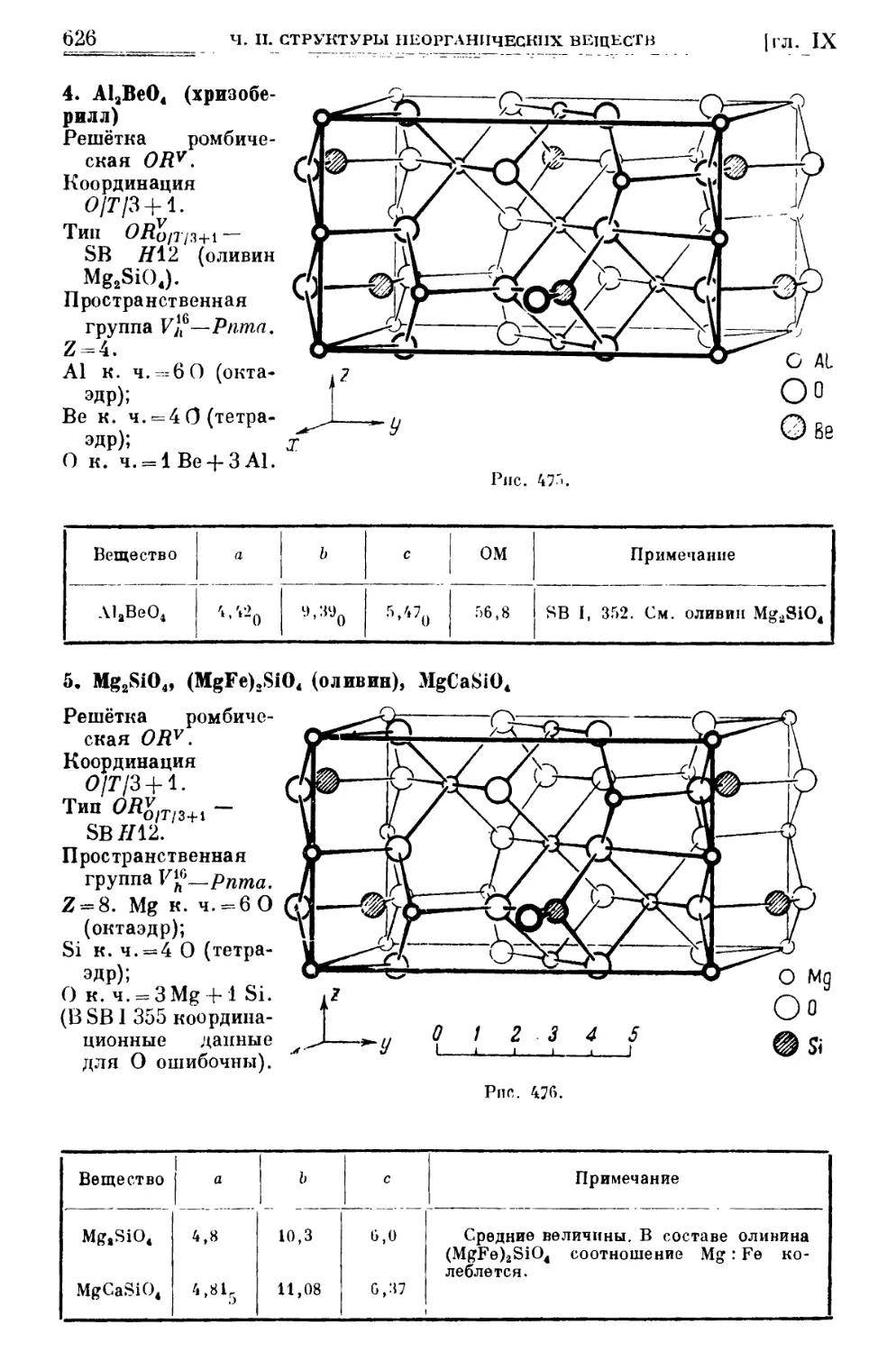
/



































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Author: Ормот Б.Ф.
Tags: химия аналитическая химия неорганическая химия государственное издательство технико-теоретической литературы
Year: 1950




Text
Б. Ф. ОРМОНТ
СТРУКТУРЫ
НЕОРГАНИЧЕСКИХ
ВЕЩЕСТВ
ГОСУДАРСТВЕННОЕ ИЗДАТЕЛЬСТВО
ТЕХНИКО-ТЕОРЕТИЧЕСКОЙ ЛИТЕРАТУРЫ
МОСКВА 19 5 0 ЛЕНИНГРАД
4
АННОТАЦИЯ
Книга представляет собой обзор структур
неорганических веществ, систематизированных на основе
периодической системы элементов и освещаемых с
позиций современной теории.
В связи с этим в первой части книги приведено
краткое изложение кристаллографической теории
симметрии и теории химической связи, дающее
возможность ориентироваться в материале
читателю-неспециалисту.
Книга охватывает большой фактический материал,
что позволяет широко использовать её в качество
справочника. В ней приведён составленный автором
наиболее полный из имеющихся регистр
исследований структур неорганических веществ.
Предназначена для студентов старших курсов,
аспирантов и научных работников, работающих в
различных областях химии, рентгенографии,
кристаллохимии и смежных дисциилин.
ОГЛАВЛЕНИЕ
Предисловие
Ч А С Т Ь НЕРВА Я
ОСНОВЫ ТЕОРИИ СТРУКТУР
Глава I. Элементы теории симметрии
§ 1. Введение JL_L__:_^_1__t_^__1_^_*_^_.-. . .
с ..^ ■ " тур
Опечатки „
вине
вине
161 7 св.
194 24 сн.
199
216 7 сн.
238 7 сн.
(табл.)
265 5 сн.
266
350
426
429
463
561
700
711
756
810
917
3 св.
12 сн.
6 св.
3 сп.
4 CD.
8 сн.
2 табл.
сверху
15 CD.
12 св.
24 сн.
2 CD.
Б. Ф. Ормонт
Напечатано
« 6,02х1028
Тх ioM
v 6,02 . 10»
Т х ~ iu>4
Координация К 12
c\h — Р21/с
Координация CcjC
С/Т
PdP
[Sr(HaO)8]OH2
[PBpJ*
(МНЛ [GeFe]
амины
SBJ8
Должно быть
[PBrJ*
(NH4), [OePJ
аммины
Kopp • •
'ИЯ
Авт. . .
Тип. • •
w-
ые
Корр.
Ред. ия
»
1И
Тип. ой
Авт
Корр.
г
Тип
2
СО
8ВВ /2а
Б 2
со2
В табл. 31 пунктирные
^стрелки,
направленные вниз, должны
быть жгрными
SB /2a
v 6.02XW28
Т ' 10й
_у_ 6,02 • 10м
4 ' W2*
Координация Я (12)
Координация С\С
с/т
Pd-P
[Sr(HaO)8] (ОН),
■и я
W-
ые
и я
/i ОГЛАВЛЕНИЕ
§ 37. Физические и химические свойства кристаллов п элементы симметрии
последних. Фигуры травления 87
§ 38. Об отклонениях от закона постоянства углов и плоско it формы граней 00
§ 39. Полиморфные превращения и формы кристаллов «Л
Б. ЭЛЕМЕНТЫ ТЕОРИИ СИММЕТРИИ ДИСКОНТИНУУМА
§ 40. Пространственная решётка кристалла. Понятие об элементарной ячейке У1
§ 41. Трансляция 92
§ 42. Двумерная трансляционная группа 93
§ 43. Трёхмерная трансляционная группа О'*
§ 44. Различные способы интерпретации процесса образования
пространственной решётки 95
§ 45. О сложных пространственных решётках 96
§ 40. Атомные полиэдры Е. С. Фёдорова 101
$ 47. 14 решёток Праве. Закон Праве 101
§ 48. Базис 10IJ
§ 4У. Действие трансляции на непараллельные её вектору элементы
симметрии 103
§ 50. Скользящая плоскость симметрии 105
$ 51. Винтовые осп-гелпкогпры 106
§ 52. О кратности, собственной симметрии и ориентировке точек,
находящихся ла скользящих плоскостях симметрии и винтовых осях. О
степенях свободы точек 110
§ 53. Координаты точек, возникающих при действии на точку различных
элементов симметрии 110
§ 54. Координаты точек, лилий и плоскостей в элементарной ячейке . . . 112
$ 55. Симметрия пространственной решётки и элементарной ячейки .... 115
$ 56. О выводе возможных пространственных групп 11ь
§ 57. Обозначения пространственных групп по IT 119
§ 58. О расчётах расстояний и объёмов в пространственных ре шоп.-ах . . . 121.
§ 5Я. Плотность упаковки. Количество частиц в элементарной ячейке . . . Ш
§ 00. Конфигурации координационных сфер. Валентные углы в
координационных сферах . . * 1*24
§ 01. Некоторые замечания о путях определения структуры кристалла
с использованием экспериментальных рентгенографических данных.
Кднипцы кХ и единицы А J2.4
§ 02. Об основных типах пространственных решёток 12S
§ ЬУЛ. Основные структуры, принадлежащие кубической епипчпш . . . 1;;1,
§ 04. О двух тинах плотнейшпх шаровых упаковок г:'*
§ 05. О структурах ZnS (вюрцита), NiAs, Cd.L, MoSa i;*'.
§ 00. Островные, цепочечные (ценные), слоистые структуры 141
$ 07. Цепи и сетки из октаэдров и тетраэдров и стехнометрнческпП с< .cj .и:
координационных сфер 14:»
§ 08. Переход от пространственной решётки к структуре вещества . . . 145
Г л а в а II. Теории химической связи л структуры веществ 148
§ 09. Введение 14S
а. период классической кристаллохимии
§ 70. Принцип М. В. Ломоносова. Представления о природе химической
связи до создании структурной теории А. М. Бутлерова \\$
S 71. Возникновение классической кристаллохимии 14*.»
5 72. Обобщённый закон кристаллохимии 151
§ 1'.). Морфотроппя. Пзотпппн % 15?
% 1\. Химическая структура веществ но А. М. Бутлерову.%Оптпч( скан
активность и структура веществ 1.V2
П. СТРУКТУРА КРИСТАЛЛА И СВЕТЕ ТЕОРИИ ХИМИЧЕСКОЙ
СВЯЗИ В ПЕРИОД ОТ СОЗДАНИЯ ТЕОРИИ АТОМА ВОРА ДО
ВОЗНИКНОВЕНИЯ ВОЛПОПОИ МЕХАНИКИ
§ 75. О различных типах химической связи и разных типах структур
кристаллов. Расчёт энергии решётки па базе электростатических
представлений * 150
$ 70 Принцип плотной упаковки анионов 161
§ 77. Соотношения ионных радиусов и структура кристалла 162
§ 78. О дальнейшем развитии ачсктростатической теории 1G&
§ 79. Дисперсионные силы, структура и энергия решётки 170
ОГЛАВЛЕНИЕ 5
§ (SO. О методе координационных полиэдров 172
§ 81. Представления о коиалентной связи и электростатический принцип
валентности. Правило эффективных атомных номеров (ЭА11) ... 173
S 82. О предположенной Паулппгом мнимой неустойчивости структуры СаО 174
§ 83. Характер связи и межатомные расстояния 175
f 84. Атомные радиусы и структуры металлов. Закон Вегарда. Концентрация
электронов и структур» фаз 177
И. О ПРИЛОЖЕНИИ КВАПТОНО-ХНМПЧЕОКПХ ТЕОРИИ К
НЕОРГАНИЧЕСКОЙ ХИМИИ 11 КРИСТАЛЛОХИМИИ
§ Н^). О волнах материн. Диффракция электронов 181
§ 8(1. Электронные уровни атома. Принцип Паули. Правило возбуждения
валентностей 182
§ 87. Нол поме ханнческая модель атома 186
$ 88. О волпомеханическнх теориях химической связи ; J88
$ 89. Энергия диссоциации молекул * 193
§ 90. О теории кгантопо-химического резонанса 194
5 91. Об установлении общих закономерностей, описывающих структуру
и устойчивость неорганических веществ 195
§ 92. Коли честно холостых w-электронол в изолированных атомах.
Температуры плавления простых веществ и максимальная валентность тех же
элементов в устойчивых соединениях 198
§ 9'). Недостатки правила эффективных атомных номеров (ЭАП).
Магнитные моменты атомов и ионов 202
$ 91. О трактовке структуры и устойчивости комплексных соединений
с помощью некоторых квантоно-хпмнческих моделей 203
$ 9Г>. Развитие вопроса о валентных конфигурациях' 204
§ 9Ь\ Об исследованиях электронной плотности в кристаллах.
Рентгенографический Фурье-анализ 206
§ 97. О зонной теории кристалла и её исходных предпосылках 210
§ 98. О /Г-иространстве и зонах Прпллюэпа 210
§ 99. Зависимость электропроводности кристаллов от характера
перекрывании зон 214
§ 100. оинная теории и структура кристаллов 215
§ J0I. О реальных твёрдых телах 218
£ 10-!. Типы реальных кристаллов 220
§10.4. О влиянии примесей на образование структуры . . , 222
§ JO'i. О применимости законов Пру и Дальтона в современной неорганической
и общей химии 223
§ 10.">. О зависимости структур комплексных соединении от
термодинамических факторов 22Г>
Ч Л С Т Ь И Т О Р Л Я
СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТИ
Г л а и а 111. Типы структур неорганических веществ 233
$ К)'). Введение. Конфигурации координационных сфер 233
$ 107. Тины структур 241
$ f()8. О задачах химической систематики структур 203
Глава IV7. Структуры простых веществ 2G4
$ 109. Об обозначениях модификаций и структурных форм 204
$110. Об обозначениях величин, принятых *в таблицах гл. IV—X и связи
. между ними 265
$ 111. 1 группа: литий, натрий, калий, рубидий, цезии, акацезпй (№ 87) 272
§ 112. II группа: бериллий, магнии, кальцин, стронций, барий, радий . . . 274
$ 113. Ill* группа: скандий, иттрий, лантан, актиний 278
$ 114. III** группа: металлы редких земель 280
§ МГ>. IV* группа: титан, цирконий, гафний, торий 284
$ 11П. V* грлпиа: ванадии, ниобий, тантал, протактиний 287
$ М7. VI* группа: хром, молибден, вольфрам, уран 288
$118. VI** группа (?): 93 (нептуний?), 94 (плутонии?), 95 (америции?),
90 (кюрий?) 297
5 1J9. VII* группа: марганец, технеции?, рений 298
$ 120. ЛТП* группа: железо, рутений, осмий 303
§ 121. IX* группа: кобальт, родий, иридии 308
6
ОГЛАВЛЕНИЕ
§ 122. X* группа: никель, палладий, платина 311
§ 123. I* группа: медь, серебро, золото 315
§ 124. И* группа: цинк, кадмий, ртуть 31S
§ 125. III группа: бор, алюминий, галлий, индий, таллий 321
§ 126. IV группа: углерод, кремний, германий, олопо, свинец 326
§ 127. V группа: азот, фосфор, мышьяк, сурьма, висмут 335
§ 128. VI группа: кислород, сера, селен, теллур, полоний 341
§ 129. I (VII): водород и VII группа: фтор, хлор, бром, иод 349
§ 130. VIII группа: гелий, неон, аргон, криптон, ксенон, радон 353
§ 131. Структуры простых веществ 355
Г л а в а V. Структуры галогенидов и гидридов 364
§ 132. Введение :*64
§ 133. I группа периодической системы: литий, натрий, калий, рубидий, цезий 3f>5
§ 134. II группа периодической системы: бериллий, магний, кальций,
стронции, барий, радий 37!
§ 135. III* группа периодической системы: скандий, иттрий, лантан .... 376
§ 136. III** группа периодической системы: металлы редких земель .... 379
§ 137. IV* группа периодической системы: титан, цирконий, гафний, торий 382
§ 138. V* группа периодической системы: ванадий, ниобий, тантал, протактиний 385
§ 139. VI* группа периодической системы: хром, молибден, вольфрам, уран 385
§140. VII* группа периодической системы: марганец, рений • 390
§ 141. VIII* группа периодической системы: железо, рутений, осмий .... 393
§ 142. IX* группа периодической системы: кобальт, родий, иридий .... 398
§ 143. X* группа периодической системы: никель, палладий, платина . . . 402
§ 144. I* группа периодической системы: медь, серебро, золото 406
§ 145. II* группа периодической системы: цинк, кадмий, ртуть 411
§ 146. III* группа периодической системы: бор, алюминий, галлий, индий,
таллий 421
§ 147. IV группа периодической системы: углерод, кремний, германий, олово,
свинец * 430
§ 148. V группа периодической системы: азот, фосфор, мышьяк, сурьма, висмут 438
§ 149. VI группа периодической системы: кислород, сера, селен, теллур,
полоний . 441
§ 150. VII группа периодической системы: водород, фтор, хлор, бром, иод 444
Глава VI. Окислы и халкогенидм (соединения О, S, Se, Те) 4'#8
§ 151. I группа периодической системы: литий, натрий, калий, рубидий, цезий 448
§ 152. II группа периодической системы: бериллий, магний, кальций,
стронций, барий, радий 450
§ 153. III* группа периодической системы: скандий, иттрий, лантан .... 452
§ 154. III** группа периодической системы: редкие земли 454
§ 155. IV* группа периодической системы: титан, цирконий, гафний, торий 458
§ 156. V* группа периодической системы: ванадий, ниобий, тантал,
протактиний 466
§ 157. VI* группа периодической системы: хром, молибден, вольфрам, уран 470
§ 158. VII* группа периодической системы: марганец, рений ....... 477
§ 159. VIII* группа периодической системы: железо, рутений, осмий ... 481
§ 160. IX* группа периодической системы: кобальт, родий, иридий .... 484
§ 161. X* группа периодической системы: никель, палладий, платина . . . 488
§ 162. I* группа периодической системы: медь, серебро, золото 494
§ 163. II* группа периодической системы: цинк, кадмий, ртуть 499
§ 164. III группа периодической системы: бор, алюминии, галлий, индий,
таллий 502
§ 165. IV группа периодической системы: углерод, кремний, германий, олово,
свинец " 507
§ 166. V группа периодической системы: азот, фосфор, мышьяк, сурьма,
висмут , ; 5-5
? 167. VI группа периодической системы: кислород, сера, селен, теллур,
полоний 532
§ 168. VII группа периодической системы: фтор, хлор, бром, иод 534
Глав а VII. Нитриды, фосфиды, арсениды и др 535
§ 169. Введение 535
§170. I группа периодической системы: водород, литий, натрий, калий,
рубидий, цезий 535
§ 171. II группа периодической системы: бериллий, магний, кальций,
стронции, барий 539
ОГЛАВЛЕНИЕ
7
§ 172. Ill* группа периодической системы: скандий, иттрий, лантан .... 542
§ 173. Ill** группа периодической системы: редкие земли 542
§174. IV* группа периодической системы: титан, цирконий, гафний, торий 543
§ 175. V* группа периодической системы: ванадий, ниобий, тантал .... 545
§ 176. VI* группа периодической системы: хром, молибден, вольфрам, уран 547
§ 177. VII* группа периодической системы: марганец, рений 551
§ 178. VIII* группа периодической системы: железо, рутений, осмий .... 554
§ 179. IX* группа периодической системы: кобальт, родий, иридий .... 557
§ 180. X* группа периодической системы: никель, палладий, платина . . . 560
§181. I* группа периодической системы: медь, серебро, золото 562
§ 182. II* группа периодической системы: цинк, кадмий, ртуть 567
§ 183. III группа периодической системы: бор, алюминий, галлий, индий,
таллий , , . , 570
§ 184. IV группа периодической системы: углерод, кремний, германий, олово,
свинец 573
§ 185. V группа периодической системы: азот, фосфор, мышьяк, сурьма,
висмут 575
§ 186. VI группа периодической системы: кислород, сера, селен, теллур,
полоний . . 575
§ 187. VII группа периодической системы: фтор, хлор, бром, иод 575
Глава VIII. Карбиды, силициды (в отдельных случаях станниды и др.)*
бориды 576
А. КАРБИДЫ, СИЛИЦИДЫ
§ 188. I группа периодической системы: литий, натрий, калий, рубидий,
цезии 576
§ 189. II группа периодической системы: бериллий, магний, кальций,
стронций, барий 576
§ 190. III* группа периодической системы: скандий, иттрий, лантан . . # 578
| 191. III** группа периодической системы: редкие земли 578
§ 192. IV* группа периодической системы: титан, цирконий, гафний, торий 570
§ 193. V* группа периодической системы: ванадий, ниобий, тантал .... 581
§ 194. VI* группа периодической системы: хром, молибден, вольфрам, уран 58:5
§ 195. VII* группа периодической системы: марганец, рений 588
§196. VIII* группа периодической системы: железо, рутений, осмий .... 591.
§ 197. IX* группа периодической системы: кобальт, родий, иридий . . . 594
§198. X* группа периодической системы: „никель, палладий, платина . . . 595
§ 199. I* группа периодической системы: карбиды, медь, серебро, золото . . 597
§ 200. II* группа периодической системы: цинк, кадмий, ртуть 599
§201. III группа периодической системы: бор, алюминий, галлий, индий,
таллий 599
§ 202. IV группа периодической системы: углерод, кремний, германий, олово,
свинец 600
§ 203. V группа периодической системы: азот, фосфор, мышьяк, сурьма,
висмут 604
§ 204. VI группа периодической системы: кислород, сера, селен, теллур,
полоний . . 604
§ 205. VII группа периодической системы: фтор, хлор, бром, под 604
Б. БОРИДЫ
§ 206. I группа периодической системы: литий, натрий, калий, рубидий,
цезий 605
§ 207. II группа периодической системы: бериллий, магний, кальций,
стронций, барий, радий 605
§ 208. III* группа периодической системы: скандий, иттрий, лантан . . . 605
§209. III** группа периодической системы 605
§ 210. IV* группа периодической системы: титан, цирконий, гафний, торий. 605
§211. V* группа периодической системы: панадин, ниобий, тантал .... 605
§ 212. VI* группа периодической системы: хром, молибден, вольфрам, уран 605
§ 213. VII** группа периодической системы: марганец, рений 606
§ 214. VIII* группа периодической системы: железо, рутений, осмий . . . 606
§ 215. IX группа периодической системы: кобальт, родий, иридий .... 609
§ 216. X группа периодической системы: никель, палладий, платина . . . 609
§ 217. I* группа периодической системы: медь, серебро, золото 610
§ 218. II* группа периодической системы: цинк, кадмий, ртуть 610
5 219. III группа периодической системы: бор, алюминии, галлий, индий,
таллий 610
8
ОГЛАВЛЕНИЕ
§ 220. IV группа периодической системы: углерод, кремний, германий, олово,
еиппец СИ
§ 221. V группа периодической системы: азот, фосфор, мьпиьнк, сурьма,
нисмут 011
§ 222. VI "группа периодической системы: кислород, сера, селен, теллур,
полонии 011
§ 22:}. VII группа периодической системы: фтор, хлор, бром, иод 011
Глина IX. Структуры сложных соединений 012
§ 224. Внеденпе 012
А. СТРУКТУРЫ СЛОЖНЫХ СОЕДИНЕНИЯ
a) В е с к о и е ч н ы е т р ё х м е р и ы е с т р у к т у р ы
§ 225. Галогенпды 010
§ 22»). Цианиды 018
§ 227. Оксигалогеыиды 621
§ 228. Окислы ЛЛВ<>2 021
§229. Окислы ЛпВО, 022
§230. Окислы А„В04: АШ)4. AaBOi, ,\8HOi ( = Мм,ХО ,) . . . ." 024
§231. Окислы АшВдО<, 035
§232. Сульфиды AXBS„, Л1Ю2-=А3В2Й4; A2BS3, A2HS,f A3BS,, ABS .... 035
§ 233. Нитриды и другие сложные соединен ил 041
б) Г) е с к о и е ч п ы е д и у м е р и ы е с т р у к т у р ы (сиш, сетки)
§ 234. PbFCl, Pl)FBr 642
л) В е с к о н е ч н ы е о д н о м:е р и ы е с т р у к т у р ы (цепи)
§ 23Г>. CoCU»2ll20 и др СМ
1) М о ;i е к у л }i р н ы е р е ш ё т к п
§ 23(). Общие замечании. Карбоннлы металлом 040
§ 237. Металлоорганическпе соединении 050
§238. COS, П3В03, Hg(ClBr)a 055
§ 239. Крпсталлосольнаты 058
В. СТРУКТУРЫ С.ЮЖПОКОМПЛЕКСПЫХ СОЕДИНЕНИИ
§ 240. Структуры с бесконечным трёхмерным ионом СаТН)3 перовскит и др. 05'J
§ 24 I. Структуры г. бесконечным диумерным ионом (слои, сетки) 003.
§ 242. Структуры с бесконечным одномерным ионом (цепи) 000
I л а и а X. Структуры комплексных соединений 00D
§ 243. Внеденпе 00<)
А. ПО/1ПСОЕДИПЕ1ШН
§244. Иолпсоедпиепин Ая[И„ |, AsIBB/z] п близкие к ним лещестиа ОСУ
§245. Полнгалогеппды Ая|На1,], А.т[НаГ, ТтЫ'/,] 070
§240. Перекиси МО, и М,,02 072
§ 247. Полисульфпды, полиселен иды, полителлурпды 07'i
§ 248. Полппптрпды (азиды MN3) 070
§249. Полифосфиды, нолпарсенпды и т. и 081
§ 250. Сульфоарсенпды и т. и 08'f
§251. Поликарбпды, тюлпенлнцпды . .' 085
§ 252. Цианиды, цианамиды 0U1
§ 253. Полпборпды 0У5
£ 2">4. Полиметалл иды 0%
В. СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ КАТИОНОМ
§ 255. Гидраты и аммиакаты солей щелочных и щёлочпо-земольпых мсталлон 0)7
§ 250. Гидраты солей хрома 700
§ 257. Аммиакаты солей марганца 702
§ 258. Аммиакаты солеи железа 702
§ 259. Аммиакаты и другие соли кобальта 703
§ 260. Аммиакаты солей никелн, палладии, платины 70'f
§201. Аммиакат]»! солей цинка и кадмия . . . . , 707
§ 202. Гидраты солен алюминии 707
§203. Соли аммония и фосфонин 70'.>
ОГЛАВЛЕНИЕ
»
В. СОЕДИНЕНИИ С КОМПЛЕКСНЫМ АНИОНОМ
£ 264. II группа периодической системы: бериллий, магний, кальций,
стронций, барий 712
§205. IV* группа периодической системы: титан, цирконий, гафний, торий 712
§ 206. V* группа периодической системы: нападий, ниобий, тантал. ... 714
§ 207. VI* группа периодической системы: хром, молибден, иольфрам, уран
(и уралиды) 715
§ 208. VII* группа периодической системы: марганец, рений 726
§ 20!). VIII* группа периодической системы: желе:ю, рутений, осмий . . . 72(>
§ 270. IX* группа периодической системы: кобальт, родий, иридий .... 732
§271. X* группа периодической системы: никель, палладий, платина. . . 737
§ 272. I* группа периодической системы: медь, серебро, золото 742
§ 273. II* группа периодической системы: цинк, кадмий, ртуть 747
§ 271. III* группа периодической системы: бор, алюминий, галлий, индии,
таллий 748
§ 275. IV группа периодической системы: углерод, кремний, германий,
ололо, сиинец 75!*
§ 270. V группа периодической системы: а:ют, фосфор, мышьяк*, сурьма,
лпсм\т 7GI
§ 277. VI группа периодической системы: кис/юрод, сера, селен, теллур,
Полоний 770
§ 278. VII группа периодической системы: фтор, хлор, бром, под 78с>
§ 279. Некоторые общие замечании к главам IV—-X 803
Ч А СТЬ ТРЕТЬЯ
РЕГИСТР ИССЛЕДОВАНИЙ СТРУКТУР НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
§ 280. Ваедеппе , . . 800
Г л а и а XI. Регистр исследовании структур неорганических нещеетв *) .... 817
§281, Актиний Ас ' 931, S17
§282. Серебро Ag 931, 8*7
§283. Алюминий А1 931, 82)
§ 284. Аргон Аг — 830
§ 285. Мышьяк As 932, 830
§ 286. Полото Ли 933, 833
§287. Нор В 933, 1135.
§288. Парий На 934, 837
§280. Бериллий Be 9,34, 840
§ 290.. Висмут Ш 931, 842
§201. Пром Вг — 844
§292. Углерод С 93t, 8-Vi
§ 203. Кальцин Са 931, 847
§204. Кадмий СА 93о, 853
§ 29Г|. Церий Се 93'>, 855
§200. Хлор (11 — 85ft
§207. Кобальт Со .9-3-5, 857
§ 208. Хром Сг 930, 8(И
§299. Цезий Cs 93(>, 863
Й 300. Медь Си 930, 865
§301. Фтор V — 870
« 302. УКелезо Fo . . . . . 930, 870
§ 303. Галлий Ga 937, 87Г>
§ 304. Германий Ge 937, 877
§ 305. Водород 1Г>, дейтерий J) — 877
$306. Гелий Не — 877
§ 307. Гафний ИГ — 877
§ 308. Ртуть Ilg 937, 878
§ 309. Индии hi 937, 880
§310. Иридий 1г 937, 880
§311. Иод .1 937, 881
§312. Калий К 938, 881
§:ПЗ. Криптон Кг — 885>
$314. Лантан Га . . 93S, 88.',
§315. Литий Тл .... !Ш, 88G
*) Для удобства читателей и содержании гл. XI куренном даны номера
страниц дополнения к регистру (гл. XII).
10
ОГЛАВЛЕНИЕ
§316. Магний Mg 938, 888
$317. Марганец Ми 938, 892
§ 318. Молибден Мо 938, 894
§ 319. Азот N2 (и аммоний (NH4)=*группа) 938, 896
$320. Натрий Na 939, 897
8 321. Ниобий Nb 939, 902
$322. Неон Ne — 902
$323. Никель Ni 939, 902
$ 324. Кислород О. — 905
$ 325. Осмий Os — 906
$326. Фосфор Р 939, 906
$327. Свинец РЬ 939, 907
$328. Палладий Pd 940, 909
$ 329. Полоний Ро — 910
§330. Платина 14 940, 910
$331. Радий Ra — 911
§332. Рубидий Rb 940, 911
$333. Рений Re 940, 912
$334. Родий Rh 940, 913
$ 335. Радон Rn — 913
$336. Рутений Ru — 913
« 337. Редкие земли 940, 913
$ 338. Сера S — 9l(i
$ 339. Сурьма Sb 940, 916
$340. Скандий Sc 940, 917
$341. Селен So 940, 917
$342. Кремний Si 941, 918
$343. Олово Sn 941, 919
$344. Стронций Sr 941, 920
$ 345. Тантал Та 941, 921
$ 346. Теллур Те 941, 922
$ 347. Тории Th — 922
$348. Титан Ti 941, 9П
$349. Таллин Т1 941, 924
$350. Уран U (ураниды) 941, 924
$351. Ванадии V 942, 926
$ 352. Вольфрам VV 942, 927
$ 353. Ксенон Хе — 928
$ 354. Иттрий Y 942, 928
$ 355. Цинк Zn 942, 928
$356. Цирконий Zr 942, 929
$ 357. Разные соединения. Общие формулы — 930
Глава XII. Дополнение к регистру исследований структур неорганических
иещестн *) 931
Именной указатель 943
Предметный указатель 946
Указатель формул веществ, упоминаемых в I и II частях 951
') См. примечание к гл. XI.
ПРЕДИСЛОВИЕ
При создании этой книги автор ставил себе целью способствовать,
по возможности, решению научных задач теории структур, а также
сделать современные структурные данные достоянием широких кругов
химиков, в особенности молодых исследователей—будущих продолжателей
возникающей в настоящее время новой химии. Книга затрагивает
теоретические представления и экспериментальные данные ряда дисциплин —
кристаллографии, кристаллохимии, неорганической химии, физики
твёрдого тела, рентгенографического и электронографического структурного
анализа, теории химической связи и т. д. Создание такой книги
представляет задачу большой трудности, в чём автор в течение многих лет работы
над рукописью полностью отдавал себе отчёт.
Эти трудности усугублялись во много раз намерением автора
подвергнуть коренному пересмотру самый принцип систематики структур, широко
распространённый в мировой литературе, а также свести воедино
разрозненные сведения о многочисленных работах по изучению структур
неорганических веществ и составить, повидимому первый в литературе, регистр
исследований подобного рода.
R самом деле, излагая в этой книге фактический структурный материал,
автор не мог мириться с тем обстоятельством, что в основе международной
«систематики)/ структур Strukturbericht (SB) лежит формалистическая
концепция группировки структур по соотношению количеств разнородных
атомов в формуле вещества.
Эта концепция помогает лить воду на мельницу теории идеального
кристалла и особенно затрудняет использование структурных данных для
интересующей нас цели—трактовки проблем современной неорганической
химии.
Отвергнув эту концепцию как непригодную для нас, мы положили
в основу систематики, принятой нами в данной книге, важнейший закон
химии —периодический закон Д. И. Менделеева.
Далее, при исследовании структур мы исходили не только из
соотношений количеств атомов, но и из представлений о возможном составе
структурных узлов. При создании систематики сложных структур мы
учитывали также роль не только структурных, но и, по возможности,
термодинамических факторов. Это повлекло за собой иное, чем в SB,
расположение и сопоставление материала, иную подачу его читателю.
Наконец, мы отказались и от характеристики типа структур,
принятой в SB, прибегающей к обозначению типов с помощью последовательности
букв алфавита и натурального ряда чисел. Такая характеристика
полностью затушёвывает физический смысл отвечающей ей структуры. Нами
введена система, основанная на указании: си агонии, к которой принадлежит
вид симметрии кристалла, типа решётки и формы координационных сфер
вокруг отдельных атомов и структурных узлов. Такая характеристика
облегчит освещение структурного материала с позиций современной теории
12
ПРЕДИСЛОВИЕ
валентности и сможет, вероятно, в свою очередь, способствовать развитию
последней.
Поскольку вся литература в области структурного анализа использует
обозначения типов по SB, в данной книге сопоставлены обе системы: наша
(по структурно-координационному принципу) и SB. При этом мы не могли
пройти мимо того обстоятельства, что в SB номенклатура типов от тома
к тому менялась и одна и та же структура обозначается в разных
томах по-разному. В § 107 нами сделано отсутствующее в SB
сопоставление типов, к которым причисляются одни и те же структуры в то .мах
1—VII SB.
Подобный пересмотр представляет собой чрезвычайно сложную научную
задачу, без решения которой, мы уверены, неорганическая химия не может
занять современных позиций. Мы убеждёны, что эта задача вполне но плечу
только советской науке.
Автор будет удовлетворён, если его многолетний труд сможет хотя бы
в некоторой мере способствовать достижению названной цели.
П е р и а я часть к н и г и является очень сжатым изложением основ
теории структур и состоит из двух глав. Первая из них посвящена
кристаллографической теории симметрии, вторая — проблемам кристаллохимии и
природы химической связи применительно к строению главным образом твёрдого
тела. Принципиальная сторона вопроса о таком освещении основ теории
структур изложена в § 1 (Введение). Оно вытекает из несогласия автора с теми
кристаллографическими школами, которые считают возможным излагать
теорию структур только с позиций теории симметрии и с теми физико-
химическими школами, которые подходят к решению этой задачи,
пренебрегая достижениями теории симметрии и опираясь только }ia схемы общей
теории химической связи. Успех дела зависит от творческого синтеза обоих
направлений, в каждом из которых отечественная наука имеет выдающиеся
достижения.
Выбор и изложение материала главы I были подчинены и второму
требованию — максимально учитывать специфику подготовки и склонности
широких кругов химиков, с тем чтобы предлагаемый их вниманию
трудный материал был усвоен с наименьшей затратой сил. В частности,
например, излагая основы теории симметрии, автор имел в виду наличие
значительных разногласий между отдельными школами кристаллографов,
а также разных и противоречивых определений понятий. Учитывая интересы
и подготовку читателя-химика, автор пытался отразить отдельные различия
в освещении проблемы, а также в терминологии, а само изложение
приспособить к образу мыслей и подготовке читателя-химика, и, следовательно,
был вынужден выбирать между строгостью выводов и доступностью
материала. Наконец, пришлось преодолеть большую трудность изложения
обширного и довольно сложного материала всего на нескольких десятках
страниц.
Глава II написана иод несколько иным углом зрения. Основы
кристаллохимии обычно хорошо известны химикам. Слабым местом этой области
пауки в её современном состоянии является некритическое применение
ряда представлений, развитых геометрической теорией кристалла,
примитивных электростатических теорий, примитивных теорий ковалептной
связи. Иногда это вызвано неразумным и вредным преклонением перед
«авторитетом» Магнуса, Гольдшмидта, Паулинга, Сиджвика и других
зарубежных исследователей. Между тем многие представления и труды
ПРЕДИСЛОВИЕ
13
этих авторов безусловно ошибочны или содержат ряд существенных
недостатков. ...
В частности, распространённая у нас книга Л. Паулинга «Природа
химической связи» (1945) (русский перевод, 1918) замалчивает иные пути
развития квантово-химичеекой теории структур (теория молекулярных орбит,
зонная теория твёрдого тела и др.), кроме тех, которые вытекают из
взглядов самого Паулинга. Более того, книга Паулинга в отдельных главах
беснритщишю пропагандирует примитивные электростатические модели
комплексных ионов и координационных структур и тем самым неправильно
ориентирует читателей в этом важном вопросе. Серьёзные ошибки были
допущены в развитой: Паулингом, Уэлаидом, Сыркипым., Днткииой и др..
теории квантово-химичоского резонанса. Хотя критическому рассмотрению
этих представлений посвящена специальная паша монография «Основы
теории структур», закапчиваемая в настоящее время, однако и в данной
книге мы вкратце касаемся некоторых слабых мест этих теорий и широко
распространившихся в литературе ошибочных взглядов особенно
зарубежных школ кристаллохимиков.
С другой стороны, мы считали необходимым коснуться различных
возможных подходов к освещению рассматриваемых проблем, с тем чтобы
привлечь к ним внимание читателя, и в частности тех, которые могут быть
основаны на общих закономерностях, не противоречащих квантовой химии,
по не сформулированных ею. Некоторые из них были выдвинуты автором
if опубликованы в советской литературе значительно ранее, чем к ним
пришли зарубежные исследователи.
При создании второй части книги, посвященной рассмотрении)
важнейших типов структур, мы положили в основу работы те принципы, о
которых говорилось в начале предисловии. Повторять их пет нужды. Здесь
надлежит отметить лишь, что громадной работы в течение нескольких
лет потребовала подготовка большого и сложного графического материала.
Дело в том, что редакции SB не придерживалась, повидимому, той точки
зрения, что для столь ответственного издания обязательна хотя бы
некоторая унификация обозначений па чертежах и рисунках структур. В
результате на одних чертежах металлы обозначены так, как на других
неметаллы. Сами способы обозначения —величина кружков, их штриховка
—произвольно меняются от чертежа к чертежу. Более того, методы изображении
разных структур совершенно различны и сами изображении недостаточно
наглядны. Кратчайшие расстояния между атомами —«связи»—показаны
•сплошь и рядом линиями одинаковой толщины, и па сложных чертежах
бывает трудно установить, какой атом, находится ближе, какой дальше
и как они расположены по отношению друг к другу.
Придя к выводу о целесообразности перестройки большой части
графического материала SB, автор считал необходимым с фотографической
точностью сохранить позиции атомов, отвечающие чертежам SB. Па базе
фотоснимков этих чертежей строились новые чертежи структур: обозначения
атомов и т. п., по возможности, регламентировались по величине и
штриховке, и равномерные линии заменялись «киеобразными»; принимались
и другие меры, чтобы усилить впечатление перспективы. В ряде случаев
пришлось создать по нескольку вариантов чертежей структур, прежде чем
отобрать лучшие. Многие чертежи структур были вообще построены
заново.
Автор счёл нужным дать рисунки и чертежи структур достаточно
крупным масштабом. В ;пом ощущалась особая необходимость, поскольку
приведённые в большинстве курсов и монографий изображения структур
неудобочитаемы ввиду малых масштабов, а иногда и плохого качества
клише.
14 предисловие
Часть третья «Регистр исследований структур неорганических
ве^цеств» является результатом ответственной и чрезвычайно трудоёмкой
работы автора, о содержании которой подробно говорится в вводном
параграфе к III части. Регистр охватывает свыше 25 000 определений (ссылок),
относящихся более чем к 5100 неорганических веществ (наименований).
Автор весьма признателен официальным рецензентам книги—члену-
йорреспонденту Академии наук СССР И. А. Казарновскому и
профессору Г. С. Жданову, а также члену-корреспонденту Академии наук СССР
Й. В. Белову за сделанные ими замечания.
Автор с искренней благодарностью отмечает помощь, оказанную ему при
Оформлении рукописи научным сотрудником Института им. Л. Я. Карш ка
В. И. Смирновой,
Москва 1949 г.
ЧАСТЬ ПЕРВАЯ
ОСНОВЫ ТЕОРИИ СТРУКТУР
ГЛАВА I
ЭЛЕМЕНТЫ ТЕОРИИ СИММЕТРИИ
§ 1. Введение
В 1946 г. химия переступила две юбилейные даты: 30 лет со дня
создания первых современных теорий химической связи и 20 лет со дня создания
первой квантово-химической теории химической связи.
Это были блестящие десятилетия научного прогресса, сопутствовавшие
великому штурму атомного ядра, который сделает человечество
властелином сказочных богатств—внутриатомной энергии.
Стремительный поток новых фактов, гипотез, теорий захлестнул старую
науку, которая чуть ли не на пороге этого периода обсуждала «волнующую»
проблему —существуют или не существуют ионы, глубоко переживала
возникновение теории квантов и теории относительности, постулировала
«исчезновение» материи.
Вместо того чтобы усиленно работать над освоением новых открытий,
над созданием новых теорий, многие физические школы отдали дань
растерянности, философской отсталости и реакционным тенденциям и взглядам,
столь глубоко и всесторонне вскрытым в замечательном труде В. И. Ленина
«Материализм и эмпириокритицизм».
Смежные дисциплины—неорганическая и общая химия,
кристаллография, минералогии и даже физическая химия—значительно
запаздывают с освоением нового материала, каждая в разной мере и по разным
причинам.
В наиболее близкой автору неорганической химии ещё в 1932—1933 гг.,
т. е. в середине рассматриваемого периода, пользовались учебниками, либо
умалчивавшими о новых теориях и фактах, либо излагавшими их в
вводных главах без связи с остальным материалом.
Нередко научно-исследовательские школы в области неорганической
химии —и у то в первую очередь относится ко многим представителям
зарубежной науки—вместо творческого освоения новых теоретических данных
с целью поднятия этой дисциплины на более высокий уровень скатывались
на позиции грубого эмпиризма, довольствуясь в ряде случаев ценными
экспериментальными работами, которые сделались возможными при новом
уровне техники эксперимента.
За истекшие с того времени 16 лет многие, тогда новые идеи
стали достоянием курсов химии и научных работ. Однако наука о строении
вещества и структурах веществ тоже не стояла на месте. Она шла
вперёд и притом быстрее, чем смежные дисциплины, и, в частности,
неорганическая химия, осваивавшие сё замечательные открытия. В
настоящее время разрыв становится вопиющим. Он не может быть более
терпим.
Сказанное выше можно подтвердить многочисленными примерами.
Ограничимся ссылкой на распространённый в США, выдержавший ряд
18 ч. i. основы теории структур
изданий учебник Снида и Майнарда, являющийся образцом беспринципной
мешанины, подновлённой «сенсационными» сведениями об отдельных
новейших открытиях.
В чём же заключается острота сложившейся ситуации?
1. Основная сложность заключается в том, что в настоящее время
основы теории структур развиваются почти независимо друг от друга
в области кристаллографической теории симметрии и в области общей
теории химической связи, причём выпускаются курсы и монографии, в
которых одна из этих точек зрения доминирует, а другая часто даже не
упоминается. В качестве примера/ и притом не самого яркого, приведём
вышедшую за рубежом книгу Э. Бранденбергера (1947) «Основы химии
материалов», в которой химии, собственно говоря, вообще нет, а речь идёт
о структурах, применяющихся в технике материалов, и этот вопрос
рассматривается только с геометрических позиций. Полнейшее пренебрежение
ценными данными современной физики и химии ведёт к однобокости текста.
В настоящее время особенно остро чувствуется необходимость
творческого синтеза обоих направлений с целью создания общей теории
структур.
2. Достижения и характер развития науки о строении вещества
всесторонне подтверждают глубокий смысл замечания Энгельса на полых
рукописи «Диалектика природы»: «Кристаллография —часть химии» (как бы
предрешавшего необходимость творческого использования в химии ряда
идей кристаллографии).
Однако современные монографии и курсы неорганической и общей
химии совершенно недостаточно используют сведения о структурах веществ,
а необходимые для понимания этого материала кристаллографические
данные излагаются зачастую даже в лучших учебниках неорганической химии
самым неудачным образом*).
Неорганическая химия совершенно недостаточно использует
результаты структурных отечественных и зарубежных исследований, в частности,
собранных в Strukturhericht (SB), фундаментальном семитомном справочнике
структур и в «Интернациональных таблицах для исследования структур
кристаллов» (IT). Этому препятствует, во-первых, то обстоятельство, что
полный комплект SB имеется лишь в немногих библиотеках, а также и то, что
SB перегружен множеством данных, важных для рентгенографа, но менее
существенных для химика и физика.
Наконец, главным препятствием для использования структурных
данных в химии является принятая в SB чуждая химикам формалистическая
система классификации и нумерации структур по соотношению атомных
концентраций. В силу этого химически столь различные вещества, как FeS2
и СО2, попадают в один и тот же тип, в данном примере С2.
С другой стороны, нельзя уклониться и от того факта, что для
понимания обширной современной литературы в области структур веществ
требуются достаточные знания теории симметрии дисконтинуума, в
большинстве случаев, к сожалению, лежащие вне круга научных интересов
даже ведущих исследователей и педагогов —химиков и физиков.
Пополнение химиками сведений в области кристаллографии
затрудняется, на наш взгляд, в частности, тем, что современные курсы
кристаллографии для химиков не содержат многих важнейших данных в области
теории симметрии, что делает их непригодными для интересующей нас
*) См., например, критическую статью автора «Кристаллография — часть химии»
в «Вестнике Высшей школы», 1947, вып. 3 и 7.
|гл 1
§ 1]
ВВЕДЕНИЕ
19
цели. Полные же курсы кристаллографии излагают соответствующие
вопросы в расчёте на читателя, специально совершенствующегося в
кристаллографии, включают многие дополнительные положения и выводы, без
которых химик мог бы обойтись, а важные с точки зрения интересующей
нас цели сведения даются столь сухо, что во многих случаях—можно не
сомневаться—материал вряд ли будет усвоен читателем-химиком.
Кроме того, русские курсы кристаллографии исключают, как
устаревшие, сведения и термины, отвечающие гемиэдрическим формам. Между
тем IT пользуются именно этой терминологией. В результате читатель, физик
или химик, должен перерыть немало курсов кристаллографии и потерять
значительное время, чтобы понять содержание даже таких заголовков
из IT, как «Пространственные группы моноклинной гемиморфной геми-
эдрии» (IT, том J, стр. 97), «Пространственные группы ромбической эиантио-
морфной гемиэдрии» (IT, том 1, стр. 118) и т. п., не говоря уже о
графической и числовой символике теории симметрии дисконтинуума, применяемой
в публикациях по структурным исследованиям, а также в SB и IT.
Наконец (впрочем, эта наша точка зрения, может быть, является
дискуссионной), мы считаем, что параллельное изложение (в ряде курсов
кристаллографии) теории симметрии континуума и дисконтинуума недостаточно
подчёркивает особенности дисконтинуума и затрудняет химикам понимание
специфических свойств последнего.
В результате, неорганическая химия, опираясь на старую трактогку
периодического закона Д. И. Менделеева, уходит от нового содержания,
которое должно быть вложено в этот величайший закон естествознания
в свете новейших достижений науки, и тем самым напрасно создаёт
«противоречия» между фактами и теорией, легко устранимые при правильной
трактовке. Неорганическая химия уверяет читателя, что существуют такие
соединения, как V2S5, тогда как рентгеновский анализ свидетельствует,
что формулы такого типа для подобных веществ являются мифическими.
Курсы неорганической химии по существу скрывают от читателя
существование соединений, подобных Си9А14 или V^N,^, и возможность любых дроб
лых соотношений, например в атомных концентрациях Ti и О в ряду
TiO—ТЮз п тысячах аналогичных веществ.
Положение в этом вопросе таково, что, как мы указывали недавно
(см. ниже, § 104), даже такие фундаментальные законы, как законы
постоянства состава и химических эквивалентов, безусловно нуждаются в иных
формулировках.
Нельзя не подчеркнуть в связи с этим и тот факт, что в
современных курсах неорганической химии, к сожалению, уделяется
недостаточное внимание созданному замечательным русским химиком Н. С. Кур-
наковым учению о бертоллидах. Если оно и излагается в курсах
неорганической химии, то не как важнейший принципиальный вопрос химки,
а в большинстве случаев не излагается вовсе (см. § 104).
Проблема механизма химической реакции часто трактуется в
неорганической химии оторванно от структур веществ, а проблема структур
веществ—независимо от влияния примесей и условий реакции (§ 103).
Правило фаз оперирует представлениями об однородной структуре
идеальных фаз, не считаясь с особенностями реальных твёрдых фаз
(см. §§ 101, 102).
Весьма печально, что даже лучшие новейшие монографии и обзорные
статьи по неорганической химии часто являются скорее мемуарами в
области давно потерявших остроту проблем, чем боевыми программами и
трактатами современной науки, пускай в отдельных вопросах спорными.
Как общий вывод, следует сказать, что современная неорганическая
химия не может более оставаться лишь на позициях химической термоди-
20
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
иамики и кинетической теории материи: она должна также стать подлинно
структурной неорганической химией.
3. Современная кристаллохимия также развивается весьма однобоко,
опираясь главным образом на электростатическую и стереометрическую
теории твёрдого тела, которые мы считаем относящимися к переломному
периоду кристаллохимии (см. §§ 75—84). Современные курсы
кристаллохимии, а также обзорные статьи недостаточно излагают выводы квантовой
химии и особенно теории реального кристалла, но зато подробно
останавливаются на геометрической и электростатической теориях, как правило,
недостаточных для трактовки структур реальных твёрдых фаз. Более того,
•стремясь представить структуру как плотную упаковку «атомов-шаров%
кристаллохимики часто не учитывают в сколько-нибудь достаточной мере
влияния химических, кинетических, термодинамических факторов, влияния
примесей, условия роста и т. п. С таким положением никак нельзя
далее мириться.
4. Наконец, постановка работ в области структурного фазового
анализа (рентгенографического и электронографического), с точки зрения
химика, страдает досадными промахами и недостатками.
Отдельные группы соединений изучены довольно подробно,
другие—незаслуженно — весьма плохо. Так, например, при наличии детальных сведений
о структуре многих сложных силикатов (этим мы обязаны
заинтересованности школ Белова, Брегга и др.)> структуры ряда простых веществ и
соединений, например, галогеиидов и окислов элементов, в состоянии не
максимальной валентности (ReCl5, OsF4, VC14, SC14 и т. д. и т. п.) изучены весьма
плохо. Это не позволяет сопоставить ряд принципиальных теоретических
взглядов различных исследователей, в том числе автора книги (на
необходимость таких экспериментальных работ автор указывал уже много лет назад,
см., например, ЖФХ XI, 274, 1938 и др.).
Нигде не опубликованы общие сводки работ но структурам веществ,
что крайне затрудняет ориентировку в уже имеющемся материале. Даже
в справочнике SB нет общей сводки типов структур и общего формульного
регистра по 7 томам. Громадное количество работ посвящено материалам, в
отношении которых сведения о химическом анализе и наличии примесей
отсутствуют, что значительно снижает ценность полученных результатов, а иногда
приводит даже выдающихся учёных к грубым ошибкам (см. стр. 750). Лишь
в последнее время в этом вопросе наметился некоторый перелом. Как мы
указывали выше, игнорирование основных положений физической химии
влечёт за собой ошибочную трактовку важнейших вопросов даже в таких
трудах, как SB.
Таким образом, даже в вопросе о систематике структур положение
оставляет желать лучшего.
Необходимость существенных изменений в основных предпосылках
и формулировках ряда дисциплин, о которых мы говорили выше,—очень
сложная задача, для решения которой требуется объединение коллективных
усилий многих исследователей.
Решение таких задач по плечу прежде всего отечественной науке,
вклад которой в рассматриваемую область знания исключительно велик.
§ 2. О роли отечественной науки в создании современной теории структур
Здесь мы имеем возможность лишь коротко остановиться на вопросе
о выдающейся роли отечественной науки в создании современной теории
структур.
Отечественной науке принадлежит высокая честь создания основ теории
симметрии. Назовём выдающиеся труды математика Эйлера (XV111 в.),
§ 2] О РОЛИ ОТЕЧЕСТВЕННОЙ! НАУКИ И СОЗДАНИИ ТЕОРИИ СТРУКТУР 21
работы А. В. Гадолипа по выводу 32 видов симметрии и позже блестящий
труд Е. С. Фёдорова по выводу 230 пространственных групп.
Нельзя не подчеркнуть, что в основном применяемые в IT
графические изображения 230 пространственных групп симметрии и др.
представляют собой воспроизведение (и притом с бесцеремонным отсутствием
ссылок на первоисточник) чертежей, опубликованных Е. С. Фёдоровым
в 1895 году. Таким образом, автором всех важнейших диаграмм IT
с относящейся к ним -характеристикой, составляющих главную ценность
для структурных исследовании (среди прочего материала IT), является
наш великий соотечественник. (См. также Н. В. Белов [13].)
Проблемам теории симметрии посвятили ряд глубоких исследований
Л. К. Болдырев, Г. В. Вульф, Б. Н. Делоне, А. В. Шубников и другие.
Е. С. Фёдорову принадлежит фундаментальная работа «Царство
кристаллов», в которой было сделано обобщение огромного экспериментального
кристаллографического материала. Им же были заложены основы кристал-
лохимического анализа и создан основной метод кристаллооптического
анализа, известный пауке под названием фёдоровского метода. Этот метод
лежит в основе многих исследований в области кристаллографии,
минералогии, петрографии.
Позже он был дополнен А. II. Заварпцким. В последние годы в области
кристаллооптического и кристаллохимического анализа ряд ценных работ
выполнен Г. Б. Бокием.
Фёдоровым была создана плодотворная теория атомных полиэдров,
нашедшая в паше время развитие в двух направлениях: а) в теории
координационных полиэдров, воплощённой в очень интересных работах
советского учёного Н. В. Белова и в ряде зарубежных исследований и б) в
к на пто во-химической теории атомных полиэдров.
Проблеме образования структур из координационных полиэдров
посвящен ряд исследований Л. В. Белова, нашедших своё обобщение
в его монографии «Структуры ионных кристаллов и металлических фаз».
В этой монографии показана, в частности, возможность существовании
ряда новых форм координационных полиэдров. Второе направление
представлено в работах Зейца («атомные полиэдры»), С. Т. Копобсевского и др.
Исключительное значение для понимания атомных сеток, образующих
грани кристалла, имеет закон Г. В. Вульфа, вложивший физический смысл
в неточный закон Браво и давший физическое обоснование закону
постоянства углов кристалла.
Г. Б. Вульфу принадлежит честь вывода, одновременно и независимо
от Брегга, основного уравнения, связывающего межплоскостиые
расстоянии в кристалле с. длиной волны и углом отражения рентгеновых лучей —
уравнения, лежащего в основе структурной рентгенографии и
электронографии (ii\ = 2dhklbU\ Ь).
Сыгравший столь большую роль в описании симметрии внешней формы
и структуры кристаллов метод стереографической проекции с применением
градусных сеток" особенно обязан своим развитием Г. В. Вульфу и А. К.
Болдыреву.
Основоположником структурной теории в химии был А. М. Бутлеров.
Его выдающиеся работы дали сильнейший толчок развитию теоретической
химии и, в частности, позволили создать современную органическую
химию.
Современная теория строения молекул и химической связи в
молекулах весьма успешно разрабатывалась в трудах наших спектроскопистов
А. Н. Тсреиина, Г. С. Ландеберга, В. II. Кондратьева и др.
Современная физическая теория твёрдого тела создавалась под
непосредственным воздействием трудов советских учёных А. Ф. Иоффе,
22
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл.1
С. Т. Коиобеевского, В. Д. Кузнецова, П. А. Ребиндера, И. Е. Тамма,
В. А. Фока, Я. И. Френкеля, причём некоторые из названных
исследователей дали толчок также работам в области физики реальных
твёрдых фаз.
Проблемы энергии кристаллической решётки нашли развитие в
работах А. Ф. Капустииского, а приложения энергетики кристаллов в
геохимии—в трудах блестящего советского исследователя А. Е. Ферсмана,
разделившего с В. И. Вернадским честь создания геохимии.
Принцип М. В. Ломоносова является руководящей идеей химии
реальных твёрдых фаз.
Систематический ряд исследований в области химии (и отчасти физики)
реальных твёрдых фаз был выполнен в лабораториях С. о. Рогннского
и автора книги.
Периодический закон Д. II. Менделеева является основным началом в деле
построения современной химической систематики структур. Данная книга
и представляет собой попытку его приложения в рассматриваемой области.
Отечественная школа исследователей явилась основоположником
современного физико-химического анализа. Н. С. Курнаков, Г. Г. Уразов
ir их сотрудники заложили основы химии фазовых переходов, играющей
столь большую роль в современной теории структур.
Физика фазовых переходов нашла развитие в трудах Л. Д. Ландау
л В. К. Семенченко.
Спектроскопические исследования твёрдого тела, и ро ведённые
Л. И. Мандельштамом и Г. С. Ландсбергом, привели к открытию эффекта
комбинационного рассеяния света (эффект Ландсберга—Мандельштама —
Рамаиа), столь важного для многих выводов о природе химической связи
и к ряду работ в этой области (Г. С. Лаидсберг, Я. К. Сыркин, А. И.
Бродский и др.).
Структурные рентгенографические исследования большой
сложности и экспериментального совершенства выполнены в лабораториях
П. В. Агеева, Н. В. Белова, Г. Б. Бокия, Г. С. Жданова, А. И.
Китайгородского, 3. Г. Пинскера, Я. С. Уманского и др.
А. К. Болдыреву и его сотрудникам принадлежит честь
опубликования первого в мировой литературе рентгенографического определителя
веществ (1938 vX
В постановке структурных исследований неорганических веществ
большую инициативу проявил И. А. Казарновский. В результате был
расшифрован ряд структур гидридов и высших окислов. При этом И. А.
Казарновскому и его сотрудникам удалось открыть новый ряд кислородных
соединений — озонидов.
В СССР за последние годы получены чрезвычайно важные результаты
также в области приложения общей теории структур к различным
специальным группам веществ. Здесь надо прежде всего назвать исследования
Н. В. Белова и его сотрудников в области строения силикатов,
Г. Б. Бокия и Г. С. Жданова в области структур комплексных соединении,
В. А. Каргина и его сотрудников в трудной области строения
высокомолекулярных веществ, А. 11. Китайгородского в области структур
органических веществ.
Несмотря на исключительно интересные результаты экспериментальных
структурных исследований, осуществлённых в СССР за последние 10 лет,
необходимо отметить, что экспериментальные работы в области
рентгенографического и олектронографического структурного и фазового анализа
в количественном отношении поставлены у нас — относительно — ещё
в недостаточной мере. Мы не сомневаемся, что в ближайшие годы и в этом
направлении будут достигнуты надлежащие сдвиги.
§ /|| ЛГРКГЛТНЫК СОСТОЯНИЯ. ЗАКОН АНИЗОТРОПИИ 23
С другой стороны, при критическом рассмотрении многочисленных,
выполненных за рубежом экспериментальных исследований в области
строения различных фаз (с чем мы столкнулись в процессе создания книги)
бросается в глаза, что многие из этих работ были выполнены с материалами
недостаточной чистоты, часто содержат грубые ошибки и могут быть
использованы лишь с серьёзными оговорками. Более того, даже редакции
таких фундаментальных трудов, как SB, допустили ряд принципиальных
и практических ошибок, что будет нами проиллюстрировано многими
примерами в следующих главах.
Обобщая наш по необходимости беглый и ни в коем случае не
претендующий на полноту обзор, можно сказать, что для отечественной науки
характерны работы над фундаментальными, основными проблемами
рассматриваемой области знания и блестящее решение многих узловых
вопросов классической и современной теории структур.
А. ЭЛЕМЕНТЫ ТЕОРИИ СИММЕТРИИ КОНТИНУУМА
§ 3. Континуум и дисконтинуум
Вещество может рассматриваться в одно и то же время и как
непрерывная среда—континуум и как прерывная—-дисконтинуум. Среда
представляется нам непрерывной при обычном визуальном или
микроскопическом изучении. Она оказывается прерывной при увеличении в 107—108 раз,
когда мы говорим о положениях отдельных атомов. Столь высокое
увеличение влечёт за собой необходимость учёта новых особенностей среды.
Рассмотрим некоторые особенности континуума.
§ 1. Агрегатные состояния. Закон анизотропии
Вещество может находиться в твёрдом, жидком и газообразном
агрегатных состояниях, различаемых по своим свойствам. В газообразном
состоянии вещество, как учила классическая физика, легко изменяет свой
объём и форму; в жидком состоянии легко изменяет форму, но с трудом
изменяет объём; в твёрдом—с трудом изменяет объём и форму. По этим
признакам более или менее однозначно определяется лишь газообразное
состояние. Твёрдое и жидкое состояния различить подобным образом в ряде
случаев нельзя. Так, «твёрдое» на вид стекло оказывается переохлаждённой
жидкостью (см. §§ 7 и 8).
Рассмотрим в лупу снежинки, медленно кружившиеся в воздухе и
упавшие на ветку дерева. Мы видим правильные звёздочки, состоящие из 6 лучей,
углы между которыми с удивительным постоянством близки к 60°. При
более сильном увеличении видны отдельные плоскогранные призмы
правильной формы. Это—кристаллы льда, твёрдой воды.
^Кристаллом называется твёрдое тело, ограниченное в силу своих
внутренних свойств плоскими поверхностями—гранями»,—писал выдающийся
русский кристаллограф Г. В. Вульф.
В настоящее время мы знаем, что подлинно твёрдое состояние является
кристаллическим. Мы живём, таким образом, в мире кристаллов.
«Многогранная форма кристалла служит лишь одним из выражений
свойств составляющего его вещества, и правильное, исчерпывающее
определение кристалла должно, очевидно, характеризовать именно те внутренние
свойства вещества, которые отличают кристаллизованное вещество от
аморфного*) и внешним выражением которого является многогранная форма
кристалла» (Вульф).
*) 05 аморфном состоянии см. §§ 7 и 8.
«н
4.1. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Изучая твёрдые тела и жидкости, следует различать векторные
(векториальные) и скалярные свойства вещества.
Скалярные свойства для любого агрегатного состояния вещества
не зависят от направления (например, теплоёмкость, скрытая теп.юта
испарения). Свойства веществ в жидком состоянии обычно не зависят от
направления. Ряд свойств вещества в твёрдом состоянии являются вск-
торными, т. е. зависят от
направления (например,
электропроводность, теплопроводность
и т. п.).
Для всех без исключении
твёрдых тел (кристаллов)
справедлив закон анизотропии:
Вещество в твёрдом пн-тон-
пии анизотрошю, т. е.
векторные свойства его в любых
внутренних точках кристалла в
одинаковых (параллельных), а также
в симметричных направлениях
одинаковы, в разных
направлениях различны (рис. 1: а—к\б,
вырезанный из изотропного
материала — стекла, Ь — к рис тал. i
белого олова). Вещество в жидком состоянии статистически изотропно*),
т. е. любые свойства его не зависят от направления.
Рис. 1. Зависимость теплоироводности^от
направления.
Плаилеиис поверхностного слоя парафина нокруг
горячей иглы.
§ 5. Твердое и жидкое состояния в свете химической термодинамики.
Закон Вульфа
Опираясь на чисто термодинамические представления, можно
предложить довольно строгий критерий, позволяющий различить тнёрдое
и жидкое состояния. Вещество в твёрдом состоянии стремится в сил}
своих внутренних свойств приобрести плоскогранную форму, в жидком
состоянии—форму шара.
Это положение можно обосновать следующим образом:
Энергетически наиболее выгодной формой конденсированной (твердой,
жидкой) фазы является такая, свободная поверхностная энергия которой
является наименьшей. Энергией элемента поверхности с/// является
dF=adij
(I)
где z—удельная свооодная поверхностная энергия.
Так как вещество в жидком состоянии изотропно, то поверхностная
энергия различных участков поверхности жидкой фазы одинакова.
Наименьшая работа образования поверхности отвечает той геометрической
форме, поверхность которой при данном объёме является минимальной.
Именно эта форма является для жидкости термодинамически наиболее
устойчивой. Из геометрии известно, что такой формой является шар.
Естественно, что наиболее устойчивой формой вещества и в твёрдом
состоянии является та, свободная поверхностная энергия которой при
одном и том же объёме вещества является наименьшей.
*)Мы говорим -статистически изотропно, ибо и в жидкости возможны мпкроитрук-
турные образования, статистически беспорядочно ориентированные ио отношению др\г
к другу.
Некоторые специальные случаи (например, жидкие кристаллы) мы лдесь но имеем
возможности рассматривать.
/
СТЕКЛОВИДНОЕ II АМОРФНОЕ СОСТОЯНИЯ
25
Но в случае вещества в твёрдом состоянии поверхностная энергия
участков поверхностей, перпендикулярных различным направлениям
(радиусам-векторам), различна. В результате вещество приобретает
плоскогранную кристаллическую форму. Согласно
принципу Гиббса-Кюри, наиболее термодинамически
устойчивой форме отвечают такие грани, для
совокупности которых соблюдается условие: суммарная
свободная поверхностная энергия
)Lzidy = ruin- (2
при г—const, где ^ означает суммирование но всем
граням (г — объём тела).
Искомый минимум свободной поверхностной энергии
кристалла при данном объеме, согласно закону Вульфа
(1895 г.), достигается при том взаимном
расположении граней кристалла, когда они удалены от одной
и той же точки на расстояния, пропорциональные их
удельным свободным поверхностным энергиям.:
д.,
(о) фа пг
Рис. 2. ^акон Вуль-
Этот закон (рис. 2) был многократно доказан также последующими авторами,
в частности Я. 11. Френкелем, М. Лауэ и др.
Закон Вульфа является крупным вкладом русской пауки в дело
создания современной кристаллографии.
§ 6. Монокристалл и поликристалл
Если, например, правильный*) в смысле внутренней структуры
кристалл (идеальный монокристалл) измельчить в порошок или подвергнуть
быстрой кристаллизации расплав того же вещества, то образуется
поликристаллическое вещество, состоящее из многих беспорядочно
ориентированных кристалликов. Каждый такой микрокристалл в отдельности
сохраняет анизотропию и структуру, присущую монокристаллу.
Хотя в иоликристаллттческом веществе векториость физических
свойств при исследовании обычными макрометодами, вследствие
статистической ориентировки кристаллов, часто не выявляется, она всё же может
быть обнаружена специальными методами, например
рентгенографически (метод «порошка»). Это, и частности, отличает поликристаллпческое
вещество от жидкостей.
§ 7. Стекловидное и аморфное состояния
Этот сложный вопрос различными исследователями трактуется
по-разному и подробно будет подвергнут нами критическому
рассмотрению в другом месте. Вкратце нам кажется целесообразным ограничиться
здесь следующими представлениями.
Осторожно п е р е о х л а ж д ё н и ы е ниже точки затвердевания жидкости
приобретают всё большую вязкость и превращаются в
«стекло»—обыкновенное оконное стекло, карамель, столярный клей. В стёклах мы не
обнаруживаем, даже в микроскоп, разрыва сплошности.
Порошкообразные вещества, большинство химических связей в
пылинках которых разорваны химическим или механическим путём и в которых
не обнаруживается кристаллическая структура даже микрометодом, пазы-
*) Как мы увидим ниже (§ 101, 102), рояльные монокристаллы далеко но
иодны от дефектом структуры.
2в
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
р\
Рис. 3. р—Г-диаграмма одиоком-
понентной системы.
ваютси аморфными. Примером таких веществ является сажа. Элементы
структуры исходных твёрдых тел в них в некоторой мере сохраняются,
однако при этом возникает и ряд особенностей, характерных для аморфного
состоянии [11].
0
§ S. Фазовые превращения в слете правила фаз. р—Т-диаграашы
В работах современных авторов часто обнаруживаются неправильные
характеристики упомянутых выше агрегатных состояний*). Нам кажется
поэтом) целесообразным иллюстрировать соотношения между последними
с помощью р—1 -диаграмм, оперируя
известным химику понятием фазы.
Физическая химия относит реакции
превращения друг в друга твёрдых
модификаций вещества, например [А]а;г^[А]р,
к двум различным типам: а) энантиотроп-
пым и б) монотропным превращениям.
В первом случае (смотри р—Г-диаграмму,
рис. 3) существует некоторая темпера-
тУРа *п (температура превращения), ниже
которой термодинамически устойчива одна
модификация (ветвь а), а выше которой —
другая (ветвь 3). В точке Ти свободная
энергия перехода ЛД=0 (рис. 4, а). Во
втором случае (смотри р — Г-диа грамму,
рис. 3) одна из форм во всём интервале
температур, начиная от абсолютного нуля,
термодинамически неустойчива (ветвь тп)9 так что для реакций
перехода в другую форму при всех температурах 1А < 0 (рис. 4,6).
Классическим примером
систем первого типа
является:
Sn серое ~^Sn белое, для
которой *п—18°С,
S ромб, T^S мо но к л., для
которой /П=9Г)°С.
Классическим примером
систол! второго типа
является:
С алмаз—>С графит.
Алмаз термодинамически
неустойчив при р=\. атм
при "всех температурах.
' Г) Области ЛТлавл — *mm
термодинамически
устойчивой является жидкая фаза
(ветвь /, рис. 3). Переохлаждённой жидкости—стеклу —отвечает ветвь #,
являющаяся продолжением влево ветви /.
Наконец, в зависимости от степени разрыва связей в твердом теле его
структура становится всё более и более дисперсной и под конец аморфной.
При этом упругость пара всё более растёт. На р— Г-диаграмме возникает
бесконечно большое количество состояний (полоса) а.
*) Например, Стилвелл (1038) в своей «Кристаллохимии» утверждает, что твёрдое
состояние может быть условно разделено на кристаллическое, стеклообразное и
аморфное состояния.
кал
500
Л00
300
200
100
-
-
.L.
а£ —
\лВ
I-J 1 J-^l L-
нал
woo
800
600
400
200 \
\-
^У
S
' |гта„|
О 50 150
в
Рис.
Значении \Е и Д. L для энантиотроиного
и монотроиного процессов.
а; Унантиотропный процесс aSii^p.Sn ГЦСр=-291 К.
Кривая АЛ пересекает ось абсцисс при 291 . Ь) Монотроиный
процесс Салм= ('граф. Кривая ал ни при каких
температурах ие пересекает осп абсцисс (ирп р — 1атм).
101
С11ММСТМ1Я. aiMMKTl'Ilfl ФОГМЫ. СЛММКТГПЯ СТРУКТУРЫ
27
ЙЧ)
т
/77
§ 9. Закон постоянства углов
В соответствии с законом анизотропии и законом Бульфа
термодинамически устойчивая внешняя форма вещества является функцией его
внутренней структуры.
Следствием этого
является ряд важных
законов кристаллографии,
в частности закон
постоянства углов.
«Кристаллы одной и той же
модификации данного вещества
могут иметь разную
величину, форму и количество
г/пи (с ii, по углы между
соответствующими
гранями при данной
температуре и давлении
остаются постоянными» (рис. Г>).
Особый интерес
представляют правильные
плоскогранные кристаллические формы, отвечающие равновесным соотношениям
/ii : п2 : л8- (3)
Правильность кристаллических форм лучше всего описывается теорией
симметрии.
Ч^>
Рис.
Углы между соответственными гранями
различных кристаллов гипса одинаковы.
10. Симметрии. Симметрия формы. Симметрия структуры.
Симметрия физического процесса
Явление симметрии в геометрических формах окружающих нас
предметов внешне хорошо известно. Например, цветку на рис. 6, а свойственна
симметрия. Комментируй 5
птот приме]) в своем
учебнике [181, К. С. Фёдоров
так определяет понятие
^симметрия»: ^Симметрия
есть свойство геометри-
"''Ct.'ux фигур... в
различных положениях
приходить в совмещение с
первоначальным положением».
Y
h
4,
-1
%
[о,
г
А
'"*
м
V7
Рис. G. Явление симметрии.
При повороте цветка вокруг оси, проходящей через его центр
перпендикулярно чертежу, он Г> раз совмещается с первоначальным положением.
28 ч. i. основы теории структур [гл. ).
Цветы на рис. 6, Ь симметричны, как предмет и его изображение в зеркале 55.
Линия SS делит пополам линии АЛ, соединяющие симметричные точки
(см. § 11).
Обычно подчёркивают, что симметрия формы кристалла находится
в прямой зависимости от симметрии его структуры. Например, кубическая
форма кристаллов каменной соли отражает соответствующее
расположение в них атомов (ионов) натрия и хлора (см. рис. 95,е). Однако такая чёткая
связь выявляется не всегда (см. § 31 и 37).
Наконец, следует считаться с симметрией физических процессов и
явлений как третьей формой проявления симметрии в природе. Симметрия
физического процесса связана с симметрией структуры и часто выявляет
последнюю. Например, в опыте, показанном па рис. J, обнаруживается
связь теплопроводности в кристалле с симметрией! последнего. Важно
отметить, что характер этой связи зависит от характера физического
процесса. Например, в кубических кристаллах скорость распространения света
во всех направлениях одинакова, т. е. обладает шаровой симметрией, гораздо
более высокой, чем симметрия куба. Приняв симметрию физического
процесса за симметрию структуры, можно также допустить серьёзную ошибку
(см., например, § 61, закон Фриделя).
§ 11. Операции симметрии и элементы симметрии
Объектом исследования является фигура, т. е. некоторая группа
(совокупность) точек. В отличие от математических точек* в теории структур
обычно принимается, что кристаллографическая точка имеет протяжённость,
собственную симметрию и ориентировку. Обоснование этому следует видеть,
в частности, в том, что с переходом от кристаллографической точки к атому
приходится учитывать строение последнего и пространственное
распределение связей.
Наличие симметрии и характер последней определяются с помощью
элементов симметрия и отвечающих им операций симметрии.
Операцией симметрии называют операцию совмещения точки (пли
части) фигуры с другой точкой (или частью) последней. Обе совмещаемые
части фигуры симметричны друг другу. Операцию симметрии осуществляют
с помощью элементов симметрии.
В теории симметрии континуума различают: элементы симметрии
I рода: плоскости симметрии, поворотные оси симметрии или гиры и центр
инверсии, и сложные элементы симметрии II рода: инверсионные оси или
гироиды, а также зеркально-поворотные оси или нлангироиды. Все они
называются элементами точечной симметрии. Буквенные и графические
обозначения элементов симметрии см. § 14, табл. 1.
Плоскостью симметрии называется плоскость зеркального
отражения, осуществляющая совмещение симметрично равных точек (рис. (3, Ь
и 7, а).
Плоскость симметрии делит пополам все перпендикулярные си
прямые, соединяющие симметричные (симметрично равные) точки (части)
фигуры.
Поворотной осью симметрии п-гп нарядна (гирои) называется
прямая (ось), при повороте вокруг которой на некоторый определённый угол
360°/я, именуемый элементарным углом, происходит совмещение
симметричных (совместимо равных) точек (соответственно частей) фигуры
(рис. 7, Ь).
Порядок оси (п) определяет, таким образом, сколько раз произойдёт
совмещение симметричной фигуры с самой собой при её повороте вокруг
оси на 360°.
Ill
ОПЕРАЦИИ СИММЕТРИИ И ЭЛЕМЕНТЫ СИММЕТРИИ
29
Если речь идёт не о кристалле, а о некоторой совокупности
кристаллографических*) точек, образующих ту или иную фигуру, или о
самостоятельной кристаллографической точке, порядок поворотной оси может быть
любым. Так, шар имеет бесконечно большое количество поворотных осей
любого, в том числе бесконечного порядка, т. е. приходит в совмещение
с исходным положением при повороте на любой, в том числе и бесконечно
.малый, угол.
Цилиндр и эллипсоид вращения имеют одну ось любого, в том числе
бесконечного, порядка и бесконечно большое количество осей 2-го порядка.
Правильные многоугольники с количеством углов от 3 до ос, в
соответствии с натуральным рядим чисел, т. е. 3, 4,
«>, 6, 7, 8, 9, 10 и т. д., имеют соответственно
поворотные оси 3, 4, 5, (j, 7, 8, 9, 10-го и
т. д. порядков. В предельном случае
окружность имеет ось бесконечного порядка.
Прямоугольники имеют поворотную ось 2-го
порядка.
Ось 1-го порядка означает, что «в данном
направлении элемент симметрии отсутствует»
\
Аэ
\
/
Рис. 7. Плоскость симметрии, поворотная ось, центр инверсии.
(формулировка IT), т. е. тело совмещается с самим собой лишь при
повороте па 300°.
13 кристалле порядок поворотных осой (гир) ограничен. В этом случае
возможны лишь поворотные осп 1, 2, 3, 4 и (>-го порядка и никакие дру-
1ие. Поряден; оси указывается приставками: дигира—поворотная ось 2-го
порядка, тригира—3-го, тетрагира—4-го, гексагира—6-го порядка (рис. 8).
Поворотные оси, порядок- которых выше чем 2, называют главными осями,
остальные—побочными.
Оба конца поворотной оси могут быть одинаковыми, например, в
случае поворотной оси 4-го порядка, проходящей через тетрагональную бипи-
рамиду, или разными, например, в случае поворотной оси 4-го порядка,
проходящей через тетрагональную пирамиду. И первом случае поворотная
ось называется биполярной, во втором—полярной. Их обозначения —
|-м. § \'\. Оба случая показаны и на рис. 8 (см., например, ось 4 и 4/т).
Центром инверсии {центром обратного равенства, центром симметрии)
низы кается точка пересечения линий, соединяющих противоположные,
*) T.j-есть имеющих протяженность и собственную симметрию.
30
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. \
параллельные, равные, но обратно направленные (антисимметричные) части
фигуры.
Соединив соответствующие части точек (рис. 7, с) прямыми,
обнаруживаем, что точка пересечения последних может быть уподоблена
фотообъективу, опрокидывающему (инвертирующему) изображение. На рис. 13
показан кристалл анортита, имеющий центр инверсии.
Таким образом, мы будем различать: симметрично равные (при
наличии плоскости симметрии), совместимо равные (при наличии гиры) и
чё2/ *fe/
О £><♦>{•>
Рис. 8. Поворотные осп 2, 3, А и г, и соответствующие'проекции
перпендикулярно оси.
а) Полярные (ось л). Ь) Пшго.шрнмс I — J. (Горизонтальная
плоскость'симметрии.)
обратно равные — антисимметричные (при наличии .'центра инверсии)
точки, соответственно, части фигуры*) (см. рис. 7).
Как видно из рис. 9, гексагональная призма как геометрическая форма
имеет 1 гексагиру, 6 дигир, 6 плоскостей симметрии, параллельных гекса-
*) Если подходить к форме и строению кристалла с точки зрения закона
анизотропии, то, очевидно, в нём должны обнаруживаться не только симметричные точки
м части, но и симметричные направления.
§ И] ОПЕРАЦИИ СИММЕТРИИ II ЭЛЕМЕНТЫ СИММЕТРИИ 31
гире, 1 плоскость симметрии, перпендикулярную гексагире, и центр
инверсии.
Тетрагональная пирамида (рис. 9, Ь) как геометрическая форма имеет
1 тетрагиру и 4 плоскости симметрии, параллельные тетрагире.
Рис. 9. Элементы симметрии гексагональной призмы и тетрагональной
пирамиды как геометрических форм.
На рис. 9, Ь плоскость симметрии (чёрная), параллельная переднему ребру основания
пирамиды, в ле и ой части рисунка не показана, чтобы не заслонять леную зад mow
иершину основания.
Фигура и её зеркальное отражение либо могут быть пространственно
совмещены друг с другом (рис. 10, а), либо не могут иначе, кате с помощью
I
J-
i
\L±
\
Рис. 10. Явление анантиоморфизма.
зеркального отражения (рис. 10, Ь). Фигуры, которые могут быть
щены только путём зеркального отражения, как, например,
и левая перчатки, называются энантиоморфными.
совме-
правая
32
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
§ 12. Плоскости симметрии и действующие совместно и нераздельно
плоскости зеркального отражения
Эффект поворотной оси с элементарным углом а эквивалентен
последовательному отражению в двух действующих совместно и нераздельно
плоскостях зеркального отражения, пересекающихся под углом а/2.
• I
I
—+-—
9
^
V
Л
/'
\
Рис. 11. Плоскость двойного отражения и две взаимно
перпендикулярные плоскости симметрии.
Рассмотрим в качестве примера дигиру (я =180°) (рис. 11, а). Точка Pi
в результате первого отражения попадёт в промежуточную позицию Si,
в результате второго отражения — в позицию Р2> т. е. один поворот на угол а
Рис. 1-. Плоскости тройного отражения и три взаимно перпендикулярные плоскости
симметрии.
эквивалентен двум последовательным отражениям в плоскостях,
пересекающихся под углом а/2. Подчеркнём, что в этом случае две плоскости
зеркального отражения не являются плоскостями симметрии данной грушпт
§ 13]
ЭЛЕМЕНТЫ СИММЕТРИИ 2-ГО РОДА
33
точек Pi, Р2у иначе мы пришли бы к фигуре рис. 11, 6, состоящей не из двух,
а из четырёх точек.
Такие две действующие совместно и нераздельно плоскости отражения
Вульф называл нереальными (плоскости двойной симметрии, двойного
отражения). Отметим, что если в фигуре действительно имеются две взаимно
перпендикулярные плоскости симметрии, то прямая, по которой они
пересекаются, является поворотной осью 2-го порядка.
Последовательное отражение в трёх действующих
совместно и нераздельно взаимно перпендикулярных
плоскостях зеркального отражения (из коих каждая,
сама по себе, не является реальной плоскостью
симметрии фигуры) равнозначно действию центра
инверсии (рис. 12, а). Напротив, в случае трёх взаимно
перпендикулярных плоскостей симметрии возникла
бы фигура не из двух, а из восьми точек
(рис. 12, 6).
Таким образом, наличие центра инверсии,
приводящего к образованию фигуры из двух точек, не
эквивалентно по действию на точку трём взаимно
перпендикулярным плоскостям симметрии,
приводящим к образованию 8 точек. Напротив, точка
пересечения трёх взаимно перпендикулярных плоскостей
симметрии есть в то же время и центр инверсии.
(На рис. 13 показан кристалл анортита, имеющий
центр инверсии.)
В связи с этим примером уместно подчеркнуть
(см. § 24), что кристаллография различает
порождающие элементы симметрии (в данном случае
плоскости симметрии) и порождённые элементы
симметрии (в данных примерах поворотная ось 2-го порядка, соответственно
центр инверсии), необходимо возникающие из данной совокупности
элементов симметрии.
Рис. 13. Кристалл
анортита. Грани
кристалла попарно
взаимно равны,
параллельны и обратно
направлены (обратно
равные грани).
§ 13. Элементы симметрии 2-го рода. Зеркально-поворотные
и инверсионные оси
В некоторых случаях для совмещения с исходным положением
необходимо осуществить не только поворот фигуры на элементарный угол, но и
отражение в действующей совместно и нераздельно вспомогательной
плоскости, перпендикулярной к оси, вокруг которой поворачивается фигура. (Эта
плоскость, как мы сейчас увидим, необязательно является плоскостью
симметрии фигуры.)
Подобные сложные оси или оси сложной симметрии называются
зеркально-поворотными осями, или плангироидами. Они относятся к
элементам симметрии 2-го рода в отличие от простых элементов симметрии
1-го рода, с которыми мы частично познакомились в предыдущих
параграфах.
Операции, выполняемые с помощью зеркально-поворотных осей, могут
быть осуществлены и с помощью инверсионных (или
инверсионно-поворотных осей), также относящихся к осям сложной симметрии.
Инверсионными осями п-го порядка называют оси, сочетающие действие
поворотной оси того же порядка и, совместно и нераздельно, — центра
инверсии. Подобные оси часто называются гироидами (дигироиды, трп-
гироиды, тетрагироиды, гексагироиды).
34
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Инверсионная ось 1-го порядка и зеркально-
поворотная ось 2-г о порядка*)
Повернём точку Рх (рис. 14, а) вокруг поворотной оси 1-го порядка
(т. е. на 360°). Она совместится сама с собой. Отразив её теперь через центр
инверсии О, переведём её в точку Р2. Очевидно, инверсионная ось 1-го
порядка эквивалентна
центру инверсии.
Теперь рассмотрим
действие
зеркально-поворотной оси 2-го
порядка.
Проведём плоскость
отражения тс и под
прямым углом к ней ди-
гиру (рис. 14, 6),
пересекающую плоскость
те в точке О.
Осуществив поворот на 180е,
переведём точку Pi
в промежуточное
положение *Si.
Последующее отражение в
плоскости те переведёт
точку в окончательное
положение /V- в
совокупности получим
зеркально-поворотную ось
2-го порядка (а =180°).
Итак, центр
инверсии, как инверсионная
ось 1-го порядка,
эквивалентен по конечному
результату зеркально-поворотной оси 2-го порядка.
На рис. 14, с показана проекция точек на плоскость, перпендикулярную
инверсионной оси. Неразличимые проекции верхней и нижней точек условно
обозначены по-разному. То же относится к рис. 15, с—18, с включительно.
Рис. 14. Инверсионная ось 1-го порядка Г. Зеркально-
ново ротная ось 2-го порядка. Плоскость « не является
плоскостью симметрии т. Центр инверсии имеется.
Инверсионная ось 2-го порядка и зеркально-
поворотная ось 1-го порядка
Очевидно, что инверсионная ось 2-го порядка равнозначна по
результату плоскости симметрии (рис. 15, а). Повернув точку Pi вокруг ди-
гиры на 180°, переведём её" в положение Si, но отражение через центр
инверсии переводит её в положение Р2, куда точка Р1 попала бы просто
в результате отражения в плоскости симметрии, перпендикулярной оси
в точке О,
Воспользуемся теперь зеркально-поворотной осью 1-го порядка
(рис. 15 ,Ь). Повернув точку Рх вокруг оси 1, приведём эту точку в исходное
положение. Последующее отражение в плоскости тс переведёт точку Pt вполо-
*) На рис. 14 —17, с кружками показаны соответствующие проекции точек.
Нижние и верхние точки отличаются графически.
§ ^_
жение Р2 и т. д.
Очевидно, что зеркально-
поворотная ось 1-го
порядка эквивалентна по
результату оси 2.
Плоскость тг является
плоскостью симметрии т.
Инверсионная
ось 3-го порядка
(3) и зеркально-
поворотная ось
6-го порядка
Инверсионная ось
3-го порядка
эквивалентна по результату
поворотной оси 3-го
порядка плюс центр
инверсии. Рассмотрим
рис. 16, а.
Точка Р\ (вверху)
поворотом на 120°
переводится в положение Р5
и через центр
инверсии— в положение Р2
(внизу). Точка Р>>
поворотом на 120° в
промежуточное положение Р±
и через центр инверсии
в положение Рз (вверху)
и т. д. В результате
получим в верхней
плоскости три положения:
Pi, Рз и Р5, в нижней
также три положения:
Р2, Ра и Рс, причём
три верхних положения
находятся над
незанятыми местами между
тремя нижними
положениями.
Совершенно
очевидно, что
зеркально-поворотная ось 6-го
порядка приводит к тому же
результату (рис. 15, Ь).
Поворотом Рг на 60° и
отражением в
плоскости ти
(перпендикулярной оси) находим точку
Р2 и т. д. Плоскость тс
не является плоскостью
симметрии фигуры.
Рис. 15. Инверсионная ось 2-го порядка -J. Зеркально-no-
поротная ось 1-го порядка. Плоскость тс является
плоскостью симметрии т. Центр инверсии отсутствует.
Рлс. 16. Инверсионная ось 3-го порядка 3.
Зеркально-поворотная ось Ого порядка. Плоскость тс не является
плоскостью симметрии т. Центр инверсии имеется.
36 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР |ГЛ. I
Инверсионная ось 4-го порядка (4) и зеркально-
поворотная ось 4-го порядка
Инверсионная ось 4-го порядка и зеркально-поворотная ось 4-го порядка
дают один и тот же конечный результат (рис. 17). S — промежуточные
положения точки /\
Из рис. 16 видно, что тетрагиройда является также дигирой. (Напротив,
Ф. дигира не всегда является тетра-
\\J гироидой.) Плоскость т: не
является плоскостью симметрии фигуры.
Инверсионная ось 6-го
порядка (6) и зеркально-
поворотная ось 3-го
порядка
Наконец, инверсионная ось
6-го порядка и
зеркально-поворотная ось 3-го порядка ведут
себя, как поворотная ось 3-го
порядка плюс перпендикулярная ей
плоскость симметрии (рис. 18).
Из рис. 18 видно, что
положения Pi, Р3 и Р$ верхней
плоскости лежат в точности над
положениями Р2у Р* и Р& нижней
плоскости, в отличие от
инверсионной оси 3-го порядка.
Обратим вновь внимание на
следующее важное обстоятельство.
Начинающие очень часто
рассматривают плоскость отражения,
являющуюся элементом
зеркально-поворотной оси, как всегда
эквивалентную плоскости симметрии фигуры, а инверсионную ось—как
признак обязательного наличия у фигуры центра инверсии, что неверно.
Из расположения точек на рис. 13—17 с очевидностью вытекает, что
фигуры, полученные: при действии на точку нечётных инверсионных осей
Ги 3 имеют центр инверсии; при действии чётных осей 2 и 6 —
горизонтальную плоскость симметрии; при действий оси 4 не имеют ни центра
инверсии, ни горизонтальной плоскости симметрии.
Как мы видели, зеркально-поворотные оси по конечному результату
их действия могут быть полностью заменены соответствующими
инверсионными и наоборот. Различные авторы отдают предпочтение тем или иным
осям. Но в мировой структурной литературе в последние годы под
влиянием IT отказываются от зеркально-поворотных осей в пользу
инверсионных, в то время как многие наши кристаллографы решительно отвергают
применение инверсионных осей и сохраняют зеркально-поворотные. Это
заставило нас рассматривать в данном параграфе действие как зеркально-
поворотных, так и инверсионных осей, чтобы облегчить читателю
ознакомление с теми и другими и создать у него представление о том, как
и в каких случаях изменяются наименования и обозначения при
переходе от одной группы осей к другой. При замене
зеркально-поворотных осей инверсионными порядок оси не меняется только в* случае
Рис. 17. Инверсионная ось 4-го порядка 4.
Зеркально-поворотная ось 4-го порядка.
Плоскость -к не является плоскостью
симметрии т. Центр инверсии отсутствует.
§ 141 ОБОЗНАЧЕНИЯ ЭЛЕМЕНТОВ СИММЕТРИИ 37
осей 4-го порядка. Оси 1 и 2-го порядка, с одной стороны, и 3 и 6-го,
с другой, меняют кратность в пределах своей пары (т. е. 3 на. 6 и 6 на 3;
1 на 2 и 2 на 1).
В настоящее время в большинстве учебников, например, гексагироидой
называют инверсионную ось 6-го порядка (6), а не зеркально-поворотную
ось 6-го порядка (3) (см., например, Доливо-Добровольский [2] (1937)), хотя
первоначально термин гексагироида относился к зеркально-поворотной
оси 6-го порядка. Отметим, что чёткое соответствие наименования гироиды
именно порядку инверсионной оси всё же во многих трудах отсутствует
(см., например, Ниггли [14]).
В широко распространённом у нас учебнике кристаллографии А. В. Шуб-
пикова, Е. Е. Флинта и Г. Б. Бокия инверсионная ось 6-го порядка обозна-
Рис. 18. Инверсионная ось 6-го порядка 6. Зеркально-
поворотная ось 3-го порядка. Плоскость * является
плоскостью симметрии т. Центр инверсии отсутствует.
чается 3, 3-го порядка—6 и т. д., что, как это, впрочем, и оговорено в тексте
названного учебника, идёт вразрез с советской и международной системой
обозначений, принятой в теории структур.
Приставкой ди-, три- и т. д. при названии гироида мы будем указынать
порядок инверсионной оси.
§ 14. Обозначения элементов симметрии
В кристаллографической литературе существует большой разнобой
в обозначении элементов симметрии. Плоскость симметрии в большинстве
случаев обозначается Р или Е (Ebene), особые плоскости, как, например,
перпендикулярные главной поворотной оси (например, оси 6-го порядка
в шестигранной призме) иногда обозначаются тт. Поворотные оси /г-го
порядка обозначаются Ln> Gn (гира), Ап и т. п. Центр инверсии —z, Сит. п.
В наиболее принятых обозначениях элементы симметрии шестигранной
38 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР |ГЛ. I
призмы запишутся так: Lq 6L2 IPC, Элементы симметрии четырёхгранной
призмы—L4,4L2 ЪРС (см. рис. 9).
Структурные исследования сделали весьма распространёнными в
последнее время две другие системы обозначений: старую—Шенфлиса и новую —
Могена—Германа. Эти системы приняты в IT*). В связи с потребностями
теории структур и старые системы обозначений претерпели видоизменения.
Начнём с этих последних. Во-первых, стали различать плоскости симметрии
вертикальные и горизонтальные. При этом, как и в системе Шенфлиса,
условно принимается, что главная ось установлена вертикально; тогда
плоскость, перпендикулярная оси, оказывается горизонтальной. Её
обозначают Ph или Eh\ плоскость, проходящая через главную ось, параллельно
ей, т. е. вертикальную,—/^соответственно Еи. Побочные оси обозначают
в иностранной литературе Ап. Между побочными осями иногда можно
провести промежуточные оси того же порядка; их, особенно в иностранной
литературе, часто обозначают Az. Например, в шестигранной призме
(рис. 9,a) 3Lo—побочные, а другие 3L2—промежуточные. Одни из них
проходят через центры граней, другие —через рёбра призмы.
В ряде курсов различают также основные и промежуточные
плоскости симметрии, например, в шестигранной призме 3PD(3EV)—основные и
3PV(3EV)—промежуточные, в связи с чем можно встретить обозначения Еп, Ez.
На рис. 19 и 20 основные плоскости симметрии показаны сплошной,
а побочные пунктирной линией. Для обозначения полярных осей иногда
применяют стрелки, например, ^2J.
В системе Могена—Германа поворотная ось и-го порядка обозначается
просто п (ось 1-го порядка—1; ось 2-го порядка—2 и т. д.), т. е. 1, 2, 3, 4, 6.
Главная ось условно принимается вертикальной. Плоскость симметрии
обозначается т (miroir—зеркало). Ось п-го порядка и проходящая через
неё (вертикальная) плоскость симметрии обозначается пт, т. е. 6т означает
поворотную ось 6-го порядка и проходящею через неё, ей параллельную
(вертикальную) плоскость. Знаком п\т или — , или п: т обозначают ось
/г-ro порядка и перпендикулярную ей (горизонтальную) плоскость т.
Например, б/т означает: поворотная ось 6-го порядка и перпендикулярная ей
(горизонтальная) плоскость симметрии. Центр инверсии в этой системе
обозначают 1 (см. § 13).
Ниже мы часто будем пользоваться подобным обозначением элементов
симметрии, однако для указания количества данного вида элементов
соответствующий индекс нам кажется удобным проставлять справа вверху.
Вышеупомянутые элементы симметрии шестигранной призмы запишем
так: 612вот5далГ-
В системе Шенфлиса знаку пт отвечает индекс при букве,
характеризующей комплекс элементов симметрии (см. ниже). Знак Спи, например Съ„,
означает: поворотная ось 6-го порядка + вертикальная плоскость симметрии
(повторенная поворотной осью 6 раз), т. е. 6/тг. Знак Слл, например См,
означает: поворотная ось 6-го порядка-^горизонтальная плоскость
симметрии, т. е. 6//и. Других обозначений этих систем мы коснёмся ниже.
В таблице 1 приведены обозначения элементов симметрии континуума.
§ 15, Кратность точки, кратность позиции точки, собственная симметрия
и ориентировка точки
При действии элемента симметрии или пространственной совокупности
элементов симметрии на точку, не лежащую на элементе симметрии,
возникает соответствующая фигура (совокупность точек). Например, при действии
*) См. § 1.
§ 15] КРАТНОСТЬ ТОЧКИ, КРАТНОСТЬ ПОЗИЦИИ ТОЧКИ 39
Таблица 1
Обозначения элементов симметрии континуума
J Наименование
I элемента
1 симметрии
1 Плоскость
1 симметрии
вертикальная
Плоскость
симметрии
горизонтальная
Поворотные
оси порядка:
1
•>
о
4
6
Инверсионные
оси порядка:
1
1 центр
I инверсии
1 ~
1 плоскость
I симметрии:
3
4
6
Классическая
русская
литература
Р, иногда
Р, иногда |
Т'1
L*
Ьа
h
h
с. С, z, Z
см. выше
V**)
V**)
Я3***)
ровский
ститут
Р
Р
Gl
G2
о,
G"
Gr,
On
<*2!
G3f
C?4/
<^
IT
m
/m \
1
2
3
4
С
T
T
T
4"
T
Графические
обозначения no IT
вертикальные
♦
▲
♦
•
о
А
Ф
•■
горизонтальные
**)
"
е л
Комбинация
элементов
симметрии по IT
n/m *)
2/m
3/m
4/wi
6/m
Обо- 1
значе- I
ние |
о
ч
♦
о
*) Вертикальная ось порядка п и перпендикулярная ей плоскость симметрии,
**) Знак 1 ставится на чертежах IT в правом верхнем углу.
***) Обозначения соответственно зеркально-поворотным осям.
40 ч. i. основы теории структур [гл. I
оси 6 на точку, не лежащую на оси, получаем фигуру, состоящую из 6 точек
(рис. 19, а), при действии оси 6 и проходящей через неё (вертикальной)
плоскости симметрии (С6и—6/гс)— фигуру из 12 точек (рис. 19, 6), при
действии оси 6 и горизонтальной плоскости симметрии, совпадающей с
плоскостью чертежа (Сел—6/т),— другую фигуру из 12 точек (рис. 19, с). Сравни
с рисунком 8.
Принято говорить, что точки, не лежащие на элементе симметрии,
находятся в общих положениях, лежащие на элементе симметрии—в
специальных, или частных, положениях.
Позиции точек, определяемые данной пространственной совокупностью
элементов симметрии, отличаются по своей кратности,
V/JV ~ /
а 5 с
Рис, 19. Действие на точку оси 6, Ьт и 6/т (проекция на плоскость чертежа,
перпендикулярную оси 6).
Кратностью положения (позиции) точки называется число р,
показывающее, сколько аналогичных положений имеется в данной группе точек.
Например, если точки располагаются в вершинах куба, кратность этой
позиции jp = 8.
Кратность положения в центрах граней куба равна 6, в центре
куба равна 1. Порядок (кратность) элемента симметрии равен количеству
точек, возникающих из одной точки, находящейся в общем положении
при действии на неё данного элемента симметрии.
Кратность дигиры, плоскости симметрии и центра инверсии равна 2,
кратность тригиры равна 3 и т. д.
Кратность точки определяется произведением кратностей исходных,
порождающих элементов симметрии, проходящих через неё (см. § 24), что
равно количеству точек в общих положениях, возникающих из данной точки
при смещении её с элемента симметрии.
Собственная симметрия и ориентировка точки отвечает совокупности
элементов симметрии, проходящих через неё.
Точки, возникающие из данной, при действии на неё совокупности
элементов (точечной) симметрии называются равнозначными, или
эквивалентными.
Рассмотрим действие на точку совокупности элементов симметрии
Cqv—6m. Если точка Pi находится в общем положении, её кратность равна 1, её
собственная симметрия в общем случае равна 1 (поворотная ось 1-го порядка),
точка может быть ориентирована по отношению к элементам симметрии как
угодно. По другую сторону плоскости симметрии возникает другая точка Р2.
Эта группа точек повторяется гексагирой 6 раз с образованием 12 точек
(рис. 20,а). Кратность позиции равна, таким образом, 12. Приближая
исходную точку Pi (а тем самым и её зеркально-симметричное изображение Р2)
к плоскости симметрии, мы не меняем её кратности и собственной симмет-
§ 161
ВИД СИММЕТРИИ. ТОЧЕЧНАЯ ГРУППА
41
рии. пока она не попадёт на плоскость симметрии. В этот момент она сольётся
с точкой Р\у кратность возникшей точки Ри станет равна 2. Её собственная
симметрия станет равна т, причём она так ориентирована по отношению к
плоскости симметрии, чтобы последняя совпала с плоскостью симметрии точки.
Одновременно гексагира создаст 6 таких двукратных точек Ри (рис. 20, 6).
Будем приближать точку Ри к гексагире. Остальные 5 двукратных точек
повторяют её движение и, совпав с гексагирой, сольются в точку Р ,
кратность которой станет равна 12 (рис. 20, с). Её собственная симметрия станет
равна 6т. Она ориентирована так, что её элементы симметрии 6т
совпадают с элементами симметрии фигуры. Таким образом, в рассматриваемом
Рис. 20. Кратность позиции (Р) точки, кратность (V) точки и собственная
симметрия (S) последней.
a) /J«12, и = 1, s-=l; b) р-»6, v = 2, s~=m; с) р = 1, v —12, s-6 /л.
случае возможны следующие группы точек: кратность точки 1, кратность
позиции 12, собственная симметрия 1; кратность точки 2, кратность
позиции 6, собственная симметрия т; кратность точки 12, кратность позиции 1,
собственная симметрия 6т.
Мы запишем следующее важное соотношение:
ро = const, (4)
т. е. а) произведение из кратности у точек данной совокупности на кратность
их позиций р есть величина постоянная и б) относительные количества р
точек разной кратности v обратно пропорциональны кратностям этих точек.
В нашем примере 1 X 12 = 2 х 6 = 12 х 1.
Очевидно, что максимальное количество позиций, образуемых данной
пространственной совокупностью элементов симметрии, отвечает наиболее
общим положениям точек.
§ 16. Вид симметрии. Точечная группа
Полная пространственная совокупность элементов симметрии данной
фигуры называется её видом симметрии.
Точка, в которой пересекаются все элементы симметрии, является
особой точкой, так как она остаётся неподвижной при действии на неё данной
совокупности элементов симметрии.
Как было показано выше, точка в этой позиции обладает максимальной
кратностью. ,
Отвечающая данному виду-симметрии совокупность операций симметрии
называется точечной группой.
Так же как тот или иной элемент симметрии отвечает определённой
операции симметрии, тот или иной вид симметрии отвечает определённой
точечной группе.
'42
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
§ 17. Виды симметрии и форма многогранников
Если плоскость параллельна (наклонна), например, тетрагире, при
действии последней на эту плоскость возникает кристаллографическая призма —
рис. 21, а (соответственно пирамида, рис. 21, 6), отличающаяся от
геометрической призмы (пирамиды) прежде всего тем, что получаются фигуры,
лишённые граней основания, т. е. боковые стороны их уходят в
бесконечность. (На рис. 21 обозначены волнистым пунктиром.)
-+р\
1
1
1
1
(
1
1
1/
/>
L—
7
/
)
\
/
/
/
/
i
/
ч
/
/,
j i
' J
W^
^
N
\/
У
":
Рис. 21. Действие тетрагиры на плоскость: а) параллельную и Ь) наклонную
к тетрагире иод углом а.
IS случае а) иоаникаст кристаллографическая призма, в случае Ь)—кристаллографическая
пирамида (если ось полярна) или бипирамида (если ось биполярна).
«Простой формой называется такой многогранник, все грани которого
выводятся из одной заданной посредством элементов симметрии
многогранника. Комбинацией простых форм называется многогранник, у которого
из одной данной грани не все остальные выводятся посредством
элементов симметрии многогранника» (А. К. Болдырев). Следовательно,
кристаллографическая призма и бипирамида являются простыми формами,
а геометрическая призма—комбинацией двух простых форм:
кристаллографической призмы (боковые грани) и пинакоида (соответственно грани
верхнего и нижнего оснований), см. ниже.
Опустив, например, из любой точки тетрагиры перпендикуляр к грани
в точке Pl9 рис. 21,а, обнаружим, что на каждой из трёх остальных граней
имеются эквивалентные точки (Р2 и др.)- Сопоставляя действие элемента
симметрии (или вида симметрии) на точку Pi и на грань (плоскость), мы
обнаруживаем далеко идущий параллелизм в количестве, пространственном
расположении и собственной симметрии точек и в количестве,
пространственном расположении и собственной симметрии граней, возникающих
при действии данной совокупности элементов симметрии на точку,
соответственно грани (рис. 22).
Если грань косо расположена по отношению к элементу симметрии,
количество возникающих граней будет максимальным. Такая форма называется
§ 17] шгды симметрии и форма многогранников 43
общей формой. Если грань расположена перпендикулярно или параллельно
улементам симметрии (а в некоторых случаях также перпендикулярно к
биссектрисе угла между одинаковыми осями или к плоскости, соединяющей две
поворотные оси), возникающая форма является частной формой.
Поэтому, исследуя пространственные совокупности точек, возникающих
из одной точки при действии на неё данной совокупности элементов
симметрии, мы тем самым характеризуем пространственные совокупности
граней, т. е. многогранников, возникающих из одной грани, при действии на неё
\
[
it
/•
\
♦
\
Рис. 22. Действие элементов симметрии на точку и на грань.
а) Действие плоскости симметрии т на точку в общем положении и на грань, наклонную к ш.
Кратность позиции точки и количество граней увеличиваются вдвое и равно 2. Ь) Действие оси 4
на точку в общем положении и на грань, наклонную к оси 4. Кратность позиции точки,
соответственно количество граней, увеличивается вчетверо и равно 4. с) Действие плоскости
симметрии на точку в специальном положении и на грань, перпендикулярную hi. Как точка;
так и грань приобретают симметрию т. Кратность точки увеличивается вдвое, d) Действие оси 4
на точку в специальном положении и на грань, перпендикулярную оси 4. Как точка, так и грань
приобретают симметрию 4, кратность точки равна 4.
той же совокупности элементов симметрии. Естественно, что аналогия имеет
место, если сопоставлять случаи, когда точка и грань находятся в положении
равной кратности, например, в общем положении.
Если точка попадает на элемент симметрии, кратность её позиции
уменьшается. В равной мере, если грань перпендикулярна элементу симметрии,
количество граней уменьшается. В соответствии с этим частные формы
содержат меньше граней, чем общие. Например, куб (шестигранник) является
частной формой по отношению к гексаоктаэдру (48-граннику) (см. ниже).
Кристаллы, имеющие одну и ту же точечную группу симметрии,
•составляют класс. Поэтому вывод возможных кристаллографических
классов и кристаллических форм сводится к определению возможных
пространственных комбинаций элементов симметрии—видов симметрии
(соответственно точечных групп), а следовательно, и возможных групп точек,
возникающих из одной точки при действии на неё различных видов симметрии.
В заключение параграфа заметим, что хотя с точки зрения теории групп
группой симметрии называется совокупность совместимых друг с другом
операций симметрии, в некоторых курсах понятия «точечная группа», «вид
*симметрии» и «класс» не различаются.
44
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Так, в курсе Доливо-Добровольского на стр. 149 дано определение:
«Видом симметрии (классом или точечной группой) называется полная
совокупность элементов симметрии той или иной фигуры».
В книге Брегга «Кристаллическое состояние» на стр. 72 читаем:
«Природа симметрии для каждого кристаллического класса определяется группой
элементов симметрии, совокупность которых принято называть точечной
группой».
§ 18. О стереографической проекции
Для описания как многогранников-кристаллов, так и структур часто
прибегают к стереографической проекции. В этом методе, в большой мере*
разработанном русскими учёными Гадолиным, Вульфом, Болдыревым и др.,
исходят из того, что «размеры, взаимное расстояние граней и рёбер
кристалла или их расстояние от какой-либо одной точки, взятой внутри его,
не являются существенными
признаками его вещества.
Они зависят от внешних
причин. Существенным
признаком для граней и рёбер
данного кристаллического
вещества является лишь
взаимный наклон. Поэтому, для
удобства изучения можно
все грани и рёбра кристалла
переносить мысленно в одну
и ту же точку пространства,
параллельно самим себе.
Совокупность всех
граней и рёбер кристалла,
перенесённых параллельно
самим себе в одну и ту же
точку О пространства, будем
Рис. 23. Построение стереографической проекции. называть кристаллическим
пучком, а точку О—центром
пучка. В тот же центр пучка переносятся параллельно самим себе все
направления и плоскости, характеризующие оптические и многие другие
физические свойства кристалла» (А. К. Болдырев [22]).
Из центра пучка О любым радиусом описывается шар, именуемый шаром
проекции. Через экватор шара проводится плоскость ti, которая называется
плоскостью проекции. Диаметр шара NS, перпендикулярный плоскости тс,
называется осью проекций. Точка S(N) называется точкой зрения. Она
совпадает с южным (северным) полюсом шара. На поверхности шара мысленно
проводится сетка меридианов и параллелей (долгот и широт).
Для построения стереографической проекции кристалла восстановим
в точке О перпендикуляр к данной грани (рис. 23) и продолжим его до
пересечения с поверхностью шара в точке Р9 именуемой полюсом грани. Для
определения положения точки Р на шаре достаточно знать полярные
координаты в градусах (0—360°) долготы и (0—90°) широты.
Соединив точку Р, лежащую в северном полушарии, с полюсом S
прямой PS, найдём в пересечении последней с плоскостью экватора точку Р'
проекцию точки Р. Проекция полюса N лежит в центре круга проекции
(в точке О); если точки лежат на экваторе, они совпадают со своими
проекциями Р'. Проекции Р'к точек Рм (лежащих в верхнем полушарии на
меридианах) находятся на прямых линиях, проходящих через О.
§19] ПОСТРОЕНИЕ СТЕРЕОГРАФИЧЕСКОЙ ПРОЕКЦИИ КРИСТАЛЛА 45
В соответствии с излагаемой в учебниках теоремой «проекция
окружности есть окружность» в частном случае, проекции точек, лежащих на
параллели, образуют круг с центром в точке О, а проекции точек, лежащих на
окружности (дуге большого круга), по которой пересекается с поверхностью
шара любая плоскость, проходящая через его центр О под углом к оси NS,
отличным от 0 и 90°, также лежат на дугах круга. Хордой этих дуг является
диаметр круга, по которому плоскость тс пересекает плоскость экватора
(рис. 24, а).
При проектировании точек южного полушария их соединяют с полюсом Лг
(метод Гадолина).
Рис, 24. Построение стереографической проекции.
л) Если плоскость пересекает сферу по диаметру экватора, проходя в те же время накловно
к оси N-S, проекция точек лежат на кривых, являющихся дугами окружности. Хордой дуги
-является один иа диаметров экватора. Ь) Проекции точек северного 'ja южного полушарий
(метод Гадолина),
Точки северного и южного полушарий обозначаются соответственно +
(или •) и О- Если две точки Рх и Ps обоих полушарий лежат на одном
меридиане и на одинаковом градусе северной, соответственно, южной
широты, их проекции совпадают, что обозначается © или (•). Если точки Рк и Ps
лежат в диаметрально противоположных частях полушария, их проекции
будут лежать на прямой, проходящей через центр (рис. 24, 6), но по
разные стороны от последнего.
§ 19. Построение стереографической проекции кристалла
На рис. 25, a, b показано построение стереографической проекции
кристалла. Об обозначениях (символах) граней (100), (110), (111) см. § 33.
Так как любое направление в кристалле может быть задано точкой,
образуемой пересечением вектора, характеризующего это направление
со сферой, то очевидно, что все горизонтальные направления, «полюса»
которых будут лежать на вертикальных гранях, проектируются на экваторе,
все вертикальные — в центре проекций и т. д.
Зная проекции полюсов граней и других направлений в кристалле,
надо уметь решать и обратную задачу, т. е. построить кристалл, найти углы
46
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. 1
Рис. 25. Построение стереографической проекции кристалла.
между его гранями. Это можно сделать очень просто, графическим путём7
используя стереографические сетки Вульфа (двухполюсную, рис. 26) или
Болдырева (однополюсную, рис. 27).
О решений соответствующих задач см. курсы кристаллографии,
например В. В. Доливо-Добровольского.
§ 20. Сипгонии, классы, простые формы
Классическая кристаллография строила систематику кристаллов на том
принципе, что все кристаллы можно отнести, по сходным углам между осями
(см. § 36), к 7 (6) сингониям «со сходными углами», две из которых,
гексагональная и тригональпая, часто рассматриваются как подсингопии одной
и той же гексагональной сингонии, так как симметрия внешней формы
соответствующих кристаллов не позволяет установить различие между этими син-
гониями (см., например, А. К. Болдырев [22]). Некоторые авторы, например
5 201
СИНГОНИИ, КЛАССЫ, ПРОСТЫЕ ФОРМЫ
47
Рис. 26. Стереографичесная сетка Вульфа.
26 г? 28
10 д 8
Рис. 27. Стереографическая сетка Лолдырева.
Таблица 2
Простые формы низших и Средних сивговий *)
Наименование
простой формы
Ромбическая
Тригональная
Призма
<•>
Пирамида
Г» и пирами да
(Дипирамнда)
У
V.
А
Дитригональ-
ная
4
г
Тетрагональная
Дитетрагональная
В
Д17
ггтъ
Гексагональная
Дигексагональная
ро=
Ids
00
Трапецоэдр
1 (правый
и левый)
Скале-
ноэдр
|Ромбоэдр
(Сфеноэдр
I
(тетраэдр)
*) Вверху даны сечония призм (бипирамид) и т. д.
**) В кристаллографии различаются два диэдра (осевой и безосев^й).
Простейшие формы
Диэдр* *)
Иинакоид
Моноэдр
//""Г7
££ПУ
Простые формы кубической оингонии
Таблица 3
куб
Октаэдр
Тетраэдр
/
/
Куб
л
/
Октаэдр
Тетраэдр
Ромбододекаэдр
Додекаэдр
Пирамидальный куб,
тетрагексаэдр
24
Тригонтриоктаэдр
24
Тригонтритотраэдр
12
Тетрагонтриоктаэдр
24
Тетрагонтритетраэдр
12
Пентагоктриоктаэдр
правый левый
Пентагонтритетраэдр
праный левый
12
Пентагондодекаэдр
112
Гексаоктаэдр
48
Гексатетраэдр
Дидодекаэдр
Примечание. В каждой клетке, в углу слева—количество граней.
ГОЛОЭДРИЧЕСКИЕ II МКРОЭДРИЧЕСКИЕ ФОРМЫ 51
А. В. Шубников, Е. Е. Флинт и Г. Б. Бокий, не без основания относят
кристаллы по «гониометрической симметрии» (но симметрии внешней формы)
к 6 системам, а по симметрии внутренней структуры к 7 сингониям.
Сингонии делятся на низшие (без главных осей, т. е. порядок которых
выше, чем 2)—сюда относятся триклинная *), моноклинная *) и ромбическая;
средние (с одной главной осью)—сюда относятся тригональная
(ромбоэдрическая), тетрагональная и гексагональная**) (с осями соответственно 3, 4 и 6);
высшую (с более чем одной главной осью)—кубическая или правильная.
Как показал русский учёный Гадолин в 1869 г., всего возможно 32 вида
симметрии, соответственно класса кристаллов, каждый из которых
относится к одной из сингонии. Наименования классов давались по
отвечающим им простым формам.
Исследование показало, что всего существует 47 простых форм, из них
32 в низших и средних сингониях (таблица 2) и 15 в кубической (таблица 3).
При этом было установлено, что в кристаллах возможны простые формы
из 1, 2, 3, 4, 6, 8, 12, 16, 24 или 48 граней и никакие другие.
Названия пирамида, призма и бипирамида (см. таблицы 2 и 3) не
нуждаются в комментариях. Названия моноэдр (однограиник), или педион
относятся к форме (грани), не имеющей элементов симметрии. Пинакоид (от слова
«пина»—доска) —форма, состоящая из двух равных и параллельных граней.
Эти грани могут быть симметрично равными, совместимо равными и обратно»
равными.
Диэдр—двуграшшк. Теория симметрии различает два типа диэдров:
осевой и безосный (см. § 25).
Название трапецоэдр происходит от «трапеца» — четырёхугольник
с двумя равными смежными сторонами (не смешивать с трапецией, т. е.
четырёхугольником с двумя неравными параллельными сторонами).
Название скаленоэдр происходит от «скалена»—разносторонний треугольник. Ди-
тригон (соответственно дитетрагон, дигексагон)—шестиугольник,
соответственно восьми- или двенадцатиугольник, у которого все стороны равны,
но углы равны попеременно. Названия простых форм кубической сингонии
и большинстве случаев не нуждаются в комментариях. Такие названия,
как тригонтриоктаэдр, иентагонтриоктаэдр и т. п., означают триоктаэдр
(24-грашшк) с треугольными, соответственно пятиугольными гранями.
Отметим, что название гироэдр присвоено пентагонтриокгаэдру, фигуре,
имеющей только поворотные оси (гиры).
Известны различные комбинации простых форм кубической сингонии.
Па рис. 25 показан кристалл, имеющий грани куба (100), октаэдра (111)
п ромбододекаэдра (НО). См. также рис. 41.
§ 21. Голоэдрические и мероэдрические формы
В классической кристаллографии при рассмотрении классов и форм
предпочитали итти от кубической сингонии к триклипной, соответственно
исходить из полногранных (голоэдрических) форм (например, в кубической
сингонии из 48-гранника и т. п.) и путём удаления в определённом порядке
части граней приходить к неиолногранным (мероэдрическим) формам —геми-
.чдрическим (с половинным количеством граней), тетартоэдрическим
(содержащим */4 часть граней) и т. п.
В современных курсах кристаллографии представления этого рода
часто опускаются как устаревшие. Однако в теории структур и в IT ими
*) С тремя наклонёнными под углом Ф 90э координатными осями, соответственно-
с одной наклонённой осью, см. ниже § 36.
**) От «тригон», «тетрагон», «гекеагон» (треугольник, четырёхугольник,
шестиугольник).
121L
52
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
часто пользуются. Это заставляет нас коротко остановиться на различных
видах гемиэдрии.
Вернёмся к табл. 2. Важнейшими простыми формами ромбической
и средних сингоний являются бипирамиды. Бипирамида — единственная
простая голоэдрическая форма этих сингоний,
образующая замкнутую фигуру без комбинации с другой
формой. Её рассмотрение позволяет наилучшим
образом охарактеризовать сингонию. Из неё выводятся
и гемиэдрические формы.
Сформулируем простые правила образования и
описания гемиэдрических форм.
1. Грани исходных бипирамид мы будем
нумеровать последовательно от 1 до п, отдельно по
верхней и по нижней пирамидам так, что грани с
одинаковым номером располагаются одна над другой
(рис. 28).
2. Удаление граней будем производить по
строгому закону, определяющему характер гемиэдрии.
Остающиеся грани, будучи продолженными, пересекаются с
образованием новых рёбер.
Рис. 28. Нумера
ция граней бипи
рамид.
§ 22. Различные типы гемиэдрии
Параморфная гемиэдрия, соответственно тетартоэдрия
Удаление заштрихованных граней гексагональной и тетрагональной
бипирамид (т. е. по закону —- ... ) ведёт к образованию двух совершенно
аналогичных форм
положительной ( + ) и
отрицательной (—), совмещаемых друг
с другом после поворота.
(В числителе номера граней
верхней, а в знаменателе —
нижней части бипирамиды.)
Цифры означают номера
сохраняемых граней, а
кресты стоят вместо номеров
удаляемых граней или
наоборот. Многоточие после
дроби означает, что в этом
порядке удаляются и
следующие грани.
Из тетрагональной
бипирамиды (рис. 29, а)
возникает, таким образом,
тетрагональный тетраэдр (сфено-
эдр) (рис. 29, 6), из
гексагональной (рис. 29, с) —
ромбоэдр (рис. 29, d, e)
(сплющенный или вытянутый
куб), отличающийся от
куба тем что углы у него не
равны 90°.
Ромбоэдр и тетрагональный ^тетраэдр являются также примерами двух
видов (гемиэдрии—тетартоэдрии), отличающихся по наличию или отсутствию
Рис.
29. Параморфная (гемиэдрия) тетарто-
1х
здрия -^ .
§ 221
РАЗЛИЧНЫЕ ТИПЫ ГЕМИЭДРИИ
53
2IS
&
се
СО
я
s &
се
(М
зЯ И
ее ,
5 -
S з
н %
g. "
Я
о
о
со
О
си
54
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
центра инверсии: 1) параллелогранная форма имеет параллельные грани
и центр инверсии, (пример — ромбоэдр) *) и 2) наклонногранная—центр
инверсии отсутствует. Пример—тетрагональный тетраэдр.
Энантиоморфная гемиэдрия
1 х
Удаление по закону тт^-- граней дитригональной (рис. 30, а), дитетра-
гональной (рис. 30, d) и дигексагональной (рис. 30, g) бипирамид ведёт
к образованию тригонального (рис. 30, b и с), тетрагонального (рис. 30, е
и /) и гексагонального (рис. 30, h и i) трапецоэдров.
Гемиэдрия этого вида часто называется трапецоэдрической. Она является
частным случаем весьма важного типа гемиэдрии— энантиоморфной геми-
Образование ромбических тет- зование скаленоэдров:
раэдров: ЛСНОГО и правого. а> j,) тетрагонального, с, d) гексагонального.
эдрии. Удаление граней по -~ или ^ влечёт за собой в этом случае удаление
всех плоскостей симметрии; возникает либо левая, либо правая форма.
По тому же закону образуется из ромбической бипирамиды ромбический
тетраэдр (табл. 2), являющийся другим частным случаем энантиоморфной
гемиэдрии. Из трёх пар взаимно противоположных рёбер рёбра каждой
пары одинаковы, принадлежащие к разным парам—различны (рис. 31).
*) По IT—гексагональная тетартоэдрия II рода.
§ 22] РАЗЛИЧНЫЕ ТИПЫ ГЕМИЭДРИИ 55
Гемиморфная гемиэдрия
12
Удаление по закону ... граней дитригона ль ной, дитетрагональ ной,
дигексагональной бипирамид ведёт к образованию пирамид, т. е. как бы
отрезает нижнюю или верхнюю часть фигуры. Такая гемиэдрия является геми-
морфной.
Гемиэдрия второго рода
Если произвести удаление гранен дитетрагональной и дигексагональной
12 К X
бипирамид по закону ~ 3, ...,то возникают соответствующие скаленоэдры
(рис. 32). Так как между гранями 1 и 2, соответственно 3 и 4, проходят
вертикальные плоскости симметрии, то в отличие от трапецоэдров у скалено-
эдров эти плоскости сохраняются.
Из простых форм кубической сингонии исходной голоэдрической
формой является гексаоктаэдр.
Энантиоморфная гемиэдрия и тетартоэдрия
Пентагонтриоктаэдры левый и правый (гироэдр) (рис. 33, а' или а")
получаются из гексаоктаэдра (рис. 33, а) таким образом, что прилегаю-
Рис. 33. Плагиэдрическая—пентагопальная—гемиэдрия (и тетартоэдрия).
щие к вершине 8 граней исключаются по закону ( —- J. Пеитагонтритет-
раэдры левый и правый (рис. 33, Ъ' и Ь") получаются из гексатетраэдра
(рис. 33, 6).
Обе формы относятся к энантиоморфной гемиэдрии, соответственно
тетартоэдрии. Для того чтобы показать, что числитель и знаменатель
относятся к одной и той же полусфере, дробь взята в скобки.
В результате удаления граней по закону — ... получаются: тетраэдр
из октаэдра (рис. 34, а и Ь), тригонтритетраэдр из тетрагонтриоктаэдра
(рис. 34, с и d)y тетрагонтритетраэдр из тригонтриоктаэдра (рис. 34, е и /),
гексатетраэдр из гексаоктаэдра (рис. 34, h и g).
56
4.1. ОСНОВЫ ТЕОРИИ СТРУКТУР
]гл. I
* 1
Рис. 34. Образование тетраэдрических форм.
6'
Рис. 35. Параллелограннан гемиэдрия.
а) Пентагинальная параллелограннан гемиэдрия. Ь) Дидоденаэдрнческая параллелограниая
гемиэдрия.
S 23]
О НАИМЕНОВАНИЯХ СИНГОНИЙ
57
Наконец, пентагондодекаэдр образуется из тетрагексаэдра путём
удаления по закону Г—« J граней, прилегающих к одной и той же вершине
пирамиды (рис. 35, а и а'). Рис. 35, Ь и 6'—образование дидодекаэдра из гекса-
октаэдра. Схематическое, условное изображение разных случаев гемиэдрии
}
1
L
Г
}
L
С
X
1
X
с
X
7
X
Г
}
X
X
1
X
X
X
X
с
)
X
X
i
X
X
X
X
7
X
X
(
X X
X X
1
/
с
Рис. 36. Схематическое изображение разных случаев гемиэдрии:
а) Голоэдрия. Ь) Гемиморфная гемиэдрия. Образование пирамид и а
соответствующих бипирамид. с) Образование бипирамид иа дибипирамид. d)
Энантиоморфная гемиэдрия. Образование трапецоэдров и т. п. е) Иараморфная
тетартоэдрия. Образование ромбоэдра и тетрагонального тетраэдра иа три-
гональных бипирамид. /) Об].ааовавие скалсноэдров. Н ирной чертой показаны
плоскости симметрии.
показано на рис. 36 (а—голоэдрия, 6—гемиморфная гемиэдрия,
с—образование бипирамид, d и е—параморфна^ и энантиоморфная гемиэдрия л
т. п., /—образование скаленоэдров.
§ 23, О наименованиях сингоний и путях вывода видов симметрии
и кристаллических форм
В § 17 мы видели, что с точки зрения теории симметрии проблема
образования многогранных форм является второй стороной проблемы
образования групп точек при действии на точку той или иной возможной
пространственной совокупности элементов симметрии (вида симметрии).
Если, например, в мире кристаллов обнаруживаются 32 класса, то это
находится в тесной связи с тем фактом, что всего в кристаллах возможны
32 вида симметрии.
Если возможны только такие простые формы, которые состоят из 1, 2,
3, 4, 6, 8, 12, 16, 24 или 48 определённым образом (см. § 30, табл. 10)
расположенных граней, то этот факт находится в тесной связи с тем, что при дои-
58 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
ствии этих 32 видов симметрии на точку возможно образование групп точек,
содержащих 1, 2, 3, 4, 6, 8, 12, 16, 24 или 48 определённым образом
расположенных точек.
Это позволяет иначе подойти к изложению вопроса.
Вместо того чтобы итти от рассмотрения сложных форм к рассмотрению
простых, от классов кубической сингонии—к классам триклинной синго-
нии, ири современном состоянии теории структур целесообразно
рассматривать действие на точку элементов симметрии всё увеличивающейся
кратности, например, осей 1, 2, 3, 4, 6, ..., и всё более сложных совокупностей
элементов симметрии и итти, соответственно, от триклинной сингонии к
кубической, от моноэдрак гексаоктаэдру, от простого к сложному. Первым шагом
к такого рода рассуждениям является рассмотрение возможных
пространственных сочетаний элементов симметрии (см. следующий параграф).
Более того, в систематике Фёдоровского института сингонии
приобретают наименование уже не по формам многогранников, а по характеру
элементов симметрии: триклинная—агирная (оси 1 или 1), моноклинная—моно-
гирная (оси 2 и 2-плоскости симметрии, причём каждый элемент симметрии
может присутствовать в единственном числе), ромбическая—дигирная (оси
2 и 2, причём хотя бы один из элементов симметрии присутствует не в
единственном числе: или более чем одна дигира или более чем одна дигироида,
т. е. плоскость симметрии), тригональная — тригирная (единственная главная
ось 3), тетрагональная — тетрагирная (единственная главная ось 4 или 4),
гексагональная—гексагирная (единственная главная ось 6 или 6),
кубическая— полигирная (несколько главных осей). Характерным для всех
видов симметрии кубической сингонии является наличие четырёх осей 3.
§ 24. О возможных пространственных сочетаниях элементов симметрии
Число возможных видов симметрии (соответственно точечных групп)
ограничено не только тем, что некоторые из элементов симметрии друг друга
взаимно исключают (например, оси 6 и 4), но и тем, что комбинируемые элементы
симметрии могут пересекаться лишь под определёнными углами друг к другу.
Геометрический анализ показывает, что возможны лишь следующие
сочетания поворотных осей.
Оси 2—под углами 90° и 180°, а при наличии главных осей 6 и 3 также
иод углами 30°, 60°, 120°, 150° (в зависимости от кратности главной оси),
как следствие шестикратного или трёхкратного повторения оси 2
перпендикулярной к ней осью 6 или 3. Угол 180° означает, что ось 2 биполярна.
Оси 3 —под углами 70°31' (угол между диагоналями куба, примыкающими
к одному ребру) и 109°28' (угол между диагоналями куба, примыкающими
к концам диагонали грани) (см. рис. 38).
Оси 4 —под углами 90° и 180°.
Оси 6—только под углом 180°, т. е. возможна только одна ось 6
полярная или биполярная.
Оси 3, 4 и 6 — под углом 90° к осям 2, а оси 3 также (см. кубическая
сипгония) под углом
Щ-~ = 35°15,5' к оси 2 (рис. 40).
Таким образом, новые виды симметрии могут возникать в различных
сингониях, например, лишь путём добавления осей 2 (соответственно 3,
4 и др.), перпендикулярных к исходным осям, и т. н.
В равной мере оси 2 (это же относится к осям 3, 4 и др.) могут быть либо
перпендикулярными к плоскости симметрии, либо лежать в этой плоскости,
|гл^1
§ 25] ПРОСТЫЕ ИСХОДНЫЕ ФОРМЫ. ВИДЫ СИММЕТ1 ИИ 59
за исключением случаев, когда оси 2 под влиянием оси л, повторяясь п раз
в одной и той же плоскости, перпендикулярной к главной оси, пересекают
плоскости симметрии под углами, равными половине элементарного угла (см.
рис. 9). Всё это весьма сокращает количество возможных видов симметрии.
Ось 2 полярная, которую мы будем обозначать 2J, пересекаясь с т, ей
перпендикулярной, даёт центр инверсии и становится, как мы знаем,
биполярной.
§ 25* Простые исходные формы. Виды симметрии триклинной и
моноклинной сингонии
Из числа простых форм выделим 5 исходных (таблица 4):
I. Моноэдр, или педион-форма, не имеющая элементов симметрии.
II. Пинакоид, состоящий из двух граней, параллельных, равпых и
обратно направленных (обратно равных), т. е. имеющий центр инверсии.
III. Диэдр (двуграиник) безосный или планальный, в литературе
обычно называемый «дома» (крыша), состоящий из двух непараллельных
граней, одинаково наклонённых к плоскости т, являющейся, таким образом,
плоскостью симметрии.
Обе грани совмещаются друг с другом при отражении в плоскости.
IV. Диэдр осевой (аксиальный), или сфеноид, состоящий из двух граней,
наклонных к дигире и совмещаемых друг с другом при повороте на 180°.
V. Призма является комбинацией форм III и IV. Она имеет и плоскость
симметрии, и перпендикулярную ей ось 2. Такая простая форма, призма
без верхнего и нижнего оснований, обладает сама по себе ещё
дополнительными элементами симметрии, а всего имеет три оси 2, и три плоскости
симметрии. Но чтобы образовать из этой простои формы геометрическую
призму, надо добавить ещё пинакоид, т. е. верхнее и нижнее основания, что
приведёт к образованию различных сложных форм, т. е. или ромбической,
или моноклинной призмы (см. ниже). Последняя (форма V) обладает
только одной осью 2 и одной плоскостью симметрии т (рис. 37).
Сопоставление формы II и формы V показывает, что форма V получается
из формы II также путём комбинации последней с дигирой. Тогда (см.
проекцию, табл. 4) точки верхнего полушария попадут в нижнее и наоборот, в
результате появится плоскость симметрии и центр инверсии.
Эти 5 исходных форм составляют в схеме вывода возможных видов
симметрии как бы 5 основных исходных ступеней:
I примитивная (полярная), II центральная, III плапальиая, IV
аксиальная, V планаксиальная.
Стереографические проекции простейших форм ступеней I—V
изображены на таблице 4 и последующих. Соответствующие первым двум из них
виды симметрии (I и II) характеризуют два класса триклинной сингонии,
следующие три (III, IV и V) характеризуют три класса моноклинной
сингонии. Наименования видов симметрии, соответственно классов, даются ниже:
а) в международной системе Фёдорова — Грота—по общей форме, отвечающей
данному виду симметрии (М); Ь) в системе Фёдоровского института —по
элементам симметрии, характерным для данной сингонии, и по ступени,
отвечающей данному классу (Ф); с) по системе IT.
Виды симметрии триклинной сингонии:
1 С\—1.Моноэдрический, педиональный (М), агирно-примитивный (Ф),
триклинная гемиэдрия (IT). Если элементы симметрии отсутствуют,
то остаётся единственный, имеющий больше формальное значение
«элемент симметрии»—поворотная ось 1. Грани такого кристалла
являются моноэдрами. Полюс такой грани, расположенный в
верхнем полушарии, спроектируется в виде одной точки.
5 видои симметрии триклинвой и моноклинной сивгоний
Таблица 4
Ступень
Примитивная I
(полярная)
Центральная II
Планальиая III
Аксиальная IV
Планаксиальная V
Исходная форма
Стереографическая проекция
Пространственная
совокупность
элементов
симметрии
I моноэдр
II пиьакоид
III диэдр
безосный
IV диэдр осевой
V призма
V
Элементы
симметрии
Вид симметрии
Сингония
2т 1
Моноэдрический;
агирно-примитив-
ный
Пинакоидальный;
агирно-центральный
с-Г
Трпклинная
Диэдрический
планальный;
моногирно-планальныи
Диэдрический
осевой; моногирно-акси-
альный
С2-2
Призматический;
моногирно-планак-
сиальный
С2—2/т
Моноклинная
Состав кристаллов
Форма кристаллов
Кислый
виннокислый стронций
[CoOH(GlI01I)2COOLRr
/Р^
^
Анортит
CaOAU)82SiO,
/га
CuS043H,0
Молочный сахар
ОиНюОиН20
Гипс
CaS042HaO
и
ш
62
4.1. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
II d—1. Пинакоидальный (М), агирно-центральный (Ф). Триклинная
голоэдрия (IT). Ступень II отвечает наличию у кристалла центра
инверсии (1). Такой кристалл образован пинакоидами с обратно
равными гранями.
Виды симметрии моноклинной сингонии.
III Cs-—m. Диэдрический безосный (М), моногирно-планальный (Ф),
моноклинная гемиэдрия (IT). Вид
симметрии ступени III
отвечает наличию плоскости
симметрии, т. е. каждой точке
одной грани отвечает
зеркально-симметричная точка
другой грани. Общая форма:
диэдр безосный; частные:
1) пинакоид, имеющий
симметрично равные грани (грань
задана параллельно
плоскости симметрии) и 2) моноэдр
(грань задана
перпендикулярно ей).
IV Сг—2. Диэдрический осевой (М),
моногирно-аксиальный (Ф),
моноклинная гемиморфная рИс. 37.
гемиэдрия (IT). Ступень IV
характеризуется поворотной
осью 2, условно изображённой горизонтально. Тогда каждая
точка северного полушария при повороте формы на 180° вокруг
оси 2 совместится с соответственной точкой южного
полушария. Общая форма: диэдр осевой. Частные формы: 1) пинакоид
с совместимо равными гранями (грани заданы параллельно
оси 2), моноэдр (грани заданы перпендикулярно к оси 2).
V C:h—2/т. Призматический (М), моногирно-планаксиальный (Ф),
моноклинная голоэдрия (IT). Хотя ступень V относится к
простейшим, она отличается от остальных тем, что является первым видом
симметрии, образованным комбинацией двух элементов
симметрии: оси 2 и ей перпендикулярной плоскости т. Общая форма:
ромбическая призма. Частная форма: пинакоид (грань задана
параллельно оси 2 или плоскости т). На рис. 5 (см. §
9)—кристаллы гипса с гранями ромбических призм и пинакоида.
Как мы уже говорили, в кристаллографии «призма» имеет лишь
боковые грани без оснований (в отличие от призмы в геометрии), т. е. является
простой формой—призмой. Для образования сложной формы моноклинной
призмы необходима комбинация призмы и нинакоида, при которой
пинакоид пересечёт призму под углом к ребру, не равным 90°.
Если угол будет равен 90°, образуется сложная форма—ромбическая
призма (рис. 37).
Моноклинная и ромбическая
призмы.
§ 26. Дополнительные элементы симметрии
При пространственных сочетаниях исходных—порождающих элементов
симметрии возникают дополнительные элементы симметрии. Так,
например, точка пересечения дигиры с перпендикулярной ей плоскостью
симметрии есть в то же время центр инверсии. Линия пересечения п
плоскостей симметрии есть в то же время гира ?г-го порядка. (Напротив, обратное
§ 27] ВИДЫ СИММЕТРИИ РОМБИЧЕСКОЙ (ДИГИРНОИ) СИНГОНИИ 63
заключение само по себе неверно: гира n-го порядка не является линией
пересечения п плоскостей симметрии, если таковые дополнительно не
присутствуют; см. рис. 19.) Поэтому кратность точки определяется кратностью
исходных, а не всех элементов симметрии, через неё проходящих (§ 15).
Например, кратность точки, лежащей в пересечении двух взаимно
перпендикулярных плоскостей симметрии (тт)у равна F = 2x2 = 4, а не 8, хотя
через точку проходит ещё дигира, лежащая в пересечении плоскостей.
При выводе полной совокупности (так называемой полной
комбинации) элементов симметрии, присущих данному виду симметрии,
приходится отличать, таким образом, полную комбинацию от минимальной
комбинации, необходимой и достаточной для образования этого вида
симметрии.
Как мы увидим далее, например, возможная пространственная
совокупность из 2 осей 3 влечёт за собой возникновение четырёх осей 3 и трёх
осей 2 (вид симметрии 71-кубичсская сингония) и т. п.
§ 27. Виды симметрии ромбической (дигирнои) сингонии
Добавив к 5 исходным видам симметрии одну из следующих
поворотных осей: дигиру, тригиру, тетрагиру, тетрагироиду, гексагиру или гек-
сагироиду, получим 24 новых вида симметрии ромбической и средних
сингонии, рассмотренных нами в данном и следующих параграфах.
В соответствии с § 24 дополнительная гира устанавливается вертикально
(перпендикулярно плоскости проекции, в которой лежит дигира, или
горизонтальная плоскость симметрии). Добавление её к ступеням I и II новых
видов симметрии не даёт, но повторяет лишь уже известные виды симметрии
С« и Съъ. моноклинной сингонии.
Добавление дигиры к ступеням III, IV и V приводит к возникновению
3 видов симметрии дигирнои (ромбической) сингонии (см. табл. 5).
В обозначениях полной совокупности элементов симметрии, присущих
данному виду симметрии, количество элементов симметрии мы будем
проставлять, как условились ранее (§ 14), справа вверху (показатель
степени). Три дигиры обозначатся соответственно: 23; три плоскости
симметрии: т9.
Ивдекс v или h справа внизу означает вертикальные, соответственно
горизонтальные, плоскости симметрии, отсутствие его в случае т? обозначает
наличие 3 взаимно перпендикулярных плоскостей симметрии. Полная
комбинация элементов симметрии указывается в скобках.
Три вида симметрии ромбической сингонии:
I Со,, — mm— ромбо-пирамидальный (М), дигирно-планальиый (Ф),
ромбическая гемиморфная гемиэдрия (IT) (2j/n2). На проекции
возникают ещё 2 полюса граней (т. е. возникает плоскость
симметрии).
Общая форма: ромбическая пирамида. Частные формы:
1) ромбическая призма (заданная грань параллельна
поворотной оси), 2) пинакоид (грань параллельна плоскости
симметрии), 3) диэдр (грань перпендикулярна плоскости симметрии),.
4) моноэдр (грань перпендикулярна поворотной оси). На
кристалле каламина — таблица 5—видны грани: ромбической
пирамиды (две соседние нижние), призмы (узкие вертикальные
над гранями пирамиды), пинакоида, моноэдра (верхняя) и,
наконец, диэдров.
II />2—222—ромбо-тетраэдрический (М), дигирно-аксиалышй (Ф),
ромбическая энантиоморфная гемиэдрия (IT), (23).
В вадй симметрии рэмбической сннгонни
Таблица 5
U24
Ступень
Примитииыая I
(полярная)
Центральная II
Нланаль-
ная III
Аксиальная IV
Планаксиаль-
ная V
Стереографическая
проекция исходной формы
Добавляется элемент
симметрии
Поворотная ось 2-го порядка (дигира)
Стереографическая
^проекция, отвечающая но-
ному виду симметрии
Ф/ I *
z3_.
Пространственная
совокупность элементов
симметрии
Элементы симметрии
Нид симметрии
Состав кристаллов
Форма кристаллов
См. табл. 4
(2 1)
Диэдрпческий
осевой—моноклинной сингонии
(см. табл. 4)
См. табл. 4
(2 т 1)
Призматический-
моноклинной
сингонии
(см. табл. 4)
2 mi
4
f
Г
23
Ромбопирамидальний, дигирно-пла-
нальный
С^0—mm
Ромботетраэдриче-
ский, дигирно-
аксиальный *)
Do—222
Каламин,
гемиморфит
Zn3(0H)2SiO8
Эпсомит, английская
соль MgS047H>()
Ol
Рвотный камень,
виннокислый
антимония калия,
[(CHOH)3(COO)J.
■K-SbO-HaO."
2WI
Ромбодипирами-
дальный дигирно-
планаксиальный
К/, =D<>h—ттт
'V//
С ера
Иод
*) Буквой /) обозначаются диэдрическне виды симметрии, имеющие более одной поворотной оси.
66 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. I
(Виды симметрии, являющиеся сочетанием нескольких
поворотных осей, называются диэдрическими и обозначаются D.)
Сочетание двух взаимно-перпендикулярных порождающих осей
2 влечёт за собой возникновение, и третьей оси 2, им
перпендикулярной.
Общая форма—ромбический тетраэдр (через середины каждой
пары противолежащих рёбер проходят поворотные оси 2).
Частные формы: 1) ромбическая призма (грань параллельна
одной из осей 2); 2) пинакоид (грань перпендикулярна к оси 2 и
параллельна другим осям 2).
На кристалле эпсомита (таблица 5) видны грани ромбического
тетраэдра, ромбической призмы и пинакоида.
Ill D*h—тптт (иногда этот класс обозначают также Vh). Ромбо-дипира-
мидальный (М), дигирно-планаксиальный (Ф), ромбическая
голоэдрия (IT) (23ml). Общая форма—ромбическая бипира-
мида. Частные формы: ромбическая призма (грань
параллельна одной из осей 2); пинакоид (грань параллельна
плоскости симметрии).
§ 28. Виды симметрии ромбоэдрической (тригональной, тригирной)
сингонии
Добавление к исходным 5 ступеням вертикальной тригиры влечёт
за собой возникновение 5 новых видов симметрии ромбоэдрической сингонии
(см. табл. 6).
I Сз—3— тригонально-пирамидальный (М), тригирно-примитивный (Ф)г
ромбоэдрическая тетартоэдрия (IT) (ось 3|).
Общая форма—тригональная пирамида. Частвые формы: 1) три-
гоиальная призма, 2) моноэдр.
Кристалл периодата натрия (таблица 6) образован тремя три-
гональиыми пирамидами и моноэдром.
II Сы—3—ромбоэдрический (М), тригирно-централышй (Ф),
гексагональная тетартоэдрия II рода (IT) (ось 3). Общая форма—ромбоэдр-.
Частные формы: 1) гексагональная призма, 2) пинакоид.
III Сз„—Зт— дитригонально-пирамидальный (М), тригирно-планальный (Ф)>
ромбоэдрическая гемиморфная гемиэдрия (IT) (3ml).
Общая форма—дитригональная пирамида. Частные формы:
1) дитригональная призма, 2) тригональная пирамида, 3)
тригональная призма, 4) гексагональная пирамида, 5)
гексагональная призма, 6) моноэдр.
IV />з—32—тригоиально-трапецоэдрический (М), тригирно-аксиальный (Ф)~
Ромбическая энантиоморфная гемиэдрия (IT) (3 |23j). Все оси
полярны. Общая форма—тригональный трапецоэдр. Частные
формы: 1) дитригональная призма, 2) тригональная дипира-
мида, 3) ромбоэдр, 4) гексагональная призма, 5) тригональная
призма, 6) пинакоид.
V D^—3m-—дитригонально-скаленоэдрический (М), тригирно-планакспаль-
ный (Ф), ромбоэдрическая голоэдрия (IT) (3 2° яг3).
Горизонтальная плоскость симметрии из-за 3 отсутствует, в отличие от
других видов симметрии ступени V. Общая форма —дитриго-
нальный скаленоэдр. Частные формы: 1) дигексагональная
призма, 2) гексагональная бипирамида, 3) ромбоэдр, 4)
гексагональная призма, 5) пинакоид.
§ 29] ВИДЫ СИММЕТРИИ ТЕТРАГОНАЛЬНОЙ (ТЕТРАГИРНОЙ) СИНГОНИИ 67
§ 29. Виды симметрии тетрагональной (тетрагирной) сингоыии
Добавление к исходным 5 ступеням вертикальной тетрагиры,
соответственно тетрагироиды, влечёт за собой возникновение 5, соответственно 2Г
а всего 7 новых видов симметрии тетрагональной сингонии (табл. 7).
Начнём с видов симметрии, возникающих в результате добавления оси 4.
I С4—4—тетрагонально-пирамидальный (М), тетрагирно-примитивный
(Ф), тетрагональная тетартоэдрия (IT) (ось 4 j ). Общая форма —
тетрагональная пирамида. Частные формы: 1) тетрагональная
призма, 2) моноэдр.
II C±h—4///г—тетрагонально-бипирамидальный, тетрагирно-централышй
(Ф), тетрагональная параморфная гемиэдрия (IT) (4тл). Общая
форма—тетрагональная бипирамида. Частные формы: 1)
тетрагональная призма, 2) пинакоид* В таблице 7 кристалл
шеелита образован четырьмя тетрагональными бипирамидами.
III C±v—4mm—дитетрагонально-пирамидалышй (М), тетрагирно-планаль-
иый (Ф), тетрагональная гемиморфная гемиэдрия (IT) (411Щ).
Общая форма—дитетрагоиальиая пирамида. Частные формы:
1) дитетрагональная призма, 2) тетрагональная пирамида,
3) тетрагональная призма, 4) моноэдр.
IV «D4—42— тетрагонально-трапецоэдрический (М), тетрагирно-аксиаль-
ный (Ф), тетрагональная энантиоморфная гемиэдрия (424).
Общая форма—тетрагональный трапецоэдр (поворотные оси 2
проходят через середины рёбер, непересекающихся с осью 4).
Частные формы: 1) тетрагональная бипирамида, 2)
дитетрагональная призма, 3) тетрагональная призма, 4) пинакоид.
Кристалл монокалийтрлхлорацетат (таблица 7) образован
тетрагональным трапецоэдром и тетрагональной бипирами-
доп.
V Z)4h—4/ттт—дитетрагонально-бипирамидалышы (М), тетрагирно-плана-
ксиальный (Ф), тетрагональная голоэдрия (IT) (4«2 • nfv mh 1).
Общая форма—дитетрагональная бипирамида. Частные
форумы: 1) дитетрагональная призма, 2) тетрагональная
бипирамида, 3) тетрагональная призма, 4) пинакоид.
Добавление оси 4 к ступеням 1 if 111 влечёт за собой
возникновение ещё 2 видов симметрии:
1* S± —7±—тетрагонально-тетраэдрический (М), тетрагироидо-примитив-
нъш (Ф), тетрагональная тетартоэдрия II рода (IT) (4).
Общая форма—тетрагональный тетраэдр. Частные формы:
1) тетрагональная призма, 2} пинакоид.
Ill* D^a—7j2 Его обозначают иногда также Vd. Тетрагоналыю-скалено-
эдрический (М), тетрагироидо-плаиальный (Ф), тетрагональ-
пая гемиэдрия II рода (IT) (i2rmi).
Общая форма—тетрагональный скаленоэдр. Частные формы:
1) тетрагональная бипирамида, 2) тетрагональный тетраэдр,
о) дитетрагональная призма, 4) тетрагональная призма,
5) пинакоид.
Обратим внимание на проекции в случаях С4, С^ь. и S*. При таком
расположении точек, казалось бы, можно провести вертикальные плоскости
симметрии. Почему таковые отсутствуют?
Это вызвано тем, что точки в общем положении могут быть асимметричны.
Гира—поворотная ось я-го порядка сама по себе не влечёт за собой
появления п вертикальных плоскостей симметрии и не является,
следовательно, линией их пересечения.
Таблица 6
(> видов Симметрии тригонатьной (ромбоэдрической) сингонии
00
Ступени
Полярная I
Центральная II
Планаль-
ная III
Аксиальная IV
Планаксиаль-
ная V
Стореографическая
проекция исходной формы
Добавляется элемент
симметрии
Стереографическая
проекция, отвечающая новому
виду симметрии
Поворотная ось 3-го порядка (тригира)
Пространственная
совокупность элементов
симметрии
Элементы симметрии
Вид симметрии
Состав кристаллов
Форма кристаллов
\
Н
Тр игона льно-пира-
мидальний;
тригирно-прими-
тдвный
с3-з
Периодат натрия
NaJ043H2O
/К
<£— )\
3= 31
Ромбоэдрический;
тригирно-централь-
ный
с3/-з
Доломит
CaMgG03
И^
з™;;
Днтригонально-
пирамидальный;
тригирно-планаль-
н ый
Cov-Zm
Турмалин
Si803,(BOH)4R3A]6>
где R = A12, Mg3
Fe3, Ke, Li6,<TsTae, He
fcK
^
32d
Тригонально-трапе-
цоэдрический;
тригирно
-аксиальный
А,-32
Низкотемпературный кварц SiOa
Ш^1
32*т?Д
Дитригонально-
скаленоэдрическнй;
тригирно-илан-
аксиальный
D.6d - 3m
Кальцит
СаСО,
Таблица 7
7 видов симметрии гетраг.шалыми сингонии.
Ступени
Стереографическая
проекция
простейшей формы
Добавляется
элемент
симметрии
Стереографическая
проекция,
отвечающая новому !
виду симмет- !
рпи
Полярная
(примитивная) I
О
Центральная И
0
1 Планаль-
ная III
а
D
Аксиальная IV
'^)
План
аксиальная V
\д
\3
в
и
Поворотная ось 4-го порядка (тетрагира)
С ♦ у
/С х
xN\
\^ х]
W|\7
1
Инверт-
ная I*
О
Планинверт-
ная III* (IV*)
а
D
Инверсионная ось 4-го порядка
(тетрагироида)
С* j
уч-
[ 4- Ж. 1
\/о\
Г\р/
1 0* 1
Пространственная
совокупность
элементов
симметрии
Элементы
симметрии
Вид симметрии
Состав
кристаллов
Формы
кристаллов
И
Тетрагональ-
но-пирами-
j дальний;
тетрагирно-
иримитивный
С4-4
Вульфенит
РЬМоО.
I
ЛГУ
1
kmh\
Тетрагонально-
бипирамидаль-
ный;
тетрагирно-
центральный
См -Ь/т
Шеелит
CaW04
4ю*
Дитетраго-
ьально-пира-
мидальный;
тетрагирно-
планальный
С±и — 4 mm
Сукциниодимид
C4H402NJ
Vi
424
Тетрагонально-
трапецоэдри-
ческий;
тетрагирно-
аксиальный
At - 42
Трихлорацетат
калия
/СС13С00К\
^СС1,С00НУ
424
Дитетрагонально
бипирамидаль-
ный;
тетрагирно-
планаксиальный
D±h — \\mmm
Касситерит
Sn00
■^
i м и
w
г»
Тетрагональ-
но-тетра-
эдрический*)
тетрагироидо-
примитивный
st-l
Пентаэритрит
С(СН2ОН)4
422т?.
Тетрагонально-
скаленоэдри-
ческий;
тетрагироидо-
планальный
Vd=DM-l2
Медный
колчедан
CuFeS0
*) По Шубннкоиу, Флинту, Гюкиюр] (1940) и др. авторам известен только 1 представитель класса S^—алюмосиликат
кальция Al2Ca2Si07 (на основании фигур травления), а пентаэритрит относится к классу С v. Проведённое на мисопоставление структурных
данных показывает, что симметрия, например пентаэритрита, определяется пространственной группой S^
Та же пространственная группа (§ 57) характеризует симметрию BP04t AsP04, [Cu(S = C(CH3)(NH2))4C1j; [Ag(S = C(CH3)(NH2))4]Cl,
Мп3Р и других веществ, которые относятся, таким образом, к виду симметрии S±.
!! IS
72 4.1. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. I
Грани пирамиды, бипирамиды, тетраэдра в видах симметрии С4, С^, 5±
не являются симметрично равными, как это видно из рисунка кристаллов
шеелита.
В случае асимметричных точек вполне возможно возникновение
вертикальных плоскостей симметрии при удвоении количества точек (рис. 19, Ь).
Поэтому, например, кристаллы дитригональной и дитетрагональной
ступеней имеют вертикальные плоскости симметрии.
Подлинно тетрагональный тетраэдр (как кристаллографическая форма),
имеющий элементы симметрии, отвечающие геометрической форме, в
частности вертикальные плоскости симметрии, попадает не в тетрагональ но-
тетраэдрический (54), а в (ди)тетрагонально-скаленоэдрический (Fd = Zbd)
вид симметрии (см. § 37).
§ 30, Виды симметрии гексагональной (гексагирной) сингонии
Добавление вертикальной оси 6 даёт пять и оси 6 ещё два, а всего 7 видов
симметрии гексагональной сингонии (табл. 8).
I Со—6 —гексагонально-пирамидальный (М), гексагирно-примитивный
(Ф), гексагональная тетартоэдрия (IT) (ось 6J).
Общая форма—гексагональная пирамида. Частные формы:
1) гексагональная призма, 2) моноэдр.
II— Cg/,—6m—гексагонально-бипирамидальный (М), гексагирно-централь-
ный (Ф), гексагональная параморфная гемиэдрия (IT)
(6m/, 1).
Общая форма—гексагональная бипирамида. Частные формы:
1) гексагональная призма, 2) пииакоид.
III Се»—6mm—дигексагонально-пирамидальный (М), гексагирно-планаль-
ный (Ф), гексагональная гемиморфная гемиэдрия (IT)
(6jmJ).
Общая форма—дигексагональная пирамида. Частные формы:
1) дигексагональная призма, 2) гексагональная пирамида,
3) гексагональная призма, 4) моноэдр.
IV Z>6—62—гексагонально-трапецоэдрический (М), гексагирно-аксиальный
(Ф), гексагональная энантиоморфная гемиэдрия (IT) (6 26).
Общая форма—гексагональный трапецоэдр. Частные формы:
1) дигексагональная призма, 2)гексагональная бипирамида,
3) гексагональная призма, 4) пинакоид.
V Д,л—штат—дигексагонально-дипирамидальный (М), гексагирно-планакси-
альный (Ф), гексагональная голоэдрия (IT) (626т£тл1).
Общая форма —дигексагональная бипирамида. Частные
формы: 1) дигексагональная призма, 2) гексагональная
бипирамида, 3) гексагональная призма, 4) пинакоид.
Добавление оси 6 к ступеням I и III даёт 2 новых вида симметрии:
I Сз/i— 6—тригонально-бипирамидальный (М), гексагироидо-примитив-
ный (Ф), тригональная параморфная гемиэдрия (IT) (ось
6 =3/m=3mh).
Общая форма—тригональная бипирамида. Частные формы:
1) тригональная призма, 2) пинакоид.
Ill Dsh — 6m2—(ди)тригонально-бипирамидальный (М), гексагйройдо-пла-
нальный (Ф), тригональная голоэдрия (IT) (6 23|m^).
Общая форма—дитригональная бипирамида. Частные
формы: 1) дитригональная призма, 2) гексагональная дипи-
рамида, 3) гексагональная призма, 4) тригональная
бипирамида, 5) тригональная призма, 6) пинакоид.
§ 31] виды симметрии кубической сингонии ___ 73
Эти два вида симметрии причислялись раньше к
тригональной—ромбоэдрической (под)сингонии, затем к гексагональной. Некоторые авторы
считают эти два вида симметрии относящимися к самостоятельной восьмой,
тригональной сингонии (в отличие от ранее рассмотренной ромбоэдрической).
§ 31. Виды симметрии кубической сингонии
Характерными элементами симметрии кубической сингонии являются
оси З4.
Как мы знаем (§ 24), между такими осями возможны лишь углы 109°28'
и 70°31', отвечающие углам между диагоналями куба. Если мы допускаем,
Рис. 38. Образование видов симметрии Т и Тд.
что количество осей 3 больше единицы, нетрудно показать, что в простейшем
случае оно равно 4, ибо всякая тригира, действуя на тригиру же, повторяет
её трижды, а всего возникнут 4 тригиры (рис. 38, а).
Так как у ступени I центра инверсии нет, все 4 тригиры полярны.
При этом 3 из них должны быть направлены остриями обратно четвёртой
(рис. 38,а). Но в таком случае через центры противоположных граней
куба проходят три биполярные оси 2 (рис. 38, 6). В результате возникает
простейший вид симметрии кубической сингонии Т.
? видов симметрии гексаговальной сивговии
Таблица 8
Ступени
Примитивная
(полярная) I
Центральная II
Планаль-
ная III
Аксиальная IV
План-
аксиальная V
I* Инвертная
Планинвертная
III* (IV)
Стереографическая
проекция
простейшей формы
Добавляется
элемент
симметрии
Стереографическая
проекция»
отвечающая виду
симметрии
Пространственная
совокупность
элементов
симметрии
Элементы
симметрии
Поворотная ось 6-го порядка (гексагира)
С2С
чу® | ®\/
щ
62emGm. Г
х I х
Инверсионная ось 6-го порядка
(гексагироида)
6 = Smh
фл
62>»=32»л:
Вид
симметрии
Состав
кристаллов
Форма
кристаллов
Гексагонально
пирами-
дальпый;
гексагирно-
примитивный
Нефелин
(только по
фигурам
травления)
к
г
(
к<
N
<*
4.
а\
*
Гексагонально-
бпппрами-
дальный;
гексагирно-
центральный
У6Л"
- f» m
Апатит
Ca(Cl,F, ОН),
ЗСа3 (Р04)3
Дигекса-
гонально-
ппрамидаль-
ный;
гексагнрно-
планальный
Сл,.
6mm
Гренокит
CdS
Дигексагонально
трапецоэдри-
ческий;
гексагирно-
аксиальный
Д:
■02
В лсокотемпера-
турный кварц
Дигексагонально
бипирамидаль-
ный; гексагирно-
планаксиальный
2>6Л — 6mmm
Тригонально- I Дитригонально
дппирамидальный; бипирамидальный;]
Берилл
Ве,А14(ЗЮ,)в
гексагироидо-
примитивный
y3h'
Нет надежных
примеров
Ag2HP04?
гексагироидо-
плапальный
D*
ЗЛ"
6т2
Г>еннтоит *)
BaTiSiaOj
*) По кристаллографическим данным (см., например, Шубников, Флинт и Бокий8 (1040), к классу D%h относится лишь один
единственный (и то не вполне достоверный) представитель — бенитоит. Проведённое автором сопоставление структурных данных
показывает, что симметрия бенитоита определяется пространственной группой D\h (§ 57); он, следовательно, относится к классу l)^h.
Пространственная группа D*J} определяет симметрию также CeFG03, LaFC03, возможно NaLiC03 и др.
76 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. I
1 Т—23 (Пентагон)—тритетраэдрический (М), полигирно-примитив-
ный (Ф), кубическая тетартоэдрия (IT) (34|23).
Установив куб так, чтобы ось 2 стояла вертикально, строим проекцию:
2 горизонтальные взаимно перпендикулярные дигиры-(-след вертикальной
дигиры в центре проекции. В каждом квадранте—тригира, причём две
направлены остриями к северному и две—к южному полушариям в шахматном
порядке. Точка в общем положении будет повторена тригирой в том же
полушарии трижды, них проекции расположатся в одном и том же квадранте
(четверти) круга.
Вертикальная дигира переводит эту тройку в диагонально
противоположный квадрант, любая же из горизонтальных дигир переведёт всю эту
группу точек в южное полушарие. Их проекции займут два других
квадранта по диагонали, в соответствии с тетраэдрическим характером вида
симметрии.
Общая форма—пентагонтритетраэдр. Частные формы: 1) тетра-
гонтритетраэдр, 2) тригонтритетраэдр, 3) тетраэдр, 4)
ромбододекаэдр, 5) пентагондодекаэдр, куб.
Добавив к ступени I центр инверсии, получаем ступень II, в которой
возникают 3 взаимно перпендикулярные плоскости т, параллельные граням
(100) и пересекающиеся в центре куба. Эти три плоскости разбивают куб на
8 октантов (см. рис. 38, с).
II Ти-т3—дидодекаэдрический (М), полигирно-центральный (Ф),
кубическая параморфная гемиэдрия (3423т31).
На проекции каждой точке северного полушария отвечает
аналогичная точка южного полушария (24 точки).
Общая форма—дидодекаэдр. Частные формы:
^пентагондодекаэдр, 2) ромбододекаэдр, 3) тетрагонтриоктаэдр, 4) три-
гонтриоктаэдр, 5) октаэдр, 6) куб.
111 Та—43т—гексатетраэдрический (М), полигирно-планальный (Ф),
кубическая гемиморфная гемиэдрия (IT) (34|23m6).
Добавление к ступени I плоскости симметрии параллельно стороне
куба не даёт новых результатов (получается ступень II). Остаётся ещё одна
возможность—если плоскость m пройдёт через 2 противоположных ребра
по диагонали грани куба (рис. 39). Такая плоскость включает в себя две
оси 3J.
Рис. 39. б плоскостей симметрии, отвечающие виду симметрии Td.
Две другие оси 3 повторят эту плоскость 6 раз. Проекции двух таких
плоскостей пройдут по диаметру, а остальных четырёх, наклонённых к
плоскости проекции под углом 45°,—по дугам.
Количество точек в каждом квадранте по сравнению со ступенью I
удвоится (24 точки).
§ 311 виды симметрии кубической сингонии 77
Обща;* форма —гексатетраэдр. Частные формы: 1)тетрагонтри-
тетраэдр, 2) тригоитритетраэдр, 3) тетраэдр, 4) тетрагексаэдр
(пирамидальный куб), 5) ромбододекаэдр, 6) куб.
IV 0—43—пентагонтритетраэдрическип или гироэдрический (М), ноли-
гирно-аксиальный (Ф), кубическая энантиоморфная геми-
эдрия (IT) (344326).
Ступень IV должна возникнуть из ступени I путём добавления
оси 2.
При этом во всех сингониях ведущие оси становились биполярными.
Как видно из рис. 40, есть только одна возможность провести ось 2:
через середины противоположных рёбер куба. Соответствующий угол между
диагоналями (осями 3), т. е. 70°32', будет разделён осью 2 пополам, и при
Рис. 40. Возникновение вида симметрии О.
повороте вокруг оси 2 на 180° элементы симметрии и, в частности, тригиры
совместятся. Характерный для кубической сингонии комплекс З4
сохраняется. Но взаимное действие этого комплекса и одной оси 2 поведёт к тому,
что тригиры повторят ось 2 шесть раз и сделают её биполярной. (Если хоть
одна ось 2 останется полярной, то оси 3 исчезнут.) В свою очередь, дигиры
делают биполярными оси 3 по аналогичным соображениям. Но при наличии
4 биполярных осей 3 и 6 биполярных осей 2, проходящих через середины
рёбер, проходящие через центры граней оси 2 превратятся в оси 4. При
действии на точку в общем положении данной пространственной совокупности
элементов симметрии возникают 24 точки.
Общая форма: пентагонтриоктаэдр. Частные формы: 1) тетра-
гонтриоктаэдр, 2) тригонтриоктаэдр, 3) октаэдр, 4)
ромбододекаэдр, 5) тетрагексаэдр (пирамидальный куб), 6) куб.
VOit—тЗт—гексаоктаэдрический (М), полигирно-планаксиальный (Ф),
кубическая голоэдрия (IT) (3* 4'* 26 т9 1).
Комбинация элементов симметрии ступеней III и IV добавит ещё центр
инверсии 1, причём получится удвоение количества (48) точек против
классов О и 7V
Общая форма—гексаоктаэдр. Частные формы: 1)тетрагонтриок-
таэдр, 2) тригонтриоктаэдр, 3) октаэдр, 4) ромбододекаэдр,
5) тетрагексаэдр, 6) куб. См. таблицу 9.
б видов симметрии кубической сингонии
' Таблица 9
Ступени
Примитивная I
Центральная II
Планальная III
Аксиальная IV
Планаксиальная V
Стереографическая
проекция простейшей формы
Добавляются элементы
симметрии
Стереографическая
проекция, отвечающая новому
виду симметрии
Пространственная
совокупность элементов
симметрии
Элементы симметрии
3423
Две оси 3 под углом 109'28' друг к другу
342ят'Т
342"т6
,,44я2б
Л
Уш»У
3*4Г2V 1
Вид симметрии
Состав кристаллов
Форма кристаллов
Пентагонтритетра-
эдрический;
полигирно-прими-
тивный
Г —23
NaCIO,
Дидодека-
эдрический;
полигирно-
цен тральный
Г*-тЧ
FeSa пирит
^^
Гексатетраэдриче-
ский;полигирно-
планальный
rd-4:*m
ZnS сфалерит
Пентагоитриокта-
эдрический или
гироэдрический;
полигирно-
аксиальный
О —43
NH4C1 *)
нашатырь
фигуры
травления
Гексаокта-
эдрический;
полигирно-
планаксиальный
0/i — т\\т
CaF,
гроссуляр
Са3А12 (Si04)3
*) В кристаллографической и минералогической литературе (см., например, Шубников, Флинт и Бокий [9] (1940), Дана [10] (1937)
к классу О относится, например, куприт, СгьО. Структурные данные заставляют причислить СиаО к классу Oh. Повидимому, влияние
иримесей на возникновение форм, отвечающих классу О, довольно сильно. Следует относиться с осторожностью и к примеру NH4C].
, ее
Триклинная
(2 вида
симметрии)
и моноклинная
(3 вида
симметрии)
Ромбическая
(3 вида
симметрии)
Тригональная
(5 видов
симметрии)
Таблица 10
Стереографические вроекцпи 32 видов еиннетрпи
полярная
примитивная I
триклинная
гемиэдрия (1)
см. С
ромбоэдрическая
тетартоэдрия (3)
центральная
П
планальная
III
триклинная
голоэдрия (2)
см. С.,
3/
моноклинная
гемиэдрия (к)
аксиальная
IV
планаксиальная
V
инвертная
I*
планинвертная
III*
моноклинная
гемиморфная
гемиэдрия (2)
^2v
ромбическая
гемиморфная
гемиэдрия (4)
гексагональная
тетартоэдрия
2-го рода (G)
ромбоэдрическая
гемиморфная
гемиэдрия (О)
ромбическая
э нашпиоА.орфна я
гемиэдрия. (4)
ромооэдрическап
гемиэдрия (6)
моноклинная
голоэдрия (4)
V/,=D2/.
ромбическая
голоэдрия (8)
ромбоэдрическая
голоэдрия (12)
Жирной чертой обведены 8 видов
симметрии, образованных только
1 элементом симметрии:
1, 2, 3, 4, 6, Т, 2(= #п), 4
Звездочкой в левом верхнем углу
отмечены 11 видов симметрии,
имеющие гхентр инверсии.
В скобках — количество наиболее
общих положений точек.
Эти числа: 1,2, 3, 4, 6, 8, 12, 16,
24, 48 образуют кристалло? рафиче
ский ряд чисел.
Наименования по IT
Гетрагональная
(7 видов
симметрии)
тетрагональная
тетарюэдрия (4)
Гексаг опальная
(7 ви;.ов
симметрии)
Кубически н
(5 видов
симметрии)
гексагональная
тетартоэдрия (6)
кубическая
тетартоэдрия
гемиэдрин (12)
4 Л
тетрагональная
пара.* орфная
гемиэдрия (8)
6/i
гексагональная
параморфиая
гемиэдрия (12)
>« *•<
кубическая
параморфиая
гемиэдрия {'24)
'4v
тетрагональная
ге.. иморфная
гемиэдрия (^)
'6/>
гексаголальная
гелмморфная
гемиэ;»рия (12)
кубическая
гемиморфная
гемиэдрия (24)
иеграгональнан
эиаппшоморфная
гемиэдрия (8)
гексагональная
энантиол.прфная
гемиэдрия (12)
'Ah
кубическая
эна нтиоморфная
гемиэдрия (24)
тетрагональная
голоэдрт, (16)
геграгоннльнаь'
1етарю>дрни
2-го ро^а \4)
D,. i
oh
гексагональная
голоэдрия (2-1)
кубическая
голоэдрия (48)
те тра гон ал ь н а я
гемиэдрия
2-го рота (8)
'3/»
лз/,
тригональнал I трнгональная
параморфиая голоэдрия (12)
гемиэдрия (6) I
I
82 ч- t. основы теории cTPV&fyfr [гл. 1
§ 32. Сопоставление проекций 32 видов симметрии
В таблице 10 для большей наглядности сопоставлены проекции всех
32 видов симметрии. Из таблицы 10 (см. стр. 80, 81) видно, что центральная
ступень отвечает параморфной, планальная — гемиморфной, аксиальная —
инантиоморфной гемипдрии (в ромбической, средних*) и высшей син-
гониях).
§ 33. Виды симметрии с одинаковой кратностью позиций точек
в общих положениях
Из таблицы 11 следует, что одни и те же кратности р позиций
точек встречаются в следующих видах симметрии:
Таблица 11
V
1
2
3
'*
Вид симметрии
с.
^i» С5» С2
с,
с2„, v-n2
c2v, cv s4
V
6
8
12
Вид симметрии
V
If)
2'i
48
Вид симметрии 1
fl4*
г,,, о
1
§ 34. О соответствии форм кристаллов 32 видам симметрии
Сопоставление форм кристаллов с формами, предсказываемыми
теорией симметрии континуума, приводит к весьма хорошим результатам.
В природе не найдены формы, которые относились бы к какому-нибудь
непредвиденному «33» виду симметрии. С другой стороны, если отдельные
виды симметрии представлены весьма богато (0/„ Ал» Ал и ДР-)> то таким,
как iS4 или Сзл, отвечает пока небольшое количество изученных объектов.
Большинство форм кристаллов, встречающихся в природе, являются обычно
комбинациями простых кристаллографических форм. На рис. 41 приведены
некоторые такие комбинации для кубической сингонии.
§ 35. Закон рациональных отношений
Для описания положения граней кристалла естественно пользоваться
приемами аналитической геометрии, а именно системой координатных осей.
Если в аналитической геометрии часто предпочитают пользоваться
декартовыми координатами, т. е. осями, угол между которыми составляет
90°, то в кристаллографии выбирают систему координат так, чтобы оси
координат были параллельны рёбрам кристалла, и если в кристалле имеются
поворотные оси,—совпадали бы с последними.
) За исключением вида симметрии С.
$щ
ЗлкОН РАЦИОНАЛЬНЫХ ОТНОШЕНИЙ
S3
Напомним, что положительным направлением в аналитической
геометрии обычно считают (рис. 42, а): для оси я—вперёд к читателю, для
оси у—вправо от читателя и для о.си z—вверх.
й=г\
Ь>~
У
WJ
П1\
Рис. 41. Некоторые комбинации простых форм кубической сингонии:
а) и Ъ) кубооктаэдры, с) комбинации куба, октаэдра и ромбододекаэдра, d)
комбинация куба с тетрагонтриокта:>дром, е, комбинация куба с тригонтриоктаэдром.
Кроме того, в некоторых случаях (гексагональная сингония)
пользуются не тремя, а четырьмя осями, из коих одна совпадает с осью 6, а 3
других лежат в плоскости, перпендикулярной оси 6, проходя через
противоположные рёбра призмы (дипирамиды) (рис. 42: 6). Положительное направление
а h
Рис. V2. Положительное и отрицательные паиранления координат:
а) всех сингонии, кроме гексагональной, Ь) гексагональной.
на рис. 42 показано стрелками на концах осей. Систему координат
данного кристалла иногда называют «крест осей».
Второй особенностью в определении положения плоскости в
кристаллографии, но сравнению с аналитической геометрией, является выбор само-
84 ч* i. Ьсноёы теорИи структур [гл. 1
стоятельного масштаба измерений по каждой оси, ибо хотя единицы измере.-
иий по разным осям (осевые единицы) и могут быть равны, но в общем
случае они не равны друг другу. Между тем, в аналитической геометрии единицы
измерения по всем трём осям обычно одинаковы.
Таким образом, для определения положения грани кристалла
необходимо знать:
3 угла между осями а, р и у)
3 осевые единицы а, 6, с, откладываемые по этим осям.
При описании внешней формы кристалла нас интересуют, собственно,
не абсолютные значения осевых единиц, а их отношение а : 6 : с.
Совокупность углов между осями и отношений осевых единиц называют
иногда «метрикой» данного вида кристаллов. Так как осевые единицы
и углы (осевой крест) меняются
с температурой (и давлением), то
метрика должна быть отнесена
к определённым условиям Т и р
(см. § 106).
Для нахождения осевых
единиц кристалла одну из граней
кристалла, которая пересекает
все три оси координат, принимают
за единичную грань. Единичной
гранью называется грань,
пересекающая оси на расстоянии
одной осевой единицы от центра.
[Грань ABC (рис. 43) является
единичной.]
Рис. ъя. Грани (Ш) и (\Щ. Положение любой грани
кристалла можно задать так
называемыми параметрами грани, однако удобнее пользоваться числами,
обратными им, называемыми индексами грани.
Индексы грани—это величины, обратно пропорциональные параметрам
грани—отрезкам, отсекаемым плоскостью грани на осях (причём за единицу
приняты отрезки, отсекаемые единичной гранью).
Если мы имеем, следовательно, плоскость, которая отсекает на осях
отрезки (параметры), равные числу осевых единиц ,г, /у, z, то вместо
отношения этих отрезков х : у : z берут отношение
±:1 : 1=A:*:Z. (5)
х у z '
Числа Л, /с, I являются индексами грани. Их совокупность называется
символом грани. Символ грани в общем случае записывается так: (hkl),
символ единичной грани (111).
Если грань пересекает отрицательные направления осей, символ
запишется (hkl). (hkl) означает, что грань пересекает положительные концы
осей х и z и отрицательный конец оси у.
Рассмотрим, например, нахождение символа грани, параметры которой
равны х=3} у=3, 2=1 (см. грань АгВгС рис. 43), т. е. х : у : г.-3 : 3 : 1.
ill
Отношение индексов граней h■ : к :1 = ~ : т{- : у .
Приведём дроби к общему знаменателю, который после того отбросим.
Получим h : к : 1=1 : 1 : 3. Следовательно, символ грани (ИЗ).
Если грань параллельна одеюй из осей, например, ху отношение её
параметров запишется: х : у : z = oo : у : z.
§ 3G| ОТНОШЕНИЯ ОСЕВЫХ ОТРЕЗКОВ И УГЛОВ КРИСТАЛЛОВ 85
Найдём отношение индексов: h : к : / -■ — : — : — .
Следовательно, символ данной грани: (ОА/).
Если грань параллельна двум осям, например, у и z, её символом будет
(М)0). Разделив на А, получаем окончательно (100).
Например, приняв для кубической системы (^а=^/р=,/у—90°)
декартовы координаты, поскольку они параллельны рёбрам куба и осям 4, найдем,
что (как видно из рис. 44) каждая грань куба перпендикулярна одной из
осей и параллельна двум другим. Символ передней грани куба будет (100),
правой (010), верхней (001), задней (100), левой (010), нижней (001).
Грани ромбического додекаэдра получат символ (110) и т. п., грани
октаэдра (111) и т. п. Символ (111) означает грань октаэдра в передней
левой нижней его части (рис. 44).
Если оси координат принимать параллельными рёбрам кристалла
и параметры граней измерять осевыми единицами (отсекаемыми единичной
гранью), то при таком методе характеристики положения граней выявляется
весьма важный закон—второй закон кристаллографии, закон рациональных
отношений:
Параметры любой грана кристалла, выраженные в осевых единицах,
относятся, как простые целые числа.
§ 36. Отношения осевых отрезков и углов кристаллов
различных сингошш
Отношения осевых отрезков и углы, характерные для различных
оингоний (рис. 45), непосредственно вытекают из конфигураций
соответствующих бипирамид (или призм). См. табл. 12.
Таблица 12
1 Сингония
Кубическая
1 Тетрагональная
Ромбическая *)
1 Ромбоэдрическая **)
Моноклинная
Триклинная
Гексагональная ***)
л, Ь, с
а = b =.- с
а—Ьфс
афЬфс
а — ') = с
афЪфс
афЬфс
ai — а^—а^фс
*. Р< 7
3b=~i»=Y=cJ0o
а=^у=90^
2.^ = Y^<)0°
а=1*у=£«Ю°
j-=Y = i)0°
$ФМ°
*Ф$ФчФ№
углы между осями 1
а1:а2:ая=110\
между осями 1
а и с=У0°
*) 13 сечениях xyi)t xOz и 0i/z ромбической бипирамиды лежат 3 ромба с разными
отношиниими осей.
**) Ромбоэдр можно представить себе как куб, вытянутый или сплюснутый вдоль
оси 3. С другой стороны, ромбоэдр мож,но рассматривать как форму, производную от
гексагональной бипирамиды. Ьинирамида сплюснутая (малые отношения с/а) даёт
сплюснутый ромбоэдр, вытянутая (большие отношения с/а)—вытянутый ромбоэдр
(рис. 29).
***) Позже мы остановимся на других способах описания форм ieKcaiопальной
сингонии, помимо сцетеады 4 осей.
86 ч. i. основы теории структур |гл. I
/
ч°
\1
/
100
i
> 001
100
/!
'001
7\
ою\
У
Рис. 44. СимвелыЗграней куба, октаэдра и ромбододекаэдра.
• ♦С
+а \ +а \
е f а
Рис. 45. Осевыо отрезки и углы в кристаллах различных сингоний:
а) кубическая, Ь) тетрагональная, с) ромбическая, d) мононлинпан, с) триклинная, /)
гексагональная, д) ромбоэдрическая.
§ 37]
ФИЗИ'ШСКИВ И ХИМИЧЕСКИЕ СВОЙСТВА КРИСТАЛЛОВ
87
§ 37. Физические и химические свойства кристаллов и элементы
симметрии последних. Фигуры травления
Изучение форм, в которых кристаллизуются вещества данной сингонии,
обнаруживает, что вещества, относящиеся к классам кристаллов с более
низкой симметрией, весьма часто кристаллизуются в формах, внешне
похожих па формы кристаллов, которые относятся к классам, обладающим
Рис. 'iG. Штриховка граней пирита.
^
J3
Рис. 47.
Комбинация куба и иента-
гондодекаэдра.
более высокой симметрией, относящимся к той же сингонии. Так, например,
пирит класс Th очень часто кристаллизуется в кубах совершенно
правильной формы, как и NaCl, класс 0\х, но иногда покрытых штриховкой (рис. 46).
Обращая внимание только на геометрическую форму, нельзя однозначно
решить, к какому из пяти классов кубической сингонии следует отнести
кристалл пирита: воввсех этих классах куб является частной формой, см. §31.
Рис. \Н. Симметрия куба н зависимости от штриховки, формы и
расположения частиц.
Если сопоставить штрихованный куб пирита с пентагондодекаэдром
(рис. 35) или комбинацией последнего с кубом (рис. 47), нетрудно
обнаружить, что штриховка граней куба параллельна длинным рёбрам Пентагон-
додекаэдра и характеризует симметрию пирита, показывая, что, например,
ось 4 у куба, образованного пиритом, отсутствует, иначе штриховка имела
бы совсем другой вид (рис. 48, а).
88 ч. i. осноны теории структур [гл. 1
Рассматривая куб как группу точек, фиксированных в его вершинах,
нетрудно представить, в каких случаях три оси 4, несмотря на
«кубическое» расположение точек, могут исчезнуть. Это будет иметь место, если,
например, точки в вершинах имеют симметрию цилиндра,
На рис. 48, Ь показан такой куб, имеющий симметрию тетрагональной
призмы, у него две оси 4 (из 3) заменены на две оси 2, т. е. эта фигура
даже иной сингонии. Вторая ось 2 на рис. 48, Ь не показана, во избежание
загромождения рисунка. Мы сейчас не рассматриваем, как в
действительности построен кристалл пирита (см. гл. IX). Для наших целей в этой главе
вполне достаточно того вывода, что, несмотря на свою внешнюю
высокосимметричную от природы геометрическую форму, кристалл может
принадлежать к классу, имеющему более низкую симметрию.
Поэтому, хотя во всех классах кубической сингонии вещества могут
кристаллизоваться в виде куба (или комбинации куба с другой формой),
геометрически совершенный куб кристалла другого класса, чем Oh, не имеет
положенного геометрическому кубу комплекса элементов симметрии
(84432,;т<)1), ибо его физические (кристаллографические) свойства не отвечают
его форме, и, например, куб в классе Т или 71/. не имеет осей 4 (сравни
рис. 46 и 48).
Очевидно, что для выяснения причин таких явлений необходимо
рассмотреть собстненную симметрию и ориентировку точек, слагающих
кристалл. Эта задача является довольно сложной. Рассмотрение таких систем
требует введения новых принципов, что и делается теорией симметрии
дисконтинуума, к изложению элементов которой мы переходим в
ближайших параграфах, а также физической теорией структур (см. главу II).
Здесь мы скажем несколько слов о кристаллографических методах
установления вида симметрии кристалла.
Основным мотодом является гониометрическое определение междугран-
ных углов. Впрочем гониометрический метод даёт однозначные результаты
не всегда. Иногда представляет трудности дажо определение сингонии.
См. [15].
Рассмотрим группу кристаллов разных веществ, имеющих вид
гексагональной призмы с углами между гранями 120°.
Если судить лишь но геометрическим данным, их всех можно было бы
отнести к одному и тому же классу. По если мы попробуем химически
воздействовать на грани кристаллов, подобрав соответствующие вещества,
то получаются своеобразные фигуры травления, форма которых, прежде
всего, зависит от внутреннего строении вещества.
На рис. 4!) сопоставлены фигуры травлении нефелина (а), апатита (Ь)
и кальцита (с). Мы ясно видим, что нефелин имеет только ось 0 и относится
к классу CY,, апатит имеет симметрию G/m и относится к классу См, а
кальцит имеет 323т?, и принадлежит к классу D-A(h Три оси 2 проходят через
ребра гексагональной.призмы. Три плоскости m—через середины граней
призмы. На рис. 50 изображены проекции верха кристаллов, имеющих
сечение гексагональной призмы: берилла (класс Zfoi), кальцита (/);»</)>
кварца (/>з) левого и правого кристаллов (энантиоморфны).
Наличие 6т£ в первом случае, 3ml во втором и оси 3 в третьем случае
чётко видно по положению «верхних» граней. (Рассмотрение кристалла
кварца в целом позволяет обнаружить ещё оси 2) (см. рис. 109).
Заметим, что исследование разных свойств кристаллов разными
методами по-разному выявляют симметрию кристалла. Например, изучение
скорости прохождения света систематически приводит к выводу о более
высокой симметрии кристалла (так, все кристаллы кубической сингонии
изотропны в отношении скорости света я неотличимы, таким образом, друг
§ 37}_ ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СНОПСТНЛ К1'ПСТЛЛЛ013 89
от друга). Травление обычно (по не всегда) позволяет открыть пониженную
(но сравнению с симметрией внешней формы) симметрию кристалла.
(7
(Г
(7
Я
а
v
Рис. Ъ9. Фигуры трдплонпн на кристаллах.
Надо помнить, что симметрия фигуры травления является функцией
не только симметрии кристалла, но и 1) симметрии (структуры) компонентов
реакции, а также 2) наличия примесей и продуктов реакции травления.
Рис. 50. Проекции верха кристаллов.
Примером влияния первого фактора является опыт Руайе (см. [17J),
показавшего, что при травлении кальцита рацемической (оптически
недеятельной) малоновой кислотой получаются симметричные «треугольники»
травления, при травлении же вращающей^малоновой кислотой (содержащей
90
Ч. I. ОСНОВЫ ШОРИИ СТРУКТУР
[гл. 1
молекулы только одной из двух эиантиоморфных форм) фигуры травлении
не имеют плоскости симметрии, вопреки структуре кальцита.
Примером влиянии второго фактора являются фигуры травления KCL
В старых курсах принималось, что КС1, в отличие от NaCl, принадлежит
к классу О (а не О/,), поскольку получались фигуры травления, повёрнутые
вокруг оси 4 (рис. 51, а). Позже
z
4
ш
V
выяснилось, что это вызвано
следами примесей, а КС1
принадлежит, как и NaCl (рис. 51, 6),
к классу 0\х.
Снимки травленых граней к\
разной степени чистоты,
полученные с помощью электронного
микроскопа, обнаруживают на
поверхности кристалла глубокий
рельеф («небоскрёбы» и
«колодцы») и показывают, что,
например, водный раствор НС1 не даёт фигур травления с гранями
(111)—октаэдра, которых требует теории, тогда как обезвоженный,
газообразный НС1 даёт такие грани и т. д.
Piic. Г>1. Фигуры» транлинии на кристаллах.
§ 38. Об отклонениях от закона постоянства углов
и плоской формы граней
Современная теория роста кристаллов приводит к необходимости
отклонений от закона постоянства углов кристалла, что и наблюдается.
Более того, как было показано Шубниковым [17], Леммлейном и другими
советскими учёными, на гранях кристаллов часто возникают пирамидки
(один из видов так называемых вицинальных граней), высота
которых зависит от
условий роста кристаллов.
Отклонения от
плоскогранной формы
особенно разительны, если
исследовать не
макрорельеф, а микрорельеф
граней, как это и
делается в электрономи-
кроскопических
работах, а также в интер-
ферометрических
исследованиях, например,
Толанского [16] (1943—
1945 гг.).
Толанский,
использовав интерферометри-
ческий метод,
позволяющий, как оказалось, различать рельеф до 5—20 А, установил, что на
поверхности кажущихся плоскими граней кристаллов кальцита, топаза,
кварца, гипса, каменной соли, слюды, сильвина, карбида кремния имеется
рельеф глубиной в разных точках от 5 до 1000 и более ангстрем (рис. 52).
Подобные данные приводят нас к формулировке следующего
положения:
«Кажущаяся плоской грань кристалла является лишь воображаемой
плоскостью, параллельной реальным микрограням, проведённой через
Г 2345*78 мм
Рис. 52. Рельеф поверхности грани кристалла гиыса.
§ 40] ПРОСТРАНСТВЕННАЯ РЕШЁТКА КРИСТАЛЛА 91
микрорёбра грани. На разных расстояниях от этой плоскости расположены
параллельные ей микрограии, образованные веществом кристалла и
пересекающиеся с боковыми; поверхностями рельефа по ломаным рёбрам, отрезки
которых параллельны рёбрам кристалла».
Таким образом, воображаемые (идеальные) рёбра и грани кристаллов
параллельны микрограням и микрорёбрам рельефа, т. е. задают их
направление. Это иоложепие устанавливает связь между макро- и
микроструктурой кристалла.
§ 39. Полиморфные прекращения и формы кристаллов
Говоря о структурных формах, которые могут быть образованы
кристаллами данного вещества, мы по можем упустить из виду явление
полиморфизма. В последующих главах это явление будет более подробно
рассмотрено. Ядесъ отметим лишь, что в классической кристаллографии под
полиморфизмом понималась способность вещества одного и того же состава
образовывать кристаллографически различные модификации.
Во всей классической литературе явление полиморфизма охватывает
лишь случаи образования одним и тем же веществом кристаллографических
форм, принадлежащих разным сингониям и, гораздо реже, к разным видам
симметрии той же сингонии.
Так, было известно, что углерод может кристаллизоваться в виде алмаза
(октаэдры, кубическая сингония) и графита (гексагональная призма,
гексагональная сингония). Углекислый кальций СаС03 образует минералы
кальцит (гексагональная сингония) и арагонит (ромбическая) и т. д.
Но, как показано теорией симметрии дисконтинуума и исследованиями
структур веществ, не только в одной и той же сингонии, но даже в одном
и том же классе данное вещество может образовать разные структуры.
Например, Fe, CsCl или RbCl образуют по меньшей мере по две
модификации, весьма различные по структуре, но тем не менее относящиеся
к классу О],.
Далее, следы примесей, как мы полагаем, гораздо чаще влияют на
выбор не только формы, но и структуры, чем это принято думать (см. § 103).
Поэтому, в более глубоком освещении, проблема полиморфизма и
условий фазовых переходов не является проблемой теории симметрии континуума
и должна быть рассмотрена с иных позиций.
В. ЭЛЕМЕНТЫ ТЕОРИИ СИММЕТРИИ ДИСКОНТИНУУМА
§ 40. Пространственная решётка кристалла. Понятие об элементарной
ячейке
Уже давно в кристаллографии была проявлена тенденция к
объяснению свойств кристаллов таким образом, что кристалл, например, кубической
формы, слагается из более мелких «кубиков», несущих в себе все элементы
его симметрии. При этом предполагалось, что дальнейшее продолжение
дробления приводит, в конце концов, к некоторому элементарному
многограннику, сохраняющему все элементы симметрии и структуры исходного
кристалла и теряющему эти свойства при дальнейшем раздроблении.
Разбив мысленно весь кристалл любой сингонии на элементарные па рал-
лелепипедальные многогранники, мы констатируем, что рёбра последних
образуют пространственную решётку (рис. 53, а).<
Теория пространственной решётки не осталась, однако, на этих
первоначальных, примитивных позициях, Она эволюционировала в сторону
92
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
представлении о пространственной решётке как бесконечной системе узлов,
составляющих бесконечные ряды плоских сеток, объединённых в
пространственную решётку (рис. 53, 6). Такая концепция, приводящая, на первый
взгляд, лишь к подчёркиванию роли узлов решётки, в действительности
позволила создать теорию симметрии дисконтинуума в её современном
виде.
Учение о пространственной решётке оперирует с системой узлов —
кристаллографических точек—т. е. позволяет сделать ряд общих выводов,
независимо от состава вещества кристалла, после чего эти выводы могут
Рис. 53. Пристрапстненнан решётка кристалла.
быть широко использованы в теории структур. Поэтому следует различать
понятия «пространственная решётка» и «структура кристалла»,
предусматривающие в первом случае определённую совокупность
кристаллографических, а во втором—материальных точек.
4—ф-
Х-
и
Х-£~-
-ш
§ 41. Трансляция
Представим себе бесконечный ряд кристаллографических точек,
расположенных по прямой линии на одном и том же расстоянии а одна от
другой, определяемый, величиной и направлением бивектора ±%и. (рис. 54).
Операции бесконечного количества смещений %а на определённое
расстояние и в определённом направлении, приводящая к образованию из
каждой данной точки бесконечного
ряда точек, называется трансляцией.
Все возникающие точки имеют
параллельно одинаковое расположение
и параллельно одинаковое
окружение и называются
параллельно-равными или идентичными.
Таким образом, идентичные точки
параллельно равны друг другу.
Расстояние а между двумя
ближайшими идентичными точками ряда
(вдоль вектора %а) называется периодом идентичности.
Соответствующий трансляции элемент симметрии дисконтинуума
называется переносом, трансляционным вектором (или бивектором). Его можно
было бы назвать вектором идентичности.
При отсутствии бесконечного ряда точек трансляция не имеет места.
В учебниках часто называют эту операцию не трансляцией, я иосту-
нанием, а соответствующий ей вектор—трансляцией. (Нам кажется, что
термин «поступание» для операции, которая приводит в совмещение
уходящий в бесконечность ряд точек, не очень удачен.)
^ТТа
~ТТПГ
Рис.
Г/t. Одномерный трансляционный
ряд.
У ?1.
ДЬУМШ'НАЯ ТРАНСЛЯЦИОННАЯ Г1 УППЛ
§ 42. Двумерная трансляционная группа
93
мы приведем
Представим теперь себе плоскую сетку, состоящую из бесконечной
совокупности узлов (точек), возникающих друг из друга в результате
смещения в одном направлении (параллельно оси .г) на расстоянии а, в другом
(параллельно оси у)—на расстоянии b друг от друга (рис. 5Г>).
Осуществив сначала смещение ± т„, затем ± ч
точку к совмещению с другой точкой
той же сетки (пунктиром показаны
смещения + ча + чь и --frt4-fb). В равной мере,
подвергнув все точки нашей бесконечной
плоской сетки такому смещению, мы как бы
совместим её с исходным положением.
Кратчайшие расстояния а и соответственно Ь
между ближайшими узлами сетки
называются периодами идентичности (в
направлении оси х и соответственно у).
Если *са и ih обозначают
соответствующие трансляции, то их пространственная
совокупность (tatb) называется <)ву мерной
трансляционной группой.
Данной трансляционной группе,
действующей на точку, отвечает
определённая плоская сетка. Данная плоская сетка
может быть образована 'бесконечным
множеством трансляционных векторов, подчиняющихся
Двумерная
трансляционная группа.
условию
■тхп
:И*а
где- т и п-
ч> (0)
-целые числа (см. рис. 56 и 57).
Очевидно,.что чем меньше период идентичности в направлении данной
оси, тем плотнее сидят узлы в данном направлении.
Иараллелограмы (ячейки плоской сетки, имеющие узлы решетки
только в вершинах) называются однократно примитивными или просто
примитивными. Так как каждый узел
принадлежит одновременно 4 соседним ларал-
лелограмам, а каждому данному на -1 ,
аждый такой параллелограм содержит
то
,-. 4=1 узел.
Плоскую сетку можно представить
себе к^к построенную из разных
примитивных параллелограмов, имеющих
одинаковое основание и высоту, а следовательно,
одинаковую площадь. С уменьшением
расстояний d\ d"y d"' между рядами
увеличивается расстояние между точками в
пределах ряда, т. е. уменьшается плотность
узлов (рис. 56).
В зависимости от выбора
трансляционной группы, элементарный
параллелограм может содержать внутри ещё 1 узел, а всего 2 узла; внутри ещё
- узла, а всего 3 узла и т. д. (см. рис. 57). Если внутри параллелограм
имеет т узлов, его кратность равна /г=да+1. Такие иараллелограмы
называются двукратно-, трехкратно- ... соответственно и-кратно-примитивными.
Рис. 55. Плоская сетка может
быть построена из различных
примитивных параллелограмов.
Площади всех однократно-при-
митинных параллелограмов сотки
одинаковы.
94 4. !• о^йовы теории структур [гл. 1
Так как данная площадь сотки F см2 содержит N узлов, то на один узел
приходится площадь
/=
N'
0)
•^^—- ОднократноприлштиВныи параллелограмм
— —»— — ДЬухкратнопримитиВный »»
Трехкратнопримитибныи п
Рис. 57. Однократно, -двукратно- и
трёхкратно-примитивные параллелограмм
площади относится, как 1:2:3.
Их
на два узла—площадь 2 / и т. д. Иначе говоря, площади элементарных
параллелограмов данной плоской
-V сетки относятся, как их
кратности. Если последние одинаковы,
то площади равны.
§ 43. Трёхмерная
трансляционная группа
Пространственная
совокупность трёх трансляций (хахьхс),
параллельных осям хуу и z,
называется трёхмерной
трансляционной группой (рис. 58).
Бесконечная совокупность
точек, возникающих при действии
на данную точку трёхмерной
трансляционной группы {хахьхс)}
называется пространственной
решёткой, а расстояния а, 6 и с
между точками (узлами решётки)—периодами идентичности
пространственной решётки.
Элементарный параллелепипед—элементарная ячейка
пространственной решётки, определяемой трансляционной группой {хахьхс), часто
называется параллелепипедом
повторяемости.
В то время как данной
трёхмерной трансляционной
группе (хахьхс), действующей
на точку, отвечает одна
единственная, строго определённая
пространственная
решётка,данная пространственная решётка
может быть образована
бесконочным множеством троек
трансляционных векторов (TjToT,),
подчиняющихся условию
х' = ±тха±пхь±рхс, (8)
т. о. могущих представлять
собой векторную сумму или
разность исходных или кратных
им векторов (т, п9 р—целые
числа) (рис. 58).
Очевидно, что в
пространственной решётке можно
выбрать трансляционные векторы, а значит и параллелепипед повторяемости
различно (рис. 56—58. Рис. 56 и 57 можно представить как проекции
пространственной решётки на плоскость, в которой лежат х и у).
Параллелепипеды, внутри которых нет узлов, называются однократно-
примитивными (или простыми): они имеют 8 узлов (вершин), каждый
Т.-Та + гч+Ъ
Рис. 58. Трёхмерная трансляционная группа
и образованная ею пространственная рещётка.
§ 44] ИНТЕРПРЕТАЦИЯ ПРОЦЕССА ОБРАЗОВАНИЯ ПРОСТРАНСТВЕННОЙ РЕШЙТКИ 95
из которых принадлежит одновременно 8 соседним параллелепипедам,
а каждому данному на 7а- Таким образом,
I О
п
1 ft _ 8
Если внутри параллелепипеда имеются ещё т узлов, его кратность будет
равна и=/ю+1.
Такие параллелепипеды называются соответственно двукратно-,
трёхкратно, «-кратно-примитивными. Если объём, занимаемый N узлами
пространственной решётки, равен V, то объём, занимаемый одним узлом, равен
о =
/V
(9),
двумя узла ми-
/: и т. д.
Иначе говоря, объёмы элементарных параллелепипедов данной
пространственной решётки относятся, как их кратности. Если последние
одинаковы, то объёмы параллелепипедов равны.
Тогда как кратчайшие расстояния между узлами решётки
определяются периодами идентичности, межшюскостные расстояния в решётке
зависит от выбора плоскостей (сеток). Это отчётливо видно из рис. 56, который,
как мы указывали выше, можно представить как проекцию трёхмерной
пространственной решётки с межплоскостными расстояниями d\ d", d'".
Зависимость между d и периодами идентичности а (соответственно Ь, с)
легко устанавливается методами геометрии.
Из §§ 41—43 вытекает, что, в отличие от операций точечной симметрии,
любые совокупности которых могут привести к возникновению 1,2, 3, 4,6,
8, 12,16, 24 и не более 48 равнозначных (эквивалентных) точек, расположенных
определённым образом (32 вида симметрии), трансляция сама по себе
приводит к образованию бесконечного количества параллельно равных
(идентичных) точек, образующих бесконечные ряды, плоские сетки или
пространственные решётки, в зависимости от характера трансляционной группы.
§ 44. Различные способы интерпретации процесса образования
пространственной решетки
Наиболее важны 4 основных способа.
Пространственная решётка может возникать при действии:
1. Трёхмерной трансляционной группы (таТ//Сс) на точку (рис. Г>8).
а с "В
Рис. 50. Различные способ:»! интерпретации образования njo-
странстненной решётки.
2. Трёхмерной трансляционной группы (хахьхс) на элементарную ячейку
(рис. 59, а). Таким образом, элементарная ячейка—это элементарный
параллелепипед, действием па который данной трёхмерной трансляционной группы
образуется рассматриваемая пространственная решетка.
96 ч. 1. основы теории структур [гл. 1
3. Трансляции 1Ъ на плоскую сетку, соответствующую двумерной
трансляционной группе (татс) (рис. 59, с).
4. Двумерной трансляционной группы (tat/;) на трансляционный ряд,
соответствующий вектору хс и т. п. (рис. 59, 6).
В одних случаях все четыре метода являются одинаково удобными,
н других целесообразно предпочесть какой-либо один из них (см. § 45).
§ 45. О («ложных проетранстпенных решетках
Рассмотрим две простые, примитивные, обозначаемые бук-вой Р
решётки, образованные одинаковой трансляционной группой (таТ/Ас)
ромбической сингопии.
Мы можем мысленно вдвинуть эти решётки одну в другую так, что
узлы одной из них окажутся в центре элементарных ячеек другой (рис. 60, а).
Мы получим в этом случае центрированную (объёмноцентрированную)
элементарную ячейку (рис. 60, 6). Такие решётки обозначаются /. Вдвинув две
/^-решётки одну в другую так, что узлы одной из них будут центрировать
пару взаимно противоположных граней элементарной ячейки другой решётки
(рис. 60, г), мы получим три типа двугранецентрировапных элементарных
ячеек: А, В к С в зависимости от того, какой из осой Лг, Y или Z
перпендикулярны центрируемые грани (рис. 60, d, e, /). Элементарные ячейки
с центрированными верхними и нижними основаниями (^-ячейки) часто
называются базоцентрированными.
Вдвинув в исходную решётку не одну, а три таких же решётки так,
чтобы узлы первой центрировали грани А> второй—грани VJ, третьей
грани С, мы получим решётку, элементарная ячейка которой будет иметь
все 6 центрированных граней. Такая ячейка называется гранецентриро-
ванной и обозначается F (рис. 60, g).
А, В и (7-элементарные ячейки являются двукратно-примитивными,
F-ячейка—четырёхкратно-примитивной. В кубических решётках но
характеру симметрии возможны только ячейки Р9 /, F (см. рис. 60, Л, /, /).
Межплоскостные расстояния в этих ячейках показаны на рис. 61 (a, b, с—
Р-рсшётка; d. е, /,—/-решётка; g, Л, iy—F-решётка).
Для подсчёта кратности элементарной ячейки мы пользовались в § 43
дробями, числитель которых показывает количество узлов
(кристаллографических точек) в элементарной ячейке, а знаменатель—какому количеству
элементарных ячеек каждый данный узел принадлежит. Такие дроби мы
будем называть структурными дробями.
В рассматриваемых и им подобных ячейках точка, находящаяся в
вершине, принадлежит 8 элементарным ячейкам, лежащая на ребре между
вершинами—4 элементарным ячейкам, лежащая на грани внутри её периметра —
2 элементарным ячейкам, лежащая внутри элементарных ячеек—только
ей одной. Поэтому кратность сложных элементарных ячеек равна сумме
структурных дробей:
для /-ячейки: п— -~ + -г~2,
о L
для Л, В и С-ячеек: п = -г+-^=2,
для F-ячейки: и-—f-^-=4.
Сложную решётку можно разбить на примитивные элементарные ячейки.
В таких решётках примитивные элементарные ячейки иногда находятся
в прямом и наглядном соответствии с симметрией самой решётки, иногда
не находятся. Примеромпервого случая является тетрагональная С-ячейка,
соответственно F-ячейка (рис. 62, а и 6). Выбрав вместо трансляционной
§ 45|
С) СЛОЖНЫХ ПГОСТРЛНСТИКННЫХ I'KJIlIiTKAX
97
д
/ \
ААМ\
♦4=71 ^И^\ ^^г\
k==F
h i I
Рис. 00. Образование ромбических Л .1, В, С и F-лчеек. Кубические Р, /, F-ячеикя.
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
•zoo dZOo= y^
<vf
d2QQ - ~f
Рис. 61. Межплоскостные расстояния в кубических Р, / и F-ячейках.
ЛЗ^~А
Рис» 62. Переход от тетрагональной С-ячейки к Р -ячейке
(а) и от. тетрагональной F-ячейки к /-ячейке (Ь)
§ 45] О СЛОЖНЫХ ПРОСТРАНСТВЕННЫХ РЕШЕТКАХ 99
группы {ха"Хь^с) трансляционную группу ("с/д^т^как показано на рисунке,
мы получаем тетрагональную же однократно-примитивную JP-ячейку,
соответственно /-ячейку. Примером второго случая является кубическая
С-ячейка, соответственно F-ячейка: аналогичной операцией мы получим
тетрагональную Р-ячейку, соответственно /-ячейку. Новая элементарная
ячейка не будет отвечать симметрии пространственной решётки.
Наконец, кубические J- и F-ячейки можно заменить ромбоэОрической,
элементарной ячейкой с углом а=109°28' (рис. 63, а) в первом случае
и 60° (рис. 63, Ь) во втором. Очевидно, однако, что описывать гс-кратно-
примитивные кубические решётки однократно-примитивными ромбоэдры-
Рис. ОН. Переход от кубических /- и F-нчеек к ромбоэдрическим
однократно-примитивным ячейкам.
ческими элементарными ячейками в большинстве случаев будет
нецелесообразно, ибо характер сингоиии будет, таким образом, виден гораздо менее
наглядно.
В подобных случаях для характеристики пространственной решётки
пользуются не однократно-, а гс-кратно-иримитивпыми элементарными
ячейками.
На рис. 64 показаны три способа выбора гексагональной элементарной
ячейки (а—трёхкратно-примитивная, fc—однократно-примитивная
(выделена жирными линиями), с—двукратно-примитивная (орторомбическая)
(выделена жирными линиями) и способ перехода от
однократно-примитивной ромбоэдрической к девятикратно-примитивной гексагональной
элементарной ячейке (рис. 64, е) или трёхкратно-примитивной (рис. O'i, /).
На рис. 64, d показана шестикратно-примитивная ячейка плотной гекса
тональной упаковки.
В § 44 мы рассматривали 4 разных метода описания пространственной
решётки.
В случае сложных элементарных ячеек особенно удобно пользоваться
вторым методом, трактующим пространственную решётку не как
совокупность вдвинутых друг в друга простых решёток, а как результат действия
трёхмерной трансляционной группы на сложную элементарную ячейку,
взятую как одно целое. Этот путь особенно важен в тех случаях, когда
в сложной элементарной ячейке имеются точки, являющиеся по отношению
друг к другу равнозначными (т. е. связанными элементами точечной
симметрии) (рис. 65). С переходом к материальным точкам (к структуре
кристалла) этот путь способствует исследованию физических сил, об у с ловли-
100 Ч. I. ОСНОВЫ ТЬОРПИ СТРУКТУР [ГЛ. I
Рис. 6'*. Три способа пыбора гексагональной элементарной ячейки и
способ перехода от [однократно-примитивной ромбоэдрической к гексаго-
вальной ячейке.
Рис. 65. Каждая из 8 элементарных
ячеек, содержит сложный структурный
(октоэдрический) узел, все точки
которого являются эквивалентными.
§_47J
14 РЕШЁТОК Ш'АВЕ
101
вающих образование подобных строгих конфигураций, ведущих себя в
структурном отношении как одно целое, которое мы будем называть структурным
узлом, и выявлению комплексных ионов, радикалов, молекул и т. п. (см.
§ 60 и следующие).
§ 46. Атомные полиэдры Е. С. Фёдорова
К. С Фёдорову принадлежит творческая идея подхода к образованию
решёток разных типов как к результату заполнения пространства атомными
полиэдрами-многогранниками, содержащими несколько нар параллельных
граней: трппараллолоэдрами (рис. GG, а), тетрапараллелоэдрами (рис. 66, Ь),
с d
Рис. г>6. Атомные полиэдры К. С. Фёдорова.
гексапараллелоэдрами (рис. 66, с), гептанараллелоэдрами (рис. 66, d).
(Последние содержат 3, соответственно 4, 6 или 7 пар таких граней.)
В результате получаются Р-> J- и /'"-кубические и Я-гексагональная
решётки, как это видно из рис. 6(5.
Эти представления нашли развитие в современной кристаллохимии.
§ 47. 14 решбток Jipniic. Закоп Браво
Браве, рассматривай точки шаровой симметрии, пришёл к выводу, что
существуют \\ типов пространственных решёток, которые распределяются
по 7 сипгониям (рис. 67 и табл. 13). На рис. 67 отмечены углы,
отличающиеся от 90': и триклинноп (1) решётке углы а, [3, у; в моноклинной (2) и
(г!) угол [В. В ромбоэдрической (11) это наглядно видно из рисунка.
При выводе возможных типов решёток обнаруживается, что некоторые
типы могут быть заменены другими, уже рассмотренными, например, как
мы видели (рис. 62), тетрагональная С-ичейка может быть представлена
как ^-ячейка, тетрагональная /"-ячейка, как' тетрагональная /-ячейка.
102
4.1. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Рлс. 67. \\ решёток Браво:
1) триклиниаи примитивная а Ф b ^ с <*>ф р Ф у Ф 90°; 2) моноклинная примитив-
пая; 3) моноклинная двугранепснтрировапная а Ф b Ф с а = т = 90 р Ф 90°; 4)
ромбическая примитивная, 5) ромбическая двугранепентрированная; 6) ромбическая
гранепентрированная; 7) ромбическая Центрированная афЬфс а. = $ — v = 90 ;
8) тетрагональная примитивная; 9) тетрагональная центрированная а — b Ф с
а=э=т = 90; 10) гексагональная ai = я2 = аз ^= г. Углы между о/ями а и с
равны 90е, между осями а—120°; 11) тригональная (ромбоэдрическая) а^Ь^с,
j. = (J =y ^z 90 ; 12) кубическая примитивная; 13) кубическая гранецентрированная;
14) кубическая пептрированная.: « » 6 = с a = JJ = *f ^ 90u.
§ 49] ДЕЙСТВИЕ ТРАНСЛЯЦИИ НА ЭЛЕМЕНТЫ СИММЕТРИИ ЮЗ
Таблица 13
1 ^
| по
1 пор.
1
1 2
3
4
5
6
1
| 1
№
1
2а
2Ъ
За
зь
Зс
3
1
Решётки
Браве
Триклинная
примитивная (1)
Моноклинная
примитивная (2/Wi)
Моноклинная
базоцентриро-
ванная ('2/т)
Ромбическая
примитивная (ттт)
Ромбическая
базоцентриро- i
ванная (ттт)
Ромбическая гра-
нсцентрирован-
па я (ттт)
Ромбическая
центрированная
(ттт)
1
значения
Tir
Гт
Гт
Го
/
Го
//
Го
///
Го
п
1
1
о
1
9 |
4
о
по
пор.
1
8
у
1)
и
12
13
14
№
4а
4Ь
5а
ПЬ
Ga
()b
Г)С
Решётки
Браве
Тетрагональная
примитивная
(4/mmw)
Тетрагональная
центрированная
(4, ттт)
Гексагональная
базоцентриро-
ванная (Ь/ттт)
Ромбоэдрическая
примитивная (3/тг)
К'убическая
примитивная
(т'Лт)
Кубическая гране-
центрированпан
(тЗ/я)
Кубическая
центрированная
(т\\т)
значения
rt
г<
г,,
ГгП
Гс
i
Ге
Гс
\
п
1
2
1
1 1
1
4
2
На рис. 61 видно, что различные плоские сетки «заселены» точками
неодинаково. Исходя из такого рода представлений, был сформулирован
так называемый закон Браве:
«Грани кристаллов образуются сетками с максимально плотным
расположением атомов».
Этот закон сам по себе недостаточен для понимания существа процесса
и может повести даже к неверным выводам. Он приобрёл реальный
физический смысл лишь после сформулирования закона Вульфа (§ 5), который,
собственно, только и позволяет ответить на вопрос, параллельно каким
сеткам должны образовывать грани термодинамически стабильных форм
кристаллов. В табл. 13 приведены обозначения и симметрия решёток Браве.
§ 48. Базис
Базисом, принадлежащим трансляциям ta, th, Тс, называется
совокупность точек, которая возникает вместо точки в решётке Браве и путём
параллельного смещения соответственно (ха Хь Тс) образует всю данную
систему точек (см. § 44). Базис описывается точным указанием
координат точек в пределах элементарной ячейки. Иногда базисом называют
именно совокупность координат всех находящихся внутри элементарной
ячейки неидентичных точек, выраженных в долях трансляции.
§ 49. Действие трансляции на непараллельные ее вектору
элементы симметрии
В теории симметрии дисконтинуума нам придётся встретиться со
знакомыми нам уже элементами точечной симметрии, а кроме того, и с новыми
элементами симметрии, характерными лишь для дисконтинуума.
Если трансляция не совпадает с направлением элемента симметрии,
например перпендикулярна поворотной оси, то под действием трансляцион-
104 ч. i. основы теории структур _ [гл. 1
ной группы происходит трансляция элементов симметрии, т. е. образуется
бесконечное множество тех же элементов симметрии, допустим поворотных
осей 6. Этот процесс имеет важную особенность (см. рис. 68).
Рис. 68. Элемонтгл симметрии, возникающие в пространственной решетке
при действии трансляции на перпендикулярные вектору х поворотные
оси 4 и 6.
а) и Ь) Образование семейств осей 4 и 2-го порядков при действии
трансляции на ось 4. с) и d) Образование семейств осей 6, 3 н 2-го порядков при действии
трансляции на ось б. Рис. d отвечает части рисе, выделенной жирной чертой.
Трансляции осей, кратность п которых может быть представлена как
произведение двух отличных от 1 целых чисел: п=тр, т. е. 4 = 2 • 2 и 6 = 2 • Л,
приводит к образованию; кроме семейства осей /г, также семейств осей
низшей кратности тар (3 и 2).
Идентичными называются элементы симметрии, возникающие друг
из друга*) в результате трансляции. Трансляция образует бесконечное
множество идентичных элементов симметрии. Эквивалентными, равнозначными,
называются элементы симметрии, совмещаемые друг с другом с помощью
операций точечной симметрии.
*) Т. е. совмещаемые друг с другом.
5 50]
СКОЛЬЗЯЩАЯ ПЛОСКОСТЬ СИММЕТРИИ
§ 50. Скользящая плоскость симметрии
105
Если трансляция параллельна плоскости симметрии, то возможно
осуществление новой операции симметрии.
Операция симметрии, являющаяся результатом совместного действия
зеркального отражения и трансляции, —параллельной зеркальной плоскости
отражения, — на расстояние, равное полупериоду идентич/юсти, называется
операцией скользящего отражения, а соответствующий ей элемент
симметрии—скользящей плоскостью симметрии*) (плоскостью скользящего
отражения).
\
Т/г
sj
р
ff
4-
1
i
I
/л
4
р'
Р2
V2
I
Р,
U
t
к
i
v
т
с
a.S
п
d
а б с d в
Рис. 60. Скользящая плоскость симметрии.
Рассмотрим рис. 69, а, Ь. Томка Рг даёт зеркальное изображение в Sl и в
результате трансляции т попадает в положение Р2. Точка Р2 даёт
зеркальное изображение в $2И в результате трансляции -^ попадает в положение Р].
На рис. 09, с, d показан случай нахождения точки на плоскости
скользящего отражения.
Вектор -^ называется компонентой скольжения.
Если компонента скольжения (а значит, и сама плоскость скользящего
отражения) параллельна оси а, соответственно 6 или с, то и плоскость
обозначается соответственно a, b или с. Плоскостью а не может быть (100),
которая перпендикулярна а, но может быть (010) или (001). Если плоскость
скользящего отражения параллельна диагонали основании, её обозначают п
и называют клипоилоскостыо скольжения. В .пом случае компонента
скольжения у равна полусумме двух векторов, т. е. -—;г^ или —т>— , или ^—~ч
т. е. половине диагонали соответствующего параллелограма.
Наконец, если компонента скольжения, параллельная пространственной
диагонали элементарной ячейки, равна 1/4 суммарного вектора Trt-г **/.-f **с»
плоскость обозначают d и называют плоскостью алмазного скольжения
(см. § 03) .
В IT для графического обозначения скользящих плоскостей симметрии
прибегают к пунктиру трёх видов (рис. (59, с): прерывистый для плоскостей
*) Подчеркнем, что наличие п решётке сколь.пщих плоскостей отражении
необязательно нлочёт л a codoii присутетпио плоскостей силшетрин.
106
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл.
а, 6, с, прерывистый с точками для плоскости п, и добавляют стрелку в
случае плоскости d, причём стрелка указывает направление, в котором верти-
кальная компонента скольжения равна + -г (значениедроби 1/4см. § 54).
§ 61. Винтовые оси-геликогиры
Совмещение фигуры в результате совместного действия вращения
вокруг поворотной оси (гиры) п-го порядка и трансляции параллельно гире
называется винтовым преобразованием, а соответствующий элемент
симметрии— винтовопово ротной
или винтовой осью (геликоги-
рой) п-го порядка.
Наименьший угол а
поворота геликогиры, который
осуществляет (при
одновременной трансляции всех
точек на величину — Л
совмещение с исходным положением,
называется элементарным
углом, а соответствующий
вектор «элементарной
трансляцией» (ходом) этой
геликогиры.
Величины а и — тесно
п
связаны между собой через
360°
величину п9 п== . При
/г= 6 совмещение произойдёт,
если элементарный ход гели-
когиры составит — х, причем
с d
Рпс. 70. Винтовые оси 6j и б5.
точка совершит путь вправо
вверх (см. рис. 70, а).
К тому же результату приведёт вращение в другую сторону на -jr- f.
Вращение же влево вверх на — т приведёт к возникновению фигуры рис. 76, 6,
которая энантиоморфна предыдущей. Если направленная к читателю точка
Р\ при повороте смещается вправо вверх, ось называется правой (6i), если
влево вверх — ось называется левой (65).
Принятый нами, в согласии с IT, способ обозначения винтовых осей,
например 6Ь позволяет сразу найти величину элементарной трансляции
в долях -:. Сделав индекс обозначения числителем, а порядок оси
знаменателем, сразу находим величину элементарной трансляции -г- X.
Помимо рассмотренных, возможны ещё геликогиры 62 и 64 ( ход равен
~ j, а также 6-/ход равен ~) (рис. 71 и 72).
В этих случаях подвергается винтовому перемещению совокупность
из 2 соответственно 3 точек, расположенных в плоскости,
перпендикулярной геликогире.
Если сумма индексов двух осей одного и того же порядка равна порядку
п оси, то получающиеся фигуры энантиоморфны. Пример: 6i и 65 (1 + 5 = 6)
§ 511
ВИНТОВЫЕ ОСИ-ГЕЛИКОПИ>Ы
107
108
4. I . ОСШШЫ ТЕОРИИ СТРУКТУР
[гл. I
или 6^ и 04. Если индекс равен —, различить левое и правое вращение по
расположению точек невозможно (63).
На рисунках 73, 74, 75 и 76 показаны винтовоповоротные оси и других
порядков: 4i, 4о, 4з, Зь 32, 2\. В нашей кристаллографической литературе
геликогиры соответствующих порядков называются дигеликогира, тригелп-
когира и т. д. В последнее в[юмя стали употреблять также наименования:
теликодигира, гсликотригира и т. д.
Рис. 75. Винтоиые оси ЬЛ1 и 32.
Рис. 7(>. Виптоиан
ось 2Х.
(Следует обратить внимание на то, что, например, в случае осой 6,
6i, tii», ti , ti4 и 6f) проекции точек на плоскость, перпендикулярную
оси, во всех случаях одинаковы и оси но этому признаку
неразличимы. Лишь изучение разреза параллельно оси позволяет установить,
с какой осью мы имеем дело. То же относится к осям других порядков
(рис. 70—75).
Таким образом, в теории симметрии дисконтинуума мы должны
различать следующие элементы симметрии: 1, 1; 2, 2\\ 3, 3j, 32; 4, 4, 4i, 4o, 4,; 6, (5,
6ь 6о, 63, 64, 65; т, а, 6, с, п, d.
В таблице 14 приведены обозначения винтовых осей по IT.
§ 51) шштовьш оси-геликогигы 109
Таблица 14
Обозначения винтовых осей пг
Порядок впнто- 1
вых осей 1
Величина 1
трансляционной
комноненты
Обозначение 1
винтовой оси
(IT)
; 1
3
4
1 б
i |
2 i
~Г- 1 :*
i |
1
Г
о
1 ^
2
т
6v
5
4i
4:{
61
б-
6з
(и
05
Характер энан-
тиоморфной
формы
Графические обозначения из IT
Вертикальные
винтовые
оси
Вертикальные
винтовые оси-f
+центр
инверсии |
Горизонтальные
винтовые
оси
! ! , 1
i к j tf !
э. ф. нет \Ш, V ^ ~—*" 1
правая
левая
Л
!
правая /^Ъ/
э.ф.нет ^Ь
лова» | ^^^Ч
правая
правая
э. ф. нот
левая
левая
и"
*
4
i
I
I
иг
t--f
Наклонён- 1
ные винто- 1
вые оси *) 1
*) Только в кубических пространственных группах.
110 ч. i. основы теории структур [гл. I
§ 52, О кратности, собственной симметрии и ориентировке
точек, находящихся на скользящих плоскостях симметрии и винтовых
осях, О степенях свободы точек
Как мы знаем из § 15, с попаданием точки на элемент точечной симметрии
её кратность п, собственная симметрия s и ориентировка должны
изменяться соответственно характеру элемента симметрии.
Если точка при аналогичном перемещении попадает на скользящую
плоскость симметрии или винтовую ось, не только её кратность и
кратность позиции, но и её собственная симметрия не меняются, равно как и не
налагаются никакие ограничения на её ориентировку (рис. 69, a, d и 77).
Подчеркнём, что наличие геликогиры, равно как и скользящей
плоскости симметрии, проходящей через «центр тяжести» точек, может быть
обнаружено лишь, если точки обладают достаточно низкой симметрией, В
противном случае обнаруживается лишь простой трансляционный ряд или
обычная плоскость симметрии и т. п. (см. рис. 78). На этом примере вновь
отчётливо видна важность учёта собственной симметрии точек при
исследовании подобных проблем.
\
VJ
х\
г
а 6
Рис. 77. Попадание точки на
скользящую плоскость симметрии или на
винтовую ось.
7
г. i
1 ^
^ ё
Рис. 78. Попадание точек на
винтовую ось 22.
Количеством степеней свободы / точек называется количество
независимых перемещений (параллельных кристаллографическим осям), при
которых кратность и собственная симметрия точек не меняются.
Так, в точечной группе D% — ттт pv =8 возможны случаи (рис. 12,6
и 79):
а) р = 8 и = 1 s = l / = 3 (рис. 79, а)
Ь)р = 4 у = 2 s = т / = 2 (рис. 79, 6)
c) /? = 2 w = 4 s = mm / = 1 (рис. 79, с)
d) р = 1 v = 8 s = ттт / = 0 (рис. 79, d).
Точка, находящаяся на пересечении трёх плоскостей симметрии
(случай 4), имеет количество степеней свободы / = 0, её перемещение вдоль
любой плоскости симметрии приводит к случаю (с), т. е. / == 1, и т. п.
§ 53. Координаты точек, возникающих при действии на точку
различных элементов симметрии
На рис. 80 и 81 показаны изменения координат точек, возникающих
при действии на точку: тетрагиры и геликотетрагиры, а также плоскости
симметрии и скользящей плоскости симметрии. В нашем примере эти
элементы симметрии перпендикулярны плоскости XY и параллельны оси Z.
§ 53] ДЕЙСТВИЕ НА ТОЧКУ РАЗЛИЧНЫХ ЭЛЕМЕНТОВ СИММЕТРИИ 1И
л
1\
и
и~
ZZV
т
7
к
\А
и
44
ш
г
\А
иг
~А
V.
V
ж
Ряс. 79. Различные положения точки относительно
плоскостей симметрии.
*
7-
УР^. 4
Рис. 80. Изменение координат точек при действии а) тетра-
гирм*и Ъ) геликотетрагирьт.
112
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
|гл. I
И случае тетрагиры и плоскости симметрии координаты z не изменяются.
Меняются лишь знаки координат или координаты х и у или и то и другое Г
И случае гсликотетрагиры координаты z изменяются, приобретая
добавочные значения
:P(xyz); Pt (У*,2 + {) ; Р, (xy.s-t™) ; Р3 (j,x,z+ty
у и т. п. обозначают отрицательные значения соответствующих коор-
\\
динат. Перемена мест х и у, например в {yx,z+ —), обозначает, что коо|)
1 J.
2
дииата х отрицательна и равна по величине у. Дроби - , -^- и т. и. отвеча-
X
ют величине вектора --, -л- и т. н.
Рис. Hi. Изменение координат точек, возникающих при действии а)
плоскости симметрии и Ь) скользящей плоскости симметрии.
В случае скользящей плоскости симметрии наблюдается та же картина,
например, P(xyz), I\fxy9z + ^ и т. д. (рис. 81).
§ 54. Координаты точек, линий и плоскостей в элементарной ячейке
Как мы знаем, вектор т' — wcx + nt2 + px3, где т} п и р—целые числа,
определяет положение в пространственной решётке любой идентичной
точки (узла) по отношению к одному из узлов, принятому за нулевую точку.
Для описания положения любой точки внутри элементарной ячейки,
т. е. ее базиса, пригодно то же уравнение, однако значения т, п и р в этом
случае будут дробными числами, характеризующими значения координат
точек по отношению к нулевой точке в долях от величин, описывающих
данную ячейку векторов а Ь с (её осей). Координаты точек элементарных ячеек
прежде проставлялись в двойных квадратных скобках [[ ]], однако в
современной литературе, как правило, пользуются круглыми.
За нулевую точку (000) чаще принимается позиция в левой нижней
передней вершине элементарной ячейки, но иногда точке (000) отвечает
левая нижняя задняя вершина.
В литературе пользуются также разными направлениями осей а Ь с.
§ Ъ\] КООРДИНАТЫ ТОЧЕК И ПЛОСКОСТЕЙ В ЭЛЕМЕНТАРНОЙ ЯЧЕЙКЕ 113
В IT и пояснениях к ним вектор а обычно уходит от читателя в глубь
ячейки, векторЬ — вправо, векторе —вверх; в SB вектор а обычно уходит
вправо от читателя, вектор ft —вглубь и вектор с —вверх. Иногда вектор
а направлен вперёд к читателю, вектор Ь—вправо, вектор с—вверх (рис. 82).
При рассмотрении рисунков элементарных ячеек надо обращать
внимание на направление осей.
Л А
f ■ ■ ■ г |
Т 1
г 1
1
V > ..У
Рис. 82. Различные способы выбора нулевой точки элементарной
ячейки и направления осей координат.
На рис.83, а (векторы направлены, как в SB) показаны точки
При этом, например, Рз(~т~о~т) означает, что для нахожде-
<> i \
ния точки Рь надо отложить —а.тгЬ и ^ с. Точка РА лежит в
центре элементарной ячейки.
Точки с тремя
одинаковыми векторами, т. е.,
/1 1 1\
например, ^ — т т) ;
U Уз;' v 4 4 4 ) '
Г 1 1 1\
[jg-j ), лежат на
диагонали элементарной
ячейки (рис. 83, 6).
Если все три
координаты точки фиксирова-
/ 1 1 1 \
ны, например (^yyyj ,
число степеней свободы
точки равно 0.
Если одна
координата может быть
переменной, например (у 2/у)» возникает одна степень свободы. Точка может
иметь любые значения г/: от 0 до 1. Следовательно, запись (^Ут)
отвечает геометрическому месту бесконечно большого количества точек,
в данном случае прямой линии, параллельной оси г/.
На рис. 84 показаны прямые
(ЫХООтН-
Прямые (#00) (0г/0) (00z) совпадают с осями а Ь с.
\ У
\1
)—
) г
Л 1
1
иЛ
Л
Рис. 83. Положения точек внутри элементарной
ячейки при различных значениях параметров.
114
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Так как обе координаты точек, лежащих на диагонали параллелограма
плоской сетки, равны, любой такой точке отвечает запись типа (ххО). В
равной мере любой точке, лежащей на диагонали элементарной ячейки,
отвечает запись типа (ххх). На рис. 85 показаны
символы диагоналей куба, часто применяемые
в литературе. (В общем случае прямая в
пространстве в соответствии с требованиями
геометрии задаётся двумя точками.)
Если две координаты могут быть
переменными, например ( у г/zЛ , возникают 2
степени свободы. Запись f — yz j отвечает плоскости.
На рис. 86 показаны плоскости Г— 2/гЛ, (хуО)
и (Ог/z), на рис. 87 —плоскости {xxz)y {zyz)
и (хуу).
Наконец, если все три координаты точки
могут быть переменными, возникают 3
степени свободы. Положение точки в
элементарной ячейке никак не фиксировано.
Если известна позиция точки в элементарной ячейке, например (0-т-^ ) >
то бесконечному количеству идентичных ей точек в решётке (вне данной эле-
7
ХОЗ/4
/
1
! |
i ;
У г-
L
—L
\ 1
А
7\
)
Рис
//« //4Z
84. Прямые 0/ау1/«).
(Х£х)
(ххО)\^-
(XXX)
Y\ Х—^Йт
(ххх)
(ххх)
(хП.т) I <0№ (ххх)
Рис. 85. Символы диагоналей куба.
ментарной ячейки) отвечает запись ( ±т, -^ ±п, ± р\ где т, n и р — целые
числа.
/, А> B> С и F-ячейкам отвечают координаты точек:
J: (000, уу 1); А: (ООО, of -£); В: (000, -| 0J); С: (ООО, 1у О);
F:(000,0T4.4°4'4f0)-
§ 55] СИММЕТРИЯ ПРОСТРАНСТВЕННОЙ РЕШЁТКИ И ЭЛЕМЕНТАРНОЙ ЯЧЕЙКИ 115
При описании координат точек, во избежание повторного выписывания
меняющихся местами одних и тех же дробей, характеризующих позиции
нескольких точек, иногда приводят координаты одной из них, после чего
Рис. 86. Плоскости (xyO)f
(Qyz)t (>Uyz).
Рис. 87. Плоскости (xxz)%
(zyz), (xyy).
ставится знак (j\ Например, положение точек в F-гранецентрирован-
ной ячейке записывается так:
(ооо, о{1 о).
§ 55. Симметрия пространственной решётки и элементарной^ ячейки
Симметрия элементарной ячейки определяется следующими факторами:
а) метрикой элементарной ячейки (т. е. осевыми отрезками я, 6, с и углами
а, р, у или отношением а : Ь : с и углами а, р, у), Ь) расположением
«центров тяжести» точек (частиц) в элементарной ячейке, с) собственной
симметрией точек, d) их ориентировкой по отношению к осям элементарных
ячеек, как это показано на рис. 88.
•Г~7Т И^М /Р^ТТ /r^vt
Vz3?
Ж?
Рис. 88. Зависимость симметрии элементарной ячейки от метрики ячейки (а),
расположения центров тяжести точек (6), собственной симметрии точки (с)
и их ориентировки по отношению к элементам симметрии (d).
Таким образом, даже при высокосимметричной метрике элементарных
ячеек, высокой собственной симметрии точек и высокосимметричном
расположении их центров тяжести неправильная ориентировка точек
резко снижает симметрию элементарных ячеек.
В учебниках физической химии, физики и т. п. иногда делается ошибка —
о степени симметрии структуры судят по метрике элементарных ячеек и по
положению в ней центров тяжести частиц, упуская из виду вопрос о
собственной симметрии последних и их ориентировке по отношению к
элементам симметрии.
116 ч. i. основы теории структур [гл. I
§ 56» О выводе возможных пространственных групп
Если возможные пространственные совокупности элементов точечной
симметрии приводят к возникновению 32 видов симметрии,
характеризующихся наличием особой точки, то возможные пространственные
совокупности элементов симметрии дисконтинуума (см. § 51) приводят к
возникновению в дисконтинууме 230 видов (разновидностей) симметрии,
соответственно 230 пространственных групп, не обладающих особыми точками *).
Теория пространственных групп была впервые создана великим
русским кристаллографом Е. С. Фёдоровым (1890).
Рис. 89. Пространственные группы, подчинённые видам симметрии Сх и С\.
Вопросу изображения кристаллографических групп симметрии, в том
числе и пространственных групп, на основе размещения в пространстве
асимметричных фигур посвящен атлас, составленный А. В. Шубниковым
[18] (1946).
Не имея возможности проследить здесь ход вывода всех 230
пространственных групп и дать им графическую характеристику, отсылаем читателя
к специальным курсам и IT. Здесь же для примера рассмотрим
пространственные группы, подчинённые точечным группам Си Ci, Cs и Сг.
В видах симметрии Ci и Ct — осей и плоскостей симметрии нет. Таким
образом, трансляция не создаёт дополнительных сочетаний элементов
симметрии, и, значит, каждому из этих видов симметрии отвечает только одна
пространственная группа, соответственно Ci—1 (рис. 89,а)и С\—1 (рис. 89,6) **).
Обозначения пространственных групп даны по международной системе:
верхний правый индекс при обозначении точечных групп соответственно
вида симметрии по Шенфлису (например, Сх) показывает порядковый номер
пространственной группы. Тире отделяет обозначение по Шенфлису от
обозначения по Могену—Герману (см. § 14), в основу которого кладутся
символы, принятые для соответствующих видов симметрии (табл. 10) с указанием
порождающих элементов симметрии. Для обозначения пространственных
групп перед символом вида симметрии проставляется один из следующих
специальных знаков: Р—примитивная, А, В, С— двугранецентрированная,
F—всесторонне гранецентрированная, /—центрированная, С или
Н—гексагональная, R—ромбоэдрическая.
В пространственной группе с\ точки образуют триклинную
пространственную решётку без элементов точечной симметрии. В пространственной
группе С\ в решётке расположены пары обратно равных точек,
подчиняющиеся центру инверсии (рис. 89,6).
Виду симметрии Cs моноклинной сингонии отвечает плоскость
симметрии т. Соответствующая пространственная решётка содержит семейство
плоскостей симметрии. Это значит, что точки, слагающие решётку, должны
*) Е. С. Фёдоров разработал, как мы указывали в § 2, также и методы их
изображения, принятые н IT.
, **) На рис. 89,6 и следующих показанч изображения пространственных групп
по IT, т. е. слева положения точек и справа положения элемевтов симметрии.
§561 о выводе возможных пространственных групп ]
С'гРт
+0О+
+®о-
//?+© о+
CJ-FQ
^+© C5+
#+® Ю+
//?+©0+
С$3-Ст
+©0+
+0О+
с:-сс
I + 0O+
+0
4+00+
0+
1+0 0+
4+0 0+
I
I
t
Рис. 90. Пространственные группы, подчинённые виду симметрии С9
118
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
|гл. I
обладать зеркальной симметрией или быть соответствующим образом
—симметрично—ориентированы (рис. 90).
Плоскости симметрии делятся на 2 группы:
Основные, которые проходят через узловые точки, и промежуточные,
расположенные в точности посередине между первыми плоскостями.
Пространственная решётка может обладать либо только зеркальными
плоскостями симметрии, либо только скользящими плоскостями симметрии,
либо и теми и другими. Первому случаю отвечает пространственная группа
С\—Рт (рис. 90,я), второму С\ — Рс (рис. 90,6), третьему С\ — Cm (рис.90,с).
О-
о+
1 о-
04-
С?-Р2
О-
о-
0+
о-
о+
о~
о+
о-
о+
о-
о-
оч-
04-
tf-cz
6-
о-
Рис. 91. Пространственные группы, подчинённые виду симметрии С%.
Кроме того, возможна пространственная группа С\—Сс, так же как
и Ci, имеющая только плоскости клиноскольжения, причём в этом случае
осуществляются 2 трансляции в разных направлениях а/2 и с/2 (рис. 90,d).
Дроби около кружков указывают высоту точки над плоскостью чертежа(+)
или под плоскостью чертежа (—). Запятая отличает левые точки от правых.
Виду симметрии Съ моноклинной сингонии отвечает ось 2.
Соответствующая пространственная решётка содержит уже семейство осей 2, причём
возможны три варианта: 1) все оси являются поворотными
(пространственная группа С\—Р2) (рис. 91,а), все оси являются винтовыми
(пространственная группа Ci—P2>i) (рис. 91, b), часть осей является поворотными, часть
винтовыми (пространственная группа С*±— С2) (рис. 91,с).
В § 57 приводятся обозначения пространственных групп по IT.
§ 571
ОБОЗНАЧЕНИЯ ПРОСТРАНСТВЕННЫХ ГРУПП ПО IT
119
§ 57. Обозначения пространственных групп по IT
Пространственные группы
1. Триклинной
гемиэдрии Сх—1
1. С\-Р1
2. Триклинной
голоэдрии d — 1
2. С]-Pi
3. Моноклинной
гемиэдрии Cs —m
3. Сг-Рт
4. Ci-Pc
5. С6-Cm
6. С*-Сс
4. Моноклинной
гемиморфной
гемиэдрии С 2—2
7. С.2-Р2
8. С!-Р21
9. С^-С2
5. Моноклинной
голоэдрии G'2h — 2//п
10. clh-P2/m
11. C*h-P21/m
12. С^л-С2/т
13. Clh-P2jc
14. Clh-P21/c
15. C|ft-C2/c
в. Ромбической
гемиморфной
гемиэдрии C2t,—
21, — mm
16. Coy-Pmm
17. C?y-Pmc
18. CJjp-Pce
19. C*-
2i>
20. C»,-
21. C§„-
22. CL-
23. C\v
24. C^-
25. СЙ-
26. C\\-
27. C«-
28. C**-
29.
СИЗО. C^-
31- C2P-
32. Cj„7-
33. C^-
34. СЦ-
35. C52-
36. C*-
37. eg-
Рта
Pea
Pnc
Pmn
c-Pba
-Pna
Pnn
Omm
Cmc
Ccc
Amm
Abm
Ama
Aba
Fmm
Fdd
Jmm
Jba
J ma
7. Ромбической
энантиоморфной
гемиэдрии
К=Д>—222
38. DJ-P222
39. 1>2-Р2221
40. Z>2--P2A2
41. Z)^-P212121
42. Dl-C222x
43. Z)£-C222
44. fl£-F222
45. /)*-/222
46. D^-72^2^,
8. Ромбической
голоэдрии
47.Z>1„-
48. /)?,„-
49. Z)3A_
DO. Z>*„ -
51. D%,-
Ь2. />|й -
53. Z>7„,-
54. Z>«ft-
55.D29„-
ЬЬ.вЦ-
57. Л»-
58. D1^-
b4.D\l-
60. D*£-
61. D»jj-
6',. D«-
65. D«-
66. 2>«-
67. Dfh-
68. DS-
Pmmm
Pnnn
Pccm
Pban
Pmma
Pnna
Pmna
Pcca
Pbam
Pccn
Pbcm
Pnnm
Pmrnn
Pbcn
Pbca
Prima
С mem
Cmca
Cmmm
Cccm
Cmma
Ccca
69. L^—Fmmm
70. D^-Fddd
71. D?>^ — Jmmm
72. a!j)|-/&am
73. ВЦ-Пса
74. Dr^- Jmma
9. Тетрагональной
тетартоэдрии _
2-го рода £4 — 4
75. ^-Р4
76. 6^-/4"
10. Тетрагональной
тетартоэдрии
С*-4
77. С}-Р4
78. С^-Р41
79. С|-Р42
80. С^-Р43
81. C\-Jk
82. C\-J^
11. Тетрагональной
параморфной
гемиэдрии С4л— 4,'w
83. C^-P4/m
84. С^-РЦт
85. C\h-Pbln
86. С*л-Р42/л
87. С^-/4/г/г
88. C$A-/Va
120
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. ?
Пространственные группы
12, Тетрагональной
гемиэдрии 2-го рода
Krf=Z)2d-42m
89. h\d-Pklm
90. i)|d-P42c
91. h\d-P\\m
P\\c
C\2m
C42c
шь
СШ
Fk2m
F42c
J42m
J42d
92. L\d
93. Dld-i
94.Z)2Y
95. B\d r
96. Z)*d-
97. d\u-
98.2)™-
99.D|J.
100. D}£
13. Тетрагональной
гемиморфной
гемиэдрии
Cav — 4mm
101. C\v-P[imm
102. C\v-P'+bm
103. C\v-Pbcm
104. C\v-Pknm
105. C^-P4cc
106. С64„-Р4лс
107. C^-P4mc
108. С|и-Р46с
109. C\v- Jbmm
110. C^-74cm
111. C\\-Jkmd
112. сЦ-Jbcd
14. Тетрагональной
энантиоморфной
гемиэдрии f)4—42
113. l)l-P42
114. Z)^-P42i
115. Z)|-
■Р442
116. 7^-P4121
117. 0*j-
118. 2>^
119. D\-
120. l)^-
121. D^-
•P422
-P4o2i
•P432
P432i
742
122. D
10
-JT4j2
15. Тетрагональной
голоэдрии
^4h "" 4mm
123. D\h-Pkjmmm
124. /)^Л-Р4/тсс
125. D\h-Pblnbm
126. 2>JA-P4/nnc
127. Dlh — Pk/mhm
US. D\h- Pk.'mnc
Vl<S.D\h-Pkinmm
m.DS4h-Pb/ncc
131. Dlh-Pk/mmc
№.D\°h-Pk/mcm
№.Dll-Pklnbc
т. ВЦ- РЬ/ппт
135./)^-Р4/тЬс
136. Z>JA-P4/mnm
137. Lj;J-P4/nmc
138. £*£-P4/ncm
139. D\7h - Jb/mmm
140. Z)^- Jkjmcm
141./)Jj}-J4/amd
142. L52-J4/acd
16. Ромбоэдрической
тетартоэдрии
Сз-3
143. C*-C3
144. С|-С31
145. Сз-С32
146. С|-Я3
17. Гексагональной
тетартоэдрии
2-го рода iSe Сз/ — 3
147. С^.-СЗ
148. С§£-ДЗ
18. Ромбоэдрической
гемиморфной
гемиэдрии Сзг — Зт
149. C\v-C*m
150. С^-7/Зт
151. С^-СЗс
Ш.С^-ЯЗс
153. с|„-ЯЪп
154- ^|и-ЛЗс
19. Ромбоэдрической
энантиоморфной
гемиэдрии />з— 32
155./)£-Я32
156. D\- С 32
157.£|-Я3.2
158.2)^-03^2
159. / jj-tf32-2
160. /-5--CV2
161. 1^- Я32
20. Ромбоэдрической
голоэдрии D' d — 3w P
162.Z)£d-H3m
163.1*d-f/3c
164.Dgd-C"Jm
165. Z)^~C3c
166. #3d - ЯЗт
167.Z)§d-ДЗс
21. Тригональной
параморфной
гемиэдрии С: /г — 6
163. С£л-С6
22. Гексагональной
тетартоэдрии Со—6
163. CJ-C6
170. C£-Cbi
171. Cg-C65
172. С|-С62
173. С^-С6А
174. С£-С63
23. Гексагональной
параморфной
гемиэдрии С6Л — 6/т
175. с£л-С6/т
176. Clh-C63/m
§ 581 О РАСЧЁТАХ РАССТОЯНИЙ И ОБЪЁМОВ В ПРОСТРАНСТВЕННЫХ РЕШЕТКАХ 121"
24. Тригональной
голоэдрии
D-ft-C6m2
177. D\h-Cbm\
178.*>§Л-С6с2
179. n\h-Hfm-l
180. r;\h-Hlcl
25. Гексагональной
гемиморфной
гемиэдрии
Ccw — 6 mm
181. Cj„ -СШт
182. Cg,, -Сбсс
183. C\u -СЬст
1S4. с£„ -C6mc
26. Гексагональной
энантиоморфной
гемиэдрии />в — 62
185. /)£-C62
Ш.Т^-Сб^
187. 7)3-C6,2
Пространственные группы
188.Л^-Сб22
189. Z>jj-C642
190. I>j}-C632
27. Гексагональной
голоэдрии
/. c/j — Ыптт
191. Z)L -CG/m-n
m
'6fi
192. hlh-Cblmcc
l№.Dlh-Cb/mcm
m.D$h-Cbfmmc
201. Tjx-Pn2
202. 7^-Fm3
203. Tl-Fd\l
204. T£-/m3
205. Г^-ГяЗ
206. Tj,-/aH
2S. Кубической
тетартоэдрии
Г - 23
195. Г1-Р23
1J6. r^-F23
197. 7^-/23
198. 7M-P2i3
199. ГЗ- /2i3
30. Кубической
гемиморфной
гемиэдрии Td — ЬЪт
29. Кубической
параморфной
гемиэдрии Тл—гаЗ
200. Т\- РтЗ
207. Ti-P43m
208. Tj-Ftfm
209. 7^-/43^
210. Т^-Р43д
211. 7^-F43c
2L2. Т*- Jfcd
31. Кубической
энантиоморфной
гемиэдрии О— 43
213. 01-Л43
214. 02-Р4оЗ
215. 03-F43
216. 0±-F4t3
217. 05-/43
218. Об-Р433
219. 07-Р^З
32. Кубической
голоэдрии
Од— игЗт
221. 0/t
222.0,-
223. Of,—
224. OjJ-
225. 0>-
226. Ol~
227. О*-
228. 0j>-
229. OjJ-
230. О*0 -
РтЪт
РпЗп
РтЗп
РпЗт
FmSm
FmZc
Fd^m
FdZc
/m3m,
- JaM
§ 68. О расчётах расстояний и объёмов в пространственных решётках
Как мы знаем, расстояния между двумя точками в пространственной
решётке могут быть выражены вектором (см. (8), стр. 94)
г = та + rib + ре.
Если за нулевую точку выбрана некоторая идентичная точка (узел)
и т, п и р целые числа, уравнение (8) определяет положения других
идентичных точек, если же т, пир дроби (>0 и < 1), то уравнение (8)
определяет положения точек в элементарной ячейке.
Для определения абсолютной величины расстояния до любой точки
надо умножить вектор г скалярно на самого себя, причём получим
функцию r2 — f(mnp) в виде квадратичной формы
г2 = т2а2 + п2Ь2 + р2е2 + ЪьрЬс cos a + 2pmca cos p + Imnab cos y. (10)
С повышением симметрии решётки формула (10) упрощается. Для
ромбической сингонии (a = p = y = 90°, cos a = cosp = cos y = 0)
r2=m2a2 + n2b2 + p2c2
(11)
122 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. 1
Для кубической сингонии (а = 6 = с, а = р = у = 90°)
г2 = а2(т2 + п2 + р2). (12)
Квадратичная функция f(mnp) = r2 характеризует сингонию
пространственной решётки.
Площадь параллелограма в плоскости (hkl) в общем случае, как
функция (абсару), выражается уравнением
F2 --= h2b2c2 sin2 a + к2а2с2 sin2 р + l2a2b2 sin2 у + 2kla2bc (cos р cos у - Cos а) +
+ 2lhb2ca (cos у cos а — cos р) + 2hkc2ab (cos a cos p — cos y). (13)
Объём элементарной ячейки в общем случае определяется выражением:
и2 = а2Ьгс2 (1 + 2 cos a cos p cos у — cos2 а - cos2 p — cos2 у). (14)
Поскольку и объём элементарной ячейки, и площадь параллелограма
её основания описываются квадратичными выражениями, целесообразно
выразить аналогичным образом и межплоскостное расстояние d как
функцию ~ .
В общем случае получается довольно сложное выражение, сильно
упрощающееся в случае кубической сингонии
* = Р+Т+Г. (15)
И
д«=* *i±*l±L\ (16)
Отсюда вытекает dhkl = y==== (15а) и, например, dllQ = ~ (15b)
(см. рис. 61).
В отличие от метода обозначения граней кристалла (см. § 35), при
описании элементарных ячеек приходится различать сетки, параллельные
друг другу, но имеющие разные (межплоскостные) расстояния, например
(НО) и (220) (см. рис. 61).
С целью упрощения задачи определения межплоскостных расстояний
и ряда других выводов пользуются представлениями об обратной решётке.
При этом исходят из того положения, что векторным произведением
векторов а и 6, т. е. с = [а6], является вектор, перпендикулярный
к плоскости векторов а и & и равный по величине площади
параллелограма, построенного на векторах а и Ь. Поэтому в качестве
трансляционных векторов принимают а = [Ьс]; Ъ = [ая] и c~[ab], непосредственно
определяющие направления нормалей к исходной сетке и величины её
иараллелограмов.
Векторы а, &, с обратно пропорциональны исходным
а = [Ьв] = -5Г-, Ь = [ас]=-|; с = [а&] = -£-. (17)
где i> — объём элементарной ячейки.
Подобная обратная решётка часто называется F-решёткой.
Можно пользоваться обратной .D-решёткой, для перехода к которой
надо ввести нормирующее условие: F = l. Тогда
(a*aj = l; (Ь*Ь) = 1; (с*о) = 1, (18)
где а*, Ь*, с*—принятые в IT обозначения векторов обратной /)-решётки.
При этом должно соблюдаться условие
(a*b) = (a*e)... = 0. (19)
§59] КОЛИЧЕСТВО ЧАСТИЦ В ЭЛЕМЕНТАРНОЙ ЯЧЕЙКЕ 123
т. е. разноимённые векторы прямой и обратной решётки должны быть
взаимно перпендикулярны (а* X a cos 90° = 0).
(Круглые скобки обозначают скалярное произведение векторов,
в отличие от векторного, обозначаемого квадратными скобками,)
§ 59. Плотность упаковки. Количество частиц в элементарной ячейке
В теории структур часто пользуются формально-геометрическими
представлениями, характеризующими «плотность упаковки» атомов,
понимаемой в смысле отношения объёма шароподобных частиц, приходящегося
на 1 элементарную ячейку, к объёму всей ячейки. Плотность упаковки
обычно выражают в процентах.
Если расстояние между центрами двух одинаковых шаров в элемен-
тарной ячейке равно R и радиус шара г = -~-, то ооъём шара t^ — — R А ,
а объём п шаров, входящих в элементарную ячейку, равен
Ю„ = |пД*А*. , (20)
Если объём элементарной ячейки равен геА.5, то плотность упаковки
/> = -^100(%) (21)
и для известных нам решёток равна
Р-кубическая: р =^)/Г- 100 = 52%,
/-кубическая: р = £ ^ . 100 = 68%,
F-кубическая: р= f-|/2 • 100=* 74%,
Я-плотная гексагональная: р= у |/"2 • 100 = 74%, причём
о
У*
= 1,633.
с
а
F-кубическая и Я-гексагональная упаковки являются плотнейшими
из известных шаровых упаковок.
Число частиц в элементарной ячейке, согласно обычно применяемой
формуле, равно
n=-J-> (22)
где ах — удельный вес, N—число Авогадро, Л —атомный вес, и—объём
элементарной ячейки в кубических сантиметрах. Например, для меди
8,93- 6,02 -1023 . 4гь7 . 1(Г24 0 пг ,
п= Ж57 =3,95^4,
поскольку п не может быть дробным числом. Используя современное
значение N, получаем
"=тда- (22а>
где у —объём элементарной ячейки в кубических ангстремах. То же
уравнение (22а) удобно использовать для расчёта плотности вещества
ох = 1,66020 -~,
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
где пА — сумма атомных весов атомов, входящих в элементарную ячейку. В
случае разнородных атомов, естественно, вместо пА следует ввести SZA
сх = 1,66020^. (22Ь>
§ 60. Конфигурации координационных сфер. Валентные углы
в координационных сферах
При переходе от пространственной решётки к структуре вещества
исследователю приходится считаться с необходимостью выяснения ряда
факторов, специфических уже не для математической системы точек, но для
физической системы многих тел.
Особое значение приобретает под этим углом зрения вопрос об
окончательной конфигурации, возникающей при действии валентных сил. Нас
особенно интересует в этом свете: форма 1-й и ближайших
координационных сфер и
величина валентных углов
® Центр и валентных
расстояний в пределах этих
• Первая координационно* координационных сфер.
>^Ег!
■СР?
О Вторая — * —
■ Третья — » —
Рис. 92. Первая, вторая и тротья координационные
сферы в решётке типа каменной соли.
Чтобы построить
для данного атома
картину расположения
соседей, например в Р-,/-
или F-кубической
решётке, представим себе,
что из данного атома,
как центра,
распространяется сферическая
волна. В момент, когда она достигает центров тяжести первых «соседей»,
зафиксируем их количество и положение на сфере (в SB—первые «соседи»). Мы
получим, таким образом, первую координационную сферу, наиболее
важную для теории структур и кристаллохимии (рис, 92). Затем
распространяющаяся сферическая волна достигнет центров тяжести второго слоя
соседей—получим вторую координационную сферу (в SB—вторые «соседи»).
Для описания положения «соседей» в теории структур часто прибегают
к стереографической проекции.
Иногда бывает важно отличать нечётные (1, 3, 5, ...)и чётные (2, 4, G, ...)
слои, особенно если решётка состоит из атомов или ионов двух типов А и В
(например, Na + Cl~). Тогда часто (но не всегда!) оказывается, что если в центре
находился ион А+, то в 1-й, 3-й, 5-й сфере находятся только ионы В-, а во
2-й, 4-й, 6-й,... сфере только ионы А+. Образуется как бы слоистый шар,
состоящий из множества концентрических разноимённых (состоящих
попеременно из атомов А и атомов В) сфер. Такие решётки называются
координационными. Они встречаются, например, у солей и веществ с солеобраз-
ной структурой, структурными узлами которых можно принять ионы
А+ и В~ (с некоторыми оговорками в соответствии с § 88).
В Р-кубической решётке в первой сфере лежат 6 атомов, во второй—12,
в третьей—8, в четвёртой—6, в пятой—24 и т.д. Приняв радиус первой сферы
за 1 или, что то же, за ]/1 , находим, что радиусы последующих сфер
соответственно равны: у 2 , }/ 3\ |^4 , ]/5 . С соображениями этого рода
связаны расчёты энергии решётки (см. § 75).
Возможные количества атомов, принадлежащих координационным
сферам в подобных пространственных решётках, определяются кристаллогра-
§
#
фическими числами (табл. 10), а сами атомы лежат в вершинах
соответствующих многогранников, форма которых представляет для теории
структур исключительный интерес (см. Б. Ф. Ормонт, Основы теории структур).
С другой стороны, возможны и наблюдаются решётки, в узлах которых
находятся целые группы атомов—молекулы (радикалы), каждая из которых
ведёт себя, как одно целое.
Центральный атом А такой молекулы, как, например, АХ7, может иметь
координационное число, не относящееся к ряду кристаллографических
чисел, в данном случае 7 (например, таково строение ионов TaF^~; NbF^';
ZrF**~), если только поле кристаллической решётки не перестроит
существенным образом структуру отдельных узлов. Так, по современным данным,
решётки РС16 и РВгб построены не из молекул типа АХБ, но из ионов [РС14]+
и [РС1в]-, соответственно [РВг4]^н Br~\
Формы координационных сфер в случае молекулярных решёток и
соединений со сложными комплексными ионами представляют также весьма
большой теоретический интерес.
В структурах типа K2PtCletfoH [PtCle]= образует октаэдрическую
конфигурацию с атомом Pt в центре и 6 атомами С1 в вершинах октаэдра.
Вся эта группа атомов тесно связана между собой. Если фиксированы
позиции четырёх из 7 атомов этого структурного узла, то число степеней
свободы иона равно нулю и позиции остальных получаются однозначно.
При этом все 6 атомов С1 совмещаются друге другом с помощью операций
точечной симметрии, т. е. являются равнозначными, а не идентичными
точками. Если фиксированы позиции двух из 7 атомов иона [PtClJ2-, ион
приходится рассматривать как одно целое, могущее поворачиваться вокруг
прямой, соединяющей эти 2 атома, и т. д. Это позволяет нам считать
подобные образования сложными структурными узлами. Таким образом, в решётке
K.2PtCle имеются не 9 самостоятельных структурных узлов (2К + lPt + 6G1),
а только три (2К + 1 [PtCl,]).
Но, как мы знаем, в справочнике SB структуры веществ
систематизированы на основании формальных признаков—количества атомов, входящих в
формулу вещества, а не на основании количества структурных узлов. Физика
и химика такая систематика не может удовлетворить как из теоретических,
так и из практических соображений, поскольку мы должны стремиться к тому,
чтобы разобраться в механизме взаимодействия силового поля сложного иона
или молекулы и силового поля кристаллической решётки.
Вкратце этого вопроса мы коснёмся в следующих параграфах.
§ 61. Некоторые замечания о путях определения структуры
кристалла с использованием экспериментальных рентгенографических
данных. Единицы кХ и единицы А
Изложение теории и методов определения структур выходит за рамки
книги. Здесь мы ограничимся немногими замечаниями.
Задача заключается в том, чтобы установить связь между результатами
изучения интерференции рентгеновских лучей, т. е., например,
рентгенограммой и той совокупностью сведений о веществе, которую мы имеем
в виду, говоря о структуре кристалла. В частности, необходимо установить
размер и форму элементарной ячейки, количество входящих в неё атомов,
пространственную группу и, наконец, координаты центров тяжести атомов.
В основе исследования лежит известное читателю из курсов физической
химии уравнение, предложенное одновременно в 1913 г. в России Г. В. Буль-
фом и в Англии Брэггом, уравнение Вульфа—Брэгга
пХ = 2dm sinOt (23)
126 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. I
~"~_— v ' =
где /ч—длина волны, d—межплоскостное расстояние, б —угол отражения
луча.
Для расчётов необходимо также точное знание химического состава,
удельного веса и других физических свойств вещества. Таким образом,
структурные определения требуют совокупности исследований
математических, химических, физических и ни в коем случае не являются лишь
математической задачей, решаемой на базе рентгенограммы.
Выбор метода исследования структуры твёрдого тела зависит от
характера кристаллов (монокристалл, поликристалл), которые могут быть
получены, от их величины, от симметрии кристаллов и их инертности в отношении
физических и химических факторов, действующих на вещество во время
подготовки к съёмке и самой съёмки.
Предохранение кристалла от самопроизвольного протекания реакций
является весьма серьёзной задачей, например, при исследовании метаста-
бильных фаз.
При проведении структурного анализа сначала производится
определение сингонии, затем лауевского класса симметрии, элементарной ячейки,
а также возможной пространственной группы, что производится по-разному,
в зависимости от качества исследуемого препарата. При наличии хотя бы
небольших монокристаллов и возможности, таким образом, получить
рентгенограмму вращения удаётся непосредственно установить вид симметрии,
возможную пространственную группу и элементарную ячейку. Возможные
11 лауевских классов симметрии отвечают тем 11 видам симметрии из 32,
которые обладают центром инверсии и в таблице 10 отмечены звёздочкой.
Простейшим путём установления лауевских классов симметрии при
наличии достаточно крупных монокристаллов является получение лауе-
граммы. Существование всего 11 лауевских классов симметрии связано с
законом Фриделя, согласно которому рентгенографическое исследование
приводит к получению рентгенограмм, соответствующих наличию в кристалле
центра инверсии, даже если кристалл таковым не обладает. Иначе говоря,
симметрия эффекта в этом случае либо совпадает с симметрией объекта (если
кристалл сам по себе имеет центр инверсии), либо оказывается выше её,
о чём мы говорили в § 10.
В случае очень мелких кристаллов (порошков) удаётся воспользоваться
лишь методом порошков, который даёт однозначные результаты, собственно,
лишь в случае кристаллов кубической сингонии.
При исследовании кристаллов средних сингонии удаётся более или менее
надёжно определить методом порошков элементарную ячейку лишь в более
простых случаях. Зная объём элементарной ячейки, удельный вес и
химический состав вещества, нетрудно рассчитать, сколько и каких атомов входит
в состав элементарной ячейки, после чего перейти к определению возможных
положений частиц.
Здесь возможны два пути. Первым является метод проб и ошибок,
заключающийся в том, что атомам приписываются условно положения в
элементарной ячейке, вытекающие из той или иной теоретической модели. После
этого данные рентгенограмм (интенсивность рефлексов), найденные
экспериментально, сопоставляют с теоретически вычисленными; изменение
положения атомов производится до тех пор, пока не будет достигнуто достаточно
удовлетворительное совпадение.
Второй путь—так называемый рентгеновский Фурье-анализ — нами
рассмотрен вкратце в § 96 (см. также Б. Ормонт [6] (1940)).
Подчеркнём, что основную роль в структурных исследованиях играет
изучение интенсивностей рассеянного веществом рентгеновского излучения,
которое позволяет решить не только вопрос о координатах атомов, но и ряд
важнейших для теории и практики проблем строения реального кристалла.
§ 61] НЕКОТОРЫЕ ЗАМЕЧАНИЯ. ЕДИНИЦЫ кХ И ЕДИНИЦЫ А 127
В заключение параграфа мы должны остановиться на вопросе о
величинах, которыми измеряются длины волн рентгеновских лучей.
В основе расчётов по уравнению (23) лежат длины волн рентгеновских
лучей. Следовательно, и межплоскостные расстояния выражены в особых
рентгенографических или спектрографических единицах.
В качестве таковых принята «единица X», равная 103 от
величины межплоскостного расстояния du.i) между плоскостями спайности
кальцита, которое принималось равным 3,02904 А при 18° С (как среднее из трёх
измерений, опубликованных Зигбаном [19] в 1919 г.).
Таким образом, 1 X принимался равным 10~11 см, а 1000 Хо—1 кХ —«один
килоикс» принимался равным 10~8 см, т. е. 1 ангстрему (1 А). Поэтому во
всей структурной и рентгенографической литературе до 1945 г. и даже до
сентября 1947 г. 1 кХ ошибочно заменялся 1 А.
В течение последних лет атомные постоянные подвергались
основательному пересмотру—в частности, и число Авогадро (6,023 -1023 вместо 6,06-1023).
Уже в 1945 г. стало окончательно очевидно, что 1 кХ не точно равен 1
ангстрему, и для перехода от 1 кХ к 1 А следует пользоваться переходным
коэффициентом, который мы обозначим у, примерно равным 1,00203. Впрочем,
в течение нескольких лет вопрос об окончательной величине переходного
коэффициента, принятой рентгенографамй, оставался открытым.
В сентябре 1947 г. было опубликовано сообщение Брэгга [20] (см. также
121]) о том, что в свете современных данныхо следует принимать
переходный коэффициент от единиц кХ к единицам А
Y = 1,00202 ±0,003%.
Этот переходный коэффициент действительно отвечает современному
уровню наших знаний и может отныне считаться общепринятым. Тем самым
для перехода от принятых до сего времени длин волн «в А», а в
действительности в кХ, к длинам волн в А требуется умножение на у-
Например, для Си КЛ19 по общепринятым ранее табличным данным,
л = 1,537395А, в действительности же л = 1,54050А.
В упомянутой статье [20] приводится таблица длин волн различных
характеристических излучений в ангстремах. Таблица сопровождается
примечанием, что впредь во всех рентгенографических работах следует
точно указывать принятые для расчёта длины волн.
Ниже приводится составленная нами таблица, в которой сопоставляются
длины волн в ангстремах по Брэггу и длины волн в килоиксах, отвечающие
прежним значениям «в ангстремах» (см. табл. 14* на стр. 128).
При расчёте значений X для Ка принималось, что вес КЛ1 вдвое больше,
г п lt 2-1,54050 + 1,54434 лмао
чем ла2; например, для Си ЛГа= j~—- = 1,5418.
Как мы знаем, при пользовании уравнением (22Ь) ах = 1,66020
для вычисления плотности величина v обозначает объём элементарной
ячейки в куб. ангстремах. Для перехода от и, выраженного в куб.
килоиксах, к v, выраженному в куб. ангстремах, надо умножить первую
величину на коэффициент ш = (l,00202)s = 1,00607, т. е. пользоваться
выражением ах= 1,65018—, где и —объём элементарной ячейки в куб.
килоиксах.
В связи с изложенным все литературные данные (до 1947 г.) о
межатомных расстояниях и т. п. характеризуют значения последних не
в ангстремах, а в килоиксах. Поэтому во второй части книги также
приняты величины, выраженные в килоиксах.
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Таблица 14*
Сг
Мп
1 Fe
Со
Ni
Си
Zn
Mo
Rh
Pd
A?
Длины волн в
ангстрем . . . ,
килоикс .
ангстрем .
килоикс .
ангстрем .
КИЛОИКС .
ангстрем ,
КИЛОИКС .
ангстрем .
КИЛОИКС .
ангстрем .
КИЛОИКС .
ангстрем .
КИЛОИКС . .
ангстрем .
КИЛОИКС .
ангстрем ,
КИЛОИКС .
ангстрем .
КИЛОИКС .
ангстрем .
КИЛОИКС .
A'ai
2,28962
2,28503
2,10174
2,0975
1,93597
1,932076
1,78890
1,7853'
1,65783
1,65450
1,54050
1,5*7395
1,43510
1,4322
0,70926
0,707831
0,61326
0,61202
0,58545
0,58427
0,55941
0,55828
К*2
2,2 3352
2,28891
2,10570
2,1015
1,93991
1,936012
1,79279
1,7892
1,66168
1,65835
1,54434
1,541232
1,43894
1,4350
0,71354
0,712805
0,61762
0,61637
0,58982
0,58863
0,56381
0,56267
к*
2,2909
2,1031
1,9373
1,7902
1,6591
1,5418
4,4334
0,7107
0,6147
0,5869
0,5609
*Pi
2,08473
2,0806
1,91016
1,9062
1,75654
1,753013
1,62073
1,6174
1,50008
1,49705
1,39217
1,38935
1,29520
1,2926
0,63225
0,630978
0,54559
0,54449
0,52052
0,51947
0/*9701
0,49601
Граница
поглощения
2,0701
2,066
1,8954
1,890
1,7429
1,740
1,6072
1,602
1,4869
1,484
1,3802
1,377
1,2831
1,280
0,6197
0,6185
0,5341
0,5330
0,5090
0,508
0,*855
0,485
1
§ 62. Об основных типах пространственных решёток
В результате структурных исследований обнаружены сотни различных
структур, из которых многие будут рассмотрены в следующих главах.
Возможен различный подход к задаче их сопоставления и
систематики. Одной из наиболее распространённых является систематика SB,
отвергнутая при изложении материала в этой книге.
Для интерпретации громадного структурного материала в области
неорганической химии мы считаем целесообразным выделить основные типы
пространственных решёток, каждый из коих присущ структурам большого
количества веществ или задаёт основной мотив более сложных структур.
В § 45 мы назвали атомы или группы атомов, фигурирующие в
структуре как одно целое, структурными узлами. Здесь необходимо отметить,
что узлы могут быть простыми (если они состоят из 1 атома, например
в NaCl узлы Na и С1) и сложными (если они состоят из нескольких
атомов). Сложные узлы могут быть однородными (т. е. состоять из
одинаковых атомов), как, например, узел [J3] в решётке KJ3, и неоднородными, т. е.
состоящими из разных атомов, как, например, узел [СОа] в решётке СаС03.
Учитывая физические особенности структур, целесообразно при
описании пространственных решёток как совокупности кристаллографических
точек выделять комплекс атомов, составляющий структурный узел.
Узлы мы будем обозначать греческими буквами а, [3, у» сохраняя
латинские буквы для обозначения атомного состава.
Например, K2PtCle отвечает химической формуле А2ВХв, но его
структурная формула будет а2р, ибо комплекс [PtClJ ведёт себя как единый
структурный узел р (см. рис. 602), Хотя в ряде случаев атомный и
структурный состав могут совпадать (например, в случае NaCl), более интересны
и важны случаи, когда они не совпадают. Учёт этого обстоятельства
требует коренного изменения систематики структур по сравнению с SB.
§ 62] ОБ ОСНОВНЫХ ТИПАХ ПРОСТРАНСТВЕННЫХ РЕШЕТОК 129
Для характеристики типов пространственных решёток, а значит,
и структур, нам представилось наиболее целесообразным различать их,
вопреки формалистической нумерации SB (в порядке букв алфавита или
натурального ряда чисел), по характеру сингоний и конфигураций
координационных сфер. Такой принцип мы назовём координационным.
О целесообразности такого подхода в свете представлений
современной теории химической связи мы уже коротко говорили.
Основные структуры кубической сингоний мы будем обозначать
символом Су гексагональной — Ну тригональной (ромбоэдрической)—Д,
тетрагональной — 7\ ромбической (орторомбической)—OR, моноклинной—М,_
триклинной — Тг.
Хотя в структурах веществ наблюдаются самые разнообразные
координационные числа и формы координационных сфер, в основных
структурах их количество сравнительно ограничено.
Мы обозначаем их индексами следующим образом (табл. 15, см. также
табл. 31).
Таблица 15
Обозначения конфигураций по координационному принципу
1 fi О
1 ЁГ «
1 rt ё
2g
1 О аз
1 ° £
1 К о
12
12
8
' 1
Конфигурация
Кубооктаэдр
Кназикубоок-
таэдр (гек-
сагональн.
аналог ку-
бооктаэдра)
Гексаэдр
(куб)
о
X
«
X
К
V
С
н 2
sr ч
On О
о я
° X
А О
6
6
4
Конфигурация
Октаэдр
Призма (три-
гональная)
Тетраэдр
о
X
<х>
«
s
|^>»
О
7>3
т
в о
Я" «
Ss
9 я
9 х
1 К о
4
3
о
Конфигурация
Квадрат
Треугольник
Линия
о
X
о
«
Я
К
?
t
1
Примечание. Индексы объёмных конфигураций обозначаются прописными,
плоских и линейных конфигураций — строчными буквами.
Кубооктаэдр и квазикубооктаэдр показаны на рис. 93, а и 6
соответственно. Индексы мы проставляем справа внизу, около обозначения сингоний. На-
Рис. 93. Кубооктаэдр и его «гексагональный аналог».
пример, Сс кубическая центрированная ячейка Na (к.ч. 8). В случае наличия в
решётке узлов двух и больше типов мы будем проставлять индексы для
130 ч. ь основы теории структур [гл. I
каждого типа узлов, отделяя их знаком дроби: например,
СС/с—кубическая центрированная ячейка типа CsCl (Cs к.ч. = 8С1, С1 к.ч. = 8 Cs).
На рис. 94 показана кубооктаэдрическая , координационная сфера
в плотнейшей кубической упаковке и направление связей в этой
сфере. *
Рис. 94. Кубооктаэдрическая координационная сфера
в кубической гранецентрированной пространственной
решётке и 12 связей от центрального атома.
Так как основные типы пространственных решёток выбраны нами
с учётом их роли в теории структур, мы будем называть эти типы
также по структуре той модификации и того вещества, которому в SB
отвечает этот тип пространственной решётки.
§ 63. Основные структуры, принадлежащие кубической сингонии
С тремя основными видами кубических элементарных
ячеек—примитивной Р-ячейки, центрированной /-ячейки и гранецентрированной
f-ячейки — мы уже знакомы (рис. 60 и рис. 95. a,b,d). В предлагаемых нами
терминах координационной систематики соответствующие структуры
обозначаются Со , Сс и Ск-
На рис. 95 эти элементарные ячейки сопоставляются с другими. Тип Сс
(рис. 95, Ь) носит название «тип вольфрама», С к—«тип меди».
Близкой к структуре Сс является структура Сс\с (тип хлористого
цезия) (рис. 95, с), в которой координационное число узла а = 8(Р),
координационное число узла р = 8 (а).
Очень важны структуры, производные от Ск- Для понимания их
взаимной связи между собой разделим элементарную ячейку С к на
8 октантов. В каждом октанте, представляющем собой куб с ребром,
равным половине исходного, из 8 вершин, заняты только 4 в тетраэдри-
ческом порядке (рис. 95, d). С другой стороны, не заняты и центры
октантов.
Наличие этих двух сортов незанятых мест в решётке Ск влечёт
за собой два разных пути построения производных структур.
Если ввести в Ск-решётку (с узлами а, занятыми атомами металла)
вторую такую же решётку с узлами (3-атомами неметалла, и притом так,
чтобы начальная точка второй С^-решётки центрировала ячейку первой
Ск-решётки, то окажется, что в каждом октанте элементарной ячейки
прежде незаполненные вершины окажутся занятыми атомами неметалла,
которые займут, таким образом, центры рёбер и цев:тр элементарной ячейки
(рис. 95, е). Возникает восьмикратно-примитивная элементарная ячейка —
§ 63] ОСНОВНЫЕ СТРУКТУРЫ, ПРИНАДЛЕЖАЩИЕ КУБИЧЕСКОЙ СИНГОНИИ 131
С0- ячейка (тип Ро)
^^
Ш
\sv
И
Сс~ячейна(типМ)
Ъ
*£&
\Л
ш.
\Z.
'F*
\/
Сс/с-ячеина (тип CsCQ
^4^^
СН'Ячеина(типСи)
d
Со/о {тип Nod)
Сс/Т(типСи20)
/
Pin-. C.J5.
132
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
Ci/T(munCu20)
Г
i4^q
Ст (тип алмаза)
о^5з2й6
СТ/Т (man ZnS) цин новой обмотни
h
CcjT(munCaF2)
i
cc/ojl^fjnBiF3)
i
Рис. 95. (Продолжение)
§ 63] ОСНОВНЫЕ СТРУКТУРЫ, ПРИНАДЛЕЖАЩИЕ КУБИЧЕСКОЙ СИНГОНИИ 133
тип поваренной соли. Таков первый способ введения атомов неметалла
в Сд-ячейку. Но, как мы знаем, этот путь не является единственным.
В ячейке Ск центры всех 8 октантов не заполнены. Их последовательное
заполнение атомами даёт 4 важных типа структур: куприта (Cu20), алмаза
(Сал.м)> цинковой обманки (ZnS) и плавикового шпата (CaF2j.
Пусть атомы металла занимают позиции в узлах 6\-решётки.
При соотношении а2р на 4 атома металла, необходимые для
заполнения узлов четырёхкратно-примитивной С^-ячейки, требуются 2 атома
О0
О
1 >
г- 1
6
г О
1 6
о
V
GO
q—^-^0
<к
о а
6 О-
^ о
,<U
05
О С
О (.
6—t
?
)0 \
0J5 7
О
\ 6
f
О О О
07/ 0Л5\
О О
о А П
ДО \°
о о
0J5
6 О О
Алмаз
ь*
ZnS
-О
*р
Ъ!
ь*
905
О С
0,25
о
0,75
A t
0,2}
о
1 0,73
О——-—t
р о
0,4
о
а «» 1
Ч А
Q25
о
«W 1
—-—6
CaF,
Q-
0,S^J U5U>
0,21
О
O.ZS
о
о о,7*а
±J!2WJ4№
ДО
О
0,75
0,25
О
0.75
8IF,
нб
Рис. 96. Проекции основных типов структур кубической сингонии на основании
элементарной ячейки:
о) тип полония (?); Ь) тип вольфрама; с) тип хлористого цезия; d) тип меди; г) тин каменной соли;
/) тип куприта, д) тип алмаза; Л) тип цинковой обманки; О тип флюорита; /) тип фтористого
висмута. Общее примечание: цифры около кружков указывают высоту позиции точки над
основанием элементарной ячейки.
неметалла. Такое соотношение наблюдается в формуле куприта (Cu20).
Атомы О занимают центры двух октантов (рис. 95, /), лежащие на одной и той
же объёмной диагонали элементарной ячейки, например позиции С-г — -г- )
(3 3 3 \
■j-TT-J. Возникает шестикратно-примитивная элементарная ячейка
(тип куприта). Обычно приводимая структура куприта (Cu20) no SB
(рис. 95, /') не выявляет связи с С^-решёткой.
При соотношении ав (в случае ZnS) в элементарную ячейку типа
Ск> узлы которой заняты атомами Zn, надо ввести 4 атома неметалла.
134
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I
При образовании восьмикратно-примитивной элементарной ячейки
цинковой обманки (Сцт) заполняются центры четырёх октантов исходной
б'к-решётки (а) в тетраэдрическом порядке (рис. 95, Л'). При этом нулевая
точка второй С/с-решётки (Р) должна быть совмещена с центром октанта
первой Ск-решётки (а).
В результате к.ч. атома Zn = 4 (S) и к.ч. атома S = 4(Zn).
Если в решётке ZnS а- и [3-узлы заместить атомами углерода, то
получается решётка алмаза Су (рис. 95, g).
Наконец, введём в решётку Ск, заполненную атомами металла (а),
две дополнительные С^-решётки, заполненные атомами неметалла (р),
и притом так, чтобы нулевая точка одной С^-ячейки р совместилась
с центром одного из октантов ячейки а {как в случае ZnS), а нулевая
точка второй Ск-ячейки р совместилась с центром одного из 4
октантов, оставшихся в решетке типа ZnS незанятыми (с таким расчётом,
чтобы все 8 октантов оказались центрированными атомами неметалла
(рис. 95, i).
В результате возникает диеиадцатикратно-примитиннан элементарная
ячейка типа CaF2 (флюорита или плавикового шпата).
Заполнив атомами неметалла (узлы Р) центры всех 8 октантов
С^-решётки, узлы которой заняты атомами металла (а), и образовав
решётку типа CaF2, мы обнаруживаем, что места, занятые атомами
неметалла при образовании решётки типа NaCl (центры рёбер и центр
элементарной ячейки), остались в решётке GaF2 свободными (рис. 95, /').
В решётке BiF3 (шестнадцатикратно-примитивной) эти позиции также
оказываются занятыми (рис. 95, /). Структура BiF3 является исходной
при образовании структур NaTl, Cu3Al и др.
Взаимная связь между всеми вышерассмотренными элементарными
ячейками очевидна также из рис. 96, где показаны проекции всех структур
на основание элементарной ячейки. Цифры около кружков обозначают
высоту данной позиции в ячейке над основанием последней в долях aw.
Нуль означает, что структурный узел лежит в основании элементарной
ячейки.
§ 64. О двух типах плотнейших шаровых упаковок
Как мы видели, структура Ск является исходной в длинном ряде
других структур, образованных разнородными узлами (а2р, ар, afJ2 и аР3).
Эта структура является важнейшей и среди структур, образованных
однородными узлами а, по той причине, что она является одной из двух
плотнейших шаровых упаковок. Второй является плотная
гексагональная упаковка (рис. 97, 6).
Существует только одно решение задачи плотнейшей упаковки
шаров одинакового диаметра в одной плоскости: вокруг центрального
шара должно быть расположено 6 шаров в вершинах правильного
шестиугольника (рис. 98, а). Другие варианты упаковок являются менее
плотными.
Более сложно обстоит дело с укладкой друг на друга нескольких
рядов, построенных подобным образом.
Упаковка, выполняемая таким образом, чтобы центры тяжести
шаров верхнего и нижнего рядов находились на одной вертикали (рис. 97, а).
не является наиболее плотной. Расстояние между 1 и 3-м рядами
определяется отношением — = 2.
а
Если шары каждого последующего ряда будут помещены в
углубления между шарами предыдущего ряда, расстояние между 1 и 3-м
$ 64| О ДВУХ ТИПАХ ПЛОТНЕЙШИХ ШАРОВЫХ УПАКОВОК 135
рядами определится отношением — = 1,633 (рис. 97, 6), что будет
отвечать наиболее плотной упаковке.
Как видно из рис. 98,я, над основанием элементарной ячейки
имеется 6 углублений, в которых надо поместить 3 шара, заняв
половину всех углублений.
Поэтому при упаковке трёх слоев возникает два варианта.
1. Три шара верхнего (3-го) ряда, обозначенные пунктиром на
рис. 98, л, лежат \\ точности над шарами нижнего 1-го ряда, по обе
стороны от показанного на чертеже среднего (2-го) ряда. Такое размещение
приводит к образованию «плотной» гексагональной упаковки (рис. 98, Ь \\d).
Её особенностью является наличие \\ решётке бесконечных незанятых
«люков» (на рис. 98, а обозначены
белым кружком). Этот способ
упаковки называется упаковкой
по гексагональному закону.
2. Три шара верхнего (3-го)
ряда занимают три углубления,
лежащие над тремя незанятыми
углублениями нижнего (1-го)
ряда (обозначенные белым
кружком), и только в четвёртом
слое три шара располагаются в
точности над шарами 1-го ряда.
Получается ромбоэдрическая
элементарная ячейка из 4 слоев
атомов (рис.98,с и е; ср. рис.64, е.),
причём -а -1,633 f 0,817-2,45.
Ось ромбоэдра совпадает с
поворотной осью 3 куба.
Такая упаковка является
илотнейшей кубической и
отвечает ячейке Ci: = С к:
--т
&
\C4.S33
a-i
Рис. 'М. Гексагональная (а) и шютная
гексагональная упаковки (Ь).
Однократно-примитивный ромбоэдр, показанный на рис. 64, е, при
соотношении c/a = 2/kb отвечает кубической элементарной ячейке (т. е.
имеет угол 60°. При других соотношениях с/а ячейка является
ромбоэдрической). Он расположен в ячейке Ск, как это показано на рис. 63.
Очевидно, что можно укладывать шары, чередуя порядок упаковки
то по гексагональному, то по кубическому закону. При этом возникают
гексагональные элементарные ячейки с различным количеством слоев —
вплоть до нескольких десятков, как, например, в структурах карбида
кремния SiC. Более того, в таких случаях мы считаем вполне
возможным и беспорядочное чередование слоев по обоим законам.
Упорядоченным чередованиям слоев посвящено интересное
исследование Г. С Жданова [5] (1946 г.). Н. В. Белов (1947 г.) вывел
возможные типы шаровых упаковок, вплоть до 30-слойных.
Обе плотнейшис основные упаковки можно рассматривать и как
состоящие из тетраэдров, в вершинах которых расположены атомы. В кубической
упаковке тетраэдры всех слоев повёрнуты в одну сторону (рис. 99, а\ Ь\ с')
(в SB обозначается рр...щ т. е. параллельно), в гексагональной —
э разные стороны (в SB обозначается аа . . . — антипараллельны)
(см. рис. 99, я, bt с.)
В гексагональной упаковке тетраэдры первого и третьего рядов (косая
штриховка) находятся друг иод другом. Их проекции совпадают (рис. 99, с).
136 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. I
Рис.98. Плотнейшие упаковки <шаров по кубическому и гексагональному
законам.
На рис. d и е показаны координационные сферы из 12 атомов вокруг
центрального, лежащего в центре шестиугоььника.
§ 64] __Р_Д11УХ ТИПАХ ПЛОТНЕЙШИХ ШАРОВЫХ УПАКОВОК 137
Рис. 99. Образование плотной гексагональной и кубической упаковок из тетраэдров:
а) гексагональная; Ь) кубическая. Если соединить нее атомы координационных сфер рис 98 d и о
с центральным, то будут чётко видны упаковки из тетраэдров рис. 99, и л а> (плоскость
шестиугольника показана пунктиром).
138 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. I
В кубической упаковке совпадают тетраэдры первого и четвёртого рядов
(косая штриховка, рис. 99, с').
Отметим, что в плотных упаковках имеются октаэдричсские
междуузлия, количество которых равно количеству атомов (п), и тетраэдри-
чсские междуузлия, количество которых равно удвоенному количеству
атомов (2п).
Места этих междуузлий в решётке Сц = С/, мы найдём без труда
(см. рис. 100, а также § 63).
Октаэдричсские междуузлия расположены в центре рёбер и в центре
элементарной ячейки (эти позиции октаэдрически окружены 6 атомами):
Тстраэдрические расположены в центрах октантов (эти позиции
тетраэдрически окружены 4 атомами).
Таким образом, образование
структур, рассмотренных в § 63, идёт по пути
замещения либо октаэдрических (тин
NaCl), либо тетраэдрических (куприт,
алмаз, ZnS, CaF2) междуузлий, либо
и тех и других (BiFJ. s
§ 65. О структурах ZnS (вюрцита),
NiAs, CdJ„ MoS2
Ещё 4 гексагональные структуры
должны быть нами здесь рассмотрены
(рис. 101).
Решётка ZnS (вюрцита) является
производной от решётки Ну. Для её
образования надо совместить две
решётки Ну таким образом, чтобы нулевая
точка одной центрировала тетраэдр
другой решётки (рис. 101, ((). Возникает
структура #7/г-
Гексагональная октаод рически-приз-
матическая никельа рселидная решётка
Hojps имеет элементарную ячейку, включающую 3 слоя узлов а,
занятых атомами Ni, и, таким образом, по высоте равную двум ячейкам
Ну (рис. 101, Ь). Вызвано это тем, что атомы As (узлы Р) образуют в
элементарной ячейке два соседних слоя, разделённых слоем узлов а так,
что центры тяжести узлов р верхнего слоя не находятся над центрами
тяжести узлов р нижнего слоя, но располагаются между ними (как
в кубической упаковке). В результате узлы а и р имеют к.ч. 6, однако
каждый узел a (Ni) окружён 6 узлами р, расположенными в вершинах
октаэдра, тогда как каждый узел р (As) окружён 6 узлами а,
расположенными в вершинах тригоналыюй призмы.
Гексагональная слоистая октаэдрическа-треуголъная решётка CdJ*,
(тип SB C6)Яо//—одна из нескольких структурных форм CdJ2.
Элементарная ячейка простирается по высоте от первой сетки атомов Cd до
второй, причём атомы металла (узлы а) расположены, как в простой
гексагональной ячейке (рис. 101, с и 97, а).
Узлы P(J) образуют две сетки внутри элементарной ячейки, причём
узлы р верхней сетки не находятся над узлами В нижней сетки. Таким
образом, в отличие от ячейки NiAs в рассматриваемой ячейке GdJ2
средняя сетка узлов а (атомы металла) отсутствует, сетки узлов р односторонне
притягиваются к сеткам узлов а, образуя трёхслойные упаковки, в которых
о Чзлы
(•) Тетраэдричесние меж^уузж*
О Цктаэдричвскиь междуузлия
Рис. 100. Октаэдрпческие и тетра-
эдрические можлуузлин и плотей
кубической упаковке.
§ 65]
О СТРУКТУРАХ ZnS, NiAs, CiJ2, MoS_.
139
d b
Рис. 101. Структуры пюрцита ZnS, NiAs, CdJ., и MoS2.
140
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. I .
узлы ос октаэдрически окружены узлами р, как и в NiAs элементарное
ячейке, узлы р имеют в 1 координационной сфере треугольник из 3 узлов а.
Гексагональная слоистая призматически-треугольная решётка
сернистого молибдена MoS2 #|3/г
Элементарная ячейка имеет сходство с двумя предыдущими
элементарными ячейками. Как и в ячейке NiAs, узлы а (атомы металла)
образуют в ней три сетки. Однако узлы р (атомы неметалла) образуют между
узлами а по 2 сетки, причём узлы верхней сетки не находятся под
узлами нижней сетки (рис. 101, d). В результате, как и в ячейке GdJ2,
образуются трёхслойные упаковки. Однако в них каждый узел а призматически
окружён 6 узлами р, т. е. в любых двух р сетках, прилегающих сверху
и снизу к а сетке, р атомы находятся друг над другом, образуя триго-
нальную призму. Зато в соседних трёхслойных упаковках рар сеток
узлы р занимают позиции, не лежащие на вертикали над позициями р узлов
предыдущего слоя рар.
Таким образом, в структуре CdJ2(SBC6) атомы металла образуют
только октаэдрические, в MoS2 —только призматические координационные
сферы.
Ниже мы приводим сводную таблицу основных кубических и
гексагональных структур (табл. 16).
Таблица 16
Основные структуры, обозначенные по координационному принципу
п/п
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
Обозначение
—
Л2
В'1
Л1
В\
Д4
£3
СЗ
а
лз
£4
£8
СО
С1
По SB
Наименование
Кубические с
примитивная
тип вольфрама
тип CsCl
тип Си (плотная
кубическая упаковка)
тип NaCl
тип алмаза С
тип сфалерита ZnS
тип куприта Cu20
тип плавикового
шпата CaF2
тип BiF3
Гек сатональные
тип Mg (плотнейшая
гексагональная упаковка)
тип вюрцита ZnS
тип NiAs
тип CdJ3
тип MoS^
По координационному принципу [
Обозначение
труктуры
Со
СС
СС1С
Ск
со/о
U т
СТ/Т
cljT
CCjT
CC + OlOtT
структу
"v
HTjT
HOlP3
IIs
11 рз//
Наименование 1
октаэдрическая |
(гексаэдрическая) 1
дважды гексаэдрическая 1
кубооктаэдрическая I
дважды октаэдрическая 1
тетраэдрическая [
дважды тетраэдрическая t
линейно-тетраэдрическая 1
куботетраэдрическая 1
— 1
Э Ы I
квазикубооктаэдрическая 1
дважды тетраэдрическая 1
октаэдрически-призмати- 1
ческая 1
слоистая октаэдрически-
треугольная 1
слоистая призматически-
треугольная
Примечание. В структурах GdJ2 (и MoS2) одностороннее распределение узлов
J и Cd с образованием слоистых структур делает целесообразным лведение общего
индекса S (слоистая структура).
§ 66] ОСТРОВНЫЕ, ЦЕПОЧЕЧНЫЕ (ЦЕПНЫЕ), СЛОИСТЫЕ СТРУКТУРЫ 141
§ 66. Островные, цепочечные (цепные), слоистые структуры
До сих пор мы рассматривали главным образом координационные
решётки (структуры) и лишь в последних примерах Ноц< H%/t коснулись
слоистых решёток.
Среди структур различных веществ, помимо координационных,
наблюдаются решётки и других типов — островные, цепочечные, слоистые, при
описании которых обыч-
6<х>9
С>—о
о—6
Рис. 102. Острова. Количество атомов ни в одном
измерении не бесконечно.
но прибегают к разным
формулировкам.
Мы
придерживаемся следующих.
Структуры, о
которых мы говорили,
отличаются от
координационных наличием
ограниченных по
протяжённости в одном или нескольких измерениях групп атомов, на которые
как бы разбивается вся решётка.
Если протяжённость такой группы атомов во всех измерениях конечна,
структура является островной.
6
6
6
о
6
0
Рис. 101). Цени. Количество атомов бесконечно в одном измерении.
Если протяжённость группы атомов в одном измерении (вдоль
некоторого вектора) бесконечна, структура является цепной, если в двух
измерениях— слоистой. Прообразом последних двух типов структур
являются изображённые на рис. 59, с и соответственно 59, Ь.
В зависимости от количества атомов в трёх измерениях островные
и цепные структуры могут быть одномерными, двумерными и
трёхмерными; слоистые—двумерными и трёхмерными (на рис. 104 показан один
слой). Их различия с очевидностью вытекают из рис. 102, 103 и 104
и не нуждаются в комментариях.
142
Ч. I. ОСНОВЫ ТЕОРИИ-СТРУКТУР
[гл. I
Рис. 104. Сетки. Количество атомов бесконечно в двух измерениях}
§ 67] ЦЕПИ И СЕТКИ ИЗ ОКТАЭДРОВ И ТЕТРАЭДРОВ 143
§ 67. Цепи и сетки из октаэдров и тетраэдров и стехиометрическии
состав координационных сфер
Допустим, что в центре тетраэдра или октаэдра находится атом А,
в вершинах —атомы В. Каков химический состав группы АВЬ
бесконечной цепи, иными словами, каково значение индекса b в зависимости
от характера цепей? Для определения Ь надо сложить структурные дроби
для атомов В в каждой позиции (рис. 105 и 106j; см. §224.
Если, например, октаэдры соединяются в цепи так, что четыре
вершины принадлежат только данной координационной сфере, а две являются
общими между данным и соседним октаэдрами (рис. 105, а), то на один
атом А приходится атомов В
6 = Y + | = 5 (24»
и координационная сфера имеет стехиометрическии состав [АВ6].
Если октаэдры соединяются в цепи противоположными рёбрами, то
(рис. 105, Ъ)
*=Т+Т=4 <25>
и координационная сфера имеет стехиометрическии состав [АВ4].
В случае октаэдров, соединённых гранями (рис. 105, а,
6 = |— 3 (26)
и координационная сфера имеет стехиометрическии состав [АВ3].
Если тетраэдры соединяются в цепи так, что две вершины принадлежат
только данной координационной сфере и две являются общими между
данным и соседним тетраэдрами (рис. 106, а), то па один атом А
приходится
6 = 4 + | = 3 (27)
атомов ^ и координационная сфера имеет стехиометрическии состав [АВ31»
Если цепи тетраэдров соединены в сетки вершинами оснований
тетраэдров (рис. 106,6, чёрные шары), так что каждый из трёх атомов
принадлежит двум тетраэдрам и лишь один атом (белый шар)—одному,
то на один атом А приходится атомов В
6 = 4+4 = 4 <28>
и координационная сфера имеет стехиометрическии состав [А2В5].
Наконец, если тетраэдры соединены в цепи противоположными
рёбрами (рис. 106, с) или образуют так называемую «пространственную вязь»,
разделяя каждую из четырёх вершин с четырьмя соседними тетраэдрами
(рис. 107, а и 6), то на один атом А приходится атомов В
Ь = ~ = 2 (29)
и координационная сфера имеет стехиометрическии состав [АВ2]. Па
рис. 107, Ь показана элементарная ячейка р-кристобалита и на рис, 107, с —
структура берилла из кремне-кислородных тетраэдров (атомы Si в центре,
тетраэдров, атомы О в вершинах).
I '!
Рис. 105. Цепи из октаэдров и состав
координационных сфер.
Рис. 106. Цепи из тетраэдров н состав
координационных сфер.
§ 681 ПЕРЕХОД ОТ ПРОСТРАНСТВЕННОЙ РЕШЕТКИ К СТРУКТУРЕ ВЕЩЕСТВА 145
Рис. 107. Пространственная вязь ил тетраэдров и октаэдров и состав
координационных сфер:
<i) и Ъ) р-кристобалит (S1O2); с) берилл (А1Л]е3 (SiOs)e). Кольца Si8Ois связаны между собой
атомами А1 и Be.
§ 68. Переход от пространственной решётки к структуре вещества
Очевидно, что структура вещества отличается от пространственной
решётки, т. е. от образованной трансляцией бесконечной системы
кристаллографических точек, по меньшей мере следующими особенностями:
1. Она является системой не кристаллографических точек, а физических
тел (атомов, молекул, ионов) и управляется в первую очередь не законами
геометрии, а законами физики, которые в ряде случаев диктуют
необходимость образования структур той или иной симметрии, не противоречащих
ограничениям,- налагаемым кристаллографической теорией симметрии.
2. Она образует не неподвижную, статическую систему, какой является
пространственная решётка, но подвижную, динамическую систему
движущихся материальных частиц.
Первая из этих особенностей ведёт к необходимости построения
современной теории структур на базе учения о природе химической связи с
распространением выводов этого учения на твёрдое тело.
146
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. 1
Достойно сожаления, что до сего времени пользуются большим
авторитетом школы кристаллохимиков, утверждающие, что структура кристаллов
определяется в основном законами симметрии (т. е. лишь геометрическими
построениями) и что рассмотрение особенностей силового поля, учёт
действия сил химической связи влекут за собой лишь несущественные поправки *).
Одна из причин распространённости таких мнений заключается в том,
что количественная физическая теория твёрдого тела, если говорить о её
историческом развитии, приобрела прежде всего термодинамический
характер и показала, что для разных структур свободная энергия перехода из
одной модификации в другую часто бывает очень невелика сравнительно
с энергией решётки **). Это затруднило использование теории твёрдого тела
в вопросах предсказания наиболее вероятных структур и привело
кристаллографов к недооценке значения в этом вопросе физической картины
твёрдого тела, к постулированию превалирующей роли принципа плотных
упаковок и принципа соотношения радиусов атомов в образовании структур.
В действительности образование атомами сплошь и рядом
высокосимметричных и плотных упаковок является лишь следствием (а не причиной)
надлежащего распределения в пространстве физических сил.
Законы взаимодействия этих сил—«химической связи»—описываются
квантовой химией и вообще физическими теориями твёрдого тела. Эти теории
приводят к выводу, что во многих случаях нельзя ожидать плотных упаковок
и присущих им высокосимметричных атомных конфигураций. В таких
случаях, как алмаз, графит, селен и т. п., должны возникать и возникают
рыхлые упаковки с пониженной симметрией, малым координационным числом
и т. п. В следующей главе, и особенно в нашей книге [11], мы поставили
себе задачу критического рассмотрения ряда распространённых
представлений и теорий современной «геометрической» кристаллохимии, идущих
вразрез с выводами современной физики.
С другой стороны, нами подвергнуты критике также отдельные попытки
специалистов квантовой химии непосредственного перенесения в область
строения кристаллов тех выводов, которые имеют прямое отношение,
собственно, к строению молекул, тогда как в кристалле большую роль при
распределении валентных сил играет поле кристаллической решётки. В
некоторой мере этих вопросов мы касаемся в главе II.
Утверждая принципиальную важность создания физической теории
структур, мы не можем не отметить, что описание исследованных
элементарных ячеек (а в некоторых случаях и предсказание возможных
структур на основе общих закономерностей) может быть сделано прежде
всего с учётом представлений теории симметрии. Собственно это и заставило
нас уделить большую часть вводной главы именно этому, обычно недостаточно
известному физикам и химикам разделу теории структур.
Вторая особенность материальной решётки—её динамический характер—
имеет два аспекта. Говоря о положении атомов в элементарной ячейке, мы
подразумеваем, собственно, положение их центров тяжести, около которых
атомы совершают колебательные и вращательные движения. Учитывая
сравнительную незначительность амплитуды колебаний в твёрдом теле, этой
особенностью можно было бы практически пренебречь и не усложнять теорию,
если бы не наличие другой стороны этого явления: хотя вследствие движения
атомов в одних условиях имеет, в других не имеет места возможность
реального изменения структуры во времени, но принципиально такой процесс всегда
может возникнуть во всяком случае при изменении температуры, давления,
состава компонентов и т. д. Это важнейшее обстоятельство влечёт за собой
*) См. например, нашу критику работы Новотного и данную там литературу [12]»
**) И по вэличине иногда не превышает точности расчёта последней.
ЛИТЕРАТУРА К ГЛАВЕ I
147
возможность существования термодинамически нестабильных структурных
форм, а в данной структурной форме—множества нарушений правильной
периодичности поля кристаллической решётки, типичных для структуры
реальных кристаллов.
Особенности последней настолько своеобразны и важны, что теория
строения реальных кристаллов вносит сегодня весьма серьёзные оговорки
в идеалистическую картину правильной периодической повторяемости
структуры, свойственную теории идеального твёрдого тела, а также в
классическую интерпретацию химией и физикой процесса фазовых переходов в
реальных телах.
ЛИТЕРАТУРА К ГЛАВЕ I
А. Общие руководства
1. Е. С. Фёдоров, Курс кристаллографии (18J7). См. также Е. С. Фёдоров,
Симметрия кристаллов (1949).
2. Г. В. Вульф, Кристаллы, их образование, вид и строение (1917).
3. Г. В. Вульф, Основы кристаллографии (1926).
4. В. В. Д о л и в о-Д о б р о в о л ь с к и й, Курс кристаллографии (1937).
5. Е. Е. Флинт, Практическое руководство по геометрической кристаллографии
(1937).
6. А. В. Шубников, Е. Е. Флинт, Г. Б. Б о к и й, Основы кристаллографии
(1940).
7. P. Niggli, Lehrbuch d. Mineralogie, I (1924).
8. H. H. Белов, Структура ионных кристалла и металлических фаз (1947).
9. Г. М. Попов и И. И. Ш а ф р а н о в с к и й, Кристаллография (1948).
Б. Летература к сноскам текста
1. М. Laue, Z. Krist. A. 105, 124 (1943).
I. N. S t r a n s k i, Z. Krist. A. 105, 91 (1943). D i n g h a s, Z. Krist, A. 105, 304 (1943).
2. В. В. Д о л и в о-Д обр о в о л ь с к и й, Курс кристаллографии (1937).
3. P. Niggli, Handb. d. Experimentalphysik, т. VII, 69 (1928); Lehrbuch d. Minera-
logie (1928).
4. П. Ниггл и, Стереохимия. ИЛ (1949).
Г>. Г. С. Жданов, ДАН СССР XI, VII 40 (1046), стр. 42.
Г. С. Жданов и 3. В. М и н е р в и н a, Journ. of Phys. IX, 244 (1945).
6. S t i 11 w e 1 1, Crystal Chemistry (1938).
7. А. В. Шубников, Е. Е. Флинт и Г. Б. Бокий, Основы кристаллографии,
стр. 122 (1940).
8. См. сноску [7], стр. 128.
9. См. сноску [7], стр. 135.
10. Дана, Описательная минералогия. ОНТИ (1937).
11. Б. Ф. Ормонт, Основы теории структур (готовится к печати).
12. Б. Ф. Ормонт, О применимости законов Иру и Дальтона в современной
неорганической и общей химии, Журн. Общ. химии XIX. 210 (1949).
13. Е. С. Фёдоров, Симметрия кристаллов. Изд. АН СССР (1949), стр. 580.
14. P. Niggli, Qrundlagen der Stereochemie, Basel (1946) стр. 46.
15. В. В. Доливо-Д оброкольский, Зап. Рос. Минер. Общ., т. 58, вып. 1 (1929).
16. Talanski, Nat. 152, 722; 163, 195(1943); Ргос. Roy Soc; L. A. 184, 41, 51 (1945).
17. А. В. Шубников, Как растут кристаллы. М.—Л. Изд. Ак. Наук (1935).
18. А. В. Шубников, Атлас Кристаллографических групп симметрии. Изд. АН СССР
(1946).
19. Siegbahn, Phil. Mag. 87. 601 (1919); Ann d. Physik 59, 56 (1919).
20. W. Bragg, Phis. Rev. 72, 436 (1947).
21. E. Armstrond—Wood, Phys. Rev. 72, 436 (1947).
22. А. К. Болдырев, Кристаллография (1931).
10*
ГЛАВА II
ТЕОРИИ ХИМИЧЕСКОЙ СВЯЗИ И СТРУКТУРЫ ВЕЩЕСТВ
§ 69. Введение
Очень важный вопрос, рассматриваемый в этой главе, удобнее всего
излагать как проблему, в развитии которой довольно чётко намечаются
три этапа:
A. Период классической кристаллохимии, охватывающий XIX и начало
XX столетия. В это время отсутствовала строгая физическая теория
строения атома и эквивалентная ей теория химической связи.
Б. Период с 1912—1916 по 1927г.—до возникновения волновой механики
(и позже), когда были созданы первая научная теория строения атома и ей
отвечающие первые теории электростатической и ковалентной связи с их
последующим развитием.
Этот период мы называем переломным.
B. Современная кристаллохимия—после возникновения волновой
механики, т. е. после 1927—1932 гг.
В таком порядке мы и будем строить изложение, отводя, как указано
в предисловии, большое место критике неправильных взглядов зарубежных
школ кристаллохимиков.
А. ПЕРИОД КЛАССИЧЕСКОЙ КРИСТАЛЛОХИМИИ
§ 70. Принцип М. В. Ломоносова. Представления о природе химической
связи до создания структурной теории А. М. Бутлерова
В 1741 году М. В. Ломоносов, гениально опередив два столетия,
высказал принцип:
«Практическая часть химии состоит в историческом познании
изменений составного тела» (курсив наш). Этот принцип должен быть признан
основой не только передовой теоретической химии, но и ведущей
современной теории твёрдого тела—недавно возникшей теории реального
кристалла. Последняя ломает многие положения созданной уже в XX веке
теории идеального кристалла и, как нам кажется, вносит существенные
поправки в ставшие классическими представления правила фаз и в ряд
законов общей химии (см. § 101 и след.). Поэтому, принцип Ломоносова
по праву должен рассматриваться не в хронологическом порядке, не как
веха давно пройденного этапа науки, а как движущая сила современного
естествознания. К нему мы вернёмся поэтому позже, в конце главы II.
Открытие электричества, явления отталкивания одноимённых и
притяжения разноимённых зарядов вызвали к жизни уже в начале XIX в.
электростатическую дуалистическую теорию химической связи Берцелиуса.
§ 71] возникновение классической кристаллохимии 149
Эта теория наделяла каждый атом или молекулу вещества
одновременно двумя зарядами—положительным и отрицательным, соотношение
количеств которых, согласно этим взглядам, было характерным для частиц
данного вещества, составляло его индивидуальную особенность,
обусловливало его химические и физические свойства.
При взаимодействии двух разнородных молекул положительный заряд,
одной и отрицательный заряд другой компенсировались; оставшиеся + и —
заряды характеризовали свойства новой молекулы.
Дуалистическая теория, утверждавшая индивидуальные свойства
атомов, способствовала развитию атомистических представлений, как и закон
Авогадро—и, несмотря на наивность многих своих положений, на
которых мы подробно остановиться ле имеем возможности, должна быть здесь
отмечена как смелая догадка выдающегося учёного. Однако неумение
или нежелание Берцелиуса модифицировать свою теорию в соответствии
с новыми фактами, открытыми нри исследовании органических веществ
в тридцатых годах прошлого столетия, повлекло за собой крушение
рассматриваемой теории и сформулирование принципа Дюма (1834):
«Химический характер соединения зависит от расположения и числа
атомов и в значительно меньшей мере от их химической природы».
Этот принцип лёг в основу теории типов, под воздействием которой
отказ от учёта индивидуальных химических свойств атомов зашёл к середине
XIX века так далеко, что за это время произошёл возврат от атомных
весов (установленных Берцелиусом) к соединительным; закон Авогадро был
забыт настолько, что даже в многотомной истории химии Коппа (1843 —
1847 гг.) имя Авогадро не упоминается.
Теория типов осуществила в это время систематику органических
соединений, разбив их на ряды, тип каждого из которых определялся первым
членом ряда. Эта систематика, принеся химии определённую практическую
пользу, уводила в то же время науку на рельсы глубокого формализма.
Лишь в результате теоретических работ автора структурной теории
замечательного русского учёного А. М. Бутлерова, экспериментальных
исследований Кашшцаро и, наконец, гениального труда Д. И. Менделеева
удалось к 80-м годам XIX в. восстановить и прочно утвердить в химии
представление о первостепенной важности индивидуальных химических
свойств атомов.
В основу не только химии, но и других областей точного
естествознания лёг один из величайших законов природы, периодический закон,
открытый Д. II. Менделеевым.
Время с 1869 по 1912 г. знаменовало собой полную перестройку общей
и неорганической химии на основе этого закона, открытие ряда новых
элементов, систематическую проверку индивидуальных свойств простых
веществ и химических соединений.
Исследования Бутлерова, Вант-Гоффа и других учёных показали
возможность составить чёткое представление о взаимной последовательности
расположения атомов в пространстве, исходя лишь из общих физических
и химических свойств веществ (см. § 74).
§ 71. Возникновение классической кристаллохимии
На рассмотренной теоретической базе выросли представления
классической кристаллохимии, т. е. науки, ставившей себе задачей нахождение
закономерностей, которые связывали бы элементы симметрии кристалла
с его химическим составом.
Уже в конце XVIII в. Аюи установил положение, часто именуемое кри-
сталлохимическим «законом» Аюи.
150 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
«Химически индивидуальное вещество характеризуется присущими ему
кристаллическими структурами, и по этим структурам может быть отличено
от других веществ».
Этот принцип находится в соответствии с основным положением теории
Берцелиуса—свойства вещества фундаментальным образом зависят от
свойств атомов, образующих это вещество.
Однако уже в 20-х годах XIX в. было обнаружено много фактов, не
находившихся в соответствии с «законом» Аюи.
В 1819 г. Митчерлих открыл явление изоморфизма в гомологических
рядах (в частности у соединений иода и мышьяка): «Вещества, разные
но химическому составу, могут иметь одинаковые кристаллические формы»,—
например, КН2Р04 и KH2As04 изоморфны,—и сформулировал в 1821 г.
«закон»:
«Формы кристалла зависят не от природы атомов, но от их количества
и способа соединения» и указал, что одинаковое количество связанных между
собой атомов (пусть химически различных) обусловливает появление
одинаковых кристаллических форм.
В 1822 г. Митчерлих открыл явление полиморфизма: «Одно и то же по
составу вещество, в зависимости от условий возникновения, может
образовать кристаллы нескольких (двух, трёх и т. п.) модификаций».
В 1823 г. было открыто в химии явление изомерии на примере циа-
новокислого и гремучего серебра: «Одно и то же по составу вещество,
в зависимости от условий возникновения, может иметь совершенно
различные химические свойства».
Очевидно, что «законы» Митчерлиха находятся в полном соответствии
со взглядами теории типов, «закон» же Аюи—с теорией Берцелиуса.
Естественно поэтому, что как теория типов выступила против теории
Берцелиуса, так и Митчерлих считал свой «закон» опровержением «закона»
Аюи.
По этому поводу В. М. Гольдшмидт (1934 г.) писал: «В настоящее время
мы знаем, что оба закона не противоречат, а дополняют друг друга».
С этой оценкой мы не считаем возможным согласиться. Во-первых, оба
эти закона в том виде, как они были сформулированы, но являются
законами природы и, во-вторых, они не дополняют и не могут в таком виде
дополнить друг друга.
Законом природы мы называем такое обобщающее положение науки,
которому подчиняется вся совокупность явлений, охватываемых этим
законом, за немногими, может быть, исключениями. Науке известно много
примеров, когда два, три и более законов дополняют друг друга, однако каждый
из них всё же охватывает всю совокупность относящихся к нему фактов.
Так, первый и второй законы термодинамики дополняют друг друга, но
и в отдельности каждый из них имеет фундаментальное значение настолько,
что никто не будет пытаться построить Perpetum mobile 1-го рода на том
основании, что первый закон дополняется вторым. То же относится к трём
законам Ньютона, к двум законам Фарадея и т. п.
Напротив, привцип Бертло, согласно которому возможно
самопроизвольное протекание лишь экзотермических реакций, не является законом
термодинамики, несмотря на то, что весьма многие химические реакции и
протекают с выделением тепла. Но допустим, что кто-либо, исходя из
возможности протекания, вопреки этому принципу, и экзотермических и
эндотермических реакций, вывел бы, «закон», гласящий, что возможность
протекания химической реакции вообще не зависит от величины и значения её
теплового эффекта. Такой закон не был бы законом и не мог бы дополнить
принцип Бертло, ибо и порознь и вместо они в таком виде ничего
не могли бы утвердить в науке, кроме путаницы.
§ 73] морфотрошш. изотопия 151
Эта аналогия полностью относится и к «законам» Аюи и Митчерлиха.
Они не могут дополнять друг друга, ибо исключений из каждого из них можно
найти не меньше, чем случаев, их подтверждающих.
В самом деле, в рядах, подобных хотя бы
/riOo-Zr02—CeOo—НСО..—ThO,
SiO /
xGe02-Sn02-Pbt)2,
изоморфизм, как правило, наблюдается лишь в отдельных случаях (см.
табл. 17). С другой стороны, не случайно, что галогениды Li, MgO, а также
TiN, TiO и др. имеют одинаковые структуры Со/о, несмотря на различие
химических свойств атомов.
§ 72. Обобщённый закон кристаллохимии
Если пытаться установить закон, отражающий то, что отвечает фактам,
служившим поводом к формулировке обоих этих «законов», и учесть при этом,
хотя бы в первом приближении, состояние современной теории химической
связи, чтобы не впасть с ней в противоречие, то правильнее было бы сказать:
«Структура вещества данного химического состава может изменяться
как в зависимости от условий её образования и существования, так и
вследствие химического замещения части атомов. В последнем случае, однако,
изменение кристаллической структуры с изменением состава имеет место
не всегда и лишь в той мере, в какой это вызывается достаточно
существенным изменением, при данных внешних условиях (р,Т): а) характера
химической связи между атомами, Ь) стерических—геометрических факторов».
Это весьма общее положение может, по нашему мнению, рассматриваться
как основной закон кристаллохимии, соответствие коему фактического
материала должно быть установлено далее с привлечением современных теорий.
§ 73. Морфотропия. Изотипия
Изменение кристаллической формы в результате химического
замещения в гомологических рядах получило название морфотропии.
Эти изменения иногда бывают незначительными, иногда же весьма
существенными. Нам кажется удобным различать 4 наиболее характерных
случая:
1. Сингония и класс кристалла, а также тип элементарной ячейки
полностью сохраняются. Это наблюдается, * например, при переходе в ряду
NaF—NaCl—NaBr— NaJ (тип NaCl C0/o).
2. Сингония, а иногда и класс кристалла у всех членов ряда
сохраняются, но с переходом к отдельным членам ряда наблюдаются резкие
изменения метрики элементарной ячейки.
В ряду CaS04—SrS04—BaS04 (ромбическая сингония, класс Vh) между
CaS04, с одной стороны, и SrS04 и BaS04—с другой, наблюдаются резкие
различия: отношение а : Ь : с = 0,893 : 1 : 1,001 дляСа804, но 1,560 : 1 : 1,276
для SrS04 и 1,630 : 1 : 1,314 для BaS04.
3. Сингония и класс сохраняются, но возникает иной структурный тип
элементарной ячейки.
Например, в ряду LiCl—NaCl—КС1—RbCl—CsCl при стандартных
услониях температуры и давления кубическая элементарная ячейка Со\о
наблюдается у первых членов ряда, а у последнего, CsCl, появляется
кубическая центрированная Сс\с элементарная ячейка.
4. В гомологическом ряду меняется класс и даже сингония кристалла
иногда в одном, а иногда и в нескольких местах ряда (см. табл. 17).
152 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
Таблцца 17
Вещество
Класс
Вещество
Класс
SiO,
Da(Oh,Dih)
8Ю2
Da (0», D,»)
Ge02
D,. Ол
TiOa
Dth, D,h
SnO, ! PbO.,
1 " 1
Dih
ZrO,
Он
Dih
CeO.
Oh
Если путём химического замещения отдельных рядов атомов на другие
в гомологических рядах наблюдаются аналогичные кристаллические формы,
тотакое явление, как мы видели, называется изоморфизмом. Но иногда
аналогичные кристаллические формы возникают у веществ, имеющих совершенно
различный состав, например NaCl и TiN. Такое явление получило название
изотипии.
*
§ 74. Химическая структура веществ по А. Ж. Бутлерову,
Оптическая активность и структура веществ
В 1858 г. Кекуле открыл четырёхвалентность углерода и правило
сцепления.
Несколько позже возникла структурная теория, которая в основном
своим развитием и расцветом обязана А. М. Бутлерову.
Под химической структурой веществ в структурной теории А. М.
Бутлерова понималась «последовательность обоюдных химических связей
различных элементарных атомов».
А. М. Бутлеров высказал чрезвычайно важное положение, которое легло
в основу построения громадного и стройного здания современной
органической химии:
«Химическая природа сложного тела об условливается природой и
количеством его элементарных составных частей и его химической структурой».
Из этого положения вытекало важное следствие,что каждому
соединению может быть присуща только одна формула, соответствующая его
химическим свойствам.
Существенный вклад в структурную теорию был сделан в
результате исследования оптически активных органических веществ, вращающих
плоскость колебания поляризованного света (рис. 108).
Если эта последняя проходит вертикально к поверхности кристалла,
совпадающей с поверхностью страницы, рис. 108, а (т. е. угол падения
равен 90°), пересекая её по линии S—S, то, пройдя кристалл, луч будет
иметь плоскость поляризации, повёрнутую либо по часовой стрелке, либо
против часовой стрелки. В первом случае кристалл называют
правовращающим и обозначают (+), во втором—левовращающим и обозначают (—).
Обладают способностью вращать плоскость поляризации далеко не все
кристаллы, а лишь не имеющие плоскостей симметрии. К таким относятся
кристаллы аксиальных и полярных классов (ступени IV и I) во всех синго-
ниях. Но в кристаллах аксиальных классов имеет место, как мы знаем, энан-
тиоморфизм. Так, например, известные нам энантиоморфные кристаллы
кварца Дя оказываются соответственно лево- или правовращающими
(рис. 109, а, 6).
§ 741
ХИМИЧЕСКАЯ СТРУКТУРА ВЕЩЕСТВ ПО БУТЛЕРОВУ
153
Таким образом, все энантиоморфные кристаллы оптически активны, на
не все оптически активные кристаллы энантноморфны.
Если оптически активный кристалл растворить или расплавить, то
возможны 2 случая:
"<*?**
1) оптическая активность не
сохраняется,
2) оптическая активность
сохраняется.
Первый случай имеет место, если
кристалл не образует в расплаве, соот-
Рис. 108. Вращение плоскости'
поляризации.
Рис. 10Э. Форма лево- и праиопращающпх
кристаллов кварца и хлората натрия.
ветственно в растворе, оптически активных групп: например, NaCI03
образует оптически неактивный расплав или раствор. Если
выкристаллизовывать NaC103 из расплава, соответственно из раствора, то получатся как
правые, так и левые
кристаллы (рис. 109, с, и).
Следовательно,
оптическая активность
кристалла связана с его
собственным строением.
На рис. 110
показан элемент структуры
правого и левого
кварца, причём цифрами 1,
2 и 3 отмечены
атомы Si,
располагающиеся на разной высоте
от плоскости проекции.
Кристалл обладает
семейством однородных винтовых осей, т. е. либо Зх, либо 32. Плоскости
симметрии в нём проведены быть не могут.
винтовые оси 3t
(правые)
Рис.
ВинтоВые оси 32
(левые)
С растворением кристалла раствор оптически
не активен
ПО. Лево- и правовращающий кварц (элемент
структуры).
154
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
г^*г^Н
Рис. 111. Структура одного из видов оптически
активных молекул. Лтом углерода соединён с 4 различными
группами. Структуры энантиоморфны.
Оптическая активность кварца обусловлена, таким образом,
расположением атомов Si по винтовым осям. С растворением кристалла, т. е. с
разрывом связей между атомами, оптическая активность должна исчезать.
Второй случай имеет место, если и в растворе сохраняются оптически
активные группы, например молекулы (рис. 111). Если оптическая
активность кристалла была вызвана только их присутствием, то после обратной
кристаллизации вещества из раствора возникают оптически активные
кристаллы, вращающие
^ ^ в ту же сторону, что и
исходные (рис. 112).
В этом случае
энантиоморфные формы, как
правило, не
выкристаллизовываются.
Наконец,
наблюдаются случаи, когда
и кристаллы веществ,
вращающих в растворе,
обнаруживают явление
энантиоморфизма.
В этих случаях
приходится считаться с
двумя причинами
вращения: с асимметрией
структуры молекул и
с особенностями расположения молекул в кристалле (энантиоморфизм).
Влияние структуры самого кристалла было рассмотрено нами ранее на
примере кварца. Нам остаётся теперь остановиться на случае вращения,
вызванного структурой
молекул. У^^~^Ь\
В 1874 г. Вант-Гофф ^нньчх
(и независимо от него Ле-
бель) осуществил попытку
установить дедуктивным
методом строение оптически
активных молекул. Согласно
этим авторам вращение
плоскости поляризации должно
иметь место, если атом
углерода связан с 4 разными
группами. По их
предположениям (позже
подтверждённым электронографиче-
ским методом) группы GR4
имеют тетраэдрическое строение: с атомом С в центре и
радикалами R в вершинах тетраэдров. В таких молекулах (группах), где все
радикалы различны, нет плоскости симметрии, и возможны энантиоморфные
образования (рис. 111).
Дальнейшие исследования подтвердили исключительную
плодотворность взглядов Вант-Гоффа и показали, кроме того, что могут быть, однако,
оптически деятельны соединения, молекулы которых не содержат углерода,
связанного с 4 различными группами. Это имеет место, например, в случае,
когда два кольца (или кольцо и двойная связь) замыкаются через один
углеродный атом, т. е. плоскости колец перпендикулярны друг другу. Если
кольца построены так, что, например, ни одно из них не является плоскостью
Рис. 112. Оптическая активность
звана лишь свойствами молекул.
кристалла вы-
Раствор такого
вещества оптически активен.
§ 741 ХИМИЧЕСКАЯ СТРУКТУРА ВЕЩЕСТВ ПО БУТЛЕРОВУ 155
симметрии молекул (см., например, рис. ИЗ), то молекула является
асимметричной.
Оптическая активность, по мнению некоторых авторов, может
ожидаться и при наличии двух двойных связей у одного атома углерода, т. е. в
соединениях, производных от
аллена, RXR8C =С = CRJR,,
когда возможно образование
♦шантиоморфных молекул
{рис. 114).
Отсутствие оптической
деятельности вещества может
быть вызвано, например,
либо отсутствием винтовых
осей в кристалле (в
молекуле), либо одновременным
наличием левых и правых
винтовых осей в кристалле, как,
например, в алмазе (рис. 115),
тде цифрами 2, 2, 3, 4
отмечены на проекции
элементарной ячейки алмаза по-
к
тЬ
1
С0{*\
HN Ajtf
/ с\
&-®
[ОС HN
L _ _
^_ mh
HN A
Г' *т—^Я
(Щг^1
СО
J /
ipo ос
NH
Hh
СО,
Рис. 1L3. Молекулы спиро Г>5' дигндаитоина (энан-
тиоморфны).
Рис. 1Г*. Структура производного алло на, могущая
вызвать оптическую активность (схема).
Рис. 115. Элемент
структуры алмаза.
СПОИ
а п с
Рис. 110. Правая, левая винные кислоты и антнвинная кислота (строение молекул).
зиции атомов, находящихся на разной высоте над плоскостью основания
{соответственно смесью разных количеств лево- и правовращающих
молекул. Такие смеси называются рацемическими и обозначаются знаком (+))•
156 Ч. I- ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
На рис. 116, а и b показаны структуры молекул, левой и правой винных
кислот. Виноградная кислота состоит из смеси молекул левой и правой
винных кислот и оптически не активна. Кроме того, на рисунке показана
структура молекул оптически неактивной антивинной кислоты (116, с), как бы
состоящих из левого и правого тетраэдров.
Б. СТРУКТУРА КРИСТАЛЛА В СВЕТЕ ТЕОРИЙ ХИМИЧЕСКОЙ СВЯЗИ В ПЕРИОД
ОТ СОЗДАНИЯ ТЕОРИИ АТОМА БОРА ДО ВОЗНИКНОВЕНИЯ
ВОЛНОВОЙ МЕХАНИКИ
§ 75. О различных типах химической связи и разных типах структур
кристаллов. Расчёт анергии решётки на базе
электростатических представлений
Широко известная сегодня из учебных курсов квантовая теория
атома, установив характер связи между строением атома и его спектром,
открыла путь к выявлению зависимости между строением атома и его
химическими свойствами и, следовательно,—к созданию новых теорий химической
связи. Такие теории были развиты в 1916 г. одновременно Косселем и Люи-
сом [1]. (Впрочем, хотя сегодня многие из этих представлений выглядят
крайне примитивно, приходится подчеркнуть, что так называемая
«современная» кристаллохимия часто использует, к сожалению, главным образом
именно эти, а ие более современные взгляды теории химической связи.)
В течение прошедшей с того времени трети столетия сложная картина
истинного строения кристалла условно заменялась для целей описания
прочности химической связи и предсказания возможной структуры
некоторыми приближёнными моделями, причём последующие модели обычно (я<>
ие всегда) претендовали на большую точность. При этом теория столкнулась
с двумя проблемами:
1. При заданной (экспериментально установленной) структуре
(элементарной ячейке) кристалла рассчитать его энергию и другие свойства.
2. При заданном химическом составе кристалла теоретически найти
наиболее устойчивую структуру (его элементарную ячейку).
Эти две задачи до последнего времени решались с разным успехом и
притом разными приёмами.
Как известно, электростатическая теория видела механизм химической
связи в том, что атомы активнейших элементов, перераспределяя электроны
внешних оболочек, приобретают строение электронной оболочки атомов
инертных элементов, несущих, однако, на себе разноимённые заряды,
т. е. являющихся ионами.
Так, атом Na с приближением к атому С1 отдаёт, а атом G1 приобретает
1 электрон, причём образуются разноимённые ионы Na+ и С1~, имеющие
электронные оболочки благородных газов.
Между ионами возникает притяжение за счёт электростатических сил,
и образуется кристаллическая решётка. Величина сил и энергия
кристаллической решётки вычислялись на этом этапе на основании закона Кулона,
причём ионы рассматривались как жёсткие шары с зарядом в центре. При
таком подходе работа удаления двух ионов друг от друга в
бесконечность равна
Я = ^, (1)
где е1 и е2—заряды ионов, г0—расстояние между зарядами, т.е. в данной
модели—между центрами ионов и е— диэлектрическая постоянная.
§ 75] О РАЗЛИЧНЫХ ТИПАХ ХИМИЧЕСКОЙ СВЯЗИ 157
- ж + ж - ж + ж - к
Рис. 117. Строение кристаллов NaCl и РС15 по Косселю.
Очевидно, что-чем меньше расстояние между ионами и чем больше
заряды, тем должна быть больше прочность химической связи, рассчитанной
по такой модели.
Электростатическая модель сразу же позволила сделать ряд общих
выводов, даже без сколько-нибудь детальных расчётов. Например, в 1916 г. Кос-
сель подчёркивал, что растворение солей в воде, имеющей огромную
диэлектрическую постоянную, связано со значительным уменьшением работы
разделения ионов, вытекающим из закона Кулона.
В 1920 г. он же отметил, что между структурой и прочностью
кристаллов NaCl и РС1б должна существовать принципиальная разница: в
кристаллической решётке NaCl каждый ион Na+ окружён 6 С1~ ионами и,
наоборот, каждый С1~ ион
окружён 6 Na+ ионами,
что обусловливает
сильное притяжение и проч*
ность peniefW (рис.
117, а). Напротив, в
кристалле РС15
отрицательно заряженные обо*
л очки РС15 - молекул
должны взаимно
отталкиваться (рис. 117, в).
В результате
должен возникнуть, в
отличие от тугоплавкого,
механически п рочного
ионного кристалла NaCl, легкоплавкий, непрочный молекулярный
кристалл РС1б. Последний имеет более или менее прочные связи внутри
молекул, но слабые связи между молекулами, что характерно для
типичных кристаллов с молекулярной решёткой.
Таким образом, исходя из одного и того же (электростатического) типа
химической связи, электростатическая теория смогла дать
удовлетворительную модель двух типов кристаллических решёток: ионной и молекулярной.
Однако эти рассуждения можно было применить лишь в тех случаях, когда
-само вещество состояло из разнородных атомов, и притом настолько
отличающихся по химическому характеру, что один из них мог фигурировать
в качестве аниона, а другой—в качестве катиона. В случае образования
химических связей между однородными атомами (например, молекул Н—Н или
С1— С1), а также между атомами, близкими по характеру, например, Вг—С1,
электростатический механизм оказывался непригодным, требуя, например,
образования полярных молекул водорода и т. д., что не отвечало
фактическим данным.
Важную роль в создании теории химической связи сыграли
представления о ко валентной связи, развитые впервые также в 1916 г. Эти
представления предусматривали, что, например, два однородных атома, участвуя
в образовании связи каждый одним электроном, образуют связывающую
пару электронов по схеме: Нф+'Н = Н : Н (электроны обозначены точками).
Такая модель предусматривает, что электроны, осуществляющие связь,
могут в равной мере принадлежать обоим атомам (так что, например,
молекула в целом также может не иметь дипольного момента), но могут
быть в той или иной мере смещены к одному из атомов, например, в молекуле
Н : С1—к атому хлора, так что молекула в целом оказывается полярной,
неся на атоме Н положительный, а на атоме С1 —отрицательный заряд. В
предельном случае такого смещения связывающей пары электронов к одному
из атомов возникает ионная связь. Смещение связывающей пары может быть
158
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл.11
вызвано внешними условиями (температура, давление), воздействием
растворителя и т. п. Иногда такое смещение может быть очень сильным.
Например, в молекуле газообразного HJ при стандартной температуре
и давлении характер связи близок к ковалентному, а в молекуле
газообразного HF —более близок к ионному. При растворении же HJ в воде
пара связывающих электронов переходит к атому иода, с образованием
иона иода, в случае же HF аналогичный процесс протекает в гораздо меньшей,
степени. В результат Ш образует более сильную кислоту, чем HF.
Поэтому серьёзную ошибку можно сделать, пытаясь судить, например,
о характере связи в безводном веществе по его поведению в растворе.
Представления о ковалентной связи позволили объяснить возможность
образования кристаллов неметаллов с атомными решётками, типа алмаза
(состоящими из однородных или близких по характеру атомов).
Необходимо подчеркнуть, что механизм ковалентной связи
первоначально объяснить не удалось, и это стало возможным лишь значительно
позже, с позиций волновой механики. х
Образование кристаллов неиолярными молекулами, типа 02, Н2,
молекулами углеводородов и т. д. за счёт сил Ван-дер-Ваальса, известных химику
из классических курсов физической химии, заставило выделить группу
кристаллических структур с молекулярными решётками в самостоятельную
группу, отличающуюся тем, что в таких кристаллах существуют
значительные внутримолекулярные силы и слабые межмолекулярные, в отличие от
ионных и атомных кристаллов, в которых границы молекул наметить не
удаётся; связи между всеми атомами кристалла в общем одинаковы *) и
различаются лишь по характеру: в ионных кристаллах связь более близка
к ионной, в атомных—к ковалентной. Мы говорим более близка потому, что,
как вытекает из современных квантовохимических представлений, она
практически никогда не бывает чисто ионной или чисто ковалентной, и между
двумя крайними случаями существует множество промежуточных
состояний, с которыми и приходится иметь дело при изучении структур веществ.
Четвёртую группу кристаллических структур составляют металлы.
Первоначально электростатическая теория сделала попытку
интерпретации этих структур таким образом, что в металлах существуют как бы
положительные ионы (остовы) атомов, а места отрицательных ионов
занимали «свободные» электроны. (Этот взгляд легко увязывался с более
ранними представлениями о существовании в металле электронного газа.)
Теория связи как в металлах, так и в кристаллах с атомными
решётками нашла своё удовлетворительное выражение лишь позже (с 1928 г.),
после создания волновой механики.
Кроме того, кристаллические структуры различаются также и по
характеру распределения в них структурных узлов. Помимо координационных
решеток (ионных и атомных), а также молекулярных, обычно наблюдаются,
слоистые (например GdJ2), сетчатые (например графит), цепные (например
селен) решётки, что вызвано спецификой пространственного распределения
связей в соответствии с характером последних.
Период с 1916 по 1930 г. явился, по существу, этапом большого размаха
работ по исследованию, на базе, главным образом, электростатических
представлений, кристаллов с ионной решёткой, к которым модели
электростатической теории были наиболее приложимьи
*) Как это вытекает из теории реального кристалла, в частности из
представлений, развитых автором в вопросе о неприменимости законов Пру и Дальтона для
интерпретации границ возможного фазового состава твёрдых тел (см. § 104), особенно
в кристаллах с ковалентными связями, возможно неравномерное распределение
электронной плотности (в частности, и связывающих электронов) между атомами одного
и того же элемента.
§75] О РАЗЛИЧНЫХ ТИПАХ ХИМИЧЕСКОЙ СВЯЗИ 159'
Методике расчёта энергии кристаллической решётки посвящен ряд
монографий (см., например, В. Кузнецов [2], М. Борни Гепперт-Мейер [3]).
Остановимся здесь лишь на схеме расчёта.
В отличие от системы двух ионов, в кристалле с координационной
(ионной) решёткой, например типа NaCl, как мы знаем (см. § 60), ион данного
знака окружает слой ионов противоположного знака, затем идёт слой
ионов того же знака, затем снова противоположного и т. д.
Например, в кристалле типа NaCl ион данного знака окружают 6 ионов
противоположного знака на расстоянии -f- = r0yl (где aw—период
идентичности), образуя I координационную сферу, затем 12 ионов того же знака на
расстоянии г0)/2 (II координационная сфера), 8 ионов противоположного
знака на расстоянии r0j/3 (III координационная сфера) и т.д.
Первый слой определяет кулоновское притяжение центрального иона,
второй—кулоновское отталкивание и т. д. до бесконечности, так что
потенциал этого иона равен
Ф=_^Л* " + « « Л=_.^. (2)
r0 Vyi у2 /з /4 / r0 K >
Член— — , стоящий перед скобками, непосредственно определяется законом
го
Кулона и не зависит от структуры решетки. Напротив, величина А,
получаемая суммированием бесконечного ряда членов, стоящих в скобках,
прямо зависит от типа решётки. Она называется постоянной Маделунга
и характеризует данный тип решётки, независимо от величины периода
идентичности.
Значения А были рассчитаны для решёток разного типа (табл. 18).
Таблица 18
Константы Маделунга, отнесенные к г0 и к объёму моля
Тип
решётки
CsCl
NaCl
ZnS
(сфалерит)
ZnS
1 (вюрцит)
А
(число для
формулы
с г0)
1,7627
1,7476
1,6381
1,639
а'
эргов *)
40,19
39,84
37,35
37,45
а"
ккал **)
563,9
515,1
666,4
669,3
Тин
решётки
Cu20
(куприт)
CaF2
(флюорит)
TiOa
(рутил)
А
(число для
формулы
с г0)
4,1155***)
5,0387***)
4,82
а'
эргов
93,83
114,83
109,9
я0
ккал
2110
2050
2164
Так как в случае изолированной ионной молекулы, например Na+Cl~,
работа удаления иона в бесконечность равна — , причём А — 1, то значе-
ние А для кристаллической решётки, равное 1,75, показывает, что с
образованием кристалла энергия отрыва иона (изменение г0 и характера связи
не учитывается) возрастает примерно на 75% .
*) На моль-1 ангстрем"1.
**) На моль-1 емг1, отнесённое к объёму моля.
***) С учётом, что заряд Си2+ соответственно О2" равен 2 (иначе надо уменьшить
вдвое, т. е. для CaF2 a=2,519).
160
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
Зная потенциал ср, т. е. работу отрыва одного иона в решётке,
нетрудно рассчитать значение энергии U решётки для одного граммоля,
взятой с обратным знаком (энергия сублимации с образованием ионон)
С/= ^ZJzANk, (3)
где Zx и Z2 — электровалентность (количество зарядов) положительного
и отрицательного ионов, N — число Аногадро и А — коэффициент,
зависящий от характера отталкивания оболочки ионон. Величина к была
рассчитана (1918 г.), причём было показано, в частности, что энергия
отталкивания подчиняется закону вида £=-^, где Ь — константа.
Однако для ряда целей удобнее пользоваться в уравнении (3) просто
коэффициентом
*-^\ (4)
где п колеблется от 6 для галогенидов Li, до 13—для галогенидов Ag.
В случае LiH n падает до 4. Для не очень малых ионов, например,
щелочных металлов, п *& 9.
В результате из (3) получается известное уравнение Борна:
U = NeiZ^A ^ , (5)
принимающее при Zx = Z2 = 1 вид
и~а***=±. (б)
Кстати, надо помнить, что константы Маделунга иногда относят не
к г0, а к а, т. е. «постоянной» решётки. Но этими неличинами нельзя
пользоваться без пересчёта, применяя уравнение (6) с г0. Поэтому напрасно,
например, в книге Брэгга «Кристаллическое состояние», стр. 168, указано без
каких-либо оговорок для решётки
(На) 'J»a (Na*)
* (Cl) i£fl ^ (CI")
I
►LI
TnnaCsCl Гдля]которогоаш = -^= т\Л
A = 2,025 вместо 1,763.
Очень удобна также формула
и = *Р(£)Щя-=±, (7)
N1*J|clzJ ЙШ т[Наа] где ^_плотность, М — молекуляр-
„ ,(л т, ^ ^ ный вес.
:Рис. 118. Круговой процесс рбразования г> „ .„ „л л™»,„« «, /д\
кристалла с ионной решёткой. Рассчитанные по формулам (6)
или (7) значения и были
сопоставлены с «экспериментальными» величинами, получаемыми на основе
кругового процесса Борна-Габера. Как видно из рис. 118,
U= +Q + SM + IM+(j-E)x ккал, (8)
где (^ — тепловой эффект образования 1 моля кристалла из простых веществ,
$м — энергия сублимации одцого грамм-атома металла, в данном примере —
натрия, с образованием атомов, /м — энергия ионизации одного грамма атома
металла с образованием ионов Na+, -х- —энергия диссоциации молекул Х2,
в данном примере — хлора, о образованием одного грамматома неметалла и
# —энергия присоединения электронов с образованием одного граммиона
анионов X".
§ 76] ПРИНЦИП ПЛОТНОП УПАКОВКИ АНИОНОВ 161
Так как значения Е и (у — Ej были известны для ряда атомов не
очень точно, обычно подчёркиваемую хорошую сходимость теоретических
и экспериментальных данных не следует переоценивать.
В одном из наиболее надёжных случаев — иодидов, например, NaJ
получаем:
^NaJ = <?XaJ + #Na "f ^Na + ~~ ~ #./ ~ t>9 KM Л f 26 КК(1Л + 118 КПП Л +
+ 26 ккал~1?) ккал — 1()6 ккал (J-т/ включена энергии сублимации /).
В качестве примера приведём рассчитанные таким образом величины
энергий решётки для галогеиидов Na:
Таблица И'
I
т.
2М) !
| ]
к. н. !
2И)
(.1
Т.
Ш
1
к. п.
18* ;
1
Т.
l;'i
В г
; к. л.
i
т.
LGU
J
! К. II.
1
1 lG()
(Т.—теоретическое, К. П.—кругопой процесс.)
§ 76. Принцип плотной упаковки анионов
Учитывая трудности точных энергетических расчётов, некоторые
авторы стали 30 лет назад на путь использования представлений теории
плотных упаковок. Это направление сохранилось и поныне.
Как было указано в §§ 59, 61 и следующих, наиболее плотными
упаковками шаров одинакового радиуса являются плотная кубическая С к и
плотная гексагональная Ну (к.ч. 12). Между шарами в этих упаковках
имеются октаэдрические и тетраэдрические промежутки — на каждые
п шаров: п октаэдрических и 2п тетраэдрических.
Их распределение очевидно из рис. 119, а.
Ряд октаэдрических промежутков (светлые кружки) приходится на
2 ряда тетраэдрических (тёмные кружки). Учитывая, что катионы как бы
«вписываются» в промежутки между анионами, ряд авторов, особенно
Брэгг, описывали с 1920 г. и до сего времени ионные решётки как
плотные упаковки анионов, в которых октаэдрические или тетраэдрические
промежутки заняты катионами. Например, катионы занимают
следующие места в случае:
NaCl: октаэдрические промежутки в решётке Ск,
NiAs: октаэдрические промежутки в решётке Ну,
ZnS (сфалерит): половину тетраэдрических промежутков в решётке СКу
ZnS (вюрцит): половину тетраэдрических промежутков в решётке Ну,
На рис. 119, Ь показано тстраэдрическое и октаэдричеекое. окружение
катиона но II. В. Белову [12|.
Структуры типа К2"♦'AT могут возникнуть из плотной упаковки по
кубическому или гексагональному законам с заполнением катионами
половины всего количества октаэдрических промежутков; структуры К+А2~ —
с заполнением катионами всех тетраэдрических промежутков.
Как указывают Шубников, Флинт и Бокий [4], этот принцип илотиейшей
упаковки подтвердил правильность предложенного Фёдоровым разделения
кристаллов па 2 типа: кубический и гексагональный.
162
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[глЛ!
Те же авторы отмечают, что в структуре CaF2, формально, места
шаров плотнейшей упаковки занимают уже катионы Са2+, а анионы F"
располагаются в тетраэдрических пустотах, тогда как все октаэдрпчоскпе
пустоты остаются свободными. Это позволяет объяснить образование
изоморфных структур типа CaF2 — YF8. Так как радиусы ионов Са2 + и Ya+
одинаковы —1,06, то катионы обоих типов могут замещать друг друга,
избыточное же количество F-ионов может располагаться в октаэдрических
пустотах, свободных в структуре CaF2 (см. рис. 95 h и 100)).
^к — Тетраэдрическив промежутки
(р\ — Октаздрические промежутки
Рис. 119. Октаэдрическио и тетраэдрическив мвждуузлия в
плотной упаковке.
Напротив, в системе Се02 — La203, кристаллизующейся в той же
структуре, имеет место нехватка ионов кислорода, по сравнению с Се02.
В результате оказывается незанятой часть тетраэдрических промежутков.
§ 77, Соотношения ионных радиусов и структура кристалла
Параллельно с расчётными работами, использовавшими для
суждения о межионных расстояниях г0 экспериментальные данные, отчётливо
выявилось стремление исследователей представить эти значения г0 как
сумму гк-\-гА радиусов катиона и аниона, что позволило бы оперировать
атомными параметрами по аддитивным схемам.
Строгое решение этой задачи, как показала волновая механика
(см. § 87), если не невозможно, то крайне затруднительно.
§ 77] СООТНОШЕНИЯ ИОННЫХ РАДИУСОВ И СТРУКТУРА КРИСТАЛЛА 163
Наилучшим критерием в этом случае являются лишь сравнительно
недавно введённые исследования электронной плотности (§ 9(3). Кроме
того, как показало исследование структур различных модификаций одного
и того же вещества, для решёток разного типа значения г0, а
следовательно, гк-\-гА различны [5].
Поэтому с современной точки зрения приходится решительно
предостерегать против переоценки значений ионных (и атомных) радиусов в
соединениях. Между тем крупнейшие исследователи отдали в течение 1920 —
1940 гг. значительную дань этой переоценке; в литературе опубликованы
тысячи работ, авторы которых
оперируют величинами г~~ для самых
разнообразных целей, вплоть до
далеко не всегда оправданных
суждений о характере химической связи
в кристаллах. Лишь некоторые из
этих выводов справедливы, хотя бы
в нервом приближении. # В
Исходным толчком для прове- Риг 120 1)тношоние рад11у,ов 1ЮН0Б, до_
дения подооиых исследований яви- пускающее образование различных коор-
лись взгляды Магнуса и Гольд- динационных сфер по Магнусу,
ш.мидта (1922 — 1925 гг.), в силу
которых строение координационной сферы является функцией
возможной «упаковки» ионов-шаров с разным соотношением радиусов.
Постулируя, что устойчивой является такая решётка, в которой имеет
место касание оболочек шаров-ионов, Магнус без труда вывел
соотношения радиусов к = —А- , при которых возможны координационные числа
8 (куб), 6 (октаэдр), \ (тетраэдр), 4 (квадрат), 3 (треугольник). Идея
вывода ясна из рис. 120. Результат приведён в таблице 20.
Т а б л и ц а 20
Коорд.
число
к
4
G
8
12
Конфигурация координационной
сферы
Треугольник АХ3
Тетраэдр АХ4
Квадрат а\4
Октаэдр АХ6
Куб АХ8
Плотная кубическая или
гексагональная упаковка АХ1а
Отношение радиусом l- — —L
г 1
0,225 > к > 0,153
0,'il5> к > 0,225
0,7.*2>*>0,'iiri
0,7:<2> А>0,Ш
1,0 >й>0,7.<2
А'^1,0
Ещё в 1938 г. мы отмечали, что вывод Магнуса противоречит
электростатической картине, на которую опирается всё это направление (рис. 121).
Исходя из модели жёстких шаров с зарядом в центре, нельзя
объяснить на базе закона Кулона, почему с переходом от большего катиона
к меньшему, с прекращением касания катиона с анионом,
координационная сфера становится неустойчивой, — ведь в этой схеме электронные
оболочки катиона и аниона осуществляют только отталкивание, которое будет
меньше в случае меньшего катиона. Физическая картина, которой отвечает
вывод Магнуса, вытекает, собственно, лишь из волновой механики, согласно
которой электронные оболочки могут осуществлять силы связи.
Так или иначе, но поскольку свободные энергии переходов
различных модификаций типа Ма^М. и др. малы и термодинамический расчёт
161
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
Радиусы ионов (по Гольдшмидту) (Г),
\ Группа и
s\ электрон.
N^ уровни
Оболочка \
II
III*
IV!
dV-
V*
VI* j VII*
dV n dVldV n dV
im
2(D
(He)
11-
1,54 Г
2,08 11
1,36 3
3 (Л/)
(Ne)
4(,V)
(Лги Сп)
L1+
0,78Г
0,60 11
0,68 3
Na+
0,98Г
0,95 П
0,98 3
K+
1,33 Г
1,33 11
1,33 3
5(0)
(KrnAg)
6(P)
(XenAu)
ВЪ+
1,49 Г
1,48 П
1,48 3
Be' +
0.34 Г
0,31 П
0,393
0,78 Г
0,65 11
0,713
Са8+
1,06 Г
0,99 П
0,98 3
Cs+
1,65 Г
1,69 П
1,673
Ва* +
1,43Г
1,35 П
1,313
87
Sr3 +
1,2 7 Г
1ДЗП
1,153
Sc3+
0,83 Г
0,81 II
0,783
Y3+
1,06Г
0,93 П
0,93 3
La3+
1,22 Г
1Д5П
1,06 3
Lal+—С|»3^
1,22—0,99
(Г)
Ra- i Ac
1,52 (Ш)1
Ti4-
0,G4 Г
0,64 II
0,62 3
Ti3+
0,69 Г
0,7G((>)
/г4+
0,87 Г
0,80 11
0,793
Ге4+
1,02 Г
1,01 П
0,89 3
V5+
^0,4 Г
0,59 11
V* +
0,61 Г
0,59 П
уз +
0,65Г
у2+
0,72(0)
Th4+
1,09(0)
Nb5+
0,69Г
0,70 11
Nb4+
0,69Г
0,67 П
ТаБ+
0,68
Ра
ов+
—0,35 Г )
0, 52 III
Сг3 ■■- |
0,64 Г
31ос+
0,62 11
31о4+
0,68 Г
0,66П
Мп7+
0,46 П
Мп4 +
0,52 Г
0,50 П
Мп3+
0,70 Г
М.па +
0,91Г
0,80 П
Тс7-г 1
0,56 П
i
W4+
0,68(0)
0,55(0)
1.05
1.14(0)
§ 771
СООТНОШЕНИЯ ИОННЫХ РАДИУСОН II СТРУКТУРА КРИСТАЛЛА 165
Таблица 21
Паулингу (П),
VIII*
А2
dV
Fe3+
0,67 Г
Fe-+
0,83Г
0,75П
Ru4+
0,65Г
0,6311
Os4+
0,67 Г
0,6511
IX*
<*V
<*V
Со2 +
0,82 Г
0,72П
Rh3b
0,68Г
1г4 +
0,66Г
0,64П
0,65(0)
1
(
Захариасену (3) (координационное число 6)
X*
dV
dK»
Ni-ь
0,78 Г
0,69 П
Р<14 +
Pd2+
0,72(0)
Pt-ь
^0,87(0)
1*
d<{sl
II*
wV
\
Си+
0,63(0)
0,96 П
Сиа+
0,51(0)
Ag+
1,13Г
1,26 11
Аи+
—
1,37П
Zn-r
0,83 Г
0,74П
^_
Cd2+
1,03 Г
0,97П
Нс:-+
I 1,12 Г
1,юп
ш
.s'V
В3)
0,20 П
0,24 3 |
А13ь
0,57 Г
0,50 II
0,55 3
Ga3+
0,62 Г
0,62 11
—.
1п3+
0,92Г
0,81 П
Т134-
1,05 Г
0,9511
TI +
1,49 Г
|1,44 П
IV
bV
V 1
у2р '
i
с4+
—0,2 Г
0,15 П
0,19 3
С4-
2,60 11
Si4+
0,39 Г
0,41 И
0,44 3
Si4"
2,71 П
Ge4r
0,44Г
0,5311
—
Ge4"
2,72 П
Sn4+
|0,74Г
,0,7111
!Sn2 +
Sn4-
2,94 П
~
|pb«+
|0,84Г
0,84 11
N6+-
~0,15 Г
0,11 II
N3"
1,7111
Р6+
—0,35 Г
0,34 11
рз-
2,12 II
As54-
0,47 Г
—
—
Ак3~
2,2211
Sb*+-
t—
0,62 II
Sb*~
2,4511
—
Bi5+
—
0,74П
VI
2 4
S p
oe+
0,09П
o2-
1,32 Г
1,4011
1,403
S*+
0,34Г
0,29 [I
s2~
1,74Г
1,8411
1,853
See-b
—0,45 Г
0,42 11
—
Se2~
1,91Г
1,9811
1,993
Tce+
—
0,56 11
Tc4+
|0,89Г
0,8111
JTe-"
!2,11Г
12,21П
2,183
Примечания: 1) Для Те2+ из электроно rpai
ных (TeCla,N r = 0,55. 2) (О) означает экспериментальные
считанные автором из более поздних структурных даннь
рассчитанные Шульцем.
VII
F7+
0,07П
F-
1,33 Г
1,36П
1,33 3
CI7+
0,2611
ci-
1,81Г
1,81П
,1,813
Br7+
0,39П
*~~
Briber
1,95П
1,96 3
J7+
—
0,50П
J-
2.20Г
2,16П
2,193
VIII
sV
H» [
— 1,22!
Ne
1,60
Ar 1
1,92
Kr 1
1,98 J
Xe 1
2,18
Rn
1 ! 1
[)ических дан-
радиусы, pac-
jx; (Ш)—то же,
166 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [гл. II
важнейших структур веществ из атомных констант оказался в ряде
случаев недостаточно точным (см. § 78), то направление Магнуса-Гольд-
шмидта, стремившееся охарактеризовать структуры ионных кристаллов
попросту как функции соотношения -— , завоевало умы исследователей
'А
в течение четверти века. Как известно, за это время опубликованы
таблицы ионных радиусов Гольдшмидта (эмпирические), Паулиига
(«теоретические»), Захариасена («эмпирикотеоретические») (см., например [Ъ] (1938)
и табл. 21). При пользовании таблицами ионных радиусов никогда не
следует упускать из виду, что «ионный радиус» отнюдь не является
константой, хотя бы даже для стандартных температуры и давления: как мы
указывали в начале параграфа, межионные расстояния г0 для различных
структур одного и того же
вещества различны. Поэтому
изменяются и ионные радиусы гк + гА = г0<
зависимости от величины
координационного числа в данной
решётке.
Уменьшение координационного
числа влечёт за собой следующее
уменьшение радиусов: с переходом
Рис. VH. Неправильность модели Магнуса. от координационного числа 12 к
Ксли бм чисто электростатическая модель координационному числу 6 в среднем
была достаточна, то модель с меньшим Un/
катионом блла б» устойчивее вслед- }Qia \2%> от координационного числа
ствие меньшего расталкивания электрон- 8 к 6—па 4у0. С переходом от коорди-
ных оболочек. национного числа 4 к 6—увеличение
на 6% (в среднем). В литературе
ионные (и атомные) радиусы для разных координационных сфер часто
различаются соответствующими индексами: г12, т*8, rs и т. п. В таблицах
ионных радиусов обычно приводятся значения для координационных
чисел 6 (тип каменной соли—NaCl).
На базе таблиц ионных радиусов опубликованы многочисленные
таблицы, устанавливающие степень соответствия структур рассчитанному
соотношению ионных радиусов.
Из этих таблиц читатель узнаёт, что в одной и той же структуре,
например, типа NaCl, кристаллизуются самые разнообразные соединения,
подчиняющиеся условию 0,73 > г1: г2 > 0,41, например:
О®О ОЭД"""
галогениды
окислы
гидриды
сульфиды
нитриды *)
карбиды
RbJ
LiF
МцЮ
Natl
Caft
VN
vc
a = 7,32
a = 4,02,
a - 4,20
a = 4,8ft
a - 5,08
a --= 4,12.1
a = 4,П
KC1 a -6,27,
A^Bra = 5,76,
NiO a- 4,17,
Lili a = 4,0«,
MgS a = 5,19,
TiN л='*,*23Г>,
TIC a -4,31.
Сопоставление, например, структур кислородных соединении как
функции соотношения радиусов ионов привело В. М. Гольдшмидта к двум
известным таблицам, характерным для многих аналогичных построений
в курсах кристаллохимии в течение 20 лет. Одну из них мы здесь
приведём (см. табл. 22).
В какой мере эти представления оказались живучими, видно,
например, из того факта, что в монографии (1945 г..) Паулинг вновь ссылается
па свою работу (1933 г.), в которой очень важный вопрос о составе и строе-
*) Нами приводятся исправленные значения а для нитридов, установленные
в нашей лаборатории Л. X. Ьрегером и В. Л. Эпельбаумом [9].
§ 771 соотношения ионных радиусов и структура кристалла 167
Таблица 22
1 Пример
СО,
ыО,
TiO,
Th02
Род рошётки
Молекулярная
решётка
Координационная
решётка
То же
» »
национное
число
2 и 1
4 и 2
6 и 3
8 и 4
Геометрическая
граница отношения
радиусов
<0,15
0,22—0,41
0,41—0,73
>0,73
нии координационных сфер кислородных кислот рассматривается, по
существу, с тех же позиций (см. § 81), иллюстрируемых таблицей 23,
данные которой лишь несколько уточняются Паулингом в 1945 г.
Таблица 23
Отнэшения унивалентных радиусов -^ по Паулингу,
1935—1945 гг.
1 Ве2+
1 0,25
1 Mg2+
1 °'48
| Zn2+
1 0,50
Gd24
I 0,65
В3+
0,20
aV
0,41
1
Ga3 +
0,46
In3"1"
0,59
0,17
0,37
Go4 +
0,43
0,55
Область ROe
N6+
0,14
p5 +
0,34
As5+
0,40
Sb5+
0,51
Область R03 1
0,31
Se6+
0,37
Teot
0,47
CI7 +
0,28
Br7+
0,35
0,44
Область ROj
Kr8+ 1
0,33
Xe84- 1
0,42 I
В той же монографии [7] (1945 г.) Паулинг ссылается также на работу
Цинтля и Моравеца (1938 г.), которые считают необходимым ввести в
вышеприведённую таблицу отдельные поправки (к.ч. Ве2+ равно 3 вместо 4;
к.ч. N5+ равно 4 вместо 3; к.ч. Sn4+ и Pb4f равно 4 вместо 6).
Подобного рода представления уязвимы не только вследствие
условности значений ионных радиусов (см. выше), но и вследствие одной из
важнейших причин этой условности; в данном случае: ионы Si41, IV>+ и т. п.
практически не существуют, ибо в соответствующих соединениях характер
связи весьма далёк от чисто ионного. Более того, если сопоставить
соотношения — даже для кристаллов солей, привлекая более широкий современный
экспериментальный материал, то обнаруживается гораздо менее чёткая
зависимость, чем это принято изображать и в недавно изданных
монографиях.
Прежде в^его, обычно замалчивается то обстоятельство, что случаев,
не подчиняющихся правилу отношения радиусов, не меньше, чем
соответствующих ему, и притом даже среди галогенидов щелочных металлов.
Ряд примеров неподчинения структур веществ этому правилу мы
приводим в таблице 24. Общее положение нам кажется возможным прибли-
168 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
Соединение
CsF
KF
RbF
RbCl
RbBr
RbJ
TIJ
NH4J
uv
LiCl
LiBr
LiJ
AgF
AgCl
AgBr
AgJ
га
1,2'*
1,00
0,90
0,82
0, 7G
0,68
0,68
0,65
0,59
0,43
0,39
0,35
0,85
0,62
0,57
0,51
Коорд
Теоретическое
8
8
8
8
8
G
G
G
6
G
4
4
8
G
6
6
число
Эксперимент.
G
G
6*)
ft
G
G
8***)
g***\
G
G
G
6
G
6
6
4
Соединение
CuF**)
CuCl
CuBr
CuJ
BaO
SrO
CaO
FeO
ZnO***)
HgS ***)
CdS***)
ZnS
MgSe
LfL
0,50 (0,71)
0,37 (0,53)
0y34 (0,49)
0,30 (0,44)
1,09 (0,92)
0,96
0,80
0,62
0,62
0,64
0,60
0,47
0,408
Таблица 2t
Коорд
Теоретическое
6 (G)
Мб)
4 (6)
8
8
8
G
6
G
G
6
4
число |
Эксперимент
4
4
4
G
6
()
6
4
4 (G)
4
4
6
жённо охарактеризовать следующим ооразом: катионы, возникшие из
атомов с наружными ^-электронами (Li+, Са2+), не проявляют тенденцию
к образованию решёток с к. ч. меньше 6 (в том числе 4). Напротив,
катионы, возникшие из атомов с наружными dsp-электронами (Zn2+, Cu+,
Ag+), проявляют тенденцию к снижению к.ч. и, в частности, к образованию
решёток с к.ч. = 4, даже при неблагоприятном отношении ионных радиусов.
В заключение отмечаем вновь, что самый факт образования одними
и теми же веществами модификаций с разным координационным числом
также свидетельствует о том, что проблема образования структур не
может быть сведена к проблеме упаковки шаров данного радиуса, вопреки
взглядам Магнуса, Гольдшмидта и их многочисленных последователей.
§ 78. О дальнейшем развитии электростатической теории
Дальнейшее развитие теории шло в двух направлениях:
1. В сторону уточнения формулы (3). Борн и Майер учли, что
электронные оболочки могут осуществлять не только кулоновские силы
отталкивания, но и ван-дер-ваальсовские силы притяжения (дисперсионное
взаимодействие), и предложили уравнение вида
*>'. = * (^А + Ъ-ВЫ-^къ),
(9)
где первый член выражает действие кулоновских сил притяжения,
второй—дисперсионное притяжение (с—константа); В(г0) — сложное экспонен-
*) При давлении ^ = 5000 атм к.ч. равно 8.
**) Экспериментальный радиус Си+ иона по Гольдшмидту отсутствует. В скобках
(столбец 2) показаны соотношения теоретических радиусов, т. е. для ионов Си+ радиус
принят равным 0,96А по Паулингу. В качестве основных цифр в этом столбце показаны
соотношения rx: r.it если принять для Си + иона радиус 0,бЗА, получаемый из
межатомного расстояния CuF (увеличенного на 6% в связи с переходом от к.ч = 4 к к.ч=6)
за вычетом г/?- = 1,33А.
***) Интересно, что при примерно одинаковом значении г2 : г2 NH4J и TJJ имеют
к.ч.=8, a ZnO, HgS, CdS к.ч.=4.
§ 78] О ДАЛЬНЕЙШЕМ РАЗВИТИИ ЭЛЕКТРОСТАТИЧЕСКОЙ ТЕОРИИ 169
циалыше выражение, которое заменяет отталкивательный член типа ^ >
ц, наконеп., — /ivm — нулевая энергия решетки при максимальных
частотах vmax колебания решётки.
Значения U0 для различных кристаллов, согласно уравнению (9), были
вычислены (1930 г.) и сопоставлены с данными, полученными по
уравнению (3). На основе результатов этой работы той же школы и таблицы [3] нами
была составлена (1939 г.) таблица 25, для чего подавляющее большинство
цифр пришлось пересчитать ввиду наличия в указанных источниках
отдельных неточностей.
Как можно установить из таблицы, с переходом к ионам больших
радиусов влияние дисперсионных сил проявляется в значительной мере,
в случае же ионов малых размеров расхождения невелики, что позволяет
пользоваться старой формулой (3).
Позже ряд исследователей показал, что образование решётки типа CsCl
обеспечивается именно проявлением довольно заметвых сил Ван-дер-Ваальса
(см. § 79).
Таблица 25
Энергии решётки галогенадов
1 Соль
LiF
LiCl
LiBr \
LiJ
NaF
NaCl
NaBr
NaJ
KF
KC1
KBr
KJ
RbF
RbCl
RbBr
RbJ
CsF
CsCl
CsBr
CsJ
Энергия притяжения
статическая
часть
Го
[
287,0
'>25,1
209,2
189,9
249,8
205,5
193,9
179,1
216,0
183,7
175,5
163,9
205,1
177,3
168,0
157,4
193,4
162,3
156,8
147,7
1 Ван-
Дер-
1 вааль-
сова
часть
с
"Я
2
1,3
3,6
3,3
3,8
2,0
2,9
2,8
3'2
3,5
3,9
3,6 !
3,8
4,2
4,6 |
4,1
4,1
6,5 1
7,7
6,9
6,7
; Всего
3
" В
288,3
228,7
212,5
193,7
251,8
208,4
196,7
182,3
220,1
187,6
179,1
167,7
209,3'
181,9
172,1
161,5
198,9
171,0
163,7
154,4
Энергии отталкивания
либо
по ур.
(3)
4
больш
48,0
33,1
29,2
31,4
25,5
15,2
22,1
26,3
20,2
18,7
18,2
20,8
18,0
13,0 1
. .
16,4
16,3
14,7
либосуммир.
по ур. (9)
ft
5
их кал
44,3
27,0
22,6
18,4
35, Г,
23,6
20,7
17,2
28,2
20,8
18,6
15,9
26,3
20,0
17,7
15,5 1
24,0
17,8
16,5
14,6
колебательная
энергия при
\Т = 0 |
6
юриях
3,9
2,5
1,6
1,2
2,9
1,7
1,4
1,2
2,2
1,4
1,2
1,0
1,4
1,2 ,
0,9
0,7
1,2
1,0
0,9
0,7
Сумма
отталки-
ватель-
ной
энергии
гр. 5 +
4-гр. 6
7
на моль
48,2
29,5
24,2
19 ДГ
38,4
25,3
22,1
18,4
30,4
22,2
19,8
16,9
27,7
21,2
18,6
16,2
25,2
18,8
17,4
15,3
1
Энергия решётки
При
подсчёте
uoyp. (3)
(гр. 1-
-гр. 4)
8
239,0
192,0
180,0
158,5
180,0
178,7
157,0
190,3
163,5
156,8
145,7
156,5
150,0
144,4
145,9
140,5
133,0
При
подсчёте
поур. (9)
(гр. 3-
-гр. 7)
9
240,1
199,2
188,3
174,1
213,4
183,1
174,6
163,9
189,7
165,4
159,3
150,8
181,6
160,7
153,5
145,3
173,7
152,2
146,3
139,1
Экспе-
рим.
найдено
10
—
—
— 1
—
— 1
— 1
1
— |
1
— г
—
153,8
1
1
— 1
— |
1
— 1
— |
141,5
170
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
2. В сторону упрощения формулы (3). Наиболее успешны в этом
направлении работы А. Ф. Капустинского.
Капустинский исходил из того, что для расчёта энергии решётки
формулы (3) и (5) требуются знания констант Маделунга для данного
типа решётки, получаемой путём сложного расчёта.
Кроме того, «мы не в состоянии заранее вычислить энергию кристалла
любого соединения, необходимо сперва получить его, а затем изучить его
свойства и строение». Анализируя соотношения между стоящими в
числителе и знаменателе различными параметрами, входящими в уравнение Борна
для решёток разных типов, Капустинский пришёл к выводу, что это
соотношение с переходом от типа к типу меняется в весьма малой мере. Этот
анализ позволил Капустинскому предложить [8] весьма простую формулу
U = 256 A^f^L ккал (Ю)
где Ел —число атомов в «химической молекуле» кристалла, Zjc и ZA —
валентность катиона и аниона, гк и гЛ — ионные радиусы по Гольд-
шмидту.
Уравнение (10) нашло применение в ряде исследований и, в
частности, в работах выдающегося советского геохимика А. Е. Ферсмана.
В 1943 г. А. Ф. Капустинский аналогичным образом, упростив
уравнение (9), получил [9] новое выражение для U:
C/ = 287,2S„r-^(1-^)' (И)
где тг)1 и т]2 —заряды, rt и г2 —радиусы ионов.
Как указывает А. Ф. Капустинский, уравнение (11) даёт хорошее
совпадение вычислений с опытом.
Рекомендуя это уравнение вниманию читателей, заметим, что в
таблице, приведённой в цитируемой работе, обнаруживаются неточности.
Так, дляРЫ2 указано £/раСч. = 441,5 ккал, U-JKCn. = 459,1 ккал, Д = 3,98%,
для GdJ2 соответственно 476,4 и 475,2,Л = —0,25%. В действительности же
энергия решётки £/Эксп. для Pb.I2 и CdJ2 гораздо выше (505 соответственно
575 ккал).
В качестве иного примера приложения электростатических
представлений в химии может служить работа Ю. В. Ходакова [10] (1934 г.).
§ 79. Дисперсионные силы, структура и энергия решётки
Как видно из таблицы 25, влияние дисперсионных сил для решёток
со «средними» по размерам ионами невелико, достигая 1 — 2% энергии
решётки (при точности самих расчётов около 1%). Отсюда ни в коем
случае нельзя сделать вывод, что влиянием дисперсионных сил на структуру
и энергию решётки можно пренебречь.
В самом деле, в ряду, например, галогенидов щелочных металлов
CsCl, в отличие от галогенидов Na, имеет к.ч. 8. Так как константа
Маделунга для типа CsCl лишь на 1% больше, чем для типа Г\аС1, она
не может компенсировать 3 — 4-процентное увеличение межионного
расстояния с переходом от к.ч. 6 к к.ч. 8, а значит и вызванное этим
3—4-процентное уменьшение энергии решётки, обусловленное кулоновским
притяжением. Поляризационная же деформация аниона Св+-ионом меньше,
чем Na+'-ионом (поляризующая способность катиона убывает с
увеличением его радиуса) и не может дать дополнительной энергии,
способствующей переходу к структуре Ccjc- С этой точки зрения образование структур
типа CsCl рассматривалось как энергетическая загадка.
§ 79] ДИСПЕРСИОННЫЕ СИЛЫ, СТРУКТУРА И ЭНЕРГИЯ РЕШЕТКИ 171
Таблица 25 проливает свет на этот вопрос.
Дисперсионные силы пропорциональны произведению поляризуемостей
аниона и катиона, увеличивающихся с объёмом иона.
Если поляризуемость катиона (например, в ряду1л+ — К+) мала,
то и произведение мало. Но у очень больших катионов поляризуемость,
а отсюда дисперсионные силы и обусловливаемый ими энергетический
эффект становятся значительными.
Дисперсионный эффект у соединений Cs достигает уже 4 — 5% от
энергии решётки, перекрывая нехватку энергии, необходимую для
образования структуры CsCl.
С той же точки зрения представляет интерес переход от SrCl2
к ВаС12.
Вместо к.ч. 8 у SrCl2, тип флюрита Сер-, — ВаС12 имеет к.ч. 9
(тип РЬС12). Такая координационная сфера может быть изображена
следующим образом: в плотной шаровой упаковке ионов С1" по
гексагональному закону (§ 62) Ва2+-ион «укладывается» в центр
между тремя шарами СГ~. Сверху и снизу этого
треугольника к нему примыкают треугольники из С1~-
ионов, ориентированные в противоположные стороны;
С1~-ионы среднего треугольника несколько
раздвинуты. Образуемая фигура с 9 вершинами является
14-гранником (рис. 122), как бы сложенным из двух
искажённых октаэдров, имеющих общую грань, в центре
которой находится Ва2+-ион.
Установив, что дисперсионные силы, несмотря на
их небольшое значение в общем балансе энергии
решётки, могут компенсировать нехватку энергии,
необходимой для образования структур с большим
координационным числом, мы приходим к важному
выводу, что при такой трактовке меняется смысл
«критического» отношения радиусов 0,73,
обусловливающего образование структур типа Сс/с-
Если в интерпретации Магнуса это отношение обусловливало
критический объём катиона, достаточный, чтобы вокруг него уместилось 8
анионов, то с рассматриваемой точки зрения речь идёт об объёме, достаточном,
чтобы значение поляризуемости могло обеспечить величину
дисперсионных сил, при которой тип структуры с большим координационным числом
становится устойчивым.
Советским исследователем 3. Г. Пинскером и его сотрудниками [11]
в 1941 — 1945 гг. проводился интересный анализ структуры и межатомных
сил в решётках типа CdJ2, CdBr2, CdCL, PbJ2, в частности,
исследовалась роль дисперсионных сил в образовании слоистой решётки. Поскольку
снаружи каждый слой примыкает к другому своими «анионами», то, как
предполагалось ранее, силы между слоями должны быть в первую очередь
дисперсионными, тогда как внутри каждого слоя в основном —
электростатическими.
3. Г. Пинскер пришёл к выводу, что внутри слоев ван-дер-ваальсова
энергия достигает у всех галогенидов Cd довольно значительных величин
37 — 38 ккал\ между слоями же ее величины растут от CdCl2 к CdJ2 от
5,05 до 7,12 ккал/моль.
В целом значение ван-дер-ваальсовой энергии достигает в этих
решётках, согласно Пинскеру, соответственно 42 — 46 ккал/молъ, а для PbJ2
65 ккал/молъ, что представляет собой довольно значительную часть энергии
решёток (Г/эксиеР.):
CdCU : 594 шал: CdBr2: 583 ккал, CdJ : 575 ккал\ PbJ2: 506 ккал.
Рис. 122.
Координационная сфера ВаСЬ.
172
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. И
§ 80. О методе координационных полиэдров
В 1929 г. Паулинг опубликовал, а в 1940 и 1945 гг. вновь
воспроизвёл в своей монографии [7] «привципы, определяющие структуру
комплексных ионных кристаллов», обычно воспроизводимые без оговорок во всех
курсах кристаллохимии.
В этой книге мы лишь коротко о них напомним, с целью отдельных
критических замечаний.
1. Каждый катион окружён полиэдром из анионов (см. рис. 119, Ьу
где показаны октаэдрическая и тетраэдрическая координационные сферы),
причём расстояние катион—анион определяется суммой их радиусов,
а координационное число катиопов— отношением радиусов.
В комментариях Паулинг опирается на подвергнутые нами выше
критике магнусовские предельные соотношения радиусов для разных
координационных чисел.
Рис. 123. Различные случае сочетания координационных полиэдров:
нерхипй ряд — тетраэдры, нижний ряд — октаэдры, имеющие общие
вершины, рёбра или грани.
2. В устойчивой координационной структуре электрический заряд
каждого аниона стремится компенсировать напряжение электростатических
валентных связей, направленных к нему от катионов, расположенных в
центрах тех полиэдров, общей вершиной которых является данный анион.
Под напряжением электростатической связи понимается
S=y, (12)
где Z —заряд центрального катиона и V — его координационное число.
Правило 2 должно удовлетворяться лишь приближённо.
3. Общие рёбра и, особенно, общие грани (рис. 123), возникающие-
у полиэдров в координационной структуре, уменьшают устойчивость
последней. Этот эффект тем больше, чем больше валентность и чем меньше
координационное число катиона. Он особенно заметен, если отношение
радиусов приближается к низшему пределу устойчивости полиэдров.
4. В кристаллах с несколькими типами катионов, катионы с большей
валентностью и малым координационным числом стремятся образовать
полиэдры, не имеющие между собой общих геометрических элементов
(граней, рёбер, вершин).
Так, в силикатах тетраэдры Si не имеют общих геометрических
элементов, если отношение О/Si равно или больше 4. (Ортосиликаты — оли-
§ 81] ПРЕДСТАВЛЕНИЯ О КОВАЛЕНТНОЙ СВЯЗИ 17.'5
вин, циркон, топаз.) Если этого требует стехиометрия, возникают общие
вершины, по не рёбра или грани.
5. Количество существенно разных типов структурных слагающих
в кристалле стремится быть возможно меньшим, например, количество
разных типов координационных сфер.
Эти принципы были комментированы (1930 г.) и (1932 г.) Брэггом, по
мнению которого наулинговские многогранники удобны как метод
описания, но не имеют иных преимуществ. С нашей точки зрения, столь
распространённые в кристалло-химической литературе принципы Паулинга
i*o многом уязвимы. Это относится к 1 принципу, на который
распространяется сказанное нами о недостатках модели Магнуса, и с тем боль
шим основанием, что в данном случае речь идёт об «ионах» типа Si4+,
существование которых в действительности не имеет места.
Неточность 2-го принципа оговорена и самим Паулингом и позже
.неоднократно подтверждалась на опыте.
Принципы 3 и 4 сформулированы без достаточного учёта требований к
структуре, вытекающих из стехиометрических соотношений и
соображений о допустимых валентных углах. Например, для Si02 образование не
октаэдров, а тетраэдров и притом с общими вершинами, вытекает из
наличия у атома Si i-х валентных .s'/>3 связей иод углами 109° 28' и из
химического состава (см. § 67, 88 и 95 ).
Принципы Паулинга неоднократно давали повод к суждению о том, что,
например, в силикатах характер связи является ионным. Эта точка
зрения не является правильной. См. § 83.
В работе Паулинга содержится ряд других слабых мест, которые
более подробно рассмотрены нами в другой работе. Некоторые замечания
нами будут сделаны в следующих параграфах.
Сопоставляя метод Паулинга (построение структур из полиэдров)
и метод Гольдшмидта, Брэгга и др. (построение структуры из шаров),
мы констатируем, что теория структур продолжает развиваться в обоих
направлениях, предусмотренных задолго до того теорией симметрии
дисконтинуума,—теорией полиэдров Фёдорова (см. § 46) и теорией
кристаллографических точек. В этом смысле теория полиэдров Паулинга является
продолжением и притом не всегда удачным работ русской школы
кристаллографов. Последняя нашла своё дальнейшее и гораздо более
глубокое развитие в трудах советского исследователя Н. В. Белова [121 (19-i7 г.).
§ 81. Представления о ковалентиой связи и электростатический принцип
валентности. Правило эффективных атомных номеров (ЭАН)
Как мы уже отмечали, в 1938 г. (см. также § 77), закономерность,
лежащая в основе магнус-гольдшмидтовской модели, противоречит
принципам электростатики и наблюдается вопреки им. Она означает, что
электронные оболочки, в соответствии с принципом квантовой химии,
могут осуществить не только силы отталкивания, но и силы притяжения.
Эта (квантовохимическая) концепция (см. § 88) имеет глубокие корни
в модели ковалентной химической связи, обусловленной парой электро-
• • •• ••
нов, как, например, в молекуле :С1 :С1: или H:J:. Рассмотрим модель HJ.
•• •• ••
В зависимости от условий реакций (/?, Г), от среды, как мы знаем, пара
электронов (из электронной оболочки т~-иона) может быть больше или
меньше смещена' к протону с возникновением самых разнообразных
состояний молекулы Н J от «предельно» ионного Н_|~: J:~, когда иара связывающих
электронов смещена к аниону, до «предельно» ковалентного H:J:, когда
пара связывающих электронов расположена между обоими ионами. (Мы
174 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ, II?
пишем «предельно» —в кавычках, ибо современная квантовая химия
признаёт этот крайний случай практически нереальным. Химическая связь,
никогда не бывает чисто ионной или чисто ковалентной.)
В соответствии с этим (как указывал ещё Брэгг (1930 г.) паулингов-
ский электростатический принцип валентности вполне может быть
интерпретирован и с помощью предельно ковалентной модели. Поэтому
неправильны часто высказываемые заключения о том, что соблюдение
указанного принципа якобы свидетельствует об электростатическом характере
связи, например, внутри кремне-кислородвых тетраэдров.
Паулинг с комментариями Брэгга был вполне согласен [7] (1940 г.).
Почему же он предпочёл для интерпретации существа дела выбор именно
предельно-электростатической модели? На этот вопрос даёт ответ
работа (1933 г.), в которой Паулинг указывает, что модель типа
i °2~ 1
Н+ О* СГ+ О2 якобы ближе отвечает фактическому характеру связи.
I. °8~ J
На эту же работу он ссылается без оговорок и в 1945 г.
Эта неправильная позиция поощрила недопустимо широкое
применение примитивных электростатических формул в подобных и других
случаях. Отметим, что в нашей книге (1934 г.) мы, в противовес подобным
воззрениям, уже тогда указывали[4], что «взаимодействие между такими
веществами, как сера, кислород, галогены, подчиняется иному механизму,
который не может быть описан какой-нибудь электростатической теорией»,
и ссылались на перспективы в этом отношении волновой механики.
Двадцать лет назад Сиджвик ввёл представления о донорноакцептор-
ной связи, показав, что пара электронов, обусловливающих ковалентную
связь, может первоначально принадлежать одному из партнёров (донор),
а после возникновения связи обмениваться между донором и вторым
партнёром (акцептором).
Одним из наиболее полезных, хотя и не лишённых слабых мест,
примеров использования этих представлений является правило эффективных
атомных номеров (ЭАН). Допустив, что атомы промежуточных элементов
больших периодов стремятся приобрести количество электронов,
отвечающих оболочке последующего инертного газа, Сиджвик указал, что,
например, Сг, Fe, Ni, которым нехватает 12, соответственно 10 и 8 электронов,
для образования оболочки инертного газа криптона, присоединяют 6,
соответственно 5 и 4 молекул СО (:С : :: 0:)— за счёт того, что каждая из них
представляет, в качестве донора, по два электрона центральному атому
металла с образованием Сг(СО)в, соответственно Fe(CO)5 или Ni (CO)4-
Например, никель (ат. № 28), приобретая от четырёх молекул СО 2 X 4 - Н
электронов в дополнение к своим 28, образует 36-электронную оболочку
криптона. Эффективный атомный номер Ni в этом случае Ъ^ = 30.
Представления этого рода были позже дополнены и в другом аспекте-
автором книги. Правило ЭАН наталкивается на ряд не подчиняющихся ему
примеров — «исключений», которых мы бегло коснёмся в § 93.
§ 82. О предположенной Паулингом мнимой
неустойчивости структуры СаО
В доказательство своего тезиса о неустойчивости структуры типа NaCI
для силикатов, вытекающего из принципа 3, Паулинг приводит тот довод,
что эти структуры якобы неустойчивы и для окислов типа СаО, MgO„
как это видно, якобы, из теплот реакции
СаО + Н20 = Са (ОН)2 + 15500 кал,
MgO + Н2Ъ = Mg (ОН), + 9000 кал.
§ 83] ХАРАКТЕР СВЯЗИ И МЕЖАТОМНЫЕ РАССТОЯНИЯ 175-
Этот довод безусловно ошибочен.
Во-первых, потому, что, например, СаО (тин NaCl) термодинамически
устойчив в отношении перехода в иную модификацию СаО, и, во-вторых»
потому, что если бы даже существовала другая, более устойчивая
модификации (при Т = 298\ р = 1 атм) СаО, то тепловой эффект реакции
СаО + Н20 характеризует с термодинамической стороны совсем иную
систему, т. е. не [СаО]а^[СаО]р, а
[СаО]-| Н20 = [Са(ОН)2].
В действительности же энергия решётки Са (ОН)3 значительно меньше,
чем энергия решётки СаО, а положительный тепловой эффект реакции
определяется в основном совсем иными обстоятельствами, как это видно
из кругового процесса образования Са(ОН)2 (представленного на рис. 124),
из которого следует
Q= Uca(OHh — UCaO—hhO + <» =~ W ~ >>1120 + '
(13)
Ca*\0" + HiO
^Ca4\20H"
Легко показать, что — (MJ + \)— величина отрицательная, порядка
180—200 ккал/молъ, именно вследствие большой прочности решётки СаО.
Поэтому положительное значение Q
возможно лишь, если значения ш могут
компенсировать нехватку энергии
порядка 180 — 200 ккал/молъ.
Но этот тепловой эффект
отвечает реакции
-и,
СаМ
02"+Н20 = 20Н" + «> кал
[СаО]
-\
+Я.
+U
Са(0Н)3
•(Mk
[Са{0Н)2]
* - Рис. 12». Круговой глюцеес образов;*-
и, таким образом, вызван неустои- ' Jmm са(ОН)*.
чивостью 02~-иона, а вовсе не
структуры [СаО]. В действительности в кристалле СаО характер связи не
отвечает идеальному ионному распределению зарядов но схеме Са+ и 0=.
§ 83. Характер связи и межатомные расстояния
В литературе за последние 10 лет можно найти много попыток
интерпретации характера химической связи на основе межатомных расстояний
в кристаллической решётке. При подобного рода построениях требуется
большая осторожность, во избежание грубых ошибок.
Ещё Гольдшмидт указывал, что, например, в периодах Си —Вг н Ag — J
Си
Аз
Zn
Cd
Ga
In
Ge
Sn
As
Sb
Se
Те
Br
Л
бинарные соединения, составленные из атомов, имеющих и сумме
одинаковое количество валентных электронов, сохраняют постоянные
межатомные расстояния, см. табл. 26.
Между тем характер связи в этих веществах различен.
Характер связи в Sn близок к металлическому, в AgJ —к ионному,
со значительным весом ковалентного состояния.
176 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР \ГЛ И
Таблица 26
Межатомные расстояния в кристаллах с разным
характером связи
Соединение
СнВг
Zn^e
GaAs
GeGe
Расстояние
в А
2,46
2,45
2,435
2,445
Тип
рошетки
Опт
С Пт
С т it
С г
Соединение
AyrJ
CdTe
InSb
SnSn
Расстояние
в А
2,81
2,7.)
2,79
2,79
Тип
решетки]
Ст/т
Ctjt
Ст/т
С?
Позже отдельные авторы отмечали даже, что если речь идёт об ионах
<•, оболочкой инертного газа, сумма ионных и сумма атомных радиусов
вообще совпадает, причём это формулировалось как закономерность. См.,
например, Ю. В. Ходаков [13]. Отметим, что приводимые в защиту этой
другой крайней точки зрения цифры также бывают далеко не
безупречны.
Например, по Ю. В. Ходакову межатомные расстояния Be—О = 1,65 А,
сумма атомных радиусов 1,05 +0,60 = 1,65А, сумма ионных 0,34 + 1,32=-
= 1,66 А. В действительности, если принять данные Паулинга и Хиг-
гинса, то сумма атомных тетраэдрических радиусов Be + О равна
1,07 + 0,66 = 1,73; если же взять сумму тетраэдрических ионных радиусов,
т. е. с поправкой 6%, то получим 0,32 + 1,24=1,56, так что
расхождение достигнет 11%.
Во всяком случае на примере изученных в лаборатории автора
нитридов ванадия и титана, состава ^ VN и TiN, можно увидеть новые
доказательства в пользу принимаемого нами положения о необходимости
большой осторожности в выводах о характере химической связи на основании
межатомных расстояний. Пересчитав приводимые Лавесом радиусы для
к.ч. 12 на к.ч. 6 тип Со/о, т. е. с поправкой 6%, получаем сумму
металлических радиусов V + N = 1,30+0,75 = 2,05и Ti + N = 1,39 + 0,75 = 2,14 А.
Найдено экспериментально для VN — 2,064; для TiN —2,117 % 2,12. Но TiN
обладает прекрасной металлической проводимостью при комнатной
температуре, значительно отличающей его от VN. Если же определить октаэдри-
ческий радиус NJ~ для VN как соли V3+N:>~, исходя из гольдшмидтов-
ского радиуса Vj[+ = 0,65, получаем для NJ~ = 1,41A. Рассчитав с тем
же радиусом заведомо близкую к металлической структуру TiN, получаем
0,69 + 1,41=2,10 вместо 2,117. Более того, если рассчитать структуру
A1N с совершенной иной решёткой (тип вюрцита) и заведомо иным
характером связи, приняв АРб+ —0,57 (по Гольдшмидту) и Nj}~ = 1,41 (из
структуры VN), получаем для A1J+ + N3- сумму г0=1,98А. Введя
поправку для перехода к к.ч. = 4, получаем г0=1,86, тогда как в SB
находим г0~-- d = 1,868^ 1,87.
Атомные радиусы, как и ионные, меняются для одного и того же
элемента, в зависимости от величины координационного числа.
Уменьшение расстояния с переходом от координационного числа 12 к другим
равно: от 12 к 8 — 3%; от 12 к 6 —4%; от 12 к'4 —почти 12%. В связи
с этим, как мы указывали, часто обозначают с помощью индекса,
например г12, к какому координационному числу относится данная величина
радиуса.
АТОМНЫЕ РАДИУСЫ II СТРУКТУРЫ МЕТАЛЛОВ
177
§ Ь4. Атомные радиусы и структуры металлов. Закон Вегарда.
Концентрация электронов и структура фаз
Количество работ, пытавшихся обт>яснить структуру металлов и их
сплавов на основе соотношений атомных радиусов (см. таблицу 27),
довольно велико. Однако, по нашему мнению, нельзя не считаться с
данными, свидетельствующими о второстепенном значении в этом случае
стерических факторов. Так, на- ^
пример, Си (г12 = 1,275А) даёт
с золотом (г12 — 1/±39 А) твёрдые
растворы при любых
соотношениях концентраций атомов,
тогда как с серебром (ri2 = 1,441 А)
лишь в очень ограниченных
соотношениях.
Си и Fe сплавляются лишь
частично, несмотря на равенство
атомных объёмов.
Кроме того, лёгкие металлы
образуют фазы сложного состава
типа CueZn8. Рассмотрим систему
Си— Zn подробнее (рис. 125).
Если растворять в меди
(тин Ск) цинк, образуется а-фаза
системы Си —Zn, могущая
содержать при комнатной температуре
до 38,05% Zn. При изменении
концентрации Zn меняются
межплоскостные расстояния в решётке а-фазы от 3,605 (0% Zn) до 3,698 (38,05% Zn).
По закону Вегарда «при образовании твёрдых растворов период
идентичности решётки является линейной функцией атомных концентраций
Сг и Со компонентов решётки, имеющих периоды идентичности ах и а2У
т. е.
(14)
Рис. 1:>Г>
Диаграмма состояния
медь цинк.
системы
- С1а1 + Гаа2»-
(Закон Вегарда обычно точно не соблюдается.)
С дальнейшим увеличением концентрации Zn образуется [}-фаза,
Р-фаза имеет структуру Сс\с, причём атомы Си и Zn беспорядочно
занимают положения (000) и \\ \ \). При высокой температуре область
существования fi-фазы широка, но с понижением температуры она резко
сужается: [3-фаза распадается с выделением кристаллов а- и у-фаз
(см. рис. 125). Ниже 450°С [3-фаза постепенно переходит в (З'-фазу
с упорядоченным расположением атомов, например, Си (000), Zn {\\\).
При комнатной температуре область существования [3-фазы лежит
между 47 — 50 атомных процентов Zn.
у-фаза имеет неожиданно очень сложную структуру. Представим себе
решётку Се/с с увеличенным втрое ребром и в 27 раз — объёмом. Она
содержит 2x27 = 54 атома. Удалив два атома и сместив остальные,
получим решётку у-фазы с 52 атомами в элементарной ячейке. Таким
образом, при химическом составе Cu5Zn8 элементарная ячейка имеет состав
20 атомов Си и 32 атома Zn.
о-фаза изучена ненадёжно.
е-фаза имеет структуру плотной гексагональной упаковки с с/а,
несколько меньшим 1,63, и состав CuZna.
178
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
Р\ Группа и
Хэлектрон.
\VpOBHH
Оболочка ^v
1 (А')
2 (L)
(Не)
3 (М)
(Ne)
4 (xV)
(Аг) и (Си)
5 (Г))
(Кг) и (А-г)
6 (Р)
(Хе) л (Аи)
7 (Q)
Rn
1
Н 0,78
Li 1,57
1,52
Ха 1,92
1,86
К 2,36
2,29
Ш) 2,5.4
2,46
Оя2,74
2,46
87
II
Be 1,13
1,10
Т 1,07
>1Я 1,60
1,55
Т 1,40
Са1,97
1,91
Sr 2,16
2,10
Ва 2,25
2,18
Ка
III*
Sc 1,65
Y 1,81
La 1,86
TR
А«
Радиусы атомов («металличе
IV*
Ti 1,45
1,41
Zr 1,60
1,55
Ш 1,59
1,53
Th 1,80
1,75
V*
d2s*
V 1,36
1,32
Xb1,47
Та 1,46
1,42
Pa
VI*
Cr 1,28
1,24
Mo 1,40
1,36
W 1,41
1,37
и i, 5*; *)
VII*
rf-'C-
>In 1,31
1,26
>Ial,34
He 1,37
1,34
TR
Ce 1,82
Pr 1,82
X<1 1,82
61 1,8
или 2,0
Sa 1,8
или 2,0
Eli 2,04
Gd 1,79
Tb 1,77
Dy 1,77
Ho 1,75
или 1,У5(?)
Ег 1,75
Tu 1,74
Yb 1,93 ! Cp 1,74
*) Размеры атомных радиусов колеблются от 1,38 до 1,68; в среднем
принимают 1,53 (см. § 117).
Примечание. Верхняя цифра в каждой клетке — металлический радиус
эдрический ковалентный радиус по Лаулингу и Хиггинсу. Буква Q—к. ч. 4 обоз
октаэдрические ковалентные радиусы (по П. и X.) в валентных состояниях II, III,
ставлена автором по данным разных исследователей.
§ 811
АТОМНЫЕ РАДИУСЫ II СТРУКТУРЫ МЕТАЛЛОВ
179
скис» и «коналептнме»)
Т а б л и ц а 27
I VIII*
| d*S*
i d7xi
IX*
d?s*
dV
X*
d*s*
dl°*°
I*
dl0sl
II*
(d10)*8
III
*'/'1
IV
s*p*
V
s'-p3
VI
s*p*
VII
5-/>6
Fe 1,27
1/2'i
Co l,2o Xi 1,24
1,22; 1,21
т — ! r —
Q 1,211 Q 1,22
R:i 1,32 Hli l,"»4
1,2<S 1,30
I ^ —
■ О 1,34
Pd 1,37
1,33
T --
Q l,32
Os 1,34 ! Ir 1,35 ' IN 1,38
1,30; 1,31
О 1,зз
1,34
О 1,32
Си 1,28
1,24
T 1,35
Q 1,21
Aff 1,44
1,40
T 1,53
Q 1,33
\u 1,44
1,40
T 1,50
Q 1,31
Zn 1,37
1,34
T 1,31
1
1
Cd 1,52
1,47 1
T 1,48
Hgl,55
1,50
T 1,48 1
1
Г. 0,95
J7 0,80
AI 1,43
1,39
71 1,2)
Ga 1,39
Г 1,25
С 0,8Г,
T 0,77
Si 1,">4
X 0,8
T 0,70
P 1,3
T 1,17 I Г 1,10
Ge 1,3ч
1,34
T 1,22
In 1,57 |Sn 1.58
1,52 j 1,53
T 1,44 I T 1,40
I
Tl 1,71 Vh 1,74
1,Г>>| 1,69
T 1,47 71 1,46
As 1,40
1,35
T 1,18
Sb 1,01
1,56
7' 1,36
Г 0,5 i
т
го,
S 01
Т 1,04 |7Т 0/J0
Se 1,0 Br
Г 1,14 i?1 1,11
i
1Н 1,82
1,77
71 1,45
JN
Те 1,7 | J
71 1,32 Г 1,28
8 о
VIII
s~p9
Tie
1,22
Xe
1,60
Ar
1,92
Kr
1,98
Xe
Г 2,18
Kn
Группа
IV*
VIII*
IX*
X*
1*
0
II
III
IV
Ti
—
1,3G
Zr
—
—
1 ,51
Fo
1,23
Ru.Os
1,33
—
1,37
Co
1,32
1,22
—
Rh,Ir
1,43
1,32
—
Xi
1,39
1,31
1,21
14, Pt
1,50
1,42
1,31
Си
Ag,Au
1,54
1,49
1,41
IV
sn i рь
—
—
1,49
—
—
1,54
VI j
Se ,
1,40
при к. ч., равном 12, следующая — npii к. ч., равном. 8. Буква Т обозначает reipa-
начает квадратный ковалентный радиус (но II. и X). Суква О—внизу таблицы—
IV. TR — металлические радиусы редких земель (к. ч. 12). Таблица со-
180
Ч. Т. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
Наконец, т]-фаза имеет искажённую плотную гексагональную
упаковку Ну с соотношением cja ==1,86, характерным для Zn.
Как ни странными кажутся структурные соотношения в этих фазах,
состоящих только из двух сортов атомов, Си и Zn, но в 1926 г. Юм-Розери
удалось сформулировать эмпирическое правило, описывающее условия
возникновения структур в подобных металлических системах.
Согласно этому правилу, «состав некоторых интерметаллических фаз
определяется концентрацией электронов».
Концентрация электронов описывается выражением
где А и В — количества атомов обоих компонентов, р и q -количества
электронов, участвующих в образовании решётки. Например, если сплав
содержит 60 атомных процентов Си и 40
атомных процентов Zn, то концентрация
(валентных) электронов равна
1 X 60 + 2 х 40
К =
100
= —- = 14.
100 '
(16)
/У^\*^7ГУУС^~Т"у*у^ I По Юм-Розери, у~Фаза возникает при
>• i v i^aA^i i \Jr\ I концентрации электронов 21:13 = 1,61.
В самом деле, CusZn8, Cu9Al4, Cu7Zn4AL
имеют концентрации 21:13 (21 электрон
на 13 атомов). Дальнейшие исследования
показали, что это же правило относится
и к возникновению других фаз. Так, р-фаза
латуни возникает при концентрации
электронов 3:2= 1,5, £-фаза при концентрации
7:4= 1,75. Аналогичные фазы наблюдаются
при тех же электронных концентрациях
и в других системах, например, (3-фазы:
CuZn, AgZn, AuZn, AgCd, s-фазы: CuZn3, Cu3Ge, Cu3Sn, AgZn3 и др.
Вместе с тем пришлось предположить, что атомы Fc, Co, Ni участвуют
в образовании у-фаз с количеством электронов, равным 0, как это
видно, например, из формул Fe,Zn2i, Co6Zn2l, Ni5Zn2l, когда приходится
принять
Рис. 126. Структура NaTl
(:)лементс'1рнан ячейка).
к =
04- 2 X 21
26
21
13 #
При одном и том же К соотношение атомов при образовании данной
«фазы меняется в зависимости от числа валентных электронов, например,
(3-фаза в системе Си — А1 имеет состав Си3А1.
Помимо р-фазы типа CsCl (Ccjc) наблюдается р-фаза структуры NaTl
и NaPb3, в которой К Ф 1,5. Образование [3-фаз этого типа сопровождается
сильным сжатием. NaTl имеет структуру, производную от решётки алмаза
Ст. Поместим в узлы последней (а) атомы Na и вставим в неё вторую
ллмазную решётку с атомом Т1 в узлах (Р) так, чтобы начальный узел
второй решётки центрировал ячейку первой. В структуре NaTl вокруг
каждого атома находятся две одинаковые по размерам тетраэдрические
•оферы I (a)Na и II ф)Т1 (рис. 126). NaTl имеет то же положение центров
тяжести атомов, что и структура Си3А1 (тип BiF3); одинаково
расположены атомы на сторонах элементарной ячейки и в её центре. Различны
расположения атомов в центрах октантов. В Си3А1 все 8 октантов
центрированы атомом одного рода (Си) (а-узлы), в NaTl 4 октанта в тетраэд-
рлческом порядке центрированы узлами а, 4—узлами р.
§ 85] О ВОЛНАХ МАТЕРИИ. ДИФФРАКЦИЯ ЭЛЕКТРОНОВ 181
Теоретическое освещение правила Юм-Розери было впервые дано
в 1935 г. с позиций волновой механики, после того как уже в 1931 г.
Бриллюэн показал путь, по которому удалось теоретически установить
границы устойчивости интерметаллических фаз для более простых
структур {CK,Cc,Hv).
В. О ПРИЛОЖЕНИИ КНАНТ01Ю-ХИМИЧЕСКИХ ТЕОРИИ К
НЕОРГАНИЧЕСКОЙ ХИМИИ И КРИСТАЛЛОХИМИИ
§ 85. О волнах материи. Диффракция электронов
В первой четверти XX в. физика переживала острый «кризис»,
философский смысл которого с непревзойдённой глубиной был вскрыт
Лениным в его гениальном труде «Материализм и эмпириокритицизм».
К 1924—1925 гг., в развитие борьбы противоречий, физика сделала
дальнейший очень важный шаг вперёд, положив начало волновгй
меланине. Автор касался ряда вопросов, в частности, взаимоотношений между
корпускулярной и волновой теориями света и т. д. в своей книге [14J
(1934).
О волнах материи
В 1924 г. де-Бройль пришёл к выводу, что движение материи может
рассматриваться как процесс одновременно и корпускулярного, и
волнового характера, причём длина волны определяется энергией частицы.
При этом
h h h tл <~\
™и /2m<g P
где X —длина волны, т и о — масса и скорость частицы, Л—квант
действия Планка, р — импульс, £ —кинетическая энергия частицы.
Из уравнения (17) следует, что для быстрых электронов катодного
луча, имеющих скорость 0,2 — 0,4 скорости света, длина волны л порядка
10~9 см. Для медленных электронов, теряемых раскалёнными
проволоками, — к порядка 10~7 см. Если пучок электронов ускоряется напряжением
V вольт, его длина волны
>.=У£°Л, (18)
при 150 в л = 1 А.
Таким образом, мы имеем дело с волновым процессом, длина вслны
которого порядка длины волн рентгеновских лучей.
Диффракция электронов
В 1925 г. был сделан важный вывод из теории де-Бройля.
Учитывая величину длины волны электронов при вышеприведённых
скоростях, можно ожидать при столкновении электронов с атомами,
особенно при низких температурах, диффракционных явлений, описываемых
уравнением Вульфа-Брэгга:
rtX = 2dsinO. (19)
В 1927 г. Дэвиссон и Джермер установили, что электроны,
отражающиеся от монокристалла никеля, действительно подвергаются диффракции,
подобно рентгеновским лучам. Советский учёный П. С. Тартаковский
и Томсон открыли диффракцию быстрых электронов на металлических
фольгах.
182 Ч. I. ОСНОШЛ ТЕОРИИ СТРУКТУР [гл. II
В отличие от рентгенографического метода, электрон ографический
требует меньших углов скольжения; электроны сильнее поглощаются
веществом кристалла, чем рентгеновские лучи. Поэтому электронографи-
ческие методы исследования нашли широкое распространение при
исследовании тонких поверхностных слоев кристаллов, а также паров и газов.
О прочих методах исследования строения вещества
Рентгенографические и электронографические . методы исследования
строения вещества приобрели за последние годы исключительно большое
значение. Однако не вызывает сомнения, что ни один физический
метод исследования сам по себе не может дать достаточно полных и
удовлетворяющих требованиям современной науки сведений о строении
вещества, но лишь комбинация разных методов.
Изучение магнитных свойств веществ, дипольных моментов,
молекулярной рефракции, спектров поглощения, комбинационного рассеяния
света, инфракрасных спектров, а также применение ряда других
физических методов, с одной стороны, и химических и электрохимических
методов—с другой; позволяют дать более глубокую и всестороннюю картину
строении веществ.
§ 86. Электронные уровни атома. Принцип Паули.
Правило возбуждения валентностей
К началу рассматриваемого периода на основе, в первую очередь,
спектроскопических методов были чрезвычайно сильно пополнены паши
знания о строении и энергетических уровнях атомов. В этом смысле
оказался особенно плодотворным 1925 г., как бы завершивший построение
квантовой модели атома, чем был подготовлен фундамент для
последующего перевода науки на более высокую ступень волновой механики.
Как это было установлено ещё квантовой теорией атома,
электронные (энергетические) уровни атома определяются прежде всего главным
и побочным квантовыми числами.
Наиболее глубоко лежащей АГ-оболочке отвечает главное квантовое
число 72 = 1, L-оболочке я = 2, Af-оболочке п — 3, TV-оболочке п = 4 и т. д.
Различные уровни этих оболочек характеризуются побочными
квантовыми числами / и обозначаются, в зависимости от величины /:
s(l = 0), /)(Z = 1), d(l =2), /(/ = 3). Количество таких возможных уровней—
электронных состояний—было определено из условий комбинирования
побочного / и магнитного т квантовых чисел из коих числу s отвечает один,
числу р — три, числу d — пять, числу / — семь энергетических уровней.
Общее количество уровней в каждой оболочке равно я2, где п —
главное квантовое число оболочки. Таким образом, в if-оболочке имеется
один .v-уровень. В L-оболочке один s- и три /^-уровня, а всего 4 (т. е. 22).
М Л/-оболочке один s , три р- и пять d-уровней, а всего 9 (т. е. 3*).
И .V-оболочке один s-, три />-, пять d- и семь /-уровней, а всего 16 (т. е. 42).
В 1925 г. было открыто явление, которое было интерпретировано
и как вращение электрона вокруг своей оси (спин), что привело к
введению четвёртого квантового числа s и открытию (1925) принципа Паули,
сыгравшего фундаментальную роль не только в спектроскопии, но и в
неорганической и общей химии.
В соответствии с этим принципом в каждом уровне может
находиться не более 2 электронов, и притом с антипараллельными спинами (f J)
(что интерпретировалось как вращение по часовой и против часовой
стрелки). Всего, таким образом, каждая оболочка может содержать 2л2 электро-
§ &G] ЭЛЕКТРОННЫЕ УРОВНИ АТОМА. ПРИНЦИП ПАУЛИ 183
нов, т. е. iiT-оболочка 2, L-оболочка 8, А/-оболочка 18, Лг-оболочка 32. Этими
цифрами определяется длина периода в периодической системе, поскольку
каждый следующий элемент имеет количество наружных электронов на один
больше, чем предыдущий. См. также В. II. Кондратьев [69].
Порядок заполнения уровней электронами регламентируется правилом
возбуждения валентностей (правилом Гунда), согласно которому при
заполнении группы уровней, относящихся к данному побочному числу
<?, р> d или /, например, пяти d-уровней, более низкому энергетическому
(основному) состоянию атома отвечает размещение в каждом из этих
уровней сначала одиночных г^-электронов (холостых), и лишь если
у атома нехватает, например, d-уровней для заселения —в одиночку —
всех d-электропов, происходит последовательное «вселение» в каждый из
этих уровней второго валентного электрона со спином, противоположным
ашну первого электрона.
Количество ^-электронов, имеющихся у атома в каждом s, p. d, /-уровне,
описывается формулой, в которой сначала указывается главное квантовое
число, затем побочное, а число электронов проставляется в показателе
справа сверху.
Таким образом, количество валентных оэлектронов вовсе не
обязательно равно количеству холостых гг-электронов.
Так, атом О имеет в основном состоянии в наружной оболочке
3$23/>5-электронов, т. е. 2 на 3s- и 5 на Зр-уровне, а всего 7/;-электронов,
j;o в основном состоянии только 1г£'-электрон (таблица 29). Наиболее
глубоко лежат ls-уровни, затем 2s2p, 3s3p, 4s, 3d (причём после элемента
Sc (скандий) уровни 3d лежат ниже, чем 4s), 4p, 5s и т. д. Общее
распределение электронов у элементов Li — Ne, Na — Ar в основных состояниях
показано в таблице 28, из которой наглядно вытекает, как именно ира-
Таблиц а 28
Распределение валентных электронов но уровням
Характеристика валент- В основном
состоящих электронов нии
Элемент iii
2-го
периода
1 Li 2si
Во 2s2
1 В 2s*2p
1
С 2^2/>-
N 2б-'2/Я
1 0 7^:>/И
1 F 2^2/>5
1 Ne 2s32p'5
Элементы
;;-го
периода
Распределение
•V
Na ^ [ [
М* 352
Al WZp
Si №3p*
P ^3,^,3
S [\s4p*
> 1
1 S
Л 1
1 s
^ !
' 1
1 4"
* 1
1 i
ci ^p> ; |
Ar Wipfi | '\1
P
L
A
1
i 1
I V
и
1 fl
i
i
и
n
i
л
i
T
ti
Количество
электронов
го
1
0
1
■)
! "
i
0
В 1-м возбужденном
Распределение
• 1
л
f
л
1
t
I
р
\
t
i
t
л
1
'*
Коли- 1
чество
ХОЛОСТЫХ
электронов го 1
1 1
2
;i
4
ктичоски осуществляется правило возбуждения валентности.
Распределение и-электронов по энергетическим уровням всех элементов показано
в таПлипе 29.
184
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
| Группа 1
Г——-^^^^ томный те]:м j
оболочка ""——_
1
ЦК)
S
4L) р 1
ЦК) I
1 г?
1 2{L) \
1 1(АГ> 1
4(.V) p
\ S
d
ЦЛГ) р
1 s
2(L)
ЦК)
40) р
1 'Ч'
\(N) d
1 р
1 ***
"'-Ij7')-".!". Г
КА)
6(Р) />
j .s'
d
5(0) р
1 *
40V)
:»(Ю
2(t)
клг)
7«?) *
t\(/J) /'
1 '^
5(0)
4(ЛГ)
3(1/)
4L)
| 1(g)
I
1
1 1
0 LI
1 3
2
<> Xa
1 11
8
2
0 К
1 19
0
0
2
8
2
0 lib
1 37
0
!
2
IS
S
2
0 V,h
1 55
0
r>
2
18
и
8
2
1 Fr
0 87
2
18
12
18
s
2
11
0
0 Be
2 4
2
2 12
8
2
о Ca
2 29
0
6
2
8
2
0 Sr
2 38
0
?,
2
IB
2
0 Ha
2 56
0
2
18
18
8
•>
2 tta
1 0 88
2
IS
Г2
8
1 2
Распределение электронов по
III* ! III**
1 1
0 Se
2 21
1
ft
2
8
2 J
0 Y 1
2 39
0 '
1
G
2
18
8
2
о La
2 57
1
6
2
18
,18
8
2
1 2 Ac
1 89
6
2
L8
m
18
8
1 2
0
2
1
1)
2
19 -32
IS
8
2
Лантаниды
Се 58
Nd69
Sm62
Gd 64
Dy 66
Ег 68
Yb70
Pr 59
Pm6l
En 63
Tb 65
Ho 67
Tu 69
Cp 711
Актиниды * --
Th —Гт
(Щ - (Щ
IV*
3F
2
0 Ti
2 22
2
»>
2
8
2
0 Zr
2 49
0
2
! Г>
2
18
8
2
I n Hf
2 72
2
6
2
32
18
8
2
■ 2 Th
«2 »(>
'•6
В 2
bis
r2
"18
Г
Ь
тхш т т ■
1 i
V* 1 VI» i
1
1
0 V
2 23
3
r>
2
S
2
0 \h
1 41
0
4eV)
67*
2
IS
8
2
и Та
2 73
3
6
2
32
18
8
1 2
; 2 Pa
3 91
f>
2
18
32
18
~"
0 Cr !
1 24 i
5 '
6 Г
■> !
1
8 !
1
2 ,
0 Mo
1 42
0
0
18
8
2
о \V
2 74 .
4
6 *>D0
1 2
32
18
8
' 2
L U
Г» 92
Г»
2
18
32
LS
8 j 8
1 2
I 2
§ 861
ЭЛЕКТРОННЫЕ УРОВНИ АТОМА. ПРИНЦИП НАУЛИ
■ik:
Таблица 2У
!
энергетическим уровням
VI**
1
VII*
в^/2
Vill *
Ч
'
I
" 1 " ! "
i
oMn
2 25
5
<>
2
0 Fe
2 26
Г»
2
IX*
V/,
_
0 Го
2 27
п
у
Г)
2
1 8 ! 8 i8
2
0 Tc
1 4:1
' 0
is
1
18
1 '*
:
Np Pu;
из 94;
■
1
в
■
Am Cm
i
95 96 i
■
l
i
1 5
2
0 Ke
2 75
f>
f>
2
2
0 Ни
1 44
0
;6f5
0
2
18
8
о
0 Он
2 76
2
32 32
18 'l8
8 | 8
2 2
2
oJih
1 45
0
8
Г>
2
18
8
2
о 1г
2 77
7
Г)
2
;2
18
8
1 2
X*
Ч
„
I*
2 „ ,
II*
и
1
'
1
0 XI
2 2S
8
Г)
2
8
2
0 Р<1
0 46
0
6
2
18
8
2
0 Pt
2 7S
8
6
2
32
18
8
1 1
0 Ги
1 29
10
Г)
2
8
2
1 47
0
10
0
2
18
8
2
0 Л и
1 70
10
(>
2
32
18
8
2
0/п
2 30
10
()
2
8
о
0 СЛ
2 48
0
10
<;
2
18
8
о
0 Нц
2 М1>
10
2
|32
18
1 8
1 2
ш
1 К
2 5
2
1 Л1
2 1;
8
2
1 Он
2 :'1
IV
0
V
VI
3'\
VII
VIII 1
0 1
1 ! Hel
! ' 1
2 Г
2 0
2
2 Si
2 14
8
2
2 tie
2 Я2
10 10
г. | г,
2 2
8 8
2 > 2
1 In 1 2 Sn
2 40 ! 2 50
0
L0
Г)
2
IS
8
2
1 Tl
2 SI
10
1)
2
32
IS
8
1 '?
о
10
f.
2
18
8
2
2 Vb
2 S2
10
С
1 2
32
18
8
2
з х 4 о
2 7 ! 2 S
Г) F 0 Xe 1
2 0 2 10 j
2 | 2 ' 2 12 J
3 P 4 S
2 15 ! 2 1«
8
2
3 As
2 M
8
2
4 Se
2 34
r> Г1 0 Ail
2 17 2 IS 1
8
2
5 Br
2 35
8 1
2 1
6 Кг|
2 36 1
ю Ilo no io 1
2 ! 2 2 2 I
8 8 J 8 j 8 j
2 ! 2 i 2 ! 2 I
3 Sb
2 51
0
10
0
2
4 To
2 52
0
10
l)
2
5 J
2 53
0
10
0
2
Г» X 1
2 54 I
0 [
10
г, 1
2 Г
18 |L8 [IS |18 [
8 ! 8 | 8
2 2 ' 2
3 IU , 4 Po 5 At
2 S3 • 2 S4 | 2 S5
10
C>
i 2
io ;io
Г, G
2 2
8 1
2 1
Г> It ill
2 SO I
10
6
2 1
132 32 !32 ,32 |
18
8
IS jl8 18 j
8
2 i 2
К ! 8 j
1-2 1 2 |
Примечание. Рядом с номером группы указан спектральный
терм атомов л основном состоянии. 1>уквы S,Pt 1) или F указывают
величину результирующего вектора Ь = S /, где /—векторы,
отвечающие отдельным внешним электронам. Индекс слева вверху
отвечу
BI
,Г
М(
01
1
ют мул
шзу у
= £ +
)М ГруЕ
чюсятс
'-уровне
ьътш1Л(
казано
s. Вел
[пы пер
я к грз
зм.
°тност
сумма
учаях:
иодичес
^ппс III
и сне]
зное ]
Nb, R
ЖОЙ С*
**, обр
ктра д
кванто]
u, Pd,
ic темы
азуя с
анногс
raoe 4i
VV тер
Элеме
эрию а
) атом
1СЛО, (
м атом
П1ТЫ IK
KTItHHJJ
a. jih,d
лтиечак
а не со
эсле 89
Ob с з;
сексом
ощее
впадае
, ПОВЩ
полня
снрав
вектор
Т С TCJ
UI.MOMN
юпшмс
а
у
>- 1
я 1
18») Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
Периодическая система, приведённая на таблице 29, нуждается
в некоторых примечаниях. До самого последнего времени элементы Th(90),
Ра (91) и U(92) относились соответственно к IV*, V* и VI* группам
периодической системы.
С открытием элементов Np(93), Pu(94) исследователи сначала
доказали, что они не являются гомологами Re, Os и других элементов
следующих групп периода, в результате чего элементы 93 — 94 («ураниды»)
были причислены к новой подгруппе |15], которую мы называли бы VI**.
С открытием элементов Am (95) и Cm (96) и исследованием их свойств-
совпал ряд работ, приходивших к выводу, что, начиная с Th (90) и до
Cm (96), псе эти элементы относятся к группе «актинидов» по аналогии с
лантанидами (III**) и что в ряду 90 — 96 имеет место постепенное
заполнение 5-го /-уровня [62] и [63].
Такое предположение весьма вероятно, хотя, может быть, его но следует
считать до конца проверенным.
Его подтверждение означало бы, что U не является гомологом W, а
Th является гомологом не Zr и Hf, но Се. (Недавно было вновь показано [64],
что аналогичная редким землям группа в последнем периоде системы
начинается скорее от Ас и Th, чем от U, который ранее принимался в качестве
начального пункта.)
Для наглядности, с целью подчеркнуть «переходное состояние»,
в котором находятся сейчас элементы №№ 90—96, переселяемые из
своих квартир в новые, мы показали в подгруппе III** семейство
актинидов (Th —Cm), а в подгруппах IV* —VI** обвели эти же
элементы рамкой со стрелкой. В случае окончательного переселения этих
элементов в квартиры актинидов подгруппа VI**, просуществовав 7 лет,
исчезнет.
Из 92 элементов, предусмотренных периодической системой, между Н
и U включительно, 86 получили химические обозначения, которые позже
не менялись. Остальным посчастливилось в меньшей мере.
71-й Lu долгое время обозначался Ср (кассиопий). 41-й Nb в
американской литературе и сейчас обозначается СЬ (Колумбии).
43-й сначала (с 1925 г.) обозначался Ма (мазурий) затем, когда его
открытие не подтвердилось, стал безымянным и, наконец, приобрёл
обозначение Тс (технеций), искусственный.
61-му в 1924 г. было предложено присвоить обозначение Fr
(Флоренции), с 1926 — II (иллиний); с 19'*1—-1943 гг. (после ряда
исследований и публикаций, опротестовывавших возможность существования
61 в природе)—Су (циклониум от циклотрон) и, наконец, с 1947 г. Рт
(промеций).
Элементу № 85 несколько раз присваивались разные названия: алаба-
мий (АЬ), англогельвеций, наконец (1947), At (астатин, нестойкий).
Элементу № 87 присваивались обозначения: Vr (виргиний), а затем Fr
(франций); не смешивать с Fr (Флоренции, предложенный в 1924 г. для
элемента № 61). См. § 118.
§ 87. Волномеханическая модель атома
В квантовой механике атом описывается как система стоячих волн,
возникших вокруг ядра, подобно тому как струна, оба конца которой
укреплены, может колебаться с определёнными характерными для неё
частотами.
Система стоячих воли, характеризующая атом, отличается тем, что
на определённом, очень малом расстоянии от ядра амплитуды этих волн
становятся незначительными, как бы определяя тем самым размер («ра-
диуо) атома (см. рис. 127).
§ 871
ИО.'ШОМЕХЛПИЧЕСКЛИ МОДЕЛЬ АТОМА
187
Пространственное распределение амплитуд волн, лежащих вокруг
ядра, может быть найдено с помощью известного пз курсов физической
химии волнового уравнения Шроднигера
причем
(20)
(21)
(оператор Лапласа).
У?—полная и V — потенциальная энергия электрона.
Отличительным свойством функции 6 является то, что | &2, определяет
вероятность нахождения электрона в той или иной точке.
Пз анализа этого уравнения вытекает, что в случае атома система
должна иметь дискретные значения полной энергии Е, и, следовательно,
0,0 Q,ZS 0,50 0,75
125 1,50 Г(А)
Рис. Ml. Функция Р дли иона Rb\ Обращаем
на себя шшманпе постепенное гглажииапис
остроты никои, отвечающих областям боропских орбит
мо мере перехода от А-урогшя к следующим, Б
области 1/i9 А от ядра попа, что отвечает
эмпирическому радиусу иона Rb*" по Гольдшмидту,
значения )) не ратш пулю. Кршшт не дает
возможности резко отграничит!» конец попа.
дискретные значения частот колеоании, т. с. характеристических частот
v -_ - - и т. д., как и в теории Бора. Однако расчёт значений амплитуде
и величии 6априводит и к принципиально новым результатам. В частности,
на рис. 127 показаны значения D = 4тгг262 для попа Rb+, определяющие
вероятность нахождения электрона на данном расстоянии г от ядра, так
называемую электронную плотность. Как видно из рисунка, на кривой
имеются максимумы. Эти максимумы лежат в области боровских Кч L
и т. д. орбит и как бы отвечают последним. Но, в отличие от
представлений теории Бора, функция является непрерывной, причём её значения
между орбитами нигде не равны пулю; орбиты как бы «размазаны». Более
того, к периферии атома кривая спадает постепенно, и, таким образом,
«конец» атома также «размыт». По вопросу об исследованиях электронной
глотности в кристаллах см. § 9G и приведённые там рисунки.
В целом вероятность нахождения электрона в точках на различвых
расстояниях от ядра не одинакова, или, как иногда неточно говорят,
заряд «размазан» в пространстве, образуя так называемое «электронное
облако».
188
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
|гл. II
Волновая механика рассматривает также характер распределения
валентных орбит в пространстве вокруг атома. С такой точки зрения
.9-орбита (т. е. s-электроны) образует вокруг атома электронное облако
Рис. 128. s-орбита; рх-, ру- и />г-орбиты.
шаровой симметрии. Напротив три ^-орбиты распределены вокруг атома
вдоль трёх осей х, у, г, а именно декартовых ординат (/^90°), так что
обычно различают р'х-, ру- и /^-орбиты (рис. 128).
§ 88. О волномеханичсских теориях химической связи
Волномеханические теории химической связи в первую очередь
занимались двуатомными, а затем многоатомными молекулами. Образование же
кристалла, как проблема расчёта очень сложной системы, стало предметом
всесторонних исследований значительно позже.
Проблема химической связи представляет самостоятельный интерес,
далеко выходящий за рамки немногих страниц, которые мы можем здесь
ей посвятить, и, кроме того, освещена в ряде специальных трудов, в
частности, в гл. I —IV и VII опубликованной и 1946 г. монографии Я. К. Сыркина
и М. Е. Дяткиной. Поэтому мы коснёмся лишь некоторых особенностей
различных методов, обратив внимание на отдельные принципиальные вопросы,
имеющие касательство к интересующим нас проблемам. Независимо
от этого, мы настоятельно рекомендуем читателю изучение вопроса
о природе химической связи (см. список литературы).
Как известно, волновая механика создала три теории химической
связи, из коих первые две весьма близки друг к другу.
1. Теория связывающих холостых электронов (Гейтлера — Лондона)
исходит из представлений о реагирующих атомах, при сближении которых
имеет место взаимодействие холостых г#-электронов (|) с образованием пары
электронов с компенсированными спинами (|\[), что даёт выигрыш энергии.
Таким путём было освещено образование молекул Н2, LiH, Li2
и необразование (неустойчивость) НеН, Не2, Ве2.
Однако интерпретация такого простого примера, как молекула Н+,
где связь осуществляется одним электроном, оказалась для этой теории
затруднительной. Теория натолкнулась на ряд других трудностей, на
которых мы здесь останавливаться не будем.
С другой стороны, надлежит остановиться на одном общем выволе.
Как это видно из таблицы 28, валентность элементов второго периода
от Li до С должна была бы изменяться так: 1, 0, 1, 2, если
исходить из числа холостых электронов в основном состоянии атома. Но
образование химической связи определяется по существу тем, являет-
§ 881 о волномеханичкских теориях химической; снязи 189
ся ли энергетически выгодным затратить энергию на декомпенсацию
антнпараллельных спинов электронов данного атома ( например, для утле-
РОДа: \ Villi
(г/4 -- 2)—>| | | | 11! , ' (//' = 4) J, с тем чтобы выиграть
энергию химической связи в результате компенсации спинов большего
количества, в данном случае четырёх, электронов рассматриваемого атома.
В случае Be, В, С это как раз и имеет место. С переводом в 1-е
возбуждённое состояние Be становится двухвалентным, В — трёхвалентным,
С — четырёхвалентным. С образованием, например, GF4 из C-|-4F затрата
энергии на возбуждение (w — 2 —> w --4) компенсируется энергией двух
добавочных химических связей С— F.
То же относится к ряду Na, Mg, Al, Si.
Но декомпенсация спинов (возбуждение атомов этих элементов)
облегчается тем, что у них ость незанятые близлежащие /ьуровни
(таблица 28).
С переходом к элементам N — Ne (см. таблицу 28) картина становится
иной. Ни одного свободного /j-уровня у этих атомов нет. Чтобы декомпен-
сировать спины 25-электронов, их надо перевести на высоко лежащий
3.9-уровемь. Эта операция оказывается очень невыгодной с энергетической
стороны, вследствие чего согласно теории Лондона N — трёхвалентен,
О —двухвалентен, F — одновалентен в соответствии с количеством холостых
электронов. Однако с переходом к ряду Р —СЛ положение меняется
вследствие большей близости As- и Зр-уровней. В результате элементы ряда
Р — (Л осуществляют более высокую валентность, чем 3, соответственно
2 и 1, например, атом S в SF6— шестивалентен.
2. Теория Слетера —Паулинга имела в числе других особенностей
важное отличие от предыдущей, а именно, она позволила непосредственно
подойти к вопросу о структуре молекулы, т. е. величине валентных углов
между связями.
Изучая взаимодействие атомов, рассматриваемая теория приняла
во внимание пространственное распределение зарядов (обусловленное
валентными электронами и определяемое квадратом их волновых функций).
Исследуя пространственное распределение электронной плотности
разных валентных уровней («орбит») и величину квантового резонанса
(см. § 90) в различных условиях, особенно в случае совпадания
(«перекрывания») областей наиболее вероятного распределения электронов
реагирующих атомов (или, как говорят коротко, «перекрывания орбите,
теория привела к выводу: «Та из двух орбит атома, которая может больше
перекрываться с орбитой другого атома, будет образовывать более сильную
связь. Более того, связь, образованная данной орбитой, будет стремиться
расположиться в том направлении, в котором будет сосредоточиваться орбита».
s-орбиты атомов имеют шаровую симметрию, тогда как, например,
р-орбиты вытянуты вдоль декартовых координат, центром которых является
центр атома (рис. 128). Вследствие сосредоточения р-орбит в
определённых направлениях их силы имеют значения в ]/3 раза больше, чем
значения сил 5-орбит, т. е. р-связь сильнее я-связи в 1,73 раза. По Паулингу,
стремление р-связей расположиться под прямыми углами в известной мере
подтверждается тем, что, например, в молекуле H2S с электронной
структурой :S: H угол между связями равен 92°.
Н
Но атомы второго малого периода (например, углерод) обыч) о
не образуют всё же р-связей под прямыми углами. Вследствие так на-ы-
190
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[глЛ1
ваемой гибридизации (т. е, линейной комбинации функций, см. ниже)
энергетически неравноценных .<?- и р-орбит возникают 4 равноценные
валентные орбиты, направляемые к вершинам тетраэдра, причём силы,
возникающие вследствие гибридизации связей, очень велики (равны 2).
Этим и объясняется тетраэдрическая структура соединений углерода.
Согласно Паулингу (1937 — 1939), однако, «невалентная пара электронов
стремится занять устойчивую s-орбиту, предоставляя р-орбитам
образовывать связи под углом 90°. В то же время общие пары электронов
стараются вызвать гибридизацию и образованно тетраэдрических связей*.
Поэтому у веществ, имеющих пары несвязывающих электронов: Н20
ЛО' Н\; NH8 fU :N: НЛ и т. п., эти два противоположных влияния влекут
^Н ) ^ Н '
за собой возникновение промежуточных величин углов (между 90° для
р-связей и 109° 28' для тетраэдрических).
В общем многие исследователи считали в 1937 — 1938 гг.
нормальными для Н20, NH8, ClaO и т. п. молекул углы 90°, а наблюдаемые
большие значения (близкие к 109°) объясняли взаимным стерическим
отталкиванием протонов и других атомов.
В 1937 г. Стюарт, опираясь на модель Паулинга, нисал [16]: «110° угол
атома кислорода (в молекуле воды и т. п.— Б. О.) не является
нормальным углом, требуемым теорией, но углом, который чаще устанавливается.
т. е. средним практическим значением».
Уже в 1937 г. автор допускал [17], что несвязывающая пара
электронов (г-электроны) в этих и многих других соединениях должна вести себя
в стерическом отношении, как связывающая, и что, следовательно,
молекула NH3—это не тригональная пирамида с атомом азота в вершине
и тремя атомами водорода в основании (с углами между связями менее
109°), а тетраэдр, к одной из вершин которого направлона несвязывающая
пара электронов. Захват протона с образованием 1\Н+ иона влечёт за собой
не уменьшение углов между остальными связями, как это должно было
бы наблюдаться в случае стерического эффекта, но некоторое увеличение1
(до 109° 28'). Иначе говоря, несвязывающая пара электронов занимает
больше места, чем связывающая (см. § 91).
Теория Паулинга также натолкнулась на затруднения в случаях, когда
связь обусловливается непарным электроном, или если связывающие
электроны принадлежат одному из атомов (донор).
3. Теория молекулярных орбит (Гунда-Мулликена), в отличие от двух
предыдущих, рассматривает молекулу как систему из и-ядер, заранее
образующих определённую конфигурацию. В эту систему вносятся
электроны, последовательно занимающие более низкие уровни, т. с. молекула
рассматривается примерно как атом с тг-ядрами. (Такая модель позволяет
легко подойти к освещению молекулы Н+ и т. п.) Задача построения
молекулы сводится к нахождению наиболео энергетически выгодных для
электронов «молекулярных орбит». Теория предусматривает наличие дгух
энергетических типов орбит. Энергетические уровни электронов,
находящиеся на орбитах 1-го типа, ниже, чем в изолированных атомах,—это
связывающие орбиты (связывающие электроны). Энергетические уровни
электронов, находящихся на орбитах 2-го типа, выше, чем в
изолированных атомах,— это разрыхляющие орбиты, разрыхляющие электроны.
Так как молскулярныо орбиты, занимаемые валентными электронами
атома, играют основную роль в решении вопроса об устойчивости
молекулы, то разделение валентных электронов на связывающие и
разрыхляющие представило большой интерес непосредственно для неорганической
химии.
§ 88] О 1ЮЛИ0МЕХАНИЧЕСКИХ ТЕ0Р1ШХ ХИМИЧЕСКОЙ СВЯЗИ 191
В заключение этого раздела коротко остановимся на: а)
представлениях о s- и 7Г-СВЯЗЯХ (подробнее см. В. 11. Кондратьев [69], а также [18])
и б) вопросе о характере химических связей.
Связи, которые дают максимальное перекрывание по прямой,
соединяющей атомы, называются а-связями*). Например, в молекуле Н, имеется
одна а-связь, образованная двумя s-электронами. Связи, образованные
^-электроном одного атома и р-электроиом другого (HCI) или двумя
б W9°28'
•< Н
Рис. 129. с*-связи (этан).
Рис. 130.^с3 тс-связы (этилен).
/^-электронами (С1С1), т. е. вообще простые связи, являются а-связями.
Если же в молекуле возникают кратные связи (двойные, тройные), то
между атомами образуется одна а-связь (по прямой линии) и т:-связи,
Рис. 131. с2 тс2-связи (ацетилен).
Рис. 132. Схема связей и С02.
перпендикулярные к а-связям. ir-связи значительно слаоее а-свнзи, поэтому
двойная или тройная связь не оказывается вдвое, соответственно втрое,
прочнее, чем одинарная.
В результате, например, атом углерода может осуществить либо
четыре а-связи (а4) под углом 109°28' (направленные к вершине тетраэдра)
(рис. 129), либо три а-связи под углом 120° и одну перпендикулярную
тс-связь \а*к) (этилен, рис. 130), либо две с-связи под углом 180° и две
гс-связи в двух взаимно перпендикулярных плоскостях (о2?:2) (ацетилен,
рис. 131). Реакционная способность, например, ацетилена объясняется
в этом свете не напряжённостью связей, но меньшей прочностью тг-связей.
Схема связей С02 показана на рис. 132. Аллен имеет распределение
связей, соответствующее не рис. 133, а (классическая модель), а рис. 133, 6.
Таким образом, с этой точки зрения в случае кратных связей,
например, атом углерода не является тетраэдрическим, как это раньше
предполагалось в органической химии и стереохимических представлениях
Вант-Гоффа.
*) Такое часто даваемое определение, выигрышное по своей простоте, не
является строгим (см. Б. Ф. Ормонт [59]).
192
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. Н
Т\
L в й
1 *
L б д
1/
^)Н
\120*
Теперь перейдём к вопросу о характере возникающих связей.
В развитие первоначальных представлений о ковалентной связи
квантовая химия принимает, что связь, вообще говоря, не является чисто
ионной или чисто ковалентной, но, как правило, является смешанной.
Этот случай в электростатике трактовался как наличие поляризационного
эффекта.
С количественной стороны, связь между двумя атомами А и В за счёт
нары электронов может быть описана на языке волновой механики
нахождением значения ф-функции как линейной комбинации собственных
функций обоих электронов.
В простейшем случае—двух
одинаковых атомов:
^ = С[фв(1)Ф»(2) +
+ «Ы1) -Ы2)], (22)
где фа(1) означает
собственную функцию электрона
1, находящегося у ндра А,
а ^ь(2)—электрона 2,
находящегося у ядра В, и т. д.
Первые два члена относятся
к случаю, когда электроны
1 и 2 находятся у разных
ядер (т. е. состояния А —В)
и называются часто
обменными членами, а вторые два
члена, когда оба электрона
находятся у одного и того же
ядра а или Ь (состояния
А-*- В" или А^ В+) —ионные
члены. Относительная
величина или, как говорят, вес
этих членов может быть в
зависимости от характера
связи неодинаков, так что
в общем случае, если атомы А и В однородны (например, С1 — G1)
в выражении
^ = С1[0Л1)^(2)+фЛ1)Фа(2)]+СЛ'|>а(1)^а(2)+^(1)^(2)], (23)
С\ ф С2.
'Если принять Cx = 0, Ci = l, связь носит чисто ковалентный
характер и «ионные» члены отсутствуют, и наоборот. *
В действительности, даже в случае таких молекул, как Н2,
величина С2 хотя и очень мала, но отлична от нуля.
Если же два атома неодинаковы, то соответственно меняются веса
отдельных членов уравнения*) (23), в частности, повышается вес ионных
членов, связь приобретает более ионный характер. Очевидно, что в
случае более ковалентной связи плотность электронного облака между
атомами (вероятность нахождения здесь электронов) более велика. С
повышением веса ионных состояний плотность электронного облака между
Рис. 133. Схема связей в аллене:
а) класссичеснан модель;
б) модель с а- и и-евяэями.
*) Вместо прежнего коэффициента С2 появляются коэффициенты С2 для первого
и С8 для второго ионных членов, стоящих в скобках.
§ 89]
ЭНЕРГИЯ ДИССОЦИАЦИИ МОЛЕКУЛ
193
атомами должна падать с образованием минимума в точке, отстоящей от
центров обоих ионов на величину их условных (эффективных) радиусов.
Легко понять, что представление о «радиусах» в подобных системах
действительно весьма условно, атомы не имеют резко очерченных границ
и электронное облако одного атома как бы проникает в электронное облако
другого, так что, практически, мы имеем в кристаллах дело лишь с
расстояниями между центрами тяжести атомов. В § 75 мы указывали,
что с изменением температуры в присутствии растворителя и т. п.
характер связи меняется, так что вещества, имеющие связи более ковалентного
характера, оказываются в растворе иной раз совершенно
диссоциированными, например HJ, тогда как вещества, гораздо более «ионные», как,
например, HF, в воде лишь частично распадаются на ионы.
§ 89. Энергия диссоциации молекул
Современные оптические и термохимические методы позволяют
установить значения энергии диссоциации молекул Z); некоторые данные
приведены в таблице 30.
Таблица 30
1 Вещество
и,
О,
/>
Оптические
данные
101
и;
' 58,5
Термохимические
данные
100
57,2
Вещестло
Вг,
1U
~ |
Оптические
данные
45,2
35,2
G6,0
Термохимические
данные
46,2
34,5
69,3
Нахождение энергий диссоциации молекул па атомы позволило
вычислить теплоты образования молекул из атомов, а не из простыл
веществ. Например, теплота образования НО из H-f-Cl:
I
Н = у Н2 + 50,5 ккал
С1 = уС12+29,3 ккал
~H2 + -J CL - ИС1 +22,0 ккал
Н + С1 = НС1 +101,8 ккал.
Если исходить из энергий образования из атомов (а не из простых
веществ), то практически все известные химии соединения являются в
стандартных условиях экзотермическими.
Если бы энергия диссоциации молекул АА или ВВ, т. е. D\
соответственно Вву была величиной аддитивной из инкрементов двух атомов,
то можно было бы ожидать, что энергия диссоциации молекул АВ
окажется средней арифметической из энергии диссоциации молекул А А и ВВ,
т. е.
Яав
что и имеет место у ряда веществ.
Лаа + Я-
ьв
(24>
194 ч. i* основы теории структур [гл. II
Так, для BrCl DBU4. = 51 ккал, Дшсл. = 52 ккал.
В действительности же часто
£>лв>
^AA + D
вв
(25)
Например, Дн2 = 101 ккал, DC\2 = 58,5 ««ал, Z>Hci (аддитивное) - 79,75 ккал,
Z>hci (экспериментальное) = 102 ««#.#. См.
а+а *-б 6-Ь рис. 134 (схема).
л
-Иде
-D
45
§ 90, О теории квантово-химичеекого
резонанса
-D.
Избыточная энергия связи молекул
ДВ вызвана по Паулингу явлением
квантово-химичеекого резонанса, наложением
ионной связи на ковалентную и
называется резонансной энергией.
Теория резонанса была позднее раз-
Рис. 134. Повышение энергии связи вита в том смысле, что если молекула мо-
молекул типа /А\в против аддитив- жет иметь несколько примерно близких в
пого значения, (схема). энергетическом отношении электронных
структур с одной и той же ядерной
конфигурацией и одним и тем же числом непарных электронов, то
нормальное состояние молекулы характеризуется не одной какой-либо из
них, но суперпозицией (наложением) всех этих структур, т. е. суперпозицией
разных валентных состояний. При этом молекула делается устойчивей,
ибо в результате резонанса имеет место понижение энергетического уровня
системы на величину Е резонансной энергии. Волновая функция,
описывающая нормальное состояние молекулы, является линейной
комбинацией волновых функций, отвечающих различным электронным структурам.
Например, в молекуле СО возможен резонанс между состояниями
с двойной и тройной связями
:0=С = 0:, :0"]=С —(')•" и :()--(]= б Г.
Существенно, что в каждой молекуле одновременно накладываются
друг на друга все возможные резонансные состояния, каждое с
присущим ему весом и в системе нет молекул, которые по своей структуре
отвечали бы только одной формуле.
Изложение теории резонанса дано в книгах Паулиига [7] (1945) и
Я. К. СыркинаиМ. Е. Дяткиной [8] (1946).
Авторы рассматриваемой теории допустили в своих исходных
предпосылках и в развиваемой теории ряд серьёзных ошибок и соскользнули на
опасный путь научного формализма. Основной факт понижения
энергетического уровня системы при отклонении характера связи от чисто ионного
или чисто ковалентного—в первую очередь в молекулах с разнородными
атомами -сам по себе никакого отношения к так называемому «явлению»
резонанса не имеет. Необходимость такого изменения характера связи,
и притом даже в одном и том же веществе, в зависимости от внешних
условий и т. п. —всегда рассматривалась в химических теориях (см., в
частности, § 75).
Вместо пользования резонансом, как .методом описания систем
(ценность которого является к тому же спорной), теория резонанса
часто говорила о нем, как о реальном явлены,и. Нарушая принципы
Бутлерова (обеспечившие создание структурной химии отказом от формул,
допускающих произвольные толкования свойств молекул), теория пользовалась
§ 91 ] СТРУКТУРА И УСТОЙЧИВОСТЬ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ 195
(см. Я. К. Сыркпп и М. Е. Дяткина, 1. с. [18]) десятками резонансных
формул одного 11 того же вещества, веса которых либо неизвестны, либо
весьма малы.
Ничего не объясняющая и, тем более, ничего подобными схемами не
предсказывающая, пользующаяся терминами и представлениями, не
имеющими ничего общего с действительной структурой (говоря, например,
о ионах Сг3 в комплексных соединениях, см. стр. 4об 1. с. [18]), такая
теория не является плодотворной. См. также дискуссию в Успехах химии
за 1949 г. и след.
Наши критические замечания в адрес теории резонанса ни в коей мере-
не затрагивают основных представлений и теорий квантовой химии (§§ 87, 88).
§ 91. Об установлении общих закономерностей, описывающих структуру
и устойчивость неорганических веществ
В ряде работ автора [19] (1935— 1945 гг.) было подчеркнуто различие
в поведении элементов, имеющих sp- и ds-валсптные электроны (мы
называем их, краткости ради, соответственно sp- и ds- элементами). К числу
первых следует отнести, например, Sn, Pb(s2p2)f к числу вторых—
Ti, Zr(dV)Cr, Fe и др.
Как это было автором сформулировано в 1935 г., ^-элементы не
образуют летучих гидридов в отличие от .ч-у^-элементов, по легко образуют
твёрдые гидриды (структуры внедрения, § 102). С завершением d-оболочки,
невидимому, резко ослабляется также способность атома к
присоединению дополнительных электронов за счёт акцепторной связи, вопреки
правилу ЭАН, что отражается, например, на процессе образования кар-
бонилов (см. Б. Ормонт [20]).
Во всех химических теориях, как мы указывали [21], в качестве критерия
устойчивости химических соединений рассматривались либо ядерная
(например, теория плотных упаковок Вернера, Косселя), либо электронная
(теория Льюиса, правило ЭАН и др.) конфигурации.
Первые считали критерием устойчивости соединения образование,
например, координационной сферы с 3, 4, G атомами (безотносительно
к количеству, квантовой характеристике и роли электронов в
образовании связей), вторые же считали таким критерием количество
электронов в оболочке (2, 8, 18 и т. п. электронов) безотносительно к форме
ядерной конфигурации.
В 1937—1938 гг. автор, опираясь на квантово-химические представления,
высказал общее для всех веществ положение [21]. согласно которому «сама
по себе устойчивая электронная структура может оказаться менее
устойчивой в комбинации с энергетически невыгодной ядерной структурой ^и па
оборот)», причём при более ионном характере связи последняя должна
быть более требовательной к соблюдению энергетически выгодных ядерных
конфигураций.
Исходя из учёта возможности стерического отталкивания,
уменьшающего устойчивость конфигурации, автор высказал положение, что (если
условно отвлечься от электронной структуры) сами по себе наиболее
устойчивы ядерные конфигурации, в которых валентные углы (между
связями) максимальны.
Из этого правила вытекает, что для соединений АХ3, АХ4 и АХ8
наиболее устойчивы треугольная (^ = 120°), тетраэдрическая (^/ = 109°28'),
октаэдрическая (/^ = 90°) конфигурации. Напротив, куб в случае АХ8
является мало выгодной конфигурацией (z^"г^3°), почти столь же
невыгодной, как и плоская ядерная конфигурапия АХб {/_ = 72°). С помощью
этого простого критерия нетрудно установить, что тригопальная бипира-
196 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР |ГЛ. II
мида —одна из наиболее выгодных ядерных конфигураций для случая АХ&
(три связи под углом 120°, две связи —90°) (рис. 135, а).
Как известно, наиболее устойчивые электронные конфигурации —
2 (гелий), 8 (неон и др.), 18 (аргон и др.).
Остановимся на некоторых выводах, сделанных нами в результате
учёта обоих критериев — электронной и ядерной конфигураций: первый
вывод касался структур молекул и координационных сфер.
Структура молекул состава АХЛ, имеющих одинаковое соотношение
количеств разнородных атомов (например, ВС1Э и РС1Я, SnCl4 и ТеС14),
может быть разной в зависимости от того, какова электронная
конфигурация системы, и, напротив, координационные сферы, имеющие разное
количество периферических атомов (например, РС13 и SnCl4, ТеС14 и РС15),
могут иметь одинаковые конфигурации и
близкие валентные углы.
Как было подчёркнуто в работе автора [21]
(1938), несвязывающие электроны могут не
только ослаблять прочность связей, как это
постулировала теория
молекулярных орбит, но и
влиять на конфигурацию
молекулы, причём
возможно либо искажение
конфигурации, которую
следовало бы ожидать
в отсутствии несвязы-
вающих электронов, либо
даже возникновение иных
конфигураций, как если
бы несвязывающая пара
вела себя как
связывающая*). Именно второй механизм, по мнению автора (1938),
наблюдается в Н20, NH3 (см. § 88), молекулы которых с этой точки зрения
являются тетраэдрическими. Такая картина означала на языке квантовой
химии возможность гибридизации s- и р-орбит (§ 88), даже если s-элек-
троны являются не связывающими.
Как принципиальный вывод, автор подчёркивал, что критикуемые
взгляды нуждаются в существенной поправке в том смысле, что
максимальное координационное число и конфигурация координационной сферы
могут определяться не только связывающими электронами, но и несвя-
зыеающими (в частности, s-элоктронами, вопреки тогдашнему состоянию
теории Паулинга), и что вообще в соединениях, в которых
центральный атом не осуществляет максимальную валентность, возможно
изменение ядерных структур и валентных углов по рассматриваемой причине
(см. рис. 136).
Поскольку представления этого рода были высказаны позже Ивенсом
и Листером [23] и, наконец, нашли отражение в книгах Паулинга [7] (1940),
(1945) и Уэллса [23] (1945) и в недавней [59] (1947) статье Паулинга, без
ссылок на советские работы, мы должны заметить, что они впервые были
опубликованы в работах автора в советской научной прессе.
Второй общий вывод позволил охарактеризовать, исходя из
совершенно общих положений, устойчивость соединений, в том числе тех,
которые до того времени описывались в химии, главным образом, пра-
а 6
Рис. 135. Тригоналыгая бипирамида.
*) В работе 1. с. мы обозначали пару электронов двумя штрихами (а не двумя
точками), что лучше отражает процесс обмена электронами.
§911
СТРУКТУРА II УСТОЙЧИВОСТЬ НЕОРГАНИЧЕСКИХ 1ШЩЕСТН
19,
вилом ЭАН (§ 81), например карбонилов (и комплексных цианидов), и тех,
свойства которых (например, цианидов) правило ЭАН объяснить не могло.
В 1938 г. автор сформулировал положение, что устойчивость
структуры определяется:
1. Электронной конфигурацией е11 (чем ближе к нулю разность
между числом электронов устойчивой электронной оболочки и эффективным
числом алсктроиоп (ЭАН) центрального атома, тем устойчивее соединения
при прочих равных условиях).
2. Ядерной конфигурацией аи (т. е. числом окружающих
центральный атом групп). При этом в общем случае конфигурация тем у стопчи-
Т] еуго.1ъиия
а
К
В ) м'
а <
Cl
Искажённый
тетраэОр
а ci
\ /(
Р , W4'
// \
Cl
Тетраодр
Cl Cl
\ /
P,'W
/ Ч
0 Cl
Тетраэдр
Cl Cl
\ <
Sn 1 W9C
// К
Cl Cl
месбязб'Зающир пара занимает
5ольшв м9ста ч?.*> ебязыбающоя
Тригоиалъная
Ьипиралшда
Тригопалъиая.
бипирамиОа
Искажённый
октаэдр
или
т ригоналъпаа.
бипирамида
Otntiaobft
/\
Cl Cl
:CL
Cl
// \
Cl Cl •
Cl
Cl
■ч
or
Mi
<?
Cl
^4
CL
Рис. 136. Зависимость форм координационных сфер как от
ядерной, так и от электронной конфигураций (Ормонт, 1937 — 1938).
все, чем больше валентные углы (углы между связями) и чем
равномернее распределены связи в пространстве.
3. Первоначальным валентным состоянием центрального атома. Чем
выше исходный заряд центрального атома, тем сильнее удерживаются
ионы (или легко поляризуемые молекулы), например, связь Са — NH;<
слабее связи Са2=—(NHJ.
Чем ниже исходный заряд центрального атома, тем слабее
удерживаются ионы и дипольные (или легко поляризуемые) молекулы и тем
относительно прочнее должны быть связи типа Me СО (Б. Ормонт [21| (1940)).
Проверка вышеизложенных правил на многочисленных соединениях
и, в частности, летучих карбонилах дала хорошие результаты [24].
В 1942 г. положение \\ было но существу подтверждено также
фактами и выводами более поздних работ [25| других исследователей.
В 1942 — 1943 гг. были открыты [20,27] соединения K4Ni(CN)4 и
K4Pd(GN)4, свойства которых уже в 1940 г., т. е. задолго до синтеза,
были впервые предсказаны автором, исходя из вышеизложенных
представлений, и которым автор (в 1940 г.) предложил присвоить название
цианилы, по аналогии с карбопилами. (См. также Б. Ормонт [28].)
198 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР (ГЛ, I!
§ 9:2. Количество холостых гоэлектронов в изолированных атомах.
Температуры плавления простых веществ и максимальная валентность
тех лее элементов в устойчивых соединениях
В 1934—1935 гг. автор поставил перед собой задачу попытаться
использовать ряд представлений квантовой химии дли решения некоторых
задач неорганической и кристаллохимии, которые, особенно в то время,
развивались на базе классических законов и едва только начинали
использовать хотя бы выводы теорий Косселя -Люиса. Сложный же
математический аппарат квантовой химии, неконкретность многих её выводов и
применение их, главным образом, лишь к простейшим молекулам: H2f,
Н.,, LL и т. и. разделяли в то время эти дисциплины высокой стеной.
См. Г/. Ормоит[19] (1935), (1936) и др.
Нельзя сказать, что эта картина сильно изменилась к лучшему
в настоящее время.
Поэтому, продолжая горячо пропагандировать необходимость быстрей-
jnero ознакомления химиков с методами и результатами самой квантовой
химии, ни в коей мере не стремясь к подмене последних (составляющих
самостоятельную дисциплину) и ставя перед собой, как видно из
сказанного, совсем другие задачи, автор в течение ряда лет сформулировал
несколько закономерностей, позволивших предвидеть возможность
образования или необразования тех или иных веществ (см., например [19, 24]),
описать свойства новых, позднее открытых соединений (циаиилы [28] (1940),
(J 915))» опротестовать и затем доказать несуществование некоторых
соединений, ранее описывавшихся в новых курсах [19]: Fe04, Ni04, BaFoOs,
BaNi05 (1936), предвидеть возможность отклонения структур веществ от
тех, которые вытекают из классических представлений (ТеС14 и др.) (см. [21]
(1938)), указать пути синтеза некоторых новых соединений (карбонилгало-
гениды и карбоиилы и цианиды t/s-мсталлов низшей валентности [28] (1938),
(1940—1946)). оценить критические энергии связен некоторых не
открытых ещё веществ, например, Zn(CO)3, Cd(CO)3, Mg(CO;3, Sn(CO).,
и др. [20] (1916) и т. д.
Опираясь на представления теории* молекулярных орбит о
связывающих и разрыхляющих электронах и взгляды теории связывающих
(холостых) электронов на валентность элементов малых периодов, автор
рассмотрел изменение количества непарных г#-электронов в пределах всех периодов
системы и провёл параллели между количеством гт-электронов, с одной
стороны, и температурами плавления простых веществ, валентностью
атомов этих веществ в соединениях, а значит — составом
координационных сфер,—с другой.
Рассмотрение ряда К —Мп и их гомологов показывает, что все
валентные d-элсктроны этих элементов легко могут стать холостыми,
например, для Mn(3tf5-Ss2):
ы
11111111 f t
4.s 1 [if
til 1 1
требуется лишь возбуждение 4s2-—>isip с относительно небольшой
затратой энергии; в результате Мп, Сг и т. д. и их гомологи могут
осуществлять максимальную валентность w = o, причём число г^-электронов
растёт к середине периода до VII* группы (Мп и др.) (где v—суммарное
количество валентных электронов).
Далее, от Fe к Zn число гг-электронов в основном и ближайшем
возбуждённом состояниях падает — в основных состояниях атома с 4 до 0.
Если же учесть, что 4.92-электроны сравнительно легко возбудить до 4ь4p,
§ 92] КОЛИЧЕСТВО ХОЛОСТЫХ ш-ЭЛЕКТ1'ОНОВ В АТОМАХ 199
то количество ^-электронов практически равно 6 для Fe и падает с 6
до 2 у Zn (см. табл. 31).
'•* Таблица 31*)
:ы
Т
1 1
1
t
т
t
т
| ;
1
1 т
!
\ 1 i
1
1 Т
1 1
1 1
1 1
*
у
Ф
1
1
1
*
л
!
1 1 t
1
i I
Т
■-
—;
Т
t
—
—
i
t
т
45
т ' т ! ■
1 ! Ы^
liH'
1 ! ku
t
t :
i
i
i •
ф
'
_
' i
1
t
.-'
!
-
4/i
Состояние |
осионное
M,'=-'i + 0=4
w«3-f n=:t
w = 2-1-0=2
/r=n f-l = l
IV =,: 0+0=0
1-е
возбуждение
G
5 1
4
л
2
1
Указав, что прочность связи атомов металла в кристаллической
решётке должна зависеть от числа гг-электронов, автор сделал [30] (1936)
вывод о том, что наиболее высокая температура плавления простых
веществ должна, в общем, наблюдаться около седьмой группы
периодической системы, что подтверждается опытом (рис. 137). На этом рисунке
показаны для 2-го и 3-го больших периодов кривые изменения
максимального количества ^'-электронов, абсолютные температуры плавления
простых веществ и максимальные валентности тех же электронов по Fe
и С1. Налицо чёткая связь в ходе этих кривых.
В 1938—1940 г., исходя из аналогичных соображений, в терминах
зонной теории кристалла (см. § 97 и след.), к тому же выводу пришли
Зейц и Джонсон.
Этой картиной опровергается вывод Паулинга [7] о том, что в Zn, Cd,
tig tv —6, чем определяется структура этих элементов. Как это вытекает
из прочности решётки Zn, Cd, их энергий сублимации и т. д., атомы
Zn пли Cd имеют гораздо меньше г^-электронов, чем атомы Сг или Fe
и т. п.; практически для Zn следует принять zv — 2.
В равной мере нельзя считать, что в Си, Ag, Au w = 7, т. е.
электроны Зс?-уровня переведены на 4/>-уровии: возбуждение 3rf104sx до 3d74s4p3
требует весьма большой затраты энергии, кроме того, такая решётка
была бы более тугоплавкой (сравни особенно Ag и Mo, Au и W).
Наконец, энергия возбуждения валентности атомов может иметь
в данном случае место лишь за счёт реакции взаимодействия атомов,
т. е., как указал автор [30] (1935—1936), должен существовать параллелиз и
между прочностью решётки простого вещества и максимальной валент-
*) Тонком пунктиром иоказаы электрон в первом возбуждённом состоянии атома,
жирным пунктиром указан электрон, переходящий на р-уровень.
200
Ч I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
ностью тех же атомов в соединениях (см. ниже). Но медь никогда не
бывает семивалентной и цинк шестивалентным, так что использование,
как это делает Паулинг, формальной максимальной валентности элемен-
'аос
2600
2400
2200
2000
1800
f600
1400
111
w
р
s
d
/\ \
' in /ч
i ^ \
/^ 8-
i 7 7
• • 7-
У'\ v-Ч <-
F-ti-криВая \-\M-
3
4
1
u
t
4
*
1
H
t
_L
5
4
tf
t
I
Jj
6
t
t
t
t
t
Jj
7
1
1
ti
t
t
t
♦
1j
№
4
1
tf
t
t
t
t
it
5
1
tf
t
t
t
\\
JJj
4
*
it
t
1
H
u
jKj
/^
1
t
tt
u
H
и
w
•
A
У1 \
II \
1» \
J 4v v
-/
13
u
1
г
Ll
4
1
н
t
jj
5
t
t
t
t
t
6
t
t
1
t
t
t
7
1
u
1
t
t
t
jj
*$
1
4
1
tt
t
t
t
♦
ti
7
1
t
t
t
t
t
u
n
4
,
i
1
i If 1
t
t
ti
tf
H
u
tl
u
H
ы
Sc Ti V Cr Mn Fe CO Ni Си Y Zr N6 Mo Ma Ru Rh Pd Aq
Рис. 137. Снизь между абсолютной температурой плавления иргстых веществ,
максимальной валентностью тех же элементов по фтору и хлору и колпчестпо.м
холостых w-электронов. Один ^-электрон (пунктир) может легко переходить на
/^-уровень (Ормонт, 1934—1937).
тов, определённой по числу возможных «клеток» с холостыми
электронами, без учёта ограничений энергетического характера, по нашему
мнению, является ошибочным.
§ 92] КОЛИЧЕСТВО ХОЛОСТЫХ /«-ЭЛЕКТРОНОВ В АТОМАХ 201
Исходя из вышеизложенных представлений (см. табл. 31), автор
(1935) опубликовал вывод о том, что максимальная валентность Fe в
химических соединениях с ковялентной связью должна быть равна числу
^'-электронов (если отсутствуют акцеиторподонорные связи), т. е. 6, а для
Ni w — 4 (причём учитывалось, что образование соединений восьмпвалент-
ного железа требует возбуждения Зс?-олектрона до уровня 4/?, т. е. должно
затрудняться). Этому выводу противоречил тот факт, что в литературе
были широко распространены утверждении *) о существовании
соединений не только восьмивалентного железа, по и никеля, например, HaNi05
(на основе работ Горалевича [31]). Проведённая в лаборатории автора
(совместно с Б. А. Петровым) проверка птнх работ Горалевича, а закже
попытки получения подобных веществ иными путями показали, что
соединения воеьмивалептного никеля и железа выделить не удаётся [32], а Горале-
вичем была допущена экспериментальная ошибка. Подобная проверка была
предпринята позднее О. Е. Звягинцевым и II. В. Гогоришвили [33] (СССР),
Крепелка и Блаболил [34] (Чехословакия), Шольдер [35] (Германия), причём
наши выводы сперва горячо оспаривались отдельными авторами [30]. а
затем были ими же и другими исследователями подтверждены.
В рядах Ru Cd и Os — Hg количество vr-олектронов также убывает
к концу ряда, так что, в общем, осуществление более высокой
валентности всё болыис затрудняется. Однако в этих периодах возбуждение kd
соответственно 5с?-электронов па уровни 5.v или Ър соответственно 6* и 6/?
(или 4/ соответственно 5/) требует меньшей энергии, вследствие чего
соединения тина OsF8 или RuF8, Os04 и Ru()4 удаётся выделить. Так или
иначе, к 1938 г. (см. [70]) максимальная валентность но кислороду при
стандартных Тпру этих элементов отвечала таблице 32**):
Т а б л л ц а :>2
Максимальна» валентность элементом V111*, IX* и X*
групп по кислороду (Ормонт)
Элемент
Валентность
Соединение
максимальной
валентности
Элемент
Валентность
Соединение
максимальной
палеи тности
Элемент
Валентность
Соединение
максимальной
валентности
Fe
6
Na.Fe04
Ru
RnQ4
8
Os04
Co
Co,),. .HLO
Rh
4 —G
RhO. (Rh03??)
lr
G
lr03
Ni
4
NilV-rlLO
Pd
4
Pd02..6-II,0
in
G — \
1Ч03:Ш.,0(?) •
mo.
*) Re вызвавшие ни и ком сомнений, поскольку FenNi относилиськ МП группе
периодической системы.
**) В 1943 г. было показано [.47], что при р0 t < 275 мм термодинамически устойчив
PtO., при 275 < jd0o < 88П .u.u-PL03; выше 85i)\n,w РЮ, а=121ШэС)| что также
подтверждает вышеизложенное.
202 ч. i. основы теории структур [гл. II
Учитывая совершенно различные энергетические требования, которые
приходится осуществлять для возбуждения валентности у Fe, Co, Ni
(и их гомологов), автор подчёркивал, что их ни в коем случае нельзя
относить к одной и той же группе периодической системы (VIII*).
На рис. 137 сопоставлены для первых двух больших периодов кривые:
количества ^'-электронов элементов больших периодов (средние),
температуры плавления простых веществ (верхние) и максимальная валентность тех
же элементов в соединениях: а) по кислороду и б) средняя по F и С1 (нижние
кривые). Рис. 137 не нуждается в комментариях, Для III большого периода
картина та же. См. Ормонт, 1. с.
Это позволило автору сделать [30] следующие общие выводы:
1. Температуры плавления простых веществ с металлической или
атомной решётками изменяются в первом приближении параллельно средним
значениям М осуществляемой максимальной валентности S тех же элементов
по фтору Sy и хлору Sq\
M=Sf+/*. (26)
2. Отнесение Fe, Со и Ni (и их гомологов) к одной и той же группе
периодической системы (VIII*) является неправильным.
Эти элементы, например, Fe и Со или Fe и Ni, отличаются друг от
друга по количеству г#-электронов, которые могут реализовать химические
связи (во всяком случае в такой же мере, как, например, Fe и Мп или
Fo и Сг). и поэтому они должны принадлежать к разным группам
периодической системы.
S 93. Недостатки правила эффективных атомных номеров (ЭАП).
Магнитные моменты атомов и ионов
Выше мы указали (§ 81), что правило эффективных атомных номеров
позволяет объяснить образование некоторых видов комплексных
соединений с ковалентной связью (Fe(CO)6 и др.).
Исключения из этого правила, о которых мы говорили в § 81,
встречаются слишком часто, например, K2Ni (CN)4 устойчив, K4Ni (CN)4 менее
устойчив (см. Б. Ормонт [28]). Как неоднократно подчёркивалось автором,
правило ЭАН в некоторых случаях осуществляется лишь формально, что
вытекает из величины магнитных моментов соответствующих соединений.
Здесь мы коснёмся этого вопроса подробнее.
Величина магнитного момента определяется в первом приближении
количеством in некомпенсированных спинов
{х = | г ^Г(^Т2) ■= /45(5+1),! (27)
где 5 -относительная величина суммарного спинмомента, в которую
1
каждый спинмомент входит значением — (абсолютная величина
спинмомента — ™ , где Л —квант действия Планка). В ионе Fe2^, например,
распределение 6 валентных электронов, согласно правилу Гупда, таково:
45
1р
т. о.
устопчмность комплексных согдпненип
203
В действительности на спинмомент налагается орбитальный момент
л в тем большей мере, чем слабее ноле кристаллической решётки. (Стопор
(1929), Б. Ф.чек (1931) н др.) В утих случаях и. приближается к
t* = /4A,(5 + "D + A(L-i-l)
(28)
V=V J (/+1),
(29)
где L —суммарное побочпое квантовое число и 7— суммарное внутреннее
квантовое число. Об их значениях см. Б. Ормонт[1], стр. 215 и след.;
В. Н. Кондратьев [69], стр. 8G и след. Впрочем, и при наличии такого
дополнительного взаимодействия магнитный момент чаще отличается от
вызванного спинмомонтом лишь на очень малую величину. Так, у FeCl2, у
Fc(H20)4Cl> nFe(NH3)eCL> ц = 5,2 вместо 4,9.
Величина jj. указывает на ионный, в основном, характер связи внутри
координационной сферы Fe (NH3)J+, ибо если бы связь была ковалентной
п правило ЭАН соблюдалось, атом Fe в последнем случае имел бы
в наружных уровнях 3d, 4s и 4/> — 18 электронов: 3e/104s24pe
w = 0,
3d _
I <
ьр
п\и\г
п, следовательно, пи одного холостого (но если w = 0, то р- = 0).
Такой магнитный момент и такая картина распределения электронов
наблюдаются у Fe (CO)5. Однако и применительно к комплексным цианидам,
карбонилам и т. д. наблюдается много отклонений, например K2Ni(CN)4,
K4Co(CN)6 и др. диамагнитны.
§ D4. О трактовке структуры и устойчивости комплексных соединений
с помощью некоторых квантово-химических моделей
По предложенной Паулингом общей схеме образования таких
соединений, как K4Fe(CN)e, к центральному иону Fe2+ переходят сначала
6 электронов от 6 CN-грунп. При этом возникает промежуточный Fe4~-itoH,
имеющий 12 валентных электронов. Заняв ими наипизшие уровни 3d104s2,
этот ион становится нульвалептным. Для образования 6 связей надо,
чтобы 1С было равно 6. Это имеет место
3d8
1*4/Г
<r = ojfj
w = 61 U
3d
If 1 fl 1 tllli
| 1 Y | 1 Y | 1 1 1
4.9
11
1
TT'"'|
111111
в состоянии 3d8 4s 4//* за счёт d'2sp* непарных электронов. Присоединив
в этом состоянии ещё но одному электрону от шести CN-групи, атом Fe
образует диамагнитную конфигурацию 3d104/r4p* с 18-электронной
оболочкой благородного газа. Как это вытекает из рассматриваемой теории,
пространственное распределение d2s/>3 волновых функций влечёт за собой
октаэдрическую конфигурацию координационной сферы.
При образовании lv2Ni(GN)4 имеет место такая картина: ион Ni2+r
присоединив 4 электрона от четырёх CN-rpyim, образует сначала
«промежуточный» ион Ni2~ с 12-валентными электронами. Для образования
четырёх связей необходимо, чтобы w было равно 4. Это возможно при двух
204
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. 1Г
конкурирующих структурах: а)
3d
||\:|м|п|п
t
4v
1
■\р
м;|
и b) 3d84s4/?3 г#=6, рассмотренной выше; в первом случае за счёт dsjr,
а во втором —за счёт .9р3-валентных электронов. Первому случаю отвечает,
согласно рассматриваемой теории, квадратная, а второму — тетраэдриче-
скан конфигурация распределения валентных орбит.
Кроме того, остаточный магнитный момент будет равен в первом
случае р- = 0 (все 4 электрона образовали связи), а во втором и = 2,8'^
(2 из 6 электронов остались холостыми), так что на основании (27)
Р = У 2 (2 -t-2) = 2,83.
Как известно, подобные соединения, например K2Ni(CN)4, дсистви-
тслыю имеют квадратную структуру координационной сферы (и
диамагнитны), что не поддаётся объяснению в рамках электростатики. Этот вывод
теории о пространственном распределении направленных валентностей,
позволяющий объяснить квадратные (плоские) координационные сферы,
является её достижением.
С другой стороны, рассматриваемая схема встретила критику с
разных позиций (см., например, [66] (1938)) по поводу самой волномехаиическоп
модели, равно как и по поводу представлений об образовании ионов Fe4~,
Ni2~. В частности, отмечалось, что вовсе не обязательно заряд должен
«сидеть» на центральном атоме и может быть распределён между ним
и периферическими группами.
Но нас гораздо больше интересует вопрос, в какой мере
рассматриваемая теория в состоянии не только объяснить свойства экспериментально
изученных соединений, но и предвидеть поведение и свойства неизученных
и неоткрытых соединений. В этом разрезе положение дела нельзя считать
достаточно удовлетворительным, не говоря о большом количестве
исключений, в своё время приведённых разными авторами, в частности для
диамагнитных соединений с нечётным количеством электронов, например
K4Co(CN)e, K3Co(CN)bCO, K2Ni(CN)3 и др.
В 1913 г. была сделана попытка [67] сформулировать, исходя из
рассматриваемой теории, общий критерий устойчивости таких соединений,
причём было высказано положение, что при их образовании центральный
атом стремится осуществить конфигурацию (тетраэдрическую, квадратную,
октаэдрическую и т. д.), которой отвечает наименьшее количество
непарных электронов. По нашему мнению, эти взгляды весьма уязвимы.
§ 95. Развитие вопроса о валентных конфигурациях
Как мы видели, в различных случаях гибридизации .9-, р- и ^-^лек-
тронов линейные комбинации dsp2-функций приводят к квадратной
конфигурации АХ4; яр3-функций — к тетраэдрической АХ4, d2sp3—K октаэдрн-
ческой АХ8, причём возникают 4 соответственно 6 с-связей.
В 1940 г. Кимбол [39], рассмотрев вопрос о различных возможных
гибридизациях ,v-, р- и d-орбит, показал, в каких случаях могут возникать
и другие структурные формы (для к. ч. от 2 до 8).
Результаты сведены в таблицу 33.
Представляется интересной осуществлённая в работе попытка
расширить наши знания о зависимости между валентной конфигурацией
центрального атома и возникающей структурой координационной
сферы. Тем не менее работа нуждается в ряде критических замечании.
§ 95] рлзнитик нопросл о валентных конфигурациях 205
вызванных не столько дефектами самих расчётов и выводов, сколько
всем подходом рассматриваемой теории к этому важнейшему вопросу.
Таблица 33
Возможные конфигурации связей
|Коор-
дина-
цион-
ное
число
2
3
4
фигурации
sp
dp
р*
ds
d<
spur?
d*s
d3
dsp
p3
d*P
s/>3
d's
dsp-
d2p2
~ p*d~
d2sp
dp*
d*
Структура
линейная
»
треугольная
»
»
треугольная
правильная
плоская 1
»
»
» 1
несимметричная плоская
трпгональная
пирамида
»
тетраэдр
»
тетрагональная
плоская, кпад-
рат
»
тригональная
бнппрампда
с незанятой
в ершиной
п осноттнитг
Коор-
дина-
цион-
ное
число
фигурация
1 d2sp
4
5
dp3
d*p
d*
dsp3
d3sp
d1sp'1
d*s
d'-p*
d*p
d*p'
d*
Структура
неправильный
тетраэдр
»
»
тетрагональная пирамида
центр, атом и
и орш и но
тригональная
бипирамида
»
тетрагональная
пирамида
»
»
»
Пентагональ-
пая плоская
пентагопальн.
пирамида(центральный атом
в вершине)
Коор-
дина-
цион-
ное
число
6
7
8
фигурация
d1spz
d*sp
d*p
d3p3
dzsp'z
d*s
dzspA
d5sp
d*sp*
d*p*
dbp>
d4sp3
d5/?3
d?spl
Структура 1
октаэдр
тригональная 1
призма 1
» 1
тригональная
антипризма
смешанная
» 1
» 1
Zr
Реконфигурация
»
ТаР?-конфи- 1
гурация
»
»
«тригональный
додекаэдр»
антипризма
«гранецентри-
рованная»
тригональная
призма
1. Основной, как нам кажется, недостаток изложенных построений
заключается в том, что не учитывается влияние структуры кристалла
в целом на конфигурацию координационной сферы.
Поэтому будет более правильным непосредственно прилагать эти
результаты к молекулам и с некоторой осторожностью к молекулярным
кристаллам. Говорить же о молекулах K4Fe(CN)e, K3ZrF7 и т. д., когда
обсуждается структура ионов в кристалле K4Fe (CN)e и т. д., вряд ли следовало
бы. Тем более не следует без оговорок применять к структуре ионов
в кристалле соображения, относящиеся, собственно, к изолированным
ионам.
2. Слабым местом теории является то, что она пытается объяснить
факты, известные к моменту сё построения, но почти не делает попыток
предсказаний относительно веществ, известных химии, но не изученных
в структурном отношении.
3. Теория допускает для каждой конфигурации АХП по нескольку
структурных вариантов. В ряде случаев одному и тому же веществу
-могут отвечать разные конфигурации. В подобных случаях соответствие
206 ч. т. основы теории структур (гл. II
экспериментально найденной структуры какому-либо из вариантов является
весьма вероятным и вряд ли сможет послужить подтверждением теории;
предсказать же, почему, например, ZrF*~ и NbF^ предпочитают разные
структуры, теория оказывается вообще не в состоянии.
4. Небезынтересно, что наиболее «сенсационные» структуры [NbF7]-~,
[ZrF7]3", а также [OsF8] (двугранецентрированная тригональная призма)
ТеС14, J02F2 и др. вполне могут быть в большинстве случаев объяснены,
исходя даже из общих соображений, изложенных нами в § 91.
Наконец, многие выводы, сделанные в работе Кимбола, сильно
выиграли бы, если бы были поддержаны данными термодинамического
характера. Например, из этой работы вытекает, что молекулы Ni(CO)4 (тстра-
эдрическая) и Zn(GO)3 (треугольная плоская) вполне могут сущостновать,
так же как и Ge(CO)2 или Sn(GO)2; между тем термодинамическая оценка
этих систем (Ормонт [20]) (1946) показывает, что это бесспорно только
в отношении Ni(CO)4, если говорить о термодинамически устойчивом
состоянии.
Эти критические замечания ни в коем случае не означают
пренебрежение или недооценку с нашей стороны научного направления, ставящего
себе задачей установление формы валентных конфигураций на основе
теории групп и т. д.
Это направление заслуживает особого внимания советских
исследователей.
В равной мерс мы нисколько не противопоставляем этим теориям,,
например, сформулированные нами ранее (см. § 91) общие положения.
§ 96. Об исследованиях электронной плотности в кристаллах.
Рентгенографический Фурье-анализ *)
Современный рентгеноструктурный анализ дает возможность
характеристики распределения в кристалле электронной плотности (см.,
например. [40-43]).
В общей форме электронная плотность в кристалле может быть
выражена как трёхмерный ряд Фурье
H*.»,*) = j 2 S S FihkDc-^^+e*, (30)
h — oo к -oo |:--:-oo
где F(AAZ) —так называемая структурная амплитуда (см. [46]), квадрату
которой пропорциональны интенсивности рефлексов рентгенограммы, V -
объём элементарной ячейки.
Ввиду большого числа измерений и особенно расчётов, необходимых
для построения трёхмерного ряда Фурье, часто прибегают к измерению
отражения всех порядков от данной плоскости кристалла, изучая
распределение рассеивающего вещества перпендикулярно выбранной плоскости.
Тогда приходится иметь дело уже с одномерным рядом Фурье (см.
приведённую литературу).
При таком способе исследования можно описать изменения
электронной плотности в точках, лежащих на прямой, соединяющей центры двух
соседних атомов. Таким образом, были исследованы [48] изменения
плотности (I (х) в решётке NaCl между центрами ионов Nal и С Г" и устано-
*) Ишгду недостатка места мы стремимся, главным образом, указать читателю
на ряд работ, которые должны привлечь его внимание, если он пожелает более
подробно ознакомиться с этим чрезвычайно важным методом структурного исследования.
§ 96] ИССЛЕДОВАНИЯ ЭЛЕКТРОННОЙ ПЛОТНОСТИ В КРИСТАЛЛАХ 201
влены изменения распределения [> (х) с температурой вдоль некоторой
координатной оси.
Наконец, очень часто пользуются «двойным» или двумерным рядом
Фурье типа*)
р(у. *) = ^ 2,2, F«icos 2* (т + ~) (31)
-от — оо
(Л —площадь плоскости проекции, Z общее число электронов),
позволяющим описать электронную плотность в той или иной точке проекции
изучаемой кристаллической структуры на некоторую плоскость (например,
перпендикулярную оси изучаемой зоны).
Полученные результаты в графической форме в виде изоэлектронных
кривых (кривых одинаковой электронной плотности) являются
чрезвычайно наглядными и позволяют не только охарактеризовать положение
атомов, обнаруживающихся в виде максимумов, но и описать расстояние
между ними, границы атомов и распределение между последними
связывающих электронов.
В последнее время одно- и двумерные диаграммы электронной
плотности приводятся в большом количестве структурных исследований.
Важным дополнением и развитием метода явились работы Паттерсона \V\\
и Харкера[45], упростившего метод Паттерсона на основе учёта элементов
симметрии кристалла и практически исследовавшего на этой базе AffgAsS.,
и Ag8SbS3.
В СССР ряд весьма интересных работ был выполнен Н. А. Агеевым [29].
Беловым [41, 43] и Г. С. Ждановым [40, 50], А. И. Китайгородским [12] и их
сотрудниками.
Таким образом, мы устанавливаем, что исследования в области
рентгенографического Фурье-анализа представляют большой интерес с двух
точек зрения.
Во-первых, при определении характера и природы химической связи
и распределения электронной плотности в кристалле.
Во-вторых, для точного установления структуры кристаллом.
Из рис. 138,а видно, что в решётке каменной соли имеется резкий
минимум электронной плотности между атомами, свидетельствующей
о характере связи, близком к ионному. С повышением температуры этот
минимум сглаживается, кривая становится более пологой, связь
приобретает более ковалентный характер [48]. Впрочем, вопреки прежним
предположениям многих авторов, было показано, что, судя по диполь-
ному моменту молекул NaBr и KJ, исследованному с применением
молекулярного пучка, даже в состоянии молекулярного пара, подобные
вещества имеют связи, всё ещё близкие к ионным (с поправкой на эффект
поляризации).
В 1939 г. тем же методом было исследовано [49] распределение
электронной плотности в кристаллах каменной соли, алмаза, гекеаметилон-
тетрамина и найдено, что электронная плотность между ионами Na +
и С1~ падает практически до нуля в весьма широком интервале
расстояний между центрами ионов Na и С1~, т. е. также был подтверждён
ионный характер связи.
*) В общей форме двумерный ряд Фурье имеет вид
20S
Ч. Т. ОСНОНЫ ТЕОРИИ СТРУКТУР
[гл. II
xja. О 0.1 0,2 0,3 0,4 0.5 0,6 0.7 0,8 0.9 1.0
1 1-, i i i I i I 1 ■ , 1 i I i i .i i . . , i i.L i ,i i J
0 < ? .?
Pur. 138. Распределение электронной платности в решётке
NaCl.
§ 96) ИССЛЕДОВАНИИ ЭЛЕКТРОННОЙ ПЛОТНОСТИ В КРИСТАЛЛАХ 209
На рис. 138, бис показано распределение заряда в решётке NaCl,
спроектированное но (101) при 100° С, и вид решётки NaCl, если смотреть по
направлению, перпендикулярному (НО), т. с. вид на плоскость проекции (110).
Пунктиром на рис. 138, Ь обведена группа узлов Nar + С1~, отвечающая рис. 138, с.
Как видно из последнего, заряд распределён почти сферически
симметрично. Цифры на горизонталях представляют электронную плотность
в электронах на квадратный ангстрем.
ь
Рис. Ш. Распределение электронной плотности п решётке
алмаза.
За толщину проектируемого слоя выбирался наименьший период
идентичности в направлении проекции, чтобы интегралы, взятые по
области скопления электронов около ионов СГ соответственно Na+, давали
бы непосредственно число электронов данного иона.
Помимо этого, мы хотели бы отметить ещё одно обстоятельство. Если
исключить области с почти нулевой плотностью электронов, то сумма
радиусов обоих ионов окажется гораздо меньшей, чем «межионные
расстояния». Иначе говоря, ионы не сближаются в решётке до касания,
отталкивание начинается гораздо раньше.
210
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
|гл. II
Исследование алмаза (проекция на (110)) показало, что, в отличие
от NaCl, в нём нет ни одной точки на оси между двумя С — С-атомами,
где не имела бы места значительная электронная плотность (рис. 139, а
и 6), что свидетельствует о роли в этом случае обменных членов и о
наличии гомеополярной ковалентной связи.
Разрез распределения заряда по линии XY показан на рис. 139,а,
В заключение отметим, что проведённые специальные исследования
минимумов плотностей дали для NaCl 0,006 электрона
° / 1 1 1 \
в Аа —точка (— J и для С (алмаз) 0,050 эле-
ктрона в А —точка ( т7- ут Ь
В 1940 г. А. X. Брегер и Г. С. Жданов [50]
опубликовали результаты исследования с по-
~) 2 j~V мощью одномерных рядов Фурье, электронной
440. Распределение плотности в графите и нитриде бора (BN), имею-
электропной плотности и пщх аналогичную слоистую структуру. Изуча-
решётко нитрида бора. лось распределение электронной плотности
перпендикулярно плоскостям сеток (рис. 140).
Подробнее о значении рентгенографического Фурье-анализа для решения
проблем общей и неорганической химии см. Б. Ормонт [51] (1940).
§ 97, О зонной теории кристалла и её исходных предпосылках
В 1931 г. вышла книга Бриллюэна [52]. выдающаяся по значению для
последующего развития рассматриваемой теории, нашедшей продолжение,
в частности, в работах С. Т. Конобеевского [53], Зейца [54], Джонса [55]
и других авторов. Эти и другие исследования повели к созданию зонной
теории твёрдого тела, впервые осуществившей единый подход к кристаллам
разных типов химической связи.
При образовании кристалла электроны атомов, из которых он состоит (это
положение справедливо независимо от типа химической связи —ионного,
атомного, металлического), делятся на две группы: а) заполненные
электронные оболочки, которые вместе с атомным ядром составляют одно целое,
атомный «остов», и б) валентные электроны, обусловливающие связи между
атомными «остовами» (или, как мы будем их ниже называть, - узлами).
Узлы правильно расположены в пространстве, как это вытекает
из теории пространственной решётки кристалла, и создают периодическое
потенциальное поле. Электроны, входящие в состав узлов, связаны с ними,
экранируют заряд ядра, влияют на потенциал поля и значительно менее
на характер связи.
Валентные электроны, напротив, определяют характер связи. В
зависимости от последнего они более или менее свободны. В металлах
вероятность пребывания электрона в той или иной точке определяется характером
поля и имеет соответствующую последнему периодичность. Подобный
кристалл рассматривается как трёхмерное периодическое поле, в котором
движутся валентные электроны.
§ 98. О Я-пространствс и зонах Бриллюэна
Ниже мы остановимся самым кратким образом на вопросе о зонах
Бриллюэна с целью дать читателю хотя бы некоторое представление
о «АГ-пространство». В числе других была предложена схема, в которой
волновая функция электрона имеет в идентичных*) позициях в каждой
*) У Зейца неправильно: эквивалентных, I.e., стр. 272 (см., однако, § 41 и след.)-
§ 98] О К-ИРОСТРАПСТНЕ II ЗОНАХ НРНЛЛЮЭНЛ 211
элементарной ячейке одну и ту же амплитуду (см. рис. 137). Эта функции
имеет вид
где г вектор положения, компоненты которого г, //, z; ушк(г) имеет
трансляционную периодичность (т. е. отвечающую периодам идентичности:
Рис Г*1. Е(К) как функция А".
решётки), К является вектором, модуль которого А'= у, имеющим
компоненты Кх, Ки, Kz.
В простейшем случае /к постоянна, так что О/. — волновая функция
«свободного» электрона, для которой зависимость энергии от К
определяется выражением
£(Щ = £п1С, (34)
где /и —масса электрона, -Б(Я) —энергия электрона, имеющего волновое
число К. Это приближение отвечает представлениям старой квантовой
теории металлического состояния, причём свободный электрон движется
в постоянном поле Фока-Хартри (см. [56], стр. 139 и след.; 227 и след.)
и соответствующая функциональная зависимость изображается
непрерывной кривой (рис. 141, а) —параболой.
212
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
|гл. II
Граница I
кристаллах
Если же Хк не является вполне постоянной (т. е. электроны*)
не совсем свободны и взаимодействуют с атомными остовами), то кривая,
отображающая величины Е{К), не является непрерывной. Будучи
непрерывной на некотором, довольно широком интервале значений К, она при
определённых значениях K(KV K2) испытывает разрывы, ширина которых
зависит от степени постоянства ук (жирная кривая, рис. 141, Ь) и может
измеряться и долями вольта, и несколькими вольтами (1 эл.-в = 2'3 югал).
Нулевая точка системы рис. 141 отвечает- (центру) if-пространства.
В направлении разных пространственных векторов Ка, Кь, Кс места
разрывов могут лежать при разных значениях К (рис. 141, cud), причём
в некоторых случаях возможно перекрывание ^(Л^-функции для отдельных
направлений (рис. 141, с) и, значит, перекрывание зон (вещество является
проводником). В других же случаях перекрывание не имеет места
(рис. 141, а) —вещество, если оно имеет
чётное количество электронов,
оказывается изолятором.
При значениях Кх, К2 и т. д.
кривая пересекает границы концентрических
А-нолиэдров (имеющих центр в нулевой
точке /^-пространства), ограничивающих
данную зону.
Таким образом, непрерывные
значения кривой Е (К) лежат между
границами А'-полиэдров, а разрывы— на
поверхности последних.
Число электронных состояний в
каждом уровне изолированного атома, в
соответствии с принципом Паули, равно 2.
Сближая изолированные атомы,
имеющие валентные электроны на
определённых уровнях, мы наблюдаем такую
картину: по мере сближения атомов и
образования кристалла, вместо каждого данного
энергетического уровня электрона,
например, 2s (Li), существовавшего в
изолированном атоме, в кристалле возникает практически непрерывный спектр
близких подуровней (количество которых N является целым кратным количества
элементарных ячеек в кристалле, т. е. для кристалла видимых размеров
близко к числу Авогадро N), образующих зону (полосу) (рис. 142). Так
как одному s-уровшо отвечает два электронных состояния, то
количество энергетических состояний в такой полосе вдвое больше, чем
количество наличных 2$-электронов. В случае же щёлочно-земельных
металлов (.г-валентные электроны) число состояний равно числу электронов.
А'-пространство имеет следующее весьма важное свойство.
Разрывы AZ? определяют запрещённые значения энергии электронов,
отвечающая которым электронная волна не может проникнуть в кристалл.
Совершенно очевидно, что значения К, определяющие границы этих
запрещённых областей, отвечают условиям, при которых имеет место
отражение электронов от кристалла, согласно уравнению Вулъфа-Брэгга.
Это следствие имеет большое значение для теории структур (§ 100).
Зоны К-пространства получили название зон Вриллюэна. При
рассмотрении встречающихся в литературе чертежей, изображающих зоны
Бриллюэна в кристаллах, надо иметь в виду, что теория Вриллюэна
У робни 8
кристалле
Уро8т\ £
изолированных атомах
Рис. 1V2. Рюзникниюнне полосы
энергетических уровней при
образовании кристалла.
*) С точки зрения модели «полусвободных» электронов.
§ 98] О К-ПРОСТРАНСТВЕ И ЗОНАХ БРИЛЛЮЭНА 213
в описании А'-пространства в основном пользуется не обычной, а обратной
решёткой (§ 58), в связи с тем, что вектор волнового числа в обратной
решётке, обозначаемый /Г, обладает тем важным свойством, что функция
типа еЫШг имеет в этом случао ту же периодичность, что и решётка.
Поэтому, вместо, например, значений а или d, решётка характеризуется
Рис. I'i3. Двумерное распределение зон и кубической />-решётке (Со)-
а о а Ь
Рис. IV». ^онм Ьрпллкюпл для Рис. 1 i5. Зоны Ириллюпна для кубнчо-
кубической Ск-решётки. ской С^-рсшётки.
в теории Бриллюэна величинами - или -т (см. рис. 143 для двумерного
распределения зон в простой кубической решётке).
Форма первых двух трёхмерных зон Бриллюэиа для Ск- и (7с-рсшёток
показана на рис. 144 и 145 (сравни с рис. 141).
214
Ч. Т. ОСНОНЫ ТЕОРИИ СТРУКТУР
[гл. II
Обратная решётка для решётки Сг является центрированной, для
решётки Сс -гранецентрировашюй. Размер первой зоны (рис. 144) Ск равен
половине объёма куба, образованного пересечением показанных на рисунке
граней куба, объём второй зоны равен объёму куба. Кстати, форма второй
зоны очень хороню выявляет элементы симметрии вида Oh.
§ 99. Зависимость электропроводности кристаллов от характера
перекрышшия зон
Рассмотрим схематически вопрос о зависимости электропроводности
кристаллов от характера перекрывания зон в кристалле на примере (рис. 146).
На рис. 146, а показано (см. § 98)
типичное распределение энергетических
уровней с образованием запрещённых областей
(не заштрихованы) между зонами.
На рис. 146, d показано перекрывание
зон в случае типичного металла.
На рис. 146, b в элементарной ячейке
имеется нечётное количество электронов,
например, ls-электрон, нижняя полоса 1
заполнена наполовину. Это даёт
возможность переноса электронов с более низких
уровней сначала в полосу 1, а затем на
более высокие уровни —в полосы 2.
Кристалл в этом случае может проводить ток, если энергетический разрыв
между полосами 1 и 2 (Е[) невелик (щелочные металлы).
На рис. 146, с нижняя полоса 1 заполнена целиком.
Если разрыв Е1 достаточно велик, как на рис. 146, с, электрон из более
низкой полосы может попасть в более высокую, чем 1-я (например, во 2-ю),
лишь преодолев энергетический разрыв между более низкой и второй
зонами. Такое вещество должно быть изолятором.
а
Рис. I/if». Яаниспмоггь
электропроводности от характера
перекрывания зон (по Зейцу и
Джонсону), схема.
1 k 2 к 3
J ^г0 для графита
r0 u г0 Зля алмаза
а В
Рис. 1';7. Услопии перекрышшия зон при сближении атомом
металла (а) и углзрода (Ь).
Если же энергетический разрыв оказывается небольшим (рис. 146, е),
то бывает достаточно тепловой энергии, чтобы осуществить возбуждение»
электрона -переход его .из полосы 1 в полосу 2. Такие вещества начинают
проводить ток при некотором нагревании.
Наконец, энергетический разрыв Е\ вообще может не иметь места
(перекрывание полос, как на рис. 142). Вещество является проводником
(щёлочно-земельные металлы) (см. также рис. 147, а).
§ 100] ЗОННАЯ ТЕОРИЯ И СТРУКТУРА КРИСТАЛЛОВ 215
При сближении изолированных атомов возможна сложная картина
перекрывания зон (рис. 147). Так, при сближении атомов углерода,
на расстоянии 2,5А зоны перекрываются, на расстоянии же 1,5 А имеет
место громадный разрыв (рис. 147, 6). Поэтому кристалл алмаза, где
межатомные расстояния равны 1,54 А и заполненные уровни
распространяются до границ запрещённой области, является изолятором. В графите
имеется два рода межатомных расстояний 3,5 и 1,45А, в среднем 2,5А.
Этому расстоянию соответствует возможность перекрывания зон, что
влечёт за собой гораздо более высокую электропроводность графита.
§ 100. Зонная теория и структура кристаллов
Предсказание структуры решётки из общих соображений пока ещё
наталкивается на понятные трудности, поскольку энергия связи
вычисляется с точностью, редко превышающей 0,2 эл.-в^Ъ ккал/мсль, тогда
как разность энергии стабильной и нестабильной формы может быть
гораздо меньше.
Однако ряд общих выводов, важных для теории структур, тем не менее
может быть сделан именно в свете зонной теории. Остановимся только
на одном из них.
Задолго до тего, как Юм-Розери сформулировал своё правило, в
основной работе Бриллюэна[52] (1931, стр. 312 и след.) было обращено внимание
на вопрос, при каких предельных электронных концентрациях в решётке
металлов (Ск, Сс и др.) возможна диффракция электронов при данных
значениях \Е (см. § 98).
Тем самым были созданы все предпосылки к теоретическому
объяснению правила Юма-Розери, как только оно было сформулировано.
В целях доступности широким кругам читателей, мы попытаемся
изложить вывод этого правила в возможно более простой форме,
исходя не из обычно принятой схемы по работе Джонса, а
непосредственно из построений Бриллюэна. Для более детального ознакомления
см. цитированную литературу.
Мы знаем (§ 85), что электрону с энергией Е= :>- соответствует
длина волны
х = А = _а (17)
т. е. чем больше энергия электрона, тем меньше длина волны.
Так как в полосе «свободные» электроны образуют спектр с
различными энергиями Е, то кристалл можно охарактеризовать теми
значениями £mav максимальной энергии «свободных» электронов, при которых
данная структура нвляется устойчивой, а этим значениям будет отвечать
^min. т. е. минимальная длина волны электрона.
В самом деле, как мы знаем (§ 85), по достижении /, достаточно
малых значений происходит отражение электронов, в соответствии с
уравнением Вульфа—Брэгга
/Л = 2d sin 6. (19>
Из уравнения (19) следует, что отражение начинается при -in 0 == 1
И rfmax, Т. С.
n\ = 2dmsixj (35)
где dmax — максимальное межплоскостное расстояние в структуре.
216 4.1. ОСНОВЫ ТЕОРИИ СТРУКТУР [гл. II
Если, таким образом, энергия электронов больше, чем Етах, а длина
волны меньше, чем Xmin, т. е. X < 2dmax, структура не может быть
устойчивой: электрон отражается от кристалла. Можно показать, что
где Ne — количество электронов в данном объёме кристалла V,
у--количество электронов в объёме w одного узла (атома), т. е. (в духе правила
Юма-Розери) электронная концентрация. Для дальнейших рассуждении
приведём уравнение (36) к другому виду.
Разделим обе части равенства (36) на 2dmax и возведём их в 3-ю степень
"max
ix zamax V ЛТ У
f *min\a kW /no.
V«max / *Mamax
Как мы указали, диффракция электронов не может иметь места, пока
/min не станет меньше, чем 2dmax, т. е. «j™-- не станет меньше единицы.
^"тах
Посмотрим, чему равно значение ^-р-- для решёток разных типов.
В кубической гранецентрированной решётке Ск элементарная ячейка
с ребром и имеет 4 атома (узла), т. е. объём одного узла
w = %. (.39)
Теперь нам надо подставить в уравнение (38) наибольшее возможное
значение duui для решётки Ск* Как мы знаем, § 58, в кубической решётке
и наибольшие значения d должны, следовательно, иметь место для
плоскостей (100), когда йюо""-"— =в9 что и наблюдается в случае
примитивной кубической Р-решётки (Со). Однако у решёток более высокой
кратности, т. е., например, двукратно-примитивной центрированной, или
четырёхкратно-примитивной гранецентрированной, вследствие наличия
в решётке дополнительных позиций атомов наибольшими являются другие
межплоскостные расстояния (см. рис. 61).
Рассмотрим кубическую центрированную решётку (рис. 61, я, с, /).
Благодаря наличию атома в центре ячейки, в направлении ребра ячейки
возникают дополнительные, занятые атомами, плоскости (200)(^свдвое меньшим
расстоянием между ними, чем в случае (100). В этой решётке наибольшим
межилоскостным расстоянием будет следующее из возможных, согласно
формуле (39а), а именно, йШ) — --— .
В кубической гранецентрированной решётке (см. рис. 6J) возникают
занятые атомами дополнительные промежуточные плоскости (200) и (220),CV
В этом случае наибольшим является межплоскостное расстояние dlir
Поскольку наибольшее расстояние dmax в случае Ск наблюдается между
плоскостями (111), то
if Ш01 30HHUI ТЕОРИЯ II СТРУКТУРА КРИСТЛЛ1ЛОВ 217
и, следовательно, упитывая (39),
я
w = ——-. (41)
Подставляя в (38), получаем:
л
f *min \ Я* те 1,362 ,/0v
Счг.;;=пйгв г • (42)
Из уравнения (42) вытекает ряд важных следствий, в частности: если
каждый узел (атом) в среднем отдает кристаллу количество электронов
у< 1,362, то дробь в уравнении (42) больше единицы, и диффракция
электронов не должна иметь места (X > 2dmax).
Если же у > 1,362, то \тц\ < 2dmax и электрон должен отражаться
решёткой.
Теперь перейдём к кубической центрированной решётке Сс
Так как в элементарной ячейке Сс с ребром а находятся только
2 атома, то
«■=4- (43)
Так как наибольшими расстояниями в ячейке Сс нкляютсн */,,„ и, значит,
а
У 2
*>.« = -"- = -Ф- (44)
то, учитывая (43),
'Л
>v =-- -*■ • 21' (45)
С
;min V *'Z « ».'•«
2rfmax У f'Y T
(46)
Из уравнения (46) следует, что если у < 1,48, то дробь в уравнении (46)
больше единицы и диффракция электронов не должна иметь места.
Если теперь, учтя эти выводы, обратиться к правилу Юма-Розери
и распространить изложенные соображения на сплавы, то из (42) сразу
<тановитея очевидным, что если мы будем добавлять к Си(у=1) атомы
Zn, увеличивая среднюю концентрацию электронов, то структура Ск
останется устойчивой, пока концентрация электронов у не станет равной 1,362.
Это значение у и таком случае является той величиной электронной
концентрации, которая определяет границы существования а-фазы.
Как мы видели (§ 84), а-фаза латуни содержит до 38,05 атома Zn,
чему соответствует значение
у = **"■'* + »*»»£! = 1-3805. (47)
В других системах у колеблется также около значения 1,362. В равной
мере, распространяя вывод (46) на сплавы, мы приходим к заключению,
что кубическая центрированная р-фаза должна быть устойчива, пока у
не станет равной 1,48, тогда как, согласно правилу Юма-Розери, у = 1,50-
Можно также показать, что зонная теория позволяет установить
границы устойчивости у-фазы (и других фаз), причём в ячейке у-фазы
на 52 атома должно быть ~88 —90 электронов, т. е. у ^ ~ .
218 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
Естественно, что у вовсе не всегда равно количеству w электронов
атома, но может быть значительно меньше, чем w (что особенно чётко
выявляется в случае переходных элементов типа Fe, Co, Ni; см. § 84).
Уравнения (42), (46) и др. дают повод к очень важному для
кристаллохимии выводу, который мы запишем следующим образом:
Тип кристаллической структуры, в частности, в сплавах, не связан
непосредственно с отношением объёмов атомов или далее с отдельно взятым
количеством валентных электронов, но с возможными соотношениями
концентрации у в сплаве.
Мы должны однако подчеркнуть, что, несомненно, тонкая структура
атомных энергетических уровней, вызывающая химическую индивидуальность
атомои в образованном соединении должна вызывать и их
индивидуальные различии при образовании простых веществ и сплавов (см. § 84).
Поэтому элементы с одинаковым объёмом атомов (v) и даже в
некоторых случаях с одинаковым количеством валентных электронов
(например, Аи и Ag) могут образовывать сплавы с третьим элементом
(например, в системах Аи —Си, Ag —Си), отличающиеся по характеру
и структуре.
Тем более не следует сводить проблемы структуры и характера
химической связи, в частности, в металлах, к соотношениям радиусов
элементов.
В заключение отметим, что большим достоинством зонной теории
является намечающаяся, особенно в работах Конобеевского и Зейца,
тенденция к освещению с вышеуказанных позиций ряда проблем теории
реальных кристаллов.
Развитию зонной теории металлов много способствовали работы
С. Т. Конобеевского*) (1936), (1938), (1944). Последняя из упомянутых
работ представляет большой интерес, в частности, поскольку в ней
не только приведены интересные теоретические соображения, но и дано
критическое рассмотрение новых экспериментальных данных, обзор
Э- и у-структур. Развитием представлений зонной теории в области теории
полупроводников и т. п. физика во многом обязана также школе
А. Ф. Иоффе.
К сожалению, более подробное рассмотрение этих интересных работ
выходит за рамки данной книги.
§ 101. О реальных твердых телах
Теория идеального кристалла рассматривает пространственную решётку
как математический прообраз структуры твёрдого тела, с той разницей,
что кристаллографические точки заменяются в теории структур атомами
и молекулами, собственную симметрию и ориентировку которых приходится
устанавливать методами физики, например, путём изучения электронной
плотности к кристалле.
Этот подход допустим, например, для расчётов энергии решётки
идеального твёрдого тела и характеризует таковое в первом приближении.
Более детальное изучение показывает, что в действительности
кристаллы редко обладают вполне идеальной структурой.
Если даже отвлечься от того факта, что атомы в кристалле не
неподвижны и колеблются около своих центров тяжести, и рассматривать только
положенно последних, то оказывается, что в реальных кристаллах, в
том числе монокристаллах, как правило, наблюдаются дефекты
структуры, зависящие от условий возникновения и дальнейшей «жизни»
*) I. v.
$ 101] О РЕАЛЬНЫХ ТВЕРДЫХ ТЕЛАХ 219
кристалла. Кристалл не остаётся в течение своего существования
неизменным, его структурная характеристика непрерывно меняется; меняется
не только концентрация дефектов и их распределение, но иногда даже тип
решётки. Мы имеем все основания утверждать, что в кристаллах, как
правило, имеют место постепенно изменяющиеся флюктуации структурного
характера, т. с. микроразличия, иногда обнаруживаемые лишь путём
специального исследования интенсивности отражённых кристаллами
рентгеновских лучей или с помощью прецизионного определения межплоскостных
расстояний (с точностью до 0,0001 А).
Именно эти особенности реальных кристаллов определяют в ряде
< лучаев их поведение и свойства при практическом использовании.
Теория и, в частности, химия реального твёрдого тела создавалась,
по существу, в течение последних 10—15 лот, в качестве своеобразного
антитезиса ко многим положениям классической химии и химической
термодинамики.
Поэтому нашедшая широкое распространение, особенно в
результате работ германской школы физиков, теория идеального кристалла,
воспроизводимая, как правило, совершенно некритически в
учебниках и монографиях, нуждается в весьма существенных оговорках
(Б. Ф. Ормоит [38]).
Тем более сильное впечатление производит тот факт, что ещё в 1741 г.
М. В. Ломоносов сформулировал замечательный принцип, которым мы
начинаем § 70. Не имея здесь возможности более подробно остановиться
па теории реального кристалла, мы вынуждены отослать читателя к нашим
статьям [57] и [68]. Ограничимся здесь лишь некоторыми замечаниями,
изложенными в этом и следующих параграфах.
В работе [57] (1936) мы подчёркиваем тот общеизвестный факт, что
мотастабильнос состояние в кристаллах часто является стабильным
вследствие большой высоты потенциальных барьеров. Из этого элементарного
положения вытекает возможность длительного сосуществования разных
структурных форм без заметного перехода их друг в друга. Общеизвестным
примером является существование при комнатной температуре и атмосфер-
ком давлении термодинамически неустойчивого в этих условиях алмаза,
возможность сосуществования кристаллов белого и серого олова и т. п.
Ещё 20 лет назад в отдельных работах предлагалось установить не только
химическую, но и физическую чистоту вещества т. е. содержание в нём
кристаллов только одной структурной формы.
Мы считаем нужными дальнейшие поправки к классическим
воззрениям химии.
Мы считаем, что поскольку в одном и том же моно- или
поликристалле, кок бы мал он ни был, возможны флюктуационные
изменения структурных форм, то термодинамическое определение понятия
«раза» (гомогенная часть гетерогенной системы, ограниченная
поверхностью раздела) применительно к твёрдым фазам нуждается в серьёзных
^говорках: при наличии флюктуации исчезает физическая гомогенность
а внутри поверхности раздела может сосуществовать множество очагов
иных структур.
Это приводит к важному следствию, что нет двух одинаковых по
структуре кристаллов данной модификации данного вещества и что, вопреки
основным установкам термодинамики, приходится интсрееоваться не только
начальными и конечными, но и промежуточными состояниями системы,
которые, вследствие упомянутых выше причин, могут оказаться
практически конечными состояниями.
Отсюда вытекает, что существование аллотропических форм и
полиморфизм следует рассматривать не как частные случаи, но как правило,
220 ч. I. основы теории структур [гл. II
которому лишь в виде исключения могут не подчиняться отдельные
вещества. И далее в случае существования одной структурной формы мы
должны в общем случае считаться с возможностью возникновения для
разных образцов вещества множества структурных «вариантсв», в общем
подчиняющихся основному структурному мотиву данной формы.
Этот процесс возникновения вариантов структурной формы связан
с образованием разного рода так называемых «дефектов», которые в
действительности должны рассматриваться как неотъемлемая особенность
реального кристалла.
Были предложены различные схемы систематики структур реальных
кристаллов. Мы неоднократно подвергали эти точки зрения критике
и в следующем параграфе приведём принятую нами простую схему.
§ 102. Типы реальных кристаллов
На рис. 148, а и Ь изображена идеальная структура монокристалла
и поликристалла. На рис. 148, а показана структура идеального
монокристалла. Пунктиром обозначены междуузлия или, как можно их назвать,
узлы условной решётки. Эти узлы в структурах смещения и внедрения,
см. ниже п.п. 5 и 7, заполняются атомами, В реальном кристалле возможны
следующие отступления от идеальной структуры:
1. Структуры разрыхления возникают в случае, если в
материальной решётке, состоящей из однородных узлов а (например, решётке
простого вещества), часть позиций не занята атомами (рис. 148, d).
2. Структуры вычитания возникают в случае, если в
материальной решётке имеется не менее двух типов структурных узлов а
и р, например, V и N в решётке нитрида ванадия (исследованного нами
совместно с В. А. Эпельбаумом [6] в 1940— 1947 гг.), причём не занята часть
узлов а или р (рис. 148,с) или тех и других (рис. 148, /). В таких
структурах, естественно, меняется соотношение узлов а и р и химический
состав вещества.
3. Структуры замещения (твёрдые растворы 1 го рода)
возникают, если в материальной решётке, состоящей из однородных узлов ос,
отдельные позиции замещены посторонними узлами р (рис. 148, h) (см.
закон Вегарда). Сюда же можно причислить структуры (состоящие из
узлов ос и р), в которых часть позиций а замещеяа узлами р (рис. 148, g).
Наконец, примером структур замещения и вычитания является
изучавшаяся автором, совместно с В. А. Эпельбаумом и А. X. Брегером, система
VjNj.Oy (где х + г/^1, например, V\ (No^sOo.fja), "° может быть больше
и меньше 1)(см. Эпельбаум и Ормонт [6] (1940—1947гг.)).
х и у могут расти за счёт друг друга во всём интервале от 0 до 1
(см. Эпельбаум и Брегер [6] (1940)).
4. Структуры деления обнаружены во многих случаях, когда
один из видов структурных узлев, например ос, содержится в формуле
вещества в количестве, недостаточном для заполнения всех
предоставленных ему позиций и вынужден распределяться по этим позициям
статистически (рис. 148, г).
Например, в ос Ag2HgJ4 три атома металла (2Ag + lHg) статистически
распределяются по 4 мостам.
5. Структуры смещения (их можно было бы назвать
структурами Френкеля) возникают в случае, если атом смещается с позиции
в узле материальной решётки в позицию в междуузлии. Позиции в между-
узлиях мы можем отнести к некоторой условной решётке, которая
окажется реально представленной, если количество смещений будет
достаточно велико (рис. 148, с).
§ 102]
ТИПЫ РЕАЛЬНЫХ КРИСТАЛЛОВ
221
+4i
222 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР |ГЛ. 11
6. Структуры чередования (переменные структуры). Как мы
знаем, возможны плотные упаковки структур по кубическому или
гексагональному законам. Несомненно, в различных кристаллах не
исключены, наряду с правильным чередованием подобных слоев,
неупорядоченные чередования.
Переменные структуры детально исследовались в СССР II. В. Бело-
ным[12], 3. Г. Пинскером (галогониды кадмия), Г.С.Ждановым (SiC) и др.
При малом количестве нарушений рентгенографическим путём их
обнаружить не удаётся. Такие нарушения особенно возможны в
структурах с более атомными (ковалентными) связями, например, ZnS
(сфалерит и вюрцит), SiC и др.
7. Структуры внедрения (твёрдые растворы 2-го рода).
Если в предыдущих двух типах структур «дефекты» условной решётки
зависели от дефектов материальной решётки, то принято утверждать, что
в структурах внедрения нарушается в основном только условная решётка.
При этом позиции узлов последней замещаются либо посторонними
структурными узлами (нормальный случай, подробно исследованный Хэггом),
(рис. 148, к), либо узлами одного из сортов, входящих в состав
материальной решётки.
Согласно всей современной литературе, структуры внедрения
образуются примесями атомов Н, N, О и т. п. вследствие малых размеров этих
атомов, могущих проникнуть в междуузлия, например, решётки
металла.
Этот механизм ещё в 1936 г. подвергался критике со стороны авторат
которым было подчёркнуто, что, пожалуй, решающую роль в этом
процессе играет квантовая характеристика валентных электронов атома
металла. Например, гидриды, имеющие структуру внедрения, образуются
^-элементами (имеющими ds-валентные электроны типа Ti, Zr и др.), но
не образуются sp-элементами (типа Sn, Pb и р) при одинаковых радиусах
атомов (сравни Sn (1,56 А) и Ti (1,48 А) или Zr (1,61 А)).
Помимо всех этих «дефектов», внутри элементарной ячейки (мы
называем их «дефектами 1-го рода) возможны дефекты 2-го рода (поры,
трещины и т. п.) (рис. 148, 6).
§ 103. О влиянии примесей на образование структуры
Так же как незанятые места, примеси оказывают различное влияние
на процесс образования структур.
Рассмотрение значительного экспериментального материала, а также
собственные экспериментальные данные приводят нас к выводу, что примеси
влияют на образование или необразование аллотропических форм и вообще
на ход полиморфных превращений и притом как в кинетическом смысле
(затормаживая или ускоряя переход метастабильных форм в стабильные),
так и в термодинамическом (изменяя знак AF для реакции перехода одной
формы в другую).
Поэтому грубо ошибочными мы считаем работы многих школ
зарубежных физиков, изучавших подробные превращения без детального
химического анализа на содержание примесей.
Кроме того, как известно наличие более значительных количеств
примесей может вызвать образование иных валентных углов, ввиду замены
одного центрального атома координационной сферы другим, и вообще,
ввиду изменения распределения электронной плотности в решётке.
Примеси влияют на характер химической связи, на
электропроводность и магнитные свойства веществ и, наконец, на величины
межплоскостных расстояний в решётке.
§ 1041 о применимости законов пру и длльтонл в неорглническ. химии 2Z\
§ 104. О применимости законов Пру и Дальтона в современной
неорганической и общей химии
Успехи современной кристаллохимии в деле исследования фаз
переменного состава, в особенности выдающиеся работы школы
основоположника физико-химического анализа Н. С. Курнакова давно показали, что
в твёрдом теле возможны дробные соотношения компонентов АЖВ/У, в
нарушение законов Пру и Дальтона (см., например, Е. С, Макаров [711).
В неорганической же химии подобные вещества обычно рассматривались
как твёрдые растворы, хотя в физическом отношении трудно было провести
границу между ними и обычными химическими соединениями.
Во всяком случае все современные курсы и монографии по
неорганической химии излагают законы Пру и Дальтона как фундаментальные законы
химии, без каких-либо поправок.
Мы считали, что такое положение не отвечает современному
состоянию физики и химии твёрдого тела, и позволили себе в 1946 —1947 гг.
сформулировать следующие положения (см. также [65]).
1. Дальтонидный или бертоллидный характер соединения определяется
не только химическим характером образующих его атомов, но и
агрегатным состоянием вещества. Во многих случаях последнее играет даже
решающую роль, меняя характер силового поля, которое формирует соелк
и структуру вещества.
2. Химические соединения (и простые вещества), находясь в
парообразном состоянии, т. е. в виде молекул, должны иметь в соответствии с
механизмом возбуждения валентности, как правило, состав (и плотность
пара), который подчиняется дальтонидным соотношениям (атомные
концентрации характеризуются целыми числами).
Например, НС1, СС14, SFe, PC15, СО и др.
Если анализ смеси даёт дробные соотношения, это обусловливается,
как правило, наличием нескольких сортов молекул в разных
концентрациях: zPCl6 + ?/PClg + zCl2 или xS8 + t/S4.
3. Химические соединения (и простые вещества), образуя реальный
кристалл, как правило, приобретают возможность менять соотношение
концентраций как основных атомов, слагающих вещество, так и
примесей (а кроме того, очень часто бывают вынуждены изменять состав
структурных узлов, см. Б. Ормонт [65] (1947)) и являются бертоллидамив
широком смысле слова.
Степень колебания соотношения атомных концентраций зависит: от
характера химической связи, от характера структурных узлов и типа решётки,
косвенно от температуры и давления. В очень большой мере она
определяется генетическим типом кристалла, т. е. условиями его
возникновения и существования. (См. Б. Ф. Ормонт (1936) [57].)
В частности, если кристалл обладает молекулярной решёткой,
соотношения концентраций основных атомов могут оставаться дальтонидными,
тогда как соотношения концентраций основных атомов, с одной стороны,
и примесей — с другой, оказываются, естественно, бертоллидными,
например, (СС14, примесь бензол). В случае типичных ионных кристаллов
соотношения концентрации основных атомов К+ : А~ также имеют
тенденции быть дальтонидными в той мере, в какой (выражаясь в терминах,
вытекающих из предельно электростатической модели) в веществе К£+А£~"
отношение зарядов ионов q/p является целым числом (ибо из формулы
вещества вытекает, что xjy = qjp).
В связи с этим, например, образуются соединения типа CaF2 или
NaF с отношением индексов х: у, весьма близким к 2
(соответственно 1).
224 Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР [ГЛ. II
Хотя электростатический, в основном, характер связи сильно затрудняет
попадание анионов в решётку катионов (и наоборот и т. и.) с
образованием структур замещения, вычитания с дробными коэффициентами, не
следует полагать, что подобные явления в типично ионных кристаллах
не могут вообще иметь места, хотя бы в весьма малой мере. Например,
с удалением части ионов одного знака, скажем, с переходом от GaF2
к составам, более близким к CaF, должно иметь место
перераспределение электронной плотности в кристалле, как целом. Этот процесс
облегчается, если один из ионов может иметь переменную валентность,
с ним особенно приходится считаться в случае tf.v-элементов.
В какой мере эти тенденции реализуются, зависит от
индивидуальных свойств системы. Ибо, если, например, характер связи становится
более ковалонтным или более металлическим, например, FeO, FeS, VN
или TiN (а такие случаи наиболее распространены), то атомы могут
занимать «отведённые для них» узлы неполностью и не в строгом порядке.
Более того, в решётке возможно существование атомов одного и того же
элемента, например, Fe, с различным зарядом. Требование целочислен-
ности отношений атомных концентраций уже не вытекает из
соотношения зарядов ионов-партнёров и тем самым не играет в такой мере
регулирующей роли. Многочисленные виды микродефектов 1-го рода
становятся возможными, а с ними —различные дробные соотношения
коэффициентов х и у, характеризующих основной состав вещества. В разных
модификациях одного и того же вещества, например, aMn^Sy (тип Со/о),
ЭМпгЯ,, (тип Сг/т)у уМп^ (тип Нт/т) существуют разные условия
для соотношения индексов х: у (а следовательно, для заряда ионов
Мп в решётке), которые в первом приближении равны единице (MnS).
4. Таким образом, образование дальтонидов должно наблюдаться,
как правило, у веществ, находящихся в парообразном состоянии (и отчасти
в виде молекулярных и чисто ионных кристаллов).
Образование бертоллидов в широком смысле слова, т. е. соединений
переменного состава с дробными отношениями атомных концентраций,
должно наблюдаться, как правило, в реальных кристаллах (особенно
не имеющих молекулярной или чисто ионной решётки). Степень
колебания концентраций зависит от условий возникновения кристалла и
характера связи в последнем (определяемой, в свою очередь, строением
атомов-партнёров и их способностью к возбуждению переменной
валентности).
При обсуждении нашего доклада на эту тему отдельными
оппонентами высказывалось мнение, что основным критерием для решения,
является ли вещество соединением или нет, должна служить
термодинамическая характеристика вещества, и предлагалось сохранить термин
«соединение» лишь за теми из них, которые являются термодинамически
устойчивыми. При такой концепции существовал бы повод не считать
соединениями многие вещества с дробными коэффициентами.
Как мы указывали в прениях, такая точка зрения должна быть
отвергнута, ибо, принципиально говоря, каждое соединение, каждая
структурная форма (модификация) имеет но отношению к каждой
возможной реакции своё термодинамическое поле устойчивости при
определённых значениях р и 7\ и притом, будучи устойчивым по отношению
к одной реакции, вполне может оказаться неустойчивым в отношении
другой, не говоря о таких веществах, как, например, NH4N03 (I) (который
в определённых условиях стабилен, например, в отношении перехода
в модификацию (II), но термодинамически совершенно нестабилен в
отношении распада на летучие компоненты (N2, H20 и т. п.). Между тем,
вряд ли кто-либо откажется считать NI^NO, химическим соединением.
§ 105| 1ШИЯНИЕ ТЕРМОДИНАМИЧЕСКИХ ФАКТОРОВ 225
Сказанное в полной мере относится и к органической химии, которая
справедливо считает соединениями даже такие вещества, как
нитроглицерин.
К сожалению, от бликованные в литературе результаты
структурных исследований были получены без учёта необходимости строгого
анализа не только возможных примесей, но и соотношения
коэффициентов в химической формуле.
Будучи вынужденными пользоваться при создании этой книги
имеющимися литературными данными, мы хотели со всей настойчивостью
подчеркнуть, что считаем их далеко не всегда удовлетворительными,
и притом не только, и даже не столько с методической:, сколько с
принципиальной стороны, что нашло выражение, в частности, в отдельных
замечаниях в тексте.
§ 105. О зависимости структур комплексных соединений
от термодинамических факторов
Чрезвычайная близость в энергетическом отношении различных
структур и роль кинетических факторов, о которой мы говорили в § 68,
привели к том}', что исследование влиянии факторов чисто
термодинамического характера на структуру
II
а
1
~[Ин,0)п]%сг
[LCI]
Рис.
-лА
♦*
• п Н?0Э
[Li{H20)n]CL
IV.>. Кругоной процесс Л« I образо-
НП1П1Н комплектного соединении.
мы вынуждены,
кристалла далеко не всегда
приводит к полезным заключениям.
В результате возникли
неправильные представления о
невозможности использования
соображений этого рода для предвидения
возможных структур веществ, особенно
сложных, комплексных соединений.
,В связи с тем, что в основу нашей
систематики комплексных
соединений (гл. ТХ и X) легли не только
структурные, но и термодинамические соображения,
хотя бы коротко, на последних остановиться.
Из наиболее часто встречающихся типов реакций образования
комплексных соединений особенный интерес представляют два тина:
1. Взаимодействие вещества с ионной (или атомной) решёткой и
вещества с молекулярной решёткой.
2. Взаимодействие двух веществ с ионными (или атомными) решётками.
Оба эти типа реакций, особенно первый, были в своё время
рассмотрены в теории комплексных соединений под иным углом зрения —
с целью выяснить связь между упругостью диссоциации соединения
и энергией расширении решётки. Нам показалось полезным
интерпретировать аналогичные процессы в разрезе, интересующем теорию структур.
Рассмотрим круговой процесс № 1 для реакции типа [LiClJ
= [Li(H2<)),l].Cl + <) кнал.
Из рис, 149 следует, что
Q = I 'к — U — пк + со :-= Кт - - пк + <•> ккал
пН,()ж =
== Q-}- А(^ + пк ккал,
(31)
где U — энергия решётки простой соли, Uk — комплексной, к — энергия
сублимации вещества с молекулярной решёткой (например, воды),
о> — энергия образования комплексного иона. А// величина всегда отри-
226
Ч. I. ОСНОВЫ ТЕОРИИ СТРУКТУР
[гл. II
цательная (так как радиус комплексного иона больше, чем исходного
простого, то Uk < U).
Если реакция протекает между конденсированными фазами, например,
твёрдыми, то в выражении свободной энергии
AF = АС/ - T±S (T = 298°), (32)
как правило, TAS< 1 ккал/гр. атом, поскольку AS обычно меньше трёх
энтропийных единиц на г р. атом (см., кстати, [58]).
Для того чтобы \F было безусловно отрицательным, надо, чтобы
реакция между твёрдыми веществами протекала с выделением тепла
большим, чем 1 ккал/гр. атом.
В нашем примере Q должно быть > 15 ккал.
Но энергия сублимации веществ с молекулярной решёткой >,, равная
в среднем 8 — 10 ккал! моль, иногда достигает 1 — 2 стал и редко—12 — \Ъккал.
При п « 4 — 6 п\ ъ* 40 ккал.
Энергия расширения At/ в большинстве случаев, как легко показать,
должна быть примерно равна 25 — 30% от энергии решётки простой соли,
т. е. ^50 ккал.
Отсюда следует, что для кристаллосольватов энергия комплексо-
образовавия о>, достаточная для образования термодинамически
устойчивого кристалла, содержащего координационные сферы комплексного иона,
примерно равна
о) ^ 40 -f 50 -1-15 ^ 105 ккал (33)
(или ^20—25 ккал/связь),
т. е. измеряется несколькими десятками калорий —до сотни калорий.
Если летучий компонент находится в парообразном состоянии, то, как
легко показать [20], при значениях Q меньших *), чем п X 10 ккал (где н —
число граммолекул Н20, NH3 и т. п.), комплексное соединение
термодинамически неустойчиво. Непонимание этого обстоятельства приводит
многих авторов к ошибочным утверждениям. Подобная ошибка была
допущена, например, в переводе классического труда А. Вернера [60], где
в качестве теилот образования соединений типа [Ca(NH3)e]J2 указаны
величины порядка 13 ккал. В действительности — это 13 ккал ва 1 моль
NH3, а для указанного соединения теплота образования достигает
80 ккал.
Таким образом, если один из компонентов находится в парообразном
состоянии, то значение АС/ остаётся тем же, величина Д@ ъ п X 10 ккалъ
^40-60 ккал, в среднем 50 ккал и
а) ^ 40 + 50 ^ 90 ккал. (34)
Как показывают расчётные и экспериментальные данные, такая
величина ш вполне достижима. И в самом деле, в структурах типа
KJPtClJ, [Ni(NH,)e]CL tiiiiSB#61, KaGeFe тип Л1Э, Са[С03], Na[NOa]
тип SBGllt Zr[P207] тип SB KQ1 — комплексные островные, изолированные
ионы реально существуют.
Рассмотрим теперь круговой процесс № 2 для реакций типа
LiCl -f NaCl = Na [LiCJ2] + Q.
*) В большинстве случаев.
§...1051
ВЛИЯНИЕ ТЕРМОДИНАМИЧЕСКИХ ФАКТОРОВ
227
■'- к» — A U + (•> кк/(л
(35)
Из рисунка 150 видно, что
Q- Uk-U'-U'
И
кч-- Q-]-\U ккал. (36)
Посмотрим, какие значения имеют величины, стоящие в правой части
уравнения (36), если кристаллическая структура продукта реакции
является устойчивой.
С термодинамической точки зрении, учитывая значения энтропии реакции,
последняя должна быть экзотермической, причём Q должно быть в среднем
больше -f 4 ккал, соответственно числу грамматомов, участвующих в
процессе. (Для МХ2- MX Q должно быть > 5 ккал.)
Если не требовать от кристалла образования координационной сферы
иона, но распределить, например, статистически оба типа
L! *CUNa4*CL
Na+*[LiCtJ"
-U'
-If
[LiCl] + [NaCl]"
►Q
N,
комплексного
ионов Li+ и Na+ в структурных
узлах катионов, или меньшие ионы
в незанятых промежутках между
большими ионами, то требование
уменьшения энергии системы на \ — 5 ккал,
т. е. на 2 -3"0 от исходной энергии
решёток, является довольно
скромным и в ряде случаев вполне
реализуемым.
Напротив, если заставить
кристалл образовать комплексную
координационную сферу, то тогда
энергия комплексообразокаиия п> должна не только обеспечить очень скром
величины Q, по прежде всего компенсировать резко отрицатель
значения А/7, достигающие весьма значительных величин
\Г ^ — ((/,. — V) U" ^ 50 ккил—НО икал ^ —220 ккал
*" [Na[LjCLj]
Рпс. 1."»(). Круговой процесс Х- 2 обра-
аишшпп комплектного соединении.
ные
ные
(37)
(о должно быть > 220 -j- Q ккил и вообще измеряться
Мы видим, что теперь обеспечить теп.твой эффект
и, следовательно,
сотнями калорий
4 — 5 ккал вовсе не так просто: надо сперва компенсировать весьма
большею разность энергий решёток А£\
Так как лишь немногие ноны способны выделить достаточно большую
энергию связи о> при образовании островных координационных сфер,
в результате реакций рассматриваемого тина, то такой процесс должен
иметь место гораздо реже, чем это до сих пор принято было думать
(см. гл. IX). При наличии к тому возможности чаще возникают сложные
соединения. Кроме того, природа использует очень интересный обходный
манёвр: в структуре образуются координационные сферы не островного,
а цепного или сеточного типа (см. гл. IX), причём; часто периферические
атомы принадлежат нескольким координационным сферам. В этом случае
«забота» о выделении надлежащей энергии связи распределяется на
несколько центральных атомов.
В ряду
Са [COJ -> Na [NOJ -» Са [S041 -* СаТЮ, -» AUIgO, -> ВР04
первые три члена имеют структуры с реальными комплексными ионами
состава, указанного в скобках. Напротив, в СаТЮя, химическом аналоге
СаС03, изолированных ТЮ3-нонов нет, но обнаружены лишь октаэдры
ТЮв, имеющие общие» вершины.
Учёт этого обстоятельства заставил автора в своё время предложить
структурно-координационные формулы комплексных соединений, см. §221.
228 i. основы tkopiih структур
ЛИТЕРАТУРА К ГЛАВЕ II
А. Общие рувоводотва
1. Э. В. Шнольский, Атомная физика (1949).
2. В. Н. Кондратьев, Структура атомов и молекул (1940).
3. Я. К. С ы р к и н и М. Е. Д я т к и н а, Химическая связь и строение молекул,
гл. I—VII (1946).
4. Я. И. Френкель, Кинетическая теория жидкостей, гл. I и II (1945).
5. С. Т. К о нобеевск и и, Уч. зап. МГУ, в. 74, 13 (1944).
G. Б, Ф, Ормонт, Химия и строение материи, ОНТП (1934); Кристалл и его
константы — Усп. физ. наук XVI, 1001 (1936); ЖФХ сборник (1947), стр. 5s.
7. Ф. .r3e/iu, Современна» теория твердого тела (1:)49).
8. Ф. Зейц, Физика металлов (1947).
9. Р. Эванс, Введение в кристаллохимию (1948).
10. Л. Паулин г, Природа химвческой связи (1948).
Б. Сноски в тексту
1. Б. Ф. Ормонт, Химия и строение материи (1934), стр. 173 и след.
Kossel, Ann. d. Phys. 4!>, 229 (1916); G. N. Lewis, J. Am, Ch. Soc. 38, 768 (1916);
Valenz mid dor Ban der Atome und Molekule (1927).
2. В. Д. Кузнецов, Физика твёрдого тола (1937), стр. 263 и след.
:i. М. Бори и Гепперт-Мейор, Теория твёрдого тела, ОНТИ (1938).
4. А. В. Ш у бн и ко в, Е. Е. Флинт и Г. Б. Боки й, Основы кристаллографии
(1940), стр. 227.
5. См. Л а вес, Успехи химии, VII, 574 (1938). Naturw., 25, 705, 721 (19*7).
6. В. А. Эпельбаум и А. X. Брегер, ЖФХ XIV, 1600, 1G02 (1940);
В. А. Эпельбаум и Б. Ф. Ормонт, ЖФХ XXI, 3 (1947).
7. Паулинг, 1. с. (I9'i8), § 48а, а также The Nature of the Chemical Bond (1945).
8. А. Ф. Капу сти некий, Z. Krist. A., 86, 389 (1933); ЖФХ V 59, 64 (1934).
9. А. Ф. Капустин с к и й, Acta physicochim., XVIII, 370, (1943).
10. Ю. В. Хода кон, Элементы электростатической химии, ОНТП (1934).
П. Z. Pinsker, Acta physicochim. XX, 121 (1945).
12. Н. В. Белов, Структура ионных кристаллов и металлических фаз. Изд. АН СССР
(1947).
13. Ю. В. Хода ков, Элементы электростатической химии, ОНТИ (1934), стр. 21
и след.
14. См. выше, Общие руководства, п. С).
15. Bedreatf, <- R- Й15, 537 (19'i2).
16. Stuart, Z. phys. Ch. (В) 38, 155(1937).
17. Б. Ормонт, ЖФХ XI, 274 (1938); Acta physicochim. XI, 839 (1938).
18. Я. К. Сыркини М. Е. Дяткина, Химическая связь и строение молекул,
(1946) стр. 9.) и след.
19. Б. Ормоит, Acta physicochim. I, 745, 752 (1935); II, 533, 547 (1935). Успехи
химии V, 793 (1936).
20. Б. Ормонт, Acta physicochim. XXI, 409, 413, 741 (1946).
21. Б. Ормонт, ЖФХ XI, 274 (1938); Acta physicochimia VIII, 811 (1938); IX, 885
(1938).
22. Уэллс, Структурна»-неорганическая химии (1949).
23. Evens a. Lister, Trans. Far. Soc. 34, 1358 (193S).
24. Б. Ормонт, ЖФХ XI,274(1938); Acta physicochim. XI, 885(1939); IX, 885(1*38).
25. Copley, Foster, В ay la г, Chem. Rev. 30, 227 (1942).
26. Eastes a. Burgess, J. A. Ch. Soc. 64, 1187 (1942).
27. Barb ago a. Fe melius, J. A. Ch. Soc. 65, 148k (1943).
28. Б. Ормонт, Acta physicochim. XII. 411 (1940); XIX, 571 (194'*); ЖФХ XIX,
167 (1945).
[гл. JI
ЛИТЕРАТУРА К ГЛАВЕ II
229
29. См., например, Н. В. Агеев л Гусева ЛАН (\) 4 (1945); К. В. Агеев,
Химии металлических сплавов, Изд. АН СССР (1947).
30. Б. Ормонт, Acta physicochim. Yl, 40Э, 427; V, 405 (1936).
31. Д. К. Горалевич-ЖРФХО 58, 1129 (1926); 62, 1166 (1930).
32. Г>. Ф. Ормонт и Б. А. Петров, ЖФХ VIII, 665 (1936); ЖОХ VII, 1690(1937);
VIII, 5>3 (1938); Г). С. М и ц е л о в с к и ii и Б. Ф. Ормонт, ЖОХ X, 161 (1940).
33. П. В. Гогор итпили, В. И. Кульгина, O.K. Звягинцев, ЖФХ IX,
1961 (1939).
34. Krepelka a. Blabolil Zbl, II, 671 (1938).
35. S с h о 1 d е г, Z. angew. Ch. 49, 255 (1936).
36. См. А. А. Гринберг, XX лет советской науки, Природа, стр. 61 (октябрь 1937).
37. Schneider u. Esch, Z. Elektroch. 49, 55 (1943).
38. Б. Ормонт, Основы теории структур (подготавливается к печати).
39. G. Kimball, J. Ch. Phys. 8, 188 (1940).
40. H. Г. С в п а с т ь я н о в, Г. С. Ж данов и М. М. Уманский, ЖФХ XXII,
1153 (1948).
41. См., например, Н. В. Белов, Г. Б. Б о к и и и Л. А. Попова, ПАН (л),
249 (U47).
42. См., например, А. И. Китайгородский, ПАИ (л), 259 (1947).
43. И. В. Белоп и В. П. Бутузов, ДАН 54, 721 (194С).
44. Patterson, Z. Krist. 9!), 517 (1935).
45. D. Harker, J. Ch. Phys. 4, 381 (1936).
46. Г. С. Жданов, Основы рептгенонского структурного анализа (J940).
47. Б. Я. Пи нес, Лекции по структурному анализу. Харьков (1937).
48. James a. Firth, Proc. Roy. Soc. (A) 117, 62 (1927).
49. Б рил ль, Гримм, I1 о р.май, Петере, Успехи химии IX, 419 (1940).
50. А. Брегер и Г. С. Ж д а и о в, ДАН 28, 630 (1940).
51. Б. Ормонт, О применении рентгенографического Фурье-анализа для решения
проблем общей и неорганической химии, Усп. Химии IX, 413 (1940).
52. Ьриллюзн, Квантовая статистика. ДНТВУ. Харьков (1934).
53. С. Т. К оно беев ск и й, Ann. d. Phys. 26, 97 (1.136). J. Inst. Met. No. 2, 165
(1938); Учёные записки МГУ, Физика 7-*, 13 (1944).
54. Winner п. Seitz, Phys. Rev. 4-, 803 (1933); 4l$, Г.09 (U34).
55. Jones, Proc. Roy. Soc. (A) 144, 225 (U34).
56. Ф. Мепц, Современная теория твердого тела (1949).
57. Ормонт, Усп. физ. наук XIV, 1001 (1936); ЖФХ, сборник (1947), стр. 58.
58. У лих, Химическая термодинамика (1933), стр. 173.
59. L. Pauling, Chem. Eng. News, 25, 2970 (1947).
01. А. В е р н е р, Новые воззрения в области неорганической химии, Химтеорсч.
(193»), стр. 256.
61. Б. В. Некрасов, Курс общей химии (1946), стр. 20.
62. Seaborg, Nat. 157, 307 (1946).
63. Seaborg a. Wahl J. Am. ch. S. 70, 1128 (1948).
64. Perrier а. В ogre, Nat. 157, 24 (1947).
65. Б. Ф. Ормонт, ЖФХ XXII, 140) (1948); ЖОХ XIV, 210 (1949).
6 i. См., например, II е и н и, В. Ф , \ с к и Ш е р м а н, Квантовая теория валентноеi и,
ГОНТИ (1938), стр. 136—137.
67. Mel lor, Chem. Rev. ИЗ, 137 (1943).
68. Б. Ф. Ормонт, ЖФХ XXI, 569 (1947).
69. В. Н. Кондратьев, Структуры атомов и'молекул (1948).
70. В. Ф. Ормонт, Akla physicoch. XI, 911 (1939); ЖОХ X, 158 (1940).
71. Б. С. Мака рои, Строение твёрдых фаз с переменным .числом атомов в
элементарной ячейке, Изд. АН СССР (1947),
ЧАСТЬ ВТОРАЯ
СТРУКТУРЫ НЕОРГАНИЧЕСКИХ
ВЕЩЕСТВ
ГЛАВА III
ТИПЫ СТРУКТУР НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
§ 106. Введение. Конфигурации координационных сфер
Принятой в («Slruklurbeiichl» ]—VII) систематике структур свойственны
но меньшей мере три крупных недостатка, мешающих широкому
использованию данных SB в неорганической химии.
Первый из них заключается в том, что структуры систематизированы
в SB но группам на основе неструктурного признака: соотношения
количества разнородных атомов, входящих в состав вещества. Простые вещества
(состав А) относятся к группе А, соединения состава АВ к группе В; состава
АВ2 - к группе С, состава AmB7J- к группе D и т. д. и порядке алфавита.
Далее, внутри данной группы, например, 6\ структуры различаются
цифрами в порядке натурального ряда чисел, причём никакой логической
связи между тинами, например, СЛ (плавикового шпата) и С2 (пирита), нет.
К одному и тому же типу относятся иногда сходные, иногда химически
совершенно различные вещества, например, к типу Г2 —пирит (FeS2) и
двуокись углерода (С02).
Такая систематика, может быть, и удобна, если подходить к структурам
веществ с позиций стереометрической теории, но противоречит принципам
современной химии и физики. По нашему мнению, в основу
«химического SB», первым шагом к созданию которого является эта книга, должен быть
положен периодический закон Менделеева, который позволит связать
структуру кристалла со строением атома и молекулы.
Наконец, третий большой недостаток SB заключается в том, что основой
для причисления вещества к тому или иному типу в SB является
соотношение количеств атомов, входящих в состав вещества, а не структурных узлов.
Но, как вами подчёркнуто зз §§ НО и 62, соотношение количеств различных
атомов, входящих в состав кристалла, не может охарактеризовать строения ни
самого кристалла, ни его материальных (структурных) узлов
(координационных сфер и т.н.). Это является причиной того, что при изучении SB
в большой мере ускользает внутренняя физическая связь между
различными типами SB.
Поэтому SB не даёт и не может, по нашему мнению, дать научно
обоснованную, рациональную систематику структур.
В соответствии с изложенным ранее (§§ 60 и 62) правильнее
рассматривать структуры кристаллов по соотношению физически различных
структурных узлов. В этом свете неправильно относить Аг, НС], С02, NH3 к разным
группам структур, как это делает SB.
Все эти молекулы образуют самостоятельные структурные узлы а, и
кристаллы этих веществ правильно относить к одной и той же группе веществ
с молекулярной решёткой. В равной мере нецелесообразно включать,
например, С02, FeS2, CaC2, CaF2 в одну и ту же группу структур (С), как это
делает SB: в структуре С02 мы имеем один тип структурных узлов а = С02>
234 ч. и. структуры неорганических веществ [гл. Ill
в структуре FeS2 или СаС2 два типа: Fe = oc и (S2) = fi; соответственно Са = ос
и (C2) = [*J, причём ( — V-= 1, как и в случае NaCl. Напротив, в CaF2 два типа
структурных узлов Са —a, F=p, но — = 2, так что CaF2 следует отнести
к иной группе структур, чем СаС2 и т. д.
Помимо этих трёх фундаментальных недостатков, систематика SB имеет
значительные дефекты, вытекающие из недостаточно чёткого плана её
построения.
Типы структур в разных томах SB имеют разные обозначения и
по-разному объединяются в группы и классы. Одно и то же вещество переходит
из группы Н в группу / или ib' и т. д.
Этот недостаток учитывался и редакцией SB. Так, в предисловии к SB
подчёркивалось, что «как разграничение отдельных типов, так и
объединение типов в группы и классы ещё ни в коей мере не опираются на единые
рациональные положения. Поэтому в открытии таких основных положений
заключается крупнейшая, ещё не разрешённая, проблема систематической
кристаллохимии».
К сожалению, редакция SB ограничилась подобной констатацией
и даже не сочла нужным, до VII тома включительно, дать общее
сопоставление типов структур, принятых в SB.
Такое сопостЁГОление сделано нами в § 107, причём обнаружены отдельные
ошибки и с очевидностью выявлены различные неувязки в этой систематике.
Ставя перед собой задачу —возможно лучше выявить взаимную
физическую связь между структурами различных веществ, и, выделив с этой
целью основные типы (§ 62 и след.) с обозначениями, указывающими на син-
гонию решётки и характер координационных сфер, мы с целью увязки новой
систематики со SB во всех таблицах гл. IV — X будем также указывать
тип по SB, согласно § 107.
Одновременно мы указываел в последней графе таблиц § 107 обозначения
типов но предлагаемой нами координационной системе для тех типов,
которые вошли в таблицы гл. IV —X.
Остальные типы будут нами охарактеризованы в другой работе.
Как было указано в таблице 15 и подробнее рассматривается в
таблице 34 в принятой нами системе тип обозначается буквой, описывающей
сингонию (С —кубическая, Я-гексагональная, Т — тетрагональная, Л
—ромбоэдрическая (тригональная), OR — орторомбическая (ромбическая), М
-моноклинная, Тг — триклинная) с индексами, характеризующими особенности
структур.
Применяются два индекса справа: внизу и вверху (нижний индекс и
верхний индекс).
Нижний индекс описывает конфигурацию координационной сферы
(например: О— октаэдр, Г —тетраэдр, t—треугольник).
При обозначении приходится считаться с особенностями,
выявляющимися при определении метрики элементарной ячейки.
Как справедливо указывал Е. С. Фёдоров, все структуры кристаллов
должны быть производными от двух основных типов —кубического и
гексагонального. Следует отметить, что часто вещества, причислявшиеся ранее,
например, к кубической сингонии, позднее оказывались принадлежащими
к тетрагональной или даже ромбической сингонии, поскольку выяснялось,
что отношение осей а : b : с слегка отклоняется от 1, т. е. равно не 1 : 1 : 1,
а например, 1:1: 1,001 или 1,005 : 1 : 1,001. В других случаях
выяснялось, что вещество, причислявшееся на основании метрики элементарной
ячейки (т. е. отношения а:Ь:с и величины углов а, $, у) к кубической
сингонии, следует отнести к ромбоэдрической, поскольку угол а равен не 90°,
§ 1061 ВВЕДЕНИЕ. КОНФИГУРАЦИЯ КООРДИНАЦИОННЫХ СФЕР 235
а например, 89°55'. В частности, такие отклонения от кубической сингонии
даёт ряд веществ типа СаТЮ3 и др.
Очень важным критерием для определения характера метрики
элементарной ячейки является температурная зависимость соотношения осей или
величин углов.
Если при некоторой температуре tx отношение осей а : Ь : с=1 : 1 : 1,
это ещё не означает, что метрика является кубической. Для этого необходимо,
чтобы при t2 = t1 + kt и ti = tl — At это отношение осей сохранилось. Тогда
мокно условно принять, что в данном интервале температур t2 — tz метрика
является кубической. (То же относится к величине ^а = 90°.) Если же
метрика приобретает отношение 1:1:1 только в одной точке t', а во всём
интервале температур остаётся тетрагональной (например, отношение — > 1,
но с нагреванием непрерывно падает, становясь равным единице при V\
и продолжает падать с повышением температуры), метрика является
тетрагональной .
Интересный пример в этом смысле представляет ВаТЮ3, который при 20°
тетрагонален а = 3,9860, с = 4,0263, с/а = 1,0101. С повышением температуры
отношение с [а приближается к единице. При 200° С ВаТЮ3 приобретает
соотношение осей с/а = 1, формально отвечающее кубической структуре;
аш = 4,0040.
Далее, как мы знаем, симметрия кристалла определяется не только
метрикой элементарной ячейки и положением центров тяжести частиц, но и их
собственной симметрией и ориентировкой.
Как видно из таблиц 15 и 34, плоские формы (например,
треугольник) обозначаются строчными буквами, наименования объёмных форм
(например, тетраэдр) -прописными.
Особый интерес представляют конфигурации вычитания (термин наш),
типа Л3~~ или Т~, в которых центральные атомы занимают позицию в центре
фигуры, имеющей одну или две не занятые периферическими атомами
вершины. Существование этих конфигураций в большом количестве
соединений является наглядной иллюстрацией к н^шим взглядам (см. § 91).
Из таблицы вытекает, что в кристаллах осуществляется гораздо
больший выбор форм координационных сфер, чем это было предусмотрено
теорией Всрнера. Современным теориям также предстоит немалая работа по
интерпретации этого экспериментального материала.
Для обозначения деформированных конфигураций применяется черта
сверху индекса, например: О — деформированный октаэдр. Для обозначения
конфигураций, определённых ненадёжно, волнистая черта сверху: О —
вероятно октаэдр. Цифрой обозначается координационное число, если
конфигурация (в геометрическом отношении) сложная или неправильная,
индексом х—если конфигурация не уточнена. Если в решётке имеется
несколько однородных узлов с разной координацией, например, al и all, их
индексы разделяются занятой, например, OR^ означает, что в ромбической
решётке из однородных узлов а часть имеет сильно деформированную
координационную сферу октаэдр, часть неправильную координационную
сферу с координационным числом 6.
Если в решётке имеются разнородные структурные узлы, например,
Na и С1, их индексы разделяются знаком дроби Со/о — тип NaCl
(координация дважды октаэдрическая); Ccjt тип CaF2 (координация кубо-тетраэдри-
ческая)*). При наличии трёх и более типов структурных узлов даётся
*) Не смешивать кубо-октаэдрическую конфигурацию координационной сферы
Cf=--Cjc (плотная кубическая упаковка, тип меди узлы в вершинах кубооктаэдра) со
структурами кубо-октаэдричеекпми (через тире) с индексом Ссю> гДе °дна группа
узлов имеет координационную сферу «куб», а другая —«октаэдр».
236
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
|ГЛ. Ш
характеристика каждого из них. Обычно первым становится индекс для
координационной сферы металла, затем неметалла.
Если атом имеет две координационные сферы с близкими радиусами,
их к. ч. в нижнем индексе соединяются знаком +• Так, ("4+2/4+2 означает,
что оба узла имеют к. ч. 4 + 2, а всего 6, из коих 2 на значительно большем
расстоянии (не смешивать со знаком -\- ПРИ нижнем индексе, например,
О*, означающем, что помимо шести атомов, образующих деформированную
октаэдрическую координационную сферу, имеется один лишний, седьмой
атом и все 7 атомов координационной сферы примерно одинаково, в
продень
лах <20--25%, удалены от центрального), опак Л+ + означает, что над
плоскостью шестиугольника и под iieii имеется по 2 дополнительных атома,
а всего 10. Знак РЛ '+ , или P33h, означает тригоиальная призма -f- 3
дополнительных атома- Знак С~1" означает, что 6 периферических атомов
занимают 6 вершин куба, оставляя не занятыми 2 вершины по объёмной
диагонали. Знак С~ означает, что из 8 вершин кубической
координационной сферы, —не заняты 2 вершины по диагонали грани.
Верхний индекс является вспомогательным. Мы применяем
обозначения: М- молекулы, К -цепи, N — сетки, S- слои, R — кольца, форма которых
обозначается но таблице нижних индексов, с применением скобок,
например, М(0)— октаодрические молекулы. Количество членов в кольце
обозначается цифрой: RS— восьмичленнос кольцо, ORn^ означает ромбическую
молекулярную решётку, в которой восьмиатомные молекулы образуют
кольца (например, aS). Координационные сферы, характеризуемые
нижними индексами приведены в таблице 34; см также § 279.
Таблица 34
Конфигурации координациоппых сфер
(э. х.—характеристика электроном, образуювхнх с-связи)
К. ч.
12
Обозначение
Наименование
Тг
Р6
Э. х.
Пример
Тип
Кубоокта-
"эдр
Закруч. ку
бо-октаэдр
Икосаэдр
а)
правильный; б) крм-
сталлохими-
чес кий
Усечённый ! Гекеагональ-
тетраэдр \ пая нризма
Конфигурации
"J
б) с
Y Х°л
м. рис.
-'•
Па
—
Си
_...
Mg
•
—
Mn
V3Si"
г
> ■—■?—■
О
—
pi
КВк
KaPt (BCN),
CJC SB ЛI
Ну—SB-13
CK/j—SB Alt
c\s+s)ij—>BAi:>
|Сг/Л~зб c\:>,Hn п—$внь
§ 1061 1ШЕДЕНПЕ. КОНФИГУРАЦИИ КООРДИНАЦИОННЫХ СФЕР
237
К. ч.
10
Обозначение
h-\-r=h
Цаимено-1
вание I
Шестиугольник *)-Ь
прямоугольник
:). х.
Пример
MoSia
Tim
Т ч
/ih + /Z'i/-l
-SBCU
W
Триграпе-
центрир.
тригон.
призма **)
Ж"
Хг1(Н,0)в(Иг(),):
"""Рьг.1,"
'"W*7t.,v,
—SBC 23
Р4
Куб
в
Л нти
призма ***)
—закрученный К\'б
Тетрагональная призма
d5p*
CsCl
СгиС€
CrV-SBB2 |
SBD84
— С
0,3,2/Д
Sn(CeH5)4
Г/ч
К. ч.
Обозн.
Наименование
Ш
Дитетра-
/>3>
/':<;
А,
i />в
Днуграпецен-
трир. триго-
ннльная
призма00)
1зазоцен-
трир. трн-
гональная
призма
Т риго на
льны й
додекаэдр
(см. рис. 580)
Гексагональная биии-
рамида
Пример
Тип
^ DilDi
— SB #25
Пример Ким-
бо.ча [TaF8j~-
иоппднмому <>т-
поелтен к
конфигурации Р'ЛТ
юн [TaF^1"
и K.,TaFB
поп
|A1iKCN)b]<-b
K4Mo(c:N)e2H,o
KBF2t
UnJt
*) Пересечение двух шестиугольников, плоскости которых
перпендикулярны друг к другу.
**) Три группы Н лежат на перпендикулярах к центрам граней РЗ.
***) Возникает из куба, если верхнее основание последнего повернуть на '»5Э
относительно нижнего.
°) Два неодинаковых октаэдра.
0о) Две группы R на перпендикулярах к центрам граней Р?>.
238 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ |гл. Ill
К. ч.
Обозначение
Наименование
0+
Одногране-
центриров.
октаэдр *)
Р3+
Одногранецен-
триров. триго-
нальная
призма •*)
ZJ5
Пентаго-
нальная
бипирамида
Шестиугольник + 1 атом
d*sp\d*P\ d5/>'
raF7— -ион
в K2TaF7
~К^04
Mxlu—SBAT6,
OR
Л+Vs
-SB/V1.
Октаэдр
d:sp3
|PtCle|--ifon
в K.PtCh
Сг/с—SBH61
К. ч.
Обозн.
Dk
РЗ
.4:$
С
,-/-
Наименование
Тетрагональная
бипирамида
Тригональ-
пая призма
Тригональ-
ная
антипризма ***)
К}'б бэз двух
вершин по
диагонали***)
Куб без двух
вершин по
диагонали
грани
Конфигурация
1AJ
Э. х.
d*spt dbp
d3/?3
Пример
Ag|Co(NH3)-2
(N03)4|
NiAs
Cr(H10)eCls
CoAss
(C04(As4)3)
Тип
-SBJ10
"о,рз-&в*8
R
ЛЗ + Л/Т
-SBB722
I
Cc-i-ic-
—SB/>Oa
cr;-/-, c/r
*) Атом против центра грани октаэдра.
**) Атом против центра грани призмы.
***) ЛЗ и С"/- могли • бы рассматриваться как частные случаи конфигурации
«искажённый октаэдр»; поскольку, однако, они допускают значительные изменения
валентных углов, их целесообразно считать самостоятельными формами
координационных сфер.
§ 106] ВВЕДЕНИЕ. КОНФИГУРАЦИЯ КООРДИНАЦИОННЫХ СФЕР 239
К. ч.
Обозначение
Наименование
«Л»
Шестиугольник
(часть более
сложной
конфигурации)
Конфигурация
Э. х.
Пример ; Сг(Н20)вСЛ3
Тип
R
АЗ+hp —
SB/2.>
т
Тригональ-
ная бипирами-
да
Ру'ь
Тетрагональная
пирамида
Пятиугольная
илоская
I [ентраль-
ный атом
лежит вне
плоскости п
dsp'A, dssp
disp'-tdxsi I
d~pdt dxp |
d»p*
Молекула
FeC05
PbrVI
центр, атом
над о;1
копанием
1 q + qJT + РиГ
- SBEO,
К. ч.
Обозн.
Наименование
Тетраэдр
Ру\
п:\-
Неправильньш
тетраэдр
(в том числе
тетрагональный тетраэдр)
Триго-
нальная
пирамида
Г»игшрамида
три
тональная без
одной
вершины
Конфигурации
Квадрат
Э. х.
sp\ d\
d'-sp% dp\
d*p
P:UL d-*p,
dp-\ dl
dap\ d*p*
Пример Алмаз
Графит
4 а и центре
осноиания
молекула
Те С I.*
Ca\V04
Тип j CV-SB.44
//
4V*-SB//4, \^BUll!Pl
q- SJv+S ~
— >B//U.
2/iO 4. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. II
К. Ч.
Обозн.
Найме*
нива нио
Конфигурация
Г), х.
Пример
Центральный атом
лежит пне
плоскости
Я
Прямоугольник
Четырёхугольник
E3J
Ы)
Тип
T({IS- SB/УН
PtlO, PIS
CaS04
ангидрид
* rJS
rrls-*UB\:
OH
Pip'
SB//o1
Треугольник
Тетраэдр
без одной а) иранильнып
вершины*) Ь)нсснммс-
тричн.
/>•', d2p
a) sp*9dps.dsstd*
])) dsp
молекулы
NII3, ND3
a) ClOj-" -ион
b) ТЮ,(рутпл)|
b)r(/)'//--SB(V1
К. ч.
Обозн.
Наименован и о
Центральный атом
пне
плоскости t °)
Четырёхугольник
без одной
вершины э")
Конфигурации
Э. х.
/j3, d2/>
dsP(?)
Пример CdC], Т.ГЛГ;1:ЮК ч
r J - li02 (апатаа)
Тин
/?" - SB64<>
о//
7 ± SBC5
близок Т +_
S4P~
**)
Тетраэдр
без двух
вершин
>
(р2, ds, d2) p2, db\ d2
молекула
Но()
молекула
Н>
Си,О
С/г/ -SBCli
*) Валентный угол между связями^ 10ЭЭ28'.
**) Валентный угол чаще близок к 90°.
***) Валентный угол близок к 180'.
°) Валентный угол между связями близ ж к 120°.
оэ) Центральный атом лежит на стороне треугольника.
§ 107]
ТИПЫ СТРУКТУР
241
В заключение отметим, что если интерпретация более хорошо
изученных простых структур на базе предлагаемой нами координационной
системы позволяет дать, как нам кажется, простую и однозначную
характеристику структурного типа, то в более сложных случаях когда параметры
атомов известны с меньшей точностью, иногда приходится воздержизаться
от указания форм, ограничиваясь, впредь до уточнения координации,
обозначением в индексе количества периферических атомов или даже
указанием неопределённости соотношением xjy. Тем самым подчёркивается
необходимость дальнейших исследований.
С другой стороны, одна и та же конфигурация узлов в сложных
структурах позволяет выделить различные координационные сферы,
включая или не включая в них более удалённые узлы, в зависимости от
экспериментальных данных и теоретических соображений. Огромный
структурный материал, собранный современной наукой и в том числе
рассматриваемый в этой книге, ещё требует дополнительных исследований,
посвященных рассмотрению и теоретической интерпретации валентных углов,
расстояний и конфигураций в целом, в координационных сферах. В этом
смысле таблица 34, хотя и рассматривает довольно широкую группу
координационных конфигураций, но ещё очень далека от завершения и
потребует дальнейшей работы над ней. В частности, значительно может
быть обогащен список возможных конфигураций за счёт деформированных
конфигураций, производных от включённых нами в таблицу, например,
деформированный тетрагональный кубооктаэдр в тетрагональной гране-
центрированной ячейке индия, ромбоэдрически деформированный октаэдр
в ромбоэдрически деформированных кубических решётках у мышьяка и др.
Включённые в таблицу 34 из теоретических соображений такие
конфигурации, как пентагональная бипирамида и правильный икосаэдр (оси 5!)
в структурах координационных решёток, естественно не встречаются. (Хотя
SBII3 указывает, что в |Щп имеются узлы в форме правильного (regel-
massig) икосаэдра.) В решётках ряда веществ образуется конфигурация
кристаллохимического икосаэдра, рис. 441а. Но казалось бы, отдельные
узлы сами по себе могли бы иметь подобную форму. Возможность
существования октаэдрических узлов при отсутствии узлов в виде пентагональ-
ной бипирамиды связано с тем, что первая конфигурация в отличие от
второй в энергетическом отношении является одной из наиболее выгодных
для данного к. ч. Гексагональная бипирамида в координационных
решётках не может конкурировать с многочисленными более выгодными
конфигурациями числа 8 и является редким исключением.
Наконец, некоторые формы, как Л, сами по себе не встречаются, но
входят в состав более сложных координационных сфер; см. также § 279.
§ 107; Типы структур.
Переходя далее к сопоставлению типов структур по SB, отмечаем,
что в последнее время найден ряд новых структурных типов, не
зарегистрированных в 7 томах SB. В частности, это относится к актинидам
(уранидам). С некоторыми из вновь открытых типов мы знакомим читателя
в гл. IV—X. Таким образом, всего в SB описано 407 типов структур
неорганических веществ. Из них некоторые дублируют друг друга, некоторые
оказались неверными; в отдельных случаях к одному и тому же типу ошибочно
отнесены совершенно разные по структуре вещества. Наконец, ряд структур
неорганических веществ в SB попал в группу органических веществ. Они
нами также включены в регистр (группа «О»), равно как и некоторые
органические вещества, например СН4, имеющие не меньшее основание для
включения в этот регистр, чем CO(J321) или СОя(С2).
Типы структур
Наименование
Алмаа
Белое
олово
VV
Щ
с
Sn
In
As
Se
Графит С
Hg
Тип
(SB)
Том
Про- j
CTp.i стРан-
г |ственная
группа
Обозначение типов
по координационно-
структурному
принципу*)
А. Простые веществе**)
Си I Л\
А1
лз
AW
АЪ
ЛС
А1
AS
/49
.410
II
VI
IV
II
13
3
3
15
3
16
3
3
19
21
23
3
25
27
28
31
737
01
ol
'6h
D
19
4Л
D
17
4Л
D
3d
I>lDl
'6Л
D
3d
CF = Ci
С j = Cc
Ct
+
TS+2 = S+
Tf = TTk (cm.
стр. 806)
*o
H
Ht+1
Структура неверна
Наименование
NaCl
CsCl
Цинковая ZnS
обманка, сфе-
лерит
Вюрцит ZnS
SiC III
SiCII
SiC I
NiAs
Киноварь HgS
Тип
(SB)
Tom
Стр.
Про- Обозначение типов
стран- по координационно-
ственная структурному
группа I принципу*)
В. Соединения АВ
1 В1
вг
S3
£4
въ
вь
В1
въ
£9
1 I
III
IV
VI
VII
I
III
IV
I
III
I
III
VI
I
I
I
1
V
I
| 72
7
6
3
3
74
7
6
76
7
78
7
4
80
82
83
84
4
87
о\ I
о\
г\
cU
Cqv
<*.
cl
t>i*
C3C3
Core
Ccfc
'TIT
Htii
2.2
^77 rl Верхние нн-
I дексы—чи-
^з.з . еловые сим-
т/т ( волы упа-
2 з I КОВКИ ПО
rtit\ Г.С.Жданову!
Я,
О/РЗ
но/о
Ga
Mn
Mn
J,
Сера ром- S
бическан
Фосфор P
чёрный
ci,
Полоний Po
U
! ли
Л12
1 Л13
Л14
' Л15
Л16
Л17
Л18
Л19
Л20
I
II
III
II
III
II
II
IV
II
VII
III
III
IV
IV
VI
738
1
3
о
3
3
5
3
6
3
4
6
3
4
3 j
7)18
Ol
D
18
2h
oi
D
24
2/1
D
18
2/i
D
16
4/i
D»
Структура неверна
OR
м
C16/13/li
OR¥ (ОД—дефор-;
к мация)
'14/J
M-R«
OR
0RS+1
TM
1 2
Мц. В 1946 г.
существование типа
Л19 поставлено под
сомнение;
предполагается примитивная
кубическая
элементарная ячейка С0 и
примитивная
ромбоэдрическая Rq.
Oi?4+8
(деформированный тип Ну)
PH4J
PbO
PbO
BN
Миллерит NiS
FeAs
FeB
GeS
Куперит PtS
Ковеллин CuS
AuCd
FeSi
CO
BIO
Bll
£12
£13
£14
Bib
£16
£17
BIS
£19
£20
' £21
1
III
I
I
I
II
II
II
II
II
II
II
II
II
89
7
94
95
740
6
7
7
8
9
10
41
13
13
< |
<
»lk
r5
cl
"It
u2h
OH
»lb
Dik
D5
u2h
Г*
ТГ
a) Tq+q/Di (PH4J)
rpS
IqJS
ь) T*gs
Hfft или Ht4l/Ul
Изменен, параметров]
"5/5 - пРуА1Рц4.
OR*f-3 CM.NiAs<£8)|
и MnP (£31)
ОДу/:г В SB III 13
FeB отнесён
к типу В 27.
ORo/o
HTtt/D3,o- Правиль
но Cu2[S • CuS,]
C7/7, искажённый
C0jQf близок к
конфигурации P3+/Pz+;
см. тип G0e(KClO8)
•3 I 1в^ } "Р~- — » «i таблице.
tlродолжение
Наименование
KSU
Agl
TIF
7 NII4Br
1 Тенорит CuO
FeB
FeSi
SnB
MgZn
MnP
NaTl
•шт^^^—ыш
Тип
B22
B23
B24
вгь
£26
£27
£28
£29
£30
£31
£32
Том
III
V
III
III
III
III
III
III
III
III
III
VI
HI
Стр.
8
4
8
9
10
11
12
13
14
16
17
4
19
странственная
группа
Z>5
—
»й
"Ir,
г6
7>16
u2h
Ti
U2h
I>ih
7>16
u2h
—
Обозначение типов
по координационно-
структурному
принципу
Ro/o~
Статистическое
распределение
атомов Ag
ORo/o
Практически
идентичен с В10
(PH4J)! Tq+q/Di
МПТ
0R2JP3
Отпадает,
идентичен с £20.
Небрежность SB
°4iu i
к
ORo/pz
См. NiAs (£8)
и FeAs (£14)
Производный от Л4
См. § 84
Наименование
CdL
1 Молибденит MoS2
Кварц SiO*
Кристобалит Si02
Тридимит SiOa
а) СаС,
б) MoSia
CaSi2
Hgl2
MgZna
Тип
(SB)
Сб
CI
cs
C9
CIO
С11
C12
С13
C14
Том
III
IV
VI
VII
III
IV
VII
IV
Стр.
161
20
8
5
7
164
166
21
169
171
174
740
8
7
175
177
180
8
странственная
группа
*>la
«6h
D% и др.
ol
*>6Л
Dll(f)
Pfd(?)
T>\1
»ih
Обозначение типов 1
по координационно-
структурному 1
принципу 1
Н— очень близок 1
OJt
типу С27, C19 и
С24. См. § 145
"PZlt
Неверно,
см. SB III 21
яу
стц
nTjl
Неверно. TD4(D4
(искаж.: C0i0)
т — т + <■ 1
1 Ю/Ру4 - * h—/Ptf4
Л6/3,4
rpS 1
H6jl2
смотри С41
§ 107]
ТИПЫ СТРУКТУР
245
СО
к^г
и. л
-ЕС се н
© сз ^ "• ■
о
Э 5
х К ts
° 2 Я 2 ьГ
5 Э Я S
Eg я« и н
О Си
сб <Л
Ни
§°
СП
• О
I-
|S-
о
о I
.со
Xе*
го 2
ы се
о ~
Л -г
О ~.
оО
Я ч
ч и
^0 Ю
ОС
ft н
се • 'о гЗ 5
н 2 й
«Дот g?
•^ s
ф
fa
s
о
fcll©
a,
О
О
И
О CQ
1Я«
гНГ1 СЧСО
о
о>
«#
WJ
Ч-»
О
*4
СМ
^
~
ОО Гч
»
ИИ)
чр4
О»
•*
гн
VO
И
О
сч
см
ми
•ч
со
а»
*#
ря4
о*
^
О
vC <г*
а> сч
**
, t
~
ОО
^
о
(N CN
•* CM
Гх
И-. НИ
~
<?>
И
О
Гч
ИИ
Он
О
<N
О
X
ел
Ы
*#
«*■»
^
"
^
сч
О
\П VC
ч*
мн
~>
l~t
с*
О
<£> 00
' чН
со
(М
о
ИИ
>
СО 1
«и»
1—4 1
** 1
<М 1
О
о
to
2
fa
00
fa
о
3
о
2
О
••и
я
х
а,
fa
Д
о;
о
ОС
О
со
0*
со
со а,
ОС
о ,
о
я
н
к
X
о
и
а>
Д
N
f
М§?
S н ^ н >» "1
^ я о и t» t.
О Ск Я "
н х я и, «
о-
«о
5ч
+
> to
3
•с «
CNC^ и^«0
COd
се*
г**
О
сХ>
>
СО
со
0Q
со
>
СО
-*
>
со
0Q
-#
VII
о
со
о
VII
со
о;
я
Я
со
О
U
ОО
-л»
чН
^.ц
4HJ
о
О 00 VT5 Г^
ем
5с:>=
о
\о
«4
^ц
СЧ
О
О
с-<
»_4
д
со
vO
«и~
ИИ
СО
О
m
\П
«И
Ъш4
<*
О .
00
vO
«И
ИИ
хЛ
О
U1
о.
о
о
д
д
о
2
ф
аз
се
U
Я
Р-
О
е
© о
■ i я
9
я
а.
Я сб
* £
Н се
>> a
Си <
Продолжение
Наименование
Тип
(SB)
Том
Стр.
странственная
группа
Обозначение типов
по координационно-
структурному
принципу
Наименование
Тип
(SB)
Том
Стр.
странственная
группа
Обозначение типов
по координационно
структурному
принципу
HgCl4
N02-Na04
CdJ2
HgCl2
SrH,
Низкотем-
дератур- SiO*
ный кри-
стобалрт
Zn(OH)2
А1В,
Тетрадимпт
Bi,Te^S
С25
С26
С27
С28
С29
сзо
С31
С32
сзз
II
II
III
III
III
III
Ш
III
III
VII
19
21
22
23
24
25
26
28
28
8
D
rs
<U
u<Zh
D
16
2/i
ИЛИ
Di
^>6/,(?)
*&<?>
OR—
O/2+l ТИП, П0ВИ-
димому, отпадает ^?),
см. С28
с»
2-я модификация
**%
ORMW
OR
1Ф.4
TU
OR.
т/г
H
Р6/ЯЗ-
Возмояшы
*V Cbh* DSh>
Возможны D\t C\t
Креннерит AuTe*
Se02
Cr2Al
ZrSi2
FdCl2
CoSn2
|Теллурит TeOs
SrBr2
Si2Ti
NHt
CoAs3,
точнее Co4(As4)8
C4G
| C47
C48
C49
1 C50
C51
C52
C53
C54
IV
V
V
V
VI
VI
VII
VII
VII
4
5
5
5
7
8
10
12
b2v
)13
D
17
2ft
)12
J2h
)18
.15
J2h
Di
16
2h
)24
}2h
D. Соединения Am В™
OR
M
4,6,4
fpK
1 //2,1
OR
8/4
OR
К
очень близок к С 23]
ОЛР38+/7\4
#1
D0t
D2
DO,
I
II
I
II
230
XXII
232
XXII
TA
Tb
l10/5
к
'C'l-JC
/i + +/5
калаверит AuTe,
CaCl*
MgNi2
Co,Si
Cu3Sb
ZrW,
CrSi,
• Fe2W
SiS2
Бадделеит Zr02
GeS3
CuGlt 2H30
С 34
C35
C36
C37
C38
C39
C40
C41
C42
C43
C44
C45
III
III
VII
III
VII
HI
III
IV
VI
III
III
III
III
IV
IV
IV
30
30
8
31
8
32
33
9
5
34
35
36
37
9
и
13
r3
b2h
ИЛИ
*2
или
r10
b2v
Dib
*й
»Sft
o\
Di
dU
Dfh
c%h
ell
»ln
М2+4Ц
ORoit
—
«жйи-оя^зц.
TT, Py4lq+Py4
(или О4, или Г*)
С12/б • Аналогичен
С15
#4+4/2+4
#б/з отпадает,
аналогичен типу С14
0RTJZ
MW
TU
0Rmq)
BiF,
CrCl3
Bil,
Тизонит LaF3
Сг03
Al (OH)3
Mo03
ReOf
wo3
Цементит Fe3C
FeF3
A1C1,
AIF,
DO,
DO,
D05
DO,
DO,
D07
D08
D09
D010
D0n
D0la
*>o13
D014
II
11
II
II
II
III
II
II
V
II
II
II
II
III
22
2J
25
27
29
38
30
31
12
32
33
34
36
40
Di
'3*
A
6A
D5
U2
°2h
D
16
2/i
o\
D
11
2ft
D
16
2/i
D
3d
D
3d
Di
'C-fO/7\ О
H*
o/z
*S
очень
близки
o/Z
its
Я11/3,4
ОД(?) „
o/z Путаница.
2 разные структуры
отнесены к одному
типу D0r
КГ2
oni
0/3, 2, 1
JQ\l
ORx близок к СО,
Точно не
исследован
ОЯ2/рз или ORZIPz
Rou
#,
ОЦ,1
4 + 1/2
очень
близок к D0V2
Продолжение
Наименование
А1С1,
NH4J8
ш
BaS3
Na8As
Ni8Sn
NiAI3
Cu8P
TiAl8
Al8Zr
Ni8Ti
SnJ4
Тип
(SB)
D0M
M*
D017
D019
DO»
D0„
DQ%1
D0t2
DO^
£>0u
Dlt
D\x
Tom
III
HI
IV
V
V
V
VI
VII
VII
VII
I
11
|Стр.
41
43
18
6
7
8 '
1
13
14
14
234
XXII
странственная
группа
или D%
или D\
1 H\
D\
»th
<
•D\t
H„
])t7
Л17
u4h
DU
Tt
Обозначение типов
по координационно-
структурному
принципу
Ms-M(o)
ОЯ4/4 см. F514
(практически DQU
и F5U один и тот
же тип)
0RP4in-r см« ВЩ
PH4J
Hf, з/»
—
—
НхЦ2
. 1
— 1
— 1
СМ(7)
Наименование
<ф-корунд»
Ala08NajO
А1а08
^-корунд
Антимонит SbjSa
Zn8Pa
Cr8C8
Валентинит Sba08
Р BiaOs
NiaAl8
Сенармонтит
Sb208
SbA-SbSbO,
А14С,
Тип
(SB)
Db%
D5T
Я58
7)5t
Д51в|
D5n
D5ia
£518
Z)61
Д6а
D7X
Tom
II
II
III
III
III
IV i
V
V
III
VII
III
Стр.
41
43
49
51
53*
20
9
10
54
15,
126
55—
56
странственная
группа
*>1ъ
0е
или
О7
7)16
U2h
j ЛЬ
n16
u2h
Dt0
u2h
*L
»l*
ol
Dh
Обозначение типов
по координационно-
структурному
принципу
—
Близок к типу 1
шпинели (#11) I
0Rfu
гр _ близок и тяну 1
1 Г/6 Mua08 <£>58) I
0RtlP3 1
0Rtlft 1 J
Tc~l~iT 1
—
Отпадает, см. D54 1
Неверно. Тип D6a I
отпадает 1
*Ш 5
§'107J
ТИПЫ СТРУКТУР
249
£
О Ь«
с*
о
о
о.
о
S
о*
ф
ш
ф
X
с
О
43
юсо
3
8ч
о
I©
•в
со<о
©
<0
О
л со
•С
^14 Гх «-^
**Х *Х
О X Гч «О
-х> —<
>
Гч
VII
М
Гч
00 00
3Q
*-т
СЧ <*
00 00
•-*
NH »-H JT4
СО «• *»
00 00 00
*-*
00
2
14
00
3
х>
•
00
>
г»
00
>
•
00
^
0»
со
3
^
е
ОсГ
>
00
л
Ь
S3
ф
с
о
я
с»
о
ф*
СЛ
53
ф
^.3 3
г^ © ч» -,
£ u *■? о
о
о £•
^^
N ОС
О Р5
X И
со
В*
(Я ft
©
о
я
о*
PQ
Ф
со
со
2.
со
ft
п
о
п
я н
f S.
X"
■?* 55
О
V
•о
«О СО
^
О
О
Гч
00
„
чН
3
\Л
<г
,_,
м>
w4
Q
Ю
^*
~
К^
Гч
СО
»>4
*■«
вЧ
Q
Гч
<f
^
*""■
сч
Cf
00
г*
СЧ
Q
Гч
СО
сч
*■*
чН
СО
-^—
и^
X
1-4
со
Q Q
о
со
сч
•—'
ЧН
■*
~*s—
*м
*-4
•*
Q С)
О
<f
СЧ
*""*
ч-н
Ю
»-н
И-4
X
и
1—и
*-•
\о
Q Q
сч
•*
сч
"^
сч
\Л
Q
**
*3"
Гч
**
j^— —-
>^
XJ 00
м-
»-Н ^ч4
»^ и-^
^сч
irt ю
^Q
со
со
я
ю
^
а>
**
^^
\п
<f
сч
*"*
^о
Q
^.
^^
X
X
~
\iZ
Q
о
**
к*
vft
Q
a
с
с
S3
• ев
X
X
О
>>
о,
о
° ч
СО СЛ
55
Продолжение
Наименование
Тип
(SB)
Том
Стр.
странственная
группа
Обозначение типов
по координационно-
структурному
принципу
Наименование
Тип
(SB)
Том
Стр.
странственная
группа
Обозначение типов
по координационно-
структурному
принципу
Е. Соединения с более чем 2 сортами атомов без
образования радикалов
PbFCl
Диаспор А100Н
CdOHCl
FeOOH
FeOCl
Манганит
Mn (OH)O
Арсенопирит
FeAsS
Халкопирит
CuFeS2
Zn (NH3)2C12
Cd(NH3),Cl2
P4N4C18
EOj
EO,
Е0г
E0A
E05
EOi
E%
F61
Eh
El2
Eh
Eh
II
III
II
III
III
III
III
IV
IV
VII
I
11
IV
IV
VII
45
64
46
64
65
66 |
67
28
30
17
279
48
31
33
17
D
4h
D
16
2Л
C6v
D
17
2Л
D
13
2h
y2h
°2Л
D
12
2d
D2&
U2h
rll
b2v
<?+9/T,Py4
OfTfT
(D^d). Неверно
v"
T/TJT
Tl
опмау
Utipz+f
OR?™
TM (Д8)
P4
Ниобит FeTsb^Og
Sr (0H)28H ,0
Sr028H20
Коппит R2X207
Трикальций
алюминат Са3А1206
Сведенборгит
4Be0 NaSb03
Fe3W8C
A16C3N
£5,
E*i
Eb,
E*i
E9X
E92
: я»,
E94
11
II
II
II
VII
II
III
III
III
55
56
57
58
24
60
69
71
73
*>&
<
»2„
ol
o\
C6v
ol
C6v
QH
lQlQ
F. Решётки с радикалами ВХ и ВХ2
Кобальтин CoAsS
COS
Hg(CN)2
К4Мо (CN)e2H>0
Fl
F0X
F02
Fll
Flx
F2X"
I
II
II
I
II
vir
269
XXII
62
270
XXII
25
1
Tl
r5
v?
»a
Сщ (тпп пнрпта)
?M-s
Ч(-Ю/Д+0//
x/tf
о#Л?
Перовскйт CaTiOj
Ильменит FeTiO,
LiI03
NH4CdCl3
NH4HgCl,
Карналлит-бром-
карналлит
|Ю^(Н.0)в(С1,Вг)3
Ag2HgI4
Метаборат кальция]
CaB,04
Бертьерит
FeSbiS4
K,HgCl4.H,0
К28пС14.Н20
Псевдобрукит
Fe2Ti05
G5
G0&
Е2Х
Gk
G04
E'22
Е2Ъ
El,
Eh
Б29
E3l
Eh
E33
£34
E%
E4X
II
II
III
I
II
III
II
VI
VII
VI
VII
II
II
IV
VI
VII
II
300
Ixxin
49
68
300
XXIII
69
49
13
19
15
19
50
51
16
22
53
o\
cl
Неверно. Как
показывают
современные исследованип,
перовскйт
является моноклинным
М1--
KIOjO
D*
D
16
2ft
c\„
'4h
D2d
Di
14
2/t
D
16
2/i
D
'2ft
16
2h
^2Л
O/OJp
8/8
T-
= г*
0/P4/P4 Л 04(p4lp
Г/Т/2+1
0RK(0)
0/6 + 2/2
Fe2 (CO),
NaHF2
и CsICl,
KHF2
CuFe82
NIJ4C102
NaN02
Вольфе бергит
CuSbS,
NH4H2P02
NH4HF,
KCNS
KAg(GN),
KNOa
F4t
F51
F5t
F52
F52
F53
^54
F5e
F57
F5e
F59
F5n
VII
I
II
III
VI
I
II
III
LB
II
II
111
III
III
IV
III
III
IV
28 I
271
XXII
75
18
276
XXII
75
Eg.
II
65
75
77
79
37
80
82
39
C6ii
D
3d
ol
D
18
4ft
D26
u2h
Jiv
.20
'2v
D!
16
Ih
D
21
2Л
D
2h
D
11
2Л
D
3d
HMiG + 0)
*o/o
TP4!P4
В этом типе
кристаллизуется азид
серебра AgN3
дотетрагональный)—ромбический
Отпадает, см. Е\х
TDijp4t почти
аналогичен F52
OR
D2/D2
0RPA/P4
0RDijDi
мо/о
близок к F5fi
Продолжение
Наименование
Тип
(SB)
Том
Стр.
странственная
группа
Обозначение типов
по координационно-
структурному
принципу
Наименование
Тип
(SB)
Том
Стр.
странственная
группа
Обозначение типов
по координационно-
структурному
принципу
AgNOa
кво,
Ш4С1Вг1
F5ia
F513
F514
IV
V
VI
41
12
18
14
-,20
J2v
D
3d
D
16
2Л
OR
D2/D2
R
x/y
OR
4/4
Нортупит
MgC08-Na2C08-
•NaCl
Азурит
Cu3 (C08)8 (OH)a
Фосген ит
PbC03PbCI2
Na2C03Ha0
G73
G\
Oh
G7,
II
II
III
IV
80
81
89
42
ol
r5
°2/i
Dl
V2v
(?) описание SB
неверно
G. Решётки с радикалами ВХ,
Н. Решётки о радикалами ВХ4
Кальцит СаС03
Арагонит СаС03
NaC103
Ильменит
FeTi03
Перовскнт СаТЮ,
КСЮ8
КВг03
G1
G0X
; G2
GO,
G3
G03
G4
G04
G5
G05
; go.
G07
I
II
I
II
III
I
II
I
II
I
II
II
II
292
XXII
295
XXII
84
297
XXIII
300
XXIII
300
XXIII
66
63
тг!6
vh
TA
C3f
o\
C2h
Ro/o
ORPb/o
'6+1/6+1
Яб/0/4АнаЛ°ГК0РуН
да. Повторение,
см. Я2а
^с/с/6 -Повторение
см. Е2Х
Л*РЗ*/РЗ+
Д6 +2/6+2 (ДеФ°Р-
мироваиный тип
CsCl)
Ангидрит CaS04
Барит BaS04
Циркон ZrSi04
Шеелит CaW04
КСЮ4
Hi
нох
Я2
яоа
яз
яо8
Six
Я4
Я04
яо,
I
II
I
II
III
I
II
III
I
II
III
VI
VII
II
340
XXIII
! 343
XXIII
90
345
XXIII
91
347
83
91
! 18
30
84
I *!
уГ
vl6
»\н
С\п
т\
°К 2+2/2+2= 0Rplp
1 777/2+2_
см. S\x
Tq+Sjq+S
NH4N03L
NH4N03II
NH4N0,III
NH4N03IV
NaHC03
Доломит
CaMg(C03)2
Pb (N02)
Nd(BrOa)39H.,0=
=[ЩН20)д][ВЮ3]8
Перилл
B*3A12 (Si03)e
Na4 S03
H3B03
Л1 (P03)3
Бастнезит
RFC03
Гамбергит
Be2B03H0
GO,
GO,
G010
G0U
G012
Gil
Glx
G21
G2X
G22
G31
G32
G5X
G53
G7,
G72
II.
II
II
II
III
I
II
I
11
MI
I
II
III
V
II
11
09
69
71
72
86
303
XXIII
304
73
29
305
74
87
15
75
78
o\
Dh
U2h
u2h
o\h
r2
°3i
/T«6
1 h
h
C6»
i>lft
c3/
»?h
(Кубический) СС{С
(Тетрагональный)
(Ромбический)
(Ромбический)
0ROilDt
K*
R6/o
Параметры неверны
Cg^.o/34 l близок к
CaFl(Cl)
^P3+ + + /f
Отпадает, см. S^
^ш/рз+оз
T s
//2 + 1
0/772
—
MgW04
BP04
T1A1F4
AgMn04
KJG]4
1 Шпинель
A],Mg04
1 Оливин Mg»Si04
1 Фенакит
BeaSi04
LiKB04
K3PtCl4
KaS04
Na2S04
Na,Cr04
яов
Я07
яо8
7/09
яо10
ЯП
HU
ни
ни
SU
Я13
Я13
Я14
ни
Я15
ни
ни
ни
ни
и
III
V
VI
VI
VI
I
II
I
II
II
I
II
II
I
II
I
II
II
III
VI
II
IV
85
92
17
18
19
20
350
XXIII
352
XXIII
XX 1\
356
XXIII
XXIV
357
XXIII
358
XXIII
86
94
21
88
45
сЬ
Ъ
DU
cL
Г5
°2Л
ol
F.e
C3i
(Л
°\ь
u2h
D24
v2h
D6
u2h
Mqjq (?). Положе- 1
ния атомов недо- 1
статочно уточнены 1
Т/Г/1 1
OjPlJr + lPyA 1
^7/7(?)
Mxly
CK ~
0/774
См. /Г-типы. I
OROiTI$ + l I
nTfTlt
Ge3N4 образует ре- I
шётку типа BeSi04—1
—nTft
— 1
TP4[r
1 OR* ~
/3
олГо/т*
'ОД9/5
Продолжение
Наименование
AgsP04
KH2P04
Сзтльванит
Cu3VS4
1 Энаргит
1 Cu3AsS4
1 Станнит
1 Cu,FeSnS4
J Zn(ZnOH)As04
ВаА1204
1 Гранат
AlaCa3(Si04)s
kai (so4)i
Тип
(SB)
Я21
Я21
Я 22
Я24
Я26
Я26
Я27
Я28
Я 31
яз2
Том
I
II
I
III
III
III
V
VI
VII
V
I
II
II
II
Стр.
360
XXIII
362
94
96
96
17
22
32
19
363
XXIII
XXIII
90
странственная
группа
т\
ИЛИ
о\
у1/
т\
&2о
u2d
и12
U2h
"б
о{°
1>1
Обозначение типов
по координационно-
структурному
принципу
если и ф -£
1
если и = —
v
—
—
0RTtTjT> см* Eii
TrjriTjT
—
n 9/4/4,3—л h-M/4/4,3
—
S
Наименование
у-Квасцы
NaAl(S04)212HaO
Н8Р(\У8О10)45Н2О
Ag2S044NH3
LiC1043H20
КАпВг42НЮ
3CdS048H20
Н3Р\У1204о29Н20
BaNi(CN)44H20
Манганлеонит
K2Mn(S04),4H40
Кианит AljjSiOs
Силлиманит
Al2Si06
Андалузит
Al,Si06
Тип
(SB)
"415
"*,«
mxl
"418
Я4И
Я420
H421
Я422
Я423
Я51
sox
Я52
so3
Я53
£02
Том
III
III
III
III
IV
IV
IV
VI
VII
I
II
II
II
II
II
II
Стр.
112
113
115
117
50
52
56
23
32
427
109
XXIII
112
XXIII
• но
XXIII
странственная
группа
ге
oi
D*2*
^6»
C2h
Г6
°2h
о\
—
Г3
b2h
о}
^
*>%
Обозначение типов
по координационно-
структурному
принципу
С-. ._
с+о/с+о/г+г
—
ТЧП
—
мр+р/р+р
—
-^б+4/g/l/l
—
Описание неверно 1
— 1
K2CuCl42H20
1 Квасцы
KA1(S04)212H20
BeS044H20
1 Соль Туттона
(NH4)aMb4S04)26H-0
NiS046H20
Гнис CaS042H20
Я41
Я4,
Я42
Я42
Я43
Я44
"45
Шш
CaS04 V2H20 j Я47
Li2S04HaO j Я4
PdlNHj^Cl.H.O Я49
CuS045H20
Mg(C104),6H40
NiS047H20
Я410
Я4и
Я412
1 a= Квасцы ■ Я413
KA1(S04)212H20|
3= Квасцы
NH3CH3AKS04)2 •
Я414
• 12H.0 ■!
1 i
I
II
I
II
II
II
и'
II
IV
VI
III
III
III
III
III
III
III
III
336
ХХШ
369
XXIll
91
9)
95
97
47
99
97
98
100
102
103
105
108
HI
D14
4/t
re
<
^2Л
*>\
r3
°2/i
°2/i
o\
VI '>
C$
»\n
c\
c1
b2v
D*
T*
t\
11
T P4[V
Неверно, см. Я41я
TSJS
—
Неверно. Должно
быть:
м%т
■—
^g + Z'4/r-i г
—
—
—
Сс-к7/с+о/7Ч-г
С —
С M0/C-I-O/7 -YT
Ставролит
Fe(OH)aAl4Si2O10
Топаз AI2F2Si04
i Тихит
Na.Mg.SO^CO,).
Апатит
Ca5F(P04)8
Сульфогалит
2Na2S04Na2ClF
Аутунит
Ca(UO>)2(P04)2
lOViH.O
Метааутунит I
Ca(U02)2(P04)2
6V.H20
KaPtCl6
K2Sn(OH)e
K2Pt(SCN)e
Ni(N03)2(NH3)6
(NH4)3A1F6
Я54
S0A
Я55
s%
Я5в
Я57
Я58
Я58
Я510
Я61
л,
Я62
Я63
Я6 4
/14
НЛ1
J2,
II
II
11
II
II
III
VI
VI
I
II
I
II
I
II
I
II
1
II
113
XXIII
113
98
99
118
24
25
429
XXIII
431
XXIII
433
XXIII
435
ХХШ
437
ХХШ
»11
*>2П
ol
с6/1
0Ъь
7)17
D\b
of,
Dld
Dh
т%
n
T7i (?)
—
—■
v2+q/0
—
—
Ctjc Cm- Jii
Rj/c Cm- Mi
HpGJPS ^*M' ^*з
С С IT См. /14
с
^C+0/7\ О ^М* ^2l
II родолжение
Наименование
Тип
(SB)
То.ч
Стр.
Про- !
стран-
стненная
группа
Обозначение типон
по координационно-
структурному
принципу
Найменонание
Тип
(SB)
Том
Стр.
Прост ран
ственная
группа
Обозначение типов
по координационно-
Структурному
принципу
K.PtCI,
K2Sn(OH)6
K2Pt(SGN)e (и
[Са(Н20)в]С12)
J. Решётки с радикалами ВХв
С
Ni(N03)2(NH,)4
KtOsO,Cl4
(NH4)4SiFe
Mg(H20)eCh
[Rh(NH3)4CI]Cl2
Я61
/1,
//62
Я63
/la
Я64
JU
JU
/17
/1,
I
III
IV
VII
I
II
I
II
1 I
II •
III
III
III
III
429 1
121
60
34
431
XXIII
433
102
435
XXIII
122
123
124
125
o5h
ь\й
Tt
r>17
*>h 1
°2h
*A
TfC
Rt
i/c
Неточно. Эта
пространственная груп
па сохранена для
Са(Н20)вС12. Для
K,Pt(SCN)e~C^
ЯР6/РЗ
С--. Близок к/1,0
Лд см. тип С6
М*цХ Решётка пере
ходная от
координационной к
молекулярной
К. Решётки со сложными радикалами
Пиросульфит К
K2S205
Дитионит К
K2S2Oe
Cs2S2Oe
CS3C0C16
TI2A1F5
NH4Pb2Br6
КН,(Н2О)2В5О10
(NH4)2S2Oe
K2S3Oe
ZrP207
K2NbF7
клг
K2,
K2Z
ки
кзг
кзг
JT34
к%
\к^
къг
кьх
#6,
11
II
II
II
II
II
III
V
V
V
III
III
III
IV
VII
XXIII
104
XXIII
106
XXIII
107
134
20
22
23
135
138
140
68
36
Г2
°2h
*i
D%
D%
b\ j
Л18
r17
^2/i i
7)16
Л
°2/i
В одном томе SB
разное
обозначение типа
«
М
4/4
J0( О
М
х/у
U°71
ТИПЫ СТРУКТУР
257
1
МО
О
С1
"^
•"■'♦
-
Гч
к
t>
со
*
С,
О
24-
и
ф
1С*
£-1
С;
rr T
^
~
-^
>
^
О
ч»^
'О
5,
'Г
<!
с
н
J2S
X
I
•о
ом
Q
т
<г
*~'
е*
Гч
к
О
-м
н
CJ
о
ее
о —
a,cj
a гз
1
С1С0
ь*,
Ю
^
•""'
ео
гч
t*
91
<
CJ
|ГЧ =
)) Я
гч а
^:Ф
±1
сл ,—
^
СО
о
>
^
О
Я
О
&
£
iS3
1
0"0
Ь,
00
о
>
1—4
к
СО
О
-<
О
СЗ
а
ч-(
CJ
<w
1С
-с
"Ч*^
О
о
сч
>
^
^
■в
л'
'Л
СЗ
Ъ
\
_
О **
^
^
к-*"
О
^
fa
^
Л
г:
5
с
X
Он
7;
мсо
*«ч
Гч
<М
>
^
Я
се
Л
1
!>■=:
11'Ч*
Q
<70
СО
х-*
»
Гч
«С
«0
о
<
р
п
S
о
со
й-
о"
й-
т=
coco
■21
..*
со
>
со
•-«5
ф
3
<Х>
с
о
ft
в
о о s
ь а н
H°S
? § «
4 Я
Л о с;
25 Я <j С
2
d
Б
О
^
„
Л
с
—
с Е
' «р
^ • &-< ^
а я
—
Гч ич Гч
СО к> <М
X
- = 5
£ <^
£:^
<
ет
«г
Я
5,
п
Л
лс
Ьн
ь
X
н
с;
^
-г.
<—'
<«-<
ь^
С
<
ф
2
|гч
со
•^
осо
^
Гч
<м
S
са
»**
с
со
/—Ч
о
X
ф
о
н
>о
ОО
<Г
^
<?-•
ь»!
'3
X
о
ф
Я
г.
О
о
Он
I
о
О
С'
с*
=
«
<м
^
■л
5
Л
a.
н
О
■f
О
с
со<
&N
v.O
О
>
^
«о
О
'Л
^
*
,.->
00
<г
нн
^
со
и
<
у—ч
со
PC
о
X
Л
А
о
00
<!<
ГГ\
«е-<
^3
р
и
о
ф
VC
*^.
I
ю -с
О
00
с<
>
>-»
CN
^
'ф'
СО
л 5
а,н
^ w
ь к
5 5
^ ф _.
Гч
со
**
о
^
к4
г:
fa
с*
^2
^;
ЮМ
О
а>
<N
>
0-I
^
<
СО
Л
S
ч
о
я
*
Л
со
Л
лс
>iS
н
^
>»
а,
ф
с
с
Гч
ОО
<г
о
сч
^
1i
Ou
I
i>.c:
•п4*
Q
•н
со
5
»->
О
я
о*
«0
г-Н
со
н
к
к
S
ф
В
ф
S
•ч
Я 00
чО
<м
о
-
ф
о
н
00 1
ос 1
** 1
»-н 1
(М 1
^
с 1
5
с< 1
О
^
О
Я
С
ее
2
1
П родолжеиие
Наименование
Тип
(SB)
Том
Стр.
ранствен ну я
группа
Обозначениз типов
по координационно-
структурному
принципу
Наименование
Тип
Том
Стр.
ранственная
группа
Обозначение типов
по координационно-
структурному
принципу
Sb,TJ7
Мартенсит Fo—G
содержит до 0% С
Кианит ALSi05
Андалузит
AT.2Si05
Силлиманит
AuSi05
Ставролит
Fe(OlI),AI4Si,O10
Топаз Л1 ,F,Si04
Титанит
CaTiSi05
Силикаты ряда
хондродита
Ms(FOII)., • п •
• М^>Ю4
L22
L'2U
488
489
I
S. Силикаты
7751
НЬЛ
so±
Я 53
so2
, #52
^о3
н:и
&0А
\ ИЬЪ
Мб
; SQ6
SO,
I
II
II
II
II
II
II
II
П
11
II
II
427
109
XXIII
110
XXIII
112
ХХШ
ИЗ
XXIII
ИЛ
117
119
°'
Z)12
u2h
I)16
u2h
I)11
U2h
J)16
u2h
b2h
Переменная
Сверхструктура
замещения к типу Al
Сверхструктура
внедрения к типу
А2
Неверно. См. SB II
Бенитоит
BaTiSi309
Канкринит
3(Na2AL04 • Si204)
Волластонит
CaSiO,
При п—чётном—
моноклинная, напр.,
пр. гр.С?/гС^л;цри
п—нечётном—ромбическая, напр.,
пр. rp. DV}t
Хабазит
CaSi4Al20146H20
Катаилеит
Na,ZrSi3092H20
£32
j ^33
S*z
S:\,
S34
II
III
IV
III
V
128
150
71
151
24
Ȥ/.
'2h
D
31
D
6ft
В SB допущена
путаница—к одному
типу отнесены: кан-|
кринит пр. гр. Cq
и волластонит пр.
гр. С|л, не
имеющие даже сходных
эле .ментов
структуры. В волласто-
ните (тин А/2/б)
образуются кольца
Si309, сближающие
его с бенитоитом
В SB допущена
путаница—к одному
типу отнесены:
хабазит пр. гр. I>3d
и катаплеит пр. гр.
Дунпит
Ali3Si->O20(OH,F)i8Cl
Элклаз
HBoAlsiO,
Цирк >н
ZrSi04
Олшшн
M.^Si()4
Фенакит
Be.SiO,
Гранат
Al J'a^iSiOj).,
'«>1МИТ1Ш
13i4Si3<>1:!
Тортноитит
Se.Si.O-
Ге мн мор фит
- ILZibSKX
BeavmiaH
ь Са10М^..Л14:
му()у4Ю1И4
r>oj)JM.r
Bc3AUSiOjie
SK
Si)«
Н'Л
//03
s\;
//12
//1,
.91,"
//15
//13
<Sb
//:n
•VU
52я
Л;;,
HI
III
14;
148
I i r.5
II 'XXIII
лм2
C%
III
I
II
II
I
II
II
I
III
У1
552
ХХП1
XXIV
856
XXIII
XXIV
:?r>:t
150 I
ih
'Ah
,-H,
<U
>io
i22 ; /;;
ii
и
12'.
125
126
C?
2Л
^20
/У
4Л
:;o:> /;5/l
Г
'/ /W/2 ; 2
0«
г
О/7/З- 1
n'I ft It
Длинен д
CaMg(Si03)a
Тремолит
JLCa,M.ir5(Si03)
;)|И'татпт
.V'i.
MtfSiO»
Хризотил
• Н4М^ь09
Бертрандит
Ве4м207(011)а
Эпидндииит
UNaBe>i3Os ;
Mvckowit I Л5Х
'KILAIgSijOj. '
Анофпллнт | Л5,
K(:a4Si,O20K-81I,U
Гардштонит
Ca,ZnSi207
Накрит
AI2SL05(OH)4 ;
.S'5,
i
i
и i vm)
II I 131
ii
Антофиллит S\i II
IUM-7(SK)3),
«5 И
S\e II
54; III
II
II
II
III
VII
In |iot]ijf.i.i]jT ЛГ
Аи<14()10(ОЦ),
154
Ш
159
141
155
145
145
146
155
37
c!
Группа хлорита ; Л"5^ \]\ \ \:
■VliM^iaO10(OH), j
2/1
°2Л
/>15
,f2h
Cr
2Л
r12
°2u
л16
Lf2h
°2Л
/>
//
.Л
3
2-V
4
Г4
L S
' 2Л
III ! i:.y
(Л
2Л
Моноклинный пм
фи бол
Ромбический ни
рок си
Мпп
0/Т/Р6
В SB III неверно.
См. SBVII
Продолжение
1 Н именование
1 Кропстедтит
1 Fe^Fel11 .
•(OIDeFe^SiaOjo
1 Анальцим
NaAlSbOJI.O
1 Содалит
Na4(AlSi04)eCl
1 Данбурит
1 GaBjSLOg
1 Скаполит
Na4C1(A1Si308)3
1 Карнегиит
NaAlSi04
Ka2CaSi04
1 Санидин
KAlSiaOe
1 Альбит
NaAlSi308
1 Гаюин
Na,(Ca, К)., .
• (AltSif024) -
Патролпт
Na,A1>Si3O10.
• 211*0
Тип
(SB)
*У57
£6 J
Sot
S63
£64
Sn.
Sbt
S67
568
So9
S(>
\
Той
VII
11
! VI
II
II
II
П
II
III
III
III
III
1
Стр.
40
148
1 34
150
153
155
158
159
161 1
1G4
166
168
ранственная
группа
Г4
°з
*>«*
(4°)
^
П16
С\н
Ti
Г4
clh
с)
d
^.1 Q
с11
1
Г Обозначение типов
по координационно-
стуктурному
принципу
—
Псевдокубический
—
—
—
—
—
—
—
—
—
Наименование
[NHsCH,LPtCle
[N(CH3)3C2H6].2 .
. SnClG
Ь\(СН3)2(СЛ15),] •
• SnCI.
[NII3C2H5]2 •
• SnC1e
[(CH3)8NH]2SnCle
I(CH3)4N]2SiFe
[(CFI3),NH2]^nCle
Тип
(SB)
0//62
Of/65
ОД 66
OHM
OJ
OJU
OJi
« i
Tom
I
I
I
I
II
III
III
Стр.
611
611
612
613
805
653
658
II. Чисто органические
CH4 1 01
1
Тетраметил метан
(СН3)4С
0
i
1 ':
с.т4 ! оз !
I
1
I
II
I
I
V
из SB)
613
805
614
614
155
Прост-
I'll!
Л5 9
I;3<J
7-16
Г6
/)3
1 3d
T6
u4h
Civ
Обозначение типов
по координацией >
структурному
принципу
Близок к Я62
Близок к H(>i
Близок к HGy
Близок к Я63
—
Близок к Я6Х
—-•
соединения (заголовок |
тЪ
ci
Old)
T}
C}t/==(T).
Кубическая гранецентр.ре-1
шётка из тетраэд- 1
рзв СН4
Как /14 (алмаз)
См
О. Нее .торые из типов, причнелевиые в SB б i егистру
органических веществ
I. «С мешанные» соединения
NH(CaHi),J
N(CH,)4J
N(Crt3)4JCI2
NH3CH8J
NH3CH3C]
<Crf,)4Si04
<свн5)Л
SbBra(CH8),
[As(CH3)aPdBr2]2
Au(C3H7)aCN
(CH,)4NC104
GH3SHgCl
K,Pt(COS)4
LN(CH3)4],PlGle
OBW
OB\Q
OBW
OB20
OBl\
ОГ23
01Л,
OGU
OG\2
OE\x
OHi\
OtfO;
OH%
ОЯ61
I
1
VII
I
II
I
II
II
VI
VI
VII
11
VII
III
I
60 b
607
215
607
803
609
803
803
199
201
216
804
219
655
611
oU
»\n
J)3
U1d
»\b
Г1
Tl(?)
D*
»L
('4Л
oL
i>L
o\h
Г6
rp2^
1 d;
Близок к В 4
Близгк к В 10
—
Близок к В 10
Близок к В 2
—
—
—
—
—
—
—
—
Близок к НЬХ
Пентаэритрит
G(GH2OH)4
Пентаэритрцт—
тетраацетат
C(GfI2OCOGII3)4
Пентаэритрит
тетранитрат
C(CH2ON02)4
и тетрафенил
метан
С(С«Н5)4
CHJ3
Метальдегид
(СН,СНО)4
Мочевина
CO(NH,)2
Тиомочевина
CS(NH2)2
Гуанидоний
бромид
C(NiL)3Br
Гуанидоний
иодид
C(NH2)3J
О'*
! 0412
05
Об
011
01а
021
02,
022
022
С24
0%ш
,
V
I
I
I
II
IV
I
III
I
11
III
III
615
140
615
015
617
805
253
617
600
619
805
661
663
V2 1
Л4 1
c\h 1
v*
c\
rfro
1 /I
*>%
cU
TP4JI>
T14iP4
ТрАЦЧ
SB I
(рис.265).|
Собствен-!
пая сим-I
метрия
мо леку л|
в типах
04, 05 и |
06: SA.
Структуры отли-j
чаются
ориенти-|
ровкой
тетраэд-I
ров
Неверн)
или KJJ, или iSj
Ион гуанидония
плоский
треугольник с атомом С
в центре
То же
Продолжение
Наименование
[Cu(8 =
UC(CH,)(NH1)JC1
(C.H«>«As • Cul
Н8(8С,Н5)г
с,н,
с2н4
с2н4ь
Ве40(С02СН3),
NaUO,(CH,COO),
ВаСа,(СгН5СОО),
Тип
(SB)
01,
027
02,
031
038
оз4
041
04,
04,
043
Том
IV
IV
V
I
III
III
I
III
III
III
Стр.
255
258
142
619
665
665
621
666
668
670
Прост-
lip
si
тз
с54
—
b\
Г5
b2h
Th
ТА
ol
Обозначение типов
по координационно-
структурному |
принципу
__
—
Идентичен В^Н6
тип D 41
или D\h
или ВЦ
Текст SB I заменить
на текст SB III
—
—
Наименование
^-щавелевая
кислjTa
(СООН),
р-щавелевая
кислота
(С00Н)*2Ла0
Оксалат калия
KdC204
(СООК)3НаО
(COONH4)2lLO
iAg(U02) (СНзСООз-
■ ?н2о
cecie
Тип
(SB)
044
046
046
047
048
04,
0410
0517
Том
III
III
III
IV
III
III
IV
IV
II
Стр.
671
672
673
2э0
674
675
252
2»4
808
ранственная
группа
l)2h
С%п
с\к
Г6
*!
V*4H
Cln
Обозначение типов 1
по координационно-1
структурному 1
принципу 1
—
Текст SB III
заменить на текст SB IV
—
—
—
—
—
I
§ 108] о задачах химической систематики структур 263
§ 108, 0 задачах химической систематики структур
Задачи, стоящие перед химической систематиком структур, по нашему
мнению, состоят из следующих^ частей:
1. Установление условий возникновении тех или иных
кристаллических форм данного вещества и термодинамических границ их устойчивости.
2. Установление зависимости между различными структурами данного
вещества, с одной стороны, его составом и строением атомов—с другой.
3. Установление рациональной связи между структурами различных
веществ, в отличие от формальной системы, принятой в SB.
При сопоставлении и подытоживании материалов в целях создания
химической систематики структур обнаруживается, что не только
отдельные простые вещества, не только соединения отдельных элементов, но и
некоторые группы периодической системы (напр., ванадий, ниобий, тантал)
и целые классы химических соединений изучены очень скудно. Более
того, в работах рснтгенографов часто наблюдается пренебрежение чисто
химическим и термодинамическим исследованием системы, изучением
наличия примесей и их влияния на возникновение структурных форм.
Недостаточно обращено внимания на установление условий возникновения
границ существования реальных фаз, а также условий фазовых
переходов, в частности химических составов, ограничивающих пределы
устойчивости фазы и отвечающих этим границам изменений элементарной ячейки
и т. п. (на необходимость иного подхода к этим вопросам неоднократно
указывалось в работах автора и его лаборатории, начиная с 1936 года).
Всё это ведёт к тому, что в нашем распоряжении далеко не всегда
имеются сведения, необходимые для построения полной и всесторонней
систематики структур на химической основе. Химики на сегодняшний день
часто вынуждены пользоваться данными, которые не являются
удовлетворительными .
Естественно, что мы не имели никакой возможности охватить
в сравнительно небольшом объёме книги все известные структуры. Мы
ограничились тремя значительными, важнейшими для неорганической
химии разделами:
Простые вещества (гл. III).
Простые соединения (гл. IV —VIII),
Сложные и комплексные соединения (гл. IX — X). В этих главах
рассмотрены некоторые из наиболее важных объектов.
Интерметаллические соединения требуют составления специального
тома, что, возможно, явится предметом работы в будущем.
Литературные данные в этой области —см. часть III.
ГЛАВА IV
СТРУКТУРЫ ПРОСТЫХ ВЕЩЕСТВ
§ 109. Об обозначениях модификаций и структурных фори
В современной литературе наблюдается большой разнобой в
обозначении различных структурных модификаций и вообще различных фаз.
Вызвано это тем, что исследования фаз проводились под разным углом
зрения и в разных отраслях наук: в неорганической химии, в физико-
химическом анализе, металлургии, рентгенографии, физике.
Во всех случаях, однако, для обозначения энантиотропных
модификаций в однокомпонентных системах, как правило, пользуются буквами
греческого алфавита.
В неорганической химии устойчивая при более низкой температуре
энантиотропная модификация (однокомпонентная твёрдая фаза) называется
а-фазой, следующая р-фазой к т. д.
Так обычно приводятся для железа следующие границы устойчивости
фаз (в скобках —точки превращения по Тамманну):
aFe - (740 780) - pFe - (900) _ 7Fe - (1411) - 8Fe.
Се Cq Ck Cc
Напротив, в ряде трудов в области теории аллотропии, изучавших
систему с позиции правила фаз, часто термодинамически устойчивая при
более высокой температуре форма обозначается «а-фаза», устойчивая при
более низкой температуре <ф-фаза» и т. д. (например, [7]).
В определении некоторых авторов, если твёрдая фаза находится во
внутреннем равновесии, она называется (см., например, Смите [7])
модификацией, если нет —то кристаллической формой.
Однако в большинстве работ, особенно физических, различие подобного
рода не проводится.
Рентгенография в подобных случаях следует обозначениям авторов-
химиков, стоявших на тех или иных позициях (см., например, SB I Fe,
Sn и др.).
В случае же, если химики не присваивают обозначения а- и
'^-модификаций структурно разным фазам, рентгенография в данное время также
воздерживается от унификации системы обозначений; например, описываются
ZnS сфалерит и ZnS вюрцит (типы SB — ВЗ и SB—JS4), но не aZnS или BZnS.
Наконец, если обнаруживают тонкие различия в структурах,
например, CSi, рентгенография вводит нумерацию фаз римскими цифрами CSi I,
CSi II, CSi III и т. д. (см. SB I, тип ВЪ и след.).
При изучении двухкомпонентных и других более сложных систем,
например Си—Zn, твёрдые фазы обозначаются разными буквами, в
зависимости от состава. ^
Наиболее богатая одним из компонентов (в данном случае Си) фаза
обозначается а, следующая [J и т. д. Для обозначения степени упорядочен-
§ 110] ОБ ОБОЗНАЧЕНИЯХ ВЕЛИЧИН, ПРИНЯТЫХ В ТАБЛИЦАХ 265
ности фазы, например при температурных переходах и т. п., пользуются
штрихом, например, (3 и р'-фазы системы Си — Аи.
Жидкие фазы обозначаются буквами из середины греческого алфавита
л, а и т. п., например, для серы SA, S^.
Предполагается возможность постепенных переходов в зависимости
от соотношения разных структурных компонентов в жидкости (цепи,
кольца и т. п.), определяемого равновесными (и кинетическими, см. гл. I)
факторами.
Нами применяются обозначения фаз, принятые в SB, с
соответствующими комментариями, вытекающими из сказанного в данном параграфе
и гл. I.
§ 110. Об обозначениях величин, принятых и таблицах гл. IV — X
и связи между ними
a, bf с —периоды элементарной ячейки.
aw — периоды элементарной ячейки в кубической сингонии.
d, е,/ит. п —межатомные расстояния.
г— радиус атома (равный для простых веществ у J .
vA — объём элементарной ячейки (из рентгенографических дан.
ных) в кХ*, за исключением случаев, где это оговорено
обозначением (А3).
V А—грамма томный объём в (см*) м. л. (миллилитрах).
zx~плотность вещества (из рентгенографических данных).
ац, aj_ — линейный коэффициент расширения параллельно,
соответственно перпендикулярно главной оси.
аа, аг —то же параллельно осям а, соответственнно с,
И — средний линейный коэффициент расширения
поликристаллических тел.
Очень часто лица, пользующиеся структурными данными, не
представляют себе чётко связи между различными расстояниями в элементарной
ячейке, даже для простейших структур Сс, Ск, Ну С этой целью мы
помещаем ниже три графика для структур Сс и Ск и три графика для
структуры Ну* нуждающиеся в следующих комментариях.
Решётка Сс- Кратчайшие межатомные расстояния
d=««y± = 0,866 аш (lgd = lga+T,93753). (1)
Таким образом, d является линейной функцией а. Объём элементарной
ячейки v = АшА3 (lgu = 31gaw). Кривая, отражающая зависимость v от аШУ
принадлежит к семейству кубических парабол.
Объём одного атома (в SB «атомный объём») равен w =^-АЛ т. е.
для Сс: w = тгА3. Грамматомный объём равен
г V 6,02 X Ю23 з л огн /о\
т. е. является линейной функцией от и и параболической от aw.
Удельный вес ох = у , где А — атомный вес элемента. (3)
Решётка Ск. Кратчайшие межатомные расстояния
d=±^ = 0,101 aw(lgd=lga+T,8i9i9). (4)
266 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гЛ. IV
Объём элементарной ячейки и = aw A8 (lg о-=3 \gaw). Объём одного атома
равен #>= ^АД т. е. для C%w = т-А3. Грамматомный объём
V 6,02 • 1023
V
1Q24
- 0,1505?; см*
(5)
также является линейной функцией v и параболической функцией aw.
Удельный вес zx -■= -^ .
йО А с\Г «чГ «sf «чГ «^ *4N *s* «nj4 *<Г *сГ *tf CL*u,8ooa
-I 1 1 I
-I 1 1 г
§ в §§
do/j 45,00 <^ S ^ ** ** «sr «sr <n* *tf <nt Nr
1 Ы707а\
6Л5
щЦ1505и\
Уем3
Vf0,301v
-*~a
«О ^ 2?» ^ » ^i Cs ^ ^ ^ .^ _ f J
^ ^ n « ^ § ^ ^ § « ^ aw6A
Csj4 Cvcf Csf <\j «Nj4 fcf ^ C^ C^ C*£ C*^ W
В соответствии с изложенным, графики для Сс и Сд;-решёток построены
следующим образом.
График (рис. 151) относится к веществам, для элементарной ячейки
которых яш = 2,5 — 3,5 А.
§110] ОБ ОБОЗНАЧЕНИЯХ ВЕЛИЧИН, ПРИНЯТЫХ В ТАБЛИЦАХ 267
График (рис. 152) aw = 3,5-—4,5 А. График (рис. 153) aw =■■= 4,5 —5,5А.
Значения аш нанесены на абсциссе —нижняя сторона прямоугольника.
Значения d — на верхней наружной стороне для решётки Сс и верхней
внутренней стороне для решётки Ск- Если aw различаются у двух веществ на
0,1 A, d у тех же веществ различаются в случае: решётки Сс — па 0,0866 А,
решётки С к— на 0,0707 А.
Значения аю и соответствующие им d соединены вертикалями.
Значения с находятся с помощью параболической кривой v = aw A3
и нанесены на левой стороне графика. Соответствующие объёмы одного
do A *>$*?*$ t^ «*>ч ** ** «•* *s c*s *>* а=цоиоа
lCc
vc=0,301v
Рис. 152.
атома для решётки Ccw =■ ~ А3, для решётки Скъи = ~к*
рассчитываются без труда.
Величины V являются линейной функцией от з;. Они нанесены на
правой стороне графика и лежат на тех же горизонталях, что и соответ-
268 ч. и. структуры неорганических веществ [гл. IV
ствующие v. Для решётки CjF = 0,301u, для решётки СкУ = 0,1505 о *)
Зд. =:■ 21 СМ •
Пример. яш = 5,10Л. Решётка Ск: d = 3,6062Л; и = 132,651';
= 33,16 Л3; V - 19,965 см\ Решётка
г# (не показан на графике) = ——f—-
э «s S «
>
ггА3
dBA
-~7Ш
S ^ J^ 5* *ч «5
^ ** v v v v v ^ ч4 v v d=0.856t
,—i t \—i г
II 5 I I I I fc I I
|frfr c*g с*£ t^T *a* *У 5t »g **f fr£
§§ £ S i
ад
Vc„s \v*(l30tv
V V* ^ V ^ ЧГ »ЧГ <С* *«* ^ Vc} UWU"
Рис. 153.
Сс: d = 4,41ee А; и = 132,65 А8; го=^^ -66,32 А8; V = 39,93 си3; зх в
обоих случаях рассчитывается по (3), г равен - . В случае плотной
гексагональной упаковки Ну d = a, Z = 2. Объём ячейки равен
v =-
: 0,866 а2с А8 = 0,866 ha2
(6)
где й= — , т. е. высота элементарной ячейки, выраженная в единицах а.
а
*) Если числовые значения v выражены в кубических ангстремах, значения V
получаем в ем3.
§ 110]
ОБ ОБОЗНАЧЕНИЯХ ВЕЛИЧИН, ПРИНЯТЫХ В ТАБЛИЦАХ
269
г ■■ V V
Из (6) легко вычисляется и атомный ооъом го = ^ = -^ »
Объём одного грамматома V =0,301 v см3 (как в случае
уравнение (2)).
Например, принимая для ".Л
1,625,
55,57
5082
56,00
46,34
24,86
С с) (см.
• W
Щ25
/
/
+
X
/
-/-
Г
/
/
Mg а- 3,20А, с/а
найдём ?;, tcy V см*:
ю = 0,866 xl,025x(3,20)3^
= 46,11 А3;
м; = 23,05А3;
V - 13,88 еж3.
Согласно (6), кривая
^ <= f (д) при /г = const
является кубической параболой.
Значения ?; = /(а) при Л =
= 1,633 —см. на рис. 154,155. ^12
С изменением h возни- '
кает семейство таких кри- w,uo
вых; чем больше //, тем боль- Д^
ше ?; и все линейно
зависящие от о величины (w, V, a 34,49
и т. д.). Hoa = d. Таким
образом, шесть межатомных 2104
расстояний d в основании 30,00
-элементарной ячейки не оп- 2783
ределяют сами по себе объём
элементарной ячейки.
При одном и том же
d( = a) объём ячейки может 22,09
быть больше или меньше иде- оппп
ального (которому отвечает ' -
к = 1,633, возникающее при
плотной укладке шаров по
гексагональному закону). *'
В этом случае (h Ф 1,633) шесть межатомных расстояний е будут либо
'больше а (с/а > 1,633), либо меньше а(с/а < 1,633), ибо
4" - " У """ 12
При h = 1,633 е = а.
Значения ?? и других величин для /г = 1,633 мы будем называть
идеальными (иид и т. п.). Соответствующая кривая на рис. 156 отмечена
стрелкой.
Величина К = ^-Г~п 0ПРеДеляет степень отклонения v от иид.
На рис. 156 представлена иИСт == / ("ид) при разных значениях с/а,
соответственно v.
Каждому значению ft, а следовательно, и К отвечает прямая, для
которой tga = K, при К=19 а = 45°. Из рисунка наглядно видно, как
увеличивается (соответственно уменьшается) v с ростом (соответственно
уменьшением) h против 1,633.
В следующих параграфах мы переходим к описанию структур простых
веществ. В ряде таблиц указаны том SB и страница, где помещены сведения
о рассматриваемом веществе. Это не означает, что комментарии к данной
V-
J I ,, I
16,73
15,30
13,95
12,68
11,49
10,38
9,34
8,38
m
6,65
stages
cs^ cv^ esj4 c^J4 «^ «^
c-* c*j»
Рис| 15't.
/t + v-«/-
270 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ (ГЛ. IV
структуре излагаются по SB. Сплошь и рядом они не отвечают взглядам
и установкам SB, как это неоднократно подчеркивалось и в предыдущих
разделах киши.
Пользуясь материалом глав IV—X, надлежит учитывать сказанное
в § 61 о связи между единицами кХ и единицами А% В этих главах всюду,
где это не будет оговорено проставлением значка Л, расстояния в
элементарных ячейках и т. п. даются в кХ.
В таблицах, вместо того чтобы писать, например, 4,257±0,002, мы
пользуемся, краткости ради, обозначением 4,257 ±2, причём число, набранное
курсивом, обозначает, чти
К..
130,00
тм
120,44
120,00
112,43
110,00
104,76
100,00
0,45
90,50
90,00
83,88
8000
71,53
71,52
70,00
65,98
6d,62
60,00
/
-h
4-
поправка выражена в
единицах, определяемых
положением последнего знака
предыдущего числа.
Как было указано в
предисловии к книге,
графический материал SB был
подвергнут нами
критическому пересмотру и
переделке.
Элементарные ячейки
строятся с применением
«киеобразных линий» п
линий различной ширины,
подчёркивающих
положения атомов на переднем
или заднем плане, и т. п.
Атомы металла, как
правило, показаны
меньшими кружками чем атомы
неметалла, разные
неметаллы (соответственно
металлы) отличаются разной
штриховкой при одной и
той же величине «кружка».
Границы
элементарной ячейки на сложных
чертежах иногда
отмечаются пунктиром. Жирные,
двойные и т. п. линии для этой цели не применяются вовсе. Лишь
в отдельных сложных структурах пришлось допустить исключения, если,
иначе, чтение чертежа могло быть затруднено.
В таблицах, при наличии соответствующих сведений, проставлена графа
«температура» для указания температур, при которых проведено
измерение. В таблицах SB, а часто и в самих исследованиях, к сожалению, такие
сведения отсутствуют; имеется в виду комнатная температура; зто иногда
наблюдается и тогда, когда размеры ячейки приводятся с точностью до
одной десятитысячной ангстрема. Подобная точность без
фиксирования температуры теряет смысл, поскольку при возможных колебаниях
«комнатной» температуры от 16 до 25°С, т. е. в пределах 10°, при обычных
порядках величин линейных коэффициентов расширения заметные
колебания и особенно в четвёртом знаке после запятой вполне воз
можны.
В следующих параграфах мы переходим к освещению структур простых
веществ. Этот очень важный раздел неорганической химии был объектом
С At"
38,78
36,25
ЗЩ
31,53
29,33
27,24
25,25
23,35
21,56
19,86
18,25
Ч
**
§
**
S3
§8
5
^
Рис. 155.
§ \ ] 0] ОБ ОБОЗНАЧЕНИЯХ ВЕЛИЧИН, ПРИНЯТЫХ В ТАБЛИЦАХ 271
многочисленных исследований, и некоторым из веществ посвящены десятки
работ. Так как советские рентгенографы в большой мере сосредоточивали
своё внимание на более трудных объектах, важных для создания общей
теории структур, -ла исследовании химических соединений, особенно на
сложных и комплексных соединениях и т. п., —то рассматриваемый гораздо
более простой в методическом отношении раздел представлен многими
10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160
Рис. 156.
V.
данными зарубежных авторов. И вот, при рассмотрении результатов
их работ почти за i десятилетия, особенно остро ощущается
бессистемный и бесплановый характер развития буржуазной науки, недостаточная
глубина большинства исследований.
Существенным недостатком большинства работ зарубежных рентгено-
графов в этой области является оторванность от тех или иных теоретических
проблем физической и неорганической химии, измерения ради измерении.
Эти работы часто трудно назвать и чисто методическими, поскольку многие
из них выполнены па невысоком экспериментальном уровне и с
недостаточно чистыми препаратами. В частности, у автора как химика, особенно
вызывает неудовлетворённость то обстоятельство, что какую бы из групп
периодической системы пи взять, почти ни в одной не решён сколько-
272
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. IV
нибудь надёжно вопрос о существовании и границах устойчивости
структурных модификаций в разных интервалах температур и давлений, а так-
как подавляющее количество исследований относится к препаратам
сомнительной чистоты, то использование этих структурных данных в столь
важной для химии и физики общей теории фазовых переходов
затрудняется, а многие исследования, за ненадёжностью, вообще не могут быть
приняты во внимание. В противовес многим таким работам зарубежных
авторов резко выделяются прецизионные исследования советского учёного
Евинса, выполненные по асимметричному методу, а также ряда других
исследователей (см. § 2 и ниже).
Проблемы строения простых веществ и условий фазовых переходов
требуют большего внимания к ним советских исследователей.
Советская наука должна в ближайшие годы с присущей ей
плановостью взяться за наведение порядка и в рассматриваемом разделе
структурной неорганической химии.
§ 111. I группа: литий Li,
натрии Na, калий К,
рубидий Kb, цезий Cs, экацезий
(№ 87) s\
1. Литий, натрий, калий,
рубидий, цезий
Решётка кубическая С
центрированная.
Координация С.
Тип Cc-SB.42.
Пространственная группа 09h.
Jm3m.
Z = 2, к.ч = 8
(расстояние d
_<^V\
) + 6
(расстояние e = aw).
Рис. 157.
Атомный
номер
3
11
19
37
55
Вещество
Литий [1]
Натрий [1]
1 Калий [1]
Рубидий [1]
Цезий [1]
t°C
—173,1
комн.
—173,1
комн.
---173,1
комн.
-173,1
1 —173,1
aw
3.4,
4,2,
4,30
5,2,
5,20
5,6.
! б,о5
d
ЗА
з,о,
3,6,
3,7а
4,5.
4,5,
4,8,
5,24
г
1,5,
1,5,
1,8,
1,8,
2,27
*.25
° 4
2,6,
о
w А3
214
ЗУ8
70,3
—
"
Уем*
13,1
23fl
24,г
43,9
44,9,
53,84
67l3
*х
0,55
0,53
0,995
0,954
0,89х
0,85
1,3
1,98
Чистота 1
Вальцован 1
под
охлаждённым толуолом
Перегнан в 1
вакууме 1
Перегнан в
вакууме
Перегнан в
вакууме
Перегнан в
вакууме
§ 111]
I ГРУППА: ЛИТИЙ, НАТРИЙ, КАЛИЙ, РУБИДИЙ, ЦЕЗИЙ
273
2. Литий
Решётка кубическая С
гранецентрированная.
Координация К.
Тип Ck — SBAI.
Пространственная группа
Obh — Fm3m.
Z = \; к. ч = 12 (
расстояние d = ^ = e-4-=}+1
(расстояние e — aw).
Атомный номер . . .
Вещество
t°C
\ а«
\ d
«х
3 1
\л
— 190
4,41
3,118 1
0,534
Рис. 157а.
Общие замечания
В 1938 г. было найдено [3] для Na (20*С) яш = 4,2820 ± 5 А.
В 1939 г. 1-4] найдено для Na (20°) *Ш = 4,29А, К (20Э):5,31А;
Rb ( — 10°) : 5,66 A; Gs ( 10') 6,13 А. Тримбл и Гингрич[7] (1938)
исследовали жидкий Na при 100° и 400 С (в капилляре из пайрекса).
При 100° восемь атомов находятся на расстоянии 3,83 А; при
400' —3.90А.
ы В интервале от температуры жидкого воздуха до комнатной других
модификаций щелочных металлов, кроме Сс, долгое время обнаружить
не удавалось. В 19-И г. был сделан [8] вывод, что интенсивности отражённых
рентгеновых лучей не могут быть объяснены на основе кубической
центрированной элементарной ячейки с 2 атомами шаровой симметрии.
В том же году Лорд |9] ]. с. ссылался на неопубликованную работу Гар-
риса, в которой Li, па основании исследования с помощью ионизационного
спектрометра, приписывалась пространственная группа Td (в 1. с. [9]
неверно—7^) с 16 атомами Li в элементарной ячейке, изолированными в пары.
В 1946 г. это сообщение не было подтверждено 110], причём было найдено
при комнатной температуре аш = 3,5023±2; для —183° К aw 3,4762 kX. Для Rb
было найдено [10] при 18,5—19,5°С аш =■= 5,698— 5,699; при 185° aw = 5,62'i.
В 1947 г. было установлено [И], что при —196° С Li образует
(^-структуру с параметрами, приведёнными в таблице. Модификация Сс при той же
температуре имеет я—3,50 kX. Выше —117°С С^-форма быстро исчезает.
Автор делает попытку интерпретации фазовых переходов в этой системе
на базе теории Донора (1947).
К сожалению, нам не удалось найти данных о возможности
образования другими щелочными металлами, кроме лития, иных модификаций,
в частности Ск, и в первую очередь при низких температурах (и высоких
давлениях).
Впрочем, следует указать, что в 1947 г. Бриджмеп[13] опубликовал
заметку, в которой отмечалось обратимое изменение объёма Gs при
р = 50000 атм примерно на 12%, и высказывалось предположение, что это
связано с образованием плотной упаковки. По поводу этой работы было
274 ч. п. структуры неорганических веществ |гл. IV
высказано и другое предположение, что внезапное сжатие вызвано
переходом бз-электронов Cs при столь большом давлении на уровень bd.
С повышением температуры и приближением её к точке плавления,
судя по некоторым позже не проверявшимся данным (см., например, [5]),
решётка кристаллических щелочных металлов, в частности Li, постепенно
переходит в «аморфную» форму.
Структурных данных о простом веществе, образуемом элементом
№ 87 —экацезием, которому в последнее время предлагалось присвоить
наименование франций (Fr), нам до конца 1948 г. найти не удалось.
Дополнительные сведения о строении жидких щелочных металлов
имеются в статье [12] (1943), а также в более ранней работе [1-1], где найдено,
что при 200°С атом Li в жидком Li окружён в сродном 9,8 соседями на
расстоянии 3,24 кХ.
ЛИТЕРАТУРА к § 111
1.
2.
3.
4.
5.
6.
1.
8.
Z. ph. Ch.
Phvs. Rev.
Z. Krist. (A)
Z. Aiior^. Ch.
Phys. Rev.
J. Ch. Phvs.
Phvs. Rev.
Phys. Rev.
m,
a»,
101),
243,
!",
4,
5 4
29,
165
352
195
69
381
23>
21S
HO
(1928).
(1927).
(1938).
(1939).
(1026).
(1933).
(Ш8).
(1941).
9.
VK
11.
12.
13.
14.
J. Ch. Phys.
Phil. Mag.
Phys. Rev.
Rev. Mod. Phvs.
Усп. химии
Phys. Rev.
J. Ch. Ph.
%
36,7 W,
72,
15,
XV,
72,
s,
700
S'i5
245
90
297
533
450
(1941)
(1946)
(1947)
(1943)
(1946)
(1947)
(1941)
§ 112. II группа: бериллий Be, магний Mg, кальции Ca,
стронции Sr, барий Ва, радий Ra (s2)
1. а-бериллий, магний,
р-кальций, "/-кальций?
Решётка гексагональная Н.
Координация V.
Тип: НУ SB A3.
Пространственная группа
Dlh — CG/mmc.
Z = 2; к. ч. в (расстояние
d = а) 4-6 (расстояние
е--=
/9+V)*»
при с/а --1,6,33 с = а,
» с/а > 1,633 е > а,
» г/а < 1,633 е< а.
Рис. 158.
•) В тексте SB I эта формула набрана с опечаткой.
§ 112] II ГРУППА: БЕРИЛЛИИ, МАГНИЙ, КАЛЬЦИИ, СТРОНЦИИ, БАРИИ, РАДИЙ 1 Jo
3
2 0<
л О
о 2
"" О
<^ i
о
а
н
z;
(У
а
/ г,
-• — ! г lw А«>
' сред.
I I
V <-.«»! х
Чисю1а
12
20
л Бе [1]
a lie [2]
а Be [3J
а Ве [4]
Mg [8] I 20
i
Mg [9j | 25
Mg[10j
i
i
Ugfiijjio
i
YCa[J7JjlOO
20 I 2,28^
20 I 2,2070 J X
20 i 2.2080 i-3
2,2810r
3.20.
3,2031.' ±1
,2fo!80±3
3,2017
3,0.
P Ca Г13/43о! 3,98
14J I
12,28.,
3.01. '1.58.,
2,23,
3,0912+J j 1,5818 I~o2"30
3,5912 : 3
3,5771.
>.!'.)
5.2002 ' 4
5,19983b
1,5817
2,2080
2,2237
1 ЛЗ '«. loi o.05l>ll ,S5>
I ,1229i8.00
1.1221)18,00
12.2810
156820 2ТГ360
l,62r
320.;
1 85
I .857
Спектр осьо-
пически
чистый, следы
Ба, С, Fe
Спектроскопически
чистый, следы
Fe, Ба, С
Следы !•>,
Ба, Са
1.12 92|s.l0| -
I ,59 , ;23.0
8
! i Fe 0,01%,
I i С 0,05%,
1 No 0,005%,
I I следы Si, Л1
13.85
1.73 99,98%
С»
I
1,730
5,1980 ! 1,0237
|3,9<1
0.4 , 1 ,0 1 ' п-
0 ,3,9о
1,97 ,43,-1
0.52 11.639
3_98
i3.99"
1 .99
44,0
20. 13И.52
11.4 8
Mg 99,995%,
Fe 0,0006%,
Si 0,00010%,
Си 0,0013%,
Al 0,0012%,
Cl 0,0007%
99,9
2. р-бериллин
Решётка гексагональная Н.
Координация?
Тип tf?-SB?
Пространственная группа?
Z = 60?
Нуждается в проверке.
Атомный
номер
Вещегтно
4 ^Вв[5]
1 i
t°C
:,оо
i
а | с
7,1
10,8
с/а
0Х
1 1
1,Г,211 | 1,91
.__ 1 I
276
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. IV
3. ос-кальций, стронций
Решётка кубическая С
гранецентрировап-
ыая.
Координация К.
Тип CK-SBA1.
Пространственная
группа Oh — Fm$m.
Z=4, к. ч. 12
(расстояние d — aw у 2) [ +
-j- 6 (расстояние е = aw)\
Атомный номер
20 I 38
Вещество
аСа
[22]
Sr[22j
г °С
v A
Vcm*
20
5,53
ДО
5,5 »5|
3, '33
1,96,
43,0
25,9
1,54
—173,1
6,05
ДО 0
6,04,
4,28
2,14
33,5
4. Барий
Решётка кубическая С
центрированная.
Координация С.
Тип CC-SB^2.
Пространственная
группа Oh — JmSm.
Z = 2, к.ч. = 8
(расстояние d=aw j/~3/2),
[ + 6 (расстояние
е = аш
Атомный
номер
53
]•
Вещество
Барий [201
» ,211
» [22]
t °С
комн.
аи>
5,01
5,04
5,009
d
4,34
1
>J
2,17
i
Рис.
Vcm*
38,0
160.
*х
3,61
3,52
о
пикнометры- ,
ческ е
3,52
3,5
Чистота I
99,9
б. Радий
В распоряжении автора сведений нет.
§ 112] II ГРУППА: БЕРПЛЛПП, МАГНИЙ, КАЛЬЦИЙ, СТРОНЦИЙ, БАРИЙ, РАДИЙ 277
О б щ и е замечания
Be. Помимо низкотемпературной формы аВе, было установлено[5] (1933)
и [3] (1935) возникновение высокотемпературной формы ЗВе. В первой работе
было найдено, что [Же (чистота не указана) образуется при нагревании
до 500 —700°С, лучше всего при ~630°С, и предположено, что ос-форма
всегда загрязнена [^формой.
Во второй работе специально исследовались аллотропические
превращения спектроскопически чистого Бе со следами Fe, Ba, Са.
Найдено, что температура превращения а—» [4 лежит между 500-600°С.
При охлаждении обратное превращение протекает медленно,что
аргументировалось [6] (1934) измерением удельной теплоёмкости и рентгенографически.
Таким образом, предположено, что термическая обработка (нагрев и
охлаждение) ведёт к образованию р-формы, влияющей на параметры
а-формы Be.
В работе [5] I.e. для аВе (неизвестной чистоты) найдено
а с с'а at
2,283 3,G'i 1,5877 1,816
В работе Л. К. Трапезникова и Г. Ф. Косолапова для Be с Fe=0,43%,
Si = 0,12%,Cu-0,62%,Mg = 0,18%,Mn=-0,2/i%, следы А1 (Кальбаум) было
найдено в частности [23)
* °С а с с fa
(18D) 2,2808 3,5735 1,5Г>68
(454°) 2,2966 3,5938 1,5СЛ8
и определены коэффициенты расширения, например, ац = 1,31 • 10~5; aj_ =
= 1,57 • 10"5 в интервале от 18° до 454°С.
Mg. В 1932 г. было найдено [12] для Mg, сублимированного в вакууме
(чистота не указана), с пересчётом на 20°С:
« = 3,202; с-5,199; с/а = 1,623.
В 1939 г. Mg был исследован [И] в| интервале 10— 597°С. Найдено,
например, для 597° С
а = 3,2599; с = 5,2960 A; cfa = 1,6246;
от 0 до 160° аа- 27,0 хЮ~в, а, = 27,9 х Ю~\
Са. В 1933 г. были изучены [15] возможные переходы модификаций Са
в* интервале от комнатной температуры и до /^450°С и выше. Переход
аСа^±ВСа происходит при /^450°.
В 1934 г. был исследовап1Ю] тот же процесс и найдено, что чистый Са
выше 450°С образует ^-модификацию, загрязнённый же —кубическую
центрированную или гексагональную.
Нейбургер[17) (1935) считает, что существуют 3 устойчивые фазы:
о-Са —ниже 300°С, ЗСа-устойчив в интервале 300 —450°С (в SB III 196
ошибочно указано 200-300°, что лишает это утверждение смысла),
уСа—гексагональный (ЛЗ) устойчив выше 450°С..
Клемм и Мика[22] (1941) приводят для аСа сг„, = 5,565 А.
Sr. По работе [18| при температуре жидкого воздуха яы; = 6,03 (1928 г.)
и при комнатной температуре 6,06 (1929) для Sr (99,9%).
По [19] (1929) aw = (yfil&'k (для Sr 99,9%). Но работе [20] (1929) <7Ш = 6,05 А.
В работе [22] (1911) приведено для Sr яю = 6,049А.
Ва. Чистота препаратов, исследованных в работах [20]и [21], не известна.
Данные являются приближёнными.
Для Ва приведено |22| ям, = о,009А.
Ii78
Ч. II. СТРУКТУРЫ ИКОРГЛНИЧЕСКИХ НЕЩЕСТР»
|гл. IV
ЛИТЕРАТУРА к § 112
1. SBI 19,40.
2. Z. Кг. (А) 85, 325 (193 0.
3. Z. Кг. (А) 92, 474 (1935).
4. Phil. Mag. (7) 20, 1155 (1935).
5. Proc. Ac. AmsL. 36, 636 (L933); SB
III 193. ifc.-«% «**
fi.Rec. Tr. Ch. Pays. Bas. 53,451 (1934).
7. Z. Kr. (A) 94, 53 (1J36).
8. SB I JG- 19, 40.
9. J. Ch. Phys. 3, 005 (1935).
10. Straumanis u. Jovins,
Z. Phys. 98, 461 (1936). Die
Praezisions-bostimmung von
Gitterkonstanlen nach dor
asyminetrischeu Mothode
(1940).
11. J. Inst. Mot. «5, 477 (1939).
12. Z. Krist. 84, 20 (1932).
13. SB III 196.
14. SB [II 3 J95.
§ 113. HI* группа:
скандий Sc, иттрий Y, лантан La,
актиний Ac (dls2)
1. а-скандий, иттрий, а-лан-
тан
Решётка гексагональная Н.
Координация V.
Тип Яг -SB A3.
Пространственная группа
Щь— CQ/гптс.
Z-—2; к.ч. = 6 (расстояние
d = a) f^/'
+ 6 Г расстояние е = ' *"—'
15. Z. Anorb'. Ch. 215, 126 (1933).
16. Z. phys. 35, 551 (1934).
17. Z. Elektroch. 41. 790 (1935).
18. Z. Ph. Ch. 1*3, 165 (J928).
Proc. Nat. Acad. 15, 695 (1929).
19. Proc. Nat. Acad. 15, 337 (L929).
20. Z. Ariorg. Ch. 179, 418 (1929).
21. Proc. Nat. Ac. 14, 617 (192S).
22. Z. Anorir. Ch. 24S, 155 (1941).
23. SB IV 79.
УЧ+ЧУ
ри cja = 1,033
» cja > 1,633
» c/a< 1,633
e = a y
e > a,
с < r/.
Pill'. 161.
ГОМНЫЙ
о мер
< =
21
39
57
!й'1Ц0 "тип
а-скандий [2J
To же [3|
Иттрий [1]
To же [2]
а-лантан
[1]
To же [2]
» » [4]
t
комн.
комн.
»
»
a
3,302±.3
3,30±/
3,66 ^
3,62 J ±4
3,7..
3,754
3,72
с
5,245 + «
5,23±Z
5,8U
5,750±~
6'°63
6,063
6,06
rt'fi
1,558
1,585
1,588
1,585
1,613
1,613
1,63
d
3,302
3,66
' 54
3,754
3,72
e
3,59
3.7!
^
3,72
r
1,644
1,81
1,797
1,87
1,87
1,86
Г см'
15,0d
—
19,87
22,4,
22,43
: Mll-
Cr 1 1
x !стота 1
I
3,00,
4
3,02±/
4,34
4'"5
6,19/t
6,194
6,3
указана
§ 1131
III* ГРУППА: СКАНДИЙ, ИТТРИП, ЛАНТАН, АКТИНИЯ
279
2. У-скандий, £-лантан
Решётка кубическая С
гранецентрированная.
Координация К.
Тип Cf=Ck-SB-41.
Пространственная группа
0J —Fm3m.
2 = 4, к.ч. = 12
( расстояние d= aw ]/2 =
„**£!) [ + 6(ра
е--аю)].
о
<Л ? </
о
эасстояние
о
tff
о
к^>
Рис. 162.
-о
У
о
Атомный
номер
21
57
Вещество
fj- скандий
1 (1
(J-лантан [5]
» » [3]
ate
4,Г>32±5
~> Ol") 19
5,294
d
3,204
3,74
3,744
г
1,002
M72
1,872
if A3
23,2
37()
37,0
V смг
H,l
22,5
22,48
ax
3,20±/
6,17
6,18
Чистота
99,6%
+ Si, Al, Mg
c,
/n -812° С
Общие замечания
Sc. В 1939 г. найдено [2], что Sc действительно диморфен и образует а)
гексагональную плотную и Ь) кубическую структуру.
В 1939 г. были исследованы кубическая и гексагональная структуры [3].
Y. В 1932 г. был исследован [1] Y, полученный электролизом расплава
хлоридов натрия и лития. Исходный продукт Y203 имел «степень чистоты»
99,5%. Анализ Y отсутствует. Рентгенограмма показала сильные линии
В работе [2] (1939) была найдена структура и данные, приведённые
в таблице.
La. В 1932 г. был исследован [1] La, полученный электролизом расплава
LaCl3-j-NaCl или KF без последующей детальной очистки. Анализ La
отсутствует.
В указателе SB II (171) ссылка на реферат этой работы пропущена.
В 1930 г. были папдепы [4] значительно отличающиеся данные
(температура не указана).
280 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ |гл. IV
В 1933 г. [5] был исследован [BLa, полученный электролизом
хлорида с добавкой 15% NaCl; катод—W; анод —графит. Перед съёмкой
продукт диспергировался в атмосфере N2 и много дней выдерживался в
высоком вакууме при 350° С.
О б щ и и в ы в о д
Sc, Y, La изучены в старых работах; степень чистоты либо не известна,
либо далека от 100%. Данные по Ас вовсе отсутствуют. Учитывая важность
III* группы в теоретическом отношении, следовало бы приветствовать
постановку проверочных исследований на современном уровне.
ЛИТЕРАТУРА к § 113
1. Z. Anorg. Ch. 208, 59 (1932); 4. Trans. R. Soc. Canada III, 2J, 33 (1930);
2. Z. ElekLroch. 45, 357 (1939). см. SB II 171.
3. Natnrw. 27, 230 (1939). 5. Z. Elektr. 89, 84 (1933).
§ 114. IIP* группа:
металлы редких земель T.R.
1. а-церий, а-празеодим,
а-неодим, гадолиний,
тербий, диспрозий,
гольмий, эрбий,
туллий,кассиопий
Решётка гексагональная//.
Координация V.
Тип //F-SB A3.
Пространственная группа
Dth — Сб/ттс.
Z = 2, к.ч. = 6 (расстояние
+ 6 ( расстояние
Рис. 163.
Атомный
номер
58
59
1 60
Вещество
а-церий
а-празеодим
а-неодим
а
3,65
3,662
3,650
с
5/J6
5,908
5,890
с/а
1,63
1,613
1,613
г
1,81
1,824
1,818
V см*
20,7 .
20,80
90.60
°1
6,78
6,77,
7,00/§
ИР
§ U4| Ш** ГРУППА: МЕТАЛЛЫ РЕДКИХ ЗЕМЕЛЬ 281
П родолжеиие
[Атомный
1 номер
64
65
66
67
68
69
1 П
Вещество
Гадолиний
Тербий
Диспрозий
Гольмий
Эрбий
Туллий
Кассиопий
(лютеций)
а
3,622
3,585
3,578
3,557
3,532
3,523
3,509
с
5,748
5,064
5,048
5,020*)
5,589
5,564
5,559
с а
1,587
1,580
1,579
1,580
1,582
1,580
1,584
г
1,794
1,773
1,709
1,759
1,748
1,742
1,737
V см*
19,79
19,11
18,97
18,05
18,23
18,12
17,95
°х
7'9'«8
8.3:52
8,56,
8,764
УД64
«,346
9,740
2. р-церий, ^-празеодим,
иттербий
Решётка кубическая С
гранецентрированная.
Координация К.
Тип С к-SB 41.
Кространстпеинаи группа
Ol—Fm3m.
Z = 4, к.ч.= 12 Грасстоя-
ние а— —-— ) .
Рис. 164.
Атомный
номер
58
59
! 70
Вещестио
р-дерий
fl-празеодим
Иттербий
°1Г
5, ПО
5,151
Г., 4^8
d
3,634
3,642
3,806
г
1,817
1,К21
1,943
V см3
20,58
20,71
24,75
°Х
0,81
6,805
7.010 |
*) В SB VII ошибочно —5,650.
Д^
о
оГ
Y/
о
D
О
-о
1/
6
Z82
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТН
|гл. IV
3. Европий
Решётка кубическая С
центрированная.
Координация С.
Тип С,. —SB A2.
Пространствснная
группа 0% — Jm'im.
Z = 2, к. ч. = 8__
/ , а V 3 '\
{ расстояние а --- --— i.
Рис. 165.
\томный
1 номер
63
Вещество
Евроиий
flw
4,573
d
4,084
г
2,042
V см*
29,00
*х
5.24,
1. Самарий
Решётка тетрагональная Т
гранецентрированная (?).
Координация К.
Тип 7\?
Z = 4, к. ч. - 12.
Атомный
номер
62
Вещество
Самарий *)
V см*
21,7
а 1
G.93 |
Г). Иллиний, он же циклоний, он иге промоций (№ 61).
!3 распоряжении автора сведений нет.
*) Надёжными данными не располагаем.
$ 114] III** ГРУППА: МЕТАЛЛЫ РЕДКИХ ЗЕМЕЛЬ 283
Таблица 35
Сводная таблица структур простых леществ, образованных элементами
III* и III** групп
Вещество и
валентные
уровни злек-
iponoR атома
х Sc ds'1
a Sc ds*
Y ds*
a La ds1
tt La ds2
i Ce flds*
ri Ce pds*
i Pr f*ds*
3 Pr /W
Nd /W
61 /4ds« (?)
Sm fbds*
Eu /e<*s2
(id /7rf*3
Tb /Bd*a
Dy /ecka
Ho /10ds*
Rr /"ds«
Tin /"<fca
Yb fl*ds*
Cp /*»ds«
Тип
решётки
гскс.
куб. rp.
гекс.
»
куб. гр.
гекс.
куб. гр.
гекс.
куб. гр.
гекс.
—
тетраг. гр.
куб. центр.
гекс.
гекс.
гекс.
гекс.
гекс.
гекс.
куп. гр.
гекс.
а
5,302
4,532
3,62Э
3,754
5,29'*
3,65
5,140
3,662
5,151
3,650
—
--
4,573
3,622
3,585
3,578
3,557
3,532
3,523
5,468
3,509
с
5,245
—
5,750
6,063
—
5,96
—
5,908
—
5,890
—
—
—
5,748
5,664
5,648
5,620
5,589
5,564
—
5,559
с!а
1,588
—
1,585
1,613
—
1,63
—
1,613
—
1,613
—
—
—
1,587
1,580
1,579
1,580
1,582
1,580
—
1,584
1,634
1,60
1,797
1,870
1,872
1,^1
1,817
1,824
1,821
1,818
—
—
2,042
1,794
1,773
1,769
1,759
1,748
1,742
1,933
1,737
V ел*3
15,01
14,1
19,87
22,43
22,48
20,7
20,58
20,80
20,71
20,00
—
21,7
29,00
19,79
19,11
18,97
18,65
18,29
18,12
24,76
17,96
°*
з,оо4
3,20
4'475
6,194
6,180
6,78
6,81
6,77б
6,805
7,00,
4
—
6,93
5,244
7,948
8,332
8,562
8,764
9,164.
9,346
7,010
9,74,
Таблицы § 114 составлены нами на основании работ, цитированных
в §§ ИЗ и 114.
В 1914 г. опубликованы работы [3], в которых указывается, что
существуют три модификации Се (а, (3, у,) и, возможно, четвёртая (о), причём
первые три формы могут существовать вместе.
ЛИТЕРАТУРА к § 114
J. Z. anorg. Ch. 231, 138 (1937); 241, 264, 272 (1939).
'2. Z. EI. 46, 357 (1939).
.3. Ann. Chim. 19, 417 (1944); С. А. 40, 3955 (1946).
284
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕШЕСТВ
|гл. IV
§ 115. IV* группа: титан Ti, цирконий Zr, гафний HI, торий ТЬ
(d2s2)
1. а-титан, а-цирконий,
а-гафний
Решётка гексагональная Н.
Координация V.
Тип Hv- SB A3.
Пространственная
группа D\h—Cd>lmmc.
Z = 2, к.ч. = 12.
6 (расстояние с? = д) + 6
(расстояние е= у -*--|-_J.
Рис; 166.
Атомный
номер
22
30
72
Вещество
а-титан [ 1 ]
а-циркошш
[3]
» [7]
»
[12]
а-гафний |6]
» И
г°С
комн.
»
»
а
2,95
3,23
3,223±2
3,227
3,32
3>200±2
с
4,69
5,14
5,123±3
5,137
5,46
5,077±5
cja
1,59
1,59
1,64
1,587
d/e
2,59
2,90
3,23
3,18
3,32
зТзз
3,30
3,14
г
1,47
1,60
1,66
1,59
V
17,7
23,2
26,1
V см*
—
13,08
сх
4,49
4,42
6,531
Чистота
99,9%
%%
§ 1151
IV* ГРУППА: ТИТАН, ЦИРКОНИИ, ГАФНИЙ, ТОРИЙ
285
2. р-титан, ^-цирконий
Решётка кубическая С
центрированная.
Координация С.
Тип CC-SB A2.
Пространственная
группа Ob—Jm3nt.
8 (расстояние
к.ч
*-*£)■
\г°С
а\в
d
г
Vx
\VcM*
1 °-
Атомный
40 | 40
номер
22 |
Вещество
В-цир-
коний
[41
867
3,61
3,126
1,56
23,5
14,27
р-цир-
КОИПИ
[71
842
3,61
3,12,
1,56
23,5
14,27
р-тптан
14]
900 1
3,32
2,875
^в
18,29J
4,31 J
3. Тории
Решётка кубическая, С
гранецентрированпая.
Координация К.
Тип СР=-- C^-SB Al.
Пространственная
группа Ol—Fm3m.
Z = 4, к.ч. ---12 ( расстоя-
• ^ ' i ' \
/
V*
Рит. 167.
и ие
Рис. 168.
Атомный
номер
90
Вещество
Торий
* [8] а)
» [8] б)
г °С
комн.
<*и>
5,077
5'08lj-2
5'074±2
d
3,59
3,60
3,59
Г
1,795
1,80
1,795
V см*
19,85
19,95
19,85
*г
11,695
11,685
11,695
Чистота
?
Примечание
Старые
данные
SB
286 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [>Л. IV
Общие замечания
Ti. Титан диморфен. Переход aTi (гекс.)—>3Ti (куб. центр.)
исследовался [1] в 1936 г., и для металла, свободного от кислорода, была найден?»
точка перехода tn = 882 ± 20° С (1155° К).
В работе [5] в 1939 г. даётся tu = 885°С ± 10°.
В 1941 г. этот процесс был исследован [10] на базе измерения магнитной
в^приимчивости и найдено tn^ 1200° К.
aTi был исследован [2] (1925), после более ранней работы [3] (1921).
Более низкое значение zx = 4,42 для aTi было дано в работе [4] (1936).
№ исследовался в работе [4] (1936).
Ti, растворяя водород, образует при составе TiHj 2 кубическую гране-
центрированную ячейку [13] (1941).
Zr. Цирконий диморфен. Переход aZrl^Zr исследовался в 1934 г. [12]
и в 1939 г. [5]. Была найдена £Ц = 862 ^ 5°С. В 1941 г. этот процесс
исследовался вновь [10]. Изменение кристаллической структуры происходит, по этим
данным, в согласии с [1] при ~ 1100° К — до 1115° К.
aZr исследован в работах [3] (1921) и [6] (1925). Данные этих работ
подверглись критике [7] (1927).
В 1934 г. были опубликованы [8] данные по исследованию коэффициента
термического расширения aZr \\L6: a,| = 2,5* 10~6;
±L9: a±= 14,3. Ю-6.
Среднее значение a = 10,4-10-6 (см. SB III 209).
pZr исследован в работе [4] (1936).
Как было показано [13], растворяя водород, Zr образует
тетрагональную гранецентрированпую решётку с с = 4,58 и а =--= 4,87 кХ при 66
атомных процентах Н; увеличение объёма достигает 8,2%. Откачивая
систему Zr — Н при 1000° К, удавалось полностью удалить водород; вновь
образовывался aZr с прежними размерами ячейки.
Hf был исследован в 1925 г. [6], а также в 1928 г. [7]. Найдена
гексагональная плотная упаковка. Предположение о существовании
кубической центрированной структуры высказывается Бранделбергером
111] (1947).
Th был изучен в работе [9] (1930). Th был получен а) термическим
разложением иодида, а также б) восстановлением хлорида натрием.
Элементарные ячейки охарактеризованы в таблице.
Проблема изменения структуры металлов IV* и V* групп
периодической системы при переходе к гидридам неоднократно дискутировалась
в общей форме и в деталях в работах советских исследователей (см., в
частности, монографию Я. С. Уманского [14]).
ЛИТЕРАТУРА к § 115
1. Ргос. Acad. Arast. 39, 515 (1936). См. также Пес, Тг. Ch. 55, 450 (1936).
2. Phys. Rev. 25, 581 (1925); 28, 56 (1925).
3. Phys. Rev. 18, 88 (1921).
4. Z. Krist. (A) 94, 299 (1936).
5. Rec. Tr. Ch. Pays-Bas 58, 973 (1939).
6. Z. Krist. 62, 255 (1925).
7. Z. Phvs. Ch. 130, 100 (1927).
8. Mem.'Coll. Sci Kyoto Imp. Univ. 17, 27(1914).
9. Z. Anorg. Ch. 193, 144 (1930).
10. J. Ch. Phys. 9, 673 (1941).
11. Bran den barger, Grnndlage:i der VVerkstoffs Chemie, Rasher-Verlag, Zurich
(1947).
12. Physica 1, 561 (1934).
13. J. Ch. Phys. 9, 678 (1941).
14. Я. С. Уманекий, Карбиды тнордых сплавов (1947);
§ 1161 V- ГРУППА: НАНАДИЙ, НИОБИЙ, ТАНТАЛ, ПРОТАКТИНИЙ 287
§ 116. V* группа:
ванадий V. ниобий Nb,
тантал Та,
протактиний Ра
(dV и d**1)
1. Ванадий (dV),
ниобий (dAsl), тантал
(dV)
Решётка кубическая С
центрированная.
Координация С.
Тип Cj~Cc-SB Л2.
Пространственная
группа 0\ — Jm\im.
Z = 2, к. ч. _ 8
f расстояние а = -^-2— )
+ 6 (растояиис е --=- аш).
Рис. 1Г,9.
Лго.м-
iiF.iii
|номер
21
41
73
Вещестпо
Ванадий [2|
Ниобий [4]
Тантал [11]
» |12|
t 9С "
2 Г,
20
комн.
»
«1/.
3,0338±3
:{,291±3
з,зо3±2
''-yj9x3
3,298-^2
<*
2,r»27,t
2.8Г»28
2,81)
2,8544
2,85Г>
г
1,:пз7
1,4264
1.430
1Л2 72
1,428
>г А3
П,%5
17,87
17,У55
F c.u3 сс | Чистота
III
8,47
10,84
10,80
С01Г(
8,569
8,50
16,65Г|
1Г)//.)
100%
(Спектроск.
следы Sn)
чистейший
Очень чистый,
свободный
от иодорода
препарат
Общие за м е ч а ни я
V. В 1930 г. был исследован [1] чистый (?) ванадий и найдено aw =3,011.
В 1936 г. был вновь исследован [2] чистый V (см. таблицу).
Nb. Данные SB 1 14 по Nb неверны (aw ■-■= 4,191 и т. п.).
В работе [7| найдено (1929) аю -3,291.
В работе [3] (1930) найдено для Nb aw - 3,31. Иойбургер, пересчитав
данные последней работы, получил аи) = 3,32. В 1931 г. он же
опубликовал данные [5] (см. таблицу) и в 1936 —более новые данные [4].
В 1932г. были найдены [6] для спектроскопически чистого Nb (следы Sn)
а = 3,299±/, <зх »= 8,575. В 1934 г. было найдено [12] ян, =■■= 3,294 ±i А.
Та. (Согласно старым данным[81 (1921), аю =3,272, /-=17,09, ^=1,142.
Согласно [9] (1927), аи, = 3,32 А, ох= 16,3. Согласно [7] (1929), яш--3,291.
Согласно 11| (1930), аш = 3,298. Согласно [6] (1932), аи) = 3,298[ ., ат = 16,69.
В 1932 г. для99,8% Та найдены [10]: 0Ш = 3,3114:Ы<ъ зг (16,5°С) = 16,49
при аэксп = 16,6. Бюргере и Бозорт[12] для Та, полученного разложением
ТаС14 в вакууме, нашли аш = 3,294^,. Пейбургер [11] (1936) —цифры,
288 ч. ii. структуры неорганических веществ [гл. IV
приведённые в таблице. Как отмечалось в предыдущем параграфе, проблема
образования твёрдых растворов внедрения элементов V группы
неоднократно освещалась в работах советских авторов.
ЛИТЕРАТУРА к § 116
1. Z. Ph. Ch. (В) 11, 4.Ю (1930).
2. Z. Krist. (A) 03, 314 (Ш6).
3. Z. Anorg. Ch. 19D, 237 (1930).
4. Z. Krist. (A) 93, 158 (1936).
5. Z. Krist. (A) 78, 164 (1931).
Z. Anorg. Ch. 197, 219 (1931).
6. Z. Anorg. Ch. 208, 257 (1932).
7. Trans. R. Soc. Canada III 23, 257
(1929).
8. Phys. Rev. 17. 571 (1921).
9. Z. Phys. 16, 165 (1927).
10. Phil. Mag. 13, 1020 (LJ32).
11. Z. Krist. (A) 93, 312 (1936).
12. Z. Anorg. Ch. 216, 223 (193i).
§ 117. VI* группа: хром Сг, молибден Мо, вольфрам W, урап U
(dV; W: rfV; U: </V(?))
а-хром (dV), молибден
(dV), а-вольфрам
(dV) H«^-ypaH»(dV)
Решётка кубическая С
центрированная.
Координация С.
Тип Cc-SB Л2.
Пространственная
группа О* — Jmllm.
= 2, к.ч.=8
,1^3
[расстояние d-~
[ + 6 (расстояние с = аш)].
>+■—t—
' !
I
I
•\
I
У
I
I
I
— Ь-
I
I
-Л>'
"\
i
i
i
Рис. 170.
Атомный
[номер
24
42
74
92
Вещество
а-хром
Молибден
л-вольфрам [15]
» [16]
» [26]
Р-уран
ГС
17
20
20
КОМН.
aw
2,8786
3,14103±4
3,14оз±10
3,1589±4
3,15837
3,1586±2
3,43
3,43
1
d
2,493
2,72
2,72
2,7357
2,97
г
1,2465
1,36
1,36 ,
1,368
1Л85
w А3
11,9
15,0
15,0
: 15,76
20,17
V см*
7 94
9,0
■ 9,0
9,49
12,2
°х
7,188
10,217
Ю,231
19,24
19,261
19,6
19,45
Чистота
Электролитически
чистый
99,5%
§ 117] VI* ГРУППА: ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ, УРАН
1. J5-XpOM
Решётка гексагональная #.
Координация V.
Тип Hv- SB A3.
Пространственная группа D^h — Ciyjmrnc.
Z = 2, к. ч. = 6 (расстояние d =* а)+6 (расстояние е
Рис. 171.
[Атомный
номер
24
Веще-
CTDO
3 |
?Сг
а
с
I ;
коми. 2,71-14,41«
1 !
cja \ d/e
1,626
2,71
2,70
г
1,35
V см*
8,57
*с
Чистота
1 Злектро-
6,07 литич.
чистый
) |
1 [о условиям образования, полученные электрохимически
модификации хрома всегда могут содержать растворённый водород. Это особенно
относится к р-хрому (см. следующую страницу).
Таким образом, по нашему мнению, вопрос о возможности и
условиях существования tt-хрома как одной из модификаций хрома остаётся
пока неясным.
Кроме того, большой интерес представило бы установление
термодинамической устойчивости (3-хрома в отношении перехода в другие формы.
289
/т+т)-
290 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IV
2. Y"XP0M*)
Решётка кубическая сложная.
Координация 16/13/12.
Тип Clj;i3/12~SB Л12.
Пространственная группа 71* —743т.
Z-58.
Рис. 172.
Атомный
номер
24
Вещество
^-хром
t°c
3
комн.
°«
8,717
d
2,444
г
V см*
1,222 6,93
°х
7,507
Из сопоставления структур а-, [4- и у~хРома вытекает, что плотная
гексагональная упаковка атомов Сг, т. е. рСг, имеет гораздо меньшую
плотность и гораздо больший атомный объём, чем две другие формы.
Поэтому мы считаем, что рСг, повидимому, не является чистым хромом,
но твёрдым раствором водорода в Сг, тем более, что он образуется в
процессе электролиза при высокой плотности тока.
*) Описание структуры см. на стр. 300, 301.
§ 117|
VI* ГРУППА: ХРОМ, МОЛИИДЕИ, ВОЛЬФРАМ, УРАН
291
3 4.
I I
О wi
©WD
Vnc. 173.
3, р-вольфрам
Решётка кубическая С.
Координация 14//.
Тип Сии — SB Л15.
Пространственная группа
()\- РтЗп.
Z -=8.
Атомный номер 74 |
Вещество |
Р-воль-
фрам
*■<:!
*„ I 5,04
1(5,038)
d ' 2,52
I (2.81,)
г |1,26(до
Ген8' 9,70
с- 1 18,97
1 !
Р-воль- (4-воль-
фрам фрам
L24] | [25]
18
5,0408^>5,0'Л^«5
1
1 |
Рис. 17 '*.
Рис. 173 по SB1; рис. 174
построен нами. Сравним с
рис. 243 и 283.
Координационная сфера
атома W1 является
интересным дополнением к табл.
34 для к. ч. = 12.
(Кристалл охимический
икосаэдр У; см. рис. 441а.)
Расстояния
VVI к. ч. = 12 WII (с = 2,82),
WII к. ч.= 2Wl(d = 2,52),
4 WI (б-= 2,82),
8 WII (/ = 3,09).
292 ч. и. структуры неорганических веществ
[гл. IV
4. а-уран
Решётка моноклинная, «С —
гранецентрированная*.
Координация?
Пространственная группа
Clh-C2lm.
Z = 2.
Описание SBI 192, см. ниже.
Эта «модификация» но-j
вейшими исследованиями
отвергнута (см. рефераты), j
б. а-уран
Решётка ромбическая OR.
Координация 4 + 8.
Тип ОЛ^-SB 420.
Пространственная группа
У)? = вГ}1-Стст.
Z = 4.
о
о
о
о
о о
оо
о
с
о
о о
1
щество
а
Ъ
1 с
*х
i-уран
2,852
5,865
4,945
18,97
Расстояние кХ
2,76
2,85
Л,27
:j,36
Примечание: По
^Уоррену [27].
Коли- |
чество
соседей 1
2 1
2
4
4
Нкобу и
i
О О
Рис. 175а.
О
Рис. 175Ь.
§ 117| VI* ГРУППА: ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ, УРАН 293
По форме координационная сфера четырёх ближайших соседей,
показанная на рис. 175Ь, близка к координационной сфере TeGi4 (рис. 135,6).
Позиции 4 атомов в элементарной ячейке определяются
координатами:
Фу1!*), (ОугП),
(7*. у+ 7.. 7«Ь CU. 7,-*.*/«).
причём
у - 0,105 ±5.
На рис. 175а -2 проекции элементарной ячейки, данные Якобом и
Рис. 175с.
Уорреном. На рис. 175Ь — построенная нами на основании указанной
работы элементарная ячейка. На рис. 175с —чертёж из SBVI3 (G ячеек).
6. р-уран Рэндла (см. ниже, ср. рис. 170).
Структура неизвестна.
294
Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТВ
[гл. IV
7. у-УРан
Решётка кубическая центрированная С.
Координация кубическая С.
Тип Сс-SB A2.
Пространственная группа Oi, — JmZm.
Z = 2.
Вещество
ТГ-уран
а1Г
*,\*
d
2,97
Примечание
По Рэидлу 128]
Рис. 17G.
у-уран Рэндла отвечает старым данным SB по [бурану (см. рис. 170).
О о щи е замечания
О. Хром, возможно, триморфен, однако характер фазовых переходов
неясен*). Описаны три модификации: аСг, [ЗСг, уО.
Из них, при комнатной температуре, судя но работе[1], ^Сг и у Сг
переходят в аСг через 40, соответственно 230 дней. При электролизе раствора
сульфата хрома при плотности тока ниже 15 а/дм2 выделяется аСгса =2,877 ±3,
о, = 7,21. При плотности тока > 18 а/дм* — гексагональный рСг с а ^ 2,717;
с/а-1,626; 03 = 6,08 или Л12са = 8,717, Z = 58, ах - 7,48. Интересно,
что устойчивый аСг имеет ах большую, чем зх для рСг (плотная упаковка Ну).
В 1935 г. было установлено[2], что электролитический хром,
полученный в отсутствии восстановителя, имеет кубическую центрированную
решётку (аСг). Из ванн, содержащих восстановитель (винная кислота
и ячменный сахар) и при условии катодной концентраций Or > 18%
выделяется (ЗСг— гексагональный. Нагретый в вакууме при 800°С в течение
1,5 часа t3(> переходит в аСг, с aw = 2,8788. Продажный аСг (99,15%)
имеет aw - 2,8781.
В более ранних работах было найдено: в[3] (1921) для аСг aw = 2,895,
ах « 7,07; в [4] (1925), а также в [5] (1925) аш = 2,875.
Джетте, и др. [6] исследовали а) электролитический Сг с 0,04% Fe и
0,017% S и следами Si и Ь) «очень чистый» Сг, полученный восстановлением
с помощью Са. Образцы перед снимками отпускались при 1000°С в
атмосфере Н2 (?!). Оба имели aw - 2,8787±10 А.
Мо. В старой работе [3] найдено дляМо, полученного восстановлением
окисла водородом aw == 3,143, d = 2,720, г = 1,360, ох = 10,16 г/см3,
В SB 1 реферирована [7] величина 3,08. Позже было найдено для
99,8% Мо аю = 3,142±,, зх = 10,2i±3 и для 98% Мо аш = 3,130.
Наконец, В. Аркель нашёл (1926) а = 3,14А.
Эти данные относятся к комнатной температуре. В SB они
прореферированы без указания температуры.
В 1930 г. было найдено [18] для Мо aw = 3,139.
В 1932 г. Оуэн и Ибелл [17] и в 1935 г. Джетте и Футе [16] нашли
данные, приведённые в таблице.
*) Не исключено, что некоторые из этих форм метастабильны или содержат
примеси. См. выше вСг.
§ 117] VI* ГРУППА: ХРОМ, МОЛИБДЕН, НОЛЬФРАМ, УРАН 295
W. Вольфрам, повидимому, диморфен. Известны две структуры aW
и pW, обе кубические. aW SB А29 тип (6'с). Р W относится в SB к типу А 15.
Он является переходным от кубической гранецентрированной с
координационным числом 12 к кубической центрированной с координационным числом
8 + 6 = 14. В решётке имеется два положения атомов W. В положении I к. ч.
атомов W = 12, причём каждый атом WI окружён 12 атомами W II, на
расстоянии е = 2,82 А, образующими конфигурацию кристаллохимического
икосаэдра (см. рис. 441а).
В положении II к. ч. атомов WII = 14, причём каждый атом WII окружён
2 атомами WII на расстоянии d = 2,52 А,+4 атомами WI на расстоянии
е = 2,82 А + 8 атомами WII на расстоянии / = 3,09 А.
Таким образом, переходный тип А 15 не похож ни на А\, ни на .42.
Подобного рода сложные структуры, возникающие при тенденции атома
образовать координационную сферу промежуточного типа, наблюдаются,
например, в случаях о^Мп (-412), у Cv (^412), у-латуии (Z>8) и др. Интересно,
что то же расположение атомов металла, что и в р W, наблюдается для атомов
урана в решётке гидрида урана (см. гидриды урана), исследованной Рэндлом
[28] (1947) рис. 243 и it решётке V3Si рис. 441Ь.
Работы по определению <xw для aW весьма многочисленны. Назовём [10]
(1917) (аю - 3,18); [И] (1921) (aw = 3,15); 112], в которой для aW 99,9995%,
99,999% и 99,997% найдено одинаковое значение а (3,155±ь зх = 19,32i2)» Для
продажного (99,58%) — аш = 3,15.
В. Аркель [13] (1926) нашёл aw = 3,154; Беккер [14] (1926) нашёл
коэффициент расширения W в интервале 18° —1380°С (5,8 х 10~6),
18°-1750°С(6,6х10-6) и 18о-2200°С (7,5x10-6).
В 1930 г. было получено [18] для W чистейшего ^=3,156. В 1931 г. [19]
а = 3,16. В 1934 г. Нейбургер [15] получил данные, приведённые в таблице.
В 1935 г. Джетте и Футе [16] также получили более точные данные
(см. таблицу).
Страуманис и Евинс [26] (1940), в результате исследований структуры
вольфрама асимметричным методом, нашли а = 3,1586±2 при 20°С.
PW изучался сравнительно немногими авторами.
Как было показано [20] (1931), при электролизе W03 в NaJP^O? и К4РЛЭ7
(с добавками NaP03 и NaCl) получается pW новая модификация, при
температуре ванны ниже 700° С в смеси с aW, а в некоторых условиях даже
только pW. Нагревание в вакууме выше 700°С переводит [3-форму,
необратимо, в a-форму. Для pWZ = 8, пространственная группа ОЦ — РтЗп;
тип SB А 15, а = 5,04, 03 = 19,0; найдено пикнометрически аПИКн = 12,з(.-±25?
В 1931 г. было показано [21], что p\V можно получить не только путём
электролиза фосфатов. Продукт, полученный электролизом при 500°С
и смеси 19 моль% K2W04, 35% Na2W04, 46% Li^WO* (при температуре
плавления 370°С) обнаруживает линии обеих модификаций. После
прокаливания до 900° С остаётся только aW.
В 1944 г. опубликованы [24] [25] данные по PW (см. таблицу).
U. Исследования U сравнительно немногочисленны. U, видимо, три-
морфен. alJ был исследован [22] в 1930 г.
В 1933 г. [23] опубликованы результаты исследования aU, в котором
была найдена моноклинная ячейка пространственной группы С?/, — С2\т
а = 2,829, Ъ ~ 4,887, с = 3,308, р = 63°26', Z = 2, ах = 19,05, олн/= 18,68.
Эти результаты в SB сопровождаются примечанием, что однозначность
определения не вытекает непосредственно из сообщения и из методики
работы, поскольку метод порошков вообще допускает неоднозначную
расшифровку снимков моноклинных и триклинных структур.
296 ч.п. структуры неорганических веществ [гл. IV
В 1937 г.было опубликовано [27] сообщение, в котором данные,
указанные выше [23], опротестовывались и устанавливалась структура
элементарной ячейки ромбической сйнгонйи, указанная в таблице.
В SB этой структуре присвоен тип А20.
В отличие от Сг, Мо и aW, уран имеет не кубическую центрированную,
но особую решётку, причём структура all сильно искажённая
гексагональная, плотная упаковка с 12 соседями на весьма неравных расстояниях.
В результате 4 ближайших соседа образуют весьма характерную
координационную сферу: две связи направлены под углом 180° параллельно
оси а (расстояние —2,85) и две связи лежат в плоскости, перпендикулярной
оси а под углом 127°. Эти 4 связи показаны на рис. 175Ь жирной линией.
Плотность урана значительно меньше, чем следовало бы ожидать,
исходя из хода кривой удельный вес — атомный номер, как это
подчёркивается в указанных работах. Отмечается также высокое
электросопротивление урана по сравнению с вольфрамом (и, добавим мы, с молибденом).
32-76x10-6 вместо 5,5-10-6 0м\м • мм\
а-модификация урана, описанная в работе [27] в 1937 г., не встречает
возражений и в современной литературе. Так, в работеРэндла [28] (1947),
посвященной структуре гидридов урана, на стр. 1721 эта структура
рассматривается без каких-либо возражений и, более того, без изменений
приводятся межатомные расстояния, налдешше в работе [27]. Из ссылки,
на той же странице, на специальную работу [29] (1947) можно заключить,
что структурные модификации урана детально исследовались Рэндлом
в период 1944 — 1947 гг. Что касается других модификаций и
исследования фазовых переходов в этой системе, то са последние 2 — 3 года
опубликован ряд работ, значительно пополнивших литературу в этой области.
Согласно работе [29] (1947), структура [3-формы точно не известна,
у-форма имеет кубическую центрированную элементарную ячейку, в которой
каждый атом имеет 8 соседей, на расстоянии 2,97 А. (Повидимому, следует
читать кХ.—Примечание наше.) Это дало нам возможность рассчитать
и другие значения, приведённые в таблице. Как отмечено в этой работе,
повидимому, yU ранее был получен случайно [22] (1930).
Область фазовых переходов, значения теплоёмкостей, теплот
переходов aU ;ZPU ^yu изучались в работах [30] (1947) и [31] 1947). На базе
этих работ нами составлена следующая небольшая таблица:
Точка перехода °К
Теплота перехода
1 (в кал/гр. атом)
1 Энтропия перех. (в кал/град.
1 ер атом)
1 Теплоёмкость, Ср.
1 (кал/град).
XJa
—
—
См. ниже
примеч.
<—
935
680
0,73
—
up
—
■—
10,38(30]
10,15 131]
^Z
1045
1165
1,11
—
ur 1
1
— 1
— 1
0,10 [30]
9,15 [31]
Примечание. Атомная теплоёмкость a-урана определяется [30] выражением:
Ср =3,15 + 8,44 • 1ГГ3Г-{-0,80 . ЮБГ~а. Теплоёмкость р и у U примерно постоянны.
ЛИТЕРАТУРА к § 117
1. SB II 190.
2. Trans. Far. Soc. 31, 1253 (1935).
3. Phys. Rev. 14, 540 (1919); 17, 571 (1921).
4. Phys. Rev. 25, 107 (1925).
5. Phys Rev. 25, 581; 26, 56 (1925).
6. SB III 219.
7. SB I 61.
8. Phys. Rev. 23, 292 (1924); 25, 753(1925).
9. Physica 6, 64 (1926).
10. Phys. Z. 18, 483 (1917).
11. Phys. Rev. 17, 571 (1921).
12. Phys. Rev. 25, 753 (1925); 26, 736(1925).
13. Physika 6, 64 (1926).
14. Naturw. 14, 10 i6 (1926).
§ 118]
VI** ГРУППА (?): 93, 91, 95, 96
297
15. Z. Anopjf. Ch. 21?, 154 (193't). 24. Nature 154, 337 (1944).
16. J. Ch. Phys. 3, 605(1935). 25. Nature 164, 337 (1944).
17. PhiJ. Mag. 13, 1020 (1932). 26. Straumanis a. Jevins, Prazisionsbeatim-
18. Z. Ph. Ch. (B) 7, 339 (1930). mung von Gitterkonslanten etc. (1940).
U». Proc. Phys. Soc. 43, 512(1931). 27. J. Л. Ch. S. 59, 2588 (1937).
20. Z. Anorg. Ch. 198, 116 (1931). 28. J. A. Ch. S. 69, 1719 (1947).
21. Rec. tr. Ch. Pays. Bas. 50, 1050 (1931). 29. Wilson a. Rundle, Metallurgical
22. Trans. R. S. Canada III 24, 1 (1930). Proj. Report, CT-1775 (19V0-
23. Physics 4, 148 (1933). 30. J. Am. Ch. S. 69, 2105(1947).
31. Phys. Rev. 72, 182 (1947).
§ 118. VI** группа (?): 93, 94, 95, 96
Физические и химические свойства элементов 93 и 94 и особенно 95 и 96
описаны в литературе ещё далеко не достаточно. Структурные данные
отсутствуют. Однако при дальнейших исследованиях в этом направлении
нельзя пройти мимо вопроса о положении этих элементов в периодической
системе. См. § 86 и табл. 29.
Развитие знании в этой области происходит столь быстро, что новейшие
сведения через несколько месяцев устаревают. Излагаемое ниже отражает
нашу попытку остановиться на наименее спорных вопросах, касающихся
группы VI**, исходя из литературных данных.
Химические свойства элемента 93 изучались в ряде работ [1—4].
Элемент 93 не осаждается сероводородом и не увлекается в осадок сульфида
рения, т. е. 93, повидимому, не является гомологом U. 13 то же время 93
обнаруживает сходство с U в том, что образует соединения 93VI и 93IV
(т. е. в 6- и 4-валентных состояниях).
В состоянии 93IV он, как и U, даёт реакции, аналогичные реакциям ТЬ
(осаждается щавелевой кислотой, сульфидом аммония, фтористоводородной
кислотой и т. п. и идет в осадок вместе с Th, Ce, La), т. е., повидимому, должен
Сыть причислен по химическому поведению к VI**, а не к VII* группе.
Это привело отдельных авторов к выводу, что в атоме 93 имеется
не менее, чем 1 электрон на 5/-уровне (/х). Кстати, и у Th+ и Th2+
действительно было обнаружено в 1941 г. нахождение электрона на 5/-уровне [5].
В 1942 г. (на основе предположения, что 94 также является уранидом)
было предложено [6] ввести фв периодическую систему XX группу для 93
и 94 элементов (в дополнение к «XI», включающей Сг, Mo, W).
Периодическая система такого типа [6] весьма не удобна с химической
точки зрения, поскольку номера групп не отвечают валентностям элементов,
уровни валентных электронов в этой таблице в ряде групп даны
неправильно; сами обозначения уровней не отвечают принятым в спектроскопии.
Таблица 29 построена нами на основе таблицы с распределением
электронов по уровням, появившейся впервые в Советском Союзе (см. [7] и
[11]) со включением элементов 93—96 (т. е. нептуния (Np), плутония (Рп),
америция (Am) и кюрия (Cm)?) в гр. VI** на основании работ [1—0] см.
§ 86. Последующие публикации [8 — 10], а также [15] и др. дают
основания к выводу, что элементы 90—96 относятся к актинидам (гр. III**
периодической системы). Однако, мы не считали возможным менять места
элементов в системе, и даже закреплять за вновь открытыми те или иные
названия впредь до решения этого вопроса авторитетными советскими
научными органами.
ЛИТЕРАТУРА к § 118
1. Phys. Rev. 55, 1104 (1939). дической системы элементов в конце
2. Phys. Rev. 57, 1185 (1940). книги.
3. Naturw. 30, 107 (1942). 8. Science 104, 379 (19'ifO.
4. Naturw. 30, 250, 877 (1942). О. Abstr. 41, 1929 (1947).
5. Z. f. Phys. 118, 581 (1941). 9. Ch. Eng. News, стр. 2897 (1947).
6. С. R. 215, 537 (1942). 10. Ch. Eng. News, стр. 2887 (1947).
Г. Б. Ормонт, Химия и строение 11. Сборник «Периодическаясистема эле-
материи, OIITI1 (1934). Таблица перио- ментов», изд. V Менделеевского съезд;»
298
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТП
(ГЛ.IV
§ 110. VII* группа: марганец Мп, технеций Тс, рений Be
(dW; Тс: d«sl (?))
1. а-марганец
Решётка кубическая С,
сложноцентрированная.
Координация 16/13/12.
Тип Ci6/i.vi2-SB A12.
Пространственная группа Та -743т.
Плотность упаковки 54,8 %.
Z = 58.
0 12 3 4 5
I 1 1 i
Рис. 177. -l Мп.
Атомный
номер
Вещество
К с „и1
25
а Мп
комн.
8,89л
2,49.
1,24-
7,36 ! 7,46,
2. р-марганец
Решётка кубическая С
Координация K/J.
Тип CK/j SB Л 13.
Простравственная группа
0'-/>4?(*ли0в-Р4?).
Плотность упаковки 67,6%,
Z = 20.
IAtom-
1 ный
[номер
Веще- :
CTRO
or; I ^-марга-
J i нец
г°
комн.
«ш
6,30
1 !
2,53
1,265J7,585
бх
7,24
§119]
3. у"маРганеЦ
Решётка тетрагональная,
гранецентрированная
TF.
Координация 12 {ТК).
Тип Тгк — SB АЪ.
Пространственная группа
Dlh — Jblmmm.
V 2 *
VII* ГРУППА: МЛРГЛИЕЦ, ТЕХНЕЦИЙ, РЕНИИ
о
299
<'-/т
е — а,
е' - с
Атомный помор
Вещество . .
г°С . ...
я
с
с/а
\ d .
\ d'
г
\vi . .
\ см1
1 сг
25
у-марганоц
комн. (?)
3,774
:*,53ч
0.936
2.67
2,58
1,33-1,20
13,3
8,00
7,21
Переход к /-ячейке с
Z=2 показан па рис. 02,6
4. Технеции (43), рений
Решётка
гексагональная И.
Координация V.
Тип Hv —SB A3.
Пространственная
группа D*h—C (yjtnmc.
Плотность упаковки —
= 73,2%.
Z = 2, к. ч. +6
(расстояние d — я)+6 (расстоя-
а
—о
Рис. 178. гШп.
Рис. 179.
[Атомный
I номер
43
75
Вещество
Технеций
Рений [ 101
» |8|
t°C
КОМН.
20
а
2,735
2,755
2,765
.
4,388
4,449
4,470
с/а
1 , (\()\
l.ftU
d
2,735
2,755
V см*
8,874
1
11,497
20,996
20,53
300 Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ |гЛ. IV
Общие замечания
Мп считается триморфным*). Описаны структуры aMn, fiMn, уМп,
с обозначениями фаз по Вестгрену, причём SB I считает, что ячейку {ЗМп
с Z = 20 следует, возможно, ещё увеличить в 8 раз.
Если у Сг одна из структурных форм имела необычный для простых
веществ сложный состав, то у Мп две основные структурные формы аМп
и jiJMn весьма сложны. Условия перехода аМп^рМп^у^11 точно не
изучены.
В 1925 г. были исследованы [1] Брэдли условия возникновения а-, р-
и у-форм, причём р-форма образовывалась при температурах от комнатной
до 800—900°, у-форма в ещё большем интервале—до 1005°, причём лишь после
900°, повидимому, без р-формы. аМп наблюдался, главным образом, при
низких температурах (до 120°?).
Электролитический Мп является а-формой.
Вестгрен и Фрагмен[2] (1925) назвали а-форму Бредли у-формой и
наоборот (их номенклатура принята нами выше, но не следует забывать об этом
смешении понятий). Поэтому по мнению этих авторов электролитический
марганец — у-форма. Она возникает лишь при низких температурах и при
нагревании переходит в [3-форму и в высокотемпературную а-форму.
(Сплавленный или дестиллированный в вакууме Мп — а-форма с Z = 58.)
В 1927 г. была установлена [3] структура а Мп и найдены 4 сорта
позиций.
Как показано в[4] (1930),чистый (?) дестиллированный в вакууме,
закалённый от температуры 760° Мп даёт линии двух модификаций а и 3,
имеющих периоды решётки, несколько более высокие, чем ранее найденные [5J
(1927) (см. таблицы).
Превращение а^р происходит при температуре 742°С,
> P^ty » > > 1191° С,
причём а-фаза [4] отвечает у-фазе Вестгрена.
Элементарная ячейка aMn (SB А 12) содержит 4 сорта позиций: Мп 1,
Мп II, Мп III и Мп IV.
Эта решётка, в изложении SB II, может рассматриваться как
производная от кубической центрированной А2 (Z = 2). Если ребро [куба А2
увеличить втрое, т. е. объём в 27 раз, то получится элементарная ячейка, имею-
/ 1 1 1 \
щая 54 атома в положениях I 000; - - — —- ) + :
] (000);
н ±(±11. IIIV* •
"1±(о|т; о11)С;
iv±(±oo)0-
Если теперь удалить половину атомов Мп II, а именно
^4.6 6 6' 6 б gA'
т. е. 8 атомов, и все 12 атомов Мп IV, а всего 20 атомов, и ввести 24 атома
IV примерно по середине между центрами тяжести удалённых атомов
/* 1 1 1 1 1 I X
(т. е. в идеальном случае в положения ± ( т То То "» Т 1212 )^ )» то П0ЛУЧИМ
структуру Л12.
*) В последнее время указывалось и на наличие четырёх модификаций [11] (1946),,
§ 119] VII* ГРУППА: МАРГАНЕЦ, ТЕХНЕЦИЙ, ГЕНИИ 301
(При этом произойдут ещё смещения атомов, см. ниже, координаты).
На рис. 177 представлены идеальные положения: 2 кубических решётки,
(взаимно центрирующие друг друга), вершины которых заняты частично
атомами I, II и III. На диагональных соединительных линиях между
незанятыми вершинами лежат атомы IV. Наглядности ради, смещения из
идеальных положений не показаны; читатель легко находит, например, Мп
\{) О ti J
В результате элементарная ячейка а Мп содержит 2Мп I, 8Мп И, 24 Мп III
/^1 1 1 \
и 24 MnlV, занимающих положения: (000) I — — -у J + .
I. (000), т. е. 8 вершин и центр элементарной ячейки, а всего
1 + 1 = 2.
II. (ххх\ ххх О)жп = 0,317, т. е. 4 диагональных положения вокруг
угловых атомов и 4 таких же положения вокруг центрального атома,
а всего
1 = 8.
III. (xxz\ xxz\ ххъ\ ххъ) (j\ хш = 0,356, zlu — 0,042, т. е. 4
положения на каждой стороне элементарной ячейки (всего 4 х 6 = 24 положения)
и аналогичные положения по отношению к центральному атому внутри
24 12
элементарной ячейки (т. е. 12 положений), а всего -у + j- = 24.
IV. 24 положения на диагоналях, соединяющих незанятые места, а всего
Ц. = 24.
1
(Координаты: как и Мп III, но +х1У -0,089; ziV = 0,27вЛ
В результате координационная сфера Mnl состоит из 16 соседей;
МпП —из 16; МпШ —из 13 и MnlV —из 12, а именно*):
Мп I 12 атомов IV (2,71) + 4 атома II (2,82);
Мп II 3 атома III (2,49)+ 6 атомов IV (2,69)+ 1 атом 1 (2,82) +
+ 3 атома IV (2,89) +3 атома III (2,96);
МпШ 1 атом IV (2,45)+ 1 атом II (2,49)+ 2 атома IV (2,51) +
+ 2 атома IV (2,66)+ 4 атома III (2,67)+ 2 атома III (2,67) +
+ 1 атом II (2,96);
MnlV 1 атом IV (2,24)+ 2 атома IV (2,38)+ 1 атом III (2,45) +
+ 2 атома III (2,51) + 2 атома III (2,66) + 2 атома II (2,69) +
+ 1 атом I (2,82) + 1 атом II (2,89).
[3 Мп представляет собой необычный тин плотной кубической структуры,
включающий позиции I и II, которые могут быть охарактеризованы
следующим образом:
*) В скобках даны межатомные расстояния в ангстремах.
302 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. IV
Положения I примерно соответствуют кубической гранецентрирован-
ной структуре С к — SB A1 с небольшими искажениями.
Положения 11, как указывает SB II4, отвечают якобы не встречающейся
в других кристаллических структурах фигуре—правильному икосаэдру*).
При этом положению 1 отвечает к. ч. 12: 3 атома I (2,36), 3 атома II (2,53)
и 6 атомов II (2,67).
Положению II—также к. ч. 12:2 атома I (2,53), 2 атома II (2,60),
4 атома И (2,66) и 4 атома I (2,67).
Структура имеет винтовые оси 4Х. Общее расположение атомов Мп в
этой структуре нуждается в уточнении.
(Щи был исследован в работе [6] (1925) и найдено яш = 6,29; о^.---7,338;
Z-20.
В работе [7] найдено для ЗМп удвоенная ячейка £>• — Ftn3C aw = 12,58;
ах = 7,29; Z = 160, из них
8-MnI, 8-МпН, 48-МпШ и 96-MnIV.
уМп имеет гранецентрированную тетрагональную решётку индия
с Z=2. Он был исследован в работе 11 (1925), где найдено а = 3,764;
с = 3,556; с/а = 0,9445. Была найдена [2] F-гранецентрированная
тетрагональная решётка: а = 3,774; с = 3,533, ^\а = 0,937, Z = 4, ох = 7,21.
Координация: атом (000) имеет соседей:
4Гу-2"0)на расстоянии d\
8 (VOyJ на расстоянии d'\
4 (100) на расстоянии е = а\
2 (001) на расстоянии е' = с.
Таким образом, к. ч. = 12; дополнительные 6 атомов уже сильно
удалены. Влиянио растворённого водорода на межнлоскостцые расстояния
аМп**) исследовалось в работе [12] (1945).
Тс. Дли технеция (Тс99) найдена (см. Acta Cr. 1? 161 (1948)) плотная
гексагональная упаковка (см. табл. на стр. 299). См.[Ю] (1947)).
Re. В работах [8] (1931) и [9] (1931) изучалась структура рения. Найдена
плотная гексагональная упаковка ЛЗ, см. таблицу.
В работе [9] найдено: а = 2,755; с = 4,450; с/а=1,615 для обезгаженного
в течение 3 часов в высоком вакууме при температурах до размягчения
кварца.
В 1932 г.[10| найдены данные, приведённые в таблице.
ЛИТЕРАТУРА к § 119
1. Phil. Mag. 50, 10G8 (1925).
2. Z. Phys. Ь\} 111 (1925).
3. Proc. R. G. L. 115, 456 (1927).
4. Metallwirtsch. 9, 825 (1930).
Г». J. Ir. Steel П5, 393 (1927).
6. Z. Krist. 61, 463 (1925).
7. Bull. Am. Phys. Soc. 9, 16 (1934); SB III 221.
8. Z. anorg. Ch. 196, Ш (1931); Naturw. 19, 108 (1931),
9. Naturw. 19, 575 (1931).
10. Nature 159, 24 (1)47).
11. U. S. Bur. Mines Tech. Paper 681, 34(1946).
12. Phys. Rev. 68, 24 (1945); С. А. 39, 4785.
*) Который и SB назван правильным. По ем. Cu8 Si стр. 597; см. также V8Si;
стр. 582.
**) Выделенного электролитическим путём.
§ 120] VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ 303
§ 120. VIII* группа: железо Ге, рутений Ru, осмий Os (d"a2; Ru; d7«i)
1. а-железо, [3-железо, о-железо)
Решётка кубическая С центрированная.
Координация С.
Тип CC-SB A2.
Пространственная группа Oh — Jm'Sm.
Z = 2
, к.ч.=8(
i/"v
расстояние d= -w у-'- )+6 (расстояние е =-- а).
Рис. 180.
Атомный
номер
Вещестио
*ЭС
V смл
26
а-железо [10]
» [14]
[15]
» [23]
[25]
р-железо [1]
Ш
[3]
Д-железо [1]
№
20
25
25
22
19
785
860
800
1400
1425
2,8611
(до 2,8617)
2,8604,.
о
2,8607,
4
2,8605±<3
2,86038.^
2,8995
2,9043
2,90
2,9347
2,93
2,4778
1,2389
7,10,
4
7,8б1
7,86,)
7,86, j
Чистота
0,007% С
0,004% Si
0,004% S
0,004% О и
другие образцы
Чистое
Fe-99,46%,
примеси С, следы Р
Fe —99,32%,
примеси С, Мп,
следы Р и S
Fe - 99,98%
Fe -99,46%
примесь С, следы Р
Fe = 99,98% в
присутствии f-фазы
304
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ЬЕЩЕСТВ
[гл. IV
2. -^-Железо.
Решётка кубическая С, гранецентрированная.
Координация К.
Тип CK-SB Л1.
Пространственная группа Оь — РтЗт.
2f=4, к. ч. 12 (расстояние = а" ^ ' Vf 6 (расстояние е = а).
Л
о
г^/
о
of
и
£>
о
-о
у
о
Рис. 181.
1
Атомный
(номер
26
Вещество
•f-железо [10]
»
t°C
20
экстра- <
полиция
от 900
г
>
900
950
aXD
3,5605
3,5548
3,5679
3,6425
3,649
d
г
2,5811,29
Vcm*
7,37
ах
7,58 |
Чистота
Чистое, переплавлено
в вакууме
С = 0,004%
То же, С = 0,01%
Спечённое
карбонильное железо, следы С
Чистое, переплавлено
в вакууме
С = 0,004%
Переход от кубической гранецентрированной ячейки к тетрагональной
центрированной (§ 45) осуществляется выбором других координатных осей
а и 6 в той же самой решётке. При этом отношение с/а равно j/2.
Таким образом для перехода в кубическую центрированную (например,
в случае железа) должно иметь место сжатие решётки в [/2=1,41 раза
по оси с или расширение её по осям а и 6, или оба процесса
одновременно. Таким образом, с геометрической точки зрения, в подобного рода
переходах, связанных с изменением координационного числа, коренная
реконструкция решётки не требуется.
§ 120]
VIII* ГРУПП \: ЖЕЛЕЗО, РУТЕНИИ, ОСМИП
:;05
3. Рутений, оемпй
Решётка гексагональная //.
Координация Г.
Тип Ну SB A3.
Пространственная группа
D\h — CQjmmc.
Z-=2, к. ч. в (расстояние
d ---•■ a) -f- 6 (расстояние
е = ) а3/3>сУ/4),
при cja = 1,633 е — ач
при с)а > 1,633 е > д,
при cla < 1,633 е< а.
1>ш\ 182.
Атомный : В;?мим-т»о : *э С.
номер! ;
г Г r.u';
4'* ■ А.11\'тенпй[211 1-'» |2,6(JS
i •
76 i Осмий [211 18
2,01)84,
i,271 .1/183.,
I
! -\6'.)8-
■2Лй\.\
4 е' ;> ''""-•■л
.\730, \\,Ш)К) \\,
1,348
4,2730. l,583V,
I 2,7 10,!
8,17
r:/i730| -■
i 8,44
12/il
22,61
Чистота
JJ» J /О
99,80/c
I
20 2.72У8 4t:ii0'i 1,r)790L| — j
13/J08
Общ и е з а м о ч а и и я
Fo. Исследование твёрдых фаз, образуемых железом, представляет
исключительный теоретический и практический интерес.
Опубликованные в литературе данные требуют тем большей
осторожности, что «химически чистое» железо и действительности содержит следы
примесей, особенно углерода, меняющего в значительной мере поле решётки
(см. §§ 99 103).
Железо образует 4 модификации: а, 3, у. £.
Как мы указывали в § 109, в химической литературе эти модификации
считаются энантиотропиыми при п ■■■•- 1 атм> и за их точки перехода
принимаются температуры:
ccFe (740-780° C)-:3Fe(900° С): :YFe(lill°C);.-- 8 Fe
С,
cL.
с.
Сс
306 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. IV
Структуры, определённые рентгенографически, показаны для каждой
формы в нижней строке.
Первая детальная рентгенографическая работа [1|, в которой делалась
попытка установить tu , относится к 1929 г. В ней исследовалось железо:
I 99,32% примеси С, Мп, следы Р и Si,
II 99,46% примеси С, следы Р,
т. е. довольно сильно загрязнённые. При этом, однако, наблюдался ряд
интересных явлений.
Переход а—->$ происходит с выделением тепла и сжатием решётки,
«совпадающим с точкой Кюри» (в которой исчезают ферромагнитные свойства
железа, см. Б. Ормонт[2| (1934); точка Кюри для железа лежит, как известно,
при 769°С). Вследствие этого точное установление температуры между 750
и 800°С невозможно.
Если принять для Fe при комнатной температуре ат =-■ 2,8700, то
оказывается
Проволока (образец) II
i
Модификация
Температур!)
°w + Ааш
аи>
а
730
1,06
2,9004
11
785
1,02
2,8995
1 1
ж i ж 1
1
830 ; 860 19
1,29 1,195
2,9071 ! 2,90'i3
т. е. сжатие при переходе а—>р весьма значительное.
Подчёркиваем, что большое содержание примесей, особенно фосфора и
углерода, резко задерживает в данном случае переход а —-»{3.
Переход (3—>у также протекает со сжатием:
Проволока
(образец)
Модификация
Температура
(а 4- Да )
I I
1
и
860
1,195%
2,9043
Т
885
1,02%
3,653
I
Т
' 900
1,08%
3,655
Примечание
Для ^-формы вместо
взято
V v
Переход у—>2 на образце I наблюдать не удалось (переход
запаздывал!—/?. О.). На образце II наблюдался переход при tu > 1370 ,
т. е. около 1400° С, и при 1400° С была найдена S-модификацц» с аш*= 2,9347,
т. е. Л -2,25%.
Эти данные интересно сопоставить с более старой работой [3] (1922)
(Fe^ 99,98%), в которой было найдено
Fe а(16°С)а,„ = 2,87; Fep(800°C) аш = 2,90;
FeY(U00 ) лш = 3,63; Fe у -fFeo(1425°) «ш = 3,68 и 2,93.
о Fe, свободное от у Fe, не удалось получить из-за близости точки
плавления. (Отсюда также можно сделать вывод, что и в очень чистом Fe
переход у—>S задерживается.)
§ 120] viii* П'уппчтжелегю, pyti-пип, осмпп 307
Поажо удалось получить свободное от у-фаз1»1 о Fc (t 1430°) и
подтвердить тип Сс (А 2),
Роберте|4] (1930), работай со спектроскопически свободным от углерода
очень чистым железом, полученным из гидрата окиси Fe путем иосстапоиле-
иин водородом при съёмке в атмосфере водорода, нашёл 3 —> у переход (в SB-
ошибочно а—>у) при 921 °С1 (запаздывание).
И 1936 г. была исследоваиа[10| ячейка у Fe при 900э и на оспине
коэффициентов расширения нутом экстраполяции рассчитаны аш дли у Fe п[)и 20"
(см. таблицу).
Определение aw дли a Fo при комнатной температуре было сделано
многими авторами.
В 1925 г. для Fe, восстановленного водородом из Fo.,03 и
электролитического (Fe ~= 99,937%, С- 0,020%, Мп-=0,001%, Р - 0,004%, S - 0,017%,
Si — 0,021 ?о), было найдено [5] а,,,-- 2,85Г,1Ь;{, ~>г--7,9.ч±з, а также [0) а11} = 2,85у±4.
В 1929 г. было исследовано [7] 40 препаратов карбонильного железа. При
22° С найдено 2,8610, как среднее из G0 независимых измерений с
максимальным отклонением 2%.
В 1931 г. [8] нашли дли Fe (из чистой окиси, восстановленной водородом)
<iw -■- 2,8614; Fe электролитический aw 2,8614; Fe прокалённый в вакууме
аш == 2,8614.
В 1932 г. было найдено для армкожелеза<7„, - 2,860Г1±1 и 116] aw - 2,861^0,
В 1933 г. дли Fe с 0.018% примесей при 18° С было найдено [ 1 7] а =2,8б07г):Ь2<;
~х = 7,8бб±и."
В работах |10 12| найдены цифры, приведённые в таблице: aw от 2,8б04
до2,8607; ат от 7,808 до 7,8б4.
Ф. Маршаки Д. В. Степанов [13] (1935) нашли для электролитического Fe
af|l- 2,863-2,864.
Джетте и Футе 114] (1935) нашли дли карбонильного Fe цифры,
приведённые в таблице (при 25'С) и в 1936 г. подтвердили [15] .лот результат.
В НЛО г. Страуманис и Кпипс опубликовали [23] результат точного
исследования при 22° С: а --- 2,8605.
В 1949 г. А. И. Жмудский (Киев) опубликовал [25] результаты
точного исследовании ocFe; найдено аи) = 2,86038., при 19° С (излучение Со).
Ru. Повидпмому, др)гие структурные формы, кроме плотной
гексагональной (тип Ну SB A3), неизвестны.
В 1921 г. было найдено 118| дли aRiirt = 2,686; с,а - 2,59; -г -.-- 12,56;
в 1926г.(19) для aRu (восстановленного) а ---- 2,695; с - 4,273; с/а 1,586.
В 1926 г.|20] а - 2,680; с = 4,261; rsr = 12,71.
В 1935 г. Оуэн и др. [21] нашли данные, приведённые в таблице,
и средний линейный коэффициент расширении Ru: о, --= 9,1 х 10_6. Более
точное определение выполнил позднее (1937) Оуэн 124].
0s. Известна только одна структурная форма—плотная гексагональная
структура Л у SB A3.
Os был изучен)22] в 1921 г., причём было найдено а = 2,714; с=4,32;
с/а -1,59.
В 1926 г. было найдено [ 191 а = 2,714; с=4,314; с/а = 1,584. ДляОя,
восстановленного водородом из окисла, было найдено [20] (1926) я — 2,714; г = 4,316;
с/а = 1,59; сх = 22,98.
В 1935 г. были найдены [21] данные, включённые в таблицу, и средний
линейный коэффициент расширения а = 6,1 • 10~6 .
В 1937 г. найдены [21] значении а-2,72980; с = 4,3104; с/а = 1,57901
при 20''С.
308 ч. и. структуры неорганических пеществ [гл. IV
ЛИТЕРАТУРА К § 120
1. Helv. Phys. Acta 2, 95 (1929). 14.
2. Ь. Ормонт, Химия и строение ма- 15.
терии (1934), стр. 316 (ОНТИ). 16.
-3. 2 Phvs. Ch. 102, 1 (1922). 17.
Nature 114, 9'» (1У24). 18.
4. Phys. Rev. 35, 1426 (1930). 1».
5. Phys. Rev. 26, 60 (1925). 20
6. J. A. Ch. S. 47, 2868 (U25). 21.
7. Z. Krist. 70, 383 (1929). 22.
8. Z. Melallk. 2t, b.) (1931). 'I'..
9. Proc. Phvs. Soc. 41, 563 (Ш2).
.10. SB III 221.
HI. SB III 2>2. 24.
12. SB III 222. 25.
13. Z. El. 41, 59) (1J35).
J. Ch. Phys. 8, 6i>5 (1935).
SB IV 22).
SB II. 1%
Phil. Mag. 15, 472 (1J33).
Phvs. Rev. 17, 571 (1921).
Z/Ph. Ch. 121, 78 (1926).
SB I 69.
Z. Krist. (A) 91, 70 (1935).
Phys. Rev. 18, 88 (1921),
Straumanisa. Jevins, Prazi-
sionsbcstimmuiig von Gitterkonstanten
etc.(19i0).
Z. Krist. (A) 96, 4)7 (L9I17).
Зап. Лабир. XV, 1055 (1949).
§ 121. IX* группа:
кобальт Со, родий Bh,
иридий Ir (r<V;Uh:r<V)
1. {^-кобальт, 3-родий,
иридий Решётка
кубическая, гранецентрирован-
ыая 6\..
Координация К.
Тип Ск-Sibil.
Пространственная группа
0'1—1чнЪт. Z = 4, к. ч.12
(расстояние d — -- -^—J + 6
(расстояние е = а).
Л^г\
о
о7
И
о
-о
1—X \-zP
Рлс. 18:?.
Атом)
ныи | Вещество
номер;
27
45
1
1
1
1
р- кобальт
р-родий [ 111
Н>одий|12]
Иридий (11]
г
комн.
18°
18°
18е
1
i
3,545
3,7966
3,79559
3,8312
2,50,.
о
2,684
2,709
г
1,25.»
1,342
1,354.
о
Уем3
6,76
8,53
i I
1 1
zt j а Чигтота 1
8,72
12,414
(расч.
для 20°)
12,418
22,65
(расчёт
для 20°)
12,5
о°с
(ICT*)
22
40°С
(ICT*)
Чистый 1
?
Спектроск.
чистый
99,8
1
*) IGT—Ir.ter.-.at. Critical Tables.
§ 1211
IX* ГРУППА: КОБАЛЬТ, РОДИЙ, ИРИДИИ
309
2. а-кобальт
Решётка
плотная гексагональная //.
Координация V.
Тип Hv-SB AS.
Пространственная группа
Din -Сб/штс.
Z-2, к. ч. - 6 (расстояние
d = a) + 6
(расстояние е).
Атомный помор
Ве*щсч:пю
t°<:
и .
с
<;]а
de . . .
Г см'6
с.г
Чистота
• ' 27
. а-кобалы
комн.
. ! 2,Г)07
' 4,07
1,02,
2. Г.07
• 1 «,72
• ! н'77
1 ЧИСТ.
О б щ не намсчан и я
Со. Кобальт, по старым данным, диморфен, но более новым — тримор-
фен. Так, в 1926 г. были установлены [1] две модификации: а Со (450 С)
и ЗСо, причём низкотемпературная яСо — гексагональная Ну\ Р Со—
кубическая CV- Для реакции а Со ^[4 Со t^=^kll при нагревании и ^400°С
при охлаждении. Точка Кюри равна 1106°С.
Со, полученный восстановлением СоО при 600° С, состоит из обеих
фаз; превалирует ЗСо(Л1). После нагревания до 1150 С и медленного
охлаждения остаётся только а Со (^43).
Эти результаты для двух энантиотропных форм, вообще говоря,
непонятны, и удивительно, что они не вызвали комментариев, в том числе SB,
перепечатавшего их полностью. При нагревании до 1150° С, при достаточной
кинетике перехода, должен был бы остаться лишь термодинамически
устойчивый в этих условиях |ЗСо, а не а Со. В 1929 г. было найдено [2] 1и (зь~_!р) ^
340°~3G0JC и в 1930 г.|3] три формы:
Форма
IV 'Шср.чтура
п<*ргхо;?а &>'.
('-тру к тура
("о
//,■-.l:t
;Со
ТСо
00-'-20 Ю1 5 — 20
SA—-11 ЩИ v:>)
Наиболее высокотемпературную форму мы обозначили у Со.
В свете работы [3] наблюдение, сделанное в работе [11, получает иное
содержание. Нагрев до 1150° С, повидимому, способствует возникновению термо-
310 Ч. II. СТРУКТУРЫ НЕ )IJI\UIH4hCKlI>C ШЛЦ1.ХТ]', [ГЛ. IV
динамически устойчивого выше 1015° С у Со (гексагонального); охлаждение
ниже —1015° сохраняет эту форму в интервале 400 — 1015°, как метастабнль-
пую, и ниже 400°, как' стабильную (?).
По наблюдениям, описанным в 1927 г. [4], свежеосаждённый
электролитический Со состоит лишь из аСо. В 1932 г. было найдено [5], что при высокой
концентрации Н -ионов в электролите образуется а Со, при низкой
концентрации Н -ионов — fiСо (плотность тока 0,01 а/см2) (измерения— в интервале рН
от 6,5 до 1,5). Зависимость структуры от плотности тока не исследована.
Аллотропические превращения Со (полученного путём восстановлении
Соо03НоО + 11.2) были исследованы в 1942 г. [8| (было найдено /п^450~)
и в 1943 г. [18]. При этом было найдено, что кубическая и гексагональная
структуры часто сосуществуют в одном и том же образце, особенно при 400"С.
При охлаждении до 300° С остаётся в основном гексагональная структура, при
нагревании до 500° С —кубическая. Отсюда авторы делают вывод, что
свободные энергии различных форм Со ниже 500° С примерно одинаковы.
Подобный вывод нельзя считать непосредственно вытекающим из
названных фактов. Торможение процесса в первую очередь определяется
кинетическими причинами. Даже при больших отрицательных значениях AF
реакции в твёрдых фазах могут не иметь места; это относится, например, к
алмазу в стандартных условиях р \\ Т.
а Со был исследован в 1921 г. \(Ц: а = 2,514; с/а = 1,633, ах-- 8,06; а.1ИТ = 8,718.
И 1927 г. [4| найдено: а - 2,498±5; с =-- 4,05,; с/а - 1,622; а г - 8,89; о.!ит = 8,8105
(для Со со значительным количеством загрязнений: Fe = 0,10%, С = 0,12%,
1' - 0,005%, S - 0,027%, Si-0,010%, следы А1, пет Ni, Мп, Си).
В 1934 г. [7| был исследован линейный коэффициент расширения а Со:
а,,= 16,1 хЮ-6; а± - 12,6 х Ю"в; а = 13,8 х Ю'6.
ЗСо был изучен в|6] (1921): л - 3,554; d-2,514; cr--8,66.
Rh. Родий детально исследован в 1931 г. [9].
Как при комнатной температуре, так и при 750, 1000 и 1400° С препараты
получались осаждением из солей гидразин-гидратом, электролитическим
выделением, кроме того, исследовалась проволока Rh.
Электролитический Rh дал богатую линиями дебаеграмму, тип Сц ---- А\\
с г/ш(20°) ^ 3,777Л, часть линий была приписана второй кубической
модификации с я--9,2ц±оо и Z--48 (?).
Точка перехода a Rh ~Z. '$ Rh была указана в интервале 1100 — 1200' С.
Нагрев до 1000" С изменял рентгенограмму не очень значительно.
[При нагреве до 1500° С граиецентрироваиная решётка «3 Rh» всё ещё
сохраняется. Проволока Rh имеет почти исключительно О,-структуру.
Оуэн и Яте [11 | и [ 12] (1933) нашли для Rh (Ск-модификация) данные,
приведённые в таблице, и опротестовали существование второй кубической
модификации с ^ —9,211.
Считать Rh диморфным пока нет оснований.
Ячейка Rh (SRh) исследована в [6] (1921): aw - 3,820; gx = 12,18; в[10|
(1925): aw = 3,79si2 (восстановленный из окисла с помощью водорода);
в[14] (1928): aw = 3,7944;
ii [151 (1932): aw = 3,7954±m (чистота 99,5%); сх (при 16,5°) 12,42ц.
В 1933 г. для спектроскопически чистого Rh было найдено и„, = 3,799Л.
1г. У 1г полиморфизм пока не обнаружен. Впрочем, детальные
исследования по этому вопросу не опубликованы.
В 1921 г. было найдено |6]: aw = 3,805; d - 2,090; zx = 23Д5.
В 116] (1923): aw = 3,823. В [10] aw = 3,823. Оуэн и ЯтсЩ] нашли
величины, приведённые в таблице, а также линейный коэффициент расширении
д = 6,5хЮ Л
* 121J
X* П'УГШЛ: НИКЕЛЬ, ПАЛЛ \ДПП, ПЛАТИНА
311
ЛИТЕРАТУРА к § 121
1. SB I 703.
2. J. Am. Ch. S 61, 324') (1929).
3. Z. Krist. 3, 37(3 (1930).
4. SB I 703.
5. Physics 2, 274 (1932).
6. Phvs. Rev. 17, 571 (1921).
7. SB* ill 222.
8. Naturw. 30, 439 (19'i2); C.
4947 (1943).
9. SB II 199.
A. 37
10 Z. Phys. Ch. 117, 478 (1925).
11. Phil. Ma-. [VII] 15, 472 (1933).
12. Phil. Mag. [YI1116, 605 (1933).
13. Phil. Ma,'. [VII] IK, 2.)4 (1933).
14. Z. Krist. 67, 235 (1928).
15. Phil. Mag. 1*. 1020 (1932).
16. Z. Krist. 59, 55 (1923).
17. Z. Ph. Ch. 117, 478 (1925).
18. J. Inst. Met. 69, 177 (1943);
Nature U8, 105 (19'. 1).
§ 122. X* группа: никель
Ni, палладий Pd. платина
Pt (f/V и Yu:tr°sb)
1. ^-никель, палладий?
платина
Решётка кубическая С,
гранецентрированная.
Координации К.
Тип Ск SB Л1.
Пространственная группа
Oh — Fm 3/n.
Z^4, к. ч. = 12 (расстояние
d = l
лП
1-
Рис. 185.
Атомный
номер
28
1
4(i
78
Вещество
^-никель [8]
|9]
» ИЧ
»
Палладий) 18)
*
Платима (17]
»
ГС
20
18
18
20
L5
LS
■.'0
"//.
3,518
3,5168
3,5172
3,516,
3,882д
3,8824
3,8817
3,9161
3,9158
:*.9158
1
1
!
)
;
i
| 2,4878
i
2,744g
1
2,769
г
1,2439
1,372,
4
1.384,.
! Vcm*
1
6,598
1
1
,9,Ю7
*х
8,900
8,895б
12,03
12,02?
(пересчитано на 20°)
12,02,
21,44-,
(пересчитано на 20°)
21,438
Чистота
Чистый
никель
99,99%
99,99%
99,98%
\ Спектро-
> СК0ПИЧ9СКИ
J чистый
Спектроскопически
чистый
Спектроскопически
чистый
312
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. IV
2. а-никель
Решётка гексагональная //.
Координация V.
Тип Hv-SB A3.
Пространственная группа
В^ — Сб/ттс.
Z—2, к. ч. =■_-.• 6 (расстояние
d — a)-\-§ (расстояние е).
Атомный
[Номер .
Вещество . .
Iе.
\ а . . .
\ с . . .
с/а . .
\d=r.e. .
г . . .
28
Никель
а[7|
КОМН.
2,60
4,15
1,60
28
Никель
[ч]
комн.
2,65
4,82
1,6*
2,65
1,325
28
Никель
|-2-2|
комн.1
2,50
4,02
1,6*
Рис. LKG.
(3 о щ и о намеча и it я
Ni. Вопрос об аллотропических формах никеля был предметом
исследования в ряде работ, приводивших к разноречивым результатам.
Наиболее хорошо изучен ^ Ni, имеющий кубическую грапецентрирован-
ную ячейку Ск, размеры которой многократно определялись с большой
точностью. Никель в виде компактного металла (жесть, проволока и т. п.)
существует обычно в форме 3 Ni между 0°С и 1200° С.
В 1927 г. были опубликованы две работы [1, 2] по исследованию
структур различных металлов, в том числе Ni в распылённом,
диспергированном состоянии, В первой работе утверждалось, что Ni,
распылённый в Н,, Аг, Не при р < 1 мм Hg, во всех случаях сохраняет Ск-ро-
шётку, но с aw на 6% большей, чем нормальная. Слои Ni немагнитны, что
рассматривалось как результат соединения Ni с Не, Аг, Н2. В о в т о р о й
работе утверждалось, что Ni, распылённый в Н2, образует гексагональную
структуру д= 2,684; с =4,282; с; - 7,04 и до 350° каталитически неактивен.
В 1929 г. была подтверждена работа [2] и найдена дли Ni структура A3
(Ну): а - 2,474 и с = 4,06.
В 1929—1931 гг. Ингерсолл и Ханауолт[4] повторили те же опыты
и нашли, что никель, распылённый в Н2, Не и Аг, образует решётку Ск,
распылённый же в N2—тетрагональную гранецентрированную структуру
Л6 (!?).
В 1932 г. было найдено [5], что Ni а) полученный электролитически,
б) карбонильный, переплавленный в вакууме и в) то же, что б), но
закалённый при 700° С, имеет только структуру Ск, аш (соответственно) равно
3,5162; 3,5170 и 3,5170.
£ 122| X* ГРУПП \: НИКЕЛЬ, ПЛГК1ЛДПП, НЛЛ'ПШЧ 313
Этой работой подтверждались прежние данные [6] (1927), что Ni
обнаруживается лишь в виде р-формы.
В 1931 г. вновь утверждалось|0|, что Ni, образующийся при распылении
химически чистой Ni-жести в 1L или N-> (pzzO.,1 мм Hg), немагнитен
и имеет гексагональную структуру Н\ (Л'Л).
Нагретый в водороде до 300С, Ni образует структуру С к с ow -3,52.
Обратный переход исследован не был.
Специальное исследование |8|(1934) показало, однако, что от 20°С
до высоких температур существует только £Ni; гексагональная
низкотемпературная модификации aNi в компактном Ni вплоть до 20°С не
образуется.
Целым рядом прецизионных исследовании |9|, | 10| и др. (1935—1930)
(см. ниже) также не обнаружено низкотемпературной a-модификацпн в
различных образцах Ni.
В результате до 1938 г. вопрос, о существовании aNi во всяком случае
как анантиотроппой формы оставался открытым. Он, возможно (?), нашёл
решение в работе Леклерк и Мишель 111) (1939), ловидимому, благодаря
случайному обстоятельству, сущность которого совершенно не вскрыта в
лишённой теоретической интерпретации упомянутой работе [J1] и несомненно
не понята также редакцией Sl> VII.
Эти авторы оставили компактный £Ni при 170°С на длительный срок
в атмосфере (10 (а не (10l>, как :>то написано |12| в SB VII, иначе сущность
дальнейшего была бы мало понятна). В результате, судя по этой работе,
оказалось, что 3Ni якобы перешёл в немагнитную гексагональную форму
с и —2,65; с =4,32; с/а --- 1,63. Обратное превращение aNi—>j3Ni эндо
термичпо, из чего авторы заключили, что р Ni должен быть при обычной
температуре метастабилеп.
Переход aNi—>pNi происходит при нагревании до 250— 300°С, причём
вновь возвращаются магнитные свойства р-формы. Каталитическая
способность а- и р-форм совершенно различна.
Таким образом, авторы якобы осуществили переход aNj^ipNi в обе
стороны, в интервале 170 250°С, при р 1 атм, так что эта реакция
эпаитиотроппа.
Мы хотим остановиться на роли (10. Если полученный авторами
препарат является чистым Ni, то наиболее вероятна, нам кажется, следующая
интерпретация: Ni образует, как известно, карбопил Ni (СО)4, границы
устойчивости которого при р — 1 атм лежат как раз около 170° С (см.
В. Ормонт[13], lO'ib'). При этой температуре, следовательно, являются
возможными медленное образование и распад карбонила, а значит, и
образование aNi в виде термодинамически устойчивой формы, т. е. СО ведёт
себя так' же, как растворитель. См. L3. Ормопт (14] (1936) (1947). Реакция
в этом с л) чае имеет вид p[Ni] 4-4C); _" Ni (Cr>)4I"la [Ni] + 4CO, и вся
система в целом должна эволюционировать при 170°С слева направо. При
более низких температурах реакции р—va задерживается по кинетическим
причинам, при более высокой, чем 170°, по термодинамическим (большая
упругость разложения Ni(CO)4). Наконец, с дальнейшим повышением
температуры до 300 С имеет место изменение знака ±F (около 250°С) и
осуществляется обратный переход а—>р.
Однако возможна и другая интерпретация упомянутых наблюдений.
Если при нагревании никеля в среде СО могло при названных выше
температурах иметь место протекание реакции* 2CO = C02hC, что с
термодинамической стороны не встречает возражений, и если никель мог
каталитически ускорить этот процесс, то не исключено образование карбида
никеля и возникновение гексагональной структуры, не характеризующей
чистый никель.
,';Kl Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ КЕЩЕСТВ [ГЛ. IV
В последнее врем» вновь отмечалось [23], что якобы Ni в плёнках,
полученных катодным распылением, бывает аморфным, гексагональным и
кубическим.
Ячейка аМ изучена плохо. Для распылённого состояния
и 1927 г. было найдено [2]: «-2,684; с = 4,282; сг-7,04;
и 1929 » » » 131: а -2,474; с-4,06; с/а = 1,64;
в 193'] » » » 17]: а — 2,66; с = 4,29; с/а = 1,64 (распылённый в Н2);
а = 2,60; г = 4,1о; с/а = 1,60 (распылённый в No).
Несомненно, что растворённые газы могли влиять на размеры ячейки.
Леклерк и Мишель [И] приводят цифры, включённые в таблицу. В J942 г. [22]
пыл подтверждён факт значительного влияния газов и методов получения.
Ячейка Р Ni изучена тщательно, в десятках работ.
В 1932 г. был исследован [5] электролитический Ni, осаждённый на меди
(/■„, = 3,5162), карбонильный Ni, переплавленный и прокалённый в вакууме
('/ —3,5170), и опилки карбонильного никеля, закалённые (после указанной
выше обработки) при 700° С (aw = 3,5170). [3Ni был исследован [8] от 20° С
(г/ 3,518) до 1200° С (я = 3,592) и найдены данные, указанные в таблице.
Джетте и др. [15) (1934) нашли данные, указанные в таблице.
В 1935 г. было найдено 116]: « = 3,5151 ± 10 (0,013% Fe, 0,11 % Со, следы
Si и S) и в[91 (1935): « = 3,5168 + 0$, ат-8,900(Ni = 99,99%).
В 1936 г. для Ni 99,99% найдено[10] нри 18° ^ = 3,5172(^ = 8,897).
При 100° яш==3,52125.
а = 14,6х1(Гв. Между 350 и 370° С обнаружена аномалия: а растёт до
20 X Ю""в. Выше 370° О а снова падает до 16 X 10~в. (Эта аномалия совпадает
для [jNi с точкой Кюри 356°С. —Б. О.)
Электронная плотность в кристалле Ni исследована в работе Н. В. Агеева
» Гусевой [24] (1948).
Наконец, для сомнительной «тетрагональной» формы Ni(?) по Ингер-
г.иллу и Хаиауолту[4] «-=3,994; е'-=3,760; гД/-0,942.
Pd. Пока известна только одна модификация — кубическая гранецентри-
рпванпая. В работе(20] было найдено, что ])екристаллизоваппые нри более
высоких температурах образцы Pd (как и Pi и lih) имеют меньшую
элементарную ячейку.
В 1932 г. найдено |19] "„,-3,884.
Оуэн и Яте [18] (1933) нашли данные, помещённые в таблице.
Ft. Пока известна только одна модификация—кубическая гранецентри-
;юванная.
В 1934 г. были найдены [171 данные, приведённые в таблице.
В 1937 г. было найдено при 20° С аш = 3,9145 А.
Интересно, что плёнки Pt и других металлов, в частности группы
платины, легко меняют период элементарной ячейки при осаждении на
«подкладки», имеющие разную структуру, и элементарные ячейки. Так,
it работе [21] было найдено, что PL имеет следующие значения aWy в
зависимости от материала подкладки: на Zn a„,=3,86; на А\ яш=4,05; на Си
//,„=4,20 (решётка не Ск)\ на Ag «„,=5,18 (!!) (ащ=3,91; аА<г = 4,06). Осадки
на А1 и Zn каталитически активны, на Си—неактивны.
(Возможно, одновременно иногда образуются сплавы. — Б. О.)
ЛИТЕРАТУРА К § 122
1. Nat. 119, 23'» (11)27). i. Phys. Rev. 34, 972 (192J).
2. Z. Ph. Ch. 12G, 41 (1927). .1. A. Ch. S. 53, 2008 (1931).
:*. Nature 123, 912 (192.)). 5. Proc. Phys. Soc. 144, 5fi3 (19^2).
$ ISA
I* ГРУППА- МКДЬ, OFlPEniY), ГЮЛОТО
315
7.
8.
9.
Ю.
11.
12.
13.
14.
13.
Natnrw. 17, 1)40 (1927).
Z. Phvs. Ch. 172 (1 "31).
I'iivsirs •">, 147 (1934); 813 III 223
J. Cil. S. 3, «".05 (1955).
Phil. Мац\ 7 21, 809 (1930)
С. к. 208, 158:5 (193.)).
SB VII 48.
Ada Phvsiroch. \\1 4I3 (194м).
Жф.\, XXI 509 (1947).
8 В 111 223.
10. KB III 2213.
17. Phil. Mag. fVII] 17, 113 (1934).
18. Phil. Alas?. fVIIJ 15, 472; 10,006 (1933).
I ». Ann. Phys. 15, 249 (1932).
20. SJJ 111 181.
21. Proc. k. S. L. (A) 128,649(1930).
22. Ck. '215, 12) (1942).
23. C. A. 3D, 3474 (1945).
24. ДАН 5И, 05 (1948).
§ 123. I* ipyuua: медь Гц, серебро Ag, золото Au (rflV)
J. Медь, серебро, золото
Решётка кубическая, гране-
центрироиаштая CF,
Координапия К.
Тин Ск- SB Л1.
Пространственная группа
Obh-Fm\\m.
Z = 4, к.ч.— 12 v расстояние
к* стояние
/9—^ :#
а
of
^
6
л
о
-о
к
6
Nic. 187.
Атом
н ы и
ПОМ'М
ный | Всчцоггно | t -С,
d I
IV.ir3
Чистота
•29
Медь[19]
» |20|,[2l!
3,6н75
3,00751 \-J 2,5509
» [I7|
' 3,0077. -!-2
47 Copeupu [17]; 18 4,0772, -2
» [Pi!, j i,077S; JV
, » [15] . 19,0 , 4,0775 2,883/,
» j| 18] 25 | 4,07784 + 3
7lJ | Золито[17|1 18 '4,0099 ±30
i » [ 13 | i 4,0099..
! I 9 I
14]' 25 | '''°'0VfcG
| » [9] i 20 ' 4,070 j2,878
1,27Г'
10,285
1,439 l10,22r
8,92(J ±/(20e)
8,»39(0°)
10,498 ± 1(20")
10,5^(0°)
(лит. В Т)*)
10.,'i'J.-, (20°)
10,489
19,29 'r 2,4(20')
I9,30y (iV)
19,287(25")
19,285
Спектроскопически]
чистый
Спектроскопически;
чистый
Снектро
сконически!
чистый
310 ч. и. структуры неорганических нЕШЕСтн |глЛУ
Общие замечания
Медь, серебро и золото были объектами многочисленных
структурных исследований; многократно определялись и размеры элементарной
ячейки.
В результате этих работ пока не удалось найти аллотропические
формы. Все три металла образуют только кубическую гранецентрирован-
ную ячейку Ск-
Большое количество работ посвящено структуре тонких слоев Cur
Ag, An, см. [16].
Си. В зависимости от загрязнений данные для aw колебались в разных
работах от 3,59 до 3,63. Как полагала редакция SB I, меньшее значение
должно отвечать более чистому материалу. В основе этой точки зрения
Vwc. 188.
лежала неправильная концепция, что примеси могут лишь расширять
решётку; это, безусловно, не так. Их поведение зависит от химического
характера примеси и типа структуры реального кристалла (см. гл. II).
Прецизионные определения более чистых образцов дали:
[1] (1931): Си = 99,949% (с 0,04% О, 0,02% Ni, 0,003% Fe, 0,004% As,
0,001% РЬ, 0,001% Са, спектроскопические следы Mg, Cs, N а) аш--~- 3,607я±ь
ся (20°) = 8,93.
[2] (1933): вю = 3,6078А.
$ 123] I* ГРУППА: МИД!», ГКРЕГ.1Ю, ПОЛОТО 317
[3] (1933): aw = 3,607e A (t° — комн.), (чистая медь?)
[4| (1934):олектролитическан медь «Мерк>> 6/^ = 3,608(20°).
[5] (1934.: яы,-3,6081 (t° -комн.).
[20] (1936) и [21] (1938): «ю - 3,60751 ±4.
|19| (1935) Страуманис и Енинс нашли: «,„-=3,6075.
В 1948 г. Н. В. Агеев и Д. А. Агеева опубликовали [22J результаты
исследования электронной плотности меди (см. рис. 188) при 20° для проекции на
(001) решётки Си и нашли, что распределение электронной плотности подобно
обнаруженному в Ni, по сильно отличается от найденного у AI.
Ag. Старые данные колеблются не меньше, чем для Си.
<7,„ = 4,03 до 4,НА.
В |(>1 (1924) для Ag--99,999% найдено: aw -4,079 + 4;
з,= 10.4.»±з и для 99,9% aw 4,05^.,А.
В работах периода J 925—1932 гг. в среднем «чистое» Ag давало аш-= 4,070.
Оуэн и Яте [17] (1933) нашли данные, приведённые в таблице.
По работе |8| (1933) ^ = 4,0774 (20ЭС).
По [9] (1933) aw -4,077 с точностью до 1°„.
По Н. В Агееву и Д. UloiixoTy[10] (1933) ^-4,077,.
По [И] (1935) а- 4,0772.
В 1936 г. было сообщено, что в тонких слоях Ag aw на 1% больше
но сравнению с рентгенографическими данными (анализ (?)/>. 6Л).
Джетте и Футе [14] (1935) для спектроскопически чистого Ag нашли
данные, приведённые в таблице.
Согласно [1 fj| (1938), afll = 4,0775 (19,6° С).
В 1940 г. Страуманис и Ешшс. [18] опубликовали сводку исследований
ячейки серебра асимметричным методом и нашли а = '1,07781^3 при 25° С.
Аи. Старые данные для а,„ (до 20 работ) колеблются от 1,06 до 4,09 А.
По] И] (1933) aw-=i,0W3±10.
По [9] (1933) я„, = 4,070.
По [5] (1934) а =4,0701.
Оуэн и Яте [13] (1933) и Джетте и Футе [ 14] (1935) для спектроскопически
чистого золота нашли данные, приведённые в таблице.
ЛИТЕРАТУРА к § 12;;
1. Phys. Rev. «7, 1696 (1931).
2. Ргос. R. бос. Ь (Л) 139, 526 (1930-
3. Naturw. 21, У82 (1933).
4. Z. Anorg. Ghemie 22f), 293 (1944).
5. Z. Krist. (A) 89, 560 (1934).
ft. Phys. Rev. 25, 753 (192i).
7. Phil. Ma*. (7) 17, Ш (1933).
8. Helv. Phys. Acta 6, 597 (U33).
9. Z. Anorg. Ch. 214, 16 (1933).
10. J. Inst. Mot. 62, 119 (Р.Ш).
11. J. Ch. Phys. 1, 753 (19 M).
12. J. Inst. Met. 67, 257 (193 >).
13. Phil. Ma,'. (7) 16, 606 (1933).
14. J. Ch. Phys. 3, 605 (1935).
15. Proc. R. Soc. L (A) 167, 25 (1938).
16. SB VII; стр. 48—58; Phys. Rev. 56, 58 (1Г39) я др.
17. Phil. Mag. [7] 16, 472 (1933).
18. Straumanis a. J e v i n s, Prazisionsboslimmung'von GUferkonstanlen etc. (1940).
19. Z. Phvs. 94, 184 (1935).
20. Ann. d. Pays. 26, 533 (1936).
21. Ann. d. Phys. 33, 737 (1938).
22. ИАН (x) 273 (1948).
:Л8
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТН
[гл. IV
§ 124. II* группа: цинк Zn, кадмий Cd, ртуть Hg (dlV)
t. Цинк, кадмий
Решётка гексагональная II.
Координация V.
Тип HV-SBA3.
Пространственная
группа Dc.h -Сб/ттс.
Z = 2, к. ч. = 6 (расстояние
d = a)-f6
( расстояниее= I/ тг+т ) •
Рис. IS).
Атомный
номер
30
48
I
Вещегтно
с/а
Vcm*
Чистота
Цинк [7] 2
» Is!
18
Кадмий [7J I 25
» 12
26
189
2,6594,, , 8 4,9 Л',8Г) ±1 1,8563! 0,7
1,<Ш1 1,^>60'
2,9::;!. 5,Г,069, ,1,882 . 12,90
1 , 4 |
2,9730,,
2,98:t7a
5,fi05.>r
5,0498,
7, ПО СпоКТрОСКОПИ-
i чески чистый
(PI) - 0,0004%,
|Fe, Co, Си следы)
'ш,18°
7,П5
7,ПУ
,(V.i7
99,9%
Спектроскопически чистый
(слабые следы
Си, Мп, Fe)
Cd - 99,94%
Zn = 0,04%
Pb-0,01%
Fe = 0,004%
§ 124]
II* ГРУППА: ЦИНК, КАДМИЙ, 1-ТУТЬ
319
;>. Ртуть
Решётка ромбоэдрическая /?.
Координация 6.
Тип /?G-SB Л10.
Пространственная группа
Dld — Ют.
Z <= 1 (примитивн.
ромбоэдр.) (см. таблицу) или
Z = 2 (центрир.-ромбоэдр.)
(см. текст, работа [\'Л\).
Гиг.. Г.и)
Атомный
номер
Вещество j t °(!
80 | Ртуть | 1Г>]
! » [!:>,]
—80
i
а 1 а
2, ТО)
3,004
70°:U',7
7o°.w
<* | г 1 **
2.WJ
1,Г)()0
К, 2'»
Общие замечания
Способность простых веществ Zn, Cd и Kg образовывать разные
аллотропические формы надёжно не исследована, несмотря на
большое количество работ. Первые два метала кристаллизуются в тине Ну—
A3 — гексагональная плотная упаковка. Ртуть (»:ак и в некоторых других
свойствах) ведёт себя своеобразно. См. ниже.
Цинк и кадмий отличаются очень высоким оччм^пением с/а.
Zn. Поиски иной модификации, кроме Ну, от комглтной температуры
и вплоть до температур плавления положительных результатов не
дали [1] (1928), хотя прежние исследования с помощью физико-химических
методов заставляли подозревать наличие нескольких модификаций. См.
также [2] (1925).
Колебания значений а в нескольких десятках исследований были в
пределах 2,64 — 2,66, колебания с — 4,90 — 4,93 с/а -1,855 — 1,862, причём
большинство работ было выполнено с металлом сомнительной чистоты.
В работе [3] (1929) найдено:
при tZoMH- a = 2,657; с-4,948; с/а -1,862;
при Сидк. воЗД: а -2,651; с-- 4,899; с/а =1,862.
Н2') Ч. II СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ГЕЩЕСТП [Гл IV
В работе [4] (1933) Zn. был исследован в интервале от 20° до 400° С:
20} а ==--=2,659; с = 4,93; г/а-1856; ?г = 15,11 (к\)\
400° « = 2,675; с -5,02; с/а~ 1,880; <г=П5,62(кХ)».
В работе [5] (1934) было найдено:
20° а =2,6590; с -4,9351; с/а 1,8560; w -----15,109 (к\)\
407° ««2,6788; с-5,0469; с/г/-1,8840; <г --15,683(к\)\
Но [6J (1933) для Zn = 99,99%:
а « 2,6590 ± г>; с - 4,9351 ± о; с/а - 1,8560 ± 7.
Джетте и Футе(71 (1935) и Оуэн л др. |8| нашли величины,
приведённые в таблице.
Среднее из многих работ периода 1933- 1936 гг.:
а - 2,659; с--- 4,935; с/а .-.-. 1,856.
Г» 1939 г. структура полированной поверхности Zn было исследована
;>лектронографически 118].
Cd. Старые физико-химические, например дилатометрические,
исследовании давали повод утверждать, что С<1 образует не менее трёх
модификаций—а, (3, у и что переход aCd^f^Co! происходит при температуре 64 С.
Бриджмеи при высоком давлении и температуре паше.;! ещё одну
модификацию (1925).
Рентгенографические исследования ;3| (1929) от температуры жидкого
воздуха до комнатной и [10] (1931) при более высоких температурах не
позволили обнаружить признаков аллотропии.
Средние значении (но старым работам):
а ■-= 2,97; с - 5,61; с/а - 1.89.
В 1. с. [3] найдено:
при tf.oMH: a = 2,965; с
при /жидк. возд: а --= 2,964; с
В работе[11] (1933) найдено:
при tZoau- а = 2,9713 ± :>о\ с -.-- 5,6016 ± yj.
Г. Ф. Косолапов и А. К. Трапезников [12] и Джетте и Футе [7| (1935)
нашли данные, приведённые в таблице.
И 1937 г. Cd исследован в работе [16]. В 1939 г. было опубликовано
исследование[19] вибрации атомов Cd.
Hg. Но вопросу об аллотропических формах ртути на сегодня, в
общем, известно, что Ня образует при р = 1 атм только одну
модификацию—ромбоэдрическую решётку Л10. Структура ртути вызвала длительную
дискуссию. Описание ее в SB 1 31 встретило возражения, причём была
предложена иная структура, которую SB I 42 считал невероятной. Однако
в SB ] 737 принимается уже эта последняя структура с ромбоэдрической
ячейкой, пространственная группа D^d-
а =- 3,00; а - 70^32'; d = 3,00; г = 23,2 А3.
- 5,598; с/а = 1,888;
- 4,564; с/а = 1,877.
§ 1251
Ш ГРУППА: БОР, АЛЮМИНИЙ, ГАЛЛИЙ, ИНДИЙ, ТАЛЛИЙ
321
Позже [13] (1929) эти данные были подтверждены: а = 3,009, а •■■=-■ 70°33'.
Рассматривая не примитивный, а гранецентрированный ромбоэдр, получаем:
а = 4,598; а = 98°14' (при t* •■■■■= —80° С). К аналогичным выводам приводит
работа[14] (1932) и (15] (1933).
Структура ртути заслуживала бы более тщательного исследования.
В последние годы большое внимание привлекала структура жидкой
ртути.
В результате исследований жидкой Hg был сделан вывод о наличии
гексагональной плотнейшей упаковки и в жидкой Hg (ближний порядок).
В 1939 г. структура жидкой Hg была исследована 120] в интервале—36 -f 250°.
ЛИТЕРАТУРА к § 124
1. Z. Ph. Ch. 133, 165 (1928).
2. SB I 41 (1925).
.4. Trans. R. S. Canada (III) 23, 255(1929).
4. Phil. Mag. (7) 16, 479 (1933).
5. Phil. Mag. (7) 17, 113 (19!4).
6. Mctallwirtsch. 12, 539 (1933).
7. J. Ch. Phys. 3, Л05 (1935).
Я. Z. Krisl. (A) 91, 70 (1935).
9. Z. Ph. Ch. 87, 409 (1914).
J. A. Ch. Soc. 44, 1.143 (1918).
10. Metallvvirtsch. 10. 221 (1931).
11. J. Ch. Phys. 1, 753 (1933).
12. Z. Krisl.(A)91,410(1935); SB 11737,750.
!3. Z. Phys. 53, 72 (1929).
14. Z. Krist. 8*, Ш (1932).
15. Z. unorg. Ch. 212, 40 (1933).
in. J. Phys. Ch. 41, 4)9 (1937).
17. SB VI 4'*.
18. ^B MI 55.
19. Proc. Phys. Soc. (A) 51, 73 (1939).
21). J. Ch. Phvs. 7, 958 (193*).
§ 125. Ill группа; бор В, алюминий А1, галлий Ga. индий In. таллий TI
1. Бор I.
(Игольчатые кристаллы).
Решётка тетрагональная 7\
Вид симметрии Dun — 4/ю.
Тип (?).
Z = 52.
(н*р*)
Атомный
НОМО[)
г
[
Вощество
Вор I [4]
комн.
а
8,93
с
5,06
с/а
0,567
V И Аа
403,77
V см*
4,675
*х
2,314
С11ШШ.
2,30—2,34
(2 7° С)
Чистота
От 99 до
100,2%В
2. Бор II.
(Пластинчатые кристаллы).
Решётка ромбическая (или моноклинная?).
Пространственная группа -■?
Z = ?
Атомный
номер
Во ще с т по
Г
а
b
с
V II А3
Чистота
Вор II [4] | комн.
10,13
'Л
17,86
137,819 2,30—2,34 От 9.) до
100,2-о В]
; (2;° с)
Структуру бира расшифрованной считать нельзя.
322
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ИЕЩЕСТВ
[ГЛ. IV
3. Алюминий, р-таллий
Решётка кубическая С гранецентриро-
ванная.
Координация К.
Тип Ск-SB А1.
Пространственная группа 0\—Fm'Sm.
Z = 4,k.4. -- 12 ( расстояние d = fl" t>
(+6 расстояние е~а).
л
О
is
о
^
А/
СЛ
о
У
6
Рис. 191.
[Атомный
|номер
13
81
| Нощостио | t К]
a to ' d
Алюминии [13| 25 1 4,0И43±2
| Ю] 1 25 ' 4,04 Wg±8
» |2<)1 19 4,03970.
I
Таллин [17] комн. 4,8'*1
2,8578
3,42;*
г
1,428е.»
1,711
1-А3
06,01
113,45
Vr.u*
9,935
17,0'Г)
".г
2,715
2,695
11,97
Ч пепла
Очоиь чистый
4. Галлий
Решётка ромбическая
OR™.
Координация 7.
Тип ОД?*-SB ЛИ
Пространственная группа
Dm —С тс а.
Z = 8, Ga к. ч. -
- 1(2,45А) + 2(2,70А) +
+ 2(2,73А)+2(2,79А) = 7.
Намечаются «молекулы»
Ga2.
Атомный
номер
1 Вещество
\ t°
\ а • •
\ь . . . .
\ с . .
\ а : Ь : с .
\d .
\ г . . . .
i;bA3 . .
V см3 . .
а, ...
Чистота .
1 1
I :Ц
| Галлий [21 ]
J Комнатная
1 4,5167±i
4,5107±i
7,6448±2
!l : 0,99868 : 1,69257
2,437
1,217
155,75
11,453
6,087
Оч. чистый 1
Hilger'a I
1 1
Рис. 192.
§ 125|
III ГРУППА: Ь01\ АЛЮМИНИИ, ГАЛЛПП, ИНДИИ, 'ГАЛЛИЙ
IV23
о. Индий
Решётка тетрагональная
Т гранецентрированиая.
Координация ТК (стр.806).
Тин Ттк -SB Aii.
Пространственная группа
Dih - Jihnmm
Z —4. Плотное if, упаковки
^68,7/V
[Атомный
1 номер
Вещсстно
\t° . .
\а . .
\с . . . .
\с/а . . .
<*....
г .
Uas . .
i(?A8
IV см* . .
~х • • ■
Чистота
* i
Иидий[-х>-:].|[цд11Й[^:{|
Комн. i 22
4,583-f^i 4,588
4/.Ш + 2 4,938
1,077+7
V-^ i
1,62
КМ, 6 !
25/JO '
1 Г>, .">«.> ;
;,:м8 !
Чистый ■
Рис. 19:*.
6. а-Та.ыий
Решётка
гексагональная Н.
Координации I7.
Тип Hv SH A3.
Пространственная группа
Dlh-'-CQfmmc.
Z 2, к. ч. ~6(расстояние
d ,-_= а) -;- () (расстояние
'-■/■?
Атомный номер
Вощсстно . .
t3
гА»
wA8
Кем3 ....
' 4 /
i и
Таллий
Комн.
3,450
5,520
1,600
50, S9
2S,44
17,ОС)
11,83
17
Рис. 194.
324 __ Ч. П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. ]V
Общие замечания
Характерной особенностью III группы является то, что все 5
элементов, в неё входящих, образуют разные решётки; таким образом
III группа охватывает не менее 5 структур.
Аллотропические формы исследованы также не слишком детально;
в отдельных случаях описаны дополнительные структуры,
В. Получение чистого бора и исследование его структуры не удавалось
до последнего десятилетия. При восстановлении бора из соединений
алюминием или магнием и т. п. возникают бориды. Старые работы по бору
поэтому вообще нельзя считать надёжными.
Не случайно, что в связи с этим, а также с большим техническим
значением бора и его соединений, в последние годы опубликован ряд
исследований, посвященных его получению и свойствам, например [1 ] (1945), [2| (1945)До]
((1946). В 1943 г. опубликована работа Гоарда(4] с сотрудниками, посвященная
приготовлению и структуре бора. В этом исследовании бор получался
восстановлением ВВГд водородом на нагретой вольфрамовой или танталовой
проволоке; при 600—800° С (аморфный бор), 900 — 1000°С (частично кристаллические
образцы) и 1000 —1300° С (кристаллические). Образцы, полученные в
интервале 1100 «1300° С, имели от 99 до 100,2% В (?) Уд. вес <г- 2,30-2,34 (27°С);
среднее значение —2,310, близко примыкающее к данным работы [12| (1931).
В результате рентгенографического исследования по методам Лауэ,
вращения, Вансенберга оказалось, что кристаллы В имеют необычайно
сложную структуру, причём у игольчатых образцов кристаллов В был найден
тетрагональный вид симметрии D±h— -4///г. Наименьшая тетрагональная
(или псевдотетрагональная) ячейка имеет я = 8,93; с-5,06. В тетрагональной
элементарной ячейке 52 атома бора. У пластинчатых кристаллов бора
предполагается элементарная ячейка, основанная на примерно или точно
ортогональных осях с а = 10,13; 6=8,93; с = 17,80. Другие данные свидетельствуют
о том, что симметрия не выше моноклинной.
Авторы заканчивают статью указанием, что полное определение
структуры В очень трудно, но не невозможно и что работа ими продолжается.
Однако за истекшие шесть лет новые публикации не последовали.
А1. Хорошо известна кубическая гранецентрироваипая структура
тин Ск — S8AI.
В работе [5] (1933) исследовался А1, полученный катодным распылением
на Pt, и было найдено (электронографически), что в очень тонком слое А1
имеет тетрагональную гранецентрированную структуру с а = 3,90; с/а =1,03
(т. е. с = 4,017. — Б. О.). В более толстом слое А1 образует кубическую
гранецентрированную структуру с я = 4,01.
Образование тетрагональной, вытянутой в длину элементарной ячейки
следует поставить в связь с тем обстоятельством, что для Pt ^ = 3,91 А.
Таким образом, А1 как бы наращивает решетку Pt; такая структура
может быть и стабильна в весьма тонких слоях, но несомненно является
метастабильной в достаточно толстом слое. Она не может считаться
модификацией А1. Таким образом, пока известна лишь одна модификация А1.
Согласно старым работам, а = 4,04 — 4,07 А.
Для А1 99.6% по 16| при 18°С а = 4,04065±2; з (20°) = 2,69^,;
но[7] (1934) я = 4,0400А;
по [8] (1935) я = 4.0770 при 368°С;
по 191 (1935) а - 4,038 ± 1 А.
Жмудский 126] (1949) опубликовал величину, приведённую в таблице.
В 1940 г. Страуманис и Евинс опубликовали большую сводку
результатов исследований алюминия асимметричным методом и нашли я = 4,04145 ±2
§ \2Ъ] Ш ГРУППА: БОР, ЛЛЮМШШП, ТАЛЛИН, ИНДИЙ, ТАЛЛИП 32Г)
при 25е С. Рассчитанные нами на основании этого результата величины прп-
недены в таблице (стр. о22).
Агеев и Агеева опубликовали [24] (1948) исследование электронной
плотности в А1 при 20°.
Ga. Аллотропические формы Ga детально не исследованы. Описание
в SB J, как на стр. 44, так и на стр. 738 (тип .411) структуры Ga оказалось
неточным. В SB II она изображена, как па рис. 192. Элементарная ячейка
ромбическая, как бы состоящая из молекул Ga., с расстоянием Ga - Ga = 2,45 А.
Т1омимо ближайшего атома на указанном расстоянии, атом Ga имеет двух
соседей d' = 2,7(), 2d" = 2,73 и 2с/"'= 2,79'. Симметрия атома: Cs — m,
симметрия «молекулы»: С,-—1. Бредли [21] (1935) нашёл величины, приведённые
в таблице, причём d-2/i37; c/, = 2,706; d" = 2,736; d'" = 2f795.
In. Известен в виде одной модификации тетрагональной гранепентри-
рованной, тип Ati. В [14] (1921) были найдены: а =-4,58; с=-4,86; с/а = 1,06;
^=3,24 (в плоскости базиса) и 3,33 (по оси 4); ат —7,24 (тин Л6).
В [15] (1932) было найдено, что электролитический, переплавленный
в вакууме In образует решётку Л6 тетрагональную гранецентрированную,
'7 = 4,5884:i;.; с---4,94ап>; ^/а = 1,078±2* ^ = 3,24 (в плоскости базиса) и г/"-3,37;
■ г — 7,28. В 1935 г. были найдены [22], [23] данные, приведённые в таблице.
При этом d-3,24; d' = 3,37; e = 4,583; t = 4,936 A (SBII1).
В работе [23] исследовался In в интервалеот — 25°С до -4-140° С и
определён коэффициент линейного расширения аа==5,6 X Ю'6; ar=1,3x'K"5.
Имеется наблюдение, что In при давлении 400 т на 1 см2 якобы
становится аморфным [27] (!). Заслуживает проверки.
Т1. По исследованиям ряда авторов, в частности [16] (1928) и [17] И930), TI
образует две формы: аТ1—тип АН (гексагональный) и {JTI — тип А\
(кубический гранецентрированный).
Для получения ВТ1 расплавленный Т1 закаливается в ледяной воде.
Отсюда следует, что [4-форма является высокотемпературной.
В [18] (1920) найдена температура перехода aTl^ рТ1 между 225 и 235°С,
что нуждается в проверке и подтверждении.
Наконец, в|19| (1923) была найдена тетрагональная грапоцентрировашшя
решётка с ^/ = 4,75; 6-5,40; г.х= 11,02.
В 1926 г. была найдена [20] тетрагональная ячейка с а -^ 5,2; с- 8,2;
Z-8 (Л). Эти данные позже были опровергнуты. В одной из работ был
также исследован жидкий Т1 и найдено наличие структуры, близкий к
гексагональной плотнейшей упаковке (ближний порядок).
ЛИТЕРАТУРА к § 125
1.
2ш
3.
4.
5.
6.
8.
9#
io".
11.
12.
13.
С. К. 221, 698(1945).
С. А. 44, 4594 (1946).
С. R. 221, 747 (19'*5).
С. А. 4'), 3956 (1916).
J. A. Ch. S. 65, 19-14 (1943).
Ргос. К. Soc. h. (A)14t, 398 (1933).
Phil. Mag. (VII) 15, 472 (1933).
Phil. Ma^. ( II) 17, 433 (1934).
Phil. Mag. (VII) 21), 761 (1935).
Z. MetaMv. 27, 126 (1935).
J. Ch. Phys. 3, 605 (1935).
SB VI 44.
С. Н. 19$, 776 (1331).
S t г a u m a n i s u. J e v i n v, Pra/isl-
onsbestimmung von Gitterkonstanten
14.
15.
16.
17.
18.
19.
20.
21.
22.
23*.
24.
25.
26.
27.
Phys Kev. 17, 266, 549, 571 (1921).
SB II 173.
Z. Phys. 60. 433 (1928).
Z. Krist. 74, 189 (1930).
Phvs. Kev. 15*38 (1920).
Z. 'Phvs. 16, 105 (1923).
Z. Krist. (A) G3, 318 (1926).
Z. Krist.. (A) 91, 302 (1935).
Z. El. 39, 81 (1933).
J. A. Ch. S. 67, 228 (1935).
Изв. АН(х) 17 (1947).
J. Appl. phys. 12, 461(1941); Ch. Zbl.
1, 458 (1942).
.Чав. Лабор. XV, 1055 (1949).
J. Appl. phys. 1.2, 4G1 (1941).
:*2G
Ч. II. СТРУКТУРЫ ПКОРГАКПММСК'ПХ ПЕЩЕСТ1',
Iгл. IV
§ 126. IYr группа: углерод С, кремний Si, германий Ge, олово Sn,
еиинец ГЬ (s-p-)
1. Углерод (алмаз),
кремний, германии, а-олово
Решётка купичоскан С
(алмазная).
Координация Т.
Тип С г -SB Л\.
Пространственная
группа Ol—Fdlim.
Z --=-- 8. Плотность \ паков-
кл 34n/.v
d - ат -,- .
Рис. Н>5
Чтомный
номер
Вещество
Г
d
О/
6 I Алмаз [2] |комн.. 3,55%. , г
32
50
I
» ум
» i'l]
Кремшш[20]
3,559-
о
2оэ г.
а ±г,
19
» [2G
Германии
з-олово
«5° С
2;*э С
5,4198^
5 /i \<т±й
20>С;5,6^±6
I8M: 6,46
1,54
'2
2/.ИГ).
2,35
~'4I6
2,80
0,77,
1117:*
1 99
1 ,—.,
1,40
3,5()
2,32,
5,31
5,8.
Примечание
Типичная «валентная»
структура с 4 связями,
направленными под те-
траадрическими углами
Распределение
электронной плотности
в алмазе—см. § \п\.
Подробнее о
структуре см. § 03.
W,84% Si; 0,1% N,;
0,027% Fe; 0,014% ('a;
0,008% Al; 0,004% (';
0.002% О2 следы IL;
0,006% Na;
0,020% Fo;0,005%Ca;
0,016% Al;
0,025% C; 0,001% Mn;
0,006% H2
§ 126) IV ГРУПП \: УГЛЕРОД, КРЕМШШ. ГЕРМАНИЯ, ОЛОИО, СМИПЕП 327
2. Графит
Решётка гексагональная (слоистая) Hs.
Координация t + 1.
Тип Ян-1 SB .49.
Пространственная группа D\h — Cti/wmc.
Z = 4. Плотность упаковки 16,9%.
Рис. 106.
Атомный |
помор |
ВО
О
Я
О
6 Графит [7]
| » |8]**)
j » М
I
О
комн.
1'«,6°С
а
с
\
2,458 j 6,701
2,455±2| 6,69
2,46 6,78—6,80
i
1
2,4562±7- 6,6943±7
с/а
2,726
2,75—2,76
2,725
die *)
1,42
2,46
1,4210
г
0,7Х
0,70
*>
*<
и
8,9
5,39
°JC
2'23
*) d -pace lomiue между двумя соседними вершинами шестиугольника, т. е. в орто-
ноложеппн, с --расстояние между двумя вершинами, в.чптымн черен одну, т. е. в мета-
положении. Расстояние между двумя соседними атомами вдоль оси 0, т. е. вдоль
г
4-j'i св»;ш атома, равно } и, значит, больше, чем между атомами в мечаположении.
**; Бемольный ij,eii.ioitciviu'i графит и очень чистый ачесссновснпй графит.
328
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ БЕЩЕСТК
[гл. IV
2а. Графит
Решётка ромбоэдрическая
(слоистая) Л" .
Координация / + 1.
Тин Лн-i-SB?
Пространственная группа
Dm -Ют.
Вещее т по ! Г графит |
/°
а . .
л . .
а
с ...
Комнатная
:*,6tt
.4<Vi(j°
2,-'|Г)6
6,6% xs/a-=W,0'i'i
Примечание. По [30]
(1942) ячейка простирается
идоль оси С на 3, а не на
2 слоя (как в обычном
графите). В исгледонанных
обр. зцах нес но ной
структуры—до l'i°f,
Рис. 1у;.
чаблпце даны значения а и -л для ромбоэдрической,
а и с для гексагональной ячеек.
3, 3-олоно
Решётка
тетрагональная Т
(деформированная
алмазная).
Координация £4-2.
Тип 7\s+2.
Прост])анственная
группа i)V/,—Jijamd.
2=4.
Рис. ЮЯ.
? / f
Рис. I'J'J.
§ 120] IV ГРУППА: УГЛЕРОД. КРЕМНИЙ, ГЕРМАНИЙ, ОЛОВО, СНИНЕЦ
riu
Л томный
номер
Вещество t V
Г.О
1
3-олоно [2|
» ш
1 » н
» [Ю]
» |20|
25
23
2 Г,
Г.,819Г,0±:Г|
5,819
5,81К8±.7
5,8191»'t
ДО
5,81970
1
3,17500±12
3,1728±2#
3,174
3,1745±4
3,17478
ДО
3,1748 8
1
0,5455
0,5455'»
7,281
|
1
j
i
i
На рис. 199 показаны 2 элементарные ячейки HSn, метрика которых
приводится в таблице. На рис. 198 показана элементарная ячейка |*JSn
с осями а и в, в качестве которых приняты диагонали основании ячейки
рис. 199. Такая ячейка является гранецентрированной. Сопоставляя еГ>
с ячейкой алмаза и «Sn, легко обнаружить их сходство и их различия.
Алмазная решётка как бы сплющивается (с =3,15, «=8,23).' Атомы,
находившиеся в центрах 4 октантов (т. е. 1, II, III и IV в тетраэдрлческом
порядке), смещаются.
Координационная сфера атома,
показанного большим кружком,
состоит теперь из 4 + 2 атомов
(приближаются V и VI), см.
рис. 198 справа внизу и
стр.333. Тетраэдрическое
расположение первых четырёх
хорошо заметно.
4. Сьинед
Решётка кубическая С гра-
нецентрированная.
Координация А\
Тип Ck-SBAI.
Пространственная группа
О^—Ь'пгАт. Z = 4, к. ч. 12
(расстояние a = -J£->— '.
Атимнып
номер
82_
Вещестьо
Сшшец | 3]
» [2G]
»> Г281
t>c
18
25
25
j «w
4t939G_h3
!
1
| 4,94006±3
1 в-
(при 0^)
Ч»стита
99,9
Очень
чистый
99,999%
ХЮ 4.II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТН [гл. IV
О б и; и е замечания
Элементы IV группы периодической системы образуют модификации,
отвечающие 4 основным структурам.
1. Алмаз Cr -SB .44, являющийся типичной валентной (гомеополяр-
ной) структурой, где каждый атом, осуществляя ^//-валентность,
направляет 4 связи к вершинам тетраэдра, в центре которого он сам находится.
2. Графят (SB 49) и
3. 3-олово (SB АЪ)} которые являются двумя промежуточными
ступенями (низкое координационное число—4, заметная или значительная
металлическая проводимость).
\. Свинец С к SB A\ - типичная металлическая структура.
Кроме того, существуют и другие структурные формы, особенно
у углерода.
Со структурной и термодинамической стороны общая характеристика
систем, подобных Сал.ч<~С1г1,аф, aSn Г ! ?Sn и др., была дана в гл. I и П.
С углерод образует две модификации: алмаз и графит, причём для процесса
СалМ1:Сг1Шф при р^\ амм ±Fm~±Hm- ■ T±Sm = 454 + 0,85x298 =
= 809 ш\л (по старым данным IF = 180+0,7X 298 - 390 кал). При р ^ 50 000 атпм
и /э около 2000° С система становится практически энантиотроиной (см. S 80).
Кроме того, углерод образует различные аморфные формы. Некоторые
из них имеют обрывки структуры графита, другие своеобразную структуру,
присущую только «аморфному» углероду.
С (алмаз). Атом углерода в алмазе имеет тстраэдрическую симметрию.
Будучи фиксирован в валентной решётке, исключающей вращение,
птом С во лшогих случаях препятствует возникновению кристаллов, например
/.масса Oh.
Однако, хотя в кристалле алмаза отсутствуют оси 4 и плоскости
симметрии, параллельные граням куОа, вместо них появляются винтовые оси
четвёртого порядка и плоскости скольжения (см. рис. 115), придающие
Л'псталлу в целом симметрию ()h (пространственная группа 0\).
Более детально о структуре алмаза см. § 63 и* 90.
В работах 1913 — 1926 гг. я = 3,55 -3,56 А.
Позже |1] (1926) было найдено: «-35595±ш и |2] (1932) а = 3,5596П;,.
Из этих данных следовало для расстояния С С d-1,54 А.
В* 1944 г. было найдено [211 для С алмаза г/ = 3,55974. В том же году было
опубликовано [22] исследование рентгенографическим путём термического
коэффициента расширения алмаза от 25 до 650° С.
Распределение электронной плотности в алмазе было изучено в работе [29]
(1939) (см. § 96).
В 194'i —1945 гг. имела место большая дискуссия |34], [35], [36] о
строении реальных кристаллов алмаза четырёх возможных структур.
С (графит). Впервые был установлен для графита тип Л9 в 1917 г. См.
также |3](1922), решётка гексагональная « = 2,47; с-=6,80; с/я *= 2,75; Z — 4;
d-^1,50 А и расстояние между сетками (0001) 3,40 А.
В 192'* г. было найдено [4|: «=2,47, с=6,79, Z^=4, а также |5|: а =
= 2,\Ь±4; г-=6,82^3; ф = 2,П; Z=4.
В 1925 —1926 гг. найдено [6]: я = 2,46; с = 6,74; Z=4; расстояние С—С
и плоскости базиса = 1,42 А; в 1928 г. [7|: а = 2,48; с=-6,78 А.
В 1915 г. Нельсон и Релей [31] нашли для С (графит) данные,
приведённые в таблице.
Вопрос об изменении элементарной ячейки в процессе перехода
|С) аморфный —> [С] графит изучался многими авторами, и было наилено.
§ 126| IV ГРУППА: УГЛЕРОД, 1П ЕМНПП, ГЕРМАНИИ, ()Ji()HO*Cj ПНКЦ 331
что глубокая рекристаллизация ведёт к получению графита со
значительными изменениями элементарной ячейки, несомненно, вследствие
наличии посторонних атомов (см. гл. -Х- Гр'афитиды).
Так, было обнаружено [9], что вместо расстояния между сетками 3,41 А
возникают расстояния 3,73 (графит из сажи горящего бензола или
нафталина) и до 4,0 для графита из СС14. Кроме того, возникают кристаллы
разных размеров.
Изменения d исследованы также в работе [10] (1935).
Общая генетическая характеристика графита дана В. С. Веселовским
и К. В. Васильевым [8| (1934). См. также Веселонский [37].
Для очень чистого ачессоновского графита и для беззолыюго
цейлонского графита с 99,3% С и 0,65% золы данные [И] приведены в таблице.
Финн и Вильман [12) (191 6) нашли электронографически данные,
приведённые в таблице.
Изменение ячейки с величиной кристаллов изучалось в [11].
В j912 г. ].<•. [30| была описана ромбоэдрическая модификация графита
(см. таблицу).
В 1915 г.(31] был измерен коэффициент линейного расширения графита
х- и a_j_ в области от 15 до 800° С и обнаружено, что имеет место
значительная анизотропия (а,, > a_j_), а в интервале до 100° С якобы
наблюдается сжатие. Это последнее наблюдение было опровергнуто (1916) [32]
и даны значения аи и а.; а X 10е равно:
ГС
! "и
1 "-L
—
1 1
0—Г>00
17,2
1,:*
П,2
Г. 00—1000
20,4
2,2
9,3
1000—1500
2'i,4
2,Г»
9,4
1500—2000
27,7 1
8,7
В 1933 г. была описана] 13] «особая форма» «кристаллического углерода»,
выделяющегося при неполном сгорании на глазурованной стенке
фарфорового тигля при G50 900°С (очень твёрдый продукт), а в 1926 г. было
найдено | И], что блестящий уголь отличается от графита величиной
кристаллов — вместо 55 Л - 38 Л .
Наконец, был исследован блестящий уголь и получена порошковая
диаграмма мелкодисперсного графита [15). Эти данные опровергают (?)
старые наблюдения Шибольда о появлении наряду с линиями графита
линий алмаза.
Углерод * с перекрещивающимися связям и»
и другие формы
В 1934 г. автор книги, совместно с Б. А. Петровым, исследовал
реакционную способность карбидного углерода и показал, что в
зависимости от типа исходной структуры карбида процесс хлорирования идёт
совершенно различно. Если карбиды типа С1Т4, имеющие 1 атом углерода
в структурном узле, образуют галогениды, например СС14, то ацетиле-
ннды типа Na2C., выделяют сажу, так же как и карбиды с атомной
решёткой типа карбида кремния. Карбид кремния, будучи подвергнут
хлорированию при температуре порядка 1000е С, даст хлористый кремний,
углерод же образует сначала сажу, постепенно переходящую, как показал
рентгенографически В. II. Касаточкин, в графит.
В 1910 г. Джибсон, Гологан и Релей [16] опубликовали сообщение об
углероде с перекрещивающимися связями. Там же даны рисунки структур
332
Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТГ
[ГЛ. IV
углерода, вытекающих из'этой работы (рис. 201,202). Эти
авторы,'подчёркивая, что углерод, полученный из молекулярных карбидов (СН4, СО
P.jr. 2П1.
и др.), имеет структуру графита, показали, что аморфный углерод,
возникающий при термическом разложении целлюлозы и других веществ, а также
Рис. 202.
при восстановлении гексахлорбензола натрием CeCle+6Na==Ce+6NaCl,
имеет своеобразную структуру графитовых пластинок" с
перекрещивающимися связями (уравнение реакции мы пишем иначе, чем указанные авторы,
чтобы привести его в соответствие с теоретическим содержанием реакции).
126) IV ГРУППА: УГЛЕ! ОД, КРЕМНИЙ, ГЕРМАНИИ, ОЛОВО, СВИНЕЦ 333
Работа эта носит экспериментальный характер, в ней рассматривается
структура кокса и не формулируются выводы более общего порядка.
Если отщепление углерода вести постепенно, то образование связей,
по стерическим соображениям, требует поворота колец друг относительно
друга (рис. 203) и возникновения в окончательном итоге метастабильной
формы аморфного углерода, могущей образовывать структуры, отличные и
от алмаза, и от графита. Подобные образования вряд ли могут повторяться
чрансляционно, даже в самом грубом приближении.
Таких форм может возникнуть очень много, но, вопреки опубликованно
му сообщению [16], нельзя называть какую-то совокупность структурных
элементов в подобного типа структурах элементарной ячейкой.
В термодинамическом смысле они также не имеют ничего общего с
нормальной кристаллической фазой, характеризуемой воспроизводимой,
хотя бы в первом приближении, кривой v = f (/).Они дают множество кривых —
полосу, характерную для аморфного
состояния § 8.
Si. Пока описана только одна
модификация Si—тип Ci — SB Л4.
В старых работах яш=от 5,31 до
5,46.
В работе [17] (1928) a = 5,U8.
В работе [18] (1933) a-5,417O±i0.
Джетте и Футе [19] (1935) и Нейбур-
гер [20] (1935) нашли величины,
приведённые в таблице.
Страуманис и Евипс |26] (1940) в
результате исследований структуры
кремния асимметричным методом нашли
«„, = 5,41975 ±5 при 23° С.
Sn — диморфен. aSnT^pSn, tu=18°C
aSn — тип алмаза Ст — SB Л4, изучен
мало. SB I, на основании старых работ
принимает а ^6,46.
|3Sn —тип АЬ, деформированный
алмазный. (См. рис. 198 и 199.)
pSn — тетрагональная структура,
изучена весьма детально. В SBI 22
приведена^ фотография элементарной ячейки
с периодами а — 5,84; с = 3,15; б'/я = 0,539;
Z = 4. Этот чертёж двух элементарных
ячеек pSn приводится во всех курсах и Рис. 203.
монографиях. Согласно описанию SB 122,
к. ч.—б. Чертёж этот, однако, плохо отображает близость структуры
>Sn к типу алмаза Ст — АА.
Если повернуть координаты на 45° вокруг оси с, то получится удвоенная
Г с 3,15 л ,10\
тетрагональная ячейка типа алмаза со сжатием но оси с ( — ^о-о^^оо J.
Это сжатие внесло существенное изменение в величину к.ч. — оно стало
равным 6. На рис. 198 центральный атом обведён кружком. Атомы I —V
помечены римскими цифрами. Атом VI не показан на рисунке. Он находится
над центральным атомом но оси с, на том же расстоянии, что атом V. Все 6
расстояний одинаковы. Координационная сфера имеет необычный вид:
центральный атом находится в центре сфеноэдра —тетрагонального тетраэдра,
Г$4 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕ1ПЕСТН [ГЛ. IV
в вершинах которого находятся 4'атома. Кроме того, по оси 4, па тех ;ке
расстояниях от центрального атома находятся ещё 2 атома.
Такая координационная сфера может быть получена следующим
образом (рис. 198 справа вверху): а) впишем в круг радиусом с = 3,1Г) А
правильный шестиугольник, Ь) поместим в центр круга центральный атом (см.
рис. 198) и сместим точки I и II с одной стороны и точки Ш и IV с другой
стороны по окружности по направлению к обозначенному пунктиром
диаметру, пока расстояние между хордами 1—11 и 111—IV не станет
равным — == 1,575 А, с) разрежем круг по пунктиру и повернём его нижнюю
часть вокруг оси с (VI — V) на 90° относительно верхней; тогда хорда 11 - J
будет отвечать верхнему ребру тетраэдра, хорда 111 -- IV — нижнему
ребру, а расположение точек 1 —VI—координационной сфере р-олова.
Все расстояния от центрального атома до периферических—одинаковы,
И работе [22] (1925) указаны а = 8,235; с==3,165.
В работе 124| (1932) даются для малой ячейки значения (Sn химически
чистый —0,01% примесей): а = 5,819.1±з; с = ЗД75з-«; с7« = 0,545б i 2-
Г. Ф. Косолапов и А. К. Трапезников [25J (1936) нашли ячейку,
приведённую в таблице.
С большой точностью определили параметры [йпСтрауманис и Евинс |2б|
(1940) (см. таблицу).
Дефекты реальных кристаллов Sn исследовались в 1943 — 1944 тт. |38].
РЬ. У РЬ описана только одна структура Ск-
По старым работам aw^4,91 —4,94.
По [18| (1933) аю = 4,9385±70.
По [27] (1934) aw=r 4,9382 с точностью нескольких единиц последнего
знака. Оуэн и Яте [28] (1933) нашли данные, приведённые в таблице.
В 1940 г. были найдены данные [26], приведённые в таблице (стр. 329).
В 1941 г. было найдено [33] для РЬ яш = 4,9400.
В 1946 г. была определена элементарная ячейка свинца, в зависимости
от наличия примесей и для очень чистого РЬ (99,999%) найдено а = 4,9408.
В заключение отметим, что (SB 1 55) РЬ был исследован также при
температуре жидкого воздуха для выяснения структуры РЬ в состоянии
сверхпроводимости и найдено, что структура кристаллической решётки РЬ (6'к)
остаётся без изменения.
ЛПТКРАТУРА к § 12:>
1. Z. Krisl. 6 J, 320 (192*>).
2. Phys. Rev. 40, 662 (1032).
3. Phvs. Rev. 10, 661 (1917); 20, 113 (1922).
4. Z.Phys. 25, 317 (1924).
f>. Proc. R. S. A 106, 749 (1924).
6. Journ. Phys. 6, 38 (1925).
7. Aim. Phvs. 85, HI (L928).
И. ЖФХ V, 082 (№3-1).
9. SB II 175.
10. C. R. 201, 1189 (1935).
11. Z. Klektroch. 42, 504 (1930).
12. Proc. R. Sh. A 155, 345 (1936).
13. В:зг. 56, 2071 (1923).
14. Her. 50, 2433 (1926).
15. Z. Krist. 83, 503 (1932).
16. J. Ch. Soc. июнь, 456 (1946).
17. Z. Krist. 67, 235 (1928).
18. J. Ch. Phys. 1, 753 (1933).
19. J. Ch. Phys. 8, 605(1935).
20. Z. Krist. (А) И2» 313 (1935).
21. Mat,. 153, 22, 587 (1944).
22. Nat. 154, 486 (1944).
23. Phys. Rev. 25, 107 (1925).
24. Z. Krist. (A) «4, 20 (1932.) *
25. Z. Krisl.. (A) 94, 53 (1936).
26. S t v a u in anis u. Jevi n s,Prazisions-
bestimmun^ von GiUerkonslanten (19'»0),
стр. 100.
27. Z. Ph. Ch. (A) 16S, J 74 (1934).
28. J. Am. Ch. Soc. 68. 1493 (1946).
29. Aim. (1. Phvs. (5) 34, 393 (1939).
30. Proc. R. Soc. 1. (A) 181, 101(1942).
31. Proc. Phvs. Soc. 57, 477 (i'45).
32. C. R. 223, 501 (1946).
33. Proc. Phys. Soc. 53, 517 (19 41).
34. С. А. 39, 449 (1945).
35. C. A. 3», 2018 (1945).
36. Nat. 156, 83 (1945).
37. Веселонг к u Ji, Технология графита.
ОПТИ (1935).
38. Physica 10, 7»5 (1943); 11, 114 (1944).
§ 127]
V ГРУППА: АЗОТ, ФОСФОР, МЫШЬЯК, СУРЬМА, ННСМУТ
§ 127. V группа: азот N, фосфор Р, мышьяк As, сурьма Sb, висмут Bi (я2//)
1. а-азот (N2).
Решётка кубическая 6'л/.
Координация К.
Тип С% — SB521 (тип СО).
Пространственная группа ТА--Р2Х3.
ос — N2.
Z — 1 молекулы N2,
ак. ч.= 12а.
(Начало координат лежит и центре шюбраячёлиоп элементарной ячейки.)
Ц£фа#£
О
Рис. 20'к (Ср. с (Jo, pur. 20S.)
[Атомный
1 номер
7
Вещество
а-азот [2]
» [7|
!
t° !
1
1
20° К
(—2r,:i, i° <;> 1
20° К
(—253,1° С)
|
°1П
Г), Г, Г,
Г,, 67
d
' 1,05
1,(Ж
Г
0,525
o,5:i
е
ViG
I
Г c.w-s
1'{,7
j
с.г
1,02
Чистота
Не шшестна
тт
Не пзпестна.
Оспобиж-
доо от к
мело j)o да
пропусканием
через
нагретую медную
сетку
Решётка состоит из узлов а = N2. образующих приблизительно
кубическую гранецентрированную решётку С-^.
336 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ (ГЛ. IV
2. £-Азот (Na)
Решётка гексагональная Н.
Координация V.
Плотнейшая упаковка N., молекул.
Тип Н v- SB A3 (Mg).
a = N2.
Z = 2 молекулы N..
Рис. :>()Г.
|Лтомный
номер ■
Взщество
7 . Д N, |2j
» Г1]
J i
t°
46° К
39° К
( -234,1°С)
а
4,039
1
с c'ta
6,670
6>587±15
1,651
1,633
V см*
14
14,0*
Сх
0,99,
Чистота
Неизвестна
Неизвестна.'
Освобождён1
от
кислорода
пропусканием
чзрез
нагретую
медную сетку
В рассматриваемой ячейке (рис. 205) кружком показано положение
центров тяжести молекул N2.
В отой группе периодической системы, начиная с фосфора, пар
простых веществ состоит из тетраэдрических молекул. Структура РА была
недавно вновь исследована в работе [21] (йЭ^б).
§ 127]
V ГРУППА: АЗОТ, ФОСФОР, МЫШЬЯК, СУРЬМА, ВИСМУТ
337
3. а-фосфор, а-мышьяк, а-сурь-
ма, а-висмут
Решётка ромбоэдрическая R.
Координация О.
Тип Ro-SbAl.
Пространственная группа
Did—ДЗт.
Размеры элементарной
ячейки даются в разных
работах либо для двукратно
примитивной ячейки (Z — 2), либо
для деформированной ячейки
типа Со/о —SB Bl (NaCl) —
(с Z = 8), либо в осях
гексагональной ячейки (для Sb и Bi).
Рис. 206.
Атомный
номер
Кп
31
51
83
Вещестно i t°
i
а-фосфор
«чёрный»[3],[4]
комн.
.
а-мышьяк [11]
» w
»
» 19}
а-сурьма [16]
» [15]
» [И]
а-висмут [15]
»
» Г16]
» [19]
» [20]
комн.
комн.
комн.
25° С
Z
2
8
2
8
2
2
2
2
гекс
8
2
гекс
о
гекс
гекс
а
1
!
5,14
5,96
4,123
5,573
4,1235
4,123 ±5
4,492 ±70
4,475
4,295 ±2
6,621
4,704
4,Г>35±2
4,749 ±7
4,53720
4,53674±4
1
1
34°6/
60°47'
54°10'
84°62'
54°4'5
»
54ПО'±<3'
5 7° 5'
56°54'
—
87°42'
| 57°0'
—
1 57°16'
—
!
i
с
1
.
—
—
—
__
—
11,247+4
~
—
1L,83G±4
—
11,83810
1i,83S3'*±4
d I
~7
—
—
—
2,87
—
—
—
—
—
3,10
—
2,6094
Приме- 1
чанне 1
Данные 1
по ста- j
рой pa-I
боте.Не-|
надёж- I
пы. 1
_ 1
— 1
— 1
—
—
—.
—
—
—
Чистый
Чистый
Чистый
Очень
чистый 1
338
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. IV
4. Фосфор чёрный
Решётка ромбическая
слоистая OR .
Координация 3 + 1.
Тип ОЛз+1 ~ SB 417.
Пр странственная группа
D&—Bmab(~-Cmca).
Z = 8.
На рисунке
изображены 4 элементарные
ячейки. Каждый из двух
параллелепипедов делится
пополам плоскостью,
параллельной осям Ь и с.
Рис. 207.
[Атомный
номер
15
Вещество
Фосфор
(чёрный)
Г
а
комн. 3,31
Ъ
4,38
с
10,50
а : Ъ : с
d I
0,7557: 1:2,397 | 12,17
1
Сх
2,69
Общие замечания
Если в III группе появились, в отличие от предыдущих групп,
валентные структуры, то, начиная с V группы, возникает новая особенность:
по свойствам простые вещества,, входящие как в эту, так и следующую
группу, не похожи друг на друга ни в смысле химического поведения
(сравним инертный азот, активный фосфор), ни по характеру кристаллической
решётки (молекулярная у азота, атомная у мышьяка, металлическая у
висмута). И всё же, исключая азот, четыре остальных элемента образуют
отдельные, сходные по расположению атомов, структуры.
№. Азот диморфен. Согласно [1] (1932), для (N2)a^(N2)3 температура
перехода гп = 35,4° К.
Ниже этой температуры устойчива кубическая аГ^2-модификация,
выше — гексагональная BNo-модификация.
§ 127] V ГРУППА: АЗОТ, ФОСФОР, МЫШЬЯК', СУРЬМ V, Ы1СМУТ 339
'^-модификация при 63,08° К плавится.
Элементарные ячейки обеих модификаций были определены Ругеманном.
Они включены в таблицы, а и ЗЛ 2 были изучены также в работе [2J (1932)
(см. таблицу).
Р. Рентгенографическое исследование модификаций фосфора
представляет большие трудности.
В течение многих лет неорганическая химй*я различала 2
модификации: жёлтый и красный фосфор; позже добавилась третья—чёрный фосфор.
В 1925 г. было указано [3|, что фосфор существует в виде трёх
модификаций: белый (кубический), красный (моноклинный) и чёрный
(ромбоэдрический).
Съёмка белого фосфора (з~ \ ,83) считалась невозможной, ибо при
действии рентгеновских лучей возникает красный фосфор; расчётным путём,
принимая Z--2, получили а-=3,83; при Z-4 а---4,82 Л.
Рентгенограмму красного фосфора в этих исследованиях интерн
ротировать не удалось. Было найдено, что чёрный фосфор имеет ромбоэдрическую
ячейку а=-Г>,96 A; а = 60°47'; Z. 8. Возможен выбор двукратно
примитивной, меньшей ячейки с Z 2, а- о,14; а = 34~()'.
В 1935 г. [5] был исследован методом Фурье-аиализа чёрный и
красный фосфор. Оказалось, что чёрный фосфор имеет ромбическую
элементарную ячейку. Структура является слоистой и каждый слой состоит
из двух сеток атомов фосфора. Тип орторомбический, слоистый САз+1 —
SB .117.
Каждый атом имеет двух соседей на ближайшем расстоянии d,
лежащих в той же сетке, и третьего соседа на расстоянии d', лежащего в
другой сетке того же слеш. Наконец, один сосед находится на гораздо
большем расстоянии d" в другом, ближайшем слое, d -2,17; d'=-2,200; d" = 3,87.
Расстояние между слоями-- .г —5,25 А.
Углы между связями Р — Р в одном и том же слое—99°, а между
связями, одна из которых направлена к 3-му атому, 103° 30'.
Красный фосфор имеет близкую структуру.
А. В. Фрост [6] (1930) исследовал фиолетовый фосфор. Было найдено,
что препараты принадлежат, новидимому, моноклинной системе, но дают
разные рентгенограммы в зависимости от метода приготовления.
В работе [7] (1935) исследован электронографически пар Р4 при
200° С. Было найдено правильное расположение в вершинах тетраэдра
с расстоянием (Р- - Р) d--=2,2\ ±2 А. См. также [21] (19ib). Вопрос о
модификациях фосфора рассматривался и 1944 г. в работе К). Ходакова [22].
As, В фундаментальных курсах различаются, как известно, 4
«модификации» мышьяка: I) металлическая серая, z = 5,72, тритональная,
проводящая ток, II) жёлтая, о = 2,06, кубическая, не проводящая тока,
111) серая, скрытно кристаллическая, a =•- 4,64, не проводящая тока, IV)
коричневая, аморфная, a =■■= 3,(57 —4,13.
Из этих «модификаций» хороню изучена «1», которую можно назвать
a As. Жёлтый As, согласно [3,4 J, при действии видимого света переходит
в серый. Из плотностей вычисляется а = 4,97 (Z = 2) соответственно 6,28
(Z- 4) (см. замечание при фосфоре).
Коричневый As получается восстановлением As203 гипофосфатом натрия
и является аморфной формой. Наконец, чёрный (серый) мышьяк
.мышьяковистого зеркала также является аморфной формой.
В 1934 г. [8] исследовалась ещё одна форма аморфного мышьяка, обо-
значенная как «металлическое стекло», видимо, близкая к «J JI».
340 ч. и. структуры неорганических веществ [гл. IV
В [10] (1932) Стилвелом была найдена постоянная точка перехода
IА8|аморФн—>{Аз]крист ta =270 — 300°С. Это «открытие» является грубой
ошибкой.
Не приходится говорить, что рассматриваемый переход монотропен
и «постоянная точка», отвечающая AF = 0, в этой системе отсутствует.
Известно, что HJ катализирует этот переход.
В [9] (1939) описано исследование р-, ^-и о-формы аморфного мышьяка
и найдено, что для всех их температура превращения в aAs лежит между
270 и 280°С.
Аморфные формы не следует, как мы указывали, считать
«модификациями» вещества. Поэтому на сегодня мы знаем надёжно лишь одну
модификацию (aAs) и ряд аморфных форм, характер структурных узлов которых
остаётся неясным.
aAs образует ромбоэдрическую структуру, тип SB A1, по старым
данным—с а = 4,142 — 4,151 и a = 54°7' —53°43' (Z = 2) или большую ячейку
из вставленных друг в друга двух гранецентрированных ромбоэдров
(Z-8) с а -5,59-5,601; a = 84°18'-84°36'; ах = 5,77.
Структуру aAs легко представить себе как производную от
элементарной ячейки NaCl, состоящей из двух гранецентрированных ячеек, вставлен-
ных друг в друга. Пусть позиции (~ + -^-)заняты^атомами As (вместо
атомов Na); остальные 4 атома As (вместо атомов хлора), занимающих пози-
/12 1Л
ции ( -7- + Т ) 9 несколько смещены (рис. 206), и вся ячейка слегка
деформирована таким образом, что угол а вместо 90° сделан равным 84°62'.
Вместо ячейки Со/о возникает ячейка, которую мы обозначим Яъ[д»
Если бы смещения 4 атомов As не было, решётка As была бы
производной от простой кубической Ср.
Наконец, тип А1 может быть интерпретирован с помощью
гексагональной ячейки (см. SB 1). К этому способу обычно прибегают только в случае Bi.
В [11] (1935) были найдены для «чистейшего» As (Кальбаум):
а = 5,573; а = 84°62' или а = 4,123 А; а = 54°10'; в [12] (1938): а = 4,1235;
а - 54°4,5'; в [9] (1939): а - 4,123 ± 8; а = 54°10' ± 3'.
В [7] (1935) был найден электронографически для пара As (500°С) состав
молекул As4 и структура: тетраэдр с атомами As в вершине и d = 2,44 ± 3.
Sb. Известны различные формы аморфной или стеклообразной Sb,
в том числе стеклообразная взрывчатая сурьма, см. [17] (1935),
стабилизируемая следами SbCl3 (см. А. Ф. Герасимов и И. С. Морозов [13] (1930). Кроме
того см. [14] (1930)). В зависимости от температуры (100 — 250°С) цереход
в a Sb происходит от 40 часов до нескольких секунд.
Для aSb найдена та же структура, что и aAs (До/б ~ SB A7).
В работе [15] (1933), [16] (1934) и [11] (1935) найдены данные,
приведённые в таблице.
Bi. Аллотропические формы Bi изучены плохо. Основная структура —
Rojo-SB AT.
Аномальный ход коэффициента расширения заставлял отдельных
авторов предполагать наличие нескольких модификаций, что
рентгенографически не подтвердилось. См. [18] (1932).
По старым данным, а ,= 6,52 — 6,56; a = 87°24' —87°34'; Z = 8, т. е.
а = 4,73; a - 57°16'; Z = 2.
В работах [15] (1933), [16] (1934) и [19] (1935) найдены результаты,
приведённые в таблице. В 1938 и 1940 гг. Страуманис и Евинс [20] исследовали Bi
асимметричным методом и нашли величины, приведённые в таблице,
а также коэффициенты линейного расширения ац «= 16,2 х Ю"в и а_^ =
= 12,0 . 10-в.
§ 1281
VI ГРУППА: КИСЛОРОД, СЕРА, СЕЛЕН, ТЕЛЛУР, ПОЛОНИЙ
341
ЛИТЕРАТУРА к § 127
1. Z. Phys. 76, 368 (1932).
2 SB I и Z. Phys. 79, 471 (1932),
3. Z. Anorg. Ch. стр. 147, 288 (192Г>).
4. СЫ. f. Mineral 107 (1926).
5. Phys. Rev. (2) 47, 808 (1935);
J. Ch. Phys. 8, 351 (1935).
ЖРФХО 62, 2235 (1930;.
J. Ch. Phys. 3, 698 (1935).
SB III 21.
Z. Anorg. Ch. 242, 138 (1939).
J. A. Ch. S. 64, 472 (1932);
Ch. Rev. 8, 335 (1932).
11. Phil. Mag. (7) 20, 913 (1935).
6
7
8
9
10
12. Z. Anorg. Ch. 238, 255 (1938).
13. ЖРФХО 62, 827 (1930).
14. Z. Phys. 63, 815 (1930).
15. J. Ch. Phys. 1, 753 (1933).
16. J. A. Ch. S. 56, 385(1934).
17. Nature 136, 299 (1935).
18. Phys. Rev. 49, 643 (1932) и там цити-
ров. работы.
19. J. Ch. Phys. 3, 605 (1935).
20. S t r a uman is u. J e v i n §, Pra-
zisionsbestimmung von Gitterkonslan-
ten etc. (1940).
21. J. Ch. Phys. 14, 351 (1946).
22. ДАН 42, 125 (19'*'*).
§ 128. VI группа: кислород О, сера S, селен Se, теллур Те,
полоний Ро (s2p*)
1. у-кислород
Решётка кубическая~См.
Координация К.
Тип C£~-SB A1.
а = 02.
Пространственная группа Tjt — РаЗ.
Z = 40..
Рис. 208 (ср. о Na рис. 204).
Атомный
номер
6
Вещество
Т0а
1
|
Примечание
50° К 1 6,83 SB IV 61
342
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТВ
[ГЛ. IV
2. а-сера, сера
ромбическая
Решётка
ромбическая
молекулярная ORM'R\
Координация 5.
Тип
Пространственная
группа Lr\ -
Fddd,
Z= 128 S- 16(S)
Sk. ч. = 5S.
ci
8
0 5 W
i ■ i > > i » i i i i
Рлс. 209.
Атомный
номер
lfi
Вещество
10,'i8
a : Ъ : с
Примечание
12,02
24,55
0,811 : 1 :1,900
d=-.2,l0;cf = 3f27;
<?=2,ll; e'=»,37;
6=:^ ,32. Атомы S
соединены в восьми-
членные кольца (i?8)
§ 128] VI ГРУППА: КИСЛОРОД, СЕРЛ, СЕЛЕН, ТЕЛЛУР, ПОЛОНЦЙ 343
3. [3-сера, сера моноклинная
Согласно [19] (1937) исследованы монокристаллы [-S методом вращения
и Вайсенберга при 103° С. Обнаружена элементарная ячейка: а --10,90;
6 = 10,96; с = 11,02; а : Ь\ с =0,9945: 1: 1,006; р=83°16';
пространственная группа С«й — Р2\(а. Молекула S8, возможно, вращается (?).
4. Селен, теллур
Решётка гексагональная цепная Нк.
Координация 2 (Z) *).
Tmi#*-SB,48 (селен).
Пространственная группа D\7 D\.
Ряс. 210.
На рис. 210 справа показан более чётко элемент «винтового хода» цепи
в структуре SB А8.
|Атом-
1 иый
I номер
48
52
Вещество \ а \ с
1 1
Se
Те
4,34
"i,44.
о
4,95
5,91
с/а
1,14
1,33
d
Se—Se
2,32
2,8G
iodA3**)
20,9
33,7
Примечание
SB I 28. Винтообразные
бесконечные цепи
Se—Se ...
соответственно
Те—Те
*) См. таблицу 34, стр. 240.
**) В данном случае мы имеем один из многочисленных примеров путаницы SB
и обозначениях объёмов. Вместо объёма 1 атома в SB даётся обозначение
«молекулярный объём».
344 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IV
5. Полоний
Решётка моноклинная М.
Координация О.
Тип Mo—SB 419 (полоний).
Пространственная rpynua C\ — C2.
Ро к. ч. = 6 (деформированный октаэдр).
Z = 12.
0 12 2 4 5 6 7 6
i i i i—-i 1 ■ ' ' '
Рис. 211.
Атомный
номер
84
Вещество
Ро
а
7,42
Ь
4,29
с
11,16
Р
92°
а : Ъ : с
1,7295 :1 : 3,287
Примечание 1
I
SB IV 4. Судя по работе 1
[181(1946), тип Л19 нэ
существует (см. ниже) 1
§ 128]
VI ГРУППА: КИСЛОРОД, СЕРА, СЕЛЕН, ТЕЛЛУР, ПОЛОНИЙ
345
6. «а»-полоний
Решётка кубическая, примитивная С.
Координация 6 (октаэдр).
Тип CP = C0-SB?
Пространственная группа?
Z = l. аш= 3,34.
Рис. 212.
7. <ф»-полоний
Решётка ромбоэдрическая R.
Координация 6?
Тип R, (?)-SB (?)
Пространственная группа?
Z- 1(?); а = 3,36; а = 98°13\
Температура перехода Род-
^75° С.
Рос
Рис. 213.
Надёжно расшифрованной структуру Ро считать нельзя. Если описанная
выше модификация Ро, тип С0 подтвердится, это будет первый случай
образования простым веществом примитивной кубической решётки.
Общие замечания
Кислород О. Простые вещества, образуемые элементом «кислород»,
различаются: числом атомов, входящих в молекулу, и структурой кристалла,
образованного из молекул данного типа.
Атомы кислорода образуют молекулы двух типов, детально
исследованных с физической и химической стороны: 02 (кислород) и 03 (озон).
Молекулы типа 04 были в последнее время предметом дискуссии. См., например,
Сыркин и Дяткина [5] (1946).
Отметим, что кислород является чуть ли не единственным элементом,
образованные которым простые вещества имеют разные наименования
в зависимости от числа атомов, входящих в молекулу пара (газа). Уже
346 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IV
в случае серы наименование простых веществ, образованных молекулами
разного состава и структуры (S8, S4, S2 и т. п.), не меняется (сера).
Кислород 02. Впервые ясность в вопрос о кристаллических
модификациях кислорода была внесена работой [1] (1932).
До этого на основании работ старых школ предполагали, что 02, в
отличие от азота, образует только низкосимметричные модификации
■(ромбическая, моноклинная, триклинная); позже были допущены
гексагональная и тетрагональная.
В 1. с. [1] показано, что кислород триморфен, причём
а 02 (23,5° К) z± 3 02 (43,5° К) £± у 02 (54,1° К) ^ жидкость.
Снимок при 16° К показал, что а02 имеет, повидимому, ромбическую
центрированную ячейку, близкую к найденной в [2] (1927), согласно которой:
а о,50; b = 3,82; с = 3,44; Z = 4 атома 0 = 2 молекулы 02; ах = 1,46;
-лит- 1,426. рОа — ромбоэдрическая элементарная ячейка. у02 была
приписана гексагональная структура тип ЛЗ. В 1936 г. была исследована
рентгенограмма монокристалла у 02 при 50° и найдена кубическая
структура пространственной группы T*h — Pa3.
Размеры ячейки указаны в таблице.
Жидкий 02 был исследован в [21] (1938).
Расчёт энергии образования иона кислорода О" был выполнен
И. А. Казарновским (см. полиоксиды, гл. X).
Озон 03. Исследования структуры кристаллического озона в
литературе нами не обнаружены.
Молекулярный озон (газ) исследовался неоднократно.
Из более поздних укажем на работу [4] (1943), согласно которой в моле-
/°
куле 03 угол между связями 0^ равен 127°, и предполагается суперпо-
Х0
зиция 4 резонансных структур:
I
0+
// \
:0 :(Ь
II
б*
/ ч
:0- 0:
III
0
/■•\
:0- :0>
IV
0 .
/"\
:0 :0-
И. А. Казарновским, Г. П. Никольским и Т. А. Абледовой (1948)
открыт новый ряд соединений кислорода—ряд озонидов, содержащих
комплексный ион 0~ (см. полиоксиды).
S. По вопросу о валентных состояниях S см. Е. Н. Гурьянова [26]
(1942). Исследование модификаций серы привлекло в своё время внимание
физико-химиков, посвятивших ряд работ термодинамической
характеристике модификаций серы, изучению диаграммы состояния (см. [6]).
Напомним, что, согласно этим работам, низкотемпературная aS
(ромбическая) переходит в [4S (моноклинную) при температуре 95,5°С; ^S
плавится при 119,25°. С другой стороны, можно перегреть ромбическую a S
значительно выше tu и довести до плавления при температуре 112,8°,
причём в этой точке и ромбическая сера, и её расплав метастабильны в
отношении моноклинной серы.
Рентгенографическими исследованиями [7] (1914), [8] (1925), [9] (1935)
были установлены ячейка и пространственная группа aS {D^ — Fddd) тип Л16.
§ 128| VI ГРУППА: КИСЛОРОД, СЕРА, СЕЛЕН, ТЕЛЛУР, ПОЛОНИЙ 347
Данные двух последних работ приведены в таблице. Согласно [9], в решётке
8 атомов связаны в 1 молекулу S8, образующую складчатое кольцо, как бы
состоящее из двух квадратов, повёрнутых на 45° друг около друга.
Плоскости квадратов параллельны оси с. Валентный угол^103°.
Согласно исследованию [10] (1934), моноклинная pS сера образуется
не только из ocS, но и при растяжении нитей эластичной серы.
Пространственная группа Cljl — P2lfm. Элементарная ячейка [JS, судя
по работе 110], имеет размеры: а = 20,4^0,1; 6 = 9,26±0,05; с= 12,32±0,05;
р = 79°15/; а=2,01; Z ^ 112; пространственная группа C:) — P2i или
С1н—Р2фп.
Борвслл [19] (1937) нашел для моноклинной серы pS ячейку,
приведённую выше, на стр. 343.
В работе |7] предполагалось, что до 200°С жидкая сера ещё содержит
кольца; при 200°С кольца разрываются, превращаясь в цепи неправильной
формы, обусловливающие большую вязкость красной серы (при температуре
200 —240°С). С другой стороны, в 1. с. [10] предположено, что в [JS (Z ^ 112)
в каждую ячейку входят 14 плоских цепей из 8 атомов S, расположенных
параллельно оси Ь. Равновесию кольца *^ цепи в жидкой сере посвящено
недавнее исследование [11] (1943).
В работе [12] (1936) изучались электронографически пары серы.
Допуская, что при 850°С образуются молекулы J32, было найдено, что при этой
температуре расстояние S — S = 1,94 ± 3 А.
В 1946 г. было опубликовано по этому вопросу исследование [26],
освещавшее равновесие 48S^ 24S2^±8Se ^±6S8.
Исследование аллотропических форм серы освещено в работах [20]
(1938), [22] (1939).
Жидкая сера была исследована в работе [23] (1939).
Se. В химии обычно различали гексагональный серый и аморфный
красный селен; кристаллографы различали а- и fi-модификации
моноклинного красного селена: aSe (а : Ъ : с) = 1,63495 : 1 : 1,6095; Zp = Ю4°2'; pSe
(а : Ь : с) - 1,5916 : 1 : 1,1352; Z? - 93°4' и др. См. также [27].
В работе [13] (1924) найдена для серого металлического Se
гексагональная элементарная ячейка (тип AS), позже подтверждённая в других
исследованиях. Высказывались также предположения, что красный селен может
быть и аморфным, и моноклинным.
В [14] (1930) и [15] (1934) подтверждалось существование двух
моноклинных модификаций красного селена aSe и [JSe.
Принятые в SB обозначения модификаций Se вряд ли целесообразны.
Учитывая, что красный селен при стоянии (медленно), при растирании
(быстро) переходит в серый (металлический) селен, повидимому, следует
считать металлический Se термодинамически более устойчивой формой при
стандартной температуре и давлении и обозначать его aSe.
Обе формы красного моноклинного солена относятся к одному и тому же
классу C>2h, образуя различные элементарные ячейки разных
пространственных групп. Их мы обозначим соответственно ^Se I и pSell, пока не
будет выяснено, что эти формы моноклинного Se устойчиво
воспроизводятся, т. е. являются подлинными модификациями и не меняют параметров
в зависимости от длины и расположения цепей Se. Таким образом,
возникает некоторая аналогия с серой.
Га aS ромбическая ~ aSe гексагональный
Р [3S моноклинная ел н pSe моноклинный
Для ромбической серы можно принять (а = 10,61; Ь = 12,87; с = 24,56;
Z = 128) другую элементарную ячейку таким образом, что ось с умень-
348 ч. и. структуры неорганических веществ [гл. IV
а'
а
с
= 8,90
= 8.99
= 9,25
с'
Ь
Ь
= 8,90
= 8,97
= 8,05
Ь'
с
а
= 10,61
= 11,52
= 12,74
Р = 92°44'
р = 91°34'
р = 93°4
шить вдвое и полудиагональ стороны be принять за ребро новой ячейки.
Тогда получится моноклинная ячейка с а' = 8,90; с' = 8,90; Ь' = 10,61;
р'= 92°44'; Z = 32. Сопоставив с fJSe, находим (обозначения модификаций,
введённые нами выше):
a So
pSel
Р Se II |
См. SB 111, 217-218.
В 1945 г. Se исследовали Г. С. Жданов и Ю. В. Шарвин [28].
Согласно [24] (1939), пар Se состоит из молекул Se2 с межатомными
расстояниями 2,21 ± 3. Согласно [26] (1946), пар Se состоит в основном из
легко диссоциирующих молекул Se2, что находится в соответствии с
меньшей энергией связи Se—Se по сравнению с серой.
Те. Так же как и в случае Se, в химии допускается, что в
стандартных условиях Те образует одну металлическую модификацию
и несколько аморфных форм. Структура металлической модификации была
установлена ещё в работе [13] (1924).
Вопрос о структурных формах Те изучен недостаточно. Лишь в 1944 г.
было опубликовано [25] исследование, посвященное строению и
аллотропическим формам Те. В нём указывалось, что прежние предположения о
существовании 2 модификаций Те, присутствующих в разных соотношениях
(в зависимости от температуры и обработки образца), не подтвердились.
В интервале от — 192°С до + 160°С наличие 2 модификаций не
обнаружено. Те, испарённый в вакууме, образует при конденсации на стекле
2 слоя: гомогенный металлический слой и чёрный мелкий порошок
одинаковой структуры.
Ро. Роллье, Гендрикс, Максвелл [17] (1936), располагая 10^7г
Ро, исследовали строение Ро электронографическим путём.
Найдена была моноклинная элементарная ячейка тип SB Л19. Спустя
10 лет Бимер и Максвелл [18] (1946) на основании также электронографиче-
ских исследований пришли к выводу, что Ро диморфен и что так называемая
моноклинная форма является смесью 2 фаз. Низкотемпературная форма
(назовём её аРо) имеет простую кубическую решётку с а = 3,34,
высокотемпературная (назовём её ЗРо) — ромбоэдрическую с а = 3,36; а = 98°13';
tn (аРо^±рРо)^75°С.
Структура Ро должна быть объектом дополнительных исследований.
В настоящее время Ро является чуть ли не единственным представителем
простых веществ, имеющих примитивную кубическую элементарную ячейку.
ЛИТЕРАТУРА к § 128
1. Z. Phys. 76, 368 (1932).
2. Phil. Mag. 3, 383 (1927).
3. Physica 3, 141 (1936).
4. J. A. Cii. So 63, 179 (1943).
5. Сыркин и Дяткина, Химическая
связь и строение молекул, стр. 305
(1946).
G. Финдлей, Правило фаз, стр. 57—01,
142 след. (1936).
7. Proc. R. Soc. A 89, 575 (191'i).
8. Z. El. 29, 36'* (1925).
9. J. Ch. Phys. 3, 6 (1935).
10. Helv. Chim. Acta 17, 1081 (1934).
11. J. A. Ch.S. 65, 648 (19W).
12. Phys. Rev. 49, 199 (1936).
13. Phil. Mag. 48, 477 (1924).
14. Z. Phys. 11, 455 (1930).
15. Z. Krist. (A) 88, 128 (193i).
16. C. A. 40, 4595 (1946).
17. J. Gh. Phys. 4, 648 (1936).
18. J. Gh. Phys. 14, 569 (1946).
19. Z. Krist. (A) 97, 123 (1937).
20. Indian J. Phys. 12, 163
SB VI 51.
21. SB VI 50 (1941).
22. Nature 143, 165 (1939).
23. Nature 143, 332 (1939).
24. Phys. Rev. 55, 238 (1939).
25. Phys. Rev. 65, 348 (194'*) (?).
26. Acta XVI, 181 (1942).
27. ЖФХ XXII, 1034 (1948).
28. ЖФХ XIX, 160 (1945).
(1938);
§ 129] I (VII): ВОДОРОД И VII ГРУППА: ФТОР, ХЛОР, БРОМ, ИОД 349
§ 129, I (VII): водород Ща1) (и VII группа: фтор F, хлор С1, бром Вг,
иод J (s2p5)
1. Водород (Н2)
Решётка плотная гексагональная Н.
Координация V'.
Тип Hv-SB AS.
Пространственная группа D^x — Сб/ттс.
Z = 21L; к. ч. -= 6 (расстояние d = я) +
-f 6 ( расстояние е = 1/ — + — ) ,
при с/а = 1,633 е=а
Рлс. 214. Положения центров тяжести молекул П2.
Атомный
номер
1
Вещостпо
Водород
\'Л](пара), а также
смесь орто и
пара в
соотношении
Я орто:1 пара
1
t° ■ а
1,6ГК
4,2°К(?)
3,75
с
0,12
с'а
1,633
V см*
п,;>1
1 1
ar i Чистота
1
1
0,088; ?
1
1
350 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. IV
2. Фтор (Ро). Нет надёжных данных.
3. Хлор (С12)
Решётка
тетрагональная молекулярная
грМ
Координация 2.
Тип Г*'-SB 418.
Пространственная
группа D^-Pi/ricm.
Z- 16. С1 = 8(С1,),
С1 к.ч. - 1 + 4.
(С1,) к. ч.»2.
Ok
8s
0 f 2 3 4 5 6 7 6
i i i i i i i i1. j
Рис. 21Г.
Атомный
1номер
17
Веще.', т по
С12 [5]
t°C
— 185
а
8,56
с
6,12
с/а
0,715
d
1,81
Сг
2,0J
1
4. Бром, иод
Решётка ромбическая гра-
нецентрированная,
молекулярная ORM.
Координация К 12.
Тип OR%- SB .414.
Пространственная группа Q-
Дм — Сс/гм.
ос = J2.
Z = 8. J = 4J„. а к. ч. =
-12а.
Рис. 216а. (По SB П.)
Оси на рисунке (SB II 5) обозначены неверно. Проходят: с--слева
направо, 6 —снизу вверх и а —спереди назад. Сам рисунок в SB (рис. 216а)
неудачен, смазывается молекулярный характер решётки, см. рис. 216b..
Центры молекул совпадают с узлами ромбической гранецентрированнон
решётки ORp.
§ 1291
I (VII): ВОДОРОД И VII ГРУППА: ФТОР, ХЛОР, 1>РОМ, ИОД
351
Атомный
номер
35
53
Веще
стпо
Вг2 [6]
Ja[8]
t JC
— 150
IS
a
b
4,48 ■ 6,67
4,795|7,25л
4,7761|7,2501
с
8,72
9,78
9,7711
a : b : с
0,672 : 1 : 1,307
0,6609:1:1,3481
0,6588:1:1,3477
i i
l
] |
2 27
2,70
3,30
3,54
1,135
1,35
4,05
4,9520
Примечание
Вг2 Моно-
крист.
-JT\
о—
0 12 3 4 5
Рис. 216Ь.
Энергии д и с с о ц и а ц и и О и м е ж а т о м н и о р а с г. т о я и п я d
газообразных водорода и г а л о г е и о и
1 1
I) к кал.
102,6
0,74
D.
105,1
0,74
F.
64
1,45
СЬ
57
1,99
Вг2
45
2,28
h
35
2,65-2,67
О б щ ие за меча л и я
Но строению и составу молекул гантельного тина водород И2 и D.,
обнаруживают далеко идущую аналогию с галогенами, заставляющую
рассматривать 112 и D2 (как простое вещество) вместе с последними.
352 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IV
Эта группа веществ представляет исключительный научный интерес,
содержа ряд членов с постепенно ослабляющимися вш/т/?нмолекулярными
силами и постепенно возрастающими л*#ж?молекулярными силами. Если
водород (Н2 и D2) (и фтор?)*) иллюстрируют типичные случаи упаковки
гантельных молекул со слабым внешним полем, то с переходом к иоду
картина быстро меняется.
Из сопоставления с инертными газами следует, что первые члены ряда —
водород Н2 и D2, как и гелий, образуют гексагональную плотную упаковку,
что заставляет предполагать в структуре Н2, благодаря крайней слабости
поля решётки, вращение или беспорядочную, статистическую ориентацию
молекул.
С ростом межмолскулярных сил симметрия падает.
У хлора наблюдается тетрагональная решётка пространственной группы
&IL У брома и иода даже ромбическая д5®.
Исследования всех этих веществ ведутся с крайним запозданием. Если
исследования Н2 и F2 представляют затруднения вследствие необходимости
работать при весьма низких температурах и громадной реакционной
способности фтора, то остальные галогены вполне доступны изучению. Тем
не менее даже иод был впервые более или менее детально изучен лишь в
работах 1927-1928 гг. [1] (1927) и [2] (1928).
1930 г. следует считать годом перелома в изучении структур простых
веществ, образованных элементами VII (и VIII) группы.
В этом году появились сообщения" [3] о структуре твёрдого Н2 и
инертных газов. Для Н2 была найдена структура #у — SB AS при температуре
1,65° К и 4,2ЭК, как у обычного водорода (в соотношении Зорто + 1 пара),
так и у чистого параводорода.
Галогены F2, Cl2 и Вг2 продолжали оставаться вне поля зрения
исследователей.
В 1933 г. прежние спектрографические исследования парообразного J2
пополнились электронографическими: в [41 (1933) было получено значение
d = 2,64 А.
Лишь в 1936 г. опубликованы сообщения о структуре хлора [5] и
брома [6]. Все эти работы заслуживали бы проверки вплоть до температур
плавления с целью, в частности, установить, существуют ли
аллотропические формы более высокой симметрии.
Газообразный фтор в 1938 г. исследовал Броквей [7].
Межатомные расстояния в молекулах рассматриваемых веществ см.
выше (таблица).
В 1943 г. была определена [8] структура кристаллического иода
прецизионным (асимметричным) методом и найден объёмный коэффициент
расширения {J = 264 X Ю~в.
Л PIT ЕР AT УРА к § 129
1. SB I 759.
2. J. Am. Gh. S. 50, 1583 (1928).
3. Proc. Acad. Amsterd. 33, 814 (1930); SB II 166.
4. J. Ch. Phys. 1, 549 (1933).
Bull. Am. Phys. Soc. 8, 13 (1933).
5. Physica 3, 237 (193fi).
6. J. Am. Ch. Soc. 58, 2459 (1936).
Phys. Rev. 50, 872 (U36).
7. J. Am. Ch. S. 6^, 1348—49 (1938).
8. Z. ph. Ch. В 63, 'Л20 (1943).
*) Свойства молекулы F2 заставляют ожидать структур, близких к N2 и в
большей мере к П2.
§ 130] VIII ГРУППА: ГЕЛИЙ, НЕОН, АРГОН, КРИПТОН, КСЕНОН, РАДОН 353
§ 130. VIII группа: гелий Не, неон Ne, аргон Аг, криптон Кг, ксенон X,
радон Rn (s*j/)
1. Гелий
Решётка плотная гексагональная Н.
Координация V.
Тип Ну -SB A?>.
Пространственная группа Вцн—Сб/ттс.
Z = 2, к. ч. = 6 (расстояние d = а) + 6 ( расстояние е = у %г- + -^- J .
Рис. 217.
Атомный
номер
2
Вещество
1
Нэ |
а
3,57
с
5,83
1
с а
1,633
d
€
3,57
а, 57
1
г
1,785
354 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл IV
2. Неон, аргон, криптон,
ксенон
Решётка кубическая гра-
нецентрированная С^.
Координация К.
Тип Ск — SB А1.
Пространственная группа
0\— РтЗт.
Z = 4, к. ч. 12
(расстояние d =
**/2
стояние е = яш).
) + 6 (рас-
Рис. 218.
[Атом-
1 ный
[номер
10
18
36
54
Ne [3j
Ar |t]
12]
Kr [4J
[5]
14
M
Xo [7J
1 tQ
40°K
20°K
(жидкий
водород)
—252,5ЭС,
(жидкий
водород)
t°
жидкого воздуха
и жидкого
азота
82°К
89°К
У2°К
—пом:
88°К
(—185,1°С)|
aw
4,52
5/*2
5/|0±6
5,59
5,78
5,684±^
5'G9'i±i7
М8±2
d
3,20
3,83
3,95
6,24±2,J4/iO|
i 1
г
1,60
1,915
1,975
2,20
wA
23,1
39,8
39,4
43,7
38,3
V см3
14,0
24,0
23,7
26,3
23,0
60,7
3>,5
cx
U'.;,
l'*b±2
1,68
3,13
•
2,S3
з,оо4
2>»\ 1
2'У86
3,64 1
(плотность
жидкого Хе
ПРИ ^пл= 1
=3,06)
3'555±40
Чистота
Данные
отсутствуют
-^98% чистоты!
N2, Оа, очи- 1
щался вымо- 1
раживанием 1
и откачкой. 1
Спектроскоии-1
чески чистый 1
(?)
Очищен по 1
Петере и Вайлы
в
3. Радоп—в распоряжении автора сведений нет.
^ 1ol| структуры простых игсщестн Я55
Общие замечания
Изучение структур инертных газов с наиболее слабыми
межмолекулярными силами представляет тот громадный интерес, что в этих случаях
силовое ноле молекулы имеет шаровую симметрию, и, следовательно, законы
плотных упаковок должны проявляться в наиболее благоприятных
условиях. К сожалению, условия фазовых переходов в этих веществах изучены
совершенно недостаточно, несмотря на достаточный уровень техники
исследований при низких температурах. Впрочем, к VII] группе относятся два
вещества, плавящиеся при чрезвычайно низкой температуре: Не—272°, 1 С^
^ + 1,1°К и Ne— 248,8°С ^ 24,4°К. Это особенно задержало их изучение.
Температуры плавления остальных членов этой группы находятся в
пределах легко осуществляемых низких температур: Аг—187,9°С; Кг—156,6°С;
Хе—-111,5'С; Rn—71е С. Последний член этого ряда радиоактивен, что,
очевидно, затруднило его исследование.
Первые работы—[1] (1924) и [2] (1926) коснулись аргона. Лишь в 1930
1931 гг. появилась большая группа сообщений, относящихся чаще к газам
невысокой или неизвестной чистоты: [3] (1930) но Ne; [4] (1930), [5] (1930),
[6] (1931) по Кг; [7] (1930) и [6] (1931) но Хе. Затем наступил весьма
длительный перерыв, в силу чего в учебной и справочной литературе до
1946 г. включительно, как правило, приводятся данные упомянутых выше
работ (1924 — 1931). Лишь в 1938 г. появились работы [8] по твёрдому
и жидкому гелию. В 1945 г. опубликована работа [9] по жидкому аргону.
Совокупность сведений о структурах инертных газов вряд ли можно
считать удовлетворительной.
ЛИТЕРАТУРА к § 130
1. Z. Phys. 25, 160 (1924). 5. Nature 125, 389 (1930).
2. Comm. Phys. Lab. Leiden 178, 19 6. Z. Phys. Ch. (B) 15, ;И9 (1931).
(1926); SB 1 71. 1. Nature 125, 457 (ly.iO).
3. Proc. Acad. Amsterdam 83, 255 (J930); 8. Physica 5, 161, 270 (ly.W).
SB II 201. 9. J. Ch. Phys. 13, 317 (1У»Г>).
4. Proc. Acad. Amsterdam 33, 447 (1930);
SB II 202.
§ 131. Структуры простых веществ
Попробуем найти некоторые общие характеристики, описывающие
образование структур простых веществ.
Первая группа. Группа из 13 — 14
элементов правой части периодической системы,
включая отнесённый сюда водород Н2, образует I N I 0
молекулярные решётки, состоящие из одноатом- I I _.
ных или двуатомных молекул, обладающих
слабым межмолекулярным полем и не имеющих
тенденции к осуществлению дополнительных
химических связей. Этот признак позволяет резко
отграничить рассматриваемую группу от соседних
слева элементов Р, S, Se, Те. К этой группе,
исходя из строения атома, лишь с большими
оговорками может быть отнесено неизученное
ещё простое вещество, образованное элементом Л!» 85, «аетатии».
На диаграмме (рис. 219) сопоставлены границы существования
твёрдых фаз и точки фазовых переходов при р - 1 атм. Из диаграммы можно
сделать следующие выводы для молекулярных решёток:
1. Если частица имеет шаровую симметрию (инертные газы), или её
шаровая симметрия достигается путём вращения (или статистически
разной ориентации (А2)), гантельных молекул и жжмолекулярпые силы
н.
Fa
С1я
Вг2
J2
85
Не
Ne
Аг
Кг
Хе
1 Rn
356 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. IV
чрезвычайно слабы—возникает плотная гексагональная упаковка типа
flV(He, H2, высокотемпературная модификация f*N2).
2. При несколько более значительных (но всё же ничтожных)
межмолекулярных силах частицы шаровой симметрии и гантельные
молекулы образуют плотную кубическую упаковку типа Ск(№е, Аг, Кг, Хе,
низкотемпературная модификация ocN2).
i
300
386*
261
ZOO
ioo V
!
I
171%
%
I
!
4
S3'
4 U ^
Z3S'
-? 8
i.
о
о
1
I
I
±
!
л
24,4%
I
4
t°L
I
8Щ
msi
CI, Br, 3
2 J2 n2
Рис. 2U.
He Ne Ar Kr Xe
3. G ростом межмолекулярных сил, как, например, в случае молекул
02, даже наиболее высокотемпературная кубическая форма (у02) имеет
пониженную симметрию (класс Th).
4. С понижением температуры, а значит, с ростом межмолекулярных
сил (и прекращением вращения) симметрия падает. Как мы видели,
pN2(#v) переходит в ocNt {Ск вид симметрии 71). В равной мере у02
кубическая (вид симметрии Th) переходит в [Ю2 (ромбоэдрическая) и,
наконец, в а02 (ромбическая, центрированная). Более сильное взаимодейт
ствие молекул 02, по сравнению с молекулами N2, влечёт за собой
образование кислородом кристаллов с более низкой симметрией.
Структура кристаллического Оя, повидимому, остаётся неисследованной.
Это редчайший пример треугольной по форме молекулы из однородных
атомов со слабым внешним полем, отличающим 03, например, от S02.
Влияние температуры на структуру кристалла не сказывается лишь
в случае частиц шаровой симметрии (инертные газы).
§ 131] СТРУКТУРЫ ПРОСТЫХ ВЕЩЕСТВ 357
Аналогично понижению температуры, на структуру кристалла,
образованного из гантельных молекул, влияет увеличение атомного номера.
С ростом последнего симметрия кристаллов падает, например, в ряду Н2 — J2
имеют Н2—гексагональную Ну — SB A3, F2 : (?), С12 —тетрагональную
структуру класс D^, Вг2 и J2 —ромбическую класс Д>Л.
Опять-таки увеличение атомного номера не влияет на структуру лишь
в случае молекул с шаровой симметрией (инертные газы).
У С12 и Вг2, к сожалению, интервал температур, примыкающий к
температуре плавления, не исследован. В этом интервале можно ожидать и
появления структур с более высокой симметрией, поскольку не исключено,
что плавлению предшествует статистическое распределение осей молекул
по отношению к осям элементарной ячейки, в результате вращения.
Сделанные выше выводы будут в следующей главе сопоставлены с
выводами о структурах кристаллов, образованных гантельными и квазишаро-
выми молекулами типа СО, HF, HC1, НВг, Ш, HCN,CH4,CF4h т. д.
Следующая, вторая группа включает молекулярио-атомные и моле-
кулярно-металлические структуры. К первым можно отнести: S, Se, Те, Р,
As, Sb (Po и Bi имеют более резко выраженные металлические свойства),
ко вторым—Po, Bi, Ga, In, Tl, Hg, т. е. всего 12 элементов.
Отличительным свойством первых 6 структур является наличие
межмолекулярного силового поля примерно столь л^е интенсивного, как и
внутримолекулярное. Уже в парах молекулы этих веществ бывают
двуатомными лишь при достаточно высокой температуре; при более низкой они
соединяются по 4 и даже по 8, образуя кольца. В жидком состоянии
комбинируются кольца и цепи. На первый взгляд может показаться
странным, что разрыв колец с образованием цепей происходит не при более
высоких, но при более низких температурах. Повидимому, в жидком состоянии
энергия разрыва колец компенсируется взаимодействием концов последних
с соседними молекулами с образованием цепей. С испарением же цепеобраз-
ных молекул, каждой из них, вследствие отдалённости от соседей, остаётся
лишь образовать кольцо, что даст выигрыш энергии. С дальнейшим
повышением температуры кольцо разрывается на небольшие части, например,
S,-»4St.
Так как образование связей возникает у атомов элементов, например,
V группы, главным образом, за счёт &3-электронов, то несмотря на
естественные различия валентных конфигураций sp2- и /А-элементов
возникает и некоторая аналогия со структурами группы элементов, симметрично
расположенных по другую сторону от вертикали С —Si —Ge —Sn- Pb,
а именно Ga — In (s/r-элементы). Решётки Po, Bi, Ga и Hg можно было бы
назвать молскулярно-мсталличсскими. Пи в одной из них пет шт плотной
упаковки атомов, свойственной металлам других групп с к. ч. 12 или 8,
ни правильной атомной структуры с к.ч. 4, свойственной группе
С— Si— Gc — Sn, последний член которой РЬ имеет кубическую гранецеи-
трированную, т. о. плотную, упаковку.
Десять из названных двенадцати элементов образуют искажённые
структурные типы, А1, А8, А\0, ЛИ, Л16, АП, Л19 и др., в которых
каждый атом чаще окружёнН соседями на разных расстояниях (к.ч. =6),
а один из них обычно находится несколько ближе, чем остальные. !
Повидимому, элементы Hg, Ga, In, Tl, с одной стороны, и 6 (8)
названных выше элементов, с другой стороны, участвуют в образовании
структур простых веществ двумя и более, но менее чем четырьмя электронами
со значительным количеством несвязывающих электронов. Этому
соответствует и их значительная легкоплавкость; см. § 92.
Как отмечено на стр. 345, заслуживает внимания, что
низкотемпературная форма Ро имеет, повидимому, примитивную кубическую решётку Со
358 ч. п. структуры неорганических веществ [гл. IV
(к.ч. 6), которая, таким образом, невидимому, впервые открыта у простого
вещества—металла; к.ч. 6 не наблюдается у подлинных металлов.
Следует подчеркнуть,что давно изученные структуры Bi, Sb, As и Up
могут быть представлены как построенные из деформированных примитивных
кубических элементарных ячеек, причём осуществляется к. ч. 6.
Учитывая изложенное выше, нам кажется целесообразным объединить
в группе II молекулярио-атомные S, P, Se, As, Те, Sb и молекулярно-метал-
лические структуры 13i, Po, Ga, In, Tl, Hg. Эти 12 элементов образуют
не менее 8 разных типов, перечисленных выше, отражающих
индивидуальные особенности строения атомов. Они отличаются также особо ярко
выраженной тенденцией к образованию своеобразных аллотропических форм
как в твёрдом, так и в жидком состоянии; в разных фазах этих веществ
возникают сложные равновесные состояния, в которых одна форма
переходит в другую, вторая —в третью. Наблюдаются колебания валентных
углов, вследствие компромисса между силовым полем решетки и внутренним
силовым полем молекулы.
Третью группу образуют 5 элементов: С, Si, Ge, Sn и В. Это
вещества с атомными и атомно-мсталлическими решётками, имеющие либо
алмазную (С, Si, Ge, Sn), либо валентную другого типа (В), либо а томно-
металлическую (fiSn) структуру. Особняком стоит атомно-металлическан
структура графита Л9. Эти вещества имеют ряд особенностей. С и Si
плавятся с трудом; температурный интервал жидкой фазы углерода крайне
узок. Вследствие большой упругости пара он сублимирует, повидимому,
в виде двуатомных молекул (С2). Как показали различные авторы,
углерод может быть расплавлен лишь под давлением.
Уже у Si интервал существования жидкой фазы М:и =- /КИц — tn:i = 1000",
интервал существования твёрдой фазы (от абсолютного нуля до точки
плавления) ЛЛ]В = 1673°, т. е. они почти сравниваются.
Напротив, для Sn ltm -=2041°, Д/тв=-505°, т. е. интервал существования
жидкой фазы необычно громаден, свидетельствуя о сравнительной
неустойчивости в случае Sn алмазной (a Sn) и искажённой алмазной структуры
(3Sn), несмотря на очень большую прочность межатомных связей олова,
отвечающую энергии сублимации 78 ккал/гр. атом. Структура жидкого Sn
заслуживает более детального исследования при разных температурах.
Все остальные элементы образуют наиболее многочисленную
четвёртую г р у ни у—металлы, для которой характерна «борьба» трёх основных
упаковок: HVi Ск и С(: с к.ч, 12, 12 и 8; см. рис. 220. #
Общие замечания по вопросу о выборе структуры.
Важнейшей проблемой теории структур является трактовка образования
данным элементом той или иной структуры и условий фазовых переходов
в однокомпонентных системах. Этому вопросу посвящена обширная
литература, по разному освещающая его с позиций различных теорий. Здесь мы
имеем возможность лишь коротко его коснуться.
Подход геометрической теории оказывается явно неприменимым: если
бы решал принцип плотной упаковки однородных атомов—биллиардных
шаров — возникали бы только плотные упаковки, что наблюдается далеко
не всегда. Кроме втого, гексагональная и кубическая плотные упаковки
были бы почти равновероятны. Между тем переходы этих форм друг
в друга вообще имеют место при стандартном давлении (1 атм) лишь в
некоторых, определённых случаях (см. ниже).
Динамическая (кинетическая) теория твёрдого тела позволяет придти
к общим выражениям, определяющим температуру, при которой разнопь
свободных энергий двух модификаций, участвующих в полиморфном
превращении, равна нулю. Однако, практическое использование этих представле-
§ 131]
СТРУКТУРЫ ПРОСТЫХ ВЕЩЕСТВ
359
ний наталкивается на затруднения, так что, например, Ф. Зейц [1] в главе
о динамике ядерного .движения в конце концов трактует фазовые переходы
различных простых веществ с позиций классической термодинамики, не
пытаясь, однако, увязать их здесь со строением атомов.
Достижения классической термодинамики хорошо известны химикам.
Затруднения возникают лишь при практическом, точном, определении обычно
очень малых теплот переходов, разностей теилоёмкостей и энтропии. Сама
необходимость точной экспериментальной характеристики каждой данной
системы ограничивает широкое использование этого рода представлений
в целях обобщений.
Отсюда вытекает естественное стремление различных авторов к
установлению связи между строением электронных оболочек атомов и типом
структуры кристалла. Например Мотт и Джонс |21 отмечали, что с заполнением
^/-оболочек в решётке возникают значительные отталкивательные силы,
благоприятствующие образованию типа С к для меди (по Фуксу, переход
Ov-тила в Сг-тии требует затраты «0,1 электроновольт па атом»). С ростом
количества незанятых мест в rf-оболочке возникают и другие структуры.
Эти выводы неполны (касаются только элементов V*—XI* групп и
обходят факт образования в ряде случаев аллотропических форм). Сами по
vcOe они весьма уязвимы. В частности, если энергия перехода Ск—Сс
может быть сколько-нибудь значительной (вследствие разницы к. ч. обеих
структур), то в случае перехода Ск—Ну—она должна быть весьма
небольшой, ибо плотность упаковки и валентные углы не меняются. Поэтому,
исходя из общих термодинамических соображений, казалось, следовало бы
ожидать возникновения структур Ну и С к у одних и тех же элементов,
с образованием аллотропических форм. Поскольку в структуре Ну
существуют бесконечные «н}стоты» (см. § 04), тогда как в Ск имеется более
равномерное распределение атомов между пустотами и наоборот, то не
исключено, что переход в неё может быть связан с увеличением энтропии, а это
означало бы, что энтропийный фактор благоприятствует возникновению
структуры С]{ у более высокотемпературной формы, хотя, конечно, его роль
не всегда может оказаться превалирующей. Но всяком случае, особенно
при малых тепловых эффектах реакции, неучёт энтропийного фактора, как
правило, приводит к ошибочным заключениям о знаке свободной энергии.
Далее, следует ожидать, исходя из принципов термодинамики, что
повышение давления и понижение температур должны способствовать
возникновению более плотных упаковок.
Обратимся к экспериментальному материалу, касающемуся примерно
00 элементов, образующих типичные металлы (см. табл. 30, рис. 220).
Таблица W
Структуры низкотемпературных и высокотемпературных аллотропических форм
типичных металлов
пературная форма
Be
Са
Sibil
Се
Рг
Структура
Ну
Ск
Ну
Л у
И у
Ну
температурная
форма
Be
Са
Sc
La
Се
Рг
Структура
гекс?
Ну
Ск
Ск
Ск
Ск
Низкотем-
, норатур-
ная форма
Ti
Zr
*, ?, Fe
Со
Ni
Tl
Структура
Ну
Н V
Се
11 у
Ну
Ну
Высоко-
темпэ-
ратуриая
форма
Ti
Zr
v Vo
Го
Ni
Tl
Структура
Се
Се
Ск*)
Ск
Ск
Ск
1
^ Но ещё более нысокоюмиоратурная форма (8)—вновь Сс.
360 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ РЕЩЕСТИ [ГЛ. IV
Из таблицы следует, что наиболее частой является действительно
комбинация Ну- и Ск-форм. Комбинации Ну и Cq или Ск и Cq
встречаются гораздо реже. Из 8 случаев «сосуществования» Ну и С а'-форм
в 7 случаях Hv отвечает низкотемпературной форме. Исключением является
Са. В обоих случаях «сосуществования» Ну- и Сс-форм, Нуу т. е.
плотная упаковка, является низкотемпературной формой, что и следовало
ожидать.
Таким образом, сказанное нами выше позволяет, казалось бы,
осуществить, хотя бы в некоторых случаях, исходя лишь из самых общих
термодинамических предпосылок, попытку интерпретации образования
структур металлов. Однако, вряд ли эти выводы можно сколько-нибудь
переоценить
Дело в том, что нельзя не ожидать также влияния квантовой
характеристики валентных электронов атома на структуру фаз, в частности, в духе
теории Бриллюэна. Вышеприведенные рассуждения Мотта и Джонса —
весьма неудачны также и с этой точки зрения.
Если выйти за пределы затронутого ими узкого круга элементов и
рассмотреть рис. 220, то следует подчеркнуть, что существует область в
периодической системе, где переход Ну и Са с образованием аллотропических
форм наблюдается и, в равной мере, существует область, где подобный
переход не наблюдается. К первой относятся группы II и Ш* периодической
системы, ко второй —■ группы X* и I*. В первом случае начинается
заполнение с/-оболочки или имеется только что заполненная s-оболочка.
Во втором—заканчивается заполнение й-оболочки или имеется заполненная
d-оболочка. Таким образом, например, медь принципиально отличается
от скандия тем, что выбирает для кристалла только С#-форму и не меняет
(или с трудом меняет) её на .ffy-форму, тогда как скандий переходит из одной
формы в другую. Иначе говоря, при одном и том же количестве холостых
электронов в ^-оболочке, наличие в ней же уровней со спаренными
электронами, повидимому, препятствует переходу С к- в Z/V-форму с уменьшением
свободной энергии.
В этом свете вопрос о термодинамической устойчивости а-никсля (см. § 122)
приобретает широкий интерес. Последний связан и с тем фактом, что
уменьшение атомного номера в пределах группы способствует возникновению
полиморфных превращений Ск—> Ну. Это наблюдается не только во II, но
даже в IX*, а может быть и в X* группах.
С с -решётки возникают в основном у элементов I подгруппы
системы (.v1) и в области V, Сг и их гомологов, имеющих наибольшие
количества холостых w-электронов в периоде (см. также § 92). Кстати, атомы
большинства элементов, образующих Cq -структуру, обладают в основном
состоянии нечетным количеством холостых электронов на а1-,
соответственно, .v-ypo шшх
d
V dz s2
Nd dA s1
Та d* s*
Cr db s1
Mo db s1
Mn db s2
t
A
t
1
A
t
t
t
t
t
t
t
t
t
t._
t
t
t
t
t
t
i
t
J
t
ti
t
t
I
§ 13J1
СТРУКТУРЫ ПРОСТЫХ ВЕЩЕСТи
362
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТИ
[ГЛ. IV
СЧЗ QQ "^ UQ UQ схэ «NJ
CNO CQ Огэ Со UQ UQ СЧЗ
C4J QQ C4J CQ C3Q Oq СЧЛ
b^w
счл со >^ Qqcjq со счл
счл со С^ со со со счл
V СУ^
СЧЗ Со С>5 Со со Со СЧЗ
C4J йо Со йо Со Со СЧЗ
C\J QQ ts^ Со Оо Со СЧЗ
I I
III
liii
C4J Qq CQ Со Со Со СЧЛ
CNJ Cq Vr^ CXD Со Со СЧЛ
Сч X* СУ} ^
«■о
счз ^ Xf со со со счз
C4J Со СУ} UQ Co Co C4J
счз со счз Со с^ со счз
счз со >-■ ср со со счз
счз со ^ со со со счз
>3 >*
> Cv?
5;
r
i
I
в
Он
s
I
C\J
? 131]
СТРУКТУРЫ ПРОСТЫХ ВЕЩЕСТИ
363
С дальнейшим увеличением или уменьшением количества электронов
в d-оболочке количество холостых г#-электронов падает, что влечёт за
собой немедленный переход к Ск или С к-Н у-структурам.
Таким образом, 6"с-область охватывают элементы типа: s1, dV, d*sl.
■Гк-/Л-область-s8, dls\ d\s\ d*s\ d7s\ dV, dV, d10s, dl0s\
Внутри последней области в данное время чётко вырисовываются
поля Ск- и С к—//у-структур (см. рис. 220).
Надлежит подчеркнуть важность постановки исследований с целью
уточнить термодинамическую и структурную характеристику Ну^Юк
перехода у металлов, в частности, где таковой до сих пор не обнаружен.
В свете изложенного представляют интерес структуры редких земель.
У этих элементов ds2-rpymia остаётся неизменной. Как влияет на их
структуру наличие холостых /-электронов?
Небольшому количеству последних -до /4 — отвечают аллотропические
Ну — Ск-структуры, в случае /5— тетрагональная. У Ей (/в) с большим
количеством ^-электронов появляется 67;-структура. Начало заполнения
/-уровней спаренными, электронами (как и в случае d-уровней) сейчас же
находит выражение в возникновении либо Ну либо Сд;-решёток (от ТЬ до
Ср, т. е. от /8 до /м). К этому полю примыкает даже Gd (/7), возможно
вследствие перехода типа fds2 —>/V (см. рис. 221).
Из данного параграфа, как нам думается, вытекает явная
зависимость структур кристаллов простых веществ от строения атомов и
совершенная недостаточность геометрических теорий для каких-либо обобщений.
Вместе с тем мы считаем, что изложенные здесь факты и соображения
дают дополнительные основания, чтобы отвергнуть концепцию Паулинга,
касающуюся строения металлов и изложенную в его книге «Природа
химической связи» (особенно, поскольку идёт речь о структурах типичных
металлов); см. § 92.
Сделанные выше замечания не претендуют на сколько-нибудь
всестороннее освещение вопроса. Они преследовали основную цель, заключающуюся
в том, чтобы показать недостаточность одностороннего подхода к вопросу об
образовании структуры металлов, проявляемого многими авторами.
Необходимы более глубокие исследования роли строения атомов и влияния
термодинамических и кинетических факторов на процесс возникновения
структур.
ЛИТЕРАТУРА к § Ш
1. Ф. Зейц, Современная теории твёрдого тола, гл. XIV (1949). Б. Ормонт,
Ada phvsicochim. (JRSS; IV, 427: V, 405; VIII, 811 (19,15—1938); Успехи химии
V, 79Л (1*936).
2. Motta. Jones, Theory of the Properties of Metals and Alloys (1936),
гтр. 174.
См. также: Я. II. Френкель, Физика металлов (1948), Я. II. Френкель,
«Статистическая физика (1948) и литературу к главе II.
ГЛАВА V
СТРУКТУРЫ ГАЛОГЕНИДОВ И ГИДРИДОВ
§ 132. Введение
Главы V — VIII посвящены структурам простых бинарных соединений,
образуемых двумя компонентами. Эта, пожалуй, наиболее изученная
химическая группа веществ, рассматривается нами в 5 разделах:
Галогениды и гидриды, т. е. соединения с F, CI, Br, J, H, D (тяжёлый
водород).
Окислы и халкогениды, т. е. соединения с О, S, Se, Те.
Нитриды, фосфиды, арсениды и т. п., т. е. соединения с N, P, As.
Карбиды и силициды, т. е. соединения с С и Si.
Бориды, т. е. соединения с В.
В порядке исключения, вместе с галогенидами рассматриваются гидро-
ксиды, сульфгидриды, селеногидриды (поскольку ОН-, SH-, SeH-группы
по своим свойствам во многом являются соответственно аналогами фтора,
хлора, брома). Впрочем, эти последние вещества изучены очень плохо, чему
способствовали их пониженная кристаллизационная способность,
склонность образовывать аморфные осадки. Кроме того, они поглощают
переменное количество воды и, с другой стороны, часто расплываются и разлагаются
на воздухе.
Материал расположен по подгруппам периодической системы элементов.
Внутри каждого из пяти упомянутых выше разделов каждой
подгруппе элементов периодической системы посвящен специальный
параграф, т. е. до двадцати параграфов (считая также подгруппы III**
и VI**).
В случае, если элемент образует соединения разной валентности, то они
приводятся в порядке убывающей валентности. Соединения типа поли-
иодидов, полисульфидов, перекисные соединения, кристаллосольваты и т. п.
вынесены в главу комплексных соединений, поскольку они содержат
комплексные радикалы ОТ и т. п., фигурирующие как единый структурный
узел.
В отличие от простых веществ для соединений в SB даются значения
не удельных весов, а молекулярных объёмов (Molekularvolum).
Во избежание явных недоразумений, предупреждаем химиков, что
так называются в SB, к сожалению, не молекулярные объёмы, под
которыми химики обычно понимают граммолекулярные объёмы в см5, а объёмы
в А3, приходящиеся на 1 молекулу (мы их будем обозначать ОМ .
Для получения граммолекулнрных объёмов (в см3) надо умножить ОМт.
выраженные в (ангстремах)3 на
' :Q^4— , т. е. на 0,602.
5 133] ГАЛОГЕНИДЫ. ПГРУППА: ЛИТИЙ, НАТРИЙ, КАЛИЙ, РУБИДИЙ, ЦЕЗИЙ 365
§ 133, I группа периодической системы: литий 1л, натрий Na, калий К,
рубидий Rb, цезий Cs
1. Галогениды, гидриды ^^^
и др. щелочных метал- \jT
лов
Решётка кубическая С
Координация октаэдричс-
ская О /О.
Тип Со/о-SB 51.
Пространственная группа
О t — Fmirn.
Z = k.
М к.ч.=6Х(0);
X к.ч.-бМ(О).
^^9
П
Г
6^
6
lb*?"
О
Ь^№"
^?
b^W^b'
о
-С\
<jr Qf— (JT Ом Q) Галоген
Рис. 222.
г~
1 Li l
1 ад
1 аЕ
d
ОМ
Na t
aR
\ аЕ
d
ОМ
1 к г
| ад
ai
ОМ
1 Rb t
I aR
ai
ом
J Cs t
aR
^
1 OM
—F
25° С
4,01808±S[«3]
4,027±5
2,0090/i±/
—
25° С
4,62345 ±5
4,641 ±4
2,3093iJ
—
—
5,356±J0
—
—
5,63
2,82
44,6
—
6,00g
з,оо4
54,2
—CI -Br
25° С —
5,12952±4[<3j
5,136±<3
2,56476 ±4
—
18° С
5,G2712±3[3]
5,669 ±5
2,81356±2
—
—
6,277±2
6,319±5
— .
—
6,54
3,27
70,0
445
-Ь5ЭС
7,02
3,51
r-
KOAIH.
7,10
3,55
89,5
5,489 ±6
5,495±S
2,7443±3
—
25° С
5, 96095±5
5,963±5
2,У807±3
—
6,58f>±2
6,630±4
—
—
6,854
3,427
80,5
—
—
—-
—
- 1
6,000 ±7
6,()19±5
3,000+3,5
—
6,462±6
6,469 ±5
3,231±.3
—
—
:7,052±3
7,078 + 8
—
—
~>325
3,663
98,6
—
—
—
— .
-H
4,085
2,043
17,02
4,88
2,44
—
5,70
2,85
—
6,04
3,02
—
—
6,37
3,18r
-D
4,065
2,032
16,78
—
—
-
—
—
—■
=
—
—
—
—
OH
_^
—
—
—
—
—
—
300QC
5,78
2,89
—
—
—
—
—
—
—
—SH
-SeH
1
высокотемпературные
р-формы 1
—
—
—
6,05
3,025
55,4
6,60
3,30
71,8
6,93
3,465
83,2
—
—
—
—
—
—
—
6,30
—
—
6,92
—
—
7,21
—
—
—
— 1
— 1
— 1
Примечание. aR—рентгенографические, аЕ— электронографические значения а„.
366 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гЛ. V
2. aRbCl, CsCl, CsBr, CsJ, CsSeH
Решётка кубическая С.
Координация кубическая С/С.
Тип С с 1С- SB B2.
Пространственная группа О^ — Рт'Лт.
Z = l.
М к.ч. = 8 X (С); X к.ч. = 8 М (С)
Рис. 223.
aRb
Cs
Р
d
ом
tDC
р
d
ом
С1
—1*30
1
3,742
3,24
52,4
комн.
1
4,П0
3,560
GO, 4
Br
комн.
1
4,28,
3,713
78,7
J
комн.
1
4,56
3,95
94,8
SH
комн.
1
4,29
3,71t)
79,0
SeH
комн.
1
4,4*7
Примечание Г
На «подкладке») Т1С1 1
яНЬСЛ (Сс,с) имеет
удельный вес сх (при
— 190 °C)=3,U43
Чистые
3. LiOH
Переходя дадее к структуре LiOH (тип В 10), мы хотим подчеркнуть,
что этому типу отвечает положение центрального атома в тетрагональной
призме, не совпадающее с позицией (72 7а lj2)* При небольшом смещении
можно пренебречь отклонением от формы кординациониой сферы Pi (Pi)
(тип Pi/Pi). Но при значительном смещении, как, например, на рисунке 224,
конфигурация Li приближается к Я + Я (причём решётка приобретает
слоистый характер), конфигурация О к S(+ S)=-Di, а тип — скорее
§ 133] ГАЛОГЕНПДЫ. 1 ГРУППА: ЛИТИЙ, НАТРИИ, КАЛИИ, РУБИДИИ^ ЦЕЗНИ_
39"
Решётка тетрагональная Т.
Координация тетрагонально-
призматическая,
q_( + q)IS( + S)
не симметрично
центрированная (атом внутри смещён
ill.
с позиции — — — ).
ТИП ^(+?)/5( + 5)-SB В10.
Пространственная группа
Z = 2,LiK.4. = 40,?( + 40(tf));
Ок.ч. = 41л.(5) + 40(£)
--=Д/.
Вегцеспю ....
\ с
с/а
\ d
*х
ион I
4,334
1,221
1,97
1,402
Рис. 224.
4. aNaSH, aNaSeH, aKSH.
aKSeH, aRbSH, aRbSeH
(низкотемпературные формы)-
Решётка ромбоэдрическая И
центрированная.
Координапия квазиоктаэдри-
ческая О/О.
Тип Rnid-SBB22 (KSH).
Пространственная группа
D\d-Шт.
Z = 4, М к.ч. = 6S(GM;
S к.ч. = 6М(0).
1 1
a j л
1 !
LNaSH . .
NaSeH . .
KSH . . .
KSeH . .
RbSH . .
RbSeH .
5,98
6,24
6,60
6,83
6,85
7,11
96° 30'
96°27'
97°0'
97°21'
97°20'
93°07'
d 1
M~S
2,99
— 1
3,30
—
^,43
—~ I
Примечание. Деформи-|
[рованный сплющенный по осп!
l.i тип NaCI. 1
Рис. 225; см. также рис. 63, в.
aNaSeH переходит в 3-форму при 150° С,
a KSeH и aRbSeH-при 180е С.
368 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
В SB III 8 описание типа 522 производится в первую очередь на базе
примитивного ромбоэдра (Z = 1), причём для KSH получается а = 4,374 А,
я = 68°51'.
Но в следующих томах SB тип В22 трактуется на базе гранецентри-
рованного ромбоэдра (Z = 4) с иными периодами идентичности. Поэтому
наша таблица построена для случая гранецентрированного ромбоэдра.
Общие замечания
1. Галогениды щелочных металлов занимают в теории структур
особое место. Применительно к ним было проведено громадное
количество расчётных и экспериментальных работ. Поэтому мы имеем
возможность подробнее остановиться на метрике элементарных ячеек,
относящихся к этим веществам.
Структуры галогенидов, гидридов, гидроксидов щелочных металлов
либо относятся к 2 кубическим формам (тип NaCl и CsCl), либо
обнаруживают деформацию ромбоэдрическую — сульфгидриды, селеногид-
риды — или тетрагональную —LiOH. Ромбическая деформация не
обнаружена.
2. При конденсации кристаллов галогенидов из пара наблюдается
значительное увеличение межатомных расстояний по сравнению с
расстояниями в молекуле.
При испарении имеет место сближение атомов. Это поведение
характерно для координационных решёток солей, в которых вес ковалентного
состояния, по сравнению с паром, понижен. (В молекулярных решётках
часто имеют место обратные соотношения.)
Сопоставление межатомных расстояний галогенидов щелочных
металлов в парообразном состоянии (молекулярный пучок) и в кристаллах
даёт:
Вещество
Пар
Кристалл
Вещеотло
Пар
Кристалл
NaCl
2,51
2,814
RbBr
3,06
3,427
КС1
2,79
3,139
CsBr
3,14
3,713
RbCl
2,89
3,27
NaJ
2,90
3,231
CsCl
3,06
3,560
KJ
3,23
3,526
NaBr
2,64
2,931
KBr
2,94
3,293
1 1
RbJ CsJ
3,26
3,663
3,41
3,9Г,0
3. Большой интерес представляет вопрос о взаимных переходах
в зависимости от температуры и давления одной структуры в другую
при разных координационных числах.
В ссЮтветствии с общими принципами термодинамики можно было
ожидать, что понижение температуры и повышение давления должны
способствовать переходу от к.ч. 6 к к.ч. 8, т. е. к уплотнению
«упаковки» атомов; повышение температуры (и понижение давления)— к
обратному процессу.
§ 133] ГЛЛОГЕНИДЫ. I ГРУППА: ЛИТИЙ. НАТРИЙ, КАЛИЙ, РУБИДИЙ. ЦЕЗИЙ 369
Влияние температуры на поведение RbCl было изучено ещё в 1936 г.,
причём была сделана попытка получить «aRbCb типа Сс/с путём
конденсации паров RbCl (чистейший) на стекле капилляра при t = —190° С.
Однако при этом образовался лишь «pRbCh типа Coio» Лишь на
подкладке aTlGl (тип Сс/с) удалось получить «aRbCb. При нагревании
до комнатной температуры «aRbCb спонтанно переходил в «pRbCb.
В таблице 38 мы приводим данные о соотношении метрики ячеек
RbCl и TICK
Таблица 38
| Температура "v>
конденсации
20
-190
-190
иссле-
доыанип
20
-190
20
KbU
а
6,53,.
6,399
°х
2,79,
4
3,04.
T1G1
а
3,830
3,752
"
°JC
7,032
7,49,
О
форма
р
a
?
RbCl на ТКЛ |
RbCl
а о£
6,53г
3,742
б,г>:»з
2.794
3,80;
2,79.
TICI 1
1 1
а j ох
3,825
—
—
7,04
По поводу исследованных структур гидридов щелочных металлов над-
лежит отметить, что впервые правильные значения экспериментальных
удельных весов их были даны в тщательной работе И. А. Казарновского
и М. А. Проскурнина Zs. f. Anorg. Ch. 170, 301, 311 (1928), что позволило
обнаружить грубые ошибки в структурных исследованиях зарубежных
авторов. К сожалению, в литературе отсутствуют надёжные данные о
современных исследованиях в этой области.
Были изучены также структуры CsGl, GsBr, GsJ (чистейших) при t°
от комнатной до температуры на 50° меньшей температуры плавления.
Переход Ccjc—^ Cojo был найден лишь у GsCl при 445 ± 5° С. При этой
температуре
яш(тип С ас) = 4,241;
яш(тип Со/о) = 7,02А.
Энергия перехода равна 1,8 ккал/молъ.
Влияние давления было изучено в 1938 г., причём был рассмотрен
теоретически и доказан экспериментально переход структур С0/о У RbCl,
RbBr, RbJ, КС1, KBr и KJ при давлении р > 3000 атм в структуру Сс/с
с к.ч. 8. Для RbJ, например, этот переход требует 4500 атм.
Сказанное выше относится к влиянию температуры на плотность
упаковки (к.ч.).
4. Влияние температуры на изменение симметрии элементарной ячейки
хорошо изучено на примере сульфгидридов и селеногидридов.
Низкотемпературная a-форма (кроме CsSeH) —ромбоэдрически искажённый тип NaCl,
сплющенный вдоль оси 3 (тип SB —/?22). Нагрев до 150—180° С вызывает
переход в кубическую р-форму (тип NaCl), механизм которого рассмотрен
нами ранее.
CsSH и CsSeH, в отличие от аналогичных соединений других щелочных
металлов, образуют элементарную ячейку типа CsCl уже при комнатной
температуре. Более слабое силовое поле Cs+ иона (при его большом радиусе)
неспособно, повидимому, преодолеть вращение SH- соответственно SeH-
ионов, повышающее симметрию решётки.
370 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. V
Вопрос о повышении с температурой симметрии тетрагонально
деформированных решёток (тип LiOH) пока остаётся, в экспериментальном
отношении, недостаточно изученным.
Низкотемпературная форма аКОН при 248° С переходит в [Ж.ОН
(тип Сор)- Из значения аш = 5,78 вытекает, что «радиус» ОН~-иона
;-e = l,56 kX.
5. В таблице стр. 365 мы, в виде исключения, сопоставили
рентгенографические ад и электронографические а% данные по аю для иллюстраций
эффекта изменения межатомных расстояний в поверхностных слоях.
Из рентгенографических данных особняком стоят точные определения
постоянной решётки LiF, LiCl, NaF, NaCl и NaBr, выполненное советским
исследователем Евинсом.
В 1940 г. была исследована структура расплавов КС1, NaCl, LiClr
LiBr. Наиболее резкую картину дал КС1. Каждый атом^ (ион) имеет
6 соседей на расстоянии 3,14 А и 12 на расстоянии 4,44 А.
7. В заключение параграфа приведём сводную таблицу типов
рассматриваемых очень важных соединений элементов I группы, а также 1*
группы и Т11.
Таблица 39
Li
1 Na
К
Rb
Cs
Си
Ag
Аи
Tl1
F
COjO
cop
COJO
GOfO
co\o
cTiT
C0(0
\0Rdlo-B2k
CI
co\o
COjO
COfO
ccic
cop
CC}C
^c\c
cTfT
cop
Ccic
Br
COJO
co\o
С с ic
G0}0
CCfC
ccic
C0}0
CCjC
J
COfO
coio
co/o
С сic
cop
CCjC
cc\c
CTJT
CTIT
HT{T
1 cc/c
|0Я7£—ЯЗ.*
OH
Г<7 (+<f)/SO £)
coio№
C0io№
coio№
Дб/oW
^c\c
SH
—
C0/o(P)
RoioiJ
CCfC
SeH H
—
crio
C0J0
C0/0
-
C0J0
Htit
H'l I'J
D
C0/0\
Структуры, входящие в данную таблицу постоянно приводятся в
качестве иллюстрации справедливости правила Магнуса (о роли
соотношения ионных радиусов в выборе типа решётки). Между тем, существует
немало отклонений от этого правила, что обычно не подчёркивается.
См. § 77 и таблицу 24.
§ 134) ГАЛОГЕНИДЫ. II ГРУППА: БЕРИЛЛИЙ-РАДИЙ 371
§ 134. II группа
периодической системы:
бериллий Be, магний Mg,
кальций Са, стронций Sr,
барий Ва, радий Ва
1. BeFa
Решётка тетрагональная Т.
Координация Tjl
(деформированная).
ТииТтц — SB? (искажённый
SB C9 — кристоба лит).
Пространственная группа?
Z = 8.
Of ов«
Рис. 226.
Вещество
1 BeF>
а
6,60
с
6,74
с/а
1,02
Примечание
Существенно, что BeF2, обладая ре- 1
шёткой Si02, образует «пространствен- 1
ную вязь». Это один из немногих фтори- 1
дов, способных к стеклообразованию, 1
как и SiOa. I
2. MgF2
Решётка
тетрагональная Тк.
Координация треуголь-
но-октаэдрическая Ojt.
Тип Го/i —SBC4 (тип
рутила).
Пространственная
группа D\\—Pilmnm.
Z = 2, Mg к.ч. -6F(6>);
F K.4.-=3Mg(0-
Рис. 227.
Вещество
MgF2
t°
комн.
a
4,64
с
3,06
cja
0,659
d
(два)
2,05
d'
(четыре)
ОМ
1,96 33,0
Примечание 1
См. ТЮа рутил I
372
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. V
3. MgCla
Решётка ромбоэдрическая слоистая
Rs
Координация O/t.
Тип ^/,-SB C\% (CdCl2).
Пространственная группа Did —R3m.
Z = l.
О Мд
Рис. 228.
Вещество
MgCla
а
6,22
а
33°30'
Примечание
Атомы ГС1 образуют почти точно кубическую
упаковку, 'в мождуузлия которой слоями
внедряются атомы магния.
В результате атомы Mg с двух сторон
окружены слоями атомов хлора.
Имеются указания (Z.Anorg. Ch. 246, 282 (1940)), что найдена новая
модификация MgCl2.
§ 134]
ГЛЛОГЕНИДЫ. II ГРУППА: БЕРИЛЛИЙ-РАДИЙ
373
4. MgBr2, MgJ2, Mg(OH)2,
CaJ2, Ca(OH)2
Решётка гокса1 опальная
слоистая И .
Координация Oft.
Тип Hf)tг-SB Cd (CdJ2).
Пространственная групп *
ъ
СЗт.
Рис.
Вещество | а
I
i
MgBre I 3,815
MgJ, | 4,14^2
i
Mg(OH)a | 3,12
CaJ2 | 4,48±2
Ca(OH)a : 3,5844
с
6,25,,
6,88±5
4,73
6,96±3
4,8062
c/a
1,64
1,66
1,516
1,55
1,3660
d
2,945'
2,09
3,12
—
OM
—
—
ЗУ, 9
—
56,0
°X
3,876 1
— J
—
—
— 1
374 ч. п. структуры неорганических веществ [гл. V
5. CaF2, SrF2, SrCl2, BaF2, RaF2
Решётка кубическая С.
Координация куботетраэдрическая С/Т.
Тип Ccit- SB CI (GaF2).
Пространственная группа Oh—Fm3tn.
Z = 4.
Ca к.ч. = 8Х(6'); F к.ч. = 4M(7,).
Вещество
CaF2
SrFa
SrCl2
t°
комн.
»
»
au>
5,451
5,78
(до 5,86)
6,98
d
2,362
2,50
з,о2
OM
40,50
48,2
85,0
Вещество
BaF2
RaF.,
t°
комн.
»
a*
6,19
(от 6,18
до 6,20)
6,368±3
3
2
d
,68
,75;
OM
59,3
64,5
6. CaCl2
Решётка ромбическая OR.
Координация O/t.
Тип ORfTft-SBСЗЬ.
Пространственная группа D^ — Priam (или Cx{v —Pnn). Z = 2.
Вещество
| CaCl,
a
6,24
b
6,43
с
4,20
a : b : с
0,971 : 1 : 0,653
Qx
2,17
Слегка деформированный тип С4 (рутила), что выражается как в
неравенстве периодов решётки, так и в повороте октаэдров [СаС1в]
вокруг оси С. См. также Chem. Abstr. 40,4580 (1946).
1341
ГАЛОГЕНИДЫ. II ГРУППА: БЕРИЛЛИИ-РАДИИ
375
7. СаН2> SrH2, BaH2
Решётка ромбическая, слоистая ORs.
Координация 11/3,4.
Тип ORfii3ti-SBC29.
Пространственная группа Dzh — Pnam.
Z-4.
М к.ч. = 5Н(1) + 6Н(11); Н(1) к.ч. = ЗМ; Н(И) к.ч. = 4М.
Вещество
СаН,
*гН,
НаН.
а
5,936±<5
6,3ft4±5
6,788±5
ь
6,838±J
7,343±5
7,829±5
с
3,600±3
3,875±3
4,167±<3
а :Ъ : с
0,867 : 1 :0,528
*х
1,90
3,27
4,15
Н-ионы образуют слегка деформированную гексагональную упаковку,
причём ось а отвечает гексагональной оси с, а ось 6 — гексагональной
а уд. Решётка очень близка к CdJa.
8г++-ион имеет 5HI на расстоянии 2d (2,35) + 2d' (3,20)
6Н II на'расстоянии 4е (2,71) + 2е' (3,09)
8. SrBr2
Решётка ромбическая OR.
Координация P3*+jT,5.
Тип 0/?P38+/T,l-SBC53.
Пространственная группа
Дй — РЪпт.
Z = 4.
Sr к.ч. = 9 Вг(Р33+).
Вг1 к.ч. = 4 Sr (тетраэдр).
Вг II к.ч. = 4 Sr
(неправильная конфигурация)
( + lSr)<*5.
о0о |о0о |о0о
о о°п о°п о°1
oOQo
о°о
ОоО
оО Q
о°о
000
;о о°Ь о°1о о°
оСПЦоО OJoO Q
0°Oi 0°0! 0°0
Г23456789Ю11
■ ■ ' ' ' ■ У ■ ■ L-J
Рис. 2П.
О Sr
О Br
Вещество
SrBr,
а
9,20
Ь
11,42
с
4,3
а : Ь : с
0,8056 :1:0,3765
Примечаний
SB VII 11 Струн-
тура близка к
С23(РЬС12» ВаСЛЛ
376
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
(ГЛ. V
9. ВаС12, BaBr2, BaJ3
Решётка ромбическая OR.
Координация Р3*'/Т, Pyi.
Тип ORpv+p ,p„4 - SB £23.
Пространственная группа
Dll-Pmnb Ba к.ч. =9 X.
Z = 4.
XI к.ч. = 4Ва
(тетраэдр).
XII к.ч.=
= 4Ва + 1Ва(^/>у4)
(неправильная
конфигурация).
^-О-
Вещество
\а . . . .
\ь . . . .
\с . . . .
ох ...
ОМ . . .
ВаС1,
4'705
7,823
9,33л
4,002
52,0
ВаВг*
4,94,
8,24?
9,838
4,886
60,8
BajJ
5,26,
8,86л
10,56J
5,236
74,7
SB VII 8.
Координационные сферы SrBr2 и
ВаВг2 (см. для ВаС12 рис.
233) настолько близки,
что наши обозначения
типов С23 и С53 почти
одинаковы.
§ 135. III* группа
периодической системы: скандий
Sc, иттрий Y, лантан La
1. YF3
Решётка кубическая С
Координация?
Тип С? SB(?)
Пространственная группа
Он — РтЗпг (или О1 ~
— Р43 или Т\- Р43т или
1п-РтЗилкТ1-Р23).
3Yb(V.V.0)O
9F статистически в
(V.oo);C; (V/.7«)
(7, 7« V«); С; (7« 7. 7 J
(7.7* 7 J 2=з.
Рис. 232.
^N.
О В£
• CI
Рис. 233 (схема).
Вэщзстно aw
YF,
5,644±3
сч.
5,36
Y—F
F—F
2,44 | 2,44 1
Надёжными данными не располагаем.
§ 135] ГЛЛОГЕН1ЩЫ. Ill* ГРУППА: СКАНДИЙ, ИТТРИЙ, ЛАНТАН 377
2. LaF3
Решётка гексагональная, слоистая Hs.
Координация l + t + P3/t,4 ^ 11/3,4.
Тип #fi/3,4-SBZ>0e.
Пространственная группа Df)h — C6/mcm.
Z = 6. La k.4. = 2F(/) + 3F(/) + 6F(/>3);
Fk.h. :3La(/); Fill к.ч. =HLa = 3La.
Рис. 234.
Вещ-сство
LaF,
Тизонпт
a
?Л\
7,124
С
7,280
7,32,
с/а
1,022
1,02.>
i
Примечание 1
SB II 27. Атомы F образуют 1
волнистые слои с двух сто- 1
рон среднего плоского слоя 1
(состоящего из атомов La и |
F) в виде шестичленных ко- 1
лец I
Тизонит — LaF3 с примвеями I
редких элементов 1
378
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. V
3. ScF3
Решётка ромбоэдрическая R
(псевдокубическая).
Координация Ojl.
Тип Rq}1 — SB? искажённый
Д09 тин (Re03).
Пространственная группа
DI-R32.
Z = 3. Sc к.ч. =6F(октаэдр).
Q-xte
Вещество
а . . . .
d (Sc - F)
Примечание. Надёжными
данными не располагаем.
4. LaCl8, LaBr„ La(0H)3,
АсС13, АсВг3
Решётка гексагональная Я.
Координация (?) 9/г/.
Тип Нщ (UG1,).
Пространственная группа
CSA-C6,/m.
Z = 2. M к.ч. = 9Х.
Рис. 235.
Вещество
LaGl3
LaBr3
La(OH)3
AcCl,
АсВг3
a
7,468±«3
7,951±3
6,510±5
7,62 ±2
8,06 ±4
с
4,366±3
4,5014:3
3,843±5
4,55 ±2
4,68 ±2
c/a
0,584
0,56g
0,59
0,598
0,586
Примечание
По работе
Захариасена Acta Gr. 1,265
(1948)
б. LaJ3
Решётка орторомбическая
OR.
Координация 8/т/.
Тип ОЛ8/у-(РиВга).
Пространственная группа
Dzh — Ccmm.
Z = 4. Мк.ч. = 8Х.
Общие замечания
Начиная с III* группы периодической системы у элементов
появляются валентные ds-электроны.
Как указывал автор*) (1935), эта особенность обусловливает
неустойчивость летучих гидридов. Поэтому элементы ряда Sc —Си и их гомологи
*) Б. ф. Ормонт, Успехи химии, V 739 (1936).
Вещество
LaJ3
а
14,1±*
Ь
4,33±5
с
1О,О5±70
Примечание
По работе
Захариасена
Acta Сг.
1,215 (1948)
§ 136] ГАЛОГЕНИДЫ. Ш** ГРУППА: МЕТАЛЛЫ РЕДКИХ ЗЕМЕЛЬ 379
образуют гидриды, в большой мере не похожие на галогениды, например,
не имеющие молекулярной решётки (сравним, например, TiCl4 и TiHn).
Решётки металла в этих случаях обычно сохраняют свои металлические
свойства с образованием гидридов. Соотношения МхНу являются резко
выраженными дробными; структура фаз сохраняется в широких пределах
соотношений атомных концентраций.
§ 136. III** группа периодической системы: металлы редких земель
Се, Рг, Nd, 61, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Cp.
1. CeF8, PrF3, NdFa, SmF3
Решётка гексагональная слоистая Hs.
Координация 11/3,4.
THnfffirt4-Z)Oe(LaF,).
Пространственная группа D\h —Сб/тст.
Z = 6.
Координация—см. стр. 377.
Рис. 2:№
Вещество
CeF3
PrF3
NdFs
| SmF,
a
7.114
7,06x
7,021
6,9«
с
7,273
7,196
7.196
7,15
c/a
1,022
1,019
1,025
1»024
Примечание
О типе D06 см. LaF3
В SB II с/а
неправильно указано 1,025 1
380 ч. п. структуры неорганических веществ (гл. V
2. СеС13, СеВг3, РгС13, РгВг3, Рг(ОН)3, NdCla, M(OH),
Решётка гексагональная Н.
Координация (?) 9/х.
Тип НЧХ (UC13).
Пространственная группа С6л — С65/т.
Z = 2. Мк.ч.-9Х.
Вещество
CeCl,
СеВг,
PrCl,
PrBrs
Pr(OH),
NdCl,
Nd(OU)3
a
7,436 ±4
7,936±3
7,41 ±1
7,92 ±1
6,47 ±3
7,381±4
6,42 ±2
с
4,304±4
4,435±3
4,25 ±1
4,38 ±1
3,76 ±3
4,231±<3
3,74 ±2
ca
0,579
0,559
0,573
0,553
0,58t
0,573
0,583
Примечание 1
По работе Захариа-1
сена Acta Cr. 1, 265 1
(1948)
3. NdBr8, SmBr8
Решётка орторомбическая OR,
Координация 8/у.
Тип OR8/y-(PuBr3).
Пространственная группа D^ — Ccmm.
Z = 4. Мк.ч. -8X.
Вещестпо
ШВг3
SmBr8
a
12,6't-b5
12,62±5
b
4,10±3
4,03±<3
с
9,15^-4
9,06±i
Примечание
По работе Захариа-
сена AclaCr. 1, 26Г> (19ri8)
В работе Захариасена полученные им данные, имеющие пока
предварительный характер (поскольку сведения об исследовании
монокристаллов не опубликованы), не комментируются с кристаллохимической
точки зрения, между тем нам кажется не лишённым интереса
следующее обстоятельство: с увеличением поляризационного эффекта (и, пови-
димому, веса ковалентного состояния в химической связи М — X)
элементарная ячейка становится более сжатой по оси с, как это легко
обнаружить сопоставлением величин с/а в рядах М(ОН)3, МС18 и МВг8
для одного и того же элемента»
В равной мере, с переходом от одного редкого элемента к другому,
с увеличением количества электронов в начале ряда (La— Се — Рг) имеет
место уменьшение величины с]а. Сказанное нам кажется полезным
иллюстрировать таблицей с/а.
1 м
Ас
La
Се
(ОН)3
0,59
—
С1,
0,598
0,584
0,579
Вг3
0,586
0,56,.
0,559
М
Рг
■Nd
(ОН),
0.58J
0,583
Cls
0,57.,
0,573
Вг, [
0,553
|
f
Этот структурный тип характерен не только для La и лантапидов*
но и для уранидов (актинидов).
§ 136] ГЛЛОГЕНИДЫ. Ш«« ГРУППА: МЕТАЛЛЫ РЕДКИХ ЗЕМЕЛЬ 381
4. EuF,
Решётка кубическая С.
Координация куботетра-
эдрическая С/Т.
Тип СС1т-SB CI (CaFa).
Пространственная группа
<А —Fm'Srn.
Z = 4. Eu k.4.=8F[(C);
F к.ч. = 4Еи(7).
1 Вещество
EuF,
aw
5,796±<S
d
2,51
OM
48,0
Рис. 237.
5. EuCl2, SmCl2
Решётка ромбическая OR,
Координация Р3*>/ТА.
Тип ORp^it 4 —
—SBC23(PbCl2).
Пространственная i группа
Dlh—Pmnb.
Z = 4.
С1(1)к.ч.=4'М(!Г);
CI (II) к.ч. = 4 M
(неправильна я координация).
Рис. 2М8.
Вещество
EuGl2
SmCl*
а
4t493
4,497
Ь
7,499
7,532
с
8,914
8,973
Ох
4,899
4,807
ОМ
45,5 1
46,0
382
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
(гл. V
6. YbJa
Решётка гексагональная
слоистая Hs. _
Координация —Ojt.
Тип Нщ-ЪВ C6"(CdJ2).
Пространственная группа
Dbd—СЗт.
Z=i.
Вещество
YbJa
а
4,48
с
6,96
с/а
1,552
О YB
О 1 2 3 4 5 Г~\ j
J L_
Рис. 239.
§ 137. IV* группа периодической системы: титан Ti, цирконий Zrr
гафний Ш, торий ТЬ
Молекулы летучих тетрагалогенидов IV* группы (МХ4) должны быть,
с точки зрения теории валентности, тетраэдрическими*) (см. § 95), что в
исследованных случаях и обнаруживается, например, у TiCl4 d (Ti — CI) =
= 2,3 А, см. М. Е. Дяткина, ЖФХ XX, 6(1946). Этот ряд изучен ещё
недостаточно.
•) С возбуждением d2s2 электронов до d4p или d*s
§ 137] ГАЛОГЕНПДЫ. IV* ГРУППА: ТИТАН, ЦИРКОНИЙ, ГАФНИЙ, ТОРИЙ 383
Структуры молекул немаксимальной валентности, например, типа
TiX , TiX2, представляют большой теоретический интерес, но, к
сожалению, не изучены.
Структуры кристаллических тетрагалогенидов также изучены в
немногих случаях.
Структуры три- и дигалогенидов не изучены.
1. ZrF4, HfF4
Решётка моноклинная, центрированная.
Тип М-SB?
Пространственная группа С^ — С2\с.
Z = 12.
Вещество
ZrF4
HfF4
а
9,46
9,45
Ъ
9,87
9,84
с
7,64
7,62
3
94°30'
94°29'
I
4,66
7,13
SB III 327. ZrF4 и HfF4 совершенно изоморфны. Решётка молекулярная.
Как указывал SB III 328, этому, якобы, противоречит склонность фтора
к образованию ионов. Не приходится говорить, что подобные свойства)
фтора нисколько не
определяют характера решётки
и, в частности, не мешают
образованию молекулярных
решёток у соединений типа
АХ4.
2. TiBr4, TiJ4, ZrCl4
Решётка кубическая,
молекулярная Сщ~т\
молекулы тетраэдрические.
Координация 1.
Тип Cf(~7)-SB DU-
Пространственная группа
T*h-Pa3.
а = МХ4.
Z=8. ак.ч. = 1а.
Ох
Рис. 240.
Вещество
TiBr4
11,25
3,406
TiJ4
12,00
ZrCI4
10,32
Примечание
SB III
305—307 ,
384 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
Гидриды Ti, Zr, Hf и Th
Исследования структур гидридных фаз названных металлов
охватывают, главным образом, Ti и Zr и имеют пятнадцатилетнюю давность.
Из обзоров надлежит назвать стр. 87 — 107 книги Я. С. Уманского (1947) *),
где также воспроизведены старые данные Хэгга. Некоторые обобщения
теоретического характера сделаны автором**).
Остановимся на этих системах вкратце.
Гидрид титана:
а-фаза. Гексагональная. Твёрдый раствор Н в титане. Металлический Ti
поглощает до 33 атомных % Н, причём а увеличивается от 2,953 до
3,11 и с от 4,723 до 5,02.
JJ-фаза. Кубическая гранецентрированная. aw растёт с концентрацией Н
от 4,397 до 4,460 (состав меняется от TiH до ТШ2).
Гидрид циркония:
В системе Zr —H обнаружено пять фаз:
а-фаза. Гексагональная. Твёрдый раствор водорода в Zr (до 5% Н) (?)♦
а = 3,229-3,247 (5% Н);
с = 5,141-5,173 (5% И); с/а ^1,59.
р-фаза. Атомы Zr образуют кубическую гранецентрированную решётку
с внедрёнными атомами Н (а' —4,664 А).
Предположено, что фаза устойчива при высоких температурах и
отвечает составу Zr4H.
f-фаза. (Zr2H) гексагональная
а = 3,335 — 3,339; с = 5,453 — 5,455А,
5-фаза. ZrH. Атомы Zr образуют кубическую гранецентрированную решётку;
Н внедрён в мсждуузлия
а = 4,765-4,768.
г-фаза. ZrH2. Тетрагональная гранецентрированная решётка
а = 4,644; с = 4,440; с\а = 0,894.
В соответствии с нашей точкой зрения (см. § 104), проявляющиеся
© работах Хэгга попытки подчинить состав фаз закону Дальтона являются
лережитком старой традиции. В этом смысле мы рассматриваем как условную
структуру «ZrH2» по Хэггу.
Очень важен вопрос о том, сохраняются ли в решётке ZrH2 молекулы
водорода («пары атомов»), т. е. структурные узлы р = Н2, как это принимает
Хэгг и воспроизводят другие авторы, и каково распределение зарядов
в решётке. Нам кажется, что образование ZrH связей за счёт s-электронов
с разрывом «пар» атомов Н более вероятно, поскольку эффект
значительного расширения решётки металла при внедрении водорода вряд ли может
быть компенсирован с энергетической стороны силами, которые могут
возникнуть между молекулами Н2 и кристаллом Zr.
Тетрагональность решётки ZrH2 вполне может быть обусловлена
образованием структурных узлов а = (ZrH2).
В связи с этим отметим, что нашей точке зрения соответствует весьма
различная магнитная восприимчивость Ti и Ti—Н, соответственно Zr и
Zr-H (J. Ch. Phys. 9, 678 (1941)).
*) Я. С. Ум ан с кий, Карбиды твёрдых сплавов. Металлу ргиз дат (1947), Москва.
**) Б. Ф. Ормонт, Успехи химии, V (1936).
§ 139| ГАЛ0ГЕН1ЩМ. VI* ГРУППА: ХРОМ, МОЛИБДЕН, ИОЛЬФРАМ, УРАН 385
§ 138. V* группа периодической системы: ванадий V, ниобий Nb,
тантал Та, протактиний Ра
Рассматриваемые соединении этих элементов изучены в структурном
отношении весьма плохо. Это относится и к структурам молекул
максимальной и не максимальной валентности и к кристаллам, в частности, гало-
генидов. В какой мерс элементы V группы и их соединения остались
вне поля зрения исследователей, видно из того, что в отдельных томах
SB, например III, соединения V и Nb в предметном указателе вообще
отсутствуют. В 1945 г. найдена тетраэдрическая структура у молекул
VC14 (J. Am. Ch. S. 67, 2019).
Система Nb И была изучена Я. С. Умапским, ЖФХ, XIV № 3 (1)
(1940). В этой системе обнаружена структура NbH, производная от
кубической центрированной элементарной ячейки (я =3,42) с ромбической
деформацией.
В системе Та — К обнаружены:
a) Та2Н гексагональная структура, плотная упаковка атомов Та.
а =3,094; с/а =1,591, атомы Н занимают тетраэдрическис междуузлия;
b) ТаН -ромбическая, производная от кубической центрированной,
«=3,41; 6=3,38; с =3,43 (см. Я. С. Умапский, Карбиды твёрдых
сплавов, Металлургиздат, 1917).
§ 139. VI* груииа периодической системы: хром Сг, молибден Мо,
вольфрам W, уран U
JJ отличие от хрома Mo, W и U образуют высшие летучие га л ore виды
MoFe, WFe, UF„ Will., МоСЛ6.
И |)азличных исследованиях их структуры описаны по-разному.
Первоначально было найдено, что молекулы А\ь не октаэдрические. WFe и UFe
(и, вероятно, MoFJ— ромбические бишт|)амиды с соотношением oceii
J : 1,12 : 1,22 и кратчайшим расстоянием
W-F=1,(S4; U —F =1,78.
Другие авторы нашли, что WCle имеет октаэдрическую конфигурацию
W — С1=2,2б. Пыркип и Дяткина (1946) (стр. 429 1. с.) не находят
теоретических доводов в пользу эквивалентности или неэквивалентности
0 связей центральных атомов Mo, W, U и отмечают, что этот вопрос
требует дальнейшего исследования.
В книге Паулинга (19-45), стр. 177-178, также указывается, что
вопрос не может быть решён a priori, исходя из тео])стических
соображений, и что необходимы дальнейшие экспериментальные исследования.
МоС15 образует тригоиальную бииирамиду с атомом Мо в центре
н расстоянием Мо — О = 2,27.
Переходя к структуре кристаллических фаз, образованных галогени-
дами элементов VI* группы, надлежит отметить, что и в этом случае
количество исследований явно недостаточно. Если некоторые из галогенидов
хрома исследованы, то галогсииды. молибдена и вольфрама в большинстве
ещё ожидают своей очереди. Лишь в самое последнее время появились
в печати сообщения о структурах галогенидов (и гидридов) \рана. Ещё
менее повезло гидридным фазам в этих системах. Очень часто
исследование гидридных фаз имело место в связи с выяснением структур
металлов, выделенных электролизом при высоких плотностях тока, как это,
очевидно, имело место и случае Н-хрома, (см. стр. 289).
38tt
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. V
1. СгС18
Решётка гексагональная слоистая Hs.
Координация 0\2 = 0\/_.
тип tf§/2 = tf§/z-sezx)4.
Пространственная группа /)*—/73 2.
Z = 6.
Сгк.ч. = 6С1 (тригонально деформированный октаэдр), С1к.ч. = 2Сг (^).
Вещество i а
CrCl, J 6,00
[ I
с
17,3
с/а
2,88
0 12 3 4 5 6
i i i i i i i
Рис. 241.
О Сг
OCL
Атомы С1 образуют почти кубическую плотную упаковку. Атомы Сг
образуют слои, с двух сторон ограниченные слоями С1; см. стр. 395.
§ 1391 ГЛЛ0ГКШ1ДЫ. VI* ГРУППА: ХРОМ, МОЛИ1ЩЕН, ЬОЛЬФРЛМ, У1>ЛЫ ,487
2. СгВг,
Решётка ромГнюдрическан слоистая Bs.
Координация 0\'1='0\/_.
Тин /f£/z-SBZ)0, (BiJ,).
11 ростра hci ионная группа С%— R'S.
Z - 2.
Ок. ч. 6Вг((9); Вг к. ч. =2Cr(Z).
ОО
Вещество
(>Вг3
7,оо
Рис
--
. чи. ьг
52° :г,'
j Примечание
Типы /л)5 и /J04 весьма
близки (см. стр. .495)
3. UF4—исследование начато в 1946 г. (см. J. A. Ch. Soc. 68, 1969 (1946).
A. UCI4 —решётка кубмческа51? а -- М,57? SB II 306.
388 ч. и. структуры неорганических веществ [гл. V
5. UCI„ UBrs
Решётка гексагональная //.
Координация (?) 9/у.
Тип #9/J/ «UC1,».
Пространственная группа Сел-- С63///г.
Z = 2. М к. ч. == 9Х.
Вещество
UC13
UBr3
а
7,926±2
с
4,:Л2±3
4,432±2
с/а
0,59,
0,56
Примечание
С Acta Ср.
1,255 (194&)
1
<>. UJa
Решётка орторомбическая OR.
Координация 8/х.
Тип ORsfx- «PuBr8».
17
Пространственная группа D^u — Canm.
Z - 4. М к. ч. = 8Х.
Вещество i a
UJ3
13,98±tf
6
4,31±3
с
9,99±5
Примечание
См. Acta Cr. 1,255
(1948)
Исследованию гидридов урана посвящен ряд вышедших на последние
годы в свет публикаций. Сюда относятся прежде всего статьи Рэндла
(1947), а также ряд статей в Nucleonics, тетради 1—3 (1949). Согласно
этим работам, в отличие от других гидридов металлов с ds-валентными
электронами, гидрид и дейтсрид урана, повидимому, представляют собой
соединения с узкой областью гомогенности, т. е. состава, близкого или
точно равного MII3 (UH3, UD3).
В тех же исследованиях указываются равновесные упругости
разложения гидрида и дейтерида урана, причём оказалось:
для UH3 lg />н2 = - ~- + 9,28,
для UD, lg 7^^-^ + 9,43, АЯ- -30,8—.
3 ° Ul Г ' ' ' ' моль
Если раньше структурный тип р-вольфрама стоял особняком, то позже
к нему добавился совершенно тождественный тип V3Si (стр.582). С
появлением цитируемых работ такое же расположение атомов металла
регистрируется и в гидридах урана.
5 \'Л9] ГЛЛОГЕН1ЩЫ. VI* ГРУППА: ХРОМ, МОЛИПДЕН, ВОЛЬФРАМ, УРАН
389
группа
ol
- 10,92; UD
СН„ UD,
Решётка кубическая С
Координация?
Тип С-SB?
11ространственная
(Ош и TlT).
UH, аю = 6,631; <
аш = 6,620.
Координаты атома U 2 а — (000),
С/ '/•> V*)-
6C=V/<072');('/27.0);(0V274);
(V* о »/s); (7* V. 0); (0 7» '/,)•
Атомы U сидят попарно в
центре граней параллельно рёбрам
куба [см. Rundle, J. Am. Ch. Soc.
69, 1719 (1947), Nucleonics,
тетради 1 и 2 (1949)].
Структура гидрида и дейтерн-
да U представляет
исключительный интерес как пока единственная описанная в литературе структура
гидрида dsr или d/s-эле мента, возникающая не по принципу структур
Рис. 243.
Рис. 244.
внедрения (т. е. тому же структурному мотиву, что и металл), по
имеющая структурный мотив, отличный от U.
VI** группа периодической системы: Np, Pu, Am, Cm
8. NpCl3, siNpBr,, PuCl3, AmCI,
Решётка гексагональная Н.
Координация (?) 9/у.
Тип Я,/у (UC1,).
Пространственная группа Сел- -С63///г.
Z - 2. М к. ч. = 9 X.
Вещсстио
NpCl3
aNpBr8
PuCl,
AmCl,
a
7,4O5±10
7,У17+«5
7,.Ч80±1
7,37 ti
с
• 4,£7Z±5
4,H82£:c5
4,238±/
4,'24±/
c/a
0,572
0,55,
0,57
(1,57
Примечание
См. Acta Cr.
1,255 (1948)
1
1
М90 ч. и. структуры неорганических веществ |гл. V
9. (ШрВг3, NpJ3, PuBr8, PuJ3, AmBr3, AmJ3
Решётка орторомбическая ОН.
Координация 8/2/.
Тип ORs/u — «PuBr3».
Пространственная группа Dzh — Ccmm.
Z - 4. М к. ч. = 8Х.
Вещество
pNpBr,
Npj8
PuBr3
PuJ3
I AmBr3
AmJ3
a
14,00±б
12,62±5
14,00±6'
12, 6±/
14,0±/
Ь
4,11-t^
4,29±<3
4,09+5
4,29-L3
4,10±4
4,30-f-S
с
9,ir>i4
9,93-1-5
9,13±4
9,90±5
9,10±5
9,9±7
Примечание
См. Acta Cr.
1,255 (1948)
§ 140. VII* группа периодической системы: марганец Мп, рений Re
1. MnF2
Решётка тетрагональная Тк,
Координация Ojt.
О Мп
Tire-. 245.
Тип Го/£ —SB C4 (рутила).
Пространственная группа D^h — Pi/mntn.
Z — 2
Мп к. ч. = 6 F(0); F к. ч. .-. 3 Мп(/).
Вощостпо
MnF2
а
4,87
С
з,з0
с/а
0,678
d
Мп—F
ОМ
2,11 39,1
1401
ГЛЛОГЕНИДЫ. VII» ГРУППА: МАРГАНЕЦ, РЕНИЙ
391
2. МпС12
Решётка ромбоэдрическая, слоистая R .
Координация Oft.
Тип Л^-SB С19 (CdCl,).
ftoJ?
Рис. 246.
Пространственная ipvnna Z)L — R3m.
Z=l.
Мп к. ч.=6 CI (О); CI к. ч. = ЗМп(;).
О Мп
OCL
Вещество
MnCl,
а
а
6,22 [ 33*30'
? 1
392
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
|ГЛ. V
3. Мп (0Н)2, МпВг», MnJ2
Решётка гексагональная, слоистая //" .
Координация ()\t.
Тип Hs6jt SB-C6 (CdJ2).
Пространствеиная группа Вза — СЗш.
Z = l. Mn к. ч. = 0 (ОН)(О); (ОН) к. ч. = ЗМп(*).
Рис. 217.
О Мп
Оон
Вещестпо
1 Мп (ОН),
MnJJi-a
1 MnJ>
а
3,^4
:'.8*0~3
4,10
с
4,68
С188
6,82
с/а
1,401
1,62
I 1,64
Мп -О
2,30
-
ОМ
4Г..2
-
— |
S 141]
ГАЛОГЕННДЫ. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ
393
§ 141. VIII* группа периодической системы: лселезо Ге, рутений Ru,
осмий Os
Среди элементов VIII* группы летучие галогениды образуют Os
и Ни: OsFe, RuF6, OsCl4, RuCl4.
Из них особенно интересны (см. § 1)1) структуры OsCl4 и RuCl4,
однако найти ссылки на исследование их нам не удалось.
Структура OsFe, новидимому, отвечает конфигурации закрученный
куб (антипризма). См. также J. Am. Ch. Soc. 65, 329 (1943).
О
1. FeF, VQJ
Решётка ромбоэдричес- С*)
кап R. _ __ ^
Координация 0/2 = 0\/_.
Тип %/z —SB D0M.
Пространственная группа
D*(, — R3c.
Z = 2. _
Fe к. ч. —6F(0) (триго-
нально
деформированный октаэдр); F к. ч. ==
= 2Fe(Z).
Bemecmo
FeF8
a
С, ЗУ
а
58°0
d
2,07
о
L
2 3
_| 1_
О Fe
Of
Рис. i!'iS\
SB II 34. Атомы F образуют слегка деформированную гексагональную
упаковку. Fe-атомы занимают часть октаэдричоских междуузлий, образуя
примерно простую кубическую решётку (чему соответствовало бы а-=60).
Ср. с рис. 225, заменив в нём оба типа узлов па Fe.
394 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
2. FeCl,
Решётка ромбоэдрическая, слоистая Ks.
Координация 0\2 = 0\/_*
Тин Д^-SB i)06.
Пространственная группа Cli — R3. Fe к. ч. = 6 С1 (тригонально
деформированный октаэдр). С1 к. 4. = 2Fe(^/).
ILLLU о О
Fe CL
P*fc. 249.
Вещество а
FeCl, 1 6,69
i
52°30'
Примечание
8ВП2Г) 1
§ 141] ГЛЛОГ^НИДЫ. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ 395
Тип /)05 близок типу D0A (например СгС13 § 139), они отличаются
упаковкой атома галогена. В случае D0A последняя является слегка
деформированной кубической, в случае D05—гексагональной. Кроме того, */2
атомов металла двух соседних слоев в структуре /Ю5 лежат по вертикали
друг над другом, тогда как в случае, например, СгС13 они взаимно
смещены и сторону. (Сравни CrCl и СгВг3, стр. 386 и 387). Добавив ещё
1 атом Fe в (000), получим тип Сб.
3. Рс(0Н)з
Электронографическое исследование плёнок Fe(OH)3 обнаружило
кубическую гранецентрированную решётку с аш = 5,70, при величине
кристаллов >200А (J. Ch. S., L (1937), стр. 483).
4. FeF,
Решётка тетрагональная Т-.
Координация Oft
Тип Тоц —SB СЬ (Ti02, рутил).
Пространственная группа Dl^ — Pkfmnm.
Fe к. ч.=6 (О); F к. ч. = 3(/)-
О Fe
Рис. а.ОО.
Вещество
FeF2
а
4,83
с
:*,36
с/а
0,696
d
Fe—F
I
-■ч
ом
УУ,2
390
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТН
[гл. V
5. FeCl2 (RuCU)
Решётка ромбоэдрическая слоистан h .
Координация 0/t.
Тип Rs0jl -SBC19(CdCl8).
Пространственная группа Dld — RSm.
Z = l
Fe к. ч. = 6 (i>)\ G1 к. ч. = 3 (/).
Рис 251.
О Fe
Oct
Вещество
FeCl,
1 1
а \ з I
6,19
3°33,5'
SB 11 245.
$ 1411 ГЛ.ЮГКПИДЫ. VIII* П'УПНЛ: ЖЕЛЕЗО, ГУТЕНИЙ, ОСМИП 397
4i. Fe(OH)2, FeBr4, F<>.T,
Решётка гексагональная, слоистая Hs.
Координация 0/t.
Тин Hi., —SB Сб.
Пространственная группа Dm — С'Лш.
2 = 1.
Fe к. ч. = 6 Oil (О); (ОН) к. ч. , 3Fo(/).
О Fe
О ОН
Вещество
Fe (ОН),
1 Felir,
1 FeJ*
а
*.**
3,7'i
4,04
с
4/.7
G,17t
0.75
с/а
1,:»8
J ,65
1,67
<*
Fe — О
~
ОМ 1
40,6 1
1
— |
398 Ч. П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
§ 142. IX* группа периодической системы: кобальт Со,
родий Шь иридий Ir
|гл. V
Hh и Ir образуют легко летучие высшие галогсииды, структуры
молекул которых, однако, не изучены.
Из кристаллических галогенидов исследованы:
nrw CSJ\ r?^L
^
^
1. CoF„, RhF,
Решётка
ромбоэдрическая R.
Координация 0/2 (&JZ.)*
Тип Лб^-SB D0lt/
Пространственная
группа D*d— R3c.
Z = 2.
M к. ч. =6F(0);
F к. ч. = 2М(^/).
9^drl4
Рис. 253.
О м
Of
1 Вещество
CoF8
RhF8
a
5,30
5,34
л
57°0'
54°20'
d
2,02
1,98
Примечание [j
1
Cm. FeF3
§ 142] ГЛЛОГЕНМДЫ. IX* ГРУППА: КОБАЛЬТ, РОДИЙ, ИРИДИЙ 399
2. Со(ОН)л.
В SB I 771 отмечено вскользь, что Со(ОЛ)., якобы всегда даст тс же
линии, что и Со03.
3. 1гС13
В работе Ханауолта, Ринна и Фревела (Ind. Enpr. Chem., Anal*
Edit. X, 457 (1938)) имеются данные о структуре IrC!s (№ 400, стр. 487)-
4. CoF,
Решётка тетрагональная Г.
Координация Ojt.
Тип Го// — SBC4 (рутил).
Пространственная группа Dlh — P&rnnm.
Z-2.
Со к. 4.=6F(0); Fk. ч.==ЗСо(/).
О Со
Рис. 25'i.
Вещество
а
CoF» | 4,70
| 1
с
cja
3,19 1 0,679
d
Со—F
2,0
ОМ 1
35,2 I
400 ч. н. структуры неорганических иещести (гл. V
S. CoCI2 (RhCl2, IrCI2)
Решётка ромбоэдрическая слоистая Rs.
Координация 0\t.
Тип Rfi,t — SB C19(CdCls).
Пространственная группа Dld -Шт.
Z = l.
Со к. ч. = 6 (О); С1 к. ч. = 3 (£).
О Со
Рис. 255. Q CL
Вещество
СоС13
а
6,13—6,16
а
33°30'
33°26'
Примечание 1
Для гексагональной ячейки
а=;>,5'|Г>; с=17/*4; с/а = '*,92
§ 142]
ГАЛОГЕНИДЫ. IX' ГРУППА: КОБАЛЬТ, РОДИЙ, ИРИДИИ
401
G. Со (ОН),, СоВг„ CoJ2.
Решётка гексагональная слоистая IIs.
Координация Ojt.
Тип Я^-SB Сб.
Пространственная группа Л'ц — С'Зги,
Z = l.
Со к. ч. = 60Н(С>)
(ОН) к. ч. = ЗСо(*)
Рис. 256
1 [Вещество
I Со(ОН),
I СоВг,
| GoJ2
а
:»,1734.3
3,08,.
3,%
с
4'G40±*
0,12
0,05
с/а
1,402
1,00
1,08
d
2,10
—
—
ОМ
40,40
— 1
— |
402 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
§ 143. X* группа периодической системы: никель Ni, палладий Pd,
платина Pt
1. PdF3
Решётка ромбоэдрическая Д. j —.
Координация 0/2--=#/,/.
Тип A^ — SBDO,*.
0/- l~ r - См* рИС' 253'
Пространственная группа D^ — Ric.
Z = 2.
Pd к. 4.=GF(tf); I 1
F к. 4. = 2Pd(^).
Решётка PdF3 аналогична RhFs. Атомы F образуют слегка
деформированную гексагональную упаковку. См. стр. 398.
2. NiF2, PdF2
Решётка тетрагональная Тк.
Координация 0/t.
Тин 7^,-SB С4 (рутил).
Пространственная группа Вм — Р'ь/тпт.
Z-=2.
Со к. 4. = 6F (О); F к. ч. = 3 Со(*).
ОМ
Рис. 257.
I Вещество
Ш2
PdFa
а
4,7Х
4,93
с
3,38
с/а
0,66
0,687
М—F
2,0
2,15
ОМ
34,5
ах
5,8
§ 143] ГАЛОГЕНИДЫ. X* ГРУППА: НИКЕЛЬ, ПАЛЛЛДИП, ПЛАТИНА
403
3. NiCI2, NiBr2, NiJ2 (PdCL, PtCU)
Решётка ромбоэдрическая слоистая Rs.
Координация Ojt.
Тип /?o7_,-SB С19 (CdCl2).
Пространственная группа D\.j—RSni..
Z = \.
Ni к. ч. = 6Х(С); CI к. ч. =• 3Ni (/).
О Ni
Ox
Рис.. 258.
1'ещестио
NiC!3
NiBr.,
NiJa
a
С ,12
G,4G.
G,92
^
:i:i°:to'
:w"'20'
:*2°4u'
1
i
3'7V>±7
j 3>»'W
(0
18,30±4
19,6Я±4
(c/a)
i
4,\>:J
Г>,04
c.r
5,25
0,3G
404 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
(гл. V
4. Ni(OH),
Решётка гексагональная слоистая IIs.
Координация 0\t.
Тип Я?,-SB Сб.
Пространственная группа Вц—С'дт.
Z = l.
Ni к. ч.=б;ОН(0); ОН к. ч. = 3 Ш(1).
вещество
Ni(OH)2
1 а
3,U7±o
Рис. 259.
1 с
| 4,595±4
cja
1,474
О Ni
Оон
l d
1 2,06
ОМ |
38,66
§ 143] ГАЛОГЕНИДЫ. X* ГРУППА: НИКЕЛЬ, ПАЛЛАДИЙ, ПЛАТИНА 405
5. PdCI2
Решётка ромбическая цепная ORK.
Координация r/2 — r/j/^.
Тип 0/tf/z-SB СЬО (PdCl2).
Пространственная группа ■/)*/, — Рптп.
2 = 2.
Pd к. ч.^4С1 (прямоугольник); G1 к. 4. = 2Pd(,/).
С
Рис. 260.
Вещество
PdCla
а
3,81
Ь
3,34
с
11,10
а :Ь :с
1,1407:1:3,2934
Атомы С1 образуют вокруг Pd почти квадратные
координационные сферы. Углы между диагоналями 87°. Прямоугольники (PdCl4)
соединены в пояса (цепи) таким образом, что каждый прямоугольник
имеет по 2 общих атома С1 с каждым из соседних квадратов. Пояса
вытянуты вдоль оси Ь; их плоскость повёрнута по отношению к осям а
и с на углы 65°35', соответственно 24°25\ Одна сторона прямоугольника
(вдоль оси цепочки) равна 3,34 кХ, другая (перпендикулярно к оси
цепочки) 3,18 кХ.
На рис. 260 показаны три элементарные ячейки, прорезаемые
бесконечными плоскими, как линейка, поясами [PdClJ.
406
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. V
§ 144. I* группа периодической системы: медь Си, серебро Ag,
золото Аи
1. CuF2
Решётка кубическая С.
Координация С/Т.
Тип С с it -SB CI.
Пространственная группа
Tl-РаЗ.
2 = 4.
Си к. 4.:=8F(C).
F к. ч.=4 Си (Т).
Вещество ....
«*
d
Си—F
ОМ
Их
CuF,
5,40.
2,34
39,5
4,24
2. СиВг2
Решётка моноклинная
цепная Мк.
Координация г/2 = г//^.
Тип M?lL.
Пространственная группа С?
или Сг/|.
Z = 2. Сик.ч.-:
- 4Вг^)(2,40А) +
+ 2Вг (3,18 А);
Вгк.ч. = 2Сп(2,40 А).
Угол между диагоналями
прямоугольника равен 87°30\
ОСа
О
Рис. 262.
i
Вещество
СиВг2
a J Ъ
7,К±2
3,46 ±1
с
7Д8 ± 1
Р
Шэ15'
Примечание
J. Am. Ch. Soc.
69, 886 (1947)
CuBr2 обнаруживает цепную структуру, близкую к PdO, но с иной
ориентировкой цепей. Цепи плоски и лежат почти в плоскости 001.
ГАЛОГЕНИДЫ. I* ГРУППА: МЕДЬ, СЕРЕБРО, ЗОЛОТО 407
3. CuF, CuCl, CuBr, CuJ,
YAgJ
Решётка кубическая С.
Координация Т/Т.
Тип Стгг—SBB3
(сфалерит).
Пространственная группа
Ta-F43m.
Z = 4.
Си к. ч. = ЩТ).
X к. ч. =4Си (Г).
О
Рис. 263.
Вещество
Си
Aff
ОМ
d
ОМ
I-1
4,255
1,84
10,3*)
—
С1
5,41
2,34
39,6
—
Вг
5,68
2,46
45,8
—
J
6,05
2,62
55,4
6,473
2,801
67,8
I) последние годы, в дополнение к большой группе советских
исследований, посвященных дефектным решёткам и колебаниям химического
состава фаз, появились заслуживающие внимания публикации зарубежных
авторов, в частности по CuJ. В работе Маурера J. Ch. Phys. 13,321 (1945)
показано, что CuJ находится в равновесии с парами иода, причём 1 г CuJ
поглощает 10 3 г избытка иода. При 132,1°С концентрация
«адсорбированного» (? В. О,) иода изменяется как корень квадратный из унругостей
пара иода в равновесии с образцом. К сожалению, рентгенографических
исследований проведено не было.
Как и следовало ожидать (см. § 104), состав М1Х1 и в этом ряду
является скорее данью классическим традициям, чем реальным фактам.
*) Был найден (Z. an. Ch. 210, 269 (1933)) исключительно большой удельный г?ес
7,07, что при молекулярном весе 82,56 даёт МО = 11,6 см н ОМ=19,3 (кХ)3.
408
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. V
4. AgF, AgCl, AgBr
Решётка кубическая С.
Координация О/О.
Тип Сою — SB Bi (каменной соли).
Пространственная группа 0\—Frn3m.
Z = 4.
Ag к. ч. = 6Х (О).
Хк. 4. = 6Ag (О).
о^
^ш
ЬЗ-&*
ел
6^
ь^
^
о
\У'
у
о
ь^
с\
ь^^—ь^
Рис. 264.
Олд
Вещество
Ag
t
а
d
ОМ
F
Комн.
4,92
2,46
29,8
С1
Комн.
5,54
2,77
42,5
Вг
Комн.
5,76
2,88
47,8
Дефекты решётки в AgCl исследовались в работе Nat. 157,19(1946);
они, повидимому, распределены в решётке параллельно осям и плоскостям
симметрии (?) кубической элементарной ячейки.
5. a AgJ
Решётка кубическая, тип SB В 23, Z = 2,aw=^ 5,034.
/1 1 1Л
2J в (000) (уут)» 2Ag статистически:
■ (>)о,(-Но)о; и G4o)o,(!4 °)c,;
III (xx — jO.f^TjCV (xx yjO c x — у • Структура деления, см. § 102.
Ag I k.~4. = 2J(2,52);
Ag II к. h. = 4J (2,86); Ag III к. 4. = 3J (2,67).
Расстояние Ag—Ag—от 2,52 до 3,21.
§ 144] ГАЛОГЕНИДЫ. I* ГРУППА: МЕДЬ, СЕРЕБРО, ЗОЛОТО 409
6. CuH, pAgJ
Решётка гексагональная Н.
Координация Т/Т.
Тип Яг/г — SB2?4 (вюрцит).
Пространственная группа С^—С^тс.
Z = 2.
М к.ч.=4Х (Т);
X к.ч.-4М (Г).
Рис. 265.
1
81
Cull
а
с
с/a i
d
н
О 01)
4,f,l
1,Г>9.
Г), 38
?JAg
*
с
с/а
d
J
1
4,580
7,49,
1,вз7
2,78—2,83
В литературе описан был также кубический гидрид меди СиН
(SB II 243), структура внедрения в исходный тип Си, Z = 4, а = 4,33.
410
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. V
7. Ag2F
Решётка гексагональная, слоистая Hs.
Координация —1\0.
Тип Я^-SB С6(?) (антн CdJ2?).
Пространственная группа Dla—СЪт.
Z = l.
OAq
Of
Рис. 266.
Вещоство
Ag3F
а
3,0
с
5,74
с/а
1,91
§ 1451 ГАЛОГЕНИДЫ. II* ГРУППА: ЦИНК, КАДМИЙ, РТУТЬ 411
Структуры галогенидов Ag и, в частности, AgJ представляют большой
теоретический интерес.
Согласно литературным данным, AgJ триморфен:
TAgJ ^Г (MgJ Zl a AgJ
SB ВЗ (гп = 137°С) SB Ж (*П«176°С) SB £23
(кубическая) (гексагональная) (кубическая)
По мнению некоторых авторов, в решётке a AgJ серебро ведёт себя,
как жидкость. С такой формулировкой мы не можем согласиться.
Статистическое распределение части атомов в кристалле по определённым
позициям не отвечает свойствам жидкого состояния.
♦
8. Галогениды золота
Надёжные данные по галогенидам золота, представляющим, вообще
говоря, теоретический интерес, к сожалению, отсутствуют.
§ 145. II* группа периодической системы: цинк Zn,
кадмий Cd, ртуть Hg
Изучался пар ZnJ2, CdJ2. Расстояние Zn - J = 2,433±2; Cd-J --= 2,596±5.
Кристаллы рассматриваемой группы веществ изучались неоднократно.
Электронографически найдены в парах расстояния Hg —X в HgCl
(2,20А), HgBra (2,40A) и HgJ2 (2,55А).
1. ZnF2
Решётка тетрагональная Тк.
Координация O/t.
Тип Го//-8ВС4 (рутил).
Пространственная группа D& — Pifmnm.
/j = Z.
Zn к. ч. = 8F; F к. ч. - 4Zn.
Рис. 267. _
О Zn
1 р !
Негцество i а
ZnFa j 4,72
с с/а ОМ
1 ! |
3,1. : 0,66.
1 \ о
35,U
412 Ч. П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕШЕСТН Ггд. \*
2. ZnCI2, CdCI2, CdBr2 (CdJ2?)
Решётка ромбоэдрическая слоистая Rs.
Координация O/t.
Тип R^it — SBC19 (хлористого
кадмия).
Пространственная группа Вм — ИЗт.
Z=l.
Gd к. ч. = 6Х(0); X к. ч. = 3Cd(/)-
О М
Ох
Рис. 208.
Вещество
ZnCl2
CdC]2
CdHr2
CdJ,
a
6,32
6,35
?
a
34°40'
36°40'
?
Примечание
Атомы галогена образуют
плотную шаровую упаковку по
кубическому закону: на рис. 268
отчётливо видно, что большие шары(Х)
первого и четвёртого слоев (по
вертикали) лежат друг над другом.
См. хотя бы 3 атома X переднего
плана (жирные кружки), а также
MgCl2.
§. 145] ГЛЛОГЕНИДЫ, II* ГРУППА: ЦИНК, КЛДМИП, РТУТЬ 413
3. Zn(OH)a
Решётка
ромбическая OR.
Координация Tj /_.
THnOi?T/z-SBC31.
Пространственная
группа D* —
Р2 2 2 .
Z = 4. Zn к. ч. -
4(ОН)
(тетраэдр); (ОН) к. ч.=
2Zn(/).
Рис. 269.
Вещество
Zn(OH),
а
5,16
Ь
8,53
с
4,92
а :Ъ : с
0,605: 1 :0,577
а 1
Zn-O
1,95 1
Интересный пример пространственной вязи из (Zn(OH)4) -тетраэдров,
имеющих общие вершины, ср. с Со(ОН)3 и др.
4. CdPt, HgP,
Решётка кубическая С
Координация куботетраэдри-
ческая С/Т.
Тип Cc/r-SB Ci.
Пространственная группа
Z = 4.
М к. 4.=8F (С);
F к. ч.=4М (Г).
Вещество
CdF3
л«
5,40
5,54
d
2,34
2,398
ОМ
39,3
42,5
Рис. 270.
414 Ч. II. СТРУКТУРЬ^НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
5. CdJ,
Решётка гексагональная
слоистая Hs. _
Координация Ojt.
Тип Я^-SB Сб.
Пространственная группа
Dld-C3m.
Z = \.
Cd к. ч. = 6 J (октаэдр).
J к. ч.-3 Cd (t).
1
Вещество а
CdJ2
с с/а
*.24|м4
ОМ
1 1
1,61.{ LOG,5
Примечание.
Атомы галогена образуют
плотную шаровую упаковку по
гексагональному закону.
Рис. 271.
Центральный атом J лежит вне треугольной координации ннш сферы.
Па рис. 171 отчётливо видно, что большие шары (J) первою и третьего
слоев (по вертикали) лежат друг над другом.
6. CdBr2
Решётка гексагональная Н.
Координация Ojt,
Тип Hf)ft —«переменная» структура с чередованием типов С6 и С19.
Z - 2.
§ 145] ГАЛОГЕНИДЫ. II* ГРУППА: ЦИНК, КАДМИЙ, РТУТЬ 41£>
Вещество
CdBr2
1
а 1 с
2,30
5,23
с/а
Примечание
Помимо обычной структуры CdBra
(тин CdCl2) образуется «п^рем&ннан
структура», невидимому, подобная
типу С27 (CdJ2) (upii растирании
вещества в ступке). При нагревании
происходит обратный процесс
7. CdJ2
Решётка гексагональная
слоистая Hs.
Координация O/t.
Тип Hl„-SuC21.
Z = 2.
Р ^
Вещество
GdJa
а
4,24
с
1*,67
С1
'J
& ё t
4 %
р « щ_р
Рис. 272.
j^ l
л
По сравнению с типом С6, ось с увеличена вдвое; вышележащий слой
повёрнут по отношению к нижележащему на 60° вокруг проходящей
через (2/3 1/3 0) оси, перпендикулярной слою. Тогда как в типе С6 атомы
галогена упакованы но гексагональному закону, в типе С19 — по
кубическому и в типе £27 атомы галогена образуют плотную шаровую
упаковку с чередованием слоев по гексагональному и кубическому законам.
Вопросы строения галогенидов кадмия, в частности CdJ2, детально
изучались советскими исследователями. Помимо работ 3. Г. Пинскера
и его сотрудников (см. § 79), следует назвать работы Г. С. Жданова
и его сотрудников, см. ЖФХ XVIII, 425 (1944).
На рис. 272 показано схематически различие описанных в литературе*
структур CdJ2 по Г. С. Жданову, Л. Л. Смирновой и А. X. Брегеру,
пришедшим, в результате тщательной рентгенографической проверки
структуры образцов CdJ2, полученных кристаллизацией из раствора и из пара
к выводу, что в изученных им кристаллах наблюдалась структура с а = 4,24,,
б» = 13,7 Z = 2, т. е. тип С 27. Нисколько не ставя под сомнение
результаты исследования именно данных кристаллов, мы считаем, что
изменением условий кристаллизации несомненно можно получить кристаллы и
других типов. Поэтому общий вывод 3. Г. Пинскера, (на основе его работ
с Л. И. Татариновой, В. А. Новиковой и др. по галогенидам кадмия) что
в зависимости от метода приготовления возникают или могут возникать,
разные структуры (С6, С19, С27 и т. п.), мы считаем правильным. Он
целиком отвечает нашей общей установке в данном вопросе (см. § 101 и УФН
XVI, 1001 (1936))—это относится ко многим структурам различных веществ,
С экспериментальной стороны в отношении данных соединений это
подтверждается также наблюдениями Хэгга (С. А. 39, 2018 (1945)).
416 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ (гл. V
8. HgCI,
Решётка ромбическая цепная
ORK.
Координация 0/2 + 1.
Тип 0fl£/2+1-SBC25(SBII).
Пространственная /~>/\
группа DfK- Pnma. (J
Hg к. ч. =6СЭД;
С1 к. 4.=3Hg (0.
Z = 4.
Вэщество
HgCl,
а
4,307
0
1
•
5,936
1 2 3
i i 1
Онд
f_f Oct
Рис. 273.
с
12,667
а \Ь : с
0,726 :1:2,134
Примечание. Атомы хлора образуют шаровую упаковку, как
атомы С в GSi III или как атомы Вг в HgBr2. Атомы Hg располагаются в
октаэдрических люках, так что решётка не является слоистой. Каждый октаэдр
[HgCl]6 имеет 4 общих ребра с соседями, образуя цепи по (0.10), состоящие
из 2 параллельных простых октаэдрических цепей. Атомы С1 I образуют
плотную упаковку по гексагональному закону, атомы C1II — по кубиче-
(1 11 1 " \
-r-yz'j j- -}— г/; -77 — - ) у
соответственно для С1 I и С1 II */Г = 0,428, zj = 0,250 и ?/ц = 0,750 zu = 0.
§ 145]
ГЛ. ЮГЕНИДЫ. II* ГРУППА: ЦИНК, КЛДМ11П, РТУТЬ
417
9. HgCl2
Решетка ромбическая ORW\
Координация //1.
Тин OR^SB 6'28 (SB III).
Пространстпеннан группа D->t — Pmnb.
Z = 4.
Hg к. ч.-2С1(/), CI к. ч. = 1 IJg.
Ь^О
.8
О 1 2 3 4 5
i i i i 1 1
Рис. 274.
Онд
О
I ' i
1 Вещество I a | Ь
HgCl,
5,963 ±5
12,735 ±5
a:b:c | Hff-ci
4,325 ±о
0,4682 : 1 :0,3395
2,25
Примечание. Молекулярная решётка с прямолинейными
молекулами. SB III никаких выводов о противоречии между структурами
HgCL, С25 и С 28 не делает. Вопрос о возможности аллотропических
форм также оставлен открытым.
Судя по отдельным литературным данным, структура С28 исключает
структуру £25.
Тем самым, возможно, тип С25 исключается из списка.
10. Hg (C1, Вг)_>
В числе рассмотренных выше галогонидов ртути данный занимает
несколько особое место; как вследствие некоторых особенностей
структуры, заставляющих отнести его к ромбической эшштиоморфной гемиздрии
(вид симметрии ])), так и но формальным причинам, он относится но
своему составу к сложным соединениям; ом. § 2о8.
418 4. II.jCTPyKTyPbI НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. V
11. HgBra
Решётка ромбическая слоистая ORs.
Координация O/t.
Тип OR?/t-SB Clb.
Пространственная группа С^—Сте.
Z=4.
Hg к. ч. = 6((?); Вг к. 4. = 3Hg(t).
Hf ? О
Рис. 275.
I Вещество
HgBr,
а
4,624
Ъ
б,798
С
12 - «5
а:Ъ : с
0,681:1:1,880
Структура очень.похожа на CQ или С19. Атомы Вг образуют
шаровую упаковку как в SiC III. Атомы Hg располагаются в октаэдрических
междуузлиях так, что образуется слоистая решётка. В решётке можно
выделить прямолинейные HgBr2 молекулы.
И*.5!
ГАЛОГЕНИДЫ. II* ГРУППА: ЦИНК, КАДМИЙ, РТУТЬ
12, HgJ2 (красный)
Решётка тетрагональная, слоистая Is.
Координация S/2 = Sj^.
Тип 7\£,Z-SBC13.
Пространственная группа Z)]* — Р i/func.
Z = 2.
Hg к.' n. = 4J(.S'); J к. 4.=2HgU).
Рис. 276.
Вещество
HgJa
1 i
о с 1 с/а
1 I
4,36 12,36
2,84
d
2,78
Kj гетке^ атомов; Hg сверху и снизу примыкают сетки атомов1 J.
Каждый атом Hg имеет координационную сферу из 4 J-атомов, образующих
тетрагональный тетраэдр.
420
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТН
[гл. V
13. HgaCl2, HgfBr2, Hg2J2
Решётка тетрагональная Т.
Координация 1 -J-4/1-L4.
Тип Ti+iji+i SB #31.
Пространственна» группа вЦ~^^ч'")1>1111-
2 = 2. Hg к. ч. = (1 + 4)Х;
X к. ч. = (1 н--/j)Hg.
Рис,
Вищестио
Hg.CI,
Н^Вг2
HgiJ,
а
4,45
4,05
4,<>2
с
10,81»
ид0
11,6L
с/а
2,447
2,387
2,2 >8
Hg-Hg
2,54
2.58
2, 00
«Г
Hg-Cl
2,52
2,57
2.04
«Г
CI-CI
a,:w
:i,40
3,55
OM
103
120
Ш
В элементарной ячейке атомы Hg попарно сближены, как бы
образуя поп (группу) Hg*+.
§ 146| ГЛЛОГЕНИДЫ. Ill* ГРУППА: БОГ, АЛЮМШШП, ГАЛЛИП. 1И1ДПП, ТАЛЛИЙ 421
§ 146. III* группа периодической системы: бор В, алюминий А1,
галлий (5а, индий In, таллий Т1
Первые члены ряда образуют легко летучие галогениды.
Бор, кроме того, образует несколько различных по составу гидридов
(бораиы).
Строение молекулярных галогснидов В и А1 характеризуется
следующими данными: согласно :>лектронографичсским исследованиям,
молекулы BF3, BC13, ВВгэ плоские, треугольные, с атомом В в центре, т. е.
с валентным углом 120°.
B-F = l,32. В-С1 = 1,76А.
С другой стороны принимается, что молекулы Al2Cle, Al2Br,, Al,Je
имеют структуру, представленную па рис. 278, т. е. 2 деформированных
Рис. 278.
тетраэдра, соединённых двумя общими вершинами (атомы С1;; в таблице
приведены расстояния между атомами; в графе п показано, сколько
раз повторяется это расстояние в молекуле. (Атомы А1: слева А1,,
справа А1>; атомы галоида: верхний—Х8, вправо по часовой стрелке Х3, Х4 и
т. д.)"
Alt— A1 а
А1,-Х5
А12-Хя
АЬ-Х6
\3-Х4
Х3- X,
хь-х.
х.-х7
х3-хв
А1Х1в
3,41 ±20
2,05 ± 4
2,21 ± 4
4,77 ±15
:i, 53 ± 4
\\, 56 ± 2
2,83 ± 10
5, 4У ± 5
6,52 ± 5
А1.Вгв
Я,Ъ\)±10
2,21 ± 4
2,33 ± 4
4,<М±./0
3,72 ± 3
3,78 ± 3
:*,20 ±10
5,76±*0
fi . 86 ± 10
A1,J.
Я,2Ь±15
2,)^ 4
2,58 ± 4
5,22 - 4
4,20^ 3
4,24 ± 2
2,90 ±15
6,24 ± 15
7,5'i ±10
п
1
4
4
4
2 1
8
1
2 1
2 1
422 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
1. В2Нв, В4Н10* BRHn
По структуре диборан В2Н6 отвечает этану *), тетраборан В4Н10 — бутану,
пентаборан В5НП — иентану.
В молекулах тетраэдрические валентные углы.
В-В-1,84. В-Н = 1,28.
2. Т1С1, Т1Вг, T1J (пар)
Межатомные расстояния в молекулах галогенидов таллия:
Т1 —С1 = 2,55 ±3; Т1- Вг = 2,68 ±3; Т1 —J = 2,87 ± 3.
Структуры кристаллических галогенидов и гидридов рассматриваемых
элементов изучены неравномерно.
Кристаллы галогенидов бора мало изучены; напротив, гидридам было
посвящено несколько работ, которые, впрочем, нельзя считать
достоверными.
3. В2Нв
Решётка гексагональная молекулярная Нм или RM(?).
Координация 6(?).
Тип tff-SB £41 (этан).
Пространственная группа (см. тип АЮ).
Н к. ч.-1В; В к. ч. = (3 + 3)Н.
Вещество 1 а
1
В,Нв | 4,54
1
с \ с/а
8,69
1,914
d
В—В
1,82
ОМ
77,5
Согласно SBI 239, положение атомов такое же, как атомов Hg в решётке
типа ^410. Работа двадцатипятилетней давности и нуждается в проверке,
тем более, что и модель структуры ртути претерпела со временем
изменения; см. стр. 320.
4. В10Н19
Решётка ромбическая ОН.
Координация ?
Тип ?
Пространственная группа DZh — Cmma.
2 = 8.
Ве'щество
а
В10Н14 14,46
1 1
Ь ! с j а : Ъ : с \
20,85
5,69 | 0,69 : 1,00 : 0,27
Эта структура подвергнута критике в J. Am. Ch. S. 70, 115 (1948).
Гам же обсуждаются структуры В.Н9 и В2Н6.
*) Согласно Z. Ph. Ch. (В) 53, 85 (1944) B.,Hti не этаноподобная (D3h или D9t£),
а этиленоподобная структура. См. также Я. К. Сыркин и М. Е. /Дяткина, /КФХ XV,
'i.Vj (1(У*1) и Acta XIV. W (lfJkl).
§ 146] ГАЛОГЕНИДЫ. III* ГРУППА: БОР, АЛЮМИНИЙ, ГАЛЛИЙ. ИНДИЙ, ТАЛЛИЙ 423
б. A1F3
Решётка ромбоэдрическая слоистая Rs.
Координация t + г/2.
Тип Rf+t/2 - SB D01V
Пространственная группа D\ — Д32.
Z= 2. А1 к. ч. 3 F на расстоянии 1,70 + 3 F на расстоянии 1,89.
F к. ч. = 2А1.
. О АХ
О
Рис. 279. Структурный тип ReOs.
Вещество
A1F3
а
5,029±5
а
58°31/
ах
3,19
Наименьшая гексагональная элементарная ячейка с Z = 6 имеет
метрику а = 4,914 ± 5; с= 12,46 ± 1; с/а = 2,534. Решётка состоит из слоев А1 и F.
В каждом слое А1 каждый атом окружён 6 Al-атомами на расстоянии
4,91А. В F-слоях каждый атом F имеет*4 соседа на расстоянии 2,53Аи2 на
расстоянии 3,66 А. Расстояние слоя АГот одного из F-слоёв равно 0,88 А,
от другого —1,20 А. Расстояние двух слоев друг от друга равно 2,08 А.
Решётка близка к типу Z)ol2(FeFs). На рис. 279 показана
идеализированная кубическая решётка (угол ромбоэдра = 60°) (типа ReO,), к которой
близка решётка A1F8. Сопоставить с рис. 225 и 248.
424
Ч.Н. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. V
6. Л1С13
Решётка гексагональная Hs.
Координация 0/t, I.
Тип Яо,_,,, -SBM»,,.
Пространственная группа D\a — СЗт.
Z = l.
А1 к. ч. = 6С1 (октаэдр).
С1 I к. ч. = 3(0.
СШк. ч. = 2(/).
Ряс. 280.
Вещество
А1С13
а
3,84
с
8,51
с/а
2,45
Атомы С1 образуют кубическую шаровую упаковку. Атомы AI
ванимают часть междуузлий так, что образуются слои А1С13 как в С6- или
С19-типах, причём атом А1 октаэдрически окружён 6 атомами С11 (к.ч.
последних = 3(/)). Остальные атомы С1 II «втиснуты» между слоями А1С12
и больше удалены от АЬатомов, располагаясь в основании элементарном
ячейки. К. ч. этих атомов С1 = 2(/).
§ 146] ГЛЛОГЕПИДЫ. III* ГРУППА: БОР, АЛЮМИНИИ, ГАЛЛИИ, ИНДИЙ, ТЛЛЛИ» 425
7. A1CI,
Решётка моноклинная /1/^~л/, слоисто-молекулярная
(псевдогексагональные оси).
Координация Ojl — Oj/-.
Тип M^-SELO».
Пространственная группа С* — С2 (соответственно /)* — //322, или Db6 —
-1ГХ2).
Z = 4 (()).
Рис. 281.
Моноклинная ячейка показана пунктиром. В ней изображены не все
атомы. Её начальная точка совладает с ( -{) --" -О J гексагональной ячейки.
Вещество
АИЛ,
Гексагональные оси
а
с
с/а
I I
5,У1±2 | 17,52±<3 ! 2,97
Моноклинные оси
а
5,91±2
Ъ 1 с
1
= 10,24 ±4
6,16±2
? 1
71°2Г±3'
Примечание. dAi-ci == 2,15; 2,38; 2,58; ЙА1-А1 = 0,64. Решётка
состоит из структурных узлов молекул А12С16, образующих слоистую
структуру. Расположение Cl-атомов, как* и в D04 типа (СгС13).
426
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. у
8. В(ОН)3 см. гл. IX
9. А1(0Н)3 (гидраргиллит)
Решётка моноклинная слоистая Ms(°h
Координация 0/2 = #/</.
Тип M^-SB Щ.
.Пространственная группа Сад— P2iic.
.Z = 8. А1 к. ч. = 60 (октаэдр);
О к. ч. = 2А1.
О AL
\ *> 01234 56Г\ПЦ
Рис. 282а.
Вещество а
А1(ОН)з
Ь
8,6236±7 ' 5,0602+6
с
9,699+4
1 ]
85°2Г/
1,7043:1:1,9108
1
SBIII 38 — 39. Решётка слоистая. Атомы О образуют несколько
искажённую плотную упаковку. Каждый атом А1 окружён 6 атомами О,
§ 146] ГАЛОГЕНИДЫ._ III* ГРУППА: БОР, АЛЮМИНИЙ, ГАЛЛИЙ, ИНДИЙ, ТАЛЛИЙ 427
ооразующими искажённый октаэдр. Каждый октаэдр имеет по одному
общему ребру с обоими соседними октаэдрами. В решётке возникают
сплошные люки в кольцах из октаэдров [А106].
U£ L-LJ-LJ-L-LJ :Оон
а
Рис. 2Ч2Ь.
10. AlBr, описан в Rec. Tr. Ch. Phys-Bas 64, 275 (1945); С. А. 40, 3956
(1945). Моноклинная решётка; пространственная группа C--h —Р21с, а —
= 10,20, 6 = 7,09, с = 7,48, ,3 = 96°.
11. 1пС12
Решётка ромбическая OR.
Координация ?
Тип OR -SB ?
Пространственная группа ?
Z = 8.
Вещество
InCIa
а
6,85±4
Ь
9,64±б
с
а: Ь : с
l"»f.Vi±/J 0,710:i:lf09.J
Предполагался изоморфизм с SnCL. Надёжными данными не
располагаем.
12. Т12С13 кристаллический был исследован недавно (С. А. 41, 2947
(1947)); однако надёжные данные, не получены. Элементарная ячейка:
a = 14,3kX; с = 25,1 кХ.
Пространственная группа Z)3ad — С31с (#3с), Z=32 Т12С1§.
/<28
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. V
13. T1F
Решётка ромбическая слоистая ORs.
Координация О/О.
Тип 0/?g/5-SB£24.
Пространственная группа D^h — F пиит.
Z = 4. T1 к. ч. = 6К; F к. ч.—6Т1.
^
в
OTL
' 0 1 2 3 4 5 6 7 8 9 ЮГ)?
-*- I 1 1 1 1 1 1 1 1 1 1 V-^
Рис. 283.
На рис. 283 показано восемь элементарных ячеек.
Вещество
T1F
1
; (1
Ъ
Г>,№±5
с
6,080±<5
: i
а : Ъ : с
0,944:1: 1,107
<*
T1-F
2d =2,59
2d7 =2,75
2<г = :з,04
Ромбически деформированная решётка Co/o(NaCl), удлинённая ось с
облегчает расщепление на слои по (001). Интересный пример слоистой
решётки у фторида состава АВ.
§ 146] ГАЛОГЕНИДЫ. III* ГРУППА: ПОР, АЛЮМИНИИ, ГАЛЛИИ, ИНДИИ, ТАЛЛИИ 429
14. T1CI, ТШг, [-JT1J. Г> Г)
Решётка кубическая 6\ ^ч
Координация Сас-
TimC(:i<:-SBB2 (GsCl).
Пространственная группа
О,1,—Рт?т.
Z = l. Tl к. ч. = 8Х(С).
X к. ч. = 8Т1(С).
|Вещз-
1 ство
Т1П
Ч TlBr
1 pTIJ
гэс
2 Г)
2 Г)
яш
\, 18
d ! ОМ 1
1 1
i,:i2
t,44
62, (
7U)
Рис. 2К
15. aTU, жёлтый
(низкотемпературный)
Решётка ромбическая
ORs.
Координация 7/7 *&!.*..
Я¥*1<1 •
Тип 0Я7/7 —ЭВДЗЗ
Пространственная группа
/>2л — Агата (Спгст).
Z-4. T1 к. ч. 7
атомов J. d — 3,36 + 4с?'
( = 3,49)+ 2d"( = 3,87).
J к. ч. — 7 атомов Т1.
Риг. 285.
Вэщостбо
1 aTU
«'
коми.
а
•V2'i±3
Ъ
4,Г)7-£2
12
с
•12 4-/ 1
а : b : с
1,147:1:2,827
Слоистая структура. Наиболее короткое расстояние Т1 — J между
двумя слоями. aTU ^жёлтый) устойчив ниже 175° С.
430 ч. п. структуры неорганических веществ [гл. V
§ 147. IV группа периодической системы: углерод С, кремний Si,
германий Ge, олово Sn, свинец РЬ
Молекулы МХ4
Молекулы тетрагалогенидов и гидридов этих элементов имеют тетра-
эдрическую структуру с расстояниями М —X:
1 1 1
с
Si
Ge
Sn
Расстояние
Расстояние расчётное
Сжатие
Расстояние - . . . .
Расстояние расчётное
Сжатие
Расстояние
Расстояние расчётное
Сжатие
Расстояние расчётное
Сжатие
F
1,36
1,41
3,7%
1,54
1,81
14,9%
—
—
С1
1,7о
1,76
0
1
2,00
2,16
7,4%
2,08
t 2,21
, 5,9%
2,30
2,39
3,8%
Вг
i,9:i
1,91 1
-1%
2,31
2,32
I 2,36
1,7%
—
J
2,12
2,10
2,50
2,48
2,55
2,7%
1 2,65
2,73
3,3%
i
f
О CH2J2 и CHJ3 см. С. А. 40, 4574 (1946).
1. Si2H„ Si2Cle
В Si2H„ Si —Si = 2,32, Si — H = l,47, тетраэдрические углы 109°27'.
В Si2Cle рассчитаны были две формы симметрии:
Се (цис) и С3* (транс); оказалась удовлетворительной вторая, но
возможна и смесь изомеров с избытком трансформы.
Si -Si =2,32; Si-CI = 2,00.
Расстояние Si —Si, как в элементарном Si.
2. Ge2H„ Ge3H8
Исследование пара германов показало, что расстояние Ge —Ge = 2,41 —
-2,42, угол Ge-~Ge-Ge = 112° (1938).
Перейдём теперь к структурам кристаллов.
3. СВг4, CJ4
Согласно SBI 614 — 615, 641 — 642, СВг4 и CJ4 образуют кубические
элементарные ячейки, пространственная группа Tld, Z = l. Структура
§j!47] ГАЛОГЕНИДЫ. IV ГРУППА: УГЛЕРОД, КРЕМНИЙ,ГЕРМАНИЙ, ОЛОВО.СВИНЕЦ 431
была зарегистрирована в SBI 614, как 03 тип; по тем же данным эта
структура устойчива при t° > 46,91° С.
Кроме того, указывалось, что ниже 46,9° С устойчив моноклинный
«СВг4, пространственная группа Ch или С£л, 2 = 8, а = 12,10; 6 = 3,41;
с -10,20; р=125°3\
В 1937 г. были найдены для СВг4 и CJ4 при комнатной температуре
моноклинные элементарные ячейки (см. приведённую далее таблицу).
Эта работа опровергает цифры, приведённые в SBI.
4. pCBr4, CJ4
Решётка простая кубическая молекулярная СМ(7^ (CJ4 образует тетра-
эдрический структурный узел = а).
Координация О.
Тип <^(r)-SB03.
Пространственная группа Т^—Р^Зт.
Z=l.
СГ4-тетраэдры параллельны друг другу.
Веществ о
£СВг4
CJ4
t °С
>46,9
аи>
С—С
5,67
5,81
d
С—X
2,26
2,32
Чистота
3,54% С, 95,7% Вг
2,20% С, 97% J
Примечание. ^СВг4 устойчива при t° > 46,91°. Ниже 46,91°
устойчива, моноклинная модификация,
б. (a?) CBr4, CJ4
Решётка псевдокубическая моноклинная М.
Координация ?
Тип?
Пространственная группа ?
Z=?
Вещество
СВг4
CJ4
а
21,12
22,39
Ь
12,26
12,93
с
24,14
25,85
Р
125°3'
125*16'
43!:
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. V
6. СН4
Решётка кубическая
молекулярная гране-
центрированная
Координация кубоокта-
эдрическая К.
Тип Ck-SBOI.
Пространственная груп-
па_Г^-^43т,илиП
Р43л, или Т* — Р2г3
(жёсткое положение
тетраэдров СН4) или
пространственная
группа 0\ (свободное
положение —
вращение тетраэдра).
Z=4(CH4 = a);
а к. ч. = 12.
--+-0--
>'
Рис. 286.
Вещество t °С
СН4
— 180
аш
6,35
d
С—С
4,4У
Чистота
('иектроскопиче-
ски чистый
Примечание
Температура между
t жидкого воздуха и
t жидкого водорода.
На рис. 286 показано
положение центров
молекул СН4.
1, SiF4
Решётка кубическая См*7>.
Координация С.
Тип CgV)—SBDit.
Пространственная группа
ТЪ-ЯЗт, a = SiF4.
Z = 2.
(Вещество!
1 i
SiF4
I
V»l
fi
Si -F
1,00
(]
F -F
2,60
О ос -51F,
l'lic. 287.
5 147] ГЛЛОГЕНИДЫ. IV ГРУППА: УГЛЕРОД, КРЕМНИЙ, ГЕРМАНИЙ, ОЛОВО,СВИНЕЦ 433
Молекулярная решётка из SiF4 = a. Центры узлов а образуют
кубическую центрированную решётку. На рис. 287 показано положение
центральных атомов (Si). Положение атомов F (ориентировка тетраэдров)
недостаточно уточнено.
Оз
Рис. 288.
Вещество
1 GeJ4
aw
11,89
d
идеальное
2,57
ОМ
210
Вещество
SnJ4
аи>
12,23
d
идеальное
2,65
—2,63
ОМ
229 1
Общие замечания
Проблема образования структур кристаллов из молекул различной
формы с образованием молекулярных решёток является одной из
интереснейших в кристаллохимии. К сожалению, большинство работ в этом
направлении было сделано с# использованием в качестве объектов лишь сложных
соединений, в частности металлорганических веществ с длинными
органическими радикалами (см. гл. IX). Исследование же тетрапроизводных,
пентапроизводных, гексапроизводных и др. более простого состава, к
сожалению, продвинуто ещё очень слабо. Между тем, именно среди этого
класса веществ можно найти очень хорошие объекты для выяснения, в
частности, вопроса возможной ориентировки структурных узлов, в данном
случае тетраэдров, по отношению к осям кристалла.
434
Ч II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. V
9. GeJa
Решётка гексагональная слоистая IIs,
Координация О/t.
Тип Я?,-SB C6(CdJ2).
Пространственная группа Df^—C'-im.
Z = l.
Вещество
GeJ,
а
4,13
с
6,79
Рис. 289
с/а
1г64
<*х
5,37
OGe
Оз
•
d
Ge-J
2,94
Примечание
Ge -ион
г = 0,98 А
§ 147] ГАЛОГЕНИДЫ. IV ГРУППА: УГЛЕРОД,КРЕМНИЙ, ГЕРМАНИЙ, ОЛОВО, СВИНЕЦ 435
10. SnCl2
Решётка ромбическая OR.
Координация х.
Тип ORx-SB?
Пространственная группа?
Z = 8.
Вощегтои
SnCl2
*°С 1 а
1
коми.
G,6L_h3
Ь
Ъ№±4
с
У,98±^
а : Ъ : с
0,708:1:1,069
11. £PbF,
Решётка кубическая С.
Координация С/Т.
ТипСс/г-SBCl.
Пространственная группа О),—РшЗлг.
Z = 4. Pb к. 4. = 8F {С); F к. ч. = 4РЬ (Г).
О Р6
Or
Рис. 290.
Веществ* ая
I
PbF, I 5,93
<*
2,5,
МО
52,1
Примечание
«; > 200°С (?)
436 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. V
12. aPbF„ PbCI„ РЬВг,
Решётка ромбическая, OR.
Координация PZ**/f, Pyi.
Тип ORpw+ir.pyi - SB С23.
Пространственная группа D*h—Pniau.
Z = 4. Pb к. ч. = 9 X (P33t).
X I к. ч. = 4РЬ (Г); X II к. ч. =5РЬ (Pyi).
Вещество
PbF2
PbClt
РЬВг2
a
„''"о
4,526
5,717
Ъ
6,4a
7,б05
8,038
с
7,6Х
9,027
9,518
°х
8,75
5,916
6,714
ОМ
47,0
54,7
13. PbJ, II форма (3. Г. Пинскер)
В результате электронографического исследования 3. Г. Пинскер, Л. И. Та-
таринова и В. А. Новикова (ЖФХ XVII, 419 (1944)) нашли, что в
зависимости от условий образования могут быть получены две формы РЫ2.
Одна из них возникает при медленной кристаллизации из водного раствора
(форма I), пространственная группа Dld, а = 4,54, с = 6,90, Z = 1. Она,
невидимому, не отличается от PbJt тип Сб. Другая (форма II) возникает при
медленной кристаллизации из пара (а = 4,54, с = 20,7, Z —3).
§ 147) ГАЛОГЕНИДЫ. IV ГРУППА:УГЛЕРОД, КРЕМНИЙ, ГЕРМАНИЙ, ОЛОВО, СВИНЕЦ 437
14. PbJ2
Решётка гексагональная слоистая Hs.
Координация Ojt.
Тип Hf)lt—SBC6~(CdJ2).
Пространственная группа D^a—R3m.
Z=l.
Вещестпо
PbJ>
а
4,59
Р
с
6,86
ис. 292
с/а
1,495
d
:*,12
О Рб
о*-
ОМ
125,2
Примечание
В разных
работах а = 4,59;
с=7,02 до 6,78
16. PbFCI см. глану IX.
438 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ f I л. V
§ 148. V группа периодической системы: авот N,
фосфор Р, мышьяк As, сурьма Sb, висмут Bi
Пар PF5 и РС15 был исследован электронографически.
Молекула РХ5 имеет структуру тригональной бипирамиды с атомом Р
в центре и расстоянием Р —F = l,59, Р — С1 = 2,05.
Пар РХ3 и других был также исследован электронографически.
Молекулы Р\3 и другие имеют строение тригоналъной пирамиды с атомом Р
и т. п. в вершине пирамиды.
Результаты приведены в таблице.
Расстояние MX .
Валентный угол
РВг,
2,23
100°
PJ8
2,52
98°
AsBr3
2,35
100°
AsJ8
2,58
100°
ebci8
2,37
104°
SbBr,
2,52
96°
SbJ,
2,75
98°
Гидриды NH3, PH3, AsH3 также рассматриваются обычно как
пирамидальные молекулы, хотя мы более склонны считать NH8 тетраэдриче-
ским. Расстояния N —Н = 1,0кХ. Валентный угол Н —N —Н=108°;
Н-Р-Н = 93°.
Строение кристаллов галогснидов и гидридов
V группы
1. NH3
Решётка кубическая
молекулярная См.
Координация примерно кубо-
октаэдрическая .К" (12).
Тип CifCJ>-SBZ>l(NH3).
Пространственная группа
a = NH,.
Z=4. к. ч. а = 12
(если принять NH8 = a, то
решётка кубическая
примерно гранецентрированная)/
0 I 2 3 4 S (~)N
1 I I I 1 V-/
Рис. 293.
Вещество i aw I N_^N
NH,
0
3,58
OM
34,1
Примечание
SB I 230 (t жидкого воздуха)
Вегард и Хиллезунд нашли при —185° С для NH3 aw — 5,2253,
для ND, aw -5,2153.
Ch.Abstr. 38, 4588 (1944).
§ А48] ГЛЮГЕНПДЫ. V ГРУППЛг'ЛЗОТ, ФОСФОР, МЫШЬЯК, СУРЬМА, ВИСМУТ 439
2. РС16, РВгБ
Г: Эти вещества, формула которых позволяет причислять их к простым
соединениям, в действительности оказываются комплексными и
рассматриваются в соответствующем разделе (гл. X).
3. AsJ3, SbJ3L Ш31
Решётка ромбоэдрическая ■*
слоистая Rs.
Координация 0/2 ^0\/_*
ТипЛ^-SBZX),.
Пространственная J ' группа
С\-№.
BiK.4. = 6 J(6>);'
J к. Ч.-2В1 (^).
Z-2.
Рис. 294.
Вещество
а а
Примечание
AsJ,
SbJ8
BiJ,
8,25
8,21
8,14
51°40'
54°15/
54°50'
Решётка состоит из слоев J (слегка I
деформированная гексагональная уна- I
ковка ь отличие от кубической и Z)04) и I
Bi. См. стр. 386, 387 ы 395.
440 ч. и. структуры неорганических веществ [гл. V
Те же структуры были описаны другими авторами с помощью
гексагональных элементарных ячеек с Z = 6.
Вещестно
AsJ8
>bJ8
BiJ3
а
7,18;±3
7,46
7,49
С
21'394
20,89
20,67
с/а
2,977
2,798
2,758
Интересно, что объём элементарной ячейки в AsJ3, SbJ3 и BiJs
мало меняется, ибо хотя а растёт от AsJ3 и BiJ3,—период а уменьшается.
4. BiF,
Решётка кубическая С.
Координация С + 0/Т,0.
Тип Cc+0/r.o-SB Z)0s.
Пространственная группа
01 — Fm3m.
Bi к. ч. = 8 FII (2,56 куб) +
• +6F 1(2,93 октаэдр);
FI к.ч. = б Bi (октаэдр)(2,93);
FII к. 4. = 4Bi (тетраэдр)
(d = 2,56). Z-4.
Вещество | aw
1 |
BiF,
5,853
5
J
о
Рис. 295.
Как указывает SBII 23, решётка BiF3 образуется из структуры CaF2
(тип С1) в результате внедрения третьего F-иона в центр элементарной
ячейки (г/2 7г 7г)- Это неверно. Помимо F-иона, внедрённого в центр элемеитар-
12
ной ячейки, дополнительно — F-ионов надо разместить в середине рёбер
последней (ср. с рис. 290).
Кроме того, можно представить структуру BiF3 как возникшую
из элементарной ячейки NaCl (тип 1?1), центры 8 октантов которой
дополнительно заняты F-иона ми (см. § 63).
§ 149] ГАЛОГЕНИДЫ. VI ГРУППА: КИСЛОРОД, СЕРА, СЕЛЕН, ТЕЛЛУР, ПОЛОНИЙ 441
§ 149, VI группа периодической системы: кислород О, сера S, селен Ser
теллур Те, полоний Ро
Структура галогсноироизводных кислорода изучена только в
газовой фазе. Молекулы OF2 и ОС12 треугольны. Угол F—О — F —105°;
С1—0-С1=111-115°; doF=l,36A;doci=l,68 —1/71A. В молекулеСЮ2
валентный угол О—G1—О =137° ± 15°; dC\o =1,53 А.
Молекулы 0F2 и 0С12, содержащие нечётное количество валентных
электронов, являются примером «нечётных» молекул.
Гидриды кислорода представляют громадный научный и практический
интерес, тем более, что один из них, Н20, играет исключительную роль
в природе и технике.
Гидриды кислорода различны:
а)Н20и Н2О2 —соединения со смесью изотопов водорода (ат. вес=1,008).
б) D2O и D2O2—соединения с тяжёлым изотопом водорода (ат.вес^2).
в) Н*0 и Щ02 — соединения с лёгким изотопом водорода (ат. вес—1).
Из них наиболее изучены соединения группы а).
Молекула воды имеет треугольную структуру с углом Н—О—Н = Ю5С.
Этот валентный угол НзО стремится сохранить и в конденсированном
состоянии. В молекуле расстояние (О —Н)=0,96.
В кристаллическом состоянии НгО образует несколько структур.
Хорошо изучен НгО гексагональный.
Недавно открыт Н20 кубический.
1. аН20 (обнаруживается
при температуре
жидкого воздуха).
Решётка кубическая С.
Координация тетраэдриче-
ски-линейная Т/1 (?)
Тип: возможно, Стц —
SB C9?
Рлс. 290. Схема (тми С 9—кристобалита).
он
О
Атомы О образуют решётку алмаза, атомы Н расположены между
атомами О. Точное положение атомов Н на прямой О—О для а-льда
неизвестно, повидимому, они смещены к атомам О; см. р-лёд.
2. рН20
Для рН20, гексагонального льда, предложены четыре основные
структуры: А) тридимита (модель Бариса), В) модель Бернала и Фоулераг
С) Паулинга и D) ротационная.
/iAV
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
(ГЛ. V
2, А. (Ш>0, {Ш20, обычный лёд (лёд 1)
Решётка гексагональная Н.
Координация тетраэдрическая Т.
Тип Hi/iX (тип р-тридимита).
Пространственная группа D%h —C&jmmc.
Z=4.
оо
он
О Кислород Ф
Рис
Вещество
[ »нао
3D, О
. 297а. Модель Бариса.
1 t°C
—66
0
—185
—66
0
4
—185
1 •
4,5085
4,5135
4,470
4,5055
4,5165
4,5175
4,470
i
с
7,338
7,3521
7,293
7,338
7,3537
7,3552
7,301
Рис. 297Ь.
Примечание
Атомы О образуют решётку
вюрцита. Между ними
располагаются атомы водорода точно
посередине расстояния О—О.
(Расстояние О—Н = 1,38. Такое
увеличение вместо 0,96 для газа
маловероятно). См. также С. А. 38 4488
(1944).
Новейшими исследованиями
опровергнуты; см. стр. 447.
2, В. |Ш20, лёд I (модель Бернала и Фоулера) см. рис. 297Ь, модель В.
Решётка гексагональная, молекулярная НМ^Т2'\
Координация Т/1.
Тип #^p->-SB?
Пространственная группа С1С — Сбст.
2 = 12.
Отличаясь величиной элементарной ячейки, модель В предусматривала
то же тетраэдрическое расположение атомов О друг около друга, что и
в А, но протоны, попарно смещённые к одному из атомов кислорода (рис.297Ь,
модель В) на расстояние 0,99 кХ. Молекулы Н20 чётко определены. Но-
йемшими исследованиями отвергнута. См. стр. 447.
Вещество
НЮ
а
7,82
с
7,36
с/а
0,941
§ 149) ГЛЛОГЕ1ШДЫ. VI ГРУППА: КИСЛОРОД, СЕРА, СЕЛЕН, ТЕЛЛУР, ПОЛОНИЙ иЗ
29 С. [ШоОи D20—модель Паулинга, рис. 297Ь, С. Допускается, что протон
может колебаться вдоль оси линии О—О и переходить от одного атома О к
другому; наиболее вероятны положения на расстояниях 0,96 кХ от обоих
ядер О и, следовательно, перемещения протона на 0,78 кХ. Этот вывод
Пыл сделан в связи с тем, что с понижением температуры энтропия льда
не стремится к нулю, но имеет остаточное значение 0,82 кал/моль-град.
Мидели Паулинга отвечает теоретическое значение энтропии R In — -■=
= 0,WX> кал/моль-град.
Новейшими исследованиями эта модель, повидимому, подтверждается,
см. стр. 147. Нам кажется, что такое колебание протонов облегчается наличием
у атома О в Ь20 четырёх, а не двух связей под углами ^-109° (см § 91).
2. D. [ШоО и ЮоО— вращательная модель (рис. 297b, D). Протоны
вращаются вокруг атомов кислорода. Новейшими исследованиями отвергнута;
см. стр. \ \1.
3. Лед II (Н30)
Лёд II образуется при высоких давлениях; при достаточно низких
температурах и обычном давлении он метастабилен. (В SBIV 116
неправильно указывается, что он в этих условиях стабилен.)
Лёд II— «ромбический», пространственная группа D\ — С 222х; Z = 8;
а -- 7,80 А; 6 = 4,50 А; с —5,56 А; а : Ь : с—1,73 : 1 : 1,24. Каждый атом
кислорода окружён 4 атомами кислорода в вершинах сильно деформированного
тетраэдра, d- 2,71 А.
С моделью Бернала и Фоулера для льда I структура льда II не
приводится в соответствие, ибо не удаётся сделать одинаковыми углы между
ОН-связями. В «ромбическом» льду II допускался «распад» молекул воды
с образованием ОН~ и Н + -ионов.
4. Лед III (ILO)
Исследование льда Ш проведено при низкой температуре ( —155°С).
Обнаружена центрированная ромбическая ячейка, пространственная
группа Щ -Jham\ «-10,20; 6 = 5,87; 0 = 7,17; а : Ь : с = 1,748 : 1: 1,221;
Z-16; с/-1.105.
В вой росе о строении льда, несмотря на большое количество исследований,
ясности нет, что связано с трудностями, вызываемыми спецификой объекта.
б. На03
Б структ\-ре молекулы II2O3 о теоретической стороны не имеется доста-
точной ясности.
Структуры кристаллов Н2Оо изучались на продуктах с 88 —90% НоОз,
а также на продуктах со 100% Н2О2.
Найдена тетрагональная решётка—пространственная группа D\— Pit2
или D\-P\£x с «4,02; с = 8,02; с/а = 1,995; Z = 4; ах = 1,73.
Из обсуждении возможной структуры молекулы Н202 в кристаллах
был сделан вьлюд, что все атомы II и О не могут лежать в одной плоскости.
Было найдено также, что Н„02 не образует твёрдых растворов ни с Н„0
шг с Н,Ой- 211.0. См. также J." Am. Ch. Soc. 63, 1507 (1941).
6. S,
S и её гомологи образуют с галогенами соединения с более высоким
валентным состоянием центрального атома, до SF, и т. п.
SFe. SeF6, TeFfi —октаэдрические молекулы. Расстояния S —F=l,56;
Sc -F-1,67; Tu-F- 1,82.
SC12 имеет валентный угол CI — S — С1 = 101 — 103°; расстояние S — С1 =
=.-.= 1,99— 2,00А. S2CI2 —зигзагообразная молекула типа CL
S~S\Ci
с расстояниями S-S^ 2,04 ± 5. S С1 = 1,98±5. Угол Cl-S —S = 103°.
44 \ ч. и. структуры неорганических веществ [гл. V
Молекула ТеС14 имеет структуру тригональной дипирамиды, к одной из
вершин основания которой как бы направлена несвязываю'щая пара
электронов Те.Форма молекул ТеСЬ, ТеВг2, TeJ2 надёжно не установлена.
Валентный угол Hal —Те —Hal равен по крайней мере 150°.
Расстояние Те-С1 = 2,36 ± 3, Т1-Вг = 2,49 ± 3; Т1- J = 2,70 ± 3.
В 1947 г. Роджерс и Спурр (J. Am. Ch. S. 69, 2102 (1947)) опубликовали
сообщение, что валентный угол Вг — Те — Вг в ТеВг2 равен 98° (!).
Расстояние Те Вг = 251кХ.
ГидридыSH2, SeH2(TeH2) треугольны, угол Н—S—Н = 92°. S — Н = 1,35;
угол H-Se-H = 90°.
Кристаллические структуры галогенидов, гидридов и гидратов
элементов VI группы почти не изучены. В кристаллическом H2S и H2Se атомы S
и Se образуют кубическую гранецентрированную элементарную ячейку Ск.
Положение атомов Н надёжно не определено. В работе С. А. 38, 3488 (1944)
допускались положения, как в типе Сс/г, в центрах октантов (см. также
SB II 281—282.)
7. а Те(0Н)в
Решётка кубическая
молекулярная CM<°h
Координация 0/1.
Тип Cg^-SB.
Пространственная группа
0%-Fd3c.
Z = 32, Те к. ч. = 60Н (окта-
эдрическая конфигурация);
О к. ч. = 1Те.
8. £Те (0Н)в
Решётка моноклинная (молекулярная?) Мм(°).
Координация 0/1.
Тип, повидимому, МЩй) ?
Пространственная группа C2U — P2JC.
2 = 4;
Вещество а„
а Те (ОН)в
15,48
сх
3,26
Примечание. Эта структура была вновь под- 1
тверждена в 1935 г.; согласно другим
данным пространственная группа Oh — Fm3m; a ~
-7,83.
Впрочем, имеются данные, подтверждающие 1
первую структуру, ох = 3,158; а = 15,68 см.
SB III 333.
| Вещество
рТе(ОН),
а
5,70
Ь
9,30
с
9,74
а : Ь : с
0,613:1:1,047
э
104°30'
См. SBIII333. Интересно, не отсутствует ли в моноклинной форме
молекулярная структура.
§ 160. VII группа периодической системы: водород Н, фтор F, хлор С1Г
бром Вг, иод J
Соединения галогенов между собой.
Соединения в высшем валентном состоянии известны только у иода.
JF7(ra3). Следующая степень наблюдается у J и Вг; JF6 и BrF5 (жидкий).
Далее известны JC13, BrF3 (жидкий) и C1F3 (газ) и, наконец, выделен
ряд веществ, молекулы которых состоят из различных галогенов: ClF,BrF,
JC1, JBr и т. п. Структуры соединений высшей валентности изучены
недостаточно. Структуры двуатомных молекул известны значительно лучше
благодаря работам спектроскопистов.
§ 150| ГАЛОГЕНИДЫ. VII ГРУППА: ВОДОРОД, ФТОР, ХЛОР^РОМ, ИОД
445
В группе комплексных соединений мы рассмотрим также структуры
комплексных ионов тип t [JC1J~, [JCh]^ и др.
Структура молекул JF7 надёжно неизвестна.
Молекуле JF6 приписывается структура идеальной тригональной бипи-
рамйды DS с атомом J в центре. С нашей точки зрения, не исключено, что
такая конфигурация JF6 является идеализированной, т. е. что пара несвя-
;швающих электронов иода может обусловить деформацию этой структурной
формы, естественной для молекулы PF6.
С этой же точки зрения, молекула типа JC13 может быть плоской,
если три атома хлора занимают вершины основания тригональной бипира-
миды, а две несвязывающие пары электронов J направлены вдоль оси 3.
Двуатомные молекулы, естественно, линейны, причём их энергий связи D
и межатомные расстояния d сравнительно мало отличаются от полусуммы
энергий связи и полусуммы расстояний в молекулах соответствующих
галогенов, что указывает на малый, в данном случае, эффект резонансной
энергий. Так, например, найдено для JC1:
^эксп (J-C1) = 2,3115 A; d - Щ^ - 1,323 + 0,922 - 2,315.
Ажсп = 2,143 электрон-вольт; D = -^—- = 2,0 электрон-вольт.
1 j IIF | HCl | DCl
Энергия связи, ккал
Межатомные
расстояния
0,864
—102
1,272
103
1,27
НВг
1,410
HJ
63
1,01-,
В кристаллическом состоянии га-
логеноводороды исследованы довольно
ПОДробнО;
1. aHCl, аНВг (?)
Решётка ромбическая молекулярная
OR™.
Координация (12),
Тип OR™.
Пространственная группа DI - ^222
или Dil — Fmmrn.
Z=4.
На рис. 298 показаны чёрными
шарами положения центров тяжести
молекул НХ.
>НС1
Рис. 298.
I Вещество
aHCl
аНВг
| Примечание
f °К
95
(—178°1С)
100
>. SB III 229
а
5,03
5,555±*0
b
5,35
">,640^0
с
5,71
6,O63±J0
а : b : с
0,94: 1 : 1,007
0,985 : 1 :1,075
Ох
1,55
2,81
446 ч. ii. структуры неорганических веществ [гл. V
2. РНС1, &HBr, £HJ (?)
Решётка кубическая гранецентрированная
молекулярная Ск .
Координация К.
Тип C%-SBA1 (?)
Пространственная группа Oh—-Fm3rn (?).
Рис, 299.
Вещестпо
3 НС1
рНВг
PHJ (?)
t°6
103,1
115,1
103,1
а»
5,466±£
5,76 -10
6,18
°х
1,48
2,76
3,59
#НС1
На рис. 299 показаны чёрными шарами положения центров тяжести
молекул НХ.
3. ocHJ
Решётка тетрагональная, молекулярная Тм.
Координация?
Тип Т?.
Пространственная группа ?
1 Вещество
ли
t°K
125
82
21
а
6,194
G,o:i7
Г„048
с/а
1,08
—
—
°х
3?'>9
3,56
3,72
Резюмируя известные из литературы разрозненные данные о фазовых
нереходах галогеноводородов, приходим к следующему выводу:
HF-?
НС1-невидимому, диморфен: аНС1 {ОЩЩНС1 {Ск){Т„ = № К).
НВг—повидимому, триморфен:
аНВг((9Д)г±(7'1,= 111-115°К)^грВг(Ск)<^(7,п= 115- 119°K)^YHBr(?>
5 150] ГАЛОГЕНИДЫ. VII ГРУППА: ВОДОРОД, ФТОР, ХЛОР, БРОМ, ИОД 447
HJ —повидимому, диморфен:
аН J (7>) ^ (Г„ = 85 - 124 К) z± pHJ {Ск)
Имеются данные о наличии двух областей перехода 65 —72° К
и 85-124° К.
Рассматривая структуры кристаллов простых веществ с
одноатомными и двуатомными молекулами и сравнительно слабым
межмолекулярным взаимодействием, мы пришли к выводу о том, что
высокотемпературные модификации должны иметь и имеют более высокую симметрию.
Это подтверждается нижеприведённой таблицей.
Вещество
HG1
НВг
HJ
NH4
Форма
- | р !т
OR»
rpM
1 к?)
°к
°к
?
?
Вощо-
ство
сн4
СВг4
SiF4
Форма
?
ьк
М(?)
Л(?)
см
°0
т
Вещество
GeJ4
SnJ4
GO,
СО
Форма |
а
С
с
С010
Ск
?
н.
г
Общие замечания к главе V но вопросу об определении
положения водорода в гидридах методом диффракции
нейтронов
До последнего времени исследования структур гидридов
рентгенографическим методом позволили локализовать положение более тяжёлых атомов
и оставляли неясным вопрос о положении водорода. На примере строения
льда 1 мы видели, что основные модели, не расходясь в вопросе о
положении кислорода, опирались на различные вспомогательные соображения для
характеристики позиций водорода, давая разные решения задачи.
Применённый в 1939-1940 г. (см. Phys. Rev. 65, 1101 (1939); 57, 97G
(1940)) для изучения степени упорядоченности в реальных кристаллах, метод
диффракции нейтронов открыл очень широкие перспективы и в изучении
положения лёгких атомов—Н и D. В работе, опубликованной Окриджской
лабораторией (Воллэн, Дэвидсон, Шелл, Phys. Rev. 75, 1348 (1949), см. также
УФН XXXIX, 140 (1949)) сообщаются результаты исследования
структуры льда, приведшие к отклонению моделей Барнса, Бернала и Фоулера
и ротационной и установившие близкое соответствие к модели Паулинга
структуры тяжёлого льда D20 при—90° С.
Несомненно, в ближайшие годы предстоит ещё немало сделать для
окончательного решения вопроса о структурах льда и других гидридов.
Это исследование приведено нами в качестве иллюстрации больших
возможностей, открываемых перед изучением подобных структур в
результате применения метода диффракции нейтронов.
ГЛАВА VI
ОКИСЛЫ II ХАЛКОГЕНИДЫ (соединения О, S, Se, To
$ 151. I группа
периодической системы: литий Li,
натрий Na, калий К,
рубидий Rb, цезий Cs
1. Li20, Na20, К20, Li2S,
Na2S, K2S, Li2Se, Na2Se,
K2Se, Li2Te, Na2Te, K2Te
Решётка кубическая С.
Координация кубо-тетраэдри-
ческая С\Т.
Тип Сс/т — SB C1.
Пространственная группа
Oh—Fm3m.
Z = 4. Li к. ч. = 4Х (Г);
Хк. ч. = 8М {С).
Рис. 300
1 Вещество
Li,—
Na2-
к,-
Rb2-
|
а
d
ОМ
а
d
ОМ
а
d
ОМ
а
d
ОМ
о !
4,619
2,00
24,5
5,55
2,405
42,8
6,43,
2,79
66,5
б,742
*> 91„
' 9
76,67
S |
5,708
2,47
46,5
6,52б
2,83
69,5
(7,35),
7,39t
3,20
101,0
7,65
3,31
2,99
111,9
Se
б,оо5
2,60
54,0
6,80 *
2,95
79,0
7,67б
3,325
112,7
—
Те |
6,504
2,82
56,4
7,313
3,17
97,7
8Д52
3,53
135,2
—
Примечании 1
В SB III 283
ошибочно вместо d
соответствующие
цифры приписаны сх\
§ 151] ОКИСЛЫ II ХАЛКОГКШ1ДЫ. I ГРУППА: ЛИТИИ, НАТРИИ, КАЛИП, ЦЕЗИИ 449
2. Cs20
Решётка ромбоэдрическая слоистая Rs.
Координация O/t.
Тип flg/t-SBC19.
Пространственная группа D\a— R3m.
Z=l.
О Cs
Вещество
CsaO
i
1 а
1 6,74
1
Рис. :Ю1.
а
:*6,93э
*х
4, 64
1 тг 1
[ Примечание |
j SB VII 7
1 '
3. Высшие окислы и перекиси щелочных металлов см. гл. X.
450
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВШЦЕСТВ
[гл. VI
§ 162. II группа периодической системы: бериллий Be, магний Mg,
кальций Са, стронций Sr, барий Ва, радий Па
1. BeO, MgTe
Решётка гексагональная /f.
Координация тетраэдриче-
ская Т/Т.
Тип H-urSB — Bi (вюрцит).
Пространственная группа
Со,. — Сбтс.
М к. ч. = 4Х(Г);
X к. м.--4М(Г).
гл
Рис. 302
Вещество | а
ВеО ! 2,6.^
MgTe j 4,Г»2
с.
с\а d. OM I
4,37 1 1,62.
1 •>
7,3? 1,62.
1,64
2,75
13,8
65,3
2. BeS, BeSe, BeTe
Решётка кубическая С.
Координация Т/Т.
Тип Ст/т —SB£3
(сфалерит).
Пространственная группа
ТЗ —МЗте.
Be к. ч. = 4Х(71);
X к. ч.=4Ве(7').
1 Вещестио
BeS
Be Se
Be Те
aw d
4,86
5,13
5,51
2,10
2,18
2,43
OM
2*,7
33,8
44,1
Рис. 303
§ 152] ОКИСЛЫ И ХАЛКОГЕШЩЫ. II ГРУПП \: БЬРИЛЛИИ, МАГНИЙ-БАРИЙ 451
3. MgO, CaO, SrO, BaO, MgS, CaS, SrS, BaS, MgSe, CaSe, SrSe, BaSe.
CaTe, SrTe, BaTe
Решётка кубическая С.
Координация октаэдрическая О/О.
Тип Со/о--SBБ1.
Пространственная группа О),—Fmcm.
М к. ч. = 6Х(0); \ к. ч. = вМ(0).
2 = 4.
^—^
о^
c&^&f'
О
]оЧ-^'
с
У\
ЙЬ<Р
O^f-^fW'
ь^
о
Рис. 30',.
[
Вещество
Mg
Са
Sr
Ва
а
d
ОМ
а
d
ОМ
а
d
ОМ
а
d
ОМ
О
4,20.
2,Ю3
18,6
4,80
2,40
27,6
5,1Г>
2, 58
34,1
•г>,53
1 2 77
42,4
S
5,19
2,60
34,9
5,68
2,84
45, S
6,01
з,01
54,3
6,37
| 3,19
64,6
Se
5,45
2,73
40,5
5,91,
2 95
51,6
6,23
3,12
60,4
6,59
3,30
71,6
To
-
6,34
3,17
63,7
6,65
3,33
73,6
6,99
3, 5U
85,4
Примечание
Средний линейный
коэффициент расширения MgO
20—1300° С ранен Г.,45-10~'
при 22 °С а„ = 4§Ш'|
В SB I, стр. 74, даны
неиерные значении для
CaTe а„ и др.
-•
4. Перекиси щСмочно-яемельных металлом см. гл. X.
452 ч. ii. структуры неорганических веществ |гл. VI
§ 153. III* группа периодической системы: скандий Sc, иттрий Y,
лантан La
1. Sc2Os, Y203
Решётка кубическая С.
Координация С ' , С /Т.
Тип cVr,c=/r-SB Z»53(Mn203)-
Пространственная группа Т\—/а.З.
Z = 16. Ml к. ч.= 60 (С'г); О к. ч.=- 4М (Т);
МП к. ч.- 60 (С).
Оо
Рис. 305.
1 Вещество
Sc203
Y203
aw | d | Примечание 1
1
9,7CJ 2,0<j
10,60 ' 2,27
1
Cm. Mn203
2. Описываемая далее структура La303 интересна в том отношении, что
координационная сфера La является существенным дополнением к таблице Н4.
В данном случае образуется конфигурация тригопальной аптипризмы с
одним дополнительным атомом против грани основания. Подобную
конфигурацию мы будем обозначать: l.'i' . Атомы 01 имеют конфигурацию триго-
налыюи антипризмы A3. Атомы Oil - - тригональпоп пирамиды JP/y«J. Таким
образом, координация: Д3~< /Ло,Р//3.
§ 153] ОКИСЛЫ И ХЛЛКОГШ-ШДЫ. Ill* ГРУППА: СКМ1ДИП, ИТТРИИ, ЛАНТАН 453
Lavj03, La202S
Решётка гексагональная Н.
Координация сложная
7/0,4 = 437^3,^3.
Тин ffA3*/A3f/>tf3-SB £52 =
Пространственная группа
D\d—Obn.
Z= 1.
La к. ч. = 7 0(ЛЗ');
О 1к.ч.= 6Ьа (.43);
О II к. ч.= 4Ьа(Рг/3).
(Близко ко второму
основанию АЗГ (и РуЗ) находятся
ещё по одному атому О
(соответственно La). С их
включением в координационную
сферу получаем: Л3+и/)3.)
Рис. 306.
1
Вещество
La203
La.O.S
а
3,93
4,03
с
6,12
6,88
с! а
1,56
1,7
4
4 (£* + <*') =
-2,42
3<Г = 2,69
2,38 (Ы)
2,41 (W)
3,06 (3<?)
ом
81,8
1 Примечание
1
\ См. SBI 744
[
! —3 атома О 1
| —1 атом О 1
i —3 атома S I
! По работе Захариасена 1
l Acta Cr. II 55 (1949)
3. La2S3, Ac2S3
Решётка кубическая С.
Координация 8/х.
Тип Cs/x (Ce2S3).
Пространственная группа T"d — Jb3d.
Идеальный состав при M3S4; Z = 4.
™«
Вещество
La>S
а
8,706±i
8,97±i
gx Примечание
5,01
6,75
По работам Захариасена
Acta Cr. I, 255 (1948)
и II 57 (1949)
Смотри Ce2S3.
154 ч. и. структуры неорганических веществ [гл. VI
§ 154. III** группа периодической системы: редкие земли
Структуры окислов и халкогенидов III группы представляют
исключительный интерес в связи с вопросом о влиянии /-электронов на характер
химической связи и, следовательно, на тин структур. Если это влияние
ври образовании структур простых веществ, несомненно, удаётся
проследить (см. § 131), то в отношении галогенидов (§ 136) и окислов до
последнего времени не было достаточного экспериментального материала.
Недавние исследования показали, что в этой группе действительно возникают
новые виды структур. Однако многие из этих структур образуются и
соединениями лантана и актиния (111* группа), хотя пока не описаны у
скандия и иттрия. Поэтому пока в ряде случаев следует воздержаться от
поспешных выводов о роли /-электронов, впредь до накопления
экспериментальных данных.
1. Сс02, Рг02
Решётка кубическая С.
Координация кубо-тетраэдрическая С/Т.
Тиц Cc/r — SB Ci.
Пространственная группа On — Fm3m.
М к. ч. = 8 0(C); О к. ч. = 4М(Г).
Оо
Рис. 307.
Вещество
СеОа
РгО,
аи>
5,40
5,36
d ОМ
2,34
2,3£
39,3
38,5
§ 1541 ОКИСЛЫ И ХЛЛКОГЕННДЫ. III** ГРУППА: РЕДКИЕ ЗЕМЛИ 455
2. Се,03, Рг203, Nd,03 (?) Ce202S
Решётка гексагональная Н.
Координация сложная —7/6,4 = ЛЗ/Л3, РуЗ.
Тип Ялз^/лз, Руз-SB D\.
Прсстранственная группа Ь-щ — Сбт.
Z-l. M к. ч. = 70 (ЛЗ*); 01 к. ч. -6М(ЛЗ); ОН к. ч. =4M(/>j/3).
О
Рис. 308.
Вещество
Се208
Рг,08*)
NdaOs**)
Ce,OaS
а
3,88
3,85
3,84
4,00
с
6,06
6,00
6,01
6,82
cja
1,56
1,56
1,56
| 1,70
d
k(d + d') -
= 2,39
3<Г=2,66
2,37 соотв.
2,63
2,36 соотв.
2,63
Се—О 2,39(14)
Се—О 2,36(34')
Се—Si 3,04 (3<?)
ОМ
79,0
77,0
*) Прокалённый 3 часа при 1000° С -тип D7u. Прокалённый 3 часа при 700° С —
тип /)5§.
**} Но работе Захариасена Acta Сг. II 55 (1949) слабо искажённый тип D 52.
456
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТ1Ч
[ГЛ. VI
3. Sra203, Eu203, Gd,0„ Tb203, Dy203> Ho20„ Er203, Tu20a, Yb20„ Cp20,
Решётка кубическая С.
Координация C~l-,Cr\T.
Тип Сс-;-, c^/r —SB Z)5, (Mn203).
Z = 16.
Ml к. ч.= 60 (С-/-); МП к. ч. -60 (С~)\ О к. ч. = 4М (Г).
1 i 1 i
В-во ;Sm,03 Ku208 0<la03
1 i 1 !
1 a
d
10f85l 10,8,. 1 10,7l}
2,32! 2,:V2 2,40
i 1
Tb>03;Dy,03
1
10,70
2,29
10,0o
2,27
Puc. 309
Ho208
10,5S
2,26
ЕгаО,|ти208
i
10,54
2,25
10,52
2,25
0 м
Oo
Yb,0,
10, »9
2,22
Cp,0,
10, з7
2,22
Примечание 1
См. MibO,
4, CeaS8, Ce3S4
Решётка кубическая С.
Координация 8/х.
Тип Сш, «Ct\Sa».
Пространственная группа Td —./4 3d.
Z = -т- • (Идеальный состав при CeaS4, Z - 4.)
М к. ч. = 8S.
1 Вещество а
Со2Ч3
Co3S4
8,61734- J
8,6075± 5
сх 1 Примечание
5,19 1 Acta Сг. I, 255 (1948) 1
! 5,675 И II 57 (1949) |
Ср. со структурой шпинели типа Ni3S4 (стр. 627, 628 и 640).
§ 154]
ОКИСЛЫ II ХЛЛКОГЫШДЫ. Ill** ГРУППА: РЕДКИЕ ЗЕМЛИ
457
Структура является хорошим примером структур деления: в
кубической элементарной ячейке в предельном случае имеется 16 атомов серы
и 12 атомов металла. Если все позиции металла полностью заняты,
вещество имеет состав М3А4. Позиции атомов: (000) {2/2 7г 7г) + 12Се
в (7*7.0)0(аЛ7. 0)Oibs в {ххх)(х + ч„ ll*-***)0[* + lU> s + 7-.
* + 74)» (74 + *. 74 — *. 74 —*) О;* = 0,083 ±0,015. Однако возможно
статистическое распределение меньшего количества атомов М по этим
местам, и в предельном (?) случае на 16 атомов серы приходится только
2
10 -у атомов металла, статистически распределённых по 12 местам, так что
1 „ -о
в среднем — всех мест оказывается незанятой. В этом случае состав
вещества М2Х3.
В зависимости от соотношения S : М, в случае сульфидов Се,
значения а и плотности вещества меняются следующим образом:
S : Се
а <к.\)
1,495 К,0173+5
1,45 S,0131±J
Плотность
(г см"6)
5,186
5,313
S : Со
1,40
1,33
а (кХ)
8,6123±5
8,6076±5
Плотность
(г см~*)
5,455
5,675
5. EuS, EuSe, EuTe, YbSe,
YbTe
Решётка кубическая С.
Координация октаэдриче-
ская О /О.
Тип Со/о-SB B\.
Пространственная группа
0\ — Fm3m.
Z = 4.
Мк.ч. = 6Х(0);
Хк.ч. = 6М(0).
Рис. :ио.
Вощсчтво EuS
d
5,956
2,978
EuSe
6,173
3,086
EuTe YbSe
6,572
3,286
5,867
2,933
YbTe
6,340
3,170
458 ч. и. структуры неорганических веществ [гл. VI
§ 155. IV* группа периодической системы: титан Ti, цирконии Zr, п
ний Ш, торий Th
1. Ti02, рутил
Решётка тетрагональная цепная Т .
Координация октйэдрически-треугольная OJL
Тип ТоП -SB СЛ (рутил).
Пространственная группа вЦ—РЩтпгп.
Z =. 2.
Ti к. ч. =GO (0)\ О к. ч. =3Ti(*).
Q: О
Рис. 311.
Вещество
Ti02
рутил
а
4,58
с
2,95
с/а
0,644
d
Ti—0
2,01
OM(kX)3 Примечание
30,9
[ТЮ„]-октаэдры
имеют по 2 общих ребра
с соседями и образуют
цепи
На рис. 311,а показано положение атомов Ti и О в элементарной
ячейке; на рис. 311, 6 —то же, плюс соседние атомы; ячейка 311, b
повёрнута на 90° по сравнению с ячейкой рис. 311, а.
§ 155] ОКИСЛЫ II ХЛЛКОГЕНИДЫ. IV* ГРУППА: ТИТАН, ЦИРКОНИЙ, ТОРИЙ 459
2. ТЮ2, анатаз
Решётка тетрагональная Т.
^±
Координация S+jt.
Пространственная вязь (V).
Тия^гд-SB СЪ.
Пространственна» группа Ли,—
— Ji/amd.
Z = k.
Ti к. ч. -■= 60 (S+); О к. ч. -
= 3Ti(0-
О
c^ra^t
Рис. 312.
Вещество
анатаз
а
8,73
С
9 Я
с/а
2,51
1 "^
Ti—0
1,95
(ОМкХ)3
32,0
Чистота
[ ТЮв] сильно
деформированные октаэдры
имеют по 4 общих
ребра и образуют
пространственную вязь
1
Конфигурация [ТЮв] в анэтазе часто называется «октаэдрической». Но
из рис. 312 видно, что это —октаэдр, претерпевший своеобразную
деформацию и внешне похожий на координационную сферу JJ Sn, рис. 198. т. е.
это- тетрагональный тетраэдр, плюс 2 атома сверху и снизу (*SV).
460 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
3. Ti02, брукит
Решётка ромбическая ORs.
Координация октаэдрически-треугольная Ojt. Сетки (N).
N
Тип OREц- SB £21.
Пространственная группа Z)'/, — РЬса.
Ti к. ч. =60((5); О к. ч. ~3Ti(/).
ь О Ti
Ufa ? f 2 f f 5 О О
• Рис. 313.
BeraecTBj
брукит
а
0,16,
о
в
5,43,.
С
5, П.
а :в :с Примечание
1 4 |
1,683: 1 : 0,944
Ti—0-«октаэдры» имеют 1
но 'Л общих ребра,т.
е.образу ют цеии, сшитые в сетки
Характер фазовых переходов в системе Ti02 неясен. Судя по некоторым,
нуждающимся в проверке данным, переход рутил 7* анатаз энантиотропен;
переходы же брукит—> анатаз и брукит -крутил — монотропны. См. также
Памфилов и Иванчева, ЖОХ X, 154 (1940), Ниггли и др. Helv. Ch Acta
27, 88 (1944) С. А. 38, 3179 (1944).
§ 1551 ОКИСЛЫ И ХЛЛКОГЕНПДЫ. IV* ГРУППА: ТИТАН, ЦИРКОНИИ, ТОРИЙ
461
4. TU),
Решётка
ромбоэдрическая R.
Координация октаэдриче-
ски-тетраэдрическая О IT.
Тип Ло/r-SB DM (ко-
РУнд).
Пространственная группа
Z = 2.
гПкГч. = 6 0(0);0 к. ч =
= 4Т1(Г).
Вещеегпо I а
TiiO,
5,42
а
50*32'
Как указывает SB I241,
решётка типа корунда
может быть представлена
как ромбоэдрически
деформированная решётка типа
NaCl (fll), оба сорта атомов
которой замещены двумя
энантиморфными сортами
молекул А1203 или в
данном случае Ti>03.
Рис 314.
о Ti
Оо
о^^=^\
б. TiO
Решётка кубическая С.
Координация октаэдриче-
ская О/О.
Тип Со/о -SB Bl (NaCl).
Пространственная группа
0а —F/гаЗ/я.
М к. ч. -60; О к. ч.= 6TL
Рис. 315.
Вощестно
TiO
4,16Г)
d Примечание
2 ogo По Орлпху. (В старых работах иа-
' "5 блюдалось также а1Г^=\,Т.\ъ). См.
| пригир Ac'ta (Шп)
462 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТП [гд. VI
Изучению фазового состава в системе Ti—О посвящена работа Эрлиха
[Z. E1 45. 362 (1939)]. В системе наблюдаются 4 фазы: а-фаза, тип рутила,
с границами устойчивости от Ti02 до ТЮьоо; (3-фаза с границами
устойчивости от ТЮ1>80 до ТЮЬ7(); двухфазная область (В, у); у-фаза, тип
корунда, с границами устойчивости от ТЮЬ6в до TiO]f46; двухфазная
область (у, о), й-фаза, тип каменной соли, с границами устойчивости
от Ti0lf25 до ТЮ0>в; двухфазная область (<*-фаза + металл).
Несмотря на большой интерес, который вызывает донная работа, одна
из наиболее обстоятельных и точных в этой области, она имеет ряд
принципиальных недостатков (более или менее типичных для постановки
подобных исследований), )страненис которых в дальнейшем важно для успеха.
в этой области.
Условия взаимной устойчивости рутила, анатаза и брукита в отношении
их перехода друг в друга с термодинамической точки зрения не
определены. В равной мере не сделана даже попытка дать термодинамические
характеристику других рассматриваемых фаз и их переходов.
В работе не сопоставлены представляющие большой интерес данные
о границах устойчивости по составу TiOx структур анатаза и брукита
по сравнению с границами, найденными для рутила.
Большим дефектом работы является отсутствие прецизионного
исследования межплоскостных расстояний и его изменения в зависимости от
состава, например для Ti02 (а-фаза) было найдено
а = 4,583 и с-2,954 кХ.
Уже при малом недостатке кислорода порядка Ti01>97 имеет место
расщепление линий, появление новых. Ввиду этого приходится поставить под
сомнение вопрос о возможности существования а-фазы при составе TiO]tft0,
(вопрекиуказываемой Эрлихом границе); подобное соотношение атомных
концентраций может быть вызвано наличием нераспавшейся фазы Ti02 и вновь
возникшей р-фазы.
Вопрос о структуре [3-фазы остался невыясненным. Эрлих указывает, что
она является искажённой структурой: рутила.
Структура у-фазы—тип корунда—не вызывает сомнений.
Наконец, структура 3-фазы, тип NaCl также не вызывает сомнений.
Для этой фазы приводится зависимость а от состава фаз. Период
идентичности а меняется от 4,183 при составе ТЮ0,58 до 4,142 при составе Ti0lf33..
При составе TiOx 00 *
а = 4,165 кХ.
К сожалению, в работе не указано, составляет ли сумма Ti и О 100%„
что позволило бы учесть, каково содержание примесей (N).
При обсуждении структуры о-фазьь указывается, что точному составу
TiOlf00 отвечает экспериментальный удельный вес 4,888, из чего делается
вывод, что заполнено лишь 85°/0 атомных узлов и приводится таблица
степени занятости мест в решётке от состава фазы.
По поводу подобных, исключительно распространённых в кристалло-хи-
мической литературе рассуждений следует подчеркнуть, что пикнометри-
ческие определения удельного веса дают колебания в зависимости не
только от наличия дефектов 1-го рода, т. е. в узлах ячейки, но и от
дефектов 2-го рода—микротрещин, пор и т. п. Концентрация же последних внхтри:
кристалла обычно идет параллельно с концентрацией внешних микропор,
наличие которых можно выявить, применяя высоковакуумную методику никно-
метрического определения удельных весов реальных кристаллов (Б. Ф. Ордшнт
и М. А. Хачванкян, ЖФХ, XXI 575 (1947)). В противном случае
нельзя быть уверенным в отсутствии грубой погрешности за счёт микропор.
§ 155] ОКИСЛЫ И ХАЛКОГЕНПДЫ. IV* ГРУППА: ТИТАН, ЦИРКОНИЙ, ТОРИЙ 463
6. Zr02 бадделеит ^-^ ^-х
Решётка моноклинная Л/. С у ~^~ ~ ~^>^)
Координация искажённая
кубо-тетраэдрическая С/Т.
Тип Mcir-SB С43.
Пространственная группа
Clh -P2 /с. _
Z-4. Zr к. ч. =--=8 О (С);
О к. ч. ^4Zr(~T).
Вещее т по
а
!'
С
а : Ь : с
р
d
ZrO, I
бад to л еит
г>,л
Г>, 26
г),^
0,УУ1 : 1 : 1,021
80°32'
1,25—2,05
Рис. 31 fa
7. ZrU2, Ш02, Th02
Решётка кубическая С.
Координация кубо-тстра-
эдрическая С/Т.
Тип Cc/r-SB C\ (CaF8).
Пространственная группа
Ой Fm3m.
М к. ч. = 8 О:
О к. ч. ^t M.
Рис. :Л7
1 Вещество
Zr02
Hf02
Th02
«*
5,08
5,5,
d
2,20
2/.1
MO
32,8
43,2
Примечание
Но литературным данным
кубическая форма Zr02 для
чистого пещестпа ноустойчи
на. Она стабильна лишь
при наличии примесей,
например '*—50/0 МсгО [
/i64
Ч II СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
ГЛ
VI
8. TiS2, ZrSs, TiSe2, ZrSe8, TiTc2
Решётка гексагональная //.
Координация Oft.
Тип #o//-SB C6 (CdJ2).
Пространственная группа
Ad — С'Зт.
М к. ч. = 6Х(0); X к. ч. = ЗМ(/).
Рис. 318
Вещество
J Ti
1
а
с
с/а
d
МО
-s,
3,40
5,69
1,675
(2/.2)
57,0
—Se,
3,53
6,00
1,70
(2,53)
64,7
—TeJ
3,79
6,45
1,70
(2,72)
80,2
Вещество
Zr
а
с
с/а
d
МО
—s.
з,б8
5.85
1,59
2,58
68,6
•
—So,
3.7q
6,1,
1,631
2,68
76,9
-TeJ
—
—
—
—
В системе Ti — S обнаружены фазы TiS3, TiS2, Ti2S3 (границы
устойчивости Ti2S3 лежат от TiSli6 до TiSlfl3) и TiS. Даже при составе TiS0>.
линии Ti не возникают.
£ 155] ОКИСЛЫ II ХАЛКОГЕНЛДЫ. IV* ГРУППА: ТИТАН, ЦИРКОНИЙ. ТОРИЙ 465
Общие замечания
О системе TiO мы подробно говорили на стр. 461, 462. Система Zr—О
изучена недостаточно.
Структура Zr02 была исследована в зависимости от содержания примесей.
В целом, повидимому, можно сделать заключение, что чистый ZrOa
даёт при низких температурах устойчивую моноклинную форму а = 5,21;
6=5,26; с ~ 5,37^; Р = 80°32' (см. Бадделент). При нагревании имеет
место возникновение модификации при температуре порядка 600°С,
сопровождающееся изменением удельного веса.
Если же получать Zr02 в присутствии примесей, в частности MgO,
или просто в трубке из MgO, возникает и сохраняется при всех
температурах кубическая структура Zr02.
Zr02, образующийся из ZrSi04, при нагревании выше 165CP (причём
Si0.2 улетучивается)— также кубический (#„,= 5,12).
Система Ti — S изучалась путём исследования распада TiSs.
TiS, внешне похож на графит. Его порошковая диаграмма отличается
от диаграмм TiS2, с которым TiS3 образует двухфазную систему.
ТцЙд-фаза имеет интервал устойчивости TiSlfl3 — TiSb50.
Граница TiS-фазы — от TiSlj04 до TiS0>?, причём и в этом случае Ti
не обнаруживается.
Все эти сведения за исключением системы Ti—О нам калсутся
ненадёжными или, во всяком случае, далеко не полными, исходя из наших взглядов
по вопросу о границах устойчивости фаз и влиянии на последнюю примесей.
Из затронутых на этой странице систем это особенно относится к системе
цирконий— кислород.
IV* группа охватывает, как известно, длинный ряд элементов, строение
электронной оболочки которых проходит, если так можно выразиться,
критические моменты: титан и цирконий—dV-элсктроны, гафний —только что
закончилось заполнение /-оболочки, что позволяет провести параллель с
церием, у которого имеет место начало заполнения /-оболочки, и наконец, торий,
у которого снова, повидимому, начинает заполняться /-оболочка. Очень
интересно, что у титана проявляется исключительная способность
образовывать фазы с широчайшим интервалом устойчивости по составу.
Исследование границ устойчивости фаз, особенно окислов, халкогени-
дог, нитридов, фосфидов и др., карбидов, силицидов этих элементов
представляет исключительный интерес.
Начало этому исследованию на чистых препаратах и под указанным
выше углом зрения было положено советской наукой работами, впервые
поставленными в 1935 году в лаборатории автора в Физико-химическом
институте им. Л. Я. Карпова, а затем и в других советских лабораториях.
Позже появились работы в этом направлении и за рубежом. Тем не менее
здесь остаётся сделать ещё многое.
Прежде всего должно быть выяснено, в какой мере незаполненность
tf-оболочки у гомологов титана также влечёт за собой, как это можно
ожидать, широкие по составу интервалы устойчивости фаз. В первую
очередь это относится к окислам и нитридам, а затем и к другим
соединениям с достаточным весом ковалентной связи, см. § 104.
Очень важно также сопоставить электропроводность и магнитные
свойства этих веществ для более чёткого суждения о характере связи, тем
более, что у нитрида титана электропроводность настолько велика, что не
уступает наблюдающейся у многих металлов.
Наконец, исследования структуры окиси циркония заставляют с
особенным интересом отнестись и к вопросу о влиянии примесей на выбор
структуры при образовании кристаллов этой группы соединений; см. также § 103.
466 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VI
§ 156. V* группа периодической системы: ванадий У, ниобий Nb,
тантал Та, протактиний Ра
1. У20.
Решётка ромбическая сетки ORN.
Координация У/2,3.
Тип OR%j2s-SB D87.
Пространственная группа Clv — Ртп.
Z = 4.
V к. ч.-40(У); 01 к. ч. = 2У; ОН к. ч.= 3V; 0111 к. 4.-2V.
Рис. 319.
Вещество а
v.o.
11,48±J
Ъ
4')60±5
С
3»550±5
а:Ь :с
2 633 :1 :0,814
d 1
V-0
Ы=1,36Г)
Ь = 1,745
1/ = 1,76
1 ' =1,8^
Атомы V тетраэдрически окружены атомами О: три из четырёх атомов
О каждого тетраэдра принадлежат к соседним тетраэдрам, образуя цепи
тетраэдров, параллельные (001), сшитые в сетки, параллельные (010).
§ 156] ОКИСЛЫ II ХАЛКОГЕНИДЫ. V* ГРУППА: ВАНАДИЙ, ТАНТАЛ 467
2. V02j Nb02
Решётка тетрагональная цепная Тк.
Координация октаэдрическая треугольная Ojt.
Тип rgjjf-SB Ci (рутил).
Пространственная группа Dl\ — P4/mnm.
Z = 2.
М к. ч. =60(0); О к.ч. =ЗМ(<).
Ом
Рис. 320.
Вэщестио
а с
VOa \ 4,54
NbO, j 4,79
2,88
•> ij
-,j6
с, а d
0,634
0,С>21
1,93
2,01
ОМ
29,7
33,7
Примечание
См. ТЮ,
В Z. Anorg. Ch. 248, 1 (1941) указывается, что структура Nb02 лишь
близка к типу рутила; а -4,84; с — 2,99.
В 1947 г. появилась работа (J. Am. Ch. S 69, 998 (1947)),
посвященная исследованию системы V —О и указывающая на существование новой
модификации V02.
Недавно опубликована работа Эрлиха (Z.Anorg. Chem. 259, 1 (1949)),
в которой, на основании также работы Клемма и Гримма (Z. Anorg. Chem.
250, 42 (1942)), подчёркивается аналогия этой системы с системой Ti—О.
В ряду кислородных систем следующих металлов (хрома, марганца»
железа и др.) системы Ti—О и V—О, действительно, дают фазы с очень
широким интервалом устойчивости по составу. В системе V—О
существует, повидимому, 5 фаз: а) ванадия, с растворённым в нём кислородом,
эта фаза устойчива до VO0,42, причём наблюдается лишь лёгкая
деформация решётки ванадия Cq\ б) VOx с коэффициентом х при О от 0,88 до
1,32; в) VO*(«VoOs>>) с коэффициентом х при О от 1,35 до 1,53; г) VOx(«VOa»)
с коэффициентом х при О от 1,85 до 2,2; д) фаза V206 с узким интервалом
гомогенности.
£68 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ- VI
3. V203
Решётка ромбоэдрическая R.
Координация октаэдрически-
тетраэдрическая О/Т.
Тип Яо/т-SB £51.
Пространственная группа
Dlt-ягс
Z = 2.
Ук!ч. = 60(0); О к. ч.=
= 4 М (Г).
Вещество
аю
а
d
V—О
ОМ
v,o3
5^3
53°53'
2,1
48,5
Примечание. См. Ть03 1
Рис. 321.
О V
Оо
4. VO, NbO
Решётка кубическая С. *^
Координация октаэдриче- Г>^__
екая О/О. Ч/
Тип Со/о-SBB1.
Пространственная группа
Ofr — Fm3m.
Z = k.
Мк.ч. = 6О(0); О к.ч.=
= 6М (О).
Рис. 322.
1 Вещестпо
1 VO
NbO
aw
—4,08
4,203
1 d
~~1,04
! Примечание
A. X. Брегер
и В. А. Эпольбаум
ЖФХ. XX, 549 (1946);
Acta physicoch. XXI,
764 (1946)
Структура NbO по Z. Anorg. Ch. 248, 1 (1941) образуется из
структуры Cqjo (Z==4) удалением одного моля, т. е. Z — 3. Атомы распределены
статистически.
§ 156]
ОКИСЛЫ И ХАЛКОГЕНИДЫ. V* ГРУППА: ВАНАДИЙ, ТАНТАЛ
469
б. VS, VSe
Решётка
гексагональная Н.
Координация октаэдри- i
чески - призматическая !
О/РЗ. !
Тип Яо/рз-SB В8 (тип |
NiAs).~ I
Пространственная группа г,
Z = 2. I
V к. ч. = 6Х (-^ октаэдр)
(О);
X к.ч. = 6У (РЗ).
Рио. 323.
Вещество
VS
а с С/'а
3,34
5,785 ! 1,73
Вещество
VSe
а
3,580
С
5,977
с/а
1,670
Общие замечания
(В дополнение к сказанному выше о системе V—О.)
Окислы Nb и Та (системы Nb — О и Та —О) изучены крайне плохо.
Система Nb—О изучалась в Z. Anorg. Ch. 248, 1 (1941). Найдены
фазы Nb205, Nb02 и NbO. В системах V — S и V — Se обнаружены:
1) ас-фазы, по составу близкие к VS и VSe, причём VSe имеет границы
устойчивости от VSei,L4 до VSeifi3 (тип SBB8); 2) р-фазы, по составу
близкие к VSi.eo и VSei.sr.
VSe1>5 имеет границы устойчивости от VSei>25 до VSei>6f.
Наконец, обнаружены у-фазы VS4 (то же и минерал патронит) и VSe2.
Область устойчивости последней: от VSe^67 до VSei>97. Структура последней
близка к типу CdJ2 (SBC6) с а = 3,348; 0 = 6,12..,; с:а=1,828.
Обращаем внимание читателей, что фазы V2S5 и V2Se5, постоянно
упоминаемые в учебниках, рентгенографически не обнаруживаются.
В системе Nb—-S обнаружены 2 основные фазы: Nb2S3, которая может
растворять ещё 5 атомов S, образуя Nb2S8, т. е. «NbS4» (фаза? Б. О.) и NbS„
которая может растворять до 1 атома Nb («NbSo.5»)-
В системе Та —S обнаружено соединение TaS2 с гексагональной
решёткой, а = 3,396; с = 5,902; r/a = lf73R; Z = l; ах = 6,905.
Решётка слоистая. Тип не определён, возможно —тип MoS2 или CdJ2.
470 ч. ii. структуры неорганических веществ [гл. VI
§ 157. VI* группа периодической системы: хром Сг, молибден Мо,
вольфрам W, уран(?)
1. СгО,
Решётка ромбическая OR.
Координация 0/2 =OjZ. (исследована неполно).
Тип ORqU- SB DO,.
Пространственная группа D\ — C222,.
Z = 4.
Сгк. ч. = 60(0); О к. ч.=2Сг.
ОСг
LJ О
Рис. 324.
Вещество
СгО,
а
8,4б
Ь
4,7,
С
5,70
а : Ъ : с 1
1,774: 1 : 1,196
Структура псевдогексагональная, атомы О образуют гексагональную
шаровую упаковку, октаэдрические промежутки которой частично заняты
атомами Сг. В SB к типу Z)07 отнесены две совершенно разные
структуры Сг08 (SB II, стр. 29), пространственная группа DI и Al (OH)8 (SB IIIr
стр. 38), пространственная группа Clh, т. е. моноклинной сингонии.
§ 157] ОКИСЛЫ II ХАЛКОГЕШЩЫ. VI* ГРУППА: ХРОМ-УРАН 471
2. Сг203
Решётка
ромбоэдрическая R.
Координация О /7\
Тип Ro/т — SB Z>5X.
Пространственная группа
Dld-R3c.
Z = 2.
Cr к.' ч. = 6 0 (О);
О к. ч.=4Сг {Т).
Рис. 325,
Вещзство
1 Сга03
а
а
5,33 | 55°9'
d
Сг—О
2,1
ОМ
47,5
ОСг
Оо
3. CrS, CrSe, (СгТе?)
Решётка гексагональная Н.
Координация О/РЗ.
Тип Яо/рз-SB В8.
Пространственная группа
Deh — C6/mmc.
Z = 2.
Сгк. ч. = 6Х(0);
X к.ч. = 6Сг(ЯЗ).
РИС. 326
1 Вощэгтво
CrS
CrSe
СгТе
а
3,44
3,59
?
с
5,67
Г., 80
?
г а
1 ,65
1 ,62
у
СМ
2",о
32,6
V
Система Cr—Se освещена в Z. Anorg. chem. 239, 369 (1946).
472
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. VI
4. МоО,
Решётка ромбическая слоистая
ORs.
Координация 0/3, 2, 1.
Тип OR^2i-SB DO*.
Пространственная группа Д?/| —
— РЬпт ( = Рпгпа).
Z = 4.
Mo к. ч. = 6 О (сильно
деформировавшей октаэдр).
О МО
k'HMff 0°
Рис. 327.
1 Вещэство
Мо08
а
3,92
Ь
13,94
С
з,б6
а : Ь : с 1
0,281: 1 :0,263
Примерно кубическая шаровая упаковка атомов О, часть октаэдриче-
ских промежутков которой занята Мо-атомами.
§ 157] ОКИСЛЫ И ХЛЛКСГЕНПДЫ. VI* ГРУППА: 'ХРОМ-ХРАП 473
5. Мо02, W0,
Решётка тетрагональная цепная Тк
Координация О It.
Тип То it — SB С • (рутил)
Пространственная группа / Ih — Pi/mnm.
Z = 2.
М к. ч. = 60 (О);
Ок. ч. = ЗМ (t).
Ом
Рис. 328.
Вещество
МоОа
wo2
а
4,86
4,86
с
2,79
2,77
с/а
0,574
0,57
d
2,00
2,00
ОМ
33,0
32,7
1
Примечание!
См. ТЮ2
474 ч. ii. структуры неорганических веществ [гл. VI
6. Мо32, молибденит, WS2
Решётка гексагональная слоистая Hs.
Координация A3//.
Тип Hpsit — SBCl.
Пространственная группа Вм — СЪ/ттс.
Z = 2. Mo к. h. = 6S (РЗ); S к.ч. = 3 Мо (/).
Рис. 329, О М
Os
1 Вэщзство
J MoSa
а
3,15
зд8
с i с/а \ d \ ОМ
12.*0
12,5
3,90
3,93
2,35
2,48
52,8
54,7
Примечание
См. § 65
§157] ОКИСЛЫ II ХАЛКОГЕНИДЫ. VI* ГРУППА: ХРОМ-УРАН 475
7. W0,
Решётка ромбическая (?) OR (или М> или Т).
Координация xjy \ производный от Re03 (£Ю9),
Тип ORX/U — SBD010 f точно не исследован.
Пространственная группа ВЦ — Pmab ( = Pbcm)y если р Ф 90е, то Clh — P2jc.
Z = 4.
Вещество
wo3
а
7,28
Ь
7,48
С
3,82
а : Ь : с
0,973: 1 : 0,510
Атомы W образуют простую кубическую решётку. Атомы О, возможно,
лежат близко к середине расстояния между двумя W-атомами.
8.W8 023
Решётка моноклинная (?), производная от триклинной решётки WOa.
Восстановление WOa активным водородом приводит к образованию
W8023 и W4(k11. При этом происходит повышение симметрии решётки
до тетрагональной (W4Oxl).
9.W40U
Решётка тетрагональная, производная от триклинной решётки WOt.
Порошковые диаграммы приводили к выводу, что W40X1 обладает
однократно-примитивной ячейкой для атомов W, вершины которой заняты
атомами W, и имеющей а — 3,85; 6 = 3,79; с = 3,85; [3 = 79,45°. (Эта ячейка
не есть истинная элементарная ячейка.)
ДО. Система уран—кислород в структурном отношении была изучена
до самого последнего времени довольно плохо. Единственным
исследованным более или менее детально объектом был уранинит (UOn)*
Соответствующую таблицу
помещаем ниже.
11. U02, уранинит
Решётка кубическая С.
Координация кубо-тетраэд
рическая С/Т.
Тип Се/г —SB CI (CaF2).
Пространственная группа
Oh-Fm3m.
Z-4.
U к. ч.-=80 (С); О к. ч. =
= 4 U (Т).
Вещество
иоа
а«
5,47
d
2,37
ОМ
40,9
476 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VI
В последние годы был опубликован ряд работ, указывающих на
сложность системы U — О в структурном отношении (С. R. 224. 1395 (1947);
J. Am. Ch. S. 69, 2105 (1947) и 70, 99 (1948), Acta Cryst. 1, 265 (1948))
и намечающих границы существования в этой системе различных фаз
(Nat. 162, 69, 70 (1948)).
Вопрос о низших ступенях окисления U в этом свете выглядит
довольно однозначно: обнаружены фазы UO и U02.
Фаза UO имеет структуру каменной соли(ШС1), Nat., 162, 69 — 70(1948);.
Cojo с а =4,92 А, фаза U02 имеет структуру плавикового шпата, что
подтверждает более старые структурные данные, представленные в таблице
(уранинит).
Чистейший U02 имеет а =5,4581 ^ 5 А, причём в зависимости от
точности соотношения U : О величина а может меняться в небольших
пределах.
При нагревании U02 с U при температуре порядка до 2000° а
увеличивается в пределе до 5,4610 А; при нагревании U02 с U03 —уменьшается до
а =5,4297 ±S (предел), образуя в первом случае, повидимому, структуру
внедрения; во втором случае вероятны структуры, переходные к типу BiF3,
т. е. со статическим размещением избытка атомов О в центрах рёбер
и центре элементарной ячейки. (Если бы имело место неподчинение
этому требованию, то это составило бы редкое исключение из чётко
выраженных в других случаях тенденций структур типа CaF2 размещать
избыток атомов неметалла в указанных выше позициях.)
Несколько менее определённо выглядит в свете тех же литературных
данных вопрос о высших окислах урана.
В тех же работах рассматриваются окислы U206, U3Oe, U03 и
«гидрат U04». U205, повидимому, имеет OR (орторомбическую) элементарную
ячейку с а =8,27 ±2\ 6=31,65±i0; с =6,72 ±2\ ^=8,35; Z=16 U206
в ячейке.
Эта ячейка может быть сведена к псевдоячейке с а =4,135; 6=3,956;
с =6,72.
Было также найдено (1948), что U3Ofi, возможно, имеет ромбическую
элементарную ячейку с а =6,703; 6=3,969 и с =4,136 кХ; оЭКСП=8,34.
Позиции атомов U : (000) (1/а 1/2 0). Структура вычитания (легко
видеть связь между элементарными ячейками, описанными в этих двух
работах).
Имеются данные (Nat. 162,69 (1948)), что при 150°С окисление UOa
идёт до образования U02,24, имеющего кубическую структуру с а =5,40 кХ
(тогда как U02 отвечает а"=5,468 кХ); саксп =10,80 г/сл£3дляи02 и 11,05
для U02,24 г/см*.
Таким образом (из вышеуказанных литературных данных),
повидимому, высшие окислы U образуют на базе U205 или U808 (а может быть
U307 по С. R. 224, 1395 (1947)), широкую серию структур вычитания,
внедрения и, может быть, деления, причём окончательную картину вряд
ли можно считать установленной.
12. Система U-S.
В системе U —S были обнаружены (Z. Anorg. Ch. 243, 307 (1940))
фазы US3, US2, U2S2 и U4S3 (US0,75). Последняя из этих фаз «субсульфид»
US (изученная на базе порошковой диаграммы) образует кристаллы
кубической сингонии; авторами предполагалась структура вычитания (на
основе ячейки типа NaCl), в которой не занят атомом неметалла центр:
(7г 7г 7г)-
Таким образом, позиции атомов U : (000) (0 7з 1/й) О; атомов S:
(7. 0 0) С-
§ 158] ОКИСЛЫ II ХАЛКОГЕНИДЫ. VII* Г1-УППА: МАРГАНЕЦ-РЕНИЙ 477
Вещэстно а
1 Pu>O..S
с
3,919±<ф,755±-*0
cfa
1,72
VI** группа периодической системы: Np, Pu, Am, Cm
1. Pu202S
Решётка гексагональная Н.
Координация 7/6,4.
Тип #7/6,4-SB Z)52.
Пространственная группа /)3</ —СЗ/и.
М к. ч. = 4 (0) + 3 (S).
2. Pu2S3, Am2S3
Решётка кубическая С.
Координация 8/х.
Тип Cs/x (Ge2S3).
Пространственная группа 7^ —/43d.
Z = — . Идеальный состав при Ce3S4.
Z = 4.
Примечание. Acta Gr. I.
(1948) и 1157 (1949).
(Ср. La202S.)
225
Вещество
PuA
ArrVjS3
a
8,4373±5
8,428 ±«3
X
8,41
8,50
Примечание. Acta Gr. I,
255 (1948) и II 57 (1949).
(Ср. Ce2S3.)
§ 158. VII* группа периодической системы: марганец Мп, рений Re
1. Мп02 (рМп02?)
Решётка тетрагональная цепная Тк.
Координация октаэдрически-треугольная Oft.
Тип 7^,,-SB С4.
Пространственная группа Dm — Pijmnm.
2 = 2.
Мп к.ч. = 60 (О); О к. ч.-^ЗМп (t).
Рис. 331
Е. Я. Роде, В. Г. Кузнецов, В. П.
Васильева и 3. С. Головлёва (ИОНХ,
11СФХА XIX, 264 (1949)) нашли,
что из упоминавшихся в литературе
а-, В-, f и 5-форм существуют лишь
«3 Мп02» Тоцу см. таблицу, и у Мп
(решётка которой может быть
рассчитана как тетрагональная центрированная с а = 10,19; с = 8,07; с:а =
= 0,792), необратимо переходящая в ^Мп02 при 325—^80° С.
Вэщестио
МпО.
а
4,31)8
с
2,807
с/а
0,6519
478
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТ15
[ГЛ. VI
2. МпаО,
Решётка кубическая С.
Координация С"'-, (? IT.
Тип С -, , ,_ -SB Z>58.
с '-'с2 /т *
Пространственная группа T\ — JaZ.
Z = 16.
Mnl к. ч. =6 0 (С г)\ МпПк. ч. = 60 (Са_); Ок. ч. =4 Мп(Г).
Рис. 332.
Вещество
Мпа08
a
9,4Х
d
2,01
Примечание
Возникает из типа SB CI n результате
удаления части анионов и небольшого
смещения катионов. 6 атомов 0 попрежнему
занимают 6 из 8 вершин куба вокруг атомов Мп
В случае Мп I атомы О занимают вершины куба, кроме двух,
соединённых его пространственной диагональю. В случае Мп II атомы О занимают
вершины куба, кроме двух, соединённых диагональю грани.
3. Мп804 = Мп2Мп04, см. сложные соединения,
4. MnS2, см. стр. 674.
§ 1581 ОКИСЛЫ И ХАЛКОГЕНИДЫ. VII* ГРУППА: МАРГАНЕЦ-РЕНИЙ
479<
5. MnO, a MnS (зелёный),
aMnSe
Решётка кубическая С. :,
Координация октаэдриче- ^>^_
екая 0\0. \Jr
Тип Со/о-SB B\.
Пространственная группа
Obh-Fm3m.
Z = 4.
Mn к.ч. = 6 X (О);
X к. ч. = 6Мп (О).
<£^?1
'q^W
о
b^-W^^T'
О мп
Рис. :ш.
Ненастно
МпО
a MnS
f°
290° К
2J9° К
комн.
att
4,4345
4,43б±2
5,212±й
Mn —X
2,2172
2,218
2,61
Нещегтнл
a MnS
a MnSe
t°
1У9° К
комн.
fl«
5,210±2
5/t4
<* 1
Mn —X
2,72
MnS образует З модификации: a (зелёная), З (красная) и у (красная).
6. £ MnS красный, £MnSe (~\ __/Л
Решётка кубическая С. s^ri S^S
Координация тетраэдриче-
ская Т/Т.
Тип Ci/т- SB 53.
Пространственная группа
r-F43m.
Z = 4. Mn к. ч. =4 X (Т)\
X к. ч. = 4 Мп (Г).
Be то г т но
fi MnS
Р MnSa
"*■
5,600±2
5,82
Mn —S
2,4'?
Рис. 334.
480 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
7. y MnS (красная), у MnSe
Решётка гексагональная
Н.
Координация тетраэдриче-
ская Т/Т.
Тип #7/7 — SB 54.
Пространственная группа
Мп к. ч. = 4 X (Г);
Хк. ч.=4Мп(Г).
Рис. 335.
Вещзстгю
Y MnS
Y MnSe
а
3,976 £2
4,12
с*
6,432±4
6,72
с/а
1,617±2
. 1,63
d | Примечание 1
2,41—2,43
Интересно, чтэ
гексагональный CdS даёт
с MnS непрерывный
ряд твёрдых рас- 1
творов, тип СТ(Т (?) I
8. Re03
Решётка кубическая С.
Координация 0/L
Тип Со и- SB DO,.
Пространственная группа
0)г-РтЗт.
Z = i.
Re к. ч. =60 (октаэдр);
О к. ч. = 2 Re (прямая).
Вещество
Re03
aw
3.734
d 1
Re-0
1,87
Примечание. Re
образует простую
кубическую элементарную
ячейку (8), середины рёбер
которой заняты атомами
Рис. 336.
§ 159] ОКИСЛЫ И ХАЛКОГЕНИДЫ. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ 481
§ 159. VIII* группа периодической системы: железо Fe, рутений Ru,
осмий Os
1. Fe20,
Решётка ромбоэдрическая R.
Координация О/Т.
Тип До/т —SB ВЫ (корунд).
Пространственная группа D-^a—R3c.
Z = 2.
Fe к. ч. =6 0(0); О к. ч. =4Fe {T).
О Fe
Рпс. 337.
Вещество
Fea03
а
5,42
1
55°17'
d и
Fe —0
2,06
d'
Fe-0
l.Sl
2. yFe203, см. сложные соединения, тип шпинели.
3. Fe304, см. сложные соединения, тип шпинели.
О процессе образования окислов и окисления железа см. стр. 627, 628,
а также В. И. Архаров, Окисление металлов (1945).
482 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
4. FeO (вюстит)
Решётка кубическая С\
Координация О/О.
Тип С о fo -SB Bl (NaCl).
Пространственная группа
0\ — Fm Зт
Z = 4.
Fok.4. = 6O(0);Ok.4.=
= 6Fe(0).
Рис. 338.
Вещество
FeO
%
содержания Fe,
(атомн. %)
47,68
47,85
48,15
48,23
48,56
48,56
«„
4,2816
4,2847
4,2900
4,292i)
4,2997
4,2010
°Х
5,622
5,639
5,667
5,678
5,706
5,701
сэксп.
5,613
5,624
5,643
5,658
—
Примечание
На примере структуры
вюстита было
показано наличие в «FeO»
состава Fex0lt где
х ъ 0,94—0,97 (часть
узлов не занята),
см. § 230, а также
G.R. 205, 912 (1937)
б. FeS2, пирит. См. стр. 674.
6. FeS2, марказит. См.
стр. 675.
7. FeS (троилит), FeSe
Решётка гексагональная Н.
Координация О/Рб.
Тип Яо/рв-SB В8 (NiAs).
Пространственная группа
Dik — CG/mmc.
Z = 2.
Fo к.' ч. = 6Х (0);Х к. ч.=
= 6Fe (РЗ).
Вещество
FeS
FeSe
а
3,433
3,6t
с
5,860
5,8?
с/а
1,707
1,62б
ОМ
29,6
33,1
Рис. 339.
§ 159] ОКИСЛЫ И ХАЛКОГЕНИДЫ. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ 483
При наименьшем содержании S = 50 ат.%, отвечающем устойчивой фазе
помимо структуры типа NiAs, наблюдаются сверхструктурные линии,
отвечающие решётке с удвоенной осью с (а = 5,946; с = 11,720; с/а = 1,971;
Z= 12). С добавлением S к FeS сверхструктура исчезает, причём образуются
незанятые позиции Fe.
8. FeS, пирротин
Решётка гексагональная, а = 3,453 ± 9; с = 5,670 ± 20;
с/а = 1,644.
Пространственная группа £>б/и CL или £>*/,.
9. Ru04, 0s04
Молекулы Ru04 и Os04 имеют структуры,
в которых два расстояния М — О (d) гораздо
длиннее, чем два других (d').
Надёжных данных о кристаллической
структуре Ru04 и Os04 не имеется; такие
данные представляли бы большой
теоретический интерес.
d
d'
Ru04
1,66
2,74
Os04
1,79
2,81
10. Ru02, OsOa
Решётка тетрагональная Т.
Координация О/t.
Тип Tojt-SB C4 (рутил).
Пространственная группа D\h — P^/fnnm
Z = 2
М к/ч. =60(0); О к. ч.=ЗМ(0.
Рис. 340.
Вещество а
Ru02
Os02
4,5,
4,5,
с
3,^
3,1у
с/а
0,690
0,707
d
1,98
1,99
ОМ
31,6
32,5
Примечание
См. TiOa
484
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. VI
§ 160. IX* группа периодической системы: кобальт Со, родий Rh,
ирридий 1г
1. Со2Оа
Решётка
ромбоэдрическая R.
Координация О/Т.
Тип Roit - SB Z>51 (?)
(корунд) (метрика?)
Получение чистого Со203
удаётся, повидимому, с
большим трудом. См.
также SB II 228. Повидимому
возникают соединения ко»
бальта в промежуточном
валентном состоянии
между 2 и 3. Возможен также
тип шпинели, см. § 230.
2. Со304, см. сложные
соединения (тип
шпинели), § 230.
3. Co3S4 см. сложные
соединения (тип
шпинели), § 232.
т Рис. 441.
T^^tf
4. СоО
Решётка кубическая С.
Координация октаэдри-
ческая О/О.
Тип Со/о — SB 51.
Пространственная
группа 0\ — Fm3m.
Z = 4
Сок. ч. = 6 0(0);
Ок. ч. = 6Со(<9).
Рис. 342.
Вещество
СоО
*«
4,25
d
2,13
ОМ
19,0
§ 160] ОКИСЛЫ II ХАЛКОГЕНИДЫ. IX* ГРУППА: КОБАЛЬТ, РОДИЙ, ИРРИДИЙ 485
5. CoS, CoSe, CoTe
Решётка гексагональная.Я.
Координация 0/PQ.
Тип Яо/pe-SB Я8 (NiAs).
Пространственная группа /)6Л — Сб/ттс.
Z = 2
Со к. ч. = 6Х frt): X к. ч. - ПО (7>3).
Ох
Рис. 343.
Вещество
CoS
CoSe
CoTe
а
з,з7
3,59
3.*9
с
м4
5,2,
5,36
с/а
1,525
1,4б8
1,378
ОМ
25,3
35,1
6. Co3S4
В SBI Go3S4 относится к типу шпинели ЯП и сопоставлен нами с
другими соединениями того же типа (стр. 640). Однако, в SB VI 9 Co3S4
причислен уже к новому типу Dl2t пространственная группа 0\. В
работе, где помещено исследование этой системы и, в частности, данного
соединения (Z. Anorg. Gh 239, 85 (1938)), указывается, что период
идентичности кубической элементарной ячейки = 9,382. 8 атомов Со окружены
тетраэдрически, каждый четырьмя атомами S на расстоянии 2,19; 16
атомов Со окружены октаэдрически шестью атомами S на расстоянии 2,40.
Возникает вопрос о принадлежности остальных сульфидов (стр. 640) к тому
же типу.
486 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
7. CotS8, а также (Ni, Fe)e S8, пентландит
Решётка кубическая С.
Координация О, Т/^}Ъ,
Тип Co.774,5-SB £>8t (CofS8).
Пространственная группа 0\ — Fm3m.
Z = 4. Col к. ч. =6S(0); SI к. ч. -4 Со;
СоП к. ч. =4S(r); SII к. ч. =5 Со;
Os
Рис. 344.
Вещество
CoeSe
1 (NiFe)9S8
а
9,907
10,02
d
Co-S
Go I: 6/=2,48
Coll: d + 3e=2,14
Атомы S образуют плотнейшую кубическую упаковку Ск- В окта-
эдрических пустотах размещаются атомы Col, в тетраэдрических CoIL
SI-атомы тетраэдрически окружены атомами Со, принадлежат одновременно
4 тетраэдрам. Атомы SII принадлежат каждый одному октаэдру и 4
тетраэдрам.
§ 160] ОКИСЛЫ И ХАЛКОГЕННДЫ. IX* ГРУППА: КОБАЛЬТ, РОДИЙ, ИРРИДИЙ 487
8.
Rh20s
Решётка ромбоэдрическая
Л.
Координация О/Т.
Тип До/г — SB DM.
Пространственная группа
£&,-R3c.
2 = 2. Rh к.ч.=6 0(0);
Ок. ч. =4 ИЬ(Г).
Вещество .
а ....
а
\d ....
ОМ ...
RhaU8
5,47
55°40'
3d-2,l
3d'=2,0
52,1
Примечание. См. 1
ALO, и Ti,0, |
О Rh
Оо
Рис. 345.
9. 1гОа
Решётка тетрагональная цепная Т .
Координация Oft.
Тип Toit — SB C4 (рутил).
Пространственная группа В\кь — РЩтппт.
Z = 2.
1г к. ч. = 6 0(0); О к. ч.-31г(/).
Вещество
1 1гОа
а
4,49
с
3>ч
Рис. 346.
с/а
0,699
d
1,97
ОМ
31,7
Примечание 1
См. Ti<)2 J
488 ч. II. структуры неорганических веществ [гл. VI
§ 161. X* группа периодической системы: никель Ni, палладий Pd,
платина Pt
Высшие окислы Ni, Pd, Pt (M03) при обычной температуре и давлении
термодинамически неустойчивы (см. § 92).
1. NiO
Решётка кубическая С.
Координация октаэдриче-
ская О/О.
Тип Со/о-SB 51.
Пространственная
группа Oh — Fm3m.
Z = 4.
NiK. ч.=6 0(0);
Ок.ч. = 6 Ni(O).
Вещество
NiO
aw
4,172
d
2,08,
OM
18,2
Рис. 347.
2. PdO, PtO
Решётка тетрагональная Т.
Координация r/S.
Тип Trls.
Пространственная группа
Dih — РЩттс.
Z = 2.
М к. ч. = 4 О (г);
О к. ч. = 4 М(5).
Вещество
а
с
с/а
PdO
3,02 ±1
5,31 ±1
1,758
PtO
3,04±J
5,34 ±5
1,757
На рис. 348 две элементарные
ячейки.
В SBI тип решётки PdO
был определён ненадёжно.
Сходство с РЬО не имеет
места. В J. Am. Ch. Soc. 63,
1392 (1941) даны иные
значения параметров атомов и
строения координационной
сферы. Позиция атомов:
Рис. 34S.
Pd (000), (-i- \ \у, ° (4 ° тУ (4 ° Ю • Атом Pd ыах°Дится в Центре
прямоугольной координационной сферы (г) с углами между диагоналями 820,
§ 161] ОКИСЛЫ II ХАЛКОГЕНПДЫ. X» ГРУППА: НИКЕЛЬ, ПАЛЛАДИЙ, ПЛАТИНА 489'
ж 98°. Атом О — в центре тетрагонального тетраэдра (S) с углами между
связями 98° и 116°. Pd-O = 2,01 и Pt-O = 2,02 kX.
3. Pt,04
Решётка кубическая С.
Координация С/С"'-.
Тип Ссс-/-.
Пространственная группа (?) Возможны Т\ Т*,0'9 О8, Т%, T]lf T%J*dl 01 0JV
Z = 2Pt804. Pt к. ч.-80(e); О к. ч. = 6 Pt(C"/-).
2^S
%
tf
<D
ЬШШ
Рис. 349а.
Вещество
Р*304
а
Pt—0
6,226 2,192
Pt-Pt
3,113
«х
8,8
' Примечание 1
По J.Ch.phys. 9,87Г»(1941)
Если структура (и состав) отвечают указанным выше данным, то
структура эта представляет интерес в том отношении, что она должна
рассматриваться как один из новых членов ряда простых кубических
структур, близких к типу флюорита (плавикового шпата).
В элементарной ячейке CaF2 при Z = 4 находится 8 атомов фтора
в центре октантов и 4 атома Са, в вершинах элементарной ячейки и в
центрах граней. В элементарной ячейке Pt304npnZ=2 находится 8
атомов кислорода и 6 атомов платины. Первые занимают центры октантов
(сравни со структурой плавикового шпата), атомы же Pt занимают
позиции в центрах рёбер (у 00)Q^ и центрах граней ГууО ) ^'
В цитированной работе даётся грубо приближённое значение важного
расстояния Pt — 0=2,2 kX. В действительности же это расстояние
я=-|-]/2, т. е. при а =6,226, х =2,192 расстояние Pt—Pt равно у,.
т. е.*3.113 kX.
490
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
1гл. VI
Координационная сфера атома Pt имеет форму куба;
координационная сфера атома О—форму ~ тригональной антипризмы СГ'-, как мы
показали на рис. 349Ь.
Pt,04 и система Pt —О заслуживают, на наш взгляд, дальнейших
исследований и проверки, в силу значительного теоретического интереса,
который они представляют; см. стр. 540, 541.
4. Из хадкогенидов типа АВ2 следует
упомянуть изоморфный пириту NiSa. г\
Он будет рассмотрен в гл. IX (комп- Г^^_^±_
лексные соединения). \^^т^^^Г
5. Ni,S2
Решётка ромбоэдрическая Д.
Координация 4/6
Тип /?4/c(Z. Anorg. Gh. 239, 82 (1938)).
Пространственная группа D\ — Д32;
а = 4,041; а = 90,3°; ах = 5,87.
6. fNiS, yNiSe
Решётка ромбоэдрическая цепная RK.
Координация Pyi/Pyi = 5/5.
Тип Д*5 = Rpyi/Pyi — SB S13 (миллерит, Рис> 349Ь.
NiS).
Рис. 350.
Пространственная группа Сз„ — ДЗяг.
-Z»=3.
Ni к. ч. = 5 X (~Ру4); X к. ч. = 5 Ni (~Рг/4).
|_161] ОКИСЛЫ II ХАЛКОГЕНИДЫ, X* ГРУППА: НИКЕЛЬ, ПАЛЛАДИЙ, ПЛАТИНА 491
] Вещество
| yNIS
1 *
1 -fNiSe
1
а
9,60
9,62
9.84
с
3,15
3,17
здн
с:а
0,32
0,32
d
2,35
yNIS получается из раствора сульфата + H2S. В короткий срок
yNiS и yNiSe превращаются в р-формы. По SB II миллерит
(ромбоэдрическая ячейка) имеет а = 5,63, а=116°35\ NiS-«uenn» соединены друг
с другом мостиками NiS, являющимися кратчайшими Ni —S-расстояниями.
Поэтому по SB II 6 решётка скорее молекулярная, чем цепочечная,
с к. ч. = 5 (Рг/4).
7. pNiS, NiSe, NiTe
Решётка гексагональная Н.
Координация О/РЗ.
Тип Яо/рз-SB £8(NiAs).
Пространственная группа D^h — C 6jmmc.
Z = 2. Ni к. ч. = 6 Х(О); X к. ч. = 6 Ni (Р'З).
о
Рис. 351.
Вещество
PNiS
NiSe
NiTe
а
v.,
з,п6
3,95
с с'а
5,30
5,33
5,3G
1 *\Г1
о
1,357
ОМ
20,8
30,9
30,2
Примечание
aNiS является аморфным
jiNiS получается из
раствора ацетата никеля-fH2S I
492 ч. и. структуры неорганических веществ [гл. VI
8. NiTea
Решётка гексагональная
слоистая Hs.
Координация Ojt.
Тип #5yj-SBC6.
Пространственная группа/)^—
— СЗт.
2 = 1.
Ni к. ч. = 6Те(0); Те к. ч.=
= 3 Ni (*).
Рис. 352.
Вещество
NiTaa
а
3,861
с
5,297
С увеличением содержания Те, NiTe—тип Яо/рз—SB 58 (см. стр. 491)
переходит в NiTe2 с постепенным изменением периодов решётки. NiTe3,
кристаллизуясь, повидимому не образует структур типа SBC18 (марказит),,
см. гл. X, в отличие от Fe, дителлурид которого образует решётку типа
SBC18, но не SBC6; см. SB VI 166.
§ 161] ОКИСЛЫ И ХАЛКОГЕНИДЫ. X* ГРУППА: НИКЕЛЬ, ПАЛЛАДИЙ, ПЛАТИНА 493
9. PtS, куперит
Решётка тетрагональная 7\
Координация r/S.
Тип Trjs-SB Bll(PtS).
Пространственная группа
£>?ь — РЩттс.
Z = 2.
Pt к. 4. = 4S(r); S к. ч.=
«4Pt(Sj.
Рис. 353.
Вещество
а
с
с/а
<*
Pt—S
| PtS Куперит | 3,47 | 6,10 | 1,758 | 2,32 1
Тип близок к В'З (Сг/т)' Однако тогда как координационная сфера серы —
тетрагональный тетраэдр, координационная сфера Pt —плоский
прямоугольник, почти квадрат.
10. PdS
Решётка тетрагональная Т.
Координация r/S.
Тип 7V-SBtf34(PdS).
Пространственная группа C\\x — Pk*\m.
Z = 8.
Pdl к. 4. = 4S (r)(/= 2,43);
Pdll к. 4. = 4S(r) (d = 2,26);
PdHI к. ч. 4S(r) (e = 2,29 и 2,34);
S к. ч. = 4 Pd(S).
Атомы S занимают вершины прямоугольников (близких к квадратам),
в центре которых атомы Pd. Atom Pd занимает вершины
деформированных тетрагональных тетраэдров, в центре которых — атомы S. Структура
подобна структуре PtS, но повидимому, менее правильная.
Вещество
PdS
а
6,43
1
с
6,63
с'а
1,03
494 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. V
§ 162 I* группа периодической системы: медь Си, серебро Ag, эолото Аи
1. СиО (тенорит)
Решётка моноклинная М.
Координация г/Т.
Тип il/r/,F-SB 526.
Пространственная группа Сгл — С2/с.
Z = 4.
Си к. ч. = 4 О (прямоугольник);
О к. ч. = 4 Си (искажённый тетраэдр).
Вещество
СиО
а
4,653±10
Ъ
3,4Ю±10
с
5,1О8±10
а : Ъ : с
1,365 :1 :1,498
Р
99°29'
°х
6,569
cuto
1,95
Каждый атом Си окружён 4 атомами О, образующими плоский
прямоугольник со сторонами 2,62 и 2,885 А. Расстояние О — Cu=l,95kX.
Каждый атом О окружён 4 атомами Си, образующими искажённый тетраэдр
с теми же расстояниями Си — О.
2. Ag2Oe
Решётка кубическая С.
Координация?
Тип С-SB?
Пространственная группа Т\~ Fi3m или 0е — F43 или О* — Fm3m. Z = 16.
Вещество '
Ag203
<*„
9,82±«3 (монокристалл)
до 9,91> , к (порошок)
Примечание 1
В литературе отмечается сходство Ag^O 1 и]
Aga03 и допускается внедрение атомов 0t в ре-|
шётку AgaO. Характер связей и структурных!
узлов не уточнён. Данные сомнительны. 1
Вопрос о структуре окислов и сульфидов серебра (и золота) пока, в целом,,
остаётся открытым.
§ 162] ОКИСЛЫ II ХАЛКОГЕНИДЫ. I* ГРУППА: МЕДЬ, СЕРЕБРО, ЗОЛОТО 495
3. АиТе2, креннерит
Решётка ромбическая молекулярная ОЯм.
Координация 4, 6, 4.
Тип (9Д4,6,4 -SBC46.
Пространственная группа C^v — Рта.
Z = 8. Auk. ч. = 2Те; Тек. ч. = 1 Аи.
Принимая [АиТе2] = а, находим, что al к. ч. = 4a; all к. ч. = 6э
аШ к. ч. = 4а.
Рис. 354.
Вещество
АиТе8
а
16,51±3
Ь
8,80±,3
с
4,45±<3
а : Ъ : с
1,876 : 1 :0,506
d
2,56—2,73
Примечание
SB IV 15
Структура исключительно интересна. В решётке три типа молекул
AuTe2:Aul (симметрия С2 — 2) имеют 2 атома Те на расстоянии 2,65.
Aull (симметрия Cs — тя) —на расстоянии 2,56 и 2,73. AuIII (симметрия
С1 — 1) — 2,64 и 2,65. В среднем d (Au — Те) = 2,65. Вопреки всем
ожиданиям, группа Те2 не является комплексной, т. е. не образует, повиди-
мому, структурного узла Те2. Угол Те —Аи —Те близок к 180°. Молекулы
AuTe примерно параллельны, что также заслуживает внимания.
Расстояния Те —Те (кратчайшие) = 3,03 А.
496 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VI
4. АиТе2, калаверит
Решётка моноклинная М.
Координация 2 + 4/6.
Тип Л/2+4/б.
Пространственная группа
С\ — С2 или C\h — С2//и.
Au к. ч. = 6 Те (2 + 4);
Те к. ч. = 6 Au.
Вещество .
\ а
\Ъ
\*л- • ' •
а: о : с . .
Примечание
АиТва 1
7,IS
4,40
5,07
90°±30'
1,632 :1 :1,523
SB III30
б. CuS, ковеллин —см. стр.
639.
6. Cu2S, высокотемператур-
^ный халькозин
Решётка гексагональная Н.
Координация 3/3 (+3).
Тип #з/з (+3)-
Пространственная I группа
Dh — C6/mmc.
Z}= 6.
Вэщзство
а ....
с .
с/а . .
Cu2S
.4,89
6,68
1,717
Примечание. По
Белову и Бутузову, ДАН
54,721 (1946)
Рис. 355Ь.
Рис. 355а по Н. В. Белову и [В. П. Бутузову. Структура содержит
два типа позиций Си: Cul и Cull. Для большей наглядности этот чертёж
нами перестроен (см. рис. 355Ь). Исключительный интерес представляет,
что к. ч. =3 у Си и S.
§ 162] ОКИСЛЫ PI ХАЛКОГЕНИДЫ. I* ГРУППА: МЕДЬ, СЕРЕБРО, ЗОЛОТО
497
7. Cu2S, низкотемпературный халькозин
Решётка ромбическая OR.
Координация?
Тип?
Пространственная группа?
Z = 96.
Вещество
Cu3S
а
11,90
. 1 .
27,28 13,41
i
Примечание
Am. Min. 27,712 (194:2)
8. aCu2Se, aAg,Te
Решётка кубическая С.
Координация сложная. .
Тип промежуточный между Ст/т — SB В'З и Сс/т — SB C1.
Z = 4.
Вещестно
aCu,Se
aAgaTe
* op _
170
250
5,840±6
6,572±W
Вещество
aAgaSe
taC
170°
*"
4,933
Если aGu2Se при комнатной температуре имеет структуру С1 с
«примесью» кубической гранецентрированной, то при более высоких
температурах наблюдается кубическая грапецентрировапная решётка ZnS (Ст/т),
в промежутках которой добавочно размещено равное количество катионов.
aAg2Te устойчив при /°>149,5"С. Кубическая гранецентрированная
решётка со статистическим распределением атомов Ag. Параметры атомов
надёжно не установлены.
9. aAg2Se
Решётка кубическая С.
Координация?
Тип, производный от Сс —SB B2('f)
Z = 2.
/1 1 1Л
Устойчива выше 133° С. Позиции (000) и (— — — J занимают стати-
стычески атомы Ag и Se. Кроме того Ag занимает узлы (— 00 JO или
a«^o+G-o4)o......
Система Ag —Те исследовалась неоднократно, причём надёжные
данные не были получены. SB VII 210 реферирует приводимые сведения по
фазам aAgTe (OR) и aAgl2Te7 (см. ниже).
10. aAgoTe «низкотемпературная»
Решётка, вероятно, OR.
Координация?
Тип?
Пространственная группа?
Z = ? а - 13,0; Ь - 12,7; с 12,2.
498 ч. н. структуры неорганических веществ [гл. VI
11. a.AgiaTe;
Решётка гексагональная Н.
Координация?
Тип?
Пространственная группа Dlh — Cfymmm или Dq — С62.
а = 13,429; с = 8,4508; с/а = 0,6297.
Z = 3 молекулы.
12. Cu20, куприт, Ag20
Решётка кубическая С.
Координация линейно-тетраэдрическая 1/Т.
Тип СцТ -SBC3. Z = 2.' Си к. ч. = 20 (Z); Ок. ч. = 4Си(Г).
Пространственная группа Oh—Pn3m.
? ? f { о-си О-о
Рис. 356.
Вещество aw d
Си20 Куприт
Ag20
4,26
1,84
2,05
ОМ
38,6
53,2
Примечание
См. § 61
Сопоставь с типом С9. СЗ как бы
состоит из 2 вставленных друг
в друга решёток С9
Да рис. 356 показано 8 элементарных ячеек, см. рис. 95 и 96.
Электронографические исследования (SB VI 61) показали, что при
окислении кристаллов Си образуется Си20 с а = 4,258 — 4,27i±4» тогда
как для природного куприта aw = 4,284 — 4,294.
5 163] ОКИСЛЫ И ХАЛКОГЕНИДЫ. И* ГРУППА: ЦИНК, КАДМИЙ, РТУТЬ 499
При рассмотрении окислов и халкогенидов I* группы особенно
бросается в глаза, что, как и в случае галогенидов, почти не изучены
соединения золота, в частности, фазы, возникающие в системах Аи—О,
Aii —S, Аи —Se.
Окислы и халкогениды Ag также исследованы недостаточно, их состав
и структура нуждаются в уточнении.
§ 163. II* группа периодической системы:
цинк Zn, кадмий Cd, ртуть Hg
1. ZnO, ZnS, вюрцит, CdS, CdSe.
Решётка гексагональная Н.
Координация Т/Т.
Тип Htjt — SB M.
Пространственная группа Съо — С6тс.
Z — 2.
М к. ч. = 4Х (Т)\ X к. ч. = Ш{Т).
о*
Рис. 357.
Вещество
ZnO
ZnS
CdS
CdSe
t°C
18
—
—
a
3f24265,.10
3,8'*
4,131±3
4,30
с
5,19'i8±<3
G,28
G,G91±7o
7,01
c/a
1,G020±2
l>*\
l,G20±fl
1,G3Q
OM
—.
40,0
50
56,3
По литературным данным, CdS тип HTfT образуется главным обра
зом при пропускании H2S в раствор галогенидов Cd.
Одна из основных структур (см. § 65).
500
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. VI
2. ZnS, сфалерит, CdS,
HgS, метациннабарит
Решётка кубическая С.
Координация Т/Т.
Тип Ctit — SB 53 (ZnS).
Пространственная группа?
rd-F43w.
Z = 4.
М к. 4. = 4S (тетраэдр).
S к. ч.=-4М (тетраэдр).
Вещество
ZnS
CdS
HgS
aw
5,42
5,81
5,83—
5,85
d 1
M-S
2,35
2,52
2,53
Pnc. 358.
Одна из основных структур, см. § 63.
По литературным данным CdS тип Ctit получается главным образом
пропусканием H2S в горячий раствор нитрата и сульфата Cd.
3. CdO
Решётка кубическая С.
Координация октаэдри-
ческая О/О.
Тип SBB1.
Пространственная
группа Of,—Fm3m.
Z = 4.
О^
?^т
ьз-р-
Рис. 359.
Вещество
*и>
4,70
CdO I
1 J 4>б99
cdio ! ом
2,35
*> 34,-.-
°х
26,0 ( —
26,0 8,1б4
SB I и II. Имеются данные, что избыток Cd в решётке CdO ведёт
к увеличению aw на величину до 0,007 А. Одновременно с увеличением aw
препарат темнеет.
§ 163] ОКИСЛЫ И ХАЛКОГЕНИДЫ. II* ГРУППА: ЦИНК, КАДМИЙ, РТУТЬ
501
4. HgO жёлтый и красный
По мнению отдельных авторов, различаются не структурой решётки, но
степенью, дисперсности (жёлтая HgO гораздо более дисперсна*)).
Решётка ромбическая OR,
Координация?
Тип?
Пространственная группа?
Z = 2.
Вещество
HgO
а
3,296
Ь
а, 513
с
Г), 504
°х
11,22
Структуру HgO нельзя считать надёжно расшифрованной. Повидимому,
как и в случае других веществ, структура окислов Hg зависит от метода
приготовления. См, также Z. Anorg. Ch. 244 133 (1940).
5. HgS, киноварь
Решётка
гексагональная Я.
Координация 0[0.
Тип //5/0-SB B9.
Пространственная группа
(Dl Я'!, С! или Сз) (?)
Z = 3.
Hg к. ч. = 6S (сильно
искажённый «октаэдр»).
S к. ч.—-6 (октаэдр, S не
в центре последнего).
Рис. 360.
*) Эта точка зрения подвергалась критике (см. Z. Ph. Ch. А ЗЗЯ, 311 (1928)).
502 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
>б. HgS, киноварь (продолжение).
i 1 i i i i
Рис. 361.
Вещество
1 HgS, киноварь
а
4'Ч + 2
с
9'49 + 5
с/а
2 2
'92 + 13
ОМ
47,0
Сильно деформированная решётка NaCl (-B1), в которой атомы Hg
занимают нормально узлы Na, а атомы S смещены (рис. 361).
§ 164. III группа периодической системы: бор В, алюминий А1,
галлий Ga, индий In, таллий Т1
1. В208
Решётка кубическая С.
Координация?
Тип?
Пространственная группа?
Z=16.
Вещество '
вао3
fl» ;
10,035
сх \ Примечание 1
1,818
Вг03 сильно полимернзован, судя по
большому интервалу жидкого состояния
(294—1860° С) и большой энтропии
испарения (32,2). Допускается наличие в
твёрдом В203 различных полиморфных форм
§ 164] ОКИСЛЫ И ХАЛКОГЕНИДЫ. Ш ГРУППА: БОР, АЛЮМИНИЙ, ТАЛЛИЙ 503
Рис. 362а.
Вещество
»А1аО,
а
5,12
а
55*17'
d
Me -О
3d=l,93
3d' = l,89
ОМ
42,7
Вещество а
а Ga203
^8
2
55°35'
с*
Me-О
3d=2,0
ОМ
46,7
Ha рис. 362a показана элементарная ячейка корунда. Главная ось —
в плоскости чертежа. На рис. 362Ь показана группа атомов,
ограниченная фигурной скобкой на рис. 362а; главная ось на рис. 362Ь почтя
лерпендикулярна плоскости чертежа. На рис. 362а выделены в SB1
504 ч. и. структуры неорганических веществ [гл. VI
«молекулы» А1203, причём правые молекулы отличаются от левых
штриховкой. На рис. 362Ь выявлены координационные соотношения (тетраэдр
из атомов А1 вокруг атома О
и октаэдр из атомов О вокруг
атома А1).
Как указывает SB I 241,
решётка корунда может быть
представлена как
ромбоэдрическая деформированная
решётка типа NaCl(fil), оба сорта
атомов которой заменены
двумя энантиморфными сортами
молекул А1203.
3. f*AI203
Решётка гексагональная Н.
Координация 4,6/2,3.
Тип #4,6/2,3—SB Z)56 ([3-корунд).
Пространственная группа Dhi —
СЩттс.
Z=12.
Рис. 363.
1 '
Вещество | а
РА1а03
5,62
с
22,5
с/а
4,0о
с*
3,31
Примечание
SB И 41—43
В структуре имеются
небольшие искажения
«следствие примеси Na20
§ 164] ОКИСЛЫ II ХАЛКОГЕНИДЫ. Ill ГРУППА: БОР. АЛЮМИНИЙ, ТАЛЛИЙ 505
4. уА1203, см. сложные соединения, тип шпинели.
5. у'АКОз
Решётка кубическая (гранецеитрированная?) С»
Вещество
?'А1205
аи>
3,95
Примечание
Образуется при электрохимическом
окислении AI-анода. В Y имеет место статистическое
распределение катионов, тогда как в у
наблюдается частичный порядок; см. § 230.
Рис. 364
в. 1п208, Т1203
Решётка кубическая С.
Координация С-'-, С*"/Т.
Тип Cc-/->c=/r-SB ВЬ3 (МпА)-
Пространственная группа Т\ — ]аЪ.
Z = 16. M I к. ч. = 6(С"/-);
М II к. ч. = 6 О {С").
О к. ч.-=4 (Г).
Вещество
1п303
Т)а03
«1Г
ю,12
10,5„
d
Примечание
2,16 J См. MibOJ
2,20
[ |
506 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VI
^8
7. TISe
Решётка тетрагональная Тв (полосы).
Координация 7\ Pi[Pi.
Тжп Тт.рцрь —SB 537.
Пространственная группа /)\л — Ji/mcm.
Z = 8.
Tl I к. 4. = 8Se (7>4); Tl II к. 4. = 4Se (T);
Se к. ч. = 8 (i>4).
Рис. 365.
Вещество
TISe
1
a
8,02
с
7,00
с/а
0,873
Атомы Tl] II (на боковых гранях элементарной ячейки) окружены
слегка деформированными тетраэдрами Se, образующими пояса вдоль
оси с. Атомы Т11 также образуют цепи вдоль оси с, проходящие через
центр и вершины элементарной ячейки, через центры рёбер которых
проходят пояса Т1II—Se. ТП-атомы лежат на параллельных оси с ребрах и на
оси 4 элементарной ячейки.
Общие замечания
А1аОя
•три- или тетраморфен (?), aAl2Os —ромбоэдрическая, (ЗА1303 —
гексагональная, yA1203 — кубическая, устойчива около 930°С (?), y'A1203 —
кубическая, оа203 — диморфея (?), auaaOs — ромбоэдрическая, ?Ga203 более
низкой симметрии —моноклинная или ромбическая (?), ^Ga2Os возникает
выше 650'С.
Окислы типа М20, (но не их фазовые переходы!) всех элементов
III группы изучены довольно хорошо; однако уже сульфиды, селениды
и т. п. практически не исследованы. Кроме того, не изучены низшие
окислы в тех случаях, когда их образование имеет место (In, Tl).
§ 165] ОКИСЛи И ХАЛКОГЕНИДЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ 507
§ 165. IV группа периодической системы: углерод С, кремний Si,
германий Ge, олово Sn, свинец РЬ
Углерод образует с кислородом летучие окислы С02, СО, субоксид
С,02, а также летучие соединения с серой и селеном CS2, CSe2.
Молекулы С02, CS2, CSe2 и, естественно, СО линейны.
Расстояния С —X и X — X показаны ниже.
Расстояния
сой
С-Х | 1,15
X—X 2,30
сяа
1,54
3,08
CSea
—
СО
1,13
Возможная структура С302 обсуждалась в 1933 г. в работе Паулинга
и Броквея; по их мнению, из двух структур
С = С
| | и 0=С=С=С=0
о-с = о
наиболее вероятная вторая. Для неё е?с-о = 1.20± 2,
С-С=1,30±2,
причём имеет место сулерпозиция состояний
:0 = С = С = С = б: , :О^С-С^С-ОГ и70-С=С-С=ьО:
1. С02
Решётка кубическая Сл/ (,).
Координация К (12).
Тип Ck(0-SB СЪ (пирита)
Пространственная группа
ТЧ-РаЗ.
а = СОг.
Z = i. С к. ч. = 2 0(1).
а к. ч. = 12 {К).
ъл
(Центры тяЖеши
молекул)
Вещество
С02
г °К
0 (экстраполяция)
20,4
35
65
83,1 (190° С.)
86
94
111
114
а
5,540
5,542
5,546
5,560
5,574 + 5
5,574
5,581
5,597
5,602
Рис
d
С-О
1,12
—
—
—
—
—
—
—
,. 366.
с*
1,708
—
—
—
1,68
—
—
"
ОМ
42,50
43,25
Точность отсчётов 1~С.
508 ч. и, структуры неорганических веществ [гл. VI
В SB III 294 для температуры 0° К, при а = 5,540, дан ОМ= 25,76
(ошибка? Эта величина отвечает граммолекулярному объёму в см3).
Ск-решётка, если считать С02 = а (см. положение атомов С
—центров тяжести линейных молекул СОо), см. также Helv. Phys. Acta 14
324 (1941).
2. аСО
Решётка кубическая гранецентрированная CMW.
Координация К (12).
Тип С к-SB £21.
Пространственная группа Т* — Р213] сс = СО.
Z-4.
о
Рис. 367.
Вещество
аСО
aw
5,63
d
С—О
1,065
*х
1,03
аСО имеет ту же структуру, что и aNa, т. е. С к, если принять
СО = а.
tn для aCCCPCO^ei^K (tn„aBJI. £СО = 68,2' К). В SB II 14 ошибочно
указано расстояние dc-o = l>51 Л.
§ 1651 ОКИСЛЫ И ХАЛКОГЕНИДЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ 509
3. &С0
Решётка гексагональная //.
Координация V (12).
Тип Як-SB A3
(узел а = СО); к. ч. для
a =12.
2 = 2.
Веще-
стла
jiCO
65° К
а
с
с/а
4,11 . 6,70 j 1,651
[) Центр тяжести СО
Рис. 368.
рСО имеет ту же структуру, что [iN3. Молекулы вращаются. Их центры
тяжести образуют примерно плотнейшую гексагональную упаковку.
С
т р у кт у р ы окислов кремния
Прежде чем перейти к изложению материалов о строении окислов
кремния, мы должны остановиться на основных чертах фазовых
переходов в этой сложной системе.
Двуокись кремния образует большое количество кристаллических
форм, исследование которых различными методами продолжается уже
несколько десятилетий. Всё же в современных монографиях состояние
вопроса освещается главным образом на базе старых работ. Более того,
даже в наименовании фаз существует расхождение между структурной
и физико-химической литературой.
В физико-химических работах обычно принимается, что существует
7 кристаллических форм, а именно: (3-кварц, а-кварц, у-тридймит, [3-три-
димит, а-тридимит, |3-кристобалит, а-кристобалит. В этом описании
наиболее высокотемпературными являются а-формы. В структурной
литературе наиболее высокотемпературными являются $-формы.
Методами физической химии и др. давно установлено, что практически
наблюдаются быстрые фазовые переходы: друг в друга различных форм
одной и той же структуры (например, р-кварца в а-кварц) и очень
медленные переходы от одной структуры к другой (например, кварца—в кристо-
•балит или трпдимит).
510 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
Поэтому в основном ряду фазовых переходов (обозначения «физико-
химические»)
р-кварц^±л-кварц^±а-тридимит^а»крисгобалит
при t° С 575 870 1470
быстрым является только переход р-кварца в а-кварц (и наоборот),
остальные же протекают крайне медленно. В связи с этим высокотемпературные
формы: кристобалит и тридимит сохраняются при более низких
температурах, не переходя, как дравило, в кварц.
Напротив, переохлаждённый высокотемпературный кристобалит
(кубический) в интервале между 198 и 275°С, или, как часто принимают,
при 230° С, переходит в низкотемпературный триклинный кристобалит.
В свою очередь, переохлаждённый высокотемпературный гексагональный
тридимит переходит при 163°С в тригональный; последний при
температуре 117° С переходит в низкотемпературную ромбическую
форму—тридимит.
В результате, общая картина фазовых переходов воспроизводится
следующей таблицей (обозначения фаз, принятые в структурных
исследованиях):
Вещество
Вещество
Температура i
перехода 1
(быстр.) °С
Вещество
1 Температура
перехода
(быстр.) °С
1 Вещество
Р-кварц
| 579
У
а-кварц
Температура
перехода
(медлен.)
<н-н>
870
Вещество
у-тридимит
1
р-тридимит
t 117
а-тридимит
Температура
перехода
(медлен.)
°С
< ►
1470
Вещество 1
3-крпстобалит|
* 230
1
а-кристобалит|
Формы, указанные в верхней части таблицы, являются наиболее
высокотемпературными формами.
В рентгеноструктурных исследованиях было показано, что все
кристаллические формы двуокиси кремния состоят из тетраэдров,
соединённых между собой различным образом. Переход, например, кварца
в тридимит требует частичного разрыва связей и вследствие большой
энергии активации протекает медленно. Напротив, различные формы одной
и той же модификации, например [3- и а-кварц, отличаются лишь
небольшими различиями в поворотах тетраэдров по отношению друг к другу.
Их взаимный переход не требует разрыва связей и протекает быстро.
Так, левовращающий (3-кварц может быть переведён в а-кварц; по.
охлаждении он вновь оказывается левовращающим вследствие
неизменности связей.
Так как в рентгеноструктурных работах чаще называются а-формами
Si02 низкотемпературные формы, ниже мы сохраняем эти рентгенострук-
турные обозначения, обращая внимание читателей на наличие
расхождений между рентгеноструктурными и физико-химическими наименованиями,
фаз в рассматриваемой системе.
§ 165] ОКИСЛЫ II ХАЛКОГЕНИДЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ 511
4. Низкотемпературный а-кварц Si02, Ge02 («растворимый в воде»)
Решётка гексагональная Hv.
Координация Tj2 = TJZ_.
Тип #t/z-SB C8.
Пространственная группа Dl — СЪХ2 или Dl — CS^l.
Z = 3. Si к. ч. = 4() (2 по 1,61 и 2 по 1,62) (Т); О к. ч. = 2 Si(Z)*
Рис. 369.
Вещество
SiCK
низкотемпературный кварц
GeO,
t
(18° С)
а
4,90,
4,98
с
5,393
5fG4
с/а
1,099
1,1,
d
Si-О
2^=1,61
2^ = 1,62
По SB III 21 (1935).
8Ю4-тетраэдры образуют пространственную вязь таким образом, что
каждый атом О принадлежит сразу двум тетраэдрам. Описания SBI 16ft
неверны.
512 Ч. П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VI
5. ВысоБОтемпературный р-кварц Si02 (и его переход в а-кварц)
Решётка гексагональная Ю\
Координация Г/2.
Тип Д?/2.
Пространственная группа?
Рис. 370.
Рпс. 371.
Вещество
/°С
единиц X
"100
единиц X
Примечание
aSi02
I
fiSiO,
ЛЯ
118
280
413
558
567
570
590
610
665
7i:0
5393,3
5398,1-
5406,8
5416 А
о
5433,8
5435,5
5446,4
5447,3
5447,0
5446,4
5445,9
4246,0
4252,з
4264,6.
4277,7'
4299,0
4302,7
4318,1
4319,6
4319,6
4319,6
4319,5
Переход aSi02 £ P Si02 (кварц)
изучался в интервале 18—730°С,
причем определялись значения С
и расстояния между сеткамиd1Q0.
Выло найдено /п (*-»р)=579°С.
Повидимому, всё же этот переход
нроисходит в размытом интервале
579—610°С. Переход р-кварц->
a-кристобалит изучался на
образце , содержащем 96,32% Si О;,. До
600°С наблюдалось сильное
расширение, от 600 до
^1100°—значительное сжатие. После 1100°
сильное расширение, увеличивающееся
с температурой. Между 1400 и
1450°С линии [i-кварца и кристоба-
лита. После двухчасового
нагрева при 1500° только кристобалит.
Ыа рис. 371 показано расположение атомов Si в р-кварце по
сравнению с a-кварцем (рис. 370). Разная штриховка показывает высоту
атома Si над плоскостью проекции.
§ 165] ОКИСЛЫ И ХАЛКОГЕНИДЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ 513
6. Низкотемпературный а-кристобалит Si02
Решётка тетрагональная Tv.
Координация тетраэдрическаи линейная T\Z_.
Тип,J?,^-SB C30.
Пространственная группа Z)* —/)4121 или D\ — Diz2r
Z = 4.
Si к. ч.=:4 0(Т); О к. ч. = 2 Si(Z)
Li! f ■ ■ ■ f oo
Piic. 372.
Вещество
Si02
{Низкотемпературный
кристобалит
a
4'%0±5
с j с/а
1
&.920±5
1,395
d j
Si—0
1.59
Псевдокубическая решётка имеет а' = 7,02o±s; с' = 6,92^±з; c'/a' =0,986.
Структура близка к С9 —высокотемпературный кристобалит, отличаясь
от последнего тем, что [8Ю4]-тетраэдры в СЗО испытывают винтовое
движение вокруг оси а' на 22° и 0,05 а'. Атомы О —Si —О не лежат
на прямой (^/ = 150°).
514
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. VI
7. Si02 меланофлогит
Минерал из
Сицилии образует
правильные кубы с
длиной ребра порядка
2 мм. Кристаллы
дают порошковую
диаграмму (Z. Krist.
(А) 99, 238 (1938)), не
поддающуюся инди-
цированию, исходя
из кубической или
тетрагональной
решётки, но
хорошо
удовлетворяющую гексагональной
структуре с а =--4,907;
с = 5,396; с/а = 1,0996
(ср. а-кварц), т. е.
структура идентична
а-кварцу рис. 373.
Очевидно, что
меланофлогит был
прежде
высокотемпературным (3-кристоба-
литом, который
постепенно
превратился в
низкотемпературный а-кварц.
8.
Высокотемпературный р-кристо-
балит Si02
Решётка кубическая
С.
Координация Tjl.
Тип СТц — SB C9.
Пространственная
группа
Ol-Fd3m.
Z = 8
Si к/ч. = 4 0(71);
О к. 4. = 2Si(/).
Рис. 373.
Рис. 374.
Qo °sf
Вещество
1 SiO.
р-кристо-
балит
*°С
20
250
400
600
а
7,08—
7,12(?)
7,1141
7,1218
7,1219
d
^1,53—
1,54
—
—
ОМ
44,8
45,1
—
Вещество
р-кристо-
балит
t °С J а
800
1000
1100
1200
1300
7,1318
7,1348
7,1341
7,1339
7,1329
d
=
—
—
ОМ 1
45,4
—
—
§ 165] ОКИСЛЫ И ХАЛКОГЕШЩЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ 515
Атомы Si образуют алмазную структуру (Ст). Посредине связей Si —Si
расположены атомы О; в целом—кубическая структура из 8Ю4-тетра-
эдров.
Р-кристобалит был получен из чистого кварца при 1300° С с добавкой
небольшого количества плавней. При 220^ наблюдается резкое изменение
'объёма элементарной ячейки; максимум при 1000° С, далее имеет место
сжатие. Интересно постоянство объёма между 400 и 600°С. Точка
перехода а-кристобалит;~^-кристобалит по данным разных авторов лежит
в пределах между 198° — 270° С.
9. ji-тридимит Si02
Решётка гексагональная Я.
Координация TJL
Тип Н1ти-SB СЮ.
Пространственная группа D\h — Ctijmmc.
Z = 4.
Si к. ч.-- 4()(Г); О к. 4.-2Si(/).
Рис. :i75.
Вещество
Si02
(З-триди.мит
а \ с с/а d
5,0:*
8,22 I 1,»ЗЧЧ
1
1,54
ОМ
45,0 1
Атомы Si образуют гексагональную структуру из 8Ю4-тетраэдров,
отличающуюся от типа С9, как вюрцит от сфалерита (см. § 62 — 63).
516
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ liEIHECTJS
[гл. VI
10. SiS,
Решётка ромбическая ORK.
Координация Т\^_.
Тип ORtu- SB C42.
Пространственная группа ВЦ — J ста.
Z = 4.
Si к. ч.= 4 0(Г); О к. 4. = 2Si(Z).
Рис. 376.
На рис. 376 представлены две элементарные ячейки.
Вещество
SiS,
а
Ъ 1 с
5,Г * 5,53
0,55
а:Ъ : с
1,013: 1: 1,727
<* 1
Si—S
2,14
SiS2 образует длинные волокна. Цепная решётка из тетраэдров [SiS4l,
соединённых общими рёбрами#в цепи, параллельные оси Ь.
§ 165] ОКИСЛЫ It ХАЛКОГЕНИДЫ. IV ГРУППА: УГЛЕРОД-СНШШЦ 517
11. GeOk>. «Нерастворимая в воде модификация».
Решётка тетрагональная Тк.
Координация Oft.
Тип Топ-
Пространственная группа D\\ — Pi/mnm.
Z = 2.
Ge к. ч. = 6 О (октаэдр); О к. ч. =3 i\e (треугольник).
Рис. з;7.
Вещество
GeOa
а
4,39
с
2>859±4
с/а
0,6513
**
6,27
Примечание
Sb III 262—263
)
Разделение в литературе двух описанных выше (стр. 511 и 517) форм
GeO;. на «растворимую» и «нерастворимую» в воде следует считать
абсолютно неудовлетворительным.
Сомнительно, чтобы растворимая форма была чистым препаратом Ge02,
например не содержащим влаги, полностью образующим
кристаллическую, видимо метастабильную (в отношении перехода в структурный тип
рутила) форму, типа а-кварца.
518 Ч. И. СТ1 УКТУРЫ НЕОРГАНИЧЕСКИХ ЬЕЩЕСТИ [гл. VI
12. GeS2
Решётка ромбическая ОИУ\
Координация TJZ.-
Тип ОЩи -SB C44.
Пространственная группа СЦ— F dd.
Ge к. ч. = 4 8(Г); S к. 4. = 2Ge(Z)-
в
\^1
а
0 12 3 4 5 6 7 8 9 10
' ' ' ' ■ i i i i 1_1
Рис. :as.
О бе
OS
Каждый атом Ge окружён тетраэдрически 4 атомами S. GeS-тетра-
эдры соединены вершинами, образуя пространственную вязь.
§ 165] ОКИСЛЫ II ХЛЛКОГЕПИДЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ 519
13. GeS
Решётка ромбическая ORN.
Координация О/О.
Тип Ofl^=-SB Я16.
Пространственная группа ВЦ — РЬпт.
Z = 4.
Ge к. ч. = 6 (О); S к. ч. = 6 (О).
Pint. :J79.
Вещество
GeS
а
4,29
Ь с
Ю,42
з.б4
а : & : с
0,412 :1 : 0,349
Me—S
Ы =2,47
2d' =2,64
1сГ =2,91
2d'" =3,00
Координационная сфера — сильно деформированный октаэдр. Структура,
сильно деформированная Со/о, содержит сетки по (010).
520 Ч. II, СТГ'УКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. VI
14. Sn02 (касситерит) РЪО.
Решётка тетрагональная Тк.
Координация О ft.
Тип Топ — SB С4 (рутил).
Пространственная группа D\*t — Pf+jrnnin.
Z = 2,
М к. ч. = 6 0(0), О к. ч.=3 М(/).
О м
Рис. 380.
Вещество
Sn02
РЬ02
а
4,72
4,96
с
з,17
з,з9
е/а
0,672
0,683
d
2,Он-2,07
2,1G
ОМ
35,3
41,7
Примечание
Ср.
TiOj
§ 165] ОКИСЛЫ И ХАЛКОГЕНПДЫ. IV ГРУППА: УГЛЕРОД-СВИПЕЦ 521
16. SnS2
Решётка гексагональная слоистая Hs.
Координация Oft.
Тип H~j,-SB СИ.
Пространственная группа D*3l/ — C'Sm.
Z = l. Sn к. 4.---(3S((>); Sk. 4. = 3Sn(i).
О Sn
Os
Рис. 381.
Вещество
SrbS2
a
з,о2
с
5,8.
«>
с/а
1,616
J OM
2,55
66,4
Примечание
см. § 65 1
522
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. VI
16. SnO, PbO красный*)
Решётка тетрагональная слоистая Ts.
Координация qjS.
Тип JTf/.s — SB"jSiO.
Пространственная группа Dl, —Pi/nmm.
2 = 2.
2
4
-Ju.
5
РИС. 382.
Вещество
SnO
а
3.80
3,790
с
4,81
4,81
с/а
1,27
1,276
ОМ
34,7
34,7
Вещество
РЬО
а
3,98 ±2
3,9 47 ±2
с
Г», 01
4,988
с/а
1,26
1,26
ОМ
39,7
39,7
По SB I 93 (верхняя строка) и J. Am. Ch. Soc. 63, 1392 (1941)
(нижняя строка).
Последние работы подтверждают слоистую структуру РЬО и SnO SB510,
в которой каждый атом кислорода тетраэдрически окружён 4 атомами
металла; атомы же металла расположены в вершине тетрагональной
пирамиды, основание которой образуют 4 атома О, d(Sn — 0) = 2,21 А;
<d(Pb-O) = 2,30A. В J. Am. Ch. Soc. 65, 1107 (1943) дано сопоставление
структур этого и других окислов свинца.
Размеры элементарной ячейки 2 обсуждавшихся типов одинаковы.
*) Имеются данные, что при окислении поверхности свинца (при 500— 700° С)
■окисленный слой состоит из двух частей. На металле лежит слой красной
тетрагональной модификации РЬО, над последним— жёлтой ромбической.
«§ 165] ОКИСЛЫ И ХАЛКОГЕНИДЫ. IV ГРУППА: УГЛЕРОД-СВИНЕЦ
17. SnO
Решётка тетрагональная?
Координация?
Вещество
SnO
а
5,360 до
5,364
с
4,825
с/а
0,901
Согласно Z. Anorg. Ch. %VAt 301 j(1933) в зависимости от метода
приготовления возникают различные по виду препараты SnO с
изменяющимися периодами решётки а. Структура этого соединения не может
считаться расшифрованной.
18. SnS
Решётка ромбическая
слоистая ORs.
Координация О/О.
Тип ОНщ-SB #29.
Пространственная
группа ВЦ—Ртсп (Pnma).
2 = 4.
Sn к. ч. = 6 S(<9);
S к. 4. = 6Sn(0).
а
-6
Р|1С. Ж1.
О Sn
Вещество
SnS
а
3,98±2
Ъ
4,33±2
с
11,18±2
а : Ъ : с
0,919: 1 : 2,582
d 1
Sn—S
1—2,62
2—2,68
2—3,27
1—3,39
Сильно деформированный тип Сою— ^г» причём возникает слоистая
решётка.
524
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТН
[гл. VI
19. РЬО (жёлтый)
Решётка ромбическая OR.
Координация ?
Тип ОД-SB?
Пространственная группа С\-0—РЬа{1).
Z = 4.
Вещество
РЬО
а
5,48
Ъ
4,74
с
5,88
d 1
Pb-0
2,20—2,18
См. С. А. 39/2018 (1945). Структура подлежит уточнению.
20. PbS, галенит, PbSe,
PbTe
Решётка кубическая С.
Координация О/О.
Тип Со/о-SB B1.
Пространственная
группа 01 —Fm Зт.
Z = 4.
Pb к. ч. =6 Х(0);
X к. ч. = 6 Pb(tf).
*£^ГТ:
и
=ЖР
-ю
Рис. 384.
Вещество
PbS
PbSe
PbTe
а»
5,91±3
6,14
6,34
d
Pb-X
2,955
3,07
- 3,17
Примечание 1
Очень чистый 1
галенит из Пши-1
брама
Общие замечания
Окислы свинца и, особенно, окислы олова представляют собой фазы,
состав которых по кислороду может в большой мере колебаться в
зависимости от условий получения и существования. Поэтому следует считать
в настоящее время совокупность структурных данных по системам Sn —О
и РЬ — О недостаточной.
§ 166]
ОКИСЛЫ II ХАЛКОГЕНИДЫ. V ГРУППА: АЗОТ-ВИСМУТ
525
§ 166. V группа периодической системы:
азот N, фосфор Р, мышьяк As, сурьма Sb, висмут Ш
С кислородом азот образует 4 вида молекул:
N20, линейная NNO или NON, расстояние между крайними атомами
равно 2,38 ± 5,
N0 — линейная, расстояние N -0 — 1,416;
N02, треугольная, dN_0 = 1,15 — 1,3 Л,
N.,04, форма молекулы не определена, dN_N= 1,6—1,7 А для модели
OaN-NOa;
N205, повидимому, равна 02N —О —N02; форма точно не определена,
dN_o^ 1,3 —1 А.
Данные по жидкому N0 и N20 —см. J. Ch. Phys. 11, 435 (1943);
N30 — линейная молекула.
Фосфор, мышьяк, сурьма образуют Р4()10иРД, соответственно As4Oie
и As4Oe; Sb4O10 и Sb4Oe.
Молекулы А40§ построены таким образом, что атомы кислорода
занимают вершины октаэдра (рис. 385). Над двумя гранями верхней половины
октаэдра и над двумя гранями нижней половины, на перпендикулярах,
восстановленных к центру грани, лежат атомы А(Рили As), образуя тетраэдр:
^ As- О - As = 126 , /О-As-O = 100", Z Р — О — Р = 127,5 ,
^0 —Р —0 = 99°. Несвязывающие пары электронов атома А как бы
вытянуты вдоль перпендикуляров. (Расстояния Р — Р — 2,95 ± 3\ Р —0=1,65;
As- As -3,20 ± 3\ As -О --1.80 ± 2.)
Рис. :ю5.
Рис. М86.
В молекулах типа Р4010 (рис. 386) эти пары электронов обусловливают
присоединение атомов О. Расстояние от этих атомов О до атома А меньше,
чем три других. Таким образом, 4-я связь Р —О отличается от первых
трёх и, повидимому, частично является двойной.
^0__р__0 = 101,5°;ZP — О-Р = 123,5Г;Р-Р- 2,84 ± 3 А; Р-0 =
= 1,62 ±2к.
В кристаллах структуры молекул А406 и А4010 часто сохраняются;
несколько изменяются межатомные расстояния.
526 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VI
1. N204
Решётка кубическая
см.
Координация q.
Тип C^-SB C266.
Пространственная
группа Т* — /23 или
T\-Jm3.
Z = 12NOa=6 N..04.
a-(N.,04).
Вещестно .
d (N—0) . .
N>04
7,77
Примечание.
Структура не N02,
как указано в SB II
21, но N,04.
о
/ 2 3
j_
О N
ОО
Рис. 387.
Принимая a = N204, обнаруживаем, что узел a расположен в центрах
граней и рёбер кубической элементарной ячейки, как атомы Pt в решётке
Р^зО« (рис. 349, а). Редко встречающаяся упаковка в молекулярной
решётке.
Решётка кубическая С*
Координация К (12).
Тип Cf-SB C2.
Пространственная группа
Т\-Ра 3.
N,0-a.
Z-4-
Вещество j aw
\ 1
N.0
5,78
Примечание
Тип С02—
молекулярная решётка
Рис. :ш*.
О N02
(Центры тяэ/сесш
молекул)
3/Р206, по старым данным, имеет ромбоэдрическую ячейку с
гексагональными осями а = 11,12; с=1,12; Zzzl2.
§ 1G61 ОКИСЛЫ II ХАЛКОГЕНИДЫ. V ГРУППА: АЗОТ-ВИСМУТ 527
4. Sb206
По старым данным, основа решётки Sb,06,- структура Sb4Oe, в
которой растворены атомы кислорода.
б. Sb204
По старым данным, Sb204 имеет кубическую элементарную ячейку.
Пространственная группа 0\ — Рс13т или 04 — F^'S; Z^l<6; a= 10,24. Эта
структура занесена в SB III как тип Z>62. В SB VII 15,126 этот тип
отвергается и структура, описанная в SBIII, признаётся неправильной
(равно как и состав вещества(?)). В Z. Anorg. Ch. 239, 64 (1938)
утверждается, что Sb>04 ромбическая OR, Пространственная группа С\ —Р?ш.
Z = 4'SbX>4; a-=4,804; 6-5,424; с — 11,76.
6, As203 = As4Oe; Sb203 = Sb4Oe (сенармонтит)
Решётка кубическая молекулярная См.
Координация Т.
Тип С?— SB Z>61=Z)54 (сенармонтит), a = [Sb40«],
соответственно [As4Oe].
Пространственная группа 0\ — Fdf6m.
Z = 16A203 = 8A4Oe.
I Вещество
As40«
Sb4Oe
au>
u,o6
11,1,
d
A—0
M =2,0L
3d'= 2,38
3d =2,22
3d'= 2,37
OM
109,1
172,8
Примечание
Кубическая алмазная С г
молекулярная решётка, состоящая из
узлов a = Sb4Oe или соответственно)
As4Oe, структура которых отве- 1
чает структуре молекул 1
528
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. VI
7. Sb,0, (валентинит) Пространственная группа Dfh — Pccn.
Решётка ромбическая, цепная 0RK. Z = 4.
Координация tjf, 1. Sb к. ч. = 3(*).._
Тип OR*f t-SB Z>5n. о I к. 4.=4Sb(r); О II к. 4. = lSb.
£
л
0 12 3 4 5 6 7
i__i i 1 1 1 i 1
1'ис. 380.
О Si
Оо
Вещество \ а \ Ь \ с
*ЬЛ03
4,92
12,',6
5,42
а: 6: с | d j
0,395 :1 :0,4€>>5 1 d=l,30
I e = l,5:^
1 '
Решётка состоит из цепей Sb,Oa типа
2V3
О О
\(y/Sb\o/Sb\)/
/°\sb/°\sb/°\
о
о
вдоль оси с.
§ 166f ' окислы и хллкогениды. v группа: азот, фосфор, висмут 529
В литературе были описаны 4 различные модификации Bi203:
а—моноклинная, устойчива при обычной температуре; р —тетрагональная,
псевдокубическая; кубическая центрированная; кубическая.
Образование кубической центрированной облегчается наличием примесей Fe203,
Si02 и др. См. SB V 69 и след.
8. aBi203
Решётка моноклинная М.
Координация?
Тип М SB-?
Пространственная группа С\ — Р2 или С\ — Р2Х. Z^4.
Вещество
aBia03
а
5,83
Ъ
8,14
с 1 3 1
7,48
67°07'
9. [Ш20,
Решётка тетрагональная Т.
Координация С'1 Т.
Тип Tc-rf--SB Z)5l:.
Пространственная группа
Z = 8.
Bi к. ч. = 6 0 (С'Г);
О к. 4. = 4Bi(f).
Рис. 390.
Вещество
1 ЭВц03
а
10,93
с
5,62
с а
0,514
Шесть атомов О занимают вершины кубической коордииацйоипой
сферы. Не заняты 2 вершины по телесной диагонали. Четыре атома Bi
образуют тетраэдр вокруг атома кислорода. Тетрагонально сжатый 'тип
/)55; см. SB V 9, 69-71.
5.40
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. VI
10. Bi203
Решётка кубическая
центрированная С.
Координация?
Тип?
Пространственная группа
7'»-J23. Z = 12 (?) (J +
b/Fe2Oa?)
ffpr= 10,16,
11. Bi203
Решётка кубическая
простая С.
Координация C~f~ /Т.
Тип Сс-/-/г —SB анти- Z>55
(тип Mg8P2).
Пространственная группа
Он—РпЗт.
Z — 2.
BiK.4.=-6 0(C"/-);
OK.4.4Bi(r).
^7^
Рис. 391
Согласно SB 111 49, вещества, отнесённые к типу Z)53, возможно, должны
быть отнесены к типу Dbb (тогда ребро ячейки удваивается).
Вещество
Bi.O,
aw
5,25 ± 5
Примечание 1
Получвн из зВь03 нагреванием
до 900° С с последующим быстрым
охлаждением
12. NS, AsS
Решётка моноклинная М.
Координация?
Тип?
Пространственная группа С\ь — P2Jn.
Z = 16.
Вещество
NS
AaS
а
8,7R
9,27
Ь
7,14
i:i,50
е
8,04.
о
6,56
Р
87°39'
73°(?)
а : Ь : с
1,229 :1: 1,211
0,0875 :1 : 0,486
'* 1
2,37.
j I
Примечание. NS-SBIV 99. AsS-SBIII 255.
Эти данные расходятся с более ранними результатами (SB II, 327), где
принимают состав N4S4 (плавится 178° С) и ромбическую элементарную
ячейку а ==8,87; й = 8,47; с = 7,20; а : Ь : с = 1,0472 :1:0,8512; простран-
ственная группа />оЛ — Рпгтт. В структурном отношении химическая
формула NS и A3S остается неясной.
§ 166] ОКИСЛЫ ИХАЛКОГЕНИДЫ. V ГРУППА: А; ОТ, ФОСФОР, ВИСМУТ 53
531
13. Sb2S3 (антимонит), Bi2S3 (иисмутоный блеск, бисмутинит)
Решётка ромбическая (пояса) OR .
Координация t\/_.
Тип OR*z- SB ПЪ8.
Пространственная группа DA Pbnm.
Z-4. Sbl к. ч. - 2SII+1S III (0;
.ЧЬИ к. ч. 2SIII+1S I (/):
S к. ч. --■ 2(Z).
Рис. 392.
На рис. 392 показаны три элементарные ячейки.
Вещество
BbS3
а
ll,20±2
11. l»±2
f/
11,28±^
11,2:^2
с
:\tm:\2
a : b : с
0.УУ2Г» : 1 : 0,:Ш5;
0,487'i : L : о, .4523
Sbl
Id =2.50
2e - 2,52
SMI
ld^2,38
2*=^2,67
Примечание. Зигзагообразные цени Sbl —S —Sbl и Sbll- S — SbJl
соединены мостиками серы в пояса iполоски) [Sb4Se]. Каждый атом Sbf
окружён тремя атомами S в форме почти равностороннего
треугольника, каждый атом SII окружён 3 атомами S в форме примерно
равнобедренного треугольника. В решётке Hi2S3 параметры Bi почти совпадают
с найденными для Sb в Sb,S3. Точные параметры для S не
определены (SB III 49).
532 ч: и: структуры неорганических веществ [гл. VI
§ 167. VI группа периодической системы: кислород О, сера S, селен Se,
теллур Те, полоний Ро
Соединения кислорода с S, Se, Те рассматриваются ниже. Соединения
S, Se, Те между собой, а также соединения Ро с О, S, Se, Те не изучены.
S, Se, Те образуют с кислородом химически метастабильные молекулы
SO, SeO, ТеО, изученные в спектроскопии, но не исследованные химией.
Молекулы S03, S02, Se03, Se02, Te08 Те02 в структурном
отношении изучены неравномерно.
Из молекул S03, Se03, Те08 более полно изучен S03.
Структурные данные молекул SOa, SeDa, Те02 отражены в таблице:
Форма
d (S—О)
1 Валентный
1 угол
О—А—О
SOa
Треугольник
1,46±2
122 ±5°
SeOs 1 Те02 1
Треугольник
1,61
Треугольник
Жидкий S02 был изучен недавно (С. А. 39, 1799 (1945)).
SOjj-молекула окружена 8 молекулами SO, (к, ч.=8). Угол О —S —О
равен 124°; S -0 = 1,46; 0-0 = 2,60.
Структуры кристаллов исследованы совершенно недостаточно.
1. SeO.
Решётка тетрагональная
пепная Тк.
Координация t/j/,1.
THH7^z,i--SBC47(Se02).
Пространственная группа
вЦ-РЩтпЬс или
Z = 8.
Se к. ч. = ЗО(0;
01 к. 4. = 2Se^Z);
0Ик.гч. = lSe.r
Рис. 393.
Вещество,
SeO,
а
8,353±5
с | cja
Примечание
5,О51±10| 0,605 SB V 4
В рещёт^е ^ва сорта атомов О. Атомы 01 соединены с двумя атомами
Se каждыц,, атрдеы, Oil только с 1 атомом Se. В первом случае
расстояние §е — 0=f 1T79A, (Jbq втором случае; 1,75А. За счёт связей Se — 01 — Se
возщщают .лдеоседе цепи, параллельные оси с.
Интересно, что в парах SeO* расстояние Se —О = 1,61 +<?А., т. е.
гораздо меньше, чем в кристалле.
§ 167]
ОКИСЛЫ II ХАЛКОГЕНИДЫ. VI ГРУППА: КИСЛОРОД-^ ТЕЛ ЛУГ
533
2. Те02
Решётка тетрагональная цепная Тк.
Координация октаэдрически-треугольная Ojt.
Тип Tfyt-SB C4 (рутил).
Пространственная группа DxA-~Pilmnm.
Z = 2.
Те к. ч. = 6 0(0);
О к. ч. = 3 Те(/).
Рис. 39'..
Вещество
с/а |
ОМ
Примечание
Те02
4,7Q
3,7,
0,78,
2,22
43,3
См. Ti02
3. Те02 (теллурит)
Решётка ромбическая
олв-ы.
Координация Ojt.
Тип ORg^-SB C52.
Пространственная
группа Dui — Pcab.
Z = 8.
Те к. ч. =60
(деформированный октаэдр);
О к. ч. = 3 Те (О
Проекция Вдоль оси с
а\
'интобап ось ^S \шУ
'ВинтоЗая - .
JPffl - Плоскость О Те
скольжения
0 си vfs
' Центр инверсии
0 12 3
\ \ \ i
5 6 7
_. 1 I
Рис. 395».
Оо
Тип близок к брукиту (Ti02) SB £21, но октаэдры (ТеОв) сильно
деформированы. Они имеют общие рёбра и образуют пояса вдоль оси с.
534 ч. п. структуры неорганических веществ [гл. VI
1 Вещество
1 Те02
а
5,50
Ь
11,7,
с
5,59
а : Ь : с 1
0,467:1:0,476
По оси а пояса сшиваются через общие рёбра октаэдров в сетки; по оси Ь
октаэдры имеют лишь общие вершины. Сетки или слои октапдров па рал-
Проекция Вдоль оси а
о Общ вершины Общие ребра.
01234567Я9
I I I I I I 1 I U—1
ОТе
Рис. 395Ь.
! _ Дбойноя цепь I
|- - Цепь -
Рис. 395 с.
лольны (010), что вызывает спайность вдоль соответствующей плоскости.
На рис. 395, с несколько октаэдров выделены, чтобы показать характер
их взаимной связи.
§ 168. VII группа периодической системы: фтор F, хлор С1, бром Вг, иод J
См. галогениды О, S, Se, Те.
ГЛАВА VII
НИТРИДЫ, ФОСФИДЫ, 1РСЕНИДЫ И ДР.
§ 169. Введение
Как и в случае окислов, вещества, относимые в химии к нитридам,
фосфидам и т. п., но содержащие структурные узлы типа [N3] и др.,
рассматриваются в разделе: комплексные соединения (гл. X).
Нитриды, фосфиды и т. п., содержащие изолированные атомы N,P и т. п.,
вместе с карбидами, силицидами, боридами, составляют тот
интереснейший класс соединений, где особенно у элементов с ds-валентными
электронами чётко проявляется неприложимость законов Пру и Дальтона к составу
химических соединений. См. Б. Ормонт *) и § 104.
Поэтому следует подчеркнуть, что формулы веществ, проставляемые
нами вслед за авторами, изучавшими структуры этих веществ, носят
условный характер и в действительности не дают представления о стехиометри-
ческой области устойчивости рассматриваемых структур.
С другой стороны, в этих группах веществ сплошь и рядом возникают
реальные структуры (вычитания, замещения и т. д.), более или менее
далёкие от идеализируемой
структуры, отображаемой
в работах авторов и в SB.
§ 170. I группа
периодической системы:
водород Н, литий Li, натрий
Na, калий К, рубидий Rb,
цезий Cs
1. H3N, (Li3N)
Решётка кубическая С^
гранецентрированная.
Координация примерно
кубооктаэдрическая
#112).
Тип Cjj-SBZM.
Пространственная группа
Т*-Р213.
ос к. ч. = 12. lH3N]^a.
*) Журнал общей химии, XIX, 210 (194^г).
0 12 3 4 5 0N
I 1 1 I L I Ч*/
536 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТН [гл. VII
Вещество
H3N
(Li3N)
aw
15,50)
d
N—N
з,53
(3,90)
ом
34,1
(41,0)
Примечание
Расположение атомов Н
соответственно Li в SB I 230 не указано.
Атомы N образуют кубическую
гранецентрированную решётку CF.
В графе d приведены расчётные значения. В
SB вместо 3,58 для NH3 указано 3,38. Данные
по Li3N неверны, см. ниже.
2. Li3N
Решётка гексагональная Я.
Координация /, t/D6
Тип tfM/Z)6-SB?
Пространственная группа Dm •
Z)!a —#&и2 или Dlh-CQm2.
Z=l.
Li I к. ч. = 2 N (/);
Li II к. ч. = 3 N (0;
N к. ч. = 8 Li(Z)6).
СЪ/ттт или
Рис. 396*
Вещество
Li3N
а
3,658
с
3,882
с/а
1,061
' °х
1,28
Примечание
Согласно Z. E1. 41, 102
(1935) этими данными
ранее описанная структура
опровергается
3. Li3P, Li3As, aLi9Sb, Na3P, Na.Ae, Na.Sb, Na.Bi, K,As, K,Sb, K,Bi
Решётка гексагональная Я.
Координация Т, 3/9.
Тип Яг,з/9-SB Z)0ie (Na,As).
Пространственная группа Вы — Сб/пгтс.
Z = 2.
М I к. ч. = 4 X (F);
М Пк. ч. = 3 X;
X к. ч.^9 М.
Вещество
Li3P
LigAs
aLigSb
Na,P
1 Na8As
a
4,264
4,3*7
4,701
4,980
5,083
с
7,579
7,810
8,309
8,797
8,982
с/а
1,770
1,780 1
1,768
1,767
1,765
•
Вещество
Na3?b . . .
Na3Bi . . .
K3As . . .
K.fcb . . .
K.Bi . . .
a
5,355
5,448
5,782
6,025
6,178
с
9,496
9,655
10,222
10,693
10,933
с/а I
1,773
1,772
1,76S
1,775
1,770
§< 170] ' ': НИТРИДЫ, ФОСФИДЫ И ДР. I ГРУППА: ЛИТИЙ, НАТРИЙ, ЦЕЗИЙ 537
4. £Li3Sb, Li8Bi
Решётка кубическая гране-
центрированная С.
Координация 8 + 6/6,4 —
= С + 01Т, О.
ТипCc+oit, о-SB Z>03(BiF3).
Пространственная группа
0l-Fm3m.
Z = 4;В1к.ч. = 8 Li (куб) +
+ 6 Li (октаэдр);
Li Ik. ч. = 6 Bi (октаэдр);
Li II к. ч.-= 4 Bi (тетраэдр).
Вещество
PLisSb
Li8Bi
*w
6,559
6,70S
с*
3,32
5,03
V
282,3
302,1
5. aLiBi
Решётка тетрагональная
центрированная Tj.
Координация
тетрагонально-призматическая Р4/Р4.
Тип Tpkipk
(тетрагонально-деформированный тип CsCl).
Пространственная группа?
Z=2.
Рис. 398.
О L'
О
Вэщество
а UBi
а
3,301
с
4,247
с/а
1,264
Примечание
При 48,6 am % Li Li—Bi = 3,19;4
Bi—Bi=:Li—Li=3,36; SB III 637—638\
538 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VII
6. NaBi
Решётка тетрагональная Т.
Тип Т-SB?
Пространственная группа Dlh — Pi[mmm.
Вещество
NaBi
а
Vi6
с
4,SO
с/а
1,39
Примечание
Bi в (000); Na (V*7a0)?
7. KBia
Решётка кубическая С.
Координация 12/6 = Tt/6.
Тип Cr//6-SBC15 (Cu2Mg).
Пространственная группа
Ol-Fd3m.
Z = 8 KBi2.
К к. 4.=--12Bi(7V);
Bi к. ч. = 6К.
Рис. 399.
Вещество
кш8
*w
9,501±5
V
857,37
Тип С15—кубическая гранецентрированная структура из атомов Bi,
50°/0 атомов которой удалены и на их место, в центры тяжести
удалённых тетраэдров, помещены атомы К. С другой стороны, можно
представить решётку так, что атомы К образуют алмазную структуру,
4 пустующих октанта элементарной ячейки которой и центр ячейки
заняты решёткой из 5 тетраэдров Bi, имеющих общие вершины.
Атом К имеет 12 соседей, принадлежащих попарно 6 ближайшим
Bi-тетраэдрам, причём каждая пара Bi-атомов лежит на одном ребре
тетраэдра. Атом Bi окружён 6 атомами К. Н. В. Белов подчеркнул, что
форма координационной сферы К близка к усечённому тетраэдру (её вид
вытекает наглядно из рис., см. табл. 34). См. Белов Н. В., Структура
ионных кристаллов и металлических фаз, стр. 184 и след., тип Cu2Mg.
См. также ниже Au2Bi.
§ 171) НИТРИДЫ, ФОСФИДЫ И ДР. II ГРУППА: БЕРИЛЛИЙ -БАРИЙ 539
§ 171. II группа периодической системы:
бериллий Ве$ магний Mg, кальций Са, стронций Sr, барий Ва
1. Be3N2, Ве3Р2, Mg3N2, Mg3P2, Mg3As2, aCa3N2
Решётка кубическая С.
Координация TJC''-, С~.
Тип C-i,c-i_. с= -- SBZ>5, (апти-Мп2Оэ).
Пространственная группа Г/, — 1аЗ.
Z-16 M3N2:
М к. ч. 4Х(Т);
XI к. ч. = 6 М{С'1-У,
XII к. ч.-6М(С").
Шзщостпо
Be3N2
2,70
Пе3Ра
10.Ь
• >
2,25
1
Mtf3Na Mff8P2
2,70.
12,0j
2,0о8
M-3As2 aCa3Na
12,4,
:М»8
11,М) -11,3S
2,64 - 2,66
Примечание. Вещестла этого типа в разных томах SB относятся
либо к структурному типу ВЪ5 (см. следующую страницу), либо к типу <D53.
В этом случае ребро элементарной ячейки удваивается. См. SB III 49,
352 и след. Следует ожидать уточнения этого вопроса.
540
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[Ь-vn
2. Mg8N2
Решётка кубическая С.
Тип С -SB?
Пространственная группа 0^— Jm3rn(?)
Z = 16.
Вещество
Mg8N2
9,9»
Применение
*,74
Эти данные идут вразрез с более ранними
(Z = 11 и сх = 2,04 — 2,06) и находятся в
соответствии с экспериментально найденным
с = 2 72
эксп. *»'А
3. MgsP2, (Mg,As2)
Решётка кубическая С.
Координация Т/С'1-.
Тип Cric-i.—SB Dbt.
Пространственная группа
0*h-Pn3m.
Z = 2MgaX„.
MgK. ч. = 4Р (Г);
Р к. 4. = 6Mg(C"/-).
Рис. 401.
См. примечание на предыдущей странице.
Вещество
(MgaAs2)
а„
5,92
6,10
d
Mg — Р
5,90
**
2,162
3,26
*
Примечанив
Атом Mg тетраэдрически окружён
4 атомами Р. Атом Р окружён
6 атомами Mg, занимающими 6
вершин к>ба (2 лежащие по
объёмной диагонали выпадают).
Состав элементарной ячейки
= Mg,P4
Мы обращаем внимание на то, что структура Mg3P2 находит
интересный аналог в структуре Pt304 (стр. 489), в которой атомы Pt также
занимают позиции в центрах граней и рёбер, атомы же кислорода — центры
8 октантов. Таким образом, структуры Mg3P2 и Pt304 относятся друг к
другу как структуры ZnS (сфалерит) и CaF2. При соответственно
одинаковом расположении атомов неметалла в центрах 4 или 8 октантов, атомы
§171] НИТРИДЫ, ФОСФИДЫ И ДР. II ГРУППА: БЕРИЛЛИЙ-БАРИЙ 541
металла в первых двух структурах занимают центры граней и рёбер, но
в остальных двух—центры граней и вершины элементарной ячейки.
Учитывая, что-в случае нехватки атомов неметалла в структурах вычитания
тю типу CaFo—в последних остаются незанятыми позиции в центрах
октантов, с приближением к типу сфалерита, было бы интересно
установить границу устойчивости но составу структуры Pt304 в случае недостатка
атомов кислорода против стехиометрического и возможность
приближения структуры фаз низших окислов платины к типу Mg3P2. В равной
Aiepe вызывает интерес, какова граница устойчивости по составу этой
структуры в случае избытка атомов кислорода, например при осторожном
окислении: занимают ли атомы [кислорода места в центре элементарной
ячейки Pt304, как это имеет место в структурах типа CaF2, когда
неметалл начинает располагаться в центре элементарной ячейки (переход к
типу BiF3), а также возможна ли при окислении перегруппировка с
занятием атомами металла позиций в вершинах элементарной ячейки (вместо
центров рёбер) и атомами
неметалла позиций в центрах
всех октантов (тип CaF2).
Заслуживает упоминания
и тот факт, что расположение
структурных узлов в центрах
граней и рёбер элементарной
ячейки без заполнения
центров октантов наблюдается в
рассмотренной нами на
стр. 526 структуре N204,
образующей молекулярную
решётку.
4. Mg3Sba, MgsBi2
Решётка гексагональная Н.
Координация — A3, РуЗ/АЗ*.
Тип Ялз,руз/лз+— SB анти-
£>52(La2Os).
Пространственная группа
Did-СЗт.
Z = l.
Mg I к. ч.=0 X (A3);
Mg 11к.Ь.=4Х (РуЗ);
X к. ^. = 7 Mg {A3').
Рис. 402
1 1
!
Вещество
Mg,Sb>
Mg,Bia
а
4,57ч
4,66,
с
1 9->
Л—9
7,401
с/а
1,581
1,58G
Примечание
SB III 48, см. La?03, стр. 453.
Нитриды Са
Описаны (см. SBIII) три нитрида Са.
А (получается ниже 600 —750°С) чёрный.
В (получается выше этой температуры) коричневый.
С (образуется между J150 — 1200° С) жёлтый.
А переходит в В, но В перевести в А не удалось.
А г- гексагональный (а = 3,553; с = 4,11; сх = 2,72).
В — кубический (а= 11,38; аг = 2,66) см. стр. 539.
Ул2
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. VII
§ 172. III * группа периодической системы: скандий Sc, иттрий Y,
лаптан La
1. ScN, LaN(?), Lal'(1),
LaAs, LaSb, LaBi ^<
Решётка кубическая С. Qf
Координация октаэдриче-
ская О/О.
Тип Со/о -SB 51.
Пространственная группа
Ofr-FmSm.
Z ,4; М к. ч. = 6 Х(0);
X к. ч. = 6 М {О).
<£=№?[
ЬЯЧ^'
(Л
^
ь*
о
Рис. 403.
Вещество
ScN
LaN
LaP
aw
M4
5,275
6,013
d
2,22
2,637
3,006
г;
87,8
14'), 8
217,4
Вещество
LaAs
LaSb
LaBi
aw
6,125
6.47,,
6,565
i ' 1
d | v Примечание
i 1
3,0:i
3,237
3,282
229,8
271,5
282,9
Cm. SBV 42—43
§ 173. Ill *• группа
периодической системы:
редкие земли
I. CeN, СеР, CeAs, CeSb,
CeBi, NdN, NdP, NdAs,
NdSb, PrN, PrP, PrAs,
PrSb, PrBi
Решётка кубическая С.
Координация октаэдриче-
ская О \0.
Тип Со/о-SB 51.
Пространственная группа
0%-Fm3m.
2 = 4.
М к. ч. =6 X (О);
X и. ч. =-6 М (О).
О
Рис. 404.
§ 174]
НИТРИДЫ, ФОСФИДЫ И ДР. IV ГРУППА: ТИТАН-ТОРИЙ
543
Вещество
<*«■
1 d
Вещество
аи>
1 d
1 CoN
I
2,05.
NrtAs
| Г,>'Г,8
J
CeP
Г>,Н9-
2,M8
NdSb
6,30,
3,154
(oAs
b,0G0
з,оя0
PrN
G,i55
2,57.,
7__
< eSb
r,'3aa
3,18,,
PrP
Гь860
2,410
CoHi
6,'.87
3,243
PrAs
Г>,997
2'91J8
1
1 i
2,07
Pr>b
G,353
3,17,
NdP
PrBi
fi.44g"
3,224
§ 174. IV * группа иериадической системы: титан Ti, цирконий* Zr,
гафний III, тории Th
О
1. TiN, ZrN
Решётка кубическая С.
Координация — О/О.
тип Сою-sb т.
Пространственная группа
0%-Рпйт (01 -F43 и
7,id-F43w).
Z = 4.
Мк. 4.1- 6N; N к. ч. = 6М.
О^
<Д^?
Ь^^
я
г
^ы^
е<Ы52ДУ"
^
Рис. 405.
Вещество 1 а„ i d
1 1
TiN
ZrN
4,22
4,g:;
2,11
2,3.,
ОМ
18, У
24,У
«г
г>,'.з
Примечание
См. SB VII 3, 79; Прспф (1939)
SB IV 11, 150
Исследование условий образования, строения и свойств TiN было
поставлено автором в Институте им. Карпова в 1935 г. Б. Ф. Ормонт,
Л. Е. Апельбаум и В. И. Касаточкип показали, что нитрид титана
образуется начиная от iOOC. A. X. Брегер, при участии Б. А. Петрова.
М. В. Павловой и др., синтезировавших различные образцы нитрида,
показал, в частности, что существовавшие ранее в литературе данные
зарубежных исследователей относительно периода решётки TiN (4,40kX)
неправильны. Повидимому, столь значительная ошибка в определении
периода решётки вызвана, прежде всего, тем, что зарубежные авторы,
не имели в своем распоряжении чистил образцов,
544 ч. и. структуры неорганических веществ [гл. VII
В той же работе рассматривалось влияние дефектов решётки на
величину межилоскостных расстоянии, и было ноказано, что в соответствии
с ожиданиями автора книги (см. Усп. Физ. Наук XVI, 1001 (1936))
в кристаллах этого типа имеют место значительные колебания периода
решётки, достигающие значений от 4,20 до 4,238 кХ. См. Брегер Жури.
Физ. Химии. 272, 1304, 1483 (1939); Acta Phys. URSS 10, 593; 11, 617 (1939),
13, 723 (1940), В этой стадии исследования в рентгенографической части
работы участвовал и Г. С. Жданов. См. стр. 516. Из новых работ
см. Z. Anorg. Ch. 259, 1 (1949).
2. Ti—P
Титан, невидимому, образует с Р соединение TiP и возможно TiaP.
3. Zr — P
Цирконий образует с Р, повидимому, ZrP2 и ZrP, а также, возможно,
-субфосфид (Zr3P?).
4. Th—P
Торий образует с фосфором несколько фаз, из коих подробно изучена
лишь одна Th3P4 (см. ниже). Эта структура представляет существенный
интерес в связи с её обнаружением у соединений трансуранидов в 1949 г.
ThP изучена менее надёжно, причём исследовавший систему Майзель
(Z. an. Ch. 240, 300 (1939) )допускает возможность существования также
соединения ТЬ4Р4 с той же структурой, что и ThP с частично незанятыми узлами Р.
5. ТЬаР4
Решётка кубическая С.
Координация дисфеноэдрическая треугольная Dijt*
Тип CDilt SB £73<Th3P4).
Пространственная группа T*d — ./43d.
-2 = 4, Th к. ч. = 8Р(Л'); Р к. ч. = ЗТЬ(/).
Рис. 406.
Вещество
ТЬ,Р4
в10
8,600±2
d \ d \
Th- -P ! Р —Р
2,08
3,20
.Примечание. Структура образует пространственную вязь из
тетрагональных тетраэдров (сфеноэдров), причём каждый атом Th сфено-
эдрически окружён 4 атомами Р; каждый атом фосфора ~ тригонально
окружён тремя атомами Th. Тетраэдры соединены так, что каждый атом
Р принадлежит одновременно 3 тетраэдрам.
§ 1751 НИТРИДЫ, ФОСФИДЫ JJ ДР. у ГРУППА: ВАНАДИЙ, 1ШОБПП, ТАНТАЛ 545
Против каждой из 4-х граней тетраэдра лежит атом фосфора,
принадлежащий другому тетраэдру и находящийся на том же расстоянии
от центрального атома ТЬ, что и 4 атома Р, входящие в 1-й тетраэдр.
Таким образом,
координационная сфера Th
состоит из 2 тетраэдров
(дисфеноэдричсская) и к.
ч. равно 8. См. SB VII 15.
На рис. 406 показаны
соединения трёх тетраэдров
в одной вершине, причём
хорошо видна треугольная
координационная сфера
фосфора (средний
чертёж) , координационная
сфера Th в одном
тетраэдре (левый чертёж) и
суммарная
координационная сфера Th (правый
чертёж).
6. ThP( + Th4P3?)
Решётка кубическая С.
Координация октаэдриче-
ская 0\0 (?).
Тип Со/о-SB 51 (?)•
Пространственная группа
0\x-Frriim (?).
Z = 4.
Th к. ч.=6Р(0);
Р к. ч.=6ТЬ(0).
§ 175. V* группа
периодической системы:
ванадий V, ниобий Nb,
тантал Та
1. VN, NbN
Решётка кубическая 6\
Координация О/О.
Тип Со/о-SB 51.
Пространственная группа
Oh — Fm3m.
Z = 4; М к. ч.=6 N(0);
N к. ч. = 6М (0).
Рлс. 407.
Вещество
ThP
aw
Г>,818±2
Вещество
VN
NbN
•W!3
Л,
2,0.».
Рис. 408.
По данным Я. С. Уманского («Карбиды твердых сплавов», Металлург-
издат (1947)), нитрид ниобия M)N имеет плотную гексагональную структуру
с отношением осей с/я = 1,85 (а =3,016 и с = 5,580±6) и прежние иссле-
546 ч. ii. структуры неорганических веществ [гл. VII
дования, приписывавшие этому нитриду кубическую решётку, неверпы.
К сожалению, полное исследование фазового состава и структуры нитридов
Nb отсутствует.
В 1935 г. под руководством автора книги в Институте им. Карпова
была поставлена группа исследований, посвященных изучению влияния
условий получения, наличия примесей и других нарушений решётки
на величину периода идентичности, шлифоспособности и других свойств
различных соединений, в том числе нитридов ванадия и титана. Эта серия
работ была поставлена в противовес господствовавшему тогда увлечению
теорией идеального кристалла и имела целью связать химию реальных
твёрдых фаз со строением последних.
См. Б. Ф. Ормонт, Успехи физических наук XVI, 1001 (1936),
Ж. Физ. Хим. XXI, 569 (1947) XXII, 1406 (1948).
Применительно к нитриду ванадия нами было показано, в частности,
что в зависимости от условий получения и последующей обработки состав
и свойства нитрида ванадия меняются следующим образом:
Изменение состава и физико-химических свойств препаратов питрида ванадии
в зависимости от температуры последующей его рекристаллизации
Номера 1
1
2
3
4
1 ,Г>
Условии
получения и
дальнейшей
рекристаллизации
теми-
в ° С
600
НОО
1000
1200
j 1400
иро-
лоляе.
нагрева в
часах
1G
2
2
•)
2
Химсостав по
анализу и расчёту
"и N
21,8
21,28
21,1
20,К
18/J
"и V
76,6
77,2
78,4
78/.)
80,6
% О
и»
1,52
0,5
0,»
0,5
Период решётки и (
стохиометрическая
формула
Vo.90 *1
X0-98 к1
Vl, 010^.1
Vo.cm £i
Vl.17 N«
период
решётки
и А
4,ПН
4,1-20
4,13
4,128
4,19
'изико-химические свойства 1
!
уд. вес
_ тивление
в г/глз ;
j OAt/AtM--M
:
5,12
5,40
5,.03
5,80
5,78
0,(»358
0,0338
0,0332
0,0326
0,0325
ШЛИфО- 1
способ- I
ность в г 1
снятого 1
вещества |
0,020
0,043
0,004
0,000
0,050
На структуру вычитания накладывается и структура замещения в
зависимости, например, от температуры обработки исходных материалов азото-
водородпой смесью:
ВвМ?"са | Формула Vx(Ny01_|/)
600 I V0 9 (N0 94°0 06>
™ Vo;L(No;9K;^)
1000 Vl,0 (N0,98°0,02>
1200 \'г 03(N0 98O0 04)
1300 \\ lt N0 98O0 02)
He лишенным интереса нам кажется и то обстоятельство, что, как
видно из таблиц, кривая шлифепособности в зависимости от периода
решётки имеет максимум, вопреки ходячему убеждению, вытекающему
из теории идеального кристалла, что связанные с ней свойства тем выше,
чем меньше период решётки. См. статьи В. А. Эпельбаум и Б. Ф. Ормонт,
ЖФХ XXI, 3 (1947), Acta Phys. XXII, 319 (1947> В. А.*Эпельбаум и А. X.
Брегер, ЖФХ 14, 1600 (1940); 14, 1602 (1940); 20, 459 (1946) и Acta 13,
600, 604 (1940); 81, 764 (1946).
Так же как и в случае TiN, этими работами были исправлены
неверные периоды решётки, установленные зарубежными авторами.
§ 176] НИТРИДЫ,ФОСФИДЫ Л ДР.VI* ГРУППЛ:ХРОМ,МОЛПБ:1ЕН,1ЮЛЬФРЛМ,УРАН 54^
2. Система Та —1\
Лишь в 19Н г. были открыты соединения Та и Р (Z. Anorg. Ch. 246
35 (1941)).
Оказалось, что их 2: ТаР2 и ТаР, причём ТаР диморфен. В работе
1941 г. структуры г| а фосфидов окончательно расшифрованы не были,
§ 176. VI* группа периодической системы: хром Сг, молибден Мо,
вольфрам W, уран U
В системе Сг N описаны структуры фазы: «р-фаза» Ci\,N и «е-иу'-фаза» CrN.
1. CrN (е-фаза)
Решётка гексагональная плотная //.
Координация?
Тип Н?
Пространственная группа Д; 6'32.
Z = 2CrN.
Вещее тло
CrN
а
2,751
с
4,'.15
с/а
1,Г)()5
°х
7,75
Примечание 1
^В 11 7УЗ; V Ш и др.
Cr2N (?) также гексагональный с периодом решётки близким к Cr]4 (SB II 792).
S. CrN
Решётка кубическая С.
Координация ?
Тип?
Пространственная
группа?
При нитровании хрома
при 550 560° С
получается, кроме «г-фа-
зы» CrN —
гексагональной, также у'-фа-
за CrN- кубическая
с а-4,14 (SB V 133).
3. CrSb
Решётка
гексагональная Н.
Координация О/РЗ.
Тин Яо/рз - SB BH
(NiAs).
Пространственная
группа Dih — C6/mmc.
Z = 2*
Сг. к! ч. = 6 Sb(0);
Sb к. ч. = 6Сг{РЗ).
Рис. 400.
Вещестио
CrRb
а
с
:>/i6
с/а
1. :••-'„
d
..я
ом
'•Од)
548 ч. и. структуры неорганических веществ [гл. VII
4. Система Mo-N*) SB II, 793.
у-фаза кубическая ^ 33 атомных процентов N, т. е. Mo2N ac^ 4,155 —
4,160.
р-фаза устойчива >600°С. Содержит ^29 атомных процентов N,
распадается при закалке на а- и уФазы- Элементарная ячейка
тетрагональная, гранецентрированная, близка к элементарной ячейке у~Фазы*
а^4,180; с = 4,016; с/а = 0,961.
б. CncTejuaW —N
SBII, 794. При 18,2 атомных процентах N, т. е. ~ W5N, а = 4Д18;
появляется кубическая гранецентрированная решётка. Границы
устойчивости — WaN(?) (33 атомных процентов N).
6. W4P и W2P
W4P, повидимому, имеет гексагональную ячейку с а = 6,18; с = 6,78;
с/а = 1,097.
При нагревании W4P распадается на W и W2P.
7.Система U—N освещена в работе Рэндла (J.Am. Ch. Soc. 70,99(1948)).
В ней существует, повидимому, ряд фаз: a) UN, тип NaCl, рис. 410а);
Рис. 410а.
aw = 4,880; ая= 14,82; б) U2N3, тип MuaO„ рис. 410b z = 16; flfw= 10,678;
ox= 11,24, с широкой областью существования (по составу). Например,
при соотношении N:M = 1,75, а = 10,58; в) UN2, тип CaF2, рис. 410с.
Этот нитрид освещен пока недостаточно. Сведения о границах его
существования в литературе отсутствуют.
8. Система U —Р
Геймбрехт, Цумбуш и Бильц (Z. Anorg. Gh. 245, 391, 402(1941))
опубликовали исследовании в области системы U-P и нашли, в частности, что
*) Система, условия фазовых переходов и структуры фаз плохо изучены.
§ 176] НИТРИДЫ, ФОСФИДЫ ПД1\ VI* П УИНА: \ЛЧШ,МО;Ш1-ЩКН, Г.ОЛЬФРАМ.УРАН 549
Рис. 4Mb.
Рис. 410с
550 ч. и. структуры неорганических нещести [гл. VII
в ней существуют 3 соединения: UP2, U3P4 и UP. Теоретические взгляды
их в этой, как и в других, работах, касающихся образования структур
фосфидов и т. п., нами в большей мере не разделяются. Впрочем, авторы
правильно подчёркивают сходство фосфидов U и Th.
В приводимой нами ниже таблице приближённо намечаются границы
существования фаз в системе уран —фосфор на базе этих литературных
данных.
Соотношение
1 количеств P/U
2,07
1,>1
1,-7
Структура
UP,
U3P4 и отчасти \}PZ
ил
Соотношение
количеств P/U
1,16
1,о:$
0,78
Структура
UP и UjPi
UP
UP и U
9. UP2 имеет сложную структуру (не расшифрована).
10. U8P4
Решётка кубическая С.
Координация Dijt.
Тип С out-SB Д7,(ТЬ„Р4).
Пространственная группа T*a--J-\3d.
Z = 4.
1°
сг^
-О
Вещество
а„.
i
U,P4 8,Ю7
d
U — Р
2,84
d
Р—Р
:!,ог>
На основании статей в Z. Anorg. Ch. 240,300 (1940), 246, 402 (1911).
См. выше ThaP4.
§ 1771 тип щи, фосфиды п др. \ц* группа: марганец, рений
551
11. UP
Решётка кубическая С.
Координация октаэдриче-
ская О/О.
Tim Cojo-SB Sl(NaCl).
Пространственная группа
01 — Fm'3m.
Z = 4.
U к. ч.=6Р(0);
Р к. ч. =6U(0).
Рис. 411*
Вещество
UP
««•
5,58,,
U - P
2,79
P —P
3,95
Па основании статей в Z. Лнигр. Ch. 210,300 (1940) и 8-15, 402 (1941).
§ 177. VII* группа периодической системы: марганец Мп, рении Re
Система Мп N
а Мп растворяет очень мало N. При :ггом а растёт от 8,894 до 8,897.
Н Мп ])астворяст значительно больше N. При этом а растёт от 6,289
до 0,305.
з-фаза Mn4N —структура у'-фазы системы Fe N. Пространственная
группа rj —P43m mm Он -Рт'Зт.
о-фаза — деформированная s-фаза.
?-фаза Mn.,N — аналогична Cr2N и Fe0N, гексагональная плотная
упаковка Яг —SB A3.
Границы гомогенности 28,4 и 34,6 атомных процентов
С переходом
изменяются:
а
с
с/а
от 28,4% X к :*5% N 1
от 2,773 до 2,825
от 4,520 до 4,528 I
от 1,630 до 1,601 1
т)-фаза Mn3N8 (?). Тетрагональная гранецентрированная, с
беспорядочным распределением атомов азота (^38 атомных процентов N).
а ^4,20; с ^4,03; с/а^0,97.
00^
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ БЕЩЕСТН
[гл. VII
1. MnP, MnAs
Решётка ромбическая цепная 0ЯК.
Координация О/РЗ.
Тин ОД ^p3~SB mi <MnP)-
Пространственная группа Dift — Pbnm.
Мп к. ч.^О As (искажённый октаэдр); As к. ч. = 6Мп (искажённая призма),
Z = 4.
Cf
■4
и о
Рис. 412.
Ох
Вещество
МпР
MnAs
а
5,905
Ь = 5,907
6,38
Ь
5,249
с-5,250
5,63
с
3,167
а = 3,171
3,62
Примечание. По С. А. 39, 2684 (1945). В работе
Новотного Z. ph. Ch. (В) 38, 356 (1937) оси взаимно заменены, как указано в
таблице.
Слегка деформированная решётка NiAs (B8)} зигзагообразные цепи Мп
и зигзагообразные цепи Р (соответственно As).
2. Мп2Р
Решётка гексагональная Н (изоморфен Fe2P, т. е. тип SB С22 (?), см. Fe2P).
а = 6,08; с = 3,45.
Новотный, 1. с. (1937),
§ 1771 Ш1Т1 ИДЫ, ФОСФИДЫ И ДР. VII» ГРУППА: МАРГАНЕЦ, РЫШИ
3. Ми8Р
Решётка тетрагональная Т.
Пространственная группа S\ — /4 .
2 = 8; а « 9,160; с - 4,599.
Новотный I. с. (1937).
4. MnaSb
Решётка тетрагональная Т.
Координация — Т, Pyk/q + Рук.
Тип Тт, Pif.i/e+/>B4 —SB C38(Cu.,Sb).
Пространственная группа Dili, — Pk/nmm.
Z = 2. Mn I к. ч. - 4Sb(r).
Mn II к. и. == 5Sb(i>4)-
Sb к. ч. = 4Mn J (q) -1- 5Mn II (РуЛ).
553
Вещестио
Mn3Sb
а с
4,08
6,56
с/а
1,62
Примечание
См. CibSb
5. MnSb
Решётка
гексагональная Я.
Координация О/РЗ.
Тип Яо/рз —SB 58.
Пространственная
группа D\h — C6jmmc.
Z = 2.
Мпк. 4. = 6Sb(0);
Sb к. ч. = 6Мгн7>3).
. гъ
'^^г^Ьг^^Г^
Рис. 413
1 Вещество
MnSb
а
4,12
С
5.78
с/а d
1/i04 2,78
ОМ
42,5
Примечание
См. § 65.
|
6. Система Re —P
В системе Re —P найдены фазы: Re2P, ReP, ReP2 ReP3.
7. Система Be —As
В системе Re —As найдено только одно соединение —ReAs2, причём индекс
при As может быть несколько больше 2.
ЬЪ\ Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ РЕЩЕСТН |ГЛ. VII
§ 178. VIII * группа периодической сиетемьг.железо Ре,
рутений Ви, осмий Os
1. В системе Fe --N найдены при обычной температуре следующие фазы:
а —чистое aFe.
y'Fe4N —кубическая гранецентрированная решётка; aw = 3,79.
е от Fe3N до Fe2N гексагональная при 25 атомных процентах N аш = 2,69;
с - 4,36; при 33-а = 2,77; с = 4,42.
llFejjN — ромбическая; а = 2,76; Ь = 4,82; с = 4,42 (почти
гексагональная шаровая упаковка). См. SB —II 782-789, SB V 133, а также И. Р.
Кричевсшш и II. Е. Хазанова, ЖФХ XXI, 19 (1947).
2. В системе Fe -P SB I, 593 обнаруживаются при обычной температуре
4 фазы:
а —чистое aFe растворяет несколько процентов Р без изменения
■ячейки (?).
г Fc3P—тетрагональный; а=^9,09; с=4,45.
Fe3P -пространственная группа S\ Z = 8. Нет заметной растворимости
для Fe или Р.
С Fe2P—гексагональный; а =-5,85; с = 3,45.
Fe2P -пространственная группа D\h или D\n Z = 3.
73 —неопределённый состав (FeP?).
По SB III 310, 625: Fe3P, FeJ\ FeP, FeP2, а также и FeaP5.
3. В системе Fe — AsSBI 594 обнаруживаются при обычной температуре
3 фазы:
a-фаза - aFe растворяет около 4 атомных процентов As с увеличением
aw от 2,86 до 2,88.
з Fe-iAs—узкая область гомогенности, тетрагональная; а = 3,63;
с --5,97; FeoAs. Пространственная группа D\h или D\u, Z = 2.
y) FoAs -ромбический; я---3,666; 6---6,016; с=5,428.
FoAs -пространственная группа V^6, Z = 4; ромбически
деформированная структура NiAs.
I. В системе Fe — Sb при обычной температуре известны 4 фазы.
a-фаза aFe растворяет до 3 атомных процентов Sb, причём aw «уве-
личивается от 2,86 до 2,89.
з FcSb, тип В8, область гомогенности 43—46 атомных % Sb; д = 2,106;
г^-5,146 (43% Sb) до a=i,07; с=5,13 (46% Sb).
По некоторым данным, наблюдается даже а ^4,12; с = 5,16; с/а = 1,25 (состав
неизвестен).
С FcSb ромбический; а=3,19; 6 = 5,82; с=6,52. Пространственная
группа /)1|, тип С18 (марказит).
y) Sb-чистый. См. SB I 597, SB II 747.
5. В системе Ru—Р найдены соединения RuPa, RuP, R112P (?).
в. В системе Os —Р найдено соединение ОэРг, близкое по структуре RuP2.
Перейдём теперь к рассмотрению структуры отдельных фаз.
При рассмотрении материала бросается в глаза большое количество
модификаций и значительные колебания в составе и параметрах фаз (по
различным литературным данным).
7. Fe2N
Тип SBL3a. Этот тип (указанный в SBII 284) в списке типов, помещённом
в SB И, вообще отсутствует.
Пространственная группа D^d — НЗпг. См. SB II 284, 789 и т. п.
§ 178] 1ШТИ1ДЫ, ФОСФИДЫ II Д1>. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИИ, ОСМИП 555
Ь. Fe3P
Решётка гексагональная Н.
Координация 4,3/9,6.
Тип #4,з/9,б SB С 22*).
Пространственная группа D) — C'S2.
Z = 3.
1.'. «к:
Рис. '.Г..
Fe 1 2d' 2,24 (Р П) + 2е --- 2,30 (Р I) к. ч.
Fe II 2Л ,, 2.19 (Р Il) + lf' - 2,34 (Р I) к. ч.
Р I Ос - 2,30 (Fe I) + &;' 2,34 (Fe II) к. ч.
Р II 3d - - 2,19 (Fo II) f 3r/' -- 2.24 (Fo 1) к. ч.
4.
3.
9.
6.
I Вещество
Fe,P
! а
5,85,
i
! с
\
1
с/а
0,5М1)
Примечание
Сильно деформированная
гексагональная плотная упаковка Fe-атомоп. В
результате внедрения Р ось с
сжалась, горизонтальные осп упелпчоны.
Fe — Fe = 2,6:*, атомы РП лежат на
пертикалях.
9, PeaAs
Решётка тетрагональная Т.
Координация 7\ /).;4/(/-г-/)1/4.
Тин 7+,/>/,/,,+ />/! -SB 6^ Ж
Пространственная группа /)(/, — Pkjwtim.
Z - 2.
Fe I к. ч. г, 4As(fi 2,40А) (тетраэдр).
Fe II к. ч. = 5As(l<? ■- 2,41 А+4А --=
^ 2,60 А) (пирамида).
As к. ч. = 4Fe I (d == 2,40) (квадрат) +
+ 5Fe II (1*- 2,41 +4ft 2,60)
(пирамида).
Вещо-
стно
1 ' 1
с \ via I Примечание
II
Fe.As :i,627 5,973
t ,,J SB II 742, 1
,blbj SB III33t34
*) Если на стр. 15 SB II указана пространственная группа #J— С32, то на
стр. 285 того же тома без комментариев приводятся работы, в которых типу С22
приписывается пространственная группа />;?/,-»/76 m 2 или Т)\1г-СЬ ml.
556
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл.УИ
10. FeP, FeAs
Решётка ромбическая, цепная ORK.
Координация OJP3.
Тип OR*lPi-SB В 31 (МпР).
Пространственная группа D-u\ — РЬпт.
Z = 4.
Fe к. ч. - GP(As); P(As) к. ч. = 6Fe.
1 Вещество
FeP
FeAs
а | Ь | с 1
5,782
6,016
5,177
5,428
3,089
3,666
Примечание. Слегка деформированная решётка NiAs. Атомы Р,
соответственно As, образуют зигзагообразные цепи. Атомы Fe также образуют
зигзагообразные цепи.
Fe: Id = 2,26; 2d' = 2,275; 2d" = 2,33; d'" = 2,34.
FeAs: ld = 2,34; W = 2,46; W = 2,40; d'" = 2,50.
11. FeAs
Решётка ромбическая, сетки ORs .
Координация О/РЗ.
TmxOfl^-SBB14.
Пространственная группа Dw — Pmcn.
Z - 4.
Fe к. ч. - 6 As(6>); As к. ч. = 6 Fe(/>3).
Примечание. Искажённый тип NiAs. Слоистая решётка из слоев
Fe (на расстоянии е=2,72), с двух сторон сопровождаемых слоями As.
В решётке намечаются молекулы FeAs. Для Fe—As ld=2,28; 2d'=2,36;
Ы"=2,59; 2rf"'=2,70.
1 Веще-
1 с тв о
а
Ъ J с
i
FeAs 3,67 J 6,02
1 1 i
5,43
а : Ъ : с
1:1,64:1,48
§ 1791 НИТРИДЫ, ФОСФИДЫ И Др. 1Х* ГРУППА: КОБАЛЬТ, РОДИИ, ИРИДИИ
12. FeSb
Решётка гексагональная Я.
Координация — О/РЗ.
Тип Яо/рз-SB В8 (NiAs).
Пространственная группа Dth. Сб/мтс.
Z=2. Fok. ч.=6 Sb(0); SbK. ч. = 6 Fc(P3).
557
./-Л
S^^«cci^rLl2^^:/,4 *^~~r~*y^
Рис. 410.
1 Вещество
FeSb
а
4,06
С
м3
с/а
d
ОМ
1 i
1,26, 2,07 | .40,6
* 1 !
Примечание
См § 05
13. FePa, FoAs2, FeSb2 —см. комплексные соединения.
14, Системы Ru —Р и Os —Р.
В системах Ru —Р и Os —P найдены фазы Ru—P.,, OsP2, RuP и Ru2P (?),
структуры которых но выяснены.
§ 179. IX * группа периодической системы: кобальт Со,
родий Rh, иридий 1г
1. Система Со —N
а Со (устойчив ниже 450D С), как и рСо (устойчив выше 450° С) даёт
ту же рентгенограмму после обработки N2, что и до обработки.
Предполагалось, что фазы COa-N,, не возникают.
В 1940 г. сообщалось о выделении Co3N, имеющего гексагональную
плотную упаковку атомов Со с внедрёнными в неё атомами N. (Z. Апоге*
Ch. 211, 145 (1940)).
2. С0Р3
В системе Со —Р описаны 3 фосфида: СоРэ, СоР, СоР(>5 = Со2Р.
Структура первого и последнего до сих пор остаётся неясной (Z. Anniv. Ch
237, 132 (1938)).
558 ч. ti. структуры неорганических веществ [гл. VII
3. СоР, CoAs
Решётка ромбическая ценная 0RK.
Координация О/РЗ.
ТипОД^рз-ЭВ В31 (iMnP).
Пространственная группа Дм — РЪпт.
Z = \.
Со к. ч. = 6Р (As) (искажённый октаэдр); Р (As) к. ч. = 6Со (искажённая
призма).
Вещество
СоР
CoAs
а Ъ
5,388
5,90
5,000
5, Г,
с
3,274
V)!
Примечание
14 = 2,17; 2d' =2,31; 2d" = 2,:$f>;
Id"'= 2,27 (см. МпР и SBIII 203)
1. CoAs3=Co4(As4)3, см. комплексные соединения.
5. CoSbo, см. комплексные соединения.
§ 179] НИТРИДЫ, ФОСФИДТЛ И ДР. IX* ГРУППА: КОБАЛЬТ, РОДИЙ, ИРИДИЙ Г; 59*
6. CoSb
Решётка
гексагональная H.
Координация О/РЗ.
Тип #o/P3-SB£8
(NiAs).
Пространственная труп
па Dth—Ce/mmc.
Z = 2.
Сок. 4.-GSb(6>);
Sb к. ч. 0-Со(/'.Ч).
Рис. 418
Вещестно
CoSb
а
:*,8;
.4, 866
с j
Г», l«:i !
i
г/а \
1,:<Ч" 1
1
2, Г18
ОМ
:w,7
Примечание |
См. § Г)5
SB VI 17Г)
7. В системе Kh—Р обнаружены фосфиды RhPs, RhP.,, RhfiP4, т. е.
RhP,lS, RhsP.
8. В системе Ir—Р найдены 1гР2 и 1г2Р.
Большая часть фаз в структурном отношении надёжно не изучена,
за немногими исключениями. >—^ ,-.
9. Rh2P, 1гаР ^w*^V\ ^ ^У^
Решётка кубическая 6\ ^ !_Л ^Vs
Координация Т/С.
Тип С tic — SB анти-Cl.
Пространственная группа
ОЙ—F/w3w.
Z = 4.
М к. 4.--=/iP(7,)f
Р к. ч. = 8М(0.
Веще-
CTDO
I Rh>P
Ir.P
1
1 i
j j
!
1 1
' 5,50.
1 5 I
5,53r
| 5
X—P
2,38
2,40
560 ч. и. структуры неорганических ^веществ [гл. VII
§ 180. X* группа периодической системы: никель Ni,
палладий Pd, платина Pt
pNi после обработки чри 300° аммиаком в течение 4 часов
обнаруживает небольшое увеличение ат с 3,517 до 3,519.
Ранее предполагалось, что нитриды Ni не образуются. В 1940 г.
сообщалось о выделении Ni3N, в котором атомы Ni образуют плотную
гексагональную упаковку (атомы N внедрены в междуузлия)*).
1. Система Ni —P
Существование фосфида Ni оставалось под вопросом долгое время.
См. SBIII 264. Позже было показано**) существование фаз: NiP3, NiP.,
NiP0,83, NiPo,5 = Ni2P, а также NiO0,4 и NiP0f33.
2. NiAs2 (раммельсбергит)
Решётка ромбическая OR.
Координация— ?
Тип ОЯ-SB?
12
Пространственная группа Ьчь — Ртпп.
Z = 2NiAs1.
Вещество
NiAs,
а
3,53
Ь
4,78
с
5,78
а*
7,06
Примечание. См. SB VII 94. Возможно тип C18(FeS2, марказит).
Тогда NiAs2 относится к комплексным соединениям, см. гл. X.
13. NiAs2 (парараммельсбергит)
Решётка ромбическая OR
(или псевдоромбическая).
Координация ?
Тип OR
Пространственная группа Dli — Pbma.
Z-8NiAs2.
Веще-
1 с тп о
NiAs2
а
5,74
b 1 с
5,81
11,405
с*
7,24
Примечание
SB VII 94—возможно
комплексное соединение с узлом (As.,)
*) Z. Anorg. Ch. 244, 145 (1940).
**) Z. Anorg. Ch. 237, 132 (1938).
§ 1801 нитриды, фосфиды и др. х* группа: никель, палладии, платина 561
4. NiAs (никвелин), NiSb (брейтгауптит), Ni(As, Sb) (арит)
Решётка гексагональная Н.
Координация О/РЗ.
Тип Яо/рз — SB 58 (тип NiAs).
Пространственная группа D\u—C/6mmc.
Z = 2.
Ni к. ч. = 6As(0); As к. 4.=6Ni(P3).
On.
О
Рис. 420.
Вещество
NiAs
NiSb
Ni(As, Sb)
J "
3,6X
3,92
3,80
с
r,,o(
Г-.Ч
5,20
c/a
1,3'*3
1,зо4
1,37
d
2,43
2,60
OM
28,4
34,2
Примечание 1
1
См. § 65 I
SB I 143, 144
6. PdP
В системе Pd —P описаны:
Р-фаза — твёрдый раствор среднего состава Pd5P;
у-фаза — соединение Pd3P (плавится при 10'47° С);
а-фаза (перитектическая) —Pd6P2.
Наконец, найдено соединение PdP2 (не тип пирита).
6» PdAs2, PdSb2, PtP2, PtAs2, PtSbo. См. комплексные соединения
(тип пирита С2).
562 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ |гл. VII
7. PbSb, PtSb.
Решётка
гексагональная Н.
Координация OfP'i.
Тип H0IP3-SBB8
(тип NiAs).
Пространственная
группа Въп—Сб/ттс.
Z = 2'
Мк. ч. = 6 8Ь(0);
Sbn. ч. = 6 М(РЗ).
—ГЛ
Вещество
\а . . . .
с . . . •
с/а . . .
«х ■ • ■ ■
<*
[Pd—Sb .
PdSb
4,070
5,582
1,371
9,4
2,732
PtSb
4'1:l0±4
•V^o^rJ
1'32Г,±2
Рис. 42J.
§ 181. I* группа периодической системы: медь Си, серебро Ag, золото Аи
В системе Си-N в 1940 г. Z. Anorg. Ch. 239, 282 (1938); 241, 177 (1939);
244, 144 (1940) был открыт чрезвычайно интересный нитрид Cu8N с
возможно ионной решёткой (?).
1. C«,N Г&Г^
Решётка кубическая С.
Координация I/O.
Тип С//0--SB ZX)9 (анти
Re03).
Пространственная группа
Ok — РтЗт.
Z =1.
N к. ч. = 6 Си (О):
Си к. ч. - 2N(Z).
2. Системы Ag —N, Au — N
надёжно не изучены.
Структуры азида AgN3 — см.
стр. 679.
3. Ag,N.
Опубликовано
сообщение (Z. Anorg. Ch. 258, 77
(1949)) о структуре Ag3N.
Решётка С к для атомов Ag.
Z=l,33 Ag,N, т. е. 4 Ag
(+ 1,33 N расположенных
статистически в октаэдрических междуузлиях), а
4. Система Си —Р
В системе Си — Р были обнаружены две фосфорсодержащие фазы: СиР2
и Си8Р. Структура второй из них установлена не была.
§ 181] НИТ1Ч1ДЫ, ФОСФИДЫ II ДР. I* ГРУППА: МЕДЬ, СЕРЕБРО, ЗОЛОТО
56Я
5. Система Си —As
Диаграмма состояния и структурные исследования системы Gu —As
свидетельствуют о наличии фаз Gu9As, CueAs, Gu.As (Cu.As,?) и CuAs
(см. также SBII 300, 739-740).
6. Cu3P, Cu3As (искусственный домейкит)
Решётка гексагональная Н.
Координация я/12.
Тип Ях/12 —SBZX)21.
Пространственная группа Dtd—СЗс.
Z- 6.
Вещество
Си,Р
1 >}
| Си3Ач
а ! с
\
6,942 1 7,098
7,070 7,1.45
7,088 7,232
с /а
1,022
1,008
1,020
Примечание
SB VI 7,Г,Г)
SB VI 7,65
7. Cu9As (уайтнеит) -f Cu6As
(альгодонит)
Решётка кубическая гранецен-
трированная С к.
Координация К (12).
Тип Ск — SBAi.
Пространственная группа 0\- -
Fm3m.
Z--4.
£>
оО^^
О
о
of
Ъ£
о-
0
о
НЭ
У
6
О cms
Рис. 423.
Вещество
Cu9As
CueAs
1
аи>
Колеблющаяся
:J,G47 i
3,651 I
Примечание 1
Обычно соприсутствуют обе
фазы—твёрдый раствор AsS. в решётке
Си и Cu3As (искусственный
домейкит).
См. SB II .400 и след.
Статистическое (?) распределение As и Си.
Альгодонит, возможно, образует также фазу с гексагональной
структурой с д = 2,598; с-4,215; с/а = 1,622.
564 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VII
8. Cu8As (домейкит)
Решётка кубическая С.
Координация x/t + t(?).
Тип Cxit+t(?)- _
Пространственная группа Та— У43с/.
Z=16(?).
Вещество
Cu3As
а*
9,592
*х
7,95
Примечание. SB VI 66. Решётка кубическая центрированная (?).
Лтомы Си образуют друг с другом тетраэдры. Атом As окружён 2
треугольниками из атомов Си, повёрнутыми друг относительно друга.
9. Система Си —Sb
Изучалась довольно детально (SB VII 207 —209), причём дана её
диаграмма состояния (1с).
Результаты исследования структуры фаз сведены нами в таблицу
(обозначения элементарной ячейки наши) в порядке алфавитного обозначения фаз.
1 Фаза
1 а
Р
Т
*
4
0
«
Формула
—
Cu5Sb,
CuaSb
Си9ЯЬа
CunSb2
CunSb4
Весов
°/
/о
100-95,3
56,71
при
t°
b'OMII,
51,08
70,14
74,17
58,95
i
Элементарная
ячейка
Ск
Tp,
Tin
и
OR
И
R
а
3,6079.,
ДО
3,6413
9,014
5,621
10,8360
9,285
5,494;
4,290
4,490
(а
ромбоэдр.)
с
—
8,574
6,074
8,6И2
8,629
8,6863
11,237
57*4'
с/а
—
0,9512
1,081
0,7947
8,179
1,5Ь0-
2,619
z
4
2п
12
12
49
15
6
1
с
8.92
ДО
8,542
8,9
8,446
8,878
8,909
8,617
G,68
Примечание
Фаза устой-1
чива выше 1
410°; при за-|
калке возни-1
кает Р'-фаза 1
(55,5—68% Си)
и др. фазы.1
См. 1.с
Сверхструк-|
тура плотней-1
шей гексаго-1
нальной упа-1
ковки 1
Деформиро-|
ванная гекса-|
тональная I
Сверхструк-1
тура плотней-!
шей гексаго- 1
нальной упа-1
ковки 1
При ком-1
натной тем-1
пературе Си 1
:* Sb не рас- 1
творяетсн 1
5 181]
НИТРИДЫ, ФОСФИДЫ И ДР. !• ГРУППА: МЕДЬ СЕРЕБРО, ЗОЛОТО
565
Обозначения фаз в порядке букв греческого алфавита не отвечают
порядку, вытекающему из стехиометрического состава:
а=100%Си; 4 = 74,17% Си;
S = Cu9Sb2 = 70,14o/o Си;
6 = 58,95% Си; £ = 56,71% Си;
у = 51,08% Си; £-0%Си.
б. Cu3Sb (е-фаза системы
Си—Sb), Ag8Sb (а-фаза
системы Ag — Sb), дискразит
Решётка гексагональная плот-
неишая Н.
Координация ^(12).
Тип Hv-SBA3.
Z=iCuSb.
QcatSb
Рис. 424.
1 Вещество
Cu3Sb
Ag.Sb
(средний состав)
а
2,777
От 2,984
до 2,985
2,98г
с
4, «67
От 4,803
до 4,816
4,81
с/а
1,572
От 1,610
ДО 1,631
Примечание. SBI 596. По SB II 741 и д., г-фазы имеют
беспорядочное (статистическое) распределение атомов Си и Sb. С изменением
содержания Sb в пределах устойчивости фазы её параметры меняются. Например,
дляСи38Ь а = 2,72-2,78; с = 4,32-4,37.
6. Cu2Sb
Решётка тетрагональная Т.
Координация: тетраэдр, пирамида (квадрат + пирамида) Т,Ру &/q-\-Pyi.
Тип ^2,ру4/а+Р[/4 —SBC38.
Пространственная группа D\,h — P^jnmm.
Z — 2
Cul к. ч. -4Sb(d-2,70) (тетраэдр);
Си II к. ч. =5Sb(le = 2,62 + 4/* = 2,83) (пирамида);
Sb к. ч. = 4 Cul (d-2,70) (квадрат)+ 5CuII(le = 2,62+ 4А = 2,83) (пирамида).
Вещество
Cu.Sb
Средний состав
а
3,99—4,02
3,992
с
6,17—6,13
6,091
с/а
1,5—1,6
1,525 1
Примечание
SB I 596
SB 11 743 1
SB III 33 I
566 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VII
7. Система Ag — As. По SB II 741 существует лишь одно соединение
Ag0As (?) р-фаза (87,8 атомных процентов Ag). (Этому атомному составу
р-фазы отвечает формула ~Ag7As.) [3-фаза—гексагональная шаровая
упаковка со статистическим распределением атомов, а = 2,891; с = 4,772;
с/а = 1,633; Z-2; оЛ = 9,94.
8. В системе Ag — Sb по SB II 744 и д., кроме а-фазы (Ag содержит до 5 % Sb)
существует:
е-фаза—гексагональная плотнейшая упаковка с 10 —16 атомных
процентов Sb. В зависимости от содержания Sb а ^ 2,92 — 2,96; с = 4,77 -4,78;
г/а «1,63.
е'-фаза— слегка искажённая гексагональная упаковка, решётка
ромбическая, 20 — 25 атомных процентов Sb. а = 2,98—3,00; b = 5,17 — 5,23;
0 = 4,80-4,83; Z=4.
$♦ \\ системе Au —Sb существует по SB II только одна промежуточная
фаза AuSb2.
10. Предполагалось, что Gu, Ag, Auc Bi не дают соединений, но лишь
эвтектики (SB II 745 и д.). Однако в SBIII описано соединение Au2Bi.
11. Ag2Sb, (гольдшмидтин)
Решётка ромбическая, центрированная OR.
Тип 0Я — SB (?).
Пространственная группа .©2 — ^222^
Z^10(?).
Вещестио ' | а | Ь 1 с \ а : Ь : с 1 сг
| Ag.Sb
7,75
12,32
8,42
0,629 : 1 : 0,083 | 6,02
12. AuSb3 (тип пирита), см. комплексные соединения.
13. Аи2Ш гл
Решётка кубическая С. «~» "^ ' С ■
Координация 77/6.
Тип C-rm -SBC15 (Cu2Mg).
Пространственная группа
Ol—Fd'Am.
Z = 8Au,Bi.
Вещество ] а ох
АиаШ
7,942
15,70
SB III 21. Положение
атомов, как в структуре
шпинели положение А1 и Mg.
Та же пространственная
группа; см. стр. 631.
Структура SB —C15
может быть описана таким
образом, что атомы Mg.
соответственно Bi, занимают позиции Рис. 425.
атомов С в решётке алмаза,
т. е. образуют кубическую гранецентрированную решётку, центры 4 октантов
которой в тетраэдрическом порядке заняты дополнительными атомами того же
рода. Таким образом, координационное число этих атомов друг относительно
I 182]
НИТРИДЫ, ФОСФИДЫ II ДР. II* ГРУППА: ЦИНК, КАДМИИ, РТУТЬ
567
друга равно 4. Не занятые центры 4 других октантов элементарной ячейки
и центр последней заполняются тетраэдрами атомов Си, соответственно Аи,
причём эти тетраэдры связаны друг с другом общими вершинами, ср. KBi2.
14. В системах Ag —P и Au —P описаны фазы: AgPs, AgP2 и AuaPs.
Последнее соединение является единственным примером фосфида,
имеющего состав М2Р3 в ряду переходных металлов середины больших периодов.
Структуры этих фаз, к сожалению, не изучены.
§ 182, II* группа периодической системы: цинк Zn, кадмий Cd, ртуть Hg
Азиды соединений II* группы рассматриваются в разделе
комплексных соединений (М (Ns)2) гл. X.
1. Zn3N2, CdsN2
Решётка кубическая С.
Координация Т/С ~, С".
Тип СТ1с~1~ус= - SB анти-£53 (Mn208).
Пространственная группа T\—Ja^.
Z = 16.
]
Вещество
"'
Рис.' 426.
Zn.N,
*'743±5
Cd,N,
Ю,79±2 1
588 _ ч. и. структуры неорганических веществ [гл. VII
2. Zn3P2, Zn3As2, Cd3P2, Cd,As2
Решётка тетрагональная Т.
Координация Т/6<
Тип rf/6-SB£59(Zn8P2).
Пространственная группа D\\ —Pi/mmc.
Z = 8. Zn к. ч,:г=4Р (искажённый тетраэдр); Р к. ч. =6Zn.
Рис. 427.
См. примечание на стр. 539.
ВещестБо
а
с
с/а
Zn3Pt
8,09-,
11,45
1,418
Zn3As2
8,316
11,76
1,418
С(13Ра
8,746
12,28
1,404
CdgAs-j Примечание
8,«45
12,65
1,418
Решётка очень
близка к Sb £>58
(Mn,03) рис. 426.
Атомы неметалла образуют плотную упаковку. Атомы металла
занимают часть тетраэдрических промежутков. 4 атома Р образуют слегка
деформированный тетраэдр; 6 атомов Zn занимают 6 из 8 вершин куба.
§ 182] нитриды, фосфиды и др. п* группа: цинк, кадмий, ртуть 569
3. Zn8P2, Zn3As„ Cd3P2, Cd3As2
Решётка кубическая С.
Координация Т/С !~.
Тип Ст/с~'— SB Dbb (Mg3PJ.
Пространственная группа Of, — РпЗт .
М к. ч. = 4Р(Г); Р к. ч. = 6М(С7-).
Ох
Рис. 428.
1 Вещество i Zn3P2
ОМ
1 с*
5,68
2,46
91,6
4,678
Zn3Asa
5,81
5,854
Cd3P2
6,0Г
о
2,02
111,27
5,956
Cd3Asa
6,29
2,73
124,42
3,851
Примечание
См. Mg3Pa
При сопоставлении
структур рис. 427
и 428, см.
примечание на стр. 539.
4. ZnP2, CdP2
Решётка тетрагональная Т.
Координация?
Тип Г -SB?
Пространственная группа В\ — Р^121 или D\ — P^32t.
Z = 8MP„.
Вещество
ZnP,
CdP2
a \ с
1
5,07
5,28
18,65
19,70
с/а
3,68
3,73
°x
1
3,51
4,19
570 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВШЦЕСТН [ГЛ. VH
б. ZnAs2
Решётка ромбическая OR\ Z = 32.
а-7,72; 6 = 7,99; с = 36,28; ах = 5,08.
6. Системы Zn — Sb и Cd — Sb. По SBII 749, SBIH644 в обеих системах
существуют соединения ZnSb и CdSb, образующие ромбическую элемен-
тарную ячейку с параметрами:
Вещество
а
ZnSb 6,17
1 CdSb 6,52
Ь
8,27
8,604
с
3,94
4,16
Z
4
Кроме того, допускалось существование гексагональной модификации
CdSb с а = 4,42; с/а = 0,78.
В обеих системах существуют, повиднмому, метастабильные фазы
Zn3Sb2 и Cd3Sb2.
У Cd3Sb2 обнаружена моноклинная ячейка с а = 7,20; 6 = 13,51; с = 6Д6;
Ji = 100e14'; Z = 4.
Наконец, в системе Zn —- Sb допускалось существование соединения
Zn4Sb3 с неуточнённой структурой.
§ 183. III группа периодической системы: бор В, алюминий AI, галий Ga,
индий In; таллий TI
1. BN
Решётка гексагональная Hs.
Координация tjt.
Тип tff„-SBfil2.
Простр анственная группа
Dth—CQ/mmc.
Z = 4.
в к. ч. = 3N(0;
N к. ч. = ЗВ(0-
Вещество
BN
а
2,51
с
6,69
Рис.
с/а
6,67
429.
л
1,45
ОМ
18,-Г)
Примечание. По SBI структура BN отвечает структуре графита,
где один атом С замещён на В, другой—соседний—на N, причём атомы N
§ 483] НИТРИДЫ, ФОСФИДЫ И ДР. III ГРУППА: БОР, АЛЮМИНИЙ, ТАЛЛИЙ 571
расположены в каждой сетке друг над другом по оси с. Они па рис. 429
соединены прямыми. Атомы же В лежат друг над другом в 1-й и 3-й сетках.
Прямые, их соединяющие (на рис. 429 не показаны), проходят через центры
колец промежуточной (2-й) сетки. Такому положению отвечает
координация tjt. Эта же структура воспроизводится в книге Уэллса*). В других
работах допускалось иное расположение—атом азота верхнего слоя над
атомом бора нижнего слоя, что отвечает координации РуЗ/РуЗ. См. также
А. X. Брегер и Г. С. Жданои. ДАН 28, ($30 (1940) и § 96.
2. AIN, GaN, InN
Решётка гексагональная Н.
Координация тетраэдрическая Т/Т.
Тип Яг/г — SB 54.
Пространственная группа Cqv —Сбгпс.
Z = 2
М к. ч. = 4N(T);
N к. ч.----4М(Г).
On
Рис. W).
Вещестпо
A1N
GaN**)
InN
а
:«fl80
С
'»,965
МО.
о
с/а
1, 000
1,025
1,611
ОМ
2 L, :*
°х
0,11
0,01
Примечание
SB III 201
SB VI 59
*) Уэллс, Строение неорганических веществ (1949).
**) По Г. С. Жданову и Г. В. Лнрман, <'h. Z. 1,4739 (1937); для GaN «-=3,11)0 + *;
с=*5.125 ± 10; «/*=!.Г>22.
572 ч. ii. структуры неорганических вкществ [гл. VII
3. А1Р, AlAs, AlSb, GaP,
GaAs, GaSb, InSb
Решётка кубическая С.
Координация Т/Т.
Тип Cj/r—SB 53.
Пространственная группа
Td—FA3m.
Z = 4.
Мк. ч. = 4Х(Г);
X к. ч.=4 М(Г).
Рис. 431
Вещестпо
A1F>
AlAs
AlSb
GaP
GaAs
GaSb
InSb
<*»
5,45
5,63
6,10
5,44
5,63
6,09
6,45
d
2,36
2,44
2,60
2,36
2,44
2,60
2,62
OM
40,5
44,6
56,8
40,3
44,6
56,5
67,1
Примечание
См. § 63.
По SB II 236a„(AlP) =
ox=2.424.
По SB II 236 ^(AIAs)-
cx=3,81
5,42;
5,62;
1
1
4. TISb, TIBi
Решётка кубическая С.
Координация кубическая С/С.
Тип CC/c-SB52.
Пространственная группа
ОЦ—РтЗт.
Z = l.
Т1к. ч. = 8Х(0;
Хк. ч. = 8Т1(С).
Рис. 432.
§ 184) нитриды, фосфиды и др. iv группа; углерод, кремний, свинец 573
Вещество
ОМ
Примечание
TISb
TIBi
3,8,
*>%
з,зл
3,45
56,6
63,0
См. CsCl. Согласно SB I 79
в SB I 76 и 141 тип TISb и TIBi
неверно причислен к типу В 2
вместо Л 2.
В системе Т1 —Sb
-4 фазы (SB I 599).
а — гексагональная,
раствор Sb в а Т1
(до 7 атомных
процентов Sb),
Р —кубическая, раствор
Sb; в рТ1 узкая
область гомогенности
при комнатной
температуре (около 11
атомных процентов
Sb).
у—кубическая
(центрированная), Tl7Sb2
со сверхструктурой
(рис. 433).
о-решётка Sb,
растворимость Т1 мала.
Т1 образует с Sb и Bi
кубические > гране-
центрированные р-фа-
зы — с 8 атомными
процентами Sb, т. е.
TleSba = 4,841,
соответственно с 2 — 4
атомными процентами Bi, т. е.
dfe^b^bi?
. Рис. 433.
Tl49Bi до Tl34Bi « = 4,844±2.
§ 184. IV группа периодической системы: углерод С, кремний Si,
германий Ge, олово Sn, свинец РЬ
1. Углерод образует с азотом соединения C2N2, т. е. (CN)2 — дициан, в
нормальных условиях—газ. По данным SB III 731, SB VII 240, расстояние
0-0 = 1,43 ±3; C-N = l,16±2.
Кристаллический (CN)2, повидимому, не исследован.
2. В нашем распоряжений нет данных о структуре фаз также в системах
С-Р, С-As, С —Sb, С-В1.
3. По системе Si—N надёжных данных нет.
4. В системе Si —P обнаружен единственный фосфид SiP, упругость
диссоциации которого р = 1 атм при 1140°С. Рентгеновская картина SiP
и GeP имеет существенные различия.
5. Система Si —As изучалась недостаточно детально, имеется указание*), что
в ней существуют одна или две интерметаллические фазы.
6. Si — Sb, повидимому, образует эвтектическую систему.
7. Si и Bi не смешивается в жидком состоянии*).
*) Z. Anorg. Ch. 244, 221 (1940).
57^
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТН
[гл. VII
8. В системе Ge —N изучена структура фазы Ge3N4. См. сложные
соединения.
9. В системе Ge —P описан единственный фосфид GeP. Структура
фосфидов SiP и GeP всё же не была расшифрована.
10. В системе Ge —As описаны соединения GeAs2 и GeAs с широкой
областью гомогенности (обозначено чертой). Структура их точно не определена.
В мышьяке (а-фаза) может раствориться до 16% германия. При этом,
периоды решётки могут изменяться от а = 3,754; с == 10,52 и cja = 2,802 (для
мышьяка) до а =-^ 3,701; с = 10,71 и cja = 2,894, т. е. имеют место сжатие а
и удлинение с. Если пересчитать на ромбоэдр, то получится: а = 5,576;
а = 84°39' (мышьяк)и а = 5,569; а = 83с18' (твёрдый раствор), т.е. ребро
ромбоэдра остаётся почти неизменным, но увеличивается отклонение угла от 90°.
В германии ф-фаза) мышьяк растворяется, вызывая значительное
увеличение удельного веса, несмотря па то, что атомные веса обоих элементов
близки и что период решётки практически не меняется: повидимому,
возникают структуры внедрения.
Ge — Sb и Ge — Bi образуют эвтектические системы.
В системе Sn —P выделены фазы SnP3, Sn8P4, Sn4P3.
В системе Sn —As (SB III 649 — 651) обнаружены фазы:
11.
12.
13.
19,22;
14. Sn5As2 —ромбоэдрическая элементарная ячейка; а = 12,23; а
Z = 7. Высказано предположение, что фазе отвечает идеальный состав Sn4As3,
но гомогенность достигается, если часть атомов As замещена на Sn.
15. SnAs, тип Со/о-SB Я1; а-5,716; ах = 6,845.
16. В системе Sn —Sb выделена фаза SnSb, относительно структуры
которой в SB без комментариев приводятся противоречивые данные.
Согласно SB I 77, SnSb имеет кубическую решётку, тип Ст/т— SB53
с дш = 6,13. Согласно SB II 752, SnSb имеет кубическую решётку, тип Со/о —
SB Bi с аш = 6,12 при 50 атомных процентах Sb.
Согласно SB III 651,
SnSb имеет не кубическую, L) ~£~)
но ромбоэдрическую
решётку с а = 6,214; а = 89,38°
при 52,4 атомных
процента Sb; Z = 8.
17. SnSb
Решётка кубическая С.
Координация Т/Т.
Тип С7 it -SB S3.
Пространственная группа
rj—F43/W.
Z-4.
Sn к. ч. = 4 Sb(T)\
Sb к. ч.=4 Sn(T).
Вещество
SnSb
1
fl*
6,13
d
3,05
ОМ
57,6
Рис. 434.
§ 187] НИТРИДЫ, ФОСФИДЫ И ДР. VII ГРУППА: ФТОР, ХЛОР, БРОМ, ИОД blh
18. В системе Sn — Bi промежуточные соединения, повидимому, не
выделены .
19. Системы Pb — N, Pb — Р, повидимому, исследованы плохо.
20. РЬ и As образуют эвтектическую систему.
21. В системе Pb —Sb промежуточные соединения, повидимому, не
обнаружены (непрерывный ряд твёрдых растворов SB III 652).
22. В системе Pb—Bi обнаружено соединение Pb2Bi с гексагональной
элементарной ячейкой.
23. Pb2Bi (?)
Решётка плотная гексагональная // (?).
Координация тетраэдрическая Т/Т (?).
Тип Нтit-$>В A3 (?).
Z — 2 атома.
Вещество
a
с
Pb2Bi 3,47-3,48 j 5,77-Я,78
1 1 1
cja
1,Г.Г>
Примечание
Надёжных данных но
имеем.
В с и/теме Pb —Bi описано (Z. Anorg. Ch. 244, 221 (1940))
единственное соединение Ri2Pb3 с широкой областью гомогенности.
§ 186. V группа периодической системы: азот N, фосфор Р, мышьяк As,
сурьма Sb, висмут Bi
Элементы V группы в отдельных случаях образуют соединения между
собой.
P3N5 и PN
Описаны два нитрида: P3N5 и PN. Последний получается термическим
разложением PaN6 между 750 — 810° и разложением в вакууме при 720°С.
При нагревании PN сублимирует.
Судя по порошковым диаграммам, оба нитрида получились аморфными.
О структуре фаз в системе As —Sb и, в частности, (З-фазы см. дискуссию
Z. Anorg."Ch. 238, 255 (1938); 240 139, 142 (1939).
§ 186. VI группа периодической системы: кислород 0, сера S, селен Se,.
теллур Те, полоний Ро
См. окислы и халкогениды.
§ 187. VII группа периодической системы: фтор F, хлор С1, бром Вг,,
иод J
См. галогениды.
ГЛАВА VIII
КАРБИДЫ, СИЛИЦИДЫ <В ОТДЕЛЬНЫХ СЛУЧАЯХ СТАННИДЫ
И ДР.), БОРИДЫ
А. КАРБИДЫ, СИЛИЦИДЫ
§ 188. I группа периодической системы: литий Li, натрий Na, калий К,
рубидий Ш>, цезий Cs
Щелочные металлы, повидимому, не образуют карбидов с
изолированными атомами углерода (известны ацетиленнды типа КагСг и NaHC2,
содержащие комплексные группы [Сг1 и [НСг]), а с атомами Si образуют,
повидимому, комплексные силициды (см. комплексные соединения гл. X).
§ 189. II группа периодической системы: бериллий Be, магний Me,
кальций Са, стронций Sr, барий Ва
Большой интерес представляет то обстоятельство, что из всех
элементов этой группы пока только у Be обнаружено соединение типа ВегС
(решётка типа Сj/с — SB анти-С1) рис. 435; при этом ионизационный потенциал
и энергия сублимации J3e значительно выше, чем у его гомологов.
Mg,Ca, Sr, Ва образуют ацетилениды MgC2, СаСг, SrC^, BaC2 (см.
комплексные соединения).
Mg образует с Si, Ge, Sn и Pb соединения типа Mg2Si (опять-таки с
решёткой типа Ctic — SB анти-С1): Mg2Ge, Mg2Sn, Mg2Pb.
Кроме того, Са образует силицид состава CaSi2 — Л6/з,4 — SBC12 с
своеобразной структурой.
§ 180| КЛРКПДЫ, СИЛИЦИДЫ. И П\: r.Kl'll. 1., МЛГП., 1П.ИЦ., СТРОШГ., ПЛШП 577
1. Вс,С
Решётка кубическая Г.
Координация Т/(\
Тип Г;/с SB апти-П .
Пространственная \\л пна
Z---4.
Рис. /.иг
1 Вощестпо 1 л,,. | ас
B'J.C
'i,:w
2/»'i
Примечание
9'i,7% Ве2С и другие более загрязнённые
препараты
2. Mg2Si, Mg2Ge, Mg2Sn,
MgaPb
Решётка кубическая С.
Координация CjT.
Тип Ссп - SB C1.
Пространственная группа
О/, — Fm3m.
Z-4.
Mg к. ч.=4 Si (тетраэдр)
Si к. ч. = 8 М#(куб).
'не /i:wi.
О Мд
Вещество |
Mg-Oe ;
MgiPb i
G,:*'J
0,S78 !
о, 7or» ;
G.83G
- 1
2,77
2,l.)
OM
<'-■',, 2 ,
77,G '
77,, !
5C
:t,:»7
Г>,2У
" " О Si
Примечание
См. SB I 1Г.0 и SB III Ш
578
1. II. СТРУКТУРЫ НКОРГЛНИЧЕСКИХ ПКЩКСТН
[гл. VIH
3. CaSi2
Решётка ромбоэдрическая Я.
Координация 6/3, 4*).
Тип flfi/3,4-SB С12.
Пространственная группа D$a—R'Jw (?)•
Z = 2.
Са к. ч. = 6Si;
Si к. ч. = ЗСа.
Вещество
\ а . . . .
1 а
/
1 Si - Si ...
d
1 Ca — Si . . ...
h
Ca-Ca ....
OM .
CaSi^
10,4
21°Л0'
2,48
2,00
3,88
66,5
0 12 3 4 5
ill111
Рис.. М7.
О са
§ l\)(). Ill* rpyuua периодической системы: скандий Sc, иттрий Y,
лантаы La
Карбиды и силициды почти не исследованы. Изучен ацетиленид
лантана LaC.» (см. комплексные соединения).
§ 191. Ill** rpyuua периодической системы: редкие земли
Карбиды и силициды не изучены.
Описаны структуры ацстиленидов отдельных металлов (см.
комплексные соединения).
*) Координационные отношения и этой структуре более сложны и отражаются
формулой лишь приблизительно.
£ 192] КАРБИДЫ, СИЛИЦИДЫ. IV* ГРУППА: ТИТАН, ЦИРКОНИЙ, ГАФНИЙ, ТОРИЙ 579*
§ 192. IV* группа периодической системы: титан Ti, цирконий Zr,
гафний Hf, тории Th
Начиная с IV группы периодической системы, широко образуются
нормальные карбиды*). Впрочем, Th образует карбид ThC2 ацетиленид-
иого типа, рассматриваемый в разделе комплексных соединений.
Силициды Ti и Zr, хотя и имеют состав TiSi2 и ZrSi2, образуют
координационные структуры. (Во всяком случае это относится к лучше
исследованному TiSi2.)
1. TiC, ZrC, HfC
Решётка кубическая С.
Координация О/О.
Тип С о/о —■ SBBi.
Пространственная группа O'h — Fmoni.
Z = 4.
М к. ч. = 6 О (oi,Tii:),(i>); С к. ч.~6 М (октаадр).
О^
63
Г\-
т
ь^-ч^"
3
<^ЧГ
X)
Рис. 438.
Вещество
тн;
ZrC
HfC
4,31-
4,687
4,4Г.78
2,i:>„ !
J t
2,3*, |
-\228ч I
ОМ
20,1
25,ЧГ>
22, И)
Примечание
l
, См. SB IV 6,100, а|
[ также Г. А. Меерсон и
! А. Г>. Липкое, '/К. Пр.
7,81 : X., 12, 1759 (1939). i
*) Их свойства рассматриваются, в частности, в монографин Я. С. Уманского,
Карбиды твёрдых сплавов (19'*7), где даютин подробные сведении о алиисимостп
периодов решётки от состава ф:к$, и л работах И. С. Уманского и С. С Хнчекелп
ЖФХ 14 (1940-19И).
580
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТК
ft л. VIII
2. TiSi2
Решётка ромбическая OR.
+ •*
Координация 10/5 ■= /Г' + /.").
Тип Oflm/5-SB С54.
Пространственная группа />гл -Fddd.
Z — 8
Ti к. ч. -=10Si: Si к. ч. = 5Т5.
•*<Э
Si
Рис. 439.
Вещество
TiSi2
а
8,236
b 1 с j я : b : с Примечание
! 1 II
4,773
i
8,52 J 1,726:1:1,786
SB VII 12
Слои из одного атома Ti (чёрный) и двух атомов Si (белый) по
принципу плотной упаковки. В этом слое Ti к. 4.=6Si; Si к. ч. = ЗгП
(рис. 439). Слои укладываются друг на друга особым образом: атом Ti
второго слоя располагается над точкой 1> третьего -над точкой 2,
четвёртого—над точкой 3. В результате каждый атом Ti приобретает ещё
по 2 соседа Si снизу и сверху, а всего 10 соседей; каждый атом Si—ещё но
одному соседу Ti сверху и снизу, а всего 5 соседей. Сравни MoSu (стр. 580).
3. ZrSi2
Решётка ромбическая, слоистая ORs. »
Координация 8/4.
Тип ОД^-SB С49 (ZrSL).
Пространственная группа D^ — Cmcm.
Z = i.
Zr к. 4. = 8Si;
Si I к. 4.—4Zr; Si II к. ч. = 4Zr.
Вещество \ a \ b \ с \ Примечание
ill I
ZrSi,
3,72 14,01 3,67 j SB V 5
§ 193] КАРБИДЫ, СИЛИЦИДЫ. V* ГРУППА: НАНАДПП, НИОБИИ, ТАНТАЛ 581
§ 193. V* группа иериодичеикоц системы: ванадий V, ниобий Nb,
тантал Та
4. VC, NbC, ТаС
Решётка кубическая С.
Координация — 0\0.
Тип Со/о-SB 51.
Пространственная группа
01 Fm3m.
Z = 4.
Мк.ч. --6С(0); С к. ч.-.-=
= {jM{0).
В системе V — С,
согласно SB II 780,
возможны (предполагаются) фазы:
а-ванадий.
р — гексагональная
плотная упаковка.
V5G (a -2,856; с -4,545;
с/а = 1,592) или V2C.
2 —кубическая гране-
центрированная V4G3 или
VG с широким интервалом
гомогенности. Рлс. 44).
В монографии Я. С.
Умай о кого (см. стр. 579) подробно цитирована литература по вопросу о
возможных карбидах ванадия, ниобия, тантала, а также результаты
собственных исследований, выполненных Я. С. Уманским и его сотрудниками.
Из .)тих последних работ вытекает, что область гомогенности карбида
ванадия с решёткой Со/о действительно простирается в сторону составов
с низким содержанием углерода «по крайней мере до составов, отвечающих
формуле V4Ct1>. Если образец изготовлен не в вакууме, дефицит углерода
восполняется замещением атомами О и N. (Вопрос о границе области
гомогенности второго карбида V2C—V5C (см. выше) ещё не уточнён.)
1 Вещестио
VC
NbC
ТаС
ТаС
**
4,15
4,40
'i,44G
4,445
d
2,075
2,20
2'223
• 2,22Г)
ОМ
1 17'Э
21,3
21,95
21,81
Примечание
SB III 7. Следы О it N
SB IV 100
С увеличением содержания связанного углерода в карбиде «VG» от
14,9% до 17,5% период решётки возрастал от 4,147 до 4,159 кХ.
В системе Nb-C Я. С. Умапскпй обнаружил (ЖФХ 14, 332 (1940))
(см. также монографию 1. с.) кроме NbC фазу Nb4C, в которой атомы С
не полностью занимают узлы неметалла решётки Со/о (структура
вычитания). Взаимная связь и условия фазовых переходов в этой системе
остаются всё же несовсем понятными. С изменением содержания С от 20
до 50 атомных процентов период решетки растёт от 4,395 до 4,46 кХ.
Работы}!. С. Уманского по карбидам ванадия, ниобия и др. полностью
подтвердили правильность наших установок о возможности широких
колебаний в подобных веществах состава в пределах структурно однородной
фазы, с изменением периода решётки, высказанных в работах автора
1936 г. и подтверждённых выполненными в нашей лаборатории работами
А. А. Брегера и В. А. Эиельбаума но нитридам титана и ванадия
(1937—1940).
о^
?-7Л>
ЬЗ-Р*
о
п
г
o^f^f^r
с\
6
b¥^Z&*
о
Ос
о
м
582
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТЬ
[гл. VIII
1 Вещество | а
1 a Ta3G
Я,091
с 1 с/а
4,93
1,59. 1
э
б. аТа2С
Решётка гексагональная Н.
Координация?
Тип (Я-SB С 6 (?)).
тт г^з ^т; «л- Примечал и с. Плотпоишая
Пространственная группа Du — tdmii гексагональная упаковка атомов тан-
Z = lTa2C. тала. См. SB III'309.
Карбид тантала ТаС тип Со\о растворяет тантал до состава Таь2С,
причём период решётки уменьшается от 4,445 до 4,39. При большем со
держании тантала возникает новая фаза: аТа2С, в которой атомы металла
образуют плотную гексагональную упаковку. Помимо этой формы (a Ta2G)
предполагается образование второй, возможно, также гексагональной
формы Та.С,достаточно надёжно не охарактеризованной.
Системы ванадий — кремний, ниобий — кремний и тантал —кремнии
изучались в работе (Z. Metallic. 33, 370 (1941)), обнаружившей, что в этих
системах образуются силициды названных металлов, общей формулой MSi2,
тип CrSi2 (SB С40). Кроме того, имеется указание (SB VII398) о том, что
в системе V—Si существует соединение V3Si со следующей характеристикой.
6, V3Si /Г 'А
Решётка кубическая С.
Координация S( + S)/J.
Тип Cs(+S)U-SB 415(pW).
Пространственная группа 0*, —РтЗл.
Z = 2.
V K.4.^4Si(S)( + 4 Si(S) = 8) (Di);
Si k.4. = 12V(/).
Расстояние d(V—JSi) = 2,637; e (Si—Si) = 4,099.
Элементарная ячейка, типа &W изображена на
рис. 173 и 174.
Рис. 441а.
На рис. 441а изображён кри-
оталлохимический
(минералогический) икосаэдр. Образуемый в этом.
как и в других случаях, икосаэдр
является неправильным в том
смысле, что в отличие от
«правильного» икосаэдра не имеет осей 5.
Это — 20-гранник, состоящий из
12 граней — равнобедренных
треугольников и 8
граней—равносторонних треугольников (t).
Вещество
V, Si
aw j 4,712 ±3
Координаты атомов
Si: (000) (i-J-i);
v<oii;0)(o4f;0)
Рис. 441b.
§ 194] КАРБИДЫ, СИЛИЦИДЫ. VI* PP.: ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ, УРАИ 583
(6-кратные). Атомы Si занимают позиции атомов WI. Атомы V — позиции
атомов W1I типа А15 (J3W), см. рис. 173 и 174, а также ридрид урана (рис. 243).
Атом V имеет к. ч. 4 атома Si в вершинах тетрагонального тетраэдра
( -f- ещё 4 атома Si в вершинах значительно большего тетрагонального
тетраэдра), что даёт в совокупности дитетраэдр(1>0- Атом Si имеет к. ч. 12 V(/).
Координационные сферы Si и V показаны на рис. 441а, соответственно 441Ь.
Общие замечания
Системы ванадий — углерод, ниобий — углерод, тантал — углерод,
а также смешанные карбиды: карбид тантала — карбид титана,
карбид тантала —карбид циркония, карбид ниобия —карбид титана,
карбид ниобия — карбид циркония, карбид вольфрама — карбид тантала —
карбид ниобия неоднократно исследовались советскими авторами. Здесь надо
назвать, в первую очередь, работы Я. С. Уманского, Уманского и Хиде-
келя, Уманского и А. Е. Ковальского (например, ЖФХ 15, 983 (1941),
14, 332 (1940); 15, 997 (1941), 20, 769, 773, 929 (1946), и др.), а также см.
монографию Я. С. Уманский, «Карбиды твёрдых сплавов», Металлургиздат
(1947) и Г. А. Меерсона и его сотрудников, например, Журнал прикладной
химии 12, 17Г)9 (1939).
§ 194. VI* группа периодической системы:
хром Сг, молибден Мо, вольфрам W, уран U
Карбиды Сг, Мо, и особенно W, приобрели за последние годы
исключительный практический интерес в связи с развитием производства твёрдых
сплавов и слецсталей.
1. Карбиды хрома. Как указано в SB I 573, при обычной температуре
известны три карбида:
^Сг4С кубический, <7Ш -= 10,6; Z ^ 24 молекулы;
~Cr7C:j тригональный, а = 14,0; с = 4,52; Z *& 8 молекул.
^-ОзС. ромбический, а -= 2,82; Ь =-- 5,52; с = ll/*e; Z^4 молекулы.
В последующих томах SB даны сведения о структурах карбидов
Сг7С3, СгаСо. О карбиде Оо3Св см. комплексные соединения, т. X.
2. Сг7С3 (часть Сг может быть замещена на Fe).
Решётка тригональная R.
Координация?
Тип Ю
Пространственная группа Сг^ — НЗс.
2 = 8; а = 13,98; с = 4,523.
3. Сг3С2
Решётка ромбическая цепная ORh.
Координация tjP'A.
Тип OR*ps — SB Db10.
Пространственная группа D^h — Pbnm (Prima),
Z - 4. Сг 111 к. ч. -"ЗС(0; С I к. ч. - 6 Сг (/>3).
1 Вещество
1 СгаСа |
а |
11,46 |
Ь
5,52
С
2,821
а : b : с
2,076:1:0,511 ■
i Примечание
| vSB III 53, 54 J
Решётка очень близка к 1ипу FoB [Bll). Каждый атом С находится
в центре тригональной призмы, образованной атомами Сг. Атомы С
образуют зигзагообразные цепи, dcr-c = 2,00 —2,07.
4. В системе Сг — Si описаны структуры соединений CrSi и CrSi,.
Ч. II. СТРУКТУРЫ Н|-:01'ГЛПМЧКСК11Х ШЛЦКОТН
||Л. VIII
Вещество
CrSi
4, 020
5. CrSi
Решётка кубическая С-
Координация 7/7.
Тин C7/7-SB #28-SB В20.
Пространственная группа
Z 4. Сг к. ч. ^ 7Si; Si к. ч. ~
-7Сг.
В решётке как бы намечаются группы CrSi. См. FeSi.
i>. CrSi2
Решётка гексагональная Н.
Координация 4 + 4/2 + 4.
Тип #4-h/2+4-SB C40.
Пространственная группа D\ — С6,2.
Z- 3. Сг к. ч. = 4 f'(4) Si; Si к. ч. = 2( + 4) Сг.
Примечание
ld = 2,32; 3<? = 2,44;
:i/ = 2,58.
Вещество
а
CrSi. 4,422
с
6,351
с/а
1,44
Примечание
SB III 35
Карбиды Mo hW известны двух
типов (но составу): МоС и Мо2С,
соответственно WC и W2C
7. МоС, WC
Решётка простая гексагональная Н
из атомов металла?
Координация Р'З/РЗ.
Тип Нрз/рз*
Пространственная группа ?
Z --1.
ОМо
Рис. 442. ® С
Вещестно
I Мое:
WG
а
2,001
4,88
2,910
2,94
с
2,7G8
f>,54
2,830
2,8fi
с/а
—
1,34
0,975
Состан
—
—
50 атомных
процентов С
■?
*л
—
—
15,67
15,52
1
Примечание
SB II 240 Mo в (OjO),
SB Ц 777
SB 11 239 J
Фазы этого типа изучены недостаточно, повидимому, из-за трудности
получения чистых препаратов. Па рис. 442 показано возможное строение
МоС (и WC).
В системах Мо —С и W - С обнаружены и другие карбиды, в том числе
(более надёжный) а Мо2С и а W2C, a также Н Мо2С и р WoC (?).
Мо2С и WjC приписывалась (Z. Anorg. Ch. 166, 27 (1926)) структура,
в которой атомы металла образуют плотную гексагональную упаковку,
а атомы С распределены в междуузлиях. Нтюжсн и тип анти-ГО.
i> НИ I КЛНЗИДМ, 1Л1. НПШДЫ. VI* П\: XI'OU, ЛЮ. IMIUE1I, НОЛЬФГЛМ, VI ЛИ 5ЯГ>
9ч
N. яМо2С. aWX
Решётка гексагональная
слоистая /Г .
Координация //С
Тип H*lf) (анти SB СО ?).
Пространственная группа
.Оз<,-Со>«.
Z - \.
М к. ч. -X (0;
С к. ч.-=0М ((У).
ОМо
Ос
Рис.
Вещество
яМо.,С
a\VX
< tf
2, *Ш
:*,004
2,9KG
2,99
с
4,725
4,725
4,712
4,71
с/а
1,579
1,57:*
1,578
1,575
Атомный
процент
С
:ш
ЗУ
:<4
«X
-
17,15
Примечание
SB 1
SB 1
SB II 240
По некоторым данным, ниже 2400°С aW2C устойчив, выше 2400 С
устойчив pWaC • pW2C даёт аналогичную рентгеновскую картину, что и
aW2C, но много линий отсутствует.
Те же элементы с Si образуют MoSi2 и WSi2.
r,m
Ч. II. Г/П'УКТУРЫ НЕОРГАНИЧЕСКИХ ВЫЦЕСТН
[гл. VIII
9. MoSi,, WSi,
Решётка тетрагональная Т.
+jt
Координация Л+ *]Pyk-
Тип Т *± -SB Cll (MoSi,).
h**IPy\
17
Пространственная группа Dni —
Л/пышп.
Z = 2. М к. ч. = 10 (Л'*"Ь;
Si к. ч. = 5(/у'|).
1 1
1 Вещистно 1 я
1 МоЙь
WSia
1 BemocTuo
3,20,
3,212
d
M -Si
MoSi, 2,620
WSi, 2,627
с
7,86t
7,88,
Si—Si
2,61.
э
2,62.
D
c/fl 1
2,457
2,453
OM
40.2
40,6
0
1
f 2 J
i i i
Рис. 444а
4
i
О мо
О Si
Координационная сфера атома Мо образована 10 атомами Si,
расположенными в вершинах двух взаимно-перпендикулярных шестиугольников,
имеющих две общие вершины (рис. 445а).
оМо
О Si
Риг. 445а.
Рис. 4451).
Центр этой фигуры и занимает атом Мо. Координационная сфера
атома Si образована пятью атомами j Мо, образующими тетрагональную
пирамиду (рис. 445Ы.
§ 194] КАРБИДЫ, СИЛИЦИДЫ. VI* ГР.: ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ, УРЛИ 587
В SB CaC. и MoSi2 причислены к одному и тому же типу. Однако
в MoSi2 параметры атомов Si таковы, что группы Si —Si как бы
растянулись. Каждый атом Si имеет уже не одного, но пять ближайших соседей
Si на одинаковом расстоянии.
В виде исключения приведём здесь данные по очень интересной
структуре, W2Zr, изученной сравнительно недавно.
10. W>Zr
Решётка кубическая С.
Координация 12/6.
Тип Ci2/6 - SB C39
(»SBC15).
Пространственная группа
07h-Fd3m (?).
Z.,8W,Zr.
W к. 4.-6Zr;
Zr к. 4.-12W.
Рис. 416.
о *
На рис. 446 положение Zr- точно. Положение W показано, как An
н типе 64.5 (рис. 425), что несколько неточно.
Атомы Zr образуют решётку алмаза Cr/т» Атомы W образуют сетку из
тетраэдров.
11. Система уран —углерод
Система уран — углерод освещена за последнее время в публикациях
Рэндла и его сотрудников (J. Am.Ch. Soc. 70, 99 (1948)) и Литца, Гарретт и
Крокстон (J.Arn.Gh. Soc. 70,1718 (1948)). В этой системе образуются 2
структуры: UC, тип Со/о с а —=4,951 кХ согласно первой работе, и а = 4,995 -
согласно второй, и UC2, тип Т^ SB 611 (карбида кальция), относящегося
в нашей систематике к ацетиленидам (см. стр. (588).
12# Системы уран —кремний, нептуний —кремний, плутоний —кремний
Системы U—Si, а также Np — Si, Pu Si представляют большой
теоретический интерес. Судя по работам Захариасена *) (1948 — 1949), в этих
системах обнаруживается ряд фаз, например USi2, USi, U3Si2, U3Si.
Если в соединениях типа МС2 углерод проявляет тенденцию к
образованию замкнутой гантельной группы —самостоятельного структурного узла
С2, то в аналогичном по составу силициде USi2 группы Si2 отсутствуют —
атомы кремния образуют пространственную вязь, как в aUSi2, или сетки,
как в pUSi2. Атомы U в этих структурах занимают междуузлия.
Соединения типа USi особенно любопытны. И отличие от карбидов стехиометри-
•) Acta Crist. I, 208 (Ш8) и 2, 9i (19'49).
588 ч. и. структуры нкорглнпчкскпх ш:щ£хтв ]гл. VI1F
ческого состава MX, образующих структуры типа каменной соли Со/о,
аналогичные силициды названных элементов образуют структуры, в
которых атомы кремния связаны друг с другом в цепи. Лишь у соединения U3Si.2
появляется гантель Si2, причём для ликвидации явной тенденции атомов
кремния образовывать цепи с себе подобными потребовалось соотношение:
3 структурных узла урана на один структурный узел Si2. Наконец, у U3Si
атомы кремния теряют способность соединяться с себе подобными. Их
координационная сфера образована 12 атомами урана.
К сожалению, физические свойства этих силицидов остались вне
сферы внимания Захариасена. Несомненно, что эти новые типы соединений,
являются ценным материалом для общей теории структур.
15, U,Si
Решётка тетрагональная центрированная 7\
Координация 4/12.
Тип Г4/12.
Пространственная группа Вц, — Л/тст.
Z = 4.
Вещество
UaSi
1 l-i 1
а 1 С \ (кХЗ) 1 °Х 1
6,017 ± 2
8,Г>79 4:3 1 .Ш
15,58 I
В структуре имеется 2 сорта атомов урана: UI к. ч.---4 (квадрат)
на расстоянии 3,01; UII к. ч. - 4 (из них 2 на расстоянии 2,92 и 2 на
расстоянии 3,17); Si к. ч. 12 (из них 4UI на расстоянии 3,01, 4UII
на расстоянии 2,92и4Ш1 на расстоянии 3,17).
Описанная Захариасеном структура характеризуется структурными
дробями: Ul({+ [-) -3 + UII (* + *.} ==8, а всего 11. Si(£) = 4,
т. е. формулой UnSi4.
ТакихМ образом, новидимому, U8Si —это структура вычитания с
недостатком атомов кремния (?). Впрочем, этот вопрос, представляющий
самостоятельный интерес, Захариасеном не освещен, равно как и осталась
невыясненной область устойчивости по составу как этой, так и других фаз
в этих системах.
§ 195, VII* группа периодической системы: марганец Мп, рений Re
1. Система Мп—С.
Карбиды Мп изучены скудно. Сначала были описаны карбиды Мп3С
и Mn4C(SBIl). Позже в этой системе были описаны карбиды Мп23Св
и Мп7С3; в последнем часть Мп может быть замещена на Fe. См. также С. А.
39, 3198 (194Г>) и нолисоединения.
2. Мп7С3 (часть Мп может быть замещена на Fe).
Решётка тригональиая R.
Координация — ?
Тип Я.
Пространственная группа б1^ — НЗс (?).
а : 13,87: с-4.53.
§ 1951
КАРБИДЫ, СИЛИЦИДЫ. VII* ГРУППА: МАРГЛПМЦ. РКНПИ
589
3. 1> системе Мп- Si, согласно SB 1U 028, существуют фазы:
а— |Шп + некоторая концентрации растворённого Si;
p~Mn3Si, класс I)bh — Ummm\ Z 4; а --(5,898; г--4,802; с/а 0,090;
зг (i/ii;
з-MnSi, тип SB #28;
у- MnSi.,—пегомогенна, тетрагональная; а Г>?51;>; с^17/»22; с/а 3,10.
Согласно SB J]] 029, вместо Mn3Si существует фаза Mn:>Si3, для
которой константы решётки, симметрии if интенсивности точно равны тем,
которые Ботен нашёл для фазы, обозначенной им как Mn3Si.
Напротив, фаза с составом Mn3Si оказалась кубической, тип Сс SB .42;
Z --■■■- 2; а = 2,85, со статистическим распределением атомов Si и Мп.
Предположено, что подобная фаза возникла вследствие присутствия
загрязнений.
Позже, см. SB IV 2i, '1.Т7, была расшифрована структура фазы Mn.Sis
и подтверждены данные SB 111029. Следовательно, р-фаза "имеет соетанМп5Йц.
4. Mn3Si
Решётка кубическая С,
Координация С.
Тип Сс —SB Л2
Пространственная группа Од — Jт-ш.
Z = 2.
К. ч. = 8 (куб).
Ги<\ кМ.
Лещегтмо
Mii4Si
i ~\<>,
IS,01
В решётке имеет место статистические распределение атомов марганца
и кремния. Интересно сопоставить :>ту структуру с рассмотренной выше
структурой V3Si. Занятие холостыми электронами Мп всех ^-уровнен
вызвало при таком же стсхиометрпчееком составе образование типично
металлической решётки. Повидимому, характер связи в MiySi является
значительно более металлическим.
590 ч. п. структуры неорганических веществ [гл. VIII
5. Mn5SI,
Решётка гексагональная Н.
Координация 0,5/4.
Тип #of5/4-SB Д8в (Mn5Si3).
Пространственная группа Dlb — Cbjmcm.
Z^2MneSi3, Mnl к. 4. = ()Si (деформированный октаэдр); МиII к. ч,- 5 Si;
Si к. ч. ^4 Mnl.
Вешэство
а
Mn5Si., 0,898
с
cja
4,802 0,096
Примечание
SB IV 24, 137
Мм I окружён 6 Si-атомами, в вершинах искажённого октаэдра.
Ми II атомы образуют октаэдрические группы, не имеющие внутри цен-
грплыюго атома.
§ 196] КАРБИДЫ, СИЛИЦИДЫ. VIIIе ГРУППА: ЖЕЛЕЗО, РУТЕНПН/ОСМПЙ
591
0. MnSi
Решётка кубическая С.
Координация 7/7.
Тип C^-SB 528-SB #20 идентичны.
Пространственная группа Т4 — Р2Х 3.
Z = 4.
Мл к. ч. 7; Si к. ч. ---.- 7.
Вещество
MnSi
а7(,
4,548
Примечание
Ы = 2,30; :*e = 2,W; .4/^2,5:,; SB 11 14 (см. FeSi)
7. Система Re —С. Данных о
структурах карбидов Re нам найти не
удалось.
8. Система Re—Si. Силицид Re: ReSi2,
согласно одной из работ, имеет
структуру MoSi2, тип Tujpyi SB СИ, рис «ч»;
см. также рис. 445.
Рис. 449
§496. VIII* группа периодической системы:
железо*Ге, рутений Ни, осмий Os
1. Система Fe—С. Карбиды железа представляют исключительный
практический интерес, являясь основой чёрной металлургии.
В системе Fe —С от 0°/0 Д^ 6,67°/0 С существует одна дистектическая
точка — соединение Fe3C —цементит (G,G7°/0C.)
Кроме того, исследована неустойчивая при комнатной температуре
фаза аустенит «у -твёрдый раствор» — раствор углерода в у-железс.
Аустенит имеет кубическую гранецентрированную решётку, а1Р которой
растёт с концентрацией С, доходя до 3,03 А при 8 атомных процентах С
(SB I 578).
Структура цементита изучена детально.
51)2
Ч. II. CTJ-.VKT.VPLI НКШ'ГЛШНШСКПХ HHHUXTH
[гл. VIII
2, Pc3C (цементит)
Решётка ромбическая OR,
Координации 2/РЗ.
Тин ОД./,* SB Z)()n.
Пространственная группа
DX2h~Pbnm(Pnma),
Fe к. ч. 2С;
С к. ч. () Fe.
Fe С - 2,0
(соответственно 1,85 кХ).
Вещество
д . . . .
\Ъ
\ с . .
\ а : Ь : с . .
Fe3G
4.517-4,513
5,07у-;>,048
r>'7V"r>,74
0.8УО : 1 :1,325
О Fe
Рис. 450
50. ОС
• С - 8 призмах
О С~ £ октаэдрах
Гпс. 451. Лтомы Fo в вершинах призм.
§ 196] КАГБВДЫ, СИЛИЦИДЫ. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ 593
Опубликованы два описания структуры:
а) с 4G в позициях (000), (оо|), (4т°)> (туу)"
Этому описанию отвечает рис. 450 со слегка деформированными окта-
эдрическими координационными сферами [GFee] (ось с элементарной ячейки
направлена вверх);
б) с 4С в позициях ± (*У-%) > (jf — ** Y + ^t) с я = °>43; У = 0,13.
Этому описанию отвечает рис. 451 (SB II 33) с координационными
сферами [GFe6] в виде тригональных призм с атомом С в центре (чёрные
кружки на рис. 451 показаны только в передних призмах) Fe — С = 2,0 кХ.
На том же рисунке белыми кружками показана часть позиций в
элементарной ячейке при выборе октаэдрической координации (структура «а»).
Сравни с рис. 450. Последние работы подтверждают призматическую
конфигурацию [GFe6] (структура «б»).
Так же как Fe замещает Мп в Мп3С, в свою очередь, Мп замещает
Fe в значительных количествах в Fe„C. С. А. 39, 3198 (1945).
3. Система Fe —Si. В системе Fe—Si (см. SB II 760) выделены
соединения FeSi2 (г-фаза—кубическая) и FeSi2 (S-фаза, тетрагональная,
пространственная группа D\h ?).
Помимо того (согласно SB III 624):
а-фаза: aFe растворяет до 30 атомных процентов Si; aw падает при этом
от 2,867 (Fe) до 2,81,.
у-фаза: область yFe с добавкой Si сильно сужается.
tt-фаза: Fe2Si или Fe3Si2. Сопутствует а-фазе. Структура неизвестна.
ii-фаза параметры: а = 2,687; с = 5,127 (в равновесии с е-фазой); а =
= 2,67,; с = 5,12 (в равновесии с ?)-фазой).
у\-фаза: Si —алмазная решётка. Не растворяет Fe.
Атомы Fe —в вершинах призм.
Несомненно, дальнейшие исследования внесут изменения в эту картину.
В частности, найдено (Nature 152, 413 (1943)), что соединение Fe6Si,
изоморфно Mn5Si3 (решётка гексагональная, тип Я с, 5/4 — SBZ)88).
4. FeSi
Решётка кубическая С.
Координация 7/7.
Тин C7/7-SB 528 (SB 111) идентичен с SB B20 (SBII), ошибочно принят
и SB как новый тип.
Пространственная группа Г4—jP2j3.
Z =4.
Fe к. ч. = 7 Si; Si к. ч. = 7Fe.
Вещзстно
FeSi
aw
4,438
d 1
Fo-Si
2,28—2,50
Координационная решётка с к. ч. = 7. Расположение атомов, как
Na и С1, в решётке NaC10„ (тип £Ю3) см. рис. 677.
В решётке как бы выделяются группы FeSi благодаря кратчайшим
расстояниям Ы - 2,28 (3d' = 2,35; 3d" = 2,50).
5. Карбиды и силициды Ru и Os
Сведениями не располагаем.
594 ч. и. структуры неорганических веществ [гл. VIII
§ 197. IX* группа периодической системы:
кобальт Со, родий Rh, иридий 1г
Система Со—С. В системе Со —С (как и в системе Fe — С) до сего
времени описывался лишь один карбид Со3С, подобный цементиту (Fe8C).
Однако недавно появились указания на существование ещё одного
карбида CoeC (J. Am. Ch. S. 69, 893 (1947)). Эти данные заслуживают проверки.
1. Со8С
Решётка ромбическая OR.
Координация 2/РЗ.
Тип OR2/pz-SBD011.
Пространственная группа L^l — Pbnm i = Pnma).
Z = 4.
Рис. 452.
+ C-tf призмах
о C-J октаэдрах
Со-в вершинах
призм
Вещество
Со.С
а
4,52
Ь
5,08
с
6,73
Примечание
Область образования—550е SB V129.
SB VI 178
См. Fe8G.
2. Система Со—Si.
Со (а и р) растворяет Si со сжатием решётки (SB III 626). Помимо
других возникающих при этом фаз, известны и изучены &- и е-фазы.
§198} КАРБИДЫ, СИЛИЦИДЫ. X» ГРУППА: НИКЕЛЬ, ПАЛЛАДИЙ. ПЛАТИНА 595
3. CoaSi (fl-фаза), переменный состав
Решётка ромбическая цепная 0RK.
Координация TJT + 7.
Тип 0Д3*г+7 - SB C37.
Пространственная группа DSt — Pbnw.
Z = 4.
Со к. ч. = 4Si (T)\ Si к. ч. = 5CoI-f 6CoII (Г+ 7).
Вещество
а
Co^Si 7,005
b
4,908
с
3,730*)
а: Ъ : с
1,446: 1:0,765
Примечание. Для сплава с 12 весовыми процентами Si эта фаза,
повидимому, стабильна при t° < 1000' С.
Как и в типе 515 (FeB) и Z>57 (Сг8С2), Si-атомы образуют
зигзагообразные цепи параллельно (001). В отличие от этих типов, в типе С37
ближайшие координационные сферы атомов X (здесь Si) имеют вид не триго-
нальной призмы (к. ч. = 6),но искажённых тетраэдров. См. SB III32, 33, 626.
4. CoSi («е-фаза»)
Решётка кубическая С.
Координация 7/7.
Тип C7/7"SB 528 = SB 520.
Пространственная группа ТА — Р213.
Z = 4.
Со к. ч. - 7Si; Si к. ч. = 7Со.
Вещество
CoSi
аи Примечание
4,438
1<*«2,28; Зе=2,33; 3/ = 2,47
См. FeSi; (§ 196) SB III 626
Б. Карбиды и силициды Rh и 1г
Сведениями о структурах карбидов и силицидов Rh и 1г не
располагаем.
§ 198. X* группа периодической системы:
никель Ni, палладий Pd, платина Pt
1. Система Ni —С
В системе Ni—С подвергалась исследованию структура одной лишь
карбидной фазы —соединения Ni3C.
По этим данным, NiaC имеет гексагональную решётку с а = 2,646;
с =* 4,329; с/а = 1,630. Более поздние работы утверждают, что NiaC
изоморфен Fe8C (см. SB VI 170), что, повидимому, правильно.
•) SB III 33. По SB III 626, а=»3,730; £ = 4,908; с= 7,095.
596 ч. и. структуры неорганических веществ [гд. VIII
a. Ni,c*)
Решётка ромбическая OR.
Координация 2/РЗ.
Тип OR2tP?. — SB D0lx (цементит).
Пространственная группа Дгл — РЬпт (=Рпта).
о С-6 октаэдрах
Hi-в вершинах
призм
Рис. 453.
Хотя в системе никель — углерод до сего времени описывался лишь
один карбид NitC, однако в работе (J. Am. Ch. S. 69, 893(1947)) имеется
предположение о возможности существования ещё одной карбидной фазы,
аналогичной Со2С. Это предположение пока следует считать
неподтверждённым (см. также § 122, никель).
3. Система Ni — Si
В системе Ni— Si, согласно SB IV 247, имеется б соединении:
Ni8Si, Ni5Si2, NiaSi, Ni3Si2, NiSi, NiSi2.
Из них подчёркнутые плавятся без разложения. Фаза NiSi, имеет
2 аллотропические формы с tu — 981°С (?).
*) См. текст к FesC (§ 1%).
§ 1991 КАРБИДЫ, СИЛИЦИДЫ. !• ГРУППА: МЕДЬ, СЕРЕБРО, ЗОЛОТО 597
4. NiSi («е-фаза»)
Решётка кубическая С.
Координация 7/7.
Тип C^-SB 528 = SB 520.
Пространственная группа Тх - Р2Х3.
Z = i.
I Веществ о
NiSi
aw
4,437 (?)
Примечание
См. F:eSi. См. SB III 627—628
5. Карбиды и силициды Р<1 и Ft
Надёжными сведениями о структурах карбидов и силицидов Pd и Pt
мы не располагаем.
§ 199. I* группа периодической системы:
Карбиды Си, Ag, Au, медь Си4, серебро Ag, золото Аи
1. Карбиды Си, Ag, Аи
Сведениями о структурах карбидных фаз Си, Ag и Аи не располагаем.
2. Силициды Си, Ag, Аи
Сведениями о структуре силицидных фаз Ag и Аи не располагаем.
3. Система Си —Si
В системе Си — Si (SB II 757, 758) изучены р'-фаза Cu6Si, тип А13
(Р Мп), а = 6,210; Z = 20 атомов. (Электронная концентрация —3:2, как
и в Ag3Al. Гомогенна при 17 атомных процентах Si), у-фаза Cu15Si4 или
Cu3lSi8 и е-фаза Cu3Si кубическая гранецентрированная, а = 9,694; Z = 76.
4. Cu.Si
Решётка кубическая С.
Координация K/J.
Тип Ски-SB A13 (рМп).
В позиции «Мп I» к. ч. = 12 (кубооктаэдр);
В позиции «Мп II» к. ч. =з 12 (икосаэдр)*).
Вещество
Cu4Si
а«
6,211
Примечание 1
Статистическое распределение Си- и Si-ато- |
мов в положениях атомов Мп в структуре 1
(4 Мп см. SB 111 332 J
*) См. рис. 441а, чертёж икосаэдра, а также стр. 236, 241, 291, 302,
598 ч. п. структуры неорганических веществ [гл. УНГ
б. Cu16Si4
Решётка кубическая С.
Координация 1/12=1/4/.
Тип Cm-SB jD84.
Пространственная группа IT'S—/43d.
Z = 4.
Si к. ч. = 12 Си (4 треугольника = it); Си I к. ч. = 1 Si.
Рис. 454.
Часть атомов, ради наглядности, на рис. не показана.
Вещество
Cu15Si4
aw
9,694
Примечание
Si имеет 3 Си II на расстоянии 2,38
3 Си II » » 2,48
3 Си II » » 2,62
3 Си I » » 2,62
Примечание. Атомы Си образуют равносторонние
треугольники, перпендикулярные осям 3. Атомы Si лежат на осях 3, в центрах
отдельных треугольников, образованных Cul, 3d (Si —Си) = 2,38, имея
вблизи себя по 4 треугольника.
§201]
КАРЛИДЫ, СИЛИЦИДЫ. Ill ГРУППА: БОР - ТАЛЛИЙ
599
§ 200. II* группа периодической системы:
цинк Zn, кадмий Cd, ртуть Hg
Надёжных данных о структуре
карбидных и силицидных фаз,
образованных Zn, Cd, Hg, в нашем
распоряжении нет.
§ 201. III группа периодической системы:
бор В, алюминий А1, галий Ga, индий In,
таллий Т1
Надёжных сведений о структуре
карбидных и силицидных фаз Ga, In,
Т1 и силицидных фаз В и А1 в нашем
распоряжении нет.
Значительно лучше обстоит дело
с исследованием структуры одного из
важнейших, с точки зрения техники,
карбидов—карбида бора, а также
карбида алюминия.
1. Карбид бора B4C^(Bl2) (C3), см.
комплексные соединения.
2. А14С3
Решётка ромбоэдрическая слоистая Rs.
Координация TfO,b.
Тип /^/5>5-SB Dlt (A14C3).
Пространственная группа Вза — ЯЗт.
Z = 1. А1 к. ч. = 4С (деформированный
тетраэдр);
С I к. ч. = 6 А1 (деформированный
октаэдр);
С II к. ч. = 5 А1.
Рис; А 55
Вещество
А1А
а*)
8,53
а
°х
1
22э28' | 2,99
Примечание
Резко выраженная слоистая решётка.
Атомы С образуют плотнейшую шаровую
упаковку. Атомы А1 занимают часть тетра-
эдрических промежутков, SB III 56
3. Система AI —Si. Имеется небольшая и недостаточная аннотация Ch.
Zentralblatt II 99 (1944).
*) Или а «3,325; с =.24,94; с'а = 7,500; z»3.
600
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩКСТВ
[гл. VIII
§ 202. IV группа периодической системы:
углерод С, кремний Si, германий Ge, олово Sn, свинец РЬ
Сведений о структуре карбидов Ge, Sn, Pb в нашем распоряжении нет.
Углерод и кремний образуют группу карбидов кремния SiC.
Карбид кремния имеет в основе плотную шаровую упаковку. Как
мы знаем (§ 64), в зависимости от того, в одну и ту же или в разные
стороны повёрнуты тетраэдры 1 —2-го и 2 —3-го рядов, возникают
гексагональная или кубическая упаковки.
В зависимости от закона, которому подчиняется ориентировка
тетраэдров разных слоев в SB, зарегистрированы 4 формы SiC: IV
(кубическая—тетраэдры параллельны р, р, />...)> III (гексагональная—второй
слой тетраэдров параллелен, третий—антипараллелен первому ра, ра, ра ...),
II (гексагональная рар, рар, рар...), I (гексагональная рарар, рарар,
рарар...).
По Н. В. Белову вместо обозначений р и а, указывающих
направления поворотов тетраэдров, применяются и и г (кубическая и гексагональная).
Г. С. Жданов применяет числовые символы, характеризующие количество
слоев упаковки по тому или иному закону, например, 2-2 или 3-3.
Подробнее смотри ДАН XXVIII, ^0 (1945). Ниже при характеристике типа
мы применяем в верхних индексах числовой символ по Жданову.
Сведения о строении этих форм отражены в SB следующими данными:
1. SiC IV («аморфный»)
Решётка кубическая С.
Координация тетраэдрическая Т/Т.
Тип Стуг-SB ВЗ.
Пространственная группа Т\ — F43/W.
Z-4.
Si к. ч.=4С (тетраэдр);
С к. 4. = 4Si (тетраэдр).
Рис. 456.
I Вещество | а„ | d | ОМ | Примечаьнэ 1
SiC
4,37 1 1,89
20,9
См. § 63
§ 202] КЛ) ВИДЫ, СИЛИЦИДЫ JIV ГРУППА: УГЛЕРОД-СВИНЕЦ 601
2. SiC III
Решётка гексагональная Н*'г.
Координация тетраэдрическая Т/Т.
Тип Яг/г-SB Bb.
Пространственная^ группа C\v — C бтс.
Z = 4.
Si к. ч.=4*С (тетраэдр);
С к. 4.=4Si (тетраэдр).
Рис. 457.
Вещество
SiC III
а
3,095
С
10,09
с/а
3,260
d
ОМ
1,89 20,9
Примечание
См. § 64
602 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ (ГЛ. VIII
3. SiC II
Решётка гексагональная Н?,г.
Координация тетраэдрическая Т/Т.
Тип #2$-SB В6.
Пространственная группа Cq»— CQ?nc.
Z-6.
Si к. ч.=4С (тетраэдр);
С к. 4. = 4Si (тетраэдр).
Рлс. 458.
1 Вещество
SiC II
1
а
з,оэ5
3,076
с
15,17
15,07
с/а | d
4,902
4,899
1,80?
ОМ
20,9х3/а
°*
3,212
Примечание
См. § 64
§ 202] КАРБИДЫ, СИЛИЦИДЫ. IV ГРУППА: УГЛЕРОД - СВИНЕЦ
603
4. SiC I
Решётка (гексагональная)
ромбоэдрическая Л2*3.
Координация тетраэдрическая TJT.
Тип /ft/x-SB Я7.
Пространственная группа Cl»—R Зт.
Z- 15.
Si к. ч.=4С (тетраэдр);
С к. ч. = 4 Si (тетраэдр).
Вещество
SiC I
а | с
3,0У5 37,95
с/а
12,25
Примечание
См. § 64
В 1943 г. исследование структуры SiC, в зависимости от условий обра*
зования последнего, было поставлено в Институте им. Карпова в нашей
лаборатории и, по нашему предложению, Г. С. Ждановым в рентгеновской
лаборатории того же Института.
604 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VIII
Нами предполагалось, что влияние на последовательность поворота
тетраэдров помимо температуры и давления могут оказывать примеси.
Это наше предположение было подтверждено после того, как в нашей
лаборатории были специально синтезированы образцы кристаллов карбида
SiC (Б. Ф. Ормонт, В. А. Эпельбаум, И. Г. Шафран и др.) с примесью бора
и без последней и отобраны образцы кристаллов SiC, полученных нами
и И. Г. Шафраном.
Исследования этих образцов в лаборатории Г. С. Жданова показали,
что тогда как в высокотемпературной зоне в отсутствии В возникает,
главным образом, SiC II, в присутствии В образуется SiC III.
В 1945 г. Г. С. Жданов и 3. В. Минервина (Journ. of Physics IX, 244
(1945)) расшифровали по порошковой диаграмме, опубликованной в работе
Отта (Sommert'eld Festschrift 208o (1928)), структуру единственного
кристалла SiC с длиной оси с = 129 А, что отвечает упаковке 51 слоя атомов.
Подобную структуру авторы приписали карборунду SiC V. При расшифровке
в основу был положен учёт числового символа плотной шаровой упаковки
по Г. С. Жданову (ДАН XXVIII, 40 (1945)).
Эти же авторы дали характеристику форм карборунда в терминах
числового символа: SiC III (2.2), SiC II (3.3), SiC I (2.3), SiC V (233333),
характеризующего толщину слоев.
В 1944 г. появились ещё 3 работы (Amer. Mineral. 29, 249, 327, 431
(1944)), посвященные структуре SiC. В первых двух из них указывается,
что существуют 2 модификации SiC: a — гексагональная и [3 — кубическая.
Гексагональная модификация встречается в виде следующих форм,
отличающихся при одинаковом значении а (а =3,073) разными значениями с.
I. с = 37,70; пространственная группа Clv—R3m.
II. c = 15,07g; пространственная группа С^ — Сбтс.
III. с = 15,079; пространственная группа С\и — Сбтс.
IV. с = 52,78; пространственная группа C\v — R3m.
V. Новая модификация, расшифрованная Г. С. Ждановым, не
найдена с = ?
VI. с = 82,94; пространственная группа C'zv — R3m.
В третьей работе также подчёркивается, что указанные значения с
отвечают количеству расстояний между слоями: 1 = 15; II — 6; 111 = 4;
IV = 21;VI=33 (d = 2,5lA).
pSiG отвечает аш = 4,349; Z = 4. Пространственная группа Т\ — F43m.
5. Система Ge— Si была изучена в 1939 г. Z. Anorg. Ch. 241, 305 (1939).
§ 203. V группа периодической системы: азот N, фосфор Р, мышьяк As,
сурьма Sb, висмут Bi
См. нитриды, фосфиды и т. п. элементов IV группы.
§ 204. VI группа периодической системы: кислород 0, сера S,
селен Se, теллур Те, полоний Ро
См. окислы, сульфиды и т. д. элементов IV группы. Сведений о
карбидах и силицидах Ро в нашем распоряжении нет.
§ 206. VII группа периодической системы: фтор Р, хлор С1,
бром Вг, иод J
См. галогениды элементов IV группы.
Общая литература:
Я. С. У м а н с к и й, Карбиды твёрдых сплавов (1947); SB I—VIk
§ 212] БОРИДЫ. VI* ГРУППА: ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ 605
Б. БОРИДЫ
Бор, вступая во взаимодействие с другими элементами, часто образует
соединения, в которых атомы В сохраняют связи между собой. В
результате возникают комплексные соединения—полибориды типа СаВ, или В4С
(правильнее В12С9) и другие. Они рассматриваются нами в разделе Шоли-
соединения» в главе X.
§ 206. I группа периодической системы: литий Li, натрий Na,
калий К, рубидий Rb, цезий Cs
Сведениями о структуре простых боридов 1 группы не располагаем.
§ 207. II группа периодической системы: бериллий Be, магний Mg,
кальций la, стронций Sr, барий Ба, радий На
Сведениями о структурах простых боридов Be и Mg не
располагаем.
Бориды Са, Sr и Ва типа МВв, см. комплексные соединения гл. X.
§ 208. III* группа периодической системы: скандий Sc, иттрий Y,
лантан La
Бориды Y, La типа МВв см. комплексные соединения гл. X.
§ 209. III** группа периодической системы
См. комплексные соединения гл. X.
§ 210. IV* группа периодической системы: титан Ti, цирконий Zr,
гафний Ш, торий Th
В системе Ti — В недавно описаны (Z. Anorg. Ch. 259, 1 (1949)) две
фазы: TiB -тип С?/7-SB ВЗ и TiB2-Tmi #P6/P3-SB С32 (А1В2) с
широкими границами существования (но составу). Системы Zr —В, Hf —В,
Th — В, к сожалению, изучены очень плохо. По некоторым данным,
существует борид циркония типа ZrB2 (?). Состав: Zr = 78,55%; В -=18,15%;
С =1,89%; Si = 0,03%; спектроскопические следы Fe. Ячейка
гексагональная; а =3,15; с = 3,53; с/а = 1,12.
Hf, повидимому, образует HfB2.
Th образует карбид ThB6, см. комплексные соединения гл. X,
§ 211. V* группа периодической системы: ванадий V, ниобий Nb,
тантал Та
Надёжными данными о структуре боридов V, Nb, Та мы не
располагаем.
§ 212. VI* группа периодической системы: хром Сг, молибден Мо,
вольфрам W, уран U
В литературе описаны 2 борида хрома СгВ и Сг8Ва.
Надёжными сведениями о структурах боридов Сг и его гомологов мы
не располагаем.
606 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. VIH
§ 213. УН* группа периодической системы:
марганец Мп, рений Be
Описана структура борида Мп.
1. МпВ
Решётка ромбическая OR.
Координация — ?
Тип ОД-SB?
Пространственная группа Dtn — Prina (?).
Z%8.
Вещество
МпВ
а
2,95
Ь
U,5
с
4,10
d
кратч.
Мп—В
2,1
кратч.
Мп—Мп
2,3
а :Ь : с 1
0,256:1:0,356 1
§ 214. VIII* группа периодической системы:
железо Fe, рутений Ru, осмий Os
В системе Fe —В (SB II 795) описаны соединения FeaB ж FeB.
Структуры этих веществ в SB охарактеризованы неоднозначно.
1. FeB
Решётка ромбическая OR,
Координация TJT.
Тип ORTjTSB £15 (FeB).
Пространственная группа ВЦ — РЬпт.
Z = 4 FeB.
Вещество
FeB
а
4,053
Ь
5,495
С
2,946
а : Ь : с
0,738: 1 : 0,536
Примечание 1
См. SB II 7
В SB II 795 даны также другие размеры ячейки: а = 5,495; Ь = 1,053;
с = 2,946; а:Ь:с= 1,36:1:0,536.
В SB III FeB присвоен уже тип SB Z?27, см. ниже (тип SB J515
исключается ? ? —Б. О.).
Согласно SB II (тии £15), каждый атом Fe окружён сильно
деформированным тетраэдром из атомов В (Id = 1,68; Id' = 1,69; 2d" = 1,70).
Каждый атом В на тех же расстояниях имеет атомы Fe, образующие
почти правильный тетраэдр.
§■ 214]
БОРИДЫ. VIII* ГРУППА: ЖЕЛЕЗО, РУТЕНИЙ, ОСМИЙ
607
2. ГеВ
Решётка ромбическая, цепная ORK.
Координация 2/РЗ.
Тип ОД^рз-SB 527 (FeB).
Пространственная группа Дал — РЬпт( — Рпта).
2 = 4.
Fe к. ч. = 2В; В к. ч. = 6Fe(/>3).
Рис. 460. (Две ячейки.)
1 Вещество
FeB
а
4,053
Ь
5,495
с
а : Ь : с
2,046 0,736:1:0,536
Согласно SB III 13 (тип 527) атомы В образуют зигзагообразные
цепи. Каждый атом В лежит в центре тригональной призмы, вершины
которой заняты атомами Fe; d(Fe — В) = 2,15 и 2,18.
На рис. 460 в правой нижней части хорошо видна выступающая
из элементарной ячейки одна из боковых граней лежащей «на боку» призмы
и обращенная к читателю верхняя треугольная грань последней.
Описан также
3. FeB (??)
Решётка тетрагональная Т'.
Координация ?
Тип T-SB ?
Z^16 FeB.
Вещество
FeB
а
7'38±5
с
5'98±5
Примечание 1
SB II 797 1
608 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. VIII
4. Fe2B
Решётка тетрагональная Т, центрированная.
Координация Т/А.
Тип Г---SB C16 (CuAl2).
Пространственная группа Z)i/J — /4/mc/n,
Z-4.
FeK. ч. = 4 В (Г);
В к. ч. = 8 Fe (A).
Рис. 461.
Вещество
Fe.B
а
5,099
е
4,240
с/а
0,832
d
2,12
•
См. SB II 286. SB III 22, 619, см. также Косолапов и Севастьянов,
ЖТФ 11, 607 (1941). См. Ni2B.
(В SB I 493 Fe2B приписана структура, тип SB С17;
пространственная группа РУ=ДЙ — / 42т; а = 5,078; с = 4,22; с/а = 0,832.
BI тетраэдрически окружён 4Fe. ВII сфеноэдрически окружён 4Fe.
Fe I занимает вершину разностороннего треугольника, во 2-й вершине
атом В I, в 3-й В II. Решётка состоит из цепей, составленных из
тетраэдров BFe4, чередующихся со сфеноэдрами BFe4 (каждый из них
имеет два общих ребра с соседями) и параллельных оси С.
В SB I 573 указана та же ячейка, но тип SB C16 (рис. 461), SB I491.
SB II 285 принимает структуру, найденную в работе (Z. ph. Ch. (В)
12, 413 (1931)). SB II 796 и SB III 22, 619 дают ячейки для изоморфных
Fe2B, Со2В и №2В.
§ 216]
БОРПДЫ. X ГРУППА: НИКЕЛЬ, ПАЛЛАДИЙ. ПЛАТИНА
609
§ 215. IX группа
периодической
системы: кобальт Со,
родий Rh, иридий 1г
1. Со2В
Решётка
тетрагональная центрированная
Т.
Координация 77 Л.
Тип Гщ-БВСМб
(анти СиА]2).
Пространственная
группа D)^ Л/тст.
Z = 4
Со к. ч. --■= 4 В
(искажённый тетраэдр);
В к. ч. = 8 Со
(искажённая атттипризма)
9^гж~у^9
Рис. 462.
О Со
О8
1 Вещее т no i a | с
СоД* Г>,01)0 4/212
i
(i
2,06
Примечание 1
SB III 22, 619. См. Кь В
Сислемы Со —В, Rh В, 1г -В| в структурном отношении изучены
слабо.
§ 216. X группа по- -f~V ^Л
риодической системы ' ч-х*^ г\ -^^
ииксль Ni,
палладий Гй, платина Vt.
1. Ni2B
Решётка
тетрагональная, центрированная
Г.
Ко(»рдинация Т/А.
Тип аг7/А—sb сю
(анти СиА1а).
Пространственная
группа D** — Л[гпст.
Z = i.
Ni к. 4.-4BJ7);
В к. ч. -8Ni(i).
Всще-
ствр
Ni2B
а | 'с
1 ■ '
4,983
4,980
4,240
4,236
с/а
0,8509
0,85
Pnc. 4GU.
.еж
Примеча ние. Слои атомов 13 окружены слоями атомоп N'.'« ' '"'
GiCf ч. и. структуры неорганических веществ [гл. VHI
Координационная сфера В близка к ашииризме (закрученный куб)
(отличие от типа анти-СаР2). Координационная сфера Ni близка к тетраэдру.
SB III 22; 619. См. также Косолапов и Севастьянов, ЖТФ 11, 607 (1941).
Системы Ni—В, Pd —В, Pt — В в структурном отношении изучены
слабо.
В системе Ni —В в данной концентрационной области [имеются
промежуточные фазы (?).
§ 217. I* группа периодической системы:
медь Си, серебро Ag, золото Аи
Системы Си —В, Ag — В, Аи —В в структурном отношении изучены
слабо.
§ 218. II* группа периодической системы:
цинк Zn, кадмий Cd, ртуть Hg
Сведениями о структурах не располагаем.
§ 219. III группа периодической системы:
бор В, алюминий AI, галий Ga, индий In, таллий Т1
В пашем распоряжении отсутствуют данные о строении боридов da,
In, Tl.
В системе А1— В изучено два борида: А1В12 и А1В2. Хотя структура
А1В12 надёжно не известна, учитывая состав вещества и то, что
большинство боридов но своей структуре относится к разделу сложнокомплекг-
ных и комплексных соединений, А1В,2 будет рассмотрен в
соответствующей главе.
1. А1В2
Решётка гексагональная Я.
Координация Р6/РЗ.
Тип Ярвурз -SB C32.
Пространственная группа Dlh — CG/mmm.
Z = l.
А1 к. ч. = 12 В(Р6); В к. ч. = 6 А1(РЗ).
Вещество
А1В4
а
3,00
с
3,24
с/а
1,08
Примечание
А1 : (000)
12 атомов В расположены в вершинах гексагональной призмы, образуя
координационную сферу А1; А1 — В = 2,37. Координационная сфера B = fi
атомов А1. (В вершинах тригональной призмы (с = 2,37).) (В SB III 28
ошибочно указано гексагональной призмы.)
Тип, производный от С6: слоистая решётка. В типе С6 в пределах
элементарной ячейки два /-слоя || (0001), в ячейке А1В2 только 1 слой В-атомов.
В SB IV 121 (по Z. ph. Ch. (В) 31, 214 (1936)) структура и параметры
подтверждаются сх = 3,17.
§ 22,41 1ЮГИДЫ. VII ГРУППА: ФТОР, ХЛОР, БРОМ, ИОД 611
§ 220. IV группа периодической системы: углерод С, кремний Si,
германий Ge, олоно Sn, екинец Pb
Карбиды бора см. гл. X.
(.ведениями о структурах боридов Si, Go, Sn, Pb мы не располагаем.
S 231. Y группа периодической системы: азот N, фосфор Р, мышьяк As,
сурьма Sb, ииемут Ы
См. нитриды III группы.
Сведениями о структурах боридов Р, As, Sb, Bi мы не располагаем.
§ 222, VI группа периодической системы: кислород О, сера S,
селен Se, теллур Те, поллонии Ро
См. окислы, халкогениды III группы периодической системы.
§ 223. VII группа периодической системы: фтор F, хлор CI,
бром Вг, иод J
См. га л о геи иды III группы периодической системы.
ГЛАВА IX
СТРУКТУРЫ СЛОЖНЫХ СОЕДИНЕНИИ
§ 224. Виеденис
Определение понятии комплексное соединение за последние полвека,
т. е. со времени создания теории Верпера, претерпело множество
изменений. О некоторых формулировках чисто физико-химического характера
мы уже упоминали |1] (1934).
Здесь нас интересуют примепителыю к кристаллам
структурно-термодинамические критерии, вытекающие из энергетических соображений,
изложенных нами в § 105, Если стоять на этого рода позициях, как нам кажется,
наиболее приемлемых для химика, то тогда и определение интересующего
нас понятия, и интерпретация структур, и их систематика должны будут
отличаться в большей пли меньшей мере как от верперовских, так и от
современных кристаллохимических, в основе которых лежат прежде всего
гтехиомотрическне соотношения.
Комплексным соединением (в кристаллическом состоянии) мы будем
называть соединение, имеющее, помимо простых структурных узлов, группы
атомов {координационные сферы), ведущие себя, как изолированный
структурный узел. Например KJ04 (в равной мере NaN03, CaC03 и др.)»
—комплексное соединение. В нём имеются чётко выраженные театраэдрические
координационные сферы [J0J, ялвяющиеся самостоятельными
структурными узлами.
Сложным соединением (в кристаллическом состоянии) мы будем
называть соединение со сложным химическим составом, не имеющее, помимо
простых структурных узлов, групп атомов (координационных сфер),
ведущих себя, как изолированный структурный узел.
BP04 (AlAs04, ZrSi04, FeTiOj, и др.) является примером сложного
соединения, где нет ни изолированных *) координационных сфер [Р04], ни
[В041, но имеет место структуры типа [Si02], в которой половина атомов Si
занята атомами В, другая — атомами Р.
Подобные соотношения возникают в ряду Л1308 — YA108 — LaA103—
—LaGa08-~FeTiOa, где оба атома металла имеют примерно равные шансы
стать центральным атомом координационной: сферы. В результате в этих
соеданениях изолированных координационных сфер нет. Mg.>Si04 является
примером соединений переходного типа. В нём хотя и имеются островные
координационные сферы [Si04], но связи в них значительно ослаблены
влиянием силового поля Mg, образующего, в свою очередь, координационные
сферы [Mg06].
* Островных.
§ 2241
ВВЕДЕНИИ
61.4
В этом соединении расстояние MgO равно примерно 2,09 А (в MgO
dUg0 = 2,10 А), расстояние Si —О равно 1,81 А (в Si02 Si —О = 1,54 А).
Поэтому мы причисляем Mgj.Si04K сложным, а не к комплексным соединениям.
Таким образом, как мы отмечали, многие вещества, формулы которых,
казалось бы, отвечают комплексным соединениям, не могут быть
причислены к этой группе —FeTi03, ZrSi04, YA103.
С другой стороны, исходя из данных выше определений, мы относим
кристаллические FeS2, CuS, РС16, РВг5, вопреки их формулам и теории Вер-
нера, не к простым, а к комплексным соединениям, поскольку
координационные сферы в них существуют как самостоятельные структурные узлы:
Fe[SJ; Cu8[CuS,]; [PG1J4 [РС1.Г; [PBrJ+ Br".
Интересно, что соединение CoAsS (тин SB 7^1) — комплексное,
(lo [AsS]; CuFeSo, напротив, только сложное. Са (ОС1)а — комплексное
соединение, содержащее изолированные группы атомов [ОСГ|. FeOCl -
сложное соединение, поскольку оно имеет структуру О -Fe--Cl.
K3Fe(CN)e —комплексное соединение с координационной сферой [Fe(CN)J;
K2Zn (CN)4 —сложное соединение (тип ншипели).
Таким образом, рассмотрение структур подобных веществ требует
внесения существенных поправок1 в общепринят)ю до сего времени верпе-
ровскую систематику.
В некоторых промежуточных случаях вряд ли следует добиваться
строгого разграничения. Ото относится, в частности, к сложным веществам,
имеющим молекулярную решетку, в которых каждая молекула является
изолированным структурным узлом, например: [P4N4C1J, [B3N3HJ,
[Fe(CO)J.
Поскольку, помимо сложных структурных узлов, обозначенных [ ],
простые структурные узлы в них отсутствуют, эти соединения, пожалуй,
удобно причислять к сложным, а не к комплексным. В самом деле, если,
например, СвНв(бензол) не комплексное соединение,то таким можно признать
и его неорганический аналог —BaN3H6.
Наконец, внушительная группа соединений образует структуры с
бесконечными комплексными ионами.
Сюда относятся вещества типа СаТЮ3, T12A1F6, T1A1F4, YA10, и др.
В некоторых из них группы атомов образуют не отдельные
координационные сферы, но, например, пространственную вязь из ионов одного знака,
в которую вкраплены ионы другого знака. Так, в СаТЮ3 деформированные
октаэдры [ТЮв] соединены ь пространственную вязь (бесконечный
отрицательный поп), в междуузлиих которой размещаются ионы Си.
В T1A1F4 октаэдры [A1FJ образуют бесконечные сетки, в ThAlF6--
бесконечные цепи, несущие суммарный отрицательный заряд. Вне этих
сеток, соответственно цепей, находятся попы Т1+. Таких структур много
среди силикатов.
Подобные вещества можно назвать (ложнокомплекепымн
соединениями. Они рассматриваются нами как промежуточные соединения
в главе IX.
Комплексные соединения состоят из конечных по размеру ионов
(радикалов) разных знаков (типов), могущих состоять также из цепей, колец
и т. п. конечного размера.
Исследованиями систематике комплексных и сложных (в смысле нашей
номенклатуры) соединений посвящены работы ряда советских учёных.
Из них надо в особенности отметить работы П. В. Белова и его
сотрудников по структурам комплексных соединений и силикатов (см., в
частности, Известия Академии наук (хим.), стр. 364 (1948), книгу «Структура
ионных кристаллов и металлических фаз» и др.)* Г- Б. Бокия (в частности
Известия Академии наук (хим.) № (5 (1944), Известия Сектора платины
614 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. IX
Института общей неорганической химии АН СССР № 21 (1948) и др.)>
а также отдельные работы Г. С. Жданова и др.
Химики в этих вопросах часто пытаются оставаться на позициях в
большой мере устаревшей теории Вернера.
Критическое рассмотрение этой теории выходит за рамки данной
книги и осуществляется нами в других местах. Однако и здесь мы не
можем пройти мимо краеугольного камня вернеровской теории —вопроса
о координационном числе в комплексных соединениях.
Как мы отмечали в 1938 г., химическая формула соединения не может
характеризовать подлинный состав координационной сферы —для этого
необходимо знание строения этой координационной сферы (например,
[MnOJ), тем более, что оно вполне может изменяться с заменой других,
находящихся в решётке, структурных узлов (например, атома натрия на
атом серебра или лития в структурах перманганатов и т. и.) и с переменой
температуры и давления, и в присутствии следов примесей и т. д.
Нам трудно поэтому согласиться и с таким мнением некоторых
исследователей комплексных соединений, согласно которому комплексные
соединения продолжают разделяться на два класса: двойных солей и
комплексных соединений, главным образом по поведению этих веществ в растворах,
причём сама дискуссия о том, причислить ли то или иное вещество
к классу двойных солей или нет, ведётся без учёта структурных и
термодинамических характеристик вещества, но лишь на базе того, что вещества,
относимые к двойным солям, «представляют собой продукты сочетания
молекул отдельных солей, порознь способных к самостоятельному
существованию»*). Подобный критерий не может, по нашему мнению, привести
к правильным заключениям, ибо он неубедителен в своей основе: нельзя
судить о свойствах продуктов реакции, исходя лишь из свойств
компонентов реакции. Именно этот уязвимый, на наш взгляд, подход и
приводит к тому, что, например, в названном труде K4Fe (CN)e попадает в класс
двойных солей, т. е. оказывается в химической монографии соседом
NH4 Fe (S04)2 или KClNaCl, на которых он ни по строению, ни по
распределению связей, ни по термодинамическим условиям образования
совершенно непохож» Такая систематика нам не кажется плодотворной.
Кроме того, как мы подчёркивали в 1938 г., для описания структуры
комплексного соединения вериеровские координационные формулы
совершенно недостаточны. Нам казалось целесообразным предложить введение
структурно-координационных формул, исходя из следующих фактов и
принципов (приводим их здесь вкратце).
Например в ряду Na8AlFe — T12A1F5 — T1A1F4 — A1F3 координационные
числа А1, если судить по химическим формулам, различны. Однако для
подобных суждений никаких оснований нет. Исследование структур этих
веществ показывает, что во всех случаях А1 образует октаэдрическую
координационную сферу, но A1F3 имеет координационные сферы, в которых
каждый из шести атомов F одновременно принадлежит двум соседним
координационным сферам, в T1A1F4 четыре из 6 атомов фтора; в T12A1F6 — 2 из 6
атомов фтора каждой координационной сферы принадлежат соседним
координационным сферам. Наконец, в Na3AlFe все 6 атомов фтора
принадлежат только данной координапионной сфере.
С целью отобразить формулой особенности строения вещества автор
it предложил придерживаться следующего.
В формуле пишется сперва катион, затем анион. (При наличии
нескольких ионов одного знака сначала пишутся комплексные ионы, затем про-
*) А. А. Гринберг, Введение в химию комплексных соединений, Госхим-
иадат (1945), стр. 17.
§.2241
ВВЕДЕНИЕ
615
стые.) Для обозначения координационных сфер применяются квадратные
скобки. Открыв скобку, пишем сперва обозначения центрального атома,
затем периферических. При этом сначала обозначаются количества атомов,
принадлежащих только данной координационной сфере, затем количества
атомов, принадлежащих данной координационной сфере, но разделяемых
ею с соседними координационными сферами, затем ставится квадратная
скобка и количества атомов, вошедших в состав данной координационной
сферы от соседних координационных сфер. В нашем примере:
Стехиометрическая
формула
Na3AlF,
T12A1F5
T1A1F4
A1F3
Структурная
формула
Na3 [A1F,]] вместо
Na,[AlF,-]~]
Te2lAlF4F]F]
TelAlF2F2lF3]
[Al-FJF,]
Формула T12A1F5, изображённая по структурно-координационному
принципу T12[A1F4F]F], гласит, что: 1) состав соединения T12A1F6 (фтор
между двумя квадратными скобками принадлежит другой координационной
сфере); 2) состав координационной сферы в кристалле fAlF4F]F], т. е. AlFe.
Центральный атом —А1. Число атомов фтора, принадлежащих только данной
координационной сфере, равно 4, число атомов фтора, принадлежащих
и другим координационным сферам, равно 2, из них 1 предоставлен данной
координационной сфере от соседней, вследствие чего структурный и
формульный составы не совпадают, и т. д.
Формулой Na3[AlFJ] мы подчёркиваем, что формульный и структурный
состав координационной сферы одинаковы.
Рассмотрение большого фактического материала по сложным
соединениям требует по сравнению с нашей старой работой введения немногих
дополнительных обозначений и некоторого изменения формул сложных
веществ. Важнейшим является случай, подобный ВР04 (или Li2BeF4).
В этом случае и В и Р образуют тетраэдрические координационные сферы
и имеют каждый к. ч. ^4. Атом же кислорода имеет к. ч. = 2.
Рациональная структурно-координационная формула ВР04 будет: В[02|02]Р.
Из нее .вытекает, что координационное число как В, так и Р равно 4 0,
стоящим внутри скобок, а координационное число каждого из атомов О
равно 2 (1 атом В и 1 атом Р, стоящие снаружи скобок).
С этой точки зрения структурной формулой для модификаций Si02
является Si [021 ()o|Si с координационными числами Si =4 О и 0 = 2Si:3TH
формулы наглядно выявляют большое сходство структур ВР04 и Si02.
В равной мере, структурно-координационная формула Li2BeF4 имеет вид
B(?[F21 FojLio или Be[F4]Li2. Из нес вытекает, что координационное число
как Be, так и каждого атома лития равно 4 0 (внутри скобок), а
координационное число каждого из атомов О равно 3 (1ВеЧ-2Ы, вне скобок).
По сравнению с ранее предложенной формулой для A1F3, формула,
подобная приведенной выше, имеет то преимущество, что все входящие
в ее состав атомы составляют и стохиометрическую формулу вещества.
С этой точки зрения удобнее для A1F3 писать, согласно
сказанному выше: A1[FS|F3]A1. Из такой формулы наглядно вытекает не
только состав вещества, но и координационные числа: А1 к. 4. = 6F;
F к. ч. = 2АЬ
Подчёркиваем: в состав вещества только в том случае не входят
все атомы, составляющие структурно-координационную формулу, если
в последней имеются две квадратные скобки, повёрнутые в одну сторону.
616
Ч- ^СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. IX
Тогда, например, T1[A1F4F|F], атомы, стоящие между такими скобками,
в данном случае [F], при рассмотрении стехиометрического состава
вещества во Внимание не принимаются.
Структурно-координационные формулы, как мы видели выше на
примере SiCK, могут быть полезны при описании не только сложных и
комплексных но и простых соединений. Например, для NaCl: Na6 [Chi; для
ZnS: Zn [S4]; для SiC: Si4[G4]; для AlaO,: Al2 [О, | О,] А1а. При этом
может быть осуществлено и описание формы координационной сферы
с помощью наших индексов, например Na6(0) [С16(0)] или Zru(7) [SM-M].
А. СТРУКТУРЫ СЛОЖНЫХ СОЕДИНЕНИЙ
a) Г>КСКОНЕЧНЫЕ ТРЁХМЕРНЫЕ СТРУКТУРЫ
§ 226. Галогеииды
1. Li.BeF,
(-="Ы,[Р4]Вс)
Решётка ром боэдри-
ческая Rv.
Координация TjTjt,
Тип R%ITJt — SB #13
(фенакит).
Пространственная
группа Cli — R3.
Z = 6.
Be к. 4.=-4F (T).
Li к. 4.-4F (77).
Fk.4. -2LiflBe(*).
Рис. 464.
Вещество
LL Be F4
a
8,13
a
107°40'
Примечание
См. BeSi04
(фенакит)
§225] СЛОЖНЫЕ СОГДИИЕНИЯ. ГЛЛОГЕНИДЫ 617
2. 8Na,S0« • NaCl • NaF • (Na.FCl (S04)2 (сульфогалит) Na,, [FC1 (SO,),] (SO*),]
Решётка кубическая С
Координация 2 + д/О.
Тип C2+9/o-SB#5e.
Пространственная группа Of, — Fm3m или 0s — F43 и.чн 7/,-~F?uo;
a = Na; 6^ (SOJ (Г); y = -f» соответственно Gl.
Z = 4.
Nan. ч.= 1 F(2,28) +1 CI(2,76) + 4S(3,57) (q)
F K.4. = 6Na(C); Gl к. ч. —6Na(0);
S к. ч.== 12 Na (3.57).
О
UtZ'J45b?B9W ШЬ С
1 i i l I i i ■ . | f X^ J
Рис. 465.
Вещество
L* Na2so4 • NaCl • NaF
Решётка типа NaCl претерпевает следующие изменения: 1) ионы F и G1
занимают места узлов С1 поочерёдно, причём ион Na смещается ближе
к фтору. Ряд: F —Na —CI — Na —F... хорошо прослеживается по ребру
элементарной ячейки (ось 4); 2) позиции четырёх остальных атомов галогена,
окружающих данный атом Na, остаются незанятыми. Вместо этого в
центрах октантов элементарной ячейки появляются тетраэдры S04.
Очень хороший пример замещения в решётке NaCl большей части
ионов одного сорта (С1~) на ионы другого сорта, в том числе и с иным
зярядом (SO^) без изменения принципа построения структуры.
Примечание
100,8
-В HI 11S
618 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IX
3. CuaHgJ4, Ag2HgJ4 =Mfi[J2| Jt] Hg
Решётка тетрагональная Tv'.
Координация T/Tfi+l.
Тип гТ/г/г-м-вВЯЗ,-
Пространственная группа Dzd — Pi2m.
Z = i.
Рис. 466.
Веще ст во
Cu*HgJ4
Ag2HgJ4
а
6,08
6,34Q
с
6,135
6,340
с/а
i,oo9
i,oo0
d
Hg-J
2,74
2,82
d'
Gu—J
Ag-J
2,56
2,85
Примечание. Согласно SB II 51, «чисто геометрически» можно
представить наличие радикалов [HgJ4].
Однако нетрудно усмотреть из рисунка наличие также и
координационных сфер AgJ4 (тетраэдр), т. с. типичную структуру сложного, а не
комплексного соединения.
Как отмечает SB II 51, атомы J образуют кубическую плотную
упаковку, тетраэдрические промежутки которой частично замещены атомами
Ag или Hg. Это определение ничего не говорит о химическом характере
структуры.
§ 226. Цианиды
1. Цианиды щелочных металлов.
Структуры сложных соединений—цианидов щелочных металлов
исследовались в ряде работ, причём было обнаружено у щелочных металлов
стремление образовывать две модификации цианидов—низкотемпературную
и высокотемпературную. Высокотемпературная модификация является
кубической, тип каменной соли Coio, что связано с вращением циан-ионов
в* решётках KCN, NaCN (см. SB I1 371).
§ 226|
СЛОЖНЫЕ СОЕДИНЕНИЯ. ЦИАНИДЫ
619
NaCN, KCN, RbCN, CsCN(-Nafl[(CN)el) (см. стр. 691-693).
Решётка кубическая С.
Координация О/О.
Тип С0{0 — SB~£1 (близок к типу NaCl).
Пространственная группа (?).
2-4.
Вещество
а
NaCN
r>,s:t
1,624
KCN
6,51
1,557
RbCN
6,82
2,32
"Й. CsCN имеет иную структуру с Z -1.
3. Цианиды серебра и золота.
Серебро и золото образуют цианиды AgCN и AuCN. В отличие от
галогенидов, цианиды серебра и золота имеют структуры не
координационные, но цепные. Имеющиеся данные критически рассмотрены,
дополнены и обобщены в работе Г. С. Жданова и Е. Шугам (Acta Phys. Ch.'
XX 253(1945)). Этими же авторами определена структура цианида золота.
Эти цепные структуры см, стр. 645.
4. Zn (CNL,Cd (CN)a = M[(CN)21 (CN)JM
Решётка кубическая С.
Координация Tjl.
Тип ~ Cr/i «анти-куирита»
(формальное сходство).
Пространственная группа
Та Pi 3m.
Z=2. a = (CN).
M" к. ч.
(CN)
= 4 (CN) (тетраэдр);
ц. = 2 М (линия).
Рис. 467.
Вещестпо aw \ d (Zn--CN—Zn)
Zn(CN),
Cd (CN).
5,80
6,32
5,1
5,47
Примечание
По работе V. С. Жданова. ДАН АН
СССР XXXI 352 (1941). Циангруппа
занимает позиции атома кислорода в
решётке куприта, причем образуется
структура, сходная с С^т
По Шугам и Жданову (Acta 20, 247 (1945)) отсутствие вращения
циан-группы приводит к образованию четырёх направленных связей под
тетраэдрическими углами от центрального атома. На этих полудпагоналях
элементарной ячейки лежит цепочка М—С—N—М.
620 Ч. И. СТРУКТУРЫ НКОРГЛНИЧЕСШ1Х ВЫЦКСТВ |ГЛ. 1\
Мы хотим затронуть вопрос, не рассмотренный в атой работе. Так как
атомы С и N в химическом отношении различны и в исходной молекуле
группа циана повёрнута атомом углерода к цинку (соответственно к Cd),
то в кристалле около каждого атома цинка (соответственно кадмия) должны
были бы находиться два атома углерода (малое расстояние) и два атома
азота (несколько большее расстояние). Повидимому, центр тяжести группы
циан должен был быть смещён от центра полудиагонали (центра
соответствующего октанта элементарной ячейки) по двум связям (с атомами С)
и сторону данного атома ц и и к а (соответственно Cd) и по
двум другим (с атомами N) в сторону от д а и и о г о а т о м а цинка
(соответственно Cd) к соседнему. Поскольку такое смещение в кристалле
не обнаружено, это может быть либо результатом вращения циан-группы
(чему препятствует её фиксация ковалентной связью металл—углерод),
либо перераспределением электронов с возникновением химической связи
металл-азот (что должно быть доказано), либо неточностью в фиксации
циан-группы.
Вопрос о характере связей металл—азот в цианидах данного тина,
с нашей точки зрения, является одним из самых интересных в
структурной химии цианидов. Он
подлежит
экспериментальному выяснению на ряде
систематически исследованных
объектов (см. комплексные
цианиды, гл. X).
5. Сложные цианиды
A.nB(CN)4 K2Zn(CN4),
K2Cd(CN)4, K2Hg(Ctf)4.
Решётка кубическая Cv.
Координация 0\T\r±%.
Тип C£t,a-SB//11.
ОТ/4,5
Пространственная группа
Ol—FdSm
Z=8.
К к. ч.=6 (GN);
Zn к. ч.-4 (CN).
(GN) к. 4. = 3K + l75Zn Mn-£ центрах тетраэдра В
о К
Рис. 408.
Вещество
KaZn (C.N)4
K2Cd(CN)4
KaIIg(CN)4
a»
12f54
12,8,
л
12,7G
OM
247
265
260
Примечание 1
См. AbMg04
Структура шпинели позволяет с успехом комбинировать тетраэдри-
ческие координационные сферы типа [Zn(CN)4] (см. структуру Zn(CN)2)
и октаэдрические координационные сферы типа |Ke(CN)e), имеющиеся в
решётках цианидов типа каменной соли (см. цианиды щелочпых металлов)..
§ 228]
СЛОЖНЫЕ СОЕДИНЕНИЯ. ОКИСЛЫ Л„В02
621
§ 227. Оксигалогениды
1. LaOF( = La4[(0,F)J)
Решётка кубическая Cv,
Координация С/Т.
Тип C£,r-SBCl(CaF2).
Пространственная группа Oft
Fm 3 т.
Z = 4.
La к. ч. = 8 (0,F);
(0,F) к. ч.=4 La
Вещество
а„ . . . .
LaOF
Г>,756±3
Примечание. La—О и La—F—
— 2.49А. В случае избытка О
решётка становится
тетрагональной с я = г>,7:{; е = Г>,84. В сл}г-
чае избытка F а уноличппается
до 5,810
§ 228. Окислы АпВ02
1. (A^-iBO^LiFeO,;
Na2CeOe: NaaPrO„
Li3Ti08( = Li3[Oc]Fe:!
и т. п.)
Решётка кубическая Су.
Координация октаэдриче-
ская О/О.
SBtfl(NaCl).
Тип Со/о
Z = 2LiFeOa. C^S_
(Li.Fe) к. ч.--6 О (октаэдр); \7
О к. ч. -6 (Li. Fe) I
Рис. 470.
Вещество
\ а . . . .
h\Tr4 . . .
LiFeO,
4,14
1,16
NaaCeOa
4,82
0,96
Na.PrO,
4,84
0,98
Li2Ti03
4,10
1,22
Примечание
Цинтль и Мораве ц
(См. Z. Anorg. Ch.
245,26 (1940))
Оба катиона
Занимают места в решётке
статистически
Попытка указанных авторов приписать возникновение подобных
структур соотношению ионных радиусов, близкому единице, на наш
взгляд, не выдерживает критики. Любопытно, что соединение NaLa02
выделено в той же работе, но, как оказалось, не имеет структуры Со/о,
622 ч. и. структуры неорганических шлцьств [i л. IX
хотя соотношение ионных радиусов такое же. В то же время у Li.2Ti()3
структура Со/о наблюдается, несмотря на отношеш е rAjrB, равное 1,22.
Хотя вес ионного состояния в химических связях подобных
структур несомненно не так уж велик, переход к более низкому
координационному числу не наблюдается, в отличие, например, от халкоинрита
(см. стр. 636). Таким образом, вопреки тому, что предполагают
некоторые авторы, решётка, «типичная» для солей, может быть образована, по-
видимому, связями, далёкими от ионных. См. также ThP и VN, стр. 545:
TiN, стр. 543.
§ 229. Окислы А„ВО,
1. MgTiO,, MnTiO,, FeTi03 (ильменит), CuTiO,, NiTIO,, (MTiO
(=Mga[Oa|OJTI,)
Решётка ромбоэдрическая Iiv.
Координация O/O/i.
Тип RV. — SBG4 —SB^22 (ильменит),
искажённый тип Dbx (корунд).
Пространственная группа Z)°d—#3t\
Z--=2. _
Ti к. ч. 6 0(0);
Fe к. ч.-О 0(0);
О к. ч.»4 (Ti, Fe).
Рис. 471*). О М»
а
а.
MgTiOs
5,54
54°39'
MnTiOa
5^62
54°16'
FeTi03
5,52
54°49'
CoTi08
5,49
54°42'
NiTi03
5,45
55э8'
CdTi03
5,82
53°36'
Примечание
SB III 69, см. такте 1
SB I 300
*) См. корунд.
§ 229] СЛОЖНЫЕ СОЕДИНЕНИЯ. ОКИСЛЫ АпВ03 623
В SBI к этому же тину причислен LiNb03, являющийся, как и Li.2Mo04
и т. п., одним из примеров отсутствия комплексных координационных
сфер, несмотря на то, что за пределами координационной сферы типа
Рис. 'i72.
[NbN08J находился бы ион щелочного металла. Причина такого
поведения Li -интенсивное силовое поле Ы+-иона (К-оболочки) — ясна. Это
заставляет вообще поставить Li —при образовании структур сложных и
комплексных соединений особняком, отдельно от других щелочных
металлов и ближе к Си+ и Ag+. Сравним Ag8P04-
Рис. 471 показывает группу атомов, отмеченную на рис. 472
фигурной скобкой. На рис, 471 ось 3 почти перпендикулярна плоскости чертежа,
на рис. 472—лежит в плоскости чертежа.
024
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. IX
§ ИЗО. Окислы А„В04: АВ04, А2В04, АаВ04 ( = ,МгаХ0„)
0^=^
1. ZrSi04 (циркон), YP04 (ксенотим), YP04, YAs04, YYG4( = Zi'l04]Si)
Решётка
тетрагональная Tv. rs ^^ y^r
Координация те- <^0 х. _ j&\ ~30
траэдрическая
TjDift + 'l.
1ИП Л'гцлрт
SB#3=-#03
(циркон).
Пространств
группа
Pi/man.
Z 4.
М к. ч. 40 (5)
(+ 40 (.V) =
_--=8 о (до;
X к. ч.-4()(Г);
О к. ч.-2 M-i 2\.
Рис. 473.
Вощостпо
ZrSi04
YP04
(ксенотим)
YP04
YAs04
YV04
a
б,Г,8
G,88
G,86#1
6,890
7,120
с
5,93
6,03
6,17^
6,26-
6,179
cja
0,904
0,877
0,900
0,908
0.8J1
kd
P-0
1,62
1,64
1.7i
1,73
1,02
ke
Y-0
2,05
9 9')
2,24
2,26
2,30
4/
Y-0
2,41
2,24
2,24
2,26
2,58
OM
C'«,2
—
—
—
78,4
Примечание
SB I 345
SB III 91
Примечание. В SBI, в соответствии с формулой ZrSi()4,
изображены координационные сферы [Si04] с атомами Zr, находящимися вне
тетраэдров, как если бы они были совсем неравноценными.
Однако в соответствии с § 105 нельзя ожидать подобной структуры,
поскольку энергии решёток Zr02 й Si02 довольно близко. В
действительности Zr также окружён 4 (+4) атомами О (т. с. Di (4Zr — 0 = 2,05 -j-
-r4Zr —0 = 2,41)* Если; в решетке j* ^ристобалита. Si — 0^= 1,54, в 3-кварце
1,59 и в решётке Zr02, Zr — 0 = 2,20, то в решётке ZrSi04 расстояние
Si — 0 увеличено,, расстояние Zr —О сильно уменьшено для 4 атомов
О, образующих несколько диформировашшй тетраэдр, и увеличено для
4 остальных О-атомов. Очевидно, что с образованием ZrSi04 как раз
упрочнена 4 связи Zr -О, а не Si —О.
Из структуры ZrSiC)^ следует; что ей не отвечает ни
координационная формула Zr[Si04], ни jZrlSiOs]Oa], но [Zr[02,02]Si], т. е. Zr[04]Si.
§ 230]
СЛОЖНЫЕ СОЕДИНЕНИИ. ОКИСЛЫ Л„В04, AjHO,, Л:.ИО,
02)
2. BP04, BAs04
Bt02|0,]X-Bl04lX
и т. д.
Решётка
тетрагональная Ту.
Координация TjT/l.
Тип Tv- -SB //0,
тип
(ВР04).
Пространственная
группа S] — Jh. vj'
Z-=2. •—£ ^-
X к. ч. = 4 0 (тетра- '/
эдр);
Ок. ч. -1В + IX
(линейная);
В к. ч.-=4 О
(деформированный
тетраэдр).
Вещество
ВР(>4
BAs04
а \ с \ v /a jj (J j p__Q ! Примечание i
4,:п2_м; ! амо±8 1 1,Ь'Ы±14
4,458 + /? ; 6,796^.3 1,525 ±J4
1 1
i 1
1,44 | 1,54 i Структура близ-
! i ка к Si02(высо-
1,149 1,66 ! коте мнератур-
ный кристобалит)!
3. VCr04, CoCr04, Nilr04, CuCr04, ZnCr04 CdCr04
Решётка ромбическая ORv\
Координация О/Т/З.
Тип ОЛ^-SB?
Пространственная группа DVh -Cmcm.
Z = i.
Cr к. ч.-бО (искажённый октаэдр);
V к. ч.-4 О (иска'.кёшшй тетраэдр).
1 Вещество
1 а
Ъ
1 с
Вещество
а
Ь
с
VCrO. CoCrO, NiCrO.
_J .... 1 ._!_.
5,568 5,Г,05 1 5,492
8,204 8,281 1 8,219
5,977 6,207 I 6,1П
1
СиГ.г04 , ZnCr04
i
5,426
8,925
5,878
5,505
8,38*
6,219
CdC,r04
5,674
8,674 |
6,893
Примечание
Координационная сфера [C-rOe] —
искажённый октаэдр. Соединяясь
вершинами, октаэдры образуют 1
цепи. Атомы V и др., образуя 1
тетраэдры [V04], сшивают цепи 1
октаэдров между собой 1
Сг-0 = 1,95 и 2,03 1
V-O-1,72 и 1,80 I
626 Ч, II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТИ [гл. IX
О к. ч. = 1Ве + ЗА1.
Рис. 47"».
Вещество
а
Л1аВе04 'i,V20
Ъ
Ч ЗУ
с
ОМ Примечание
5,47() Г)6,8 SB I, 352. См. оливин Mg,Si04
5. Mg2Si04, (MgFe)2Si
Решётка
ромбическая ORv.
Координация
0/773 + 1.
Тип ORlmw -
SB #12.
Пространственная
группа V^—pnma.
Z = 8. Mg к. ч.-бО
(октаэдр);
Si к. ч. = 4 О
(тетраэдр);
О к. 4. = 3Mg + l Si.
(В SB I 355
координационные данные
для О ошибочны).
Вещество
Mg.SiO,
MgCaSiO,
1
а
4,8
4'815
Ь
10,3
11,08
с
0,0
0,37
Примечание
Средние величины. В составе оливина
(MgFe)2Si04 соотношение Mg: Fe
колеблется.
4 (оливин)} MgCaSi04
Рис. 470.
§ 230] сложнь.к соединения, иншнклп 627
Шпинели
Ниже мы рассматриваем большую группу важных в теоретическом
и практическом отношении структур, носящих общее название шпинель.
Их своеобразный и разнообразный состав и довольно сложная структура
наставляют нас предпослать этому разделу ряд замечаний.
Как мы знаем, в характерной для металлов кубической плотной
упаковке Ск имеются как октаэдрические, так и тетраэдричеекие междуузлия
(§ 63 и 64, рис. 100). Их заполнение атомами неметалла приводит к
образованию в первом случае структуры каменной соли Со/о, а во втором —к
структуре плавикового шпата Сс/т- Уже из § 63 мы знаем, что вовсе не всегда
имеет место заполнение всех междуузлий данного типа. Это особенно
относится к тетраэдрическим междуузлиям: заполнение в строго
определённом порядке двух центров октантов в кубической гранецентрированной
ячейке ведёт к* типу куприта Сщл заполнение четырёх —к типу
сфалерита CV/7- Мы знаем также, что в ряде случаев, при наличии избытка
атомов неметалла, но заполнении всех тетраэдрических промежутков
заполняются и октаэдрические, что ведёт в пределе к структуре
фтористого висмута Сс+ор\ Оу при нехватке же атомов неметалла, по
сравнению с Сс/т часть тетраэдрических междуузлий в статистическом беспорядке
остаётся незанятой (см. § 104). Во всех этих примерах узлы кубической
гранецентрированной ячейки занимали атомы металла (Си, Ъх\ и т. п.),
колебалась же степень заполнения междуузлий атомами неметалла (F,0).
Шпинели удобно рассматривать как группу структур, у которых узлы
кубической плотной упаковки занимают атомы кислорода (или другого
неметалла, а также группы атомов, например CN), междуузлия же
в той или иной мере заполняются атомами металла.
Представим себе плотную кубическую упаковку в ячейке с удвоенным
ребром. Кратность позиций в такой ячейке будет в восемь раз больше,
чем в обычной, т. е. равна 32. В ней имеется, следовательно, 32 октаэд-
рических междуузлия и 64 тетраэдрических (рис. 100), первые — в
центрах рёбер и в центре каждой из восьми исходных нормальных
элементарных ячеек (являющихся теперь только октантами новой ячейки),
вторые—в центрах октантов исходных нормальных ячеек. Эти междуузлия
и должны заполняться атомами металла. Совершенно очевидно, что если
все 32 октаэдрические междуузлия большой ячейки окажутся
заполненными—возникнет знакомая нам структура Со/о, если все 64
тетраэдрические—также знакомая нам аитифлюоритиая структура Cj/с- Примером
первой является решётка MgO, примером второй —решётка К20 (см. гл. VI).
В этом случае пет оснований к удвоению ребра ячейки.
Но упорядоченное занятие либо октаэдрических, либо тетраэдрических
междуузлий не всегда происходит, даже если занимаются позиции только
одного из этих двух типов —не всегда они занимаются полностью-.
Возникают структуры вычитания и деления, картина осложняется. Тут возможны
три характерных случая, которые, по нашему мнению, могут рассматриваться
как структурные варианты при классификации шпинелей: в междуузлия
внедряются а) атомы одного элемента с разной валентностью, например II
и III (например Fe), б) атомы разных элементов, например Mg (II) и А1 (III)
с разной валентностью, в) атомы одного элемента с постоянной
валентностью, главным образом равной III (например А1).
Важнейшей системой первого типа является система Fe —О.
Предельное заполнение атомами Fe всех октаэдрических междуузлий в ячейке
с удвоенным ребром повело бы к образованию структуры Cojo состава FeO,
в которой атом О окружён 6 Fe (в октаэдрических междуузлиях). В
действительности часть позиций Fe остаётся незанятой: вмести 32 атомов в ячейку
628 Ч- II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. XI
примерно входит 31 и т. п. Ребро такой ячейки равно 4,30 X 2 = 8,6 к X.
При окислении FeO происходит в окончательном результате (механизм
мы здесь не рассматриваем) не внедрение новых атомов кислорода
в ячейку, как это часто полагают, но, напротив, извлечение части
атомов железа (и образование нового объёма занятого веществом окисла).
Оставшиеся на месте 32 атома кислорода будут удерживаться теперь не
31 атомом железа, а при составе, например, Fe304, только 24 атомами.
Из них 1G, т. с. но 3 соседа Fe вокруг атома О останутся в
октаэдрических промежутках, а 8, т. е. по I1/* соседа Fe на атом О, займут тетра-
эдрические промежутки, в порядке, типичном для ншинелыюн структуры
и представленном на рис. 477 и следующих, того же типа. Детали шпи-
нельной структуры хорошо видны на рис. 478 и след. При этом важно, чти
в октаэдрических междуузлиях останутся не атомы Fell, но атомы Fe III,
первые же должны совершить миграцию в тетраэдрические междуузлия.
Поэтому механизм возникновения шпинели вовсе не является таким
простым, 1<ак это освещается, например, в монографии Уэллса; он связан
с существенными изменениями в распределении электронной плотности
в решётке. Дальнейшее окисление влечёт за собой образование у Fe203
с 211/;! атомами Fe, как принято писать —статистически распределёнными
между 2\ позициями. Подчеркнем, что в 16 октаэдрических междуузлиях
положение атомов Fe III будет с энергетической стороны нормальным и в
идеальном случае эти позиции должны были бы быть заняты полностью
(т. е. по 3 атома Fe III вокруг атома О), в 8 же тетраэдрических
промежутках преимущественно следовало бы ожидать статистического распределения
Г)х/3 остальных атомов Fe (структура деления), т. е. 1 Fe II на атом О.
С этой точки зрения шпинели, подобные Fe304, являются типичными,
естественно, что соединения типа MgAl204 (у которых при Z = 8 на 32
атома кислорода приходится 16 атомов А1Ш и 8 атомов MgH, no числу
занимаемых
октаэдрических и тетраэдрических
промежутков) должны
легко образовывать
шпинельные
структуры, что и наблюдается.
Вещества,
подобные у А1203, могут
образовывать лишь
шпинельные структуры
деления, подобно у Feo03.
6. Fe2Mg04f Fe,Mn04,
Fe2Fe04 = Fe304;Fe2Co04,
Fe2Ni04, FeXu04,
Fe?Zn04, Fe2Cd04
Решётка кубическаяСг.
Координация 0/774,15.
Тип Co/r/O-SB #11
(шпинель).
Пространственная грун-
ua Ol—Fd3m.
Z = 8.
Fe III к. ч.--6 О (О);
М II к. ч. 4 0 (Т). ,• 0 о
Ок. 4.^3FeIIM-iMlI оМш М-Вцентрах тетрил
(линейная). Гпс. Viz
§ 2301
СЛОЖНЫЕ СОЕДИНЕНИИ. ШПИНЕЛИ
Ь29
Всщестно
ОМ
Вещество
«1Л
| ОМ
Fe,M?04
8,S4
72,5
Fe,Ni04
8/.1
~1Г 1
Pit
Fe.,Mn04
8,57
78,7
Fe.CuO,
8,Vi
75,2
'. 478.
FeaFe()4
8,41
74,4
Fo,Zn04
8,40
74,1
Fe.ToO,
8, '\\)
Fc2C<]04
8,74
84,2
Примечание
SB 1 450 1
7. Co804-Co2Co04
Решётка кубическая Cv.
Координация О/Г/4,0.
Тип CM _ -SB ЯП (шпи-
0/7 /4,5
ноль).
Пространственная группа
Ol—Fd'Am.
2 = 8.
Со III к. ч. в О
(деформированный октаэдр);
Со II к. ч. Л О (тетраэдр);
О к. ч.-З Со III + 1^5 Со II,
Сош Соь tf центрах тетроздроЬ
Рис. 470.
(530
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТЬ
8. у Fe203, у А1203
Решётка кубическая Cv.
Координация О/ЗГ/4.
Тип Cv-~-SB ЯП
0/7/4
(шпинель).
Пространственная
группа 0l—Fd3m.
Z = 8.
М 111 к. ч. -6 О
(деформированный октаэдр);
М II к. ч.=---4 О
(деформированный тетраэдр);
Ок.ч.-ЗМШ И МИ
(рис. 480).
Описание структуры
(тип Z)57, SB 11 43)
последующими работами
опровергнуто. yAL03
и у Fe.,03—тип
шпинели //!,, рис. 481.
ом Оо
Рис. Ш.
Вещестпо
я„
Объём
элементарной
ячейки . .
TFe203
8,322
TAL03
7,89
576,3 | 491,1
Примечание. Возникает при
окислении FO3O4 (тип
шпинели), для которого аи = 8,380
и О : Fe -- 1,333. с
приближением О : Fe к 1,5
наблюдается, таким образом,
сжатие решётки. Одновременно
образец светлеет
оМ*
MD -В центрах твтраэдроВ
Рис. Ш.
Подробное изложение вопросов окисления железа и возникающих
структур окислов см. В. И. Архаров, Окисление металлов (1945).
§ 230] СЛОМ.НЬ.Е СОЕДИНЕНИИ. ШПИНЕЛИ 631
9. Al2Mg04, Al.,Mn04, A1 ,Fe04,
А13Со04, Al.,Ni04, AKCuO»
Al2Zn04
Решётка кубическая Cv.
Координация (9/Г/4ДЗ.
Тип СУ.- ~ — SB ЯП (шпи-
0/-1 /4,5 v
нель) (Al,MgO,).
Пространственная rj)vnna
Ol—Fd3m.
Z = 8.
А1 к. ч.«6()(0);
MgK. ч.--=-4() (У);
О к. ч. - 3 А1 -:- О Mg (••»-
нейпая).
оМи
М -S центрах тетраэдров
1 Вмц.>-
(ТМО
1
ОМ
Ah\U<)4
8,0.»
66,2
ALMn0.t
8,26
70,4
Al,Fo04
8,12
66,9
Л12Со04
8,0,.
6Г),Г>
Рис. 482.
Al,vNi04
8,04
ог.,о
А]3ОЮ4
8,°7
6>,7
Al2Zn04
8,10 1
66,4
П |) и м е ч а и и е. SB 1 '■ Г>0.
10.Cr,MnO„ Cr,Fe04, Cr,Co04,
Cr3Zn04
Решётка кубическая Cv.
Координация б/Т/4,5.
Тип С1'- - -SB ЯП (шпи-
0/7 /4,0 V
ноль).
Пространственная группа
01—Fd dm.
Z = 8.
Сг к. ч. = 6 О (О);
М к. Ч.-.-.4 О (Г);
О к. ч.-,3 О+ 1,5 М.
Вещество
Сг2Мп04
CraFe04
Сг2Со04
Cr»Zn04
ОМ
8,49
8,36
8,32
8,32
76, П
73,0
72,0
72,0
• о
о Сг
центрах тетраэдроВ
PilC. \$?i
632 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ |ГЛ. IX
11, Мп304
Решётка тетрагональная Ту.
Координация ВЦТ\Ь~Ь (?)
Тип шпинели тетрагонально искажённый, T\L .
Пространственная группа (?).
Z = 8.
Вещество а
Мп304
—
1 1
с | с/а I Примечание 1
1 ! 1
—
- Phys. Z 39,208 (1У38)
12. Ag2Mo04
Решётка кубическая Су.
Координация 0\Т\С,Ъ.
Тип Су_ „ -SBЯП (шпинель).
0/1 /4,5 N '
Пространственная группа Ol — Fd3m.
Z = 8. AgK. ч. = 6 О (О); Мо к. ч. = 4 О (Т); О к. ч.- 3 Ac ИЗ Мо.
rf(Ag-'0)-2/i2±2, d'(MoO) = lf83±3.
Мо - 5 центрах тетраэдроЬ
Рис. ',8V
Вещество
А£,Мо04
а„
У, 26
ОМ
У У, 2
Примечание 1
См. J. Am. Ch.S 69,222 (1У47). Расстояния Ag-0
и Мо—О короче суммы ионных радиусов (2,46),]
соответственно (2,00) 1
§ 230] С"lOlhHL E СОЕДИНЕНИЯ. ЛоН1)4 633
13. Li2Mo04, Li2W04
Решётка
ромбоэдрическая Яу.
Координация TjT/t.
ТипЛЗг/г/i—SB»13
(фенакит).
Пространственная
группа Си —ЯЗ.
Z = 6 (см. фенакит).
Вещест-1
во . . |Ы.МоО
\а . . . 8,77
а ... | 108в10'
4Li2VVOj
i 8'77
| 108°10'
Примечание. По SB
356, разницы в
метрике обоих соседей нет.
См. Be2Si04. I
14. Be2Si04 (фенакит),
Zn2Si04(Zn, Mn)2Si04,
Boa[04lSl
Решётка
ромбоэдрическая Rv.
Координация T/T/t.
Тип Rrpit SB #13
(фенакит).
Пространственная
группа См —ЯЗ.
Z- 6; Si к. ч. = 40 (У7;;
Be к. ч. - 4 О (Г);
Ок.ч.-2Ве 4-1 Si(/).
1
а
d
ОМ
Вещество |
Be3Si04
7,68
108°1'
61,0
Zn,Si04
Ми),
SioJ
8,69 8,81
107°4Г —108°
89,1 92,9
Примечание. SB I 356.
Как и в кварце
или тридимите (SiOj,
координационное число
Si равно 4 О;
координационное число Во
также равно 4 0, но
недостаток атомов
кислорода по сравнению с
решёткой Si02 повлёк па
собой то, что атом
кислорода имеет
координационное число, равное
2 Be 4-1 Si = 3, вместо 2
в Si.
Рис. ',86.
634
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. \1
15. ВаА120,
Решётка гексагональная Ну.
Координация 9/4/4,3.
Тип tfJ^/M-SB #2, (ВаА1204).
Пространственная группа £>§ —
-С6Э2.
Ва к. ч. = 3 01 (3,01)+ 6 О II
(3,30) = 90 = /< + *;
А1 к. ч. = 10Г(1,75) + ЗОИ
(1,79)-4 О;
О I к. ч. = 1 А1 + ЗВа;
О II к. ч. = 2 А1+1Ва.
1 и 1
Вещество ■ а
ВаА1а04
5,201»
]
с ; с/а
8,761 1,682
1 1
Примечание.SBV19,20.
Замена в шпинели (Al2Mg04)
Mg на Ва привела к возникио- р^ /8;
вению нового типа, однако
характер решётки сохранён.
Интересно, что ослабление силового поля М И иона не вызвало
образования островных координационных сфер [А10п].
Напротив, к. ч. А1 уменьшилось, а М И — увеличилось; ср. BaCl...
16. Ag5P04, Ag5As04
Решётка кубическая Cv.
Координация искажённая ,
кубо-квадратная С/д/к.
Тин C^/4-SB- #21.
Пространственная группа
П-Р
\Лп[ иф
1\
— J или
0\ — РтЪп(и =
Z= 2.
Р (см. центр ячейки) к. ч. =
-80(C); Agк. ч. = 4 0(7);
О к. ч. = 3 Ag -|- 1 Р-4.
Параметры: Р (000, А ~ ^)
\ - - *• /
Ag:±(0i-5-;O)
О : (www; uuu О)»
1,1 1
У + Ы. у + И> 2" + И
-$ + и, -2—и;С)
+ w,
Рис. 488
§ 232]
сложный соединения, сульфиды ли»
635
Вощсстно
Ag3P04
aw
5,УУ
6,12
d*)
Ag--0
2,12
2,16
, *)
P—0
(As -0)
2,5У
2,65
ОМ
107,5
114,6
Степень искажении координационных сфер в структуре AgaP04 зависит
от того, на сколько и отличается от 1/4.
Небезынтересно подчеркнуть следующую особенность этой структуры:
атомы Ag и Р занимают позиции атомов V и Si в структуре V3Si
(тин (3 W). Атомы Р занимают позиции в вершинах и центре
координационной сферы, атомы же Ag—располагаются на гранях куба, образуя крн-
сталлохимический икосаэдр (рис. 411а) вокруг атома фосфора.
Атомы кислорода располагаются примерно в центрах октантов, по-
1
СКОЛЬКУ П ОЛИЗКО К -- .
Вещество
А1(Р03)3
а"'
1^,63
§ 231. Окислы АтВ„0„
I. А1(Р0„), ~А1*0,ЗР,0(
Решётка кубическая Cv.
Координация OjTj'l.
Тип CrV/2-SBG5s (А1(Р03)3).
Пространственная группа Та — Л'М-
Z = 16 А1(Р03)3.
А1 к. ч. -60 (1,80-1,83) (октаэдр);
Рк. ч. = 40 (1,39; 1,55; 1,56; 1,60) (тетраэдр);
01 к. ч. = 2Р; 011 к. ч. = 1Р+1 А1; ОШ к. ч.=
= 1Р+1А1.
Решётка состоит из почти правильных АЮ,-октаэдров и Р04-тстра-
эдров, их коих последние группируются по 4, образуя кольца [P40i2].
Каждый атом О принадлежит двум полиэдрам. Из 12 атомов О 4
принадлежат двум тетраэдрам каждый, 8—одному тетраэдру Р04 и одному
октаэдру АЮ, каждый. Каждый (А10,)-октаэдр имеет 6 общих вершин
с 6 разными Р40,.,-комш1ексами (SBV16.)
§ 232. Сульфиды AXBS„
ABS2 = AaB8S4, A,BS„ ABS4,
A3BS4, ABS
1. NaBiS., KBiS,
Решётка кубическая С .
Координация OJO.
Тип Со/о -SB Bi (NaCl).
Z = 2ABS, =4(A,B),Sl-
Вещеетпо
NaBiS,
KBiSa
"tn
5,76
6,01
Примечание
Радиус
Bi3^l,l--1,2 A
Рис. 489.
*) Для w^VV
636
Ч. II. СТРУКТУРЫ НКОРГЛНИЧЕСКНХ ВЕЩЕСТВ
[гл. IX
2. CuFeS2 (халькопирит), AgFeS2
Решётка тетрагональная Tv.
Координация тетраэдричсская Т/Т/Т.
Тип T*f-SB Е1г.
Пространственная группа Da J'lld.
Z = 4.
Си к. ч. = 4 S; сГ = 2,32 (тетраэдр);
Fe к. ч..-=4 S; d ■= 2,20 (тетраэдр);
S к. ч. = 2 Fe (d == 2,20)+ 2 Си(сГ = 2,32) (слегка искажённый тетраэдр).
Вещество
CuFeS,
AgFe?^
а \
5,24
5,6Г>
Рис.
с 1
10,30
Ю,:>о
490.
с'а
1,820
Примечание
Идеальнал решётка (SBII 48)
возникла бы при с/а=2,
решётка сфалерита, в которой
атомы 2п заменены
попеременно на атомы Си и Fe
Исходя из структуры халькопирита, мы считаем необходимым
подчеркнуть, что это, издавна присвоенное ему минералогами, название
является крайне неудачным. В решётке CuFeS2 нет структурного мотива
пирита, нет и полисульфидной группы S2, которая влечёт за собой
отнесение пирита к иному классу соединений, чем «халькопирита».
§ 232|
СЛОЖНЫЕ СОЕДИНЕНИЯ. СУЛЬФИДЫ A.rHS,,
Ш
3. Cu2FeSnS4 (станнит)
Решётка тетрагональная Т/Т/Т/Т.
Координация тетраэдрическая Т.
Тип Т tit ml- SB #2e.
Пространственная группа D\vfl — J^2m.
Z = 2
Си к,# ч.-4 S (Г)
Fe к. ч. =4 S (>)
Sn к. ч.-4 S (Т)
S к. ч. =--.2 Си-; 1 Ге-j- 1 Sn (7).
Рис. 491.
1 Вещество
а
с
1
CiiaFeSnS, j Г>,40 10,725
с/а
1,964
Примечание
Сравни с CuKeS* 1
Атомы Си занимают слой параллельный (001); атомы Fe и Sn также
образуют параллельный (001) слой, в котором, судя по SB, Fe и Sn
строго чередуются, а не расположены статистически. Между слоями
атомов металла располагаются слои атомов серы. Атомы металлов
(железа, олова, меди) окружены каждый только атомами S,
образующими тетраэдрическую координационную сферу, с расстояниями
(Sn-S)d = 2,425; (Fe —S) е-2,36 и (Си —S) / = 2,31. Напротив, каждый
атом серы окружён 2 атомами меди, 1 атомом железа и 1 атомом олова
на указанных выше расстояниях. Таким образом, координационная сфера
атомов серы представляет собой не совсем правильный тетраэдр.
В этой модели для нас остаётся неясным вопрос о возможности
статистического, а не упорядоченного распределения атомов железа и
олова. Аналогичная картина, повидимому, имеет место, например, у эыаргита
(см. ниже).
638
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. IX
?v-s
4. PbSnS2 (теалит)
Решётка ромбическая ORx
Координация квазиоктаэдрическая OjO.
Тип OR-"J-SB 529 (SnS).
о/о v '
Пространственная группа Вы — Ртсп.
Z = 2PbSnS0.
РЬ к. 4. = 6S;
Sn к. ч. -= 6S;
S к. ч. = 6 (Sn + Pb).
а
OPb,Sn
Os
Во. к ест по о
Pb'SilSa
г
4,04
Ь
4,28
с | а : Ь : с
1
11,34
0,944 : 1 : 2,047
Примечание |
Сильно деформированный тип 1
NaCl.
Распределены ли атомы Sn
и РЬ статистически или упоря-
доченно—не определено;
предполагается статистическое
распределение.
I 232] СЛО'ЛШЫЬ: СОЕДИНЕНИЯ- СУЛЬФИДЫ A.^HS,,
(Ш
б. CuS (ковеллин) = Cu2CuS, -
= (CuI)a [SI CuII(SII)2]
Решётка гексагональная Н .
Координация Г/г/ЯЗД
T*nHYitiDbtJJ-SBBi8.
Пространственная группа
В\ь — СЩттс.
Z = 6.
Cul к. 4. = 3SI (равносторонний
плоский треугольник);
Си II к. 4.-SI + 3 S II =-=4
(правильный тетраэдр);
SI к. ч.=ЗСи1 (равносторонний
плоский треугольник) + 2 Си II
(ДЗ); S II к. ч. = ЗСи(0.
I'm-. '.9.'!.
Вешестно
CuS (ковеллин)
а с
\
:i,80
10,4
г/а
4,32
Примечание
По существу CuS
-комплексное соединение Cua CuS3—
= CuaS. CuS2 (см. § t>24)
Атомы S II соединяются в
радикалы S2 (d = 2,05) с к. ч.=
-6 Си (О).
Таким образом, в решётке существуют и атомы S(S I) и радикалы
(SII),.
В результате возникает набор координационных сфер: J у Cul; T у Cull;
7)3 у SI; О у групп (S2).
Стремление атомов Си образовывать с серой треугольную
координационную сферу является характерным и для других сульфидов меди.
Как это было обнаружено Н. В. Беловым и В. П. Вутузовым
(ДАН СССР 54, 721 (1946)), в 3Cu2S также наблюдается подобная
координация, встречающаяся и в тетраэдрите и других минералах меди
(см. рис. 355).
640
ч. ii. структуры нкорглничнеких инщисть
|гл. IX
6. Co2CuS„ Co2CoS4*), Ni2NiS4
Решётка кубическая Сх.
Координация О/Т/4,5.
Тип C^-SB ЯН.
Пространственная группа
Ol — Fd'An.
Z :,- 8.
Mill к. [ч.=60
(искажённый октаэдр);
МП к. ч. =40 (тетраэдр);
О к. ч.-3 М 111 + С5 М II
(линейная).
1 Веще-
стпо.
flw. . . .
ОМ..
Co4CuS4
O/Hi
105,8
Примечание
шпинели.
Со, c0s4
8,4
10't
Ni3NiS4
9,5
107
». См. иыше,
оМ,
М^-ё центрах тетраэдров
Рис. 494.
7. €u3AsS4 (энаргит)
(Cu3[S4lAs)
Решётка ромбическаяORy •
Координация тстраэдри-
ческая Г/Г/Г-
Тип ORtjtiT SBtf25.
Пространственная группа
Z = 2.
Си к. ч. = 4S(T);
As к. ч. - 4S(7');
S к. ч.3 = Си-hi As (Г).
Вещестно .
\ а . .
\ Ъ .
с
\ а: Ъ : с
Cu3AsS4
6,46
6,18
0,871 : 1 :0,832
Рис. 495.
Примечание. Тип вюртцита J54, в котором - атомов Zn заме-
3
щена на атомы As и —- атомов Zn на атомы Си. Координационные
сферы Си, As и S — тетраэдры.
*) См. стр. 485 (!).
§ 233|
СЛОЖНЫЕ СОЕДИНЕНИЯ. НИТРИДЫ
641
8. CuZnAs
Решётка кубическая С.
Координация TjTjx.
Тип Стр/х-
2 = 4.
Вещегпю
CuZnAs
«»
5,872
Примечание
Атомы цинка и мышьяка образуют
структуру цинковой обманки (ZnS) *). Атомы Си
расположены и промежутках*) (?), подобно
структуре a—Agl. См. Metallforschuiitf 1.38(1946)1
Рассматриваемое соединение заслуживает внимания в том
отношении, что при данном составе атомов не исключалось бы образование
антифлюоритной структуры, т. е. элементарной ячейки, в вершинах
и центрах граней которой расположены атомы мышьяка, центры 4
октантов из 8 заняты атомами цинка (как это вытекает из структуры
цинковой обманки), в центрах же остальных 4 октантов располагались
бы атомы Си.
§ 233. Нитриды и другие сложные соединения
1. GeaN4- = Ge2GeN4 =
-Ge2[NJGe
Решётка
ромбоэдрическая Rv.
Координация тетра-
эдрически -
треугольная TIT ft.
Тин Л г/77/ --SBS1, -
= Я18 (фенакит).
Пространственная
группа Caj — R 3.
Z = 6.
GelK.4.=4N(71);
Gell к. ч. =4N(r).
N к. ч.=20еИ +
+ К;еН(0.
Вещество
а . . .
Ge3N4
8,57±i
107°4s'
Примечание. См.
фенакит Be2Si04
Рис. 49G.
Гексагональная элементарная ячейка имеет периоды: а --=-- 13,84;
с- 9,25; с/а = 0,668; Z = 18.
*) Тип Ст/Т .
7«1 Б. Ф. Ормонт
642 ч. ii. структуры неорганических веществ [гл. IX
б) БЕСКОНЕЧНЫЕ ДВУМЕРНЫЕ СТРУКТУРЫ (СЛОИ, СЕТКИ)
§ 234. PbFCl, PbFBr
Рис. 497.
1 Вещество
PbFCl
PbFBr
а
4,09
• 4,18
с
7,21
7,59
с/а
1,7Г>3
1,81.
0
d
Pb—F
2,52
2,50
d
Pb—CI,
(Pb—Br)
3,07
3,21
Слоистая решётка —5 слоев: CI, Pb, FF, Pb, CI. 4 атома F зани-
1 2 -v- 4 5
3
мают 4 соседние вершины куба, образуя квадрат. 4 атома С1 также
подходят к атому РЬ, образуя второй квадрат. Между этими квадратами
и расположены атомы РЬ.! Так как верхний и нижний квадраты
повёрнуты друг относительно друга—возникает искажённая антипризма А.
Как и в решётке PbF2 (тип С1), каждый атом F окружён 4 атомами РЬ
(тетраэдр).
§ 235]
СЛОЖНЫЕ СОЕДИНЕНИЯ СоС122Н20
643
в) БЕСКОНЕЧНЫЕ ОДНОМЕРНЫЕ СТРУКТУРЫ (ЦЕПИ)
§ 235. СоС122Н20 и др.
1. СоС122Н20
Решётка моноклинная цепная Мк.
Координация 4 + Ц/_ = q-\- Ц/_.
ThiiJHW =M*+lu.
Пространственная группа С\ь—С21т.
Z = 2.
Со к. ч. г= 4С1 (?) + 2Н.0--■ 6 (<5); С1 к. ч. = 2Со(/).
-*~а
Со Qci ОН20
Рис. 498.
1 Вещество
СоС]32Н20
а
7-315±10
Ъ
8'544±10
С
Й,Г.81±5
В
97°30'
сх
2,468
Согласно работе Б. К. Вайнштейиа (ДАН 68, 310 (1С49), атомы хлора
образуют вокруг центральных атомов кобальта квадратную
координационную сферу с расстояниями 2,53 и 2,54 от центрального атома Со и 3,58
и 3,57 друг от друга. В каждую координационную сферу центрального
атома входят также по 2 молекулы Н20 с расстоянием Со —0 = 1,93.
Образующиеся, в результате, «октаэдрические» деформированные
координационные сферы соединяются в цепи по обоим рёбрам. Интересный
пример электронографического исследования структуры кристалла,
основанного на применении Фурье-анализа для характеристики распределения
потенциала кристаллической решётки, см. ДАН 54, 48 (1949). (Вайнштейн
и Пинскер) и др.
644 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ |ГЛ. IX
2. [Cd (NH3)2 Cl2], [Cd (NHa)2 Br2] ([Cd (NH3) X2] X,])
Решётка ромбическая цепная ORK(°K
Координация 4 -f 2/2 = г -f-1\/_ •
Тип ORK$L-SBEl2.
Пространственная группа СЦ — Стт.
Z = 2. a = [Cd(NH3)2X4].
Рис. 498*.
Вещество
Cd (NH8)t С1а
J Cd (NH3), Вг2
а
8,18
8,55
Ь
8,29
8,55
с а :Ь: с Примечание
3,96
4,13
0,987 : 1 :0,478
1 : 1 :0,483
SB IV 33
Структура Cd(NH3)2Gl2 отличается как от Zn(NH3)2Cl2, так и от
Gu (NHS)2C12(C 45); в последних (стр. 658) имеются изолированные
координационные сферы: тетраэдрическая (соответственно квадратная) —
в Cd(NH3)2 Cl2,—сплюснутые по оси 4 октаэдрические координационные
сферы iCd(NH3)2Cl4l, имеющие общие рёбра G1 — С1 и образующие
октаэдрические цепи параллельно оси с.
Таким образом, атом Cd проявил очевидную тенденцию к
увеличению координационного числа до 6; при этом имеет место весьма
незначительное изменение расстояний М —NH8 по сравнению с Zn, но сильное
увеличение расстояния М —С1. Так, Cd--Cl = 2,71 (2,31); Cd — NH8 =
2,07 (2,04); Cd-Br = 2,87 (2,38); Cd-NH3 = 2,14 (2,16). В скобках даны
цифры для соединения Zn. Таким образом, с образованием цепи атомы
С1, являющиеся общими для 2 координационных сфер, удаляются на
значительные расстояния от центрального атома, молекулы же NH3 остаются
на прежнем расстоянии от центрального атома, или даже приближаются
к нему.
§ 235]
СЛОЖНЫЕ СОЕДИНЕНИЯ AuCN, ФОСГЕНИТ
645
3. AuCN
Решётка гексагональная примитивная,
цепная Нк.
Координация Ifl.
Тип Я^.
Пространственная группа Вм — Сб/ттм
(или Clv — C6mm?).
Z = l, a = Au, p = CN.
Рис. 499.
Вещество а
AuCN
5,09
с
3,40
с,'а
Gx
1,50 1 7,30
Примечание
Г. С. Жданов и Е. А. Шугам,
Acta XX 253 (1945). См.
также стр. 693 |
Атомы золота и группы CN образуют последовательно чередующиеся
сетки, перпендикулярные оси с. Расстояние Au —N принимается равным
расстоянию Аи —С = 1,97, что значительно меньше аналогичных
расстояний в цианиде серебра (2,06). Эта особенность решётки цианида золота
вызвана, как указывают авторы работы, иовидимому, большим весом
ковалентной связи. Этим же вызвано и второе отличие этих решёток:
в решётко цианида серебра (ромбоэдрической) атомы серебра и группы CN
соседних цепей лежат, попеременно чередуясь, в одной плоскости (как
атомы натрия и хлора в соответственно деформированной решётке
каменной соли), ср. рис. 225 (NaScH).
Неясен в этой схеме вопрос о связях M-N и равенстве расстояпий
М —С и М — N (см. стр. 619, 620, а также 691 и след.).
В заключение этого раздела остановимся на структуре фосгенита,
описание которой в SB III является типичным для SB примером
совершенно недостаточного привлечения физических и химических критериев
при описании структур.
4. РЬСОз-PbCL (фосгенит)
Решётка тетрагональная Т.
Координация х\у.
Тип 7^ —SBG7B.
Пространственная группа D\ — Pb2.
Z = 2.
Вещество
РЬС03.РЬ(.1,
а
8,12
с
4,4
с/а
0,541
Примечание 1
SB III 89
Соединения типа фосгенита представляют большой интерес для
неорганической химии и теории строения вещества.
Как было давно установлено автором, совместно с Фёдоровым,
поглощение фосгена СаО идёт по уравнению:
2СаО + СОС1, = Са2С1аС03,
646 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IX
что отвечает формально СаС08 • СаС12, аналогично фосгениту. Мы знаем,
что в сульфогалите Na2S04, NaFNaCl группы S04 внедрены в решётку
NaCl. Но в элементарной ячейке фосгенита, по SB III 416, групп С03 нет,
а координационное число атома С равно 5 (Рг/4). Id (С — О) = 1,28;
4d(C—С1) = 2,88; группа С—С14—квадратное основание пирамиды.
Такая модель с к. ч. 5 для углерода и атомами на указанных
расстояниях не отвечает современным представлениям .теории химической связи.
Поэтому напрасно SB III 90 опубликовал такую структуру без оговорок.
г) МОЛЕКУЛЯРНЫЕ РЕШЁТКИ
§ 236. Общие замечания. Карбон или металлов
Проблема образования кристалла веществами, в парообразном
состоянии обладающими сильным внутримолекулярным, но слабым внешнемоле-
куляриым полем, исследована незаслуженно мало, особенно если учесть
тот теоретический интерес, который она представляет.
Здесь возможны два варианта: а) при сближении молекул
межмолекулярное силовое поле не влечёт перестройки структурных узлов—молекулы
сохраняют свой состав и симметрию, в частности —валентные углы
остаются неизменными, и б) межмолекулярное силовое поле вызывает
перестройку структурных узлов, меняя их конфигурацию при неизменном составе
или даже одновременно меняя их состав.
В первом случае возникает интереснейшая возможность
исследовать влияние формы молекул на их упаковку, поскольку можно выбирать
молекулы линейные, треугольные, тетраэдрические и т. д.
Переходя от упаковки гидридов к галогенидам, карбонильным
соединениям и т. п., можно при одной и той же исходной форме молекул
менять их внешнее силовое поле, их поляризуемость и тем самым учитывать
роль физических факторов при образовании структур этого типа.
Во втором случае силовое поле кристалла возникает за счёт
серьёзной перестройки структурных узлов. Иногда последние
дезагрегируют, как, например, РВг6 на [РВг4]+ и Вг". Иногда происходит
перераспределение их компонент, как, например, у РС16, образующего в
кристалле ионы [РС14]+ и [РС1в]~. Иногда возникает «сшивание» исходных
структурных узлов в цепи, сетки и даже «пространственную вязь». Очень
характерно в этом смысле поведение химического аналога двуокиси
углерода—молекулы COS. Тогда как С02 упаковывается в типичную
молекулярную решётку, COS за счёт добавочных валентностей серы образует
сетки, сшиваемые, возможно, и между собой. Аналогичные сетки образует
борная кислота В(ОН)3.
В зависимости от характера взаимодействия решётка перестаёт быть
типично молекулярной.
Проблема полимеризации веществ и образование пластиков
различного типа зюпосредственно примыкает к рассматриваемой более широкой
проблеме, заключающейся в изучении структур, возникающих при
сближении молекул, в характере распределения и возможности перераспределения
силового ноля. В таких веществах, как сера, всё богатство свойств
продуктов конденсации пара зависит в первую очередь от того, сохранятся ли
кольцевые или возникнут ценные структуры за счёт разрыва колец и их
замыкания в цепи.
К сожалению, структурные исследования в рассматриваемой области
чрезвычайно разрознены. Они посвящены отдельным химическим
группам веществ, йрнчём выбор объектов не позволяет сделать надлежащих
обобщений.
§ 236] СЛОЖНЫЕ СОЕДИНЕНИЯ. КАРБОНИЛЫ МЕТАЛЛОВ 647
Отметим, что в вышедшей недавно (1949) монографии 3. Г. Пинскера
«Диффракция электронов» дана, в частности, сводка весьма значительного
экспериментального материала по электронографическим исследованиям
структур молекул в парах.
Структуры многих кристаллов с типично молекулярной решёткой
рассматривались нами в разделе «Простые вещества» (N202 и т. д., а также
инертные газы), «галогениды», «гидриды» и «окислы» и т. п.
Здесь мы останавливаемся лишь на некоторых сложных соединениях.
Карбонилы металлов
С теоретической стороны проблемы устойчивости и структуры
карбонилов металлов рассмотрены автором в ряде работ —ЖФХ XI, 274 (1938);
XII, 223, 259 (1938); XIX, 167 (1945); Acta Phys. URSS VIII, 811, 848 (1938);
XI,585 (1939); XII, 159, 411, 759 (1940); XIX, 571 (1944); XXI, 409, 413 (1946),
в которых подчёркивается (1938 — 1939), что легколетучие карбонилы должны
образовывать элементы чётных групппе риодической системы. Структуры
молекул карбонилов исследованы главным образом в газовой фазе
электронографическим методом.
1. Cr(C0)e, Mo(CO)c, W(C0)e
Октаэдрические молекулы Сг — С = 1,92; С —О = 1,15;
Мо-С = 2,08; С-0 = 1,15;
W-С = 2,06; С-О =1,15 (см. рис. 502)
2- Fe(CO)5
Молекулы имеют конфигурацию тригональной бипирамиды 2)3, рис. 500;
Fe-C=l,84; С-0 = 1Д5.
О
Рис. 500.
Рис. 501.
3. Ni(C0)4
Молекулы имеют тетраэдрическую конфигурацию Т, рис. 501
Ni-C = l,82±3; С-0 = 1,15.
«Структуры кристаллических карбонилов изучены недостаточно.
648 ч. и. структуры неорганических веществ [гл. IX
4. Карбонилы Сг(СО)в,
Mo(CO)e, W(CO)e
Решётка ромбическая
молекулярная 0ДМ.
Координация х.
Тип OR*£.
Пространственная
Clv — Pria.
Z = 4. сь = М(СП),.
группа
Вещество
Г.г(СО),|Мо(СО)в| W(CO)t
11,72
0,2,
0,81'
1,77
12, "о
6/,8
11,23
1,96
И,90
6,42
И,27
Примечание. Параметры
атомов металла: (1/8, 1/16,
1/4); (3/8,9/16,3/4); (5/8,7/16,
1/4); (7/8, 15/16, 3/4)
Рис. 502.
Параметры остальных атомов не определены
(см. Z. Phys. Ch. (В) 28,351 (1935)).
5. Fe2 (СО),
Решётка гексагональная #л/(°+°).
Координация 12 = 7.
Тип Н$ - SB Fit (Fe2 (СО),).
Пространственная группа Clh — C6Jm. (^\
a = Fe., (CO)9, два октаэдра с общей гранью. ^^'
Z = 2/
а к. ч. = 12а.
Вещество
Fe> (CO).
а
6,45
1 /
с с/а
15,98
2,4?75
SB VII 28, 131
Положения центров узлов а отвечают
плотной гексагональной упаковке.
Рис 503,
§ 236]
СЛОЖНЫЕ СОЕДИНЕНИЯ. КАРБОН11ЛЫ МЕТАЛЛОВ
649
Молекула Fe2 (C0)9 имеет горизонтальную плоскость симметрии,
на которой лежат под углом 120° друг к другу три СО-молекулы. Над
плоскостью симметрии и под ней лежат атомы Fe, с каждым из которых
связаны ещё по 3 молекулы СО. В целом, вокруг каждого атома Fe
возникает почти октаэдрическая координационная сфера из 6 атомов
углерода.
Расстояния в пределах молекулы: Fe — Fe = 2,46; Fe — С = 1,19*
Fe- С=1,8; С -0 = 1,15; С —0 = 1,3.
Валентные углы: Fe-C - Fe = 87°; О —С —Fe = 94°; С-Fe —С = 78°.
Бросаются в глаза исключительно малые углы С — Fe- С (78°).
6. Fe(C0)4
Решётка моноклинная М (псевдотетрагональная).
Тип JI/-SB-?
Пространственная группа СоЛ ~-С2/с.
Z=12.
Вещество
Fe (C0)4
а
13,00
Ь
ll,U
с
р
и /a 85°35;i
I
Примечание
SB II 373.
Возможно соединение
3 молекул [Fe (C0)4],
7. Fe(C0)4CI2, Fi(C0)4Br2, Fe(CO)4J2
Исходя из термодинамических и спектроскопических данных, автор
рассмотрел возможные структуры координационных сфер в решётке
карбонилгалогепидов и пришёл к
выводу, что предложенные зарубежными
исследователями структуры тина
[Fe (СО)4]2^ Х2 термодинамически
неустойчивы; гораздо выгоднее с
термодинамической стороны структуры типа
[Fe(CO)4X.,] (рис. 504). См. Acta XII,
411(1940).
. Транс
8. Fe(C0)4H2
Допускается тетраэдрическое
расположение молекул СО вокруг
центрального атома и наличие групп «СОН». При
этом Fe-СО-1,84; Fe- СОН =1,79;
С-0 = 1,15. (См. Со(СО)4Н.)
цис
о
Fe
Cl
Со
Рис. 504.
9. Со(СО)4Н
Допускается тетраэдрическое расположение СО вокруг центрального'
атома и наличие групп СОН. Отсюда вытекают расстояния Со -СО = 1,83;
Со-СОН = 1,73; С-0=1,15.
10 Fe(CO)4Hj. и Со(С0)4Н (см. Trans. Far. Soc. 35,681 (1939)). Трактовка
структур Fe(C0)«H„ и Со(СО)4Н требует обсуждения.
Из этой трактовки вытекают формулы Fe (СО)а (СОН)2 и Со (СО), (СОН),
как если бы к центральному атому металла присоединялись группы—С—Н
II
О
или С-О-Н.
Первый вариант, повидимому, отпадает, ибо центральный атом не смог
бы заполнить при этом все свои валентные dsp-уровни, получив от групп
650 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IX
СОН слишком мало электронов. Второй вариант более приемлем. Не
исключена, однако, структура, в которой атом Н связан непосредственно с Fe.
Впрочем, учитывая свойства этих веществ в растворах, можно полагать
(но не обязательно, см. § 75 и 90), что допустимы формулы с заметным
весом ионного состояния IFe(CO)4]H+H [Со(СО)4]~Н+.
§ 237. Металлсорганичеекис соединения
Остановимся на структурах некоторых металлоорганических
соединений.
Структуры молекул
1. А1(СН3)з
Структура является объектом полемики. Рассматриваются структуры
типа (Al(CHt)t)a. См. Nat. 156, 601 (1945).
2. Si(CH8)4, Ge(CH3)4, Sn(CH3)4, Pb(CH8)4
Электронографическое исследование показало, что молекулы тетраэдри-
ческие. В свою очередь 3 атома Н окружают атомы С, по видимому, также
под тетраэдрическими углами.
Расстояние Si-C = lf93; Ge-C = l,98; Sn-C = 2,18; РЬ-С-?
3. N (СН8)8
Электронографическое исследование паров N (СН8)а показало, что
молекула «пирамидальна» (т. е., с нашей точки зрения, тетраэдрическая, см. § 91).
Угол C-N = 108°±4°.
Расстояние C-N = l,47±2.
4. As(CH3)8
Электронографические данные позволяют считать, что угол С —As —С
равен 96°.
5. 0(CH8)2, S(CH3)2
УголС-О-С равен 111±4°; С-0 = 1,42±ЗА.
Угол C-S-C лежит между 100° и 110°. C-S = 1,82 ± <3 А.
Структура кристаллов некоторых металлоорганических
соединении
Ранее нам приходилось сталкиваться со случаями образования
кристаллических структур тетраэдрическими молекулами. СН4, как и SnJ4,
образовывают кубические структуры, причём во втором случае вращение молекул,
повидимому, совершенно исключается.
Среди комплексных соединений также имеется ряд примеров
образования веществ тетраэдрическими ионами (см. гл. X). Наряду с образованием
тетрагональных, ромбических и других структур, что связано с
определённой ориентировкой ионов, известны и случай возникновения кубических
структур, как, например, [NHJ+ [ClOJ^, тип Со/о,что обычно связано либо
с вращением, либо со статистически различной ориентировкой ионов
по отношению к осям кристалла.
Так как возникающие кристаллические модификации зависят от
способности силового поля кристалла помешать вращению структурных узлов,
то особый интерес приобретает исследование зависимости структуры от
температурного фактора, от давления, а также от распределения зарядов
внутри структурных узлов и т. п.
В этом отношении особый интерес вызывают металлоорганические
соединения. Среди них имеется, в частности, широкий класс соединений с
тетраэдрическими молекулами, начинающийся соответствующими соедине-
§ 237] СЛОЖНЫЕ МЕТАЛЛООРГЛНИЧЕСКИЕ СОЕДИНЕНИЯ 651
ниями углерода. Многие из этих веществ вполне устойчивы, чтобы быть
использованными для рентгеносъёмки.
С теоретической стороны наиболее интересно сопоставление структур
соединений разных элементов —углерода, кремния и т. п. с наиболее
короткой боковой цепью — с радикалами СН3, С2Нб и т. п., поскольку значительное
нагромождение этого рода групп должно затушёвывать специфику,
вытекающую из свойств центральных атомов этих веществ. В действительности
же основной имеющийся в нашем распоряжении материал, к сожалению,
охватывает только свойства фенильных и ещё более сложных соединений
(за немногими ис-
типа
ключенинми,
С(СН,)4).
1. С(СН3)4 (тетраме-
тилметан)
Решётка кубическая
молекулярная См.
Координация Т.
Тип С?*-SB 02.
Пространственная
группа 07h — Fd3m.
(?) (по SB I614,
алмазная решётка из
молекул [С(СН3)4].
Z — 8
а~[С(СН,)4].
аш=11,2у.
2. [С(С,Н,)4],
[Si(C,H$)4],
[Ge(C,H6)4],
[Sn(C6H5)«l,
[Pb(C.H6)4j
Решётка
тетрагональная Tf
центрированная.
Координация Р±.
Тип TPi-SB Об.
Пространственная
группа i>|d—P4 2/.
а=С(С6НБ)4 и т. д.
Симметрии молекул
С:(000;11|).
Молекула, проекция которой помещена в центре, занимает позиции-^-^ -^
й«ш
0 12 3 4 5 6 7
1 I ' : I ! L
8 9 W 11 12
• М
Ос"
Рис. 505. По Г. С. Жданову и И. Г. Исмаилзаде.
Si (тетраэдрическая конфигурация). Z = 2.
ill
\ а . . .
\ с . . .
10,8б
0,60,
Si(C6H5)4*
11,30
7,08
0,620
Ое(С6Н5)4
и,б0
6,8.
0,590
<VI,)4*
11,85
6,65
0,562
РЬ(СвН6)4*|
12,03
6,55
0,544
Звёздочкой ' отмечены соединения, по которым приводятся новые
данные, по работе Г. С. Жданова и И. Г. Исмаилзаде (ДАН 68, 95(1949).
652 ч. ii. структуры нкорглнических вещкстб [гл. IX
В этой работе, существенно дополнившей и исправившей старые
данные SB I 615 и ел., были определены также углы ср поворота молекул
M(R)4 по отношению к осям х (или у), а также углы ty поворота фениль-
ного кольца около связи М —С:
Вещество
Si (CeH5)4
Sn (СвН5)4
РЬ(С8Н,)4
?
8°
8°
5°30
+
—37°
—42°
—50*
ш*>
Этими данными опровергаются предположения (см. SBI 615) о том,
что для всех тетрафенильных соединений угол <р = 45°.
Кроме .того,
авторы пришли к
выводу, что не исключена
вероятность того, что
углы S\( равны
104°.
3. [Sn(C.H,CH,)J,
[Sn(C,HI0CH,)4]
Решётка
тетрагональная Т,
центрированная.
Координация Pi.
Тип Тра.
Пространственная
группа SJ—7~4 (?)
Z = 2
a = Su(R)A.
\1*
0 1 2 3 4 5 6 7 В 9 10 11 12 13 14 Г\ р
; I I м i i i I I i i i ■ i W °
Рис. 506 (по Г. С. Жданову и И. Г. Исмаилзаде).
Вещестпо
Sn(CeH6CH3)4
Sn(CeH5OCH3)4
а
13,47
14,3
с
6,35
6,5
Н
1,372
1,356
*
14°30
* 1
—42°
Узлы а образуют тетрагональную центрированную элементарную
ячейку. Эти соединения были синтезированы в лаборатории К. А. Коче-
шкова и исследованы в структурном отношении Г. С. Ждановым и,
И. Г. Исмаилзаде (ДАН 68, 95 (1949)).
§ 237] СЛОЖНЫЕ МЬ:ТАЛЛООРГЛНПЧ1'ХКН1£ СОЕДИНЕНИЯ 653
4 (С«Н4ОС2Н,)4
Решётка моноклинная М.
Координация ?
Тип Л/?
2 = 4.
Вещество
(С,Н,ОС,Н,)4
а
19,8
Ь
13,9
с
В
1
13,1 !123,5Р
°х
1,352
(По работе Г. С. Жданова и И. Г. Исмаилзаде). Структура
окончательно не установлена.
5. [С(СН2ОН)4], (нента-
эритрит)
Решётка тетрагональная
Т, центрированная.
Коордивация Pi.
Тип 7>4-SB 04.
Пространственная группа
Z = 2.
л-[С(СН2ОН)4].
D
С*
Рис. 507.
Вещестно i a \ с
С (СНаОН)4
6,0t j 8,7,
cfa
1,46
Примечание
По SBI.
Ранее это соединение неправильно считалось относящимся к иному
виду симметрии.
6. As(C.H6)3, Sb(C6H6)3, Bi(C,H6)3
Первые две структуры относятся к триклинной сингонии класс С{.
Bi (СвН5)3 в соответствии с пространственной группой C*2h относится
к моноклинной сингонии (Z. Krist. 102, 305 (1942)).
654 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IX
7. (AsCH3), (арсенометан) и (AsCHa)5
С давних пор предполагалось, что арсенометан образует структуру
типа H3CAs = AsCH3. Недавно арсенометан подвергся рентгенографическому
и электронографйческому исследованию, причём было обнаружено, что могут
быть выделены две «модификации»: жёлтая маслянистая (вязко жидкая)
и вторая—красно-коричневая (твёрдая), см. J. Am. Ch. S. 67, 2014 (1945).
Первой отвечает состав, близкий к (AsCH3)3, второй—близкий к (AsCH3)6.
Расстояния As—As равны 2,42, расстояния As—С равны 1,98. Углы
As— As —As равны примерно 90°.
В пентаполимере возникают складчатые пятичленные кольца, структура
которых в работе не приводится. Образовать пятичленное кольцо со строгим
соблюдением равных межатомных расстояний и углов, равных 90°, трудно, и,
вероятйо, имеют место небольшие нарушения. Напротив,шестичленное коль-
цо легко укладывается в схему.
С каждым из атомов As
связана I группа СН3.
Таким образом, структура
типа H3CAs = AsCH3 не
подтверждается.
8. [Pt(CH3)3CH3]4 = Pt(CH3)4,
[Pt(CH3)3Cl]4 = Pt(CH3)3Cl
Решётка кубическая С.
Координация С.
Тип Сс.
Пространственная группа
П— Шт.
Z = 2.
а*= [Pt (CH3)4]4,
соответственно [Pt(CH3)3Cl]4.
а к. ч. =8.
Рпс. 508.
На рис. 508 структурный ззел—тетрамер ( = а). Элементарная ячейка
/11 1 \
содержит такие узлы в (000), ( — -у) , т. е. относится к тину Сс.
Вещество
Примечание
Pt(CH3)4
Pt(CHa)8Cl
10,45
10,55
По работе Рэндла и Стэрдиванта,
J. Air.. Ch. S. 69, 1561 (1947).
Оба вещества являются тетрамерами. Молекула тетрамера образует
несколько искажённый куб, вершины которого в тетраэдрическом
порядке попеременно заняты узлами [Pt(CH3)3] и [СН3], соответственна
[G1]. В первом из этих соединений мы имеем, судя по данной работе,
единственный пока найденный пример группы [СН8], углерод которой
связан более чем с одним атомом, находящимся вне группы СН3. Этот
атом углерода имеет 6 октазд рических связей под углами, близкими к 90°
друг к другу, как TiG (тин С0/0). В хлориде расстояния в молекуле
тетрамера равны: Pt-Pt = 3,73; Pt-Gl = 2,48; Gl—CI = 3,28; угол Pt-Cl-Pt=99°.
В тетраметилнлатипе расстояние Pt — Pt = 3,44. Эта работа является,
с нашей точки зрения, лишь началом серии исследований, которые
должны быть предприняты с целью проверки и объяснения структурных
особенностей этого рода веществ. Рассуждения же но этому поводу,.
§ 2381 СЛОЖНЫЕ СОЕДИНЕНИЯ. COS, Ы3ВОз, llg (CI Br)2 655
изложенные в статьях Паулинга и Рэндла(СЬ. Eng. News (1947), J. Am.
Ch. S. 69,1327(1947)), нам кажутся преждевременными (впредь до синтеза
и исследования аналогов этих соединений с иными радикалами и
центральными атомами Pt — Pt и других элементов), и в большой мере
спорными. С точки зрения теории химической связи, скорейшее
накопление фактического материала по подобным веществам является, на
наш взгляд, очень существенным.
Заканчивая раздел, посвященный металлоорганическим соединениям,
мы должны вновь подчеркнуть, что исследователи структур этих веществ,
делая крен в область веществ сфенильными и более крупными радикалами,
содержащими 10-15 атомов в каждом, напрасно обходят соединения с малыми
радикалами, которые позволили бы выявить влияние строения центральных
атомов в гомологических рядах (углерод —кремний— германий и т. д. или
азот—фосфор —мышьяк и т. д.) на свойства молекул металлоорганических
соединений и на структуру образуемого ими кристалла. В фенилышх
соединениях свойства центральных атомов будут проявляться гораздо менее чётко.
Переход к метильпым, этильным и тому подобным соединениям требует
в отдельных случаях преодоления значительных экспериментальных
трудностей, не могущих, впрочем, явиться препятствием при современном
состоянии техники эксперимента.
1. COS
Решётка
ромбоэдрическая,
молекулярная, цепная
(слоистая) RM~S.
Координация
/( + *)//( + * 4-1)//.
Тип Aft7/f-SBF0a.
Пространственная
группа Civ — R3m.
• с
ч , 2 ? f ? О
Рис. 509.
С к. ч. = IS (1,97) + 10(1 ДО)(/) (+ 3S(3,57) (t) = l( + t);
S к. ч. = - 1С (1,97) +10(2,79)(/) (+ 3С (3,57) (t) + 10 (3,07) = I ( +1 4-1);
О к. ч. =: 1С (1,10) + 1S (2,79) (/) ( +1 S (3,07)).
1 Вещество
COS
а
4,08
а
98°58'
Примечание
SB II 62—63
i
§ 238. COS, H3B03) Hg(ClBr);
,656 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. IX
Решётка состоит из линейных молекул OCS(dC-o = i ,10; dc-s = l»97).
Эти молекулы сшиты в линейные цепи связями S —О: OCS. , .OCS.. . OCS.
Jso = 2,79kX. Цепи параллельны друг другу, причём атомы С одной
цепи как бы соединены с атомами S трёх соседних цепей. В результате
получаются перпендикулярные осям цепей горизонтальные сетки из
атомов С и S
)C-S
s< >c-s
yes'
соединённые с 2 соседними сетками, «как в BN» — замечает SB II 63.
По этому поводу нам кажется уместным подчеркнуть, что роль атома S
в сшивании молекул в цепи и отчасти цепей в структуру вполне понятна;
однако перегрузка в представленной SB II схеме атома С тремя кова-
лентными связями в плоскости колец и двумя связями по оси цепи (а
всего 5 связями) бросается в глаза. При этом 3 расстояния /iC-s = 3,57kX
в плоскости сетки слишком велики для химической связи C-S и на 1,6кХ
превышают прочие расстояния С — S в структуре. Поэтому аналогия с BN
не обоснована.
2. 113В0Э (борная кислота)
Решётка триклинная Тг .
Координация tj2-\-1.
Тип Trfw-SBGWPO,). ч
Пространственная группа С\ — Pi.
Z = 4.
В к. ч. = 30 (равносторонний треугольник).
О к. ч.-=2Н + 1В.
Вещество
н3во3
а
7,04±4
Ь
7№±4
с
6,50 ±4
а
92о30'
Р
101°10'
т
120°
а : Ь : с
1,00: 1,00 :0,9318
SB III 87. В пределах: элементарной ячейки атомы лежат в 2
плоскостях, параллельных (001); расстояние между плоскостями равно 3,18 кХ
(рис. 510). Атом В окружён в данной плоскости 3 атомами О. Атомы
кислорода, принадлежащие разным группам (В03), соединены друг с
другом чороз атомы Н, лежащие посредине линии связи О —О на расстоянии
1,35 от атомов О (рис. 511).
Сетки (плоскости) соединены друг с другом силами Ван-дер Ваальса
do_H = 1,355А, еВ-о = 1,36А; /в-о (в разных сетках) = 3,19А.
3. Hg(Cl, Вг)2
Имеются указания (Z. Кг. А. 103, 415 (1941)), что ото вещество имеет
ромбическую молекулярную ячейку ORMW с линейными молекулами
Hr-Hg-Gl; а = 6,78; 6-13,17; с = 4,10; а : Ь : с = 0,515 : 1: 0,301; Z = 4.
Пространственная группа ^2^2,; Hg—Cl = 2,24; Hg—Вг — 2,44,
§ 238) СЛОЖНЫ!; COI ДШШШШ. COS, Н3В03, Hg(ClBr)2 657
Pm\ ПИ.
Положение протонов по середине значительного расстояния О — О —
----=3-71, к X, принятое Захариасеном, нам представляется дискуссионным.
Моишо было бы ожидать смещения протона к атомам О, в духе модели
<чВ» «С» для [Ш20 (стр. 441 и след.).
658 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТИ [ГЛ. IX
§ 239. Кристаллосольваты
1. СиС12 2Н20
Решётка ромбическая
молекулярная ORM&.
Координация /.
Тип 0Д-у(?)—SBC45.
а = [СиС12>2Н20].
Пространственная группа Dm —
— Pbrnn.
Z = 2.
Вещее тв и
а
Ъ
I с
а : b : с
[СиС1,2НаО]
7,33
8,04
3,72
0,918 : 1 :0,462
Рис. 512.
Си С1 2,31; Си -Н,0 = 2,01.
2.[Zo(NH,)1ClJ;[Zn(NHi), Br2]
Решётка ромбическая
молекулярная ORM(f)-
Координация РЗ + г.
Тип OR^l-SB El2.
Пространственная группа
Dl\~Jmma.
Узлы a = [M(NHj,)2 X2]
(изолированные тетраэдры, не
имеющие общих элементов).
Z = 4.
I Вещество
[Zn(NH3)2Cl2]
[Zn(xNH3),Br2|
а
7,78
8,12
Ъ
8,50
8,81
с
8,08
8,41
Рис. 513.
а : b : с
0,915: 1 :0,051
0,922 : 1 :0,954
Примечание 1
SB IV 31 1
Сравни с
С(1(Ш3ЬС]2,
стр. 644 |
Хлорид Zn-NH, 2d = 2,04; Zn-Cl 2е = 2,30.
Бромид Zn — NH, 2d = 2,16; Zn —Br 2e = 2,38.
Позиции Zn:(000); (i- -| j) + 4Zn в (о y{), (О, j-y, |) C2/Zn = 0,139.
§ 240] СЛОЖНОКОМПЛЕКСН. СТРУКТУРЫ С БЕСКОНЕЧН. ТРЕХМЕРНЫМ ИОНОМ 659
3. P4N4C18
Решётка тетрагональная
молекулярная TMJmK
Координация />4.
Тип Tf4(m)~-SBE1<{P4N4C16).
Пространственная группа С\и —
-P4Jn.
a = [P4N4Gl8].
Z = 2.
як. ч. -.8я(.Р4)(?).
Рис. 514.
Вещество а
P«N4C18
10,82
с
5,95
с/а
0,550
Примечание 1
SB VII 17,128 1
Структура близка к типу Тр4 и состоит из 8-атомных складчатых колец с
чередующимися атомами Р и N. Атомы Р сами по себе расположены в
вершинах тетрагонального тетраэдра. Атомы N также расположены в
вершинах тетрагонального тетраэдра (рис. 514, на котором показана одна молекула
P4N4G18). Каждый атом фосфора соединён, кроме того, с 2 атомами
С1. Угол G1-P—С1 = 105°30'; угол N—Р—N = 117°; угол Р—N—Р=123°.
Следует учесть, что атомы Р в данном окружении должны стремиться
к осуществлению угла 109° 28', а атомы N—угла 120°, так что
реализуемые углы являются компромиссными.
Б. СТРУКТУРЫ СЛОЖНОКОМШШССНЫХ СОЕДИНЕНИИ*)
§ 240. Структуры с бесконечным трёхмерным ионом
СаТЮ3 перовскит и др.
В SB I 300 дано подробное описание типа перовскита СаТЮ3 (G5 — E2i),
чрезвычайно важного и очень распространённого структурного типа,
охватывающего также иные группы соединений, например: NaNbOs, YA103,
CaSn03, KJ08, KMgF8> KZnF3, CsCdCl3 и очень много других. Типу
приписывалась кубическая решётка; пространственная группа 0\ — РтЗт; Z = l.
Са(0 0 0), Ti (V, V. V.). О (V. V. ОС)-
Эта структура может быть интерпретирована таким образом,
что октаэдры [ТЮв] всеми своими вершинами соединены друг с другом,
образуя бесконечный трёхмерный анион, в междуузлиях которого
расположены атомы Са, имеющие, таким образом, кубооктаэдрическую коорди-
*) Напоминаем, что в отличие от аналогичных структур сложных соединений, у
сложнокомплексных имеются изолированные структурные узлы—ионы—вне
бесконечных цепей, сеток и т. п. Например, в структуре Cd(NH3)2Cl2 (стр. 644) существуют
только бесконечные цепи (сложное соединение). В структуре NH4CdCl8 (стр. 666)—
ионы [NH]4+ лежат вне бесконечных цепей из октаэдров [СаС1]в.
660 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гл. IX
национную сферу из атомов кислорода (рис. 515). Эта же структура может
быть, но лишь чисто формально, представлена как плотная кубическая
упаковка из атомов кислорода и кальция, в октаэдрических междуузлиях
которой расположены атомы титана. Та же структура часто отображается
с атомами Са в центре ячейки. В этом случае хорошо видна
координационная сфера из 12 кислородных атомов вокруг атома Са (рис. 516). К. ч. атома
кислорода = 2 Ti + 4Са = 6
(октаэдр). Таким образом,
координация: К\0\0 и тип
можно было бы
обозначить Ск/о/о. Так как связи
не являются чисто кова-
лентными, то часто
говорят об ионах названных
элементов и, в частности,
беконечная группировка
из октаэдров [Ti06], неси
на себе заряд, является
ионом. Удалив атомы из
центру ячейки рис. 516,
получаем структуру ReO.^
(рис. 336).
В последующих томах
SB сохранилась
интерпретация типа СаТЮ3 как
кубической структуры,
объединяющей множество веществ (см. SB III 68). В SB VII,
последнем вышедшем томе серии, прореферирована без комментариев
редакции работа, дискутирующая вопрос о необходимости удвоения
ребра кубической ячейки СаТЮ8 (7,645 A SB VII 120 вместо 3,80 А
SB 1303). В той же работе
высказывается предположение, что не-
ровскит имеет псевдокубическую, в
действительности же ромбическую
или даже моноклинную структуру
(см. рис. на стр. 121 SB VII).
В последние годы, однако (см. Nat.
155, 484 (1945) и Ргос. Phys.
Soc. 58, 133 (1946)) было показано,
что СаТЮа и ряд других веществ,
отнесённых ранее к кубическому
типу перовскита, не являются
кубическими. При этой ревизии
положение очень быстро менялось. Так,
сначала было найдено, что СаТЮ3
является моноклинным (а ^ Ь ъ с ъ*
^7,60А), так же как и PbZrO,,
CdSnOa, PbSnOa, РЬТЮ3, но ВаТЮ8
(яш = 3,98), BaSnOs (aw = 4,10),
BaZr03 (4,19) —оставались среди рис. 516.
кубических.
Позже в работах, цитированных выше, было указано, что
структуры, относящиеся к этому типу, являются лишь псевдоизоморфными.
Например, SrTiO, кубический, а ВаТЮ8 тетрагональный а« 3,9866.
с - 4,0195 при 22°.
§ 210J СЛОЖНОКОМПЛЕКСН. СТРУКТУРЫ С БЕСКОНЕЧН. ТРЁХМЕРНЫМ ИОНОМ 661
Наконец, было показано (Nat. 155, 484 (1945)), что ВаТЮ3
тетрагонален при 20°С, а = 3,9860; с = 4,0263; с/а -1,0101, но при 200JC ВаТЮ3
является «кубическим» с а = 4,040.
В соответствии с этим тип иеровскита, повидимому, распадается наряд
структур: 1)кубичсская$тТ103, SrZr03, BaZr03, SrSn03, BaSnOs; 2)
тетрагональная BaTi03, РЬТЮ3, PbZr03; З) ромбическая CaTi03 (?), CdTi03,
CaZr03, CaSn03 (особенно при высоких температурах); 4) моноклинная,
повидимому, часть ромбических соединений п. 3) при низких температурах.
Вопрос нуждается в дальнейшем уточнении, причём пам кажется
очень важным исследование температурной зависимости метрики ячейки
(в SB VII дано удвоенное значение «аш»).
Сказанное не меняет основного характера структуры — бесконечный
трёхмерный анион, из правильных или деформированных ТЮв-тетраэдров
с вкрапленными в пего ионами Са2+.
Ilcei доизомонфные соединения тииа ие[ овекита
Группа
|пери>ди-
I ческой
Соединении
1 системы |
И ; KM^F8
X KNiF3
II** ! KZ11F3
i
III**
BaC103
ВаРгО,
IV*
V*
СаТЮ3
SrTi03
BaTiO.,
CdTi03
CaZr03
SrZrO,
BaZr03
SrHf03
BaThO,
NaNb03
KNb03
air
4,0Q
4,008
4,05„
i |
' i
4,377 '
4,354 !
3,80
3,890
3,971
3,75
3,99
f
1 f
j-т
) и
I*
4,088
4,177
4,069
4,480
3,89
4'°1
Примечание
Группа
периодической
системы
Соединении
«w
Примечание
Галоген иды 1
SB I 00
»
»
[1**
»
CsCdCl3
CsIIgCl,
5'20
5/*n
' 0
S \ I 300
» 1
Окислы 1
SB 1 ::oo
Bill О*
етраго-
альная
= 3,9860
-4,0263
III
IV
YA103
I-aAlOj
IaGa03
Га5п08
Ca-rO.,
' *,ы
*'7« ; |
3,s9 '
3,92
4.025
i !
VII
NH4J03 ! 4,5L
NaJ08
KJ03
RbJ03
CsJ03
'<\
f'A, 1
4,662
1
.'SB I 300, I
\ 339
I SB II 333
( Ромби- 1
ческая; 1
а = 5,75;
6=6,3;
j 0=4,05.
^ Простран* 1
1 ственная 1
группа ПЦ
{ —Сттпъ 1
1
В связи с I
соедине- 1
ниями 1
M[J03|
см. гл. X. 1
662 Ч- П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТИ |гл. IX
LiJ03( = Li2[OJJL)
Решётка гексагональная Ilv~.
Координация О /О/р.
Тип Яо/оур- SBE23 (Li J О,).
Пространственная группа Z>e —С632.
Z = 2.
Li к. ч. = 60 (почти правильный октаэдр) + 3J (равносторонний треугольник);
J к. ч. = 60 (почти правильный октаэдр)+ 3 Li (плоский треугольник).
О к. 4.=2Li + 2J-4(/>).
0 12 3 4 5 О О
Рис. 517.
Вещество
LiJO,
а
с
5,4б9 5,15.
с,'а
0,942;
Близок к типу перовскита СаТЮ3. В отличие от
кристаллизующегося в типе перовскита KJ08, у которого К имеет к. ч. = 120(К), в
структуре LiJ03 для Li к. ч.=60 (О). В равной мере отличаются к. ч. для О.
В случае KJ03 оно равно 6, в случае LiJ03—4. Никаких
изолированных [JOJ координационных сфер в решётке нет, см. SB II 40.
Пример сложнокомплексных структур, в которых октаэдры [J06]
образуют бесконечный анион (пространственную вязь), в междуузлиях
которой внедрены катионы Li+. Сравни с иодатами К, Rb, Cs.
§ 241] СЛ0ЖН0К0МПЛЕКСН. СТРУКТУРЫ С БЕСКОНЕЧН. ДВУМЕРНЫМ ИОНОМ 663
§ 241. Структуры с бесконечным двумерным ионом (слои, сетки)
1. NH4HgCI3
Решётка тетрагональная, сетки Ts.
Координация Di/Pi/Pi
Тип Tf>4/P4ph - SB Е2Ъ (NH4 Hg Cla).
Пространственная группа Cib — Pbjm (?)
Z-1.
Hg к-ч. = 4С1(2,96) + 2С1(2,34)(сжатый по оси с октаэдр— бипирамида D'A.
NH4 к. ч. = 8Cl(3f38A)(/>4)-f8Hg(/>4).
jNHJ к. 4.-,8[HgC]6l (P4); iHgCIJ к. ч. -----8[NH4] (7>4).
Рис. .')18.
Вещество
NHtHgCl,
"
4,19
с
7,94
с/а
1,895
Примечание
SB VI 15.
(Сжатые [HgCle] октаэдры (т. е.
тетрагональные бипирамиды, центры
которых (Hg-атомы) совпадают 1
с нершинамп элементарной ячей- 1
ки, образуют сплошные сетки,
соединяясь 4 рёбрами. В центре
элементарной ячейки находятся
NlIrHOI!bI С К. ;т.~*. j
i
Представляет интерес сопоставление этой структуры со структурой
Nll.CdCl, (стр. 666). ' " '
664 ч. и. структуры неорганических веществ 11 л. IX
2. T1A1F4, KAIF4, BbAlF4, NH4A1F4
Решётка тетрагональная слоистая Ts.
Координация OjPi/r^/Pyi.
Тип Tojpbir+pyi — SBHOe(TlAlF4).
Пространственная группа D\h — Pb/mmm.
Z = l.
А1 к. 4. = 6F (октаэдр);
М к. 4. = 8F(P4)( + 8F(/>4) = 16F) (тетрагональная призма + топ -рагпигкчъ-
ная призма больших размеров);
01 к. ч. =4М (прямоугольная) + 2 А1 (линейная)=6(г+).
ОН к. 4. = 4M(j) + lAl = 5(Py4).
Рлс. 519.
Вещество
T1AIF4
KAIF4
RbAlF4
NH4A1F4
а
3,61
3,550
3,615
3,587
с
6,37
6,139
6,261
6,346
Примечание
См. § 224 и Б. Ормонт, ЖФХ ХШ
646 (1939), Acta XI, 87 (1939) SB V 17,
SB VI 18,95
Октаэдры [AlFe] имеют по 4 общих
вершины и образуют сетки,
параллельные основанию призмы
§ 241] СЛОЖ1ЮКОМ11ЛЕКСН. СТРУКТУРЫ С БЕСКОНЕЧН. ДВУМЕРНЫМ ИОНОМ 665
3. KH2Al3Si3Ol2(KAl2[Si3 + Al]010[OH]a), (мусковит)
Решётка моноклинная слоистая Ms.
Координация O/TjPG.
Тип Д/о/7/Рб—SBS&! (мусковит).
Пространственная группа С^х -
Z = 4.
А1 к. ч, = 6 0(2,15) (октаэдр)+ 6Si (3,75) (октаэдр);
Si к. ч. = 4 О (1,60) (тетраэдр);
К к. ч.= 12 О (3,15) (гексагональная причма).
-С2/С
Вещество
КН,.\1,Я|,Оц
а
5,18 [
Ъ
У, 02
1
20,0. ;
-
')5°;Ю
а :b:c I
0,5743 : I : 2,217
SB II 143. Сложпокоор-
динационная решётка.
Атом К образует
координационные сферы
[К012], т. е. Р6,
находясь между двумя гас-
стичленными кольцами
(рёбрами) тетраэдров
ISiOj.Тетраэдры [Si04]
сшигы вершинами в
плоские сетки, соеди- Q
ценные между собой
сетками К и А1. Атомы А1,
в свою очередь,
образуют октаэдры [А10в],
так что один слой
октаэдров (А10в) и два слоя
тетраэдров (Si04)
образуют как бы
бесконечный трёхмерный пояс
параллельно осям anb
и ограниченный 6А по
оси с. Интервал
кратчайшего межатомного
расстояния А1 —О и
Si—О в мусковите
равен: еЛ1-о = 2,05А;
dsi-o= 1,60А (против
А1-0- 1,93 (А1,Оэ)
и Si —0= 1,59 (Si02)
кварц),т. е. связь А1 — О
ослаблена у слюд
довольно значительно.
Структурная
формула отличается от
химической тем, что часть
атомов Si беспорядочно
замещена на атомы А1,
т. е. КА12 [Si, 4- А1]
О10(ОН)2.
fr/J>? л&
666
ч. п. структуры нкорглнпчнеких шещкетп
[гл. IX
§ 242. Структуры с бесконечным одномерным ионом (цепи)
1. NH4CdCl,
Решётка ромбическая
Координация 8/8.
Тип ORt^ .
Пространственная группа
Dfh —Pnma.
Cd к. ч. = 6С1 (2,65)
(октаэдр).
a = [NH4]
а к. ч. = 4С1 (3,27) +.
+ 5С1(3,34-3,82).
Cd к. ч.^8а(8);
а к. ч. ^8Cd.;
а
0 123456789
I L_J __.l_J I l__l L i
О щ
CL-FwpiLLWb/ сшаздроВ
Рис. 521.
Вещество
NH4CdCl3
a
8/J6
b
14,87
с
3,97
a : Ь : с
0,60) : 1 : 0,267
Примечание
1
Октаэдры
соединены друг с
другом
противоположными рёбрами,
образуя столбы
вдоль оси с%
связываемые друг с
другом
[Шпионами.
SB VI 13—15.
Сравни с NH4HgCl3, стр. 663.
§ 242] СЛОЖНОКОМПЛЕКСН. СТРУКТУРЫ С БЕСКОНЕЧН. ОДНОМЕРНЫМ ИОНОМ 667
2. Т1аА1РБ
Решётка ромбическая ORK(°\
Координация 0/6J8.
Тип <?*£,$ -SBtf3,(TltAlF,).
Пространственная группа Д> — С222 .
Z = 4.
А1 к. ч. = 6К (слегка деформированный октаэдр) ( + 8 Т1);
Т1 к. 4. = 6F( + 8A1);
F к. ч. = 2АЦ-4Т1 = 0.
Ряс. Г>2.\
Вещество
T1,.\IF5
*
а
10,0G
ъ
8,2'*
с
7,46
d
А1 — К
Tl—F
4<*=1,КУ~ j 2e --=2,К8
•' 2,'=2,90
2d' = l,94 i2-" = 2,93
i
Примечание
ЬВ V20—21 1
См § 224 и Б. Ормовт,
ЖФХ XIII, 046 (1939),
Acta XI, 87 (1939)
Октаэдры [AlFe]
имеют по 2 общих
вершины и образуют цеии
пдоль оси с
Позиции 4А1 в (Ь): (00|), (00^), ({- 1 -1) и (-1 ~ -J). 8 Т1 в (С): (000;
На рис. 522 (SB V) границы элементарной ячейки не показаны.
5 «столбов» из двух октаэдров каждый расположены таким образом,
что центры октаэдров—атомы А1—лежат (у 4 периферических столбов)
на рёбрах элементарной ячейки на высоте 7* н 3/4 от е^ основания, пятый
же, средний, столб проходит в центре ячейки, причём атомы А1 также
668
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. IX
лежат на высоте 74 и */* от основания, и также строго друг над другом.
Все столбы тянутся вдоль оси с (см. рис. 522).
Атомы Т1 расположены попарно на гранях основания элементарной
ячейки, а также в плоскости, проходящей посередине последней,
параллельно основанию. Если в плоскости основания расстояние между парой
атомов Т1 равно 0,6 от оси 6, то у лежащей над нею пары атомов Т1
в средней плоскости—только 0,40 и наоборот. В результате, атомы Т1
образуют в пределах ячейки 4 зигзагообразные цепи, тянущиеся вдоль оси
с, по "обо стороны от центрального столба из октаэдров [A1FJ. Парис. Ъ21
показано по 2 звена каждой такой цепи—Т1—Т1—-Т1 —.
3. K2SnCl4H20
Решётка ромбическая
ценная 0ЯК<°>._
Координация 6>/6/2.
Тип ОД*™-SBЯЗ..
Пространственная группа
/4- Pbim.
Z = 4.
Sn к. ч. =6С1 (октаэдр)
(2,95 — 3.15);
К к. Ч.-6С1 + 2Н.О;
И./) к. ч.-2К.
SnCL
Взщоотпо
K>S. П14Ц>()
а
К.21
h
12,05
с
9,10
а : Ь : г
0,64 : 1 :0,
755 i
1
Примечание I
^В VII 22
Октаэдры (SnCl6) связаны общими рёбрами в цепи, параллельные
оси с и проходящие через рёбра и центр ячейки. Цепи, проходящие
через рёбра, слегка повёрнуты по часовой стрелке; проходящие через
центр -против часовой стрелки. Атомы К и молекулы воды распределены
между цепями.
ГЛАВА X
СТРУКТУРЫ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ
§ 243. Введение
Из рассматриваемого большого класса веществ мы выделили
самостоятельную группу А—комплексных полисоединений по причине, указанной в
следующем параграфе.
Все остальные вещества этого класса делятся нами на Б—соединения с
комплексным катионом и В—соединения с комплексным анионом,
группируемые по положению центрального атома в периодической системе.
При ознакомлении с материалом не следует забывать, что вещества,
фактически не имеющие изолированных координационных сфер, отнесены
нами к сложным, соответственно сложнокомплексным соединениям, вопреки
классической систематике Вернера, которая в этих случаях не отвечает
экспериментальным данным о строении веществ.
Вещества, содержащие и комплексный анион и комплексный катион,
отнесены к разделу соединений с комплексным анионом (например, NH4N03).
А. ПОЛИСОЕДИНЕНИЯ
§ 244. Полисоединения АХ[ВП], Ад.[ВВ^] и близкие к ним вещества
Для химика структуры полисоединений представляют исключительный
интерес. В самом деле, однородные атомы соединяются в агрегат в 2 случаях:
при образовании простых веществ (это свойство всех элементов, поскольку
и благородные газы образуют, при определённых температурах и давлениях,
кристаллы) и при образовании полисоединений (это свойство обнаруживается,
главным образом, у атомов неметаллов, однако отдельные структуры
карбидов, нитридов, фосфидов и др. имеют полиатомные комплексы металлов).
Во втором случае комплексный радикал обычно несёт на себе заряд, т. е.
является ионом.
Полисоединения делятся нами на подгруппы и соответственно будут
рассмотрены ниже в следующем порядке:
| Группа
I периоли-
1 ческой
I системы
VII
VI
»
V
»
»
VI—V
Подгруппа
Полигалогениды,
а также
Перекиси
Иолисульфиды, полиселениды
и т. д. , а также
Азиды
Полифосфиды
Полиарсениды и т. п.
Сульфоарсениды
Формула
Mx[Jn], МЯ[С1П], Мх[Вгп]
Мх\ЛЛп], Mx[JBrn] и т. ц.
МЛО,]
МЛГ8Л], Mx[Son], Mx[Te„]
MxrSSe„], Мд.ГЬТеп]
Mx[N3]
}мл[Р,], МГАз,]
М [SAs] |
(Продолжение таблицы см. на стр. 670).
670
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. X
Продолжение
| Группа
периодической системы
IV
с
V—IV
III
1 Разные
\ группы
Подгруппа
Поликарбиды:
а) графитиды
Ь) ацетилениды
с) прочие
Некоторые цианиды, цианамиды
Полибориды
Полиметаллические комплексы
Формула
К[С1в]идр.
К [Ни,] и др.
С3[В]а]
Ca[LN,l
a) МХ[В12], например, [A1[BU],
b) Мх[Вв], например, Са[В6]
^га5Св
Уже
§ 245. Полигалогениды Ae[Haln], А^НаГ, НаГ„]
в труде Вернера мы находим многочисленные примеры поли-
галогенидов, образованных однородными атомами, т. е. Лх[На1л], например
CsJ3, CsJ6, CsBr3, CsBr5идaжeKJ7, описанный в 1906 г., а кроме того, ZnJ7,
UgJe, PBr7 и т. д. Состав многих из них заслуживал бы проверки.
В случае комплексных катионов возникают полииодиды типа
[Ni(NH3)e][Jn]. Очень обширен класс полииодидов, образованных
«органическими» соединениями типа (СвН5)з С [JJn], [N (GH3)4] [J9] и т. п.
Учитывая взаимную химическую близость галогенов, мы
рассматриваем в этой же группе смешанные полигалогениды, среди которых в
неорганической химии известны
соединения типа Cs[JCl2], Cs[BrJ2],Cs[JCl4],
Са [JC14]2 8H20, Na (JC14) 8НЛ)
и множество других.
Весь этот обширный класс
химических соединений в
структурном отношении изучен весьма слабо.
Исследованы, и не всегда наделаю,
лишь немногие вещества.
Присоединение молекул
галогена к J'-ifony до [ JX2] влечёт за собой
снижение симметрии кристалла,
главным образом, либо до
ромбоэдрической (изменение угла а), либо до
ромбической (изменение
соотношения осей), как в случае
нелинейности, так и в случае строгой
линейности комплексного полигалогенид-
ного иона (располагающегося под
острым углом к оси с, в отличие,
например, от НС~-иона и от азидов
(N7) рубидия и калия.
1. CsJCl2
а) Решётка ромбоэдрическая R.
Координация О/О
Тип Д5/5 —SBF5x(NaHF2).
Пространственная группа D\a—СНт.
Z=l. 0 = 5,46; а = 70°42\
Группы G1JC1 — линейны.
Ср. рис. 225 и 532.
Рис. 52'».
f 2451
КО.МИЛККСНЬШ СОЕДПНЬШШ. ПОЛПГЛЛОГЕНИДЫ
671
2. [NHJ[J8],
[NH4] [CUBrJ,
Cs[JBrJ?CsJ8?
Решётка
ромбическая OR.
Координация
4/4.
Тип ОНф SB
D0U и F5l4
(см.ниже). к
Пространствен- "^\^Г\
пая группа \J
Dlh—Pmcn.
Z=4.
I'llC.
Вощестпо
• NH4J3*)
NH4CUBr*)
CsJBr, (?)**)
GsJ3 (?)***)
a
6,04
6,14
6,57
6,82
Ь
9,66
8,58
9,18
9,95
с
10,82
10,U3
10,66
11,02
3. NH4CUBr
Решётка ромбическая'^R.
Координация 4/4 (?).
Тип OR4tf)
Пространственная группа D~t—
Рте п.
Z=-'i.
а--6,23; Ъ -8,50; с -9,94;
группа С1—J—Вг линейна.
Особой ясности в вопросе о
структурах тригалогенидов —
лет. Описаны три типа: 1
ромбоэдрический и 2 ромбических.
За исключением, может быть,
лишь случая трииодида,
комплексная группа линейна. Это
отвечает предложенной нами в
1937 г. (см. § 91) схеме, в
которой несвязывающие
электроны занимают валентные на-
Рис. 526.
С1
i
правления. В данном случае имеем: > J—конфигурацию D3 с 3 незанятыми
i
Вг
вершинами.
JII-
*) По SB ,1П 43^ тип У01в группа J—J—J не строго линейна
-ЛШ=2,82А. ClJBr группа строго линейна, CI—J — 2,41бА, J-
**) Группа JBra линейна.
***) SB I 288.
_, л—jn=3tioA.
Вг=2,52А.
;G72 ч. ii. структуры неорганических веществ [гл. X
То же относится (см. ниже) к квадратной координационной сфере в
KJC14 (механизм образования которой отличается от такового в
случае атомов Pt (см. § 95)). Её можно представить, как октаэдр с 2 неза-
С1 | С1
витыми вершинами: ) J ^
CV | >С1
4. KJC14
Решётка моноклинная Л/.
Координация х/у.
Тип Mx/y~SBHOlo.
Пространственная группа C*h —P2Jn.
-Z = 4. B^[JGI4] {q).
Рис. 527. SB VI 20 21.
Нэщзетво
K.!C14
a
М.ОУ
b
14,18
с
4,20
a
95,7°
a : Ъ : с
0,921 : 1 : 0,246
Примечание. JC14-ион — слегка искажённый квадрат, с
атомом J в центре, квадраты наклонены и лежат друг над другом по оси с.
§ 246. Перекиси М02 и М£Оа
Состав и строение перекисей щелочных металлов длительное время были
объектом дискуссий. Предполагались соединения типа Cs2Oe, K208, К2Оа
(по аналогии с Н202) и др.
Фундаментальную работу в области наведения порядка в классе
перекисей (главным образом, щелочных металлов), в установлении стро-
§ 246]
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. ПЕРЕКИСИ
073
ении и свойств «порокисных» кислородных ионов удалось осуществить
лишь в СССР (школа И. А. Казарновского). См. Касаточкин и Котов,
J.Ch.Ph. 4, 458 (1936). II. Л. Казарновский и С. И. Рейхштейн - ЖФХ
21, 2'i5 (1947) и там цитированную литературу.
Как было выяснено, 07-жшы эллиптически вытянуты вдоль оси с.
Ранее в литературе указывалось, что КЬ2Оя и Cs203 = M4 (02)8
существуют и по структуре относятся к типу анти-ТЬ3Р4.
Казарновский и Рейхштейн приходят к выводу, что помимо К20
существуют лишь К2()2 и КО.,, по не К203. II. А. Казарновский недавно
рассчитал значение энергии присоединения электрона к молекуле 02 и
нашёл Ео2 = 22±\0 ккал (ДАН, 59, 67 (1948)). В последнее время И. А.
Казарновским и его сотрудниками было опубликовано сообщение о
выделении озонида калия К03, содержащего ион ()~ (см. JI. А. Казарновский,
Г. П. Никольский и Т. А. Аблецова, ДАН СССР, 64, 69 (1949)).
Элементарная ячейка этого вещества была определена Г. С.
Ждановым и 3. В. Звонковой. Повидимому, обнаруживается структурная
аналогии между озоиидом и азидом калия, однако, в связи с физическими
различиями структурных узлов О" и N" вопрос о структурном сходстве
обеих решёток пока следует признать открытым.
1. КО,, Rb02, Cs02, Sr02? Ba02
Решётка тетрагональная Т.
Координация О/О (Z>4/£4).
Тип yD4/o4-SBCll(CaG9).
Пространственная группа
Z = i.
Мк. ч.-б •* (D'i);
i к. ч. -6М(Д4).
#
О
f
О
°°ГЫ
ЩЩ
->^J
Р/1С 528.
1 Вещество
ко,
КЬО,
CsO,
Sr ',
ИаО,.
*
5, 70
0,00
Г,, 28
Г), 02
5.34
с
6,72—0,7:)
7,03
7,20
0,55
Г,, 77
с/а
1,178—1,182
1,171
1,145
1,30
1,27
°JB
2,14
з,оо
3,80
4,77
5,68
ОМ
218,3
—
—
—
Примечании
SB III 292. 293. См. также
SB IV 8, 118; SB Vll 7, 84
•
2. Li,Os, Na,0,
Решётка тетрагональная Т.
Координация х/у.
Тин Тх,у.
Пространственная группа?
2 = 8.
Вещество
Ы,0.
Na.O,
а ; с сэкс
5,48
j G,G5
7,74
9,91
2,14
2,47
4.IL СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ИЫЦКСТН
[ГЛ. \
§ 247. Полисульфиды, полиселениды, полителлуриды
Основная группа исследованных в структурном отношении веществ —
дисульфиды, диселениды, дителлуриды типа пирита и отчасти переходного
к простым соединениям типа марказита.
Между тем в литературе по неорганической химии описываются
«пентасульфиды» типа Na2Su, ZnS5, T1S6, гсксасульфид Cu.,S„
комплексные соединения тина Cr(NH3)5ClS5 и т. д.
1. MnS.„ MnTe2, FeS2, RuS2,
RuSe,, RuTe2
OsS2, OsSe2, OsTc2, CoS2, CoSe2,
RhS2, NiS2, NiSe2
Решётка кубическая С.
Координация О/О.
Тип С(-7/Г> —SBC2 (пирит).
p = S.> (соответственно Se2 или
Те,).
Пространственная группа Tft —
РпА.
Z-4. М к. ч. = 6(3);
(^ к. ч.-=6 М.
Молекулы S2, Se2, Te2 изобра-
жены не в масштабе. В
действительности они
значительно «длиннее»
(сигароподобные).
Вещество
*«
<*s->
°х
ОМ
MnS.
б'097
2,086
—
56,6
МпТеа
6,943
—
6,15
—
FeS2
5,404
2 „10
—
39,4
RuS2
5,57
1,93
6,93
43,4
RuSe2
5,921
—
—
—
RuToa
о,зб0
—
—
OsS2
5,607.
1,9„
44,1
OsSe,
5,933
~~*
—
—
Вещество
*»
ds-s
Cjc
ОМ
OsTe2
6,369
—
—
—
•
CoSa
*.б4
2,0,
—
44,8
CoSe2
5,85.
4
—
7,18
—
RhSa
5,57.
4
—
—
—
NiS,
5,74
>•%
47,:t
NiSo,
I
6,02
6,69
—
§ 247] колшлексн. сокд.полпсульфпды, ноллсклшшды, гюлптк/мурлди 675
Структура содержит самостоятельные комплексные группы S>,
образующие структурный узел [j в решётке типа NaCl, т. е. Со/о.
Структура СО.м отнесённая в SB к тому же типу, с нашей точки зрения
принципиально отличается от FeSo, не содержа узлов |* = 02, но сложные
структурные узлы С02, имеющие линейную форму, см. гл. VI. Данные
Фурье-анализа приводят к выводу, что атомы Fe почти олектропейтралыш.
2. FeS.: (марказит)
Решётка ромбическая ОН.
Координация (jjt + t.
Тип ORblt+t-SB С18.
Пространственна я группа
Dfh ^Pmnn.
3 = S,.
Fe к. ч.-6S, (<>>;;
S2 к. ч.-,..(> Fe (/+/).
f Г 1 f OFeQs
Рис. 530.
Вещество
FeS,
(марказит)
a
3,3.
3,37
l
b \ с
|
V.0
4,44
5,:i5
Г..39
!
a : b : с j c^
1
0,759: 1 : 1,214
4,91
OM
39,5
Примечание
SB I 497
природный марказит.
SB 11 272
Как указывает SB I, решётка (718 очень близка к решётке рутила
(С4). Но нам кажется, однако, целесообразным больше подчеркнуть
различия между ними и, в частности, то, что S-j-гругша в марказите
гораздо более обособлена, чем в рутиле. Атом S в FeSo-марказите имеет
1 атом S на расстоянии, вдвое меньшем, чем другие атомы S, тогда как
атом О в рутиле имеет 9 соседей О на расстояниях ей/, отличающихся
в пределах 10% (2,46-2,78 А). Ср. рис. 311 и 530.
676 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
3. BaS,
Решётка ромбическая OR.
Координация 8/8 --•= Pijr + г.
Тип ORp4r+r-SBDQ17{BaSu).
Пространственная группа D* — P2 2 2.
-Z = 4.
Рис. 531.
Вещество | а
1
; ВаЗД
8,32
Ь
0,64
с j а : Ь : с
4,82 | 0,863:1:0,500
Группа S3 —треугольная с углом ^S-= 103°.
Атомы Ва сами по себе образуют примитивную ромбическую решётку.
Если четыре такие примитивные ячейки объединяются (см. рис. 531),
то получается контур одной элементарной ячейки BaS8l включающей
£*. + £ + |), а всего: 4 атома Ва: (000) (д 00) (о j О) Q-» о) . В этой
ячейке размещены четыре группы S3.
Сравни с J310 —(PH4J), рис. 572.
На рис. 531 показаны 8 элементарных ячеек BaS3. В каждой из них
4 узла p=[S8], расположенные внутри ячейки в тетраэдрическом порядке.
§ 248. Полинитриды (азиды MN3)
Тогда как атомы кислорода соединяются либо по 2 (молекулы кислорода),
либо по 3 (треугольная молекула озона) с образованием соответствующих
простых веществ 02 и 09, атомы N соединяются в этом случае лишь по 2 (N2).
Однако, присоединив один электрон от других атомов (металла), три
§ 248]
КОМПЛЫКСНЫК СОЕДИНЕНИЯ. ПОЛШШТЕПДЫ (АЗИДЫ)
(577
атома N способны образовать за счёт /;3-электронов линейный ион N~ с
возможной суперпозицией простой, двойной и тройной связей. Образование
вещества типа а|3 с одним сигароподобным структурным узлом N8 ведёт
к одному из двух видов деформации: а) ромбоэдрическому или б)
тетрагональному (в предельном случае— ромбическому). К первому относится NaN3,
ко второму -Kj\3; у AgN8 и XFI4N3 наблюдается даже ромбическая
элементарная я чей к'а.
1. NaX8
Решётка ромбоэдрическая R.
Координация О/О.
Тип fl^-SBFj^NaHFJ.
Пространственная группа D\a — Н'Лт.
Na к. ч.=6 ?; 3 к. ч. (>Na.
Рис. 532. Сцятщ цсптриропднпьк' p<'\io<>:>;ipi>i, риг. 225 и Г>24.
Вэщоство
OAJ
<*х-ч
dN«-X
Примечание
NaNa
5,48х
5,488
38°43'
38°43'
5G,G
1,17
1,10
1,20
2,50
—
SB I 276, SB IV I'l
(M'—N" равно N"—N'").
SB VII (J7оспарикается
равенство расстоянии
атомов н N3: N'--N" = 1,10,
N"—N'" = 1,26, а не
одинаковы (1Д65— 1,17).
См "тр. '>кп уотгтлазпд.
G78 ч.п. структуры икорглиичгхшгх вкщкстн [гл. X
2. KN„ Rb\3
Решётка тетрагональная 7'.
Координация Pi/Pi.
Тип 7p4/P4-SBF52(KHFa).
Пространственная группа D^n-- Ji/mcm.
Z = 4.
Кк,ч. = 83; р к. ч. = 8 К.
О М
Рис. WM. C_)N
Нияшоу к верхнее основания ячепки и средняя плоскость,
параллельная основанию, зачернены. Иногда выбирают ячейку удвоенного объёма
с осями, п:жёрпутыми па '\Ъ° (пунктир), т. о. cja уменьшается в У 2 раза.
BiMiujcth ) | а
i
KN3 ! Г>,094
1
HbN3 | 6,%
i
с | с ''а
7,056
7,41
1,1Г,8
1,165
ОМ | Примечание
65,5
ЬВ I 279
SB IV 151
SB II 375—376
Тетрагонально деформированный тип CsCl.
§ 248J коми. iKKciiNi; ст-диннпин, ткпгшгп-пдм (лзпдгл) 079
3. NH4NS
Решётка 1)омбическая OR.
Координация DijDl.
Тип OR miDi — ShF5H.
» = INH41; ? = IN7].
Пространственная группа Djt — Pmna.
Z = 'i.
|NH4]k.4.-4N3(.S') + 4N;j(^)=-^; (N„ к. ч.=4|М14](Л) + 4|М14](Л) -- Di.
\s" О Ч 2 3 4 5 В 7 5^\mu
b'i/o. 5:i'i.
ВеЩ9('ТВО
NIJ.N,
a b
8,9:10
8,042
с
8,800
a : b : с 1 Примечаний
1,(Ш: 1 :0,4397
SB III 79 1
SH IV 37
На рис. 534 показаны две элементарные ячейки.
4. AgK,
Решётка ромбическая OR (псевдотетрагональыая).
Координация Р4/Р4.
Тип ORp^ipi, — SBF52 (KHF.) деформированный.
Пространственная группа D^ — Jbam. j3 = N3.
Z = l; структура искажённая (см. рис. 533).
Вещогтно
AsrN,
а
Ъ
5,58 | 5,93
с
0,04
a : Ъ : с
ОМ
0,941:1:1,019 ' 49,95
|
Примечание
SB III 75
Хотя KHF2 образует тетрагональную элементарную ячейку, редакция
SB приписала AgN3 к тому же структурному типу, вопреки принятой
в SB системе и без каких-либо комментариев.
680
П. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
|.-л. X
б. Ba(N3)2, SPb(N3)2
Решётка моноклинная 71/.
Координация xjy.
Тип Mxlv.
Класс С*2л-
Пространственная группа?
Z для Ba(N8),%10(?);
Z для 3Pb(N3)2^8(?).
Исследование ненадёжно.
Вещество
а
i
Ba(N,)2 f.,22
ftPb(N,)3 5,10
1 '
Ъ
29,29
8,83
c \ . я : 6 : c \ определи Примечание
j ние) ; |
7,02
17,00
0,212'i : 1 : 0,2397
ior,°i/*'
90э49'
SB II 376—377
6. aPb(N,)a
Решётка ромбическая OR.
Координация xjy.
Тип ORxhJ.
Класс /)оЛ.
Пространственная группа?
Z--12.
Исследование ненадёжно.
Во постно
aPbCN,),
1
a j b \ с \ Примечание
О//*
11,34
10,25
*В 1[ 377
7. CH3N3 (метилазид)
Электронографическое
исследование пара (SB III 712) обнаруживает
структуру
в которой
//V
КС
d-1,10; <Г = 1,26;
е --1,17; а--=135°± 15°.
Ср. со строением Г\3-гругп!ы в NaNs
(стр. 077), где характер связи гораздо
более мошна it.
Рис. 535.
§ 249] КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. ПО. 1ПФОСФ11ДЫ, ПОЛПЛРСЕНПДЫ II Т. И. 681
§ 249. Нолифосфиды, нолиареениды и т. п.
1. ГеР25 FeAs2 (лёллингит) FcSb2
Решётка ромбическая OR.
Координация Ojt+t,
Тип 0/?5// + <- -SBC18 (марказит).
Пространственная группа D}*h-- Pmnn.
Z = 2.
Гек.ч. ()(\2){0)\
(Х,)к. ч. -О Ye (t rt).
О Fe
Ox
Рис. 53f>.
В.ЧЦ.Ч'ТПО |
FeP2
FeAs2 j
FeAs,
1 (лёллингит)
FeSb,
| I
a
2 72.
"y "5
3,17
2,86
3,19
. 1
о
4,86
5,20
5,82
с
5,65_,
5, SO
5,92
6,52
a : h : r
0,550 : L : 1,139
OM
;;s,34
39,5
63,3
1Три.М1?ч1!1ие
SB Г11 12, 310
SU 1 497
SB II 273
SB I 497
Cm. FfiS., (марка.чит).
682
Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. X
2. GoAs3==Co4(As4)8 (скуттерудит)
Структуре больше отвечает формула Co4(As4)3.
Решётка кубическая С
Координация C'l'/C.
Пространственная группа Tn — Jm3.
Тип С С-/-/С SB D2.
? = (As)4.
Z-8. CoAs, = 2Co4(As4)a.
Co к. 4. = 6(As)4 в форме (С"'-); (As)4 к. ч. = 8Со(С).
Вещестло
Co4(As4),
а*
8,18
d
Со—As
2,35
d
As — As
ОМ 1
CoAss
2,45 68,4
Рис. .037. Одна оломсчггагнти мчопкп.
Структура, изображённая без комментариев на рис. 537 (SB1233), нам
представляется довольно поучительной. 4 атома As образуют кольцо As4
с валентным углом ^90°. В пределах кольца атом As осуществляет две
связи. Две другие связи он осуществляет с двумя атомами Со
(искажённый тетраэдр). Если, как нам представляется удобным, рассматривать
группу (As4) как структурный узел, формула вещества а4 (33 приводит
к кубической центрированной ячейке, причём из 8 октантов могут
содержать узел р только 6 октантов. Остальные остаются незанятыми. Кольца
(As4)равномерно распределены между тремя ориентировками, параллельно
(100), (010) и (001).
§ 249] комплексный соединения, по/шфосфпды, по.'шлрсенпды и т. п, 683
3. PdP.,, PdAs2, PdSb2, PtP2,
PtAs.,, PtAs2 (гнеррилит),
PtSb2, AuSb2
Решётка кубическая С.
Координация О/О.
Тип Го/o-SBCo (пирит).
Пространственная группа
Т%-РаЗ.
3 = Х„.
Z = 4MX2.
М к. ч. = 6 (Х2) (октаэдр);
(Х.2) к. ч.--6М (октаэдр).
Х2-групиы не в масштабе.
В действительности они
должны быть сильно увеличены
в длину.
О м
Рис. 538. ( J X
Вищо- P(lpJ pjAsJPdSb.,
('ТИП " i
"tr —
d
Г), 92
-Г>,97()
12,07,
207, Г) !
Pi IV,
6/i39 | 5,fi8,j
!•
2(>7,L Its;;,:;
Pl\J PtA^
-j (сиоррилит)
5, УГ|
211,2
Г», 93
0,00
10,89—11,0
208,5
PtSb,
G,V28
265,8
AuSb,
G,f>36
292,1
Примечание
SB I 153,780—781
SB II 271
4. ZnP., Cdl»a
Решётка тотрагииальная Т.
Координация?
Тин У —SB?
Пространственная группа D\ -Pi 2 или D\--Pit2l.
2 = 8.
Вгщсн'ТШ)
Znl\
слр2
1
1 °
1
| 5,07
! 5,28
!
с
18, Go
19,70
с а
3,Г)8
3,7i
°х
3,51
4,19
1
Примечании
SB III 311
б. ZnAs2
Решётка ромбическая OR.
Координация?
Тип OR -SB?
Пространственная группа?
Z-=32.
Вещество
Z;iAs. | 7,72
Примечание
7,93 36,28 1 5,08 j SB П1 311
684
Ч. И- СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
|гл. X
§ 250. Сульфоарсениды и т. и.
1. FeAsS (арсенопирит), FeSbS (гудмундит)
Решётка моноклинная М.
Координация 3 + 3/3 + V3 + 1 (^OjTft).
Тип M:^ = -SB E07.
о/т/т 7
Пространственная группа С-м — В2Jd(P2 1с).
Z = 8.
Fe к. 4.=3S (расстояния: 2,23 + 2,27 + 2,27) +
+ 3 As (расстояния: 2,32+2,37 + 2,37) искажённый октаэдр (О)',
As к. 4. = 3Fo (2,32+2,37+2,37) +IS (2,30) искажённый тетраэдр {Т)\_
S к. 4.=3Fe (2,23 + 2,27 + 2,27) + lAs (2,30) искажённый тетраэдр (Т).
Рис. 539.
Вещестпо
FeAsS
FeSbS
1 1
а Ь \ с а : Ъ : с ,->
! i
9,51
10,04
5,65
5,93
6,42
6,68
1,683: 1: 1,136
1,692: L : 1,126
90°
90°
Интересно, что v обоих соединений при комнатной температуре угол
р=90э (см. § 106).
§ 251] КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. 1ЮЛИКАРЫ1ДЫ, НОЛПСШШЦПДЫ
685
2. Co[AsS] (кобальтин), Ni[AsS],
Ni[SbS]
Решётка кубическая С.
Координация 010.
ТипС^-SBFl.
Пространственная
Р =
группа
rAsSJ.
4.
1>ещест1ш j u}r \ ОМ j
1 i
Co[AsS]
(кобальтин)
NifAsSl *
Ni[6bS]
5,f>0
5,70
5,00
45,1
45,S
51,
о 1
Примечание. SB I 2СУ,
илилок к иприту.
О Со
O(AS.S)
Рис. .ViO.
§ 251. Неликарбиды, по шсилициды
Полпкарбиды, по нашему мнению, следует разбить на две резко
отличные друг от друга группы:
Поликарбиды ароматического ряда, производные от графита. Их можно
назвать «графитиды».
Они образуются, например, при воздействии щелочных металлов на
графит. Так возникают С1вК, С8К и другие вещества, сохраняющие
в своей решётке элементы структуры (кольца и сетки) графита.
Внедрение атомов металла происходит с разрывом связей, параллельных оси 6, и
значительным удлинением межплоскостного расстояния между кольцами
(значит, и величины оси с). Формулы подобных веществ мы будем писать
МСгр (если числовой индекс при С не проставляется).
Поликарбиды алифатического ряда. К сожалению, до сих пор не
была поставлена задача систематического изучения поликарбидов
алифатического ряда.
В литературе мы находим лишь несколько разрозненных исследований,
касающихся немногочисленных представителей этого ряда.
Сюда следует отнести ацетиле//иды, которые являются первым членом
ряда, содержащие группу -кратчайшую цепь- из 2 атомов С. Они имеют
состав NalKlo, СаС2 и др. В первом случае в решётке имеются группы (НСг),
во втором (Сг), служащие структурными узлами. Важный для
интерпретации этих структур расчёт константы Мадслупга для решёток этого
типа был выполнен Л. И. Казарповской (ЖФХ, XX, 1403 (1946)).
При разложении подобных веществ кислотами и водой выделяется
ацетилен.
Карбиды с более длинной цепью Сл специально не изучались.
Неожиданным исключением, может быть, является карбид бора. В поставленном
нами исследовании системы В - С, где были выделены карбиды бора типа В4С,
возник вопрос об определении структуры этого вещества. К этой работе была
привлечена рентгеновская лаборатория Института им. Карпова (Г. С. Жданов
и Н. Г. Севастьянов); см. ниже.
686
Ч..Н. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТН
[гл. X
Строение ароматических поликарбидов
(графитидов)
1. КС8> НЬСв, CsC8, «коричневый 2. KClf, HbCie, CsClf, «чёрный гра-
графит калий» и др.
Решётка гексагональная
слоистая Hs.
Координация?
Тип Hs.
Пространственная группа
Z-4KC8.
фит калий» и т. д.
Решётка гексагональная
слоистая Hs.
Координация?
Тип Hs.
Пространственная группа?
Z = 2 KCU.
1 Вещестпо ас
| К(в
RbC8
J CsCe
4,94
4,94
4,94
2it:*4r
22,73
23, 76
I Вощсч'.тно
кс1§
ш>с1в
CsCie
4,94
4,94
4,94
с
17,4S
17, УГ>
18,51
Ha рис. 541 (Z. Ph. Gh. (B) 18, 1 (1932) показаны границы
элементарной ячейки КС8 (с удвоенной по отношению к графиту осью и
удвоенным количеством (4) сеток Сп по высоте ячейки, т. е. по количеству
атомов (32) эквивалентной 8 ячейкам графита (а= 4,94; с — 21,34). Внедрение
атомов К не меняет основания ячейки, которая увеличивается по оси с.
Построение по тому же типу, что и КС8, с соответственно меньшим
количеством сеток калия.
Для понимания рис. 541 надо добавить следующее. Графитндм МС8
и MG]f (см. рис. 541) обнаруживают структурное сходство в том, что они
§ 251]
КОМПЛЕКСНЫЕ СОЕДИНЕНИИ. ПОЛНКЛРБИДЫ, ГЮЛНСПЛПЦПДЫ
687
являются продуктами внедрения калия в решётку графита. При этом
атомы металла располагаются между слоями атомов С, образуя слои,
перпендикулярные оси 6, и раздвигая слои атомов углерода. В результате
высота элементарной ячейки —по оси С —определяется соотношением
количества слоев С—С, не разделённых слоями металла (т. е. с межплоскостным
расстоянием 3,40 кХ), и слоев С С, содержащих прослойку атомов металла.
В цитируемой работе принималось, что для графита с 0,79 (в
действительности 0,70). Расстояние между сетками — — 3,395. Таким образом с
внедрением между сетками С атомов К расстояние между сетками
увеличивается до 21,3/f : 4-=5,335, т. е. на 1,94 кХ. Значит, и ячейке KC,S ось
с ■-■- 21,34, в ячейке КС16, содержащей половинное количество слоев калия, есь с
должна быть равна: 5,335x2 + 3,395x2-10,070 4-0,79 17,10. что, в
общем, и наблюдается. Принимая для графита современное значение —=■-■:3,35,
находим увеличение за
счёт внедрения К 1,985
кХ при диаметре атома
К 4,50 (в решётке К
к. ч. -= 8) и при диаметре
иона К-2,00 к\.
Отсюда вытекает
серьёзный вопрос: если
значения — определены
правильно (точность
определения значительно
выше величины
расхождения 0,05 КХ), то иоьы
К+, несомненно
существующие в решётке, по-,
видимому, не только
должны быть вдавлены
в кольца графита, по и
сплющены в
направлении оси 0, что само но
себе представляется
сомнительным. Поэтому в
данной модели
структуры МС8 и МС1в
остаются неясные узлы.
3. CF фторграфит
Фторграфит
получается, например, при
действии фтора на
графит при 420°С при
760./WAf(Z.Anoro;.Ch.2t7,
1(1934)). При этом
образуется
структура,показанная на рис. 542
(каждое деление масштаба
соответствует 1 кХ). Рис. 542.
Как и в случае гра-
фитидов калия, расстояние между соседними слоями атомов углерода,
разделёнными слоями фтора, увеличивается на этот раз до 8,17 кХ.
(588
Ч.1К СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ЕШЩКСТВ
[гл. X
4. FcmC„ графит
Упорядоченное расположение атомов Fe в графите между слоями,
состоящими из колец графита. По Конобеевскому, Z. Кг. 72, 381 (1929),
для графита, после введения Fe, a = 2,48, с = 6,72, но сверхструктурные
линии свидетельствуют об удвоении периода а (см. КС16).
Ароматические поликарбиды относятся скорее к сложнокомнлексным
соединениям с бесконечным двумерным ионом.
5. Графитиды серной кислоты, хлорной кислоты и т« п.
При действии концентрированных кислот (серной, хлорной и т. п.)
на графит возникают графитиды серной кислоты, хлорной и т. п.
При этом получается набор структур, промежуточных между
структурой графита и структурой, содержащей один слой С— С, с
расстоянием 3,40 кХ, и один слой С —R—С с расстоянием, увеличенным на
4,55кХ в случае серной или хлорной кислот, на 4,44 соответственно
4,85 г случае азотной и селеновой кислот.
Строение алифатических поликарбидов
1. NaHC, КИС, СаС„ 8гС„
ВаС„ LaCo, CcCo, РгС2,
NdC2, UC2
Решётка тетрагональная Т,
Координация Z)4/Z)4.
Тип rD4/D4-SBCH (CaC2).
Пространственная группа
Р = (НС2).
Z--4.
Ом
I&. ° °i°i°
Рис. 543.
1 Груцна
периодической
системы
1
11
III*
Ш**
I VI*
Вещество
NaHC2
К НС,
СаС,
8гС.
ИаС,
LaC,
CeC. •
PrC,
NdC,
1C2
a
3,89
.4,28
3,88
4,11
4,40
3,92
3,88
:i, 85
:;,90
-
с
8,17
I 8,42
6,37
6,68
7,06
6,55
6,48
6,38
6,23
—
с fa
2,14
1,97
1,641
1,626
1,60J
1,67
1,67
1,65
1,63
1,20
*x
1,33
1,37
2,21
3,26
3,90
5,35
5,56
5,75
6,08
—
Примечание 1
SB I 740, SB II 275,
деформированный тиа NaCI. При отсут- I
ствии вращения узла Ь = С2 или
НС2, являющегося эллипсоидом,
ось с значительно удлиняется
(тетрагональная метрика). В SB I
приведена ячейка, повёрнутая
на 45° с Z = :>
Эта же структура найдена согласно Эгансу, а также новым работам, 1. с.
стр. 587, У UC2 (?) (Введение в кристаллохимию, стр. 148 (1948).)
§ 2511 комплексные соединения, поликарбиды, гюлпсилшдиды 689
Пространственная групна Z>i*— {^Лг
Ji[mcrn. \~J
Z — i (8) (в зависимости от выбора оси а элементарной ячейки, см. стр.678)
рис. 544Ь.
Вещестио
а
a Y'l
TliC, | 4,14 5,85
1 !_,__ 1
с
с 'а
Г),28 J 1,:ш
_ 1
с'а Y'l
0, *.»();{
а*
Примечание
9,3 | SB II 27Г>. См. KHF,
i
Вопреки этой модели, согласно ряду авторов «дпкарбиды» ThC2 и ZrC2
имеют ту же структуру, что и UC.» (Tdaida), но гантельные группы С>
расположены не параллельно оси 4, а перпендикулярно ей, в плоскости (001)
под углом ф 90° к осям а п Ь. Отношение осей с/а подобной, также
тетрагональной, структуры меньше 1 и дли ТпС2 = 0,903. Однако тип —
тетрагонально-деформированный Со/о, т. е. Toi/D-h М к. ч.=6 3 (Z>4V
Р к. ч. = 6 М (М); рис. 544Ь.
3, В4С = [В12] [С3] (?) По литературным данным, в системе В —С имеются
3 фазы: В, В4С и С. Диаграмму состояния системы В - С детально
исследовали В. Ф. Ормонт, И. Г. Шафран, А. И. Золотаревский (1936 — 1940)
в Институте им. Карпова.
Структуру карбида бора В4С (а не ВвС, как полагали ранее)
исследовал Лавес. По Лавесу, решётка тригональная. Пространственная
группа Ви-Шт; а = 5,62 ±2; с = 12,12 ±5. См. SB HI 329.
По Лавесу, В4С имеет ромбоэдрическую симметрию, пространственная
группа D^d —Rom) а ^ 5,62 ±2; с = 12,12 ±3; Z = 3,06 (не исключается
также В7С с Z = 1,93).
В 1938 г. по нашему предложению Г. С. Жданов поставил в
рентгеновской лаборатории Института им. Карпова исследование структуры фаз
карбидов бора, не удавшееся Лавесу. В нашей лаборатории И. Г. Шафраном
был проведён синтез монокристаллов карбида бора. В результате большой
и тщательной работы по исследованию сложной структуры карбида бора
Г. С. Жданов и II. Г. Севастьянов (ЖФХ 17, 326(1943)) пришли к
структуре, представленной на рис, 545.
Результаты их исследования сведены в таблицу.
690 ч.и. структуры неорганических веществ [гл. X
В 1943 г. к аналогичным данным пришли Клерк и Гоард (J. Am.
Gh. Soc. 65, 2115, (1943)), имевшие до того в своих руках опубликованную
работу Жданова и Севастьянова.
Успешная работа Жданова и Севастьянова представляет собой
интересный пример изучения сложных структур. Указанными авторами путём
исследования методом Фурье-анализа (1 проекция) было установлено, что
атомы В образуют группы В12. По утверждению Г. С. Жданова и Н. Г.
Севастьянова, 3 атома С лежат на одной прямой.
Однако мы должны повторить здесь замечания, сделанные нами
(в докладе в Институте им. Карпова (май 1945) о структурах реальных
кристаллов), по существу полученных указанными
авторами, как и Клерком и Гоардом,
результатов. Линейная конфигурация С —С — С,
найденная указанными авторами, — ими не комментируется,
как если бы она отвечала какой-либо известной
валентной модели. Нельзя не отметить, что как
известно, цепи атомов углерода обычно строятся
либо по тетраэдрическому — алмазному— закону
(валентные углы 109°28') (за счёт а-связи), либо
по графитному (валентные углы 90° и 120°), в
соответствии с представлениями квантовой химии
(см. § 95), линейная же цепь атомов обнаруживается ,
например, у соединений алленового ряда,
вследствие наличия «двойных» (зтс) связей, см. рис. 133.
Рис. 545.
С I—средний, С
II—крайние, В I—белые, В II—за-
штрихсжанные.
Но в модели карбида бора, предложенной
Ждановым и Севастьяновым, расстояние СI — С II =
= 1,45 кХ, что не отвечает расстояниям С —С
в случае двойной связи (делающей понятной
линейную конфигурацию), ординарная же а-овязь
обусловливает зигзагообразную цепь С — С — С.
Гоард и Клерк, воспроизведя, по существу,
работу советских исследователей, внесли, однако,
ту поправку, что расстояние CI — СИ = 1,39 кХ.
Это расстояние больше отвечает «двойной связи»
и делает данную модель в структурном отношении более правдоподобной.
Но тогда возникает затруднение химического характера: двойные связи
между атомами углерода сравнительно легко разрываются, например при
окислении. Между тем, как известно, карбид бора чрезвычайно инертен
по отношению к оильным окислителям.
В 1947 г. Н. В. Белов и В. П. Бутузов (ДАН, 57, 819 (1947)) нашли,
что, исходя из экспериментальных данных Жданова и Севастьянова,
расстояние CI—СИ следует принять равным 1,34 кХ, а но 1,45.
Если расстояние 1,45 заставляло делать догадки насчёт возможности
попадания атомов В или О (за счёт загрязнения кислородом) в позицию
CI, то поправка Белова и Бутузова наталкивает, по нашему мнению, на
вопрос о распределении зарядов в группе С — С — С, в связи с
химической неактивностью карбида, том более, что атомы СИ находятся в
непосредственной близости (1,63 кХ) к атомам В (на рис. 545 —белые кружки),
осуществляя три связи с последними, под углами ^120°, типичными для
структуры графита, тогда как четвёртая связь (CI —СИ) оказывается в два
с половиной раза короче 4-й связи в графите. К этому вопросу мы
вернёмся в специальной статье.
§ 2521
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. Ш1АН1ЩЫ, ЦИАНАМИДЫ
691
4. В4С=[В12][С,](?)
Решётка ромбоэдрическая R.
Координация О/О.
Тип Д5/у —SB?
Пространственная группа Х>з<* — R3m.
а — В^; P = G3 или скорее a —(BII)e;
P = (BI),C,.
Z = l.
Вещество
B4G
a
5,17
я
65°45'
Примечание
Деформированная структура NaCl. Куб,
сжатый по диагонали, так чтобы отношение
с/а= У6 = 2,45 для кубической решётки
сделалось равным отношению с/а = 2,16;
cfB_B = l,74—1,80 А, в том числе ^Ш_В1 = 1>78»
^BI-CII^1»63 А
Полисилициды
Примеры истинных полисилицидов практически изучены весьма
плохо. Как правило, структуре MSi2 отвечает слоистая решётка.
Наиболее близок ацетиленидам (СаС2) структурный тип MoSi2
(SB СИ), где атомы Si всё же образуют гексагональные сетки. Атом Мо
имеет 10 атомов Si в координационной сфере; атом Si — 5Si на
одинаковых расстояниях, так что о группах [Si2] в подобных веществах
говорить не приходится (см. рис. 444 и \\Ь),
§ 252. Цианиды, цианамиды
CN^-ион является аналогом группы N2, самостоятельно в
структурах пока не обнаруженной.
Некоторое сходство мы наблюдаем,
однако, между цианидами и ацетилени-
дами, ср. СаС2 и NaCN; между
цианамидами и азидами, ср. CaCN2 и NaNs.
Цианиды щелочных металлов по своим
свойствам и структуре могут
рассматриваться и как сложные и как комплексные
соединения (Ср. также §§ 226 и 235).
1. aNa[CN]
Решётка ромбическая OR.
Координация D2/D2.
Тип ORoofDo — SB? (Деформированный
тип NaCl.) p = GN.
Пространственная группа? С^, — Jm2m.
Z = 4.
• Ncl
Рис. 546. Ср. рис. 544а.
Вещество
a NaCN
а
3,74
Ь
с
4,71 1 5,61
d
C-N
1,05
Примечание
SB VI 225
44*
692 ч. п. структуры неорганических веществ [гл. X
2. pNa[CN], K[CN], 1Rb[CN]
Решётка кубическая С.
Координапия О/О.
Тип Сб/о —SBBl(NaCl)iwniSBFl
(кобальтин), или SBC2 (пирит)(?).
Пространственная группа (?).
Z-4.
Рис. 547.
с^
^Ш
<г—^>
ЬЧ^'
с1
п
с^Ч£Я-^
о
Рис. 548.
Вещество
?Na[GN]
K[CN]
Rb[CN]
а„
5,83—5,87
6,51—6,55
6,82
d
С—N
1,06
°*
1,624
1,557
2,32
Примечание
SB II 371
SB VI .225 |
CN-группа, вращаясь, приобретает шаровую симметрию.
С повышением температуры поэтому структура кристалла изменяется,
жереходя от изображённой на рис, 546 к структурам рис. 547 и, наконец,
рже. 548.
§ 252]
КОМПЛЕКСНЫЙ СОЕДННКШШ. ЦИАНИДЫ, ЦИАНАМИДЫ
693
3. CsCN
Решётка кубическая С.
Координация CJC.
Тип Ccic -SB52(CsCl).
Пространственная группа Т'а —
/43т или Г* —/2,3.
P = (CN).
Z = 2
Вощество
CsCN
"»
сх
Примечание!
4,2Г)0 U,93 SB II 371
Рис. 549.
4. [NH4]+[CN]-
Решётка тетрагональная Т.
Координация 8/8.
Тип r8/8-SB? a = [NH4]+. P = [CN]-.
Пространственная группа Z>4/,— Pi/ntcm.
Z = 8.
вещество
[NI1-4][CN]
tJ
Комнат.
a
4,1G
с Примечание
7,01
Тетрагонально-деформированная решётка
CsCl. Группа CN осциллирует в плоскостях
(lid) и (U0). С нагреванием от —80° до 35°
размер с растёт с 7,45 до 7,04, размер а
неизменен
5. aAgCN (см. стр. 645).
Решётка ромбоэдрическая цепная RK.
Координация?
Тип Як.
Пространственная группа С&,—R3m.
Z = 2(?) «-4,60; а- 8П4'; зт--4,72.
Z = l. a-3,88; a=101°ll/. См. SB III 236-237.
aAgCN должен быть причислен скорее к сложным соединениям с
цепной решёткой. Мы рассматриваем его здесь для сопоставления с
комплексным соединением pAgCN, проводя аналогию с полисоединениями.
Соотношение между а- и И-формамл схематически может быть,
невидимому, представлено рис. 22.') и 222.
6. frAgCN
Решётка кубическая С] Z=-4; « = 5,69А; 3^ = 4,80. SB III 236.
7. AuCN (см. стр. 645).
8. Zn(CN)2> Cd(CN)2 (см. стр. 619).
694
Ч.П. СТРУКТУРЫ ННОРГАШ1ЧЕСКНХ ВЕЩЕСТВ
1гл. X
9. Hg(CN),
Решётка тетрагональная Т.
Координация х/у.
Тип Txiy -SBF11 [Hg (CN)&]. _
Пространственная группа Dw—Jb2d.
Z = 8.
Исходя из анализа интеисивностей по данным
работы Ханауолта, Ринна и Фревела (см. часть III),
сопоставленным с данными работы SB I 270, в
последние годы было уточнено положение атомов, в
частности CN-групп в вышеописанной ячейке,
оси которой, пространственная группами Z
остались без изменения (см. ниже Г. С. Жданов и
Е. А. Шугам). Наиболее согласуется с этими^данпыми следующее:
структура Hg(GN)2 состоит из линейных молекул, складывающихся в
зигзагообразные цепи. Последние образуют ленты, вытянутые вдоль оси с.
Плоскости лент в соседних цепях взаимно перпендикулярны. Молекулы лежат
не в самой плоскости ленты, но несколько смещены в разные стороны
относительно последней. (Г. С.Жданов иЕ. А/Шугам ЖФХ XIX, 433 (1945).)
Вещество
1 с/а ....
ОМ ... .
Hsr(CN),
9,67
8,92
0,922
104,3
Примечание: SBI270— |
271, точное
CN-групп не
положение
дано.
10. CaCN2 (циашшид кальция)
Решётка ромбоэдрическая R.
Координация О/О.
Тип R- , -SBF51.
о/о
Пространственная группа D <j — R3m.
P = (CN2).
Z = 1. Са к. ч. = 6? (О); ? к. ч. = 6Са (О).
Вещество .
а
а
ОМ . . .
CaCN,
5,11
43а50'
56,2
Примечание. SB I 276
Сравни со структурой NaNs рис. 532.
Рис. 550.
§ 253] КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. ПОЛПБОРИДЫ 695
§ 253. Полибориды
Сопоставление экспериментального материала по полисоединениям пока-
зывает, что с переходом от галогенов к бору тенденция к образованию
полисоединений усиливается. Особенно резко это проявляется в случае бора,
образующего комплексные группы в большинстве соединений бора с другими
элементами. Это явление находится в связи с нехваткой валентных
электронов у В, при наличии значительного сродства к электрону.
При этом наблюдается два типа В-групп:
а) группа В12, как, например, в С3В12(?), (см. карбид бора) или в А1В1а;
б) группа Вв, как, например, в CaBf.
Группы В2, как, например, в А1В2, в решётке обычно не существуют,
образуя слой (см. А1В2, бориды, стр. 610).
1. АШ1а «графнтоподобный»
Решётка моноклинная М (?) гранецентрированная.
Координация?
Тип?
Пространственная группа Сгл —С2//п, Сч — С2, Cs — Cm.
Z = 52 атома = 4 AlBia. a = 8,50t; 6 = 10,98;
с = 9,40; р = 110°54'.
См. SB VII 110-111, SB IV 126-128.
2. А1В12 «адмазоподобный», «кристаллический бор» (?); «ЗА1В122В4С»
А1В12 с С8В1а образует соединение указанного выше ориентировочного
состава. Наблюдается тетрагональная ячейка с а = 10,28; с =14,30А;
Z= 196 атомов (вместо 208), 12 узлов свободны (структура вычитания
пли деления? Б. О.) (Пространственная группа С\ (или />{, или D\).
Ненадёжность анали-
тических данных не *ч^| т г^^\
позволяет, по наше- ^^ -
му мнению, внести
ясность в истинное
соотношение
составов и структур в
рассматриваемых фазах
(см. SB VII110-111,
SB IV 126-128).
3. С3В12, борид
углерода — см. карбид Лы
бора. ^
CaBe, SrBfl
YB„ LaBe,
BaBe,
СеВ„
PrB„ NdB„ GdBe,
ЕгВ„ YbBe, ThB,
Решётка кубическая
С.
Координация С/С.
Тип Cc/c-SBZ)27.
Пространственная
группа О^ — РтЗт.
Z=l.
М к. ч. = 8р(С);
3 к. ч.=-8М(С).
Рис. 551. 8 ячеек СаНй.
о Си
ОВ
696 Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
| Группа
Вещество
ОМ
II
СаВв
4,15
71,47
£гВв
4,19
73,56
ВаВв
4,28
78,40
III*
YBe
4,07
67,42
LaHe
4,145
71,25
III**
<eBd
*'Х~9
70,4
Примечание
SB II 37, 308
| Группа
Вещество
им
РгВв
4,12£
70,0
III** |
NJBe
4,П8
69,5
GdBe
4Д2(?)
69,9
ErBe YbBe ThB,
4,Ю2
69,0
4,13 4'32
70,44 80,62
§ 264. Полиметаллиды
При внедрении в решётку металла атомов некоторых элементов
(С, N, P, As) в исходной структуре металла иногда возникают комплексы
атомов, соединённых в группы (координационные сферы).
В качестве яркого примера приведём Сг23Св и Мп23Св, где атомы Сг
(соответственно Мп) образуют комплексы в форме кубооктаодра — К
О Сг
ОС
^ х\ х\
\Z±
хРп
iz:
Рис. 552.
(к. ч.= 12) и куба —С (к. ч. = 8), внутри которых нет атомов углерода, и А
(антипризмы), содержащие атом углерода (см. рис. 552).
1. Сг2яСв (Мп28С,)
Решётка кубическая С.
Координация К/О/3,2/А.
Тип Сяуоуз,2/л —
Пространственная группа Oh — Fm3m.
Z-4.
Сг 1к.ч. = 6С(0); ОН к. ч. = 12С(А');
Сг III к. h. = 2C(Z); CpIVk.4. = 3C;
С к. ч. = 8Сг(Л).
Вещество
aw
Г.г.8Св Ю,0Л8
Мп28Св ?
Примечание
SB II. 59,368
§ 255] совдинкння с комплексным катионом, i группа 897
На рис. 552 слева—схема ячейки из 8 октантов. Справа—частичное
распределение атомов в двух октантах. Буквами С и К отмечены позиции
кубов и кубооктагщров на верхней грани.
Для понимания структуры Сг2зСв рассмотрим тип BiF3 (хотя Сы по
рис. 560). В вершинах и центрах граней ячейки поместим атомы Crl
. ( а всего —-f-^ ^=4 J. С этими же позициями совместим центры кубоокта-
эдров СгШ (при том же количестве позиций, что и Сг I, всего разместим
в 12 раз больше, т. е. 48 атомов СгШ). В центрах октантов поместим атомы
С И (8). Наконец, в центрах рёбер и в центре ячейки ( —+ - = 4 ) кубы С IV
(8 х 4 —32 атома). Таким образом, все позиции в ячейке BiF3 атомов
как металла, так и неметалла займут структурные узлы из атомов
Сг (4 + 48 + 8+32-92).
24 атома углерода расположены между кубооктаэдрами и кубами Сг
и образуют октаэдры вокруг куиооктаэдров СгШ и лежащих в центре
последних атомов CrI.
В свою очередь, атомы Сг образуют в результате вокруг атомов углерода
координационную сферу в виде закрученного куба (антипризмы): г^ань
куба + грань кубооктаэдра.
Б. СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ КАТИОНОМ
§ 255. Гидраты и аммиакаты солейГщелочных и щёлочноземельных
металлов
1. LiOH НО
Решётка моноклинная М'.
Координация T/Z.-
Тип Mju.
Пространственная группа
C\h-C2\m.
Z = 4.
Вещестпо .
\ а
\ Ь
а : Ь : с
?
LiOH-H2.)
7,:J7
8,2G
3,19
0,89J : I :0,386
lions7
Примечание. SB VII 4
Атомы лития имеют
координационную сферу,
содержащую 4 атома О:
2 (группы ОН) на
расстоянии 1,95 кХи 2 (группы ОП2)
Рис. 553.
698 ч.н. структуры неорганических веществ [гл. X
на расстоянии 1,97 кХ, образуя искажённые тетраэдры (Т). Последние
соединены попарно общим ребром. Кроме того, общими вершинами они
соединены с парами тетраэдров из соседних ячеек, образуя бесконечные
цепи вдоль оси с. Тетраэдры связываются в цепи через группы ОН.
0(ц2о)-атом тетраэдра образует с О(он)-атомами угол в 100°.
00н"атом образует с 0И2о-атомами углы в 80°.
2. Mg(HaO)eCl2, Mg(HaO).Br2
Решётка моноклинная Л/.
Координация Z/1.
Тип Mm~SBJl7.
Пространственная группа C\h — C2\m.
3 = [Mg(HaO),].
2 = 2.
Ms к.ч. = 2С1(3,99) + 6С1(4*' = 4,61;2»' = 4,81);
CI к. 4.= lMg(3,99)( + lMg(4,8lA)).
Рис. 554.
Вещество
М.ъ'(НаО)вСЛ2
М^(Н,0),Вг,
а
9,90
10,25
Ъ
7,15
7,40
с
6,10
6,30
а : Ь : с
1,390 : 1 :0,856
1,386: 1:0,852
Р
94°
9S°30'
Примечание
SB III 491
SB III 124
Mg(H.,0)e -почти правильный октаэдр. Два ближайших атома С1 лежат
над серединой граней октаэдра, так что решётка как бы сложена из
молекул |[Mg(HoO)eCL]|. Иными словами, молекулы воды не полностью
экранируют Mg' + -HOH.
Ср. с [Mg(NHs)e]X2, где возникает координационная решётка типа
Ссгг или с [Ga(H20)e]Glt типа #рб/рз- На рис. 554 сопоставляются
элементы структур MgCla6H20 и MgCla.
§ 25">|
СОКДПЬЕНИН <' КОМПЛЕКСНЫМ КАТИОНОМ. II ГРУППА
Ь99
3. Mg(NH9).Cl2, Mg(MI3)cBra,
Mg(NU8)A
Решётка кубическая С.
Координация С/Т.
Тип Ccir-SBH6l.
Пространственная группа
Oft—Fm'im или Tfi.
Z = 4.
1 Вещество
Mg NH8)eCb
Mg(NH8)eBr,
M-(^II3)tiJ,
ate
10,158
10,468
1D,978
Примечание. SB III 475
Структура вполпо
аналогична структуре CaF2 (Сс/т)-
4. [Са(Н20),]С12,
[Ca(H20)e]Br2,[Ca(H20)e]J2,
[Sr(H,0),]Cl„
[Sr(H20),]Bra, [Sr(H20),]J2,
[Ba(H20)e]J2
Решётка гексагональная
II.
Координация 12/6 = P6/P3.
Тип HP6ip3-SBJi9 = H63
(деформированный).
Э=[М(Н,0),].
Пространственная группа
Did -C3m (вместо
Clt — C3 для /13).
2 = 1.
Э к.ч. = т2Х(Р6);
Хк.ч. = 6|)(М).
Щ[Щ
Рис. 555.
ЙРЧЗ^
l'nc. 55(1.
О Hcll
Вещество
I a
с
1 Л * П
[Са(Н20),]СЦ 1 [Са(11,0),]Иг,
7,86
3,905
0/.07
7,97
3,97
0/OS
1Са(Н,0)6].Ь
8,4
4,25
0,-1
[Sr(IIaO)e]C]2
7,906
4,07
о *г
Примечание
SB II 102
KaPt(3CN)e
700
Ч.Н. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ 1ШЩКСТВ
[гл. X
Вещество
а
с
с/а
[Sp(HjO).]Bp1
8,212
4,146
0,505
[Sr(H20)e]J,
8,51
4,29
0,50't
[Ва(Н20)в],Ь
8,9
4,6
0,Г*
Вещество
[Sr(H,OVjOH2
а
6,41
с
5.R1
с/а
Примечание
0,906 SB II 56
б. [Sr(H.20)8](OH)2
Решётка тетрагональная Т.
Координация qjl.
Тип Tin-SBE^.
Пространственная группа
D\h — Pkjmmm.
p = [Sr(H20)8](/>4).
Z = l.
p к.ч. = 4(ОН) (квадрат);
ОН к.ч. = 2 р (линейная).
Решётка состоит из слоев, перпендикулярных оси 4. В этих слоях
атомы Sr образуют квадратную сотку. Середины сторон квадратов
заняты группами ОН. Молекулы Н20 образуют вокруг атома Sr
тетрагонально-призматическую координационную сферу.
6. [Sr(H20)e] [OJ
Решётка тетрагональная Т.
Координация qjq.
Тип Т^/я—SBE62.
Пространственная группа
D\h — РЬ/ттт.
а = 02; p = Sr(HtO)e;
Z = l.
Sr к. ч.=8Н20 (/>4).
ак.ч.^43 (квадрат);
р к.ч. = 4а (квадрат).
Как и в случае Sr(H20)8(OH)2, решётка состоит из слоев,
перпендикулярных оси 4. В этих слоях атомы Sr образуют квадратную сотку.
Они окружены 8 атомами О ( = 402). Центры квадратов заняты центрами
групп 02. Таким образом, при соотношении структурных узлов а^ мы
имеем редкий пример квадратных координатных сфер у иона с
оболочкой, имеющей шаровую симметрию (Sr2+) за счёт экранирования 8
молекулами воды.
§ 256. Гидраты солей хрома
1. [Сг(Н20)в]С13
Решётка ромбоэдрическая R.
Координация АЗ + h/T.
Тип /U3+/«/T-SB/22J
Простр. гр. Dld — R3c.
Вещество
[Sr(H20)8]L)a
а
6,32
с
5,56
с/а
0,88
Примечание
SB II 57
P = [Cr(HaO)e]. p к.ч = 6С1(4,ЗА) (тригональная
(шестиугольник) {АЗ + h); Сг —Н20=1.90.
G1 к.ч. = 43(7т).
Z = 2.
SB III 127-128. Октаэдры [Сг(НаО)а]
расположены по одной оси с треугольниками С13,
образующими тригональные антипризмы.
Сравни с типом tf 63[K2Pt (SCN)] и Li3N рис. 396*.
См. А1(Н,0)6С13.
антипризма) + 6С1(4,5А)
Вещестно
а
[Сг(Н,0),]С], 7,95
л
97°
§ 250]
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ КАТИОНОМ. IV* П'УНПА
701
702
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. X
§ 257. Аммиакаты солей марганца
1. [Mn (NH3).] С12,
[Mn(NH,),]Br2,
[Mn(NH,),]Ja
Решётка кубическая С.
Координация С /Т.
Тип Ctc/t-SB//61 . ..
Пространственная группа
Ot-FmZm, (или Т?,).
P = [Mn(NH3)e].
Z = 4.
р к. ч. = 8Х (куб);
X к. ч. = 4 ^ (тетраэдр).
Вещество
[Mn(NlI8)elCla
[Mn(NII3)e]Br2
[Mn(NH3)e]Ja
fl«
io,w8 ,
«,519 '
11,0 *7
Примечание. SB III 475
См. замечание к рис. 555.
Рис. 558.
§ 258. Аммиакаты солей железа
1. [Fe(NH3)e]CI2,
[Fe(NH3),]Br2,
[Fe(NH3).]J2
Решётка кубическая С.
Координация С]Т.
Тип Cc/r-SB#61 (SBC1).
Пространственная группа
Oh— Fm3m (или T'i,).
P = [M(NH3),].
Z-4.
рк.ч. = 8С1(С);
€1к.ч. = 4р(Г).
Вещество
[Fe(NH,),]Cl2
[Ре(МН,),]Вгг
[Fe(NH3),]Ja
о„
10,148
10,468
10,9б5
Примечание. SB III 475
Hal
FefNH,)6
Рис. 559.
§ 259] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ КАТИОНОМ. IX* ГРУППА 703
§ 269. Аммиакаты и другие соли кобальта
1. [Co(NH,)e]J.
Решётка кубическая С.
Координация С+ 0/0, Т.
Тип Сслою, т -SB DO, и
НИ.
<x = J; ?=[Co(NH,),].
Пространственная группа
Oh — Fm3m (или T2d).
Z = 4.
\i к. ч. --= 8а11(куб) + 6а1
(октаэдр);
al к. ч. = 6р (октаэдр);
all к.ч. = 4|3 (тетраэдр).
Вещество
а»
ОМ
[Co(NH,)t]Je|
10,89(10,88)
323
Примечание. См. (NH4)t
AlFeSB I437 HBiF3SBU23.
В SB III 475 ошибочно
отнесён к типу С\ (Я61)
вместо #71. Период
идентичности по SB III—в
скобках
о
Рис. 560.
№
О з
Рис. 561.
Co(NH3)6
Элементарная ячейка Со (NH8)eJ3 (рис. 561) построена по типу
элементарной ячейки BiF3 рис. 560, в которой |Co(NH3)„]d+ -ион занял позиции
онов Bi3+, т. е. узлы решётки Ск.
04
/U'*
Ч. II СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
2. ICo(NH,),]Cl2,
[Co(NH,),]Bra, [Co(NH,)e] J2,
[Co(NH2CH,)«] J2
Решётка кубическая С.
Координация CjT.
Тип Сс/г-SBC1 и #61.
Пространственная группа
0^, — Fm'dm или Т\.
3=|Co(NH3),].
Z==4.
Як.ч. = 8X(C);Xк.ч. = 4S(7,).
Вещество
[Co(NII,)e]a.
[<"о(МН8),]Вгя
[<<o(NH3)6]Ja
[r,ou\H,CHa)e]J2 j 12,0Г
pCo-N
10,10
о
10,389
i 10,91,
2,5|
2,7
Примечание. SB III 475, см.
также SB I 429
Рис. 502
§ 260. Аммиакаты солей никеля, палладия, платины
1. [Ni(NH,).]Cl„
[Ni(NH3),]Br2, [Ni(NB,).]J„
[Ni(NH2CH3),]J*
Решётка кубическая С.
Координация С/Т.
Тип Ccit-SB CI; #61; /1..
3 = Ni(NHt)..
Пространственная группа
Oh — Fm3m или (Та).
Z = i.
Зк.ч. = 8М(С);Мк.ч. = 43(7').
Вещество
[Ni(NH,),lCl4
[Ni(NH,)f]Brt
|Ni(NH,),]Ji
[Ni(NHiCH,)e]J;!
aw
10,064
10,340
10,875
12,027
Примечание. SB I 429, SB
III 475
Рмс. 563.
§ 260]
СОВДПНННПЯ С КОМПЛЕКСНЫМ КАТИОНОМ. X* ГРУППА
705
2. [Pd(NH3)4]Cl2 Н20
Решётка тетрагональная Т.
Координация q + Pijq + Pijr +r^q + Pijr + r.
Тип Tq+Pitr+r-SBHi».
Пространственная группа Dtn — Pb/mbm.
a = [H20]; p-[Pd(NHe)4] (квадрат).
Z-2.
ак.ч.-43 (квадрат) + 8С1 (призма);
p к.ч. = 4а (квадрат) + 8Gl (призма);
CI к. ч. = 4а + 4Й (r + r).
Вещество
IPd(NH8)4|CL-H,U
a
10,302
с с n
1
4, .4'» 1 0/i2i:i
Примечание j
sn in loo
Центры квадратных ионов [Pd(NH3)4]2+ и ионы С1— занимают в ячейке
позиции, в точности отвечающие позициям центров ионов [PtCl4]2"" и ионов
К+ в решётке K2PtCl4 (тип //15) (рис. 604). Большой ])азмер комплексного
с^#=о
J'in*, fiC/j. Дно илгмсптлрииг ичгпкм.
иона вызвал, однако, возможность размещения дополнительных молекул
воды. Интересно сопоставить [Pd(NH3)4] Cl2-Н20 с квасцами (рис. 663).
Напишем ряд:
[K(!l,O)0][Al(H,O)el[SO4], и H,0[Pd(NlI:,),]cL
В элементарной ячейке первого вещества и двух элементарных ячейках
второго положение групп S04 и атомов О (в центрах октантов) примерно
аналогично. Но если в квасцах узлы а и р лежали на осях 4, чередуясь
(а -3 — a — [J), как в типе NaCl, то в Pd(NH3)4Cl2H20 узлы д лежат друг
над другом; узлы Я также лежат друг над другом.
706
Ч.И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
3. Pt(NH8)aCl4 (транс-форма),
соль Жерара
Решётка тетрагональная 7\
Координация Pi.
Тип Тр^.
Пространственная группа Ал—
Pi/mmm.
Z-2.
Вещество . . .
Pt(NH8)2Cl4
5,72
10,37
1,81
Примечание. Н. В. Пелоп,
Г. Ij. Покий, Л. Л. IJonona.
ИЛИ (х) 1947 г., стр. 2V.).
5
U
12 3 4
Эта структура представляет
большой интерес.
Тетрагональная центрированная
элементарная ячейка содержит
структурные узлы, состав которых
отвечает формуле соединения.
Атомы хлора образуют вокруг
атома платины прямоугольную
(не квадратную)
координационную группу со сторонами 3,41
и 3,12 (рис. 565*). Расстояние
Pt —G1 равно 2,31 кХ и
валентные углы равны 85° и 95°.
Хотя у многих аммиакатов ^_<^
расстояние Me — ГШ3 равно (^WH (/h (][ О Pt
2,02 — 2,05, в рассматриваемом v—^ 3 ^^
соединении оно оказалось
равным 2,35 кХ.
Вопрос о причинах
искажения формы координационной
сферы в работе не
рассматривается. Нам кажется
возможным предположить,
что это вызвано в
определенной мере
необходимостью разместить в
вершинах бипирамиды
(основание которой занято
атомами хлора) не шаровой,
а тетраэдрический
радикал, каким является в
данном случае группа
NH3, образовавшая связь
с атомом платины. Если
основания тетраэдров -
равносторонние
треугольники—расположены
антисимметрично и притом так,
что одна из сторон тро-
6 7 8
10
:>г>ь.
§ 262]
сокдпнкнпя с комплексным катионом, и* группа
707
угольника параллельна основанию бипирамиды, образованному атомами
хлора (рис. 565*), а такое расположение наиболее выгодно в стерическом
отношении, то одна из сторон квадрата должна удлиниться не менее, чем
на 0,16-0,2 к\. Такое искажение должно было бы исчезнуть, если бы
оказалось возможным вращение молекул Nll3 вокруг связи, соединяющей
их с центральным атомом.
§ 261. Аммиакаты солей цинка и кадмии
l.[Zn(NHa)e]J2,[Cd(NII:1)e]J,
Решётка кубическая Г.
Координация С\Т.
Тип -Сс/т- SB//61.
11ространственная группа
Ob—FmUm (или Та).
?-[M(NH3)e].
Z-4. Я к. ч. ,-_ 8 J (Г);
J K. Ч.:--7!? (Г).
2. [Zn(NII3)X1l2], см. § 658.
3. |('<l(NH,)Xlo], см. § (>'i'i.
I
[Zn(NH3)6J.I > [(M(N!l3)e]J2 | Примечание
. _____ ^
№,%'_
ll.OV, . SH HI W~
J'lie. .>(><
§ 262. Гидраты солей алюминия
[Al (ILO).] CI,
Решётка ромбоэдрическая R.
Координация _13 + h/T.
Тип IiA.4hlf-S\iJ22.
Пространственная группа D^a Rlir.
MAI(H2())e] {()).'
3 к.ч.-.■GCH + 6C1II (Л3~)-й); ('<м- Р"«'- Г;>(>~ »'» •■тр. "08.
ГЛк.ч. ^(f).
1 Нощестно
|А1(П.О)в|(-.1;,
1
! И
7,К.
1
SB III !•_>«
\pимочanno
129, см. <-r(H2())6(:]3
Октаэдры Al(IJ,0)e окружены 12 CI; шестью Cl: образующими три-
гональную антипризму и расположенными по три сверху и снизу
октаэдра, и шестью, образующими шестиугольники в плоскости, проходящен
через атом А]. Атом С1 окружают \ октаэдра, см. стр. 700, 701.
708
Ч.П. СТРУКТУРЫ НКОРГАНИЧКСКПХ ВЕЩЕСТВ
1гл. X
сэ
§ 263]
С01;ДШ11.1ШЯ С КО.Л1Г.Л1 KC1IUM КАТИОНОМ. V 1ТУ1П1Л
709
§ '^03. Соли аммония и фосфоншг
1. [MI J Г, [ND4]P
Решётка гексагональная
Я.
Координация Т/1'.
Тип #г/7—SB54.
Пространственная г[>уппа
Cti> — СЬтс.
Z---1.
1 Вещество
1 а
1 с
с/а
d
М'
1 ом
i
Примечание. ^
UND4]F
'.,Н9
7,02
1,59,
-',№
2,70
Г>9,7
*И 1 79, 107.
2. a[NH4]Cl, a[NH4]Br,
a[NH4]J, a[ND4]Cl,
a[ND4]Br, a[ND4]J
Решётка кубическая С.
Координация OjO.
Тип Co/o-SBfil.
Пространственная группа
Ои — FmSm.
P=[NH4].
Z = 4.
Рис. 568.
С^
9-zp9
b^Mo7
И
г
^и
<^^
6
6^—Ъ^
О NH*
О Hat
Рис. 569.
Вещество
ОМ
e[N114]C]
0,53
69,G
a[NH4]Br
0,90
82,1
a[NH4]J
7,2'i
9'i,9
«[NDJC1
—
a[ND4]Br
—
a[ND4]J
—
Примечание
SBI 73,108
устойчивы выше 184,3°,
соответственно 137,8°]
и—17,6°С 1
710
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
ГЛ. X
3. aprajCl, p[NH4]Br, p[NH4]J, p[ND4]Cl, p[ND4]Br, p[ND4]J
Решётка кубическая С.
Координация С/С.
Тип С с/с -SBB2.
Пространственная группа
0Хн—РтЗт.
P = [NH4].
Z = l.
Рис. Г.70.
Вещество
t°C
<lw
ОМ
t4\
U1P
t°C
°w
t\:
aic
P[NH4]C1
ниже
184, 3
3, 80
57,5
18
3,8G84
3[NH4]Br
ниже
147,8
4,05
6G,4
—78 j
з,83',3
—185
3,8206
S$[NH4]J
ниже
-17, G
4,37
91,3
„ .
1
—
18
3,868,
-78
3,8375
— 185
3,8H>0
■--
—
i 1
[i[ND4]J Примечание
i |
--
—
.__
SBI 75, 108, три-
морфны (?). Ниже
указанных
температур устойчивы
ячейки Сс,с Иыше этих
температур ячейки
по Ch. Abslr.
38 4488 (lUVfc)
1 'Коэффициент jiii-l
I нейного расширения
i между - 78 и - 185°С
1 для NH4C1=3,5-Ю-*
j | 1гдляЫ04С1=5.5.Ш-5.|
4. Y[NH4]Br, y[ND4]Br, NH3CH3C1
Решётка тетрагональная Т.
Координация q + q/Di.
Тип Тч+1„„ (или~7,ш/о«)-ВВВ25.
(Y[NH4fBr);
Пространственная группа Din — Pi/nmrn,
P = [NH4].
Z = 2.
J'nc. Г>71.
®NH4
• Br
$ 2631
сокдпшшпя с комплексным катионом, v группа
И
Вещестпо
v[NH4]Br,
v|ND4]Br
| NH8CH,C1
-юо
-71,5
-145
комн.
a
6,007
5,713
5,697
6,0'»
с
I
4,035
4,055
4,046
5,05
с/а
0,707
0,7098
0,7102
0,836
Примечание
8В III 10,231, t перехода
?7±Т = -39°С 1
^ SB IV 95, деформированный
/тип CsCl
На рис. 571 ионы Br изображены чёрными и маленькими, чтобы
лучше подчеркнуть небольшое смещение их центров тяжести от ребра.
Параметр z =+0,0г30. Если принять за основание ячейки рис. 571
среднюю плоскость, л которой расположены NH4-ионы, то легко обнаружить
сходство с ограниченной пунктиром ячейкой рис. 572.
5. PBr-[PBr4]Br
Решётка ромбическая, « = 5,0; 6--=10,9; с—8,3. Пространственная группа
Щ--РЬст\ Z^i; р = [РВг4]*; а--=В7.
6. [PH4]J, NH4SH
Решётка тетрагональная Т.
Координация q-\-q\Di.
Тип 7\+,/м-ЬШЛ0. р- [РН4].
Пространственная группа Din* Pi/nmm.
Z = 2. р к. ч..-=8 J (призма); J к. ч.-8 р (призма).
р »н
Рис. 572.
Вещество
[PHJJ
[NHJSH
«
6,34
6,011
с с/а
4,62 ! 0,729
4,099 0,667
| ОМ
92,9
72,4
| Примечание
SB 1 93
SB III 7
Сопоставление рис. 571 и части элементарной ячейки рис. 572,
ограниченной пунктиром, показывает, что, по существу, структуры NH4SH,
отнесённая SBI к типу PH4J(i?10), и yNH4Br, отнесённая SB III к
новому типу 525, настолько близки, что типы PH4J и yNH4Br могли бы
быть объединены. Лишний пример, по нашему мнению, недостаточно
критического отношения редакции SB к собственной нумерации типов.
7111
4. U. СТРУКТУРЫ 1Ш0РГЛ1ШЧ1ХКПХ ШЛЦ1ХТН
I гл. X
Н. СОЕДИНЕНИИ С КОМПЛЕКСНЫМ АНИОНОМ
§ 264. II группа периодической системы: бериллий Be, магний Mg,
кальций Са, стронций Sr, барий Ва
1. BaBeF4 изоморфен
BaS04, см. SB II 486;
2. [NH4]2[BeF4], Na,
[BeF4] (?).
Решётка ромбическая
слоистая ORs.
Координация Л-j- 1/3.
Тип ORZ+i~-SBHlt.
Пространственная
группа Dlh—Pmcn.
a = [NH4]; p-[BeF4J.
Z = 4.
Рис. 57:i.
Na2BeF4?
a
b
с
a : b : с
j dbe-V
4,9
10,9
fi fi
0,448 : 1 : 0,606
(NH4LBeK4
5,8 1
10,2
7,5
0,569 : 1 : 0,7353
1,61
По SB III 436-437, (NH4)2BeF4 изоморфен K2BeF4, но не NaaBeF4.
В SB VII 118 приводятся без комментариев данные работы Г. С. Жданова
м И. Г. Севастьянова, относящиеся к Na>BeF4. Класс /)0/|, а = 10,9;
Л-=6,6; с = 4,9.
Вышеприведённая таблица показывает, что изоморфизм обоих
соединений не исключён. Оси для Na2BeF4 изменены: а ---- Ь\ £ = <:; с— а.
Структура элементарной ячейки см. K2S04.
§ 265. IV* группа периодической системы: титан Ti, цирконий Zr,
гафний Ш, торий Tli
1. (NH4)3 [ZrF7]; K3[ZrF7]
Решётка кубическая С.
Координация С+д/7\д.
Тип Сс ч о/Г.о — SB nTl (720.
Пространственная группа 0\- - Fm'im.
a = [NHJ; 3 = [ZrJ'V|.
2051
СОКДШШППЯ С КОМПЛЕКСНЫМ ДНПОНО.М. IV* П УШ1А
71Я
Вещество
й„ . . .
(NH4)3ZrF7
9,365
K8ZrF7
9,365
Примечание. Статистическая
ориентировка во всех
направлениях группы ZrF73' (рис. 575),
имеющей конфигурацию Ог.
8. [Mg(HaO),][TiF,J;[Zn(HaO)6
[TiP.l; [Zn(Ha0),][ZrP.]
Решётка ромбоэдрическая R.
Координация RfR.
Тип Яд/я -SB/6,
Пространственная группа
Clt-Rb.
a = [M(H2G).]; ? = [AF,1.
Z = l;a:(000);3: (»/, Ъ '/*)
о к. ч. = 8 [i (ромбоэдр,
Р к. ч. = 8 а (ромбоэдр, R).
Я);
Вещество . .
Mg(H2Oe)TiFe
Zn(H20)eTiFe
Zn(H20)e ZrF6
96°4'
96°2'
SH II 10/j, ( м. M£(H2L)e£iFe
М^НД
Рлс. 570. Ромбоэдрический деформированный тип.
сс/с-
В центрированных ромбоэдрах, имеющих
угол, меньший 60° или близкий к нему
(отвечающий ромбоэдрически искажённой
решётке Nad), к. ч.=6, а не 8 (см.
532): к центральному узлу близки только 6 узлов, два же,
находящиеся в вершинах ромбоэдра, через которые проходит ось 3, сильно
удалены. С увеличением угла ромбоэдра два эти узла все более
приближаются к центральному (см. рис. 527j). Углу 90° отвечает к. ч. 8. При
ещё больших значениях углтж к. ч. .8 переходит н к. ч. 2.
рис.
9,91
714 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩНСТИ
[гл. X
3. Rb, [TiCl,], Cs2 [TiCIJ,
Rb, [ZrCI,], Cs2 [ZrCI,]
Решётка кубическая С.
Координация TjC.
Тип Cric — (анти-SBCl)-
-SBtf6l и Л,.
Пространственная
руина
О.1,—Fm3m (или Г).
? = [ТКИ.|.
2 = 4.
Вещее т но
Rb,TiC1e
oaTic:ie
Rb.Zrcie
Cs,ZrC1e
a
<) 9;>
10,219
10,178
10,40;
Примечание. SB
См. K,PtCle.
OM
244
2<i7
263
2S2
III 121
Pjic. 577
§ 266. V* группа периодической системы: ванадий V,
ниобии Nb, тантал Та
1. M3Ta*VTatV —ион, - дву-
гранецентрированная *)
призма i>32+.
2. K2[NbF7], K2[TaP7]
Решётка моноклинная М.
Координация х\у.
Тип Мх,у- SBtf62(K2NbF7).
Пространственная группа
C\h-P2Jc.
H^[NbF7].
Z = 4.
Вещество
а
Ь
с
р
а\Ътс
K,NbF7
5,8Г>
12,67
8,50
90°
I
K>TaF7
5,85
12,67
8,50
90°
0>'iGiU:0,G710f'if>:J:l:0,G7l
Примечание. ^B VII об.
Рис. 578.
Случай соиершешю одинаковых размеров элементарной ячейки у двух
гомологов Координационная сфера NbF*~ имеет форму тригональной
призмы, против одной из граней которой расположен седьмой атом F (P'S+).
<]\т. рис.. 578, внизу. Вверху—проекция.
*) См. J.A.Ch.S. в*>, 329 (1943). См. таблицу ?»',.
§ 267|
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ ЛШШПОМ. VI* Г1-У11НЛ
715
3. K,[\b<U K,[TaOJ, Rb,[TaO,], Cs, [TaOJ
Решётка тетрагональная Т.
Координация х/у.
Тип Tjcjy — SB? Пространственная группа Dm —J Vim.
Р - INbO.].
Z ----- 2.
U иачппо K3[NbOJ
6,78
7,86
KaLTaUj
Ub3[Ta()J | Cs3[TaOs]
6,78
7,88
7, Of»
8,05
7,:*7
8,3'»
Примечание
Ионы [MOM'J тотра-
эдрические. Ь перши-
пах тетраэдра раеиоло-
жены группы 02
§ 267. VI* группа периодической системы: хром Сг, молибден Мо,
вольфрам W, уран U (и ураниды)
1. [Co(NH,)1][Cr(CK)e]
Решётка ромбоэдрическая R.
Координация R/R.
Тип /?/?//? — SB У6Х.
Пространственная группа С^
ЯЗ.
« [Co(NII3)e]; p = lCr(CN).]-
Z = 1 аЗ. а к. ч. - 8 3
(ромбоэдр, R)\ J3 к. ч. = 8а
(ромбоэдр, R).
_. ^^$®щ
ColNHJ, Ct(CN),
Рис. 570. См. примечание на стр. 713.
Вощостио
[<'o(NH,),] [Cr(CN),]
а
6,71
X
97°55/
Примечание
S4 II Ю'». см. [M^IIjO),] [SiF,]
716 Ч.П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
2. KJMo (CN)J 2Н20
Решётка ромбическая OR1 °.
Координация сложная xjyjz.
Тип UR^yiz-SBF-Zt.
Пространственная группа D^h — Рпша.
й-= [Mo(CN)4]; т-=[Н8')].
Z -= 4.
Рис. 580.
1 Вещество
K4[Mo(CN)8] 2Н.О
а
Ь
16,55 11,70
с
а : b : с 1
8,68 ! 0,7073:1:0,3709
См. SB VII 25, 132.
В структуре представляет особый интерес р-узел: [Mo(CN)8]4 -ион.
К. ч. — 8 выразилось в данном случае в образовании
координационной сферы в виде весьма редкой конфигурации—(чуть ли не
единственный пример среди комплексных ионов) тритонального додекаэдра (Do).
Расстояние Мо —С равно в среднем 2,15. Мо — N = 3,29. С — N = 1,14-
(тройная связь). CN-группы лежат на прямых, проходящих через центр
координационной сферы. Взаимное расположение атомов весьма
своеобразно: атомы лежат в двух взаимно перпендикулярных плоскостях.
Группы CN расположены линейно на радиусах, идущих от атомов Мо.
§ 267]
СОЕДИНЕНИЯ С КОМПЛККСНЫМ АНИОНОМ. VI* ГРУППА
717
3. [NHJ3[Mo08FJ
Решётка кубическая С.
Координация О, Т/С + О.
Тип С0% т/с+о - SB анти НИ.
Пространственная группа fj —F-Ydm (Tl—F2\\ или Т1 —РЧ$)Ч причём, если
О- и F-атомы распределены пс статистически, ^пространственная группа
отпадает, повидимому, имеет место Т\
a = [NHJ; ^[Mo03F8].
Z - 4.
a I к. ч. =: 6 [4 (октаэдр); a II к. ч. - 4 3 (тетраэдр);
р = 8 a II (куб)+ 6 a I (октаэдр).
О 1 2 3 4 5 6 7 в 9 Ю г\ кш
I : 1 1 1 1—-1 1 ' « ' \J NH4
MaO,R
з'з
Рис. 581.
Вещестпо
[NHJ3 [Мо03Г,]
ахо
9,10
ОМ
188
Примечание
См. [NlI4]3[A!Pe]f стр. 750.
4. KJCrO.]-K,[CrlO,)J
Решётка тетрагональная Т,
Координация?
Тип Г -SB.
Пространственная группа Du~ J^-m.
р = [СЮ8] = [Сг(02)11.
Z = 2.
См. K3Nr>08, нозможно один н тот же тип.
Вещестно
а
К,СгОв 6,70
с
7,00
718 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
о. Na2Cr04
Решётка ромбическая OR.
Координация 9/5.
Тип 0fl9/:>-SB#le.
Пространственная группа D\u — Pbnn.
Z =- 4; а - Na; p == [СК)4].
Риг. Г,82.
Вещестпо
Na,Cr04
Л
5,91
Ь
9,21*
с
а : Ь : с
7,20 ; 0,Ш : 1 : 0 ,780
Примечание
SB IV 45
Атомы О образуют вокруг атомов Сг островные координационные
сферы (О04)" в виде почти правильных тетраэдров. Узлы 3
окружены 3 узлами Na I: 1 на расстоянии 3,26 и 2 на расстоянии 3,63 и 6 Na II
на расстоянии 2 — 3,68 кХ и 4 3,74 кХ. Узлы Nal окружены 5 узлами [;.
Внутри тетраэдров (Сг04) 2 расстояния Сг —О, равные 1,58 кХ, и 2
расстояния Сг —О, равные 1,62 кХ.
Ячейка lioimoii решётки Na.2Cr04 с нонами Na+ вне тетраэдров |(1г04|
заслуживает сопоставления с ячейкой молекулярной решётки Zn(NI 1.,)Д'1.
рис. Г) 13.
§ 267] СОКДПНКШШ С КОМПЛЕКСНЫМ АНИОНОМ. VI* ГРУППА 719
6. СаМо04, ВаМо04, РЬМо04
Решётка тетрагональная Т.
Координация q + Sjq-\-S.
Тип Tq+siq+s — SB#4 (тин шеелита).
Пространственная группа Си, -~J4Ja.
Z = 4. H - [Мо()4] (тетраэдр).
М к. ч. = ip (квадрат) + 4р (тетрагональный тетраэдр);
р к. ч.—4 М (квадрат) [4 М (тетрагональный тетраэдр).
© Мо
Рис. 58:!.
Вещество
СаМо04
ВаМо04
РЬМо04
-
i
5,2,
5,5G
г.,4
С
".'-,,
1^,70
12,08
c/tf
2,1'.)
:,;;и
2/.>з
ом
78,2
98, Г»
88, Г»
Примечание
1 г HI МУ, см. KJO,
!
i
720
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. X
7. CaW04 (шеелит), BaW04, PbW04
Решётка тетрагональная Т.
Координация q -f Sfq + S
Тип T4+siq+s -8ВЯ4 (CaW04, шеелит).
Пространственная группа С\ь —Л/а.
3 = [W04] (тетраэдр).
Z = 4.
М к. ч. --4 р (квадрат) + 4 3 (тетрагональный тетраэдр);
р к. ч. ^4 М (квадрат) + 4 М (тетрагональный тетраэдр).
Рис. 584.
1
Вещество
i CaW04
1 BaW04
PbW04
а
5,24
5,60
5,44
с
11,38
12,69
12,01
■
с/а
■
2,17
2,28
2,21
ОМ
Примечание
78.1 I SB1 349, см. К-Ю4
99.2 |
8S,9 I
§ 267]
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VI* ГРУППА
721
8. Mg[W04], Mn[W04], Fe[W04], Co[W04], Ni[W04], Zn[W04]
Решётка моноклинная М.
Координация 6/6 (?).
Тип Мт (?) - SBtfO, (MgW04).
Пространственная группа С{\х — Р2\с,
■} = [W04] (тетраэдр).
г*2.
Puc. 585
Вещсспш
1
Ь
с
Й
*х
Вещестно
1
Ь
1 п
сс
Mg[W04]
4,68
5,66
4,92
80°40'
6,88
Cu\V04
4,Г>6
5,69
4, (J8
90э
7,76
1
xMn[\V04l
4,84
Г), 76
4,97
89°7'
7,18
NiVV04
! 4,69
j 5,66
! 4,93
1 89^40'
7,78
1
Fe[VVO]4
4,70
5,69
4,9:i
90D
7,61
Zn\V04
4,68
i 5,73
1 4,95
89^30'
7,79
19 Примечание 1
rfw-o = *>':*A- 1
dMg-0=2'25A
(срапнн с 2,10s ii MgO) 1
В каждой ячейке (рис. 585)
2 атома Mg ( -- -[- — J и 2 атома
ЧтЧ)
В работе были определены точно лишь позиции атомов металла.
Положение атомов кислорода дано в S13II на основании общих соображений.
Если бы они были правильными, можно было бы сделать заключение, что
л решётке сущестиуют координационные сферы с арочными связями
[W04] —тетраэдр. Хотя формально Mg образует вокруг себя октаэдры [MgOe]
Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
рис. 585), соединяющиеся в цепи по (001), однако большие расстояния Mg — О
говорили бы о наличии переходного типа, невидимому, более близкого
комплексным соединениям типа Mg[W04]. Впрочем, для подобных выводов
положения атомов недостаточно уточнены.
9. Структуры сложных и комплексных соединений урана, уранидов
(а также близких к ним новых соединений редких земель и тория).
В самое последнее время в литературе появились данные,
относящиеся к строению сложных и комплексных соединений пазванных выше
элементов. Назовём, например, статьи Захариасена J. Am. Ch. Soc. 70^
2147 (1948), Acta Grist. 1, 265, 268 (1948). Acta Grist. 2, 57(1949).
Несомненно, опубликованные сведения неполны и не во всех случаях
структуры могут считаться надёжно установленными. Некоторые из них
заслуживают дискуссии. Тем не менее с целью ознакомления читателя с
некоторыми из этих данных мы приводим здесь материалы из названных
выше сообщений, В связи с необходимостью более строгого анализа
координационных соотношений, мы воздерживаемся здесь от указания
координации. Считать эти соединения комплексными можно в настоящее
время лишь условно.
10. £2K;UFe, №2UF0, NaPuFe
Решётка гексагональная Н.
Координация х/у.
Тип Hxjy, структурный тип.
Пространственная группа D\ — С32.
ptNa,ThFe.
z = i.
Вещество
P2K,UFe
tl3Na2UFe
NaPuFe
a
6,53 ±2
5,94 ±1
6,117 ±6
с
4,04 ± 1
3,74 ±1
3,746 ±4
Примечание I
Для последнего из приведённых 1
здесь веществ Z = 3/.> 1
11. &,K,UF.
Решётка гексагональная Н.
Координация х/у.
Тип ff.fc-p^UF..
Пространственная группа D%x — C62 т.
Z-1.
12. yNa2UFe
Решётка ромбическая OR.
Координация х/у.
Тип 0^/y-YNa2UFe.
Пространственная группа ВЦ — Jmmrn.
Z = 2.
Вещество
TNaaUFe
а
5,54 ± 1
Ь
с
4,014-7 11,67 ± 2
Примечание
Na — F = 2,38, U—F = 2,38
722
In 1 1 1
Вещество \ а \ с \
PiK,UFt
6,53 ± 1
3,77 ::: 1
§ 2671
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VI» ГРУППА
723
13. Na2UF7
Решётка тетрагональнан Т.
Координация xjy.
Тип. Г*/.,-
Пространственная группа Z)4/'; - J^jmmm.
Z = 2.
Вещество
Na,LF7
а
5,448 ± 7
с
i 10,8% -j
1
14 \
Примечание
Na — 7F = 2,У0 kX U — 7F =
2,:>ii
14. Cs2PuCIe
Решётка тритона льная (ромбоэдрическая) И.
Координация xjy.
Тип Bxjy (Ka(ieFe).
Пространственная группа Д^ — ГЗ/я.
Z = l.
Вещество
Cs2P«Cl6
1
а с
i
Примечание
7/|-)4-1 | 6,o:i i , Рп-атом окружен октаэдрической координа-
1 ! цлоннойвсферой из атомов хлора на расстон-
, 1 или 2,62 А. Каждый атом цезия имеет 6 ато- !
; мои хлорана расстоянии 3,70 А и 6 на расстои-
| | шш .4,72 А, т. е. по отношению к С] к. ч. = 12
i ! 1
15. CaU04, SrU04
Решётка ромбическая R.
Коордииа ция xjy.
Тип Дг/у.
Пространственная группа.Did — B'Srn.
Вещостно
CaUO,
Sri О,
6,254^1
G, 53 -— 3
36°2'
г>в;*-2'
Примечание
Структура содержит линейные группы
U '2 0 = 1,91 А,
Са -SO = 2/1Г1 А,
U --(И) « 2,2У А
Обращает на себя внимание небрежность в описании этой, да и других
структур в работе Захариасена. В частности, периоды идентичности даны в
единицах кХ, межатомные расстояния даются в ангстремах.
724 Ч. И. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
16- U02F2
Решётка ромбоэдрическая слоистая Rs.
Координация х/у.
Тип R%y.
Пространственная группа /)з« — R'Sm.
Z = l.
Вещество
UOsFa
а
Ь,Ш±1
а
4243'
Примечание
U —6F = 2,50A
U — 20 = 1,91А
Кроме того, приводим некоторые данные о двойных фторидах К и Na
с ураном и уранидами с помощью принятых нами обозначений сиигоний
(и типов) и с указанием пространственных групп.
Вещество
KU§FSI
KU,F„
KU,F9
K'Np2F9
KPusF0
NaUF5
KLT5
Сингония и
пространственная группа
Я
D.fh—C6/mmc
OR
иЦ—Ртст
OR
пЦ—Рпат
OR
D}J--Pnam
OR
пЦ—Рпат
R
R
Элементарная
ячейка
a= 8,18±Jf
c = 16,42 ±2
а = 8,03±3
a^ 8,08 ii
Ь= 7,02 ±1
c = Jl,4,i±4
a= 8,64.4:5
b= 7,01±J
c= 11,43 ± 7
a^ 8,56
b== G,95
c= 11,33
а = 9,08±1
a-107°56/
а = 9,387 ±2
a=107D15' ±2'
Я
2
2
4
4
6
G
1
6,73
6,64
6,49
6,54
6,73
5,81
5,38
§ 267] соединения с комплексным анионом, vi* группа 725
1 Вещество
NaPuF5
KP11F5
RbPuF5
a NaaUFe
* K,UFe
Ь K,UF,
pa Na2UF6
PiK.UF,
p, NaPuF4
TNa,UF6
aK3UF7
«iK,LF7
Na8UF7
Сингония и
пространственная группа
ft
C*-Rl
R
R
С C('iT
0l~-Fm\hn
С С (jT
O^—FiriMn
If
DZh—Cblm
11
/>§—C32
11
H
b\~ С 32
OR
/>2л—Jfnmm
С
T
D\l—J\lamd
T
Элементарная
ячейка
£1 = 8,934:3
a=107°28' ±10'
a -9,27 ±3
*-=\{)ТУ ± W
a = 9,40 ±3
a=106°50' ± 10'
a = 5,565 ±4
ci = 5,934 ±1
c-3,7G±i
a -5,94 4 1
o = 3,74 ±1
e = f>,53±2
c = 4,U4±i
a = G, 12 -t- ^
c = 3,75 4 1
a«5,56±2
c-4,01±i
a = 9,2141
a= 9,20-^ 2
0=18,40-^6
a = 5,448 ±7 |
c= 10,890 ±14
Продолжение
A
0
1 G
V,
/ 3
1
1
t
7*
2
4 |
1
1
8
2
i .
i a<-
1 Г., 03
5,60
1 Г|,88
i 1
5,08
4,53
5,Ю
5,74
4,77 1
6,87 j
5,06
4,12
4,13
*,49
726
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
§ 268. VII* группа периодической системы: марганец Мп, рений Re
1. KMn04
Решётка ромбическая
OR. __ _
Координация РЗ+/РЗ+.
Тип 07?P3+/P3+-SB#2
(барит BaS04).
Пространственная
группа V™ —Рпта.
8 = [Мп04].
Z = 4.
Мп к. ч. = 40
(искажённый тетраэдр).
К к. ч.=7р(РЗ^);
? к. ч. = 7 К (Р3+).
Pin-. 58G. Положения О-атомон не точны.
Pirc. 587.
1 1
Веще ст но i a
КМц04 | 9,10
i
Ъ | с
5,00
ОМ
7,40 9'*,2
Примечание
См. BaS04,
стр. 778
На рис. 587 показано расположение в элементарной ячейке узлов К и Мп
(т. е. центров [Мп04]).
§ 268] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VII* ГРУППА 727
2. AgMn04
Решётка моноклинная М.
Координация 7/7 (?).
Тип Min-SBH09.
Пространственная группа C^ — Р21/п.
^Z~4 AgMn04. 3 = [Mn04] — сильно искажённый тетраэдр.
Риг. 588.
Вещество
Ag[MnOJ
а
5,665
Ъ
8,27
1 1
i |
7,127
92*29,5'
Структура близка"к Я2-типу (BaS()4). На плоскостях Г— yzЛ n( — yzj
лежат по 2 атома Ag, 2Mn и 40. По обе стороны каждой из этих
плоскостей выступает по 1 атому О, а всего по два атома О от каждого из
тетраэдров [Мп04].
4'(Мп-0) равны 1,48; 1,51; 1,59; 1,86.
6 (Ag-O) равны 2,19; 2,23; 2,58; 2,02; 2,69; 2,72.
3 расстояния Мп—-О (1,48—1,59) исключительно малы; в частности,
они значительно меньше суммы паулинговских ионных радиусов для Мп7+
и О2"*. 2 расстояния Ag—О (2,19 и 2,23) также относительно малы,
оставляя повод для сомнений и проверки.
728 ч. ii. структуры неорганических веществ ]гл. X
3. NH4Re04, NaRe04, RbRe04, AgRe04, TlRe04 = MRe04
Решётка тетрагональная Т.
Координация q-\- Sjq-\- S,
Тип Tq+siq+s — SBHOA (шеелит). p = [Re04] —тетраэдр.
Пространственная группа C\h—J&Ja.
Z = 4.
M к. ч. = 4р (квадрат)-(-4 |3 (тетрагональный тетраэдр);
к. ч. = 4М (квадрат) + 4 М (тетрагональный тетраэдр).
Рис. 589.
[Вещество
а
с
ОМ
d
(Re - 0)
М--0
NH4Re04
5,87,
12,У49
111,5
1,87
2,83
NaRe04
5,36.,
11,718
84,6
1,68
2,57
RbRe04
5,803
13,16;
110,8
1,83
2,86
AgRe04
5,349
11,916
85,3
—
—
TlRe04
5,76,
13,33
110,5
1,83
2,87
Примечание
SB III 91
§ 269] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VIII* ГРУППА 729
§ 269. VIII* группа периодической системы: железо Ге, рутений Ru,
осмий Os
1. [NH4]3 • [FeF6j
Решётка кубическая С.
Координация С |- О/О, Т.
Тип Сс+о/о, г —
SB #71.
Пространственная груп-
, па Ta—F£&n (Г—F23
или 7м—Р2ХЪ).
Z = 4.
рк. ч.=8а II (куб)+ 61
(октаэдр);
а I к. ч. = 6[3 (октаэдр);
а II к. ч. = 4 р
(тетраэдр).
О NH
Рис. 590.
Вещестло
[NH4]3[FcFe]
aw
У, 10
ОМ I Примечание
188
См. [NH4],tAlF,]n
BiF3
1
2. К3 [Fe(CN)6]
Решётка моноклинпая М.
Координация?
Тип М.
Пространственная группа Сгл—РЧ\с (или Сы—РЧ^с).
Z-4.
Вещество
K3[Fe(CN)e]
а Ъ
\
16,9
13,1
с
5,20
?
Примечание
I 1
90°6' | SB I 450
! I
730 ч.и. структуры неорганических веществ [гл.
3. K4Po(CN)6 3H20, K4Ru(CN)e ЗН20
Решётка моноклинная слоистая М .
Координация ?
Тип Ms.
П ространствепная
группа C\h — C2\c.
Z = 4.
1 Вощостно
K4Fo (CN)e • ЗНлО
}K4Ru((N)e .311,0
а
b
^9,3 ! 16,8
^9,3 j ъ 16,8
с
?
-L-9,3 1 ^90'
^9,3 | ^90э
а : b : с
^0,556: 1 :~ 0,556
^0,556 : 1 : ^0,556
Он,о
Рис. C9J.
О О R О О ж
о о
о о
о о
CE^XID^QD-CIZD
^^~о ооо о
(} 0-гп>ф<ТГУЛ ^>
OqO О
СО><Ю#<Ю-<Т]Т)
о
О О 9 О
OqO
О
/v|r)-r]fyyiT>-fi ' п
о ооо
о
Рис. 592.
Рис. 593.
§ 269] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VIII* ГРУППА 731
В 1946 г. Г. С. Жданов и В. Поспелов (см. ЖФХ XXI, 521, 879
(L947)) закончили обстоятельное и интересное исследование структур
K4Fe (CN)e«3H20 и K4Ru (CN)6*3H20. Обнаружена склонность к
образованию, в частности, моноклинных и тетрагональных форм.
Группа Fc(CN)e—октаэдрична. Расстоянием— С = 1,92 А,С — N = 1,15 А.
Дли тетрагональных кристаллов этих веществ найдена
пространственная группа — C£/i —/4х/г/; а~9,35\ с = 33,63.
Один слой представляет собой квадратную взаимно центрированную
сетку октаэдров M(GN)e. Расстояние между ближайшими плоскостями,
проходящими через центры атомов Ru = 2,42A.
Сетка октаэдров расположена между двумя плоскостями из атомов К,
которые образуют почти правильные квадраты со стороной 4,2 А. В центры
квадратов К+ попадают атомы N из соседних координационных сфер. Октаэдр
повёрнут вокруг оси у на 18°.
Таким образом, в плоскости сетки из атомов К- и CN-групп образуется
структура, подобно сетке NaCl, на грани куба. См. план (рис. 591) и
разрез структуры (рис. 592 и 593) —из работы Жданова и Поспелова.
4,[Co(NH3)6H20][Fe(CN)6]
Решётка ромбоэдрическая R.
Координация RJR.
Тип RR/R-SBJ6,.
Пространственная группа C3i — -R3.
a-[Co(NH8).]; p = [Fe(CN).l.
2 = 1.
a к. ч.=83(Д); ? к. ч. .= 8а(Д).
Co(NH3)5H20 Fq(CN)6
Рис. 5<J'i. (См. рис. 57G.)
Вещество
[Co(NH3)5H2O][Fe((:N)0]
6,46
Эб'бЯ'
Примечание
sB II 104
См. [М»(Н,0)6] SiF,
732
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. X
5. [Co(NH3)e][Co(CN)9],
[Co(NH,)§H,0][Co(CN).b
[Со(М1э)4(Н20)2][Со((Ж)в]
Решётка ромбоэдрическая R.
Координация С/С.
Тип fl(-./6-SB/6,.
Пространственная
группа
си -яз.
*-lC°#J; N[Co(GN),].
Z = l.
л к. ч. = 8 Э; Р к. ч. = 8 а.
1 Вещество | о ' а 1
![Co(NH,)] [
Г (NH3)5-|
LCo(H,0)2.
Co(CN)f]
[Co(CN,e]
[Co(CN)6]
6,54
6,46
С,42
97U6'
96°51'
95°50'
Примечание: SB II 104. См.
[Mg(HaO).USiFe]. |
Cq(H20,NH3)6 Co{CN)&
Pin-. Г.'.С).
§ 270. IX* группа периодической системы: кобальт Со, родий Rh
иридий Ir '
1. (NH4)3 [Co.(N02).],
Ks[Co(N02)e],
Rb3[Co(N02),],
Cs3[Co(N02)e],
Tl3[Co(N02)e],
(NH4)3 [Rh (N02),],
K3[Rh (N0,),],
Rb3[Rh(N02),],
Cs3[Rh(N02)e],
Tl3 [Rh(N02),],
(NH4)3[Ir(N02),],
К,[1г(Щ).]
Rb3[Ir(NOa)6],
Cs3 [Ir (NO,).],
Tl3[Ir(N02)e]
Решётка кубическая С.
Координация О,Т/С + О.
Тип Co,tic+o —
SB(#71; /2.) /24.
Пространственная
группа Tah-Fm3.
a = (NH4) или М;
P = [M™(NO,).].
Z = /4.
О м,
Mn(N02)fi
Рис. Г)%.
5 2701 СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. IX* ГРУППА 733
(Вещество*)
1 а
ОМ
Веще-
ство
а
ОМ
Вещество
а
ОМ
(NH4)8[Co(NOs)f]
10,81 (10,78)
316 (313)
(NH4),[Rh(NOa)J
10,91
32Г>
(NH4),LIr(NO.)J
10,73
309
К3 [Со (NO,)J
10,44 (10,46)
285 (286)
K3[Rh(N02)e]
10,63
300
K,[lP(NOa)e]
10,59
295
Rb3[Co(N02)e]
10,73
309
Rb3 [Rh (N03)J
10,83
317
Rb3[.Ir(NO,)e]
10,77
312
Cs3 [Co (NOa)J
11,15
346
Cs3[Rh(N02)e]
11,30
361
Csa [Ir(NOa),]
11,17
342
Tl3[Co(NOa)e]|
10,72
308
Tl3[Rh(N02)6]
10,91
325
Tla[Ir(NOa)§]
10,73
309
K3Co(N02)ti тип (а,3) образует структуру анти- BiF8; [M(N02)e]3~-Hon
имеет октаэдричоскую форму. Атомы N обращены к центральному
атому М. См. ниже K2CaLNi(NO.)e] и рис. G08 и 609.
2. Ba3[Co(N02).]i, Pb3[Co(N02)e]2
Решётка кубическая С.
Координация C^jT.
Тип Сс-р-
Пространственная группа ?.
а^Ва, РЬ и т. п.
p = [Co(N02)e].
Z = 2.
Рис 597. Рис. 598.
Если с ионом Co(N02)6 соединяются двувалентные катионы,
соотношение структурных узлов становится равным 2 : 3 (вместо 1 : 3, как это имеет
место в предыдущем примере, например, KJCo (N02)el).
*) SB III 127, SB IV 65.
734 ч. и. структуры неорганических веществ [гл. X
В SB VII возникающая в этом случае структура иллюстрируется
рис. 598. На рис. 597 в интерпретации SBVII структура Ме2Ме [(Go (NO.,)c],
имеющая одновалентные катионы двух типов. Для понимания рис. 597
и 598 сопоставим их с рис. 596, В соответствии с § 63 (см. образование
типов алмаза и сфалерита), 4 октаэдрических структурных узла,
расположенных в центрах 4 октантов, образуют кубическую граиецентрирован-
/1 1 1\
ную решётку. Если октаэдр в позиции (туу) перевести в позицию (000),
то ячейка (рис. 597) примет вид рис. 596, в котором узлы кубической
гранецентрированпой ячейки будут попрежнему заняты октаэдрами,
позиции в центрах рёбер и в центре ячейки (октаздрические между-
узлия)—чёрными шарами и, наконец, центры октантов—(тетраэдриче-
ские междуузлия) —белыми шарами (сравни с рис. 608). Ячейка (рис.
598) рассматриваемых нами соединений типа Ba3[Co(N02)e]2 примет вид
рис. 596, в котором узлы кубической гранецентрирэванной ячейки будут
также заняты октаэдрами, октаэдрическис междуузлия окажутся пустыми,
а из 8 тетраэдрических междуузлий в центрах октантов окажутся
занятыми только 6 (в сфалерите заняты 4, в CaF2—8), образуя куб с двумя
незанятыми вершинами по объёмной диагонали. Структура является
интересным членом ряда (в скобках количество центрированных октантов);
Gu20 (2); ZnS (4); Ba,[Co(N02)e](> (6); Ba2[Ni(NO,)e](8); (CaF. (8))"(см. § 63).
3. Ag+[Co(NH3)2(N02)4r
Решётка тетрагональная Т.
Координация DijDi.
ТИП 7z)4/Z)4-SB/l9.
Пространственная группа D\h — РЩппс.
* = Ag; 3 = [Co(NH3)2[N02]4.
Z = 2.
Рис. 599.
§ 270]
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. IX* ГРУППА
735
Вещество
Ag[Co(NH3)2(NOi)J
а
6,07
Рис. 600.
с
ю, 4:*
cja
1,406
1
Примечание 1
SB IV GO .
По SB узлы 3 образуют центрированную ячейку, узлы а занимают
центры 4 рёбер и середины 4 граней (рис. 600). Эта неожиданная на
первый взгляд структура соли а'[Гск. ч. =-6/6 интерпретируется автором
исследования Уэллсом (Z. Krist. A. 95, 74 (1936)) лишь как «продукт
внедрения А^+-ионов в промежутки плотной кубической упаковки,
образованной комплексными ионами». Здесь Уэллс допускает неточность,
забыв отметить, что речь идёт лишь об окггшэдричестх промежутках
(плотной упаковки), причём все они оказываются занятыми, так что
ячейку можно представить как имеющую слегка деформированную структуру
NaCl (рис. 599). Для этого надо выбрать в качестве оси диагональ
грани элементарной ячейки (см. пунктир рис. 599). Возникает
тетрагональная элемеьтарная ячейка с Z —2. Положения в ней октаэдров (узлов
3) показаны квадратами.
736 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
4. (NH4)2Na[Rh(N02)4]
Решётка кубическая С.
Координация OJjC + O.
Тип Co,r/c+o--SBtf 71 и J2X (см. тип /24, см. рис. 608).
Пространственная группа Т%— Fm'S.
oc = [NH4]+ (тетраэдр); [3 = [Rh(N02)e]8- (октаэдр).
£-4.
Рис. 001.
Вещество
а Примечание
(NH4)2 Na [Rh (N02)e] ! 10,52
Г. Б. Бокий и Л. А. Попова ИАН(х)
стр. 88 (1945)
Октаэдры lRh(NOa)e]3~ занимают вершины и центры граней
элементарной ячейки, образуя структуру, аналогичную aHTH-BiF3. Тетраэдры
[NH4]+-hohob занимают .центры октантов. Na^-ионы занимают центры
рёбер и центр элементарной ячейки типа MeJ'Me*"[Co(N02)e] (см. рис. 597)
или K2Ca[N(N02)e]. Сравни с (NH4)3 Со(1М02)/(рис. 596 и 608).
Отмечая, что в данной структуре ионы натрия занимают октаэдри-
ческие междуузлия, образованные плотной упаковкой ионов [Rh(N02)e]3^,
а ионы аммония —тетраэдрические, имеющиеся, как известно (см. § 63),
в удвоенном количестве, авторы высказывают предположение, что не-
§ 271] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. X* ГРУППА 737
образование соединения [NH4] Na2 [Rh (N02)e] вызвано энергетическими
затруднениями, связанными с необходимостью размещения больших ионов
натрия в меньших -тетраэдрических-—междуузлиях, а меньших ионов
аммония в больших - октаэдричоских — междуузлиях. Такое объяснение
нам кажется не совсем убедительным потому, что, вообще говоря, иному
составу вещества может отвечать иная структура, а не обязательно
та же самая, что и рассматриваемому соединению. Кроме того, в
рассматриваемой ячейке саш = 10,52 (против аш = 5,45 для GaF2) междуузлия
достаточно велики для размещения Na-1-ионов, тем более, что
структурный тип фтористого висмута легко допускает образование структур
замещения и вычитания в широких пределах изменения состава фазы и
соотношения ионных радиусов.
§ 271. X* группа периодической системы: никель Ni, палладий Pd,
платина Pt
1. RbJUCle, Rb2PdBre,
(NH4)2PtCle, К,Р№1,,
Rb2PtCle, CsPtCl,,
Tl2PtCle
Решётка кубическая С.
Координация С/Т.
Тип CC/r-SB#61;
JU [КаР1С1.].
Пространственная группа
Obh — F?n3m (или
П - F43m).
a = [NH4] или М;
~3 = [МХ,].
Z-4.
? к. ч. = 8М (С);
М к. Ч.-4В (Т).
MnCL6
Рие. 002.
а
ОМ
Rb.PdCl I Rb,PdBr6*)
1
10,0.> 10,0.,
1
252 ; 252
i
(NH4)2-
-PtCl.
9,834
238**)
K3Pt( I,
0,72.
230
Rb3PtCIe
9,88,
2425
Cs.2Ptr.]e
10,19.,
264
Tl2PtCIe
9,755
232
Примечание
SB I 429
SB III,A2JL
*) Cm. SB I 447. ox - 4,95.
**) В SB I ошибочно указано ОМ равно 298.
л. :\i)
738
Ч. Н. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[ГЛ. X
2. (NH4)2[Pt(SCN),],
KJPMSCN),], Bba[Pt(SCN),]
Решётка гексагональная Н.
Координация 12/6 = Р6/РЗ.
Тип Яр6/рз-5ВЯ63--= SB71,.
Пространственная группа
DU—НЪт.
P = [Pt(SCN),].
Z--=l. К к. ч. =6 jiJ (тригональ-
ная призма РЗ);
(3 к. ч. = 12 К (гексагональная
призма Р6).
О м
Рис. 603.
Pt(SCN)e
Ьещоство
(NH4)2[Pt(SCN)e]
K2[Pt(SCN)e]
Bba[Pt(SCN)J
а
6,77
6,73
6,75
с
10,45
10,26
10,47
с/а
1,543
1,525
1,551
ОМ
415
403
413
Примечание
sb I vri
3. (NH4)JPdCU
KJPdClJ, KJPtCl.]
Решётка тетрагональная
Т.
Координация Pi/r.
Тип Tpur-SBHlb
(K,PtGl4).
Пространственная группа
D\h— PUmmm.
HPtci4.
8 к. ч. = 8М (призма Pi);
М к. ч. =4В
(прямоугольник г).
Вещество
(NH^PdCl,
K2PdCl4
K2PtCl3
а
7,21
7,04
6,99
с
4,26
4,10
4,13
ом|
2,21
203
202
M„CL.
1ЬЧ
Рис. 604. 2 ячейки.
§ 2711
СОЬДПНЬНПЯ С КОМИЛЬШ'.НЫМ АНИОНОМ. X* ГРУППА
739
4. Ba[Ni((JN)J4H20, l*a[Pd(CN)«J-4H30, «a ЦЧЦЗД 4Н,0
Решётка моноклинная Л/.
Координация 6 -Ajq/ij'L
Тип Л/е+wi/i-SB//42it.
Пространственная группа
Ch-C2/v.
Z = 4.
a = H20;
Ва к. ч.
Ni к. ч.
;j=cn.
= 6a + 4fJ;
= 4Р(?).
~аИ
s
^
-О
«фо
О 1 2 3 4 5 В 7 8 9 10
L . . .-^ .—-_t_J
Рис. 605.
ОоО О Ва
' 9 N.
СНо
CN
►N/
Рис. G0G.
Вещегтно
BaNi(CN), • 'iH><)
BjiPd(CN)l.,iiLO
BuF-Ч (CN)4.'»1L()
а
11,71
11, W
1LS4
Ъ
i
14,82 Г,
i
l'i,HS G
l
C !
i
;,s mi
■".'J ; It)
>
'■4>'
;1><S'
JtjHiAKMiniiiie
Ni м (Ooo), (о i)(- )
H-H"
G- ' ') J
740 ч. п. структуры неоргадическых веществ [гл. X
Каждый атом никеля окружён 4 циангруппами, находящимися в
вершинах квадратной координационной сферы рис. 605. Атомы Ni расположены
лдоль рёбер элементарной ячейки, параллельно оси с, а также вдоль
линии, параллельной оси с и проходящей через центр элементарной
ячейки рис. "006.
i
б. SrJNKNO,).], BaalNi(NOs).], PbJNi(NO,),]
.Решётка кубическая С.
Координация Т/С.
Тип Ctic - SB#61 - SB/1».
Пространственная группа 0),-~1гтЗт (или Т%).
;i = [Ni(NOa).l.
Z~4, 3 к. ч.--.8М(6'); М к. ч.---■>$ {Т).
Ni(NQ2)6
Рис G07.
Вещество j Sr2 [Ni (NO,)e]
1 aie
ОМ
1
10,54
29I1.
Ba.> [Ni (NOa)J
10,67
:Ю4
Pb, [Ni (NOa)J
10,55
294
Примечание
SB III 122,
см. CaF2
§ 271] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. X* ГРУШ'Л 741
6. K2Ca[Ni(N02)6] ,•"-
Решётка кубическая С.
Координация О, Т/С ---О.
Л^ип С0,т/с+о - SB/24 (K3 Co (CN),).
Пространственная группа Tu—FmW.
i^ = [Ni(NOj,].
Z = 4.
К к. ч. -=6 Н(0);
Са к. ч. : = 4 $(Т);
р к. ч. = 8 Са(С)-г6 К (О).
О о о J Ni(NQ2)s К Си
О N Ni
Рис. GUIS.
Вещество
K.CaNi (NOa)6
aw
10,34
ОМ
274
Примечание
SB IV 05, см. К3С:о(КОа)в
742 ч. и. структуры неорганических веществ [гл. X
В SB IV 65 дан чертёж структуры К2Са [Ni(N02)e], приведённый нами
на рис 609. Вследствие большой сложности он мало понятен. Мы сочли
более разумным изобразить эту же структуру, как на рис. 608. Это
делалось нами и ранее (рис. 596 (К0Со (N02)6).
Рис. 609,
§ 272. I* группа периодической системы:
медь Си, серебро Ag, золото Аи
1. (NH4)2 [СиС14] 2ELO, К2 [СиС14] 2ELO, Bb2 [CuClJ 2H20
Решётка тетрагональная Т.
Координация д/^4.
Тип Tq!P% -SB//41 (K2CuCl4 2HaO).
Пространственная группа DW—Pilmnm.
a'= [CuCl4] (квадрат) ;J/ = HLO, но удобнее
<х=[СиС142Н20] (октаэдр)Г В = М«К или Rb пли (NH4) М к. ч. = 4а(д);
Z = 2 ' ~
а к. ч. = 8р(Р4).
§ JJ72] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. I» ГРУППА 743
Oct
Фнго
Рис. 610.
I Вещество
(NH4)2 СиС142НаО
К, СиС142НаО
Rb2CuCl42H*0
а
7,58
7,45
7,81
с
7,96
7,88
8,00
с/а
1,050
1,058
1,025
ОМ
229
219
247
Как отмечает SB I 41, квадратная конфигурация [СиС14]~-иона отвечает
представлениям теории направленных валентностей (см. §§ 94, 95), но,
с другой стороны, две молекулы воды втянуты в координационную сферу
узлов СмС14, так что формула соединения скорее имеет вид: К2 [СиС14(Н20)2],
причём вокруг атома меди координационная сфера имеет конфигурацию
октаэдра. Узлы а образуют тетрагональную центрированную ячейку,
узлы р образуют призматическую координационную сферу вокруг узлов а,
довёрнутую на 45° по отношению к оси ячейки.
744
Ч. II- СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
2. Cs., [AuClo] [AuClJ,
Cs2[AgC!2][AuCl4]
Решётка тетрагональная
Т.
Координация Z)4 -\~ PijS +
+ S.
Тип TD±+D -1/54-5 — SBiHe.
Пространственная группа
D\7h—Jilmmm.
a = [AuCL] или
[AgClJ; p = [AuGl4].
Z = 2.
a к. ч. = 6? (#4) + 8у¥(/>4);
fJ к. ч. = 6* (Z)4) + 8Л/(/>4);
М к. ч.---=4х(5)+43(5).
-*.«
Рис. 611.
Вещество
а с
Cs2AuAuCl, 7,49
CRaAgAuCl9 i 7,38
i
10,87
11,01
с/а
1,452
1,491
Примечание
В SB "VI ошибочно показаны
Аи II в позициях Ди I.
Квадратные [АиС14]-группы в вершинах и центре элементарной ячейки.
Гантельные [АиС12]-группы центрируют: а) рёбра, параллельные оси 4,
и Ь) грани основания ( 2 Аи I в (0 0 0) и f у Т Т )' ^ Аи II вГОО — j и
( — — 0 ) \ Атомы серебра в производном Ag занимают позиции Аи II.
Координационные числа: Аи I штрихованный: 4 С1 II штрихованный — на
расстоянии 2,41 (квадрат);
Аи II чёрный: 2 G1 I белый — на расстоянии 2,30
(линия-гантель);
Аи I: 2 G1 I—на расстоянии 3,13;
Аи II: 4 G1II —на расстоянии 2,88.
Из сопоставления рис. 611 и 610 следует, что в обоих случаях
квадратные координационные сферы занимают вершины и центр элементарной
ячейки, атомы Cs, соответственно К, образуют Pi вокруг квадратного иона;
ион же [AuClo] занимает позиции, аналогичные двум молекулам воды.
§ 272] соединения с комплексным анионом. I* группа 745*
3. К [ AuBrJ • 2Н20
Решётка моноклинная М.
Координация р 4- р/р + р/1 + 1 --- 8/8 '2.
Тин Л/8/8/2 -SB#419.
Пространственная группа C^h — Plja.
MAuBM; Г = [Н20]. ^ #
Z = 2.
К к. ч, =-83 (два четырёхугольника);
р к. ч.^8К (два четырёхугольника).
К к. ч.--- 1а -г l.JJ#
Рис. 61.2. (2 элементарные ячейки).
I Вещество
К[АиВг4] • 2Н-0
а
9,51
Ь
11,9J
с
4,23*)
.*
94°24'
1 1
a :h :c 1 Примечание
0,797 : 1 : 0,:1Г>9
SB IV 50. Согласно
примечанию SB
истинная! элементарная
ячейка имеет с^=8,46
и Z-4.
11 11 11
Положения узлов: Аи: 0 0 0; — — 0; К: - 0 — ; 0-^~w .
Атомы золота занимают, таким образом, вершины элементарной
ячейки и центры верхнего и нижнего оснований. Центры 4 остальных
граней занимают атомы К; 2о?(АиВг)~2,50 А; 2d' —2,57. Координационная
сфера АиВг4 — четырёхугольник.
*) В SB IV 50 ошибочно указано 4,28.
746
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВБЩЬСТВ
|гл. X
4. KAg(CN)2
Решётка гексагональная слоистая молекулярная HS~M.
Координация 6/6 ^ О/О.
Тип H?--M-SBF5.0 (KAg(CN),).
Пространственная группа Did — C'Slc.
P = Ag(GN)2. ♦
Z = 6.
Рис.
Вэтцегтво \ а \ с
KAs(CN),
7,:W'i
17,5Г>
613.
Примечание 1
SB IIf 83. В решётке атомы К обра-1
зуют слои, за которыми следуют слои!
линейных ионов [NCAgCN]
§ 273] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ- II* ГРУППА 747
б. K?Pb[Cu(N0>)6]
Решётка кубическая С.
Координация OyTjC ~t О.
Тип Co,r/c+o-SB./LV
Пространственная группа 77. — Fm3 [K3Co (N02)e].
О N Си J
Рис. 614.
Вещество
KaPb [Си (N0 ,)в]
а»
10,65
ом
302
Примечание
SB IV 65. См. KjCofNOa),
§ 273, II* группа периодической системы: цинк Zn, кадмий Cd, ртуть Hg
Комплексные соединения типа Мх [ZnXn] в структурном отношении
почти не изучены. Большинство исследованных веществ оказываются
сложными или сложнокомплексными соединениями, не имеющими
изолированных координационных сфер.
Из изученных веществ, повидимому, относящихся к комплексным
соединениям, следует назвать (NH4)3ZnCl6 и K„ZnCl4 (J. A. Ch. S. 66,
1056 (1944) и 67, 878(1945)).
748
Ч* II. СТРУКТУРЫ- НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[тл.;Х
1. (NH4)3ZnCl5==(NH4UZnCl4]Cl
Решётка ромбическая OR.
Координация x/tj.
Тип ORx!ir
Пространственная группа D-A — Рпта.
? = lZnCl4l (Г); *-[С\(Ж\4)0)(д).
Z = 4.
1 Вещество j а | Ь 1 с | Примечание 1
1 (NH4)3ZnCl5
8,74
9,84
12,61
Zn—C1 = 2,25A 1
5-й атом О не входит в координационную сферу [ZnCl4], он «октаэдри-
чески» окружён 1ЧН+-ионами на расстоянии ^ 3,41 А. В отличие otTLAIF^
и т. п. в данном случае образуется ионная решётка без каких-либо цепей —
в состав комплексного иона [ZnCl4] входит меньше ионов С1, чем
«позволила бы» химическая формула.
2. K,ZnCI4
Решётка ромбическая ОН.
Координация?
Тин ОЮ
Пространственная группа C%v — Pma.
Z - 12.
1 Вещество | а 1 Ь 1 с 1 а : b: с 1
1 K3ZnCl4
26,70
12,26
7,28
2,178:1:0,5935
§ 274. Ill* группа
периодической системы: бор В,
алюминий А1, галлий Ga,
индий In, таллий Т1
1. [NH4][Br4],
RbBF4( + KBF4?)
Решётка ромбическая OR,
Координация РЗ^/РУ.
Тип ORp3+fp3+ —
SB#03(BaS()4).
Пространственная группа
D-zh—Рпта.
а = [NH4] (тетраэдр);
р= [BF4] (тетраэдр).
2 = 4. _
а к. ч.--7р(^3+);
р к. ч. ---7а(РЗ+).
О NH.
Вещество
1 NH,BF4
RbBF4
KBF4
а
9,06
9,07
7,84
Ъ
5,64
5,60
5,68
с
7,23
7,23
7,38
а : b : с
Примечание
. 1,606: 1 : 1,282 1 SB III 90
1,619 : 1 : 1,291
1,382 : 1 : 1,297 | SB II 48о
NH4BF4, KBF4 и др. с повышением температуры переходят в
кубическую модификацию за счёт вращения тетраэдров. Тп для KBF4^300°C,
аю -7,26; 7^ для NH4BF4 = 260° С, а = 7,25 A (SB IV 164).
§ 274]
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. III» П'УИПЛ
749
*-». [Co(NH3),](BF4)2;
[Ni(NH3),](BF4)2
Рошётка кубическая С.
Координация С/Т.
Тин Сер—SBH61.
Пространственная группа
0\ — Fm3m (или
T'i — F43m).
a==[BF4l; Р-[М(М1,).|.
Z==4.
рк.ч.-8а(С);*к.ч.-4Р(Г).
В еще ст ни
йхв
lCo(NH,),] (131<\), |ll,2i».
[Ni(NH8),l (ВРД. 111,26'
I 7
Примечание ;S15 (I 50:1,
SB 111 475
Рис. 616.
3. [Со(РГН,),](ВР4),
Решётка кубическая С.
Координация С г О/Т, О'.
Тип Сс+о/г.п —
SBaiiTH#7l^
«((NH4),A1F.).
Пространственная группа
Of, — Fm3in (или Та —
—F43m(?)).
* = [Со(ШШ;
P = (BF4).
Z = 4.
ак.ч.==8рП(С) + 6р](0);
pi к. ч. =6а(0);
311 к. ч. =4а(Г).
Вещество
[Co(NH3)e] <BF4)S
Примечание. SB
i *»
: tt.2l1
i [
III 475
О BF4
Co(NH
3/6
Рис. 617.
1Ы) ч.и. структуры неорганических веществ [гл. X
4. [NH4]3[A1F,]
Решётка кубическая С.
Координация О, Т/С-t- О.
Тип Со, >ц<:+о - SB#71 - SBJ2,.
Пространственная группа 7'J ~F43rn(T2 или 74).
a^|NH4]; MAlFe].
Z--4. а I к. ч.---6р (октаэдр); а II к. ч. --= 43 (тетраэдр);
р к. ч. = 8а II (куб) + 6л I (октаэдр).
О NH4
AIF6
Рис. 618.
Вещество
flti
[NHJaCAlF.] I 8t904
**
1,8»-,
Тип Я71 образуется из типа /761 путём внедрения третьей группы:
(NH3) в центр элементарной ячейки и в середину рёбер.
Расположение центров групп в решётке (NH4)8 AlFe в точности
отвечает структуре BiFs (тип SB iX)8)d(Al —F) = 1,6бА.
Паулинг в 1924 г. получил другие значения aw, что было вызвано
грубой ошибкой в изготовлении препарата. Повидимому, выпаривая раствор
в фарфоровой чашке, Паулинг тюлучил и без анализа исследовал.
(NH4):SiF. вместо (NH4)3AlFe (см. SBI 793).
§ 274]
СОКДПНШПШ С КОМПЛЕКСНЫМ АНИОНОМ. III* ГРУППА
751
5. Na3AlF6 (криолит) JNaI(Na II)2AlFe
Решётка моноклинная (исевдоромбическая) М.
Координация Ojx.
Тип Mnlx-SBJ26.
Пространственная группа Со^ — P2Jn.
P4A1F6].
Z = 2.
Nal к. ч. -.-= 6(3 {О); Na II к. ч. х%.
Рис. 610.
•Viti
:>,<•>!
7,80
я : i>: г
90ЧГ ; 0,»»>2: I : 1,:<90
I
Примечание
>В VI 29.
Г легка дефор-
миропанные
октаэдры [AlFe]
образуют
центрированную
элементарную ячейку
Два иона Nal занимают позиции (О 0— i и Г— — 0 j и октаэдрически
окружены узлами р. Четыре иона Na II+ находятся в общих положениях.
Два из них расположены па левой и правой гранях элементарной ячейки
и по одному около передне!! и задней граней. Их к. ч. близко к в.
752 ч. и. структуры неорганических веществ
6. КВ08~Кз(В,0,)9 NaBO^Nfl.fBA)
Решётка ромбоэдрическая R.
Координация xjy (нуждается в уточнении).
Тип"Rx!v-SBFbl3.
Пространственная группа Вы—КЗ?-
я = [В.О.].
Z = 6.
Рис. 620.
Вещество
КВО,
NaBO,
а \ -4
1
7,7;»
7,22
11Р29'
Примечание
Гексагональная ячейка имеет
а=12,75 и с~7,33(КО.>)
Соответственно 11,91 и 6,45 (Na02)
В,03-атомы образуют шестичленное кольцо, вне которого
соединённые с атомами В расположены ещё 3 атома кислорода. Комплексная
группа В3Ов образует самостоятельный структурный узел. Каждый атом В
расположен в центре почти правильного треугольника. Расстояния В —О
равны: 1,33, 1,38 и 1, 38 кХ.
Сравни с рис. 510 (Н8В03).
§ 27S1
Соединений с Комплексным анионом, iv группа
753
§ 275. IV группа периодической системы: углерод С, кремний
германий Ge, олово Sn, свинец РЬ
1. (NH4USiF,]F
Решётка тетрагональная Т.
Координация / + <//2 -f- 4/g + PijD'i + Pi = к.
ТИП ?'/ + Qf/iJ+,i/«-l-P4/D.i+/>4 = 3ГА'.
Пространственная группа /?4л- Р*/мЬт.
Z = 2. K'^|NH4|+(l),a" = [NH4]+(II), p^SiFJ .
Si,
Рис. 621.
Oni/4
Октаэдр SiFe ф f
Риг. 622.
I Вещество
. (NH«),[SiFJF
a
8,04
с
5,845
с/а
0,727
Примечание
По J. Am. Gh. S. 64, 63J (1942)
В работе даётся лишь проекция элементарной ячейки на основание (рис. 621)
с указанием высоты позиции над основанием. По этим данным мы построили
ячейку (NH4)JSiFe]F, рис 622. Рассматриваемое соединение
представляет теоретический интерес в том отношении, что, вопреки прежним
представлениям, не содержит комплексного иона с к. ч., равным семи.
Структурные узлы а образованы ионами [NH4]1 , структурные узлы р —
ионами [SiF^. Кроме того, в решётке существуют самостоятельные
структурные узлы—ионы фтора.
В основании элементарной ячейки расположены ионы аммония и
ионы i [SiFJ. В плоскости (ху~}Л лежат ионы аммония и ионы фтора.
Координация: [NH4]+I к. ч. = 2F~(/) + 'ipfo);
[NH4]+II к. ч. =2F"+4p;
[SiF,]-K. ч. ^4a(<7) + 4F-(P4);
F" к. ч. —2a (расстояние ■= 2,92) + 4a
(расстояние = 2,89) = 6a (7)4)+8р (Pi); S1-F(b SiFe) = J.,71;
F~F(b SiFc)-2,42.
754 ч. и. структуры неорганических вещестё [гя. X
2. (NH4)JSiF,], KJSiF,], RbJSiF.J, Cs2[SiF,], TlJSiF,], CsJGeF,],
(NH,)2 [SnCl,], K2 [SnCl,], Rb2 [SnCl,], Cs„ [SnCl.J, Tl2 [SnCl,], (NH4)2 [PbCl,],
Rb2 [PbCl,], Cs2 [PbCl,]
Решётка кубическая С.
Координация Т\С.
Тип Ctic -SB#61 (KsPtCl,).
Пространственная группа
Ol — FinSm (или Т\ и др.).
a = (NH4) или М; р = [МХ,].
?к. ч. = 8М (С); М к. ч.=
■~Ц{Т).
Z = 4.
Рис. 623
мпх6
1 Вещество
°" '
ОМ
dSt-F
Вещество
в|Р
ОМ
(NH4),SiP€*)
8,38
147
1,72
8,зз?
145
—
RbaSnCl,
Ю,099
256,5
10,100
257
1 •"
1 u
10,34,
273
fa
8,168
136
—
•
fa
•*
1 а
8,446
151
—
Tl4SxiCle
10,348
275
9,970
248
S3
1 СО
о
8,867
174
—
fa
8,56о
157
—
(NH4)aPbClf
*м4
261
10,135
261
1 *"
ф
О
со
CJ
8,99
182
■--
(NH4)4SnCle
ю,о5
254
-
КЬ,РЬС1в
10,198
265
10,195
265
ю,оз8
253
—
• 1
со 1
9Л
247
"~
Сз„РЬС1в
10,415
282
10,416
282,6
См. SB I, 429 и SB III, 121.
*) Ниже 5" С гексагональная, выше тип Я61.
§ 215] соединения с комплексным анионом, iv группа 75Й
3. [NHJJSiF,]
Решётка ромбоэдрическая (гексагональная) слоистая IV .
Координация О/t.
Тип R?)jt SB./!, (блнлок к SBC0).
Пространственная группа 1)'а — СЗт.
e = |MM;p = lSiF,l. '
Z = \.
Si к. ч. = Са (С); а к. ч.-- 3 Si (/).
Вещостпо
[NHJitSiF,]
а
5,76
с
4,77
с/а
0,828
Примечание
SB 111 123. SiF„— октаодры.
d(Si--F)=l,65., тип CdJ, (Cti).
Вместо I'd ~ -[SiFJ. Слоистая ре- 1
шётка [
Ячейка типа CsCl (из октаэдрических узлов а и р), слегка
деформированная по оси с (угол не равен 90°).
4.[Mg(H.20),][SiF,j,
[Mn(Ha0).][SiF.],
[Fe(H80).][SiF.],
[Co(H20.)][SiF.],
[Ni(H80).][SiF4],
[Zn(H20)e][SiF,],
[Mg(Ha0),][SnF,J,
[Zn(H20),][SnFe]
Решётка ромбоэдрическая Н.
Координация R/R.
Тип Яд/R-SB/e,
(Ni(H20),SnCl,).
Пространственная группа C-Jt
ЯЗ.
a = [M(H20),]; p = [AF.l.
Z=l. ак.ч. = 8р (Л);
рк.ч. = 8а (R).
1 Ще-
| ство
а
1 а
[Mg(H20)e][SiFe]
5,75
96°4'
Вещество
а
а
[Mn(H20)e][SiFfl]
5,80
96°53'
[Ni(H20)e][SiFe]
5,57
96°18'
[Fe(H,0)6][SiFe]
5,78
97°0'
[Zn[H,0)e][SiFe]
96°6'
[Co(ILO)e][SiFJ Примечание
5,61
95°50'
[Mg(H20)e][SnFe]
5,88
96°20'
SB II 104 1
[Zn(HaO)e][SnFe] I
5,87
95°50'
М,(НД MDF6
756 ч. п. структуры неорганических веществ [гл. X
5. (NH,)2 [GePJ, KJGePJ
Решётка гексагональная //.
Координация Р6/РЗ.
Тип Ярс/рз - SB 7113(K2GeFJ.
Пространственная группа Dld — CSm
P = lGeF.].
Z = l.
К к. ч. = 6р(^3);
Зк. ч.==12К(Р6).
Рис. 625.
Вещество а
K,[GeFJ
(MHJaCOePJ
5,62
5,85
с
4,65
4,775
с/а
0,827
0,816
Примечание
SB VII 34, 154
Ge—F-1,76
Октаэдры [GeF,] образуют примитивную ячейку, в обеих трйгональных
призмах которой расположены ионы К+ в позициях (-г г); Г-q-"?2 )
при 2 = 0,7. Ср. K2Pt (SCN)„ тип #63, рис. 603.
6. KJSn(OH),]
Решётка ромбоэдрическая Д.
Координация Т/С.
Тип Rtic - SB #62.
Пространственная группа D\u—Rbm.
j3 = [Sn(OH),].
Z=»l.
Вощество
K2[Sn(OH),]
а
5,67
а
70°!'
ОМ
156
Примечание
SB I 431u^0,26.
Приа«60°и и=74-
SB^Gi переходит; I
в SB //61, рис. 61>3
Близок к* структуре, изображённой на рис. 623.
§ 275) соединения с комплексным анионом, iv группа 757
7. MgCO,, CaCO,,
MnCO,, FeCO„
ZnCO,
Решётка
ромбоэдрическая R.
Тип flo7o-SBGl =
= SBG0j (кальцит).
Пространственная
группа Z)«j-C3,2.
Z = 2.
Мк.ч. = 6р(0);
рк.ч. = 6М(0).
Вещество
MgC03'
СаСОа
МпС03
FeCO,
ZnCO,
CdCOs
а
5,61
6,361
5,84
5,82
5,62
5,U2
а
48*1^
46*7'
47*20'
47°45
48*20'
47*24'
ОМ
45,2
61,0
49,4
49,8
45,5
1
(искажённая
решётка типа NaCl).
Рис. 626.
са
Рис. 627. * Рис. 628.
Рис. 626 и 627 по SB I; координация более чётко видна из рис. 628, ср, рис. 627.
758 ч.и. структуры неорганических веществ [гл. X
8. CaC03, SrC03, BaC03, РЬС08
Решётка ромбическая OR.
Координация РЗ/О.
Тип OR P3/0 — SBG2 (арагонит).
Пространственная группа Dw—Pnma.
Z^4. Са к. ч. = (3 + 3)С (искажённая призма); С к. ч. = (3 + 3) Са
(искажённый октаэдр).
Рис. 629.
Вещество Caf,Oa SrC03
а
Ь
с
ом
7,9't
:•, 72
м>,1
8Д>
6,10
66,0
ВаС03
Г., 20
К,88
РЬСОэ
5.14
8,4,
6,10
Примечание
Искажённый тип #8 (NiAs), Са
вместо As и С03 нместо Ni
вследствие наличия треугольных С032_-
групи увеличиваются периоды а
и Ъ. SB I 2J5, 339
9. CaMg(C08)2 (доломит)
Решётка ромбоэдрическая /?.
Координация О (б. См Р
Тип Rojo — SBG11.
Пространственная группа C\i — /?3.
Z = l.
Вещестпо
СаМ8(СО,),
——■> ■ i ■ ————
а л
6,0()
47э3'
ОМ
107, Г)
Примечание
Тип кальцита, но с несколько
изменёнными параметрами
положений атомов (SB I 303)
§ 275] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. IV ГРУППА 75У
о са,мд
Оо
Рис. 031. (Идеализированный тип кальцита.)
760 Ч. П- СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
10. Na[HC03]
Решётка моноклинная слоистая Ms.
Координация xjy.
Тип М$в - SBG012 (NaHCO,).
Пространственная группа С|л — P2iln( — P2Jc).
э=[нсо,]=«.
Z = 4.
Вещество
NaHC.O,
а
7,51
Ь
9,70
с
3,53
Р
а : Ь : с 1
93°19' 0,774: 1 : 0,364
SB III 86. С03-группы образуют плоские равносторонние треугольники
с С в центре. Они параллельны (101) и соединены в бесконечные сетки,
параллельные (101). Посередине между атомами лежат атомы Н, на
расстоянии О ~-Н = 1,275а. Переход к сложнокомплексным соединениям.
11. Ca0-Si02 = CaSiO3 (воддастонит)
В соответствии со структурой правильнее формула Cae[SisO,]
Решётка моноклинная М.
Координация 2/6.
Тип Д/й/А-вВЯЗ,.
Пространственная группа С^л — Р2\[а.
Z = 12 CaSiOs = 4Ca, [Si.O,].
P = [Si.Ot].
р к. ч.=6Са;
Са к. ч. = 23.
Рис. 632.
Вещество | а
C'aSi03 1Г),33
Ъ
7,28
с
Р
7,07 J 95°24,5'
1
а : Ь : с
2,106:1:0,971
3 [Si04] тетраэдра соединены вершинами в трёхчленное кольцо. В
элементарной ячейке 4 таких кольца и 12 ионов Са.
§ 2761 соединения с комплексным анионом, v группа 761
§ 276. V группа периодической системы: азот N,
фосфор Р, мышьяк As, cypbMa^Sb, висмут Bi
1. [Co(NH8)e][PFe]2,
[Ni(NH3)e][PFe]2
Решётка кубическая С.
Координация Т\С.
Тип Ctic —
SB#61.
Пространственная группа
Oi- Fm3m (П - F43m).
2 = 4.
« = [PF.|; p = |Ni(NH,),l.
В к. ч. = 8<х (С) а к. ч. =
= 48 (Т).
Нещттно
[Co(NH3)0] fPF.b
[Ni(NH3)s][PF,.]2
aw
и, *С
Примечание. SB III 475
Рис. 633.
2. РС16 = [РС14]+[РС1.Г
Решётка тетрагональная 7\
Координация 8/8.
Тип Г8/8.
Пространственная группа
dh-Pi/n.
2-4.
а = [РС14]; В = [РС1,].
ас к. ч. =8 В;
В к. ч. :-8а.
Вещестно
[РС14] * [PCI,]"
а
У, 22
с
7,V»
Тетрагональная решётка РС1б является искажённой структурой CsCl, в
которой имеет место не только тетрагональная деформация ячейки, но и
смещение узлов р, находящихся внутри ячейки, попеременно ближе к
верхнему или нижнему основанию, примерно как в решётке РЬО (см. рис. 382).
Представляет интерес на наш взгляд следующий факт: в молекуле РС1Б
(тригональная бинирамида) расстояния Р—G1 равны: в основании бипира-
миды 2,10, но оси 3—2,25 kX (SB V1 70). В кристалле же в тетраэдрах—
1,98 кХ, в октаэдрах -в среднем 2,06 кХ, т. е. в обоих случаях гораздо
меньше, чем в молекуле.
Структура РС15 заслуживает специального рассмотрения, к которому
мы вернёмся в другой работе.
762 ч. п. структуры неорганических вещестц [гл. X
Рис. GM.
3. NaNO,
Решётка ромбоэдрическая R.
Координация 0\0.
Тип До/o-SBGl.
Пространственная группа D\—C3a2.
Z-2.
? = N04.
Na к. ч. = 6р (О);
Э к. 4. = 6Na(0).
Вещестло
NaNO,
LINO,
а
0,32
а
47в1'
Г)
ОМ
62,1
См. СаСОв—кальцит.
Рис. 635.
§ 276] СОВДШШН11Я С КОМПЛЕКСНЫМ АНИОНОМ. V ГРУППА 763
4. KNO,
Решётка ромбическая ОЯ.
Координация РЗ/О.
Тип ORtio -SBG2.
Пространственная группа Dm- P/wut.
Z = 4.
?-N08.
К к. ч.^б Н (P\i) (точнее t-ht).
[i к. ч.-б К (О).
Гис. 63Г,.
Вещэстло
KN4)3
а
г,/,6
ь
?'14
Г
«,'•!
Примечание
См. СаС03 (араюнит)
5. RbNO,
Решётка ромбоэдрическая Я.
Координация х/у.
Тип Д,/, —SB ?; SB I339.
Пространственная группа С^ — Я'Ит.
Z = i.
Вещегтпо
RbNO,
а \ а
1
7,3G
109°28'
764 ч- и. структуры неорганических веществ [гл. X
6. NH4N03I
Решётка кубическая С.
Координация CJC.
Тип Сс/с -[SBM (CsCl)]- SBG08.
Пространственная группа O^ — Pin'Sm.
Z = l.
* = INH4]; p = [NO,l.
а к. ч. = 8р(6'); р к. ч.=8* (С).
Вещество
NH4N03 I
«••
'i,40
Примечание
SB II 69
Для возникновения
кубической симметрии необходимо
вращение ионов или
беспорядочно статистическая
ориентировка. Термодинамически
устойчив (при р = 1 атм)
между точкой плавления ( + 169°)
и +125,2>С.
Рис. 637.
7. NH4N0,II
Решётка тетрагональная Т.
Координация Dl\Di (или Р4/Р4).
Тип TDiiDi-SBG09.
Пространственная группа D'd—P42 га (?)
Z = 2
a = [NH4]; p = lNO,l.
а к. ч. = 8р (дисфеноэдр Di); p к. ч. = 8а
(дисфеноэдр Z)j).
(7
1_
2 J
J L.
О NHv,
• N
00
Рис. 638.
Pitc, 639.
Вещество
ГЧН4Ш II
с/а
Примечание
5,74 | 5,00 | 0,872 | SB IT 69
§ 2?б] с!оедийени#|С Комплексным анйойом. V ГрУппА _7б5
Тетрагональная деформация ячейки рис. 637, показанная на рис. 639,
не даёт ещё элементарной ячейки NH4N02II, вследствие взаимно
перпендикулярного положения соседних NO^-hohob. Необходимо удвоение осей
а и Ь (Z = 4). Рекомендуем читателю сравнить не очень наглядный рисунок
638 (SBII) с рисунком 682 структуры NH4C10a. Различие обеих структур
в основном в форме узла P([N1I..]-hoh — равносторонний треугольник dN_o==
= 1, 32А). Ячейка с Z = 2 получается, как и в случае NH4G102 (см. рис. 683),
другим выбором осей а. Термодинамически устойчив (при р -\ атм)
между 125,2°С и 84,2°С. Ниже 84,2°С переходит в NHJ40, Ш.
8- NH4NOaIII
Решётка ромбическая OR.
Координация О/РЗ.
Тип 0fl6/P3~SBGOlo.
Пространственная группа Dtfh — Pbnm.
a = [NH4]; p = lNO,].
Рис. Г.40.
1 Вещество
1 NH4NO,
a
7,06
b
7,66
с
5,88
a : b : с
0,922 :l : 0,788
Примечание
SB II 71 1
Расположение центров тяжести
ионов изменилось. Оно
отвечает теперь не структуре CsCl,
а скорее структуре Ni As (тип В 8).
В Sfe II указано расстояние
N-О, равное 1,28г А 1
Изучение перехода NH4N08I1I~.: NH4N )3IV показало влияние добавок
на температуру перехода, которая для чистого вещества равна 32,2°С.
766 4. ft. СТРУКТУРЫ НЕЦРГАНИЧЕСКИХ ВЕЩЕСТВ |гЛ. X
9. NH4N03IV
Решётка ромбическая OR.
Координация Di/Di.
Тип ORDUDt-SBGO^.
Пространственная г{>упна
Z>V/t - - - Pmmn.
a-NH4; [t NOa.
Z = 2.
% к. 4.=--8$(Di)\
p к. ч. = 8а(Д/).
Вещество
а
Ь
с
а : b : с
NH^NO, IV
5,75
5,45
4,%
1,053: 1 :0,909
Рис. r.'il.
SB II 72. Образуется из ячейки NH4NOeII таким образом, что ионы
N0^, не меняя положения центра тяжести, поворачиваются, ориентируясь
параллельно оси а, что удлиняет ось а и укорачивает ось 6. Расстояние
N — 0 = 1,24 — 1,26 А. Таким образом, в низкосимметричных модификациях
расстояние N — О меньше, чем в высокосимметричных, энергия которых
больше. Около — 18° С и около — 00° С зарегистрированы точки фазовых переходов,
нуждающиеся в проверке.
10. Ca(N03)2I Sr(N0s)2,
Ba(N0,)2, Pb(N0,)2
Решётка кубическая С.
Координация 6 + 2/3 +1.
Тип Ce+2/3+i —
SBG21(Pb(NO,)I).
Пространственная группа
П-РаЗ.
P = [NO,l.
2 = 4.
М к. ч.=63; jj к. ч.=
= ЗМ( + 1).
Вещество
Ca(N08)a
Sr(N08)2
Ba(N08)a
Pb(N03)a
au>
7,60
7,84
8,11
7,84
Примечание. SB II 73
Рис. 642
SB II 73 опровергает описание SB I 304. Атомы Са (и т. п.)
образуют структуру Ск- Сравни рис. 643а и б. При этом, всё же решётка
§ 276] Соединений с Комплексным лйиоНом. УсГрУпгтА 767
больше приближается к типу
например, из 8 расстояний РЬ
Треугольная
(плоская) форма (NOs)~-mo-
нов снижает
симметрию до Т)г. На чертеже
показаны положения
атомов М
(кубическая гранецентриро-
ванная решётка) и
положения атомов
N, которые
находятся не точно в
центрах 8 октантов, но
сильно смещены.
11. IJNi (NH,),] lNOJ2
Решёткакубическая С.
Координация С/Т.
Тип Cc,/-SB#64.
Пространственная
группа Т\—Ра&.
a = [Ni (NH,),1; fi =
-=|NO,l.
Z = 4.
ак.ч.-8р (6');
рк. ч. =4а(71).
CaFj, чем к типу пирита. Однако,
— N 6 одинаковы = 3,20 и 2 = 4,64.
Ni(NH3)e
Вещество
[Ni(NH3)el [N08]a
а»
10,96
ОМ
329
(Идеализщшамое -*\J 3
лолоэ/смие цешдо*
тяЖм/nuj
Рис. 6436.
Ni(NH3)6
SBI 435. Симметрия решётки #61 снижается с заменой К+-иона
на NOT-ион.
768 4.At. структуры нёоргайи^ёски^ веществ [гл. X
Близок к типу Pb(NOa)2 (G2J, как если бы атом РЬ был заменён
на группу Ni(NHa)e без изменения симметрии.
12, [U02eiIaO] [N0J2 (?)
Решётка ромбическая OR.
Координация xjy.
Класс OR-SB?
Пространственная группа
V^—Cmcm илиК/,*—Сттт.
г = 4.
Вещество
[UO,6H,0] [NOJe
а
11,^2
ъ
13,15
с
8,02
Примечание
SB I 455
13. Ag,NOu
Решётка кубическая
С.
Координация C\q\/2q.
ТИП CciqUl2q-
Пространственная
группа Оь—
Fm3m{?)
Z = 4.
Вещество
Ag7NOu
а 1
1
9,87
Примечание. По
работе Г. С. Жданова
и 3. В. Звонковой, 1
ЖФХ XXIL 1283
(1948) *)
О Agl
© ДдП
Qm3 О о
Рис. 644.
Ag I к. ч. =80 (С); NOsK.4.=4AgI (q) 4 Ag ll{q) = 2q\
Ag II к. ч.= 40(g); Ок. 4.^1Ag I+lAg II^2(Z).
В основу рис. 644 положен чертёж работы Г. С. Жданова и 3. В.
Звонковой, несколько перестроенный нами для большей ясности, но с
сохранением позиций к Ag I и Ag II по подлиннику. В действительности же
Ag II—в позициях Ag I, и наоборот.
*) В подлиннике статьи на чертеже перепутаны Ag I и Ag II и неточно даны
параметры Ag II (0 1/2 V*) вместо (О Х/А */4); то же относится к приведённому там рисунку.
§ 276] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ» V ГРУППА 769
Допускается свободное вращение N03-rpynnbi. Узлы Agl, Agll, N03 и 0
расположены в элементарной ячейке, состоящей из 8 кубических гранецент-
рировашшх ячеек (образованных атомами Agll, из которых отсутствует
74 часть, т. е. из 32 осталось лишь 24). В каждой из последних атомы О
занимают 4 из 8 тетраэдрических междуузлий. Атомы Agl и группы N03 занимают
позиции отсутствующих атомов Agll, так что возникает сверхструктура
AgI(N03) типа каменной соли. Вокруг атомов Agll расположен
деформированный квадрат (прямоугольник) атомов О. Вокруг атомов Agl образуется
координационная сфера из 8 атомов О (куб), невидимому, за счёт ионных
связей Agl—О равно 2,26 kX; Ag II—О равно 2,12 кХ.
14. Na [N02]
Решётка ромбическая OR,
Координация D2/D2.
Тип 0fli>2/D2-SBF5B.
Пространственная группа C^v — Jm2m.
2 = 2.
P = [NOa].
Na к. ч.^бр (сильно деформированный октаэдр, бипирамида с
прямоугольником в основании);
8 к. ч. = 6 Na (сильно деформированный октаэдр, бипирамида с
прямоугольником в основании).
2 3 4 5 О О
J i I l ^-^
Рис. 645.
Ось а направлена назад, ось b -вверх, ось с—вправо.
1
Вещество
NaN02
а
3;г>5
Ъ
V
5,56
с
5,38
а : Ь : с
0,6399 : 1:0,9670
Примечание
SB II 65
Решётка типа NaCl, в которой ион С1^ замещён на ион NO~ с углом
О—N — 0 = 132°, повёрнутым в направлении (НО) решётки NaCl. Это
вызвало ромбическую деформацию решётки. Сравни с рис. 599. с?м_о = 1,13.
770 ч.п. структуры неорганических веществ [гл. X
16. KN02
Решётка моноклинная М.
Координация 5/5 (или 6/6).
Тип il/5/5-SBF5u.
Пространственная g группа
С\ — Лт( — Cm).
? = [NO,l.
Z = 2.
К к. ч.=5р;
? к. ч.=5К.
| Вещество \ а \ Ь
1 KNOa 1 4,45 1 4,99
Деформированный тип N*
Рис. 646.
с | р | о:Ъ:с
7,31
*NOa, уг
114°50'
ол О — ]
0,894:1:1,465
4-0 = 132°.
Примечание 1
SB IV 39
16. AgN02
Решётка ромбическая OR.
Координация D2/D2.
Тип 0tfD2/D2-SBF512.
Пространственная группа
СЪ-РЬа.
P = [NO,l.
Z = 2
Ag к.' ч. = 63(£2);
? к. 4.=6Ag(Z)2).
о^ттл-^
О
1 Вещество
A?NO>
-
3,505
1 ь
6,14
•
5,16
Рис. 647.
а : Ь :с
0,5706 :1 : 0,840
Примечание 1
SB IV 41 1
Ю2-ион, в отличие от NaN02, смещён к одному из атомов Ag, так что
теперь атомы О находятся на равных расстояниях от двух соседних
атомов металла, в решётке же NaNOa атомы азота находились на равных
расстояниях от двух атомов натрия.
§ 276] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. V ГРУППА 771
17. NH4 [Н2Р02] = (NH4)2 [Н4Ра04]
Решётка ромбическая OR.
Координация Pi/Pi.
Тип (?^4/Pi-SBF57(NH4H2P0,).
Пространственная группа Dm — Астт.
a=NH4; p = [H,PO,].
Z = 4.
а к. ч. = 4р(г); р к. ч.^-4а(г); позиции: (000) (о±±') +
+ 4N:(o|0; о| о) 4Р : («о| ; хО|Лс *Р-0,542.
Рис. 648.
Ось я направлена вверх, 6— назад, с—вправо.
Вещество
NH4 [H2P04]
а
:i,9s
ъ
7,57
с
11,47
Примечание
SB III 77
1
Р02Н2-группа —деформированный тетраэдр. Атомы Р лежат на
плоскости x-t> z (проходящей посередине ячейки, параллельно оси z) и на передней
(обращенной к читателю) и задней сторонах (плоскостях) ячейки. Р—0 = 1,52;
Р Н = 1,50; Н—Н = 2,16; 0 — 0-2,62; О —H = 2/i9. Интересный случай
ионной решётки, по характеру не являющейся координационной. Такая
структура заставляет предполагать возможность О — Н-связеи между NH+-
и Н2РОТ-ионами.
772
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
18- YP042H,0 ((Y, Ег) Р04 Ш,0 (вайншецкит))
Решётка моноклинная слоистая Ms.
Координация 6/2/1.
Тип M*Sm-SBHA..
Пространственная группа Сгл — C2/c (?).
Z = 4.
Рис. 6'i9.
Ось а направлена вправо, 6 —вверх, с —влево; один узел Y находится
за левым передним нижним тетраэдром [Р04]= и не виден.
Вещестио я
1
YP042ILO((Y, Ег)Р04 211,0 ! 6.4S
(вайншенкит)) |
Ь
с
(i
s 1 • 1
15,12 , 6,2Я 129°24/
Имеются данные о наличии изотипии. YP04 2Н„0 аналогичен гипсу,
Nalurvv 30, 64 (1942) (ср. GaS042H20, гипс стр. 780).
§276] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. V ГРУППА 773
19. Ti[P207], Zr[P207], Ш[Р207], U[Pa07], SI[Pa07], Sn[P207]
Решётка кубическая С.
Координация О/О.
Тип С0}о - SB KG^ZrPfi,).
Пространственная группа Т% — Ра 3.
е=[р,от].
Z = 4.
М к. ч.=63 (О);
,3 к. ч. = 6М (О).
Рии. 650.
Вещество
а
TiP207
7,80
ZrP.C^
8,20
HfP,07
8,18
UP,07*)
8,Г>1
SiP207
7,46
SnPa07
7,89
Примечание. SB III 140. Тип NaCl, в котором ион С1" заменён группой
(ионом) Р207, ион Na+—атомом (ионом) Zr и т. п. Группа Р.,07—два тетраэдра с общей
вершиной.
Интереснейший случай сохранения типа Со/О при наличии асимметричной
группы очень больших относительных размеров.
*) SB IV 68.
774 ч. п. структуры неорганических веществ
20. Na[Sb(0H)$], A«[Sb(0H)$]
Решётка тетрагональная Т.
Координация О/О.
Тип Гб/е-А»-
Пространственная группа C\h — Pi2/n*
? = [Sb(0H),J.
Z = k.
Sb к. ч. = 60 (1,87 А) (октаэдр);
M к. ч.=бр (б);
Э к. ч.=6М (О).
Рис. 651.
1 Вещество ас с/а
NaSb(OH),
AgSb(OH)§
8,01
8,12
7.88
7,91
0,984
0.974
1
Примечание 1
SB VI 2G. Несмотря на очень малое
соотношение г1/г.г, вопреки правилу
1 ольдшмидта, слегка деформированная
решётка NaCl с октаэдрами [Sb(OH),]~
вместо С1~-ионов
§ 276] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. V ГРУППА 775
21. Na[SbF4(OH)J
Решётка гексагональная Н.
Координация б/РЗ.
Тип H3/p3-SBJltt.
Пространственная группа Dlt — HZc.
H = [SbF4(0H),].
Z = 2.
Na к. ч. =6р(0);
р к. 4. = 6Na(/>3).
1 I I I I 1 L
Рис. 652.
I Вещество а
1 (
Na [SbF4 (OH)2]
с
5,227 I 9,98
1
e/a
1,909
Примечание
* SB VI 27. Мы хотели бы подчеркнуть,
аналогию с решёткой типа J98 (NiAe),
в которой половина мест занята ионами
JNa, другая—октаэдрическим и
комплексными ионами (J (вместо As)
776
Ч. ДЬ ^Р.УКТХВД ННрРГДН1]ЧНСВД1Х.ЧЫДД.СТф .
[гл. X
§ 277. VI группа периодической системы: кислород ;Q, (jepa S,
селен Se, теллур Те, полоний Ро ., . .,
1. [NHJJSeClJ, RbJSeClJ, CsJSeClJ, KJSeBrJ, [NH^JTeClJ,
Rb2[TeCl6], CsJTeClJ, Tl2[TeCle]
Решётка кубическая С.
Координация Т/С.
Тип Ctic -SB #61 - и /14.
Пространственная группа 06h — Fm3m.
|3=[МС1,].
Z=4.
м к. ч.=4? (Л;
р к. ч. = 8М (С).
Рис. 653.
[Sex6]
Вещество
ОМ
(NH4),SeCle
9,935
Вещество ., .
ОМ
RbjSeCle
9,97g
248
. (NH^TaCl, ,
2
10,178
53,5
Cs^SeCle
10,266
271 (
K2SeBre
10,363
278
R^TeGlf
10,23а
(10,221)
257,5
(267)
Примечание] If
SB III 121
Cs2TeCle
10,4
(10,4
285
45
V
Tl>TeCle
io,io7
257
1
§ 277] соединения с комплексным анионом, vi п-уипл 777
Комплексные сое динения/АВ04
2. CaS04, ангидрит \ ^
Решётка ромбическая OR.
Координация 4/4.
Тип OR 4/4 - SB tfl= SB H 01.
Пространственная группа VlH — 2)^ — <?Ш(?//?.
Z = 4. S04 = P (тетраэдр).
Са к. ч. = 2+-2(?);
[4 к. ч. = 2 + 2(Са).
Рис. 654.
Вещество
CaS04
г а. 6 с
6,22
6,9'.
6,97
kd (S^O)
1,40
4с? (Га—О) Примечание
2,16
Структура нсевдоте-
трагональная, тетраэд-
рические группы-[S04].
SB I 340
778 ч. и. структуры неорганических веществ [гл. X
3. SrS04, BaSO, (барит), PbS04
Рис. 655.
1 Вещество
SrS04
1 BaS04
I PbSO,
i
Л
a 1 &
i
i
8,36
8'85 .
8,45
5,36
5.44
5,3g
С
Г>,8,
4
7,1,
6'»3
OM
7J,6
83,4
78,7
Примечание
SB I 343. Положения О-атомов
в элементарной ячейке точно не
установлены.
См. рис. 586 и 587.
§ 277]
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VI ГРУППА
779
4. [Be(Ha0)J[SO4]
Решётка тетрагональная Т.
Координация SjS.
Тип ТslsSB Hi,. *
Пространственная группа
DH-j\c2-{Fl2c).
а = [Ве(Н20)4] (тетраэдр);
Э — fSOJ (тетраэдр).
2 = 4.
а к. 4. = 4P(S); р к. 4.-4a(S)
(сфеноэдр).
Вещество
а
с
с/а
Be (H.OJ.SO, *
8,02J
' 10'75'
1,340
Примечание. SB II У1. Тетраэдры
[Be (OH)J и [S04] образуют
тетрагонально-деформированную
решетку сфалерита.
Ве(Н20)4
Рис. 656.
б. NH4A1 (S04)„ NH4Fe (S04)2,
KA1(S04)2, KCr(S04)2 (безводные
квасцы)
Решётка гексагональная
слоистая Я*.
Координация б/РЗ/t + t.
Тип#(?/Р*/1+_, -SB^32(KA1(S04)2).
Пространственная группа D\—С32.
P = fSOJ.
£ к. ч. = ЗА1(*) + ЗК.(*);
К и. ч.=60 и 6р(0);
А1 к. ч. =60(/>3);
А1 к. ч. = 68 (О).
1 Вещество
NH4A1(S04)>
NH4Fe (S04)2
K\1(S04),
KCr(S04)2
a
4,724
4,825
4,706
4,737
с
8,225
8,310
7,960
8,030
с/а
1,740
1,723
1,692
1,696
Примечание: 3 "*» H 90. 1
Pec. 657.
780
4.II. СТРУКТУРЫчИЕОРГ.ЛНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
Тетраэдры S04 образуют слой по гексагональному закону (плотная
упаковка). По этому же закону образуются слои А1 и слои К. Слои
чередуются в порядке: К, [S04], Al. i
Атомы К. лежат над атомами А1 (гексагональный закон). Тетраэдры
двух соседних слоев [SOJ повёрнуты основанием в. разные стороны.
6. Ca[S04]2H20 (гипс)
Решётка моноклинная слоистая Ms.
Координация 6/2/1.
Тин ilff/2/1-SBtf4f.
Пространственная группа См — СХ/с.
а = Са; ?-[SOJ; Y = [H20].
Z = 4.
а к. ч. ==2y + 6k3; p к. ч. -бСа;
Y к. ч. = 1Са.
г\
Рис. 658
Ось а направлена вправо, Ь—вверх, с —влево. Один узел Са ho о;лпся
за передним левым нижним тетраэдром [S04] и не виден.
1 Вещество | а
CaS042II2O
10,47
Ь | с
15,15
6,51
р | а:Ь:с
15ГЗЗ',
0,693:1:0,4293
Примечание 1
SB IV 47
см. SB VI 99
Решётка гипса слоистая. Слои молекул Н20 чередуются , со слоями
ионов Са2+ и SOJ~. Плоскость спайности проходит между слоями Н20 — НДХ
параллельно последним.
§ 277] СОЕДИНЕНИЯП КОМПЛЕКСНЫМ АНИОНОМ. VI ГРУППА 781
4d(S-0) = l,49 А (тетраэдре 2d-(Ca-Н20) = 2,44. Каждый ион Са
имеет 2 соседние молекулы Н20. Каждая молекула Н20 имеет 1
соседний ион Са2+, угол Н —О —Н в молекулах Н2О = 108°. >
В рентгенографически найденной ячейке [001] направление отвечает
кристаллографическому [101] направлению. Размеры
кристаллографической ячейки: а = 10,47; 6=15,15; с = 6,28, (3 = 90°58'; Z = 8. Но, согласно
SB VI, элементарная ячейка гипса имеет периоды: а = 5,63; 6=15,15;
с = 6,23 А; f* = 113°50'; Z = 4.
См. также YP042HaO (Nalurw. 30, 64 (1942)).
7. Na2S04, Ag2S04, Ag2Se04
Решётка ромбическая OR.
Координация 10/7+ (или 6/3).
Тип ОЯд/7+ (или <9/?6/3)-SBtfl7(Na2S04).
Пространственная группа D"h — Fddd.
p-[S04](F); S-0 = l,49.
Z = 8.
а к. 4.^10Na или 6Na (2 no 3,03+4 no 3,45);
Na к. ч.«5р или 3Na (1 ш> 3,03 + 2 no 3,45).
Рис. 659.
1 Вещестно
1 a
b
1 c
Na>S()4
5,85
42,29
9,75
Al',S04
5,82
12fr,5
10,25
A^.-o04
M)7
12,81
10,21
Примечание
SB И-8
782 Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [ГЛ. X
8. (NH4),Sft4, K2S04, Bb2S04f CssS04, K,Se04
Решётка ромбическая слоистая ОЯ*.
Координация (ближайшая) Л + 1/3.
TBn0tfh?l/?-~SB#MK2SO4).
Пространственная группа Dlh — Pmcn = Pnma.
P=[SOJ.
К к. ч. » 3 (6) (3; р к. ч. ^ h + 1 (ближайшие) (12). (Координация намечается
довольно неопределённо и охарактеризована нами неточно.)
о м
so.
Рис. 660/
Вещество
<Ш4)а S04
k2so4
RbaS04
СзлА04
K2Se04
а \ Ъ \ с
5,97
5'76
5,97
6,24
6,02
10,60
ю,о5
10,43
10,93
10,40
7,76
'}\
7,81
8,21
7,60
а : b :с
0,563:1:0,732
0,573:1 :0,742
0,572:1:0,748
0,571 :1:0,753
0,579:1 :0,731
Примечание 1
SB II 86. Структура построена
из тетраэдрических ионов SO}~
и К* по псевдогексагональному
закону; <*s_o=l,50 4-1
Klx
Структуру можно
сравнить со структурой
графита (Л9). В плоскости сеток—
кольца из атомов KI и
тетраэдров S04, чередующихся
попеременно. Тетраэдр SO*"
имеет сверху 4-го соседа К+ I,
чем обеспечивается связь
между сетками. Таким образом,
каждая вершина тетраэдра
как бы фиксирует по 1
иону К+. На рис. 661
обозначении, кроме того, ионы K+II,
расположенные в центрах шести-
членных колец. Хотя сетки искривлены и описанное «идеальное» положение
нарушается, характер координации, в основном, остаётся (см. рис. 661 (схема)).
х///
Рис. 661.
§ 277] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VI ГРУППА 783-
9. AgaS04 4NH,*)
Решётка тетрагональная Т.
Координация qjL
Тип Tqfl-SB /7417.
Пространственная группа Z>2<f — Pi2xC.
« = Ag(NH,),; (3 = S04.
Z = 2.
ок. ч. = 2ji (линия); £1 к. ч. = 4a (квадрат).
a-p = 4,21§kX.
Рис. 662.
Вещество
[А8(КН,),],804
a
8,43
с
6,35
с/а
0,7533
Примечание
SB III 115
Узел а имеет на более далёком расстоянии (5,2 кХ) ещё 4р (д), а всего
6JJ (jf>4). Узел р имеет на таких же расстояниях (5,2 кХ) ещё 8а, а всего 12а
(тетрагонально деформированный кубооктаэдр). Решётка имеет в большой
мере ионный характер [Ag (NH3)2]+ [S04]=. Координация DijTK—редкая
для ионных решёток.
*) Эта формула не отвечает строению. Правильнее было бы [Ag(NH3)2] [S04].
m-'i
4. Ii. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
[гл. X
10. NH4A1 (S04)212На0, KAl(S04)a12H20, RbAl (S04)212H20,
T1A1 (S04)212H20, KA1 (Se04)212Ha0, KCr (S04)212Ha0 (a - квасцы)
Мы бы предложили более отвечающую структуре квасцов формулу типа:
[K(H20).][Al(H20)e][S04]e
Решётка кубическая С.
Координация C'+O/C + OjT + T.
Тип Сс+7;/с+0/7 +г — SB #413.
Пространственная группа Th — РаЗ.
a = [К(Н20)в]; [* = [А](Н20)в]; Т = [SOJ.
z = ^-
К к. ч. = 60 (точнее, 6Н20) (2,94 А) (большие октаэдгоподобные группы);
А1к. ч. = 6 О (точнее, 6Н20) (1,98 А) (октаэдр);
S'k. ч. = 4 О (Зе - 1,54, Id = 1,47) (тетраэдр);
а*к. ч. = 8y-f 6?; Э». ч. - 8у + 6а; у к. ч. =4а + 4?.
Рис. 663.
§ 277]
СОЕДИНЕНИЙ С КОМПЛЕКСНЫМ АНИОНОМ. VI ГРУППА
785
| В-во | NH4 Al (S04)212Н20 | KAl (S04)212 Н ,0 | Rb Al (S04), 1211,0
a„
В-во
ati
12,215
T1A1(S04)>12H,0
12,207
12, ш \ 12,220
i
KAl (Se04), 1211,0 | KCr (SO,), L2H.0
12,351
_
12,171
1
11. CsAi(S04)2 12H20, NH3CH3Al(S04)2r2[L0 ([^-квасцы)*) (метиламмошш-
ные квасцы). Мы бы предложили более отвечающую структуре квасцов
формулу типа: [Cs(H20)e] [Al (H20)e] [S04]a
Решётка кубическая С. _
Координация С + О/С + О/Т + Т.
Тип Сс+о/с+о/г+'7 — SB#418.
Пространственная группа Т\ --- Раб.
e-Cs(H10)i(0);P-Al(HaO)i(0);T-S04(71).
Z-4.
Cs к. ч. = 60 (точнее, 6H2O)-f0O (от
групп S04);_
а к. ч.=8у(С) + 6р(7,)-г-6 атомов О от
тетраэдров S04; __
рк. ч.=8у(С) + 6х(Г); у к. ч. = 4а(У) +
Вещество
CsAl(S04).l.2H,0
NH3CH3A1(S04)a12H3()
12, :Ш
12,47У
12. NaAl (S04)a 12 Н20 (у-квасцы)
Мы бы предложили более отвечающую структуре формулу:
Na(H20)eAl(Ha0)e(S04)2
Решётка кубическая С.
Координация С + О/С + 0\Т + Т.
Тип Сс+о/с+о/7+~/— SB//418.
Пространственная группа T*h — Pa'S.
a-Na(H2())e(0); р = А1(НаО).(0); Т-(S()4) (У1).
Z = 4.
Na к. ч.---6 О (2,45 А) (точнее, 6 Н20) (октаэдр);
А1 к. ч. = 6 0 (1,92 А) (октаэдр);
S к. ч.-=4 0 (1,48 А) (тетраэдр).
а к. ч.=8у (С) + 6? (О);
Р к. ч.-8у (С) + 6а (О);
у к. ч.=4а (Г) + 4р (Г).
Вещество
NaAl(^04),.12Ha0
12,19
*) Построенный для а-кпасцов, рис. 003 отображает расположение структурных
узлов также и в ji- и в ^-квасцах. Во всех трёх случаях, решётка слагается тремя
типами узлов: а например [K(H,0)J+; ^—[Al (Н2О)0]а + и у—lS04l"\ Причём В-узлы
располагаются в вершинах и центрах граней элементарной ячейки решётки BiF3.
а-узлы занимают центры рёбер и центр ячейки, образуя вместе с узлами |э ячейку С0/0.
Внутрь октантов внедряются ^-узлы. Однако, они занимают не точно центр!.!
октантов, но несколько смещены. Величина смещения и определяет собой одну из трёх
структур: 8 S занимают позиции: ± ( (ххл)> ( ^7 + ^» "*>—х> х )0 \ причём для х-квае-
цов #s = 0,31, для р-квасцов—0,33, для 7-^»асцов-0,205.
В результате fi-квасцы отличаются от а-квасцов и тем, что вследствие большого
размера Cs^-иона, молекулы воды отходят дальше. Это вызывает смещение <> S()4-rpynn
вдоль оси 3 и их втягивание в координационную сферу этого иона, с повышением
его к. ч. до 12.
786
ч. п. структуры' пшитлйпчгхкпк ш:Щ1:с1'в
[гл. К
В числе многих других веществ квасцы являются ярким примером,
опровергающим так называемый закон Грота, согласно которому
сложный состав влечёт за собой низкую симметрию кристалла. Дело по
к химическом составе (т. е. не в соотношении концентрации атомов), а
в составе и строении структурных узлов. Подобные соображения и
заставили нас отказаться от принятой в SB систематики по химическому составу.
13. [Со№),]£ол, [Co(]m,).]?Roir [Co(NH,).]fSe04r [Co^J g^ ,
lt0(NH,).i ISO,]
Решётка кубическая С.
Координация С/Т.
Тип Cr.tr- SB//61.
Пространственная группа U'), — Fm'Sm или T%t — lf\<\m.
а-=Вг, соответственно J, [S04], [Se04];
p = [C(i(NH,)§l, соответственно [Со(Ш„),ОН];
Z=4.
рк.ч.--8а(С); ан.ч. = 4р(7').
Рис.. Г.С4.
Co(NH3,0H)e
Вещество
[CoCNH^lisul]
1Со(№18)§]Г8Й4]
[Co(NH3)e][s4i
^•0(NH»)6I [S04]
llj0(NHa)5J [SO4]
а»
10,51
10,71
10,79
10,45
10,02
ОМ
290
307
314
285
' 299
Примечание 1
SB I 429
Группа Co(NH3)5OH сама по себе не 1
имеет кубической симметрии.
Расположение ОН-групп и кристалле является
статистическим.
Сохранение кзгбической метрики при
столь разных величинах ионов является
наглядным примером влияния
слабого силового ноля большого катиона,
1 допускающего вращение ионов в решётке |
§ 2l1\ СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ ЛНЛОНОМ. VI ГРУППА 787
14. (NH4)., S20„ (персульфат аммония), Cs.>S20g (персульфат цезия)
Решётка моноклинная М.
Координация 4/4.
Тип М41л SBKAy.
Пространственная группа Cbh — P2ljc.
? = [NH4l-t;
? = [S04|".
Z - 2.
а к. ч. М (/?);
Рис. G65.
Вещество
(NII4)sS3Os
Cs2S,08
1
a
7,83
8,13
b
8,04
8,33
с
6,13
6,48
a : & : с
0,9774 : I : 0,7Г»71
0,97:i2 : 1 : 0,7333
?
95°9'
9r>°t9'
В SB III 136 даны приведённые выше значения периодов элементарной
ячейки и их отношения. Проверочные расчёты показывают, что эти цифры
находятся в не очень точном соответствии друг с другом. Во всяком
случае, не следовало приводить а:Ь:с с точностью до четвёртого знака.
В решётке существуют тетраэдры S04 (S- 0-1,44—1,52), соединённые
мостиками через атомы кислорода с расстоянием О —О связи, равным
1,46 кХ, и углами между связями 122°.
/" 1 1 1 \
Положение атомов: ± (xyz) i-j + ^j ~^ — у> it + z ) ПРИ *nii4 =0,144;
?/nh4 = 0,125; zNh4 =-.,0,250; zs-0,136; 2/s - 0,350; "rs-0,709. Очень nine*
ресна структура с последовательным чередованием двух узлов а и двух
узлов 3.
788
П. it. структуры 1ШорглПТ'1чр:с1{их пкщкств
(гл. X
16- Na2S03
Решётка
гексагональная Я.
Координация -^ />3/Р3 +Д
Тип //^.ш/рзьш"-SBG3*
(Na,S09).
Пространственная
группа си-сз.
P = [SOJ.
Ma к. 4. = ,r)Ii (.03);
p к. 4.-.= GNa(/'H)-|-
-1-5 Na (Ш).
Рис fi6f>a
Вощеетио
Na,S03
a
5,44,
С
r„n3
rja
1,127
Примечание
SB II 74. S(Vrpynna—
тетраэдр, л одной из иер-
шин которого атом о0 отсут-
етпует; S -0=1,:19 Л
Рис. 666а, SB II74,
отвечает
гексагональной призме,
состоящей из 3
элементарных ячеек. Так как
чертёж SB не
нагляден, сопровождаем
его рис. 606b, где
показано
расположение центров тяжести
узлов а и [3 в одной
элементарной ячейке,
выделенной жирным и
линиями, и показаны
контуры всех трёх
элементарных ячеек,
отвечающих рис. 066а.
Тетраэдры S03
опрокинуты основаниями
внутрь ячейки.
Рис. сббЬ.
§ 278]
СОКДПШЛПШ С КОМПЛЕКСНЫМ АНИОНОМ. VII П'УППЛ
780
§ 278, VII группа периодической системы: фтор F, хлор CI, бром Вг,
иод J
KJC14, и др. (см. полисоединопия)
1. NaHF2
Решётка ромбоэдрическая R.
Координация 6/6 ^ О/О.
Тип R6jb -SBFb^NMF,).
Пространственная группа D*d— R3m.
P = |HF2]
Z = l.
Na к. ч. = 6? (О);
3 к. ч. = 6Na (О).
Вещестио
NailF,
а
5,00
Рис.
а°
40°2'
Г>07.
ОМ
4:'.,з
Примечал цо
SB 12 П. Груииы |F-
линейны, h (Na -II) -=
d (F—11) = 1,2Г)А.
-11 -F]
;j,ogA; j
790 ч. ii. cTi'yjrryi'i.i шдл глшгчкекнх шшц-хтн |1Л. X
2. KHF2
Решётка тетрагональная Т.
Координация PijP-i.
Тин rP4IPi-SBF5,.
П[)Ост1)анстиеиная группа Мл — Pi/nan.
f. [FHFj.
Z-4.
К к. ч. .--8 $ (/>',);
?, к. ч.=8К(/»1).
О К
ч г f f f f Of
Рис. 668.
Вещество i a
l
KHF,
5,67
с
6,81
ОМ
54,7
Примечание 1
SB I 276
Примечание. Группы F — H — F линейны i (К —Н) —ЗД А
rf(F-H)-l,l2(?).
Деформированная решётка CsCL Вследствие разной ориентировки HF2-
групп приходится пользоваться элементарной ячейкой, ограниченной
затушёванными верхним и нижним основаниями.
5 278| сокд1ПШШ1Я с комаикопим лимоном, vu ггуиил 7'Л
3. NH4HF2
Решётка ромбическая OR.
Координация DijDi (или PijPi).
Тип ORmiiH-SBF5t.
Прострапствсннан группа Dlil~ Pman.
Z-.-Л.
a = |NH4j-i-;
У- IHF.J-;
a к. ч. = 8{J(Z>t) (Pi);
? к. ч. -8a (/Л) (Pi).
I'm. 000.
Вощестио
NH4HF2
8,:w
ft
•
:i,r>8
Примечание
SB III 79. F1IF линейна
792
Ч. II. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ НЕЩЕСТП
|гл. X
4. NH4C104, NaC104, KC104, RbCl04, CsC104, AgC104, T1CI04
Решётка кубическая С.
Координация О/О.
Тип Сою - (SB Bl) -SBtfO, (КСЮ_4).
Пространственная группа Т% — F43w.
Р = СЮ4.
Z=4.
М к. ч. = 6 ЦО);
[>, к. ч. = 6М(0).
Ом
OCL04
Рис. 670.
Вещество
I УСТОД'ШН
1 выше t3 С
*°С
С"
NH4C104
240
243
7,67
NaQ04
:Ю8
ш
7,08
КС104
200,5
зю
7,5,
иьгю4
270
300
7,70
CsC104
210
230
7,98
AgC104
155
160
7,00
Т)С104
266
280
7'70
См. SB II84. Тип NaCl [Bl] тстраэдрическис радикалы [C104ldci-o = 1,6А
группа [С104] вращается.
Ниже указанных в таблице температур —переходят в OR
модификацию (стр. 794), с увеличением координационного числа до 7.
§ 2781
СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ ЛШЮНОМ. VII ГРУППА
793
б. NH4J04, NaJ04,
KJ04, AgJ04
Решётка
тетрагональная Т.
Координации
q+Slq + S.
ТИП Tq+Siq+S —
-SBtf4 = tf04
(шеелит).
Пространственная
группа CI h —Jl J a.
а = [N Н4],
соответственно К; p = [J:)4].
Z = 4.
а к. ч.=4р квадрат
+ 4р
тетрагональный тетраэдр;
Р к. ч. = 4а квадрат
+ 4а
тетрагональный тетраэдр.
В SB I348 -в
обозначениях к рис. 166
К и О перепутаны
(К вместо О и
наоборот).
Рис. 671.
1 Вещестпо
Nll4J04
NaJ04
KJ04
At?JO,
a
.ri,0.fi,
5,:i>
5,75
r>,*.fl
' 68
с
12,80,
i
11,0 1
12 ,6 <
12 ,0, ,
' i:{
rja
2,15,
2,2'i
2,20
2,248
ОМ
—
8'.,'.
10', ,5
Примечание
См. SB I :М7, SB II Kit,
J04 —тетраэдр
J—O«sl,50A.
Очень интересна форма
координационной сферы я, соответственно
(3 (квадрат+тетрагональш.ш
тетраэдр) (к. ч.=8).
Орашш с решёткой циркона
ZrSi04, где наблюдается та же
конфигурация
Решётка К,Ю4 как бы состоит из сеток, параллельных основанию
элементарной ячейки. Каждая сетка образована квадратами, вершины
которых заняты тетраэдрами J04, а я центре—атомы К (и наоборот).
Элементарная ячейка охватывает 5 сеток. Тетраэдры первой и пятой лежат в точ-
704 ч.и. структуры нкогглничшлшх нкщисти [гл. X
иости друг над другом. Тетраэдр третьей- над атомами К первой сетки.
Тетраэдры второй и четвёртой — на гранях элементарной ячейки.
Расстояние J -О 4d = l,80 (в KJ()4). Расстояние К —J равно \
но 4,19 (q) и 4 но 4,35 (S).
Расстояние К-О 4е = 2,7(5 (в KJOJ;
4е' = 2,81(в KJ04).
tt. NH4CI04, KC104f KbCI04, CsC104, Т1С10,
Решётка ромбическая OH.
Координация Р'*¥/Р3*.
Тип ОДрз7рз*-8ВЯ2.
Пространственная группа V\? = 1)^ — Prima.
a = |NH4] соответственно К и др.
Р = |С1041.
Z=4.
CI к. ч.=4,0._
а к. ч.~1${РЗ*);
3 к. ч. = 7а (РЗЛ).
а
b
с
ОМ
N1I4CI04
9,-2,
:>,80
7,'|2
У9,1
ксю/
8,85
, 5'б6
| ?'\
00,7
RbC104
9,2,
5,81
! 7»^
1101,4
CsC1041
9,82
б,о0
7,7,
|И'#,7
Т1С104
9А>
5,88
7,;,0
Ш,У
Примечание 1
См. BaS04, стр. 778.
При достаточно высоких температурах переходят в кубическую моди-
фикацию, см. стр. 792.
§ 2?8|
СОКДМПЫПШ С КОЛ1М. IKKCIIMM Л1ШОИОЛ1. VII 1ТУ1111Л
795
7. Mg (С104), 6 Н20 [Ms (ILO)J [С104]о
Решётка ромбическая Oil.
Координация 8/i.
Тип Olis/Z -SJW/4,,. »
Пространственная группа (11п Ртп.
a--[Mg(HsO.)r;
Р=[СЮ4]-.
Z--2.
а к. 4.sk8('j.;
Н к. ч.^4 а.
О
О HjO
Рис. 673.
Вещество
Mg(Cl'04)aGH20
а
7,76
£ с
IViG
5,2 G
а : ft : с
0,Г»7Г>Г> : 1 : 0,3908
Решётка состоит из двух типов сложных структурных узлов: тетраэдров
[СЮ4] и октаэдров [Mg(H40)e]. Расстояние С1-—О равно в среднем 1,48,
расстояние Mg — О равно 2,14кХ.
В SB III 452 рассматриваются также соединении ряда Мс (С104)вН20
следующих металлов: марганец, железо, кобальт, никель, цинк, кадмий,
ртуть. Как правило, все они изоморфны друг другу.
70G Ч.И. СТРУКТУРЫ ИНОРГЛШРЧЕСКНХ 1ШЩ1ХТН [ГЛЛ X
8. [Mg (NH3). (С104)2, [Mn (NIL.),] (C104)„ [Fe (NIL.).] (C104)2, [Co (NH3).] (C104)2,
[Ni (NIL,),] (C104)2, [Zn (Nil,).] (C104)2, [Cd (NH3),] (CI04)3
Решётка кубическая С.
Координация CjT. *
Тин CC/r-SB#61.
Простраиствспная группа Of, — Fm 3 т или Та — I' 43/м.
3=[M(NHa),]; а = (СЮ4)8.
Z = 4.
3 к. ч.-8х (6*); а к. ч.=43 (7').
Рис. 674.
Веще- |
ство \
аи>
[Mg(NiU,].
•(СЮ«).
11,5:»1
[Mn(NH,),]-
(СШ4),
11,578
Вето- [Ni(NH,),|-
1 стпо (С104)2
«И
11, «6
[Fe(NH3),|-
•(СЮ4),
11,51^
[Zn(NH,).l-
•(СЮ4)г
Ю,250
[С<> (N11,),].
•(СЮ«),
11,«9
Примечание
SB III 475
[Cd(NHa).]-
•(СЮ4),
Н,о'.2
§ 278] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VJT П'УППА 797
ft. [Co(NH3)e][CK>4]a
Решётка кубическая С.
Координация С-'^О/О, Т.
Тип С^+о/о, r-SBJ)(\ SB//71. _
Пространственная группа Т;{ — F Ь'Лm (Т'1 пли У4).
а-[С104Г; ^lCo(NH3)§r.
Z -i. [j к. ч. = 8аII (куб) [-Gal (октаэдр);
al к. ч. - G3 (октапдр); all к. ч.^=\'} (тетраэдр).
О CL04
Рис. 075.
Вещество
Co(NH3)flC104I8
ОМ
1
il,:*9(ii,:isG)
:to9
Примечание
SB I W. См. также BiFv
См. |MI4|3 A1F6, В SB III 47П
ошибочно отнесены к тину С1 (//ГЛ) вместо //71.
aw no SB III —d скобках.
ы
t-l. tr. СТРУКТУГЫ НЕОРГЛНПЧЁСКГГк BlilUKCTTi
[гл. X
10. Na€10;!, Na«r03
Решётка кубическая С.
Координация 6 И/6 И.
Тип Cc+i/o.-i-i —SB С: J
(хлорат натрия).
Пространственна)!
группа 2,4--Р21;{.
3 = СЮ3.
Z = 4.
3 к. ч. :-6Na + lNa;
Na к. ч.-вЗ |--|р.
^QL-o
Vw. f>7G.
Вещество
NaC103
NaBrU-з
а„
6,50
0,72
ОМ
70,6
75,9
1 Примечание. Искажён-
I пая решётка NaCl. Лто-
1 мы Na и С1 соответ-
1 ственно Na и В г смещены
1 (по сравнению с NaCl) в
1 глубь элементарной ячей-
1 ки, как бы образовав
1 4 молекулы. См. рис.676.
Октанты ячейки NaCl
деформируются,
превращаясь в совокупность из
вытянутого и сжатого
ромбоэдров (рис. 677),
причём оси ромбоэдров
равновероятно
ориентированы в одном из 4
направлений под углами
тетраэдра (109°28')
соответственно виду
симметрии Т.
Рис. 677.
i 278]
cob.wrimvmn с комплексным лппоном. Vii группа
7§9
п. ксю8
Решётка моноклинная М.
Координация ~7/7 ~ РЗ"1 /7*3+.
Тип Мрз+1рз+ —
Пространственная группа С'-ч,
-P2Jm.
3 = [СЮ3].
Z = 2.
К к. "ч. = 7Р; ? к. ч. = 7К.
Вещестпо
а . . .
6 . . . .
с
? . . . .
КС1()3
гьС47
5,585
109°:*'
Примечание. SB II №
Риг. G78.
Вещество
АяСЮ,
1 *
8/*7fj
с
7 9
/,J0
Приме»
чание
SB I :m
Ионы GlOj имеют асимметричную форму Id (Cl — О) = 1,60 А;
2d' (С1— 0) = 1,42 А. Координационная сфера обоих узлов имеет форму,
схематически представленную на рис. 678.
12. Ag C10,
Решётка тетрагональная Т.
Координация xjy.
Тип Тх1а. ц п
Пространственная группа VD = D2<i
J Kim (?)
2 = 8.
Эти данные мало надёжны.
13. КВг08
Решётка
ромбоэдрическая Я.
Координация RJR.
Тип Яд/л-SBGO,.
Пространственная
группа DU —R3m.
3 = ВгО,.
Z-1. К к. ч. = 8? (Я);
Зк. ч. = 8К(Д).
Искажённый тип GsGl
с пирамидами ВгСГ
вместо СГ-ионов.
Рис. 079.
800 Ч. П. С'П-УКТУГЫ НК01'ГАШ№СкИХ ПНЩЬХТП [ГЛ. X
Деформированная решётка тина GsCl (6 расстояний К—Вг 3,90±
±2 расстояния = 4,07) из ионов К+ и BrOg.
Все три расстояния Вг —О в ионе ВгО~ одинаковы (1,68А), с углами
близкими к 109°28'.
li. NaJ03
NaJOg длительное время относился к типу перовскита {Е2Х) или
антитипу перовскита. (Соответствующие данные были нами приведены на
с/г р. 661.) В работе J. Am. Gh. S. 69, 1281 (1947) они подвергнуты
сильной критике; даны иные периоды элементарной ячейки: а =-- 5,75; 6 = 6,37;
с.-,-=8,10; а : Ь: с = 0,903 : 1 :1,272; Z — 4 и иная пространственная группа
Vj? — Pbnm. Более того, утверждается, что в этой структуре содержится
ион J03", так что она является переходной «от тииа перовскита к типам
грушш G». В работе делается предположение, что и LiJ03 содержит
группы J03, с 3 атомами О в форме равностороннего треугольника.
Расстояния На1-0 равны: в КС103 --1,42 А, КВг08 —1,68 и в NaJ03—2,05-2,08.
Эти выводы не находятся для солей Na, К и далее до Cs в
противоречии со сказанным в § 105. Новидимому, эта группа иодатов относится
скорее к комплексным соединениям.
15. HJ03
Решётка ромбическая OR
Тип OR.
Вид симметрии Z>2 — 222.
Z==4.
16. [Zn(H2O)0][BrO3]2
Решётка кубическая С.
Координация 2 (+ 6)/7\
'['ми C2(+G)/r-SBtf04.
Пространственная группа
П РаЗ.
a = [Zn(H,0).]; ? = [ВгО,1
Z = 4.
а к. ч.=,2р( + 63)^8?(Г);
$ к. 4.==4Zn (T).
Оо
Рис. С80.
Вещество а„
|Zn(II,U)J |Ur03|,
1(),:л
ОМ
•27/.
Примечание
SB I 435, более достоверно
описание SB II 73,74
' См. INi(NH,)e| |NO,l,.
Необходимо уточнение
структуры
Вещество
а
1
Н.Ь'з .",,55
Ь
•V>2
с
7, 1 „
Примечание
sb I :m,
SB II 332
®[Zn(H20)6]
§ 278] СОЕДИНЕНИЯ С КОМПЛЕКСНЫМ АНИОНОМ. VII ГРУППА 801
17. Nd(Br03)3. »H.,0--|Nd(U:!0)B]fl»rOJa
Решётка гексагональная 11.
Координация P'3:]+/t.
Тип Hirf+ц -SBGL'B.
Пространственная группа Г,4;(1 —Ci\mc.
Z - 2.
а = (HrOJ - (у-); 3 =■- [Nd (H..OU -- ЛЧ*+.
? к. ч. =i 9а (Л;р*-); а к. ч. = ЬЧ (/).
Непрост но
К<1(ИЮ3) УН.О
а
11,7:*
с
г,,7Г>
о/а
of fj7o:t
Примечание
ЯН VII 29,14S
Редкий случай структуры эниеа-гидрата с ионом (Nd (ILO),,]. Этот
ион имеет форму тригональиой призмы, против центров Л граней которой
расположено по 1 молекуле Н20 (к. ч.=9).
Такая структура обеспечивает максимальные углы между связями
(см. § 91).
Группы Вг03, в соответствии с нашими представлениями (см. § 91),
не плоски, но тетраодричны. В 4-м направлении атом О отсутствует.
Каждый узел $ сверху и снизу призматически окружён 0 группами
1Вг03|, против центра узла [1 расположены ещё 3 группы |ВК)3].
802 Ч.П. СТРУКТУРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ [гЛ. X
18. [NH41[C102]
Решётка тетрагональная Т.
Координация DijP\.
Тип TDi,Ph -SBF54 (NH4C102).
Пространственная группа C\v — P4.bm.
Z „2. a=lNH4l; 3 = |C1CX].
a к. ч.-4^(3,68 A) (Z)*); p - 4a (3,68 A) (P-i).
Край четвёртого (левого заднего) атома G1 (за атомом О) на чертеже1 не
показан. См. рис. 683.
Рис. 683.
| Вещем но
| М114СЮа
■~ \
6,г,3 |
С
*'\
1 ф 1
0,594 |
Примечание 1
SB II 64
Решётка N[I4C102 имеет сходство с решёткой KHF2 • [NIIJ'-ионы
образуют простую тетрагональную решётку. Комплексные анионы
центрируют эту элементарную ячейку. В отличие от иона HF2, ион
С1СЬ—нелинейный, что снижает симметрию. На рис. 682 показана структура решётки
но SB 1I64. Из этого не очень понятного чертежа трудно усмотреть границы
ячейки cZ=2. Мы представляем вниманию читателя рис. 683, где справа
показана проекция элементарной ячейки с Z ==• 2.и подчёркнуто взаимно-
цернеидикулярное положение групп СЮа.
§ 279] HIiKOTOl-ЪШ ЗЛМЬЧАНИЯ И ВЫВОДЫ К ГЛАВАМ IV-X 803
§ 279. Некоторые общие замечания к главам IV —X
Наша попытка перестройки на базе структурно-координационного
принципа и закона Менделеева материала о структурах неорганических веществ
является лишь первым шагом к созданию физико-химической теории
структур, в противовес нашедшей яркое выражение в SB формальной
систематике. Хотя ещё многое надлежит сделать, для того чтобы эта задача нашла
достаточно удовлетворительное решение, нам кажется, что уже сейчас
полезность рассмотрения структурного материала под данным углом зрения
вряд ли может вызвать сомнения.
Здесь мы ограничимся немногими дополнительными замечаниями (см.
также § 280).
1. Несомненно, в истекшем тридцатилетии выбор объектов структурных
исследований отличался хаотичностью. С точки зрения неорганической
химии приходится, например, считать большим пробелом то, что, тогда
как отдельным группам соединений, например галогенидам щелочных
металлов, посвящены сотни работ, другим, не менее важным объектам, как,
например, боридам или карбидам,—единичные исследования.
Можно указать на целые группы периодической системы элементов,
относящиеся к которым соединения в очень большой мере остались вне поля
зрения структурных работ. Для примера назовём хотя бы элементы III*,
JV*, V* и VI* групп периодической системы.
Совсем недостаточно изучены соединения Аи, отчасти Ag, S, Se, Те и т. д.
2. С точки зрения теории химической связи очень важно определить
возможные конфигурации координационных сфер. На многочисленных
примерах мы видели, что возникающие в кристаллах конфигурации
координационных сфер весьма разнообразны. Они во многом выходят за рамки
предпосылок как теории Вернера (1906), так и теории Кимбола (1940) и др.
(см. также стр. 806 п. 12).
Не приходится сомневаться, что дальнейшая обработка структурного
материала позволит значительно расширить наши сведения о новых
видах конфигураций, выявить новые особенности структур. К сожалению,
однако, большое количество важных в теоретическом отношении веществ
(особенно соединений немаксимальной валентности) вообще со структурной
точки зрения не исследованы. С другой стороны, характер приведённых
в SB и в оригинальных работах структурных данных не всегда позволяет
использовать их непосредственно в интересующем нас разрезе (см. § 108).
Например, в разделе комплексных соединений типа Ат[ВлОл] часто нет
данных о расстояниях А —В и конфигурациях координационных сфер АВЛ
соответственно ВАП, но приводятся лишь данные о расстояниях А —О и
В —О и конфигурациях координационных сфер АОт и ВОт.
Это требует специального рассмотрения позиций структурных узлов
А и В и соответствующих расчётов.
3. Принятая нами структурно-координационная систематика структур
показывает со всей чёткостью, что удобная для рентгенографа систематика,
принятая в SB, скорее дезориентирует читателя-химика. Достаточно взять
любую группу типов по SB, например G-соединения.
Как известно (см. стр. 252), по SB—это соединения с радикалом ВХ3.
Действительно ли это так? Рассмотрим для примера первые же типы
этой группы. Обе формы CaC03 (SBGOi и G02) —комплексные соединения,
имеющие треугольный (i) комплексный радикал СОз = [3 с деформированными
октаэдрическимм координационными сферами Са(Зв и [ЗСав.
Nd(H20)9 (Br03)3 (SB /Х)3) — комплексное соединение с изолированными
координационными сферами, имеющими конфигурации Р33+
соответственно Т~.
804 ч.п. структуры hi:oj глнпчнских ныцкстн [гл. \
FeTi03 (SJ.J G()4), ильменит, — сложное соединение, никаких радикалов
ИХ3 в своей структуре не содержащее и являющееся структурным аналогом
корунда ALOj,.
Наконец, перовскит (!аТК)3 (SH (К)6)~--сложиокомилекепое соединение,
не содержащее никаких радикалов НХ3, но октаздры ТК)в, соединенные
вершинами' в бесконечный ион.
В нашей систематике CaC03, FcTi()3 и CaTi()3 попадают в различные
разделы вместе с соединениями, близкими им по структуре.
4. .13 выборе и интерпретации материала мы старались иллюстрировать
и тот принцип, что простои химический состан вовсе не означает образование
nportnou структуры или структуры с простыми структурными узлами
(например,(л1Яч<оксллин скорее отвечает* ni./lnS3; РС1й отвечает (РС14] |РС1<5|).
Простой химический состав не означает также возникновение бездефектной
структуры. Например, соединение Fe^O,, (вюстит) имеет при соотношениях
х : у как близких к 1, так и равных единице, дефектные структуры.
И равной мере сложный химический состав не только не обязательно
влечёт за собой образование сложной структуры, но, вопреки закону Грота,
часто облегчает образование простой структуры (например, К2 [Pt(Ue|,
Na [Sb(011)e] и Др.), если сложному химическому составу отвечает простой
структурный состав, в данных примерах a^-i соответствует 3$.
Решающую роль в атом вопросе играют химические тш,
обусловливающие образование или необразованно изолированных сложных узлов
(координационных сфер). Учёт их влияния требует использования как
структурных, так и термодинамических критериев (см. § Wo), определяющих
в больше]'! мерс состав и конфигурацию структурных узлов.
И самом деле, при взаимодействии разнородных атомов могут возникать
изолированные комплексные координационные сферы как самостоятельные
структурные узлы (например, CO., в('а(Х)д) или не возникать (как в FeTiOs
или СаТЮ3). «)тл узлы могут иметь разный состав. Формально рассуждая,
можно выделить ряды комплексных соединений, например (в результате
реакции х NH4F -f у SiF4)
NH4SiFs. (NHt)3|SiFe|, (NH4)3SiF7 и т. п.
Однако образование тех или иных ионов, в данном примере иона SiFT,
не имеет места, возникает ион SiFfl, тогда как в аналогичном ряду с ZrF4
как раз образуется ион [ZrF7]3".
11а состав и конфигурацию структурных узлов значительное влияние
оказывает поле кристаллической решётки. 11 а приме]), в возникновении
структур Na, [A1F.I, (NIT4)3 [Л1Р61, Na [Sb (OII)J решающую роль
сыграла термодинамическая возможность образования изолированного
структурного узла |AlFe]3", [Sh (OH)e|"", что привело к структуре типа
комплексного соединения. Поле решётки вносит само по себе существенные
различия, вытекающие из характера иона-партнера, например,
большой [Nfl4]!-ион образовывает с [А1Рв]3"-иоиом высокосимметричную
структуру, близкую к HiF3 (Cc\oio,t)\ напротив, Na'-ион деформирует октаэдры
[AlFe|; последние образуют центрированную ячейку, в которой Na (1)+-ион
1 11
занимает позиции 00-- и -,, — 0 с к. ч.~(>; два дополнительных иона Na !
распределены в общих положениях с к. ч., также близким к в.
Наконец, в Na [Sh (OII)e], Na-* -ион образовывает с lKb(()Jl)e]~ решетку
типа NaCI, посмотрят болыпую разницу в величине обоих ионов, что лишний
раз иллюстрирует сказанное нами ранее (§ 77) о явном несоблюдении правила
ГольдшмЦдта в значительном количестве случаев.
Г). Границы изолированной группы (координационной сферы) иногда
бывают очерчены весьма чётко, иногда они более размыты и расплывчаты.
§ 27(JJ нккотогьш злмьчлппн и шлгюди к гллнлм iv-x 805
Например, л случае (±NII.,)2 [SiFJ конфигурация чётко отвечает структуре
f-Y/c (аптл GaFo тли Саг). -В случае Iv,S04 —граница координационной
сферы, например узла fi—SO.A, определяется менее чётко.
Наконец, в некоторых случаях координационная сфера может быть
установлена в известной мере произвольно (см. хотя бы тот же Na.r\lF0 (сфера
Na (11)+-понов)), хотя в ряде случаев приведённые нами соотношении,
возможно, придётся пересмотреть на базе новых работ или тех или иных
физических соображении.
Характер координационной сферы, но.юмсенныи нами в основу система-
тики у во многом определяет физические особенности вещества и имеет
практическое значение при подборе веществ с заданными свойствами.
6. При образовании простых веществ, как мы видели, выбор
элементарной ячейки может быть охарактеризован немногими общими положениями.
Позпиклонение той пли иной структуры элементарной ячейки и случае
сложных веществ и химических соединении тоже подчиняется, немногим
общим закономерностям, которые мы воспроизводим здесь лишь частично.
1\ этому вопросу мы вернёмся в другом месте.
7. Каждое вещество в определённом интервале температур, давлений
п концентрации примесей может образовывать различные модификации.
Частые утверждения, что наиболее плотным структурам должен
отвечать минимум свободной энергия—в корне ошибочны. Из рисунка 'Л
(стр. 2()) следует, что выше точки перехода термодинамически устойчива
х-форма, ниже—S-форма. Хотя одна из ниv имеет меньшую плотность,
чем другая, она является термодинамически более устойчивой при данных
Р и Т. Для пизкосимметричпых форм повышение температуры обычно
создаёт возможность достижения более высокой симметрии. Повышение
давления приближает систему к упаковкам с большим координационным числом,
а при том же координационном числе к более плотной модификации
(графит—алмаз). Осуществляется ли практически переход- зависит от
термодинамических и кинетических факторов.
Состав вещества, а также концентрации примесей, несомненно, сильно
влияют на возникновение модификаций. К сожалению, мы имеем крайне
мало достоверных данных в этом отношении, поскольку прекрасные рентге-
пографы часто оказывались плохими химиками, или, вообще, не освещали
этот вопрос столь подробно, как он того заслуживает.
Безусловно, что присутствие примесей влечёт за собой как изменение
границ термодинамической устойчивости тех или иных форм, так и
различную скорость образования термодинамически устойчивых форм, а также
изменяет условия существования реальных твёрдых фаз. Структурные
исследования должны обязательно выявлять зависимость структур от
температуры, давления и состава.
8. При упаковке молекул типа [ЛХЛ] а выбор типа структуры должен
определяться характером взаимодействия между узлами д. Ксли силы
взаимодействия очень невелики (например 1Г,,1\.,,Лг) и возможно вращение
узла, следует считать вероятным, что центры узлов образуют плотную
упаковку С & (или Ну).
Остановка вращения означает необходимость симметричного
расположении не только узла в целом, но и его периферических групп, а значит,
соответствующей ориентировки узла. Коли этого не удаётся достигнуть,
симметрия будет снижена (О*, CL и др.)- При более интенсивном
внешнем силовом поле характер упаковки часто зависит от формы
структурных узлов, например тетрагональные молекулы такого типа часто
образуют структуры Сс. С другой стороны, кристаллы, образованные тетра-
эдрическими молекулами типа II20, NJJa и др., с. парами несвлзыиающих
электронов часто образуют тетраэдрические узлы д; однако теперь тетра-
806 Ч.Н. СТРУКТУРЫ 1Ш01 ГАШ1ЧЕСКПХ ВЕЩЬСТВ [ГЛ. X
эдры соединяются общими вершинами в цени сетки или даже образуют
пространственную вязь далеко не с самой плотной упаковкой, поскольку
тетраэдрам приходится иметь общие вершины, и т. п.
В случае, если узел имеет состав АХв или АХ4, можно в известной
мере формально представить себе такой путь построения структуры
симметрии: периферические атомы X располагаются в узлах некоторой плотной
упаковки, а центральные атомы А центрируют октаэдрические (в случае АХв)
или тетраэдрические (в случае АХ4) дыры. При этом центрирована 1/в часть
всех октаэдрических дыр (их общее число равно п=--6), соответственно */8
часть тетраэдрических (их общее число равно 2^ = 8).
9. Структура химических соединений оф, ajia, aj33 определяется
характером химических связей между узлами и свойствами самих узлов. Вопреки
многим ходячим представлениям в случае соединений с комплексными
ионами обычно возникают координационные решётки, имеющие простые
структуры типа GsCl, NaCl, CaF2 или BiF3 и производные от них, а также
2—"3 из основных простых типов гексагональных структур.
Напротив, в случае простых соединений (и продуктов их реакций между
собой, когда образование изолированных координационных сфер по
термодинамическим причинам невозможно) наличие значительно более сильного
взаимодействия узлов с наложением достаточного веса ковалентных связей,
влечёт за собой уход от координационных структур рядаСзСЛ — BiF3 и
возникновение сложных и сложнокомплексных решёток типа Si02, BP04 и т. д.,
рассмотрение которых позволяет хорошо проследить влияние характера
химической связи на отклонения от принципа плотных упаковок.
10. В соответствии с законом пределов Фёдорова громадное
большинство структур является производными от небольшого количества основных
структур. Изменения касаются либо метрики, либо ориентировки частиц,
либо чередования мотивов 2 и более структур, либо, наконец,
упорядоченных или неупорядоченных замещений позиций узлов в решётке.
11. Структуры реальных твёрдых тел (реальных кристаллов) включают
множество нарушений порядка, задаваемого трансляционными группами,
который, таким образом, даёт лишь идеализированную картину
структуры твёрдого тела. Поэтому важной задачей теории структур остаётся
исследование реальных кристаллов.
12. В дополнение к сопоставленным нами в табл. 34 формам
координационных сфер во II части книги рассмотрено много других форм.
Назовём некоторые: (к. ч. 12): тетрагонально искажённый кубооктаэдр ТК,
например индий; кристаллохимический икосаэдр /, встречающийся вместо
правильного икосаэдра с осями 5 (см. табл. 34), например V3Si и др.;
(к. ч. 11): P3 + D3, например NaaS08; (к. ч. 10): h + r в разных
комбинациях, например, TiSi2, Zn (NH3)2C12 и т. п.; (к. ч. 8): гексагональная
бипирамида DQ в Zi8N; квадрат + тетрагональный тетраэдр (q f S),
например тип шеелита CaW04; ромбоэдр, сжатый R (например lMg(H.jO)e][SiFe]);
(к. ч. 7): тригональная антипризма с одним дополнительным атомом
±
(i43+)Za203; (к. ч. 6): тетрагональный тетраэдр полюс 2 атома (S+) анатаз
или р = олово; бипирамида с прямоугольником в основании (D2)—нитрит
натрия или серебра и много других конфигураций.
Обобщённая и дополненная таблица конфигураций координационных
сфер подготавливается нами к печати в последующих работах.
ЧАСТЬ ТРЕТЬЯ
РЕГИСТР ИССЛЕДОВАНИЙ СТРУКТУР
НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
280. Введение
О о щ и е з а м и ч а н и и
Ла несколько десятилетии существовании рентгенографического и злек-
тронографического анализа структур веществ в литературе описаны
результаты многих тысяч подобных работ.
В картотеке, собранной автором книги, более чем на 13 400 карточках
зарегистрировано более 25 000 таких исследований различных
неорганических веществ, причём автор далёк от мысли, что сведения о всех
публикациях картотекой охвачены. Однако уже и собранная в настоящее время
картотека является значительно более полной но количеству наименований
исследованных неорганических веществ и количеству литературных ссылок,
чем, например, разрозненные по томам указатели наиболее
фундаментального справочника SB (томы I —VII). Она включает более 5100
наименований неорганических веществ, большинство из которых изучалось
неоднократно и освещено в ряде работ, на которые имеются ссылки в регистре.
Одно ато обстоятельство вполне оправдало бы опубликование картотеки,
с тем чтобы сделать её доступной широким кругам исследователей, темболе-е,
что в самом SB не проделана важная работа- составление общего
предметного указателя но I VJ1 томам, так что читатель лишён возможности сразу
найти данные по интересующим его объектам.
Работа по пополнению картотеки новыми сведениями автором
продолжается. Предположено в дальнейшем выпустить дополнительный указатель
работ, посвященных исследованию структур неорганических веществ, что
явится следующим шагом но упорядочению и систематике фактического
материала, накопленного в этой важной области точного естествознания.
Однако не только ату цель ставил перед собой автор при составлении
картотеки. О принципиальной стороне дела, заключающейся в подготовке
материала, совершенно необходимого для перестройки неорганической
химии на базе современного учения о строении вещества, автор говорил
по введении. Кроме того, имелась в виду необходимость приобщить к
овладению богатым структурным материалом возможно более широкий круг
читателей и, наконец, по возможности, устранить ошибки, обнаруженные
и регистре и тексте SB.
При подготовке материалов автор стремился сделать сводку возможно
более компактной, чтобы облегчить пользование ею и иметь возможность
включить сводку в книгу.
С этой целью различные структурные формы простых веществ и
химических соединений одного и того же состава объединены^ и материал
по ним приводится вслед за химической формулой вещества.
Исключения составляют отдельные сложные соединения, где одному и тому же
составу могут отвечать совершенно различные структуры. В равной мере,
и регистре объединены сведения, относящиеся к структуре веществ в разных-
агрегатных состояниях. Каждое соединение (за немногими исключениями)
810 ч. ш. 1»г:гист1* исследований структур неорганических веществ
приводится в сводке только один раз но наименованию элемента металла,
стоящего первым по алфавиту. В тех случаях, когда работа прореферирована
в SB, мы указываем том SB и страницу, а не отдельные журналы, поскольку
па одной странице SB часто прореферировано по нескольку статей (из разных
журналов), относящихся к данному объекту. Заказав в крупных
библиотеках фотокопии соответствующих страниц SB, читатель получит рефераты
но всем соответствующим работам, с указанием фамилий авторов, журвала.
тома, страницы, года, когда работа опубликована, и сведения о
содержании и результатах работы (комплекты томов SB имеются в Москве,
например, в научной библиотеке Института им. Карпова, в Институте
кристаллографии АН СССР, в Государственной научной библиотеке Министерства
высшего образования и др.).
В связи с этим мы должны обратить внимание на то, что тщательный
просмотр и проверка формульного и предметного регистров семи томов
SB привели автора к неожиданному обнаружению многих десятков грубых
ошибок и пропусков, свидетельствующих о недостаточном внимании к
химическому составу объектов, структура которых описывалась, и о небрежном
составлении регистра SB.
Рассмотрим для примера хотя бы том SB III. В тексте стр. 455 и
в указателе (стр. 895) даны 7 неправильных формул квасцов, например,
КСг (S04)3 12Н20. Соединение мочевины и AgNO, в указателе SB III, стр.
892 занесено в регистр как AgN03Co(NH 2)2.
Сульванит Cu3VS4 фигурирует в регистре как CuVS4 (стр. 894); LiD
(соединение Li с тяжёлым водородом) как LiO (стр. 895). Соединение Li2Fc204
как Li,Fe04 (соль железной кислоты (I), стр. 895), (NH3)4 PtCl2 (стр. 897)
указателя —в действительности отвечает в тексте (стр. 702) двум
соединениям: (NH3),PtCl4 и (Nfl3)4PlOUI20 и является «редакционным» гибридом
Pt11 и Ptiv. Na r\V03 (где х < 1) фигурирует в указателе как Na4\V03 (стр. 896).
На стр. 485 и в указателе стр. 897 напрасно воспроизведены путаные,
химически неправильные формулы, вроде Rb,Cd[Ni, Cd(N02)6]. Лучше было бы,
например, xRbXd [Ni(N02)6]yRbaCd[C(i(N08)f].
В списки органических соединений зачислены комплексные амины Cd,
Pt и Pd, в том числе и упомянутые выше соединения Pt (стр. 701 703), не
имеющие ничего общего с органическими аминами и не содержащие даже
атомов углерода в своём составе. Эти вещества не становятся органическими,
даже если, как это делает SB, именовать их аминами (стр. 702).
Даже после приведённых выше примеров нельзя не удивиться
включению в группу органических алифатических веществ таких соединений, как
B4Hfi (SB IV 280).
Пропущены в указателе SB III такие вещества, как дициан (C3N,),
прореферированный в тексте на стр. 731. Множество минералов, по которым
в тексте SB имеются рефераты, в формульном регистре вообще пропущено,
как если бы составители SB не знали их формул. Это было бы понятно в
отношении сложных минералов неопределённого состава. Однако трудно понять,
почему пропущена в этом указателе SB V, в разделе SiO2
высокотемпературная модификация кристобалита (Hochkristobalit) (реферировано па стр. 110)
и в разделе Ми группа соединений Мп, в том числе даже пиролюзит
(реферированный на стр. 101). В формульном указателе пропущены также многие
комплексные соединения с органическими аддендами, например, оксалат
кальция (стр. 727), кислые оксалаты калия и рубидия (стр. 728), сукциыат
бария (стр. 729) и т. д. и т. п.
Исследователь, удостоверившийся в отсутствии интересующих его
соединений в формульном регистре SB и даже в предметном указателе,
ошибется, если не займётся постраничной проверкой текста в соответствующих
разделах.
S 280]
ШШДЕН1Ш
.811
Напротив, указатель SB содержит многократные повторения одних
и тех же формул, во-первых, если вещество состоит из нескольких
элементов (например, вещество, состоящее из 6 элементов, иногда
повторяется 4—5 раз, что очень сильно увеличивает объём указателя), во-вторых,
без всякой видимой причины. Например SB 1, стр. 810, третий столбец:
Cu20, затем ЗСиОА1203, затем снова Cu20 и опять 3GuOAI203 и т. д.
В SB II, например, система А1 — Си — Mg — Fe— Si приводится в регистре
на стр. 951, 954, 956 и 959, в том числе на стр. 954 дважды.
Наконец, немалую путаницу мы находим в SB в столь важном вопросе,
как причисление вещества к тому или иному типу, и в самой номенклатуре
типов. Как отмечалось в § 107, в разных томах SB принята разная система
классификации. Одно и то же вещество попадает, таким образом, в разных
томах SB в разные группы, переходит, например, из группы Н в группу J
и т. п. и в пределах одной и той же группы претерпевает изменения в пуме-
рации. Это заставило нас составить отсутствующую в SB сводную таблицу
типов (§ 107), позволяющую, особенно лицам, мало знакомым с SB, быстрее
разобраться в этом деле.
Но при всем этом указатель SB ещё и от себя путает типы.
Например, (NH4)2SnCle сначала относился к типу Я61 (SB I, 431), затем
к типу Jix (SB III, 121), в указателе же SB III, 89G находим /1в и т. д.
Не только одно и то же вещество с одной и той же структурой
причисляется к разным типам, но и разные по структуре вещества
зачисляются в один и тот же тип (см. § 107).
Мы остановились на упомянутых недостатках SB с тем, чтобы
предупредить читателя о необходимости учитывать это при пользовании SB.
С другой стороны, в самом тексте SB имеет место ряд существенных
недостатков и ошибок физико-химического характера, происходящих из
некритического воспроизведения этой стороны работ отдельных авторов.На
подобных ошибках мы здесь останавливаться не будем.
После внесения ряда исправлений и пересмотра регистров семи томов SB
автором были составлены около 3000 карточек (охватывающих каждая
от 1 до 20сносок и в результате—до 20 000 определений). Кроме того,
было составлено до 1500 карточек по новым работам, не вошедшим в SB.
Эти 4500 карточек явились первой ступенью к составлению картотеки.
О порядке составления регистра
Все 4500 карточек были написаны автором от руки, как во избежание
описок при перепечатке, так и с целью выявления ошибок в регистре SB,
поскольку при этом автором переписывалась и просматривалась каждая
химическая формула. В результате все карточки были сведены на 400 листов
и затем в регистр, занимающий минимальный объём. Для достижения
компактности, в частности, был введён шифр (см. ниже), позволяющий свести
название журнала, том, страницу и год к немногим печатным знакам, а весь
регистр—к нескольким десяткам страниц. По окончании этой работы (к
середине 1947 г.) до середины 1949 г. в перепечатанный регистр были внесены
автором ещё свыше 2500 указаний на различные работы, в том числе и
опубликованные в 1947—1948 гг. Наконец, в 1949 г. в значительной мере
в корректуре в регистр было внесено несколько тысяч ссылок на сводные
труды различных исследователей по рентгенометрии неорганических
веществ (Болдырев, Ханауолт, Ринн, Фревел, Седлецкий и др.), а также
и данную книгу. В этой последней части работы нами была использована
также картотека, составленная в Лаборатории комплексных и твёрдых
соединений Института им. Л. Я. Карпова, по указаниям автора. Все
карточки были лично проверены автором (сопоставлены с оригинальными
812* 4. 111. РЕГИСТР ИССЛЕДОВАНИЙ СТРУКТУР НЕ0Р1;Л1ШЧЬ£КПХ ВЕЩЕСТ13
работами. При :.»том также был обнаружен и последних ряд ошибок*,
главным образом, я формулах веществ) и лично им «писаны я регистр1).
И окончательном итоге в регистр яключены данные свыше 13 400
карточек.
Только сознание невозможности дальше мириться с отсутствием
регистра структур, столь необходимого для развития широкой отрасли
естествознания, могло способствовать выполнению автором этой трудоёмкой
работы.
Отметим, я частности, что уже из простого просмотра регистра можно
сделать ряд интересных общих выводов, о которых мы говорили частично
во введении.
Громадное количество структурных работ касается алюмосиликатов,
а также соединении алюминия, кальция, магния, железа, входящих
в состав минералов, наиболее распространённых или имеющих
практическое значение. Хуже изучены интерметаллические системы и очень
плохо— галогениды, халкогепиды, а также другие соединения очень многих
элементов - металлов и неметаллов, даже таких, как олово, сурьма или сера.
13прочем, надо предостеречь читателя от того, чтобы судить о количество
исследований по объёму того или иного параграфа. Так как регистр составлен
в алфавитном порядке, то параграфы влемеитои, наименования которых
начинаются первыми буквами алфавита, значительно более объёмисты
(система Ag Sn попадает B§Ag, а не в§ Sn). Главная часть соединении,
образованных неметаллами, например S, попадает я параграфы,
относящиеся к соответствующим металлам, например FeS в параграф Fe, CuS
в параграф Си.
Остаётся фактом и то, что неорганическая химия и химия
комплексных соединений до сих пор значительно меньше вдохновляли рент-
генографов и ещё меньше сами использовали методы структурного и
фазового рентгеновского анализа для освещения структур простых и
комплексных соединений и даже простых веществ, чем л то было сделано
минералогией в отношении не только простых, но даже весьма сложных
минералов.
Особенно плохо, как мы отмечали в предыдущих главах книги,
освещена проблема фазовых переходов в случае простейших химических
соединений и даже простых веществ.
Очень плохо изучены конфигурации, возникающие в соединениях
максимальной и, особенно, немаксимальной валентности разных элементов.
Отличительной особенностью советских структурных школ является
целеустремлённая работа но систематическому освещению целых рядов
веществ, как, например, силикатов, комплексных соединений и др.
Надо надеяться, что в ближайшее десятилетие проводимые советскими
учёными исследования структур неорганических веществ не только более
равномерно охватят периодическую систему, но и позволят правильно
интерпретировать ряд проблем теории валентности и, в окончательном итоге,
будут гораздо более широко использованы также в неорганической химии.
II о р я д о к и о л ь з о ваий я р е г и с т р о м
1. J] основу регистра положен алфавитный принцип. При этом
элементы редких земель (за исключением Се, № 58, включённого в
самостоятельный параграф регистра) помещены в общую группу (редкие земли).
Элементы №№93 96 включены в параграф «Уран и трансураниды».
х) В с шиш со пключепием и регистр рентгенометрических данных нами были
внесены и пег.) подобные спедения и относительно более простых органических
соединений, упоминавшихся в I части книги, см. § 107.
$ 2Й6|
Ьпедеймг:
813
2, Все элементы делятся условно на 2 группы: металлы и неметаллы.
К последним условно отнесены элементы таблицы:
I i
\е
Кг
I Z\
! „
I ,ln
I |
т. е., исключая благородные газы, следующие элементы в порядке алфавита:
С, CI, F, .), N, О, 1\ S, Se и П.
о. Если вещество состоит из атомов металла (например, CiuMnAl),
искать его следует и роди соединении того элемента, который является
первым но алфавиту, т. о. Л1, и среди соединений А1 опять-таки в порядке
алфавита, т. о, между системами Л1 -Си — M# и А1 Си—Мо.
4, Если вещество состоит ил атомов металла и неметалла, искать его
следует среди соединений металла, т. е., например, Hi./).,— среди соединений
Hi между Hi-- Ni и Hi — Os.
5. Если соединение состоит из атомов нескольких металлов и
нескольких неметаллов, его следует искать среди соединений того из металлов,
который является первым но алфавиту, а между ними—в порядке
алфавита всех входящих в его состав металлов. Так, соединение SbHi03 следует
искать в списке соединений Hi между Hi — Sb и BiSbS3. Однако Ш203
следует искать (и. 4) между Hi —Ni и Hi()3. Значит, получаем ряд: Hi —Ni,
Hi-S, Hi- 0, Hi Sb, Bi-Sb-O, Hi—Sb-P, Hi-Sb-S, Bi-Sn,
Hi-Sn-O, Hi-Sn- S и т. д.
(i. Если данные относятся к химической системе, записанной иным
образом, например, вместо Hi —Sb С): Hi«08—- Sba03, вид формул мы часто
оставляем без изменения (поскольку они не являются неправильными),
искать же соответствующие данные следует там же, где и для системы
Hi —Sb О. В самом деле, в ряде случаев мы не переписывали в алфавитном
порядке формулы многих соединений, которые принято в химии или
минералогии писать иначе. Например, Na3AlFe мы не переписывали, как AINa3Fe;
однако искать подобное вещество следует среди соединений Л1, в списке
между Л1- -Na и А1- Na-О.
7. Формулы сложных комплексных соединений типа
[CofNHAUOfCNU
мы также не изменяем, однако искать данное вещество следует среди
соединений Со, в порядке: Со —Сг С—N.
8. В связи с тем, что водород в большинстве соединений фигурирует
в виде воды (и групп ОН), гидроксиды и гидроксильные соединения, в порядке
исключения, помещаются вслед за оксидами, например, А1аОа, АЮОН,
Ala()8~Jl,0 или SbaOs, Sb,<V~JLO.
i С I N О
! р
CI
s<-
_
Иг
.1
814: ч. tn. pEi-Hcf p исследований структур Неорганических веществ
В гидридах же типа СН4, LiH,Pd—H водород естественно
рассматривается как неметалл, и соответствующее соединение помещается в
согласии с п. 4, т. е., например, SbF3, SbH3, Sbl3.
9. Если один элемент замещает другой, особенно в минералах, названия
обоих ставятся в скобках и отделяются занятой. В этом случае
последовательность элементов внутри скобок определяется основным компонентом.
Например, (Zn, Fe)S означает, что основной состав: ZnS, но небольшая
часть цинка замещена на железо. (Fe, Zn)S означает, что основной состав
FeS, но часть атомов Fe замещена на цинк.
10. Сокращённые названия (шифр) журналов приводятся ниже. При
обозначении журнала сначала даётся шифром его наименование, затем
жирным шрифтом том и обычным шрифтом страница и в скобках год. Напр.,
Acta XX, 737 (1945)читается: Acta physicochimica URSS, том XX, страница
737, год 1945. Буква, проставленная в скобках между шифром журнала
и номером тома, означает серию, например P. R. S.(A) 132, 266 (1931) читается:
Proceed, of Royal Soc. London, серия А, том 132, стр. 266, год 1931.
Обозначение в скобках вида (А6:24)63 отвечает, как и в SB,
указанному типу (AQ) и странице соответствующего тома.
11. Ссылки на книги и монографические работы.
В 1947 г. вышла монография Н. В. Белова «Структуры ионных
кристаллов и металлических фаз». В этой книге широко представлен полиэдрический
метод интерпретации структур. Ссылки на эту монографию делаются
и регистре шифром: Б (1947).
В 1938 году советскими учёными А. К. Болдыревым, В. И. Михеевым,
Р. А. Ковалёвым и В. Н. Дубининой (при участии Е. Е. Флинт, А. Н. Лями-
ной и А. И. Любимцева) был опубликован первый в мировой литературе
рентгенометрический определитель минералов (Записки Ленинградского
Горного института, том XI, стр. 1, 1938 г.) на основе порошковой
рентгенограммы. Этим трудом положено начало важнейшей для неорганической
химии и минералогии отрасли рентгенографии (соответствующие сноски
обозначаем по фамилиям основных авторов БМКД (1938)). Несколько позже
была опубликована статья Ханауолта, Ринна и Фревеля (Ind. Eng. Che in.
An., Ed. 10, 457 (1938)), содержащая данные о рентгенографической
характеристике фазового состава 1000 веществ. В целом статья может быть полезна
при идентификации фаз. В 1939 г. Болдырев и его сотрудники
выпустили часть И рентгенометрического определителя минералов (Записки
Ленинградского горного института, т. XIII, вып. 1, стр. 1 (1939)). Ссылки
обозначаются БМКД (1939). В 1942 и 1947 гг. были опубликованы статьи
Фревеля (Ind. Eng. Ch. Anal., Ed. 14, 687 (1942) и Ind. Eng. Ch. Anal.,
Ed. 18, 83 (1946)), содержащие аналогичные данные по расшифровке
нескольких сот структур веществ кубической и тетрагональной сингоний.
Сноски обозначаем: HRF (1938), F (1942) и F(1946).
В.1939 г. опубликована монография И. Д. Седлецкого «Почвенная
рентгенография», посвященная рентгенографической характеристике минералов,
встречающихся в почвах (Изд. Акад наук СССР). В регистре нами
приводятся ссылки на группы минералов, характеризуемые в ней шифром: С (1939).
Вещества, обсуждаемые в данной монографии, обозначаются в регистре
ссылкой на текст данной книги: О (1950). Чтобы найти номер страницы,
читатель должен обратиться к предметному указателю, имеющемуся в конце
книги. В сомнительных случаях наименование и состав минералов
проверялись нами по книге Э. С. Дана «Описательная минералогия»,
переведённой на русский язык и дополненной большой группой советских учёных
под общей редакцией акад. А. Е. Ферсмана и О. М. Шубниковой (ОНТИ,
1937); при этом был устранён и исправлен ряд недостатков книги и дан
список наименований минералов.
§ 280]
вйеДейик
815
Таблица 1
Сокращённые обозначения журналов, принятые в регистре*)
ОСозначе-
1 ние
1 Acta
А. Сг.
A. Min.
Ann.
Б(1947)
БМКД
(1938) и
(1939)
Бег.
Bull.
С. А.
С. R.
Ch.
Ch.E.N.
Ch. Ph.
Ch. R.
Ch. S.
Ch. Z.
ДАН
F (1942)
или (1.946)
FS.
J. Am.
Oaz
Ж. П. X. 1
Ж. Т. Ф.
Ж. Ф. X. 1
Ж. Э. Ф.
J.I.M.
J. ph. J
H. Ch.
H. Ph.
HRF(1938)
3. Л.
I. J. ph.
I. E.
I. S.
ИАН(х)
ИОНХ
KZ
M j
МГУФиз.
N
NW
0 (1950)
1
Полное паименонлппо журнала
Acta physicochimica URSS
Acta crystallographica
American Mineralogist
Annalen dor Physik
1 Монография Н. В. Белова
Рентгенометрический определитель А. К.
Болдырева, В. И. Михеева, Г. А. Ковалёва it В. И.
Дубининой, ч. I (1938 г.) и ч. 11 (1939 г.)
Berichte'der Deutschen Chemischen (lesellschafl
Bulletin de Societe Chimique de France
Chemical Abstracts
ComptesRendus del'Academie dos Sciences
Chemie
Chemical Engineering News
Journal of Chemical Physics
Chemical Review
Journal of Chemical Society London
Chemisches Zentralblatt
Доклады Академии паук СССР
Статьи Фревели
Transactions of Faraday Society London
Journal of American Chemical Society
(iazetta Chimica Italiana
Журнал прикладной химии
Журнал технической физики
Журнал физической химии
Журнал экспериментальной и теоротич. физики
Journal Inst. Metals
Jornal of Physics
Helvetica Chimica Acta 1
Helvetica Phys. Acta J
Статья Ханауолта, Ринна, Фревели
Заводская лаборатория
Indian Journ. of Physics
Industrial Engineering Chemistry, Anal. Ed.
Iron and Steel
Известия Академии наук СССР, отд. хим. наук
Изв. Сектора ф.-х. анализа ИОПХ А\\ СССР
Kolloid Zeitschrift
Metalloforschung
Учёные записки Моск. госуд. унин., часть физ.
Nature
Naturwissenschaften
Данная книга (Б. Ф. Ормонт)
Страна
' СССР
Англия
США
Германия
СССР
СССР
Германия
Франция
США
Франция 1
Германия
США
США
США
Англия
Германия 1
СССР
США
Англия
США
Италия
СССР
СССР
СССР
СССР 1
Англия
СССР
Швейцария 1
США
СССР
Индия
США
США
СССР
СССР
Германия
Германия 1
СССР
Англия 1
Германия 1
СССР
*) Ванду того, что обработка части картотеки была накопчена лишь к коррек
гуре книги, н гл. XII помещён дополнительный регистр.
816 4. 111. PL<:rilCf P ИССЛЕДОВАНИЙ СТРУКТУР 1*ЕОРГАНПЧЬХКНХ ВЕЩ1ХТВ
Т а б л и ц а 1 (продолжение)
Сокращённые обозначения журнилок, принятые и регистре
Обозначение
P. R. S.
P. Ph.
Phvs.
Ph.Rad.
Ph. Ch.
Vh. M.
Ph. R.
Ph. Z.
IMA
Roc.
R. M.
С(Ш9)
SB I I
SB Hole|
Труды UK
Z. An.
Z. Kl.
Z. Kr.
Z. Met.
Z. Ph.
Z. ph. Ch.
Усн.
Усп.Ф.Н.
Полное папмепонание журнала
Proceeding of Royal Society London '
Proc. Phys. Soe. |
Physica I
Journ. do Phys. ot Radium
Journal of physical Chemistry
Philosophical Magazine
Physical Review
Physikalischo Zeitschrift
Proceeding of Indian Acad.
Recueil des Travaux Chimiques do Pays-В as
Review of Modern Physics
Монография И. Д. Седлецкого
Strukturbcrieht, т. 1, 11 и т. п.
Труды Института кристаллографии
Zeitschrift f. anorganische u. allgem. Chomie
Zeitschrift f. Eloktrochomio
Zeitschrift f. Kristallographic
Zeitschrift f. Metallkundo
Zeitschrift f. Physik
Zeitschrift f. physikalische Chomie
1 Успехи химии
Успехи физич. наук
(/грана 1
Англия
Англия
Голландия 1
Франц if я
США
Англия 1
США
Германия
Лидия
Голландия
США
СССР
Германия
СССР
Германия
Германия
Германия
Германия
Германия
Германия
СССР
СССР
ГЛАВ Л XI
РЕГИСТР ИССЛЕДОВАНИЙ СТРУКТУР НЕОРГАНИЧЕСКИХ
ВЕЩЕСТВ
§ 281. Актиний Ас
1.АсВг3 А.Сг. 1, 265 (1948); 0(1950).
АсС13 А.Сг. 1, 265 (1948): 0(1950).
Ас(0Н)3 А.Сг. 1, 265 (1918); О (1950).
AcsS3 А.Сг. 1, 265 (1948); 0(1950).
Актшшды, см. торий, уран, ураниды и § 84.
§ 282. Серебро Ag
Ag SB I (41: 14) 36, 748; SB II 163, 165, 166, 167, 607-615, 623,
624, 626-629, 663, 677, 678, 693, 698, 701, 704, 705, 717, 718, 741,
744-746, 797, 798; SB III 181, 184, 186, 187, 189-192; SB IV
77, 87, 223; SB V 32; SB VI 41-43; SB VII 45, 49-51, 199;
HBF (1938); БМКД (1939); F (l942);Ch. Z. I, 1162 (1944); О (1950).
Ag-AI SB I 502, 557; SB III 605; SB IV 233; Ph. Bad. |8], 3
124 (1942); Ch. Z. I, 13 (1942); Ch. Z. I, 709 (1943); P. B. S. (A)
193, 1 (1948).
Ag3Al SB I 557; SB II 677, 678; SB III 326, 605; SB IV 124, 234.
Ag4Na2CaAleSieO,8S, SB II 563-565.
lO.Ag.Al.Si.O^S, SB II 563.
Ag2Al2Si30102H.,0 SB III (S6ia : 168) 530.
AgJVa2SrAleSi0O28Sa SB II 563.
AgeAs SB II 741.
Ag3AsO< SB I (//2X :362) 395; HRF (1938); F(1942); 0(1950).
Ag3AsS3, прустит SB V 75; Б (1947).
AgJAs(C2H6), SB IV 285; F (1946).
Ag-Au SB I 502, 510, 765; SB III 574.
Ag.Au SB I 508.
Ag,Au2 SB I 511.
20. AgAu SB I 508.
Ag2Au„ SB I 511.
Ag3Au7 SB I 511.
AgAu3 SB I 508.
Cs„Ag [AuClJ; Cs[AgCl2]Cs[AuCl4] SB III 513; SB V 103; SB
VI (A7,: 34) 126; F (1946); О (1950).
AgAuTe, SB V 99; Am. Min. (1949) 26, 457 (1941); Ch. Z. I, 140(1943).
Ag-B О (1949).
Ag-Be SB IV 240; SB VI 162.
28.AgBe2 SB IV (C15:8)240; F (1942); ДАН 40, 262 (1943).
5£ Б. Ф. Ормонт
?18 29z_AgC82[Bi(N02)e]-75. Ag-Ga-Zn .1^X1
29.AgCs..[Bi(N02),] SB IV 193; F (1942).
ЬО. AgK2 [Bi (N02),] SB IV 193; F (1942).
AgRb2[Bi(N02),] SB IV 193; F (1942).
AgTl2[Bi(N02),] SB IV 193; F (1942).
(NH4)2Ag[Bi(N02),] SB IV 193; F (1942).
AgBiS2 SB IV 148.
4gBr SB I (JS1: 73) 37, 111; SB III 230; SB IV 95; F (1942); О (1950).
AgBr-AgCl F (1942).
AgBr-AgJ F (1942).
A.gBrO, SB I 312; HRF (1938); F (1946).
Ag(CH3CSNH2)4Cl F (1946).
40. AgCN SB 1282; SB HI 236; HRF (1938); Ch. Ph. 10,199(1942); О (1950).
Ag2COs HRF (1938V
Ag(CH,COO)2 HRF (1938).
Ag,C„04 Ch. Ph. 11, 499 (1943).
As, тартрат HRF (1938).
Ag, цитрат HRF (1938).
Ag-Ca SB III 604.
Ag.Ca SB III 604: F (1946).
AgCa SB III 604.
(Ag, Mg)2 Ca M. 1, 31 (1946).
50.Ag-Cd SB VI 160; SB VII 196, 199; Ch. Z. I, 985 (1940).
AgCd SB I (L20:488) 501, 553, 555; SB IV 223; F (1942).
Ag2Cd3 SB I 555.
Ag.Cd, SB I (D8: 499) 554; SB VII 198.
Ag-Ce С. А. 38, 3597 (1944).
AgCl SB I (51: 73) 111; SB И 211, 213, 214; SB III 230; HRF (1938);
F (1942); ИАН (x) 382 (1944); С A. 39, 3988 (1945); О (1950).
AgC102 SB II 378; F (1946).
AgCIO, SB I 311, 339; Z. Кг. A. 104, 28 (1942); F (1946); О (1950);
AgC104 SB II 84, 411, 412; F (1942); О (1950);
AgC104H20 HRF (1938).
/ /CHa \
60. AgCl S == C< SB IV (02,: 255) 256, 257.
\ xnhJ4
AgCo (NH,)2 (N0„)4 SB IV (Л,:60) 191; F (1946); О (1950).
Ag2Cr04 HRF (1938).
Ag-Cu SB I 504; SB II 607-615, 693; SB III 568.
Ag- Cu-Mn-Zn Z. M. 35, 27 (1943); С. А. 38, 49 (1944).
Ag-Cu-Ni Ch. Z. 1, 1104 (1942).
4AgJ lCuJ SB I, 112; БМКД (1938).
AgaF SB I 784; SB II 276, 277; О (1950).
AgF SB I (Bl : 73) 111; F (1942); О (1950).
AgFe02 SB VI 87.
70.AgFeS2 «аргентопирит» Rec. 63, 69 (1944); С. А. 39, 4276 (1945);
F (1946); О (1950).
AgFe.S, SB V 76.
AgsH(Fe02)4 SB IV 171.
Ag-Ga SB IV 234; Ch. Z. 1260 (1947).
Ag,Ga SB VII 199.
75. Ag-Ga-Zn SB VII 199.
§,282] 76. AgJELJO,- 12pJ^|?S0^NHS:===== =819
76. Ag2HsJO, H. Ph. 18, 267 (1945); С. А. 40, 258 (1946).
Ag-Hg SB I 557; SB II 706; SB III 611; SB V 123; Ж. Ф. X.
XV, 96 (1941); Acta XIV, 193 (1941); ИАН, стр. 683 (1941).
Ag10Hgls SB II 706.
Ag3Hg4 SB II 704, 706; SB III 361.
80. Ag2HgJ4 SB 1Г 50, 331; SB III (#3:7) 235; F (1942); F (1946);
О (1950).
Ag-In SB I 558; SB III 609; SB IV 223; SB VII 199; Ch. Z.
1260 (1947).
Ag,In SB I 559; SB III 609.
Ag5In, SB HI 610.
Agin, SB III 610.
Agl, иодирит SB I 112, 767; SB II 211, 212, 214, 331; SB III
(523 : 8), 232 (M: 7), 233; SB VI 55; HRF (1938); Ph. R. 64, 325,
468 (1938); БМКД (1938); БМКД (1939); F (1942); О (1950).
AgJ04 SB II 84, 417; F (191(5); О (1950).
KAgCO, J. Am. 66, 295 (1944).
KAg(CN)a SB III (F510:82) 400; HRF (1938).
• Ag-La Z. An. 252, 62 (1943); Ch. Z. I, 518 (1945); О (1950).
90.Ag-Li SB III 604.
AgLi SB II 663; SB III (Л20:175) 265; F (1942).
AgLi, SB II 663.
AgMg SB I (/,20:488) 551; F (1942).
AgMn04 SB II 411, 416; SB VI 19, 94, 97; О (1950).
Ag2Mo04 SB I (Я1,:352) 379; HRF (1938); F (1942); J. Am. 69,
222 (1947); О (1950).
AgN, SB III (F52: 75) 393; О (1950).
AgjNO,, Ж. Ф. X. XXII, 1284 (1948); О (1950).
AgNOa SB IV (F5,a:41) 153; HRF (1938); О (1950).
AgNO, SB I 339; SB II 385; HRF (1938).
100. AgNO,Hg(CN)22H20 SB IV 159.
AgNO, (CONH2)2 SB III 731.
AgNO,2NH, SB III 417.
AgO SB I 115.
Ag20, SB III 326; О (1950).
Ag.,0 SB I (C3:155) 222; SB II 280: HRF (1938); F (1942); О (1950).
Ag>04 SB I (Я2:362) 395; SB IV 175; HRF (1938); F (1942); О (1950).
Ag-Pb SB VI 193; Ch. Z. II, 2663 (1940); Z. E. 49, 89 (1943).
Ag3PbaSb,S, SB VI 131.
Ag6Pb2SbsS,2, фрайслобснит SB VI 131.
"410. Ag-Pd SB I 519.
Ag-Pd- II SB I 604; SB III 575.
Ag-Pt Z. E. 49, 72 (1943).
AgPt SB II 629.
AgPt, SB II 629.
AgRe04 SB III (Я04:91) 421; F (1946); О (1950).
Ag-Bh SB III 575.
AgaS SB I 224; SB II 283; SB IV 110; SB VII 91; HRF (4938).
8(Ag, Cu)aS-Sb2S, SB III 351.
AgaS04 SB II 89, 425, 426; HRF (1938); О (1950).
120. AgtS044NH, = [Ag(NH,)2l8-[S04l ЗВШ(Я417:115) 467; F(1946); О (1950).
820 12L Ag-Sb —160. Al^B.Si.O^O [гл. XI
121. Ag-Sb SB I 596; SB IV 223; SB VII 199; 0(1950).
Ag,Sb, дискразит SB I 596; БМКД (1939).
Ag2Sb, гольдшмидтин SB VI 65; SB VII 95; 0(1950).
AgSbO, SB VI 118, 120, 132; SB VII 24.
AgSb(OH), SB VI 27, 118; F (1946); 0 (1950).
Ag(Sb, Bi)S2, арамайоит SB I 291; SB VII 123.
Ag5SbS4 SB II 348.
Ag,SbS„ нираргирит SB I 787; SB V 75; Б (1947).
AgSbS2, миаргирит SB VI 83.
130. Ag2Se SB IV 114; 0 (1950).
Ag2Se04 SB II 89, 426; 0 (1950).
Ag-Si SB III 567.
Ag-8n SB I 502, 559; SB IV 223; SB VII 199; Ж. Ф. X. XIV,
846 (1940).
Ag,Sn SB I 559.
Ag-Te SB VII 210; 0 (1950).
Agl2Te, SB VII 210; 0 (1950).
Ag2Te, гессит SB II284; SB IV115; SB VII210; БМКД (1939); 0 (1950).
Ag-Th Z. El. 49, 487 (1943).
AgU02 (CH3COO), 2H20 SB IV (04U : 264) 287.
140. Ag4V20, HRF (1938).
Ag-Zn SB I 501, 502, 552; SB III 606; SB VI 160, 161; P. R. S.
(A) 177, 149 (1941); M. 1, 56 (1946).
AgZn SB I (£2: 76); (L20: 488) 147, 501, 552; SB VII 199; F (1942).
AgsZn, SB I (Z>82: 499) 552.
§ 283. Алюминий Al
Al SB I (41:14) 43; SB II 163, 165, 166, 167, 170, 171, 668-
690, 693, 722-724, 730-733, 761, 762, 795; SB III 181, 184,
186, 187, 201; SB IV 82; SB V 32; SB VII 49, 52; HRF (1938);
БМКД (1939); F (1942); NW 31, 302(1943); NW 32, 33 (1944);
Ch. Z. II 263 (1944); P. R. S. (A) 193, 1 (1948); ИАН (х),
стр. I*7 (1948); О (1950).
AlAs SB I (£3:7',/ 141, 773; SB II 236; Z. An. 244, 146 (1940);
F (1942); О (1950).
AlAs04 SB II 465; SB III 428; SB IV 166, 204; О (1950).
Al (AlOH)2 Mn6 (As, V) Si50242H20 SB II 579.
NaAlF (AsOJ, дурангит SB V 95; SB VI 110-112.
Al2Au SB II 679; SB III (Cl : 20) 314; F (1942).
150. AIAu SB II 679; J. I. M. 71, 213 (1945); С. А. 39, 3198 (1945).
AlAu2 SB II 679.
AIAu, SB III 326.
A1B, SB III (C32:28) 311; SB IV 121; О (1950).
AlBtl SB IV 127; SB VII 110, 111; О (1950).
3AlBt22B4C = Al,B48C2 SB II 170; SB IV 127.
A1B0„ еремееьит SB III 41'.; SB VI 94.
HMgCa,AltB(Si04)4 SB II 580.
Al2Ca2B H(Fe, Mn)(SI04)4, аксинит SB V 118.
Al,(B0H)2H,Si40l((, турмалин SB V 119.
160. AiliBaSi,Oa,HaO SB II 581.
§ 2831 161. А14Ва —205. Ca4MgiAl2Ee?Ol5 821
161. AI4Ba SB III (Z>13:45) 330; F (1046).
Ba06Al,0, SB VI 75.
Al20JBa0 = AIJBa04 SB V (#2„:19) 97; Б (1947); О (1950).
Al2BaSi208 SB I 465; SB III 550.
BaAl2Si30104H20 SB III (S610:168) 529, 531.
BaAlaSi.OI,6H20, гармотом SB V 120*.
Na.Ba.Al.S.O^S, SB II 563.
Al-Bc-Cu Металлург 12, 81 (1938) (?).
Al2Be04 SB I (Я1.,:355) 400, 415; О (1950).
170. KCa2Be2Al(Sil2030), миларит ИАН(.\), стр. 364 (1948).
Ве04СаО Al20,9Si02 И20 - AI,0,BcO 4CaO 9Si02 H20 SB III 560.
Al4Be,Lt4(Na, K)2B10O„ родизит SB VI 94.
(На, Са) Be (Si, А1)2 (О, ОН, Р), SB II 542, 543.
Be3Al2(SiO,),, берилл SB I (G3,: 307) 329; SB III 522; БМКД (1939);
0(1956).
AlBeHSiO. = AlBe (OH)Si04, эвклаз SB II 585; SB III (50.: 148)
516.
Al-Bi-Pb-Sn Cb. Z. 1, 459 (1942).
AlBi03 G. A. 41, 6102 (1947).
Al,Br, SB VI 76; HRF (1938); Rec. 64, 275 (19451; С А. 40,
3956 (1945); 0(1950).
A1(CH3), N. 156, 601 (1945); О (1950).
180. [A1(R3)]2 (R-CH3...) J. Am. 69, 1327 (1947); см. J. Am. 63, 3287
(1941); N. 156, 601 (1945).
A14C, SB III (.07,: 56) 360; HRF (1938); 0(1950).
A14C3A1N SB III (£94: 73) 383.
Al (CH2CHCH2CHCH3)3 SB I 680-682.
Al2Cl2Ol218H.,0, меллит SB III 779; F (1946).
Al-Ca SB VII 203; HRF (1938).
Al2Ca SB VII (C15: 7) 203; F (1942); ДАН 40, 262 (1943).
A120, - CaO SB V 40.
xCaO yAl20 SB I 478.
6Al203CaO SB VI 75.
190. 16Al2033CaO SB V 40.
Ca02Al203 SB V 40.
Ca3AlI0Ol8 SB I 478; SB II 320, 323, 344.
CaAl204 SB I 478; SB II 323; SB V 40.
7Al2Os12CaO SB IV (Klt: 68) 198; SB V 40.
Ca5Al,014 SB II 321, 323, 591; HRF (1938).
Ca3Al20, SB II 60, 321, 323, 344, 595, 596; SB V 40; SB VI 135 —
137; HRF (1938).
4CaOAl20312H,0 SB III 390.
5CaOAl30„ SB I 478.
СаДА1(ОН),]2 SB III (/2:129) 494.
200. mCa (OH)2 nAI (OH), pH20 SB III 495.
Ca,Al, (P04)e 20H20 SB VI 105.
Ca4Al2Fe2O10 == Al2034CaOFe20„ SB II 321, 345, 596; SB VI 87, 136.
(Al2Os) r (CaO) „ (Fe2Os), (MgO) p (H20) t SB II 320.
H2(Mg, Fe, Al)6 (Ca, Na, K)2.3(Si03)3 SB II 13'., 533.
205. Ca4Mg2Al2Fe.,Or, SB II 321.
*) По Дача (Ва. К.) Al2SUO,,611,0.
822_206. (Al, Fe™, F"),.(Fe", Mg, Ca, Mi),„„-252. Ca8AlaSiaQt. [гл. XI
206. (Al, Геш, Fe")It(Fe", Mg, Ca, Mn)iw, (Si, A1)X60.2 15,61 HaO, даф-
ндт SB VII 176.
Ca2Al2(Al, Fe)OHSi04 SB II 582.
:|Ca10(Mg, Fe)Al4(OH)8$i,034 SB II 525.
Cal0Al4(Mg, Fe)„(0H)4Si,034 SB II 525.
210. (Na, Ca)(Mg, Al, Fe) (Si, A1)20, • SB II 527.
Ca2Al2(AI, Fe)0HSi04 SB II 582.
A12(A1, Fe)OH Ca2(Si04)„ эпидот SB II 582.
Ca —Mg —Na—Al, силикаты SB I 466.
Ca8Na2Al,Ol3 SB II 323.
Ca3Na4Al10O20 SB II 323.
(Na, Ca)2(Mg, Al)(SiAl)20„ мелилит SB II 148, 541; F (1946).
Na2(Ca, K)2AleSi,024(S04)2 SB III (S6,: 166) 546.
HK Ca2AlaSil2080 = HK Ca„Al2 (Si206). SB I 476; SB II 586.
Al,CaKSi(Oia6HkO, христианит SB VI 151.
220. Al2(Ca, K2, Na,)Si100247H20, морденит SB VI 152.
Al2(Ca, Na2, K2) Si10Oa47H20, морденит (птилолит) SB VII 177.
(CaS04)(NaAlSi04)3 SB I 470.
(Na, Ca)4_8(AlSi04),(S04),_2, гаюин SB II 152, 562.
(Ca, Mg) OAl20,5SiO, nH20, n = 5 —7, монтмориллонит SB VII 166
БМКД (1938); БМКД (1939).
Ca10Mg2Al4(0H)4Si„0a4, везувианит SB II 126, 525; F (1946).
Al2(Ca, Na2)Si40126H20, хабазит SB I 474.
NaAlSi308Al2CaSi2083CaO = Ca.NaAlSi.O,,, сарколит SB I 791; F (1946).
Ca8Na4Al2„Si,,04024H.,0 SB HI 528.
Na2Ca2Al,Sie0808H20" SB III (56:168) 529.
230. (Na, Ca)3(Al, Si)l0O.,„7H2O SB II 573.
(Na2Ca)2Al2COeSi209, канкринит*) SB II 568, 569.
Na2Ca2Al3Sis02o6H„0 SB III (S6l0:158) 529.
NaoAl,042Si022CaC03 SB III (S33:150) 524.
(Ca, Na) Al„0HSi08 SB II 546.
NaCa2AlsSi662„6H20 SB II 572, 573.
Na,CaAl,(Si04)4 SB II 568.
(Na, Ca)A10HSi05 SB II 546.
(Na, Ca)4.e (AlSi04), (SO,),., SB II 152, 562.
Al20,2CaOSi02, геленит SB VI 138; F (1946).
240. AL03Ca02Si02, анортит SB VI 138; БМКД (1938); G (1939).
CaSi„Al2082H20, лаусонит SB V 117.
CaAl2Sia084H20, жизмондин SB VII 178, 179.
Са2АШО, SB II 148, 543.
CaAl.,Si4Ol,6H20 SB II 575, 576.
Ca4Al,Si,024Cl2 SB II 560.
Ca, (A100H)2 Si303 SB II 588.
Al2CaSi30103H,0 SB I 475; SB III 529; SB IV 214.
Ca2Al4Si,02„6H20 SB III 528.
CaAl2Si40,o6H20 SB III (S3,:151) 526.
250. Ca4Al,Si,02eS3 SB II 563.
Ca4Al.Cla (Si04). SB II 560.
252. Ca,Al2Sl2Ol5 SB II 595, 596.
*) Формула неверна. Не соответствует ни .~>В II 568, ни Дана, стр. 185.
|_283] _ 253JXAi2(Sj04)3-m Al4Cu, 823
2.13. Ca,Al2 (Si04)3, гранат, гроссулярит, гроссуляр SB I (ЯЗ.: 366) 411;
БМКД (1938); F (1942); Б (1947).
Ca2Si04Al203 SB I 791.
Al2CaSi,0ie6H20 SB III 527.
3CaOAl2032Si022H20, плазолит SB V 111.
Al2CaSi,Oie 6V2H20 SB I 475.
Al2CaSi2Oe4H20 SB VII 178, 179.
Ca2Al3 (OH) (Si04), SB III 553.
260. H4CaAl2Si2010 SB II 586.
A1C13 = A1,C1, SB II 36, 291; SB III (£>0l5:41) 319; SB VI 76;
HRF (1938); О (1950).
A1(H,0),C1. SB III (/2.: 127) 493; HRF (1938); О (1950).
Al-Cd-ltg SB VI 181.
(Al, Mg)Cd SB VI 181.
Al -Co SB VI 175; SB VII 196; J. I. M. 64, 81 ^1939).
AlCo-Co. Z. Met. 33, 381 (1941); Ch. Z. 11, 10 (1942).
AJ,Co2 SB VII 196.
Al18Co, SB VII 196.
Alx3Co4 SB VII 196.
270. Al3Co SB VII 196.
Al6Co2 SB VI (Z)8U: 11) 175; SB VII 196.
AICo SB II 683; SB III 196.
4(Fe, Co, N1, Mg)09(Al, Fe)2Os 8Si02H20 SB III 558.
Al2Co04 SB I (Hi: 352) 417; SB II 468, 471, 472, 474; SB IV 167;
F(1942); О (1950).
Al-Cr SB VI 183; Z. Met. 33, 369 (1941).
Al„Cr6 SB V Ш810:11) 65.
AlCr2 SB V (C48:5) 53; F (1946).
(AL03, Cr,0,)FeO HRF (1938).
Al263Cr2032MgO SB III 243, 340, 342.
280. CsAl (S04)212H20 SB I (Я42:370i 390; SB III (Я414:112) 455; Hi F
(1938); F (1942); О (1950).
CsAlSi2Oe SB II 588.
Al,03Cs20 4SiOJI20 SB VI 150.
CsAlSi20, Н20, подлупит SB VI (56^34) 149; ВМКД (1939).
Al Cu SB I 501, 502, 538; SB II 668-677, 693; SB III 589; SB IV
236; SB VI 159, 181; SB VII 195; P. R. S. (A) 177, 149 (1941);
Ch. Z. I, 13 (1942); Ph. Rad. [81 3, 124 (1942); Ch. Z. I, 709
(1943); Ch. Z. I, 798 (1944); P. R. S. (A) 193, 1 (1948).
Al.Cu SB I (C16:493) 229, 504, 539; SB II 668, 669, 676,
' 677; SB III 590; SB IV 243; SB VI 181; SB VII 194; Ch. Z. 1
1104 (1942); P. Ph. 62. 701 (1940); Ch. Z. 1, 1472 (1942); Z. Met.
35, 218 (1943); С A. 39. 1095 (1945); F (1946); Б (1947).
AlCu SB I 504; SB II 668, 674, 675, 687; SB IV 243; SB VII 194.
AL,Cu..3 SB II 674, 675.
All3Cu,4 = AI3Cu4 SB II 674, 675; SB VII 194.
Al.Cu/ SB II 668.
290. AileCu32 SB VII 194.
AlCu, SB II 668, 669.
292. Al4Cu9 SB I (Z)8:499) 498, 542; SB II 668, 676; SB III (Z>83: f>7)
590; SB VII 194.
824 293. Al2Cu3-340. AlFc3 [гл. XI
293. Al2Cu, SB VII 194.
AlCu3 SB I 503, 539; SB II 668, 670, 676; SB III 589.
Al-Cu-Fc SB VII 192; Ch. Z. 1, 459 (1942).
Al7Cu2Fe SB VII 195.
AleCu„Fe SB VII 195.
All8Cu10Fe SB VII 195.
Al10Cu10Fe SB VII 194.
300. Al—Cu-Fc—Mg—Si SB II 672.
Al-Cu-Mg SB IV 242; Ch. Z. 1, 1104 (1942); Ж. Ф. X. XX,
387, 1475 (1946).
AlCuMg SB III 312; SB IV 243.
Al,CuMg4 SB III 391; SB IV 243.
Al6Cu2Mg, SB IV 243.
All0Cu,Mg, SB IV 243.
Al2CuMg С. А. 38, 5707 (1944); Ж. Ф. X. XX, 1475 (1946).
Al2CuMg4 SB IV 243.
AlCuMg ДАН 40, 262 (1943).
AlCu2Mg, SB IV 243.
310. Al2Cu2Mg3Ni2 SB III 312.
Al-Cu-Mg-Si SB II 668, 670, 671, 673.
AlCuMg-MgZn2 SB III 313.
AI-Cu-Mn SB I 549.
Al(Cu, Mn) SB II 688.
AlCu2Mn SB I (/.21 :488) 551; SB II 689; SB III 599.
Cu,(Mn, Cu)Al SB III 598.
Al(Cu, Mn)3 SB II 688, 689.
Al4 (Cu, Mn), SB III 598.
Al-Cu-Ш SB VI 158, Ch. Z. 1, 459 (1942).
320. Al,Cu3Ni Z. Mot. 33, 278 (19il); Ch. Z. 1, 1223 (1942).
Al-Cu--Pb Ch. Z. 1, 13 (1912).
Ag3Cu8Pb4Cl.,lO(19H20 SB II 365-367.
Al-Cu-Si Ch. Z. 1, 459 (1942).
Al-Cu-Ti SB III 592.
Al-Cu-Zn SB I 5i4; Z. Met. 33, 278-289 (1941); Ch. Z. 1,
1223 (1942).
AL03-CuO SB III 342; SB IV 167.
ALCu04 SB I (#1^352) 417; SB II 469, 472, 473; HRF (1938);
F (1942); О (1950).
3CuOAl203 SB I 478.
A1F3 SB II 288, 291; SB HI (Z)0l4:40) 318; О (1950).
330. A1F33H20 J. Am. 67, 64 (1945).
A1F33,/,H„0 HRF (1938).
A1F4(N"H4) SB VI (Я0:18) 95; F (1946); О (1950).
AlFe(NH4)„ SB I (#7^437) 448, 793; SB VI 95.
Al-Fc 'SB III 613.
Al,Fc, SB VII 194.
ALFe" SR II 679, 761, 762; SB III 324, 325, 613; HRF (1938).
Al6Fe2 SB II 679; SB VII 194; HRF (1938).
Al2Fe SB II 679; SB VII 194.
AlFe SB II 679, 680, 683; SB VII 194; HRF (1938).
340. AlFe3 SB II 679, 680, 682; SB VII 194.
§ 283] 341. Al-Fe-C-383. Al3KH2(Si04)3 825
341. AI-Fe-C SB III 614.
AlKFe(CN), SB VII 155.
(Na, K)(A1, Fe) Si308 SB I 463.
Al-Fe-Ni SB HI 583; SB VI 167; J. I. M. 66, 53 (1940).
Al-FeNi Ch. Z. II, 198 (1944).
Al2Fe04 SB I (Я1:352) 416; SB II 470, 471, 472; SB IV 167;
F (1942) ; О (1950).
Al2Fe20, SB II 477.
(Mg, Fe)3 AI, (Si04), SB I (ЯЗ : 366) 411.
Al4Fe„033 " SB Й 477.
350. Al-Fe-Sl Ж. Ф. X. XX, 597 (1946).
Al4Fe (0H)2 Si20l0 SB II 113, 514; Б. (1947).
Al,Fe,H(SiOe)4 SB I 452.
(AI, Fe)aH4Si,0„ фаратсихит SB VII 167-168.
(AI, R)aK(OH,F)2(AI, Si)4010, слюды (It = Mg,Fc, Li,Ti и др.) SB VII
168-171; HRF (1938).
5Al,O3Fe20322Mg022Si0240H,O, вермикулит SB VI 146^148; БКМД
(1939).
AlK(Mn, Fe, Mg), Sia010 (F, ОН)2, биотит SB VI 151; БМКД (1938);
С (1939).
(NH4)2(A1, Fe)4Mn5(S04),,l8H20 SB II 443.
(Ca, K2, Na2)Al2Sil00247H20 SB VI 152.
HK2(A1, Fe)3 Fe4 (S04)I013H,0 SB II 443.
360. (AI, Fe, Mg, Mn)10KSil2030 (OH)l2, стилпномелан SB VII 175.
K(Fe, Mg),(Fe, Al)5.e (OH)2oSi,e039.4„ стилпномелан SB V 112.
(K, Na, Ca)„(Fe, Mg, Al),9(OH)x,Si3o00313H20, стилпномелан SB V
112; БМКД (1939).
(NH4), Mn5 (AI, Fe)4(S04)1>l8n20 SB II 443.
AI-Ge SB VII 212; "Z. An. 241, 309 (1939); P. R. S. (A) 193,
1 (1948).
Al-Mg,Ge Z. El. 49, 198 (1943).
A1,J, SB VI 76; HRF (1938); 0 (1950).
KAIF4 SB VI (Я08:18)95; F (1946); 0 (1950).
K2NaAlFe SB II 498.
(Na, K)AlSi04( нефелин PI АН (х), стр. 364 (1948).
370. A1203 - K20 SB III 292, 430.
11A1203 K,0 SB IV 128; SB V 72; SB VI 75.
K.,Al2o03l " SB II 436.
ЛГ,03К,0 SB III 430.
KA1(804), SB I 789; SB II 90; Б (1947); 0 (1950).
KA1 (S04)2" 12H20 SB I (Я42:370) 390; SB III (Я413:108) 454;
SB V 91; HRF (1938); F (1942); 0 (1950).
KA1 (S04)212D20 SB V 91.
KA13(S04)2(0H),, алунит SB V 92, 93; БМКД (1938).
K203A12034S036H,0 SB II 436.
KA1 (Se04)212H„0" SB III (Я413:110) 455; 0 (1950).
330. AlLi2KSi4010F2, полилитионит SB VI 151.
Al„Si30I0KLi (OH, F)2; К (Li, Al)3 (Si, A1)4 010 (0Н)8, лепидолит SB V114.
Al,K4Li6Al4Sil2O40 (F, 0H)e, литий-мусковит (?) SB VI 151.
383. Al3KH2(Si04)3: AI203KAlSi307 (OH. F)„ мусковит SB I 412; SB V 114;
SB VI 151, 152; SB VII 166; HRF (1938); БМКД (1938); БМКД
(1939); С (1939); 0(1950).
^_= 384' КА^1зОз-425, AUMk.SI.0,, (ОН), Jrn. XI
H8i. KAlSi,0„ ортоклаз, адуляр SB 1464; SB III (56,: 161) 547; HRF (1938);
С (1939); БМКД (1939).
AlKSi20„ лейцит SB IV 211; SB V 116; SB VI 153; Z. Kr. 104,
'39(1942); F(1946).
KA13 (0H)2 Si3010 SB II 143, 546, 547; 0 (1950).
Al-La SB III 644.
Al-Li P. R. S. (A) 193, 1(1948).
Al4La SB III 644; F(1946).
Л90. Al2La NW 29, 654 (1941); Z. Kr. A. 104, 257 (1942); F (1942); Ch. Z. I,
127 (1943).
LaAlO, SB I (G5 : 303) 334; F(1942); О (1950).
Al-Li SB III 635.
AlLi SB II 722; SB III (£32: 19) 266, 635, 636; F(1942).
Na3Li3Al2Fl2, криолитионит SB I 366, 449; SB II 498; БМКД (1939);
F (1942).
Al2LiF03 SB II 365, 485.
5Al203Li20 SB III 435.
AlLi [P04] (F, ОН), амблигоиит SB VII 142.
AlmLi„SUV. SB II 593.
LiAl (Si03)2 SB I 328; SB II 131, 527; F(1942).
400. AlLiSi4010 SB II 588.
Al6LiSi3Ol0(OH)8 = LiAl4Xi3A10.0(OH)e, кукеит SB V 113, 114.
Li2Al,Si3O102H2O SB III (S610:168) 530.
A)-Mg SB I 566; SB IV 248; SB VI 181, 182; SB VII 204; Ch.
Z. I, 798 (1944); N. 154, 606 ^1944); С. А. 40, 4273 (1946).
Al3Mg SB II 762.
Al2Mg SB IV 242.
Al8Mg, SB IV 248.
Al-Mg P. R. S. 193, 1 (1948).
Al3Mg2 SB II 722, 762; SB IV 249; SB VI 181; SB VII205; HRF (1938).
AlMg SB IV 248; N. 154, 606 (1944).
410. Al3Mg4 SB II 722; SB IV 248.
Al2Mg3 SB II 722: SB HI (412:3) 358; HRF (1938).
Al,03-Mg0 SB HI 242, 340, 431; SB II 170.
Al,Mg04 = Al203MgO, шпинель SB I (Я1,: 352) 350, 417; SB II 321,
468, 470, 471, 473, 474, 484, 592; SB IV 167, 170; SB VI 115;
HRF (1938); БМКД (1938); F (1942); Б (1947); Ch. Ph. 15, 174
(1947); 0(1950).
(Al, Mg)2 Na0.35 [(H2O)0,875 (F, ОН),], ральстонит SB VII (£8,: 24) 127.
AlMg2Na (Si03)4 SB I 791; SB II 536.
Al -Mg.Pb Z. El. 49, 198 (1943).
Al-Mg-Si SB VII 206.
Al-Mg2Si Z. El. 49, 198 (1943).
Al203-MgO-Si02 С. А. 38, 5707 (1944).
420. Al4Mg2Si010 = Mg2Si042Al203 SB I 791; SB II 589.
3Al,034Si0aMg SB I 791*).
Al.Mg.Si.O,, SB II 588, 589.
Al4Mg2Si,Ol3 SB I 791; SB II 581.
Al2Mg6Si,0„ SB II 553.
425. Al2Mg5Si30l0(0H)8, хлорит SB V 114; С (1939).
*) Формулы по SB I 791, очевидно, неверны. Минералу прпзмотин, по Дана,
отвечает состап примерно MgAl2SiOe.
§ 283] 426. Al1Mg1Si,01.(OH).-467. NaAl(Si03), 827
426. Al2Mg,Sis0l0(0H)8 SB III (S5 : 156) 537.
Al2Mg,(OH)eSi3Oie SB II 553.
Al.,Mg1...1Si,_,Oie_2JHg_]()(H20)„_4, палигорскит SB V 118.
a.(Al4/3Mg2)H2Si,0l0H4(0H)2 (?), аттапульгит формула не верна д. б.
б. (Ah/.Mg), H2Si,010H4 (ОН), SB VI 151.
4о0. Al-Mg.,Sn Z. El. 49, 198(1943).
Al-Mg-Zn SB IV 248; SB VII 205; Ch. Z. I, 13 (1942).
AL.Mg,Zn, SB III 391.
Al-MgZn, Z. El. 49, 218 (1943).
Al-Mn SB II 684, 687.
Al7Mn SB II 686.
Al.Mn SB II 684.
A14M» SB II 684.
Al,Mn SB II 684, 686.
AlMn3 SB II 686, 687.
440. Al2Mn04 SB I(tfl :352j 417; SB II 469, 471, 472; SB IV 167; HRF
(1938); F (1942); О (1950).
Al (N03)3 • 9H20 HRF (1938).
AlNaCl4 HRF (1938).
Na2Al4Mn2Si,0>8S, SB II 563.
MnsAl2(Si04)3," епессартин SB I (Я3:366) 411; БМКД (1939);
F (1942).
A1N SB I (£4:79)140; SB III 261; Z. An. 244, 146 (1940); О (1950).
(NH4), A1F, SB I (Я71 : 793); F (1942); Б (1947); О (1950).
NH4A1(S04)212H20 SB I (Я42:370) 390; SB II 434, 435; SB III
(Я4„ : 110) 455; HRF (1938); F (1942); О (1950).
NH3CH3A1(S01),12H,0 SB II 435; SB III (Я414:111) 455; F (1942);
О (1950).
NH4A1(S04)2 SB I 789; SB II 91; 0(1950).
450. (NH4)2 A12S13010 SB III (S610:168) 530.
Al3Fl4Na5, хиолит SB IV 201; SB VI {Klb :31) 32, 121; БМКД (1939);
F (1946).
Na3AlF„ криолиг SB I 793; SB VI (/2: 29) 30, 1-20, 121; SB VII 153;
HRF (1938); О (1950).
AINa P04 (OH. F), фрсмоитит SB VII 142.
Al203Na,0 SB II 323; SB III 430; SB VII 85.
HAL03Na20 SB IV 128; SB V 72; SB VI 75; HRF (1938).
Na3A'l (S04)3 SB II 440.
NaAl(S04)2 SB II 440.
NaAl (S04)212H20 SB III (Я415:112) 456; HRF (1938); О (1950).
Na2S04(NaAlSi04), SB I 470.
460. NaSb(A103);! SB I 334.
AlNaSi4 M 2, 127 (1947).
ALO,Na20 4S10.H20. анальцим SB VI 150; БМКД (1939).
JVa4(AlSi04)3Cl (содалит) SB I 470; SB II 150, 562; F (1942);
Б (1947).
JVa2Al4Pb,Si,0.)4S, SB II 563.
Na4AlClSi3Oe " SB II 560*).
JVaAlSi04, карютиит SB I 469; SB II 158, 553-555; БМКД (1939).
467. NaAl(Si03)„ жадеит SB I 327; SB II 528; БМКД (1938).
*) См. также «Na,C.l(AlSi.,Os).4» (?) SB II 155, 559—561.
828 468^Na4Al4Si,O2„4H2O-505. RbAlF4 [гл. XI
468. Na4Al4Si,O204H2O SB III (56 :168) 528.
Na2Al2Sis0102H20 SB II 575-578; SB III 529.
470. NaAISi20,H20 SB I 473, 792; SB II 148, 569-571; F (1942).
NaAlSi,08, альбит SB III (56:164^ 546; HRF (1938); БМКД (1938).
Na- (AlSi04), S04 SB II 152, 562.
Na,Al4Si,024S, SB II 563.
Na2Al.ZnsSi,028S, SB II 563.
Al-Nb SB VI 183; SB VII 100.
ALNb SB VI 183; SB VII (D0M: 13) 100; Z. An. 242, 1 (1939); Z.
El. 49, 208 (1943); F (1946).
AlNd SB III (£2:7) 273; F (1942).
JU-Nl SB I 564; SB V 127; SB VI 159.
AINi-Ni Z. Met. 33, 347 (1941).
480. AlaNi SB II 683; SB V (DQ2t: 8) 60.
AlaNi SB II 683.
Al,Nia SB V (D5„: 10) 67, 129.
AINi SB I (B2 : 76) 147; SB I (L20:488) 564; SB II 683; 684; SB V 128;
F (1942).
A12N104 SB I (Hi,: 352) 417; SB II 471, 472; SB IV 167; HRF
(1938); F (1942); О (1950).
Al-NiSi Ch. Z. II, 198 (1914).
AI20„ корунд SB I (Ub1 : 242) 114, 257, 777; SB II 41, 43, 176, 196, 310,
311-313, 318, 320, 322, 323, 350- 352, 472, 473, 592, 593, 594, 596;
SB III 338, 339, 592; SB IV 128, 129; SB VI 72, 138; SB VII
153, 161; HRF (1938); БМКД (1938); I. E. Ch. 37, 158 (1945);
Б (1947); О (1950).
«Р-окись алюминия» 11А120, • Na20, см. А12Оэ.
А120, х НаО, бсмит SB II 350-353; SB III 340; С (1939).
А12034Н20 SB III 341.
490. А1203ЗН20, гидраргилит, байерит SB III 340; БМКД (1938); С (1939).
А120, • 2Н20, «боксит» HRF (1938)*); БМКД (1939).
А12ОаН„0, диаспор, бемит SB IV 129, 130; БМКД (1938).
А12033,14Н20, байерит С. А. 38, 9 (1944).
А12Оэ, аэрогель SB IV 130.
А1,0„ золь SB IV 130.
А100Н, диаспор SB II 46, 320, 350-353, 360; SB III (Е02:64)371;
SB IV 129, 130; HRF (1938); С (1939); Z. Кг. (А) 103, 73 (1940);
104, 11 (1942): Z. El. 49, 269 (1943); Б (1947).
A1(0H)3, гидраргиллит SB 1253, 254; SB II 320, 350-353; SB III
(Z>07: 38) 322; Z. El. 49, 269 (1943); Б. (1947); О (1950).
A1P SB I (S3: 77) 140, 772, 787; SB II 236; Z. An. 244, 146(1940);
F (1942); О (1950).
A1P04 SB III 423; F (1946).
500. A1(P03), SB I 315; SB V (G53:15) 80; О (1950).
A1P042H20, метаварискит, варискит SB VII 143.
Al (OH)3 P2Os5H20, вавеллит SB III 457.
4A1P042A1(0H)39H20 SB III 457.
Al2Pt SB V 54; F (1942).
505. BbAlF4 SB VI (#08:18) 95; F (1946); О (1950).
*) Согласно Дана, «Описательная минералогия», стр. 115 (1937), боксит
содержит переменное количе-тво волы: А1^ия(1 — 3) Н20.
§ 283] 506. RbAb (SOt)2 12H20 - 546. Al-Ta 829
506. RbAb(S04)212П.0 SB I (H42: 370) 392; SB III (H4lt: 110) 455; HRF
(1938); F (1942); О (1950).
AlRbSi04 SB II 555.
А1Д HRF (1938).
A12(S04), SB II 436, 440; HRF (1938).
510. Al2 (SO,), 18H20 HRF (1938).
A120,2S02 R\0 SB IV 162.
Al-Sb SB I 599.
AlSb SB I (53:77) 141, 599; Z. An. 244, 146 (1940); F (1942);
О (1950).
AlSb04 C. A. 39, 2684 (1945); F (1946).
Al-Si SB V 134; Ch. Z. II 99 (1944); P. R. S. (A) 193, 1 (1948);
О (1950).
Al2(SiF,)3 HRF (1938).
Al2F2SiO, SB I 400, 410; SB II 116, 516.
A12(F, OH)28i04, Топаз Б (19'i7).
AluSi,02o(OH, F)lBCl, вуниит SB III (50,: 147) 517; F (1942).
520. A12S106, андалузит, силлимонит SB I (#5,: 428) 438, 792; SB II109,
110, 112, 511-513; SB III 515, 516; HRF (1938); БМКД (1938);
БМКД (1939); Б (1947).
Al,Si2018, муллит SB II 513; БМКД (1939).
A120,2S1022H20 SB I 476; SB III 539; HRF (1938).
H4Al2Si20„ каолинит SB VII 166, 175; БМКД (1938); С (1939);
Ж. Ф. X. XXII, 1017 (1948).
AlaSi206 (ОН)4, метагалоизит, мстагаллуазит SB VI 141; С (1939);
БМКД (1939).
Al2(0H)4Si20s = Al2032Si022H20, накрит SB II 548-550; SB III
(S54:155) 540, 541; SB V 114; SB VII (554:37) 38-40; 166;
БМКД (1938); С (1939).
Al4Si4010 (ОН)8 - A12SLH409 = Al2Oa2Sl022H20, диккит SB • V 114;
БМКД (1938); С (1939).
g A12S14010 (0H)2, пирофиллит SB III (55,: 159) 534; SB V 114; SB VT
148-149; БМКД (1938); БМКД (1939); С (1939).
Al4Sie(OH)402o SB IV 205.
Al20,4Si02H20 SB III 542.
530. Бентонит HRF (1938).
Al4Si,0246H20 SB II 552.
2Al20,8SiO,2H,0, монтмориллонит SB V 115.
Tl,AI4Si20„4S, SB II 563.
AlHSiWl264o28H20 SB IV 212.
Al-Sr SB VII 203.
Al4Sr SB VII (Z)l,:15) 204; F (1946).
AlSr SB VII 204.
Al20,-SrO SB V 40.
16Al2033SrO SB V 40.
540. 6Al20,SrO SB VI 75.
2Al20,SrO SB V 40.
Al20,3SrO SB V 40.
Sr, [Al(OH),]2 SB HI (J2 :129) 494.
Al,Sr(0H),P20„ гамлинит SB V 93.
SrAl (OH)2 P20, (OH) SB V 93.
546. Al-Ta SB VI 183; SB VII 100.
830
5_4_?'_AJ.?a-575. BAs04
[гл. XI
547. Al,Ta SB VI 183; SB VII (/*),,: 13) 100; Z. El. 49, 208 (1943); F (1946).
Al-Th SB VI 183; Z. El. 48, 418 (1942); Z. El. 49, 208 (1943).
T1A1F4 SB V (Я08:17) 97; SB VI 95, 122; F (1946); О (1950).
550. T12A1F6 SB V (K3t: 20) 97; SB VI 122; О (1950).
Al Ti SB VI 183; SB VII 100.
Al,Ti SB II 762; SB VI 183; SB VII (£022:13) 100; Z. An. 242,1 (1939);
Z. El. 49, 208 (1943); F (1946); Б (1947).
T1A1(S04)2 12H.0 SB I (#42: 369, 370) 392; SB III (Hi..: 110) 455;
F (1942); О (1950).
Al4Tl4Si,020xII20 SB III 531.
Al4Tl,Si2021S, SB II 563.
A14U Z. El. 49, 208 (1943).
Al-V Z. Met. 32, 356 (1940).
Al.V Z. El. 49, 208 (1943); F (1946).
YA103 SB I (Gb: 303) 334; F (1942); О (1950).
560. Al- Zn SB I 567; SB III 640; SB VII 206; P. R. S. (A) 193, 1 (1948);
Ж. Ф. X. XXI, 785 (1947).
Al2Zn, SB II 730, 731.
Al2Zn04 8В1(Я1г:352) 417; SB II 469, 470-473; HRF (1938);
F (1942); О (1950).
Al2OsZnO SB IV 167.
Al-Zr SB VI 183; SB VII 100.
Al,Zr SB VI 183; SB VII (D0„t: 14) 100; Z. An. 242, 1 (1939);
F (1946); Б (1947).
Общие формулы
K(A1, Д)а(ОН, P),(A1, Si)4010, слюды (R = Mg, Fe, Li, Ti и др.)
SB VII 171.
KR3(OH, F)2(A1, Si)40l0, слюды (R - Mg, Fe, Li, Ti и др.) SB VII
168-174.
Сопоставление накрит, диккит и каолинит SB VII 37 —40.
sc(Ca, Fe, Mn)0 (Fe, А1)20, (cc + 4f/)Si02 SB I 328.
570. se(Ca, Mg, Fe, H2)0 у [(Na2Fe)0 (Al, Fe)20,] (ae + 4y) Si02 SB I 325.
§ 284. Apron Ar
Ar SB 1 (Ai:lb) 71; Ph. R. 57, 1054 (1940); F (1942); RM. 16,
90 (1943); Z. El. 49, 308 (1943); Ch. Ph. 13 317 (1945); Усп. 16,
297 (1946); Ж. Ф. X. XX, 1414 (1946); ДАН 62, 231 (1948):
О (1950).
§ 285. Мышьяк As
As SB I (Al: 27) 57; SB II 184, 739 - 741; SB III 210, 211; SB IV 87;
SB VII 63; HRF (1938); Z. An. 242, 138 (1939); НМКД (1939)j
О (1950).
Арсениды, см. также Z. El. 49, 254 (1943).
AsAu J. I. M. 71, 213 (1945); С. А. 39, 3198 (1945).
575. BAs04 SB III (Я0,:93) 426, 428; F (1946); О (1950).
§285] 576. AsBr,—616^AsF8 831
576. AsBr, SB V 57; SB VI 68; Ж. Ф. X. XX, 6 (1946); 0 (1950).
AsiCH3), SB VI 234; J. Am. 67, 2014 (1945); О (1950).
CaHAs04 2H20, фармаколит (SB : Ca As03OH 2H20 неверна) SB V 91.
R(As04) (ОН)Б, синадельфит (It =■-= Mn + Ca, Mg, K, JVa, Fe. AlV
SB V 96.
580. Ca,(As04)a HRF (1938).
ApceuHT кальция HRF (1938).
CaCu As04OH, хиггинсит SB II 466; SB VII 151.
CaMg As04F, тилазит SB V 96; SB VI 111, 112; БМКД (1939).
CaMg As040H, аделит SB II 466; SB III 428; SB VII 150, 151.
(Ca, Na)„(Mg, Mn)2(As04)3, борцелиит SB III (SI,: 150) 521; БМКД.
(1939)*); F (1942).
NaCa2Mn2As3Ol2 SB II 465.
(CaSr),(F, OH, 0)2(P, As),0O4Ca4, ферморит SB VI 104.
CaZn[As04]OH, аустинит SB VII 151.
As2Cd, SB I 786; SB II 41, 326; SB III (Z>50:53) 354; F (1946);
О (1950).
590. AsCe SB V 44; F (1942); О (1950).
AsCl, SB IV 123; Ж. Ф. X. XX, 6 (1946).
CoAs SB HI (531:18) 264; О (1950).
CoAs„ скуттерудит SB I (£2:234) 250; БМКД (1939); Б (1947);
О (1950).
CoAsS, кобальтин SB I (Fl: 269) 283; БМКД (1938); F (1942);
О (1950).
Co,(As04)28H20 = Co3As,,038H20, эритрин SB V 94; HRF (1938).
Ae-Cr SB VI 189.
AsCr SB I 142; SB VI (£31:4) 189, 190.
As,Cr, SB VI 189, 190.
AsCra SB VI (C38:5) 189, 190; F (1946).
600. Cs,As2CU SB III (Kl3: 145) 506; Б (1947).
As-Cu SB VII 201; О (1950).
A82Cu, SB II 739.
AsCu,, доиейкит, SB II 300, 301, 739; SB VI (Z>02l: 7) 8, 65; БМКД
(1938); О (1950).
AsCu,, альгодонит SB II 300, 309, 739; БМКД (1938); О (1950).
AsCu,, уайтнеит SB II 300, 739; О (1950).
Cu2(As64)OII, оливенит SB VI (#2,: 22) 109.
CuPb (As04) ОН, дифтит SB VII 151.
CuAsS SB I 282.
(CuOAs20,) Cu (CH3COO), HRF (1938).
610. CuAsS (?), лаутит SB VII 123.
Cu.AsS4, эпаргит SB II 347; SB III (#2.: 96) 438; БМКД (1938);
БМКД (1939); Б ^1947); О (1950).
CuJAs(C2H6). SB IV (02,: 258) 259, 260, 285.
CuJAs(P4H,)„ SB IV 285 (не аналогична CuJAs(C2H6),).
(Cu, Fe)l2As4Slt SB III 413.
AsCuZn M 1, 38 (1946); О (1950).
616. AsF, SBIV 123; Ж. Ф. X. X, VI (1946).
*) В работе БМКД дана неправильная формула берцелиита.
832 617. Asг:?.е.—Щ• *h4?i9? Л11*0 |гл- XI
617.As-Fe SB I 594; 0 (1950).
AsFe SB 1 490, 595, 596; SB II 7; SB III (531:16; 263, 264;
О (1950).
As2Fe, лёллинтит SB I (C18:497) 217; SB II 273; SB IV 112;
О (1950).
620.AsFe2 SB I 594, 595; SB III (C38:34) 288; F (1946); О (1950).
Fe[AsOJ(H20)2, скородит SB IV 112.
Fe3 [As04] 6 (H20)2 HRF (1938).
Fe [(CH,).,As02], HRF (1938).
FeAsS, арсенопирит SB I 283; SB IV (E07:30) 142, 143; БМКД
(1938); О (1950).
AsGa SB I (53:77) 141; Z. An. 244, 146 (1940); F (1942);
О (1950).
As-Ge Z. An. 244, 205 (1940); О (1950).
As2Ge Z. An. 244, 205 (1940).
AsGe Z. An. 244, 205 (1940).
AsII3 SB II 288; О (1950).
•630. AsJ, SB II 294, 295; SBV 57; SB VI 68; HRF (1938); Ж. Ф. X. XX,
6 (1946); О (1950).
AsJ33S3 SB II 369; SB V 152.
K,As SB V (D0l8: 6) 60; О (1950).
KH£As04 N. 156, 20 (1945); F (1946).
Арсенат калия HRF (1938k
Арсенит калия HRF (1938).
K,As04 12MoO, 4H20 SB VII 149.
K,As04 12W0, 4H20 SB VII 149.
AsLa SB V 44; F (1942); О (1950).
AsLi3 SBV (Z>0l8:8) 80; О (1950).
640. As2Mg SB II 326.
MgsAs2 SB I 786; SB III (Z>53:49) 353, 356; F(1942); О (1950).
2MgHAs04 13H20 HRF (1938).
NH4MgAsO, 6H20 SB II 465; SB III 647.
AsMn SB I 765; SB III (531:18) 264; О (1950).
Mn3As206 = As20.3MnO SB II 410; SB III 510.
Mn2(AsO«)OII, саркинит SB VI 109.
(NII4)a As04 12MoO, 4H20 SB VII 149.
Tl3As04 12MoOs 4H20 SB VII 149.
(NH4)3 As04 3H20 HRF (1938).
650. (NHt)3 As04 12W03 4H20 SB VII 149.
Na3As SB V (00.,: 6) 59; О (1950).
NaHAs04 7H26 HRF (1938).
NaHAs03 HRF (1938).
CH3As0(0Na)a 6Н HRF (1938),
(CH3)2 AsO(ONa) 3H20, какодилат Na HRF (1938).
As-Nd SB V 43.
As2Ni, раммельсбергит, парараммельсбсргит SB VII 94; О (1950).
AsNi, никелин, «купферникель» SB I (58:87) 143, 490; БМКД (1939);
ИАН (х) 29, Hi, 201 (1944); Б (1947); О (1950).
AsNiS SB I (Fl:269) 125, 284; F (1942); О (1950).
660.№As2Ot Ch. Z. 1, 342 (1944); С. А. 39, 2019 (1945); F (1946).
661.Ni3As208 8П20, аннабергит SB V 94.
1_Ш] ^62-^A.=_As«pio-70i- AuBc5 833
662. AsaO6 = As4Ol0 hrf (1938); 0 (1950).
As2Os —As40„ арсенолит, клаудетит SBI (Ж,: 246) 264; SB II 315;
SBV 66; SB VI 77; БМКД (1938); HRF (1938); Ch. Z. 11, 9(1942);
F (1942); О (1950).
Pb5(AsOs),Cl SB I 335.
Pb5(As04)3Cl SB I 399; SB II 101, 458.
PbHAs04 HRF (1938).
Pb4As«S13 SB III 508.
Pb3As4S„ SB III 508.
PbAs2S4, сарторит SB III 508; SB VII 157.
670. Pb2As2S5 SB III 508.
Pbl4As7S,4 SB VI 131.
Pb3As„Se SB III 508.
Pb4As*S, SB III 508; SB VI 131.
Pb-Sb + As SB VI 193.
PbZn[(V, As)Ot]OH, ареокесп SB VII 151.
PdAs„ SBI 781; F (1942); О (1950).
? [PdBr, (CII3)3As], SB VI (OG12:201), 221; F (1946).
? [PdClBr(CH3)3As]., SB VI 222.
? [PdCl„(CH,)3As]/ SB VI (6>G1,:203), 220; F (1946).
680. AsPr " SB V 14."
Pt.As„, сперрилит, SB I (C2 : 153) 125, 217, 780, 781; SB II 274;
F (1942); О (1950).
As2Re SB VII 94.
AsS, реальгар, SB III 255; БМКД (1938); О (1950).
As,S3, аурипишент БМКД (1.938); HRF (1938); Б (1917).
As-Sb* SB VI 192; SB VII 211; О (1950).
SbAs04 HRF (1938).
Sb, арсенит HRF (1938).
As.,Se, SB II 324.
As-Sn SB III 649; О (1950).
690. AsSn SB III 649; F (1942).
As,Sn8 SB III 6i9, 650.
TLAs04 12WO, 4H20 SB VII 149.
YAs04 SB I'll (Я03: 91) 429; F (1946); О (1950).
ZnAs. SB III 311; О (1950).
Zn3As., SBI 786; SB II 325; SB III (£5,: 63) 353; F (1946); О (1949).
Zn(As04)., 8Н..0 HRF (1938).
ZnAs04Zn"OH," адамип SB IV 167; SBV (//27:17) 95; SB VI (#27:22)
109, HO; БМКД (1939).
Znl4 (As04), OH 12HaO SB II 467.
§ 28C. Золото Au
Au SB I (Л1 : 14) 38, 748; SB II 163-165, 167, 168, 615-623, 627,
630-632, 679, 701, 702, 704, 718, 719, 745-747; SB III 181,
182, 184, 186, 187, 192, 193; SB IV 77, 89, 227; SB VI 42, 43;
SB VII 45, 48, 49, 52; HRF (19?8); БМКД (1939); F (1942);
О (1950).
700. Ац-Ве J. I. M. 71, 213 (1945); С. Л. 39, 3198 (1945); 41,4021(1947;.
701. AuBe. SB III 330.
834 702. Au-Bi-739. Au-Sb lгл. XI
702Au-Bi J.I.M. 71, 213 (1945); C. A. 39, 3198 (1945).
Au2Bi SB И 747; SB III (£15:21) 315; F (1942); О (1950).
AuCN HRF (1938); Ж. Ф. X. XIX, 433, 519 (1945); Acta XX, 253
(1945); О (1950).
Au(C,H7)2CN SB VII (OE1,: 216) 232.
Au-Cd SB I 561; SB II 11, 701, 702; Ch Z. I, 206 (1944); J. I. M.
71, 213 (1945); С A. 39, 3198 (1945).
Au3Cd SB II 701.
AuCd SB II 11, 701, 702; F (1942).
AuCd, SB II 701, 702.
HO. CsAuCl2CsAuCl4 SB III 513; SB V 103; SB VI (A'7,: 33) 126; F (1946);
Ch. E. N. 26, 2970 (1947); О (1950).
Au-Cu SB I 502, 505; SB HI 570; SB VI 165; SB VII 184.
CuAu, SB I 508.
AuCu SB I (/Л0:485)*) 505, 507; SB II 615-618, 620-622; Ch.
Z. 1, 165 (1942); F (1946).
Au2Cu, SB II 615, 620-622.
AuCu3 SB I (L12 :486) 505, 507, 508; SB II 615, 619-622; Ch. Ph. 8,
860 (1940); Ж. Т. Ф. XI, 711 (1941); С. А. 39, 852 (1945).
Au-Pe SB III 577.
Au-Ga J. 1. M. 71, 213 (1945); С A. 39, 3198 (1945); Ch. Z.
1260 (J947).
AuGa2 SB V 54; F (1942).
Au-Ge J.I.M. 71, 213 (1945); С A. 39, 3198 (1945).
720.Au-Hg SB I 561; SB III 611; ИАН (x) 683 (1941); J. I. M. 71.
213 (1945); С. А. 39, 3198 (1945).
Au-In SB VI 163, 165; J. I. M. 71, 213 (1945); С. А. 39, 319K
(1945); Ch. Z. 1260 (1947).
Auln2 SB V 54; F (1942).
KAuBr42H20 SB IV (Я4 : 50) 180; О (J950).
KAu(CN)2 HRF (1938).
Au-La Z. An., 252, 62 (1943); Ch. Z. I, 518 (1945).
AuMg SB IV '52:6) 101; F (1942).
Au Mn SB III 578.
Au-Na SB VI 157.
Au2Na SB V 54, 55; SB VI 157; F (1942); ДАН 40, 262 (1943).
730. AuNa2 SB V 55.
NaAu(CN)2 Труды ИК (4), 179 (1948).
Au-Ni SB IV 227.
Au-Pb SB III 612; J. I. M. 71, 213 (1945); С. А. 39, 3198
(1945).
Au2Pb SB III (C15:21) 612; F (1942); ДАН 40, 262 (1943).
AuPb2 SB III 612; С. А. 36, 2764 (1941); Z. Met. 36, 218 (1943);
С. А. 39, 1095 (1945).
AuTl26Pb(S, Tl)2, нагиагит SB III 391; F (1946).
AuPd SB I 519.
Au-Rh SB III 578.
739. Au-Sb J.I.M. 71, 213 (1945); С A. 39, 3198 (1945); О (1950).
*) В работе F (194">) в разных местах указаны отношения с/в для Аи(д|, рапные
0,935 и 1,32 (тип L10 и обоих случаях).
§ 287] 7i°^AuSb^:_7§i^(M4-<?Lk_- 8^ъ
740.AnSb2 SB I 780; SB II 271, 745; F (1942); О (1950).
An-Si SB III 567.
Au-Sn SB I 562; SB II 718, 719; J. I. M. 71, 213 (1945); C. A. 39,
3198 (1945).
AuSn SB I 490; SB II 718, 719.
AuSiu SB II 718, 719.
AuSn4 SB II 718, 719.
AuTe2, калавсрит, креннерит SB III (C34 :30) 315; SB lVi<.'46:15)
115; О (1950).
Au-Tli Z. El. 49, 487 (19'i3).
Au-Zn SB VI 161; J. I. M. 71, 213 (1945); С. А. 39, 3198 (1945).
AuZn SB I (Б2: 76), (L20:488) 147, 559, 560; F (1942)
750.Au,Zne SB I (£8.,: 499) 560.
§ 287. Бор В
В SB II 170, 795- 797; SB IV 126; J. Am. 66, 1924 (1943); О (1950).
Бориды, см. также Z. El. 49, 254 (1943); О (1950).
BaB, SB И 38, 308; F (1942); О (1950).
Борат Ва «ВаДВОД,,» HRF (1938).
Боровольфрамат Ва HRF (1938).
BaB40, Ch. S. 742 (1941).
ВеВ2Н8 J. Am. 68, 312 (1946).
CBe SB II 309.
Ве,В030И SB II 78, 404.
760. ВВг, Ж. Ф. X. XX, 6 (1946); U (1900).
В4С SB HI 329; HRF (1938); rd. Л. 8, 1317 (1939); Ж. Ф. X. XVII,
326 (1943); J. Am. 66, 2115 (1943); ДАН 67, 819 (1947); Б (1947);
Ch. E. N. 26, 2970 (1947); О (1950).
B.,(C1I3)«1I2 Ch. E. N. 25, 2970 (1947).
СаВ, SB II 37, 308; SB III 334; F (1942); Б (1947); Ch. E. N, 25,
2970 (1947); О (1950).
Ca,(B.OH).Cl44HaO SB V 106.
Са8В1ЧОазС144Н„0, парагЕлгардит SB VI 130.
€aB„04 SB II 51, 405.
Ca.,B,Ou2H„0, нитшит SB VI 128.
СаЛ,Ои5Н.О, колеманит Z. Kr. (A) 103, 431 (19^1).
CaX°,i7H20, мейсргофферит SB VI 129.
770. II, (Mn, Mg, Zn) Ca (B03)2, рокгит SB V 86.
CaB.,Si„08, данбурит SB II 153, 558; Б (1947).
CaB0HSi04 SB II 581; SB IV 169, 202.
Борат кальция HRF (1938).
CaSn (B03)o SB II 403; SB III 406.
CeB6 SB"II 38, 208; F (1942); О (1950).
BC13 SB V 59; Ж. Ф. X. XX, 6 (1946); О (1950).
Co В SB III 619; 0(1950).
CoB SB III (527:12) 619.
Co,B SB III (£16:22) 619; Z. An. 242, 279 (1939); Z. Mot. 35, 218
" (1943); С A. 39, 1095 (1945): F (1946); О (1950).
780. Co (NH3),(BF4)3 SB III 475; F (1942); О (1950).
781.Co(NH,),(BF4)2 SB II f/>3; SB III 475; F (1942); О (1950).
836 782. БОРАТ ХРОМА-822. В№ [гл. XI
782. Борат хрома HRF (1938).
CsBF4 SB V 87.
Cs3H2BWl204onH20 SB III 466.
Борат меди HRF (1938).
CuB,04CuCUH20, бандилит SB VI 132; F (1946).
ErB, SB" II 38, 308; F (1942); О (1950).
BF3 SB V 58; Ж. Ф. X. XX, 6 (1946); О (1950).
NH4BF4 SB III (Я0,:90) 419; SB IV 164; HRF (1938); О (1950).
790. BF32H20 SB III 324".
BF3 -Me,0 J. Am 67, 339 (1945); G. A. 39, 1794 (1945).
N0BF4 SB V 88.
B-Fe SB I 573; SB III 619; О (1950).
FcB SB II 7, 241, 795, 796; SB III (527:12) 619; Б. (1947);
О (1950).
Fe2B = Fe4B2 SB I (6*17:494) 573; SB II 283, 795, 796; SB III
(C16:22) 619; Ж. Т. Ф. XI, 607 (1941); Z. Met. 35, 218 (1943);
С A. 39, 1095 (1945); F (1946); О (1950).
Fe -FeS-FeB С. А. 39, 2448 (1945).
В,„НЛ Ж. Ф. X. XV, 459(1941); Acta XIV, 547 (1941); ДАН 35, 200
(1942); Ch. S. 250 (1943); С. А. 37, 5919 (1943); Ch. E. N. 25,
2970 (1947).
B,H6 SB I (£4^240) 257; Z. ph. Ch. (B) 53, 85 (1943); Ch. S. 250
(19431; С. А. 37, 5919 (1943); J. Am. 69, 1327 (1947); 70, 115
(1948); О (1950).
B2H,2NH3 SB VI 129.
800.B4H10 SB VI 78; О (1950).
B6H, SB IV 280; J. Am. 70, 115 (1948).
BsHn SB VI 78; О (1950).
Bl0Hn SB II 327; J. Am. 70, 115 (1948); О (1950).
InBO, SB II 403.
BJ5 Ж. Ф. X. XX, 6 (1946).
K6BWl204018H20 SB IV 213.
KBF4 SB II 486; SB IV 164; SB VI 97; О (1950).
K2 [B2H4(0H)2] SB III 388.
K2(B2H.) SB III 388.
810. КН2(Н,0)„В6010 = Ко05В 0,811.0 SB V (КЗ,: 23) 108; SB VI 128;
Ph. R". 53, 202^ 203 (1938).
K206B203 SB IV 198.
2KBO = K,B204 SB II 405; SB III 389; SB V (F5lt: 12) 74.
K2B40. • 5H20 HRF (1938).
LaB, SB II 38, 171, 308; F (1942); О (1950).
LaBO, SB II 403.
LiBH4 J. Am. 69, 1231 (1947).
Mg,B1402eCl2, борацит SB 1477; SB II 407; SB III 511; БМКД (1939).
Mg, борат HRF (1938).
H (Mg, Mn) BOs = 2MgOB.03H,0, индсрашарит, камссллит SB II405;
SB VII 134.
S20. Mn, борат HRF (1938).
MnB SB IV 101; О (1950).
S22. BN SB I (B12 :95) 139; Z. An. 244, 146 (1940); ДАН 28, 630 (1940);
О (1950).
§ 288] ^^ 823^B,,N;,HB-867. BcBaF4 _ 837
823. B3N3H, SB VI 129; О (1950).
B„NH7 SB VI 129.
NH4, борат HRF (1938).
NaB2 SB II 309.
NaBF4 SB IV 164; SB V 87.
NaOHBF3 SB V 58.
NaBH4 J. Am. 69, 987 (1947); С. А. 41, 4348 (1947).
830. B203 - Na20 SB III 291.
4B203Na,0 SB III 291.
3B,0,Na",0 SB III 291.
Na2B107 = 2B.,03Na„0 SB II 406; SB III 291.
Na„B407lHO" SB II 406, 597; SB III 389.
NalB4075H2'0 SB III 389; HRF (1938).
Na„B40,10H„0 SB III 389; HRF (1938).
NaB0„ = «Na, (Bs0> SB III 389; SB V 72 - 74; SB VI (F518:18) 90;
HRF (1938).
NaBOa8H„0 SB III 389; HRF (1938).
NdB, SB II 38, 308; F (1942); О (1950).
840.В-Ш SB III 619; О (1950).
NiB SB III 619.
Ni2B SB III (C16 : 22) 619; Ж. Т. Ф. XI, 607 (1941); Z. Met. 35, 218
(1943); С A. 39, 1095 (1945); F (1946); О (1950).
Ni(NH3),(BF4)2 SB II 503; F (1942); О (1950).
B2Oa Ch. S. 742 (1941).
B203 SB II 310; SB III 337; SB IV 218; SB VII 161; О (1950).
B406 Ch. S. 742 (1941).
H.BO, SB I 334; SB III (G5,:87) 411; HRF (1938); О (1950).
BP04 SB III (#0,:92) 426; F (1946); О (1950).
PrB, SB II 38, 308; F (1942); О (1950).
850. RbBF4 SB III (H02: 90) 419; О (1950).
ScBO, SB II 403.
SrB, SB II 38, 308; F (1942); О (1950).
TaB2 SB II 309.
Ti-B Z. An 259, 1 (1949).
T1BF4 SB V 87.
ThB4 SB II 797.
ThB, SB II 38, 308, 797; F (1942); О (1950).
(NH4)6BW1„04026H20 SB III 466; F (1946).
Hs [B (W3010)4]5H20 SB III (//4ie: 115) 465.
860. H,BW1204014H,0 SB VII 148.
HsBWxO^lHaO SB III 466.
YB, SB II 38, 308; F (1942); О (1950).
JfBO, SB II 403.
IbB, SB II 38, 308; F (1942); О (1950).
ZrB2 SB IV 122; О (1950).
§ 288. Барий Ва
Ba SB I 749; SB II 168; HRF (1938); Z. An. 248, 155 (1941);
F (1942); Ch. Z. I, 978 (1942); С. Л. 37, 2634 (1943); О (1950).
867. BeFaF4 SB II 486; О (1950).
838 = = ^g^BaBiOgBr- 912. BaJJO,), HjO [гл. XI
868.BaBi02Br Z. An. 248, 135 (1941).
BaBiO,Cl Z. An. 248, 135 (1941).
870.Ba-B!-O-J Z. An. 250, 173 (1942); Ch. Z. I, 488 (1943).
BaBr2 SB VII (C23:8) 80; Z, An. 241, 248(1939); (C 23 тип);
О (1950).
BaCa SB I (СИ: 741) 782; SB II 275; F (1946); О (1950).
Ba(CN)2 HRF (1938).
Ba(CNS)2 2HO HRF (1938).
BaCO, SB I (G0a: 297) 325. 339; SB II 391, 397; SB III (G02:85)
407; SB V 79; HRF (1938); О (1950).
Ва-ацетат моногидрат HRF (1938k
Ва-калсршшат HRF (1938).
Ва-оксалат HRF (1938).
Ва-формват HRF (1938).
880. Ва-фмшл-сульфоиат HRF (1938).
Ва-тартрат HRF (1938).
Ва-цнтрат HRF (1938).
Ba-Ca Z. An. 248, 155 (1941); Ch. Z. 1, 978 (1942); С А. 37,
2634 (1913).
CaBa(CO,), SB II 393, 394.
BaCdCI44H,0 SB IV 181.
BaO03 SB III (Я2,:68) 378; F (1942); О (1950).
Bad., SB VII (£23:8) 80; HRF (1938); Z. An. 241, 248 (1939);
ДАН 60, 1169 (1948); О (1950).
BaCUHO SB V 49; HRF (1938); С. А. 41, 19 (1947).
BaCU(Nfi.CILCOOH), SB II 876.
890. Ba(C104)33H26 SB III 450; HRF (1938).
Ba(C103)2 HRF (1938).
Ba(C103)2 HO HRF (1938).
Ba3 [Co(NO.,)e]2 SB VII 152; О (1950).
К„ВаСоШ02)6 SB II 486; F (1942).
BaCr04 SB II 445; HRF (1938).
BaCu[Fe(CN)J SB VII 155.
BaF, SB I (CI : 150) 187; SB II 248, 249; HRF (1938); F (1942);
О (1950).
Ba2[Fc(CN)e] SB VII 155.
BaC03—Fe,0, C. A. 41, 22, 24 (1947).
900. BaFe04 SB VI 97.
BaFc204 SB II 477.
Ba06Fi'.,03 SB VI 75.
BaK, [Fe (CN),] SB VII 155.
K.,Ba" [Fc (N02)„] SB III 483; F (1942).
(NH4)oBa[Fe(NO..)6] SB III 483; F (1942).
Tl,BaFe(NO,), 'SB III 483; F (1942).
BaGeV, Б (1947).
BaTiOe.O, SB II 467.
Ball, SB III (C29 : 25) 307; О (1950).
9l0.BaJ2 SB VII (C23:8) 80; О (1950).
. BaJ26H20 SB II 102, 499; О (1950).
912.Ba(J03),H20 HRF (1938).
§_288J ^ = __^ .913^%а1^У08)^-952. BaSeQ4 839
913. «Ba,Ir(N02)e» (в действительности Ba3 [Ir(NO,)„], SB III 482*).
ILBaNi (NO,), SB II 496; F (1942).
BaMg., Ch. Z. И 270 (1944).
BaF2 - MgFa SB III 276.
BaBfn04 SB VI 97; HRF (1938).
Ba(Mn04), HRF (1938).
BaMo04 SB I (Я4-.349) 386, 790; SB II 83; Ch. Z. 1, 270(1944);
С A. 39, 2021 (1945); F (1946); О (1950).
920. Ba3N2 SB III 353.
Ba(N,)2 SB II 376; О (1950).
BaNH SB III 387; F (1942).
Ba(N03), SB I (G2,: 305) 313, 787; SB II 74, 385-387; HRF (1938);
F (1942); О (1950).
Ba(NO,)3 H20 HRF (1938).
Ba(N03)2-BaS04 SB III 419.
NaBa(Ti, Fe)tSi401§ SB II 585.
BaNaPOt J. Am. 63, 2533 (1941); F(1946).
BaNi(CN)44H20 SB III 458; SB VI (#422:23) 113; 0(1950).
Ba2Ni(N02)& SB III (Лх: 121) 480; F (1942); О (1950).
930. Ba-0 Ch. Z. 1, 306 (1942).
BaO SB I (51: 74) 119: SB II169, 217; HRF (1938); F (1942); О (1950).
Ba02 SB III 292; HRF (1938); Ж. Ф. X. XX 1403 (1946); F (1946)
Б. (1947); О (1950).
BaO x H20 SB II 217, 256.
Ba(0H)2 SB II 217.
Ba(OH)28H20 HRF (1938).
Ba,P2 SB III 353.
Ba3(P04)2 HRF (1938); A. Cr. 1, 263 (1948).
Ba10(PO4),(OH),, барий-гидроксил-апатит SB VI 105.
Ba, фосфит "HRF (1938).
940. Ba (HaP02)2 H20 HRF (1938).
BaPd(CN)44H20 SB III 457; SB VI i#422:24) 113; О (1950).
BaPrO, SB III (£2,: 68) 378; F (1942); О (1950).
BaPt(CN)44H,0 SB III 458; SB VI (#422:24), 113; HRF (1938);
О (1950).
«Ba3Rh(N02)e»**) = Ba3[Rh(N02)e]2 SB III 483; F (1942).
BaS SB I (51:74) 125, 127; HRF (1938); F (1942); ИАН(х) стр. 382
(1944); С А. 39, 3988 (1945); О (1950).
BaS, SB IV (DO,,: 18) 123; О (1950).
BaS04, барит SB I (Я2 :344) 382; SB II 432, 438, 439, 442; SB V 90;
SB VI 97; HRF (1938); О (1950).
BaS03 HRF (1938).
BaS203H20 HRF (1938).
950. Ba[Sb(0H),]22H,0 SB IV 193.
BaSe SB I (51:74) 135; F (1942); О (1950).
952.BaSe04 SB VI 97.
*) В SB III 4S2—483 реферируются работы но структуре Ba3Rh(N02)e и
BasJr(NOi)e (а также Pb3Rh(N02)e)." Эти же формулы воспроизводятся в регистре
SB III. Эти формулы неверны; им не соответствуют также приводимые в SB III
аналитические данные о составе иридиевой соли: BaO : Jr303: N30„ = 3,019 : l : 5,914,
которому отвечала бы формула Ba, [Jr(NOa),]3.
**) См. выше примечание к «Ba»Jr(NOje».
840 J53, BaSiF«-992. (BeSiO.), (FeJIIn^S _ ^ jra .XI
953.BaSiF, HRF (1938).
BaaSiWl204024H20 SB IV 213.
BaTi(Si03)3 SB I 328; SB II 128, 467, 526.
BaSn03 P. Ph. 58, 133 (1946); C. A. 40, 3957 (1946); О (1950).
Ba-Sr Z. An. 248 155 (1941); Ch. Z. 1, 978 (1942); С. А. 37,
2634 (1943).
BaO-SrO SB III 243.
BaTe SB I (51:74) 135, 766; SB II 231; F (1942); О (1950).
960.Ва'ГеО4 SB VI 97.
BaTh03 SB III (E2l: 68) 378; F (1942); О (1950).
BafiFe HRF (1938).
BaTi03 SB I (G5:303) 333; F (1942); N. 155, 484 (1945); N 156,
177 (1945); P. Ph. 58, 133 (1946); С. А. 40, 3957 (1946); F (1946);
A. Cr. 1, 330 (1948); О (1950).
Ba (UO), (P04)210H20 SB VI 106.
BaW04 SB I (Я4:349) 386; SB II 83, 452, 453; Ch. Z. 1, 270
(1944); С. А. 39, 2021 (1945); N. 157, 548 (1946); F (1946);
О (1950).
BaZrO, SB III (£2^68) 378; F (1942); P. Ph. 58, 133 (1946);
С A. 40, 3957 (1946); О (1950).
§ 389. Бериллий Be
Be SB I (43:19) 40; SB II 168, 664, 666, 722; SB III 185, 193,
194; SB IV 79; SB VII 52; HRF (1938); О (1950).
Be2C SB III (CI: 20) 309; F (1942); О (1949).
BeC034H20 SB VII 133.
970BeO3Be(CH3COO), SB I (041:676); SB HI (04. : 666) 716; N. 152,
447 (1943). "
BeO4Ct.Hu SB VII 235.
(Ca, Na)2BeSi,(0, OH, F)„ лейкофанит SB II 543; F (1946).
(Ca, Na)2Be(Si, А1)Д0, F), мелифанит SB II 542.
BeO-CaO SB III 241.
Be (Ca, Mn)Si04 SB I 407; SB III 425.
(Na, Ca)2Be(Sl, Al)2 (0> OH, F), SB II 543, 549.
BeCa„Si207 SB II 542, 543.
Be-Go SB IV 240.
BeCo SB IV (52:6) 240; F (1942).
980.Co(NH4)2 (BeF4)2 6H20 С. А. 39, 3988 (1945).
Be-Cr SB IV 240.
BeXr SB IV (C14:8) 240.
Bc-Cu SB I (L20:488) 529; SB III 588; SB IV 235; HRF (1938).
Bc3Cu SB I 529.
Be2Cu SB III 588; F (1942); ДАН 40, 262 (1943).
BcF„ ВеР2-стекло SB II 248; SB III 274; О (1950).
(NH4)2BeF4 SB III (Я1в:94) 436; О (1950).
Be-Fe SB III 612.
Be.Fe SB III 613; Ж. Т. Ф. XI, 607 (1941).
Be,Fe SB III 613; Ж. Т. Ф. XI, 607 (1941); ДАН 40, 262 (1943).
990.4BeOFe203 SB III 344.
BeFe204 SB II 477.
992. (BeSi03),(Fe, Mn)4S SB II 152, 562, 566.
I 2891 993. (BcSi04)3 (Mn, Pe, Zn)4S -1039. Bc.,Si04 841
993. (BcSi04)3 (Mn, Fc, Zn)4 S SB I 470.
BcGeO, SB II 468; SB IV 173.
Be-Ir SB IV 240.
K2BcF4 SB III 437.
K.Ni (BeF4) S046H.,0 С. А. 39, 3988 (1945).
Li„BcF4 SB I (#1, :356) 423; 0 (1950).
100O.BeF2-MgF2 Ch. Z. 1, 1114(1942).
BcO-MgO SB III 241.
Be-Mn SB IV 240.
Be,Mn SB IV Cl'i:8\ 240.
(BeSi04)3Mn4S SB II 152, 562, 566.
Be-Mo SB IV 210.
Be2Mo SB IV «714:8) 240.
Be,N, SB II (Db,: 49) 353; F (1942); О (1950).
BeF.-NaF ЖОХ 14, 385 (1944).
NaBeF, ЖОХ 14, 385 (1944).
1010.Na2BcP4 SB VII 118; ЖОХ 14, 385 (1944); О (1950).
Be4NaSb07 SB II 339.
4BeONaSb03, сведенборгит SB III (£9,: 69) 381; Б (1947).
NaBe4Sb07 SB III 382.
NaBe(OH)Si307 SB II 582, 583'.
HNaBeSi308 SB III (54.: 153) 533.
Be-Ni SB III 617.
BenNi6 SB III 617.
BeNi SB III 617; F (1942).
(NH4)2Nl(BeP4)26H,0 SB III 442; С. А. 39, 3988 (1945).
1020. BeO, броиеплит SB I (54:79) 40, 115; SB II 215, 268, 269, 320;
SB III 240; HRF (1938); БМКД (1939); Z. An. 244, 146 (1940);
NW 29, 336 (1941); С A. 36, 696 (1942); О (1950).
Be(OH)2 SB I 253, 254; SB II 215, 255.
BeO x H20 SB II 215.
Be-Os SB IV 240.
Be,P2 SB III (Z>53:49) 353; F (1942); О (1950).
Be-Pd SB IV 240.
BesPd SB III 330.
BePd SB IV (52:6) 240; F (1942).
Be-Pt SB IV 240.
Be21Pt6 SB III 617.
1030. Be-Re SB IV 240.
BeJte SB IV (C14 : 8) 240.
Be - Kb. SB IV 240.
Be-Ru SB IV 240.
BeS SB I (B3:ll) 126; Z. An. 244, 146 (1940); F (1942); ИАН(х)
стр. 382 (1944); С. А. 39, 3988 (1945); О (1950).
BeS04 HRF (1938).
BeS044H20 SB I 788; SB II 91, 427, 428; HRF (1938); Б (1947);
О (1950).
BeSe SB I (53:77) 134; Z. An. 244, 146 (1940); F (1942); О (1950).
Be^iW1.Ow31H.O SB III 462.
1039. Be,,S10;, фенакит SB I (Я13:356) 402; SB II 517: SB IV 174;
"БМКД (1939); О (1950).
842 1040. H2Be4Si2O,-1083. Bi-Fe [гл. XI
1040. H2Be4Si20, SB II 539.
BeSiO, SB I 326.
Be-Та SB IV 240.
BeTc SB I (53:77) 134; Z. An. 344, 146(1940); F (1942); О (1950).
Be Ti SB IV 240; Z. An. 269, 1 (1949).
BeoTi SB IV (C15-.8) 240; F (1942); Z. An. 259, 1 (1949).
Be-V SB IV 240.
Be,V SB IV (C14:8) 240; ДАН 40, 262 (1943).
Be-W SB IV 240.
Be,W SB IV (£14:8) 240; ДАН 40, 262(1943).
1050.(NH4)2Zn(BcF1).,6H2O С. А. 39, 3988(1945).
Be-Zr SB IV 240.
Be3Zr207 SB II 268.
§ 290. Висмут Bi
Bi SB I (.47:27) 58; SB II 185, 186, 203, 204, 294, 745-749, 751,
754-756; SB III 184, 210, 212; SB IV 78; SB VI 50; HRF (1938);
О (1950).
BiBr, Ж. Ф. X. XX, 6 (1946).
BiOBr SB III (£0,:64) 370; HRF (1938); Z. An. 342, 46 (1939);
F (1946).
Bi(C(H(),Cl. SB II 819.
Bi-ацетат HRF (1938).
Bi-бензоат HRF (1938).
Bi-еубкарбонат Bi203C02H..O HRF (1938).
1060. Bi-амионийцитрат HRF (1938).
Bi-лавтат HRF (1938).
Р-нафтол HRF (1938).
Bi-оксалаг HRF (1938).
Bi-салпцилат HRF (1938).
Ca-Bi-O-Br Z. An. 248, 121 (1941).
Ca-Bi-O-Cl Z. An. 248, 121 (1941).
Bi-Cd SB IV 226; SB V 134.
Bi20,CdBr»CdO=Cd,Bi20,Br> SB VII 159, 160; Z. An. 242,46(1939);
F (1946).
Bi-Ce SB V 45.
1070.Bid, Ж. Ф. X. XX, 6 (1946).
BiOCl SB III (£0,: 64) 370; HRF (1938); Z. An. 242, 46 (1939);
F (1946).
BiO(OH, CI) SB III 371.
BiCl,3 [CS (NH,)2] SB I 639.
BiCo(CN),6[Sfc(NH2)2] SB I 640.
(BiO).CrO, HRF (1938).
BiCr63 G. A. 41, 6102 (1947).
Cs8Bi(N02)„ SB IV, 193; F (1942).
Cs2Li Bi (N02), SB IV 193; F (1942).
Cs.,NaBi(NO,)e SB IV 193; F (1942).
1080. CiiBiS, SB III (F5,: 77) 393.
CuBi»S4, эмплектит SB VII 157; БМКД (1938).
BiF3" SB II 22, 290; Ж. Ф. X. XX, 6 (1946); Б (1947); О (1950).
1083. Bi-Fe SB I 564.
$ 290] 1084. Bi-Fc-Si-1128. Bi,Ni 843
1084. Bi-Fe-Si Ch. Z. I, 13 (1942).
Bi-Ge Z. An. 244, 205 (1940); О (1950).
Biln ДАН 69, 899 (1948).
BiJ3 SB II 25, 294, 295; Ж. Ф. X. XX, 6 (1946); Б (1947); О (1950).
BiOJ SB III (£0,:64) 370; Z. An. 242, 46 (1939); F (1946).
KBi, SB II 271; F (1942); ДАН 40, 262 (1943); О (1950).
1090. BiK3 SB V (£>018:6) 60; О (1950).
K2Li Bi(NO,)e SB IV 193; F (1942).
BiK(Mo04)3" Ch. Z. 1, 271 (1944); С. А. 39, 2019 (1945); F (1946).
KaNaBi(i\02)e SB IV 193; F (1942).
BiKS2 Rec. 63, 32 (1944); С A. 39, 1583 (1945); О (1950).
BiLa SB V 45; F (1942); О (1950).
Bi-Li SB III 637.
ВШ SB III 638; F (1946); О (1950).
BiLi3 SB III 637; О (1950).
BiLi(Mo04)., Ch. Z. 1, 271 (1944); С. А. 39, 2019 (1945); F (1946).
1100.Bi3LiO4Brs:=2Bi2Os2BiOBr-2LiBr SB VII 159, 160; Z. An. 242,
41 (1939); F (1946).
Bi3Li04Cl2 = 2Bi20,2BiOCl 2LICI SB VI 131; SB VII 159, 160; Z.
An. 242, 46 (1939); F (1946).
Bi3Li04J, = 2BL032BiOJ2LiJ SB VII 159, 160; Z. An. 242, 46 (1939);
F (1*916).
(NH4),LiBi(NOo)e SB IV 193; F (1942).
Rb2LiBi(N02)e " SB IV 193; F (1942).
TI8LiBi(NO„), SB IV 193; F(1942).
BLMg, SB III (£>52:48) 356; 0(1950).
Bi-Mn SB VI 191; SB VII 210.
BioMn SB VI 192; F(1946).
BiMu SB VII {B8 :4) 210.
1110. BiMn, SB VI 191.
(Bi306H2)Mn04 Z. An. 248, 77 (1941^; Ch. Z. 1, 1115 (1942).
(Bi202OH) (Mn04) l,5H,0 Z. An. 248, 77(1941); Ch. Z. 1, 1115(1942).
BL03-Mo03 Ch. Z. I, 1363 (1944).
BiON03H20 HRF (1938).
BiNa SB II 237; F(1942); F(1946); 0(1950).
NasBi SB V {DO,,: 16) 60; 0(1950).
BiNa(Mo04)2 Ch. Z. 1, 271 (1944); С. А. 39, 2019 (1945); F(1946).
BiNa5(Mo04)4 Ch. Z. 1, 271 (1941); С. А. 39, 2019 (1945).
(NH4)2NaBi(N02)6 SB IV 193; F(1942).
1120. Rb2NaBi(N02)„ SB IV 193; F(1942).
BiNa5(W04)4 Ch. Z. 1, 271 (1944); G. A. 39, 2019 (1945).
BLNa04Br2 = 2BL032BiOBr2NaBr SB VII 159, 160; Z. An. 242, 46
(1939); F(1946).
Bi3Na04Cl2 = 2Bi2032BiOCI2NaCI SB VII 159, 160; Z. An. 242, 46
(1939); F (1946).
Bi3Na04J,= 2Bi20,2BiOJ2NaJ SB VII 159, 160; Z. An. 243, 43
(1939); F(1946).
BiNa03 HRF (1938).
BiNaS2 Rec. 63, 32 (1944); С A. 39, 1583 (1915); О (1950).
BiNi SB II 749.
H28.Bi,Ni SB II 749.
844
1129. Bi203-1169. С
[гл. XI
1129. Bi,0„ бисмит SB I 262; SB II 354; SB V (Z)5,„: 9) 69- 71; SB YII
104, 106; HRF (1938); БМКД (1938); О (1950).'
U30.Bi,O3xH2O SB II 354.
Bi204 Н20, тетраоксид HRF (1938).
Bi(0H)3 SB II 354; HRF (1938).
Bi, осмиат HRF (1938).
BiP04 HRF (1938).
Bi-Pb SB IV 226.
BLPb SB II 755; О (1950).
BiF, -PbF2 Z.An. 242, 79 (1939).
В^РЬД, SB VI 130.
BiPr SB V 45; F (1942); О (1950).
1140.Bi.Pt Ch Z. I 980(1944).
Bi2S„ висмутинит SB III (Z)58: 51) 349; SB VI 86; О (1950).
Bi,(S04)3 HRF (1938).
Bi-Sb SB III 652.
Bi-Se SB II 203.
BiaSe3 SB II 203, 324.
Bi-Si SB III 567; О (1950).
Bi4Si3Oia, эилитин SB I 792; SB II 122, 522; БМКД (1939).
Bi-Sn SB IV 226.
Bi-Sr-0 - J Z. An. 250, 173 (1942); Ch. Z. I 488 (1943).
H50.BiTa2O,F SB VII 24, 126.
Bi-Te SB VII 109, 110.
Bi„Te3 SB VII (C33:8) 109, 110.
Bi„Te,S, тетрадимит SB III (£33:28) 287; SB VI 86; БМКД (1939).
' Б. (1947).
BieTeS4, оруэтит SB III 441; SB VI 86.
Bi3Te(S, Se), жозеит SB VI 86.
Bi4TeSe3, грюолингит SB VI 86.
Bi.,Ti3 SB II 204, 324.
Bi'-Tl SB I 573; SB III 648.
BiTl SB I (52:76) 141; F (1942); О (1950).
1160.BiTl7 SB III 648.
BiV04 SB IV 176.
(Bi, W)8.nOt., SB VII 108.
Bi.,03-W,0," Ch. Z. I, 1363 (1944).
BUVW03, (Bi, W)8-„0]2, русселит SB VI 75; F (1946).
§ 291. Бром Вг
Br, SB IV (AH : 3) 83; О (1950).
HBr SB II 207; SB III 229; Ж. Ф. X. XV, 597 (1941); 0(1950).
DBr 0(1950).
Бромиды J. Am. 63, 37 (1941); О (1950).
§ 292. Углерод С
1169. С алмаз, графит и др. SB I (A4: 21) 46; (А9:30) 47, 751; SB II
163, 174; 175-177; SB III 203-207; SB IV 79-81; SB VI
45, 231; SB VII 58, 59; БМКД (1938). HRF (1938); ДАН 28, 630
(1940); Ph. R. (2) 60, 617 (1941); F(1942); P. R. S. 181, 101
(1942); С A. 39, 449 (1945); Ch. Z. 1, 458 (1942); PJA 18, 251
(1943); 19, 189, 394 (1944); N. 153, 587 (1944); N. 154, 486 (1944);
I 292] _ 1170. КАРБИДЫ-1215. CF 845
С. А. 39, 460, 851 2018, 2683, 4538(1945); N. 166, 144 (1945); N. 166,
83 (1945); N. 158, 752 (1946); С. А. 41, 2297 (1947); О (1950).
1170. Карбиды, см. также Z. E1. 49, 254 (1943).
С (графит) «соли» SB VI 46; О (1950).
С8Вг, бромграфит Z. An. 245, 383 (1941); С. А. 39, 2917 (1945).
С„ (графит)ад1бСв) SB II 179, 181; О (1.950).
С8 (графит)С* (<;8€s) SB II 179. 180: О (1950).
С (графит)Г SB III 228; О (1950).
С (графит)Рс SB II 179; О (1950).
С (графпт)ГеС!., Z. An. 245, 121 (1940).
С1в (графит)К (С„К) SB II 179, 181; О (1950).
С8 (графит)К (С,Ю SB II 179, 180; О (1950)
ПЯО.Графит-алотная кислота SB VII 60; О (1950).
Графитовая кислота SB II 177, 178.
С„ (графит) Rb (C„Rb) SB II 179, 181; О (1950).
С8 (графит) Kb (C.ttb) SB II 179, 180; О (1950).
C„(HS04),, графитссрная кислота SB VII 61, 62; Acta XVIII, 351
(1943"); О (1950).
С (графит)-бисульфат SB VI 46, 47. 48.
CBr4 SB I (03:615) 641, 642; SB V 62, 63; F (1942); Cb. Z. 1,
330 (1942); С. А. 38. 3194 (1944); О (1950).
CHBr3 SB III 706.
CH,Br„ SB III 706.
C(CH„X)4, (X = U1, Br, J) SB V 156.
1190. C(CH.Br)4 SB V 156.
C,H,Br2 SB VI 231.
C2H3Br SB VI 231.
C„HBrs SB III 706.
C.H,Br. SB III 706.
CHCBr" SB VI 231.
CPBr, SB VI 82.
BrCN SB VII 130.
C(CN)3Br SB VI 228.
CBr(NH2)3, гуанидонии-бромдд SB III (024:661) 733, 734.
1200. CC14 SB III 328; SB IV 125; Ch. Z. 1, 330 (1942); О (1950).
153, 154.
CHmFnClD (m-i-n
CHCC1
SB VI
C(CH2C1)4 SB
C2CI,
C2CI4
C2HC13
CH2CI,
CH3Cf
CHC1,
210. CFCL
SB III '
SB III 1
SB III
SB III
SB III
SB III
SB VI
+ p=
231.
V 15C
710.
'06.
706.
706;
705.
706;
82.
= 4) SB 1
SB IV 27!
SB IV 275
(ch3)4ncu sb vii (сш1:215) 216, 231.
cicn sb vii 130.
C(NH,)3C1 SB Ц 880.
CF4 " SB III 327; SB IV 125; О (1950).
1215. CF SB III 228; О (1950).
846
1216. СН4-1265. (СООНЬ
[гл. XI
1216. СН4 SB I (01:613) 640; SB II (01:805) 819, 820; SB VII 238, 239.
C3HS SB I (.04,:240) 257; SB I (031:619) 658, 659.
C2H2, ацетилен SB II 846.
CH2 = CH„ этилен SB II 846; SB III (03a: 665) 708; SB VI 230.
1220.C(CH3)4 SB I (02:614) 641; О (1950).
С (C,H5)4 SB I (06 : 615) 616, 649-651; F (1946); 0 (1950).
C2HsHal SB III 708.
C2Be(R = Hal,CH3, OH), SB 1659-662.
C2HmHaln (m + n = 6) SB VII 241 - 243.
CJ4 SB I (03:614) 641; SB II 821; SB V 62, 63; F (1942); О (1950).
СШ3 SB I [OH : 617) 651; SB II805, 820, 821; С. A. 40, 4574 (1946).
CHJ,3SS SB II 386.
CH,J3S3 SB V 152.
С (CH„J)4 SB V 156.
1230.CH2J2 SB III 706; С А. 40, 4574 (1946).
C2HJ2 SB III (034:665) 709, 711.
C2H2J2 SB III 711; SB VI 231.
NHB3J (R = CH„) SB I (054 : 606).
JCN SB VII 129.
C(NH2)3J, гуанидоний иодид SB III (02. : 663) 734. 735; Б (1947).
N(CH3)3 SB IV 277.
NH3R (J) (Br), (CI), (R = CH3) SB I (0520:607) 634; SB II 812.
N (CH,)4 (J) (Br), (CI) SB I (0510: 607) 627 - 629.
NH,(CH3)C1 SB I (0521 :609) 610; J. Am. 68, 1405, 1970 (1940).
1240. CH3CN SB VII 240.
CH9N, SB III 712; 0(1950).
[C(NH)2]„ SB I 664.
NH3CsHnCl SB II 811.
(CHS)4NC104 SB II (Otf05: 804) 815.
(CN), SB III 731; SB VII 240; О (1950).
ОС (NH„)2 SB I (02t: 617) 618, 652; SB II 881; SB III (02,: 660) 731.
CO(NH2)(NHCH3) SB III (O23:660) 732.
(NH4)„ (COO). H„0 SB IV (04,: 262) 263, 264, 291.
С (N02)4 S"B I 642.
1250. С (CH2ON02)4 SB I (06:615) 616, 646; F (1946).
(CO),(NH)3 SB VI (187 : 213) 226.
CH3NO, SB IV 276.
(CO)2(NH)2(CH„)2 SB VI (08,: 212) 213, 253.
(NH2)2(CH„).2-H„S04 SB I 662.
HCHO SB VII 249.
CHsOH SB V 158; SB VI 234.
CH3OH SB III 712.
CH2CO SB VI 234.
CH,0» SB I 693.
1260. C„H4(C00H)2, фталевая кислота SB VII 252.
C,H5COOH (H II и D). SB VII 252.
С (CH2OCOCH3)4 SB I (05:615) 616, 647; F (1946).
C„H4(OH)2, резорцин SB VII 252, 253.
C(CH.OH)4, пентааритрит SB I (04:615), 616, 043 «46; SB V
(O4l2:140) 155; F (1946); 0 (1950).
1265. (COOH), SB I 662; В III (044:671)(04s :G72) 723.
§ 293] 1266. (СООН),2Н20 -1308. Ca10CQ3(P04)„ 847
1266.(СООН)22Н20 SB I 662; SB III (04,: 673) 724; SB IV 260-262,
290; SB VII 250, 253.
C,lf4 (OJ))2 SB VI 244.
(COOD),2D20 SB VII 250.
C(N02), SB I 642; SB VII 240.
1270. SC (NH2)2 SB I (6>22: 619) 653, 654; SB- II (02 : 805) 882.
(CH,),S SB VI 233.
(C,H5COS)2 SB II 813.
(C,H6CH2S)2 SB II 813.
(C,H6S)a SB II 803. 813.
(CaH5Se)2 SB II 803, 813.
(C,H,CH2Se)2 SB II 813.
CO SB II 13, 229; SB III 245; О (1950).
C02 SB I (C2:153) 256; SB II 270; SB III 293; Ch. Z. 1, 718
(1942); H. Ph. 14, 324 (1941); Б. (1947); 0(1950).
C30, SB III 341; О (1950).
1280. COS SB II 373; SB III 386; Z. Ph. Ch. (B) 51, 126 (1942); О (1950).
CS2 SB I 227; SB III 286; F (1946); О (1950).
§ 293. Кальций Си
Ca SB I 1,11:14) 41, 749; SB II 248; SB III (Л3:3) 195, 196;
HRF (1938); Z. An. 248, 155 (1941); F (1942); Ch. Z. 1, 978 (1942);
С. А. 37, 2634 (1943); F (1942); О (1950).
CaBr., SB VII (C35 : 8) 80; Z. An. 241, 244 (1939); HRF (1938).
CaBr„6H20 SB II 102, 499; О (1950).
CaC2 SB I (Cll : 175) 174, 218; (CH : 741) 782; SB II 275; HRF (1938);
Ж. Ф. X. XX, 1403 (1946); F (1946); О (19501.
CaCN2 SB I (F5:276) 290; SB VI 86; HRF (1938); О (1950).
Ca (CN)2 CaO SB VI 86.
Ca(CN)2 SB VI 86; HRF (1938).
Ca(CN)22HCN SB VI 86.
1290. Ca (CN)2 2NII3 SB VI 86.
Ca(0H)CN SB VI 86.
Ca(CNS), • 3H..0 HRF (1938).
CaCO,, кальцит, арагонит SB I (G\ : 295) 317; (G2: 297) 317, 318;
SB II388-390; SB III 406; HRF (1938); БМКД (1938): БМКД (1939);
С (1939); Z. Ph. 116. 194(1940). Б (1947); О (1050).
Ca(COO), SB III 727; HRF (1938).
<a(OCOH)2 SB I 657: HRF (1938).
Са-бензоат HRF (1938t.
Са-гликолат HRF (1938).
Са-гликолат гидрат HRF (1938).
Са-гликолат тригидрат HRF (1931-).
1300. Са-гиппурат HRF 1938).
Са-салицилат HRF (1938).
€а-тартрат HRF (1938).
Оа-фенолеульфонат HRF (1938).
Са-урат HRF <1938).
Са-цитрат HRF (1938).
Ca (CeF)2 (C03)2, еинхязит SB I 326.
Ca (CeF). (СОэ)„ паризит SB I 326; БМКД (1939).
1308. CaJOC<UP04);, подолит SB VII 146, 147.
348_ 1309. Calf,C03H»0 (Р04), -1349. Ca, (SiOJ, (F, ОН) [гл. XI
1309. Ca10C03H,0(P04)6 SB II 102, 455, 458.
4310.CaFCa4(Pb4)3; (Ca3F)2(P,C),(0,OH,F)24(Ca,C)4, франколит*) SB VI 104;
см. также штаффелит SB VII 146, 147.
Ca, (Oil), (P, C), 024 (Ca, C)4, даллит SB VI 104.
(Ce, Ca, JVa)6 [(Si, P)04]3 (F, ОН), бритолит SB VII 146.
(Ca, Ce, Na) (Nb, Ti) 03, лапарит SB II 334; БМКД (1938); F 11942).
EP C03CaCO, (R - Cc. La...) SB II 77, 401.
Ca„(Cl, F, 0, OH)a (S, Si, P, С), 024(Ca, С)4) эллестадит SB VI 104.
CaCI. SB II 246; SB III (C35:30) 278; HRF (19384.; Z. An. 241, 244
(1939); ИАН(х) 382 (1944); С. А. 39, 3988 (1945); С. А. 40, 4580
;1946); 0(1950).
CaCl,xH20 SB II 244; С. А. 40, 4580 (1946).
CaCl„H,0 HRF (1938).
CaCI, • 4H20 HRF (1938).
1320.CaCl,CaF,-CaFCl HRF (1938); F (1946).
CaCl26HsO SB II 102, 499; HRF (1938); Б. .1947): 0(1950).
CaCI Ca (OH), H20 SB III 279.
Ca(C10.), SB II 378.
Ca (OCI), ■ 2Ca (OH)., SB III 279.
Ca (OC1), 3H..0 SB III 279; F (19'i(>).
Ca(0Cl),4H,0 HRF (1938).
Ca5Cl(P04), SB II 99, 455, 458.
Ca6(P04)3 (F, Cl), апатит SB I 398; Б. (1947).
Группа апатита SB VII 145, 147.
1330.Са,.(РО4),Х8 SB VII 146.
CaCO,- СоСОэ SB IV 156.
Ca,Cr04J,0, SB II 449.
CaCr04 SB II 447; HRF (1938); F (1946).
CaCr04 H,0 SB II 448.
CaCr04 2H',0 SB II 448; HRF (1938).
CaCr20, "HRF (1938).
Ca,Cr, (Si04)3, уваровит SB I (H'6l : 366) 411; БМКД (1939); F(19'i2).
CaCu5 Z. An. 244, 17 (1940).
CaCu Fc(CN), SB VII 155.
1340. CaCu V04 ОН, фольбортит или тангеит SB VII 151.
CaF, SB I (Cl:150) 150, 185. 186; SB II 236. 244, 248, 320, 463,
"598; SB III 275; SB VII 81; HRF (1938); F (1942); Б (1947); О (1950).
Ca5F(P04)3, = (Ca,F)Ca4(P01)31 апатит SB II 99, 455-458; SB VII
146; БМКД (1939).
Ca5(P04)3F, остеолит SB VII 146.
Ca4F,(P04), SB II 463.
(Ca3F), (PO"4)e Ca4, фторанатит SB VI 104.
Группа апатита, в том числе моиит, цсйгит, сподиозит, апатит,
* SB VII 145, 147.
Cal(1(F,Cl),(P04)e; хлорапатит SB VII 146; БМКД (1939).
Ca (F, Cl, OH) (Ca, C)4 (P, С) (О, ОН, F)4, франколит SB V 107;
см. также пышс и SB VI 104.
1349. Ca5 (Si04)3 (P, OH) SB VII 146.
*) По Дана франколит (стр. 2Щ — разновидность обычного фтор-апатнта.
Карбонатные апатиты —курскит, ппл! еит, штаффелит (стр. 288).
§';293] 1350. Ca2Fe(CN),-1397. CaMg(P04)F 84d
1350. Ca2Fe(CN)6 SB VII 155.
Ca2Fe(CN),12IU> HRF (1938).
CaK2Fe(CN)6 SB VII 155; HRF (1938),
K2CaFe(NO.). SB III 483; F (1942).
Ca4MgFe20; SB II 321.
Ca2MgFe20. SB II 321.
(Fe, Mn, Ca)3 (P04)2, графтонит, реп ос о нт SB VII 142.
(Mn.Fe.CaMg) (Si03)8 SB I 328.
Ca(Mg, Fe^Si.O,, SB II 533.
CaMgFe, (Si03)4 ' SB II 537.
1360. H2 (Mg", Fe)6 Ca2 (Si03)8 SB II 134.
Ca^Jttn^Fe^P.O^OH),, варулит, мангавгидроксоапатит Ch. Z. 1,
1116 (1942).
(Ca, Na, Mn)3 (Fe, Ti, Zr) Si208F, розенбушит SB V 119.
(Na, Ca, Fe), ZrSie018 (OH, CI) SB II 584.
(HN4)2Ca Fe(NO,), SB III 483; F (1942).
4CaO Fe20,7H20 SB IV 172.
4CaOFe„0314H20 SB IV 172.
Ca,Fe2Of SB II 321.
CaFe2(FeOH) (Si04)2 SB IV 217; Ch. E. N. 25, 2970 (1947).
CaFe"(Fei"OH) (Si04)2 SB II 585.
1370. Ca8Fe2 (SiO,)s, андрадит SB I (#3,: 366) 411; БМКД (1939)
F (1942).
CaFe8(Si0»)i SB II 537.
CaFe(SiOs)2 SB I 327; SB II 528.
CaSi2Fe20, (FeOH) (?), лиеврит SB V 117.
Tl,CaFe(N02)e SB III 483; F (1942).
Ca"Ga2 NW 31, 145 (1943); С. А. 38, 9 (1944).
CaMg(Ge03)n SB II 467.
CaH„ SB "III (C29:25) 306; HRF (1938); О (1950).
Caln204 SB II 475; F (1946).
CaJ2 SB III (C6:20) 281; 0(1950).
1380. CaJ2 • 4H20 HRF (1938).
CaJ2 6H20 SB II 102. 499; О (1950).
Ca(J03)2 SB II 333; SB V 85; HRF (1938).
K„Ca(C03)2 SB II 396.
CaO-K20-P205-CO, SB VI 107.
CaS04 - MgS04 - I^SO; SB III 430.
K2Ca Ni(N02), SB II 496; SB IV (/24:65) 192; F (1942); О (1950).
CaO-K20-P203 SB VI 107.
CaKP04 SB VI 107; J. Am. 63, 2533 (1941).
Ca4KH7 (Si03)8 4'/2 H20, апофиллит SB I 331, 472.
1390. Ca8La2 (P04),02 SB II 464.
CaLaSi„08 SB II 555.
CaLi2 " Ch. Z. II 270 (1944).
CaMg2 SB V 53; HRF (1938); ДАН 40, 262 (1943).
CaMg(C03)2, доломит SB I (Git: 304) 317, 324; SB II 393; HRF
(1938); БМКД (1938); О (1950).
CaO-MgO SB III 241.
(Pb, Mn, Ca, Mg)3 (P04)„ каришшт SB VII 142.
1397. CaMg(P04)F, крифиолит SB VI 112.
8j0 13j)8.JX(P04)2,^-MgSi03-1441. 6(Ca,Na)(0, F)2 (Zr, Ti) 4Si02 [гл. X]
1398.Ca,(P04)2-MgSi03 G. A. 39, 1341 (1945).
Cas(P04),-MgSi04 Ch. Z. I, 1068 (1944).
1400.MnSiO3Mn4CaSi4O1, SB I 791.
3 MgSi03CaSi03 = Mg3Ca(Si03)4) грамматит SB I 794; БМКД (1939).
CaMg(Si03)„ диопсид SB I 327, 328; SB II 130, 134, 467, 527, 528;
БМКД (1938).
Ca2MgsSi,0,, SB II 535.
CaMgSiO, " SB I (Я12:355) 400, 406; О (19УЗ).
Ca10(Si04),(F, ОН),, эллестадит, SB VII 146.
Ca2Mg, (Si,Ou) (OH, F), тремолит SB VII 164.
Ca2Mg6Si702l SB II 535.
ЗСаО MgO 2SI0„ мервшгат N. 152, 35C (1943); С. A. 38, 9 (1944).
CaMg3Si4012 SB II 532.
1410. Ca3MgSl20, SB II 541.
H2Mg6Ca2 (Si03)8, тремолит (амфибол) SB II 131, 533, 535; Б (1947).
Ca(Mn04)2-4H20 HRF (1938).
(Na, H)2 (Ca, Mn)2 (Si03)3 SB I 328.
(Ca, Mn)2 NaHSi303, шизолит SB VII 163.
ЗСаОМпО 2SiO, N. 152, 356 (1943); С. А. 38, 9 (1944).
(Ca, Mn) Si03 SB I 328.
CaMnSiO, = (Ca, Mn)2 Si04, глаукохроит SB II 519; Z. Kr. 105, 273
(1944); С. А. 39, 5148 (1945).
(CaMn), F,(P04), Са4, манганапатит SB VI 104.
CaMo04 SB I (ff4:349) 385, 790; SB II 83; Ch. Z. I, 270
(1944); С A. 39, 2021 (1945); F (1946); О (1950).
1420.Ca.Nj SB III (Д5,: 49) 353; F (1942); О (1950).
CaNH SB III 387; F (1942).
Ca(N0,)2 SB I (G2,: 305) 313; SB II74, 385-387; HRF (1938); F (1942);
О (1950).
Ca (N0a)2 4H20 HRF (1938).
Ca(N02)2H20 HRF (1938).
Na2Ca(C03), SB II 396.
Ca —Na, глицерофосфат HRF (1938).
(Ca, Na), (OH). (P04), (Ca, C)4, дернит SB VI 104.
(Ca, K, Na), (0H)2 (P04), (Ca, C)4, левистонит SB VI 104.
CaNaNb20,F SB VI 132.
1430. CaTi03NaNb03, дизаналит SB VII 119-122.
CaNaP04 SB VI 107; J. Am. 63, 2533 (1941).
Ca(NaHPO,)., HRF (1938).
CaS04-Na2S04 J. Am. 63, 2533 (1941).
Na.,Ca2 (Si03)3 SB I 330.
Na4Ca(Si03)3 SB I 330.
(Na, H)2Ca2(Si03)3 SB I 328; SB II 528, 532.
Na2CaSi04 SB I 330, 407; SB II 158, 553-555.
CaeNa4Si2o08o24ILO = NaCa,Si60206H20 SB III 528.
Ca2Na(Ce, Ti)F(Si04)2 SB III 555, 559.
1440.Ca2Na(ZrF) (S104)„ SB III 554, 555.
1441. 6 (Ca, Na) (0, F) 2 (Zr, Ti) 4Si02, розенбушит*) SB V 119.
*) По Дана стр. к>8.
§2931 1442. CaNaTa20, (ОН, F)—1486. CaS 851
1442. CaNaTa,0,(OH, F), микролит SB VI 132.
CaNb207 SB II 340.
CaNd.81.0, SB II 555.
CaNi(CN)45H.,0 SB III 460.
CaNiSioO, Cii. Z. I, 163(1942).
CaO SB I (51:73) 41, 118, 765, 769; SB II 216, 227, 269, 320, 323
334, 462,463,464, 593, 595, 596; SB VI 138; HRF (1938); F (1942V
О (1950).
Ca02 Ж. Ф. X. VII, 1057 (1937) ??; JJRF (1938); F (1946); О (1950).
CaOxHO SB II 2216.
1450. Ca (OH), SB I (C6:163) 194, 775; SB II 216, 256, 258; SB III 279,
303; HRF (1938); 0(1950).
(Ca3OH)2(PO4)0Ca4 Ca10(OH)„ (Р04)„ гидроксилапатит SB II 102,
458, 464; SB IV 187; SB VI 104.
Гидроксилапатит и стронций, гидроксилапатит SB VII 146, 147.
Ca3P, SB III 353; HRF (1938).
Ca3(P04)., SB II 460-464; HRF (1938); I. E. 17, 491 (1945).
Са3(Р04)Л1,0 HRF (1938).
Ca,P207 SB II 464; I. E. 17, 491 (1945).
CaP,Oe = Ca(P03), SB II 464; SB IV 160.
Ca4P20, SB II 461, 464.
Ca(H2P04)., SB IV 165; HRF (1938); I. E. 17, 491 (1945).
1460. Ca (H2P04)JI20 SB IV 165; HRF (1938); I. E. 17, 491 (1945).
CaHP04 SB II 460; SB IV 165; SB VII 147; HRF(1938); I. E. 17,
491 (1945).
CaHP042H,0 SB II 431, 455, 462, 464; SB IV 165; HRF (1938);
1. E. 17, 491 (1945).
Са-хлорогидрофосфат HRF (1938).
Ca100(P04)6 SB II 102, 459, 461, 464.
Ca10(P04)e (OH)., 1. E. 17, 491 (1945).
Ca, (H20)2 (P04), SB II 102, 458.
CaH2P207 I. E. 17, 491 (1945).
Ca2P207 HRF (1938).
Ca2HP04S044H„0 SB II 432, 455.
1470. Са-глицерофосфат HRF (1938).
Са-лактофосфат HRF (1938).
Ca(H2P02)2 HRF (1938).
CaS04CO(NH,)„ SB III 445.
CaS04HP04 SB II 432, 455.
Ca.Si04Ca3 (P04), J. Am. 63, 2533 (1941).
Ca2"SiO. 1/2 Ca, (P04)2 J. Am. 63, 2533 (1941).
CaaP2Si,014 SB II 462.
Ca,P2Si,020 SB II 464.
Ca,P2SiOl2 SB II 463, 464.
1480. CaIP2BSI20,+BB+2i SB II 462-464.
Ca-Pb SB III 639; SB VI 193.
CaPb3 SB III (LI 2: 174) 639.
CaPb(C03)2, плюмбокальцит SB VI 92, 93.
CaPd (CN)45H20 SB III 461.
CaPt(CN)45H20 SB III 460.
1486. CaS SB I (51 : 74) 125, 126, 765, 769; SB II 231, 236; HRF (1938);
F (1942); ИАН (х), стр. 382 (1944); G. A. 39, 3988 (1945); О (1950),
852 1487. CaS04-1526. Ca (Zr, Ti)2 Os [гл. XI
1487. CaS04, ангидрит SB I (HI: 343) 380; SB II 429, 432; SB IV 165,177;
SB V91; J. Am. 63, 2533 (1941); HRF (1938): Б. (1947); 0(1950).
CaS041/2 HS0 SB II 429, 432; SB III (#4.: 97) 443; SB IV 165,
177; HRF (1938).
CaS042H20 SB I 388; SB II 97, 429 - 432; SB III 442; SB IV (Hi.: 47)
165, 179; SB VI 99; HRF (1938); Б. (1947); О (1950).
1490. CaSO, HRF (1938).
CaS04 — M„S04 (М- щелочной элемент) J. Am. 63, 2533 (1941).
Ca2Sb207 SB III (Z>62: 54) 358; SB IV 200.
CaOSb20(3H20, гидроромеит SB III 359; БМКД (1939).
Ca2Sb20,4H20, гидроромеит SB III 359; БМКД (1939).
CaScSi„Oe SB II 555.
CaSe " SB I (51:74) 134, 765, 769; F (1942); О (1950).
CaSi2 SB I (C12:177) 218; HRF (1938); Б. (1947); О (1950).
Ca,(Cl, F, OH, 0)2(P, S, Si, C), 0J4(Ca, С)4, вилкеит SB VI 104.
CaSiF, HRF (1938).
1500. CaSiF, • 2H20 HRF (1938).
CaO-SiO, SB III 532.
Cau (OH, F)22Si50X0 SB II 580.
CaO • Si02 = CaSiO,, волластонит SB I 328; SB II 528, 531, 532;
SB III 533; SB IV(S33: 71) 207; БМКД (1938); HRF (1938); О (1950).
2Ca0Si02 = Ca2Si04 SB II 464, 519, 532, 595-597; SB III 522;
HRF (1938).
ЗСаО Si02 = Ca„SiOs SB II 464, 532, 595-597; SB VI 136, 137;
HRF (1938).
ЗСаО • 2Si02 = Ca3Si20, SB II 464; SB III 532.
Ca02SiO, SB III 532.
Ca8Si40ie SB II 595.
CaOSi02xH20 SB IV 203.
1510. Ca2Si04H20 SB II 585.
Ca,Si20,3H20 SB II 578.
CaCO,CaS04CaSiOa15H„0 SB II 590; SB III 555.
Ca5Si30„2H20 SB II 585.
CaOTiSi04=CaOSi02TiO„ = CaTiSiO(i, титанит, сфее SB II 117, 515.
593; SB V 96; SB VI 111, 112; Б. (1947).
Ca2SiW1204,26H.O SB IV 213.
CaY2Si20, SB II 555.
Ca8ZnSi20„ гардистонит SB II 143, 542; F (1946).
Ca-Sn SB III 638.
CaSn, SB III (L12 :174) 638.
1520. CaSnO, SB I (Gb: 303) 333; F (1942); N. 155, 484 (1945^; P. Ph.
58, 133 (1946); С A. 40, 3957 (1946); О (1950).
Ca-Sr Z. An. 248, 155 (1941); С. А. 37, 2634 (1943).
CaSr(C03)2 C. R. 216, 844 (1943).
CaTe SB I (51:74) 135, 765, 769; F (1942); О (1950).
CaTiO,, перовскит SB I (G5:303) 331; SB II 49, 340, 515, 593;
SB VII 119-122; F (1942); NW 31, 202, 466 (1943); С. А. 38, 9
(1944); N. 155, 484 (1945); P. Ph. 58, 133 (1946); С. А. 40, 3957
(1946); Б. (1947); О (1950).
CaTh03 С. А. 41, 6102 (1947).
1526. Ca (Zr, Ti). 05, улигит SB VII 119-122.
§294]
1527. Ca—TI—1565. CdCr04
853
1527. Ca-Tl SB III 639.
CaTl SB III (£20:175) 271; F (1942).
Ca'fl, SB III (112:174) 639.
1530.Ca(UO2)O2 A. Cr. 1, 265 (1948); О (1950).
Ca (U02), (P04)2101/, H20, аутунит SB VI 24, 105, 106; F (1946).
Ca(U02),(P04)2nH,0 аутунит SB VI 105, 106.
Ca(U02)2(P04)2672H20, метааутунит SB VI 25, 105, 106; F (1946).
CaW04, шеелит SB I (Я4: 349); SB II 83, 452; HRF (1938); Ch. Z. I,
270 (1944); С. А. 39, 2021 (1945); N. 157, 548 (1946); F (1946);
Б (1947); О (1950).
СаД,(Р04),02 SB II 464.
CaF2'-YF3 Z. An. 240, 150 (1939).
CaZn5 Z. An. 244, 17 (1940); B. (1947).
CaC0,-ZnC0a SB IV 156.
CaZr63 SB I (G5 : 303) 333; SB II 334; F (1942); P. Ph. 58, 133 (1946);
С A. 40, 3957 (1946); О (1950).
§ 294. Кадмий Cd
1540. Cd SB I (A3 :19) 42, 750; SB II 164, 169, 699 - 702, 710, 727 - 729,
733, 736-738, 749, 750; SB III 184, 186, 187, 199, 200; SB V
31; SB VI 57; SB VII 50, 56; HRF (1938); О (1950).
Cd-Bi-0-J Z. An. 250, 173 (1942); Ch. Z. I, 488 (1943).
CdBr2 SB II 246; SB HI 280; Ж.Ф.Х. 16, 1 (1942); Acta XX,
121 (1945); Ж. Ф. X. XX. 6 (1946); О (1950).
CdBr2 • CdJ2 Ж. Ф. X. 21, 777 (1947).
Cd(NH3)2Br2 SB III 388; SB IV (FAa: 33) 145; О (1950).
Cd(Br03)„H20 HRF (1938).
Cd(CN), " Ж. Ф. X. XIX, 433, 515 (1945); Acta XX, 247, 253 (1945);
О (1950).
CdCO, SB I 339; HRF (1938); С. А. 10, 409 (1946).
Cd(CH3C00)2 2H20 HRF (1938).
Cd, салицилат HRF (1938).
1550. Cd (C204) HRF (1938).
Cd(C,04)3H20 HRF (1938).
CdCe' SB V 43; F (1942).
CdCl2 SB I (C19.-743) 189, 773; SB II 246, 249; HRF (1938); Acta
XIV, 737 (1941); С. А. 38, 4173 (1944); Acta XX, 121 (1945);
Ж. Ф. X. XX, 6 (1946); Б (1947); О (1950).
GdCL2V.Ht0 HRF (1938).
Cd(NH.)tCl. SB III 388; SB IV (£1:33) 145; Б. (1947); 0(1950).
Cd (NHt), (C104)2 SB III 475; F (1942); О (1950).
NH4CdCl, SB VI (£24:13) 14, 79; SB VII (Я24:19) 115; о (1950).
Cd(0H)Cl SB III (£03 :65), 373; SB V 49.
3CdCl2 • 5Cd (0H)2 SB V 49.
1560. CdCl24Cd (OH), SB V 50.
Cd(C102)22H20 SB II 378.
Cd(C104)26H20 SB III 452.
CdC08-CoCOs SB IV 155.
CdCr204 = CdOCr2Oa SB II 480; SB IV 167; F (1942).
1565. CdCr04 Ch. Z. I, 410 (1944); C. A. 39, 2020 (1945); О (1950).
854 1566. CdCrA-1610. NH4Cd(NO.), (гл. XI
1566.CdCr„S4 SB II 485; F (1942).
Cd-Cs SB VI 157.
Cd13Cs SB VI (Z)2S:8) 9, 157.
CsCdBr, SB II 329, 330; F (1942).
1570. CsCdC*l3 SB I (G5 : 303) 337; F (1942); 0 (1950).
CsCd(NO.,)3 SB III 402.
Cs.,Cd [Si, Cd(N0„)e] SB HI 485; F (1942).
Cd-Cu SB III 601.
CdeCu, SB II 699; SB III 602.
CdF, SB I (CI :150) 188; F (1942); О (1950).
Cd„ [Fe(CN),]2 SB VI 29, 116.
(NH4)2CdFe(N02)e SB HI 483; F (1942).
CdK2Fe(CN), SB VII 155.
K2CdFe(N02), SB HI 483; F (1942).
1580. Fe2Cd04 SBI(F11 : 352) 417; SB II 476, 477; SB IV 167; HRF (1938);
О (1950).
CdaHg SB II 737; F (1946).
CdHg SB I 568; F (1946).
CdHg(CNS)4- ZnHg(CNS). Z. ph. Ch. 193, 121 (1944).
CdS-HgS Ph. Ch. 47, 537 (1943).
Cd-In SB VI 179; Z. Met. 36, 1425 (1943); Ch. Z. II, 4 (1944).
Cdln204 SB II 475; F (1946).
CdJ„ SB I (£6:163) 189; SB HI (C27:22) 282; SB V 50; SB VI 61;
"Ж. Ф. X. XV, 559 (1941); Acta XIV, 255, 276, 503 (1941); Ж. Ф.
X. XVIII, 308, 425(1944); ИАН (х), стр. 114 (1944); Ch. Z. 1,270
(1944); С. А. 38, 4173 (1944); С. А. 39, 2018(1945); С. А. 39, 3189
(1945);Acta XX, 121 (1945); Ж. Ф. X. XX, 6 (1946); Б (1947); О (1950).
Cd(NH„), J2 SB HI 475; F (1942); О (1950).
Cd13K SB V 65; SB VI (Z)23: 8) 9, 157.
1590. K2Cd(CN)4 SB I (tfl,: 352) 425; F (1942); О (1950).
KCd(N02), SB III 402.
K2CdJ42H20 HRF (1938).
K2CdNi(N02), SB III 484, 485; F (1942).
CdLa SB V 43; F (1942).
Cd-Li SB III 636.
Cd,Li SB III 636.
CdLi SB III (532:19) 266, 636; F (1942).
LisCd SB III 636.
Cd-Mg SB I 565; Ph. R. [2], 68, 1121 (1940); Acta XXI, 749(1946).
1600.CdaMg SB II 727; SB V (D0lt:8) 62.
MgCd2 SB I 565.
CdMg SB II 727.
CdMg2 SB VI 181.
CdMg3 SB II 727; SB V (D0lt: 8) 62; Acta XXI, 749 (1946); ИАН (v),
стр. 143 (1946).
Cd(Mg, Al), SB VI 181.
CdMo04 "SB II 450; F (1946).
Cd(N„)2 SB V 66.
Cd„N2, типанти-Я5,-Мпа03 Z. An.244, 125(1940); F (1942); 0(1950).
Cd(NO,)24H20 HRF (1938).
1610. NH4Cd(NO„), SB III 402.
§2951 16Н. (NH4)2Cd(S0.1h6H.0-lt;49. CeC, 855
16H.(NH4)2Cd(S04)26H20 SB II 94, 436.
Cd-Na SB I 564.
NaCd2 SB I 229, 564.
CdNa3P,0JO = 2Cd03Na203P2Os SB V 106.
(NH4)2 CdNi (N02), SB III 485; F (1942).
RbjCfl (Ni, Cd) (N02), SB III 485;F (1942) *).
Tl2CdNi(N02). SB III 485; F (1942).
CdO SB I (51:74) 120; SB II 218, 219, 227, 228; SB TI 57; HRF
(1938); БМКД (1939); Z. An. 244, 146 (1940): F (1942); О (1950).
Cd(OH), SB I (C6:163) 195, 775; SB II 219, 256, 258; SB TI 80;
HRF (1938).
1620. CdOxH20 SB II 218.
CdaP2 SB 1786; SB II 41, 325; SB III (D\ : 53) 353; F (1946); О (1950).
CdP2 SB III 311; F (1946); О (1950).
Cd,(P04)2 HRF (1938).
CdPr SB T 43; F (1942).
Cd-Bb SB TI 157.
Cd:3Rb SB TI (Z>2,:8) 9, 157.
RbCdCl, SB TII (£24:19) 115.
RbCd (N02), SB III 402.
Cd (NH3)4 (Re04)2 SB III 701.
1630. CdS. гринокит aCdS, ftCdS SB 1(53 : 77) (54 : 79) 129; SB II 233;
SB III 250, 251; БМКД (1939); Z. An. 244, 146 (1940); F (1942);
О ^1950).
3CdS048H20 SB II 433; SB IT (Я42о:52) 182; HRF (1938).
CdS-CdSe SB III 252.
Cd-Sb SB III 645; О (1950).
Cd,Sb2 SB II 749-751; SB III 645; О (1950).
CdSb SB II 749-751; SB III 272; О (1950).
CdsSb207 SB IT 200.
CdSe SB I (53:77) (54:79) 136; Z. An. 244, 146 (1940); F (1942);
О (1950).
Cd-Si SB III 567.
Cd-Sn SB T 134.
1640. CdTe SB I (53:77) 136; Z. An. 244, 146 (1940); F (1942).
CdTi03 SB II (E2„: 69) 379; F (1942); P. Ph. 68, 133(1946); С. А. 40,
3957 (1946); О (1950).
TlCd(NO,)3 SB III 402.
CdW04 " HRF (1938).
Cd-Zn SB HI 644.
CdC03-ZnCO, SB IT 156.
CdZrO, С. А. 41, 6102 (1947).
§ 296. Церий Се
Се SB I (Al : 14) (ЛЗ: 19) 44; SB II 172; HRF (1938); F (1942);
G. A. 40, 3955 (1946); О (1950).
CeBr. A. Cr. 1, 265 (1948); О (1950).
1649. CeC, SB I (Cll: 741) 783; SB II, 275; F (1946); О (1950).
♦) В работе F (1942) дана неверная формула Rb2CdNi, Cd (N02)4.
856^ ^ 1650. СеС13^~1693. ХЛОРИДЫ [гл. XI
1б50.СеС13 HRF (1938); А. Сг. 1, 265 (1948); 0(1950).
Се-Со М. 2, 97 (1947).
Се„ (С204)39Н20 HRF (1938).
Се-Си Z. An. 252, 62 (1943); Ch. Z. I, 518 (1945).
(CeF)2 (C03)s SB I 326.
CeF3 SB II 28, 289; 0 (1950);
CeGa, NW 31, 145 (1943); С. А. 38, 9 (1944).
CeHg SB V 45.
pKCeF4 A. Cr. 1, 265 (1948).
CeK(W04)., Ch. Z. I, 271 (1944); С. А. 39, 2019 (1945); F (1946).
1660.CeO2~-LaO3, тип CI Z. An. 242, 79 (1939).
(La, Ce, Y)P04, монацит SB VII 141.
CeMg, SB Ш (532: 19) 326; F (1942).
MgCe(N03),8H20 SB II 388.
Ce2(Mo04)8 SB I 790; F (1946).
CeN SB V 42; 0(1950).
Ce(N03)36H20 HRF (1938).
NaCeF4 A. Cr. 1, 265 (1948).
CeNa(W04)„ Ch. Z. I, 271 (1944); С. А. 39, 2019 (1945); F (1946).
CeNa203, тин Bl Z. An. 242, 79 (1939); О (1950).
1670.Ce-Ni M. 2, 97 (1947).
CeO, SB I (CI: 150) 197; SB II 265, 267, 269; SB VII88; HRF
(1938); F (1942); О (1950).
Ce203 SB I (Z>52: 244) (Z)5,: 745) 261; SB II 314; SB VII 102; Z. An.
241, 278 (1939); О (1950)"
Ce20„S A. Cr. 1, 265 (1948); О (1950).
CeP SB V 43; О (1950).
C«P04, монацит, турнернт — монацит SB VII 142.
Ce-Pb SB III 647.
CePb3 SB III (L12 :174) 647.
Ce2S, SB II 323; A. Cr. 1, 265 (1948); О (1950).
Ce,S4 A. Cr. 1, 265 (1948); О (1950).
1680.Ce2(SO4)s HRF (1938).
Ce(S04)24H20 HRF (1938).
CeSb SB V 44; F (1942); О (1950).
Ce -Sn SB III 647.
CeSn3 SB III (L12 :174) 647.
SrCeO, SB III 378.
Ce-Zn Z. Met. 33, 358 (1941).
CeZn SB V 44; F (1942).
§ 296. Хлор С)
CI2 SB IV (Ai8:3) 83; SB VII 65; F (1946); О (1950).
HCI SB I 96; SB II 207; SB III 229; Ж.Ф.Х. XV, 597 (1941);
О (1950).
1690. DC1 0(1950).
C120 SB III 279; SB IV 118.
CI02 SB III 303.
1693. Хлориды J. Am. 63, 37 (1941); О (1950).
§ 297) 1694. Со-1733. Co(NH3)« CI 0,80, _____ 857
§ 297. Кобальт Со
1694. Co SB I (A\ : 15) (.43:19) 68, 760, 763; SB II193, 196, 197, 632-
634, 655, 656, 659, 660, 683, 707-710, 775, 780; SB III 187,222;
SB IV 99; SB V 33; SB VII 49, 50; HRF (1938); N. 148, 165
(1941); C. A. 35, 7260 (1941); F (1942); NW 30, 439 (1942); С. Л. 37,
3650, 4947 (1943); JIM 69, 177 (1943); О (1950).
CoBr2 SB II 246; HRF (1938).
Co(NH3), Br„ SB HI 475; F (1942).
CoBr, 3Co (OH)„ SB IV 108.
4Co(OH),CoBrOH SB III 305.
CoBr9Co(OH)2 SB IV 108.
1700. Co (NH3)e S04Br SB I (Я6_ : 431) 460; SB II 500; F (1942);
О (1950).
Co (NH3)6 H2OS04Br SB I (Я6_: 431) 460; SB II 500; F (1942); О (1950).
Co (NHs)e BrSe04 SB II 500; F (1942).
Co (NH3), BrSe043H20 SB II 500.
Co-С SB V 129; J. Am. 69, 893 (1947); О (1950).
Co3C SB V 130; SB VI 178; О (1950).
Co2C J. Am. 69, 893 (1947); О (1950).
Co(CHsCOO)>4H20 HRF (1938).
Co(C204) HRF (1938).
Co (NH3), Co (CN), SB II 104, 504; О (1950).
1710.Co(NH3)6(H„0)Co(CN), SB II 104, 504; О (1950).
Co(CO)4H SB VII 130; О (1950).
Co(NH3)6(H20)Co(CN), SB II 104, 504.
CoC03 SB II 392; HRF (1938).
CoCO, (NH3)4 CI04 SB III 510.
[CoC03 (NH3)4]2 S043H20 SB III 511.
CoCl2 SB I (C19 : 743) 774; SB II 215, 245, 246; SB III (C19 : 22) 279;
HRF (1938); 0(1950).
CoCls(NHs)2(C,Hl2)3 SB V 150.
CoCl, (CeH_„)3 4H,0 SB V 150.
[Co(NH3)6Cl]CL,' SB III (/18:127) 492; HRF (1938).
1720. Co (NH3). Cl3 " HRF (1938).
Co(NH.)0Cl2 SB I (Я61:431) 457; SB III 475; F (1942); О (1950).
СоП-СйН8[1, 2](NH8)2],C183H„0 SB II 503.
[Co(NH3)6Cl]J2 SB III (Л,:127) 492.
Co (NHS). C1S043H20 SB II 501.
CoCl22H20 SB II 215; О (1950).
CoCl26H20 HRF (1938).
CoCl2SCoOS 6H20 SB II 257.
CoCl23Co (OH), SB III 306; SB IV 108.
CoC10H-4Co"(0H)24H„O SB III 306.
1730. (CoCl2)x (CoO)y (H.O)z SB II 365.
Co (NH.), (C104), SB II 503; SB III 475; F (1942); О (1950).
Co(NH3"),(C104)3" SB I (Я71:437) 459; SB II 501, 503; SB III 475;
F (1942); О (1950).
1733.Co(NH,),C103S04 SB II 502.
858 1734. Co(WH,),C104S04—1781. Co,Mn04 [гл._.XI
1734.Co(NH,),C104S04 SB II 502.
Co(NH3)4(H20),C104S043H20 SB II 502.
Co (C104), 6H20" SB HI 452.
Co-Cr SB I 528.
CoCr SB II 655, 656.
Co2Cr3 SB II 655, 656.
1740.Co(NH3),BrCrO43H2O SB II 500.
Co (NH3), Cr (CN), SB II 104, 504; О (1950).
Co(NH3),JCr04 SB II 501.
CoCr204 = Cr2Co04 - CoOCr208 SB II468; SB IV 167; F (1942); О (1950).
CoCr04 HRF(1938)Ch. Z.I,410(1944);C. A. 39, 2020(1945); О (1950).
Cs.CoCl, J. Am. 57, 359 (1935).
CssCoCl6 SB III (#3,: 134) 498; F (1946); Б. (,1947).
CsCo(N02), SB III (J2l: 127) 481; F (1942); О (1950).
NaCs2Co(N02), SB III 482; F (1942).
Co-Cu SB I 512.
1750.Cu3[Co(CN)0]2 SB VII 155.
(Co, Cu, Ni)4S5 SB I 421.
Co2Cu04 = CuCo204 SB II 479; HRF (1938).
Co„CuS4, карролит SB I (HI. : 352) 422; БМКД (1939); F (1942);
" 0(1950).
CoF, SB I (C4 :158) 192; F (1946); О (1950).
CoF3 SB II 35, 290, 291; О (1950).
Co-Fe SB I 520.
Co, [Fe (CN),]* SB VI (/2,: 28) 29.
Co-Fe-W " SB V 130.
Co2Fe(CN), SB VII 155.
1760.Co(NH1)4(HiO)Fe(CN), SB II 104, 504; 0(1950).
CoK2Fe(CN), SB VII 155.
CoM2Fe(CN), (M—щелочной металл) SB VII 155.
Co2Fe40, SB II 478.
Fe,Co04 SB I (Hi,: 352) 417; SB II 468, 478; SB IV 167; F (1942);
" 0(1950).
Co2Ge Z. Met. 35, 218 (1943); G. A. 39, 1095 (1945).
CoHg(CNS)4-ZnHg(CNS)4 Z. ph. Ch. 193, 121 (1944).
CoJ, SB II 247.
Co (NH3), ,T2 SB I (#6, : 431) 457; SB III 475; F (1942); О (1950).
Co(NH3),Js SB I (Я7,:437) 457, 458, 793; SB II 500; SB III 475;
F (1942); О (1950).
1770. Co (NH2CH3), J2 SB III 475.
CoJ3(NH3)5H20 SB I 458, 793; SB II 500, 501; F (1942).
Co (NH3), S04J SB I (Я6,: 431) 460; SB II 500; F (1942); О (1950).
Co (NH3)5 H2OS04J SB I (Я6,: 431) 460; SB II 500; F (1942); О (1950).
Co(NH3),Se04J SB I (Я6,: 431) 460; SB II 500; F (1942); О (1950).
K,Co(CN.) Ж. Ф. X. XVIII, 437, 445 (1944).
K,Co(N02), SB II 496; SB III (/2, :127) 481; F (1942); О (1950).
K,Co,(N02).2H20 SB II 496.
K2PbCo(N02), SB II 496; F (1942).
KjSrCo (N02), SB II 496; F (1942).
1780.CoCO,-MnCO, SB IV 155.
1781.Co2Mn04(Co-Mn шпинель) SB II 470; HRF (1938).
§ 297] _ J782. CoMn.204-1824. CoS 859
1782. CoMn„04 (Co —Mn гапинель) HRF (1938).
С-Co-Mo SB III 623.
Co(NH3),JMo04 SB II 501.
Co(NOa)26H20 HRF (1938).
(NH4)aCo (NO.,), SB III (J2t: 127) 481; SB IV (/2,: 65) 192; F (1942);
О (1950).
Co(H20,)S04(NH4),S04 SB II 435.
Co(N03)26Co(OH)2" SB V 84.
Na2Co(CNS),8HoO, жюльснит SB III 461; БМКД(1939).
1790. NaRb2Co (NO„)," SB III 481; F (1942).
NaTl2Co(N0„), SB HI 481; F (1942).
Co2Nb Z."Kr. (A) 103, 391 (1941); Ch. Z. I, 1472 (1942).
CoNb.O, С. А. 39, 2684 (1945).
Co,N104 SB II 479.
(Co, Ni)3S4 SB I 417; F (1942).
Co-О Ch. Z. I, 306 (1942).
Co20 SB I 771.
CoO SB I (51:74) 114, 123, 771; SB II 196, 222, 223, 225-228,
359, 479, HRF (1938); F (1942); О (1950).
Co203 SB I 267, 771; SB II 228; 0(1950).
1800. Co304 HRF (1938); F (1942); О (1950).
Co(OH)2 SB I (C6:163) 195, 771, 775; SB II 223, 257, 258, SB IV
(C6:8) 120; SB VI 89; HRF (1938); О (1950).
Co(OH)3 SB I 771; SB II 359; 0(1950).
CoOxH20 SB II 223.
Co203xH20 SB II 223.
CoOOH SB II 359, 360.
Co„03H20 SB HI 348.
Co-P SB VII ИЗ; О (1950).
Co2P SB VII 113.
CoP SB III (Я31: 18) 263, 264; SB VII 113; О (1950).
1810. CoP, SB VII 143; Z. An. 241, 349 (1939).
Co (NH3), (PF,)3 SB HI 475; F (1942)*).
Co(NH3), (PF,), SB III 475; F (1942); О (1950).
Co3(P04)28H2O" HRF (1938).
Co(H2P02)26H,0 SB V 78.
Pbs [Co (NO„),]2" SB VII 152; О (1950).
Co-Pt Ch. Z. I, 166 (1942).
Rb2CoCIs J. Am. 359 (1935).
Rb,CoCl, SB HI (КЗ.: 134) 498; F (1946).
Rb3Co(N02), SB III (/2,: 127) 481; F (1942); О (1950).
1820. Co-S SB VI 174.
Co4S3 SB II 234; SB IV 99.
Co.S. SB IV 99.
Co.S, SB IV iD8: 26) 137; SB VI 174; О (1950).
1824. CoS SB I (58 : 86) 132, 134; SB II 233; SB IV 99; SB VI174, llAH(x),
стр. 382 (1944); С. А. 39, 3988 (1945); О (1950).
*) В работе F (1942) нспраоильпая формула Co(NH3)e(PFs).
860 1825. Co3S4-1872. Co2Zn04 |гл: XI
1825. Co3S4 SB I (Я1,: 352) 421; SB II 485; SB VI (Dl2: 9) 10, 174; F (1942);
О (1950).
CoS2 SB I (C2:153) 217; SB VI 174; F (1942).'
CoS04 SB II 433; HRF (4938).
CoS04H20 SB I 389.
CoS047H20 SB I 389.
1830. 2CoS043Co (0H)2 5H20 SB III 473.
CoS043Co(0H)24H20 SB III 472.
CoS04 (NH4)2 S04 • 6H20 HRF (1938).
CoSO36H20 SB IV 160.
Co—Sb SB VI 175; Ch. Z. I, 1104 (1942).
CoSb SB I (58:87) 142, 765; SB VI 175; О (1950).
CoSb., SB VI 175; О (1950).
CoSbl04 Ch. Z. I, 342 (1944); С. А. 39, 2019 (1945); F (1946).
Co—Se SB VI 166.
CoSc SB I (58:86) 138, 765; О (1950).
1840.CoSe2 SB I 780; SB VI 166; F (1946); О (1950).
Co—Si SB III 626; О (1950).
Co2Si SB III (C37 : 32) 310, 626; О (1950).
CoSi SB III (525:14) 626; F (1942); О (1950).
CoSiP,6H„0 SB II 104, 504; О (1950).
Co—Sn " SB VI 177; Ch. Z. 1271 (1944).
CoSn SB VI (535:4) 177, 178..
CoSiu SB VI (C51 : 7) 177, 178; Z. Met. 35, 218 (1943); С. А. 39,
1095 (1945); F (1946).
CoSnO, SB II 483.
Co2Sn04 SB II 482, 483; F (1942).
1850. Станнат кобальта HRF (1938).
Co2Ta Z. Kr. (A) 103, 391 (1941); Ch. Z. I, 1472 (1942).
Co—Те SB VI 166; NW 26, 429 (1938); Z. An. 239, 126 (1938).
CoTe SB I (58:86) 138, 765; SB VI 166; О (1950).
CoTe, SB VI 166.
Co—Ti SB VII 211.
Co2Ti SB VII (C15:7) (C36-.8) 211.
CoTi SB VII 211.
CoTi03 SB III (£2„: 69) 379; О (1950).
Co2Ti04 SB II 484," 485; F (1942).
I860. Co—W SB VI 176.
Co,W2 SB II 659.
Co3W SB VI 176.
Co,W, SB VI 176.
CoW SB II 659, 660.
C—Co—W SB III 623; Ж. Ф. X. XVII, 108 (1943).
W,Co„C SB II 780.
Co4W;C SB III 623.
CoW04 SB II 86, 450; О (1950).
Co—Zn SB VI 187; Z. Met. 33, 375 (1941); Ch. Z. II, 10 (1942).
1870. CobZn2l SB II 708.
CoC03-ZnCOs SB IV 154.
1872.Co2Zn04 SB I 792; SB II 478, 479; HRF (1938).
§298) __ 1873. (Co, ZnKOHa)—1913. Cr,FeS4 861
1873. (Co, Zn) (OH), SB VI 89.
Co2Zr Z. Кг. (А) 103, 391 (1941): Ch. Z.fl, 1472 (1942).
§ 298. Хром Сг
Сг SB I (Л2.-16) (Л3:19) 61. 755; SB II 190, 191. 652-657,
775-777, 791-792, 798; SB III 187.218; SB IV 85; SB V 34; SB
VII 49, 50; HRF (1938); Ann. 38, 97 (1940); F (1942); О (1950).
CrBrs SB II 294; О (1950).
СгВг36Н20 HRF (1938).
Сг (en)3Br33H20 (en—этилендиамин) SB VII 235.
Cr(NH3)5(H20)BrS04 SB II 502.
1880. Cr-C SB I 573; О (1950).-
Cr4C SB I 573; SB II 775, 776; О (1950).
Cr23C, SB III (Z)84: 59) 367; Б. (1947); О (1950).
Cr7Cs SB I 573; SB II 775, 776; SB III 363; Б (1947); О (1950).
Cr.C, SB I 573; SB II 775, 776; SB III (Z>510:53) 351; Б (1947);
О (1950).
CrC SB II 776.
Cr(CO), SB HI 334; SB VI 88; J. Am. 69, 1723 (1947); О (1950).
Ст„ (C204)3 H„0 HRF (1938).
CrCla SB I 248; SB II 23, 292; О (1950).
Cr(en),Cl33H„0 (en —этилендиамин) SB VII 235.
1890. NH2 (CH,), NH2 SB VII 235.
[Cr (NH3)6 CI] Cl2 SB III (/1,: 127) 492.
CrCl,6H20 SB III (/22:127) 493; О (1950).
Cr02Cl2 SB VI 82.
Cr(NH3),(C104)3 SB II 501, 502.
Cr(NH9)5(H20)(C104)3 SB II 501, 502.
CsCr03F SB VII (Я04:30) 136; F (1946).
CsJCrOJ SB VI (#1,:21) 101.
Cu2Cr(CN), SB VII 155.
Cr203-CuO SB III 342; SB IV 167.
1900.Cr,CuO4 SB I (Я1Х:352) 417; SB III 623; F (1942)*).
CuCr04 Ch. Z. I, 410 (1944); C. A. 39, 2020 '1945); О (1950).
(NH4KCrF, SB II 493; F (1942).
CuCr642Cu02H20 HRF (1938).
CuCr2072H„0 HRF (1938).
CtF34H„0 " HRF (1938).
Сг-Fe" SB I 501, 525.
CrFe SB II 652, 653; SB III 586.
Cr-Fe-C SB I 583; SB IV 243.
Cr-Fe-N SB III 586.
1910.Cr-Fe-Si SB IV 244.
Cr20,-FeO SB IV 167.
Cr„Fe04 SB I (#1,: 352) 416; SB II 470, 480, 481; F (1942); О (1950).
1913. Cr2FeS4, добреелит С. А. 39, 2685 (1945).
*) В работе F(l%2)—ошибочная формула Си.. Сг2 04, не отвечающая типу //1
(шпинель).
862
1914. Cr4H—1955. Cr-P
|гл. XI
1914. Cr4H SB II 798.
Cr(NH,)eJCrO< SB II 501.
K,Cr(CN)4 SB II 495.
KCr03F SB VI (Я04:18) 101; F (1946).
KCr(S04). SB I 789; SB II 91; О (1950).
* K2Cr04 SB I 379; SB II 88, 446.
1920.K2Cr207 SB II 509.
K3CrOe С A. 36, 308 (1942); Ch. Z. I, 11 (1942); F (1946); О (1950).
KCr(S04),12H,0 SB I (Я4.,:370) 390; SB HI (#4l3: 110) 455;
SB V 91;"HRF (1938); F (1942); О (1950).
K2Cr(S04),12D20 SB V 91.
MgCr204=Cr,03 MgO SB II 468,470, 480; SB III 243, 342; SB IV
167; F (1942).
Cr,Mn04 SB I (Я1,:352) 417; SB II 460; SB IV 167; F (1942)*);
О (1950).
31gCr04 SB I 389.
MgCr04m20 SB I 389.
Cr.,Mn84 SB II 485; F (1942).
Cr-Mo SB VI 188.
1930. 5PbCrO43PbMoO410PbSO4 SB II 445; F (1946).
Cr-N SB V 133; О (1950).
Cr.N SB II 792; SB III 586.
CrN SB II 793; SB V 133; F (1942); О (1950).
Cr(N03)3-9H,0 HRF (1938).
H2Cr042NH3 (не идентична (NH4), Cr04) SB IV 187.
(NH4),Cr04 SB II 446; HRF (1938).
(NHj; Cr207 SB II 509; HRF (1938).
Cr (NH4) (S04)2 < 12H20 HRF (1938).
Cr(NH4)(S04)212H,0 HRF (1938).
1940. Na2Cr04 SB IV (Я1.:45) 172; HRF (1938); О (1950).
Na,Cr04 4H20 HRF (1938).
Na2Cr,07 2H20 HRF (1938).
CrNa2P30106H20 SB V 106.
CrNbO. С. А. 39, 2684 (1945); F (1946).
Cr-Nl SB I 528; SB III 585; SB IV 230; Z.El. 45, 87, 838(1939);
48, 377 (1942).
Cr,Ni SB II 793; F (1942).
Cr„N104=Cr203NiO SB II 481; SB IV 167; F (1946).
CrN104 Ch. Z. I, 410 (1944); С A. 39, 2020 (1945); Б (1947); О (1950).
Cr203 SB I (Z)5,:242) 196, 197, 266; SB II 310, 318, 322, 323,
481; HRF (1938); О (1950).
1950. Cr03 SB II 29, 296; 316; HRF (1938); О (1950).
Cr203 —CrO SB II 319. (Приведён в указателе SB, как СгО.
Структура неизвестна, состав не исследован).
Cr20sxH20 SB II 355.
CrOOH SB II 355.
Cr(OH)s SB I 254.
1955.Cr-P SB VI 189; Z. An. 248, 209 (1941).
*) В работе F(1'J42) к типу Н\г реферированы различные периоды ячейки
Cr2Mn04: 8/.3 и 8,49 к X.
§ 299]
1956. Cr3P-1997. CsCN
863
1956. Cr,P SB VI 189; Z. An. 248, 209 (1941); F (1946).
Cr2P Z. An. 218, 209 (1941).
CrP SB VI 189; Z. An. 248, 209 (1941).
CrP, Z. An. 248, 209 (1941).
1960. PbCr04, крокоит SB II 440, 445, 448, 449; SB VII 142; HRF (1938).
PbCr04PbO SB II 445.
PbCr04Pb(0H)2 SB II 445.
Cr3Pd2 SB IV 230.
CrPd SB IV 230.
Cr-S SB V 131.
Cr-Pt SB III 587.
Cr3Pt SB III 587.
Rb,CrFsH,0 SB II 494; F (1942).
Cr-S Z. An. 234, 372 (1937).
1970. CrS SB I (58:795) 131; О (1950).
Cr-Sb Gh. Z. I, 1104 (1942); О (1950).
CrSb SB I (58:87) 142, 765; О (1950).
CrSb04 С. А. 39, 2684 (1945); F (194(5).
£r ge gg у| i9o_
CrSe SB I (58:795) 138; О (1950).
Cr-Si SB III 628; О (1950).
Cr3Si SB III 628.
CrSi SB HI (528:14) 628; F(1942); О (1950).
CrSi2 SB III (£40:35) 628; О (1950).
1980. CrHSiW.,0,,,24 (?) H.,0 SB IV 212.
CrHSiWl2O1028H,O SB IV 212.
CrSr04 SB II" 445, 448, 449.
CrTa04 G. A. 39, 2684 (1945); F (1946).
CrTe SB I 765; О (1950).
Tl2CrF6H20 SB II 493: F (1942).
CrV04 Gh. Z. I, 410 (1944); G. A. 39, 2020 (1945); О (1950).
Cr-W С SB III 623.
CrnW2C, SB III (£84: 60).
Cr"Zn04 = Cr,0,ZnO SB I (Я1,:352) 417; SB II 469, 470, 481;
SB III 435; SB IV 167; F (1942); О (1950).
1990.ZnCrO4 SB II 445; HRF (1938); Ch. Z. I, 410 (1944); C. A. 39,
2020 (1945); О (1950).
(ZnCr04),(ZnO).2H20 SB II 445.
ZnCr2S4 = Cr2SsZnS SB II 485; SB HI 435; F (1942).
§ 299. Цезий Cs
Cs SB I 747; SB VII 202; F (1942); R. M. 15, 90 (1943); Усп. 15,
90 (1946); О (1950).
CsBr SB I (52:75) 97, 98, 107; SB IV 92; SB V 38; F (1942);
О (1950).
CsJBr2 SB I 287; О (1950).
CsC, SB II 130; 0(1950).
1997. CsCN SB II 371; F (1942); О (1950).
864
1998. CsCI—2042. CsSH
[гл. XI
1998. CsCI SB I (52:75) 97, 98, 99, 107; SB III (51: 7) 231; SB IV 92;
SB V 37; HRF (1938); F (1942); ИАН(х), стр. 382 (1944); С А.
39, 3988 (1945); Б (1947); О (1950).
CsJCU SB I (F5,:276) 287; HRF (1938); О (1950).
2000. CsCIO, SB II 378.
CsC104 SB I (Я2:344) 372; F (1942); О (1950)
CsCuCl, J. Am. 68, 1133 (1946).
Cs2CuCl4 SB VII 152.
CsF SB I (51:73)97, 98, 107; F (1942); О (1950).
CssFeF, SB V 100.
CsGa (S04),12BLO J. Am. 65, 2071 (1943).
Cs2GeF, " SB I (Я6,:431) 443; SB IV 174; F (1942); О (1950).
Cs..Ge04 SB IV 173.
CsH SB II 242; F (1942); О (1950).
2010.CsHgBr3 SB II 329, 330; F (1942).
CsHgClBr.. SB II 330.
CsHgCl2Br SB II 330.
Cs2Hg2Cl3Br, SB II 330.
CsHgCl, SB I (G5:303) 338; F (1942); О (1950).
CsHg(N03)2 SB III 401.
Cs,Hg[Hg, Ni(N02),] SB III 486.
CsJr(CN), SB II 495.
Cs3IrCl,NH4NO, Ж. Ф. X. XVIII, 160 (1944).
CsJ SB I (52:75) 97, 98, 107; SB IV 92; SB V 38; SB VII 70;
HRF (1938); F (1942); О (1950).
2020. CsJ3 SB I 288; О (1950).
CssJ,(N02), SB III (/2,: 127) 482.
CsJO, SB I 339; F (1942); О (1950).
CsJ04 SB III 422; SB V 89.
Cs-K SB VII 202.
CsMn04 SB VI 97.
CsNO, SB III 414; SB V 83; HRF (1938).
Cs-Na SB VII 202.
CsnO SB VII (£19:7) 85; Z. An. 242, 33 (1939); О (1950).
Cs20„ анти тип Th3P4 Z. An. 242, 201 (1939); О (1950).
2030. CsO, SB VI 63; SB VII (Cll : 7) 84; Z. An. 241, 97 (1939);
Ж. Ф. X. XX, 1403 (1946); F (1946); ДАН 59, 67 (1948); О (1950).
CsOs ОД SB II 419.
CsPF, SB II 491.
CsH2P04 SB I 394.
Cs2PbCl, SB III (Л. :121) 477, 479; F (1942); О (1950).
Cs2PtCl, SB II 497; SB III (/1,: 121) 477, 479; F (1942); О (1950).
Cs-Rb SB VII 202.
CsRbCl2=CsCl RbCl HRF (1938).
CsRe04 SB III 422; SB V 90.
CssRhCl,NH4N03 Ж. Ф. X. XVIII, 160 (1944).
2040.Cs3Rh(NO,), SB III (У2,: 127) 482; F (1942); О (1950).
CsRh (S04)212H20 SB II 435.
2042.CsSH SB III (52:7) 261; SB VII 78; F (1942); О (1950).
§_300J__ 2043-. Cs,S04-2081.Cu 865
2043.Cs2SO4 SB I 374; SB II 88, 420, 424; HRF (1938); О (1950).
Cs2S„06 SB II 107, 508.
Cs.s"0, SB III (ЛГ4,: 136) 499; О (1950).
CsSbF, Ch. Z. I, 1450 (1943); С. Л. 39, 2239 (1945).
Cs2SeCI, SB III (/1^121) 479; F (1942); О (1950)..
CsSell SB VII (52:4) 78; О (1950).
Cs„SiFt SB III (Л,:122) 479, 480; SB IV 174; F (1942); О (1950).
20.X). Cs3HSiW1204anH20 SB ill 466.
Cs.SnBr, SB V 103; SB VI 121: F (1942).
Cs2"SnCl, SB III (Л,:121) 476, 479; F (1942); О (1950).
Cs.SnIs SB VII (Л,:34) 154.
СвДаО, Ch. Z. I, 11 (1942); F (1946); О (1950).
Cs,TeCl, SB II 493, 497; SB III (/l1:12l) 479; F (1942); О (1950).
CsJTiCl, SB III (/lt:121) 479; F (1942); О (1950).
Cs.,TlCl6 SB III (Л, см. 121) 476.
Cs.TlCl, SB III (A'7,: 144) 505; Б (1947).
Cs2W,Cl, SB III (K77 :142) 505.
20C0. Csll3H2Wl204(,nH.,0 SB III 466.
♦
Cs.PW. ,040nH,0 SB III 465.
CSjZrCl," SB III (Л,:121) 479; F (1942); О (1950).
§ 300. Медь Си
Си SB I (Л1:14) 35, 747; SB II 164-168, 218, 243, 607-623,
625-627, 638-641, 663-666, 668-677, 684, 686-699, 702,
703, 713-717, 720, 739-743, 745, 756-759; SB III 181, 187-189;
SB IV 223; SB V 31; SB VI 40, 43; SB VII 45, 49, 50, 52, 53;
БМКД (1938); HRF (1938); ДАН 24, 554 (1939); P. R. S. (A) 177,
149 (1941); UAH (x), стр. 273 (1948); О (1950).
CuBr. HRF (1938); J. Am. 69, 886 (1947)*); О (1950).
CuBr" SB I (A3: 77) HO; SB VII 69; HRF (1938^; F (1942); Ж. Ф. X.
XX, 6 (1946); О (1950).
(NH4).,CuBr4 ~'NH3 SB V 87.
(NH4), CuBr42H.,0 SB IV (/7^:47) 179; SB V 87; F (1946).
CuCN HRF (1938).
CuCNS HRF (1938).
2070. Cu3(C03)2(OH)2. лазурит SB II 81. 398; HRF (1938); БМКД (1938).
Cu,C03(0H)„ малахит SB II 397; HRF (1938); БМКД (1938).
8Cu01C0..5H20 SB IV 159.
2CuOC02i,5H20 SB V 79.
(CuC03)x(CuO)j,(H,0). SB II 254.
Cu(HCOO)2 HRF "(1938).
Cu(HCOO)22H20 SB II 842, 843.
Cu(HC00)24H20 SB II 843.
Cu(COO)2 SB II 254.
Cu-бензоат HRF (1938).
2080. Си-фснолсульфонат HRF (1938).
2081. Си-тартрат HRF (1938).
*) Хотя в работе J. Am. 69, 886 (1947) речь идёт о структуре Cupric bromide,
CuBr.,, в резюме этой же работы гоиоритсн о структуре cuprous bromide, CuBr.
866 2082. Си—2123. CuFcoS, |Гл. XI
2082. Си-цитрат HRF (1938).
Cu(C,H4C0S02N), 6H,0 SB V 174.
Си 01 SB 1(53 : 77) 110; SB VII 69, 70; HRF (1938); F (1942); ИАН(х),
стр. 382 (1944); С. А. 39, 3988 (1945); Ж. Ф. X. XX, 6 (1946); О (1950).
СиС12 HRF (1938).
СиС122Н,0, антофагазит SB III 388; SB IV (£45:13) 104, 105; SB
VI (C45-.60); HRF (1938); О (1950).
Cu(C104)26H„0 SB III 453.
(NH4)2 CuCl42H.,0 SB I (#4,-.369) 425; SB III 441; HRF (1938);
F (1946); О (1950).
(NH4)2CuCl46H,0 SB II 489.
2090. N (CHS)4 CuCl4 SB VII 231.
CuCl S = C( I SB IV (02,: 255) 256, 257; F (1946).
\ 4NH2 /4
CuF SB III (53:7) 226; F (1942); Ж. Ф. X. XX, 6 (1946); О (1950).
CuF2 2H20 HRF (1938).
(NH4)2 CuF4 • 2H..0 HRF (1938).
CuFe2 SB III (CI : 20) 274.
Cu-Fe-Ni' Gh. Z. I, 1104 (1942).
Cu8F*(CN), SB VII 156.
Cu2Fe(CN), 7H,0 HRF (1938).
CuFe(CN)4 SB V 99.
2100. CuFe„(CN), SB V 101.
Cu3Fe2 (CN)l2 SB V 101.
Cu3[Fe(CN),]2 SB VI (/2,: 28) И6.
Си" М Fe"1 (CN), SB IV 191.
CuM2Fe(CN), (M—щелочной металл) SB VII 155; F (1942).
CuCl2 2KC1 2H„0 HRF (1938).
CuK2 Fe(CN), "SB V 101; HRF (1938).
CuLL Fe (CN), SB V 100; F (1942).
CuMg Fe (CN), SB VII 155.
(CuFeMoSn), (S. As, Те), 4 SB III 253.
2110. Cu(NH4)2 Fe(CN), SB V 100.
CuNa2 Fe(CN), SB V 100.
CuRb, Fe (CN), SB V 100.
Cu2SnFeS4 = Cu,FeSnS4, станнит SB I 291; SB III (#2G:96) 440;
БМКД (1938); F (1946); Б (1947); О (1950).
CuSr Fe (CN), SB VII 155.
CuTl2 Fe (CN), .SB V 101.
CuFeO., = Cu,Fe204> делафосснт SB III (i-'51 :75) 392; SB VI 86;
БМКД (1938).
CuFe204 = CuO Fe„0, SB I (Я1,: 352) 417; SB II 477; SB IV 167, 169;
F (1942); F (1946); О (1950).
CuFeS„. халькопирит SB I (F6X: 280) 290; SB II 48, 346; SB III
38"5; БМКД (1938); HRF (1938); Rec. 63, 69, (1944); С. А. 39, 4276
(1945); F (1946); Б (1947); О (1950)
CuFe2S3, кубанит (чальмсрзит) SB IV 148; БМКД (1938).
2120. Cu,FeS3 SB I 422; SB II 234, 346.
Cu2Fe4S., валерит SB VII 157, 158.
Cu.FeS4.' борнит SB I 422; SB IV 149; БМКД (1938).
2123. CuFe.S,. SB V 76; Ch. E. N. 25, 2970 (1947).
§3001 _ 2124. Cu—Ga-2169. Na4CuO,_ 867
2124.Cu-Ga SB III 600; SB IV 223, 237; SB Vll 201; P. R. S. (A) 177,
149 (1941); Ch. Z. 1260 (1947).
Cu3Ga SB III 600.
CuGa, SB IV 238; F (1946).
Cu-Ge SB I 544; SB IV 223; SB VII 201.
Cu3Ge SB I 544.
Cull SB I 147; SB II 243; О (1950).
2130. Си-Hg SB I 537.
Cu4Hg3 SB I 538.
CuHg SB III 268.
CuHg (CNS)4 SB IV 165.
Cu,HgJ, SB III (A3: 7) 235; F (1942); F (1946); О (1950).
Си" In SB III 572; P. R. S. (A) 177, 149 (1941): Ch. Z. 1260 (1947).
Cu4In SB III 573.
Cu.In SB III 574.
Cuj, маршит SBl(/tt:77)110, 112; SB II 214; Si? VII69; VA KU (1938);
HRF (1938); F (1942); Ch. Ph. 13, 321 (1945); Ж. Ф. \". XX, 6
(1946); О (1950).
CuJ030H, еалезит SB VII 136.
2140. Cujr(C4H9)3 SB IV 286, поиидимому, аналогична CuJAs (C2Ht18.
KCuBr3 SB VII 114.
K1Cu(CN)4 SB IV 176; HRF (1938).
K„CuCI42H,0 SB I (#4:369) 425; SB 111 441; F(l94t,); О (1950).
KCuPMNO.), SB II 496; SB IV (724: 65) '192; F (1942); О (1950).
K;Cu(S04)„6H.,0 SB II 254; HRF (1938).
K„Cu(S04)28HaO SB V 91.
Cu—La Z. An. 252, 62 (1943); Ch. Z. I, 518 (1945).
CmMg SB I (C15:490) 531; SB II 661-666; SB IV 242; SB VI 180;
" HRF (1938); F (1942); Б. (1947).
CuMr., SB I 531; SB II 664-666; HRF (1938); Б (1947).
2150. CuJUg - MgNiZn SB III 313.
t'u-Mg-Si SB VI 180; SB VII 210.
Cu,Mg8i SB VI 180.
Cu,Mg.,$i ДАН 40, 262 (1943).
Cu1(Hg,Si, SB VI 181.
CuMgZn ДАН 40, 262 (1943).
CuO-MgO SB III 240.
Cu-Mn SB I 501, 520, 758; SB VII 186.
Cu2Mn SB II 687.
CuMn SB II 687.
2160. Cu - Mn Sin Sli I 549.
Cu2MnSn SB I 551; SB II 721.
Cu2 Mn (Ci\), SB VII 156.
2CuMo04 Cu(OU). SB HI 471.
Cu3N, тип аити ВеО.( (ХЮ.) SB VI 67; Z. An. 241, 177 (1939); 248,
118 (1941); О (1950).
Cu(N,)., Z. An. 251, 315 (1943); Ch. Z. I, 518 (1944).
Cu(NH3)4 S04H.,0 SB II 427.
Cu(\03)., 3H..O" HRF (1938).
(Cu(N03),)r (CuO) (H.,0). SB II 254.
2169.Na4CuO, SB III 417.*
868 2170. CuiYa (SO,), OH H,0-22Q8. CuS04 x_IM> 1™JU
2170. CuNa (S04)2 OH ILO, натрохалкит SB VII 137.
NatCu(S04). SHgO. крёнкит SB VII 137, 138; БМКД (1938).
Cu—Ni SB I 502, 513; SB III 571.
Cu—Ni—Sb SB V 123.
Cu—Ni—Sn SB V 123.
Cu—Ni—Zn SB IV 241.
CuO, тенорит, парамслаконит SB I 36, 114; SB II 215, 218, 243, 279,
440; SB III (£26:11) 239; SB VI 56; HRF (1938); БМКД (1938);
БМКД (1939); A. Min 26, 657(1941); О (1950).
Cu20 SB I (6'3 :155) 36; 222, SB II 167, 243, 279; SB III 567; SB IV
119; SB V 47; SB VI 61; HRF (1938); БМКД (1938); БМКД (1939);
Ж. Э. Ф. 10. 1450 (1940); F (1942); О (1950).
CuOxH20 SB II 215, 254, 255.
Cu(OH)2 SB II 215, 254; SB V 47; SB VI 56.
2180. CuP, SB VII 99.
Cu3P SB VI (/)(),, : 7) 65; SB VII 99.
Cu3(P04)„ 3HO HRF (1938).
CuOHCuP04, либетенит SB IV 167; SB VII (Я27: 32) 143; HRF (1938);
БМКД (1939).
Cu2Pb6 (S04)3 (C03) (ОН),, каледонит SB VII 141.
Cu(OH)2 PbCl,. кумеигеит SB 1 477; F (1946).
Cu4Pb,Cle045H„0 SB II 365-367.
Cu4Pb6Cl0046H,0, псевдоболеит SB II 365-367; F (1916).
CuPbSbS3, бурцоиит SB II 346; БМКД (1938).
CuPb(V04)0II SB VII 151.
2190.Pb(Zn, Cu)(V04) ОН, десклоизит SB VII 151.
Pb(Cu, Zn)(V04) ОН, моттрамит SB III 473; SB VII 151.
Cu—Pd SB I 515; SB III 574; SB VII 187.
Cu3Pd SB I (LI2:486) 515; SB II 626; SB VII 187; F (19Ш).
CuPd SB I (52:76) 147; (/.20:488) 487, 515; SB II 626;
F (1942).
Cu—Pt SB I 517; С. А. 40, 4943 (1946).
CuPt SB I (111:485) (L13:487) 485, 486, 517.
CujPt SB I (L12:486) 517.
Rb,CuCl43H20 SB I (Я4Х:36Э) 42Л; F (1946); О (1950).
CuS, коиеллин SB I 126, 771; SB II 10, 229. 230; SB VII 91;
БМКД (1938); Ph. Ch. 47, 496 (1943); HAH (x), стр. 382 (1944); С. А.
3», 3988 (U4f>); Б (1947); О (1950).
2200. Cu2S, халькозин SB I (CI : 150) 224; SB II 282; SB IV 109; SB VII
91; БМКД (1938); БМКД (1939); F (1942); Ph. Ch. 47, 496 (1943);
ДАН 54, 721 (1946); О (1950).
Cu,S. SB VII 91.
CuS04 SB II 426, 427; HRF (1938).
CuS04 (NH4)2 S04 • 6H20 HRF (1938).
(CuS04)x(CuO)(,(H20). SB II 440.
Cu, S04(OH)4-CuS042Cu(OH)2, антлерит SB II 440; SB VII 138;
БМКД (1938).
Cu4 S04 (0H), = CuS04 3Cu(OH)2, брошантит SB II 440; SB VII
138, 139; БМКД (1938); БМКД (1939).
2CuS04 Cu(0H),4H20 SB II 440.
2208. CuS04xH„0 SB II 427.
§ ;;00| 2209. CuSO45H,0 2253. CuS—ZnS 8t9
2209. CuS0451LO, ха.п.кантит SB II 427; SB III (//410 : 102) 449; SB IV
181; БМКД (1938); I1HF (1938).
2210. CuS043II.,0 SB II 427.
CuS04HO SB II 427: HRF (1938).
4CuO S033H..O SB IV 185.
4CuO—SO—4ИО SB HI 471; SB IV 181. 185.
3CuOSO,2H,0 " SB IV 181, 185.
2CuO S03 " SB IV. 181.
Cu^H.O HRF (1938).
Cu—Sb " SB I 596; SB VII 207; О (1950).
CunSb. SB VII 207; О (1950).
Cu„Sb,' SB VII 207; О (1950).
•-'220. CuuSb4 SB VII 207; О (1950).
Cu3Sb SB I 596; SB II 300, 301, 742, 743; SB VII 207; О (1950).
Cu6Sb. SB VII 207, 209; О (1950).
Cu„Sb SB I 596: SB II 742, 743; SB III (C38:33) 288; SB VII
* 207; F (1946); О (1950).
Cu3SbS, SB I 335.
Cu,SbS4 SB II 347.
CuSbS, SB III (Fb, : 75) 393.
Cu.,Sb2S4, халкогтибит, вольсборгит, SB VII 157; БМКД (1938).
Cu2Se SB I (CI : 150) 225; SB IV 113; F (1942); О (1950).
CuSeO.SH.O SB V 80.
2230.Cu„Si4 " SB II 758; SB III (D8t: 62) 332, 366: О (1950).
Cu6Si SB II 758; SB III 332; О (1950).
Cu2SiWl204027H„0 SB IV 213.
CuH,Si04 SB I 407.
Cu—Sn SB I 501, 502, 545; SB III 602; SB IV 238; P. R. S.
(A) 177, 149 (1941).
Cu6Sn SB I 503, 545.
Cu4Sn SB II 714, 715.
CuslSn, SB I (Z)84:499) 545.
Cu2oSn, SB II 717.
Cut0SnI( SB II 715.
2240. Cu3Sn SB II 713-716; SB II 602.
CuSn SB I 490, 501, 545.
Cu,Sn, SB II 713.
Cu2Sn04 SB II 484.
CuSn03 SB II 484.
Cu2Te M. 1, 40 (1946); С. А. 41, 637 (1947).
Cu—Th Z. El. 48, 418 (1942); Z. El. 49, 487 (1943).
CuTi03 SB II 484.
CusVS4, сульианит SB II 347; SB III (//24:94) 437; SB IV 17t3;
БМКД (1938). См. также ДАН 32, 427 (1941); F (1912): О (1950).
Cu—Zn SB I 501, 502. 532; SB III 593; SB IV 223; SB VI 158;
SB VII 201, 202; Ж. Э. Ф. 10, 103 (1940); P. R. S. (A) 177, 149
(1941).
2250. CuZn SB I (52:70) 147 (1.20:488), 501, 503, 533; F (1942).
CusZn( SB I (DSt: 499) 533; SB II 693, 697.
CuO—ZnO SB III 240
2253. CuS—ZnS Ann. 42 (5). 561; Ch. Z. I. 1362(1944).
870 _ 2254. F,-2290. Fe [гл. XI
§ 301. фтор F
2254.F. SB VI 52; О (1950).
HF" SB VII 65, 66; Z. ph. Gh. (B) 43, 229 (1939); Ж. Ф. X. XV,
597 (1941); О (1950).
F.,0 SB III 276.
Фториды J. Am. 63, 37 (1941).
§ 302. Железо Fe *
Fe SB I (.41:15) (.12:16) 66, 760, 761; SB II 165, 167, 193-196,
317, 624, 632-638, 641 — 654, 657-659, 661, 662, 666, 679-683.
706, 707, 710, 720, 747, 759 -776, 779, 780, 782-789, 792, 795;
SB III 181. 187, 221; SB IV 229; SB VI 43, 52, 53. 54; SB VII
45, 49; HRF (1938); БМКД (1938); ДАН 24, 554 (1939); F (1942):
Ch. Z. II, 989 (1943) и Ж. Ф. X. XXII, 956 (1948); О (1950).
FeBr„ SB II 246.
2260. Fe(NH3)„ Br, SB III 475; F (1942); О (1950).
NH4FeBr46H..O SB V 56.
Fe—С ' SB I 576; SB HI 619; Ch. Z. I, 5 (1945); F (1946);
О (1950).
Fe3C SB I 577, SB II 33, 194, 302-304, 763-765, 767-769, 772,
776, 779, 782; SB III 620; SB IV 243; SB V 61; Б. (1947); О (1950).
Fe2C SB II 769.
Fe(CN), SB I 451.
Fei"Fc"i(CN), SB IV 190.
Fe4[Fe(CN),]3 HRF (1938).
(NH4)4Fe(CN)e SB I 451.
FeCO, SB I {СЛ : 295) 326; SB II 392; О (1950).
2270. Fe(CO)s SB VII 131; О (1950).
Fe.(0OK SB I 285; SB VII (FV28), 29, 131; Ch. E. N. 25, 2970
(1947); О (1950).
Fc(C0)4 SB II 373; О (1950).
Fe(CO)4H2 SB VII 130; О (1950).
Fe(CO),X,(X (l,Br, J) Acta XII, 411 (1940); О (1950).
Fe (HCOO)3H20 HRF (1938).
Fe(C,04) 2НД) HRF (1938).
(NH4),Fe(C,04)3 HRF (1938).
Fe, лактат' HRF (1938).
Fo. тартрат HRF (1938.)
2280. Fe. фенолсульфонат HRF (1938).
Fe, са.шцилат HRF (1938).
Fed, SB I (£19: 743) 773; SB II 245, 246; ИАН (х), стр. 382 (1944);
С. А. 39, 3988 (1945); О (1950).
FeCl, SB II 215, 293; HRF (1938); О (1950).
Fe(AH3),Cl, SB III 475; К (19-12); О (1950).
Fe (П,0)(С1,* HRF (1938).
Fo(H*,0)4Cl, HRF (1938).
Fe(H.;0),n. HRF (1938).
(NII4V,FeCl5H.,0 SB II 216; J. Am. 70. 3064 (1948).
FeOCl SB III (£0,:67) 376; SB IV 142; О (19:0).
2290. Fc-аммоиийхлорид HRF (1938).
§J02| __^ 2291. FeCI33N,0-2:!38. (Fe,03)„(MeO)., 871
2291.FeCl,3H20 SB II 215.
Fe(NHs),(C104), SB III 475; F (1942); 0 (1950).
Fe(C104)26H,0 SB III 452.
(Fe, Co. Ni)3P SB II 302.
(Fe, Cr)4C SB IV 243.
(Fe, Cr),C, SB IV 243.
(Fe, Cr)3Si2 SB IV 245.
FeF3 SB II 34, 290, 291; О (1950).
FeF2 SB I (C4 : 158) 192; F (1940); О (1950).
2300. FeF3 • 4l/, H,0 HRF (1938).
(NH4)3FeFe SB I (//7,: 437) 449; SB V 100; F (1942); Б (1947);
О (1950).
Fe2Ge Z. Met. 35, 218 (1943); С. А. 39, 1095 (1945).
FeGe, Z. Met. 35, 218 (1943); С. А. 39, 1095 (1945): F (194(3).
K,HgFe(\0,)B SB III 483; F (1942).
FeJ„ SB II 247.
Fe(NH,),J. SB III 475; F (1942); О (1950).
Fe11 (CN)„Fe»'K SB I 452.
Fe1» (0N),Fei[K SB I 452.
K3Fe(CN)e SB I 450: SB II 495; HRF (1938): О (1950).
2310. K4Fe(CN)e3H,0 SB I 450: HRF (1938); Ж. Ф. X. XXI, 405, 521, 87!)
(1947); О (1950).
K3FeF, SB V 100: F (1942).
K3NaFeCl6 SB II 494.
Xa4 (KFe2) ОН (804),5НЛ), унгемахит SB VI 100.
K2NiFe(CN), SB VII 155.
K3Fe(C204)33H„0 HRF (1938).
11 Fe„03K20 SB VI 75.
K2Fea64 SB III 431.
K,PbFe(NO,), SB III 483.
KFeS2 SB III 385.
2320. KFe(S04)2 J. Am. 67, 1490 (1945).
K3Fe(S04), J. Am. 67, 1490 (1945).
KaFe(S04j, J. Am. 67, 1490 (1945).
NasFe(S04)3 J. Am. 67, 1490 (1945).
Fe2 (FeO) K6 (S04), 9H20, мотавольтин SB II 443; SB IV 18S.
(K, Na) (Fe, Mn)4 (OH, F), (Ti, Zr) (Si,07)2 SB III 557.
K„SrFe (N02), SB III 483; F (1942).
K;ZnFe(CN), SB VII 155.
Li3FeF, SB V 100; F (1942).
LiaFea04—MgO SB III 409, 410.
2330. Li=Fe,04—MgO—Li2Ti03 SB III 409, 410.
(Li, Mn)FeP04, ферриеиклерит SB VI 107.
Li(Fe, Mn)P04 SB II 455.
Li,FeN„ SB II 368.
LiFeO/ SB II 225, 332; О (1950).
Li2Fe204—Li2Ti03 SB III 409, 410.
FeM„Fe (CN), (M—щелочной металл) SB VII 15.';.
FeMeO, SB I (#1,:352) 417.
2338. (FelO,),, (MgO)r SB II 321.
872 2339. FPtMeP«~_Fc.0,%qr-2383. Na4Fe(CN). _ [гл. XI
2339. Fe.,Mg04 = Fe303MgO, магнезиоферрит SB II 321, 469 -471, 476,
"477; SB IV 167, 169; HRF (1938); ВМКД (1939); F (1942); О (1950).
2310. Fe2Mg,0915H20 SB II 361.
MgMn (Fe.,04)., SB II 478; F (1942).
(Mg, Fe). Si04, оливин SB I (tf 1,: 355) 400, 405; БМКД (1938); О (1950).
(Fe, Mg)Si03, чиперстен SB II 137, 529, 530; БМКД (1938).
(Fe, Mg), (OH)2 (Si^O,,).,, антофиллит, кум мин попит SB IV 216;
БМКД (1939').
Fe—Mn SB III 578; SB IV 244.
Fe6Mn SB II 649.
Fe—Mn—С SB I 579; SB IV 244; С. А. 39, 3198 (1945).
Fe,MnC SB II 34.
(Fe, Mn)3C SB II 34, 302, 303; О (1950).
2350. Mn3 [Fe (CN)e]2 SB VI (J25: 28) 29, 116.
(Fe, Mn)Nb,0, SB II 55, 337.
Fe—Mn—О, вреденбургит Ch. Z. I, 637 (1945).
Fe203—Mn.,04 SB III 348.
(Fe, Mn),03, биксбиит SB II 39; БМКД (1939); F (1942).
FeMn03 SB I 785.
Fe2Mn04 SB I (tf 1,: 352) 417; SB II 469, 470, 477; SB IV 167; F (1942);
О (1950).
(Mn, Fe). (ОН) P04, триплоидит SB VI 109.
(Mn, Fe)*P04, гетерозит SB VI 107.
(Mn, Fe)NaP04 SB VI 108.
2300. (Fe, Mn)4 (C!, OH)6 Si30, SB II 589.
MnOmFeO„ (SiOa)m+„, пироксмангит SB V 112.
«Железородонит» (близок к пироксмантиту) SB V 112.
(Fe, Mn)Ta2Oe SB II 337.
(Fe, Mn) \V04, вольфрамит Ж. Ф. X. XIV, 844 (1941); Ch. Z. I,
585 (1942).
Fe—Mo SB I 502, 526.
Fe3Mo2 SB I 526; SB II 657, 658.
Fe,Mo0 SB III (DS,:62) :64.
FeMo SB II 657, 658.
Fe—Mo—С SB III 623.
2370. Fe2lMosC, SB III (DSt: 60).
Fe3MoaC SB I 586; SB III 623.
Fe-N SB I 590; SB V 133; О (1950).
Fe,N SB II 783.
Fe4N SB 1(7/10:487) 590; SB II 783-787, 789; SB V Ш; HRF (1938).
Fe.N SB I 590; SB II 284, 302, 785, 787, 789; HRF (1938).
Fe2N SB I 590; SB II 284, 783, 785, 787; О (1950).
FeN SB V 133.
(NH4)2Fe(S04), SB II 95.
NH4Fe(S04), " SB I 789; SB II 91; О (1950).
2380. (NH4)2Fc(S04).,6II20 SB II 436, 437; HRF (1938).
NH4Fe (S04) „Ш1.0 SB I Ш42:370) 390; SB III 455; HRF (1938);
F (1942).
NH4Fe(S04)., < 1Ш..0 HRF (1938).
2383. Na4Fe(0N)", HRF (1938).
§302) 3384. Xa4Fe(CN), • 10Ц.О-2419. Fe.P 873
2384. Na4Fe(CN), • 101I2O HRF (1938).
Ла.Ге (CJV), • NO • 2H„0 HRF (1938).
2Na3Fe(O,04)39H20 HRF (1938).
NarFeF, SB V 100; F (1942).
NaFeO, --- Fe.,03Na,0 SB III (F5, : 75) 392; SB VII 85.
NaFe(SiO,)„ акмит—эгирип SB I 327; SB II 527; БМКД (1938).
2390. Na2(Ti, Fe)Si4On, нарсареукит SB III 558; F (1946).
Na.Ft'SioO.Si.TiO, SB I 791.
Fe,Nb Z. Kr. (A) 103, 391 (1941); Ch. Z. I, 1472 (1942).
FeNb04 С. Л. 39, 2684 (1945); F (J946).
FeNb.O,, колумбит С. A. 39, 2684 (lf'45); Б. (1947).
Fe—Ni SB I 502, 521; SB III 582; SB IV 228; SB V 124, 126;
SB VI 169, 170; SB VII 189; Ph. R. (2) 58, 1031 (1940).
Исследование с помощью нейтронов. Ch. Z. I, 982 (1944).
Fe8Ni SB II 634.
Fe3Ni, SB II 634.
FeNi, SB II 634.
FeNi^ SB II 6: 4; SB VI 169, 170; SB VII 190.
2400. FeNi4 SB II 634.
Ni3[Fe(CN)e]., SB VI (J2§: 28) 29, 116.
NiFe(CN), SB VII 155.
Fe.,Ni04 SB I (tfl,: 352) 417; SB II 468, 477; SB IV 167, 169; V (1942);
О (1950).
(Fe, Ni)9S8 до (Fe, Ni)S, иентландит SB IV (Я8,: 28) 137; Б (1947);
О (1950).
Fe—Ni— V SB III 585.
Fe2n+103„tl SB II 477.
Fe—О Ж. Ф. X. XV, 101 (1941); В. И. Архаров, Окисление металлов
(194S);J.Am.67, 1398(1945); 68, 798(1946); Ж. Ф. X.XX1I.956 (1948).
FeO, шоСтит SB I (#1 : 74) 122; SB II 222-225, 315; SB III 248,
249; SB IV 97; SB V 40, 48; SB VII 72; HRF (1938); F (1942); О (1950).
Fe304. магнетит SB I (Я1,:352) 114, 417, 418; SB II 44, 223, 224,
317, 470, 476, 477, 769; SB III 433; SB IV 132, 167, 169, 170; SB
V48, 68; SB VI 116; HRF (1938); БМКД (1938); F(1942); Ch. Z. II,
989 (1943); Ch. E. N. 25, 2970 (1947); О (1950).
2410. Fe.,0.(, гематит SB I (Drol: 242) 114, 196, 266; SB II 45, 196, 224,
"310, 314, 317, 318, 320, 322, 323, 356-358, 476, 477, 591: SB III
343; SB IV 131; SB V 48, 68; SB VI 72, 73, 87; SB VII 102,
103, 104; ДАН 1, 458(1935); HRF (1938); БМКД (1938); ДАН 24,
554 (1939); С (1939); Ж. Ф. X. XV. 101 (1941); Z. ph. Ch. {В) 50,
13 (1941); С. А. 39, 2441 (1945); Б (1947); О (1950).
Fe,0,xH,0 SB II 224, 321, 356--359; Ж. Ф. X. XXII, 953 (1948).
FeOOH - Fe.03H,0. гетит SB II 224, 352, 356 - 360, 411, 477; SB
HI (£0.: 64)" 372 (EQ,: 66) 375, 347; SB IV 131, 141, 142; SB VI 87,
88; HRF (1938); С (1939); Z. ph. Ch. (B) 50, 13 (1941); Ж. Ф. X.
XV.101 (1941); Z. El. 49, 269 (1943).
Fe(OH), SB I (6'6: 163) 196, 775; SB II 224; SB V 56; (J (1950).
Fe(OH)3 SB I 253; SB II 357; SB VI 87.
Fe(OH)., Fe(0H)3 Ch. E. N. 25, 2970 (1947).
Fe(OH)3H..O SB II 357.
Ферриты С А. 39, 2441 (1945).
Fe—P SB I 593; SB HI 625; 0(1950).
2419. Fe,P SB I 593, 594; SB HI 625; F (1946).
87/f 2420. Fe4P—24>3. Fe—Se [гл. XI
2420. РсЛ» SB I 593. 594; SB II15, 284; SB III 625; HRF (1938); 0 (1950).
FeP SB I 593; SB III (531 : 18) 263, 264, 265; 0 (1950).
FeP2 SB III (£18:22) 310, 625; О (1950).
Fe„P5(FeP2,5V) Z. An. 242, 233 (1939).
FePO, SB III 425.
FeP042H,0, фосфосидерит SB VI 102; SB VII 143.
FePO«(H„0)2, етренгит SB VI 112; SB VII 143, 144.
Fe2P„Oft8II.,0, вивианит SB V 94.
Fe(H2P0.,)"a HRF (1938).
PbFe(CN)4 SB V 99.
2Ш. Pb.Fe (CN), SB VII 155.
Pb„Fe(CN), 311,0 HRF (1938).
(NH4), PbFe(N0,), SB III 483; F (1942).
Pb5Fe4(Cl, 0Н)Д0, гоматофанит SB II 368; БМКД (1939); F (1946).
6Fe203Pb0, магнетоплумбит SB VI 74, 75.
PbFea04 SB I 417; SB II 477.
F>4PbO, SB П 338.
Fe2PbeSb..,S,8. джемсонит SB VII158, 159.
Tl.PbFe (NO,), SB III 483, 484; F (1942).
Fe—Pd SB VI165, 166; SB VII188.
li'i'iO.FePt SB II638.
Fe"M..Fe"i (CN), (AI—щелочной металл) SB IV 190.
MFe3(S04), (OH) (M = K, Na, Ag, NH4, Pb), яроаит SBV92.
Fe^MFe» (CN)6 (M—щелочной металл) SB IV 190.
Rb3FeFe SB V 100.
Rb3Fe (CN), SB III 480.
11 Fe,03Rb,0 SB VI75.
РешМНии (CN), SB IV 191.
Fe—S Z. An. 246, 169, 195 (1941).
FeS. пирротин, троилит SB 1(58: 86) 132, 133. 134; SB II233, 272;
SB III 256; SB V 41; SB VII 73; HRF (1938); ИАН (х), стр. 382
(1944); С. А. 39, 3988 (1945); 0(1950).
2450. FeS,. пирит, марказит SB I(C2:153) 215, 780; SB I (CI 8 : 497) 216,
781; SB II272, 273; SB V 52; HRF (1938); БМКД (1938); F (1942);
ДАН 57. 821 (1947); Б (1947); О (1950).
FeS04 HRF (1938).
FeS04H20 SB 1389; HRF (1938).
FeS047H,0 SB 1389; HRF (1938).
Fe4 (S04)ДОН), 16 H20, копиапит*) SB VI101.
[Fe,(S04),]r (Fe.O,), (H20). SBII441.
Fe,(SO,)aH,0 HRF (1938).
Fe—Sb SB I 597; О (1950).
FeSb SB I (58:87) 142, 597, 598, 765; SBII747; 0(1950).
FeSb, SBI(C18:497) 597, 598; 0(1950).
!i4(i0. FeSbS, гудмундит SB IV (£0,: 30) 142, 143; SB VI 84; SR VII
(E0,:17) 122; О (1950).
FeSbO, С. А. 39, 2684 (1945); F (1946).
FeSb,04 SBIV(#33:35) 147; Ch.Z. I, 342 (1941); C. A. 39,2019
(1945); F (1946).
2463. Fe—Se SB III 625; SB VI166.
*) По Дана и формуле копиаиита 18Н20.
jj021_ __ 2464. FeSc—2508. FoV 875
2461. FeSe SB I (#8 : 86) 125, J38; SB HI 625; F (1946); 0 (1950).
FeSe, SB VI166.
Fe—Si SBI587; SBIII624; N. 163, 413 (1943); 0(1950).
Fe,Si SB I (/.21 :488) 588; SB III 625.
Fe>i, SB II760.
Pe'Si SB I 587; SB II 13, 241, 759, 760; SB 111(528: 13) 624; HRF
(1938); F (1942); К (1947); A. Cr. 1, 212 (1948); 0 (1950).
2470. FeSL SB 1229, 587; SB II 760; HRF (1938); F (1946); 0(1950).
Fe—Si с доб. Л1... Ж. Ф. X. XX, 597 (1946).
Fe (НО), SIP, SB II104, 504; 0(1950).
FeSiO, SB II 518, 533.
Fe,SI04 SB II 518.
Fe,Si40l0(0H),. нонтронит SB VI141; БМКД (1939) *); G (1939).
H,Fe7(Si03)s " SB II134, 533.
9 FeOFeoO, 8 Si02 S 11,0. гриналит SB IV 217; БМКД (1939).
Fe^Fe»1 (6H),,Si.,0.,, 211,0 SB IV 217.
H.Fe,Si,0„ 4H.0' "" SB II 587.
2/iS0.Pe,Si8OIO(OII)e, крою-тедтнт SB VII (55,: 40) 41, 167.
Силикат желе»», ферростилниоме.иш SB VII175.
Fe5HSi ,W240e4 53 H.,0 SB VII149.
FeHSlW^O*. 30ILO SB IV 212.
FeHSiWJ204028H.,0 SB IV 212.
FeHSlW,,04,34(?j HO SB IV 212.
FeHSiW, 0„,30H,0 SBV 107.
Fe—Sn SBIII615; SB VI177; M. 1, 17 (1946).
Fe,Sn G. A. 39, 2683 (1945).
FeSn SB II 720; SB III 615; SB VI (£35:4) 5, 178.
2490 FeSn., SB III 616; Z. Met. 35, 218 (1943); C. A. 39, 1095 (1945);
C."A. 39, 2683 (1945); F (1946).
Fe6Sntl,09 SB II 477.
Fe4SnCl,0, SB II 577.
6Fe203SrO SB VI75.
(NH4)2SrFe(NO,), SB III 483; F(1942).
Fe2Sr04 SB II477.
TI„SrFc (N0„)4 SB III 483; F (1942).
ГеЛа Z."Kr. (A) 103. 391 (1941); Ch. Z. I, 1472 (1942).
FeTa04 C. A. 39. 2684 (1945); F (1946).
Fe—Те SF;VI166.
2Л00. FeTe SB I 765.
FeTe, SB VI166.
Fe—Ti SB VI167.
Fe2Ti SB VI167; ДАН 10, 262 (1941).
Fe,Ti SBIV 124; F (1946).
FelTiO,, пеевдобрукит SB 1442; SBII53, 336.
FeTi03, ильменит SB I 300; SB III (£2,: 69) 380; БМКД (1939);
Б. (1947); 0(1950).
Ff/nSi.O., SB II 579.
2508. FeV SB II661, 662.
*) По БМКД (ШУ), С (1WJ) и "и Дана нонтрониту отнечаот иной состав:
876 2509. FeVs04—2551. GaSb [l^A1
2509. FeV204 SB II480; F (1942).
7510. Pe—W SB I 502, 526, 586.
Fe3W SB II659.
Fe,W SB I 526; SB II 659; SB III (C41:36) 317; ДАН 40, 262
" (1941).
Fe3W2 SB I 526. 586.
Fe,W, SB III (08,: 61) 364; SB VI176. 177; Б (1947).
Fe—W—С SB 1585.
Fe4W2C SB 1586; SB II779, 780.
Fe2,W2C, SB 111(08,: 60).
Fe3W3C SB 1586; SB II779; SB HI (£9,: 71) 382, 623.
FeWO„ фсрбсрит SBII86, 450; БМКД (1938); О (1950).
2520. Fe—Zn SB I 562; SB VI 184, 185; SB VII 198; Z. Met. 33, 375
(1941); Ch. Z. II, 10 (1942); J. S. 16, 543 (1943); Ch. Z. I, 1271
(1944).
Fe2Zn Z. Kr. (A) 103, 391 (1941); Ch. Z. I, 1472 (1942).
Fe3Zn10 SB I (D8l : 499) 562: SB II 707.
Fe^n,. SB II706, 707.
FeZnl3" SB VII198.
Zn3 [Fe (CN)J., SB VI (/25: 28) 29, 116.
Fe20 — ZnO Z. El. 49, 301 (1943).
Fe2Zn04 SB I (Я1,: 352) 417; SBII469, 470, 476; SBIV 167; HRF
(1938); F(1942); 0(1950).
(Zn, Fe)S SB VII74.
Zn2 FcS, SB 1337.
§ 303. Галлий Ga
2530.Ga SBI(,111:740) 44, 750; SBII172; SB III (All :3) 201, 202;
О (1950).
GaF3 SB IV 125.
GaK(S04)212H20 J. Am. 65, 2071 (1943).
GaLi SB III (532 :19) 267; F (1942).
Ga—Mg SB VI180.
GaMg SB VI180.
GaMg, SB VI180.
Ga2M& SB VI180.
Ga2Mg04 SB II469, 471, 474; F (1942).
GaN SBVI(54:4) 59; Z. An. 244, 111, 146 (1940); 0(1950).
2540.GaNH4(SO1)212Hi.O J. Am. 65, 2071 (1943).
Ga—Ni Ch. Z. 1260 (1947).
Ga203 SB I (Z>5,: 242) 260; SB II310; 0(1950).
GaP SBI(53:77) 141; Z. An. 244, 146 (1940); F (1942); 0(1950).
Ga—Pd Ch. Z. 1260 (1947).
Ga—Pt Ch. Z. 1260 (1947).
Ga„Pt SBV54; F (1942).
GaRb(S04), 12H„0 J. Am. 05, 2071 (1943).
Ga2S SBII23"2, 284.
GaS SB II232.
2550. Ga2S, SB II232, 324.
2551.GaSb SBI(£3:77) 141; Z. An. 244, 146 (1940); F (1942); 0(1950).
§ Ж)7] 2552. GaSb04 -2591. (NH,), Hf F, 877
2552,GaSb04 С. А. 39, 2684 (1945); F (1946).
Ga,Ti Z. El. 49, 208 (1943); F (1946).
GaTl(S04)212H20 J. Am. 65, 2071 (1943).
Ga3Zr Z. El. 49, 208 (1943); F (1946).
§ 304. Германий Ge
Ge SBI(44:21) 54; Z. An. 244, 205 (1940); F (1942); 0(1950).
GeBr4 SBVI70; Ж.Ф. X. XX 6 (1946); 0(1950).
Ge(CH,)4 SB IV 277; 0(1950).
Gc(C,Hs)4 SB I (06: 615) 616, 649-651; F (1946); 0(1950).
25 ;0. GeCl4 SB III 328; SB IV 125; Ж.Ф.Х. XX, 6 (1946); 0(1950).
GeF4 Ж.Ф.Х. XX, 6 (1946); 0(1950),
(NH4)GeF, SB VII(/1,. : 34) 35, 36, 154; 0(1950).
Ge„H, SB VI 78: 0(1950).
Ge*H8 SB VI78; 0(1950).
GeJ2 SB VI (6*6 : 5) 62; О (1950).
GeJ4 SB l{D\l: 236) 251; SB VI68; F (1942); Ж. Ф. X. XX, 6 (1946);
0(1950).
K,GeFs SBVII(71„: 34) 35, 36, 151; О (1950).
Ga2La NW 31, 145 (1943); С A. 3S, 9 (1941).
Ge—Mj? Z. An. 245, 365 (1941).
2570. GeMg, SBIII(Cl : 20) 314; F (1942); 0(1950).
Ge.,Mg()4 SBII167.
Ge3'N4 SB VII 111, 112; Z. An. 24-1, 111-124,125-147(1940); 0(1950).
Ni,Ge04 SB II468.
GeO, SB I (C8: 168) 209; SB И 263; HRF (1938); F (1946); О (1950).
Ge—P Z. An. 242, 237 (1939); О (1950).
GeS SB II8. 232; 0(1950).
GeS, SBIII285; SBIV(C4'i : 11) 112; 0(1950).
Ge—Sb Z. An. 244, 205 (1940); 0(1950).
Ge2SeO, SB II467.
2580. Ge—Si SBVII212; Z. An. 241, 309 (1939); 0(1950).
Ge—Sn SB VII212.
Ge—Zn Z. An. 241, 309 (1939).
Ge02 2 ZnO = Zn2Ge04 SB II 4(57; SB IV 173.
§ 305. Водород Н2, дейтерии I)
H2 SBII166, 797-800; SB VI37; ДАН 62, 231 (1948); О (1950).
1), О (1950).
Гидриды см. соответствующие элементы, а также J. Am. 63, 37
(1911); Z. El. 49, 254 (1913).
§ 306. Гелий Не
Не SB II 797; SB V 29; SB VI (.43:3) 37, 38; ДАН 62, 231 (1948);
О (1950).
§ 307. Гафнии HI
Н! SB I (43:19) 53, 752; 0(1950).
HIC SB II 237; SB IV (SI: 6) 100; F (1942); О (1950).
2590. H!F4 SB III 328; О (1950).
2591. (NH,), HIP, SB I 453.
878__ 2592. ШО.-2629. Hg,J;HgJ |гл. XI
2592. HI02 SB II 265, 267, 434; О (1950).
H!P207 SB III (#6,: 140) 503; F (1942); О (1950).
H!(S04)2 SB II 434.
SrH!03 SB III (£2,: 68) 378; F (1942); О (J950).
§ 308. Ртуть Hg
Hg SB I (410:32) (410: 737) 42.750; SB II 169, 170. 702-706,
710, 736; SB III 183, 200. 201; SB VI 44; SB VII 56; Ch. Ph.
7, 958 (1939); Ch. Z. I, 1315 (1940); Ж. Ф. X. XX, 1414 (1946);
О (1950).
Hg2Br2 SB I (Z)3.:239) 113, 256; SB II 309; HRF (1938); F (1946);
0(1950).
HgBr2 SB II 18, 250. 251, 441, 442; SB III 278; SB V 51; HRF
(1938); Ж. Ф. X. XX, 6 (1946); О (1950).
Hg(Cl, Br)2 Z. Кг. A. 103, 415 (1941).
2600. HgBrClC2H4 ИАН (x), стр. 264 (1947).
Hg(NHa)2Br2 SB III 388; SB IV 147.
Hg(CHs)2 SB IV 276; SB V 151.
Hg(C,Hs)2 ИАН (x), стр. 262 (1948).
Hg(CHsCOO)2 HRF (1938).
Hg(CH3C0O) HRF (1938).
Hg, бевзоат HRF (1938).
Hg, сукцинат HRF (1938).
HgClSCH. SB VII (0tfO,:219) 220, 237.
HgClSC2Hs SB VII {OHO,: 219) 220, 238.
2561. Hg(CN)2 SB I (Fl, : 271) 270, 282, 774; SB II 372; HRF (1938); ДАН
45, 312 (1944); Ж. Ф. X. XIX, 433 (1945); F (1946); 0(1950).
Hg(CNO), 7..H..0 SB II 880.
HgCNS " HRF (1938).
Hg(SC2H6), SB V (02,: 142) 151.
Hg(SC,H7)2 SB V 143, 152.
Hg(SC4H,)„ SB V 152.
Bg(SC,Hu)t SB V 143, 152.
Hg(SC,Hl3), SB V 143, 152.
Hg(SC,H16), SB V 143, 152.
Hg(SC.Hl7); SB V 152.
2620. H2C12; HgCI SB I (03,: 239) 113, 237, 255; SB II 309; SB VII 70;
HRF (1938); F (1946); О (1950).
HgClj SB II 19, 249, 250, 441; SB HI (C28:23) 277, 278; SB V 51;
HRF (1938); Ж. Ф. X., XX, 6 (1946); Б (1947); 0(1950).
Hg(NH,),Cl2 SB III 388; SB IV 147.
NH4—Hg, хлорид HRF (1У38).
NH4HgCl, SB VI (А'2Б:15) 16, 79; F (1946); Б (1947); О (1950).
Hg(C104)2 HRF (1938).
Hg(CI04)26H20 SB III 452.
HgCI2 • 3HgO HRF (1938); БМКД (1939).
HgF2 SB III (CI: 20) 276; см. Ж. Ф. X. XX, 6 (1946); F (1942);
О (1950).
2029.Hg.,J2-HgJ SB I (D:\ : 239) 256; SB II; HRF (1938); F (1946);
О (1950).
§ 308| 2630. HeJ,—2674. HgS—ZnS 87»
2630. HgJ, SB I (C13: 180) 177. 189; SB II 251, 441; SB III 278; SB V 51;
HRF (1938); Ж. Ф. X. XX, 6 (19-10); F (11)1(3); О (1950).
Hg,J2S04 SB II 441.
Hg—К SB VI 179.
Hg2K SB III 362; SB VI 179.
K2HgI43H20 HRF (1938).
K,Hg(CN)4 SB I (Я1.Г352) 425; F (1942); О (1950).
K2HgCI4H,0 SB VI (£34:16) 17, 80.
HgNO, H,0 HRF (1938).
K2HgNi(N02), SB III 486; F (1942).
(NH4)>HgNi(NO,), SB III 486; F (1942).
2640. Rb2HgNi (NO.,), SB III 486; F (1942).
Tl2HgNi(NO,), SB III 485; F (1912).
HgLa SB V 45.
Hg—Li SB III 632.
Hg3Li SB III 632.
Hg.,Li SB III 632.
Hgti SB III (/.20:175) 265, 633; F (1942).
HgLi2 SB III 632.
HgLi, SB III 632.
HgMg SB IV (52:6) 101; F (1942).
2650. HgO, монтронднт SB I 120. 769; SB II 219, 220; HRF (1938);
БМКД (1939); Z. An. 244, 133, 144 (1940); О (1950).
Hg3(P04)2 HRF (1938).
Hg3P04 HRF (1938).
Hg—Pb SB I 571.
HgPr SB V 45.
Hg—Pt Acta XIII 90 (1940).
HgPt Acta XIII 90 (1940).
HgPt, Acta XIII 90 (1940).
RbHg(N03)2 SB III 401.
HgS, киноварь, мегациннабарнт SB I (£3:77) 129, 772; SB I
(£9:89) 129; HRF (1938); БМКД (1939); Z. An. 244, 133, 116
(1910); F (1942); О (1950).
2660.HgSO4 SB II 441; HRF (1938).
HgS042HgO SB II 441; HRF (1938).
(MgS04)x (HgO)„ (H20), SB II 441.
Hg2S04 HRF (1938).
HgSb4S7 SB IV 150.
HgSe, тиманнит SB I (£3:77) 136; Z. An. 244, 133, 116 (1940);
БМКД (1939); F (1942).
Hg—Sn SB I 570; SB III 645.
HgTc SB I (£3:77) 137; Z. An. 244, 133, 146 (1940); F (1942).
Hg—Tl SB III 645; SB VI 179.
HgsTI„ SB III 362; SB IV 136; SB VI 179.
2670. Hg4TI2 SB III 362.
TlHg(NO,)3 SB III 401.
Hg—Zn SB I 567.
HgZn(CNS)4 SB IV 165; F (1946).
2674. HgS—ZnS SB IV 98.
880 _ 2675. Jn-2716. _TI,Ir (NO*), | гл. XI
§ 309. Индий In
2675. In SB I (46:24) 44; SB II 173; SB III (.46:3) 202*; HRF (1938);
Ch. Z. I, 458 (1942); С A. 37, 4948 (1943); F (1946); О (1950).
InCl, HRF (1938).
InCl2 SB IV 103; О (1950).
(NH4)>InCl6H„0 J. Am. 70, 3064 (1948).
InF, " SB IV 125.
2680. InF, SB IV 125.
InLi SB III (532:19) 267; F (1942).
In—Mg SB VI 180.
In2Mg SB VI 180.
InMg SB VI 180; F (1946).
InMg„ ' SB VI 180.
In,Mg6 SB VI 180.
InlMg04 SB II 471; F(1942).
InN SB VI (54:4) 59; Z. An. 244, HI, 133, 146 (1940); О (1950).
InNa SB III 268; F (1942).
v,,!J0. In—M Ch. Z. 1260 (1947).
InNL, ПАИ (х) 39, 114, 201 (1944); С. А. 39, 1583 (1945).
In03 SB I 261; SB II 39, 310; HRF (1938); F (1942); О (1950).
ln.!o,3H.O In (Oil), SB IV 132.
In— Pb SB IV 249; SB VI 194.
In-Pd Ch. Z. 1260 (1947).
In-Pt Ch. Z. 1260 (1947).
In2Pt SB V 54; F (1942).
InSb SB I (#3: 77) 141; Z. An. 244, 133, 146 (1940); F (1942); 0(1950).
In—Zn Z. Met. 35, 1425 (1943); Ch. Z. II, 4 (1944).
§ 310. Иридии Ir
2700. Ir SB I (41:15) 70, 764; SB II 164, 165, 200, 266. 638; SB III
181, 224; HRF (1938); БМКД (1939); F (1942); О (1950).
IrCl2 SB I 193, 773; О (1950).
IrCl3 HRF (1938).
(NH4)3 IrCJeNH4N03 Ж. Ф. X. XVIII, 160 (1944).
IrF, SB II 307.
K,Ir(CN), SB II 495.
KJr(3V02)e SB HI (/2,: 127) 482; F (1942); Б (1947); О (1950).
<NH4)2 IrCl. ДАН 26, 789 (1940).
(NH4)3 Ir (N02), SB III (/2t: 127) 482; F (1942); О (1950).
Ir02 SB I (C4:158) 213; SB II 266; F (1946); О (1950).
2710.1r—Os SB I 525; SB VI 195.
IrmOsnRu„, рутеносмиридий SB IV 144; БМКД (1939).
Rb,Ir(NO,), SB III (/2^127) 482; F (1942); О (1950).
Ir-S SB V 132.
Ir2S, SB V 132.
Ir—Sn M. 1, 137 (1946).
2 46. Tl,Ir(N02)„ SB III (/2,: 127) 482; F (1942); О (1950).
>• 312|
2717. J, Js-2759. KCI
881
§ 311. Под J
2717. J, J, SB I 759; SB II 5; SB III 221; HRF (1938); Z. ph. Ch. (B)
5i, 219 (19'i2); 53, 320(1943); Ж. Ф. X. XX, 1414 (1946); О (1950).
JF, SB V 83.
HJ SB II 207, 208; Ж. Ф. X. XV, 597 (1941).
-720. HJ03 SB I 339; SB II 332; О (1950).
J.,0. HRF (1938).
(NH4).H,JO, H. Ph. 18, 267 (1945); G. A. 40, 258 (1946).
Нодиды J. Am. 63, 37 (1941); О (1950).
§ 312. Ка.шй К
К SB I (Л2 : 16) 34, 35, 746, 747; SB II 166; SB VI 40; SB VII 202;
HRF (1938); F (1942); R. M. 15, 90 (1943); Усп. 15, 297 (1946);
О (1950).
KBi SB I (B\ : 73) 97, 98, 99, 106; SB II 211, 213, 214, 329; SB IV
91; SB V 37, 38; SB VI 55; HRF (1938); F (1942); 0(1950).
KBrO, SB I 311, 339; SB II 68, 408; HRF (1938); О (1950).
K€8, графит—калий SB II 180; О (1950).
KC,„ графит—калий SB II 181; О (1950).
КНС. SB I 783; SB II 275; F (1946); О (1950).
2730. KCJf SB I (Ft: 269) 281; SB II 371; HRF (1938); F (1942); О (1950).
KCNO SB I (F5.,:279) 289; HRF (1938); F (1946).
KCNS SB III (РЬЩ:Щ 396; HRF (1938).
KCNSe HRF (1938).
K2C0, HRF (1938).
KHCO, SB IV 158; HRF (1938).
K(CH,COO) HRF (1938).
KHC.,04 SB III (6>4.: 674) 728.
KHC*0« 1/2 11,0 HRF (1938).
KaC,0. ' HRF (1938).
1740. (СООК).Н20 SB III (04, : 675) 726; HRF (1938).
K(CCH6C00)3H20 HRF (193S).
K, <CeH507) H,0, лимоннокислый калий 11RF (1938).
K(C6H4OHSOJ HRF (1938).
C,H60K, фенолят калия HRF (1938).
C„H4(C0)3NK, фшлимид К HRF (1938).
CeH4C00HC00K HRF (1938).
C,H40HC00K HRF (1938).
P-CH,CeH5S0,K HRF (1938).
C6HrS0,K HRF (1938).
2750.CsH,S'OtK HRF (1938).
2(rH3)S04KH,0 HRF (1938).
(:4H400K2 HRF (1938).
C4H4OeK(NH4) HRF (1938).
C4H40,KH HRF (1938).
C5H.,N403KII, уреат калия HRF (1938).
0.1H6"C0S.,K HRF (1938).
K20S, HRF (1938).
P,KCeF4, A. Cr. 1, 265 (1948).
2759. KCI SB I (51:73) 97, 98, 99, 105; SB II 210, 213, 214, 329;
SP IV 91, 92; SB V 37; SB VI 55; SB VII 69; HRF (1938); I (1942);
882 2760. KJC14 -2801.KJIoU4 [гл. XI
Hi. В. 66,1'. i (1911); ИА11 (x), стр. 382 (1914); С. А. 39, 3988(1945);
39, 4275 (J945); 0 (1950).
2760. KJCI4 SB VI (//010:20) 21, 95; 0 (1950).
K0104 SB I {112 : 341) 372; SB II8'., 411 - 113; SB IV 163: 1IRF (1938):
]< (1942); О (!950).
KC103 SB I 311. 339; SB II 66, 408; 1IRF (1938);'Z. Kr.(A) 104.
375 (1942); О (1950).
KC102 SB II 378.
ICCrO, HRF (1938).
КД5г40, HRF (1938).
KP SB I (7il : 73) 97, 98, 105; SB II 210; SB IV 91; HRF (1938):
F (1942); О (1950).
KIIF2 SB I (F5,:279) 286; SB VII 132; F (1946); О (1950).
KP • 2 HO HRF (1938).
Kl»F„ SB II 491.
2770. KH SB II 242; F (1912); О (1950).
К I) 0(1950).
KJ SB 1 (ZM :73) 97, 98, 99, 106; SB IV 91; SB V 37, 38; SB VI
55; HRF (1938); F (1942); О (1950).
KJ, SB I 287.
KJ04 SB I (//4 : 349) 373; SB II 83; HRF (1938); F (1916); О (1950).
KJ03 SB I (,.5 :303) 311; SB II 49; HRF (1938); F (1912): О (1950).
KJ03HJ0, HRF (1938).
KF— LaF, J. Am. 70, 2147 (1948).
<xKLaF4 ' J. Am. 70, 2147 (1948).
(i'KLaF4 J. Am. 70, 2117 (1948); A. Cr. 1. 265 (1948).
2780.KLa(MoO4)2 Ch. Z. I, 271 (1914); С A. 39, 2019 (1945); F (191(5).
KLaSiO, SB II 155.
KLa(W04), Ch.Z. I, 271 (1944); С. А. 39, 2019 (1945); V (1916).
К1ЛС03 SB II 396.
KLiMg,Si4010F„, тайниолит SB VI 152.
LiKS04 SB "I (ffl4: 358) 376.
КМ?(Н,0)6Вгч SB VII 117.
KMg (H20)„ (Br, Cl)a SB VII (Е26:19) 20. 21, 115-117; F (1916).
K„Mg(C03), SB II 396.
К.ОВМйОбСОбНО SB V 82.
2790. KX03MgC034H,6 SB V 82.
KMgF3 SB I (G5:303) 337; F (1942); О (1950).
KMgCl,6H,0 SB I 338; SB II 329; SB VII 117.
K.,Mg.,(S04'),, лангбсйнит SB II 442; БМКД (1939); F (1942)*)
KJUnfCN), SB II 195.
KMn04 SB I {HI:. 344) 373; SB II411. 115, 439; SB VI 97; HRF (1938):
О (1950).
KMn [SO,] 411,0, манганлеонит SB VII (Я4,3:32), 33, 34, 1.0.
K4Mo (CN)", 2H20 SB VII (F2,:25) 26, 27. 132; О (1950).
K3MoC16 Ch. E.N. 25, 2970 (1947).
К.МоДеО^Н.О J. Am. 70, 1291 (1948).
2800. K3P0412Mo0941I,0 SB VII 149.
2801. K3Mo04 HRF (1938).
*) В работе ¥ (1»VJ) ошибочная формула: К, Мц<(304)3.
§ 3121 2802. KN—2847. К, I4(CN)4-3IU)
2802. KN, SB I (7"52:279) 289; SB IV 151; IIRF (1938); F (1940);
Б (1947); О (1950).
К-аммонин фосфат ПНР (1938).
KNO, SB I 339; SB II 38!; SB VII 133; 11RF (1938); О (1950).
KNO, SB IV (F5U:39) 152; HRF (1938): О (1950).
К—Na SB VII 202.
KNaC03 IIRF (1938).
KNa,,(?i04)„ (CO,). CI, ганскнт SB И 141 п след.; SB VI 98; SB VII
"136. 137.
ХаКдЯО,)., SB I 378.
2810.K,SO4-Na.SO4 Hi. Cli. 4~. 587 (1913); С. Л. 38, 90, (1914).
(К, Xa).,S04 J. Am. 03. 2533 (1911).
(NaK),f3O103H,O SB II 315.
KNbO, SB 1 (C;5:303) 310; SB II 335; F (1912); О (1950).
KsNbOs Gii. Z. 1. 11 (1912); С. А. 36, 308 (1912); F (194(5); О (1950).
Ko.N'bF, SB VII (A0, :30) 37, 159; О (1950).
KJNbOF, J. Am. «1," 1139 (1942).
KNdSi04 SB II 555.
K..Xi(CN)43H40 SB V 99.
KJSi(CoS)4 "SB III (Ctf0s: 655) 693.
2820. KNiFs SB I (<75:803) 337; F (19-42); О (1930).
K4Ni(NO)0 SB II 196; F (1942).
K.PbXi (NO.,). SB II 196.
(K,(XII4>,Tl),S04(Zn, Xi)S046H,0, шенит SB VI 97.
KN|>,F„ J. Am 70, 2117 (1918); О (1950).
K0.K,04 SB IV (CT1:8) 118; SB VI. 63; SB VII (Г11 : 7) 84;
Ж. Ф. X. IX. 932(1937); Ж. Э. Ф. VII, 1418 (1937); Z. An. 211. 97
(1939); ДАН 47, 199 (1915); С. Л. -10, 3948(1916): Ж. Ф. X. XX,
1103 (1916); F (1946); Ж. Ф. \. XXI, 215 (1917); О (1950).
КО, Ж. Ф. X. XXI. 245 (1917); ДАН 5!). 67 (1948); О (1950).
1U> SB III (П : 20) 283; FH912); О (1950).
КОН SB VII Tl; HRF (I938); О (1950).
K..0slir( SB IV (./1^-60) 189; F (1942).
2830.K,OsC!6 SB IV (.71,: 60) 189; F (1912).
K„Os\Cl5 SB III 507.
KOsO.N SB II S;<, ',18; F (1946).
K,OsOtCl4 SB III (./!.: 122) 487; F( 191(5).
K,Os042II,0 IIRF: (19*38).
к;НР04 MRF (1938).
K.IIP04- 311,0 IIRF (1938).
Kil,P04 SB I (/72.,: 363) 393, 391; SB II 454; IIRF (1938); N. 166,
20 (1945); F (191(1).
К.РО-ЗИО HRF (1938).
KHP0, HRF (1938).
2840. K2 (COO), Pb (COO),. (?) SB V 158.
KPbCl3 SB II 329.
(KPbCl,)3II,0 SB II 496.
K„Pd(C0S)4 SB Ш (У//0В:05Г>) 693.
ICPdCI4 SB I (7/1r:360) 121; SB V 98: F (1946): О (1950).
K".Pd(N0,)4 ДАН 58. 603 (1917).
K„"PtBr, " SB II 496, 497; F (1912).
2847. K2Pt (CN)4 • 3II..0 HRF (1938).
884 2848. K2Pt (SCN)„-2892.K2SnCl42ILO [гл. XI
2848.KtPt(SCN), SB I (Я6,: 435) 447; SB II 102; О (1950).
K2Pt(COS)4 SB III (ОЯ08:655) 693.
2850. K„PtCl, SB I (Я6Х: 431) 445, 793; SB II 496, 497; SB III (/1. : 121)
479; HRF (1938); F (1942); Б (1947); О (1950).
K„PtCI4 SB I (Я18:360) 424; HRF (1938); F (1946); Б'(1947);
0(1950).
K2Pt(N02)4 Ch. Z. II, 197 (1944).
KPu2F, J. Am. 70, 2147 (1948); О (1950).
KPuF, J. Am. 70, 2147 (1948); О (1950).
К—Bb SB VII 202.
K2ReBr, Z. An. 262, 317 (1944); С. А. 40, 4268 (1946).
K2ReCl, SB IV (71,: 60) 189: F (1942); Z. An. 262, 317 (1941); С. А.
40, 4268 (1946).
K2ReF, SB III 478.
KRe04 SB II 83, 416; F (1946).
2860. K,Rh [(COO)s], H„0 SB 1 664.
K3RhCl„ Ch. E.N. 25, 2970 (1947).
K,Rh(N0.)e SB III (/2t:127) 482; F (1942); О (1950).
K4Ru(CN), • 3H,0 Ж. Ф. X. XXI, 405, 879 (1947); О (1950).
K2S SB III (CI: 20) 283; SB VI 98; Z. An. 241. 281 (1939); F (1942);
О (1950).
КББ SB III (£22: 8) 258, 259 (£1 : 7) 258, 259; SB VII (B22 : 4) 78;
F (1942); О (1950).
K2S04 SB I 374, 376; SB II 86, 420, 423, 436, 438; SB IV 173; SB
VI 98; HRF (1938); О (1950).
K2SO, 2H20 HRF (1938).
K.S.O, HRF (1938).
3K.,S,OfH„0 HRF (1938).
2870.KHSO4 SB VI 97; HRF (1938).
K2S206 SB II 104, 505; HRF (1938).
K2SnO, SB IV 201.
K.S.0, SB II 108, 506-508; SB VI (#1^30) 31, 123, 124.
K2S"0. SB III (#5,: 138) 502; SB IV 201,
K„S40, SB IV 201.
КД0„ SB IV 201.
K:s,Oe SB I 479; SB III (£4,: 136) 501; HRF (1938).
KsSb SB V (Z>018:8)60; О (1950).
K„S-Sb,S3 HRF (1938).
2880. К(SbO) (C4H40e) 1/2 H20 HRF (1938).
K2Sc SB III (CI: 20) 283; Z. An. 241, 281 (1939); F (1942); О (1950).
KSeH SB VII (£22:4) (£3:3) 4, 78; О (1950).
KJSeBr. SB III (Л,:121) 477; F (1942); О (1950).
K;Se04 SB I 378; SB II 88; SB IV 173; HRF (1938); О (1950).
K2ScO, HRF (1938).
K2SiF„ SB III (У1Ж: 121) 479; HRF (1938); F (1942); О (1950).
K2OS—i02 SB VI 134.
K4SiWx,04018H„0 SB IV 213.
K,SnBr," SB'V 103; SB VI 121; F (1942).
2890. K2SnCla SB I (Я6,: 431) 443; SB III (/lt: 121) 479; F(1942); О 1950.
K,SnCUU) SB VII (£35: 22) 23, 118; О (1950).
2892. K2SnCl42H20 SB V 98; VII 119.
2893. KaSn(OH),—2930. Кг 885
2893.K„Sn(OH)f SB I (Я6„:432) 444; С. А. 30, 697 (1942); О (1950).
KSrM (NO.,) SB II 496; F (1942).
K..TaF. SB VII (A"6,:30) 37, 159; О (1950i.
KTaF, HRF (19Я8)Г
K9Ta08 Ch. Z. I, И (1942); С. A. 36. 308 (1942); F (1946); О (1950).
KTa03 SB II 335; F (1912).
КоТе SB III (CI :20) 283: Z. An. 2J1. 281 (1939); F(1942); О (1950).
2900. KjTeCI, SB III 479; F (1942).
K„Te04 SB IV 173.
K.,Tc042HO SB IV 173.
K,Te044,5H„0 SB IV 173.
K„TeO, HRF (1938).
KF—ThF4 J. Am. 70, 21П (19',8).
KTh4F», J. Am. 70, 2147 (1918).
KTiuF, J. Am. 70, 2147 (1948).
KThF, J. Am. 70, 2147 (1948).
aK,TbF, J- Am. 70, 2117 (1948).
2910. ^K2ThFf J. Am. 70, 2147 (1918); A. Cr. 1, 265 (1948).
K6ThF, J. Am. 70, 2147 (1948).
K,T1C1,2H.,0 SB III (J3,: 131) 495; F (1946); О (J950).
K„TiF,H„0" HRF (1938).
KlTi-,0, " C. A. 39, 2441 (1915).
КД'б, HRF (1938).
К./П0, HRF (1938).
KF—UF4 J. Am. 70. 2147 (1948).
KU„F„5 J. Am. 70, 2147 (1948); О (1950).
KUJP„ J. Am. 70, 2147 (1948); О (1950).
2920. KUJF8 J. Am. 70, 2147 (1948); О (1950).
KUF6 J. Am. 70, 2147 (1948); О (1950).
аКДТ, J. Am. 70, 2147 (1948); О (1950).
3,К.иР, J. Am. 70, 2147 (1948); A. Cr. 1, 265 (1948); О (1950).
З.КДТ, J. Am. 70, 2147 (1948); A. Cr. 1, 265 (1948); О (1950).
aK,PF, J. Am. 70, 2147 (1948) ;0 (1950).
a'KJT. J. Am. 70, 2147 (1948); О (1950).
KUO, (CII,CO0), SB III 718; F (1946).
KVO3 HRF (1938).
К W042H,0 HRF (1938).
2930. K3W.CI," SB III (Kll: 142) G0'(; Б (1947); Ch. E. N. 25, 2970 (1947).
К,Р041'ЖО,4П.О SB VII 149.
K—Zn SB VI 157.
KZn , SB V G5; SB VI {D23: 8) 9, 157.
K..Zn(€N)4 SB I (1П,: 352) 424; F (1942); О (1950).
K.,S04ZnS04OII..O HRF (1938).
K2ZnCl< J. Am. 67, 878 (1945).
KZnF3 SB I (G5 :303) 337: F (1942); О (1950).
K,ZrF7 SB VI 126; О (1950).
§ 313. Крицтон Кг
2939. Кг SB II 202, 203; F (1942); ДЛН 62, 231 (1948); О (1950).
«86 29'.0. Га—2**82. Li-ЛЛКТЛТ |гл. XI
§ 314. Лантан La
2940.La SB II 171: SB III (Al:3) 203; Z. El. 45, 337 (1939): F (1942);
О (1950).
La«r3 A. Cr. 1, 265 (19'iS), О (1950).
LaC. SB I (П1:741) 783; SB II 275; F (1940); О (1930),
LaCl, A. Cr. 1, 265 (1948); О (1930).
LaF3 SB II 27, 28, 288. 289: SB VII 82; О (1950).
La—0—F Z. An. 24S, 167 (1941); О (1950).
LaOF Z. An. 248, 167 (1941); О (1950).
LaGaO, SB I (Go : 303) 334.
I.aJ, A. Cr. 1, 265 (1948): О (1950).
LaLi (Mo04), Ch. Z. I, 271 (1944): C. A. 39, 2019 (1945); F(1946).
2950. LaLi(W04)2 Ch. Z. I, 271 (1944); С. А. 39, 2019 (1945); F (1946).
LaMg, SB III (Й32 :19) 326.
La, (MoOA, SB I 790; Ch. Z. I, 271 (1944); С. А. 39, 2019 (1945);
F (1916).
LaN SB V 42: О (1950).
LaF3—JfaF J. Am. 70. 2147 (1948).
NaLaF4 A. Cr. 1, 265 (1948).
p.NaLaF4 J. Am. 70, 2147 (1948).
LaXa(MoO<), Ch. Z. I, 271 (1944); С. А. 39, 2019 (1945); F (1946).
NaLaSlO, SB II 555.
LaNa5(W04)4 Ch. Z. I, 271 (1944); С. А. 39, 2019 (1945).
29(H). LaXa(WO«)2 Ch. Z. 1, 271 (1944); С. А. 39, 2019 (1945); F (1946).
La-Ni M. 2, 97 (1917).
l.a.,0., SBI (Z>5,: 244) 261 (Do.,: 745) 785; SB II 314,354; SB III
" 342; SB VII 88, 102; Б (1947); О (1950).
La,0,xH,0 SB II 354.
La(OH), SB I 253; SB II 351; A. Cr. 1, 265 (1948); О (1950).
LaOOH SB II 354.
LaP SB V 43; О (1950).
La-Pb SB III 646.
LaPb, SB III (/.12:174) 646.
La.Sa SB II 323: A. Cr. 1, 265 (1948); О (1950).
2970.LaSb SB V 44; F (1942); О (1950).
La—Sn SB III 646.
LaSn, SB III (112 :174) 646.
LaF,-SrF2 SB V 51.
LaTi, SB III 327.
La—Zn Z. Met. 33, 358 (1941).
LaZn SB V 44; F (1942).
§ 315. Литий Li
Li SB I (42: 1С) 32, 35, 747; SB II 166, 663, 722, 735; SB VII 202;
IIRF (1938); Ch. Ph. 9, 700 (1941); F (1942); R. M. 15, 90 (1943) к
Уси. 15, 297 (1946); N. (1915); Ph. R. 72, 245 (1947); С. А. 41,
6100 (1947); V (1942); О (1950).
LiBr SBI (#1:73) 97, 98, 101; SB IV 91, 92; HRF (1938); F (1942); О (U'50\
LiBr2H»0 HRF (1938).
2980. Li2C03" HRF (1938).
Ls-бешоат HRF (1938).
2M82. Li-аактат HRF (193b).
« Ш] 2983. Li-ОКСАЛЛТ—3026. Li.Se 887
2983. Li-оксалат I whj? mqq«\
Ll-i-йдпциаат HRF (1938).
Li-TapTjtaT моногидрат HRF (1938).
L\™>;paT } HRF (1938).
LiCl SB I (Bl:73) 97, 98, 100; SB IV 91, 92; SB VI 55; SB VII
69; HRF (1938); 1IAI1(.\), стр. 382 (1944); С А. 39, 3988 (1945):
F (1912); О (1950).
LiClH.O SB I 100, 766; HRF (1938).
LiC1043H,0 SB III (//4„:117) 468; В (1917).
2990. LiXrO. HRF (1938).
Li,Cr04m,0 HRF (1938).
U,(Y,0-m.O HRF (1938).
LiF SB I (Bl : 73) 97, 98, 99, 100, 184; SB II 236; SB IV 91; SB V
37; SB VII 67; HRF (1938); F (1942); О (1950).
(Li (Mn, Fe)) P04 трнфилии SB VI 107.
LiH SB I (H\ :74)146;SBII242;SB11I(/J1:7)238F(I942);0(1950).
LiD SB III («1 :7) 238; F (1942); О (1950).
LiJ SB I («1:73) 97, 98, 101; SB IV 91; HRF (1938); F (1942);
О (1950).
LiJ3H20 SB I 766; SB III 237; HRF (1938); Б (1947).
LiJ03 SB II 49, 332; Б. (1947); О (1950).
3000. Li—31? SB III 633; SB IV 247.
LlMs. SB III 633.
Ш1.\*(М- Мр и Д|». металлы) MV 33 121 (1946).
LLTiO,—MsO SB III 409, 410; F (1942).
LiMn04 SB VI 97.
LiMn043II,0 SB III 447.
Li.,Mo04 SB I (tfl3:356) 380; О (1950).
Li3N SB I (Z>1 : 231) 248; О (1950).
LLNO, SB I 339; HRF (1938).
Li—Na SB VII 202.
ЗОЮ.ЫХаСО, SB II 77, 394, 396.
LiNbO, SB I (9i :300) 315; О (1950).
Li.,0 SB I (Cl: 150) 221; SB III (CI: 20) 283; F (1942); Б (1947);.
О (1950).
Li.,0, SB VI 65; F (1946); О (1950).
LiOH SB II 215; SB III 238; HRF (1938); F (1946); Б (1947); U(1950).
LiOHH.,0 SB VII (#36:4) 5, 6, 70; 0(1950).
Li3P ' SB V (Z)014:6) 60; О (1950).
Li3P04 SB II 454.
Li—Pb SB VI 193; Z. Met. 33, 388 (1941).
Li.0Pb, (тин Cu.Al,), SB VII (7)8, : 16) 113.
;.020. LI.S SB I (Cl:150) 224; SB 111 (Cl : 20) 283; F (1942); О (1950).
Li.S04 SB I 376; SB II 420; HRF (1938).
I.i.;S04H.,0 SB III (/74,: 98) 446; HRF (1938).
Lis'Sb "SB V (ZH)lt:6) 60; О (1950).
LiSbO, SB VI 118, 120.
LiSb(OH), SB VI 118.
X026. Li,Se SB III (Cl:20) 283; F (1942); О (1950).
888 3027. TJHSiW„O<o24H.O-3070. Mr (HC0O>2.2H20 [гл. XI
3027. LIHSIWlOuS^O SB IV 213.
LijHSiW.oO^SeH.O SB IV 213.
Li4Sn SB II 735.
3030.Li,Sn2 SB II 735.
Li.Sn, SB II 735.
Li2Sn SB II 735.
Li,Sno SB II 735.
LiSn SB II 735.
LiSn, SB II 735.
LiSn4 SB II 735.
Li2Te SB III (CI: 20) 283: F (1942): О (1950).
1ЛТ1 SB III (L20: 175) 268; F (1942).
Li.TiO, SB HI 408; F (1942); С A. 39, 2441 (1945); О (19"0).
3040.Li/WO4 SB I (Я1а:356) 380; О (1950).
Li—Zn SB III 633.
LiZn SB III (#32 :19) 265; F (1942).
LiZn, SB III 634.
LiZn3 SB III 633.
LiZn4 SB III 633.
§ 316. Магний Mg
Mg SB I (.43 : 19) 40; SB II 164, 168, 664 667; 673, 722-724,
726 729. 732, 762; SB III 185-187, 195; SB V 32; SB VI 43. 44;
SB VII 52, 54, 55; Z. ph. Ch. (B) 40, 347, 356 (1938); HRF
(1938); J. 1. M. 67, 173 (1941); С A. 35, 6227 (1941); О (1950).
MgBr, SB II 246; О (1950).
Mg(NH3),Bi\ SB III 475; F (1942); О (1950).
MgBr26H20 = Mg(H20),Br> SBIII(/17: 124)489; HRF (1938): О (1950).
3050. M&C, HRF (1938).
MgC03 SB I (СЛ : 295) 317; SB II 388; SB V 82; ПМКД (1938): О (1950).
MgCl2MgC037H20 SB V 81.
MgCO,x H20 SB II 388.
MgC03 H„0 SB II 388.
MgC033H,0 SB II 388, 389; SB III 415; SB IV 158; SB V 82:
HRF" (1938).
MgCO,5H20 SB II 388, 389; SB IV 158.
xMgWjMg(OH),zH.O SB I 318; SB II 399.
4MgCO,Mg(OH),4H20 SB II 399; SB IV 158.
4MgCO,Mg(OH)25H„0 SB II 399.
3060.4MgCb3Mg(OH),2H.,O SB II 399.
MgC03 Mg (OH)2 3H20 SB IV 158; SB V 82, см. артиннт.
Mg..(CO,)(OH),3H.O, артшгат SB VII 134.
5Mg04CO,5H>0, гидромагнозит SB V 81, 82; HRF (1938).
4Mg03COo3H,0 SB V 82.
4Mg02C0,S0,711,0 SB IV 158, 159; SB V 82.
Mg(CH3C00), HRF (1938).
Mg(CH?C00)2.4H.,0 HRF (1938).
Mg (СДС00)2 . 3li20 HRF (1938).
Mg3(C,HsO,)*. 14H20 HRF (1938).
3070. Mg (HCOO)2 • 2H..0 HRF (1938).
«3101 3071. Mg (СЯН,0,)4.ЗН«0-3117. MgNLSb 889
3071. Mg(C,H403), • 3H0 HRF (1938).
Mg(C,04)„ • 2HO ' HRF (1938).
Mg(C6H4OHS03)2 HRF (1938).
MgC4H40e • 511,0 HRF (1938).
MgC,H..N40„, vpear HRF (1938).
MeCl. SB I (£19:743) 185, 189, 192, 773, 774; HRF (1938):
I1AH (x), стр. 382 (1944); С. A. 39, 3988 (1945); 0(1950).
Mg(NH,),Cl. SB III 475; F (1942); 0(1950).
*JH4Mg(H20)6Cl3 SB VII 117; HRF (1938).
MgC16ILO = Mg(H,0)(iC], SB III (71,: 124) 489; HRF (1938):
0(1950).
3080. Mg4Cl2 (OH)6 911,0 SB II 363, 596.
(MgCI2)x(MgO)v(H,0)z SB II 363. 364.
MgCUMgOl-JltO SB II 363. 364.
Mg(Cl04), SB II 415; HRF (1938).
Mg(NH3),(C104), SB III 475; F('I942); 0(1930).
Mg(C104)„xH20 SB II 415.
Mg(C104),2H20 SB II 415.
Mg(C104)23H20 HRF (1938).
Mg(C104),4H20 SB II 415.
Mg(C104)2 611,0 SB III (#4n:103) 451, 452; HRF (19:8); 0 (1950).
3090. Mg (C102)2 6H20 SBV 77; F(1946).
MgF„ SB I (C4: 158) 184; SB VII 67; HRF(19;.8); F(t940); 0(1949).
Mg2(P04) F, вагнерит SB VII 144.
Mg —Hg M. 2, 81 (1947).
Mg,Hg M. 2, 81 (1947)
Mg6Hg3 M. 2, 81 (1947).
Mg —In F. S. 44, 15 (1948).
Mg2Hg HRF (1938).
MgJ, SB HI (£6:20) 281; 0(1950).
Mgj;SH20 HRF (1938).
3100. Mg(MI3)e.T„ SB III 475; F(1942); 0(1950).
Mg —Mn SB III 618.
(Mn,Mg)6Mn8(P04)e (ОН)1015H0, бгрманит SB IV 184.
Mfr..SiMo1,04,)30H20 SB III 462.
MssN\ SB II 32-4, 325; SB III (Do.,: 49) 353; MRF (1938); Б (1947):
F (1942); 0(1.950).
Mg (N03), 6H,0 HRF (1938).
MgNII4P046H„0 HRF (1938).
(NH4),Mg(S04)26H,0 SB II 93, 43G.
KasMgBr(CO.)., SB II 81, 400.
Na„Mg(C03)2 SB II 395, 396.
3110.Ka8MgCl(COs)2 SB II 80, 399, 400.
>'a,Mg„ (CO,)., SO, SB II 98, 400, 444.
MgNCO, C. A. 39, 2684 fl945).
Mk,NI SB III 617.
MgNi SB III 617.
MgXi, SB III (C36:31)312, 316, 617; В (1947); ДАН 40,262(1943).
Mg —Ni —Sb SB V 124.
3J17. MgNi2Sh SB V 124.
890 3118. MgNillSSi,,,,-3159. Mg (H40), Sb (0H)6 [гл. \[
31J8.MeXli.8Si..,2 ДАН 40, 262 (1943).
Mg — Ni — Sn SB V 124.
.'Л20. Mg\i2Sn SB V 124.
Mg — Ni — Zn SB III 312.
MgNiZn F (1942); ДАН 40, 262 (1943).
MgO. иериклая SB I (51 : 73), 97, 114, 117, 765; SB II 216, 225-228,
255, 269, 321, 388, 476, 528-530, 533. 534. 538, 546. 592; SB III
211, 568; SB VI 115; ДАН 2, 165 (1934); HRF (1938); БМКД
(1939); Z. An. 244, 133, 146 (1940); F (1942); Б (1947); 0 (1950).
MgO xll.,0 SB II 216, 255.
Mg(OH)„ орусит, пирохроит SB I (£6:163) 193, 253,254,775;
SB II 216, 255, 258, 363, 364, 399; SB III 303; SB V 82; HRF
(1938); Б (1947); БМКД (1939); 0(1950).
Msr.P» SB I 786; SB II 40, 325; SB III (Db,: 49) 314, 353, 356;
F (19i2); 0 (1950).
Mg,(P04)., • 4Н..0 HRF (1938).
Mg.PO^HO, бобисрит SB V 94.
МхШ'04ЗН.,0 SB IV 185; HRF (1938).
:i.O MgHP047ILO. фогфорссслорит SB VII 145.
Mg<HP04>, I1RF (1938).
Mg(II,PO,),8H>0 SB V 78.
Mg (II PC), 6И..0 HRF (1938).
Mg.(PO,), i/3 Mg (Oil), 9H.0 SB IV 186.
Мц^РО.Й'З Mg(OH), 511,0 SB IV 186.
Mg2P.,07 J1RF (1938).
MgPb SB I 567.
Mg.Ph SB I (6:1:150) 229, 567; SB HI (CI : 20) 314; HRF (1938);
'" F(I942); 0(1950).
(Pb,Mg),Si04H,0 SB I 415.
:r.(».Mg3Pr " SB I'll (B32:19) 326; F(1942).
MgPr SB III 272: F(1942).
MsPt«'\)4<H,0 SB I 426; SB II 486; F(19iG).
3Ie—Pt, цианид HRF (1938).
Мц«Ь(Н,0)в(1, SB VII 117.
MgS SB 1 (/i I : 74) 125, 126; HRF (1938); HAH (x), стр. 382 (1944); С. А.
3!), 3988 (19J5); F(1942); 0(1950).
MgS04 SB 1389; HRF (1938).
MgS04xlI,0 SB VI 99.
6MgOS038H,0 SB V 82.
MgSOJLO, "кизерит SB VI 100; SB VII 139; HRF (1938).
:;i:.0. MgSO^ILO SB VI 99; HRF (1938).
MgS047II,0 SB I 389; SB II 429; SB VI 100; HRF (1938).
MgOSO,lilI,0 SB V 82.
MgS036H„0 SB III 415; SB IV 160; HRF (1938).
MgS 0,6H,0 HRF (1938).
Mg,Sb', 'SB III (jD5,:48) 356; 0 (1950).
MgSb,04 Ch. Z. I, 342 (1944); F(1946).
Mg£il\6H,0 = Mg<H0)eSiFe SB I 461 SB II 404, 504; 1IRF(I9.:8);
0(1950).
Mg.,„(F,OH),(SiO«).u SB II 119.
:; 150. Mg (H ,0)„ Sb (OH), SB IV 195.
: :-Щ 31fi0. Mg:,Sb,—Xti,SI>,, 319Г). Mg/П, 891
■'■!(iO. Mg,Sb2 —ZibSbo SB III (Z>5,: 48) 357.
MgSc SB I (B\ : 74) 134: Z. Ли. ',4-1, 133, 14C (1940); F (1942); 0(1950).
(XH4),Mg(Se04) ОНО SB 11 95, 430.
Jig —Si SB 1 586.
Ms..Si SB I (6*1:150) 228, 586; SB II 670, 673, 762; HHF (1938);
" F(1942); 0(1950). '
4 Mg, Si04 Mg (F, OH)., SB 1 '.00; SB II 121.
3Mg,Si04 Mg (F, OH), " SB I 400; SB II 121.
SMs.SiO, Mg (F, OH), SB I 400; SB II 121.
Mg,Si04 Mg (F. ОН),," норбсргит SB 1 400; SB II 121. 516; SB VI
112; Б (1917).
Mg„Si04, олишш SB I 401: SB II 121. 518, 534; 11RF (1938); Б.
(1У4.7): 0(1950).
!T0.MgSi03, стеатит и др. SB 1 327: SB II 134, 529, 530; SB IV 204;
SB VI 138 140; SB VII 164; IIRF (1938).
.*'Mg0ySi0.,mО SB VI 110.
3Mg04SiO.,H.,O-H,Mg.,(Si03)4, тальк SB V 114; SB VI 148-149:
SB VII 165; ВМКД(ПШ).
Mg^Si.O,,, (Oil),, белделит SB III (Л\56:159) 534, 542; SB VI 141,
146, 152; SB VII 165.
HMga Si40l2 SB II 545.
3M»'0:JSiO, *Л1,0= H43lg.,Si„0,, «Асбест»*), серпентин антнгорит,
благородные ьчеекик, гшкролит, хршнмпл SB II 139, 540; SB IV
206: SB V lli;JIRF (1938); BMK Д(1938); БМКД (1939); С (1939).
H,Mg-(Si0.X SB II 134, 137. 533, 537.
>Ig-S)\0,.(0H),, антофиллит SB VII 164, 165.
Mg,Si„O"0"(OII)" 1И.О, аттанулыит SB VII 177.
Mg(OII), • 2Mg..Si04, хондродит Б (1917).
••14>. Mg(OII), • 3Mg:Si04, io.viut (гуммт) Б (1947).
Mg(OH), 4Mg.Ki04, hvnuKiHtmiT Б (1947).
Слюды " SB V 115.
•JMgO 3SiO, Л10, еениолит SB V 116.
Mg — Sn "SB "I 567.
Mg Sn SB I (Cl : 150) 229, 567; SB (CI: 20) 314; IIRF (1938); F (1942);
0(1950).
Mg (H..0),SnF( SB II 104, 504; О (1950).
M«,$tiO, SB II 483: F (1942).
Mi,v4r Cli. Z. II, 270 (1944).
Mg-Te SB VI 180.
.!i'0. MgTe SB I («4:79) 134; SB II 231; Z. An. 244, 133, 146 (1940);
F(1942); 0(1950).
:»lgO — TiO„ SB V 39.
M-TiO.,, гснкнлнт SB I (<71 : 300) 331; SB III (H2t: 69) 379;
БМКД (1939); О (1950).
Mg,Ti04 SB II 184; F(1942).
Mg(H,O)0TiFB SB II 101, 504; 0(1950).
31'. 5. Mg5TI„ SB VI 1S'0.
*) Хаиауолт, Pmni n «]>pcucti неточно именуют минерал итого состава сегшентин
■Л' ■'< стпм (i(OTup!>iii может иметь ралнын гчстап).
892 3196.J«g Т1~323()._Мп(>*Н4)([804)г_611аО [гл. XI
3196.Mg2Tl SB VI 180.
MgTI SB III (L20:175) 271; SB VI 180.
Tl2Mg(S04)6H,0 SB II 95, 436.
Mg —вападий HRF (1938).
3200.MgWO4 SB I 790; SB II 85, 451; Ж. Ф. X. XIV, 844 (1940); N. Шг
548 (1946); F. S. 42, 705 (1946); С. А. 41, 3340 (1947); 0(1950).
Mg —Zn SB I 564.
MgZn SB II 726; SB III (£30:16) 269, 270.
MgZn, SB I (C14:183) 228, 564; SB II 720, 732. 733; SB III 311.
312.
MgZn5 SB II 726; SB III (Z)2,:47) 331.
MgO — ZrO„ SB III 301; SB V 39; F(1942).
MgaZrsOe * SB I 778; SB III 301.
MgZrO, SB I 778; F(1942).
Mg4ZrSiO„ SB II 592.
§ 317. Марганец Mn
Mn SB I (Л6:24) 63, 756; SB II 2, 3, 192. 193. 638-652, 067.
684-690, 711, 712, 720, 761, 789, 790; SB III 187, 221, 579; SB
IV 244, 246; SB VI 52; HRF (1938); F(19it>); 0(1950).
3210. MnBr2 SB II 246.
Mn(NH3), Br, SB III 475; F (1942); 0(1950).
Mn4C SB II 307; 0(1950).
Mn,8C, SB III (Z)84:60); С. A. 39, 3198 (1945); 0(1950).
Mn30 SB II 307; 0 (1950).
Mn,Cs SB HI 363; C. A. 39, 3198 (1945); 0(1950).
MnC03, родохрозит SB I (91 :295) 326: SB II 391, 397; HRF (1938):
БМКД(1938); 0(1950).
Mn (CH3C00),4H,0, ацетат HRF (1938).
Mn (C„H.C00),3H,0, бензоат HRF (1938).
Mn(C04), океалат HRF (1938).
3220. Mn(C.,04) 2 J H20 HRF (1938).
Mn (C4H40J, тартрат HRF (1938).
MnCt SB I (C19:743) 189, 192, 773; SB II 255.246; HRF (19c£);
ЙАН (х), стр. 382 (1944); С. А. 39, 3988 (1945); 0(1950).
Mn(NH0,Cl. SB III 475; F(1942); 0(1950).
MnCl.. 211,0 HRF (1938).
MnCL 411,0 HRF (1938).
Mn(NH,),"(CI04)., SB III 475; F (194.2); 0(1950).
Mn (C104), 6H,0 SB III 452.
MnF, SB I (C4:158) 192; HRF (1938); FtlO'iO): О (1950).
(Fc,"Mn) W04, вольфрамит Ж. Ф. X. XIV, 845 (1941); Ch. Z. 1.
585 (1942).
3230. Mnl, SB II 217.
Mn(NH,)f J„ SB HI 475; F(1942); 0(1950).
Mn4N ' SB II 791.
Mn2N SB II 791.
NH4Mn04 SB VI 97.
N(CH,)4 Mn04 SB II 411. 816; F(1946).
3236. Mn (\H4) №X вН.,0 HRF (1938).
§ 3171 3237. УаМп04-ЗН,0-3272. MnSc ___893
3237. >aMn04 • 3H„0 HRF (1938).
MnNal'O, SB VI 97; Ch. Z. 1, 342 (1914); С. А. 39, 2020 (1945).
MnJNb Z. Kr. (A) 103. 391 (1941); Ch. Z. 1, 1172 (1942).
3240. Mn>b20„ С. А. 39, 2684 (1915).
Mn —Ni SB I 758; SB II 193, 651.
MnNi SB II 651; F(1916).
MnNi, SB II 652.
Mn„Ni04 HRF (1938); F(1942).
Mn"—0 Ch. Z. I, 306 (1942).
MnO„ пиролюзит и др. SB I (Г4 : 158) 212; SB II 266, 356; SB
III 246, 568; SB V 111; SB VII 90; HRF (1938); БМКД(1938);
Бег. 72, 1879 (1939); F (1946); 0 (1950).
Mib03 SB 1 785, SB II 38, 266, 310, 316, 322; SB III 246, 349; SB IV
171; SB V 111; SB VI 72; БМКД (1938)*); F (1942); 0 (1950).
MnaO„ гауемашшт SB I 417, 418; SB JI 266; SB III 246, 432 ;SB IV
109, 171; SB V 111; SB VI 116; HRF (1938); БМКД (1938);
F (1946); О (1950).
xMnO., yMnO, лгндсмигал, над SB V 111; БМКД (1938)**).
3250. MnO, шпгишмит SB I (#1:74) 122. 765; SB II 222, 227, 228;
SB III 246; HRF (1938); БМКД (1939); F (1942); О (1950).
Mn20,xII..O SB II 355.
MnOIUb MnOOH, манганит SB 1 285; SB И 355, 360; SB III
372; SB IV (F0,:28) 141; SB V 111; ПМКД (1938); С (1939).
Mn (OH)., SB I (C6:1G3) 195, 775; SB II 257, 258; 0 (1950).
Mn —P SB V 134.
MnP SB III (531:17) 263, 264; С. А. 39. 2684 (1945); 0(1950).
MnP3 SB V 71.
Mn3(PO,), 7H0 HRF (1938).
Mn(H,PO.,).lL0 HRF (19381.
MnPbV04(OH), инршЪчншпт SB VII 151.
3260.Mii—Pd SB IV 231; SB VII 191; Z. t.l. 15, 776 (1939).
MnPd SB IV 231.
Mn.Pd, SB IV 232.
RbMn04 SB VI 97.
MnS, альбаидии SB I ЦП : 74) 132: SB II 233. 272; SB III 255;
SB VII 75; HRF (1938); ИАП(х). стр. 382 (1944); С. А. 39, 3988
(1955); БМКД (1938); F(1942); 0(1950).
MnS2 SB I (C2:153) 215, 216; SB II 272; SB III (C2:20) 287;
SB IV 111; F(19'i2); 0(1950).
MnSO, ■ 4H..0 HRF (1938).
Mn,Sb SB IV (6'38:9) 122; F(1946); 0(1950).
MnSb SB I (/38:87) 142, 765; 0(1950).'
MnSb.04 Ch. Z. I, 342 (1944); С. А. 39, 2019 (1945); F(1946).
3270.MnSb;O, С. А. 39, 2684 (1945).
(Mn, Zn)I(i Sb.,Si402<i, иетманит SB VI 152.
3272.Mn.Se SB I (M : 74) 138: SB VI 58; F (19-12); 0(1950).
*) В БМКД (1938) МгцОз пазили «ирауннт», что неверно (см. Дама) Брауннт—
см. ниже.
**) Но КМКД (1938) псиломелан и над содержат и НЮ, причём у роиен от 0 до t.
См. также Дана, стр 117.
894 3273. Mn—Si-33.10. Мо,Д1, (ОН),.1411,0 [гл. XI
3273. Mn —Si SB III 628; SB IV 246; 0(1950).
Mn3Si SB III 629; 0(1950).
Mn,Si SB II 761.
Mn5Si, SB III 629; SB IV (Z>88 :24) 137, 240; N. 152, 4l3(l9i3):
0(1950;.
MnSi SB III (528: 14) 629; F(1942); 0(1950).
MnSi., SB III 629; F(1940).
Mn5ll7Cl(Si04)4 SB I 415.
3280. MnSiF6 6IL0 SB I 461; SB И 104, 504; 0(1030).
2Mn,Si04 Mn (OH, F), SB III 519.
Mn,Si04, тофроит SB II 519; КМКД (1939); Z. Kr. 105, 273 (1944);
С. А. 39, 5148 (1945).
. MnSiO, SB 1 328.
От MnO-SiO, до Мп6Са(^03)в, родонит SB V 113.
MnrSi,09 SB II 579.
3MnO 4SiO.. 411,0, jiapi'OT'iviii'iiT SB VII 175.
3Mn,0.,MnSiO.„ браупит SB V HI; SB VI 153; F(I940).
(Zn, Mn),Si04 SB I '#1,: 356) 404; 0(1950).
Mn — Sn Ch. Z. 1, 1104 (1942); M. 1, 17 (1946).
3290. MnnSn3 M. 1. 17 (1946).
MnSn M. 1, 17 (1940).
MnSn, M. 1, 17 (1940); F(1946).
Mn.,Ta Z. Kr. (A) 103, 391 (1941); Ch. Z. I, 1472 (1942).
MnTa..O, C. A. 39, 2084 (1945).
MnTe SB I 765.
MnTo, SB I 780; F(1942); 0(1950).
Mn2Ti Ch. Z. 1, 1472 (1942); Z. Kr. Л. ЮЗ, 391 (1941).
MnTiO, SB III (£2,: 69) 379; 13МКД (Ш9); 0 (1950).
MnTi04 SB II 484;" F(1942).
33C0.C — Mn — W SB III 623.
MnW04, побиерит SB II 86. 450; Б.МКД (1938); 0(1950).
MnZn.3 SB II 711, 712.
Mn,Znn SB II 713.
MnZn/ SB II 711.
MnZn, SB Ц 711, 712.
MnZn,, SB II 711.
MnCO — ZnC03 SB IV 156.
MnaZr Ch. Z. I, 1472 (1942); Z. Kr. (A) 103, 391 (1941).
§ 318. Молибден Mo
Mo SB I (Л2:16) 61; SB II 163, 191, 240, 657, 058, GOO. 674.
777, 793, 795; SB III 180, 187; HRF (1938); F (1942): 0(1950).
3310. Мол но даты со структур»»» шеелита Z. techn. ph. 24, 8 (1943); Ch.
Z. 1, 270 (1944)'.
С —Mo SB I 575; 0(1950).
Mo,C SB II 237. 239, 240, 777; SB III 023; HBF(1938); O(HIJO).
MoC SB I 144; SB II 237. 240, 777; Б. (1947); 0(1950).
Mo (CO), SB III 334: SB VI 88; 0(1950).
Mo —океалат HRF (1938).
33№.MofeUOH)4 • 141I20 Ch. E. N. 25, 2970 (1947).
§_318] _3317. Moll,—ЗЖЛ1о—W
3317. MoCl, SB VI 71; 0(1950).
MoCI,- [МовС18](Л4 Ch. E. N. 25, 2970 (1917).
II, fMo.ClJ CI. 2H.0 Ch. K. N. 25, 2970 (1917).
3320. (NH4), [Mo.WJ CI,HsO Ch. E. N. 25, 2970 (1917).
MoF, SB V 04; 0(1950).
(NH4)3Mo03F3 SB I (//7,: 437) 450; F(1912); 0(1950).
Mo.Fe, SB I 52G.
Mo.N SB II 794.
MoN SB II 79-1.
(NH4),Mo04 SB II 450.
<NH4)3P04 I2Mo0, ЗИО HRF (1938).
(NU4)I(Mo,..Or1 ISHO SH II 150.
(NH4)eMo.O,424H,0" SB V 105; HRF (1938).
3330. (NH«), Mo,Te024 711,0 J. Am. 70, 1291 (1918).
NazMovO. SB II 335; HRF (1938).
Na,Mo04 211.0 HRF (1938).
Nd"„(Mo04), SB I 790; F (1910).
Л di'Mo.O,„3011,0 SB 111 462.
Mo —Ni SB VI 171, 172.
MoNJ, SB VI 172.
MoNi4 SB VI 172; F (HK.6).
Mo — Ni — С SB III G23.
NijSIMOj.O,,, 31H..0 SB III 1G2.
3310. Mo-О С A. 39, 5118 (1915); Ch. E. N. 25, 2970 (1917).
Mo03 SB I 249; SB II 30, 297, 335; SB VII 97; HRF (19,58): Б (1917);
0(1950).
H,Mo04 SH 1 219; HRF (1938).
MoO, SB 1 (CI. : 158) 211: Ch. E. N. 25, 2970(1917); F (1910): 0(1950).
Mo,0, HRF (1938).
Mo —P Z. An. 248, 209 (1911).
Mo2P Z. An. 2-18, 209 (1911).
MoP Z. An. 2-18, 209 (1911).
MoP, Z. An. 248, 209 (1911).
H,P(Mo.O,), SB II 510.
3350. 12МоОП3Р04 I4IL0 HPMo, ,04„I4IU) SB VII 148.
12Mo03H3PO, 30110 II,PMo1.04o 30110 SB III 162.
WMoOJI3P04 2411,0 203IoO, 2II.P0, 4811,0 HRF (1938).
MoO, — PbO Ch. Z. I, 1363 (1914).
PbMo04 SB I (//i:3'.P) 385, 386, 790; SH II 83, 115, 450; Ch. Z. 1,
270 (1941): С A. 39, 2021 (1915); F(194t>); O(!950).
Pr,(Mo04)3 SB I 790; E(1946).
Mo — Pt Ch. Z. 1, 106 (1912).
MoS,, молибденит SB I (<1 : 1(50) 211; SB II 210: I> (1917); 0(1950).
SmPMo,,04o30II,0 SB III 402.
MoSi, SB I (fll:7-'il) 219. 783; HRF (19.".8); см. Z. Met. 33.
378 (1941): F(19-'j<>); 0(1950).
3360. H4SiMo„0.10l4H.,0 SB VII 148.
Sm,(Mo04), SB I 790; F(1946).
SrMo04 SB I 790; SB II 450; F(1910).
Tl3P0412Mo03 4H..0 SB Vll 149.
3304. Mo —W SB I 502, 528.
896 ^^^g.5-,_y3zj407:_NaH.CI1- [гл. X
§ 319. Азот N2 (и аммоний (NH4)-rpynna)
3365. -\, SB I 55, 753; SB II 14, 182, 183. 782-795; SB VI 48; Ch. Ph. 11,
435 (1943); ДАН 62, 231 (1948); 0(1950).
Нитриды, см. cooгиетстиующие металлы, а также Z. E1. 49, 254
(1943); 0(1950).
NII,Br SB I (51 : 73) (52: 75) 99, 108; SB ll 211; SB III (525:10)
231; SB IV 94; HRF (1938); Z. ph. Ch. (5) 52, 222 (1942); F (1942);
F(1946); 0(1950).
M>4Br Z. ph.- Ch. (B) 49, 13 (1941); 52, 222 (1942); 0(1950).
NH,CH,Br SB I (0520:609)631; F(1946).
3370. N (CH3)4Br SB I (0510:607) 627; F(1946).
NH3C2H\Br SB I 632.
NH(C.H6),Bi SB I (054:606) 635.
N1I4ClBrJ SB III (Z>0ie:45) 322; SB V (Folt: 14) 76; 0(1950).
N"H(CH3), SB VI 233.
NH[(CN,)nCH,], SB VI 232.
N [(CH„)„ CH3]3 SB VI 233.
N(CH,j, SB IV 277; 0(1950).
1Ш,СН,С1 SB I (0521:610) 631; F(1946); 0(1950).
NH3(C6Hn)Cl SB II 811; F(1946).
3380. NH, (C]8H37) CI SB II 812.
Nil (СЛ1,,), CI SB I (054 : 606) 635.
N(CH,)4C1 SB I (0510:607) 627; F(1946).
N(CH3)4CI04 SB II 411, 804, 815; F(1946).
NH,(C„H.„.i)J SB II 803, 812, 813.
NH3CH3J SB I (0520:609) 631; F (1946).
NU3C,H.J SB I 632.
NiI((UfsM SB I (054:606) 635.
iV <CH3)4J SB I (0510:607) 627; F(1946).
N{C.H4)4J SB I 630; F(1946).
3390. NH4CN Rec. 63, 39 (1944); F(1946); 0(1950).
MI4CNS HRF (1938).
N(CII,i\'0.,)3 SB I 658.
(NH4)"S COs HRF (1948).
NH,HC03 SB II 394; HRF (1938).
NH4(CH3C00) HRF (1938).
NH„ (CeH6COO) HRF (1938).
NH4(COONH„) HRF (1938).
(NH4), (HCcH607) HRF (1938).
NH4(HCOO) HRF (1938).
ЗЮ0. NH4 (C,H6N N00) HRF (1938).
NH4 [C,H2 (N02)3 0] H20 HRF (1938).
NH4 [CeH4 (OH) COO] HRF (1938).
[CH„COO (NH4)]2 HRF (1938).
<СНбН)„ (C00NH4)2 HRF (1938).
ND4C1 " Z. ph. Ch. (B) 52, 222 (1942); Ch Z.I, 483 (1943); 0(1950).
NH4C1 SB I (51:73) (52:75)99, 107; SB I 211, 215: SB III 229;
HRF (1938); БМКД (1939); ИАН (х), стр. 382 (1944); С. А. 39, 3988
(1945); F(1942); 0(1950).
3407.N,H,C12 SB I 479.
§ 3201 3408. NH4CI04-3446.Na 897
3408.NH4CK)4 SB I (//2:344) 372; SB II 85, 411-413; SB IV 163;
HRF (1938); F(1942); 0(1950).
N0C104 SB V 88.
3410. NH4C102 SB II 64, 377, 379; F(1946); 0(1950).
NH3 SB I (D\ :231) 247; Ж.Ф.Х. XV, 597 (1941); 0(1950).
NH,K, SB IV (/?5e:37)151; 0(1950).
NH4F SB I (54:79) 107, 767; HRF (1938); 0(1950).
ND4F 0(1950).
NH4HF, SB II 374; SB III (Fb,: 79) 396; 0(1950).
(H4NH4F,)2H,0 SB II 375.
>TH4J . SB I (51:73) (52:75) 99, 109; SB II 211; SB III
(525:10) 232; HRF (1938); Z. ph. Ch. (5) 52, 222(1942); F(19i2);
F(1946); 0(1950).
NDJ Z. ph. Ch. (5) 52, 222 (1942); 0(1950).
NH4J3 SB HI (5-0,,: 43) 321; 0(1950).
3420. NHJ04 SB II 83, 417; SB III (#04:9i) 421; SB T 88; F (1946);
0(1950).
NH4J03 SB II 333; F(1942); 0(1950).
(NH4)„ H3JO, H. Ph. 18, 267 (1945); C. A. 40, 258 (1946).
N,06 SB III 302; О (1950).
N ,04 SB II 269, 270, 315; SB II 302; N. 153, 408 (1944); 0(1950).
NO.. SB II 21, 269, 270; SB HI 302; 0(1950).
N,0, SB II 31
NO Ch. Ph. 11, 435 (1943); 0(1950).
N,0 SB I (C2:153) 228; SB II 270; SB HI 302. Ch. Ph. 11,
435 (1943): 0(1950).
H,iNO« SB HI 417.
3430. NH4N03 SB II 69. 71, 72, 382-385; SB V 83; HRF (1938); F (1946);
0(1950).
NH4N02 HRF (1938).
NH4H2P04 SB I (Я2,: 363) 394; SB HI (Fb7: 77) 395; HRF (1938);
F(1946).
(NH4)aHP04 HRF (1938).
NH4H2P02 HRF (1938); 0(1950).
N4S4 = 4«NS» SB II 327; SB IV 99; 0(1950).
HNS SB II 328
NH4SH SB III (510:7) 260; F(1946); 0(1950).
(NH4)2S04 2NH4N03 SB I 387.
(NH4)2 S04 SB I 374; SB II 88, 420, 425, 438; HRF (1938); 0(1950).
3440. NH4HS04 HRF (1938).
(NH«). S208 SB III (#4,: 136) 499; HRF (1938); 0(1950).
(NH4bS203 HRF (1938).
(NH4)2 S03H„0 HRF (1938b
(NH4)2 SeCl„ " SB HI (71,: 121)479.
(NH4)2S204 HRF (1938).
§ 320. Натрий Na
3446. Na SBI(A2:16) 33, 35, 747; SB II 166, 237, 733, 734: SB IV 81:
SB VI 39; SB VII 202; HRF (1938); F(1942); R. M. 15,90 (WW;
Ж.Ф.Х. XX, 1414 (1946); и Усп. 15, 297 (1946); 0(1950).
898 3447. NaBr-3489. NaC104H;0 [гл. Х]
3447. NaBr SB I (51:73) 97, 98, 99, 105; SB II 209, 372; SB IV 91;
SB V 37, 38; SB VI 55; SB VII 69; HRF (1938); ИОНХ 16, 82
(1943); 0(1950).
NaBr2HsO SB II 209.
NaBrO, SB I (G,: 300) 310; SB VI 93; HRF (1938); F (1942); О (1950).
3450.NaHC2 SB I 783; SB II 275; F(1946); 0(1950).
NaCN SB II 371, 372; SB VI 225; HRF (1938); F(1942); 0(1950).
NaCNS HRF (1938).
NaCICo (NH2)2H20 SB VI 229.
NaCNO SB VI (Fbx: 18) 227.
(NaC2Ns), 3H20 SB VI 211, 252.
NaHCO, SB III (G0X2: 86) 404; SB VII 134; HRF (1938); О (1950).
NaDCO, SB VII 134.
Na2CO, HRF (1938).
Na2CO„H20 SB III 416; SB IV (G7,:42) 157; HRF (1938).
3460. HCOONa SB VI 226; HRF (1938).
NaH (CH,COO)2 SB I 676.
Na (CH3COO) HRF (1938).
Na (CHsCOO) • 3HaO HRF (1938).
Na (CH,CH2CHaC00) HRF (1938).
Na (C.H4CH:CHC00) HRF (1938).
2Na, (C6H60,) 11HS0 HRF (1938).
NaOC2H, HRF (1938).
Na-яафтионат HRF (1938).
Na2C204 HRF (1938).
3470. NaHC204H20 HRF (1938).
Na (pC.H4OHSO.) 2H20 HRF (1938).
Na-нитрофенилат HRF (1938).
Na-динитрофенилат HRF (1938).
Na (C,H4OHCOO) HRF (1938).
Na (Cl7H„COO) HRF (1938).
[Na (CH2COO)]2 • 6H20 HRF (1938).
Na (C14H,02SOs) HRF (1938).
Na (Cl0H7S0,) HRF (1938).
Na2 [C10H, (SO.)2] HRF (1938).
3480. Na2C4H40. • 2H20 HRF (1938).
NaHC4H40, • H20 HRF (1938).
Na2CtH20,N4 • H20 HRF (1938).
Na-аспарагинат HRF (1938).
Na-«6ap6HTaab» HRF (1938).
NaCcF4 A. Cr. 1, 265 (1948).
NaCI, галит SB I (51:73) 97, 98, 103, 490; SB II 209, 213, 214,
329, 372; SB IV 91, 92; SB V 37; SB VII 67, 69; БМКД (1938);
HRF (1938); БМКД (1939); Z. Ph. 116, 194 (1940); F (1942); ИОНХ
16,82(1943); ИАН (х), стр. 382 (1944); С А. 39, 3988; 4275(1945);
N. 166, 567 (1945); Б (1947); О (1950).
2Na,S04NaCINaF, сульфогалит SB III (НЪ,: 118) 470; БМКД (1939);
*F (1942); О (1950).
NaC104 SB II 85, 411, 412. 414, 415; SB IV 163; HRF (1938);
F(1942); 0(1950).
§320]. 3490. NaUlO,--3530. NaH,P04 899
3490. NaC103 SB I (G'S: 300) 216, 388; SB 11407; IJRF (1938); F (1942);
0(1930).
NaCIO, 3H20 SB II 377.
NaF. виллиомит SB I (Ш : 73) 97, 98, f02; SB II 208, 598; SB
IV 91; SB VII 70; HRF (1938); I1U1X 16, 82 (1943); БМКД
(1939); F (1942); 0 (1950).
NaHF2 SB I (F5t:276) 285; В (1947); 0 (1950).
2Na.,P04NaF19H20*) (?) SB III 474.
Nail SB II 242; F (1942); 0 (1950).
NaJ SB I (Bl : 73)97, 98, 105; SB II 209, 372; SB IV 91; SB V 37,
38; SB VI 55; HRF (1938); F (1942); ИОНХ 16, 82 (1943); 0 (1950).
NaJ2H20 SB II 209.
NaJ 4H20 SB II 210.
NaJ04 SB I (#4:349) 372; SB VI 96; F (1946); 0(1950).
3500. NaJ04 3H20 SB I 373, 788.
NaJ03 SB II 49, 333; HRF (1938); J. Am. 69, 1280 (1947); 0(1950).
Na2H3J0a HRF (1938).
NaLaF4 A. Cr. 1, 265 (1948).
NaN3 56 1(^:276) 288; SB IV 151; SB VII 97; HRF (1938);
0 (1950).
Na,NF, SB V 103; Z. An. 235, 77 (1937).
NaNH2 HRF (1938).
Na (NH4)nP04. 411,0 HRF (1938).
Na3N04 SB III 417.
NaN03 SB I (Gl : 295) 313; SI? II 380; HRF (1938); Rec. 64, 174 (1945);
G. A. 10, 4274(1946); 0 (1950).
3510. Na3N03 SB III 417; 0 (1950).
' NaNO* SJi II 65, 377; HRF (1938); С A. 40, 395'J (1946); 0 (1950).
Na (N0)t SI: III 386.
NaNbO, SB I (G5 : 303) 315; SB II 335; F (1942); 0 (1950).
NaN<lSi04 SB H 555.
Na.O., SB VI 65; HRF (1938); F (1946); 0 (1950).
Na~0~ SB II 279, 323; SB III (CI : 20) 283; SB VII 161; F (1942);
" 0 (1950).
NaOH HRF (1938); 0 (1950).
NaaP SB V (D0H : 6) 60; 0 (1950).
Na3P04 HRF (1938).
3520. Na4 (P03)4 4H,0 SB V 94.
Na4P207 SB II 453; HRF (1938).
Na4P.0710H20 SB II 453; HRF (1938).
NaHP04 HRF (1938).
Na,UP04 • H,0 SB II 453.
NaHP04 • 2H> HRF (1938).
Na,HP04 • S2H..0 HRF (1938).
NaHP042H,0, SB II 453.
Na6P3010 "SB VI 103.
Na6F30106H,0 SB V 106.
3530. NalLP04 " HRF (1938).
*) Ошибка SB: 2 Na.>P04NaF19HaU d действительности должна быть кислой солью
2Na,HP04NaF 19H20 или 2 NaH,P04NaF 19H20.
900 3531. NaH2P04H,0-3579. Na3SbS49H..Q [гл. XI
3531.NaH2P04H20 HRF (1938).
Na.P.O.lOHsO HRF (1938).
NaPO, HRF (1938).
NaH2P0„H20 HRE(1938).
Na—Pb" SB III 638; SB VI 193.
Na4Pb SB II 734.
Na3lPb, SB III 638.
Na„Pb4 SB IV (2)8,: 22) 138.
NasPb2 SB II 734.
3540. NaPb SB II 734.
Na4Pb, SB II 734.
Na4Pb, yNH, SB II 734.
Na4Pb, SB II734.
Na4Pb,yNH, SB II 734, 735.
Na2Pb6 SB II 734, 735.
NaPb3 SB II 735.
Na2Pb03 SB II 335.
Na2Pb03 3H20 SB II 335; HRF (1938).
Na2Pr03, тип 51 Z. An. 246, 26 (1940); F (1942); 0 (1950).
3550. Na—Pt—хлорид HRF (1938).
NaPuF, J. Am. 70, 2147 (1948); 0(1950).
S'NaPuF4 J. Am. 70, 2147 (1948); 0(1950).
NaPuF4 A. Cr. 1, 265 (1948); 0(1950).
Na-Rb SB VII 202.
NaRe04 SB HI (Я04:91) 421; SB V 89; F (1946); 0(1950).
Na (NH4)2 Rh (N02), ИАН(х), стр. 87 (1945); С. А. 39, 5140 (1945);
О (1950).
Na2S SB I {СЛ : 150) 224; SB III (Ci : 20) 283; Z. An. 241, 281 (1939);
F (1942); ИАН (х), стр. 382 (1944); С. А. 39, 3988 (1945); 0 (1950).
Na.S • 9H20 HRF (1938).
NaSH SB III (B22 : 8) 258, (51: 7) 258; SB VII 78; F (1942); 0 (1950).
3560.Na2SO4 SB II 88, 421-423,440; БМКД (1939). J. Am. 63, 2533
(1941); HRF (1938); Б (1947); 0(1950).
Na2S0410H,0 SB II 422; HRF (1938).
NaHS04 HRF (1938).
NaHS04H20 HRF (1938).
Na„S03 SB II 76, 409; HRF (1938); 0 (19501.
Na^SO, 7H20 HRF (1938).
N8,8,0, HRF (1938).
Na2S204 • 2H20 HRF (1938).
Na2S„06 HRF (1938).
Na3S20, HRF (1938).
3570. Na2S203 5H20 HRF (1938).
Na,Sb SB V (ZX)lf:6) 60; 0(1950).
NaSb SB II 237.
NaSbF, SB VI 118.
NaSbF4(0H)2 SB VI (Ла:27) 28, 118; Б (1947); 0(1950).
NasSb7NH3 SB II 327.
Na3Sb76NH3 SB II 327.
NaSbO, SB VI 118, 132; SB VII 24.
NaSb(OH), SB VI (Ли : 26) 27, 118; F (1946); 0 (1950).
3579. Na3SbS4 911,0 SB III 469.
$ 320] 3580. Na2Se-3630._Na5ZnO4 901
3580. Na2Se SB III (Cl : 20) 283; Z. An. 241, 281 (1939); О (1950).
NaSeH SB VII (Б22:4) (Б1.-3) 4, 78; 0(1950).
Na2Se04 HRF (1938).
Na2SeO, HRF (1938,.
NaSi2 M. 2, 127 (1947).
NajSi2Ti2Oe, размаит Б (1947).
Na,SiF, HRF (1938).
Na2S103 HRF (1938).
Ha,SiOs • 9H,0 HRF (1938).
NaaY8i04 'SB II 555.
3590. Na2ZrSisO„ каташюит SB V (S3,: 24) 119.
Na,ZrSi,0,2HaO SB III 555.
NauSn4 SB IV 139.
Na,SnO, HRF (1938).
NaSrP04 J. Am. 63, 2533 (1941); F (1946).
NaTaO, SB И 335; F (1942).
Na2Te Z. An. 241, 281 (1939); F (1942); 0(1950).
Na2TeO, HRF (1938).
Na-Tb Z. El. 48, 418 (1942).
NaF—ThF4 J. Am. 70, 2147 (1948).
3600.NaTh2F, J. Am. 70, 2147 (1948); A. Cr. 1, 265 (1948).
&2Na2ThF4 J- Am- 70, 2147 (1948); A. Cr. 1, 26.1 (1948); 0(1950).
8Na2ThF, J. Am. 70, 2147 (1948).
Na4ThF, J. Am. 70, 2147 (1948).
Ha.TiO, C. A. 39, 2441 (1945).
Na2Ti,0T C. A. 39, 2441 (1945).
Na2TiS4On SB II 586.
NaTl SB II 733; SB III (£32:19); F (1942).
NaF UF4 J. Am. 70, 2147 (1948).
aNa.,UF, J. Am. 70, 2147 (1948); 0(1950).
3610. NaUF, J. Am. 70, 2147 (1948); 0(1950).
P,Na2UF. J. Am. 70, 2147 (1948); A. Cr. 1, 265, 268 (1948); О (1950).
YNa2UF, J. Am. 70, 2147 (1948); A. Cr. 1, 265, 268 (1948); О (1950).
Na,UF7 J. Am. 70, 2147 (1948); A. Cr. 1, 265, 268 (1948); 0(1950).
Na,UFT J. A. 70, 2147 (1948); A. Cr. 1, 265, 268 (1948); О (1950).
NaUOs(CHsCOO), SB II 844; SB III (04,: 668) 717.
Na(U02)Zn(CH,COO) HRF (1938).
Na,V0416H20 HRF (1938).
NaVO, Ch. Z. I, 206, 1069 (1944); С. А. 39, 2020 (1945).
NaVO,H20 HRF (1938).
3620.Ka.(WO.), SB П 336; SB HI 377.
Na2W20, Ch. E. N. 26, 2970 (1947).
Na2W,0, Ch. E. N. 26, 2970 (1947).
Na,W04 HRF (1938).
Na,WO„ где x< 1, SB Щ 377,
(Na,H)WO, SB II 336.
NaWO, SB II 336; F (1946).
Na^WO,). SB II 336.
NaZn SB VI 157.
NaZnlt SB VI (Z>2,:8) 9, 157.
3630. Ua,Zn04 SB III 417,
902
363J. Nb-3665.NiBr,
[гл. XI
§ 321. Ниобий Nb
3631. Nb SB I (Al: 14) 56, 754; SB II 187; SB III 212; SB IV (A2 : 3) 87;
IIHF (1938); F(1942); 0(1950).
NbBr, Ж. Ф. X. XX, 6 (1946).
Nb—С Ж. Ф. X. XIV, 332 (1940); Ch. Z. II, 3 (1944): 0(1950).
Nbt' SB I (SI : 74) 144; Ж. Ф. X. XIV, 332 (1940); Ch. Z. II, 3 (1944);
F (1942); 0(1930).
Nb.C Ж. Ф. X. XIV, 332 (1940); Ch. Z. II, 3 (1944); 0(1950).
NM:!, Ж. <J). X. XX 0(1946).
Nb-H Ж. Ф. X. XIV, 332 (1940); Ch. Z. 11,3 (1944); 0 (1950).
NbJ, Ж. Ф. X. XX, 6 (1946).
Nb-N Ж. Ф. X. XIV, 332 (1940); Ch. Z. II, 3 (1944).
3<>'j0.NbN SB I (Bi :74) 139; F (1942); О (1950).
Nb.,N Ж. Ф. X. XIV, 332 (1940); Ch. Z. II, 3 (1944); 0(1950).
Nb-Ni SB VII 190; Z. El. 45, 881 (1939): IIOHX 16, 158 (1943).
Ni.\b,Os C. A. 39, 2684 (1945).
.Nb-0 Z. An. 248, 1 (1941); 0(1950).
Nb.,08 SB I 196; SB II 360; SB VII 89; Z. An. 248, 1 (.1941); Z. El.
•15, 885 (1939).
«Nb,06-f-H,» Z. El. 45, 885 (1939).
Nb,0„xH30 SB II 360.
NbO SB I (C4:158) 211; SB VII 89; Z. An. 248, 1 (1941); V (1946);
0 (1950).
NbO SB VII 89; Z. An. 248, 1 (19'd); О (1950).
.:6;>0. Nb20 SB VII 89.
Rh.VbO, С A. 39, 2684 (1945); F (1946).
Nb-S Z. An. 241, 337 (1939); О (1950).
Nb,S SB VI 58.
NbS SB VI 58.
NbSi, Z. Met. 33, 378 (1941).
NbC-TaC—WC Ж. Ф. X. XX, 773 (1946); О (1950).
\ (Nb, Ta)0, SB I 397; F (1946).
XbC—TIC Ж. Ф. X. XX, 769 (1946); 0(1950).
NbC-TiC-WC Ж. Ф. X. XX, 929 (1946).
ЗС.1Ю. YNb04 SB I 397; F (1946).
ZnNb.,0. C. A. 39, 2684 (1945).
XbC-ZrC Ж. Ф. X. XX, 769 (1916); 0(1950).
§ 322. Неон No
>e SB II 201; ДАН 62, 231 (1948); F (1942); 0(1950).
§ 323. Никель Ni
Ni SB I (Л1: 15) 68, 760, 763; SB II 164, 193, 197-199, 624-626,
634 637, 651, 652, 657, 683, 684, 690, 708-710, 747-749, 777,
789. 792: SB III 186, 187, 223; SB IV 77, 86, 228, 229; SB V 33;
SB VI 43; SB VII 45, 48-53; HRF (1938); С R. 208, 1583 (1939):
БМКД (1939); С R. 215, 129 (1942); F (1942); Ann. 19, 272 (1944);
С A. 38, 4174 (1944); 39, 3474 (1945); J. Am. 69, 893 (1947);
ДЛН 59, 65 (1948); 0(1950).
3665. XiBr.. SB II 246; SB III (C19: 22) 280; О (1950).
§323] 3666. Ni(NH,).Br,—3711. NiO 903
3666. iVi (NH.), Br, SB I (Я61:431) 456; SB III 475; F (1942); О (1950).
NiBr26NH, С. А. 39, 2917 (1945).
NiBr2 2Ni (0H)2 4H ,0 SB VII 83.
NiBr23Ni(OH)2 SB IV 106, 107; SB VII 83.
3670. NiBr2 5Ni (OH), 7H20 SB VII 83.
NiBr., 5Ni (0H)2 8H20 SB VII 83.
NiBr„ 7Ni (0H)2 8H20 SB VII 83.
NiBr, 7Ni (0H)2xH20 SB VII 83.
Ni-C SB III 627; SB VI 170; Z. El. 49, 200 (1943); 0 (1950).
NiaC SB III 327, 627; SB VI 170; 0(1950).
NiC SB II 237.
Ni(CN), .4H,0 HRF (1938).
NiCO, " SB'II 392.
Ni,(C0,)(0H)44H,0 SB II 392.
3680. Ni (CH,COO)2 HRF (1938).
Ni(CH,COO)24H20 SB III 719.
Ni(HCOO)2 • 2H20 HRF (1938).
Ni (C00)2 • 2H20 HRF (1938).
Ni—,тартрат HRF (1938).
Ni(CO), SB III 329; 0(1950).
Ni—,фталоцианин SB V 175, 176; см. также SB IV 270.
NiCl„ SB I (C19: 743) 193, 774; SB II 215; HRF (1938); ИАН (х),
"стр. 382 (1944); С. А. 39, 3988 (1945); О (1950).
Ni(NH,)eCl2 SB I (Я6 :431) 456; SB III 475; G. A. 39, 2917 (1945);
F(1942); 0(1950).
NiCl2Ni(OH), SB IV 106, 107; SB VII 83.
3690. NiCl, 8Ni (OH). SB IV 106; SB VII 83.
NiCl23—4Ni(OH),xH20 SB VII 83.
NiCl, 6—7Ni (OH), xH20 SB VII 83.
NiCl2 2Ni (0H)3 3 - 4H,0 SB VII 83.
NiCl, 2H20 SB II 215; HRF (1938).
NiCL-4H20 HRF (1938).
NiCl* . 6H20 HRF (1938).
Ni (NH8), (C104), SB II 502; SB III 475; F (1942); 0(1950).
Ni (C104)2 6H20 SB III 452.
NiF, SB I {СЛ : 158) 192; F (1946); 0 (1950).
3700. Ni-H SB I 604; SB III 581; 0 (1950).
Ni—D Z. An. 247, 131 (1941).
NiJ. SB II 247; SB III (C19: 22) 282; 0 (1950).
Ni(NH,)„J, SB I (ff6,:431) 456; SB III 475; F (1942); С. А. 39,
2917 (1945) 0 (1950).
M[NiJO,]xH20(M = К или Na) N. 167, 627 (1946).
Ni(NH2CH,), J2 SB Ш 475; F (1942); 0 (1950).
Ni (NHS), (NO,), SB I (Я64: 437) 456; SB VI 90; 0 (1950).
Ni (NO,), (H20), HRF (1938).
(NH4)2 Ni (S04)2 6H20 SB III 442; HRF (1938).
Ni20, SB I 268; SB II 223, 359.
3710. Ni,04 SB I 417.
3711. NiO, бунзенит SB I (51:74) 114, 123; SB II 222-224, 226, 227,
359, 484; SB III 249; SB IV 97; HRF (1938); БМКД (1939);
F (1942); N. 162, 304 (1943); Ch. Z. I, 1364 (1944); A. Cr. 1, 226
M948); О (1950).
904 3712. N1,0—3754. Ni—Si [гл. XI
3712. Ni20 SB I 123.
Ni,0,xH20 SB II 359.
Ni80,2H20 SB III 308.
Ni,0,H20 SB III 308.
NiOOH SB II 359.
Ni,042H20 SB III 308.
Ni(OH), SB II 359.
1/2 Ni,0,3H,0 SB III 308.
3720. NiOxH.O " SB II 223.
Ni(OH), SB I (C6: 163) 195, 775; SB II 223, 257, 258; SB III 308;
SB"IV (C6:8) 120; SB VI 89; HRF (1938).
Ni—P SB VI 173, 174; 0 (1950).
NiP, SB VI 174.
NiP„ SB VI 174.
NiP" SB III 264; SB VI 174.
NiaP SB VI 173.
Ni7P, SB VI 173.
Ni.P, SB VI 173.
Ni,P SB VI 173; V (1946).
3730.Ni(NH,),(PF.)2 SB III 475; F (1942); 0 (1950).
Ni(H2P0,)26H20 SB V 78.
Pb2Ni(N0,). SB III (Л,: 121) 480; F (1942); 0(1950).
Ni-Pd " SB VII 191.
(Ni, Pd, Pt)S SB V 42; F (1946).
Ni—Pr M. 2, 97 (1947).
N1—Pt SB VI 170.
Ni,Pt SB VI 171.
NiS, SB I (C2:153) 217; F (1942); 0(1950).
Ni,S4 SB I (#1^352) 421, 422; F(1942)*); 0(1950).
3740. NiS. миллерит SB I (£8: 86) (£13 : 740) 133, 772; SB II 6, 234,;
SB III 257; БМКД (1939); ИАН(х), стр. 382 (1944); С. A. 39, 3988
(1945); Б (1947); О (1950).
Ni,S2 SB VI 75; Б (1947); О (1950).
NiS04nH,0 Z. An. 242, 386 (1939).
NiS047H20 SB I 389, 390; SB III (ff4„ : 105) 453; HRF (1938).
NiS046Hf0 SB II 95, 433; HRF (1938).
NiSO, 6H.0 SB IV 160.
Ni-Sb SB V 124; SB VI 172; Ch. Z. I, 1104 (1942).
NiSb, брсйтгауптит SB I (£8:87) 143, 765; 0(1950).
Ni(H20), [Sb(OH).]2 SB IV 195.
NiSb204 Gh. Z. I, 342 (1944); G. A. 39, 2019 (1945); F (1946).
3750. NiSbS SB I (fl : 269) 125, 284; F (1942); 0 (1950).
Ni-Se SB VI 166.
NiSe, SB I 780; F (1942); 0(1950).
NiSe SB I ;£8:86) 138; SB III 257; 0(1950).
3754.Ni-Si SB III 627; SB IV 247; SB III (£28:14) 628; SB IV 247;
0 (1950).
*) В работе F (1942) несомненно ошибочно среди соединений типа НХ^ помещено
соединение NiN3S4 с а„ = 9,5 кХ. Повидимому, речь должна итти о соединении
NiNi2S4, тип шпинели с а„ = 9,5 кХ.
§ 324J __3755. NiSi,-3794.R0.2 905
3755. NISI, SB IV 247.
NiSi SB IV 247; F (1942); О (1950).
Ni2Si SB IV 247.
Ni,Si2 SB IV 247.
Ni.Si SB IV 247.
376().Ni,Si2 SB IV 247.
NiSiF, 6H20 SB II 104, 504; О (1950).
Ш (NH,), (Si0,)2 SB II 503.
т2тА sb ii 5i9.
NiSiO, SB II 519.
Ni—Sn SB III 616; SB VI 177; Ch. Z. I, 1104 (1942); I. S. 16. 543
(1943); Ch. Z. I, 1271 (1944); M. 1, 23(1946).
Ni.Sn SB V (.DO,,: 7) 61, 62.
NiSn SB I 765.
Ni, Sn4 M. 1, 23 (1947).
NiSnCl, 6H,0 SB II 102-104, 504; Б (1947).
3770. Sr2Ni (tf02j, SB III (/!,: 121) 480; F (1942); 0(1950).
Ni-Te SB VI 166.
NiTe2 SB VI 166; Ch. Z. 1171 (1947); 0(1950)..
NiTe SB I (#8:87) 138, 765; 0(1950).
NiTe SB VI 166.
Ni,Ti SB VII (DOit: 14) 15, 98.
NiTiO, SB II 333; SB III (£2„:69) 380; О (1950)
Ni—W SB I 528.
Ni—W—С SB III 62.!.
Ni,W,C SB III 623.
3780. NiWO, SB II 86, 450; 0(1950).
Ni—Zn SB IV 240; SB VI 186.
NiZn SB II 708, 709; SB IV 241; F (1946).
NiZn, SB II 708, 709; SB IV 241.
Ni,Zn2l SB II 708.
NiaZn16 SB II 709.
(Ni, Zn) (OH)2 SB VI 89.
§ 324. Кислород 02
02 SB I 60, 756; SB II 188; SB IV 81; SB VI 50; ДАН 59, 67
(1948); 62, 231 (1948); 0(1950).
Oa J. Am. 65, 179 (1943); Ch. Ph. 14, 505, 531, 641 (1946); C. A. 41,.
1900 (1947); Ch.E. N. 25, 2970 (1947); 0(1950).
OX2 (X = P, CI, Br, J) Ж. Ф. X. XX, 6 (1946); см. также соотв.
элементы; О (1950).
3790. Н.,0 SB I (С 10: 174) 219; SB II277, 278; SB III 289 - 291; SB VI 59;
" Ж. Ф. X. XV, 597 (1941); Z. Кг. 106, 279 (1944); N. 166, 335
(1945»; С. А. 39, 1798, 5148 (1945); Ж. Ф. X. XX, 1414 (1946);.
F. S. 44, 556 (1948); 0(1950).
I)a0 SB III 289, 291; 0(1950).
Н,Ог SB IV 118; SB V 55; Ch. Ph. 14, 504 (1946); F (1946).
H О 2Н О SB IY 118
3794. R02 (R- щелочной металл) Ж. Ф. X. IX, 932 (1937); Ж. Э. Ф. VII,
1418 (1937); ДАН 47, 199 (1945).
906
3795. Os-3836. PH,
[гл. XI
§ 325. Оеинй Ов
3795. Os SB I (A3: 19) 70,764; SB II 638; SB III 185; SB V 35; HRF
(1938); Z. El. 49, 222 (1943); О (1950).
OsC12H,8 SB VII 237.
OsO,OC4(CHs)8 SB VII 237; F (1946).
OsF, SB Ш 335; 0(1950).
NH40s03N SB II 419.
3800. Os04 SB VI 70; 0(1950).
Os02 SB I (C4:158) 213; F (1946); 0(1950).
Os-P SB VII 91; 0(1950).
OsP., SB VII 91.
Os-Pt Z. El. 49, 222 (1943).
RbOsO.N SB II 419.
Os-Re Z. El. 49, 222 (1943).
OsS2 SB I (C2:153) 217, 780; SB III 288; F (1942); О (1950).
OsSe2 SB I 781; F (1942); 0(1950).
OsTe2 SB I 781; F (1942); О (1950).
3810. T10s03N SB II 419.
§ 326. Фосфор Р
P SB I (.47: 27) 56, 754; SB II 184, 795; SB III (417:6), 209; SB
VI 49; Ж.Р.Ф.Х.О. 62, 2235 (1930); БМКД (1939); Gh. Ph. 14,
351 (1946); О (1950).
фосфиды см. соотв. металлы, а также Z. E1. 49, 254 (1943); О (1950).
РВг„ N. 145, 971 (19iO); Roc. 62, 167 (1943); Ch. Z. И, 1076(1943);
'О (1950).
PBr3 SB V 56; Ж.Ф. X. XX, 6 (1946): О (1950).
PSF2 Br J. Am. 66, 1941 (1944).
PSF Вг„ J. Am. 66, 1941 (1944).
POBr3 J. Am. 66, 1941 (1944).
PSBr3 SB V 73; J. Am. 66, 1941 (1944).
P(CH3), SB VI 234.
3820. PC16 SB VI 70; HRF (1938); N. 145, 149 (1940); Ch. S. 642 (1942);
F(1946); 0(1950).
PC13 SB IV 123; SB VI 69; Ж.Ф.Х. XX, 6 (1946); 0(1950).
PF3C12 SB VI 69.
PFC12 SB VI 69.
P0FC12 SB VI 69.
P0F2C1 SB VI 69.
(PNC12)4 - P4N4C18 SBII 369; SB VII(Eit: 17) 128; F (1946); 0(1950).
[(PNC12)3]„ SB III 387; Ch. E. N. 25, 2554 (1947).
(PNC12)3 SB II 370.
P0C13 SB VI 69.
.3830. PSC13 SB VI 83.
PF, SB V 63; SB VI 69; О (1950).
PF, SB IV 123; SB VI 69; Ж.Ф.Х. XX, 6 (1946).
NH4PFe SB II 489.
POF, SB VI 69.
PSF, SB VII 114.
:3836.PH, SB II 287; 0(1950).
§ 327] 3837. PJ3-3872. РЬД (Р04), 01 = Pb,n (РОЛ <1, 907
3837. PJ3 SB V 56; SB VI 68; Ж.Ф.Х. XX, 6 (1946); 0(1950).
PH4J SB I (510:93); 109; F (194■,); 0(1950).
PN SB IV 101; 0(1950).
3840. PSN, SB IV 101, 136; 0(1950).
(PN0)„ SB V 72.
P.O, = P,Oie SB II 326, 464, 593; SB V 66; SB VI 7S; HRF (1938);
Rec. 60, 413 (1941); С A. 33, 6853 (1941); 0(1950).
H8P,010 SB V 106.
P408 SB V 66.
P,08- P40e SB V 66; SB VI 78; 0 (1950).
P40,S4 SB VII 124.
P2S6 HRF (1938).
§ 327. Свинец Pb
I'b SB 1(41: 14) 55, 753; SB II 163, 164, 165, 167, 182, 222,
280, 734, 738, 753; 755; Si] III 181, 183, 208; SB IV 78; SB V 31;
SB VI 193; HRF (1938); БМКД (1939); P. Ph. 53, 517 (1941-;
F (1942); J. Am. 68, 1493 (1946); 0 (1950).
PbBr, SB II 18, 253, 254; SB VII (C23 : 8) SO; HRF (1938); Ж.Ф. X.
XX, 6 (1946); 0 (1950).
3850. (PbBr), CO, SB II 254.
(PbBi)., (COO), SB II 254; F (1946).
PbFBr SB II 45, 363; F (1946); О (1930).
NH4Pb,Br, SB V (A'34:22) 104; F (1946).
РЬ(СНЛ SB IV 277.
Pb(C.H,)« SB I (U6:615) 616, 049-651; SB VI 230; F (1946);
О (1У50).
PbX4 (X, = Hul.) Ж. Ф. X. XX, 6 (1946).
ГЫ'Л)3, цоруееит SB I 339; SB II 391; SB III (G0t: 84) 407;
SB VI 91-93; HRF (1938); Ch. Z. II, 7 (1944); О (1950).
2РЬСО,РЬ(ОН). HRF (1938).
Pb(HCOO), SB I 656-658; SB II 844; HRF (1938).
3860.Pb(CH3COO), 3H,0 HRF (1938).
Pb,(OH). (СП.СОО), ILO HRF (1938).
Pba0(0H),(CHtC00), " SB V 162.
PbC204 HRF (1938).
Pb(C,H40HC00), SB V 168.
(PbCl). COs-РЬС0,РЬС1„ фосгенит SB II 254; SB Ш(С7,:89)
416; F (1946); О (19"50).
(PbCl),(C00), SB II 254.
PbCUCS (NH.). SB V 158.
PbCU, котупййт SB II 16, 251, 252, 254; SB VII «723: 8) 80; HRF
(1938); БМКД (1939); Z. ph. Ch. (B) 51, 219(1942); Ж. Ф. X. XX,
6 (1946); О (1950).
Pb(C102)2 SB II 378; F (1946).
3870. Pb (OH) CI, лаурионит SB VII 125.
PbFCI SB II 45, 362; SB III {ЕО.-.Щ 369; SB VII 125; F (1946);
О (1950).
3872. Pb5 (РОЛ CI =Pbl0 (РОЛ CL SB I 399; SB II 101, 458; SB VI 104.
908 3873. (NH4)2 PbCle-3912. Pb3 (Sb04b _[гл^ XI
3873. (NH4)., PbCl, SB I (Я6Х: 431) 445; SB III (/1,: 121) 479; F (1942);
О "(1950).
NH4Pb2Cl5 SB II 331.
Pb5(P04)3(F, Cl), пироморфит SB VII 146.
PbF, SB I (Cl: 150) 190; SB II 251; HRF (1938); F (1942); О (1950).
PbJ2" SB I (6'6:163) 191; SB II 248; HRF (1938); Acta 18, 378 (1943);
Ж.Ф.Х. XVIII, 419 (1944); Acta 20, 121 (1945); Ж. Ф. X. XX,
6 (1946); О (1950).
Pb(N3), SB II 376.
Pb(NOs)., SB I (G2,:305) 313, 787; SB II 73, 74; 385; HRF (1938);
F (1942); О (1950).
3880. Pb-0 Ch. Z. I, 306 (1942); J. Am. 66, 1107 (1943); Ch. Z. I, 343
(1944); C. A. 39, 2018 (1945); О (1950).
PbO, SB I [СЛ: 158) 211; SB II 221, 222, 264; HRF (1938); J. Am.
66, 1107 (1943); F (1946); О (1950).
Pb20, SB II 221; J. Am. 66, 1107 (1943); С. А. 41, 5356 (1947).
Pb,04 SB II 221, 222; HRF (1938); С. А. 38, 5207 (1944); F (1946);
О (1950).
Pb.O, SB II 221.
PbO SB I (B10: 93) (511:95) 121,769; SB II 221, 280; SB IV 97;
SB VI 57; SB VII 71, 104; HRF (1938); J. Am. 66, 1107 (1943);
F (1946); О (1950).
PbaO SB I (CA: 155) 223; SB II 222, 280.
Pb02xH„0 SB II 264.
PbaO,H20 SB II 222.
PbOxfl О SB II 221.
3890. бРЬО 2Н20 SB VII 71.
PbO 1/2H,0 SB II 221, 222.
Pb(0H)2 SB V 47.
Pb10 (P04), (OH)2 SB VI 104.
Pb3(P04), SB I 399; HRF (1938).
Pb—Pd M. 1, 137 (1946).
Pb2Pd Z. Met. 36, 218 (1943\; C. A. 39, 1095 (1945); F (1946).
Pb—Pr SB III 648.
Pb,Pr SB III (/.12:174) 648.
Pb—Pt M. 1. 137 (1946).
3900. Rb2PbCl. SB III (/1,: 121) 477, 479; F (1942).
Pb2Rh Z. Met. 35, 218 (1943); С. А. 39, 1095 (1945); F (1946).
PbS, галенит SB I (BI : 74) 125, 131; SB II 233; SB III 258; SB
IV 148; HRF (1938); БМКД (1939); Б (1947); F (1942); О (1950).
PbS04 SB I (#2:344) 384; SB II 440, 445; HRF (1938); О (1950).
Pb2S05 SB VI 130.
2 PbO PbS04 SB IV 187.
3 PbO • PbS04 SB IV 187.
4 PbO • PbS04 SB IV 187.
PbS8Os HRF (1938).
Pb—Sb SB III 652; SB IV 250; SB VI 193, см. также Z. Met.
30, 47 (1938); О (1950).
3910. Pb2Sb20, = Pb„0 Sb,0„ биндгеймит SB III (Z)6,; 54) 359; SB IV 200;
SB V 106."
Pb,SbaO,8H20 SB III 359.
3912. Pb3 (Sb04), HRF (1938).
§3281
3913. Pb4Sb2S,—3954. MP,
909
3913. Pb4Sb2S„ менегинит SB VI 85, 131.
Pb„Sb7S,,, менегинит новая формула SB VI 131.
Pb,Sb2S„ фалькманит SB VII 12 i.
Pb5Sb2S8, геокронит SB VI 85.
Pb6Sb4Sn, буланжерит SB VI 85.
Pb2 Sb.8,, джем<онит SB VI 85.
PbuSb.aS.,,, гетероморфит SB VI 85.
3920. Pb,SbuSS0, нлагионит SB VI 85.
PbSb,S4, цинкенит (старая формула) SB III 395; SB VI 85; SB VII
157.
Pb.SbuS.„, цинкенит SB VII 156.
PbSe, кшустнлмт SB I (#1: 74) 125, 137; БМКД (1939); F (1942);
О (1950).
Pb~Si SB III 567.
PbSiO, SB I 328.
PbSiF,2H20 HRF (1938).
PbO UO,Si02H20 SB IV 217.
Pb-Sn SB I 573; SB III 646; SB IV 226; SB VI 193.
Pb2Sn04 С. А. 38, 5207 (1944); F (1946).
3930. PbSnO, С A. 41, 6102 (1947); О (1950).
PbSnS.,, теалит SB HI (7i29 :15) 253; О (1950).
Pb-Sr SB III 639.
Pb.Sr SB III (1Л2: 174) 639; F (1916).
Pb—Те SB VI 193.
PbTe SB I (fil: 74) 125, 138; F (1942); О (1950).
PbThO, С. А. 41, 6102 (1947).
PbT103 SB V 85; NW 31, 202, 466 (1943); P. Ph. 68, 133 (1946);
С А. 40, 3957 (1946); F(1946); 0(1950).
Pb—Tl SB I 571.
Pb6Cl(V04), SB II 102, 458.
3940. PbZnV04OH, десклоизит SB II 466; SB VII 150.
PbO—WO, Ch. Z. I, 1363 (1944).
PbWO, SB I {Hi: 349) 386; SB II 83, 450, 453; Ch. Z. I, 270
(1944); C. A. 39, 2021 (1945); F (1946); О (1950).
PbZrO, SB III 378; P. Ph. 58, 133 (1946); С. А. 40, 3957 (1946).
§ 328. Палладий Pd
Pd SB I (Л1:15) 70, 764; SB II 164, 165. 201, 626, 627, 694, 711;
797, 798: SB III 181, 182; SB IV 77; SB VII 47, 49, 52; HRF
(1938); БМКД (1939); F (1942); 0(1950).
(НаР)2РагС13Сг04(К, = бутил-грушм) SB VI 222.
Pd(NH,)2C,04 SB III 703.
PdCl, SB I 193, 773; SB VI (C50:5) 61; HRF (1938); Б (1947);
0 (1950).
Pd(NH,)2Cl, SB III 703; F (1946).
Pd(NHa)4Pd"Cl4 SB II 489.
3950. (NH4)2 PdCI4 SB I (HI, : 360) 424; F (1946); 0(1950).
Pd(NH,)4CJ2H20 SB III (ff49:100) 447, 702; F(1946); 0(1950).
Pd(NH3)4H2OCl3 SB III 702.
PdF, SB II 35, 165, 290, 291.
3954. PdF, SB II 254, 291; F (1946); О (1950).
МЦ^ _^-..^-...-3J551^^H^993^Ft[(C<H,)1Sj1 CI, |гл. XI
3955. Pd—H SB I 601, 604; SB III 630; F (1942); P. ph. 56, 52(1944);
Ch. Z. II, 300 (1944).
Pd2H SB I 601.
Pd(NH3),J, SB III 704; F (1946).
Pd(NHa)"(N02)> SB III 704.
[Pd(NH3)4j[Pd"(NO,)4] SB III 704.
3960. Pd(NO,)2 HRF (1938).
Pd02 SB I 213.
PdO SB I (510: 93) (fill: 95) 124, 771; SB VII 7?; J. Am. 63, 1392
(1941); F (1946); О (1950).
Pd20 SB I 124.
Pd—P SB IV 245; 0(1950).
Pd3P SB IV 245.
Pd4P2 SB IV 245.
PdP„ SB IV 245: 0(1950).
Pd—Pt SB I 765.
(Pd, Pt)S SB II 235.
3970. Rb2PdBr, SB I (#6,: 431) 447; F (1942); 0(1950).
PdS2 SB IV 113.
PdS SB II 235; SB V (£34 : 3) 41; F (1946); О (1950).
PdSb SB I 781; О (1950).
PdSb, SB I 781; F (1942): О (1950).
Pd—Sn M. 1, 137 (1946)!
PdTe SB I 781.
PdTe2 SB I 781.
Pd,Zn21 SB II 711.
§ 329. Полоний Ро
Po SB IV (A 19:4) 90; Ch. Ph. 4, 648 (1936); Ch. Ph. 14, 56.)
(1946); 0(1950).
§ 330. Платина Pt
3980. Pt SB I (Aii lb) 71, 764; SB II 163-165, 168, 200, 201,627-632,
638, 720, 797; SB III 181, 182, 225; SB IV 77; SB V 35; HRF
(1938); БМКД(1939); F (1942); 0(1950).
Pt(CH,),Cl SB III 697; J. Am. 69, 1561 (1947); О (1950).
Pt(CH,)4 J. Am. 69, 1327, 1561 (1947); Ch. E. N. 25, 2970 (1947);
О (1950).
(NH,CH,)a PtCl. SB I (0#62:611) 637; Б. (1947).
Pt(NH,)C2H4Cl2 ИАН (x), стр. 413 (1942); F (1946).
(NH,C2H,)a PtCl. SB I (0№7 : 613) 638.
[N (CH,)4]2 PtCl, SB I ((Жб^бИ) 635; F (1942).
[N (CH3)» C2HJ2 PtCl, SB I (6>#6,: 612) 611.
Pt(C,H,N),Cl2 SB II 489.
Pt (C2H,N2)2C12 SB III 702.
3990.PtCl3 R„ (?) (R — циклопснтандиамин и циклогександиамин) SB
V 175.
(NH4)aPt(SCN)„ SB I (Я6, :435) 447; 0(1950).
Pt[(CH,),S]2Cl.. SB II 696.
3993.Pt[(C,H,)8S]2012 SB III 696.
§ 332] 3994. PtCl2-4034. RbF _ 911
3994. PtCl3 SB I 193, 773.
H—Pt SB I 604.
(NH,)aPtCl, SB I (#6, :431) 446; SB 111 (Л,: 121) 479; HRF (1938);
F (1942); О (1950).
Pt(NH,)2Cl4 SB III 702; Б. (1947); ИАН (х), стр. 249 (1947);
F(1946); О (1950).
Pt(NHs)4Cl2 SB II 488.
Pt(NH,)4PtCl4 SB II 487, 488; ДАН 38, 96 (1943); F (1946).
4000.Pt(NH„)<PtCl,NH2 ДАН 38, 96 (1943).
Pt(NH„)4Cl2H,0 SB II 488; SB III 701, 702; F (1946).
Pt,04 Ch. Ph. 9, 875 (1941); 0(1950).
Pt—P SB III 630.
PtP, SB I 781; SB III 630; F (1942); О (1950).
Pt2oP7 SB III 630.
Rb„Pt(SCN)4 SB I (#6S:435) 447; 0(1950).
Rb2PtCl, SB II 497; SB III (Лх :121s 477, 479; F (1942); 0(1950).
PtS, куперит SB II 9, 234, 235; Б (1947); F (1946); О (1950).
PtS2 SB I 781; SB II 235.
4010. PtSb SB I 781; 0(1950).
PtSb2 SB I 781; F (1942); О (1950).
PtSe2 SB I 781.
PtSn SB I 765; SB II 720.
Pt2Sn, SB II 720.
PtSn2 Ch. Z. I, 980 (1944).
Pt,Sn, SB II 720.
PtSn, SB II 720.
SrPt(CN)46H20 SB IV 182.
PtTe2 SB I 781.
4020. Pt—Tl SB III 618.
PtTl SB III 618: SB VI (535: 4) 5, 178.
Tl2PtCl, SB III (Л1:121) 479; F (1942); 0(1950).
Pt—W Ch. Z. I, 166 (1942).
Pt4Zn21 SB II 711.
§ 331. Радий Ra
RaF, SB IV (CI: 8) 103; F (1942); О (1950).
§ 332. Рубидий Rb
Rb SB I 747; SB VII 202; F (1942); Усп. 16, 90 (1946); R. M. 15y
90 (1947); 0(1950).
RbBr SB I {Bl: 73) 97, 98, 99, 107; SB V 38; SB VI 55; HRF
(1938); F (1942); О (1950).
RbCN SB II 371; F (1942); О (1950).
Rb2(COO)2 SB III (04e:676) 726.
4030. RbHC204 SB III (Ok,: 675) 728.
RbCl SB I (Bl: 73) 97, 98, 99, 106; SB IV (B2: 6) 94; SB V 37; SB
VI 55; HRF (1938); F (1942); ИАН(х), стр. 382(1944); С. А. 39, 3988
(1945); 0(1950).
RbC104 SB I (Я2:344) 372; SB II 85, 411, 412; F (1942); 0(1950).
RbCIO SB II 378
4034. RbF ' SB I (Bl: 73) 97, 98, 106; F (1942); О (1950).
912
4035. RbH—4076. ReP
[гл. XI
4035. RbH SB II 242; F (1942); О (1950).
RbD О (1950).
RbJ SB I (51: 73) 97, 98, 99, 107; SB V 38; SB TI 55; HRF (1938);
Ph. R. 54, 325, 468 (1938); F (1942); 0(1950).
RbJO, SB I (G5:303) 311; F (1942); 0(1950).
RbJ04 SB III (#04:91) 421; SB V 88; F (1946).
4040. RbN, SB II 375; F (1946); О (1950).
RbN03 SB I 339; SB II 381; SB III 405; SB V 83; HRF (1938);
0(1950).
RbC SB VI 63; SB VII (СИ : 7) 84; Z. An. 241, 97 (1939); Ж.Ф.Х.
XX, 1403 (1946); F (1946); ДАН 59, 67 (1948); 0(1950).
Rb.,03 Z. An. 242, 201 (1939); О (1950).
Rb.,0 SB VII (Cl :7) 85; Z. An. 242, 33 (1939); О (1950).
ItbOH 0(1950).
RbH„P04 SB I 394.
RbRe04 SB III (Я04:91) 421; SB V 89; F (1946); 0(1950).
Rb3Rh(N0,), SB III (/2,: 127) 482; F (1942); О (1950).
RbS* Z.~An. 241, 281 (1939).
4050. Rb2S SB IV (Cl: 8) 110; О (1950).
RbSH SB 111(522:8) 260, (51:7) 261; SB VII 78; F (1942);
О (1950).
Rb,S04 SB I 374; SB II 88. 420; HRF (1938); О (1950).
Rb'Sa0. SB II 107, 506-508; SB VI (K\1: 30) 31, 123, 124.
RbSbF6 Gh. Z. I, 1450 (1943); G. A. 39, 2239 (1945).
Rb.SeCl, SB HI (/1,: 121) 479; F (1942); О (1950).
RbSeH SB VII (522:4) (53:1) 78; О (1950).
Rb.SiF, SB III (Jlx:121) 479; F (1942); 0(1950).
Rb'SnBr, SB V 103; SB VI 121; F (1942).
Rb.SnCl, SB III (Лх: 121) 476, 479; F (1942); О (1950).
4060. RkSnJ, SB VII (Л,: 34) 154.
Rb3Ta08 Ch. Z. I, 11 (1942); G. A. 36, 308 (1942); F (1946); 0(1950).
RbTeCl, SB III (71^121) 476, 479; F (1942); 0(1950).
Rb/Ti20. G. A. 39, 2441 (1945).
Rb3TlBr. 8/7 H20 SB III (/3»: 131) 495; F (1946).
RbVF,H20 SB II 492; F (1942).
Rb.W,Cl, SB III (#7,: 142) 505.
Rb2ZrCl, SB HI (Лг:121) 479; F (1942); О (1950).
§ 333. Рений Re
Re SB I 759; SB II 164, 193, 660; HRF (1938); О (1950).
ReCla SB II 293.
4070.(NH4)KeOt SB III (Я0, :9l)421; SB V 89; F (1946); О (1950).
ReO, SB II 31, 299; Б (1947); 0(1950).
Re206 SB II 299.
P—Re SB III 632; О (1950).
ReP, SB III 632.
RcP2 SB III 632.
4076. ReP SB III 632.
§3371
4077. Rc2P-4112. RFCO,
913
4077. Rc2P SB III 632.
ReSia Z. Met. 33, 370, 378 (1941); 0 (1950).
TlRe04 SB HI (#04: 91)421; SB V 90; F (1946); 0 (1950).
4080. Re,W2 SB II 660.
§ 334. Родий Rh
Rh SB I (41: 15)69, 764; SB II 163-165, 199, 266, 318, 711; SB III
181, 182, 224; SB VI 54; SB VII 53; HRF(1938); F(1942); 0(1950).
RhCI2 SB I 193, 773; 0(1950).
d-[Rh(l-CsHe[l,2](NH2)o]sCls3H,0 SB II 503.
d-lHh(l-C6H8 [1,2] (NH2bJ3-(U104)a 12IL0)
d-[Rh(l-C6He[l,2](NH2)2)a].(C10,), 12H20 SB II 503.
(Rh (en), CI, 3H-0 (en—э!Илендиамин=М2 (CH2)2 NHo) SB VII 235.
Kh(NH,)5ClCl.* SB III (/1,: 125) 492.
(NH4),RhCl„NH4N03, сольВильма Ж.Ф.Х. XVIII, 160(1944); Б (19i7).
Rh (NH3)6 C1J, SB III (J\t: 127) 492.
4090. RhF» SB II 35; О (1950).
(NHJ, Rh (N02), SB III (/2,: 127) 482; F (1942); О (1950).
Rh02 SB II 266.
Rh203 SB I (ОГ)^ 242) 268; SB II 310, 318; О (1950).
RhS, SB I 781; F (1942); 0(1950).
RhSb04 С. А. 39, 2684 (1945); F (1946).
RhTa04 С. А. 39, 2684 (1945); F (1946).
Tl,Rh (N02)e SB III (/2,: 127) 482; F (1942); О (1950).
Rh.Zn ! SB II 711.
§ 335. Радон Rn
Rn ДАН, 62, 231 (1948).
§ 336. Рутении Ru
4100. Ru SB 1(43: 19) 69, 764; SB II 274; SB III 185; SB V 35;HRF
(1938); 0(1950).
RuCl2 SB I 193, 773; 0(1950).
Ru04 SB VI 70; 0(1950).
Ru02 SB I ((74:158)213; SB II 274; F (1946); 0(1950).
P—Ru SB VII 91; 0(1950).
Ru2P SB VII 91.
RnP SB VII 91.
RuP2 SB VII 91.
RuS, SB I {CI: 153)217, 780; SB II 274; F (1942); О (1950).
RuSe, SB I 781: F(1942); 0(1950).
4110. Rufe, SB I 781; F (1942); О (1950).
§ 337. Редкие земли (лантаниды) *): кассиопий (Ср) (лутеций (Lu)),
диспрозий (Di), эрбий (Ег), европий (En), гадолиний (Gd); хольмий^ (Но),
неодим (Nd), празеодим (Рг), промеций (Pm), самарий (Sm), тербий (ТЬ),
туллий (Тт), ите'рбий (lb), церий (Се)**)
Лантаниды: ДАН 60, 371 (1948).
4112. RFCO, (R-CV, La,...) SB II 75, 400.
*) См. тангке соединения других металлов.
**) См § 295.
-914
4113. (BFC0,),-4146. Nd
[гл. XI
4113. (RFCO,)a BaCO, (R-Ce, Xa,...) SB II 77, 402.
M (C2HsS04)3 -9HaO (M = La, €e, Pr, Nd, Sm, Gd, l)y, Y) SB V 149.
Me20, (Me—элементы редких эемель) SB VII 101.
Стеенструпин — фторосшшкат редких металлов Ch. Z. II, 19(1942);
I, 414 (1944).
Ср кассиопий.
Ср 0(1950).
Срп+ Ch. E. N. 25, 2970 (1947).
Cp20,*) SB I 261; SB II 40, 310; SB VII 101; Z. An. 24]. 278 (1939);
F (1942); 0 (1950).
Dy диспрозии.
4120. Dv 0(1950).
Dy203 SB I 261; SB II 40, 310; SB VII 101; Z. An. -241, 278
(1939); F (1942); О (1950).
Dy3S, SB II 323.
Er эрбий.
Er SB II 172; 0(1950).
ErCl,6H20 HRF (1938).
Ег.О, SB I 261; SB II 40; SB VII 101; HRF (1938); Z. An. 241, 278
(1939); F (1942); 0(1950).
EraS8 SB II 323.
1'u европий.
Eu О (1950).
Eu"* Ch. E. N. 25, 2970 (1947).
EuBr, SB VII 80.
4130. EuCU SB VII (C23:8)80; Z. An. 241, 248 (1939); 0(1950).
EuF, " SB VI (CI: 5) 57; SB VII 80; Z. An. 241, 243 (1939); F (1942);
О (1950).
EuJ„ SB VII 80; Z. An. 241, 249 (1939).
Eu263 SB I 261; SB II 39; SB VI 101; Z. An. 241, 278 (1939);
F (1942); О (1950).
EuS SB VI (fil:3) 57, 58; SB VII {Bl: 3) 77; Z. An. 211. 259, 263
(1939); F (1942); 0(1950).
EuSe SB VII (Bl : 3) 77; Z. An. 241, 259 (1939); О (1950).
EuTe SB VII (B\ :3) 77; Z. An. 241, 259 (1939); 0(1950).
Gd гадолиний.
Gd О (1950).
Gdn+ Ch. E. N. 25, 2970 (1947).
GdFe. Z. An. 252, 377 (1944); С. А. 40, 4273 (1946).
4l40.GdMn2 Z. An. 252, 377 (1944); С A. 40, 4273 (1946).
GdPMol,04130H,0 SB III 462.
GdNi, Z. An." 252, 64 (1943); Ch. Z. I, 518 (1945).
Gd,0„ SB I 261; SB VII 101; Z. An. 241, 278 (1939); К (1942);
" О (1950).
Но гольмий.
Но SB VII (ЛЗ: 3) 65; О (1950).
Но203 SB I 261; SB II 40; Z. An. 241, 278 (1939); F (1942); 0(1950).
•Vd неодим.
4146. Nd SB II 172; О (1950).
*) В SB 11 помещены без нзаимных ссылок данные о <'1>^Оэ (стр. VI) и ч Lu.,0,
(стр. :510), хотя это окислы одного и того жо элемента.
§_::37| 4147. N(161,-4189. ТиьО» 91Г)
4147.>'dBr, А. Сг. 1, 265 (1948); О (1950).
NdCl,6H20 IIRF (1938).
Nd (ВгО,)э 9Н20 SB VII (G2.: 29) 30, 31, 135; 0(1950).
4150. NdC, SB I (Г11:741) 783; SB II 275; F (1946); 0(1950).
NdCI, IIRF (1938); A. O. 1, 265 (1948); 0(1950).
NdF, SB II 28, 289; 0(1950).
NdN SB V 43; F (1942); О (1950).
(NH4),Nd(N03)„ IIRF (1938).
Nd,03 SB I (Db.,: 244) (Z)5„ : 745)261; SB II 314; SB III 341; SB VII
" 101; Z. An. 241, 278 (1939); F(1942); 0(1950).
Nd(0H3) SB I 253; A. Cr. 1, 265 (1948); 0(1950).
NdP SB V 43; F(1942); 0(1950).
Nd,Ss SB II 323.
NdSb SB V 43; F(1912); 0(1950).
Pr празеодим.
4160. Pr SB II 171: 0(1950).
PrBr3 A. Cr. 1, 265 (1948): 0(1950).
PrC, SB I (Cll : 741) 783; SB II 275; F (1946); 0(1950).
PrCl3 A. Cr. 1, 265 (1948); О (1950).
PrF3 SB II 28, 289; О (1950).
PrN SB V 42; 0(1950).
PrO, SB I (CI : 150) 197; SB 11 259; F(1912); 0(1950).
Pr40u Ch. E. N. 25, 2970 (1917).
Pre0„ SB I 197, 263.
Pr203 SB I (Л5,: 244) (7J5,: 745) 261; SB II 314; SB VII 102; Z. An.
241, 278(1939); 0(1950).
4170.Pr(OH)3 A. Cr. 1, 265 (1948); 0(1950).
PrP SB V 43; 0(1950).
Pr2Ss SB II 323.
PrSb SB V 44; F(1942); 0(1950).
PrSn3 SB III (/.12:174) 647.
PrZn SB V 44; F (1912).
Sin самарий.
Sm О (1950).
SmBr3 A. Cr. 1, 265 (1948); О (1950).
SmBr, SB VII 80; Z. An. 24A, 249 (1939).
SmCI.," SB VII (С23:8)80; Z. An. 241, 248 (1939); 0(1950).
4180. SmF3 SB II 28, 289; О (1950).
SmJ., SB VII 80; Z. An. 241, 249 (1939).
Sm„63 SB I 261; SB II 39; SB VII 101; Z. An. 241, 278 (1939);
F(1942); 0(1950).
Sm.,S3 SB II 323.
Sm.SC^ 8H.0 SB III 454.
Tb тербий.
Tb О (1950).
Tb..03 SB I 261; SB II 40; Z. An. 241, 278 (1939); F (1942); 0(1950).
Tb407 SB I 197, 263.
Tm туллий.
Tm 0(1950).
4189.Tm.,0, SB1 261: SB II10; SB VII 101; Z. An. 241, 278(1939); F (1942);
О (1950).
916
4190. Yb-4224. SbF
[гл. XI
Yb иттербий.
4190. Yb 0(1950).
Yb"' Ch. E. N. 25, 2970 (1947).
YbBr., Z. An. 241, 249 (1939).
YbCl.," SB VII 80; Z. An. 241, 249 (1939).
Ybl," SB VII (C6:7) 80; 0(1950).
Yb263 SB II 40; SB VII101; Z. An. 241,278 (1939); F (1942); О (1950).
YbaS, SB II 323.
YbSe SB VII (51: 3) 79; О (1950).
YbTft SB VII (fil : 3) 79; О (1950);
§ 338. Сера S*)
S SB I 60; SB II 189, 205; SB III (Л16 : 4) 213-215; SB IV 82, 83;
SB V 29-31; SB VI 51; SB VII 63, 64; HRF (1938); Acta XVI,
181 (1942); J. Am. 65, 648 (1943); С A. 40, 4595 (1946); О (1950).
4200. SBr2 Ж.Ф.Х. XX, 6 (1946).
S(CH3)2 SB IV 276; 0(1950).
SC12 SB VI 63; Ж. Ф. X. XX, 6 (1946); 0(1950).
SaCl2 SB IV 96; SB VI 63; О (1950).
S02C1„ SB VI 63.
SOC1, SB VI 63.
SP, " SB III 332; О (1950).
SO F SB VII 119.
H2S * SB II 270, 281; C. A. 38, 6154 (1944); 0(1950).
D2S С A. 38, 6154 (1944); О (1950).
4210. H2S2 SB VI 71.
S03 SB VI 63: J.Am. 60, 2360 (1938); С A. 36, 695 (1942); Ch. Z.
I, 10 (1942); 0(1950).
S30, Rec. 60, 794 (1941); Ch. Z. I, 10 (1942).
SO, SB III 302; SB IV 119; J. Am. 62, 1240 (1940); С. А. 39, 1799
" (1945); 0(1950).
SX2 (X = Hal) Ж.Ф.Х. XX, 6 (1946).
§ 339. Сурьма Sb
Sb SB I (A7-.27) 58; SB II 184, 185, 237, 741-745, 747, 749,
751-753, 756; SB III 186, 187, 210, 211; SB V 31, 34; SB VI 42,
49, HRF (1938); Z. An. 244, 205 (1940); О (19.50).
SbBr, SB V 57; SB VI 68; HRF (1938); Ж. Ф. X. XX, 6 (1946);
0(1950).
Sb(CH3)3Br2 SB VI (6>G1,:199) 219.
SMCH^Cl, SB VI (0G1,: 199) 200, 201, 219.
Sb(CHa)8J.' SB VI (OGli:199) 200, 201, 219.
ч220. SbCla SB II 184; SB V 57; HRF (1938); Ж. Ф. X. XX, 6 (1946);
О (1950).
Sb-тартрат гексагидрат HRF (1938).
(NH4)2SbCl, Ch. E. N. 25, 2970 (1947).
SbOCl HRF (1938).
4224. SbF, HRF (1938); Ch. Z. 1,342 (1944); С A. 39, 2019(1945);
Ж.Ф.Х. XX, 6 (1946).
*) См. сульфиды металлов.
§ 341]
4225. NH4SbF„-4258. Se
917
4225. NH.SbF. Ch. Z. I, 1450 (1943); С. А. 39, 2239 (1945).
SBJa SB II 294; SB V 57; SB VI 68; HRF (1938); Ж. Ф. X. XX,
6(1946); 0(1950).
Sb20s SB I 264; SB II 598; HRF (1938); О (1950).
Sb2OsH20 SB IV 133.
Sb.OIS SB I 264.
4230. Sb204 SB I 264; SB III (Z)62: 54) 358; SB VI 113; SB VII (DQ,: 15)
126; SB VII 126, 127; 0(1950).
SbsO,OH SB VII 24.
Sb..04H20, стибиконит SB III 359; БМКД (1938); БМКД (1939).
Sb20s = Sb40„ валентинит, сенармонтит SB 1(2)6,: 246) 264; SB IV
(£5„:20) 132; SB VII 109; БМКД (1938); HRF (1938); Ch. Z. II,
9 (1942); F (1942); Б (1947); О (1950).
Sb,S„ антимонит SB I 268; SB II 324; SB III (Z)5,:49) 349; HRF
(1938); Б (1947); О (1950).
Sb2(S04), HRF (1938).
Sb—Si SB III 567; О (1950).
Sb-Sn SB I 77, 142, 601; SB III 651; HRF (1938); 0(1950).
SbSn SB I (£3 : 77) 142, 601; SB II 751 - 753; SB III 651; F (1942);
О (1950).
Sb2Sns SB II 751, 752.
4240. SbTa04, стибиотанталит SB VI 113.
Sb„Tes SB II 324.
Sb-Tl SB I 599; О (1950).
SbTl SB I (52:76) 141, 599: Z. An. 244, 146 (1940); F (1942);
0(1950).
Sb2Tl, SB I 599; SB III (L22: 175)362; SB I (L22:489) 488.
Sb-Zn SB III 644; О (1950).
SbZn SB II 749; SB III 644.
SbaZn4 SB II 749; О (1950).
Sb2Zn, SB II 749; SB III 644; 0(1950).
Zn(SbOs), SB V 86.
4250. Sb2Zn04 Ch. Z. I, 342 (1944^; С. А. 39, 2019 (1945); F (1946).
§ 340. Скандий Sc
Sc SB VII (41:3) (43:3) 57, 58; NW 27, 230 (1939); Z. El. 45,
357 (1939); О (1950).
SeF3 SB VII (Z)0„: 12) 96 (тип чуть искажён); О (1950).
ScN SB I (51: 74) 139; F (1942); О (1950).
Sc20, SB I 196, 197, 261, 333; SB II 39, 310; F (1942); 0(1950).
Sc (OH)3 SB I 253.
Sc2S, SB II 323.
Sc2Si20„ тортвейтит SB II 124, 467, 523; Б (1947).
§ 341. Селен Se*)
4258. Se SB I (48:28) 63; SB II 189, 190, 203, 205; SB III 245-218;
SB V 31; SB VII 64, 65; HRF (1938); БМКД (1939); Ж. Ф. X.
XIX, 160 (1945); Ж. Ф. X. XXII, 1034 (1948): О (1950).
*) См. селонпды металлом.
918 4259. (Cf,H,),ScBr,-4294. ThSiW,,O4«30H,O fгл. XI
4259.(C,H5)2SeBr2 J. Am. 63, 803 (1941).
42(50. (NHOsSeBr, SB II 492.
SeF, SB III 332; 0(1950).
H2Se SB II 281; С. А. 38, 6154 (1944); 0(1950).
D,Se С. А. 38, 6154 (1944).
SeO. SB V (C47:4) 46; SB VI 64; F (19iG); О (1950).
H2SeO, HRF (1938).
§ 342. Кремний Si
Si SB I (/14:21) 52, 752; SB II 163, 181, 673, 756-762; SB III
186, 187, 207; HRF (1938); БМКД (1939); F (1942); 0(1950).
SiBr4 J. Am. 64, 62 (1942); Ж. Ф. X. XX, 6 (1946); О (1950).
SiBr.F. J. Am. 64, 62 (1942).
SiC SB I (#3 : 77) (55 : 81) (M : 82) (Bl: 83) 144; SB II 238; SB III
261, 262; SB V 109; HRF (1938); F (1942); A. Min. 29, 249, 327.
431 (1944); 30, 519 (1945); С. А. 39, 2440, 2441 (1945): Acta
XX, 386 (1945); J. ph. IX, 244 (1945); Ж. Ф. X. XVIII, 192(1945);
Ж. Э. Ф. 15, 655 (1945); Б (1947); О (1950).
427().Si(CH3)« SB IV 277; 0(1950).
Si(C,H,)4 SB I (СЛ):615) 616, 649-651; SB VI 230; F (19'iG):
О (1950).
(CH3)4Si04 SB II (07)2,: 803) 818.
SiCl4 SB IV 125; SB VI 81; Ж. Ф. X. XX, 6 (1926); 0(1950).
81,01, SB VI 82; О (1950).
SiHCL, SB VI 81, 82; SB VII 117.
SiH3Cl2 SB VI 81.
SiH3Cl SB VI 81.
SiF4 SB II 37. 305; SB IV 125; SB V 63; Ж. Ф. X. XX, 6 (1926).
(NH4)3SiF7 J. Am. 64. 633 (1942); О (1950).
4280. (NH,),SiF, SB I (#6, : 431) 442; SB III (71,: 121) 479 (71, : 123)
488; HRF (1938); F (1942); Б (1947); 0(1950).
[N(CH3)4],SiFe SB III (0/^:656) 699; F (1916).
Si„H„ SB VI 82; 0(1950).
Si,t4 SB II 306; Ж. Ф.Х. XX, 6 (1946); F (1942); 0(1950).
SiO,, кнарц, криетобалит, тридимит (халцедон), меланофлогит SB
'I (С8: 168) (С9: 171) (СЮ: 174) 198, 199, 202, 203, 776; SB II 248.
259-261, 352, 591-596; SB III (£8:21); 294 - 300 (£30: 25); 296
299; SB IV 202, 204. 218; SB V 109, 110; SB VI 133,-138; SB VII
68, 161, 166; HRF (1938); БМКД (1938); БМКД (1939); С (1939);
Z. Kr. A. 104, 261 (1942); F (1946); Б (1917); О (1950).
SiO.„ стекло Ж. Ф. X. XIV, 235 (1940).
Si.O, SB V 110.
Sib Mazda Kenkyu Ziho 15, 305 (1940); С. А. 36, 4001 (1942).
Si —P Z. An. 242, 237 (1939); О (1950).
SiP207 SB III (£6,: 140) 503; F (1942); 0(1950).
4*290.818, SB III (C42.-37) 286; Б (1947); О (1950).
Si-Sn SB III 567.
ThSi04, торит SB I 410; БМКД (1938).
ThSiW,,04(,27H2O SB IV 213.
429',. ThSiW.AoSOH.O SB IV 213.
§ 3131
i2P- .*bJl —J?7®1.- SnClt5H?0
919
4293. Si8Ti SB VII (C54: 12) 95; 0(1950).
TUSiF, SB III Ш,:121) 478, 479; P(19i2); 0(1950).
Si2V Z. Met. 33, 378 (1941), тип С40.
SiV, SB VII (415 : 3) 98.
(H16)a;(Sl01)jr(W01).(H20)m SB II 510.
4300. HJSi (W3010)4] 5H20 SB HI (Я41в:115) 465.
H4SiW1204014H20 SB VII 148.
H4SiW12O4031H„O SB III 466; F(19i6).
Zn2SiW1204„27IL0 SB IV 213.
Si—Zn SB III 567.
ZnSiF,6H20 SB II 104, 504; О (1950).
Zn,Si04. ииллемит SB I (Я13:356) 404; SB II 518; SB IV 174;
" БМКД (1939); Z. Krist. 105, 273 (1944); С. А. 39, 5148 (1945);
О (1950).
Zn2Si04H20 SB II 524.
H2Zn4SiO, SB II 125, 524.
Zn4Si207(OH)2 SB II 524.
4310.Zn4(OH)2Si2O;H2O, гсмиморфит (каламин) SB II 524; Б (1947).
ZnSi03 SB II 518.
ZrSi2 SB II 276; SB V (C49: 5) 53; 0 (1950).
ZrSi04, циркон SB I (//3:347)408; SB II 592; SB V HO; SB VII
162; БМКД (1938); I-JRF (1938); F (1946); Б (1947); О (1950).
Силикаты ИАН(х), стр. 364 (1918).
Силикатные стёкла И АН (х), стр. 691 (1941).
§ 343. Олово Sn
Sn SB I (44:21) (45:23) 54; SB II 164, 182, 219, 713-720, 735,
737, 751-754; SB III 183, 184, 186, 187, 208; SB IV 78, 88;
SB VI 48; SB VII 62, 63; HRF (1938); F (1912); Ж. Ф. X. XX,
1414 (1946); F (1946); О (1950).
SnBr4 Ж. Ф. X. XX, 6 (1946); О (1950).
SnBr, Ж. Ф. X. XX, 6 (1946).
(NH4)2SnBr, SB V 103; SB VI 121; F (1912).
4320. (NH.CH.), SnBr, SB I 637.
Sn(CHs)4 SB IV 277; 0(1950).
Sn(C,H5)4 SB I (06 :615) 616, 649-651; SB VI 230; F(1916); 0(1950).
[NH2 (CH,),] SnCl, SB III (0/1:658) (071.: 694).
[NH(CH3),]*2 SnCl, SB II 805, 816; F (1942").
[N (CH3)4]2 SnCl, SB I (0#6x:611); SB II 816; F(1912).
(NH,CH3)2 SnCl, SB I (0#6.,:611) 637.
[N(C2H5)3(CH3)]2SnCle SB 1*(0Я6,:612) 636; SB II 817; F (1942).
[N(CH,),C2HJ2SnCl, SB I (0Я6,:612) 636; F (1942).
[N (CHa)2 (C.,H5)2]2 SnCle SB I (0Я6,: 612) 636; F (1946).
4330. (NH,C,H,)*4 SnCl, SB I (0Я67: 613) 612, 638.
SnC204 = Sn-oKcanaT HRF (1938).
Sn-тартрат HRF (1938).
[P(CH,)(C2H6)s]8SnCl, SB II 818; F (1942).
[(CH8),S]2SnCl, SB II 817; F (1942).
Sn[SC(CH„),]4 SB VII 233; F (1946).
SnCI4 SB IV 125; SB VI 69; Ж. Ф. X. XX, 6 (1946); 0 (1930)
4337. SnCl4GH„0 HRF (1938).
920 4338. SnCl2-4375. SrCl86H,0 [гл. XI
4338. SnCl2 SB IV 103; Ж. Ф. X. XX, 6 (1946); О (1950).
SnCl2-2H20 HRF (1938).
4340. (NH4). toiCl, SB I(ff6,: 431) 444; SB 111(71,: 121) 479; HRF (1938);
F (1942); О (1950).
SnJ, SB I (2)11:236) 251; SB VI 68; HRF (1938); F (1942); Ж. Ф. X.
XX, 6 (1946); Б (1947); О (1950).
Sn«F2 Ж. Ф. X. XX, 6 (1946).
Sn02, касситерит, «деревянистое олово» SB I (C4 :158) 39, 114, 210;
SB II 263, 264, 483, 484, 598; SB IV 120; БМКД (1938); HRF (1938);
F (1946); Б (1947); О (1950).
Sn02xH20 SB II 263, 264.
Sn02H20 SB II 263.
Sn022H20 SB II 263.
SnO SB I (£10:93) (£11:95) 120; SB II 220; SB III 250; HRF
(1938); N. 154, 643 (1944); F (1946); О (1950).
SnOl/2H20 SB II 220.
Sns(P04)2 HRF (1938).
4350.SnP,0, SB III (#6,: 140) 503; F (1942); 0(1950).
SnS.. SB I (C6:163) 214, 780; HRF (1938); 0(1950).
SnS* SB III (529:14) 253; HRF (1938); 0(1950).
Sn(S04)2 • 2H20 HRF (1938).
SnS04 HRF (1938).
SrSnO, SB III (£2,168) 378; F (1942); P. Ph. 58, 133 (1946); C. A.
40, 3957 (1946).
SnTe SB I (£1: 74) 137; F (1942).
Tl2SnCl, SB III (Лх: 121) 479; F (1942); О (1950).
Sn—Zn SB IV 226; SB V 134.
ZnSnF,6H20 SB II 104, 504; О (1950).
4360.Zn2SnO4 SB II 483; F (1942).
ZnSnO, SB II 483.
§ 344. Стронций Sr
Sr SB I 748, 749; HRF (1938); Z. An. 248, 155 (1941); F (1942);
Ch. Z. I, 978 (1942); С A. 37, 2634 (1943); О (1950).
SrBr2 SB VII (C53:10) 11, 82; Z. An. 241, 249 (1939); О (1950).
SrBr„ • 6H20 SB II 102, 499; HRF (1938); О (1950).
SrC2 SB I (Cll:741) 782; SB II 275; F (1946); 0(1950).
SrCOs SB I (G2: 297) 325, 339; SB II 216, 390, 397; HRF (1938);
О (1950).
Sr (CH3COO)2 V, H20 HRF (1938).
Sr (COO)2 2 -J- H20 SB V 159.
Sr(HCOO)2 " HRF (1938).
4370. Sr (HCOO)2 • 2H20 HRF (1938).
Sr-лактат тригидрат HRF (1938).
Sr-салицилат дигидрат HRF (1938).
Sr-тартрат тетрагидрат HRF (1938).
SrCl, SB I (CI: 150) 187; SB II 246; F (1942); ИАП(х), стр. 382 (1944);
С. А. 39, 3988 (1945); О (1950).
4375.SrCK6H20 SB II 102, 246, 499; HRF (1938); 0(1950).
§ 345) 4376. Sr(C103)2- 8H20-4414. _TaCI, 921
4376. Sr (C10s)2 • 8H20 HRF (1938).
SrF2 SB I (CI: 150) 186; SB II 236, 244, 248; SB VII 82; HRF
(1938); F (1942); О (1950).
SrH2 SB III (C29: 24) 307; О (1950).
SrJ2 Z. An. 241, 249 (1939).
4380. SrJ2 6H20 SB II 102, 499; HRF (1938); О (1950).
SrMo04 Ch. Z. I, 270 (1944); С. А. 39, 2021 (1945).
Sr,N2 SB III 353.
SrNH SB III 387; F (1942).
Sr(N03)2 SB I (G2i:305) 313, 787; SB II 74, 385-387; HRF (1938);
F (1942); О (1950).
SrNi (CN)4 5H20 Ch. Z. I, 1363 (1914); С. А. 39, 3190 (1945).
Sr—О Ch. Z. I, 306 (1942).
Sr02 SB III 292; HRF (1938); Ж. Ф. X. XX, 1403 (1946); F (1946);
О (1950).
SrO SB I (51:73) 118; SB II 216; SB VII 106; F(1942); О (1950).
Sr048H20 SB II 57, 216, 349; F (1946); 0(1950).
4390. SrOxHO SB II 216.
Sr(0H)2 SB II 216.
Sr(OH)2H20 SB II 216.
Sr(OH)28H20 SB I 776; SB II 56, 216, 256, 349; SB VII 90; JIRF
(1938); F (1946); О (1950).
Sr,P2 SB III 353.
Srs(P04)2 A. Cr. 1, 263 (1948).
Sr,(P04)3OH SB VII 147.
SrHP04 HRF (1938).
SrS SB I (51:74) 125, 127; SB II 231, 236; Z. An. 241, 263 (1939);
F (1942); О (1950).
SrS04 SB I (#2:344) 382; HRF (1938); О (1950).
4400. SrSe SB I (SI: 74) 135; Z. An. 241, 263 (1939); F (1942); О (1950).
SrSiO gg jtt 370
SrTe " SB I (Bl: 74) 135; Z. An. 241, 263 (1939); F (1942); О (1950).
SrThO С. А. 41 6102 (1947).
SrTiO,' SB I '(G5':303) 333; SB III (Е21:Щ 378,F (1912) P. Ph. 58,
133 (1946); С A. 40, 3957 (1946); О (1950).
SrTl SB III (£20:175) 272; F (1942).
SrU04 A. Cr. 1, 265 (1948); О (1950).
SrW04 SB II 450; Ch. Z. I, 270 (1914); С. А. 39, 2021 (1945); N. 157,
548 (1946); F (1946).
SrZrO, SB III (£2,: 68) 379; F (1942); P. Ph. 58, 133 (1946);
С A. 40, 3957 (1946); О (1950).
§ 345. Тантал Та
Та SB I (А2:Щ 57; SB II 163, 187, 188, 662, 674, 798; SB III 212;
SB IV (Л2:3) 88; HRF (1938); F (1942); 0(1950).
4410. TaBr5 Ж. Ф. X. XX, 6 (1946).
Ta,Brl4 Ch. E. N. 25, 2970 (1917).
TaC SB I (51:74) 144, 146; SB II 237, 238; SB III (51 : 7) 262;
SB IV (51: 6) 100; F (1942); О (1950).
Ta.,C SB II 276; SB III 309; О (1950).
4414. TaCl. Ж. Ф. X. XX, 6 (1940).
922 4415. Ta,Cl4-4454. Th(K0,)44H,0 I гл. XI
4415.Ta6CI4 Ch. E. N. 25, 2970 (1947).
TaF5 Ж. Ф. X. XX, 6 (1946).
Та—II SB II 799; О (1950).
TaJ5 Ж. Ф. X. XX, 6 (1946).
TaNr SB I 139.
4420. TaN SB I 139.
Ta.O, SB I 196; SB II 360; HRF (1938).
Ta,0,xH20 SB II 360.
'ia-S Z. An. 241, 337 (1939); 0(1950).
TaS, SB VI 64.
TaSi, Z. Met. 33, 378 (1941).
Ta.,To SB III {CI : 20) 283.
Ta(J—TiC . Ж. Ф. X. XX, 769 (1946); 0(1950)'.
YTa04 SB I 397; F (1946); О (1950).
ZnTa„Oe G. A. 39, 2684 (1945).
4430. TaZ—ZrC Ж. Ф. X. XX, 769 (1946); 0(1950).
§ 346. Теллур Те
Те SB (-48: 28) 63; SB II 190, 204; SB III 183; IIRF (1938); I. J. ph.
15,233(1946); БМКД (1939); 0(1950).
TeRr, SB IV 104: Ж. Ф. X. XX, 6 (1946); J. Am. 69, 2102 (1947);
0(1950).
Te('l4 Acta XX, 407 (1945); Ж. Ф. X. XX, 6 (1946); 0(1950).
TeCl., SB IV 104; HRF (1938); Ж. Ф. X. XX, 6 (1946); 0(1950).
(NHJ„TcCl, SB III (/l^ 121) 479; F (1942); 0(1950).
TeF6 SB III 332; 0(1950).
TeF, Ж. Ф. X. XX, 6 (1946).
Tej; SB IV 104; Ж. Ф. X. XX, 6 (1946); О (1950).
Те-нитрат HRF (1938).
4 440. 4Te02N206 1 J- H ,0 HRF (1938).
ТеО„. толлурит SB 1(64:158) 211; SB VII (652: 8) 9. 10, 89; HRF
"(1938); F(1946); О (1950).
H.,Te04 HRF (1938).
Te'(0H)6 SB I 254; SB III 332; HRF (1938); 0(1950).
Te2Ti SB I (643: 163) 213, 780; О (1950)
Tl„TeCI6 SB III (/1, : 121) 479; F (1942); О (1950).
ZnTe Z. An. 244, 146 (1940); F(1942).
§ 347. Торий Th
Th SB I (-41:14) 54; SB II 182, 266, 690, 797: HRF (1938);
F (1942); О (1950).
ThC SB I 783; SB II 275; F(1946); 0(1950).
Th-ацетат HRF (1938).
450. Th (Cs04)s HRF (1938).
ThCl4 HRF (1938).
ThF4 SB VII 81.
Th (N0,)412H,0 HRF (1938).
4454. Th(NO3)44H.,0 SB 111 418.
§ 3481 _ 4155. ThOo - 4192. TiO, - ZrO, 923
4155. TliO., SB I (6'1:150) 197, 208; SB II 205-207, 269; HRF (1938);
F(1942): 0(1950).
Th(OII)4 SB I 253, 25'i.
Th—P SB VII 112; 0(1950).
ThP SB VII Z. An. 245, 391, 402 (1941); 0(1950).
ТМ4 SB VI 76; SB VII (2)7,: 15) 16, 112; Z. An. 210, 300 (1939^:
245, 391, 402 (1941); 0(1950).
1160.Th(SO4), IIRF (1938).
TbZne M. 1, 31 (1946).
§ 318. Тптан Ti
Ti SB I (Л3:19) 53; SB II 181, 759, 762, 799; SB IV (/12:3) 84;
HRF (1938); Ch. Ph. 9, 673 (1941); F (1942); О (1950).
TiBr4 SB II 306; Ж. Ф. -\. XX, 6 (1946); 0(1950).
TiC SB I (51 : 74) 144; SB II 238; SB III (BI : 7) 262; SB IV (Bi : (>)
100; HRF (1938); Ж.П. X. 12,1759(1939); Z. An. 244, 191 (1940);
Acta XIII, 723(1940); Ж.Ф. л. XV, 983 (1941); F (1912); 0(1950).
TiC • TiN SB 773; SB II 238.
4TiN • TiC SB 238; F (1912».
TiCl, Ж. Ф. X. XX, 6 (1946); 0(1950).
TiF4 HRF (1938); Ж. Ф. X. XV, 6 (1946).
(NH4)3TiOFB Gaz. 71, 620 (1911); С. Л. 36, 6934 (1942).
4470.Ti—H "Ch. Ph. 9, 678 (1941); F(1912); 0(1950).
TiJ4 SB II 307; Ж. Ф. X. XX, 6 (1916); F(1942); 0(1950).
TiN SB I (Bi : 74) 139; SB II 238; SB IV (BI : 6) 100; SB VII (Bi : 3)
• 4, 79; Z. An. 244, 191 (1940); Acta XIII 723(1940); F (1942); О (1950).
Ti—О Z. El. 45, 362 (1939); Ch. Z. II, 989 (1943); 0(1950).
Ti02, рутил, анатаз, брукит SB I (6*4 :158) 196, 204; SB I 778:
SB'lI 265, 314, 320. 442. 484, 593; SB II (C21: 14) 265; SB III
305; SB VI 64; SB VII 72, 87; IIRF (1938); БМКД (1938); Z. El.
45, 362 (1939); Ж. O. X X 156 (1910); Z. Кг. (А) Ю4, 358(1912);
Ch. Z. I, 488 (1943); II. Ch. 27. 88(1911); С Л. 38, 3179 (1911);
G. A. 39, 2141 (1945); F (1916); Б (1917); О (1950).
Ti02xH20 SB III 305.
Ti203 SB I (D5t : 242) 263; SB II 310, 311; SB VII 87; 0(1950).
TiO SB II 220; SB VII 87; Acta XIII, 723 (1940); F (1912); О (1950).
MTi03 C. A. 38, 3179 (1944); 39, 2441 (1945).
Ti—P SB VI 192; О (1950).
4480. TiP SB VI 192.
TiP20. SB HI (#6,: 140) 503; F (1912); 0(1950).
TiS, ' SB V 71, 72.
TiS, SB I (6*6:163) 213, 780; 0(1950).
Ti2S3 SB V 72.
TiSes SB I (C6:163) 213, 780; 0(1950).
Ti—Zn Z. Met. 33, 355 (1941).
ZnTiF,6H20 SB II 104, 504; О (1950).
Zn2Ti04 SB II 484, 485; SB VI 116; F (19'.2).
Zn2Ti04Ti02 SB VI 116.
4490. ZnTiO, SB II 484.
TiZr SB VII 211.
4492. Ti02—ZrO, SB V 48.
924
4493. Tl-4530. UC13
[гл. XI
§ 349. Таллий Tl
4493. Tl SB I {A3 :19); (AQ: 24) 45, 751; SB II 173, 733, 738, 751; SB VI
44; HRF (1938); F (1912); О (1950).
TIBr SB I (52:75) 113, 768; SB IV 92; F (1942); О (1950).
Tl (CH,)2 Br SB III 695; F (1946).
T1(CH3)2C1 SB III 695; F (1946).
T1(CH,)2J SB III 695; F (1946).
T1CNS SB III (F5,: 82) 397.
T1N034CS (NH2)2 SB VI 229.
4500. T1C7H302 SB VI 229.
TIC„H„02 SB VI 230.
Т1гС13 С. А. 41, 2947(1947); 0(1950).
TlCl SB I (52:75) 113; SB III 231; SB IV 92; SB V 38; HRF (1938);
F(1942); 0(1950).
TICK), SB I (#2:344) 372; SB II 85, 411, 412; F (1942);
О (1950).
TIF SB III (524:9) 226; 0(1950).
T1HF, SB II 374, 375.
(H„T1F3)2H20 SB II 375.
TIJ SB I (52:76) 113, 768; SB IV (533:6) 92, 96; F (1942);
Б (1947); О (1950).
T1(N03)3 -3H20 HRF (1938).
4510. T1N0, SB V 83.
Т1Д, SB I 261, 333; SB II 39, 310, 354; HRF (1938); F (1942);
О (1950).
Tl20,xH20 SB II 354.
Tl2S SB VII 92.
Tl2SO, HRF (1938).
11,8,0, SB IV 198.
TISbF, Ch. Z. I, 1450 (1943); С. А. 39, 2239 (1945).
TISe SB VII (537:6) 7, 77; F (1946); Б (1947); 0(1950).
T1./VF6H20 SB II 492; F (1942).
T18W2C1, SB III (Я7,: 142) 505.
4520. T13P0412W034H„0 SB VII 149.
§ 350. Уран U
U SB I 62; SB II 192; SB VI (Л20:3) 51; J. Am. 59, 2588 (1937);
J. Am. 69, 1719, 2105 (1947); О (1950).
UBr, A. Cr. 1, 265 (1948); О (1950).
U—С J. Am. 70, 99 (1948); 0(1950).
UC J. Am. 70, 99, 1718 (1948); 0(1950).
UC, SB II 276; J. Am. 70, 99, 1718 (1948); F (1946); О (1950).
NH4U02 (CH3C00)3 SB III 717; F (1946).
U02 (CH3COO)2 2H20 HRF (1938).
UC1, A. Cr. 1, 265 (1948).
UC14 SB II 306; О (1950).
4530. UC1, A. Cr. 1, 265 (1948); 0(1950).
§350|
4531. UF.-4568. NpJ,
92E
4531. UF, SB V 64; 0(1950).
aUF, A. Cr. 1, 265 (1948).
U2F, A. Cr. 1, 265 (1948).
UF202 A. Cr. 1, 265 (1948); О (1950).
UH, J. Am. 69, 1719 Ц947); 0 (1950).
UD3 J. Am. 69, 1719 (1947); О (1950).
UJ3 A. Cr. 1, 265 (1948); О (1950).
U—N J. Am. 70, 99 (1948).
UN J. Am. 70, 99 (1948).
4540. U2N3 J. Am. 70, 99 (1948).
U02(N03)„ • 6H„0 SB I 455; SB VI 105; IIRF (1938); О (1950).
U—0 J. Am*. 70, 99 (1948); О (1950).
U03 SB I 249; J. Am. 69, 2105 (1947); J. Am. 70, 99 (1948); A. Cr.
1, 265 (1948); О (1950).
UA SB I 249; Ch. E. N. 25, 2970 (1947); J. Am. 70, 99 (1948),
N. 162, 70(1948); О (1950).
L80- О (1950).
U.O. J. Am. 70, 99 (1948); 0(1950).
U02, уранинит SB I (CI: 150) 219; SB III 302; J. Am. 69, 2105
(1947); J. Am. 70, 99(1948); F (1942); 0(1950).
(UO,) (группа) F. S. 34, 1483 (1942).
UO" 0(1950).
4550. U—P Z. An. 240, 300 (1939); Z. An. 245, 391 (1941); 0(1950).
UP Z. An. 240, 300 (1939); Z. An. 245, 402 (1941); 0(1950).
U,P, Z. An. 245, 402 (1941); 0(1950).
UP2 Z. An. 245, 391 (1941); О (1950).
UP207 SB IV (#6,: 68) 200; F (1942); О (1950).
U—S Z. An. 243, 313, 322 (1940); 0(1950).
US0l76 О (1950).
U—Si 0(1950).
UaSi A. Cr. 1, 265 (1948).
U3Si2 A. Cr. 1, 265 (1948).
Ураниды (или актиниды"1)
Америций (Am) 95; кюрий (Cm) 96; нептуний (JVp) 93; илутоний
(Pu) 9t.
Am
4560.AmBra A. Cr. 1, 265 (1948); 0(1950).
AmCl3 A. Cr. 1, 265 (1948); 0(1950).
AmJ, A. Cr. 1, 265 (1948); О (1950).
Am2S3 A. Cr. 1, 265 (1948); 0(1950).
Cm — да и ыы х nor.
Np
GtNpBr, A. Cr. 1, 265 (1948); 0(1950).
jlNpBr, A. Cr. 1, 265 (1948); 0(1950).
NpCl3 A. Cr. 1, 265 (1948); 0(1950).
'<568. NpJ3 A. Cr. 1, 265 (1948); О (19-30J.
*) !l порядке исключения для уранпдои даются повторно формулы комплексных
соединен ни с другими металлами, п частности щелочными. Эти формулы приведены
выше соответственно алфавитному порядку.
926 4569. КУрД1. —4609. V — So [гл. XI
456H.KNp,F, J. Am. 70, 2147 (1948); 0(1950).
Pu
4570. PuBr, A. Cr. 1, 265(1948); 0(1950).
PuCI., A. Cr. 1, 265 (1948); 0(1950).
CU'uCl, A. Cr. 1, 268 (1948).
PuJ3 A. Cr. 1, 265 (1948); 0(1950).
KPu,F, J. Am. 70, 2147 (1948); 0(1950).
KPuP, J. Am. 70, 2147 (1948); 0(1950).
NaPuF5 J. Am. 70, 2147 (1948); 0(1950).
NaPuF4 A. Cr. 1, 265 (1948); 0(1950).
P,NaPuF4 J. Am. 70, 2147 (1948); 0(1950).
KbPuF6 J. Am. 70, 2147 (1948); 0(1950).
4580. PiijO.S A. Cr. 1, 265 (1948); 0(1950).
Pu2Ss A. Cr. 1, 265 (1948): 0(1950).
§ 351. Ванадий V
V SB I (42:16) 56; SB II 187, 316, 661, 662, 780- 782, 798; SB IV
(/12:3) 84; HRF (1938); F(1942); О (1950).
VC SB 1(51:74)144; SB II 780-782; HRF (1938); Z. An. 344, 190
(1940); Acta XIV, 297(1941); F (1912); 0(1950).
V4C3 SB II 780-782.
V2C SB II 780-782.
V5C SB II 781, 782.
V-хлорид HRF (1938); 0(1950).
V0C1, SB VI 83.
(IIN4)SVF. SB II 491; F (1942).
4590. (NH4), VFBH40 SB II 192; F (1942).
VN SB I (51: 74) 139; Ж. Ф. X. XIV, 1599, 1602 (1940); Z. An. 344,
190 (1940); Acta XIV, 297, 1600, 1602(1941); F(1942); Ж. Ф. X.
XXI, 3(1947); Acta XXII, 319(1947); Z. An. 258, 58 (1949); 0(1950).
VN—VO Ж. Ф. X. XX, 459 (1946); Acta XXI, 764 (1946); 0(1950).
NH4VO, HRF (1938).
V—0 Z. An. 343, 63 (1939); Acta XIV, 297 (1941); Ж. Ф. X. XX,
459 (1946); Acta XXI, 764 (1946); О (1950).
Y,0, SB II 315, 360; SB IV (Z>8,: 22) 134, 135; HRF (1938); О (1950).
VO," SB I(6'4:158) 211; SB II 266, 315; J. Am. 69, 998 (1947);
F (1946); О (1950).
V,03 SB I (£5:242) 264; SB II 310, 315; HRF (1938); О (1950).
V304 SB II 315.
VO SB II 222, 315, 662; Acta XXI, 764 (1946); F (1942); О (1950).
4600. V20 SB II 315.
V,06xH20 SB II 360.
V206H20 SB II 360.
V—S Z. An. 241, 324 (1939); 342, 60 (1939); О (1950).
VXS„ SB IV 140.
VS4 SB VII 75; Z. An. 241, 324 (1939).
V2S3 SB VII 75; Z. An. 241, 324 (1939).
VS SB. VII (58:4) 75; Z. An. 241, 324 (1939); 242,60(1939).
(V0)2 (S04)„ • 16H..0 HRF (1938).
4609. V—4e Z. An" 242, 49 (1939); О (1950).
L352! ._ /jG1°- VScg —4(Ш. ВОЛЬФРАМОВАЯ ClfHI» 927
4610. VSe2 SB VI 59; SB VII 76.
VSe SB VI 59; SB VII (//8:4) 76; 0 (1950).
VV04 SB III (#03:91) 423; F (1946).
§ 352. Вольфрам W
\V SB I (42:16)61, 755; SB II 6, 163, 164, 191, 192, 239, 658- 660.
662, 674, 690, 777-780, 794; SB III 186, 187, 219, 220; SB V 36:
HRF (1938); F (1942); N. 154, 337, 338 (1944); 0 (1950).
Вольфраматы со структурой шеелита Z. tcchn. Phys. 24, 8(1943);
Ch. Z. I, 270 (1944).
■yy q gg I 575
WC SB I 144, 575; SB II 237, 239, 777- 780; SB III 623; О (1950)
W3C2 SB II 778.
W2C SB I 225, 575, 576; SB II 237, 239, 777- 780; О (1950).
W(CO), SB III 334; SB VI 88; О (1950).
4620. WC1, SB VI 71; Rec. 62, 197 (1943); О (1950).
(NH4)3 W2C19 SB III (K7t: 142) 505.
WF„ SB V 64; О (1950).
W—О С. А. 39, 5148 (1945); Ch. F. N. 25, 2970 (1947).
WO„ туигстенит SB I 219; SB II 32, 298. 299; HRF (1938);
БМКД (1938); О (1950).
W03xH20 SB II 298.
W,02a SB III 364; О (1950).
W40u SB III 364; F (1946); О (1950).
WO, SB I (C4 : 158) 212; F (1946); Ch. E. N. 2Й, 2970 (1947); О (19Г.О).
W„0M (OH)., SB III 366; F (1946).
4630. WtO10(OH)2 SB III 366.
H,W«Ot. HRF (1938).
H2W04 SB I 249; SB II 298.
H4W0s SB 1 249; SB II 298.
He[H2 (W?Oj0)<1 5H,0 SB III (#410: 115) 465.
(NH4),WO, nH„0 " HRF (1938).
(Nn^Po.iawd^H.o sb vn 149.
3(NH4)20. P2066W6,9H,0-2(MI4)3P04 • GW039II,0 HRF (19>).
H3[P(WS010)4]5H.0 SB III (#416: 113) 463. 465.
H3PW„O4014H2O " SB VII 148.
4650. 1LPW,,04024H,0 SB IV 213.
H3PW, ,04p29ILO SB IV (Я4.,:50) 183, 213.
H,PW1,O4(,30H,O SB III 463.
W—P Z. An. 248, 209 (1941).
W4P Z. An. 248, 209 (1941); О (1950).
W.P SB IV 121; О (1950).
WP Z. An. 248, 209 (1941).
WP. Z. An. 248, 209 (1941).
WS.. SB I (C7:166) 215; О (1950).
WSI, SB I (Cll:741) 219, 783; О (1950).
4650. W2Zn ДАН 40, 262 (1943):
ZnVV04 SB II 86, 450; О (1950).
W„Zr SB III (£39:34) 316; F (1942); О (1950).
4653. Вольфрамовая синь Z. An. 252, 160(1913); Ch. Z. II, 403 (1944).
928 4654._Xe-4695. Zn(MnQ4)2 • 6Н.0 |гл. XI
§ 353. Ксенон Хе
4654. Xc SB II 203; F (1942); Ж. Ф. X. XX, 1414 (1946); ДАН 62.
231 (1948); О (1950).
§ 364. Иттрий Т
Y SB II 171; SB VII (A3: 3)58; О (1950).
YF, SB I 186; SB VI 69; SB VII 81; О (1950).
Y (N03)8 • 6H20 HRF (1938).
Y20s SB 1261,333; SB II39,171,310, 314; HRF (1938); F (1942); О (1950).
Y(0fl)3 SB I 253; С. А. 41, 5357 (1947).
4660. YP04, ксенотим SB I (ЯЗ: 347) 396; SB III (Я0,: 91) 429; F (1946);
О (1950).
Y2S, SB II 323.
§ 355. Цинк Zn
Zn SB I (A3:19) 41, 750; SB II 164, 169, 218, 691-699, 706-712,
726, 730-732, 736, 749; SB III 184-187, 196-198; SB VII 50,
55, 206; HRF (1938); О (1950).
Zn (NHs)2Br„ SB IV (£12: 31) 144; О (1950).
(ZnBr2)I(ZnO)(/(H20)2 SB II 364.
ZnBr2Zn03H20 SB II 364.
ZnBr24Zn04H20 SB II 364.
Zn(H20), (BrO,)2 SB I (Я64: 437) 454; SB IV (Jll0: 63) 194; О (1950).
Zn(CN)2 HRF (1938); ДАН 31, 350 (1941); О (1950).
ZnCO, SB I (Gl: 295) 317, 326; SB II 391; О (1950).
4670. Zn03Zn (CH3COO)2 SB I (041 :677).
ZnC204xH,0 SB II 857.
Zn(CH3COO)2 HRF (1938).
Zn(CHaCOO)<, 3H20 HRF (1938).
Zn-бензоат HRF (1938).
Zn-лактат тригидрат HRF (1938).
Zn-фенолсульфонат HRF (1938).
Zn-салицилат HRF (1938).
Zn-валерат дигидрат HRF (1938).
ZnC204 • 2HoO HRF (1938).
4680. ZnCl2 SB I (C19: 743) 188, 1£9, 773; SB II 246; HRF (1938); О (1950).
Zn(NH3)2Cl2 SB III 388; SB IV (£12:31) 144; Б (1947); О (1950).
(NH4),ZnCI5 J. Am. 66, 1056 (1944); О (1950).
(ZnCl2)I(ZnO)„ (H,0)2 SB II 364.
ZnCl2ZnOH20 SB II 364.
ZnCl24Zn05H,0 SB II 364.
Zn(NH,UCl64)a SB III 475; F (1942); О (1950).
Zn (C102)s 2HaO SB II 378.
Zn(C104)26H20 SB III 452; HRF (1938).
4690.ZnCrO4 HRF (1938).
ZnF„ SB I (C4:158) 188; HRF (1938); F (1946); О (1950).
ZnF^H.O HRF (1938).
ZnJe SB VI 61; HRF (1938); Ж. Ф. X. XX, 1401 (1946); О (195Q.
Zn(NH,),J., SB III 475; F (1942); О (1950).
(ZnJ2U (Ln0)№(a20)z SB II 365.
4695.Zn(Mn04)2-6H20 HRF (1938).
§3561 4696. Zn,N_—4733. ZrO.H.O _____ 929
4(i96.Zn3Xs Z. An. 244, 125 (1940); F (1942); О (1950).
[Zn(No3)j-(ZnO)„(ii.,o), sb ii 387.
Zn(N03)24Zn04H,0 SB II 387.
Zn (N03)_ (ZnO) ЗН',0 SB II 387.
4700. ZnO, цинкит SB I (B4 : 79) 114, 119, 750; SB II 217, 218, 256, 472,
473, 479, 481, 483; SB III 244, 567; HRF (1938); БМКД (1939);
J. Am. 65, 872 (1943); О (1950).
Zn(OH), SB I 194, 253, 254; SB II 256-258, 364, 365, 387, 440;
SB "III (£31: 26) 303; SB VI 89; О (1950).
ZnP„ SB III 311; F (1946); О (1950).
Zn-P, SB 1 786; SB II 41, 325; SB III (Z>59:51) 353, 354;
F (1916); О (1950)
гп,фоефат HRF (1938).
Zn2P04(0H), та];буттит SB VI 102.
Zn3(P04).,4H„0, гопсит SB VI 103; HRF (1938).
ZnllPO^'/IbO HRF (1938).
Zn (II2PO.)2 • H20 HRF (1938).
ZnS, цннко1Ш[ обманка, сфалерит, шорцит SB I (53:77) 127, 772;
SB I (54:79) 128; SB II 217, 231, 232, 233; SB III 252; SB IV
98; SB VII 74, 75; HRF (1938); БМКЦ (1939); Z. An. 244, 146 (1940);
Z. ph. Ch. (A) 188. 109 (1941); F (1912); ИЛИ (х) 382 (1944); С. А. 39,
3988 (1945); Б. (1947); О (1950).
4710.ZnS04, цинкоаит SB III 420; БМКД (1939).
ZnS04(NH4)2S046H20 HRF (1938).
ZnS043Zn (0H)2 SB II 440.
ZnS04H20 HRF (1938).
ZnS047H20 SB 1 389; HRF (1938).
ZnS032£H20 HRF (1938).
ZnSt> SB I (/.3:77) 135; Z. An. 244, .46 (1'JiO); F (1942).
ZnTe SB I (53 : 77) 135.
Zn—Zr Z. Mot. 33, 355 (1944).
ZnZrF.eiLO SB II 104, 504; О (1950).
§ 356. Цирконий Zr
4720.Zr SB I (A3: 19)53, 752: SB II 181, 800: SB III 209; HRF (1938);
Ch. Ph. 9, 673 (1941); F (1942); О (1950).
ZrC SB I (51:74) 144; SB II 237; SB III (Bl:7) 262; F (1942);
О (1950).
ZrCl4 SB II 305.; F (1942); О (1950).
ZrOCt, HRF (1938).
ZrP4 SB III 327; О (1950).
(NI_4).ZtF7 SB I 453; SB VI 126; О (1950).
Zr—If SB II 800; Ch. ph. 9, 678 (1941); F (1942); О (1950)
Zr4H SB II 800.
Zrll SB II 800.
Zrll, SB II 800; F (1946).
4730.ZrN SB I (51:74) 139; F (1942); О (1950).
Zr (NO,)4 • 5H..0 HRF (1938).
г1-0..(!ядделевт SB I 196, 197, 777; SB II 265, 267-269, 334, 434, 592;
SBIV(C43 : 9) 119; SB VII 87; HRF (1938); F(1942); F(1946); 0(1950).
4733. ZrOJI.O SB II 265.
93j)= __4733;_Zr(ON)4—4775. MB" 0, |гл. XI
4733.Zr(OH)4 SB I 253.
Zr—P SB VI 192; 0 (1950).
ZrP SB VI 192.
ZrP2 SB VI 192.
ZrP20, SB III (K6, : 140) 503; Б (1947); F (1942); О (1950).
Zr—S Z • An. 242, 249, 2(37 (1939).
4740ZrS2 SB I (C6:163) 214; О (1950).
Zr(S04)2 SB II 434.
Цнрконнлсульфат I1RF (1938).
ZrSe, SB I (C6 : 163) 214; О (1950).
§ 357. Разные соединения. Общие формулы
Ме"А1,04 SB II 471 и след.
A,B(CN)e, (А =-■ щелочной металл; B = Cr, Mn, Fe, Ir) SB II 494.
МеС. SB I 782.
Me„C SB II 285.
(HCOO)„Me„ HO, (Me = Li, Ca, Sr, Ba, Pb; m - 0, 1, 2; n = 1, 2)
SB I 656-658.
Ме(СНХОСН.,СОСНэ)3, ацстилацетонсоединснин (Me —- Al, Co, Cr, Fe, Ga,
In, Mn, Sc) SB I 680-682.
47oO.MeCl, SB I 773.
MellCo.,04 SB II 478 и след.
Me"Gr,04 SB II 480 и след.
Me"Fe„04 SB II 476 и след.
MeIMe»PeJI1(S04)1.18H,0 (Me1» К, Rb, NH, Tl; Me»=Fe, Mn, Zn, Cd)
SB III 473.
R(NH3)Hal (JB = C„H2Jltl) SB II, 803, 812.
A2BR(NO.,)e, (А= щелочной металл, B = Pb, щелочной или
щёлочноземельный металл).
MUNI, Co, Cu SB II 495.
M«i(Nb, Ta)04 SB I 398.
Ме(ОН)2 SB II 775.
4760.Me»MeinS4 SB II 485 и след.
M»M»r(S04)2 SB I 789.
МеШш(804)26Н20 SB II 93, 436.
MiJftHi (S04)212H,0 SB I 390.
Me'Me" (Se04)> 6H20 SB II 93, 436.
MeOMesO,2Si02 SB II 554.
(Si04), MPUMJ1 SB I 411; SB II 520, 521.
Me^SnO/SB II 483 и след.
MnTi04 SB I 398: SB II 484 и след.
М4П(ТЮ4), SB I 398.
4770.AmB„Op SB II 598.
АВг.зХв (A = Ni, Co, Си; В-галоген C10„C104, S04 и т. д.; X-NH„
Н20) SB II 500-503.
АВХ3 (А = щелочной металл; В = Zn, Cd, Hg; Х = галоген) SB И 329.
M,QX, (М = Са, Се, Na, К, Y, Tb, U, Mn; Q = Kh, Fe, Ti, Та, Sb; X О,
ОН, F) SB II 58, 340-344.
MG.LR, (G и R —комплексные группы) SB I 461; SB II 504.
4775 MB1V08, полеиые шпаты (М—щелочные и щёлочноземельные мета кш;
B-Si, Al) SB VII 177-178.
Г.1Л I! Л -\II
ДОПОЛНЕНИЕ К РЕГИСТРУ ИССЛЕДОВАНИИ СТРУКТУР
НЕОРГАНИЧЕСКИX ВЕЩЕСТВ
Актиний Ас
-/i77G.Ae(0]N)3 А.Сг. 1, 266 (1948); О (1950).
Серебро Ag
Ag—As О (1950).
(См, Ag, Zn, Ft', PI>, Hg)3 (Sb, As, Bi) (S, 8b, As)4, блсилан руда
(«фальорц») БМКД (1939).
AsS3 (Си, Ag, i'V, Zn)3, теннантит БМКД (19.48).
478().(Cu, Ag)1(, • (Zn, Fc), (So, As)4 S13 F (19i2).
Ag—В О (1950).
Ag Br—AgCl F (1942).
AgBr—AgJ F (1942).
Ag (CH3 CSNH,)4 CI F (1946).
SbS3 (Cu, Ag, IV, Zn),,, тетраэдрит БМКД (1938.).
9PbCl*-8Cu (Oil). 3AgCI НО, болеит БМКД (1938); F (1946).
Ag.,ScCu.,Se, эккайрит БМКД (1938).
75% AgJ • 25% CuJ = 3AgJCuJ, майорсит—маршит БМКД (1938).
38% CnJ • 62% AgJ, майсрсит—маршит БМКД (1938).
4790.50% AgJ -50% CuJ г AgJ . CuJ, майсрсит—маршит БМКД (1938).
AgFO, F (1946).
Ag—H О (1950).
Ag—N О (1950).
Ag3i\ Z. An. 258, 77 (1950).
AgUO, (CH3COO)3 xILO F (1946).
Алюминий Al
(Cn, Na), Be (Al, Si)., (OF),, мышфанит F (1946).
AI4Ca F (1916).
n [Si03], Ca (Mr, i>)m (Al, F«')aO„ аигнт БМКД (1938).
(SiAl),0.,., (OH, F). (JVa, Ca, K, Mn),_3 (Mg, Fe, Ti, Mn, Al)., рогоная
обманки БМКД (1938).
4800.(A1, Fc)2 Ca3 (Si04)3, гессопит F (1942).
(Al, F»)s (Ca, Ft', Mg), (Si04)„ гранат БМКД (1939).
Na, К (Ca, Mg, Mn) Al4Si50,68H,0. ашкрофтпп F (1946).
CaNa4Alx< (P04)8 (OH)l8 6H.O, вардит F (1946).
50% Si,0,NaAl + 50% Si.O.CaAL., Лабрадор БМКД (1938).
Ca4AleSi,0o4 (S04, C03), мейонит F (1946).
4806.3CaOAl.,O3 3 (SiO„ CO,) • 2П.О, плазолит БМКД (1939).
.VJ*
932 _4807. А1.Сс_-4841. As(C„H5)3_ [гл. XII
4807.А1.Се F (1942).
(Сг, А1),04 (М«, Fe), хромпикотит БМКД (1938).
3 (Р04) А1 (Мк, Fe) [ОН]., лазулит БМКД (1938).
4810. Al, (Mg, Fe), (SiO.),, пирон F (1942).
(K„Mg, Fe) (Fe, Al)., Si4Ot„ • 211,0, глауконит БМКД (1939).
93,52 [П. (Mg, Fe)4 Al4SLOl8] + 6,48 [H8 (Mg, Fe)6Si4Ol8], дафнит
БМКД (1938).
Al.,Fe3 (Si()4);„ альмандин БМКД (1938); F (1942).
OH, (Al, Fe") „ [SI,..,, (Al, Fe"*),] 04. • mH.,0, нонтропит
БМКД (1939).
7 (i\i, Mg, Fe)0 • 2 (Al, Fe)303 • 5SiO> • 7Я,0*), шухардит
БМКД (1938).
Na3Li3Al,Fl2, криолитионит БМКД (1938); F (1942).
KAlSi30e, ортоклаз С (1939).
KAlSi3Oe, адуляр БМКД (1939).
Na4Al3Si„034Ci, мариалит F (1946).
4820.211,0 • 2Mg0 • A1,03 • Si02, амезит БМКД (1939).
x [211,0 3MgO 2SIO,] у [2НО • 2MgO • AI203SiO,], хлорит
ЬМКД (1939).
3 1211 О • 3Mg() • 2Si02] 7 [211,0 • 2MgO • A1203 • SiO,], прохлорит
СМКД (1939).
2[H.MgaAl.Si,0,. -3HOJ, гидробиотит БМКД (1939).
Al,Mg6Si30]0 [Oli]„ ьлинохлор БМКД (1939).
Mg,AI.8i,0,., пирон БМКД (1939).
Si64 • Al (H, К), серицит БМКД (1939).
AI..0, • MgO • 4SiO„ • H,0 + nH.,0, монтмориллонит С (1939).
Na4Al3Sia02«Cl F (19'i6).
9Mg0 • A1,03 • HSi02 • 15—161LO, сапонит БМКД (1939).
4830.«K»0„ «Si4» 1«04» «OH,»] «Mgs», флогопит - (»БМКД (1938).
Al—О О (1950).
A1,03 • 3SiO, xH.O, байдслит С (1939).
AI.,0, • 2SiO, • 4H20 = H4Al,Si20„ • 2H20, галлуазит, галлоизпт
БМКД (1938); С (1939); БМКД (1939).
А12Оч • SiO, • 5Н О, аллофан С (1939).
(Fe, CO, Al) ..О, • Н,0, стенисрит БМКД (1939).
Общие формулы
2К20 • ЗМО SK..O, • 24SiO, . 12Н»0, иллит (М = Fe, Mg, Ca; R = Al, Fe)
БМКД (1939).
* Пермутит С (1939).
Гумат алюминия (гумалит) С (1939).
Аллититы (окиси и гидроокиси AI) С (1939).
Мышьяк As
4840.BiAsO4 F (1946); А. Сг. 1. 163 (1948).
4841.As (C0H5)a 0 (1950).
*) Формула не отпечаст Дана.
4842. AsJ(C0lL)4-4SS4.T!i— В 933
4842. AsJ (C.H,), F (1916).
(CoAs,, смальтин БМКД (1938); БМКД (1939).
(Си, Fe),. As4S,„ биинит F (1942).
(Си, IV, Sn, Mo, Zn), (S, As, Fe, 8b)., „ колуеит, БМКД (1938).
(Cu, Fe, Mo, Sn)4 (S, As, Fe)3 „ кусит F (1942).
(Kb, As),S3 • 3 (CuHg) S (?) (<• 11k от 6 до 17%), шиатцит
БМКД (1938).
Y (Ni, Fe) AsS, нлсесит, горсдорфит БМКД (1939); F (1952).
Asia F (1942).
4850.Mn2As F (194(>).
NHJLAsO, F (1940).
AsNd F (1942); О (1950).
>'i (As, Sb), арит О (1950).
Xi (As, Sb) S, корииит F (1942).
As—Pb О (1950).
1'rAs F (1942).
Re—As О (1950).
As—Si О (1950).
Au—В О (1950).
48G0.Au—N О (1950).
Золото An
Вор It
Ca2BeOn • 13H.O, инвоит БМКД (1938).
CaMgBeO„ • 6HUO, гидроборацит БМКД (1938).
CaNaB60, • 8Н.6, улексит (n БМКД опечатка «улевсит»); БМКД (1938).
Cd (NH3), (BF4):! F (1942).
CrB О (1950).
Cr3B. О (1950).
Си—В О (1950).
Fe (NH3)8 (BF,h F (1942).
GdB, F (1942).
4870. Ш—В О (1950).
Ш—В. О (1950).
В—Ir О (1950).
КВ02 О (1950).
Мк (NH,), (BF4)S F (1942).
2MgO • 3B,03 • 15II..O, индерит БМКД (1938).
2MgO • В. О, • НО, камселлит БМКД (1939).
2МкО • В.,Оп • НО, ашарит БМКД (1939).
2 (Мп, Мк) 0В,0;Н О, магнеаиоеуссекеит БМКД (1939).
2 (Mn, Mg, Zn) О В.0,11.0, сусеексит БМКД (1939).
4680.Мп (NII3j6 (BF,) К (1942).
Pd—В О (1950).
Ft—В О (1950).
Rh—В О (1950).
4884.Th—В О (I960).
931 j4885, Ti—В—4921. W.CajMg, Ft»), [г.-ь XH
4885. Ti—В О (1930).
TiB Z. An. 259. 1 (1949).
TiB, Z. An. 259. 1 (1949).
B,0, • tUWO, 66H 0 F (1946).
Zr—В О (1950).
Барни Ba
4890.Ва (('II., (JOO),*) F (1916).
BaFtl F (1946).
BaFJ F (1946).
BaFcSi4Oin, джилленеит F (1916).
KBaP04 F (1916).
Воридлин Во
BcCu F (1942).
Be—(W, Mo) F (1916).
Висмут Bi
Bi (C.H,). ° (1950).
Bi.O,CdJ,,CdO = Cd..BL04J. F (1946).
BiCc F (1942): О (1950).
1900.Tl.NaBi (NOa). F (1912).
Ni (Sb, Bi) S, каллилит F (1912).
Bi,03—РЬО F (1916).
Углерод С
С (графит) (СЮ,)„ О (1950).
€ (графит) (Se04)n О (1950).
(СН3СНО)4 F (1946).
С2 (СН,)4 Br, F (1916).
С4 (СН,)2 Вр4 F (1946).
С (С.Н.), [П„] О (1950).
(СН,СО), NJ F (1916).
i910.CDH,NH,J F (1946).
C—N О (1950).
О (СИ,). О (1950).
С (СН,ОС,Н,)« F (1916).
С (СООСП,)« F (1916).
CSo, О (1950).
Кальции Са
€а (N11,), Br. F (1912).
(Zr, La, Di) F (CO,) CaC03, синхизит БМКД (1939)
(Ca, О, Na, К), (Nb, Fc) О, (О, ОН, F), коппит БМКД (1939).
Ca (JC14), • 811 6 О (1950).
4920.K,CaCo (NO,), F (1942).
1921.H,Ca., (Mg, Fc), (Si0.,)8 = [Si40J, Ca, (Mg, Fe)5 (ОН),, актиполит,
" нефрит БМКД (1938).
•) В работе F (1046): <.Ba (СН2СОО)у>.
4922. (Са. Na, Fc^baO0(OH)-4962. jCo._Nj)gbS_
4922.(Са, Na, Fe)2 Sb.O, (ОН), шнеебергиг БМКД (1939).
(Са, Fe, Na), (Sb, Ti)., (ООН),, лыоиеит*) БМКД (1939).
Са (NH,),J3 F (1942).
К. (Са, Mg) (S03)3**) F (1942).
KCa4Si802„F8H,0 F (1946).
(Са, Mn, Na). Sb (OHF)., ромоит, атоиит БМКД (1939).
CaO • Si02 • TiO,—MnO • TiO.. SB IV 209.
Ca—N О (1950).
4930.(Pb, Ca)a SIM), • SILO, биндгонмит БМКД (1939).
CaSrO, О (1950).
91CaF, • 9ThF4 F (1942).
66CaF,, • 33YF3 F (1942).
Ca2ZnSbO„ гардистанит F (1946).
Кадмии Cd
Cd (NH3),«r2 F (1942).
[Cd (NH3),] S03F F (1942).
Cd [Hg (CNS)4] F (1946).
Cd—К SB VI 157.
CdSnO, О (1950).
Церий Сс
4940.95ШО, • 5CeO., F (1942).
(Ce, La...) FCO„ бастнезит БМКД (1939).
Се (0И)3 О (1950).
Сс—Si Л.Сг. 2, 94 (1949).
CeSi, Л.Сг. 2. 94 (1949).
957гО. • 5СеО, F (1942).
Кобальт Со
1-Со (x\H2CH,CH.,NH.,)3 BrsII20 F (194G).
Co(NH,CH3)«J., F (1942); О (1950).
Co(NH3)4(H,0),Co(CN)e О (1950).
[Co(NH3)5H20](C104)3 F (1942); О (1950).
4950.(Си, Ni, Co, Fe)(S, Щ, F (1942).
3CuSe . 2PbS<\, • 5(Ni, Co)Se,(?), пенрозеит БМКД (1938).
Co(NH,).(SO,F)2 F (1942).
(Fe, Ni, Co)3 P F (1946).
53,8% NiS; 39,1",, FeS,; 7,1% CoS„ бравоит F (1942).
Oo—H О (1950)
Co|Hg(CNS)4] F (1946).
KCoF, F (1942).
MgCo,04 F (1942).
(Mn, Co)(Co, Mn),04(Co—Мп-шнинель) F (194.°).
4960.CO—X***) F(19i2); 0(1950).
(Ni. Co) (Co, Ni)., 04 F (1942)
4962.(Co, \i)SbS, 1шлы1мит F (1942).
*) В работе БМКД (1939) приведена формула Лыоисита, тнлыю отличи
от указанной Дана (1. f. ctj>. 313).
**) Формула неверна (/>. О.).
***) Раствор N л Си.
936 4963. СоТа.О, 5000. Fe(M>, ТаЦ)(| __ [гл. XII
4963.CoTa2Oe F (1946).
Tl,[Co(NO..),] F (1942); О (1950).
(Zn, Со)Со<)4 F (1942).
Сг—Н О (1950).
Н«,Сг04 HRF (1938).
Сгб. F (1946).
Хром Сг
Цезии Cs
CsCI6 О (1950).
4970.CsFSO3 F (1946).
Csl> О (1950).
Cs3Ir(N0,), F (1942); О (1950).
Cs.,0., О (1950).
Cs(OH) О (1950).
€s,PuCl, О (1950).
Медь Си
Си, апонрснат С (1939).
СиС1, ЗСи(ОН),, атакомит БМКД (1938).
SCuCliCnS04 • 1вСи (ОН), ЗН,0, коннеллит БЫКД (10W).
CnCl(S-:=C<^)4 F(1946).
4980.CUF./ F (1942)'; 6 (1950).
Cu3(Fe, Ce)S4, германит SB II 348; БМКД (1938); БМКД (1939):
Б (1947).
Си—Н О (1950).
CuN3 Л.О 1,11 Г) (1948).
СиС1о2РЬ(ОН)>, диаболеит F (1946).
(Pb,Cu) |ОН]., (РЬ, Си) SO,, линарит БМКД (1938).
5PbS • 9Cu,S • 7Sb.S3, бентонит БМКД (1938).
3Cu,S • Sb.S„ фаматинит БМКД (1938).
CuSo • Cu.,Se, умангит БМКД (1938).
Си-Si О (1950).
4990.CuSi03 • ЗН.О, хризоколла БМКД (1938).
СиДе„ риккардит БМКД (1938).
('«(ШУ, (РО,), 8JL0, торбернит F (1946).
Железо Fc
Fe(C0,) «г., О (1950).
Го, графитид О (1950).
IV"RFc" (СХ), (R - Na, К, Kb, NH4) F (1942).
Ft—II О (1950).
03f,LFe.O4; 3<1ЛаТЮ, F (1942).
Fe.04Mj>F< ,0.(Mn,Fe), магноякобсит БМКД (1939).
MnCO,; (Mn,Fe)C03, родохрозит С (1939).
5000.1>(ХЬ, Та)., 0„ носеит *) F (1946).
*) Но Дана мосопт имеет состав FeNbX)c, т. е. япляется чистым колумбитом.
Припедённая п работе F< IO-'iG) формула отвечает промежуточному члену* и ряду
колумбит (ГеМь' >6) — чаптолит (FeTa/)e).
5001. FeTa,0„ 5033. JF5
937
5001.FcTa.,OB, танталит, тапполит F (194(5).
Fe[(Nb, Та) О.]. • 6TiOo, ильменорутил БМКД (1938).
(57"„ Ре; 40,8",, Ni; 0,5%Р), тенит F (1942).
(Fc, Ш), тэнит, камасит БМКД (1938).
(Fe, Ni)S,(6,5'0 N1) F (1942).
(NH4),FeCl4 ■ 2ПЛ) F(1946).
Fc(NH,),(SO,F), F (1912).
Феррилиты (окислы и гидроокиси железа) С (1939).
Fc,03 • Н,0, ленидокрокит, рубиновая елюдка С (1919).
5010.Fe,O3 • НО • nH,0, гпдрогётит БМКД (1938).
Fc.,63-nHoO, гидрогематит БМКД (1938).
ГеРа F (1946).
РЬО • 2Fe20„ илюмбоферрит БМКД (1939).
FcS04.3H.,6 HRF (1938).
Fe.Sis N. 152, 413 (1943); О (19Г>0.
Fc°Si A. (Jr. 1, 212 (1948).
Fc, (Tc03)3 • хН.О, марколит F (1946).
riFe„04 F (1942).
Калий Ga
LaGa03 F (1942); К (19.10).
5020.Ga.ZnO, К (1942).
Германий Ge
Ge—X С) (19ГЮ).
Ge((JS(CN3)3]4 F (1946).
Ртуть Ни
ЩХ[.0, клсйнит *) БМКД (1939).
Ug,C10, терлингуаит БМКД (1939).
Hr.CI О, эглестонит БМКД (1939).
HgaFa F (194(5).
Водный хлорид (и сульфат) Hg и ]VH3, мозезит (Гииаок m> счета ну
к клейииту) ' БМКД (1939).
Индий In
j„_P F (1942).
Pbln2_3 F (19'i6).
Иридий Ir
503().Jr(CSN..H4)4(JUH-211,0 C.A. 35, 7786 (1941).
Jr_l» о (1950).
Ir,P О (1950).
Иод J
5033. JF6 О (1.950).
*) По Г.МКД (Ш9) и Дана состав клони и та Ug4CI203 нлп ШХЬО.
lli^'LO,, см. регистр.
938 5034. КО,—5070.^ 1Ш,С«Ц,С1 l£n_i2^1
Калий К
Г>034.КО„ озонид калия О (1950).
KPb.Br, F (1946).
K,PbNi(NO,)e F (1942).
K,SrP04 F (1946).
Лантан La
La.MoO, F (1916).
NaLaO. О (1950).
r>040.La..O,S О (1950).
66SrF2 • 33LaF3 F (1942).
LiMo04 О (1950).
LiSH О (1950).
LiSeTI О (1950).
Литий Li
Магний Ms
M-Co О (1950).
2SI0, • 3(NI, M$?)0 • H.,0, пепуит БМКД (1938).
MgTa.O, F (1946).
3ZrO, • MgO F (1942).
4MgO • 3SiO • 611.0, девсйлит БМКД (1938).
Марганец Мп
5050. Mn-C О (1950).
Mn(NH3)6(S03F)2 F (1942).
Mn—H О (1950).
Mn—N О (1950).
JriMn-SN F (1946).
62MU-3SX F (1946).
Mn,P О (1950).
Mn3P F (19'i6); О (1950).
<>6Mn 4Pd F (1916).
Mn,Zn04 F (1916).
."ОбО.оМпО • SSiO. • НО или 2Mn,Si04 • Mn(0II,F)3, аллеганит БМКД
(1939).
Si-0 H10Mn, бементпт БМКД (1938).
Mo—N О (1950).
7«Mo • 30N F( 1946).
С.Н.-.ХП.Вг F (1946).
СЛ1и-ЧН,Вг F (1946).
C4II,NIIaBr F (4946).
C.H7NH,Br F (1916).
(XII4),SeBre F (1942).
NH,C,H,C1 F (1946).
:.O70.Nli3C4N„Cl F (1946).
Молибден Mo
Азот N
5071. NH,C«Ht,ei—5107. Pb Tl2
5071.XH1C,II„C1
.\H,C7II14Cl
X(CH,)t [JJ,
.\(CH,)4JCI.
<',H7NII,J
С4НоЛНзЛ
l5HuNH3J
CeHi3^H3J
G,II1SNH,J
5080.CSHU-NH3J
l10H,>!M
CUH, NIF3J
C.J.NIIjJ
F (19-iil).
F (1946).
,] 0 (1950).
F (19i6).
F (191(3).
F (1946).
F (1946).
F (1916).
F (1946).
F (1916).
F (1946).
F (1956).
F (1946).
Натрии Na
Na (e03 F (1912).
NII4Na, [Bh(NO.),l О (1950).
Xa-UF- О (19>)0).
\u(,,,_(,,)W03 F (1946).
Ниобий Nb
M,_Si О (1950).
Никель Ni
5090.Ni.C О (1950).
Ni (C1JJ • 8H..0 О (1950).
M(CH,)4 [J,] О (1950).
\1(М1Л rJ„l О (1И50).
\i_N F (1946): О (1950).
NiN.,S4*) F (194'i).
Ni3Sb4 F (1946).
Ni,Sb F (1946).
Ni(NH3)0 (S03F), F (1942).
NiTa-O, F (1946).
Фосфор P
l»_X о (1950).
Свинец Pb
510O.Pb—N О (1950).
Pb(N,), О (1950).
Pb—p" О (1950).
RbPb.Br, F (1940).
(Th, V)0> • nUO, • PbO, торианит БМКД (1938).
(V, Th)0., • (О 0,5) U03 • mPbO, уранинит БМКД (1938).
(V, Tli) 0, (0,5-3) 1ГО, • mPbO, настуран БМКД (1938).
.")107.Pb-TI.. SB I 571.
*) В работе V (19-V2) рефергропаиы данные о веществе NiN2S4, что яв
несомненной ошибкой, (.м. примечание к соединению NOUUO(Ni,S4 = NiNi,S4).
940 Г>108. ВЬ.РДП,—5130. Set), [гл. XII
Палладий Р<1
5i08.Rb.,PdCl, О (1950).
Платина Pt
(С,Н4 [1,2J О СН = N0H), Pt F (1946).
5110.Pt—0 0(1950).
PtO F(1946)
Рубидий Rb
RbC„ 0(1950).
RbC„ 0(1950).
RbZrO, 0(1950).
RbaTiCl, F(1942); 0(1950).
Rb3Se 0(1950).
Rb2Te 0(1950).
Rb202 0(1950);
RbS„ F(1942).
Рений Re
5120.Rc—С 0(1950).
Re_Si 0(1950).
Родий Rh
RhVO, F(1946).
Rh—P Z. An. 244, 317 (1940); 0(1950).
RlbP 0(1950).
Редкие земли
Самарин Sm
S:nC2 F(1946).
Эрбий Ег
(¥,Er)P04 2H.0, вайпшенкит 0(1950).
Сурьма Sb
Sb(CcIIs)3 0(1950).
Скандий Sc
Sc—H 0(1950).
Селен Se
(NH4)2ScCl. F(1942); 0(1950).
МЗО.ВеО, 0(1950).
5131. f(CH3)aSiO]a—5160. TICN 941
Кремний Si
5131.[(CH3),KiO]9 F(1946).
Si[SC(CH,)1)4 F(1946).
(CH3)2 CHSSi ISC (CII3)3]3 F (1946).
Si—N 0(1950).
Si02 опал (гель) С (1939).
SiO,, гель С (1939).
ThSi, F(1946); Л. О. 2, 94 (1949). •
Si—U Л. Сг. 2, 94 (1949).
Ш, А. Сг. 2, 94 (1949).
5140.USI Л. Сг. 2. 91 (1949).
SIU, Л. Сг. 1. 205 (1948).
WSi, F (194в).
Олово Sn
SnF4 0(1950).
Sn—P 0(1950).
Sn (С#114СН,)4 0(1950).
Sn(C,H40CII,). 0(1950).
Sn(C.H4OCH04 0(1950).
[S(CH3M)JI5bSnCls F(1942).
Стронций Sr
SrFCI F(1946).
Тантал Та
5150.Ta—С 0(1950).
М8ТаР8 (М—щелочной металл) 0(1950).
Та—О 0(1950).
Та—Р Z. Ли. 24Г). 35 (1941);0 (1950).
Та—Si 0(1950).
ТаС—TiC 0(1950).
Технеций Тс
Тс Л. Сг. I, 161 (1948); О (1950).
Теллур Тс
ТсН2 0(1950).
Те03 0(1950).
Ti—Ч 0(1950).
5160.TICN Г (1942)
Титан Ti
Таллии Т1
Ураниды (Np, Pu, Am, C/n)
Np—Si Л. Cr. 2, 94 (1949).
Pa—Si Л. Cr. 2, 94 (1949).
942 5161. V—С-5189. Beyerite
Ванадий V
5161.V—С F(1942); 0(1950).
V—Н 0(1950).
V—N F(1942); 0(1950).
V—Si 0(1950).
VsSi 0(1950).
W-N 0(1950).
YP04-2H30 0(1950).
5170.YO4 О (1950).
Zn(NII3)eBr2 F(1942).
Вольфрам AV
Иттрий Y
Цинк Zn
Цирконий Zr
Zr—0 0(1950).
ZrOS A. Gr. 1, 287 (1948).
Полисоединения О (1950).
Полиметаллические комплексы 0(1950).
Полна рсениды О (1950).
Сульфоарсениды О (1950).
Полибориды 0(1950).
Поликарбиды О (1950).
5180.Ацетилсниды О (1950).
Графитиды 0(1950).
Циапиды 0(1950).
Цианамиды О (1950).
Азиды 0(1950).
Высшие окислы и перекиси металлов (полиоксиды) 0(1950).
Полифосфиды О (1950).
Нолисульфиды О (1950).
Полисслениды О (1950).
5189. Beyerite*) F(1946).
*) Неопределённого состава (Б. О.).
ИМЕННОЙ УКАЗАТЕЛЬ
Аблецова Т. A. 316, 673
Авогадро 149, 1(Ю
Агеев Н. В. 22, 207, 229, .417, 324
Агеева Д. А. 317, 324
Апельбаум Л. Е. 543
Архаров В. И. 481, 630
Аюи 149, 150, 151
Байлар 228
Барджесс 228
Варне 441, 447
Бедреаг 228
Веккер 295
Белов Н. В. 20—22,
228, 229, 496, 538,
706, 814
Борбодж 228
Верна л 441, 442, 443,
Бертло 150
Берцеллус 148—150
Бильц 548
Блаболил 201, 229
Бозорт 287
Еокий Г. В. 21
228, 229, 613
.2, 37, 51, 71, 75, 79,
Болдырев А.
814
Вор 187
Ворвелл 347
Борн М. 159,
Брава 21, 101
22
706, 73(3
21, 22, 42
. 46, 47, 811
161
811
160,
103
168, 228
праьц б± , j.ui , ли.j
Бранденбергер 0. 18, 286
Брегг 20, 21, 44, 125, 127, ,
181, 212, 215
Брегер А. X. 166, 210, 220, 228,
461, 543, 514, 546, 571, 581
Бредли 300, 325
Бриджмен 273
Врилль 229
60, 173,
229,
174,
415,
.00
148,
496,
149,
639,
152
риллюэн 181, 210—218, 229
Бродский А. И. 22
Ди-Бройль 181.
Б[)0квей 352, 507
Бутлеров А. х\1. 21,
Бутузов В. И. 229, 496, 639, 690
Бюргере 287
Вайль 354
Вайнштейн Б. К. 643
Вайсенберг 324, 343
Ван-Аркель 294, 295
Ван-дер-Ваальс 158
Влнт-Гофф 149, 191
Васильев К. В. 331, 334
Васильева В. ]"[. 477
Вегард 117, 438
Вернадский В. И. 22
Вернер А. 195, 226, 229,
Веселовский В. С. 331,
Вестгрен 300
Вигнер 229
Вильсон 297
Воллен 447
Вульф Г. В. 21
55, 669, 803
125, 181, 212, 215
23, 2'i, 25, 33, 44, 46, 103
135,
600,
447
161,
613,
173, 222,
639, 690,
I
Г
Г
г
р
J
г
21, 44, 51
159, 22S
340
229
'абер 160
'адолин А. В.
'арретт 587
аррис 273
'еимбрехт 548
ейтлер 188
еидрикс 3)48
'ешюрт-Мейер
ерасимов А. Ф
ерман 38, 116,
пббе В. 25
ингрич 273
оард 324, 690
огоршивилп Л.
оловлёва 3. С,
'ологап 331
'пльдшмидт 12,
176, 801
оралевач Д. К.
римм 229, 467
ринберг А. А.
рот 59, 786, 804
унд 190, 202
урыпюиа Б. II. 346
Чсева Л. \\. 229
В. 201,
477
229
150,
201,
229
163-
229
166, 173, 1"
53,:
Дальтон 158, 223, .-
Дана 814
Делоне Б. 11. 21
Джемс 229
Джермер 181
Джотто 294, 295, 307, 3J4 и след.
Джибсон 331
Джонс 210, 229, 359, 360, 363
Джонсон 13, 199
Доливо-Добровольскип В. В. 31, yi4,
Дубинина В. II. 814
Дэвидсон 447
Дэви ее он 181
Дюма 149
Днткипа М. Б. 13, IKK, 191, 345, ЗЬ2.
422
94i
ИМЕННОЙ УКАЗАТЕЛЬ
Eiiiuir A. 272, 278, 295, 297, 307, 308,
317, 324, 325, 333, 334, 340, 341
Ждшов Г. С. 22, 135, 207, 210, 222, 229,
348, 415, 544, 571, 600, 603, 604, 614,
619, 651— 653, 673, 685, 689, 690, 694,
712, 731, 7И8
Жмудский А. 3. 307, 32i
Заварицкий А. П. 21
Захариаеен 164, 165, 380, 4Г>3, 455, 587,
588, 657. 722, 723
Звон копа 3. В. 673, 768
Звягинцев О. К. 201, 229
Зейц 19, 199, 210, 218, 228, 229, 359,
363
Зиборг 229
Зигбан 127
Золотаревскнй А. И. 689
Ибелл 294
Иванчоиа Е. Г. 460
Пвею 196, 228
Пнгергол 312, ЗИ
Иоффе А. Ф. 22, 218
Измаил заде И. Г. 651, 652, 053
Ист 228
Казарновекан Л. П. 685
Кааарновский И. А. 22, 346, 369, 673
Канпицаро 149
Капустиными А. Ф. 22, 170, 228
Карпш В. А. 22
Касаточкин В. 11. 331, 543, 673
J lOiiVJie 152
Кимбол 229, 803
Китайгородский А. И. 22, 207, 229
Клемм 277, 467
Клерк 690
Ковалёв Г. А. 8 К»
Ковальский А. К. 58J
Кондратьев В. П. 21, 183, 203, 228,
229
Конобеевский С. Т. 22, 210, 218, 228, 229,
687
lion л ей 228
Коли 149
Косолапов Г. Ф. 277, 320, 334, 608,
610
Коссель 156, 157, 195, 198
Котов В. П. 673
Кочешков К. А. 652
Крепелка 201, 229
Кричевский И. Р. 554
Крокстон 587
Кузнецов В. Г. 477
Кузнецов В. Д. 22, 159, 228
Кулон 156, 157
Кульгина В. И. 229
Курнаков Н. С. 19, 22, 223
Кюри П. 25, 306, 309
Лавес 228, 689
Ландау Л. Д. 22
Ландсберг Г. С. 22
Лаплас 187
Лауэ М. 25, 324
Леклерк 313, 314
Леммлейн Г. Г. 90 S)
Ленин В. И. 17, 181
Липкес А. Б. 579
Лирман Г. В. 571
Листер 196
Литц 587
Ломоносов М. В. 22, 148, 2J9
Лондон 188 и след.
Лорд 273
Льюис 26, 156, 195, 198
Любимцев А. И. 814
Лнмнна А. 11. 814
Магнус 12, 163, 171, 173, 370
Маделунг 159, 160, 1)85
Манер" 168
Майзель 544
Майнгрд 18
Макаров Е. С. 223
Максвелл 348
Мандельштам Л. П. 22
Маршак Ф. 307
Маурер 407
Меерсон Г. А. 579, 581
Меллор 229
Менделеев Д. И. 11, 17, 20, 14'j
Мика 277
Минервпна 3. В. 604
Митчерлих 150, 151
Михеев В. И. 814
Мицеловский 3. С. 229
Мишель 313, 314
Моген 38, 116
Моравец 167, 621
Морозов И. С. 340
Мотт 359, 360, 363
Мулликен 190
Нейбургер 277, 287, 295, 33:,
Некрасов Б. В. 229
Нельсон 330
Нштли 460
Никольский 1\ II. 346, 673
Новикова В. А. 415, 436
Новотный 552
Ньютон 150
Ормонт Б. Ф. 125, 126, 164, 165, 190, 191,
195—202, 210, 219 и след. 297, 306, 208,
313, "331, 378, 384, 41)2, 465,535, 543,
546, 604, 664, 667, 689
Оуэн 294, 307, 310, 317, 320, 334
Павлова М. В. 543
Панфилов А. В. 460
Паттерсон 207, 229
Паулинг 12, 13, 19, 164—167, 172—174,
182, 189 и след., 194, 196, 228, 229, 385,
441, 443, 445, 507, 655, 750
Пенни 229
Перье 229
Петере 229, 354
Петров Б. А. 201, 229, 331, 543
Иинес Б. Я. 229
Иинскер 3. Г. 22, 171, 222, 228, 415, 436,
643, 647
Попова Л. А. 229, 706, 736
Поспелов В. А. 731
Проскурнин М. А. 369
Иру 158, 223, 535
ИМЕННОЙ УКАЗАТЕЛЬ
945
Ребиндер П. А. 22
Рейхштейн С. И. 073
Релей 330, 331
Ринн 399, 694, 810, 814
Роберте 307
Рогинский С. ГЛ. 22
Роде Е. Я. 477
Роллг.е 348
Pvre.ua mi 339
Рандалл (1':>1гд.'1) 20, 293-- 21)7, 54К, 5К7,
054, 055
(.VimcTi.Himii N. Г. 22!), 00N, 010, 085, 090,
712
Согре 22»
Ссдленкпй И. Д. S1 !, КГ»
Семелчеики П. К. 2*2
Снджлнк 12, 173
Счете]) IK9
Смирнова .-I. А. 4 I Г»
Смите, 20'i
Спид IS
Стена ион Д. 15. .'107
Стилвелл 20,. 340
Сишер 203
Страуманне 27<S и след.
Стярдлналт 054.
Стюарт 190, 228
Сыркип if. 1С 13, 22, IKK, li)4, 345, 385,
422
Тамм И. К. 22
Таммап 2154
Та рта коне кий II. С. 1NI
Татарпнова «I. И. 415, 430
Теренин Л. Н. 21
Толанекий 90
Томеон 181
Трапезников Л. К. 227, 320, 334
Тримбл 273
Улнх 229
Умайский М. М. 229
Уманскпй Я. С. 22, 280, 384, 385, 545, 579,
581, 583, 1504
Уоррен 292, 293
Уразов Г. Г. 22
У э ланд 13
Уэллс 19(5, 22cS, 571, 02.S, 735
Фарадей 150
Фёдоров Е. С. 21, 27, 59, 101, lit), 101, 17:5,
234, 800
Фёдоров 045
Фернелиус 228
Ферсман Л. К. 22, 170, 814
Финдлей 348
Фирс 229
Флек 203, 229
Флинт Е. Е. 37, 51,71, 75, 79, НИ, 228, 8(4
Фок В. Л. 22, 21 1
Фостер 228
Фоулер 441, 442, 443, 447
Фрагмен 300
Френел 3)99, 094, «10, 814
Френкели Я. 11. 22, 25, 220, 228, 303
Фрмдель 2S, 120
Фрост Д. Pi. 3,3,9
<1>\к<- 359
Ф\'р|.е 12Г|, 201)
Ф\и' 2!)i, 2!15, 3,07, 3,17, 320, 333
Xa:»anoiu II. К. 5.Vi
Хапауолт 312, 314, 399, 094, ,'ЛО, JJK
Харкер 207, 229
Хартрп 21 I
Хачиапкни М. A. 402
Хпдекель С. (I. 579, 583,
Хил лену ид 43N
Хода Knit К). Н. 170, 170, 22«, 3,3,9
Хпгг 3N'i, 415
Цене]) 27.')
Цилт.м, 11)7, 021
Цумбуш 548
Шарвпп К). И. 3,48
Шафран И. Г. (104, 1)89
Шелл 447
Шепфлие. 3,7, 3,<S, 1 Hi
Шерман 229
Шибо.м.Д 331
Шнайдер 229
Шойчет Д. 317
Шольдер 201, 229
IN нолье к пи 0, И. 228
Шубников Л. И. 21, 37, 51, 71, 75, 7*3, 90,
ПН, МП, 228
Шубнпкопа О. M. 814
Шуга.\[ И. А. (Я 9, 094
Эване 228, 088
Эйлер Л. 21
Энгельс Ф. IS
Эпельбаум В. А. 100, 220, 228, 54Г., 581,
004
Орлих 401, 402, 407
!)ш 229
10м-Розерп 180, 215
Якоб 292, 293
Яте 310, 317, 3)3)4
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Лкпшность оптическая 152 156
Аллотропии 2 19
Анализ фпзнко-химический 22
Атома волномеханическая модель 18(1
и след.
Базис 103
Ьертолиды 223—224
Валентность максимальная элементов и
количество холостых электронов 200—
1^02
Иалентные \ глы 124
Зоктор идентичности 92
трансляционный 92
Неса ионной и ковалентной связи 192
Вещества физически чистые 219
Вид симметрии 41
Видов симметрии проекции 80--82
Волновая механика \i\'.\
Волны материи 181
Геликогнры 108
Гемиадрии 11 рода 55
— - гемиморфная 55
— иаклонногранная 54
— параллелогранная 54
- иараморфная 52
— плагиэдрическая 55
— трапецоэдрическая оЗ
— энантиоморфная 54
Гидридов летучих условия неустойчивости
378—379
Гпра 28, 29
Гироида 28
Грани индексы 94
— параметры 84
— символ 84
— форма 90
Грань единичная 84
Графики зависимости межатомных
расстояний и объёмов от периода
идентичности ячеек: Сс\ Ск\ Ну 265—272
Группа точечная 41
— трансляционная двумерная 93
трёхмерная 94
Группы пространственные 116—121
Дальтониды 223, 224
— и бертоллиды и агрегатное состояние
вещества 223, 224
Диаграммы р—Т 26
Дисконтинуум 23
Единицы кХ 125 и след.
Закон Авогадро 149
— анизотропии 23, 24, 27
«Закон Аюи» 149, 151
Закон Браве 101, 103
— Вегарда 177
— Вульфа 24, 27, 103
— Грота (неправильность) 804
— Дальтона 158, 223
-- кристаллохимии- обобщённый 151
«Закон Митчерлиха» 150, 151
Закон периодический (Д. И. Менделеева)
22, 149
— постоянства состава 19
углов 27, 90
-- Пру 158, 223
— рациональных отношений 82
— симметрии и структура кристаллов Мб
— Фриделя 28, 126
— химических эквивалентов 19
Зонная теория кристалла 210 и след.
Зоны Бриллюэна 210 и след.
Изомерия 150
Изоморфизм 150, 151
Изотипия 151, 152
К-иространство 210 и след.
Классы кристаллов 46
— симметрии лауевские 126
Комионента скольжения 105
Континуум 23
Конфигураций обозначения по
координационному принципу 129
Координаты линий 112—115
— плоскостей 112—115
Координационные сферы 124—125
Координационных сфер конфигурации
124—125, 233—241, 803
стехиометрический состав 143
—- — структурный состав 612—616
(комплексных) условия устойчивости
226—227
формы 806
Коэффициент переходный от единиц кХ
к единицам А. 127
Кристалл 23
— реальный и химическая
термодинамика 219
Кристалла реального дефекты и их
выявление с помощью высоковакуумной
методики определения удельных весов 462
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
947
Кристаллический пучок 44
Кристаллы левовращающие 152
— правовращающие 152
— реальные 218—224
Магнитные моменты атомов и ионов 202
Между уз лия октаэдр ические и тетраэдр н-
ческие 161—162
Метод координационных полиэдров 172—
173
— проб и ошибок 126
— стереографической проекции 21
Метрика кристалла и температурная
зависимость соотношения осей (или
величин углов) 235
Монокристалл 25
Монокристаллы реальные 25
Морфотрония 151
М ул ьтпнл ет и ост ь 185
Направления симметричные 30
Нейтронов диффракция и определение
структур 447
Обозначения величин, характеризуемых
при описании структур 265
Операции симметрии 28
Определитель рентгенографический
веществ 22
Ориентировка тетраэдров в плотны v
упаковках 600
Осевые отрезки, отношения 85
Оси винтовые (винтовоповоротные) J 06
, обозначения 109
— зеркально-поворотные (плангироиды)
28, 33—37
— инверсионные (гироиды), 28, 33-37
— поворотные биполярные 29
главные 29
побочные 29
полярные 29
— порядок 28
Ось новоротная симметрии 28
Отношение ионных радиусов 162—168
— с/а и вес ковалентного состояния
380
Параллелепипед л-кратноиримитнвный 95
— однократнопримитивный 94, 95
— повторяемости 94
— элементарный 94
объём 95
Параллелограммы и-кратнопрнмитивпые 93
— примитивные (однократнопримитивные)
93
— элементарные, площадь 94
Иараллелоэдры (гекса-, гепта-, тотра- три-,
параллелоэдры) 101
Перенос 92
Период идентичности 92
Периодическая система элементов 185
Плавления абсолютная температура и
количество холостых электронов 200
Плангироиды 28
Плоскость алмазного скольжения 105
— двойного отражения 33
— двойной симметрии 33
— зеркального отражения 32
— симметрии 28
Плоскость скольжения (клиноплоскоеть)
105
— скользящая и её обозначение 105
— скользящего отражения 105
Плотность вещества рентгенографическая
123
— упаковки 123
— упаковки и свободная энергия 805
Поликристалл 25
Полиморфизм 150, 219
Полиэдры атомные (октаэдр, тетраэдр)
161 — 162
Фёдорова 10J
— координационные 21
Полюс грани 44
Постоянная Маделунга 159
Правило возбуждения валентностей t82—
183
Правило Гольдшмндта (и Магнуса)
(несоблюдение) 804
— Гунда 183, 202
— сцепления 152
— эффективныv атомных номеров (ЭЛИ)
174, 202
Превращения полиморфные 91
— фазовые 26
Примеси, влияние на образование
структуры 222
—, — на последовательность иоворотон
тетраэдров в плотных упаковках 604
—, — на устойчивость кубической формы
Zr03 463, 465
Принцип Бертло 150
— Гиббса-Кюрн 25
— Дюма 149
— координационный систематики типов
структур 129
— Ломоносова 22, J 48, 219
Принципы плотной упаковки анионов 16J
— Паулннга 172—173, 182-183
Проекции ось 44
— стереографической построение \~>
— шар 44
Проекция окружности 45
— стереографическая 44 и след.
Простые формы кристаллов 40
Процессы (превращения) энанпютроиные^б
Радиусы атомные 146, 176, 177 и след.
— — и структура металлов 177 и след.
-- ионные 146, 164—165, 17Г»
-■- ---, соотношение 1 62
— —, -- и структ\ра крипа.ма 1(>2-~
1 68
Расстояния межплоскостные 95, 122
— межатомные н характер связей 17:)—
176
Резонанс квднтовохимическнй 193, 194,
195
Рентгеновское характеристическое
излучение, длины волн 128
Решётки атомные 158
— Браве 10J —103
— ионные 157
— координационные I2'i--I25, 158
— металлов 158
кубической гранецентрпрованной
устойчивость 216—217
центрированной—216—217
948
предметный указатель
Решётки молекулярные 124—125, 157
— обратные 122
— пространственные 91, 92, 94—96, 145—-
147
— —, периоды идентичности 94
сложные 96—10L
— сетчатые 158
— слоистые 158
— солевые и характер связи 622
— структурные узчы 125
— ценные 158
Решёток пространственных основные тины
128 и след.
Ряд трансляционный 92
Свойства вещества скалярные 24
— — векториальные (искторные) 24
Связи ионные 173
— ковалептные 157, 158, 173
--- металлические 158
■■- химические спи 1У1, 192
■--- химической характер (зависимость от
внешних условий) 156—158, 192
— - электростатические 156, 157
— электростатической напряжение 172
Связь между кратностью точек и
кратностью н\ позиций 41.
Сетка стереографическая Болдырева
(однополюсная) 46
Вульфа (двупол юс на я) 46
Силы Пан -дер-Ваал ьса 158, 169
•— дисперсионные 170, 171
— — и структура решетки 170, 171
Символы буквенные по 11. П. Велову,
характеризующие ориентировку
тетраэдров в плотных упаковках 600
по SB — 600
— - числовые по Г. С. Жданову 600
Симметрии изменение (влияние
температуры) 369, 370
Симметрия 27
--- внешней формы 27, 28, 51
— внутренней структуры 27, 51
- кристаллов и фигуры травления 87—90
- физического процесса 27, 28
Сннгоинн 46
Сипгонип наименования 58
Сингонпн высшая 51
— низшая 51
средняя 51
Систематика c/i руктур етруктурпо-коор
днпацнонпап 11, 16—18, 234—263,
so;;
Сштимы кристаллов 51
Сложность химического состава и
сложность структуры 804
Смеси рацемические 155
Соединение комплексное 612—615
— сложное 612—615
— сложно-комплексное 613—615
— химическое и дробное отношения
атомных концентраций 223—225
н термодинамическая устойчивость
224, 225
Соединений комплексных образование 806
— сложно-комплексных—806
— сложных—806
Состав стехиометрический и структурный
614—615
Состав химический кристалла иодида
меди как функция упругости пара иода
407
Состояние аморфное 25
— жидкое 24—26
— стекловидное 25
— твёрдое 24—26
Состоянии агрегатные 23
Стекло 23
Строение молекулярных решёток 433
Структур зависимость от
термодинамических факторов 225—227
— закономерности образования 805
Структур образование, влияние давления
805
, — взаимодействия структурных
узлов 805, 806
— —, — примесей 805
, — температуры 805
— окислов металлов образование 628
■-- систематика лимическая 263
(Структура вещества 145—147
■— координационной сферы и подбор
веществ с заданными свойствами 805
— кристалла 92
— кристаллов и зонная теория 215—218
реальных- 147
— химическая 152
— цезии в зависимости от изменения
давления 273—274
Структурная формула вещества 128
Структурно-координационные формулы 227
Структурные дроби.96
— исследовании, выбор объектов 803
— узлы неоднородные 128
— ■■- однородные 128
— ••- простые 128
сложные 128
Структурный и химический
(стехиометрический) состав, несоответствие 612
- состав химических- соединений 806
- узел 99—101, 128
Структурных модификаций варианты 220
— и форм обозначения 264, 265
~- типов систематика по SB 233—262
1>. Ф. Ормопту 231—262
— узлов обозначения 128
Структуры внедрении 222
— — п квантовая характеристика
электронных уровней 222
- вычитании 220
— - у-фаз и устойчивость 217, 218
—- деления ' 220
— зтмещенпя 220
— кристалла, дефекты 1-го рода 220—•
222
2-го рода 222
— кристаллической тин и отношение
объёмов атомов 128
— кристаллов, электропографичеекпе
исследования 643
-- молекулярных решёток 646—659
—, определение методом диффракции
нейтронов 447
- основные гексагональные 138
— — кубические 130
и производные от них. Соответствие
закону пределов Фёдорова* 806 ..
— островные 141
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
949
Структуры переменные 222
— полисоединений 669—697
— разрыхления 220
— реальных кристаллов 806
— с бесконечным одномерным ионом 666—
668
— - с двумерным ионом 663—665
— с комплексным анионом 711—802
катионом 697—711
■--<■ трёхмерным ионом 659—662
— слоистые 141
- смещения 220
— цепочечные (цепные) 141
— чередования 222
Теории химической связи волномеханиче-
ские и их приложение в химии 188—
206
Теория атомньп- полиэдров 21
— квантовая, атома 156
— квантовохимического резонанса 193—
195
— молекулярных орбит
(Гунда-Мудлинена) 190
— реального кристалла 148
— связывающих холостых электронов 188,
189
— симметрии 17, 18, 21
дисконтинуума 91, 103
— — континуума 23
— Слетера-Паулинга 189—190
— структурная (А. М. Бутлерова) 1 '*9
— твёрдого тела 22
— типов 149, 150
— химической связи 17, 18, 156
дуалистическая 148
Точек координаты \ 10—11Г>
— кратность 110
— ориентировка НО
— положения общие 40
специальные частные 40
— собственная симметрия НО
— степени свободы 110
Точка Кюри 306, 309
Точки антисимметричные 30
— идентичные 92
— кратность 38
позиции 38
— кристаллографической «протяжённость»,
собственная симметрия, ориентировка
28
— обратно равные 30
— ориентировка 38
— параллельно равные 92, 95
— равнозначные (эквивалентные) 40, 95
— симметрично равные 28, 30
— собственная симметрия 38
— совместимо равные 28, 30
Трансляция точек 92
— элементов симметрии 104
Угол элементарный 28, 106
Упаковка плотная гексагональная 134—
138
кубическая 134—138
Упаковки плотной междуузлия октаэдри-
ческие 130—138
тетраэдрические 130—138
Уравнение Ьорна 160
Уравнение Вульфа-Брегга 21, 125—126,
181,215—216
Уравнения Капустинского 170
Фаза, определение понятия 219
Фигуры травления 87
— энантиоморфные 31
Формулы стехиометрические и
структурные (структурно-координационные) 614—
616
Формы гемиэдрические 51
— голоэдрические (полнограниые) 51
— мероэдрические (неполногранные) 51
— общие и частные кристаллов
гексагональной сингонии 72
кубической — 76, 77
моноклинной—62
ромбической — 63, 66
— ромбоэдрической — 66
тетрагональной — 67
■ триклинной—59,62
— простые исходные 59
— тетартоэдрические 51
Фурье-анализ 126, 206, 210, 643
(1)~электронов влияние на характер
химической связи и тип структур 454
Химия структурная неорганическая 20
Центр инверсии 28
— обратного раненства 29
— симметрии 29
Числа кристаллографические 80, 82, 124,
125
— квантовые 182—185
Число Лвогадро 123, 160
Шпинели тип, образование 627, 628
, три основных варианта 627, 628
Электронная плотность 186 н след., 206
и след.
Электронные d- и /-оболочки, заполнение
и образование фаз с широким
интервалом устойчивости по составу 465
— уровни атомов 182—185
Электронов диффракция 181
— концентрация 177 и след.
и структура фаз 177 и след.
— несвнзывлощих пары и стерический
эффект 190
Электроны, длина волны 181
— разрыхляющие (развязывающие) 190
— связывающие 157, 190
—, скорость 181
— холостые 182—183
Электропроводность кристаллоп и
характер перекрывания зон 214, 215
Элемент симметрии, кратность (порядок) 40
Элементы симметрии 28
дисконтинуума 103—112
— — дополнительные 62
идентичные 104
, обозначения 37, 38
, полная комбинация 63
порождающие 33
порождённые 33
, пространственные сочетания 58
950
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Элементы симметрии I рода 28
11 рода 28
— симметрии, семейство 104
точечной 28
эквивалентные, равнозначные 104
— химические, недавно открытые 186
Энергии решётки галогенидов 161, 169
— связи и межатомные расстояния гало-
геноводородов 445
Энергия диссоциации 160, 193
и межатомные расстояния Н2 и
галогенов 351
— ионизации 160
— кристаллической решётки 22
Энергия поверхностная 24
кристаллов 25
— решётки 146, 156, 160
— сублимации 160
Эффект комбинационного рассеяния света
22
Ячейка элементарная 91, 92, 94, 124
, базоцентрированная 96
, гранецентрированная 96
, двугранецентрированная 96
, количество частиц 123
, метрика 115
, центрированная 96
Ас, актиний
Ac 278, 280
ЛсВгз 378, 380
АсСЬ 378, 380
Ac2S3 453
Ag, серебро
Ag 128, 315, 317
Ag—As 566
AgeAs (?) 566
Ag8 As04 634, 635
Ag8AsS3 207
Ag—Au 177
Ag—В 610
AgBr 166, 168, 408
AgCN 619
aAgCN 693
pAgCN 693
AgCd 180
AgCl 168, 408
AgClOs 799
AgC104 792
Ag[Co(NH3)2 (N02)J 257, 734—735
Cs2 [AgGUl [AuClJ 744
Ag[S-C(CH3) (NH2)J CI 71
Ag—Cu 177, 218
Ag2F 410
AgP 168, 408
AgFeS2 636
Ag-галогениды 160
Ag2HgJ4 251, 618
AgJ 168, 175, 176, 244, 410
aAgJ 408, 411
PAgJ 409, 410
vAgJ 407, 410
AgJ04 793—794
KAg(CN)2 251, 746
AgMn04 253, 727
Ag.Mo04 632
Ag—N 562
AgNs 562, 677, 679
Ag7NOn 768—769
УКАЗАТЕЛЬ ФОРМУЛ*) ВЕЩЕСТВ,
УПОМИНАЕМЫХ В I и II ЧАСТЯХ**)
[AgtNHskh [S04] 254, 783
AgNo2 252, 770
Ag20 494, 498
Ag203 494
Ag—P 567
Ag3P04 254, 634, 635
Ag2HP04 75
AgRe04 728
Ag2S04 781
Ag—Sb 566
Ag.Sb, гольдшмидтин 566
Ag[Sb(OH)e] 774
Ag3SbS3 207
aAg2Se 497
Ag2Se04 781
aAgsjTe 497
aAgi2Te7 498
AgTlJCh) (СН8СОО)з?Н20 262
AgZn 180
AgZn3 180
Al, алюминий
AlAs 572
AlAs04 612
Al—В 610
A1B2 246, 610, 695
AIB12 610, 670, 695
Si808e(BOH)4.R8Ale, турмалин
(R-A12, Mg3, Fea, Ke Lie, Nae,He) 69
ALBa 249
BaAl204 254, 634
Al2Be04, шпинель, 253, 627 и след.
АЬВеО*, хризоберилл 626
Be2Ah(Si03)e, берилл 75, 88, 89, 143, 145,
253 259
HBeAlSi05, эвклаз 259
AlaBre (А1Вг3) 421, 427
А14С3 248, 599
А1(СН3)з 650
A16C3N 250
Са3А12Ов, трикальций, алюминат 250
Са3[А1(0Н)вЬ 257
*) Вещества приводятся в указателе формул в порядке алфавита их химических
обозначении. В случае сложных веществ, в состав которых входит несколько
металлов, их следует искать среди соединений металла, название которого стоит первым
по алфавиту, независимо от того, в каком порядке названия этих металлов
приводятся в химических формулах (например, MgAla04 следует искать среди соединений
Al; KFe(CN)e—среди соединений железа и т. д.).
В случае общих наименований следует обращаться к концу списка соединений
соответствующего элемента.
**) См. также III часть «Регистр исследований структур неорганических веществ».
952
УКАЗАТЕЛЬ ФОРМУЛ
12СаО-7А1203 257
Na2(Ga,K)2(AleSie024)(S04)2, гаюин 260
везувиан 259
АЬСагЙЮ,, алюмосиликат кальция 71
Ca3Alt(Si04h, гроссуляр 79
Ca3Al2(Si04)3, гранат 254, 259
CaOAUO -2Si02, анортит, 33, 61
CaSi4AlaO]2-6H20, хабазит 258
А1С13(тип Z)0,3) 247, 424
AICU (тип Z)016) 248, 425
Al2GL, молекула 421
[А1(Н2ОдС1з 707
Со2А1б 219
Al2CoU4 631
СгаА1 246
СгБА]8 249
GsA](S04)2-12H20, р-квасцы 785
CuAU 245
Gu9Al4 17, 180, 249
Cu3Al 134, 180
Ai2Cu04 631
Cu2AlMn 257
Gu7Al2Zn4 180
AIF8 247, 423, 614, 615
NH4A1F4 664
(NH4bAlFe 255, 257, 750, 804
Al2Fe()4 631
Al4Si2O10Fe(OH)2, ставролит 255, 258
A!2Je 421
KA1F4 664
N'»8 К iAl8Sie034, нефелин 75, 88
KAL(S04)2 254, 779—780
KAi(SCY2-12H20, квасцы 255
KAl(S04b 12Н20,а-квасцы 255, 705, 784—
785
KAl(Se0^212H20 784—785
KH2Al8Si80i2 или KAl2[Si3-bAl]O10(OH)2,
мусковит 90, 259, 665
KAlSi308, санидин 260
ЬШОз 612, 661
Al2Mg04 227, 628, 631
Al2Mcf5Si3O10(OH)8, группа хлорита 259
AhMn04 631
AlN 176, 571
NUaCHaAl(S04)2-12HaOf 3-квасцы255, 785
NH4A1(S04)2 779—780
NH4AL(S04)a 12H20 784—785
Na3[AlFe), криолит 257, 614, 615, 751,
804, 805
N46A13F4, хиолит 257
NaAl2Os 248
NaAl(S04)212H20, у-квасцы 254, 785
Na4(AlSi04bCl, содалит 260
Na4Cl(AlSis08)s, скаполит 260
NaAlSi04, карнегиит 260
NaAlSi3U8, альбит 260
3[Na2Ab04,Si204], канкрпнит 258
NiAlSioOgtbO, анальцим 260
Ni2Al2Si3O102H2O, натролит 260
Al3Ni 248
AI3N12 248
AlNi04 631
А120з 612, 616
aAl208. а-корунд, 249, 503, 504, 506, 804
fJAl3Os, р-корунд 504, 506
YAlaOs, Г-корунд 248, 506, 628, 630
T'At203, -^корунд 505, 506
А1(ОН)8 247, 470
А1(ОИ)з, гидраргиллит 426
АЮОН, диаспор 250
А1Р 572
А1(РОз)з=А12Оз-ЗР205 635
А1(Р04)з 253
RbAlF4 664
RbAl(S04)2-12H20 784—785
AlSb 572
Al—Si 599
Ali3Si5Oao(OH,F)i8Cl, цуниит 259
(AlF2)aSi04, топаз 90, 255, 258
Al2Si05, кианит 254, 258
Al2Si05, силлиманит 254, 258
Al2Si06, андалузит 254, 258
Al2Sia05(OH)4, накрит 259
Al2Si4O10(OH)2, пирофиллит 259
Al3Ti 248
T12A1F5256, 613, 614, 615, 667
T1A1F4 253, 613—615, 664
T1AI(S04)212H20 784—785
YAIO3 612, 613, 659, 661
Al2Zn04 631
Al3Zr 248
Am, америций
Am 297
AmBr3 390
АтСЛз 389
AmJ8 390
Am2S3 477
Ar, аргон
Ar 233, 353—355
As, мышьяк
As 242, 335, 338, 339
a As, металлический серый 337, 339, 340
As, жёлтый 339, 340
As, коричневый аморфный, 339, 340
As, серый скрытометаллический 339, 340
BAs04 625
AsBra 438
As(CH3)3 650, 654
As(CH3)6 654
As(CeH6)3 653
Gd3As2 (тип D59) 568
Gd3As2 (тип £56) 569
GeAs 542, 543
GoAs 558
GoAs3=Go4(As4)s, скуттерудит 246, 558, 6S2
GoAsS, кобальтин 250, 613, 685
Gs3As3G]9 257
Gu—As 563
Gu3As, искусственный домейкит 563
Gu3As, домейкит 564
Gu9As, уайтнеит 563
GueAs, альгодонит 563
(GaHBbAsGuJ 262
GuaAsS4, энаргит 254, 640
CuZuAs 641
Fe—As 554
FeAs (тип В31) 554, 556
FeAs (тип £14) 243, 554, 556
FeAs2, лёллингит 557, 681
Fe2As 554, 555
FeAsS, арсенопирит 250, 684
GaAs 175, 176, 572
Ge—As 574
УКАЗАТЕЛЬ ФОРМУЛ
As H8 438
AsJ3 438, 439, 4'»0
КзАз 536
KlhAsCU 150
LaAs 542
Li3As 536
Mg3As2 (тип Z>53) r>;U)
Mg3As2 (тип Z)55) VtO
MnAs 552
Na3As 248, 536
NdAs 542, 543
NiAs (никкелин) 138, МО, ПН, 2ill, .Mil
NiAsg, раммельсбергит 560
NiAsa, пара раммельсбергит ГиИ)
NiAsS 685
Ni(As, Sb), арит 561
Zn3Asa (тип D 55) 569
Zn3Asa (тип D5e) 568
Zn(ZnOH) As04 254
Иолиарсениды, 669
Сульфоарсениды 669
Аи, золото
Au 315, 317
Au—В 610
Au2Bi 566
Au(G3H7)2CN 261
AuCN 619, 645
AuCd 243
Gs2lAuCl2][AuCl4] 257, 74i
Au—Cu 177, 218, 265
AuGu 257
AuGus 257
K[AuBr4]2H20 254, 745
Au—N 562
Au—P 567
Au—Sb 566
AuSb2 566, 683
AuTe2, креннерит 246, 495
AuTe2, калаверит 247, 496
AuZn 180
В, бор
В 321 32't
BI 321
B1I 321
BaBe 695, 696
Be2B03OH, гамбергит 253
BBr 421
BiG^lB^] IG3] (?) 599, 605 689, 690, 691,
695
GaBe 249, 605, 670, 695, 696
GaB204, метаборат кальция 251
GaB2Si208, дан бурит 260
CeB6 695, 696
BGb 196, 197, 421
Go—В 609 *
Go,В 609
[Co(NH3)e] (BF4)3 749
[Go(NH3)J (BF4)2 749
Gr—В 606
Cr3B2 606
CrB 606
Си—В 610
ErB6 695, 696
BF3 421
[NH4] [BF4] 748
Fe—В 606
FeB (?) 607
FeB (тип #15) 243, 606
FeB (тип #27) 244, 607
Fe2B 245, 608
GdB6 695, 696
B2He 249, 422
\Ull10 422
B5HU 422
B10Hl4 422
Hf—В 605
HfBa 605
Ir— В 609
KBFt 748
Kli4(ll2O)>B5()10 256
KIU)3 252, 752
L;iBe 695, 696
Mn В 606
BN 210, 243, 570, 571
B3N3He 613
NdBe 695, 696
Ni—В 610
NLaB 609
[Ni(NH3)e] [BF4]2 749
B203 502
B(OH)3 426, 646
JI8B03, борная кислота 253, 656
BP04 71, 227, 253, 612, 615, 625
Pd—В 610
РгВ6 695, 696
Pt—В 610
Rh—В 609
RbBF4 748
SpBc 695, 696
Th—В 605
ThBe 605, 695, 696
Ti—В 605
TiB 605
TiB2 605
YB6 695, 696
YbB6 695, 696
Zp—B 605
ZrB2 (?) 605
Полмбориды 670
Ba, барий
Ba 274, 276, 277
BaBeF4 712
BaBr2 376
BaC2 576, 688
BaCa2(C2H5COO)6 262
ВаСОз 758
BaC103 661
BaCl2 171, 376
Ba3[Co(NOa)eh733f 734
BaFa 376
BaFe05 198
BaH2 375
BaJ2 376
fBa(H20)e]Ja 699, 700
BaMo04 719
Ba(N3)2 680
Ba(NOa)2 766—767
Ba[Ni(CN)4].4H20 254, 739
Ba2[Ni(N02)e] 734, 740
BaNiOB 198, 201
Ba02 673
BaO 168, 451
Ba[Pd(GN)4]-4H20 739
954
УКАЗАТЕЛЬ ФОРМУЛ
ВаРгОз 661
Ba[Pt(CN)4b4H20 739
BaS3 248, 676
BaS 451
BaS04, барит 151, 252, 778
BaSe 451
BaTiSi3Oe, бенитоит 75, 258
BaSn03 660, 661.
BaTe 451
BaTh03 661
ВаТЮз 235, 660, 661
BaWCU 720
BaZK)3 660, 661
Be, бериллий
Be 274, 277
aBe 274, 275, 277
ЙВе 275, 277
BeaG 576, 577
Be20(C02CH3)e 262
BeF2 371
[NH4]2 [BeF4]7l2
KaBeF4 712
bi2BeF4 615, 616
Be3N2 539
Na2[BeF4J 712
4BeONaSb03, сведенборгит 250
HNaBeSi308, эпидидимит 259
BeO 176, 4;j0
Be3P2 539
BeS 450
[Be(H20)4] [S04] 255, 779
BeSe 450
Be2Si04, фенакит 253, 259, 633
BeSi207(OH)2, бертраидит 259
BeTe 450
Bi, висмут
Bi 335
aBi 337, 338, 340
Bi(CeH5)3 653
CeBi 542, 543
BiF3 133, 134, 138, 140, 180, 247, 440,
804. 806
Ge—Bi 574
BiJ3 247, 439, 440
KBi2 236, 538
K3Bi 536
KBiS2 635
LaBi 542
aLiBi 537.
Li3Bi 537
Mg8Bi2 541
NaBi 538
Na3Bi 536
NaBiS2 635
BUO3 (кубическая простая) 530
Bi203 (кубическая центрированная) 530
aBi208 529
pBi2Os 248, 529
Pb—Bi 575
Pb2Bi (?) 575
PrBi 542, 543
Bi2S3, бисмутинит 531
Bi4Si301.>, эвлитин259
Sn—Bi 574
Bi2Fe2S, тетрадимит 246
TeBi 572, 573
Br, бром
Br 193, 349, 351, 352
BrCl 157, 194
BrF3 444
BrF5 444
BrF 444
Полигалогениды 669
С, углерод
С, алмаз 26, 91, 133, 134, 138, 140, 146,
155, 180, 207, 210, 239, 242, 326, 329,
330 333
С, графитов, 91, 146, 242, 330—333
С,—гексагональный 327—331
С,—ромбоэдрический 328, 331
С, кокс 333
С, сажа 26, 331
С, углерод 158, 210, 326—333
С,—аморфный 330, 332, 333
С. — «с перекрещивающимися связями» 331,
СВг4 430
?аСВг4 (моноклинная) 431, 447
ВСВг4 431, 447
C(NH2)3Br 261
[Crpl [С104]л, графитиды хлорной кислоты
688
[Сгр] [S04]ra, графитиды серной кислоты
688
CCU 223, 331, 430
Св01в 262
CF4 430
CFj., фторграфит 687
(СН3СНО)4, метальдегид 261
С2Н4, этилен 191, 261
СН4 241, 260, 331, 432, 447, 650
С2Нб, этан 191, 261
СвНв 613
С3Н4, аллен 155, 191
С(СН3)4, тетраметил метан 260, 651
С2Н2, ацетилен 191
С(СвН6)4, тетрафенил метан 261, 651
GHJ3 261, 430
C2H4J2 262, 430
G(NH2)3J 261
C(CeH5)3 [Jjn] 670
С(СН2ОН)4, пентаэритрит 71, 261, 653
[С(СН20СОСН3)4], пентаэритрит тетрааце-
тат 261
C4H402NJ, сукциниодимид 71
G(GH2ON02)4, пентаэритрит тетранитрат
261
G12H22OnH20, молочный сахар 61
0(СН3)2 650
CH3N02, нитрометан 194
CJ4 кубическая 260, 430, 431, 447
GJ4 моноклинная 431, 447
CJ4 тетраэдрическая 430
G2N2=(GN)2 573, 810
CH3N5, метилазид 680
СО 174, 194, 223, 241, 243, 507
аСО 447, 508
ВСО 447, 509
С02 167, 191, 233, 241, 245, 447, 507, 508,
646
С302 507
(СООН)2 а-щавелевая кислота 262
fi-щавелевая кислота 262
УКАЗАТЕЛЬ ФОРМУЛ
955
<СООН)2.2Н20 262
(COONH4)2H20 262
GO (NH 2)21 мочевина 261
C,S(NH2h, тиомочсвина 261
COS 250, 646, 655, 656
CS2 507
CSea 507
Антивинная кислота 155
Ацетилениды 670
Винная кислота 155
Гексаметилентетрамин 207
Графитиды 670
Глщро 55' дигидантоин
Цианамиды 670
Цианиды 670
Га, кальций
Са 274, 277
аСа 276, 277
[1Са 274, 275, 277
Са 274, 275, 277
1Са(Н20)в]Вг2 699
СаСа 233, 234, 244, 576, 685, 688, 691
CaCN2 670, 691, 694
СаСОз(ромбоэдрическая) 91, 128, 227, 612,
757
СаСОз (ромбическая) 758
СаС03, арагонит 91, 252, 803, 804
СаСОз, кальцит 69, 88, 89, 90, У!, 127,
226, 252, 803, 804
(Ca,Cl,Na,K)2(Nb,Fe)2(0,OH,F)7, копнит
250
СаС12 247, 374
(Са(Н20)в]СЬ 256, 698, 699
Ca(JCl4)2SH20 670
Са(ОС1)2 613
CaF 224, 540
CaF2, флюорит, плавиковый шпат 79, 133,
134, 138, 140, 159, 162, 224, 233—235,
245, 374, 489, 734, 806
СаН2 375
CaJ2 373
Ca(NH3)eJ2 226
fCa(H20)e]j2 699
K2Ca[Ni(NOa)e] 741, 742
KCa4Si8020F8H20, анофиллит 259
CaMg(C03)2, доломит 69, 253, 758
MgCaSi04 626
CaMg(Si03)'2, днопсид 259
II3Ga2Mge(SiO,)> тремолит 259
CaMo04 719
uCa3N2 539
Ca(N08)2 766, 767
Na2CaSi04 260
CaO 168, 174, 175, 451
Ca(OH)2 175, 373
Ca(Cl,F,OH)a. 3Ca2!P04), апатит 75, 88, 89
Ca5F(P04)3 255
CaS 166, 451
CaS04, ангидрит 151, 227, 252, 777
CaS04 ya H20 255
CaS04.2H20, гипс 27, 61, 90,255,780,781
CaSe 451
CaSi4 244, 576, 578
<CaO-Si02=CaSi03, волластонпт 258, 760
CaTiSi05, титанит 258
Ci2ZnSi207, гардистонит 259
<CaSn03 659, 661
CaSrOa 661
CaTe 451
CaTi03, перовскит 251, 252, 659—661, 804
CaTiOa 227, 235, 613
CaU04=Ca(U02)02 723
Ca(U02)2 (P04)2-6V2H20, метааутунит I 255
Са(иО2)2(РО4)2-101/3Н2О, метааутунит 255
CaW04, шеелит 71, 252, 720
CaZr03 661 нитриды
Кальций 541
Cd, кадмий
Cd 318—320
aCd 320
{4Cd 320
vCd(?) 320
CdBr> (тип CdCl2) 171, 412
CdBr2 «переменной структуры» 414, 415
[Cd(NH3)2Br2] 644
Cd(CN)2 619
Cd(CO)3 198
CdCl2 171, 245, 4J2
CdOlTCl 250
[Cd(NH3)2Cl2] 250, 644, 707
NH4CdCl3 251, 666
[Cd(NH3)e] (C104)2 796
CdCr04 625
CsCdCh 659, 661
CdF> 413
Fe2Cd04 628, 629
CdJ2 411, 415
CdJ2? (тли C19) 412
CdJ2 (тип С6) 138—140, 158, 170, 171, 244,
414
CdJ2 (тип С27) 246, 415
[Cd(NH3)e]J2 707
K2Cd(CN)4 620
Cd3N2 567
CdO 500
Cd3P2 (тип Z)59) 568
Cd3P2 (тип D55) 569
CdP2 569, 683
CdS, гренокит 75
CdS (тип £4) 168, 499
CdS (тип Bz) 500
3CdS04, 8il20 254
Cd—Sb 570
CdSb 570
Cd3Sb2 570
CdSe 499
CdSn03 660
CdTe 175, 176
CdTi03 612, 661
€e, церий
aCe 280, 283
fiCe 281, 283
fCe 283
dCe 283
СеВга 380
CeC2 688
GeCU 380
CeF3 379
CeFC03 75
RFC03(R-Ce,La,...), бастнезит 253
CeN 542, 543
Na2Ce03 621
CeOo 162, 454
956
УКАЗАТЕЛЬ ФОРМУЛ
Сеа03 455
Ge202S 455
СеР 542, 543
Ce2S3 456
Ce3S4 456
CeSb 542, 543
С1, хлор
СЬ 157, 173, 193, 223, 242, 349—352
C1F 444
C1F3 444
Полигалогениды 669
Cm, кюрий
Cm 297
Со, кобальт
Со 128, S08-310
аСо 309, 310
jJCo 308—310
ТСо 310
СоВг2 401
[Co(NH3)e]Br2 704
[Co(NH8)jf54 786
lb0(NH3)6IsO4
?86
Co—С 594
C03C 594
Co2C 594
[Co(NH3)6][Co(CN)e] 732
[СоШЩ)Б1[Со(СК)в] 732
[Co((gb°^][Co(CN)e] 732
Co(CO)4H 649
[Co(NH3^e] СЬ 704
CoCla 400
CoCla-2HaO 643
[Co(NH3)e][C104]2 796
[Co(NH3)e][C104]3 797
fCo(NH3)e][Cr(CN)e] 715
Cr2Co04 631
СоСг04 62b
Cs3CoCl5256
Cs3[Co(N02)e] 732, 733
CoCuS4 640
C0F3 398
C0F2 399
IGofNH3)5][Fe(CN)e] 731
Fe2CoOj 628, 629
K4Co(CN)6 203, 204
K3Co(CN)6CO204
C0J2 401
[Co(NH3)elJ2 704
[Co(NHs)e]Js 703
[Co(NH2CH3)e]J2 704
[Go{NH,)e]fs04] Ж
[Co(g&°,).]fso4] ™
[Co(NH3)ejfSe04] 786
K3[Co(N02)]e257, 732, 733
Co—N 557
Co8N 557
(NH4)3lCo(NOa)e] 732, 733
Co203 484
Со304=Со2Со04 484, 629
CoO 484
Co02xH20 201
Co(OH)s 399, 413
Co(OH)a 401
Co—P 557
C0P3 557
CoP 557, 558
[Co(NH3)e] [PFe]2 761
Pb3[Co(N02)eh 733
Rb3[Co(N02)el 732 733
CoS2 674
CoS 485
Co9S8 249, 486
Co2CoS4=CosS4 249, 484, 485, 640
CoSb2 558
CoSb i)59
CoSe3 674
CoSe 485
Co—Si 594
CoSi 595
Co2Si 247, 595
[Co(H20)J [SiFeJ 755
CoSn 245
CoSno 246
CoTe 485
CoTiOs 622
Tl8[Co(NOa)e] 732, 73:;
CoW04 721
Co5Zn2i 180
Cp, кассиопий
Cp 280, 281, 283
Cp203 456
Cr, хром
Or 128, 288, 290, 294, 296
aCr 288, 290, 294
]3Cr 289, 290, 294
vCr 290, 294, 295
Crl3r3 387, 395
Cr4C 583
Cr23Ce 236,
Cr7C8 583
Cr8Ca 248,
249, 583, 670, 696, 697
583
Cr(CO)e 174, 647, 648
CrCla 247, 386, 395, 425
|Cr(HaO)e]Cl8 257 700
CuCr04 625
Cr2Fe()4 631
KCr(S04)2, безводные квасцы 779, 780-
Кз[Сг08]=Кз[Сг(Оа)4] 717
KCr(S04)2 • 12НаО 784, 785
Cr2Mn04 631
CrN (кубическая) 547
CrN (гексагональная) 547
Na2Cr04 253, 718
NiCrCU 625
СгОз 247, 399, 470
Сг203471
CrS 471
CrSb 547
Cr—Se 471
CrSe 471
Cr—Si 583
CrSi 584
CrSi2 247, 584
CrTe (?) 471
VCrCU 625
УКАЗАТЕЛЬ ФОРМУЛ
957
ZnCrCU 625
CraZnCh 631
Cs, цевий
Cs 272—274
CsBr 169, 366, 368, 369
CsBr3 670
CsBr6 670
CsJBi^ 670
GsC8 686, 671
CsGie 686
CsGN 619, 693
CsCl 91, 129, 130, 133, 140, 151, 159, 160,
169—171, 180, 236, 242, 365, 366, 368,
369, 806
CsJCl, 251, 670
GsJGU 670
CsCK)4 (кубическая) 792
CsClCU (ромбическая) 794
CsF 168, 169, 365
Csa[GeFeJ 754
Csil 365
CsHgCla 661
CsJ 169, 366, 368, 369
CsJs 670
CsJ5 670
CsJOj, 661
Gs[lr(N02)e] 732, 733
Cs02 673
CS2O3 672, 673
Cs.O 449
Gs2[PbCle! 754
Cs2PtGle 737
Cs2PuCle 723
Gs3[Rh(N02)e] 732, 733
GsSH 369, 370
CS2SO4 782
CszSaOi 256
CS2S30» 787
CsalSeCI.] 776
GsSeH 366, 369, 370
CsalSiFeJ 754
Cs2f SnCie 1 754
Cs8|TaO*J 715
CsalTeClel 776
Cs2[TiGle] 714
Cs3Tl2Gl9 257
Cs2[ZrGls] 714
Си, медь
Cul27, 128, 130, 133, J40, 236, 242, 315—.",17
CuBr2 406
CuBr 168, 175, 176, 407
CuGl 168, 407
CuCl2 - 2H20 247, 658
Cu3(G03)2 (011)3, азурит 252
Gu(NH8)2Gl2 644
(NHi)2 (C11CJ4) • 2H20 742, 743
[Gu[S=C(CII3)(NH2)]4JG1 71, 262
CuF2 406
CuF 168, 407
Cu—Fe 177
Cu3[Fe(CN)eh 257
Fe2Cu04 628, 629
CuFeS2, медный колчедан халкопирпт 71,
250, 251, 613, 636
Cu2FeSnS4, станнит 254, 637
Cu3Ge 180
CuH 409
Gu2HgJ4 618
CuJ 168, 407
K2[GuGh].2H20 255, 742, 743
KaPb[CulN02)el 747
Gu Mg 245
Gu—N 562
Gu3N 562
GuO, тенорит 244, 494
Gu20, куприт 79, 133, 138, 140, 159,240,
245, 498, 734
Gu—P 562
Gu3P 248, 563
PdGu 257
GuPt (кубическая) 257
GuPt (ромбоэдрическая) 257
Rb2[CuGl4]2H20 742, 743
CuS=Cu2GuS3> ковеллин 243, 496, 613,
639, 804
Gu2S, высокотемпературный халькозин 496
Gu2S, низкотемпературный халькозин 497
Gu>S04- 3H20 61
GuSCU - 5ll20 255
Gu—Sb 564
GunSb2 564
Cu9Sb, 564
CunSb4 564
Gu3Sb 565
(.luBSb 564
GuoSb 247, 565
<zGu2So 497
Gu—Si 597
CiiaSi 597
Gu6Si 597
CuuSi 249, 597, 598
Gu3Sn 180
(JI3VS4, сульваннт 254
Gu—Zn 177, 217, 264
GuZn, 177, 180
у-латунь (Gu—Zn) 295
Gu5Zn8 177, 180, 2'»U
Gu Zn3 177, 180
1>у, диспрозий
Dy, 280, 281, 283
DyaOs 456
D2, тяжёлый водород
Dd 351, 352
DG1 445
Er, эрбий
Er 280, 281, 283
Er203 456
(V, Kr)P(>4 • 2И2О, вайншенкит 772.
Eu, европий
Eu 282, 283
EuGh 381
EuF2 381
Eu203 456
EuS 457
EuSo 457
EuTe 457
F2, фтор
F2 349, 350, 351, 352
Полигалогениды 669
958
УКАЗАТЕЛЬ ФОРМУЛ
Fe, железо
Fe 91, 128, 303, 305
aFe 264, 304—307
[iFe 264, 303, 305—307
^Fc 264, 304—307
iFe 264, 303, 305—307
FeBr2 397
Fe(CO)4Br2 649
Fe(NH8)e Br2 702
Fe—С 591
Fe—С, мартенсит (содержит до 6% С) 258
FemGrt, графит 687
Fe3G, цементит 247, 591, 592, 593
FeC08 757
Fe(CO)5 174, 202, 203, 238, 613, 647
Fe2(GO)fl 251, 648, 649
Fe(CO)4 649
Fe(CO)4H2 649
Fe(CO)4Cl2 649
Fe(CO)4J2 649
FeGl2 203, 396
Fe(H20)4Gl2 203
FeGU 394
[Fe(NH8)e]Gl2 203, 702
FeOGl 250, 613
Fe(NH3)e (C104)o 796
FeF2 247, 393, 423
FeF.> 395
(NH4)3 ^eF6) 729
FeJ2 397
Fe(NH3)eJa 702
K3Fe(GN)e 613, 729
K4Fe(CN)e 203, 205, 614
K4Fe(GN)e • 3H20 730, 731
LiFe02 621
Fe2Mg04 628, 629
(MgFe)2Si04, оливин 626
Fe2Mn04 628, 629
Fe—N 554
от Fe3N до Fe2N 554
Fe>N 554
FiuN 257, 554
NH4Fe(S04)2 614, 779—780
Na2Fe04 201
FeNb2Oe 250
Fe2Ni04 628, 629
(Ni, Fe)e S8, пентландит 486
Fe—О 627
Fe 04 198
Fe203 481
«VFeaOs 481, 628, 630
Fe3G4 = Fe2Fe04 481, 628, 629
FeO, вюстит 168, 224, 482, 627
Fe(OH)3 395
Fe(OH)2 397
FeOOIl 250
Fe—P 554
FeP2 057, 681
FeP 554, 556
Fe8P 554
Fe2P 245, 554, 555
FeS2, пирит 79,87,233,234, 245, 482, 613,
674
FeS2, марказит 245, 482, 613, 675
FeS 224
FeS, троилит 482
FeS, пирротин 483
Fe—Sb 554
FeSb2 557, 681
FeSb (тип CI 8) 554
FeSb (тип £8) 554, 557
FeSbS, гудмундит 684
FeSb2S4, бартьерит 251
FeSe 482
Fe^Si 593
FeSi2 (кубический) 593
FeSia (тетрагональный) 593
Fe6Si8 593
FeSi 243, 244, 593
Fe(H20)e (SiFe) 755 •
Fe^Fe*11 (OH)8Fe*nSi2O10, кронстедтит 260
FeTi08, ильменит 251, 252, 622, 804
FeTiOa 612, 613
Fe2TiOe, псеидобрукит 251
Fe2W 247
Fe7W6 249
Fe3W8C 250
FeWO* 721
Fe6Zn2i 180
Fe3Zn10 249
Fe2Zn04 628; 629
Fr, франций (экацезий)
Fr 272, 274
G, галлий
Ga 243, 322, 325
LaGaOa 612; 661
GaN 571
<xGa203 503, 506
{*Ga203 506
GaP 572
GaSb 572
Gd, гадолиний
Gd 280, 281, 283
Gd203 456
Ge, германий
Ge 175, 176, 326
GeBr4 430
Ge(CH8)4 650
Ge(CeHfi)4 651
Ge(GO)2 206
GeCU 430
GeF4 447
(NH4)2[GeFe] 756
Ge2He 430
Ge2H8 430
GeU 430, 433
Gel2 434
K2lGeFe] 257, 756
Mg2Ge 577
Ge—N 574
Ge3N4=Ge2GeN4 574, 641
Ge02, «растворимый в воде» 151, 152, 511
Ge02, нерастворимый в воде 151, 152
517
Ge—P 574
GeS2 247, 518
GeS 243, 519
Ge—Sb 574
Ge—Si 604
УКАЗАТЕЛЬ ФОРМУЛ
959
Н2, водород
Н2 157, 158, 192, 193, 349, 351, 352
НВг 445
зНВг 445, 446, 447
(ШВг 446, 447
ТНВг (?) 446
НС1 193, 233, 445
аНС1 445, 446, 447
[ШС1 446, 447
ИС104 174
HF 158, 193, 445
HJ 158, 173, 174, 193, 445, 447
iHJ 446, 447
{Ш 446, 447
Гидриды металлов 195
Не, галий
Не 353—355
Ш, гафний
Hf, 284, 286
aHf 284
HfG 579
HfF4 383
Hf02 151
Hf[P207] 773
SrHfOs 661
Hg, ртуть
Hg 242, 319—32J
Hg2Br2 420
Hg(Cl, Br)2 417, 656
HgBr2 245, 411, 418
CH3SHgCl 261
Hg(SC2H5)2 262
Hg(CN)2 250, 694
HgCl2 (тип С25) 246, 416
HgCh (тип C28) 246, 417
Hg2Ch 249, 420
HgF2 413
HgJ2 (красный) 419
HgJ2 (тип (713) 244
Hg2J2 420
HgJe 670
K2Hg(GN)4 620
NH4HgCl3 251, 663
HgO (жёлтый и красный) 501
UgS 168
HgS, метациннабарит 500
HgS, киноварь 242, 501, 502
Не, гольмий
Но 280, 281, 283
НооОз 456
Jj, ИОД
Ji 65, 193, 243, 349, 350, 351
JBr 444
JCl 444, 445
JC12 400, 445
JGI3 399, 444, 445
JCU 445
JF± 444, 445
JF>5 444, 445
HJ03 800
J02F2 206
In, индий
In 242, 323, 325
InCh 427
InN 571
ln203 505
InSb 175, 17G, 572
Ir, иридий
lr 308, 310
K3Ir(NOo)6 732, 733
(NH4)3 [Ir(N02)e 732—733
Ir02 487
Ir03 201
Rb3[lr(N02)6J 732—733
Ir—P 559
lr2P 559
Tl8[Ir(N02)a] 732—733
К, калий
К 272, 273
КВг 169, 365, 368—370
КВг03 252, 799—800
КС8 685, 686
KCie 685, 686, 670
КНС2 688, 670
K(CN) 618, 619, 692
KHG2O4 262
(СООК)2Н20 262
КС1, сильвин 90, 151, 166, 169, 365, 368—
370
КС104 (кубическая) 252, 792
КС104 (ромбическая) 794
КСЮз 237, 252, 799
СС13 СООН\ кислый
GGl3COOK J трихлорацетат калия 71
KGNS 251
KF 168, 169, 365
KHF2 251, 790
КН 365
K2HgCLi- II20 251
KJ7 670
KJ3 128
К J 169, 207, 365, 368—370
KJCU 253, 672
KJO4 612, 793, 794
KJ03 659, 661
LiKS04 253
KMg(H20)e (Cl, Вг)3, карналлит-бромкар-
наллнт 251
KMgF3 659, 661
KMn04 726
K2Mn(S04)2 • 4H20, манганлеонит 254
K4[Mo(CN)8] • 2H20 237, 250, 716
KN3 677, 678
KN03 763
KNO2 251, 770
KC1 • NaCl 614
KNb03 661
K3(Nb08) 715
K2[NbF7J 206, 256, 714
K4Ni(CN)4, Ni-цианил калия 197, 202
K2Ni(CN)4 202, 203
K2Ni(CN)3 204
KNiF3 661
KNp2F9 724
K03 673
КО2 673
960
УКАЗАТЕЛЬ ФОРМУЛ
КаО» 672, 673
К2Оа 672, 673
КаО 448, 627, 673
аКОН 370
РКОН 365, 370
К20502С14 256
КН2Р04 150, 254
K4Pd(CN)4, Pd-цианил калия 197
K2[PdCl4] 738
К2 [PtCle] 125, 128, 226, 255, 256, 737, 804
K2[PtCl4] 253, 738
K2[Pt(SCN)e] 255, 256, 738
K2Pt(COS)4 261
KPuF5 725
KPuaFe 724
K3[Rh(N02)J 732—733
K4Ru(GN)e 3H20 730, 731
K2S 448
KSH 244, 365, 370
«KSH 367, 368
K2S04 237, 253, 782, 805
K2S3Oe 256
KaS208, пиросульфит калия 256
K2SaOt, дитионит калия 257
K8Sb 536
C4H40e(SbO)Klf20, рвотный камень 6L
K2Se 448
K2[SeBrf] 776
KSeH 365, 370
aKSeH 367
KaSe04 782
K2[SiFe] 7.V*
K2[SnCle] 754
K2SnCl4H20 251, 668
K2Sn(OH)e 255, 256, 756
K3[TaOe] 715
K2[Ta07] 714
K2Te 448
КзТ1С1в2Н20 257
M^UFe 722
KUF6 724
aK2UF0 725
piK2UFe 722, 725
fJ2K2UFe 725
<xK3UF7 72л
aiK3UF7 725
KU2Fd 724
KU8F13 724
KUeF25 724
K3W2C18 257
K2Zn(GN)4 613, 620
KZnF3 659, 661
K2ZnCl4 747, 748
K3ZrF4 205, 206, 712, 713
Кг, криптон
Кг 353—355
La, даитаи
La 278, 279, 280
aLa 278, 283
pLa 279, 280, 283
LaBr3 378, 380
LaCa 578, 688
LaCl8 378, 380
LaF3, тизонит 247, 377
LaOF 621
LaFG03 75
LaJ8 378
LaN (?) 542
NaLaCh 621
La208 162, 249, 452, 453
La(OH)3 378, 380
La202S 453
LaP (?) 542
La2S3 453
LaSb 542
Li, литий
Li 272, 273
LiBr 168, 169, 365, 370
LiGl 151, 168, 169, 225—227, 365, 370
Li(H20)n CI 225
LiC104.3H20 254
LiF 166, 168, 169, 365
LiH 160, 166, 365, 370
LiD 365, 370
Li-галоген иды 160
Li J 365, 370
LiJOa 251, 662
Li2Mo04 623, 633
Li3N (гексагональная) 237, 53*>
Li3N (кубическая) (?) 535, 536
NaLiC08 75
LiNaCl 226, 227
LiNb03 623
Li202 673
Li20 448
LiOH 367, 368, 370
LiOH . H20 245, 697—698
Li3P 536
Li2S 448
LiSH 365
LiaS04H20 255
aLi3Sb 536
BLi3Sb 537
Li2Se 448
LiSeH 365
Li2Te 448
Li2TiOs 621, 622
Li2W04 633
Mg, магний
Mg 140, 236, 242, 269, 274, 275, 277
MgBr2 373
[Mg(NH3)e]Bra 699
[Mg(H20)e]Br2 698
MgC2 576
MgC03 757
Mg(GO)3 198
MgCL, 372, 698
MgCb • 6H20 698
[Mg(NH3)e]Cl2 699
[Mg(H20)eJCl2 256, 698
[Mg(NH3)e](C104)a 796
Mg(C104)26HaO 255, 795
MgF2 371
Mg2Ge 576
MgJ2 373
[Mg(NH3)e]Ja 699
Mg3N2 (тип Mn203) 539
Mg8N2 (кубическая) 540
(NH4)2Mg(S04)26HaO 255
MgC03Na2C03NaCl, иортупит 252
NaeMg2S04(G08),2 тихит 255
MgNi2 247
УКАЗАТЕЛЬ ФОРМУЛ
961
MgO 166, 174, 175, 451, 627
Mg(OH)a 175, 373
Mg3Pa (тип Я5Б) 540, 541
Mg3P2 (тип D53) 249, 539
Mg2Pb 576, 577
MgS 166, 451
MgSCU • 7H20, английская соль 65
Mg„Sb2 541
MgSe 168, 451
MgaSi 576, 577
MgSiOs, энстатит 259
Mg2Si04, оливин 253, 259, 612, 613, 626
Mg(FOH)anMg2Si04, силикаты ряда хонд-
родита 258
[Mg(H20)e][SiFe] 755, 806
HaMg7(Si03)8, антофиллит 259
H4Mg3Si2Oe, хризотил 259
MgaSn 576, 577
[Mg(HaO)e][SnFe] 755
MgFe 450
MgTi03 622
[Mg(H20)e]lTiFe] 713
MgWCU 253, 721, 722
MgZn 244
MgZna 244
MgZn5 249
Hn, марганец
Mn 128, 298, 300
aMn 243, 295, 298, 300—302
jiMn 243, 298, 300—302
yMn 299, 300—302
MnBra 392
[Mn(NH3)eJBr2 702
Mn—G 588
Mn4C 588
Mn23Ce 588, 696, 697
Mn3G 588
Mn7G 588
МаСОз 757
MnGU 390
[Mn(NH3)elGl2 702
lMn(NH3)e](C104)2 796
MnFa 390
Mi) J 2 392
[Mn(Nli3)e]j2 702
Mn—N 551
Mn4N 551
M.iaN 551
ivln8N (?) 551
jiMnOa (?) 477
f.vlnOa 477
Miw08478, 632
МП3О4 249, 478
MnO 479
Mn(OH)0 250
Mn(OH)2 392
MnP 244, 552
MruP 553
Mn3P 552
Mnr Sw 224
aMiiS ^зелёная) 224, 479
fblnS (красная) 224, 479
-pUnS (красная) 224, 480
MnSi 478, 674
MnSb 553
MnaSb 553
nMnSe 479
jiMnSe 479
YMnSe 480
Mn—Si 589
MnSi2 589
MnSi 589, 591
Mn6Si3 249, 589, 590
Mn3Si 589
[Mn(HaO).J[SiFe]755
(Zn, Mn)2 S1O4 633
MnTea 674
MnTi03 622
M11WO4 721
Mo, молибден
Mo 128, 288, 294, 296
Mo—G 584
aMo2G 585
MoG 584
Mo(CO)e 647, 648
M0GI5 385
MoFe 385
[NH4]3[Mo03F3l717
Mo—N 548
Mo03 247, 472
МоОа 473
PbMoCU, вульфенит 71, 7iJ
MoS2, молибденит 138—140, 244, 474
MoSi2 (тип С И) 236, 244, 586
MoSia(TiraC20)245
N, азот
N2335, 338
a-N2 335, 338, 339
p-Na 336, 338,339
a[Nll4]Br 709
ji[NH4]Br 710
YlNIUlBr 244, 710, 711
a[ND4]Br 709
§[ND4]Br 710
TlNDi]Br710, 711
lNH4][GUBr]252, 671, 672
N (СН3)з 650
NH3CH3CI26I, 710, 711
IN(GII3)4] [J9] 670
[NH4] (GN) 693
a[ND4]Cl 709
p[ND4]Cl 710
N114 Gl, шшатырь 79
a[NH4lG1709
p[NH4] Gl 710
(СИ8)4 NG104 261
NH4 LIO4 (кубическая) 650, 792
Nix4 GIO4 (ромбическая) 794
[Nll4] [G102] 251, 802, 805
NH8190, 226, 233, 246, 438, 535, 53l>
ND3 438
NH4N3 677, 679
<NH4)F 709
(N1U)F 709
NH4IIF2 251, 791
N(CH8)4JCla 260
N(GH3)4J 261
NU3CH3J 261
N11 (G2HB)3J 261
NH4J 168
o[NU.|]J 709
§[NH4JJ 710
a[ND4JJ 709
962
УКАЗАТЕЛЬ ФОРМУЛ
0[ND4]J 710
(NH4)J3248
NH4J04 793, 794
NH4JO3 661
NH4N08224, 669
NH4N03 I 253, 764
NH4NO3II 253, 764, 765
NH4NO3 HI 253, 765
NH4NO3 IV 253, 766
N2O б молекула 525
N2O4 молекула 525
N204-N02 (кристалл) 246, 526, 541
N02 молекула 525
N0 молекула 525
N2O молекула 525, 526
N20 448
NH4[H2P02]-(NH4)2[H4P204] 251, 771
NS 530
(NH4)SH711
(NH4)2S208256, 787
(NH4)2S04 782
Азиды 669
Na, натрий
Na 129 272 273
NaBrlbt, 161,169, 206, 365, 368, 370
NaBr03798
Na2G2331, 576
NaHG2576, 685, 688
NaGN 618, 619, 691
aNa(GN) 691
pNa(CN) 692
Na2G03H20 252
Na[HC03] 252, 760
NaC128, 87, 90, 124, 128, 133, 134, 138, 140,
151, 156, 157, 159, 161, 169, 174, 206, 209,
210, 226, 227, 234, 235, 237, 242, 365, 368,
370, 616, 806
NaC104 792
NaGlOs 79, 153, 252, 798
2Na2 S04-NaCbNaF, сульфогалит 255, 617
Na( JGI4) • 8H2O 670
NaF 151, 161, 169, 365, 370
NaHF2 251, 789
NaH365
Na-галогениды 170
NaJ 365, 368, 370
NaJ04793, 794
NaJ08 661, 800
NaJ04 • 3H20, периодат натрия, тригидрат
69
NaN8 677, 691
NaN08226, 227,612,792
NaN02251 , 769
NaNbOa 659, 661
Na202 673
Na20 448
NaOH 370
Na8P 536
NaPb8180
Na2Pr08 621
NaPuF, 722
p2 NaPuF4 725
NaPuFe 725
NaRe04 728
(NH4)2Na[Rh(N02)e] 736, 737
Na2S 448
aNaSH 367
NaSH 365, 370
Na2S08 253, 788, 806
Na2S04 253, 781
NasSb 536
Na[Sb(0H)e]257, 774, 804
Na[SbF4(0H)2] 257, 775
Na2Se 448
aNaSeH 367
NaSeH 365, 370
Na2ZrSi8Og»2H20, катаплеит
Na2Te 448
Na Tl 134, 180, 244
NaU02 (GH3 GOO)8 262
NaUF5 724
aNa2[UFe]725
[i2Na2UFe722, 725
YNa2UFe 722, 725
Na8 UF7 725
Na2 UF7 723
NaZn18 249
Nb, ниобий
Nb287
Nb—G581, 583
NbG 581
Nb4G 581
[NhF7]'.HOH 125
Nb—H 385
NbH 385
NbN 545
Nb—О 469
Nb02 467, 469
Nb206 469
NbO 468, 469
Nb—S 469
NbG—TaC—WG 583
NbG—TiC 583
NbG—ZrC 583
Nd, неодим
aNd 280, 283
NdBr8 380
Nd(Br08)8. 9H20=Nd(H20)
801, 803
NdC2 688
NdCl8 380
NdF3 379
NdN 542, 543
Nd208 455
Nd(OH)3380
NdP 542, 543
NdSb 542, 543
Ne, неон
Ne 353—355'
N1, нивель
Ni 128, 311—314, 317
a-Ni 312—314
P-Ni 311—314
NiBr2 403
[Ni(NH8)elBr2704
Ni—G 595
Ni8C 595, 596
Ni(GO)4 174, 206, 647
NiGh 403
lNi(NH8)€] Gh^26, 704
УКАЗАТЕЛЬ ФОРМУЛ
963
[Ni (NH8)el (G104)2 796
NiF2 402
NiJ2 403
[Ni (NH3)e J2 704
[Ni(NH2 CH3)e]J2 704
[Ni (NH8)eJ [N03]2 255, 256, 767
Ni(NH8)e[In]670
Ni—N 560
Ni3N 560
N1O4 198
NiO 166, 488
Ni02xH2O 201
Ni (OH)2 404
Ni—P 560
!Ni(NH3)J [PFe]2 761
Pb2[Ni(N02)e] 740
NiS, миллерит 243, 491
p NiS 491
^NiS 490, 491
NiS2 490, 674
Ni3S2 490
Ni2NiS4 640
NiS04-6H20 255
NiS04«7H20 255
NiSb, брейтгауптит 561
NiSbS 685
NiSe 491
rNiSe 490, 491
NiSe2 674
Ni—Si 596
NiSi 596, 597
[Ni(H20)e] [SiFe] 755
Ni3Sn 248
NiSnGle • 6H2O 257
Sr2[Ni(N02)e] 740
NiTe2 492
NlTe 491, 492
Ni3Ti 248
NiTiOg 622
Ni(W04) 721
Ni6Zn2i 180
Np, нептуний
Np 273
aNpBr8 389
NpGb 389
NpJ8 390
PNpBr8 390
Np—Si 587
O2, кислород
02 158, 193, 341, 345, 346
a02 346
(*02 346
f02 341, 346
Os, овен
08 345, 346
О 4 345
0C12 190, 441
OF2 441
H20 175, 196, 225, 226, 441, 805
aH20 441
aD20 441, 447
b H20 441
H202 441, 443 '
D202 441
Лёд I(J • HtO ж D20 442, 443
Лёд II (H20) 443
Лёд III (H20) 443
Перекиси 449, 451, 669
Os, осмий
Os 305, 307
OsGl4 393
OsF4 20
OsF8201. 206, 393
Os02 483
Os04 201, 483
Os—P 554, 557
OsS2 674
OsSe2 674
OsTe2 674
P, фосфор
P 335, 339
P жёлтый 339
P красный (моноклинный) 339
P белый (кубический) 339
a-P чёрный (ромбоэдрический) 337, 33£
Р чёрный (ромбический) 243, 338, 339
Р фиолетовый (моноклинный) 339
РВг3 438
РВгБ 125, 438, 439, 613, 646, 711
РВг7 670
PCU 125, 157, 196, 197, 223, 438, 439, 613,
646, 761, 804
РС13 196, 197, 223
P4N4C18 250, 613, 659
POCI3 197
PF5 445
РН8 438
PJ3 438
PH4J 243, 711
PN 575
P3N6 575
Р20Б=Р40ю525, 526
P4Oe 525
Полифосфотиды 669
Ра, протактиний
Ра 287
Pb, свинец
Pb 326, 329, 330, 334
PbBr2 436
PbFBr 642
NH4Pb2Br5 256
PbG03 758
Pb(GH8)4 650
Pb(GeH5)4 651, 652
PbG08.PbGh, фосгенит 252, 645, 646
PbCl2 245, 436
PbFGl 250, 437, 642
(NH4)2[PbCle] 754
aPbF2 436
|iPbF2 435
PbJ2 170, 171, 437
PbJ2 (I форма) 436
PbJ2 (II форма) 436
Pb—N 574
aPb(N8)2 680
pPb(N8)2 680
Pb(N08)2 766, 767
PbN02 253
964
УКАЗАТЕЛЬ ФОРМУЛ
Pb—О 524
РЬ02 151, 152, 520
РЬО (тип В 10) 243, 522
РЬО (жёлтый) 524
РЬО (тип В И) 243
Pb—P 574
Rb2 [РЬСЦ] 754
PbS, галенит 524
PbS04 778
Pb—Sb 575
PbSb 562
PbSe 524
PbSnOa 660
PbSnS2, теалит 638
PbTe 524
РЬТЮз 660, 661
PbW04 720
PbZrOa 660, 661
Pd, палладий
Pd 128, 311, 314
PdClo 246, 403, 405
PdCU 405
(NTll4)2[Pda4] 738
[Pd(Nlia)4]Cl2. H20 255, 705
PdF3 402
PdF2 402
PdO 406, 488
Pd—P 561
PdP2 683
Rb2PdBre 737
Rb2PdCle 737
PdS 245, 493
PdSb2 561, 683
Pm, промецин (?) (циблоний иллиний)
Pm 282, 283
Po, полоний
Po 133, 140, 243, 341, 34 \
xPo 345, 348
jJPo 345, 348
Pr, празеодим
i Pr 280, 283
jiPr 281, 283
РгНгз 380
PrC2 688
РгСЬ 380
PrF3 379
PrO2 454
Pr203 455
Рг(011)з 380
PrN 542, 543
PrP 542, 543
PrSb 542, 543
Pt, платина
Pt 311, 314
(РЦСНзЬСНзЬ 654, 655
[Pt(CH3bCl]4 654, 655
PtCh 403
[N(CH3)4]2 PtGle 261
[NH3ClI3|2PtCle 260
L\H4)2[Pt(SCN)6] 738
f\H4hPtCle 737
iPt(NH3)2]Gl4, соль Жерара 706
Pt—О 490
Pt04 201
РЮ8 201
Pt02 201
Pt304 489, 490, 540, 541
PtO 488
PtOs.3H20 201
PtP2 561, 683
Rb2PtCle 737
Rb2[Pt(SCN)e] 738
PtS, куперит 243, 493
PtSb2561, 683
PtSb 562
Tl2PtCle 737
Pu« плутоний
Pu 297
PuBrs 390
РиСЛз 389
PuJ3 390
Pu202S 477
PlPuF8 725
Pu2Ss 477
Pu—Si 587
Полиметаллические комплексы 670
Ra, радий
Ra 274, 276
RaF2 374
ВЪ, рубидий
Rb 272, 273
RbBr 168, 169, 365, 368, 370
RbC8 686
RbCu 686
RbCN 619, 692
a RbCl 366, 369
P RbCl 369
RbCl 91, 151, 168, 169, 365, 368—370
RbC104 (кубическая) 792
RbC104 (ромбическая) 794
RbF 168, 169, 365
RbH 365
RbJ 166, 168, 169, 365, 368, 370
RbJ03 661
RbN3 678
RbN03 763
Rb02 673
Rb203 673
Rb20 448
RbRe04 728
Rb8[Rh(N02)e] 732, 733
Rb2S 448
oRbSH 367
RbSH 365
Rb2S04 782
Rb2[SeCle] 776
a RbSeH 367
RbSeH 365
Rb2[SiFe] 754
Rb2[SnGlel 754
Rb3[TaOe] 715
Rb2lTeCle] 776
Rb2[TiCle] 714
Rb2[ZrCle] 714
Be, рений
Re 298, 299, 302
утзлтнль формул
965
Re-С 591
ReCI5 20, 197
[NIli] R0O1 728
НеОз 2yi7, 423, 480, (ИИ)
Re—p 553
He—Si 591
ReSi-. 591
TIReOi 728
Rh. родии
Rh 128, 308
zRh 310
(JRIi 308, 310
RliCli 398
IRIiINIIbUUI Cl> 250
Rlil's 398, '102
(\4-l4)-(J|Rl»(N()•»)«I 732, 735
Rh()3 201
RliOa 201
Rha08 487
Rh—I» 559
Rli-P Г>59
RhSo i)7/,
TURhtNOa),,! 7:>.2, 7:;:;
Kn, радой
Rn 353—555
Rh, рутении
Ru :H)."i, 507
allii :;o5, :;o7
RuCh 393
RuCl,> 390
RuKe 201
Rii Kb Ж\
RuOi 201, 183
Run..» -'!»:•>
Rul» 554, 557
UnS_, r,74
RuSi»... 074
RuTc. (',74
S. сора
s 311, :\\\\
a S(|)OMuii4iM4,aa) 20, 05, 213, 342, 310,
■i»S (.Moiioi.viiiMH \n) 20, 343, 540,
SM 223, 313, :;'i(i, 517
s, 22:;, з'о;
s.» :,,/i7
SiCI|:{)j 050
(<;«H5bS, 2<;i
s.ci2 ww
SCla 4 43
SCI i 20
SFe 189, 197, 22.;, 113
S!|. 189, 2iO, ul
SO2 5.52
Soa 5;;2
По.И1су.1ьф||Д1.| 009
SS, гурьмя
Sh :•,:;;,
aS|) 337, 510
Sb (аморфным) 510
Sh (г/геклоибранлыи) 3'i-l
SbHi-:i 438
Sbl3r2(CU3)a 201
Sb(Ceii5)a озз
SbCl;, 438
S|)J:, 438--1Ю
Sbj()r> 527
Sb,()„ 525
SbiOio .r)25
Sb.O] -SbSbO, 218, 527
Sbi()e (тип 05.i) 248
Sb-jO.,-. .ShiOti, сеиармпичиг 248, 527
SbiO;,, млсмгпшит 248, 528
SbjOa (тин Ш1) 249
SbiS;,. атидимшт 218, 551
Si-Sb 575
Sn—S»> 571
SnSb .574
Ti— si) 57:;
TISb 572, 57:;
Sb..Tl7 258, 57:;
/ih"-Sl> 570
Zn:lS|)2 570
ZniSb;. 570
Si\ скандии
S:- 278—280
a S,- 278 285
ri So 279, 28:5
Sri-1;} ;;78
ScN 512
S-.'-jO;, 452
S"jSi^()7, тортиоптпт 259 *
So, годен
So llo, 158, 212, 31I, :;i:i, 317
a Sr (rn.rarojri.u.iii.iii) 517, 348
■i I Sr (mdiiok.'ihinibiii) 347, ;>ls
■til Se 517, :;is
s,- (naj)) 348
lNrllil2 ISeClJ 770
Sei-(1 v.;;
Soil. 144
Si'()2 21(i, 552
Si.(>:, 5:;2
ll(».tllce,lt'iNl;U.i 009
: ; 1'/
317 Si, кремний
Si :;20, :;:;:;
Si r,r, 1;;о
SiC 90 135, 222, ;.')(), 001, OR
Sic l ((ил И1) 212. ооз
SiC 11 (тип 1Щ 212, tH)2, 001
SiC III (inii />5) 242, 418, 001, 00'
Sic I V «i.\io|iil)]ii,iii)> 000
SiC V 001
Si (CM;.) i 1)50
Si(C,.|j;>)i 051, 052
(CII.Oi SiO» 201
Si-,CI, 430
Sicii 4:;o
si к i 219, i;w, i:;2, 4:;:;, 417
|(CII;i)iN|-» Sil'\; 200
iNllib Sir'7 755, 801
iNII 1)2 ISIKJ (куПичегкал) У50, T
■Nil ib ISiFj (ромио^дрлчсч:!;.;4/!) 75
805
MJ.1SM4 «01
Si3Ile 430
Si J.j 430
966
УКАЗАТЕЛЬ ФОРМУЛ
Si—N 573
SiOa G15, 806
Si О о, а-кварц низкотемпературный 69,
88—90, 151. 152—154, 167, 244, 510—512
SiOa. p-Kmipu высокотемпературный 75,
510, 5)2
SiOa, а-кристобалпт низкотемпературный
151, 152, 246, 510, 513
SiO-ъ В-крпстобалнт
высокотемпературный, ИЗ, 145, 244, 510, 514, 515
SiG2, з-тридимнт 510
Si02, ji-триднмит 131, 152, 244, 510. 515
S1O2, f"Tl)n^,IMirT "^
Si—P 573
SiP 573
Si[I\>07| 77:;
Si—Та 582
Ti2[SiFe] 75'i
TiSi2 246, 579, 580, 806
[Zn(H20)e] [SiFj 755
Zn2Si04 633
2ZiiO.Si()2-H20-:Zii2(OH)aSi03, гемнмор-
фпт, каламин 65, 259
ZrSis 246, 579, 580
ZrSiOi, циркон 252, 259, 612, 613, 62'i
Sm, самарий
Sm 282, 283
SmBr3 380
SmCh 381 •
SmF3 379
Sh^O» 456
Sn, олово
Sii 175, 176, 326, 333, 334
aSn 26, 326, 329, 330, 333
tfSn 24, 26, 242, 328—330, 333, 334,
Sn(GH3)4 650
Sri(CeH5)4 651, 652
Sn(CeH6CH3)4 652
[М(СН3)зС2Н5]2 SnCl6 260
fN(CH3h (C2H5)21 SnCle 260
[NH3CaHeb SnCle 260
i(CII3)3NHh SnCle 260
KUHabNlI-la SnCI(i 260
Sn(CeH4OCH3)i 652
Sn((:eri40C2H5)-i 65;;
S!i(CO)a 198, 206
SiiCU 196, 197, 430
SnCh 435
(Nll4)2 [ftnClel 754
S11J1 2iS, 430, 433,, 447, 650
Sn -() 524.
Sn() 522, 523
S11O2, касситерит 71, 520
Sn02 151, 152
Sn—P 574
Sn[P207] 773
SnS 244, 323
SnS2 521
SnSr03 661-
Tl*[SnCle) 754
lZn(H20)e] [SnFj 735
Sr, стронций
Sr 274, 276, 277
SrBrft 246, 375
[Sr(H20)e]Br2 699
SrC2 b76 688
SrC03 758
[GOOH(CiHOH)aGOO]2Srf кислый,
виннокислый Sr 61
SrCb 171, 374
[Sr(H20)G]U2 699, 700
SrF2 374
SrH2 246, 375
[Sp(H20)eJj2 699, 700
Sp(N03)a 766, 767
SrO 168, 451
Sr()2 673
[Sr(HaO)eI [02] 250, 700
[Sr(H20)8] (OH)3 250, 700
SrS 451
SrS04 151, 778
SrSe 451
SrTe 451
SrTi03 660, 661
SrU04 723
SpZpOb 6(51
Та, тантал
Та 287
Та—С 583
ТаС 581, 582
аТа2С 582
TaF7=HOH 125
Та—Н 385
ТаН 385
Та2Н 385
Та—О 469
Та—Р 546
Та—S 469
TaS2 469
ТаС—TiC. 583
ТаС—ZrC 583
Tb, тербии
Tb 280, 281, 283
ТЬ20з 456
Тс, технеций
Тс 298, 299, 302
Те, теллур
Те 341, 34:;, 348
ТеВгг 444
ToCh 4'»4
TeCU 19(5--198, 206, 293, 444
[NH4]2 [TeClJ 776
TeFe 443
ТеJ2 444
ToHa 444
Te02 532, 533
Te02, теллурит 246, 533, 534
ТеОз 532
аТе(ОН)в 444
[iTe(OH)e 444
Tl2[TeCle] 776
TiTe3 464
Т1ь торий
Ти 284—286
TI1C2 579, 689
Th02 151, 152, 167, 463
Th—P 544
ThP 544, 545
УКАЗАТЕЛЬ фбр&УЛ
ThsP4 (тип D 73) 249, 544
TluPa (тип Bi) 545
Ti, тятан
Ti 284, 280
aTi 284, 281»
pTi 285, 280
TiBr4 383
TiOU 379, 382
Ti—И 379, IW
TiHi,3 280
TiJ4 383
TijN 100, 170, 224, 543, 54
Ti -0 19, 402, 405, 407
TiO 401
TiOa, рутил 151, 152,
402
TiO a, a на таз 151, 152,
Ti02, брукит 151, l.»2,
TiiiOa 461
Ti—P 544
THP2O7J 773
Ti—S 464, 405
TiS2 464
TiSe-2 404
[Zn(H2U)J |TiFj 713
TI, таллии
TI 322, 325
aTl 323, 325
pTl 322, 325
TIBr 422, 429
a T1C1 309
TICl 309, 422, 429
'П2013 427
TICIO4 (кубическая) 792
T1C104 (ромбическая) 794
TIF 244, 428
a TIJ (низкотемпературный,
p TIJ 429
TIJ 168, 245, 422
TI2O3 505
TISe 245, 506
Tu (Tm), туллий
Tu 280, 281, 283
TuaOa 450
U, уран
U 243, 288, 290, 297
a U (моноклинный) 292, 295
a U (ромбический) 292, 290
р U 288, 296
(Ш Рэидла 293
ТU 293, 290
UBr3 388
U—С 587
UG 587
UC2 587, 088, 689
UCls 388
UCU 387
UF4 387
UFe 385
UO2F2 724
UII3 295, 388
UD8 388, 389
UJ8 388, 389
U—N 548
159, 107, 215, 458,
459, 402
4(H), 402
жёлтый) 429
UN 548
U2N3 548
UNa 548
[UO2 6Н2О] [NChk 708
U—О 475, 470
Ш)з 470
иэ08 470
UaOe 470
U3()7 470
U(>2, Уранинит 475
ПО, 476
НО 470
(I—P 548
UP2 550
U3P4 55L)
LIP 550, 551
UUMhl 773
M--S '*70
OS0f7B 470
U----Si 587
LJ Si 587, 588
U3Si 587, 588
UsSia 587, 588
IJ Si, 587, 588
V, ванадии
V 287
V-C 581, 5K3
VO 100, 5SI
VOl, 20
VN 100, 1.70, 220, 224, 545
VNi.i 19
V—N—О 220
V -О 407
VO 408
VO* 407
VaOs 249, 406, 407
V2O3 467, 408
V—S 409
VS 409
V2Se 19, 469
V—Se 469
VSe 469
VSe2 469
V2Se6 469
V—Si 582
VaSi 295, 388, 582, 583
\V, вольфрам
W 288, 295
a\V 130, 133, 140, 242,
296
p\V 243, 291, 295
VVG 584
aW2G 585
W(CO)e 647, 648
WCle 385
WFe 385
W—N 548
WOa 247, 475
W4O11 475
W802a 475
WO 2 473
W2P 548
W4P 548
ll3P(W3Oio)4-5H20 254
H3PWi304o-29H20 254
WSa 474
968
УКАЗАТЕЛЬ ФОРМУЛ
WSi2 586
Zn(W04) 721
W2Zr 247, 587
X, ксенон
\ :uv-.">55
Y, иттрии
Y 278 -280, 2N:1.
YT9 102, :W>
V2()., 27», 452
Yl*<) i, icc(MjoTn\r 624
VIM)., C,24
Yf4).,.2ll><> 772
YV<h 62 4
Yl>« irrrepoiiii
Yl» 2S|, 2X2,
YI1J2 :'mS2
YWb 45li
Yb-jKo 457
Yb'IV 457
/п, цинк
Zn 128, 318—:'»20
/nlNlls)2Br2 658
rZn(ll>(.))(J |ИЮ31а 257, SIX)
/n(CN)2 619, 620
ZnC,()3 7Г)7
Zii(C0)3 198, 206
Z11W3 112
Zii(NIlJ,)2(,il2 2r»0l l»-'i.,if t>58, 707, HOC»
(NlUbZnCl, ,- (Nll-i)2 iZnChlCl 717,
74 8
Zn(NII:0B(ClO.i)» 790
ZnF2 411
ZnJ2 411
ZnJ7 670
Zn(NH3)oJ2 707
Zn3N2 567
ZnO 168, 499
Zn(()H)2 246, 4П
ZnPa 569, 685
Ziial's (тип />5Г>) Г>69
Ziinl*2 (тип />59) 24S, 5(iS
Z11S 108, 61 <), 7:54
ZiiS, iiypmiT (пюрцит, шортцит) l'>8, 140,
159, ' Mil, 222, 242, 264, 4У9
Zntt, цппкппаи ofi.uimua сфалерит 79, 152,,
Ш, 158, 140, 159, 161, 222, 242, 2U4,
5)0, 540, 541
ZnSo 175, 176
|Xn(IU>)J [ZrFol 71:*., 755
Zr, цирконии
Zr 2S4, 280
aZr 281, 28Г,
jiZr 285, 286
ZrC 579
ZrC» liS»)
Zrcii :ш
ZrlM 585
|ZrF?|=-no]! 125
(Nlbb fZi'K7| 712, 71:;
Zr—Jl 286, 584
ZrN 54:5
Zr—О 465
Zr(b (тип CI) 4(>5, 405
ZrOo, баддо.чсчп 247, 462, 405, 465.
Zr -P 544
Zi'UVbl 226, 25li, 77:$
ZrS2 404
Zrftoa 464
Редакторы: £>. К. BaiimitmeUH и E» Б, Кузнецова^
Техн. редактор С. Я. Ал ламой.
Т-00236. Подписано к печати 22/Ш 1950 г. 60,5 печ. л.+ 5 вклеек. 82,86 уч.-изд. л. 52 500
тип. зн. и печ. л. Тираж 3 000 экз. Цепа книги 49 руб. 70 ьоп. Переплет 3 руб. Заказ № 1659
10-я типография Гланполиграфиздата при Сонете Министров СССР. Москва, Трёхпрудный пер , 9