/































































































































Text
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ ИМ. В. И. ВЕРНАДСКОГО
СЕРИЯ: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ*
АНАЛИТИЧЕСКАЯ ХИМИЯ
КАДМИЯ
Д.П.Щербов, М.А.Матвеец
1
ИЗДАТЕЛЬСТВО «НАУКА»
МОСКВА 1973
546.48:543
Серия: «Аналитическая химия элементов»
Главный редактор академик А. Л. Виноградов
Редакционная коллегия:
И. П. Алимарин, А. И. Бусев, А. П. Виноградов, А. Н. Ермаков, Ю. А. Золотов, А. В. Карякин, П. Н. Палей, С. В. Саввин, И. В. Тананаев, М. П. Волынец (ученый секретарь)
Редактор тома «Аналитическая химия кадмия»
Ю. Ю. Лурье
Адрес редколлегии:
117334. Москва, Воробьевское шоссе, 47а
Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
m 0255-0674
Щ 042(02)-73 497’73
© Издательство «Наука»! 1973 г.
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов»— составит около пятидесяти томов. Потребность в подобного рода издании давно назрела. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом, возникло настоящее издание — серия «Аналитическая химия элементов», которое осуществляется впервые. Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазонов концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы как физические, так и физико-химические и химические, излагаются применительно для количественного определения данного химического элемента, начиная с анализов сырья, далее — типичных полупродуктов производства и, наконец, конечной продукции — металлов или сплавов, окисей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей. Монографии содержат обширную библиографию, доведенную до последних лет;
3
они рассчитаны на широкий круг химиков, в первую очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на химиков-преподавателей и студентов химических высших учебных заведений. К составлению монографии привлечены наши крупнейшие специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии аналитической химии элементов будут выходить самостоятельно, по мере их подготовки. Вышли в свет монографии, посвященные торию, урану, рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, плутонию, бериллию, никелю, редкоземельным элементам и иттрию, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, алюминию, селену и теллуру, нептунию и трансплутониевым элементам, платиновым металлам, кремнию, магнию, германию, золоту. Готовятся к печати монографии по аналитической химии рения, марганца, кальция, ртути, лития, фосфора.
Посвящается светлой памяти Михаила Тихоновича Козловского, учителя и наставника химиков-аналитиков Казахстана
ПРЕДИСЛОВИЕ
Кадмий — относительно новый для техники металл, области применения которого непрерывно расширяются. Диапазон его содержаний в природных и промышленных объектах анализа очень велик и находится в пределах от миллионных долей до десятков процентов. Поэтому для определения кадмия необходимы самые разнообразные методы, позволяющие проводить анализ в присутствии почти любых элементов Периодической системы и при самых различных количественных соотношениях. Избирательных реакций на кадмий нет, вследствие чего большое значение имеют методы его отделения от других компонентов пробы, мешающих выполнению определений, и пути устранения их влияния.
В связи со все более широким использованием кадмия трудно предусмотреть все разнообразие возможных сочетаний элементов в реальных объектах анализа; поэтому во многих случаях необходим индивидуальный подход и творческое комбинирование существующих методов разделения и определения. В этих целях авторы монографии стремились возможно полнее осветить основные известные реакции кадмия, в том числе и многие опубликованные в старых работах. Краткая характеристика большинства реакций представлена в форме таблиц; подробнее рассмотрены некоторые новые реакции и методы, уже рекомендованные или перспективные для практических целей.
Для удобства все реагенты, соединения и элементы — как в таблицах, так и в тексте — расположены в большинстве случаев в алфавитном порядке (символы элементов — по латинскому алфавиту). Общие справочные данные по свойствам соединений, ионов и т. д. вынесены в Приложения.
При написании монографии использована основная литература по 1970 г. включительно и наиболее существенные работы, опубликованные позднее. Для уменьшения числа цитируемых источников ссылки даны по возможности не на отдельные работы и журнальные статьи, а на сборники и руководства (с указани
5
ем соответствующих страниц), обобщающие материал периодической и другой литературы.
Работа над монографией, под общей редакцией Д. П. Щербова, была распределена следующим образом. М. А. Матвеец собрала основной библиографический материал, описала электрохимические методы в гл. IV, гл. V, VI и вместе с Д. П. Щербовым — фотометрические методы в гл. IV. А. В. Пивоваров описал рентгеноспектральный анализ и совместно с Д. П. Щербовым — радиохимический и радиометрический методы; А. С. Бажов — спектральный анализ и фотометрию пламени в гл. IV и VI; Д. П. Щер-бов — гл. I—III, VII и химические методы анализа в гл. IV. Приложения составили авторы соответствующих глав.
Выражаем искреннюю признательность рецензентам — докторам химических наук Н. А. Филипповой и В. П. Живописцеву — за высказанные ими замечания и глубокую благодарность научному редактору профессору Ю. Ю. Лурье за ряд ценных указаний, сделанных им в процессе редактирования. Приносим также благодарность кандидату химических наук Р. Н. Плотниковой за помощь при подготовке рукописи к печати.
Авторы
Глава I
ОБЩИЕ СВЕДЕНИЯ О КАДМИИ
Краткие исторические данные
Кадмий открыт в 1817 г. Фридрихом Штромейером, профессором химии и фармации в Геттингенском университете [96; 199; 456, стр. 5].
При инспекции аптек Штромейер обратил внимание на то, что при прокаливании некоторых образцов карбоната цинка образуется не белая, а оранжево-желтая или коричневая окись, которая, однако, не содержит ни железа, ни свинца. Он предположил, что такая необычная окраска вызвана присутствием окиси какого-то постороннего элемента. Специальной обработкой Штромейер отделил неизвестную окись от соединений меди и пинка, а затем восстановил ее до металла [354, 773].
Независимо от Штромейера и почти в то же время кадмий в соединениях цинка открыли К. Херман из Шенебека и И. Ролофф из Магдебурга (обнаружившие новый элемент в окиси цинка, раствор которой давал с сероводородом вместо белого — желтый осадок), а также В. Мейсснер из Галле и С. Карстен из Берлина [199, 773].
Более поздние исследования подтвердили наличие неизвестного элемента (кадмия) во многих образцах металлического цинка и его соединений и позволили получить из них окись искомого металла и выделить его в чистом виде [456, стр. 5]. По свидетельству Гомера, цинковую руду, добавляемую к меди при выплавке латуни, именовали в Древней Греции к а д м е й я (xaSpeia) (у Плиния — cadmia) [354]. Пыль цинкодистилляционных печей (из которой новый металл начали добывать с тридцатых годов прошлого столетия [431]) назвали cadmia fornacum [619, стр. 173]. Поэтому Штромейер открытому в соединениях цинка новому элементу дал название «кадмий» [773]. В русской химической литературе первой половины XIX в. кадмий нередко называли кадмом (Страхов); за рубежом для него в первое время предлагали и другие названия: меллиний (Карстен, 1818), клапротий (Джон, 1821) и юноний (Гильберт) [388, 432].
7
Положение кадмия в Периодической системе элементов
В Периодической системе элементов Д. И. Менделеева кадмий находится в побочной подгруппе II группы (5-й период) между цинком и ртутью; его соседи по периоду — серебро и индий. Строение электронных оболочек этих элементов приведено в табл. 1 [383, стр. 321].
Таблица 1
Строение электронных оболочек кадмия и соседних с ним элементов
Элемент Атомный номер Группа Период & Распределение электронов в атомах
Zn 30 II 4 5
Ag 47 I 5 7 ls22s22/?63s23/?63tZ104524p64(Z105s1
Cd 48 II 5 7 Is22s22p63s23pe3di°4s24pe4d1«5s2
In 49 III 5 7 ls22s22p63s23p63dlt’4s24p64d1(l6s25pl
Hg 80 II 6 9 ls22s22p63e23p63d104s24p64d105s25pfbdl 6s2
Для элементов II группы характерна отдача двух внешних электронов, в результате чего образуются катионы с двумя положительными зарядами; строение их внешних электронных оболочек и некоторых других ионов, близких по размеру и конфигурации к Cd2+ [233], представлено в табл. 2. Исходя из ее данных,
Таблица 2
Внешние электронные оболочки и радиусы ионов Cd2+ и его электронных аналогов [383]
Ион Ионные радиусы, A Распределение электронов
по Белову и Бокию по Гольдшмидту по Полингу
Au+ (1,37) 1,37 5s25 p^Sd1 ’
Ag+ 1,13 1,13 1,26 4s24pG4d1'1
Hg2+ 1,12 1,12 1,10
TP+ 1,05 1,05 0,95 б^бр^бй11
Cd2+ 0,99 1,03 0,97 4s24p'!4d11
Cu+ 0,98 — 0,96 3$23рв3(й’>
In2+ 0,92 0,92 0,81 4s24p64d1 *
Zn2+ 0,83 0,83 0,74 3.ч23рв3.'1>
Sn4+ 0,67 0,74 0,71 4j24p64d11
Ga3+ 0,62 0,62 0,62 3s23pW>
8
Таблица 3
Изотопы кадмия [383]
Стабильные изотопы Радиоактивные изотопы Стабильные изотопы Радиоактивные изотопы
массовое число содержание в природной смеси, % массовое число период полураспада массовое число содержание в природной смеси, % массовое число период полураспада
106 1,21 104 59 мин. из 12,26 117* 3,0 часа
108 0,88 105 54,7 мин. 114 28,86 117 50 мин.
107 6,7 часа 116 7,58 118 50 мин.
НО 12,39 109 470 дней 119 10 мин.
111 12,75 111 * 48,7 мин. 119 2,9 мин.
112 24,07 ИЗ* 5,1 года
115 2,25 дня
* Возбужденные изомерные состояния ядер.
можно ожидать, что кроме ближайших аналогов кадмия — цинка и ртути — подобные его реакциям химические взаимодействия вероятны также еще и для ионов Сн+, 1п3+ и Т13+, быть может,— для ртути и серебра.
Атомный вес природного кадмия 112,40. Содержание в нем стабильных изотопов и периоды полураспада искусственных радиоактивных изотопов приведены в табл. 3 [383].
Нахождение кадмия в природе
Кадмий — типичный рассеянный элемент; его распространенность в природе и весовое отношение к цинку, которому он постоянно сопутствует, характеризуется следующими данными [67]:
Содержание Cd, вес. % Zn -. Cd
Литосфера 1,5-10-5 750
Г раниты 1-10-5 500
Основные породы 2-10'5 1000
Почвы 10-6—10-5 500—1000
Метеориты [79] 1-Ю-6—3-10-3
Кадмий образует ограниченное число природных соединений, да и те встречаются редко. Основной его минерал — гринокит, гексагональный CdS (77,6% Cd) открыл в 1841 г. Кеткарт, лорд Гринок [773]. Гринокит не образует естественных скоплений и встречается обычно лишь в виде землистой корочки на цинковых минералах, особенно на сфалерите [104]. В самом сфалерите кадмий часто присутствует в виде изоморфной примеси (иногда до 2—5%) и придает ему характерный желтый цвет; красная
9
разновидность сфалерита, включающая —5% Cd, носит название пршибрамита [104, 456, стр. 86]. Гринокит находят в зоне окисления некоторых сульфидных месторождений Центрального Казахстана, Средней Азии и Южного Урала [39]. Известны еще два сульфида кадмия — кубический—хоулиит и аморфный, содержащий воду, ксантохроит [79]. Очень редки другие минералы кадмия: монтепонит — CdO (87,5% Cd), отавит — CdCO3 (65,3% Cd) и кадмоселит — CdSe (47,00% Cd) [38, 39, 79, 199]. Кадмий содержится в галените, метациннабарите, тимманите, торианите и примерно в 50 других минералах [79, 375]. В различных типах изверженных пород и пегматитов присутствует 1-10~6—6*10'6% кадмия [79].
Гидротермальные цинковые и свинцово-цинковые руды обычно содержат п 10~3% кадмия (0,02—2,5% от количества цинка), находящегося главным образом в сульфидных минералах [199, 371]. Наиболее частое содержание кадмия в промышленных полиметаллических рудах находится в пределах 0,01 —0,05% (в месторождениях Кавказа —0,01—0,02%, Урала —0,02—0,03% , Алтая — 0,04—0,05%). Подобно цинку, кадмий обнаружен в морской воде, минеральных источниках, золе каменных углей и др. [456, стр. 87]. а
Присутствие кадмия в растениях было обнаружено еще в середине XIX в. (содержание — га• 10-4% от веса сухого вещества). В животных организмах его открыли в 1931 г. (содержание в губках, червях, иглокожих 4-10"6—3-10“3%; в моллюсках — до 1—4-10-а%, а в их печени— 5*10-2 — 2-10-1% сухого вещества). Организм человека содержит —10~4% Cd, который концентрируется главным образом в печени [456, стр. 6].
Получение кадмия
Основное сырье для получения кадмия — отходы цинкового, свинцового и медного производств: пыли при обжиге цинковых концентратов, пыли свинцовоплавильных печей, медно-кадмие-вые кеки цинковых электролитных заводов, остатки литопонного производства. Содержание кадмия в этих продуктах изменяется от тг-10"1 до 30—40% (например, в кадмиевой фракции при ректификации цинка). Наряду с кадмием цинковые и свинцовые концентраты содержат тг-10~3% Ga, Ge, In, TI, Se и Те; содержание последних двух элементов в свинцовых концентратах достигает сотых (селена — даже десятых) долей процента. Для извлечения кадмия из перечисленных продуктов на цинкоэлектролитных и свинцовых заводах СССР имеются кадмиевые цехи [371; 456, стр. 85].
Первые упоминания о промышленном производстве кадмия (в Верхней Силезии) относятся к 1829 г.; до 1870 г. оно не превышало 125 кг в год. В начале XX в. был разработан гидроэлектро-металлургический способ получения кадмия, и к 1925 г. его вы
10
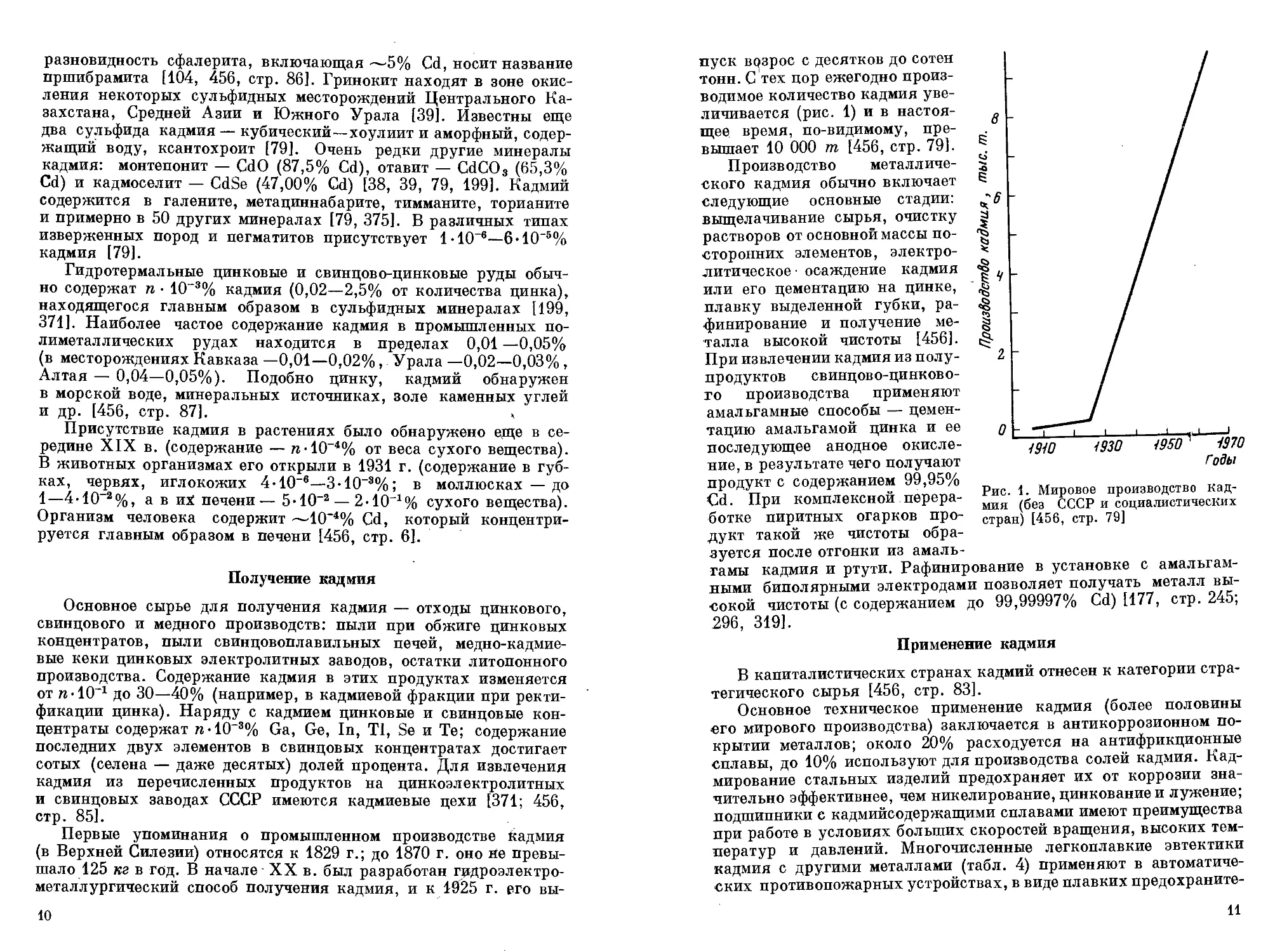
пуск возрос с десятков до сотен тонн. С тех пор ежегодно производимое количество кадмия увеличивается (рис. 1) и в настоящее время, по-видимому, превышает 10 000 т [456, стр. 79].
Производство металлического кадмия обычно включает следующие основные стадии: выщелачивание сырья, очистку растворов от основной массы посторонних элементов, электролитическое • осаждение кадмия или его цементацию на цинке, плавку выделенной губки, рафинирование и получение металла высокой чистоты [456]. При извлечении кадмия из полупродуктов свинцово-цинково-то производства применяют амальгамные способы — цементацию амальгамой цинка и ее последующее анодное окисление, в результате чего получают продукт с содержанием 99,95% Cd. При комплексной переработке пиритных огарков продукт такой же чистоты обра-
Рис. 1. Мировое производство кадмия (без СССР и социалистических стран) [456, стр. 79]
зуется после отгонки из амаль-
гамы кадмия и ртути. Рафинирование в установке с амальгамными биполярными электродами позволяет получать металл высокой чистоты (с содержанием до 99,99997% Cd) [177, стр. 245; 296, 319].
Применение кадмия
В капиталистических странах кадмий отнесен к категории стратегического сырья [456, стр. 83].
Основное техническое применение кадмия (более половины его мирового производства) заключается в антикоррозионном покрытии металлов; около 20% расходуется на антифрикционные сплавы, до 10% используют для производства солей кадмия. Кадмирование стальных изделий предохраняет их от коррозии значительно эффективнее, чем никелирование, цинкование и лужение; подшипники с кадмийсодержащими сплавами имеют преимущества при работе в условиях больших скоростей вращения, высоких температур и давлений. Многочисленные легкоплавкие эвтектики кадмия с другими металлами (табл. 4) применяют в автоматических противопожарных устройствах, в виде плавких предохраните-
11
лей и ж качестве припоев; ими можно паять железо, латунь, нейзильбер, олово, свинец и цинк [371; 456, стр. 71].
Добавление кадмия в медные сплавы также повышает их механическую прочность, жароустойчивость и сопротивление коррозии; введение циркония увеличивает и твердость; такие сплавы используют в линиях высоковольтных передач. В платино-железных сплавах кадмий служит для изготовления часовых пружинок, а со свинцом и оловом — для типографских клише [456, стр. 73].
Наряду с бором, гафнием, золотом и некоторыми редкоземельными элементами металлический кадмий высокой чистоты применяют в атомной промышленности в качестве поглотителя тепловых нейтронов. Употребление в ювелирном деле основано на способности кадмия придавать изделиям из драгоценных металлов различные оттенки («зеленое» золото); он входит также в некоторые составы монетного серебра [371; 456, стр. 74]. Кадмиевые электроды используют в нормальных элементах Вестона и в кислотных аккумуляторах, а с железом и никелем — в щелочных. Металлический кадмий широко применяется в военном деле, входит в состав смесей для дымовых завес и добавляется к металлам цериевой группы в пирофорных сплавах [456, стр. 73].
Соли кадмия также широко применяют в медицине, в стеклоделии, в фотографии, в производстве люминофоров и силиконовых каучуков, в качестве катализаторов в органическом синтезе, а также красок различных цветов (от цвета слоновой кости до малинового). Антимониды, арсениды, селениды, сульфиды и теллуриды кадмия (для синтеза используют 99,99999%-ный Cd) обладают полупроводниковыми свойствами и применяются в фотоэлектрических и электронно-оптических приборах, — в качестве материала для фототранзисторов и фотоэлементов [456, стр. 76].
Токсичность кадмия и его соединений
Металлический кадмий не обладает токсическими свойствами. Соединения же кадмия, независимо от их агрегатного состояния (пыль, дым окиси кадмия, пары, туман), ядовиты. Отравления кадмием могут происходить при нагревании металла или его сплавов, плавке руд и при производстве и применении красок и сплавов, в состав которых он входит. По своей токсичности кадмий аналогичен ртути или мышьяку [456, стр. 222; 619, стр. 175]. Менее растворимые соединения его действуют в первую очередь на дыхательные пути и желудочно-кишечный тракт, а более растворимые — после всасывания в кровь — поражают центральную нервную систему (сильное отравление), вызывают дегенеративные изменения во внутренних органах (главным образом — в печени и почках) и нарушают фосфорно-кальциевый обмен. Симптомы отравления кадмием зависят от пути его поступления в организм [72а].
13
Характерными ранними симптомами хронического отравления малыми дозами кадмия (обычно после двух лет работы^ с ним) являются снижение обоняния (вплоть до полной его цотери), золотистое окрашивание десен в области зубных шеек («кадмиевая кайма»), головокружение, головная боль, нарушение Аппетита и сна. В городах, атмосфера которых содержит относительно больше кадмия, смертность среди страдающих сердечными заболеваниями выше [341а, стр. 287].
Для острых отравлений обычен более или менее длительный (10—36 час., иногда — 30 мин. 2 час.) скрытый период. Начальные симптомы — сухость слизистых оболочек, неприятное вяжущее сладковатое ощущение на губах, медный вкус во рту, горловые спазмы, бледность, слабость, головокружение и головная боль (в области лба), тошнота и боль в подложечной области. Возможны дерматиты и изъязвление кожи; в тяжелых случаях наблюдаются боли в грудной клетке и брюшной полости, с последующей рвотой [619, стр. 175; 293, стр. 345]. Затем развиваются трахеит, бронхит с приступами болезненного судорожного кашля и сильной одышкой, повышается температура [72а].
Для человека смертельной дозой является вдыхание в течение 1 мин. воздуха с содержанием 2500 мг/м3 окиси кадмия (или 30 сек. при концентрации 5000 мг/м3) [456, стр. 22, 222]. Максимально допустимое количество CdO в воздухе — 0,1 мг/м3 [321, стр. 262]; для пыли кадмиевого сплава — 0,3—0,4 мг/м3 [72а].
В качестве первой помощи при остром кадмиевом отравлении рекомендуется свежий воздух, полный покой, предотвращение охлаждения. При раздражении дыхательных путей — теплое молоко с содой, ингаляции 2%-ным раствором NaHCO3. При упорном кашле — кодеин, дионин, горчичники на грудную клетку; необходима врачебная помощь. Противоядием при отравлении, вызванном приемом во внутрь кадмиевых солей, служит альбумин с карбонатом натрия. Специфическое лечение кадмиевых отравлений — применение комплексообразующих препаратов (2,3-димеркапто-пропанол-1, двойной этилендиаминтетрацетат кальция и натрия); но клинический опыт их использования еще недостаточен. Выделение кадмия из организма (через почки и кишечник) происходит чрезвычайно медленно; введение 2,3-димеркаптопропанола-1 увеличивает его примерно в 3 раза. Механизм токсического действия кадмия заключается, по-видимому, в связывании карбоксильных, аминных и особенно сульфгидрильных групп белковых молекул, в результате чего угнетается активность ферментных систем. В связи с ядовитостью кадмия и его соединений их контакт с пищевыми продуктами недопустим [72а; 293, стр. 345; 619, стр. 175].
Г лава II
ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА КАДМИЯ И ЕГО СОЕДИНЕНИЙ
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА КАДМИЯ
Кадмий — серебристо-белый, с синеватым отливом, металл, по .цвету ближе к стали, чем к олову, имеет несколько желтоватый оттенок. На воздухе кадмий быстро тускнеет из-за образования тонкой окисной пленки, но сохраняет металлический блеск; излом его острый, лучистый. Тонкая кадмиевая фольга в проходящем свете имеет синевато-фиолетовую окраску. Пары кадмия оранжево-желтые; подобно большинству других металлов, состоят преимущественно из отдельных атомов (их молекулярный вес 114,1 при атомном весе кадмия 112,4). Плотность паров кадмия по отношению к воздуху —4. В вакууме кадмий возгоняется уже при 164°, кипит при 450° С. Температура возгонки и кипения при различных давлениях представлена на рис. 2. Плотность металлического кадмия при 0° К равняется 9,65 г/см3- в нормальных условиях для литого кадмия она составляет 8,604, у кованого — 8,690 г/см3, плотность металла несколько ниже точки его плавления — 8,37 г/см3 [456, стр. 9; 354, стр. 477]. Плотность и вязкость жидкого кадмия при различных температурах представлены на рис. 3.
Кадмий кристаллизуется в виде пирамид гексагональной сингонии; в токе водорода могут быть получены кристаллы величиной 6—8 мм. Его решетка отличается от идеальной тем, что ось с несколько более вытянута по отношению к оси а (с/а = 1,8856 вместо 1,6333). Кадмий существует в трех различных модификациях, неразличимых структурно при рентгенометрическом исследовании, но различающихся коэффициентами расширения и величиной электролитического потенциала растворения (но лишь на тысячные доли вольта). Данные об его аллотропных превращениях не вполне достоверны. Одно из них происходит при 65° С (а -> (3), второе около 95 °C (|3 г-> у). Хорошо изучена только низкотемпературная a-модификация, образующая гексагональные кристал-
15
Рис. 2. Температура кипения и возгонки кадмия при различных давлениях [383, стр. 594]
Рис. 3. Вязкость т; п плотность d кадмия при различных температурах [383, стр. 983; 456, стр. 9]
1 — вязкость; 2 — плотность
лы. Взаимный переход модификаций происходит чрезвычайно медленно, поэтому обычно имеется смесь всех трех форм [354, стр. 452; 456, стр. 10].
Чистый кадмий при сгибании подобно олову издает хрустящий звук; при наличии в нем даже незначительного количества примесей треска не слышно. Твердость чистого кадмия невысока, по шкале Мооса она равна 2,0; твердость литого металла по Бринелю (под грузом 500 к/’) соответствует 16—20 кГ!мм2, в отожженном состоянии — 21,3—25,5 кГ/мм2. Кадмий тверже олова, но мягче цинка; он режется ножом, хорошо куется, катается в листы и протягивается в проволоку. Его модуль Юнга (продольной упругости) равен 6,93-1011 дин!см2. Нагретый до 80° С кадмий делается хрупким и может быть истолчен в порошок. При 300° С два кадмиевых листа, наложенных один на другой, приплавляются при слабом давлении [456, стр. 10].
Теплоты реакций (АН) и свободные энергии (AH’) плавления, возгонки и испарения металлического кадмия выражаются следующими формулами:
С(1(тв) — Сй(ж)
АН = 902 + 1,67 • Т — 1,233 • Ю’3?12;
\F°T = 902 — 1,67г • In Т + 1,233 • 10-3?2 + 8,415Г;
АН298 = 1290 кал/г-атпом', AFws = 685 кал/г-атом.
С(1(тв) — Cd(ra3)
АН = 26919 - 0,467 - 1,233 • 10-3Z’a;
АЕт = 26919 Ц-0,467’-In ГЦ- 1,233-10 3Г2- 31,368Г;
АН298 = 2 66 1 7 кал/г-атом',
16
AF298 = 18460 кал/г-атом.
Cd (ж) — Cd(ras) ХН = 26017 — 2,13Г;
XFт = 26017 + 2,1ЗТ • In Г — 39,783г.
Зависимость теплоемкости, тепло- и электропроводности кадмия от температуры представлена на рис. 4. Некоторые другие физические свойства кадмия представлены ниже.
Атомный вес
Атомный радиус, А
Ионный радиус, А
Атомный объем, см3/г-атом Параметры решетки, А Т. пл., °C
Т. кип., °C
Теплота плавления (при Т. пл.)
Теплота испарения (при Т. кип.)
Теплота сгорания
Теплопроводность, кал/см-сек-град Коэффициент линейного расширения, град~г
Расширение при плавлении
Поверхностное натяжение, дин/см
Прочность на разрыв, к Г/мм2
Удлинение, %
Магнитная восприимчивость
Упругость, кГ/мм2
Модуль сдвига, кГ/мм2
112,41
1,5680
1,03; 0,99; 0,97 [383, стр. 381;
363, стр. 37]
13,01
а = 2,9728; b = 5,63; с = 5,6054
320,9 (в вакууме сублимируется при 164 °C)
767 (в вакууме кипит при 450 °C) 12,9 кал/г', 1,46 ккал/г-атом
212 кал/г’, 26,28 ккал/г-атом
65,2 ккал/г-атом
0,2213 (0° С); 0,2045 (100 °C)
0,00002879
0,0064 см3/г', 0,73 см3/моль
570 (330 °C); 597 (400 °C); 585
(600° С)
6,4 (0° С); 2,45 (130° С); 0,55 (237° С)
17 (0° С); 34 (130° С); 45 (237° С)
-0,185-10-в
модуль — 5090—5240; предел —
2,73
2260
Данные, приводимые разными авторами, нередко существенно различаются между собой. По-видимому, основная причина этих различий заключается в неодинаковых степени чистоты, кристаллической структуре (моно- и поли-), механической обработке (литой — кованый) или других «тонких» свойствах исследованных образцов металла.
При распылении в пламени электрической дуги (5—10 а, 30—40 в) под водой, не содержащей воздуха, образуется темно-коричневый гидрозоль кадмия; при добавлении электролитов он становится сине-зеленым и коагулирует. Под действием воздуха золь быстро окисляется; без него в присутствии желатина — устойчив достаточно долгое время. При распылении кадмия в органических растворителях высокочастотным разрядом можно получить органозоли, содержащие 0,01—0,07% металла; устойчи-
17
Рис. 4. Теплоемкость (Ср), теплопроводность (X) /и электропроводность (х) Кадмия при различных температурах [371, стр. 185; 383, стр. 741 и 932; 456, стр. 12]
1 — теплоемкость; 2 и з — теплопроводность моно- и поликристал-лического металла; 4 — электропроводность общая; 5 и в — то же, соответственно, моно- и поликри-сталлического металла
вые золи получаются в этилацетате и в ацетоне (в последнем — в присутствии стабилизатора — бромбензола) [456, стр. 12].
Некоторые электрические свойства кадмия приведены ниже [383, 385], а значения стандартных электродных потенциалов — в табл. 5.
Удельное сопротивление, о.и-с.и-10 6
Температурный коэффициент, град~1 (Т = О— 100° С)
Ионизационный потенциал, в
Работа выхода электронов, эв
Перенапряжение водорода (при 20 °C и плотности тока I, а/см2)
Электрохимический эквивалент иона Cd2+
Электронная поляризуемость иона Cd2+, А3 кристалл водный раствор
6,83 (0° С); 7,4 (20° С) 0,0043
1-й — 8,991; 2-й — 16,904; 3-й — 44,5
4,04
.1,40 + 0,120-lg I
0,5824 мг!кулон\
2,096 г/а~час
1,8
0,92
Зависимость электропроводности кадмия от его кристаллической структуры и от температуры приведена на рис. 4; влияние температуры на удельное сопротивление рт {ом-см) выражается следующими данными [383, стр. 932]:
Т, °C —253 —192 —78 0 100 200
рт 0,14 1,73 4,74 6,83 9,73 12,9
рт/ря 0,021 0,253 0,693 1,0 1,424 1,886
Электродный потенциал реакции Cd = Cd2+ + 2е“ в эквимолярном расплаве КС1 — NaCl относительно электродного потенциала серебра имеет следующие значения: при 700° С — 0,620 в, при 800° С — 0,580 в [385, стр. 838]. Энергия возбуждения атома кадмия дана в работе [293, стр. 340].
Кадмий хорошо растворим в ртути; теплота его растворения — 0,05 ккал! миль. При 0° С насыщенная амальгама содержит 3,3 вес.% кадмия, при 20° С — 5,9% , при 100° С — 22% , при 200° С — 52% , а при 300° С — 88% . Путем электролиза можно получить амальгаму кашицеобразной консистенции, содержащую
18
Таблица 5
Стандартные электродные потенциалы кадмия [385, стр. 745 , 826; 222, стр. 177]
Электродный процесс Среда E0, «
CdH = Cd[+ Н+ + е~ H2O —2,417
Cd + S1 2 *~ = CdS|+ 2е“ H2O —1,24
Cd + 4CN~ = [Cd(CN)4]2~ + 2e" H2O —1,09
Cd + 2OH- = Cd(0H)4+ 2e“ H2O —0,81
Cd + CO32~ = CdCO3],+ 2e~ H2O —0,74
Cd + 4NH3 = [Cd(NH3)4]2+ + 2e~ H2O —0,61
Cd = Cd2+.+ 2e“ HCOOH -0,75
CH3CN -0,47
CH3OH —0,43
H2O -0,403
C2H6OH —0,38
NH3 -0,20
N2H4 —0,10
CdS = Cd2+ + S + 2e~ H2O 0,32
900 г Cd'л Hg. Вязкость 0,025%-ной амальгамы соответствует 1,5819 спз. Для коэффициента диффузии кадмия в ртути при 20—25° С разные авторы приводят значения от 1,45 до 2,7-• 10~6 смЧсек. Поверхностное натяжение амальгамы, содержащей 0,82 г-атп Cd!л Hg, при 20° С имеет величину 406 дин/см (для чистой ртути — 410 дин1см~). Потенциал системы Cd | Cd2+ | Cd(Hg) при 20° С 51,0 мв. Нижний предел применимости уравнения Нернста для разбавленных кадмиевых амальгам—2-10"6 г-атп CdAiHg [176, 177]. Данные о влиянии ряда факторов на потенциал кадмиевых амальгам и о их аналитическом применении см. [176, 177].
Кадмий легко образует сплавы с другими металлами и многочисленные соединения от Me3Cd до MeCd13 (Me — щелочные, щелочноземельные металлы, As,Sb и др.). Часть этих соединений (с золотом, медью и серебром) подчиняется правилу Юм-Розери х, в эти соединения кадмий вступает как «металл II рода» [354, стр. 39; 455]. Изучены диаграммы состояния бинарных систем кадмия более, чем с тридцатью элементами [456, стр. 58]; сплав кадмия с ртутью имеет состав Cd3Hg, теплота его образования 0,52 ккал/г-am, при 188° С он разлагается [177, стр. 43, 125].
1 Это ^правило связывает кристаллическую структуру образующейся ин-
терметаллической фазы и отношение общего числа валентных электронов
к общему числу атомов металлов [354, стр. 33].
19
I
При комнатной температуре кадмий устойчив и лишь незначительно тускнеет под действием воздуха и воды; поэтому его широко применяют в качестве антикоррозионных покрытий. Однако из-за мягкости кадмия эти покрытия не противостоят механическому износу [619, стр. 177]. При сильном нагревании на воздухе кадмий сгорает красным пламенем с образованием коричневого дыма окиси CdO (при этом образуются также следы перекиси СсЮ2); теплота сгорания — 65,2 ккал!г-атом Cd. При нагревании кадмий легко соединяется с галогенами, но не реагирует ни с азотом, ни с водородом [354, стр. 477]. Высокодисперсный металл (величина частиц йС 10 мкм) чрезвычайно легко воспламеняется [72а].
Способность кадмия растворять газы определяется его свойствами и условиями насыщения. Состав этих газов, зависящий от способа получения и рафинирования кадмия, приведен в табл. 6.
Таблица 6
Содержание газов в кадмии [456, стр. 15]
Кадмий Содержание газов в 100 г металла, нем3 Состав газа, объемы. %
Н2 n2 со со2
Катодный Кадмий 0,324 86,1 5,3 — 8,6
марки Кд-0 0,048 9,6 8,8 2,9 77,7
марки Кд-0, обработанный паром 0,156 10,4 10,8 — 78,8
марки Кд-1 Кадмий очищенный 0,035 12,4 9,2 3,5 74,9
дистилляцией в атмосфере азота 0,083 7,3 18,1 — 74,6
вакуумной дистилляцией 0,002 10,2 7,4 3,1 79,3
то же, после переплавки в атмосфере азота 0,004 55,1 3,9 1,6 39,4
Воду кадмий при комнатной температуре не разлагает. При перемешивании порошкообразного кадмия в воде в присутствии воздуха образуется водород и некоторое количество пергидроля. Сухой хлористый водород реагирует с кадмием при 440° С; сухой сернистый газ при нагревании образует с кадмием сульфид и некоторое количество сульфата. Сероводород не действует на кадмий при комнатной температуре даже при длительном контакте в присутствии воздуха.
Разбавленные серная и соляная кислоты при нагревании медленно растворяют кадмий с выделением водорода. Растворение в серной кислоте при 160° С сопровождается выделением не водорода, а сернистого газа. Константа скорости растворения в серной
20
кислоте возрастает с повышением концентрации деполяризаторов — пергидроля, бромата, хлората или перманганата калия^ При растворении кадмия в азотной кислоте образуется аммиак, количество которого зависит от концентрации кислоты (максимальное количество аммиака выделяется при действии 27,5%-ной кислоты). Горячая разбавленная азотная кислота энергично реагирует с выделением окислов азота; более медленное растворение кадмия в серной и соляной кислотах по сравнению с азотной объясняется высоким перенапряжением водорода [456, стр. 14; 619, стр. 178].
Кадмий легко растворяется в насыщенном растворе нитрата аммония с образованием нитритов (без выделения газа). При действии на кадмий раствора сернистого газа получается желтая жидкость, из которой постепенно выделяются сера и сульфид кадмия (последний выделяется и при кипячении с соляной кислотой). Растворяется кадмий и в растворах щелочных иодидов,. СиС12 и солей Fe (III). При действии растворов персульфата калия и аммония кадмий покрывается порошкообразным налетом основного сульфата [456, стр. 14].
Кадмий нерастворим в едких щелочах, но при нагревании легко соединяется с фосфором, серой, селеном и теллуром. Он образует твердые растворы с сульфидом железа; в полученных кристаллах (типа пирротина) содержится до одного атома кадмия на 6 молекул сульфида железа.
Алюминий, цинк и железо осаждают кадмий из растворов его солей; сам он выделяет медь и другие более благородные металлы из их растворов (однако из концентрированного раствора комплексного цианида кадмий осаждается медью). Порошок железа при действии нейтральных растворов нитрата, сульфата, хлорида,, бромида или иодида (но не хлората) кадмия образует соответствующие растворимые соли железа. Цинк полностью осаждает кадмий из хлоридных растворов за 10—15 мин., также и в присутствии Fe (II); из азотнокислой среды кадмий выделяется цинком в виде дендритов. Алюминий энергично вытесняет кадмий из расплавленных солей и водных растворов; из этих последних,, содержащих следы нитрата хрома — количественно. При действии магния на водный раствор соли кадмия, последний осаждается в виде гидроокиси с выделением водорода [456, стр. 15].
Об устойчивости тех или иных состояний системы кадмий — вода и возможности протекания соответствующих реакций можно судить по диаграмме Пурбэ [693, стр. 414], приведенной на рис. 5 (представлены графики зависимости электродных потенциалов Е (в) от pH раствора для реально устанавливающихся равновесий между различными формами кадмия; вертикальными прямыми показаны значения pH образования гидратов).
На диаграмме видны области устойчивости тех или иных форм кадмия, разграниченные между собой линиями равновесия Cd/Cd2+, Cd2+/Cd(OH)2. и т. д. Для жидких фаз положение границы зависит от активности не только ионов водорода, но и тех форм кадмия, которые участвуют в установлении
21:
Рис. 5. Диаграмма Пурбэ для системы кадмий — вода при 25° С [385, стр. 780] Область между пунктирами а и б — устойчивое состояние воды; ниже а она восстанавливается, выше б — окисляется.
7 _ Cd*4- + 2Н2О = HCdOj- + ЗН+, 1g = _ 33,34 + зрН;
Г—Cd2-/HCdO,-, pH 11,17;
2 — Cd +2НгО = Cd (ОН)2 + 2Н+ +2е-, Е = 0,005 — 0,0591рН;
3 _ Cd2+ + 2Н2О = Cd (ОН)2 + 2Н+, 1g {Cd2+} = 13,81 — 2рН;
4 — Cd (ОН), = HCdO2- + Н+, 1g {HCdO2-} = —19,54 +рН;
5 _ Cd = Cd2+ + 2e~, E = —0,403 + 0,0295 1g {Cd2+};
в — Cd + 2H2O = HCdO2- + 3H+ + 2e~, E = 0,583 — 0,0886 pH + 0,0295 1g {HCdO2~}; .7 — CdH = Cd + H+ + e- E = — 2,417 — 0,0591 pH — 0,0591 1g PCdH
равновесия в растворе. Для них на диаграмме вместо одной границы раздела нанесено семейство линий; для каждой из них указано значение логарифма активности растворенной формы кадмия. Уравнения, отвечающие линиям границ разделов, приведены в подписи к рис. 5; пунктир а и б ограничивает область устойчивости воды.
Пунктирная вертикаль 1', соответствующая разности активностей ионов Cd2+ и HCdOj, разделяет диаграмму на поля («области преобладания») преимущественного сосуществования Cd (ОН)2 с ионами Cd2+ — влево от нее, и Cd(OH)2 с ионами HCdO2 — с ее правой стороны [386, стр. 780]. На рис. 5 22
видно, что, например, при Е > —0,4 в и pH < 6 в растворе будут находиться ионы Cd2"’’. При том же потенциале и pH 10 основная масса кадмия выделяется в виде осадка Cd(OH)2. В растворе (в соответствии с произведением растворимости рПР ~ 10~14) будут преобладать ионы Cd2+ по сравнению с HCdOj. Зная потенциал какого-либо металла при заданной кислотности среды, по диаграмме можно определить, будет ли он (и в какой именно форме) выделять кадмий из раствора. Диаграмма иллюстрирует также влияние активности (концентрации) ионов Cd2+ в растворе на потенциал системы Cd/Cd2+ (снижение Е до —0,580 в при изменении логарифма активности от О до —6) и на осаждение Cd(OH)2 (повышение pH ее выделения от 6,9 до 9,9 при уменьшении активности в тех же пределах). Очевидно, что если в растворе находятся посторонние вещества (особенно комплексообразователи), то положения равновесия соответственно сместятся.
В водных растворах кадмий находится в двухвалентном состоянии. Однако имеются данные, свидетельствующие о существовании соединений Cd2O и Cd2Cl2, в которых кадмий одновалентный 1222, стр. 1801.
При растворении металлического кадмия в расплавленном CdCl2 образуется очень темный красно-черный плав. Его окраска, по-видимому, обусловлена существованием Cd+ и Cd2+, связанных галогенными мостиками. Если к этому расплаву добавить А1С13, то он становится желто-зеленым и фазовое изучение его (так же как и соответствующей бромидной системы) показывает присутствие в нем Cd+. Спектры комбинационного рассеяния позволили установить в расплаве наличие ионов Cd2+, аналогичных-Hgl+-Спектр выделенного из расплава твердого желтого CdAICl4 подтверждает присутствие иона А1СЦ; так как это соединение диамагнитно, то его правильнее представить в виде (Cd2)2+(A1C14)2-При его взаимодействии с водой образуется металлический кадмий и Cd2+, поэтому неудивительно, что в водных растворах не-удалось обнаружить присутствия ионов Cd+ [197, стр. 475].
СОЕДИНЕНИЯ КАДМИЯ
Ниже рассмотрены свойства соединений кадмия, аналити' ческое использование которых описано в последующих главах. Краткая характеристика некоторых комплексных соединений, неприменяемых пока в анализе, дана в Приложениях.
Неорганические соединения кадмия
Окись кадмия CdO получается при нагревании металла на воздухе или при прокаливании его гидроокиси, карбоната, нитрата или сульфида. Цвет окиси кадмия зависит от температуры: прокаливание гидроокиси при 350—370° С дает зеленовато-желтую окись, при нагревании до 800° С получается соединение густого сине-черного цвета; последнее образуется также при длительном кипячении ^гидроокиси с очень концентрированным
23
раствором КОН. При прокаливании карбоната кадмия получается аморфная коричневая, из нитрата кадмия — кристаллическая черная окись. Продукты разного цвета различаются лишь величиной частиц. При нагревании на воздухе СсЮ очень устойчива, около 700° С, не плавясь, начинает возгоняться, при более высокой температуре (до 1000° С) отщепляет кислород. Но в токе водорода легко восстанавливается уже при 270—300° С, а углеродом или его окисью — при 700° С. Нагреванием в токе хлора СсЮ можно перевести в хлорид CdCl2. На воздухе коричневая СсЮ поглощает СО2 и постепенно белеет, переходя в карбонат. СсЮ нерастворима в воде и щелочах, в кислотах растворяется с образованием ионов Cd2+, а в аммиаке — комплексных катионов [Cd(NH3)J2+. Окись кадмия использовали в гравиметрии в виде весовой формы после прокаливания CdS или CdCt)3; но необходимо учитывать ее способность легко восстанавливаться газами пламени и углем фильтровальной бумаги, что может привести к заниженным результатам [165, 354, 459, 565, 619].
Гидроокись кадмия Cd(OH)2 выделяется из растворов солей кадмия при действии едких щелочей в виде белого студенистого осадка, практически нерастворимого в избытке ре. агента, но легко растворяющегося в кислотах, цианидах и аммиаке. Растворимость при 25° С в воде равна 1,16-10'5 молъ1л (ПР свежеосажденной гидроокиси равно 2,2-Ю'14, рПР=13,66; после старения ПР = 5,9-10~15, рПР = 14,23). При добавлении щелочи растворимость сначала понижается, а затем возрастает и в 5 7V NaOH достигает 9,0-10~5 молъ]л [354]. Начало осаждения зависит от исходной активности ионов Cd2+ в растворе [(Cd2+)] и определяется уравнением Пурбэ
pH = 6,9 — 0,51g [Cd2+] (рис. 5, уравнение 3).
Опубликованные значения pH для разных концентраций Cd2+ несколько различаются: для 1 М растворов — 6,7 [82] и 7,2 [233]; для 0,01 М - 7,3 [459] и 8,2 [233]; для 10 6 М - 8,8 [459]; полнота осаждения достигается при pH 9,7 [233]. В присутствии избытка аммонийных солей, лимонной и винной кислот осадок гидроокиси не выделяется. При нагревании до 170° С Cd(OH)2 не разлагается, при 371° С образуется CdO [42, 619].
Карбонат кадмия CdCO3, выделяющийся в виде белого осадка карбонатами щелочных металлов, обычно загрязнен основной солью Cd2(OH)2CO3. Соли аммония препятствуют выделению, но при кипячении аммиак удаляется в виде газа и происходит полное осаждение. При действии NaHCO3 осаждается не основная, а средняя соль; при кипячении с водой гидролитическое разложение карбоната увеличивается. ПР’= 5,2-1СГ12, рПР = 11,3. Карбонат кадмия разлагается при температуре -—500° С на CdO и СО2. Он растворим в разбавленных кислотах, растворах цианида и аммиака [42, 324, 416, 619].
24
Оксалат кадмия CdC2O4-3H2O осаждается при действии оксалатов или щавелевой кислоты. Труднорастворим в воде (при 18° С-33,7 мг/л; ПР = 1,5-10’8; рПР = 7,8), легче -в концентрированных растворах оксалатов щелочных металлов с выделением двойной соли, например K2[Cd(C2O4)2]-2Н2О. Оксалат кадмия растворим также в концентрированных растворах хлоридов щелочных металлов с образованием К4[Сй2С12(С2О4)3] • 6Н2О [354, 565].
Молибдат кадмия CdMoO4 — белый, мелкокристаллический порошок, выпадающий при действии молибдатов на раствор Cd2+; при высушивании приобретает слабо-розовую окраску. Малорастворим в воде и разбавленной СН3СООН, растворяется в кислотах, аммиаке, концентрированных щелочах и растворах КС1. Обезвоживается при 82° С, может быть высушен без разложения при 120° С; при 500° С становится коричневым и теряет в весе.
Нитрат кадмия Cd(NO3)2-4H2O — расплывающиеся лучистые иглы, получаются при растворении металла или карбоната в азотной кислоте. При 59,3° С плавится в своей крис±аллизацион-ной воде; при 132° С расплав начинает кипеть, оставаясь прозрачным до удаления 75% имеющейся воды. Осторожное обезвоживание тетрагидрата при 75—80° С (или в эксикаторе над H2SO4) дает безводную соль, плавящуюся при 360° С; при более высокой температуре разлагается с выделением CdO [165, 354].
Сульфат кадмия 3CdSO4-8H2O получают кристаллизацией раствора кадмия, его окиси или карбоната в разбавленной серной кислоте; при температурах ниже 70° С нерастворим в этаноле. При нагревании соли между 80 и 120° С образуется моногидрат, а при 320° С — безводная соль, устойчивая до 906° С. Кадмий образует двойные соли с сульфатами щелочных металлов, меди и Fe(II). Из водных растворов, содержащих аммиак, кристаллизуется в виде аммиаката [Cd(OH2)2(NH3)4]SO4- Сульфат кадмия — одна из наиболее распространенных весовых форм при определении кадмия, которую высушивают при 350—400° С. Если исходят из CdS, то его сначала растворяют в соляной кислоте и выпаривают с серной; полное удаление ее происходит с большим трудом, поэтому прокаленный сульфат растворяют в воде, снова выпаривают, прокаливают, взвешивают и повторяют эти операции до достижения постоянного веса [82, 165, 354, 459, 565, 619].’
Сульфид кадмия CdS' образуется при действии на растворы кадмия растворимых сульфидов или газообразного сероводорода. Цвет осадка зависит от условий выделения и меняется от зеленовато-желтого через оранжевый до красного. Желтый сульфид получается'при его осаждении из холодного щелочного раствора, красный — из кислого при нагревании. Желтая форма при нагревании становится оранжево-красной, по охлаждении первоначальный желтый цвет восстанавливается; изменения цвета
25
можно достигнуть также трением и давлением. Концентрированные кислоты, разбавленная теплая HNO3 и кипящая H2SO4 (1 :-5) растворяют GdS; при действии HNO3 выделяется сера, которая частично вновь окисляется. Свежеосажденный сульфид немного растворим в разбавленных кислотах с выделением сероводорода (в соляной значительно легче, чем в серной), растворяется в концентрированном растворе хлорида аммония, но нерастворим в растворах щелочных сульфидов и очень мало — в растворе сульфида аммония (по Фрезениусу, при 60° С — 0,0706 г/л, до Дитте — 2 г/л, по Нойесу и Брею — не растворяется [565, стр. 265]). В отсутствие сильных кислот легко пептизируется сероводородом и образует коллоидные растворы. Прокаленный CdS подвергается лишь поверхностному действию разбавленных кислот. При умеренном нагревании на воздухе слегка окисляется; осажденный на холоду из слабокислой среды легко окисляется при 350° С, но кристаллическая форма почти не изменяется и при 450—480° С. До сравнительно недавнего времени CdS, благодаря своей малой растворимости (ПР = 7,9-10“27, рПР = 26,10), служил основой для отделения кадмия от других элементов и использовался при его гравиметрическом, титриметрическом и колориметрическом определении. В настоящее время для этой цели используют образование других соединений кадмия [82, 354, 565, 619].
Ферроцианид кадмия Cd2[Fe(CN)6]. При действии ферроцианида калия на растворы солей кадмия осаждается не нормальная соль, а осадки переменного состава; из теплого аммиачного раствора, содержащего аммонийные соли, выделяется кристаллический осадок Cd(NH4)2[Fe(CN)6], который можно сушить при 100° С. Cd2[Fe(CN)6] растворим в кислотах и концентрированном аммиаке; при встряхивании из раствора выпадают крупные белые кристаллы [Cd(NH3)4]2 [Fe(CN)6]. Известны ферроцианиды кадмия и другого состава.
Феррицианид кадмия Cd3[Fe(CN)6]2 — тонкий желтый осадок, состав которого зависит от степени гидролиза, выделяется из разбавленного раствора Cd(NO3)2.
Фосфаты кадмия. Cd3(PO4)2 выделяется в виде белого осадка при действии фосфатов на холодный слабокислый раствор . соли кадмия, свободный от большого избытка аммонийных солей. Растворим в минеральных и уксусной кислотах и в аммиаке, прокаливание при 800—900° С переводит его в Cd2P2O7. Cd(NH4)PO4-H2O можно высушивать без разложения при 105— 110° С; при кипячении раствора с осадком он отщепляет воду и аммиак, при прокаливании эта соль тоже количественно переходит в пирофосфат [42, 82, 324, 565].
Аммиакат кадмия [Cd(NH3)4]2+ получается при действии аммиака на Cd(OH)2 и соли кадмия. В зависимости от условий в растворах могут существовать и другие аммиачные комплексы, содержащие от 1 до 6 молекул NH3. При пропускании газооб-26
разного аммиака сухие соли кадмия поглощают его в большем количестве: сульфат до 6, хлорид — до 10, а бромид — до 12 молекул NH3 [354]. Аналитическое применение аммиаката кадмия , основано на е?о способности давать осадки с крупными неорга-/ ническими аниэнами.
Галогениды кадмия [165, 197, 354]. Хлорид кадмия CdCl2 получают растворением металла в НС1. В зависимости от условий опыта кристаллизуются гидраты с 4 (< 5,6° С), 2,5 (5,6° С) и 2 (33,8° С) молекулами воды; в безводную соль они превращаются после нагревания до 120° С. Хлорид хорошо растворяется в воде, немного растворим в этаноле (1,5 г/100 мл), метаноле, зтилацетате и ацетоне; в диэтиловом эфире практически
Таблица 7
Растворимость некоторых солей кадмия (г безводной соли/100 г воды) при различных температурах [233]
Соль кадмия N * Температура, °C
0 10 20 30 40 50
савга 4 H2O 56,2 75,4 98,8 128,8 151,9
CdCl2 2,5 H2O 90,01 122,8
H2O 135,1 134,5 135,3
CdJ2 79,8 83,2 86,2 89,7 93,8 97,4
Cd(NO3)2 9 H2O 4 H2O 106 153 199
CdSO4 8/3 H2O 75,4 76,1 77,7 78,6
H2O 77,1
Таблица 7 (окончание)
Соль кадмия N * Температура, °C
60 70 80 90 100
CdBr2 4 H2O 152,9 155,1 160,8
CdCl2 2,5 H2O
H2O 136,5 140,5 147,0
CdJ2 100,4 110,0 124,9
Cd(NOs)2 9 H2O
4 H2O
619 646 682.
CdSO4 8/3 H2O
H2O 70,3 67,6 64,5 58,4
(твердой .фазе), находящемся в равновесии
* Число молекул воды в кристаллогидрате с насыщенным раствором соли.
27
нерастворим. В спиртах наблюдается заметная диссоциация. В зависимости от концентрации соли и от pH в растворе могут быть различные формы хлорида, в частности — и основные соли, например Cd(OH)Cl.
Бромид кадмия CdBr2 образуется при обработке металла кипящей бромной водой; по свойствам очень близок к хлориду, но в водных растворах немного более ассоциирован. Диэтиловый эфир экстрагирует из 1—6 М НВг менее 1% Cd, метилпропил-кетон из 4,5 М НВг — 36,6% Cd [386, стр. 75].
Иодид кадмия CdJ2 получают растворением CdO или CdCO3 в разбавленной HJ. Кроме диссоциации, в этих растворах наблюдается образование аутокомплексов: 3CdJ2 = Cd8+ + + 2[CdJ3]~. Из 6,9 М HJ или 1,5 М KJ + 1,5 М H2SO4 кадмий полностью экстрагируется диэтиловым эфиром. Растворимость галогенидов и некоторых других солей кадмия в воде приведена в табл. 7.
Галогениды кадмия диссоциируют в растворах неполностью и образуют ионы [Cd(Hal)]+. Это явление осложняется комплексообразованием, в результате чего образуются анионные комплексы типа [Cd(Hal)3]“ и [Cd(Hal)4]2 . Относительное содержание различных ионов в зависимости от концентрации раствора показано в табл. 8 [619, стр. 179, 182; 197, стр. 472] (соответствующие значения рТС, см. в Приложении 2).
Таблица 8
Соотношение различных ионных форм кадмия в растворах его галогенидов [619, стр. 182; 197, стр. 472]
Ионная форма Содержание ионных форм, %
0,01 M CdCl2 CdBr2 0,01 М CdJ2
0,01 м 0,5 M
Cd2+ 41,0 32,8 2,6 23,1
Cd (Hal)+ 56,3 60,6 51,8 66,4
CdHah 3,9 6,5 32,8 6,9
Cd (Halted (Hal)p- } 0,05 } 0,17 8,6 4,2 } 0,47 )
При добавлении большого избытка галогенида щелочного металла образуются труднорастворимые комплексы типа Rb4[CdCl6] и Cs4[CdCl6], которые используют для чувствительного микро-кристаллоскопического открытия Cd.
Роданид кадмия Cd(SCN)2 получают растворением CdCO3 в роданистоводородной кислоте; выделяется при упаривании раствора в виде бесцветных корочек. При избытке роданида в растворе образуется комплексный анион [Cd(SCN)4]2-. В присут
28
ствии роданйдов щелочных и других металлов получаются хорошо кристаллизующиеся двойные соли, большинство из них типа Me2[Cd(SCN)4 (Н2О)2] [10, 354]. В анализе используют комплексные роданиды типа Cd[Hg(SCN)4] и Cd[Zn(SCN)4]. Из солянокислых растворов, 1—7 М по NH4SCN, экстракция кадмия диэтиловым эфиром не превышает 0,1—0,2%; это может быть использовано для его отделения от цинка, роданид которого хорошо экстрагируется в этих условиях [386, стр. 76].
Цианид кадмия Cd(CN)2 — белый студенистый осадок, выделяющийся при осторожном добавлении к насыщенному раствору CdSO4 концентрированного раствора KCN. Растворяется в его избытке (с образованием комплексного аниона [Cd(CN)4]2') и в сильных кислотах. Щелочные растворы комплексного цианида используют при электролитическом выделении кадмия; действием сероводорода из них можно осадить CdS и тем самым отделить от меди (хотя ПР у CdS больше, чем у CuS, но прочность цианида кадмия меньше, чем цианида меди). Переведение кадмия в цианид используют для его маскировки (в некоторых методах — с последующей демаскировкой) и в ряде других аналитических разделений [354, 416, 565, 619].
Комплексные анионы галогенидов и роданида кадмия в соответствующих условиях образуют кристаллические или окрашенные осадки (ассоциаты) с акридином, антипирином, бруцином, ди-антипирилметаном, Р-нафтохинолином, тетрафениларсонием, уротропином, хинином, с катионами трифенилметановых красителей, а также с некоторыми комплексными катионами, например железо—дипиридил, медь—этилендиамин и др.; некоторые из них экстрагируются органическими растворителями.
Основные свойства некоторых неорганических соединений кадмия сведены в Приложении 1; константы нестойкости его не-рганических комплексов представлены в Приложении 2.
Органические соединения кадмия
Главную роль в аналитической химии кадмия играют его органические комплексы. Как и другие ионы с заполненными d-уровнями, Cd2+ предпочтительно взаимодействует с легкополяри-ризующимися лигандами.
Для определения кадмия часто используются соединения, содержащие серу [323, стр. 19],. аминогруппы или гетероциклические атомы азота. Соединения с координирующим атомом кислорода для кадмия не типичны [323].
Для открытия и количественного определения кадмия используются алифатические и ароматические амины и аминокислоты, 5- и 6-членные гетероциклические соединения, содержащие азот, формазаны, диазо-, о-окси-азо- и азометиновые реагенты, трифенилметановые и ксантеновые красители, производные антипи
29
рина, алифатические и ароматические тиосоединения, алкалоиды и др.
Органические соединения кадмия, содержащие соответствующие функциональные группировки [209, стр. 77; 207, 283, стр.10], можно классифицировать следующим образом.
1. Двухзарядные катионные комплексы кадмия с нейтральными азотсодержащими лигандами.
2. Серусодержащие соединения, в которых кадмий замыкает
—N (S) = С — SH
4-членный цикл с группой I , где заместителем Б
Б
могут быть атомы С, N, О или S; известны соединения с 4-членным кольцом, в которых место центрального углерода занимают атомы фосфора или мышьяка.
3. Комплексы кадмия с азосоединениями; в них возможны 4-членные циклы (для диазосоединений) и 5-членные кольца (для о-окси-азопроизводных).
4. Замыкаемые кадмием 5-членные циклы с азот- или (и) серусодержащими реагентами, в которые входит атомная груп-
—С—С—
пировка . Солеобразующими группами (Сл) служат ОН,
NH или SH; комплексообразующими (Кс) — О, N, NH, NH2 или SH. Вместо углерода в основной цепи могут находиться атомы азота.
5. Комплексы кадмия, в которых он образует 6-членный цикл с азотсодержащими реагентами, содержащими группу
—С—С—С—
> I I ; крайние атомы углерода в некото-
(или NH)OH М(или NHa)
рых соединениях (формазанах) замещены азотом.
6. Ионные или иные ассоциаты катионных или анионных комплексов кадмия с нейтральными или несущими соответствующий заряд органическими или металлоорганическими вещества-: ми.
При такой классификации в 1-ю группу входят комплексы кадмия с анилином, 2,2'-дипиридилом, пиридином и его производными, с 1,10-фенантролином, хинолином, этилендиамином, некоторыми алкалоидами; число лигандов в них может колебаться от 1 до 4.
Ко 2-й группе принадлежат комплексы с висмутолом, п-ди-метиламинобензилиденроданином, диэтилдитиокарбаминатом, диэтилдитиофосфатом, меркаптобензотиазолом, 4-окси-З-нитрофе-ниларсоновой кислотой, тиомочевиной, тиоацетамидом.
К 3-й группе относятся комплексы с бромбензтиазо, с кадио-нами, а также соединения, которые образуют с кадмием многочисленные металлиндикаторы—азокрасители, ПАН, ПАР, сульф-арсазен.
4-ю группу представляют соединения кадмия с 8-окси-, 8-меркапто- и 8-(бензолсульфаниламино)хинолином, с глиоксаль-
30
бис-(2-оксианил)ом, глицином, дитизоном; с гидроксамовой, ру-беановодородной, тиобарбитуровой и тиогликолевой кислотами; с комплексоном III, ксиленоловым оранжевым, купфероном, пирокатехиновым фиолетовым, тионалидом.
5-я группа включает в себя соединения с антраниловой кислотой, формазанами, с 2-(о-оксифенил)бензоксазолом и -бензотиазолом, с салицилальдоксимом.
В 6-й группе находятся ассоциаты комплексных анионов или катионов кадмия с производными антипирина, с бруцином, галоидопроизводными флуоресцеина, тетрафениларсонием или фосфонием, с трифенилметановыми красителями, с хинином; с комплексами железо — дипиридил и медь — этилендиамин.
Константы нестойкости ряда органических комплексов и соединений кадмия приведены в Приложении 3.
Свойства некоторых комплексов, представляющих указанные группы и наиболее часто использующиеся или рекомендованные в последнее время для определения кадмия, приведены ниже; вероятное строение их дано в Приложении 4. О структуре некоторых из них литературные данные разноречивы и в качестве координирующих указаны различные группы атомов; эти разногласия здесь не обсуждаются.
Комплекс кадмия с пиридином [Cd(C5H5N)4] (SCN)2 —-тонкие белые кристаллы, выделяющиеся из раствора кадмия при действии роданида и избытка пиридина; на холоду медленно, при 40—45° С через 1—2 часа отщепляет 2 молекулы пиридина и переходит в [Cd(C6H6N)2] -(SCN)2. Такой же комплекс образуется и при недостатке пиридина. Подобно пиридиновым комплексам других металлов, экстрагируется хлороформов [281, 386, 565].
Комплекс кадмия с 1,10-ф е н а н т р о л ин о м [GdC12H8N2]2+ может содержать 1, 2 или 3 молекулы лиганда. Максимум поглощения водного раствора с pH 5,5 находится в области 225—270 нм (е — 36000). Образует экстрагирующиеся органическими растворителями ассоциаты с крупными органическими анионами [481, 647, 741].
Диэтилдитиокарбаминат кадмия Cd[(C2H6)2NCS2]2 — бледно-желтое нерастворимое в воде соединение, экстрагирующееся органическими растворителями при pH 5-11 [281, 386 , 741].
Диэтилдитиофосфат кадм и я Cd[(C2H6O)2PS2] — труднорастворимый в воде осадок, выделяющийся из слабо сернокислых растворов при действии диэтилдитиофосфата никеля [55, 509].
Меркаптобензтиазолат кадмия Cd(C7H4NS2)2 получается высушиванием при НО—120° С белого кристаллического осадка [Cd(NH3)2](C7H4NS2)2, выпадающего из аммиачного раствора при действии меркаптобензтиазола [565, стр. 292]. Экстрагируется хлороформом из аммиачно-тартратной среды; при встряхивании экстракта с 6 N НС1 переходит в водную фазу
31
[363, стр. 313] (по другим данным меркаптобензтиазолат — простая соль с двумя координированными молекулами аммиака [209, стр. 36]).
Комплекс с кадионом CdC18H13N6NO2 — красное соеди-динение, образующееся в щелочной среде. Аналогичный комплекс с кадионом 2В вместо n-нитрофенильной группы содержит 4-нитронафталиновую, а в соединении с кадионом ИРЕ А— крайние фенильные группы сульфированы [209, 233, 323].
Комплекс кадмия с бромбензтиазо Cd(C17HgBrN3OS)2 при экстракции ксилолом из щелочной среды окрашивает органический слой в фиолетовый цвет (в отсутствие кадмия экстракт реагента оранжевый). Окраска этого комплекса устойчивее, чем дитизоната кадмия [98, 325].
Комплекс кадмия с кислотным хром теми о-с и н и м Cd(C16HuOgN2S2)2 имеет красную окраску; служит окрашенной формой металлоиндикатора при комплексометрическом определении кадмия, используется также в его спектрофотометрии [228, 325, 464]. В условиях титрования вероятный состав комплекса 1:1 [283].
Хинальдинат кадмия Cd(C10H6NO2)2 — белые кристаллы, труднорастворимые в горячей воде (с частичным гидролизом), легко — в кислотах и аммиаке; в холодной воде растворимость менее 1 : 10~4 [565, стр. 286; 233].
8-0 ксихинолинат кадмия Gd(CgH6ON)2-H2O — желтое кристаллическое вещество, получающееся при высушивании на воздухе. При 100° С теряет ]/2 Н2О, при длительном нагревании до 130° С обезвоживается. В уксуснокислой среде растворимость < 2,5 мгк]мл, в тартратно-щелочном растворе — мгк/мл [565, стр. 283]. Начинает осаждаться при pH > 5,5, полнота выделения достигается при pH 5,7—14,6; ПР — 1022 (рПР 22,0) [459, стр. 88]. Экстрагируется хлороформом из водной фазы в области pH 5,5—9,5; в присутствии w-бутиламина — до pH И —11,5 [363, 389, 647]. Аналогичное строение имеет комплекс кадмия с 8-м еркаптохинолином [29] и с 8-(б е н золсульфаниламин о)х инолином [45, 364].
Дитизонат кадмия Cd(C13HnN4S)2 — количественно экстрагируется хлороформом при pH 7—14; Хтах экстракта находится при 520 нм, е ~ 86000. Устойчив в сильнощелочных тартратных и цитратных растворах [150, 363, 389].
Купфе ропат кадмия Cd[C6H6N(NO)O]2 — белые кристаллы. Поскольку связь с кадмием осуществляется в нем через атомы кислорода, прочность соединения незначительна. Экстрагируется эфиром из нейтральной среды и хлороформом из водной фазы с pH > 4,5 [82, 459].
Антранилат кадмия Cd(C7H6O2N)2 — тонкий белый кристаллический порошок. Растворимость в воде < 1 : 10е, легко растворим в кислотах и разбавленном аммиаке; щелочи разлагают соединение с выделением осадка CdO [647, 565, стр. 287].
32
2-(о-О ксифенил) бензоксазолат кадмия _ Cd(C13H8O2N)2 (Приложение 4) в тартратном растворе начинает осаждаться при pH 6, полностью выделяется при pH И —12; устойчив при нагревании до 275° С. Хорошо растворим (с разложением) в ледяной уксусной кислоте, очень мало — в ацетоне, бензоле, гексане, метаноле, этаноле, хлороформе и четыреххлористом углероде [553, 771, 772].
Дифенилкарбазонат кадмия Cd(C13HuN4O)2 — фиолетовое соединение, образующееся в слабокислой среде; экстрагируется бензолом, хлороформом и другими растворителями. Максимум поглощения экстракта в толуоле находится при 483 нм, е = 36700. При взаимодействии с кадмием дифенилкарб-азид, по всей вероятности, сначала окисляется до дифенилкарба-зона [42, 323, 759].
Р-Н афтохинолин-иодидный комплекс кадмия H2[CdJ4] (C13H8N)2 — желтый кристаллический осадок, выделяющийся из сернокислых растворов [10, 323, 565].
Желез о-д ипиридил-иодидный комплекс кадмия [CdJJ [Fe(C10HgN2)3] (Приложение 4) — красно-фиолетовый осадок, выпадающий из насыщенного реагентом слабокислого, нейтрального или аммиачного раствора кадмия [323, 619].
Ассоциат фенантролината кадмия с эозином [Cd(C12H8N2)2] (С20Н8О5Вг4) (Приложение 4)— красный осадок, растворяется в хлороформе без изменения окраски; при добавлении ацетона экстракт флуоресцирует желтым светом. Аналогично ведут себя комплексы кадмия с пиридином и 2,2'-дипиридилом с другими галоидопроизводными флуоресценина (эритрозином, бенгальской розовой и др.) [256, 257].
Строение комплекса кадмия сдальзином (диаллилдитио-карбамидогидразином) представлено в Приложении 4 [323, 325]. Ассоциату иодида кадмия с 1,3-д и х и н о л и л-2-п р о п е н о м приписывают 8-членное кольцо [741]. Однако практического применения в аналитической химии кадмия подобные соединения, по-видимому, не имеют. Можно ожидать, что комплекс кадмия с тетрафенилпорфином [741] будет использован для его люминесцентного определения (по аналогии с люминесцентным определением Си и Zn в виде соединений с этиопорфирином [376, 377]).
2 Щербов Д. П
Глава III
КАЧЕСТВЕННОЕ ОБНАРУЖЕНИЕ КАДМИЯ
О выражении чувствительности определений
В качественном анализе чувствительность реакций выражают обычно -понятием «открываемый минимум» (ОМ), «уверенно открываемый минимум» (УОМ) (в микрограммах искомого вещества, иногда — в мг/мл) и «предельное разбавление» (Р) («предельная концентрация, «минимальная концентрация») — весовое отношение определяемого вещества к растворителю (1 : 10п) или отрицательный логарифм этого отношения, показатель рЬ [5, 6, 13, 191, 350, 386, 428]. Открываемый минимум (мкг) и предельное разбавление (1 : В, где В — объем раствора, мл, соответствующий предельной концентрации искомого вещества), связаны между собой следующими зависимостями:
ОМ = ; В =
Величина ОМ сильно зависит от присутствия посторонних веществ и от условий проведения реакции (в частности, объема анализируемого раствора).
В количественном анализе выражение чувствительности и пределов определения еще разнообразнее — как по критерию порога, так и по форме представления определяемых концентраций [22, 363, 386, 470]. Их приводят в процентах, молГл, мг/л, мг/мл, мкг/мл, мкг/см2 (поперечного сечения кюветы), мкг в конечном объеме анализируемого раствора. В абсорбциометрии чувствительность цветной реакции нередко характеризуют молярным коэффициентом погашения е. Поэтому сопоставление чувствительности различных методов анализа по опубликованным данным очень затруднительно. В частности, для ряда качественных реакций (использующих малые объемы анализируемых растворов), величина ОМ (в мкг) создает ложное представление об их очень высокой чувствительности (на самом деле концентрация испытуемого раствора должна быть достаточно велика — нередко намного больше, чем доступная определению обычными количественными методами). Не менее обманчивым, с первого взгляда, бывает и впечатление от выражения порога чувствительности в мом/л, часто приводимое в полярографических способах анализа.
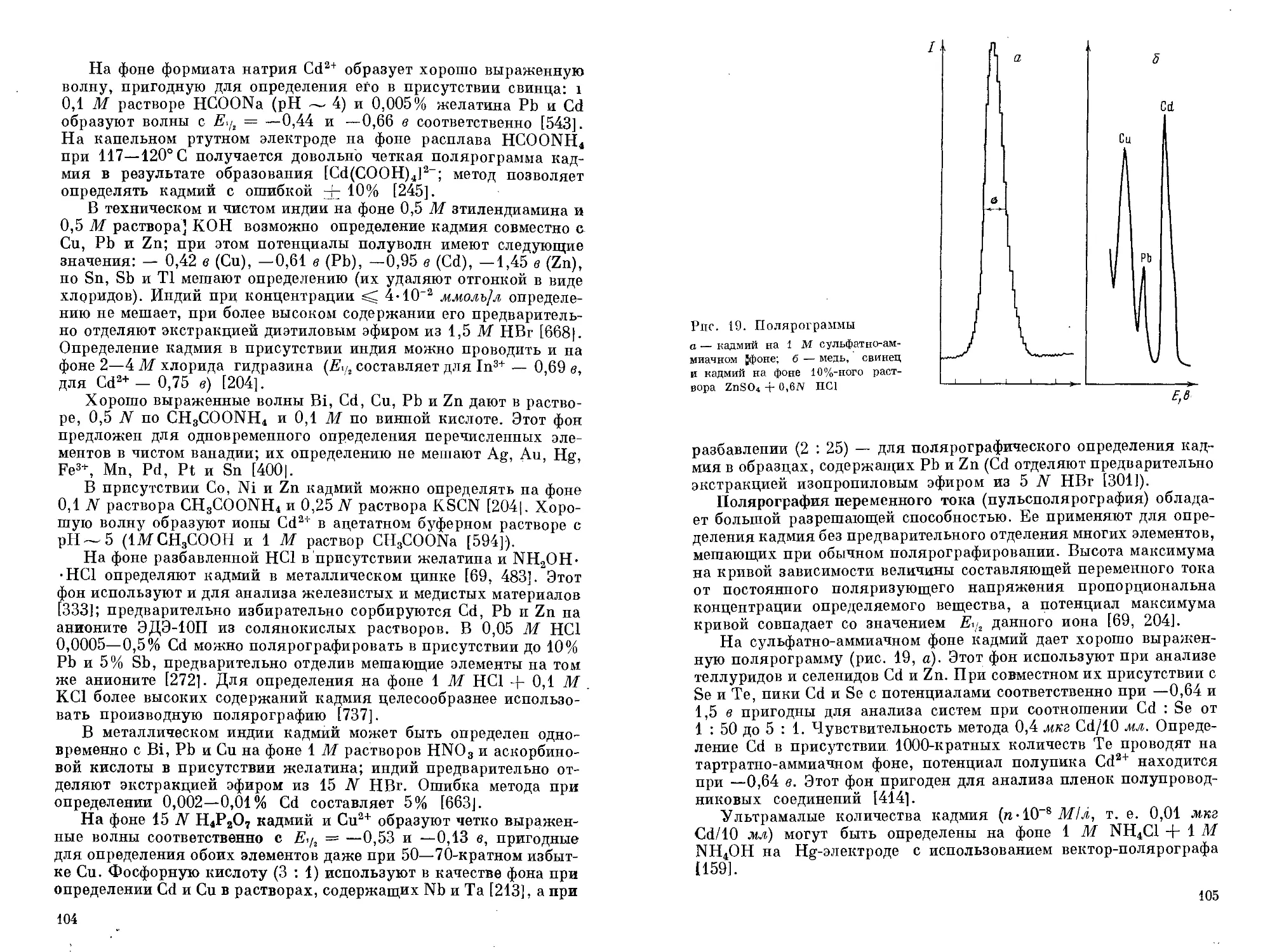
В связи со сказанным, в последующем изложении, наряду с оригинальными данными, по возможности приводится расчетная концентрация С, мкг/мл и мкг/г (для твердых проб). Доступный определению интервал содержаний кадмия выражается в его весовом количестве, мг или мкг в эффективной навеске пробы или в конечном объеме раствора.
34
ОБЩАЯ ХАРАКТЕРИСТИКА ГРУППЫ
Кадмий принадлежит к аналитической группе сероводорода; по русской классификации качественного анализа он входит во 2-ю подгруппу IV группы (подгруппу меди). Катионы этой группы осаждаются сероводородом при pH 0,5 в виде труднорастворимых сульфидов; из них несколько более растворим GdS (ПР = = 7,9-10~27). От катионов III аналитической группы (сернистого аммония) кадмий отличается очень малой растворимостью сульфида в кислотах, от элементов V группы (образующих тиосоли) — большей устойчивостью по отношению к сульфидам щелочных металлов и к едким щелочам [42].
Лучшим растворителем для сульфидов катионов IV группы служит горячая 2 N HNO3; при этом ион S2~ окисляется до элементной серы, а катион переходит в раствор. Катионы этой группы легко поляризуются сами и обладают сильным поляризующим действием; поэтому они образуют комплексные анионы с галогенидами, роданидами и цианидами. Поэтому сульфид кадмия (в отличие от остальных элементов IV группы) сравнительно легко растворим в концентрированной НС1 или насыщенном растворе Nad с образованием комплексного аниона [CdCl4]2-. Из раствора, содержащего KCN, сульфид кадмия можно осадить в присутствии меди, остающейся в растворе в виде более устойчивого комплексного цианида.
Действием едких щелочей катионы IV группы осаждаются в виде гидроокисей. Их карбонаты, фосфаты, ферро- и феррицианиды труднорастворимы. Для многих элементов группы характерно образование достаточно устойчивых комплексов с аммиаком [Cd(NH3)4]X2 и со многими азотсодержащими лигандами. Поэтому гидроокись, основной карбонат и фосфат, так же как и оксисоли кадмия, растворяются в избытке аммиака (особенно легко в присутствии аммонийных солей):
Cd (ОН)2 + 2NH.OH + 2NH.C1 [Cd (ИНф] Ch + 4ЩО.
Как уже было указано, избирательных реакций на кадмий нет. Например, группировка )>С— ОН, характерная для ионов Fe3+ [209], входит в состав большинства реагентов на кадмий; поэтому все они взаимодействуют с железом. Многие серусодержащие соединения взаимодействуют и с остальными элементами сероводородной группы, особенно — с ионами серебра, висмута, меди, ртути и свинца, с представителями группы сульфида аммония и др. Поэтому избирательность по отношению к кадмию достигается лишь при изменении кислотности среды и использовании подходящих комплексообразующих агентов.
2*
35
РЕАКЦИИ ДЛЯ ОБНАРУЖЕНИЯ КАДМИЯ
Для обнаружения кадмия описаны многочисленные реагенты, включенные в систематический ход «классического» качественного анализа (с использованием макропробирок) или предложенные для капельных, микрокристаллоскопических, люминесцентных и некоторых других реакций.
Реакции в макропробирках
Неорганические реагенты
Сероводород осаждает из 0,3 М растворов минеральных кислот ярко-желтый сульфид CdS, растворяющийся при кипячении с разбавленными НС1 и H2SO4 (отличие от меди), но нерастворимый в аммиаке. Из — 0,6 N растворов выделяется оранжевый осадок, содержащий примесь Cd2SCl2 или Cd2S(SO4). Вместе с кадмием осаждаются As, Bi, Си, Pb, Sb и Sn; при указанной кислотности Со, Fe, Мп, Ni и Zn выделяются только в том случае, если их концентрация в растворе очень велика (особенно это относится к цинку, имеющему тенденцию соосаждаться с металлами сероводородной группы) [619, стр. 194].
Сульфиды Cd, Bi, Си, Pb и Hg нерастворимы в горячем растворе желтого сульфида аммония или 2 М раствора КОН (способ отделения осажденных с ними сероводородом Sn, As и Sb). Для отделения от ртути сульфиды Cd, Bi, Си и РЬ растворяют разбавленной азотной кислотой, из которой затем осаждают РЬ действием серной кислоты и этанола, a Bi — избытком аммиака. В оставшийся раствор, содержащий [Cd(NH3)4]2+ и [Cu(NH3)4]2+, добавляют до его обесцвечивания KCN и осаждают кадмий в виде сульфида [619, стр. 195].
Кадмий и медь разделяют также тиомочевиной (связывающей медь), затем осаждают CdS при pH </ 3 [330]. Иногда перед пропусканием H2S к исходному раствору добавляют 1/5 объема НС1, в результате чего произведение растворимости CdS не достигается [3, стр. 323] и кадмий остается в растворе. В другом варианте раствор насыщают хлоридом натрия и тогда образуются комплексные анионы [CdCl4]2 [42, стр. 413]. Можно также предварительно выделить медь действием Na2S2O3 [6, стр. 77], салицилальдоксима в уксуснокислом растворе [435, стр. 74] или NaOH в тартратной среде после восстановления Си2+ до Си+ декстрозой [722].
При действии (NH4)2S или H2S на разбавленные нейтральные растворы солей кадмия осадок не выделяется, а раствор окрашивается в характерный желтый цвет коллоидного .CdS; при подкислении раствора сульфид выделяется в осадок [42, стр. 413]. Однако при обработке сероводородом аммиачного раствора, содержащего медь и KCN, желтое окрашивание осадка после стояния не служит доказательством присутствия кадмия (такой раст
36
вор в присутствии меди становится темно-желтым и из него может 'выделиться оранжево-красный кристаллический осадок (CSNH2)2 («рубеановый водород») [307, стр. 131].
Открываемый минимум при осаждении CdS, по данным разных авторов: 100 мкг/мл [13, стр. 15], 10 мкг/мл [386, стр. 200; 307, стр. 354] и 0,1 мкг (объем не указан) [619, стр. 193]. В присутствии посторонних катионов уверенно открываемый минимум 500 мкг]мл повышается еще в 2—3 раза [13, стр. 31].
Тиоацетамид. Сероводород заменяют нетоксичным тиоацетамидом, который при нагревании раствора гидролизуется:
CH3CSNH2 + 2Н2О — CH3COONH4 + H2S.
Постепенно освобождающийся сероводород выделяет крупнозернистые, легко фильтрующиеся и промывающиеся осадки сульфидов. Осаждение протекает значительно быстрее, чем при использовании H2S. Для осаждения сульфида кадмия из 6 N H2SO4 необходим 50%-ный избыток реагента, из 1,0 N НС1 — 100-кратный. В отсутствие хлоридов количественное выделение CdS возможно из 2 М НС1О4. Осаждению кадмия тиоацетамидом мешают ацетаты и комплексон III [475].
1-Амидино-2-тиомочевина выделяет сульфид кадмия из щелочной среды:
Cd2+ + C2H6N4S + 2ОН- CdS + C2H6N4O + H2O.
Аналогично взаимодействуют и ионы Cu2+, Hg2+ и РЬ2+ [665].
Тиокарбонат калия K2CS3 в аммиачной среде в присутствии комплексона III выделяет оранжевый осадок CdCS3; при наличии меди (необходим избыток реагента) раствор окрашивается в красный цвет, Со2+ и Ni2+ открытию кадмия не мешают [607].
Едкие щелочи выделяют белый студенистый осадок гидроокиси кадмия, нерастворимый в избытке реагента, но легко растворяющийся в кислотах, аммиаке и цианиде калия или натрия; хлорид аммония, лимонная и винная кислоты препятствуют осаждению. Открываемый минимум 100 мкг Cd/мл, уверенно открываемый минимум — 500 мкг Cd/мл [13, стр. 15]. В присутствии ионов Bi, Си и РЬ к раствору добавляют глицерин, образующий с ними прочные комплексы, устойчивые по отношению к едким щелочам; глицерат меди темно-синий, остальные — бесцветны. Кадмий с глицерином не реагирует и осаждается в его присутствии в виде Cd(OH)2 [3, стр. 337], чувствительность реакции та же, что и без глицерина.
Аммиак при осторожном прибавлении осаждает из кислых и нейтральных растворов гидроокись кадмия, растворимую в избытке реагента (отличие от Bi и РЬ); чувствительность реакции такая же, как при действии едких щелочей [13, 42]. Уменьшение устойчивости аммиаката кадмия в щелочной среде при пониженной температуре позволяет открывать его в присутствии меди.
37
К охлажденному раствору аммиакатов обоих элементов добавляют равный объем 4 М NH4OH, затем равный или больший объем 2— 2,5 N раствора NaOH; выделяется белый Cd(OH)2. Открываемый минимум 50 мкг СА]мл [313].
Карбонаты калия и натрия выделяют белый осадок основных солей CdCO3-n Cd(OH)2, нерастворимый в их избытке, но переходящий в раствор при действии карбоната аммония или аммиака. Свежеприготовленная взвесь ВаСО3 в воде осаждает при стоянии на холоду CdCO3 [42].
Ферроцианиды выделяют белый аморфный осадок, растворимый в минеральных кислотах и концентрированном аммиаке; однако при встряхивании последнего сразу же выпадают крупные белые кристаллы состава [Cd(NH3)4]2 [Fe(CN)6], Феррицианид выделяет желтый аморфный Cd3[Fe (CN)6]2 [42].
Гидрофосфат натрия осаждает белый фосфат кадмия Cd3(PO4)2, растворимый в уксусной и минеральных кислотах, а также в аммиаке [42, 324].
Перхлорат аммония выделяет из аммиачных растворов белый кристаллический осадок [Cd(NH3)4] (С1О4)2 (в отличие от меди) [42].
Роданомеркуриат аммония осаждает белый < кристаллический Cd[Hg(SCN)4[; осадки дают также Ag+, Pb2+, Zn2+ (белые), Со24~ (синий), Cu24~, Rh3+ (желтые), Fe3+ (красный), Fe2+(желто-зеленый) и Ni2+(грязно-зеленый). Кристаллы соединений, содержащих по два элемента, имеют другую окраску: Bi3+ 4--J- Zn2+ — розовую, Со2+ 4- Fe3+ — пурпурно-черную, Cd2+ 4- Со2+ и Со2+ 4- Zn2+ — голубую, Cd2+ 4- Cu2+, Cd2+ J- Fe3+, Zn2+ 4--]- Сп2+ и Zn24~ J- Fe3+ — фиолетовую, Cd2+ 4- Ni2+ и Zn24~ 4-Ni2+— грязно-зеленую, Fe2+ 4- Fe3+ — серо-голубую или серофиолетовую [42, стр. 415; 191, стр. 165].
В отличие от остальных катионов подгруппы меди, кадмий не осаждается при действии иодидов и тиосульфатов [42, 324].
Реакции окисления — восстановления. Металлический кадмий восстанавливает все металлы, потенциалы которых в ряду напряжений положительнее водорода: Ag, Au, Hg, Си, Pt, а также Bi, Со, Pb, Sn. Алюминий, магний и цинк выделяют-металлический кадмий из растворов его солей; в отличие от меди, он не восстанавливается металлическим железом. Это можно использовать в целях разделения (в раствор вносят железную стружку; при этом выделяются красновато-коричневые хлопья восстановленной меди, а ионы Cd2+ остаются в растворе) [42, стр. 417].
Органические реагенты
Бруцин4-бромид выделяют из нейтральных ацетатных растворов кадмия белый кристаллический осадок [42] вероятного состава [СйВг4]-бруцин2. Открываемый минимум 100 мкг Cd/ /мл. Мешают определению Bi, Hg, V, W, Au, Mo, Pt [386, стр. 200].
38
' Динитродифенилкарбазид окрашивает осадок Cd(OH)2 в бурый цвет, переходящий в сине-зеленый в присутствии формальдегида. Для маскировки меди вводят цианид. К анализируемому нейтральному или слабокислому раствору прибавляют NaOH, реагент и НСНО. В присутствии кадмия появляется зеленое окрашивание, а затем выпадает синий осадок. Открываемый минимум 100 мкг Cd/мл. Мешают определению Со, Pt, в меньшей степени Fe, Те [42; 386, стр. 200].
При выполнении реакции на пластинке на нее помещают каплю испытуемого раствора, прибавляют по одной капле 10%-ного раствора NaOH, 5%-ного раствора KCN, 0,1%-ного спиртового раствора реагента и 3—4 капли 40%-ного раствора НСНО. Сам реагент в щелочном растворе красного цвета, который переходит при добавлении НСНО в фиолетовый. Синевато-зеленое окрашивание появляется в присутствии 0,8 мкг Cd (С = 30 мкг/мл)', при наличии 400-кратного количества меди можно обнаружить 4 мкг Cd (С=140 мкг/мл) [350,416]. Ионы Со2+образуют бурый осадок, Pt — желтый; допустим 10-кратный избыток As и Hg\ 100-крат-ный - Ag, Au, Bi, Си, Hg2+, Mo, Pb, Sb, Se, Sn, Tl, V и W [350].
Дифенилкарбазид в нейтральных (ацетатных) растворах дает с кадмием сине-фиолетовое окрашивание или краснофиолетовый осадок. Открываемый минимум — 100 мкг Cd/мл, влияние Си, Hg2+ и РЬ устраняют введением иодида и роданида [42; 386, стр. 200].
К а д и о н при добавлении к нейтральному раствору кадмия и последующем подщелачивании 2 N КОН дает фиолетово-розовое окрашивание. Открываемый минимум — 10 мкг Cd/мл, мешают Ag, Со, Cr3+, Си, Fe3+, Hg, Mg, Ni, NH4 [386, стр. 200].
Куприэнат (комплекс меди с этилендиамино'м — [Си-•еп2]2+) образует с анионным комплексом [CdJ4]2- труднорастворимое соединение, выделяющееся в виде крупных кристаллов. Для выполнения реакции к раствору соли кадмия прибавляют избыток KJ и затем куприэнат [42, стр. 416].
Тионалид и хинальдиновая кислота в нейтральных или щелочных растворах осаждают кадмий в виде белых кристаллов. Подобный же осадок образуется при добавлении к раствору кадмия NH4CNS и затем пиридина [42, стр. 415].
Кристаллический фиолетовый образует с иодидным комплексом кадмия ассоциат. К 2 мл нейтрального испытуемого раствора прибавляют по 1 капле НС1 (уд. вес 1,12), 0,06%-ного раствора красителя и 25 %-ного раствора KJ. В присутствии кадмия появляется синее или фиолетовое окрашивание. Предельное разбавление — 4 000 000, открываемый минимум — 5 мкг Cd (С = 0,25 мкг/мл)-, 3000-кратный избыток цинка не мешает [206].
Некоторые производные 3-аминопиридина [188], аценафтенхи-нон-2-(окси)анил [683], бром- и иодпройзводные триоксифлуоро-нов [279], родизонат натрия в растворе, содержащем ацетон и же
39
латин [527], тиокапролактам в присутствии роданида [489J, 2', 3', 4'-триоксихалкон [747]; N-фенилацетилфенилгидроксиламин [108], диметил- и диэтилдиселенокарбаминаты [171], хинолин-8-селенол [727] и многие другие реагенты [100, 320, 441, 478, 489, 547, 726] образуют с кадмием (и прочими элементами) нерастворимые, окрашенные или (и) экстрагирующиеся соединения.
Капельные реакции на фильтровальной бумаге
Дифенилкарбазид. Фильтровальную бумагу смачивают насыщенным на холоду раствором реагента, к которому добавлен до насыщения роданид калия и кристаллик KJ. На высушенную бумагу наносят каплю испытуемого раствора и держат ее 1—2 мин. над открытой склянкой с аммиаком. Появление синефиолетового окрашивания указывает на присутствие кадмия. Открываемый минимум — 4 мкг Cd, в присутствии умеренных количеств Bi, Си, Hg и РЬ — 8 мкг Cd [324, 416, 428] (С = 80 и соответственно — 160 мкг]мл). Реакция применена для обнаружения кадмия в минералах и горных породах [210, стр. 255]. Капельная реакция с дифенилкарбазоном в присутствии кобальтицианида* калия использована в анализе сплавов на свинцовой основе [210, стр. 206]. Значительно чувствительнее реакция кадмия с ди-0-нафтилкарбазоном в щелочном растворе, содержащем KCN и НСНО. Образующаяся фиолетовая окраска позволяет открывать в присутствии 5000-кратного избытка меди —0,1 мкг Cd2+ (С = — 2 мкг]мл) [619, стр. 193].
Тетраиодовисмутиат калия KBiJ4 при действии на его раствор солей кадмия выделяет черный осадок BiJ3. На фильтровальную бумагу наносят каплю раствора реагента и затем — каплю испытуемого раствора. Образующееся черное пятно исчезает при обработке растворами KJ, Na2S2O3 или As2O3 (в присутствии NaHCO3) [324; 405, стр. 73].
Железо-дипиридил-иодид в насыщенном им слабокислом, нейтральном или аммиачном растворе дает с солями кадмия красно-фиолетовый осадок Fe (2,2'-flHnHpHflHn)3-CdJ4. Это одна из самых чувствительных реакций, позволяющая обнаружить 0,05 мкг Cd (С = 1 мкг/мл). На фильтровальную бумагу наносят каплю исследуемого раствора и, прежде чем она впитается,— каплю раствора реагента. В присутствии кадмия образуется красное пятно или кольцо [619, стр. 195; 556]. Для устранения влияния Ag, Bi, Си, Hg, Pb, Sb, Sn и Ti к исходному раствору прибавляют разбавленную НС1 и отфильтровывают осадки хлоридов Ag, Hg+, TI и большей части РЬ. Фильтрат обрабатывают аммиаком для удаления Bi, Hg2+, Sb и Sn и снова фильтруют. Обнаружению кадмия в фильтрате не мешают 5000-кратные количества меди (открываемый минимум — 0,08 мкг Cd2+; С = 1,6 мкг[мл) или цинка (открываемый минимум — 0,1 мкг/, С = 2 мкг]мл) [619].
40
Реакция использована для обнаружения следов кадмия в металлических меди и цинке [210, стр. 165].
Приготовление раствора реагента. Растворяют 0,25а 2,2'-дипири-дила и 0,146 г FeSO4-7HaO в 50 мл воды, добавляют 10 г KJ, встряхивают ~ 30 мин. и отфильтровывают осадок Ее(2,2'-дипиридил)3-J2.
К а д и о н в капельном варианте позволяет открывать кадмий (открываемый минимум 0,1 мкг, С — 3,2 мкг]мл\ уверенно открываемый минимум — 0,3 мкг, С — 10 мкг]мл) в присутствии 100-кратных количеств As, Au, Bi, Mo, Pb, Pt, Sb, Se, Sn, Те, TI, V, W и Zn. Ионы Co, Сг, Си и Ni дают синеватую окраску, Mg — синюю, a Fe — желтую, что несколько затрудняет обнаружение кадмия; но их можно замаскировать тартратом. В этом случае на фильтровальную бумагу помещают каплю нейтрального или слабо уксуснокислого испытуемого раствора и прибавляют по капле 10%-ного раствора сегнетовой соли, 0,02%-ного спиртового, содержащего 0,02 N КОН, раствор реагента и 2 N раствор КОН. В присутствии кадмия появляется розовое пятно, без него бумага окрашивается в фиолетовый цвет. Ионы Ag, Hg2+ и Hg2+ дают буро-оранжевые осадки и мешают определению [350].
Сульфид кадмия — единственный сульфид желтого цвета, нерастворимый в растворе Na2S — тоже используется для открытия кадмия капельным методом [404, 405, 392, стр. 93], в частности, в сплавах на магниевой и цинковой основах [210, стр. 203, 210] и для установления подлинности кадмиевых покрытий [210, стр. 229].
Микрокристаллоскопические реакции
Метод основан на образовании характерных кристаллических осадков, которые рассматривают под микроскопом; о присутствии определяемых ионов судят по внешнему виду кристаллов. Для выполнения анализа используют 1—2 мкл исследуемого раствора, к которому на предметном стекле прибавляют на острие оттянутой стеклянной палочки сухой реагент или, при помощи капиллярной трубочки, его раствор. Для наблюдения можно использовать любой микроскоп с увеличением в 40—200 раз (в частности — М-10, МБИ-1); при работе с ультрафиолетовым излучением рекомендован микроскоп МУФ-2. Техника работы описана в руководстве [191]. При наблюдении в УФ из-за поглощения кристаллами коротковолнового излучения с длиной волны 365 нм и менее тень от них на флуоресцирующем экране из тонкого уранового стекла становится красной на фоне зеленой флуоресценции (окраска вызывается красной линией ртутного спектра, пропускаемой стеклом УФС-1, испытуемым осадком и урановым стеклом) [392, стр. 71].
Краткие данные о микрокристаллоскопических реакциях, предложенных для открытия кадмия, сведены в табл. 9; схемати-
41
Таблица 9
Микрокристаллоскопические реакции для открытия кадмия
Открывав- —
Реагент Среда Образующееся соединение Разбавление мый минимум, мкг мкг/мл Литература
Акридин + роданид Кисля [Cd(SCN)4]H2(C13H9N)2 ]191, стр. 208]
Анилин + роданид » 1 : 1300 3,0 3000 [191, стр. 197]
Антипирин + бромид » (CuH12ON2)H2[CdBr4]. • 2CuH12ON [191, стр. 202]
Бруцин + бромид » HjCdBrJ (C23H26O4N2)23* 1 : 16000 0,12 60 [191, стр. 200]
Виолуровая кислота * 1 : 2000 15,0 500 [634]
Гидразин + роданид [Cd(N2H4)2] (SCN)2 1 : 450000 0,07 2,5 [191, стр. 396]
Купферон Аммиачная [C6H6N(NO)O]2Cd [191, стр. 397]
Морфолин + роданид 1 : 10000 0,1 100 [191, стр. 396]
2-Нафтиламин -|- роданид 1 : 8000 0,12 125 [191, стр. 396]
Никотин + роданид 1 : 2500 0,4 400 [191, стр. 396]
Нитропруссид натрия * Cd[Fe(CN)5NO]2* 1 : 3000000 0,01 0,35 [392, стр. 94]
8-Оксихинолин + иодид Кислая 1 : 800 1,2 1250 [191, стр. 397]
Пикриновая кислота Аммиачная [Cd(NH3)4] [C6H2(NO3)3O]2 1 : 1000 1,0 1000 [191, стр. 198]
Пирамидон ф- роданид Кислая [191, стр. 396]
Пиридин (пары) + + бихромат [Cd(C5H5N)4]Cr2O7 1 : 6500 0,3 150 [191, стр. 185]
Пиридин 4- бромид Cd(C5H5N)2]Br24* 1 : 20000 0,05 50 [191, стр. 166 и 200]
Пиридин + роданид [Cd(C5H5N)2](SCN)24* 1 : 20000 0,05 50 [191, стр. 166 и 200]
Роданомеркуриат аммония Cd[Hg(SCN)4]5* 1 : 1000 1,0 1000 [191, стр. 165 и 202]
Таблица 9 (окончание)
Реагент Среда Образующееся соединение Разбавление Открываемый минимум, мкг с, мкг/мл Литература
Родапоцинкат аммония Cd[Zn(SCN)4]6* 1 : 8000 0,25 125 [191, стр. 184]
Хлорид рубидия* Bb4[CdCl6]7* 1 : 100000 0,01 10 [191, стр. 203]
Селеноцианомеркуриат калия Cd[Hg(SeCN)4] 1 :1000 1,0 1000 [191, стр. 395]
Сульфид натрия CdS2* 1 : 1500000 0,02 0,7 [392, стр. 93]
Хлорид тетрафенилфос-фония 1 : 6500 0,3 150 [191, стр. 397]
Тиомочевина + пикрат [Cd(CSN2H4)4](CeH2N3O7)2 1 : 100000 0,03 10 [191, стр. 397]
Тиомочевина + соль Рейнеке 1 : 50000 0,02 20 [191, стр. 397]
Уротропин + иодид Кислая 1 : 1000 1,0 1000 [191, стр. 202]
Тиомочевина + роданид [Cd(C6H12N4)2] (SCN)2-6H2O 1 : 1000 2,0 1000 [191, стр. 202]
Фосфат натрия Cm.2* 1 : 150000 0,2 7 [392, стр. 93]
Хинолин + иодид » 1 : 10000 0,17 100 [191, стр. 397]
Хинолин + роданид » 1 : 330 3,0 3000 [191, стр. 201]
Хлорид цезия * Cs[CdCl6] ’* 1 : 100000 0,01 10 [191, стр. 203]
Щавелевая кислота CdC2O4-3H2O 8* 1 : 3000 0,35 350 [191, стр. 204]
. Упаривание с раствором реагента. ™ Красная окраска в УФ. См. Приложение 5. « См. Приложение 6. » См. Приложение 7.
•* См. Приложение 8. ’• См. Приложение 9. »• См. Приложение 10.
ческое изображение образующихся микрокристаллов [191] приведено в Приложениях 5—10. Для облегчения идентификации соединений кадмия представлены и опубликованные данные (рисунки) о форме кристаллов, образуемых рассмотренными реагентами с другими ионами.
Акридин в присутствии роданида образует с Cd2+ желтый мелкокристаллический осадок; реакция малочувствительна, также ведут себя Au3+, Со2+, Cu2+, In3+, Ir3+, Pd2+, Pt4+.
Анилин при добавлении роданида дает с Gd2+ в сравнительно концентрированных растворах бесцветный осадок, малые количества Си2+ окрашивают его в желтый цвет. Открытию Cd2+ мешают Sb3+ и Sn2+.
Антипирин, 5%-ный раствор в 10%-ном растворе КВт, в сернокислой среде позволяет открывать кадмий в присутствии Cu2+, Zn2+ и малых количеств Bi3+, Pb2+, Sn2+ и Sb3+.
Бруцин. К капле раствора соли кадмия прибавляют насыщенный раствор реагента в разбавленной H2SO4 или СН3СООН и затем каплю 5 %-ного раствора КВг. При этом тотчас же образуется мелкокристаллический белый осадок, переходящий затем в розетки из игл и ромбов (Приложение 5). В тех же условиях реагируют Bi3+, Hg2+ и Sb3+.
Виолуровая (5-изонитрозобарбитуровая) кислота дает осадки и с Со2+, Pb2+, Tl+, UO2+, Zn2+, щелочноземельными и некоторыми щелочными металлами [634].
Гидразин в присутствии роданида реагирует с ионами Cd2+ и Си2+.
Купферон, кроме бесцветного осадка с Cd2+, образует коричнево-красные кристаллы с Fe3+, светло-желтые с UO2+ и зеленовато-желтые с Си2+; осаждаются и щелочноземельные металлы.
Морфолин в присутствии роданида реагирует, кроме Gd2+, с ионами Cu2+, Hg2+, Ni2+, Pb2+ и Zn2+.
2-Н афтиламин и роданид дают реакцию и с Cu2+r которая примерно в 20 раз менее чувствительна, чем с кадмием.
Никотин и роданид реагируют также с ионами Со2+, Cu2+, Fe2+, Ми2+, Ni2+ и Zn2+.
Нитропруссид натрия. Каплю исследуемого раствора выпаривают почти досуха и добавляют каплю 1 %-ного раствора нитропруссида натрия. Образовавшиеся кристаллы обладают сильным поглощением в области 280—365 нм и под ультрафиолетовым микроскопом их тень на урановом стекле окрашивается в красный цвет.
8-0 ксихинолин и иодид калия в азотнокислой среде, кроме кадмия, реагируют с ионами Bi3+, Cu2+ и Sb3+.
Пикриновая кислота (насыщенный раствор) в нейтральной среде осаждает, кроме кадмия, NH15 К+, Pb+, Cs+ и Т1+г а в аммиачной среде, также следующие ионы:
44
Ион Открываемый минимум», мкг Ион Открываемый минимум, мкг
Ag+ 20... • --«.йлЗг Hg2+ 0,15
Aus+ ' О', 2 NP+ 0,1
Co2+ 0,3 Zn2+ 2,0
Cu2+ 0,05
Пирамидон и роданидв азотнокислой среде, кроме кадмия (шестиугольники, дендриты), осаждают Go, Си, Fe и Zn.
Пиридин. Каплю испытуемого раствора смешивают с насыщенным раствором бихромата и подвергают действию паров пиридина. Стекло, на котором находится капля, потирают стеклянной палочкой и повторно обрабатывают пиридином; в присутствии кадмия образуются мелкие желтые кристаллы. Такую же реакцию дают Си2+, Со2+ и Ni2+, аморфные осадки — Ag+, Fe3+ и Pb2+; Hg2+ реагирует и без трения стеклянной палочкой. Для более чувствительного открытия к капле раствора кадмия прибавляют пиридин и вводят кристаллик КВт или NH4SCN (Приложение 6); в этих условиях кристаллические осадки дают и Аи3+, Со2+, Cu2+, Hg2+, Ni2+, VO3, Zn2\
Роданомеркуриат кадмия — бесцветные кристаллы, выделяющиеся при добавлении капли роданомеркуриата аммония к нейтральному или слабокислому раствору соли кадмия (Приложение 7); в тех же условиях кристаллические осадки образуют Со2+ (синий), Си2+ (желтый), Fe3+ (желтовато-зеленый), Ni2+ (бледно-зеленый), Pb2+, Zn2+ (белые), Rh3+ (желтый) и Ag+ (аморфный белый). При этом получаются с кадмием смешанные соединения и цвет или форма его кристаллов изменяется.
Роданоцинкат кадмия получается при добавле
нии капли исследуемого раствора к концентрированному раствору ZnSO4 и NH4CNS (Приложение 8); аналогичную реакцию дают ионы Ag+, Hg2+.
Хлориды рубидия или цезия (насыщенные растворы) прибавляют к сухому остатку, полученному после выпаривания исследуемого раствора на предметном стекле (Приложение 9). Определению кадмия мешают ионы Bi3+, In3+, Pb2+, Pt4+, Sb3+ и Sn2+.
Селеноцианомеркуриат образует осадки также с ионами Go2+, Fe2+ и Zn2+.
Сульфид натрия. При использовании линии Hg 366 нм, по «красной окраске» тени на урановом стекле под ультрафиолетовым микроскопом можно открывать Cd2+ в присутствии цинка. Для этого в каплю исследуемого раствора вводят кристаллик Na2S и перемешивают осадок стеклянной палочкой.
Тетрафенилфосфоний реагирует, кроме кадмия, и с ионами Zn2+ и МпО4-.
Тиомочевина в нейтральной или слабокислой среде в присутствии пикрата или соли Рейнеке дает кристаллические осадки и с Ag+, Bi3+, Cu2+, Hg2+ и Pb2+.
45
Уротропини роданид, кроме кадмия, реагирует с Со24, Си24, Fe34, Hg2+ и Zn24. В отсутствие роданида утротропин образует характерные кристаллы с Bi3Jr, Hg24, Sb34 и Sn24. При замене роданида на иодид кадмий можно открывать в присутствии Bi3+, Си24, Hg24, Pb2+, Sb34 и Sn24.
Фосфат кадмия под ультрафиолетовым микроскопом дает красную окраску. Большие количества цинка частично удаляют едким натром в виде цинката, а осадок, содержащий кадмий, растворяют в НС1, нейтрализуют и добавляют фосфат.
Хинолин в присутствии роданида осаждает, кроме кадмия, ионы Bi34, Со2+, Си24, Fe3+, Ti44', Zn24 и Zr44. При замене роданида иодидом чувствительность открытия кадмия повышается.
Щавелевая кислота реагирует со многими ионами (Приложение 10), особенно мешают Bi34, Мп24, Sn24, Pb24, Zn2+, щелочно- и редкоземельные элементы.
Из табл. 9 видно, что чувствительность микрокристаллоскопи-ческих реакций невелика, а низкий открываемый минимум связан лишь с очень малым объемом раствора, нужным для выполнения анализа. Наиболее чувствительны (открываемый минимум <( 1 мкг Ccl/.ил) реакции с нитропруссидом и сульфидом, а также с фосфатом (открываемый минимум 10 мкг СЛ/мл) в УФ и микрокристалл оскопическая реакдия с гидразином в присутствии роданида. Хлориды рубидия и цезия и тиомочевина позволяют открывать 10—20 мкг СА/мл, а бруцин и пиридин — 50—60 мкг СА/мл.
Таким образом, обнаружение кадмия в растворах, содержащих медь и цинк, возможно при использовании бруцина, роданоцин-ката и, при небольшом избытке меди, 2-нафтиламина с роданидом. В присутствии цинка можно открывать кадмий при помощи хлоридов рубидия или цезия, тиомочевины с пикратом или солью Рейнеке и, в особенности — под ультрафиолетовым микроскопом по реакциям с нитропруссидом или сульфидом.
Люминесцентные реакции
Диметилглиоксим дает с ионами Cd24 осадок, обладающий сильной желтой флуоресценцией; открываемый минимум — 0,1 мкг Cd (С — 3 мкг/мл). Открытию кадмия мешают многие катионы [579; 470, стр. 161].
Морин в спиртовом растворе при pH 7 в присутствии кадмия дает желто-зеленую флуоресценцию; открываемый минимум — 0,1 мкг Cd. Реакция не избирательна [579, 681].
4-0 ксибензтиазол в аммиачном растворе образует с кадмием соединение, дающее синюю флуоресценцию. После отделения методом хроматографии на бумаге, пропитанной этим реагентом, в УФ открываемый минимум 0,1 мкг [592, стр.. 196].
8-0 ксихинолин при pH 6—9 дает с ионами Cd24 осадок с желтой флуоресценцией; разбавление — 1 : 500000, открываемый минимум — 0,1 мкг Cd (С — 3 мкг/мл). Подобные осадки вы-46
деляются и из растворов Al, Be, Са, Mg и Zr [242, стр. 169; 393, стр. 236]. В отсутствие цинка эту реакцию можно выполнять на фильтровальной бумаге [393, стр. 356]. При охлаждении до 80° К (—193° С) выход флуоресценции с максимумом излучения около 500 нм достигает значения 0,21 (у комплексов Al, In и Zn — 0,30) [678].
8-М еркаптохинолин тоже образует с кадмием ярко флуоресцирующее соединение [29].
Пиридин с иодидом калия в нейтральной среде выделяет осадок состава [Gd(G5H5N)2]J^, обладающий голубой флуоресценцией, разбавление — 5-107, открываемый минимум 0,0002 мкг Cd (С = 0,02 мкг/мл), аналогичный осадок с ионами РЬ2+ дает желто-коричневую флуоресценцию [45, стр. 271; 393, стр. 236]. По другим данным, открываемый минимум для Cd — 16 мкг GA/мл [539; 470, стр. 161].
Хинин реагирует с комплексным иодидом кадмия. Для приготовления реагента насыщенный раствор хлоргидрата хинина смешивают с равным объемом 20%-ного раствора KJ и отфильтровывают. В присутствии кадмия выпадает белый осадок, растворимый в кислотах, щелочах и большом количестве воды; такую же реакцию дают ионы Ag, Bi, Си, Fe3+, РЬ и Sb. При капельном выполнении в чистом нейтральном растворе открываемый минимум — 0,02 мкг Cd (С — около 0,5 мкг/мл) [178].
2-Т иогидантоин предложен для капельного открытия кадмия. На пропитанную реагентом бумагу наносят каплю исследуемого раствора; флуоресценция полученного пятна позволяет открывать 0,002 мкг Cd2+ (0,02 мкг/мл) [45, стр. 272] (по другим данным — 0,7 мкг GA/мл [423; 470, стр. 161]). Открытию кадмия мешают ионы Ag+, Au3+, Cu2+, Hg2+, Pd2+ и Pt4+ [393, стр. 236].
Отмечена флуоресценция сульфида кадмия, осажденного в микропробирке в присутствии цианида; открываемый минимум 0,02 мкг GA/мл, как при реакции с пиридином [393, стр. 236]. Предложено определение CdS в присутствии меди и по вызываемому им тушению свечения флуоресцеина на фильтровальной бумаге; при соотношении Си : Cd = 100 : 1 чувствительность обнаружения последнего соответствует pZ) около 5,2 (С = 6 мкг/мл) [392, стр. 195]. На бумажных хроматограммах кадмий можно открывать по люминесценции в присутствии других катионов смесью 8-оксихи-нолина, кверцетина и салицилаламина [106] или 8-оксихинолина с койевой кислотой (открываемый минимум — 0,05 мкг) [45, стр. 148]; используют также хроматографирование на бумаге, пропитанной одним 8-оксихинолином [149]. Из других люминесцентных реакций описано открытие от 0,01 мкг Cd в кристаллофосфорах на основе ThO2 при их облучении конденсированной искрой между вольфрамовыми электродами [45, стр. 138]. Вольфрамат кадмия дает ярко-желтую, а его нитрат фиолетово-синюю флуоресценцию [539].
47
Прочие реакции
Оксалат натрия при нагревании восстанавливает соли кадмия с образованием металлического зеркала, обычно окаймленного бурым кольцом (отличие от зеркала As, Hg и Zn); при сплавлении с серой оно переходит в желтый CdS.
Каплю анализируемого раствора выпаривают досуха на предметном стекле, остаток смешивают с несколькими кристаллами Na2C2O4, переносят смесь в маленькую трубку из тугоплавкого стекла и нагревают до красного каления. Открываемый минимум — 3 мкг Cd (С = 100 мкг!мл) [350, стр. 37].
Этот процесс:
СсЮ -р Na2C2O4 — Cd -1 - Na2CO3 + СО2, Cd + S^CdS,
протекающий при температуре выше 400° С, использован для открытия кадмия в минералах сухим путем. As, Hg и Sb тоже возгоняются, но влияние их устраняют в ходе анализа [287, стр. 60]; открываемый минимум в этом случае 5 мкг Cd.
1—10 мг растертого минерала обжигают в фарфоровом тигле на пламени спиртовой горелки в течение 3—4 мин. Охлажденный остаток смешивают с двойным (по объему) количеством оксалата натрия, помещают в трубку (длина 60—80 .пл, внутренний диаметр 2—3 мм), постукивая ею так, чтобы все вещество собралось в запаянном конце. Трубку нагревают на пламени спиртовой горелкп до красного каления несколько выше того места, где находится смесь, и затем медленно передвигают пламя так, чтобы оно постепенно охватило часть трубки со смесью. Смесь должна разлагаться не сразу, в противном случае она сильно вспучивается и может передвинуться туда, где уже началось отложение кадмия. После полного разложения (прекращение вспучивания) продолжают нагревание до красного каления еще 2—3 мин. и дают немного остыть. Если проба содержит кадмий, то выше места нагрева в трубке образуется черное или черно-бурое кольцо. После некоторого охлаждения в трубку вносят 1—2 кусочка серы (размер 1—2 мм3) и нагревают до образования ее паров. Место образования черного кольца продолжают нагревать для отгонки избытка серы к открытому концу трубки. В присутствии кадмия остается кольцо, имеющее в нагретом состоянии киноварнокрасный цвет, переходящий при охлаждении в капареечно-желтый.
Открываемый минимум в цинковой обманке — 0,05% Cd [287, стр. 60].
Сульфиды As и Hg летучи и удаляются при отгонке серы; при соотношении Cd : Hg = 1:10 ртуть должна быть предварительно отогнана (обычно это легко происходит при обжиге минерала). Сурьма также должна быть удалена прокаливанием, так как она частично возгоняется и ее черный сульфид маскирует окраску CdS. В смеси гринокита и антимонита открываемый минимум 0,5% Cd. Слабое белое кольцо ZnS обычно располагается ниже кольца CdS и бывает заметно при соотношении Cd : Zn ss 1 : 1000. В этом случае желтый CdS даже в горячем состоянии не изменяет окраски или становится лишь оранжево-желтым. Реакция может быть использована для ориентировочной оценки содержания 0,05—0,1%; 0,1—0,5%; 0,5—1,0%; 1—2% и )> 2% Cd путем сравнения со стандартными налетами Cd [287, стр. 62].
48
Из других реакций для открытия кадмия в минералах и рудах сухим путем, пригодных для выполнения в полевых условиях, применяют метод растирания порошков [72, 152].
Ферроцианид калия в присутствии тартрата окрашивает смесь аммиачных комплексов Cd2+ и Си2+ в малиновый цвет.
Для приготовления реагента смесь CuSO4, K*[Fe(CN)e] и KNaC*HeOe (1:10: 20) тщательно растирают в ступке или фарфоровой чашке и высушивают на теплом открытом месте. Маленький кусочек минерала растирают в фарфоровой кюветке с несколькими кристалликами KHSO* и увлажняют смесь дыханием. Прибавляют несколько крупинок реагента, повторяют растирание и обрабатывают каплей разбавленного NH4OH. Слабо нагревают. В присутствии Cd2+ красно-бурая окраска смеси переходит в сиреневую, усиливающуюся при слабом нагревании [152, стр. 89].
В качестве реагента можно использовать смесь CuSO4-5H2O, (NH4)2CO3 и Н2С2О4-2Н2О (1 : 10 : 10).
Пробу разлагают нагреванием с (NH4)2SO4, добавляют двойной избыток реагента, тщательно растирают, прибавляют столько же K4[Fe(CN)e], снова растирают и увлажняют дыханием или каплей воды. В присутствии Cd2+ смесь окрашивается в розовый цвет [72, стр. 132].
Открытие на хроматограммах. Предложены способы обнаружения кадмия после разделения смеси ионов органическими растворителями на фильтровальной бумаге в кольцевой бане [5Y5], круговой бумажной хроматографией [536], электрофорезом на бумаге, пропитанной фосфатом Sn4+ [700], методом тонкослойной хроматографии на силикагеле [606] и другие [185, 617, 689, 784].
Электрографическое открытие основано на избирательном анодном окислении кадмия с поверхности твердых образцов (например, шлифов) и его сорбции желатинизированной бумагой, пропитанной соответствующим электролитом. При последующей обработке характерным реагентом на бумаге образуется окрашенное пятно [274]. (Источником электроэнергии может служить батарейка от карманного фонаря.)
Электролит
50%-ный раствор
NH4C1
5%-ный раствор KCN, подкисленный H2SO4
0,1 7V раствор K2SO4 или KNO3
Реагент Мешающие элементы
Насыщенный спиртовый Со раствор дифенилкарб-азида H2S * Элементы группы
сероводорода
2-Нитрозо-1-нафтол Pb, Zn
Имеются указания об обнаружении микроколичеств Cd (и других элементов) по их угнетающему влиянию на жизнедеятельность культур некоторых организмов и тканей [419, 420].
Глава IV
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ КАДМИЯ
ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Гравиметрические методы
Весовыми формами для определения кадмия служат его неорганические соединения (окись, соли), внутрикомплексные соединения с органическими реагентами, тройные комплексы с неорганическими и органическими соединениями и выделенный электролитически металл. Распространенный ранее электрогравиметрический метод, позволяющий определять до 500 мг Cd, для получения точных результатов требует длительного электролиза. Обычно электролитом служит раствор цианида. Ускоренные варианты этого метода менее надежны. Для массовой работы наиболее пригодны методы, основанные на выделении соединений кадмия различными неорганическими и, особенно, органическими осадителями. Обычно их используют для определения м-101 — м-102 мг Cd, реже — п -10° мг Cd и менее.
Осаждение труднорастворимых соединений кадмия
Гравиметрические методы определения кадмия после его осаждения химическим путем сведены в табл. 10. Выбор того или иного метода зависит от сопутствующих элементов. Более подробно основные или рекомендованные за последнее время методы описываются ниже.
Сульфат кадмия — одна из его наиболее использующихся весовых форм. Предварительно кадмий отделяют от элементов, дающих остаток после выпаривания с серной кислотой и прокаливания. Недостаток метода — трудность полного удаления из остатка серной кислоты. Поэтому прокаленный CdSO4 растворяют в воде, раствор снова упаривают, остаток прокаливают, взвешивают. Эти операции повторяют до получения постоянного веса [82, стр. 272].
50
Если кадмий связан с какой-либо летучей кислотой, то раствор переносят во взвешенный платиновый тигель, обрабатывают H2SOi (1 : 1) и выпаривают, насколько возможно, на водяной бане. Нагревают сначала осторожно на воздушной бане до прекращения дымления, затем приблизительно при 500 °C. Охлаждают, растворяют остаток в небольшом количестве воды, осторожно выпаривают и снова нагревают. Взвешивают по охлаждении и повторяют эти операции до достижения постоянного веса.
Сульфат кадмия должен быть белого цвета и давать прозрачный водный раствор. Если в результате частичного разложения сульфата прокаленный остаток имеет желтый или коричневый цвет, то его смачивают разбавленной серной кислотой и повторяют прокаливание при 350—400 °C [459, стр. 249, 638].
Если кадмий был предварительно выделен в виде CdS, то осадок по возможности полно переносят в маленький стакан, а фильтр с оставшимися на нем частицами осадка сохраняют. Стакан накрывают часовым стеклом, прибавляют НС1 (1 : 3), нагревают до растворения осадка и прекращения выделения H2S; снимают часовое стекло и споласкивают, промывные воды присоединяют к раствору. Воронку с фильтром помещают над стаканом, растворяют остаток CdS горячей НС1 (1 : 3) и промывают фильтр горячей водой. Раствор в стакане выпаривают до малого объема, переносят во взвешенный платиновый тигель и поступают далее так, как описано выше, начиная с обработки H2SOi (1 : 1) [619, стр. 215; 82, стр. 273].
При анализе минерального сырья после разложения пробы объем раствора не должен превышать 100 at л (включая 2—3 мл H2SO<). Сульфиды кадмия и меди осаждают H2S. Осадок отфильтровывают, смывают в стакан при помощи H2SO< (1 : 5) и нагревают до прекращения запаха H2S. Осадок CuS отфильтровывают и промывают водой, подкисленной H2SO<, а фильтрат разбавляют водой с таким расчетом, чтобы концентрация кислоты стала 5 : 100 и вновь осаждают кадмий H2S. Фильтр с осадком озоляют, растворяют остаток в разбавленной H2SOi и определяют кадмий, как указано выше [10, стр. 488].
Для осаждения CdS вместо сероводорода можно использовать тиосульфат натрия: соляно- или сернокислый раствор нейтрализуют избытком аммиака, подкисляют тонным избытком СН3СООН, нагревают до кипения и вносят в кипящий раствор порошкообразный Na2S2O3. После кипячения в течение 30 мин. кадмий и элементная сера переходят в осадок [565, стр. 269].
Из органических реагентов для осаждения CdS предложены тиоацетамид и тиокапролактам. При pH 1 — 2 тиоацетамид гидролизуется с образованием H2S, который осаждает CdS; при pH 8 сначала образуются комплексы типа [CdTA]2+, [Cd(TA)2]2+ и
[Cd(TA)4]2+, разлагающиеся при 90° С с выделением CdS. Кадмий осаждают в аммиачном растворе при кипячении; для маскирования железа вводят тартрат [475]. Тиокапролактам осаждает CdS в результате гидролиза при нагревании в щелочной среде (см. стр. 58 [731]).
С бруцином C23H2eO4N2-4H2O при добавлении бромида калия можно определять кадмий в присутствии Al, Си, Cr, Fe, Mg, Мп, РЬ, Zn и некоторых других элементов. Метод использован для определения кадмия (от 0,2 мг) в магниевых сплавах.
0,3 — 0,7 г сплава растворяют в 10—15 мл H2SO4 (1 : 5), раствор упаривают до 5—8 млн фильтруют, если выделяется осадок SiO2. В фильтрате нейтрализуют большую часть кислоты раствором NaOH, прибавляют 5 мл 1%-ного
51
Гравиметрические методы определения кадмия
Осадитель Среда Условия осаждения Продолжительность отстаивания осадка Промывная жидкость, количество промываний
п-Анизидин Нейтральная Кипячение 30 мин. Этанол
Антранилат натрия pH 5,2 То же После охлаждения — 1 час Разбавленный водой раствор осадителя
Бруцин — бромид H2SO4 (1 •• 5) Без нагревания 1 час Разбавленный водой раствор осадителя, 5—6 раз, затем этанол + эфир (1:7) — 8—10 раз
Висмутол II Слабокислая Нагревание 2—3 мин.» по охлаждении — 1 час Холодная вода
Гидразид изоникоти- Нейтраль- Вез нагре- Несколько Разбавленный водой раствор
новой кислоты + бензоат ная вания минут осадителя, затем —• то же с добавлением этанола — 4 раза и эфир — 5 раз
Гидразин + иодид То же Нагревание До охлаждения Разбавленный раствор осадителя, затем этанол
Диантипирилметан -|-+бромид H2SO4 (1 : 40) Кипячение 1,5—3 часа Разбавленный водой раствор осадителя/ затем то же в 0,1 N НС1 — 4—5 раз
Диантипирил-о-окси-фенилметан H2SO4 Без нагревания 2 часа Разбавленный водой раствор осадителя — 3—4 раза, затем этанол + эфир (1:7)
Диацетил-бис-(5-бромсалицил гидразон) Нейтральная Кипячение Фильтрование без отстаива-. НИЯ Холодная вода
Д иацетил-бис-(са л и-цилгидразон) То же То же То же » »
2,2'-Дипиридил + + роданид pH 3-5 Нагревание До охлаждения Разбавленный водой раствор осадителя, затем абсолютный этанол + эфир (1:4) — 4—5 раз и абсолютный эфир — 3 раза
Дифенилдитиофос-форная кислота 1 N H2SO4 Без нагревания Несколько минут Насыщенный раствор комплекса кадмия
Диэтилдитиофосфат никеля H2SO4 (1 =5) То же 1 час То же
Купраен + иодид Нейтральная Кипячение До охлаждения Разбавленный водой раствор осадителя, затем этанол — 4—6 раз и эфир — 3—4 раза
Карбонат калия То же То же Длительное на водяной бане Горячая вода
п-(Меркаптоацетами-до) фенетол pH 5,5—6,5 Нагревание 30 мин. Горячий этанол — 4 раза
Меркаптобензоттазол (каптакс) nh4oh Без нагревания До образования кристаллического осадка Вода с добавлением NH4OH
Молибдат аммония СНзСООН Кипячение 2 часа Теплая вода
Р-Нафтохинолин + + иодид 2 N H2SO4 + + so2 Охлаждение водой со льдом 20—30 мин. Разбавленный водой раствор осадителя — 2—3 раза
52
Таблица 10
Условия высушивания осадка Весовая форма Молекулярный вес Фактор пересчета Литература
температура, °C продолжительность
105-110 20 мин. Cd (NH2CfH4OCH3)2SO4J 454,60 0,2473 [31]
105-110 30 мин. Cd (CeH4NH2COO)2 384,66 0,2922 [565, стр. 288]
130—150 30 мин. H2[tCdBn]. • [G21HEoO2N2(CH3O)2}2 1223,1 0,0919 [190; 565 619] стр. 332;
110-120 Cd (CeHyNaSaJs 562,91 0,1997 [645]
Вакуум * [Cd (C6H.ON:,)] (CeH3COO)2 491,79 0,2286 [582]
110 1 час [Cd (N2H4)2J J2 430,34 0,2612 [565, стр. 333; 619]
110-120 H2 [CdBrJ (С»НаО2Х4)а 1210,95 0,0928 [116; 325]
110-120 105 До постоянного веса 2 часа Нг [CdBrJ (СгвНгвОjN4)2 CdC18H20O4N6Br2 1395,36 656,62 0,0806 0,1712 [211] [724]
105 2 часа CdC18H22O4Ne 498,82 0,2254 [724]
Вакуум * 5—10 мин. [Cd (C,„H«N2)] (SCN), 384,74 0,2921 [688]
80—90 До постоянного веса То же Cd [(C6H5O)2PSS]2 Cd [(C2H3O)2PSS]2 675,02 482,85 0,1665 0,2254 [58] [55; 325]
Вакуум » 5—10 мин. [CdJ4] [Cu (NH2CH2CH2NH2)2] 803,86 0,1395 [189; 565 619] стр. 299,
900-1000 110 До постоянного веса 60 мин. CdO 128,40 0,8754 0,2111 [565, стр. [512] 307]
110-120 30—60 мин. Cd (C,H4NS2)2 444,89 0,2527 [565, стр. 292]
120 70-80 До постоянного веса То же CdMoO. H2[CdJ4] (C13h,N)2 273,36 980,53 0,4127 0,1147 [565, стр. [Ю] 309; 619]
53
Осадитель Среда Условия осаждения Продолжительность отстаивания осадка Промывная жидкость, количество промываний
Оксалат аммония Нейтральная 50% этанола 15 мин. 50%-ный этанол — 6—8 раз, затем 95%-ный этанол, потом — эфир
2-Оксинафтилаль-2-аминопиридин pH 3,8—4 То же 35 мин. Холодная вода — 2 раза, 25%-ный этанол, затем эфир
2-(о-Оксифенил)бенз-оксазол pH 9 60° С. затем pH Ц При 60° С 15 мин. и до охлаждения 50%-ный этанол + NH.OH
8-Окс'ихинолин 0,5% сщсоон 60° с До коагуляции Теплая вода, затем холодная вода
Пиридин + роданид Нейтраль- Кипяче- До охлажде- Разбавленный раствор оса-
(1 .МЛ) ная ние ния дителя 4—5 раз; абсолютный этанол -4- пиридин — 1—2 раза и эфир Д- пиридин — 5—6 раз
Пиридин (10—15лы) + + роданид То же То же То же То же
Пиридин + хлорид » » Нагревание 24 часа Этанол + эфир (3 : 1), затем их смесь (1:1) и один эфир
Ртуть (II) хлорид-р + иодид nh4oh Без нагревания 10 мин. Этанол — 4—5 раз, эфир — 2—3 раза
Салицилгидроксамо-вая кислота Слабокислая Кипячение, затем nh4oh Водяная баня и до охлаждения Холодная вода
Серная кислота НгйО4 (1 : 1) Выпаривание на водяной бане, затем на воздушной до прекращения дымления
Тиокапролактам Нейтральная Кипячение, затем NaOH 1Q мин. на водяной бане (100° С) Вода, затем этанол
Тиомочевина И- соль 1 N Без нагре- 30—60 мин. Охлажденный льдом 1%-ный
Рейнеке кислота вания в воде со льдом раствор тиомочевины, затем абсолютный этанол — 3—4 раза
Тиосульфат натрия СНзСООН Кипячение 30 мин. при кипячении Образовавшийся осадок CdS растворяют в НС1 (1:3) и выпаривают с H,SO. (1 : 1)
Трифенилметиларсо-НИЙИОдИд Нейтральная То же 1 час остывания и еще 30 мин. 0,5%-ный раствор KJ
1,10-Фенантролин -г + роданид л1-Фенилен-ди-(1-тет- разолин-5-тион) pH 3-6 pH 5,5-7,0 » » 15 мин. при нагревании и еще 5—10 мин. 5 мин. на водяной бане Разбавленный раствор осадителя 5—6 раз, затем этанол 4- эфир (1 : 3) — 3—4 раза и абсолютный эфир — 3 раза Вода (100 мл), затем ацетон (25 мл)
Ферроцианид калия NH.OH 80° С 4 часа при 80° С 2,5%-ный раствор NH.OH
Фосфат аммония Нейтральная То же Без нагревания То же До следующего дня То же 1%-ный раствор осадителя, затем 60%-ный этанол То же
Хинальдиновая кислота » » Нагревание, затем NH.OH До охлаждения Холодная вода
* Высушивание в вакуум-эксикаторе при комнатной температуре.
** Эмпирический поправочный коэффициент.
54
Таблица 10 (окончание}
Условия высушивания осадка Весовая форма Молекулярный вес Фактор пересчета Литература
температура, °C продолжительность
Вакуум * 2—3 мин. сас2о4-зн2о 254,48 0,4418 [565, стр. 310]
104 20—25 мин. CdCl2 (CWH12ON2)2 679,88 0,1653 [92]
130-140 2 часа Cd (CnHaOaNJs 500,82 0,2109 [619, 771]
130 Cd (C,H«ON)« 400,71 0,2805 [565, стр. 283; 619]
Вакуум • 10 мин. [Cd (C5HsN)2] (SCN)z 386,77 0,2906 [565, стр. 294 ; 740]
Вакуум * 20 мин. [Cd(C5H5N),] (SCN)2 544,97 0,2062 [565, стр. 296 ; 740]
110-120 . 3—5 час. [CdC5H3N] Cla 262,41 0,4284 [565, стр. 312; 619]
Вакуум * 5—10 мин. [Cd (NH,)J [HgJ3]2 1343,27 0,08375 [565, стр. 299]
110 CdC7H5O3N 263,48 0,4266 [501]
500 До посто- Cd.~O4 208,46 0,5392 [82; 459; 565, стр. 234;
янного 619]
веса
110-И 20 То же CdS 144,46 См.’* [731]
110-120 [Cd (SCN2H4)2] • 901,42 0,1247 [565, стр. 297, 619]
• [Cr (SCN)4(NH3)2]2
500 » » CdS04 208,46 0,5392 [565, стр. 269]
105-110 [CdJ4] [A.sCH3 (CBH5)3]2 1262,29 0,0890 [552]
Вакуум * 10 мин. [Cd (СтгНвКг)^] (SCN)2 590,83 0,1909 [687]
110-150 4Ь мин. Cd (C,H4N8S2) 388,70 0,2892 [605]
100-110 [Cd (NH,),] [Fe (CN),] 360,44 0,3118 [565, стр. 313; 619J
1С0-103 До посто- CdNH4PO4-H2O 243,43 0,4617 [565, стр. 281; 619]
янного
веса
800-900 Cd2P2O, 398,74 0,5638 [565, стр. 281; 619]
125 То же Cd (C,H„NCOO)2 456,73 0,2461 [565, стр. 286; 619}
55
раствора бруцина в H2SO« (1 : 5) (1 мл достаточно для выделения 0,5 мг Cd), вводят 10 мл 10%-ного раствора КВг и перемешивают стеклянной палочкой до выделения белого осадка. Спустя час его отфильтровывают через тигель с пористым дном и промывают 5—6 раз по 3—5 мл промывной жидкости (40 мл 1%-ного сернокислого раствора бруцина, 30 мл 10%-ного раствора КВг и 80 мл воды), затем 8—10 раз смесью 1 ч. этанола с 7 ч. диэтилового эфира (по 3 мл). Одновременно споласкивают стенки тигля, чтобы удалить с них остатки H2SO<. Тигель высушивают] 30 мин. в сушильном шкафу при 130—150° С и после охлаждения взвешивают на микровесах [190; 298; 565, стр. 332].
Диантипирилметан C23H24O2N4 в кислой среде образует с галогенидными анионными комплексами кадмия осадок, труднорастворимый в воде [2]; Gu, Zn, Al, Со, Ni, Мп и Сг подобной реакции не дают, поэтому ее используют в анализе сплавов на медной основе. •
0,1—0,2 г сплава растворяют в HNO3, добавляют 3—4 мл H2SO< и для удаления мешающей определению HNO3 упаривают до дымления. По охлаждении прибавляют 70—80л1л воды, вносят 0,5 г диантипирилметаиа, нагревают почти до кипения и быстро прибавляют 15—20 мл предварительно нагретого 10%-ного раствора КВг. В зависимости от количества кадмия сразу или через несколько минут начинает выделяться осадок в виде блестящих шелковистых игл. Для полноты его выделения раствор оставляют стоять 1,5— 2 часа и холодным фильтруют через пористый тигель № 3 или № 4. В присутствии небольших количеств Fe(III) выделение осадка замедляется, поэтому раствор выдерживают 2—3 часа. Осадок отфильтровывают, промывают 0,5 N H2SO<, содержащей 1,5% КВг и 0,5% диантипирилметаиа, затем 4—5 раз (по 3—4 мл) 0,1 N НС1, которая содержит 0,5% КВг и 0,2% диантипи-рилметана, и высушивают при 110—120 °C.
Цвет полученного осадка должен быть белым или слегка желтоватым. Относительная ошибка определения ± 3% [116, 325].
Дифенилдитиофосфорная кислота
(C6H5O)2PSSH осаждает кадмий из слабокислых, нейтральных и слабощелочных растворов в виде белого кристаллического осадка, растворимого в концентрированных кислотах, аммиаке и щелочах; растворимость в воде при 18—20° С составляет 2,2-10~4 молъ!л.
Допустимо изменение в широких пределах концентрации кислоты и присутствие значительных количеств Al, Be, Ga, Со, Cr, Ga, Mg, Мп, Ni, Sr, Zn и небольших количеств железа. Мешают ионы тяжелых металлов, осаждающихся реагентом в кислой среде [54, 58].
К 20—100 мл сернокислого ~17V раствора, содержащего 20—100 мг Cd, а также Zn и другие металлы, прибавляют порциями при тщательном перемешивании 2—3-кратный избыток 0,05 М фильтрованного раствора реагента. Осадку дают отстояться несколько минут, отфильтровывают через взвешенный стеклянный фильтр № 3, промывают несколько раз небольшими порциями насыщенного водного раствора дифенилдитиофосфата кадмия, высушивают до постоянного веса при 80—90° С и взвешивают.
Диэтилдитиофосфат никеля [(C2H5O)2PSS]2Ni осаждает кадмий из кислой среды. Al, Ge, Zn, Zr и некоторые другие металлы определению не мешают. Реакция применена для определения до 0,05% Cd в магниевых сплавах [55; 325, стр. 20].
56
0,2—0,5 г сплава растворяют при нагревании в 15 мл H2SO< (1 : 5) и добавлении нескольких капель HNO3 (1 : 1) (если сплав содержит большие количества Zr, добавляют 2—3 мл HNO3). По охлаждении приливают 15 мл 0,05 7V раствора реагента, тщательно перемешивают и через час фильтруют под вакуумом через взвешенный стеклянный пористый фильтр № 3. Осадок промывают 5—6 раз небольшими порциями насыщенного водного раствора диэтилдитиофосфата кадмия и высушивают до постоянного веса.
|3-Н афтохинолин G13HeN реагирует с ионами [GdJJ2~ в 2 N H2SO4, поэтому возможно осаждение их даже в присутствии больших количеств Zn; мешают определению As, Bi, Gu, Pb, Sb, Sn и высокое содержание Fe (малые количества его можно замаскировать винной кислотой). Этот реагент применялся в анализе минерального сырья.
В зависимости от характера пробу разлагают той или иной смесью кислот, выпаривают с H2SO4 и отфильтровывают нерастворимый остаток вместе с PbSOi. Из фильтрата выделяют медь с помощью Na2S2O3, окисляют его избыток (так же как и Fe2+) добавлением 5—10 капель Н2О2 и кипятят раствор в течение 15—20 мин. При высоком содержании железо отделяют 2-кратным осаждением избытком NH4OH, при незначительном — вводят 5—10 мл 10% -ного раствора тартрата щелочного металла или винной кислоты. Нейтрализуют по лакмусовой бумаге и на каждые 100 мл жидкости прибавляют точно 12 мл H2SO< (1 : 1). Раствор охлаждают водой со льдом и вводят 5—6 капель насыщенного раствора H2SOg, 10 мл 10%-ного раствора KJ и 5 мл 2,5%_ ного свежеприготовленного раствора Р-нафтохинолина в 0,5 7V H2SO4. Жидкость перемешивают и оставляют на холоду в течение 20—30 мин. (длительное стоячие может привести к неправильным результатам вследствие осмо-лечия осадка). Осадок отфильтровывают через взвешенный стеклянный тигель с пористым дном и промывают 2—3 раза смесью 90 мл воды, 5 мл Р-нафтохиполина, 2—3 капель раствора H2SO3 хи 5 мл 10%-ного раствора KJ, Промывную жидкость отсасывают, тигель с осадком высушивают при 70— 80° С (при более высокой температуре осадок осмоляется), охлаждают в эксикаторе и взвешивают.
При малом содержании кадмий определяют колориметрическим методом 10, стр. 486].
2-( о-О к с и ф е н и л)б ен з окса зол G13H9O2N осаждает кадмий в щелочном растворе, содержащем тартрат. Go, Си и Ni осаждаются в ацетатной среде при pH — 4 и могут быть удалены таким путем. Допустимо присутствие Ag, Al, Bi, As, Gr, Fe, Hg, Mg, Mn, Pb, Zn, щелочных и щелочноземельных металлов.
К анализируемому раствору, содержащему 1—80 мг Cd, не более 100 мг Си и 20 мг Со и Ni, прибавляют 3 г тартрата аммония и нагревают до 60° С. Если присутствует Са, то выпадает кристаллический осадок его тартрата, который отфильтровывают и вводят в фильтрат еще 1 г тартрата аммония. К раствору прибавляют 3 М СН3СООН до pH ~3,5, вновь нагревают до 60 °C и приливают небольшой избыток свежеприготовленного спиртового раствора бензоксазола, нужный для полного осаждения Со, Си и Ni. С помощью 3 М CH3COONa доводят pH до 4,0, нагревают жидкость в течение 15 мин. при 60° С, охлаждают до комнатной температуры, фильтруют через стеклянный тигель средней пористости и промывают его несколько раз водой, которую прибавляют к фильтрату.
К раствору, содержащему кадмий и тартратные комплексы остальных металлов, прибавляют 1 М NaOH до pH около 9, нагревают до 60° С и при-
1 Воду насыщают газообразным SO...
57
ливают 5—10%-ный избыток 1 %-ного спиртового раствора бензоксазола. Доводят 1 М раствором NaOH до pH 11, выдерживают жидкость 15 мин. при 60 °C и охлаждают до комнатной температуры. Осадок отфильтровывают через взвешенный стеклянный тигель средней пористости, промывают несколько раз 50%-ным раствором этанола, к которому добавлен аммиак, высушивают 2 часа при 130—140 °C и взвешивают.
Избыток Со и Ni предварительно осаждают диметилглиоксимом и 1-нитрозо-2-нафтолом. При определении 5—50 мг Cd погрешность составляет +0,2 мг, в присутствии малых количеств Со и Ni — +0,4 мг Cd [771; 619, стр. 217].
8-Оксихинолин C9H7ON в нейтральной или слабокислой среде реагирует со многими ионами, поэтому кадмий должен быть предварительно отделен от них (кроме ионов щелочных и щелочноземельных металлов).
К слабокислому анализируемому раствору добавляют Na2CO3 до помутнения, которое устраняют несколькими каплями СН3СООН. Раствор нагревают до 60° С и осаждают кадмий небольшим избытком 3%-ного свежеприготовленного спиртового раствора 8-оксихинолипа. После непродолжительного нагревания осадок отфильтровывают через стеклянный фильтр-тигель, промывают сначала теплой, а затем холодной водой и высушивают при 130° С до постоянного веса.
Описан микровариант метода, позволяющий выделять при pH 6—7 от 1 до 3 мг Cd. Возможно также его осаждение из тартратного раствора, 100 мл которого содержат 10—12 мл 2 N раствора NaOH [565, стр. 283].
Тиокапролактам C6HUNS.
К 100 мл нагретого до кипения раствора, содержащего 20—300 мг Cd, при энергичном перемешивании приливают 15—20 мл 1%-ного раствора реагента в 50%-ном этаноле, затем по каплям — 20 мл 0,5 N NaOH и на 10 мин. оставляют на кипящей водяной бане. Осадок CdS отфильтровывают через стеклянный фильтр, промывают водой, затем этанолом и высушивают при 110-120 °C [731].
В другом способе в присутствии роданида осаждают тройной комплекс (CeHuSNH)2[Cd(SCN)4] (разлагается при нагревании выше 80° С). Этим путем можно определять 10—200 мг Cd [488].
Тиомочевина (NH2)2CS и соль Рейнеке NH4[Cr(NH3)2(SCN)4]-2 Н2О осаждают из 1 N минеральной кислоты кадмий; цинк остается в растворе.
К анализируемому раствору, содержащему 10—50 мг Cd, прибавляют такое количество фильтрованного 5%-ного раствора тиомочевины, чтобы ее концентрация в конечном объеме стала ~1%-пой. Приливают в небольшом избытке раствор соли Рейнеке в 1%-ном растворе тиомочевины. Сосуд с раствором помещают на 30—60 мин. в ледяную воду, временами перемешивают его содержимое, после чего осадок отфильтровывают через стеклянный тигель с пористым дном. Сосуд и осадок промывают охлажденным в ледяной воде 1%-ным раствором тиомочевины, затем вытесняют ее из смачивающей осадок промывной жидкости пропусканием (3—4 раза) небольших количеств охлажденного в ледяной воде абсолютного этанола. Высушивание тигля с осадком производят при 110—120° С [565, стр. 297].
Ag, Bi, Си, Hg и РЬ мешают [179, стр. 401; 386, стр. 242].
58
Разработаны гравиметрические (а также фотометрические и кондуктометрические) методы определения кадмия в виде аналогичных четверных соединений, в которых катионный комплекс кадмия с дипиридилом, пиперазином, фенантролином или этилендиамином ассоциирован с анионом [Cr(SCN)4(C6H5NH2)2]. В условиях определения кадмия осаждаются также Ag+, Au+, Cu+, Cu2+, Hg+, Ni2+, Tl+ и Zn2+. Наибольшая точность достигается при выделении из нейтральной или слабощелочной среды осадка [Cd(C2H8N2)2]-[Cr(SCN)4(CeH5NH2)2]2. Ионы Al3+, La3+, Th4+, Zr1+, щелочных и щелочноземельных металлов определению не мешают [572].
1, 10-Ф енантролин C12H8N2-H2O и роданид осаждают кадмий из слабокислых растворов; аналогичные осадки в присутствии роданида дают антипирин, пиперазин, пирамидон и пиридин. Определению кадмия мешают Со, Си, Ми, Ni, Zn и некоторые другие элементы; Fe можно замаскировать комплексоном III [687].
При замене роданида иодидом осадок [Cd(C12H8N2)2]J2, образующийся при pH 4,5—5,0, не адсорбирует цинк [695].
Предложен метод определения кадмия в присутствии Си и Zn, основанный на различной устойчивости их пирофосфатов.
К раствору сульфатов прибавляют по каплям 5%-ный раствор Na4P2O7 до растворения образующегося вначале осадка и нагревают почти до кипения. Через 15 мин. выделившийся белый осадок соли кадмия отфильтровывают, промывают и растворяют в разбавленной НС1. Из раствора осаждают CdNH4PO4 и прокаливают его до Cd2P2O7 [734].
Описано также выделение кадмия из растворов при pH 4—7 действием 2-х инолилфосфорной кислоты. Осадок CdC9HeO3NP-H2O при высушивании до 220—225° С теряет воду, а при 800—900° С переходит в пирофосфат [748].
Предложено определение кадмия 1,2-ди-(2-пиридил)этиленом в присутствии 100-кратных количеств цинка при pH 1,3—2,7 в виде труднорастворимого осадка [G12H10N2H2] [CdJ4] [487] и осаждение 10—50 мг Cd при pH 4,0—7,3 хиноксалин-2-карбоновой кислотой или ее производными [551] и 0,5—50 мг Cd при pH 5,5— 7,0 — ?и-фениленди-(1-тетразолин-5-тион)ом [605].
Электрогравиметрия
Кадмий может быть выделен электролитическим путем на ртутных, танталовых и платиновых катодах с различными электролитическими покрытиями и взвешен в виде металла. В качестве электролитов описаны молочная, муравьиная, серная, соляная, уксусная, фосфорная и щавелевая кислоты, аммиачные, щелочно-тартратные и цианидные растворы [619, стр. 197]; сульфат аммония и этилендиамин, комплексон III в аммиачной среде; сульфосалициловая кислота в слабо сернокислой среде и ацетилацетон в
59
присутствии диоксана [580]. Детальное описание предложенных методов с указанием оригинальной литературы дано в [565, стр. 235-247].
Точное электролитическое определение кадмия лучше всего производить с неподвижными электродами в слабощелочном растворе, содержащем количество KCN, необходимое только для переведения кадмия в растворимый комплекс. Менее точное, но более быстрое определение может быть проведено при большей плотности тока с вращающимся катодом в сильно разбавленном сернокислом растворе или с вращающимся анодом — в серно-, муравьино- или уксуснокислом растворах. Осаждению кадмия из сернокислого раствора препятствуют Ag, As, Au, Bi, Cu, Hg, Mo, Pb, Sb, Sn, Те, платиновые и некоторые другие металлы; даже в цианиднощелочном растворе мешают Hg, Zn и Ag. Поэтому кадмий должен быть предварительно отделен практически от всех катионов [619, стр. 197; 82, стр. 273]. Некоторые варианты определения кадмия с наиболее часто употребляемыми платиновыми и некоторыми другими электродами приведены в табл. 11.
Точное определение кадмия в цианидно-щелочной среде. Готовят сернокислый раствор, свободный от всех мешающих элементов, выпаривают его до дымления, охлаждают, разбавляют водой и в случае необходимости фильтруют. Прибавляют каплю фенолфталеина, затем КОН или NaOH до неисчезающей розовой окраски и по каплям при непрерывном перемешивании 10%-ный раствор KCN до растворения выделившейся при нейтрализации Cd(OH)2. Избытка KCN следует избегать. Раствор разбавляют водой до 100— 150 мл, погружают в него взвешенный сетчатый платиновый катод и платиновый анод и проводят электролиз холодного раствора при напряжении 4,8— 5,0 в и силе тока 0,5—0,7 а. Спустя 5—6 час. увеличивают силу тока до 1,0— 1,2 а и продолжают электролиз еще час. Споласкивают водой стенки сосуда (вследствие чего уровень электролита повышается) и ведут электролиз еще 15 мин.
Если вновь погрузившаяся в раствор поверхность электрода осталась чистой, то, не выключая тока, опускают сосуд с электролитом и сразу же хорошо промывают катод водой [635, стр. 289]. После этого его вынимают, промывают этанолом, затем диэтиловым эфиром, высушивают при 100° С, охлаждают и взвешивают (продолжительного нагревания отложившегося металлического кадмия следует избегать). Для проверки полноты выделения кадмия электролит насыщают сероводородом; желтое окрашивание жидкости показывает, что в растворе остались лишь его следы, образование желтого осадка свидетельствует о недостаточно полном выделении [82, стр. 273; 619, стр. 216].
Наибольшее количество кадмия, которое можно выделить этим способом, ~0,5 г. Процесс электролиза длителен, но сократить его продолжительность нельзя, так как при увеличении силы тока получается губчатый металл, который легко смывается с электрода [619, стр. 216].
Ускоренное определение в слабо сернокислой среде. К 150 мл нейтрального раствора сульфата кадмия прибавляют 3 мл 10%-ной (по объему) H2SO«, нагревают раствор до кипения и проводят электролиз с сетчатым платиновым катодом и вращающимся (около 600 об/мин.) платиновым анодом при напряжении 8—9 в и плотности тока 5 а/100 см2. Для выделения 0,5 г Cd обычно достаточно 15 мин. Определение заканчивают так же, как при работе в цианидно-щелочной среде. По точности результатов ускоренный метод значительно уступает описанному выше [619, стр. 216].
СМ
+ "в о
С/9
•ч
Я
Z
м
S и: СО о
а и** 1
о
.. —1
и + О ГЛ
мл Н • НС1 О СЛ м К "01 о СЛ Я О СО м Я м к л ч
«К ч-н О fe; ч ф и сб
и СО й
1 к зё 0,5 0,5 СМ
Л
а § « о О О о ю
1 1 о t 1 см 1
.£ о . СМ ’’"sj 1
5 Оа? ю со
О
я со СО 00
8 - о О o'
s’ я 1 (М CM
я я • . 1 *ч 1
5 s СМ о 1Л ю
° 1=С •ч о
Й 5 о •ч
а о о
о «. Я О й „ +я оо 'е‘* W + О и Я g * 2 £? * JS т СО + СРя °°-о о «я .о ^4 сл ^7 м «+ 3 г CH3COONa + 0,25 мл СН8СООН 0,5 г CH3COONa + 1 г K2SO4 1 — 1,5 г NaOH + 0,5—1 г KCN Нейтрализация разбавленным раствором NaOH до помутнения и еще 2— 3 мл + KCN до растворения осадка +
2 мл NaOH
СМ и
О О
см, 1 ю 00 i LQ ( О
о । о
см о о
00
см о
о о о ОО
j ЧТИ 1 1 о СМ
см О .—4 LQ о о*
о
о о
Горячий раствор
60
Определение с контролем катодного потенциала. Этот метод объединяет в себе отделение и определение кадмия в присутствии Bi, Си, Sn, РЬ и Zn.
Готовят раствор кадмия, содержащий указанные металлы, в HNOa (1:2) или в НС1 с минимальным добавлением HNO3. Вводят 1 г мочевины для восстановления окислов азота и на каждые 100 мг присутствующей в растворе меди — 50 мл 1 М тартрата натрия, 2 г хлорида гидразина и 1 г янтарной кислоты. Разбавляют раствор до 200 мл и прибавлением NaOH устанавливают pH 5,8—6,0 (контроль на потенциометре со стеклянным электродом). Электролиз с платиновыми электродами проводят при температуре не выше 25° С, потенциал устанавливают по отношению к нас. к. э. При — 0,30 в выделяется Си, при — 0,40 в — Bi, при — 0,60 в — РЬ. Для выделения каждого металла требуется около 45 мин., после чего в каждом случае сила тока снижается до некоторого постоянного минимального значения [635, стр. 326].
После полного выделения свинца использовавшийся катод заменяют свежим (взвешенным), прибавляют в раствор 25 мл концентрированного NlhOIl, 1 г хлорида гидразина и 20 мл 0,1%-ного раствора желатина. Электролиз продолжают при —1,00 в, к концу процесса выделения кадмия потенциал доводят до —1,15 в (до—1,20 в Zn не выделяется). По окончании электролиза (для выделения 500 мг Cd требуется — 45 мин.) катод промывают, высушивают и взвешивают [619, стр. 217; 635, стр. 311] .
Определение методом внутреннего электролиза. Этот способ в свое время был рекомендован в качестве контрольного [429, стр. 689]. Количественное выделение кадмия (15 мг) происходит при pH 4,6-5,6, поэтому используют буферный раствор, содержащий 1,65 мл 80%-ной СН3СООН и 5,9 г CH3COONa в 250 мл (pH 5,2). Выделенный из такого раствора кадмий при промывании водой может частично раствориться, поэтому используют воду, подкисленную СН3СООН и содержащую некоторое количество (NH4)2SO4. В такой промывной жидкости, нагретой до 70—80° С, соединенные зажимом электроды оставляют на 20—30 мин. Если в момент их погружения кадмий частично перешел в раствор, то за это время он снова выделится на катоде. Затем кадмий промывают в 95 %-ном этаноле (в разбавленном он частично растворяется). Определению кадмия методом внутреннего электролиза мешают Ag, As, Au, Bi, Co, Cu, Fe, Hg, Ni, Pb, Pt, Sb и Sn.
Если для промывания электродов используют дистиллированную воду, недостаточно полно освобожденную от ионов хлора, то осажденный на электроде кадмий может частично вновь перейти в раствор. Поэтому воду перед употреблением следует прокипятить или добавить в нее несколько капель 0,1 N раствора Na2S2O3 [234а]. При работе с чистыми солями для увеличения электропроводности в анализируемый раствор добавляют небольшое количество (NH4)2SO4 [233а]. При использовании метода внутреннего электролиза для анализа цинковых концентратов кадмий предварительно отделяют от As, Bi, Си, Fe, Pb и Sb [429, стр. 690]. Метод может быть применен при определении кадмия в Zn и его сплавах [435а] и в кадмиевых бронзах [146а].
62
В первом случае Cd вместе с Zn предварительно осаждают в виде сульфидов 2%-ным раствором Na2S, затем осадок растворяют в HNO3, прибавляют 3 мл H2SO4 и упаривают до густых белых паров. Сухой остаток растворяют в 100 мл воды, нейтрализуют аммиаком, добавляют 1,41 мл 99,8%-ный СН8 СООН и проводят электролиз [435а]. При анализе кадмиевых бронз электролиз проводят в 0,1 N СН3СООН, содержащей 5,92 г CH3COONa в течение 40—50 мин. при 85—90 °C [146а].
Внутренним электролизом можно определить Cd в присутствии Си и Zn.
К раствору, содержащему Cd, Си и Zn в виде сульфатов, добавляют 3— 4 г (NH4)2SO4, разбавляют водой до 70 мл, нейтрализуют аммиаком, прибавляют 2 капли H2SO4 (1 : 1) и разбавляют водой до 250 мл. Выделяют Си с Fe-анодом при 50—60° С. Затем в оставшийся электролит, содержащий Cd, Zn и небольшие количества Fe, прибавляют 15—20 капель H2SO4 (1 : 1), 0,5 г (NH4)2S2O8, нагревают раствор до кипения п осаждают Fe аммиаком. Фильтрат после отделения гидроокиси железа упаривают до 150 мл, подкисляют H2SO4 (1 : 1), прибавляют 1,(55 мл 85%-ной СН3СООН, 5,9 г CH3COONa, нагревают раствор до 75—80° С и выделяют Cd с Zn-анодом в течение 30— 40 мин. [234в].
Тщательно контролируя условия электролиза, можно отделить Cd от Ag, Bi и Си и затем количественно его определить.
К раствору, содержащему указанные элементы, добавляют 10-кратный избыток ЭДТА, цианид и разбавляют до 250 мл водой. Из этого раствора с pH 9,0 выделяют электролизом Ag и Bi. Оставшийся электролит, в котором содержатсяСй и Си, упаривают с H2SO4 для удаления цианида. Разбавляют раствор до 250 мл и осаждают Си при pH 9,0 с Zn-анодом. Затем повышают кислотность того же раствора до pH 4,0 л выделяют Cd также с Zn-аподом 1641].
Титриметрические методы
Эти методы определения основаны на предварительном выделении труднорастворимых простых или комплексных солей кадмия и на образовании им прочных, хорошо растворимых в воде или органических растворителях комплексных соединений.
В первой группе методов отделенный от раствора осадок растворяют, либо непосредственно титруют, для чего используют методы нейтрализации (ацидиметрия или алкалиметрия,) окисления — восстановления (броматометрия, иодатометрия, иодометрия, перманганатометрия и др.) или осаждения (аргентометрия и пр.). В частном случае титрования диэтилдитиокарбаматом образующийся осадок соединения кадмия служит индикатором для гетерометрического установления конечной точки.
Вторая группа методов включает различные варианты комплексонометрического титрования с металлоиндикаторами. К этой же группе можно отнести экстракционное и фотометрическое титрование с образованием комплексов кадмия, растворимых в органических экстрагентах.
В большинстве титриметрических методов точку эквивалентности устанавливают визуальным путем, но некоторые авторы ис-
63
Таблица 12
Титры 0,1 У растворов по кадмию при различных методах титрования
Метод титрования Титруемое соединение кадмия Титр, мг СЛ/мл
Алкалиметрия Антипирин-бромидный комплекс 5,620
Ионы Cda+ 5,620
Аргентометрия Бруцин-иодидный комплекс 2,810
Пиридин-роданидный комплекс 5,620
Сульфид 5,620
Ацидиметрия Гидроокись 5., 620
Броматометрия 8-Ок сихино линат 1,405
Иодатометрия ₽-Нафтохинолин-иодидный комплекс 5,620
Сульфид 2,810
Иодометрия Антипирин-бромидный комплекс 1,405
Арсенат 5,620
Дифенилдитиофосфат 1,405
Диэтилдитиофосфат 5,620
Хромат * 1,8735
Сульфид 5,620
Комплексонометрия Ионы Cd2+ 11,240
Перманганатометрия Оксалат 5,620
Титрование ферроцианидом2* Ионы Cd2+ 5,620
То же3* То же 4,496
* Образуется после разложения комплекса кадмия с солью Рейнеке и тиомочевиной. Индикатор FeCl3.
•» Индикатор K3[Fe (CN),] + 3,3'-диметилнафтидин.
пользуют для этой цели фотометрическую аппаратуру того или иного типа (спектрофотометрическое титрование и др.). Визуальными методами обычно определяют п-10° — п-101 мг, в некоторых микровариантах — п-Ю^мг Cd; фотометрическое титрование позволяет определять п-103 — п-10~3мг Cd.
Титры рабочих растворов при различных методах определения зависят от используемых реакций и меняются в довольно широких пределах; для основных методов титрования они даны в табл. 12.
Титрование труднорастворимых соединений кадмия
Краткая характеристика методов, основанных на выделении труднорастворимых соединений кадмия, представлена в табл. 13. Наиболее надежные или перспективные методы приведены ниже.
Йодометрическое титрование антипирин-бромидного комплекса. К кислому раствору (5—25 мг Cd) прибавляют соответственно 5—25 мл раствора реагента (5 г антипирина -f-10 г KJ в 85 мл 0,25 N H2SOa) и 15—20 сек.
64
Таблица 13
Титриметрические методы определения кадмия, основанные на образовании осадков
Осадок Реакции титрования * Индикатор Литература
Антипирин-бромидный комплекс кадмия Антранилат кадмия Арсенат кадмия Бруцин-ИОДНДНЫЙ комплекс кадмия Гидроокись кадмия Дифенпл-дитиофосфат кадмия [CdBr4] ICUH1SON2H]2.2 c„h12on2 + + 4J2 H2 [CdBrJ + 4 HJ + + 4 CnHuOJV; J2 + 2 Na2S2O3 Na2S4Oe 4- 2 Na J избыток Cd(C7HeO2N)2 + 8 HCl + 6 KBr 4-+ 2 KBrO3 CdBr2 + 8 KC1 -Д-+ 2 C7H4O2NBr2 H- 6 H2O; KBrO3 + 6 KJ 4- 6 HCl 3 J2 + избыток + KBr + 6 KCl + 3 H2O; J2 -{- 2 Na2S2O3 Na2S4Oe 4- 2 NaJ CdSO4 4- KH2AsO4 CdHAsO4 + + KHSO4; KH2AsO4 4- 2 KJ 4- H2SO4 избыток -» KH2AsO3 4- H2o 4- K2SO4 + J2; J2 4- 2 Na2S2O3 Na2S4Oe 4- 2 NaJ избыток [C21H20O2N2(CH3O)2]2[CdJ4]H2 4- 4- 4 AgNO3 -» Cd(NO3)2 4- 4- 2 C21H20O2N2(CH8O)2 4- 4 AgJ 4-4- 2 HNO3; AgNOg 4- NH4SCN -> AgSCN 4-избыток 4- NH4NO3 CdSO4 4- 2 NaOH -> Cd(OH)2 4- 4- Na2SO4; 2 NaOH 4- H2SO4 -> Na2SO4 4- H2O избыток CdSO4 4- 2 NaOH -» Cd(OH)2 4- 4- Na2SO4 Cd [(CeH5O)2PSS]2 4- 2 NaOH -» Cd(OH)2 4- 2 Na[(CeHeO)2PSSJ; Крахмал Индигокармин 4~ три-иитрорезор-цпп Крахмал » Эозин AgNOs Крезолфта-лени [212] [179, стр-. 678; 565, стр. 124. 290; 567] [179, стр. 375, 565, ctp. 335| [619, стр. 219] [565, стр. 333] [542]
3 Щербов Д. П.
65
Таблица 13 (продолжение)
Осадок Реакции титрования * Индикатор Литература
Диэтилдитиофосфат кадмия Карбонат кадмия Молибдат кадмия |-Нафтохино-лин-иодидный комплекс кадмия (CeH5O)2PSSNa + 20 NaOH + + 8 J2 -» (CeH5O)2POONa + 16 NaJ 4- 2 Na2SO4 + 10 H2O; J2 4- 2 Na2S2O3 Na2S4Oe 4- NaJ избыток Cd[(C2H5O)2PSS]2 + 4 nh4oh + + 2 HC1 [Cd(NH3)4]Cl2 + + 2 (C2H5O)2PSSH + 4 H2O; 2 (C2H5O)2PSSH -|-J2-,(C211-1O)2 PSS-- SSP (C2H5O)2 + 2 HI; J2 + 2 Na2S2O3 Na2S4Oe 4- 2 NaJ избыток CdCO3 + H2SO4 -» CdSO4 + H2O + 4- CO2[; H2SO4 4- 2 NaOH Na2SO4 4- 2 H2O избыток Cd(CH3COO)2 + (NH4)2MoO4 -» CdMoO4 4- 2 CH3COO(NH4) Cd(CH3COO)2 4- (NH4)2MoO4 CdMoO4 4- 2 CH3COO (NH4); 2 MoO42~ 4- 3 Zn°(Hg)3* 4- 16 H+ избыток 2 Mo3+ 4- 3 Zn2+ + 8 H2O; Mo3+ 4- 2 Fe3+ Mos+ 4- 2 Fe2+; 5 Mo5T 4- 10 Fe2+ + 3 MnO7 + 4- 24 H+ 5 Mo«+ 4- 10 Fe3+ 4- 4- 3 Mn2+ 4- 12 H2O (C13H9N)2H2[CdJ4] 4- 4 NaOH 2 C13H9N + Cd(OH)2 4- 2 H2O 4- 4- 4 NaJ; 2 NaJ 4- KJO3 4- 3 KCN 4- + 3 H2SO4 3 JCN 4- Na2SO4 4- 4- 2 K2SO4 4- H2O; (C13H9N)2H2[CdJ4] 4- 4 NaOH ^>2 C13H9N 4- Cd(OH)2 4 2 H2O 4-+ 4 NaJ; Крахмал » » Метиловый оранжевый Пирогаллол 2* КМпО4 Крахмал [58] [55] [656, стр. 334] [565, стр. 309] [179, стр. 112; 399; 751] [179, стр. 546; 619, стр. 218; 680]
66
Таблица 13 (продолжение)
Осадок Реакция титрования * Индикатор Литература
g-Нафтохшю-лин-иодидный комплекс кадмия Нитропруссид кадмия Оксалат кадмия 2-Оксинафти-лал-2-ампнопи- ридиновып комплекс кадмия 2-(о-Окспфе-нил)бензокса-золовый комплекс кадмия 8-Оксихино-линат кадмия Парапериодат кадмия 2 NaJ + KJ Оз + 3 (СН3)2СО + + 3 НС1 3 CH3CH2JCO + + 3 HjO + 2 NaCl + КС1 Cd[Fe(CN)5NO] + 2 NH4OH -» -» Cd(OH)2 + (NH4)2[Fe(CN)5NO]; Cd(OH)2 - 2 1IC1-, CdCl3+ + Na2S -» CdS + 2 NaCl Cd (NO3)2 4~ Na2C2O4 —-> CdC2O4 4" + 2 NaNO3; 5 Na2C2O4 + 2 KMnO4 4- 8 H2SO4 избыток 2 MnSO4 4 K2SO4 4 8 H2O 4 4- 5 Na2SO4 + 10 CO2 (CleH12N2O)2CdCl2 4- 2 HNO3 4- 4- 2 H2O 2 C10He(OH)CHO 4- 4- 2 C5H4N- NH2-HNO3 4- CdCl2; HNO3 4 KOH KNO3 + H2O избыток Cd(C13H8O2N)2 4- 2 CH3COOH -» Cd(CH3COO)2 4- 2 C13H9O2N; 3 C13H9O2N --- 4 KBr 4- 2 KBrO3 4-+ 6 CH3COOH 3 C13H,O2NBr2 4-4- 6 CH3COOK + 6 H2O; KBrO3 4- 6 KJ 4- 6 HC1 3 J2 4-избыток 4- KBr 4- 6 KC1 + 3 H2O; J2 + 2 Na2S2O3 Na2S4Oe4- 2 NaJ 3 Cd(C9HeON)2 4 18 HC1 4- 14 KBr 4- Крахмал Нитропруссид KMnO4 Метиловый красный 4-4- метиленовый голубой Крахмал Метиловый красный или индигокармин Крахмал Дифениламин [179, стр. 568; 565,' стр. 291] [565, стр. 331] [179, стр. 125; 565, стр. 311] [92] [772] [179; стр, 667; 565, стр. 111, 285; 619, стр. 220] [179,-стр. 597]
+ 4 KBrO3 6 C9H5ONBr2 4- 4- 3 CdBr2 4- 18 KC1 4- 12 H2O; KBrO3 4- 6 KJ 4- 6 HC1 -» 3 J2 + избыток 4- KBr 4- 6 KC1 4 3 H2O; J2 + 2 Na2S2O3 Na2S4Oe + 2 NaJ Cd5(JOe)2 + 7 H2SO4 + 4 FeSO4 -» -» 5 CdSO4 4- 2 HJO3 + 4- 2 Fe2(SO4)3 + 6 H2O
3’ &
Таблица 13 (продолжение)
Осадок Реакции титрования * Индикатор Литература
Пиридин-роданидный комплекс кадмия Пиридин-хло-ридный комплекс кадмия Репнекат-тио-мочевинный комплекс кадмия Сульфид кадмия CdCl2 + 2 KSCN + 2 C5H5N -» -» [Cd(C5H5N)2] (SCN)2 + 2 KC1; KSCN + AgNO3 -> AgSCN + KNOS избыток CdCl2 + 2 KSCN + 2 CsH5N -» -> [Cd (C5H5N)2](SCN)2 + 2 KC1; • 5 KSCN + 6 KMnO4 + 13 HCl -» избыток -» KC1 + 6 MnCl2 + 5 HCN + + 5 K2SO4 + 4 H2O [Cd (C5H5N)2]C12 + 2 HCl -> —> CdCl2 4" 2 C5H5N-HC1; HCl + KOH KC1 + H2O [Cd(CSN2H4)2] [Cr(SCN)4NH3)2]2 прокаливание . —> CjQO клГ2Оз, _ __ „ сплавление -T ~ ~ , Cr2O3 + 3 Na2O2 > Na2Cr3O7+ + 2 Na2O; Na2Cr2O, + 6 KJ + 7 H2SO4 -» - > Cr2(SO4)3 + Na2SO4 + 3 K2SO4+ + 7 H2O + 3 J2; J2 + 2 Na2S2O3 —» 2 NaJ -j- Na2S,jOe CdS + 2 HCl + J2 CdCl2 + + S + 2 HJ; J2 + 2 Na2S2O3 -» 2 NaJ + Na2S4Oe избыток 3 CdS + з KJO3 + 12 HCl -> 3 CdCl2 4- 2S + H2SO4 + 3JC1 + + 5 H2O; KJO3 + 5 KJ + 6 HCl -» 3 J2 4-избыток + 6 KC1 4- з H2O; J2 + 2 Na2S2O3 2 NaJ + Na2S4Oe; CdS + 2 AgNO3 Ag2S 4- Cd(NO3)2; AgNO3 + NH4SCN AgSCN 4-избыток 4- NH4NO3; Дпфенил-карбазон KMnOi Патент-синий Крахмал » [565, стр. 295] [636; 637] [565, стр. 312] [179, стр. 401; 565, стр. 297; 610] [565, стр. 269] [179, стр. 563] [181, стр. 357; 565, стр. 270]
S8;
Таблица 13 (окончание)
Осадок Реакции титрования * Индикатор Литература
Сульфид CdS + Fe2(SO4)3 -» CdSO4 + [565,
кадмия + S + 2FeSO4; 10 FeSO4 + 2 KMnO4 + 8 H2SO4 -» -» 5 Fe2(SO4)3 + K2SO4 + 2 MnSO4+ + 8 H2O KMnO4 стр. 270]
Ферроцианид CdSO4 + Ki[Fe(CN)e] -» FeCls [565,
кадмия -^K3Cd[Fe(CN)s] + K2SO4, стр. 313]
5 CdSO4 + 4 K4 [Fe(CN)e] K3 [Fe(CN)e] + [179, стр
-> Cd5Ke[Fe(CN)e]4 + 5K2SO4 -p 3,3-диме-тилнафти- Дин 805; 493]
2 CdSO4 + Li4 [Fe (CN)e] —> Cd2[Fe(CN)e] + 2 Li2SO4; 10 Li4[Fe(CN)e] + 2KMnO4 + избыток + 8 H2SO4 10 Li3 [Fe (CN)e] + + 2 MnSO4 + 6 K2SO4 + 5Li2SO4 + + 4H2O KMnO4 [179, стр. 180; 490]
Фосфат кадмия CdSO4 + N2HPO4 -> CdHPO4 + Метиловый [181,
+ Na2SO4; красный стр. 243]
CdSO4 + Na2HPO4 -» CdHPO4 + Хлорфено- [565,
-[- Na2SO4; CdNH4PO4 + H2SO4 CdSO4 + + NH4H2PO4; ловый красный стр. 334]
H2SO4 + 2 NaOH -> Na2SO4 +2 H2O Метиловый [565,
избыток оранжевый стр. 283]
* Титрант подчеркнут. Внешний индикатор. 34 Амальгамированный цинк.
потирают стенки стакана стеклянной палочкой. Через 30—35 мин. осадок отфильтровывают через стеклянный фильтр-тигель № 4 и промывают осадок 1—2 раза разбавленным раствором реагента (1 : 1). Влажный осадок смывают 25—30 мл воды в склянку с притертой пробкой, после его растворения прибавляют 3 г CH3C00Na (для связывания выделяющейся HJ) и 25—30 мл 0,1 N раствора J2; образуется темно-бурый осадок. Через 30—40 мин. для его растворения вводят 25—50 мл этанола и оттитровывают избыток иода 0,1 N раствором Na2S2O3.
1 мл 0,1 N раствора J2 эквивалентен 1,405 мг Cd.
Ионы Al, Си, Fe, Mg, Мп и Zn определению не мешают.
69
По другому варианту, осадок из тигля смывают 50 мл воды в колбу емкостью 250—300 мл, слабо нагревают до его полного растворения, разбавляют водой до 150.M.UI титруют по метиловому оранжевому 0,1 N раствором NaOH.
1 мл такого раствора соответствует 5,620 мг Cd.
В этом случае определению не мешают Со, Си, Mg и Ni [212].
Йодометрическое титрование арсената (микровариант). К 2,5 мл нейтрального раствора (по фенолфталеину), содержащего 0,15—0,25 мг Cd, прибавляют 0,1 мл 2 N СН3СООН, нагревают до кипения и по каплям при непрерывном помешивании приливают нагретый до кипения раствор осадителя (1 мл 0,5 М арсената натрия нейтрализуют 2 N СН3СООН, прибавляют еще 0,1 мл этой же кислоты и разбавляют водой до 2,5 мл). Горячий раствор с осадком выдерживают 10—15 мин., фильтруют через плотный небольшой фильтр, промывают горячей водой и растворяют осадок в 5 мл H2SOa (2 : 5). К полученному раствору приливают 5 мл СвНв, 0,5 мл 2 N раствора KJ, удаляют воздух током СО2, перемешивают 15—20 сек., разбавляют 5 мл воды и титруют выделившийся иод 0,005 N раствором Na2S2O3 до слаборозовой окраски бензольного слоя [462].
Аргентометрическое титрование бруцин-иодидного комплекса кадмия возможно в присутствии 1000-кратного избытка Zn, но Си, Fe, РЬ и Sb должны быть удалены; мешают свободные HNO3 и nh4oh.
К 50 мл нейтрального или слабокислого раствора, содержащего 20— 50 мг Cd, прибавляют 1,5 мл свежеприготовленного 1%-ного раствора бруцина в 5%-ной H2SO4 (на каждый миллиграмм Cd), 1,5 мл 10%-ного раствора KJ и перемешивают. Спустя 10 мин. фильтруют под вакуумом через воронку Бюхнера с тонкой фильтровальной бумагой. Осадок промывают 3 раза свежеприготовленной смесью (1 : 1) растворов сульфата бруцина и KJ и затем, для удаления иодида, 3 раза смесью этанола с толуолом (1 : 4). Смывают водой осадок и фильтровальную бумагу в колбу, разбавляют до 100 мл и нагревают до растворения осадка. Прибавляют 5 мл 0,5%-ного раствора эозина Y и титруют 0,03 М раствором AgNO3 при сильном взбалтывании до красного окрашивания осадка AgJ [619, стр. 219].
Алкалиметрическое титрование комплекса с диантипирил-о-ок-оксифенилметаном можно производить в присутствии А1, Со, Сг, Си, Fe, Mg, Zn, щелочных и щелочноземельных металлов.
К сернокислому раствору кадмия прибавляют осадитель (смесь равных объемов 1%-ного спиртового раствора реагента, 5%-ного раствора КВг и IN H2SO«), перемешивают и оставляют на 1,5 часа. Осадок отфильтровывают через фильтр-тигель № 4, промывают 3—4 раза разбавленным (1 : 4) раствором осадителя и подсушивают. При помощи 30—35 мл этанола осадок из; тигля переносят в колбу емкостью 300—350 мл, нагревают до растворения осадка, прибавляют 150 мл воды, 4—6 капель метилового оранжевого и титруют 0,1 N раствором NaOH до перехода окраски [211].
•а
Йодометрическое титрование дифенилдитиофосфата кадмия основано на его окислении иодом в сильнощелочной среде. Для небольших количеств кадмия оно удобнее гравиметрического определения.
Отфильтрованный через стеклянный фильтр № 4 и промытый осадок дифенилдитиофосфата (полученный так же как для гравиметрического определения) смачивают несколькими каплями СНС13 или CCU и обрабатывают на фильтре 6 N раствором NaOH, в результате чего комплекс разрушается. Щелочи берут столько, чтобы после прибавления раствора J2 ее концентрация 70
в растворе составила 0,5—1,5 N. Добавляют избыток титрованного раствора J2, через 5—10 мин. приливают H2SOi до слабощелочной реакции по универсальной индикаторной бумаге и титруют избыток иода тиосульфатом [58]. Осадок дифенилдитиофосфата кадмия можно экстрагировать СНС13.
Йодометрическое титрование диэтилдитиофосфата кад мия аналогично предыдущему; точность его такая же, как гравиметрического определения, но значительно быстрее и проще по технике выполнения.
Кадмий осаждают 0,05 N раствором чистого перекристаллизованного диэтилдитиофосфата никеля. Осадок отфильтровывают через плотный бумажный фильтр, промывают 4—5 раз насыщенным раствором диэтилдитиофосфата кадмия, смывают осадок струей воды в коническую колбу и растворяют при добавлении небольших порций аммиака. Раствор нейтрализуют чистой НС1 по метиловому красному до появления розового окрашивания и прибавляют еще 1 мл кислоты в избыток. Затем титруют 0,05 N раствором иода в присутствии крахмала до неисчезающей синей окраски. Вблизи конечной точки титрование следует производить медленно при энергичном перемешивании. Соотношение растворов диэтилдитиофосфата никеля и иода устанавливают в таких же условиях; при вычислении пользуются теоретическим титром диэтилдитиофосфата никеля по кадмию [55].
Иодатометрическое титрование Р-нафтохинолин-иодидного комплекса кадмия позволяет определять кадмий в присутствии А1, Со, Сг, Fe, Mg, Мп и Ni; Sn и Sb маскируют тартратом или цитратом, влияние As, Bi, Си и РЬ устраняют восстановлением металлическим железом. Ниже описан ход анализа растворов, содержащих цинк и элементы группы сероводорода.
К 50 мл раствора, 1 М по H2SO«, содержащего указанные металлы и не более 50 мг Cd, прибавляют несколько капель H2SO« и нагревают до кипения. Вносят несколько кусочков железной проволоки и продолжают нагревание около часа при слабом кипении, периодически добавляя воду, чтобы сохранить объем раствора постоянным. Отфильтровывают осадок металлов и PbSO« через стеклянный фильтрующий тигель, содержащий кусочек железной проволоки, тщательно промывают осадок холодной водой и присоединяют промывные воды к фильтрату.
К раствору объемом 100—120 мл прибавляют 5 г тартрата калия — натрия, несколько капель H2SO3, 40—50 мл 0,2 М раствора KJ и достаточное для осаждения кадмия количество 2,5%-ного раствора (3-нафтохинолина в 0,25 М H2SO«. Образующийся осадок выдерживают в течение часа, фильтруют через слой асбеста на стеклянном фильтрующем тигле или тигле Гуча. Дают жидкости стечь с осадка возможно полнее и тщательно промывают свежеприготовленной смесью, содержащей по 10 мл растворов (3-нафтохинолина и KJ, несколько капель H2SO3 и 80 мл воды. В первые 2—3 порции промывного раствора прибавляют еще по 2—3 мл раствора KJ. Отсасывают насколько возможно жидкость с осадка и переносят его вместе с асбестом в колбу, в которой производили осаждение. Приливают 20 мл раствора NH«OH, тщательно перемешивают для разложения комплекса, фильтруют асбест с остатком через тигель Гуча и хорошо промывают 2 М NH«OH и водой. Фильтрат переносят в коническую колбу и смывают стенки сосуда (в котором проводили фильтрование) таким количеством 2 М НС1, чтобы ее концентрация в титруемом растворе была 2—2,5 N. Прибавляют 5 мл 10%-ного раствора KJ, раствор крахмала и титруют 0,025 М раствором KJO3 до перехода синей окраски в красно-фиолетовую и затем до обесцвечивания.
1 мл 0,025 М раствора KJO3 эквивалентен 1,405 мг Cd (0,1 .У — 5,620 мг •Cd) [619, стр. 218; 179, стр. 568].
71
По иодно-ацетоновому методу Берга [179, стр. 569] осадок комплекса кадмия растворяют в 20 мл 2 N раствора NaOH, подкисляют H2SO«, прибавляют 20—30 мл ацетона и столько H2SO« (1 : 1), чтобы ее концентрация в 100 мл объема в конце титрования была 2—2,5 N. Прибавляют крахмал и титруют 0,1 N (при малом количестве кадмия — более слабым) раствором KJO3, прибавляя его по каплям с такой скоростью, чтобы коричневатая иодо-крахмальная окраска к концу титрования стала чисто-синей и обесцветилась в конечной точке.
По иодно-циановому методу Ланга [179, стр. 565] вместо ацетона в титруемую жидкость вводят раствор KCN и тоже титруют йодатом до исчезновения синей окраски. Конец титрования более четок, если в качестве индикатора вместо крахмала применять органический растворитель [565, стр. 291; 179, стр. 569].
Йодометрическое титрование осадка кадмия с 2-(о-оксифенил)бенз-оксазолом [772]. К 25—50 мл раствора, содержащего 2—80 мг Cd и свободного от мешающих ионов, добавляют равный объем 96%-ного этанола, нагревают до 60° С. Устанавливают pH ~ 11, осаждают кадмий избытком реагента, как при весовом определении, и фильтруют через стеклянный фильтр средней плотности. Осадок промывают 50%-ным этанолом для удаления следов аммиака, растворяют в 50 мл горячей ледяной СН3СООН и разбавляют 20 мл воды. Раствор переносят в «бромную» или «иодную» колбу [179, стр. 37], вводят 35 мл 0,1 N бромид-броматной смеси, немедленно закупоривают колбу и наливают в ее резервуар 2 мл раствора KJ. Через 60—75 мин. добавляют — 1,5 г KJ и титруют выделившийся иод 0,1 N раствором Na2S2O3.
Содержание кадмия рассчитывают по формуле
х = 1,53 (А — Ао) мг Cd,
где 1,53 — эмпирический фактор; А и Ао — объем раствора титранта (в мл), израсходованного на титрование растворов пробы и холостого опыта соответственно [771, 772, 553].
Б рома то метрическое титрование 8-оксихинолината кадмия возможно после его отделения от элементов, реагирующих с 8-оксихинолином. Осаждение кадмия начинается при pH 4,0—4,5, полнота его достигается при pH 5,5—14,5. В этих условиях осадки образуют Al, Bi, Со, Си, Fe, Мп, Ni, Pb, Th, Ti, Zn и, частично, некоторые другие металлы.
К нейтральному или слабокислому раствору, содержащему 2—100 мг Cd, прибавляют раствор Na2CO3 до образования мути, которую растворяют приливанием по каплям 0,5%-ной СН3СООН. Вводят 3—5 г CH3COONa, растворенного в небольшом количестве воды. Осаждают кадмий небольшим избытком 2%-ного спиртового или 4%-ного ацетонового раствора 8-окси-. хинолина. Нагревают до начала кипения и дают постоять до коагуляции осадка. Фильтруют через стеклянный пористый тигель № 3—5 и тщательно промывают сначала горячей, а затем холодной водой. Тигель с осадком переносят в колбу, в которой производили осаждение, растворяют осадок в 10— 15 мл горячей 3 N НС1, вынимают тигель и споласкивают водой. Разбавляют раствор до 400 мл, прибавляют несколько капель 1%-ного раствора индигокармина (или 0,2%-ного раствора метилового оранжевого), 0,5 г КВт и титруют по каплям 0,1—0,2 N раствором КВгО3 до перехода синей (красной) окраски в желтую. Прибавляют еще 2—3 мл последнего и накрывают часовым стеклом. Через минуту приливают 15 мл 20%-ного раствора KJ и титруют тиосульфатом натрия шоколадно-коричневый раствор до слабо-желтой окраски. Прибавляют несколько капель 1%-ного раствора крахмала и титруют до обесцвечивания раствора [619, стр. 219; 565, стр. 284; 179, стр. 657, 667].
72
Пиридин-роданидный комплекс кадмия можно титровать несколькими методами — аргентометрией, меркуриметрией и пер-манганатометрией. При использовании Hg(NO3)2 определению кадмия не мешают Al3+, Bi3+, Сг3+, Sb3+, Sn2+, Sn4+.
К азотнокислому раствору 5—25 мг Cd прибавляют 0,2 N раствор KSCN и 0,5 мл пиридина. Раствор разбавляют до 25 мл, нагревают на водяной бане (60—70° С) в течение 1—2 мин. и филируют через 5 мин. К 5 мл фильтрата приливают 10—15 мл воды, устанавливают pH ~ 3,1 разбавленной НСООН. Вводят 1—2 капли насыщенного спиртового раствора дифенилкарбазона и титруют 0,1 N раствором Hg(NO3)2 до светло-фиолетовой окраски [705.]
В перманганатометрии к 100 мл раствора, содержащего 2,5—25 мг Cd, прибавляют 1 г KSCN, 1 мл пиридина и через 10 мин. отфильтровывают осадок через плотный бумажный фильтр, на который нанесен слой крахмальной суспензии. Промывают 10 мл раствора, 1%-ному по обоим осадителям, затем 3 мл смеси этанола с эфиром (1 : 1) и отсасывают. Осадок на фильтре растворяют в 50—75 мл 2 N НС1, приливают 5 мл ССЦ, 5 капель 0,5 М солянокислого раствора JC1 и титруют 0,1 N раствором КМпО< до исчезновения фиолетовой окраски СС1« и появления розовой у водного слоя.
Кроме пиридина, для осаждения комплексного роданида кадмия можно использовать 2,2'-дипиридил и 1,10-фенантролин, а вместо роданида — комплексные анионы типа [Hg(SCN)a]2-, [Cr(SCN)e]3~ и другие [636].
Йодометрическое титрование рейнекат-тиомочевинного комплекса кадмия позволяет определять его в присутствий цинка.
В условиях осаждения кадмия Ag, Bi, Си, Hg и Pb образуют нерастворимые комплексы [565, стр. 297; 179, стр. 397].
Осадок комплекса, содержащий 1—10 мг Cd и полученный описанным в гравиметрии путем, фильтруют через беззольный фильтр, промывают 1%-ным раствором тиомочевины, переносят вместе с фильтром в маленький никелевый тигель, озоляют и слабо прокаливают для разрушения органического вещества. Остаток сплавляют с небольшим количеством Na2O2 и плав выщелачивают водой. Для разложения избытка перекиси разбавляют до 60—70 мл и кипятят 15 мин. Прибавляют 5 мл 1 N раствора KJ, 15 мл 4 N НС1 (пли H2SOa) и титруют выделившийся иод 0,1 N раствором Na2S2O3.
Описаны и другие варианты метода: осаждение рейнеката пи-ридил-2-альдоксимом с броматометрическим титрованием роданида в отфильтрованном осадке [544]; титрование раствором КМпО4 по индикатору JC1 [637]; окисление тиомочевинного-комплекса кадмия хлорамином Т, избыток которого титруют иодометрически [676].
Известно два варианта титрования комплекса кадмия с тиокапролактамом—ацидиметрический и аргентометрический. По первому варианту осаждение проводят так же, как при гравиметрическом определении, но вводят точное количество 0,5 N раствора NaOH, избыток которого (после отделения фильтрованием осадка Cd) оттитровывают по метиловому оранжевому 0,1 N НС1 [731]. Во втором варианте отфильтрованный осадок [Cd(SCN)J(CeH11SNH)2 промывают, растворяют в этаноле и в полученном растворе титруют роданид 0,1 N раствором AgNОз с индикатором эозином [488].
73
Визуальная комплексонометрия
Теоретические основы метода и детали его практического применения изложены в работах [344, 381, 464]. Для массовых анализов при определении кадмия в качестве титранта чаще всего используют комплексон III. Константа нестойкости его комплекса с кадмием — 3,46-10-17 (рК = 16,46). В чистых растворах комплексообразование начинается при pH’—1, при pH —4,5 99,9% Cd переходит в комплекс. Большинство других элементов тоже образует комплексы с ЭДТА; значения рА для них приведены в табл. 14, а значения pH для области начала и завершения комплексообразования — на рис. 6 [380].
При комплексонометрическом определении кадмия применяют как прямое, так и обратное титрование.
Индикаторы для комплексонометрического определения кадмия и pH среды, в которой они применяются, приведены ниже [381, 464, стр. 40, 268].
Индикатор pH
Метилтимоловый синий 11,5—12,5
Галлеин, глиоксаль-бис-(2-оксианил), глицинтимоловый синий, 9—10 нафтоловый фиолетовый, пиридил-азо-резорин, пирокатехиновый фиолетовый, сульфарсазен, фталеиновый пурпуровый, эриохром черный Т; цинкон (при обратном титровании сульфатом цинка)
Мурексид 8,0—8,5
Бромпирогаллоловый красный 7,0—8,0
Азоксин, диметилнафтидин (в присутствии ферроцианида), ди- 5,0—6,0 фенилкарбазид, ксиленоловый оранжевый,метилтимоловый синий, а-нафтилазоксиндисульфокислота, пиридил-азо-нафтол
Дитизон (в CCh) 5,3
Дитизон (в 60%-ном этаноле); диметилнафтидин, бензидин или 4,5 вариаминовый синий (в присутствии ферроцианида при обратном титровании сульфатом цинка)
Пирогаллоловый красный (при обратном титровании нитратом 5 олова)
Пирогаллоловый красный (при обратном титровании нитратом 2—3
висмута)
Кроме них описаны и следующие металлоиндикаторы: арс-азен, глициннафтоловый фиолетовый, калькой, кислотный ализариновый черный SN, кислотный хром синий К, а-нафтилазоксин-5-сульфокислота, пиридил-азо-диоксинафталинсульфокислота, тиа-золил-азо-орсин, тиазолил-азо-резорцин, хром прочный синий RLL, эрио В, эрио SE, эриохром темно-синий BRL, хром фиолетовый, эриохром черный PW [464, стр. 47, 56]; азоимидазолы [783], гематеин и гематоксилин [735], кальмагит [618], производные 2,2'-диаминодифенил-М,М,М',К'-тетраацетата [615], 2-тиазолцлазо-4-метоксифенол и 2-тиазолилазо-5-метоксифенол [529] и эриохром красный В [535]. В качестве индикаторов для титрования кадмия с комплексоном III рекомендованы эриохром черный Т и сульфарсазен [325].
74
Рис. 6. Интервалы минимальных значений pH начала (0,1; 1,0 и 5%) и завершения (95,0; 99,0 п 99,9%) образования комплексонатов в зависимости от рК
Избирательности титрования кадмия способствует использование некоторых маскирующих комплексообразователей. Иодид (умеренные количества) связывает Hg, тиосемикарбазид — Ag и Hg, триэтаноламин—А1, фторид —А1, Са и Mg, цитрат — Sn, Th и Zr. Для маскировки самого кадмия применяют [3-аминометил-меркаптан, диэтилдитиокарбаминат, полиамины (в частности, тетраэтилпентамин), цистеин. При определении Са, Mg и Ni вводят димеркаптопропанол, при титровании In — 1,10-фенантролин; избыток иодида (до 50 г/100 мл раствора) позволяет титровать малые количества Zn в присутствии почти 3000-кратного количества Cd. Цианидом связывают Со, Си, Hg, Ni, Cd и Zn, а затем демаскируют последние два элемента формальдегидом или хлоралгидратом и титруют их по эрихром черному Т [464, стр. 137]. Дитиокарбаминоацетат маскирует Cd, Си, Hg и РЬ, что дает возможность в их присутствии титровать Со, Мп, Ni и Zn [520].
Эриохром черный Т (кислотный хром черный специальный, хром черный специальный ЕТ 00, хромоген черный специальный ЕТ 00) — кристаллы черного или коричневого цвета, растворимые в воде и этаноле с винно-красной окраской и в щелочах — с темно-синей. При pH 9,5—10,0 образует с кадмием красно-фиолетовое соединение, которое разрушается комплексоном III; поэтому в точке эквивалентности окраска раствора становится синей. Применяется в виде сухой смеси (0,25 г индикатора и 25 г КС1 или NaCl) [233, стр. 206; 325, стр. 23].
К 100 мл нейтрального раствора, содержащего 0,5—50 мг Cd, прибавляют 2 мл буферного раствора (50 г NH4CI растворяют в 200 мл воды, прили-
75
Таблица ,14
рК комплексов некоторых ионов с комплексоном III
Ион PK Ион pK Ион pK
Na+ 1,66 Cd2+ 16,46 (19,1) *
Li+ 2,79 Zn2+ 16,50 Tu3+ 19,'32
ТВ 5,8 Nd3+ 16,61 Yb3+ 19,51
Ra3+ 7,1 Sm3+ 17,14 Lu3+ 19,83
Ag+ 7,2 TiO22+ 17,3 Ga34" 20,3
Eu2+ 7,7 Eu3+ 17,35 Ti3+ 21,3
Ba2+ 7,76 Gd3+ 17,37 Hg2+ 21,80
Sr2+ 8,63 Tb3+ 17,93 Sn2+ (22,1)*
Mg2+ 8,69 pb2+ 18,04 Sc3+ 23,1
Be2+ 9,3 уз+ 18,09 Th4+ 23,2
Ca2+ 10,96 Pu3+ 18,1 TP+ 23,2
V2+ 12,07 VOa+ 18,1 Cr3+ 23—24
Mn2+ 14,04 Am3+ 18,2 In34" 24,9
Fe2+ 14,33 Dy3+ 18,30 Fe3^ 25,1
UO?+ 14,40 pd2+ 18,5 IB 25,62
La3T 15,50 Ni2+ 18,62 V3+ 25,9
Ce3+ 15,98 Ho3+ 18,74 Bi3+ 27,94
Al3+ 16,13 VO2+ 18,77 Zr4+ 29,5
Co2+ 16,31 Cu2+ 18,80 Co3+ 40,7
Pr3+ 16,40 Er3+ 18,85
* Приближенное значение.
вают 350 мл 25%-ного NH4OH и разбавляют водой до 1 л), 0,1 г индикаторной смеси и титруют 0,05 М раствором комплексона III [344, стр. 304]. Кислые растворы предварительно нейтрализуют NaOH. Если присутствуют трехвалентные ионы металлов, их сначала выделяют ацетатным гидролизом, при наличии Mg вводят фторид и титруют 0,01 М раствором комплексона III при pH 7 [186, етр. 109; 464, стр. 264].
При определении кадмия в его солях 160—180 мг соли растворяют в воде, прибавляют 5 мл буферного раствора, 0,1 г индикатора и титруют 0,05 М раствором комплексона III до перехода красно-фиолетовой окраски в синюю.
При определении кадмия в цианистых электролитах 1 мл его помещают в колбу емкостью 250 мл, приливают 20—30 мл воды, 5 мл буферного раствора и 1—2 мл 10%-ного раствора формальдегида (уд. в. 1,027—1,034). Вводят индикатор и титруют 0,05 М раствором комплексона III [325, стр. 23].
В присутствии Со, Си и Ni (Са и Mg) предварительно определяют их содержание.
К 100 м.г кислого анализируемого раствора приливают избыток 0,01 М раствора комплексона III (А мл), 2 мл 1 М раствора NH4CI и устанавливают pH 10 прибавлением NH4OH (посинение тимолфталеиновой бумаги). Вводят индикатор и титруют 0,01 М раствором MgSC>4 до появления красной
76
окрАски (расход титранта — В мл). Разность А — В соответствует общему содержанию присутствующих металлов.
Затем на каждые 100 мл титруемого раствора прибавляют по 2 мл 1 М растйора KCN, который связывает в комплексы Со, Си, Ni, Cd и тем самым освобождает эквивалентное количество комплексона III. Вводят новую порцию индикатора и титруют раствором MgSOa, расход которого (С мл) пропорционален содержанию тяжелых металлов.
Исходя из этих данных, для основного титрования берут столько анализируемого раствора, чтобы он содержал 1—5-10-10 г-атомов тяжелых металлов, осторожно (во избежание выделения осадка) нейтрализуют раствором NaOH, приливают 2 мл i М раствора KCN и разбавляют до 100 мл. Если в растворе присутствуют щелочноземельные элементы (А — В — С > 0), то прибавляют индикатор и титруют раствором комплексона III до перехода красной окраски в синюю. Приливают 4 мл НСНО, через несколько секунд восстанавливают прежнее значение pH введением 8 мл раствора NHaCl и нескольких капель NH4OH. При необходимости добавляют еще индикатора и опять титруют раствором комплексона III до синего окрашивания раствора. Последний расход титранта соответствует содержанию кадмпя.
Для демаскирования кадмия можно вместо НСНО вводить 6 мл 1 М расувора хлоралгидрата, но тогда перед обратным титрованием выжидают 1 мин. [186, стр. НО; 344, стр. 417; 464, стр. 265].
В присутствии si 120 мг А1 и небольших количеств Fe и Мп в качестве маскирующего агента применяют триэтаноламин. При этом А1 медленно реагирует с индикатором, но при охлаждении до температуры 5° С реакция прекращается.
Анализируемый раствор разбавляют до 200 мл, прибавляют (в зависимости от содержания AI) 5—20лм триэтаноламина, тщательно перемешивают и вводят несколько миллилитров буферного раствора (см. стр. 76). Когда раствор станет прозрачным, его охлаждают в ледяной воде, вносят индикатор и титруют при энергичном перемешивании раствором ЭДТА до темно-синей окраски. Если раствор содержит Mg, то кадмий связывают добавлением KCN и вводят комплексон III до перехода окраски индикатора; затем демаскируют Cd посредством НСНО и заканчивают титрование как обычно [344, стр. 420; 464, стр. 140; 694, стр. 89].
В присутствии РЬ в слабокислый раствор вводят винную кислоту, нейтрализуют по метиловому красному аммиаком, прибавляют 10—15 мл буферного раствора, KCN, индикатор и титруют свинец раствором комплексона III. Добавляют НСНО и вновь титруют Cd комплексоном III до перехода окраски [344, стр. 416; 694, стр. 82].
Сульфарсазен (плюмбон ИРЕ А). Желтый 0,05 %-ный раствор сульфарсазена в 0,05 М растворе Na2B4O7 устойчив в течение двух недель; окраска в 1 N NaOH — сине-фиолетовая. В минеральных кислотах реагент разлагается. При pH 7—10 сульфарсазен образует розовые соединения с Cd, Hg, РЬ и Zn; кроме того, он реагирует с ионами Со, Си, La, Мп, Ni и U. В растворах с pH 9,5—10 розовый комплекс Cd разрушается комплексоном III и раствор приобретает лимонно-желтую окраску. Индикатор применяют в виде 0,05 %-ного раствора в слабоаммиачной воде или в 0,05 М растворе Na2B4O7 [233, стр. 214; 317; 325, стр. 21; 372].
77
При определении кадмия в его солях пробу, содержащую 160— 180 .мг Cd, растворяют в 100 мл воды, прибавляют 5 мл буферного раствора с pH 9,5—10, затем 0,4 мл индикатора и титруют 0,05 71/ раствором комплексона III до перехода розовой окраски в лимонно-желтую. При титровании в цитратно-аммиачной среде при том же pH железо оказывает меньшее влияние [325, стр. 22].
Пирокатехиновый фиолетовый (пирокатехинсульфофталеин, бренцкатехинвиолет). Обычно применяют 0,1 %-ный водный раствор, устойчивый в течение неограниченного времени.
К 100 мл раствора, содержащего не более 40 мг Cd, приливают хлоридпо-аммпачный буферный раствор, затем индикатор и титруют до перехода окраски от зелено-сппей в красно-фиолетовую [344, 694].
Ксиленоловый оранжевый используют при титровании кадмия в слабокислой среде, при этом щелочноземельные элементы не мешают.
К кислому анализируемому раствору прибавляют уротропин до pH 6, вводят индикатор и титруют допереходаокраскиизкраспо-фиолетовой в ярко-желтую.
Если в растворе присутствует висмут, то его можно предварительно оттитровать при pH 7^ 2, после чего нейтрализовать раствор уротропином [694].
1 - (2-П и р и д и л-а з о)-2'-н афт о'л (ПАН) в слабокислом растворе образует с кадмием комплекс, труднорастворимый в воде. В некоторых случаях для повышения растворимости комплекса в раствор вводят спирты, диоксан, диметилформамид или др. [694]. Влияние ионов Sn2+, Th4+ и Zr1+ устраняют цитратом [386].
К анализируемому раствору, содержащему 5—25 мг 'Cd, прибавляют CHsCOONa до pH 6, вводят 3—4 капли 0,1 %-пого метанольного раствора ПАН й титруют до перехода розовой окраски жидкости в чисто-желтую [344].
Описано определение Cd и обратным титрованием с ПАН раствором сульфата меди [464, стр. 268].
В связи с малой избирательностью определения кадмия с комплексоном III предложены методы с его предварительным выделением. Для отделения от As, Bi, Со, Мн, Ni, Sb, Sn, Zn и некоторых других элементов кадмий осаждают в присутствии тартратов или цитратов висмутиолом (как при гравиметрическом определении), промытый осадок переносят в коническую колбу избытком титро-4 ванного раствора комплексона III и создают pH — 10 хлоридно-аммиачным буферным раствором. После растворения осадка оставшийся несвязанным избыток комплексона III титруют раствором MgSO4 по эриохром черному Т [645].
Несколько способов основано на образовании бромидом или иодидом кадмия комплекса с диантипирилметаном.
Тигель Шотта с осадком комплекса бромида Cd с диантипирилметаном (выделенного как при гравиметрическом определении) переносят в коническую колбу, заливают 100—150 мл воды и осторожным вращением переводят
78
основную массу осадка в раствор. Приливают 2—3 мл х лори дно-аммиачного буферного раствора, 1—2 мл NH4OH и титруют с индикатором эриохром черным (Г [116].
В вариантах с иодидом комплекс кадмия отделяют от цинка 2-кратной экстракцией с СНС13 [122]. Иногда Cd экстрагируют вместе с Zn и удаляют последний из органической фазы промыванием раствором KJ [124]. В обоих случаях кадмий реэкстрагируют из СНС13 разбавленным NH4OH и титруют по эриохром черному Т. Можно прибавлять избыток комплексона III и проводить обратное титрование раствором ZnSO4 [124]. По-видимому, можно также осаждать кадмий тиоацетамидом и по растворении отфильтрованного CdS в горячей НС1 титровать его раствором комплексона III [675].
Описаны методы, основанные на суммарном титровании раствором комплексона III кадмия и некоторых других элементов, на-пример-Zn или Мп. В первом случае кадмий затем связывают иодидом калия [561], а — во втором 2,3-димеркаптопропанолом [532], При этом освобождается эквивалентное количество комплексона III.
Описано ультрамикроопределение 0,4 мкг Cd2+ в присутствии 40-кратного количества Си2+ (маскируют дитиооксамидом) после их выделения методом кольцевой бани. Раствор (— 0,5 мл, pH 6) титруют 0,001 М раствором комплексона III; индикатором служит смесь ксиленолового оранжевого с метиленовым синим [574].
Для титрования кадмия используют и другие комплексоны: нитрилотриуксусную кислоту с индикатором пирокатехиновым фиолетовым при pH 10,2; тетраэтиленпентамин с индикаторами эриохром черный Т или цинк + цинкон при pH 9—9,5 [386], диэтилентриаминпентауксусную кислоту и 0,0'-диаминодиэтилгликолевый эфир N,N,N',]\Г-тетрауксусной кислоты [464]; диамино-циклогексан-N, N, N', N'-тетрауксусную кислоту по ксиленоловому оранжевому (в присутствии 0-меркаптопропионовой кислоты, маскирующей медь) [696] и этилен-бис-(оксиэтиленамин)тетраук-сусную кислоту (обратное титрование раствором СаС12, с фталеин-комплексоном в качестве индикатора) [697].
Экстракционное и фотометрическое титрование
Для экстракционного (косвенного) титрования кадмия описаны методы с дитизоном и 4-диметиламинофенил-4~метилбензиламино-фенилантипирилкарбинолом-
Экстракт дитизоната кадмия в ССЦ, содержащий 1—25 мкг Cd (экстракция из щелочной среды, как для колориметрического определения), промывают от следов свободного дитизона 0,5 N раствором NaOH и разлагают встряхиванием с 1%-ной HNO3 или 1 N H2SO4. Кадмий переходит в водный слой; в ССЦ остается эквивалентное ему количество дитизона. Органическую фазу обрабатывают известным избытком слабо азотнокислого 0,025 мМ раствора AgN03, который (после отделения органической фазы) титруют ра
79
створом дитизона такой же молярности. При этом водный раствор, серебра последовательно встряхивают со все уменьшающимися порциями дитизона— в точке эквивалентности желтый цвет очередной порции дитизоната серебра приобретает зеленоватый оттенок.
1 мл 0,025 мМ раствора дитизона (с его содержанием 0,64 мг в 100 мл СС14) соответствует 1,405 мкг Cd [150, стр. НО, 265; 565, стр. 143, 303].
При использовании 4-д и метилам и и офени л -4-м ети л бенз и л-аминофенплантипирилкарбипола (дающего с анионным комплексом CdJa окрашенный ассоциат) к слабокислому или нейтральному раствору, содержащему 2—12 мг Cd, приливают 2,5—4 мл 50%-ного раствора KJ, 1 мл 10%-ного раствора аскорбиновой кислоты и 4 A H2SO4 (ее концентрация в растворе должна быть 0,5—1 АГ). Прибавляют 2—3 мл бензина и титруют 0,02 М раствором реагента в 1 N H2SO4 до розового окрашивания органического слоя.
Титр реагента устанавливают в тех же условиях ио растворам кадмия известной концентрации [127].
Д и ф енилдитиофосфорная кислота. К раствору 0,1—20 мг Cd в 0,05 N H2SO4 прибавляют 2—3-кратный избыток 0,02 М раствора реагента, разбавляют до 100 мл и 3 раза экстрагируют по 2 мин. порциями хлороформа по 25, 25 и 10 мл. Объединенные экстракты встряхивают с 1,25%-ным раствором CuSO4 и желтую органическую фазу титруют 0,01 или 0,001 N раствором HgCl2 до обесцвечивания.
Метод может быть использован для определения кадмия в продуктах его производства [522].
Для спектрофотометрического титрования кадмия используют дитизон, пиридил-азо-нафтол и комплексон III с различными ме-таллоиндикаторами. Описано и гетерометрическое определение кадмия с диэтилдитиокарбаминатом натрия.
С дитизоном (0,1 мМ раствор в СС14) прямое титрование 1—5 мкг Cd выполняют в специальной кювете без отделения органического слоя; конечную точку находят графически.
Отмечена возможность использования диэтилдитиокарбамина-та, купферона, 8-оксихинолина, пиридил-азо-нафтола и пиридил-азо-резорцина [570].
При добавлении 1-(2-п иридилаз о)-2-н а ф т о л а кадмий экстрагируется СНС13 из водной фазы при pH 13,5.
В сосуд помещают 50 мл слабокислого раствора, содержащего 2—10 мкг Cd, 5 — 10 мл 10 М раствора NaOH и 10—15 мл СНС13. Во второй сосуд вносят такие же количества указанных растворов, но без кадмия. Жидкости в обоих сосудах титруют 0,3 мМ раствором ПАН в этаноле, взбалтывая каждый раз по 1 мин. и давая отстояться по 20 сек. Затем хлороформные фазы спектро-фотометрируют при 550 нм-, эквивалентную точку находят по графику (по изменению хода разности оптических плотностей) [571].
При комплексонометрическом титровании кадмия используют азоксин, эриохром черный Т, «самоиндикацию» в ультрафиолетовой области спектра (различие в оптических плотностях растворов комплексона III и его комплекса с Cd при 220 нм). В присутствии цинка вводят в качестве фотометрического индикатора медь, комплекс которой с комплексоном III окрашен [464, стр. 104].
80
Рис. 7. Кривые спектрофотометрического титрования 0,1 мкг Cd/мл комплексоном III с различными индикаторами
1 — 4-(2-пиридилазо)рсзорцин; 2 — ксиленоловый оранжевый; 3 — сульфарсазен; ' — арсазен; 5 — эриохром черный Т
Из ряда индикаторов (рис. 7) наиболее четкий переход окраски для 2,5—100 мкг Cd наблюдается при использовании ПАР и ксиленолового оранжевого [296, 297, 651].
Пробу помещают в мерную колбу емкостью 50 мл, приливают 0,5 мл аммиачной буферной смеси с pH 10,2, 0,2—0,3 мл 1-10-3 М раствора ПАР и разбавляют до метки. Аликвотную часть раствора 5 мл титруют комплексоном III при 495 нм.
При определении с ксиленоловым оранжевым к пробе, помещенной в мерную колбу емкостью 50 мл, приливают 5 мл 20%-ного раствора уротропина, 1—2 капли 6 N HNO3, 0,2—0,5 мл 1 10-3Л7 раствора индикатора и титруют аликвотную часть 5 мл раствором комплексона III при 575 нм [296, 297].
Точки эквивалентности рассчитывают по уравнению
_ (ч3 — щ) — Х1 (газ — п2)
Хп----------------------- ,
^2 — Гц ’
где ха — эквивалентный объем титранта, мкл; и х2 — соответствующие объемы титранта до достижения точки эквивалентности, мкл; пхи п2— показания гальванометра, соответствующие объемам добавленного титранта, мка; п3 — то же после достижения точки эквивалентности, мка.
Для создания требуемой кислотности среды уротропин заслуживает предпочтения по сравнению с ацетатным буферным раствором [651]. Ксиленоловый оранжевый и уротропин использованы для обратного титрования 4—20 мкг Cd на фотометрическом титраторе [492].
Для последовательного определения 0,3—2,8 мг Cd и 0,1— 0,6 мг Zn в одном растворе применено титрование 0, 0'-диаминодиэтилгликолевым эфиром N, N, N', N'-тетрауксусной кислоты по мурексиду при 450 нм [562]. Для определения цинка в присутст
81
вии 3300-кратного избытка Cd (маскируют иодидом калия KJ) предложено титрование диэтилентринитрилопентауксусной кислотой по ксиленоловому оранжевому (на фототитраторе при 570 нм) [560].
Для титрования комплекса кадмия (а также Mg и Zn) с 8-оксихинолин-5-сульфокислотой, флуоресцирующего при 495— 515 нм, предложен метод, сочетающий фотолиз с флуориметрией. Он основан на фотохимической генерации ионов Со2+ (из Со2(С2О4)з), вытесняющих кадмий из его комплекса с образованием нефлуоресцирующего соединения. Для фотолиза и возбуждения флуоресценции использован один и тот же источник ультрафиолетового излучения [491].
Микрогетерометрическое титрование диэтилдитиокарбамина-том в ацетатной среде по реакции
CdSO4 + 2 (С2Н6)2 NCS2Na Cd [(С2Н5)2 NCS2]2 + NaaSOj
позволяет определять 0,5 мг Cd в 20 мл раствора. Конечную точку находят по изменению степени помутнения жидкости. При введении тартратов, цитратов, комплексона III и других комплексообразующих агентов можно анализировать пробы, содержащие до 99,9% Al, Ba, Са, Cr, Fe, Mg, Мп, Pb, Sb и Zn [508].
При титровании в диметилформамиде и в диметилсульфоксиде достигается точность -4- 0,7%.
К 0,5 мл раствора, содержащего 1—12 мкг Cd (Мп, Zn), прибавляют 0,03 мл 0,02%-ного раствора индикатора (ПАН, эриохром черный Т) и титруют 2-10“4 М раствором комплексона Ш (или питрилотриуксусной кислоты) в тех же растворителях до перехода окраски [616].
В другой работе раствор дитизоната Cd или его смеси с дитизонатами Си и Zn в СНС13 титруют 2,4-10-3 М раствором диэтилдитиокарбамината натрия в этаноле. Точку эквивалентности устанавливают по изменению оптической плотности жидкости при максимуме поглощения дитизона (615 нм) или ди-тпзонатов (530 нм).
Метод позволяет определять 1—13 мкг С<Умл [584].
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Фотометрические методы
Кроме очень немногих неорганических соединений для фотометрического определения кадмия используют главным образом его органические комплексы, принадлежащие к указанным в гл. II основным группам. Методы, основанные на светопоглощении растворов этих соединений, позволяют определять п-10'1 — п-102 мкг Cd, большинство флуориметрических способов — от п-10'3 до целых единиц микрограммов Cd. Обзор литературных данных по фотометрическому определению кадмия с некоторыми органическими реагентами приведен в работе [372а].
82
Абсорбциометрия
Из неорганических соединений для определения кадмия были использованы сульфиды и соль Рейнеке; основные органические реагенты, предложенные для этой цели, сведены в табл. 15.
Неорганические реагенты
Сероводород, образующий в разбавленных слабокислых (и аммиачных) растворах солей кадмия желтый коллоидный сульфид, по-видимому, первый реагент, использованный для колориметрического определения 2—10 мг Cd [588]. Фотометрирование при облучении "ртутно-кварцевой лампой позволило снизить определяемый предел до 0,02 мг Cd в 50 мл раствора [554]; при анализе органических веществ, после их разложения и повторного 3-кратного соосаждения с сульфидом меди, кадмий колориметрировали в присутствии KCN. Измерение на фотометрах типа Пульфриха при 430 нм растворов, содержащих защитный коллоид (желатин), применяли для определения от 0,2 мг Cd [565, стр. 274]. Эта реакция была использована в анализе минерального сырья. Медь предварительно выделяли с Na2S2O3, кадмий осаждали в виде ассоциата метилового фиолетового с иодокадматом и после озоления и растворения осадка колориметрировали визуально со шкалой эталонов в пределах 0,01—0,1 мг Cd, просматриваемых по вертикальной оси пробирок (эталоны готовили из CdS или растворов бихромата аммония и хлорида кобальта, имитирующих его окраску) [340, 341].
Соль Рейнеке дает с тиомочевинным комплексом кадмия розовый осадок, легко растворимый в кетонах с образованием красной окраски.
Осадок комплекса (полученный так же, как для гравиметрического определения) отфильтровывают через стеклянный фильтр, промывают и еще влажным растворяют при добавлении тиомочевины и по каплям — метил-этилкетона, содержащего 2% тиомочевины. При измерении интенсивности окраски на фотометрах Лейтца или Цейса при 530 нм можно определять 1-10-1 — га-101 мг Cd [565, стр. 298].
Возможно определять кадмий фотометрически в виде соединений, аналогичных соли Рейнеке: Gd[Gr(SGN)4 (анилин)2]2, [Сй(еп)2][Сг(ЗСК)4(анилин)2]2 [Сй(пиперазин)][Сг(ЗСК)4(анилин)2]2, [Сй(дипиридил)2][Сг(ЗСК)4(анилин)2]2 и [Сй(1,10-фенантролин)2] • •1Сг(ЗСН)4(анилин)2]2 [572].
Органические реагенты
2,2 ' - бис-Б ен зо ти азо л ил образует с кадмием комплекс, окраска которого устойчива 25 мин. При использовании маскирующих реагентов можно определять кадмий в присутствии значительных количеств элементов, указанных в табл. 15. Метод применен в анализе металлического цинка [101].
83
Абсорбциометрические
Реагент pH Экстрагент « й и к к Константа нестойкости комплекса
2,2'-бис-Бензотиазолил <шс-(4-Натрийтетразо-лпл-азо-5)-этилацетат Бромбензтиазо 13,2—14,6 8 3% NaOH СНС13 + + C5H5N (4 : 1) С0Н4(СН3)2 1 : 1 1 : 1 1 : 2 1,54-10-1°
Бромпирогаллоловый красный 7,8—8,1
Глиоксаль-бис-(2-окси-анил) 2,2'-Дипиридил + FeSO4 1 ^-Дипиридил-З-^-ти-азолил) формазан Дитизон 10,6—13,6 6,5, 0,1 М KJ 8,5 5% NaOH СНС13 + + C5H5N (5:1) Дихлорэтан СНС13 1 : 2
Дпэтилдитиокарбаминат Иодэозин 5,6% NH4OH + 4- 0,6% NH4C1 5-11 8,5 СНС13 СС14 СНС13
Кадион 0,2# КОН
Кадион ИРЕА 9,4, (СН3)2СО
Кислотный хром темносиний Кристаллический фиолетовый Ксиленоловый оранжевый3* 10—11 o,im h2so4+ ф0,0Ш KJ 6,2 1 : 1 1,7-10-8 2-10-4
N-М етиланабазин-а'-азоазатол N-Метиланабазин-а'-азо-а-нафтол 8,5—9 9—11 СНС13 СНС13 1 : 3 1 : 4 6,012-Ю-18
+Метиланабазин-(а'-азо-7)-8-оксихинолин 5—6 2 : 1 1,032-Ю-1
Метиловый фиолетовый 0,1# НС1 + + 22,5% KJ
84
Таблица 15
методы определения кадмия
- Окраска комплекса Л, нм еЛ Конечный объем раствора, мл Определяемое количество Cd, мкг Мешающие 'элементы Литература
Краснофиолетовая 370, 570 500 580 3 800 10 000 18 200 10 4 4—120 0,1—5,0 Ag, Со, Си, Hg, Mg Со, Си, Ni, Zn [ioi] [439] [51, 98, 469]
Синяя » 620 610 526 10 000 25 10 10 20,0—240,0 1,0—40,0 4,0—28,0 Al, Со, Си, Fe (III), Мп (И) Pb, Zn Со, Ni Hg [102] [308, 672J [516, 782]
625 . 57 500 (Ю) (0,6—22,0) Со, Си, Ni, Zn [427]
Розовокрасная 520 260 262 80 000 32 70G 15 50 0,25—10,0 10,0—100,0 Al, Со, Си, Сг, Fe, Мп, Ni, Zn Многие [66, 150, 280, 290, 661] [510, 716] [510]
Красная Оранжево-красная Красная 540 460 410 555 87 600 48 000 6 25 5 0,25—8,0 0,05—0,7 0,5—8,0 Сг, Ее, Ga, Hg, Мп, Ni, Pb, Zn Ag, Co, Си, Cr, Hg (I), Hg(II), Mg, Ni Al, As,Bi,Co,Cr, Cu(II), Fe(HI), Mn, Ni, Zn Mg, Zn [256, 257] [397] [232] [228]
Синефиолетовая Фиолетовая Красная 580 580 570 550, 590 130 000 27 500 6 400 21 150 10 25 25 10 0,1—3,0 5,6—56,0 5,0—30,0 0,5—7,0 Bi, Sn (IV) Си, Fe (II), Fe (III), Ga, Pb, Tl, Zn [533] [25, 639] [9, 474] [9, 474]
» Сине-зеленая 540 11 360 25 2 5,0—50,0 0,05—1,0 * Al, Си, Fe (II), Fe (III), Ni, Pb, Zn [9, 474] [195, 461]
85.
Реагент pH Экстрагент Состав комплекса Константа нестойкости комплекса
1-(2-Пиридилазо)-2-нафтол 4-(2-Пиридилазо)резор-цин 70% этанола + + 0,2% бутил- амина 9,8—10,4 Дихлорэтан 1 : 2
Пирролидиндитиокар-баминат аммония Родамин С Сульфарсазен Хромпиразол II Эозин Ацетатная среда 0,3AH2SO4 + + 0,1 N KJ 9,5—10,0 2 N НС1 + + 10% KBr 1—2 N H2SO4+ + 0,5% KJ 8,0 CHC13 C0H0 СНС13 1 : 2 1 : 1
Эритрозин 8,5 СНС13
Бромбензтиазо [6-бромбензтиазо-(2-азо-1)-2-нафтол] реагирует с кадмием, образуя комплекс, устойчивый в течение многих часов. Кривые светопоглощения комплекса и реагента приведены на рис. 8.
К слабокислому или нейтральному раствору кадмия прибавляют 4 мл ксилола, 0,2 мл 0,0001 М раствора бромбензтиазо в ксилоле, 20 мл 10%-ного раствора NaOH, встряхивают 1—2 мин. и фотометрируют по разделении фаз.
При определении 1 мкг Cd в присутствии 0,5 мкг А1 и Hg2+ в раствор добавляют 0,5 мл 20%-ного раствора Na2S3O3. Влияние Mg устраняют введением 0,5 мл раствора сегнетовой соли. В присутствии 250-кратных количеств Со определение кадмия возможно при добавлении нитрозо-Р-соли. Влияние 1000-кратных количеств Сг3+ устраняют прибавлением 0,1 лиг 25 %-ного раствора цитрата. Влияние Со, Си, Ni и Zn устраняют пропусканием 0,5 N солянокислого раствора через колонку с анионитом ЭДЭ-10П, кадмий при этом сорбируется на колонке [469].
Определение кадмия возможно в присутствии цинка при их соотношении 1 : 200; в рекомендуемых условиях определению кадмия не мешают 500—1000-кратные количества As3+, Be, Bi, Са, Cr, Fe3+, La, Mn2+, Mo, Pb, Pt, Sb3+, Sc, Th, Ti, U6+, V4+, W и 250-кратные— Ba, Ga, Ge, Pd, Sb5+ и Sn4+ [51, 98].
89
Таблица 15 (окончание}
- Окраска комплекса Л, нм Ел 1 "Конечный объем* ра-i створа, мл Определяемое количество Cd, мкг Мешающие элементы Литература
555 50 5,0—30,0 [447, 497}
495 84 000 Со, Си, Fe(III), Ga, Hg, In, Mn, Ni, Pb, [172]
Sn (IV), Y, Zn
262 35 000 25 0,01—0,15 Многие [610]
540—550 74 000 (610 нм) 25 - 0,2—15,0 Bi, Си (II), Hg(II), Sb [219]
510 51000 5 0,2—15,0 Hg, Zn [316, 372]
100 10,0—100,0 Многие [119, 125]
25 5,0—40,0 » [119, 125]
Красная 530 89 900 6 0,25—6,0 Cr, Fe, Ga, Hg, Mn, Ni, Pb, Zn [256, 257]
» 530 87 600 6 0,32—8,0 Cr, Fe, Ga, Hg, Mn, Ni, [256, 257]
Pb, Zn
Бромпирогаллоловый красный реагирует в присутствии 1,10-фенантролина с ионами Gd2+.
К раствору кадмия прибавляют 2 мл 2%-ного раствора аравийской камеди, 1 мл 0,05%-ного раствора 1,10-фенантролина, 10 мл 0,01%-ного раствора реагента, разбавляют водой до 25 мл и спектрофотометрируют через 15 мин. [102].
Глиоксаль -б и с-(2-о ксианил) в присутствии пиридина образует экстрагирующийся комплекс. При содержании мешающих элементов до 1 мг их маскируют прибавлением 1лм5%-ного-раствора KGN с последующим демаскированием Gd 1 мл 10%-ног» формалина. Реакция использована при анализе металлического' цинка [672, 308].
п, п'-Д иметилдитизонол при pH 3 образует с кадмием комплекс с максимумом поглощения при 525 им [755].
д и-0-H афтилтиокарбазон по своим аналитическим, свойствам очень близок к дитизону, но не растворяется в водных растворах щелочей и поэтому может быть использован, после экстракции кадмиевого комплекса из раствора NaOH, для его колориметрирования по методу смешанной окраски; присутствие
«7
Рис. 8. Спектры поглощения бромбензтиазо в ксилоле (I) и его комплекса
-с кадмием (2)
Рпс. 9. Спектры поглощения дитизона в четыреххлористом углероде (I) и его комплекса с кадмием (2)
реагента в органической фазе на конечной стадии определения повышает устойчивость ее окраски [363].
2,2'-Д ипириди л 4- сульфат железа (II) в присутствии иодида образуют с кадмием ионный ассоциат следующего состава: [CdJ4][Fe(G10H8N2)3].
К раствору кадмия прибавляют по 2 мл 0,008 М раствора реагента, 0,002 М раствора соли Мора и 3 М раствора CH3COONa, затем 7 мл 0,3 М раствора KJ и устанавливают нужное значение pH. Разбавляют до 25 мл и экстрагируют 10 мл 1,2-дихлорэтана в течение 4 мин, После расслаивания (~20 мин.) экстракт обезвоживают добавлением 1 г Na2SO4 и фотометрируют по отношению к раствору холостого опыта в кювете с толщиной окрашенного слоя 1 см [621, 782].
1,5-Д ипириди л-3-(2-т иазолил) формазан предложен для фотометрического определения кадмия в присутствии многих металлов [427].
Дитизон (1,5-дифенилтиокарбазон) образует с кадмием в-области от pH 6—8 (в зависимости от присутствия комплексообра-зователей) и до pH 14 розово-красный комплекс, экстрагирующийся органическими растворителями (рис. 9). Это наиболее распространенная реакция для определения малых количеств кадмия. В нейтральной среде с дитизоном реагирует более десятка элементов (см. стр. 148).
В СНС13 растворимость дитизоната кадмия выше, чем в СС14 (1,3 -10~4 и 1,3• 10-8 молъ[л соответственно); в первом растворителе больше и его устойчивость на свету (несколько дней; во втором — всего 30 мин.) [150, 363, 459]. Исследована экстракция дитизоната Cd (а также РЬ и Zn) и другими экстрагентами [17].
При определении кадмия в чистых растворах или в присутствии Al, As,. Bi, Fe, Мп, Pb, Sb, Sn, Zn, щелочноземельных и щелочных металлов, к 3— 30 мкг Cd в 5—10 мл почти нейтрального раствора, содержащего достаточное-количество тартрата, приливают равный объем 10%-ного раствора NaOH. Экстрагируют малыми порциями 0,005 %-ного раствора дитизона в ССЦ до тех пор, пока последняя порция экстрагента после встряхивания в течение 30 сек. не станет вместо красной — бесцветной. Объединенные экстракты разбавляют до определенного объема ССЬ, дважды промывают половинными объемами 2%-ного раствора NaOH, один раз водой и сразу же фотометрируют при 520 нм.
В связи с недостаточной устойчивостью дитизоната кадмия в ССЦ можно разрушить этот комплекс (с освобождением эквивалентного кадмию количества дитизона) встряхиванием экстракта с 0,5 N НС1 и фотометрировать его при 620 нм [150, 509].
В присутствии Ag, Bi, Со, Си, Hg, Ni, Pb, Т1 и Zn к 25 мл слабокислого или нейтрального раствора, содержащего не более 1000 мкг Cd, прибавляют при перемешивании 1 мл 2,5%-ного раствора тартрата калия, 5 мл 1%-ного раствора KCN в 40%-пом растворе NaOH, 1 мл 20%-ного раствора хлорида Iгдроксиламина и экстрагируют 1 мин. с 15л«л 0,008%-ного раствора дитизона в СНС13. Экстракт сливают в делительную воронку, содержащую точно 25 мл 2%-ного раствора винной кислоты. Оставшийся водный слой вновь взбалтывают с 10 мл дитизона, который присоединяют к первому экстракту. Следят за тем, чтобы водная фаза (которая должна иметь желтый цвет) не попала в растворы дитизоната. Объединенные экстракты взбалтывают 2 мин. с виннокислым раствором и после отделения промывают этот последний (в который перешел кадмий) 5 мл СНС13. Аликвотную часть водной фазы (или всю, если она содержит <J10 мкг Cd) разбавляют до 25 мл 2%-ным раствором винной кислоты, прибавляют 0,25 мл раствора гидроксиламина, 15,0 мл 0,0008%-ного раствора дитизона, 5 мл 0,05%-ного раствора KCN в 40 %-ном растворе NaOH и взбалтывают 1 мин. После разделения фаз переливают экстракт (если он содержит воду, то через комочек ваты) в кювету и измеряют оптическую плотность при 520 нм [459; 619, стр. 221].
При определении кадмия ц растворах, содержащих по 10 мг-каждого из 70 наиболее распространенных элементов Периодической системы, его предварительно осаждают из аммиачно-цитрат-ного раствора бензотриазолом (с Ni в качестве коллектора). Солянокислый раствор осадка пропускают через колонку с ионитом и в элюате определяют кадмий с хлороформным раствором дитизона-1589]. При экстракции трибензиламином кадмий отделяется от А1; Со, Cr, Си, Fe, Мп и Ni [66]. Добавление 10 мл 20%-ного раствора тартрата устраняет влияние 50 мг Mg [280].
При анализе металлического индия кадмий отделяют экстракцией в виде пиридин-роданидного комплекса хлороформом [290]. Определение кадмия в таллии проводят после предварительного осаждения последнего роданидом и последующей экстракции кадмия в виде пиридин-роданидного комплекса [289], в металлическом хроме — после предварительного отделения мешающих элементов на анионите [390[. Определение окиси кадмия и свободного металла в его селениде проводят экстракцией дитизоната из 2,5 N раствора NaOH [422]. При анализе платино-родиевых сплавов, мешающие элементы сорбируют на катионите Амберлит IR-120 [649]. Дитизон применен для определения кадмия в сульфиде цинка высокой чистоты [166], металлическом висмуте [124], едком нат
89
ре [578| и многих других объектах [21, 150, 281, 288, 452, 648, 650, 661]. Широко используют дитизон для определения кадмия и в природных водах [216, 248, 249]. В щелочной среде кадмий (а также Hg2+ и Zn) образует с ди-(п-сульфофенил)тиокарбазоном малиновое соединение, которое можно использовать для фотометри-рования и в качестве мет'аллохромного индикатора [329].
Дифенилдитиофосфат кадмия, осажденный так же, как для гравиметрического определения, растворяют в СНС13 и обрабатывают раствором CuSO4, которая вытесняет кадмий из органического слоя. Интенсивность окраски последнего пропорциональна первоначальному содержанию кадмия [58].
При добавлении в раствор по 1 а тартрата и цитрата можно определять кадмий с диэтилдитиокарбаминатом в присутствии железа и некоторых других элементов. Ag, Au, Bi, Со, Си, Мп, Ni, Sn и Zn мешают определению.
Экстрагируют сначала 25 мл, затем 15 мл СНС13. Разбавляют объединенные органические фазы до 50 мл и спектрофотометрируют в кювете с I = 1 см (при таком объеме экстракта, очевидно, можно использовать и более толстые кюветы и тем самым снизить предел чувствительности определения) [41]. В СС14 комплекс кадмия извлекается из водной фазы в области pH 5—И, реэкстрагировать его можно 1 N НС1 [510]. При косвенном определении кадмия бесцветный экстракт в СНС13 обрабатывают раствором CuSO4 и образующийся в органической фазе коричневый комплекс меди (в количестве, пропорциональном содержанию кадмия) фотометрируют при 535 нм (полоса поглощения диэтилдитиокарбамината меди находится в области 300—800 нм, Хтах—440 нм) [761].
Диэтилдитиофосфат натрия взаимодействует с кадмием при pH 4—6 с образований красного комплекса, экстрагирующегося СС14. Эта реакция позволяет определять микроколичества Cd в присутствии 104 — 107-кратных количеств Zn [509].
К а д и о н (n-нитрофенилдиазоаминоазобензол) образует с ионами Gd2+ лак, спектр поглощения которого представлен на рис. 10, а; окраска развивается в течение 15—20 мин. и остается неизменной 30—40 мин. Ионы Ag+, Hg+, Hg2+ дают в условиях определения кадмия оранжево-бурые осадки; синюю окраску, вызываемую ионами Go, Cr, Си, Mg и Ni, можно предотвратить добавлением тартрата калия — натрия.
Для определения кадмия к растворам прибавляют 2 мл 0,2 %-ного желатина, 0,75 мл 0,02%-ного раствора кадиона в 0,02 N растворе КОН, 7 мл 2 N раствора КОН, 6—8 мл воды, переносят раствор в мерную колбу на 25 мл и доводят до метки водой [252, 347, 406].
К а д и о н ИРЕА (4-нитробензол-1",4-диазоамино-1,Г-азо-бензол-2",4-дисульфокислота) реагирует с кадмием в 0,05 N растворе карбоната и 0,025 А тетрабората натрия, содержащего ацетон; спектр поглощения комплекса приведен на рис. 10, б [232, 444]. Окрашенные соединения с кадионом ИРЕА образуют также 90
Рис. 10. Спектры поглощения
а — водный раствор кадиона (г) и его комплекса с кадмием (2); б — водный раствор кадиона ИРЕА (Г) и его комплекса с кадмием (г)
указанные в табл. 15 мешающие элементы при их содержании 1— 5 мкг. Влияние А1 устраняют введением в раствор фторида, a As5+ и Сн2+ (до 100 мкг) — Na2S2O3. В присутствии железа определение кадмия с кадионом ИРЕА без предварительного отделения невозможно. В условиях определения кадмия не образуют окрашенных соединений Ba, Be, Hg, К, Mg, Mo, Na, Pb, Tl, W, Zr; мешают определению хлориды, сульфаты и нитраты.
К 1—2 мл раствора кадмия прибавляют 1,5 мл ацетона, затем раствор разбавляют буфером с pH 9,4 до 4,5 мл. После перемешивания добавляют 0,5 мл 0,02%-ного раствора кадиона ИРЕА и через 5 мин. измеряют интенсивность окраски [232].
Кислотный хром темно-синий [Г,8-диокси-2-(2-оксиазобензол)-3,6-нафталиндисульфонат натрия] образует с кадмием комплекс, максимальное развитие окраски которого происходит через 1—1,5 часа после прибавления реагента.
Для образования комплекса к 2—3 мл раствора кадмия прибавляют 5 мл хлоридно-аммиачного буфера, 10 мл 0,016%-ного раствора реагента и фотометрируют полученный раствор [228].
Кристаллический фиолетовый взаимодействует с иодидом кадмия с образованием осадка, флотируемого изопропилоксидом. Сине-фиолетовый раствор осадка в ацетоне используют для спектрофотометрического определения кадмия. Проведение этой реакции возможно в присутствии 15-кратных количеств Си, 500-кратных А1, 2500-кратных Fe2+ и Fe3+ и 15000-кратных Zn; Bi3+ и Sn2+ должны отсутствовать.
91
К 15 мл нейтрального раствора кадмия прибавляют 1 мл 1 TV H2SO4, 1 мл 0,1 N раствора KJ и 1 мл 0,04%-ного раствора реагента, разбавляют водой до 10 мл, приливают 10 мл изопропилоксида и встряхивают в течение 5 мин. После тщательной декантации водного слоя осадок кадмия растворяют в 10 мл ацетона и фотометрируют по отношению к раствору холостого опыта в кювете с I = 1 см [533J.
С ксиленоловым оранжевым [3,3'-d«c-[N, N'-ди-(карбоксиметил)аминометил]-о-крезолсульфофталеином (спектр поглощения его комплекса с кадмием дан на рис. И ) возможно определение кадмия в присутствии 20 ммолей цитратов, 500 ммолей сульфатов и хлоридов, 100 ммолей роданида, 40 ммолей фосфатов и фторидов. При содержании 60 ммолей фторида калия в качестве маскирующего агента можно определять 50 мкг Cd в присутствии 0,5 ммолей РЗЭ и 0,04 ммолей Th4+ и Zr1+. Не мешают определению Ag, Сг3+, Мо и W при их соотношении с кадмием 50 : 1, a Bi3* и 1п3+ — при соотношении 2:1.
Рис. 11. Спектры поглощения водных растворов ксиленолового оранжевого (1, 3) и его комплекса с кадмием (2, 4) при pH 6,3 (1, 2) и 3,6 (3,
В мерные колбы емкостью 25 мл, содержащие раствор 5—50 мкг Cd, вводят 10 мл 20%-ного раствора уротропина, подкисляют до pH 6,3 и прибавляют 2 мл 0,1%-ного раствора реагента. Содержимое колб доводят до метки водой и через 15 мин. измеряют оптическую плотность.
Окраска раствора устойчива в течение двух часов и не изменяется при нагревании до 70° С. Пирофосфат, комплексон III и оксалат в соизмеримых количествах обесцвечивают комплекс. Присутствие в растворе нитратов снижает оптическую плотность кадмиевого комплекса [25, 639].
N-М етиланабазин -а'-а з о-a з а т о л «ПА» взаимодействует с кадмием, образуя соединение, окраска которого развивается в течение 10—20 сек., после чего остается неизменной.
С N-м етиланабази н-а'-а з о-а-н а ф т о л о м кадмий реагирует практически мгновенно и в течение часа окраска комплекса остается неизменной.
Определению не мешают (предельно допустимые отношения к кадмию даны в скобках) А1 (920), Ag (55), As (175), Be (214), Cr (84), Mg (1000), 400-кратный избыток фторидов, 700-кратный оксалатов и 100-кратный янтарной кислоты; комплексон III полностью маскирует кадмий.
92
В делительную воронку, содержащую кадмий, приливают 1 мл 0,1 %-ного раствора реагента в этаноле, 1,5 мл буферного раствора с pH 11 (смесь 0,1 М раствора борной кислоты и 0,1 М раствора Na2CO3), 10 мл СНС1з, встряхивают в течение 1 мин. и фотометрируют экстракт [9].
N-М етиланабази н-(а'-а з о-7)-8-о ксихинолин образует с кадмием соединение, окраска которого развивается сразу после сливания растворов и устойчива в течение 20 мин.
При определении в отсутствие мешающих ионов к растворам кадмия добавляют 2 мл 0,2%-ного раствора красителя, 10 мл буферного раствора с pH 5,3, разбавляют водой до 25 мл и фотометрируют. Влияние Си устраняют тиомочевиной, А1 — фторидом [9, 474].
Метиловый фиолетовый. При определении кадмия в его сульфиде к раствору прибавляют 0,2 мл 0,06%-ного раствора реагента, 0,3 мл 1 N НС1, 0,2 мл 25%-ного раствора KJ. Доводят объем до 2 мл водой и через 15—20 мин. фотометрируют взвесь образовавшегося комплекса.
Ошибка определения кадмия 20% [461].
Моноацетат 2-(2-о к с и-5-н итрофени л-а з о)-4,5-дифенилимидазол при pH 5,5 взаимодействует с ионами кадмия, образуя хелаты состава Me : Р = 1 : 2 с максимумом поглощения при 630 нм', продукт реакции экстрагируется изопентанолом и хлороформом. Сам реагент при pH 3,6—4,6 имеет максимальное поглощение около 470 нм [657].
1-(2-П и р и д и л-а з о)-2-н а ф т о л (ПАН) используют после экстракции кадмия диантипирилметаном в дихлорэтане.
Пробу, содержащую 5—20 мкг Cd, растворяют в НС1, раствор упаривают до 4—5 мл и прибавляют 40 мл 20%-ного раствора LiCl. Экстрагируют 1 — 2 мин. 10 мл 0,1 М раствора диантипирилметаиа в дихлорэтане. После разделения фаз к органическому слою прибавляют 5 мл 2%-ного спиртового раствора бутиламина н 2 мл 0,2%-ного раствора реагента, разбавляют этанолом до 50 мл и измеряют оптическую плотность [252, 447].
Ошибка определения кадмия в чистых растворах 3,5% [699].
4-(2-П иридилаз о)р е з о р ц и н (ПАР), 4-(2-т и а золи л а з о)-2-н итрорезорцин и 4-(5-с у л ь ф о-2-т и а-з о л и л а з о)р е з о р ц и н [1, 591] изучены в качестве спектрофотометрических реагентов на кадмий.
Родамин С (тетраэтилдиамино-о-карбоксифенилксантенил-хлорид) взаимодействует с иодидным комплексом кадмия с образованием ассоциата [219]. Спектры поглощения взвеси образующегося соединения — [CdJ4](P)2 — и реагента приведены на рис. .12. Наибольшая разница оптических плотностей между комплексом и родамином С наблюдается при 610 нм. При повышении температуры светопоглощение растворов уменьшается, что связано с изменением растворимости соединения. В присутствии винной, лимонной и щавелевой кислот, гидроксцламина, тиомочевины и тиосульфата чувствительность реакции снижается. Со, Ni и Zn мешают определению в количествах более 10 мг; ионы Bi3+, Cu2+, Hg2+ и Sb3+ завышают результаты, их отделяют экстракцией дихлорэтаном из щелочных или слабокислых растворов в виде ди-
93
Рис. 12. Спектры поглощения родамина С (1) и суспензии его комплекса с иодидом кадмия (2)
Рис. 13. Спектры поглощения водных растворов сульфарсазена (1) и его комплекса с кадмием (2)
этилдитиокарбаминатов. При проведении реакции с родамином С необходимо строго соблюдать порядок сливания растворов.
К 10 мл нейтрального раствора кадмия приливают 2 лиг 3,6 N H2SO«, добавляют 2 мл 0,08%-ного раствора родамина С, 2,5 мл 1 N К J, разбавляют водой до 25 мл и через 10 мин. фотоколориметрируют в кюветах с I ~ 50 мм.
Метод применен при определении кадмия в алюминии, магнии и хроме [219]. |
Сульфарсазен (4-нитробензол-1", 4-диазоамино-1, Г-азобензол-2'-арсоно-4"-сульфонат натрия). В цитратно-аммиачном буферном растворе реагирует с кадмием; спектры поглощения комплекса и реагента даны на рис. 13. Определению 5 мкг Cd не мешают 2 мкг Al, Со, Си, Ni, Mn, Pb, Sn и U; 10 мкг Ag; 25 мкг Fe3+, Ti, Zr; 50 мкг Be и La; 500 мкг Мо6+, V5+ и V1+. В при-' сутствии sg; 1000 мкг Zn кадмий предварительно экстрагируют дитизоном [316, 372].
К 2,75 мл нейтрального или слабокислого раствора кадмия прибавляют 0,5 мл 4%-ного раствора лимонной кислоты, 0,75 мл 0,05%-ного раствора сульфарсазена, 1 мл 2%-ного NH4OH и спектрофотометрируют при 520 нм. Оранжево-желтая окраска раствора устойчива в течение многих часов. Аналогичную цветную реакцию с сульфарсазеном дают Hg и Zn.
Тиотеноилтрифторацето н, растворенный в СС14, извлекает кадмий из водной фазы с pH 3 и образует с ним окрашенный комплекс с максимумом поглощения около 430 нм;
94
окраска экстракта устойчива в течение 1—3 суток [496, 730].
Хромпиразол II [бнс-(4-метилбензиламинофенил)анти-пирилкарбинол] используют для определения кадмия в двух вариантах — «бромидном» и «иодидном» [119, 120, 125, 126, 532]. В бромидном варианте осадок кадмий-бромидного комплекса с реагентом растворяют в соляной кислоте и измеряют его оптическую плотность. Иодидный метод основан на фотометрировании бензольного экстракта избытка реагента, окраска которого уменьшается с увеличением количества кадмия (образующего с ним нерастворимый в бензоле комплекс). Определению мешают Bi, Hg, Sb, Sn и большие количества Zn. В присутствии меди применяют бромидный вариант, в присутствии цинка — «иодидный». По обоим вариантам анализ возможен в растворах, содержащих Al, Be, Со, Cr, Си, Fe, Mg, Мп и Ni [335].
При определении кадмия по бромидному варианту к 20 мл анализируемого раствора, 1—2 jV ио H2SOa, медленно при перемешивании приливают 15 мл смеси равных объемов 0,1 %-ного раствора хромпиразола II в 4 jV H2SO« и 20%-ного раствора КВг и оставляют стоять на 40—60 мин. Осадок отфильтровывают через воронку с пористым фильтром № 3 или № 4, врзможно полнее отсасывают маточный раствор и, не промывая осадка, растворяют его в 5—10 мл ацетона или этанола. Раствор отсасывают, фильтрат количественно переносят 2 N НС1 в мерную колбу емкостью 100 мл, доводят до метки такой же кислотой и измеряют оптическую плотность с зеленым светофильтром в кювете с Z = 20 мм [126, 335].
В иодидном варианте к 0,5—1 jV сернокислому раствору кадмия прибавляют KJ до его концентрации 0,5%, несколько кристалликов аскорбиновой кислоты (для предотвращения окисления иодида) и приливают при помешивании 4 мл 0,02%-ного раствора хромпиразола II в 2 N H2SOa. Через 3— 4 мин. вводят 25 мл бензола, взбалтывают смесь влечение 7 —10 мин. на механической качалке, дают отстояться и измеряют оптическую плотность бензольного слоя [335]. В обоих вариантах калибровочные графики строят для каждого типа анализируемых объектов (А1-, Mg- и других сплавов) отдельно [126, 335].
Имеются указания на образование окрашенных комплексов кадмия с 1-(2-т и а з о л и л-а з о)-2-о к с и-3-н афтой ной кислотой при pH ~ 9 [59] и с динитрофениламинороданином [188].
Галоидопроизводные флуоресцеина (эозин— тетрабром-, иодэозин — тетраиодпроизводное, эритрозин — его натриевая соль) в присутствии 1,10-фенатролина реагируют с кадмием. С уменьшением кислотности водной фазы оптическая плотность экстрактов сначала увеличивается, а затем начинает снижаться (рис. 14); максимальное развитие окраски наблюдается при pH 8-9 [256, 257].
К растворам кадмия с pH 8—8,5 прибавляют 1—3 мл (в зависимости от содержания элемента) 0,2%-ного раствора 1,10-фенантролина, 0,2 мл 0,01 %-ного раствора реагента, разбавляют водой до 10 мл, экстрагируют 6 мл хлороформа и сиектрофотометрируют [257].
95
Рис. 14. Влияние кислотности водной фазы на оптическую плотность экстрактов холостого опыта (1) и фенантролпната кадмия с эозином (2)
Рис. 15. Спектр флуоресценции комплекса кадмия с 8-(бепзолсульфаниламп-но)-хинолином (.7 — относительная интенсивность флуоресценции)
Флуориметрия
Флуоресцентных реакций на кадмий значительно меньше, чем цветных. Предложенные реагенты относятся преимущественно к азотсодержащим гетероциклическим соединениям. Некоторые оксиксантеновые красители также дают с кадмием ярко флуоресцирующие соединения, которые можно использовать в аналитических целях. Краткая характеристика флуоресцентных реакций кадмия приведена в табл. 16.
8-(Б ензолсульфониламин о)х и н о л и н образует с кадмием комплекс, спектр флуоресценции которого представляет бесструктурную полосу в пределах длин волн 440—600 нм (рис. 15). Наибольшая яркость свечения достигается через 3—5 мин. и сохраняется неизменной в течение 15—30 час. Соединение кадмия извлекается многими органическими растворителями, но при экстракции хлороформом интенсивность флуоресценции максимальна. Со, Си, Ni при их содержаниях, равных кадмию, снижают яркость флуоресценции его комплекса; мешают проведению реакции 10-кратные количества Cr, Fe, Hg, Sb, Sc и 100-кратные — Al,Be, Ce, In и Zr [335].
К фосфатному буферному раствору кадмия с pH 8 добавляют 0,2 мл 0,01 %-ного ацетонового раствора реагента и измеряют интенсивность зеленой флуоресценции водного раствора или хлороформного экстракта.
Можно определять 1-10"7% Cd в HNO3 в присутствии 10-кратного избытка цинка [364].
3,5-6 и с-(Д икарбоксиметиламиномети л)-4,4'-д и о к с и-ш joawc-стильбен образует с кадмием, флуоресцирующий комплекс. Для избирательного определения кадмий предварительно экстрагируют диэтилдитиокарбаминатом натрия из винокислого раствора с pH И и затем реэкстрагируют 0,2 N НС1. Интенсивность флуоресценции устойчива в течение трех ча-
96
Л
Д' я и ю л ь
Флуориметрические методы определения кадмия
1 Литера- тура ।—1—, I—. lO i—i i—i ।—। ।—। lO i/Э s!< C5 CM CO Г- Г- CD M СЧ CD CD чН CO -чН CO CO Ю CD lO> i—“i F” l—“l *d CD l—“l l—“l uO 1/3 1/3 СЧ СЧ <M
Мешающие элементы Co, (fcu, Ni, Zn и др. Pb, Tl, Zn, РЗЭ Cr, Fe, Ga, Hg, Mn, Ni, Pb i Co, Cu, Ni и др. Ag, Al, Ca, Ce, Co, Fe, Ga, Mg, Mn, Ni, Pb, Zr, U Al, Ca, Ce, La, Mg, Sn, Th, Zn | Многие Cr, Fe, Ga, Hg, Mn, Ni, Pb, Zn Cr, Fe, Ga, Hg, Mn, Ni, Pb, Zn
Определяемое количество кадмия, мкг от 0,005 от 0,001 (0,5—10,0) 1 0,05—1,0 ; 100—2500 0,03—2,0 0,01-2,0 0,1—4,0 0,05—1,0 0,1—1,5
| Конечный объем раствора, мл i/3 -ч-i l/JT CD О i/Э О lO CD CD
1 я i/ЭО О О О l/З О О чН ОД t> I D О V- V- UOi/Э 1/3 1 1/3 1/3 1/3 1/3 ОО О О CD CD СО О О СОоО CD CD tD СО СО СО СЧ <М СО 1/3 со со СО 1/3 1/3
Цвет флуоресценции 3 о =Я § 1И § =я £ « й й К 3 о Д Й Д Я ± Я а Ф А ДО фЯфЙ н S Й м «5 сб 3 Д Л з О со и О СО О Я Е-1
pH 1/3 сч N N 'М м # # * сю 00 * * О О ОЗ 1/3 О | j О 1/3 со 00 Г- 00 со О О 00 00 t— 00
Реагент 8-(Бензолсульфаниламино)- хинолин*1 3,5'-бис-(Дикарбоксиметил-аминометид)-4,4'-диокси -транс-стильбен*1 Иодэозин 2-(о-Оксифенил)бензоксазол *3 1 -(8-Окси-2-хино лин)-3,5-ди-метилпиразол 8-Оксихинолин-5-сульфокпс-лота 8-(и-Тосиламино)^инолин Эозин Эритрозин
•« Состав комплекса 1 : 2, *2 Экстрагент — СНС1а. 3* Образовавшийся осадок комплекса кадмия растворяют в ледяной СНаСООН.
сов. Определению 10 мкг Cd мешают 2 мкг РЬ, 10 мкг Т1, 40 мкг ЭДТА. В присутствии до 6 мкг Zn кадмий можно определять без предварительного отделения с ошибкой, не превышающей 5% [519].
2-(о-О к с и ф е н и л)б ензоксазол («люмоген светло-зеленый») .
0,1—2,0 мг Cd осаждают так, как это описано при титриметрическом определении, промытый осадок высушивают 30 мин. при 130° С, растворяют в 50 мл ледяной СН3СООН и измеряют интенсивность голубой флуоресценции в УФ. При повышении температуры раствора яркость его свечения снижается [533].
1-(8-0 к си-2-хин ол ин)- 3,5- дим ети л пи р а зо л. В условиях взаимодействия с кадмием цинк — постоянный его спутник —не образует флуоресцирующих соединений [47]. Изменение концентрации КОН от 1,5 до 10% не отражается на интенсивности флуоресценции комплекса кадмия, экстракция его соединения хлороформом увеличивает интенсивность флуоресценции в несколько раз. Спектры поглощения и флуоресценции кадмиевого комплекса приведены на рис. 16. Максимальное свечение растворов с содержанием 0,03—2,0 мкг Cd в 5 мл развивается через 10—20 мин. и остается постоянным несколько часов. При облучении ртутно-кварцевой лампой со светофильтром УФС-3 за первые 5 мин. интенсивность свечения снижается на 1—2%, через 15 мин.— на 5—7%, а через 30 мин.— на 40%. При возбуждении лампой накаливания с первичным светофильтром из цветного стекла марок СС-5 ф- СЗС-22 яркость флуоресценции практически неизменна в течение 2 час.
Гасят флуоресценцию равные количества Се4+, Cr6+, Fe3+ и Zr1+, десятикратные: Ag+, Со2+, Fe2+ и Мп2+; остальные элементы в сто- и тысячекратных количествах. Mg образует с реагентом растворимое флуоресцирующее оранжевым светом соединение, а Са (при концентрации более 60 .и кг/5 мл) — флуоресцирующий зеленым цветом осадок.
К 5 мл раствора кадмия, 3%-пого по КОН, прибавляют 0,3 мл 0,01% -ного ацетонового раствора реагента, экстрагируют 3,5 мл СНС13 и флуори-метрпруют после разделения фаз.
Возможно определение 5-10”% Cd в хлоридах лития, натрия, калия и в других солях щелочных металлов, а также 5-10-5 — 5-10” % Cd в солях цинка [47].
Галоидопроизводныефлуоресцеина (эозин, иодэозин и эритрозин) образуют с кадмием в присутствии 1,10-фенантролина тройные комплексы, способные флуоресцировать в органических растворителях. Наиболее яркой флуоресценцией обладают хлороформные экстракты; интенсивность их свечения увеличивается с добавлением ацетона. Спектры флуоресценции комплексов приведены на рис. 17; наиболее яркое свечение, 98
Рис. 16. Спектры поглощения хлороформных экстрактов 1-8(-окси-2-хинолии)-3,5-диметилпиразола (7) и его комплекса с кадмием (2); спектр флуоресценции этого комплекса (3); спектры поглощения первичного и вторичного светофильтров (4, б)
Рис. 17. Спектры поглощения (I) и флуоресценция (2—5) экстрактов холостого опыта (2, 4) и фенантролината кадмия с эозином (3, 5), с добавлением ацетона (4, 5) и без него (2, 3) (спектры поглощения в присутствии и в отсутствие ацетона совпадают)
как и окраска, наблюдается при pH 8—9. Коэффициент распределения кадмия (с эозином) между водной и органической фазами имеет величину около 35. Как и в случае фотоколориметрического определения, эта реакция не специфична [256, 257].
Другие методы
Нефелометрия. Комплексный иодид кадмия образует с трифе-нилметиларсонием белый осадок [GdJ4][(G6H5)3CH3As]2. Содержание кадмия определяют по степени помутнения раствора. Нерастворимые иодиды или подобные же комплексы дают Ag, As, Bi, Си, Hg, Pb и Sb; для их удаления раствор предварительно кипятят с железной проволокой и фильтруют.
При определении 10—100 мкг Cd к 10 мл его нейтрального раствора прибавляют при перемешивании 2 капли раствора желатина, 5 мл раствора реагента и измеряют степень помутнения на нефелометре с нейтрально-серым светофильтром. Если в том же объеме содержится 100—1000 мкг Cd, то берут из него 2 мл, прибавляют желатин, 1 мл реагента и пефеломет-рируют в кювете с меньшей толщиной слоя.
Допустимо присутствие 100-кратного избытка Zn [552, 576].
Отмечена возможность нефелометрического определения кадмия ис ферроцианидом калия [363, стр. 320].
Поляриметрия. При осаждении K2[CdJ4| раствором сульфата хинина образуется оптически активный комплекс; его растворяют в 50 мл ацетона, измеряют угол (ж°) вращения плоскости поляризации и рассчитывают содержание кадмия (г/, мг) по эмпирическому уравнению [684]
у = 31,62 -х° -2,7628.
99
Комплексы Cd, Си и Zn с D-(—)-1,2-пропилендиаминтетраук-сусной кислотой обладают различным молекулярным вращением. Это использовано для установления точки эквивалентности при последовательном титровании (pH 10) компонентов смесей Cd + + Си и Cd + Zn. Угол вращения поляризации измеряют в ходе титрования при 365 нм на фотоэлектрическом поляриметре; ошибка < 0,37% [677].
Каталитические способы. Диэтилдитиокарбаминат меди ускоряет протекание иод-азидной реакции, приводящей к ослаблению окраски раствора иода в метаноле.
При определении 0,1—0,8 мкг Cd его сначала экстрагируют раствором диэтилдитиокарбамината в СНС13 и взбалтыванием с раствором CuSOi замещают эквивалентным количеством меди. К 2 мл экстракта медного комплекса добавляют следующие метанольные растворы: 4 мл 0,1 М NaN3, 0,2 мл 0,2 М KJ и 0,6 мл 0,02 М J2, затем 0,2 лслСН3ОН и через 5 мин. фотоколориметри-руют при 420 нм в кювете с I = 10 мм [20].
Гидролиз этилового эфира цистеина в щелочном растворе ускоряется ионами Cd; скорость этой реакции определяют по изменению pH во времени.
В отростки смесителя[480, стр. 46] вносят следующие растворы’.в первый— 5 мл 0,01 М эфира цистеина; во второй — 5 мл 0,04 М NaOH; в третий — анализируемый раствор, 2 мл 0,01 М Са (NO3)2, 5 мл 0,1 М КВг. Разбавляют бидистиллятом до 25 мл. После термостатирования при 25° С в течение 10 мин. растворы смешивают, помещают в термостатированную (25° С) ячейку и через каждые 3 мин. (в течение 30 мин.) измеряют pH на.рН-метрах типа ЛПУ-01 или ЛП-58 со стеклянным электродом.
По полученным данным строят график в координатах (lg?3(j + pH) — время, где <7Эф = С®ф + Сон_ — (С^ф и CqH — начальные концентрации соответственно эфира и щелочи; ?он — концентрация гидроксильных ионов в данный момент времени). Концентрация кадмия пропорциональна тангенсу углов наклона прямых (lg(7^ + pH) — время.
Чувствительность метода — 10 мкг СЛ/мл [480, стр. 190].
Электрохимические методы
Электрохимические методы определения кадмия можно разделить на следующие три группы [177].
1. Методы выделения металлического кадмия при восстановлении электрическим током с его последующим определением гравиметрическим или другими способами (см. стр. 59).
2. Полярографические методы (о содержании элемента судят по силе диффузионного тока).
3. Амперометрия, потенциометрия и кондуктометрия.
Полярография
Для определения тг-10-7% и до десятков процентов кадмия, кроме «классической» полярографии, используют переменнотоковую, осциллографическую и амальгамную полярографию с на
100
коплением. Чувствительность этих методов приведена ниже:
Вид полярографии
«Классическая»
Переменнотоковая Осциллографическая Амальгамная с накоплением
Содержание Cd
n-10-2% (п-102 мкг/г)
п.10-з% (п-КАмкг/г)*
1.10-4% (1 мкг/г)
10-8 моль!л (1-Ю-2 лекг/10 мл) **
1-Ю-9 молъ)л (1 • 10-1 лскг/25 мл) (п-10-8 лскг/10 мл) **
* При обогащении. ** Предел обнаружения.
Теоретические основы этих вариантов полярографии изложены в [69, 78, 94, 180, 204], аппаратура описана в [449].
«Классическая» полярография. Ионы кадмия легко восстанавливаются на ртутном электроде в нейтральных, щелочных и кислых растворах, используемых в качестве индифферентного электролита («фона»); при этом в известных пределах содержаний кадмия диффузионный ток прямо пропорционален концентрации его ионов. Потенциал начала восстановления кадмия при увеличении его содержаний в растворе сдвигается в сторону положительных значений, но потенциал полуволны Е1/г при неизменном электролите остается постоянным:
[Cd!+], ммоль'л 6
0,2 —0,594
0,5 —0,593
1,0 —0,594
[Cd^+J, •илнмь'л
2,0
5,0
10,0
—0,601
—0,598
—0,605
и возрастает с увеличением концентрации раствора фона (н.к.э.; 25° С) [180]:
Фон Концентрация раствора фена, N Фон Концентрация раствора фона, N
KNO3 0,02 —0,572 KCI 0,02 —0,578
0,1 —0,578 0,1 —0,599
1,0 —0,586 1,0 —0,642
Величина Е1г зависит и от природы раствора (см. Приложение 11) [204].
Полярографические «спектры» ионов некоторых элементов на фоне различных растворов приведены на рис. 18. Из них видно, что путем выбора подходящего состава инертного электролита кадмий можно полярографировать без отделения некоторых элементов, мешающих определению на фоне других растворов [471]. ‘Ниже дана краткая характеристика определения кадмия на фоне различных растворов (нейтральные электролиты, аммонийные, щелочные и кислые).
Хорошо выраженную полярографическую волну кадмий дает в присутствии хлорида или нитрата калия. В растворах сульфатов и хлоридов на его волне образуется максимум; в растворах же
101
О 0,8 -1,6 ЕВ
Рис. 18. Полярографический «спектр» некоторых ионов па различных фонах, построенный по данным Лингейна ([Sn] — анодная волна)
1 — IN НС1; 2 — IN H2SO4; 3 — IN HNO3; 4 — IN раствор NaOH;
5 — раствор тартрата, pH 4,5; 6 — раствор тартрата, pH 8,8; 7 — раствор тартрата, pH 13; 8 — 1N раствор NH4CI -J-1N NH4OH
комплексообразующих агентов (NH4OH, KCN и др.) появляется отрицательный максимум, который можно легко подавить поверхностно-активными веществами [204]. Потенциалы полуволн кадмия и 24 других ионов были измерены на фоне 2 и 4 М растворов КС1, KBr, KJ, KSCN в присутствии 2 М НС1 и без нее [545].
Обычно изменение концентрации индифферентного электролита не влияет на высоту волны кадмия, однако при использовании в качестве фона сульфата натрия она увеличивается с уменьшением его содержания; это объясняется изменением коэффициента диффузии ионов Cd2+. По той же причине полярографическая волна одного и того же количества кадмия на фоне хлорида и сульфата цинка имеет меньшую высоту, чем на фоне хлорида и сульфата натрия; при этом с увеличением концентрации солей цинка высота волны Cd2+ возрастает. В качестве фона предложен и раствор СаС12, на котором Ei/, для кадмия равен —0,66 в и не зависит от pH раствора [204].
Хорошо выражена волна кадмия (£\а = —0,8 в) на сульфатно-аммиачном фоне; его используют для анализа теллуровой губки и медных концентратов. Основную массу теллура предварительно 102
отделяют тиосульфатом натрия из 5%-ной H2SO4, а медь экстрагируют в виде диэтилдитиокарбамината при pH — 5 [137, 600]. Насыщенный сульфитом и содержащий тилозу концентрированный раствор аммиака применен для определения кадмия в металлическом таллии и его солях; таллий предварительно отделяют осаждением в виде бромида [204]. Изучена кинетика выделения кадмия на ртутном (и амальгамном) капающем электроде из буферных растворов с pH 1,5—12 в присутствии нитрилотриуксусной, гексаме-тилендиаминтетрауксусной кислот и комплексона III [89].
При определении кадмия в рудах и продуктах металлургического производства используют хлоридно-аммиачный фон (Z?i/2 = —0,85 в). При увеличении концентрации аммиака в растворе фона сдвигается в сторону более отрицательных значений, а величина диффузионного тока, хотя и незначительно, но уменьшается [204]. На этом фоне определяют 0,02—5,0% Cd в свинцово-цинко-вых рудах, сплавах на цинковой основе (содержащих Cr, Т1 и до 20% Fe), в сульфате цинка и др. [272, 440, 573].
При определении в чистых солях к 5—10 мл нейтрального раствора, содержащего 0,25 — 50,0 мг Cd, приливают 20 мл раствора фона (100 г NH4C1 и 100 г Na2SO3-7 Н2О растворяют в 500 мл воды, добавляют 150 мл 25%-ного NH4OH, 50 мл 1%-ного раствора желатина и разбавляют водой до 1 л), доводят объем водой до50мл и тщательно перемешивают. Через 20—30 мин. (после полного восстановления растворенного в жидкости кислорода) полярографи-руют при 0,7—1,0 в (н. к. э.) и рассчитывают содержание кадмия по калибровочному графику, построенному в идентичных условиях [271, 272].
Этот метод применяют также для определения кадмия в медном концентрате, кеке, сульфате меди [204, 599]; основную массу меди предварительно осаждают с помощью Na2S2O3 или NH4SCN [204].
Описан метод определения кадмия в медном концентрате после предварительного отделения его экстракцией в виде тройного комплекса с бромидом и диантипирилметаном; метод позволяет отделить 0,01 мг Cd от 200 мг Си и 300 мг Fe [599].
Cd, Pb и Sn в воде определяют на фоне 1,2 М КС1 -ф- 4 М NH4C1; все три элемента предварительно осаждают 1 N раствором-Na2CO3, растворяют осадок непосредственно в инертном электролите и полярографируют при потенциалах между —0,3 и —0,9 в. Относительная ошибка определения при содержании 0,005 мг]л Cd составляет 5,6% [766].
Определение кадмия в присутствии сурьмы на фоне NH40H + -ф- NH4C1 затруднено возникновением волны Sb3+ —» Sb°, которая налагается на волну кадмия. Для устранения влияния сурьмы её соосаждают с гидроокисью железа в виде Sb(OH)3 [276].
При определении кадмия в оловянных концентратах Sn предварительно отгоняют в виде тетрахлорида, присутствующую в пробе Си осаждают на металлическом железе; кадмий же осаждают в виде сульфида из уксуснокислого раствора при pH 4,0, растворяют осадок в НС1, раствор нейтрализуют NH4OH добавляют хлоридно-аммиачный буфер, сульфит и полярографируют [170].
103
На фоне формиата натрия Cd2+ образует хорошо выраженную волну, пригодную для определения его в присутствии свинца: i 0,1 М растворе HCOONa (pH ~ 4) и 0,005% желатина РЬ и Cd образуют волны с Е</2 = —0,44 и —0,66 в соответственно [543]. На капельном ртутном электроде на фоне расплава HCOONH4 при 117—120° С получается довольно четкая полярограмма кадмия в результате образования [Cd(COOH)4]2-; метод позволяет определять кадмий с ошибкой + 10% [245].
В техническом и чистом индии на фоне 0,5 М зтилендиамина и 0,5 М раствора’ КОН возможно определение кадмия совместно и Си, РЬ и Zn; при этом потенциалы полуволн имеют следующие значения: — 0,42 в (Си), —0,61 в (РЬ), —0,95 в (Cd), —1,45 в (Zn), но Sn, Sb и Т1 мешают определению (их удаляют отгонкой в виде хлоридов). Индий при концентрации 4*10-2 ммолъ]л определению не мешает, при более высоком содержании его предварительно отделяют экстракцией диэтиловым эфиром из 1,5 М НВт [668]. Определение кадмия в присутствии индия можно проводить и на фоне 2—4 М хлорида гидразина (Ечг составляет для 1п3+ — 0,69 в, для Cd2+ - 0,75 в) [204].
Хорошо выраженные волны Bi, Cd, Си, РЬ и Zn дают в растворе, 0,5 N по CH3COONH4 и 0,1 М по винной кислоте. Этот фон предложен для одновременного определения перечисленных элементов в чистом ванадии; их определению не мешают Ag, Au, Hg, Fe3+, Mn, Pd, Pt и Sn [400|.
В присутствии Co, Ni и Zn кадмий можно определять на фоне 0,1 N раствора CH3COONH4 и 0,25 N раствора KSCN [204]. Хорошую волну образуют ионы Cd2+ в ацетатном буферном растворе с рН~5 (1МСН3С00Н и 1 М раствор CH3COONa [594]).
На фоне разбавленной НС1 в присутствии желатина и NH2OH-•НС1 определяют кадмий в металлическом цинке [69, 483]. Этот фон используют и для анализа железистых и медистых материалов [333]; предварительно избирательно сорбируются Cd, РЬ и Zn на анионите ЭДЭ-10П из солянокислых растворов. В 0,05 М НС1 0,0005—0,5% Cd можно полярографировать в присутствии до 10% РЬ и 5% Sb, предварительно отделив мешающие элементы на том же анионите [272]. Для определения на фоне 1 М НС1 + 0,1 М КС1 более высоких содержаний кадмия целесообразнее использовать производную полярографию [737].
В металлическом индии кадмий может быть определен одновременно с Bi, Pb и Си на фоне 1 М растворов HNO3 и аскорбиновой кислоты в присутствии желатина; индий предварительно отделяют экстракцией эфиром из 15 N НВг. Ошибка метода при определении 0,002—0,01 % Cd составляет 5% [663].
На фоне 15 N Н4Р2О7 кадмий и Си2+ образуют четко выраженные волны соответственно с Ку, = —0,53 и —0,13 в, пригодные для определения обоих элементов даже при 50—70-кратном избытке Си. Фосфорную кислоту (3 : 1) используют в качестве фона при определении Cd и Си в растворах, содержащих Nb и Та [213], а при 104
Рпс. 19. Полярограммы
а — кадмий на 1 М сульфатно-ам-миачном [фоне; б — медь, свинец и кадмий на фоне 10%-ного раствора ZnS04 + 0,6N НС1
разбавлении (2 : 25) — для полярографического определения кадмия в образцах, содержащих РЬ и Zn (Cd отделяют предварительно экстракцией изопропиловым эфиром из 5 У НВт (ЗОН).
Полярография переменного тока (пульсполярография) обладает большой разрешающей способностью. Ее применяют для определения кадмия без предварительного отделения многих элементов, мешающих при обычном полярографировании. Высота максимума на кривой зависимости величины составляющей переменного тока от постоянного поляризующего напряжения пропорциональна концентрации определяемого вещества, а потенциал максимума кривой совпадает со значением Е^2 данного иона [69, 204].
На сульфатно-аммиачном фоне кадмий дает хорошо выраженную полярограмму (рис. 19, а). Этот фон используют при анализе теллуридов и селенидов Cd и Zn. При совместном их присутствии с Se и Те, пики Cd и Se с потенциалами соответственно при —0,64 и 1,5 в пригодны для анализа систем при соотношении Cd : Se от 1 : 50 до 5 : 1. Чувствительность метода 0,4 мкг Cd/Ю мл. Определение Cd в присутствии 1000-кратных количеств Те проводят на тартратно-аммиачном фоне, потенциал полупика Cd2+ находится при —0,64 в. Этот фон пригоден для анализа пленок полупроводниковых соединений [414].
Ультрамалые количества кадмия (ге-10-8 М1Л, т. е. 0,01 мкг Cd/Ю мл) могут быть определены на фоне 1 М NH4C1 + 1 М NH4OH на Hg-электроде с использованием вектор-полярографа [159].
105
В 1 мл 1 М раствора КОН, содержащего 0,6 М As, при Еуг =• = — 0,77 в можно определять 1 мкг Cd, а в 0,5 М растворе КОН в присутствии 0,25 М As — 0,6 мкг Cd при Еу, = — 1,06 в [500].
Предложено одновременное определение Cd, Си и Pb в сульфате цинка с чувствительностью 0,0005% на фоне ZnSO4 + 0,5 N HCl; эти элементы дают хорошо выраженные пики при потенциалах — 0,35 в (Си), — 0,45 в (РЬ) и — 0,65 в (Cd) (рис. 19, б) [86].
В присутствии меди в продуктах свинцового производства кадмий определяют на фоне 0,5 М НС1 +1 М H2SO4. Высота образующегося пика при — 0,65 в пропорциональна концентрации кадмия в интервале его содержаний 1—20 мг!л [425]. Исследование пульсполярографического поведения Cd, In, Sb и Sn на 10 кислотных и буферных фонах показало, что количественное определение этих элементов следует производить в 6 А НС1 [246].
Переменнотоковая полярография применена для определения 7-10~6% Cd в цинке на фоне Zn (NO3)2 и 0,1—0,3 М HNO3. На этом фоне Eih относительно Hg-электрода составляет для Си, Мо, РЬ и Cd соответственно —0,25; —0,56; — 0,72 и—0,92 в [261]. Благодаря значительной разнице их потенциалов, одновременно с кадмием возможно определение и остальных трех элементов. На фоне 0,2 М раствора лимонной кислоты (pH 3,5—3,9), содержащего 0,03% додецилдифенилтердисульфоната натрия, можно определять 1—250 мкг Cd в присутствии 10000-кратного количества In; As, Bi, Со, Си, Fe3+, Мп, Ni, Pb, Sn, Т1 и Zn не мешают [597].
Ускоренное определение Cd в сульфате цинка, содержащем Си и РЬ, предложено на фоне 1 М Н3РО4 + 0,5 М НС1О4. Потенциалы пиков для Си, РЬ и Cd равны соответственно: — 0,32 в; — 0,42 в и — 0,76 в. As, Мп и Sb не мешают [65].
Для определения микропримеси кадмия в металлическом цинке используют хлорно-фосфорнокислый фон 0,5А по НС1О4 и 1N по Н3РО4. Одновременно с кадмием в этих условиях возможно определение Си и РЬ. Потенциалы пиков Си, РЬ, и Cd соответствуют - 0,1; - 0,45 и - 0,73 в [394 .
Небольшие количества кадмия в цементе определяют на фоне 1 М Н3РО4 + H2SO4 (1 : 200), раствор полярографируют при потенциалах от—0,7 до — 1,2 в. Метод позволяет определять 0,001 % Cd [153].
В концентрированной Н3РО4 (77,6% Р2О5) при 130° С Cd и Си образуют четко выраженные пики при — 0,85 и — 0,17 в относительно платинового анода [365].
Быстрый и точный метод определения 0,05 моль ОЛ/л в AgBr предложен на фоне 10%-ного раствора KSCN; полярографирова-ние раствора проводят при потенциалах от—0,5 до—0,76 в [679].
В осциллографической полярографии на электроды ячейки тоже налагают переменное напряжение; индикатором тока служит электронно-лучевая трубка осциллографа, на горизонтальные пластины которой падает напряжение, пропорциональное заданному на электродах ячейки, а на вертикальные — напряжение, пропор-
106
Рис. 20. Осциллограммы
а — Т1+, Cd2+ и Zn2+ на фоне раствора IN NH4OH 4-IN NH4C1; б — In3+, Cd2+ и РЬ2+
в цинковой руде на"'фоне 2М КОН + 2М KCN
циональное протекающему в ней току. О природе иона судят по потенциалу максимума пика на кривой зависимости тока от напряжения, а о его количестве — по высоте этого пика [69, 204]. Осциллограмма ионов Tl+, Cd2+ и Zn2+ на аммиачно-хлоридном фоне имеет вид, представленный на рис. 20,а [204]. На этом фоне пики кадмия и некоторых сопутствующих ионов располагаются в следующем порядке: NH|> Mn2+, Fe2+, Zn2+, Cd2+, Tl+ [204]. Ос-циллополярографическиа характеристики хлорокомплексов Cd, Си, Ni, Pb и Zn были исследованы в кислых и нейтральных средах [236].
В присутствии растворенного кислорода значения максимального тока на осциллограммах Cd2+ в нейтральной и слабокислой среде снижаются вследствие образования гидроокиси Cd (ОН)2, которая осаждается на электродах и препятствует проникновению к ним свободных ионов металла. В разбавленных растворах при pH 4—6 это влияние уменьшается [355]. В присутствии кислорода определение можно проводить на фоне IN NH4C1 (Ек = — 0,76 в, Еа = - 0,72 в) и 42V NH4OH (Ек = - 0,82 в, Еа = - 0,66 в); в интервале содержаний 5—25 мкг Cd/мл высота пиков пропорциональна его концентрации [74]. Без удаления кислорода возможно определение 1—100 мкг Cd/мл по анодному пику на фоне 1М раствора KNO3 [75].
Осциллографический метод применен для определениям-10~6 % кадмия в солях КС1, NaCl, NH4C1, K2SO4, KNO3, KJ, KSCN, К2Сг2О7; ошибка определения 26% [71]. На фоне 2М раствора КОН и 2М раствора KCN, совместно присутствующие Cd2+, In3+ и Pb2+ дают хорошо выраженную осциллограмму (рис. 20,6).
Следы (0,001—0,1 мкг/мл) Cd, Си, РЬ и Zn могут быть определе- I ны осциллографическим методом в металлических Be и Mg (и в ; самой HF) на фоне iM HF [495]. j
107
Рис. 21. Дифференциальная осциллограмма 0,11 мг Си/л и 2,38 мг Cd/л в растворе очищенного цинкового электролита (фон 140 г/л ZnSO4 + 0,1 N НС1)
В очищенном цинковом электролите и сульфате цинка кадмий определяют в присутствии Си и РЬ на фоне 0,1А НС1 + ZnS04 (140 г!л). Их дифференциальная осциллограмма представлена на рис. 21. Чувствительность определения Cd и Си в электролите составляет 0,1 мг, в ZnSO4 — 0,2 мг/л [227, 331]. С увеличением концентрации последнего чувствительность определения Cd2+ несколько уменьшается: его минимально определяемая концентрация в 0,5 М растворе ZnSO4 составляет 3 мкг Cd/мл, а в 2 М растворе ZnSO4 — 5 мкг Cd/мл. При увеличении концентрации H2SO4 от 0,5 до 2 М анодный пик Cd2+ на фоне ZnSO4 хорошо воспроизводим, тогда как катодный пик не воспроизводится уже в 0,05 М H2SO4. Методика позволяет определять 5 мкг Cd/мл в технических растворах сульфата цинка. РЬ и Sn2+ не мешают [585].
При анализе минерального сырья волну кадмия на фоне растворов 0,1 М NH4SCN + 0,25 М уротропин + 0,5 N НС1О4 + 1,2%-ный пирокатехин + тайрон измеряют при потенциале — 0,60 в. От Си, Zn, Ni и Мп кадмий отделяют экстракцией СНС13 при pH 11; ошибка метода 6% [774].
На фоне аммиачного буферного раствора на амальгамно-сер еб-ряном электроде анодный пик Cd2+ выражен четко и его высота пропорциональна концентрации в интервале 10—100 мкг Cd/мл и в 2,5 раза больше высоты катодного пика [334].
В жаропрочных сталях на хлоридно-аммиачном фоне кадмий определяют после отделения его экстракцией раствором диэтил-дитиокарбамината в СНС13 при pH 8,5. Вместе с кадмием в экстракт переходят Bi, Си и РЬ, которые не мешают определению. Влияние Al, Cr, Fe, Nb, Та, Ti, W и Zr устраняют добавлением винной кислоты или ее солей. Ошибка метода 10—15%. Большие количества Сг (> 10%) удаляют отгонкой в виде СгО2С12 [284].
Для ускоренного определения Cd, Си, РЬ и Zn при анализе продуктов цветной металлургии используют 1 М аммиачный буфер jb присутствии сульфата натрия [332].
В электролитах, содержащих фосфатный буфер с pH 7, 10"2 М Na2S2O3 и 10-2 М раствор купферона, Cd2+ дает хорошо выраженные кривые; на этом фоне можно определять 0,2—5,0 мкг Cd/мл. Мешают даже небольшие количества многих элементов [548].
108
На фоне KJ, KBr, КС1, KF и KSCN возможно определение 7-IO-2 % Cd; мешают Cu, In, Pb и Sn [746].
Амальгамная полярография с накоплением часто используется для определения субмикрограммовых количеств кадмия, особенно <— в материалах высокой чистоты. Она основана на электролизе анализируемого раствора со стационарным ртутным микроэлектродом (в частности — с лежащей каплей ртути) и последующем анодном полярографировании — растворении металла из по« лученной амальгамы. Положение пиков на такой полярограмме характеризует определяемый ион, а их глубина — его содержание в растворе [69, 204]
В электролитическую ячейку с выдавливаемой каплей ртути (рис. 22) помещают 25 мл исследуемого раствора, 0,1М по КС1 (очищенного от тяжелых металлов хроматографическим путем) и продувают азот (освобожденный от кислорода) 30 мин. Затем поворотом винта выдавливают капли ртути; первые две сбрасывают, третью оставляют для работы. На электроды ячейки подают напряжение, на 0,2 в большее, чем 2?1/г определяемого иона (для Cd — 0,8 в) и ведут электролитическое обогащение ртутной капли кадмием при перемешивании раствора током азота в течение 30 мин. Затем перемешивание прекращают и спустя 1—2 мин., после успокоения раствора, с постоянной скоростью (400 мв/мин). понижают напряжение до —0,2 в, регистрируя при этом кривую анодного растворения амальгамы кадмия; его концентрацию рассчитывают по методу добавок.
Характерная форма кривых при низких содержаниях Cd приведена на рис. 23. Этим способом в 25 мл раствора можно обнаружить 0,003 мкг Cd; минимально определяемое количество—0,1 мкг Cd [70].
При использовании 0,05 N растворов бромидов и нитратов Li, К, Mg и Ва зависимость высоты анодного пика кадмия от природы
Рис. 22. Электролитическая ячейка
1 — сосуд (емкостью 50 .чл); 2 — капилляр (диаметр 0,03—0,06 .ч.ч) со ртутью; 3 — стальной винт; 4 — тефлоновая пробка; 5 — шлиф; 6 — трубка для продувания азота; 7 — отверстие для вливания раствора и отвода азота; 8 — электролитический мостик от каломелевого электрода; 9 — рубашка для термостати-рования сосуда 1
Рис. 23. Анодные полярограммы для малых концентраций кадмия
1 — фон 0,1 моль КС1/л; 2 — ЗИО-8 моль Cd/л; 3— 1,3*10—8 моль Cd/л
109
катиона фона проявляется более четко в случае нитратов. На бромидном фоне вследствие комплексообразования это влияние менее заметно [161]. На фоне 0,5 М раствора КВг с выделением на аноде (стационарной ртутной капле) при — 0,62 в можно определять 0,05 мкг Cd в 20 мл раствора; РЬ выделяется при — 0,34 в, In при — 0,55 в. Этот метод использован для определения 10-4 — 10~5% Cd в чистых Se и SeO2 [61]. На фоне 0,1—0,4 М раствора BeSO4 при pH 1-3,5 порядок выделения элементов следующий (в скобках указан потенциал в в отн. н. к. э.): Си (+ 0,14), Bi (+0,40), Pb (— 0,36), In (—0,49), Cd (— 0,56), Zn (— 0,94). Определению 4 мкг Cd не мешает 50-кратный избыток Си, 10-кратный Bi, равное количество In и 5-кратный избыток РЬ [61].
Максимальная чувствительность определения кадмия на фоне 0,1 М раствора КС1 составляет 0,001 мкг/мл; присутствие Си, РЬ и Zn не влияет на высоту зубца пика. Совместное определение Cd, Си и РЬ проводят при pH 2—5 [370, 654].
На фоне 0,09 М раствора NH4C1 глубина пика Cd2+ пропорциональна его концентрации в интервале 0,1—0,5 мкг!мл. При добавлении 96,8%-ного этанола она уменьшается на 25%, потенциал же пика при этом почти не изменяется [291]. Определение Cd (и Zn) методом высокочастотной полярографии на хлоридно-ам-миачном фоне после накопления при — 0,8 в (соответственно — 1,0 в) в течение 5 мин. (1 мин. для Zn) использовано для определения этих элементов в теллуре высокой чистоты [64], а в присутствии цитрата — микрограммовых количеств Cd, Си и РЬ в чистых реактивах [454].
Определение 5-10“6% Cd и )> 1-10“6% РЬ при совместном их присутствии в металлическом индии проводят на фоне 0,2 М раствора КОН или смеси 0,5 М КОН и 0,25 М этилендиамина. Кадмий в этом случае определяют по разности, предварительно определив свинец. Ошибка метода 12—20% [367]. На первом фоне определяют 1-10~6% Cd (а также Си и Zn) в солях индия [366].
Смесь солей и минеральных кислот, нейтрализованная аммиаком до pH 8—9, применена для определения кадмия совместно с Zn, РЬ и Си в фосфоритах. В этих условиях Еу2 анодных пиков для Си, Pb, Cd и Zn имеют соответственно следующие значения: — 0,47; — 0,65; — 0,82 и — 1, 0 в. Точность метода + 10 отн. % [275]. Для определения 1 • 10-5% Cd в фосфоре полярографирование проводят в азотнокислой среде при pH 4,2—4,6 [368].
На фоне 15%-ной НС1 в присутствии пиридина одновременно определены следы Cd, Си, РЬ, и Zn; метод предусматривает предварительное растворение амальгамы в азотной кислоте и экстракцию ртути циклогексаноном [596]. На солянокислом фоне проводят определение 10"5—10-6% Cd в металлическом алюминии [68].
При определении микроэлементов в почвах и растениях накопление Cd, Си и РЬ из слабокислого раствора (без их предварительного разделения) проводят на лежащей капле при — 0,8 в в течение 5—10 мин.; для ~ 0,7 мг Cd/кг пробы ошибка 10% [251].
НО
Хорошо выраженные анодные пики Cd2+ образуются на фоне бромистоводородной кислоты [62, 158]; 2 М НВг применена в качестве фона при определении 0,004 мкг Cd/мл в чистом галлии в присутствии In и РЬ [62], а 0,05 М НВг — для его определения в кремнии. Мешают In и Т1; присутствие небольших количеств As, Bi, Си, Fe, Ge, Sb, Snn Zr на определении кадмия не отражается [158К, Разработан метод одновременного определения 10~7 — 10-8% Cd, Си, РЬ и Zn в азотной кислоте особой чистоты на фоне 0,1А раствора винной кислоты. Полярографируют при Eyt от—1,2 до — 0,2 в. Минимально определяемая концентрация кадмия — 0,004 мкг/мл [134, 418].
Предложено определение 1-10“6% некоторых элементов в GeCl4 и SiCl4. Концентрирование проводят на стационарном Hg-электроде при 50° Си — 2,8 в, полярографируют на фоне 0,05 М раствора бромида тетраметиламмония в метаноле. £\/2 для Си, Pb, Tl, Cd и Zn: —0,25; —0,43, — 0,45, — 0,60в и —1,04 в соответственно [60]
Для определения ~ 0,001 мкг Cd необходимо тщательное соблюдение постоянства температуры, скорости перемешивания, длительности электролиза и некоторых других условий выполнения анализа с предварительным накоплением на Hg-капле [653]. Анодное окисление амальгамы кадмия в присутствии ионов UO22+ при постоянном токе (с его ступенчатым изменением) и осциллографическая (хронопотенциометрическая) регистрация этого процесса позволяют повысить чувствительность определения до п-•10~5 мкг Cd в 5 мл раствора [764]. При определении ~ 0,1—1,0 мкг Cd исследован метод «обратных бросковых» токов [369].
Для предварительного накопления кадмия использованы и другие электроды: золотой стационарный микроэлектрод (0,3— 30 мкг Cd/мл определяют разностной осциллографией на фоне 1 М раствора NaClO4) [550] и графитовый, пропитанный под вакуумом смесью парафина с полиэтиленом. Применение для анодного процесса вектор-полярографа позволяет определить 0,01 мкг Cd/мл [353].
Теоретические вопросы амальгамной полярографии см. [135, 157], высокочастотной полярографии кадмия на галогенидных фонах [244], его поведения на стационарном платиновом электроде на фоне расплава КС1 — LiCl [95]. Для выбора оптимальных условий определения и-10"7% Cd (и некоторых других элементов) из пробы весом 1 г использованы одно- и многофакторный дисперсионный анализ [160].
Области применения рассмотренных вариантов полярографического метода анализа представлены в табл. 17.
111
Применение полярографических методов для определения кадмия в различных объектах *
Фон Полярографический
«Классический» Переменно
объект содержание Cd объект
Аммиачный Промышленные продукты, металлы (100 мкг/г)
Аммиачно-хлорид-пый Минеральное сырье, промышленные продукты 2-10-2—5% (200—50 000 мкг/г)
Аммиачно-сульфат-пый Промышленные продукты Реактивы
Хлоридный Минеральное сырье
Хлоридно-сульфат-пый Реактивы
Хлоридно-фосфат-пый Металлы, сплавы
Фосфатно-сульфат-ный Промышленные продукты Промышленные продукты
Азотнокислый Металлы, сплавы 2-10~3—1-10~2% (20—100 мкг/г) ** Металлы, сплавы
Бромидный
Солянокислый Металлы, сплавы 5 • 10-1— 5-10-1% (5—5000 мкг/г) **
Тартратный Реактивы
Фторидный
Щелочной Металлы, сплавы
* Минеральное сырье — руды, почвы, природные воды; промышленные продукты — Концентраты и промежуточные продукты металлургической переработки.
Ампериометрия
Для установления точки эквивалентности при амперометрическом (полярометрическом) титровании измеряют величину диффузионного тока, изменяющегося в процессе титрования вследствие взаимодействия определяемого вещества с титрантом. По результатам этих измерений строят кривые титрования в координатах:
142
Таблица 17
метод
ТОКОВЫЙ Осциллографический Амальгамный с накоплением
содержание Cd объект содержание Cd объект содержание Cd
4-10-’ моль /л (1,0 .мкг/25 1 • 10-4% (1 мкг/г) 1 ИО"3 % (10 мкг/г) и Промышленные продукты Металлы, сплавы То же мл) Реактивы 1-10-4% (1 мкг/г) Металлы, сплавы Металлы, сплавы Реактивы Металлы, сплавы То же 1-10~8 моль/л (0,03 мкг/25 мл) 1-10-6% (0,1 мкг/г) 4-10~8 молъ/л (0,1 мкг/25 мл) 1.10-в-5- IO"6 %
- Реактивы (0,01 мкг/г) 1-10-8 молъ/л
Металлы, сплавы Ю-s—10-е моль/л (2,5 мкг/25 мл} Металлы (0,03 мкг/25 мл) 5-10-6% (0,05 мкг/г)
** С предварительным отделением и концентрированием.
величина предельного тока (/) — объем добавленного титранта {мл). Точка эквивалентности соответствует перегибу этих кривых; ее находят графически на пересечении обеих ветвей кривой (их прямолинейных отрезков или их продолжений). В зависимости от потенциала, выбранного для выполнения измерений, и поведения на индикаторном электроде реагирующих веществ или продуктов
ИЗ
Рис. 24. Кривые амперометрического титрования
Электродную реакцию дают: а — определяемое вещество; б — ионы титранта; в — определяемое вещество и ионы титранта; г — индикаторные ионы, восстанавливающиеся в условиях, при которых остальные компоненты раствора не дают диффузионного тока; А — точка эквивалентности
реакции, кривые титрования кадмия могут иметь одну из форм, представленных на рис. 24.
Для амперометрического титрования кадмия используют реакции его осаждения и комплексообразования, в качестве титрантов — преимущественно органические реагенты. Индикаторными электродами служат ртутный капельный, вращающиеся платиновый или танталовый, два станционарных платиновых электрода. Амперометрический метод характеризуется высокой точностью титрования малых количеств кадмия (п-10-1— 10-гел«гС(1)ив ряде случаев позволяет проводить анализ в присутствии большого количества посторонних элементов [69, 204, 378, 379].
Условия амперометрического титрования кадмия некоторыми реагентами приведены в табл. 18, дополнительные данные по их применению приводятся ниже. Очевидно, могут быть использованы и многие другие реакции, представленные ранее в табл. 13.
К-бпс-(Р-Аминоэтил)дитиокарбаминовой кислотой в условиях определения кадмия можно также титровать Bi, Pb и Т1; при pH 7,5—8,5 и —0,55 в — Ni [528]. Антрахиноновой кислотой при pH 3,7 сначала определяют Си, затем устанавливают pH 6,0 и титруют Cd [379]. 0,1 М НВг можно определять Cd в присутствии Ba, Bi, Са, Си, Mg, Ni и Zn; метод использован при анализе серебряного припоя [200[. N, N'-Дитиокарбамоилгидразином и его диалилпроизводным (дальзином), кроме 1,4—8,4 мг Cd, можно также титровать Си и Zn [540]. Выполнению определения Cd с диэтилдитиокарбаминатом мешает Zn [379|. Титрование иодидом в присутствии пирамидона применено для определения 20—90% Cd в его сплавах с Zn и в Cd-губке; As, Ni, Pb, Zn и до 0,8% TI не мешают, Си предварительно цементируют на железной проволоке [786].
Во многих работах рассматривается титрование кадмия раствором комплексона III. В присутствии Zn десятые доли и целые миллиграммы Cd можно определять на фоне 1 М раствора NH4OH [73].
114
СО Литература [528] [379] [20] [464] [379] [786] О 05 г- о СО «гЧ [306]
сб !=Г Я К хо сб Ь форма кривой титрования * V© е VO vo е
о титрования кадмия Электродная реакция Окисление реагента Восстановление Cd Окисление В г Окисление реагента | Восстановление Cd То же Окисление реагента Восстановление Cd
Напряжение, в г‘о- +1,0 00 о 1 СО о” + 1 СО о" + -1,0 О 1 + 00
О и © © S’ а © в ©-© в Электрод i Hg Hg £ I Hg Pt Hg Hg Та Pt
pH от—в- СО СО 4,2 5—5,5 О
Условия ам Среда 0,02—0,15 М 1 KNO3 + тартрат | Этанол СН3СООН безводная o' nh4ci + nh4oh 0,1 М НС1 1,2 М Na2SO4 + пирамидон СН3СООН CH3COONa «si о са О са й
Титрант 'к' § л ' i СО. й ю дитио ка рбаминовая кислота Антрахиноновая кислота Бромистоводородная кислота I N, NДитиокарбамоил-гидразин Диэтилдитиокарбаминат натрия Иодид калия Комплексон III
115
Последовательное титрование (по току окисления комплексона III) Bi и Cd в одном и том же растворе при + 1,2 в (н. к. э.) проводят сначала в 0,2 N HNO3 (титруется сумма Bi и Cd), затем добавляют ацетатно-буферную смесь до pH 4,0 и титруют при том же потенциале Cd. Метод позволяет определять по 0,05—9 мг обоих элементов. Титрованию кадмия мешают Со, Си, Cr, Fe, Мп, Ni, Nb, Pb, Zn, лимонная, сульфосалициловая и щавелевая кислоты [114[. При 1,2 в можно последовательно титровать также In (при pH 2—3) и Cd (при pH 4,5—5,5); In определяют в азотнокислом растворе, удалив предварительно окисли азота, затем добавляют сухой CH3COONa до pH 4,5—5,5 и титруют Cd [111].
Описано титрование комплексоном III бинарных смесей Cd с Hg и некоторыми другими элементами по току окисления реагента на танталовом электроде. На фоне 0,25 N HNO3 при +1,2 в сначала титруют ртуть, затем сухим CH3COONa устанавливают pH 5—5,5, снижают потенциал электрода до +1,1 ей определяют Cd [110]. Подобный метод может быть применен и к смесям Cd + Ga + + In [112], Cd + Mg и Cd + Mg + Bi [109], Bi + Cd + Ag [113]. При титровании на Hg-электроде на оксалатном фоне возможно’ последовательное определение 6 мкг Cd/мл и по 3 мкг/мл Мп и Zn [306]. Вместо комплексона III для титрования кадмия предложена оксиэтилендиаминтриуксусная кислота; pH 4,6 создают прибавлением уротропина [379]. С 8-меркаптохинолином определяют даже п-10~2 мг Cd, но вместе с ним осаждаются и другие ионы. Однако разработан метод дифференциального определения Cd, Fe и Си, при котором по ступенчатой кривой титрования можно рассчитать содержание всех трех элементов [379].
При использовании р-нафтохинолина к раствору 1—2 5 мг Cd в 10 мл 0,5 N H2SOi, из которого удален кислород, прибавляют KJ и постепенно титруют 0,01—0,1 М раствором реагента в 0,1—0,5 N H2SOi. После прибавления каждой порции реагента раствор продувают азотом в течение 3 мин.
Определению не мешают Al, Ca, Со, Сг3+, Fe2+, Mg, Mn2+, Ni и Ti4+; Fe3+восстанавливают раствором NH2OH; Ag, Bi, Cu, Hg, Pb и Sb цементируют на железной проволоке из горячего раствора. При невысокой концентрации кислоты и иодида миллиграммовые количества кадмия можно определить в 2 М растворе ZnSO4 [204,. 619].
Титрование кадмия 8-оксихинолином возможно практически лишь в чистых растворах, так как Zn и многие другие элементы осаждаются реагентом в тех же условиях [379]. 0,2 М раствором роданида в присутствии изохинолина и 0,01% желатина можно последовательно тцтровать Ni (pH 2,8); Zn (pH 3,7) и Cd (pH 5,3). Допустимо наличие Al, Bi, Cr, Fe3+, Mn и Pb; мешают Ag, Hg,. ионы галогенидов [706]. Подобное же титрование роданидом при добавлении пиридина использовано для определения кадмия в-гальванических ваннах [379]. При применении в качестве титранта тиоацетамида допустимы 100-кратные количества Zn, но для
116
117
получения воспроизводимых результатов необходимо соблюдать указанные в табл. 18 пределы pH [379].
В ацетатной среде с ферроцианидом калия образуется осадок двойной соли K6Cd5[Fe (CN)6]4, в аммиачной — простая соль Cd2 [Fe (CN)e], В первом случае чувствительность определения ~ 0,5 мг Cd, во втором — (из-за большей растворимости осадка) в 4 раза ниже. В 1 М растворе НС1 или КС1 кадмий не осаждается; это использовано для последовательного титрования растворов проб губчатого кадмия, содержащих 1—10% Zn и 62—70% Cd.
Титрование проводят с двумя платиновыми электродами при напряжении 0,05 в. К 50 мл раствора пробы в 1 N НС1 (после экстракционного удаления железа) добавляют 5 капель 0,6%-ного раствора K3[Fe(CN)6] и титруют 0,05 М раствором K4[Fe(CN)6] до возрастания силы тока (что указывает на полное связывание Zn). Затем нейтрализуют раствор аммиаком до pH 5—6, выжидают некоторое время для понижения силы тока до первоначальной величины и вновь титруют — уже кадмий — до вторичного возрастания силы тока [379].
При определении 2,9—6,4 мг Cd хинальдинатом натрия ошибка ~ 2% [669]. При использовании этиленгликоль-бис-(аминоэтил)-тетрауксусной кислоты и ионов Си2+ в качестве индикатора, можно титровать Cd в присутствии Zn. Диффузионный ток Си при — 0,3 в начинает снижаться лишь по окончании титрования Cd, который не восстанавливается при этом потенциале [379]. Показана также возможность титрования Cd на Pt-электроде, при — 0,6 в относительно Ag/AgJ-электрода, электрогенерированными ионами S2- на фоне расплава KSCN -]- NaSCN (при 70° С) [526]. При амперометрическом титровании раствора CdSO4 поливанадатами состав образующихся осадков при pH 6,5—7,5 соответствует формуле CdO-V2O5, а при pH 7,5—8,5 — 3CdO-V2O5 [719].
Другие методы
Потенциометрическое титрование [182] для определения кадмия применяют сравнительно редко. Опубликованные способы основаны на образовании осадков комплексов или солей кадмия. Иногда определение проводят в неводных средах. Краткая характеристика некоторых методов потенциометрического титрования кадмия приведена в табл. 19.
Диантипирилмета н-б ромидный комплекс кадмия осаждают известным избытком КВг, который титруют затем нитратом серебра [117, 118]. При осаждении кадмия избытком 0,1 N раствора Na2C2O4 осадок отфильтровывают и аликвотную часть фильтрата титруют 0,1 А раствором HgNO3; амальгамирование платинового электрода следует обновлять [565, стр. 311].
В солянокислом, содержащем 2,5 М NaCl растворе CdCI2, хлорид тетра-фениларсония образует белый кристаллический осадок (несколько растворимый в чистой воде), который выдерживают 3 часа, фильтруют и промывают концентрированным раствором NaCl. Избыток реагента в фильтрате и промыв-
118
Методы потенциометрического титрования кадмия
О
сб !=Г Н И \о сб
Н
Me(Hg) — амальгамированный электрод.
119
ных водах титруют иодометрически, причем выделяется коричневый осадок (CeH6)«AsJ3 [565, стр. 331].
Последовательное титрование РЬ и Cd (при их соотношении от 1:1 до 5:1) диэтилдитиокарбаминатом натрия возможно при pH 1—12, но с повышением pH воспроизводимость результатов несколько ухудшается [77]. Последовательное титрование Си и Cd в растворе, содержащем этанол, диэтиловый эфир и большое количество KNOS, проводят с Hg2Cl2- и Ag/AgP-электродами (Р — анион реагента); два четких перегиба на кривой титрования позволяют определять оба элемента в одном растворе [505]. Титрование молибдатом натрия выполняют при 95° С [565, стр. 310], а определение 0,1 А раствором Na2S 55—110мг Cd в нейтральной среде (содержащей CH3COONa) — на холоду [565, стр. 270]. В другом способе титрование Na2S проводят в слабокислом растворе тартрата, с электродной парой Hg/Ag в 0,001 N растворе KJ [182]. Для титрования кадмия тиомочевиной использовано и образование при pH 3 прочного комплекса Cd(CSN2H4)3 [76].
Титрование ферроцианидом калия (см. табл. 19) производят при 75° С. Метод используется для концентраций кадмия <; 0,01 N [565, стр. 314].
При анализе фосфора в солянокислом растворе (pH 1—3) при 60—70° С сначала титруют Zn, а затем повышают pH до 4,0 и находят сумму Cd -|- Zn [682].
При анализе магниевых сплавов в раствор пробы bO,5N кислоте, содержащей 50 мг Cd, вводят 30 мл насыщенного раствора K2SO«, при титровании выделяется осадок 5 Cd2[Fe(CN)e] • 4K4[Fe(CN)e], магний в этих условиях не осаждается [409].
Описано потенциометрическое определение Cd и Си относительно нулевой точки с использованием пары полуэлементов: ферроцианид — феррицианид.
В две потенциометрические ячейки вводят смесь (1:1) 0,1 М растворов [Fe(CN)e]4- и [Fe(CN)e]3- и ацетатный буфер с pH 4,5—5,5, соединяют полуэлементы и устанавливают нулевое значение ЭДС. Затем в ячейку с ферроцианидом вводят анализируемый раствор, а в ячейку с феррицианидом — 0,5 М стандартный раствор кадмия до достижения нулевого значения ЭДС. Ионы А1з+, NH4, С1~, СгО4“, NO~ и SO2- мешают; допустимо присутствие ионов Fe2+, Fe3+ и РЬ2+ [656].
Для прямого потенциометрического титрования Cd2+ (и других ионов) комплексоном III в боратном буферном растворе в качестве индикаторного применяют Ag-электрод в присутствии 10“® М Ag+. Определению мешают ионы NH4 и анионы, образующие с Ag+ более прочные комплексы, чем комплексон III [743]. В другой работе раздельное или совместное титрование смесей Cd, Си и Zn с другими тяжелыми металлами производят при помощи регистрирующего титратора [401]. При определении кадмия в медных сплавах используют обратное титрование избытка комплексона III (или циклогександиаминтетрацетата) солями Hg2+.
120
К 10 мл анализируемого раствора прибавляют избыток 0,05 М раствора комплексона III, 40 мл уротропинового буферного раствора с pH 9 и оттитро-вывают избыток реагента 0,05 М раствором соли Hg2+. Ионы Ni титруются вместе с Cd [613]. Более избирательно титрование тетраэтиленпентамином с Hg-электродом [220, стр. 271].
Раздельно бромиды Cd и Zn, растворенные в смеси метанола с ацетоном (1 : 3), можно титровать бензольно-метанольным раствором тетраэтиламмония [201]. Растворы иодида кадмия (и других его галогенидов, а также солей некоторых металлов) в диметилсульфоксиде титруют 0,1 N раствором метилата натрия в том же растворителе; электродная пара — Sb/н. к. э. [602]. Растворы 8-оксихинолинатов кадмия и многих других элементов в ледяной СН3СООН титруют 0,1 N раствором НС1О4 в диоксане со стеклянным электродом и н. к. э., а в растворе ацетонитрила или пиридина — метилатом натрия с электродной парой Sb/н. к. э-[601].
Кондуктометрическое титрование [32, 243] для определения кадмия применяется очень редко. Отмечена возможность титрования раствором K3[Fe(CN)e], осаждающего (в противоположность K4[Fe(CN)6]) нормальную соль кадмия. Определение можно производить в присутствии РЬ [565, стр. 335]. Другой способ основан на количественном осаждении кадмия (в присутствии до 3-кратного количества цинка) в аммиачной среде анилидом тиогликолевой кислоты. Высокочастотным титрованием определяют 0,1—11 мг Cd в 20 мл раствора; оксалаты, тартраты и цитраты не мешают [173]. Очень разбавленные растворы Cd2+ (0,02—0,5 мг в 40 мл) предложено титровать сероводородной водой в токе азота [565, стр. 271]. Можно титровать кадмий и роданидом в присутствии пиридина, при этом Си маскируют тиосульфатом, Ni — диметил глиоксимом. Ag, Au, Со, РЬ и Zn должны быть удалены, a Al, As, Bi, Сг, Fe, Sn, платиновые и щелочноземельные металлы определению не мешают [707]. Комплексы кадмия с аналогами соли Рейнеке (см. стр. 59, 83) могут быть использованы для его кондуктометрического титрования [572].
Кулонометрия. Электролиз ведут при постоянном потенциале (для кадмия — 1,0 в), окончание выделения элемента определяют по резкому уменьшению силы тока [23, 94, 243]. В присутствии до 1000-кратных количеств Bi, 0,03—0,5 мг Cd выделяются на Hg-электроде при— 1,0 0 из 0,1 М НС1О4. Затем окисляют полученную амальгаму при — 0,33 в [312]. Можно производить кулонометрическое определение кадмия после его выделения при — 0,90 в (относительно Ag/AgCl-электрода) из раствора с pH 9, содержащего 0,5 М тартрата [780]. Предложено кулоностатическое титрование кадмия раствором комплексона III с капельным Hg-электродом [738].
121
Радиохимические методы
Для определения кадмия применяют методы радиометрического титрования и изотопного разбавления. В первом из них используют способы, приводящие к образованию двухфазных систем и добавляют к исследуемому раствору радиоактивный индикатор; в ходе титрования измеряют активность растворов, строят на графике ее зависимость от прибавленного объема титранта и находят точку эквивалентности по излому полученной кривой; чувствительность п-10-2 мкг Cd. В методе изотопного разбавления в раствор пробы вводят радиоактивный изотоп 109Cd или 116Cd (с известной активностью), который при этом «разбавляется» находящимися в пробе другими природными (нерадиоактивными) изотопами этого же элемента; после выделения из раствора некоторого количества кадмия измеряют его активность и по ее уменьшению (относительно введенной) рассчитывают количество «разбавителя», т. е. искомое содержание кадмия в растворе пробы; чувствительность 1 • 10-3 мкг Cd. Теория и практика радиохимического анализа описаны в работах [7, 50, 128, 255, 294, 382, 433, 515, 716, 742].
Радиометрическое титрование
Для разделения фаз в ходе титрования используют реакции осаждения, экстракцию, флотацию, ионный обмен; в качестве индикаторов — радиоактивный 115Cd [Ti/2 = 53 часа; энергия [3-излучения: 1,11 Мэв (58%) и 0,58 Мэв (42%)] и изотопы некоторых других элементов — так называемые «неизотопные» (по отношению к определяемому элементу — кадмию) индикаторы.
Для отделения осадков при титровании можно применять прибор, схема которого дана на рис. 25,а. Из сосуда 1 титруемый раствор засасывают шприцем 2 в резервуар 3, где определяют его активность; затем жидкость возвращают в сосуд 1 и прибавляют порцию осадителя. Эту операцию повторяют несколько раз и строят кривую титрования в координатах активность — объем титранта (с учетом эффекта разбавления раствора).
Схема прибора другого типа приведена на рис. 25,6. При осаж- . дении 115Cd (или другого его радиоактивного изотопа) нерадиоактивным реагентом график титрования имеет вид кривой а на рис. 26. Если радиоактивный изотоп содержится в осадителе, то кривая титрования приобретает форму б. Для определения этим путем Cd (и Zn) используют 1-дитиокарбокси-5-метилпиразолинат натрия, образующий с ним при pH 14 осадок (Cd : Р = 1 : 2). При pH 9 подобное соединение дает и Zn, не мешают определению обоих элементов Al, Ca, Ga, Mg и др.
При последовательном титровании миллиграммовых количеств Cd и Zn индикатором служит e5Zn (Ti/2 = 245 дней, энергия 13-излучения — 0,32 Мэв), при определении одного кадмия — 115Cd [53]. Аналогично можно титровать оба элемента и раствором 122
a
б
Рис. 25. Схема прибора для радиометрического титрования методом осаждения
а — первый вариант конструкции: 1 — сосуд для титровайия; 2 — шприц; 3 — резервуар для измерения активности; 4 — свинцовый блок; 5 — торцовый счетчик; в — микрофильтрующая трубка; 7 — капиллярные трубки
б — второй вариант конструкции; 1 — счетчик Гейгера — Мюллера; 2 — стеклянная рубашка; з — трубка со стеклянным фильтром; 4 — бюретка; 3 — двухходовой кран; в — сосуд с титруемым раствором; 7 — трубка для подачи сжатого воздуха; 8 — трубка к водоструйному насосу
1-дитиокарбокси-3-метил-5-фенилпиразолината натрия [255, стр. 58]. Вместо фильтрования.при осадительном титровании можно использовать флотацию образующихся осадков, например [Cd (C5H5N)J (SCN)2, на границе раздела вода — органический растворитель (бутанол, диэтиловый эфир, СС14). При установлении точки эквивалентности по измерению активности водной фазы целесообразно применять органический растворитель более тяжелый, чем вода (в частности, СС14) [255, стр. 53].
Для титрования растворов, содержащих радиоактивные изотопы Cd, по-видимому, можно использовать многие соединения, приведенные в табл. 13 для нерадиоактивного кадмия — меченые арсенат, иодид (в присутствии пиридина), молибдат, фосфат и другие анионы, с которыми для выделения осадка не требуется введения избытка реагента (в частности, феррицианид, содержащий 59Fe [255, стр. 134]).
При экстракционном титровании применяют как 11BCd, так и неизотопный индикатор — 131J (7\г =8,1 дня; энергия 7-излу-
123
Рис. 26. Кривые радиометрического титрования
а — нерадиоактивный осадитель; б — радиоактивный осадитель; в — экстракционное титрование дитизоном Ag, Си и Cd, индикаторы ,10Ag и 115Cd: 1 — водная фаза; 2 — экстракт.
А — точка эквивалентности
чения — 0,36 Мэв, |3-излучения: 0,61 Мэв (87%), 0,33 Мэв (9,3%. При анализе дюралюминия, латуней и Zn-сплавов в качестве экстрагента использован раствор дитизона в СНС13, индикатор — 115Cd. Метод позволяет без предварительного разделения последовательно определять н-10~2 мг Си + Cd или Hg + Cd [194].
В несколько пробирок с притертыми пробками вводят по 0,5 мл анализируемого раствора, по 0,5 мл раствора известной концентрации 115Cd (удельная активность 35 имп!мин- мкг) и по 1 мл буферного раствора, pH 4,7. Добавляют различные количества раствора дитизона в СНС13 и столько чистого CHClg, чтобы объем неводной фазы во всех пробирках стал равным 2 мл. Содержимое пробирок встряхивают 15 мин., центрифугируют 1—2 мип. и измеряют активность всех водных и хлороформных слоев. Сумма активностей равных объемов обеих фаз во всех точках титрования должна быть постоянной. При постепенном увеличении объема дитизона сначала экстрагируется только Си (или Hg), активность каждой фазы сохраняется постоянной. Когда начинает экстрагироваться Cd, активность водного слоя уменьшается, а хлороформного возрастает. После его полной экстракции на графике для обеих фаз снова наблюдается перелом.
Подобным же образом можно последовательно определять в одном растворе Ag, Си, и Cd. В этом случае кроме 115Cd вводят и известное количество 110Ag (Ti/a = 253 дня; р-активность: 0,54 Мэв (43%); 0,09 Мэв (55%), кривые титрования имеют вид, представленный на рис. 26,в [193; 142, стр. 209].
При экстракционном титровании дитизоном эквивалентную точку находят и по изменению степени поглощения медленных нейтронов; по мере экстракции дитизоната кадмия, имеющего большое сечение захвата, поглощение нейтронов водной фазой снижается, на основании чего строят кривую титрования [255, стр. 120].
При работе с 131J кадмий экстрагируют в виде иодид-пиридино-вого комплекса.
К 10 мл раствора пробы (10—2000 мкг Cd) в 0,02—0,3 М НС1 или HNO3, 1%-ной по аскорбиновой кислоте и 0,02 М по K.J (131J — 3,33 мккюри!мл), прибавляют 10 мл 5%-ного раствора пиридина в СвНв и встряхивают 2 мин. Жидкость центрифугируют, отбирают 5,0 мл экстракта, измеряют его у-ак-тивность и строят кривую титрования.
124
Определению мешают Ag+, Cu2+, Hg, Pb2+ и Zn2+; допустимо присутствие Al3+, Bi3+, Go2+, Gr3+, Fe3+, La, Mg, Mn2+, Ni2+, Th, ТВ и UO1+ [658].
Заслуживает внимания оригинальное осадительное титрование Cd2+ (и Са2+) фторидом с индикатором—радиоактивным крип-тонатом кварцевого стекла [получают бомбардировкой стекол ускоренными ионами 8бКг (Т1/г = 10,4 года, энергия [3-активности 0,67 Мэв (99%), энергия у-иэлучения 0,52 Мэв)}.
Раствор (рН7) титруют 0,1 М раствором NaF, после приливания каждой «го порции и перемешивания в течение 1 мин. индикаторное стекло вынимают из жидкости, споласкивают дистиллированной водой, сушат и измеряют остаточную радиоактивность. После достижения точки эквивалентности избыток ионов F- разрушает поверхность стекла, вследствие чего освобождается соответствующее количество 85Кг и на графике титрования образуется излом [757].
В методе ионного обмена используют пеницилламин, который образует с Gd (а также с Hg, Ni и Pb) гораздо более прочные комплексы, чем с Со.
Готовят серию растворов проб (равные объемы) с pH 8,8—12, в каждую прибавляют равные количества 10-5 М раствора в0Со =5,2 года, энергия (3-излучения 0,32 Мэв, у-излучения — 1,33 и 1,17 Мэв) и 10-5 М раствор пеницилламина (постепенно возрастающее количество, лл)- Для удаления избытка непрореагировавшего еоСо, растворы обрабатывают катионитом, измеряют их радиоактивность и находят эквивалентную точку по перегибу графика титрования.
Чувствительность определения 0,01—0,1 мкг Cd/1 мл раствора [403].
Предложено косвенное микрорадиометрическое определение 0,05—2 мкг Cd (и As, Со, Си, Ni, Pb и Zn) на фильтровальной бумаге с 110Ag.
В центр кружка фильтровальной бумаги диаметром 5,5 см наносят капил Лярной пипеткой 5 мкл раствора пробы, обрабатывают газообразным H2S, помещают бумагу с осадком на кольцевую баню и отмывают от избытка осадителя. Фильтр высушивают, наносят в его центр 10 мкл раствора, меченного AgNO3 (50 мкг Ag с активностью 1 мккюри), выдерживают 5—10 мин. в камере с влажным воздухом и переносят на кольцевую баню. Отмывают избыток AgNO3 с помощью 200 мкл NHjOH (1 : 1). После высушивания из фильтра вырезают центральную зону диаметром 17 мм, измеряют ее радиоактивность на торцевом счетчике Гейгера — Мюллера и рассчитывают содержание кадмия по калибровочному графику [622].
Изотопное разбавление
К исследуемому раствору добавляют определенное количество W\, радиоактивного изотопа кадмия с известной активностью Аг (или удельной активностью SJ. Выделяют соединение кадмия (содержание W2) и измеряют его активность А2 (или удельную активность д2).
При использовании радиоактивного изотопа без носителя содержание кадмия вычисляют по формуле
иг = ГК» —
125
Если введенный индикатор содержит весомое количество кадмия, то расчет ведут по формуле [40, 142]
В этом случае нет необходимости определять выделенное количество вещества.
При определении кадмия обычно применяют субстехиометрический вариант метода изотопного разбавления. Выделение изотопов (чаще — экстракционное) производят заведомо недостаточным для полной экстракции элемента количеством реагента. Необходимо лишь, чтобы из растворов с разной концентрацией кадмия были выделены его совершенно одинаковые количества. Это достигается идентичностью условий экстракции для всех растворов и полным связыванием используемого реагента. Субстехиометрию применяют также в активационном и других радиометрических методах анализа.
Теоретический вывод условий, необходимых для выделения кадмия (и других элементов) экстракцией дитизоном, дан в работе [715]. Для экстракции кадмия применяют аммиачно-хлоридный буферный раствор, концентрация дитизона — 1 мкг!мл [625].
В две делительные воронки вводят по 100 мкл анализируемого и стандартного растворов (0,1—0,5 мкг Cd), приливают по 300 мкл реагента (10 мл 20%-ного раствора тартрата калия-натрия, 5 мл 10%-ного раствора хлорида гидроксиламина, 15 мл буферного раствора с pH 10 и 0,025 мл раствора 109Cd без носителя и разбавляют водой до 50 мл), перемешивают 2 мин., экстрагируют раствором дитизона в СНС13 (2 мкг/мл) в течение 2—3 мин. Отбирают по 50 мкл экстрактов и измеряют их радиоактивность.
Для выделенйя кадмия применена кулонометрия с контролируемым потенциалом [627].
Работу проводят в двух последовательно соединенных идентичных ячейках с ртутным катодом площадью около 7 см2, анолом из платиновой проволоки, стеклянной мешалкой и н.к.э. сравнения. В обе ячейки вводят по 3,00 мл 0,1 N раствора KNO3 и равные известные количества 109Cd (У,^ = 470 дней; энергия у-излучения — 0,087 и 0,058 Мэе). В одну из ячеек помещают раствор пробы, в другую — стандартный раствор кадмия и проводят электролиз в атмосфере азота при —0,75 в (н.к.э.) в течение 5 —10 мин. Не прекращая электролиза, из обеих ячеек одновременно отбирают по 1 мл раствора, измеряют их радиоактивность и по калибровочному графику рассчитывают содержание кадмия в пробе [627].
Описано определение ~ 3 мкг Cd в присутствии Ag, Bi, Со, Си, Hg, Мн, Ni, Pb, Т1 и Zn (по 500 мкг).
К исследуемому раствору прибавляют стандартный раствор с loeCd. Ag, Си и Hg удаляют экстракцией дитизоном при pH 2. Маскируют Со и Ni (тартратом, тиомочевиной и диметилг ли оксимом), экстрагируют Cd из 1 М раствора NaOH и реэкстрагируют его водным раствором с pH 2—3. Устанавливают pH 9 реэкстракта и экстрагируют кадмий субстехиометрическим количеством (1 мл) раствора дитизона в СНС13, 0,6 мл экстракта упаривают на алюминиевом диске и измеряют радиоактивность остатка на торцевом счетчике
126
Гейгера — Мюллера. Параллельно проводят экстракцию и измерение стандартного раствора, содержащего 109Cd [612].
Подобный метод применен для определения 0,16 мкг Cd/г говяжьей печени, 1,56 мкг Cd/г почек и при анализе других биологических объектов [506, 612].
ФИЗИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Спектральный анализ
Для определения кадмия используют эмиссионную спектрографию, пламенную фотометрию (эмиссионную, атомно-абсорбционную и атомно-флуоресцентную). Чувствительность прямого спектрографического определения — п-10'в — п-10-7 г Cd — может быть повышена его предварительным концентрированием. Чувствительность эмиссионного пламенно-фотометрического определения соответствует п-10-1 мкг Cd в 1 мл раствора, атомноабсорбционного — п-10-2 мкг, а атомно-флуоресцентного — п-10"3 мкг Cd и менее.
Эмиссионная спектрография
Аналитические линии кадмия и некоторых мешающих элементов (и чувствительность его определения при использовании угольной дуги) представлены в табл. 20 [131, 215, 229, 517], более полные данные (около 70 спектральных линий) приведены в Приложении 12 [517].
Таблица 20
Аналитические линии кадмия <.
?-са- а Энергия, эе Чувствительность определения Cd, % Мешающие элементы
нижнего уровня верхнего уровня элемент X, A Содержание, %
2288,0 0,00 5,39 l-10-з As 2288,1 >l-10-2
3261,0 0,00 3,78 0,3—1-10-3 Fe 3261,3 >1-10-1
Co 3260,8 >1-10-1
Ti 3260,2 >1-10-1
V 3261,8 >1
w 3261,2 >1
3403,6 3,72 7,34 1-10-1 Mo 3402,8 >1-10'1
4799,9 3,78 6,36 1-10-1 Ni 3403,4 >1
3133,2 ' 3,78 7,72 1-3-10-1 V 3133,3 >3-10-2
2836,9 3,72 8,07 3-10-1—1
2868,3 3,93 8,23 1-3
127
Наиболее чувствительна линия 2288,0 А, но из-за сильного самообращения ее используют лишь при определении низких содержаний кадмия, в основном — в анализе чистых веществ; в большинстве случаев определение ведут по линии 3261,0 А.
Кадмий и большинство его соединений обладают относительно низкой температурой кипения и расположены в начале ряда летучести при испарении в угольной дуге (Hg, As, Cd, Zn, Те, Sb, Bi, в окислах — Hg, As, Cd, Zn, Bi, Sb, B, в сульфидах — Hg, As, Ge, Cd, Pb, Sb, Bi, Zn, ...; в фосфатах — Cd, Zn, Bi, Sn, Pb, Na...) [131]. Поэтому его возбуждают в низкотемпературной дуге постоянного или переменного тока [358].
Спектры кадмия регистрируют на фотопластинках, чувствительных к ультрафиолетовой области (тип СП I, СП III) с помощью спектрографов средней дисперсии (ИСП-28). Применение диф-фракционных приборов (ДСФ-8, ДФС-13) на порядок повышает чувствительность определения [156]. При непосредственном спектральном анализе порошкообразных проб (минералы, руды, продукты их переработки) ~ 30 мг образца в большинстве случаев вводят в плазму дуги испарением из канала угольного электрода. Для стабилизации температуры к пробам и стандартным образцам добавляют буферные смеси (в основном соли щелочных металлов). Внутренним стандартом служат Ag, Mn, Sb, Zn и некоторые другие элементы. Этим путем можно анализировать пробы, содержащие 3-103— 1-10~2% Cd.
Некоторое увеличение воспроизводимости определения ряда элементов в минеральном сырье дает метод вдувания порошкообразной пробы потоком воздуха в плазму горизонтальной дуги. В этих условиях чувствительность определения по линии 3261,0 А повышается и составляет п-Ю“3% Cd при ошибке + 5,6% [360] (по другим данным, 1-10"5, ошибка + 4% [140]).
Для определения кадмия используют и линию 5085,8 А- Возбуждение производят разрядом в горячем полом цилиндрическом катоде при продувании гелием. Для регистрации спектра на пластинках «микро» (22 ед. ГОСТ) применяют спектрограф ИСП-51 с камерой / == 720мм\ при анализе германия коэффициент вариации составляет 25% [247].
Предложены методы определения кадмия в растворах. Хотя на анализ требуется больше времени (в связи с переведением пробы в раствор), воспроизводимость результатов выше, чем при прямом анализе порошков. В некоторых способах для возбуждения спектра используют искру, раствор пробы подают в разрядный промежуток с помощью фульгуратора; интервал определяемых содержаний 3• 10-2 — 1,0% Cd, средняя квадратичная ошибка + 6,8% [357, 472]. При введении раствора в искровой промежуток методом пористого электрода чувствительность определения 0,01 % Cd при средней квадратичной ошибке + 5% [557].
Снижения пределов определения достигают при введении пробы в разряд в виде тонкой пленки сухого остатка на торцах элект-
128
родов. При использовании медных электродов и искрового разряда чувствительность определения 1 •10-4% Cd (1 мкг/г) при коэффициенте вариации + 5% [674]. Введение пробы в дугу переменного тока на торце угольных электродов позволяет определять 1 мкг СйЛил, коэффициент вариации + 9% [141].
Использование фотоэлектрической регистрации спектров значительно повышает производительность анализа. Так, при определении примесей в марочных свинце и цинке (и определении загрязнений самого кадмия) на квантометре ДФС-10 производительность в 3 раза выше, чем при обычной фотографической регистрации спектров [217].
Однако для контроля веществ высокой чистоты чувствительность спектрального анализа недостаточна. В этих случаях применяют методы физического и химического обогащения. Физическое концентрирование основано на термической отгонке легколетучих форм кадмия из труднолетучей основы. Конденсат анализируют спектральным методом. Обычно эти два процесса совмещают, т. е. отгонку определяемого вещества производят непосредственно в зону разряда [14, 133, 628].
При химическом концентрировании устраняется влияние основы, повышаются чувствительность и, особенно, точность анализа.
Фотометрия пламени
В эмиссионной фотометрии анализируемый раствор распыляют в высокотемпературное пламя и фотометрируют излучение линии Cd 3261,0 А. Сами пламена сильно излучают в этой области спектра, поэтому необходимо выбирать такое пламя, при котором отношение интенсивности линии к излучению фона имеет наибольшую величину. Это достигается в смеси водорода с воздухом. При использовании комбинированной горелки-распылителя (кисло-родно-ацетиленовое пламя) чувствительность определения составляет 0,5 мкг СЛ/мл [336].
Метод атомно-абсорбционной фотометрии пламени обеспечивает высокую чувствительность, избирательность и точность определения кадмия [238].
Определение проводят по поглощению в пламени резонансной линии Cd 2288,0 А, источником света служат высокочастотные безэлектродные лампы с парами кадмия или лампы с полым Cd-катодом, реже — дуговые парометаллические лампы. Растворы проб распыляют при помощи углового или концентрического распылителя и в смеси с горючим газом вводят в протяженное пламя длиной ~ 10 см атомно-абсорбционной горелки. Искомое содержание рассчитывают по калибровочным графикам в координатах: оптическая плотность пламени при длине волны аналитической линии Cd — его концентрация в эталонных растворах (мкг/мл водных растворов чистых солей кадмия); реже исполь-
5 Щербов Д. П.
129
зуют метод добавок. При работе с трубкой с полым катодом (ток 5 ма) чувствительность достигает 0,07 мкг ОЛ/мл. Ca, Си, Fe, К, Mg, Na, Sr и Zr при концентрации 2 мг!мл определению кадмия не мешают. При концентрации до 2 .У не влияют НС1, HNO3, H2SO4 и СН3СООН; но Н3РО4 снижает оптическую плотность паров кадмия примерно на 40% [336]. В биологических жидкостях можно определять от 0,13 мкг ОЛ/мл с помощью спектрофотометра переменного тока и лампы с полым катодом. Анализируемый раствор распыляют непосредственно в пламя светильный газ — воздух протяженностью 10 см [147].
Использование спектрофотометра Перкин—Эльмер (модель 303) с Т-образным адаптером (длина 15 см, диаметр 3,5 см) позволяет с высокой воспроизводимостью определять в природных водах Cd, Си и Zn без их предварительного концентрирования (кислородноводородное и кислородно-ацетиленовое пламена). При расходе раствора 4 мл/мин чувствительность определения 0,002 мкг СА/мл. Из 50 исследованных катионов и анионов занижают результаты определения лишь арсенаты, карбонаты, силикаты и тетрабораты; введение 400 мг/л комплексона III устраняет их влияние [703].
Для повышения чувствительности определения фотометрируют отходящие газы пламени, которые пропускают через длинную трубку (до 100 см), в результате такого удлинения поглощаемого слоя чувствительность определения Cd может быть повышена от 0,025 до 0,0004 мкг!мл [237]. Другой способ снижения порога определения — экстракция органическими растворителями, преимущественно не изменяющими режима горения пламени. Извлечение иодида кадмия метилизобутилкетоном повышает чувствительность в 2,5 раза по сравнению с фотометрированием водного раствора и поволяет определять 0,04—0,8 мкг СА'мл [187, 785]. При экстракции раствором дитизона в этилпропионате и введении экстракта в воздушно-ацетиленовое пламя чувствительность определения кадмия возрастает до 0,001 мкг!мл [718]. При анализе почв Cd (Pb и Zn) извлекают раствором дитизона в СС14 [155], а при определении в водах — их концентрируют пропусканием через ионит Дауэкс-50 W [205]. В анализе вод использована экстракция изопентанолом комплексов кадмия с тиазолилазонафтолом и. тиазолилазорезорцином, что позволяет определять кадмий при его содержании в исходных пробах от 0,1—0,2 мкг/л [667].
Особенно большое повышение чувствительности достигается при импульсной термической атомизации твердых проб — из навески 15 мг можно определять в горных породах 2-10~7%Cd (0,00003 мкг ). Пробу, смешанную с угольным порошком, испаряют при 1850° С в электроконтактном нагревателе типа испарителя; нерезонансное поглощение учитывают по линии кадмия 2265,0 А [34, 35].
Опубликованы работы по атомно-абсорбционному определению кадмия с применением различных атомизаторов: свободно горящих пламен [148, 484, 659], водородно-кислородного пламени с 130
кюветой [568], графитовой кюветы [168, 169]. Введение твердой пробы в плймя использовано также в [239, 343].
Метод’ атомной флуоресценции основан на резонансом воз* буждении атомов кадмия в пламени при его облучении интенсив* ным источником света. В определенных условиях яркость флуо* ресценции пропорциональна концентрации атомов кадмия в пла* мени [345, 778, 779]. Чувствительность определения кадмия в значительной степени зависит от аппаратуры, особенно — от применяемого источника возбуждения. Например, при использовании ксеноновой дуговой лампы с непрерывным спектром испускания, предел чувствительности соответствует 0,1 мкг Cd/мл, а с разрядными лампами фирм Осрам и Филипс—0,0001—0,0002 мкг Cd/мл [778].
При выполнении на одной и той же аппаратуре атомно-абсорбционных (лампа с полым катодом) и атомно-флуоресцентных измерений (возбуждение кварцевой кадмиевой лампой Осрам-lSW с вырезанным окном во внешней стеклянной колбе) влияние 41 катиона, 18 анионов и ряда органических растворителей на определение кадмия одинаково, но чувствительность второго метода на порядок выше (соответственно 0,1 и 0,01 мкг Cd/мл) [538].
Водородно-воздушное пламя дает более высокую чувствительность атомно-флуоресцентного определения Cd (0,002 мкг/мл), чем ацетилено-воздушное [577]. Отмечена возможность определения 0,001 мкг Cd/мл и с использованием вместо спектральных приборов светофильтров; распыление раствора производили непосредственно в водородно-воздушное пламя [763]. Высокая чувствительность — 0,0002 мкг Cd/мл — реализована также при применении кислородно-водородного пламени с помощью горелки-распылителя [646]. В турбулентном пламени водород — воздух в комбинированной горелке-распылителе интенсивность атомной флуоресценции легко атомизируемых металлов (в том числе и Cd) в 2—3 раза выше, чем при использовании такого же, но предварительно смешанного пламени в горелке с камерой распыления [514].
Как и в атомной абсорбции, импульсная атомизация твердых проб посредством дугового нагрева намного повышает чувствительность атомно-флуоресцентного определения кадмия. Оптимальная длительность импульса составляет 1,5—2,5 сек. и связана с формой рюмочного электрода (в который помещают пробу), весом пробы и током дугового разряда. Флуоресценцию возбуждают модулированным резонансным излучением безэлектродной высокочастотной лампы, чувствительность определения в чистом графите по линии 2288,0 А составляет 3• 10-8% Cd, ошибка — 30— 40%; для содержаний порядка 10-7Cd% она снижается до 20— 30% [36]. Этот способ применен для определения кадмия в стеклоуглероде и графитовом порошке. Чувствительность атомно-абсорбционного анализа их на порядок, а эмиссионного спектрального — на 3 порядка ниже флуоресцентного [214]. В другой работе
5*
431
атомизатором служила графитовая кювета, нагреваемая в атмосфере аргона до 2600° С. Достигнутая в этих условиях абсолютная чувствительность абсорбционного фотометрирования составила 0,0000008 мкг, а флуоресцентного — 0,00000004 мкг Cd [655]. Из табл. 21 (в которой пересчет содержаний из процентов в микрограммы произведен в расчете на вес пробы 1 г) видно, что атомная абсорбция и атомная флуоресценция, особенно — при импульсной атомизации твердых проб, наиболее чувствительные из всех методов определения кадмия. В частности, они значительно превосходят такие высокочувствительные способы, как амальгамная полярография с накоплением (до п-10-4 мкг/мл) и нейтроноактивационный анализ (до 0,005 мкг Cd) [713].
Рентгеноспектральный анализ
По сравнению с оптическими, рентгеновские спектры (особенно АГ-серии) имеют незначительное число линий; поэтому, за редкими исключениями, отсутствуют погрешности, вызываемые их взаимным наложением.
Недостаток метода — сравнительно низкая чувствительность, сильная зависимость интенсивности аналитических линий от химического состава пробы, сложность аппаратуры. Метод применяют в основном в тех случаях, когда необходим экспрессный анализ состава однотипных продуктов (например, в технологических процессах) или для элементов, определение которых другими методами затруднительно. Вследствие этого число работ по определению кадмия рентгеноспектральным флуоресцентным методом невелико (например, [226, 436]).
Потенциалы возбуждения линий рентгеновского спектра кадмия приведены ниже [387] (полный спектр его дан в Приложении 13):
Серия К L М N
Е, кв 26,7 4,07 0,77 0,11
Аналитические линии /Г-серии относятся к крайней границе коротковолновых спектров, используемых в этом методе. Анализ, проводят, как правило, при двухкратном и выше превышении потенциала возбуждения, поэтому для определения кадмия по К-серии необходима аппаратура с напряжением на трубке порядка 50 кв и более.
Определение кадмия по линиям L-серии во многом уступает анализу по /Г-серии. Это вызывается тем, что линии L-серии расположены в длинноволновой области и для уменьшения их большого поглощения воздухом при прохождении от излучателя до детектора необходима вакуумная аппаратура. Кроме того, выход флуоресценции линий L-серии (Р7ь) значительно нижб, чем для линий ЛГ-серии (IVк). Найденные экспериментально значения
132
ев Д' а ч
КО “W
8 я л
§
о £
я к я л со Я в м
к я №
£
Я
Я § g g s
:трода
133
Я
g. д о
S- ф н Cl, н-г tl
W Д й О &>©<§ S S
со 5
S co И О Д
&*§§<
s 2 со
5*5®
CO
134
Wk для кадмия следующие: 0,819 [581], 0,827 + 0,015 [711], 0,846 [518]. Теоретическое значение WK = 0,843 [524].
Для L-серии кадмия выход флуоресценции найден экспериментально только при переходе атомов К —> L, для которого Wkl ~ 0,055 + 0,014 [608]. Общий выход флуоресценции Wl должен мало отличаться от этого значения [559].
Интенсивность основных линий A-серии кадмия по отношению к самой яркой линии (принятой за 100) выражается следующими числами [378]: Ка, — 100; К^, — 53,8; K?Jl — 26,1; —
4,18. Относительную интенсивность линий L- и других серий кадмия не определяли. Однако с достаточной вероятностью можно принять, что их отношения у кадмия такое же, как и у соседнего с ним серебра, у которого для L-серии они имеют следующие значения [387]: аг — 100; а2 — 12; — 59; — 21; |33 — 9,4
р4 - 5,8; Y1 - 12; Z - 4,1; ц - 2,2.
Спектр рентгеновских линий (диаграммных и недиаграммных) и краев поглощения кадмия дан в Приложении 6. Помимо длин волн испускания и краев поглощения, в ней приведены соответствующие значения частот (в ридбергах) (y/R) и величины ]/v/R, часто используемые в расчетах. Значение постоянной Ридберга Roo принято равным 109737,303 смГ1.
Значения массовых коэффициентов поглощения различных длин волн приводятся в работах [226, 710] (для кадмия см. табл. 22). При переходе от X 0,45 к 0,50 А поглощение кадмия резко уменьшается вследствие его «А-скачка поглощения». Это свойство используют, в частности, в рентгеноструктурном анализе; так, в паре с кадмием серебро пропускает область от 0,46 до 0,486 А (Лср около 0,475 А) [723]. Величину «А-скачка», выражаемую отношением коэффициентов поглощения по обе стороны от границы пропускания того или иного уровня атомов (A, L, ...) для кадмия экспериментально не определяли. Интерполяция между измеренными значениями А-скачка поглощения серебра и олова дает для кадмия величину 6,85 [327].
Один из наиболее удобных методов рентгеновского флуоресцентного определения кадмия — метод внешнего стандарта с введением поправок на массовые коэффициенты поглощения. Теория метода изложена в работах [43, 44, 230, 231, 326, 328], его применение для определения кадмия — в [436, 662, 776]. Экспериментально показано, что предел обнаружения кадмия в растворах по линии Аа, в оптимальных условиях «насыщенного» излучающего слоя составляет 5,4 мг/л [776]. Для этого применяли плоский кристалл-анализатор LiF толщиной 0,02 дюйма (0,508 мм), коллиматор Соллера и сцинтилляционный счетчик; подаваемое на трубку напряжение 57 кв. При использовании аналитической линии Lai предел обнаружения 10 мг Cd/л; в этом случае для регистрации применяли пропорциональный счетчик с гелиевым наполнителем [776]. Предел обнаружения кадмия в порошковых пробах
135
Таблица 22
Массовые коэффициенты поглощения кадмия
о X, А см,*, г X, А р.» сл<»/г О X, А pt, см1/г
0,10 0,87 1,1 102 2,89 изо
0,15 2,45 1,15 114 3,05 1320
0,20 5,60 1,20 154 3,60 360
0,25 11,0 1,25 143 3,74 380
0,30 17,5 1,30 157 3,95 440
0,35 25,0 1,35 169 4,15 505
0,40 35,5 1,40 189 5,40 575
0,45 49,0 1,50 226 5,59 635
0,50 11,50 1,60 263 4,73 635
0,55 15,0 1,66 286 5,18 820
0,60 18,50 1,79 349 5,40 935
0,65 23,50 1,93 414 5,77 1035
0,70 29,0 2,10 495 6,07 1190
0,76 35,0 2,20 571 6,21 1260
0,80 42,0 2,28 604 6,45 1360
0,85 49,0 2,37 706 6,86 1730
0,90 57,5 2,46 760 7,08 1830
0,95 77 2,54 830 8,34 2900
1,0 76 2,68 940 9,89 4080
1,05 86 2,76 1020 11,9 6250
при оптимальном времени регистрации линии Kai во втором порядке отражения— 2,5-10~3% (25 мкг/г) [436]. Предварительное химическое обогащение снижает предел чувствительности определения до 0,я мкг [37].
Прямой метод внешнего стандарта использован при определении состава красителей для текстильных материалов (в частности, хлопка). Метод основан на абсолютном счете интенсивности флуоресценции; предварительно строят график зависимости интенсивности линий от концентрации: J2i = / (Cd, %). Для указанных материалов кадмий в соединениях Cd—Se и Gd—S—Se можно определять при его содержании 1-10~2%. Выцветание окрасок тканей вызывалось улетучиванием кадмия под влиянием атмосферных условий [662].
Рентгенофлуоресцентный метод предложен и для анализа промышленных отходов.
В 20—30 мл раствора с pH 3—4, содержащего 1—200 мкг Cd, вводят ионообменную мембрану и встряхивают в течение 80 мин. После ее высушивания определяют кадмий по линии La. Чувствительность обнаружения 0,3 мкг Cd; для содержаний 0,5—50 мкг стандартное отклонение 6% [751а].
136
Радиометрический анализ
Для определения кадмия используют абсорбционный метод и активационный анализ. В первом из них измеряют при помощи соответствующих детекторов ослабление потока нейтронов (испускаемого ампулой с подходящим радиоактивным веществом) при прохождении через испытуемый раствор [50]; поперечное сечение захвата нейтронов в естественной смеси изотопов кадмия 2450 барн, чувствительность метода порядка 100- лг мкг Cd. Активационный анализ основан на облучении пробы в реакторе потоком нейтронов; при этом природные стабильные изотопы 114Cd и 116Cd (имеющие достаточно большие сечения активации) переходят в радиоактивные 108Cd и 116Cd. По у-излучению последних определяют содержание (т) элемента в пробе; для расчетов служит формула
At = -6,02- Ю33-/^ (1 - е~0’^Т),
где At — число распадов в 1 сек. в момент конца обучения; М — атомный вес; / — интенсивность потока нейтронов; оак — атомное сечение активации нейтронами; t — продолжительность облучения; Т — период полураспада образующегося изотопа. При / = 1013 нейтронов/см2 • сек и t = 1 — 2 суток пз пробы весом в 1 г определению доступно 10~®—10~7% Cd (10~2—10-3м,кг).
Обычно анализ выполняют в радиохимическом варианте, в котором после облучения в реакторе производят химическое выделение кадмия (с добавлением его нерадиоактивных изотопов-носителей или без них) и измеряют активность выделенного препарата [373].
Основы этих методов изложены в работах [50, 373, 433, 756], обзор по применению в них субстехиометрии — в [742]. О методе отражения |3-излучений, испытанном на растворах чистых солей, см. [294[.
Абсорбция нейтронов
Для определения 0,01—60% Cd в порошкообразных и жидких промышленных материалах (сырье, полупродукты, отходы) использованы источники тепловых нейтронов — Йа—Be (20 мкюри) и Ри-Ве (0,5 мкюри). Калибровочные графики строят по аналогичным материалам с известным содержанием кадмия; по точности метод сопоставим с классическими способами анализа, чувствительность 100 мкг/г, продолжительность определения 5 мин. [624].
Помещенный в парафиновый цилиндр Рн—Be-источник с выходом 4,6-10® нейтрон/сек применен для непрерывного определения концентрации кадмия в потоке растворов. Детектором нейтронов служит счетчик СНМ-29. Чувствительность определения 0,25 г Cd/л. Элементы, сопутствующие кадмию в технологических растворах, оказывают мало влияния на результаты анализа.
137
При определении 4—20 г Cd/л средняя относительная ошибка находится в пределах + 5% [163, 164].
Использование ампулы с Be + RaTh (0,3 кюри) с замедлением излучаемых ею нейтронов в слое трансформаторного масла позволяет определять кадмий в сплавах с другими металлами. При содержаниях 2% Cd точность анализа ~ 5% [349]. Этим же путем можно надежно измерять толщину кадмиевых покрытий при условии, что она > 0,01 мм [50].
Нейтроно-активационный анализ
При определении Cd (Bi, Hg и Т1) в горных породах 1 г пробы облучают 3 дня в реакторе с / = 6-1013 нейтрон/см2-сек и выдерживают 7 дней. Затем пробу в присутствии 30 мг носителей разлагают HF с добавлением H2SO4. Раствор выпаривают почти досуха, растворяют в 0,5 М НС1 и сорбируют кадмий наколонке с анионитом|Дауэкс-1 в С1~-форме. Затем кадмий элюируют дистиллированной водой и определяют активность 115Cd (7'1/г = 53 часа; у-пик при 0,53 Мэв), чувствительность определения 0,001 мкг Cd/г [652].
В другой работе Cd (Cs, In, К, Pb, Y и РЗЭ) облучают 4—6 час. в реакторе с f == 1,4-1013 нейтрон/см2-сек и выдерживают 2 дня. Добавляют носитель, сплавляют с Na2O2, выщелачивают разбавленной НС1 и проводят измерения. Химический выход кадмия находят повторной активацией выделенного из облученной пробы носителя [708].
При определении кадмия в металлических А1 и Zn 0,1—0,8 г пробы и стандарт кадмия облучают 130 час. в реакторе с / = 4-1013 нейтрон/см2-сек. Пробу растворяют (с добавлением 400 мкг Cd-носителя) в небольшом количестве НС1, раствор выпаривают досуха. К остатку прибавляют 5 мл 0,1 М раствора тартрата, 15 мл 10 М раствора NaOH, пб 1 мл 20%-ного раствора NH2OH и 0,1 М KCN. Экстрагируют 2 раза (по 2 мин.) избытком раствора дитизона в ССД. Объединенные экстракты промывают 2М раствором NaOH и ре-экстрагируют кадмий 10 мл 0,1 М HNO3. Добавляют к водной фазе раствор NaOH (содержащий NH2OH и тартрат) с таким расчетом, чтобы он стал 1 М по NaOH и экстрагируют в течение 1—2 мин. 1 мл 10-3 М раствора дитизона в ССД. Измеряют радиоактивность (a) 115Cd.
Содержание кадмия в пробе рассчитывают по формуле
= Мст ~ > “ст
где МСТ — количество мкг Cd в объеме стандарта, взятого для измерения! аот — его радиоактивность [623].
Аналогичный метод использован для определения кадмия в арсениде галлия [742].
Для определения кадмия в уране пробу растворяют в HNO3. Раствор выпаривают с НС1 и отделяют Cd от U пропусканием его через колонку (диаметром 4 мм), содержащую 0,5 мл анионита Дауэкс-1Х4. Промытую колонку с сорбированным кадмием высушивают, запаивают с обоих концов и облучают 70 час. вместе с холостой пробой и Cd-стандартом в реакторе с / = 1 • 1013 нейтрон/см2 сек. После 6-часовой выдержки прибавляют 10 мг Cd-носителя и производят ряд химических разделений, включающий 4-кратное осаждение CdS, соосаждение с Fe (ОН)3, осаждение кадмия 2-меркаптобензтиазолом (химический выход кадмия определяют гравиометрически) и измерение активности 115Cd по у-пикам 0,49 и 0,52 Мэв. При содержании 0,005—0,02 мкг Cd стандартное отклонение составляет ±84%, для 0,05—2,5 мкг — 15% [713].
138
Для отделения Cd и Си при их активационном определении в иттрии использована экстракционная хроматография на пористом фторопласте ПФ-4 с раствором диэтилдитиокарбамината цинка в СНС13. При облучении в течении 20 час. потоком 1,2-1013 нейтрон! /см2-сек чувствительность определения 6-10~5% Cd (0,06 мкг из 100 мг) [476].
Нейтронная активация применена и для определения Cd (и 12 других элементов) в цинке 99,9999 %-ной чистоты [626], в электролитическом сульфате цинка [633], в полупроводниковом кремнии высокой чистоты [282] и в биологических материалах [485, 513].
Ряд работ посвящен выделению кадмия в радиоактивно чистом состоянии путем многократного осаждения сульфидом [218], ионообменному разделению радиоактивных изотопов кадмия и других элементов [105], разделению Cd и Zn на бумажных хроматограммах [128], получению 108Cd без носителя из циклотронных мишеней (четкое разделение Cd и Zn достигнуто при их соотношении от 30000 : 1 до 1 : 1000) [744]. Радиоактивный изотоп 108Cd выпускается нашей промышленностью в виде раствора его солей — Cd (NO3)2 и CdCl!2— с удельной активностью 1—10 мкюри/г или мкюри/мл.
Тип распада — электронный захват, сопровождающийся УС-из-лучением серебра (0,022 мэв), Tt/2 = 470 дней, основные у-линии— 0,022 и 0,088 Мэв [225].
Отражение Р-излучений
Интенсивность отраженного p-излучения возрастает пропорционально порядковому номеру элемента и его концентрации (в бинарных системах — с увеличением в смеси доли элемента с более высоким порядковым номером). Для определения кадмия в растворах смеси чистых солей Cd2+ + Со2+ и Cd2+ 4- Hg2+ в качестве источника p-излучений использован 204Т1 (Ту2 = 3,56 года, энергия р-излучения 0,76 Мэв). Содержание кадмия находят по калибровочному графику, построенному по стандартным растворам в идентичных условиях [525].
Г лава V
МЕТОДЫ ОТДЕЛЕНИЯ КАДМИЯ ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
В природных и большинстве промышленных объектов кадмий встречается в незначительных количествах и отделение его от сопутствующих элементов является необходимой и трудной задачей. Наиболее часто приходится отделять кадмий от его спутника— цинка, очень сходного с ним по химическим свойствам. Для отделения от элементов, мешающих определению, используют методы осаждения и особенно — экстракции и хроматографии.
МЕТОДЫ ОСАЖДЕНИЯ И СООСАЖДЕНИЯ
Эти методы используют для отделения относительно больших количеств кадмия и, как правило, при определении его в концентратах, сплавах и некоторых других продуктах.
От цинка кадмий отделяют тиоацетамидом [84, 85, 475], сульфидом натрия [457] или насыщенным раствором соли Рейнеке [714]. Осаждение тиоацетамидом выполняют в условиях, описанных в гл. III. При анализе сталей проводят групповое осаждение Bi, Cd, Си и РЬ 2%-ным раствором тиоацетамида из раствора с pH 7,5—8,0 при нагревании до 90—95° С [361]. Отделение насыщенным раствором соли Рейнеке в присутствии тиомочевины осуществляют, как описано в гравиметрии. Образующийся при этом осадок имеет состав [Cd(CSN2H4)2] [Cr(CSN)4(NH3)2]2. Сульфид натрия осаждает кадмий при pH 3,5 [166].
Алюминий отделяется при соосаждении кадмия с Fe(OH)3 в 1М растворе KNO3 при добавлении NaOH при pH 11—13,5 [303].
От Al, Be и РЬ кадмий может быть отделен осаждением в виде Cd (ОНу2 5%-ным раствором NaOH. Образующийся при этом осадок сорбирует на своей поверхности ~ 1,5% отделяемых элементов, степень соосаждения уменьшается в ряду Be А1 РЬ [192]. В присутствии галогенидов pH начала осаждения увеличивается в порядке J" Вт- С1_, что совпадает с константой устой-140
чивости галогенидных комплексов кадмия [553, 720, 721]. Этот метод отделения в аналитической практике не нашел широкого применения.
Описано отделение кадмия от бериллия соосаждением первого с Fe(OH)3. При последующем переосаждении Fe(OH)3 из 1 И раствора NH4NO3 при pH 3—4 достигается отделение кадмия от носителя [304].
Разделение малых количеств индия и кадмия проводят путем последовательного соосаждения с Fe (ОН)3 из 1И раствора NH4NO3 пиридином, первого — при pH 4,5—6,5, второго — при pH 7,5—8,5. Для отделения кадмия от носителя осадок растворяют в небольшом количестве кислоты и вновь осаждают Fe(OH)3 пиридином в 1 И растворе NH4NO3 при.pH 6 [305].
При анализе металлического молибдена и его сплавов кадмий осаждают тиоацетамидом из аммиачного раствора с pH 8 на коллекторе — сульфиде ртути или меди [475]. Отделение кадмия от основы при анализе металлического ниобия проводят осаждением тиоацетамидом при pH 8,5 в присутствии тартрата аммония [475].
От многих элементов (Со, Си, Cr, Ni, Mn, Zn и др.) кадмий отделяют диантипирилметаном в присутствии ионов Вг_, как описано в гравиметрии; еще более труднорастворимый осадок образуется в присутствии ионов J-, но при этом вместе с Cd осаждается и Си [116].
Для отделения кадмия используют осаждение его иодидного комплекса с метиловым фиолетовым [339, 341], а также соосажде-ние на Са3 (РО4)2 [132] или Bi (ОН)3 в среде КОН [356].Выделение оксихинолином, [3-нафтохинолином, меркаптобензотиазолом описано в гл. IV.
Из других реагентов для осаждения кадмия описаны 2-мер-капто-8-анилин-1,3,4-тиодиазол [691], 2-амино-1,3,4-тиодиазол-5-тиол [546], монотиомочевинофталевая кислота [583], диэтилдитио-карбаминат натрия [19] и этилендиамин [273, 410].
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Отделение кадмия от некоторых элементов можно успешно провести электролизом водных растворов, используя комплексообразование и разницу в потенциалах выделения. Этот метод рекомендован особенно для отделения от Си, РЬ и Zn. Краткая характеристика условий электролиза с платиновым электродом приведена в табл. 23. Из слабокислых растворов, содержащих Zn, кадмий можно выделить на 99,99% [234; 619, стр. 184].
Достаточно полно кадмий отделяют от Bi, Fe и Pb при последовательном электролизе раствора: сначала из тартратной среды на платиновом электроде выделяют Bi, Fe и Pb, а затем (после подщелачивания аммиаком) при 1,00—1,15 в количественно осаждают кадмий. Подобным путем можно последовательно выделить Cd, Sn и Zn. Образование рыхлого осадка кадмия предотвращается до-
141
Таблица 23
Электролитическое отделение кадмия от других элементов на платиновом электроде [565, стр. 250; 619, стр. 188 ]
Отделяемый элемент Состав электролита Температура, °C Напряжение, в Плотность тока, а/100 см2
Al+Mg H2SO41* 5,0 5,0
As KC№* 2,6-2,7
Au KCN 60 7,0
Bi HNO3 + винная кислота 80 1,7
Co+Fe (NH4)2SO4 + H2SO43* 2,8—2,9
Cr h3po4 65 7,0
Cu KCN4* 2,6—2,7
Fe KCN + NaOH6* 5,0 5,0
Mn H2SO4s* 65 2,6 0,08
Mo nh4oh —
Ni KCN + KOH 40 2,25-3,00 0,03-0,04
Pb HNO3 65 1,9-2,2
Sb Тартрат аммония 50 1,8-2,0 0,1
Sn HC1 + NH2OH 70 0,7(2,0)
w nh4oh + (NH4)2SO4 2,6
Zn HC1 + NH2OH 0,8
h2so43* 2,6 0,08
CH3COONa + CH3COOH 70 2,2 0,10
N &2C2O4 80-85 2,4 0,1—0,3
«* Разбавленная горячая IIzSO,.2* Небольшой избыток. •* Разбавленная II2SO4. 4* Избыток. 6* Горячий раствор.
бавлением к раствору деполяризатора—гидразина и 0,01 % -ного-раствора желатина [619, 184, стр. 217].
С ртутным электродом кадмий отделяют от незначительных количеств цинка из хлоридных или сульфатных растворов. Электролиз проводят при потенциале —- 0,85 в, который на 0,25 в более отрицателен, чем потенциал выделения цинка (при этом кадмий остается в растворе) [619, стр. 184; 635, стр. 201].
Разработан метод разделения Ag, Cd, Си, Bi и Sb при внутреннем электролизе с цинковым анодом и сетчатым платиновым катодом из растворов, содержащих ионы Fe3+ и комплексон III [641, 642].
Для определения кадмия в чистых металлах и сплавах используют метод электролитического выделения основного компонента [24].
Детальное описание условий электролитического отделения кадмия от Ag, Al, As, Au, Bi, Co, Cr, Cu, Fe, Hg, Mn, Mo, Ni, Pb, Sb, Sn, W и Zn изложено в работе [565,стр. 250 —264].
142
ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Экстракционное выделение кадмия, отличающееся высокой избирательностью, используется при анализе различных объектов как эффективный способ разделения и концентрирования. Кадмий экстрагируется органическими растворителями в виде простых га-логенидных комплексов, внутрикомплексных соединений или тройных комплексов. Краткая характеристика экстракционных методов отделения кадмия приведена в табл. 24.
Экстракция простых галогенидных комплексов
Иодид кадмия количественно экстрагируется из 6,93/ HJ (или из смеси 1,5 N раствора KJ и 1,5 N H2SO4), эфиром. При этом Al, Ba, Be, Ca, Со, Cr, Fe (II), Ga, К, Mn, Ni, Pb, Ti, TI, U, V и платиновые металлы не экстрагируются; Bi, In, Mo(IV), Te(IV) и Zn экстрагируются частично. В органическую фазу вместе с кадмием переходят Au, Hg, Sb и Sn(II) (в случае 6,9М HJ), In (из 1,5М раствора KJ).
Наиболее полно кадмий экстрагируется диэтиловым эфиром из 1,6 до 2,0Л/ HJ. При дальнейшем повышении кислотности расслоение ухудшается. При экстракции диэтиловым эфиром из растворов сисходной концентрацией кадмия 0,4371/ и при концентрации кислоты вводной фазе 1М образуются две органические фазы, кадмий и кислота при этом находятся в более тяжелой органической фазе.
При экстракции изоамиловым спиртом двух органических фаз не образуется 1208]. Изоамиловый спирт позволяет достаточно полно отделять кадмий от Al, Со, Cr, Fe, Mg, Mn, Ni, Ti и Zn.
Иодидка дмия количественно экстрагируется диэтиловым эфиром из 1,5Л/ раствора KJ, 1,5Л/ по HaSO4 [281, 619].
Из кислых растворов, содержащих иодид калия, кадмий может быть извлечен раствором Амберлита-La-l в ксилоле. Степень экстракции иодида кадмия из сернокислых растворов значительно выше, чем из бромистоводородных и при концентрации иодида калия ~ 0,5Л/ достигает 99% [396].
Роданидный комплекс кадмия экстрагируется вместе с цинком из слабощелочных растворов, содержащих 15% NH4SCN, смесью (1 : 2) и-амилметилкетона и н-бутилфос-фата и может быть отделен таким способом от Cu, Ni и Ag. При промывании органического слоя кислым раствором кадмий переходит в водную фазу. При экстракции из 15 %-ного раствора NH4SCN (0,6Л/ по НС1) смесью амилового спирта и диэтилового эфира (1 : 1) экстрагируется только цинк, а кадмий остается в водном растворе [619].
143
Таблица 24
Экстракционное отделение кадмия от других элементов
Экстракционный реагент Среда Экстрагент Отделяемые элементы IgK РИ1/, Литература
Экстракция простых галогенидных комплексов
Иодид 1,6-2 М HJ Изопентанол Al, Со, Cr, Fe, Mg, Mn, Ni, Ti, Zn [208]
6,9 М HJ Диэтиловый эфир Al, Ba, Be, Ca, Co, Cr, Fe (II), Ga, K, Mn, Ni, Pb, TI, Ti, Zr [208]
H2SO4 + 0,5 М KJ Амберлит LA-1 в ксилоле [396]
6N H2SO4 + O,lAf KJ Метилизобутил кетон [151] [187]
Роданид Щелочная+ + 15%NH4SCN н-Ами лмети л кетон + и-бутил-фосфат (1 : 2) [142]
Экстракция внутрикомплексных соединений
Бензоил ацетон pH 9,5-11 СвНв Си, Hg, Zn —14,92 8,93 [389]
pH 9,5-11 СНС13 Си, Hg, Zn —15,83 8,48 [389]
Дибензоилметан pH 9-11 СвНв Си, Hg, Zn —13,98 8,0 [389]
Ди-и-бутилтиофосфорная кислота 0,01-1 N НС1 или HaSOj СС14 —4,0 [389]
Ди-(Р-нафтил)тиокарбазон Щелочная СНС13 Zn, Pb, Bi 1,6 [389]
Дитизон . pH 7-14 СНС13 Zn, Pb, Bi, In 2,14 2,9 [195, 248,
249, 253, 254, 725]
Диэтил дитиокарбаминат диэтиламмония pH 1-12 СС14 [389, 400]
Таблица 24 (окончание)
Экстракционный реагент Среда Экстрагент Отделяемые элементы IgK Литература
Диэтилдитиокарбаминат натрия pH 5 СС14 [389]
Ди-2-этилгексилфосфорная 0,03А НС1 СвН5СН3 [389]
кислота
Диэтилдитиофосфорная кислота 0,5—1А НС1 СС14 [33, 389]
Р-Изопиразолон pH 8-10 5,05 [389]
7-[а-(о-Карбометоксианилинобен-зил]-8-оксихинолин СНС13 6,3 [389]
8-Оксихинальдин pH 10 СНС13 [389]
8-Оксихинолин pH 5,5-9,5 СНС13 —5,29 4,66 [389]
1-Фенил-3-метил-4-бензоилпира-золон-5 pH 7—7,5 СНС13 Mo, Ti, W [145]
Экстракция тройных комплексов с органическими основаниями
Диантипирилметан НС1 (1 : 1) СС14 Zn [93, 253]
Дифеиилгуанидин + иодид калия Кислая СС14 Zn [253]
Пиридин + роданид Щелочная СНС13 Ag, Си, Hg, TI [281, 289]
Трибензоилметан 0,3—0,5 М НВт Дихлорэтан Co, Си, Ni, Zn [66]
0,1—0,2А HJ » Al, Fe, Mg, Mn [66]
Три-ле-октил амин ел HJ или НВг СвНв Al, Fe, Mg, Mn [66]
Экстракция внутрикомплексных соединений
Бензоилацето н, 0,1 М раствор в хлороформе или бензоле, экстрагирует кадмий на 98%. Влияние кислотности раствора на степень экстракции кадмия приведено на рис. 27. Регулируя pH среды, можно отделить кадмий от Gu, Hg и Zn, которые экстрагируются из более кислых растворов [389].
Дибенэоилметан используют как экстрагент кадмия из щелочных растворов; его 0,1 М раствор в бензоле экстрагирует кадмий практически.полностью (рис. 28), при этом Си, Hg и Zn остаются в растворе [389].
Д и-и-б утилфосфорная кислота в четыреххлористом углероде экстрагирует кадмий на 99% [389]. Д и-н-о к-тилсульфид, д и-н-о ктилсульфоксид и д и-н-октилсульфон почти не экстрагируют кадмий иэ азотно-и солянокислых растворов (коэффициент распределения 10-2 — IO"3) [299].
Д и-(р-н а ф т и л)т иокарбазон в хлороформе и четыреххлористом углероде экстрагирует кадмий из сильнощелочной среды в присутствии тартрата. Метод используют для отделения кадмия от Bi, РЬ и Zn, которые в указанных условиях не экстрагируются [530].
Дитизон. Наибольшее применение нашла экстракция кадмия в виде его дитизоната [88, 136, 195, 254, 281, 686, 725]. Разделение основано на различии кислотности водной фазы, из которой экстрагируются дитизонаты тяжелых металлов, и их неодинаковая устойчивость по отношению к маскирующим комплексооб-разователям, кислотам и щелочам [150, 281, 686].
Значения кислотности среды для экстракций дитизонатов схематически приведены на рис. 29. В сильнокислых растворах до кадмия взаимодействуют с дитизоном Hg и Ag, в слабокислых Au, Си и Bi. В сильнощелочных растворах (содержащих тартраты или цитраты) с дитизоном реагируют Ag, Cd, Со, Ni, Т1, и Zn.
Данные по кислотности растворов, разлагающих экстракты дитизонатов различных металлов, приведены в табл. 25. Из 1—2N растворов NaOH комплекс кадмия с дитизоном хорошо извлекается хлороформом. 0,2 А кислоты легко разлагают его дитизонат и переводят кадмий в водную фазу. Hg, Au и Си экстрагируются вместе с кадмием из щелочной среды. При обработке органического слоя 0,2 А кислым раствором эти элементы остаются в экстракте. Таким путем можно отделить кадмий от 100-кратных количеств указанных металлов. От 1000-кратных количеств ионов Bi3+, РЬ2+ и Т1+ кадмий отделяют экстракцией раствором дитизона в СС14 при pH 14. Препятствуют экстракции кадмия ионы S2-, CN~ в сильнощелочной среде, цитраты и тартраты — в нейтральной.
Для отделения Cd от Со, Ni и Zn к солянокислому раствору добавляют аммиак до pH 8—9 и экстрагируют все четыре элемента 0,002%-ным раствором дитизона в СС]4 в течение 2 мин. После промывания водой экстракт встряхи-
146
Е,°/о
Рис. 27. Влияние pH водной фазы на экстракцию некоторых элементов 0,1 М раствором бензоилацетона в бензоле [389, стр. 94]
Рис. 28. Влияние pH водной фазы на экстракцию некоторых элементов 0,1 М раствором дибензоилметана в бензоле [389, стр. 104]
Концентрация кислоты, К
13 7 /
т---1---1---1---1---1---1--
нд
Pd
Pl ____________________________________________________
Au
Cu
Bi
In Sn Pb Zn Cd Co Ni Fe TI
^w\wssx
^\\\\\\>v^
ZXxXx,.
_____I____I___I_____I___I_____I____I
О 4 8 /2 pH
Рис. 29. Пределы кислотности для экстракции дитизонатов некоторых элементов четыреххлористым углеродом
вают 30 сек. с 10 мл разбавленной НС1 с pH 2,5. В солянокиблый реэкстракт переходят Cd и Zn, а в органическом слое остаются Со и Ni. Водную фазу выпаривают до объема 5 мл, переносят в делительную воронку, добавляют 2,5 мл 2 N раствора КОН и экстрагируют кадмий 0,001 %-ным раствором дитизона в CCU [254].
При анализе Nb и Та высокой чистоты для отделения Cd применена его экстракция хлороформным раствором дитизона из тартратной среды. При pH 6 экстрагируется до 90% кадмия, при pH 9-100% [88].
Лучшим экстрагентом для кадмия служит этилпропионат, который при pH > 7,5 за одну экстракцию практически полностью экстрагирует его дитизонат [718].
Д и-2-э тилгексилфосфорная кислота, 1,5 М раствор в толуоле, экстрагирует из солянокислых растворов 10% кадмия [389].
0,01—0,ОЗМ раствор диэтилдитиокарбамината натрия в СС14 используют как экстрагент кадмия. Раствори-
148
Таблица 25
Условия для полной экстракции и реэкстракции дитизонатов металлов [150]
Элемент 4 Кислотность водной фазы Примечание
для экстракци для реэкстракции
Ртуть От 107V кислоты До 4,57V H2SO4 + Устойчив от
Серебро pH 4 От 5N кислоты До + галогениды 0,17V H2SO4 + 127V кислоты до 2 TV NaOH
Платина pH 7 От ЗУ НС1 до pH 4 + галогениды Устойчив к
Палладий От 107V кислоты смесь НС1 + HNO3 кислотам и щелочам Устойчив от
Золото От 2N кислоты До См.* 127V кислоты до 27V NaOH
Медь pH 0,5 От 1,07М кислоты До 6—77V H2SO4 или Устойчив в
Висмут Индий Цинк pH 4,0 pH 2-10 pH 5-6 pH 5,5-8,5 НС1 0,157V минеральные кислоты 0,017V NH4OH 0,017V HC1 3,07V H2SO4 Устойчив при
Кадмий Никель ’ Свинец Железо (II) Марганец (II) pH 6,5-14,5 pH 6,0-8,5 pH 7—10 pH 7-9 pH 10 0,27V HC1 pH 3,5 pH 4,5 Не разлагается кислотами Малоустойчив Неустойчив
Таллий pH И pH 6—7
* В литературе приведены разноречивые данные.
мость его комплекса с кадмием в хлороформе (2 г/100 мл) значительно выше, чем в СС14 (8 тиг/ЮО мл). При pH 5 в органический слой совместно с кадмием переходят Go, Mn, Ni, РЬ и V [142], но при обработке экстракта 12VHC1 кадмий реэкстрагируется. Константа экстракции диэтилдитиокарбамината кадмия равна 5,81 [143,389]. Описана экстракция кадмия раствором реагента в СС14при pH 3,7 [447]. В щелочной среде, содержащей тартрат, с этим экстрагентом можно разделить кадмий и медь [203].
а-Д итионафтойная кислота образует с кадмием комплекс 1 : 2, который экстрагируется СС14 [81].
Диэтилдитиофосфорная кислота образует с кадмием хелат, экстрагирующийся СС14 из солянокислых растворов [33].
149
Рис. 30. Влияние pH водной фазы на экстракцию некоторых элементов 0,1 М раствором 8-оксихинолина в хлороформе [389, стр. 127]
1—з— экстракция кадмия при 0 , 18 и 50° С соответственно [142, стр. 71]
Таблица 26
Зависимость экстракции 8-оксихинолина (1§.РНА) и оксихинолината кадмия (lg J*MA) от температуры [142]
Температура, °C lgpHA РКНА 1g «ех РН5о lgpMA 1g ₽МА
0 2,7 9,9 -4,4 4,2 4,9 21,2
18 2,7 9,8 -5,6 4,8 4,5 20,1
25 2,6 9,7 -6,1 5,0 4,1 19,5
30 2,6 9,7 -6,4 5,2 3,9 19,4
40 2,5 9,6 -7,2 5,6 3,7 18,6
50 2,5 9,5 -7,6 5,8 з,о 18,1
К°/с
1 00°J°
Fzzzz/xWI
-bzzzzzzzzzzzzl
- Pzz/zz/zl
~tzzz////z^zzzzzW4 '
Рис. 31. Пределы кислотности для экстракции некоторых элементов раствором 1-фенил-З-метил-4-бензоилпиразолона-5 в хлороформе
E^zzzzWzWz31 Wzzzzzz///zWaZ Vzzzzzzzz//z//ZWT~ lzzzzz//zWW~
IzzzzW/^F-
—i—i____i_i i ।
Мо(И) Ti V(?)
Fe(HI)
Си Pb
Ni
Co
Mn Cd
в pH
О
P-И зопиразолон экстрагирует кадмий из щелочных растворов на 99% [389].
0,1М раствор 8-о ксихинолина в хлороформе полностью экстрагирует кадмий в виде комплекса типа CdA2(HA)2, а в присутствии полярных растворителей (спирты, алкиламины) в виде CdA2(POH)2, CdA2(H2O)(PNH2) или CdA2(H2O)(P2NH). При применении менее концентрированных растворов 8-оксихи-нолина (0,001—0,0147) кадмий экстрагируется на 70—97%. Степень экстракции комплекса зависит от кислотности раствора (рис. 30) [389]. С увеличением температуры степень экстракции оксихинолината кадмия уменьшается, значение рН50 возрастает (см. рис. 30). Значения соответствующих констант представлены в табл. 26 [142].
1-Ф е н и л-3-м е т и л-4-б ензоилпиразоло н-5 в хлороформе извлекает кадмий из ацетатно-аммиачного раствора. Интервалы pH полной экстракции элементов схематически представлены на рис. 31 [145].
Экстракция тройных комплексов с органическими основаниями
Для разделения цинка и кадмия часто используют образование тройных комплексов, содержащих ионы металла, электроотрицательный лиганд и органическое основание. В основе разделения лежат различная устойчивость галогенидных и роданидных комплексов металлов. В качестве органических оснований используют хлороформные растворы реагентов, указанных в табл. 25. При этом более слабые основания (рК 9) образуют экстрагируемые соединения с иодидным комплексом CdJj- в кислой среде. Цинк в этих условиях не экстрагируется из-за различия в устойчивости этих комплексов. Сильные органические основания ipA <( 9) взаимодействуют в слабощелочной среде и с кадмием и с цинком (рис. 32, а).
В кислой среде хлоридные комплексы кадмия и цинка слабыми органическими основаниями (антипирин и трибензиламин) не экст-
151
Рис. 32. Влияние pH на экстракцию галогенидов кадмия (сплошная линия) и цинка (пунктир)
а, б, в —иодвдные, хлоридные и роданидные комплексы, соответственно
1, 1а — антипирин; 2, 2а — диантипирил-метан; з, за — трибензиламин; 4, 4а— дифенилгуанидин; 5, За — 1,10-фенантро-лин; 6,6а — пиридин; 1, Та — 2,2-ди-пиридил
рагируются, а в слабокислой и щелочной средах — экстрагируются совместно (рис. 32, б) [121, 253].
Устойчивость роданидных комплексов цинка и кадмия типа Me(SCN)f близка: Кнесг = 1,6-1(Г2 для Cd(SCN)t и 5,0-10~2 -для Zn(SCN)!-- В кислой среде слабыми органическими основаниями экстрагируется преимущественно роданидный комплекс цинка (кадмий при этом экстрагируется незначительно). В отличие от иодидных систем разделение кадмия и цинка в присутствии роданида протекает значительно хуже (рис. 32, в) [93, 123].
Экстракция хлоридного комплекса кадмия хлороформным раствором диантицирилметана используется для отделения его от цинка при определении в никелевых сплавах [124]. Оба элемента экстрагируются из. ЗА НС1, затем цинк реэкстраги-руется 2А H2SO4 в присутствии аскорбиновой кислоты и иодида калия, а кадмий при этом остается в экстракте.
2,2'-Д ипиридил образует с иодидным комплексом кадмия в присутствии ионов Fe2+ ионный ассоциат в 0,25 М растворе NaCH3COO, экстрагирующийся дихлорэтаном [621].
Дифенилгуанидин образует тройные комплексы с кадмием в кислой и щелочной среде. Цинк экстрагируется количественно лишь при pH 6 [253].
Пиридин в присутствии роданида образует с кадмием растворимый комплекс Cd(Py)2(SCN)2, который экстрагируется хлороформом. Это свойство используют для отделения кадмия от Ag, Hg и Си [619]. Экстракцией роданидного комплекса кадмия хлороформным раствором пиридина отделяют его от больших количеств таллия (предварительно последний осаждают избытком
152
KSCN) [2891. При экстракции кадмия из аммиачно-пиридиновых растворов коэффициент распределения комплекса с увеличением концентрации NH3 уменьшается (составляет в отсутствие NH3 и при его концентрации 0,04 М — 11,6 и 1,7 соответственно), что объясняется образованием координированных молекул NH3, число которых в данном случае равно 5 [4381.
При экстракции трибензоилметаном в дихлорэтане или три-н-о ктиламином в бензоле кадмий отделяется от Al, Со, Cr, Си, Fe, Mo, Mn, Ni и Zn в присутствии галогенидов [66].
Групповое экстракционное концентрирование кадмия и других элементов для их последующего определения различными методами дано в Приложениях 14 и 15. Библиографический указатель работ по экстракционным методам разделения и выделения кадмия, опубликованных в 1945—1967 гг., см. в [26, 27], с 1968 г.— в [471а].
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Для отделения и выделения кадмия используют ионообменную, адсорбционную и распределительную хроматографию [362, 466].
Условия отделения кадмия от других элементов суммированы в табл. 27.
Для отделения кадмия чаще всего применяют сильнокислотные катиониты - КУ-2, СДВ-3, ЭДЭ-10П, АН-9, АН-31, АВ-17 и Дауэкс-1 и S8TM, Дауэкс-50 и Амберлит IR-120, а также аниониты.
Катионит КУ-2 используют для отделения кадмия от Си, Ga и Zn.
+
В колонку диаметром 10 мм вводят 5 г катионита в Н-форме и промывают 0,001 М раствором тартрата, цитрата или триоксиглутарата. Затем через колонку пропускают растворы сульфатов Cd и Си (0,1 мл/мин) и вымывают медь 0,1 М раствором указанных выше солей. Кадмий затем элюируют 2 М НС1 [146].
Для отделения Cd от Си и Zn колонку промывают раствором моноэтаноламина, который полностью элюирует Си и Zn (кадмий остается на катионите) [346]. В присутствии цитрата, натриевой соли яблочной или п-аминосалициловой кислоты галлий образует анионные комплексы, а кадмий — катионные комплексы, которые сорбируются катионитом. Для его десорбции используют 2 М НС1; вместе с кадмием на колонке осаждается и цинк [398].
КатионитСДВ-3 сорбирует кадмий и медь. Последнюю элюируют щелочным раствором глицерина, образующим с ней растворимый комплекс. Кадмий затем снимают с катионита 2 N НС1.
Через колонку, содержащую 10 г катионита, пропускают 10 мл исследуемого раствора (1 мл/мин), промывают 20 мл воды и десорбируют медь 50— 70 мл щелочного раствора глицерина. После ее полного удаления (контроль
.153
Таблица 27
Ионообменное отделение кадмия
Ионит форма Среда Элюант Отделяемые элементы Литература
Катиониты
Амберлит инг 3%-ный раствор Hg, Bi, Pb. [33]
IR-120 NH4SCN Мп, U
Амберлит Na+ 0,05 М раствор [644]
Н+ Na2S2O3 0,4 М HJ и
НС1 (pH 1— 1,5) 4М НС1 Pt, Rh [649]
Na+ 2%-ный раствор Bi, Со, [502]
NaNO3 Мп, Ni, Zn
Дауэкс-50 Н+ О,1ЛГ 0,5 М раствор In [130]
CH3COONH4 в 0,14 М СНзСООН CH3COONH4
Н+ HF, H2SO4 — Al, Sb [458]
Солянокислая Дитизон + тетра- Ag, Bi, Co, [673]
гидрофуран + Cu, Fe, Pb,
+ HNO3 (1:9: 1) Th, U, Zn
nh4+ 0,1 М раствор NH4SCN Zn [685]
н+ Нейтральная 0,5 N HCl Zn [660, 754]
0,4 N НС1 н20 Cu [521]
КУ-2 н+ Моноэтаноламин Cu, Zn [346]
н+ Нейтральная Растворы солей оксикислот Cu [146]
н+ Растворы солей оксикислот 2 A HC1 Ga [398]
СДВ-3 н+ Щелочная 2 N НС1 Cu [311]
Аниониты
AH-9 AH-31 СГ 0,7 A H2SO4+ + 0,3 M раствор KJ 2 M HCl Н2О Zn [348] [302]
AB-17 0,7 N H2SO4 + 4- о,з м раствор KJ Zn [348]
Дауэкс-1 10% HCl 3 М HNO3 Co, Cu, Ni [590]
NO3- 0,5 N HNO3 в этаноле 1 N HNO3 Al, Zn [620]
СГ S2Oa2' 2M HCl 1 M раствор Na3S2O3 1 М HNO3 Cu In, Pb [534] [395]
ЭДЭ-10П 2 М HCl 0,05 М HCl Cr, Ni, Те, Fe [272]
154
Таблица 27 (окончание)
Ионит Форма Среда Элюант Отделяемые элементы Литература
ЭДЭ-10П 0,5 N НС1 0,005 N НС1 Со, Cu, Ni, Zn [469]
0,7 N H2SO4+ + 0,3 N раствор К J Zn [348]
S8TM С1- 1А НС1 0,01 N СН3СООН Al, Си, Ni [733]
с K.4[Fe(CN)e]) колонку промывают 20 мл воды и элюируют кадмий 80 мл 2 N НС1; полноту его извлечения проверяют сульфидом [311].
Амберлит IR-120 сорбирует Cd вместе с Bi, Со, Мп Ni и Zn на колонке диаметром И мм и высотой 10 см. Затем кадмий элюируют 2%-ным раствором NaNO2 (3 мл/мин)-, на вымывание 200 мг Cd требуется 200 мл элю-анта. После удаления кадмия Со, Мп, Ni и Zn десорбируют 5%-ным раствором NaNO2, a Bi — 2,5 М НС1 [502].
При анализе платино-родиевых сплавов из солянокислых растворов с pH 1—1,5 кадмий вместе с другими примесями сорбируют на Амберлите IR-120 и элюируют его 4 М НС1 [649].
При разделении смесей кадмия с Bi, Hg, Мп, Pb или U используют различную устойчивость роданидных комплексов этих элементов. Анализируемый раствор, содержащий ~ 10 мг каждого элемента, пропускают через колонку со скоростью 2—3 мл]мин, а для элюирования применяют растворы роданида аммония различной концентрации (для Cd 3%-ный раствор) [643].
Для элюирования кадмия с Амберлита IR-100 лучше всего использовать 0,05 М раствор Na2S2O3 [644].
Д а у э к с-50. При совместном определении Cd и Zn в светящихся составах пробу растворяют в небольшом количестве НС1 и выпаривают раствор до полного удаления ионов S2~. Остаток растворяют в воде, раствор нейтрализуют едким натром и пропускают через колонку с катионитом. Затем элюируют кадмий 100 мл 0,5 N НС1, а цинк — 60 мл 2,5 N НС1. Количественное разделение элементов достигается при их соотношении от 1 : 100 до 100 : 1 [660].
В другом методе разделения зтих элементов для кадмия в качестве элю анта используют 0,1 М NH4SCN в 90 %-ном метаноле, а для цинка — 0,1 М раствор NH4SCN в 80 %-ном этаноле. Метод обеспечивает эффективное разделение при отношении компонентов от 1 : 10 до 10 : 1 [6851.
Для разделения Cd и Си 0,01 М раствор сульфатов обоих элементов в 0,4 N НС1 пропускают через колонку с катионитом и элюируют кадмий водой (1 мл!мин), затем Сп2+ — 4 N НС1 (0,5 мл/мин) [521].
При отделении кадмия от In пробу растворяют в 0,14 N СН3СООН, 0,1 М CH3COONH4 и пропускают через колонку. Сорбированный кадмий вымывают .0,5 М раствором CH3COONH4 [130].
155
Из сильнокислых растворов кадмий вместе с Ag, Bi, Go, Cu, Fe, Pb, Th, U, Zn и РЗЭ сорбируется на колонке, с которой его элюируют смесью дитизона с тетрагидрофураном и HNO3 (1:9:1) [673]. От больших количеств А1 и Sb кадмий может быть отделен из фторидных или сернокислых растворов [458]. Изучено концентрирование следовых количеств кадмия совместно с Ag, Al, Со, Cu, In, Mn, Ni и Zn с последующим элюированием кадмия вместе с примесями 12,2 А НС1 [754].
Иминодиацетатный катионит IDE использован для отделения кадмия от Мп, РЬ и U. При пропускании через колонку раствора, 1 М по NH4NO3, 5 М по NH4OH и 1 М по KCN, Pb, V сорбируются катионитом, а кадмий вместе с Ag, Со, Hg и Zn проходят в фильтрат [587]. Марганец тоже сорбируется и может быть десорбирован 0,3—0,5 М НСЮ4 или 0,5 М HNO3, нагретой до 50° С [586].
Анионит ЭДЭ-10 П используют при определении малых содержаний кадмия в сульфидных свинцово-цинковых рудах, содержащих Cr, Ni, Т1 и до 20% Fe (все они сорбируются из 2 М НС1). Кадмий десорбируют 0,05 М НС1 [272].
При анализе продуктов цветной металлургии кадмий отделяют от Ni, Си, Со и Zn пропусканием раствора в 0,05 N НС1 через колонку. Кадмий элюируют 0,05 М НС1 [469].
Для разделения Cd и Zn из растворов, 0,3 М по KJ и 0,7 N по H2SO4, кроме ЭДЭ-10П, используют также аниониты АН-9 и АВ-17. С увеличением концентрации H2SO4 от 0,1 до 1,5 А коэффициенты распределения Cd и Zn понижаются в ряду АВ-17 — ЭДЭ-10П — АН-9. При 0,75 М H2SO4 коэффициент распределения кадмия достигает максимального значения при молярном отношении J" : Cd2+ = 4 [348].
Предложен бесколоночный метод отделения кадмия при анализе железистых и медистых материалов. Метод основан на избирательном поглощении кадмия анионитом ЭДЭ-10П из солянокислого раствора и десорбции его 0,01 N НС1.
Пробу разлагают смесью НС1 и HNO3 и добавляют 20 мл 2,5 N HCI. Нагревают до кипения, охлаждают и отфильтровывают нерастворимый остаток. В фильтрат помещают анионит и взбалтывают в течение 2 мин. Затем сорбент с Cd, Fe и Си отфильтровывают и промывают 0,5 TV" HCI. Fe, Си и другие примеси при этом переходят в раствор, а кадмий элюируют с анионита. 0,01 N НС1[533].
Хлоридные комплексы кадмия хорошо сорбируются на анионите АВ-17 даже из разбавленной НС1; для анионита АН-2Ф концентрация НС1 должна быть >6 М [198]. Для анализа сталей и сплавов применяют анионит АН-31.
Пробу растворяют в смеси НС1 и HNO3. Раствор выпаривают досуха, сухой остаток растворяют в 12,5 мл 8 М НС1 и фильтруют. Фильтрат разбавляют до 50 мл водой и 25 мл полученного раствора пропускают через колонку с анионитом, предварительно промытую 50 мл 2 М НС1. Затем элюируют с колонки все примеси 50 мл 2 М НС1, после чего десорбируют кадмий вместе с РЬ и Zn 50 мл воды [302].
Для отделения Cd от А1 или Zn из азотнокислых растворов, содержащих органические растворители, используют Д а у э к с 1.
156
0,5—1,0 г пробы растворяют в конц. HNO3, раствор выпаривают досуха, остаток растворяют в 5 мл HNO3, добавляют 45 мл этанола и пропускают через колонку с анионитом, предварительно промытую смесью 5 N HNO3 и этанола (1 : 9). Cd, 2п(или А1) осаждаются на анионите. Кадмий затем десорбируют 1A HNO3.
Метод рекомендован для отделения миллиграммовых количеств кадмия от значительно превосходящих количеств Zn и AI [620].
При анализе металлической меди 1—2 г пробы растворяют в 15 мл разбавленной НС1 в присутствии 1,2 мл 30%-ной Н2О2. Раствор нагревают до удаления избытка Н2О2, разбавляют раствор до 2 М по НС1 и сорбируют Cd, Sn и Zn на анионите Дауэкс-1. Затем колонку промывают 20 мл 2 М НС1 и-десор-бируют Cd вместе с Sn и Zn 1 М HNO3 [534].
При пропускании 10 %-ной НС1 через этот же анионит Cd nZn можно отделить от Со, Си и Ni, для элюирования первых двух элементов используют 3 М HNO3 [590]. Те же два элемента отделяют от In и РЬ из 1 М раствора Na2S2O3, последние два сорбируются анионитом, a Cd и Zn остаются в растворе [395].
Микроколичества Cd и Zn отделяют на анионите S8TM от 104—108-кратных количеств А1, Си и Ni.
60—80 анализируемого раствора, 1 N по НС1, пропускают через колонку высотой 6 см, диаметром 0,45 см, предварительно промытую 30—50 .мл 1 N НС1. В фильтрат проходят А1, Со и Ni, a Cd и Zn сорбируются. Кадмий элюируют 40 мл 0,01 N CH3GOOH, a Zn — 30 мл 0,001 N НС1 [733].
Групповое разделение Ag, Ba, Ca, Cd, Со, Си, К, Mg, Mn, Ni, Pb, Sn и Zn исследовано на смоле Sel-K2 [632].
Адсорбционная хроматография. В качестве сорбентов используют некоторые неорганические соединения и целлюлозу.
Al, Ag, Ba, Cd, Со, Cr, Cu, Fe, Mn, Ni и Zn разделяют на колонке, заполненной сорбентом Fe(OH)3. Раствор исследуемой смеси пропускают через колонку, на которой указанные катионы располагаются по увеличению их сорбционной способности: Ba, Ni, Со, Mn, Cd, Zn, Cu, Ag, Pb, Cr, Al и Fe. Для вымывания катионов с колонки ее промывают водой, а затем 0,01 М HNO3 [630].
Изучено адсорбционно-хроматографическое разделение дити-зонатов Cd и Zn, Cd и РЬ, Cd и Bi с использованием в качестве сорбентов КНСО3 и трехзамещенного цитрата калия [57]. Разработан метод отделения кадмия от мешающих элементов с помощью минерального ионообменника — фосфата кальция [221].
Кадмий от галлия отделяют на колонке с целлюлозой. Комплекс галлия с n-аминосалициловой кислотой избирательно сорбируется [397].
Cd, Zn и Hg разделяют на модифицированной порошкообразной целлюлозе в присутствии их радиоактивных изотопов e8Zn, 108Cd, 2o3Hg. Следовые количества Cd и Zn прочно удерживаются функциональными группами модифицированной целлюлозы (фосфат, карбоксиметил, аминоэтил и др.); Hg в этих условиях полностью проходит в фильтрат. Этим путем можно отделить >0,01 мкг Cd и Zn от 3 г Hg [644].
157
Распределительная хроматография. Для отделения кадмия применяется редко. Носителями служат бумага, силикагель, окись алюминия и другие сорбенты, в качестве подвижной фазы используют органические и неорганические растворители. Разделенные на хроматограммах зоны проявляют по образованию цветных реакций с соответствующими органическими или неорганическими реагентами, а затем обрабатывают эти зоны для последующего количественного определения.
При разделении Cd, Ga, In и Zn методом хроматографии на бумаге подвижной фазой служит смесь 45 мл «-бутанола, 12 мл воды, 0,02 г комплексона III и 0,92 мл НС1 (уд. в. 1,19). Хроматограмму проявляют опрыскиванием 0,05%-ным раствором дитизона в СС14 [310].
При разделении дитизонатов Cd и Со в качестве подвижных фаз изучены растворы дитизона в СНС13, СС14, эфирах, спиртах и других смесях; лучшие результаты получены с СНС13 и СС14.
Разделение на бумажных дисках диаметром 15 см, имеющих надрезы по радиусу длиной 2—3 см и шириной 1—3 мм, проводят следующим путем: 0,03 мл раствора СНС13, содержащего 20 мкг дитизоната каждого элемента, наносят в центр диска, подсушивают и помещают в закрытые камеры, содержащие насыщенные пары растворителя. О положении зон Cd и Со судят по окрашенным кольцам дитизонатов [503].
Для разделения Ag, Bi, Cd, Со, Си, Ni и Zn предложена бумага, пропитанная дитизоном. Лучшим проявителем является смесь 3 N СН3СООН и ацетона (1 ! 1,2). Порядок расположения полос на кружках фильтровальной бумаги находится в прямой зависимости от констант устойчивости дитизонатов разделяемых металлов [286].
Описано разделение Cd, Со, Си, Ni и Zn методом радиальной (на кружках бумаги Шлейхер-Шюлль № 583) и восходящей (на полосках бумаги Ватман № 426) хроматографии.
Анализируемый раствор, содержащий по 10—30 мкг указанных элементов, наносят на бумагу и обрабатывают смесью 2 мл 96%-ного этанола, 4 мл НС! (уд. в. 1,19), 7 мл воды и 87 мл ацетона. Полученные хроматограммы высушивают под инфракрасной лампой и опрыскивают раствором, содержащим 50 мл 1 %-ного этанольного раствора диметилглиоксима и 5 мл концентрированного аммиака: зона Ni окрашивается в красный цвет, Со—в коричневый, Си—в зеленый. Для проявления Cd и Zn хроматограммы обрабатывают 1%-ным этанольным раствором 8-оксихинолина; их зоны становятся желтыми и флуоресцируют при облучении в УФ [765].
Для открытия Cd, Bi и Pb на полосах бумаги Ватман используют 2-меркапто-5-анилино-1,3,4-тио диазол.
Подвижным растворителем служит смесь н-бутанола, кислот (для Cd — СНзСООН, для Bi и Pb — HNO3) и воды (4:1: 5). Проявляют хроматограммы 1%-ным этанольным раствором указанного реагента. Ионы Cda+ образуют белое пятно с желтой каймой, Bi31" — светло-желтое, РЬ’+ — жел-
Чувствительность открытия Cd—30,7, Bi—0,l, Pb—0,4 мкг [692].
158
Для количественного разделения Cd и Си применен ацетил-ацетон, образующий в ацетатном буфере нерастворимый комплекс с медью и растворимый ацетилацетонат кадмия. Их разделение возможно при соотношениях от 50 : 1 до 1 : 50. Полученную при 25—27° С хроматограмму опрыскивают 1 М раствором (NH4)2S; ионы Cd при этом дают желтое окрашивание, а Си — черное [494].
В качестве подвижной фазы при разделении и определении Cd, Со, Си, Fe, Hg и Ni использована смесь бутанол — ацетон — НС1 (уд. в. 1,19)— вода (38 : 18 : 22 : 7). Хроматографирование проводят на двух одинаковых полосках бумаги из двух отдельных аликвотных частей пробы. Одну хроматограмму опрыскивают растворами реагентов, дающих с разделяемыми ионами цветные реакции и соответственно полученным на ней окрашенным зонам, на второй хроматограмме очерчивают карандашом соответственно зоны отдельных ионов. Затем эти зоны вырезают и вымывают содержащиеся в них ионы металлов. Полученные злюаты всасывают полоской бумаги, пропитанной соответствующим раствором. Для кадмия используют Zn2[Fe(CN)e], Площади окрашенных пятен пропорциональны содержанию определяемых ионов [631].
Показана возможность хроматографического выделения кадмия из смеси катионов на бумаге, пропитанной фосфатом олова. Для ее приготовления полоски бумаги сначала опускают в горячий 17,6%-ный раствор SnCl4-5H2O, удаляют его избыток и затем на несколько секунд опускают в горячий раствор 10 %-ной Н3РО4. Подвижной фазой служат растворы НС1 различной концентрации, с увеличением которой коэффициент распределения большинства ионов металлов увеличивается (для кадмия он составляет 0,87). Развитие хроматограммы осуществляют восходящим способом [701 ].
Описан метод отделения и косвенного определения микроколичеств кадмия, в присутствии большого числа посторонних ионов с использованием хроматографической бумаги, обработанной раствором иодида свинца. Метод основан на осаждении Cd2+ определенным количеством раствора карбоната в специальном капилляре и хроматографировании избытка ионов СО|~ на бумаге Ватман №1, пропитанной раствором PbJ2. Количество кадмия определяют по степени интенсивности окрашенного пятна [175].
При разделении Cd, Bi и Си на фильтровальной бумаге Ватман № 40, пропитанной смесями минеральных кислот и органических растворителей, используют 48 мл СНС13, 4 мл НС1 (уд. в. 1,19), 24лел этанола и 24л«л ацетона (12 : 1: 6 : 6). Коэффициенты распределения возрастают в ряду Си Bi Cd [504]. Растворители, содержащие хлороформ, были испытаны для смесей ионов Cd2+, Cu2+, Hg2+, Bi3+ и Pb2+; наилучшее! разделение компонентов достигается при использовании смеси СНС13—СН3ОН — ацетон — изопентанол — НСООН (1 : 1 : 1 : 1 : 0,5) [690].
Подвижной фазой для разделения Cd, Си, Ni и Zn на бумаге могут служить 10—90%-ные растворы н-пропанола в воде, в кото
159
рых коэффициенты распределения уменьшаются в ряду Ni > Си > >Cd>Zn [609].
Описано отделение Cda+ от Ti на бумаге Ватман № 1 при использовании смеси НСООН — НС1 — ацетон (3:5:2) [702].
Поведение Cd, Cu, Bi, Se, Те и благородных металлов изучено методом хроматографии на бумаге. Применение 1 М раствора тиомочевины в 4 М НС1 позволяет разделить смесь Se — Bi — Cd— Те - Cu [446].
Предложено разделение ~ 30 элементов методом хроматографии с обращенными фазами на бумаге, предварительно пропитанной 2%-ным этанольным раствором фенил бензогидроксамо-вой кислоты в смеси (40 : 60) октана и этанола. Для обнаружения Ag, Au, As, Bi, Cd, Co, Cu, Fe, Hg, Ni, Pt, Sb, SnnV хроматограмму опрыскивают раствором (NH4)2S, а затем соляной кислотой [566, 729].
Хроматографическое разделение на бумаге, пропитанной послойно три-н-октиламином и АмберлитомЬа-А, с подвижной фазой— растворами НСООН, СН3СООН и СН2С1СООН — применено к смесям Al, Be, Cd, Со, In, La, Mn, Ni, Pb и TI. В 0,05—6 Mкислотах их коэффициент распределения >0,8 [698]. В качестве подвижной фазы были испытаны также 0,5—1 М НС1 и 0,5—4 М растворы NaCl, для Cd2+, Zn2+, Hg2+, Sn4+, Au3+ и Pt4+ значения коэффициента распределения невелики и мало зависят от изменения концентрации НС1 и NaCl в указанных пределах [769].
Тонкослойная распределительная хроматография. Предложено разделение и открытие Cd, Си, Hg, Ni и Zn методом тонкослойной хроматографии на силикагеле, пропитанном дитизоном.
Тонкий слой готовят смешиванием 30 мл 0,3%-ного раствора дитизона в СНС13 с 10 г силикагеля марки Wakogel-О. Для развития хроматограммы применяют смесь 1—2 М СНзСООН и ацетона (1 : 1) или 2—4 М раствора CH3COONa и ацетона (1 : 0,66). После проявления на хроматограмме образуются окрашенные концентрические кольца. Хроматограммы на тонком слое более четки, чем полученные тем же путем на бумаге, пропитанной дитизоном [103].
Cd, Pb и Zn разделяют на пластинке, покрытой тонким слоем, силикагеля (0,2 мм).
Хлороформные растворы дитизонатов указанных металлов наносят на пластинку. Для развития хроматограммы (15—35 мин.) используют смесь СвН6 и СН2С12 (5 : 1), коэффициенты распределения Cd, Pb и Zn равны 0,13; 0,34 и 0,50 соответственно [445].
Cd, Be и Са разделяют в виде оксихинолинатов методом тонкослойной хроматографии. Сорбентом служит смесь силикагеля и порошка целлюлозы (1 : 1). Для развития хроматограммы используют смеси 2 %-ной СН3СООН, СН3ОН и 2-пропанола (6 :3 : 1). При облучении в УФ (360 нм) по интенсивности флуоресценции хроматограммы можно открыть 0,025—1,0 мкг металла [402].
160
Cd, Cu, As, Hg, Bi, Pb, Sb и Sn также могут быть разделаны на пластинках, покрытых тонким слоем целлюлозы или силикагеля. В качестве подвижной фазы при этом используются смеси этанола, гексана, ацетилацетона с кислотами (HCI, HNO3, СН3СООН) различной концентрации. Подсушенную пластинку с нанесенным испытуемым раствором опрыскивают 2%-ным этанольным раствором 8-оксихинолина и выдерживают в атмосфере NHS. Указанные элементы обнаруживают по люминесценции зон в УФ [629].
Cd от Cu, Со, Ni и Zn в виде роданидных комплексов отделяют на силикагеле марки G. Проявитель—0,5%-ный этанольный раствор 8-оксихинолина. В качестве подвижной фазы можно использовать смеси различных составов (с обязательным наличием в них иона SCN~) : 80 мл С2Н6ОН (или СН3ОН, и-бутанол, изопропанол), 10 мл 10%-ного раствора (CH3)4NOH (или NH4OH, NaOH) и 4 г NH4SCN; 60 мл С2Н5ОН, 40 мл метилэтилкетона, 10 мл (CH3)4NOH, 10 мл сс-пиколина и 4 г NH4SCN. Кадмий определяют по желтой флуоресценции в УФ-свете [762].
Разработан способ разделения диэтилдитиокарбаминатов кадмия, Hg, Pb, Си и Bi на тонком слое силикагеля марки «Д». Подвижной фазой служит смесь н-гексана, СНС13 и диэтиламина (20 : 2 : 1). После разделения элементов пластинку подсушивают для удаления диэтиламина и опрыскивают 5%-ным раствором CuSO4; диэтилдитиокарбаминаты металлов при этом окрашиваются в коричневый цвет. Коэффициенты распределения диэтилдитиокарбаминатов Cd, Pb, Cu, Hg равны 0,34; 0,27; 0,44 и 0,56 соответственно [728].
Предложено разделение кадмия, Cu, Fe3+, Bi, Pb и Ni на тонких слоях силикагеля, нанесенного на фильтровальную бумагу; в качестве подвижной фазы применен 0,01 М раствор Na2HAsO4. Для обнаружения Cd, Cu, Bi, Fe3+ и Pb хроматограмму опрыскивают раствором (NH4)2S, а для обнаружения Ni — диметилглиокси-* мом [595].
Смола Челекс-100 использована при хроматографическом разделении Cd, Cu, Hg и РЬ.
Длн приготовления тонкого слоя применяют смесь 30 г смолы с 5 г целлюлозы М № 300 и 60 мл воды. В качестве подвижной фазы используют 0^25]М раствор LiCl. Проявляют хроматограмму щелочным раствором Na2S
Исследовано хроматографическое разделение Cd, Со, Cu, Fe, Hg, Mn, Ni и Zn в виде их комплексов с пиридил-2-альдегид-2-хинолингидразоном на тонких слоях окиси алюминия [564],
6 Щербов Д. П,
Глава VI
ОПРЕДЕЛЕНИЕ КАДМИЯ
В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
Наиболее часто применяемые растворы при анализе
Стандартные растворы кадмия. 1. И з металлического гранулированного кадмия (ГОСТ 1467—42). Точную навеску 1 г растворяют в смеси НС1 и HNO3 и раствор упаривают почти досуха. Для полного удаления HNO3 выпаривают еще 3 раза с 5—10 мл НС1. Остаток CdCl2 высушивают досуха, растворяют в горячей воде, по охлаждении переносят в мерную колбу емкостью 1 л и разбавляют водой до метки [196, 258]. Полученный раствор с концентрацией 1 мг Cd/мл разбавляют по мере надобности до содержаний 0,05 и 0,01 мг Cd/мл (50 и 10 мкг Cd/.чл) или других, предусмотренных методикой определения.
2. Из сульфата кадмия (3CdSO<-8H2O, х.ч. ГОСТ 4456—48). Для контроля качества препарата его точную навеску (Н) ~ 1 г сушат в фарфоровом тигле в течение 30 мин. при 100—105° С, затем постепенно повышают температуру до 600—800° С и прокаливают до постоянного веса (В).
Содержание чистой соли находят по формуле
% 3 CdSO4 • 8Н2О = -23^5'В .
2,2820 г 100%-ного препарата растворяют в воде и разбавляют в мерной колбе водой до 1 л; 1 мл такого раствора содержит 1 мг Cd [196, 258].
Хлоридно-аммиачный полярографический фон. В 500 мл воды растворяют 100 г NHiCl и 100 г Na2SO3-7H2O (или 50 г Na2SO3), приливают 150 мл 25%-ного NH4OH, 50 мл 1 %-ного раствора желатина и разбавляют водой до 1 л. Работать следует со свежеприготовленным раствором. В некоторых методиках в качестве подавителя максимумов вместо желатина применяют агар-агар или столярный клей; иногда используют в 2 раза менее концентрированный фон.
Титрованный 0,05 М раствор комплексона III. Точную навеску 18,61 г чистого препарата растворяют в воде и раствор разбавляют в мерной колбе до 1 л. При недостаточной чистоте препарата титр устанавливают по металлическому цинку, 0,05 М раствор которого готовят из электролитного металла. Для этого 3,2690 г Zn растворяют при слабом нагревании в 20—30 мл НС1 (1 : 1) и разбавляют бидистиллятом до 1 л. При помощи бюретки или точной пицетки отбирают 25 мл раствора цинка в мерную колбу емкостью 250 мл, разбавляют водой до 50 мл, прибавляют 10 мл хлоридно-аммиачного буферного раствора с pH 10, на кончике шпателя — сухую смесь индикатора кислотный хром черный ЕТ-00 с NaCl (1 : 100) и титруют комплексоном III винно-красный раствор до перехода его окраски в сине-фиолетовую.
Очистка загрязненного препарата описана в [196].
Подробно приготовление растворов других реагентов дано в работе [196, 258].
162
АНАЛИЗ МИНЕРАЛЬНОГО СЫРЬЯ
Методы разложения
Разложение проб, содержащих кадмий, обычно не представляет затруднения; для этого чаще всего применяют кислотный способ вскрытия. Сульфидные руды и минералы разлагают HCl (1 : 1) [10]. Окисленные, полиметаллические и другие руды более сложного состава вскрывают смесью концентрированных НС1 и HNOS (3 : 1) [82, 97, 768] или HCl, HNO3 и H2SO4 (1:1: 0,5) [10]. Применяют и нагревание при 240—250° С с сухой смесью (1 : 1) нитрата и хлорида аммония [97; 429, стр. 611].
Восстановление в токе водорода при 300° С использовано в анализе бокситов, хромитов и касситеритов; восстановившийся до металла кадмий отгоняют затем в токе азота при температуре ~ 800° С. По выходе из печи металл осаждается на холодной поверхности прибора и его растворяют смесью НС1 и HNO3 (3 : 1) [97].
Методы разделения
В зависимости от применяемого метода определения необходима та или иная степень отделения кадмия от элементов, мешающих выполнению анализа. Наиболее тщательное отделение необходимо при гравиметрическом определении, меньшее — в фотометрических и полярографических методах.
При осаждении кадмия с 0-нафтохинолином медь отделяют тиосульфатом натрия, РЬ — сульфатом; большие количества железа осаждают в виде гидроокисей избытком аммиака, вместе с ним выделяются As, Sb и Bi [10].
Определению кадмия методом внутреннего электролиза мешают As, Cu, Fe, Pb и Sb. Поэтому Pb отделяют в ходе анализа в виде сульфата, Си — в виде роданида после ее восстановления до Cu(I) действием SO2. Железо окисляют Н2О2 и выделяют (вместе с As, Bi, Sb и остатком Pb) аммиаком [429, стр. 690].
При полярографическом определении, как правило, не требуется отделения кадмия от сопутствующих элементов. Однако большие количества меди осаждают тиосульфатом натрия из горячей 3—5 %-ной H2SO4 [429, стр. 609] или цементируют на свинцовой пластинке из горячей HCl (1 : 40) [272]. Мешающие элементы могут быть отделены также на колонке с анионитом ЭДЭ-10П из 2 М НС1. В этих условиях вместе с кадмием на колонке осаждаются Bi, Pb и Zn, которые затем десорбируют НС1 различных концентраций [272]:
Металл Pb Cd Zn Bi
HCl, M 0,01 0,05 0,1 0,7
При полярографическом определении кадмия рекомендуют отделять его от Al, Fe, Мп и других элементов осаждением горя-
6*
163
чим 0,05 % -ным раствором рубеановодородной кислоты из слабощелочной среды. Осадок, содержащий также Со, Си, № и Zn, обрабатывают в кварцевой чашке H2SO4, прокаливают при 500— 650° С и растворяют в НС1.
Благодаря разнице в значениях Еу„ умеренные количества Со, Си, Ni и Zn определению кадмия не мешают (их тоже можно полярографически определять в этом растворе) [2711.
Методы определения
Краткая характеристика методов определения кадмия в минеральном сырье приведена в табл. 28.
Полярография. Для определения кадмия в природных объектах чаще всего используют полярографию.
При анализе свинцово-цинковых и сульфидных (неокисленных) медных руд, содержащих от 0,02 до 5,0% Cd, следы Со, Ni и Те и 20% Fe, применяют упрощенный (унифицированный) метод, основанный на том, что практически все минералы, содержащие Cd, разлагаются в НС1, тогда как пирит и сульфиды меди в ней почти нерастворимы. Поэтому чаще всего разделений не производят, если количество Си в полярографируемом растворе не превышает содержания Cd более, чем в 2 раза.
0,1—2,0 г тонко истертой пробы помещают в стакан емкостью 100—150 мл, добавляют 25 мл НС1 (1 : 1) и кипятят 20—25 мин. Нерастворимый остаток отфильтровывают и несколько раз промывают на фильтре горячей водой, подкисленной НС1. Фильтрат и промывные воды кипятят несколько минут с 5—6 каплями 30%-ной Н2О2 до полного окисления перешедшего в раствор железа, после чего раствор упаривают до объема 5—10.в л. Охлаждают и количественно переносят в мерную колбу емкостью 50 мл. Раствор нейтрализуют 25%-ным NHiOH до появления гидроокисей или явного запаха аммиака, добавляют 20 мл раствора хлоридно-аммиачного фона. Разбавляют до метки водой и тщательно перемешивают. Для полного восстановления растворенного кислорода раствор оставляют на 20—30 мин., после чего прозрачный раствор сливают в электролизер и поляр огр афир уют в пределах напряжения поляризации от 0,7 до 0,9 в (н.к.э.).
Содержание кадмия находят по градуировочному графику, построенному по данным полярографирования серии эталонных растворов, содержащих от 0,25 до 25,0 мг Cd и проведенных через ход анализа одновременно с пробами и рассчитывают по формуле:
% Cd — Н-1000 • ЮО = ,
где а — найденное по калибровочному графику количество Cd, мг; Н — вес пробы, а.
Если ожидаемое содержание кадмия составляет доли процента, а в анализируемом материале находится более 5% Fe, то во все эталонные растворы вводят по 5—100 лег FeCl3.
При высоком содержании Си, растворимой в НС1, солянокислый фильтрат выпаривают досуха. После охлаждения к остатку приливают.20 мл НС1 (1 : 40) и нагревают до растворения солей. В полученный раствор помещают свернутую в спираль свинцовую пластинку (толщина 1 мм, ширина 10—15 мм и длина 100—150 л«ж) и продолжают нагревание в течение 20—25 мин., следя за Чем, чтобы раствор кипел не слишком сильно. Затем спираль с осевшей на ней медью вынимают и споласкивают водой. При большом количестве Си
S о П
& 0) § й о R 05 Сб
ОО ОО I-
«е О о о о
сП СП * £ СП СП
е» О О N
Д К ® д
+ + И Ч. + +
СО СО о о со о + + О о
Z к Д О О Д д
и к и д д д д
+ о + + Ей ЕЙ _|. + + + +
о о о ООО о
к яи к Д К К д
Содержание кадми: % Г о up f lol Т Г 7 о о со о . Е-* • Г-Ч Ф чН
Анализируемый объект й Ф Я Я ® 3 s В, _ © а & В ф § g «я ф И S и § ® S>e<' ® « § g S и «я Е й я ® ₽ и Ви В ф и я 2 В. Я ® « о к Й F о И S 3 И PS и о g В & к g & §«> i f и и я в а й S® В-g Ft 5 W Я *5 О g Р.Й йО Р С Ри
А Рч ф & ф Я Я
о о ф о я, 5 ° В. и g* § § А й Рч Q Ф Я Я Я Я Е-*
Я й Ф ч я & 3 а й Я й Ф ф ЕС Ф О Я я
Я О Д н - я —, о К И § я >2 о а л о Рч Я хр а
н: ₽8 2 Ф Ж Ft
Я но
Рч о Д я Рн еФ Р^
164
цементацию сначала проводят на холоду. Как только поверхность свинца покроется красным осадком, спираль вынимают, споласкивают водой и заменяют новой. В случае ее омеднения цементацию продолжают уже при нагревании до прекращения заметного выделения Си. По окончании процесса продолжают операции основного хода анализа.
Определение низких содержаний кадмия в рудах сложного состава проводят после его отделения на анионите ЭДЭ-10П. Метод рекомендован для горных пород, содержащих 0,0005— 0,5% Cd, до 10% РЬ и до 5% Sb.
0,5—2,0 а тонко истертой пробы помещают в стакан емкостью 100 мл, добавляют 15—20 мл HCI (1 : 1), накрывают стеклом и кипятят до прекращения выделения H2S. Осторожно приливают 5—10 капель 30%-ной Н2О2 и продолжают кипячение еще 5—10 мин. Снимают стекло, споласкивают его водой и выпаривают раствор досуха. К охлажденному остатку прибавляют 20—30 мл HCI (1 : 5), нагревают, кипятят 1—2 мин. Отфильтровывают нерастворимый остаток, промывают 4—5 раз HCI (1 : 5) и отбрасывают.
Фильтрат и промывную жидкость охлаждают и количественно переносят в колонку с анионитом ЭДЭ-10П (диаметр 0,8—1,0 см, высота слоя анионита 12—15 см), предварительно промытым 200 мл HCI (1 : 250), и переведенным в С1~-форму 50 мл HCI (1 : 5).
После пропускания раствора через смолу колонку промывают несколько раз 25 мл HCI (1 : 5) и затем 100—150 мл HCI (1 : 10) до отрицательной реакции на Fe с роданидом. Элюат, содержащий Al, Cu, Fe, Mn, Ni и другие элементы, отбрасывают, a Cd десорбируют 250 мл HCI (1 : 250), собирая раствор в стакан емкостью 500 мл. Элюат упаривают досуха, избегая перегревания. К сухому остатку прибавляют 2—3 капли НС1 (уд. в. 1,19) и, в зависимости от ожидаемого содержания Cd, 10 или 25 мл раствора хлоридно-аммиачного фона (разбавленного в 2 раза, приготовление см. стр. 162); раствор перемешивают и через 10—15 мин. после охлаждения до комнатной температуры полярографируют в пределах напряжения поляризации от 0,7 до 0,9 в (н.к.э).
Содержание кадмия находят по градуировочному графику, полученному полярографированием эталонных растворов 0,01—0,5 мг Cd и рассчитывают по формуле, приведенной выше.
Другие химические способы. Из фотометрических методов определения применяли колориметрирование по желтой окраске CdS. Для отделения от мешающих элементов кадмий предварительно осаждали с метиловым фиолетовым [340] или р-нафтохинолином 110). Для определения микрограммовых количеств кадмия в рудах и силикатных породах чаще используют колириметрирование с дитизоном [363, 686].
0,5 г породы разлагают в платиновой чашке смесью НС1О« и HF (1:5) и раствор выпаривают досуха. Остаток смачивают 1 мл НС1 (уд. в. 1,19), добавляют 5 мл воды и нагревают почти до кипения (до растворения солей). Добавляют 10 мл 10%-ного раствора цитрата натрия, 0,1 г солянокислого гидроксиламина, нейтрализуют по лакмусовой бумаге и добавляют еще 2 капли его избытка. Если в растворе появилась муть, ее отфильтровывают через бумажный фильтр, промывают небольшим количеством воды, озоляют фильтр с осадком при низкой температуре и доплавляют в платиновом тигле. Плав выщелачивают, проводят все описанные выше операции и объединяют с полученным ранее фильтратом. Жидкость экстрагируют 5 мл 0,02%-ного раствора дитизона в СС1« в течение 0,5—1 мин. Дают органическому слою отстояться и сливают его в другую делительную воронку. Если слой ССЦ не окрашен в зеленый цвет, приливают к основному раствору еще 2—3 мл дитизона той же концентрации, встряхивают, сливают органическую фазу и пов
166
торяют экстракцию до тех пор, пока очередная порция СС14 после встряхивания в течение 1 мин. останется зеленоватой. Дитизоновые экстракты объединяют, промывают водой и встряхивают в течение 2 мин. с 5 мл 0,01 N НС1. Отделяют слой СС14 и повторяют встряхивание с 5 мл НС1 той же концентрации. Для удаления мелких окрашенных капель объединенные солянокислые реэкстракты встряхивают с небольшим количеством СС14, затем переносят в стеклянную пробирку с притертой пробкой, добавляют по 2,5 мл 25%-ного раствора NaOH, 6 мл 0,001 %-ного раствора дитизона в СС14 и встряхивают в течение ЗОсек.Окрашенный в красно-розовый цвет органический слой спектро-фотометрируют при 520 нм или фотоколориметрируют с зеленым светофильтром. Для приготовления стандартных растворов отбирают 0,05 — 0,25 мкг Cd, прибавляют 0,01 N НС1 до объема исследуемого раствора и экстрагируют, как указано выше [363].
В практике анализа минерального сырья гравиметрия применяется редко. Определение кадмия с 0-нафтохинолином описано в гл. IV. Мешающие элементы предварительно отделяют методами, рассмотренными в начале этой главы.
Эмиссионная спектроскопия используется для определения кадмия во многих видах минерального сырья. В сульфидных минералах (сфалерите, галените и др.) испарение материала производят в дуге постоянного тока, катодом служит угольный электрод с пробой. Для стабилизации температуры дуги пробу и эталоны смешивают с альбитом (Na[AlSi3Og]) [48], либо с Ы2СО3 в отношении (1 : 3) или с ZnS. При высоком содержании железа прибавляют угольный порошок. Определение проводят по аналитической паре линий Cd 3261,0 — Zn 3282,3 А. Чувствительность метода 1-10“3%, ошибка определения ± 5% [704].
При анализе карбонатов кальция и магния, смитсонита и цинковых обманок используют горизонтальную дугу переменного тока (8а) между угольными электродами, наполненными NaCl. Пробу смешивают с Na2CO3 и NaNO3 и вводят в дугу на полосках бумаги. Аналитической парой линий служит Cd 3261,0 — Sb 3232,5 А. Метод применим в интервале концентраций 0,02— 0,05% Cd, средняя квадратичная ошибка 11—17% [359]. При совместном определении Cd и Zn в рудах и технологических продуктах на дифракционном спектрографе ДФС-13 (при дисперсии 1 К/мм) линия кадмия 3261,0 А полностью отделяется от линий железа даже при анализе железных руд. Для идентичности форм нахождения кадмия и цинка в пробах и эталонах последние готовят разбавлением пустой породой цинкового концентрата с известным содержанием обоих элементов. Эталоны и пробы разбавляют этой смесью в отношении 1 : 4 и набивают в угольные электроды. Спектры возбуждают в дуге постоянного тока (15а) и фотографируют на фотопластинках типа СП-3 или СП-2 в течение 30 сек. Ширина щели спектрографа 0,030— 0,035 мм. При анализе проб с содержанием кадмия )> 0,1% спектры фотографируют через трехступенчатый ослабитель. Определение производят по линиям Cd — 3261,0 (Jan) — Ge 3260,5 А (JCp); градуировочные кривые строят в координатах 1g (^ан/АСр) — IgC с учетом фона вблизи линий кадмия. Интервал определямых концентраций
167
6,0005—3% Cd; средняя арифметическая ошибка 3,9%. Сходимость с данными химического анализа удовлетворительна [87J.
Атомно-абсорбционное определение кадмия проводят обычно на установках, смонтированных на основе монохроматора СФ-4, с ФЭУ-18 и светострелочным гальванометром М-95 или с регистрирующим потенциометром ПС-1-02 (с катодным повторителем). Пробы руд и минералов разлагают смесями HCl с HNO3, иногда с последующим выпариванием с H2SO4; силикатные породы — смесями HF с H2SO4. Растворы распыляют в пропано-воздушное пламя (иногда — другие пламена) различной протяженности, источником света служат высокочастотные безэлектродные кадмиевые лампы или парометаллические лампы типа ДКдС-20 [139, 337].
При анализе цинковой руды 2,5—5,0 г пробы растворяют в НС1 или ее смеси в HNO3 (3 : 1), упаривают до небольшого объема и разбавляют до 50 мл. Три порции раствора смешивают с 0,5 мл бутанола, в две из них добавляют 20 и 50 мкг Cd. Все три раствора разбавляют до 10 мл и фотометрируют в пламени протяженностью 7 см с лампой типа ДКдС-20 при токе 0,5—0,55а.
Содержание Cd рассчитывают путем линейной экстраполяции; калибровочный график прямолинеен до 10 мкг Cd/мл.
Метод позволяет определять от 0,0004 до 0,2% Cd с ошибкой 1—2% [336, 337].
В других способах растворы проб распыляют в пропан-бутан-воздушное пламя протяженностью 12 см, источником света служит высокочастотная кадмиевая лампа (большинство катионов при их содержании до 1 мг/мл не мешает) [28] или в воздушно-ацетиленовое пламя такой же протяженности. Источник света— лампа типа ВКдС-2 или разборная лампа с полым катодом. Чувствительность определения 0,05 мкг Cd/мл, коэффициент вариации' 2%. Определению мешают ~ 40% Са и Р [408]. В одном из вариантов метода пробы разлагают в HNO3 (1 : 1), а затем обрабатывают НС1 и НСЮ4. Фотометрируют в описанных выше условиях [486].
Значительно повышает чувствительность анализа руд фото-метрирование отходящих газов пламени [138, 139, 568].
Анализируемый раствор распыляют в стеклянную горелку с пламенем в 2 см, над которой установлена газовая кювета — открытая с обоих концов кварцевая трубка длиной 30 см, диаметром 2,2 см. Газы пламени вводят в боковое отверстие, расположенное в середине трубки и фотометрируют с высокочастотной кадмиевой лампой. Чувствительность определения возрастает в 14 раз и достигает 5-10~6—!• 10~4% в расчете на сухую пробу; ошибка не превышает • ±0,5—5% [139].
Использование водородно-воздушного пламени и газовой кюветы длиной 45 см, диаметром 1,7 см снижает порог определения кадмия в сульфидных минералах до 3-10“6% [712].
При анализе силикатных пород (биотит, щелочные полевые шпаты и др.) 0,25 г пробы растворяют в 50 мл HF и 20 мл HNO3. Раствор выпаривают досуха, прибавляют 2 мл НС1 (уд. в. 1,19) и разбавляют водой. В растворы вводят по 0,5 мл NH«OH, для устранения влияния алюминия — 10-кратный (по отношению к А1) избыток соли лантана. Растворы разбавляют водой до 50 м л и фотометрируют [483].
168
В другом методе пробу разлагают смесью HNOs и HF (1 : 1), раствор выпаривают досуха и приливают к остатку 47%-ную НВг. Затем кадмий экстрагируют в течение 5 мин. 5%-ным раствором Амберлита LA-1 в ксилоле. Экстракт 2 раза промывают 0,2 N НВг и реэкстрагируют кадмий 0,6 N НС1О«. Реэкстракт вводят при помощи концентрического распылителя в водородновоздушное пламя горелки, представляющей собой полое кольцо с внутренним диаметром 6 мм и наружным — 10 мм, с 16 отверстиями для выхода Н2. Газы пламени направляют в кварцевую трубку, в которой фотометрируют, используя лампу с полым катодом.
Калибровочный график прямолинеен в пределах 0,35—25 мкг Cd/л (0,00035—0,025 мкг/мл) [479].
При анализе гранита, нефелиновых сиенитов и дунита импульсная термическая атомизация твердых проб (см. гл. IV) позволяет определять 10~7—10~6% Cd.
15 г образца смешивают с угольным порошком (1 : 1), помещают в тигель и проводят испарение при токе 140а, конечная температура тигля достигает 1850 °C. Коэффициент вариации для нижнего предела содержаний Cd составляет 30% [34].
АНАЛИЗ ПРОМЫШЛЕННЫХ ПРОДУКТОВ
Методы разложения и отделения
При анализе металлов, сплавов, концентратов и других продуктов металлургического производства используют различные варианты кислотного разложения, указанные в табл. 29.
Для отделения кадмия от основы пробы и других мешающих элементов используют его осаждение в виде CdS [262, 477, 697, 767], экстракцию его дизтилдитиокарбамината этилацетатом [11] или хлороформом [749], экстракцию иодидного комплекса метил-изобутилкетоном [187, 638], ионообменное отделение [64, 429, 569, 614]. Медь, мешающую при полярографировании, осаждают тиосульфатом (15, 454] или путем электролиза [292, 498]. Большие количества In отделяют экстракцией изопропиловым. эфиром [611], Ан — хлорексом [300], a Sb, Sn и Т1 отгоняют в виде бромидов [15, 4551.
Методы определения
Как и в анализе минерального сырья, для определения кадмия широко применяют полярографию [11, 12, 15, 49, 63, 64, 162, 174, 285, 292, 309, 318, 465, 569, 749, 750]; фотоколориметрирование с дитизоном используют при анализе металлических In [767], Pb [6031, Re [640] nZn [391], цинковых концентратов [544] и электролита [262].
Химико-спектральным методом определяют кадмий в металлических Ag [739], Al [261], Au [300], Bi [30, 2781, Cr [477], Ga [235], Hg [412], In [261], Pb [261, 450], Sb [261], T1 [261], а также в жаропрочных сплавах [628].
169
Таблица 29
Методы определения кадмия в промышленных продуктах
Объект анализа Содержание кадмия, % Разложение проб Методы отделения кадмия или других элементов Метод определения Литература
Вольфрам 1.10~4-5.10~5 Cd экстрагируют диэтилди-тиокарбаминатом в СНС13 Полярографический
Золото Индий 5.10-4—2-10“7 0,008-0,03 НС1 и HNO3 (3:1) НВг Au экстрагируют хлорексом In маскируют цитратом In экстрагируют изопропиловым эфиром; Cd осаждают в виде CdS Спектро химический Турбидиметрический Колориметрирование с дитизоном [300] [767] [611]
Медь Свинец Селен Сурьма от 1-10~5 Ю-e—10~7 HNO3 НС1 и HNO3 (3 : 1)+НВг Си осаждают электролизом РЬ осаждают в виде сульфата Sb, Sn отгоняют в виде бромидов; Со осаждают 1-нит-розо-2-нафтолом Полярографический с накоплением Полярографический Полярографический с накоплением По лярогр афический » [292] [309] [162] [63] [15]
Таллий Теллур От 0,05 HNO3 Т1 отгоняют в виде бромида » » [455] [64, 750]
Титан 1.10-3—1-10-4 HF (1|:5)+HNO3 Cd экстрагируют диэтилди-тиокарбаминатом в этилацетате » [11]
Хром 1- IO-2-!• 10~4 HCI Cd осаждают в виде CdS Колориметрический с дитизоном [477]
Таблица 29 (окончание)
Объект анализа Содержание кадмия, % Разложение проб Методы отделения кадмия или других элементов Метод определения Литература
Цинк Электролит цинковый Сплавы алюминиевые Сплавы жаропрочные Сплавы легкоплавкие Сплавы Ni — Sn— - Cd Сплавы цветных и черных металлов Сплавы цинковые Концентрат цинковый Купорос цинковый Электролит цинковый 7-10-* 1,25-10-* 1-10~5 От 1,0 п-IO"3 НС1 (1:1)+КВгО3 HNO3 НС1 НС1 (1:1) + + H2SO4 (1 : 4) HNO3 H2SO4 (1 : 9) HC1O4 Sn отгоняют в виде хлорида Cd отделяют ионным обменом [Cd Ji]- экстрагируют ме-тилизобутилкетоном Си отделяют цементацией Си осаждают Na2S2O3 Cd отделяют ионным обменом То же Cd осаждают в виде CdS на ZnS Полярографический Колориметрический с дитизоном Полярографический Колориметрический с дитизоном Полярографический Спектрографический Фо то турбидиметрическое титрование Комплексонометрический Колориметрический Полярографический Электролитический Комплексонометрический Колориметрирование с дитизоном Полярографический Колориметрирование с дитизоном [391] [391] [262] [558] [285, 569] [91] [223] [549] [638] [391] [544] [544] [544] [12] [262]
Таблица 30
Методы спектрального определения кадмия в металлах и некоторых других продуктах
Объект анализа Содержание кадмия, % Предварительная подготовка Литература
Алюминий 8-10-’ [261, стр. 302J
Висмут 5-10-’ Bi осаждают иодидом [30, 278, 450]
Вольфрам 5-10-» Cd осаждают сульфидом [270, стр. 529J
Галлий — Ga экстрагируют бутилацетатом [235J
Золото 5-10~з— 2-10-’ Au экстрагируют хлорексом [300]
Индий 2.10-6 In экстрагируют диэтиловым эфиром [261, стр. 193]
Молибден 1.10~з Прямой метод [270, Стр. 564]
Ниобий 5-10-5 Cd осаждают сульфидом [270, стр. 529 ]
Ртуть — Hg экстрагируют изоамиловым спиртом [412]
Селен 2-10-’— 3-Ю-5 Прямой метод [83, 261, стр. 462]
Серебро — Ag осаждают хлоридом [739]
Свинец 3-10-’ РЬ осаждают сульфатом [261, стр. 315]
Сурьма 3-Ю"7 Sb экстрагируют бутилацетатом [261, стр. 242]
Таллий 2-10-5 Т1 экстрагируют диэтиловым эфиром [261, стр. 216, 270, стр. 225]
Тантал 5-10-5 Cd осаждают сульфидом [270, стр. 529]
Теллур l-10-з Прямой метод [473]
Уран* — U экстрагируют смесью трибутилфосфата и керосина (1 : 10) [745]
Церий 5-10-5 Прямой метод [270, стр. 179]
Цинк* — Cd экстрагируют метил-изобутилкетоном в виде CdJ4 [187]
Цинк 2-10-5 Прямой метод [270, стр. 345, 463]
Сплавы жаропрочные 1-ю-* » » [91, 628]
Стали* 3-10-3 Разложение пробы смесью НС1 и HNO3 [777]
Штейны * от 2,5-10'3 То же [115]
Графит* 1-10-’ [1671
* Атомно-абсорбционный метод определения.
172
Стали [777], графит [167], штейны [115] и металлический цинк [187] анализируют методом атомной абсорбции. Прямая эмиссионная спектрография применена при анализе Мо [14], Se [83, 261], Те [473], Zn [463], сталей [91] и жаропрочных сплавов [91].
Химические методы определения кадмия в продуктах металлургического производства приведены в табл. 29, спектральные — в табл. 30.
Для определения кадмия в металлах и сплавах чаще всего используют вариант полярографического метода, описанный на стр. 164; предварительно отделяют основу пробы или выделяют кадмий.
Из проб металлического титана микрограммовые количества кадмия выделяют экстракцией диэтилдитиокарбаминатом в этилацетате.
1—2 г титана растворяют в платиновой чашке в 20 мл HF (1 : 5). После прекращения бурной реакции нагревают до полного растворения и прибавляют по каплям HNOs до обесцвечивания раствора. Затем упаривают до 8—10 мл, охлаждают, добавляют 0,2—0,3 г НзВО'з, 5—10 мл воды, 10 мл 15%-ной винной кислоты, перемешивают и раствор осторожно нейтрализуют аммиаком до pH 3—3,5. По охлаждении раствор переносят в делительную воронку и споласкивают чашку тонкой струей воды так, чтобы общий объем раствора не превышал 40—50 мл. К раствору приливают 30 мл 2 %-ного раствора диэтилдитиокарбамината натрия, 10 мл этилацетата и встряхивают в течение 1 мин. После расслаивания водную фазу сливают в другую делительную воронку и повторяют экстракцию этилацетатом. Объединенные экстракты промывают 10 мл HNOa (1:20) и затем дважды реэкстра-гируют кадмий смесью 10 мл HNO3 (1 : 1) и 1 мл Н2О2. Реэкстракт упаривают до 3—5 мл, приливают 2 мл H2SOt (1 : 1), несколько капель HNO3 или Н2О2 и нагревают до появления паров. Остаток растворяют при нагревании в 3 мл НС1 (1 : 1), нейтрализуют кислоту аммиаком и прибавляют 1 мл его избытка; по охлаждении разбавляют до 10 мл водой. При полярографировании прибавляют 0,1—0,2 г Na2SO3, 2 капли 1%-ного столярного клея.
В том же растворе при соответствующих потенциалах можно полярогра-фировать также Cu, Ni, Zn.
Содержание кадмия рассчитывают по формуле, указанной на стр. 164 [11].
При определении кадмия в металлическом цинке, кроме полярографии, используют и колориметрирование с дитизоном (после предварительного осаждения его вместе с Cu, Fe и РЬ сульфидом натрия).
5 г цинка растворяют в 50 .илННО3 (1 : 1), разбавляют водой до 100 мл, прибавляют NHaOH до растворения осадка Zn (ОН)2 и затем по каплям, при сильном перемешивании,— 5 мл 2%-ного раствора Na2S. Раствор нагревают до коагуляции осадка и отфильтровывают. Осадок растворяют на фильтре горячей HCl (1 : 1), фильтр промывают для полного растворения CuS с помощью HNO3 (1 : 1), раствор разбавляют до 100 мл и перемешивают. К 10 мл этого раствора прибавляют 15 мл воды, 2 капли фенолфталеина, NH4OH до слабо-розовой окраски и 1 мл HCl (1 : 3). Затем экстрагируют медь 0,001%-ным раствором дитизона в ССД; органический слой отбрасывают. К кислому водному раствору добавляют 1 мл 50%-ного раствора цитрата аммония, 1 мл 20%-ного раствора NH2OH-HC1, 25 мл 10%-ного раствора NaOH, 10 мл 0,05%-ного раствора дитизона в ССП и экстрагируют кадмий в течение 30 сек. Экстракцию кадмия дитизоном (по 2 мл) повторяют еще 2 раза. Объединенные экстракты промывают дважды 2%-ным раствором NaOH, затем 2 раза водой. Для разрушения дитизоната кадмия к промытому экстракту прибавляют 1 мл
173
HCI (1 : 3), встряхивают 30 сек. и колориметрируют освободившийся дитизон (количество его пропорционально содержанию кадмия) с красным светофильтром [391].
Для определения кадмия в цинковых концентратах используют комплексонометрию.
1—3 г пробы разлагают при нагревании H2SO4 с добавлением HNO3 и раствор упаривают досуха. К остатку приливают 100 мл 0,7 A H2SO4, нагревают до растворения солей и охлаждают до 50—60° С. Для цементации меди добавляют5 г металлического железа (восстановленного водородом) и оставляют на водяной бане на 15 мин., после чего фильтруют и промывают 0,7AH2SO4. К фильтрату добавляют 7,5 мл 6 УШ и пропускают через колонку с катионитом Дауэкс-1 со скоростью? мл/мин. Затем кадмий элюируют 180— 200 мл раствора 2,5%-ной HJ в H2SO4 (1 : 250). К элюату прибавляют 0,5 г, NH4CI, NH4OH до pH 8—10, нагревают до 90° С, прибавляют 0,05 мл 0,5 %-ного спиртового раствора эриохром черного, содержащего ИНгОН-НС!, и титруют 0,05 М раствором комплексона III. В отсутствие больших количеств Си и Fe операцию восстановления металлическим железом опускают, но в этом случае из иодпстоводородной кислоты выделяется иод, для восстановления которого добавляют сульфит. Осадок иодида меди отфильтровывают перед хроматографированием [429].
В металлическом хроме кадмий совместно с Bi, Sb и Sn определяют спектральным методом. Предварительно проводят химическое обогащение.
Металлический хром растворяют в 30—40 мл НС1 (уд. в. 1,19), раствор упаривают до сиропообразного состояния, приливают 80—100 мл горячей воды и нагревают до полного растворения солей. Добавляют 20 мл 50%-ной лимонной кислоты, 5 мл НС1 и 3 мл раствора Cu(NO3)2 (коллектор для Cd, Си, Sb и Sn). Разбавляют водой до 150—180 мл и пропускают в течение 20 мин. H2S со скоростью 80—100 пузырьков в минуту. Отстоявшийся осадок фильтруют, промывают HCI (1 : 100), через которую также предварительно был пропущен H2S. Осадки помещают в фарфоровый тигель, высушивают, озоляют и прокаливают при 550—600° С.
Одновременно с пробами готовят серию эталонов с содержанием от 0,3 до 300 мкг Cd, Cu, Sb и Sn, которые проводят через весь ход анализа, начиная с осаждения микропримесей на коллекторе. Прокаленные осадки проб и эталонов тщательно смешивают с угольным порошком в отношении (1 : 1) и сжигают в угольных электродах в дуге переменного тока (12а); спектрограф ИСП-22.
Фотографирование спектров проводят без предварительного обыскри-вания в течение 30 сек. на спектральных фотопластинках типа I. Внутренним стандартом служит введенная в качестве коллектора медь. Аналитическая пара линий: Cd 2288,0 А — Си 2276,2 А [477].
При атомно-абсорбционном определении кадмия в сталях 1 г пробы растворяют в смеси 10 мл НС1 и 2 мл HNO3. Раствор разбавляют водой до 100 мл и распыляют в пламя светильного газа с воздухом протяженностью 10 мл; источник света — лампа с полым катодом. Чувствительность определения кадмия 0,03 мкг/мл (3-10“4%), ошибка < 10%. Не мешают определению до 5% Al, Mo, Pb, Ti, V, до 10% Со, Си и до 20% Cr, Ni и Мп [777].
Медьсодержащие штейны (0,5—2,0 г) растворяют в смеси НС1 и HNOS и после упаривания до влажных солей тоже разбавляют водой до 100 мл. Полученный раствор фильтруют, вводят в пламя пропан-бутан—воздух и фотометрируют с высокочастотной безэлектродной лампой на установке, собранной на основе спектрофотометра СФ-4 с усилителем постоянного тока и самописцем.
Чувствительность метода 0,05 мкг Cd/.ил (2,5-10~4%) [115].
174
Фазовый анализ на соединения кадмия
Исследование растворителей для определения форм вхождения кадмия в продукты гидро- и пирометаллургической переработки (кеки, вельцокислы, возгоны) описано в [448], а разработанная на этой основе схема анализа дана в работе [434[.
Растворимость соединений кадмия в различных растворителях представлена в табл. 31 [434].
Подобрать селективный растворитель, который растворял бы окись кадмия и не действовал на силикат кадмия, не удалось, так как оба эти соединения очень близки по степени растворимости. В качестве растворителя для окислов и силикатов кадмия была выбрана 0,1 N H2SO4, в которой сульфид и феррит кадмия нерастворимы даже в присутствии некоторых количеств ионов Си2+ и Fe3 , которые могут из исследуемого материала, перейти в раствор.
Анализ пылей и огарков вельцокисей и кеков на соединения кадмия [434]
Проба
. *
Перемешивание со 100 мл воды, 20° С, 2 часа
1
Фильтрование, промывание
Фильтрат CdSOi
Фильтрат
CdO, CdO-CdSO4, 2CdO-SiO2, CdO-SiO2
Остаток
Перемешивание со 100—150 мл 0,1 N H2SO4, 18—20° С, 2 часа
Фильтрование, промывание
Остато к
I
Перемешивание со 100 мл 3,9%-ного раствора CuSO4-5H2O в 0,1 N H2SO4, 90—95° С, 2 часа
Фильтрование, промывание
Фильтрат Остаток
CdS, Cd-АЪОз 1
Перемешивание с 10 мл 1 N H2SO4j
90—95° С, 2 часа
_____________________I
Фильтрование, промывание
Фильтрат Остаток —
CdO-Fe2O3 труднорастворимые соединения
п CdO-mZnO-Fe2O3
175
Растворимость соединений кадмия в различных растворителях, отн.% [434] I I I Соединения кадмия
Сернокислый раствор сульфата меди при 90—95° С разлагает сульфид и почти весь алюминат кадмия (феррит кадмия растворяется только при обработке 1 N H2SO4 при 90—95° С).
Последовательность стадий анализа показана на приведенной выше схеме. Определение кадмия во всех фильтратах выполняют полярографическим методом на аммиачном фоне [4341.
АНАЛИЗ ПРИРОДНЫХ и сточных вод, ВОЗДУХА ПРОМЫШЛЕННЫХ ПРЕДПРИЯТИЙ
Анализ воды
Содержание микрокомпонентов, в том числе и кадмия, в природных, сточных и промышленных водах весьма незначительно, поэтому при их определении требуется предварительное обогащение анализируемых проб. Для этого применяют различные способы концентрирования: выпаривание до сухого остатка, экстракцию, соосаждение на коллекторе или адсорбцию на активированном угле.
Предложено выделение следов кадмия экстракцией раствором дитизона в хлороформе [234, 351, 736], осаждение диэтилдитиокар-баминатом натрия [758] или рубеановодородной кислотой [351], а также соосаждение на коллекторах А1(ОЙ)3 [260], Bi2S3 и СаСО3 [507]. Описано концентрирование кадмия на катионитах [205, 709].
При определении кадмия методом эмиссионной спектрографии чаще всего применяют упаривание проб воды, частичное или до сухого остатка [107].
Содержание кадмия в природных, морских и сточных водах определяют колориметрическим, полярографическим или спектральным методами (см. табл. 32, а также стр. 164—174).
Анализ воздуха
Предельно допустимая концентрация CdO в воздухе соответствует 0,1 мг/м3 (0,1 мкг!л) [321, 322]. Для выделения CdO из воздуха, пылей и дымов промышленных предприятий используют фильтры со стеклянной ватой «шерсть» [4], патроны с бумажными или перхлорвиниловыми фильтрами [322] и водяные или электростатические пылеосадители [619, стр. 213].
Через фильтр протягивают со скоростью 5 л/мин 400—500 л (дм3) исследуемого воздуха. Фильтр переносят в стакан, наливают в него 3 мл 5 % -ной H«SO< и после кратковременного нагревания на водяной бане раствор вместе с фильтром переносят на воронку со стеклянной пористой пластинкой и отсасывают под вакуумом. Фильтр помещают в тот же стакан, обрабатывают 5 мл горячей воды и раствор снова отсасывают. Такую обработку фильтра повторяют 3 раза. Основной раствор и промывную жидкость объединяют, разбавляют водой до 30 мл, центрифугируют или фильтруют. К 10 мл прозрачного раство-
177
I
aS
СГ
S ч ю
Н
Методы определения кадмия в водах
Литература [352] [352] [351, 424, 736] [351] [107, 260] ' [709] [234] [758] [507] [205] [269]
Метод определения Колориметрия с дитизоном в СС14 Амальгамная полярография с накоплением (фон 0,1 N НС1 + + NH4C1) Полярография (фон 10%-ный раствор тартрата калия-натрия) Полярография (фон хлоридно-аммиачный) Эмиссионная спектрография Атомная абсорбция, 0,1 N HNO3 (элюат) + ацетон (1 : 9) Колориметрия с дитизоном в СС14 Титрование избытка комплексона Атомная абсорбция, раствор 0,8 N HNO3 (элюат) Атомная абсорбция, раствор НС1 Полярография (pH 9; в токе аргона)
Метод выделения кадмия или отделения мешающих элементов Си экстрагируют диэтилдитиокарба-минатом свинца в СС14 Fe восстанавливают аскорбиновой кислотой Cd экстрагируют дитизоном в СС14 Cd осаждают рубеановодородной кислотой Выпаривание проб досуха Cd отделяют ионным обменом на смоле Хелекс-100 Cd экстрагируют дитизоном в СС14 Cd осаждают диэтилдитиокарбами- натом Cd отделяют ионным обменом на Дауэкс-А-1 Cd отделяют ионным обменом на Дауэкс-500 W Прямой метод
Содержание кадмия, мкг!л от 2,0 0,05 1,0 0,05 o' 10,0
сб го Я § §
К 4> tA
Ю О
К К
ра прибавляют NH4OH до нейтральной или слабощелочной реакции, воду до 15 мл, 0,5 мл 20%-ного раствора Na2S и перемешивают. Через 5—10 мин. сравнивают с одновременно приготовленной стандартной шкалой, содержащей от 0,02 До 0,1 мг CdO. В присутствии Fe и Zn во все растворы вводят по 0,5—1,0 мл 10%-ного раствора KCN [4,322].
Колориметрирование с дитизоном или ди-|3-нафтилтиокарбазо-ном более надежно [619, стр. 213]. При определении кадмия в воздушных пылях и дымах используют и полярографический метод [732]. Литература по анализу воздуха дана также в работах [72а, 219а].
Применение атомной абсорбции позволяет определять непосредственно кадмий в атмосфере. Пробу воздуха вводят в атомизатор со скоростью 1,2 л/мин и определяют поглощение при 228,8 нм. Предел обнаружения кадмия 0,02 мкг/м3’, пары многих органических растворителей определению не мешают [528а].'
АНАЛИЗ РЕАКТИВОВ
И ОСОБОЧИСТЫХ ВЕЩЕСТВ
Для определения микропримесей различных металлов, в том числе и кадмия, в реактивах и веществах особой чистоты используют высокочувствительные флуоресцентные [325, 364], колориметрические [263, 265, 325, 335], полярографические [266], атомноабсорбционные [167, 238, 241, 407], а также спектрохимические методы [261, стр. 166, 207, 254, 513; 319, 426; 430; 437; 442; 453; 555].
Методы определения примеси кадмия в некоторых реактивах и веществах особой чистоты приведены в табл. 33.
Для определения кадмия в HNO3 5 г пробы выпаривают в платиновой чашке досуха, остаток растворяют в 5 мл фосфатного буферного раствора с pH 8,0, вводят 0,2 мл 0,01%-ного ацетонового раствора 8-(бензолсульфонилами-но)хинолина и сравнивают интенсивность зеленой флуоресценции анализируемого и эталонного раствора [325, 364].
Тетрахлориды германия и кремния, содержащие 1-10"6% и более кадмия, анализируют методом амальгамной полярографии с накоплением.
В кварцевую делительную воронку помещают 10 мл препарата, 1 мл 1-метил-3,5-дипропил-4-этилпиразола и встряхивают в течение 30 мин. После отстаивания органическую фазу отделяют и выдерживают в вакуум-эксика-торе над твердой щелочью для отделения остатков GeCl4 или SiCl4. Затем полученный экстракт смешивают с равным объемом тетраметиламмония, помещают в полярографическую ячейку и проводят электролитическое концентрирование микропримеси кадмия на стационарном ртутном электроде при потенциале — 2,8 в и 50° С, пропуская через испытуемый раствор азот. После предварительного концентрирования снимают полярограмму и рассчитывают содержание кадмия по высоте полученных пиков.
Анализ проводят методом добавок [266].
179
Методы определения кадмия в реактивах и особочистых веществах
Таблица
Е Объект анализа Содержание кадмия, % Вес пробы, г Подготовка проб к определению Метод определения Литератур
Азотная кислота 1-ю-в 20 Упаривание досуха Колориметрирование [263, 325]
с бромбензтиазо в
ксилоле
1-10-’ 5 Упаривание досуха Флуориметрирование 1364]
с 8-(бензолсульфонил-
амино) хинолином
Алюминиевые МО"5— 1-2 Пробу растворяют в Н2О; экстра- Колориметрирование [325]
£•11) 4 гируют диэтилдитиокарбаминатом, с дитизоном в СС14
упаривают досуха, экстрагируют
дитизоном в СС14 и реэкстрагируют НС1
Висмут (высокой от 10-s— Пробу растворяют в HNOa; Cd То же [325]
чистоты) Ю-6 экстрагируют дитизоном в СС14
Арсенат галлия Прямой метод Спектральный [261, стр. 159]
Фосфид галлия l-10-e Cd экстрагируют диэтилдитиокарбаминатом в СНС13 » [261, стр. 166]
Тетрахлорид гер- l-10-e 10 мл Cd экстрагируют мети л-3,5-дипро- Амальгамная поляро- [266]
пил-4-этилпиразолом графия с накоплением
Антимонид индия 2-10-5 In экстрагируют диэтиловым эфиром из НВг Спектральный [261, стр. 203]
• 5-10-s Прямой метод » [83, 704]
Ниобат калия от 3-10~4 0,2 Пробу растворяют в смеси HF и HNOa (1 : 2) Атомно-абсорбционный [407]
Таблица 33 (окончание)
Объект анализа Содержание кадмия, % Вес пробы, г Подготовка проб к определению Метод определения Литература
Сульфат калия l-10-s 20 Пробу растворяют в Н3О Колориметрирование с [263, 325]
(особой чистоты) Тетрахлорид МО"6 10 мл Cd экстрагируют метил-3,5-дипро- бромбензтиазо в ксилоле Амальгамная поляро- [266]
кремния Ниобат рубидия от 3-10-4 0,2 пил-4-этилпиразолом Пробу растворяют в смеси HF и графия с накоплением Атомно-абсорбционный [407]
Галогениды сереб- 1-Ю-4 1 HNOa (1 : 2) Пробу растворяют в 4,5 М KJ, Cd Колориметрирование с [531]
ра Сульфид свинца Титан — стронций — кадмий (сплав) Пентахлорид фосфора Окись циркония Соли разных металлов Хлорид цинка (радиоактивный препарат) Сера (высокой чистоты) 1-10-5 1-10-’ ОТ 1—6-Ю-5 от 10~13— Ю-Г2 5-Ю-о 0,25—0,30 экстрагируют дитизоном Cd отделяют ионным обменом на катионите КУ-2 Пробу растворяют в HCI, Ti экстрагируют купфероном в изопентаноле Cd экстрагируют диэтилдитиокарбаминатом в СНС1а Пробу растворяют в HCI, Cd экстрагируют диантипирилметаном в дихлорэтане Пробы растворяют в HNOa Прямой метод дитизоном Спектральный Титрование комплексоном III Спектральный Атомно-абсо рбционный Колориметрирование с 1,2-пиридилазо-2-наф-толом Атомно-абсорбционный Спектральный [442] [264] [261, стр. 254] [407] [265] [240, 24.1] [431]
Определение примеси кадмия в алюминиевых квасцах проводят следующим путем.
1—2 г квасцов растворяют в небольшом объеме воды, раствор переносят в делительную воронку, добавляют 8—10 .илСС14, 2 мл 2—3%-ного раствора диэтилдитиокарбамината натрия и встряхивают в течение 1 мин. После расслоения экстракт переносят в другую делительную воронку и повторяют экстракцию из водной фазы. Из объединенных органических растворов кадмий реэкстрагируют HNOa (1 : 1). Азотнокислые реэкстракты упаривают досуха. Остаток растворяют в 10 мл воды, содержащей несколько капель НС1 и добавляют гидроксиламин, 0,5 мл 20% -ного раствора лимонной кислоты. Нейтрализуют раствор по лакмусу 25%-ным NH4OH и добавляют его избыток (3 мл). Полученный раствор встряхивают с 5 мл 0,05 %-ного дитизона в СС14. Органический слой отделяют, а экстракцию из водной фазы повторяют несколько раз до прекращения изменения окраски дитизона.
Для перевода кадмия в водную фазу объединенные экстракты обрабатывают дважды порциями по 4 мл 0,01 N НС1, встряхивая каждый раз в течение 3 мин. Реэкстракты обрабатывают СС14 и переносят в цилиндры для колориметрирования. Доводят объем раствора до 10 мл 0,01 N НС1. Добавляют 5 мл 25 %-ного раствора NaOH, 5 мл 0,005 %-ного раствора дитизона в СС14 и встряхивают в течение 30 сек. После разделения проводят сравнение окрасок экстрактов испытуемых растворов со стандартными [325].
В ниобатах калия и рубидия определение кадмия проводят методом атомной абсорбции.
0,2 г ниобата растворяют во фторопластовой чашке 6—8 мл смеси HF и HNO3 (1 : 2) при нагревании. Раствор выпаривают до небольшого объема, переносят в мерную колбу и доводят до метки 5%-ным раствором винной кислоты.
Определение кадмия проводят методом добавок: анализируемый раствор распыляют с помощью углового распылителя в пламя светильный газ — воздух, протяженной (12 см) щелевой горелки; источником света служит без-электродная лампа с парами кадмия.
Чувствительность метода 0,1 мкг СЛ/мл, при добавлении в раствор 30% этанола — 0,05 мкг!мл. Использование горизонтального пламени ацетона снижает предел чувствительности до 0,02 мкг Cd/.м.г [147].
АНАЛИЗ БИОЛОГИЧЕСКИХ ОБЪЕКТОВ
Для определения кадмия в биологических объектах (кровь, моча, печень и др.), растениях и пищевых продуктах используют фотометрические [16, 363, 760], полярографические [482, 666] и спектральные методы [16, 770] (табл. 34).
Разработан метод фотоколориметрического определения следов кадмия с ди-р-нафтилтиокарбазоном [363].
Пробу (5—100 мл мочи, 5— 20 г ткани, крови т. д.) разлагают смесью H2SO4 и HNOa (1 : 2) и раствор упаривают до появления «дыма» SO3. Раствор охлаждают, переносят в делительную воронку, добавляют 15 мл аммиачно-цитратного раствора (400 г лимонной кислоты в 600 мл воды подщелачивают аммиаком и разбавляют водой до 1 л) и разбавляют водой до 50 мл. Нейтрализуют раствор аммиаком до pH ~ 8,3 по индикатору феноловому красному и экстрагируют кадмий вместе с другими тяжелыми металлами 5 мл 0,02%-ного раствора ди-р-пафтилтиокарбазопа в СНС1а. Затем реэкстрагируют тяжелые металлы (в том числе и кадмий) 50 мл 0,2 N НС1.
182
со
ев ЕГ К
Ч
\о «В Ен
Методы определения кадмия в биологических объектах
Литература СМ о Ю СО С© СМ о йЭ Ч-Ч СС> ч-н 00 см ь- 1—1со ।—1
Метод определения J Осаждение CdS Колориметрирование с кадионом 2В Колориметрирование с дитизоном Колориметрирование с ди-Р-нафтилтиокарбазо-ном Спектрографический Поля рог рафический
Подготовка проб к определению Cd экстрагируют диэтилдитиокарба-минатом в СНС13 и реэкстрагируют НС1 Пробу переводят в раствор смесью H2SO4 и HN03; Cd экстрагируют дитизоном в СНС13; реэкстрагируют НС1 и упаривают досуха Пробу растворяют смесью H2SO4 и HN03; Cd экстрагируют дитизоном в СНС13; реэкстрагируют НС1 Пробу растворяют в смеси H2SO4 и HN03; Cd экстрагируют ди-р-нафтилтиокарбазоном в СНС13; реэк- страгируют НС1 Прямой метод
Вее пробы, г о сч S м । S 1 II 1
Содержание кадмия, % Г 7 Г Г о о о о 3 1 3 1 3 3 А А А А
Объект анализа ' Биологический материал (кровь, моча, ткань, печень и др.) Пищевые продукты
Для отделения кадмия от Pb, Zn и др. элементов в солянокислый реэкстракт добавляют 5 мл 25%-ного раствора тартрата натрия, 20 мл 25%-ного раствора NaOH, разбавляют водой до 100 мл и экстрагируют последовательно порциями по 5 мл 0,01% -ного раствора дитизона в СНС13. Реэкстракцию кадмия из органического слоя проводят 50 мл 0,2 N НС1. К солянокислому раствору, содержащему кадмий, прибавляют 50 мл 5%-ного NH«OH и встряхивают с 10 мл 0,002%-ного раствора ди-Р-нафтилтиокарбазона в СНС13. Полученные экстракты фотометрируют при 540 нм [363].
Описано определение кадмия в органических и биологических материалах с использованием экстракции его дитизоном с последующим фотометрированием дитизоната кадмия [16].
Разработан метод фотометрического определения кадмия с кадионом 2В.
Для устранения влияния Ag, Ca, Hg и Mg проводят предварительную экстракцию кадмия дитизоном и реэкстракцию его HCI (1 : 100). Солянокислый реэкстракт выпаривают досуха, растворяют в воде, добавляют растворы сег-нетовой соли, NaOH, кадиона 2В и измеряют оптическую плотность при 530 нм [760].
Для анализа растений и других биологических объектов успешно применяют эмиссионный спектральный анализ.
Испытуемое вещество высушивают до постоянного веса в платиновой или кварцевой чашке и подвергают сухому озолению в муфельной печи при 450° С, для ускорения озоления добавляют несколько капель перегнанной HNO3. Затем 20 мг золы помещают в угольный электрод и сжигают в дуге постоянного тока. Для определения кадмия используют линию 2288,0 А [16].
Библиография по определению кадмия в организме приведена в работах [72а, 219а].
Глава VII
АНАЛИЗ КАДМИЯ И ЕГО СОЕДИНЕНИЙ
Большинство предложенных методов предназначено для определения малых количеств примесей в металлическом кадмии, его сульфиде и некоторых других соединениях высокой чистоты и для нахождения различных его форм в чистых веществах. Меньшее число методов описано для анализа технических продуктов — гальванических ванн кадмирования, сырья для стекольной промышленности, пигментов, сплавов и др. Первая группа методов включает определение следующих 36 элементов: Ag, Al, As, Au, Ba, Bi,Br, Ga, Gl, Go, Gr, Gu, Fe, Ga, Ge, Hg, J, In, K, Li, Mg, Mn, Mo, Na, Ni, Pb, S, Sb, Se, Sn, Sr, Те, Ti, TI, V, Zn; для их концентрирования или отделения от основной массы кадмия используют соосаждение с различными коллекторами, экстракцию органическими растворителями, отгонку летучих соединений, ионный обмен, в спектральных методах — и физическое обогащение. Определение этих элементов выполняют преимущественно эмиссионной спектрографией и абсорбционными методами (визуальная колориметрия, фотоколориметрия и спектрофотометрия). В меньшей степени применяют полярографию и еще реже — другие методы анализа.
Работы по определению примесей в кадмии до 1965 г. рассмотрены в обзоре [261]. Спектральные и химико-спектральные методы анализа кадмия даны в табл. 35, а химические и физико-химические методы — в табл. 36.
В качестве типичных примеров приведены прямое спектральное определение 14 микропримесей в халькогенидах кадмия и определение 10 примесей после экстракционного отделения основы в высокочистом хлориде кадмия [267].
Измельченную в мелкодисперсный порошок пробу халькогенида делят на четыре части и смешивают с «нулевым» эталоном — чистым графитовым порошком — и эталонами, содержащими соответственно 8-1СГ4, 4-10-3 и 8-10-3% определяемых элементов-примесей. Все эталоны должны содержать
185
Спектральные и химико-спектральные методы определения примесей в кадмии и его соединениях
Объект анализа Определяемые примеси Чувствительность определения, % Bee пробы, г Метод концентрирования
Cd CdO CdS CdTe Ba, Tl Cu, Pb, Tl, Zn Ag, Al, Bi, Cu, Mn, Ni Ag, Bi, Co, Cu, Ni, Pb, Sb, Sn, Tl, Zn Ag, Bi, Co, Cu, Ni, Pb, Sb, Sn, Tl, Zn Ag, Bi, Ni, Pb, Sn, Tl, Zn Ag, Bi, Cu, Ga, In, Sn Ag, As, Bi, Cu, Fe, Ge, In, Pb, Sb, Sn, Tl As, Bi, Ga, In, Ni, Pb, Sb, Sn, Те, Ti As, Bi, Cr, Ge, In, Mn, Pb, Sb, Sn, Те, Ti, V, Co, In, Sn Ag, Al, As, Bi, Cu, Fe, Ni, Pb, Sb, Sn, Tl, Zn K, Li, Na Ag, Al, Bi, Cr, Cu, Fe, Mg, Mn, Ni, Pb, Si, Sn, Ti, Zn Al, Co, Cr, Fe, Mg, Mn, Ni, Ti, Zn Ga, In 2-Ю-®—5- 10“Б 5.10-3—5.io-« 10-6—10-e 2-10-1-5-IO"6 МО-4—1,5.10-6 1.10-1-3-10-’ 2-10-6—2-10-6 5-10-3-3-10-6 10-1—Ю-3 10-3—10-6 IO-5 1,3-10-3—4-10-3 1-10-3—5-10-3 1-10-1—5-10-6 IO"6— Ю-7 ~2.10~2 1,2 2 0,15— 0,25 0,15— 0,25 0,1 1 1 1 1 0,1 0,15 0,2 1 0,003 Прямой метод » » Возгон Cd — при 600— 620° С в токе азота Предварительная отгонка Cd в дуге 30 сек. То же Возгон Cd при 400° С, 0,05 мм Hg Соосаждение с диэтилди тиокарбаминатом кадмия Цементация на Zn, pH 6, NH2OH-HC1, 80° С Соосаждение с МпО3, pH 6 Соосаждение с А1(ОН)3, pH 9-10 Экстракция роданидных комплексов СНС1а Растворение Cd в HNOa и прокаливание до CdO Прямой метод » » Экстракция Cd эфиром из HJ Раствор в 4А НС1, чашечный электрод
186
Таблица 35
Спектрограф Дуга Г, а Экспозиция, сек. Коэффициент вариации, % Литература
Дифракцион- Постоянного 9 60 25 [267, стр. 25]
ный тока
ИСП-22 Переменного 2,5—3,0 25—30 3—5 1342]
тока
ИСП-28 То же 9 15—40 8-16 [80]
» 2—10 60—70 10—20 [259]
ИСП-28 Постоянного 5-14 30—60 20 [261. стр. 400]
тока
(для Т1) Переменного 2 70
тока
ИСП-28 Постоянного 12 25 10—15 [261, стр. 402]
тока
ИСП-22 Переменного 18 20 25 [90]
тока
Q-24 То же 12 40 25 [537]
ИСП-28 » 8 30 7—30 [413]
» » 8 30 7—30 [413]
ИСП-22 » 12 60 [18]
» Постоянного 10 20 [261, стр. 382;
тока 467]
ИСП-30 То же 8 60 20—25 [267, стр. 41]
» » 15 50 15-20 [267, стр. 57]
» » 14 50 20—35 [208]
Дифракцион- Искра 2 6 [671]
ный (600 штрихов/мм)
187
Объект анализа
Определяемые примеси Чувствительность определения, % Вес пробы, г Метод концентрирования
l-10~6— 7-IO-7
Соли Ag, Al, Au, Bi,
кад- Co, Cr, Cu, Fe,
мая In, Ni, Pb, Sn,
Ti, Zn
Al, Ca, Co, Cr, Fe, Mg, Mn, Ni,
Ti, Zn
1.10-4-2-IO-7
Соосаждение диметил-глиоксимом + диэтил-дитиокарбаминатом ± + 1-нитрозо-2-нафтолом на угольном порошке, pH 6
Экстракция Cd эфиром из HJ
10% NaCl. Полученные смеси плотно набивают в кратеры угольных анодов глубиной 6 мм и диаметром 4 мм; катодом служит электрод, заточенный на полусферу. Для фотографирования спектров используют кварцевый спектрограф ИСП-30 (или ИСП-28) с трехлинзовой системой освещения щели и трехступенчатым ослабителем. Для поджига дуги постоянного тока (15 а) служит генератор ДГ-2. Область спектра 2000—2500 А регистрируют на фотопластинках типа УФШ, область 2500—4500 А — на спектральных пластинках тип II. Пробы и эталоны фотографируют с экспозицией 50 сек. по 3 раза, каждый раз меняя верхний электрод; пластинки проявляют, фиксируют, промывают и сушат. Фотометрирование производят на микрофотометре МФ-2 по средней (логарифмической) шкале. Измеряют суммарное почернение аналитических линий и фона (5л+ф) и одного фона (5ф), и переводят их в относительные интенсивности по формуле
^Л/^Ф = Р= - 1,
где A S = 5л+ф — 5ф; у — коэффициент контрастности фотослоя, который находят (для каждой используемой линии) измерением в нескольких спектрах разности почернений между двумя соседними ступеньками и делением среднего из найденных значений на разность логарифмов пропускания для данных ступенек ослабителя [267, стр. 16].
По результатам фотометрирования пластинок строят калибровочный график в координатах: содержание искомых элементов в эталонах, % (ось абсцисс) — соответствующие им значения относительной интенсивности Р (ось ординат) и рассчитывают содержание примесей в пробах. Аналитические линии и место измерения для каждой из них сведены в табл. 37. Нижний предел определяемых концентраций приведенных в ней элементов—1-10-4— 5.10-6%, относительные ошибки ±15—20% [267, стр. 57].
При анализе хлорида кадмия высокой чистоты 1 г его растворяют в 7 мл 3,4 N HJ, приливают 7 мл диэтилового эфира и встряхивают в течение 5 мин. Экстракцию новыми порциями экстрагента повторяют еще 5 раз. Водный слой переносят во фторопластовый тигель, добавляют 20 мг графитового порошка и выпаривают досуха. Затем приливают 2 мл HNOa и вновь выпаривают досуха. Остаток смешивают с 10 мг графитового порошка, содержащего 10% NaCl, и сразу же набивают в кратер угольного электрода. Концентрат пробы содержит 10—15 мг гигроскопичного Cd (NOa)2, поэтому электрод помещают в эксикатор с осушающей смесью.
Для приготовления эталонов навески по 1 г Cd С1г проводят через все стадии анализа до приливания HNOa. К полученным сухим остаткам прибав-
188
Таблица 35 (окончание)
Спектрограф Дуга I, а Экспозиция, сек Коэффициент вариации, % Литература
Постоянного тока 12 60 15—25 [250]
ИСП-30 То же 20—30 [267, стр. 44]
ляют раствор, содержащий необходимые количества искомых примесей, и выпаривают досуха. В первый эталон вводят 5 мкг Zn, по 2,5 мкг А] и Fe, по 2,0 мкг Са и Mg, по 1,0 мкг Со, Cr, Ni и Ti и 0,5 мкг Мп; во второй эталон вносят г/ю часть этих количеств, третий оставляют без добавок. Ко всем эталонам приливают по 2 мл HNOa и далее обрабатывают так же, как и пробу.
Концентраты пробы, контрольного опыта и эталонов испаряют в дуге постоянного тока; содержание искомых элементов рассчитывают по методу добавок, как и в предыдущем способе.
По критерию Кайзера максимальная чувствительность определения соответствует: для А1 и Fe — 1-10~6, для Са 1-10~4, для Со, Ni и Ti 8‘10~в, для Cr 2-10~7, для Mg — 5-Ю-6, для Mn 8М0-7 и для Zn 4-10-в%. Средняя квадратичная ошибка определения отдельных элементов 20—30% [267, стр. 44].
Определение макроколичеств элементов в препаратах кадмия производят обычно титриметрическим методом.
Для раздельного определения целых процентов Ва, Са и Sr в солях кадмия из их водного раствора с pH 7 их сумму экстрагируют трибутилфосфатом (ТБФ) в присутствии хлората и реэкстрагируют 2 N НС1. Полученную водную фазу нейтрализуют, доводят до 0,05 N по NaOH и трижды экстрагируют кальций раствором азо-азокси в смеси Ch с ТБФ. Водную фазу делают 1N по NaOH и дважды экстрагируют тем же экстрагентом стронций. В оставшемся водном слое титруют комплексоном III барий, а в реэкстрактах из органических фаз кальций и стронций [267, стр. 227].
При определении в соосажденном Ва — Cd-стеарате барий титруют 0,05 N H2SO4 по индикатору хлорфосфоназо III [268, стр. 153]. Можно использовать также метод внутреннего электролиза. Для определения цинка в металлическом кадмии пробу растворяют в смеси соляной и азотной кислот, затем основную массу кадмия выделяют осаждением на алюминиевой фольге, а оставшиеся небольшие количества его — осаждением сероводородом; в фильтрате определяют цинк [2336, 258а].
Медь в металлическом кадмии определяют методом внутреннего электролиза из 250 мл водного раствора, содержащего 3—4 г (NHi)2SO4 и несколько капель H2SO4 (1 : 1). Наиболее эффективно электролиз проходит при нагревании анализируемого раствора до 75—80° С [434а].
189
Химические и физико-химические методы анализа кадмия и его соединений высокой чистоты
Объект анализа Определяемый элемент Содержание, % Bee пробы, a Метод выдсления^элемента или отделения мешающих примесей
Cd Ag 1-10-5 2 Без отделения
Al 1-ю-* 1 То же
As 4-10-e 5 Отгонка AsH3
1-10-5 1 Экстракция диэтилдитиокарбами-
натом
Си 2-10-e n-10-e 10 Без отделения
1-10-5 1 » »
Си, Мо n-10-6 10 » »
Fe 4-10-e 1 Осаждение Bi и Си суспензией ZnS, pH 4
3-10-4 2,5 Без отделения, маскирование комплексоном III
1-10-4 1,5 Без отделения
n-10-6 5 Экстракция купфероном в СНС13
4-10-5 1 Без отделения
4-10-e 5 Осаждение на МпО3
Ni 2-10-e 10 Экстракция диметилглиоксимом в СНС13
Pb 5-10-4 5 Без отделения
1-10-5 5 Соосаждение с SrSO4
Sb 4-10-7 Соосаждение с МпО3
МО'5 1 Соосаждение с Fe(OH)3
5-10-5 10 То же, pH 5
Sn 6-10-’ Соосаждение с МпО3, отгонка с НС1 + НВг
1-10-5 5 Экстракция с диэтилдитиокарбами-натом в СНС1з, реэкстракция с
КМпО4
TI 1-10-5 6 Соосаждение с Fe(OH)3, избыток NH4OH
4-10-4 1 Экстракция эфиром из 1 М НВг
1-10-4 5 То же
190
Таблица 36
Метод определения Реагент, среда или условия определения Литература
Кинетический, аб-сорбциометрическое окончание Абсорбциометрия Се4+(-|- НС1) -» Се3+, 420 нм Хромоксан фиолетовый Р [511] [261, стр. 384]
» Молибденовая синь в изопентаноле* [261, стр. 393]
» То же [261, стр. 384]
» Диэтилдитиокарбаминат в СНС13 [261, стр. 391]
» » 2,2'-Бицинхониновая кислота Дитизон в СНС13, pH 1 [374] [261, стр. 398]
Полярография пере- 0,1 N HNO3, продувание азотом; [261, стр. 400]
меннотоковая Абсорбциометрия = —0,25 и —0,50 в 72 ’ ’ Роданид + диантипирилметан, [261, стр. 388]
» СНС13 1,10-Фенантролин, р!Г8 [261, стр. 384]
» Хромоксан фиолетовый Р [261, стр. 384]
» Роданид в изопентаноле [261, стр. 389]
» Салициловая кислота, pH 2—3 [261, стр. 399]
» » 1,10-Фенантролин, pH 6—7 Диметилглиоксим в NH4OH + [411] [261, стр. 391]
Полярография осциллографическая Абсорбциометрия + окислитель 0,1 N НС1, Et/t = —0,55 в Дитизон в СНС13, pH 9,5 [154] [261, стр. 396]
Полярография переменнотоковая Абсорбциометрия » Метиловый фиолетовый в СвН5СН3 Кристаллический фиолетовый в [753] [259] [261, стр. 394]
Полярография переменнотоковая Абсорбциометрия СвН6СН3 n-Нитрофенилфлуорон, H3SO4 [753] [261, стр. 392]
» Кристаллический фиолетовый в [261, стр. 394]
» Полярография переменнотоковая СвН5СН3 Малахитовый зеленый в диэтиловом эфире 1 М НС1, —0,25—(-0,75) в [184] [183]
191
Объект анализа Опреде4-ляемый элемент Содержание, % Bee пробы, 8 Метод выделения элемента или отделения мешающих примесей
Cd Tl 2-IO-3 2,8 Без отделения, маскирование Gd 1 М ЭДТА
Экстракция эфиром из 1 М НВг
Zn nIO-3 5 Экстракция роданидом в эфире из 0,4 N H2SO4
n.lCT3 1 Без отделения, маскирование KJ + тартрат
1MCdCl2 Cd n -IO-13
CdO Cd I-IO-5 4 Cd окисляют К,Сг2О? в кислой
--- среде
CdS Br 1-10-3 2 Сжигание, поглощение Вг в Н2О
Cl 1-10-5 2 Сжигание, поглощение С1 в Н2О + НСООН
2-IO-2 0,5 HNOa, отгонка, поглощение 0,1 N КОН
Cd 0,01 Cd отгоняют в токе азота
CdO 0,01 CdO растворяют ледяной СН3СООН
Co IO"3 50 Экстракция 1-нитрозо-2-нафтолом в СНС13
Cu 1 • IO-3 0,04 Без отделения
2,5-10“4 1 Без отделения, маскирование цитратом
n-10~e 50 Без отделения
n-lO-6 50 » »
5-10"’ 2 Экстракция диэтилдитиокарбами-натом в СНС13
Fe n-10-8 50 Соосаждение с А1(ОН)3 или экстракция купфероном в СНС13
Ga 1-10-® 0,5 Без отделения '
Hg 5-IO-5 1
J З.Ю-3 2 Сжигание, поглощение Н2О + Вт
In 1-10-e 1
К 1.10-4 Без отделения
Na 2-IO-4 » »
Ni n-lO-6 50 Экстракция 1-нитрозо-2-нафтолом
в СНС13, реэкстракция 0,005 М НС1
Sb 5-Ю-3 1- Т1 — экстракция: КВт + бриллиантовый зеленый в СвН5СН3
Zn 5-10-5 0,5 Хроматографирование на КУ-2 в присутствии KI
192
Таблица 36 (продолжение)
Метод определения Реагент, среда или условия определения Литература
Полярография пере-меппотоковая pH 4,25, от —0,3 в (н.к.э.) 1752]
Полярография NH4OH, £1/а = —0,50 в [56]
» NH4OH, принудительный отрыв (417]
капель
Абсорбциометрия 1-(2'-Пиридилазо)-2-нафтол в СНС13, pH 6,8 [563]
Атомная абсорбция В графитовой кювете [240]
Абсорбциометрия Определение избытка К2Сг2О7 дифенилкарбазидом [670]
» Флуоресцеин [541]
Нефелометрия HNO3 + AgNO3 [541]
Амперометрическое титрование Hg2(NO3)2 [331]
Абсорбциометрия Дитизон в СС14 [421]
То же [421]
Нитро зо-Р-соль [593]
Атомная абсорбция Лампа с полым катодом, 324,7 нм [523]
Абсорбциометрия Купризон, pH 8,5, 600 нм [593]
» Диэтилдитиокарбаминат свинца, в СНС13, pH 4,5—6,0, 426 нм [593]
о-Метиловый эфир 2-изатиноксима в СНС13, pH 5,6, 540 нм [593]
Кинетический 1,5-Дифенил-З-аминопиразолин + + Н2О2, pH 7,3—7,6 [315]
Абсорбциометрия 1,10-Фенантролин+ NH2OH+NaClO4, экстракция СНС13 + изобутанол [593]
Флуориметрия Люмогаллион ИРЕА, pH 2,2—2,7 [224]
Абсо рбциометрия Дитизон [315]
» ^(образовавшийся из JO3 + J~) экстрагируют СНС13 [541]
Флоуриметрия 5,7-Дибром-8-оксихинолин 1315]
Фотометрия пламени [52]
То же 152]
Абсорбциометрия Диацетилдиоксим [593]
» Бриллиантовый зеленый [315]
» Дитизон [315]
У Щербов Д. П.
193
Объект анализа Определяемый искомый элемент Содержание, % Bee пробы, г Метод выделения элемента или отделения мешающих примесей
CdSe Cd 0,002 Без отделения
Se 0,002 » »
Cd—Hg—Те Соли Cd Cd Те Tl As 5.10-3 3-10-’ 0,03 0,03 10 Те — осаждение NH3OH, Hg — отгонка Cd и Hg — маскирование комплексоном III Без отделения Отгонка с HCl + N2H4-H2SO4
Fe Tl 6-10-e 10 Без отделения » »
Zii 1 • 10-4 1 Cd — маскирование 1,2-диамин-циклогексантетраацетатом Экстракция роданидом в изопентаноле
5-10-’ 100 Частичное (~ 0,7%) осаждение Cd(OH)2, маскирование KJ тиомочевиной
* Колориметрируемый продукт реакции.
Таблица 37
Спектральное определение микропримесей в халькогенидах кадмия
I Эле- । 1 мент | Аналитическая o линия, А Измерения фона в минимуме, ближайшем к аналитической линии Элемент Аналитическая о линия,А, Измерение фона в минимуме, ближайшем к аналитической линии
Ag 3280,68 С коротковолновой стороны Мп 2798,27 С длинноволновой стороны
Al 3092,71 Между линиями А1 и Ti Ni 3050,82 То же
Bi 3067,72 С длинноволновой стороны РЬ 2801,99 С коротковолновой стороны
Сг 4254,33 То же Si 2514,32 С длинноволновой стороны
Cu 3273,96 » Sn 3175,05 То же
Fe 3020,64 Скоротковолновой стороны Ti 3088,03 Между линиями А1 и Ti
Mg 2802,70 То же Zn 2138,56 По абсолютной интен-
сивности
В техническом анализе, при контроле ванн кадмирования, применяют методы определения их компонентов [338], сведенные в табл. 38. В бесцианистых электролитах содержание CH3COONH4 находят по разности между суммарным содержанием NH4 и количеством NH4C1. При определении в цианистых электролитах Fe, Cd (амперометрическим титрованием), Cu, Ni и сульфатов цианиды предварительно разрушают подкислением пробы и последующим 5-минутным кипячением. Комплексонометрическое титрование
194
Таблица 36 (окончание)
Метод определения Реагент, среда или условия определения Литература
М икро спектро фо томе-трическое титрование Комплексон III, pH 10,2, индикатор ПАР, 495 нм. [295]
То же HCl, KJ, индикатор крахмал, Na3S3O3, 585 нм [295]
Полярография NH4OH + NH4NO3 (метод добавок) [8]
» NH4OH + NH4NO3, £1/, -0,72 и —0,91 в [8]
Атомная абсорбция Лампа ВСБ-Т1, 277 нм [268]
Полярография переменнотоковая Метод добавок [753]
Абсорбциометрия Фталексон S, pH 1, 530 нм [451]
Амперометрическое титрование К3СгО4, Hg-электрод, —0,56 в (н.к.э.) [598]
Полярография (pH > 5) [598]
Абсорбциометрия Кристаллический фиолетовый + + роданид, образовавшийся осадок растворяют в этаноле [460]
Колориметрическое титрование Титрование дитизоном в СС14 [99, 468]
Cd можно производить и после связывания CN' формальдегидом. При титровании карбонатов и щелочей CN' маскируют нитратом серебра [338].
В стекольном производстве при анализе CdS в нем определяют влагу, нерастворимый остаток, сульфаты (гравиметрия BaSO4), карбонаты (алкалиметрия избытка H2SO4, взятой для разложения пробы), FeS (осаждение Fe(OH)3 или колориметрическое титрование с роданидом) и свободную серу (кипячение с сульфидом и йодометрическое титрование образовавшегося тиосульфата) [314].
Для определения Cd в пигментах (смеси CdS, ZnS и BaSO4) и в присутствии РЬ предложено введение избытка этилен-бис-(оксиэтиленамин)тетрауксусной кислоты и обратное титрование ее раствором СаС12 по фталеинкомплексону [697]. Таллий из олово-кадмиевых сплавов извлекают диизопропиловым эфиром и колориметрируют с родамином С [781]. При анализе электрохимических порошков металлического Cd, содержащих CdCO8, вместо метода цементации [415] рекомендован газоволюметрический способ; из газов, выделяющихся при кислотном разложении навески, СО2 удаляют раствором КОН и по объему оставшегося Н2 находят содержание металлического кадмия [129]. Описано и полярографическое определение In в Cd-содержащих цинковых концентратах и в ZnO [277].
7*
195
r„„ , Таблица^
Определение компонентов электролитов кадмирования [338]
Компонент Метод определения Реагент, титрант Индикатор, условия определения
А1»+ Титриметрия NaF Раствор Fe(SCN)3 в изопропанбле
во|~ Алкалиметрия Глицерин (маннит), NaOH Фенолфталеин
Cd2+ Комплексонометрия Амперометрия Комплексон III Вг~ + диантипирилметан K3[Fe(CN)6] 8-0кси хинолин Хром темно-синий, эриохром черный —0,8 в —0,8 в —0,8 в
CN" Аргентометрия AgNO3 KJ
Cu2+ Колориметрия РЬ-диэтилдитиокарбаминат в СНС13 Шкала эталонов
Fe2* Колориметрическое титрование Перманганатометрия (NH4)2SO4- Fe2(SO4)3 SnCl2, KMnO4 Сульфосалицилат КМпО4
Na2CO3 + NaOH Ацидиметрия ВаС12 , HC1 AgNO3 Фенолфталеин
NH4C1 Аргентометрия К2СгО4
NHj (сумма) Ацидиметрия HCHO + NaOH, его избыток титруют HC1 Феноловый красный
Ni2+ Колориметрия Диметилглиоксим + (NH4)2S2Os Шкала эталонов
g2- Иодометрия J2, Na2S2O3 Пиридин + Cd2+, его избыток титруют комплексоном III Na2SO3 + Cu2+, ее избыток + KJ и титрование раствором Na2S2O3 Крахмал
SON" Комплексонометрия Иодометрия Хром темно-синий Крахмал
SO42" Клей Амперометрия Титриметрия Комплексонометрия Нефелометрия РЬ(СНзСОО)а или Pb(NO3)2 ВаС12 Pb(NO3)2, его избыток титруют комплексоном III —0,8 —1,0 в Ализариновый красный С Мурексид Колориметр-нефелометр ФЭКН-57, красный светофильтр
Тиомочевина Флуоресцеин Иодометрия 2* Колориметрия J2, Na2S2O3 Крахмал Шкала эталонов
* В одной аликвоте титруют NaOtl + Na2CO3, в другой после осаждения ВаСОз — NaOH и по разности находят содержание Na2CO3. »* Титруют CdS, образовавшийся при взаимодействии ионов Cd2+ и тиомочевины при нагревании щелочного раствора электролита.
Свойства некоторых соединений кадмия [384, 443]
Принятые сокращения: ам.—аморфный; ац.— ацетон; бв.—безводный; бел.—белый; бц.— бесцветный; в.р.—весьма растворим; гекс.— гексагональный; желт.— желтый; зел,— зеленый; иг.—иглы; к.— кислота; кб.— кубический; конц.—концентрированный; кор.— коричневый; крист.— кристаллы; мет.— метанол; мн.— моноклинный; н.р.— нерастворим;
Молеку- Цвет, кристал-
Название Формула лярный лическая Плотность
вес форма
Арсенат, о-двузаме- CdHAsO4 ,Н2О 270,34 4,161
щенный
Бромид CdBr2 272,22 бел., гекс., тб. 5,19-5,28
Бромид, моногидрат CdBr2- Н2О 344,28
Вольфрамат CdWO4 360,25 желт., крист.
Гидрат окиси Cd (ОН)3 146,41 бц., гекс, или 4,79
триг.
Иодид Cdl2 366,21 кор., гекс. 5,670
Карбонат CdCO3 172,41 бел., гекс, или 4,258
триг.
Молибдат CdMoO4 272,34 тетр. 5,347
Нитрат Cd (NO3)2 236,41 бц., крист.
Нитрат, тетрагидрат Cd (NO3)2-4H2O 308,47 бц., крист. 2,455
Окись CdO 128,39 кор., ам. 6,95
CdO 128,39 кор., кб. 8,15
Окись (I) Cd2O 240,80 зел., ам. 8,192
Оксалат Cd2C2O4 200,42 бц., кб. 3,32
Оксалат, моногидрат Cd2C2O4- H2O 254,47 бц., крист.
Роданид Cd(SCN)3 228,56 бц., крист.
Сульфат CdSO4 208,46 бц., ромб. 4,691
Сульфат, моногидрат CdSO4-H2O 226,48 бц., пг. 3,79
Сульфат, гидрат 3CdSO4-8H2O 769,50 бц., мн. 3,03
Сульфид CdS 144,46 желт, ор., кб. 4,82
или гекс.
Фосфат, о-однозаме- Cd(H3PO4)3.2H3O 342,40 бц., трикл. 2,74
щенный
Фосфат, о-трехзаме- Cd3(PO4)3 527,14 бц., ам. или
щенный гекс.
Пирофосфат Cd2P2O,-2H2O 434,77 бц., пор. 4,965
Фторид CdF3 150,40 бц., кб. 6,64
Хлорид CdCl2 183,31 бц., триг. 4,047
Хлорид, гидрат CdCl3.2,5H3O 228,34 бц., мн. 3,327
Хлорид основной Cd(OH) Cl 164,86 бц., гекс. пр. 4,57
Цианид Cd(CN)3 164,44 бц., крист. 2,226
198
Приложение 1
op.— оранжевый; пор.— порошок; пр.— призмы; р.— растворим; pear.— реагирует; ромб.— ромбический; сл.р.—слабо растворим; сп.—этанол; тб.— таблички; тетр,— тетрагональный; триг,—тригональный; трикл. —триклинный; щ.—щелочи; эф.—диэтиловый эфир.
Т. пл. Т. кип. Растворимость, а/100 г
В холодной воде в горячей воде в прочих растворителях
°C
>120 567; 568 —Н2О, 130—200 388+1 ~400, разлагается 350 59,5 >900, разлагается >900, разлагается Разлагается 340, разлагается Разлагается 1000 (1750) 100, разлагается 1500 900 (бв.) 1100 564; 568 —1,5-ШО 34 >200, разлагается 863 900; 918 132 1747; 1758 968;975 75 120 0,05 0,00026 78,7 2,76-10-е сл. р. 142 327 н. р. н. р. 0,00337 0,00305 Р- 76,7 Р- Р- 8,67-10-14 н. р. 4,35 90,0 189 1,7 162 350 125 н. р. 682 в. р. н. р. н. р. 0,009 61 Р. Р- 147 287 р.сп. (26,6), эф. (0,4), ац. (1,56) р.сп. (25), ац.; сл.р.эф. р. NH4OH pear.к.; р.солях NH4 р.сп. (102), мет. (176), эф., NH4OH; сл.р. ац. реаг.к.; р. солях NH4, KCN; н.р. NH3 р.к., NH4OH, KCN р.сп., NH3 реаг.к.; р. солях NH4; н.р.щ. То же реаг.к., щ. реаг.к.; и.р.сп. pear, к.; р. NH4OH; н.р.сп. р.сп., NH3 н.р.,сп., ац., NH3 н.р.сп. pear. конц. к.; сл.р. NH4OH pear. НС1; н.р.сп., эф. реаг.к.; р. солях NH4 р.к., HF; н.р.сц,, NH3 р.сп. (1.52), мет.; н.р. ац., эф. р.мет. (2,05); сл.р.сп. реаг.к.; р. KCN, NH4OH
199
Приложение. 2
Неорганические комплексные соедкнения кадмия (25°С) [233, 481]
Лиганд Комплексный ион Среда, ионная сила рК, РК, PKf! рКз P^l.2.8 Р*« PK1>S>S>4
Аммиак 0 2,51 4,47 5,77 6,56
1 M NH4CIO4 2,54 2,24 1,30 1,18 7,25
CdNH32+ 2* 0,5—5,0 2,65
Cd(NH3)22+2* 0,5-5,0 2,10 4,75
Cd(NHs)32+ 2* 0,5—5,0 1,44 6,19
Cd(NH3)t2+ 2* 0,5—5,0 0,93 7,12
Cd(NH3)32+ 2* 0,5—5,0 -0,32
Cd(NH3)e2+2* 0,5-5,0 —1,66
Бромид • 0 2,23 3,00* 2,83 2,93
3 М NaCICU 1,65 0,75 0,88 0,22
CdBr+ 3,0 1,75
CdBr2 3,0 0,59 2,34
С(ШГз~ 3,0 0,98 3,32
CdBrt2- 3,0 0,38 3,70
Гидразин 1 2,25 2,40 2,78 3,89
Гидроксил ? 4,17 8,33* 9,02 8,6
CdOH+ 2* 0,1 2,30
Иодид 0 2,28 3,92* 5,00 6,10
3 М NaC104 2,08 0,77 2,15 1,48
Cd2J®+ 0,2—9,0 2,49
CdJ+ 0 2,28
CdJ+ 3,0 2,08
CdJ2 0 1,64 1,64 3,92
CdJ2 з,о (О,62) (0,62) (2,70) -
CdJs” 0 (1,08) (1,08) (5,00)
CdJ3 3,0 2,30 2,30 5,00
CdJi2- 0 1,10 1,10 6,10
CdJ«2- 3,0 1,49 1,49 6,49
CdJ34~ 0,05—2,5 6,0
Приложение 2 (продолжение)
Лиганд Комплексный ион Среда, ионная сила рК, pKt P^1.2 РК3 рК1*2»8 pKi pKl,2,3*4
Нитрат CdNO3+ 0 0 0,40 0,40
CdNO3+ 3,0 0,11
Нитрит 3,0 3 M NaCICk 1,80 1,80 1,21 3,01 * 0,80 3,81 0,7 3,1
Селеноцианат Cd(SeCN)?- 0,3 3,60 4,18
Гидрофосфат CdPaO?2- CdP2O,2~ 3,5 5,6 5,57
Роданид 0 3 М NaC104 1,74 1,39 0,59 2,33* 0,60 2 3
CdSCN+ 2* 2,0 1,04
CdSCN+ 3,0 1,39 0,71 1,75
Cd (SCN)a 2* 2,0
Cd (SCN)2 Cd (SCN)3~ 2* Cd (SCN)3- 3,0 2,0 3,0 0,59 1,98 (-0,97) 0,60 (0,78) 2,58 1,0 1,78
Cd (SCN)^ 2* 2,0 -0,08
Cd (SCN)e4- 0,5—5,0
Селенат 0 2,27 *
Селенит 1,0 ? 5,15
Сульфат 0 0 2,31 * 2,29
CdSO4 0 2,31
3,0 0,85
Сульфит 1,0 1 М NaNOs ? 4,19 4,19
Приложение 2 (окончание)
8
Лиганд Комплексный ион Среда, ионная сила pKi РК> рк„2 рКз рК1»*»з рК< рКъми
Тиомочевина Cd (CSN2H4)2+ 0,2 1,58
Cd (CSN2H4)22+ 0,2 1,05 2,63
Cd (CSN2H4)32+ ОД 2,92
Тиосульфат CdS2O3 CdS2O33* 0 0 0,11 3,94* 3,92 2,90 2,52 6,48
Cd(S2O3)22~ 0' 2,52 6,44
Cd(S2O3)34- 0,3—0,9 6,33
Фторид 1,0 0,46 0,53*
1 М NaC104 0,46 0,07
Хлорид 0 2,05 2,60* 2,4 2,9
CdCl+ 3 М NaC104 1,0 1,54 1,35 1,54 0,66 0,09
CdCl+ 3,0
CdCl2 CdCl2 CdCl3-CdCU2- 3* 1,0 3,0 3,0 1,0—1,6 0 ? 0,43 0,52 1,78 2,06 0,40 2,46 (2,0) (2,58) 17,11
Цианид CdCl64~ 5,18 9,60* 13,92
CdCN+ 3 М NaC104 3,0 5,48 5,54 5,14 4,56 3,58 18,'76
Cd(CN)2 3,0 5,06 10,60
Cd(CN)3~ 3,0 4,70 15,30
Cd(CN)42- з,о 3,55 18,85
. Р„и м еч а н и е: В скобках даны менее надежные данные.
* Нейтральные молекулы в растворе. 2* 30°С. 3* 18°С,
Органические комплексные соединения кадмия (25°С) [233, 481]
Йриложейие 3
Лиганд Комплексный ион Среда, ионная сила РК, рК2 РК1.2 рКз pKj,2i3 РКз Р&1,2,8>4
Амины
2,2'-Дипиридил Cd(P)a2+ 0,1 M KNO3 ОД 4,5 3,5 8,0 10,5 10,47
Диэтилентетрамин Cd(P)2+2* Cd(P)22+ 2* o,i 0,1 8,45 5,4 13,85
Имидазол Cd(P)2+ Cd(P)22+ 0,15 0,15 2,80 2,10 4,90
Cd(P)32+ 0,15 1,55 6,45
Cd(P)i2+ 0,15 1,13 7,58
Пиридин 1,0 0,1 М KNO3 1,27 2,14 2,14 2,50
Cd(P)22+ 0,1 - 2,14
Cd(P)?+ o,i 2,49
П ропилендиамин Cd(P)2+ 2* Cd(P)22+ 3* Cd(P)32+ 3* 0,5 0,5 0,5 5,42 4,55 9,97 2,15 12,12
1,2,3-Т риаминоп ре- Cd(P)2+ 2* 0,1 6,45
пан
Метиламин Cd(P)2+ Cd(P)22+ Cd(P)32+ Cd(P)4«*- 1,0 1,0 1,0 1,0 2,745 2,063 4,808 1,131 5,939 0,611 6,550
Триаминотриэтил- Cd(P)2+ 2* ОД 12,3
амин
Приложение 3 (продолжение)
Лиганд Комплексный ион Среда, ионная сила pKi pK, рКм рК3 рКъм P-К.
Т риэтилентетрамин Cd(P)2+ 0,1 10,75
Cd(P)22+ 0,3 13,9
1,10-Фенантролин 0,1 6,4 11,6 15,8
к 0,1 M KNOa 6,4 5,2 4,2 15,79
Cd(P)22+ 0,03 13,16
Cd(P)32+ 0,02 15,20
Этилендиамин 1 М KNO3 5,63 4,59 10,22 2,07 12,29
Cd(P)2+ 3* 0,5 5,47
Cd(P)22+ s* 0,5 4,55 10,02
Cd(P)32+ 3* 0,5 2,07 12,09
Анионы органических
кислот
Ацетат 3,0 1,30 2,28 * 2,42 2,00
3 М NaClOi 1,30 0,98 2,28 0,14 0,42 2,00
Cd(P)+ 1,77
Cd(P)2 1,0 2,77
Койевая кислота Cd(P)+ 3* 6,6
Cd(P)2 3* 4,7 11,3
Малонат Cd(P) 0 3,29
Нитро ацетат Cd(P)+ 0,6 0,19
Оксалат 0 4,0* 5,77
0 3,52 1,77
Cd(P) 0 3,52
Cd(P)22- 0 1,85 5,37
Цитрат Cd(P) 0,1 4,22
Cd(POH)2- 0,1 5,05 9,30
Приложение 3 (продолжение)
Лиганд Комплексный ион Среда, ионная сила рл'1 рК2 рК,,г РК3 Р^1»«»8 рК, pKi^a 4
Тартрат Cd(P) 2,7
Аминокислоты
Аминобарбитурат Cd(P)24- 0,01 6,7
Аспарагин Cd(P)2 2* 0,01 6,8
N-Оксиэтилэтилен-диаминотриацетат Cd(P) 3* 0,1 13,0
Аспарагинат Cd(P) 3* Cd(P)22- 3* 0,1 0,1 4,37 3,11 7,48
Р-Оксиэтилимино-ди ацетат Глицин Cd(P) 3* Cd(P)22- 3* Cd(P)2 2* 0,1 0,1 0,01 7,12 5,12 12,04 8,1
N, N-Диоксиэтилгли-цин Cd(P)+ 3* Cd(P)2 3* 0,1 0,1 4,78 3,37 8,15
1,2-Диаминоциклогексантетраацетат Cd(P)2- 2* 0,1 19,23
Иминодип ропион ат Cd(P) 3* 0,1 3,51
Иминодиацетат Cd(P) 3* Cd(P)22- 3* 0,1 0,1 5,35 4,18 9,53
Иминопропионо-ацетат Cd(P) 3* Cd(P)22- 3* 0,1 0,1 4,52 3,16 7,68
Нитрилоди ацетопропион ат Cd(P)“ 3* 0,1 7,5
Иминодип ропионо-ацетат Cd(P)~ 3* 0,1 5,6
Приложение 3 (окончание)
206
Лиганд Комплексный ион Среда, ионная сила РК1 рК. Рк1.» рК, рК, ,, 1 * рк< рК‘А’.‘
Нитрилотрипропинат Cd(P)~3* 0,1 3,4
Нитрилотриацетат См. 2* 0,1 М КС1 9,54
Cd(P)~ 2* 0,1 9,54
Cd(P)24~ 0,001 5,7
Этилендиаминтетра- 0,1 16,59
ацетат См.2* 0,1 М КС1 16,59
Cd(P)2-2* 0,1 16,48
Дикетоны, альдегиды
Ацетилацетон См.3* 0 3,83 2,76 6,59
Cd(P)~ 2* 0 3,84
Cd(P)2 2* 0 2,83 6,72
Салицилальдегид Cd(P)2 2* 0,1 7,8
Другие органические лиганды
Диметил глиоксим 50%-ный раствор диоксана 5,7 5,0 12,7
8-Оксихинолин 0,01 7,2 13,4*
50%-ный раствор диоксана 9,43 7,68 17,11
Cd(P)-Cd(P)2 9,43 7,68 17,11
8-Окси-2-метилхи- 50%-ный раст- 9,0 16,00
иолин вор диоксана
Примечание: Р — молекула лиганда.
* Нейтральные молекулы. г* 20° С. 3* 30° С. *• 18° С.
Прилож ение 4
to
-4
Приложение 4 (окончание)
Вероятное строение комплексов кадмия с органическими лигандами
I — пиридин; II — 1,10-фенантролин; III — диэтилдитиокарбаминат; /V — диэтилдитиофосфат; V — меркаптобензтиазол; VI — кадион; VII — бромбензтиазо; VIII — кислотный хром темносиний; IX — хинальдиновая кислота; X — 8-оксихинолин; XI — 8-меркаптохинолин; XII — 8-(бензолсульфониламино)хинолин; XIII — дитизон; XIV — купферон; XV — антраниловая кислота; XVI — 2-(о-оксифенил)-бензоксазол; XVII — дифенилкарбазон; XVIII — иодид + 0-нафтохинолин; XIX — иодид + железо(П)-дипиридил; XX — 1,10-фенантролин + эозин; XXI — дальзин; XXII — 1,3-дихинолил-3-пропен
Кристаллы комплексов кадмия (а) и цинка (6) с пиридином и бромидом или роданидом
И а и ы о £ ф и и ф
сл
Приложение 7
Кристаллы тетрароданомеркуриатов кадмия (а); свинца (б); цинка (в); кобальта (а); железа (Э); меди (е)
210
Приложение 8
Кристаллы тетрароданоцинката кадмия (а); ртути (при избытке цинка) (б); серебра (в)
Приложение 9
Кристаллы гексахлоркадмоата рубидия (а); цезия (б)
211
Приложение 10
Кристаллы оксалатов кадмия (а); кальция (6); олова (в); стронция’^)-висмута (д); цинка (е) F °
212
П р и л о же н и е И
Значения потенциалов полуволн кадмия на различных фонах [204]
Деполяризатор Состав раствора Е./,. в
Cd2+-H2O 1 Л/ НС1О4, 0,01% желатин —0,62
0,1 М KNO3 -0,578
0,1 М KNO3 + 0,01% желатин —0,582
0,9 М NH4NO3, 20% CjHgOH —0,580
1 М HNO3, 0,01% желатин -0,59
1 М KNO3, 0,01% желатин -0,59
1 М KNOS -0,63
Cd (H2SO4)X2+ 0,5 М H2SO4 —0,59
10,5 М H2SO4 —0,66
Cd2+-H2O 0,4 М CHgCOO-, pH 4,7 -0,61
0,5 М Na2F2, pH 3,1-6,7 -0,63
Cd (Cl)x<2-*)+ 0,1 М NaCl, 2,9 М NaC104 -0,57
0,1 М КС1, 0,01% желатин —0,60-
1 М КС1, 0,1 М KNO3 —0,64
0,9 М NH4G1, 20% С2НБОН —0,64
2 М NaCl -0,65
30% СаС12 —0,66
4 М NaCl —0,69
3 Л/ HCl —0,70>
6 М НС1 -0,79
45% С2НБОН -0,65-
65% С2НБОН -0,67
CdBr/2-*)* 0,1 М NaBr, NaC104 —0,58-
0,5 М НВг —0,65
0,5 М КВг —0,65
2 М НВг -0,70
3 М КВг -0,70
4 М НВг —0,76
CdJ?- 0,1 М HJ -0,64
0,1 М KJ —0,65
0,5 М KJ —0,70'
0,5 HJ -0,71
0,9 М KJ, 20% С2НБОН —0,76
2 М KJ -0,78
2 М HJ —0,79
3 М KJ -0,80
45 % С2НБОН -0,78
Gd (CNS)X<2“ ж>+ 65% С2НБОН 0,1 М KCNS, KN03 -0,81 —0,585.
0,5 М KCNS —0,614
213=
П р и л о'ж е н и е 11 (продолжение)
Деполяризатор Состав раствора е,/2> в
са(си5)^-ж>+ 0,5 М NH4CNS —0,640
1 М KCNS —0,634
1 М NH4CNS -0,66
2 М KCNS —0,664
2 М NH4CNS -0,68
5 М NH4CNS -0,74
Cd (S2O3)|- 0,1 М KNO3, 1 М Na2S2O3, 0,01% желатин -0,78
Cd (ацетат) 2+ 100% СН3СООН, 0,1 MLiCl -0,76
Cd (цитрат)" 0,042 М Na3C6H5O7 —0,664
0,4 М Na3C6H5O7, pH > 6 -0,704
1 М Na3C6H5O7, 0,1 М KNO3, 0,01% желатин -0,71
Cd2+ (тартрат) 0,5 М NaKC4H4O6, 0,01% желатин, pH 4,5-9 —0,64
1 М Na2C4H4O6, 0,1 М KNO3, 0,01% желатин -0,70
0,5 М NaKC4H4O6, 0,1 М NaOH, 0,01% желатин -0,79
Cd (NH3)x2+ 0,1 М NH3, 0,1 М NH4NO3 -0,674
0,9 М NH3 -0,783
1 М NH3, 1 М NH4C1 -0,81
2,4 М NH3 -0,847
4,7 М NH3 -0,883
Cd(/f)32+ 1 М метиламин; 1 М метил3 N -0,700
Cd2+(K) IM N(CH2CH2OH)3, 0,1 М КС1 -0,732
1 М NH2CH2CH2OH, 0,1 М КС1 -0,785
0,4 М N(CH2CH2OH)3, 0,1 М КОН —0,88
С6(метилимидазол)32+ 0,105 М 1-метилимидазол, 0,15 М KNO3 —0,688
Cd(nnpHflHH)22+ 0,1 М пиридин, 0,1 М KNO3, 0,01% желатин -0,592
Cd(nnpiifliiH)42+ 2 М пиридин, 0,1 М KNO3, 0,01% желатин -0,680
С6(этилендиамин)32+ 0,1 М этилендиамин, 0,1 М KNO3, 0,01% желатин -0,843
0,5 М этилендиамин -0,901
2 М этилендиамин -0,964
СЩпропиленди- 0,1 М пропилендиамин, 0,1 М KNO3, —0,848
амин)32+ 0,01% желатин
0,5 М пропилендиамин -0,910
2 М пропилендиамин —0,966
Cd(flH3THneHTpn- 0,1 М диэтилентриамин, 0,1 М KNO3, -0,929
амин)32+ 0,01% желатин
0,5 М диэтилентриамин -0,969
1 М диэтилентриамин —0,989
214
Приложение 11 (окончание)
Деполяризатор Состав раствора Е\/2’ в
Cd (триэтиленди- 0,1 M триэтилендиамин, ОДЛ/ KNO3, -0,949*
амин)з2+ 0,01% желатин
2 М триэтилендиамин -0,986
Cd(flnnnpiiflHn)g2+ 0,04 М дипиридил, 0,1 М KNO3, 0,01% желатин -0,766
С8(1,10-фенантро- 0,02 М 1,10-фенантролин, 0,01 М KNO3, —0,878
ЛИН)з2+ 0,01% желатин
Cd2+(X) (?) 0,6 М этилендиамин, тартрат, ОДЛ/ Na4P2O7 -0,77
Cd2+(X) 0,4 М ацетат, 0,1 М комплексон I —0,87
Cd2+(X) 0,5 М СН3СООЦ, 0,5 М этилендиамин, тартрат, 0,01 М комплексон II —1,02
CdW 0,4 М СН3СООН, pH 2,7, комплексон IV -1,04
Cd2+(X) 0,5 М СН3СООН, 0,5 М этилендиамин, тартрат, 0,01 М комплексон IV —1,35
Cd2+(X) 0,2 М NHg, NH4C1, 0,05 М тайрон -0,79
Cd(CN)?- 0,1 М KNO3, 1 М KCN, 0,01% желатин —1,16
Cd2+ 0,1 М бутил4Ш, метилцианид +0,34
Cd2+ 30% LiNO3, 17% NaNO3, 53% KNO3 (расплав), 160° С -0,55
Cd (амальгама) 0,1 М KNOg -0,58
0,1 М КС1 -0,60
1 М KJ —0,74
0,5 М NaKC4H4O6 -0,64
1 М NH3, 1 М NH4C1 -0,79
1 М KCN -0,06
30% LiNOg, 17% NaNOg, 53% KNO3 (расплав), 160° С -0,54
Примечание: X — неопределенный состав раствора; К — символ для комплексообразующего вещества»
215
Приложение 12
Наиболее интенсивные спектральные линии кадмия [156, 358, 517]
Длина волны, А Потенциал возбуждения, Эв Интенсивность V, WWW, UHJ Чувствительность определения Cd • в угольной Дуге, %
Дуга Искра
2144,4 ]1 5,78 50 200 7? 0,03
2194,6 11,07 5 100
2239,9 9,23 80 30 о,з
2265,0 II 5,45 25 - £00 0,003
2267,5 9,23 20 30 о,з
2288,0 I 5,39 1500 7? 300 Я 0,003
2306,6 9,13 20 30 0,1
2312,8 11,09 1 200
2321,2 II 11,07 1000
2329,3 9,23 50 60 0,1
2544,7 I 8,57 50 0,3
2580,3 I 8,57 50 0,1
2629,1 I 8,48 50 10
2639,5 I 8,39 75 15 0,3
2660,4 I 8,57 50 0,3
2677,6 I 8,39 100 25 0,1
2712,6 I 8,48 75 20
2733,9 I 8,23 50 25
2748,6 II 10,25 5 200
2756,8 I 8,45 50
2763,9 I 8,39 100 50 0,1
2764,1 8,39 50 25 0,3
2775,0 I 8,23 50 20 0,3
2836,9 I 8,07 200 80 0,1
2862,3 I 8,13 15 10
2868,3 I 8,23 100 80 0,3
2880,8 I 8,07 200 R 125 0,03
2881,2 I 8,07 50 Л зо - 0,1
2929,3 П 15,35 50
2961,5 I 8,13 20 15
2980,6 I 8,07 1000 7? 500 0,03
2981,3 I 8,07 2007? 40 0,1
2981,9 I 8,10 50 10
3005,4 I 8,07 25 4
3080,8 I 7,76 150 100 0,1
3082,7 I 7,76 50
3133,2 I 7,76 200 300 0,1
3250,2 II 9,28 100
3252,5 I 7,76 300 200 0,1
3261,1 I 3,80 300 300 0,01
216
Приложение 12 (окончание)
Длина волны, А Потенциал возбуждения, ав Интенсивность Чувствительность определения Cd в угольной дуге, %
Дуга Искра
3403,6 I 7,37 800 500 0,01
3466,2 I - 7,37 1000 500 0,01
3467,7 I 7,37 800 400 0,01
3500,0 I 7,37 25 15
3610,5 I 7,37 1000 500 0,03
3612,9 I 7,37 800 500 0,1
3614,5 I 7,37 60 100
3649,6 I 7,34 20 15
3729,1 I 7,26 15 Я
4414,6 II 8,59 200
4678,2 I 6,39 200 200 0,1
4799,9 I 6,39 300 300 0,03
5085,8 I 6,39 1000 500 0,03
5378,0 II 13,44 5 50
6031,4 I 8,45 30
60Э9,2 I 8,42 300
6111,5 I 8,42 100
6116,2 I 8,42 50
6325,2 I 7,37 100
6330,0 I 7,37 30
6359,9 II 15,40 10 50
6438,4 I 7,34 2000 1000 0,1
6465,0 II 13,66 5 50
6725,8 II 13,66 100
6778,1 I 8,43 30
7132,3 I 8,13 30
7346,2 I 8,08 1000
7383,9 8,08 1000
7385,3 I 8,08 800
7393,0 70
7399,2 I 8,06 70
217
Приложение 13
Рентгеновский спектр испускания кадмия, края его поглощения и значения частот в (ридбергах) (V/B) [387, стр. 170]
Серия Обозначение линий Переход Длина волны, X R Vi
Диаграммные линии
К Край поглощения 463,15 1967,55 44,357
к ~~ ш 464,37 1962,36 22,298
31 X — мш 474,12 1922,02 43,841
Зз 474,75 1919,49 43,812
ctl к — Lni 533,90 1706,82 41,314
«2 К —Ьц 538,32 1692,80 41,144
L Край пог лощения Lj 3078,30 296,03 17,206
Т2,3 ^1 ~~ ^П, ш 3131,20 291,03 17,060
Край поглощения Ln 3319,00 274,56 16,570
Ti Lu ~ ^iv 3328,80 273,75 16,545
Тз Lu ~~ Ni 3418,50 266,57 16,327
Зз I^-My 3423,10 266,21 16,316
Зю bj —^iv 3429,60 265,71 16,301
Край поглощения 3496,70 260,61 16,143
32 — NV 3506,99 259,84 16,120
Зе LIII ~ 3607,20 252,63 15,894
Зз Ll ~ MIU 3637,40 250,53 15,828
31 Li — Mn 3674,35 248,01 15,748
31 Ljj — UIV 3730,54 244,27 15,629
СИ £nI - - l/v 3948,30 230,80 15,192
аг ^III ~ MIV 3956,83 230,30 15,176
Т] Ln-Mr 4184,50 217,77 14,757
1 Ьш - Мг 4470,95 203,82 14,277
М MII ~ NIV 19,39* 47,00 6,856
т MIII — Nv 20,46 * 44,54 6,674
Мц-N! 22,90 * 39,79 6,308
Mlll ~ NI . 24,43 * 37,30 6,107
Край погло щения MIV_ v 28,13* 32,41 5,693
MlV — 0ц 30,35* 30,02 5,479
Mv — °ш 30,79 * 29,60 5,440
M4 — Na, ш 36,66* 24,86 . 4,986
MTL ~~ MIV 51,74* 17,61 4,197
MUI ~ Mv 58,46* 15,59 3,939
218
Приложение 13 (окончание)
Серия Обозначение линий Переход Длина волны, X R
Недиаграммные линии
L Ti 3296,0 276,48 16,628
32с) 3462,2 263,21 16,224
3(2Ь) 3472,6 262,42 16,199
31 3477,3 262,06 16,188
31о) 3501,0 260,29 16,134
зГ 3696,4 246,53 15,701
з}11 3703,9 246,03 15,685
311 3712,5 245,46 15,667
3} 3720,04 244,93 15,651
0t9 3910,0 233,06 15,266
0(8 3914,5 232,79 15,258
0(7 3917,8 232,59 15,251
0(6 3922,9 232,29 15,241
0(б' 3926,5 232,08 15,234
0(5 3929,6 231,89 15,228
0(4 3933,8 231,65 15,220 .
0(3 3937,2 231,45 15,213
0(2 3939,7 231,30 15,209
* Длина волны в ангстремах.
219
Приложение 14
Групповое концентрирование экстракцией кадмия и других элементов для последующего определения различными методами [144]
(ПДТК — пирролидиндитиокарбаминат аммония (кадмия); ДЭДТК — диэтилдитиокарбаминат натрия)
Объект анализа Экстракционный реагент Экстрагент pH, водная фаза Метод определения мик ропримесей Чувствительность определения, %
Алюминий особой ПДКТ (дитизон) Хлороформ 3,5—7,9 Спектральный Ю-З—ю-5
ЧИСТОТЫ 2,5— 1
Диантипирилметан » 3 М НС1 »
ДЭДТК » 8,5 Полярографический —10-4
Барий » 5—6 Спектральный Ю-4—ю в
Бериллий » » 1; 3 и 10— Активационный
12 М НС1
» » 8,5 По ля рог рафический
Бериллий особой ДЭДТК Этилацетат 4,5 » 2-10-3
ЧИСТОТЫ
Ванадий ДЭДТК (8-оксихино- Хлороформ или четы- 7,5—8,0 Спектральный 10-4—Ю-з
лин) реххлористый углерод
Дитизон Четыреххлористый уг- Полярографический ~10"1
Галлий фосфид ДЭДТК Хлороформ или четы- 6,0 Спектральный 5-10-6—2-
реххлористый углерод • IO-8
Магний » Хлороформ 8,5 Полярографический ~10-4
Оксихинолин (дитизон) » Спектральный 10-4—10 5
Ниобий ДЭДТК Хлороформ или четы- 6,0—6,5 » 10 4—10 5
реххлористый углерод
ДЭДТК (дитизон) Хлороформ »
Селен особой чисто- 8-Оксихинолин (дити- Циклогексанон 2,5 М НС1 » 10-3—10 7
ТЫ Тантал ДЭДТК Хлороформ или четы- 6.0—6,5 » 10-4—10-5
реххлористый углерод »
ДЭДТК (дитизон) , Хлороформ »
Приложение 14 (продолжение)
Объект анализа Экстракционный реагент Экстрагент pH, водная фаза Метод определения микропримесей Чувствитель* ность определения, %
Теллур особой чис- ПДКТ (дитизон или Хлороформ 3,9-5,7 Спектральный l-10-з
ТОТЫ ДЭДТК)
Титан ДЭДТК Хлороформ или четы- 6,0—6,5 » 10-4—10-5
» реххлористый углерод Хлороформ 3,0—3,5 Полярографический Ю-3—10-4
» » 8,5 » —IO’4
» » Этилацетат 3,0—3,5 » 10-3—10-4
Титан особой чистоты ПДКТ (дитизон) Хлороформ 3,5—7,9 Спектральный IO"5
Уран Дитизон Бензол 8,5—9,0 Полярографический
Фосфор хлорид Хром ДЭДТК Четыреххлористый углерод 8,0 Спектральный 10-6—10-3
» ' Хлороформ 4—5 Фотометрический или спектральный 10-4—10-5
Цирконий
» То же 8,5 Поля рографический ~10“4
» » 9,0 »
Цирконий и его соединения ПДКТ (дитизон) ДЭДТК » 3,5—7,9 Спектральный IO"5
Щелочные металлы Хлороформ 7,5 4,5 Фотометрический 10-3—10"7
Алифатические монокарбоновые кислоты Спектральный 10-6—Ю"7
Дитизон (8-оксихино-лин) ДЭДТК (8-оксихино- » 5-6 10-5—10-3
2,5; 10 » 10-4—10-3
лин)
То же » 2,5—3,0; » 10-5— Ю-7
5; 7
» Смесь хлороформа и изо- 5,5—6,0 » 10-6—10-8
to to пентанола
Приложение 14 (продолжение)
Объект анализа Экстракционный реагент * Экстрагент pH, водная фаза Метод определения микропримесей Чувствительность определения, %
Щелочные металлы ДЭДТК (8-оксихино-лин) Трибутил фосфат Дитизон 8-0ксихинолин Хлороформ Спектральный Ю-s—10-7
Бензол Хлороформ » H8SO4 » Полярографический ю-»—ю-® —10-5—10-е 10-5—10-е
Сплавы жаропрочные ДЭДТК » 6,2; 8,5 » 10-4
Висмутиол » 2.0; 6,0 » 10-«
Стали жаропрочные » » 2,0; 6,0 » 10 4
Концентраты медные ДЭДТК » 5,0 » 10 2
Руды железные » » ~5 » 10-2
Породы силикатные ДЭДТК (дитизон) » 9,0; 5,6 Спектральный 10-5—10 в
» » Хлороформ или четыреххлористый углерод 8-9 Полярографический 10~2
Почвы пдтк » Хлороформ » 4,8 4,7—5,3 Спектральный » 10 3—10 5
ПДТК (дитизон) » 3,5—4,0; 8 9 » 10-1—10-5
ПДТК (купферон) • » 2,0; 4,6—5,0 10-5
Воды природные 1,5-Ди-(Р-нафтил)тио-карбазон Дитизон » Четыреххлористый уг- 10,5 Полярографический » 10-»
ДЭДТК (дитизон, 8-оксихинолин) Хлороформ 3,5; 7,9 Спектральный 10-’—10-9
Водные растворы » » 2—13 Полярографический 10-в—10-’
Диэтилдитиофосфо рная кислота Четыреххлористый углерод 0,5—1,5 М Спектральный
Приложение 14 (окончание)
Объект анализа Экстракционный реагент Экстрагент pH, водная фаза Метод определения микропримесей Чувствительность определения, %
Водные растворы 1-Фенил-3-метил-4-бен-зоилпиразолон Смесь хлороформа и изопентанола 2,0—2,5; 7,0—7,5 Спектральный
ДЭДТК Хлороформ 2—13 Полярографический
Солянокислый 1-фенил-3,5-диами л-4-бутил-1-пиразол + солянокислый 1-метил-3,5-дипро-пил-4-этилпиразол Диметилформамид 10-е—10-7
Пиридин (роданид) Толуол 0,9 7V H2SO4 »
Воды лечебные ДЭДТК (дитизон) Хлороформ 3—9 Спектральный 10-4
Кислоты Дитизон (8-оксихино-лин) » 5—6; 7; 9 »
Растения ДЭДТК (8-оксихинолин, дитизон) » » 10-4—10-5
Биологические материалы Дитизон » 8,3 Полярографический 7,5-10-«
Рубеановодородная кислота (8-оксихинолин) » * Спектральный 10-4—10-5
Приложение 15
Групповое концентрирование кадмия и других элементов путем экстракции основы [144]
Объект анализа Среда Экстрагент Метод определения Чувствительность определения, %
Бериллий и его окись 4 М НС1 Хлороформ Спектральный 10-4—iQ-e
Галлий 6 М НС1 6 М НС1 8 М НС1 ~5 М НС1 6 М НС1 НС1 Диэтиловый эфир » » Диизопропиловый эфир Диэтиловый эфир Диизопропиловый эфир Бутилацетат Полярографический Спектральный Активационный Спектральный IO-’—Ю-8 io-» 10-4—10-в 10-s—10-’ 10-4—10-9
Галлий особой чисто- 6—7 М НС1 Диизопропиловый эфг^р » 10-4
8,5 М НС1 Бутилацетат Полярографический или спектральный 10-4—10-7
Галлий арсенит 6—8 М НС1 Диизопропиловый эфир Спектральный 10-4—10-4
Галлий арсенит 6-7 М НС1 Диэтиловый эфир Полярографический 10-4—10 ’
Галлий антимонид 10—12 М НС1 Р,Р'-Дихлордиэтиловый эфир Спектральный 10-5—10-’
Галлий хлорид 8—8,5 М НС1 6 М НС1 10—11 М НС1 Бутилацетат » 3,8'-Дихлордиэтиловый эфир » » Активационный О О С 2 J. 2 1 1 1 7?^
Железо 6,5 М НС1 Диэтиловый эфир Полярографический или спектральный —
Золото 3 М НВг 1—5 М НВг » » Полярографический Фотометрический 10-5—10-4 10-3—10-4
Индий 5 М НВг Диизопропиловый эфир Полярографический 10-4
НВг Диэтиловый эфир Спектральный 10-4—10 4
5 М НВг НВг 4,5 М НВг НВг 40%-ная НВг » » » » » » З,р'-Дихлордиэтиловый эфир Диэтиловый эфир » Полярографический Спектральный О О ф ф 2 2. 2 2 kki IL фф * фф it II ММ Ы О
Индий особой чистоты 5 М НВг Диизопропиловый эфир Полярографический —
Щербов Д.
КЗ N3
СЛ
Приложение 15 (окончание)
Объект анализа Среда Экстрагент Метод определения Чувствительность определения, %
Индий антимонид Индий фосфид Иттрий Кадмий Кадмий сульфид Кадмий хлорид Молибден Молибден и его соединения Ниобий Палладий Плутоний Ртуть Сурьма Сурьма особой чистоты Таллий Таллий хлорид Теллур особой чистоты Уран Цирконий 5 М НВг НВг 5 М НВг 5 М НВг 8 М НВг 13 М HNOs HJ 3,4 М HJ 3,4 М HJ HC1 + HNO3 (3:1) 6 М НС1 И М НС1 H2SO4 (1,84) 0,3—0,4 и 5-6 М НВг НС1 HJ (Hg : J > 1 : 2) HJ (Hg : J = 1 : 2) HJ (Mg : J = 1 : 2) 2-3 M HCl 10-12 M HCl 6 M HCl HBr 10 M HCl 6 M HCl 6 M HCl 7 M HCl HCl + HClOj винная HNOs Диэтиловый эфир Диизопропиловый эфир Диэтиловый эфир » » » » Трибутилфосфат Трибутилфосфат Диэтиловый эфир » » а-Бензоиноксим Диэтиловый эфир Амилацетат Трибутилфосфат или бензол Циклогексанон Диэтиловый эфир и хлороформ Циклогексанон » » Изоамиловый спирт ЗД'-Дихлордиэтиловый эфир Бутилацетат З.Р'-Дихлордиэтиловый эфир Бутилацетат Диэтиловый эфир » » Мети лизоб ути лкетон Трибутилфосфат, хлороформ Изоамиловый спирт Полярографический Фотометрический Спектральный » » » Полярографический или фотомет ричес кий Спектральный • » » » » » Полярографический и фотометрический Спектральный Фотометрический » Полярографический Спектральный » » Спектральный и полярографический Спектральный Полярографический » Спектральный 10-5—10-в 10-4—10-5 10-5—10-4 10-4 10-3—10-4 10-4—10-5 10-4—10-4 Ю-4—10-5 10-4—10-4 10-4—Ю-4 10-4—10-4 10-3—10-5 10-3—10-5 10-5—10-4 10-5—10-4 10-4—10-7 10-5—Ю-7 10-4—10-4 10-5—Ю-’ 10-5—10-’ 10-4—Ю-4 10-4—10-4 10-4—10-7 10-5—10-4 10-4—10-4 10-3—10-5
ЛИТЕРАТУРА
1. Адамович Л'П., Гершунс А. Л., Олейник А. А., Шкабара Н. М. Ж. аналит. химии, 26, 548 (1971).
2. Айтова В. X., Живописцев В. П. Уч. записки Пермского ун-та, № 111, 151 (1964).
3. Алексеев В. Н. Курс качественного химического полумикроанализа. М.—Л., Госхимиздат, 1952.
4. Алексеева М. В., Андронов Б. Е., Гурвич С. С., Жидкова А. С. Определение вредных веществ в воздухе производственных помещений. Госхимиздат, 1954, стр. 153.
5. Алимарин И. П. Усп. химии, 4, 854 (1935).
6. Алимарин И. П., Архангельская В. Н. Качественный полумикроанализ. М.—Л., Госхимиздат, 1949.
7. Алимарин И. П., Билимович Г. Н. Хим. наука и промышл., 1, 75 (1956).
8. Альбота Л. А., Заверач М. М. Изв. вузов. Химия и хим. технол., 14, 42 (1971).
9. Амирханова Т- Б., Подгорнова В. С-, Шестерова И П. Труды Ташкентского ун-та, вып. 288, 109 (1967).
10. Анализ минерального сырья. Л., Госхимиздат, 1956.
И. Анализ руд цветных металлов и продуктов их переработки. Сборник научных трудов Гинцветмет, № 14. М., Металлургиздат, 1958, стр. 66, 138.
12. Анализ руд цветных металлов и продуктов их переработки. Сборник научных трудов Гинцветмет, № 28. М., «Химия», 1968, стр. 16.
13. Андреева О. В., Васильев В. В., Терещенко Н. Б. Чувствительность качественных реакций. Изд-во ЛГУ, 1962.
14. Аникеева Н. П. Сб. «Спектральный анализ в цветной металлургии». М., Металлургиздат, 1960, стр. 179.
15. Арефьева Т. В., Нац Р. Г. Обогащение, металлургия цветных металлов и методы анализа. Сборник № 10. М., Металлургиздат, 1955, стр. 325,. 353.
16. Асатиани В. С. Биохимическая фотометрия. М., Изд-во АН СССР, 1957.
17. Асидвава Т., Сугидааки С., Асахи X., Сакаи X., Сибата К., Бунсэки кагаку, 19, 1333 (1970); РЖХим, 1971, 13Г45.
18. Бабко А. К., Данилов В. Н., Каплан М. Л. Зав. лаб., 34, 280, (1968).
19. Бабко А. К., Кузьмин Н. М., Лисецкая Г. С., Овруцкий М. И., Фре-гер С. В. Методы анализа химических реактивов и препаратов, вып. 15. М., изд. ИРЕА, 1968, стр. 79.
20. Бабко А. К., Маркова Л. В. Методы анализа химических' реактивов и препаратов, вып. 13, М., изд. ИРЕА, 1966, стр. 117.
21. Бабко А. К., Марченко П. В. Зав. лаб., 25, 1047 (1959).
22. Бабко А. К., Пилипенко А. Т. Фотометрический анализ. М., «Химия», 1968, стр. 219.
226
23. Бабко А. К., Пилипенко А. Т., Пятницкий И. В., Рябушко О. П. Физико-химические методы анализа. М., «Высшая школа», 1968, стр. 269.
24. Багдасаров К. Н., Коваленко П. Н., Владимирова В. Ф., Назаренко Л. В., Маврина А. С., Османов X. А., Шемякина М. А. Сб. «Определение микропримесей». № 1. М., 1968, стр. 29; РЖХим, 1969, ЗГ120.
25. Багдасаров К. Н., Коваленко П. И., Шемякина М. А. Ж. аналит. химии, 23, 515 (1968).
26. Багреев В. В., Золотов Ю. А., Курилина Н. А., Калинина Г. Ф. Экстракция неорганических соединений, 1945—1962 гг. М., «Наука», 1971.
27. Багреев В. В., Золотов Ю. А., Курилина Н. А., Калинина Г. Фг Экстракция неорганических соединений, 1963—1967 гг. М., «Наука», 1971.
28. Бажов А. С., Кока П. А. Сб. «Вопросы общей и прикладной физики». Алма-Ата, «Наука», 1969, стр. 244.
29. Банковский Ю. А. Докторская диссертация. Рига, Ин-т неорганич. химии АН ЛатвССР, 1968.
30. Баранова Л. Л., Солодовник С. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 336.
31. Бардин М. Б., Шапиро В. И. Уч. записки Кишиневского ун-та, 14, (хим.), 123 (1954).
32. Барковский В. Ф., Горелик С. М., Городенцева Т- Б. Физико-химические методы анализа. М., «Высшая школа», 1972, сгр. 164.
33. Белоглазова А. Д., Крупнов В. К., Сафаева Ф. 3. Зав. лаб., 34, 1057 (1968).
34. Беляев Ю. И. Ж. аналит. химии, 26, 492 (1971).
35. БеляевЮ. И., Иванцов Л. МКарякин А. В., Фа Хунг Фи, Шемет В. В. Ж. аналит. химии, 23, 508 (1968).
36. Беляев Ю. И., Карякин А. В., Пчелинцев А. М. Ж. аналит. химии, 25, 852 (1970).
37. Беляева И. А. Сб. «Аппаратура и методы рентгеновского анализа», вып. II. Л., Изд. СКВ РА, 1967, стр. 55.
38. Бетехтин А. Г. Минералогия. М., Госгеолиздат, 1950.
39. Бетехтин А. Г. Курс минералогии. М., Госгеолиздат, 1951.
40. Билимович Г. Н., Алимарин И. П. Ж. аналит. химии, 12, 685 (1957).
41. Бланк А. Б., Сизоненко И. Т., Булгакова А. М. Ж. аналит. химии, 18, 1046 (1963).
42. Блок Н. И. Качественный химический анализ. М.— Л., Госхимиздат, 1952.
43. Блохин М.А. Физика рентгеновских лучей. М., Гостехтеориздат, 1957.
44. Блохин М. А. Методы рентгеноспектральных исследований. М., Физ-матиздат, 1959.
45. Божеволънов Е. А. Люминесцентный анализ неорганических веществ. М., «Химия», 1966.
46. Божеволънов Е.А., Серебрякова Г. В. Химические реактивы и препараты, вып. 24. М., Госхимиздат, 1961, стр. 36.
47. Божеволънов Е. А., Федорова Л. Ф., Красавин И. А., Дзиомко В. М. Ж. аналит. химии, 25, 1722 (1970).
48. Боровик-Романова Т. Ф., БеляевЮ. И., Куценко Ю. И., Павленко Л. И., Савинова Е. И., Фарафонов М. М. Спектральное определение редких и рассеянных элементов. М., Изд-во АН СССР, 1962, стр. 146.
49. Борчева Т. А., Николаева Т. Ф., Харламов И. П. Сб. «Определение микропримесей», № 1. М., 1968, стр. 58; РЖХим, 1969, ЗГ117.
50. Бродский А. И. Химия изотопов. М., Изд-во АН СССР, 1957, стр. 435, 443.
51. Брудзъ В. Г., Драпкина Л. А., Смирнова К. А., Дорошина Н. И. Методы анализа химических реактивов и препаратов, вып. 9. М., изд. ИРЕА, 1964, стр. 33.
8*
227
52. Бурриелъ-Марти Ф., Рамирес-Муньос X. Фотометрия пламени. М., ИЛ, 1962, стр. 438.
53. Русев А. И., Быръко В. М. Труды Комиссии по аналит. химии АН СССР, 9(12), 200 (1958).
5>4. Бусев А. И., Иванютин М. И. Труды Комиссии по аналит. химш АН СССР, 11, 172 (1960).
55. Бусев А. И., Поляк Л. Я. Зав. лаб., 25, 668 (1959).
56. Бусев А. И., Типцова В. Г., Иванов В. М. Практическое руководство по аналитической химии редких элементов. М., «Химия», 1966, стр. 304.
57. Бусев А. И., Харламов И. П., Смирнова О. В. Зав. лаб., 35, 1301 (1969).
58. Бусев А. И., Шишков А. Н. Ж. аналит. химии, 22, 20 (1967).
59. Вада X., Накагава Г. Нихон кагаку дзасси, 89, 951, А57 (1968); РЖХим, 1969, 18Г15.
60. Вайнштейн Ю. П., Крузина Н. Г. Сб. «Методы анализа химических реактивов и препаратов», вып. 16. М., «Химия», 1969, стр. 222.
61. Ван Шу, Чжан Шу-юнъ, Ван Го-ли и др. Кэсюэ тунбао, № 3, 255 (1965); РЖХим, 1966, 7Г151.
62. Ван Шу, Чжан Шу-юнъ, Чжу Цзяо, Чан Пу. Хуасюэ Сюэбао, 31, 526 (1965); РЖХим, 1966, 15Г143.
63. Ван Шу, Чжан Шу-юнъ, Чэн Пу. Ланьчжоу дасюэ сюэбао. Цзыжань кэсюэ, № 1, 53 (1965); РЖХим, 1966, 13Г169.
64. Васильева Л. Н., Лукашенкова Н. В., Краснобаева Л. В., Юстус 3. Л. Зав. лаб., 36, 1436 (1970).
65. Васильева Л. Н., Пац Р. Г., Лутченко Н. Н., Юстус 3. Л. Анализ руд цветных металлов и продуктов их переработки, сборник № 28. М., «Химия», 1968, стр. 16.
66. Васютинский А. И., Кисель Н. А., Матвеева Е. Н. Ж. аналит. химии, 23, 1847 (1968).
67. Виноградов А. П. Геохимия редких и рассеянных химических элементов в почвах. М., Изд-во АН СССР, 1957, стр. 172.
68. Виноградова Е. И., Васильева Л. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 296.
69. Виноградова Е. Н., Галлай 3. А., Финогенова 3. М. Методы полярографического и амперометрического анализа. Изд-во МГУ, 1963, стр. 248.
70. Виноградова Е. Н., Игнатьев Ю. Н., Васильева Л. Н. Методы анализа химических реактивов и препаратов, вып. 5-6. М., изд. ИРЕА, 1963, стр. 62.
71. Виноградова Е. И., Каменев А. И., Лисенкова Н. В. Зав. лаб., 31, 1180 (1965).
72. Воскресенский П. И. Аналитические реакции между твердыми веществами и полевой химический анализ. М., Госгеолтехиздат, 1963.
72а. Вредные вещества в промышленности, ч. II. Неорганические и эле-менторганические соединения. Л., «Химия», 1971, стр. 364.
73. Габович А. М. Труды Кишиневского сельскохоз. ин-та, 43, 125 (1966).
74. Габович А. М. Там же, стр. 133.
75. Габович А. М. Там же, стр. 155.
76. Галкина Е. И., Иванова 3. А. Сборник научных сообщений Дагестанского ун-та. Кафедра химии, вып. 7, 128 (1971); РЖХим, 1971, 14Г68.
77. Галкина Е. И., Коваленко П. Н., Иванова 3. И. Изв. вузов. Химия и хим. технол., 12, 712 (1969); РЖХим, 1969, 23Г77.
78. Гейровский Я., Кута Я. Основы полярографии. М., «Мир», 1965.
79. Геохимия, минералогия и генетические типы месторождений редких элементов, т. 1. М., «Наука», 1964, стр. 403.
80. Геркен Е. Б. Сборник трудов Гинцветмет, № 19, М., Металлургиздат, 1962, стр. 800.
81. Гертнер М. Д., Янсон Э. Ю., Поммере В. Р. Уч. зап. Латвийского ун-та, 88, 59 (1967).
82. Гиллебранд В. Ф., Ленделъ Г. Э., Брайт Г. А., Гофман Д. И. Практи
228
ческое руководство по неорганическому анализу. М., Госхимиздат 1957, стр. 270.
83. Гинзбург В. Л., Лернер Л. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 459.
84. Гладышева К. Ф., Зиновьева Л. Д. Сборник научных трудов ВНИИ-цветмета, № 9, 49 (1965).
85. Гладышева К. Ф., Царева К. X. Зав. лаб., 33, 30 (1967).
86. Глотка Е. Д., Гейнрихс К. Я., Имангазиева С. К., Рогова Ю. Г. Сборник научных трудов ВНИИц ветмета, № 13, 41 (1968).
87. Горовых О. М. Сб. «Спектральный анализ в геологии». М., изд. ВИМС, 1971, стр. 89.
88. Грекова И. М., Назаренко В. А. Зав. лаб., 35, 537 (1969).
89. Громик Л. И., Лошкарев М.А. Химическая технология. Республиканский межведомственный научно-технический сборник, вып. 3, 63 (1965).
90. Грушина Н. В., Цевун В. И., Храпченкова Г. В., Ерденбаева М. И., Козин Л. Ф. Ж. аналит. химии, 21, 980 (1966).
91. Гулютин А. В. Сб. «Прикладная спектроскопия», т. 1. М., «Наука», 1969, стр. 298.
92. Гусев С. И., Несис А. С. Ж. аналит. химии, 17, 844 (1962).
93. Данилова В. Н., Лузина Л. Н. Укр. хим. ж., 32, 290 (1966).
94. Деляхей П. Новые приборы и методы в электрохимии. М., ИЛ, 1957.
95. Делимарский Ю. К., Туманова Н. X., Приходько М. У. Укр. хим. ж., 36, 1167 (1970).
96. Джуа М. История химии. М., «Мир», 1966, стр. 38, 149, 246.
97. Долежал Я., Повондра П., Шульцек 3. Методы разложения горных пород и минералов. М., «Мир», 1968.
98. Драпкина Д. А., Брудзь В. Г., Смирнова К. А., Дорошина Н. И. Ж. аналит. химии, 17, 940 (1962).
99. Дьяченко Н. П., Чуйко В. Т. Зав. лаб., 29, 522 (1963).
100. Дэгути М. Бунсэки кагаку, 17, 1124 (1968); РЖХим, 1969, 11Г75.
101. Дэзути М. Бунсэки кагаку, 18, 159 (1969); РЖХим, 1969, 20Г103.
102. Дэзути М., Танно К., Сакаи К. Бунсэки кагаку, 19, 1291 (1970); РЖХим, 1971, 8Г80.
103. Дэзути Т. Бунсэки кагаку, 15, 352 (1966); РЖХим, 1967, 4Г47.
104. Дэна Дж. Д., Дзна Э. С., Пзлач Ч., Берман Г., Фрондель К. Система минералогии, т. I (1). М., ИЛ, 1950, стр. 252, 272.
105. Егоров Е. В., Макарова С. Б. Ионный обмен в радиохимии. М., Атом-издат, 1971.
106. Елисеева Г. Д. Труды Комиссии по аналит. химии АН СССР, 6(9), 439 (1955).
107. Еременко В. Я. Спектрографическое определение микроэлементов в природных водах. М., Изд-во АН СССР, 1960.
108. Жаровский Ф. Г., Островская М. С. Укр. хим. ж., 32, 893 (1966).
109. Жданов А. К., Акентьева Н. А., Лукьяненко И. Л. Докл. АН УзССР, № 12, 28 (1970).
110. Жданов А. К., Капица Н. В. Труды Тапгкенского ун-та, вып. 323, 139 (1968).
111. Жданов А. К., Капица Н. В. Докл. АН УзССР, № 1, 29 (1969).
112. Жданов А. К., Капица Н. В., Ахмедов Г. Труды Тапгкенского ун-та, вып. 288, 31 (1967).
113. Жданов А. К., Мархабаев И., Ленченко Т. А. Ж. прикл. химии, 44, 456 (1971).
114. Жданов А. К., Ятрудакис С. М., Андреева Т. И. Научные труды Ташкентского ун-та, вып. 264, 73 (1964).
115. Жеребенка А. В. Сборник трудов Казахского ун-та. Физика. Алма-Ата, 1970, стр. 204.
116. Живописцев В. Н. Зав. лаб., 16, 1186 (1950).
117. Живописцев В. П. Уч. записки Пермского ун-та, 8, 141 (1953).
118. Живописцев В. П. Зав. лаб., 31, 1043 (1965).
229
119. Живописцев В. П, Уч. записки Пермского ун-та, 25, 89 (1963).
120. Живописцев В. П. Уч. записки Пермского ун-та, 26, 111 (1964).
121. Живописцев В. П. Труды Комиссии по аналит. химии АН СССР, 17, 309 (1969).
122. Живописцев В. Е., Айтова В. X., Селезнева Е. А. Изв. вузов. Химия и хим.технол., № 5, 739 (1963).
123. Живописцев В. П., Петров Б. И., Селезнева Е. А., Сибиряков Н. Ф. Труды Комиссии по аналит. химии АН СССР, 17, 301 (1969).
124. Живописцев В. П., Селезнева Е. А., Халапсин А. Д. Уч. записки Пермского ун-та, № 141, (химия), 222 (1966).
125. Живописцев В. П., Челнокова М. Н. Уч. записки Пермского ун-та, 19, 93 (1961).
126. Живописцев В. П., Челнокова М. Н. Ж. аналит. химии, 18, 716 (1963).
127. Живописцев В. П., Челнокова М. Н. Уч. записки Пепмского ун-та, № 111, 165 (1964).
128. Заборенко Е. Б., Иофа Б. 3., Лукьянов В. Б., Богатырев И. О. Метод радиоактивных индикаторов в химии. М., «Высшая школа», 1964.
129. Завгородная Е. Ф., Подольская Н. В. Зав. лаб., 35, 925 (1969).
130. Загорчев Б., Топалова Е., Мощева П. Годишник хим.-технол. ин-та (София), 13, 29, 1966, (1968); РЖХим, 1970, ЗГ76.
131. Зайдель А. И. Основы спектрального анализа. М., «Наука», 1965.
132. Зайцев В. Н. Изв. СО АН СССР, серия хим. наук, вып. 3, № 11, 69 (1965).
133. Захаров Л. С., Айдаров Т. К. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 201.
134. Захаров М. С., Трушина Л. Ф. Ж. аналит. химии, 22, 1219 (1967).
135. Захарчук И. Ф., Зебрева А. И., Павловский М. Т. Ж. аналит. химии, 26, 246 (1971).
136. Захарчук Н. Ф., Сынкова Д. И., Гладышев В. П. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 290.
137. Зеленина Т. П. Сборник научных трудов ВНИИцветмета, № 9, 75 (1965).
138. Зелюкова Ю. В., Полуэктов Н. С. Ж. аналит. химии, 18, 435 (1963).
139. Зелюкова Ю. В., Полуэктов Н. С., Никонова М. П. Зав. лаб., 33,436 (1967).
140. Зилъберминц С- М., Шаврин А. М. Уч. записки Пермского ун-та № 111, 110 (1964).
141. Зилъберштейн Х.И. Ж. техн, физики, 25, 1491 (1955).
142. Золотов Ю. А. Экстракция внутрикомплексных соединений. М., «Наука», (1968).
143. Золотов Ю. А., Алимарин И. П., Спиваков Б. Я. Изв. АН СССР, серия хим., № 3, 579 (1969).
144. Золотов Ю. А., КузьминН. М. Экстракционное концентрирование. М., «Химия», 1971.
145. Золотов К). А., Сизоненко Н. Т., Золотовицкая Э. С., Яковенко Е. И. Ж. аналит. химии, 24, 20 (1969).
146. Золотухин В. К., Олексишин С. М. В1сник Льв1вськ. ун-ту, cepin xiM., вып. 8, 51 (1965).
146а. Зотова Н. И. Зав. лаб. 7, 870 (1938).
147. Иванов Н. П., Козырев Н. А. Методы анализа химических реактивов и препаратов, вып. 10, М., изд. ИРЕА, 1965, стр. 151.
148. Иванов Н. П., Минервина Л. В., Баранов С. Б. Химические реактивы и препараты, вып. 27, М., изд. ИРЕА, 1965, стр. 280.
149. Иванова М. С. Труды Комиссии по аналит. химии АН СССР, 7(10), 77 (1956).
150. Иванчев Г. Дитизон и его применение. М., ИЛ, 1961.
151. Иида Т., Ясамаки К. Бунсэки кагаку, 18, 1088 (1969); РЖХим, 1970, 6Г148.
230
152. Исаков П. М. Качественный химический анализ руд и минералов методом растирания порошков. М., Госгеолтехиздат, 1955.
153. Исии X., Эйнага X., Сува Т. Бунсэки кагаку, 14, 24 (1965): РЖХим, 1966, 5Г156.
154. Ицуки К., Хомма Э., Сува Т. Бунсэки кагаку, 17, 1022 (1968); РЖХим, 1968, 24Г139.
155. Кавасаки Г., Сгнда И., Магда С., Нисино X., Араи М., Кубота К. Нихон эйсэйгаку дзасси, 25, 230 (1970); РЖХим, 1971, 12Г192.
156. Калинин С. К., Файн Э. К. Спектральный анализ минерального сырья. Алма-Ата, Изд-во АН КазССР, 1962, стр. 144.
157. Каменев А. И., Виноградова Е. Н., Лисенкова Н. В. Вести. МГУ, серия хим., № 2, 76 (1966).
158. Каменев А. И., Виноградова Е. И., Фигуровская В. Н. Вести. МГУ, серия хим., № 6, 113 (1966).
159. Каменев А. И., Грановский Ю. В., Виноградова Е. Н. Зав. лаб., 34, 1041 (1968).
160. Каменев А. И., Лукьянов В. Б. Сб. «Проблемы планирования эксперимента». М., «Наука», 1969, стр. 198.
161. Каменев А. И., Лукьянов В. Б., Фигуровская В. И., Виноградова Е. И. Ж. аналит. химии, 21, 535 (1966).
162. Каплин А. А., Катюхин В. Е., Стромберг А. Г. Зав. лаб. 36, 18 (1970),
163. Карташев Е. Р., Штань А. С. Труды ВНИИ радиац. техн., вып. 2, 145 (1968); РЖХим, 1969, 5Г147.
164. Карташев Е. Р., Штань А. С. Труды ВНИИ радиац. техн., вып. 3, 200 (1969); РЖХим, 1969, 15Г140.
165. Карякин Ю. В., Ангелов И. И. Чистые химические реактивы. М., Госхимиздат, 1955, стр. 158.
166. Касюра К., Марченко 3. Ж. аналит. химии, 22, 1389 (1967).
167. Кацков Д. А., Львов Б. В. Ж. прикл. спектроскопии, 10, 382 (1969).
168. Кацков Д. А., Львов Б. В. Там же, стр. 867.
169. Кацков Д. А., Львов Б. В. Сб. «Последние достижения в области атомноабсорбционного анализа», ч. 1. Л., изд. Ин-та научно-техн, информации, 1969, стр. 53.
170. Кипарисова Л. С. Уч. записки ЦНИИ оловянной пром-ти, К» 1, 29 (1965); РЖХим, 10Г130 (1966).
171. Кирспуу X. К., Бу сев А. И. Ж. аналит. химии, 23, 354 (1968).
172. Китано М., Узда Л. Нихон кагаку дзасси, 91, 760, А42 (1970); РЖХим, 1971, 9Г83.
173. Киш И. И., Шестидесятная Н. Л., Желтвай И. И. Изв. вузов, Химия и хим. технол. 12, 1451 (1969); РЖХим, 1970, 12Г92.
174. Коваленко И. Н., Мусаелянц Л. Н. Укр. хим. ж., 30, 753 (1964).
175. Коев К. Тр. Висш. ин-т народно стопанство (Варна), 38, 143 (1966); РЖХим, 1967, 17Г90.
176. Козловский М. Т. Ртуть и амальгамы в электрохимических методах анализа. Алма-Ата, Изд-во АН КазССР, 1956, стр. 19, 42.
177. Козловский М. Г., Зебрева А. И., Гладышев В. П. Амальгамы и их применение. Алма-Ата, «Наука», 1971.
178. Козловский М. Т., Меерсон Л. А., Шиханов И. М. Зав. лаб., 12, 549 (1946).
179. Кольтгоф И. М., Белчер Р., Стенгер В. А. Матсуяма Дж. Объемный анализ, т. III. М., Госхимиздат, 1961.
180. Кольтгоф И. М., Лингейн Дж. Дж. Полярография. М.—Л., Госхимиздат, 1948.
181. Кольтгоф И. М., Стенгер В. А. Объемный анализ, т. II, М.—Л., Госхимиздат, 1952.
182. Кольтгоф II., Фурман Н. Потенциометрическое титрование Л., ОНТИ — Химтеорет, 1935, стр. 155.
183. Коминами Б. Бунсэки кагаку, 18, 253 (1969); РЖХим, 1969, 14Г123.
184. Коминами Б., Оно Ф. Там же, стр. 578; РЖХим, 1969, 21Г168.
231
185. Комлев О. Й., СюцарН. А. Вкник Льв1вськ. ун-ту, cepin xiM. 79 (1964), РЖХим, 1965, 17Г72.
186. Комплексометрия. М., Госхимиздат, 1958.
187. Коно Т., Кобаяси С. Бунсэки кагаку, 19, 1491 (1970); РЖХим, 1971, 12Г103.
188. Концевич Л. С., Владз1м1рсъка О. В. Фармацевт, ж., 23, 38 (1968).
189. КоренманИ. М. Количественный микрохимический анализ. Л., ОНТИ— Химтеорет, 1936, стр. 193.
190. Коренман И. М. Количественный микрохимический анализ. М.—Л., Госхимиздат, 1949, стр. 97.
191. Коренман И. М. Микрокристаллоскопия. М.—Л., Госхимиздат, 1947»
192. Коренман И. М., Вербицкая Т. Д. Труды по химии и хим. технол., вып. 2(10), 248 (1964).
193. Коренман И. М., Шеянова Ф. Р. Труды Комиссии по аналит. химии АН СССР, 9(12), 205 (1958).
194. Коренман И. М., Шеянова Ф. Р., Горбунова Л. В., Романова Н. К. Труды по химии и хим. технол., вып. 1, 109 (1958).
195. Коренман И. М., Шеянова Ф. Р., Масленников С. Н. Труды по химии и хим. технол., вып. 2(13), 174 (1965).
196. Коростелев П. П. Приготовление растворов для химико-аналитических работ. М., «Наука», 1964.
197. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия, ч. 2. М., «Мир», 1969, стр. 465, 468.
198. Красюкова Н. Г., Хализова В. А. Сб. «Минералого-петрографические очерки Забайкалья». Улан-Удэ, 1968, стр. 159; РЖХим, 1969, 4Г66.
199. Краткая химическая энциклопедия, т. 2. М., «Советская энциклопедия», 1963, стр. 342.
200. Крешков А. П., Ворк В. А., Федухина Г. П. Зав. лаб., 34, 265 (1968).
201. Крешков А. П., Яровенко А. Н., Милаев С. М. Изв. вузов. Химия и хим. технол., 10, 620 (1967); РЖХим, 1968, ЗГ64.
202. Крылова А. Н. Судебно-медицинская экспертиза, 11, 40 (1968); РЖХим, 1968, 18Г171.
203. Крылова А. Н. Сб. «Вопросы судебной медицины». М., 1968, стр. 322.
204. Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ. М., Госхимиздат, 1959, стр. 242.
205. Кубота К., Араи М., Нисино X., Мазда С., Сэнда И., Кавасаки Г. Нихон эйсэйгаку дзасси, 25, 223 (1970); РЖХим, 1971, 12Г196.
206. Кузнецов В. И. ДАН СССР, 41, 113, (1943).
207. Кузнецов В. И. Труды Комиссии по аналит. химии АН СССР, 6(9), 249 (1955).
208. Кузьмин Н. М., Соломатин В. С., Галактионова А. Н., Кузовлев И. А, Ж. аналит. химии, 24, 725 (1969).
209. Кульберг Л. М. Органические реактивы в аналитической химии. М.—Л., Госхимиздат, 1950.
210. Кульберг Л. М., Алътерзон Г. С., Велътман Р. П. Капельный анализ. М.—Л., Госхимиздат, 1951.
211. Кумов В. И. Ж. аналит. химии, 7, 301 (1952).
212. Кумов В. И. Ж. аналит. химии, 9, 229 (1954).
213. Курбатов Д. И., Павлова С. А. Труды Ин-та химии Уральского фил. АН СССР, вып. 10, 87 (1966); РЖХим, 1966, 21Г101.
214. Кутейников А. Ф., Егорова В. А., Конькова Е. С., Заваруева И. Д., Беляев Ю. И., Карякин А. В., Пчелинцев А. М. Ж. аналит. химии, 25, 1999 (1970)
215. Кэй Д., Лэби Т. Справочник физика-экспериментатора. М., ИЛ, 1949, стр. 197.
216. Кюрегян Э. А., Эксузян Ц. О. Изв. АН Арм.ССР. Наука о земле, 1968, 6; РЖХим, 1969, 7Г165.
217. Лаврова Е. А., Кузнецова А. В. Зав. лаб., 31, 50 (1965).
218. Лаврухина А. К., Малышева Т. В., Павлоцкая Ф. И. Радиохимический апализ. М., Изд-во АН СССР, 1963, стр. 199.
232
219. Лазарев А. И. Зав. лаб. 25, 783 (1959).
219а. Лазарев Н. В. Химически вредные вещества в промышленности, ч. II, Л.—М., ГНТИХЛ, 1951, стр. 324.
220. Лайтинен Г. А. Химический анализ. М., «Химия», 1966, стр. 271.
221. Лакомкин И. Г., Алексеевский Н. В. Изв. вузов. Химия и хим. технол., 10, 1286 (1967).
222. Латимер В. Окислительные состояния элементов и их потенциалы в водных растворах. М., ИЛ, 1954,
223. Лебедева М. И., Исаева Б. И. Зав. лаб. 37, 410 (1971).
224. Лебедь Н. Б., Панталер Р. П. Ж. аналит. химии. 20, 59 (1965).
225. Леман Е. П., Болотова И. Г. Сб. «Вопросы разведочной геофизики», вып. 11. 1969, стр. 3.
226. Либхавски X. А., Пфейфер Г. Г., Уинслоу Э. Г., Земани П. Д. Применение поглощения и испускания рентгеновских лучей. М., «Металлургия», 1964.
227. Лисицына Е. В., Лысенко В. И. Сборник научных трудов ВНИИцвет-мета, № 13, 37 (1968).
228. Ломакин Г. Г., Толмачев В. Н. Изв. вузов. Химия и хим. технол., 3, 819 (1960).
229. Лонцих С. В., Педлер В. В., Райхбаум Я. Д. Спектальный анализ металлометрических проб. М., Госгеолтехиздат, 1959, стр. 69.
230. Лосев Н. Ф. Изв. АН СССР, серия физич., 24, 476 (I960).
231. Лосев Н. Ф. Количественный рентгеноспектральный анализ, М., «Наука», 1969.
232. Лукин А. М., Баранович Г. Г., Петрова Г. С. Труды ИРЕА, вып. 23, 55 (1959).
233. Лурье Ю. Ю. Справочник по аналитической химии. М., «Химия», 1971.
233а. Лурье Ю. Ю. Зав. лаб., 6, 1040 (1937).
2336. Лурье Ю. Ю., Неклютина В. Ф. Зав. лаб., 5, 587 (1936).
234. Лурье Ю. Ю., Рыбникова А. И. Химический анализ производственных сточных вод. М., Госхимиздат, 1963.
234а. Лурье Ю. Ю., Типцова И. И. Зав. лаб., 6, 507 (1937).
2346. Лурье Ю. Ю., Троицкая М. И. Зав. лаб., 5, 1425 (1936).
234в. Лурье Ю. Ю., Троицкая М. И. Зав. лаб., 7, 675 (1938).
235. Лысенко В. И., Ким А. Г. Труды Комиссии по аналит. химии АН СССР, 15, 200 (1965).
236. Лысенко В. И., Лисицына Е. В., 4-е Всесоюзное совещание по полярографии. Тезисы докладов. Алма-Ата, 1969, стр. 38.
237. Львов Б. В. Атомно-абсорбционный спектральный анализ, М., «Наука», 1966.
238. Львов Б. В. Ж. аналит. химии, 26, 590 (1971).
239. Львов Б. В., Кацков Д. А., Плющ Г. В. Сб. «Последние достижения в области атомно-абсорбционного анализа», ч. I. Л., изд. Ин-та научнотехн. информации, 1969, стр. 35.
240. Львов Б. В., Харцызов А. Д. Ж. аналит. химии, 24, 936 (1969).
241. Львов Б. В., Харцызов А. Д. Сб. «Последние достижения в области атомно-абсорбционного анализа», ч. II. Л., изд. Ин-та научно-техн, информации, 1969, стр. 35.
242. Люминесцентный анализ. М., Физматиздат, 1961, стр. 169.
243. Ляликов Ю. С. Физико-химические методы анализа. М., Госхимиздат, 1960, стр. 270, 290.
244. Ляликов Ю. С., Васильева Л. Н., Чернега Л. П. Ж. аналит. химии, 26, 278 (1971).
245. Ляликов Ю. С., Новик Р. М., Вассерштейн III. Е. Изв. АН МолдССР, № 9, 222 (1963); РХим, 1965, 1Г66.
246. Ляликов Ю. С., Чернега Л. П., БодюВ. И. Труды Кишиневского политехи. ин-та, вып. 13, 167 (1968); РЖХим, 1970, ЗГ90.
247. Максимов Д. Е., Рудневский И. К. Труды по химии и хим. технол., вып. 2 (20), 56 (1968).
233
248. Мальков Е. М., Косарева В. Г., Федосеева А. Г. Зав. лаб., 31, 1327 (1965).
249. Мальков Е. М., Косарева Е. Г., Федосеева А. Г. Сб. «Новые методы анализа химического состава подземных вод». М., 1967, стр. 43; РЖХим, 1967, 21Г161.
250. Манова Т. Г., Рогинская Л. К., Певцов Г. А., Павлова Л. А. Химические реактивы и препараты, вып. 32. М., изд. ИРЕА, 1970, стр. 137.
251. Маркова И. В., Синякова С. И. Агрохимия, № 12, 118, (1966); РЖХим, 1967, 14Г141.
252. Марченко 3. Фотометрическое определение элементов. М., «Мир», 1971.
253. Марченко П. В., Воронина А. И. Укр. хим. ж., 35, 652 (1969).
254 Марченко 3., Мо-'ский М., Касюра К. Ж. аналит. химии, 22,1805 (1967).
255. Марьянов В. М. Радиометрическое титрование. М., Атомиздат, 1971.
256. Матвеец М. А., Щербов Д. П. Конференция по химии экстракции. Тезисы докладов. М., изд-во АН СССР, 1969, стр. 164.
257. Матвеец М. А., Щербов Д. П. Ж. аналит. химии, 26, 823 (1971).
258. Мейке В. А. Руководство для препаратов химико-аналитических лабораторий. М., Госгеолтехиздат, 1956, стр. 112.
258а. Мельников В. В. Зав. лаб., 8, 1172 (1939).
259. Металлургия цветных металлов и методы их анализа. Сборник трудов, № 7. М., Металлургиздат, 1962, стр. 343, 345, 380.
260. Методическое руководство по определению микрокомпонентов в природных водах при поисках рудных месторождений. М., Госгеолтехиздат, 1961, стр. 208.
261. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965.
262. Методы анализа руд и продуктов цветной металлургии. Сборник трудов, № 5. М., Металлургиздат, 1959, стр. 107, 192.
263. Методы анализа химических реактивов и препаратов, вып. 9. М., изд. ИРЕА, 1964, стр. 35.
264. Методы анализа химических реактивов и препаратов, вып. 14. М., изд. ИРЕА, 1967, стр. 86.
265. Методы анализа химических реактивов и препаратов, вып. 15. М., изд. ИРЕА, 1968, стр. 92.
266. Методы анализа химических реактивов и препаратов, вып. 16. М., «Химия», 1969, стр. 222.
267. Методы анализа химических реактивов и препаратов, вып. 17. М., изд. ИРЕА, 1970, стр. 25, 41, 57, 227.
268. Методы анализа химических реактивов и препаратов, вып. 18. М., изд. ИРЕА, 1971, стр. 9, 107, 145, 153.
269. Методы анализа химических реактивов и препаратов, вып. 19. М., изд. ИРЕА, 1971, стр. 46.
270. Сб., «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 179.
271. Методы химического анализа минерального сырья, вып. 6, М., Гос-’ геолтехиздат, 1960, стр. 12.
272. Методы химического анализа минерального сырья, вып. 10. М., «Недра», 1966, стр. 26, 29.
273. Мечос X. 3., Гутникова Р. И,, Жданов А. К., Букина В. К., Теодорович И. Л. Узо. хим. ж. АН УзбССР Деп. редколлегией. Ташкент, 1969; РЖХим, 1969, 18Г59.
274. Милин В. П. Электрографический метод анализа. Изд-во Саратовского ун-та, 1966, стр. 99, 108.
275. Мищенко Л. В., Чайкина М. В., Лазлова Г. С. Изв. СО АН СССР, серия хим. наук, вып. 2, № 4, 94 (1968).
276. Младеновид. С. Н. Гласник Хем. друштва (Београд), 30, 13 (1965). РЖХим, 11Г82 (1967).
277. МладеновиП, С. Н., ВесковиП, Н., ФилиповиК Б. Гласник Хем. друштва (Београд), 33, 333 (1968); РЖХим, 1970, 13Г119.
234
278. Молева В. С., Пейзулаев ТЦ. И. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 331.
279. Мори И. Якугаку дзасси, 85, 679 (1965); РЖХим, 1966, 4Г26.
280. Морицугу М. Бунсэки кагаку, 13, 64 (1964); РЖХим, 1965, 8Г72.
281. Моррисон Дж., Фрейзер Г. Экстракция в аналитической химии. Л., Госхимиздат, 1960.
282. Мотодзима Й., Банто С., Накаяма Р. Буисэки кагаку, 6, 567 (1968), РЖХим; 1969. 17Г161.
283. Мустафин И. С., Молот Л. А. Органические реактивы, ч. 1. Изд-во Саратовского ун-та, 1967.
284. Мухина 3. С., Котова Г. С., Кузьмичева Р. А. Ж. аналит. химии, 20, 785 (1965).
285. Мухина 3. С-, Котова Г. С., Кузьмичева Р. А., Кондукова Н. С. Сб. «Физико-химические методы анализа металлов и сплавов», Б. М., 1969, стр. 106; РЖХим, 1970, 15Г195.
286. Нагаи X., Дзгути Т. Нихон кагаку дзасси, 86, 516 (1965); РЖХим, 1966, 14Г44.
287. Назаренко В. А., Полуэктов Н. С. Полумикрохимический анализ минералов и-руд. М.—Л., Госхимиздат, 1950, стр. 59.
288. Назаренко В. А., Флянтикова Г. В. Зав. лаб., 24, 801 (1958).
289. Назаренко В. А., Флянтикова Г. В., Фуга Н. А., Эстерлис К. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 210.
290. Назаренко В. А., Флянтикова Г. В., Шустова М. Б., Фуга Н. А., Эстерлис К. А. Там же, стр. 179.
291. Наумова А. С. Изв. Томского политехи, ин-та, 128, 71 (1964); РЖХим, 1965, 20Г75.
292. Нейман Е. Я., Долгополова Г. М. Зав. лаб., 35, 1040 (1969).
293. Некрасов Б. В. Основы общей химии, т. 2. М., «Химия», 1967, стр. 335, 342, 345.
294. Нефедов В. Д., Торопова М. А., Кривохатская И. В., Синотова Е. Н. Радиоактивные изотопы в химических исследованиях. Л., «Химия», 1965.
295. Нечипоренко А. И., Калинкин И. П., Алесковский В. Б. Ж. аналит. химии, 26, 570 (1971).
296. Нечипоренко А. П., Калинкин И. П., Веитов Н. Г., Алесковский В. Б. Изв. вузов. Химия и хим. технол., 12, 1645 (1969).
297. Нечипоренко А. П., Калинкин И. П., Веитов Н. Г., Алесковский В. Б. Зав. лаб., 35, 432 (1969).
298. Никитина Е. И. Зав. лаб., 7, 409 (1938).
299. Николаев А. В., Торгов В. Г., Гильберт Э. Н., Михайлов В. А., Пронин В. А., Стадникова Л. Г., Котляревский И. Л. Изв. СО АН СССР, серия хим. наук, № 14, вып. 6, 120, (1967).
300. Николаев А. В., Юделевич И. Г., Буянова Л. М., Валл Г. А., Чучали-на Л. С., Пещевицкий Б. И., Храпай В. П. Изв. СО АН СССР, серия хим. наук, №14, вып. 6, 78 (1970).
301. Нисимура К., Имаи Т. Бунсэки кагаку, 13, 423 (1964); РЖХим, 1965, 7Г147.
302. Новак В. П., Мальцев В. Ф., Мартынов А. П. Сб. «Ионообменные материалы в науке и технике». М., 1969, стр. 131; РЖХим, 1969, 23Г129.
303. Новиков А. И., Егорова Л. А., Веселаго Л. С., Докл. АН ТаджССР, 11, 33 (1968); РЖХим, 1969, 12Г62.
304. Новиков А. И., Рузанкин В. И., Хамидов Б. О. Докл. АН ТаджССР, 12, 22 (1969).
305. Новиков А. И., Пирогова Т. А. Докл АН ТаджССР, 9, 28 (1966); РЖХим, 1967, 16Г55.
306. Нодзаки Т., Косиба К., Накаи С., Цумура Т., Йосимура К. Бунсэки кагаку, 18, 1394 (1969); РЖХим, 1970, 12Г71.
307. Нойес А., Брэй В. Качественный анализ редких элементов. М., ОНТИ ГИХЛ, 1936.
235
308. Ou И. Бунсэки кагаку, 9, 770 (1960); РЖХим, 1961, 14Д57.
309. Окасита X. Бунсэки кагаку, 13, 61 (1964); РЖХим, 1965, 8Г124.
310. Оленович И. Л., Уфимцев С. Н., Рогачко М. М. Ж. аналит. химии, 20, 1368 (1965).
311. Олъшанова К. М., Потапова М. А., Морозова Н. М. Практикум по хроматографическому анализу. М., «Высшая школа», 1970.
312. Омарова К. Д. Сб. «Химия и химическая технология», т. 2, Алма-Ата, 1964, стр. 108; РЖХим, 1965, 13Г72.
313. Орочко А. И., Тягай В. Я. Изв. вузов. Химия и хим. технол. 10, 357 (1967).
314. Панасюк В. И. Химический контроль производства стекла. М., Гиз-легпром, 1952, стр. 252.
315. Панталер Р. П., Лебедь Н. Б., Семенова Л. Н., Колесова Л. А. Труды по химии и хим. технол., вып. 3 (24), 140 (1969).
316. Парташникова М. 3., Шафран И. Г. Труды ИРЕА, вып. 25, 258 (1963).
317. Парташникова М. 3., Шафран И. Г. Ж. аналит. химии, 20, 313 (1965).
318. Пац Р. Г., Семочкина Т. В. Сборник научных трудов Гинцветмет, № 19, М., Металлургиздат, 1962, стр. 808.
319. Певцов Г. А., Макова Т. Г., Широкова М. Д. Труды ИРЕА, вып. 27, 67 (1965).
320. Пейнберг М. Я., Янсон Э. Ю., Каган П. Ф. Уч. записки Латвийского ун-та, 117, 180 (1970).
321. Перегуд Е. А., Баховская М. С., Гернет Е. В. Быстрые методы определения вредных веществ в воздухе. М., Госхимиздат, 1962, стр. 262.
322. Перегуд Е. А., Гернет Е. В. Химический анализ воздуха промышленных предприятий. М.—Л., «Химия», 1965, стр. 302.
323. Перрин Д. Органические аналитические реагенты. М., «Мир», 1967.
324. Петрашенъ В. И. Качественный химический анализ. М.—Л., Госхимиздат, 1948, стр. 337.
325. Петрова Г. С., Лукин А. М. Ассортимент реактивов на кадмий. М., изд. НИИТЭХим, 1968.
326. Пивоваров А. В. Зав. лаб., 31, 1081 (1960).
327. Пивоваров А. В. Кандидатская диссертация. Алма-Ата, 1965.
328. Пивоваров А. В., Рубанов И. А. Зав. лаб., 33, 164 (1967).
329. Пилипенко А. Т., Арендарюк Е. Н. Укр. хим. ж. 37, 50 (1971).
330. Пилипенко А. Т., Лисецкая Г. Укр. хим. ж., 19, 87 (1953).
331. Пискарев А. Ю., Хедреярв X. X. Труды Таллинского политехи, ин-та,. А, № 263, 81 (1968); РЖХим, 1969, 7Г155.
332. Плотникова О. М., Лысенко В. И. Сборник научных трудов ВНИИ-цветмета, № 9, 69 (1965).
333. Плотникова О. М., Лысенко В. И., Машуков А. Я. Там же, стр. 127.
334. Повхан М. Ф. Сб. «Роль удобрений и других факторов в повышении продуктивности растений». Киев, «Урожай», 1964, стр. 225; РЖХим, 1965, 11Г46.
335. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 290.
336. Полуэктов Н. С. Методы анализа по фотометрии пламени. М., «Химия», 1967, стр. 254.
337. Полуэктов Н. С., Виткун Р. А. Ж. аналит. химии, 17, 935 (1962).
338. Поппель А. А. Ускоренные методы анализа электролитов гальванических ванн. Изд-во Казанского ун-та, 1962, стр. 46.
339. Попов М. А. Зав. лаб., 13, 618 (1947).
340. Попов М. А. Ж. аналит. химии, 3, 167 (1948).
341. Попов М. А. Полевые методы химического анализа. М., Госгеолиздат, 1953, стр. 56.
341а. Популярная библиотека химических элементов. Марганец—олово. М., «Наука», 1972, стр. 287.
342. Порхунова Н. А. Обогащение и металлургия цветных металлов. Сборник трудов, № 1. М., Металлургиздат, 1956, стр. 197.
343. Прудников Е. Д. Сб. «Последние достижения в области атомно-адсорб
236
ционного анализа», ч. 1. Л., изд. ин-та научно-техн, информации, 1969, стр. 51.
344. ПршибилР. Комплексоны в химическом анализе. М., ИЛ, 1960.
345. Пунгор Э. Венг. исслед. оборудование, 1967, ноябрь, стр. 13; РЖХим, 1968, 8Г32.
346. Пушняк А. Н., Мигаль П. К. Уч. записки Кишиневского ун-та, 85, 24, (1965).
347. Разумова В. П. Труды ЛПИ, аналит. химия, № 901. Л., Госхимиздат, 1959.
348. РазумоваВ. П. Изв. вузов. Химия и хим. технол., 8, 192 (1965); РЖХим, 1966, ЗГ64.
349. Ратнер А. Л., Рик Г. Р., Шебашев А. А. Сб. «Применение меченых атомов в аналитической химии». М., Изд-во АН СССР, 1955, стр. 70.
350. Реакции и реактивы для качественного анализа неорганических соединений. М.—Л., Госхимиздат, 1950, стр. 34.
351. Резников А. А., Муликовская Е. И. Методы анализа природных вод. М., Госгеолтехиздат, 1954, стр. 117.
352. Резников А. А., Муликовская Е. П., Соколов И. Ю. Методы анализа природных вод. М., «Недра», 1970, стр. 387.
353. Рейзенблат Е. М., Брайнина X. 3. Зав. лаб., 32, 1450 (1966).
354. Реми Г. Курс неорганической химии, т. II. М., «Мир», 1966, стр. 476.
355. Ропот Ё. М., Чикрызова Е. Г. Ж. аналит химии, 20, 1196 (1965).
356. Руднев Н. А., Павленко Л. И., Малафеева Г. И., Симонова Л. В. Ж. аналит. химии, 24, 1223 (1969).
357. Русанов А. К. Изв. АН СССР, серия физич., 4, 195 (1940).
358. Русанов А. К. Спектральный анализ руд и минералов. М., Госгеолиздат, 1948.
359. Русанов А. К., Алексеева В. М. Зав. лаб., 11, 181 (1945).
360. Русанов А. К. Хитрое В. Г. Зав. лаб., 23, 175 (1956).
361. Рыбина Т.Ф. «Определение микропримесей, № 1». М., 1968, стр. 65; РЖХим, 1969, ЗГ115.
362. Самуэльсон О. Ионообменные разделения в аналитической химии. М.—Л., «Химия», 1966.
363. Сендел Е. Колориметрические методы определения следов металлов. М., «Мир», 1964.
364. Серебрякова Г. В., Красавин И. А., Божеволънов Е. А., Дзиомко В. М. Труды ИРЕА, вып. 26, 97 (1964).
365. Синагава М., Янаги Т., Гото М. Бунсэки кагаку, 15, 149 (1966); РЖХим, 1966, 18Г26.
366. Синякова С. И., Дударева А. Г., Маркова И. В., Талалаева П. Н. Методы анализа химических реактивов и препаратов, вып. 5-6. М., изд. ИРЕА, 1963, стр. 58.
367. Синякова С. И., Захаров М. С., Дударева А. Г., Маркова И. В., Месяц И. А., Каплин А. А., Талалаева П. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 189.
368. Синякова С. И., Маркова И. В., Пахомова Т. Я. Там же, стр. 253.
369. Скобец Е. М., Карнаухов А. И. Укр. хим. ж., 31, 408 (1965).
370. Скривеле А., Синякова С. Изв. АН Латв ССР, № 2, 69 (1966).
371. Славинский М. П. Физико-химические свойства элементов. М., Металлургиздат, 1952, стр. 182.
372. Смирнова К. А., Лукин А. М., Петрова Г. С., ВысоковаН. Н. Труды ИРЕА, вып. 31, 7 (1969).
372а. Смирнова Н. Б., Холевинская Л. В. Труды ВНИИ стапд. образцов, 7, 28, (1971).
373. Современные методы анализа. М., «Наука», 1965, стр. 7.
374. Современные методы анализа материалов. М., «Металлургия», 1969, стр. 25.
375. Соловьев П. П. Справочник по минералогии. Л.—М., Металлургиздат, 1948, стр. 96, 242, 354, 358.
237
376. Соловьев Е. А., Божеволънов Е. А., Лебедева Н. А., Миронов А. Ф., Евстигнеева Р. П. Ж. аналит. химии, 24, 231 (1969).
377. Соловьев Е. А., Божеволънов Е. А., Лебедева И. А. Органические реагенты в аналитической химии. 3-я Всесоюзная конференция. Тезисы докладов. М., «Наука», 1971, стр. 298.
378. Сангина О. А. Амперометрическое титрование в анализе минерального сырья. М., Госгеолтехиздат, 1957.
379. Сангина О. А. Амперометрическое титрование. М., «Химия», 1967.
380. Сочеванов В. Г. Зав. лаб., 29, 531 (1963).
381. Сочеванова М. М. Ускоренный анализ осадочных горных пород с применением комплексометрии. М., «Наука», 1969.
382. Спицын В. И., Кодочигов П. Н., Голутвина М. М., Кузина А. Ф., Соколова 3. А. Методы работы с применением радиоактивных индикаторов. М., Изд-во АН СССР, 1955.
383. Справочник химика, т. I. Л.—М., Госхимиздат, 1962.
384. Справочник химика, т. II. Л.—М., «Химия», 1964.
385. Справочник химика, т. III. М,—Л., «Химия», 1964.
386. Справочник химика, т. IV. М.—Л., «Химия», 1965.
387. Справочные таблицы по рентгеновской спектроскопии. М., Изд-во АН СССР, 1953.
388. Станцо В. В. Химия и жизнь, 6, 17 (1970).
389. Стары И. Экстракция хелатов. М., «Мир», 1966.
390. Степин В. В., Поносов В. Ц., Новиков Е. В. Труды ВН11И стандарта, образцов и спектр, эталонов, 2, 14 (1965); РЖХим, 1966, 20Г134.
391. Степин В. В., Силаева Е. В., Курбатова В. И., Ханова Т. Ф., Барбаш, Т. Л., Поносов В. И. Анализ цветных металлов и сплавов. М., «Металлургия», 1965.
392. Столяров К. П. Методы микрохимического анализа. Изд-во ЛГУ, 1960, стр. 93.
393. Столяров К. П., Григорьев Н. Н. Введение в люминесцентный анализ неорганических веществ. Л., «Химия», 1967.
394. Студенская Л. С., Ворожбицкая К. Ф., Камаева Л. В., Городенце-ва Т. Б. Труды ВНИИ стандарт, образцов и спектр, эталонов, 4, 167
395. Сугисита Р., Кимата К. Бунсэки кагаку, 15, 974 (1966); РЖХим, 1967, 11Г62.
396. Судзуки Т., Сотобаяси Т. Бунсэки кагаку, 14, 420 (1965); РЖХим, 1966, 11Г24.
397. Суранова 3. П., Ермоленко И. Н., Блох Т. Ф. Зав. лаб., 35, 287 (1969).
398. Суранова 3. П., Морозов А. А., Грабчук О. А. Труды НИИ хроматографии Воронежского ун-та, вып. 2, 128 (1968); РЖХим, 1969, 18Г64.
399. Сырокомский В. С., Клименко Ю. В. Ванадатометрия. Свердловск — М., Металлургиздат, 1950, стр. 62.
400. СючА. Chem. Analit., 10, 1093 (1965); РЖХим, 1966, 11Г134.
401. Такахаси Т. Бунсэки кагаку, 18, 1253 (1969); РЖХим, 1970, 10Г85.
402. Такихтани С., Хата И., Судзуки М. Там же, стр. 626; РЖХим, 1970, ЗГ69.
403. Танака X., Сугиура Ю., Сэки К., Сакаеути К., Йокояма А. Бунсэки кагаку, 17, 1309 (1968), РЖХим, 1969, 7Г70.
404. Тананаев Н. А. Дробный анализ. М.—Л., Госхимиздат, 1950, стр. 101.
405. Тананаев Н. А. Капельный анализ. М.—Л., Госхимиздат, 1954, стр. 72.
406. Тао Хунь Чень, Жэнь Ши Сюанъ, Вань Най Цзинь. Acta chimica sinica, 29, 344 (1963) (цит. по 325).
407. Тарасевич Н. И., Козарева Г. В., Кумина Д. Н. Ж. аналит. химии, 23, 1048 (1968).
408. Таскаева Т. П., Вайнштейн Э. Е. Ж. аналит. химии, 22, 852 (1967):
409. Теодорович И. Л. Применение аналитически определенных осадков в анализе. Ташкент, «ФАН», 1967, стр. 71.
410. Теодорович И. Л., Мечос X. 3., Гутникова Р. М. Ж. аналит. химии, 25, 526 (1970).
238
411. Типцова В. Г., Конкина О. И. Изв. вузов. Химия и хим. технол., 11, 364 (1968).
412. Типцова В. Г., Малкина Э. И., Анисимова З^А. Ж. аналит. химии, 21, 459 (1966).
413. Типцова-Яковлева В. Г., Дворцан А. Г., Семенова И. Б. Ж. аналит. химии, 25, 686 (1970).
414. Типцова-Яковлева В. Г., Фигелъсон Ю. А. Ж. аналит. химии, 23, 1415 (1968).
415. Трацевицкая Б. Я. Зав. лаб., 17, 939 (1951).
416. Тредвел Ф. П., ГоллВ. Т. Качественный анализ. М.— Л., Госхимиздат, 1946.
417. Троицкая М. И.,^Пац Р. Г., Позднякова А. А. Обогащение, металлургия цветных металлов и методы анализа. Сборник научных трудов, № 10. М., Металлургиздат, 1955, стр. 345.
418. Трушина Л. Ф., Захаров М. С. Изв. Томского политехи, ин-та, 167, 95 (1967); РЖХим, 1967, 22Г132.
419. Туманов А. А., Носова Л. С., Малкина Л. А., Осипова Н. И. Труды по химии и хим. технол., вып. 1 (19), 156 (1968).
420. Туманов А. А., Осипова Н. И. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 238.
421. Туманов А. А., Шахверди И. М. Зав. лаб., 36, 918 (1970).
422. Туманов А. А., Шахверди Я. М., Глазунова 3. И. Зав. лаб., 33, 8 (1967).
423. Туркевич Б. М. Фарм. ж., № 5, 12 (1959); № 6, 9 (1959).
424. Унифицированные методы анализа воды. М., «Химия», 1971, стр. 286.
425. Устимов А. М., Утепкалиева Е. Л., Булакова 3. Л. Сборник научных трудов ВНИИцветмета, № 9, 66 (1965).
426. Устимов А. М., Чалкова Н. Я. Зав. лаб., 35, 177 (1969).
427. Утиуми А. Нихон кагаку дзасси, 90, 1133 (1969); РЖХим, 1970, 13Г34.
428. Файгль Ф. Капельный анализ. М., ОНТИ, 1937, стр. 215.
429. Файнберг С. Ю., Филиппова Я. А. Анализ руд цветных металлов. М., Металлургиздат, 1963.
430. Федяшина А. Ф., Юделевич И. Г. Изв. СО АН СССР, серия хим. паук, вып. 1, 83 (1966).
431. Фигуровский Я. А. Очерк общей истории Химии. М., «Наука», 1969, стр. 90, 398.
432. Фигуровский Н. А. Открытие химических элементов п происхождение их названий. М., «Наука», 1970, стр. 75, 116.
433. Физические методы анализа следов элементов. М., «Мир», 1967, стр. 241, 267, 271.
434. Филиппова Я. А. Фазовый анализ руд цветных металлов и продуктов их переработки. М., Гостехиздат, 1963, стр. 173.
434а. Фишер Ф. К., Самаркина К. М. Зав. лаб., 8, 989 (1939).
435. Флэгг Дж. Ф., Лайн В. Р. Полумикрометод качественного анализа. М., ИЛ, 1947.
435а. Фогельсон Е. И., Калмыкова И. В., Зав. лаб., 6, 884 (1937).
436. Фраткин 3. Г., Коварский В. А. Тезисы докладов 9-го совещания по рентгеновской спектроскопии, Ивано-Франковск. М., «Наука», 1971, стр. 133.
437. Фраткин 3. Г., Поливанова И. Г. Методы анализа химических реактивов и препаратов, вып. 12, М., изд. ИРЕА, 1966, стр. 21.
438. Фридман Я. Д., Сарбаев Д. С., Левина М. Г. Материалы научной конференции, посвященной 100-летию Периодического закона Д. И. Менделеева. Фрунзе, изд. Республ. правления ВХО им. Д. И. Менделеева. 1970, стр. 48; РЖХим, 1971, ЗГ19.
439. Фрумина Я. С., Горюнова Я. Я., Мустафин И. С. Применение органических реактивов в анализе. Изд-во Саратовского ун-та, 1967 (1968), стр. 22; РЖХим, 1968, 23Г11.
440. Ханова Т. В., Кирьянова Л. А. Труды ВНИИ стандарта, образцов и спектр, эталонов, 2, 56 (1965).
239
441. Холевинская А. В., Липунова Г. Н., Беднягина Н. П. Ж. аналит. химии, 24, 1756 (1969).
442. Хедреярв X. X. Труды Таллинского политехи, ин-та А, К» 228, 141 (1965).
443. Химические реактивы и препараты. М.—Л., ГНТИХЛ, 1953, стр. 61.
444. Химические реактивы и препараты, вып. 3. М., изд. ИРЕА, 1961, стр. 31.
445. ХранисавпевиК-J аковлвий М., Пе)ковиИ-Тадий И., ГаковлевиН К. М., М и.ъковий-Сто]ановиК J. Гласник Хем. друштва (Београд), 29, 115 (1964); РЖХим, 1966, 20Г50.
446. Ху Чжи-дэ. Кэсюэ тунбао, 17, 23, (1966); РЖХим, 1966, 17Г59.
447. Целинский Ю. К., Ланицкая Е. В. Методы анализа химических реактивов и препаратов, вып. 15. М., изд. ИРЕА, 1968, стр. 92.
448. Цефт Л. Л., Кабанова Л. М. Фазовый (рациональный) анализ руд цветных металлов, продуктов их обогащения и металлургической переработки. Материалы совещания. Орджоникидзе, 1959. М., изд. ЦНИИНЦветмет, 1961, стр. 64.
449. Цфасман С. Б. Электронные полярографы. М., Металлургиздат, 1960.
450. Чалков Н. Я., Устинов А. М. Зав. лаб., 37, 149 (1971).
451. Черкесов А. А., Краснов А. И. Фталексоны. Изд. Саратовского пед. ин-та, 1970, стр. 194.
452. Чернихов Ю. А., Добкина Б. М. Зав. лаб., 15, 906 (1949).
453. Чернихов Ю. А., Черкашина Т. В., Ноткина М. А., Петрова Е. И., Меньшова Н. П., Луговская В. И., Горянская Г. П. Ж. аналит. химии, 21, 714 (1966).
454. Чжоу Шуанъ-бао, Хэ Бао-чжэнъ, Юй Вэн-чан. Шаньдун дасюэ сюбао. Цзыжань кэсюэбань, № 4, 50 (1965); РЖХим, 1967, 8Г83.
455. Чжоу Coy-пин, Чжоу Хун-сюнъ, Чжу Ю-юй. Синьсян шифань сюэюань сюэбао, 6, 21 (1965); РЖХим, 1966, 18Г140.
456. Чижиков Д. М. Кадмий. М., «Наука», 1967.
457. Чуйко В. Т., Шпикула В. М. Труды по химии и хим. технол., вып. 3 (11), 426 (1964).
458. Чучалина Л. С., Юделевич И. Г., Буянова Л. М., Старшинова Н. Н., Валл Г. А. Ж. аналит. химии, 24, 905 (1969).
459. Шарле Г. Методы аналитической химии. М.—Л., «Химия», 1965, стр. 636.
460. Шафран И. Г., Зеленова Т. К. Химические реактивы и препараты, вып. 24. М., Госхимиздат, 1961, стр. 210.
461. ШахвердиН. М., Туманов А. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 418.
462. Шахтахтинский Г. Б., Валиев Б. С., Асланов Г. А., Маматкулов М.М. Мед. ж. Узбекистана, № 4, 76, (1970); РЖХим, 1970, 21Г81.
463. Шварц Д. М., Каперский Л. Н., Нилова И. С. Сб. «Спектральный анализ в цветной металлургии». М., «Металлургиздат», 1960, стр. 113.
464. Шварценбах Г. Ш., Флашка Г. Комплексометрическое титрование. М., «Химия», 1970.
465. Шемякина М. А.Богдасаров К. НКоваленко П. НВладимирова В. Ф. Сб. «Современные методы химической технологии и контроля производства». Ростов-на-Дону, 1968, стр. 127; РЖХим, 1969, 9Г184.
466. Шемякин Ф. М., Степин В. В. Ионообменный хроматографический анализ металлов. М., «Металлургия», 1970.
467. Шкловер Л. П. Зав. лаб. 28, 686 (1962).
468. Шкробот Э. П. Анализ руд цветных металлов и продуктов их переработки. Сборник научных трудов, № 27. М., «Химия», 1967, стр. 5.
469. Шкробот Э. П., Бакиновская Л. М., Зав. лаб., 32, 1452 (1966).
470. Щербов Д. П. Флуориметрия в химическом анализе минерального сырья. М., «Недра», 1965.
471. Щербов Д. П., Сагалович И. И. Методы полярографического анализа минерального сырья. М., Госгеолтехиздат, 1960, стр. 65.
240
471а. Экстракция. Ионный обмен. Текущий указатель литературы. Новосибирск, ГПНТБ СО АН СССР (издается с 1968 г.).
472. Эфендиев Ф. М. Изв. АН СССР, серия физич., 11, 313 (1947).
473. Юркин Я., Сильвестров Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 457.
474. Юсупов М., Талипов Ш. Т., Джиянбаева Р. X., Татарская А. 3. Труды Ташкентского ун-та, вып. 288, 74 (1967).
475. Яковлев П. Я., Разумова Г. П. Тиоацетамид — заменитель сероводорода. М., Металлургиздат, 1963.
476. Яковлев Ю. В., Степанец О. В. Ж. аналит. химии, 25, 578 (1970).
477. Яковлев Я. Я., Яковлева Е. Ф. Технический анализ в металлургии. М., Металлургиздат, 1963, стр. 198.
478. Якубович В. И. Фармацевт, ж., № 1, 22 (1969); РЖХим, 1969, 16Г9.
479. Ямада Д., ИидаТ., Ямасаки К. Бунсэки кагаку, 19, 125 9(1970); РЖХим, 1971, 6Г151.
480. Яцимирский К. Б. Кинетические методы анализа. М., «Химия», 1967.
481. Яцимирский К. Б., Васильев В. Я. Константы нестойкости комплексных соединений. М., Изд-во АН СССР, 1959.
482. Aguas da Silva М. Т., Gabriel da Crus M. J. Rev. portug. quim., 10, N 206, 111 (1968), РЖХим, 1970, 14Г186.
483. Aguas da Silva M. T., Valente Soares M. I. Rev. portug. quim., 8, 8,
111 (1966); РЖХим, 1967, 12Г155.
484. Allan J. A. Spectrochim. Acta, 18, 259, (1962).
485. Ana Chueca, Worwood M., Taylor D. M. Intern. J. Appl. Radiation and Isotopes, 20, 335 (1969).
486. Angino E. E., Billings G. K. Atomic absorption spectrometry in geology. Amsterdam—London—New York, 1967.
487. Asmus E., Werner W. Z. anal. Chem., 228, 334 (1967).
488. Basinska H., Puzanowska-T arasiewicz H. Chem. analit., 14, 165 (1969).
489. Basinska H., Puzanowska-Tarasiewicz H. Studia Soc. scienct. Torun, B7, 10 (1969); РЖХим, 1970, 7Г77.
490. Basinski A., Lamanska H. Przem. Chem., 9, 241 (1953); РЖХим, (1953), 9115.
491. Beck J. L., Fitzgerald J. M., Bishop J. A. Analyt. chim. acta, 51, 191 (1970).
492. Belcher R., Cressland B., Fennel T. R. Taianta, 16, 1335 (1969).
493. Belcher R., Nutten A. J.t Stephen W. J. J. Chem. Soc., 1951, 3444.
494. Ben-Bassat A. N., Adato I, Yariv S. Chim. analyt., 48, 609 (1966).
495. Beran P., Dolezal J., Pacak P. Chem. zvesti, 18, 330 (1964).
496. Berg E. W., Reed К. P. Analyt. chim. acta, 36, 372 (1966).
497. Berger W., Elwers H. Z. anal. Chem., 171, 255 (1959).
498. Berger W., Elwers H. Z. anal. Chem., 199, 166 (1964).
499. Berger J. A., Meyniel G., Petit J. Bull. Soc. chim. France, № 12, 3176 (1964).
500. Bersier P., Finger K., Sturm F. Z. anal. Chem., 216, 189 (1966).
501. Bhaduri A. S. Z. anal. Chem., 151, 109 (1956).
502. Bhatnagar R. P., Arora R. C. Indian J. Chem., 3, 89 (1965); РЖХим, 1966, ЗГ65.
503. Bhatnagar R. P., Sharma K. D., Varshey R. P. Indian J. Chem., 4, 47 (1966); РЖХим, 1966, 15Г56.
504. Bhatnagar R. P., Siddique S., Rao К. V. K. Indian J. Chem., 2, 205 (1964); РЖХим, 1965, ЗГ54.
505. Bhatt J. H., Soni К. P. Indian. J. Appl. Chem., 32,139 (1969); РЖХим, 1970, 9Г64.
506. Bibr B., Lener J., Zeman A. J. Radional. Chem., 3, 181 (1966).
507. Biechler D. G. Analyt. Chem., 37, 1054 (1965).
508. Bobtelsky M., Rafailoff R. Analyt. Chim. acta, 17, 267 (1957).
509. Bode H., Wulff K. Z. anal. Chem., 219, 32 (1966).
510. BoltzD. F., Havlena E. J. Analyt. chim. acta, 30, 565(1964).
511. Bontchev P. R., Alexiev A. A. Mikrochim. acta, 1968, 875.
241
512. Boparai К. S., Rao A. B., Bhagwat W. V. Indian J. Appl. Chem., 32, 343 (1969); РЖХим, 1971, 1Г93.
513. Bowen H. J. M. Analyst, 92, 118 (1967).
514. Bratzel M. P-, Dagnail R. M. Analyt. Chem., 41, 713 (1969).
515. Braun T., Tolgyessy J. Radiometric titration. Budapest, 1967.
516. Brazier J. N. Taianta, 16, 949 (1969).
517. Brode W. R. Chemical spectroscopy. New York—London, 1958, p. 452.
518. Broyles C. ,Thomas D., Haynes S. Phys. Rev., 89, 715 (1953).
519. Budesinsky B., West T. S. Analyst, 94, 182 (1969).
520. Budevsky O., Russeva E., Mesrob B. Taianta, 13, 277 (1966).
521. Burriel Marti F., Alvarez Herrero C. An. Real. Soc. Esp. fis. у quim., A62, 45 (1966); РЖХим, 1967, 5Г53.
522. Bussev A. I., Schischkov A., Nikolov N. K. Natura Ecole normale super. Plovdiv, 2, 55 (1968); РЖХим, 1970, 1Г90.
523. Cabane-Brouty F. Analyt. chim. acta, 47, 511 (1969).
524. Callan E. J. Bull. Amer. Phys. Soc., 7, 416 (1962).
525. Calu N., Cecal A. An. stiint. univ. Iasi, 13, 129 (1967); РЖХим, 1969, 1Г60.
526. Cescon P., Pucciarelli F., Fiorant M. Taianta, 17, 647 (1970).
527. Chalmers R. A., Telling G. M. Mikrochim. acta, 1967, 1135.
528. Chattopadhyay S. S. Indian J. Chem., 8, 1142 (1970); РХЖим, 1971, 14Г71.
528a. Christian С. M., Robinson J. W. Analyt. chim. acta, 56, 466 (1971).
529. Chromy V., Sommer L. Taianta, 14, 393 (1966).
530. Me Clellan В. E., Freiser H. Analyt. Chem., 36, 2262 (1964).
531. Concialini V., Lanza P., Lippolis M. T. Analyt. chim. acta, 52, 529 (1970).
532. Cormos A., Ivancin O. Rev. chim., 17, 503 (1966); РЖХим, 1967, 10Г131.
533. Courtot-Copuez J., Guerder P. Bull. Soc. chim. France, 1961, 1942.
534. Л/с Crackan J- D., Vecchione M. C., Longo S. L. Atomic Absorpt. Newsletter, 8, 102 (1969); РЖХим, 1970, 7Г60.
535. Crisan J. A., Tdnase P. Studia univ. Babes-Bolyai, Ser. chem., 13, 129 (1968); РЖХим., 1969, 8Г76.
536. Crouzoulon P. Chim. analyt., 50, 483 (1968).
537. Czakow J. Chem. analit., 12, 981 (1967).
538. Dagnail R. M., West T. S., Young P. Taianta, 13, 803 (1966).
539. Dankwortt P. W., Eisenbrand J. Lumineszenz-Analyse im filtrierten Ultravioletten Licht. Leipzig, 1964, S. 117.
540. DeP. K., Dutt N. K. Indian. J. Appl. Chem., 30, 173 (1967); РЖХим, 1968, 23Г44.
541. DeBoer F. J., Visser J. Analyst., 93, 56 (1968).
542. Denk G. Z. Z. anal. Chem., 136, 336 (1952).
543. Deshmukh G. S., Rao A. L. Indian J. Chem., 3, 319 (1965); РЖХим, 1966, 7Г68.
544. Doadrio A., Arroyo M. An. Real. Soc. Esp. fis. y. quim., B60, 403 (1964); РЖХим, 1967, 7Г35.
545. Dolezal J., Povondra P., Stulik K. Chem. listy, 59, 852 (1965).
546. Domagalina E., Przyborowski L. Z. anal. Chem., 207, 411 (1965).
547. Domagalina E., Zarfba S. Chem. analit., 15, 769 (1970).
548. Donoso G. N., Santa Ana M. A. Analyt. chim. acta, 44, 211 (1969).
549. Dordevii S., Dunhic G. Technika, 23, № 10 (1968); РЖХим, 1969, 5Г134.
550. Ducauze Ch., Вуё J. Bull. Soc. chim. France, № 12, 4541 (1970).
551. Dutt N. K., Sanayal G. S., Nag K. Analyt. chim. acta, 41, 331 (1968).
552. Dwyer F. P., Gibson N. A. Analyst, 75, 201 (1950).
553. Evcim N., Reber L. A. Analyt. Chem., 26, 936 (1954).
554. Fairhall L. T., Prodan К. J. Amer. Chem. Soc., 53, 1321 (1931).
555. Farguhar M. C., Hill J. H., English M. M. Analyt. Chem., 38, 208 (1966).
556. Feigl F., Miranda L. I. Ind. Eng. Chem., Analyt. Ed., 16, 141 (1944).
557. Feldman C. Analyt. Chem., 21, 1041 (1949).
558. Filipov D., Natchev J., Natchev С. Докл. Волг. АН, 18, 537 (1965), РЖХим, 1966, 8Г140.
242
559. Fink R. W., Jopson R. С., Марк H., Swift C. D. Rev. of modern. Phvs.
38, 513 (1966). ’
560. Flaschka H., Butcher J. Taianta, 11, 1067 (1964).
561. Flaschka H., Butcher J. Chemist-Analyst, 54, 36 (1965).
562. Flaschka H., Carley F. B. Taianta, 11, 423 (1964).
563. Flaschka H., Weiss R. Microchim. J., 14, 318 (1969).
564. Frei R. W., Ryan D. E., Stockton C. A. Analyt, chim. acta, 42, 59 (1968).
565. Fresenius R., Jander G. Handbuch*71er analytische Chemie. Teil III, Bd II. Berlin, 1945.
566. Fritz S., Sherma J. J. Chromatogr., 25, 153 (1966); РЖХим, 1967, 8Г65.
567. Funk N., Ditt M. Z. anal. Chem., 91, 332 (1933).
568. Fuwa K., Vallee B. L. Analyt. Chem., 35, 942 (1963).
569. Gagliardo E., Locatellt V., Gustin N. Metallurgia ital., 56, 316 (1964); РЖХим, 1965, 22Г62.
570. Galik A. Taianta, 13, 109 (1966).
571. Galik A. Taianta, 16, 201 (1969).
572. Ganescu I., Vdrhelyi C., Popescu A. Stud. univ. Babes-Balyai, Ser. Chem., 15, 145 (1970); РЖХим, 1971, 2Г99.
573. Garcia D. J. Rev. Metalurgia, 2, 283 (1966); РЖХим, 1967, 6Г94.
574. Gerner A., Grdinic V., Pavisic D. Mikrochim. acta, 1969, 796.
575. Ghose A., Dey-Arum K. Analyst, 95, 698 (1970).
576. Gibson JV. A. Rev. Pure Appl. Chem., 4, 101 (1954).
577. Goodfellow G. I. Atomic Weapons Res. Establ. U. K. Atomic Energy Author (Rept), № 87/66 (1967); РЖХим, 1968, 8Г36.
578. Gorczynska K., Gluzinska M., Ciecierska-Stoklosa. Chem. analit., 14, 591 (1969).
579. Gotd H. Science Repts Tohoku Imp. Univ., 29, 204 (1940); C. A., 35, N 6, 1720, 1721, 1722 (1941).
580. Graciani E., Bernaly E., Pino F. Inform, quim. analit., 19, 45 (1965); РЖХим, 1966, 5Г73.
581. Gray P. R. Phys. Rev., 101, 1306 (1956).
582. Grecu J., Curea E., Pitis M. Studii ?i cercetAri chim. Acad. RPR Fil. Cluj, 13, 219 (1962); РЖХим, 1965, 8Г6.
583. Gregorowicz Z., Ciba J. Chem. analit., 14, 943 (1969).
584. Grey P., Cave G. С. В. Canad. J. Chem., 47, 4543 (1969); РЖХим, 1970, 14Г86.
585. Griitzner G. Neue Hiitte, 12, 305 (1967); РЖХим, 1967, 22Г137.
586. Hering R., Fritsch L., Bistri W. Wiss. Z. Pad agog. Inst. Giistrow R. Biol.-Chem.-Polytechn., 5, 43 (1966—1967); РЖХим, 1969, ЗГ46.
587. Hering R., Sattler J. Wiss. Z. Padagog. Inst. Giistrow R. Biol.-Chem.-Polytechn., 5, 47 (1966—1967); РЖХим, 1969, ЗГ48.
588, Hessel G. Biochem. Z., 177, 146 (192б)г.
589. Hibbits J. O., Kailman S. Taianta, 11, 443 (1964).
590. Hibbits J. O., Kailman S., Oberthin H., Oberthin J. Taianta, 8, 104 (1961).
591. Hniliikova M., Sommer J. Taianta, 13, 667 (1966).
592. Holzbecher Z. Luminiscencni analysa. Praha, CSAV, 1957.
593. Holzbecher Z., Divis L., Nov&k J., F elcmanova-Rauchova D., Reznidek J. Sbornik v у soke Skoly Chemicko-Technologickfi v Praze, N 3, Analyticka Chemie, 83 (1968).
594. Ibarz J., Estape A., Costa J. An. Real. Soc. Esp. fis. у quim., B63, 269 (1967); РЖХим, 1967, 18Г158.
595. Ikeda N., Oguma К. J. Chromatogr., 31, 289 (1967).
596. Jackwerth E. Z. anal. Chem., 206, 269 (1964).
597. Jacobsen E., Tandberg G. Analyt. chim. acta, 47, 285 (1969).
598. Jain D. S., Gauer I. K. Z. anal. Chem., 223, 109 (1968).
599. Jankovsky I. Z. anal. Chem., 214, 332 (1965).
600. Jankovsky I. Chem. listy, 60, 1227 (1966).
601. Jasinski T., Smagowski H. Chem. analit., 14, 829 (1969).
602. Jasinski T., Stefanik K. Chem. analit., 10, 983 (1965).
603. Jfdrzejewska H., Malusecka M. Chem. analit., 12, 579 (1968).
243
804. Jedrzefewski S., Jedrzejewska H. Chem. analit., 15, 729 (1970).
605. Johar G. S., A g ar w ala U. Current Sci. (India), 38, 592 (1969): РЖХим,
1970, 18Г57.
606. Johri K. N., Mehra H. C., Kaushik JV. K. Chromatographia, К» 7, 347 (1970); РЖХим, 1971, 4Г52.
607. Johri K. N. Singh K. Current. Sci. (India), 34, 78 (1965); РЖХим, 1967, 7Г55.
608. Jopson R. S., Marc H., Swift C. D., Williamson M. A. Phys. Rev., 136, A69, (1964).
609. Jursik F. J. Chromatogr., 19, 448 (1965); РЖХим, 1966, 11Г55.
610. Kalt M. B., Boltz D. F. Analyt. Chem., 40, 1086 (1968).
611. Kasiura K. Chem. analit., 11, 141 (1966).
612. Kasparec I., Zeman A., Prasilova J. J. Radioanalyt. Chem., 2, 281 (1969);
РЖХим, 1969, 15Г72.
613. Khalifa H., El-Barbary I. Microchem. J., 14, 216 (1969).
614. KhasnobisS. K.,Dey B.R. Indian. J. Appl. Chem. 32,307(1969); РЖХим, 1971, 1Г179.
615. Kirkbright G. F., Stephen W. I. Analyt. chim. acta, 32, 544 (1965).
616. Kiss T. A., Zsigrai I. J., Girin V. S., Krizsan R. L. Microchem. J., 14, 573 (1969); РЖХим, 1970, 13Г113.
617. Klimes I., Perinkova O., Janak I. Coll. Czech. Chem. Comm. 32 1273 (1967).
618. Kodama M., Miyamoto K. Bull. Chem. Soc. Japan, 42, 835 (1969).
619. Kolhoff I. M., Elving P. J., Treatise on Analytical Chemistry, Part II, Vol. 3. New York—London, 1961.
620. Korkisch I., Feik F. Analyt. chim. acta, 32, 110 (1965).
621. Kotsufl K. Bull. Chem. Soc. Japan, 38, 988 (1965).
622. Krivdn V., Weicz H. Mikrochim. acta, 1969, 89.
623. Kudo K., Yamamoto K., Kuriyama S. Badioisotopes, 16, 514 (1967); РЖХим, 1968 10Г113.
624. Kweicihski S. Rudy i metale niezel., 14, 275 (1969); РЖХим, 1969, 23Г126.
625. Landgrebe A. R., Me Clendon L. T., Devoe I. R. Trans. Amer. Nucl. Soc., 8, 315 (1965); РЖХим, 1966, 12Г72.
626. Lanfranco G., Bianchini A. Chim.e Ind., 50, 332 (1968); РЖХим, 1968, 18Г156.
627. Langrebe A. P., Me Lendon L. T., Devoe I. R., Pella P. A., Purdy W. C. Analyt. chim. acta, 39, 151 (1967); РЖХим, 1968, 6Г71.
628. Lazansky J., Belohlavek O. Huth. Listy, 21, 272 (1966).
629. Lesigang-Buchtela M. Mikrochim. Acta, 1966, 414.
630. Lewandowski A., Czemplik W., Dzik A. Zesz. nauk. univ. Poznaniu, N 28, 3 (1960); РЖХим, 1965, 23Г61.
631. Lewandowski A., Owoc M. Ibid., p. 9; РЖХим, 1965, 23Г70.
632. Lewandowski A., Szczepaniak W. Prace Komis, mat-przyrodn. poznan. towarz przyjaciol nauk, 12, 3 (1965); РЖХим, 1966, 14Г51.
633. Lissens J. L., Dams R., Hoste J. Analyt. chim. acta, 45, 213 (1969).
634. Ligten I. W. L., Velthuyzen H. Mikrochim. acta, 1964, 759.
635. Lingane J. J. Electroanalytical Chemistry. New York—London, 1953.
636. Liteanu C., Rostds I. Studia univ. Babes-Bolyai, Ser. Chem., 8, 215 (1963); РЖХим, 1965, 10Г74.
637. Liteanu C., Rusu V. Studia univ. Babes-Bolyai, Ser. Chem., 10, 99 (1965); РЖХим, 1966, 10Г70.
638. Luke C. L. Analyt. chim. acta, 39, 447 (1967).
639. Macoto O. Bull. Chem. Soc. Japan, 37, 504 (1964).
640. Mahr C., Ohle H. Z. anal. Chem., 109, 1 (1937).
641. Majumdar A. K., Bhowal S. G. Analyt. chim. acta, 35, 206 (1966).
642. Majumdar A. K., Bhowal S. G. Analyt. chim. acta, 36, 399 (1966).
643. MafumdarA. K., Mitra В. K. Z. anal. Chem.., 208, 1 (1965).
644. Majumdar A. K., Mitra В. K. Z. anal. Chem 223, 108 (1966).
645. MafumdarA. K., Singh В. R. Z. anal. Chem., 161, 257 (1958).
244
646. Mansfield J. M., Winefordner J. D. Analyt. Chem., 37, 1049 (1965).
647. Marczenko Z. Odczynniki organiczne w analizie nieorganicznej. Warszawa,. 1959.
648. Marczenko Z. Mikrochim. acta, ll)f>5, 281.
649. Marczenko Z., Kasiura K., Krasiejko M. Mikrochim. acta, 1969, 625.
650. Marczenko Z., Mojski M. Chem. analit., 12, 1155 (1967).
651. Marinenko G. Taianta, 16, 1335 (1969).
652. Marowsky G. Z. anal. Chem., 253, 267 (1971).
653. Martin E., Monnier D., Haerdi W. Analyt. chim. acta, 34, 64 (1966).
654. Martin E., Monnier D., Haerdi W., Selz I. Chimia, 18, 251 (1964); РЖХим, 1965, 11Г65.
655. Massmann H. Spectrochim. acta, B23, 215 (1968).
656. Mathur К. K., Narang С. K. Indian J. Chem., 3, 226 (1965); РЖХим, 1966, 9Г60.
657. Mattison L. E., Metaxas J. M., O'Dell C. S. Analyt. Chem., 41, 1690' (1969); РЖХим, 1970, 4П2.
658. Mattison L. E., Wolford J. C. Analyt. Chem., 38, 1675 (1966).
659. Mensis A. C. A. Analyt. Chem., 32, 898 (1960).
660. Meyer H. G. Z. anal. Chem., 244, 394 (1969).
661. Miller T., Lamb B., Prater K. Analyt. Chem., 36, 415 (1964).
662. Mitcham D., Piccolo B., Tripp V. W., O'Conner P. T. Advances in X-ray analysis, 8, 456 (1964).
663. Musil I., Kopanica M. Chemist-Analyst, 55, 9 (1966); РЖХим, 1966, 15Г135.
664. Muzzarelli R. A. Taianta, 13, 809 (1966).
665. Nadkarni R. A., Haidar В. C. Omagiu acad. prof. Raluca Ripan. Rucu-resti, 1966, p. 375; РЖХим, 1967, 8Г12.
666. NangniotP. J. Polarogr. Soc., 11, 45 (1965); РЖХим, 1967, 1Г153.
667. Navratil 0., Frei R. W. Analyt. chim. acta, 52, 221 (1970).
668. Necia S., Aurelian M. Rev. chim., 17, 357 (1966); РЖХим, 1967, 8Г163.
669. Negoiu D., Gagescu D. Studii $i cercetiri chim., Acad. RSR, 13, 1257 (1965); РЖХим, 1967, 2Г59.
670. Norman V. S. Analyst, 91, 593 (1966).
671. Ohtsuka S. Appl. Spectrosc., 23, 115 (1969).
672. Oi N. Japan Analyst, 9, 770 (1960).
673. Orlandi K. A., KoFkish J. Analyt. chim. acta, 43, 459 (1968).
674. Oshry H. I., Ballard I. W., Schrenk H. H. J. Opt. Soc. Amer., 31, 635-(1941).
675. Owens D. V., Swift E. H., Smith D. M. Taianta, 11, 1521 (1964).
676. Padma D. K., Murthy A. R., Vasudeva. Mikrochim. acta, 1970, 647; РЖХим, 1971, 6Г75.
677. Palma R. J., Reinbold P. E., Pearson К. H. Analyt. chim. acta, 51, 329 (1970).
678. Parker C. A. Photoluminescence of Solutions. Amsterdam—London— New York, 1968, p. 476.
679. Parry E. P., Johnston T. H., Goldner H. I. J. Electroanalyt. Chem., 13, 177 (1967); РЖХим, 1967, 14Г113.
680. Pass A., Ward A. M. Analyst, 58, 667 (1933).
681. Patrovsky V. Chem. listy, 47, 676 (1953).
682. Pearson L. A. Proc. 1-st Austral. Conf. Electrochem, Sidney—Hobart, 1963. Oxford—London, 1965, p. 340; РЖХим, 1966, 5Г151.
683. Pelczar T. Mikrochim. acta, 1968, 1295.
684. PertiO.N.,PrakashI. Proc. Nat. Acad. Sci. India, A34, 1 (1964); РЖХим, 1965, 11Г55.
685. Pietrzyk D. I., Kiser D. L. Analyt. Chem., 37, 233 (1965).
686. Pinta M., Recherche et Dosage des Elements Traces. Paris, 1962, p. 211,
687. Pirtea J. Z. anal. Chem., 184, 252 (1961).
688. Pirtea J. Rev. chim., 16, 288 (1965); РЖХим, 1966, 6Г57.
689. Polard E. H., Warrender R. И7., Nickless G. Some gen. probl. paper chromatogr. Prague, 1962, p. 175; РЖХим, 1965, 23Г68.
245
690. Poonia N. S. Indian J. Chem., 4, 511 (1966); РЖХим, 1967, 18Г88.
691. Popper E.,, Crdciuneanu R., Florean E. Omagiu acad. prof. Raluca Ri-pan. 1966, S. 467; РЖХим, 1967, 7Г45.
692. Popper E., Florean E., Maron P., Sosa E. Rev. roumaine chim., 11, 283 (1966); РЖХим, 1966, 21Г96.
693. Pourbaix M. Atlas d’equilibres electrochimiques. Paris, 1963, p. 414.
694. Pfibil R. Chelometry. Basic determinations. Praha, 1961.
695. Pfibil R., Vesely V. Taianta, 11, 1613 (1964).
696. Pfibil R., Vesely V. Taianta, 12, 475 (1965).
697. Pfibil R., Vesely. Chemist-Analyst, 55, 4 (1966).
698. Przeszlakowski S., Soczewinski E., Flieger A. Chem. analit., 13, 841 (1968).
699. Piishel R. Z. anal. Chem., 221, 132 (1966).
700. Qureshi M., Israill A. H. Analyt. chim. acta, 41, 523. (1968).
701. Qureshi M., Qureshi S. Z. J. Chromatogr., 22, 198 (1966).
702. Qureshi M., Rawat J. P., Khan F. J. Chromatogr., 34, 237 (1968).
703. Ramakrishna T. V., Robinson J. W., West W. Anal. chim. acta, 37, 20 (1967).
704. Rao С. B. Proc. Nat. Inst. Sci. India, АЗО, 1 (1964).
705. Rao A. L. J., Puri В. K. Z. anal. Chem., 235, 176 (1968).
706. Rao A. K. J., Puri В. K. Z. anal. Chem., 247, 18 (1969).
707. Rao A. L. J., Puri В. K. Z. anal. Chem., 252, 375 (1970).
708. Rey P., Wakita H., Schmitt R. A. Analyt. chim. acta, 51, 163 (1970).
709. Riley I. P., Taylor D. Analyt. chim. acta, 40, 479 (1968).
710. Roger T. Quantitative Electron Microprobe Analysis. New-York, 1965.
711. Roos С. E. Phys. Rev., 93, 401 (1954); 100, 1267 (1955); 105, 931 (1957).
712. Rubeska J. Anal. chim. acta, 40, 187 (1968).
713. Ruf H. Z. Anal. Chem., 247, 297 (1969).
714. Rulfs C., Przybylowicz E., Scinner C. Analyt. Chem., 26, 408 (1954).
715. Ruzicka J., Stary J. Taianta, 8, 228 (1961).
716. Rhziika J., Stary J. Substoichiometry in radiochemical analysis. Oxford, 1968.
717. Ryan D. E., Pitts A. E., Cassidy R. M. Analyt. chim. acta, 34,491 (1966).
718. Sachdev S.L., West P. W. Analyt. chim. acta, 44, 301 (1969).
719. Saxena R.-S., Jain M. C. Monatsh. Chem., 100, 28 (1969); РЖХим, 1969, 21Г105.
720. Saxena A. K., Srivastava M. H. N., Saxena В. B. L. Proc. Nat. Acad. Sci. India, A37, 156 (1967); РЖХим, 1969, 14ГЗ.
721. Saxena A. K., Srivastava M. H. N., Saxena В. B. L. Proc. Nat. Acad. Sci. India, A37, 177 (1967); РЖХим, 1969, 14Г4.
721a. Schleicher A. Elekctroanalytische Schnellmethoden. Stuttgart, 1947, S. 111.
722. Scholl A. W., Crumrine D. S. Chemist-Analyst, 56, 22 (1967).
723. Schossberger F. Advances in X-ray analysis, 1, 73 (1957).
724. Schwartz W., Dieckmann U. Wiss. Z. Techn. Hochschule Otto von Guerick Magdeburg, 13, 173 (1969); РЖХим, 1970, 4Г91.
725. Schwarzbach E., Miinzel H. Radiochimica acta, 10, 20 (1968).
726. Schweppe H. Z. anal., Chem., 244, 312 (1969).
727. Sekido E., Fernando L., Freiser H. Analyt. Chem., 37, 1556 (1965).
728. Senf H.-I. J. Chromatogr., 21, 363 (1966).
729. Sherma I. Analyt. chim. acta, 36, 138 (1966).
730. Shinde V. M. Chem. and Ind., № 42, 1785 (1967); РЖХим, 1968, 9Г17.
731. Sikorska-Tomicka H., Puzanowska H., Tarasiewicz M. Chem. analit., 12, 175 (1967).
732. Silverman L. Chemist-Analyst. 35, 53 (1946).
733. Sime к M. Chem. listy, 60, 74 (1966).
734. Singh G. Current Sci., 37, 197 (1968); РЖХим, 1968, 24Г69.
735. Singhal G. K., Tandon K. N. Taianta, 15, 707 (1968).
246
736. Sinko I., Dolezal J. J. Electroanalyt>Chem., 25, 299 (1970).
737. Sinko I., LeskovSes D. Vesth. Slov. kem. drusta, 16, 13 (1969); РЖХим, 1970, 14Г81.
738. Sorensen R. W., Sympson R. F. Analyt. Chem., 39, 1238 (1967).
739. Sosa I., Merodio I., Russo N. Determinacion espectroquimica de impu-rezas en plata. LEMIT, № 2, 53 (1969); РЖХим, 1971, 6Г163.
740. Spacu G., Antonesky E. Comun. Akad. R. P. Romine, 5, 865 (1955); РЖХим, 1956, 7091.
741. Spectrophotometric Data for Colorimetric Analysis. London, 1963, p. 74.
742. Stary J., RAiicka J. Taianta, 18, 1 (1971).
743. Strdfelda F. Coll. Czech. Chem. Comm., 30, 2320 (1965).
744. Strelow F. W. E., Louw W. J., Weinhert С. H. S. W. Analyt. Chem., 40, 2021 (1968).
745. Stupar J., Podobnik B., Korosin I. Croat, chem. acta, 37, 141 (1965).
746. Sun-Pao-Cuan, Doletai V., Zijka V. Coll. Czech. Chem. Comm., 30, 1785 (1965).
747. Syamasunder K. Current Sci., 34, 18 (1965); РЖХим, 1966, 23Г18.
748. Sympson R. E., Ingham R. K., Rohl D. R. Analyt. Chem., 39, 262 (1967).
749. Sztics P., Klug O. Magyar kem. lapja, 24, 323 (1964); РЖХим, 1970, 2Г162.
750. SzAcs A. J., Klug O. N. Chem. analit., 12, 459 (1967).
751. Takagi V. К. J. Chem. Soc. Japan, Pure Chem. Sect., 74, 291 (1953); РЖХим, № 7 11857 (1955).
751a. Tanaka H., Yamamoto T., Akamatsu M., Motoyama M., Hashizume G. Japan Analyst, 20, 784 (1971); РЖХим, 1972, 1Г151.
752. Temmerman E., Verbeek F. J. Electroanalyt. Chem., N 4, 423 (1968).
753. Temmerman E., Verbeek F. Analyt. chim. acta, 43, 263 (1968).
754. Tera F., Ruch R. R., Morrison G. Analyt. Chem., 37, 358 (1965).
755. Than E., Molch D., Eckert H.-J., Konig H. Z. Chem., 8, 348 (1968); РЖХим, 1969, 5Г61.
756. Tolgyessy J. Jadrove ziarenic v chemickey analyze. Praha, 1962.
757. Tolgyessy J., Siles B., Jesenak V., Varga S. Isotopenpraxis, 4, 383 (1968); РЖХим, 1969, 11Г74.
758. Trautner A. Metalloberflache, 21, 339 (1967); РЖХим, 1968, 8Г171.
759. Trtilek J., Hofmann V. Diarylkarbazidy a diarylkabazony v analyticke chemii. Brno, 1962.
760. Truffert L., Favart M., Le Gall Y. Ann. Falsific. et expert, chim., 60, 275 (1967); РЖХим, 1968, 18Г172.
761. VaSdk V., Machalek V. Chem. listy, 47, 850 (1953).
762. Verma M. R., Rai L. Current Sci., 35, 478 (1966); РЖХим, 1967, 12Г71.
763. Vickers T. I., Vought R. M. Analyt. Chem., 41, 1476 (1969).
764. Vincent H. A., Wise E. N. J. Electroanalyt. Chem., 11, 54 (1966).
765. Virf L., Makai V. Studia univ. Babes-Bolyai., Ser. Chem., 8, 225 (1963); РЖХим, 1965, 10Г52.
766. Visintin B., Monteriolo S. Giuseppi S., Atti Giorn. Studio analyt. e chim. fis. acque natur. Livorno. Parma, 1963, S. 102; РЖХим, 1966, 1Г207.
767. Vydra F., Stulik K. Chemist-Analyst, 54, 77 (1965).
768. Wahler W. Neues Jahrb. Mineral. Abhandl., 108, 36 (1968); РЖХим, 1968, 20Г96.
769. Waksmundzki A., Przeszlakowski S. Chem. analit., 11, 159 (1966).
770. Wall H., Rhodes C. Canad. Spectrosc., 13, 51 (1968); РЖХим, 1968, 19Г99.
771. Walter J. L., Freiser H. Analyt. Chem., 24, 984 (1952).
772. Walter J. L., Freiser H. Ibid., p. 1985.
773. Weeks M. E., Leicester H. M. Discovery of the Elements. Easton, 1968, । p. 502.
774. Weiss D., Fidler J. Rudy, 12, 412 (1964); РЖХим, 1965, 13Г123.
247
~7b. Willard Н. Н. Ind. Eng. Chem., Analyt. Ed., 11, 269 (1939).
776. William J., Campbell W. J. Advances in X-ray analysis, 1, 73 (1957).
777. Wilson L. Analyt. chim. acta, 35, 123 (1966).
778. Winefordner J. D., Mansfield J. M. In «Fluorescence. Theory, instrumentation and practice». New-York, 1967, p. 565.
779. Winefordner J. D., Mansfield J. M. Appl. Spectr. Rev., 1, 1 (1967); РЖХим, 1968, 8ГЗЗ.
780. Wise W. M., Campbell D. E. Analyt. Chem., 38, 1079, (1966).
781. Woolley J. D. Analyst, 83, 477 (1958).
782. Yamamoto Y., Kotsuji K. Bull. Chem. Soc. Japan, 37, 594 (1964).
783. Yamauchi O., Tanaka H., Uno T. Taianta, 15, 177 (1968).
784. YarivS. Israel J. Chem., 2, 127 (1964); РЖХим, 1965, 5Г47.
785. Ymada J., lida E., Yamasaki K. Japan Analyst, 18, 1088 (1969).
'786. Ziemba S. Chem. analit., 15, 829 (1970).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбциометрия
с неорганическими реагентами 83, 165, 166, 179, 183, 191, 193 органическими реагентами 83, 84, 166, 170, 171, 173, 178—180, 182— 184, 191, 193, 196
Абсорбция
атомная 129, 165, 168, 172, 174, 178—180, 182, 193, 195
нейтронов 137
Акридин 29, 44
Активация нейтронами 138
2-Амино-1,3,4-тиодиазол-5-тиол 141 М-бис-(Р-Аминоэтил)дитиокарбами-
новая кислота 114, 115 га-Анизидин 52
Анилин 30, 44
Антипирин 29, 31, 44, 64, 65
Антранилат 31, 32, 52, 65
Арсазен 75, 84
Арсенат 65
Бензоат 52
Бензоилацетон 144, 146
8-(Бензолсульфониламино)хинолин 30, 32, 96, 97, 180
2,2-Бензотиазолил 83, 84
Бромбензтиазо 30, 32, 84, 86, 180
Бромпирогалловый красный 74, 84, 87
Бруцин 29, 31, 38, 44, 51, 65, 70, 209
Виолуровая кислота 44
Висмутиол II 30, 52
Гидразид изоникотиновой кислоты 52
Гидразин 44, 52, 142
Гидрат окиси железа 140, 141
Глиоксаль-бис-(2-оксианил) 30, 74, 84, 87
Гравиметрия 50, 52, 57, 165, 183
Гринокит 9
Дальзин 33, 114
Диаграмма Пурбэ 22
Диантипирилметан 29, 52, 56, 78, 79, 118, 141, 145, 152
Диантипирил-о-оксифенилметан, 52 70
Диацетил-б14с-(5-бромсалицилгидра-зон) 52
Диацетил-бис-(салицилгидразон) 52
Дибензоилметан 144, 146
Ди-в-бутилфосфорная кислота 146
3,5-бис-(Дикарбоксиметил аминометил)-!,41диокси-травс-стильбен 96, 97
4-Диметиламинофенил-4-метилбен-зиламинофенилантипирилкарби-нол 80
Диметилглиоксим 46, 158 га,тг'-Диметилдитизонол 87 Ди-0-нафтилкарбазон 40 Ди-0-нафтилтиокарбазон 87, 144, 146, 183
Динитродифенилкарбазид 39
2,2'-Дипиридил 30, 33, 52, 84, 88, 152
1,3-Дипиридил-3-(2-тиазолил)фор-мазан 84, 88
Дитизон 31, 32, 75, 79, 80, 84, 88,
124, 126, 144, 146, 166, 178, 180
М,М'-Дитиокарбамоилгидразин 114, 115
а-Дитионафтойная кислота 149
Дифенилгуанидин 145, 152
Дифенилдитиофосфат 52, 56, 65, 70, 80, 90
Дифенилкарбазид 33, 39, 40, 49, 75
Дифенилкарбазон 33 1,3-Дихинолил-2-пропен 33 Ди-2-этилгексилфосфорная кислота 145, 148
Диэтилдитиокарбаминатдиэтил аммония 144
Диэтилдитиокарбаминат натрия 30, 82, 84, 90, 100, 115, 120, 141, 145, 220
Диэтилдитиофосфат никеля 31, 52, 56, 66, 71, 149
249
Железо-дипиридил-иодид 33, 40, 84, 88
Р-Изопиразолои 145, 150 Изотопное разбавление 124 Индикаторы металлохромиые 74, 76,
Иодэозин 84, 95, 97, 98
Кадионы 30, 32, 39, 84, 90, 183 Кадмий, валентность 8, 23 изотопы 9 источники получения 10, 23 комплексы неорганические 26, 28, 35, 200, 210—212 комплексы органические 29, 203, 207, 209 минералы 9, 10 нахождение в природе 8 потенциалы полуволн 101, 102, 213 потенциалы электродные 18 применение 11 растворимость металла 19, 20 растворимость соединений (в фазовом анализе) 175 растворимость солей 27, 198 свойства
коллоидные 17, 26
физические 20 химические 20 электрические 18, 19 электрохимические 18 соединения
растворимость 27, 198 свойства 23, 198, 203 сплавы 12, 13, 19 токсичность 13
Кадмия аммиакаты 26, 35 арсенат 65, 70 бромид 27, 38, 44, 49, 64, 78, 95, 118, 209 галогениды 27 гидрат окиси 24, 37, 65 иодид 27, 39, 44, 46, 47, 57, 70, 71, 79, 91, 93, 95, 99, 124, 143, 144 карбоиат 24, 38, 66 молибдат 26, 6 6 нитрат 25, 47 окись 23, 186, 192 оксалат 25, 48, 67, 212 парапериодат 67 пирофосфат 26 рейнекат 68 роданид 28, 44, 73, 89, 143, 144, 152, 210, 211 сульфат 25, 50 сульфид 25, 41, 45, 47, 68, 83, 186, 192 феррицианид 26, 38, 121
ферроцианид 26, 38, 49, 69, 99, 118, 120
фосфат 26, 46, 69
хлорид 27, 192
цианид 29
Кадмоселит 10
Каптакс 52
7-[а-(о-Карбометокси)анилинобен-зил]-8-оксихинолин 145
Карбонат 52, 66
Каталитические методы 100
Кислотный хром темно-синий 32 84, 91
Комплексонаты, показатели нестойкости 75
Комплексоны 31, 74, 80, 114, 115
Кристаллический фиолетовый 39, 84, 91
Ксантохроит 10
Ксиленоловый оранжевый 31, 75, 78, 81, 84, 92
Кулонометрия 121, 126
Купраен (куприенат) 39, 52
Купферон 31, 32, 44
л-(Меркаптоацетамид)фенетол 52
Меркаптобензотиазол 30, 31, 52
2-М еркапто-5-ани лино-1,3,4-тиодиазол 141, 158
8-Меркаптохинолин 30, 32, 47, 116
N-М етилаиабазин-а'-азо-азатол
«ПА» 84, 92
N-Метиланабазин-а'-азо-а-нафтол 84, 92
К-Метиланабазин-(а'-азо-7)-8-окси-хинолин 84, 93
Метиловый фиолетовый 84, 93, 141
Молибдат 52, 66
Монотиомочевинофталевая кислота 141
Монтепоиит 10
Морин 46
Морфолин 44
<шс-(4-Натрийтетразолилазо-5)этил-ацетат 84
2-Нафтиламин 44
р-Нафтохинолин 29, 33, 52, 57, 66,
71, 117
Нефелометрия 99. 196
Никотин 44
2-Нитрозо-1-нафтол 49
Нитропруссид 44, 67
Оксалат 54, 67, 212
4-Оксибензтиазол 46
2-Оксииафтилал-2-аминопиридин 54, 67
2-(2-Окси-5-нитрофенил-азо)-4,5-ди-фенилимидазол моноацетат 93
250
4-Окси-З-иитрофениларсоновая кислота 30
2-(о-Оксифенил)бензоксазол 31, 33, 54, 57, 67, 72, 97, 98
8-Оксихинальдин 145 8-Оксихинолин 30, 32, 44, 46, 54, 58, 67, 72, 117, 145, 150
1-(8-Окси-2-хинолин)-3,5-диметил-пиразол 97, 98
8-Оксихинолин-5-сульфокислота 82, 97
Определение кадмия в воде 177 воздухе 177 концентратах 174 металлах 173, 174 особо чистых веществах 179 пищевых продуктах 188 растениях 182 реактивах 179 рудах атомно-абсорбционное 168 полярографическое 164 спектрографическое 167 фотометрическое 48, 166 сплавах 51, 56, 174 электролитах 171, 196
Отавит 10
Отделение осаждением 140, 163, 165, 169, 172—174, 177, 189
Отражение р-излучений 139
Парапериодат 67
Пеницилламин 125
Перхлорат 38
Пикриновая кислота 44
Пирамидон 45, 115 1-(2-Пиридилазо)-2'-нафтол 30, 78, 80, 86, 93, 181
4-(2-Пиридилазо) резорцин 30, 74, 81, 84, 86, 93
Пиридил-2-альдоксим 73
Пиридин 37, 31, 45, 47, 54, 68, 73, 87, 89, 124, _ 145, е52, 209
Пирокатехиновый фиолетовый 31, 74, 78
Пирролидиндитиокарбаминат 86, 220
Плюмбон 77
Поляриметрия 99
Полярография 100, 112, 164, 165, 170, 171, 173, 177—179, 183, 195 амальгамная с накоплением 109, 170, 179, 180 осциллографическая 106, 191 переменнотоковая 105, 191, 193, 195
Порфирины 33
Пршибрамит 10
Разложение проб
минерального сырья 163
промышленных продуктов 169
Реакции для открытия кадмия в макропробирках 36 капельные 40 люминесцентные 46 микрокристайлоскопические 41 на хроматограммах 49 окислительно-восстановительные 21, 38, 48 твердофазные (растирание порошков) 49 электрографические 49
Рейнеке соль 45, 54, 58, 83, 140
Рейнекат 68
Родамин С 86, 93
Роданомеркуриат 38, 45, 210
Роданоцинкат 45v 211
Ртути(П) хлорид 45, 54, 210
Рубеановодородная кислота 31, 164» 178
Рубидий хлористый 45, 211
Салицилгидроксамовая кислота 54
Селенид 13
Селеноцианомеркуриат 45
Серная кислота 54
Спектрография эмиссионная 127,167?> 170—172, 174, 177—180, 183, 184^. 186, 194, 216
Сульфарсазен 30, 74, 77, 86, 94
Сульфат 50, 163
Сульфид 13, 35, 36, 45, 68, 83, 140
Тетраиодовисмутиат калия 40
Тетрафениларсоний 29, 31, 118
Тетрафенилфосфоний 31, 45
Тетраэтиленпентамин 76, 7!Г
Тиоацетамид 30, 37, 51, 79, 116, 141
2-Тиогидантоин 47
Тиокапролактам 39, 51, 54, 58, 73
Тиокарбонат 37
Тиомочевина 30, 45, 54, 58, 68
Тионалид 31, 39
Тиосульфат натрия 51, 54
Тиотеноилтрифторацетон 94
Титриметрические методы 64, 65,.
163
алкалиметрия 70, 196
аргентометрия 70, 73, 119, 196
ацидиметрия 73, 189, 196
броматометрия 72
иодатометрия 71
иодометрия 64, 70—73, 119, 196 комплексонометрия 74, 80, 115, 121, 171, 174, 178, 181, 189, 196-меркуриметрия 73, 119 перманганатометрия 73, 196
Титрование
251
амперометрическое 112, 115, 193, 1'95, 196
в неводных средах 82, 121 гетерометрическое 82 кондуктометрическое 121 кулоностатическое 121 потенциометрическое 118 радиометрическое 122 фотометрическое 80, 171, 195 экстракционное 79, 124
8-(п-Тосиламин о) хинолин 97 Трибензоилметан 145, 153 Три-л-октиламин 145, 153 Трифенилметиларсоний 54, 99
Ультрафиолетовый микроскоп 41, 44—46
Уротропин 29, 46
Фазовый анализ 176
?астворимость соединений кадмия 75
схема 176
1,10-Фенантролин 30, 31, 33, 54, 59, 87
.и-Фенил ен-ди-( 1 -тетразолин-5-тион) 54, 59
1-Фенил-3-метил-4-бензоилпиразо-лон-5 145, 150
Феррицианид 26, 38, 121 Ферроцианид 49, 54, 69, 99 Флуоресцеина галоидопроизводные 31, 33, 95, 98
Флуоресценция атомная 131 оптическая 46, 96, 97, 158, 160, 161, 179, 180, 193 рентгеновская 132, 218
Фосфат 54, 69
Фотометрия пламени 129, 193
Хинальдиновая кислота 32, 39, 54, 118
Хинин 29, 31, 47, 99
Хиноксалин-2-карбоновая кислота
59
2-Хинолилфосфорная кислота 59
Хинолин 30, 46 Хинолин-8-селенол 40 Хоулиит 10 Хроматография адсорбционная 157 бумажная 125, 158 ионообменная
на анионитах 156, 163, 166, 169 катионитах 125, 153, 169, 174, 177
распределительная 158 тонкослойная 160
Хромпиразол II 86, 95
Цезия хлорид 45, 211
Цементация 163, 174, 189
Чувствительность определений открываемый минимум (ОМ) 34 предельное разбавление (Р) 34 спектрального анализа 133 уверенно открываемый минимум (УОМ) 34
Щавелевая кислота 46, 48
Экстракция внутрикомплексных соединений 146, 169, 173, 177, 182 галогенидных комплексов простых 143, 169, 172 реэкстракция дитизонатов 149 тройных комплексов с органическими основаниями 151
Электролитические методы 61, 141,-171 электрогравиметрия 59, 61 электролиз внутренний62, 163, 189 с платиновым катодом 60, 61, 141, 142, 169 ртутным катодом 142
Эозин 33, 86, 95, 97, 98
Эриохром черный Т 74, 76, 80
Эритрозин 33, 86, 95, 97, 98
Этилендиамин 30
Этилендиаминтетраацетат 74, 80, 114, 117, 120
Эффективная навеска 34
ОГЛ АВЛЕНИЕ
От редколлегии........................................... . . 3
Предисловие...................................................... 5
Глава I
Общие сведения о кадмии
Краткие исторические данные................................. 7
Положение кадмия в Периодической системе элементов .... 8
Нахождение кадмия в природе............................... 9
Получение кадмия ............................................ 10
Применение кадмия ........................................... 11
Токсичность кадмия й его соединений........................ 13
Глава II
Химико-аналитическая характеристика кадмия и его соединений
Физические и химические свойства кадмия......................... 15
Соединения кадмия............................................... 23
Неорганические соединения кадмия............................ 23
Органические соединения кадмия.............................. 29
Глава III
Качественное обнаружение кадмия
О выражении чувствительности определений.................... 34
Общая характеристика группы .................................... 35
Реакции для обнаружения кадмия.................................. 36
Реакции в макропробирках.................................... 36
Капельные реакции на фильтровальной бумаге.................. 40
Микрокристаллоскопические реакции........................... 41
Люминесцентные реакции...................................... 46
Прочие реакции.............................................. 48
Глава IV
Количественное определение кадмия
Химические методы анализа....................................... 50
Гравиметрические методы..................................... 50
Титриметрические методы..................................... 63
Физико-химические методы анализа................................ 82
Фотометрические методы...................................... 82
Электрохимические методы................................... 100
Радиохимические методы....................................... 122
253
Физические методы анализа..................................... 127
Спектральный анализ....................................... 127
Рентгеноспектральный анализ............................... 132
Радиометрический анализ................................... 137
Глава V
Методы отделения кадмия от Сопутствующих элементов
Методы осаждения и соосаждения................................ 140
Электрохимические методы...................................... 141
Экстракционные методы......................................... 143
Хроматографические методы..................................... 153
Глава VI
Определение кадмия в природных и промышленных объектах
Анализ минерального сырья..................................... 163
Методы разложения..................................... 163
Методы разделения....................................... 163
Методы определения...................................... 164
Анализ промышленных продуктов................................. 169
Методы разложения и отделения........................... 169
Методы определения...........'............................ 169
Анализ природных и сточных вод, воздуха промышленных предприятий ....................................................... 177
Анализ воды............................................... 177
Анализ воздуха..........:................................. 177
Анализ реактивов и особочистых веществ ....................... 179
Анализ биологических объектов................................. 182
Глава VII
Анализ кадмия и его соединений................................ 185
Приложения.................................................. 198
Литература.................................................... 226
Предметный указатель . . :.................................... 249
Аналитическая химия кадмия. Щербов Д. П., Матвеец М. А. «Наука», 1973.
В монографии, являющейся очередным томом серии «Аналитическая химия элементов» приведены общие сведения о кадмии, его распространенности в природе, формах нахождения, применения, физических, химических и физико-химических свойствах. Дается^характеристика важнейших неорганических и органических соединений кадмия используемых в аналитической химии. Приведены методы отделения и определения’кадмия (химические, физические и физико-химические), а также методы определения примесей в нем. Наиболее современные и надежиы$,методы представлены в виде конкретных прописей. ___
Монография рассчитана на широкий круг химиков-аналитиков и научных работников, преподавателей, аспирантов и студентов вузов.
Таблиц 45. Иллюстраций 39. Библ. 786 назв.
Дмитрий Павлович Щербов, Мария Афанасьевна Матвеец
Аналитическая химия кадмия (Серия: «Аналитическая химия элементов»)
Утверждено к печати
Ордена Ленина Институтом геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
Редактор Е. И. Корчемная
Редактор издательства Н. Г. Явкина Художественный редактор Н. Н. Власик Технические редакторы Т . С. Жарикова и И. С. Кашина
Сдано в набор 7/VI-1973 г. Подписано к печати 12/Х-1973 г.
Формат 60 x90*/»,. Бумага К» 2.
Уел. печ. л. 16. Уч.-изд. л. 17,6 Тираж 1900. Тип. зак. 2459. Т-14278. Цена 1 р. 42 к.
Издательство «Наука»
1037ГСП Москва, К-62, Подсосенский пер., 21
2-н типография издательства «Наука»
121099 Москва, Г-99, Шубинский пер., 10