается этилацетатом из 5,0 N НС1; Те (VI) практически не извлекается. Четырехвалентный теллур не экстрагируется бутил-, амил- и изоамилацетатом из 1,0—8,0 N НС1. Четырехвалентный селен не экстрагируется эфирами из 0,1—8АНС1. В 0,1 — 6 N НО золото количественно экстрагируется этилацетатом. Другие эфиры экстрагируют Au значительно хуже. На основании различия в поведении Se, Те и Au при экстракции сложными эфирами были разработаны методы разделения этих элементов [375].
152
Рис. 30. Зависимость коэффициента распределения селена от концентрации соляной кислоты при экстракции различными органическими растворителями
1— ди-н. бутиловый эфир; 2 — дн-н. амиловый э’ ир; 3 — н. октиловый спирт; 4 — бензол; 5 — нзооктан; 6 — четыреххлористый углерод
Рис. 31. Экстракция теллура (IV) спиртами
1 — н. амиловый спирт (Сте(1У) = 4’10~4 Af); 2 — *н. гексиловый спирт (^Te(IV) в == 4 ЧО"4 М)\ 3 — н. гексиловый спирт (Суецу) » 2 *10“2 М); 4 — н. октиловый спирт (CTe(lV) “4 • 10~4 М); 5 — н. октиловый спирт =*5 *10"4 М)
*р /
200-
У50 -
100-
2! /
Рис. 32. Экстракция теллура (IV) кетонами
I —диэтилкетон (Сфе(1У) = 4-IO-4 Af); 2—дипропилкетон (Срецу) =?4 ‘10~* Л1)
Рис. 33. Экстракция теллура простыми и сложными эфирами (СТе(1у^=24-10-4 Л1) I — и. бутиловый эфир; 2 — динзопропиловый эфир; 3 — амилацетат; 4 — беизилацетат; 3 — этилацетат; 6 — ди-н. бутиловый эфир (CfedV) = S’l0-' AfJ
Рис. 34. Экстракция селена кетонами в зависимости от кислотности среды
1 — метилэтилкетои; 2 — диэтил-кетон; 3 — метилпропилкетои; 4 — м з г I ( [ ’• и > ’ I ! .г : :» [. > - ( •
изобутилкетои
Вначале при pH 10 этилацетатом отделяют золото,затем экстрагируют теллур из 5 А НО. Селен остается в водной фазе.
Этиловый эфир применялся для количественного отделения Te(IV) от Al, Ag, Bi, Са, Cd, Cr, Со, Cu, Zn, Mg, Mn, Ni, Pb, Pd, Ti, TR при получении теллура высокой чистоты [987].
В солянокислом растворе четырехвалентный селен образует с алифатическими насыщенными монокетонами органоселеновые соединения, растворимые в хлороформе и СС14 [692, 775, 776]. Экстракция селена кетонами в зависимости от кислотности раствора представлена на рис. 34. Экстракция селена начинается при 4—5/V НО. Для всех кетонов, за исключением диэтил-и метилизопропил-кетона, соотношение Se : кетон равно 1. Кетоны экстрагируют из солянокислого раствора и другие элементы — Те, Au, Fe, Sb, Mo.
Экстракция хлороформом соединения Se(IV)- с метилэтилкетоном из 6,5 N солянокислого раствора использована для количественного отделения селена от Pb, Cu, Ag и других элементов при анализе медных концентратов, сплавов, черновой меди и других материалов [774, 775]. При экстракции 10—100 мкг Se могут присутствовать в растворе до 100 мкг Cu(II), 50 мг Fe(III), Al (III), Mn(II), Cd(II), Zn(II), 1 мг Cr(III) и небольшие количества Tl, In, Ga, Ge, W, Th, а также до 0,6 M. H2SO4 и 0,5 M. HNO3.
Экстракция диизопропилкетоном из 7,5 А НО позволяет также количественно отделить селен от мешающих элементов [692].
Теллур экстрагируется метилэтилкетоном из 3,5—6А солянокислого раствора. Коэффициент распределения при равном объеме экстрагента для 0,02 мкг Те равен 247 [494]. В этих условиях экстрагируются также Au(111), As(111) и As(V), Sb(III) и Sb(V), Cu(II), Fe(III), Se(IV). При добавлении бензола к метилэтилкетону (МЭК: : бензол = 2 : 1) подавляется экстракция Си(П) и Fe(II). Добавление тиокарбамида позволяет отделять теллур в присутствии 5 мг Cu(II); аскорбиновая кислота восстанавливает Fe(III) дб Fe(II), которое не экстрагируется метилэтилкетоном.
Для отделения следов теллура от Al, Bi, Or, Со, Cu, Ni и Se
154
применяли экстракцию теллура метилизобутилкетоном [589, 1112, 1113]. Коэффициенты распределения при экстракции из GN НС1 для Te(lV), Bi, Cu(II), Al, Ni, Co, Cr(lll), Se(IV) составляли соответственно 360 ; 3,П10“4; 5,15-IO"2; 2,5-10“4; 2,5-10“4; 2,5-10“2; 1,25-10-3 и 1,25. С уменьшением концентрации HCI в водном растворе коэффициенты распределения снижаются и для 4,5 N НО равны для селена и теллура соответственно 4,1-10~4 и 3,3-102. Таким образом, для отделения теллура от селена рекомендуется проводить экстракцию метилизобутилкетоном из 4,5А НО, а при отделении от других элементов — из 6 А НО [794].
При экстракции метилизобутилкетоном из солянокислых растворов извлекаются (в %): Fe(HI)— на 99,9, Sb(V) — 100, Sb(III) — 68,* Sn(IV) — 99, Al(III) — 91, As(V) — 28, Se(IV) — 99, Te(IV)— 96, Ge — 98, Cr(VI) — 95, V — 87, Mo(VI) — 95, Mn(VII) — 34 [718, 720]. Экстракция теллура изобутилкетоном позволяет отделить 30—300 мкг Те от Bi, Cu, Al, Ni, Со, Сг и Se. Фактор разделения для теллура в 6N НО равен 1 —1,5-106 для Bi, 7-103 — для Си, 1,5-105 — для А1, 1,5-106 — для Ni, 2,4-105 — для Со, 2,9-105— для Сг, 2,9-102 — для Se [794].
При определении теллура в присутствии очень больших количеств железа первоначально Fe(III) экстрагировали из 6 A НС1 н.бутилацетатом в присутствии К2Сг2О7 или КМпО4. В этих условиях Te(IV) окисляется до Te(VI). Затем Te(VI) восстанавливали до Те (IV) и экстрагировали метилизобутилкетоном из 4,5—6АНС1 и реэкстрагировали из органической фазы водой [827, 1126].
Экстракция иодидных комплексов теллура метилизобутилкетоном в сернокислом растворе не зависит от концентрации серной кислоты при постоянной концентрации KJ (0,5 и 1,5 М) и составляет 99,4%. Подобно ведет себя сурьма.
Процент извлечения меди, висмута и кадмия возрастает с увеличением концентрации в растворе H2SO4 и в 4—8AH2SO4 составляет 99%. Процент извлечения свинца растет с увеличением концентрации НС1 в 1,5 М растворе KJ и уменьшается в 0,5 М растворе KJ [721].
Извлечение меди, висмута, кадмия и сурьмы уменьшается с увеличением концентрации К-!, а в случае свинца наблюдается максимум в 0,75 М KJ (99,3% в 2А НС1) [721].
Экстракционное отделение селена от Cu, Pb, Ag и других элементов метил-этилкетоном [775]. 0,5—3,0 г пробы растворяют в 5—20 мл HNO3, выпаривают с 2 мл H2SO4, растворяют в 3—4 мл воды и снова выпаривают. Остаток растворяют в 20 мл 7N HCI и фильтруют в 50-миллилитровую мерную колбу. Приливают 7 мл HCI и доводят до метки водой. Аликвотную часть раствора (10 мл) встряхивают в делительной воронке с 0,3 мл метилэтилкетона и дают 15 мин. отстояться. Затем экстрагируют 3 мин. 10 мл хлороформа, переводят органическую фазу в 50-миллилитровую мерную колбу, промывают 2—3 мл хлороформа, добавляют 2 мл воды, 0,5 мл конц. HNO3 и выпаривают хлороформ на водяной бане. Нагревают затем до полного разложения метилэтилкетона. Далее определяют селен любым методом.
155
,.J>.
Экстракция внутрикомплексных соединений селена и теллура
Экстракция внутрикомплексных соединений селена и теллура — дитизонатов, диэтилдитиокарбаматов, купферонатов — применяется только в групповых методах разделения элементов.
Экстракция четырехвалентного теллура проводится раствором дитизона в СС14 из кислых 0,1—3 М растворов [896].
Есть указания на возможность восстановления Te(IV) до Те(П) при экстракции четырехвалентного теллура дитизоном в СС14 из 1 М НС1О4 [907].
На коэффициенты распределения сильно влияют температура,* природа кислоты в водном растворе, присутствие ионов F-, С1”, Br~, J-, NOg, HSO7> фосфорной, борной, кремневой и других кислот [882, 1150]. Количественно Te(IV) экстрагируется из 37VHC1 раствором дитизона в СС14; при этом возможно отделение Te(IV) от больших количеств Sb(V) [1150].
Селен образует дитизонаты в широком интервале кислотности — 1—82V НС1 и 1—14 N H2SO4. Дитизонаты селена хорошо извлекаются из кислых растворов СС14.
Экстракция диэтилдитиокарбаматов селена и теллура хлороформом и СС14 позволяет разделять селен и теллур [357, 1159]. Экстракция селена в виде диэтилдитиокарбамата при помощи СС14 проходит при pH 4,0—6,2, экстракция теллура — при pH 8,5— 8,7; в присутствии KCN и комплексона III устраняется мешающее влияние платиновых металлов, Си, Ni, Со, РЬ. Эти металлы экстрагируются в виде диэтилдитиокарбаматов при pH 4—11. В присутствии KCN при pH 8—11 свинец экстрагируется в виде диэтилдитиокарбамата, медь не экстрагируется.
Для отделения селена и теллура от мешающих определению элементов (Си, Ni, Со, РЬ и др.) проводилась экстракция диэтилдитиокарбаматов из 4 N H2SO4 [544, 890] или из слабокислого раствора [355]. Селен, по-видимому, образует с диэтилдитиокарбаматом комплексы состава Se(DDTC)4 и Se(DDTC) [544].
При использовании трибутилфосфата в качестве экстрагента комплекс Se(IV) с диэтилдитиокарбаматом количественно экстрагируется в широком интервале концентраций НО [766]. С ростом концентрации НС1 коэффициент распределения для Se(IV) практически не меняется (— 102), в то время как для Se(VI) коэффициент распределения быстро возрастает от 10-3 до 10-1. На этом основан метод разделения Se(IV) и Se(VI). Из органической фазы Se(IV) на 99% реэкстрагируется 10%-ным водным раствором НС1О4 в присутствии Н2О2 [1151].
Экстракция теллура тетраэтилтиурамдисульфидом позволяет количественно отделять следы теллура от многих мешающих определению элементов [1173].
156
Экстракция аминами
Большое практическое значение приобрели методы экстракции селена и теллура высокомолекулярными аминами. При этом в качестве экстрагента используют раствор амина в том или ином органическом растворителе (хлороформе, керосине, ксилоле и др.).
Селен экстрагируется аминами при высоких концентрациях НС1 в растворе, когда возможно образование соединений типа HSeCl5 и H2SeCle [48]. Из &N НО селен экстрагируется аминами (диэтил-амином, циклогексаэтиламином, диметиланилином, диэтиланилином, анилином) на 30—40%. Из8А НС1 селен количественно переходит в фазу органического растворителя. Из 12N H2SO4 селен экстрагируется аминами на 28%. Из нейтральных растворов селен не экстрагируется, что используется при отделении от селена таких элементов, как Ni, Со, Fe, Pt, Pd, Ir, Rh, Ru, Au [117, 141].
Сравнение экстракционных возможностей некоторых аминов и фосфорсодержащих экстрагентов показано в табл. 19. Из 0,2— Ш H2SO4 Se(IV), Se(VI), Te(IV) и Te(VI) не экстрагируются фосфорсодержащими растворителями и аминами. Повышение концентрации серной кислоты и добавление соляной кислоты способствует экстракции селена.
Таблица 49
Величилы коэффициентов распределения при экстракции четырехвалентнога ceaeia из серно- или соляюкислых растворов
Кондеитрация кислоты, М Экстрагент
H2SO4 J НС1 Смесь н. октиламииа и ди-и. октиламииа Три-и. октил-амин (ТОА) Трибутилфосфат (ТБФ) Диизо-амилме-тнлфосфат (ДАМФ) Триоктнл-фосфин-окснд (ТОФО)
3 5 0,37 — 0,004 0,004
3 6 1,1 94,4 0,02 0,004 6,0
3 7 4,13 — 47,1 41,8 —
5 6 — 59,0 — — 3,0
Таким образом, экстракционная способность по отношению к селену аминов и фосфорсодержащих реагентов снижается по ряду:
ТОД >ТОРО >смесь аминов > ТБФ > ДАМФ.
При повышении концентрации серной кислоты до 5Л4 экстракционные свойства ТОА и ТОФО снижаются в два раза.
Условия экстракции селена из этанольных и метанольных растворов тридодециламином и трибутилфосфатом были изучены Элиа-ном и сотр. [487].
157
Наиболее перспективным является применение высокомолекулярных аминов для экстракции селена и теллура. Из 9 А НО четырехвалентный селен количественно экстрагируется додецил-триалкилметиламином в ксилоле и менее эффективно — трибензиламином в СНС13. Из экстракта в ксилоле селен обратно не извлекается водой, соляной кислотой, азотной кислотой или раствором NaOH, но из экстракта в СНС13 извлекается водой и разбавленной соляной кислотой. Четырехвалентный теллур количественно экстрагируется при помощи упомянутых выше экстрагентов из солянокислого раствора (> 6N НС1) и реэкстрагируется из органической фазы водой. Полное отделение селена от теллура возможно из 4—6N НО при использовании в качестве экстрагента трибензиламина в СНС13 [700—702).
Экстракция тройных комплексов
Успешно используются экстракционные методы разделения селена и теллура и отделения их от других элементов в виде тройных комплексов — соединений с органическими основаниями (производными антипирина, арилметановых красителей) — и ацидокомп-лексов селена и теллура [366]. Возможность варьирования лиганда и использование различных оснований и органических растворителей, изменение условий реакции образования комплекса создают большие возможности для избирательной экстракции металлов в виде тройных комплексов.
Бусев с сотрудниками предложили метод экстракционного отделения теллура от Se, Au, Си, Pb, Bi и других элементов на основе реакции гексахлоро- и гексабромотеллуритов с производными пи-разолона — антипирином, диантипирилметаном, диантипирилме-тилметаном [56, 57, 62, 63]. Тройные комплексы теллура хорошо-растворимы в дихлорэтане. Селен не образует комплексов и не экстрагируется дихлорэтаном из солянокислого раствора. Из раствора бромистоводородной кислоты селен частично экстрагируется. Кадмий, таллий, висмут, сурьма осаждаются производными антипирина в присутствии бромидов, но их мешающее влияние может быть устранено добавлением раствора комплексона III и винной кислоты.
Диантипирилпропилметан образует комплексы с теллуром в 5— 7N НО, которые количественно экстрагируются дихлорэтаном. Для экстракции можно использовать также хлороформ. Теллур экстрагируется и из 6N H2SO4, содержащей бромид (5,7- 10~2 г-моль1л) или хлорид (0,33 г-моль/л).
Комплексы железа с диантипирином образуются в слабокислом растворе при pH 2,2—2,5. Разделение теллура и золота осуществляется реэкстракцией теллура водой из органической фазы; золото при этом не реэкстрагируется. Свинец, цинк, кадмий, медь, индий не экстрагируются дихлорэтаном из 5—6 А НО.
158
Экстракционное отделение теллура от селена, золота, меди и других элементов {при анализе металлической меди и полупродуктов медного производства). Навеску, разлагают в смеси HNO3 + НС1, упаривают до влажных солей с соляной кислотой. Если материал содержит много посторонних примесей, проводят предварительное концентрирование теллура осаждением при помощи SnCl2.
Аликвотную порцию раствора переносят в делительную воронку, добавляют -соляной кислоты до общего объема 25 мл (до 6 N НС1), 2 мл 1%-ного раствора диантипирилпропилметана в уксусной кислоте (1:1) и 10 мл дихлорэтана. Встряхивают 1 мин.
Органический слой сливают в чистую делительную воронку, а водную фазу выбрасывают (или оставляют для определения селена). Экстракты дважды промывают 10 мл НС1 (1 : 1). К промытому соляной кислотой дихлорэтановому слою добавляют 10 мл воды и встряхивают 1 мин. После разделения и просветления фаз органический слой сливают в другую делительную воронку, а водный сйой оставляют в делительной воронке для дальнейшего определения теллура или сливают в мерную колбу.
Для отделения теллура от селена, меди и других элементов был предложен экстракционный метод, основанный на образовании тройных комплексов с 4,4-метилендиантипиреном, хорошо растворимых в 1,1,2,2-тетрахлорэтане [991]. В этом методе происходит более быстрое и четкое разделение фаз по сравнению с разделением при экстракции дихлорэтаном комплексов с диантипирилпропил-метаном. Практическое использование метода экстракции с 1,1,2,2-тетрахлорэтаном будет ограниченно, по-видимому, из-за большой токсичности используемых реагентов. Однако этот метод позволяет очень быстро и количественно отделить теллур от селена, меди, золота и других мешающих дальнейшему определению теллура элементов. Поэтому здесь приводится методика экстракционного разделения.
Выполнение определения теллура в меди. Раствор, содержащий теллур, помещают в 100-миллилитровый стакан, добавляют 5 мл H2SO4 (1 : 1) и выпаривают до паров SO3. Охлаждают, споласкивают стенки стакана НС1 (1 : 1), доводя объем раствора примерно до 25 мл. Переводят раствор в делительную воронку емкостью 125 мл, споласкивают стакан НС1 (1 : 1).
Добавляют 2 мл раствора 4,4-метилендиантипирена и оставляют на 1 мин. Затем добавляют 10 мл 1,1,2,2-тетрахлорэтана и встряхивают 90 сек. Оставляют до разделения слоев и переводят органический слой во вторую делительную воронку. Повторяют экстракцию при помощи 10 мл тетрахлорэтана и собирают органический экстракт во вторую делительную воронку. Выбрасывают водный слой, добавляют 20 мл смеси [25 мл метилендиантипирена и 400 мл НС1 (1 : 1)] в органический слой и встряхивают 30 сек.
После разделения фаз органический слой переливают в первую делительную воронку. Водный слой выбрасывают. Повторяют промывание 20 мл раствора. Добавляют к органическому слою 15 мл воды и встряхивают 90 сек. После разделения слоев переводят органическую фазу в другую делительную воронку. Водную фазу собирают в мерную колбу, и далее в аликвотной порции спектрофотометрически определяют теллур с висмутолом II (см. стр. 108).
Этим методом теллур определили в металлической меди, содержащей (в %): Си — основа, РЬ — 0,23, Sb— 0,049, Se — 0,026, Bi — 0,056, Fe — 0,033, Те — 0,02, Ni — 0,056, As — 0,039.
159
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
При разделении селена и теллура и отделении их от сопутствующих элементов широко применяются хроматографические методы. В основном это ионообменная хроматография и методы бумажной хроматографии. Предложены также методы хроматографии на бумаге, пропитанной ионообменными смолами. Перспективными являются быстро развивающиеся методы тонкослойной хроматографии.
Поведение селена и теллура в процессах ионного обмена определяется ионным состоянием этих элементов в различных условиях. Если в щелочном и нейтральном растворах четырехвалентные селен и теллур существуют в виде анионов ТеОз~ и SeO2-, то в слабокислых растворах обнаруживается различие в поведении этих элементов. Четырехвалентный теллур образует в слабокислой среде катионы (ионы теллурила ТеО2+) по следующим реакциям [107]:
ТеО2~ 4- 2Н+ ТеО (ОН)2;
ТеО (ОН)г + 2НС1 ТеОС12 + 2Н2О.
В этих же условиях (в слабокислой среде) селен продолжает существовать как анион. В сильнокислой среде образуются теллурсодержащие анионы:
ТеОС12 +2НС1=ТеОС12- + 2Н+.
При дальнейшем повышении кислотности (>6Л' НО) образуются ацидокомплексы селена и теллура: [SeCl6]“, [SeCl6]2-, [ТеС1613-[1047]. В нейтральных растворах, содержащих умеренные количества NaNO3, существуют однозарядные анионы, в сильнощелочных растворах обнаружены только двухзарядные анионы [765].
Изучение поведения Te(IV) в солянокислых растворах методом ионообменной хроматографии показало, что в 1 N НС1 теллур существует в виде ТеС14, в<0,8МНС1 —в виде катиона [ТеС13]+, при более высоких концентрациях НС1 (8—12./V) — в виде аниона [ТеС16]2- [1020].
При исследовании спектров поглощения Te(lV) в 6,9—12 N растворах НС1 были обнаружены два максимума — при 269 и 376 нм [745]. Максимум при 370—376 нм соответствует комплексу [ТеС1в]2--Спектры поглощения Te(IV) в растворах соляной кислоты различной концентрации представлены на рис. 35 [175]. По аналогии с ионами сурьмы и олова [134, 176] можно считать, что в солянокислом растворе существуют следующие комплексные ионы Te(lV) [ТеС15-Н2О]“ (с максимумом при 268 нм), [ТеС14(ОН)2]2-или [ТеС14-•(ОН)-(Н2О)] (при 294—296 нм), [ТеС14-(Н2О)2] (при 250—252 нм).
На кривых светопоглощения растворов комплексов в 8—12 М НС1 имеются два максимума, которые принадлежат комплексам [ТеС16]2-
160
Рис. 35. Спектры поглощения теллура (IV) в растворах соляной кислоты
[HC1J; Л': 1 — 12; 2—9,4; 3 — 9,0;
4 — 7,0; 5 — 5,0; 6— 3,5; 7—2,5 [Те(1V)] = 5-Ю-5 М
и [ТеС15-Н2О]“. При уменьшении концентрации от 7,0 до 3,5А НС1 происходит сдвиг максимума (от 268 до 250—252 нм), соответствующего комплексу [ТеС14-(Н2О)21, и появляется новый максимум (при 294—296 нм), соответствующий комплексу [ТеС14(ОН) (Н2О)]“.
При концентрациях <3,5 N НС1 не наблюдается никаких максимумов в интервале длин волн 210—450 нм.
Таким образом, в сильносолянокислых растворах существуют по крайней мере четыре комплексных иона Te(IV), области существования которых зависят от концентраций НС1 и теллура.
Существование нескольких типов ионов для Se(IV) и Te(IV) в кислых растворах было подтверждено также методом электрофореза на бумаге [108].
Исследование комплексообразования в системе Te(IV) — С1“ — Нт — Н2О методом распределения с экстракцией хлоридного комплекса теллура растворами н.гексанола в бензоле при 25°С и ионной силе растворов, равной 7, показало, что чем меньше концентрация кислоты, тем больше видов ионов существует в растворе; чем больше кислотность, тем больше доминирует ион [ТеС1вР" [260].
Возможность существования соединения ТеС14«2Н2О в солянокислом растворе была показана также в работе Шихеевой [446].
Ионообменные методы
Разделение Se(IV) и Te(IV) на катионитах и анионитах было проведено Гайбакян и сотр. [107].
В нейтральных и слабокислых растворах HCI Se(lV) образует анионы 5еОз~ и очень слабо сорбируется катионитами КУ-2 и КУ-1 [107, 159, 3561 (рис. 36).
Аниониты АН-1, ЭДЭ-10П и АВ-18 хорошо сорбируют четырехвалентный селен из слабосолянокислых растворов (0,1 N НС1). При,
Л«
б Зак. № 1476
3
Рис. 36. Зависимость сорбции селена (/, 2) и теллура (3, 4) от концентрации НС1 на катионитах КУ-1 (2, 4) и КУ-2 (/, 3)
повышении концентрации соляной кислоты от 0,5 до 4,0 N сорбция селена анионитами ухудшается. В сильносолянокислом растворе O=6tV НС1) селен хорошо сорбируется анионитами АН-1 и АВ-18.
При получении селена высокой чистоты проводилась очистка на катионите Дауэкс-50 и анионите Дауэкс-2 [491], а также катионите КУ-2 и анионите АН-1 [331]. Использование азотнокислых растворов позволяет отделять от селена примеси Те, Pb, Hg, Sn, Bi, Ag, Sb, Si, Cu, Fe.
В слабокислых растворах (при pH 0—4) можно отделить селен от примесей Na, Си, Те, Si, применяя анионообменную смолу Дау-экс-3 и Амберлит IR-4B. Сорбированный селен затем элюируют 1— 10АНС1 и получают чистый препарат SeO2 [424]. Очистка селена ионообменным методом описана также в работе Шумана [1067].
Отделение селена от С1~ и Вг~ основано на избирательном поглощении галогенов анионитом АВ-17 и их последующей десорбции раствором NH4NO3 [178].
Анионит Амберлит IRA-400 в ацетатной форме не сорбирует Te(IV), но количественно сорбирует Se(VI) и Se(IV) из раствора с pH 2,7—2,8. На этом основан метод очистки теллуровой кислоты от следов селена [1129]. На анионите Амберлит IRA-400 проводили отделение следов примесей (теллура и других элементов) из полупроводникового селена [840]. При использовании НС1 различных концентраций и растворов, содержащих комплексон III, возможно отделение от селена Те, Sb, Си, S.
Ионообменная очистка теллуровой кислоты от следов селена [1129]. В растворе теллуровой кислоты с примесью селена устанавливают pH 2,7—2,8 добавлением уксусной кислоты и пропускают раствор через колонку со смолой Амберлит IRA-400 в ацетатной форме (5 мм X 22 см) со скоростью 10 мл/час. Вытекающий из колонки раствор теллуровой кислоты содержит СО,2 мкг Se, тогда как начальное содержание составляло 70 мкг Se на 1 г Те. Колонка может служить для очистки теллуровой кислоты в течение нескольких месяцев.
Теллур сорбируется катионитами КУ-1, КУ-2 и СДВ-2 из растворов НС1 с pH 1—-5 на 70—80% (см. рис. 36) [107], катионитом КУ-1 в Н-форме при pH 2,7—8,5 [160]. При pH 1,5—3,7 количе
162
ственно сорбируются также Al, Си, Fe, Pb, Zn, а из раствора, содержащего комплексон III,— только Те и РЬ [159].
С повышением кислотности вследствие конкурирующего влияния Н+ сорбция уменьшается. Катионит Амберлит IR-120 в Н-форме количественно сорбирует теллур из <; 0,3 А НО; шестивалентный теллур не сорбируется [1127]. ИзО,ЗА НС1 теллур сорбируется также катионитами Амберлит IRA-120, KPS-120, СДВ-3. Из растворов 1,0—2,07V НО анионит ЭДЭ-10П плохо сорбирует селен и теллур (рис. 37); в сильнокислых растворах теллур хорошо сорбируется анионитами. Из 10.V НС1 ЭДЭ-10П сорбирует 90% Те и 20% Se.
Сильноосновной анионит Амберлит IRA-400 сорбирует четырехвалентный теллур даже из сильнокислых (3—12 N) растворов НС1 [831].
Сорбция теллура различными анионитами из солянокислых растворов увеличивается с повышением концентрации НС1 от 1 до ЗЛ'. Для растворов в ЗЛ' НС1 емкость анионитов по теллуру составляет (мг/г):
Вофатит SBW-112 95
Вофатит F .............109
Вофатит L-150 . .......107
ЭДЭ-10П ................ 6
ЭДЭ-10-27...............45
Наилучшее поглощение теллура происходит при использовании колонки d = 0,785 см и h = 3,6 см, заполненной вофатитом SBW-112, при пропускании раствора со скоростью 0,2 мл'1мин. Селен не поглощается из 3 N НС1, теллур элюируется 1N НО. При этом основная масса теллура попадает в первые 25 мл элюента [778].
Для большинства металлов коэффициент распределения между анионитом АмберлитCR-413 вС1_-форме и солянокислым раствором возрастает с ростом концентрации кислоты. Для элементов, образующих наиболее устойчивые хлорокомплексы [Pd2+, Re(VII),
Рис. 37. Зависимость сорбции селена (/) и теллура (2) от концентрации НС! в растворе на анионите ЭДЭ-10П
б*
163
Pt(IV), Au, Hg2+, Bi3+], наблюдается обратная зависимость. Четырехвалентный селен на анионите Амберлит CR-413 и Дауэкс-50 в 9N НС1 восстанавливается до элементного состояния [849].
Различные смолы восстанавливающего действия были использованы для отделения теллура от иода [950].
Сорбция теллура на анионите из 6АНС1 применена для очистки радиоактивного Те132, содержащегося в смеси продуктов деления [638]. Химический выход составлял 90—100% с коэффициентом очистки — 106.
Для отделения теллура от висмута использован анионит ЭДЭ-10. Катиониты не применяют для разделения Те и Bi [1381.
Из щелочных растворов селен и теллур хорошо сорбируются только анионитом АВ-18, но с увеличением концентрации щелочи сорбция снижается из-за конкурирующего действия ОН_-ионов. Поэтому концентрированные растворы щелочи могут быть хорошими десорбентами селена и теллура. Если вместе с теллуром были сорбированы также и другие элементы (Al, Fe, Си, Zn), то вымывание теллура 3—5%-ным раствором щелочи не происходит количественно. Присутствие цитрата аммония и лимонной кислоты несколько улучшает вымывание теллура [81].
Вымывание теллура с катионита КУ-1 раствором NH4OH происходит на 70—8596 [1591. Катионит КУ-1 может быть регенерирован промыванием водой, 5%-ным раствором НС1 и снова водой до нейтральной реакции по метиловому оранжевому.
При определении селена и теллура в рудах для отделения мешающих элементов (Fe, Си и др.) пропускают 10 лглО,ЗА азотнокислого раствора образца через колонку с 15 г катионита КУ-2. Селен и теллур полностью вымывают 300 мл 0,3 N HNOS; фильтрат выпаривают до 1 мл и дальше определяют селен и теллур обычными методами.
Разделение селенита и теллурита может быть проведено в 1 • 10~5 — 1-Ю'2 N НС1 на катионите КУ-2. При этом теллур полностью задерживается катионитом, а селен проходит в фильтрат [1071. В растворах 3—6 N НО селен и теллур могут быть разделены на анионите АН-1.
На основании количественного поглощения теллура катионитом КУ-2 из 0,01—0,1 А растворов соляной кислоты [356] предложен метод отделения от платиновых металлов. Хлоридные комплексы платины и палладия при этой кислотности переходят в фильтрат. При пропускании аммиачных растворов через катионит селен и теллур проходят в фильтрат, a Pt, Pd, Си, Ni поглощаются катионитом. Из 0,12 N солянокислого раствора Си, Ni, Fe, Pb количественно поглощаются катионитом КУ-2 в Н-форме на колонке 2 см X 25 см при скорости пропускания раствора 3—4 мл!мин [356].
Известен метод разделения на анионите алюминия и теллура [358].
164
Рис. 38. Зависимость сорбции селена (/), теллура (2) и золота (3) от концентрации НС1 в растворе на анионите НО
Рис. 39. Зависимость сорбции селена (/), теллура (2) и золота (3) от концентрации НС1 в растворена анионите АВ-16
В 0,1 N растворах органических кислот (щавелевой и винной) теллур количественно сорбируется смолой КУ-2 [136]. При увеличении концентрации щавелевой, винной и лимонной кислот выше 0,1 N наблюдается уменьшение сорбции теллура. Это может быть объяснено комплексообразованием. По-видимому, катион ТеО2+ образует в этих условиях комплексные анионы с органическими кислотами. Комплексообразующая способность Кислот по отношению к теллуру уменьшается по следующему ряду: лимонная кислота > винная > щавелевая. Таким образом, отделение 1000 мкг теллура от такого же количества селена возможно в 0,1 А растворах щавелевой и винной кислот.
Для отделения селена и теллура от рения, золота и некоторых Других элементов применяли анионит АВ-18 в Cl-форме. В 1 N НС1 рений количественно сорбируется, а селен, теллур и молибден проходят в фильтрат [135, 137, 1391. На анионите ЭДЭ-10П в 0,05—0,1 N NaOH возможно отделение рения от селена и теллура. Ионообменное отделение рения от молибдена, селена и теллура было успешно использовано при анализе Мо-концентратов.
Аниониты НО, АН-2Ф, АВ-16, АД-17, АД-27 в Cl-формах при pH 1—6 на 90% сорбируют золото (рис. 38, 39). В этих условиях Существует комплекс [AuClJ-. Селен и теллур не сорбируются. Постепенное уменьшение сорбции происходит из-за конкурирующего действия С1~-ионов. Возможно, таким образом, разделение Se, Те и Au по двум схемам.
В первом варианте разделение проводят на анионитах НО или АН-2Ф из растворов с pH 2. Часть теллура при этом проходит в фильтрат, а остальное количество его вымывают 0,01 N НС1.
165
Селен извлекают 6N НО, а для извлечения золота анионит сжигают.
Во втором варианте разделение проводят на анионитах АВ-17 или АВ-27 в 6 А НС1. Часть селена при этом проходит в фильтрат, остальное количество его вымывают 6 N НС1. Теллур извлекают 1А НС1, а для извлечения золота анионит сжигают.
Последний вариант применен для разделения Se, Те и Ап при анализе золотосодержащей руды.
Выполнение разделения. Пробу руды растворяют на холоду в ~50 мл HNO3, прибавляют несколько миллилитров конц. НС1 и раствор выпаривают до небольшого объема. К остатку добавляют 25 мл H2SO4 (1 : 1) и выпаривают до паров SO3, разбавляют водой, вводят конц. НС1 и осаждают Se, Те и Au раствором SnCI2.
Осадок обрабатывают 12,5 мл конц. НС1 и несколькими каплями HNO3, раствор разбавляют до 25 мл- и пропускают через анионит АВ-27 в Cl-форме со скоростью 4 мл/мин. Селен проходит в фильтрат, а теллур извлекают IN НС1.
На сильноосновном анионите ЭДЭ-10П могут быть разделены Sn(IV), Sb(V) и Te(IV). Sb(V) элюируют ЗА НС1, Te(IV) — 1 А НС1 и Sn(IV) — 1,8 А НС1О4. Смесь Te(IV) и Sb(V) можно разделить также на анионитах АСД-2 или Дауэкс 1-Х8 [324].
Ионообменное разделение индия, олова, сурьмы и теллура контролировали с применением радиоактивных индикаторов [1097].
Выделение теллура из смеси продуктов деления (отделение от Си, Bi, Fe) было проведено катионообменным методом после окисления Te(IV) до Te(VI) бихроматом в солянокислом растворе. Если ОДА по НС1 раствор, содержащий Te(VI), Cu(II), Fe(III), ВЦП I) и бихромат-ион, пропустить через катионит Амберлит IR-120 в Н-форме, то Te(VI) и бихромат количественно перейдут в. фильтрат, а все другие катионы останутся на колонке [1127]. Для дальнейшего определения шестивалентный теллур восстанавливают до Te(IV) действием Н2О2 в слабокислом растворе при нагревании или выпаривании раствора. Четырехвалентный теллур из ОДА НС1 количественно сорбируют катионитом Амберлит IR-120. Десорбцию теллура проводят 4 А НС1.
На катионите КУ-2 также можно количественно разделить Bi(III) и Te(VI) в 0,1 А НС1 [1041].
Отделение теллура от урана и других продуктов деления основано на сорбции U(VI), Mo(VI), Nb(V), Zr(IV), Np(IV) на смоле Дауэкс-2 в РО4-форме [1157]. В 0,1 А Н3РО4 теллур переходит в фильтрат на 96,7%. Коэффициент распределения для Te(IV) имеет наибольшее значение (1,3) в 0,1АН3РО4. С повышением концентрации Н3РО4 до 16 А коэффициент распределения медленно уменьшается до 0,4. Коэффициенты распределения для Mo(VI), U(VI), Nb(V), Zr(IV), Np(V) в 0,1—1 A H3PO4 составляют > 1000, для Cs, Се (III), Се (IV), Sr < 10.
Для отделения Te(IV) от U(VI) анализируемый раствор в 0,1 А Н3РО4 вводят в колонку размером ОД см X 15 см со смолой Дау-166
экс-2 в РО4-форме со скоростью 0,25 мл!мин. Теллур при этом количественно переходит в фильтрат.
Разделение Те и Ра, а также Np, Zr, Nb, Мо и U при анализе продуктов деления выполняли методом ионообменной хроматографии на колонке со смолой Лефатит М-500 [909].
При разделении J“, Te(IV) и Te(VI) использовали разницу в поведении этих элементов в 47V НС1 на колонке со смолой Амберлит IRA-400 в Cl-форме. Te(IV) и J" количественно сорбируются, а Te(VI) проходит в фильтрат. Te(IV) затем элюируют 0,1 А НС1. Контроль чистоты разделения осуществлялся при помощи радиоактивных индикаторов Те127т, Те129т и J1 1 [771].
Интересно поведение селенита в слабокислом растворе на катионите Дауэкс 50 W-X8 в присутствии солей Fe (III). При достаточно высоких концентрациях Fe(III) 70 мг Fe(III)] и при pH > 1,5 резко повышается сорбируемость селенита, который в этих условиях, по-видимому, образует комплексный катион FeHSeOg+ [ 1163]. Этот метод был использован при определении селена в сульфидах железа. Раствор с pH 1,8—2,0 пропускают через колонку с катионитом Дауэкс 50W-X8. Затем селен элюируют 50 мл 0,5 N НС1 и в фильтрате определяют селен обычными методами (с 3,3'-диаминобензидином), a Fe(III) иСо(П) десорбируют 6N НС1.
На смоле Дауэкс 1-Х8 с использованием 3MNH4OH и 0,5 М NaOH в качестве элюирующих растворов ЭО^-ионы были отделены от SeOg- и TeOjT [765].
Поглощение Se(IV) и Te(IV) анионитами зависит от природы Se- и Те-содержащего раствора. Так, в случае сорбции их из нейтральных растворов по поглотительной способности аниониты можно расположить в следующий ряд: ВП-1 = АЭ-1 >ЭДЭ-10П> ВП-1 А-= АМП > АВ-17 вофатит N. В карбонатных растворах порядок существенно изменяется: ВП-1А > ВП-1 = ЭДЭ-10П = АЭ-1 > > АВ-17 > AM > вофатит N [380, 382]. Из карбонатных растворов Te(IV) не поглощается анионитами. Таким образом, селен и теллур могут быть разделены на анионитах при использовании карбонатных растворов.
Для сернокислых растворов (500 г!л H2SO4) были получены следующие значения емкости анионитов (в лгг/г) по Se(IV) и Se(VI)
Se (IV) Se (VI)
АВ-17 . . 75 80
ЭДЭ-10П ... 75 19
Вофатит L-150 . . . . . .48 21
ВП-1 . . .48 31
Вофатит N . . . . . . .100 75
Наибольшее поглощение Te(IV) анионитами средней основности (ЭДЭ-10П и ВП-1) наблюдается из 0,2 М. H2SO4 [383]. С повышением
167
и понижением кислотности (до pH 2) сорбция понижается. Сильноосновные аниониты АМП, AM, ВП-1А из 0,2 М H2SO4 не сорбируют теллура.
Максимальное поглощение теллура анионитами ВП-1 и ЭДЭ-10П происходит из 0,53 М. щелочных растворов. При высоком содержании щелочи в растворе (1,3 моль/л) целесообразно использовать сильноосновные аниониты пиридинового типа ВП-1 (содержат группу СНз—где наблюдается наибольшая поглотительная способность по отношению к теллуру. Здесь процесс сорбции протекает, по-видимому, по сложному механизму комплексообразования.
По поглотительной способности из щелочных растворов по отношению к теллуру аниониты располагаются в следующие ряды: ВП-1 > ЭДЭ-10П > ВП- 1А = АМП > АЭ-1 > AM (для 0,53 М растворов), ВП-1 А > ВП-1 >АМП > AM > ЭДЭ-10П > АЭ-1 (для 1,3 М растворов).
Распределительная хроматография
Распределительная хроматография на порошкообразном фтор-пласте, пропитанном трибутилфосфатом, была применена для выделения селена из мишени SrO. Селен и теллур сорбируются на колонке. При пропускании 5 N НС1, насыщенной С12, элюируют селен; 1N НО вымывает теллур. Этим методом количественно отделяли микроколичества селена. Чистоту фракций контролировали на у-спектрометре [242].
Использование распределительной хроматографии на порошко- образном политетрафторэтилене, пропитанном три-н.октиламином в качестве неподвижной жидкой фазы, позволило отделить радиоактивный теллур от Sb, Ni, Со, Cu, Cd, Zn, Pb, Hf, U, Am, Pu [932, 933]. Разделение проводилось на колонке размером 80 X 2,5 мм с политетрафторэтиленом (d = 0,06-ь 0,1 мм). В качестве элюентов использовали растворы НС1 различной концентрации.
Распределительную хроматографию с обращенными фазами (неподвижная фаза ТБФ) применяли также для разделения олова, теллура и сурьмы [934].
Бумажная хроматография
Бумажная хроматография является, пожалуй, наиболее эффективным и распространенным в настоящее время методом разделения селена и теллура при анализе различных, часто сложных по составу материалов. В качестве элюирующих растворов широко используют спиртовые смеси, часто с добавкой солей щелочных металлов. Применяют также смеси и других кислородсодержащих растворителей — кетонов, эфиров.
168
Особое место занимают методы бумажной хроматографии с обращенными фазами, в которых используются свойства селена и теллура образовывать сольваты с трибутилфосфатом в солянокислой среде. Интересные работы по использованию хроматографии на бумаге, пропитанной ионообменными смолами, для изучения закономерностей разделения селена и теллура в различных условиях были опубликованы Ледерером [867].
Изучение влияния различных кислот в качестве подвижных растворителей на разделение селена и теллура методом восходящей хроматографии на бумаге показало, что при использовании H2SO4, HNO3, НС1О4, щавелевой, лимонной и винной кислот не удается разделить селен и теллур [412]. Для разделения подходят только растворы соляной кислоты. В 0,1 N растворе НС1, содержащем 0,01— 0,05 М NaCl, при pH 1 значения Rf для селена и теллура равны. При pH 3 величина Rf для теллура резко падает, а для селена остается постоянной. Оптимальной концентрацией для разделения является 0,05 М НС1. Температура и влажность не влияют на разделение. При pH 2 (0,017И НС1 + 0,01Af NaCl) величина Rf для селена равна 0,96, а для теллура — 0,70. Чувствительность метода 0,05 мкг Те при соотношении Se : Те — 100 : 1.
В этих условиях не мешают определению As, Sb, Sn, Cu, Pb, Zn, Ni, Co, Fe, Ti, Al, Cr, Mn, Be, SO4-, NO4, NO3, Mo, W. Ведут себя аналогично теллуру ванадий, ртуть, золото. Однако <20 мкг ванадия и ртути посредством SnCl2 не проявляются и потому не мешают определению [412].
Для разделения смеси Au, Pt, Se, Pd, Те была использована смесь н.бутанола и HNO3 (70 : 30). Для разделения Au, Pt, Se, Те и Au, Pt, Pd, Те хорошие результаты были получены в 3—3,5N НО [532] (рис. 40).
Гельман [113] при выборе растворителей для разделения селена и теллура на бумаге марок Б и М изучила простые эфиры (этиловый), кетоны (ацетон, метилэтилкетон, ацетил ацетон), органические кислоты (муравьиную и уксусную) и спирты (этиловыу, метиловый, н.бутиловый) с добавками различных кислот и солей. Лучший разделительный эффект был получен с н.бутанолом, насыщенным водой с добавкой 4% NH4NO3(выcaливaтeль) и 3—3,5%
Рис. 40. Зависимость Rf от концентрации НО в элюирующей жидкости
У — Se; 2 — Те; 3 — Pt; 4 — Au
169
HNO3. Величины Rf для теллура и селена равны 0,11 и 0,57 соответственно. Азотная кислота увеличивает сродство воды к целлюлозе и тем самым влияет на величину Rf [416].
Большие количества Си, Со, Ni и Sb мешают хроматографическому определению селена, так как попадают в его зону. Обнаружить селен в их присутствии можно, если обработать зону азотной кислотой и затем вновь восстанавливать селен SnCl2. Для проявления зоны теллура также применяют SnCl2. Чувствительность визуального обнаружения 1 мкг Se и 0,5 мкг Те. Платина и золото не мешают хроматографическому разделению. Мешает палладий, но его влияние устраняют предварительным выделением селена и теллура Fe(OH)3 [286].
Для разделения селена и теллура на бумаге марки Б была использована смесь н.бутанола, насыщенного 10%-ным NaNO3, а также смесь н.бутанола и метанола [566], смесь н.бутанола, насыщенного 2N НВг, и смесь н.бутанола, насыщенного- 20%-ной Н3РО4 [300]. В последнем случае теллур проявляется сразу же в виде бромида (желтое окрашивание), а для проявления селена используют тиомочевину.
Для количественного определения теллура вырезанную полоску при нагревании обрабатывают H2SO4 (1 : 9), а для извлечения селена — раствором НС1 + HNO3; далее проводят количественное определение селена и теллура обычными методами.
Для изучения образования и устойчивости соединений четырехвалентного теллура (в солянокислых, щелочных и нейтральных растворах) применяли в качестве растворителя н.бутанол, насыщенный ЗА НО [1055, 975]. В щелочной среде значения Rf для Te(VI) равны 0,08 -ь- 0,15, а для Te(IV) — 0,51 ~ 0,61. С увеличением концентрации щелочи значения Rf уменьшаются. Было отмечено, что Te(VI) восстанавливается до Te(IV) не только при кипячении растворов, но и при высушивании капли раствора на фильтровальной бумаге.
При применении бумаги Ватман № 1 и смеси растворителей этанол — метанол — 1,94 A HNO3 (30 : 70 : 100) были получены следующие значения Rf [ИЗО]: для Se(VI) — 0,92; для Se(IV) — 0,79; для Te(VI) — 0; для Te(IV) — 0,67.
При определении селена в минералах, почвах, сульфосолях, содержащих также уран и свинец, в качестве подвижного растворителя использовали смесь 45 мл метанола, 45 мл этанола, 12 мл воды, 3 мл HF (уд. в. 1,1) и 0,5 мл HNO3 (уд. в. 1,33) [482]. Для обнаружения селена применяли KJ и 3,3'-диаминобензидин. Чувствительность метода п-10~3%. Исходный раствор — 0,2N Na2CO3.
Для разделения SeOf, SeO4“ и SO4~ на бумаге Arches № 302 в качестве подвижного растворителя использовали смесь: 95%-ный этанол — вода — ледяная уксусная кислота (75 : 20 : 5) [500]. Для разделения SeO3~ и SeO4~ применяли также следующие смеси 170
подвижных растворителей [825]: пиридин — н.пропанол — вода (1:2:1) (№ 1); этанол — метанол — 2N НО (3:7:5) (№ 2); диоксан — 2N NH4OH (1 ; 1) (№3). Были получены следующие значения Rf для селенита и селената соответственно: 0,39 и 0,23 (с подвижной фазой № 1); 0,78 и 0,93 (№ 2); 0,46 и 0,66 (№ 3).При определении селенита в присутствии больших количеств селената наилучшей является смесь № 1, так как Se(VI) остается практически на старте.
Для обнаружения Se(IV) и Se(VI) хроматограммы обрабатывали насыщенным раствором (NH4)6Mo7O24 в 1 N H2SO4 и SnCl2. При восстановлении гетерополисоединений Se(IV) и Se(VI) образуются пятна синего и оранжево-красного цвета [825]. Чувствительность определения составляет 0,2 мкг Se(IV) и 2 мкг Se(VI).
Для отделения полония от селена и теллура на бумаге Ватман № 1 была использована система растворителей: С4НвОН — С3Н7ОН (1 : 1), предварительно насыщенных смесью 7N LiNO3 и 2N HNO3. Были получены следующие значения Rf [878]: RaE — 0,62; RaD — 0,27; Se — 0,72; Те — 0,47; Po — 0,97.
Количественное определение селена и теллура проводили после проявления раствором SnCl2 с использованием оптического денситометра, а определение радиоактивных элементов — на у-спект-рометре [878].
Предложен метод разделения теллура и полония с применением бутанола, насыщенного ЗУ НС1, или смеси 75 мг изопропанола, 5 г СС12СООН и 25 мл воды. Наилучшее разделение получено в сильнокислых растворах [189].
Разделения ядерных изотопов Те127 и Те129 было проведено на бумаге Ватман № 1. Подвижным растворителем был бутанол, насыщенный ЗУ НО. При этом Те127 перемещался с фронтом растворителя, а величина Rf для Те129 равнялась 0,60 [629].
Отделение -и определение теллура в присутствии больших количеств висмута оказалось возможным на бумаге Ватман № 1 с применением смеси подвижных растворителей: 2.и.г этанола, 1 мл изопропилового эфира, 0,5 мл конц. НС1. Обнаружение и определение элементов проводили после обработки хроматограмм раствором SnCl2 в 2N НС1 (Те) и щелочным раствором SnCI2 с добавкой в качестве катализатора РЬС12 (Bi). Чувствительность — 0,15мкг Те в присутствии 150 мкг Bi [713, 715].
Для разделения As(III), Te(VI) и был применен метод восходящей хроматографии на бумаге Ватман № 1 с использованием смеси подвижных растворителей: метанол—вода (9: 1). Полное разделение компонентов достигалось при pH < 2 и > 12. Обнаружение зон проводили в УФ-свете после обработки хроматограмм аммиачным раствором AgNO3 [989]. После обработки вырезанного пятна теллура 5—10 мл HNO3 (1 : 1) и выпаривания раствора с 2 мл H2SO4 (1:1) теллур определяли количественно с диэтилдитиокарбаматом.
171
Этот метод использовали при контроле чистоты иода J131 [611]. Для разделения радиоактивных Те132 и J131 в качестве подвижного растворителя применяли абсолютный этиловый спирт [704, 705].
Для разделения системы J~, JO3, ТеО3~были изучены различные спиртовые системы растворителей и получены следующие значения Rf [906] (табл. 20).
Таблица 20
Величины Rf
Система растворителей J- jo- TeOf
Пропиловый спирт — коиц. NH4OH (4:1) 0,920 0,126 0,152
Пропиловый спирт — пиридин (4:1) 0,147 0,0 0
Пропиловый спирт — пиридин — коиц. NH4OH (4:2:1) 0,855 0,005 0,040
Бутиловый спирт, насыщенный коиц. NH4OH 0,260 0,0 0,032
Бутиловый спирт — пиридин (3 :1) • 0,736 0,0 0,110
Амиловый спирт — конц. NH4OH (4 : 1) 0,0 • 0,0 0,002
Амиловый спирт — пиридин (2 :1) 0,094 0,0 0,061
Амиловый спирт — пиридин — конц. NH4OH (2:1:1) 0,200 0,0 0,0
Для разделения и определения селена и теллура в присутствии Cu, Ag, Bi, Pb, Zn, Ni, Fe, Co, Ca, Mg, As, Sb, Sn и благородных металлов был использован метод восходящей бумажной хроматографии с подвижным растворителем N(CH3)3— н.С4Н9ОН— НС1 (60 : 24 : 16). Разница значений Rf при этом составляла 0,58. Из других ионов мешали определению Ag+ и SO4"- Применение более простой смеси (N(CH3)3 — НС1) давало лучшее разделение (разница в значениях Rf равна 0,70), однако пятна селена и теллура были в этом случае сильно размыты [759].
Полное разделение смеси радиоактивных изотопов Se75, Те127: 129,_ Ро210, Bi210 на бумаге Ватман № 3 было достигнуто с подвижным растворителем, состоящим из смеси 60г49%-ной HFи 100мл метилэтилкетона. В этом случае селен двигался с фронтом растворителя, Bi оставался на старте, а величина Rf для Те составляла 0,6, для полония 0,5. Продолжительность разделения 2—3 часа. Разделить смесь в растворах НО не удалось [607]. Зоны идентифицировали радиоавтографически на фотопленке.
Для разделения и определения селена и теллура и отделения их от платиноидов и других элементов была успешно использована бумажная хроматография с обращенными фазами. При разделении с применением трибутилфосфата в качестве неподвижной фазы и 172
растворов НВг и NaBr — в качестве подвижной фазы [758] значения для4 селена и теллура уменьшаются с увеличением концентрации НВг; с увеличением концентрации ISJaBr величина Rf для селена практически не меняется, в то время как значение Rf для теллура уменьшается.
Метод позволяет разделять 3—60 мкг Se и 0,5—10 мкг Те с использованием 1—2 М НВг и 8Л1 NaBr [758].
При разделении и определении селена и теллура в присутствии платиноидов применяли трибутилфосфат как неподвижную фазу и HCI, HNO3, НС1О4 как подвижные фазы [760, 761]. Смесь Au, Ag, Pt, Pd, Rh можно разделить в 2—6N НО. Растворы HNO3 различной концентрации используют для отделения Au от платиновых металлов, селена и теллура, причем значения Rf для Ru, Pd, Os и Pt возрастают с увеличением концентрации кислоты, в то время как величины Rf для Rh, Ag и Au остаются постоянными.
Смесь Au, Ag, Pd, Rh можно разделить также с применением растворов HNO3 различной концентрации. При некоторых концентрациях НС1 величина Rf для Au равна 0. Смесь, содержащую Au, Pt, Pd, Rh или Os, Pd, Rh, можно разделить при помощи 2—IOtV НО; при этом золото остается на старте. Селен отделяется от теллура в солянокислом растворе (2N НО), так как теллур образует при этом комплекс НТеС16, растворимый в ТБФ. Величина Rf для теллура уменьшается с увеличением концентрации О", величина Rf для селена остается постоянной. Азотная и хлорная кислоты не подходят для разделения селена и теллура [760, 761].
При использовании в качестве подвижного растворителя смеси ТБФ —бензол (1 : 9), насыщенной НО, можно разделить селен и теллур с ARf = 0,40. Au, Os, Pd, Ru, Pt и Rh разделяют вымыванием 0,5—4 N NaBr, если подвижной фазой служит смесь ТБФ — бензол (3 : 7). В 0,5— 1 N NaBr рутений образует два пятна, которые при повышении концентрации NaBr до 2—4 N сливаются. Применение 1М раствора тиомочевины в 4АНС1 (неподвижная фаза — 20% ТБФ в бензоле) позволяет разделять следующие смеси: Au, Ru, Os, Pd, Ir; Au, Ru, Os, Pd, Rh; Se, Ru, Os, Те; Rh, Au, Ru, Те, Ir; Se, Bi, Cd, Те, Cu. Благородные металлы по возрастанию значения Rf можно расположить в следующий ряд: Au < Ru <Os < <Pt <Pd < Rh < Ir [759].
Методом восходящей хроматографии на бумаге марки Б была изучена возможность определения селена и теллура в присутствии Au, Pt и Pd. В качестве растворителей использован н.бутанол, насыщенный 10%-ным раствором NaNO3 [286]. Для предварительного отделения селена и теллура от других элементов применяли соосаждение с Fe(OH)3 раствором NH4OH при pH 9,5.
Для разделения Se, Те и Au использована также кольцевая бумажная хроматография. Неподвижной фазой была смесь ТБФ — бензол. Элюирующими растворами служили HCI, H2SO4, NaCl, NH4C1 различной концентрации. Золото оставалось на старте, се
173
лен перемещался больше всего. При многократном промывании 1—2АНС1 кольцо теллура не смещалось, но после промывания водой оно переходило на место селена. Примеси Cu2+, Pb2+, Bi3+, As3+, As5+ двигались вместе с селеном, но не окрашивались раствором SnCl2 и не мешали анализу [429].
Для разделения Se(IV) и Te(IV) методом круговой хроматографии на бумаге Синь-Хуа № 1 в качестве подвижного растворителя служила смесь (7:2:1) ацетон — вода — кислота (H2SO4, HNO3, НС1, НВг, HJ) различных концентраций или щелочь NaOH. Наилучшее разделение было получено в щелочных растворах (0,1—0,5 А NaOH) [904].
Применение хроматографии на бумаге, импрегнированной вольфраматом четырехвалентного олова, позволило отделить селен от следов Fe, Си, Zn, Pb, Bi и Те. При хроматографировании пятно селена перемещалось вместе с растворителем, а остальные . ионы почти не перемещались. При определении 20 мкг Se ошибка составляла 5% [1007].
Для приготовления бумаги, импрегнированной вольфраматом четырехвалентного олова, полосы бумаги Ватман № 1 размером 3 X 14 см опускают на 3—5 сек. в 8,8% -ный раствор SnCI4, избыток раствора удаляют фильтровальной бумагой, погружают полосы на 5 сек. в горячий 3,3%-ный раствор NaNO2, избыток раствора отжимают, и полосы сушат при 40—45° С, а затем отмывают избыток реагентов водой.
Полосы снова сушат при 40—45° С, промывают 30 мл 5%-ной HNO3 и водой до полного удаления HNO3 и сушат.
Анализируемый раствор наносят на полоску бумаги и через 10 мин. хроматографируют восходящим методом, применяя 0,1 М раствор тартрата аммония в 4 М NH4OH. Одновременно готовят контрольную хроматограмму. Для элюирования селена пятно вырезают и обрабатывают несколькими порциями воды общим объемом 30 мл. Полученный раствор упаривают на водяной бане до ~1 мл, добавляют 9 мл 5N H2SO4, фильтруют и определяют селен обычными фотометрическими методами (здесь — иодкрахмальным методом) [1007].
Применение хроматографии на бумаге, импрегнированной ионообменными смолами (Дауэкс-50), для разделения селенита и теллурита было описано в работе Ледерера [485, 867]. Результаты определения значений Rf в условиях различных элюирующих сред могут иметь значение как в бумажной хроматографии, так и в ионном обмене.
В качестве элюентов были изучены HCI, НВг, НС1О4, HNO3, H2SO4 и органические кислоты — муравьиная, уксусная, лимонная, молочная, пропионовая.
Значения Rf для SeO3” и ТеО3~ при различных концентрациях минеральных кислот, используемых в качестве подвижных фаз, приведены в табл. 21.
В табл. 22 приведены значения Rf при различных концентрациях органических кислот в подвижных фазах (уксусной, лимонной, муравьиной).
174
Таблица 21
Значения Rf
Концентрация кислоты, N
0,005 0,01 0,02 0,05 0,1 0,2 0,5
Кислота 1 са м О 1 са м О са (5 I са м о 1 са СО о 1 са СО о । О 1 са СО о 1 са СО о । %, О 1 са СО о 1 са СО о са СО о 1 са СО о
со со н Й н со н СО н СО н со н
НС1 0,82 0,01 0,87 0,02 0,80 0,03 0,81 0,06 0,82 0,16 0,81 0,43 0,80 0,64
НВг 0,82 0,02 0,84 0,08 0,84 0,06 0,84 0,30 0,86 0,32 0,84 0,63 0,85 0,66
HNOs 0,81 0,03 0,81 0,05 0,82 0,06 0,81 0,13 0,86 0,23 0,80 0,34 — —
H2SO4 0,78 0,02 0,82 0,04 0,81 0,10 0,81 0,21 0,83 0,32 0,81 0,46 0,86 0,77
HCIO4 0,83 0,04 0,83 0,02 0,80 0,08 0,82 0,18 0,82 0,26 0,86 0,-57 0,85 0,71
Т аблица 22
Значения Rf
Концентрация, Кислота
уксусная лимонная му равьиная
SeO32- ТеОз2- SeOs2- ТеОз2- SeOs2- ТеОз2-
0,005 0,86 0,01 0,88 0,02
0,01 0,92 0,01 0,89 0,06 0,86 0,06
0,05 0,94 0,01 0,85 0,30 0,79 0,04
0,1 • 0,87 0,01 0,89 0,39 0,78 0,05
0,2 0,90 0,01 0,78 0,52 0,76 0,02
0,5 0,86 0,01 0,87 0,65 0,78 0,06
1 0,88 0,01 0,84 0,74 0,77 0,12
2 0,85 0,04 — — — —
4 0,82 0,09 — — — —
6 0,84 0,09 — — — —
8 0,80 0,04 — — — —
Для 4, 6 и 8 N молочной кислоты значения Rf для селенита равны соответственно 0,81, 0,83; 0,85, а для теллурита — 0,62; 0,74 и 0,85.
Таким образом, селенит ведет себя одинаково во всех кислотах и движется с фронтом растворителя. Теллур в кислых растворах существует в виде катиона [645, 867] и прочно сорбируется катионитом на старте. В разбавленных кислотах селенит не образует
175
катионов. На этом различии в поведении селенита и теллурита в слабокислых растворах основаны ионообменные методы разделения селена и теллура.
Определение ультрамикроколичеств селена газовой хроматографией основано на измерении пика селена, полученного при подаче в электроннозахватывающий детектор газового хроматографа толуолового экстракта соединения селенистой кислоты с 4-хлор-о-фенилендиамином. Чувствительность метода 0,04 мкг Se [959].
Тонкослойная хроматография
Основные принципы метода тонкослойной хроматографии и условия работы были изложены в работе Рябчикова и Волынец [325].
Метод тонкослойной хроматографии на пластинках, покрытых слоем силикагеля, был применен для отделения больших количеств ртути от селена и теллура. В качестве подвижного растворителя использовали смесь ТБФ—бензол (3:7), исходным раствором была 0,1 N НО. При этом происходило количественное отделение селена и теллура от Hg, Au, Fe, Sb, Bi. В зоне селена и теллура оставались частично палладий и платина и целиком Со, Ni, Pb, Rh. Этот метод был использован при анализе киновари (минерала HgS) [263].
Разделение и определение селена, теллура и хрома в различных валентных состояниях проводились на двух видах силикагелевых пластинок с применением в качестве элюентов метилизобутилке-тона, этилацетата, н.бутилацетата, н.бутанола, насыщенного 2N НС1, и ацетона. Было достигнуто хорошее разделение Te(IV) и Te(VI), Se(IV) и Se(VI), Cr(VI) и Сг(Ш), Те(1У)-и Se(IV). Для Te(IV) в 6 N НС1 было обнаружено существование нескольких типов комплексных ионов [826].
При разделении селена и теллура методом тонкослойной хроматографии на силикагеле применены в качестве подвижной фазы смесь ТБФ — бензол, насыщенная конц. НС1, смесь ТБФ — бензол, насыщенная 1—ЗА НО или НВг, и тиомочевина, насыщенная 6N НО. Для первой системы получено хорошее разделение, но неустойчивые значения Rf, во втором случае Rf для теллура было равно 0, в третьем случае Rf для селена оказалось равным 0. Разделение проводилось в течение 2 час. при 12—15° С. Ошибка метода составляла ± 5% [762, 7631.
Применение смеси амилацетата и концентрированной НС1 в качестве подвижной фазы позволило разделить селенит и теллурит на слое силикагеля [1011]. Разделение селенитов и теллуритов на тонком слое А12О3 возможно при использовании в качестве подвижной фазы смеси концентрированной НО и алифатических спиртов (метилового, этилового, пропилового и изопропилового) [106] 176
МЕТОДЫ ОСАЖДЕНИЯ И СООСАЖДЕНИЯ СЕЛЕНА И ТЕЛЛУРА
Осаждение селенидов
При восстановлении растворов, содержащих селенит, в присутствии тяжелых металлов могут образовываться селениды меди [543, 575, 653, 804, 949, 967, 990, 1050, 1156], ртути [550], серебра [736], свинца [757], висмута [804] или одновалентного таллия [737].
Для выделения микрограммовых количеств селена в виде селенида меди к 50 мл раствора, содержащего Сп2+ и селенит, в присутствии 300 мг виннокислого аммония, нагретого до 50°С, добавляют 400 мг восстановителя (NH2OH-HC1) и после 2—3 час. стояния отфильтровывают через фильтр 0,6 мк из слабокислого или нейтрального раствора. Таким образом можно выделить 2 мкг Se.
Соответствующее осаждение в присутствии РЬ2+ и Ag+ проходит значительно хуже: выделяется всего 50—60% селена, а с Т1+ и Hg+ — только 10% селена.
Отделение селена и теллура от висмута, кадмия и индия возможно в слабосолянокислом растворе, 2 N по Na2S [1044, 1087, 1104].
Соосаждение селена и теллура с гидроокисями металлов
Соосаждение селенит- и теллурит-ионов зависит от природы гидроокиси и возрастает по мере увеличения ионного радиуса и валентности металла, образующего гидроокись. Четырехвалентные селен и теллур соосаждаются с гидроокисями — Fe, Al, Cr, Ti, Bi, Pb и другими, причем теллур лучше соосаждается, чем селен. Те (IV) и Те (VI) сообаждаются одинаково хорошо [304, 305].
Четырехвалентный селен количественно соосаждается с Ti(OH)4, А1(ОН)3при рН^б, cPb(OH)2, Bi(OH)3, Be(OH).2,Zn(OH)2— при pH 9 [105, 248, 305, 308, 309, 922, 1102], с основным карбонатом цинка [936, 1176]. Показана возможность разделения малых количеств Se(IV) и Hg(II) при помощи гидроокиси лантана [310].
Соосаждение Se(IV) с гидроокисью железа изучали многие авторы [16, 115, 153, 303, 307, 706, 779, 780, 824, 1102, 1148]. Изучение процесса соосаждения селена и теллура с гидроокисью железа в условиях их аналитического определения в рудах в присутствии значительных количеств посторонних элементов (Си, Pb, Cd, Zn, Мо) проводилось Тараян и сотр. [370, 371, 375]. Четырехвалентный селен количественно соосаждается с гидроокисью железа при pH 6—8. В присутствии до 200—300 мг железа практически полностью соосаждается 0,05—0,4 мг селена при однократном выделении осадка (при добавлении по каплям концентрированного
177
Рис. 41. Влияние pH на соосаждение селена и теллура с Fe (0Н)3 о— 2“ 2— 9—
1 — SeO3 ; 2 — ТеО3 ; 3 — SeO3 + TeOj
раствора аммиака). С повышением температуры количество соосаж-денного селена уменьшается. Поэтому осаждение необходимо проводить при комнатной температуре. До 0,5 мг теллура количественно соосаждается с Fe(OH)3 при pH 9,4—9,7 (рис. 41). В присутствии селена интервал pH значительно расширяется. При совместном присутствии селен и теллур одновременно и полностью соосаждаются с гидроокисью железа при pH 6,0—9,7.
Если железа больше, чем 400 мг, то осадок гидроокиси трудно фильтруется и легко пептизируется. В растворах, содержащих макроколичества селена и теллура, в результате химического взаимодействия с трехвалентным железом образуются соответствующие малорастворимые селениты и теллуриты. При совместном соосаж-дении микроколичеств селена и теллура с гидроокисью железа происходит изоморфное замещение [371]. Возможен, по-видимому, и адсорбционный механизм соосаждения Se(IV) с Fe(OH)3 [306, 375].
Осаждение возможно из азотнокислых растворов (не более 3%-ных по объему). Метод применяется для выделения суммы селена и теллура при анализе сульфидных руд.
0,5—3 г руды выдерживают в HNO3 без нагревания в течение нескольких часов..Затем нагревают постепенно на водяной бане до полного разложения пробы. Жидкость упаривают до 1—2 мл, приливают 20 мл горячей воды, отфильтровывают нерастворимый остаток. Промывают осадок ~10 мл воды. Добавляют 25 мл 0,5М раствора NHjCl, 300 мг Fe31" в виде раствора. Осаждают гидроокись железа, добавляя по каплям конц. NH4OH до pH 6—9,7.
Через 5—10 мин. осадок отфильтровывают, переносят водой в стакан емкостью 150 мл и растворяют в 5 мл НС1, добавляя несколько капель HNO3. Далее выделяют селен солянокислым гидразином с катализатором монохлориодом и теллур — раствором SnCl2. Определяют селен и теллур обычными методами.
На процесс соосаждения селена и теллура с гидроокисью железа не влияет присутствие до 0,3 aZn, Cd, Мо. Медь в больших количествах при pH 7 почти полностью осаждается с гидроокисью железа и затрудняет фильтрование и промывание осадка. При pH 8 медь переходит в фильтрат в виде аммиачного комплекса [16, 115].
178
Совместно с теллуром частично соосаждается свинец. Для отделения селена и теллура (а также Sb, As, Sn и Bi) от платиновых металлов наилучшее соосаждение с Fe(OH)3 происходит при pH 2,3—2,4 [18, 19, 317] в присутствии цинка и достаточного количества нитратов.
Соосаждение микрограммовых количеств Se(IV) с Fe(OH)3 возможно также при Использовании раствора NaOH. При 90°С и pH 6—6,5 селен выделяется количественно через 15 мин., при комнатной температуре для количественного выделения требуются 4—5 час. [533].
Осаждение селена и теллура в элементном состоянии
Количественное осаждение селена и теллура в виде элементных происходит при содержании их 100—200 мкг в 100 мл раствора [342, 401]. При более низких содержаниях (20—50 мкг) элементы осаждаются лишь на 65—80%. Данные об условиях осаждения элементных селена и теллура противоречивы [16, 150, 389, 560]. Блюм и сотр. [45, 46] провели систематическое изучение условий осаждения селена и теллура гидразином и хлористым оловом. При осаждении селена солянокислым гидразином из солянокислых и азотнокислых растворов полного осаждения никогда не достигалось. Потери селена составляли 10—30%. Результаты, полученные для азотнокислых и солянокислых растворов, мало чем различались.
На основании различия в реальных окислительно-восстановительных потенциалах систем Fe(III)—Fe(II) (+0,77 в), Se(IV) — Se° (+ 0,74в), Te(IV) — Те0 (+0,56в) оказалось возможным разделить селен и теллур в присутствии трехвалентного железа. Селен легко восстанавливается в присутствии трехвалентного железа, а восстановление теллура начинается только после количественного восстановления железа до двухвалентного состояния. Для количественного отделения селена от теллура необходимо сохранение хотя бы части железа в трехвалентном состоянии. Для этой цели используют осаждение селена гидразином из 25%-ного раствора НС1 или из 5%-ного раствора HNO3.
Если в растворе одновременно присутствует Au(III), то оно количественно восстанавливается и осаждается вместе с селеном [372]. Для отделения золота от селена рекомендуется экстракция золота этиловым эфиром из 6А НО.
Для осаждения теллура применяют хлористое олово или треххлористый титан. Опыты показали [45], что при содержании селена, значительно превышающем содержание теллура, селен может частично попадать в осадок теллура. Процесс выделения и коагуляции селена при восстановлении разбавленных растворов селенистой кислоты гидразином протекает медленно. В качестве ката
179
лизатора был применен монохлориод. В присутствии катализатора восстановление гидразином и коагуляция селена в солянокислом и азотнокислом растворах количественно завершаются за 30 мин. Этот метод был применен при определении селена в сульфидных рудах.
Последовательное осаждение элементных селена солянокислым гидразином (в присутствии катализатора — монохлориода) и теллура гидразином и SnCl2. От 1,0 до 3,0 г руды выдерживают без нагревания в течение нескольких часов с 25 мл конц. HNO3, а затем постепенно нагревают иа водяной бане до полного разложения пробы. Жидкость упаривают до объема 5 мл, приливают 40—50 мл горячей воды и отфильтровывают нерастворимый остаток. Промывают фильтр с остатком водой до тех пор, пока объем фильтрата не достигнет 80—90 мл. К фильтрату, нагретому до 40°С, добавляют 10 мл монохлорида иода ’, немного фильтробумажной массы, 15 мл 10%-ного раствора солянокислого гидразина, перемешивают и через 1 час отфильтровывают осадок селена, а фильтр с осадком промывают 3—4 раза малыми порциями теплой воды. Воронку с фильтром, содержащим осадок селена, устанавливают на ’мерную колбу емкостью 25 мл, и растворяют осадок в 5—7 мл горячей НС1, к которой добавлено несколько капель конц. HNO3. Промывают фильтр горячей водой. Далее определяют селей обычными методами.
В фильтрат от селена вводят немного фильтробумажной массы, нагревают до кипения, приливают 10 мл гидразина и постепенно при перемешивании горячий раствор хлористого олова до полного восстановления железа и избыток его 5 мл. Кипятят 2—3 мин. и оставляют на 1—2 часа в теплом месте. Осадок теллура отфильтровывают, промывают 3—4 раза теплой водой и далее определяют теллур обычными методами.
Большие количества теллура могут быть отделены от свинца, сурьмы и других элементов восстановлением из винно-азотнокислого раствора при помощи SnCl2 и молочного сахара [279]. Этот метод был использован при анализе свинцово-сурьмяно-теллуровых сплавов.
В щелочной и слабощелочной средах теллурит в присутствии свинца образует устойчивый свинцово-теллуритный комплекс [279]. Для количественного выделения теллура из сплавов с сурьмой и свинцом в качестве восстановителя используют глюкозу, лактозу со SnCl2, галактозу, солянокислый гидразин, аскорбиновую кислоту [342]. При определении селена и теллура в меди осаждение раствором SnCl2 проводили в азотнокислом растворе [967]. Из содовых и щелочных растворов в присутствии Se(IV) и Se(VI) тел-
1 0,279 г КЗОз растворяют в 125 мл воды, а в другом стакане — 0,178 г KJ в 125 мл воды. В колбу емкостью 0,5 л добавляют 250 мл конц. НС1 и оба реактива. Раствор охлаждают и разбавляют водой до метки.
•Для проверки реактива на годность в делительную воронку приливают раствор катализатора и добавляют 10—15 мл хлороформа. Если после экстракции хлороформный слой будет очень сильно окрашен, то добавляют несколько капель разбавленного раствора KJO3. Если хлороформ будет очень слабо окрашен, то добавляют несколько капель разбавленного раствора KJ. Нормальный цвет хлороформного слоя должен быть светло-розовым.
180
лур выделяли метабисульфитом натрия при pH 6. Теллур восстанавливается в этих условиях до элементного, а селен образует растворимый селенсульфат [234]. Чувствительность реакции 0,3—0,5jws Те в 100лмраствора. Метод позволяет отделять 1—200мг-Те от 0,2—1 г Se. Присутствие меди и свинца не мешает.
Соосаждение элементных селена и теллура с мышьяком
Для полного выделения из растворов малых количеств селена, и теллура рекомендуется соосаждение с мышьяком при восстановлении гипофосфитом натрия из 6АНС1. Можно для соосаждения селена применять теллур, который в данном случае служит коллектором, а для соосаждения теллура — селен. Одновременно с селеном и теллуром восстанавливаются до металлов Hg, Au, Bi, Sb, Pt.. В осадок с мышьяком попадают также Ag, Си [280, 604, 875, 891]. Шестивалентный селен гипофосфитом не восстанавливается. Известны также методы концентрирования селена и теллура путем соосаждения с серой [34, 824].
При соосаждении селена с теллуром наблюдается изоморфное замещение кристаллической решетки теллура селеном. При выделении селена на мышьяке смешанных кристаллов не образуется. Мышьяк выделяется в рентгеноаморфной модификации и, имея большую поверхность, соосаждает селен более полно, чем теллур [102]. Аморфный мышьяк, обладая большой поверхностной активностью, способен сорбировать значительные количества Си и Fe. Сорбция резко увеличивается, если анализируемый материал содержит большие количества мышьяка [100, 102]. Выделение малых количеств теллура гипофосфитом катализируется солями меди’ [854].
Количественное восстановление селенистой кислоты гипофосфитом проходит в 5—7 N НС1 при 50—60°С через 30 мин., а при кипячении раствора — через 15—20 мин. [26].
Теллуристая кислота восстанавливается в 0,5—4,0ЛгНС1 при температуре выше 70°С. Дробное восстановление селенистой кислоты в присутствии теллуристой кислоты протекает в 6—7 N НСГ при нагревании до 50—60° С. В 6Л' НС1 реакция восстановления теллуристой кислоты гипофосфитом катализируется Си2+ даже при-комнатной температуре. При совместном присутствии селена, теллура и золота последнее восстанавливается и с селенистой, и с теллуристой кислотами.
На рис. 42—44 приведена зависимость восстановления селенистой и теллуристой кислот гипофосфитом от кислотности раствора (при комнатной температуре) и от температуры. Медь катализирует процесс восстановления теллуристой кислоты в 6—7N НС1, и в присутствии 1—10 мг Си2+ теллур количественно восстанавли-
181
Рис. 42. Восстановление Se и Те (по 0,2 мг) в зависимости от кислотности (при комнатной температуре)
1 — Se через 24 часа; 2 — Те через 24 часа; 3 — Se через 2 часа; 4 — Те через 2 часа
Рис. 4 3. Зависимость полноты восстановления селена от температуры
Время стояния осадка 2 часа, температура (°C): 1 —30 — 40; 2 — 40 — 50; 3 — 50—60; 4 — до начала кипения; 5 — 100 (3 мин.).
Рис. 44. Зависимость полноты восстановления теллура от температуры
Время стояния осадка 2 часа, тем-пература(°С); 1 —60 — 70; 2—70 — 80; 3 — 100 (3 мин.)
вается через 15—30 мин. Двухвалентная медь восстанавливается гипофосфитом до одновалентной, и уже Си+ является катализатором для восстановления ТеО|”-иона до элементного состояния [8541.
Адсорбция селена на активированном угле
Элементный селен, образующийся при добавлении к раствору восстановителей (NaH2PO2), хорошо адсорбируется активированным углем. Однако для дальнейшего определения селена уголь надо сжигать, поэтому данный метод не очень удобен для выделения следов селена.
182
Соосаждение селена и теллура с солями свинца, кадмия, магния
Из слабокислых растворов Se(IV) селективно соосаждается суспензией PbSO4 вследствие адсорбции его положительно заряженными частицами сульфата, содержащего избыток РЬа+-ионов. В нейтральных растворах происходит также соосаждение Se(IV) в. виде PbSeO3. Теллур при этом практически не соосаждается.
Метод концентрирования следовых количеств Se(IV) соосаж-дением cPbSO4 применен для его выделения при анализе металлического свинца и меди [247].
Избирательное осаждение ионов SeO4“, SeO3~, ТеО4~ и ТеО3~ в виде кадмиевых солей позволяет провести отделение от и ReO4-Совместно с селеном и теллуром осаждается арсенат кадмия [190].
МЕТОДЫ ОТГОНКИ СЕЛЕНА
Селен может быть отогнан в виде хлорида, бромида, фторида, окиси или селеноводорода из водных растворов. Проверка различных методов отгонки в специальном аппарате показала возможность потерь селена по двум причинам: всегда значительные количества селена остаются на стенках холодильника и между колбой и холодильником; часть селена может, не удерживаясь в приемнике, улетучиваться с током воздуха. Использование аппарата, представленного на рис. 45, устраняло эти потери [1062].
Отгонка бромидов
Отгонка селена в виде бромидов была хорошо изучена [596, 633, 673, 693, 712, 823, 845, 919, 921, 1026, 1081, 1141, 1143, 1155]. Мешают отгонке большие количества железа [777].
Рис. 45. Аппарат для отгонки селена
/ — колба для дистилляции (ЮО-ил); 2 — капельная воронка; 3 — введение азота; 4 — холодильник.
5 — конденсационная колба; 6 — ловушкн
183
Проверку проводили в аппарате (см. рис. 45) с 20 мл 48%-ной НВг в присутствии 0,5мл Вг2. Объем доводили до 30 мл. После добавления 20 мл H2SO4 отгонку проводили в токе азота (25 мл/мин) при выпаривании раствора до паров SO3 (20 мин.). Колбу приемника •охлаждали в ледяной воде, разбавляли смесью 40 мл воды и 5 мл НВг (48%) и снова повторяли отгонку. После отгонки определяли содержание селена. Опыты показали 100%-ную отгонку.
Отгонку бромидов чаще всего применяют для отделения селена от мешающих элементов [519, 936, 1076, 1077]. При отгонке в растворе должен присутствовать элементный бром, иначе возможно образование Se<,Br.2, а также элементного селена. Избыток брома в дистилляте должен быть восстановлен.
Таким образом можно отделить селен от большого избытка ионов Cu2+, F", РО1~, WO4~ и Fe3+. В присутствии Fe3+ и других окислителей следует увеличить концентрацию КВг.
SeBr4 в водном растворе практически сразу же гидролизуется. Если присутствуют восстановители, повышают концентрацию брома. S2O3~ даже в 106-кратном избытке не мешает. As в присутствии Вг2 не отгоняется и не мешает определению.
SeC>4 переходит в концентрированной серной кислоте, особенно в присутствии КВг и Вг2, в соединения Se(IV).
Отгонка хлоридов
Отгонка хлорида селена была описана впервые Ленером 1871] и значительно улучшена Гейльманом [699] и другими авторами 1633, 1132].
Опыты проводили в том же аппарате (см. рис. .45). Добавляли НС1 и воду до — 7 N НС1. Затем добавляли 20 мл конц. H2SO4, и смесь нагревали 40 мин. до паров SO3.
Опыты показали, что отгонка селена с НС1 протекает также количественно, но несколько медленнее, чем в бромистоводородном растворе.
Отгонка H2Se
Четырехвалентный селен под действием очень сильных восстановителей (Н3РО2) может перейти в H2Se [735]. Эта реакция позднее была проделана с использованием в качестве восстановителей SnCl2 и НдРО2 [829] или цинковой пыли 1499]. При проведении отгонки с HJ + NaH2PO2 в сернокислом растворе при выпаривании до паров SO3 отгонялось 100% селена.
Отгонка SeO2
Отгонку SeO2 в токе кислорода проводили много раз 1187., 696, 910]. Опыты показали 1533], что при концентрации 45% H2SO4 из 2,3 мкг взятого селена отгоняется селена (в %) : 47 (за 20 мин.),
184
70 (за 40 мин.), 95 (за 80 мин.), 98 (за 90 мин.). Отгонку селена в виде SeOa в токе кислорода при 100° С применяют для отделения селена от меди [696}.
Отгонкой селена в технической серной кислоте было определено 5.1О;,о/о Se [519].
При отгонке селена с НВг, по-видимому, некоторое количество теллура улетучивается. В остатке после отгонки селена присутствует 78,2% Те [641]. Наши данные [263] также свидетельствуют о некоторой летучести теллура: с повышением температуры растворов при отгонке селена потери теллура возрастают (табл. 23).
Таблица 23
Поведение Те при отгонке с НВг
Концентрация H2SO4 Температура раствора, °C Количество отогнанного Те. %
Концентрированная 200—250 64
1:1 80—100 32
11 N 80—100 30
На возможность потери до 30% теллура при отгонке мышьяка в виде бромидов из сернокислого теллурсодержащего раствора было указано в работе Беликова, Каплан и Ширяевой [34].
Г лава 6
ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В ГОРНЫХ ПОРОДАХ, РУДАХ И МИНЕРАЛАХ
При использовании высокочувствительных методов определения селена и теллура для анализа сложных геологических образцов должна быть дана критическая оценка всех предварительных операций, связанных с подготовкой проб (разложение, выпаривание, концентрирование). Как показали многие исследования [79, 173, 271, 533, 716, 743], на всех стадиях анализа селен может легко теряться из-за высокой летучести частично восстановленных соединений. При выпаривании солянокислых растворов селен улетучивается в виде оксихлоридов селена. В случае выпаривания на водяной бане 250 мл 0,2 N солянокислого раствора до объема 100 мл потери селена составляют 10%, а при выпариваний на плитке — увеличиваются до 80% [578]. Чтобы предупредить потери селена при разложении образцов и выпаривании селенсодержащих растворов, необходимо избегать введения соляной кислоты.
Селен может улетучиваться из растворов, содержащих восстановители, но не окислители. Присутствие в растворах азотной кислоты препятствует потерям селена.
Разложение руд, пород и минералов
Для разложения сульфидных руд и минералов обычно используют азотную кислоту. Разложение азотной кислотой сульфидов необходимо начинать на холоду или разбавленной кислотой, иначе выделяется элементная сера, которая окклюдирует значительные количества определяемых компонентов. Азотная кислота хорошо разлагает галенит и пирит [916, 1056]. Для более быстрого и полного разложения сульфидов добавляют несколько миллиграммов иодида калия [1005], который катализирует окисление сульфидов и способствует количественному окислению серы до сульфатной. Смесь азотной и винной кислот применяют для разложения суль
186
фидов, содержащих сурьму и олово [742]. При выпаривании растворов, содержащих азотную кислоту, потерь селена не происходит.
Они могут наблюдаться только при выпаривании растворов досуха [533, 560].
После полного разложения образца для дальнейшего проведения анализа на селен и теллур следует обычно удалить остатки азотной кислоты из раствора. Для этого выпаривают раствор с серной кислотой [121]. Некоторые авторы, однако, указывают на возможность потерь значительных количеств селена при выпаривании растворов до паров SO3 [271,533,578,673, 1060]. Необходимо заметить, что продажная серная кислота всегда содержит некоторые количества селена [150, 723].
Применение денитрации растворов формалином также приводит к большим ошибкам при определении селена и теллура [150, 271]. Для денитрации растворов перед осаждением раствором SnCl2 применяют этанол [230], а также мочевину [5991.
Хлорная кислота имеет несомненные преимущества перед серной кислотой прежде всего потому, что при выпаривании до паров хлорной кислоты потерь селена не происходит [533] и удаление избытка хлорной кислоты выпариванием идет значительно быстрее. Хлорная кислота обладает сильными окислительными свойствами, и смесь азотной и хлорной кислот гораздо быстрее, чем одна азотная кислота, разлагает любые минералы и руды [711], не содержащие кремнекислоты. Легко разлагаются и сжигаются в этой смеси примеси органического вещества в породах и почвах. При работе с хлорной кислотой необходимо не выпаривать растворы досуха, иначе возможны потери селена.
При длительном нагревании (около 4 час.) в кипящей НС1О4 происходит заметное окисление селенитов до селенатов. Для удаления следов азотной кислоты проводят повторное выпаривание до появления паров хлорной кислоты. При этом окисления селенитов до селенатов практически не происходит.
Несмотря на то, что при определении селена и теллура в различных материалах до сих пор еще практикуется разложение царской водкой, а также удаление HNOS выпариванием растворов с серной кислотой или добавлением мочевины, самым быстрым и универсальным методом разложения, позволяющим легко отогнать избыток азотной кислоты, является метод разложения азотной и хлорной кислотами. При таком разложении легко разрушаются примеси органического вещества, бумажная масса. Необходимо, однако, при этом соблюдать все правила техники безопасности для работы с хлорной кислотой [142]. Для полного разрушения органического вещества в растворе должен присутствовать избыток азотной кислоты. Обычно для разложения применяют смесь кислот HNO3 и НС1О4 (2 : 1).
Есть указания на возможность применения фтористоводородной кислоты в смеси с азотной кислотой для разложения селенсодержа
187
щих материалов [533]. Это особенно важно при разложении силикатных пород и минералов, так как методы спекания гораздо более длительны и трудоемки.
В результате изучения летучести селена на материалах глубоководных морских отложений, в которые был добавлен Se75, установлено, что при разложении фтористоводородной и азотной кислотами селен не теряется [578].
Разложение при помощи HF. К 1—2 г породы приливают 20 мл 40%-ной HF •и 20 мл конц. HNO3, нагревают на водяной бане, затем раствор выпаривают до ’влажных солей. После добавления нескольких миллилитров HF и 20жл HNO3 выпаривание повторяют.
. Для разложения пород, руд и минералов (при анализе на селен) довольно часто применяют методы спекания и сплавления со ще-лочно-карбонатными смесями [330, 533, 1134]. При тщательно контролируемой температуре спекания эти методы удобны для разложения силикатных материалов и образцов, содержащих органические примеси. Однако при сплавлении проб в открытых тиглях <о щелочами, Na2O2 и щелочно-карбонатными смесями с ZnO и MgO в большинстве случаев имеют место некоторые потери селена [533]. Чтобы свести к минимуму потери селена на этой стадии анализа, необходимо проводить спекание проб при возможно более низкой температуре или сплавление веществ — в закрытых аппаратах [533].
Белопольская [35—38] рекомендует разлагать материалы спеканием со смесями типа Эшка (2 вес. ч. MgO и 1 ч. Na2CO3). В процессе спекания кислород воздуха окисляет сульфидную серу до сульфатной, образуются нерастворимые окислы и карбонаты, которые почти полностью остаются в остатке после выщелачивания. При разложении таким методом сульфидов и углей количественное выщелачивание селена происходит только при достаточном количестве соды. В противном случае часть селената щелочного металла может сорбироваться гидроокислами железа. При достаточном количестве соды селен практически количественно переходит в водную вытяжку в виде селената: количество селена, остающееся в нерастворимой части спека, не превосходит 1—10 отн.% при общем содержании п-10-1 — п-10-3% Se.
Метод разложения спеканием со смесью Эшка был проверен при определении селена в пирите, пирротине, галените, сфалерите, халькопирите [230].
Теллур при спекании образует ортотеллурат магния и количественно сохраняется в нерастворимом остатке.
Для разложения органических примесей, углистых веществ, углей к смеси Эшка добавляют окислители — хлорат, нитрат или перманганат. Для проб с высоким содержанием кремнезема применяют четырехкомпонентную смесь КМпО4, МпО2, ZnO, Na2CO3 (1:4:1: 3).
188
0,5—1,0 г силикатных пород тщательно сминввают в:фарфоровом или корундовом тигле с 8—10 г смеси Эшка, насыпают сверху 2—3 г этой же смеси и спекают в муфеле прн 800—850° С в течение 1—1,5 часа [191].
В случае анализа ископаемых углей, лигнитов и горных пород, содержащих углефицированное органическое вещество, навеску 0,5—1,0 г тщательно смешивают в корундовом тигле с 14—16 г трехкомпонентной или четырехкомпоиентной смеси и насыпают сверху 2—3 г той же смеси. Тигель помещают в холодный муфель, дно которого покрыто асбестом толщиной 5 мм. Муфель нагревают со скоростью 100—130° С в час до 300° С, далее — со скоростью 200° С в час до 800— 850°С и выдерживают при этой температуре в течение 1—1,5 часа. Муфель выключают, оставляя в нем пробу до охлаждения (обычно на ночь).
Охлажденный спёк переносят в фарфоровый стакан емкостью 300 мл, измельчают стеклянным пестиком, заливают 100—150 мл горячей воды, кладут 3—4 стеклянные бусины и при периодическом перемешивании кипятят иа закрытой плитке 20 мин. После охлаждения содержимое стакана переносят в мерную колбу емкостью 250 мл, доливают до метки водой и хорошо перемешивают. Раствор фильтруют через сухой фильтр в сухую колбу и отбирают в стакан емкостью 100—300 мл аликвотную часть фильтрата от 5 до 200 мл, в зависимости от предполагаемого содержания селена в пробе.
Аликвотную часть раствора выпаривают до объема 5—7 мл в стаканчике и нейтрализуют НС1 (уд. вес 1,19) до прекращения выделения СО2. Добавляют 5 мл раствора сульфосалициловой кислоты, затем 10 мл НС1 (уд. вес 1,19), сливая каждый раствор в пробирку.
Общий объем раствора в пробирке доводят водой до 30 мл, прибавляют 2 г КВг и, закрыв пробирку притертой пробкой, перемешивают до растворения соли. Пробирку с раствором помещают в нагретый до 85—86° С термостат и выдерживают при этой температуре в течение 1 часа. Остывший раствор переносят из пробирки в стакан емкостью 100 мл, и далее проводят обычное определение селена с 3,3'-диамииобензидином.
Для разложения сульфидных руд, главным образом содержащих HgS, в восстановительных условиях применяют смесь порошкообразного железа и окиси цинка. В результате образуется сульфид железа и ртуть количественно отгоняется.
После спекания проб селен в водной вытяжке присутствует в виде селената. Восстановление шестивалентного селена до четырехвалентного происходит при помощи конц. НС1 на холоду, а в 6 N НО — при 100° С. Восстановление шестивалентного теллура до четырехвалентного происходит при нагревании солянокислого раствора (> 9% по НС1) до 80—100°С [79]. Нагревание и кипячение солянокислых растворов следует проводить в колбе с обратным холодильником. Для полного восстановления селена до четырехвалентного после выпаривания растворов до паров хлорной кислоты добавляют каплю концентрированной НС1 [1134].
Отделение селена и теллура от мешающих элементов
Большинство методов определения селена и теллура требует предварительного отделения мешающих элементов. Наиболее распространены методы осаждения селена и теллура в элементном состоянии (см. гл. 5, стр. 179) и методы соосаждения элементных селена и теллура с мышьяком.
189
Для материалов, содержащих большие количества меди, золота, серебра, платиноидов, используют соосаждение селена и теллура аммиаком с гидроокисью железа (см. гл. 5, стр. 177).' В некоторых случаях применяют метод отгонки селена в виде SeBr4.
Определение селёна и теллура
За последние годы произошли существенные изменения в химии селена и теллура. Если раньше большинство методик анализа руд и минералов кончалось фотометрическим определением селена и теллура в форме золей, то в настоящее время аналитическая химия селена и теллура располагает гораздо более селективными и чувствительными методами определения этих элементов.
Применение о-диаминов для фотометрического и флуориметрического определения селена целиком решило задачу определения этого элемента в самых различных по составу объектах. Наиболее распространены фотометрические методы определения селена в рудах и минералах с 3,3'-диаминобензидином [53, 127, 172, 443, 520, 842, 923]. Однако исследования последних лет показали, что более чувствительным и селективным реагентом на селен является 2,3-диаминонафталин. Применение этого реагента позволяет в большинстве случаев проводить прямое определение селена без предварительного отделения мешающих элементов.
В качестве наиболее совершенного и универсального метода определения селена здесь предлагается метод прямого флуориметрического определения селена с 2,3-диаминонафталином, позволяющий определять до !• 1(Г6% Se из навески 1 г (см. стр. 89). Для более высоких содержаний селена используют меньшую навеску образца. Поскольку флуориметрический метод с 2,3-диаминонаф-талином обладает высокой точностью, можно из небольших навесок (0,05—0,1 г) проводить прямое определение селена в образцах, содержащих до 0,01% селена. Определению 0,1 мкг селена по этой методике не мешают 20 мг Fe, 20 мг Си, 10 мг Hg, 10 мг As, 10 мг Sb, 1 мг Bi (см. также главу 4, стр. 88). Мешают определению сильные окислители (HNO3) и восстановители (Sn2+).
Поскольку наиболее распространенным в настоящее время реагентом на селен является 3,3'-диаминобензидин, здесь предлагается вариант фотометрического определения селена с этим реагентом после предварительного отделения мешающих элементов со-осаждением на мышьяке при восстановлении гипофосфитом натрия. Фотометрический вариант метода пригоден для определения селена в широком интервале концентраций, точность метода для интервала 0,07—0,01% составляет ± 1,5% [842].
Определение селена с 3,3'-диаминобензидином. 0,25—2,0 г образца (в зависимости от ожидаемого содержания селена и от метода его определения) Помещают в стакан емкостью 200—250 мл, приливают небольшими порциями 10—20 мл HNO3 (уд. вес 1,40), добавляют 50—100 мг KJ (при анализе сульфидных руд)
190
« оставляют до следующего дня. Азотнокислый раствор нагревают под стеклом на водяной бане до прекращения выделения бурых окислов азота, снимают стекло м упаривают раствор (при перемешивании) на водяной бане до объема около о мл. Приливают 5—10 мл НС1О4 (1 : 1), упаривают на умеренно нагретой плитке до начала выделения паров iHC104 и продолжают нагревание еще 1 мин.
При анализе углефицированных проб в стакан с раствором, нагретым до выделения паров HCIO4, прибавляют небольшими порциями NH4NO3 до просветления раствора. Частицы угля, приставшие к стенкам стакана, счищают стеклянной палочкой, смывают водой, раствор вновь упаривают до выделения паров НСЮ4 и продолжают добавлять NH4NO3 до полного сжигания углистого вещества Ч Сдвигают стекло, и раствор упаривают еще в течение 1 мин. при выделении паров НС 1О4.
Раствору дают остыть, ополаскивают стекло и стенки стакана 10 мл воды, вновь нагревают до выделения паров НСЮ4,после чего нагревание продолжают еще 1 мин. Обработку водой и упаривание повторяют 2—3 раза.
К хлорнокислому раствору, из которого при упаривании были удалены окислы азота, приливают 20 мл воды и нагревают до кипения и растворения солей. После отстаивания раствор фильтруют (небольшой фильтр с синей лентой), нерастворимый остаток промывают 4—5 раз 2%-ным (по объему) раствором НС1О4 и 4—5 раз — водой.
Если исследуемый материал содержит серебро, то фильтрат после отделения •нерастворимого остатка (объем 40 мл) нагревают до 70—80° С, прибавляют по каплям 5%-ный раствор НС1 и 3—4 капли избытка; раствор оставляют на ночь в темном месте, затем фильтруют (маленький фильтр с синей лентой), осадок промывают небольшими порциями 0,01 А НС1.
К фильтрату (50 мл) после отделения нерастворимого остатка или серебра прибавляют 50 мл НС1 (уд. вес 1,19), 1 мл раствора мышьяковокислого натрия и 0,1 г CuSO4 1 2. Раствор нагревают до 80—90° С, прибавляют при перемешивании небольшими порциями гипофосфит натрия до восстановления трехвалентного железа и 1—2 г избытка. Добавляют фильтробумажную массу, кипятят раствор в течение 10 мин. и оставляют на ночь.
Раствор фильтруют, и осадок промывают 3—4 раза горячим 1 %-ным раствором гипофосфита натрия в НС1 (1 : 9) и 3—4 раза — водой. Осадок вместе с фильтром переносят в стакан, в котором велось осаждение, приливают 10 мл НС1 (уд. вес 1,19),3—5 капель HNO3 (уд. вес 1,40), перемешивают содержимое стакана стеклянной палочкой и, закрыв стакан часовым стеклом, оставляют на несколько минут.
Затем стакан осторожно нагревают на водяной бане при перемешивании до побеления волокон фильтра. Приливают 10 мл воды, раствор фильтруют и промывают бумажную массу теплой водой, собирая фильтрат в мерную колбу емкостью 50 мл. Раствор в колбе доливают до метки водой и перемешивают. Отбирают пипеткой в стакан емкостью 100 мл аликвотную часть раствора, не более 25 мл, с таким расчетом, чтобы содержание селена в ней не превышало 50 мкг при фотометрическом определении и 5 мкг — при флуориметрическом определении 3 * 5.
К подготовленному таким образом раствору прибавляют 2 мл 50% -ного раствора мочевины и оставляют на 15—20 мин. Добавляют 25%-ный раствор NH4OH
1 При содержании в руде до 1% углерода расходуется обычно 1—2 г NH4NO3. Если нет прямых указаний на присутствие в материале органического вещества, рекомендуется прибавлять до 0,5 г NH4NO3.
2 Если объем фильтрата больше 50 мл, то прибавляют равный объем НС1 (уд. вес 1,19) и соответственно увеличивают объем раствора мышьяковокислого натрия и количество сульфата меди.
’ При крайне низком содержании селена (10м %) для определения используют
весь раствор. В таком случае для растворения осадка селена и теллура берут
5 мл НС1 (уд. вес 1,19) и 2—3 капли HNO3 (уд. вес 1,40), а после растворения добавляют 5 мл воды. Раствор фильтруют в стакан емкостью 100 мл. Общий объем фильтрата и промывных вод не должен превышать 25—26 мл.
191
до значения pH раствора 1 (по универсальному индикатору), 2 мл 2,5 N раствора муравьинокислого натрия, 3 мл 0,1 М раствора комплексона III, доводят объем раствора до 35 мл водой и прибавляют разбавленный раствор NH4OH (1 : 1) до установления величины pH 2—3. Прибавляют1 2 мл свежеприготовленного 0,5%-ного раствора 3,3'-диаминобензидина (очищенного в случае флуориметри-ческого определения) и оставляют стоять в течение часа, затем нейтрализуют раствором NH4OH (1 : 1) до pH 8 2, переносят раствор в делительную воронку, обмывают стенки стакана водой и доводят объем раствора до 50 мл. Приливают толуол (10 мл при фотометрическом окончании или 6 мл при флуориметрическом окончании) и энергично встряхивают в течение минуты. После расслоения жидкости толуольный слой сливают через сухой фильтр в сухую пробирку.
Фотометрическое определение селена. Через 20 мин. или более (окраска устойчива в течение 2—3 час.) измеряют оптическую плотность толуольного экстракта на фотоколориметре ФЭК-Н-57 со светофильтром № 2 в кювете с толщиной слоя 1 см по отношению к одновременно приготовляемому нулевому раствору.
Для приготовления нулевого раствора в два стакана наливают по 5 мл НС1 (уд. вес 1,19), по 2—3 капли HNO3 (уд. вес 1,40), 2 мл 50%-ного раствора мочевины; через 15—20 мин. нейтрализуют аммиаком до pH 1,0 и далее поступают, как с испытуемым раствором. Экстракты соединяют и перемешивают, получая таким образом достаточное количество раствора для заполнения двух кювет.
Проводят холостой опыт с использованием тех же реактивов.
Содержание селена в растворе определяют по калибровочному графику.
Для построения калибровочного графика в стаканы емкостью 100 мл отбирают пипеткой 0,5; 1,0; 1,5; 2,0; 2,5; 3,0; 4,0; 5,0 мл стандартного раствора, содержащего 10 мкг Se/мл (5; 10; 15; 20; 25; 30; 40; 50 мкг селена). Прибавляют по 2 мл НС1 (уд. вес 1,19), 2—3 капли HNO3 (уд. вес 1,40), 2 мл 50%-ного раствора мочевины, оставляют на 15—20 мин. и далее поступают, как в ходе анализа.
Оптическую плотность полученных толуольных экстрактов измеряют по отношению к нулевому раствору.
По полученным данным строят калибровочный график, отклыдывая по оси абсцисс содержание селена в микрограммах, по оси ординат — величину оптической плотности.
Флуориметрическое определение селена. Через час или более после отделения толуольного экстракта измеряют интенсивность его флуоресценции на флуориметре ФАС-2 в камере с цилиндрическими кюветами 3 с первичным светофильтром X = 436 нм и вторичным светофильтром № 05.
Содержание селена в растворе определяют по калибровочным графикам.
Для построения калибровочных графиков в стаканы емкостью 100 мл отмеривают пипеткой такие количества стандартного раствора, содержащего 1 мкг Se/мл, чтобы получить две серии растворов: первая — с содержанием 0; 0,5; 1,0; 2,0; 3,0; 4,0; 5,0 мкг селена н вторая — с содержанием 0; 0,2; 0,4; 0,6; 0,8; 1,0 мкг селена. Прибавляют по 2 мл НО (уд. вес 1,19), 2—3 капли HNO3 (уд. вес 1,40), 2 мл 50%-ного раствора мочевины, оставляют на 15—20 мин. и далее поступают, как в ходе анализа.
Отделяют толуольные слон и через час замеряют интенсивность флуоресценции растворов при чувствительности 10, причем регулируют диафрагмы УФО и ФЭУ таким образом, чтобы для растворов первой серии содержанию 5 мкг селена соответствовал отсчет 90 на шкале гальванометра, а для растворов второй серии содержанию 1 мкг селена — отсчет 40.
1 При флуориметрическом определении селена все последующие операции выполняют при электрическом освещении, избегая действия дневного света.
2 Универсальная индикаторная бумага может показать ошибочную -величину pH раствора. Вновь полученную бумагу надо проверить, сравнив ее показания с показаниями рН-метра.
3 Кювета должна быть заполнена раствором возможно полнее.
192
Строят графики, откладывая по оси абсцисс содержание селена в микрограммах, по оси ординат — интенсивность флуоресценции, выраженную в показаниях шкалы гальванометра (за вычетом показаний для нулевого раствора).
При производственных определениях прибор настраивают (положение диафрагм УФО и ФЭУ) на отсчет 90 или 40 по толуольным экстрактам стандартных растворов, содержащим 5 и 1 мкг селена. Одновременно готовят нулевой раствор; отсчеты на шкале гальванометра для нулевого раствора по каждой серии должны быть постоянными. Режим работы прибора время от времени контролируют по стандартным растворам.
Для определения теллура в сульфидных рудах и минералах применяют экстракционно-фотометрические методы с бутилродамином С [173]. В настоящее время Блюм рекомендует [47] вместо бутилродамина С использовать этилродамин С. Применение этил-родамина С основано на экстрагировании бромтеллурита этилро-дамина С бензолом из сернокислого раствора и измерении поглощения (или флуоресценции) экстракта. Максимум поглощения соединения при 550—560 нм, флуоресценции при 585—590 нм.
В условиях определения вместе с теллуром в большей или меньшей степени экстрагируются родаминаты бромидных комплексов индия, ртути, таллия (I) и (III), олова (II) и (IV), золота (III), меди (I), железа (III), а также, независимо от присутствия в растворе бром-иона, соединение этилродамина С с платиной.
Из перечисленных элементов в осадок от гипофосфита попадают полностью или почти полностью Hg, Au, Pt; в качестве адсорбированных примесей —Fe, Си, Sn, Sb (0,5% от содержания кислоторастворимого олова в навеске) и Sb (0,04% от содержания в навеске) .
Ртуть, сурьму и олово отгоняют в виде бромидов из аликвотной части раствора, предназначенной для определения теллура. Золото (до 100 мкг} восстанавливают до элементного при дымлении сернокислых растворов.
Для отделения в Fe, Си, Pt, а также от оставшихся в растворе следов Hg, Au, Sn применяют экстракционное концентрирование теллура в составе бромтеллурита этилродамина С. Эта операция обеспечивает отделение 1 мкг Те от 20 мг Fe и Си, 100 мкг Au, Pt, Sn, 10 мкг Hg.
Чувствительность фотометрического определения теллура с этилродамином С равна 1 мкг, флуориметрического — 0,1 мкг. Однако последняя чувствительность при принятом способе подготовки к определению не может быть достигнута, так как помехи, обусловленные неполным отделением мешающих элементов, в неблагоприятных случаях могут достигать величин, соответствующих 0,2—0,3 мкг Те. Поэтому анализ заканчивают измерением оптической плотности экстракта; определяемый минимум содержания элемента составляет Ь10-4% при навеске 2,5 г.
Определение теллура с этилродамином С. 2,5 г или меньше образца помещают в стакан или коническую колбу емкостью 250 мл, заливают 30—40 мл конц. HNO3, добавляют 4—6 мл конц. НС1 и оставляют до следующего дня. Если содержание
7 Зак. № 1476
193
золота и платины в навеске, используемой для определения теллура, превышает 1 мг, навеску вскрывают азотной кислотой без добавления соляной. В этом случае перед осаждением теллура гипофосфитом натрия отфильтровывают нерастворимый остаток.
Помещают растворы на слабонагретую песчаную или асбестовую баню и медленно упаривают до объема 5—10 мл\ добавляют 25 мл (при навеске 2,5 г) HjSO4 (1 : 1) и нагревают до начала дымления серной кислоты. Если проба содержит органические включения, их разрушают, осторожно, по капле добавляя пергидроль в горячий сернокислый раствор до обесцвечивания последнего. Обмывают стенки стакана водой и вновь нагревают до паров SO3.
К холодному раствору добавляют 25 мл воды, нагревают раствор на горячей плитке до кипения и кипятят до растворения сернокислых солей (объем раствора при этом уменьшается до 20—25 мл).
Не отфильтровывай нерастворимый остаток (во избежание потерь селена, который мог частично восстановиться до элементного состояния при дымлении сернокислых растворов), добавляют по 1 мл 0,5%-ного водного раствора арсенита натрия, 2%-ный водный раствор CuSO4, 100 мл реактива для осаждения (8%-ный раствор гипофосфита натрия в 6 N НС1). Нагревают до кипения, выдерживают раствор на песчаной или асбестовой бане 10—15 мин. и оставляют на 1—2 часа или до следующего дня. На дне стаканов должен быть виден коричневый осадок элементного мышьяка. Если его нет, добавляют новую порцию гипофосфита (сухой соли).
Отфильтровывают осадок через фильтр «белая лента», промывают 4—5 раз горячей HCI (1 : 9), и растворяют элементы, осажденные гипофосфитом, в 6—7 мг горячей смеси кислот [к 100 мл НС1 (уд. вес 1,19) добавляют 2—3 мл HNO3 (уд. вес 1,4)]. Промывают фильтр горячей водой до объема фильтрата 20 мл (2—3 раза), охлаждают фильтрат и доводят объем водой в мерной колбе до 25 мл.
Для определения теллура поступают далее так, как описано на стр. 119.
Предложен метод определения до 10-4% теллура в сульфидных рудах с помощью 5-меркапто-3-(нафтил-2)-1,3,4-тиадизолтиона-2 (далее нафтилвисмутола) в присутствии селена без предварительного отделения последнего [68, 69J.
Реагент образует с четырехвалентными теллуром и селеном окрашенные желто-оранжевые комплексные соединения, практически нерастворимые в воде, хорошо экстрагируемые хлороформом, бензолом. Реакция с теллуром более чувствительна и протекает в широком интервале кислотности — от 18 A’ H2SO4 до pH 6,0. Селен (IV) образует соединение в более узком интервале — до pH 2,0. При pH 4,8—4,9 экстрагируется только комплекс теллура и возможно его экстракционно-фотометрическое определение в присутствии 200-кратных количеств селена.
Навеску руды разлагают смесью концентрированных НС1 и HNO3. В зависимости от содержания пирита в руде изменяют соотношение кислот в смеси: при содержании пирита около 20% добавляют 25 мл НС1 (уд. вес 1,19) и 15 лм HNO3 (уд. вес 1,4), при больших содержаниях — 15 мл НС1 и 25 мл HNO3 [142]. При достаточном содержании селена в сульфидных рудах (Ю"2 — 10~3 %) нет необходимости введения его в качестве носителя.
Выполнение определения с иафтилвисмутолом. 3—4 г тщательно растертой руды помещают в стакан емкостью 250—300 мл, смачивают водой и добавляют 15 или 25 мл смеси HNO3 -ф НС1 (2 :1), накрывают стакан часовым стеклом и кипятят на песчаной бане 3—5 мин. Охлаждают, добавляют необходимое количество HNO3, 194
нагревают смесь и осторожно кипятят 10 мин. до прекращения обильного выделения окислов азота. Ополаскивают стекло водой, и раствор упаривают до небольшого объема.
Добавлиют 5—7 мл воды и продолжают упаривать до влажного остатка, избегая перегрева. К остатку приливают 25 мл НС1 (уд. вес 1,19), нагревают при помешивании до 60—80° С, добавляют 60 мл воды, нагревают до кипения и отфильтровывают нерастворимый остаток через бумажный фильтр «синяя лента». Фильтр промывают несколько раз горячей водой, фильтрат и промывные воды собирают в стакан емкостью 250—300 мл.
Для осаждения суммы элементных селена и теллура к фильтрату добавляют 2 г солянокислого гидразина, немного фильтробумажной массы, кипятят 5 мин. Затем добавляют 2—4 мл 40%-кого раствора SnClj в конц. НС1 до восстановления Fe(III), небольшой избыток хлорида олова и нагревают на водяной бане в течение часа.
После отстаивания осадка раствор фильтруют через бумажный фильтр «синяя лента», уплотненный фильтробумажной массой. Осадок на фильтре промывают 4—5 раз горячим 10%-ным раствором НС1, содержащим в 100 мл раствора 0,5 г солянокислого гидразина, и несколько раз — теплой водой. Осадок вместе с фильтром переносят обратно в стакан и растворяют в горячей смеси кислот [10 мл НС1 (уд. вес 1,19) и 3—4 капли HNO3 (уд. вес 1,4)], накрывают стакан часовым стеклом и нагревают на водяной бане до побеления волокон фильтра. Добавляют 15 мл воды, и раствор фильтруют через бумажный фильтр «белая лента», собирая фильтраты в мерную колбу емкостью 50 мл. Фильтр промывают 4—5 раз горячей водой, объем в колбе доводят до метки водой.
Для определения теллура отбирают аликвотную часть (5—25 мл) в мерный цилиндр емкостью 100 мл с притертой пробкой, добавляют 3 мл 0,1 М раствора комплексона III, нейтрализуют 2 .V раствором КОН по метиловому оранжевому до перехода окраски индикатора в желтую (рН~4,2), добавляют 15 мл буферного раствора с pH 4,9 и 3 мл 0,1%-ного свежеприготовленного водного раствора реагента.
Через 20 мин. приливают 10 мл хлороформа и экстрагируют окрашенный комплекс 2—3 мин. После разделения фаз хлороформный экстракт фильтруют через сухой бумажный фильтр в сухую кювету с толщиной слоя 2 см и измеряют оптическую плотность на фотоэлектроколориметре (светофильтр № 2) относительно нулевого раствора.
Содержание теллура находят по калибровочному графику, построенному при pH 4,8—4,9 в интервале концентраций теллура от 10 до 80 мкг в 10 мл с интервалом 10 мкг.
В некоторых случаях, однако, определяют селен и теллур по измерению поглощения коллоидных растворов. Метод определения селена и теллура по окраске золей в свое время был практически единственным фотометрическим методом определения селена и теллура [43—45, 97, 98, 164, 172, 269, 296, 417, 423, 455, 723, 1078]. В настоящее время он все еще применяется при анализе относительно «простых» образцов с высокими содержаниями селена и теллура (десятые, сотые доли процента). Мешают этому определению золото, серебро, мышьяк. При более сложном составе образцов проводят предварительное выделение селена и теллура соосаждением с Fe(OH)3 1330]. Здесь предлагается вариант метода, разработанный Тараян и Авакян [369]. Предварительное выделение селена проводят из азотнокислого раствора солянокислым гидразином в присутствии катализатора — монохлориода (см. стр. 180). Теллур выделяют раствором SnCl2 и гидразином. Далее определяют селен и теллур
7*
195
по окраске коллоидных растворов в присутствии желатины и солей меди.
Выполнение определения. 1,0 г руды выдерживают без нагревания в течение нескольких часов с 25 мл конц. HNO3, затем постепенно нагревают на водяной бане до полного разложения пробы. Жидкость упаривают до объема 25 мл, приливают 40—50 мл горячей воды и отфильтровывают нерастворимый остаток. Промывают фильтр с осадком водой до тех пор, пока объем фильтрата не достигнет 80—90 мл.
К фильтрату, нагретому до 40° С, добавляют 10 мл монохлорида иода, немного фильтровальной массы, 15 мл 10%-ногО раствора солянокислого гидразина, перемешивают и, спустя час, отфильтровывают осадок селена, а фильтр с осадком промывают 3—4 раза малыми порциями теплой воды.
Воронку с фильтром, содержащим осадок селена, устанавливают на мерную колбу емкостью 25 мл_и растворяют осадок в 5—7 мл горячей НС1, к которой прибавлены 3 капли конц. HNO3. Промывают фильтр горячей водой до тех пор, пока объем раствора в колбе не достигнет 15 мл, охлаждают, вводят в колбу 2 капли 2%-ного раствора меди, 2 мл 1%-ного раствора желатины и постепенно при интенсивном перемешивании — 2 мл раствора SnCl2. Объем жидкости доводят водой до метки и через 15—20 мин. измеряют ее светопоглощение.
В фильтрат от селена вводят немного фильтровальной массы, нагревают до кипения, приливают 10 мл гидразина и постепенно при перемешивании — горячий раствор SnCl2 до полного восстановления и еще избыток его 5 мл. Кипятят 2—3 мин. и оставляют на 1—2 часа в теплом месте. Осадок теллура отфильтровывают, промывают 2—4 раза теплой водой и далее определяют теллур, как это описано для селена.
ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В СТАЛЯХ, СПЛАВАХ, ПОЛУПРОВОДНИКОВЫХ МАТЕРИАЛАХ
Разложение материалов
Сплавы, стали, полупроводниковые соединения разлагают HNO3 или царской водкой. Удаляют HNO3 из раствора выпариванием с НС1О4 [497, 955]. При обработке азотной кислотой сплавов или других материалов, содержащих большие количества сурьмы, образуется осадок метасурьмяной кислоты, которая адсорбирует теллуристую кислоту. Для удержания сурьмы в растворе добавляют винную кислоту. Углерод не растворяется в HNO3 и может быть отделен фильтрованием. Стали, содержащие вольфрам, растворяют в царской водке с добавлением нескольких миллилитров Н3РО4 [955].
Для растворения сталей применяют также H2SO4 (1 : 4) [244,' 364]. Селен и теллур при этом остаются в нерастворимом остатке вместе с карбидами, и потерь селена при разложении разбавленной H2SO4 не наблюдается [364]. Остаток, содержащий селен и теллур, затем растворяют в конц. HNO3 с несколькими каплями НО.
Иногда применяют обработку сталей раствором СиС12-2КС1. Селен и теллур вместе с серой и углеродом остаются в нерастворимом остатке, который затем растворяют в НС1О4 [1080]..
Применяют также метод растворения сталей в конц. НС1 в специальном аппарате, позволяющем количественно улавливать летучий SeCl4 [598].
196
Для разложения сплавов с теллуром применяют смесь конц. НС1 с бромом [1183]. При разложении сплавов теллура с неодимом возникают некоторые трудности при растворении в HNO3. Сплавы, в которых соотношение Nd : Те = 1 : 1, легко растворимы в разбавленной HNO3. Образцы, в которых превалирует теллур, растворяются только при длительном кипячении с царской водкой.
Отделение селена и теллура
При анализе сплавов последовательно выделяют селен и теллур осаждением SO2 при различной кислотности [115]. В сурьмяных сплавах, содержащих большие количества свинца, теллур восстанавливается неколичественно, поскольку в присутствии свинца образуется свинцово-теллуритный комплекс. Для связывания свинца и сурьмы добавляют винную кислоту. Для предупреждения загрязнения теллура сурьмой при выделении теллура гидразином из растворов, содержащих сурьму, осадок необходимо быстро фильтровать и хорошо промывать [597].
Количественно отделяют теллур от свинца, сурьмы и других элементов раствором SnCl2 из винно-азотнокислого раствора [279].
Для отделения теллура от меди и селена, а также от железа, мышьяка и висмута восстановление проводят молочным сахаром в слабощелочной среде в присутствии ферроцианида калия [278]. Если олово присутствует в больших количествах, оно выделяется вместе с теллуром. В качестве восстановителей теллура могут быть использованы также глюкоза, лактоза со SnCl2, галактоза, N2H4-•2НС1, аскорбиновая кислота, гипофосфит натрия [342, 521].
Селен отделяют от мышьяка осаждением сернистым ангидридом из 9%-ной НС1 или из 30%-ной H2SO4 [180]. Теллур отделяют от железа хроматографическим методом: раствор образца в HNO3 (2 : 100) пропускают со скоростью одну каплю в секунду через колонку со смолой Дауэкс 50W-X8 (50—100 меш); колонку промывают водой. В фильтрате фотометрически определяют теллур [1039].
Определение селена и теллура
При анализе сплавов, сталей и других материалов с высокими содержаниями селена и теллура часто применяют весовые методы.
Осаждение селена и теллура сернистым ангидридом и солянокислым гидразином с последующим взвешиванием в виде элементных применимо в тех случаях, когда анализируемый раствор не содержит сильных окислителей. Этот метод использовали при анализе систем Bi—Sb — Те — Se [1017] сплавов Bi—As — Se [321], сплавов теллура с редкоземельными элементами [338] ив других случаях [180, 299, 848).
197
Гравиметрический метод определения теллура в форме ТеОа [270, 338, 585] пригоден для определения теллура в азотнокислых растворах и в присутствии других окислителей. Для связывания висмута, сурьмы, редкоземельных и других мешающих элементов добавляют комплексон III. Выполнение определения см. на стр. 64.
Титриметрические методы широко применяют при определении больших содержаний селена и теллура. Бихро-матный метод используют при определении теллура в сплавах РЬ — Те, гетерополисоединениях [439, 495]. Для сурьмяных сплавов необходимо предварительно отделять теллур от сурьмы [278, 342]. Теллур в стали определяют титрованием иодотеллуритного комплекса тиосульфатом [244, 425]. Определению теллура этим методом не мешают Fe2+, Cu2+, Bi3+. Мешает Se. Ошибка метода 0,11—0,13%.
В сталях и некоторых соединениях селена последний определяют йодометрическим титрованием [186, 257, 364, 497, 911].
Метод потенциометрического титрованием селенистой кислоты перманганатом калия был применен в анализе простых и легированных селенистых сталей [17]. Ошибка титрования 0,15—0,17%.
Определение теллура в сталях методом йодометрического титровании. 0,5— 1,0 г стали (в зависимости от содержания теллура) растворяют при умеренном нагревании в 50—100 мл разбавленной H2SO4 (1 : 5). После полного растворения стружки нерастворимый остаток отфильтровывают и промывают несколько раз горячей водой. Фильтр с осадком помещают в колбу, в которой проводилось растворение навески, добавляют 10 мл конц. НС1 и 5—6 капель конц. HNO3 и нагревают 2—3 мин., время от времени встряхивая колбу, до полного разложения нерастворимого остатка. Добавляют 25—30 мл горячей воды и отфильтровывают фильтробумажную массу, которую промывают несколько раз горячим раствором разбавленной НС1 (1 : 20). Фильтрат нагревают до кипения, добавляют 3 г мочевины, и раствор SnCI2 сначала до обесцвечивания и затем ещё 5 мл избытка, кипятят 10 мин. для коагуляции осадка элементного теллура. Добавляют немного фильтробумажной массы, доводят раствор до кипения и дают ему 10—15 мии. постоять в теплом месте, после чего осадок отфильтровывают через фильтр «белая лента» и промывают горячим раствором НС1 (1 : 20).
Фильтр с осадком помещают в колбу, в которой велось осаждение, добавляют 10 мл концентрированной НС1 и 5—6 капель HNO3, раствор нагревают. К нему добавляют ~60 мл дистиллированной воды и 3 г мочевины. Раствор кипятят, затем охлаждают и после добавления 10 мл 2 М раствора KJ образовавшийся иодотеллуристый комплекс H2TeJ6 титруют 0,03 М раствором тиосульфата в присутствии крахмала.
При анализе сплавов, сталей, полупроводниковых материалов применяют фотометрические методы определения селена и теллура. Фотометрическое определение селена проводят с 3,3'-диаминобензидином [1039] и 2,3-диаминонафталином [282]. Определение селена в мышьяке, галлии и арсениде галлия возможно также с асимметричным дифенилгидразином [568].
Для фотометрического определения теллура в стали применяют иодидный метод. При этом определению теллура мешают окислители (Fe3+, Cu2+ и SeOg'), реагирующие в кислой среде с выделением свободного иода, и ион Bi3+, образующий с избытком иодида в 198
кислой среде ион [BiJ4]“, окрашенный в желтый цвет. Для отделения от селена использовано различное отношение этих элементов к иодиду калия [247]. Оптимальные условия следующие: 1А НС1, 0,2 М K.J, продолжительность стояния раствора 10 мин.
Фотометрический метод определения теллура в виде иодида [244]. Раствор, содержащий теллур, полученный после растворения в HNO3 и НС1 осадка, выделенного SnCl2, переводят в мерную колбу, добавляют 3 г мочевины, 10 мл 2 М раствора KJ и разбавляют дистиллированной водой до метки, хорошо перемешивают. После 20-минутного выдерживания в темном месте измеряют оптическую плотность раствора.
Холостой раствор в этом случае готовят следующим образом: в мерную колбу емкостью 100 мл добавляют 25—30 мл дистиллированной воды, 10 мл конц. НС1 и 2 капли концентрированной HNO3, вносят 3 г мочевины, прибавляют 10 мл 2 М раствора KJ, разбавляют водой и перемешивают.
Для построения калибровочной кривой в мерные колбы емкостью 100 мл добавляют отмеренные пипеткой различные объемы стандартного раствора теллура, содержащего 10 мг Те. Добавляют 25 мл дистиллированной воды, 10 мл коиц. НС1, 10 мл 2 М раствора KJ и 3 г мочевины. Хорошо перемешивают и оставляют в темном месте 20 мии. Измеряют оптическую плотность на ФЭК-М в кювете с толщиной слоя 0,5 см с синим светофильтром.
Для определения теллура в полупроводниковых системах Sb — In — Те применяют фотометрический тиомочевинный метод [197, 198, 225]. Сурьму связывают раствором NaF, который образует с ней прочный бесцветный комплекс.
Определение теллура в сплавах, сталях и других материалах возможно также измерением оптической плотности коллоидных растворов [91, 268, 1173].
ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В ЭЛЕМЕНТНОЙ СЕРЕ, ЧЕРНОВОЙ МЕДИ, ШЛАМАХ, АГГЛОМЕРАТАХ, ПЫЛЯХ, КОНЦЕНТРАТАХ И ДРУГИХ ПОЛУПРОДУКТАХ
Разложение материалов
Шламы растворяют в конц. HNO3. Азотную кислоту после полного разложения образца выпаривают с HaSO4 до паров SO3 (а лучше с НС1О4). Для удаления окислов азота раствор кипятят с мочевиной [251]. Во избежание потерь селена растворы не рекоменду-ется выпаривать досуха.
При растворении шламов в концентрированной HNO3 некоторые благородные металлы (Ir, Rh, Ru, Os) остаются в нерастворимом остатке, Au, Pt, Pd переходят в раствор вместе с селеном [249].
Для образцов, не разлагаемых кислотами,— некоторых пылей, электролитных шламов и других продуктов свинцового, цинкового и медного производств — возможно применение методов сплавления с NaaOa и содой в железном тигле при 750—800°С [226, 454]. В водную вытяжку количественно переходит селен, а теллур частично остается в нерастворимом остатке.
199
Для разложения материалов, содержащих значительные количества сурьмы и олова, применяют смесь азотной и винной кислот [278].
Колчеданы, огарки, элементную серу и шламы сернокислотных производств разлагают сжиганием пробы в трубчатой печи при 750°Свтоке воздуха или кислорода [13, 187]. Двуокись селена и двуокись теллура поглощают раствором НС1 (1 : 1), селен улавливают ватным тампоном. Этот метод удобно применять для анализа образцов с высоким содержанием железа.
Разложение продуктов, содержащих платиновые металлы, проводили методом хлорирования [214].
При анализе возгонов, шламов и пылей селен отделяют от серы сплавлением с KCN при 200°С в течение 10—15 мин. После растворения сплава в воде получают KSeCN и KSCN. В солянокислом растворе KSCN устойчив, a KSeCN разлагается с выделением элементного селена [144]. Этот метод использовали для удаления селена из металлургических пылей и шламов.
Для растворения элементной серы применяют смесь разбавленной азотной кислоты с бромом [200]. Из серы, содержащей значительные количества битумов, селен выделяли обработкой 10%-ным раствором NaOH. Битумы при этом не растворяются [13].
Отделение селена и теллура от мешающих элементов
Применяют соосаждение с гидроокислами железа и свинца и осаждение элементных селена и теллура в солянокислом растворе солянокислым гидроксиламином, гидразином, раствором SnCl2 или аскорбиновой кислотой (см. главу 5). Для отделения селена от Au, Pt и Pd восстановление солянокислым гидроксиламином проводят в щелочной среде, селен при этом не восстанавливается [249]. Прямое осаждение селена гидразином в сильносолянокислом растворе позволяет отделить теллур и мышьяк [239]. При определении селена в анодной меди было использовано катионообменное отделение селена от меди [163]. Для выделения селена из шламов и других полупродуктов использовали экстракцию диизопропилкетоном [692], метилизобутилкетоном [819].
Определение селена и теллура
Содержание селена в анодных шламах колеблется в пределах 8—20%, теллура — 0,3—3%. В пылях сернокислотного производства содержание селена достигает нескольких десятков процентов; в горячих электрофильтрах улавливается «•10-1% Se.
Для шламов, огарков, полупродуктов сернокислотного производств и других подобных продуктов самыми распространенными являются титриметрические методы определения селена и теллура [163, 187, 188, 200, 239, 249, 250, 251, 314, 523, 692, 1150] 200
(см. главу 4). Ускоренный титриметрический метод определения теллура в шламах цветной металлургии [248] заключается в со-осаждении селена и теллура с гидроокисью свинца, в восстановлении селена и теллура до элементных двухлористым оловом и в последующем йодометрическом титровании.
Выполнение определения. 0,5—1,0 г шлама обрабатывают при нагревании конц. HNO3. Затем к раствору объемом 50 мл добавляют 1,5 г азотнокислого свинца, предварительно растворенного в небольшом количестве воды, и избыток NH4OH (до выделения осадка).
После 2—3-минутного кипячения осадок отфильтровывают, промывают горячей водой и обрабатывают непосредственно на фильтре горячим раствором смеси NaCl н НС1 [1л насыщенного раствора NaCl + 300 мл НС1 (1 : 1)], собирая фильтрат в чистую колбу. Затем кипятят раствор до полного растворения выпавшего при охлаждении хлорида свинца, после чего Se и Те осаждают SnCl2 (сухой солью). Нагревают раствор до полной коагуляции осадка и просветления раствора.
Осадок селена и теллура отфильтровывают через бумажный фильтр и промывают горячим раствором НС1 (1 : 20). Осадок с фильтром помещают в колбу, в которой ранее производили осаждение, и обрабатывают его при осторожном нагревании смесью концентрированных кислот — 10 мл НС1 и 5—6 капель HNO3. После растворения осадка и превращения фильтра в кашицеобразную массу раствор разбавляют водой до 200 мл, добавляют 5 г мочевины и раздельно определяют селен и теллур.
Аликвотную порцию раствора кипятят в течение 2—3 мин. Затем к горячему раствору добавляют 2—3 г KJ и кипятят еще 15—20 мин.
По охлаждении раствора до 60—70° С осадок селена отфильтровывают через бумажный фильтр и промывают горячим раствором разбавленной НС1 (1 : 20), после чего осадок селена вместе с фильтром обрабатывают при слабом нагревании конц. НС1 (10 мл) и 3—4 каплями HNO3 в той же колбе, в которой проводили осаждение. Вслед за растворением осадка селена и превращением фильтра в кашицу раствор разбавляют дистиллированной водой до 150 мл, добавляют 3—5 г мочевины, кипятят, охлаждают, прибавляют отмеренный объем титрованного раствора тиосульфата; избыток его оттитровывают иодом в присутствии крахмала (индикатор). В фильтрате после отделения селена хлористым оловом осаждают теллур •— к раствору добавляют 10 мл конц. НО и после доведения раствора до кипения восстанавливают теллур хлористым оловом (сухой солью).
Восстановленный элементный теллур быстро коагулирует и после кипячения в течение 10—15 мин. выпадает в осадок. Затем осадок теллура отфильтровывают через бумажный фильтр и промывают горячим раствором разбавленной НС1 (1 : 20), после чего осадок теллура вместе с фильтром обрабатывают так же, как и селен, смесью конц. HNO3 и НО. Далее раствор разбавляют дистиллированной водой до объема 100 мл, добавляют 4—5 г мочевины и кипятят 2—3 мин. По охлаждении раствора добавляют 10 мл 30%-ного раствора KJ, и выделившийся иод оттитровывают тиосульфатом в присутствии крахмала. Одновременно проводят холостой опыт.
Определение селена и теллура в пылях сернокислотного производства, огарках сернистых руд методом последовательного титрования [192] основано на окислении четырехвалентного теллура избытком раствора бихромата калия в солянокислой среде с последующим оттитровыванием бихромата солью Мора в присутствии фенилантраниловой кислоты в качестве индикатора. После определения теллура в этом же растворе определяют селен тиосульфатным методом. Определение селена и теллура возможно в присутствии Fe, Си, Pb, Ag, Zn, Ba, Са, Mg, Cr, Mn, Al, Si, S, As. Чувствительность метода 0,001%. Относительная ошибка 0,5—1%.
201'
Выполнение определения. 0,5—10 г анализируемого материала (из расчета, чтобы в пробе содержалось по 5 мг Se н Те) разлагают 25—30 мл царской водки в стакане или фарфоровой чашке под стеклом при нагревании до превращения выделения окислов азота. К раствору добавляют 50—100 мл горячей воды и на> гревают до растворения азотнокислых солей. Нерастворимый остаток отфильтровывают через плотный и двойной фильтр, промывают водой и отбрасывают. Если содержание железа в пробе невелико, то в раствор добавляют любую соль трехвалентного железа в количестве, превышающем предполагаемое количество селена и теллура в 40—50 раз.
Приливают 20—25% -ный раствор NH4OH до запаха и образовавшийся осадок гидроокисей отфильтровывают через плотный фильтр, промывают водой и растворяют в горячей HCI (1 : 1). Раствор нагревают до кипения, и для осаждения селена и теллура прибавляют небольшими порциями при постоянном перемешивании 25%-ный раствор SnCl2 в 20%-ной НС1 до полного восстановления железа и, кроме того, — избыток 3—5 мл или 1,5—2 г сухого SnCl2. Раствор кипятят до его просветления и коагуляции осадка. Осадок отфильтровывают, промывают горячей HCI (1 : 20) и растворяют в 1О.и.?НС1с5—7 каплями HNO3 при нагревании вместе с фильтром. Раствор разбавляют водой до 100 мл и нагревают 1—2 мин. с 5 г мочевины. После охлаждения раствора селен и теллур определяют методом последовательного титрования.
Для этого в раствор добавляют 20 мл 0,01 N К2Сг2О?, 10 мл H2SO4 (1 : 1) и оставляют на 20—30 мин. (не больше). Прибавляют 2—3 капли фенилантраниловой кислоты и после появления фиолетового окрашивания избыток К2Сг2О? оттитровывают 0,01 N раствором соли Мора до появления зеленой окраски. Параллельно проводят холостой опыт. Эквивалент теллура равен 63,8.
После определения теллура добавляют 100 мл воды и 5 г сухого NH4F. Через 2—3 мин. к раствору приливают 50 мл 0,0\N раствора Na2S2O3, 2—3 мл 0,1% -ного раствора крахмала, и непрореагировавший тиосульфат оттитровывают 0,01 N раствором нода до появления слабо-синего окрашивания. Одновременно проводят холостой опыт. Эквивалент селена равен 19,74.
Для фотометрического определения микрограммовых количеств селена в настоящее время широко применяют о-диамины. 3,3'-Диаминобензидин — наиболее распространенный и изученный реагент из этой серии. Его используют при определении селена в сере [13, 801, серной кислоте 1616, 1006, 1109], пылях [1185], а также металлической меди и свинца [891]. Из других о-диаминов для фотометрического определения селена предложено использовать о-фенилендиамин [449, 451, 452]. Экстрагируе-мость соединения селена с о-фенилендиамином из кислой среды делает этот реагент более селективным по сравнению с 3,3'-ди-аминобензидином. Многие элементы, мешающие вследствие гидролиза определению селена с 3,3'-диаминобензидином, не мешают его определению по реакции с о-фенилендиамином. Влияние железа и висмута легко устраняется связыванием их в кислой среде (pH 1—2) в комплексы с фосфорной кислотой и комплексоном III. Это позволяет определять селен в сложных по составу продуктах цветной металлургии без его предварительного отделения.
Фотометрическое определение селена в продуктах цветной металлургии с о-феиилендиамином [451]. 0,25—1 г образца (в зависимости от содержания селе на) помещают в коническую колбу емкостью 100 мл, приливают 20 мл HNO (уд. вес 1,4) и оставляют на холоду (в случае сульфидных продуктов). Затем на гревают до разложения навески, упаривают раствор до 4—5 мл, приливают 10—
202
20 мл H2SO4 (1 : 1) (либо 5 мл НС1О4) и нагревают до выделения паров верного (хлорного) ангидрида. Приливают 5 мл воды и снова нагревают до появления паров. После вторичного выпаривания приливают 15—20 мл воды и нагревают раствор до кипения. По охлаждении раствор фильтруют в мерную колбу. Промывают осадок сначала 2%-ным раствором H2SO4 (2%-ным раствором НС1О4), а затем водой. Доводят фильтрат до метки и тщательно перемешивают.
Аликвотную часть раствора переносят в коническую колбу емкостью 100 мл, приливают необходимое количество воды (так, чтобы конечный объем не превышал 30—35 мл), 1 мл 85%-ной муравьиной кислоты, 5 мл концентрированной фосфорной кислоты, 0,5 мл 0,1 М раствора комплексона 111 и устанавливают pH 1, прибавляя по каплям NH4OH; приливают 3 мл 1%-ного раствора о-фенилендиамина, оставляют стоять 20 мин., после чего переносят раствор в делительную воронку, добавляют 5 мл бензола и экстрагируют в течение 2 мин. Экстракт сливают в сухую пробирку и измеряют оптическую плотность на спектрофотометре СФ-4 при 335 нм и толщине слоя 1 см.
Для построения калибровочной кривой в конические колбы емкостью 100 мл отмеривают типовой раствор селена (0,5—15 мкг), приливают воду (так, чтобы конечный объем составлял 30—35 мл) и далее поступают так, как описано в методике определения селена.
Большие содержания селена и теллура можно определять в е-совыми методами [103, 1068].
При определении селена и теллура в черновой меди применяли п о*л ярографический метод [898]. Колориметрическое определение селена и теллура в пылях свинцового производства было проведено Гладышевым [119] с тиомочевиной.
Для определения теллура в самородной сере предложен экстракционно-фотометрический метод с применением нафтилвисмутола [5-меркапто-(3-нафтил-2)-1,3,4-тиадиазолтиона] [68].
Выполнение определения. 0,3—0,9 г тонкор а стертой серы смачивают водой в стакане емкостью 100 мл. Прибавляют 5—10 мл HNO3 (уд. вес 1,4) и осторожно добавляют несколько капель брома. Нагревают на водяной бане и добавляют бром до полного разложения навески. Прибавляют 2—3 мл H2SO4 (уд. в. 1,86) и нагревают до появления белых паров. Охлаждают, остаток переносят в мерную колбу емкостью 50 мл и доводят водой до метки.
В стакан емкостью 100 мл отбирают аликвотную часть (20 мл) и при помощи буферного раствора устанавливают pH 4,8—5,0, контролируют по рН-метру. Переносят в делительную воронку, добавляют 5 мл 10%-ного раствора комплексона 111, 2 мл 0,1%-ного раствора реагента и экстрагируют 10 мл хлороформа в течение 2—3 мин.
Оптическую плотность экстракта измеряют на фотоэлектроколориметре (светофильтр № 2) относительно хлороформа. Экстракт предварительно фильтруют через сухой бумажный фильтр в сухую кювету с толщиной слоя 1,0 см.
Содержание теллура в образце находят по калибровочному графику, построенному, как указано на стр. 109, но при pH 4,8.
ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В ВОДАХ
В большинстве вод содержание селена составляет — 1 —10 мкг/л [334], поэтому для его определения требуется предварительное к онцентрирование.
203
Для концентрирования селена из морской воды (из 5 л) применен метод соосаждения его с Fe(OH)3 при pH 3—6,5 1333, 578]. Выделение селена составляет 94,7 ±0,17%. Контроль выхода селена проводили методом изотопного разбавления по изотопу Se75 [578].
При выпаривании больших объемов вод наблюдаются значительные потери селена — до 30% от общего его содержания [334]. Для уменьшения потерь при выпаривании больших объемов вод пробу в 10 л анализируемой воды обрабатывают при помощи Na2O2 и упаривают почти досуха [530]. Для уменьшения потерь селена при выпаривании в пробу вводят раствор NaOH до pH 8—9 [334],
Для отделения селена от других элементов был применен метод отгонки селена в виде бромида из остатка, полученного после выпаривания воды [530]. Отделение селена от железа и других элементов проводилось при помощи ионного обмена [333, 578].
Критический обзор методов определения селена и теллура в нефтяных водах был приведен в работе Коллинса [601]. Определение селена обычно проводят фотометрическим методом с 3,3"-ди-аминобензидином [219, 333, 578, 601, 1034, 1138]. Пятикратные количества U, Fe, Ni, Си, Со и Мп и стократные количества V, Мо и As не влияют на определение селена [578].
Однако применение 3,3'-диаминобензидина для определения селена в ряде случаев затруднительно, так как этот реактив не устойчив к окислителям и восстановителям, к яркому освещению и т. п. Несомненные преимущества имеет аналог 3,3'-диаминобен-зидина — 2,3-диаминонафталин. Этот реагент позволяет определять селен с более высокой чувствительностью — до 0,01 мкг в аликвотной порции раствора; поэтому в большинстве случаев отпадает необходимость выпаривания больших объемов вод, что неизбежно связано с потерей части селена.
Определение селена флуориметрическим методом с 2,3-диамино-нафталином возможно после выпаривания небольшого объема воды со смесью конц. HNO3 + НС1О4, прямым методом без предварительного отделения мешающих элементов (см. стр. 88).
В связи с тем, что 3,3'-диаминобензидин в ряде случаев не позволял добиться хорошо воспроизводимых результатов, для определения селена в водах до сих пор применяют метод измерения окраски коллоидных растворов. В этом случае восстанавливают селен до элементного состояния посредством SO2 и определяют Se количественно по интенсивности окраски красной аморфной формы Se [601]. Те восстанавливают до элементного состояния фосфорно-ватистой кислотой и определяют фотометрированием его красного гидрозоля при 400 нм. Метод позволяет определять 0,1 жгМТе [601].
При содержании в водах свыше 1 мкг/л селена возможно фотометрическое определение его после восстановления солянокислым гидразином в присутствии монохлориода как катализатора и окончательное определение восстановлением раствором SnCl2 в присутствии солей висмута и желатины [334].
204
Определение селена в водах по окраске коллоидных растворов [334]. 0,5—1 л воды осторожно выпаривают до объема 60—80 мл. Для уменьшения потерь селена при выпаривании в пробу вводят раствор NaOH до установления pH 8—9. Соли, образующиеся при выпаривании, обрабатывают конц. HNO3 до прекращения вскипания, после чего вводят 4 мл избытка HNO3.
Нерастворимый остаток отфильтровывают и промывают горячей подкисленной водой. Затем к нагретому фильтрату добавляют 10 мл раствора монохлориода, немного тонкой мацерированной бумаги и 10 мл 20%-ного раствора солянокислого гидразина. Спустя 1 час растворы фильтруют через фильтры, обработанные 5% -ной содой, промывают водой, подкисленной НС1,и несколько раз водой (окраска селена заметна при содержании не менее 10 мкг).
Затем селен на фильтре растворяют нагретой смесью кислот (10 мл конц. НО и 2 капли HNO3), смывают в стаканы емкостью 50 мл и промывают водой до объема 25 мл. Если осадок селена очень заметный, то для растворения следует взять большее количество смеси и дальше брать аликвотную часть раствора, сохраняя кислотность.
Одновременно готовят стандартную шкалу: 0; 0,5; 1; 2; 4; 8; 10; 12; 15л<кг/л<л из свежеприготовленного стандартного раствора с содержанием селена 1 мкг/мл. Во все стандарты одновременно вводят по 1 мл 1%-ного азотнокислого висмута, по 2 мл 1%-ного раствора желатины и по 2 мл свежеприготовленного 20%-ного раствора SnCl2, тщательно перемешивая растворы. Через 10—15 мин. образующиеся золи колориметрируют.
При анализе проб воды, содержащих более 10 мкг/л, можно исключить операцию выпаривания, пользуясь меньшим объемом воды.
Определение следовых количеств селена в воде возможно также с применением кольцевой бумажной хроматографии [530].
ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ, БИОЛОГИЧЕСКИХ МАТЕРИАЛАХ, ЖИВОТНЫХ ОРГАНИЗМАХ, ПОЧВАХ И ДРУГИХ ОБРАЗЦАХ
Разложение материалов
При определении селена в биологических объектах очень важен способ разложения образца. При обработке органических материалов следует учитывать возможность потерь селена в восстановительных условиях. Имеются данные о потерях селена во время просушивания травы при 50—60° С в течение 48 час. [1016]. Чтобы избежать потери селена, необходимо при разложении создавать сильно окислительные условия.
Было предложено несколько различных способов разложения органических веществ. Однако в большинстве случаев разрушение органики проходит медленно. Методом определения, который не требует полного разложения образца, является нейтронно-активационный метод, давно применяемый для определения малых содержаний селена и теллура в биологических материалах [552, 1092, 1145].
Пробы с большим количеством органических веществ трудно поддаются кислотному разложению. Так, при действии на расте-
205
ния смеси концентрированных HNO3 и H2SO4 разложение протекает медленно, требуется нагревание до 200° С, что приводит к потерям селена.
Смесь концентрированных HNO3 и НС1О4 (3 : 1) или (4 : 1) позволяет быстро разрушить растительные материалы, органические вещества, ткани и пр. [329, 605, 716, 724, 823, 1015, 1085, 10881. Эта смесь кислот является лучшей для разложения органических материалов [740].
При микроопределении селена в органических соединениях (в 5—30 мкг вещества) Файгль использовал окислительное разложение горячей конц. НС1О4 [661]. Иногда применяют тройную смесь концентрированных кислот — HNO3 + НС1О4 + H2SO4 [673, 1100]. Известен метод разложения органического вещества конц. H2SO4 в присутствии КС1О4 [844].
Для разложения органических веществ успешно используют метод сжигания в закрытой бомбе [490, 609, 639], в колбе Шёнигера или в колбе, наполненной кислородом [213, 609, 1134], в токе кислорода в специальном аппарате [747]. Различные варианты конструкции колбы Шёнигера описаны в работе Дей [639].
Сравнение методов кислотного разложения (HNO3 4- НС1О4) и сжигания в колбе с кислородом было проведено Уоткинсоном [1134]. Потери селена во время сжигания составляли всего 2%. Результаты опытов с применением различных методов разложения образцов (сжигание в колбе с кислородом и кислотное разложение) хорошо совпадают. Однако в случае применения метода сжигания в колбе с кислородом значительно затрудняется дальнейшее флуо-риметрическое определение селена. Получаются сильно завышенные невоспроизводимые результаты из-за флуоресценции несгоревших органических частичек. Для проведения флуориметрического определения селена в этом случае требуется дополнительная экстракция цинк-дитиолом [1134].
Для разложения образцов предложена также смесь НС1О4 и H2SO4 с Na2MoO4 [195, 610]. Эта смесь, обладая сильными окислительными свойствами, имеет сравнительно низкую температуру кипения. При нагревании с этой смесью 5 г образца окисление протекает довольно быстро (~ 1 час. 30 мин.), причем температура смеси обычно не превышает 132—140°С. Разложение образца в этом случае проводят в колбе Кьельдаля с обратным холодильником.
Отделение селена и теллура от мешающих элементов
При анализе биологических образцов применяют метод отгонки селена в виде бромида [643, 823]. После этого теллур восстанавливают в остатке от дистилляции селена раствором SnCl2. Определяют теллур обычными методами.
Наиболее распространенным является метод соосаждения селена и теллура в виде элементных с мышьяком при восстановлении 206
гипофосфитом натрия (см. стр. 181). В некоторых случаях, при больших содержаниях селена, последний восстанавливают до элементного посредством NH2OH-HC1 [1100] или насыщенным раствором SO2 в конц. НС1 [298].
Экстракция от 0,2 до 100 мкг Se в виде соединения с цинк-дитио-лом из сильнокислых растворов четыреххлористым углеродом или смесью СС14 и дихлорэтана протекает достаточно полно. По данным Уоткинсона [1133], можно удовлетворительно экстрагировать до 0,02 мкг Se. Использование экстракции цинк-дитиолом обеспечивает отделение ряда элементов, мешающих определению селена с 3,3'-диаминобензидином.
Определение селена
В биологических образах селен определяют фотометрическим или флуориметрическим методом с 3,3'-диаминобензидином [550, 639, 1088, 1096], 2,3-диаминонафталином [609, 656, 740, 940, 1134, 1140] и о-фенилендиамином [1119]. Наиболее чувствительным и селективным из этих реагентов является 2,3-диаминофталин, который позволяет проводить определение селена без отделения мешающих элементов. Не мешают определению селена с 2,3-диаминонафталином (в присутствии 5 мл 0,04 М раствора комплексона III и 1 мл 0,02 М KMnO4), lOOjHKa Cu2+, 250жкаУ4+, 14 мг Fe3’ [1134].
Возможно титриметрическое йодометрическое определение селена после сжигания образцов в токе кислорода в специальном аппарате [147, 747, 918]. При анализе органических соединений, которые могут быть разложены по методу Кариуса, возможно титриметрическое определение селена в нейтральном растворе в виде селенита серебра [553].
Флуориметрическое определение селена с 3,3'-диаминобензидином [329]. 1 г пробы смачивают небольшим количеством воды и добавляют 10 мл конц. HNOS н 5 мл НС1О4 (для навески 1 г) или 3 мл конц. HNO3 и 3 мл НС1О4 (для навески 0,1—0,2 г). Стакан закрывают часовым стеклом и нагревают на плитке. Если после полного удаления HNO3 и появления паров НС1О проба разложилась не полностью, добавляют еще немного HNO3 и выпаривают раствор снова до паров НС1О4. Нельзя выпаривать раствор досуха, так как в этом случае неизбежны потери селена.
После полного разложения пробы (раствор становится бесцветным) в стакан добавляют немного воды и снова выпаривают до паров НС1О4 (для удаления следов HNO3).
В стакан добавляют 10—15 мл воды и кипятят раствор 1—2 мин. После охлаждения отфильтровывают осадок через фильтр с «синей лентой» и промывают его несколькими небольшими порциями разбавленной НС1, а затем водой. К фильтрату (~30 мл) добавляют конц. НС1 до концентрации 6 N, 2 мл раствора соединения мышьяка (1 мг/мл) и 1 г твердого гипофосфита натрия. Раствор медленно нагревают на плитке до появления темного осадка элементного мышьяка. Раствор с выпавшим осадком кипятят 2—5 мин. до коагуляции осадка, через 1—2 часа осадок отфильтровывают и промывают сначала разбавленной НС1, а затем водой.
Фильтр с осадком переносят в стакан, где проводилось осаждение, и добавляют 10—15 мл разбавленной HNO3 (1 : 1). Раствор вместе с фильтром кипятят
207
2—5 мин., охлаждают и отфильтровывают от волокон фильтра в мерную колбу емкостью 50 мл.
Аликвотную порцию раствора помещают в колбу емкостью 100 мл, добавляют 3 мл НС1О4 и выпаривают раствор до паров НС1О4. Затем флуориметрически определяют селен с 3,3'-диамннобензидином, экстрагируя комплекс селена при помощи 5 мл толуола.
Метод применим для определения селена в образцах грибов, растений, почв и дает хорошо воспроизводимые результаты. Высокая чувствительность флуориметрического метода дает возможность определять селен из небольших навесок (0,020—0,300 г) с достаточно высокой точностью. Флуориметр-абсорбциометр ФАС-2 позволяет измерять флуоресценцию растворов, содержащих 0,1 мкг Se и более с относительной ошибкой ± 25%
Флуориметрическое определение селена в растениях с 2,3-днаминонафталином (1134].' Растение высушивают при 40° С и измельчают. Навеску 1 г отвешивают в стаканчики из боросиликатного стекла размером 16 X 2 см и добавляют 5 мл HNO3. В стаканчики вставляют воздушный холодильник 12 X 1,6 см и оставляют на 1 час, а лучше — на ночь. Затем стаканчики ставят на плитку и осторожно нагревают. Добавляют 2 мл 72%-ной НС1О4 через воздушный холодильник, трубочки-холодильники снимают, раствор нагревают до паров НС1О4 и сильно кипятят 15 мин.
1 г образца разрушают таким образом в течение 3 час. Добавляют 2 мл воды и стаканчик помещают на водяную баню на 1 час. Добавляют затем 2 мл 10% -ной НС1 и оставляют на кипящей водяной бане на 5 мин. для полного перехода шестивалентного селена в четырехвалентное состояние.
К остывшему раствору добавляют 2 мл 0,04 М раствора комплексона 111, 1 каплю 0,02%-ного индикатора крезолкрасного (водного) и 7 М NH4OH до pH 1. Далее раствор разбавляют до 50 мл 0,1 А НС1 и добавляют 5 мл 0,1%-ного очищенного раствора 2,3-диаминонафталина. Стаканчик помещают на водяную баню при 50° С на 20 мин.
После охлаждения раствор переводят в делительную воронку, в которую добавлено 10 мл циклогексана, и воронку встряхивают 1 мнн. Фазу циклогексана промывают два раза 25 мл 0,lN НС1. Циклогексановый слой центрифугируют 2 мин. и измеряют флуоресценцию при облучении образца светом с длиной волны Зббнм (первичный светофильтр), применяя вторичный светофильтр А=580—590 нм.
Г лава 7
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В ВЫСОКОЧИСТЫХ СЕЛЕНЕ И ТЕЛЛУРЕ
Получение высокочистых селена и теллура является важной задачей в связи со все растущей потребностью народного хозяйства в этих металлах [204, 205, 272, 273]. Элементный селен широко используется для изготовления выпрямителей, фотоэлементов, в электронографии. Селениды и теллуриды нашли применение в производстве фотосопротивлений, люминофоров, кристаллических счетчиков. На основе селена и теллура получены сплавы с высокими термо- и фотоэлектрическими характеристиками. Наибольшее значение имеет использование Se и Те в полупроводниковой промышленности. Однако микропримеси различных металлов, а также кислорода и галогенов оказывают большое влияние на свойства получаемых на основе селена и теллура полупроводниковых материалов. Так, микропримеси кадмия изменяют электропроводность селена. Таллий очень сильно влияет на кристаллизацию селена: чем больше таллия, тем более крупнозернистым получается селен. Наличие таллия сказывается также на теплопроводности и электропроводности селена. Примеси кислорода в селене в количествах 10~4—10-30о изменяют проводимость селена. Сильное влияние оказывают также следы влаги. Известно, что галогены изменяют электрические свойства металлического селена при содержании их порядка 1СГ2—10~4%.
Таким образом, разработка технологических схем получения полупроводниковых материалов, содержащих селен и теллур, всегда должна сопровождаться контролем степени чистоты исходных металлов с применением различных методов определения микропримесей в них. Обзор методов анализа высокочастотных селена и теллура был сделан Рябчиковым и Назаренко [327], методы анализа металлического теллура подробно рассматриваются в работах Юаса Тэру [1177, 1178, 1180].
В настоящее время для создания надежных полупроводниковых приборов необходимо контролировать содержание примесей в элементных селене и теллуре с чувствительностью до 10-6—10~9%. Определение следов элементов требует значительного усовершенст
1Д8 Зак. № 1476
209
вования существующих методов анализа и применения новых высокочувствительных и специфических реакций на элементы-примеси, которые позволили бы определять элементы при концентрациях до 10~10%. Основным методам анализа веществ особой чистоты была посвящена работа Яковлева [472].
Одним из наиболее чувствительных методов определения следов элементов является метод радиоактивационного анализа, с успехом применяемый для определения многих элементов (As, Си, Sb, Cd, Ga, W, Mo, P, S, Sn, Ni, TI, Zn, Au, Hg, Co, Cr, Ag) в высокочистых селене и теллуре. Однако активационный анализ не обладает необходимой универсальностью и простотой, требует дорогостоящего и сложного оборудования.
Широко распространен в производственной практике метод спектрального анализа. Но прямое определение примесей в высокочистых веществах ограничено недостаточной чувствительностью этого метода (10-4—10~5%). Для повышения чувствительности широко используется концентрирование примесей (экстракцией, отгонкой основного компонента и другими методами). В этом случае можно определить 10-6—10~7°6 элемента-примеси.
Методы спектрофотометрии и полярографии характеризуются относительно невысокой чувствительностью определения элементов-примесей (до п-10-6%).
РАДИОАКТИВАЦИОННЫЙ МЕТОД
Анализ образцов селена (или теллура) начинают с облучения в реакторе определенной навески вещества и эталона. Затем пробу промывают горячей разбавленной соляной кислотой для удаления механических примесей и растворяют в концентрированной азотной кислоте.
В ряде методов определения примесей в селене [156, 157, 433, 435, 1072, 1153] вначале выделяют основной компонент— селен (или теллур) — гидразином из солянокислого раствора (во второй и третий раз добавляют по 20 мг носителей — селенистой и теллуристой кислот для возможно более полного удаления из раствора активных селена и теллура), а затем проводят последовательное выделение элементов-примесей на носителях.
Известен также радиоактивационный метод определения Cd, Си, Ni, Те, Zn в чистом селене, по которому вначале выделяют примеси на неактивных носителях из селенсодержащего раствора. При этом каждый элемент-примесь определяют из отдельной навески. После радиохимической очистки отдельного элемента определяют химический выход и измеряют активность. Количество примесей рассчитывают путем сравнения с активностью эталонов.
Применяют также экстракционное разделение определяемых элементов на группы, удобные для у-спектрометрических измерений [119].
210
Для определения примесей As, Sb, Ni, Си, Cd, Au, Hg, Zn, Co, Cr и Ag в высокочистом теллуре Звягинцев и Шамаев предложили радиоактивационный метод [157, 433, 434]. В отдельной пробе определяют элементы, дающие сравнительно короткоживущие изотопы (As, Sb, Ni, Си, Cd и Аи), из другой навески определяют элементы, имеющие долгоживущие изотопы (Са, Hg, In, Со, Cr, Ag, Se). Активационному определению микроколичеств мышьяка в селене мешают реакции (п, р), идущие на ядрах Se74, Se76, Se77. При очень малом содержании мышьяка необходимо предварительно отделять селен осаждением при помощи SO2 [1029, 1030].
При определении микроколичеств хлора и брома в селене их отгоняют при растворении облученной пробы, поглощая НО и НВг раствором AgNO3 [507, 509, 510].
Активационное определение фосфора, серы и хлора в селене возможно с чувствительностью соответственно 0,0003, 0,002, 0,008 мкг [1031].
Таким образом, к настоящему времени разработаны активацион-•ные методы анализа высокочистых селена и теллура на элементы-примеси As, Си, Sb, Cd, Ga, W, Mo, P, S, Sn, Ni, Те, Au, Hg, Co,Cr,Ag. Однако для других элементов, у которых периоды полураспада исчисляются несколькими минутами и меньше (К, Al, Mg, Ti, U, Nb), метод нейтронно-активационного анализа мало применим.
СПЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Методом прямого спектрального анализа с применением обычных источников возбуждения спектра (дуга, искра, пламя) можно обнаружить большинство элементов в количествах 10~2—10-5% [130, 258, 315, 399, 470, 636].
Для повышения чувствительности определения примесей в настоящее время часто применяют предварительное концентрирование, что позволяет повысить чувствительность в 10—100 раз.
Для более рационального использования вещества, которое попадает в зону возбуждения спектра, предложено добавлять к пробе соли щелочных металлов, которые, снижая эффективный ионизационный потенциал, повышают интенсивность спектральных линий. Этим методом была повышена чувствительность определения примесей Pb, Си, Bi, Ti и некоторых других элементов в селене [119]. Следует заметить, однако, что добавки NaCl снижают в 2—5 раз чувствительность определения Те, Hg, As, Cd.
Метод вдувания исследуемой пробы в разряд током воздуха был применен Давлетшиным и Айдаровым [132] для определения микропримесей Ag, Си, Ni, Al и Fe в чистых селене и теллуре. При этом обеспечивалось наиболее эффективное и равномерное сжигание образца, что позволило определить примеси при концентрациях 5-10~6%.
!/29 Зак. № 1476
211
При определении ряда элементов (Ag, Al, Bi, Со, Cd, Cu, Fe, Sn, Те, Ga, Hg, In, Mn, Ni, Pb, Sb) в чистом селене по методу трех эталонов максимальная чувствительность определения примесей равнялась 10~5% [407]. Оптимальные условия спектрального определения Ag, Sr, Pb, Sb, Mn, Hg, As, Zn, Те, Cd, Ni, Bi, Tl, Cu в селене высокой чистоты были подобраны с применением дуги постоянного тока [258]. Добавление буферных веществ (порошка А1 или Li2CO3) дает положительные результаты при определении Sn, Pb и Т1.
Чувствительность определения Zn и Cd мало зависит от нали-чия буфера, а определение Pb, As, Hg и Fe рекомендуется проводить без буфера.
Влияние формы существования примеси в селене и теллуре высокой чистоты на интенсивность спектральных линий было изучено в работе Петрова и Айдарова [301].
, При изготовлении эталонов примеси в виде металлов вводят в порошкообразные селен и теллур и сплавляют под вакуумом.
Для концентрирования примесей при анализе высокочистого теллура авторы [150, 839] применили метод последовательной экстракции 8-оксихинолином(0,1- %-ный раствор в хлороформе) и дитизоном (0,01%-ный раствор в хлороформе) при различных значениях pH (3, 5, 7 и, наконец, 10). При этом в органическую фазу переходит большинство элементов, а селен и теллур не экстрагируются. Из объединенного экстракта отгоняют хлороформ, и в остатке определяют примеси спектральным методом. Чувствительность определения микропримесей из навески 10—20 г равна 10~6—10-7%. Однако при последующем определении таких элементов, как Cu, Fe, Al и Mg, наблюдаются очень высокие значения поправок холостого опыта.
' Повысить чувствительность спектрального метода можно также отгонкой селена в виде SeO2 [49, 118, 408, 409]. Примеси концентрируют на угольном порошке с добавлением хлорида натрия и хлорида калия для стабилизации температуры разряда и повышения чувствительности. Порошок высушивают и помещают в кратер угольного электрода. Спектры возбуждают в дуге переменного тока при I = 12а. При этом возможно определение большинства элементов в количествах до 10~5—10“®%.
Проверка полноты концентрирования элементов при отгонке селена по результатам опытов «введено — найдено» установила, что для Au, Mg, Sn, Sb, Bi, Те, Al, Cu и Ag потери составляют 20—30%, для Cd, As, Tl, Pb, Ti, Mn, — 50—60% и для Cu и Ni — 80% [118].
“ | Концентрирование примесей после отгонки SeO2 в специальной трубчатой печи возможно также проводить на подложке из сульфата стронция.
' При последующем спектральном определении элементов были достигнуты следующие значения чувствительности (в %):
212
Си (по линии 3279,9 А) .5-10 7
Ag (3280,7 А)..........., 2-10-’
Al (3082,2А)..............2-10-е
Bi (3067,7 А)...........5-10;в
Ni (3050,8 А)............З-10-e
Fe (3020,6 А).............1-106
Ga (2943,6 А)............3.10-6
Мп (2801,1 А).............4-10-’
Mg (2779,8А)............1-10-»
Sb (2598,1 А)..........3-10~5
Cd (2288,0 А)...........5-Ю-’
Те (2385,8 А)..........2-10~5
As (2349,8 А)..........5-10“5
In (3256,1 А)..........5-10-»
Со (2407,2А)...........5-10-’
ПОЛЯРОГРАФИЧЕСКИЕ И КУЛОНОМЕТРИЧЕСКИЕ МЕТОДЫ
Полярографическим методом можно определить микропримеси с чувствительностью (КГ5—КГ’%). Этим методом были определены в высокочистом селене примеси Cu, Cd, Те, Pb, Т1 и Fe [567]. После отгонки SeBr4 из раствора, содержащего бромистоводородную кислоту, остаток растворяют в специально очищенной щавелевой кислоте, которая в дальнейшем служит фоном при полярографировании.
Применение специальной микроячейки (на 1 мл раствора) позволяет определять весьма малые количества элементов-примесей в селене: 2-10-5% Си, Ы(Г6% Cd, 2-КГ6 % РЬ, 5-10-®% Те, 1,5.10-’% Т1, 4-10~5% Fe.
Примеси Си и РЬ в высокочистом селене определяют методом квадратно-волновой полярографии [796]. Селен отгоняют из сернокислого раствора, а в остатке определяют медь и свинец на фоне
НС1О4.
Известны полярографические методы определения иода и селена в теллуре [228, 499].
Разработан кулонометрический метод определения микропримесей железа и алюминия (10“4%) в селене высокой чистоты. Метод основан на электрогенерировании брома окислением бромирован-ных 8-оксихинолинатов металлов [201]. Для определения Си, РЬ, Cd, Ni и Zn в теллуре высокой чистоты проводят предварительное отделение теллура экстракцией метилизобутилкетоном из 6—7N НС1 или из смеси 7N НО и 0,5 N HNO3 [818, 1101]. Для последующего определения Си и РЬ применен метод квадратно-волновой полярографии [818], а также метод дифференциальной полярографии [958].
СПЕКТРОФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Чувствительность определения микропримесей при применении спектрофотометрических методов относительно невысока — 10~3— КГ4 %. Однако эти методы все чаще находят широкое применение при контроле технологически? процессов и изучении свойств полупроводниковых селена и теллура.
9*
213
Определение микропримесей в селене
Образцы селена обычно растворяют в концентрированной азотной кислоте, и раствор выпаривают досуха. Возможно растворение образцов в смеси соляной и азотной кислот.
Для отделения основной массы селена последний отгоняют в виде SeBr4 из раствора, содержащего НВг; отгонку основной массы селена проводят и из сернокислого раствора. После разложения образца в HNO3 селен можно осадить при помощи SO2 или гидразина.
Существуют методы, в которых сразу после разложения образца примеси концентрируют экстракцией или осаждением. Методом ионного обмена могут быть отделены макроколичества селена от Си, Cd, Fe, Al, Hg, Mg, Bi, Те, СГ, Вг“,5ОГ[177, 1059]. Послехро-матографического концентрирования может быть достигнута чувствительность определения металлов до п-10“®%.
При пламенно-фотометрическом определении щелочей и щелочноземельных элементов и галлия селен отгоняют в виде SeBr4; чувствительность определения Ь10“®% Na, 1-10"5%, К, Са, Li [1059].
Для отделения примесей Си, Ni, Со и РЬ от селена их экстрагируют в форме карбаматов четыреххлористым углеродом [357]. В реэкстракте каждый элемент определяют обычными фотометрическими методами.
При определении алюминия применяют экстракцию оксихинолином в хлороформе после отделения примесей в виде диэтцлдитиокарбаматов. Метод позволяет определять 10~5% А1 в селене [930]-
Для отделения таллия от селена и других элементов экстра, гируют таллий изопропиловым эфиром. Окончательно определяют микропримеси (>10“5%) спектрофотометрически с родамином С [988].
Небольшие количества ртути в металлическом селене определяют фотометрически после экстракционного отделения хлороформом комплекса ртути с дитизоном [838, 992]. При использовании производного дитизона—1,5-ди-(Р)-нафтилтиокарбазона — в присутствии комплексона III и KSCN чувствительность определения ртути может быть повышена до 4-10“®% из навески 5 г. Ошибка определения 5—25% [385].
Определение мышьяка в селене основано на экстракционном отделении мышьяка в виде карбамата и определении в виде молибденовой сини. Ошибка для количества^ 1,5 мкг Se составляет < 10% [640, 914].
Серу в селене определяют измерением светопоглощения суспензии BaSO4 после отгонки селена в виде бромида. Чувствительность метода 5-10-4% [481, 1166]. Известен метод определения серы в селене, который основан на предварительном ионообменном разделении серы и селена в колонке на смоле Дауэкс 1-Х8 в С1-форме [931]. Серу в виде H2SO4 элюируют \N НС1, раствор обрабатывают суспен. 214
зией ВаСгО4, и выделившийся в эквивалентном количестве СгО42-определяют спектрофотометрически с дифенилкарбазидом. Чувствительность метода 5-10~5% из навески 2 г.
Небольшие количества хлора в селене (10“4%) могут быть выделены избытком раствора AgNO3. Осадок AgCl восстанавливают в щелочном растворе гидразином, серебро потенциометрически оттитровывают роданидом. Бром определяют в виде трибромфлуоресцеи-на в фильтрате после восстановления серебра [986]. Известны методы определения хлора и брома в селене после отделения в виде AgCl и AgBr и экстракции серебра дитизоном. Расчет содержания хлора и брома проводят по количеству серебра, которое определяют фотометрическим методом с п-диметиламинобензилиденроданином [841, 908].
Бусевым и сотрудниками [64] разработан метод определения следов теллура (10-5—10'6%) в полупроводниковом селене. Метод основан на образовании соединения теллура с 3,5-дифенилпиразо-.лин-1-дитиокарбаматом натрия, которое экстрагируется хлороформом в широком интервале кислотности — от 12 А НО до pH 9,0. Селен и следы сопутствующих элементов маскируют цианидом, комплексоном III, тартратом в щелочной среде при pH 8,0—9,0.
Экстракционно-фотометрический метод определения теллура в виде бромидного комплекса с диантипирилпропилметаном в дихлорэтане позволяет определять теллур в селене после экстракционного отделения хлоридного комплекса теллура с этим реагентом [61]. Для количеств 10-1—10~3% Те в полупроводниковом селене применяют также методы, основанные на образовании окрашенных золей теллура [284, 1159]. Однако в этом случае необходимо тщательное отделение следов селена.
Определение микропримесей в теллуре
Образец теллура в зависимости от схемы последующих операций можно растворять в концентрированной серной кислоте с добавлением небольшого количества концентрированной азотной кислоты или в смеси соляной и азотной кислот. Основную массу теллура отделяют в виде теллуровой кислоты, если проводят разложение пробы концентрированной азотной кислотой.
Для определения следов селена (10~4—10~5%) в металлическом теллуре предложен 3,3'-диаминобензидин [1178]. Основную массу теллура предварительно отделяют в виде Н2ТеО3 при pH 3—4 или осаждают следы селена с небольшой примесью теллура гидразином в 2—2,5 N НС1 [851, 1128].
Определение меди (104—1(Г5%) в металлическом теллуре основано на экстрагировании хлороформом комплекса с диэтилдитио-карбаМатом (1180) или неокупроином [783, 969]. При очень малых содержаниях меди навеску увеличивают до 10 а и теллур предварительно отделяют в виде теллуровой кислоты. Применение эфирной
экстракции для железа позволило определить с 8-оксихинолином 0,001% Fe 111791.
Микроколичества сурьмы (10"5%) в теллуре высокой чистоты можно определить по реакции с родамином С. Для отделения от теллура и других элементов вначале отгоняют сурьму в виде SbBr3, а из дистиллята экстрагируют хлороформом соединение сурьмы с диэтилдитиокарбаматом диэтиламмония. После разрушения органики пятивалентную сурьму экстрагируют изопропиловым эфиром и спектрофотометрируют при 562 нм [783]. Для определения сурьмы предложен 4,4-бис-(М-метил-М-бензиламинофенил)антипи-рилкарбинол [71].
В случае определения малых количеств свинца (КГ5%) основную массу теллура выделяют в виде ТеО2, а затем комплекс свинца с дитизоном экстрагируют бензолом или хлороформом при pH 10. Окончательное определение проводят спектрофотометрически или поля-рографически [782].
Экстракцию комплекса висмута с дитизоном применяют для отделения этого элемента от больших количеств теллура при pH 10. Спектрофотометрическим методом с дитизоном определяют 10~5% висмута [782].
Для отделения мышьяка и олова используют метод дистилляции их хлорнокислого раствора. Мышьяк в дистилляте восстанавливают до трехвалентного, и экстрагируют хлороформом комплекс с диэтилдитиокарбаматом диэтиламмония. Мышьяк определяют в виде синего мышьяковомолибденового комплекса, а олово — фотометрически фенилфлуороном [781]. Этим методом можно определить КГ5% мышьяка и олова в образцах металлического теллура. Определение серы в теллуре основано на отделении теллура экстракцией трибутилфосфатом из 12 N НО и окончательном определении серы в форме BaSO4 [367].
Микропримеси алюминия в теллуре определяют спектрофото-метрированием комплекса с 8-оксихинолином после экстракции его четыреххлористым углеродом [1181].
Разработан экстракционно-фотометрический метод определения иода в теллуре на основе образования окрашенного комплекса с меркаптобензтиазолом в присутствии бмс-бензтиазолил-2-дисуль-фида и ацетонитрила [850].
ЛИТЕРАТУРА
t. Агасян Л. Б., Агасян П. К-, Николаева Е. Р. Вести. МГУ, 4, 96 (1966).
2. Агасян И. К., Денисова А. Н., Агасян Л. Б., Николаева Е. Р. Зав. лаб., 34, 129 (1968).
3. Агасян П. К., Николаева Е. Р. Зав. лаб., 29, 773 (1963).
4. Агасян Л. Б., Николаева Е. Р., Агасян П. К. Ж. аналит. химии, 22, 904 (1967).
5. Агасян Л. Б., Николаева Е. Р., Агасян П. К., Лебедева 3- М. Изв. вузов, Химия и хим. технол. 10, 1315 (1967).
6. Агасян Л. Б., Юрченко А. Г., Агасян П. К. Ж. аналит. химии, 22, 229 (1967).
7. Аксененко В. М., Онуфриенок И. П., Новикова Е. С. Сб. «Теория и практика полярографического анализа». Кишинев, «Штиинца», 1962, стр. 209.
8. Алекперов А. И. Азерб. хим. ж., 1961, 133.
9. Алекперов А. И., Азерб. хим. ж., 1963, 73.
10. Алекперов А. И., Мирзоева Л. А. Азерб. хим. ж., 1962, 133.
11. Алекперов А. И., Новрузова Ф. С. Азерб. хим. ж., 1966, 117.
12. Алекперов А. И., Новрузова Ф. С. Сб. «Методы анализа веществ высокой
чистоты». М., «Наука», 1965, стр. 452.
13. Алексеева А. Н., Красножен В. А. Авт. свид. СССР 160026 (1964); Бюлл. изобр., № 2, 65 (1964).
14. Алексеева А. Н., Семенова Л. В., Шушканова Н. М. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 426.
15. Алиев А. И., Дрынкин В. И., Лейптунская Л. И., Касаткин В. А. Ядерно-физические константы для нейтронного активационного анализа. М., Атом-издат, 1969.
16. Анализ минерального сырья. Л., Госхимиздат, 1956, стр. 840.
17. Андреев А. С. Труды Ленингр. политехи, ин-та им. М. И. Калинина, 201, 58 (1959).
18. Анисимов С. М., Савальский, С. Л., Осипов А. П. Изв. вузов. Цвет, мет., 1, 101 (1961).
19. Анисимов С. М., Шулаков П. Г. Сб. «Методы анализа платиноидов, золота и серебра», М., Металлургиздат, 1960, стр. 183.
20. Аницкий И. В. Ж. неорган. химии, 2, 1342 (1957).
21. Арефьева Т. В., Васильева Л. Н. Труды Гинцветмет, 19, 669 (1962).
22. Арефьева Т. В., Позднякова А. А. Труды Гинцветмет, 8, 137,(1957).
23. Арефьева Т. В., Юстус 3. Л. Труды Гинцветмет, 19, 730. (1962).
24. Аришкевич А. М., Усатенко Ю. И., Волошина В. В. Ж- аналит. химии 24, 1069 (1969).
25. Артюхин П. И., Гильберт Э. И., Пронин В. А. Изв. СО АН СССР, серия хим., 3, 138 (1966).
26. Арстамян Ж- М., Тараян В. М. Арм. хим. ж., 19, 590 (1965).
27. Бабина Ф. Л.,Карабаш А. Г., Пейзулаев Ш. И., Семенова Е. Ф. Ж.аналит. химии, 20, 501 (1965).
28. Бабко А. К., Жаровский Ф. Г. Зав. лаб., 25, 42 (1959); 28, 1287 (1962).
29. Бабко А. К., Митюрева Т. Т. Ж- неорган. химии, 3, 2082 (1958).
30. Бабко А. К., Митюрева Т. Т. Ж. неорган. химии, 6, 421 (1961).
217
31. Багбанлы И. Л., Гусейнов И. К,. Докл. АН Азерб.ССР, 18, 27 (1962).
32. Багбанлы И. Л., Гусейнов И. К.., Албендов А. А., Багбанлы Р. И., Уч. зап. Азерб. гос. ун-та, серия хим., 1, 18 (1966).
33. Баталин А. X. Ж. аналит. химии, 15, 507(1960).
34. Беликова Т. Е., Каплан Б. Д., Ширяева О. А. Зав. лаб., 33, 1365 (1967).
35. Белопольская Т. Л. Материалы по геологии и полезным ископаемым Северо-Востока СССР (Магадан), 14, 165 (1960).
36. Белопольская Т. Л. Методы химического анализа минерального сырья 11. 35 (1968).
37. Белопольская Т. Л. Тезисы ВСЕГЕИ, новая серия, 117, 85 (1964).
38. Белопольская Т. Л. Тезисы ВСЕГЕИ, новая серия, 125, 62 (1966).
39. Белопольская Т. Л., Сериков И. В. Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, Изд-во МИТК, 1968, стр. 37.
40. Белый М. У., Кушниренко И. Д. Ж- прикл. спектроскоп., 9, 442 (1968).
41. Беляев А. В., Птицын Б. В. Изв. СО АН СССР, серия хим., 11, 144 (1965).
42. Блох М. А. Хронология важнейших событий в области химии. М.— Л., Госхимиздат, 1940.
43. Блюм И. А., Глазкова А. Ф. Бюлл. ВИМС, 5 (181), 10 (1958).
44. Блюм И. А., Глазкова А. Ф. Сб. «Химические, физико-химические и спект-тральные методы исследования руд редких и рассеянных элементов». М., Госгеолтехиздат, 1961, стр. 13.
45. Блюм И. А., Глазкова А. Ф., Сысоева Г. Л. Бюлл. ВИМС, 8 (184), 1 (1958).
46. Блюм И. А., Глазкова А. Ф., Сысоева Г. Л. Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 594.
47. Блюм И. А., Чувилева А. И. Зав. лаб., 35, 1153 (1969).
48. Бобиков П. И., Борбат В. Ф., Бугаев А. В., Долгих В. И. Цветные металлы, 1, 54 (1963).
49. Богоявленская А. Н., Геркен Е. Б., Полякова В. В. Труды Гинцветмет, 28, 138 (1968). -
50. Божевольнов Е. А., Николаева К- И., Мамедов Ф. М. Сб. «Методы анализа химических реактивов и препаратов», вып. 11. М., изд. ИРЕА, 1965, стр. 77.
51. Божевельнов Е. А., Николаева К- И., Мамедов Ф. Л-1.С6. «Методы анализа химических реактивов и препаратов», вып. 16. М., «Химия», 1969, стр. НО.
52. Боровик С. А. Докл. АН СССР, 65, 316 (1949).
53. Бояджиева Р. РЖХим., 21Г116 (1963).
54. Бронштейн А. Н. Бюлл. научно-технической информации Министерства геологии и охраны недр СССР, 5—6 (39—40), 123 (1962).
55. Бусев А. И. Применение соединений двухвалентного хрома в аналитической химии. М., ВИНИТИ, 1960.
56. Бусев А. И. Taianta, 11, 485, (1964).
57. Бусев А. И., Бабенко Н. Л. Ж. аналит. химии, 18, 972 (1963).
58. Бусев А. И., Бабенко Н. Л. Ж. аналит. химии, 19, 926 (1964).
59. Бусев А. И., Бабенко И. Л. Сб. «Методы анализа веществ высокой чистоты».
М., «Наука», 1965, стр. 451.
60. Бусев А. И., Бабенко Н. Л., Хоанг Минь-тяу. Ж. аналит. химии, 18, 1094 (1963).
61. Бусев А. И., Бабенко Н. Л., Чепик М. Н. Авт. свид. СССР 163006 (1964); Бюлл. изобр., № 11, 60 (1964).
62. Бусев А. И., Бабенко Н. Л., Чепик М. Н. Ж. аналит. химии, 19, 871 (1964).
63. Бусев А. И., Бабенко Н. ЛЧепик М. Н. Ж. аналит. химии, 19, 1057 (1964).
64. Бусев А. И., Бырько В. М., Прохорова Л. Н. Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 626.
65. Бусев А. И., Гусейнов И. К.., Багбанлы И. Л. Азерб. хим. ж., 6, 92 (1966).
66. Бусев А. И., Иванютин М. И. Труды Комиссии по аналит. химии АН СССР, 11, 172 (1960).
67. Бусев А. И., Симонова Л. Н. Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, изд-во МИТК, 1968, стр. 3.
218
68. Бусев А. И., Симонова Л. Н. Ж. аналит. химии, 22, 1728, 1850 (1967).
69. Бусев А. И., Симонова Л. Н., Заюкова И. Д. Труды Комиссии по аналит. химии АН СССР, 17, 218 (1969).
70. Бусев А. И., Симонова Л. И., Заюкова Н. Д. Вести. МГУ, серия хим., 1, 119 (1968).
71. Бусев А. И., Типцова В. Г., Богданова Е. С., Андрейчук А. /И. Ж. аналит. химии, 23, 812 (1968).
72. Бусев А. И., Хоанг Минь-тяу. Ж. аналит. химии, 17, 1091 (1962).
73. Бусев А. И., Хоанг Минь-тяу. Ж. аналит. химии, 18, 360 (1963).
74. Бусев А. И., Хоанг Минь-тяу. Ж. аналит. химии, 18, 1370 (1963).
75. Бусев А. И., Хоанг Минь-тяу. Ж. неорган. химии, 7, 88 (1962).
76. Быков И. Е., Горшкова Л. С. Зав. лаб., 25, 674 (1959).
77. Быков И. Е., Горшкова Л. С. Изв. СО АН СССР, серия хим., 4, 62 (1958).
78. Быков И. Е., Зелянская А. И., Горшкова Л. С. Труды Ин-та металлургии УФАН СССР, 2, 275 (1958).
79. Быков И. Е., Плетнева И. А. Труды Ин-та металлургии. УФАН СССР, 8, 111 (1963).
80. Ван Ин-цзянь, Тун Фэн-цинь. РЖХим., 11, 42433 (1960).
81. Варанд В. Л. Научные труды Иркутск, ин-та редких металлов, 10, 97 (1961).
82. Васильев П. И., Воронкова М. А. Бюлл. научно-технической информации • Министерства геологии и охраны недр. СССР, 1 (56), 14 (1965).
83. Васильев П. И., Воронкова М. А. Бюлл. научно-технической информации Министерства геологии и охраны недр СССР, 1 (56), 17 (1965).
84. Васильев П. И., Воронкова М. А. Сб. «Л1етоды химического анализа минерального сырья». М., «Недра», 8, 171 (1965).
85. Васильев П. И., Воронкова М. А. Сб. «Методы химического анализа минерального сырья». М.. «Недра», 8, 177 (1965).
86. Васильев П. И., Воронкова М. А., Белая Ф. Д. Бюлл. научно-технической информации Министерства геологии и охраны недр СССР, 1 (56), 21 (1965).
87. Васильева Л. И., Юстус 3. Л. Зав. лаб., 34, 1 (1968).
88 Верховод Б. Н., Хижняк Б. И. Сб. «Спектральный анализ в геологии и геохимии». М., «Наука», 1967, стр. 165.
89. Вершинина Ф. И., Вихарев Л. И. Сборник научных трудов Всес. и.-и. горно-металлург. ин-та цветных металлов, 9, 186 (1965).
90. Веселого Л. И., Герцева Н. С. Труды Комиссии по аналит. химии АН СССР,
16, 150 (1968).
91. Весене Т. Б. Зав. лаб., 35, 32 (1969).
92. Виноградов А. П. Геохимия редких и рассеянных элементов в почвах. М . Изд-во АН СССР, 1957.
93. Владимирова В. М., Давидович И. К., Кучмистая Г. И., Разумова Л. С. Зав. лаб., 29, 1419 (1963).
94. Владимирова В. М., Кучмистая Г. И. Зав. лаб., 30, 528 (1964).
95. Владимирова В. М., Кучмистая Г. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 139.
96. Волков С. Т. Зав. лаб., 5, 1429 (1936).
97. Волков С. Т. Определение селена в серных рудах и свободной сере. М. ОНТИ 1936.
98. Волков С. Т. Определение селена и теллура в рудах и концентратах, содержащих золото. М., Госгеолиздат, 1945.
99. Воронкова М. А. Методы химического анализа минерального сырья 11 51 (1968). ’
100. Воронкова М. А. Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, Изд-во МИТК 1968, стр. 44.
101. Воронкова М. А., Буткина Т. А. Бюлл. научно-технической информации Министерства геологии и охраны недр СССР, 3, 29 (1967).
102. Воронкова М. А., Сидоренко Г. А. Ж. аналит. химии, 7, 1085 (1967)
103. Вылчев М. Ц. Зав. лаб., 34 , 393 (1968).
104. Габович А. М. Труды Комиссии по аналит. химии АН СССР, 16, 155 (1968).
105. Гаев А. И. Изв. Уральск, политехи, ин-та, 3, 95 (1929).
219
106. Гайбакян Д. С., Атурян М. М. Арм. хим. ж., 21, 1015 (1968).
107. Гайбакян Д. С., Дарбинян М. В. Изв. АН АрмССР, хим. н. 16, 211 (1933).
108. Гайбакян Д. С., Дарбинян М. В. Изв. АН АрмССР, хим. н., 17, 501 (1964).
109. Ганелина Е. Ш. Ж. аналит. химии, 18, 551 (1963).
ПО. Ганелина Е. Ш. Ж- неорган. химии, 7, 1570 (1962).
111. Ганелина Е. Ш., Неревяткина Н. И. Ж. неорган. химии, 10, 894 (1965).
112. Ганиев А. Г., Сильванович Ю. А., Долматова Т. Сб. «Активационный анализ элементного состава геологических объектов». Ташкент, «Фан», 1967, стр. 121.
113. Гельман Е. М. Ж- аналит. химии, 23, 736 (1968).
114. Гиганов Г. П., Спутнова И. А., Вершинина Ф. И. Ж. неорган. химии, 12, 755 (1967).
115. Гиллебранд В- Р., Лендель Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М., «Химия», 1966, стр. 147, 390.
116. Гильберт Э. Н., Пронин В. А. Изв. СО АН СССР, 4, 168 (1969).
117. Гиндин Л. М., Бобиков П. И., Кауба Э. Ф. Изв. СО АН СССР, серия хим., 10, 84 (1961).
118. Гинзбург В. Л., Глуховецкая Н. П., Данилова И. Н. Ж. аналит. химии, 17, 1096 (1962).
119. Гинзбург В. Л., Глуховецкая Н. П., Лернер Л. А. Зав. лаб., 28, 682 (1962).
120. Гладышев В. И. Зав. лаб., 24, 275 (1958).
121. Гладышев В. ПСангина О. А. Изв. АН КазССР, серия хим., 1 (15), 14 (1959).
122. Глазов В. М., Коршунов И. А. Труды по химии и хим. технол., 1 (19), 107 (1968).
123. Гольдшмидт В. М. Сборник статей по геохимии редких элементов. М.— Л., ГОНТИ, 1933, стр. 163, 168.
124. Горюшина В. Г., Бирюкова Е. Д. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965. стр. 153.
125. Горюшина В. Г., Бирюкова Е. Д., Аргонова Г. А. Научные труды Гиред-мета, 1961 — 1962 гг., В, 293 (1964).
126. Граудиня Л. ДПелекис Л. Л. Сб. «Ядерная спектроскопия и нейтронноактивационный анализ». Рига, «Зинатне», 1965, стр. 49.
127. Гурьев С. Д., Бляхман А. А., Дуценко Н. Н. Труды Гинцветмет, 19, 661 (1962).
128. Гусейнов И. К-, Багбанлы Р. И. Азерб. хим. ж., 1, 105 (1966).
129. Гусейнов И. К-, Багбанлы И. Л. Сб. «Исследования в области неорганической и физической химии». Баку, Изд-во АН АзербССР, 1966, стр. 48.
130. Гутько А. Д., Ткачева Г. В., Панкратова Н. И., Возяева 3. Н., Воробьев Н. К- Сб. «Анализ благородных металлов». М., Металлургиздат, 1965, стр. 114.
131. Давлетшин Э. Ю., Айдаров М. К. Ж. прикл. спектроскоп., 1, 73 (1964).
132. Давлетшин Э. Ю., Айдаров М. К- Труды по химии и хим. технол., 1, 92 (1962).
133. Даймонд Р. М., Так Д. Экстракция неорганических соединений. М., Атом-издат, 1962.
134. Дакар Г. М., Иофа Б.З., Несмеянов А. Н. Радиохимия, 5, 428 (1963).
135. Дарбинян М. В., Гайбакян Д. С. Изв. АН АрмССР, хим. н., 15, 511 (1962).
136. Дарбинян М. В., Гайбакян Д. С. Изв. АН АрмССР, хим. н., 16, 443 (1963).
137. Дарбинян М. В., Гайбакян Д.С. Сб. «Рений». М., «Наука», 1964, стр. 50.
138. Дарбинян М. В., Капанцян Э. Е. Арм. хим. ж., 21, 103 (1968).
139. Дарбинян М. В., Капанцян Э. Е. Изв. АН АрмССР, хим. н., 18, 18 (1965).
140. Демьянчук А. С. Инж.-физ. ж. АН БССР, 1, 88 (1958); РЖХим., 7, 23105 (1959).
141. Долгих Н. И., Бобиков П. И., Борбат В. Ф., Герберт М. Б., Гиндин Л. М. Цветные металлы, 11, 85 (1963).
142. Долежал Д., Повондра П., Шульцек 3. Методы разложения горных пород и минералов. М., «Мир», 1968, стр. 248.
143. Егоров В. В., Жигаловская Т. Н. Материалы 23-го гидрохимического совещания. Новочеркасск, 1969, стр. 5; РЖХим., 16Г157 (1969).
220
144. Ерденбаева М. И., Драговцева Н. А. Изв. АН КазССР, серия хим., 1 (17), 59, 63 (1960).
145. Ермаков В. В., Ковальский В. В. Ж. аналит. химии, 21, 447 (1966).
146. Житенева Г. М., Румянцев Ю. В. Труды Восточно-Сибирского филиала АН СССР, 41, 155 (1962).
147. Забродина А. С., Хлыстова А. П. Вести. МГУ, серия хим., 15, 69 (1960).
148. Зайдель А. И., Прокофьев В. К-, Райский С. М., Шрейдер Е. Д. Таблицы спектральных линий. М., Изд-во физ.-мат. лит-ры, 1962.
149. Зайковский Ф. В. Бюлл. научно-технической информации Министерства геологии и охраны недр СССР, 6 (23), 72 (1959).
150. Зайковский Ф. В. Сб. «Современные методы анализа в металлургии». М., Металлургиздат, 1955, стр. 142.
151. Захария И. Ф., Карпенко Л. И., Церкаеевич К- В., Назарова Г. Ф. Бюлл. научно-технической информации Министерства геологии и охраны недр СССР, 5—6 (39—40), 125 (1962).
152. Захария Н. Ф., Турулина О. П., Карпенко Л. И., Волощенко И. А. Зав. лаб. 29, 683 (1963).
153. Захаров А. А. Ж- аналит. химии, 15, 614 (1960).
154. Захарова В. Ф., Симонова Л. Н. Вест. МГУ, серия хим., 6, 115 (1966).
155. Звягинцев О. Е., Кулак А. И. Ж- неорган. химии, 2, 1687 (1957).
156. Звягинцев О. Е., Шамаев В. И. Ж. аналит. химии, 14, 603 (1959).
157. Звягинцев О. Е., Шамаев В. И., Радиохимия, 1, 717 (1959).
158. Зелионкайте В. И., Дницкий И. В. Труды АН ЛитССР, 4 (20), 71 (1959).
159. Зелянская А. И., Быков И. Е., Горшкова Л. С. Труды Ин-та металлургии УФАН СССР, 1, 151 (1957).
160. Зелянская А. И., Быков И. Е., Горшкова Л. С. Труды Ии-та металлургии УФАН СССР, 1, 155 (1957).
161. Зелянская А. И., Быков И. Е., Горшкова Л. С. Труды Ин-та металлургии УФАН СССР, 1, 161 (1957).
162. Зелянская А. И., Быков И. Е., Горшкова Л. С., Петунина Н. И. Методы полярографического анализа минерального сырья. М., Госгеолтехиздат, 1960, стр. 96.
163. Зелянская А. И., Горшкова Л. С. Труды Ин-та металлургии УФАН СССР, 5, 137 (1960).
164. Земмель В. К- Зав. лаб., 5, 1433 (1936).
165. Зильберштейн X. И., Семенов М. П. Ж. прикл. спектроскоп., 2, 16 (1965).
166. Золотарева Л. В., Коваленко П. Н. Ж- аналит. химии, 19, 73 (1964).
167. Золотарева Л. В., Коваленко П. Н. Ж. аналит. химии, 19, 731 (1964).
168. Золотов Ю. А. Ж- аналит. химии, 22, 1644 (1967).
169. Золотов Ю. А., Петрухин О. М. Ж. Всес. хим. об-ва им. Д. И. Менделеева, 9, 145 (1964).
170. Иванкова А. И. Сб. «Исследование и разработка фотометрических методов для определении микроколичеств элементов в минеральном сырье». Алма-Ата, ОНТИ Каз.ИМС, 1967, стр. 98.
171. Иванкова А. И. Труды Казахского ин-та минерального сырья, 3, 328 (1960).
172. ИванковаА. И., Блюм И. А. Бюлл. ВИМС, 6 (206), 3 (1960).
173. Иванкова А. И., Блюм И. А. Зав. лаб., 27, 371 (1961).
174. Иванчев Г. Дитизон и его применение. М., ИЛ, 1961.
175. Иофа Б. 3., Ван Вань-син, Ридван М. Радиохимия, 8, 14 (1966).
176. Иофа Б. 3., Митрофанов К- П. и др. Радиохимия, 6, 419 (1964).
177. Иоффе В. П. Канд. дисс. М., Гинцветмет, 1969.
178. Иоффе В. П. Труды Гинцветмета, 28, 33 (1968).
179. Каганович С. Д., Смиягин Л. В. Производство селена и теллура в капиталистических странах. М., Цветметинформация, 1962.
180. Калюжная Г. А., Халонин А. С. Зав. лаб., 27, 261 (1961).
181. Каплан Б. Д., Резакова А. С. Ж- аналит. химии, 21, 1268 (1966).
182. Каплан Б. Д., Сороковская И. А. Зав. лаб., 30, 783 (1964).
183. Каплан Б. ДСороковская И. А., Ширяева О. А. Зав. лаб., 36, 659 (1964).
10 Зак. № 1476
221
184. Карабаш А. Г., Пейзулаев С. И., Слюсарева Р. Л., Липатова В. М. Ж. аналит. химии, 14, 94 (1959).
185. Каримов М. Г. Сборник трудов аспирантов и соискателей. Министерство высшего образования КазССР. Матем. и физ., 2, 187 (1966); РЖХим., 21Г142 (1967).
186. Кельман Ф. Н. Зав. лаб., 18, 790 (1952).
187. Кельман Ф. Н. Зав. лаб., 24, 1061 (1958).
188. Кельман Ф. Н. РЖХим., 42, 111 (1959).
189. Кирин И. С., Зайцев В. М. Радиохимия, 10, 42 (1968).
190. Князева Р. Н. Канд. днсс. Свердловск, Политехи, ин-т, 1950.
191. Князева Р. Н., Клейман В. Л- Зав. лаб., 31, 410 (1965).
192. Князева Р. Н., Флоринская А. А., Ластухина И. Л- Сб. «Методы определения и анализа редких элементов». М., «Наука», 1961, стр. 605.
193. Князева Р. Н., Чернова Г. Н. Изв. вузов. Химия и хим. технол., 9, 867 (1966).
194. Коваленко П. Н., Золотарева Л. В. Материалы 2-го совещания по полярографии. Изд-во Казанск. ун-та, 1962, стр. 64.
195. Ковальский В. В., Ермаков В. В. Ж- аналит. химии, 21, 447 (1966).
196. Колесникова Н. М., Иофа Б. 3. Ж. неорган. химнн, 11, 2165 (1966).
197. Копайская Л. С. Тезисы докладов на Молдавском республиканском совещании по общей и прикладной химии. Кишинев, 1961, стр. 45.
198. Копайская Л. С., Ляликов Ю. С. Изв. АН МолдССР, 10, 31 (1962).
199. Коренман И. М., Ганина В. Г., Курина И. В. Труды по химии и хим. технол., 2, 369 (1959).
200. Коренман И. М., Глазунова 3. И. Труды по химии и хим. технол., 2, 368 (1962).
201. Костромин А. И., Круглов А. М. Материалы 2-го совещания по полярографии. Изд-во Казанск. ун-та, 1962, стр. 68.
202. Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ. М., Госхнмиздат, 1959, стр. 398.
203. Кудрявцев А. А. Химия и технология селена н теллура. М., «Металлургия», 1968.
204. Кудрявцев А. А. и др. Авт. свид. СССР 169733 (1964); Бюлл. изобр., 7, 104 (1965).
205. Кудрявцев А. А., Карапетьянц М. X., Устюгов Г. П., Рябова Р. И. Тезисы докладов научно-технической конференции Московского химико-технологического ин-та им. Д. И. Менделеева. М., 1966, стр. 82.
206. Кузнецов В. И. Ж- аналит. химии, 1, 259 (1946).
207. Кузнецов В. И. Усп. химии, 23, 654 (1954).
208. Кузнецов В. И., Большакова Л. И. Ж- аналит. химии, 15, 523 (1960).
209. Кузнецов В. И., Воронкова М. А., Мясоедова Г. В. Авт. свид. СССР 177671 (1966); Бюлл. нзобр., № 1, 103 (1966).
210. Кузьмина И. И., Сангина О. А. Ж- аналит. химии, 17, 495 (1962).
211. Ку Тхань-лонг, Судаков Ф. ГГ, ШаховаЗ. Ф. Ж- аналит. химии, 19, 734 (1964).
212. Ку Тхань-лонг, Судаков Ф. П., ШаховаЗ. Ф. Ж. аналнт. хнмин, 19, 968 (1964).
213. Куус X. Я-, Липп Л. А. Ж- аналит. химии, 21, 1266 (1966).
214. Лабковская Д. Б., Братчикова Н. И., Лазарева Г. А. Анализ благородных металлов. М., Металлургиздат, 1965, стр. 211.
215. Латимер В. М. Окислительно-восстановительные потенциалы. Галль — Нью-Йорк, 1952. '
216. Лебедь Н. Б., Паталер Р. П., Семенова Л. Н. Труды Комиссии по аналит. химии АН СССР, 16, 223 (1968).
217. Ли Чон-тхэк. РЖХим., 9Г104 (1966).
218. Лобанов Е. М., Ганиев А. Г., Сильванович Ю. А., Худайбергенов У. Сб. «Активационный анализ чистых материалов». Ташкент, «Фан», 1968, стр. 33.
219. Лушников В. В., Кондратьева Э. Н. Сб. «Новые методы анализа химического состава подземных вод». М., Изд-во ВСЕГИНГЕО, 1967, стр. 84.
220. Лю Шао-лин, Чжан Мин-ин. РЖХим., 13, 45582 (1959).
221. Лысенко В. И., Лисицина Е. В. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 13, 42 (1968).
222
222. Ляликов Ю. С., Копайская Л. С. Изв. АН МолдССР, 12 (90), 47 (1967).
223. Ляликов Ю. С,, Копанская Л. С. Укр. хим. ж., 30, 91 (1964).
224. Ляликов Ю. С., Копанская Л. С., Кулева 3. П. Зав. лаб., 33, 1493 (1967).
225. Ляликов Ю. С., Копанская Л. С., Сафронкова Н. Н. Химические и физико-химические методы определения индня, сурьмы и теллура в полупроводниковых сплавах. Л., «Физика», 1962, стр. 26.
226. Максай Л. И., Милаев С. М. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 5, 46 (1959).
227. Малахов В. В. Зав. лаб., 28, 408 (1962).
228. Малахов В. В. Зав. лаб. 29, 419 (1963).
229. Мамаев В. Е., Протасов К- Т. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 9, 163 (1965).
230. Маркова Н. В. Сб. «Химические, физико-химические и спектральные методы исследования руд редких и рассеянных элементов». М., Госгеолтехиздат, 1961, стр. 75.
231. Маркова Н. В., Шуваева Т. М., Полотырхина А. К- Бюлл. ВИМС, 11 (199) (1959).
232. Маркова И. В., Шуваева Т. М., Полотырхина А. К- Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 598.
233. Масленников Б. М., Портнова В. Н., Романова Л. В. Труды н.-и. ин-та горно-хим. сырья, 4, 239 (1958); РЖХим., 10, 34623 (1959).
234. Махметов -М. Ш., Полукаров А. Н. Труды химико-металлургического ин-та АН КазССР, 3, 52 (1967).
235. Машуков А. Л- Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 599.
236. Машуков А. Я-, Гладышева К- Ф- Труды Всес. н.-и. горно-металлург. ин-та цветных металлов, 5, 56 (1959).
237. Машуков А. Я-, Маслакова Т. Г. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 5, 98 (1959).
238. Мизецкая И. Б., Матат Л. М., Раевская Л. И. Труды Комиссии по аналит. химии АН СССР, 16, 10 (1968).
239. Милаев С. М., Выгоняйло А. И. Металлургия и химическая промышленность Казахстана, 2 (12), 87 (1961); РЖХим., 4Д131 (1962).
240. Митюрева Т. Ш. Автореф. канд. дисс. Киев, ИОНХ АН УССР, 1960; РЖХим. 21365 (1962).
241. Моррисон Дж., Фрейзер Г., Экстракция в аналитической химии. Л., Гос-хнмиздат, 1960.
242. Москвин Л. И. Радиохимия, 1, ПО (1964).
243. Музгин В. Н., Золотавин В. Л., Гаврилов Ф. Ф., Балаев В. П. Зав. лаб., 30, 697 (1964).
244. Мурашова В. И. Ж- аналнт. химии, 17, 80 (1962).
245. Мурашова В. И. Ж- аналит. химии, 21, 345 (1966).
246. Мурашова В. И. Зав. лаб., 18 , 790 (1952).
247. Мурашова В. И. Канд. дисс. Свердловск. Уральский политехи, ин-т, 1951.
248. Мурашова В. И. Методы анализа черных н цветных металлов. М., Металлург-издат, 1953, стр. 112, 118.
249. Мурашова В. И. Труды Уральск, политехи, ин-та, 94, 161 (1960).
250. Мурашова В. ИБольшакова Л. П. Труды Уральск, политехи, ин-та, 94, 158 (1960).
251. Мурашова В. И., Долгова А. Л. Зав. лаб., 17, 814 (1951).
252. Мурашова В. И., Сушкова С. Г. Авт. свид. СССР193135(1965); Бюлл. нзобр., № 6, 114 (1967).
253. Мурашова В. И., Сушкова С. Г. Ж- аналит. химии, 19, 1503 (1964).
254. Мурашова В. И., Сушкова С. Г. Ж. аналит. химии, 24 , 729 (1969).
255. Мурашова В. И., Сушкова С. Г., Бакунина Л. И. Зав. лаб., 33, 280 (1967).
256. Мурашова В. И., Челышева Н. Л., Холодкова М. В. Труды ин-та Цнипромедь,
3, 240 (1958).
257. Мурашова В. И., Ячменева Т. М., Давыдова А. Е. Изв. вузов, Химия и хим. технол., 6, 577 (1963).
10*
223
258. Мюнке М. Зав. лаб., 34, 165 (1968).
259. Нагибин В. С. Зав. лаб., 30, 1450 (1964).
260. Назаренко В. А., Шитарева Г. Т. Тезисы докладов на Всесоюзном симпозиуме^ аналитической химии селена, теллура и рения. Ереван, Изд-во МИТК,
261. Назаренко Н.П. Сб. «Новые методические исследования по анализу редкометальных минералов, руд и горных пород». М., «Наука», 1970, стр. 41.
262. Назаренко И. И. Сб. «Редкометальные месторождения, их генезис и методы исследования». М., изд. ИМГРЭ, 1971.
263. Назаренко И. И., Волынец М. П. Ж. аналит. химии, 24, 1209 (1969).
264. Назаренко И. И., Кислов А. М., Кислова И. В., Малевский А. Ю. Ж- аналит. химии, 25, 1135 (1970).
265. Недлер В. В., Фришберг А. А. Труды н.-и. горно-разведочного ин-та, серия 2, 15, 45 (1960).
266. Неймарк Л. Э., Юделевич к. Г. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, i960, стр. 289.
267. Некрасов Б. В. Основы о щей химии, т. 1. М., «Химия», 1969, стр. 352.
268. Непеина Л. А. Зав. лаб?, 28, 166 (1962).
269. Непеина Л. А., Кузнецова М. К- Уч. зап. центр, н.-и. ин-та оловянной промышленности, 1, 92 (1965); РЖХим., 10Г123 (1966).
270. Нестерова Ю. С., Арапова Г. Т. Сб. «Методы химического анализа и химического состава минералов». М., «Наука», 1967, стр. 106.
271. Никонова Л. И. Сборник научных трудов Иркутского н-и. ин-та редких металлов, 9, 21 (1961).
272. Новоселова А. В. Ж. неорган. химии, 7, 960 (1962).
273. Новоселова А. В., Пашинкин А. С., Поповкин Б. А. Ж- Всес. хим. об-ва им. Д. И. Менделеева, 5, 557 (1960).
274. Ноткина М. А., Петрова Е. И., Геркалина Т. В., Чернихов Ю. А. Труды Комиссии по аналит. химии АН СССР, 15, 80 (1965).
275. Овсепян Е. Н., Тараян В. М., Шапошникова Г. Н. Арм. хим. ж., 20, 184 (1967).
276. Овсепян Е. Н., Тараян В. М., Шапошникова Г. Н. Изв. АН АрмССР, хим. н., 18, 225 (1965).
277. Овсепян Е. Н., Шапошникова Г. Н., Галфаян Н. Г. Ж- неорган. химии, 12, 2411 (1967).
278. Онуфриенок Н. П., Аксененко В. М. Ж- аналит. химии, 13, 119 (1958).
279. Онуфриенок И. П., Аксененко В. М. Ж- аналит. химии, 14, 637 (1959).
280. Онуфриенок И. П., Аксененко В. М., Новикова Е. С. Изв. Томск, политехи, ин-та, 11, 115 (1961).
281. Онуфриенок И. П., Аксененко В. М., Новикова Е. С. Сб. «Физико-химический анализ». Новосибирск, Изд-во СО АН СССР, 1963, стр. 326.
282. Орлова Е. С., Назаренко И. И., Баренбаум М. Е. Зав. лаб., 36, 926 (1970).
283. Ошман В. А., МельчековаЗ. Е. Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 596.
284. Павлова В. Н. Зав. лаб., 28, 166 (1962).
285. Павлова В. Н., Васильева Н. Г., Кашлинская С. Е. Зав. лаб., 27, 965 (1961).
286. Павлова В. Н., Мурашова В. И. Зав. лаб., 34, 392 (1968).
287. Пац Р. Г., Васильева Л. Н., Заглодина Т. В., Шувалова Е. Д. Зав. лаб., 29, 928 (1963).
288. Пац Р. Г., Васильева Л. Н., Семочкина Т. В. Ж- аналит. химии, 23, 241 (1968).
289. Пац Р. Г., Семочкина Т. В. Зав. лаб., 28, 800 (1962).
290. Пац Р. Г., Семочкина Т. В. Зав. лаб., 33, 1491 (1967).
291. Пац Р. Г., Семочкина Т. В. Труды Гинцветмет, 27, 50 (1967).
292. Пац Р. Г., Семочкина Т. В. Труды Гинцветмет, 28, 12 (1968). -
293. Пац Р. Г., Цфасман С. Б., Семочкина Т. В. Зав. лаб., 30, 140 (1964).
294. Пацук В. В. Ж. аналит. химии, 12 , 509 (1957).
295. Пацук В. В. Ж. аналит. химии, 12 , 230 (1957).
296. Пацук В. В. Труды Свердловск, горного ин-та, 36, 81, 88 (1960).
224
297. Пацук В. В., Субботина Т. С. Сборник научных трудов Свердловск, филиала Моск, ин-та народного хоз-ва, 1, 42 (1967); РЖХим., 10Г84 (1968).
298. Пацук В. В., Тикунова Н. И., Косихина Г. И. Сборник научных трудов Свердловск, филиала Моск, ин-та народного хоз-ва, 1, 50 (1967).
299. Пашинкин А. С., Новоселова А. В. Ж- неорган. химии, 4, 2657 (1959).
300. Пенсионерова В. М., Панкова В. Е. Бюлл. научно-технической информации Министерства геологии СССР, 3, 37 (1967).
301. Петров А. Г., Айдаров Т. К- Изв. АН СССР, серия физ., 26, 899 (1962).
302. Пилипенко А. Т., Капустин А. И. Укр. хим. ж., 30, 9 (1964).
303. Плотников В. И. Ж. аналит. химии, 14, 595 (1958).
304. Плотников В. И. Ж- неорган. химии, 3, 1761 (1958).
305. Плотников В. И. Ж- неорган. химии, 5, 731 (1960).
306. Плотников В. И. Ж- неорган. химии, 9, 451 (1964).
307. Плотника В. И. Зав. лаб., 25, 666 (1959).
308. Плотникове В. И. Труды Всес. н.-и. горно-металлург. ин-та цветных металлов, 5, 65 (1959).
309. Плотников В. И. Хим. наука и пром., 4, 676 (1959).
310. Плотников В. И., Новиков В. П., Кочетков В. Л., Гибова Э. Г. Ж- аналит. химии, 24, 944 (1969).
311. Пливанова Н. Г., Филиппова Т. А. Ж- аналит. химии, 22, 290 (1967).
312. Полотебнова Н. А., Деркач Л. В. Труды Комиссии по аналит. химии АН СССР, 16, 30 (1968).
313. Полотебнова Н. А., Кокорин А. И. Тезисы докладов на совещании по общей и прикладной химии. Кишинев, Изд-во АН МолдССР, 1961.
314. Полукаров А. Н. Зав. лаб., 25, 905 (1959).
315. Полякова В. В., Федорова В. В. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, стр. 172.
316. Попова Е. М., Дергачева Е. Н., Капилевич С. В. РЖХим., 10Г181 (1965).
317. Прошкович М. Ф., Фалеев Р. В. Бюлл. Ин-та механизированной обработки полезных ископаемых, 2, 2 (1957).
318. Раманаускас Э. И., Тупгкувене В. Е. Ж- Всес. хим. об-ва им. Д. И. Менделеева, 11, 355 (1966).
319. Раманаускас Э. И., Туткувене В. Е. Научные труды вузов ЛитССР. Химия и хим. технол. 7, 31 (1965); РЖХим., 23Г100 (1966).
320. Раманаускас Э. И., Шулюнене А. К-Ж- аналит. химии, 23, 1859 (1968).
321. Рещикова А. А., Шорина Е. В. Изв. АН МолдССР, серия биол. и хим. и., 11, 76 (1964).
322. Ригин В. И., Мельниченко Н. Н., Дницкий В. К- Зав. лаб., 33, 1370 (1967).
323. Рудневский Н. К-, Максимов Д. Е., Домарин В. Т., Балдово Н. И. Ж. прикл. спектроскоп.,, 9, 156 (1968).
324. Рыбаков В. Н., Стронский И. И. Ж- неорган. химии, 4, 2449 (1959).
325. Рябчиков Д. И., Волынец М. П. Ж- аналит. химии, 21, 1348 (1966).
326. Рябчиков Д. И., Ермаков А. Н., Калиниченко Н. Б. Сб. «Разделение близких по свойствам элементов». Труды Межвузовской конференции. М., Металлургиздат, 1962.
327. Рябчиков Д. И., Назаренко И. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 445.
328. Рябчиков Д. И., Назаренко И. Н. Усп. химии, 33, 108 (1964).
329. Рябчиков Д. И., Назаренко И. И., Аникина Л. И. Ж- аналит. химии, 23, 1242 (1968).
330. Сахаров А. А. Ж- аналит. химии, 14, 614 (1960).
331. Селезнева Н. А. Изв.АН КазССР, серия хим., 4, 31, 1966.
332. Семенов И. П. Ж. эксп. и теор. физ., 38, 1654 (1960).
333. Сидельникова В. Д. Сб. «Химический анализ минералов и их химический состав». М., «Наука», 1964, стр. 75.
334. Сидельникова В. Д., Леонтьева А. А. Сб. «Методы химического анализа и химический состав минералов». М., «Наука», 1967, стр. 101.
335. Синдеева Н. Д. Геохимия, минералогия и генетические типы месторождений редких элементов, т. 1. М., «Наука», 1964, стр. 557, 586.
225
336. Синдеева Н. Д., Курбанова Н. 3. Докл. АН СССР, 120, 353 (1958).
337. Синякова С. И., Кроль Г. Д. Труды Комиссии по аналит.химии АН СССР, 12, 206 (1960).
338. Скляренко Ю. С., Назаренко И. И., Разгон Е. С., Ж- аналит. химии, 23, 91 (1968).
339. Скребкова П., Торопова В. Ф. Сборник студенческих научных работ Казанского гос. ун-та им. В. И. Ульянова. Казань, 1961, стр. 125.
340. Смородина Т. И., Сеткина О. Н. Изв. вузов. Химия и хим. технол., 4, 565 (1961).
341. Соколов В. А., Левин В. И. Ж- неорган. химии, 9, 742 (1964).
342. Соколович В. Б., Онуфриенок И. П. Изв. Томск, политехи, ин-та, 102, 157 (1959).
343. Сангина О. А., Войлошникова А. П. Зав. лаб., 24, 1331 (1958).
344. Сангина О. А., Тойбаев В. К- Изв. АН КазССР, серия хим., 22, 53 (1962).
345. Сосенкова Л. И., Крейнгольд Е. У., Божевольнов Е. А: Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, изд-во МИТК, 1968, стр. 19.
346. Софронкова Н. Н., Ляликов Ю. С. Зав. лаб., 27, 21 (1961).
347. Сочеванов В. Г., Шмаков Н. В. Бюлл. ВИМС, 8, 184 (1958).
348. Сперанская Е. Ф. Ж- аналит. химии, 17, 347 (1962).
349. Сперанская Е. Ф. Ж- аналит. химии, 18, 9 (1963).
350. Станчев П. РЖХим., 12Г134 (1967).
351. Стары И. Экстракция хелатов. М., «Мир», 1966.
352. Статистический справочник «Редкие металлы капиталистических стран». М., Центральный ин-т информации цветной металлургии, 1958.
353. Степин В. В., Поносов В. И., Новикова Е. В., Сташкова В. В., Сушкова С. Г., Мурашова В. И., Емашова Т. Н. Труды Всес. н.-и. ин-та стандартных образцов и спектральных эталонов, 3, 109 (1967).
354. Стрельникова Н. П. Зав. лаб., 28, 1193 (1962).
355. Стрельникова Н. П. Труды Комиссии по аналит. химии АН СССР, 14, 305 (1963).
356. Стрельникова Н. П., Лысцова Г. Г. Зав. лаб., 26, 142 (1960).
357. Стрельникова Н. П., Лысцова Г. Г., Долгорукова Г. С. Зав. лаб., 28, 1319 (1962).
358. Стрельникова Н. П., Павлова В. Н. Зав. лаб., 26, 425 (1960).
359. Сурмий А. М., Аришкевич А. М., Усатенко Ю. И. Труды Комиссии по аналит. химии АН СССР, 16, 114 (1968).
360. Сушкова С. Г., Мурашова В. И. Ж- аналит. химии, 21, 1475 (1966).
361. Сушкова С. Г., Мурашова В. И. Труды Уральск, политехи, ин-та, 163, 35 (1967).
362. Сырокомский В. С., Князева Р. Н. Зав. лаб.. 15, 1149 (1949).
363. Сырокомский В. С., Князева Р. Н. Зав. лаб., 16, 1041 (1960).
364. Тананаев Н. А., Мурашова В. И. Ж- аналит. химии, 3, 3 (1948).
365. Тананаев Н. А., Мурашова В. И. Зав. лаб., 17, 405 (1951).
366. Тананайко М. М. Труды Комиссии по аналит. химии АН СССР, 14, 114 (1963).
367. Тараян М. Г. Труды Гинцветмет, 19, 740 (1962).
368. Тараян В. М., Авакян Т. Т. Докл. АН АрмССР, серия хим., 30, 231 (1960).
369. Тараян В. М., Авакян Т. Т. Зав. лаб., 27, 967 (1961).
370. Тараян В. М., Арстамян Ж- М. Изв. АН АрмССР, серия хим., 15, 331, 415 (1962).
371. Тараян В. М., Арстамян Ж- М. Изв. АН АрмССР, серия хим., 17, 38 (1964).
372. Тараян В. М., Арстамян Ж- М. Изв. АН АрмССР, серия хим., 17, 623 (1964).
373. Тараян В. М., Арстамян Ж- М. , Микаелян Д. А. Зав. лаб., 34, 1284 (1968).
374. Тараян В. М., Арстамян Е. М., Микаелян Д. А. Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, изд-во МИТК, 1968, стр. 25.
375. Тараян В. М., Арстамян Ж- 7И., Шапошникова Г. Н. Изв. АН АрмССР, серия хим., 14, 551 (1961).
226
376. Тараян В. М., Арстамян Е. М., Шапошникова Г. Н. Сб. «Современные методы химического и спектрального анализа материалов». М., Металлургия, 1967, стр. 33.
377. Тараян В. М., Саркисян А. А. Ж- аналит. химии, 20, 2684 (1965).
378. Тенчева Р., Салчева М. Бюлл. научно-технической информации Нипроруда, 2, 26 (1967); РЖХим., 11Г108 (1968).
379. Терземан Л. И. Труды ин-та металлургии и обогащения АН КазССР, 29, 56 (1969).
380. Тимофеева В. К. Труды Гинцветмета, 27, 22 (1967).
381. Тимофеева В. К. Цветные металлы, 7, 74 (1965).
382. Тимофеева В. К-, Жарова С. П. Цветные металлы, 11, 93 (1965).
383. Тимофеева В. К-, Ласкорин Б. Н. Цветные металлы, 11, 81 (1961).
384. Тимофеева Т. Г., Лисицкая К- В. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 9, 57 (1965).
385. Типцова В. Г., Андрейчук А. М., Бажанова Л. А. Ж- аналит. химии, 20, 1200 (1965).
386. Типцова-Яковлева В. Г., Фогельсон Ю. А. Ж. аналит. химии, 23, 1415 (1968).
387. Торопова В. Ф. Ж. аналит. химии, 11, 599 (1956).
388. Торопова В. Ф., Медведева Л. И., Поляков Ю. Н. Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, изд-во МИТК 1968, стр. 7.
389. Тредвелл Ф. П., Голл В. Т. Качественный анализ. М.— Л., Госхимиздат, 1956.
390. Трифонов А. С., Иорданов Б. РЖХим., 14Д13 (1962).
391. Троицкая М. И. Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 580.
392. Тренев В. Г., Беляков И. М. Ж- неорган. химии, 4, 1932 (1959).
393. Тренев, В. Г., Григорович А. Н. Ж- неорган. химии, 2 , 2400 (1957).
394. Тулюпа Ф. М., Баркалов В. С., Усатенко Ю. И. Ж- аналит. химии, 22, 399 (1967).
395. Туманов А. А., Шахверди Н. М. Сб. «Современные методы химического и спектрального анализа материалов». М., Металлургиздат, 1967, стр. 184.
396. Туткувене Е. Е., Раманаускас Э. И. Ж- аналит. химии, 21, 564 (1966).
397. Туткувене В. Е., Раманаускас Э. И., Аришкевич А. М., Усатенко Ю. И. Научные труды вузов ЛитССР. Химия и хим. технол., 9, 55 (1968).
398. У Го-хуэй, Хуа Сюэ-цинцзе. РЖХим., 39389 (1958).
399. Устимов А. М., Чалков Н. Я- Зав. лаб., 35, 117 (1969).
400. Файгль Ф. Капельный анализ. М.— Л., Госхимиздат, 1937, стр. 404.
401. Файнберг Р. Ю., Филиппова Н. А. Анализ руд цветных металлов. М., Металлургиздат, 1963, стр. 197.
402. Федоров А. А., Линкова Ф. В. Труды центр, н.-и. ин-та черной металлургии.
24, 147 (1962); РЖХим., 24Д153 (1962).
403. Ферсман А. Е. Геохимия, т. 4. Л., ГОНТИ, 1959.
404. Ферьянчич Ф.А. Труды Комиссии по аналит. химии АН СССР, 16, 106 (1968).
405. Флетчер Дж. М. Извлечение и очистка редких металлов. М., Атомиздат. 1960, стр. 22.
406. Фомин В. В. Химия экстракционных процессов. М., Атомиздат, 1960.
407. Фраткин 3. Г., Поливанова Н. Г. Зав. лаб., 26, 1372 (1960).
408. Фраткин 3. Г., Поливанова Н. Г. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 462.
409. Фраткин 3. Г., Поливанова Н. Г. Труды Всес. ин-та хим. реактивов, 23, 113 (1959).
410. Фришберг А. А., Недлер В. Я- Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, стр. 250.
411. Хакимова В. Д., Агасян П. К. Узб. хим. ж., 6, 21 (1960).
412. Хо Изя-лун, Ло Цзынь-фан, Жан Фэн-би. Кэхуо-Тунбао, 4, 333 (1964); РЖХим., 8Г53 (1965).
413. Хасимото Иосикадзу, Янагисаво Сабуре. РЖХим., 22Г139 (1968).
227
414. Химические основы экстракционного метода разделения элементов. М, «Наука», 1966.
415. Храпай В. Р. Сб. «Анализ благородных металлов АН СССР». М., Ин-т общей и неорганической химии им. Н. С. Курнакова, 1959, стр. 128.
416. Хроматография на бумаге. Под ред. И. М. Хайса и К- Мацека. М., ИЛ, 1962.
417. Хрдщов Н. А., Круглова В. Г., Пенсионерова В. М., Панкова В. Е., Радова Г. В. Минеральное сырье, 1, 86 (1960).
418. Ху Чжи-дэ, Сюнь Бао-чуй. Кэхуе Тунбао, 4, 1004 (1964); РЖХим., 19Г49 (1965).
419. Церковницкая И. А., Епимахов В. Н. Зав. лаб., 31, 1178 (1965).
420. Церковницкая И. А., Епимахов В. Н. Труды Комиссии по аналит. химии АН СССР, 16, 145 (1968).
421. Цзо Цзун-ци, Ли Ху-линь. РЖХим., 5Г102 (1965).
422. Цзо Цзун-ци, Цн Синь-линь. РЖХим., 18Г99 (1966).
423. Цзынь Дин-сюнь. РЖХим., 16Г108 (1966).
424. Цудзи Юкио. Япон. пат. 14754 (1960); РЖХим., 14Л69п (1963).
425. Челышева Н. Л., Хохолкова М. В., Мурашова В. И. Труды Уральского института медной промышленности. Свердловск, Металлургиздат, 1958.
426. Чепик М. Н., Бусев А. И., Бабенко Н. Л. Авт. свид. СССР 163006; Бюлл. изобр., № 11, 60 (1964).
427. Чесноков О. Ф., Сухневич В. Р. Зав. лаб., 26, 1372 (1960).
428. Чжан Пей-хуа. РЖХим., 12Г115 (1966).
429. Чжан-Туан, Чжан Му-дзюань. Кэхуе Тунбао, 5, 644 (1965); РЖХим., 6Г52 (1966).
430. Чижиков Д. М., Счастливый В. П. Селен и селениды. М., «Наука», 1964.
431. Чижиков Д. М., Счастливый В. П. Теллур и теллуриды. М., «Наука», 1966.
432. Чикрызова Е. Т., КопанскаяЛ. С. Ж. аналит. химии, 23, 394 (1968).
433. Шамаев В. И. Радиохимия, 2 , 624 (1960).
434. Шамаев В. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 436, 464.
435. Шамаев В. И., Саункин О. Ф. Изв. вузов. Химия и хим. технол., 3, 66 (1960).
436. Шахов А. С. Зав. лаб., 11, 893 (1954).
437. Шахова 3. Ф., Гаврилова С. А., Захарова В. Ф. Вести. МГУ, серия хим., 6, 79 (1965).
438. Шахова 3. Ф., Гаврилова С. А., Захарова В. Ф. Вести. МГУ серия хим., 88 (1966).
439. Шахова 3. Ф., Ку Тхань-лонг. Вести. МГУ, серия хим., 1, 72 (1964).
440. Шахова 3. Ф., Ку Тхань-лонг. Ж- неорган. химии, 9, 1848 (1964).
441. Шелпакова И. Р., Закс И. В. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 9, 189 (1965).
442. Шергина Ю. П., Кашлинская А. Б., Миллер А. Д. Фотометрический метод количественного определения селена и теллура в горных породах и почвах. Л., ОНТИ, 1960, стр. 18.
443. Шитарева Г. Г. VIII Менделеевский съезд. Секция аналит. химии. М., «Химия», 1958, стр. 67.
444. Шитарева Г. Г. Зав. лаб., 27, 1196 (1961).
445. Шитарева Г. Г., Назаренко В. А. Укр. хим. ж., 26, 368 (1960).
446. Шихеева Л. В. Ж- неорган. химии, 12, 3323 (1968).
447. Шихеева Л. В., Ж- неорган. химии, 13, 2967 (1968).
448. Шкробот Э. П., Тараян М. Г., Бляхман А. А. Зав. лаб., 32, 18 (1966).
449. Шкробот Э. П., Шебаршина Н. И. Зав. лаб., 35, 417 (1969).
450. Шкробот Э. П., Шебаршина Н. И. Сб. «Спектрофотометрические методы анализа материалов». М., Дом научно-техн, пропаганды, 1966, стр. 56.
451. Шкробот Э. П., Шебаршина Н. И. Труды Гинцветмет, 28, 18 (1968).
452. Шокол А. А., Кочерова С. А. Укр. хим. ж., 21, 89 (1955).
453. Шубин М. И. Зав. лаб., 8, 677 (1934).
454. Шустер Т. Л., Гехт И. И. Бюлл. ВИМС, 1955, 145.
455. Щербина В. В. Изв. АН СССР, серия геол., 5, 965 (1937).
228
456. Щербов Д. П. Флуориметрия в химическом анализе минерального сырья. М., «Недра», 1965.
457. Щербов Д. П., Астафьева И. Н., Плотникова Р. Н. Тезисы докладов на Всесоюзном симпозиуме по аналитической химии селена, теллура и рения. Ереван, изд-во МИТК, 1968, стр. И.
458. Щербов Д. П., Иванкова А. И. Зав. лаб., 24, 667 (1958).
459. Щербов Д. ПИванкова А. И., Гладышева Г. П. Зав. лаб., 33, 683 (1967).
460. Щербов Д. П., Иванкова А. И., Гладышева Г. П. Сб. «Исследование и разработка фотометрических методов для определения микроколичеств элементов в минеральном сырье». Алма-Ата, ОНТИ КазИМС, 1968, стр. 17.
461. Щербов Д. ПСоловьян И. Г., Дробаченко А. В. Труды КазИМС, 1, 188 (1959)
462. Экстракция в аналитической химии и радиохимии. М., ИЛ, 1961.
463. Экстракция, теория, применение, аппаратура. Сборник статей, вып. 1, 2. М., Атомиздат, 1962.
464. Юделевич И. Г., Сосновская Г. И., Вершинина Ф. И. Спектроскопия. М. «Наука», 1964, стр. 87.
465. Юделевич И. Г., Шелпакова И. Р. Ж- аналит. химии, 17, 174 (1962).
466. Юделевич И. Г., Шелпакова И. Р., Авсейко Е. М./F.. аналит. химии, 18, 634 (1963).
467. Юделевич И. Г., Шелпакова И. Р., Сосновская Г. И. Сборник научных трудов Всес. н.-и. горно-металлург. ин-та цветных металлов, 7, 360 (1962).
468. Юделевич И. Г., Шокарев М. М. Рудный Алтай, 6—7, 53 (1958); РЖХим., № 3, 8922 (1960).
469. Юделевич И. Г., Шокарев М. М., Сосновская Г. И., Станевич В. В., Алонце-ва Н. И. Сб. «Некоторые вопросы эмиссионной и молекулярной спектроскопии». Красноярск, 1960, стр. 126; РЖХим., 13Д150 (1962).
470. Юркин П., Сильвестров Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 457.
471. Юхтанов Д. М. Производство селена и теллура. М., Металлургиздат, 1955.
472. Яковлев Ю. В. Зав. лаб., 28, 643 (1962).
473. Яковлев Ю. В., Догадкин Н. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 162.
474. Яковлев Ю. В., Догадкин Н. Н. Там же, стр. 233.
475. Яковлев Ю. В., Догадкин И. Н. Там же, стр. 258.
476. Яковлев Ю. В., Догадкин Н. Н. Там же, стр. 320.
477. Яковлев Ю. В.. Догадкин Н. Н. Там же, стр. 380.
478. Яковлева В. С., Троицкий В. П. Уч. зап. Ленингр. гос. пед. ин-та им. А. И. Герцена, 140, 79 (1957).
479. Яницкий И. В., Зелионкайте В. И. Ж. неорган. химии, 3, 1755 (1958).
480. Aaremae A., Assarsson G. О. Analyt. Chem., 27, 1155 (1955).
481. Acs L., Barabas S. Analyt. Chem., 36, 1825 (1964).
482. Agrinier H. C. r. Acad, sci., 254, 1850 (1962).
483. Akira K. Japan Analyst., 11, 1162 (1962).
484. Akira K-, Yoshida I., Iwadsy U. J. Chem. Soc. Japan, Pure Chem. Sect., 85. 647 (1964).
485. Albert G., Coglioti V., Lederer M. J. Chromatogr., 7, 242 (1962).
486. Algright С. H., Burke Keith E., Yanak M. M. Taianta, 16, 309 (1969).
487. Allan A., Saud W., Khalifa H. Taianta, 15, 249 (1968).
488. Allan J. E. Spectrochim. acta, 18, 259 (1962).
489. Allan J. E. 4-th Australian Spectroscopy Conference. Canberra, 1963.
490. Allaway W. H., Cary E. E. Analyt. Chem., 36, 1359 (1964).
491. Almassy G., Kossis E., Kossls T. Acta chim. Acad. sci. hung., 33, 187 (1962).
492. Anderson W. L., Peterson H. E. Bur. Mines Rept Invest., N 6201, 9 (1963);
C. A., 59, 1090c (1963).
493. Arend A. G. Glass, 35, 564 (№58).
494. Aritoshi H., KiniwaM. Taianta, 5, 112 (1960).
495. Articolo 0. J. U. S. Atom. Energy Commiss. Кар. L-M-OYA-2. 11 (1960);
C. A., 57, 6604 (1962).
496. Asian F. W. Proc. Roy. Soc. London, 132, 487 (1931).
229
497. A. S. T. M. Methods of Chemical Analysis of Metals, 1945, 84.
498. Atanassowa B. W. Mikrochim. acta, 1966, 970.
499. Athavale H. Analyt. Chem., 33, 311 (1961).
500. Ayca Ertugrul. Istanbul, tekn. univ. bul., 17, 87 (1964); РЖХим., 24Г92 (1965)
501. Aynsley E., Campbell W. J. Chem. Soc., 1957, 832.
502. Aynsley E., Campbell IF. J. Chem. Soc., 1958, 3290.
503. Aynsley E., Hatherington G. J. Chem. Soc., 1953, 2802.
504. Babadag Tilrkan. Z. anal. Chem., 207, 328 (1965).
505. Bacon J. F., Popoff V. Analyt. Chem., 27, 319 (1955).
506. Bagnall K- W. The chemistry of selenium, tellurium, and polonium. N. Y., Elsevier Publ., Co., 1966.
507. Ballaux C., Dams R., Hoste J. Analyt. chim. acta, 37, 164 (1967).
508. Ballaux C., Dams R., Hoste J. Analyt. chim. acta, 41, 147 (1968).
509. Ballaux C., Dams R., Hoste J. Analyt. chim. acta, 43, 1 (1968).
510. Ballaux C., Dams R., Hoste J. Analyt. chim. acta, 45, 337 (1969).
511. Balloffet G., Romand J. J. phys. et radium, 20, 509 (1959).
512. Barabas S., Bennett P. IF. Analyt. Chem., 35, 135 (1963).
513. Barabas S., Kooper W. C. Analyt. Chem., 28, 129 (1956).
514. Barcza L. Magyar, kem. folyorat, 69, 248 (1963); РЖХим., 7Г104 (1964).
515. Barcza L. Mikrochim. acta, 1964, 136.
516. Barcza L. Mikrochim. acta, 1964, 967.
517. Barcza L., Schulek E. Magyar tud. akad. Kem. tud. oszt. kozl., 13, 295 (1960) РЖХим., 1Д100 (1961).
518. Barcza L., Schulek E. Mikrochim. acta, 1960, 261.
519. Barcza L., Sommer L. Z. anal. Chem., 192, 304 (1963).
520. Barcza L., Zsindely S. Z. anal. Chem., 199, 117 (1964).
521. Beresel D., Corinth U., Stahr А. Пат. ФРГ 120842 (1969).
522. Barnets IF. B., Kahn H. L. Atom. Absorpt. Newsletter. 8, 21 (1969).
523. Bastins H. Neue Hiitte, 4, 178 (1959).
524. Beattie J. R., Chunzynska H. Chem. Ind., 46, 1842 (1963); C. A., 60, 2530 (1964)
525. Becker D. A., Smith G. IT. Proc. Internal. Conf. Mod. Trends Activat. Analysis. College Station Texas, 1965, p. 230—235; РЖХим., 7Г97 (1967).
526. Bennett P. W., Barabas S. Analyt. Chem., 35, 139 (1963).
527. Bera В. C., Chakrabarthy M. M. Analyst, 93, 50 (1968).
528. Berg R., Teitelbaum M. Mikrochemie Festschr. Emich, 1930, 23.
529. Betteridge D. Res. Group U. K. Atomic Energy Author., 1965, N AERE-R 4881, 13pp.; РЖХим., 5Г160 (1966).
530. Biswas S. D., Dey A. K. Analyst, 90, 56 (1965).
531. Blanka B. Coll. Czech. Chem. Comm., 27, 495 (1962).
532. Blasius E., Fischer M. Z. anal. Chem., 178, 28 (1960).
533, Bock R., Jacob D. Z. anal. Chem., 200, 118 (1964).
534. Bock R., Kan H. Z. anal. Chem., 188, 28 (1962).
535. Bode H. Z. anal. Chem., 134, 100 (1951).
536. Bode H. Z. anal. Chem., 143, 182 (1954).
537. Bode H. Z. anal. Chem., 144, 90 (1955).
538. Bode H. Z. anal. Chem., 144, 165 (1954).
539. Bode H. Z. Anal. Chem., 153, 335 (1956).
540. Bode H. Z. anal. Chem., 155, 96 (1957).
541. Bode H., Arnswald W. Z. anal. Chem., 165, 179 (1958).
542. Bode H., Hettwer E. Z. anal. Chem., 173, 285 (1960).
543. Bode H., Mdseuthin H. Z. anal. Chem., 164, 232 (1958).
544. Bode H., Neuman F. Z. anal. Chem., 172, 1 (I960).
545. Bode H., Tusche K- Z. anal. Chem., 157, 414 (1957).
546. Bode H., Tusche K., Wahrhausen H. Z. anal. Chem., 190, 48 (1962).
547. Boef G., Rolf-Nugteen J., Laar B. Z. anal. Chem., 166, 422 (1959).
548. Bognar J., Sarosi S. Mikrochim. acta, 1969, 361.
549. Boltz G., Wiedemann H., Kurella W. Metall, 10, 821 (1956).
550. Bonhorst C. W., Mattice J. Analyt. Chem., 31, 2106 (1959).
551. Bordea A., Moroi G., Rozmarin M. РЖХим., 7Г110 (1965).
230
552. Bowen H. J. M., Cawse P. A., Analyst, 88, 721 (1963).
553. Brady IT. £., Lyons R. E. J. Am. Chem. Soc., 48, 2642 (1926}.
554. Brasted R. C. Comprehensive inorganic chemistry, v. 8. Sulfur, selenium, tellurium, polonium and oxygen. N. Y. 1961.
555. Broad IT. C., Barnard A. J. Chemist-Analyst, 50, 124 (1961).
556. Brookshier R. K., Freund H. Analyt. Chem., 23, 1110 (1951).
557. Browing P. E., Flint W. R. Am. J. Sci., 28, 112 (1909).
558. Brown E. G. Analyst, 78, 623 (1953).
559. Brown E. G. Analyst, 79, 50 (1954).
560. Bruckner K. Z. anal. Chem., 94, 305 (1933).
561. Brukl A., Maxymowicz W. Z. anal., Chem., 68, 16 (1926).
562. Brunfelt A. 0., Steinnes E. Geochim. et cosmochim. acta, 31, 283 (1967).
563. Burke К. E., Yanak M. M., Albright С. H. Analyt. Chem., 39, 14 (1967).
564. Burnel D. Chim. analyt., 51, 83 (1969).
565. Burriel M. F., Alvarez H. L. Inform, quim. analit., 16, 68 (1962).
566. Burstall F. H., Davies G. R., LinsteadR. B., Wells B. A. J. Chem. Soc., 1950, 524.
567. Bush E. L. Analyst, 88, 614 (1963).
568. Bush E. L„ Cornish E. H. C. A., 57, 6608 (1962).
569. Caley E. R., Henderson B. Analyt. Chem., 32, 975 (1960).
570. Candler C. Atomic Spectra Hinger and Watts. London, 1964.
571. Carter T., Parker A. Res. Group U. K. AtomicEnergyAuthor., 1965, N AERE-R 5293; РЖХим., 17Г167 (1967).
572. Cathro K. J. Austral. J. Appl. Sci., 9, 255 (1958).
573. Chaktabarti Chuni L. Analyt. chim. acta, 39, 293 (1967).
574. Chakrabarti Chuni L. Analyt. chim. acta, 42, 379 (1968).
575. Challis H. I. G. Analyst, 67, 186 (1942).
576. Chan F. L. Advances X-ray Analysis, 1964, p. 542.
577. Chan F. L. Taianta, 11, 1019 (1964).
578. Chan F. L., Riley J. P. Analyt. chim. acta, 33, 36 (1965).
579. Chaodarova R., Sheytanov Ch. Dokl. Bulgar Akad. Nauk, 20, 565 (1967).
580. Cheng K. L. Analyt. Chem., 28, 1738 (1956).
581. Cheng K. L. Analyt. Chem., 32, 981 (1960).
582. Cheng K- L. Analyt. Chem., 33, 761 (1961).
583. Cheng K. L. Taianta, 8, 301 (1961).
584. Cheng K- L. Taianta, 9 , 501 (1962).
585. Cheng K. L., Goydisch B. L. Analyt. Chem., 35, 1965 (1963).
586. Cheng K- L., Goydisch B. L. Taianta, 13, 1210 (1966).
587. Chinaglia B., Cin L., Fasolu G. B., Malvano R. Energia nucl., 9, 503 (1962).
588. Christian G. D., Knoblock E. C. Analyt. Chem., 35, 1128 (1963).
589. Claasen A., Bastings L. Z. anal. Chem., 160, 403 (1958).
590. Clark R. E. Analyst, 82, 182 (1957).
591. Clark R. E. Analyst, 83, 396 (1958).
592. Claude WaringL., Worthing H. W„ Hazel К. V. Analyt. Chem., 30, 1504 (1958).
593. Claude Le Peintre. C. r. Acad, sci., 252, 1968 (1961).
594. Claude Le Peintre. C. r. Acad, sci., 253, 1696 (1961).
595. Clauder О. E. Z. anal. Chem., 89, 270 (1932).
596. Close P., Raggon F. C., Smith W. E. J. Am. Ceram. Soc., 33, 345 (1950).
597. Cludey H. J., Proffitt P. M. C. Analyst, 85, 815 (1960).
598. Colleman W. C., Me Grosky C. R. Ind. Eng. Chem., Anal. Ed., 28, 19 6 (1936).
599. Colleman W. C., Me Crosky C. R. Ind. Eng. Chem., Anal. Ed., 29, 431 (1937).
600. Colleman W. C., Me Crosky C. R. J. Am. Chem. Soc., 59, 1458 (1937).
601. Collins A. G., Waters C. J., Pearson C. A. Bur. Mines, U. S. Dept Interior, N 6474, 19 (1964).
602. Cornwell J. C., Cheng K. L. Analyt. chim. acta, 42, 189 (1968).
603. Conrad F. L., Кеппа В. T. Analyt. Chem., 39, 1001 (1967).
604. Cousins F. B. Austral. J. Exptl Biol, and Med. Sci., 38, 11 (1960).
605. Cousins F. B., Cairhey J. M. Austral. J. Agric. Res., 12, 927 (1961).
231
606. Cresser M. S., West T. S. Taianta, 16, 416 (1969).
607. Crouthamel С. E., Taianta, 1, 39 (1958),
608. Crossley P. B. Analyst, 69, 206 (1944).
609. Cukor P. Analyt. chim. acta, 30, 473 (1964).
610. Cummins L. M., Martin I. L., Maky G. W., Macg D. D. Analyt. Chem., 36, 382 (1964).
611. Cvjetieanin N., Jovanovie V. Bull. «В. Kidrich» Inst. Nucl. Sci., 13, 43 (1962). РЖХим, 11Г54 (1963).
612. Dagnail R. H., Thompson К. C., West T. S. Taianta, 14, 557 (1967).
613. Dahl I. J., Langmyhr F. J. Analyt. chim. acta, 35, 24 (1966).
614. Dams R., Hoste J. Analyt. chim. acta, 41, 205 (1968).
615. Danksas K., Naruskevieins L. C. A., 54, 11831 (1960).
616. Danzuka T., Ucuo K. Analyt. Chem., 30, 1370 (1958).
617. Dastoor M. I., Haidar В. C. Indian J. Chem., 5, 335 (1967).
618. Date Kendzi. Japan Analyst, 17, 981 (1968); РЖХим., 24Г149 (1968).
619. Daugwall D., Williams W. D. Z. anal. Chem., 186, 374 (1962).
620. Deau J. A., Simms J. C. Analyt. Chem., 35, 699 (1963).
621. Demco R. H. Analyt. Chem., 20, 488 (1948).
622. Demeyere D., Hoste J. Analyt. chim. acta, 27, 288 (1962).
623. DeshmukhG. S., Anand V. D., Vishwanath A. K. Ann. chim. (Rome), 50, 439 (1960).
624. Deshmukh G. S., Asthana О. P. Naturwissenschaften, 48, 477 (1961).
625. Deshmuck G. S., Bapat M. T. Z. anal. Chem., 170, 381 (1959).
626. Deshmukh G. S., Sankare Rao V. S. Current Sci., 37, 584 (1968); РЖХим., 13Г127 (1969).
627. Deshmukh G. S., Sankara Rao V. S. Z. anal. Chem., 240, 322 (1968),
628. Dickey D. W., Wiersma J. H., Barnekow R. G.t Lott P. F. Mikrochim. acta, 1969, 605.
629. Dobici F., Salvetti F. J. Inorg. and Nucl. Chem., 26, 911 (1964).
630. Dobrowolski J. Roczn. chem., 33, 1459 (1959).
631. Dobrowolski J. Roczn. chem., 35, 1555 (1961).
632. Dobrowolski J., Prejzner J., Skarzeynska T. Roczn. chem., 35, 37 (1961).
633. Dolique R., Perahia S. Bull. Soc. chim. France, 13, [5] 44, (1946).
634. Dostal K., Zborilova L. Chem. Ges., 9, 761 (1962).
635. Dowson W. M. Mikrochim. acta, 1969, 202.
636. Dryer H. T., Xerocostas M. Analyt. Chem., 27, 327 (1953).
637. Du Frense A. Geochim. et cosmochim. acta 20, 141 (1960).
638. Dupnis M. Note С. E. A„ N 394, 5 (1962); РЖХим., 2Б329 (1964).
639. Dye W. В., BretthauerE., SeimH. J,, Blincoc.C. Analyt.Chem., 35, 1687 (1963).
640. Ebner E. Z. anal. Chem., 206, 106 (1964).
641. Echegaray R. M., Quispe P. L. Bol. Soc. quim. Peru, 25, 179 (1959); РЖХим., № 23, 92081 (1960).
642. Edwards I. A., Laferrvere A. L. Chemist-Analyst, 45, 12 (1956).
643. Edwards A. J., Monty M. A. J. Chem. Soc., 1964, 4087.
644. Eiss M., Giesecke P. Engng and Mining J., 160, 102 (1959); 15, 61101 (1960).
645. Emeleus H. J., Anderson J. S. Modern aspects of Inorganic chemistry. London, .
1940, p. 299.
646. Engelbrecht A., Clementi F. Monatshefte, 92, 555 (1961).
647. Engelbrecht A., Sladky F. Angew. Chem., 76, 379 (1964).
648. Engelbrecht A., Sladky F. Nucl. Chem., 1, 15 (1965); РЖХим., 15B31 (1966).
649. Eredey L., Gimesi 0., Rady G. Taianta, 11, 461 (1964).
650. Eredey L. Magyar kem. folyoirat., 56, 265 (1950).
651. Etten N., Muschaweck J. Z. analyt. Chem., 206, 17 (1964).
652. Evans B. S. Analyst, 63, 874 (1938).
653. Evans B. S. Analyst, 67, 346 (1942).
654. Evans B. S. Analyst, 67, 387 (1942),
655. Evans B. S. C. A., 32, 8299 (1938).
656. Ewan R.C., Banmann C.A., Pope A. L.J. Agric. and FoodChem., 16, 212 (1968).
657. Falciola P. Ann. Chem. AppL, 17, 357 (1927).
232
658. Falciola P. Z. anal. Chem., 75, 410 (1927).
659. Falkner P. R. Mikrochim. acta, 1968, 1273.
660. Farkas U. К. РЖХим., 8Г150 (1966).
661. Feigl F. Analyt. chim. acta, 24, 501 (1961).
662. Feigl F. Spot tests in inorganic analysis. N. Y. Elsevier, 1958, p. 345, 349, 375, 378, 492.
663. Feigl F. Del-Acgua A. Chemist-Analyst, 55, 15 (1966).
664. Feigl F., Demant V. Mikrochim. acta, 1, 322 (1937).
665. Feigl F„ Uzel R. Mikrochemie, 19, 133 (1936).
666. Feigl F„ West P. W. Analyt. Chem., 19, 351 (1947).
667. Feldman L. J. Opt. Soc. America, 35, 180 (1945).
668. Filder I. Chem. Listy, 46, 221 (1952).
669. Fineman J., Lfnuggren K-, Forsberg H. T., Erwall L. G. Internet. J. Appl. Radiat. and Isotopes, 5, 280 (1959); РЖХим., № 9, 34534 (1960).
670. Fischer WBastins H. Neue Hiitte, 9, 434 (1964).
671. Fleishman D. M., Guinn V. P. Trans. Amer. Nucl. Soc., 7, 327 (1964).
672. Fletcher N. W„ Wardle R., Analyst, 82, 743 (1957).
673. Fogg P. N., Wilkinson N. T. Analyst, 81, 525 (1956).
674. Foss 0. Acta chem. scand., 3, 435, 708 (1949).
675. Foss 0. Acta chem. scand., 4, 1560 (1950).
676. Foss. 0. Acta chem. scand., 5, 115 (1951).
677. Foss 0. Acta chem. scand., 6, 521 (1952).
678. Foss 0. Acta chem. scand., 9, 1013 (1955).
679. Foss 0. Acta chem. scand., 15, 1939 (1961).
680. Foss 0. Acta chem. scand., 16, 779 (1962).
681. Foss 0., Fossen S. Acta chem. scand., 15, 1618 (1961).
682. Foss 0., Hauge S. Acta chem. scand., 13, 1252 (1959).
683. Foss 0., Hauge S. Acta chem. scand., 15, 1615, 1623 (1961).
684. Foss 0., Johanessen W. Acta chem. scand., 15, 1940, 1941 (1961).
685. Foss 0., Larssen 0. Acta chem. scand., 8, 1042 (1954).
686. Foss 0., Mary K- Acta chem. scand., 15, 1943, 1945, 1947 (1961).
687. Foss 0., Qyum P. Acta chem. scand., 9, 1012 (1955).
688. Foss 0., Vihovde E. H. Acta chem. scand., 8, 1032 (1954).
689. Frigyes Solymosi. Magyar kem. folydirat., 63, 313 (1957).
690. Fritsch R. Z. physiol. Chem., 104, 59 (1919).
691. Furman N,F., Mason W. B., Pekola B. Analyt. Chem., 21, 1325 (1949).
692. Futekov L., lordanov N. Taianta, 13, 763 (1966).
693. Gaittet J., Albert P. C. r. Acad, sci., 247, 1861 (1958).
694. Gareia F. H. C. r. Acad, sci., 258, 2579 (1964).
695. Gary Ch. D., Knoblock E. C., Purdy W. Analyt. Chem., 35, 1128 (1963).
696. Gebauhr W., Sprang A. Z. anal. Chem., 175, 175 (1960).
697. Geiersberger K. Z. anal. Chem., 135, 11 (1952).
698. Geiersberger K- Z. anal. Chem., 135, 18 (1952).
699. Geilmann V., WriggeF. W. Z. anorg. allgem. Chem., 210, 357 (1933).
700. Genkichi Nakagawa. J. Chem. Soc. Japan, Pure Chem. Sect., 81, 1255 (1960).
701. Genkichi Nakagawa. J. Chem. Soc. Japan, Pure Chem. Sect., 81, 446 (I960).
702. Genkichi Nakagawa, Nakanitsu M. Японск. пат. 15645 (1965); РЖМет.,7Г188 (1967).
703. Gerd M. Z. Erzbergbau und Metall, 18, 116 (1965); Metall. Abstr., 12, 1879 (1967).
704. Getoff N., Barker W., Atompraxis, 9, 175 (1963); РЖХим., 2Г327 (1964).
705. Getoff N„ Barker W. Nature, 197, 278 (1963).
706. Ghosh К- P. J. Indian Chem. Soc., Ind. News Ed., 11, 51 (1948).
707. Gilberston L. I., King G. B. J. Am. Chem., Soc., 58, 180 (1936).
708. Girnesi 0., Pady G. Acta chim. Acad. sci. hung., 33, 381 (1962); РЖХим., 15Г125 (1963).
709. Glen K-, Schwab R. Angew. Chem., 62, 320 (1950).
710. Gmelins Handbuch der anorganischen Chemie, Bd. 11, Tellur. Berlin, 1940, S. 334, 343, 349.
233
711. Goetz Ch. A., Debbrecnt F. G. Analyt. Chem., 27, 1927 (1955).
712. Gooch F. A., Pierce A. W. Am. J. Sci., [4] 1, 181 (1896).
713. Goren S. Israel Atom. Energy Commiss, N 639, 4 (1961); РЖХим., 12Д128 (1962).
714. Goren S., Mantel M., Stiller M. Israel Atom. Energy Commiss., N 724, 18 (1962); РЖХим., 5Г109 (1963).
715. Goren S., Stiller M., Mantel M. Israel Atom. Energy Commiss., N 620, 62 (1961); РЖХим., 8Д133, (1962).
716. Gorsuch T. T. Analyst, 84, 135 (1959).
717. Goto H. Sci. Repts Res. Inst. Tohoku Univ., 29, 204 (1940); C. A., 35, 1720 (1941).
718. Goto H. Sci. Repts Res. Inst. Tohoku Univ., All, 1, (1959); РЖХим., № 5, 17479 (1960).
719. Goto H., Ikeda S., Harayama T. Sci. Repts Res. Inst. Tohoku Univ., A5, 34 (1953).
720. Goto H., Kakita I. J. Chem. Soc. Japan, Pure Chem. Sect., 79, 1513 (1958); РЖХим., № 19, 676321 (1959).
721. GotoH., Kakita I. J. Chem. Soc. Japan, PureChem. Sect., 82, 1212, A-77(1961); РЖХим., 9Д13 (1962).
722. Goto H., Kakita I. Sci. Repts Res. Inst. Tohoku Univ., A7, 365 (1955); C. A., 506252a (1956).
723. Grabe P. Papierfabrikant, 8, 668 (1910).
724. Grant A. B. N. Z. J. Sci., 6, 577 (1963).
725. Grooker A. M., Joshi I. N. J. Opt. Soc. America, 54, 553 (1964).
726. Gubser H. Chimica, 13, 245 (1949); Z. anal. Chem., 175, 58 (1960).
727. Guilbault G. G., Melurdy W. H. Analyt. chim. acta, 24, 214 (1961).
728. Gullis J. Analyt. chim. acta, 8, 97 (1953).
729. Gutbier A., Engeroff E. Kolloid-Z., 15,- 193 (1914).
730. Gutbier A., Engeroff E. Z. Anal. Chem., 54, 193 (1915).
731. Gutbier A., Flury F. J. prakt. Chem., 83, 145 (1911).
732. Gutbier A., Flury F. J. prakt. Chem., 86, 150 (1912).
733. Gutbier A., Flury F. Z. anorg. allgem. Chem., 86, 169 (1914).
734. Gutbier A., Grunewald W. J. prakt. Chem., 85, 321 (1912).
735. Gutbier A., Rohn E. Z. anorg. allgem. Chem., 34, 448 (1903).
736. Hahn H., Bartels H. Mikrochim. acta, 1961, 250.
737. Hahn H., Kleinwort W. 7. anal. Chem., 151, 40, 98 (1956).
738. Haissinsky M., Raitz A. Analyt. chim. acta 45, 143 (1969).
739. Haisty R. W. Thesis Univ. Illinois, Urbana, 1955.
740. Hall R. J., Gupta P. L. Analyst, 94, 292 (1969).
741. Hall A. J., Young R. S. Analyst, 71, 419 (1946).
742. Натре W. Chem. Ztg., 15, 443 (1891).
743. Handley R. Analyt. Chem., 32, 1719 (1960).
744. Handley R., Johnsow С. M. Analyt. Chem., 31, 2105 (1959).
745. Hanson M. W., Bradbury W. C., Carlton J. K- Analyt. Chem., 29, 4, 490 (1957).
746. Hecht F., Johw L. Z. anorg. allgem. Chem., 251, 14 (1943).
747. Heller K., Wagner U. 7. anal. Chem., 178, 428 (1961).
748. Herr W., Wolflle R. 7. anal. Chem., 209, 213 (1965).
749. Hikitne S. Bull. Chem. Soc. Japan, 33, 761 (1960); РЖХим., 11Д161 (1961).
750. Hikime S., Yoshida H., Uzumasa Y. Japan Analyst, 8, 531 (1959); РЖХим., 69106 (1960).
751. Hikime S., Yoshida H., Yamamoto M. Japan Analyst, 10, 112 (1961).
752. Hinsberg 0. Ber., 22, 2895 (1889).
753. Hinsberg 0. Ber., 52, 21 (1919).
754. Hiroshi Matsuo. J. Sci. Hiroshima Univ., Ser. A-22, 51 (1958).
755. Hoste J. Analyt. chim. acta, 2, 402 (1948).
756. Hoste J., Gillis J. Analyt. chim. acta, 12, 158 (1955).
757. Hovorka V. Coll. Czech. Chem. Comm., 4, 400 (1932)
758. Hu Zhi-tei. Acta chim. sinica, 30, 426 (1964); РЖХим., 2Г58 (1966).
759. Hu Zhi-tei. Kexue tongbao, 17, 23 (1966).
234
760. Hu Zhi-tei et al. Acta chim. sinika, 30, 352 (1964); РЖХим., 14Г49 (1965).
761. Hu Zhi-tei et al. Kexue tongbao, 8, 54 (1963); РЖХим., 2Г62 (1965).
762. Hu Zhi-tei, Lin Cheng-li. Kexue tongbao, 12, 1103 (1964); РЖХим., 18Г38 (1965).
763. Hu Zhi-tei, Lin Cheng-li. Scientia sinica, 14, 1235 (1965).
764. Ihn W., Hesse G., Neuland P. Microchim. acta, 1962, 628.
765. Iguchi Akira. Bull. Chem. Soc. Japan, 31, 748 (1959); РЖХим., 12, 42053 (1959).
766. Inarida Mariko. Bull. Chem. Soc. Japan, 339, 403 (1966).
767. Inarida Mariko. Japan Analyst, 7, 76, 171, 174, 449 (1958); РЖХим., 21, 67698 (1959).
768. Inarido Mariko. J. Chem. Soc. Japan, Pure Chem. Sect., 79, 696 (1968).
769. Inarida Mariko. J. Chem. Soc. Japan, Pure Chem. Sect., 79, 721 (1958); РЖХим., 7, 22988 (1959).
770. Inarida Mariko. J. Chem. Soc. Japan, Pure Chem. Sect., 79, 968 (1955);
РЖХим., 21, 67698 (1959).
771. InaridaMariko. J. Chem. Soc. Japan, Pure Chem. Sect., 80, 399 (1959); РЖХим.
4, 13066 (1960).
772. Inarida Mariko. J. Chem. Soc. Japan, Pure Chem. Sect., 80, 273, 280 (1959),
81, 1955 (1960); РЖХим., 2 , 4561, 4562 (1960).
773. Ingamells C. 0., Sandell E. B. Microchem. J., 3, 3 (1959); РЖХим., 17, 60553 (1959).
774. lordanov N. Z. anal. Chem., 228, 62 (1967).
775. lordanov N., Futekov L. Taianta, 12, 371 (1965).
776. lordanov N., Futekov L. Taianta, 13, 163 (1966).
777. lordanov N., Futekov L. Taianta, 15, 850 (1968).
778. lordanov С. V., Perkova E. N. Neue Hiitte, 12, 335 (1967).
779. Ishibashi M. et al. J. Chem. Soc. Japan, Pure Chem. Sect., 88, 76 (1967); РЖХим., 9Г168 (1968).
780. Ishibashi M., Shigematsu T., Nakagawa Y. Res. Oceanogr. works Japan, 2, 44 (1953).
781. Ishihara I. Japan Analyst, 12, 380 (1963).
782. Ishihara I., Kishi H., Komuro H. Japan Analyst, 11, 932 (1962).
783. Ishihara I., Koga M. Japan Analyst, 11, 566 (1962).
784. Ishimori J., Watanaba K-, Nakamura K. Bull. Chem. Soc. Japan, 33, 636 (1960).
785. Issa I. M., Allarn M. G. Z. anal. Chem., 172, 87 (1960).
786. Issa I. M., Awad S. A. Analyst, 78, 487 (1953).
787. Issa I. M., Harndy M. Z. anal. Chem., 172, 162 (1960).
788. Issa I. M., Handly M. Z. anal. Chem., 172 , 94 (1960).
789. Issa I. M., Issa R. M. Analyt. chim. acta, 11, 275 (1954).
790. Issa I. M., Issa R. M., Allarn M. G. Analyt. chim. acta, 23, 196 (1960).
791. Issa I. M., Issa R. M., Azitn A. Analyt. chim. acta, 11, 512 (1954).
792. Issa R. M., Nabey B. A. Z. anal. Chem., 231, 339 (1967).
793. Isao Muroki. J. Chem. Soc. Japan. Pure Chem. Sect., 76, 193 (1955).
794. Ito Saburo, Kenjiro Hayashi. Japan Analyst, 12, 257 (1963).
795. Itsuki K., Ide A., Minehira A. Japan Analyst, 9, 840 (1960).
796. Itsuki K., Kaji T. Japan Analyst, 8, 703 (1959).
797. Iwasaki J., Kishioka A., Joshida J., Kagaku B. Japan Analyst, 10, 479 (1961); РЖХим., 24Д104 (1963).
798. Jackwerth Ewald. Z. anal. Chem., 235, 235 (1968).
799. Jander J., Doetsch V. Angew. Chem., 70, 704 (1958).
800. Jander J., Doetsch V. Ber., 93, 561 (1960).
801. Jankovsky J., Ksir 0. Taianta, 5, 238 (1960).
802. Jenkins J. J. Am. Chem., Soc., 74, 1353 (1952).
803. Jensen R. Chim. analyt., 41, 394 (1959).
804. Jilek A., Lukas J. Chem. listy, 21, 576 (1927).
805. Jilek A., Vrestal J. Chem. zwesti, 6, 497 (1957).
806. Jilek A., Vrestal J. Chem. zwesti, 7, 33 (1953).
807. Jilek A., Vrestal J. Chem. zwesti, 7, 625 (1953).
235
808. Jilek A., Vrestal J., Havir J. Chem. zwesti, 10, 110 (1956).
809. Jilek A., Vrestal J., Havir J. Z. anal. Chem., 154, 364 (1957).
810. Jimenez-Soeo J. L., BurrielF., Sistiaga J. M. An. Real. Soc. esp. fis. у quim., B58, N 11, 669 (1962).
811. Johnson R. A. Analyt. Chem., 25, 1013 (1953).
812. Johnson R. A., Andersen R. Analyt. Chem., 27, 120 (1950).
813. Johnson R. A., Fredrickson D. Analyt. Chem., 24, 866 (1952).
814. Johnson R. A., Kwan F. P. Analyt. Chem., 23, 651 (1951).
815. Johnson R. A., Kwan F. P. Z. anal. Chem., 135, 11 (1952).
816. Johnson R. A., Kwan F. P., Westlake D. Analyt. Chem., 25, 1017 (1953).
817. Ka J., Lasa J., Ostronski K. Z. Krajowe simpoz zartosow izotop techn. Szczecin,
1966, p. 87; РЖХим., 24Г51 (1966).
818. Kakumoto Susumo. Japan Analyst, 13, 1016 (1964); РЖХим., 13Г144 (1965).
819. Kamoru Ochiko, Ono Akichiro. Japan Analyst, 15, 290 (1966); РЖХим., 4Г109 (1967).
820. Kaneko H. C. A., 55, 17369e (1961).
821. Katazato F., Sacki Y. Japan Analyst, 8, 422 (1959); C. A., 54, 20639 (1966).
822. Kawashima Takuji, Tanaka Motoharu. Analyt. chim. acta, 40, 137 (1968).
823. Kelleher W- H., Johnson M. J. Analyt. Chem., 33, 1429 (1961).
824. Keller E. J. Am. Chem. Soc., 22, 241 (1900).
825. Kempe G. Z. anal. Chem., 217, 169 (1966).
826. Kendjiro H., Toshio 0. Japan Analyst, 14, 1146 (1965); РЖХим., 24Г61 (1966).
827. Kenjiro H., Toshio 0. Japan Analyst, 15, 1120 (1966); РЖХим., 13Г121 (1967).
828. Keshava K-, Haidar В. C. Nature, 207, 187 (1965).
829. Kiba T., Akaza IHachino H. Bull. Chem. Soc. Japan, 32 4 54 (1959); РЖХим.,
7, 26336 (1960).
830. Kimura K. Bull. Chem. Soc. Japan, 34, 63 (1961); C. A., 55, 17338d (1961).
831. Kimura K-, Ikeda Nagao. Japan Analyst, 7, 174 (1958); РЖХим., 1, 974 (1959).
832. Kirihide Kedzi, Oroei Jsuaki. Anal, and Instrum., 7, 3 (1969).
833. Kirkbright G. F. Analyt. chim. acta, 35, 116 (1966).
834. Kirkbright G. F., Yoe J. H. Analyt. Chem., 35, 808 (1963).
835. Kirkbright G. F., Sargentili, West T. S. Atom. Absorpt. Newsletter, 8, 34 (1969).
836. Kisaburo Tabashi, Makoto Otomo. Sci. Rept, 2, 1, 6 (1955), C. A., 53, 13888b (1959).
837. Klason M. Ber. d. Ver. Zellstoff. u. Papierenken, 1911, 81.
838. Kaduo Kadaku. J. Chem. Soc. Japan, Industr. Chem. Sect., 63, 1580 (1960).
839. Koch G. Mikrochim. acta, 1958, 402.
840. Koch H., Schulze K- Kerntechnik, 7, 9 (1965); C. A., 62, 13814a (1965).
841. Koch H., Schulze K. Z. anal. Chem., 210, 90 (1965).
842. Kolditz L., Fitz J. J. Chem., 4, 350 (1964).
843. Kotarski A. Chim. analyt., 10, 161 (1965); РЖХим., 27101 (1966).
844. Kotarski A. Chim. analyt.. 10, 321 (1965).
845. Kotarski A., Marezenko L. Chim. analyt., 5, 235 (1960).
846. Kotowska M., Szczepkowska-Mamczarezyk I. Rudy i Metalle niezel., 13, 410 (1968).
847. Kotowska M., Szczepkowska-Mamezarezyk I. Techn. poszur, 6, 59 (1967).
848. Kriege О. H., Theodore M. L. Taianta, 13, 265 (1966).
849. Kuroda Rokuro, Ishida Koji, Kiriyama Tetsuya. Analyt. Chem., 40, 1502 (1968).
850. Kujawa R. Mikrochim. acta, 1969, 193.
851. Kujawa R. Z. phys. Chem., 221, 269 (1962).
852. Kyoji Toci, Keiko Ito. Taianta, 12, 773 (1965).
853. Lakin H. W., Thompson С. E. Science, N 3575, 141 (1963).
854. Lakin H. W., Thompson С. E. U. S. Geol. Surv. Profess. Paper, N 450-E, 128 (1962); РЖХим., ЗГ108 (1964).
855. Lanese J. G., Jaselskis B. Analyt. chim. acta, 29, 415 (1963).
856. Lanese J. G., Jaselskis B. Analyt. chim. acta, 29, 415 (1963).
857. Lang R. Z. anal. Chem., 128, 484 (1948).
858. Lang R. Z. anorg. allgem. Chem., 42, 280 (1925).
859. Lang R.t Fande E. Z. anal. Chem., 108, 258 (1937).
236
860. Langmyhr F. J., Dahl I. J. Analyt. chim. acta, 29, 377 (1963).
861. Langmyhr F. J., Dalh I. J. Analyt. Chem., 37, 200 (1965).
862. Langmyhr F. J., Myhrstad L. A. Analyt. chim. acta, 35, 212 (1966).
863. Langmyhr F. J., Norheim G. Analyt. chim. acta, 41, 341 (1968).
864. Langmyhr F. J., Omang S. H. Analyt. chim. acta, 23, 565 (1960).
865. Lansche A. M. Inform. Circ. Bur. Mines. U. S. Dept Interior. N 8340, 56 (1967).
866. Leddicotte G. N., Reynolds S. A. Nucleonics, 8, 62 (1951).
867. Lederer M. Kerles S. Analyt. chim. acta, 15, 226 (1956).
868. Lenher V. J. Am. Chem. Soc., 22, 136 (1908).
869. Lenher V., Hornberger A. W. J. Am. Chem. Soc., 30, 387 (1908).
870. Lenher V., Kao С. H. J. Am. Chem. Soc., 47, 769 (1925).
871. Lenher V., Smith D. P. Ind. Eng. Chem., 16, 837 (1924).
872. Lenher V., Titus W. J. Am. Chem. Soc., 25, 730 (1903).
873. Lenher V., Wakefield F. J. Am. Chem. Soc., 45, 1423 (1923).
874. Leontowitch N. Chim. analyt., 41, 56 (1959); РЖХим., 15, 53163 (1959).
875. Leontowitch N. Chim. analyt., 42, 329 (1960); РЖХим., ЗД117 (1961).
876. Leontowitch N. Chim. analyt., 42, 549 (1960); РЖХим., 11Д160 (1961).
877. Leontowitch N. Chim. analyt., 43, 391 (1961); РЖХим., 8Д134 (1962).
878. Levi M. C., Danon J. J. Chromatogr., 6, 584 (1960).
879. Levine V. E. C. A., 31, 8617 (1937).
880. Lewis I. T. Analyt. Chem., 32, 1807 (1960).
881. Lihl F., Paten P., Soraktin H. Z. anal. Chem., 221, 176 (1966).
882. Lingane J. J., Niedrach L. W. J. Am. Chem. Soc., 70, 4115 (1948).
883. Lingane J. J., Niedrach L. W. J. Am. Chem. Soc., 70, 1997 (1948).
884. Lingane J. J., Niedrach L. IJ7. J. Am. Chem. Soc., 76, 196 (1949).
885. Ljung H. A. Ind. Eng. Chem. Anal., Ed., 9, 328 (1937).
886. Lotte P. F., Cukor P., Moriber G., Solga J. Analyt. Chem., 35, 1159 (1963).
887. Lowy A., Dunbrook R. J. Am. Chem. Soc., 44, 614 (1922).
888. Lowy A., Dunbrook R. J. Am. Chem. Soc., 22, 136 (1900).
889. Lucas J., Jilek A. Chem. listy, 20, 396 (1926).
890. Luke C. L. Analyt. Chem., 28, 1276 (1956).
891. Luke C. L. Analyt. Chem., 31, 572 (1959).
892. Lupant F. A., Sollner K. Z. anal. Chem., 195, 393 (1963).
893. Luzzati V. Acta crystallogr., 32, 1807 (1960).
894. Maassen G. Z. Erbzergbau and Metallhiittenwes, 18, 116 (1965); РЖХим., 19Г112 (1965).
895. Mabuchi H. Bull. Chem. Soc. Japan, 29, 842 (1956).
896. Mabuchi H., Isao 0. Bull. Chem. Soc. Japan, 38, 1478 (1963), 29, 842 (1956); РЖХим., 12B3245 (1966).
897. Mabuchi H., Nakahara H. Bull. Chem. Soc. Japan, 36, 151 (1963); C. A., 59, 9626 (1963).
898. Macharo Nobumasa, Nakamura Yasucu. Japan Analyst, 12, 584 (1963).
899. Maddock R. S., Meinke W. W. Univ. Michigan Progress Rept, 8, 94 (1959).
900. Maddock R. S., Meinke W. W. Analyst, 88, 721 (1963).
901. Maienthal E. J., Taylor J. Analyt. Chem., 37, 1516 (1965).
902. Makoto Munemori. Bull. Univ. Osaka Prefect., Ser. A, 10, 57 (1962); C. A., 57, 9707 (1962).
903. Mandravel A., Gutul M. Rev. chim. (RSR), 19, 734 (1968); РЖХим., 18Г108 (1969).
904. Mao Chzi-San. Acta chim. sinica, 31, 432 (1965); РЖХим., 22Г51 (1966).
905. Marcec M. V., Kinson K-, Belcher С. B. Analyt. chim. acta, 41, 447 (1968).
906. Marcu C., Tolea F. Studia Univ. Babes-Bolyai, Sect. Chem., 88, 101 (1963);
C. A., 61, 10012 (1964).
907. Marheuke K-, Sandell E. B. Analyt. chim. acta, 38, 421 (1967).
908. Marinari P. U. Chimica, 38, 129 (1962); РЖХим., ЗГ109 (1963).
909. Marki I., Bobleter 0. Z. anal. Chem., 219, 160 (1966).
910. Marvin G. G., Schumb W. C. Ind. Eng. Chem. Anal. Ed., 7, 423 (1935).
911. Marvin G. G., Schumb W. C. Ind. Eng. Chem., Anal. Ed., 8, 108 (1936).
g12. Masao Tanaka, Japan Analyst, 12 (10), 897 (1963); C. A., 60, 3483 (1964).
237
913. Masao Tanaka, Kawashima T. Taianta, 12, 211 (1965).
914. Masao Tanaka, Shoidci Shirakawa. Japan Analyst, 12, 961 (1963).
915. Mathews F. C., Turner H. C. Trans. Am. Electrochem. Soc., 54, 293 (1928).
916. Maxwell J. A. Rock and mineral analytics. New York — London — Sydney!» Toronto. Interscience, 1968.
917. McGee, Lynch J., Boswell G. G. Taianta, 15, 1435 (1968).
918. Meier E., Shaltiel N. Mikrochim. acta, 1960, 580; C. A., 16280d (1961).
919. Me Nulty I. S. Analyt. Chem., 19, 809 (1947).
920. Me Nulty I. S. Analyt. Chem., 25, 123 (1951).
921. Me Nulty I. S., Center E. I., Macintosh R. M. Analyt. Chem., 23, 123 (1951).
922. Mecke Z. Z. anal. Chem., 39, 468 (1900).
923. Merle J. Eiss Eng. Mining J., 160, 102 (1959).
924. Metzner R. C. r. Acad. Sci., 124, 32 (1959).
925. Metzner R. C. r. Acad, sci., 125, 23 (1959).
926. Meyer J., Garn W. Z. anal. Chem., 53, 29 (1914).
927. Meyer J., Wurm R. Z. anorg. allgem. Chem., 190, 191 (1930).
928. Michal J., Zyka I. Coll. Czech. Chem. Comm., 20, 305 (1955).
929. Mihalka J. Acta chim. Acad. sci. hung., 30, 359 (1962).
930. Mijamoto M. Japan Analyst, 10, 98 (1961).
931. Mijamoto M. Japan Analyst, 10, 211 (1961).
932. Mikulski L., Nukleonika, 11, 57 (1966); C. A., 3278e (1966).
933. Mikulski J., Stronski I. Nature, 207, 749 (1965).
934. Mikulski J., Stronski I., Nukleonika, 6, 775 (1961); РЖХим., 20B1 (1962).
935. Milazzo G., Meri F. Rend. Inst. Super sanita, 26, 52 (1963); РЖХим., 15Г94 (1964). Analyt. Chem., 52, 858 (1962); РЖХим., 15Г126 (1963).
936. Minori Saito, Iwate Daigaky. Kogaku bu Kenkyn Hokoku, 13, 257 (1960); РЖХим., 11Д159 (1961).
937. Misra G. I., Tandon I. P. Indian J. Chem., 5, 560 (1967).
938. Mittal R. K.., Mehrotra R. C. Z. anal. Chem., 196, 169 (1963).
939. Miura Toshio, Ken-Ichi Tsutsumi. Japan Analyst, 13, 768 (1964); РЖХим., 4Г86 (1965).
940. Molloy I. P. J. Agric. Res., 6, 133 (1967).
941. Monde Ch., Patil R. N., Indian J. Pure and Appl. Phys., 1, 435 (1963).
942. Monde Ch., Patil R. M., Nigavekar A. S. Indian J. Pure and Appl. Phys.
3, 483 (1965).
943. Monde Ch., Patil R. N., Nigavekar A. E. Nature, 211, 518 (1966).
944. Morimoto Suzuki. J. Elektrochem. Japan, 23, 544 (1955); РЖХим., 19, 61864 (1956).
945. Morris D. F. C., Hill W. Metallurgia, 71, 99 (1965).
946. Morris D. F. C., Killick R. A. Taianta, 10, 279 (1963).
947. Moser L., Miksdi R. M. Chem. Ztg., 44, 360 (1924).
948. Muehlber G. W., Lanheger V. J. Am. Chem. Soc., 47, 1842 (1925).
949. Muller E. Z. phys. Chem., 100, 346 (1922).
950. Munze Rudolf. J. prakt. Chem., 7, 262 (1959).
951. Muraki J. J. Chem. Soc. Japan, 76, 193 (1950).
952. Murthy A. S. Indian J. Chem., 3, 298 (1965).
953. Musa Soitiro, Munemoru Makoto. J. Chem. Soc. Japan, Pure Chem. Sect., 89, 495 (1968).
954. Muthmann W., Schmid A. Ber., 26, 1008 (1893).
955. Nagano Norumhitru. Hitachi hyoron, 42, 30 (1961); РЖХим., 11Д130 (1962).
956. Nakagawa H. M., Thompson С. E. Geol. Surv. Res., 600-13 (1968).
957. Nakashima S., Toei Kyoji. Taianta, 15, 1475 (1968).
958. Nakhamura Hiroshi. Japan Analyst, 13, 1273 (1964); РЖХим., 7Г1521 (1966).
959. Nakashima Susumu, Kjoji Toei. Taianta, 15, 1475 (1968).
960. Navratil 0., Sorfa J. Coll. Czech. Chem. Comm., 34, 975 (1969). •
961. Niebuhr P. E., Macmillan A. H. Rept Invest. U. S. Bur. Mines, N 6006, 6 (1962); РЖХим., 1Г78 (1963).
962. Nielsch W. Z. anal. Chem., 144, 191 (1955).
963. Nielsch W. Z. Anal. Chem., 155, 401 (1957).
238
964. Nielsch. W., Giefer G. Z. anal. Chem., 145, 347 (1955).
965. NieseS. Internal. Sympos. Reinstoffe Wiss. und Techn. Dresden, 1965, Teil 2. Berlin, 1966, S. 447—453; РЖХим., 7K87 (1967).
966. Niviere Rierre. Chim. analyt., 47, 125 (1965).
967. Noakes F. D. Z. Analyst, 76, 542 (1951).
968. Nobuhisa Matano. Japan Analyst, 11, 346 (1962).
969. Nonova D., Tuzsuzova A. Mikrochim. acta, 1964, 784.
970. Norris J. S., Mommers R. Am. Chem. J., 23, 486 (1900).
971. Norton I. Am. J. Sci., 7, 287 (1899).
972. Norwitz G. Analyt. chim. acta, 5, 109 (1951).
973. Okada Minoru. Nature, 187, 594 (1960).
974. Okada Minoru. Repts Govt Chem. Industr. Res. Inst. Tokyo, 58, 11 (1963);
975. Ordogh M., Schneer A. Magyar akad. Kozp. fiz. kutato int. kozl., 8, 39 (1960); РЖХим., 6Д166 (1961).
976. Osava Husao. Japan Analyst, 17, 457 (1968); РЖХим., 23Г20 (1968).
977. Oswald U. A. Analyt. Chem., 33, 1766 (1961).
978. Ota Kazuo. J. Japan Inst. Metalls, 23, 581 (1959); РЖХим., № 19, 77090 (1960).
979. Otvinovskii V., Yakovlev I. V. Acta chim. Acad. sci. hung., 26, 243 (1961).
980. Parker P. A., Harvey L. C. Analyst., 86, 54 (1961).
981. Parker P. A., Harvey L. C. Analyst, 87, 558 (1962).
982. Pen Li-huang, Chan Hsia-chang, Chung Shan-Ta. Hsu >h Hsuch Dao., N 2, 29 (1957); C. A., 54, 181901 (1960).
983. Petzold W. Z. anorg. Chem., 209, 267 (1932).
984. Pien L., Desirant J., Orsini F. Ann. falsif. et fraudes, 53, 83 (1960); C. A., 54, 13978 a (1960).
985. Pluskal H. Z. anal. Chem., 153, 214 (1956).
986. Pohl F. A. Mikrochim. acta, 1956, 414.
987. Pohl F. A., Hanskrecht T., Reese M. Пат. ФРГ, 1077645 (106Э); РЖХим., 16K86 (1962).
988. Pohl F. A., Kokes K. Mikrochim. acta, 1957, 318.
989. Pollard F. H. J. Chem. Soc., 1951, 470.
990. Pollard W. B. Analyst, 71, 221 (1946).
991. Pollock E. N. Analyt. chim. acta, 40, 285 (1968).
992. Pollock E. N. Taianta, 11, 1548 (1964).
993. Poluektoff W. Mikrochemie, 15, 32 (1934).
994. Poonia N. S. Mikrochim. acta, 1969, 217.
995. Popper R. Chem. listy, 59, 1476 (1965).
996. Postowsky J. J., Lugowkin В. P., Mardryk G. T. Ber., 69, 1913 (1936).
997. Prasad S., Khandelwal B. L. J. Indian. Chem. Soc., 37. 645 (1960).
998. Prasad S., KJiandelwal B. L. J. Indian Chem. Sac., 38, 107 (1961).
999. Prasad S., KJiandelwal B. L. J. Indian Chem. Soc., 38, 837 (1961).
1000. Prasad S., Khandelwal B. L. J. Indian Chem. Soc.,39, 67 (1962).
1001. Prasad S., Pathak Keshawa Chaundra. J. Indian Chem. Soc., 42, 373 (1965) РЖХим., 2B94 (1966); 10B131 (1966).
1002. Prasad S., Pathan Keshawa Chaundra. J. Indian Chem. Soc., 42, 698 (1965);
РЖХим., 14B193 (1966).
1003. Pribil R. Coll. Czech. Chenr Comm., 18, 780 (1953).
1004. Prideaux E., Millot 0. J. Chem. Soc., 1929, 2703.
1005. Pristavka D. Chem. zwesti, 12, 682 (1958).
1006. Pvocan J. Sulfuric Acid. Assoc. Japan, 13, 98 (1960); РЖХим., 1K24 (1961).
1007. Qureshi Monsin, Mathur K. N. Analyt. chim. acta, 41, 560 (1968); РЖХим., 20Г45 (1968).
1008. Qyum P., Foss O. Acta chem. scand., 9, 1012 (1955).
1009. Qyum P., Foss 0. Acta chem. scand., 10, 279 (1956).
1010. Raasch H., Salamon A. Taianta, 15, 975 (1968).
1011. Rai J., Kukreja V. P. Chromatographia, 1, 18 (1969).
1012. RajeicD., Dordevie S. Bull. Sci. Conseil Acad. RSFY, A/12, N 34, 65 (1967); РЖХим., 21Г106 (1967).
239
1013. Ramirez-Munoz. J. Rev. Soc. quim. Mexico, 12, 25B (1968); РЖХим., 16Г67 (1968).
1014. Ranu C. S., Hambly A. N. Analyt. chim. acta, 32, 346 (1965).
1015. Ray E. M. U. S. Atom. Energy Commiss, GAT-372 (1961); C. A., 55, 18444g (1961).
1016. Reath E. G. Analyt. Chem., 32, 981 (1960).
1017. Reed J. F. Analyt. Chem., 32, 662 (1960).
1018. Ripan R., Calu N. Studia Univ. Babes-Bolyai, Ser. Chim., 10, 135 (1965); РЖХим., 6B76 (1966).
1019. Ripan R., Calu N., Tantu G. Chim. analyt., 47, 299 (1965).
1020. Ripan R„ Maria M. РЖХим., 12B71 (1965).
1021. Ripan R., Marku G. Comm. Acad. R.P.R., 8, 467 (1958); РЖХим., 23091 (1959).
1022. Ripan 'R., Pallade R. C. A., 43, 4167h (1949).
1023. Ripan R., Pop G. Rev. chim. (R. P. R.), 14 , 413 (1963).
1024. Ripan R., Raseu N., Maren G. Z. anal, chem., 188, 131 (1960); РЖХим., 5Д115 (1962).
1025. Ripan R., Vericeanu G. Studia Univ. Babes-Bolyai, Ser. Chim., 13, 14 (1968); РЖХим., 9Г119 (1969).
1026. Robinson W'. O., Dydley H. C., Williams K. T., Byers H. G. Ind. Eng. Chem., Anal. Ed., 6, 274 (1934).
1027. Rockenbauer W. Osterr. Acad. Wiss. Math.-naturwiss. KI. Anz., 92, 192 (1955).
1028. Roguebert J., Vitte G. Bull. Soc. pharm. Bordeaux, 101, 29 (1962).
1029. Bohnsch W. Kernenergie, 7, 543 (1964).
1030. Rohnsch W. Microchim. acta, 1964, 1164.
1031. Rohnsch W. Mikrochim. acta, 1965, 10.
1032. Rosengeim A., Traube A. Z. anorg. allgem. Chem., 91, 75 (1915).
1033. Rosengeim A., Weinheber M. Z. anorg. allgem. Chem., 69, 266 (1911).
1034. Rossum J. R., Villarur J. Am. Water Works Assoc., 54, 746 (1962).
1035. Roth D. J., Bohl D. R., Sellers D. E. Atom. Absorption Newsletter, 7, 87 (1968).
1036. Rowly K., Swift E. H. Analyt. Chem., 27, 818 (1955).
1037. Rudnickie R. Chim. analyt., 9, 795 (1964).
1038. Rudnickie R., Koscielny J. Chim. analyt., 10, 425 (1965); РЖХим., 4Г49 (1966).
1039. Russell B. G., Luble W. V., Wilson A., Elaim J. Taianta, 14, 957 (1967).
1040. Sachs F. Ann., 365, 150 (1909).
1041. Sagortschew B., Bojkova D., Todorova N. Докл. Волг. АН, 18, 247 (1965);
РЖХим., 8Г47 (1966); 4Г50 (1967).
1042. Saito М., Jamasoki A. Bull. Chem. Soc. Japan, 25, 1055 (1963).
1043. Saito M. Iwate Daigaku Kogakaba Kenkyw Hakoku, 13, 257 (1960); C. A.,
55, 16280 (1961).
1044. Salaria G. B. S. Analyt. chim. acta, 18, 520 (1958).
1045. Salaria G. B. S. Analyt. chim. acta, 19, 604 (1918).
1046. Sanders A. P. J. Phys. Chem., 4, 423 (1900).
1047. Sasaki J. Bull. Chem. Soc. Japan, 28, 89, 615 (1955); РЖХим., № 23, 55397 (1955).
1048. Savencu S., Bordea A., Morioi G., Rosmarin M., Matel F. Bui. Inst. Politechn. Jasi, 12, 157 (1966); РЖХим., 2Г79 (1968).
1049. Sawicki E. Analyt. Chem., 29, 1376 (1957).
1050. Sawicki E., Carr A. J. Org. Chem., 22, 503 (1957).
1051. Saxeny О. C. Microchim. acta, 1967, 1123.
1052. Scherhaufer A. M. Z. anal. Chem., 164, 327 (1958).
1053. Schindewoff U. Geochim. et cosmochim. acta, 19, 134 (1960).
1054. Schmidt E. Arch. Pharmaz. Ber. Dtsch. pharmaz. Ges., 252, 161 (1914).
1055. Schneer A., Grdogh M. J. Chromatogr., 4, 319 (I960).
1056. Schoeller W. R. Analyst, 64, 318 (1939).
1057. Schrenk W. T., Browning B. L. J. Am. Chem. Soc., 48, 139 (1926).
1058. Schrenk W. T., Browning B. L. J. Am. Chem. Soc., 48 , 2550 (1926).
240
1059. Schriiber E. Z. anal. Chem., 210, 93 (1965).
1060. Schulek E., Barcza L. Acta chim. Acad. sci. hung., 37 (4), 351 (1963).
1061, Schulek E., Barcza L. Taianta, 3, 23, 27, 31 (1959).
1062. Schulek E., Foti Gy. Analyt. chim. acta, 3, 665 (1949).
1063. Schulek E., Koros E. J. Inorg. Nucl. Chem., 13, 58 (1960).
1064. Schulek E., Koros E. Magyar tud. akad. Kern, tudoszt kozl., 12, 175 (1959).
1065. Schulek E., Koros E. Z. anal. Chem., 139 , 20 (1953).
1066. Schulek E., Koros E., Barcza L. Ann. Univ. Sci. Budapest, Sect, chim., 2,
205 (1960); РЖХим., 11Д158 (1961).
1067. Schumann Horst. Пат. ГДР 32681 (1964); РЖХим., 5Л137 (1966).
1068. Schumann H., Koelling W. Z. Chem., 1, 371 (1961).
1069. Schwaer L., Suchy K. Coll. Czech. Chem. Comm., 7, 25 (1935).
1070. Seath J., Beamisch F. E. Ind. Eng. Chem. Anal. Ed., 9, 373 (1937).
1071. Seeley F. G., Crouse D. J. J. Chem. and Data, 3, 424 (1966).
1072. Seishi Jojima, Inchiro Kamemoto, Koreynki Shibo, Yoshihiro Onodo. J. Chem. Soc. Japan, Pure Chem. Sect., 82, 343 (1961); C. A., 16268g (1961).
1073. Scotte L. Г., Leonard G. IP. Analyt. Chem., 26, 445 (1954).
1074. Serlin I. Analyt. Chem., 35, 2221 (1963).
1075. Shao-Lin, Ming ling-chang. Hua Hsuch Hsuch Pao, 24, 306 (1958); C. A., 53, 14835d (1959).
1076. Shiraishi Jichi, Saito Minoru. Rept Technol. Iwate Univ., 10, 46 (1957); РЖХим., 3, 8921 (1960).
1077. Shni Yina-Chu. C. A., 54, 22159c (1960).
1078. Shui Chu-ying. РЖХим., 4, 13121 (I960).
1079. Sill C. N., Rept. Invest. U. S. Bur. Mines, 5047, 9p (1959); C. A., 48, 9860 (1954).
1080. Silverman. L. Industr. and Engng Chem., Analyt. Ed., 8, 132 (1936).
1081. Silverthorn R. W. Chemist-Analyst, 30, 52, 62 (1941).
1082. Simon V. Chem. listy, 48, 1774 (1954).
1083. Singh B., Sood К. C. Analyt. chim. acta, 13, 301 (1955).
1084. SkaarO. B., Laugmyhr F. J. Analyt. chim. acta, 23, 175 (1960).
1085. Sluzewska Leonia. Roczn. Panstw. zakl. hig., 15, 303 (1964); РЖХим., ЗГ163 (1965).
1086. Spraque S., Manning D. C., Slavin V. Atom. Absorption Newsletter, N 20 (1964).
1087. Srivastava M. N. Analyt. chim. acta, 20, 514 (1959).
1088. Stanton R. E., Alison I., McDonald A. Analyst, 90, 497 (1965).
1089. Stantscheff P. Z. anal. Chem., 220, 33 (1966).
1090. Stantscheff P. Z. anal. Chem., 233, 404 (1968).
1091. Stefanac Z., Rakovic Z. Microchem. J., 9, 442 (1965).
1092. Steinnes E. Internal. J. Appl. Radiat. and Isotopes, 18, 731 (1967).
1093. Strecker IP., Claus L. Ber., 56, 362 (1923).
1094. Strecker VF„ Ebert VF. Ber., 58, 2527 (1925).
1095. Strecker VF., Mahr C. Z. anorg. allgem. Chem., 221, 199 (1935).
1096. Strenge К. C. A., 55, 239g (1961).
1097. Stronski Iguacy. Roczn. chem., 34, 709 (1960).
1098. Suseda B. Z. analyt. Chem., 147, 13 (1955).
1099. Suzuki M. J. Chem. Soc. Japan, Industr. Chem. Sect., 56, 323 (1953).
1100. Suzuki Jukio, Nisijamo Keimaro. Shikoku acta med., 23, 137 (1967); РЖХим., 21Г143 (1968).
1101. SzuesA. I. Chem. analyt., 12, 459 (1967); РЖХим., 2Г116 (1968).
1102. Tabonry M. F., Quenille I. C. r. Acad, sci., 217, 150 (1943).
1103. Taimni I. K-, Agarwal R. P. Analyt. chim. acta, 9, 121 (1953).
1104. Taimni I. K-, Rakshpal Ram. J. prakt. Chem., 28, 134 (1965); РЖХим., 5Г57 (1966).
1105. Taimni I. K-, Tandon S. N. Analyt. chim. acta, 21, 110 (1953).
1106. Taimni I. K-, Tandon S. N. Analyt. chim. acta, 22, 553 (I960).
1107. Takano C. J. Sulluric. Acids Assoc. Japan, 19, 199 (1966); РЖХим., 13Г162
(1967).
241
1108. Takano C., Kaneko H. J. Sulfuric Acids Assoc. Japan, 13, 98 (1960); РЖХим., 1K24 (1961).
1109. Takano C., Miyazu T., Kaneko H. Sulfuric Acids Assoc. Japan, 13, 269 (1960); РЖХим., 12Д142 (1961).
1110. Tanaka K. Bull. Chem. Soc. Japan, 37, 1085 (1964); РЖХим., 6Г103 (1965).
1111. Tanaka K- Japan Analyst, 18, 315 (1969).
1112. Tanaka K. Report of the Govt Ind., Res. Inst. Nagoya, 8, 799 (1959); РЖХим., 56152 (1960).
1113. Tanaka K-, Kawashima J. Taianta, 12, 211 (1965).
1114. Tanaka K-, Takagu N. a. oth. Japan Analyst, 18 , 319 (1969).
1115. Tandon J. P., Mchrotra R. C. Z. anal. Chem., 162, 33 (1958).
1116. Tesuro Л4., Eidsen J. J. Chem. Soc. Japan, Industr. Chem. Sec., 66, 1952 (1963); РЖХим., 18г (1964).
1117. Tesuro Л4., Eidsen J. J. Chem. Soc. Japan, Industr. Chem. Sec., 68, 1865 (1965); РЖХим., 20Г125 (1966).
1118. Thompson С. E., Lakin H. II7. US. Geol. Surv. Profess. Paper, N 475—B, 164 (1963).
1119. Throop Lewis. Analyt. Chem., 32, 1807 (1960).
1120. Tilek A., Kota J. Coll. Czech. Chem. Comm., 6, 398 (1934).
1121. Tomicek 0. Rec. trav. chim. Pays-Bas, 43, 793 (1924).
1122. Tomicek 0. Z. anal. Chem., 77, 364 (1929).
1123. Tomicek 0., Procke О. I., Paoelka V.Coll. Czech. Chem. Comm., 11, 449 (1939).
1124. Topelman H. Z. Anal. Chem., 82, 284 (1930).
1125. Trandafirsen E., Yordachesen J., Draconesen G., Creanga S. E. Farmacia, 7, 135 (1959).
1126. Uzumasa Yasumitsu, Hayashi Keujiro, Ito Saburo. Bull. Chem. Soc. Japan, 36, 301 (1963); РЖХим., 22Г31 (1963).
1127. Uzumasa Yasumitsu, Hikime S., Hayashi K-, Yoshida H. Japan Analyst, 11, 78 (1962).
1128. Veale C. R. Analyst, 85, 130 (1960).
1129. Veale C. R. J. Inorg. and Nucl. Chem., 10, 333 (1959).
ИЗО. Vesely F., Suirous F., Viprek-Siska I. Chem. listy, 49, 1661 (1955); Z. anal.
Chem., 15, 362 (1956).
1131. Vrestal J. Coll. Czech. Chem. Comm., 25, 443 (1960).
1132. Wagenmann K., Triebel H. Metall und Ers, 27, 231 (1930).
1133. Watkinson J. H. Analyt. Chem., 32, 981 (1960).
1134. Watkinson J. H. Analyt. Chem., 38, 92 (1966).
1135. Wawrzyezek W., Wisniewski IF.Chem. analyt. (Polska), 8, 395 (1963); РЖХим., 4Г98 (1964).
1136. Wawrzyezek W., Wisniewski W. Chem. analyt. (Polska), 8, 543 (1963).
1137. Wawrzyezek W., Wisniewski W. J. analyt. Chem., 194, 346 (1963).
1138. Welcher F. J. Standard methods of chemical analysis, v. 2, pt B. 6-th ed. Toronto — London — New York., 1963.
1139. Wells H„ Willis J. Am. J. Sci., 12, 190 (1901).
1140. Wells N. N. Z. J. Geol. and Geophys., 10, 198 (1967).
1141. Wells R. C. J. Washington Acad. Sci., 18, 127 (1928).
1142. Werner 0. Metalls, 16, 1062 (1962).
1143. Wernimont G., Hopkinson F. J. Ind. Eng. Chem., Anal. Ed., 12, 308 (1940).
1144. West Philip W., Ramakrishna T. V. Analyt. Chem., 40, 966 (1968).
1145. Wester P. 0., Brune D., Samsah С. K- Internal. J. Appl. Radiat. and Isotopes, 15, 59 (1964).
1146. Wheeler H. i. anorg. Chem., 3, 428 (1893).
1147. White Weimar W., Sabo I. G. Taianta, 16, 452 (1969).
1148. Whitehead C. J. Am. Chem. Soc., 17, 280 (1895).
1149. Whitnack G. L„ Donovan T. M„ Ritchie M. H. РЖХим., 24Г150 (1967).
1150. Wild F. E. Atom. Energy Res. Establ., C/R, 2666, 1959, p. 12.
1151. Willard H. HPerkins L. R. Analyt. Chem., 25, 1634 (1953).
1152. Willard H. H., Young P. J. Am. Chem. Soc., 52, 553 (1930).
1153. Williams A. I. Analyst, 86, 172 (1961).
242
1154. Williams R., Haskett R. Analyt. Chem., 41, 1138 (1969).
1155. Williams К. T., Lakin H. W. Ind. Eng. Chem. Anal. Ed., 7, 409 (1935)
1156. Willis U. F. Analyst, 67, 219 (1942).
1157. Wish. L. Analyt. Chem., 32, 920 (1960).
1158. Wiiekowa S., Witek T., Paryjezak T. J. Chern., 2, 55 (1962).
1159. Wiikowska I. Prace Przemysl. inst. elektron, 6, 45 (1965); РЖХим., 13Г170 (1966).
1160. Wlotzka F. Z. anal. Chem., 215, 81 (1966).
1161. Worton E., Stenner K., MedaliaA. J. Am. Chem. Soc., 75, 1827 (1953).
1162. Wu James J., Drop Henry A., Lott Peter F. Atom. Absorpt. Newsletter, 7, 90 (1968); РЖХим., 8Г115 (1969).
1163. Yamamoto M., Sakai H. Analyt. chim. acta, 32, 370 (1965).
1164. Ying Chun-Wang, Feng Chiin-T'Vng. C. A., 54, 2089b (1960).
1165. Yojima S., Kamemoto Y., Shiba K-, Onoda Y. J. Chem. Soc. Japan, Pure Chem. Sect., 82, 343 (1961).
1166. Yokosuka S. C. A., 54, 7418 (1960).
1167. Yokosuka S., Katayama H. Nippon Kinzoku Gaukaishi, 19, 417 (1955); C. A., 54, 2090a (1960).
1168. Yokoyama J., Mabuchi H., Shoji H., Saito N. Bull. Chem. Soc. Japan, 36, 352 (1963).
1169. Yoshida H. Japan Analyst, 11, 549 (1962).
1170. Yoshida H., Hlkime S., J. Spectroscop. Soc. Japan, 14, 131 (1966).
1171. Yoshida H., Nikime S. Taianta, 11, 1349 (1964); РЖХим., 4Г87 (1965).
1172. Yoshida H., Taga M. Japan Analyst, 14, 1109 (1965); РЖХим., 24Г128 (1966).
1173. Yoshida H., TagaM., Hikime S. Japan Analyst. 16, 605 (1967).
1174. Yoshida H., TagaM., Hikime S. Taianta, 13, 185 (1966).
1175. Yoshida H., Yamamoto M., Nikime S. Japan Analyst, 11, 197 (1962).
1176. Yoshino Y. J. Chem. Soc. Japan, Pure Chem. Sec., 71, 577 (1950).
1177. Yuasa Teru. Electr. Rev., 81, 71 (1963); РЖХим., 4Г143 (1964).
1178. Yuasa T. Japan Analyst, 10, 965 (1961).
1179. Yuasa Teru. Japan Analyst, 10, 1292 (1961); РЖХим., 16Д75 (1962).
1180. Yuasa Teru. Japan Analyst, 11, 359 (1962).
1181. Yuasa Teru. Japan Analyst, 11, 1269 (1962).
1182. Yukio Suruki, Keitaro Nishiyama. C. A., 54, 1930H (1960).
1183. Zagorski Z., Cyrankowska Mr. M . Taianta, 2, 367, 380 (1959); C. A., 54,7492 (1960).
1184. Ztgie M., Zivanovic B. Kemija u industrji, 15, 345 (1966); РЖХим., 4Г138 (1967).
1185. Zoenndren W. Z. Chem., 3, 151 (1963); C. A., 59, 6984 (1965).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адсорбция селена 182
Акридин 122
Алифатические насыщенные моноке-
тоны, экстракция Se и Те 154
Алкалиметрическое титрование 72
Аллотропные модификации Se 19
Алюминий, примесь в Se и Те 213, 214
216
Амилацетат, экстракция Se н Те 152
Амины 27
соединения с Те 27
экстракция Se и Те 157
Аммиакаты 27—29
Амперометрическое титрование Se и
Те 68, 131
Антипирин
соединения с Те НО
экстракция Те 158
Антразо, обнаружение Те 55
Аскорбиновая кислота, восстановитель
62, 76
Атомная абсорбция 140
Атомная масса 20
Бензол, экстракция 152
Бихромат калня, титрование 70
Бриллиантовый зеленый 98
Бутилацетат 152, 155
Бутнлродамин С 112, 113, 115
Валентные состояния 8
Валентные электроны 8
Виктория голубая 98
Висмут, определение в Те 216
Висмутол II
комплексные соединения Se н Те 40,
41
определение Se 93
определение Те 106
производные 108
Восстановление 46, 58—63, 68, 179—
181
Галогеннды Se и Те 22, 23
Галогеноводородные кислоты Se и Те 25
Гексагалогеноселениты 26
Гексагалогенотеллурнты 26
Гетерополисоедннения Se и Те 43—45.
51, 72, 120
Гидразин, определение 76
Гидроксиламин, обнаружение Se 47
Гипофосфит, восстановление Se и Те
61, 73, 181
Глюкоза, восстановление Se и Те 62 Гравиметрические методы определения
58
Дназнны 28
Дн-п-амнловый эфир 152
3,3'-Диаминобензиднн
обнаружение Se 52
определение Se 78, 190, 202, 204, 207
синтез 82
соединение с Se 29
1,2-Диаминонафталнн 32
1,8-Днаминонафталин 31, 85
2,3-Днаминонафталнн
обнаружение Se 52
определение весовое 64
определение фотометрическое и флуориметрнческое 85—90, 190, 202, 207, 208
соединение с Se 32
4,5-Диамино-6-тнопирнмндин 96, 143
Диантипнрилметан
определение Те НО
экстракция 158
Днантипнрилметилметан
определение Те НО
экстракция 158
Днантнпнрилпропилметан
определение Те НО
экстракция 158, 215
244
1,Г-Диантримид 32
2,2'-ДиантримиД 31, 32
определение Se 94
Диацетилдитнол, определение 90
Ди-п-бутнловый эфир 152
Диизоамилметилфосфат, экстракция
151
Диизопропилкетон, экстракция 154
Димеркантопирон, определение Те
106
4-Диметиламинофенилендиамин
обнаружение Se 52
определение Se 64
Дипиазоселенол 29
Диспропорционирование 22
Дитизон
определение Se 90
определение Те 101
Экстракция 155
о-Днфениламннокарбоновая кислота
94
обнаружение Se 49
определение Se 94, 95
Дифенилгидразин 95
Дифенилкарбазнд, определение Se 96
3,5-Днфеннлпнразолнн-1-дитиокарбамат 40
определение Те 104
1,4-Днфеннлсемнкарбазид, определение Se 96
1,4-Дифенилтнокарбазид, определение Se 96
1,4-Днфеннлтносемнкарбазнд, определение Se 97
Дихлорэтан НО
Диэтиламин, экстракция 157
Диэтиланилин, экстракция 157
Дн-2-этилгекснлфосфат, экстракция
151
Диэтилдитиокарбамат
определение Se 90
определение Те 101, 103
экстракция 156
Диэтилкетон, экстракция 154
Додецнлтриалкилметиламнн, экстракция 158
Железо, примесь в Se и Те 213
Изоамилацетат, экстракция 152
Изобутилкетон, экстракция 155
Изотопный состав 216
Иод примесь в Те 216
Иодат, титриметрическое определение
71
Иодид
обнаружение Se 48
определение
титриметрическое Se 68
фотометрическое Те 99
Кадмий, примесь в Se и Те 213 Каталитические методы определения
селена 123
теллура 122
Качественное обнаружение
селена
бромидом 51
в минералах, сере, теллуре 50
в присутствии благородных металлов 4
гидрокснламином 47
3,3'-диаминобензидином 52
2,3-диаминонафталином 52 днфеннлгидразнном 49 иодндом 48
метиленовым голубым 50
молибдатом аммония на бумажных хроматограммах 51
пирролом 49
роданнстоводородной кислотой 48 тномочевнной 47
п-этокснхрнзондином 51
теллура
антразо 55
в рудах и минералах 55
двухлорнстым оловом 52
пиролизом с Hg (CN)2 56 полисульфидом аммония 56 солями Си2+ и Fe2+ 53
станнитом 52
тиомочевнной и солью Рейнеке454
Кетоны, экстракция 152
Кислота
селенистая 22, 23
селеновая 22, 23
теллуристая 22, 23
теллуровая 22, 23
Кларк Se и Те 12
Кобальт, примесь в Se и Те 214
Кодеин, определение Те 94
Количественное определение селена и теллура
амперометрическое титрование 131-133
атомная абсорбция 140
гравиметрические методы 58—65 каталитические методы 122—124 кулонометрическое титрование
133—134
пламенная фотометрия 139
полярографическое определение селена 128—131
теллура 124—128
потенциометрическое титрование 131-133
радноактивацнонные методы 144 рентгенофлуоресцентные методы
143
спектральные методы 134—139
245
титриметрические методы 65—72 фотометрические и флуориметриче-ские методы 72—122
Комплексные соединения Se и Те с висмутолом II 40 галогеноводородными кислотами 25 гетерополикислотами 43 3,3'-диаминобензидином и другими о-диаминами 29 диэтилдитиокарбаматом 39 оксикислотами 45 органическими основаниями и аммиаком 27
тиомочевиной и ее производными 33 тиосульфатом и его производными 38 Комплексонометрическое титрование 72
Концентраты промышленные 15 Купферонаты Se и Те, экстракция 95, 156
«Люминесцентное определение Se и Те 98
Медь, примесь в Se и Те 213—215 N-Меркаптоацетил-п-анизидин 93 N-Меркаптоацетил-п-толуидин 93 2-Меркаптобензимидозол 90, 92 2-Меркаптобензтиазол 92, 106 5-Меркапто-3-(нафтил-2)-1,3,4-тиадиа-золтион-2 (нафтилвисмутол) 108 5-Меркапто-3-фенил-1,3,4-тнадиазол-тион-2 (висмутол II) 106
4,4-Метилендиантипирен 159
Метиленовый голубой (метиленовая синь) 50, 94
Метилизобутилкетон, экстракция Se и Те 143, 155
Метилизопропилкетон, экстракция Se и Те 154
4-Метилтио-1,2-фенилендиамин 52, 84 4-Метилфенилендиамин 84 Метилэтилкетон, экстракция 154, 155 Методы отделения Se и Те от сопутствующих элементов
осаждением, соосаждением 177 отгонкой 183 хроматографические 160 экстракционные 149
Минералы Se и Те 11
Молибденовоселенистая кислота 51
Молочный сахар 62 Монопиазоселенол 78 Морфин 94
Мышьяк, примесь в Se и Те 214, 216
1,8-Нафталиндиамин 52 а-Нафтафлавон 122 Нафтилвисмутол
определение Те 108 синтез 110
Нахождение в природе 12
Никель, примесь в Se и Те 213, 214 Нитрат теллура, основной 21 Нитробензол, экстракция 152 4-Нитрофенилендиамин 84
Общие сведения о селене и теллуре 7—18
Окислы селена и теллура 21—23 Окись алюминия 156 Оксигалогениды 25 Оксикислоты 45 Оксифториды 25
З-и-Октиловый спирт, экстракция 52 Олово, примесь в Те 216 Определение примесей в высокочистых селене и теллуре 209
Определение селена в аггломератах 199 аммиачных водах 204 арсениде галлия 103, 198 биологических материалах 83, 205 водах 83, 209 воздухе 83, 205 горных породах 186 животных организмах 205 концентратах 65, 67, 199 крови и волосах 83, 147 меди 83 минералах 83, 123, 190, 193 мышьяке 103, 198 никотиновой кислоте 206 органических веществах 83, 85 полупроводниковых материалах 65, 83, 109, 193 породах 186 почвах 83 пылях 65, 67, 83, 199 растениях 205 рудах 67, 83, 100, 109, 119, 186, 193 свинце 83 селенидах кадмия и циика 85 сере 105, 109, 199 серной кислоте 83, 85, 202 сплавах 65, 196 сталях 65, 83, 100, 103, 196 сульфидах кадмия и цинка 85 теллуре 83, 202 углефицированных рудах 83, 188 фосфоре 105 циркониевых сплавах 83, 196 черновой меди 199, 203 шлаках 199 шламах 199
Определение теллура в арсениде галлия 196 железе 102
246
латуни 104, 199, 202 меди 104, ill, 196 мышьяке 196 полупроводниковых материалах 196
рудах 108, 109, 119, 186, 193 свинце 104. 202 сере 109, 199, 203 сплавах 196 чугуне 102
Органические основания 27 Органоселеновые соединения 154 Осаждение селена
Z-аскорбиновой кислотой 62, 180 в виде селенидов 177 гидразином 179, 180 гидроксиламином 60 гипофосфитом натрия 61, 181 глюкозой 62, 180 2,3-диаминонафталином 64 диэтилдитиокарбаматом 63 меркаптобензимидазолом 63 молочным сахаром 62, 180 сернистой кислотой 58 солями Сг2+, Fe2+, Cu+, \2+ 60 тетраэтилтиурамдисульфидом 63 тиомочевиной и ее производными 61, 62
тиосемикарбазидом 62 элементного 58—63 Осаждение теллура в виде двуокиси 63 в виде теллуридов 177 гидразином и SO2 59 гипофосфитом натрия 61, 181 солямиСг2+, Fe2+, Си, V2+ 60 тетрафениларсонием 64 тиомочевиной и солью Рейнеке 62, 64
тиосемикарбазидом 62 элементного 58—63 Отгонка селена бромидов 183 двуокиси селена 184 селеноводорода 184 хлоридов 184
Очистка реагентов 3,3'-диаминобензидина 83 2,3-диаминонафталина 88
Пентахлоротеллуриты 25 Пентафторотеллуриты 25 Период полураспада радиоактивных изотопов 10, 145
Перманганат калия, титрование Se и Те 69
Пиазоселенол 64, 77 Пиридин 27, 63 Пиролиз, обнаружение Se и Те 56
Пиррол 46, 49, 94
Пламенная фотометрия 139
Плотность 20
Полиселениды 22
Полителлуриды 22
Положение Se и Те в периодической системе 7
Полупроводниковые свойства Se и Те 20
Полярографическое определение селена 128 теллура 124
Порядковые номера Se и Те 7
Потенциалы
возбуждения спектральных линий 135
ионизационные 8
окислительно-восстановительные 9, 66
Потенциометрическое титрование 68, 133
Применение Se и Те 16
Примеси в чистых Se и Те, определение полярографическое 213 радиоактивационное 210 спектрофотометрическое 213 спектрохимическое 211
Пропилентиомочевина 36
Простые эфиры, экстракция 152
Радиоактивационное определение селена и теллура 144 примесей в Se и Те 210
Радиохимический анализ 146, 147
Радиусы
атомов 8
ионов 20
Разделение селена и теллура экстракционное 149 хроматографическое 160 осаждением 177
Разложение биологических материалов 205 минералов 186 органических материалов 205 полупроводников 196 полупродуктов 199 пород 186 пылей 199
РУД 186
сплавов 196
сталей 196
шламов 199
Рентгенофлуоресцентное определение Se и Те 143
Родамин 6Ж, определение 112
Родамин С, определение 112
Ртуть, примесь в Se и Те 214
247
Свинец, примесь в Se и Те 210, 214, 216
Селендиметантиосульфонат 39
Селендисульфинат 39
Селениды 16, 21, 22
Селениты 23
Селеноводород 22
Селенопентатионат 38
Сера, примесь в Se 214
Сернистая кислота, весовое определение Se и Те 58
Сечения активации 145
Силикагель 176
Си.и.и-диЬенилтиомочевина, определение Те 102
Синтез реагентов для определения Se и Те
бутилродамина С 115
3,3'-диаминобензидина 82
2,3-диаминонафталина 88 диантипирилпропилметана 111 нафтилвисмутола ПО
Сложные эфиры, экстракция 152
Содержание в земной коре, метеоритах 10
Соли меди, обнаружение Se 53
Соли Fe2+, Sn2+, Сг2+, Cu+, V2+, весовое определение Se и Те 60
Соль Рейнеке 37, 64, 102
Соосаждение Se и Те с
As в виде элементных 115, 179
гидроокисями 177
солями Pb, Cd, Mg 183
Спекание 188
Спектральное определение Se и Те 134
Спектрофотометрическое определение см. фотометрическое определение 72
Сплавы Se и Те 17, 196
Станнит 52
Сурьма, примесь в Те 215
Таллий, примесь в Те 213, 214
Твердость 20
Теллур, примесь в Se 213, 215
Теллур диметантиосульфонат 39
Теллуриды 16, 21, 22
Теллуриты 23
Теллуроводород 22
Теллуропентатионат 38
Температура
кипения 20, 134
плавления 20, 134
Теплота
парообразования 20
плавления 20
Теплоемкость 20
Теплопроводность 20
Термометрическое титрование 68
248
Тетраметилтиурамдисульфид 105, 157 1,1,2,2-Тетрахлорэтаи, экстракция
159
Тетраэтилтиурамдисульфид 105, 156
Тиогликолевая кислота 93
Тиомочевина 47 , 54 , 61, 64 , 94
Тиосемикарбазид, восстановление
Se и Те 62
Тиосульфат натрия, титрование Se 67
Тиосульфинаты Se и Те 39
Титриметрические методы
алкалиметрия 72
бихроматный метод определения Те
70
иодатометрия 71
иодометрия 68
комплексонометрия 72
перманганатометрия 69
по образованию труднорастворимых соединений 71
тиосульфатный метод 67
Токсичность 18
Толуол-3,4-дитил 90
Трибензиламин, экстракция 158
Трибутилфосфат 149, 156, 157
Тридодециламин 157
Триоктилфосфиноксид 152
о-Фенантролин 94
Фенилантраниловая кислота 94
о-Фенилендиамин 84
Фенилгидразин-п-сульфоновая кислота 94
Фенилтиомочевина 33
1-Фенилтиосемикарбазид 96
Флуориметрическое определение
селена с
3,3'-диаминобензидином 78
2,3-диаминонафталином 85
теллура с
бутилродамином С 112
родаминовыми красителями 112
этилродамином С 116
Фотометрическое определение
селена с
висмутолом II 93
3,3'-диаминобензидином 78
1,8-диаминонафталином 85
2,3-диаминонафталином 85 дитизоном 90
1,1'-дифенилгидразином 95 дифенилгидразин-п-сульфоновой кислотой 94
1,4-дифенилсемикарбазидом 99
1,4-дифенилтиокарбазидом 96
1,4-дифенилтиосемикарбазидом
96
2-меркаптобензимидазолом 92
2-меркаптобензтиазолом 92
по окраске коллоидных растворов 72
тиогликолевой кислотой 93
трифенилметановыми красителями 98
о-фенилендиамином 84
теллура
в виде гетерополисоединений 120
внемутолом II 106
димеркаптотиопироном 106 диэтилдитиокарбаматом 103 3,5-дифенилпиразолин-1 -дитио-карбаматом 104
иодидом 9
меркаптобензтиазолом 106 иафтилвисмутолом 108
по окраске коллоидных растворов 72
производными пиразолона НО родаминовыми красителями 112 симм- дифенилтиомочевиной 102 тетраэтилтиурамдисульфидом 105 тиомочевиной 101
Химические свойства 21
Хлор, примесь в Se 214
Хлориды 23
Хлороформ, экстракция 154
Хроматографические методы разделения 160
бумажная хроматография 168
ионообменная хроматография 161 распределительная хроматография
168
тонкослойная хроматография 176
Цклогексаэтиламин, экстракция 157 Цинк 213
Шламы анодные 16 сернокислотные 16
Щелочные и щелочноземельные металлы, примеси в Se и Те 214
Экстракция селена и теллура аминами 157 тройных комплексов 158 внутрикомплексных соединений 15
кислородсодержащими растворителями 152
трибутилфосфатом и другими фосфорсодержащими растворителями 149
Электрогравиметрические методы определения Se и Те 65
Электропроводность 20
Электрохимические методы определения Se и Те
амперометрическое титрование 131 кулонометрическое и потенциометрическое титрование 133 полярография Se 128 полярография Те 124
Этилацетат 152
Этилендиамин 27
Этилентиомочевина 36
Этилродамин С 112
ОГЛАВЛЕНИЕ
От редколлегии...................................................... 3
Предисловие......................................................... 5
Глава 1
Общие сведения о селене и теллуре................................... 7
Положение в периодической системе элементов..................... 7
Валентные состояния............................................. 8
Изотопный состав селена и теллура............................... 9
Содержание в земной коре. Главнейшие минералы и руды........... 10
Промышленные концентраты....................................... 15
Применение селена и теллура.................................... 16
Токсичность селена, теллура и их соединений.................... 18
Глава 2
Химико-аналитическая характеристика селена и теллура и их соединений 19
Физико-химические свойства..................................... 19
Полупроводниковые свойства..................................... 20
Основные химические соединения............................... 21
Комплексные соединения......................................... 23
Глава 3
Качественное обнаружение селена и теллура......................... 46
Реакции на селен................................................ 46
Реакции на теллур............................................... 52
Глава 4
Количественное определение селена и теллура.......................... 57
Гравиметрические методы.......................................... 58
Титриметрические методы.......................................... 65
Фотометрические и флуориметрические методы....................... 72
Определение селена и теллура в форме золей.................... 72
Методы определения селена..................................... 77
Методы определения теллура.................................... 99
Каталитические методы........................................... 122
Определение теллура........................................ 122
Определение селена......................................... 123
Электрохимические методы....................................... 124
Полярографическое определение теллура....................: 124
Полярографическое определение селена....................... 128
Амперометрическое титрование............................... 131
Кулонометрическое и потенциометрическое титрование......... 133
Спектральные методы............................................ 134
Пламенно-фотометрические, атомно-абсорбционные и рентгенофлуо-ресцеитиые методы............................................. 139
Пламенная фотометрия...................................... 139
Атомная абсорбция......................................... 140
Рентгеновская флуоресценция............................... 143
Радиоактивационные методы..................................... 144
Глава 5
Методы отделения селена и теллура от сопутствующих элементов . . . 149
Экстракционные методы......................................... 149
Хроматографические методы.................................... 160
Методы осаждения и соосаждения селена и теллура............... 177
Методы отгонки селена......................................... 183
Глава 6
Определение селена и теллура в природных и промышленных объектах . . 186
Определение селена и теллура в горных породах, рудах и минералах 186 Определение селена и теллура в сталях, сплавах, полупроводниковых материалах.................................................... 196
Определение селена и теллура в элементной сере, черновой меди, шламах, аггломератах, пылях, концентратах и других полупродуктах 199 Определение селена и теллура в водах.......................... 203
Определение селена и теллура в органических соединениях, биологических материалах, животных организмах, почвах и других образцах 205
Глава 7
Определение примесей в высокочистых селене и теллуре . . . ....... 209
Радиоактивационный метод...................................... 210
Спектрохимические методы...................................... 211
Полярографические и кулонометрические методы.................. 213
Спектрофотометрические методы................................. 213
Литература....................................................... 217
Предметный указатель.............................................. 244
Аналитическая химия селена и теллура. (Серия «Аналитическая химия элементов*), Назаренко И. И., Ермаков А. Н. Изд-во «Наука*, 1971 г., стр. 251.
В монографии обобщен многолетний опыт работы авторов в области аналитической химии селена и теллура. Дана общая характеристика химического поведения, подробно изложены методы качественного обнаружения и количественного определения этих элементов. Большое внимание уделено новейшим физико-химическим методам определения селена и теллура (радиоактивационному, атомно-адсорбцноиному, флуориметри-ческому, спектральному и др.). Приведены методы определения селена и теллура в природных и промышленных объектах, а также в биологических образцах. Рассмотрен обширный малоизвестный аналитикам материал по свойствам комплексных соединений селена и теллура, который может способствовать разработке новых аналитических методов.
Монография рассчитана на спецналнсто-в, работающих в научно-исследовательских и производственных лабораториях, связанных с неорганической и аналитической химией селена и теллура. Представляет интерес для преподавателей, аспирантов и студентов вузов.
Таблиц 22. Иллюстраций 45. Бнбл. 1185 назв.
Ирина Ивановна Назаренко, Анатолий Николаевич Ермаков
Аналитическая химия селена и теллура
(Серия «Аналитическая химия элементов»)
У тверждено к печати
Ордена Ленина Институтом геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
Редактор М. П. Волынец
Технические редакторы В. Г. Лаут, Э. Л. Кунина
Сдано в набор 11/XI 1970 г. Подп. к печ. 3/1II 1971 г. Формат 60Х907л Бумага Ns 2 Усл. печ. л. 15,75. Уч.-изд. л. 16,73. Тираж 1700. Т-04913. Тип. зак. 1476
Цена 1 р. 36 к.
Издательство «Наука». Москва К-62, Подсосенский пер., 21
2-я типография издательства «Наука» Москва Г-99, Шубинский пер., 10