/

















































































































































































































































































































































































































































Author: Будникова Ю.Г.
Tags: химия физическая химия химическая физика
ISBN: 978-5-16-011761-4
Year: 2016




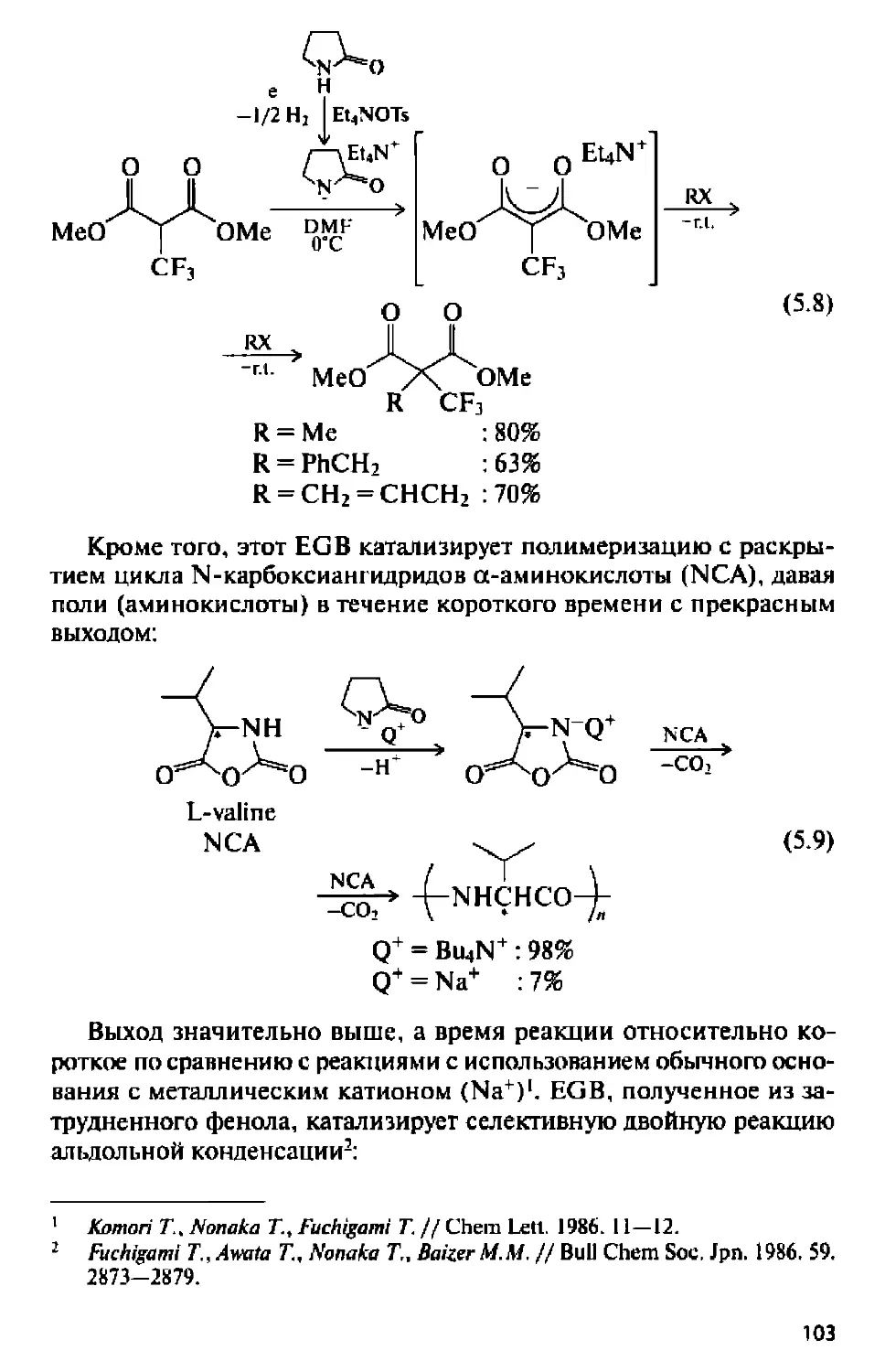
Text
I НАУЧНАЯ МЫСЛЬ!
I СЕРИЯ ОСНОВАНА В 2006 ГОДУ I
Ю.Г. БУДНИКОВА
СОВРЕМЕННЫЙ
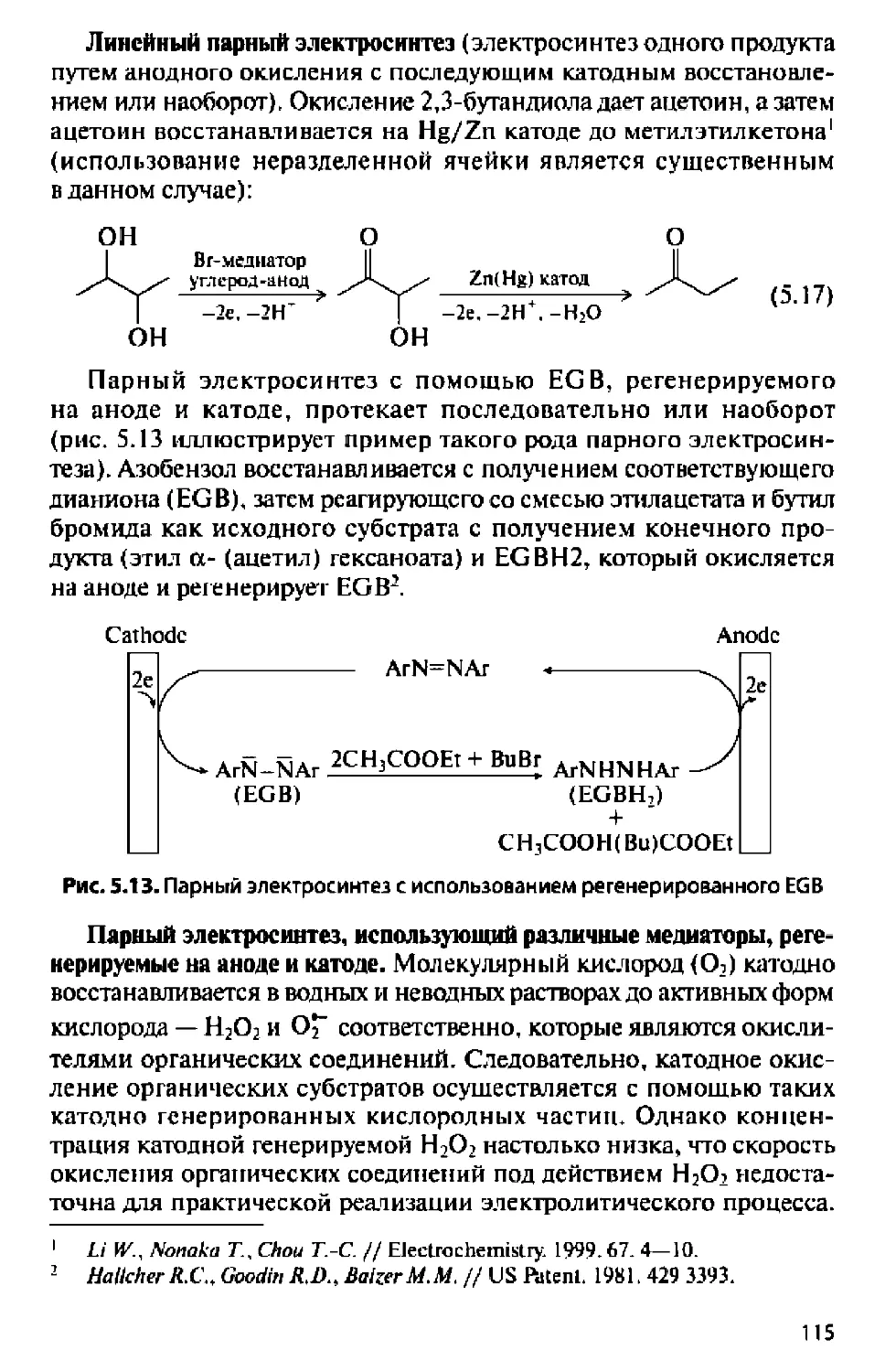
ОРГАНИЧЕСКИЙ
ЭЛЕКТРОСИНТЕЗ
ПРИНЦИПЫ, МЕТОДЫ ИССЛЕДОВАНИЯ
И ПРАКТИЧЕСКИЕ ПРИЛОЖЕНИЯ
МОНОГРАФИЯ
zrianfum.com
Москва
ИНФРА-М
2016
ФЗ
№436-Ф3
Издание не подлежит маркировке
в соответствии с п. 1 ч. 2 ст. 1
УДК 541-138/138.3(075.4)
ББК 24.57
Б90
Подготовка монографии поддержана
грантом Российского научного фонда № 14-23-00016
Автор:
Будникова Ю.Г. — доктор химических наук, заведующая
лабораторией электрохимического синтеза Федерального государственного
бюджетного учреждения науки Института органической и физической
химии им. А.Е. Арбузова Казанского научного центра Российской
академии наук
Будникова ЮТ.
Б90 Современный органический электросинтез. Принципы,
методы исследования и практические приложения : монография. — М. :
ИНФРА-М, 2016. -440с. - (Научнаямысль).
ISBN 978-5-16-011761-4 (print)
ISBN 978-5-16-104171 -0 (online)
В книге приведены систематизированные достижения в
электрохимическом органическом синтезе и смежных с ним областях за последние
десятилетия. Кратко изложены основные принципы электрохимии,
электрохимических измерений и органического электросинтеза, причем
наибольшее внимание уделено новым методам и подходам. Так. значительное
место отведено развитию металлокомллексного катализа в органическом
электросинтезе, методам оценки эффективности гомогенных
электрокатализаторов, ноным области — электролизу с использованием твердых
полимерных электролитов, применению органической электрохимии
в органических электронных устройствах, СI-химии, электрохимической
конверсии биомассы в ценные материалы и т.д.
Издание может представлять интерес дня студентов, аспирантов,
исследователей и инженеров.
УД К 541.138/138.3(075.4)
ББК 24.57
ISBN 97К-5-16-011761-4 (print)
ISBN 978-5-16-104171-0 (online) © Будникова Ю.Г., 2016
Подписано в печать 31,03.2016.
Фор мат 60x90/16. Бумага офсетная. Гарнитура Newton.
Печать цифровая. Усл. псч. л. 27,5,
Тираж 100 экз. Заказ № 00000
ТК 243000-542678-310316
Отпечатано в типографии ООО «Научио-иадательский центр ИНФРА-М»
127282. Москва, ул. Полярная, д. 31В, стр. 1
Тел.: (495) 280-15-96. 280-33-86. Факс: (495) 280-36-29
Предисловие
Мы живем в период интеграции разных областей наук, в
результате которой происходит их взаимное обогащение. Методология
классической синтетической органической химии все в большей
степени развивается за счет новых подходов и принципов,
использования реагентов in situ, новых материалов и технологических
решений. Интеграционные процессы нашли отражение и в
области электрохимии органических соединений (ЭХОС), которая,
хотя и имеет заметную историю, успешно стала проявлять себя
на практике лишь в наше время. Данная книга отражает основные
тенденции в развитии ЭХОС, которые проявились в последние
10—15 лет. Исчерпывающего рассмотрения принципов и
практических сторон ЭХОС здесь нет, так как ставилась совсем другая
задача. Сейчас химия как передовая наука призвана обеспечить
устойчивое развитие общества благодаря использованию различных
методов и подходов, которые должны следовать требованиям зеленой
химии. И здесь ЭХОС нашла свое место, чем и объясняется интерес
к ней со стороны исследователей и инженеров-практиков.
В книге рассмотрены фундаментальные принципы электрохимии
органических молекул, методы исследований, проводимых в
органической электрохимии для электрохимической оценки свойств
органических молекул, электродные реакции разных классов
соединений, медиаторные элекгрокаталитические реакции. Особое
внимание уделено активно развивающейся области металлокомплекс-
ного катализа в органическом электросинтезе, в том числе лиганд-
направленной электрохимической функционализации C(sp2)—H
связей в присутствии соединений палладия и никеля и оценке
гомогенных электрокатализаторов методом циклической вольтампе-
рометрии, в которых использованы наработки, полученные в
собственных исследованиях, поддержанных фангами различных
российских фондов, в том числе Российским фондом фундаментальных
исследований (РФФИ) и Российским научным фондом (РНФ).
В тексте изложена новая методология органического
электрохимического синтеза, описаны электролиз с использованием твердых
полимерных электролитов, электролитические системы с
использованием твердых оснований и кислот, медиаторы на твердой
подложке, двухфазные электролитические системы, показаны метод ка-
тионного пула, электролиз в сверхкритических жидкостях, в ионных
жидкостях, электрохимическое фторирование и фосфор ил ирование,
электрополимеризация, тонкослойные электролизеры,
электрохимические микропоточные системы, электролиз в ультразвуке, а также
з
с использованием специфических электродных материалов, фото-
электролиз и фотокатализ. Также уделено внимание перспективным
областям, связанным с органической электрохимией, применению
в органических электронных устройствах органической
электролюминесценции, фотовольтаическим ячейкам, органическим
транзисторам, электрохромным устройствам, а также некоторым другим
актуальным практическим приложениям.
Таким образом, органическая электрохимия в настоящее время
развивается как интегрирующая область, включающая не только
органический электросинтез, но и химию материалов,
каталитическую химию, биохимию, медицинскую химию и химию окружающей
среды. В нашей повседневной жизни органические и полимерные
материалы играют важную роль в технологических приложениях,
таких как биосенсоры, проводящие полимеры, жидкие кристаллы,
электролюминесцентные материалы, сенсибилизированные
красителем солнечные элементы и т.д.
Чтобы понять эти технологии, нужно изучить основы как
органической химии, так и электрохимии. В XXI в. области интересов
разнообразны, поэтому студенты, а особенно аспиранты, не могут
заниматься разработками передовых технологий, если они не
понимают принципов и подходов различных наук, таких как органическая
химия, неорганическая химия и физико-химия.
Кроме того, органическая электрохимия исследует проблемы
переноса электронов с участием органических молекул и материалов
с использованием электрической энергии. Таким образом,
электрохимический процесс похож на фотоэлектронный перенос, который
является важной областью органической фотохимии, использующей
энергию света. Несмотря на то что были опубликованы
основополагающие книги по органической фотохимии, практически
отсутствуют книги, в которых обсуждались бы основные аспекты
переноса электронов на электродах с участием органических соединений
и приложения в новых областях исследований.
В этой книге представлены успехи органической электрохимии,
изложенные для аспирантов, исследователей и инженеров, показаны
основные принципы электрохимии, электрохимических измерений
и органического электросинтеза, в том числе его новых методологий.
Также описаны в деталях некоторые экспериментальные примеры
органического электросинтеза.
Введение
Электрохимия органических соединений (ЭХОС) — область
науки, находящаяся на стыке электрохимии и органической химии.
Она изучает превращение органических веществ в растворах под
действием электрического тока.
Принципы электрохимии широко используют в органическом
синтезе. Органическая электрохимия включает активацию
органических молекул путем переноса электронов от поверхности
электрода или на нее. При этом можно получать разнообразные
новые продукты, изучать редокс-свойства веществ и рсализовывать
разнообразные превращения, в которых окисление или
восстановление являются ключевыми стадиями.
Важный аспект электрохимической методологии заключается
в том, что многие электрохимические процессы отвечают
требованиям экологической чистоты. Действительно, электрохимические
реакции имеют ряд преимуществ: мягкие условия протекания,
высокие скорости, селективность процесса, а также удобный
операционный контроль с использованием таких параметров, как плотность
тока и потенциал. Контроль электрохимических процессов легко
автоматизировать. Электрохимические методы можно рекомендовать
для превентивной защиты окружающей среды, поскольку не
требуются специальные реагенты.
Органическая электрохимия, в частности органический
электросинтез, развивались путем объединения с новыми способами
проведения органических реакций и органическим синтезом.
Наступивший XXI в. иногда называют веком экологии, а органический
электросинтез является заманчивым в плане требований зеленой
химии в рамках концепции устойчивого развития, поскольку он
не требует каких-либо опасных реагентов и производит меньше
отходов, чем любой другой химический синтез.
В последние годы электрохимические процессы зарекомендовали
себя как путь к малоотходному, малотоннажному, высокочистому
производству значимых соединений. Их преимущества — нетермнче-
ская активация и чистые (минимум реагентов) условия; а электрон,
как известно, относится к реагентам, не загрязняющим окружающую
среду. На рисунке показана схема прямой взаимосвязи
органического электросинтеза и требований (критериями) зеленой химии:
а) использование в электросинтезе «зеленых» растворителей
ионных жидкостей или микроэмульсий;
б) использование медиаторов-каталитизаторов при сокращении
потребления энергии и уменьшении количества химических отходов;
5
яльк'ичклшты
Прямой, непрямой
it парный Алек фолм
I
Медиаторы
Аминокислоты
Сахара
Лигнин
Комнатная
температура
Парный электролиз
Мелиаторы
Растпорителн:
*
Электролиз! it 1
I
)лсктрогенери-
руемыс реагенты
Медиаторы
ЭЛЕКТРОСИНТЕЗ
Рисунок. Схема прямой взаимосвязи органического электросинтеза
с требованиями (крунитериями) зеленой химии (Green Chem. 2010. No 12.
P. 2099-2119)
6
в) возможность выполнения прямого, непрямого или парного
электролиза, что может существенно улучшить атом-экономию;
г) использование возобновляемых исходных материалов, что
полностью соответствует методологии зеленой химии;
д) энергический выигрыш: если используют процессы или
условия, которые позволяют экономить электроэнергию (парный
электросинтез, медиаторы-катализаторы, комнатная температура);
е) мониторинг электролиза в реальном времени, который можно
легко проводить благодаря использованию методов
электроаналитического контроля;
ж) повышение безопасности и снижение вероятности аварий
за счет генерирования или регенерирования in situ реакционных
частиц или опасных токсичных реагентов;
з) избежание отходов, если реагенты генерируются в
электрохимической ячейке стехиометрически.
Недавно новый парный электросинтез фталида и п-трет-бутил
бензальдегида был разрабоган и реализован в промышленных
масштабах фирмой BASF (Германия). Немецкие ученые считают
электросинтез наиболее перспективным «зеленым» синтетическим
процессом. Эти факты побудили специалистов по органической
электрохимии, а также химиков-органиков приложить усилия для
разработки новых систем органического электросинтеза для
практического достижения «зеленых* химических процессов.
Действительно, на сегодняшний день, как показано в этой кните,
разработано много новых успешных «зеленых» органических
электролитических систем.
Концепция органической электрохимии является относительно
новой, хотя она имеет давнюю историю. В 1800 г. итальянский физик
Вольта изобрел так называемый вольтов столб. Начало изучению
соотношений и связи между химическими и электрическими
явлениями, исследованию действия гальванического тока на
различные вещества в растворах было положено работой английских
ученых В. Никольсона и А. Карлейля, которая вызвала огромный
интерес, гак как в ней нетрудно было увидеть принципиально новый
метод изучения химических явлении. Поэтому уже вскоре многие
ученые в различных странах начали конструировать вольтовы
батареи различной емкости и изучать химическое действие
гальванического электричества и причины его образования в вольтовом
столбе. Исследователи Крюнкшанк, Бруньятели, Лампадиус, Вол-
ластон, Петров, Гротгус, Риттер, Гильберт, Симон, Эрман. Био и др.
изучали действие гальванического тока. В России об изобретении
вольтова столба стало известно через несколько месяцев после его
демонстрации Никольсоном и Карлейлем в Англии. Уже в октябре
1800 г. Петербургская академия наук получила от русского послан-
7
ника в Гааге Д.А. Голицына письмо с подробным описанием нового
источника тока. В сентябре 1801 г, академик А.А. Мусин-Пушкин
впервые продемонстрировал вольтов столб и его действие перед
Конференцией Академии паук. В 1802 г. русский физико-химик
В.В. Петров построил батарею — горизонтальный столб, состоявший
из 4200 медных и цинковых кружков. Батарея В.В. Петрова в свое
время была одним из самых мощных источников тока в мире.
Результаты своих многочисленных опытов В,В, Петров изложил в 1803 г.
в книге «Известие о гальвани-вольтовских опытах», изданной в
Пете рбурге.
В 1803 г. В. В. Петров опубликовал статью по электролизу спиртов
и алифатических масел. Год спустя ученый Гроттхусс из Литвы,
который предложил ионный механизм электролиза, обнаружил, что
разбавленный раствор индиго белого может быть легко
электрохимически окислен до индито синего. В 1833 г. Фарадей открыл закон,
названный законом Фарадей, а год спустя он обнаружил, что
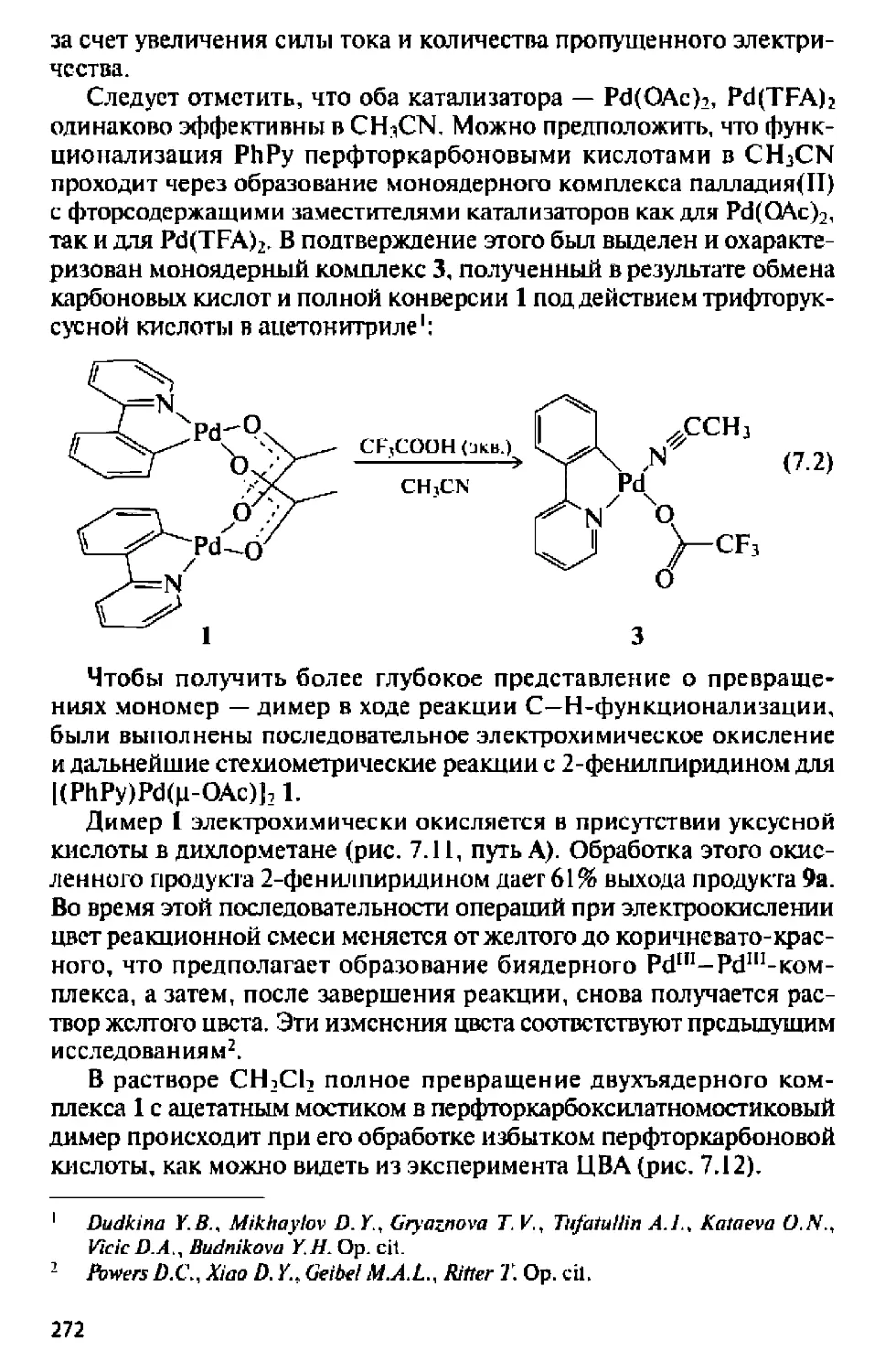
углеводороды можно образовать путем электролиза водного раствора соли
уксусной кислоты.
К сожалению, он не смог идентифицировать продукты. В 1849 г.
ученик Велера — Кольбе открыл электрохимическое окисление
карбоновой кислоты (RCOOH) в димерный алкан (R— R) и С02,
известное как электролиз Кольбе'. Следовательно, Фарадей и Кольбе
являются пионерами в исследовании органических
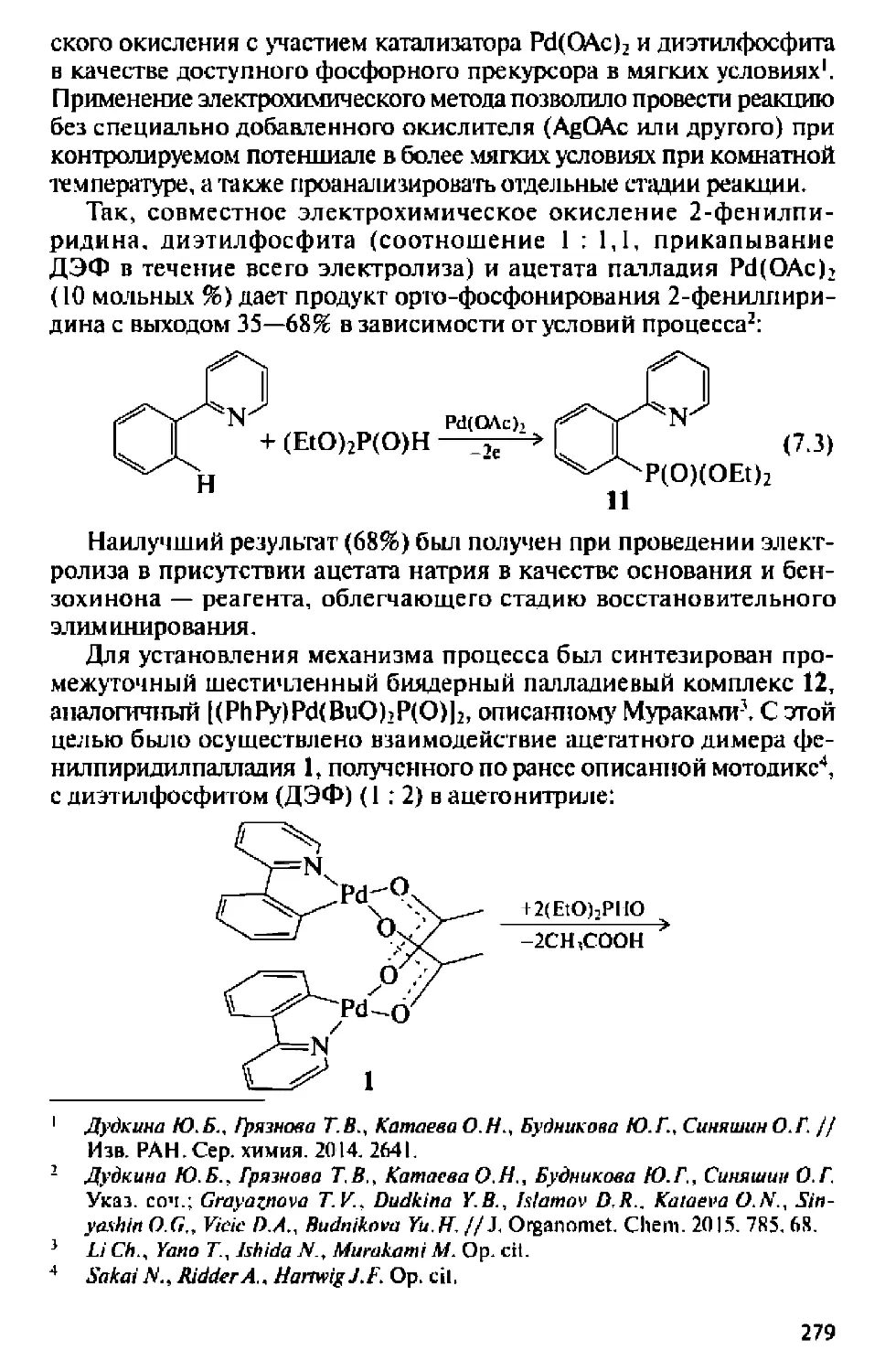
электрохимических процессов. С конца XIX в. до начала XX в. электрохимические
процессы окисления и восстаноштсиия различных соединений
изучались довольно интенсивно. Таким образом, применение электролиза
для получения органических соединений продолжилось и в первой
половине XX в. Типичным примером является электрохимический
процесс восстановления нитробензола в анилин. Важно отметить,
что органическая электрохимия развивалась вместе с новыми
электроаналитическими методами, такими как полярография, которая
была разработана Я. Гейровским, а затем и Тачи в начале 1920-х гг.2
Однако исследования органического электросинтеза были
полностью прекращены во время Второй мировой войны. В 1964 г.
М. Байзер разработал способ электрохимической гидродимеризации
акрилонитрила, который является весьма полезным промышленным
способом получения адипонитрила. Это изобретение вновь
стимулировало исследования в области органического электросинтеза
многими электрохимиками и химиками-органиками.
1 Vljih А.К., Conway B.E. Electrode Kinetic Aspects of the Kolbc Reaction // Chcm.
Rev. 1967. No 67. P. 623-664.
2 Zuman P. Hevrvvskf J, Japan, and Organic Polarographv // The Chem, Records.
2012. No 12. P. 46-62.
8
С тех пор развитие органической электрохимии, в частности
органического электросинтеза, ознаменовалось появлением новых видов
органических реакций и современных органических синтезов. Кроме
того, были разработаны реакции с различными апрогонными
полярными органическими растворителями, что позволило обнаружить
электрогенерированные неустойчивые интермедиа™. Кроме того,
циклическая вольтамперометрия и связанные с ней
электроаналитические методы помогли понять кинетику и механизмы многих
органических электродных процессов.
Точкой отсчета в истории органической электрохимии в России
можно считать публикацию А.М. Бутлерова в Chem. Вег.1 по
электролизу раствора валерьяновой кислоты. Это подчеркивает роль
Казани, где работал этот выдающийся ученый, в становлении
новой области науки. Среди других специалистов, внесших
заметный вклад в развитие и становление ЭХОС в Казани, можно
выделить Ю.П. Китаева, Ю.М. Каргина, В.Н. Никулина и
некоторых других.
В конце 1950-х гг. обозначился интерес к изучению превращений
органических веществ в растворах с применением полярографии
(Ю.П. Китаев с сотрудниками, ИОФХ им. А.Е. Арбузова). Затем
интенсивные исследования по ЭХОС с использованием полярографии
и вольтамперометрии на различных электродах стали проводиться
в Казанском университете (Ю.М. Каргин с сотрудниками). В те же
годы В.Н. Никулин с сотрудниками (Казанский
химико-технологический институт (КХТИ)) изучал превращения органических
веществ на электродах-монокристаллах с различными
кристаллографическими характеристиками. Аббревиатура ЭХОС принадлежит
Ю.М. Картину, она появилась впервые при подготовке и проведении
совещания в Казани в 1971 г. После этого Казань была признана
ведущим центром в области ЭХОС в нашей стране и за рубежом.
Аналитические аспекты ЭХОС развивали В.М. Гороховский, В.И.
Гороховская и Г. К. Будников. Развитию ЭХОС в Казани способствовали
научные контакты с зарубежными учеными до время стажировок
(Ю.П. Китаев, ГК. Будников, Ю.М. Каргин, ЮТ. Будникова,
В.В. Жуиков), а также общие усилия электрохимиков, химиков
и физиков-специалистов в области электронного паромагнитного
резонанса (ЭПР)2.
Основные достижения казанских исследователей по ЭХОС
связаны с выявлением роли отдельных стадий в механизме электродных
1 ButkrowA.M. Elektrolyse der \faleransaure (correspondenzen) // Chem, Вег 1870.
Bd. 3. S. 95-96.
2 Карги» Ю.М., Будников Г. К. Очерки истории электрохимииорганических
соединений н Казани. Казань, 2006.
9
реакций в различных средах, включая химические реакции, стадии
элиминирования, протежирования, каталитические процессы, ме-
диаторные процессы с применением переносчиков электрона. При
их изучении удачным явилось использование метода электронного
парамагнитного резонанса (А. В. Ильясов, М.К. Кадиров), что
позволило обнаружить промежуточное образование частиц
радикальной природы.
Объектами исследования в плане ЭХОС являлись многие классы
органических соединений, содержащие электрохимически активные
группировки (нуклеофильные и электрофильные функциональные
группы): альдегиды, кетоны, нитросоеди нения (прежде всего
ароматические).
Синтетический аспект — важная составляющая ЭХОС в
Казани и основа ее успехов: результаты исследований по нитрованию,
электросинтезу фосфорорганических соединений, хлорированию
высших олефинов...
В настоящее время исследования по ЭХОС в Казани в основном
проводятся в ИОФХ им. А.Е. Арбузова, где в последние годы
разработаны новые эффективные методы синтеза фосфорорганических
соединений (ФОС) в элекфокаталитических условиях, развито новое
научное направление — изучение процессов электрохимической
активации и трансформации элементного фосфора под действием
органических и металлоорганических соединений, созданы научные
основы высокоэффективной, ресурсосберегающей и экологически
безопасной технологии электросинтеза важнейших классов ФОС —
триалкил-, триарил, триамидо- и диал кил фосфатов, третичных
фосфинов и фосфониевых солей из белого фосфора в условиях ме-
таллокомплексного катализа, разработаны электрокаталитические
процессы с участием переходных металлов, выяснены их
закономерности, установлены факторы, определяющие реакционную
способность катализаторов, созданы новые эффективные методы синтеза
разнообразных соединений со связями Р-Е (Е = С, О, N, Si),
разработаны новые подходы к исследованию механизма раскрытия
тетраэдра белого фосфора в условиях металлокомплексного катализа,
детектирования интемедиатов и прогнозирования выбора наиболее
эффективных реагентов методами электрохимии (Ю.Г. Будникова,
Ю.М. Каргин, О. Г. Синяшин).
Работа казанских электрохимиков (ИОФХ им. А.Е. Арбузова)
«Новые высокоэффективные экологически чистые способы
получения фосфорорганических соединений из белого фосфора» была
удостоена бронзовой медали и диплома на V Московском
международном салоне инноваций и инвестиций 2005 года, серебряной
медали и диплома на международном салоне «Архимед-2005», премии
республиканского конкурса «Лучшее изобретение 2005 года», премии
ю
конкурса «50 лучших инновационных идей Республики Татарстан —
2005». Работа «Разработка высокоэффективной технологии галоге-
нирования высших альфа-олефинов» удостоена премии конкурсов
«50 лучших инновационных идей Республики Татарстан — 2006»
и «Идея-1000».
Работа «Направленный синтез и разработка высокоэффективных
технологий получения фосфорорганических соединений на основе
элементного фосфора» (О. Г. Синяшин, Ю.Г. Будникова, Э.С. Ба-
тыева, Б.Е. Иванов) в 2007 г. была удостоена Государственной
премии Республики Татарстан в области науки и техники, в том
числе за электрохимические исследования в этой области.
Авторами впервые в мире разработан концептуально новый комплексный
подход к активации элементного фосфора, основанный на
применении каталитических сред и реагентов, а также введении белого
фосфора в координационную сферу переходного металла с
последующим (или предшествующим) электрохимическим переносом
электрона. Это открыло нетрадиционные пути к формированию связей
фосфор — углерод, фосфор — азот, фосфор — водород, фосфор —
металл, фосфор — кислород и др. и позволило осуществлять прямое
введение атома фосфора в разнообразные доступные органические
соединения, такие как спирты, амины, органогалогениды, олефины
и др. В результате оказалось возможным легкое, наиболее простое
и атом-экономное получение ценных, ранее не известных или
труднодоступных фосфорорганических продуктов, в том числе фосфинов
и неорганических кислот фосфора и их производных, органических
производных кислот фосфора, функционалилированных
фосфорорганических олигомеров и полимеров и др. Продемонстрированы
богатые потенциальные возможности интенсивно развиваемого
направления электрохимии элементного фосфора и преимущества
электрохимических методов синтеза фосфорорганических
соединений, получение которых обычными химическими способами или
невозможно, или экспериментально сложно, или имеет
экологические ограничения. В результате проведенных исследований
заложен фундамент реализации разработанных процессов получения
фосфорорганических соединений из элементного фосфора на
технологическом уровне.
Интересные работы проводятся в группе В. В. Янилкина —
исследование электрохимических реакций индатизинов на пути создания
систем молекулярного распознавания, изучение термодинамики
электродных реакций наноразмерных супрамолекулярных систем
на основе калике [4] аренов и комплексов металлов, электропере-
ключаемого связывания комплексов металлов п-сульфонато (тиа)
калике [4| аренами, а также ионов металлов бииндолизиновыми ге-
тсроциклофанами и их ациклическими аналогами.
11
Идеи и результаты исследований казанских электрохимиков в
области электрохимии органических и элементоорганических
соединений, элементного фосфора являются базой для создания новых
крупных направлений, объединяющих работы нескольких групп
ученых в различных странах, прежде всего в США, Франции,
Германии, Италии.
Органическая электрохимия в последние годы развивалась как
интегрирующая область, включающая не только органический
электросинтез, но и химию материалов, каталитическую химию,
биохимию, медицинскую химию и химию окружающей среды1.
Можно предположить, что передовые разработки в органической
электрохимии будут достигнуты путем интеграции с другими
областями науки, как уже упоминалось выше.
Fuchigami T.,Atobe Л/., Jnagi S. Fundamenials ... далее зарезано.
Глава 1. НЕСКОЛЬКО ОБЩИХ ВОПРОСОВ
ОРГАНИЧЕСКОЙ ЭЛЕКТРОХИМИИ
Хотя имеется множество опубликованных трудов по
органической электрохимии, рассмотрение некоторых фундаментальных
сторон протекания реакций на электродах необходимо. Химики
часто сталкиваются с ситуациями, когда реакция не протекает
при нужной скорости в изначально выбранных условиях. В химии
энергия активации определяется как минимальная энергия,
необходимая, чтобы начать химическую реакцию, а следовательно,
энергия активации должна быть введена в химическую систему
для того, чтобы химическая реакция произошла. Чтобы уменьшить
энергию активации, часто используются катализаторы, однако для
протекания реакции с заметной скоростью обычно требуется
высокая температура. Электрохимические реакции в этом отношении
в общем случае могут быть осуществлены в мягких условиях (при
комнатной температуре и нормальном давлении). В
электрохимических реакциях существует дополнительный экспериментальный
параметр — электродный потенциал, который участвует в
управлении скоростями этих реакций. Скорости переноса электрона
можно легко варьировать на много порядков при одной температуре
путем точного контроля электродного потенциала. Действительно,
потенциал электрода является настолько определяющим параметром
для регулирования скорости электрохимических реакций, что
большинство реакций можно осуществить при комнатной или близкой
к ней температуре.
Понимание характера зависимости скорости переноса электрона
от потенциала имеет важное значение для понимания электродных
процессов и составляет центральную тему данной главы. Поскольку
перенос электрона на поверхность электрода является неизбежно
гетерогенным процессом, необходимо кратко рассмотреть структуру
раздела электрод — раствор и ее влияние на ход электрохимической
реакции. Недостаточно, однако, просто вывести соотношение между
скоростью переноса электронов и электродным потенциалом. Это
объясняется тем, что происходит резкое изменение этих скоростей
в зависимости от потенциала. Установлено, что при определенных
величинах потенциала перенос электронов происходит настолько
быстро, что весь процесс фактически ограничивается массопе-
реносом субстрата из объема раствора на поверхность электрода.
Существуют различные способы массоперетюса, они отличаются
по эффективности, поэтому необходимо рассмотреть каждое из этих
воздействий.
13
1.1. ОБРАЗОВАНИЕ ДВОЙНОГО ЭЛЕКТРИЧЕСКОГО СЛОЯ
Когда электроды поляризуются в растворе электролита, очень
важен заряд, который поддерживается на электродах. Для того чтобы
нейтрализовать дисбаланс заряда на границе раздела электрод —
раствор, перегруппировка заряженных частиц, таких как ионы в
растворе вблизи поверхности электрода, будет происходить в течение
нескольких сотых долей секунды и наконец приведет к сильным
взаимодействиям между ионами в растворе и поверхностью
электрода. В результате появится двойной электрический слой,
толщина которого, как правило, составляет от 1 до 10 им (рис. 1.1)[.
nm порядок
+
nm порядок
+
+
+
+
+
+
+
Анод
О
о
\э
о
о
в
J39
е
©
Нет градиента потенциала
в объеме раствора
■у/—? //
\
Кривая потенциала
— Приложенный
— потенциал
Катод
Ч£? — катионная часть электролита
© — анионная часть электролита
Рис. 1.1. Электрическая модель двойного слоя и распределение потенциала
в двойном слое
Существует градиент потенциала в двойном электрическом слое.
Разность потенциалов между поверхностью электрода и объемом
раствора, показанная на рис. 1.1, может составить 1 В и более. Для
довольно небольшой толщины двойного слоя это чрезвычайно
крутой градиент — порядка 106 В • см-1 или более, что является
электрическим полем значительной интенсивности.
Это является движущей силой для электрохимической реакции
на границе раздела электрод — раствор, поэтому когда поляризация
между анодом и катодом постепенно увеличивается, градиент
потенциала в непосредственной близости от анода и катода также
увеличивается, а следовательно, наиболее окисляемые и
восстанавливаемые частицы в системе подвергаются реакции переноса
1 Bard A.J.. FaulknerL.R. Electrochemical Methods, Fundamentals and
Applications. 2nd edn. N. Y.: John WDey & Sons. Inc., 2001. Ch. 13.
14
электрона на аноде и катоде соответственно. Поскольку после
реакции переноса электрона происходит дисбаланс заряда в
непосредственной близости от электрода, ионы переносятся к границе
раздела электрод — раствор, чтобы нейтрализовать дисбаланс, а значит,
наблюдается постоянный фарадеевский ток. Таким образом,
электролит в растворе важен для формирования двойного электрического
слоя и нейтрализации дисбаланса заряда после электролиза.
1.2. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОТЕНЦИАЛЫ
Во всех электрохимических экспериментах интересующие
реакции происходят на поверхности рабочего электрода, поэтому
существует интерес в контроле падения потенциала на границе
раздела между поверхностью рабочего электрода и раствором. Но
невозможно контролировать или измерять этот граничный потенциал
без помещения другого электрода в раствор. Таким образом, нужно
рассматривать два межфазных потенциала, ни один из которых
не может быть измерен независимо. Таким образом, одно из
требований к противоположному электроду — его поверхностный
потенциал должен оставаться постоянным, чтобы любые изменения
в напряжении на ячейке приводили к идентичным изменениям
потенциала рабочего электрода на границе раздела фаз.
Электрод, потенциал которого не меняется с изменением тока,
относится к типу идеального неполяризуемого электрода, но не
существует электродов, которые ведут себя таким образом.
Следовательно, поверхностный потенциал противоположного электрода
в двухэлектродной системе, описанной выше, варьируется при
прохождении тока через ячейку. Эта проблема преодолевается с
помощью системы из трех электродов, в которой функции
противоположного электрода разделены между электродом сравнения и
вспомогательным электродом (рис. 1.2)'. Это гарантирует, что потенциал
между рабочим электродом и электродом сравнения контролируется,
а ток проходит между рабочим и вспомогательным электродами. Ток,
протекающий через электрод сравнения, дополнительно
уменьшается при использовании импедансного операционного усилителя
высокого сигнала на входе электрода сравнения.
При использовании грехэлектродной системы можно
контролировать или измерять потенциал рабочего электрода. Рассмотрим,
в чем заключается контроль потенциала рабочего электрода с
помощью следующей простой редокс-пары:
Red^±Ox+/«r (|.|)
где Red и Ох— восстановленная и окисленная формы частиц.
BardAJ., Faulkner LR, Op. cit. Ch. 2.
15
Потенциал контролируется между рабочим электродом
и электродом сращения
Потсишюстат
U1
IJJMOI
г01
Потенциал
ко нтрол и русте я
между электродами
рабочим
и ердннения
Венрмо] 1С.1Ы1Ы11
эле
Электрод рабочий
сравнении злектрол
Электролитически!! раствор
Рис. 1.2. Схема экспериментальной установки для системы из трех электродов
Полезно обратить внимание на энергию электронов в рабочем
металлическом электроде и восстановленных частиц Red в
растворе электролита, как показано на рис. 1.3. Поведение электронов
в металле электрода можно частично понять, рассматривая уровень
Ферми (£/)'. Металлы состоят из плотно упакованных атомов,
которые имеют сильное перекрывание орбиталей. Фрагмент металла,
следовательно, не обладает индивидуальными четко определенными
уровнями энергии электронов, которые можно было бы обнаружить
в отдельном атоме того же материала.
Вместо этого существует континуум уровней с доступными
электронами, заполняющими состояния снизу вверх. Уровень Ферми
соответствует энергии верхней занятой орбитали (ВЗМО). Этот уровень
не является фиксированным и может перемещаться при наложении
электрической энергии (рис. 1.3). Поэтому можно изменить энергию
уровня Ферми путем наложения потенциала на электрод (когда
прикладывается отрицательный потенциал, уровень Ферми сдвигается
к более высокой энергии, а когда прикладывается положительный
потенциал, он смещается к более низкой энергии).
В зависимости от положения уровня Ферми вероятно
термодинамически можно восстановить/окислить частицы в растворе.
На рис. 1.3 показан уровень Ферми в металле наряду с орбитальными
энергиями (ВЗМО и нижней связывающей молекулярной орбитали
(НСМО)) молекулы (Red) в растворе.
1 Compton R.G.. SandersG.H.W. Electrode Potentials. Oxford: Oxford University
Press, 1996. Ch. I.
16
Наложение положительных, потенциален
Незаполненные
электронные
_ состояния ^
5
§ Уровень
х Ферми <£/т)
с
С
+ ^ J
Заполненные
электронные
состояния
а б в
Рис. 13. Уровень Ферми в металле наряду с орбитальными энергиями
{ВЗМО и НСМО) молекулы (Red) в растворе
Как показано на рис. L3, я, уровень Ферми имеет большее
значение, чем ВЗМО восстановленной формы Red. Поэтому для
электронов термодинамически невыгодно перепрыгивать с ВЗМО
на электрод. Тем не менее, как показано на рис. 1.3, в, когда
уровень Ферми ниже ВЗМО Red, перенос электрона термодинамически
выгоден, он происходит, и можно наблюдать ток окисления Red.
Критический потенциал, при котором происходит процесс переноса
электронов, определяет стандартный потенциал £"0 окислительно-
восстановительной пары Red/Ox (рис. 1.3, б).
1.3. ЭНЕРГИЯ АКТИВАЦИИ И ПЕРЕНАПРЯЖЕНИЕ
Как уже упоминалось в разд. 1.2, в зависимости от
относительного положения уровня Ферми к орбитальным энергиям (ВЗМО
и НСМО) молекулы субстрата в растворе становится
термодинамически возможным восстановить/окислить молекулу. Тем
не менее в целом электрохимические реакции обладают
энергетическими барьерами, которые необходимо преодолеть
реагирующим частицам. Этот энергетический барьер называется энергией
активации (рис. 1.4). Таким образом, для получения тока обычно
требуется разность потенциалов выше равновесного значения
(стандартного потенциала £°). Разность потенциалов между
стандартным потенциалом и потенциалом, при котором редокс-про-
цесс осуществляют экспериментально, называют
перенапряжением1.
huisu К. Electrochemistry in Nonaqueous Solutions. Weinheim: Wiley-VCH Verlag
GmbH, 2009. Ch. 5.
17
Незаполненные
электронные
состояния """
Уровень
Ферми (Ef)
Заполненные
электронные
состояния
Наложение положительных потенциалов
Энергия активации
НСМО
ВЗМО
'Г
Рис. 1.4. Энергия активации в процессе переноса электрона на электроде
1.4. ТОКИ, УПРАВЛЯЕМЫЕ ПЕРЕНОСОМ ЭЛЕКТРОНА
И МАССОПЕРЕНОСОМ
Хотя потенциал электрода является чрезвычайно важным
параметром для экспериментального управления скоростью
электрохимических реакций, другие параметры, такие как массоперенос, также
могут влиять на эту скорость. Рассмотрим теперь простейшую
электрохимическую модель, которая состоит из процесса переноса
электронов и процесса массопереноса, как показано на рис. 1.5. В этом
случае в зависимости от потенциала электрода лимитирующей
стадией может быть либо скорость переноса электронов, либо скорость
массопереноса субстрата к поверхности электрода. Для изучения
количественных и полуколичественных взаимосвязей между
потенциалом, скоростями электрохимических реакций и массопереноса
обычно используют различные варианты вольтамперометрии1.
Электрод Процесс
массопереноса
Red ■■—j\r\r\r\r\s\r\r Red
Процесс
переноса
электрона
Ox ir<j\i\jy/\nj\r—* Ох
Рис. 1.5. Электрохимическая модель, показывающая процесс переноса
электронов и процесс массопереноса
FryAJ. Synthetic Organic Electrochemistry. N. Y.: John Wiley & Sons, 1989. Ch. 2.
18
Теперь рассмотрим факторы, влияющие на относительные высоты
и формы вольтамперометрограмм, такие, как показано на рис. 1.6.
При сканировании потенциала электрода во время записи вольтампе-
рограммы (к более отрицательным потенциатам при восстановлении
или к более положительным потенциалам при окислении) скорости
переноса электронов резко возрастают и вольтамперомстрическая
кривая проходит через смешанную область, в которой скорости мас-
сопереноса и переноса электронов ограничены током электролиза.
.
I В перемешивающемся растворе (6)
f Ограниченный массопереносом
В неподвижном
растворе {а)
Потенциал
Рис. 1.6. Вольтамперометрические волны в неподвижном
и перемешивающемся растворе
Наконец скорость переноса электронов в конечном итоге
достигает достаточно высокого значения, так что токи
лимитируются только массопереносом, а вольтамперометрические токи пика
и плато, ограниченные массопереносом, наблюдаются
соответственно в неподвижном и перемешиваемом растворе. По этой
причине уравнения, относящиеся к току электролиза, различаются для
каждой скорость определяющей стадии.
Когда электрохимическая реакция контра!ируется стадией
переноса электрона, плотность чистого тока электролиза (/)
представляется уравнением Батлера-Фольмера1:
i = i0-ic=in[exp{anF^RT)-exp[-{l-a)nFy]/RT}], (1.2)
где /„ и /V — индивидуальные анодными и катодными плотностями
тока; /'о — плотность тока обмена; а — коэффициент переноса
заряда (его значение находится между 0 и 1, часто приблизительно
0,5 — при более низких перенапряжениях); п — число электронов,
участвующих в электродной реакции; F— постоянная Фарадея; г) —
перенапряжение.
1 Izutsu К. Electrochemistry in Nonaqueous Solutions. Wfeinheim: Wiley-VCH Vfcrlag
GmbH, 2009. Ch. 5.
19
Как указывает уравнение Батлера-Фолъмера, фактическая
плотность тока представляет собой разницу между катодной и анодной
плотностью тока. Кроме того, плотность тока обмена — это ток в
отсутствие фактического электролиза и при нулевом перенапряжении.
Это уравнение описывает, как плотность тока (/) на электроде
зависит от перенапряжения (г|), считая, что и катодная и анодная
реакции происходят на одном и том же электроде.
Когда электрохимическая реакция контролируется стадией мас-
сопереноса, плотность тока электролиза связана с величиной
градиента концентрации молекулы субстрата на поверхности электрода,
представленной формулой1
i = nFDidc/dx)^,
(1.3)
где п — число электронов, участвующих в электродной реакции; F—
постоянная Фарадея; D — коэффициент диффузии (константа
скорости движения вещества через данную среду путем диффузии); С —
концентрация молекул субстрата; х — расстояние от поверхности
электрод, а следовательно, (dc/dx) x = 0 представляет собой градиент
концентрации молекулы субстрата у поверхности электрода.
Концентрационные профили для субстрата и продукта в
неподвижном растворе показаны на рис. 1.7. Поскольку толщина
диффузионного слоя, а значит, градиент концентрации изменяются
при протекании электролиза в неподвижном растворе, уменьшение
тока электролиза наблюдается на вольтамперограмме, как показано
на рис. 1.6, кривая а.
Объемная
концентрация
т
Д иффузионны й
слой
Восстановленные
ости мы
Окисленные
частицы
Расстояние
от поверхности
электрода
Диффузионный
слой
Диффузионный
слой
Рис. 1.7. Изменение толщины диффузионного слоя со временем электролиза
При наличии конвекции, например, при перемешивании,
изменение диффузионного слоя со временем подавляется, и градиент
концентрации остается постоянным. В этом случае будет наблю-
' Rifi M.R., Covitz F.H, Introduction to Organic Electrochemistry. N. Y; Marcel
Dekker, 1974. Ch. 2.
20
даться ограничение плотности тока (/rf) от толщины диффузионного
слоя (5)':
id = nFDc/b. (1.4)
Уравнение 1.4 описывает плотность тока на плато вольтам-
перограммы, измеренной в перемешиваемом растворе (рис. 1.6,
кривая 6) при условии, что потенциал быстро сканируется, так что
концентрация субстрата в объеме раствора незначительно
исчерпывается в течение времени, необходимого для измерения вольтам-
нерограммы. Кроме того, поскольку 5 уменьшается за счет более
эффективного перемешивания, предельные плотности тока в
перемешиваемом растворе увеличиваются со скоростью перемешивания.
Они, конечно, намного больше, чем плотности тока в неподвижном
растворе, поскольку конвекция гораздо более эффективна, чем
диффузия при доставке молекулы субстрата к поверхности электрода.
i
FryAJ. Synthetic Organic Elecirochemistiy. N. Y.: John Wiley & Sons, 1989. Ch. 2.
Глава 2. ПРИНЦИПЫ ОРГАНИЧЕСКОЙ
ЭЛЕКТРОХИМИИ. НЕКОТОРЫЕ
ФУНДАМЕНТАЛЬНЫЕ ВОПРОСЫ
ЭЛЕКТРОХИМИИ ОРГАНИЧЕСКИХ МОЛЕКУЛ
К одной из задач настоящей книги относится попытка объяснить,
как знание механизма органических электрохимических реакций
позволяет управлять их протеканием и помогает наиболее эффективно
достигать целей. К настоящему времени накоплен большой опыт
получения ряда конкретных электрохимических параметров. Они дают
необходимую информацию о мехатгизмах органических
электрохимических реакций. Среди методов ее получения вольтамперометрия
является одной из наиболее популярных, поскольку она обеспечивает
достаточно полную картину механизма электродного процесса, чтобы
использовать этот процесс обоснованно в практических целях.
В этом разделе описывается, как вольтамлерометрические методы
можно применить для получения механистической информации
об органических электрохимических реакциях. Поскольку успех
или неудача при проведении вольтамлерометрических измерений
в значительной степени зависит от правильного выбора рабочих
условий и соответствующих экспериментальных аксессуаров, таких
как электрохимические ячейки, электроды, растворители и
электролиты, ниже обсуждаются некоторые проблемные вопросы деталей
эксперимента применительно к вольтамперометрии. Кроме того,
в этом разделе показано, как вольтамперометрия может
использоваться для получения информации о механизме протекания новой
электродной реакции с участием органического соединения.
2.1. РАБОЧИЕ ЭЛЕКТРОДЫ
Вольтамперометрия — это группа электрохимических методов,
в которых ток измеряется как отклик потенциала рабочего электрода.
Опыты, как правило, проводятся с помощью системы из трех
электродов, в которой потенциал между электродами рабочим и
сравнения контролируется, а ток протекает между рабочим и
вспомогательным электродами1. Поскольку измеряемая электрохимическая
реакция происходит на рабочем электроде, выбор материала
рабочего электрода имеет решающее значение для экспериментального
успеха применения вольтамперометрии.
1 Bard A.J., Faulkner L.R, Electrochemical Methods, Fundamentals and
Applications. 2nd edn. N. Y.: John WUey & Sons Inc., 2UU1. Ch. 2.
22
Электролиты могут быть использованы без заметной деградации
только а предельных диапазонах электрического потенциала. Это
окно потенциалов должно быть как можно более широким, чтобы
обеспечить в наибольшей степени характеризацию изучаемых
образцов1. Верхние и нижние пределы потенциалов определяются
раствором не только электролита, но и материалом электрода.
В водных электролитах реакции выделения кислорода и водорода
ограничивают окно потенциалов, а следовательно, окно, как
правило, уже, чем в неводных электролитах.
Чтобы преодолеть эту проблему, в качестве рабочих электродных
материалов для вольтамперометрии в водных электролитах, как
правило, используются катодные материалы с высоким
перенапряжением водорода и анодные материалы с высоким
перенапряжением кислорода. Платина и золото — хорошие материалы анода
из-за их высокого перенапряжения кислорода, в то время как ртуть,
цинк и свинец являются хорошими кандидатами для использования
в катодных материалах из-за их высокого перенапряжения водорода.
Токсичность ртути привела к ее ограниченному использованию.
А неводные электролиты стабильны и их окна потенциалов, как
правило, больше, чем водных электролитов, поэтому существуют
некоторые ограничения в выборе материалов рабочего электрода.
Чаще всего используемыми рабочими материалами электродов для
вольтамперометрии в неводных электролитах являются платина,
золото и углерод. Хотя почти все драгоценные металлы могут быть
использованы в качестве как анодного, так и катодного материала,
неблагородные металлы непригодны для анодных материалов
из-за их растворения в условиях анодной поляризации.
Используются различные формы (например, диски, проволоки,
тарелки) и размеры (например, электродные пластины — несколько
квадратных сантиметров, диаметр диска — от нескольких
микрометров до нескольких сантиметров) твердых электродов для воль-
тамперометрических измерений (рис. 2Л). Твердые электроды,
используемые для волыамперометрических измерений, чаще всего
изготавливают путем инкапсуляции электродного материала в
непроводящую оболочку из стекла или инертного полимерного
материала, такого как тефлон, Кель-Ф (полихлоротрифтороэтилен)
или РЕЕК (полиэфиркетон). Чаще всего открытая часть материала
электрода имеет форму диска (рис. 2.1, а и 6). Обычные коммерчески
доступные диаметры диска — от 1 мкм до 1 см. Металлические
электроды в виде пластин обычно соединены с проводом, а часть провода
заключена в непроводящую оболочку из стекла или инертного
полимерного материала (рис. 2.1, в).
1 Izutsu К. Electrochemistry in Nonaqueous Solutions. Wfeinheim: Wiley-VCH Vcrlag
GmbH, 2009. Ch. 4.
23
Контактная
проволока
(медная и т.п.)
Стеклянная
* или тефлоновая
трубка
Эпоксидная
смола для
закрепления
электрода-стержня
^Проводящая
серебряная паста
t~~~ Металлический
или графитовый
прут{1 цм - I см
в диаметре)
/
— Контактная
проволока
(медная и т.п.)
Стеклянная
или тефлоновая
трубка
v
Тер моусад очная
у1 трубка
Проводящая
серебряная паста
Металлический
или графитовый
прут(1 цм - 1 см
в диаметре)
- Контактная
проволока
(медная и т.п.)
Стеклянная
f или тефлоновая
трубка
JL--7 Точечная сварка
^ ^-Платиновая
:\4 проволока
• Металлическая
или стекло-
углеродная
пластинка
[ -
Рис 2.1. Примеры рабочих электродов
В идеале рабочий электрод должен вести себя одинаково каждый
раз, когда он используется. Факторы, которые влияют на
электрохимическое поведение поверхности электрода, — это его чистота,
вид и степень химической функциональности {в том числе оксидов,
которые присутствуют на поверхности металла), а также
микроструктура самого материала электрода. Как правило, стадия или
несколько стадий предварительной обработки проводятся перед
каждым экспериментом, чтобы гарантировать, что состояние
поверхности электрода может быть воспроизведено от эксперимента
к эксперименту. Эти стадии могут быть простыми (механическая
полировка), а могут включать предварительное сканирование
через определенный диапазон потенциалов или воздействие
растворителем либо химическими веществами, чтобы активировать
электрод.
Конкретные процедуры предварительной обработки различных
электродных материалов могут быть найдены в указанных работах
ученых1.
BardA J.t Faulkner LR, Op. cii. Ch. 2; Izutsu К. Op. cit. Ch. 4.
24
2.2. ЭЛЕКТРОДЫ СРАВНЕНИЯ
Потенциал электрода сравнения должен быть стабильным
и воспроизводимым. Метод измерения электродных потенциалов
в водных растворах был отработан надежно1. Стандартный
водородный электрод <НВЭ) является первичным электродом сравнения,
а его потенциал определяется как нулевой при всех температурах.
В практических измерениях используют электроды сравнения, с
которыми легко обращаться. Самые популярные из них — это нас. к. э.
(НКЭ) (рис. 2.2, й) и хлорсеребряный (Ag/AgCl) электрод (рис. 2.2, б).
В отличие от водных систем в неводных растворах метод измерения
электродного потенциала не был отработан столь надежно. Наиболее
серьезной проблемой является электрод сравнения, поскольку нет
первичного электрода сравнения, такого как НВЭ для неводных
электролитов, и электрода сравнения такого же надежного, как водный
Ag/AgCl электрод. Постепенно эта проблема находит свое решение.
Электродный
конта£>1едная
проволока
Г Стеклянная
трубка
Электродный
контактСере6рянная
^ проволока
Электродный
контакт
~А К солевому
-|— мостику
*|— Насыщенный
водный раствор
I KC1
(3^Hg+H&2CI2
vH#-Hg
Г Стеклянная
трубка
~г\ К солевому
■ мостику
X
W
-Ag проволока,
покрытая AgCl
Насыщенный
водный раствор
КС! содержащий
AgCl (нас.)
1 -Стеклянная
/
трубка
-Серсбряпная
проволока
0,1 М Bu4NC]04
+ 0J MAgNOj
/MeCN
Спечённое
стекло
Рис 2.2. Примеры электродов сравнения:
а—насыщенный каломельный электрод; б—Ад/АдО электрод е — Ад/Ад" электрод
Izutsu К. Op. cil. Ch. 6,
25
Электроды сравнения, используемые в неводных системах, могут
быть классифицированы на два типа. Один сил представляет собой
водный электрод сравнения, как правило, водный Ag/AgCl электрод
или НКЭ.
Однако водный электрод сравнения не должен быть погружен
непосредственно в неводный изучаемый раствор, поскольку
раствор загрязняется водой и электролитом (обычно КО). Чтобы
предотвратить это, электрод должен быть помещен в отдельный
отсек и должен использовать солевой мостик для ион-проводящей
связи между отсеками рабочего электрода и электрода сравнения
(рис. 2.2, в). Кончик солевого мостика, который заполняется
исследуемым неводным электролитом, погружают в неводный раствор.
Когда используются такие водные электроды сравнения, следует
учитывать контактную разность потенциалов между водным и
неводным растворами. Чтобы избежать несоответствий, Комиссия
по электрохимии IUPAC предложила измерять в той же системе
потенциал пары Fc^ /FC, а потенциал электрода приводить
относительно кажущегося стандартного потенциала этой системы.
В другом методе тот же растворитель, что и в исследуемом растворе,
используют в качестве внутреннего растворителя электрода
сравнения. Ag/Ag* электрод сравнения является самым популярным для
неводных растворов, поэтому его можно использовать в различных
растворителях (рис. 2.2, в),
2.3. ВСПОМОГАТЕЛЬНЫЕ ЭЛЕКТРОДЫ
Вспомогательный электрод (рис. 2.3), часто называемый также
противоположным электродом, является электродом, используемым
в трехэлектродной электрохимической ячейке для вольтампероме-
трии. в которой электрический ток протекает между рабочим и
вспомогательным электродами. Вспомогательный электрод поэтому
действует как катод, когда рабочий электрод работает в качестве анода,
и наоборот. Вспомогательный электрод часто имеет площадь
поверхности гораздо большую, чем у рабочего
^^ электрода, чтобы быть уверенным, что полу-
Wr реакция, протекающая на вспомогательном
4^ электроде, может происходить достаточно
быстро, чтобы не ограничивать процесс
на рабочем электроде1. Платина (например,
Рис. 2.3. Р* проволока, Pt пластинка) из-за ее высокой
Вспомогательные стабильности является хорошим материалом
электроды фирмы BAS1 для вспомогательного электрода.
Bard A J., Faulkner L.R. Op. cil. Ch. 1.
26
2.4. РАСТВОРИТЕЛИ И ФОНОВЫЕ ЭЛЕКТРОЛИТЫ
К важным характеристикам растворителей в вольтампероме-
трии относятся так называемое окно потенциалов, растворимость
изучаемых молекул субстрата и донорные или солъватарующие
физико-химические свойства'. В качестве растворителя для вольтампе-
рометрии часто используется водная среда, а многие органические
субстраты являются нерастворимыми в воде или только умеренно
растворимыми. Кроме того, реакции выделения кислорода и
водорода ограничивают окно потенциалов, когда вода используется
в качестве растворителя для вольтамперометрии. Таким образом,
йольтамперные эксперименты с органическими субстратами, как
правило, проводят в полярных органических растворителях, в
которых фоновый электролит может диссоциировать на ионы. Аде-
тонитрил (диэлектрическая проницаемость в = 38) является одним
из наиболее часто используемых растворителей, так как его высокие
верхние и нижние пределы потенциалов позволяют использовать его
в качестве растворителя для электрохимической реакции как
окисления, так и восстановления. Другие часто используемые
растворители для электрохимического окисления — это дихлорметан (е = 9),
нитрометан (е = 37), пропиленкарбонат (е = 64) и 1,2-диметоксиэтан
(е = 3). Другие часто используемые растворители для
электрохимического восстановления — это N, N- диметилформамид (ДМФА;
с = 37), диметилсульфоксид (ДМСО; е = 47), тетрагидрофуран (ТГФ;
г = 7) и бешонитрил (е = 26). Хотя гексаметилфосфорамид (НМРА;
£ = 30) также используется в качестве растворителя для
электрохимического восстановления, обращаться с ним следует с крайней
осторожностью из-за его токсичности.
Фоновый электролит, используемый в вольтамперометрии,
должен удовлетворять следующим условиям: 1) растворяться в
исследуемом растворителе и диссоциировать на ионы, чтобы создать
достаточную проводимость в растворе; 2) быть устойчивым к
окислению и восстановлению и обеспечивать широкое окно
потенциалов; 3) не оказывать неблагоприятного воздействия на
измеряемую электродную реакцию. Кроме того, следует принять во
внимание взаимодействие между диссоциированными ионами
и интермедиатами, образующимися в результате электродных
реакций. Для органических растворителей обычно используемыми
электролитами являются соли тетраалкил аммония. Итак, ионы те-
траэтиламмония (Et^N') и тетрабутиламмония (Bu^N4) часто
используются в качестве катионной части соли тетраал килам мония,
в то время как перхлорат-ион (СЮ4), тозилат-ион (TsO~), тетра-
lzutsu К. Op, cit. Ch. l.
27
фторборат-ион (BF4 ) и гексафторфосфат-ион (PF6) часто
используются в качестве анионной части. Поскольку галогенид-ион (Х~)
может окисляться с образованием иона галония <Х+), необходимо
быть осторожным при его использовании в качестве анионной части
фонового электролита. При этом для водных систем неорганические
соли, такие как NaCI и KCI, хотя они и не сани тетраалкиламмония,
могут быть использованы в качестве фоновых электролитов.
На практике концентрация фонового электролита предпочтительно
должна быть выше 0,1 М.
2.5. ЯЧЕЙКИ И ИСТОЧНИКИ ПИТАНИЯ
При планировании вольтамперометрических экспериментов
и проведении электросинтеза необходимо принять решение: ис-
пользоватьли неразделенную ячейку, в которой рабочий,
вспомогательный и сравнения электроды погружены в один отсек (рис. 2.4, а),
или разделенную, в которой рабочий и вспомогательные электроды
находятся в отдельных отсеках (рис. 2.4, б)1.
Причем при принятии этого решения надо учитывать возможное
влияние продуктов электролиза на вспомогательном электроде на
результаты вольтамперометрических измерений. Если оно возможно,
то следует использовать разделенную ячейку. Когда водные
электроды сравнения, такие как Ag/AgCl или НКЭ, используются в
неводных системах, электрод сравнения располагается в отдельном
отсеке, чтобы предотвратить загрязнение водой (рис. 2.4, е), как
указано в разд. 2.2. В этом случае солевой мостик используется
для ион-проводящей связи между отсеками и рабочим электродом
и электродом сравнения, а кончик солевого мостика погружают
в неводный раствор. Кроме того, кончик должен быть расположен
как можно ближе к рабочему электроду, чтобы свести к минимуму
омическое падение потенциала между кончиком солевого мостика
и электродом, т.е. /'/?.
Потенциостат используется как источник питания, а его
характеристики имеют большое значение при проведении
вольтамперометрических экспериментов с трехэлектродными системами2.
Кроме того, генератор импульсов необходим для нестационарных
измерений, например, в условиях циклической вольтамперометрии
(ДВА). Прибор для ДВА, состоящий из генератора импульсов и по-
тенциостата, входящих в одну установку, изготавливает ряд
производителей. Полученные вольтамперограммы выводятся на X— К-двух-
координатный самописец или ПК.
1 FiyAJ. Synthetic Organic Electrochemistry. N. Y: John Wiley & Sons, 1989. Ch. 10.
2 Ibid.
2B
Электрод
г- сравнения
Газоподводяшая r i
трубка Х_ 1
Прогшюэл ектрод
Выход газа—^. I
с
Электрод
Газолодводящая сравнения
Рабочий „ трубка \^ \ Рабочие
электрод Противо-
электрод Выход
газа
=~Ч \ электрод
Стеклянный
фильтр
Газоподводяшая "абочии Солевой
трубка _ электрод
мостик
Противоэлектрод Выходе?
газа
Лу
пни капилляр
Электрод
сравнения
V
Стеклянный
фильтр
йУ/s = скорость развертки (В/с)
ДК>
i Time (s) f
Функция возбуждения (форма волны
потенциала) для LSV эксперимента
Рис. 2 А Примеры и схемы конструкций вольтамперометрических ячеек
29
2.6. ПОЛЯРИЗАЦИОННЫЕ КРИВЫЕ СТАЦИОНАРНОГО
И НЕСТАЦИОНАРНОГО СОСТОЯНИЙ
Зависимость плотности тока от потенциала электрода в
соответствии с набором постоянных рабочих условий, известная как
поляризационная кривая, я&чяется стандартной электрохимической
характеристикой электродной реакции'.
Стационарная поляризационная кривая описывает
взаимосвязь между потенциалом электрода и плотностью тока, которая
регистрируется при наложении электродного потенциала и записи
постоянного тока электролиза. При интенсивном перемешивании
стабильный отклик тока можно также получить путем измерения
потенциала сканирования, причем в этом случае диффузионный
предельный ток зависит от скорости сканирования потенциала.
Вольтамперограмма, полученная в данном случае, соответствует
кривой в условиях так называемой стационарной поляризации.
Кривая нестационарной поляризации может быть получена при
высокой скорости сканирования потенциала в отсутствие какой-либо
конвекции. Однако кривая стационарной поляризации может быть
получена даже с помошью медленного сканирования потенциала,
поскольку влияние тепловой конвекции в этом случае нельзя
проигнорировать.
Чтобы оценить предельную плотность тока, обычно измеряются
кривая стационарной поляризации и коэффициент диффузии (£),
толщина диффузионного слоя (8) или число электронов,
участвующих в электродной реакции (ет), которые могут быть получены
из величины измеренной плотности тока и уравнения (1.4).
Чтобы получить информацию о механизме органических
электрохимических реакций, обычно измеряются кривые нестационарной
поляризации —вольтамперограммы с циклической и линейной
разверткой. ЦВА — метод, чаше всего используемый в органической
электрохимии, поэтому ниже рассмотрим его более подробно.
В ЦВА потенциал стационарного электрода изменяется линейно
в сторону более отрицательного или положительного направления,
пока не произойдет интересующий электродный процесс — либо
восстановление, либо окисление, затем же направление
сканирования потенциала изменяется в обратную сторону. Этот способ
измерений способен предоставить полезную информацию об
окислительно-восстановительном потенциале исследуемых органических
молекул и относительных скоростях переноса электрона, массопере-
носе и каких-либо химических реакциях, происходящих на
поверхности или в реакционном слое у электрода.
1 Lund И., Hammerich О. (eds) Organic Electrochemistry. 4lh edn. N. Y: Marcel
DekkeOOOl.Ch. 1.
30
Рассмотрим простую ЦВА окислительно-восстановительной
реакции ферроцена (Fc) (рис. 2.5). Редокс-реакция ферроцена
представлена уравнением
Fe" ■ • Fe"'
^> С ^
(2.1)
Fc Fc+
Уравнение Рэндлса-Шевчика (при 25°С):
ip = (2,69 х 105)лзд/Ш1'2СУ/2,
(vs. Ag/AgC1)
£рс = +0,48 V, Ера = +0,42 V,
A£=60mV, £1/2 = 0,450 V,
где ip — ток в пике; и — число электронов; А
С — концентрация; v — скорость развертки.
площадь электрона;
—tive
Е{У)
+live
AV/s = scan rate (Y/s)
Fur\iird Rirven>e
£j кап sciin E/
Гч^ j^I
: : ^4» i
Time (s)
/
0,8 0,7 0,6 0,5 0,4 0,3 0.2 0,1
Pbtential (V) vs. Ag/AgCI
Рис. 2.5. ЦВА 1 мМ раствора ферроцена (Pt диск диаметром 2 мм; 0,1 М Bu4NBF6
в ацетонитриле; Pt проволока; Ag/AgCI ЗМ водный КО)
Поскольку Fc является восстановленным состоянием в этой ре-
докс-реакниит потенциал должен быть развернут в первую очередь
в положительном направлении (анодное сканирование). В этом случае
потенциал развертки следует начинать с начального потенциала, где
нет тока (остаточный потенциал). Кроме того, скорость
сканирования, как правило, устанавливают в диапазоне от 50 до 200 мВ • с '.
Как упоминалось в разд. 2.2, при изменении потенциала электрода
энергия уровня Ферми также изменяется направленно. Когда потен-
31
циал разворачивается в положительном направлении по отношению
остаточному потенциалу (процесс а—Ь на рис. 2.6; уровень Ферми
смещается к более низкой энергии), скорость переноса электронов
от ВЗМО ферроцена на Ферми-уровень электрода резко возрастает
и потенциал проходит через смешанную область, в которой обе
скорости массопереноса и переноса электронов ограничивают ток.
Окисление
О
& I
:Г *7Z
ь^О О ВЗМО уровень
I
ферроцена(Fc)
а
Ъ>
I
^оо
с?
~=* О
Восстановление
Рис. 2.6. ЦВА ферроцене (Fc) и уровень Ферми (£» в металлическом электроде
вместе с уровнем ВЗМО Fc в растворе
Когда скорость переноса электронов является достаточно
высокой, токи ограничиваются массопереносом и наблюдается
вольтамперометрический пик, ограниченный массопереносом
(рис. 2.6, 6). Когда достигается конечный потенциал (рис. 2.6, в),
направление сканирования потенциала меняется на
противоположное (процесс в—г на рис. 2.6). Так как уровень Ферми
перемещается к наиболее высокой энергии при этом обратном сканировании,
должен происходить перенос электронов от уровня Ферми на ВЗМО
иона ферроцения (Fc") (в этом случае наблюдается ток
восстановления Fc+). Когда большая часть Fc+ в непосредственной близости
от поверхности электрода превращается в Fc, ток восстановления
уменьшается (процесс г— а на рис. 2.6).
Циклическая вольтамограмма окислительно-восстановительной
реакции Fc по форме представляет собой типичный обратимый про-
32
цесс. Ток в пике окисления (ip) обратимого процесса, такого как ре-
докс-реакция ферроцена, можно представить в виде:
/J = 0,4463«Fcrt^2(/ifv//? Л'/3, (2.2)
где п — число электронов, участвующих в электродной реакции (1 —
в случае окислительно-восстановительной реакции ферроцена); c*Fc
и Drc — объемная концентрация и коэффициент диффузии
ферроцена; v — скорость сканирования потенциала. Таким образом, для
обратимой ЦВА-волны ток пика пропорционален обьемной
концентрации субстрата и квадратному корню из скорости сканирования
потенциала.
Однако обратимая окислительно-восстановительная реакция
является достаточной редкостью в органической электрохимии, так как
многие органические электрохимические реакции включают быструю
химическую реакцию, следующую за переносом электронов
(ЕС-процесс: электродная стадия Е и химическая стадия С). В этом случае
пики ре-окисления или ре-восстановления могут полностью
исчезнуть при сканировании потенциала в противоположную сторону.
ЦВА является широко используемой методологией получения
значений энергии ВЗМО- и НСМО-уровней для оценки
характеристик органических фотоэлементов и редокс-свойств органических
веществ. Для окислительно-восстановительных пар,
характеризующихся быстрым переносом электрона, которые, как правило,
называют нернстовскими, или обратимыми, парами, предполагая, что два
состояния окисления (Ox/Red) являются стабильными в масштабе
времени эксперимента, максимальные (пиковые) токи в этих двух
связанных пиках должны быть примерно одинаковыми* Кроме того,
разность потенциалов между катодным и анодным пиком IF^— E^
должна составлять 57 мВ/л (л — число электронов, переносимых
в процессе). Среднее значение из двух потенциалов пиков
определяет потенциал полуволны {Е1/2), который берут в качестве
отличного приближения формального потенциала (£°'), соответствующего
окислителт>но-восстановителт>ной ларе, так как коэффициенты
диффузии окисленной (D0) и восстановленной (DR) частиц должны быть
примерно равны1:
Е[/2 = 1/2(F^+ Е„) = £°' + (RT/nF)ln(DR/DQWl
Формальный потенциал отражает относительную стабильность
Ох и Red, на которые, в спою очередь, шгиятот эффекты среды и
сольватации. Если мы предположим, что DR = D0, то £|/2 = ЕУ.
1 Bard A. J.. Faulkner L.R. Electrochemical Methods: Fundamentals and
Applications. N. Y.: Wiley, 2001.
33
Первые исследования, которые проводились для органических
соединений, показали корреляцию между энергией, необходимой
для присоединения одного электрона к молекуле, стандартным
потенциалом восстановления и энергией НСМО1. Энергия НСМО
может быть аппроксимирована как сродство к электрону, которое
является энергией, необходимой для присоединения электрона
к атому или молекуле в газовой фазе. Аналогично корреляции были
обнаружены для органических молекул между стандартными
потенциалами окисления и потенциалами ионизации2. Последние
связаны с энергией ВЗМО, поскольку она связана с минимальной
энергией, необходимой для удаления электрона из атома или
молекулы, находящихся в газовой фазе. Есть несколько значений шкал
энергии Ферми, которые коррелировали с формальным потенциалом
электрода сравнения НВЭ. Предполагалось, что энергии
молекулярных орбиталей можно вычислить непосредственно из
электрохимических результатов, калиброванных по ферроцену со значением
4,8 эВ, как показано ниже:
^номо = — (Щ\/2,и* vs. Ft+/Ft] + 4,8) (eV);
4имо = -(£ [i/j.mi v, Ft+/ft] + 4,8) (eV)J.
2.7. ПОТЕНЦИАЛЫ В ЭЛЕКТРОХИМИЧЕСКИХ ИЗМЕРЕНИЯХ
В статьях и книгах, посвященных органическим электродным
реакциям в понятийном плане, описано несколько видов
потенциалов4. Следующие шесть видов потенциалов часто используются,
а значит, они и будут обсуждаться:
1 Ramsay W.. Foster R. // Nature. 1949. No 163. P. 178-179.
2 Ballard R.E. // Chera. Plivs. Lett. 1976. No 42. P. 97-98: МШег L.L.. Nord-
blom G.D., Mayeda EJL //1 Org. Chem. 1972. No 37. P. 916-918; Neikam W.C>,
DimeterG.R., Desmond MM. //J. Electrochem. Soc. 1964. No 111. P. 1190— 1192.
1 DadvandA., Cicoira F., Chemichenko K.Yu., Balenkova E. S., Osuna R. M., Rosei F.,
Nenajdenko V.G., Perepkhka D.F. // Chem. Commun. 2008. 5354—5356; Car-
dona СМ., U W., KaiferA.E., Sittckdale D., Bazan G.C. // Adv. Mater. 2011,
No 23. P. 2367-2371; Liang K. Xu Z, Xia J., Tsai S. Г., Wu Y, UG., Ray C,
Yu L. И Adv. Mater. 2010. No 22. E135 - El 38; Huo L.J., HouJ.H., Zhang S.Q.,
Chen H.Y., Yang Y// Angew. Chem. Int. Ed. 2010. No49. R 1500—1503; Hsiang-
YuC, Jianhui H., ShaoqingZ,, Yongye L., Guanwen Y.r Yang K, Luping K, Yue W.,
Gang L, // Nat. Photon. 2009. No 3. P. 649-653; Hang Y„ Feng D., Wu Y,
TsaiS.-T, UG., Ray C, YuL.// J. Am. Chem, Soc. 2009. No 131. P. 7792-7799;
Liang Y, Wu Г., Feng D., TsaiS.-T, Son H.-J., LiG., Yu L. //J.Am. Chem. Soc.
2009. No 131. P. 56-57; и т.д.
4 Fry A.J. Synthetic Organic Electrochemistry, N. Y,; John Wiley & Sons, 1989;
Campion R.G.. BanbC. Understanding Nfoltammetry. Singapore: \Мэгк$ Scientific
Publishing, 2000.
34
1 > стандартный электродный потенциал;
2) формальный потенциал;
3) потенциал пика (пиковый);
4) потенциал полупика;
5) потенциал полуволны;
6) потенциал разложения (начальный потенциал).
Если известно изменение стандартной свободной энергии AG0 для
химического процесса, то можно вычислить стандартный электродный
потенциал Еа для электрохимической реакции, основанной на этом
процессе, используя соотношения между Д6,с и £°. Таким образом, £°
не экспериментальное значение, а расчетная величина. Поэтому, даже
если электродный потенциал измеряется с помощью НВЭ в
эквивалентном состоянии, измеренное значение будет незначительно
отличаться от реальной величины £°. Значение, измеряемое с помощью
НВЭ в эквивалентном состоянии, называется формальным
потенциалом (£°). Отношение между £° и £° представлено в уравнении
^=£° + ЛГ1пУо^ (2,3)
nF yR
где уо и yR — коэффициенты активности для окисленных и
восстановленных форм изучаемых частиц.
Однако отклонение £° от £° очень мало в разбавленном растворе
и, как правило, даже в концентрированном растворе меньше 300 мВ.
Потенциал на пике ЦВА-кривых называется потенциалом пика.
Для обратимой редокс-реакции потенциал анодного пика (Ел) и
потенциал катодного пика (£Л.) не зависят от скорости сканирования
и концентрации, поэтому потенциалы пиков предоставляют
информацию о природе данного вида субстрата и термодинамическом
показателе для окисления/восстановления изучаемых частиц* Кроме
того, среднее значение анодного и катодного потенциалов пиков
соответствует формальному потенциалу восстановления (£° ) для
обратимой пары (рис, 2.7).
Red<=»Gx+we~ (2.4)
Ох—£-»/>, (2.5)
При этом, когда относительно быстрая химическая реакция,
следующая за переносом электрона, участвует в электродном процессе,
как показано в уравнениях (2.4) и (2.5) (ЕС-процесс), пиковый
потенциал сдвигается в зависимости от величины ее константы
скорости (к), а величина обратного пика при обратном сканирования
должна уменьшиться. В этом случае пиковые потенциалы не могут
быть использованы в качестве термодинамического индекса для
окисления/восстановления исследуемых частиц.
35
Ток ,A£gi
Потенциал
Рис. 2.7. Циклическая вольтамперограммз для обратимого процесса
Вышеописанное обсуждение касается вольтамперограмм,
полученных при фиксированной скорости сканирования потенциала.
Однако, если скорость сканирования изменяется, вероятно, будет
наблюдаться изменение форм вольтамперограмм. Например, когда
ЦВА регистрируются со значительно более высокой скоростью
сканирования (более 1 В/с), обратимый отклик на ЦВА можно
наблюдать во времени, необходимом для записи вольтамперограммьг.
Если скорость сканирования потенциала достаточно велика, то,
возможно, Ох, образованный в соответствии с уравнением (2.4),
не успевает химически прореагировать во время записи вольтамперо-
граммы, т.е. электролиза. В этом случае можно оценить
термодинамический потенциал, например, формальный потенциал полученной
обратимой вольтамперограммы.
Хотя потенциал полупика (Ер/2) часто смешивают с потенциалом
полуволны (Е]/2), он определяется как потенциал, где ток составляет
половину от максимального тока пика на ЦВА. Кроме того,
взаимосвязь между Ер и ЕрП представлена формулой
^-£,/2 = <М>57/л, (2.6)
где п — число электронов, участвующих в
окислительно-восстановительной электродной реакции.
Заметим, что Epf2 не относится к основным термодинамическим
параметрам, а значит, он используется нечасто.
Потенциал полуволны {Е\/т) — это потенциал, при котором ток
волны в стационарной вольтамперомстрии равен половине
диффузионного предельного тока (рис. 2.8, а). Е{/2 равен формальному по-
36
тенциалу исследуемой электродной реакции, когда реакция является
обратимым окиаштельно-воссгановительным процессом и
коэффициенты диффузии окисленной и восстановленной форм изучаемых
частиц равны между собой. Однако, если процесс включает
химическую реакцию, следующую за переносом электронов, £i/2 больше
не равна формальному потенциалу (рис. 2.6, б).
п t\
Величина сдвига
зависит от константы
скорости последующей
химической реакции
_к j „ | ^"i t i „
Ei/2 E E|/2 ^1/2 £
Сдвиг потенциала полуволны
q — обратимый б — редокс процесс, включающий
редокс процесс химическую реакциию, следующую
за переносом электрона
Рис. 2.8. Стационарные вольтамперограммы простой редокс-системы без (а)
и с (б) последующей химической реакцией. Две сигмоидальные кривые
в случае (б) отображают сдвиг потенциала полуволны, который зависит
от константы скорости химической реакции
Потенциал разложения (Е^) является потенциалом, при котором
на волътамперограмме начинает наблюдаться фарадеевский ток» также
он называется начальным потенциалом (iw,). Хотя Е^. часто
используется как термодинамический индекс для окисления/восстаноатения
исследуемых частиц, это не совсем термодинамический параметр.
2.8. ИСПОЛЬЗОВАНИЕ ВОЛЬТАМПЕРОМЕТРИИ ДЛЯ ИЗУЧЕНИЯ
ЭЛЕКТРООРГАНИЧЕСКОГО СИНТЕЗА
2.8.1. Метод вольтамперометрии в селективном электросинтезе
Многие органические электродные процессы в принципе
протекают в разных направлениях, а следовательно, при электролизе при
постоянном токе, как правило, получается много разных продуктов
реакции. Например, электролиз при постоянном токе
восстановления ароматических альдегидов или кетонов в кислых условиях
дает два продукта — спирты и пинаколы одновременно (см.
уравнение (2.7))1.
1 Cheng Р.-С, Nonaka, Т. // J. Elcctroanal. Chem. 1989. No 269. P. 223-230;
ArobeM., MatsudaK., Nonaka Т. // Elearoarialysis. 1996. No 8. P. 784-788.
±J
37
Рассмотрим вольтамперометрию с линейной разверткой (LSV)
для этого восстановления при механическом перемешивании
(рис. 2.9). Известно, что первая волна соответствует одноэлектрон-
ному восстановлению протонированного субстрата с образованием
радикального интермедиата, в то время как вторая волна
соответствует второму одноэлектронному восстаноачению промежуточного
радикала с образованием спирта, как показано ниже:
Ar-C-R-
II
О
н
Ar-C-R-
I
ОН
.Ar-C-R-^^^±ArCR-CRAr (2.7)
i l
ОН
е I/Г,"
Аг—С —R
I I
ОН ОН
piniiLol
-* Аг—CHR
I
ОН
I
ОН
alcohol
Итак, когда потенциал рабочего электрода поддерживают при
конкретном потенциале (известном как электролиз при постоянном
потенциале), одноэлектронное восстановление карбонильного
соединения происходит четко через радикальный интермедиат, а значит,
пинакольный продукт может быть получен селективно. Если на
катоде поддерживать потенциал, при котором будет происходить двух-
электронное восстановление, то получится смесь двух продуктов.
Таким образом, LSV в этом процессе дает информацию о
механизме реакции, что позволяет использовать этот процесс в
селективном синтезе.
Ток восстановления
Вторая полна одно-
электронного восстановления /
радикального интермедиата У
Волна восстановления
разряда либо растворителя,
либо фонового электролита
Первая волна одно-
электронного
восстановления
протонирован ного
субстрата
Е?
Потенциал
восстановления
Рис 2.9. Вольтамперограмма с линейной разверткой для восстановления
ароматического карбонильного соединения при механическом перемешивании
38
2.8.2. Уточнение механизма реакции
В общем случае органические электродные реакции включают
несколько типов связанных химических реакций, а следовательно,
их механизм, как правило, более сложен по сравнению с
неорганическими электродными реакциями. Когда мы пытаемся создать новую
органическую электродную реакцию, желательно проводить
измерения ЦВА заранее, чтобы понять ее механизм.
Эксперимент с ЦВА, проведенный Элвингом, является
примером, позволяющим выяснить сложный механизм реакции*. Было
использовано отсечение (обрезание) волн и добавление донора
протона (фенола), чтобы получить информацию о промежуточных
соединениях при электрохимическом восстановлении бензофенона-1.
Как показано на рис. 2.10, а, ЦВА восстановления дает два
последовательных катодных пика lt и I lt при -1,8 и -2,0 В (относительно
электрода сравнения Ag/AgNOj) и один анодный пик 10, если
потенциал сканируется от 0 до —2,3 В и обратно до —1,0 В. Отсечение
волны, т.е. изменение направления сканирования при потенциале
(—1,9 В) между пиками I, и \\с, вызывает сохранение пика 1„, как
показано на рис. 2.10, 6. Это доказывает, что пик 1д, связанный с
окислением частиц, образованных на первой стадии восстановления
(1С), и восстановление при пике \с образуют частицы, долгоживущие
в примененной временной шкале ЦВА.
1<
Start /I
"Л]
olo 1,0 2Д) "И Г5 2Л)
Е £
а б
Рис. 2.10. ЦВА восстановления бензофенона:
а — потенциал сканируется от О до -2,3 В и обратно до -1,0 В; б — потенциал
сканируется от 0 до -1,9 В и обратно до -1,0 В
При этом пик Нс должен соответствовать образованию
реакционных частиц, потому что он полностью необратим. Добавление
увеличивающегося количества фенола в раствор увеличивает высоту
1 МШеШ Л./:. EMngP.J. jl J. Am. Chem. Sot. 1968. No 90. P. 1989-1995.
Start
39
пика 1с и снижает пики Пс и 1а до тех пор. пока наконец 1с достигнет
максимальной двухкратной высоты, a IIt. и [а отсутствуют.
Эти результаты согласуются со следующим механизмом:
(С6Н5)2СО + е ->Г(С6Н3)2СОТ (2.8)
2+е-^Г(С6Н5)2С012" (2.9)
С6Н5ОН+2^С6Н5О_+(С6Н07СОН (2.10)
4
"(QH5)2COf -^(С(1Н5)2СОН (2.11)
3 J 5
4 + е->5 (2.12)
5—^ (С6Н5)7СНОН (2.13)
6
Пара 1С— \а связана с обратимым образованием кстила бсизофс-
нона 2 (см. (2.8)). Пик Нг должен быть связан с восстановлением 2
в реакционноспособныЙ дианион 3 (см. (2.9)). В присугствии фенола 2
протонируется (см. (2.10)), а полученный радикал 4 затем
дополнительно восстанавливается с образованием моноаниона 5 (см. (2.12)).
Наконец, 5 протонируется с образованием спирта б (см. (2.13)).
Как видно в приведенном выше примере, 1ДВА обеспечивает
предварительную информацию о механизме неизвестной
электродной реакции. Для дальнейшего выяснения механизма можно
провести электрохимическую реакцию в достаточно большом
масштабе, чтобы выделить и идентифицировать продукты и интерме-
диаты. ЦВА измерения выделенных продуктов и интермедиатов
электролиза препаративного масштаба также могут помочь понять
механизм изученной электрохимической реакции. Таким образом,
как использование ЦВА, так и проведение электролиза
препаративного масштаба весьма необходимы для уточнения сложного
механизма неизвестной органической электродной реакции.
2.83. Применение вольтамперометрии при выборе медиатора
Большинство органических электродных реакций проводят путем
прямого электролиза, т.е. переноса электрона между органическим
субстратом и электродом. А при добавлении в среду окислительно-
восстановительного медиатора можно осуществить
электрохимическую реакцию даже при потенциале, при котором интересующее
нас вещество неэлектроактивно. В этой системе, как показано
40
на рис. 2,11 (пример для процесса окисления, но аналогичное
поведение можно наблюдать в восстановительном процессе), обмен
электронов между медиатором MRcti (катализатор) и электродом
генерирует вещество М0х, которое, в свою очередь, может обмениваться
электроном с молекулой субстрата с получением продукта. Такая
система называется опосредованной электрокаталитической
реакцией, а реакция становится непрямым электролизом'.
'Л
1^
MRed -*>ч. ^*- Продукт
VI ^*- М0х — ^— Субстрат
(М: Редокс-медиатор)
Рис 2.11. Принцип редокс-медиаторной реакции (пример для процесса окисления)
Медиаторные электрокаталитические реакции имеют свои
особенности. Так, каталитическое количество медиатора должно быть
достаточно, чтобы осуществить непрямой электролиз. Далее субстрат
претерпевает редокс-превращение при потенциале более низком,
чем требуется для его эффективного прямого электролиза.
Можно избежать пассивации поверхности электрода в результате
образования непроводящих полимеров в процессе прямого
электролиза. Можно осуществить достаточно селективное и эффективное
редокс-превращение.
Часто используемые медиаторы для непрямого электролиза — это
ионы мультивалситных металлов, галогенид-ионы,
полициклические ароматические углеводороды, такие как нафталин и антрацен,
триариламины и нитроксильные радикалы. Кроме того, многие
медиаторы были синтезированы для конкретных непрямых электро-
лизов. В этом случае ЦВА-измерения, как правило, используются
для выбора подходящего медиатора в исследуемой
электрохимической реакции.
Так как прямое электрохимическое окисление спиртов в
карбонильные соединения осуществляется с большим трудом, их
окисление было любимым тестом для новых электрокаталитических
систем. Триариламины хорошо известны как отличные посредники
для непрямого электрохимического окисления различных спиртов.
Например, трис (п-бромфенил) амин проявляет типичный
обратимый редокс-отклик на ЦВА (£■"'= 0,8 В отн. Ag/AgNOj), как по-
FiyAJ. SyniheLic OnianicElectrochenmtry. N. Y.: John Wiley & Sons, 1989. Ch. 9.
41
казано на рис. 2.12, в1. При добавлении же 4-метоксибензилового
спирта в качестве субстрата анодный пик увеличивается, а катодный
пик становится меньше или исчезает (рис. 2.12, б). Это поведение
наводит на мысль, что трис (п-бромфенил) амин может играть рань
посредника, и прямое электрохимическое окисление субстрата может
происходить в мягких условиях.
Восстановление
Окисление
1,0 0,5 0 1,0 0,5
£(B)oTll.Ag/AgNO,
а б
Рис 2.12. ЦВА окисления трис-(п-бромфенил)амина при {а) отсутствии и (6)
в присутствии 4-метоксибензилового спирта
Однако, когда бензиловый спирт добавляют к раствору трис (п-
бромфенил) амина, такое поведение медиатора никогда не
наблюдается на ЦВА, и обратимый редокс-отклик амина сохраняется,
потому что потенциал окисления бензилового спирта (Ер = 2,0 В отн.
Ag/AgN03) гораздо выше, чем 4-метоксибензилового спирта
(Ер = 1,2 В отн. Ag/AgNOO- Для того чтобы наблюдать за
поведением медиатора окисления бензилового спирта, требуется более
мощный посредник, например, трис(2,4-дибромфснил)амин
(Е° = 1,2 В огн. Ag/AgN03). Таким образом, ЦВА-исследование
является эффективным инструментом выбора подходящего медиатора
в этой непрямой электрохимической реакции.
BrinkhausK.-H.a. Steekhan £., Schmidt W. //Acta Chem. Scand. 1983. B. 37. P 499-507.
42
2.6.4. Использование вольтамперометрии при выборе материала
электродов
Как уже упоминалось в разд. 2.1, верхний и нижний пределы окна
потенциалов определяются материалом рабочего электрода, а
следовательно, выбор материала рабочего электрода имеет решающее
значение не только для успеха вольтамлерометрического эксперимента,
но и для осуществления желаемого электролиза в препаративном
масштабе с ожидаемым выходом.
Хотя электрохимическое окисление фурана в растворе метанола,
как правило, дает соответствующий продукт метоксилирования:
-t- I
\ МеОН,
сг"
МеОН,
ОМе
^*.(
х0х
-Hf
МеО
ОМе
(2Л4)
- е
(Г ^ОМе МеСГ "О" ^ОМе.
окисление на платиновом электроде протекает неэффективно. При
этом отличный выход продукта может быть получен с
использованием рабочего стеклоуглеродного углерода (СУ). Влияние рабочего
электродного материала на эффективность электрохимической
реакции также может быть подтверждено исследованиями с
применением ЦВА1.
Рисунки 2ЛЗТ а и 2.13, б показывают формы ЦВА для окисления
фурана в растворе метанола, записанные на СУ и Pt анодах. Пик
окисления фурана может наблюдаться при очень положительном
потенциале 1,9 В отн. НКЭ при использовании СУ-пластины
5 тА
н 1 i 1 1 1 1
О 0,5 1,0 1,5 2,0 0 0,5 1,0 1,5 2,0
£(V) vs. SCE £(V) vs. SCE
a 6
Рис 2.13. ЦВА окисления фурана в растворе метанола, записанные на СУ аноде
(а) и Pt аноде (б). Штриховые кривые показывают отклик в отсутствие фурана
1 HoriiD., Atobe M„ Fuchigami Т., Магкеп F. //J. Elcctrochem. Soc. 2006. No 153.
D143-D147.
43
в качестве анода. В отличие от этого, когда Pt пластина используется
в качестве материала анода, раствори!ель метанол разряжается более
легко, записывается высокий фоновый ток, поэтому ник окислении
фурана не наблюдается. Значит, на основании ЦВА-экспериментов
можно утверждать что СУ — лучший анодный материал для
окисления фурана.
Как следует из приведенного выше примера, вольтамперограммьт,
полученные для этого случая, дают предварительную информацию
о выборе подходящего электродного материала, что позволит
использовать процесс синтеза более эффективно.
Глава 3. МЕТОДЫ ИССЛЕДОВАНИЯ
В ОРГАНИЧЕСКОЙ ЭЛЕКТРОХИМИИ:
ЭЛЕКТРОХИМИЧЕСКАЯ ОЦЕНКА РЕАКЦИОННОЙ
СПОСОБНОСТИ ОРГАНИЧЕСКИХ МОЛЕКУЛ
Органический электросинтез, как правило, зависит отряда более
сложных факторов, чем обычный органический синтез, поэтому'для
успешной реализации процесса необходимо выбрать подходящие
электролизеры (разделенные или неразделенные),
электролитические методы (постоянный ток или постоянный потенциал),
электроды, фоновые электролиты, растворители и т.д.1 В этом разделе
детали описываются так, чтобы даже новички смогли осуществить
органический электросинтез,
3.1. ВЫБОР ЭЛЕКТРОЛИТИЧЕСКИХ ЯЧЕЕК
Правильный выбор конструкции ячейки играет важную роль
в успешном проведении желаемой электрохимической реакции.
Органические электрохимические реакции могут реализовыватъея
в лабораторном масштабе с использованием неразделенной ячейки.
На рис. 3.I показана простая конструкция ячейки. Для проведения
электролиза в безводных условиях или в инертной газовой
атмосфере, например, в атмосфере азота, рекомендуют конструкцию
ячейки в виде цилиндра, как показано на рис. 3.2. В атмосфере
инертного газа растворитель и субстрат вводят с помощью шприца
в ячейку через резиновую перегородку — септу
Если частицы, восстаношгенные на катоде, окисляются на аноде
и наоборот — то следует использовать ячейку с двумя отсеками,
а именно — разделенную ячейку с диафрагмой (из спеченного
стекла или с ионообменной мембраной) для предотвращения сме-
' Bard A.J., Stratmann M. Electrochemistry applied to organic synthesis: Principles
and main achievements // Encyclopedia of Electrochemistry / eds D.D. Mac-
donald, P. Schmuki. Weinheim: Wiley-VCH \ferlag GmbH, 2007. Mil. 5. Ch. 6;
Bard A.J\, Stratmann M. Methods to investigate mechanisms of electroorganic
reactions (Ch. I) and Practical aspects of preparative scale electrolysis (Ch. 2) //
Encyclopedia of Electrochemistry / ed. H.J. Schafer. Weinheim: Wiley-VCH
Vfcrlag GmbH, 2002. Vol. 8; Lund H., Hammerich O, Organic
Electrochemistry. 4lh edn. N. Y.: Marcel Dekker, Inc., 2001. Ch. 1 and 2; Hammerich 0,,
Speiser B. Organic Electrochemistry. 5th edn. CRC/Taylor & Francis, 2014;
Grimsha J. Electrochemical Reactions and Mechanisms in Organic Chemistry.
Amsterdam: Elsevier. 2000; Fry A.J. Synthetic Organic Electrochemistry. N. Y:
Wiley Interscience. 1989.
45
шения анодного и катодного растворов. Ячейка Н-типа, разделенная
стеклянной диафрагмой (размер пор — 5—10 мкм), как показано
на рис. 3.3, удобна для синтеза.
Термометр
Ф
е
_ Барботср
(инертный газ)
^v
Мешалка
Рис. 3.1. Ячейка в виде стакана
Инертный газ
Трехходовой кран
Вход для субстрата
s и растворителя
Резиновая септа
V^/Мешалка
Рис 3,2. Неразделенная ячейка для проведения электролиза в отсутствие
следов воды
Ф
э
ч^-/
-fW
Стеклянная мембрана Мешалка
Рис. 3.3. Ячейка Н-типа с диафрагмой из стеклянного фильтра
46
Подходящий объем для каждой камеры составляет около 10—
200 см3, а диаметр диафрагмы должен быть как можно больше, чтобы
уменьшить сопрогинление ячейки. Когда исходный субстрат и/или
продукты мигрируют в противоположный отсек через стеклянную
диафрагму, вместо диафрагмы должна быть использована
ионообменная мембрана. В этом случае рекомендуется разделенная ячейка,
в которой мембрана зажата между отсеками при помощи винтов.
Такие разъемные разделенные ячейки являются коммерчески
доступными. При использовании диафрагмы сопротивление ячейки
обычно возрастает, а напряжение на ячейке (напряжение между
анодом и катодом) увеличивается. Для уменьшения напряжения
на ячейке расстояние между электродами должно быть как можно
меньше.
Даже когда для катодного восстановления необходима
разделенная ячейка, можно добиться желаемого восстановления с
применением неразделенной ячейки следующим образом.
Использование растворимых анодов, таких как Mg и Zn, или добавление
расходуемых органических соединений, таких как щавелевая или
уксусная кислоты, в раствор электролита позволяет избежать рео-
кислеиия катодных продуктов. В первом случае металлы как аноды
растворяются с образованием соответствующих ионов в растворе
в процессе электролиза, а в последнем случае кислоты анодно
окисляются с образованием С02 и этана. Такие расходуемые
электролитические системы в ряде случаев позволяют избежать окисления
катодных продуктов и исходного материала на аноде.
Причем в случае анодного окисления, требующего разделенной
ячейки, в качестве расходуемого вещества в раствор электролита
должен быть добавлен источник протонов и должны быть
использованы катодные материалы с низким перенапряжением
водорода. В этом случае происходит преимущественное восстановление
протонов на катоде, которое позволяет избежать восстановления
анодных продуктов и исходного материала на катоде» Следовательно,
можно достичь желаемого анодного окисления избирательно даже
в неразделенной ячейке.
Тигли из проводящих материалов, таких как графит и стекло-
углерод, можно использовать в качестве рабочих электродов. В этом
случае можно уменьшить объем раствора электролига, а площадь
поверхности рабочего электрода увеличить. Для масштабирования для
коммерциализации рекомендуется так называемая фильтр-прессная
проточная ячейка, как показано на рис. 3.4. В этом случае объем
раствора электролита не имеет предела, поскольку раствор циркулирует
с помощью насоса, и массоперенос из объема к поверхности
рабочего электрода способствует повышению эффективности
электролитической реакции*
47
Сепаратор
©i
L
Теплообменник
t
>е
Насос
Резервуар
Рис. 3.4. Проточная ячейка фильтр-прессного типа
3.2. ЭЛЕКТРОЛИЗ ПРИ ПОСТОЯННОМ ТОКЕ ИЛИ
ПРИ ПОСТОЯННОМ ПОТЕНЦИАЛЕ
Органический ачектросинтез рекомендуется проводить при
постоянном токе прежде всего потому, что в этом случае установка
электрохимической системы и работа источника питания достаточно просты.
Селективность и выход продукта можно улучшить путем
регулирования плотности тока и количества пропущенного электричества. Так,
плотность тока коррелируете приложенным потенциалом, изменение
плотности тока создаст сдвиг потенциала. Плотность тока следует
выбирать в зависимости от концентрации исходного субстрата. Когда
концентрация субстрата низка, плотность тока должна быть низкой,
и наоборот. Пропущенное электричество легко вычислить по
следующей формуле: ток (А) х время (с) = электричество (К). Например,
если желаемая электрохимическая реакция является двухэлектронной,
теоретическое количество электричества 2 Ф (2 х 96 480 К).
Когда это количество электричества делится на приложенный
ток (А), можно легко рассчитать, сколько часов следует проводить
электролиз. Как показано на рис. 3.5, й, потенциал электрода
изменяется с расходом исходного субстрата (положительный сдвиг
в случае окисления или отрицательный сдвиг в случае
восстановления). Таким образом, селективность продукта и выход по току
иногда уменьшаются, особенно на поздней стадии электролиза.
Но можно достичь весьма избирательного и эффективного
органического электросинтеза, часто даже в условиях электролиза при
постоянном токе, что важно для осуществления коммерциализованных
электродных процессов. Однако электролиз при постоянном
потенциале (рис. 3.5, 6) удобен для достижения высокой селективности
и выяснения механизмов реакции.
48
Ток (О
.„_ Эффективность потоку
Концент-\
Потенциал (£)
рация (О \
L Ъ~\-'"
Время электролиза
а
Время электролиза
б
Рис. 3.5. Профиль, показывающий изменение параметров электролиза
(ток, эффективность по току, потенциал, концентрация субстрата) в течение
электролиза при постоянном токе (а) и постоянном потенциале электролиза (б)
Кроме того, на основе данных кулонометрии при постоянном
потенциале можно легко найти число электронов, участвующих
в электрохимической реакции. Для того чтобы осуществить
электролиз при постоянном потенциале, потенциал окисления или
восстановления субстрата нужно измерить заранее, но можно оценить
приложенный потенциал, при котором наблюдается быстрое
увеличение тока из кривой ток — потенциал субстрата. Как показано
на рис. 3.6, солевой мостик, заканчивающийся капилляром Луггина
или затычкой из пористого стекла типа Vycor, помещают как можно
Насыщенный
раствор КС1
Hg + Hg2CI2
Hg-
Электрод сравнения
(насыщенный
каломельный электрод)
О
Мотенциостат
□ □□□□
DDDDD
Солетюн mi
стнк
Термометр
Рабочий электрод — - - I
Луггин-капиллятор
Ci
- Барботер
(инертный газ)
- П ротивоэле ктрод
Мешалка
Рис. 3.6. Схема установки для электролиза при постоянном потенциале
49
ближе к рабочему электроду, а соответствующий постоянный
потенциал относительно электрода сравнения, такого как водный НКЭ,
накладывается с помощью потеншгастата> Количество
пропущенного электричества измеряется кулонометром.
3.3. ПРЯМОЙ И НЕПРЯМОЙ ЭЛЕКТРОЛИЗ
Электролиз можно классифицировать на прямой и непрямой.
Первый тип основан на прямом переносе электронов между
молекулой субстрата и электродом, что является простым и общим
электрохимическим методом (рис. 3.7, а). Второй тип основан
на переносе электронов с использованием
окислительно-восстановительных медиаторов, растворенных в электрохимической ячейке
в объеме, как показано на рис. 3.7, 6 (непрямое катодное
восстановление). Обратить внимание: непрямое анодное окисление было
рассмотрено в гл. 2 (см., например, рис. 2.9). Непрямой электролиз
также подразделяется на электролиз внутри ячейки, когда он
проводится в присутствии субстрата и медиатора, и вне ячейки, когда он
используется только для регенерации медиатора. В первом случае
окиаттигелъно-восстановителъный потенциал медиатора должен быть
ниже, чем субстрата. Если окислительно-восстановительный потен-
циал медиатора выше, чем субстрата, используется последний тип
электролиза. В первом случае должно быть достаточно
каталитического количества медиатора, в то время как в последнем случае
необходимо эквивалентное или избыточное количество медиатора. Когда
гетерогенный перенос электронов между электродом и субстратом,
а также редокс-реакция с субстратом протекают достаточно быстро
(рис. 3.7, 6), можно получить значительное увеличение
каталитического тока за счет снижения потенциала электролиза (снижения
энергии активации), как показано на рис. 3.8. Другими словами,
можно получить большой электрокаталитический ток.
Катод Катод
Субстрат
Медленно
Продукт ~"* Mr^j ""*■ Продукт
6
Рис. 3.7. Схема непрямого электролиза с использованием медиатора:
а — прямой перенос электрона между субстратом и катодом с образованием
продукта (это медленный процесс); б — непрямой перенос электрона с
использованием редокс-медиаторов, которые катализируют превращение субстрата в продукт
(это быстрый процесс)
50
Субстрат и медиатор
Медиатор!
Субстрат
Потенциал
Рис. 3.8. Кривые ток-потенциал для прямого и непрямого электролиза
Характеристики и правильный выбор медиаторов и их
синтетические приложения описаны в гл. 2 и 4. Сейчас многие типы
медиаторов доступны, а выбор окислительно-восстановительных
потенциалов медиаторов можно проводить на основе знания их
молекулярной структуры. Более тою, некоторые медиа горы можно
применять в электрохимическом асимметрическом синтезе'.
3.4. ЭЛЕКТРОДНЫЕ МАТЕРИАЛЫ И ЭЛЕКТРОДЫ СРАВНЕНИЯ
Выбор материала электрода является одним из наиболее важных
факторов проведения успешного электролиза, поскольку этот
материал не только является границей раздела фаз, на которой
происходит перенос электрона на молекулу субстрата, но может
действовать как электрокатализатор. Если не удалось выбрать
правильный материал электрода, желаемая электрохимическая реакция
протекать не будет, поэтому подходящий электродный материал
следует подбирать тщательно. В водном растворе газообразные кислород
и водород генерируются конкурентно благодаря разряду воды в ходе
электрохимической реакции. Чтобы избежать таких нежелательных
реакций, следует использовать катодные материалы с высоким
перенапряжением водорода или анодные материалы с высоким
перенапряжением кислорода. В зависимости от материала электрода
наблюдаются следующий порядок перенапряжения кислорода: Аи > Pt,
Pd, Cd, Ag > Pb02 > Си > Fe > Co > Ni и порядок перенапряжения
водорода: Hg > Zn, Pb, Cd > графит > Си > Fe, Ni > Ag, Co > Pt, Pd.
Ргугный катод часто использовали в восстановительном
органическом синтезе. В настоящее время для этих целей его больше
Тапака //., Kawakami К. Goto К., Kuroboshi M. //Tetrahedron Lett, 200L No 42.
R 445—448; Kashiwagi Y.t Kurashima F., Chiba S., Anzai J., Osa 7"., Bobitt T.M.
// Chem. Commun, 2003. 1124—1125; Shiigi //., Tanaka //,, Demizu K., On-
omum 0. // Tetrahedron Lett. 2008.49. 5247-5251; Demku Y„ Shiigi //,. Mori H.,
Matsumoto K., Onomura O. // Tetrahedron Asymmetry'. 2008. 19. 2659—2665.
51
практически не применяют из-за токсичности ртути (исключение
составляют электрохимические измерения). Выбор платинового
электрода является первоочередным при использовании aiфотонных
органических растворителей. Хотя доступны различные виды
катодных материалов, выбор используемых анодных материалов
ограничен. Большинство металлов легко окисляется, кроме благородных,
таких как платина и золото. Углерод, графит и металлы с оксидом
на поверхности, такие как РЬ/РЬ02, широко используются в качестве
анодов.
Углеродные электроды обычно включают следовые количества
металлов, таких как железо, поэтому их поверхность парамагнитна.
Следовательно, такие углеродные аноды легко захватывают анодно
генерируемые радикалы для дальнейшей активации окисления
с образованием катионных интермедиатов. При этом платиновые
аноды обычно склонны к селективной генерации радикальных
интермедиатов. Соответственно платиновые аноды являются наиболее
подходящими для электролиза по Кольбе. Следует отметить, что,
когда углеродный анод используется в растворе, содержащем LiC104,
часто происходит разрушение поверхности электрода (его коллапс)
из-за окисления связующего, такого как угольный пек в
углеродном электроде. В отличие от углерода стеклоуглеродные (СУ)
электроды имеют достаточную прочность для анодного окисления,
и пассивации анода не происходит, что довольно сильно отличает
их от других углеродных электродов.
Однако электроды из СУ являются дорогостоящими, как и
платиновые электроды. Хотя электроды из неблагородных металлов
не подходят в качестве анодных материалов, Mg, Zn, А1 и Си часто
используются в качестве реакционноспособных электродов или
растворимых анодов, как описано в разд. 3.1.
На следующие моменты следует обратить внимание при
осуществлении органического электросинтеза. В отличие от
электрохимических измерений электрохимический синтез требует
относительно большого тока 10—100 мА (-10 мА ■ см-2), даже в
лабораторном масштабе. Использование большей площади поверхности
электрода рекомендуется, чтобы завершить электролиз в течение
более короткого времени. Обычно используются рабочие электроды
в форме пластины, прута (стержня) и сетки. Площадь поверхности
противоположного электрода должна быть такой же, как у рабочего
электрода. Предварительная обработка поверхности электрода
зависит от его типа, но в целом поверхность полируется абразивной
или наждачной бумагой.
Электроды сравнения необходимы, когда осуществляется
измерение кривых ток-потенциала и проводится электролиз при
постоянном потенциале. В водных и протонных электрохимических
52
растворителях, используют электроды типа НКЭ (см. рис. 2.2, а)
и Ag/AgCl (см. рис. 2.2, 6). Для электролиза в неводном растворе
удобен электрод сравнения Ag/Ag+, так как он может быть
размещен вблизи поверхности рабочего электрода без солевого мостика.
Об электродах сравнения подробнее см. в гл. 2.
3.5. ЭЛЕКТРОЛИТИЧЕСКИЕ РАСТВОРИТЕЛИ И ФОНОВЫЕ
ЭЛЕКТРОЛИТЫ
Электролитические растворители должны быть полярными,
чтобы в них можно было растворить фоновые соли и обеспечить
достаточную ионную проводимость растворов за счет диссоциации
ионов. Индекс полярности растворителя отражается его
диэлектрической проницаемостью. Кроме того, электрохимические
растворители должны иметь низкую вязкость и токсичность. В частности,
следует отметить, что вязкость растворителя сильно влияет на
диффузию субстрата из объема раствора к поверхности электрода.
3.6. ПЕРЕМЕШИВАНИЕ
Так как электрохимические реакции, как правило, гетерогенны,
перемешивание раствора электролита контролирует массоперенос
субстрата, который в значительной степени влияет на выход по току
и селективность продукта. Даже если раствор энергично
перемешивается, перенос субстрата из объема к поверхности электрода
не всегда улучшается. Как показано на рис. 3.4, циркуляция раствора
электролита через проточную ячейку, в частности фильтр-прессного
типа, является гораздо более эффективной по сравнению с ячейкой
обычного типа стакана, поэтому именно фильтр-прессные
электролизеры обычно используют в промышленном электролизе.
3.7. КОНТРОЛЬ РЕАГЕНТА И ПРОДУКТА
В органическом синтезе очень важна эффективная конверсия
субстрата в требуемый продукт. В случае электролиза при
постоянном токе разряд растворителя и/или фонового электролита
происходит одновременно на более поздней стадии электролиза.
Теоретическое количество пропущенного заряда не всегда достаточно для
полной конверсии субстрата, следовательно, контроль электролиза
(расход исходного субстрата и количество продукта) следует
проводить с помощью ряда методов, например, тонкослойного метода,
высокоэффективного метода жидкостной хроматографии, методов
газовой хроматографии и масс-спектрометрии, чтобы подтвердить
количественно конверсию субстрата.
53
3.8. РАЗРАБОТКА, ВЫДЕЛЕНИЕ И УСТАНОВЛЕНИЕ ПРИРОДЫ
ПРОДУКТОВ
Разработка собственно электролиза аналогична разработке
обычного органического синтеза, но при этом необходимо
учитывать следующие моменты. Поскольку анолит становится кислым,
а католит — основным, после электролиза с использованием
разделенной ячейки необходимо нейтрализовать желаемый
электрохимический раствор. Так как в электрохимическом растворе
содержится большое количество фонового электролита, его следует
удалить в первую очередь. Например, когда электрохимический
растворитель водорастворим, его удаляют выпариванием при
пониженном давлении. Полученный остаток или нерастворимый в воде
электрохимический раствор смешивается с водой или рассолом
(насыщенным водным раствором хлорида натрия), если необходимо,
осуществляется нейтрализация, а продукт экстрагируется
соответствующими органическими растворителями, такими как диэтиловый
эфир. Следует отмстить, что некоторые гидрофобные фоновые
электролиты (например, Bu4NBF.1) нерастворимы в воде, но растворимы
в органических растворителях (например, в СН2С12).
Методы выделения и идентификации аналогичны тем, которые
используют в обычном органическом синтезе. Выделение продукта
осуществляется с помощью перегонки, перекристаллизации или
различных типов хроматографии, а идентификация выполняется
с использованием различных методов спектроскопического анализа,
таких как [Н ЯМР, ИК и МС.
3.9. ВЫХОД ПО ТОКУ И ВЛИЯНИЕ ИСТОЧНИКА ТОКА
Выход по току япляется наиболее важным параметром оценки
результатов электрохимического синтеза. Например, рассмотрим
следующее двухэлектронное восстановление ацетона:
Н3С Н3С
\^0 -^^-* VoH (3.1)
Н3С НзС
Количество электричества 2 х 96480 К требуется для образования
1 моль изопропилового спирта. Когда и моль изопропилового спирта
образуется из 1 моль ацетона после прохождения Q кулонов, выход
по току (%) составляет (96480 х 2л/Q) х 100.
Выход по току, как правило, ниже 100%, так как растворитель
и/или фоновая соль разряжаются одновременно во время
электрохимической реакции. Если потребление энергии в электросинтезе
54
обычно игнорируют при проведении фундаментальных
исследований, то этот фактор важен в случае осуществления
электрохимических процессов в промышленных масштабах. Потребление
энергии для производства желаемого органического соединения
выражают в кВт/ч ■ кг ', оно в значительной степени зависит от
напряжения на электролизере, поэтому рекомендуется иметь как
можно меньшее расстояние между электродами, чтобы уменьшить
сопротивление ячейки, т.е. омическое падение потенциала на
элементах цепи.
Глава 4. ОРГАНИЧЕСКИЕ ЭЛЕКТРОДНЫЕ
РЕАКЦИИ
В данной главе обсуждаются общие характеристики
электрохимических реакций и различия между электрохимическими и
обычными химическими реакциями, рассматриваются типичные
характеристики, такие как umpolung (обращение полярности), контроль
селективности совместно с контролем переноса электрона, а также
типы реакций и типы различных электрохимически генерируемых
активных частиц1,
4.1. ОБЩАЯ ХАРАКТЕРИСТИКА ЭЛЕКТРОДНЫХ РЕАКЦИЙ
Органические электродные реакции имеют следующие
характерные особенности, выделяющие их среди других синтетических
процессов:
— электродные реакции являются обычно гетерогенными, а
области реакции — специфичными, поскольку окисление и
восстановление происходят отдельно в различных пространствах;
— амполунг (инверсия полярности) выполняется легко без
использования каких-либо реагентов; селективность электродных
и обычных органических реакций часто отличаются;
— поскольку в качестве реагента используются электроны,
применения опасных реагентов можно избежать, т.е. электродные реакции
отличаются низким уровнем выбросов, протекают они в мягких
условиях, таких как комнатная температура и нормальное давление,
и могут быть запущены или остановлены легко
включением-выключением источника питания, т.е. их легко контролировать.
Эффект масштабирования, как правило, мал. Электродные
реакции — это окислительно-восстановительные реакции с переносом
электрона между молекулой субстрата и электродом. Основной
областью реакции является поверхность электрода (раздач поверхности
твердое тело — жидкость) или поверхность вблизи электрода,
которая имеет чрезвычайно большое электрическое поле, что довольно
Torii S. Electroorganic RcductionSynthesis. Vol. I and 2. Wcinhcim: Kudiinsha
and Wiley-VCH Vcrlag GmbH, 2006; Bard A J., Stnttmann M. Organic
electrochemistry // Encyclopedia of Electrochemistry. Vol. 8 / ed. H J. Schafer. Weinheim:
Wiley-VCH VerlagGmbH, 2002; Lund //., Hammerich O. Organic Electrochemistry.
4th edn. N. Y.: MarcelDekker, Inc., 2001; GrimshawJ, Electrochemical Reactions
and Mechansims in Organic Chemistry. Amsterdam: Elsevier, 2000; Fry A.J.
Synthetic Organic Electrochemistry. N. Y.: Wiley Inierscience, 1989; Shono T. Elcc-
iroorganic Chemislryas a Tool in Organic Synthesis. Berlin: Springer-VMag, 1984.
56
сильно отличается от обычных окислительно-восстановительных
реакций на гетерогенных катализаторах. Электродные реакции,
следовательно, протекают в уникальных полях.
Различие между обычной химической и электродной реакциями
будет подробно описано на примере химической редокс-реакции.
На рис. 4.1, а показана схема восстановления субстрата
восстановленным реагентом А. Когда образуется активированный комплекс
или А и В сближаются достаточно близко для переноса электрона,
происходит перенос электронов от А на В. Далее восстановитель А
превращается в окисленный С, а субстрат В преобразуется в
восстановленный продукт D.
+ В
Jf?
Активированный комплекс
(а
©
©
®
Ч<§
Рис. 4.1. Схема различия между химической реакцией {а)
и электрохимической реакцией (б):
а — реакция между субстратами А и В, восстановление субстрата В
восстанавливающим реагентом А, образование активированного комплекса, превращение
восстановителя А в окислитель С и субстрата В в восстановленный продукт D;
5—окисление и восстановление соответственно на аноде и катоде
Первый перенос электронов через комплекс называется вну-
трисферным переносом электронов или связанным переносом
электронов, в то время как последний перенос без какого-либо
комплекса называется внепшесферным переносом электронов или
несвязанным переносом электронов. Таким образом, как окисление,
так и восстановление происходят в одном и том же месте в случае
обычной химической реакции, в то время в случае
электрохимической реакции как окисление, так и восстановление происходят
57
в разных местах, таких как анод и катод (рис. 4.1, б). Другими
слонами, процессы переноса электронов на электродах (аноде и
катоде) происходят отдельно из-за существования электродных
интерфейсов, что является важным характерным признаком, отличным
от признака обычных химических реакций.
4.2. МЕХАНИЗМ ОРГАНИЧЕСКИХ ЭЛЕКТРОДНЫХ РЕАКЦИЙ
В случае органических электродных реакций перенос электрона
обычно не происходит в чистом виде, а предшествующие и/или
последующие реакции, как правило, сопровождают перенос. Это
заметное отличие от большинства неорганических электродных
реакций. Органическая электродная реакция включает стадию
переноса электрона, а также несколько химических и физических стадий.
Рисунок 4,2 иллюстрирует каждую элементарную стадию реакции
субстрата S, образующего продукт Р через интермедиат /. На стадии а
массопсрснос S из объема раствора электролита на поверхность
электрода происходит пугем диффузии, гидродинамики или
миграции. На стадии Ь предшествующие реакции, такие как десольва-
тация, диссоциация и/или денротонирование S, происходят с
образованием промежуточного /. Но такие предварительные реакции
происходят не всегда. На стадии с интермедиат /адсорбируется на
поверхности апсктрода, образуя интермедиат Ia(i. На стадии d перенос
электронов между Iasi и электродом генерирует промежуточный /^.
На стадии е протекает десорбция 1^ с последующей химической
реакцией/, давая продукт Р, который диффундирует в объем раствора
электролита, и тогда последовательная реакция завершается.
(а) Массоперенос
< S
{Ь) Химическая реакция
Объем раствора
) Химическая реакция
*► Р
(g) Массоперенос
Рис. 4.2. Схема элементарных процессов электродных реакций
Электрод
(с) Адсорбция
1« < /
1
(</) Перенос электрона
/:
ad
(е) Десорбция
-* Г
<
58
Интермедиа! /может подвергаться реакции переноса электрона без
стадии адсорбции с. Порядок последовательных стадии е и/может быть
изменен, тс, lid подвергается последующей реакции, а затем
происходит десорбция результирующего продукта. Таким образом,
электродная реакция характерна для гетерогенной системы, в которой
участвуют стадии массопереноса а и g, а также стадии адсорбции и
десорбции с и е, что заметно отличает эту реакцию от гомогенных реакций.
Если электродный процесс (процесс переноса электронов)
обозначить через £, а химический процесс — через С, то органическую
электродную реакцию можно описать с помощью этих сокращений.
Например, электрохимическая реакция, схематически показанная
на рис. 4.2, может быть описана как последовательность СЕС
(предшествующая химическая реакция -> собственно перенос электрона
на электроде —> последующая химическая реакция), а стадии
адсорбции и десорбции обычно не учитываются, если они не важны.
Чтобы выяснить механизм реакции, необходимо использовать
методы электрохимического анализа (кулонометрию и волътампероме-
трию н дополнение к обычным органическим механистическим
исследованиям) для получения информации о числе переносимых
электронов, редокс-потенциалах и выявления промежуточных продуктов
реакции. Детали электрохимического анализа описаны в гл. 2 и 3.
4.3. ХАРАКТЕРИСТИКИ ОРГАНИЧЕСКИХ ЭЛЕКТРОХИМИЧЕСКИХ
РЕАКЦИЙ
4.3.1. Обращение полярности
Инверсия полярности химической связи может быть легко
проведена в ходе электролитических реакций1. Другими словами:
электрофилы можно превратить электрохимически в нуклеофилы
без использования каких-либо реагентов, и наоборот. Такое
обращение полярности широко используется в органическом синтезе.
Например, алкил галоген иды по своей природе электрофильные
реагенты. Чтобы преобразовать их в нуклеофильные реагенты, их
следует превратить в реактивы Гриньяра или литиевые соединения с
использованием металлических Mg или Li.
Однако такая инверсия полярности может быть легко достигнута
в одну стадию катодным восстановлением алкилгалогенидов:
2е, -Х- Е+
*R-
R—X—
Mg E
—* RMgX —'
R—Е (4.1)
1 Shono Т. Op. cit.
59
Когда основные продукты выделяют после реакции Гриньяра,
разработка может быть сложной. Тогда может быть довольно трудно
выделить продукт из водного щелочного раствора из-за
нерастворимого Mg(OH)2. Напротив, в случае электролитических реакций
выделение продукта часто является довольно легким и серьезной
проблемы с отходами не возникает, потому что металлы
используются не всегда. Кроме того, электрохимические реакции не требуют
легковоспламеняемых эфирных растворителей, а альтернативные
растворители (например ацетонитрил) можно использовать в
качестве электролитических растворителей.
Когда алкильные заместители вводятся в фосфин, соединения
кремния и олова, обычно используются их соответствующие
хлорные производные (RjZCl, R2PCI) и алкильные реактивы Гри-
ньяра. Однако при электрохимическом восстановлении этих
хлорных соединений генерируются анионные интермедиаты,
которые могут легко взаимодействовать с алкилгалогенидами (R'X)
с образованием соответствующих алкил-замещенных продуктов:
R3Z-R' R'MgX >R3Z-X 2e'"X" >R3Z~ R>x >R3Z-R'
(Z = SitSn) (4.2)
R2P-R' R'MgX )R2P-X 2e"X" )R2P" R'X )R2P-R'
Аллилсиланы обычно реагируют с электрофилами, а их
электрохимическое окисление генерирует аллильные катионы, которые
вступают в реакцию с иуклеофилами:
■ SiMe? ~2е „ ^>^ Nu~» -^v^Nu (4.3)
(Nu = OR,Q\c)
Электроннонасышенные производные бензола, как правило,
реагируют с электрофилами, в то время как они могут реагировать
с иуклеофилами в условиях электрохимического окисления:
-е
—»>
Nu — —
(4.4)
60
Кроме того, промышленная электровосстановительная гидро-
димеризация акрилонитрила также является типичным примером
амполунга с помощью катодного восстановления:
<^N:n
<^cn
^^CN с.2Н^ - >_ CN ,л -v
4.3.2. Селективность
Селективность электролитических реакций контролируется
многими факторами: материалом электрода, приложенным
потенциалом, плотностью тока, природой электролитических растворителей
и фоновых электролитов, электрическим полем, адсорбционной
ориентацией субстрата или промежуточных частиц на поверхности
электрода и т.д.1 Поэтому гетерогенные электрохимические
реакции часто проявляют селективность, отличную от селективности
обычной гомогенной химической реакции. В частности, когда
электрохимически генерируемые активные частицы или интермедиаты
реагируют с реагентами, прежде чем они диффундируют от электрода
в раствор, стерео- и региоселективность продукта часто весьма
отличаются от таковых для обычных химических реакций. Типичные
примеры селективности электродных реакций описаны в следующих
разделах.
Хемоселективность. В обычных химических реакциях она
контролируется выбором реагентов, но достаточно сложно достичь
высокой хемоселективности, когда в одной молекуле присутствуют
многочисленные похожие функциональные группы. При этом
хемоселективности можно достичь посредством контроля приложенного
потенциала, основываясь на различиях в редокс-потенциалах для
соответствующих функциональных групп. Поскольку приложенный
потенциал точно регулируется с помощью потенциостата, высокая
селективность может быть достигнута. Например, даже если фенили-
мино- и алкиламино-группы существуют в одной и той же молекуле,
фенилимино-группа может быть преимущественно восстановлена
путем электролиза при постоянном потенциале:
NR NPh + NR NHPh
2с2Н+ || | (4шб)
Это происходит потому, что фенилимино-гругила более легко
восстанавливается, чем алкиламино-группа, в результате электронно-
акцепторности первой. Однако такое селективное восстановление
1 GrimshawJ, Op. clt.; Fry A.J. Op. cit.; Shono T, Op. cit.; Fuchigami 71, Nonaka 71,
Schafer H.J. // Encyclopedia of Elccirochemisiry. \W- 8 / eds AJ. Bard and
M. Stnitmaim. Vteinheim; Wiley-VCH, Nferlag GmbH, 2O03.
61
не может быть достигнуто с помощью обычных восстанавливающих
реагентов.
Аналогично в случае молекулы с тремя атомами галогена: атом
галогена в а-положении к карбонильной группе является наиболее
легко восстанашгиваемым, таким образом, этот галоген можно
преимущественно восстановить в условиях электролиза при постоянном
потенциале:
(4.7)
Селективность пути реакции. Известно, что путь реакции сильно
меняется в зависимости от приложенных потенциалов. Можно
избирательно получить продукты од но электронного и двухэлектронного
восстановления в зависимости от применяемых катодных
потенциалов:
CONH3
-l,2V
2е
1,8 V
CONH2
CONH2
H2NOC
(4.8)
CONH2
н^
Н Н
X",CONH2
Me
62
Региоселективность
Кинетически и термодинамически контролируемая
региоселективность. Региоселективность часто контролируется либо
стабильностью реакционноспособного интермедиата (термодинамический
контроль), либо скоростью реакции интермедиата с реагентом,
а также скоростью элиминирования уходящей группы
(кинетический контроль). Типичным примером первого случая является
анодное бсизильнос замещение, которое протекает легко и
относится к реакциям со стабильными бензильными катионными ин-
термедиатамн.
Однако когда N,N-диметил бензилам ин анодно окисляется в
метаноле, метоксигруппа избирательно присоединяется к метильной
группе, как показано на рис. 4.3'. Эта региоселективность не
контролируется стабильностью катионного интермедиата, т.е. эта реакция
не контролируется термодинамически. Поскольку анодно
генерируемый промежуточный катион-радикал, как кажется, адсорбируется
на аноде, депротонирование происходит преимущественно от менее
затрудненной метильной группы. Следовательно, эта реакция
является кинетически контролируемой.
СгЬСГ,
/СНз Г~\ ( +/СНз
PhCH2N ~^*\ VCH2N-^CH3
Л
-|Г,-е
+ + + +
Анод
-Н\-е
сн2 снюсн,
/ CH-tO" /
PhCH2N * > PhCH2N
4CHj Yrl,
Основной
уСН, СНз
PhCHN *—* PhCHN
Yrh | ЧСН,
ОСН3
Рис. 4.3. Схема региоселективного анодного метоксилирования
1 Fussing /., Hammerich О., Hussain A.,Nielsen M.F.. Vtiey J.H.P. // Acta Chem.
Scand. 1998. No 52. P. 328-171.
63
Аналогично анодное метоксилирование N-этил-М-метилани-
лина, соответствующего карбамата и амидных производных
происходит селективно по метильной ipynne':
СН
Y—N
-2е,
МсОН \
сн2осн3
(4.9)
СН2СН3 СН2СНз
(Y=Ph, ROCO, RCO)
Однако региоселективность также может быть объяснена рахти-
чиями в скоростях депротонирования катион-радикалытых интерме-
диатов (так называемой кинетической кислотностью2). Например,
в случае анодного метоксилирования производного анилина,
имеющего N-фторалкил (Rp) и N-алкильную фуппу, как
показано на рис. 4.4, метоксилирование происходит преимущественно
в соседнее к группе RF положение. Региоселективность возрастает
с увеличением электронно-акцепторной способности группы Rp,
т.е. селективность повышается в следующем порядке: СН3 < CH2F <
< CHF2 < CF3-\ Механизм этого проявления региоселективности
относят к процессу с кинетическим контролем.
CH3R
XCH2R
ArN
4CH>Rf
CH2R fci
4CH,Rf c ^
„CH2R
ArN MU >ArN
4CHRf NCHRf
OMe
Стабильность: В < С
Скорость депротонирования A: Rf = CF3, CHF?: A| » k2
Rf=CHjF: Jt|>b
Рис. 4.4. Схема механизма региоселективного анодного метоксилирования
(кинетический контроль)
Региоселективность электролитического фторирования также
контролируется кинетической кислотностью. Например, в случае
электрохимического фторирования гетероциклических соединений
процесс протекает преимущественно через неустойчивый проме-
1 Fuchigami Т., khiknwa S., Копп А. // J.Oig. Chem. 1994. No 59. P. 607—615.
1 Nelsen S.f., Jppoliti J. T. // J. Am. Chem. Soc. No 108. P. 4879-4881; Yoon КС,
Mariano P.S.//Ace. Chem. Res. 1992. No25. P. 233-
Fuchigami 7'., Ichikawa S.MonnoA. Op. cil.
-240.
64
жуточный катион, связанный скарбонильной группой, а не через
стабильный бснзилъный катион:
^л (4Л0)
R OR О N
<X-N.O,S)
Это может быть объяснено с точки зрения облегчения депрото-
нирования анодно генерируемого катион- радикала с помощью
электронно-акцепторной карбонильной группы1.
Региоселективность, контролируемая селективной адсорбцией
субстрата на электроде из-за его дипольного момента. Как показано
на рис. 4.5, тетрахлорпиколиновая кислота региоселективно
дехлорируется катодным восстановлением. Высокая региоселекгивность
связана с контролируемой ориентацией субстрата на поверхности
катода за счет дипольного момента молекулы2,
Катод
О С\
> Дилольиыи момент
Рис. 4.5. Региоселективное катодное дехлорирование
Стереоселективность
Стереоселективность, контролируемая селективной адсорбцией
реакционноспособного интермедиата на электроде. Как показано
на рис. 4.6, литиевая соль производного ненасыщенного амина
адсорбируется на аноде из-за кулоновских взаимодействий. Затем
происходит одноэлсктронный перенос и генерируется соответствующий
промежуточный аминильный радикал, который подвергается
последующей внутримолекулярной циклизации в адсорбированном
состоянии или вблизи анода, что приводит к преимущественному
Nelsen S.F., IppoliiiJ.T. Op. cit.; Yoon V.C, Mariano PS. Op. cit.; Fuchigami Т.,
Narizuka 5., Konno A. //J. Org. Chem. 1992. No 57. P. 3755-3757: Hif>ashiya S.,
Narizitka &. KonnoA., Fuchigami J. // J. Org. Chem. 1992. No 57. P. 3755-3757.
Edamunt F., Kvrivacou D., Love J. US Patent 4217185. 1980; Chem. Abstr. 1981.
No 94. P. 22193.
65
образованию термодинамически менее стабильной дис-формы
продукт1. Интересно, что в этой реакции термодинамически выгодная
транс-форма продукта не образуется.
t> a U
8 s * S
о х у Э
3 ^ т ■=:
52% 17%
чг
Ph^/N\
\ / 0% 35»
Рис. 4.6. Стереоселективная анодная циклизация
При этом подобная реакция с использованием химического
окислителя (HgC12) обеспечивает главным образом
термодинамически выгодный транс-продукт. Такая высокая стереоселектив-
ность в электрохимических реакциях в основном связана с
адсорбционным эффектом. Таким образом, можно констатировать, что
электрод в значительной степени способствует стереоконтролю.
В случаях катодного гидрирования и анодного присоединения к
циклических олефинам образуется преимущественно цис-форма
продукта. Это также относится к присоединению протонов и нуклео-
филов к анионным и катионным интермедиатам, адсорбированным
на электроде.
Стереоселективность, контролируемая стерическими
затруднениями между субстратом и электродом. Простой стереоконтроль
достигается за счет стерических затруднений между субстратом
и электродом. Как видно на рис. 4.7, ориентация бициклического
гем-дибромоциклопропана на поверхности катода (а) является
предпочтительной по сравнению с (б) за счет стерического отталкивания
между катодом и 6- или 8-членным кольцом, конденсирующимся
в циклопропан. Таким образом, экзо-бромид вблизи катода легче
восстанавливается, чем эндо-бромид, а следовательно, в основном
образуется эндо-бромидный продукт2.
1 Tokuda M., Yamada Y„ Takagi Г, Sugiwme N.. Furusaki A. // Tetrahedron Lett.
1985. No 26. P. 6086-6089: lidem. // Tetrahedron Lett. 1987. No 43. P. 281-296.
2 Fry A.J.. MoorR.H. //J. Org. Chem. 1968. No 33. P. 1283-1284; Erickson R.E.,
Amino Я.. SambrM.D., Zon G. // J. Am. Chem. Soc. 1969. No 91. P. 1767-1770.
66
Основной продукт
^ШГ* (CHj)l><Br
(л = 4,6)
Рис. 4.7.Сгереоселективное восстановление, основанное на ориентации
субстрата на катоде
Селективность, зависящая от материала электрода. Существуют
многочисленные примеры электродных материалов, реакции на
которых определяют в значительной степени селективность и стерео-
селективность продукта.
Селективность продукта. Ацетон восстанавливается на
свинцовом катоде в кислом водном растворе с получением
соответствующего изопропилового спирта, тогда как восстановление с
помощью цинкового и медного катодов даст соответствующий алкан,
пропан1.
Зависимость хода электролиза по Кольбе от анодного материала.
Известно, что при электролизе по Кольбе на платиновом аноде
генерируется радикальный интермеддат, в то время как на углеродном
аноде — катиотшьга интермедиат2.
Стереоселектиемость. Восстановление 2-метилциклогексанона
на цинковых и медных катодах в водном растворе NaOH в основном
дает соответствующий транс-спирт, в то время как оловянный катод
обеспечивает образование в качестве основного продукта иис-
спирта1.
4.4. МОЛЕКУЛЯРНЫЕ ОРБИТАЛИ И ЭЛЕКТРОНЫ, СВЯЗАННЫЕ
С ПЕРЕНОСОМ ЭЛЕКТРОНА
Электроны, участвующие в электродном переносе, сами по себе
не яшшются характеристикой электролиза Представления о
молекулярных орбиталях использую г для объяснения процесса окисления
как переноса электрона с верхней занятой молекулярной орбитали
1 Sekine Т., Yamum A., Sugino К. // J. Electmchem. Soc. 1965. No 112, P. 439-443.
2 Torii S. Op. cit.
} Nonaka /'., Wachi S., Fuchigami 7. // Chem. Lett. 1977. No 6. P. 47-50.
<CH2)„ ^XJ^
Катод
<снО<
67
(ВЗМО) в молекуле субстрата на анод, в то время как процесс
восстановления объясняется переносом электронов от катода на низшую
вакантную молекулярную орбиталь (НВМО) субстрата, как показано
нарис.4.81.
НВМО НВМО
ВЗМО —Ц— -=£- ВЗМО —f
-м- -ь-
-ъ- -к-
Субстрат Катион-радикал
Рис. АЛ. Молекулярно-орбитальная схема переноса электрона
У углеводорода перенос электрона происходит с ненасыщенных
связей и ст-связей напряженных соединений, таких как
циклопропан или ионные частицы. Однако обычные насыщенные
углеводороды редко подвергаются окислительно-восстановительным
реакциям. Даже окисление насыщенных гетероатомных соединений,
содержащих гетероатомы N, S и О, протекает довольно легко, как
и передача электронов от неподсленной электронной пары гсте-
роатома. Кроме того, в зависимости от природы гетероатомома Z
можно восстановительно расщепить С—Z- и Z—Z-связи.
Изолированные олефины, как правило, трудно окислить, но олефины,
напрямую связанные с гетероатомом, например, енамины, енольные
эфиры и силильные енольные эфиры (рис. 4.9), являются
электронно-насыщенными олефинами, поэтому их потенциалы
окисления значительно уменьшаются и они легко окисляются. Кроме
того, гетероатомы в значительной степени способствуют
стабильности электрохимически генерируемых соседних
peaкционнеспособных интермедиатов, как показано на рис. 4.10. Эти эффекты
являются важными характерными свойствами гетероатомных
соединений.
Рис. 4.9. Примеры фрагментов молекул легко окисляемых электронно-
насыщенных олефинов
1 Yoshida J., Kataoka К., Horcajada Я„ Nagaki A. // Chem. Rev. 2008. No 108.
P. 2265-2299; Yoshida J„ NishiwakiK. //J. Chem. Soc. Dalton Trans. 1998. 2589-
2596; Yoshida J., Maekawa Т., Murata Т., Matsunaga S.. hoe S. // J. Am. Chem.
Soc. 1990. No 112. P. 1962-1970.
68
» —с=о— > —c=s—
—C=SnMe4 —C=SiMc?
— С—X —С—N— —С—О—
—С—S— —С—SnMe.i —С—SiMei
Рис. 4.10. Схемы эффектов стабилизации гетероатомами
электрогенерируемых реакционных частиц
4.5. ЭЛЕКТРОВСПОМОГАТЕЛЬНЫЕ ГРУППЫ
Электровспомогательными являются функциональные группы,
которые облегчают перенос электронов и контролируют пути
реакции электрогенерированных активных частиц, чтобы обеспечить
селективное образование желаемых продуктов. Синтетические
приложения электровспомогательных групп в анодном окислении
распространены широко, в то время как для катодного восстановления
они редки1.
4.5.1. Электровспомогательные группы, основанные
на молекулярных орбитальных взаимодействиях
Орбитальное взаимодействие является очень эффективным для
повышения уровня ВЗМО. В принципе взаимодействие ВЗМО с
заполненной орбиталью с высокой энергией увеличивает ее
энергетический уровень. Например, уровень энергии а-орбитали С—Si,
как правило, гораздо выше уровня энергии ст-орбиталей С-Н
и С—С, поэтому о-орбиталъ С—Si эффективно взаимодействует
с несвязывающей /?-орбиталью гетероатома, например, N, О или
S (о—п-взаимодействия), если две орбитали выровнять в одной
и той же плоскости (рис. 4.11)2. Соответственно силильная группа
в а-положении активирует гетероатомное соединение к анодному
окислению.
1 Yoshida J.. Kataaka К., Horcajada Я., Nagaki A. Op. с it.; Yoshida J., Nishiwaki K.
Op, cit.; Yoshida J„ Maekawa T„Murata 7"„ Mafsunaga S„ /««? S. Op. cit.
1 Yoshida J., Kataaka K., Horcajada R., Nagaki A, Op. cit.; Yoshida J., Nishiwaki K.
Op. cit.; Yoshida J., Maekawa T.,Murata Т., Matsunaga S., hoe S. Op. cit.;
Koizumi Г., Fuchigami 1\ Nonaka T. // Bui. Chem. Soc. Jpn. 1989. No 62. P. 219-225.
—C=N—
—c=s—
69
с\
I
Si
Cb
^4 Я
С О Антисвязывающее взаимодействие
*0
si
О
_1L/ Чг-»
Q\
:"+•'■
V!
О Связывающее взаимодействие
Рис. 4.11. Схема взаимодействия между s-орбиталью С—Si и несвязывающей
2р орбиталью атома кислорода, повышающего уровень ВЗМО
Одноэлектронное окисление дает катион-радикал, в котором
о-орбиталь С—Si взаимодействует с наполовину вакантной р-ор-
биталью гетероатома, стабилизируя систему. Такое
взаимодействие также ослабляет связь С—Si, а следовательно, С—Si связь
расщепляется селективно. Полученный углеродный радикал
подвергается дальнейшему окислению с получением карбкатиона,
который захватывается нуклеофилом с получением желаемого
продукта:
R5Si
У
R
RiSi
У*
R
+SiR3
-Л
R
Nu
NuH
У
Y (4.11)
Y= N, О, S, я-система
Таким образом, силильная фуппа не только активирует субстраты
в отношении окисления, но и контролирует пути реакции".
В общем, электровспомогательный эффект силмльной группы
увеличивается в ряду S < N < О. и окислительный потенциал си-
лильного соединения с а-гетероатомом уменьшается в том же
порядке, как показано на рис. 4.12. Другими словами: эффект
кремния на окислительные потенциалы кислородных соединений
намного больше, чем на потенциалы органических соединении
азота и серы. Это согласуется с ббльшим перекрытием несвязыва-
lbid
70
ющей /i-орбитали кислорода с ст-орбиталью С—Si, чем с орбиталыо
азота и серы1.
Y = H: £,= 1.98V Y=H: £„ = 2,0V
Y = SiMe3: £"P=I,38V Y = SiMe3: Ep = 1,65V
..n r, u _ Ph^ ^\vr^COOMe
Me°YC'Hl5 MeO^Y ^^>Г
Y ^Y
Y=H: £",>2,5V Y = SiMe3: Ep = 1,6V Y = H: £,= 1,95V
Y=SiMe3: £„=l,72V Y = SnBu3: £; = 0,9V Y = SiMe3: £,= 1,45V
C7H,5^ ^SPh
О
x
CgHio Y
Y
Y=H: Ep= 1,35 V Y=H: fp>2,5V
Y=SiMe3: Ep= 1,25V Y = SiMe3: £,= 1,45 V
Рис. 4.12. Примеры уменьшения потенциалов окисления вследствие
молекулярных орбитальных взаимодействий
Взаимодействие ст-орбитали С—Si также эффективно для
повышения уровня энергии соседних я-систем (а—л-взаимодействия),
а С—Si-связь расщепляется селективно. Силильная группа,
следовательно, служит электровспомогательной для окисления я-систем. Сган-
нильная группа также выступает электровспомогательной для
электрохимического окисления гетероатомных соединений и л-систем2.
Анодное цианирование N-бензил-замещенных пиперазинов
обычно дает смесь двух региоизомерных продуктов. Однако введение
с ил ильной группы в качестве электровспомогательной направляет
путь реакции на образование одного продукта3:
Yoshida /.. Кашока К., Horcajada R., NagakiA. Op. cit.; Yoshida J., Nishiwaki K,
Op. cit.; Yoshida J., Mackawa Т., Murala Т., Matsunaga S., Isoe S. Op. cit,;
Yoshida J. Electrochemical Reactions of Organosilicon Compounds// Topics in
Current Chemistry, 170. Electrochemistry V. Berlin: Springer-Nferlag, 1994. S, 39—
82: Fuchigami T. Electrochemistry of Organosilicon Compounds//The Chemistry
of Organic Silicon Compounds. \Ы. 2 / Z. Rappoport, Y. Apeloig., Chichester: John
Wiley &Sons. Lid, 1998. Ch. 20.
Yoshida J., Kataoka K., Horcajada R„ NagakiA, Op. cit.: Yoshida J., Nishiwaki K.
Op. cit.
GaliE.L.BurvoisJ.P,t SinbandhirS. // Eur. J. Org. Chem. 1999. 2645-2653.
71
R2
l
ry\
r^An^
DK^
^R'
^SiMe^
-2e, -Me,Si"
NaCN
R2
I
RY\
r^An^
Рк-"^
/R'
^CN
(4.12)
Причем силильная группа также уменьшает окислительный
потенциал амина.
4.5.2. Электровспомогательные группы на основе
функциональных групп с легким переносом электронов
Легко окисляемые функциональные группы, например, арилтио
(ArS), работают какэлекгровсиомогательные, т.е. введение группы
ArS в ct-положение эфиров и карбаматов уменьшает их потенциалы
окисления. Важно отметить, что окисление a-ArS-замеще иного ге-
тероатомного соединения приводит к селективному расщеплению
С—S-связи, генерируя катионный интермедиат, стабилизированный
соседним гетероатомом. Полученный катионный интермедиат
реагирует с нуклеофилом с селективным получением продукта.
Использование группы ArS в качестве электровспомогательной
расширяет диапазон нуклеофилов, что позволяет in situ применять
углеродные нуклеофилы (например аллилсилан), даже если они легко
окисляются1:
О-
I
ССЬМе
SPh
Анодное окисление
I
С02Ме
89%
(4.13)
ОМе
ОМе
ОМе
ОМе
LiCKVMeNOi
SPh
PhS'
SPh
-SiMci
(4.14)
В этих случаях благодаря группе ArS происходит селективное
расщепление С—С-связи и аллильная группа вводится к а- или бензиль-
ному углероду селективно.
1 Ют S„ HayashiК, Kitano К, СЫЬа К. // Oig. bet. 2002. No4. R 3735-3737; ШЬаК,
Vchiyama 7., KimS.,Kitano У., TadaM.//Org. Lett 2001. No3. P. 1245-1248.
72
Причем известно, что электровспомогательный фрагмент
и реакдмонноспособный участок отличаются в случае катодного
восстановления. Например, первый перенос электрона
происходит на легко принимающую электроны карбонильную группу,
а затем селективно расщепляется соседняя связь углерод — гс-
тероатом1:
0 г о-^ 1 + о
кДч^-У^ КЛ^У "^КД^ (4.15)
<Y=OR', SR'^RTl")
Основываясь на этом принципе, может быть легко достигнуто
селективное расщепление связи С—О в бензильной и алл ильной
позициях:
OCOR 2е,н+
CHi
(4.16)
Хотя C-F-связь не так легко расщепляется при восстановлении
из-за ее большей энергии связи по сравнению с С-Н-связью, фтор,
присоединенный к бензильному положению или гх-карбонильной
и иминогруппам, легко удаляется катодным восстановлением. Это
очень похоже на случаи, приведенные ниже:
X
х
F3C-^^Ar
2e.-F~
Me,SiCl
XSiMc3
X,
F3C^ ^Ar
(X = O, NR)
(4.17)
Как видно из (4.17), перенос электрона происходит сначала
на ароматическое кольцо или карбонильную и иминогруппу с
последующим [i-элиминированием фторид-иона2. Когда катодная
реакция, показанная в (4.17), осуществляется в отсутствие MejSiCl,
происходит дальнейшее катодное восстановление, приводящее к
последовательному элиминированию атомов фтора, дающему сложные
продукты.
' KandeetZ.. Nmaka T.,Fuchigami Т. // Bull. Chem. Soc. Jpn. 1986. No 59. P. 338-340.
1 Uneyama K., Kato T. // Tetrahedron Lett. 1998. No 39. P. 587-590; Vneyama K.,
Maeda K., Kato 7\, Katagiri T. //Tetrahedron Lett. 1998. No 39. P. 3741-3744.
73
4.5.3. Электровспомогательные группы, основанные
на межмолекулярных координационных эффектах
Этот эффект очень важен с точки зрения электронного переноса.
Реакция переноса электрона в растворе, как правило, облегчается
стабилизацией полученного ион-радикального и ионного интерме-
диатов путем координации молекулы растворителя или противо-
ионов. Например, восстановительный потенциал катиона металла,
как правило, становится более отрицательным в растворителях
с большими донорными числами, поскольку положительная
природа катиона металла уменьшается при сольватации в таких
растворителях. Известно, что потенциалы восстановления кетонов,
иминов, циано- и нитро-соединений сдвигаются в положительном
направлении при добавлении протонов. Полярографическое
восстановление нитросоединений интенсивно изучалось ранее. Когда рН
раствора сдвигается в более кислую сторону на одну единицу,
восстановительный потенциал становится более положительным примерно
на 58 мВ. Положительный сдвиг потенциала восстановления из-за
протонирования ненасыщенных функциональных групп приводит
к положительно заряженной частице, как показано на рис. 4.13.
Z -^ \=ZH R—С =N -^-* R— C=NH
(2 = 0,NR)
/ОН
ArNOi _Л_* Ar—N
<R> (R) \>
Рис.4.13. Примеры протонирования функциональных групп, приводящего
к анодному сдвигу потенциала восстановления
Кроме того, потенциалы восстановления кетонов часто сдвита-
ются к положительным величинам в присутствии кислот Льюиса.
Это связано с координацией кислоты Льюиса по атому кислорода
карбонильной группы, приводящей к уменьшению электронной
плотности карбонильной группы или координации кислоты Льюиса
с анион-радикальным интермеди атом, генерируемым путем одно-
электронного восстановления карбонильной группы, влекущем
стабилизацию интермедиата, как показано на рис. 4,14.
Известно также, что вторые потенциалы восстановления ди-
кетонов типа антрахинона и динитробензолов сильно сдвигаются
в положительном направлении в приеугствии спиртов. Это связано
с образованием водородных связей между дианионными интерме-
диатами, генерируемыми путем двухэлектронного восстановления,
и спиртами.
>
74
RO—H "О—(v /)—(Г-Н—OR
Рис. 4.14. Примеры взаимодействий, вызывающих анодный сдвиг потенциала
восстановления вследствие координации с кислотой Льюиса и присоединения
водорода
4.5.4. Электровспомогательные группы, основанные
на внутримолекулярных координационных эффектах
Если молекула субстрата имеет определенное координирующее
место (сайт), например, функциональную группу или гетероатом,
чтобы стабилизировать электрогенерированный ионный интерме-
диат, перенос электрона усиливается за счет внутримолекулярной
координации, как показано на рис. 4.15. Так как такие
функциональные группы, как пиридил, карбонильные и эфирные группы,
являются эффективными, они заметно уменьшают потенциал
окисления. Такая координация будет способствовать последующим
химическим стадиям типа разрыва связи. Когда разрыв связи
сопровождает перенос электрона, как показано на рис* 4.15,
потенциал окисления значительно уменьшается. Это довольно новая
концепция и методология управления переносом электрона'.
Комбинация орбитального взаимодействия и внутримолекулярная
координация являются весьма эффективными для управления как
потенциалом окисления, так и путем реакции.
г
Product
Interamolecular assistance /=N
(Y=RCO, RO,^J-)
Рис.4.15. Внутримолекулярное содействие за счет координации
с подходящей функциональной группой Y, приводящее к катодному сдвигу
потенциала окисления
1
YosMda J., Izawa M. //J. Am. Chem. Soc. 1997. 119. 9361—9365; Wannabe M.,
Suga S.< YostiidaJ. // Bull. Chcm. Soc. Jpn. 2000.73. 243-247; YoshidaJ., SugaS.,
Fuke K.. Watanobe M. //Chem. LelL 1999. 251-252.
75
4.6. СТРУКТУРА ОРГАНИЧЕСКИХ ЭЛЕКТРОДНЫХ РЕАКЦИЙ
Хотя а1ектродныс реакции ограничены окислением и
восстановлением, различные последующие химические реакции часто
происходят после органического переноса электрона. Следовательно,
органические электродные реакции отличаются от реакций
неорганических соединений. С синтетической точки зрения органические
электродные реакции подразделяются на несколько типов. Ниже
приведены некоторые примеры катодного восстановления и
анодного окисления различных органических молекул.
4.6.1. Типы превращений функциональных групп
Катодное восстановление'.
R-N02
2е,2Н+
2^H^R_NQ 2*Ж ,
-н2о
> R-NHOH 2е<2*Г > R-NH,
(4.18)
R
чс=о
R'
/
2с ^Н4 \
1 > СН—ОН
R
R'
(4.19)
Ar-COOR-
(Н)
4е,4Н1
->Ar-CH2OH + RON
(4.20)
R-CONR2
R—COv
NR'
4е,4Нч
->R-CH2NR'2+H20
R—CO
Sc,8H' R СНз
R—CH2
NR'+2HiO
(4.21)
(4.22)
Ar-N^CI" 4e4H+ )Ar-NH2 + HCl
R
R
\
4C=N-R' 2е2|Г> СН—NH-R'
R/ Rx
R
V
C=N—OH
4e. 4H+ R\
R
/
» CH—NH2 + H20
Rx
R-C=N 2е,2>Г >[R-CH= NH] 2e,2Fr >R-CH2NH2
(4.23)
(4.24)
(4.25)
(4.26)
76
Ar-SO,H 4c'4H+ >Ar-SH+2H20 (4.27)
_C=C 2e>2H+ >CH=CH 2e,2H+ )CH2_CH2_ (428)
R-02^R-Oil^R-Oi^R4C> <4-29)
Анодное окисление:
R-CH2OH ~2e'-2HH )R-CHQ -2c--2QH° )R-COOH + H20 (4.30)
^\ -2e, -2H+ R\
CHOH : » C=0 (4.31)
/ /
R' R'
Ar-CH, ^e-"2H+ )Ar-CHO 2e"HJ >Ar-COOH (4.32)
3 +20H" +OH" V '
Аг-NH-NH-Ar ~2e,"2H > Ar-N=N-Ar ~2e'"2H >
+20H
-2e.-2H+, A Kr v, . (4.33)
О
О
R—S—R "2^,7H>R—S—R ~^;"H>R—S—R (4.34)
+OH || +OH || '
О О
R-CH2NH2 ^tc'-4H+ )R-C=N (4.35)
R-SH ~e~H^ >1/2R-S-S-R (4.36)
В уравнениях 4.18—4.36 использованы стехиометрические
формулы, включая элементарные стадии реакций, но показаны только
конечные продукты, а элементарные стадии реакции опушены.
Следует также отметить, что реагенты, указанные над стрелками,
взаимодействуют с электрохимически генерируемыми интермедиатами,
но они не подвергаются прямым реакциям переноса электронов.
4.6.2. Тип присоединения
Катодное восстановление:
СООН
R—С—R' 2С'2"» R—С—R' (4.37)
II °3 I
о он
77
R
2e.2H+ I I I
R—C—R' -r—j* R—С— С— СН (4.38)
О ' x OH
H2C=CH-CH=CH1 2e'2H+ >
* 2CO,
(4.39)
2e, 2H ^HOoC-CH-CH2-CH=CH-CH2-COOH
2C02
R2CX2 2e, -2X \ /
(X: halogen) ^^ (4.40)
Анодное окисление:
Q^K«~Q-VR (4-4,)
W-2e. -2hT I I
>R— COO— С—С—ООС—R (4.42)
|-+ >=( > I—С—С—NHCOCH3 (4.43)
/ \ H20/CH3CN I I
4.6.3. Тип включения
Катодное восстановление:
О
Y-NCb 2C'^' > Г ^1 (4.44)
(Y = ROCO, ArS02) M kO>v-N^Y
Н
Анодное окисление:
R R
N—NH, ~^~?Г> N—NH—NH—R' (4.45)
R/ " *-nh2 R/
4.6.4. Тип замещения
Катодное восстановление:
R-X 2c- * >R-H
КГ (4.46)
(Х = 1,Вг,С1)
78
_^_н^^^_^_у
Y+
(4.47)
Анодное окисление:
R
R-H "2e'~H* >R-X
X"
(X = LBr,CI,F)
(4.48)
(R = NH2, OR')(Y= RCOO, NCS, CN, RO, NCO, halogen)
-2e, -2H+
R—C—N—CH
О R'
R'COOH
(4.49)
- R—C—N—CH2—OOCR" (4.50)
-2е,-*Г
Ar—N—CH3
R
RO
-2e, -IT
О R'
-> Ar—N—CH2—OR'
R
RCOO
-2c, ^
-> Ar—N—CH2—OOCR' (4.51)
R
CN"
-> Ar—N—СЖ—CN
I
R
4.6.5. Тип заместительного обмена
Катодное восстановление:
\ /
N0,
6с, 6Н
ОН
*НО—^ Ь— NH2+X- (4.52)
(X = halogen, OR)
Согласно (4.52) также происходит превращение функциональной
группы.
Анодное окисление:
R-COO
-2е
*R-Y + CO,
(Y = R'COO, R'O, N03, НО)
О
R—X
(X = l, Br)
-C-I/2X, И
^ R—NHC—CHi
HiO/CH,CN
(4.53)
(4.54)
79
4.6.6. Тип элиминирования
Катодное восстановление:
—С—С ^-> —С=С— + Х- + Y- (1,2-elimination) (4.55)
X Y
(X, Y = halogen, RCOO, RS03, RS, HS, HO; X = Y = O-CO-O)
"T^Y" 2c~2X> R"-A^R'(ij-eiimination) (4.56)
X X
Анодное окисление:
I I -2e. -2H" I I
—С—С >—C=C— + 2C02( 1,2-elimination) (4,57)
HOOC COOH
4.6.7. Тип димеризации
Катодное восстановление:
R R R
C=0 -£liL-> 1/2 R— С—С—R' (4.58)
/ II
R' он он
R R R
\ ,. H+ I I
C=NR" ±J2-^ 1/2 R'— С—С—R' (4.59)
R' R"HN NHR"
C=C -^U 1/2 Y—CH—C—C—CH—Y (4.60)
/ V 1 1 1 1
(Y = CN, COOR, CONR2, RCO)
R-X—^->1/2R-R+X" (4.61)
(X = halogen)
Ar-N03 3.e^H^l/2Ar-N=N-Ar *' "* )
-J/2H1O 1 -1/2H?0
О
■ пГ1> У2 Ar—N=N—Ar <462>
eH -*l/2Ar—NH—NH—Ar
-1/2HjO
80
Анодное окисление:
R-СОСГ ~2g > 1/2R-R+ С02 (4.63)
R (4.64)
\ / ^н+ I I I I
С=С + ROH — > 1/2 R0—С—С—С—С—OR (4.65)
/ \ I I I I
R2N R2N NRt
\ _е _н+ N /
С=СН2 > 1/2 С=СН—СН=С (4.66)
/ / \
R2N R2N NR2
4.6,8. Кросс-димеризация
R R i H
\ \ / 2c 2Hh III
C=0 + C=C ' > R— С—С—С—Y (4.67)
/ / \ III
Y OH ' '
(Y= COOR, CN, CONH2)
Согласно уравнению (4.67) отношение выхода продукта кросс-ди-
мермзации к выходу продукта не перекрестной димеризации можно
увеличить, но это не так легко для уравнения (4.68):
R-COOH + R-COOH "2е2ьГ >R-R/ (4.68)
-2С02
4.6.9. Циклизация
R'
R-C-R'-CH=CH-R',i£^R^CH—CH2—R"<4.69)
^CH2^ _2 2Н- ^CFL^
(СН2)« (СН,)„ '-=^> (CH2)ffl | (СН2)„ (4.70)
4.6.10. Образование полиморфизма
CS2-^l/6 V \=/S"jf" (4.71)
81
4.6.11. Тип полимеризации
CH3=CH-Y (CataliticCamount)> Mn -CH2-CH-+- (4.72)
Y
(Y= COOR, CN, CONH2, Ph, AcO, aLkyl)
Реакция, описанная уравнением (4.72), инициируется катодным
восстановлением активированного олефина. Достаточно
каталитического количества электричества, чтобы облегчить полимеризацию
по механизму цепной реакции, в го время как последующая реакция
анодной окислительной полимеризации потребляет стехиометриче-
ского количества электроэнергии:
О -
(Y=NH,S)
-2лсг —2лН
» \/п
(4.73)
4.6.12, Тип расщепления
2с. 2Н"
R-S-S-R
■* 2R-SH
X Y
(X,Y=NR2, OR)
/
(4.74)
(4.75)
4.6.13. Тип металлирования
Металл, используемый в качестве электрода, включается в
продукт:
R R R
С=0 + 1/2 М (Cathode) 3en^H > 1/2 СН—М—СН (476)
R'
У
-ОН
(M = Hg,Pb)
RMgX + 1/4Pb( Anode) -
(X = halogen)
R'
./
\
К
-IMgXf
-H/4R4Pb
(4.77)
4.6.14. Тип ассиметрического синтеза
R
R'
R
C=Z ' > R—C—ZH
(X = 0?NR") н
(4.78)
82
R—S—R' '? > R—S* —К (4.79)
-H ||
0
^c=c/ 2e.2H\ R-_C._CH (4 gQ)
/ \ II
к н '
В качестве хирального источника используются хиральные
фоновые соли, хиральные растворители, хиральные адсорбенты и
хиральные модифицированные электроды.
4.7. ЭЛЕКТРОХИМИЧЕСКИ ГЕНЕРИРУЕМЫЕ
РЕАКЦИОННОСПОСОБНЫЕ ЧАСТИЦЫ
Электрохкмически генерируемые реакционн ©способные частицы
часто подвергаются последующим реакциям в адсорбированном со-
стоянии или вблизи поверхности электрода, давая определенные
продукты. Так как природа этих активных частиц зависит от самого
электрода и силы электрического поля (примерно 107—108 В см-1),
их реакционная способность и поведение часто весьма отличаются
от тех же покааателей активных форм, генерируемых другими
способами. Полезные синтетические реакции могут быть разработаны
с использованием таких уникальных электрогенерированных
активных частиц. Большое число ионных и радикальных частиц
генерируется путем электролиза. Электрохимически генерируемые
активные частицы подразделяют на углеродные и гетероатомные.
Методы их генерации рассмотрены ниже.
4.7.1. Углеродные частицы
Анодно генерируемые углеродные частицы
Окисление карбоновой кислоты. Анодное окислительное декар-
боксилированис карбоновой кислоты генерирует алкильный
радикал и/или алкильный катион1. Карбоновая кислота с линейной
алкильной цепью окисляется на Pt аноде в слабокислом растворе,
генерируя в основном соответствующий алкильный радикал. Причем
анодное окисление карбоновой кислоты с а-разветвленной
алкильной цепью на углеродном аноде в нейтральном или щелочном
растворе генерирует в основном соответствующий алкильный
катион:
1 Schafer H. Recent Contributions of Kolbe Electrolysis to Organic Synthesis //
Topics in Current Chemistry, 152. Electrochemistry, IV/cd. E. Stcckhan. Berlin:
Springer-\ferlag, 1990. P. 91-151.
83
R-COOH ~C'"H' >R-COO'—^^-»R*^^-»R+ (4.81)
Окисление карбаниона и активных соединений водорода.
Потенциалы окисления карбанионов чрезвычайно низки, а их одноэлек-
тронное окисление генерирует радикалы:
R ^^->R" (4.82)
X-CH2-Y ~е~И* )X-CH-Y^UX-CH-Y (4 g3)
(X, Y = CN, COR, COOR, N02)
Окисление алкилгалогенидов. Алкилиодиды легче восстановлива-
ются и окисляются по сравнению с соответствующими бромидами.
Их одноэлектронное окисление генерирует катион-радикальный ин-
термедиат, затем следует элиминирование галогена с образованием
алкильного катиона:
R-X^-HR-ХГ "1/2Хз >R" (484)
<Х = Вг,1)
Окисление ояефииов, кетонов, иминов, напряженных и каркасных
соединений.
Потенциал окисления кстона, как правило, высок, но даже
алифатический кетон окисляем. Хотя алкан трудно окислить,
напряженные циклопропаны и адамантаны каркасного типа могут быть
окислены сравнительно легко. Одноэлектронное окисление этих
соединений генерирует катион-рад и калы:
\ -е \+ ■
C=Z =—> С—Z (4.85)
/ /
(Z = C,0,N)
CwC ^> АЯ (4.86)
/ \ / \
Окисление ароматических соединений. Первоначально перенос
одного электрона от ароматического кольца происходит с
образованием катион-радикала кольца. Когда у ароматического кольца
существует ал кил ьная цепь, элиминирование «-протона изалкильной
цепи приводит к образованию соответствующего бензил-радикала,
который подвергается дальнейшему одноэлектронному окислению,
образуя бензил-катион. В случае затрудненного элиминирования
гх-протона или ароматики, лишенной а-протона, ароматический
катион-радикал атакуется нуклеофилами, в результате происходит
84
нуклеофильное замещение в кольце. В отсутствие нуклеофилов
происходит гомо-сочетание катион радикалов или радикальных интер-
медиатов:
Ar-CH2-R —^-4 Ar-CH2-R ~H+ >
-н+
^Ar-CH-R
->Ar-CH-R
Аг-OR
->Ar-OR
н+
(4.87)
^ Аг-OR—^Ar-OR (4.88)
Катодно генерируемые углеродные частицы
Восстановление алкилгалогенидов. Легкость восстановления ал-
килгалогенидов связана с энергией связи С—X, а следовательно,
йодидньтс соединения наиболее легко восстанавливаются, в то время
как хлоридные соединения восстановить наиболее трудно.
Одноэлектронное и двухэлектронное восстановление
алкилгалогенидов дает соответственно алкильные радикалы и алкильные
анионы:
R-X
с-х-
->R'
->R~
(4.89)
(X = C1sBrJ)
Восстановление кетона и амина. В случае катодного
восстановления кетона и имина в водном растворе природа образующихся
активных частиц, а также механизм реакции меняются в зависимости
от рН раствора. В кислом растворе атом кислорода кетона и атом
азота имина протонируются, поэтому их потенциалы восстановления
смещаются в положительную сторону, а их одноэлектронное
восстановление дает нейтральные радикалы. Наоборот, в щелочном
растворе протон и рования кетона и имина не происходит из-за низкой
концентрации протонов. В этом случае анион-радикал генерируется
первым, а затем образуется дианион:
\
C=Z
/
(Z
-* С—Z—|
/
0,N)
-^-* С—ZH
/
\- -
■*С—Z
/
11+
-н> С— ZH(4.90)
Восстановление активированного олефина и сопряженного оле-
фина. Активированный олефин легко восстанавливается, поскольку
электроноакцепторная группа, присоединенная к двойной связи,
уменьшает электронную плотность на ней. Изолированные олефины
85
не так легко восстановить, но сопряженный олефин
восстанавливается. Одноэлектронное восстановление олефина даст анион-радикал:
\ / е
С=С —^ —С—С—Y (4.91)
/ V м
(Y = CN, COR, COOR, ^-с')
Восстановление соединений активного водорода. Одноэлектронное
восстаноыение соединения с активным водородом генерирует
соответствующий анион, при этом выделяется активный атом водорода
в виде газообразного водорода:
X-CH2-Y C'/2H2 >х_ён-у (4.92)
(X,Y = CN, COR, COOR)
Восстановление гем- и виц-дигалосоединений. При двухэлек-
тронном восстановлении этих соединений образуются карбен и де-
гидробензол:
СХ2 " ' ^> С: (4.93)
(Х=С1, Вг, I) (Карбен)
X
2е, -2Х~
(4.94)
(X = О, Вг, I) (Дегидробензол)
4.7.2. Гетероатомные частицы
В отличие от углеводородов гетероатомные соединения
окисляются, так как они имеют неподеленные пары электронов на гете-
роатоме, от которых происходит перенос электронов. В целом
легкость окисления меняется в ряду N>S>0,ripH этом образуются
различные азотсодержащие активные частицы1.
Азотсодержащие частицы. Одноэлектронное окисление амина
генерирует катион-радикал на атоме азота. Катион-радикал
ароматического амина является относительно стабильным, а
алифатического амина — настолько неустойчивым, что а-протон немедленно
элиминирует, а затем активный участок переходит на а-углерод.
У алифатических аминов иминиевые ионы настолько неустойчивы,
что расщепление С—Н-связи происходит преимущественно. Причем
иминиевые ионы ароматических аминов, карбаматов и амидов яв-
Fuchigami Т.. Nonaka Т, // Electrochemistry. 1984. No 52. P. 19—25; Fuchlgami Т.,
Sato Г., Nonaka Т. //J. Org. Chem. 19S6. No 51. P. 366-369.
86
ляются стабильными. Следовательно, реакции нуклеофильного
замещения (метоксилирование, ацетоксилирование и цианирование)
происходят эффективно по а-положению к атому азота1:
\ _е V+ -Н+ \ -е
N—СН2 -* N—СН2 ^ N—СН—
/
/
/
—> NH +
/
сно*
HjO
-н+
н2о
N=CH— «■
/
\ +
■* N—СН—
/
> N-
/
Nu
I
СН
(4.95)
(Nu = MeO, AcO, CN, etc.)
Анодные реакции замещения являются одними из
характеристичных электрохимических реакций. Они полезны для создания
связи С—С в соседнем к атому азота положении. Анодное замещение
в соседнее к атому азота ими нов положение также возможно2:
Nu
-2е.-Н\
Nu"
■N
(4.96)
(Nu = MeO,AcO, F)
В зависимости от молекулярной структуры аминов активный сайт
сохраняется при исходном азоте, приводя к генерации различных
активных азотных частиц. В результате реакция протекает по атому
азота, как показано на рис. 4.16J. Двухэлсктронтгос восстановление
N, N-дигалосоединений дает соответствующие нитрены4, в то время
как при двухэлектронном окислении азиридина и среднеразмерных
циклических аминов с последующим депротонированием NH-
фрагмента образуются ионы нитрения5. Анодное окисление гидра-
зиновых производных приводит к образованию аминонитренов6.
В частности, анодное окисление N-фталимида в присутствии оле-
фииа приводит к образованию соответствующих азириднновых
производных с высокими выходами, как показано на рис. 4.167. Реакция
протекает через анодногенерируемый аминонитрен:
Shono Т. Ор. с it.
Baba D., Fuchigami T. // Tetrahedron Lett. 2003. No 44. P. 3133—.1136.
Fuchigami Т., Nonaka T. Op. cit.; Fuchigami Т., Sato Г., N опака Т. Op. cit.
Fuchigami T.,Jwata K., Nonaka T.//i. Chem. Soc., Chem. Commun. 1976. P. 951-952.
Gaxtmw P.O., Ni&iguchil., Yamamrto////J.Am, Chem, Soc. I975.No97, R 1600—1602.
Fuchigami Т., Sato Т.. Nonaka T. //Electrochim. Acta. 1986. No 31. P. 365—369.
Watson LP.. Yu L., YudinA.K. // Ace. Chem. Res. 2006. No 39. P. 194-206.
87
R2
R1—N—R3
2e, -2X-
<R- = R3=X)
-2c
-* R'-N ^ >
(Нитрен)
-* R-.N— N
ex
NHR
(RJ=R,N,R^RJ=H) (Аминонн^сн)
-> R—R, R2N—N=N—NR2
+N2
-2c, -W
<R3 = H)
R2
R1—N+
(Ион иитрсния)
R2
-* R1—N—R3
(ЛММИКИУМИЛ раЛНКа.1)
R2
NH
• +
NH
-H'
-c,-Hf
О
(RJ=Li,M&Br)
2e,-l/2H:
(Амннилрмиш) V^NR'R2 or R'R^-NR'R2
>3 _
(R1 = Li)
►з^
<R* = MgBr)
<RJ=H)
R2
. I
R1—N"
1d2vtb4
-> R'R'NR
(Амид анион) ^rJ or r2. элекТрОНоакцепторная группа)
Рис. 4.16. Схемы электрохимической генерирации peaкционнеспособных азотсодержащих частиц
о
R1
N—NH2 +
0
(4.97)
Кроме того, анодное окисление бициклических аминов и
алифатических аминов, лишенных а-протонов, эффективно генерирует
аммнильные радикалы. В первом случае анодное элиминирование
ct-протона не может произойти по правилу Бредта:
н+
/^
NH
N
/-BuNH2 —с' Н* >r-BuNH
(4.98)
(4.99)
Кислородные частицы
Окисление спирта и карбоновой кислоты. В основном из этих
соединений генерируется радикальные частицы, но также могут быть
получены и ка] ионные час гиды:
RO"
-+RCT
-е,-Н+
*ROH
(4.100)
R—COO
R—COOH
-е,-Нн
->R—COO- -z^R—COO+(4.101)
Окисление эфира. Аналогично азоторганическим соединениям
анодное окисление эфиров дает ot-катион по отношению к атому
кислорода, который подвергается реакции нуклеофильного
замещения. Однако сфера его применения ограничена из-за высокого
потенциала окисления эфиров:
-2е. -Н+
RO—CH2—R' 2е' Н >
RO—СН—R'«—>RO=CH—R'
NiT
Nu'
> RO—CH—К
l
Nu
(4.102)
(Nu - MeO, AcO, F)
Восстановление спирта и карбоновой кислоты. Одноэлектронное
восстановление этих соединений генерирует соответствующие
анионы, выделяя молекулу водорода:
89
YOH—6l 1/2Нд >YO" (4.103)
(Y = Ar,R,RCO)
Восстановление дикислорода. Одноэлектронное восстановление
дикислорода приводит к образованию супероксид-иона, который
называют активной формой кислорода и который работает как
окислитель или восстановитель, а также основание и радикал (см. разд. 5.2).
02—с-^ОТ (4.104)
4.7.3. Халькогенные (сера, селен, теллур) частицы
Халькогенные соединения легко окисляются. Окисление
соответствующего аниона дает радикалы, которые далее окисляются
до катионов:
RZ~ г^ RZ* r^ RZ+ (4.105)
е е
(Z = S,Se,Te)
Эти процессы окисления, как правило, относятся к обратимым
электронным переносам, хотя условия реакции и состав электролита
влияют на обратимость.
В случае сульфидов анодно генерируемый катион-радикал при
атоме серы атакуется нуклеофилами (нуклеофильное
присоединение). Однако у сульфида с сильно кислым а-водородом
преимущественно происходит а-депротонирование катион-радикала
и в конечном итоге селективное а-нуклеофильное замещение. Среди
реакций замещения в синтетической органической химии
фторирование имеет большое значение. Однако селениды, даже имеющие
электропоакцептортгую группу, не всегда подвергаются пуклеофиль-
ному замещению, а нуклеофильное присоединение также
происходит по атому селена. В противоположность этому анодное
замещение органотеллурндов неизвестно, а в основном происходит
нуклеофильное присоединение по атому теллура:
RZCH,—Е WG ~2е,~Н > RZ=CH— EWG -^-»
—н
(EWG: электроно-акцепторная группа)
(Z=S, Se)
■^p^RZCH—EWG (4.106)
Nu
(Nu = MeO, AcO, F)
90
Галогенилные частицы. Галоген легко окисляется и легко
восстанавливается. Поскольку окислительно-восстановительная реакция
галогенид-иона является обратимой, он широко используется в
качестве медиатора непрямого электрохимического окисления (см.
гл. 3 и 5):
— "e^X'rzL"»X+ (4.107)
е с
(Z = Cl,Br,l)
Производные элементов 14 н 15-й групп. Активные формы
образуются при электронном переносе преимущественно с участием ге-
тероа гомон:
R3Y-X * >R3Y~ (4108}
(Y = Si,Sn)
R2Z-X 2e'~X" ) R2Y" (4.109)
r3Z^->R3Z+
(Z = P,As,Sb)
Глава 5. ОРГАНИЧЕСКИЙ
ЭЛЕКТРОСИНТЕЗ
Разработка новой методологии создания высокоселективных
реакций является одной из наиболее важных областей
органической синтетической химии. Как уже отмечалось выше,
электродные реакции имеют специфические особенности, позволяющие
управлять селективностью, поэтому учет как электрохимических,
так и обычных химических факторов усложняет управление
электрохимическими реакциями. Природа электродов имеет значение
как для собственно переноса электронов на границе раздела фаз,
так и самой реакции. Как было описано выше, электрод обладает
свойством, позволяющим управлять химической реакцией через
адсорбцию и ориентацию молекулы субстрата по отношению
к поверхности электрода. Хотя перенапряжение водорода и
кислорода может быть критерием выбора подходящих электродных
материалов для достижения желаемой электрохимической реакции
в водном растворе, в апротонных растворах это перенапряжение
не «работает*. Следовательно, не так легко предсказать, какие
электродные материалы подходят для требуемой
электрохимической реакции в апротонных растворителях. Чтобы достичь
высокой селективности желаемых реакций, были разработаны новые
электрохимические подходы. Ниже будет подробно описана
относительно новая электрохимическая методология, которая нашла
широкое применение. Хотя уже известно множество применений
таких электрохимических реакций в органическом синтезе, в этой
главе приведено ограниченное количество примеров. Поэтому
можно рекомендовать обратиться к другим источникам
информации1.
Hammerich C.Speiser В. Organic Electrochemistry. 5lh edn. CRC / Taylor &.
Francis. 2014: Torii S. Eleclroorganic Reduction Synthesis. Vol. I, 2. Kodansha
and Wiley-VCH >№inheim GmbH, 2006; Bard A.J., Strarmann M. Encyclopedia
of Electrochemistry // Electrochemistry Organic. \fal. 8. / H.J. Schaefer. Wfein-
heim: John VWJey & Sons., 2004; Lund H., Hammerich O. Organic
Electrochemistry. 4th cdn. N. Y.: Marcel Dekker, 2001: Grimshaw J. Electrochemical
Reactions and Mechansims in Organic Chemistry. Amsterdam: Elsevier, 2000; Fry A.J.
Synthetic Organic Electrochemistry. N. Y: Wiley Interscience, 1989; Sliotio T.
Elect morgan ic Chemistry' as a Tool in Organic Synthesis. Berlin: Springer-Vferlag,
1984; Rtfi M.R., Covitz FM. Introduction to Organic Electrochemistry Techniques
and Applications in Organic Synthesis. N. Y: Marcel Dekker, 1974; Mann C.K„
Barnes K.K. Electrochemical Reactions in Nonaqueous Systems. N. Y: Marcel
Dekker, 1970.
92
5.1. ЭЛЕКТРОКАТАЛИЗ
Хотя непрямые электрохимические реакции, использующие
медиаторы, т.е. электрокаталитические реакции, уже были показаны
в гл. 2, в этой главе описаны различные примеры их синтетических
приложений1. Некоторые другие такие примеры также рассмотрены
н гл. 6 и 7.
5.1.1. Классификация и типы медиаторов
Медиаторы делятся тга две группы (табл. 5.1);
• внешнесферные медиаторы, включающие перенос электронов
между посредником и молекулой субстрата:
R-X-
T^S
-»R. +X
(5.1)
внутресферные медиаторы, включающие
окислительно-восстановительные реакции, протекающие по типу обычных химических
реакций:
I <
R
[ -2ej Rl
снон птг^
Я2 R2
R
СН01 W* С=°
R2
(5.2)
Таблица 5.1
Классификация типов медиаторов
Медиатор
Вне шнесферный
Поливалентный ион металла
Триариламин
Виологен
Полиииклический ароматический углеводород
Внутрисферный
Галоген
N-оксильное соединение
Сульфид
SaveantJ.M. //Лес. Chem. Res. 1980. No 13. P. 323-329; Steckhan E. // Angew.
Chem. 1980. No 98. P. 681-699; Torii S. // Synthesis. 1986. P. 873-886; Lund H.,
HanunerichO. Organic Electrochemistry. 4th edn. N. Y: Marcel Dekker, 2001. Ch, 29;
Steckhan E. Topics in Current Chemistry 142. Electrochemistry 1. Berlin: Springcr-
Verlag, 1987. P. 1-69.
93
Медиаторы также классифицируются на окислительные и
восстановительные и на органические и неорганические. Известны
следующие редоке-медиаторы: I) неорганические соединения (ионы
поливалентных металлов, комплексы переходных металлов и гало-
гснид-ионы); 2) органические соединения (полициклическис
ароматические соединения, триариламины и 2,2,6,6-тетрамстил
пиперидин нитроксилы (TEMPO)); 3) соединения с аномальной
валентностью; 4) гипервалентные соединения.
Недавно новые медиаторы — карбораны, содержащие атомы
бора, были разработаны для высокоэффективного катодного
восстановительного дегалогенирования1. Триарилимидазольные
медиаторы также недавно были предложены для селективного анодного
окисления2. Можно привести следующие примеры окислительных
и восстановительных медиаторов:
окислительные медиаторы: Ru^/Ru3"1", Со*+/Со2+, IO^/IOj,
MnJ7Mn2+,Ni?7Ni2+,Ce47Ce3+, s2027S02+, Cr^/Cr^, 0sN70s"\
галоген (X+/X), N03/NOJ, CAN, Ar3N7Ar3N, производные N-ок-
сила (R,R2N = О), сульфиды (R,SR2);
восстановительные медиаторы: Ni2+/Ni<(>', Co2+/Co+, Cr-1+/Cr2+,
Мо2+/Мо<°>, Ti3+/T\2+, Sn^/Sn2+, Sn27Sn(U), Mg27Mg*0\ Pd27Pd<">,
нафталин, антрацен, фенангрен, пирен, фуллерен С6(), бенэони-
трил, фталонитрил, антрахинон, нитробензол, виологен,
супероксид-ион.
Потенциалы восстановления ароматических медиаторов
непрямого катодного восстановления приведены в табл. 5.2.
Таблица 5.2
Потенциалы восстановления ароматических медиаторов,
используемых для непрямого катодного восстановления
Соединение
Фтало нитрил
4-М етоксибе нэофенон
Антрацен
Мстилбснэоат
Бензонитрил
Хризен
Нафталин
^(Вотн. НКЭ)
-1,7
-1,8
-2,0
-2,2
-2,2
-2,5
-2,5
1 Hanoi K,> fnagi S,, Kubo T,. Fuchigami Т. // Chem. Commun. 2011.47. 8632—8634.
2 francke R., Little &D. // J. Am. Chem. Soc. 2014. 136.427-435.
94
5.1.2. Органические электролитические реакции
с использованием медиаторов
Электросинтез, использующий медиаторы поливалентных ионов
металлов. Ионы поливалентных металлов использовались в течение
длительного времени, а некоторые из них до сих пор применяются
в промышленном электролизе. Ниже приведены синтетические
примеры, исполыуюшие такие медиаторы:
rO~chj *
> С ^—сно+ f4 /^соон
Прямое анодное окисление Ч ft \ ff
Ce4+/CeJ\Mn37Mn2+. R*^\
-> С /)—сно
(5.3)
orS2Os:+/SO^ ' \ /
Непрямое анодное окисление
Crft+/Cr^ ? R' ,
Непрямое анодное окисление \ // ^-^^п
Прямой электролиз производных толуола дает смесь альдегида и
производных карбоновой кислоты, в то время как каждый продукт может
быть получен селективно путем подбора соответствующего медиатора.
Электросинтез с использованием медиаторов галогенов. Анодное
окисление галогенид-ионов (Х-) генерирует различные виды реакционно-
способных частиц, например, X4, OX-, Х2, Xj и т.д., которые широко
используются в качестве посредников в различных окислительных
молекулярных превращениях (рис. 5.1). Окислительная сила
увеличивается в ряду 1 < Вг < С1, а положительно заряженный С1 является
слишком сильным окислителем, чтобы обеспечить хорошую
селективность. Следовательно, йодид- и бромид-ионы в основном используются
в качестве посредников для селективного непрямого окисления.
Синтетические применения таких электрогенерированных
активных галогенных частиц в качестве посредников были
продемонстрированы для процессов высокоселективной функционализации
олефинов, например: эпоксидирования, галогидроксилирования,
1,2-дигалогенирования и хлорирования ен-типа, формирования связи
гетероатом — гетероатом и расщепления связи углерод — гетероатом1.
Torii S., Uneyama К.У Tanaka #,, Yamanaka Y., Yasuda Т., Ono Л/., Kohmoto Y. //
J. Oi£. Chem. 1981. 48. 3312-3315; Torii 5"., Tanaka //., Saitoh N.. Siroi Т.,
Sasaoka M., Nokani J.//Tetrahedron Lett. 1981. 22.3193—3196; ToriiS., Tanaka H.,
Saitoh N„ Siroi Т., Sasaoka M., Nokam J. // Tetrahedron Lett. 1982. 23. 2187-2188;
ToriiS., UneyamaK., Nakai Т., Yasuda T. //Tetrahedron Lett. 1981.22.2291-2294.
95
R' = K2 = Jb"~ 93%
R'^QHn, R2 = Me:99%
R1 = R2 = Cyctohexyl: 74%
R1 = Ph, R2 = Mc: 100%
Рис. 5.1. Схема непрямого анодного окисления спиртов с использованием
йодного медиатора
Электросинтез с использованием медиатора триариламина. Триари-
ламины, которые хорошо известны как реагенты внешнесферного
электронного переноса, сначала были использованы Стекханом в
качестве посредников при анодном окислительном снятии защитных
групп, подобных дитиолану для карбонильной группы1:
S S Na.CO^MeCN > RCHO + W <54>
Окислительная способность этого медиатора может быть
настроена путем замещения электроноакцепторными группами,
например, атомом брома (табл. 5.3). Триариламинные медиаторы могут
быть применены к окислению спиртов, аминов и боковой цепи
ароматических соединений вместе с фтородесульфиризацией. Таким
образом, они являются очень полезными и широко применяемыми
медиаторами.
Электросинтез с использованием мультимедиаторных систем.
Были разработаны различные комбинации нескольких типов
медиаторов, чтобы расширить сферу применения электросинтеза.
Как показано на рис. 5.2, двойная медиаторная система, состоящая
из сульфид- и бромид-ионов, приводит к окислению спиртов при
значительно более низком потенциале окисления, чем у самого
спирта2.
1 Saveant J.M. Op. ей.; Steckhan E. Op. cit.; Torii S. Op. cil.; Lund //., Ham-
merich O. Op. cit.; Steckhan E. Op. cil.
3 Shono Т., Manumura Y.. HayashiJ., MizpguchiM. //Tetrahedron Lett. 1980. 21.
1867-1870.
96
Таблица 5.3
Стандартные потенциалы окисления триа рил аминов
M}n
Триариламин
А
6
С
Заместитель
X
Вг
Вг
Вг
Y
Н
Вг
Вг
Z
н
н
Вг
Еп(Вотп. НВЭ)
1,30
1,74
1,96
w-CiaH|7SMe
ВГ <■
т
о
R'
Вг
I
w-Ci8Hi7SMe
■г
о
Рис. 5.2. Схема непрямого анодного окисления спиртов с использованием
двойного медиатора, состоящего из сульфид- и бромид-ионов
Электросинтез с использованием в качестве медиаторов гилер-
валентных соединений. Анодное окисление л-метоксииодобен-
зола в присутствии фторид-ионов дает гипервалентное дифто-
риодное производное, которое является удобным фторирующим
реагентом. Таким образом, л-метоксииодбензол является
высокоэффективным медиатором анодной фтородесульфиризации ди-
тиоацеталей (рис. 5.3)1. Это один из первых успешных примеров
каталитического использования гипервалентных соединений для
органического синтеза вообще, а также для органического
электросинтеза.
Анодное сс-фторирование а-, у-дикетонов и а-кетоэфиров может
быть также достигнуто с использованием в качестве медиатора р-
йодтолуола2.
1 Рфа Т., Fuchigami Т. // J. Ощ. Chem. 1994. 59. 7190-7192.
1 Нага S., Sekiguchi A/., OhmoriA.,Fukuhara Т., Yaneda N. // Chem. Commun. 1996.
1899-1900.
97
Anode /=\ /F i^^N
=2e
^Me0 , v ,
-90%
Рис. 53. Схема электрохимической фтородесульфиризации с использованием
медиатора гипервалентного производного йодбензола
Электросинтез с использованием комплексов переходных
металлов как медиаторов. Ниже будет рассмотрено только несколько
примеров электросинтеза с участием металлокомплексоп —
катализаторов. Подробнее достижения в этой области описаны далее
в отдельной главе. Медиаторы на основе комплексов переходных
металлов имеют различную реакционную способность и много
преимуществ. Их окислительно-восстановительные потенциалы и
селективность желаемой реакции можно регулировать путем выбора
лиганда. Их синтетическое приложение главным образом основано
на реакционной способности в низко валентном состоянии,
генерируемом катодным восстановлением медиаторов1. Как правило,
Со(П1)-комплекс (витамин В12) легко восстанавливается, оптически
активен, нетоксичный и недорог. Он восстанавливается при -0,9 В
отн. НКЭ с образованием Со(1)-комплекса, который подвергается
окислительному присоединению к алкилгалогениду с образованием
алкильпого Со(Ш)-комплекса в качестве шттермедиата.
Полученный интермедиат восстанавливается при более
отрицательном потенциале (—1,5 В отн. НКЭ), образуя алкильный радикал
или анион, одновременно регенерируется Со(1)-комплекс
(восстановительное элиминирование). Полученный алкил-радикал или
анион может эффективно присоединиться по Михаэлю к олефину
в нейтральных условиях (рис. 5.4)2. Эта медиаторная реакция широко
применима к различным галогенсодержаигим соединениям (аллилга-
логенидам, винилгалогенидам, ос-галоэфирам и т.д.).
Можно использовать даже галогенсодержащие соединения с
незащищенными гидроксильными и карбонильными 1руппами, что
является одним из преимуществ этой медиаторной системы3:
i
SaveantJM. Op. cit.; Sleekhen E. Op, cil.; ToriiS. Op. cit.; Lund H.. Hammerich O.
Op. cit.; Steckhan E. Op. cil.
SiheffbMR., Dike M., DikeS., Hemlii Т., WilderL. //J. Am. Chem. Soc. 1980. 102.
3642-3644.
ScheffoldR., RytzG., Wilder L., Orlinskf R. // Pure Appl. Chem. 1983. 55. 1791—
1797.
98
X
Рис. 5.4. Электросинтез с использованием Со (111) -комплекса — витамина В12
в качестве медиатора
(CH2UMc 2c
Vitamin B12
(5.5)
Vitamin В12 \-^\^\/ (СН2)4Ме
ОН
При этом Р-бромодиэфир и ш-бромалкилакрилат подвергаются
1,2-пере группировке и внутримолекулярной циклизации с
образованием больших циклических лактонов путем их катодного
восстановления с помощью медиатора (гидрофобного витамина В12) под
действием УФ-облучения1.
Продукты гомо-сочетания получают из ароматических галоге-
нидов с использованием в качестве медиаторов Pd(0)-комплекса
и N подкомплекса2. Выходы и оборот РоЧО)-комттлекса, как
правило, превосходят полученные с Ni(0)-комплексом. Предложенный
механизм показан на рис. 5.5. Ароматический галогеннд реагирует
с Р<1(0)-комплексом, генерируя арил-Рс1-интермелиат, который
восстанавливается катодно с последующей реакцией с еще одной
молекулой арилгалогснида с образованием диарильного комплекса Pd,
лающего в результате восстановительного элиминирования продукт
гомо-сочетания, Когда эта реакция проводится в присутствии Ct\,
получаются ароматические карбоновые кислоты с высоким выходом3.
ShimakoshiH., Nakazato A., Hayashi Г., Tachi К, Naruta К, Hisaeda К //J. Elec-
troanal. Chem. 2001.507. 170— 176.
ToriiS., TanakaH., MorisakiK. //Tetrahedron Lett. 1985. 26. 1655-1658.
Torii S., Tanaka H., Hamatani H., Morisaki K., Jutand A., Pfluger F., Fau-
varqueJ.F. //Chem. Leu. 1986.169-170.
99
Аг—X [Аг—PdXl
Pd(0) [Ar—PdX]'-
Ar—Ar
"'Аг
X-
Рис. S.5. Схема гомо-сочетания арилгалогенидов помощью Pd (0) -комплекса
Электросинтез с использованием медиатора, иммобилизованного
на твердой подложке. N-оксильные радикалы, примером которых
является 2,2,3,3-тетраметилпиперидинил-Ь1-оксил (TEMPO),
считаются подходящими медиаторами для эффективного окисления
спиртов и аминов. Медиаторы ранее никогда не использовали
повторно после электролиза, но недавно начали разрабатывать реци-
клизуемые медиаторы, исходя из так называемой атом-экономии.
Медиаторы, иммобилизованные на поверхности силикагеля через
с плановый каплер (соединитель), легко выделяют и рециклизуют
после электролиза. Так, комбинация галогенид-ионов и ТЕМПО,
иммобилизованного на силикагеле или полимерных частицах в
качестве дисперсной фазы, обеспечивает опосредованную
электрокаталитическую реакцию в водном NaBr— NaHC03 в качестве дисперсной
среды1. Асимметричное окисление спиртов также возможно с
использованием хирального N-оксильного медиатора2.
5.2. ЭЛЕКТРОГЕНЕРИРОВАННЫЕ КИСЛОТЫ И ОСНОВАНИЯ
5.2.1. Электрогенерированные основания
Кислоты и основания играют очень важзгую роль в органическом
синтезе. Хорошо известно, что, когда водный раствор, содержащий
нейтральный фоновый электролит, подвергают электролизу, в
разделенной ячейке анолит становится кислым, а католит
—щелочным. Это происходит потому, что гидроксид-ионы потребляются
на аноде, а ионы водорода расходуются на катоде. Аналогично
некоторые кислоты образуются в аиолите, а некоторые основания
Tanaka Н., Chou J., Mine Л/., Kurohoxhi К. // Bull. Chem. Soc. Jpn. 2004. 77.
1745-1755: Kubota J„ /do T, Kuroboshi A/.T Tanaka //., Uchida T, Shima-
mura K. // Tetrahedron. 2006. 62.4769-4773.
Tanaka H„ Kawakami Y., Goto, K., Kuroboshi M. // Tetrahedron Lett. 2001. 42.
445-448.
100
генерируются в католите в процессе электролиза в органическом
растворителе. Даже в неразделенной ячейке в процессе электролиза
пространство около анода становится кислым, а около катода —
основным.
Анионные частицы, генерируемые на катоде, действуют не только
как нуклеофилы, но и как основания и обладают интересными
реакционными свойствами в органическом синтезе. Изобретатель
катодного процесса гидродимеризации акрилонитрила — Байзер показал,
что катодно генерируемый анион-радикал стерически затрудненного
азобензола (рис. 5.6) может быть полезным основанием в различных
органических синтезах. Он назвал такие основания «элекгрогенери-
рованными основаниями» (EGBs)1.
Рис 5.6. Структура электрогенерированного основания стерически
затрудненного азобензола
Существуют два основных метода генерации EGBs:
Y=Z s- »[ Y=Zr R4N+
R4N*X~
Y-H el/2H2 ) Y~R4N+ (5.6)
R4N X"
(Y,Z = C,0,S,N,P)
Первый метод включает катодное восстановление соединений
с ненасыщенным остатком в апротонных растворителях,
содержащих соль четвертичного аммония в качестве фонового
электролита, для генерации анион-радикалов или дианионов, а второй
является катодным восстановительным депротонированием
активных водородных соединений для генерации соответствующих
анионов.
Катодное восстановительное депротонирование 2-пирролидона,
затрудненного фенола типа 2,6-трет-бутил-4-метилфенола, и трифе-
нилметана генерирует соответствующие анионы. Поскольку EGBs
имеют катионы четвертичного аммония (О/), анионы,
образовавши-А/Ж //Tetrahedron. 1984.45.935—969.
101
шиеся в результате обработки EGB, имеют высокую реакционную
способность (рис. 5.7), а следовательно, эта методика является
весьма полезной для органическою синтеза.
Антиионные" ,
Рис. 5.7. Реакцион неспособный анион, полученный
изэлектрогенерированногооснования (Q" = EtiN4)
В частности, EG В, полученное из 2-пирролидонат является
универсальным основанием и может быть применено к
органическому синтезу (перегруппировке Стивенса, селективному
а-моноалкилированию а-(арил)ацетатных эфиров и С-моноалкили-
рованию 1,3-дикетонов)1. Это EGB, как было продемонстрировано,
является высокоэффективным основанием в синтезе фторорганиче-
ских соединений. Например, известно, что трифторметил-анион
настолько нестабилен, что подвергается а-элиминированию фторид-
аниона. В результате генерирется фторокарбен, однако стабильный
трифторметил-анион можно получить обработкой фтороформа этим
EGB, а следовательно, осуществить трифторметилирование
ароматических альдегидов и кетонов с образованием трифторометилиро-
ванных спиртов2:
^N-^O
9 - и*+ НО CF<
Г +CHF3 *^U V (
Ar-^Ar' Ar-^Ar' <5-7)
(H) (H)
-84%
Как правило, достаточно сложно генерировать енолят-анион
с a-CF3-rpynnoti, поскольку енолят-анион легко разлагается,
а именно (З-группа элиминирует фторид-ион. Однако пирро-
лидон — производное EG В позволяет генерировать стабильный
трифторметилированный енолят-анион, а апеллирование можно
выполнить с хорошим выходом без элиминирования атома
фтора3:
Shono Т.. Kashimura S„ Ishizaki К., IshigeO. // Chem. Letl. 1983. 1311-1312;
Shono Т., Ishifune A/., Jshige O., Uyama H., Kashimura £. // Tetrahedron Lelt. 1990.
31.7181—7184.
Shono Г., Ishifune Л/., Okada Т., Kashimura S. // J. Org. Chem. 1991. 56. 2—4.
Fuchigami У., Nakagawa У. //J. Org, Chem. 1987. 52. 5276-5277.
102
ex
e H
-1/2
О О
MeO
Hj |Et4NOT
CF,
OMe DQMF
RX
О 0Et«N+
МеО'у'ХЭМе
CF3
RX
-ti.
О О
(5.8)
~rl MeO' /\ OMe
R CF3
R = Me
R = PhCH2
R = CH2 = CHCH2 :70%
Кроме того, этот EG В катализирует полимеризацию с
раскрытием цикла N-карбоксиангидридов а-аминокислоты (NCA), давая
поли (аминокислоты) в течение короткого времени с прекрасным
выходом:
y-NH Qr°3 yN-Q+
L-valine
NCA ^ s (5.9)
NCA
-CO,
-*£♦ -(-NHCHCO^h
Q+ = ВщЫ+ : 98%
Q+ = Na+ : 7%
Выход значительно выше, а время реакции относительно
короткое по сравнению с реакциями с использованием обычного
основания с металлическим катионом (Na+)'. EGB, полученное из
затрудненного фенола, катализирует селективную двойную реакцию
альдольной конденсации3:
1 Komori Т.. Nonaka Т. t Fuchigami T.jj Chem Lett. 1986 Л1—12.
2 Fuchigami T.,Awata Т., Nonaka T„ BaizerM.M. // Bull Chem Soc. Jpn. 1986. 59.
2873-2879.
103
о о
2 X + JL
о
ELiN
Аг^^СНз Аг
О Аг' О
"агАЛЛаг <5|°>
Хорошо известно, что сама молекула субстрата также служит в
качестве EGB. Например, каталитическое количество EGB
эффективно катализирует альдольную конденсацию:
сно
DMF
,СНО
= EGB
-СНО
EG В
.СНО
(5.11)
СНО
хно °
* ' Т
НО ОН
(EGB)
Yield : 76%
Current efficiency : 9 х 103%
В этой реакции сам альдегид катодно восстанавливается,
генерируя следовые количества его анион-радикала, который действует
как основание для отрыва а-протона от непрореагировавшего
альдегида, а алыюлытая конденсация протекает успешно1. Наконец,
в цепной реакции элиминированный гидроксил-ион будет
действовать аналогично в качестве EG В.
Обработка спиртов металлическим натрием генерирует
газообразный водород, также образуется алкоголят натрия. Это может
быть объяснено следующим образом. Периферический электрон
металла натрия передается на спирт, генерируя радикал водорода,
образующего газообразный водород, а спирт превращается в
алкоголят. Другими словами, металлический натрий действует как
восстанавливающий реагент для спирта. Когда спирт
восстанавливается на катоде с низким перенапряжением водорода, например,
на платине, выделяется газообразный водород, а соответствующий
алкокоголят легко образуется без восстановителя. Таким образом,
можно считать, что катод будет действовать как металлический
натрий. Это вполне безопасный способ получения ал кокс ид а металла,
Shono Г., Kashimura S\, lshizaki К. // Elecirochim. Acta. 1984. 29. 603.
104
поскольку высокореакционноспособный опасный металлический
натрий не требуется. Кроме того, при катодном восстановлении кар-
боновой кислоты, фенола, тиофенола и спирта в присутствии соли
четвертичного аммония в качестве фонового электролита образуются
высокореакционные анионы четвертичного катиона аммония (Q+).
С помощью таких реактивных анионов может быть достигнута
высокая селективность этерификации, сложноэфирной этерификации
и карбонилирования при комнатной температуре1:
RCOOH С"1/2Н2 ) RCOO Q+ R>x > RCOOR'
-96» (Q+ = Et4N+)
ArYH c~1/2"2 ) ArY О/ Ar<X ) ArYAr'
(Y = 0,5) (5.12)
RON е"'/2Нг-> OR Q+ Ге(С0)*. > ROCO" Rr* > ROCOR'
Катодное восстановление молекулярного кислорода генерирует
супероксид-ион (О^-). Супероксид-ион действует как нуклеофил,
окислитель, восстановитель, радикал и основание (EGB), поэтому эти
активные формы являются весьма полезными для органического синтеза.
Хотя супероксид-ионы доступны из К02 и NaO?, эти оксиды
нерастворимы в апротонных растворителях. Чтобы растворить эти соли в апро-
тонных растворителях, требуются дорогостоящие краутт-эфиры,
следовательно, их синтетические приложения ограничены. При этом
электролитический метод позволяет генерировать супероксид-ион in situ,
что лучше, чем при использовании обычного химического метода.
Электрогенерированный супероксид-ион выступает в качестве EGB,
элиминируя а-протон производного малонового эфира с последующим
взаимодействием с 02, давая а-гидрокси продукт с хорошим выходом2:
CH3CH(COOEt)2 —^—>CH,C(OH)(COOEt)2 (95%) (5.13)
Высокий выход по току предполагает, что эта реакция может
включать цепную реакцию.
5.2.2. Электрогенерированные кислоты
В противоположность EGBs околоанодное пространство в
процессе электролиза становится кислым. Полученную таким образом
кислоту называют электрогенерированной кислотой (EGA). Она
1 Awata Т., Baiter MM., Nonaka Г., Fuchigami Т. // Chem Lett. 1985. 371-374;
Hashiba 5., Fuchigami Г., Nonaka Т. //J. Org. Chem. 1989. 54. 2475.
2 AfferPM..tiessU.,FooteCS.,BaizerM.M.//SbTi.Comm\m. 1982. 12. 123-129.
105
имеет уникальную реакционную способность по сравнению с
обычными химическими кислотами. Например, электролиз раствора,
содержащего LiC104 и следовые количества воды, приводит к
образованию кислоты, НСЮц (рис. 5.8)1.
^■сю;
1
н,о
Н+-Те
LIC104
Aprotic solvent
Рис. 5.8. Принцип электрогенерирования кислоты
EGA, генерируемая в этом случае, как предполагается, — это
безводная НСЮ4, которая является более сильной кислотой, чем
коммерчески доступная водная НСЮ4. Такая EGA действует как своего
рода кислота Льюиса и широко применима к различным
органическим реакциям, например, изомеризации, трансформации
функциональных групп, формированию углерод — углеродной связи, в том
числе реакциям Дильса-Алъдера, и т.д.2 Комбинация фоновых солей
и растворителей дает три вида продуктов, которые получаются
селективно из одного и того же исходного материала3:
EGA
LiCICVCHjCln
X
R
о
EGA
Mg(CI04)2/(CHj)jCO
EGA
E^NOTs + NaOTs
/CICHjCHjCl
R
°X°
(5.14)
R
OH
Кроме того, EGA весьма эффективны для молекулярного
превращения фторорганических соединений. Например, гх-катион,
присоединенный к СРз-группе, каталитически генерируется при обработке
1 Шеуата К., Nishiyame N.T Torii S. // Tetrahedron Lett. 1984. 25. 4137—4138,
2 Inokuchi Т., Tanigam 5., Torii S. // J. Org. Cliem, 1990. 55. 3958-3961.
J Шеуата К., Nishiyama TV., Torii S. Op. cit.
106
трифторометилированного О, S-аиеталя EGA, а далее углеродные
нуклсофилы легко вводятся в ос-положсние1:
PhSCHCH2CF} ь, Е^* > PhSCHCH2CF,
ОАс Nu (5.15)
Nu = CN : 56%
Nu = CH2CH=CH2:88%
Однако использование других обычных кислот Льюиса не
приводит к образованию целевого продукта.
5.3. ЭЛЕКТРОХИМИЧЕСКИЙ АСИММЕТРИЧЕСКИЙ СИНТЕЗ
Асимметрический синтез яа!ястся весьма важной областью
органической химии. В последнее время был разработан ряд новых
асимметрических каталитических синтезов. Однако асимметрический
синтез с применением средств электрохимик все-таки еще не вполне
«зрел» по сравнению с устоявшимися химическими,
каталитическими и ферментативными методами. Известны различные методы
электрохимического асимметрического синтеза2: I)
внутримолекулярная асимметрическая индукция; 2) использование хиральных
растворителей; 3) применение хиральных фоновых солей; 4)
использование хиральной адсорбции на электроде; 5) использование
электродов, химически модифицированных хиральными веществами;
6) использование хиральных электродов, модифицированных
полимерами; 7) использование хиральных медиаторов. Среди них 1-й
и 2-й методы не являются уникальными и не характерными для
электрохимии. Однако существуют публикации, выполненные в
соответствии с этим подходом. Хотя 3-й метод характерен для электролиза,
требуется большое количество хиральной фоновой соли, поэтому он
не очень удобен. Ниже обсуждаются методы 4—6-й.
В 1967 г. Гримшоу первым сообщил об асимметричном
восстановлении 4-метилкумарина с использованием оптически активных
алкалоидов, а максимальный асимметрический выход (17%) был получен
на электроде с использованием в качестве хирального адсорбента
небольшого количества спартеина (1 ммоль)3. Через 10 лет Миллер
получил 48% асимметричного продукта при катодном восстанов-
1 Fuchigami Г., Yamamoto К., Yano Н. // J. Org. Chem. 1992. 57. 2946-2950.
2 Nonaka 7'., Fuchigami T. Stereochemistry ofOfganic Electrode Processes in Organic
Electrochemistry. 4th edn / H. Lund, O. Hammerich. N. Y: Marcel Dekker, 2001.
Ch. 26; Fuchigami Г., htagi S. Stereochemistry of Organic Electrode Processes in
Organic Electrochemistry. 5th edn, / O. Hammerich, B, Speiser, Taylor & Francis,
2014. Ch. 27; GrimshawJ. Electrochemical Reactions Mechanisms in Organic
Chemistry. Amsterdam: Elsevier, 2000. P. 80—83, 268—269.
J Gaurtey R.M. Grimshaw J.. Miliar P.G. // Chem. Commun. 1967. 1278-1279.
107
лении 2-ацетилпиридина до соответствующего спирта в присутствии
0,5 мМ соли стрихнина1. Асимметрическая индукция, как кажется,
связана с хиральными областями реакции, созданными физической
адсорбцией оптически активных алк&тоидов на поверхности катода.
Однако такой асимметрический электросинтез очень чувствителен
к электролитическим условиям — перемешиванию, концентрации,
растворителю, катодному потенциалу и т.д.
Миллер и его коллеги обнаружили, что прохиральные
карбонильные соединения, такие как этилфенилглиоксилат,
восстанавливались в хиральиый спирт с 10%-м асимметрическим выходом на
катоде химически модифицированным (5)-фенилглицином2. Он также
сообщил о возможности асимметричного окисления w-толилметил-
сульфида до сульфона с 2,5% ее при использовании аналогичного
электрода, модифицированного (+)-камфорной кислотой3. Это были
пионерские работы по химически модифицированным электродам.
Низкие асимметрические выходы, видимо, были обусловлены низкой
плотностью хирального соединения на поверхности электрода.
Некоторые исследователи отмечают, что результаты Миллера не всегда
воспроизводимы. В дальнейшем, чтобы решить проблемы, связанные
с нестабильностью электродов, модифицированных хиральным
адсорбентом, и низкой плотностью модифицирующих хиральных
соединений, были разработаны хиралыше модифицированные
полимером электроды. Например, Нонака, Фучигами с соавторами
получили различные электроды, покрытые оптически активными поли
(аминокислотами), и применили их для асимметрического
восстановления олефинов. Они получили 43% ее при восстановлении 4-ме-
тилкумарина на катоде с покрытием из поли (L-валина) (рис. 5.9)4.
Рис. 5.9. Схема асимметрического восстановления с помощью хирального
электрода, модифицированного полимером
1 Kopilov J., Kariv E., Miller L. L.//J. Am. Chem. Soc. ] 977.99. 3450—3454.
2 Watkim B.F., Behling J.R., Kariv £.. Miller L.L. // J. Am. Chem. Soc. 1975. 97.
3549—3550.
} Firth B.E., Miller L.L.., Milam A/., RogersT., Lennox J.. Murray R. W. // J. Am.
Chem. Soc. 1976. 98. 8271-8272.
4 Abe S., Nonaka Т., Fuchigami T. //I.Am. Chem. Soc. 19H3. 105.3630-3632.
108
Впоследствии был разработан электрод из графитового войлока,
модифицированного 2,2,6,6-тетраметилпиперидин-1 -илоксидом
(TEMPO). Он применен для энантиоселективного окислительного
сочетания 2-нафтола, 2-метоксинафталина и 10-гидроксифенан-
трена в присутствии (-)-спартеина как хиралъного основания, Энан-
тиоселективность продуктов сочетания очень высока (98%)'. Был
сделан следующий вывод: нельзя ожидать высокого
асимметрического выхода, так как электрохимические реакции, как правило,
требуют полярных растворителей. Поэтому следует отметить, что такие
прекрасные результаты асимметрического электросинтеза были
достигнуты даже в полярных растворителях. Комбинация ферментов
(дегидрогеназа молочной кислоты и медиаторов или хиральных
медиаторов) также эффективна для электрохимического
асимметрического синтеза и кинетического разделения, направленных на
получение продуктов с высоким асимметрическим выходом2.
5А МОДИФИЦИРОВАННЫЕ ЭЛЕКТРОДЫ
Как было описано ранее, и электродные поверхности, и
окружающее пространство электродов являются основными областями
электродных реакций. Если поверхность электрода
модифицировать подходящими функциональными веществами, хиральными
соединениями и катализаторами, то можно реализовать требуемую
селективность и эффективность, провести асимметрический синтез.
Такие электроды называются модифицированными, а их
использование позволяет снизить количество необходимых функциональных
материалов и упростить разработку электрохимических растворов3.
Необходимо, чтобы модифицированные электроды для электро-
1 Osa Т.. Kashiwagi К, Yanagisawa К, Bobbin J.М. // I. Chem. Soc, Chem.
Commun. 1994. 2535—2537; Osa T. Construction of New Mediator Systems in
New Challenges in Organic Electrochemistry/cd, T.Osa. Amsterdam: Gordon &
Breach, 1998. P. 183-219.
г Steckhan E. // Topics in Current Chemistry 170. Electrochemistry V Springer-
Vcrlag. 1994. 84-111; Hiillrigl V., Otto К., Schmid А. // Adv. Synth. Catal. 2007.
349, 1337-1340; Huang J., Fu X., WangG., Ge Г., Miao Q. // J. Mol. Catalysis
A: Chemical. 2012. 357.162—173; Kashiwagi K, Kurashima F., Chiba S., AttzaiJ.,
Osa Г., Babbitt T.M. // Chem. Commun. 2003. 114—115; Shiigi H.Janaka Т.,
Demizu Г., Onomuru O. // Tetrahedron Leu. 2008. 49. 5247-5251; Demizu Y.,
Shiigi H., Mori H.. Matsumolo K., Onomura O. // Tetrahedron Asymmetry. 2008.
19. 2659—2665; Minafo D.,Arimofo //., Nagasue K, Demizu K, OnomuraO.
//Tetrahedron. 2008. 64. 6675-6683.
} Merz A. Chemically Modified Electrodes in Topics in Current Chemistry', 152.
Electrochemistry IV/ed. E. Steckhan. Berlin: Springer-Nferlag, 1989. P. 49—90;
MurrayR.W. //Ace. Chem. Res. 1980. 13. 135-141; Nonaka Т., Fuchigami T. //
J. Org. Synth. Chem. 1985.43. 565-574.
109
синтеза имели отличную прочность, так как при электросинтезе
используются большой электролитический ток и длительное время
электролиза, что отличается от требований к электродам,
модифицированных для сенсоров. Модифицированные электроды
классифицируются следующим образом в зависимости от способов
модификации и природы модификаторов.
5.4.1. Электроды, модифицированные адсорбентами
Когда электрод погружают в раствор, содержащий редокс-ак-
тивные соединения (флавины и феназины), а затем вынимают его
из раствора, то легко получается электрод, модифицированный ре-
докс-соединениями. Модификация не очень прочна, долговечность
модифицированного электрода невелика, а плотность распределения
частиц модификаторов низка. Причем электрод,
модифицированный гомогенными сильно адгезированными пленками с
окислительно-восстановительными катализаторами с использованием
метода Ленгмюра-Блоджстта, превосходит модифицированный
адсорбентом электрод, так как плотность модификации (плотность
нанесения частиц редокс-катализатора) и долговечность пленки
Ленгмюра-Блоджетта модифицированного электрода значительно
выше. Опираясь на гадрсфильно/шдрофобньгй баланс поверхности
электрода, с помощью пленки Ленгмюра-Блоджетта, содержащей
соль четвертичного аммония, можно достичь контроля
селективности продукта1. Электроды с пленочным покрытием Ленгмюра-
Блоджетта также позволяют проводить электрокаталитические
реакции2.
5.4.2. Модифицированные электроды с инородными адатомами
металла
Базовые электроды можно модифицировать путем осаждения
адатомов инородного металла при потенциалах, на несколько сотен
милливольт более положительных, чем обратимый потенциал,
необходимый для осаждения металла. Субмомослой адатомов
покрывает поверхность базовот электрода равномерно, превращая ею
в электрод, модифицированный адатомами3. Такие электроды весьма
полезны для элсктрокаталиэа восстановления кислорода и
окисления метанола, поэтому в настоящее время они имеют большое
значение для разработки топливных элементов. Эти электроды также
позволяют достичь интересной селективности продукта следующим
образом. Катодное восстановление динитробензолов на серебренном
катоде в кислом растворе метанола в основном дает диаминобен-
1 Kuttugi Y., Fuchigami Г., Tien ff,-J., Nohaka Т. // Chem. Lett 19Я9. 75.1—756.
2 FacaJ.S., Falcino PA, GoidJ.M. // Langmuir. 1986. 2. 732-738.
} Kokkinidis G. // J. Electroaiuil. Chem. 1986. 201. 217-236.
110
золы, в то время как использование серебренного катода,
модифицированного путем осаждения адатомов свинца, приводит к
селективному образованию фенилендигидроксиаминов1:
N02 NHOH NH2
8с, НН (Г ^*l 4c, 4H1 „ ,
■* I J > I XJ (5-16)
NHOH ^NH2
Pb-Ag cathode Ag cathode
Считается, что это происходит за счет ингибирования адсорбции
промежуточных фенилендигидроксиаминов на поверхности
электрода осажденными атомами свинца, что препятствует их
дальнейшему восстановлению.
5.43. Химически модифицированные электроды
Как описано выше, работа Миллера по модифицированным хи-
ральными соединениями электродам стимулировала интенсивные
и плодотворные исследования в области химически
модифицированных электродов. Функциональные вещества иммобилизуются
на базовых электродах через ковалентные связи — сложно эфирные,
простоэфирные и амидные с желаемыми функциональными
веществами.
Когда электроды из благородных металлов (например, золота
и платины) погружают в раствор тиола или дисульфида на
некоторое время, поверхность электрода легко модифицируется
самоорганизующимся тиольным монослоем. Поскольку различные
функциональные группы могут быть иммобилизованы на поверхности
электрода, а способ модификации является универсальным, этот
метод широко используется, но известно только несколько примеров
его применения в электросинтезе2.
Пинсон, Савьян с сотрудниками разработали эффективную
и общую методику ковалентного связывания арильных групп
с электродом путем катодного восстановления иона арилдиазония.
Арильные радикалы, генерируемые таким образом, связываются
эффективно с поверхностью электрода. Полученный
модифицированный электрод обладает высокой стабильностью. Следовательно,
с помощью этой процедуры различные арильные группы могут быть
присоединены к поверхностям углеродных, кремниевых и метал-
1 JannakaudakkA.D., Kokkmidis G. // Eltxlroehim, Acta. 1982. 27. 1199-1205.
1 Banna N.. Nakantshi Т.. Matsunaga M., Asahi Т., Osaka Т. // J. Am. Chem.
Soc. 2004. 126. 428—429; Nakantshi Т.. Matsunaga M., Nagasaka M., Asahi Т.,
Osaka T. // J.Am. Chem. Soc. 2006.128.13322-13323.
111
лических электродов1. Такие модифицированные электроды имеют
широкое применение, например, в сенсорах и электрокатализаторах,
но их применение в электросинтезе до сих пор было ограничено2.
5.4.4. Электроды, модифицированные (покрытые) полимером
Электроды, модифицированные (покрытые) полимером, имеют
преимущество, которое заключается в том, что большое количество
функциональных веществ — оптически активных и электроактивных
соединений, редокс-катализаторов может быть включено в
полимерную матрицу, покрывающую поверхность электрода, как
показано на рис. 5.103.
Редокс- кптяли затор
Электрод
Рис. 5.10. Электрод, модифицированный полимером с частицами редокс-
катал и затора
Полимер, используемый для покрытия, должен быть растворим
в каком-то растворителе и нерастворим в растворителе, содержащем
фоновый электролит, используемом для электроаналитических
исследований и/или электролитических реакций. Способы
модификации приведены ниже: простое окунание и последующая сушка,
полимерное покрытие из мономеров пугем химически или
электрохимически инициированной полимеризации, покрытие проводящим
полимером и подтвержденное химическое связывание, подобное
амидной связи полимерного слоя с использованием анкерных групп
поверхности электрода.
Следующие полимерные материалы используются для
модификации электродов: полимер с окислительно-восстановительными
свойствами (лоливиологен, поливинил ферроцен и т.д.), полимеры
типа поли-4-винилпиридин, поливинилсульфоновая кислота, по-
лиакрилонитрил и т.д., которые могут образовывать металлоком-
плексы и удерживать (иммобилизовывать) редокс-активные частицы
за счет кулоновского взаимодействия, полимеры типа поликарбонат,
полиуретан и т.д., которые обладают селективной ионно/газопро-
1 Pinswt J., Padvorica F. // Chem. Soc. Rev. 2005. 429—439.
2 Mayers B,T,< FryA.J. //Org. Lett. 2006. 8.41-414.
-1 Men. A. Op. cil.; Murray R. W. Op. cil.; Nonaka Т.. Fuchigami'/". Op. cil.
112
никаюшей способностью, ионопроводящие полимеры. Хотя
неорганических покрытых полимером электродов немного, берлинская
лазурь, гетероиоликислоты. пленки из некоторых типов глин и т.д.
могут быть использованы в качестве модифицирующих веществ.
Полимерные модифицированные электроды имеют широкое
применение в топливных элементах, конденсаторах, материалах дисплеев,
биосенсорах, ион/газовых сенсорах и в асимметрическом синтезе.
В настоящее время они интенсивно изучаются.
5.5. ПАРНЫЙ ЭЛЕКТРОСИНТЕЗ
Электросинтез включает одновременное протекание как
восстановления на катоде, так и окисления на аноде. В типичном
электролизе водного раствора хлорида натрия на катоде
образуется гидроксид натрия, а газообразный хлор образуется на аноде.
Обе электродные реакции эффективно используются па практике.
В органическом электролизе синтез целевых продуктов
осуществляется либо в катодной, либо анодной реакции. Следовательно,
в большинстве случаев продукты реакции на противоположном
электроде могут остаться бесполезными. Поэтому одновременное
синхронное использование обеих катодных и анодных реакций
для эффективного синтеза целевых продуктов является
предпочтительным с практической точки зрения. Парный электросинтез
основан на этой методологической концепции. Байзер впервые
сформулировал и показал подходы к вышеописанной концепции
и ввел термин парного электросинтеза1. До 200% выхода по току
можно ожидать в идеальном случае парного электросинтеза с
использованием катодных и анодных процессов, приводящих к
одному и тому же продукту.
Недавно компания BASF (Германия) впервые
коммерциализировала парный электросинтез. Фталид и диметилацеталь трет-бутил-
бензальдегида производятся одновременно в неразделенном
электролизере (подробнее см. в гл. 8).
Парный электросинтез классифицируют следующим образом
на основе режимов реакции2.
Параллельный парный электросинтез (электросинтез различных
продуктов из различных исходных субстратов). Типичным примером
является электросинтез дигидрофталиевой кислоты и ацетиленди-
карбоновой кислоты из фталевой кислоты и бутиндиола
соответственно на катоде и аноде3.
1 Bai&rM.M. Op. cit.; BaizerM.M., HalkherR.C. //J. Electrochem. Soc. 1976. 123.
809—8I.1.
2 Ibid.
J Baizer M.Л/., BailcherJLC Op. cil.
113
Дивергентный парный электросинтез (электросинтез различных
продуктов из одного и того же субстрата). Детали и пример
приведены в гл. 7 (рис. 7.8).
Конвергентный парный электросинтез. Это электросинтез одного
продукта из разных исходных субстратов, например, электросинтез
пропиленоксида через хлоргидрин из пропилена и хлорид-иона,
а также электросинтез сулырениминов из амина и дисульфида с
использованием медиатора — бромид-ионов'. Как показано на рис. 5. I1,
алифатические кислоты восстанавливаются реакционноспособным
металлическим Mg катодно образующимся из иона Mg2', при этом
образуется и алкоксид-ион* Тетрагидрофуран как растворитель
окисляется на аноде, генерируя соответствующий катиоиный интерме-
диат, которой реагирует с алкоксид-ионом, получаемым на катоде,
образуя а-алкокситетрагидрофуран2. Здесь и анодная, и катодная
реакции участвуют в получении единого продукта, таким образом,
общий выход по току не превышает 100%. Электролитическая реакция
(рис. 5.12) является еше одним примером на эту тему3. Трифтормети-
лирование фумаронитрила требует как анодной, так и катодной
реакций, следовательно, эта реакция является парным электросинтезом.
Pt анод
Pt катод
*СГ OCH2R
Рис. 5.11. Конвергентный парный электросинтез [а)
На аноде:
CF3COO
На катоде:
CN / 65%
NC
1)+е
CF3
NC
CN
NC
Рис. 5.12. Конвергентный парный электросинтез (б)
1 ToriiS., Тапака И., Vkida М. // J. Org. Chem. 1979. 44. 1554-1557.
2 Ishifune M., Yamashlta h,, Marsuda M., Jshlda H.. Yamashita Л1,, Kera Y.,
Kashimura 5., Masuda #., Murase H. // Elettrochim. Acta. 2001.46. 3259—3264.
J Uneyama K., Watanab S. //J. Org. Chem. 1990. 55. 3909-3912.
114
Линейный парный электросинтез (электросинтез одного продукта
путем анодного окисления с последующим катодным
восстановлением или наоборот), Окисление 2,3-бутанлиола дает ацетоин, а затем
ацетоин восстанавливается на Hg/Zn катоде до метилэтилкетона1
(использование неразделенной ячейки является существенным
в данном случае):
ОН
О
о
Бг-медиатор
углерод-анод
-2е, -2НГ
Zn(Hg) катод
-2е.-2Н+.-Н,0
(5.17)
ОН
ОН
Парный электросинтез с помощью EG В, регенерируемого
на аноде и катоде, протекает последовательно или наоборот
(рис. 5.13 иллюстрирует пример такого рода парного
электросинтеза). Азобензол восстанавливается с получением соответствующего
дианиона (EGВ), затем реагирующего со смесью этилацетата и бутил
бромида как исходного субстрата с получением конечного
продукта (этил а- (ацетил) гексаноата) и EGBH2, который окисляется
на аноде и регенерирует EGB-.
Cathode
Anode
ArN=NAr
ArR_^jAr 2CH3COOEt+BuBr MNmHAT ^У
(EGB) (EGBH,)
+
CHjCOOHtBuJCOOEt
2e
Рис. 5.13. Парный электросинтез с использованием регенерированного EG В
Парный электросинтез, использующий различные медиаторы,
регенерируемые на аноде и катоде. Молекулярный кислород (О?) катодно
восстанавливается в водных и неводных растворах до активных форм
кислорода — Н202 и ОГ соответственно, которые являются
окислителями органических соединений. Следовательно, катодное
окисление органических субстратов осуществляется с помощью таких
катодно генерированных кислородных частиц. Однако
концентрация катодной генерируемой Н202 настолько низка, что скорость
окисления органических соединений под действием Н2От
недостаточна для практической реализации электролитического процесса.
1 Li И/., Nonaka Т., Chou Т.-С. // Electrochemistry 1999.67. 4—10.
2 Haikher AC, Goodin R.D., BaizerM.M. // US ftiteiU. 1981. 429 3393.
115
Следовательно, необходимы катализаторы для практического
катодного окисления в паре с анодным окислением. Нонака и его коллеги
разработали уникальную систему катодного окисления с
использованием окислительно-восстановительного медиатора воль-
фрамат/псрвольфрамат и применили его в парном электросинтезе,
как показано на рис. 5.144 Производное N-гидрокс ил амина в виде
единственного исходного субстрата непрямым образом окисляется
до нитрона в качестве единственного продукта с участием HWOJ,
полученного из HWO4 и Н202 в катодном отсеке, и галогена (Х2),
анодно полученного изХ~ в анодной камере. Суммарные выходы
нитрона по катодному и анодному току были очень высокими
(190%).
Cathode
Н,0
Anode
2е
О,
НА
HWO
HWO,
Рис. 5.14. Парный электросинтез с помощью катодно генерированного
перо кс и да водорода в качестве окислителя
Кроме того, парный электросинтез был разработан на основе
комбинации анодного окисления и катодного восстановления с
помощью гидроксильного радикала, возникающего из Н202 и
различных ионов металлов редокс-медиаторных катализаторов —
Fe2+/Fe3\ V4+/V3+, Cu+/CuJ+ и Ni2+/Ni3+,
Параллельная парная электрохимическая реакция полимера.
Совсем недавно Инаги и Фучигами с сотрудниками достигли
успеха в реализации первой парной электрохимической реакдии
проводящих полимеров, таких как чередующийся сополимер
9-флюоренола и 9,9-диоктилфлюоренона, адсорбированный
на аноде и катоде с образованием фрагмента 9-флуоренона
на аноде и флуоренового фрагмента на катоде2. Поскольку реакции
идут в твердой фазе, выделение продукта происходит сравнительно
легко.
Li W., Sonako Т. // J. Ekctrochem. Soc. 1999. I46.592-599; Shen K, Atobe M.,
Li W„ Nonaka T. // Eleclrochim. Acta. 2003.48. 1041-1046.
Inagi 5., Nagai //.. Tomita /., Fuchigami T. // Angcw. Chcm. Im. Ed. 2013. 52.
6616-6619.
116
5.6. РЕАКЦИОННОСПОСОБНЫЕ ЭЛЕКТРОДЫ
Чтобы избежать разложения продукта или интермедиата, как
только он образовался, на противоположном электроде, требуется
разделенная ячейка. Однако разделенная конструкция требует
увеличения напряжения на ячейке (напряжения между анодом и катодом),
что приводит к усложнению эксплуатации системы.
Когда желаемой реакцией является катодное восстановление,
то анодно растворимые металлические электроды можно
использовать в качестве анодов, чтобы как избежать разложения продукта
или интермедиата на аноде, так и уменьшить напряжение на
электролизере. Такие электроды называются разрушаемыми, или
растворимыми, электродами, растворимыми металлическими анодами1.
Катодное карбоксилирование органических галогенидов с
использованием магниевого анода даст карбоновую кислоту селективно
с высоким выходом-:
Cathode:
R-X + 2e->R- + X"
R +C02->RCOCr
Anode:
Mg-»Mg^ + 2e <5',8)
Total:
R-X+C02 + Mg -> RCOOMgX
or
(RCOO)2Mg+X_
B этом случае образованный карбоксилат-анион захватывается
ионом магния и выпадает в осадок. Следовательно, можно избежать
как анодного окисления образовавшегося карбоксилат-иона, так
и его этерификации нспрорсагировавшим органилгалогенидом.
В таких катодных реакциях карбоксилирования часто используются
алюминиевые, магниевые и цинковые аноды.
Недавно растворимые металлические аноды были использованы
в электросинтезе. Ионы металлов, полученные из анода, например.
Mg, Zn, A1 и Си, часто играют важную роль в электрохимических
реакциях. Эти ионы металлов и катодные генерируемые реакпи-
1 ChaussardJ., FotestJ.C, NedekcJ. К, PerichottJ., SibtiteS., Troupe M. // Synthesis,
1990.369-381.
г SitvestiiG.,GamhinoS., FilardoG., GuhttaA.//Angpw.Chem. Int. Ed. Engl. 1984.
23. 979-980; Yamauchi К, Ham S„ Settboku H. // Tetrahedron. 2010. 66. 473-
479; Ohkashi, XL, MkhinishU., Hara S., Senboku S.H. // Tetrahedron. 2010. 66.
7732-7737.
117
онноспособные частицы образуют весьма реакционноспособные
промежуточные соединения новых реагентов или захватывают га-
логенид-ионы, генерируемые при катодном восстановлении силил-
хлоридов в полисшганы <см. разд. 5.8.6)'. Кроме того, такие анодно
растворяемые ионы металлов существенно влияют на рспгохимию,
стереохимию и селективность продукта благодаря их координации
и каталитическим эффектам, а также формирование новых реагентов
in situ2. Например, как показано на рис. 5.15, ион цинка, полученный
из анода, и трифторметил анион, катодно генерируемый из CF3Br,
образуют металлоорганическое соединение в качестве интермедиата,
которое подвергается реакции Реформатского с альдегидами и ке-
тонами, давая трифторметилированные спирты. В
противоположность этому катодное восстановление CF3Br в присутствии ионов
цинка не приводит к желаемому продукту3. Следовательно, анодно
генерируемые ионы цинка очень специфичны и весьма отличаются
от обычных ионов цинка.
2Ч
icF^-вг
CFiBr 4
v У ""
ъ<
Znanode
RCOR'
Cathode
R
1
-* F3C—С—К
1
OH
R = Ph, R'=H:
R = PhCH2, R'=Me:90%
Рис. 5.15. Схема электросинтеза, использующего реакционноспособный электрод
Другие типичные примеры включают реакции кросс-сочетания
активированных галогенированных соединений и карбонильных
соединений, а также между негалогенированными соединениями4.
5.7. ЭЛЕКТРОХИМИЧЕСКОЕ ФТОРИРОВАНИЕ
Фторорганические соединения классифицируются на две группы:
перфорированные соединения и частично фторированные
соединения. Соединения первого класса широко используются в качестве
1 Shono Г., Kashimura S., Ishifitne A/., Nishida R. //J. Chem. Soc. Chem. Comm.
1990. 1160-1161.
2 Lehmkuht H. // SynLhcsis. 1973. 377-396; Tuck D.G. // Pure Appl. Chem. 1979.
51.2005-2018.
} Sibilie S„d'ItK(ift E„ Lcporf L., Peichon J. // Tetrahedron Lctl, 1986, 27. 3129-
3132.
4 Yamamoto ¥., Goda S.t Maekawa //., Nishiguchi I. // Org. Lctl. 2003. 15. 2755-
2758.
118
функциональных материалов, а представители последней группы
находят биологическое применение в качестве лекарственных
препаратов и агрохимикатов. Перфторсоединения получают путем
превращения всех связей С-Н в С—F с использованием
электрохимического фторирования в безводном жидком HF в качестве растворителя
с никелевым анодом. Этот процесс называется электрохимическим
перфторированием. Он уже коммерциализирован (см. гл. 8)т.
Электрохимическое перфторирование в KF—2HF расплаве
на угольном аноде было также разработано для получения перфтори-
рованных органических соединений с низким молекулярным весом2.
Электрохимическое частичное фторирование, или селективное
электрохимическое фторирование, является новым методом3. Так
как потенциал разряда фторид-ионов чрезвычайно высок (>2,9 В
отн. НКЭ на Pt аноде в MeCN), фторирование протекает через
(радикал) катионный интермедиат. который является обшим на пути
анодных нуклеофилъных замещений:
■R —с-^ +R
RH^^RHt ut ,R—F (5.19)
R —^-» +R
При использовании этого метода газообразный фтор не
генерируется и никаких опасных реагентов не требуется, поэтому такое
электрохимическое фторирование намного безопаснее, чем обычный
химический способ, который часто требует опасных и/или трудных
в обращении реагентов. Селективного электрохимического фтори-
SimonsJ.Ff,//S. Electrochem. Soc. 1949.95. 47—67; Rudge AJt Electrochemical
Fluorination in Industrial Electrochemical Processes/ed. AT. Kuhn. London:
Elsevier, 1971. P. 71—88; TasakaA. Electrochemical Perfluorination in Current Topics
in Electrochemistry. Vol. 10. Berlin: Springer-Verlag, 2004. P. 1—36; RazhkovI.N. //
Russ. Chem. Rev. 1976. 45. 615—629; Rozhkov l.N. Organic Electrochemistry. 2nd
edn./ eds M.M. Baizer, И. Lund.N. Y: Marcel Dekker, 1983. Ch. 24;Chitds W.V.,
Chmtensen L., Klink F.W., Koipin C.F. Anodic Fluorination in Organic
Electrochemistry. 3rd edn. / eds H. Lund, M.M. Baizer. N. Y: Marcel Dekker, 1991.
Ch. 26.
Chitds W. V., Christensen L., Klink F. W., Koipin C.F. Op. eit.
Chitds W. V,, Christensen L., Klink F. W., Koipin C.F. Op. cit.; Fuchixami T.
Electrochemistry applied to the synthesis of fluorinated organic substances // Advances in
Electron Transfer Chemistry. Vol. 6. / ed. P.S. Mariano. Greenwich CT JAI Press
1999; Fuchigami T. Electrochemical partial fluorination//Organic
Electrochemistry. 4th edn. / eds H. Lund, O. Hammerich. N. Y: Marcel Dekker, 2000:
Fuchigami 7'.. Jnagi S. // Chem. Commun. 2011. 47. 10211-10223.
119
рования можно обычно достичь в апротонных растворителях — аце-
тонитриле (MeCN), дихлорметане, димстоксиэтане (DME), нитро-
метане и сульфолане, содержащих ионы фтора, чтобы обеспечить
получение преимущественно моно- и/или дифторированньгх
продуктов1. Электролиз проводят при постоянном потенциале, значение
которого немного выше, чем первый окислительный потенциал
субстрата с использованием платинового или графитового анода.
Электролиз при постоянном токе во многих случаях также эффективен для
селективного фторирования. Выбор комбинации фоновой фторидной
соли и электролитического растворителя важен для достижения
эффективного селективного фторирования, поскольку конкурирующая
анодная пассивация (формирование непроводящей полимерной
пленки на поверхности анода, которая подавляет фарадеевский ток)
происходит в течение электролиза очень часто. Импульсный
электролиз во многих случаях эффективен для того, чтобы избежать такой
пассивации, поэтому трудно окисляемые фторидные соли, которые
не вызывают пассивации анода и имеют сильно нуклеофильный F-,
как правило, рекомендуются в качестве фоновых фторидных солей.
Таким образом, чаще всего используются расплавленные при
комнатной температуре соли — R3N-/»HF (N = 3—5), R^NF-wHF
(N = 3—5) и пиридиниевая поли (фтористоводородная) соль (Ру-
//HF), а в некоторых случаях эффективны даже соли R4NBF4
и R4NPF,,2. В частности, когда используются HF-фоновые соли
и катоды с низким перенапряжением водорода (такие как плагина),
в процессе электролиза на катоде преимущественно происходит
восстановление протонов (выделение водорода). Разделенная ячейка,
следовательно, не всегда нужна для фторирования в таких условиях.
В алротонном растворителе F" становится более нуклеофильным,
но его реакционная способность весьма чувствительна к содержанию
воды в системе электролиза, так как гидратированный F~ является
слабым нуклеофилом. Поэтому необходимо удаление следов воды
из растворителя и электролита, чтобы оптимизировать образование
фторированных продуктов.
Несколько примеров приведено ниже, но в литературе описаны
и другие примеры.
5.7.1. Электрохимическое фторирование ароматических колец
Ароматические соединения, такие как бензол, замещенные
бензолы и нафталин, селективно фторируются путем анодного
окисления при постоянном потенциале в Ft4N-3HF/MeCN или Et4NF-
4HF без растворителя^:
1 Fuchigami T,, Inagi S. Op. eit,
2 Fuchigami T. Op. ciL; Fuchigami T. Op. cit.; Fuchigami Т., SnagiS. Op. ciL
} Rozhkov l.N. Op. tit.; Childs W. K. Christensen L., Kiink F. W.. Koipin C.F. Op. tit.
120
F
F
El,NF-3IIF/MeCN
UVvs. SCE
-2e'-H ^ (5.20)
27% 3%
Фторирование протекает через присоединение двух фторид-
ионов с последующим элиминированием Н F — так обеспечивается
образование монофторированных ароматических соединений.
Примечательно, что ипсо-замещение фтором также происходит с
высоким выходом:
Y^VJ^ B4NF/McCN ' ~H^H (5-2,)
80%
5.7.2. Электрохимическое фторирование олефинов
Электрохимическое фторирование олефинов приводит к моно-
и/или дифторированным продуктам. Селективность зависит от
молекулярной структуры субстрата и электролитического растворителя1:
Rl4 _& R'. Л2
->
X.
V.7 F/MeCNorAcOH / \ (5.22)
R" г Y
Y=F, NHCOMe, OAc
Из га-ацетоксистирола и 1-ацетокси—3,4-дигидронафталина
образуются соответствующие а-фторкетоны2:
л, ,„, Et,N-3HF/MeCN^ „ , ,
АсО R1 О R1
HRI <5*>
63%
Fuchigami Т., Inagi S. Op. cit.; Andres D.F., Laurent E.G., Marquet B,Sr,
Benotmane //., Bemadat A. If Tetrahedron, 1995. 51. 2605-2618;
Laurent E.G., Tardive! Д., Benotmane H., Bemadal A. //Bull. Soc. Chirn, Fr. 1990, 127.
468-475; Dmowski W., Kozlowski T. // Electrochim. Acta. 1997. 42. 513-523;
Laurent E.G., Tardive! R.. Ttiiebault H. // Tetrahedron Lett. 1983. 24. 903;
Ventolin F.M., Faure R., Laurent E.G., Marquet B.S. // Tetrahedron: Asymmetry. 1994.
5. 1909-1912.
Fuchigami Т., Inagi S. Op. cit,; Laurent E.G., Tardive! R.7 Thiehault И. //
Tetrahedron Lett, 1983. 24. 903, Ventahn F.M., Faure Я.. Laurent E.G., Marquet B.S. //
Tetrahedron: Asymmetry. 1994. 5. 1909-1912.
121
5.7.3. Бензильное электрохимическое фторирование
Хотя анодные реакции бензил ьного замещения протекают легко,
анодное бензильное фторирование происходит не всегда. Основной
конкурентной реакцией является ацетамидирование, когда MeCN
используется в качестве растворителя. Например,
электрохимическое бензильное фторирование в MeCN протекает селективно, когда
//-положение фенильной группы замещено электронодоыорной
группой1:
СОМе -2е. -н+
F /MeCN
СОМе
NHCOMe
СОМе (5-24>
Х=Н
Х=МеО
7%
69%
34%
<1%
Наоборот, сх- ацетамидирование становится основной
реакцией, когда фенильная группа не имеет электронодонорного
заместителя.
Моно- и дифторирование может быть выполнено селективно
в зависимости от приложенного потенциала.
5.7.4. Электрохимическое фторирование сульфидов
Фучигами с сотрудниками обнаружил, что анодное фторирование
сульфидов, имеющих ос-электроноакцепторные группы, протекает
эффективно, давая соответствующие а-фторированные продукты
с хорошими выходами2:
R'S'
R2
-2с, -H1
f^/MeCNorDME
-> R'S
R2
R1 = Ar, Alkyl, Bzl, 2-Thiazolyl, 2-Pyridyl, 2-Pyrimidyl
R2 = CF3, CN, COR, COOR, CONR2, PO(OEt)2, C = CH
(5.25)
1 Laurent E.G., Marquet В.. Taniiwt R., Thiebault N. // Tetrahedron Lett, 1987. 28.
2359-2362.
2 Fuchigami Т., InagiS. Op. cit.; Fuchigami T,, Shimojo M., KonnoA,, Nakagawa K. //
J. Org. Chem. 1990. 55. 6074-6075.
122
Фторирование протекает по механизму Пуммерера через катион
фторсульфония (А)1:
Н
PhS—CHiR-F^PhS—CHR—т=*
F" л -HF
F (5.26)
* PhS=CHR-^PhS—CHFR
-HF
Таким образом, когда R — электронно-акцепторная группа, де-
протонирование А существенно облегчается, а следовательно,
фторирование протекает эффективно.
5.7.5. Электрохимическое фторирование гетероциклических
соединений
Многие гетероциклические соединения обладают специфической
биологической активностью, в то время как известно, что введение
атома (ов) фтора в органические молекулы резко меняет или
повышает их биологическую активность. Однако селективное
фторирование гетероциклических соединений с использованием обычных
реагентов фторирования осуществить не легко. На сегодняшний
день описаны многие успешные примеры селективного анодного
фторирования гетероциклов, содержащих серу, азот, кислород и фос-
финовый заместитель2. Некоторые примеры приведены в табл. 5.4.
Селективность фторирования также сильно зависит от фоновых
фторидных солей1:
-2е
О
I H
rVf'c -*
lLs^kivV* F EL*N F"4HF/McCH
Ph
68%
(cis/trans = 2)
=* »Г
El5N-3HF/MeCN 4
О
k*A(K
0
II
58%
11
k
P]
/F
^Ph
EljN-3HF/McCN
1 (5.27)
Kornto A., Nakagawa K., Fuchigami T. // J. Chem, Soc., Chem. Commun. 1991.
1027—1029; Fuchigami 71, Konno Л., Nakagawa K.t Shimojo M. jj J. Org. Chem.
1994. 59- 5937-594 L
Fuchigami 71., InagiS. Op.cit.; DawoodM. //Tetrahedron. 2004.60. 1435—1451.
Нои Г., Higashiya £, Fuchigami T. //J. Org. Chem. 1999. 64. 3346-3349.
123
Таблица 5.4
Примеры электрохимического фторирования
гетероциклических соединений
Субстрат
„ COOEt
f-Pr
оУ
оУ"
«Я\
Соль/растворитель
Et3N-3HF/MeCN
EtjN-3HF/MeCN
Et4NF-4HF/DME
Et4NF-4HF/DME
Et4NF-4HF/DME
Продукт
c COOEt
afe
сбс
сбс™
1"^
Выход, %
74 (translcis =
= 74/26)
88
66
72
81
Поскольку Et3N-3HF содержит свободное основание Et3N, диф-
торированный продукт, как только образуется, дегидрофгорируется
в монофторированный продукт.
Кроме того, известно анодное дифторированис, которое
сопровождается расщеплением двойной связи С—С1:
-4е
N^Ar
S'is Y^ "^*-3hf/dme
(5.28)
Ar=Ph:67%
=/>-BrC6H4: 62%
1 Shaaban M.R., Jnagl S., Fuchigami T. // Electrochim. Acta. 2009. 54. 2635-2639.
124
Анодное фторирование гетероциклических соединений,
полученных из оптически активных а-аминокмслот, протекает с
хорошими выходами и с высокой диастереоселективностью:
СООМе F СООМе
W
S^ NCOR с ~2лс'"" дс > S^ NCOR (5 29)
R =j?-Tol, Ph, Me, H 52-91% yield, 59-95% de
Me, СООМе Меь F% COOMe
/ \ _->c _H( Г\
Ov NCOPh . VIE ' .ц „M > 0> NCOPh (5.30)
73% yield, 81% de
Следует отметить, что фторирование происходит не в бензильном
положении, а преимущественно в а-положение к карбонильной
группе1:
/syR -2е,-н+ ^ ^ч^у R
L-X El3N-3HF/McCN \_Х
О О
(trans major) (5.31)
R = Ph, 2-Naphthyl, Mesityl, Et, и-Рг
X = NH, MeN, /-PrN, PhN, BzlN, O, S
Yield: 58-86%
5.7.6. Электрохимическое фторирование
гетероциклических соединений с PhS-группой в качестве
электровспомогательной
Гетероциклические соединения, имеющие фенилсульфенильную
группу, также анодно фторируются с хорошими выходами2:
PhS4 f
-2е, -Н
-N EliNOHF/MeCN ' J-N (5.32)
(f VBu о VBu
92%
1 Fuchigami Т., Narizuka S.. КоппоА. // J. Org. Chem. 1992. 57. 3755—3757.
3 Narizuka 5., Fuchigami T. // J. Org. Chem. 58. 1993. 4200-4201; Cao Y„ Hi-
dakaA., Tajima Т., fuchigami T. //J. Ощ. Chem. 2005. 70. 9614-9617.
125
ArS
\ У \ Et4NF-3HF/DME \ /U 4
58%
Следует отметить, что селективность продуктов фторирования
резко меняется в зависимости от природы электролитических
растворителей1:
F /j-CIC6H4S
°v_P ^EUNF^HF/DME °S/° Et^NF-SHF/CHiCl^
Y T
о о
F (5.34)
E^NFSHF/CHjCb °Х/°
О
96%
Как упоминалось ранее, часто происходит пассивация анода, что
приводит к снижению выхода и низкой эффективности по току. Чтобы
избежать такой пассивации, можно использовать различные
медиаторы — ионы галогенидов, триариламины и арилиодиды {рис. 5.1 б)2.
Кроме того, эфирные растворители, такие как ДМЭ, гораздо
более подходят, чем MeCN, для анодного фторирования различных
гетероциклических сульфидов3:
[ 1 ^Ш> Ш (5'35)
N s C02Me N S X02Me
in MeCN: 0%
DME: 92%
Ishii H., Yamada /V., Fuchigamt T. // Chem. Commun. 2000. 1617—1618.
Fuchigami Г., Sano M. //J. Electroanal. Chcm. 1996. 414. 81-84; Fuchigami Т.,
Tetsu A/., Tajima T. // Synlett. 2001. 1269—1271; Fuchigami Т., Fujita T. // J. Org.
Chem. 1994. 59. 1269—1271; Fujita Т., Fuchigami T. //Tetrahedron Lett. 1996. 37.
4725-4728.
Damod K.M., Ishii //., Fuchigami T, //J. Org. Chem. 1999, 64. 7935-7939;
Shaaban Л/.Д., Ishii H„ Fuchigami T. // J. Org. Chem. 2000. 65. 8685-8689;
Hau Г., Fuchigami 1\//J. Elecirochem. Soc. 2000.147.4567-4572.
126
Et3N-3HF
2F~
SPh
>Y +
PhSF
ОТ V
Ar=2,4-Br2QH3
R1 = H.R2 = QH5CH2:83%
R1 = H, R2=/)-BrCbH4CH2:100%
R1 - McSiOCH -T R1 = СьНзСЬЬ: 66%
I I
t-Ъи Me
Рис. 5.16. Электрокаталитическое
фтородесульфирование fl-лактамов
Выраженный эффект растворителя ДМЭ можно объяснить
с точки зрения значительного повышения нуклеофильности фторид-
ионов, а также подавления анодного пассивирования и
переокисления фторированных продуктов.
5.7.7. Электрохимическое фторирование с использованием
неорганических фторидных солей
Неорганические фторидные соли — фториды щелочных металлов
(MFs) устойчивы, просты в обращении и недороги. Поэтому они
выступают в качестве главных кандидатов в реагенты нуклеофиль-
ного фторирования, а также фоновых электролитов в химическом
и электрохимическом фторировании. Задача состоит в том, чтобы
преодолеть такие проблемы, как плохая растворимость и низкая
нуклеофильность MFs в органических растворителях. Межфазные
катализаторы (краун-эфиры и соли четвертичного аммония или фос-
фония), как известно, снижают кулоновское взаимодействие MFs
и обычно используются для этой цели. Фучигами с сотрудниками
сообщил об успешном анодном фторировании с использованием
системы поли (этиленгликоль)/МР, где MF — либо KF, либо CsF
(рис. 5.17)1.
1 Strwamura Т.. Takaheshi К.. Inagi $., Fuchigami Т. //Angew. Chem. Int. Ed. 2012.
51.4413-4416.
127
АНОД КАТОД
, Субстрат
Интермедиат
СП
и1
^UVWUVWW
Фторированный
продукт
Рис. S.17. Схема анодного фторирования с использованием KF
и полиэтиленгликоля с двумя концевыми гидроксильными группами как
многофункциональной добавки
5.8. ЭЛЕКТРОХИМИЧЕСКАЯ ПОЛИМЕРИЗАЦИЯ
Электрохимическая полимеризация использует для
полимеризации генерированные субстраты, такие как мономер или
инициатор. В первом случае электрогенерированные ароматические
мономерные пары в процессе поликонденсации дают гс-сопряженный
полимер, в котором р-орбитали ароматики перекрываются по всей
полимерной основной цепи. Такие сопряженные полимеры
обладают внутренне присущей проводимостью и поэтому называются
проводящими полимерами1. Как правило, они являются
полупроводниковыми материалами, но химическое или электрохимическое
допирование придает им электропроводность. Проводящие
полимеры получают на поверхности рабочего электрода в виде пленок,
так как перенос электрона от мономера и его реакция сочетания
протекают вблизи поверхности электрода, а нерастворимый
полимерный продукт выпадает на нем. Хотя химический перенос
электронов с участием мономеров в растворе также возможен при
получении соответствующих проводящих полимеров, полученный
нерастворимый полимерный порошок трудно обрабатывать в целях
дальнейшего применения. Легкое образование пленки при
электрохимической полимеризации используют в электрохимических
устройствах — сенсорах и дисплеях2. Эта глава посвящена
электроокислительной полимеризации, электровосстановительной полиме-
Skotheim T.A., Reynolds J.R. Handbook of Conducting Polymers. 3rd edn. CRC
Press, Boca Raton. FL 2007; InzeltG. Conducting Polymers, Heidelberg: Springer.
2008.
HeinzeJ., Frontana-Uribe ВЛ, LudwigsS. // Chcm. Rev. 2010.110. 4724-4771;
Beaujuge P.M.. Reynolds J.R. // Chem. Rev. 2010. 110. 268-320.
128
ризаиии и электросинтезу полисиланов. Полимеризация виниловых
мономеров с использованием системы электрогенерированного
инициатора обобщена в другом месте1, хотя недавние достижения,
связанные с этим процессом, включены в эту главу.
5.8.1. Электроокислительная полимеризация ароматических
мономеров
Электроннонасыщенные ароматические и гетероароматические
мономеры, показанные на рис. 5.18, могут быть легко окислены
на поверхности анода с образованием катион-радикального
состояния. Катион-радикалы сочетаются с образованием С-С-связсй,
а последующее дспротонированис приводит к нейтральному димеру:
О
х х
X = 0,S,NR " (5.зб)
Q Q 9 О О™'
н
Тиофен Фуран Пиррол Бензол Анилин
Рис. 5.18. Ароматические мономеры для окислительной полимеризации
Образованный сопряженный димер имеет более низкий
потенциал окисления, чем мономер, так что дальнейшее окисление димера
приводит к олигомеризации и полимеризации.
Высоко сопряженные полимеры уже не растворимы в
электролитической среде, а следовательно, осаждаются на поверхности
анода в виде пленки. Осажденный полимер окислительно допиру-
ется при наложении потенциала для полимеризации, таким образом,
формируемая пленка является не пассивной, а достаточно
проводящей, чтобы проводить непрерывную электрохимическую реакцию
на электроде. При использовании этого метода позиция сочетания
мономера может быть предсказана по распределению спиновой
Bhadani S.N., Parravano G* Electrochemical Polymerization in Organic
Electrochemistry, 2ndedn. Marcel Dekker; 1983. Ch, 13; Funt B.L. Electrochemical
Polymerization in Organic Electrochemistry. 3rd edn. N. Y: Marcel Dekker, 1990.
Ch. 32.
Г\ \ к
-2HT
129
плотности в ароматическом кольце1. Введение электровспомога-
тельных групп, например силнльной, может обеспечить
предпочтительное сочетание в определенном положении2.
5,8*2» Электрохимическая полимеризация
Существует несколько методов электрохимической
полимеризации, например, развертка потенциала, потеншюстатические (при
постоянном потенциале) и гальваностатические (при постоянном
токе) методы- Подходящий способ следует выбирать в зависимости
от типа мономеров и используемых условий. Измерения
характеристик мономера с помощью циклической вольтамперометрии
(ИЩА) на трехэлектродной установке дает информацию о потенциале
окисления для электрохимической полимеризации. Чтобы
предотвратить переокисление полимерного продукта, следует избегать
применения более высокого потенциала на рабочем электроде в
течение длительного периода, поскольку полимерный продукт с
расширенным л-сопряжением окисляется легче по сравнению с
соответствующим мономером. Метод развертки потенциала и потенцио-
статический метод требуют более мягких условий полимеризации,
чем гальваностатический метод.
Используя метод развертки потенциала, полимеризацию
проводят с применением ЦВА-анализатора. Рабочий электрод, проти-
воэлектрод и электрод сравнения находятся в электролитической
ячейке, содержащей электролит и мономер. Повторяя развертку
потенциала в диапазоне потенциалов, определенном измерениями
ЦВА, получают продукт в виде полимерной пленки на рабочем
электроде, и этот процесс переноса электронов можно
контролировать с помощью ЦВА (рис. 5.19), которая показывает типичные
циклические вольтамперограммы пиррола в ходе
электрополимеризации. Первое сканирование представляет собой ток окисления
мономерного пиррола, а затем полимеризация протекает на
поверхности рабочего электрода, что приводит к необратимой вольтам-
перограмме. В течение осаждения полипиррольного продукта при
повторных сканированиях анодный и катодный токи, возникающие
при редокс-процессе полипиррола в более низком диапазоне
потенциалов, постепенно увеличиваются.
В основе потенциостатического метода лежит трехэлектродная
система, а приложенный к рабочему электроду потенциал
контролируется потенниостатом с мониторингом перенесенного
заряда. В гачьваностатическом методе используется двухэлектродная
система, состоящая из рабочего электрода и лротивоэлектрода,
1 Ando S.Meda M. // Synth. Met. 2002.129. 207-213.
3 Lemaire M., Biichner W.< Garreau /J., Hoa K.A., GuyardA,, Roncali J. // J. Elec-
troanal. Chem. 1990. 281. 293-298.
130
а между ними пропускается постоянный ток с помощью источника
питания постоянного тока. Этот процесс не может контролировать
потенциал на рабочем электроде, что может вызвать такие проблемы,
как переокисление полимерного продукта.
рМоп
15st cycle
еГ01
si cycle
I 1 1 1 1 1 1
-1,2 -0,8 -0,4 0,0 0,4 0,8 1,2
E(vs.Ae/AgCl)/V
Рис 5.19. Типичные циклические вольтамперограммы пиррольного мономера
5.8.3. Условия электрохимической полимеризации
Концентрация мономера является одним из важных факторов
электрохимической полимеризации. Если полимеризация не
протекает на поверхности электрода, то использование большого
количества мономера может эффективно способствовать формированию
пленки проводящих полимеров. Выбор материала для рабочего
электрода также имеет большое значение с точки зрения не только
перенапряжения, но и совместимости с полимерными пленками.
В дополнение к обычным электролитическим растворам {фоновая
соль/растворитель) ионные жидкости при комнатной температуре,
как известно, пригодны в качестве среды для полимеризации1. Есть
несколько преимуществ электрополимеризации в ионных
жидкостях: 1) электрополимеризация протекает быстро; 2) существует
хорошая совместимость продукта-пленки с поверхностью электрода;
3) полимерный продукт имеет плотную и гладкую поверхность,
высокую электропроводность, высокую электрохимическую емкость
и хорошую обратимость окислительно-восстановительных циклов.
Недавно было показано, что эфирный комплекс трифторида бора
является привлекательным в качестве полимеризационной среды2.
1 Sekiguchi К., Atobe М., Fuchigami Т. // Electrochem. Commun. 2002. 4. 881-8S5.
2 ShiG., Jin S.,Xue G.. Li С // Prog. Polym. Sci. 2005. 30. 783-811.
131
Трифторид бора действует как кислота Льюиса, образуя комплекс
с ароматическим мономером, что уменьшает потенциал окисления
мономера. Поэтому данный способ эффективен для полимеризации
ароматических мономеров с относительно высокими потенциалами
окисления. Снижение потенциала полимеризации позволяет
избежать переокисления полимерных продуктов.
5.8.4. Электрохимическое допирование
Если полимерная пленка на рабочем электроде получается в
результате электрополимеризации, электрод должен быть помешен
в электролитический раствор без мономера (свободный от мономера
электролит), чтобы изучить редокс-поведение проводящего полимера.
Обычно на вольтамперограмме наблюдается пара широких
пиков — редокс-откликов. Добавление или удаление электронов
на или от проводящих полимеров приводит к образованию поля-
ронов и биполяронов в повторяющейся структуре (обычно известное
как допирование), что приводит к значительным изменениям
физических свойств самого полимера и приданию ему таких свойств,
как резкое изменение цвета и электропроводности'. Таким образом,
заряды, генерированные вдоль полимера, должны быть
компенсированы путем добавления соседних ионов (или допантов), а введение
или удаление таких допантов может вызвать изменение объема
сопряженного полимера. Отношение допирования можно оценить
но величине заряда, перенесенного в проводящих полимерах.
5.8.5. Электровосстановительная полимеризация ароматических
мономеров
Ароматические дигалогениды являются примерами мономеров для
электровосстановительной полимеризации. Катодное восстановление
ароматического дигалогенидд генерирует анион-радикал, затем
следует полимеризация, сопровождаемая элиминированием галогеттида:
Вг-Аг-Вг +2с-"2Вг> -(-Аг-4- (5-37)
Образование связи происходит в определенном положении
мономера, в котором замещен атом галогена.
При элсктровосстановитсльной полимеризации 1,4-дибромпири-
дина эффективно добавление каталитического количества комплекса
никеля (11), а комплекс работает в качестве медиатора. Электроге-
нерированный никель (0) способствует дегалогенированию диб-
ромпиридинас его последующей полимеризацией2. Окислительная
1 Skotheim T.A., Reynolds J Я. (eds). Op. cil; Inzelt G. Op. cit.
3 Saito N.. Kanbara Т.. Nakamura Y, Yamamoto, Т., Kubota K. // Macromoleculcs.
1994.27.756-761.
132
электрополимеризация пиридина затруднена из-за его высокого
потенциала окисления, но восстановительная полимеризация этим
путем позволяет получить полипиридины.
Поли (л-фениленвитгилен) также синтезируется электровосста-
новитсльной полимеризацией. Электрохимическое восстановление
с^осо^а'-тетрабром-я-ксилола генерирует хинодиметановый интер-
медиат, последующая полимеризация дает поли (ксилилен), а далее
он восстанавливается до поли (п-фенилен винилена)1:
(5.38)
5.8.6. Применение проводящих полимеров
Физические свойства проводящих полимеров существенно
изменяются в процессе допирования. В табл. 5.5 суммировано
применение проводящих полимеров. Недопированное (нейтральное)
состояние проводящих полимеров, как правило, является
полупроводниковым. Его можно применить в качестве материала для
транзисторов, фотоэлементов и электролюминесцентных устройств.
Причем электропроводность проявляется в допированном состоянии
проводящих полимеров, что является полезным свойством
проводящих материалов, антистатиков и материалов конденсаторов.
Процесс допирования/дедопирования проводящих полимеров также
полезен для приложений в электрохромных устройствах, сенсорах
и преобразователях. Применение проводящих полимеров для
электроники описано в гл. 7.
Таблица 5.5
Применения проводящих полимерных пленок
Свойство
Проводимость (лопированное
состояние)
Процесс допинга/дедопиро-
вания
Полупроводниковые свойства
(нелегированное состояние)
Приложение
Проводящие материалы, антистатики,
конденсаторные материалы
Электрохромные материалы, датчики,
преобразователи, палить
Транзисторы, фотовольтаическис
ячейки, электролюминесцентные материалы
UtieyJ.H.P., GruberJ. //J. Mater. Chem. 2002.12. 1613-1624.
133
5.8.7. Электрохимический синтез полисиланов
Полисиланы, состоящие из линейно связанных цепочек Si—Si,
являются классом о-сопряженных полимеров. Они показывают
уникальные оптические свойства1. Метод Киппинга (хорошо известный
синтетический способ получения полисиланов из дихлорсилана)
является мощным, но использование металлического Na незаменимо.
Электрохимическое восстановление хлортриметилсилана дает гек-
саметилдисилан без использования восстановителя. Хотя
полимеризация дихлорсиланов путем электрохимического восстановления
достаточно сложна, использование магнийрастворимого анода
приводит к эффективному образованию полисиланов2:
Мс
Мс
2 Мс—Si—CI +C1—Si—CI
Me
Ph
THF/LiC104
Mg-Mg
Me Мс Me
I I I
>Me-Si-Si-Si-Mc (5.39)
I I I
Me Ph Me
Предложенный механизм заключается в том, что Mg2+, как только
он анодно возникает, восстанавливается на катоде, давая реакнион-
носпособный Mg. Он способствует образованию Si-Si связей
мономеров. Си и А1 также доступны в качестве анодных материалов для
восстановительной полимеризации полисиланов.
Восстановительная полимеризация дихлорсиланов дала
различные сополимеры — случайные сополимеры силановых
мономеров, поли (карбосиланы) и статистические сополимеры силано-
вого мономера и германиевого мономера (рис. 5.20)3.
/ Me Me
\ Me Me /
OCH^OCHi
Рис. 5.20. Структуры сополимеров на основе силана и германа, полученные
путем восстановительной полимеризации
Miller R.D., MlchlJ. // Chcm. Rev. 1989. 89. 1359-1410.
Shono Т., Kashimura S., hhifiim M., Nishida R. //J. Chem. Soc, Chem. Commun.
1990. 1160-1161.
Kashimura S., Ishifime M.yYamashita N., Bu H.B., Takebayashi A/., Kitajima S.,
Yoshizawa 0., Kataoka KT Nishida /Г., Kawasaki S.^ Mumse H., Shono T, //J. Org,
Chem. 1999. 64.6615-6621; Shono Г„ Kashimura S., Murase H. //J. Chcm. Soc.
Chem. Commun. 1992. 896-897.
134
5.8.8. Цепная полимеризация, инициируемая
электрогенерированными активными частицами
Электрогенерированные активные частицы малых молекул могут
работать в качестве инициаторов ионной и радикальной
полимеризации виниловых мономеров1. Например, метил-радикал,
генерируемый электролизом ацетата по Кольбе, может быть инициатором
радикальной полимеризации олефинов. Неоднородность
концентрации инициатора в электрохимической установке затрудняет
получение высокомолекулярных полимеров.
Прогресс в изучении контролируемой радикальной
полимеризации с использованием катализаторов переходных металлов был
заметен как в академических, так и промышленных кругах2. Равновесие
(kjkja) между исходными частицами (А) и активными частицами (В),
контролируемое окислительно-восстановительной реакцией
катализатора из переходного металла, может контролировать скорость
потребления мономера, приводя к регулируемой полимеризации.
Внешние стимулы электрохимическим окислением и
восстановлением металлического катализатора изменяют равновесие, а
следовательно, возможно более точное управление полимеризацией'1:
Присоединение
Анодное окисление
мономера
Р„—Вг + Cu1 — Вг <г=±- Си11—Вп + Р„- (5.40)
(А) к у (В)
Катодное восстановление
5.9. ЭЛЕКТРОКАТАЛИТИЧЕСКИЙ СИНТЕЗ
ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Нельзя обойтись без рассмотрения некоторых новых подходов
к синтезу ряда фосфорорганических соединений (ФОС), основанных
на гомогенных электрокаталитических реакциях. Решения,
предложенные в этой области, наиболее ярко демонстрируют
экологическую чистоту электрохимических синтезов. Прогресс в области
функдионализашш белого фосфора, наблюдающийся в последнее
десятилетие, обусловлен использованием либо комбинаций
реагентов (нуклеофил-электрофил), либо катализаторов, что позволяет
1 Bhadani S.N., Purravano G. Op. cit.; Funt B.L. Op. cil.
1 Maryjaszewski K.. Xia J. // Chem. Rev 2001. 101, 2921-2990; Kamigaito M.,
Ando Т., Sammoio M. // Chem. Rev. 2001. 101. 36K9-3745.
J Magenau A.J.D., Strandwitz N.C., Gennaro A. Matyjaszev/ski K. // Science. 2011.
332. 8.
135
достигать хороших результатов в мягких условиях. Широкие
перспективы открывает применение нетрадиционных методов синтеза
ФОС из Р4, прежде всего электрохимических, которые сочетают
достоинства различных подходов. Интерес к электросинтезу с участием
Р4 вызван рядом факторов, среди которых следует выделить мягкие
условия проведения процесса (невысокая температура, нормальное
давление), возможность его проведения в практически замкнутой
системе с минимальным количеством циклически регенерируемого
реагента-катализатора. Принципиально новые по сравнению с
традиционными подходы к синтезу ФОС из белого фосфора,
комбинируя преимущества обычной гомогенной химии в объеме раствора
и электрохимии, в случае использования которой реагенты
генерируются на электродах непосредственно в реакционной системе,
предложены относительно недавно1. С целью эффективного
вовлечения белого фосфора в реакцию было предложено использовать
одновременно процессы, протекающие в неразделенной ячейке:
1 Ромахин АС, Будникова Ю.Г., Зарипов И.М., Каргин Ю.М., Никитин Е.В.,
Томшов А. П., Игнатьев Ю.А., Смирнов ВВ. // Изв, АН СССР, Сер, хим, 1992.
N° 6. С. 1322; Ромахин А.С., Зарипов ИМ., Будникова Ю.Г., Каргин Ю.М.,
Никитин ЕВ., ТомшовА.П., Игнатьев Ю.А. // Иш АН СССР. Сер. хим, 1992.
№ 6. С. 1328; Будникова Ю.Г., Каргин ЮМ., Зарипов ИМ., Ромахин А.С.,
Игнатьев ЮЛ., Никитин Е.В., Тамилов А. П., Смирнов В. В. // Изв. АН СССР. Сер.
хим. 1992, № 9. С. 2033; Ромахин А.С., ЗариповИМ., Будникова ЮГ..
Игнатьев Ю.А., Никитин ЕВ., Каргин ЮМ., Томилов AM. А. С. СССР JVfe 1494492;
Ромахин А.С., Зарипов ИМ., Будникова Ю.Г., Игнатьев Ю.А., Смирнов В.В.,
Никитин Е.В., Каргин Ю.М., Томилов А.П. А. С. СССР № 159495I;
Ромахин АС, Зарипов ИМ., Будникова Ю.Г., Игнатьев Ю.А. // Электрохимия,.
1989. Т. 25. С. 780; Будникова Ю.Г., Каргин ЮМ. // Ж-л. общ. химии. 1995.
Т. 65. В. 4. С. 566; Budnikava Y.H., Kargin Yu.M., Sinyashin ОС // Phosphorus
and Sulfur. 1999. Vbl. 144-146. PL 1. R 565; Budnikova Y.H., Kargin Yu.M.,
Sinyashin O.G. И Phosphorus and Sulfur 1999. Vbl. 144—146. P. 51; Kargin Yu.M.,
Budnikova Yu.H.,MartynovB.J., Turygin V.V., TomitovAP.// S. Eleclroanal. Chem.
2001. Vbl. 507. № 1-2. P. 157; Будникова Ю.Г., Каргин Ю.М., Зарипов И. М.,
Ромахин А.С., Игнатьев ЮЛ., Никитин Е.В., Томилов А.П. Ц Изв. АН СССР.
Сер. хим. 1992. № 9. С. 2039; Ромахин АС, Зарипов ИМ., Будникова Ю.Г.,
Игнатьев Ю.А., Смирнов В.В., Никитин Е.В., Каргин ЮМ., Тактов А. П.
А. С. СССР № 1582580; Ромахин АС. Зарипов ИМ., Будникова Ю.Г.,
Игнатьев Ю.А., Смирнов В.В., Никитин Е.В., Каргин ЮМ., Томилов А.Н. А. С.
СССР № 1602015; Будникова Ю.Г., Каргин ЮМ. // Ж-л общ. химии. 1995.
Т. 65. В. 10. С. 1663; Budnikova Yu.H., Yakhvarov DC, Kargin YuM. // Mendeleev
Commun. 1997. P. 67; Budnikova Yu.H.. PerichonJ., Yakhvarov D.G., Kargin YuM.,
Sinvashin O.G. //J. Organomel. Chem. 2001. Vbl. 630. P. 185; Каргин ЮМ.,
Будникова ЮЛ. //Ж-л общ. химии. 2001. Т. 71. В. 9. С. 1472; ЯхваровД.Г.,
Будникова Ю.Г., Tan-ев Д.И., Каргин ЮМ., Синншин О.Г. // Шв. АН СССР. 2002.
Т. 51. Мр 11. С. 1903; Будникова Ю.Г., ЯхваровД.Г.. Каргин ЮМ. // Ж-л обш.
химии. 1998. Т. 68. В. 4. С. 603; Будникова Ю.Г., Каргин ЮМ., Ромахин АС, Си-
няшин О.Г. Патент РФ № 2199545; Будникова Ю.Г., ЯхваровД.Г., СиняшинО.Г.
Патент РФ №2221805.
136
на катоде — образование таких нуклеофилов, как алкоголят-, ги-
дроксид-, фенолят-, амид- и т.д. ионов, а на аноде — окисление га-
логенид-ионов, например, иодид-иона до иода1.
Последний совместно с нуклеофилом действует на Р4? приводя
к образованию различных производных кислот фосфора в
зависимости от условий электролиза. Таким образом, электросинтез в безди-
афрагменном электролизере, использующий одновременно реагенты,
генерируемые как на аноде, так и на катоде, является воплощением
наиболее перспективного подхода, заключающегося в совместном
действии нуклеофильных и электрофильных реагентов на
молекулу белого фосфора. Проведение электролиза в водно-спиртовых
растворах в оптимальных условиях и при соотношении реагентов
М(Р): М(Н20): M(ROH) = 1: (1+1.1): (2(^-50) позволяет получать три-
алкнлфосфаты практически с количественным выходом как по
фосфору, так и по току. Общая схема процесса в этом случае следующая2:
P4 + 12ROH+4H2O-^^4(RO)3PO+10H2T (5.41)
Предложенный метод получения триалкилфосфатов является
практически безотходным. Побочным продуктом в этом случае яв-
1 Ромахин А.С, Будникова Ю.Г., Зарипов И.М., Каргин Ю.М., Никитин Е.В.,
Томилов А.П.. Игнатьев Ю.А., Смирнов В.В. Указ. соч.; Ромахин А.С,
Зарипов И.М.. Будникова Ю.Г., Каргин Ю.М., Никитин Е.В.. Томилов А.П.,
Игнатьев Ю.А. Указ. соч.; Будникова Ю.Г., Каргин Ю.М., Зарипов ИМ.,
Ромахин А.С, Игнатьев Ю.А., Никитин Е.В., Тамилов А.П., Смирнов В.В.
Указ. соч.; Ромахин А.С.Г Зарипов И.М., Будникова ЮГ., Игнатьев Ю.А.,
Никитин Е.В., Каргин Ю.М., Томилов А.П. А. С. СССР № 1494492;
Ромахин А.С., Зарипов И.М., Будникова Ю.Г., Игнатьев Ю.А., Смирнов В.В.,
Никитин Е.В.. Каргин Ю.М., Томилов AJJ. А. С. СССР № 1594951; Ромахин АС,
Зарипов И.М., Будникова Ю.Г., Игнатьев Ю.А. Указ. соч.; Будникова Ю.Г.,
Каргин ЮМ. // Ж-л. общ. химии. 1995. Т. 65. В. 4. С. 566; Budnikova Y.H.,
Kargin Yu.M., SinyashinO.G./f Phosphorus and Sulftir. 1999. Vol. 144—146. Pt. I.
R 565; Budnikova Y.H., Kargin Yu.M., Sinyashin O.G. // Phosphorus and Sulfur.
1999. Vol. 144-146. P. 51; Kargin Yu.M., Budnikova YuH., Martynav B.J., Tu-
rygin V.V., TomilovA.P.Указ. соч.; Будникова Ю.Г., Каргин Ю.М., Зарипов ИМ.,
Ромахин А.С, Игнатьев Ю.А., Никитин ЕВ., Томияов А.П. Указ, соч.;
Ромахин А.С., Зарипов И.М., Будникова Ю.Г., Игнатьев Ю.А., Смирнов В.В.,
Никитин Е.В., Каргин Ю.М., ТомшовАП. А. С. СССР № I5825S0; Ромахин А.С.,
Зарипов И.М., Будникова Ю.Г., Игнатьев Ю.А., Смирнов В.В., Никитин Е.В.,
Каргин Ю.М., Томилов А. П. А. С. СССР N° 1602015; Будникова ЮГ.,
Каргин ЮМ. Ц Ж-л общ. химии. 1995. Т. 65. В. 10. С. 1663.
2 Ромахин А.С., Зарипов И.М., Будникова Ю.Г., Игнатьев Ю.А., Никитин Е.В.,
Каргин Ю.М., Томилов А.П. А. С. СССР Кя 1494492; Ромахин АС,
Зарипов И.М., Будникова Ю.Г., Игнатьев Ю.А. Указ. соч.; Budnikova Y.H.,
Kargin Yu.M., Sinyashin O.G. Op. cit.; Budnikova Y.H., Kargin Yu.M., Sin-
Yashin 0.G. Op, cil,; Будникова Ю.Г., Каргин Ю. М.. Ромахин А.С., Синяшип О. Г.
'Патент РФ №2199545.
137
ляется только водород. В изучаемых системах катодно-генерируемый
нуклеофил действует как инициатор раскрытия фосфорных циклов,
а продукт окисления иодид-иона способствует замещению протона
Р—Н связи, регенерируясь в ходе электролиза и завершая цикл.
Высокая селективность и большая скорость электросинтеза позволяют
проводить последний с высокой конверсией фосфора в целевые фос-
форорганические продукты:
+ 2е
-2е
(5.42)
ROH
(Н20)
(RO)3PO 90%
(RO)2PHO 60%
[{i-RO)2P(0)]20 80% и т.д.
Таким образом, на примере простой системы фосфор — спирт —
вода видно, что методы электрохимии являются эффективными
в синтезе, имеющими ряд существенных преимуществ перед
классическими методами органической химии: мягкие условия процесса,
его высокая скорость и селективность; экологическая безопасность
(электрофильный компонент реакции циклически регенерируется
на аноде); целенаправленный синтез требуемых продуктов за счет
варьирования условий электролиза.
Накопленный опыт был использован для разработки
электросинтеза диалкилфосфитов, триарилфосфатов и триарилфосфитов',
а также триамидофосфатов и триамидотиофосфатов из белого
фосфора по следующим реакциям2:
Kargin Yu,\f., Budnikova YuJJ., Martynov В.I., Turygin V.V., Tomilov A. P.
Указ. соч.; Будиикова Ю.Г., Каргин Ю,М., Зарипов И.М., Ромахин А.С,
Игнатьев Ю.А., Никитин Е.В., Томилов А. П. Указ. соч.; Ромахин А.С, За-
рипов И.М., Будиикова Ю.Г., Игнатьев Ю.А., Смирнов В.В., Никитин Е.В.,
Каргин Ю.М.,~ТомиювА.П. А. С. СССР№ 15Я25НО.
Kargin Yu.M,, Budnikova Yu.H., Martynov B.I., Turygin KK, Tomilov A.P. Op. cit.;
Ромахин А.С, Заршюв И.М.7 Будникава Ю.Г., Игнатьев Ю.А., Смирнов В. В.,
Никитин Е.В., Каргин Ю.М., Томилов AM. А. С. СССР № 1602015; Будни-
кова Ю.Г., Каргин ЮМ. //Ж-л общ. химии. 1995. Т. 65. В. 10. С. 1663.
138
P4+8ROH+4HzO ±12c'HX' >4(КО)2Р(0)Н + 6Нд
65%
Р4 +16 АЮН _^>1_> ц АЮЬ РО+4АгН +10Н2
90%
Р4 +12АЮН ** > Ч АЮ)3 Р + 6Н, (5.43)
75%
Р4 + 12R2NH+4H20 ±ЗХк )4(R2N)3PO+10H2
P4 + 16R2NH+4CS2^^->4<R2N)3PS+4R2NC(S)H + 6H2
68%
Описанные выше способы, основанные на совместном действии
на белый фосфор нуклеофильных и электрофильнътх реагентов в без-
диафрагменном электролизере, oi-крывают новый подход к синтезу
в мягких условиях соответствующих эфиров или амидов кислот
фосфора, однако они позволяют получать только эфиры кислот
фосфора, но не соединения со связью фосфор-углерод.
Соединения со связями фосфор — углерод являются наиболее
важными в химии фосфора. Мы предположили, что удастся воачечь
Р4 в реакцию с алкил- и арилгалогенидами в условиях, когда метал-
локомплексный катализатор циклически генерируется на электроде.
Действительно, впервые был предложен новый эффективный способ
селективного электросинтеза соединений с Р-С-связями из белого
фосфора в присутствии электрохимически генерированных
катализаторов — комплексов нульвалентного никеля'. Последний
образуется in situ из комплекса двухвалентного никеля, который в ходе
электролиза регенерируется. Активация белого фосфора и разрыв
связей Р—Р происходят в реакционной системе, обеспечивающей
циктическую регенерацию катализатора на электроде2:
Budnikova Уи.И. Yakhvarov D.G., Kargin Yu.M. Op. cil.; Budnikova Yu.H., Peri-
chonJ., Yakhvarov D.G.. Kargin Yu.M., Sinyashin O.G. Op. cit.; Каргин Ю.М.,
Будникова ЮГ. //Ж-л общ. химии. 2001. Т. 71. В. 9, С. 1472; ЯхваровД.Г., Буд-
никова Ю.Г., ТазеевД.И., Каргин Ю.М., Синяшин О.Г. Изв. АН, 2002, Т. 51,
№ И, С. 1903; Будникова ЮГ., Яхваров Д. Г., Каргин ЮМ. Указ, соч.;
Будникова Ю.Г., Каргин Ю.М., РомахинА.С, СинншипО.Г. Патент РФ N 2199545;
Будникова Ю.Г., Яхваров ДГ, Синяшин О.Г. Патент РФ № 2221805; Budnikova Yu.H.,
Tazeev Г)J., Trofimov В.A., Sinyashin O.G. // Eleclrochem. Commitn. 2004. VjI. 6.
N? 7. P. 700; Budnikova Yu.H., Sinyashin O.G. //). Otganomet. Chem. 2005, Nbl. 690.
№ 10. P 2416—2425; Peruzzini Л/., Abdreimova R,R.< Budnikova K. Romerosa A.,
SchererOJ.. Sitman H. //J. Oiganomct, Chem. 2004. \bl. 689. № 24. P. 4319.
Budnikova Yu,H. Yakhvarov D.G., Kargin Yu.M. Op. cit.; Budnikova Yu.H,, Peri-
chonJ., Yakhvarov D.G.. Kargin Yu. Л/., Sinyashin O.G. Op. cit.; Каргин Ю.М., Буд-
никова Ю.Г. //Ж-л общ. химии. 2001. Т. 71. В. 9. С. 1472; ЯхваровД.Г, Будни-
кош Ю.Г.+ ТазеевД.И., Каргин Ю.М., Синяшин О.Г. Указ. соч.; Будникова /О.Г.,
Яхваров Д.Г., Каргин Ю.М. Указ. соч.: Будникова Ю.Г.Л Яхваров Д.Г.,
139
Катод:
Анод:
P4+RX
Ni"L3 + 2e^Ni0L2 + L,
п = 2,3;
М-ие->М ,
*>P-R
L = bpy
M = Mg, Zn, A]
0)
(2) (5.44)
Ni L2
Показано, что на природу и выход конечных продуктов
электрохимического фосфорилирования органических галогенидов белым фосфором
существенное влияние оказывает материал расгворимого анода.
Использование цинкового анода приводит к появлению продуктов трехкоорди-
нированного фосфора — триорганилфосфинов, атюминиевого анода —
триорганилфосфююксидов, а натичие в реакционной смеси ионов Mg"
обеспечивает превращение Р4 в циклические фосфины (PhP)g:
Zn' or
Zn анод
PPh
(5.45)
Синхронное воздействие на белый фосфор катодно и анодно
генерируемых реагентов приводит к селективному превращению
тстрафосфора в трифенилфосфин. Использование в качестве
катализаторов комплексов никеля с сигма-донорными лнгандами типа
2,2'-бипиридила позволяет осуществлять каталитическое
превращение белого фосфора в трифенилфосфин:
Катод Анод
Э
(5.46)
+2е
-2е
Сипяшин О.Г, Патент РФ № 2221805; Budnikova Yu.H., Tazeev Л,ЛТ Tro-
Jimov В.Л., Sinyashin O.G. // Electrochem. Commun. 2004. \bl. 6. № 7. P 700;
Budmkova Yu.H., Sinyashin O.G. //J. Organomet. Chem. 2005. \bl. 690. Ns 10.
P. 2416—2425; Peruzzini A/"., Abdreimava R.R., Budnikova Y., Rumerosa A.,
SchererOJ., Sitman H. // J. Oiganomet. Chem. 2004. \bl. 689. Ms 24. P. 4319.
140
При этом происходит катодное восстановление комплекса
Ni(ll)L в каталитически активную форму Ni(0)L. Последующие
реакции носят гомогенный характер. Так, Ni(0)L реагирует с арилга-
логенидом по реакции окислительного присоединения с
образованием сигма-комплекса ArNiXL, сигма-комплекс атакует молекулу
белого фосфора с разрывом связей Р—Р и образованием связей
Р—С, в конечном счете давая соединение трехкоординированного
фосфора. При этом происходит регенерация катализатора и цикл
повторяется.
Недавно описан новый перспективный подход: третичные
фоефины получены электрохимическим путем, сочетанием моно-
и дихлорфосфиттов с ароматическими и гетероароматическими га-
логенидами, катализируемым комплексом никеля с Ыру в
неразделенной ячейке, снабженной растворимым магниевым или цинковым
анодом1:
АгХ +■ Ph„PCI3_„ *-Шт_**» ? Аг3 „Ph„P,п = 1,2
Mg или Zn анод
—^ \ СН3—f^N—СН^
R= CH31 СН30, (CH3)2N, CN, C02Et, CH3QO)
Для превращения арилгалогенидов с донорными заместителями
в кольце следует использовать анод из Mg, а с акцепторными — анод
из Zn. Предложенный метод электросинтеза третичных фосфинов
эффективен в отношении как ароматических галогенидов с
акцепторными и донорными заместителями в кольце, так и гетеро-
ароматических пиридин-, тиофен-, пиримидин- и пиразол-галогенидов.
Преимуществами его также являются одностадийность и мягкие
условия (комнатная температура) проведения процесса.
Предложена общая схема циклической регенерации катализатора2. Важной
Budnikova Yu.H., Kargin Yu.M., PerichonJ., NedeleeJ.-Y. //J. Onganomet. Chem.
1999. \bl. 575. P. 63: Budnikova Yu.ll., Kargin Yu.M.,$toyashm O.G. // Mendeleev
Comm. 1999. P. 143; Budnikova Yu.H., PerichonJ., NedetecJ.-Y., Kargin Yu.M. //
Phosphorus and Sulfur. 1999. Vol. 144-146. P. 881; Будпикоаа Ю.Г.,
Каргин Ю.М., Синяшин О. Г. Указ. соч.; Будникова Ю.Г., Карги и Ю.М. // Ж-л
общ. хим. 1995. Т. 65. С. 1660.
Там же.
141
реакцией, приводящей к целевым соединениям, является
восстановительное элиминирование из никельарилфосфидного комплекса
ArPh2PNiXbipy. Общую схему циклической регенерации
катализатора можно представить в следующем виде:
Ph2PCI
Ni(0) Ph2PCl
илие
ArNi1
РЬР-Ar
PfoP-Ar
ArX
На начальных этапах электролиза продукты образуются по
реакциям, изображенным в правом цикле, а по мере расходования
NiBr2bipy начинают преобладать реакции левого цикла.
Фосфорорганические соединения, содержащие перфторал-
кильные группы, привлекают особое внимание химиков-синтетиков
прежде всего благодаря потенциальному использованию в качестве
лигандов в металлорганической химии, а особенно в области фтор-
ного бифазного катализа. Фосфины, функционализированные пер-
фторал«сильными группами, проявляют термоморфные свойства,
заключающиеся в значительном увеличении растворимости,
наблюдаемом при повышении температуры. Наконец, введение атомов
фтора и фторалкильных групп в соединения фосфора
увеличивает биоабсорбцию и метаболическую стабильность биологически
важных соединений.
Была продемонстрирована возможность прямого получения
соединений со связями Р-С из белого фосфора и фторорганических
галогенидов под воздействием электрохимически генерированных
комплексов никеля в низких степенях окисления. Совместный
электролиз фторорганических галогенидов и эмульсии белого фосфора
в ДМФА в присутствии комплекса (bpy) NiBr2 позволил осуществить
активацию и трансформацию белою фосфора с высокой конверсией
в продукты со связями P-RFJ:
1 Mikhaylov D.Y., Gryaznova Т. У., Dudkina Y>B., Polyancev F.M.,
Latypov Sh.K., Sinyashin O.G., Budnikova Y.H. // J. Fluor. Chcm. 2013.
Vol. 153. P. 178-182.
142
»^ь,
,N4
NiBr2
N „ иль
-> P—RF —^^»
I2C/P4 D /
дмфа kf (5.49)
^"ОД Rp = CH2CH2(CF2)3CF3
Rf = CfiFn
Rf n
ha > \/
R^ NRF
RF = CH2CH2(CF2)3CF3
Rf = CeFn
Электролиз протекал в неразделенной электрохимической ячейке
с использованием платинового катода и растворимого цинкового
анода в отсутствие вспомогательного электролита. Во всех случаях
основными продуктами в реакционной смеси являются третичные
фосфины и некоторое количество фосфиноксшюв, образование
которых доказывают спектры ЯМР 31R Однако, так как большинство
третичных фосфинов является нестабильным, было решено
провести дальнейшее окисление фосфинов до фосфиноксидов, которые
стабильны и удобны для выделения. Тем более что целью данных
экспериментов было принципиальное доказательство применения
электрокаталитического метода для получения фторорганических
производных фосфора. Другим важным аспектом является тот что
все электросинтезы проводились без вспомогательных электролитов,
что упрошаст методики выделения продуктов и тем самым
расширяет потенциальные границы применения найденного метода. Стоит
отметить, что электросинтез в отсутствие никелевых катализаторов
с фоновым электролитом дает только следовые количества
соответствующих фосфинов (по спектру ЯМР -мР-реакционных смесей)
с преобладающим образованием неидентифицируем ых смесей
фосфорных производных, полифосфидов и других побочных продуктов.
Возможно предположить, что белый фосфор участвует в реакции
с перфторортаническим галогенидом в условиях каталитической
генерации металлокомплексного катализатора на электроде. По
аналогии с процессом функшгонализации белого фосфора с помощью
реактивов Гриньяра ключевой стадией данного процесса является
взаимодействие никельорганического интермедиата [RFNi"XLHJ
с молекулой белого фосфора:
143
+2с
RrX
Анод
RfNiXf
-2c (5.50)
Ранее были описаны реакции электрохимического сочетания
моно- и ди-хлорфенилфосфина с арил галогенидами, катализиру-
мыми комплексами1. Основываясь на результатах данных
экспериментов, было решено использовать дифенилхлорфосфин в реакциях
электрокаталитического фторалкилирования:
Ph
\
RFI + Р—С1
Pli
2e7RFI
ДМ<М
Zn анод
Rr = (CF2)5CF2H
RF=(CF2)5CF3
(5.51)
Ph О
НгОг> \ '/
Ph' Ч
RF = (CF2)3CF2H
RF = <CF2)5CF3
Таким образом, была показана возможность получения
различных соединений с Р—С связями (фосфинов и фосфиноксидов)
напрямую из белого фосфора либо из дифенилхлорфосфина
в условиях электрохимической генерации комплексов никеля.
Высокая селективность электросинтеза, мягкие реакционные условия
позволяют проводить белый фосфор в целевые продукты с высокой
конверсией даже в отсутствие фоновых электролитов. Более того,
найденный метод выигрышен относительно всех существующих
Budnikova Уи.Н,, Kargin Yu.M,, Perichon /., Nedetec J,-Y. Op. cit,;
Budnikova Yu.H., Kargin Yu.M., Sinyashin Q.G> Op. cit,; Budnikova Yu.H.,
Perichon J„ Nedekc J.-Y., Kargin YuM. Op, cit,; Будникова Ю.Г., Каргин ЮМ., Cu-
няшин О.Г. Указ. соч.; Будникова Ю.Г., Каргин Ю.М. // Ж-л общ. хим. 1995.
X 65. С. 1660.
144
с позиций атомной экономии. Удалось избежать использования
опасного фосфина и неустойчивых металлорганических
производных.
В заключение можно сказать, что метод электролиза с успехом
можно использовать для синтеза из белого фосфора ряда
соединений со связями Р—О, Р—N, Р—С, а, что самое главное,
электрохимические методы позволяют управлять этим сложным процессом,
достигая в ряде случаев высокой селективности. Преимущества
электрохимического синтеза по сравнению с обычными химическими
методами заключаются в том, что они не требуют дополнительных
реагентов, позволяют в ряде случаев проводить реакцию в
практически замкнутой системе с минимальным количеством реагентов,
которые можно регенерировать циклически, достигая высокой
селективности и выходов. Не менее важны возможность сокращения
или полного исключения отходов, регенерирования потенциальных
загрязнителей окружающей среды при снижении капитальных и
трудозатрат на процессы и, конечно, мягкие условия реакции.
Глава 6. МЕТАЛЛОКОМПЛЕКСНЫЙ КАТАЛИЗ
В ОРГАНИЧЕСКОМ ЭЛЕКТРОСИНТЕЗЕ
6.1. ПРЕИМУЩЕСТВА МЕТАЛЛОКОМПЛЕКСНОГО КАТАЛИЗА,
ИНДУЦИРОВАННОГО ЭЛЕКТРОХИМИЧЕСКИ
Разработка экологически чистых и ресурсосберегающих
процессов химии и химической технологии нагнется одним из
стратегических направлений развития науки и техники, В этой связи все
больше используется гомогенный катализ, который имеет в ряде
случаев преимущества перед гетерогенным, особенно сточки зрения
селективности и эффективности. Действительно, комплексы
переходных металлов в низких степенях окисления (Ni, Pd или Со) могут
реагировать со многими функциональными группами, катализируя
образование связей С-С, С-Р, С—Si и др. Механизмы действия
каталитических систем, как правило, малоизучены, что затрудняет
усовершенствование технологии применения металлокомплексных
катализаторов- Электрохимия связана с этой областью по многим
направлениям. Например, электрохимические методы позволяют
синтезировать многие металлоорганические соединения,
исследовать реакционную способность координационных соединений
и прояснять возможные механизмы реакций.
Использование электрохимически генерированных катализаторов
в органических реакциях в последние годы приобретает все большее
значение в плане как новых возможностей для органического
синтеза, так и более глубокого изучения реакций переноса электрона,
разрыва связей, замещения, присоединения и др. Это направление
интенсивно развивается, а результаты используются в целом ряде
областей физической, органической и аналитической химии. В
предлагаемом обзоре систематизированы литературные данные о реакциях,
протекающих в условиях апектрохимического металлокомплексного
катализа, их применении в органическом синтезе. В последние годы
произошел существенный скачок в применении электрохимических
реакций в промышленности, особенно в малотоннажном синтезе.
С точки зрения промышленной реализации электрохимические
реакции имеют ряд достоинств: можно легко контролировать
конверсию, скорость процесса, а в некоторых пределах и селективность
с помощью таких параметров, как плотность тока и потенциал1.
Degner D. // Topics in Current Chem. / ed. E. Steckhan. Berlin Heidelberg:
Springer-\ferlag. 1988, P. 3: Abbott A. // Chem. Soc. Rev. 1997. 26, iii; UtieyJ. //
Chem. Soc. Rev. 1997. 26. 157; ToniS. //Synthesis. 1986. 873.
146
Непрямое восстановление и окисление органических
соединений, когда образование и регенерация редокс-реагентов
происходят на электроде, в настоящее время приобретает все большее
значение среди современных методов органического синтеза. Это
обусловлено широкими, а в ряде случаев уникальными
возможностями осуществления разнообразных превращений органических
соединений, создания экономичных и экологически чистых
процессов1. Стоимость электроэнергии, как уже сейчас можно
предвидеть, будет расти медленнее, чем стоимость химических реагентов,
а в настоящее время в общей себестоимости продукта она составляет
незначительную долю2; электрон не загрязняет окружающую среду;
контроль электрохимических процессов легко автоматизировать.
При этом может быть достигнута более высокая селективность
процесса при одновременном снижении затрат энергии и достижении
более высоких плотностей тока. Кроме того, интерес к таким
реакциям вызван рядом других факторов: мягкими условиями (невысокая
температура, нормальное давление), возможностью их проведения
в практически замкнутой системе с минимальным количеством
циклически регенерируемого реагента-катализатора. При этом
достигается высокая экологическая чистота синтеза, особенно в
сравнении с традиционными методами органической химии. Следует
отметить, что кроме практического значения медиаторных процессов
существует уникальная возможность теоретического изучения
механизмов восстановления и окисления различных субстратов,
сопровождающихся химическими реакциями, с помощью
электрохимических методов. Для наиболее простых случаев разработан
математический аппарат, позволяющий по аппроксимирующим
формулам достаточно успешно определять значения констант
скорости отдельных стадий суммарного процесса образования,
расходования и регенерации медиатора, получать информацию об
энергетических характеристиках этих стадий, о влиянии на них различных
внешних факторов. Для этого используются зависимости тока (его
каталитический прирост как мера скорости процесса) и потенциала
от условий электролиза.
В настоящее время резко возросло количество исследований,
направленных на замену традиционных катализаторов с тяжелыми
токсичными металлами (Pd, Pt. Pb) более экологически чистыми
и несравненно более дешевыми соединениями никеля, пинка и др.
Именно в этом направлении в последние годы наблюдается
переориентация исследований в развитых странах.
1 ToriiS.Qp.cit.
1 Bersier Л A/., Carisson L., BersierJ, Electrochemistry for a Better Environment //
Topics in Current Chem. / ed. E. Steckhan, Berlin Heidelberg: Springer-Vfcrlag,
1994. 170. P. 113.
147
Все вышеизложенное определяет актуальность и важность задачи
установления деталей механизма реагирования субстратов в условиях
гомогенного электрохимического катализа, их кинетических и
термодинамических закономерностей с целью развития новых методов
органического синтеза, расширения возможностей их приложения,
а также прогнозирования реакционной способности катализаторов
и субстратов в тех или иных условиях. В последние годы в этом
направлении достигнут существенный прогресс.
6.2. ОБЩИЕ ПРИНЦИПЫ МЕТАЛЛОКОМПЛЕКСНОГО
ЭЛЕКТРОКАТАЛИЗА И МЕТОДЫ ИССЛЕДОВАНИЯ МЕХАНИЗМА
РЕАКЦИЙ
С общей точки зрения реакции с участием комплексов
переходных металлов можно разделить на два класса. Первый класс
включает нередокс-реашии, подобные изомеризации, димеризации
или олигомеризации ненасыщенных соединений, в которых роль
катализатора заключается в контроле кинетики я селективности
термодинамически возможных процессов. Электрохимические методы,
связанные с катализом переходными металлами, были впервые
использованы как подходящая альтернатива обычным методам
генерирования низковалентных частиц, которые непросто получаются
или/и трудны в обращении1. Однако эти реакции не являются
собственно электрохимическими процессами, так как они не требуют
расхода электричества и не будут здесь обсуждаться.
Реакции, в которых участвуют и катализаторы — комплексы
переходных металлов, и стехиометрическое количество электронов (либо
от восстановителя, либо от электрохимическою источника), относятся
ко второму классу Этот класс может быть подразделен на две группы
согласно типу действия катализатора. Действительно, комплекс
переходного металла может функционировать как переносчик электронов
от источника электронов на реагент. Подобный процесс обычно
протекает по так называемому внешнесферному механизму без образования
промежугочното комплекса между субстратом (А) и катализатором (ре-
докс-пара P/Q), а медиатор регенерируется электрохимически2:
P + e^Q
Q+A^P+B
В->С
1 Lemkuhl //. // Synthesis. 1973. 377.
- Майрановский В.Г. // Электросинтез мономеров / под ред. Л. Г. Феоктистова.
М.: Наука, 1980. С. 244; SaveantJ.-M. //Асе. Chetn. Res. 1993. 26.455; Wendt И.
// Electrochim. Acta. 1984. 29. 1513; Lund И, // J. Mol. Catal. 1986. 38. 203;
QniciuZ.Jftaru M. Silbet^J.A. // Rev Roum. Chem 1989, 34. 537; Kyriacou D.K.,
lannakoudakis С Electrocatalysis for Organic Synthesis. N. Y,% 1986.
148
Функции редокс-катализатора (медиатора) заключаются в
переносе электрона от электрода на субстрат, после чего первичный
продукт восстановлении субстрата В уже в ходе химической реакции
претерпевает превращение в конечный продукт С. В случае быстрого
и необратимого превращения В в С восстановление субстрата может
происходить в условиях гомогенного переноса электрона в
направлении против перепада стандартного потенциала (£p/Q > Еь/в)\ что
значительно расширяет возможности редокс-катализа. Очевидный
выигрыш при использовании редокс-катализаторов достигается
только в том случае, когда они способны быстро и с высокими
выходами по току регенерироваться на электроде при потенциалах
более положительных, чем потенциалы прямого
электровосстановления субстрата, или менее положительных, чем потенциалы
прямого электроокисления субстрата, быть стабильными и быстро
взаимодействовать с субстратом. Таким образом, этот вид металлоком-
плексного катализа целесообразно использовать, если переход
к гомогенному переносу электрона приводит к увеличению скорости
процесса в целом, т.е. снижению кинетического барьера
электрохимического процесса и приближению величины потенциала к
термодинамическому значению за счет вывода его в объем раствора и
снижения перенапряжения электрохимической реакции, а также если
в процессе участвуют субстраты, для которых гомогенные редокс-
ре акции по тем или иным причинам протекают с более высокими
скоростями, чем гетерогенные.
Вторую группу фарадеевских и каталитических реакций
составляют реакции, протекающие по внутрисферному механизму, т.е.
включающие образование различных координационных соединений (РА
или QA) в качестве промежуточных соединений в ходе реакции. Такой
подход был впервые описан 15 лет назад и представлен в обзорах2.
Внутрисферный перенос электрона имеет ряд преимуществ
по сравнению с внешнесферным по следующим причинам:
1) редокс-процесс не зависит от разности потеициштов медиатора
и субстрата, поэтому возможно значительно большее снижение
пере напряжения;
2) внутрисферный перенос имеет меньший активационный
барьер;
1 Майрановский В.Г. Указ. соч.; SaveantJ.-M. Op, cit.
2 Ефимов О.Н., Стрелец В. В. // Усп. хим. 1988. 57. 238; Хенрици-Оливэ Г.,
Оливэ С. Координация и катализ. М.: Мир, 1980: Байзер Л/. Электрохимия
органических соединений. М.: Мир, 1976; Troupe! М. // Ann. Chim. 1986. 76.,
151; AstrucD. J/Angew.Chem. 1988. 100. 662; Shono T. ElectroorganicChemistry
as a New Tool in Organic Sinihesis. Springer-Vfcrlag. 1984; Будпикова Ю.Г., Буд-
ииков Г.К. // Журн. общ. хим. 1995. 65. 1517.
149
3) если во внешнесферном процессе возможен перенос только
одного электрона, то с помощью внутрисферного механизма можно
передавать два и более электронов. Это особенно важно в
биокатализе и при активации малых молекул.
За последние 10 лет были сделаны попытки развить фундаментальные
аспекты этих процессов и расширить круг катализируемых реакций,
однако многие механистические проблемы до сих пор не решены.
Механизм действия этих катализаторов можно представить
следующим образом:
Р + А «=* РА
или
РА^-*РВ »Р + С
Р + С
Основную роль в каталитическом цикле приобретают
превращения субстрата в координационной сфере металлокомплекса,
поэтому такой вид катализа определяют как координационный
электрокатализ1. Сопряжение электрохимической стадии и химической
реакции субстрата происходит с участием комплекса катализатор —
субстрат, а функция катализатора заключается в связывании и
активации субстрата в координационной сфере металлокомплекса,
в результате чего реакционная способность субстрата в реакции
электронного обмена с электродом возрастает. В отличие от редокс-
катализа Р или Q при координационном электрокатализе должны
быть координационно ненасыщенными или содержать кинетически
лабильные в реакциях замещения лиганды. Это необходимо для
образования промежуточных комплексов РА и QA.
Различные подходы к изучению медиаторных процессов были
впервые описаны около 20 лет назад и представлены в обзорах2.
Однако большой интерес представляют результаты исследований
последних лет, в которых описываются не только различные новые
синтетические применения металлокомплексного электрокатализа
(МКЭ), но и некоторые фундаментальные аспекты.
Выбор конкретного металлокомплекса для координационного
МКЭ в значительной степени облегчается благодаря наличию
обширной информации по комплексообразованию и активации
кинетически инертных в ре доке-реакциях субстратов3. Выбрав подхо-
ЕфимовО.Н., Стрелец В. В. Указ. соч.
Майрановский В.Г. Указ. соч.; Wendt H. Op. cit.; Lund H. Op. cit.; Oniciu Z, Ji-
iaru M., SiIbergJ.A. Op. cit.; Kyriacou D.K.. lannakaudakis С Op. cit.; ЕфимовО.Н.,
Стрыец В,В, Указ. соч.; Xenpuw-Оливэ Г., Олию С Указ. соч.; Troupe) M. Op. cit.;
Astnic D. Op. cit.; Shono T. Op. cit.; Ьудникова Ю,Г„ Будтков Т.К. Указ. соч.
Хенрици-Оливэ Г., Оливэ С. Указ. соч.; Мастере М. Гомогенный катализ
переходными металлами. М.; Мир, 1983
150
дящий металлокомплекс, который связывает субстрат и активирует его
в своей координационной сфере, можно приступить к решению
практических вопросов сопряжения электродной и химической реакций.
Имеется искушение преувеличить преимущества
электрохимического метода. Будет неправильным полагать, что все описанные ниже
реакции предпочтительнее проводить электрохимически, а не
обычными химическими методами. Однако можно отметить, что, во-
первых, электрон является «дешевым», легкодоступным «реагентом*,
редокс-потенциал которого можно регулировать в широких пределах.
Стоимость электроэнергии растет более медленными темпами, чем
стоимость химических реактивов. Во-вторых, электрохимические
методы позволяют генерировать необычные низковалентные интер-
медиаты, которые недостижимы или с трудом получаются при
использовании химических восстановителей. В-третьих, согласно
механистическим исследованиям, во многих случаях необходимо
восстановление интермедиатов каталитического цикла, которое может быть
легко достигнуто регулированием потенциала рабочего 'электрода.
Все эти преимущества будут проиллюстрированы ниже примерами,
демонстрирующими либо улучшение существующего химического
процесса, либо открытие принципиально новых реакций.
Эти заключения должны убедить, что сейчас электрохимия
становится реальным синтетическим методом. Обычно говорят, что
электрохимический метод требует сложного и дорогого оборудования.
Однако технический прогресс привел к его значительному
упрощению и удешевлению. Большое количество электрохимических
реакций проводятся теперь при постоянном токе с использованием
очень простого по конструкции источника тока. Кроме того, все
больше используют ячейки без разделения анодного и катодного
пространства, таким образом избегая диафрагм, которые не очень
эффективны в полярных апротонных растворителях и требуют
использования большого количества фонового электролита- В этом
отношении весьма перспективны процессы с растворимыми анодами*
Кроме увеличения электропроводности генеруемые на аноде ионы
металлов могут влиять на реакционную способность химических
интермедиатов. Растворимые аноды используются для прямых и
каталитических реакций1.
Данные по электрохимическому и химическому поведению
наиболее простых комплексов переходных металлов можно найти в
обзорах и статьях2. Ниже будут приведены два класса никелевых ката-
1 Chassani J.. Folest J. С., NeMec J. Г., herichon J., Si bilk S., Trvupel M. // Synt hesis,
1990. 369; ToMiLioeA.n. // Электрохимия. 1996. 32. 30.
Тамилов AM. Указ, соч.; Walder L. Organoelemental and coordination
compounds // Organic electrochemistry / eds. H. Lund, M.M. Baizer. 3rd edn.
1ST
литических предшественников: один включает комплексы Ni(ll),
образующие соответствующие частицы Ni(0), другой — частицы
Ni(l). Первая группа — это комплексы Ni(Il), имеющие в качестве
лигандов 2,2' бипиридил (bipy), фенантролин (phen), трифенил-
фосфин или бисдифснилфосфино-этан (dppe). Восстановление
азотсодержащих комплексов с бидентатными лигандами (bipy или phen)
при —1,2 В (НКЭ) приводит к появлению соответствующих частиц
Ni(0). Стабильные частицы Ni(0) обычно имеют восемь
дополнительных электронов, поступающих с 2 bipy или 2 phen. Однако
желательнее образование координационно ненасыщенных комплексов,
так как они более реакционно способны, поскольку нестабильны.
Это усиливает интерес к электрохимическому подходу, так как такие
активные частицы могут образовываться непосредственно (in situ)
и реагировать одновременно с органическими субстратами. Вторая
группа включает комплексы никеля, содержащие тетрадентатные
лиганды типа циклама или салена. Восстановление этих соединений
при —1,6 В (НКЭ) приводит к образованию соответствующего Ni(l)-
интермедиата, который дальше восстанавливается только при очень
отрицательном потенциале. Эти два класса никелевых соединений
обнаруживают различное поведение по отношению к органическим
галогенидам.
Соли Со(Ш) или Со(Н), связанные с азотными лигандами,
кислородсодержащие полидентатные молекулы В)2 и др. также
являются кобальтовыми каталитическими предшественниками.
Восстановление Со(Ш)/Со(Н) осуществляется около О В отн. (НКЭ).
Дальнейшее восстановление при ~ -1 В (НКЭ) соответствует
образованию комплексов Со(1), которые и являются реакционно
способными частицами. Однако по сравнению с комплексами никеля
кобальтовые изучены в значительно меньшей степени как в
теоретическом, так и практическом отношении. Абсолютное большинство
работ по электрохимическим и электрокаталитическим
свойствам соединений Со относится к витамину В12 или его аналогам
Co[C2(DO)(DOH)/?„]. Ставилась цель прояснить механизм его
биологического действия, основанный на реакциях переноса электрона.
Dckker; N. Y; Basel; Hong Kong, 1991. P. 809; Budnikova Yu.H., Yakhvarov D.G.,
Kargin Уи.М. II J. Organomet. Chem. 1997. 536—537. 265; Будникова Ю.Г.,
Петрухина О.Е., Каргин Ю.М. 11 Журн. общ. химии. 1996. 66. 610;
Будникова Ю.Г.. Петрухина О.Е., Гудина Н.И., Каргин Ю.М. // Журн. обш. химии.
1996. 66. 605; Будникова ЮГ., Каргин Ю.М. // Журн. общ. химию 1995. 65.
1655; Будникова Ю.Г., Петрухина О.Е., Каргин Ю.М. // Жури. общ. химии.
1997. 67. 275; Будникова Ю.Г., Яхваров Д.Г., Каргин Ю.М. // Журн. общ.
химии. 1998. 68. 1123; Будникова Ю. Г Петрухина О. Е. Каргин ЮМ. // Журн.
общ. химии. 1996. 66. 1876; Будникова Ю.Г., Там же. 1688; Будникова Ю.Г.,
Каргин Ю.М. // Журн. общ. химии. 1995.65. 1536.
152
Сл_о^ CofC2(DO){DOH>,nJ
\\T//
Из соединений Pd были исследованы только комплексы с РРп.г
и некоторыми другими фосфинами. Электрохимическое
восстановление Pd"X2(PPh3)2 является двухэлектронным при = —1 В (НКЭ)
и приводит к появлению реакционных частиц Pd".
6.3. МЕТОДЫ ИССЛЕДОВАНИЯ МЕХАНИЗМА РЕАКЦИЙ,
КАТАЛИЗИРУЕМЫХ ПЕРЕХОДНЫМИ МЕТАЛЛАМИ
Реакции, катализируемые переходными металлами, протекают
через каталитические циклы, включающие комплексы металлов
в различных степенях окисления. Большинство металлокомплексов
является либо окисленными, либо восстановленными, либо и теми
и другими. Следовательно, они легко детектируются и
характеризуются методами электрохимии (вольтамперомегрией, ампероме-
трией, хроноамперометрией и т.д,) и их реакционная способность
контролируется этой же техникой, обладающей рядом преимуществ
в определении концентрации частиц, которая пропорциональна
токам их восстановления или окисления.
В настоящее время наиболее доступны два метода:
1) стационарная вольтамперометрия. Она используется как
аналитическая техника для характеристики комплекса металла,
уже присутствующего в растворе. Он характеризуется своим
потенциалом восстановления/окисления, и его концентрация
определяется величиной его тока восстановления/окисления. Его
реакционная способность по отношению к субстрату может быть
исследована путем мониторинга эволюции его тока
восстановления/окисления как функции времени, например, метолом
вращающегося дискового электрода, когда предельный ток
пропорционален концентрации частиц. Кроме того, если новый элсктро-
активный комплекс металла образуется в ходе реакции, он также
может быть обнаружен по его пику восстановления/окисления,
а его выход в реакции определяется величиной его тока
восстановления/окисления. Таким образом, можно охарактеризовать
реагенты и продукты химической стадии, а также их кинетику.
Этот стационарный метод в принципе аналогичен методам
спектроскопии и позволяет получать кинетические данные для
химических реакций с временами полуреакции /|/2 выше, чем несколько
десятков секунд;
1S3
2) нестационарная (или переходная) вольтамперометрия.
Она используется для доказательства существования равновесия
между различными комплексами по относительному изменению
тока в пике восстановления/окисления различных частиц при
изменении скорости развертки потенциала. Скорость развертки
потенциала фиксирует временную шкалу эксперимента.
Восстановление/окисление одной частицы на поверхности электрода
вызывает сдвиг равновесия в диффузионном слое, а концентрация
частиц, измеряемая по току их восстановления/окисления,
отражает не их действительную концентрацию, а динамическую
концентрацию (СЕ-механизм). Для все более коротких периодов
времени равновесие все менее легко сдвигается, а в пределе при
очень высокой скорости развертки динамическое равновесие
замораживается, хотя оно весьма лабильно при больших периодах
времени (низкая скорость развертки). Это позволяет определить
равновесные концентрации различных комплексов, а затем
константу равновесия. Термодинамика и кинетика равновесия могут
быть в этих случаях охарактеризованы нестационарной вольтампе-
рометрией или хроноамперометрией со ступенчатым изменением
потенциала при времени О на волнах восстановления/окисления
различных частиц. В этом контексте электрохимические методы
более близки кЯМР-спектросконии, так как прогрессивное
замораживание равновесия при увеличении скорости развертки
(вольтамперометрия) или сокращении времени импульса 0 (хроноам-
перомстрия) аналогично коалесценции в ЯМР-спсктроскопии.
В случае равновесных реакций частица, наблюдаемая в качестве
основной в растворе, не обязательно является реально реакцион-
носпособной частицей, поскольку последняя может участвовать
в эндергонном равновесии. Поскольку классические методы
способны контролировать реакцию, эффективные реакционноспо-
собные частицы будут идентифицироваться только путем детальных
исследований кинетики.
Циклическая вольтамперометрия также может быть
использована для генерации стабильных или короткоживущих органометал-
лических частиц путем последовательных электронных переносов
и химических стадий при восстановлении/окислении комплексов
металлов. Знание абсолютного числа электронов, участвующих
в любом электрохимическом процессе, позволяет определять
состояния окисления каскада комплексов, которые могут
генерироваться из данного стабильного комплекса. Также кинетика
электрогенерированных частиц с выбранным субстратом может
контролироваться увеличением скорости развертки в циклической
вольтамперометрии (или сокращением времени импульса в хроно-
амперомстрии), т.е. уменьшением временной шкалы химической
154
реакции. Изменение вольтамперограммы как функция скорости
развертки (или времени) позволяет получать кинетические данные
для химических реакций с временами полуреакций /^ от 0,1
с до 10 не. Промежуточные частицы, таким образом, могут быть
генерированы и/или определены, а их реакционная способность
охарактеризована.
Однако, хотя электрохимические методы позволяют проводить
сравнительно тонкое исследование механистических
закономерностей (кинетики и термодинамики), они не дают структурной
информации. Действительно, из величин потенциала и тока в пике
восстановления/окисления и морфологии кривой невозможно
точно определить структуру комплекса, если не проводить
сравнение с аутентичными образцами. Когда аутентичные образцы
недоступны, информацию о структуре можно изатечь комбинированием
электрохимических результатов с другими методами — ]Н- и ?1Р-
спектроскопии. Действительно, ЯМР-спсктроскотшя обеспечивает
детальную структурную информацию, но не позволяет проводить
тонких исследований быстрой кинетики. Таким образом,
взаимодополняющее использование ЯМР-спектроскопии и
электрохимических методов является важным и существенным для исследования
механизмов реакций. Кроме того, другие спектроскопические
методы могут быть скомбинированы с электрохимическими для
получения детального кинетического и структурного обзора сложной
реакционной системы. Эффективность этого двойственного
подхода для механистических исследований была проиллюстрирована
во многих работах по исследованию ряда элементарных стадий —
окислительного присоединения, лигандного обмена,
восстановительного элиминирования, а также в серии работ, которые были
определяющими для разработки не классических, а более
реалистических каталитических циклов.
6.4. РЕАКЦИИ ГОМОСОЧЕТАНИЯ ОРГАНИЧЕСКИХ ГАЛОГЕНИДОВ
6.4.1. Гомосочетание органических галогенидов
Гомосочетанис органических моногалогенидов. Восстановительная
димеризация органических галогенидов — это первая известная
реакция, включающая как фарадеевское восстановление, так и катализ
металлокомплексами переходных металлов. Действительно, в 1976 г.
Дженнингс сообщил об электродимеризации алкилбромидов в
растворе ДМФА путем их электролиза между двумя металлическими
электродами (Ni, A1 или Си) в присутствии ацегилацетонатов Fe
или Ni и трифенилфосфина1. Позднее было сообщено о некоторых
1 Jennings P.fV.fPUsbmyD.G., HaltJ.L.,Brice КГ.//J. Org. Chem. 1976,41.719.
1S5
других примерах1. Результаты, опубликованные в последние годы,
приведены в табл. 6.1. Хотя выходы могут быть высокими,
синтетический интерес к этим реакциям ограничен, так как большинство
из них могут быть проведены либо химически, либо
электрохимически в отсутствии катализатора2. Многие из этих работ были
посвящены исследованию электрохимических свойств
предшественника катализатора и механизма реакции.
Синтез биарилов из арилгалогенидов представляет больший
интерес из-за практической значимости этих соединений и определенных
трудностей получения их химическими методами. Первые
электросинтезы были проведены в 1980-е гг. одновременно тремя группами
исследователей5, которые использовали NiCUPPh^ (I—20%) в качестве
каталитического предшественника в присутствии избытка РРп3. Позднее
были использованы никелевые комплексы с Ыру, бидентатными фос-
финами -dppe и 1,2-бис(ди-2-алкил-фосфино)бензолом4.
Комплекс Ni(ll) с Ыру, восстанавливающийся в комплекс Ni(0),
также оказался эффективным катализатором димеризации арильных
соединений5, В комбинации с растворимыми анодами этот процесс
удается провести в неразделенной ячейке в очень простых
экспериментальных условиях:
2ArX + 2e >»Br,bipy(7%)+2bipy+2X- )М_М + 1Х-
DMF, Мбзнод
Аг = Ph, 3-МеСбН4,4-МеСОСбН4, NaphtyL..
X = L,Br,Cl
1 Будникта Ю.Г., Юсупов A.M., Каргин Ю.М. //Жури. обш. химии. 1994. 64.
1153: Jennings P. W., Pilsbury D.G., HallJ.L., Brice V.T, Op. cit.; Smith WJi.,
Kuo Y.-M. // J. Electroanal. Chem. 1985. 188. 189; Mabrouk S., Pellegrini S„
Folest J.C., Rollin Y, Perichon J. // J. Organomet Chem. 1986. 301. 391;
Kamau G.N., Rusting J.F. // J. Electroanal. Chem. 1988. 240. 217; Rusting J.F. //
Ace. Chem. Res. 1991. 75; Ozaki S., Urano K, Ohmori HA. // Electrochimica
Acta. 1997. 42. 2153; FryAJ., Sirisoma U.N. // J. Org. Chem. 1993. 58. 4919;
Fry A.J., Sirisoma, Singh A.N., Ugtioro A., Lee A., Kaufman S.t Phanijphand T. ff
Novel trends in elect roorganic synthesis / ed. S. Torii. Tokyo: Kodansha. 1995. P.
83; Mubarak M.S., Peters D.G, //}. Electroanal. Chem. 1995.388.195; Peters D.G.,
Dahm C.F., Bhettacharya D., Butler A.L., Mubarak M.S. // Novel trends in elec-
troorganic synthesis / ed. S. Torii. Tokyo: Kodansha, 1995. P. 67.
2 NedeleeJ.-Y., Folest J.C., Perichon J. // J. Chem. Res. (s). 1989. 394.
? Troupel M., Rollin Y., Sibilte S., Fauvarque J. F., Perichon /. // J. Organomet. Chem.
1980. 202.435; Mori M., Hashimoto Y, Ban Y. // Tetrahedron Lett. 1980. 21.631; SW-
avon G.. Bontempeili G.fComin B. //J. Chem. Soc., Dal ion Trans. 1981. 1074; Bydtm-
коеа Ю.Г., Каргин Ю.М.,Янилкт В.В. // Изв. АН СССР. Сер. хим. 1992. 1674.
4 Sock О.. Troupe/ М., Perichon J., Chevrvt С, JutandA. // J, Electroanal. Chem, 1985.
183. 237; Fax MA.. Chandler DA., Chang/in L. //J. Org. Chem. 1991. 56. 3246.
* Rollin Y.. Troupel A/.. Tuck D.G., Perichon J. // J. Organomet. Chem. 1986. 303.
UUMeyerG., Rollin У.. Perichon J. Op. ciL
156
Таблица б, 1
Продукты электровосстановительного сочетания, катализируемого соединениями N1 и Со
Субстрат
EtBr
n-QH,,Br
л-С5Н,,Вг
MeOC2H4Br
МеО2СС5Н10Вг
С1(СН2)4Вг
СН2=СНСН2С1
PhCHCl2
Х(СН2)ЯС1
(X = Br, [; и = 2-6)
Металл—лиганд
Ni — phen*
Ni — bipy
Ni — bipy
Ni — bipy
Ni — bipy
Ni —bipy
Co — bipy
Ni - <alen"*
Co — salen""
Ni — salen***
Растворитель
DMF
DM F или N MP"
DM F или N MP"
DMF или NMP"
DMF или N MP"
DMF или NMP"
H20
DMF
DMF
Продукт
C4Hiu
СиНгц
C|«H22
(MeOC2H4)2
McO2CC,0Hi0CO2Me
C1(CH2)SCI
(CH2=CH-CH2)2
PhCH(Cl)CH(CI)Ph +
+ PhCH = CHPh
PhCH2CH2Ph +
+ PhCH = CHPh
ci(CH2)2na
Выход,
%
85
89
60
65
38
80-90
Ссылки
35
36
36
36
36
36
37
40
41,42
43,44
' Phen — 1,10-фенантролин.
" NMP — ^метил-2-пирролидрн.
"" salen — дианион бис (салииилиден) этнлендиашша.
Интересно, что замена ДМФА на спирты (метанол или этанол)
не приводит к существенному снижению выхода биарилов при
использовании растворимого анода из железа или алюминия1.
Катализатором может выступать комплекс никеля с 2,2'-bipy2 либо 2,2'-би-
пиридиламином3. Этот результат имеет принципиальное значение
для разработки основ экологически безопасных и недорогих
технологий, а также показывает возможность образования устойчивых
в протонных растворителях никельорганических интермедиатов,
дающих биарилы. В этих же условиях осуществлено эффективное
арилирование олефинов4.
Электросинтез биарилов можно также проводить в присутствии
PdCl2(PPh;i)2 как предшественника катализатора. Исходным
реагентом может быть либо бром-, либо иодарильное соединение5, или
арилтрифлат-фенольное6 соединение, но процесс идет при более
высокой температуре:
2ArX + 2e PdChZPvnf%) >Ar-Ar+2X- 50-98%
DMF, RT
Ar = Ph, 4-t-BuC6H4,4-MeCOC6H4t 4-Me2NC6H4 „.
X=I, Br
2ArOTf+2e PdQ^^(?%) ) Аг-Аг + 2ТЮ- 50-70%
At = Ph, 4-CN-C«H4,4CF3C6H4,4-ClCftH4...
Эти реакции гомосочетания, катализируемые Ni или Pd, можно
проводить в присутствии цинка как восстановителя, получая обычно
сравнимые результаты7.
Каталитическое восстановление шгклогексилкарботтил хлорида
в присутствии электрогенерированного комплекса Ni(I)salen
приводит к образованию тетрамеров — 1,2-дициклогенсилэтен-1,2-диол
дициклогексаноата8. Первой стадией является перенос электрона
с Ni(T)saJen на ацил хлорид с образованием ацил радикала. Процесс
можно описать следующей схемой:
1 Courtois V., Barhdadi R„ Troupe! Д/., Perichon J. // Tetrahedron. 1997. 53. 11569;
Courtois V., Barhdadi /?., Condon S„ Troupe! Д/, //Tetrahedron Lett. 1999.40. 5993.
2 Courtois K, Barhdadi Л., Condon S.t Troupel M. Op. cit.
1 Ibid.
4 Ibid.
5 TariiS.. Tanaka H., Morisaki K.//Tetrahedron Lett. 1985.26. 1655.
* JutandA., Negri 5., Mosfeh A. // J. Chem. Soc, Chem. Commun. 1992. 1729;
Jutand A., Mosleh A., Negri 5. // Novel trends in electroorganic synthesis / ed.
S. Torii. Tokyo: Kodansha, 1995. P. 21.
7 JutandA., Mosleh A., Negri S. Op. cit.
* Bhattacharya D., Samide M.J., PetersD.G. // J. Electroanal. Chem. 1998.441. 103.
158
3 + Ni{I)salen
4 + Ni(II)salen
О
4+1
> С—Ov R ,
-C\- / N / +c
ы R С—С
Q
С—О
R
R
/+ NN
\>
R
N /
C=C
R 6 °"
6+i
—> с—о
fT 4 7
w
.R
Ni(Il)salen + e ■ Ni(I)salen
R—C—C) + Ni(I)salen > R-C+ + Ni(II)salen + Cl"
0 1
2 О
2 + 2
3 + e
С—С
R' \>
<\~ /R
■- с—с
R О
R =
О
Однако есть предположение, что роль Ni(I)salen более сложная, чем
роль простого внешнесферного переносчика электронов для
образования ацил-радикалов. Гетерогенное восстановление ацилхлорида,
также протекающее через ацил-радикал, дает не тетрамер, а альдегид.
ГЬмосочетание органических дигалогенидов. Электровосстатюви-
тельное сочетание органических дигалогенидов яааяется интересным
подходом к синтезу полимеров. Получение гюлиинов из дииодоаце-
тилена было недавно проведено в присутствии комплекса Ni с dppe1:
NiNdppc(5%)
DMF Э-ЬНГ+2лГ
I — = — I + 2we
Больше внимания было уделено сочетанию ароматических и ге-
тероароматических диталогенидов2. Результаты последних
исследований приведены в табл. 6.23.
Kijima M.,Naka^tto К., Sato Т. // Chem. Lett. 1994. 347.
Ibid.
Fox M.A., Chandler D.A., Changjin L. Op. cit.; Aboulkassim A., Chevrot C. //
Polymer. 1993. 34. 401; Tomat A., Zecchin S., Schiavon G.,Zotti G. // J. Elec-
159
Таблица 6.2
Восстановительная электрополимеризация арилдигалогенов
с использованием никелевого катализатора
Субстрат
с,^О^с|
~оо*
вг£хУг
Ф
NHCO-^\—Br
ф
CONH-^\—Br
Вг^ ^*\ ^\ ^Вг
R
(R — Alk, аминоалкил-
дисилоксан)
Металл-лиганд
Ni — dppe
Ni — dppe
Ni - PPh3
Ni — bipy
Ni — bipy
Ni — bipy
Растворитель
DMSO
DMA'
MeCN
DMA*
DMA*
DMA"
Ссылки
51
62
63
64
64
65-67
iroanal. Chcm. 1998. 252. 215; Chevrot C, Benazzi 7\ BarjM. Ц Polymer. 1995.
36. 631; Siave A., Ades D., N'Gbilo E., Chevrot С // Synlh. Mclals. 1990. 38.
331; Helary G, Chevrot C.t Sauvet C, Slave A. // Polym. Bull. 1991. 26. 131;
Abaulkassim A., Paid K., Siove A. // Macromol. Chem. 1993. 194.29; Schiavon G.,
Zotti C, BoHtempeUi C.t La Coco F. // Synth. Metals. 1988. 25. 365; lidem. //
J. Electroanal Chem. 1988. 242. 131; Zotti G, Schiavon G., Comisso M. Berlin /4.,
PaganiG, // Synth. Metals. 1990. 36. 337; Aboulkossim A., Paid K., Chevrot C, //
J. Appl. Polym. 1994. 52. 1569; Paid K„ Ades D., Stove A.. Chevrot С // Synth.
Metals 1994. 63. 89,
160
Окончание таблицы 6.2
Субстрат
хтвг
Вг"%Г
.-Q-.
"ХХХУ.
_ R
+вг-СьСКвг
(R = А1к, аминоалкил-
лисилоксан)
Металл -лмганд
N1 — Ыру + PPhj
Ni — Ыру
Ni - Ыру
Растворитель
McCN
McCN
DMA*
Ссылки
68,69
70
71,72
' DMA — N, N-диметилацетамид.
Все эти реакции проведены в присутствии комплексов Ni
с Ыру или фосфинами. В зависимости от экспериментальных
условий полимеры либо выпадают в осадок в течение электролиза
из объема раствора, либо осаждаются в виде пленок на
поверхности электрода. Этот метод подходит для получения сополимеров
из смеси двух арилдигалогенидов. Недавно были описаны
исследования механизма полимеризации, катализируемой комплексом
Ni — bipy1.
Гомогенное каталитическое восстановление алкил а, ш-дигало-
генидов под действием электрогенерированных комплексов Ni(l)
с саленовыми лигандами протекает по радикальному пути2. Так,
из 1 -бромо-4-хлоро- и 1-хлоро-4-иодбутана получен 1,8-дихлоро-
октан с (выход более 88%).
Электрохимическое восстановление гем-дигалогенидов в
присутствии комплекса меди с 1,10-фенантролином и медным
растворимым анодом приводит к образованию циклопропанов3.
Циклопропаны были получены только в случае диметилдибромомалоната,
тогда как с другими гем-дигалогенидами продукты присоединения
образуются но ценному механизму присоединения:
1 Mubarak М. S.. Peters D. С. Op. с it.
2 Mubarak M.S., PetersD.G. Op. cit.; Dahm C.E., PetersD.G. //J. Eleclroanal. Chem.
1996.406. 119.
J Leone/ £., Dolem E., Devaud M., Paugam P., Nedelec J. - Y. // Electrochimica Acta.
1997.42.2125.
161
CuBriphen. e
— >
Си анод
со2сн3
C02CH3
Исследования методами циклической вольтамперометрии
и хроноамперометрии показали, что редокс-катализ кинетически
контролируется скоростью концертных переноса электрона и
разрыва связи углерод — бром. В присутствии стирола образование
циклопропана происходит по радикальному пути, за которым
следуют концертные процессы переноса электрона и разрыва связи1.
Возможность образования никелькарбеновых комплексов была
продемонстрирована при восстановлении дибромметана под
действием комплекса Ni с PPhy2. В присутствии активированных
олефинов они дают циклопропаны. В случае использования
комплексов Ni с bipy в аналогичных условиях происходит
последовательное отщепление атомов брома, а циклические структуры
не образуются.
6.4.2. Реакции кросс-сочетания органических галогенидов
Кросс-сочетание арилгалогенидов. Несимметричные биарилы,
особенно те, которые имеют электронодонорные и электроноак-
цепторные группы в ароматическом ядре, проявляют интересные
физические свойства и находят применение в нелинейной оптике.
Их синтез сложен, но известны попытки их получения из смеси двух
различных арилгалогенидов комбинацией методов электрохимии
и катализа переходными металлами — комплексами Ni или Pd.
Основной недостаток такого подхода связан с низкой селективностью
процесса из-за невозможности конкурентности между реакциями
кросс- и гомо-сочетания. Отмечено, что если два арилгалогенида
имеют близкую реакционную способность на стадии
окислительного присоединения, то получается статистическое распределение
трех возможных продуктов (2:1:1) — соответственно
несимметричного и двух симметричных. Если реакционная способность двух
соединений значительно различается, то удовлетворительный выход
продуктов может быть получен, только если регулировать
концентрационное соотношение реагентов путем медленного добавления
наиболее реакционного арилгалогенида в ходе электролиза3.
1 LeonelE., DolemЕ., DevaudЛ/., PattgamP., NedelecJ.-Y.//ElectrochimicaAcla.
1997.42.2125.
2 Будникова Ю.Г., Петрухина О. Е., Каргин Ю.М, Указ. соч.
J Meyer G.. Troupel M., Perichon J.//L Organomelal. Chem. 1990. 393. 137.
162
R1
NiBr,bipy(4-1845)
x+r4 Vci^^
NiBr-.bipv(4-]8%)
+2bi^c a Ri_/~~\/~~У_ R2
NMP. Мванод \ / \ /
15-17%
X = Br, CI
R1 = MeO, Me2N, MeS, R2 = F, CN, CF3
Может быть проведено внутримолекулярное кросс-сочетание,
катализируемое комплексом Ni с 1,2-бис-(диизопропилфосфино)
бензолом (L)':
30%
До недавних пор высокая селективность в электрохимическом
синтезе несимметричных биарилов достигалась только в двухста-
дийных процессах. Это было продемонстрировано, например, в
реакции с комплексом Pd-PPhj, взятом в стехиометрическом
соотношении2. Генерируемый на электроде комплекс Pd11 сначала реагирует
с молекулой одного арилгалогенида, а электровосетановление
образующегося ет-арил-палладиевого комплекса в присутствии другого
арилгалогенида приводит к несимметричным биарилам с хорошим
выходом:
PdCl2L2 2ePPh3 >Pd°L2^^ArlPdXL2 Д*2** >Аг1Аг2 +Pd°L2
L = PPh3 76%
Еще один двухступенчатый синтез биарилов был осуществлен
с участием каталитической системы Ni — Ыру При элсктровосста-
новлении арилгалогенида катализируемым комплексом Ni — bipy
в присутствии избытка bipy и Zn2+ и растворимого анода из цинка
образуются не биарилы, а арилцинковое соединение, которое
стабильно в реакционной среде3. Добавление другого арилгало-
1 Fax MA., Chandler D.A., Changfin L. Op. cit.
2 Torii S„ Tunaka H„ Mvrisaki K. Op. cil.; Amoiore С Op. cil.; Armifotv C, Carre £.,
JutandA, Tamka H., Tbrii S., Chkuvtto J., СапШI // ElectrochimicaActa. 1997.42.2143.
J Sibille S., Ratovelomanana V., Perichon J. // J. Chem. Soc,, Chem. Commun. 1992.
283.
163
генида и каталитического количества PdCI2(PPhj)2 в этой смеси
легко дает несимметричный биарил с очень хорошим выходом
(табл. 6.3)1.
Таблица 6.3
Двухстадийное кросс-сочетание арил галогенидов,
катализируемое комплексами Ni, а затем Pd
e,Ni(HF4>2bipy.1(7%)
л ly +2biPy + ZnBr, !\rlZnX Ar2x iAr'Ar2
DMF,ZnaHon PdCI2(PPh3)2 (0,5-2%)
Ar'X
4-ClC6H*CF3
4-CIC6H*CF3
4-BrC(,H4OCHj
4-BrC6H4OCHj
^ClQH^OjMe
4-BrC(lH4NMc2
4-BrC6H4NMe,
2-ВгС6Н4ОСН>
AHX
4-BrC6RtOCH3
4-BrC6H4CN
4-Brq.RtCN
4-BrC6H4N02
4-BrC6H4OCH3
4-BrC6H4N02
4-BrC6H4CN
2-BrCfiH4CN
Выход, %
83
83
90
84
84
84
83
55
Метод также применим к реакциям сочетания между арилгало-
генидом и аллилхлоридом2. Важное преимущество этого метода
заключается в его большой терпимости к различным функциональным
заместителям в ароматическом кольце.
Кросс-сочетание между арил- и активированными алкилгалогенм-
дами. Арилуксусная или арилupoiтоновая кислоты, а также бензил-
кетоны являются важными полупродуктами в синтезе лекарственных
препаратов, агрохимикатов или душистых веществ, которые могут
быть получены электрохимическими методами. Было показано, что
а-галоэфиры могут реагировать с арилникелевым соединением,
генерируемым электрохимически в разделенной ячейке*. Основной
недостаток этого двустадийного метода — использование большого
избытка PPh3, необходимого, чтобы предотвратить реакцию гомосо-
четания:
1 Sibille 5., Ratovelomanana V., Nedelec J. У., Perichon J. // Synlelt. 1993.425.
2 Siblile S., Ratovelomanana У., Perichon J. Op. cil.
3 FoJestJ.C.n Perichon J., Fauvarque J.F., Jutand A. //J. Organomcial. Cliem. 1988.
341 259.
164
PhX + 2e + NiCl2 РРЬз >PhNiX(PPh3) x4:H<R>co2Et >PhCH(R)C02Et
X,X' = Cl,Br R = H,Me 30-85%
Недавно описан более эффективный метод, сочетающий
использование растворимого анода и катализ комплексами никеля.
Были найдены благоприятные условия для сведения к минимуму
нежелательного процесса гомо-сочетания путем медленного
добавления наиболее реакционно способного реагента, т.е.
активированного алкилгалогенида при 60—80°С. Этод способ был
применен для получения продуктов кросс-сочетания с хорошими
выходами между арилгалогенидами и ос-хлорэфирами (табл. 6.4)1,
а-хлоркетонами (табл. 6.5 )2, аллильными и винильными
производными (табл. 6.б)3.
Таблица 6.4
Электровосстановительное кросс-сочетание между арилгалогенидами
и а-хлорэфирами, катализируемое комплексом NiBr2 (bipy)
ArX+C1CHRC02Me ^(Ыру)<5-10%) )ArCH(R)Co Me
1 DMb,Zn или А1 анод L
ArX
4-FC(lR,Br
4-NCC6H4Br
4-Me2NQH4Br
4-FCbH4Br
S-CFjQH+Br
4-CF,QH4Br
4-CNChH4Br
4-MeOCfeH4Br
4-MeOQH4l
2-Бром-6-метоксинафталин
R
H
H
H
Me
Me
Mc
Me
Me
Me
Me
Выход, %
75
60
67
65
59
66
70
51
85
55
1 Conan A., Sibille S., D'Jncar E.t Pirtchon J. // J. Chcm. Sot., Chcm. Commun.
1990.48; Durandetti M., Nedelec J. Y., Perichon J. // Novel trends in cleclroorganic
synthesis / ed. S. Torii. Tokyo: Kodansha, 1995. P. 209.
2 Durandetti M., Nedelec J. y', Perichon J. Op. eil.; lidem. //J. Ощ. Chcm. 1996.61.
1748; Durandetti A/., Sibille S., Nedelec J. Y., Perichon J. // Synth. Commun. 1994.
24. 145.
} Durandetti M., Nedelec J.Y., Perichon J. Op. cit.
165
Таблица 6.5
Электровосстановительное кросс-сочетание между арилгалоген идами
и а-хлорэфирами, катализируемое комплексом NIBr2 (bipy)
ArX+ClCHd^COR2 ^^(bipy)(5-10%) ^^i^
DMF,Znn?mAlaH(u '
ArX
4-FQH4Br
4-NCQH4Br
4-Me2NC6H4Br
4-FQH4Br
3-CF,CeH4Br
4-CF3Ce,H4Br
4-CNC6H4Br
4-MeOQH4Br
4-MeOCbH4I
2-Бром-6-метоксинафталин
R1
H
H
H
H
H
H
Me
Me
Me
Me
R2
Me
Me
Me
Me
Ph
Ph
Me
Me
Me
Me
Выход, %
56
80
52
65
52
63
70
70
53
45
Таблица 6.6
Электровосстановительное кросс-сочетание между
арилгапогенидамм и винильмыми или алл ильными
производными, катализируемое комплексом NlBr2 (Ыру)
АгХ + RX '■NHffhWCj-'O») ,ArR
DMF, Zn или Al аиол
ArX
3-CF3QH4Br
4-CNQH4Br
4-MeOC6H4Br
3-CF,CbH4Br
4-CF,ChH4Br
4-Me2OChH4Br
4-MeIOC6H4Br
4-MeOC„H4I
RX
BrCH=CHMe(Z:£=50:50)
BrCH=CHMe (Z: £ = 50 : 50)
BrCH=CHMe (Z: £ = 50 : 50)
AeOCH2HC=CH2
AeOCH2HC=CH3
AeOCH2HC=CH3
AeOCH2HC=CHMe
MeCH (CI) CH=CH2
Выход ArR, %
(соотношение Z и £-изометров)
66(20:80)
66 (20:80)
44(25:75)
56
38
52
63'
56"
* Получена смесь ArCH=CHEt и АгСН(Ме)СН=СН2 в соотношении 86 : 12.
" Получена смесь ArCH=CHEt и АгСН<Ме)СН=СН2 в соотношении 96 :4.
166
С использованием этого метода удалось осуществить ассимит-
ричный синтез 2-арилпрохтионовых кислот1. Биологическая
активность этих соединений выше у (БЬэнантиомеров (например,
в 2-(3-феноксифенил)пропионовой кислоте (фенопрофене) или
2-(6-метокси-2-нафтил)пропионовой кислоте (1а) (напроксене)),
однако классические методы синтеза обычно приводят к рацематам.
Электрохимический синтез позволяет провести ассиметричное
арилирование производных а-хлорпропионовой кислоты с
использованием хиральных вспомогательных остатков, связанных с
карбоксильной группой. Эта реакция была применена для синтеза (S)-
фторбипрофена из соединения lb. Соединения la, b были получены
с хорошими выходами и диастереомерным избытком с
использованием одного эквивалента а-хлоримида 22 по схеме:
RC(0)CHMe
I +
2 С1 МеО
C(0)R
C(0)R
-N N
R= \ / ;a)e.NiBr2(bipy), 5-10%, DMEAI анод.
Me Ph
Соединение
la
lb
Выход, %
62
58
ее
85
82
de
93
92
Основная конфигурация
(S)
(S)
Недавно было показано, что кросс-сочетание некоторых арил
и гстероарил галогенидов протекает с высокими выходами под
действием комплекса Ni(II) с bipy, восстанавливающимся
электрохимически до Ni(0)bipy\ например:
1 Dutandetti M.t Periehon J., NedelecJ.Y. //J. Oigf Chem. 1997.62.7914.
2 Ibid.
J GosminiC, Lasrv S.. Nedelec J.-Г., Periehon J. // Tetrahedron. 5 1998. 4. 1289;
Durandetti M., Periehon /, Nedelec J. Y, // Tetrahedron Lett. 1997. 38. 8683: Буд-
167
Продукты реакций кросс-сочетания арилгалогенидов
приведены в табл. 6.7. Комплекс никеля(П) в ходе электролиза
регенерируется, а его содержание в электролите составляет не более
10% мольного содержания Аг'Х. В приведенных примерах кросс-
сочетания различные органические галогениды брались в
эквивалентных количествах, а основным продуктом
электросинтеза во всех случаях являлся несимметричный биарил. В случае
электроноакцепторных заместителей в ароматическом ядре более
благоприятным для кросс-сочетания является цинковый анод.
Вероятно, это связано с известной активностью магния и его
соединений по отношению к акцепторным функциональным
группам, приводящей к побочным реакциям трансформации
последних.
Механизм кросс-сочетания детально изучен и обсужден ниже'.
Таблица 6.7
Продукты кросс-сочетания органических галогенов под
действием электрохимически генерированных комплексов
Ni(0)(bipy). Q = 2,0—2,5 е на 1 молекулу АгХ
Аг'Х
4-MeOQH4Br
4-Me,NQH4Br
PhBr
4-FQ,H4Br
4-MeOC(0)QH4CI
2-МеОС6Н4Вг
4-CNC(,H4Br
О-
Br
4
Аг'Х или RX
2-ClC5H«N
2-CIC*H«N
2-ClC5H,N
2-ClC5H,N
2-ClC5H4N
2-CIC5H4N
2-С1С5Н4К
2-BrC5H4N
2-BrC5H4N
2-BrCjH4N
2-B1C5H4N
Выход, %
77
67
70
66
33
76
58
60
78
50
50
Анод
Mg
Mg
Mg
Zn
Mg
Zn
Mg
Zn
Mg
Mg
Zn
Ссылки
87
87
87, 89
87
87
87
87
87
88
88
88
никова Ю.Г., Каргин Ю.М„ Сшшшин О.Г. //Журн, общ. хим. 2000. 70. 123.
Будникова Ю.Г, Каргин Ю.М., Синяшин О.Г Указ. соч.
168
Окончание табл. 6.7
Аг'Х
Вг
d
Br
ci
Br
d
Br
d
PhBr
At'XhjihRX
2-BrC5H4N
0
Cl^X^Me
Me
MeCHCN
CI
McCH=C(Mc)Br
McSiCl
Выход, %
60
59
41
80
65
Анод
Mg
Al
Al
Al
Mg
Ссылки
87
88
88
88
89
'Z:E=25:75
6.5. СИНТЕЗ СОЕДИНЕНИЙ СО СВЯЗЬЮ МЕТАЛЛ — УГЛЕРОД
Для различных хелатных комплексов переходных металлов,
образующих достаточно устойчивые соединения с ст-связью алкил —
металл, например, для кобаламиновых, фгшшцианиновых, саленовых,
дитиокарбаматных комплексов Со, Fe, Ru, Rh, Ni, Cr\ характерно
катодное алкмлирование катализатора при восстановлении алкил-
галогенидов. Такие соединения содержат металл, который может
находиться в трех последовательных степенях окисления (например,
+3, +2, +1 для Со) и имеют внешнюю л-систему лиганда,
способствующую стабилизации анионного комплекса. Вторым важным
фактором, необходимым для алкилирования, является потенциал
образования иуклеофильного аниона, взаимодействующего с алкил-
галогенидами. Если этот потенциал отрицательнее потенциала
восстановления продукта алкилирования, то последний не образуется,
Craig P.J. // Organometallic Compounds in the Enviroment / ed. P.J. Craig.
Longman, Harlow, 1986. Ch. 8; РахимовД.Р.. Miuaeea E.P.. Бутин KM. // Изв.
РАН. Сер. хим. 1998.289.
169
поскольку происходит его необратимое восстановлениее с разрывом
связи металл — алкил1.
Значительная часть исследований в последние гады
сфокусирована на электрохимии витамина BI2 и на взаимодействии его
восстановленной формы В12, с органическими галогенидами2.
Реакции алкилгалогенидов с витамином В12„ кобалоксимов(Т)
и других хелатов кобалъта(1) (включая Co(I)salen) были рассмотрены
Шраузером3. Обычно эти процессы протекают по механизму SN2
и приводят к алкилированию металлоорганического комплекса,
а частицы Со(1) действуют как потенциальный нуклеофил. Было
показано4, что алкилкобальтовые частицы расщепляются либо фо-
толитически, либо пиролитически, образуя алкильные радикалы.
Раслинг5 изучил электрокаталитическое восстановление 1,2-дибро-
моэтана и 1,2-дибромобутана под действием витамина В123 и
определил, что эти вицинальные дибромиды превращаются валкены без
промежуточного образования комплекса алкилкобальта(Н1). Позже
было проведено сравнение6 каталитических реакций с участием В12,т
и Co(T)salen и обнаружено, что скорость восстановления данного ал-
килбромида зависит от формального потенциала пары Со(][)/Со(1).
Кроме того, было показано7, что восстановление бензилбромида под
действием Co(I)salen дает либо бензильные радикалы, либо карба-
нионы — в зависимосги от потенциала, при котором промежуточный
комплекс бензилкобалъта( III) подвергается электролизу.
Электрохимическое восстановление саленовых комплексов
органокобальта(Ш), а также каталитические реакции между
органическими галогенидами и электрогенерированными частицами
Co(I)salcn исследованы Коста8. Метил- и этилкобальт(П1) салены
были электрохимически восстановлены до Co(I)salen и алкил-ра-
дикала9. Показано, что Co(l)salen подвергается медленной ката-
1 Рахимов Д.Р.> Милаева Е.Р., Бутии К. П. Указ. соч.
г Kwiatek J. // Catalysis Reviews / ed. H. Heinemann. \bl. 1. N. Y: Marcel Dekker,
1968, P. 37; Scheffold Д.. Rytz G., Watder L. // Modern Synthetic Methods / ed.
R. Scheffold. \Ы. 3. N. Y: John Wiley; 1983. R 355; Lata D., SaveantJ.-M. //Ace.
Chem. res. 1983. 16.235.
3 SchrauzerC.N., Deutsch £., Windgassen RJ. // J. Am. Chcm. Soc. 1968. 90. 2441;
SchrauzerG.N., Deutsch E. //J. Am. Chem. Soc. 1969. 91. 3341; SchrauzerG.N.,
SibertJ. W„ Windgassen RJ. // J. Am. Chem. Soc. 1968. 90.6681.
4 SchrauzerG.N.. SibertJ. W., Windgassen RJ. Op. cit.
5 Connors Т.Е., Arena J. K, Rusting J. //J. Phys. Chem. 1988. 92, 2810.
* Zhou D.-U Gao J., Rusling J. /j J. Am. Chem. Soc. 1995.117. 1227.
7 Zhou D.L., H. Carrero H., Rusling J. // Langmuir. 1996. 12. 3067.
* Casta G., Puxeddu A., Reisenhofer E. Ц J. Chem. Soc, DaJion Trans. 1972. 1519;
lidem. // J. Chem. Soc, Dalton Trans. 1973. 2034; Puxeddu A., Costa GJ., Mar-
sich J. //J. Chem. Soc.t Dalian Trans. 1980. 1489.
4 Costa G.. Puxeddu A., Reisenhofer E. Op. cit.
170
логической реакции с трет-бутилбромидом или хлоридом с
образованием 2-метилпропена и молекулярного водорода. Очевидно,
из-за объемности трет-бутильного остатка обнаруживается только
нестабильный интермедиат, который разлагается на Co(Il)salen и ре-
аккионностюсобный трст-бутильный радикал, а не производные
органокобальта(Ш)1. Наиболее детальное исследование,
расширяющее все более ранние работы, было проведено Петерсом2. Так,
были идентифицированы и охарактеризованы продукты реакции
Co(Il)salen с иодэтаном, измерены кинетические и
термодинамические параметры предложенной схемы реакции, получен УФ-спектр
интермедиата EtCosalen, охарактеризовано электрохимическое
восстановление этого иитермедиата и осуществлено математическое
моделирование механизма. Циклическая вольтамперограмма
комплекса Co(II)salen представляет одну обратимую одно-электронную
волну. При добавлении иодэтана наблюдаются сильные изменения,
которые заключаются в исчезновении обратимости волны, ее
сильному сдвигу в анодном направлении (на 130 мВ) и появлению новой
волны при более отрицательных потенциалах, каталитически
увеличивающейся с ростом C{EtI). Другие алкилгалогениды менее
активны, хотя их влияние схоже. Препаративный электролиз показал
потребление 1е на каждую молекулу иодэтана. Распределение
продуктов зависит от условий, в основном от растворителя — в ацетони-
триле преобладает димер н-бутан, а в ДМФА — этан (при небольшом
количестве этилена во всех случаях). Объяснение этих фактов
делается на основе предложенной схемы реагирования:
Co(II)salen + e^[Co(I)salenr (1)
|Co(I)salenl + C2H5 -> H5OCo(III)salcn + Г (2)
H5C2Co(]H)salen + e -> [HsC2Co(H)salen1- (3)
[H,C2Co(ll)salenl- -»[Co(I)salenl- + «CaH, (4)
2.С2Н5->С4Н10 (5)
2«С2Н5 -» С2Н6 + С2Н4 (6)
•C2Hj + SH -» С2Н6 4- S«, SH - растворитель (7)
Выдвинуто предположение, что два растворителя влияют на
относительные скорости реакций (5—7) различными путями, В аце-
тонитриле радикальное спаривание четко преобладает над
радикальным диспропорциейированием (отношение выходов я-бутана
к этилену 7,2), в ДМФА это соотношение составляет 3—4. Выход
этана всегда больше, чем этилена, что обусловлено легким отрывом
атомов водорода этильными радикалами, причем от ДМФА — легче,
1 PuxedduA.t Costa С, Marsich J, Op. cit.
2 Alteman K.S., PetersD.G. // J. Electroanal. Chem. 1998.451. 121.
171
чем от CHjCN (реакция 7). Для подтверждения предложенного
механизма было осуществлено отдельное восстановление при
контролируемом потенциале EtCo(III)salen, полученного по реакции
электрохимически генерированного Co(I)salen с EtI. Показано, что
в этом процессе потребляется 1 е на молекулу с-комплекса и
образуются Co(I)salen зеленого цвета и этильный радикал (реакции
3—4). Константа скорости второго порядка (2) для EtI составляет
5 • 10(,л • моль-' ■ с"1 вДМФА1, адля BuBr2 • 102 л - моль"1 - с4 2.
В этих же условиях было изучено восстановление а, л-дигадоге-
нида 1 ^-дииодооктана3. В этом случае в качестве интермедиатов
зафиксированы (наблюдаются две новые волны как
электрохимически, так и методом УФ-спектроскопии) два ст-сигма комплекса:
8-иодооктилкобальта(Ш)сален и ц-(1,8-н-октил)-бис[(сален)
кобальта(Ш)], а относительные количества этих частиц зависят
от отношения субстрата к катализатору.
Аналогичные исследования катализа восстановления алкилгало-
генидов комплексом Co(II)salen на микроэлектродах подтвердили
предложенные выше механизмы, допустив существование
дополнительных стадий4:
RCo(ll)salen- + RI -> R-R + Co(ll)IsaIen-
Co(ll)lsalerr -> Cosalen + h
Электрохимическое алкилирование комплексов кобальта под
действием диметилглиоксиматного комплекса Со'1 исследовано
в работе Бутана5. Показано, что при изменении направления
поляризации электрода с катодного на анодный из металлической ртути
и л-бутил бромида получают соли бутилртути в присутствии
катализатора — моноаииотптого диметилглиоксиматного комплекса Со+\
который образуется из соответствующего нейтрального комплекса
Со'2 при катодной поляризации электрода. Известно, что первая
стадия этого процесса (катодное алкилирование катализатора)
характерна для различных хелатных комплексов переходных
металлов, образующих достаточно устойчивые соединения с о-связью
алкил — металл, например, для кобаламиновых, фталоцианиновых,
саленовых, дитиокарбаматных комплексов Со, Fe, Ru, Rh, Ni, Cr6.
Такие соединения содержат металл, который может находиться
1 Alleman K.S., Peters D.G. // J. Electroanal. Chem. 1998.451. 121.
2 Zhou D.-L., Gao J., Rusting J. Op. cil.
y Alleman K.S., Peters D.G. // J. Electroanal. Chem. 1999.460. 207.
4 Fletcher D.t Tompson H. // J. Electroanal. Chem. 1999.464. 168.
J Рахимов Р.Д., Мияаева Е.В., Полякова К.П., Бутин KJI. // Изв. РАН. Сер. хим.
1994. 309.
* Craig PJ.Qp.cil
172
в трех последовательных степенях окисления (например, +3, +2, +1
для Со) и имеют внешнюю л-систему лиганда, способствующую
стабилизации анионного комплекса. Однако способность металла
существовать в трех степенях окисления (л + 1), п и (п — I) и наличие
хслатирующего лиганда не всегда являются достаточными условиями
для протекания алкилирования металлокомплексных анионов.
Например, диметилглиоксиматный комплекс I г химически и
электрохимически не алкилируется1 в отличие от аналогичных комплексов
Со и Rh. Важным фактором этого процесса является потенциал
образования нуклеофильного аниона, взаимодействующего с ал-
килгалогенидами. Если этот потенциал отрицательнее потенциала
восстановления продукта алкилирования, то последний не
образуется, поскольку происходит его необратимое восстановление с
разрывом связи метал — алкил. Величины потенциалов образования
металлокомплексного аниона и восстановления продукта
алкилирования зависят от природы металла, хелатирующего лиганда,
растворителя и а-связанной органической группы2. Например, известно,
что большинство процессов восстановления металлопорфиринов,
содержащих о-связанную с металлом органическую группу,
происходит при более отрицательных потенциалах, чем в случае
восстановления металлопорфиринов с координированными лигандами.
Однако некоторые порфирины Fe со связью Fe—Alk
восстанавливаются легче соответствующих комплексов с ионными лигандами3.
Были изучены электрохимические свойства некоторых замещенных
фталоцианиновых комплексов Со+2 в растворах ДМФА и сделано
предположение, что в присутствии л-бутилбромида генерируемые
электрохимически моноанионньте комплексы алкилируются по
металлу4.
На катодной ветви поляризационной кривой комплекса РсСо"
(Pt — замещенный фталоцианин) присутствуют два пика
восстановления, первый из которых с Е* = —1,04 В соответствует обратимому
процессу ( ££ = -0,96 В, iJiK = 1) и относится к хорошо известному
переходу5:
РсСо"^=±(РсСоГ,
1 Weber J.H., Schrauser G.H.J. // J. Am. Chem. Soc. 1979. 92. 726.
2 Kadish K.M. (( Redox Chemistry and Interfacial Behaviour of Biological
Molecules / eds. G. Dryhurst and K. Niki. N. Y: Plenium, 1988. 27.
3 Lexa /)., Saveant J.-M., Li WangD. // Onjanometallics. 1986. 5. 1428.
4 Рахимов Р.Д., Милаева Е.В., Бутии K.fl. Указ. соч.
* Lever А. В. P., Miiaeva £,/?., SpeierG. // Phthalocyanines. Properties and
Applications / eds. A.B.P. Leverand C.C. LeznofT. N. Y.: VCH Publ., 1993. 3. 1.
173
а второй — необратим (££ = —1,54 В) и в 3,6 раза выше первого
пика. Последнее можно объяснить тем, что при этом потенциале
происходит перенос (обратимый) одного электрона на макроцикли-
ческий лиганд с образованием дианиона, как это обычно бывает
с фталоцианиновыми комплексами, а необратимость связана с
отщеплением анионов С1~ от бензольных колец лиганда. При
добавлении к раствору комплекса РсСо" л-бутилбромида на кривой ЦВА
появляется новый катодный пик при -1,88 В и одновременно
уменьшается высота анодного пика, соответствующего переходу
нейтральной формы в моноанион. Видимо, этот катодный пик
соответствует образованию комплекса с о-связью Со-А1кт но доказательств
и состава его нет. Предложен каталитический цикл, который назван
«химическим катализом электрохимического восстановления алки-
лталогенидов»:
[PcCo,nL]°
Iе
[РсСо11!.]0
IL
I РсСо"]0
[PcCo"-Rl0
111 nit)
RBr
lPcCo'"-R|
[PcCo'j20
RBr
Данная схема является общей для многих хелатных соединений
переходных металлов и редкоземельных элементов. Видно, что
электролиз при потенциале более катодном, чем потенциал
восстановления связи Со—Alk, приводит к регенерации реакционноепособного
аниона [РсСо]~, который быстро алкилируется присутствующим
в растворе ВиВг, а затем восстанавливается, возобновляя цепочку
превращений1.
Металлоэнзимы представляют собой натуральные катализаторы,
в которых ионы металлов имеют различные типы координации.
Рахимов Р.Д., Mwweea E.B., Бутии КМ. Указ. соч.
174
Эншмы, зависимые от витамина В12, включающие кобальтовые
частицы в качестве каталитического центра, катализируют
различные реакции изомеризации, сопровождающиеся скелетными
перегруппировками1. В работах японских авторов были
предложены новые модели энзиматического катализа, используюшис
гидрофобный витамин В12, гептаметил кобиринат перхлорат
[СоЬ(П)7С|Эфир1СЮ42, который имеет метоксикарбонильные
структурные группы на перферических амидных остатках природного
витамина В12. Также было обнаружено, что этот комплекс эффективно
катализирует перегруппировки в углеродном скелете.
Среди других каталитических процессов описана
электрохимическая конверсия нитрата в аммиак в воде под действием тетраазома-
кроциклического комплекса Со(1), полученного из Со(П)\
Также были рассмотрены некоторые теоретические и прикладные
вопросы использования комплексов кобальта с лигандами,
способными стабилизировать низковалентные состояния металла, в
электрокаталитических процессах дегалогенирования, гомо- и кросс-
сочетания, функционализации белого фосфора4. Так, показано, что
электрохимическое восстановление органических галогенидов
происходит под действием координационно ненасыщенных по bipy
комплексов Co+,bipy (при потенциалах первой волны) и
координационно насыщенных комплексов Со' bipy^ (при потенциалах второй
волны). IgA;Jtj} уменьшаются с увеличением разности потенциалов
1 Ohno Г., Nishioka Х( Murakami У. //J. Mol. Struct. (Theochem). I994. 308. 207;
Golding В. Т., Rao D> /V. // Enzyme Mechanisms / ecL M. I. Plage and A. Williams. L:
The Royal Society of Chemistry. 1987. P. 404-428; Pratt J.M. // В12 / ed, D.
Dolphin. N. Y.: John Wilev, 1982. Vol. 1. P. 325; Murakami Y.. Hisaeda X, Kajihara A. //
Bull, Chem. Soc. Jpn. 1983. 56. 3642; Murakami X. Hisaeda Xt KajiharaA, //
Bull. Chem. Soc. Jpn. 1984. 57. 405; Murakami Y, Hisaeda Г., Ohno T. // Bull.
Chem. Soc. Jpn. 1984. 57. 2091; Murakami X, Hisaeda Y. // Bull. Chem. Soc. Jpn.
1985. 58. 2652; Murakami Y., Hisaeda Y. // Pure & Appl. Chem. 1988. 60. 1363;
Murakami X. Hisaeda X, Tashiro Т., Matsuda Y. // Chem. Leti. 1985. 1813;
Murakami Y.t Hisaeda X, Tashiro Т., Matsuda Y. (( Chem. Lett. 1986.555; Matsuda X,
Hisaeda X, Ozaki T.r Tashiro Т.. Ohm Т.. Tani Y. Matsuda Y. // Bull. Chem. Soc,
Jpn. 1987. 60. 311; Dowd P., Choi S.-C. //J. Am. Chem. Soc. 1987. 108. 3493;
Hisaeda X. Takenaka J., Murakami Y. // Elect roc himica Acta. 1997. 42. 2165.
2 Murakami Y., Hisaeda X, Kajihara A. Op. cit.; Murakami Y. Hisaeda Y, Kajihara A.
Op. cit.: Murakami Y., Hisaeda Y, Ohno T. Op. cit.; Murakami X, Hisaeda Y. Op.
cit.; Murakami Y., Hisaeda Y. Op. cit.; Murakami X, Hisaeda X, Tashiro 7".,
Matsuda Y. H Chem. Lett. 1985. 1813; Murakami X, Hisaeda X, Tashiro Т.,
Matsuda Y. II Chem. Lett. 1986. 555; Matsuda X, Hisaeda Y., Ozaki Т., Tashiro Т.,
Ohno T, Tani X, Matsuda Y. Op. cit.; Dowd P.r Choi S.-C. Op cit.
} Xiang Y.t Thou De-L., RuslingJ.F. // J. ElectroanaJ. Chem, 1997.424. 1.
4 Будникова Ю.Г., Кафияту.ыинаА.Г,, Каргин Ю.М., Сипяшип О.Г. //Жури. обш.
химии. 2000. 70.1538; Оннже.//Журн. обш.химии. 2001. 71. 258.
175
восстановления субстрата и катализатора Д£"^_кх. Впервые выделены
и охарактеризованы интермедиаты каталитического цикла —
а-органил комплексы кобальта, исследованы их электрохимические
и химические свойства. Предложенный механизм реакции гомо-со-
четания ароматических галогенидов под действием Со(1)Ыру
включает стадии окислительного присоединения АгХ к комплексам
кобальта и восстановительного элиминирования а-органилкобаль-
товых частиц. Показано, что арилированис (алкилированис) белого
фосфора происходит при одновременном воздействии
электрохимически генерированных нуклеофильных (Co(I)bipy, RCoX2bipy)
и электрофильных (Мя+, RX) реагентов. Функционализация
элементного фосфора протекает по двум направлениям —
взаимодействия Р4 с ArCoX2bipy либо восстановления белого фосфора
комплексом Co(l)bipy до 5-Р?-циклотрифосфорного комплекса кобальта,
реагирующего, в свою очередь, с органическими галогенидами
с образованием соединений со связью Р—С.
Использование электрохимии позволило значительно
продвинуться в области разработки новых синтетических реакций,
протекающих через а-комплексы Ni89. Преимущества электрохимического
подхода для их синтеза и исследования заключаются в том, что орга-
ноникелевые а-комплексы легко синтезируются электрохимически
в мягких условиях, легко вступают в различные реакции, являются
интермедиатами электрокаталитических циклов реакций
кросс-сочетания, функционализации олефинов, альдегидов с №(0)-катали-
затором. Это малоизученные о-комплексы металлов, которые могут
быть выделены в чистом виде в некоторых случаях и изучены
самостоятельно, а также как модельные соединения для понимания
механизмов мсталлоорганического синтеза1. Недавно обнаружена
возможность существования нескольких а-комплексов Ni в различных
степенях окисления, например, RNiXbipy, R2NiXbipy, R2Nibipy,
обладающих разной реакционной способностью2.
6,6, ПРИСОЕДИНЕНИЕ ОРГАНИЧЕСКИХ ГАЛОГЕНИДОВ
К НЕНАСЫЩЕННЫМ ГРУППАМ
6.6.1. Реакции присоединения к двойным и тройным связям С, С
Реакции электровосстановительного присоединения
органических галогенидов к двойным или тройным связям углерод —
углерод исследуются с 1980-х гг. Было предложено два основных
метода, включающих реакцию радикального присоединения R к не-
1 Будникова Ю.Г., Каргии Ю.М., Синяшин О. Г. Указ. соч.
2 Там же.
176
насыщенному углеводороду как ключевую стадию. В некоторых
случаях использовали витамин В12 или близкие комплексы кобальта как
медиаторы непрямого электровосстановления, иногда в комбинации
с фотолизом1. Для этой цели также был использован никель,
связанный с макрошгклическими тстрадентатными лигандами, которые
могут стабилизировать частицы Ni12. Исследования, проведенные
в течение последних 10—15 лет, посвящены следующим аспектам:
— изучению различных Ni- или Co-органических соединений,
их редокс-свойств и реакционной способности в зависимости
от природы лиганда, связанного с металлом;
— повторному исследованию ранее описанных реакций с целью
оптимизации условий и повышения их эффективности, в частности,
с использованием комплексов Ni—Cyclam;
— генерированию аналогов карбанионов с помощью комплексов
Ni° в присутствии акцепторов Михаэля.
Реакции, рассмотренные в этом обзоре, можно разделить на два
класса: реакции, включающие алкены или алкины, богатые
электронами и протекающие по радикальному пути, и реакции радикального
или карбанионого типа с олефинами, бедными электронами
(акцепторами Михаэля).
Присоединение к электрононасыщенным двойным и тройным связям
С, С. Большинство результатов, опубликованных в этой области
за последнее время, связано со свободно радикальными
циклизациями, которые удобны для получения в основном пятичленных
колец с определенной стехиометрией3:
<, Rr
^*j f* с, каташтюр
X = С, О, N Основной
продукт
Электрохимические процессы с участием комплексов кобальта
исследовались подробно4. В последние годы появились новые
результаты исследований с комплексами кобальта, отличными от
результатов исследований с витамином Bi2, а также никеля, связанного
с тетрадентатными макроциклическими лигандами. Оба типа
комплексов приводят к образованию радикальных частиц,
1 Scheffold R., AbrechtS., Orlhiski R., RufH.R., Stamouii P., Tinembart P., Wilder L.,
Weymeuth C. // Pure Appl. Chetn. 1987. 59. 363; Scheffold R. // Electroorganic
Synthesis /cd. M.M. Baizcr.N.Y: Dckkcr. 1991. P. 317.
1 Gosden G., Pletcher D.//1. Organomet. Chem. 1980. 186. 401.
} Beckwith L.J., Kawrence Т., SeretisA.K. // J. Chem. Soc, Cliem. Com. 1980. 484.
4 Л,. Abrech t S.. (Minsk! R.. Ruf H. R., Stamouii P.. Tinembart P., Walter L.,
Weymeuth C. Op. cii.; Wafderl.. OrlimkiR. // Onjanomet. 1987. 6. 1606.
J
177
Прямая циклизация бромоацеталей через комплексы кобалок-
сима (I) была описана еще в 1985 п1 В то время реакции
проводились в разделенной ячейке в присутствии основания (40%-й водной
NaOH) и 50%-го хлоропиридина кобалоксима(1Н) в качестве
предшественника катализатора. Недавно было найдено, что количество
катализатора может быть уменьшено до 5% (число каталитических
циклов ~50), а основание не нужно, когда реакции проводятся в
разделенной ячейке в присутствии цинкового анода2.
Этот метод был недавно применен для получения
конденсированных бициклических производных в присутствии кобалоксима
или CofCitDOKDOH)^] из различных этиленовых и ацетиленовых
соединениий (табл. 6.8, строка I). Циклический продукт может быть
либо насыщенным, либо ненасыщенным — в зависимости от
количества используемого катализатора, потенциала катода и
присутствия донора водорода, например. RSH (табл. 6.8, строка 2). Было
обнаружено, что электрохимический метод с некоторыми
модельными реакциями является более селективным и эффективным, чем
химический путь, использующий Zn в качестве восстановителя'.
Соединения никеля, например комплексы Ni-циклам4, также
можно использовать в качестве генераторов радикалов в электро-
восстановительных процессах, в том числе в реакциях свободно
радикальной циклизации. Ni-циклам, Ni(CR) и Ni(tet а) являются
эффективными катализаторами в ДМФА в присутствии NR4CIO4
как источника протонов5. Частицы Ni1, генерируемые
электрохимически, быстро реагируют с органическими галогенидами с
образованием алкил, алкенил или арилрадикалов, которые внутримо-
лекулярно присоединяются к двойной или тройной связи, давая
циклопснтаноиды (табл. 6.8, строки 3—7а). Эти реакции могут
осуществляться в неразделенной ячейке, что было
продемонстрировано в случае арилгалогенидов, связанных с ненасыщенной
углеводородной цепью (табл, 6.8, строки 76). Наиболее подходящими
для этих реакций являются цикламные и друше гетрадентатные до-
норные литанды, связанные с никелем, а также магниевый анод6.
1 ToriiS., Inokuchi Т., Yukawa Т. //J. Org. Chem. 1985. 50. 5875,
1 Inokuchi Т., Kawafuchi //., Aoki AT., Yoshida A., Tori/ S. // Bull. Chem. Soc. Jpn.
1994. 67. 595; Inokuchi T. // Novel trends in electroorganic synthesis/ cd. S. Torii.
Tokyo: Kodansha, 1995. P. 223.
3 Giese В., Erdmann P., Gobet Т., Springer R. // Tetrahedron Lett. 1992. 33. 4545.
4 Gosden G.. Ptetchrr D. Op. cit.
5 OzakiS., Matsushita H„ Ohmori H. //J. Chem. Soc, Chem. Com. 1992.1120; lidem.//
J. Chem. Soc,. Perkin Trans. 1993. I. 2339; Ozaki S.T Horiguchi /., Matsushita //.,
Ohmori II. К Tetrahedron Lett. 1994.35.725; OzakiS., Miloh S., Urano K, Ohmori II. //
Novel trends in electroorganic synthesis/ed. S. Torii. Tokyo: Kodiinsha, 1995. P. 185.
h Oiivetv 5., Dunach E. // Synlett. 1994. 531; Olivero S.T Clirtet J.C., Dunach E. //
Tetrahedron Lett. 1995. 36. 4429; ClinetJ.C, Dunach E. // J. Organomet. Chem.
1995. 503. P. 48-50.
178
Таблица 6,8
Реакции электровосстановительной циклизации, катализируемой Со и N1
№
строки
]
т
3
Субстрат
R
я =1,2
X = 0,N-C02E
R = H,Et,C2H,,,SiMe3
R1
нВиСГ^О^
R' = H,Me,Ph
R2=H,Me
koAv
Rl = H,R2 = RJ = C6H5
Rl.R2 = -<CH2)„-R3=H
Катализатор
:o-N4f js-o^
vo->T'>j-or
Кобалоксим (5%)
Co[C3(DO)(DOHV]
5 или 40%
Ni(cyclam) ^Х""*^
Ni 1
V V
\^ (20%)
Продукт, выход %
* 1 Г^
11 J
Sc-^CK 54-80%
R1
hBuO °
42-96%
40"^R3
50-86%
Условия
реакции
МеОН
55-60°C
Zn анод
CH3CN
ДМФА
Продолжение табл. 6.8
N°
строки
4
5"
6
Субстрат
rOv /Br
R
R=H,Ts,Allylt benzyl
R1, R2 = H, Me
R
R'
R2~^ ВГу**
R\R2 = H, Me
E = C02R
Катализатор
Ni(CR)
T г Т
N---Ni -N
(20%)
Ni (CR) (20%)
Ni (CR)
(20%)
Продукт, выход %
R
11-67%
R
7-33%
R1
у
E^E
32-81%
Условия
реакции
ДМ ФА или
Cri.CN
ДМ ФА или
CH^CN
ДМФА
Окончание табл. 6,8
N°
строки
7
8
Субстрат
A = 0,NH
Х = [, Вг
Ph = (СН2)Д-СН;-Х
Катализатор
Ni(teta)
а) Г V J
(20%)
Ni(cyclam)
б) [ Ni J
N N
(10%)
Ni (salen)
(10%)
Продукт, выход %
а или б: 60-90%
PhyH
cS 84%
Условия
реакции
а) ДМФА
б) ДМФА
Mg анод
ДМФА
' насыщенный продукт получен cCoL(5%) и RSH (I : 25).
"L = C2(DO)(DOHV
» "" R идентичен R из ряда 4.
Комплекс Ni с саленовым лигандом является хорошей
альтернативой комплексу с цикламным лигандом для генерирования
интермедиатов Ni1. Действительно, циклизация 6-фенил-1-гало-
пент-5-ина протекает очень эффективно по сравнению с прямым
элсктровосстано&чснисм реагента в присутствии 10% катализатора
(табл. 6.8, строка 8)1. Следует упомянуть один пример электровосста-
новителъной циклизации М-алкенил-2-бромоанилиновс
использованием Pd-PPh3 в качестве катализатора2. Описано
электрокаталитическое дегалогенирование а-галогенуксусных кислот под действием
восстановленной формы витамина В12*.
Присоединение к электронобедным олефинам. Для осуществление!
реакций присоединения к акцепторам Михаэля можно использовать
два типа интермедиатов, т.е. радикалы, или карбанионы, или
металл оорганические эквиваленты. Свободно радикальный путь был
исследован ранее с никелевыми или кобальтовыми комплексами
в качестве катализаторов4. Эти работы были недавно пересмотрены
с целью улучшения числа каталитических циклов или/и
использования более легко получаемых дешевых комплексов.
Хорошей иллюстрацией тонкого органического синтеза,
приводящего к получению натуральных продуктов, является реакция,
катализируемая витамином В12 и инициируемая видимым светом. Один
типичный пример — это синтез соединения, выделенного из маток
медоносных пчел (Apis mellifera), включающий две ключевые
электрохимические стадии образования связи С, С-присосдинсния к
сопряженным двойным и тройным связям5:
О
C03Et
95%
е, В12 — свет
DMF
ss—CQ2Et
е, В12 — свет
DMF
Mubarak МЛ, Peters D.G. //J. Electroanal. Chem, 1992. 332. 127.
Tanaka H., Ren О., ToriiO. // Novel trends in electroorganic synthesis / ed. S. Torii.
Tokyo: Kodansha, 1995. P. 195.
RustingJ.F., MiawC.L., Couture E.C. // Inonj. Chem. 1990. 29. 2025,
Rusato 5.. Tmemburt O., ZhannZ.D., ScheffbldA. // Tetrahedron. 1990. 46. 3155;
Biiiato S., Scheflbld R. // Helv. Chim. Acta. 1994. 77. 92; Orlinski Я., Stankie-
wicz T. //Tetrahedron Lett. 1988. 29. 1601.
Scheffbld X.Ov.ciL
182
Синтез соединения, выделенного из Apis mellifera. Другие
примеры — это синтез феромона красной калифорнийской щитовки1,
просюгландина PGF2ot2, феромона домашней мыши3 и жасмонатов4.
Этот метод был также применен к синтезу необычных аминокислот
(табл. 6.9, Y=NHAc)-\
Таблица 6.9
Алкилирование или ацилирование активированных
алкемов, катализируемое витамином В12
RX
Ме(СН2)3Вг
Ме(СН-,)3Вг
Ас20
Ас20
МеС02(СН2ЬВг
Y
NHAc
Н
NHAc
Н
NHAc
Продукт реакции
Ме<СН2)4СНС02Ме
NHAc
Me(CH3J5C03Me
АсСН2СНС02Ме
NHAc
Ас(СН2)2С02Ме
МеС02(СН2)4СНС02Ме
NHAc
Выход, %
70
75
67
70
72
В отдельных случаях промежуточный радикал, образующийся
на стадии присоединения, может проявлять диастереомерную
селективность в последующей стадии протонирования. Это
наблюдалось в реакции присоединения t-BuBr к диэтилмезаконату,
катализируемому витамином В126. Однако химический метод (B12/Zn)
оказывается более эффективным, поскольку стереоселективность
определяется только присоединением аминов:
C02Et
t-BuBr + /=>
EtCbC
Me
с, В12(25%)
40% (GC)
e,BI2(2SS)
40% (GC)
tBu.
tBu C02Et _. ¥
• И + И
Et02C Me Et02C
84:16
£02Et
Ш
AuerL., Weymuth C, Scheffbld R. // Helv. Chim. Acta 1993. 76. 810.
Busato S., Tinembart 0., ZhangZ.D., ScheffbldR. Op. cit.
ScheffoldR. Op.cit.
Ruxato 5., Scheffold R. Op. cit.
Orlinski R., Stankiewicz T. Op. cit.
^nmnnP.,Sch^-J..Sprin^-R,Zei^H.G.,GieseS.//Hel\.aam.AiAa. mi. 75.638.
183
Ni-циклам и родственные с ним комплексы также можно
использовать, хотя было показано, что число каталитических циклов для
Ni/(tet а) в ацетонитриле мало1. Этот процесс был в последнее время
пересмотрен и было продемонстрировано, что Ni/(tet а) эффективен
в каталитических количествах (2%) в ДМФА, содержащем NH4C104
в качестве источника протонов, для осуществления алкилирования
ненасыщенных эфиров, кетонов или нитрилов3. Если концевым
заместителем при двойной связи является водород (R' = Н), то
достигаются хорошие выходы продукта.
RX + RCH—CRR DMP;NH4C,04> ) (
R2 R4
R'X
ВиВг
Ph(CH2)3Br
Ph(CH2)3CHMeBr
R2
H
H
Me
H
H
H
H
H
R1
H
H
(CH2)4CO
H
Me
H
H
H
H
R*
C02CH,Ph
CO:H
C02Me
CCbMe
C02Me
CN
C02Me
C02Me
Выход, %
53
32
I4
13
53
57
72
37
60
Реакции нуклеофильного присоединения можно реализовать
в присутствии комплексов Nin, которые превращаются в комплексы
Ni(>npH катодном восстановлении. Комплексы Ni(>, а именно Ni(rbipy,
быстро реагируют с алкил-, алкенил- и арилгалогенидами. Однако
в присутствии активированных олефинов никельорганический ин-
термедиат, образующийся в результате окислительного
присоединения, не приводит к соответствующему продукту присоединения.
Более подходящим в этом случае является менее связанный лиганд,
подобный пиридину, который позволяет антивированному олефину
координироваться с металлом. Таким образом, было осуществлено
эффективное присоединение арильных групп в очень простых
и мягких условиях. Реакции проводились при 60°С в неразделенной
1 Heafy K.P., Fletcher D. ff J. Organomet. Chem. 1978. 161. 109.
2 OzakiS., Matsushita H.. OhmoriИ. //J. Chem. Soc, Perkin Trans. 1993.1. 649.
184
ячейке с растворимым анодом из алюминия или железа. Выходы
продуктов арилирования составляют 20—63% и сравнимы с
выходами химического метода, использующего арил-медь или купраты.
Реакции являются региоселективными, поскольку 1,2-присоеди-
нсния не происходит1. Выше уже упоминалось, что замена ДМФА
на спирт типа метанола или этанола не приводит к существенному
снижению выхода продуктов (катализатор — комплекс никеля
с 2,2-бипиридиламином2).
ArBr+R1CH=CR2R3
Аг
Ph
4-NCQ.R,
2-нафтил
4-MeOC6H<
4-MeCOQH4
2-нафтил
2-нафтил
2-нафтил
2-нафтил
R1
Н
Н
Н
С02Е1
COjEt
Н
Me
Н
e.NiBr,(5-IO%) ^\ /Кз
DMF-Pyfy:!),5 / \
AI или Fc анод R R
R-'
Н
Н
Н
Н
Н
Me
Н
Н
(СН2)4СО
R3
COiEt
COnEt
C02Et
COnEL
CO,El
C02Et
CO,Et
CN
Выход, %
63
48
50
46
31
27
20
61
20
Внутримолекулярное присоединение может также
осуществляться по радикальному или карбанионному путям3. Синтез би-
циклических кетонов ранее был проведен в присутствии витамина
В12 <5%) с выходами от хороших до высоких4.
Вг
О
Me
DMSO 0*^7чг
Ni-PPh3
+
40% Me
О
Me 13%
Zhang P., Zhang W., Zhang Г., WangZ.t Zhou W. //J. Chem. Soc, Chem. Comtn.
1991.491.
Courtois K, Barhdadi R., Condon £.r Troupel ,W. Op. cit.
Fox M.A., Chandler D.A.t Chang/in L. Op. cit.; Qzpki S., Matsushita //., Ohmori H.
Op. cit.; Ozaki S., Horiguchi /., Matsushita //., Ohmori H. Op. cit.; Ozairi S., Na-
kanishi Г.. Sugiyama A/., Miyamoto C, Ohmori И. // Chem. Pharm. Bull. 1991,39.
31.
ScheffbldR.,AbrechtS„ Orlinsh R., RufH.R.%StamouliP., TinembartO., WalderL,
Weymeuth C. Op. cil.
185
о
Me
О
ОМе
Ni(tet а), 20% > Q/^j ^J
DMF "kiJL J
Me 23%
(|| ?Г Ni<tei a)
V^^^A^ DMF '
CQ2Me <f 86%
У
C02Me
Недавно появилось сообщено, что другие комплексы Ni и Со
катализируют такие же реакции циклизации, хотя с относительно низким
числом каталитических циклов. Наиболее эффективными среди
изученных являются Ni(cyclam)(CI04)2, Ni(CR)((C104)2 и Co(bpp)Oac'.
О
JL/(CH2)nBr
la,b
Катализатор
Ni(cyclam)(C104)2
Ni(CR){Cl04)2
Co(bpp)AcO
bpp-C5H5NC(0)N-'(C
0 0
A^ X/(CH2)flH
2a,b 3a,b
n = 4(a), 5(b)
Субстрат
la
lb
la
lb
la
lb
H2)3N-'C(0)C5F
Выход продуктов, %
2a
70
60
52
15N>
3a
6
1
5
2b
33
42
28
3b
18
14
21
4a, b
(CH2)„Br
(CH2)„_2
5a, b
n = 4 (a), 5(b)
6a, b
(CH2)„H
OzflkiS., Nakanishi Т., Sugiyama M., Miyamoto C„ OhmoriU. Op. cit.
186
Катализатор
N9(cyclam)(C104)2
Ni<CR)(CI04)3
Co(bpp)AcO
Субстрат
4a
4b
4a
4b
4a
4b
Выход продуктов, %
2a
30
42
47
За
4
4
11
2b
65
19
40
3b
21
19
13
Внутримолекулярное арилирование ненасыщенных амидов,
приводящее к лактамам, протекает по радикальному или карбанион-
ному путям. Последний приводит к смеси 7- и 5-лактамам в
отношении примерно 3:1, тогда как радикальный путь дает селективно
5-лактам, хотя и с удовлетворительным выходом.
Электровосстановительное присоединение (Z)- или (Е)-алкенил
галогенидов к электронодифшштным олефинам в присутствии
растворимого железного анода протекает с полным сохранением
стехиометрии алкетгилыюй части и позволяет функштонализировать
изомерно чистые (Z)- или (Е)-олсфины с высоким выходом1. Условия
реакции были оптимизированы для мстилвинил кстона (2, А=СОМс)
и (Z)- или (Е)-1-галогепт-1-ена 1 (X = О, Вг, 1) — модельных
соединений:
С5Нц^ NiBr23H20, 10%. е С5Нц
\ ==\ 60-80'С
Обнаружено, что достижению высоких выходов <до 84%)
благоприятствует предэлектролиз, заключающийся а окислении
железного анода и восстановлении 1,2-дибромэтана, за которым следует
электролиз реакционной смеси 1 и 2, Роль ионов железа не совсем
ясна, вероятно, они выступают кислотами Льюиса в активации элек-
тронодефицитных олефинов и благоприятствуют образованию связи
С—С. Продукты являются чистыми стереоизомерами. Механизм
процесса остается неясным, возможно, он включает координацию
активированного олефина с электрохимически генерированными
частицами Ni<0). В результате окислительного присоединения винил
галогенида к катализатору образуется никельорганический
комплекс, который протонируется остаточной водой (путь А), образуя
1 Condon-GueugnotS., Dupre/)..iSedelecJ.Y., PerichonJ.//Synthesis. 1997. 1457.
187
продукт присоединения, либо трансметаллируется ионами железа
(путь Б), высвобождая Ni(II):
Ni'^Ni0
X
Ln—Nin
R
Путь А
R
О
RX.
О
UNr1
Ln X
I
X
Ln-Ni"
R
MX„
MX„_,
R
О
|H+|
R
О
R— Ni"
I
X
+ NiX2L„
О
Путь Б
OMX^,
О
Предложенный метод синтеза характеризуется высокой регео-
и стсрсоселективностью, мягкими условиями и одностадийностыо.
Совсем недавно обнаружено, что бромид кобальта, свободный
или в комплексе с 2,2-бипиридилом, катализирует
электрохимическое сочетание арил галогенидов и активированных оле-
финов с электроноакцепторными группами в смеси растворителей
ДМФА/пиридин или ацетонитрил/пиридин1:
Ч_[/
R
Х +
30-70%
1) DMF/PyH, кат. СоВг2 + bipy (1:2), анод: Fe, катод: Ni, 70"С
2) CHjCN/PyH, кат. СоВг2, анод: Fe, катод: Ni, 60°C
R = w-COOEt, я-СОМе, n-CF3, /i-CN, л-СНО, Н. л-ОМе, о-СОМе
Х = Вг
Отмечается, что необходимо использовать только растворимый
анод из железа, тогда как с другими анодами (2n, A1 или Fe)
продукты присоединения не обнаруживаются.
1 Gomes P., Gosmini С Nedelec J.-Y.< Perichon J. jj Tetrahedron Letters 2000. 41.
3385.
188
6.6.2. Реакции присоединения к карбонильным соединениям
К настоящему времени исследованы два основных типа
процессов, которые относятся к основным синтетическим реакциям: ал-
лилирование карбонильных соединений и реакция Реформатского.
С некоторыми карбонильными соединениями возможно прямое
электрохимическое аллилирование, однако сочетания,
катализируемые переходными металлами, более эффективны, особенно с ке-
тонами. Было предложено два метода. Один использует катализатор
PdCl2(PPh3)2 вместе с солью цинка (ZnCl2) для аллилирования
карбонильных соединений и позволяет превращать аллилацетат в
соответствующий спирте хорошим выходом1. С сопряженными кар-
бонилами осуществляется только 1,2-присоединение. Из двух
изомеров, образование которых возможно в этой реакции, преобладает
разветвленный изомер:
Pd"(5%). ZrtCb, с
.OAc + PhCHO-
ДМФА
Ph
Pd"(JJ%),ZnCl;,e ^
ДМФА *
А В
R = Me: 56% (А/В = 91 : 9)
R = Ph: 71 % <А/В = 83 : 17)
Механистическое изучение этой реакции высветило ключевую
роль ZnCl22. Это наиболее легко восстанавливаемые частицы, а
образующийся Zn° может быть восстановителем образованного я-аллил
паладиевого интермедиата, приводящего к аллильным соединениям,
как показано на схеме для аллил ацетата:
Zn2+ + 2е -> Zn°
R' R2
p,i° л zJ1 / \ r'r2c = o V
Никелевый катализатор используется в альтернативном методе3.
Было обнаружено, что эта реакция, проводимая в неразделенной
ячейке, протекает эффективно только в присутствии растворимого
Qiu W., WangZ. //J. Chem. Soc, Chem. Comm. 1989. 356.
Zhang P., Zhang W., Zhang Т., WangZ.. Zhou W. Op. oil.
Conan A., Sibille S., D'lncar £"., Perichon J. Op. cit; Sibille Sr, d'fnear £r, Lepori l„
Massebiau M.C., Perichon J. // Tetrahedron Lett. 1987. 28, 55; Durandetti A/..
SibilleS., Perichon J. //}. Org. Chem. 1989. 542198.
189
цинкового анода. Соответствующие гомоаллильные спирты
получаются с хорошими выходами из ароматических и алифатических
альдегидов или кетонов. Однако в случае разветвленных кетонов типа
ди-изопропилкетона выходы более низкие. С ос, Р-ненасыщенными
карбонильными соединениями наблюдается исключительно 1,2-при-
соединение. Каталитическая система Pd/Zn также эффективна в этих
реакциях. Авторы предположили протекание процесса трансметалл и-
рования между аллил-никелевым комплексом, образующимся путем
окислительного присоединения аллилгалогенида (ацетата) к
комплексу Ni°, и анодно генерированными частицами цинка(11)'.
Каталитические реакции с участием комплексов как палладия,
так и никеля используют аштилацетат в качестве исходного реагента.
В обоих случаях ключевым интермедиатом предполагается цинкор-
ганическое соединение, хотя его образование было показано только
косвенно. Оба метода позволяют осуществить перенос аллильной
группы. Авторы полагают, что реакции электрохимического алли-
лирования очень близки к реакциям химического аллилирования
с участием аллил цинковых интермедиатов.
Реакция Реформатского также может быть осуществлена
электрохимически либо непосредственно, либо с участием медиатора.
Катализ соединениями никеля представляет собой эффективный
путь к продукту с {3-гидрокси эфирной или нитрильной группой
из соответствующих а-хлоросоединений2:
ClCH<R')C02Me + R2RJC=0 NiB*b™>;^n
__ tf он
R'
Н
Н
Н
Me
Me
Me
w
Et
Ph
РГ
Ph
анод ^
R3 СНССЬМе
(CH,)j
СН=СН(СНгЬ
R3
Et
Ph
Me
H
Выход, %
77
86
77
64
80
50
1 DumrtdettiД/,т SibiUe J., Perichon J. Op. cit; SibiUe S„ Nedeiec J,-Y„ Perichon/ //
Electroorgank Synthesis / ed. MM Baker.. N. Y.: Wfeinberg N.L. Dekker, 1989.
P. 361.
2 Conan A., SibiUe S„ Perichon J. // J.Org. Chem. 1990. 56.2018.
190
Здесь также первой стадией является окислительное
присоединение катодно генерированного Ni°bipy к галогенорганическому
соединению. Природа растворимого анода играет важную роль
в этой реакции, хотя образование цинкорганического интермедиата
не было полностью доказано.
Интересным примером, демонстрирующим возможности метода,
является реакция с метил хлородифтороацетатом1, который
невозможно активировать традиционными химическими методами:
ClF2C02Me+R1R2C=
R1
Ph
(СН=)5
Ph
Ме(СН2)2
Ме-^ч0/—
NiBr2(bipy), с,
_п гпанод
CH,CU-DMF
R2
Me
Н
Н
Н
R' ОН
R2 CF2C02Me
Выход, %
72
63
70
77
45
Самые лучшие результаты были достигнуты в смеси СН2С12—
ДМФА (90 : 10) в качестве растворителя вместо ДМФА. Анализ
реакционной смеси методом спектроскопии на ядрах l9F показал
протекание трансметаллирования.
Сочетание метил дихлороацетата с ацетофеноном или циклогек-
саноном дает высокие выходы (75—80%) и приводит к образованию
эпоксидов в аналогичных условиях реакции2:
_ _ _ , .. NiBnbipy, с, Zn анод "\/ ч
С12СНС02Ме + R'R2C=0 nvlF Д. r, nMF> X^C02Me
DM F или CH2CI2— DMF y
R^Ph.CftHnn-Bu 54-86%
R2 = H, Me, Ph
R',R2 = -(CH2)5-
Этот процесс можно распространить на реакцию сочетания хло-
роацетонитрила или а-хлоропролионитрила3:
1 Mcharek S., SibilleS., Nedelec /.-К, Perichon J. //1 Organomet. Chem. 1991.401.
211,
2 Canon j4., SJbille S., Perichon J. Op. cit.
} Ibid.
191
о 1 ОН
rchcicn + r'r*c=o NiBr;bi^;z,iaHOS R X
M R2/4CH-CN
R'-Ph.CeH^t-Bu R/
R2 = H, Me, Ph
R1, R2 = -<CH2>5-
Внутримолекулярное присоединение к карбонильной группе
недостаточно исследовано. Единственным примером является
расширение кольца а-бромометил-циклоалканонов, проводимое в
присутствии Co[C2(DO)(DOH),JCl2 как катализатора, при 55—бО'С1.
Предполагается, что реакция идет по радикальному пути в соответствии
с обычным поведением алкил кобальтовых интермедиатов. Число
каталитических циклов и выходы были улучшены совместным
использованием кобальтового катализатора и растворимого цинкового
анода2
.2.
О
VrhBr комплекс кобальта {5%)т
с5н„
МеОНт2панод
с
—»
Комплекс кобальта (535), е
МеОН, гпанол
-Э-
С5Н11 CjHn
54-68% 19=24%
Предлагается традиционная схема синтеза при электролизе:
модель витамина В12 в состоянии Со(П)[СоЬ(11)7С1эфир]СЮ4
легко восстанавливается до частиц Со(1) высоко нуклеофильного
характера в электрохимических условиях; частицы Со(1) реагируют
с алкил галогенидом с образованием алкилированного комплекса
со связью Со—С, который, в свою очередь, приводит к конечному
продукту по электрохимической реакции или при фотолизе. Особое
внимание было сфокусировано на применении этой
каталитической системы в различных органических синтезах, особенно в
реакциях с расширением цикла, которые интересны для синтеза
натуральных продуктов. Так, Тории исследовал реакции с расширением
кольца 2-алкил-2-(бромометил)циклоалканонов, которые имеют
Inokuchi Т., Kavtafuchi //., Aoki X"., Yoshida A.. Toril S. Op. cit.; Inokuchi Т.,
Tsuji A/.. Kawafuchi H.. Ibrii S. Op. cit.
Inokuchi Т., Kawafuchi JJ., Aoki K., Yoshida A., 'Ibrii S. Op. ciL
192
5- и 6-членные циклы, медиированные кобалоксимом в метаноле
при 60"С в гальваностатическом режиме при облучении видимым
светом1. Мураками2 использовал [СоЬ(Н)7С1эфир]С104 для
катализа реакций расширения кольца в потенциостатическом режиме
по реакции
О
U>co2c2h5 B12 X-JC
n = 1,2,3,4
СО2С2Н5
+
О
iy—С02ОС2Н5
* ^COiC2Hs '"
В С
Продукт расширения кольца В является основным (выход
до 56%) при различных режимах электролиза, который
контролируется также электронной и ЭПР-спектроскопией, которые
позволили определить интермедиаты, образующиеся при
различных потенциалах при освещении и в темноте. Сделан вывод,
что катализ предпочтительно проводить при —1,5 В отн. НКЭ или
более катодных потенциалах по реакционному циклу,
протекающему по схеме: комплекс Со(П) электрохимически
восстанавливается до супернуклеофилышх частиц Со(1), которые реагируют
с субстратом до соответствующего алкилированного комплекса.
Последний разлагается при электролизе (а не фотохимически)
до конечного продукта, а комплекс кобальта повторно действует
как медиатор. Молярное отношение продукта с расширением
цикла к продукту без расширения связано с энергией напряжения
цикла алициклических молекул. Так, наибольший выход
наблюдался при превращении 5-членного цикла в 6-членный
циклический кетон.
Электровосстановительное сочетание органических галогенидов
и альдегидов, катализируемое комплексом никеля(О) с 2,2 -бипири-
дилом, протекает с хорошими выходами при определенных условиях
(табл. 6.10)3.
1 Inokuchi Т., Tsuji A/., Kawafitchi //., Tari't S, Op, cit.
2 Hisaeda У., Takenaka J., Murakami Y. Op. cit.
J БудпиковаЮ.Г., Кешнер Т.Д., КаргинЮМ. //Журн.общ.химии. 2001. 71.490.
193
Таблица 6.10
Сочетание между органическими галогенами и карбонильными соединениями! катализируемое
комплексом NtBr^bipy) (1 ммоль) (АгХ —15 ммоль, R1R2CO — 7,5 ммоль, фоновый
электролит— Bu4NBF (10~2 моль • п~х),1 = 0,1 А, катод из никелевого мусса, 20°С)
АгХ
2-ВгС6Н4Ме
R'R3CO
PhCHO
2-МеОСьН4СНО
о=°
С8Н17СНО
Продукт реакции
Me
MeO Me
Me
OH 1
Me
Анод
Mg
Zn
Ni
Fe
1ПОХ
Ni
inox"*
Ni
inox""
Выход, %
(см*)
14(75)"
18
61
63
80
60
71
50
58
Окончание табл. б. 10
АгХ
2-BrCbH4NH2
2-Br-CftH4OMe
4-С1С6НцС02Ме
МеС(Вг)=СНМе
R'R2CO
PhCHO
PhCHO
PhCHO
PhCHO
Продукт реакции
Of-O
ОН 1
NH2
H3CO
/~~\-СН-ЛЛ-С02Ме
_ OH _
/^VcH—CMe=CHMe
_ OH
Анод
inox'"
Ni
inox"'
inox*"
Выход, %
(см')
42
85
50""
52
' Приведены выходы в расчете на PhCHO.
** 7 ммоль NiBrj(bipy).
*** inox — анод из нержавеющей стали.
*"* Выход в расчете на ArCl (7,5 ммоль), PhCHO — 30 ммоль.
Видно, что синтез идет с высоким выходом спирта при
использовании как никелевого, так и железного анодов и анода из
нержавеющей стали (inox), причем в реакцию вступают и другие
ароматические альдегиды с донорными группами в кольце (СН^О),
а также алифатические альдегиды и циклогексанон. В качестве
арилируюших агентов могут выступать 2-Вг-анилин и 2-Вг-анизол,
но только с орто-положением донорного заместителя, что,
вероятно, связано со стабильностью ключевого реагента ArNi'bipy.
В случае арилгалогенидов с мета- и пара-заместителями
образуются в основном биарилы Аг^, а присоединение по карбонильной
группе в заметной степени не идет. Однако были получены
удовлетворительные выходы спирта с акцепторным заместителем
С02Ме в кольце при замене X = Вг на C1. Это позволило
замедлить реакцию гомосочетания ArNi'bipy с АгХ, направив процесс
в сторону функционапизации альдегида, однако в этом случае
необходимо использовать 3—4-кратный избыток альдегида. То, что
в реакцию практически не вступают (были зафиксированы только
следовые количества спирта) ароматические альдегиды с
акцепторными заместителями в кольце, которые должны
благоприятствовать л-связыванию, а также то, что обсуждаемый процесс идет
и с алифатическими альдегидами, указывают на преобладание
о-характера связи Ni-органического комплекса с С-О группой
альдегида.
Известно, что селективное восстановление арил пропаргило-
вого эфира в соответствующие фенолы может протекать в мягких
условиях и с хорошими выходами в присутствии электрогенери-
руемых комплексов никеля1. На основе этих наблюдений были
предложены методы снятия защитных групп — аллильных и про-
паргильных. Так, описано электрохимическое внутримолекулярное
аллилирование карбонильной группы в аллиловом эфире,
катализируемое комплексами никеля2. В предложенной схеме процесса элек-
трогенерированные комплексы Ni{0) являются активными
каталитическими частицами, способными реагировать с аллильной частью
эфира, образуя комплекс никеля 7г-аллильного типа.
Внутримолекулярный перенос аллильной группы на карбонильную функцию
приводит к промежуточному алкоголнту феноляту никеля(Н).
Возможно, что ионы никеля способны замещаться в этом комплексе
на ионы Mg2+, при этом освобождается комплекс Ni(ll) и цикл
замыкается. После гидролиза алкоголята получается конечный
продукт:
1 Olivero 5., Dunach E. // Tetrahedron Letters. 1997. 38. 6193.
2 Franco D., Olivero S.. Dunach £. // Electrochimica Acta. 1997.42. 2159.
196
2е,-1,2В/н.к-э.
Регенерация
катализатора
Аллилирование
карбонильной группы
Образование
(к-аллильнпгл) комплекса Ni
L — bipy
Позднее электрохимическое внутримолекулярное карбонильное
пропаргилирование было рассмотрено на примере о-замещеннъгх
аридальдешдов1;
ОН
СНО ^ JL J^
t^^><^
I) e. Ni"bipyj
DMFT Mgnnoa
2)гидролиз
33-713
Механизм процесса включает реакцию пропаргильного переноса
с превращением ар ил пропаргиловых эфиров в альдегиды с
первоначальным расщеплением связи С—О пропаргилового эфира, за которым
следует присоединение С — 3 фрагмента к карбонильной группе.
Электрохимическое восстановительное снятие защитных групп
в аллил карбаматах с несколькими функциональными группами
в присутствии каталитических количеств комплекса Wbipy^
приводит к соответствующим аминам с выходом 40—99%2:
1 Franco Z>., Dunach Е, // Tetrahedron Letters. 40. 295J.
2 Franco D., Dunach E. // Tetrahedron Letters. 41. 7333.
197
О е. Ni'Tripft
XZn анод n X1 и
~ п. О^^ DMF 3 R~NH2
-со2, -с3н6
Реакция идет при комнатной температуре в мягких условиях
в ячейке без разделения пространства с растворимым цинковым
анодом. Аллильные арил карбаматы селективно расщепляются
с выходом 70—99%. Эфирные, кетоновые, ацетальные и нитрильные
группы сохраняются в этих условиях реакции.
6.7. ЭЛЕКТРОКАТАЛИТИЧЕСКОЕ ВОССТАНОВЛЕНИЕ ДИОКСИДА
УГЛЕРОДА
Опубликован ряд обзоров, например1, посвященных
электрокаталитическому восстановлению азота и его оксидов, оксидов углерода,
а также молекул с тройными связями (ацетилен, цианид-ион). Все
вышеперечисленные соединения объединяет то, что они
электрохимически неактивны и содержат очень прочные связи, которые
достаточно трудно поддаются расщеплению. Поэтому активация таких
молекул путем их внедрения в координационную сферу металла с
последующим (или предшествующим) электрохимическим переносом
электрона является наиболее перспективным способом вовлечения
мало реакционно способных молекул в различные химические
превращения. Помимо этого еще одним важным фактором,
обусловливающим большое внимание к электрокаталитическим процессам
с участием металлокомплексов, является стремление использовать
огромные запасы N2, С02 и 02, находящихся в биосфере, в качестве
дешевого и доступного сырья для различных химических процессов.
Прямое восстановление С02 на металлическом катоде, например,
ртути, требует высокого перенапряжения (С02/С02~ =—2,21 Both.
НКЭ в ДМ ФА). Другие металлические катоды, как, например, медь
или золото, способны восстанавливать С02 в водном растворе при
несколько менее катодных потенциалах (от —1,3 до —1,7 В), однако
и эти значения слишком велики для практического использования
данного процесса, поскольку при этих потенциалах происходит
также восстановление воды с выделением водорода. Использование
неводного растворителя позволяет этого избежать, однако порождает
большие энергетические затраты и возможность побочных реакций.
Наиболее перспективным представляется использование
различных металлокомплексов в качестве катализаторов
селективного электровосетановления С02. Как правило, это комплексы
' Ефимов О.Н., Стрелец В.В, Указ. соч.; Vasudevan P., Phoogat N., ShuklaA.K, //
Appl. Organomei. Chem, 1996. 10. 591.
198
с азотсодержащими макроциклическими лигандами, порфирины
и их аналоги, хорошо известные как биокатализаторы1. В отличие
от природных катализаторов искуственно синтезированные ме-
таллокомплексы с азотсодержащими макроциклическими
лигандами (фталоиианины, цикламы) обладают еще большей цепью
л-сопряжения, а их каталитическая активность не уступает
природным аналогам. Показано, что катализаторами восстановления
ССЬ могут служить следующие соединения: фталоцианины металлов2
и металлопорфирины Со(П)-\ цикламы (1,4,8,11-тетрааза-цикло-
тетрадекан Ni(II)4, бис- и полипиридильные комплексы Rh(ll)^,
Re(I)\ Cu(II)7, Co(II), Fe(II) и Ni(II)s, комплексы железа и кобальта
с4,5-дигидроксибензо-1,3-дисульфонатными и 2-гидрокси-1-нигро-
зонафталин-3?6-дисульфонатнымя9лигандами. Многие из
вышеперечисленных комплексов являются эффективными катализаторами
1 Kraeutler й, // Chimia. 41. 277.
1 Furuya N., Matsui К. // J. Electroanal. Chem. 1989. 271. 181; Lieber СМ.,
Lewis /V.J. // J. Am. Chem. Soc. 1984, 106. 5033; Kapusm S„ Hacherman N. //
J. Eleeiroehem. Soc. 1984.131.1511; Tanabe H.f Oohno K. // Eleetrochimiat Ada.
1987. 32. 1121; Mahmood M.N., Masheder D„ HartyCJ. //J. Appl. Elecirochcm.
1987. 17. 1233; Savinova E.R., Yashnik S.A., Savinov E.N., Parmon V.N. // React.
Kinet. Catal. Lett. 1992.46. 249; MashederD,, Williams K.PJ. //J. Raman Spectrasc.
1987. 18. 391; Omstensen P., HamnettA., MuirA. V.G. //J. Electroanal. Chem. 1988.
241. 361; Meshitsuka S., khikawa M.. Tarnaru K. //J. Chem, Soc. Chem. Commun.
1974. 158; Zagat J.H. //Coord. Chem. Rev. 1992.119. 85; Радушкина K.A., Мерен-
кова М.В.. Тарасевич M.P., Гальперин М.Г., К)>древич СВ., Новожилова И.Г. //
Электрохимия. 1992.28.85; Yoshida 7'., Катаю К., Tsukamoto AL.Jida Т., Schhlett-
wein D.t Woerte D., Kaneko M. // J. Electroanal. Chem. 1995. 385. 209.
i Craig P.J. Op. cit.
4 Beley M.. Collin J.P., Ruppert R., Sauvage J.P. //J. Chem. Soc. Chem. Commun.
1984. 1315; Beley A/., Collin J.P., Ruppert R., Sauvage J.P. // J. Am. Chem. Soc.
19S6. 108. 7461; Collin J. P., JouaitiA., Sauvage J.P. // Inorg Chem. 1988. 27. 1986;
Fujihira M., Hirata Y, Suga K. //J. Electroanal. Chem. 1990.292. 199; BalaszCB.,
Anson F.C. //J. Electroanal. Chem. 1992. 322. 325; Abba F„ Sands G.De., Fab-
brizzi L., Liccchelli Д/., Lanfredi A.M. A/,, PaUavicini P., Poggi A.. Ugozzoti P. //
lnorg. Chem. 1994.33, 1366.
5 SauthierM.N.C.D., DeronzierA., Ziessei R. // Inorg. Chem. 1994. 33. 2961.
* Rasmussen SC Rhhhr M.M., Yim E., Place H., Brewer K.J. // Inorg. Chem. 1990.
29. 3926.
7 Haines /?./., Wittrig R.E., Kubiak C. P. // Inorg. Chem. 1994. 33. 4723.
14 Yoshida 7"., lida Г., Shirasaji Т., Lin /?., Kaneko M. // J. Electroanal. Chem.
1993. 344. 355; Arana C„ Van S., Keshavarz KM., Pom K.T., Abruna H.D, //
Inorg. Chem. 31, 3680 (1992); Arana С Yon S„ Keshavarz KM., Potts K.T.,
Abruna LID. // Inorg. Chim. Acta. 1994. 225. 285; Chmtemen P., Higgins S. //
J. Electroanal. Chem. 1995. 387. 127: Sende J.A,R., Arana C, Hernandez L„
Potts К. Т., Keshavarz KM., Abruna H.D. // Inorg. Chem. 1995. 34. 3339.
4 Ogura K., Sugihara H., Yano J., Hlgasa M. // J. Elecirochcm. Soc. 1994. 141.419;
Ogura K, Higasa M., Yano J.. Endo N. //J. Electroanal. Chem. 1994. 379. 343.
199
восстановления С02 только в неводных средах, поскольку в воде
восстановление протонов предшествует восстановлению С02. Только
фталоцианин кобальта (РсСо)1 и комплекс никеля с цикламом2
оказались эффективными электрокатализаторами восстановления
С02 в воде или водном ацстонитриле, причем последний оказался
активным только на ртутном катоде, на котором перенапряжение
выделения водорода больше, чем на Pt.
Существует предположение^, что большая селективность
электровосстановления С02 по сравнению с Н20 обусловлена большим
размером макроциклического лиганда и наличием N—Н-групп
(в цикламе никеля). Следует сказать, что важным преимуществом
использования металлокомплексов в качестве катализаторов
является возможность изменения их реакционной способности путем
модификации макроциклического лиганда с помощью донорных
или акцепторных заместителей. Так, например, производное ци-
клама никеля(Н), содержащее две метильные группы в макроцикле
(3,10-диметил-1,3,5,8,10,12-гексаазациклодекан)т проявляет гораздо
большую каталитическую активность, чем незамещенный циклам
никеля(П)4. Удивительно, но введение а-акцепторных заместителей,
например фтора, в азотсодержащее кольцо также увеличивает
каталитическую активность и селективность образования СО (по
сравнению с выделением водорода). При этом наиболее эффективным
оказался 3,3,10,10-тетрафторциклам никеля(П). Дальнейшее
увеличение числа атомов фтора в макрокольце приводит к снижению
активности катализатора5.
Замечено, что металлопорфирины также способны проявлять
каталитическую активность при восстановлении СОт в СО в водных
и неводных средах6. Однако порфирины быстро расходуются в
процессе электролиза, а катализ прекращается после небольшого числа
1 Furuya i\., Matsui К. Op. cit.; LieberCM., Lewis N.S. Op. cit.; Tanabe //,, Oohno K.
Op. cit.; Mahmood Л/.Л'., Masheder D., Harty C.J. Op. cit,; Savinova E.R.,
Yashnik S.A., Savinov E.N., Parmon V.N. Op. cit.; Masheder Д, Williams K.PJ. Op.
cit.; Christensen P., HamnettA., MuirA.V.G. Op. cit.; Meshitsuka S., Ichikawa A/.,
Tamaru K, Op, cit.; K. Ogura K.. Higasa A/., YanoJ., Endo Л'. Op. cit,; Kusuda K.,
Ishihara R., Yamaguihi H. // Elecirochimica Acta. 1986. 31. 657.
1 Reley M.. Collin J.P.. Ruppert /?., Sauvage J.P. Op, cit.; Aram C, Yan S., Kes-
havarz K.M., Polls К, Т., Abruna H.D. Op. cit.; Sasaki H, //J. Am. Chem. Soc. 1992.
114. 2055; BataszC.B., Anson F.C. //J. Elcciroanal. Chem. 1993. 361. 149.
3 Веку M., Collin J.P,, Ruppert A, Sauvage J.P. // J, Chem. Soc. Chem. Commun,
1984. 1315; Befey M„ Collin J.P., Ruppert Л., Sauvage J.P. // J. Am. Chem. Soc.
1986. 108. 7461.
4 Smith С/., Craystone J.A., Hay R. W./fi. Chem.Soc. Dalton Trans. 1993. 3267.
5 Shionoya M., Kimura £.. litaka Y. // J. Am. Chem. Soc. 1990. 112. 9237.
* Atoguchi Т., Aramato A., Kozusaka A., Ennyo M. if I. Chem. Soc. Chem. Commun.
1991.156.
200
циклов. Отравление катализатора происходит за счет карбоксилиро-
вания и гидрогенизации порфиринов1.
Еще один способ избежания нежелательных катодных процессов —
это использование газофазных реакций С02 с электроактивными
пленками на твердых электролитах2. Известно много примеров
гетерогенного каталитического восстановления С02, когда активный
катализатор, например металлопорфирин или фгалоцианин, наносится
на твердый электрод — графит и покрывается проводящей пленкой
(Nafion), препятствующей его переходу в раствор3, но рассмотрение
таких систем не входит в область интересов данного обзора.
Авторы работ4 предлагают следующий механизм образования
продуктов восстановления:
С02 + РсМ -> [Рс - С02]
1. Механизм образования окиси углерода (СО):
2Н* + 2е + [РсМ - C02J -» Н20 4- [РсМ • • COJ
[РсМ ■• СО]-> СО + РсМ
2. Механизм образования муравьиной кислоты (НСООН):
Н+ + 2е + [РсМ - С02] -> [РсМ - ООСН]
Н+ + е + [РсМ ■■ ООСН[ -> [РсМ •■■ НООСН1
[РсМ - НООСН] -> НСООН + РсМ
3. Механизм образования метана (С НО:
2Н+ + 2е + [РсМ ■■ ОС1 -> Н20 + [РсМ ■•■ С]
4Н4 + 4е + [РсМ ••■ С] -> [РсМ - СН4]
(РсМ ■• СН4] -> СИ, 4- РсМ
Следует сказать, что практически во всех работах, посвященных
каталитическому связыванию С02 с помощью комплексов
переходных металлов, в качестве ключевой стадии постулируется
образование комплекса «мсталл-С02»5. В большинстве случаев в пользу
1 CosfamagnaJ., FerraudiG.XanalesJ., Vargas J. //Coord. Chein. Rev. 1996. 148.221.
2 Liu /, Weppner W. 11 Appl. Phys. A. 1992. 55. 250.
3 Furuya N., Matsui K. Op. cit.; Lieber СМ., Lewis N.S. Op. cit.; Tanabe Я.,
Oohno K. Op. cit.; Mahmood M.N., MashederD., Harty C.J. Op. cit.; Savinova E.R.,
Yashnik S.A., Savinov E.N., Parmon V.N. Op. cit,; Masheder D., Williams K.PJ. Op.
cit.; Christensen P., HamnettA.. MuirA. V.G. Op. cit.; Meshilsuka S., Ichikawa M.,
Tamaru K. Op. cit.; Atoguchi Г., AramataA., KazusakaA., Enyo M. //J. Electroanal.
Chem. 1991.318. 309.
4 Furuya N.t Matsui K. Op. cit.; LieberC.M., Lewis N.S. Op. cit.
* BraunsteinP., ManD., Nobel D. // Inorg.Chem. 1998. 27. 661.
201
этого свидетельствуют только различные косвенные доказательства,
полученные с помощью И К- или ЯМР-спектроскопии, поскштьку,
во-первых, время жизни таких интермедиатов мало, а во-вторых,
присутствие молекул растворителя, также способного к
координации, затрудняет детальный анализ структуры комплекса.
Для выяснения механизма реакции применялись также
полуэмпирические расчеты. Например, была предпринята попытка связать
каталитическую активность фталоцианинов с энергией граничных
орбиталей комплекса'. Оказалось, что каталитическая активность
фталоцианиновых комплексов коррелирует с их способностью к
координации дополнительного лиганда у атома металла, что, в свою
очередь, зависит от разности энергий (/-орбиталей фталоцианина
металла и граничных орбиталей координирующегося лиганда.
Одними из немногих работ, в которых предлагается механизм
электрокаталитического восстановления С02, являются статьи2,
показывающие, как происходит восстановление С02 в водном
растворе на графитовом электроде, на который нанесен РсСо, связанный
с поли-4-винилпиридином (PVP), в качестве донорного аксиального
лиганда. Показано, что наличие последнего сильно увеличивает
каталитическую активность РсСо, видимо, за счет увеличения
электронной плотности на металле. Хотя в данной каталитической системе
также происходит конкурентное восстановление воды, однако
селективность восстановленя С02 в ней гораздо выше, чем в случае чистого
РсСо, возможно, за счет увеличения локальной концентрации С02
п околомембранном слое за счет гидрофобного взаимодействия С0>
и PVP3. Схема реакции выглядит следующим образом4:
Со<И)Рс2- —> [Со(1)Рс2-]- ±J±^ Со(1)Рс2^Н+ —> [Со(1)Рс3-Н+Г
СО^
О
III
с
I ,
Со(П)Рс2-^-
Н20 Н+
1 Braunstein P.* Matt D., Nobel D. Op. cit.
2 Masheder D., Williams K.P.J. Op. cit.; Радушкина К.А.. Меренкова M.B., Ta-
расевич М.Р., Гмьперин М.Г., Кудревич СВ., Новожилова И. Г. Указ. соч.;
Yoshida Т., Катаю К., Tsukamoto M„ /Ida Т., Schhlettwein /)., Woerie D.,
Kaneko M. Op. cil.
J Mahmood M, IV., Masheder D„ liarty С J. Op. cit,
4 Радушкина К.А., Меренкова M.B., Тарасевич M.P., Гальперин М.Г.,
Кудревич СВ., Новожилова И.Г. Указ. соч.; Yoshida Т.. Катаю К., Tsukamoto M.,
Ша Т., Schhlettwein /)., Woerie D., Kaneko M. Op. cil.
H
<4 ®°
с
I
Co(lI)Pc2-
l
Co(H)Pc3^H+
202
Можно видеть, что каталитический процесс начинается только
на второй волне восстановления РсСо, поскольку за первой стадией
восстановления следует быстрое присоединение fT по
периферическим атомам азота фталоциан и нового кольца, и только на второй
волне генерируется активная частица, способная восстанавливать
как Н", так и С02, с образованием Н2 и СО соответственно. Однако,
тем не менее не удалось непосредственно наблюдать аддукт «РсСо-
COj»,
6.8. СИНТЕЗ КАРБОНОВЫХ КИСЛОТ
6.8.1. Карбоксил ирование органических галоген и дов
В начале 1980-х гп было показано, что электровосстановление
арилгалогенидов, катализируемое комплексами Ni-PPh3ljtriH Ni-
dppe2, в присутствии С02 в основном приводит к арилкарбоксилату
вместо биарила. На основе электроаналитического исследования
системы Ni-dppe был предложен каталитический цикл3,
Электрокарбоксилирование арилиодидов и бромидов также
катализируется комплексом Pd-PPh34:
RC6H4X + CQ2 + 2e ^(PPh^ + PPh, ,RQH4C0- +
axil =H,4-t-Bu,4-MeO 50-90%
X = I,Br
Было предположено, что в этой реакции СО? реагирует как
электрофил с [ArPd°(PPhi)2j , образующимся при восстановлении
комплекса о-арил-палладня(11)5. Арил хлориды реагируют с Pd°
слишком медленно, чтобы обеспечить эффективное карбоксили-
рование. При этом арилтрифлат и арил бромид имеют одинаковую
реакционную способность. Синтез арил карбоксильных кислот,
таким образом, можно проводить через соответствующий
арилтрифлат6:
1 Trvupcl М., Roffin К, ftrhhonJ.t FauvarqucJ.F. // Nouv. J, Chim. 1981. 5. 621.
2 Fauvarque J.F., Chevrot C, Jurand A., Francois M., Perichon J. jj J. Organomet.
Chem. 1984, 264. 273.
y Amatare C.t Jufand A. //J.Am. Chem. Soc. 1991. 113. 2819; AmatoreC, JutandA.,
MottierL. //J. Elcctroanai. Chcm. 1991,306. 141.
4 Torii S., Tanaka //., Morisaki AT., Juland A., Pfluger F., Fauvanque J. F. // Chem.
Lett. 1986. 169.
5 AmatoreC, JutandA ., KhalilF., Nielsen M.F. //J. Am. Chem. Soc. 1992. 114. 7076.
* JutandA., Negri S., MostetiA. // Op. cit.; JutandA., MoslehA., NegriS. Op. eit.
203
АЮТГ+ C02+ 2e-^^^ArCOi+ТЮ-
Аг = R-C6H< 52-95%
cR = 4-С1,4-CF3,4-C02Et, 4-F, 4-CH3
Ar = 1- или 2-нафтил
Нестероидальнъте противовоспалительные а-арилттропионовые
кислоты получены из соответствующих бензил хлоридов и С02
с катализатором Ni-dppc или Ni-dppp в присутствии COD1.
Использование катализатора в этой реакции не обязательно, но он
ограничивает реакцию гомосочетаиия, которая становится основным
процессом при высокой концентрации бензил галогенида и низком
давлении С022:
NiCI2(dpp)(IO%)
АгСН(Ме)С1+С02 SSAe > АгСН(Ме)С02Н
1 п г, Н М г А
Продукт
PhCH<Me)C02H
Me
Me
)>—C02H
Название
Напроксен
Ибупрофен
Фенопрофен
Выход, %
89
81
80
76
Катализатор необязателен также в случае электрокарбоксилиро-
вания арил галогенидов или различных бензильных соединений в
неразделенной ячейке и в присутствии растворимого анода из алюминия*
1 FauvarqueJ.F., JutandA, Francois M. // Nov. J. Chim. 1986. 10. 119-
2 FauvarqueJ.F., JutandA., FrancoisM,//J.Appl, Electrothem. 1988.18. 109.
> Silvestri G„ Gambino S., FilardoG, GulottaA. // Angew. Chcm., Int. Ed. Engl. 1984.
23. 979.
204
или магния1. Тем не менее оба метода, т.е. использование
катализатора или растворимого анода, могут быть в конечном счете
объединены для проведения электрокарбоксилирования органических
галогенидов, имеющих функциональные группы, которые не
позволяют использовать метод прямого электрохимического
восстановления.
Комплексы переходных металлов с тетраазамакроциклами
привлекают значительный интерес из-за применения в электрокатализе
и близости к природным фотосинтетическим системам2. Механизм
восстановления COs, катализируемого тетраазамакроциклическичи
комплексами Ni(TI) и Со(П), изучен методами циклической воль-
тамперометрии и препаративного электролиза. Продуктом
восстановления является СО, который далее реагирует с Ni(l) — формой
комплекса с образованием карбонильных соединений Ni(0). Таким
образом происходит отравление катализатора.
Электрокаталитическое восстановление COs под действием
трехядерных никелевых кластерных радикалов было изучено
методами электрохимии и спектроэлектрохимии3.
6.8.2. Карбоксилирование алкенильных и алкинильных
соединений
Прямое электрокарбоксилирование ненасыщенных соединений
возможно4, однако процесс ограничивается восстанавливаемыми
алкенами типа арилированных или активированных олефинов, а для
алкинов известно только несколько примеров такой реакции5.
Непрямое восстановительное сочетание между алкинами и СОт
приводит к замещенным акриловым кислотам в результате
гидрокарбоксил ирования6:
1 Chassard J., Folest /,С, Nedekc J. У.л Perichnn J., Sihilh £, Trvupe! M. Op. cit.;
GalJ., FutestJ.C, Trvupel M*. Moingeon И.О., Chassard J.//"NJ. Chcm, 1995.19,
401.
2 Bujno It. Bilewicz Л-, Siegfried I.., Kaden T. // Electrochim. Acta. 1997.42. 1201.
i Wittrig RE., Ferrence G.Mti Washing /, Kubiak C.P. ff Inorg. Cliim. Acta. 1998.
273. 111.
1 Shono T. Op. cit.; Baizer M. M. // Tetrahedron. 1984.40. 944,
5 Wawzonek S., WeaningD. //J. Am. Chem. Sac. 1959. 81. 2067; Dunach E.r Peri-
chonJ. //J. Organomet. Chem, 1988. 352, 239; Dunach £„ Derien S.t PerichonJ. //
J. Organomet. Chem. 1989. 364. C33; Dunach E. Op. cit.; Labbe £., Dunach £.,
Perichon J. //J. Onjanomet. Chem. 1988. 353. C51.
6 Kijima M., Nakazato A*., Sato T. Op. cit.; Wawzonek 5., Wearring D. Op. cit.;
Dunach £., Perichon J. //J. Organomet. Chem. 1988. 352. 239; Dunach £., De-
rien S., Perichon J. Op. cit.; Dunach £., Perichon J. // Synlett. 1990. 143; Labbe £.,
Dunach £., PerichonJ. Op. cit,; Yamamoto Т., Wakabayashi S., Osakada A-//J.
Organomet. Chem. 1992. 428. 228: Troupel M., Rollin Y., SibilleS., Fauwrque J.F..
ftrichonJ. //J. Chem. Res. (s). L980. 24.
205
R1 R2 R1 R2
но2с н н со2н
А В
a) Ni(bipy)j(BF4)2 (10%), c, DMR Mg анод; b) гидролиз.
R1
"-QH,j
Pr°
Ph
Ph
Ph
R2
H
Pra
H
Me
C02Et
Г, °C
Выход, %
65
93
55
72
75
Соотношение А: В
90: 10
71 :29
38:62
Электролиз проводился в неразделенной ячейке в ДМФА,
содержащем каталитические количества комплекса никеля, либо NiBr2dme +
2PMDTA — в зависимости от субстрата в присутствии растворимого
магниевого анода при нормальном или несколько повышенном (5 атм)
давлении С02. Реакция протекает при 20—8СГС. Высокая температура
Ьчагоприятствуеткарбоксилированию алкинов с электронодонорньгми
группами, а комнатная — с электроноакцепторными. Реакция
протекает с хорошей региоселективностью с введением С02 в положение
2 терминальных алкинов и стереоселективностыо — в основном как
цис-присоединение. Монокарбокилирование происходит только в
отсутствие электроноакцепторных групп. Выходы, полученные из
расчета на 30—85% потребление алкина, хорошие.
Этот метод можно сравнить главным образом с химическим
методом, развитым Хобергом, который использовал стехиометрические
количества комплекса никеля (0), в основном чувствительного к
воздуху Ni(COD)2l. Вместо этого электрохимический метод использует
легкодоступный комплекс Ni(II).
Здесь наиболее отчетливо видно преимущество использования
растворимого анода. Наиболее подходящим является магний, при
окислении которого образуются ионы Mg2+, которые могут вступать
в каталитический цикл, расщепляя никелациклический интермедиат
и освобождая Ni для дальнейших превращений (схема). Этот
механизм был подтвержден на основе образования никелацикла и его
характеристики методом циклической вольтамперометрии. В
отсутствие ионов Mg2" (реакции проводились в разделенной ячейке в
присутствии ионов аммония) никалацикл не разрушается и реакция
останавливается, когда все исходное никелевое соединение прореа-
1 Hoberg И., Schaefer D., Burkhart D., Kruger С, Romao Л/. Op. cit.; Hoberg H.,
BarhausenD. //J. Oiganomei, Chem. 1989. 379. C7.
206
гирует. При добавлении MgBr2 к раствору никелацикла, полученного
электрохимически, Ni(II) регенерируется1. Схему
электрохимического карбоксилирования алкинов, катализируемого комплексами
никеля, можно представить следующим образом:
R
R
Н С02" 2Mg
,2+
Н+г ( л
R ' R
NiL-2+
LNJ0^0
Ni(0)L2
R = R
+C02
LNi—
Для построения полициклических соединений используются диа-
цетилены. Так, в присутствии С02 и стехиометрического количества
Ni(0) были получены битшклические пироны2, С помощью этого
метода электрокарбоксилирования в зависимости от природы лиганда,
связанного с никелем, были синтезированы линейные или циклические
монокарбоновые кислоты как основные продукты из несопряженных
диацетиленов3. Образование цикла протекает с участием комплекса
никеля с билиридилом при нормальном давлении С02; при использовании
лиганда PMDTA и давлении С02 5 атм, образуются линейные аддукты:
II Win
jf И 1) ZC
L = bipy
L = PMDTA
. NiL:\ MgaHoa ^ /
2) H30 * \
Выход, %
35
30
V\Aco2H
a
a
59
9
— 11
со2н
6
6
4,5
45
Derien S., Dunach E., Perichon J. //J. Am. Chem, Soc. 1991. 113. 8447.
Tsuda T.t Morikawa S.t Hasegawa N., Saegusa // J. Oi)j. Chcm, 1990. 55, 2978.
Derien S., Dunach £., Perichon J. //J. Organomet. Chem. 1990. 385. C43; Derien $.,
CtinetJ.C.t Dunach £., Perichon J.//J. Org. Chem. 1993. 58. 2578.
207
Различные диацетиленовые соединения, таким образом, могут
быть превращены в соответствующие монокарбоновые кислоты. Диа-
цетилены, имеющие как терминальную, гак и инфернальную тройную
связь, образуют продукт присоединения С02 к терминальной связи
в основном в 2-положснии. Элсктрокарбоксилированис 1,3-диацсти-
ленов в присутствии катализатора Ni-PMDTA, приводящее к
образованию (Е)-2-винилиден-3-ин карбоновых кислот, регао- и стереосе-
лектнвно1. Процесс идет по следующей схеме:
<г
+С
L
Ыру
PMDTA
l)2e.NiL2~,
01 2)Н,0 > \J<
<
Выход, %
35
30
• —
^со2н со2н
5 6
Соотношение 5: б
59:4,5
9:45
При полной конверсии выходы хорошие (70—100%).
Обнаружено, что двойные связи в этом процессе менее реакци-
онноспособны, чем тройные связи (10—40%)г. В несопряженных,
а также в сопряженных алкен-ацетиленах только алкиновый остаток
карбоксилируется с регео- и стереоселективностыо, аналогичной
той, что наблюдается для ацетиленов3.
В этих процессах также исследовались 1,2-диены4. Были
получены монокарбоновые кислоты с хорошим выходом с введением С02
в основном при Сг для R = алкил, циклоалкил и при С* для R = арил
(L = PMDTA; PCOj = 5 атм) согласно следующим схемам:
^°= + со>тйо—" Ч и+ М
со2н н
Аг ,. С02Н Аг
\ 1)2е, NiL2+ I \ /
2)Н20 Аг v ^СОгН
1 Derien S., ClinetJ.C, Durtach £., function J. // J. Org. Chem. 1993. 58. 2578; De-
Hen S., ClinetJ.C, Dunacft E„ Periehon Л // J. Chem. Soc, Chem. Comm. 199L
549.
1 Derien 5., ClineiJC, Durtach E., Periehon J. // Tetrahedron. 1992. 48. 5235.
J Derien S., Clinet J.C\, Dunach £., Periehon J. // J. Organomel. Chem. 1992. 424.
213.
4 Derien S., Ciinet J. C, Dunach E., Periehon J. // Sinlett. 1990.1. 361.
208
Описаны некоторые другие примеры гомогенной
электрохимической активации С02, приводящей к синтезу лактонов путем тело-
меризации бутадиена и С02', бициклических лактонов с участием
комплексов никеля и кобальта2, реакций транскарбокилирования3
и включения С02 в эпоксиды под действием Ni(cyclam)Br24.
6.9. КАРБОНИЛИРОВАНИЕ ОРГАНИЧЕСКИХ ГАЛОГЕНИДОВ
Известно, что монооксид углерода и переходные металлы
в низких степенях окисления дают различные, хорошо описанные
комплексы. Однако из-за сильной координации с СО эти карбо-
нилы металлов обладают невысокой реакционной способностью
по отношению к связям углерод — галоген. Так, карбонилирование
органических галогенидов остается сложной проблемой, поскольку
присутствие СО приводит к дезактивации каталитической системы.
Отдельные попытки преодолеть это препятствие рассмотрены ниже.
Так, было показано, что некоторые карбонилы металлов можно
активировать путем электрохимического восстановления с
генерацией активных анионных частиц. Не вдаваясь в детали, стоит
отметить, что синтез альдегидов можно осуществить в условиях
электролиза стехиометрической смеси алкил галогенидов и пентакарбо-
нила железа5:
RX + Fe(CO)5 'К2)Н' > RCHO
ДМФАилиСНдСТЧ 30—70%
R = H-CsHII,C2H5,PhCH2
R = I,Br
В качестве альтернативы С02 может быть использован как
источник СО. Хорошо известно, что комплексы переходных
металлов в низких степенях окисления катализируют химическое или
электрохимическое восстановление С03 в СО. Этот подход был
использован для генерирования смешанного комплекса никеля
Ni°bipy{CO)2 при электрохимическом восстановлении Nibipy2"" в N-
метилпирролидоне или Д МФА в присутствии С02. Восстановленный
комплекс может реагировать с алкил-, бензил- и алл ил галоген идам и
с образованием симметричного кетона с регенерацией Nibipy2+.
1 Braunstein P.. Matt D., Nobel D. Op. cit.
2 Tascedda P., Dunach E. // J. Chem. Sou., Chcm. Com. 1995. 43.
3 Ozaki S.. Toshikazu N.. Mart S.. Chie M.. Hidenobu O. // Chem. and Pharm. Bull.
1991.39.31.
4 Walther Л // Coord. Chem. Rev. 1987. 79. 135.
* Vanhoye Z>., Bedfoui F., Mortreux A.. Petit F. // Tetrahedron Lett. 1988. 29. 6441;
Yoshida K.t Kunugita E.L Kobayashi M.,Amano SJ. //Tetrahedron Lett. 1989.30.6371.
209
Таким образом, был предложен двухстадийный метод, включающий
электровосстановление и химическое сочетание, приводящий
к образованию кетона1.
Относительно недавно было показано, что реакции карбон или-
рования также можно осуществлять электролизом раствора
органического галогенида под действием СО, содержащего
каталитические количества bipy в неразделенной ячейке, снабженной анодом
из нержавеющей стали. При этом образуются симметричные кетоны
с высокими выходами2. Анодная реакция дает комплексы Ni(ll)bipy
и Fe(II)bipy, которые восстанавливаются на катоде, в конечном счете
катализируя синтез кетонов. Реакционными частицами
определенно являются комплексы никеля, связанные как с bipy, так и с СО,
но и комплексы железа, вероятно, играют свою синергетическую
роль. Схема процесса следующая:
2RX+CO+2e-^^^^RCOR+2X-
DMF или MeCN
RX
BnCl
2-MeC,,H4CH2Br
4-МеС,,Н4СН2Вг
2-C!C6H4CH2CI
MeCH=CHCH2Q
Ме(СН2)5Вг
4-CF3ChH^Br
Выход, %
80
70
62
75
65
62
50
Кетоны были успешно получены путем
электровосстановительного сочетания органических галогенидов и СО с участием
комплекса Nibipy2+ в качестве катализатора, где СО-гругпта вводится либо
из пробулькиваемого в раствор СО3, либо из карбонила металла4.
Электровосстановление С02 протекает также под действием порфи-
ринов жслсза{0) в присутствии анодно генерируемых ионов Mg2+f.
1 Gamier £.T Rotlin Г., Perichon У. // J. New J. Chem. 1989. 13. 53; Gamier L„
Rollin K, Perichon J. ff}, Oi^ganomet. Chem. 1989. 367. 347.
2 Oca/rain A/., De\>aud A/., Troupel At., Perichon J. // J. Chem. Soc, Chem. Comm.
1995. No 22. P. 2331-2332.
J Oca/rain A/., Devaud M., Troupel M., Perichon J. Op. cit.; Ocafrain M., Dolhem E„
NedetecJ.Y., Troupel M.J. // Organomet. Chem. 1998. 571. 37; Ocafrain M.,
Devaud M.t NedelecJ. K, Troupel M. //J. Oiganomet. Chem. 1998. 571.37.
4 Dolhem E., Ocafrain M., NedeiecJ.Y., TmupelM. // Tetrahedron LeH. 1997.53. 17089.
5 Hammouche M., LexaD., MomenteauA/.. SaveantJ.-M. //J, Am. Chem. Soc. 1991.
113.8455.
210
Известны и некоторые другие реакции, отличные от электрокар-
бонилирования органических галогенидов, катализируемые
комплексами никеля и приводящие к кетонам. Так. несимметричные
кетоны образуются путем электровосстановителъного сочетания
ацилхлоридов и алкилгалогснидов, катализируемого NiBr2bipyl.
Ац и л хлориды превращаются в симметричные кетоны в
неразделенной ячейке, снабженной анодом из никеля или нержавеющей
стали. В этой реакции металлические частицы образуются путем
электровосстаноапения Ni2~, генерируемого на аноде, в отсутствие
добавленного лиганда2 по схеме:
2RCOCl + 2e "i(bipy),MeCN ► RCOR +2СГ+СО
анод — нержавеющая сталь ,45—Я0%\
R = Bn, Ph, 3-FC6H4, 3-МеС6Н4, 4-МеС6Н4, 4-FC6H4,
(~\^ г 4-ВгСбН4, 2,4-F2C6H3, 1-нафтил
Арилцинковые соединения, электрохимически генерированные
в присутствие комплекса Ni-bipy и солей цинка, также могут
реагировать с (CFjCOhO с образованием соответствующих арилтрифто-
рометилкетонов3. Реакций, описывающих электровосстановительное
образование связи углерод — гетероатом в условиях катализа
переходными металлами, немного. Так, можно отметить
электрохимическое силилирование аллил ацетатов в присутствии Pd—PPhV,
электросинтез арилтиоэфиров из тиофенола и арил галогенидов5.
На основе электрохимической активации солей и комплексов
некоторых переходных металлов разработаны каталитические системы
для карбонилирования метанола в диметилкарбонат6. Синтез был
проведен при пробулькивании СО в метанольную суспензию или
раствор комплекса при атмосферном давлении и комнатной
температуре в разделенной ячейке с ионообменной мембраной. Ме-
таллокомплексы катализируют окислительное карбонилирование
метанола по следующей реакции:
2СН3ОН + СО + 2М<я+|)+ -> (CH-jOhCO + 2Н+ + 2М<">+
1 Marzouk #., Ratlin У.. Potest J.С, Nedelec J. У., Perichon J. // J. Organomclal.
Chem. 1989.369.47.
2 PotestJ.C.,Perira-Martins£.. IboupeiM., Perichon J.// Tetrahedron Letl. 1993.34.7571.
J Sibille S., Ratovebmanana V., Perichon J. Op. cit.
4 ToriiS., Tanaka //., KnUth Т., Morisaki K. // Tetrahedron Lett 19R4. 25. 3207.
5 Meyer G., Troupei M. // J. Organometal. Chem. 1988. 354. 249.
6 Filardo G., Galia A., Rivetti P., Scialdone C, Silvestri G. // Electrochimica Acta.
1997.42.1961.
211
тогда как анодный процесс регенерирует металл в его более высокой
степени окисления:
М<">+ -> М,п+|,+ + е
В качестве катализаторов тестировались редокс-пары, уже
известные в химической реакции карбонилирования, промотируемой
кислородом — Cu(I/II), Pd(0/II) и Со(Н/Ш), однако
электрохимические условия оказались более мяткими, чем в обычных химических
реакциях. Комплексы в ряде случаев проявили большую
каталитическую активность. Эксперименты проводились с использованием
в качестве лигандов 2,2-бипиридила(Ыру), 1,6-бис(2-оксифенил)-
2,5-диаза-1,5-гексадиена(салена), 2,4-пентадионата (асас) и PPh3.
Самые лучшие фарадссвскис выходы димстикарбоната (%)
наблюдались для следующих катализаторов: CuCl(bipy) — 84,8, PdCl2(bipy) —
64,0, СоС12 - 26,0, RhCI3 - 25,0, AgBF4(bipy) - 10,2. Соединения
VCl3, Mn(salen), Mn(acac)2, ReCI3(bipy), Cr(acac)3, NiCI2bipy,
RuCl2(PPh02 проявили или очень низкую активность, или никакую.
Только комплекс PdCI2(bipy) позволяет протекать более чем одному
редокс-циклу. Известно, что многие переходные металлы
катализируют окисление СО в С02 в воде, и именно они благоприятны для
окислительного карбонилирования спиртов в диалкилкарбонаты.
Механизмы, предложенные в литературе как по химии, так и
электрохимии1, включают образование карбометоксиметаллических
интермедиатов (М—СООСН^, с М=Си, Со или Pd)r стабильность
и реакционная способность которых сильно зависит от электронной
структуры комплекса и от природы лигандов. Так, сам CuCI начинает
действовать только при 70°С, а в присутствии лиганда -bipy — уже
при комнатной температуре. Среди изученных соединений
электроактивация CuCI, PdCl2, PtCl2 и AgBF., возможна только в присутствии
bipy. Наоборот, добавление bipy к СоС12, NiCb и АиС1я, а также
лигандов асас, salen и PPh3 ухудшает эффективность процесса.
6.10. ЭЛЕКТРОСИНТЕЗ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ИЗ ХЛОРОФОСФИНОВ И БЕЛОГО ФОСФОРА
Одна из важных задач в препаративной органической химии
ФОС — это развитие новых синтетических подходов получения
соединений с Р-С связями в мягких условиях на базе белого фосфора
или хлоридов фосфора. Особое внимание здесь уделяется синтезу
третичных фосфинов, широко используемых в металлоорганической
и координационной химии. Недавно описан новый перспективный
подход, согласно которому третичные фосфины получены электро-
1 Otsuka К., Yagi Т., Yamanaka I. //J. Elcctrochcm. Soc. 1994.142. 130; Otsuka К.,
Yagi V., Yamanaka 1. // Eleclrochim. Acta. 1994. 39 2019.
212
химическим путем, сочетанием моно- и дихлорфосфинов с
ароматическими и гетероароматическими галогенидами, катализируемым
комплексом никеля с bipy в неразделенной ячейке, снабженной
растворимым магниевым или цинковым анодом (табл. 6.3)':
АгХ + Рп„РС]з_„ c'Nityipy _»at Ph p n = i2
R = СНз, CHjO, (CH3)2N, CN, CH2Et, CH3C(0) ^
Для преврашения арилгалогенидов с донорными заместителями
а кольце следует использовать Mg анод, а с акцепторными — Zn анод.
Предложенный метод электросинтеза третичных фосфинов
эффективен как с ароматическими галогенидами с акцепторными и
донорными заместителями в кольце, так и с гетероароматическими пиридин-,
тиофсн-, пиримидин- и пиразол-галогенидами. Преимуществами его
также являются одностадийность и мягкие условия (комнатная
температура) проведения процесса. Предложена общая схема циклической
регенерации катализатора, которая будет обсуждена ниже2.
Проблема селективного раскрытия тетраэдра белого фосфора Р4
и его непосредственной функционализации приобретает все большее
значение в связи с поиском новых экологически чистых путей
синтеза фосфорорганических соединений. Под действием
электрохимически генерированных катализаторов — комплексов Ni(0)bipy
и органических галогеггидов удается превратить белый фосфор
в соединения с Р—С связями — фосфины и фосфиноксиды (70%
Ph,P в ДМФА (Zn и Mg аноды), 59% Ph3PO и 47% С6Н13РО в CH3CN
(А1 анод))'. При этом протекают следующие реакции:
Катод: Ni(II)L3 + 2e^Ni(0)L2 + L; L = bipy
Анод: А1-Зе->А1(Ш)
P4 + RX Ni(Q)L2 )>P-R
-х"
1 Budnikova Yu.H.. Korgn Yu.M., PerichonJ., ,Verfefec/-K//J.Oiganomet.Chem. 1999.
575. 63; Budnikova Yu.H., Kargin Yu.M., Sinyashin O.G. // Mendeleev Comm. 1999.
143; Budnikova Yu,H..PbrichonJ.. NedetetJ.-Y., Kargin Yu.M.// Phosphorus ans Sulfur.
1999. 144-146. 881; Будникова ЮГ., Каргин Ю.М., Синяшип О.Г. //Жури. обш.
хим. 2000.70.562; Будникова Ю.Г.. Каргин Ю.М. // Ж>рн. общ. хим. 1995.65.1660.
2 Ibid.
i Budnikova Yu.H., Yakhvarov D.G., Kargin Yu.M. // Mendeleev Commun. 1997.
67; Budnikova Yu.H., Kargin Yu.M., Sinyashin O.G. f/ Phosphorus ans Sulfur. 1999.
144—146, 565; Budnikova Yu.H., Kargin Yu.M., Sinyashin O.G. // Phosphorus ans
Sulfur. 1999.144-146,879.
213
Таблица 6.3
Электрохимическое сочетание арил- или гетероарил-галогенидов
и хлорофосфинов, катализируемое комплексом никеля
AiX
Продукт
Выход
препаративный, % по хлоро-
фосфину
Ph2PCI
PhBr
2-ВгС6Н4СН3
4-BrCftH4CH3
4-CH>0-C4HsBr
4-Mc2N-CftH4Br
4-C02Et-C6H4Br
3-C02Et-C6H4Br
2-Cl-C5H4N
3-Cl-CsH4N
2-Cl-C4H3S
2-С1-6-СН30-С*Н3Н
2-Cl-C4H,N,
3-CN-C*H4Br
5-CI-1.3(CH3)2C3HN2
Ph3P
Р1т,РС6Н4<СН3-орто)
Ph3PCfiH4{CH3-napa)
Ph,PQH4 (CH30-napa)
Ph2PQH4 (NMc2)2-napa)
Ph2PC6H4 {C02Et-napa)
Р1ъРС6Н4(ССШ-мета)
Ph3P(C5R,N-2)
Ph,P (C,H4N-3)
Ph2P (C4H3S-2)
Ph3P(6-CH,0-2-CiH3N)
Ph,P(C4HiNr2)
Ph2PC6H4 (CN-мета)
Ph,P(5-C,HN2-l,3(CH3)2)
87"
54"
56'
52'
70"
63"
66"
80"
63"
65*
66'
50'
45"
25"
PhPCU
PhBr
2-Cl-C5H4N
Ph3P
PhPfCsH^N)!
72'
68'
Примечание. * — Mg анод; " — Zn анод.
Ключевой стадией является взаимодействие никельорганического
соединения RNLIlXLm с белым фосфором по следующей схеме:
Ni(0)L„,+RBr -> RNi(II)LfflBr—^->>P-R+ Ni(II)Lw
Показано, что раскрытие тетраэдра Р4 и разрыв остальных
связей Р—Р может происходить под действием комплексов Ni(0).
При этом образуется биядерный комплекс(1), который быстро
реагирует с органическими галогенидами с функционализациеЙ
цикло-Рз — фрагмента, превращаясь в моноядерный комплекс
с РЬ3Р]|-лигандом:
214
eN
Ni-
SN
"P;:Ni
A*'
Br2
PhHal
P-Ph
n+
А В
Такой путь образования связей Р—Ph конкурирует с
взаимодействием с-арилкомплекса PhNiXc P4. Таким образом,
электрохимическим методом удается осуществить функционал изацию белого
фосфора с образованием Р-С соединений в мягких условиях.
Некоторые последние примеры электрохимического
каталитического синтеза фосфорорганических соединений приведены
в разд. 5.9.
6.11. ЭЛЕКТРОХИМИЧЕСКИЙ МЕТАЛЛОКОМПЛЕКСНЫЙ КАТАЛИЗ
В РЕАКЦИЯХ ПЕРФТОРАЛКИЛИРОВАНИЯ
6.11.1. Электрохимическое восстановление
перфторалкилгалогенмдов под действием комплексов никеля
и платины в низких степенях окисления
Многочисленные исследования последних лет показали, что
комплексы никеля с различными лигандами являются
эффективными катализаторами электрохимических и химических реакции,
таких как кросс-сочетание или функционализация олефинов.
Недавно было обнаружено, что комплексы никеля и платины в низких
степенях окисления выступают эффективными катализаторами
введения групп Rp. Так, разработан простой универсальный
одностадийный элсктрокаталитический метод присоединения
различных фторалкильных групп (линейных, разветвленных) к оле-
финам, содержащим различные заместители (Me, Ph, Ру. АсО, NpT
4-Me-Ph, -CH2-DM 1С), позволяющий получать какмоиомертгые,
так и димерные продукты с одним или двумя хиральными атомами
углерода1:
Mikhaylov D.Y., Budnikova Y.H., Gryaznova Т.. Sinyashin O.G. //J. Опзапотс!.
Chem. 2009, 694. 3840; Mikhaylov D. Y., Budnikova Y.H., Gryaznova Г.,
Sinyashin O.G. II ECS Transactions. 2010. 25. 67; Budnikova Y.H., Mikhaylov D. K,
Gryaznova Т., Sinyashin O.G. ff ECS Transactions. 2010. 25. 105; МихаСиювД.Ю.,
Вудникова ЮГ., Грязнова Т. В., Синяшин О. Г. // Изв. РАН. Сер. Химия.
2010. 59. 1868; Mikhaylov D,Y„ Gryaznova Г., Dudkina К, Khrizanphorov М.,
Laiypov Sh., Kaiaeva О., Vide D.A., Sinyashin O.G,, Budnikova Y. 11 Da It on Trans.
2012.41. 165.
215
to
Rf
BibSnH
/-♦ LNiNBr2
^R2 'RfV
Ri = H,Me,OH,CN;
R2 = Ph, Py, AcO, Np;
RF = CuF|3, C6Fi2H, /-C5F11, etc.
Реакция протекает по необычному пути присоединения-димери-
зации в мягких условиях с каталитической регенерацией доступных
комплексов никеля с ос-дииминами {2,2-бипиридин, 2,2':6',2"-тер-
пиридин, (S, 5)-2,6-бис(4-фенил-2-оксазолин-2-ил)-пиридин).
Установлено, что стереоселективному образованию meso-формы
димерного продукта благоприятствует линейная структура перфто-
ралкильного заместителя, a d, 1-форме — изоструктура заместителей.
Предложена схема реакции электрокаталитического перфторалкили-
рования олефинов, в которой активной формой катализатора, гене-
рирумой и регенерируемой на электроде, является Ni(I)L. Впервые
выделен и охарактеризован комплекс (tpy) NiBr, доказана его ме-
талло-центрированная электронная структура.
Образование комплексов LNi1 с выбранными лигандами было
подтверждено с помощью методов ЦВА и ЭПР. Было обнаружено, что при
восстановлении комплексов контролируемом методом ЭПР
наблюдается появление сигнала с g-фактором, характерным для LNi1 (2,1391,
2,1403 и 2,1845 для Ni(J)bpy, Ni(I)tpy и Ni(I)pybox соответственно).
В последние годы каталитические свойства комплексов платины
сравнивают с комплексами никеля в аналогичных реакциях, притом
последние не всегда показывают худшие результаты. Обнаружено,
что комплекс платины(Н) с 4,4',4"-три(трет-бутил)-2,2':6',2"-терпи-
ридином (tpy') также восстанавливается до LPi', который успешно
регенерируется на электроде и приводит к образованию тех же
продуктов, что и никелевые комплексы, в реакциях каталитического
перфторалкилирования олефинов.
Исследования механизма перфторалкилирования позволили
получить важную информацию о свойствах каталитических систем
никеля, которые могут прояснить и пути реагирования других субстратов
в условиях электрохимического сине за, В то время как для комплекса
никеля с лигандом Ьру на кривых ЦВА не происходит разделения
пиков восстановления, а наблюдается суммарно двухэлсктротпгый
обратимый перенос Niir/Ni°, у комплексов никеля и платины с три-
денгантными лигандами (tpy, pybox) происходят постадийные
обратимые однозлектронные переносы М'^М'/М0. В присутствии
возрастающих количеств перфторалкилгалогенидов на ЦВА комплексов
наблюдаются значительные приросты тока первого пика восстановления
для комплексов с тридентантными лигандами. Активной
каталитической формой является комплекс LM1. Редокс-свойства комплексов
по отношению к перфторалкилгапогенидам позволяют предположить
их потенциальную каталитическую активность в реакциях
перфторалкилирования субстратов, например олефинов, которые с этой целью
должны быть введены в реакционную смесь.
Было обнаружено, что при 10% мольном содержании
катализатора достигаются хоропше вьсходы продуктов реакции как с перф-
торгексил йодидом, так и с бН-перфторгексил бромидом:
217
RFX +
RFX=C6F13J, RF = C6Fl3l,70%
H(CF2)6Br RF = H(CF2)6Bi; 49%
Особенно интересно, что конечный продукт реакции является
димером, содержащим по два олефиновых и перфорированных
остатка', что ранее в литературе не описывалось2.
Структуры 2,3-диметил-2,3-дифенил-1,4-бис(перфторгексил)бу-
тана и 2 3-диметил-2,3-дифенил- ] ,4-бис(6Н-перфторгексил)бутана
были подтверждены рентгеноструктурным анализом. Обе молекулы
находятся в специальном положении в центре симметрии в асиме-
трической элементарной ячейке, представляя тем самым два
фрагмента, содержащие хиральные центры. Вследствие совпадения
молекулярных центров с центрами инверсии каждая молекула является
meso-формой (рис. 6.1). Структура соединений димеров была
независимо установлена с помощью комбинации методов одномерной
и двумерной ЯМР-спектроскопии. В частности, в случае соединения
2,3-диметил-2,3-дифенил-1,4-бис(6Н-перфторгексил)бутана
комбинация ID ('Н, »H{19F}, l3C, l9F) и 2D (COSY 'Н-'Н, HSQC/HMBC
'Н—|3С) ЯМР-методовл позволяет однозначно установить структуру
цепи вплоть до атома углерода С2.
Было отмечено, что влияние пространственной структуры
заместителей влияет на образование различных диастереомерных форм
(табл. 6.11).
Ri
NiBr3L ^2\ y^l
Z^HaRpF5 RF'
Rf NR2
Mikhaybv D.Y.. Budnikova Y.H., Gryaznova Т.. Sinyashin O.G. //J. Organomct.
Chem. 2009. 694. 3840; Mikhaybv D.Y., Budnikova Y.H., Gryaznova Г.. Sin-
yashin O.G, // ECS Transactions, 2010, 25. 67; Budnikova Y.H., Mikhaybv 0. K,
Gryaznova Т., Sinyashin O.G. // ECS Transactions. 2010. 25. 105; Михайлов Д.Ю.,
Будникова Ю.Г.. Грнзнова T.B,t Синмшин О.Г. // Изв. РАН. Сер. Химия. 2010.
59.1868; Mikhaybv D. К, Gryaznova Г., Dudkina К, Khri&jnphorov А/.+ LatypovSh.,
Kataeva О., Иск D.A., Sinyashin O.G., Budnikova Y. // Dalton Trans, 2012.41.165.
Ozaki S., Matsushita #., Ohmori H. Op. cit.; Ozaki S., Horiguchi /., Matsushita H.,
Ohmori H. Op, cit.; PeHmutter P. Conjugate Addition Reactions in Organic Synthesis.
Oxford: Pergamon Press, 1992; Nedelec J. K, PerichonJ., Troupe/M. //In Topics in
Current Chemistry/eds. E. Stcckhan. Berlin: Springer-Vferlag, 1997. 185. P. 141.
DemmeA.E. Modem NMRTechniquesforChemistry Research, Pergamon, 1988; Atta-
ur-Rahman. One and Two Dimensional NMR Spectroscopy AmstenJam: Elsevier, 1989.
218
т
* т £>
• Л*' ■*•* # .
л *с1а
Рис. 6.1. Молекулярная структура 2,3-диметил-2,3-дифенил-1 А
бис(перфторгексил)бутана и 2,3-Диметил-2,3-Дифенил-1,4-бис(6Н-
перфторгексил)бутана
С помощью скрининга различных условий и реа]тентов реакции
было обнаружено, что наилучшим катализатором изучаемых
реакций является комплекс (bpy)NiBr2 в ДМФА. Комплексы (tpy)NiBb
и (pybox)NiBr2 катализировали реакции присоединения-димери-
зации (реакции 5 и 6), однако выходы продуктов оставались
средними, поэтому в дальнейшем было решено их не использовать. При
использовании комплекса платины (эксперимент 7) был достигнут
хороший выход продукта реакции и был получен лишь один meso-
стереоизомер с высокой стереоселективностью, однако комплексы
никеля более доступны и дают высокие выходы. Стоит отметить, что
использование в качестве катализатора дибромида никеля в смеси
ДМФА: пиридин (9: 1), позволяет получить целевой продукт, однако
с очень низким выходом. Из табл. 6.11 видно, что наилучший общий
выход продукта был достигнут в эксперименте 12 с использованием
разветвленного перфторизопентил йодида.
Рассмотрено ачияние различных факторов на образование
различных диастереомерных форм получаемых димерных продуктов.
Все эксперименты проводились в электролизере с разделением
анодного и катодного пространства, на платиновых катоде и аноде.
В таблицу не вошли результаты экспериментов, проведенных без
разделения с использованием растворимого Zn, Mg и Си анода.
Причиной этого послужили два фактора, во-первых, то, что выходы
продуктов были невелики (37,28 и 45% для анодов Zn, Mg и Си
соответственно), а во-вторых, то, что реакции с растворимыми анодами
проходили нестереоселективно (особенно для меди — соотношение
диастереомерных форм — 1 :0,3 (meso : dl)). Большинство синтезов
проходило стереоселсктивно с образованием стереоизомерных meso-
форм.
Использование протонодонорнъгх растворителей, иногда успешно
выступающих в качестве источника атомов водорода вместо
перхлората аммония, в данном случае не позволило предотвратить димери-
зацию. Трибугилгидрид олова в качестве источника атома водорода
219
Таблица б. J1
Перфторалкилирование олефинов при различных условиях
№
эксперимента
1
2
3
4
5
6
7
8
9
10
11
12
13
14
КрХ
QF.jl
H(CF2)6Br
QF.jl
H(CF2)6I
QF.jJ
QF.-J
H(CF2)6I
QF„]
H(CF2)6Br
H(CF2)6I
C6F,j]
(CF,)2CF(CF2)I
H(CF2)6[
H(CF2)6I
Rj
Me
Me
Me
Me
Me
Me
Me
Me
H
H
H
Me
H
Me
Ri
Ph
Ph
Ph
Ph
Ph
Ph
Ph
Ph
AcO
Py
Py
Ph
Np
(4-Mc-Ph)
Продукт*
1
2
1(1 :0.15)
2(1:0.12)
1
1
2
1
3(1:0.4)
4
5
6(1:0.8)
7
8(0,23:1)
Катализатор
(10%)
(bpy)NiBr2
(bpy)NiBr2
(bpy)NiBr2
(bpy)NiBr2
(pybox)NiBr2
(tpy)NiBrj
(tpy')PtCl2
NiBr2
(bpy)NiBb
(tpy)NiBr,
(bpy)NiBrj
(bpy)NiBr2
(bpy)NiBr2
(bpy)NiBr3
Растворитель
DMF
DMF
CH.,CN
CH3CN
DMF
DMF
DMF
DM F: пиридин
(9:1)
DMF
DMF
DMF
DMF
DMF
DMF
Выход,
%
70
49
53
39
34
46
58
14
51
48
61
72
68
65
* В скобках указано соотношение диастереомерных форм (meso : dl).
оказался эффективным реагентом и привел к селективному образо-
ванию мономера1:
(bpy)NiBr3
Ph К) мол. %
ДМФА РЬ
48%
который, однако, трудно выделить в чистом виде из реакционной
смеси, что обусловило сравнительно невысокий выход продукта.
Таким образом, на основании полученных результатов по
каталитической активности выбранных комплексов никеля в реакциях
электровосстановления перфторалкилгалогенидов был разработан
и успешно реализован метод электрокаталитического фторалкили-
рования олефинов. Впервые были получены продукты реакции
присоединения-димеризации в отличие от известных случаев функци-
онализации олефинов, описанных в литературе. Была произведена
оценка влияния различных факторов на выходы продуктов реакции
и стереоселективность и установлено, что наиболее эффективным
катализатором изучаемых реакций при ведении синтеза в разделенном
электролизере с платиновыми катодом и анодом является комплекс
(bpy)NiBr3. С помощью комплекса физико-химических методов было
установлено, что возможно влиять на образование различных стерео-
изомерных форм, меняя структуры исходных субстратов —
перфторалкилгалогенидов и олефинов. Впервые удалось осуществить
контролируемое присоединение перфторалтеильных групп к олефинам
с образованием либо димерного, либо мономерного продукта.
Впервые выделены сигма-комплексы RFNiBrL (L = 4,4'-ди(трет-
бутил)-2,2-бипирин, 2t2': 6\2"-терпиридин; RF = CAF)2H, C5Fn)
как ключевые интермедиаты каталитического цикла и установлена
лигандо-центрированная радикальная природа продукта их
восстановления, объясняющая характер реакции перфторалкилирования
олефинов.
На основании данных ДВА-экспериментов, препаративного
электролиза, ЭПР и тл. предположена следующая схема
каталитического процесса, включающая стации окислительного присоединения
и восстановительного элиминирования, в которой активной формой
катализатора, генерируемой и регенерируемой на электроде,
выступает комплекс Ni'L, а ключевым перфторалкилируюпшм агентом
является о-комплекс RpNiL. проявляющий свойства лиганд-цент-
рированного радикала:
1 Mikhaylov D. К, Gryaznova Т., Dudkina К, Khrizanphorov A/., Latypov Sh., Ka-
taeva О.. VicicDA., Sinyashin O.G., Budnikova У. Op. ril.
221
NitTX:L-f с" — Ni'XL-l X-
Ni'XLH с"-— NinL + X-
Ni'l + Ni'%L — IS'i[XL + Br
RhKiX2L
Ph
Rp
V Ni'XL 0,5RF'
Ph'
Таким образом, совокупность методов препаративного
электросинтеза, ЭПР и циклической вольтамперометрии позволила получить
доказательства образования принципиально важных интермедиатов
каталитического цикла, установить отдельные стадии, приводящие
к желаемым продуктам перфторалкилирования, что в конечном итоге
позволило предположить схему каталитического цикла.
Показана возможность применения разработанного метода фтор-
ал килирования для функционализации более сложных субстратов,
содержащих ненасыщенные связи С-С Было обнаружено, что в
реакции электрокаталитического перфторалкилирования образуется
димерный продукт присоединения, что аналогично реакциям,
рассмотренным выше1.
+ H(CF2)6I 'T_('yNIBr; )
' +2е , Pt катод
кат. (bpy)NiBr2 ^ Н(Р3С)бч \ /
+2е~. Pt катод 7 \^-/гпг л и
О Н R (CFz)6H
R= J 1 <-CH,-DMIC)
Me
i
Дудкшш Ю.Б., Михаилов Д. Ю., Грязнова Т.В., ФаттахоеС.Г., Будникова Ю.Г..
СиняшинО.Г. (I Изв. РАН. Сер. хим. 2QI3. № 11. С 2362-2366.
222
Изоциануровые производные, обладающие биологической
активностью, а также содержащие фторалкилъные заместители,
полученные согласно этому методу, могут представлять практический
интерес.
Недавно предложен одностадийный каталитический метод
ароматического перфторалкилирования при участии комплексов
металлов, восстановливающихся и регенерирущихся электрохимически
в мягких условиях, причем при решающей роли иона металла
растворимого анода1. Кросс-сочетание протекает успешно с бром- и йод-
бензолом и перфторалкилйодидом под действием ряда комплексов
кобальта и никеля в низких степенях окисления:
PhBr+RrI MX^2F )Rp_Ph
Анод: Си, Zn, Mg M = Ni, Co
L: 2,2'-бипиридин; терпиридин
2,9-димстил-1,10-фснантролин
4,4'-ди-трет-бутил-2,2'-бипиридин
1,2-бис(дифенилфосфино)этан
6.11.2. Реакции фторалкилирования олефинов с использованием
иммобилизованного катализатора
Проблема «зеленого» и устойчивого развития требует создания
наиболее эффективных химических превращений с меньшими
выбросами в окружающую среду. Один из важнейших промышленных
недостатков реализации гомогенных каталитических реакций
заключается в сложности отделения катализатора от продукта и
повторного использования дорогих катализаторов, особенно когда
применяются благородные и/или токсичные комплексы металлов.
Эти проблемы создают значительные экологические и
экономические трудности в органическом синтезе. Считается, что перевод
гомогенного катализа в гетерогенный для превращения различных
органических субстратов является перспективным решением в
направлении «зеленого* и устойчивого развития химической
промышленности. Гетерогенирование существующих гомогенных Ni и Pd
катализаторов могло бы стать привлекательным решением этой
проблемы благодаря их легкому отделению и простому повторному
использованию.
1 Khrizanjbmv Л/., Gryaznova 71, Sinyashin О., Budnikova Y.//S, OtTganomci. Chcm.
2012.718, 101.
223
Так, недавно было реализовано Ni-катализируемое перфторал-
килирование олсфинов в электрохимических восстановительных
условиях с образованием как мономерных, так и димерных
продуктов в зависимости от реакционной среды. Реииклизаиия
катализатора может быть достигнута путем иммобилизации комплекса
(bpy) NiBr2 на силикатных наночастицах, покрытых закрепленными
аминогруппами. Переключение гомогенных и гетерогенных
катализаторов оказывается еще одним фактором, регулирующим
соотношение продуктов. Этот каталитический метод можно отнести к
области зеленой химии и считать атом-экономным, объединяющим
преимущества наногетерогенного катализа и электрокатализа1.
Переход от гомогенного катализа к гетерогенному возможен
за счет иммобилизации каталитически активных комплексов
никеля на силикатных наночастицах. Для закрепления никелевого
комплекса на поверхности силикатных наночастиц можно использовать
координационные связи или неспецифическую адсорбцию.
Координационная ненасыщенность и в то же время отсутствие заряда
комплекса никеля (0) обусловили выбор в пользу координационных
связей.
Для этого с помощью метода обратной микроэмульсии типа
«вода в масле» были получены силикатные наночастиды размером
35 ± 5 нм, а на их поверхность ввели амингруппы посредством
обработки частиц 3-[2-(2-аминоэтршамино)этиламино]пропилтриме-
токсисиланом {AEPTS}2. Оценка среднего количества введенных
аминогрупп была проведена по флуорсскаминовой методике. Она
показала наличие 3500 первичных аминогрупп на каждую наноча-
стицу. Схематичное изображение силикатных наночастиц до и после
модификации представлено на рис. 6.2.
Комплекс (bpy)NiBr2 был иммобилизован на частицах путем
смешения его водного раствора и водной дисперсии комплекса.
Количество иммобилизованного катализатора рассчитывалось из разницы
в концентрации комплекса никеля в растворах до и после
иммобилизации и составило 3700 молекул на каждую наночастицу, что сви-
дельствует о том, что практически все аминоэтиламино-этиламино-
пропил-группы на поверхности участвуют в координации комплекса
никеля. Размер частиц после модификации их поверхности и
иммобилизации катализатора составил 55 ± 5 нм (рис. 6.3)\
1 Dudkina Yu.B., Gryaznova T.V., Osin Yu.N., Salnikov V.V., Davydov N.A., Fe-
dorenko S.V., MustafmaA.R., Vicie D.A., Sinyashin O.G., Budnikova Yu.H. // Dallon
Trans. 2015. Vbl. 44. P. 8833-8838.
1 Balasz СВ., Anson F.C. // J. EleclroanaJ. Chem. 1993. 361. 149: Smith C.I., Cray-
stone J,A., Hay R. W. Op. cil.
J Dudkina Yu.B., Gryaznova Т.У., Osin Yu.N., Salnikov V.V.. Davydov S.A., Fe-
dorenko S. V., Mustafina A.R., Vicic D.A., Sinyashin O.G., Budnikova Yu.H. Op. cit.
224
но. „
Si'
о' о
О"
-o^ он
Si
-Л x0
НО
Si
\
?
HOSi-0
t
о f
-si
-О
Si-OH
о
но
Si
он
Si—
I
о
\
Si
b.9-\ 0,0'
Si-
OH
OH
о
OH
но о
o'sl он
\M, -
Рис. 6.2. Схематическое изображение силикатных наночастиц (Si02),
поверхность которых модифицирована аминогруппами (Si02-AEPTS)
Рис. 6.3. Изображение силикатных наночастиц с иммобилизованным
катализатором, полученное на сканирующем электронном микроскопе (SEM)
Переход от гомогенного к гетерогенному катализу
сопровождается изменением ряда условий реакции, а для сравнения
активности и селективности гомогенных и гетерогенных каталитических
реакций необходима оценка влияния соотношения исходных
реагентов, количества и фазового состояния катализатора, состава
реакционной среды и воды, вносимой наночастицами.
225
Каталитическая активность иммобилизованного комплекса
исследовалась в реакциях фгоралкилирования олефинов на примере
реакций а, 4-диметилстирола и а-метилстирола с 6-Я-нерфтор-
гексил йодидом, результаты которых приведены в табл. 6.12'.
Таблица 6.12
Реакции фторалкмлироаания олефинов в условиях
гомогенного и гетерогенного катализа
R
R = /j-Tol R=p-Tol
R=Me R=Me
№
реакции
1
2
3
4
5
6
7
8
9
10
11
R
/>-Tol
/>-Tol
/>-Tol
P-Tol
Me
Me
Me
Me
Me
Me
Me
Олефин:
H(CF2)ftI
I :1
I :1
I :1
1:1
1: 1
1 :1
1:1
1 :1
1: 1
1:1
1 :1
1 : 1
Реакционная
среда
ДМФА
ДМФА
ДМФА: Н20
(4:1)
ДМФА: Н20
(4:1)
ДМФА: Н20
(4:1)
ДМФА: Н20
(8:1)
ДМФА: Н20
(4:1)
ДМФА: Н20
(2:1)
н2о
ДМФА
ДМФА: Н20
(4:1)
Катализатор
(bpy)NiBr2{10%)
(bpy)NiBr2(l%)
(bpy)NiBr2(l%)
(bpy)NiBr2(1%)
на Si02-AEPTS
(bpy)NiBr2(l%)
на SiCb-AEPTS
(bpy)NiBr;(l$)
на Si02-AEPTS
(bpy)NiBr2
(1%)+добав;|.
Si02-AEPTS
(bpy)NiBr2
(1%) +добавь.
Si02-AEPTS
(bpy)NiBr2 (10%)
(bpy)NiBr2 (1%)
на Si02-AEPTS
(bpy)NiBr2(l%)
Ди-
мер/моно-
мср,1/2
1,0:0,0
1,0:0,0
1,0:0,8
1,0:0,45
3/4
1,0: 1,4
1,0:1,7
1,0:2,7
1,0:4,1
Выход,
%
71
60
48
44
45
48
46
29
—
1,0:0.8
1,0:3,8
53
44
1 Ibid.
226
Окончание табл. 6.12
№
реакции
12
13
14
R
Me
Мс
Me
Олефиы:
H(CF2)6I
I: I
1: 1
1 :4
Реакционная
среда
ДМФА: Н:0
(4:1)
CH,CN : Н20
(4:1)
ДМФА : Н,0
(4tl)
Катализатор
(bpy)NiBMIft),
BuNH2(lO%)
(bpy)NiBr2(l%)
на SiOrAEPTS
(bpy)NiBr1(10%)
Диме
p/мономер, 1/2
1,0:3,5
1.0: 1,1
1,0: 1,1
Выход,
%
48
41
94
Совместное электрохимическое восстановление олефина и фтор-
алкил галоген ила в среде безволного ДМФА в присутствии
каталитических количеств комплексов никеля приводит к образованию
продукта присоединения-димеризации. Так, при совместном
электровосстановлении а, 4-диметилстирола, 6-Н-перфгоргексил йодида
и 10% комплекса (bpy)NiBr2 образуется димерный продукт с выходом
71% (реакция 1). Уменьшение количества катализатора до 1% ведет
к уменьшению выхода на 10% (реакция 2).
В реакциях, проводимых в смесях ДМФА : вода, наблюдается
образование как димерных продуктов, так и мономерных (реакции
3—8, 11, 12, 14). Увеличение содержания воды в реакционной смеси
ведет к уменьшению суммарного выхода продуктов, однако
увеличивает долю мономерного продукта, что проиллюстрировано
реакциями 5, 6 (80 и 89% ДМФА соответственно) и 7, 8 (80 и 67%
ДМФА соответственно), В водной среде фторалкилирование
олефина не идет вообще (реакция 9), а оптимальное соотношение
ДМФА: вода составляет 4:1. Несмотря на то что в
водно-органических смесях выход продуктов ниже, использование воды для синтеза
мономерных продуктов оправданно, так как в данном случае вода
является наиболее доступной, удобной и экологичной альтернативой
трибутилгидриду олова, который использовался ранее1.
Снижение выхода продуктов в реакциях, проводимых в водно-
органических смесях, видимо, связано с частичной дезактивацией
водой активных форм катализатора, что описано2 для реакций
в протодонорных средах с участием комплексов никеля.
Повышения выхода продуктов можно добиться увеличением количества
исходного перфторалкил галогенида. Так, совместный электролиз
а-мстилстирола и 6-//-псрфторгсксил йодида в соотношении 1 : 4
1 Mikhaytov D.Y., Gryayiova Т., Dudkina У., Khrizanphorov M., Latypov Sh., Ka-
taeva О., Vtcic DA., Sinyashin O.G., Budnikova Y. Op. cit.
1 Derien S., ClmetJ.C, Dunach E. // J. Perichon. Tetrahedron. 1992.48. 5235.
227
и в присутствии 10% комплекса (Ьру)МВг2 в среде ДМФА: вода
(4 : 1) приводит к образованию продуктов в соотношении 1,0 : 1,1
с общим выходом 94% (реакции 14).
Анализ продуктов реакций, проводимых с гетерогенным и
гомогенным катализатором, показывает, что при прочих равных условиях
(соотношение ДМФА : вода, количество катали затора) фазовое
состояние катализатора влияет на соотношение мономерного и ди-
мерного продуктов, но не влияет на их общий выход (реакции 7
и 8): в реакциях с использованием иммобилизованного
катализатора доля мономера меньше. Однако мономер образуется в реакции
в сухом ДМФА с иммобилизованным катализатором (реакция 10),
но в меньших количествах, чем в водно-органической смеси
(реакции 5 и 6). Содержание мономера в реакции 3, в которой
использовался гомогенный катализатор и дополнительно вводились
модифицированные силикатные наночастицы, является
промежуточным между значениями, полученными в условиях «чисто»
гомогенного (реакция 11) и гетерогенного (реакция 5) катализа при
прочих равных условиях. Таким образом, иммобилизованный
катализатор способствует реакции димеризации. Возможное объяснение
этому — затрудненный доступ молекул воды к каталитическим
центрам из-за гидрофобных AEPTS-групп, на которых иммобилизован
никелевый комплекс, что приводит в случае иммобилизованного
катализатора к более низкой скорости образования мономерного
продукта. Из реакции 12, в которой в условиях гомогенного
катализа в реакционную смесь ввели бутиламин. следует, что присутствие
аминогрупп не оказывает влияния на каталитическую реакцию.
Таким образом, изданных, представленных в табл. 6.12, следует,
что иммобилизация катализатора является фактором, влияющим
на соотношение мономерного и димерного продуктов в реакции
электрокаталитического фторалкилирования олефинов. Это также
подтверждает роль координационных связей в иммобилизации
комплекса, а не его специфической адсорбции на наночастицах.
Внешнесферная координация никеля с AEPTS- группам и, которыми
модифицирована поверхность силикатных наночастиц, затрудняет
доступ воды к реакционным центрам, что является причиной,
влияющей на соотношение мономер : димер. Важно отметить, что
реакция фторалкилирования протекает на никелевых центрах, при
этом активной формой катализатора являются комплексы никеля
в низких степенях окисления (0, +1), а не частицы Ni(0) без лиганда.
На снимке частиц после каталитической реакции (рис. 6.4)
отсутствуют следы восстановления комплекса до частиц Ni(0).
Молекула димерного продукт симметрична и содержит два ас-
симетричных атома углерода, что делает возможным существование
такого соединения в форме трех изомеров: R, S (mcso-форма), R,
228
R и S, S (не различимы в спектрах 1Н Я MP» d, 1-форма). Согласно
данным 'Н ЯМР-спектроскопии оба димерных продукта
представлены meso- и d, 1-формами, соотношение которых составляет 1: 1
для всех исследуемых реакций (см, табл. 6,12) и не зависит от
состава среды и фазового состояния катализатора. Изомеры
соединения были выделены из реакционной смеси и охарактеризованы
методом РСА (рис. 6.5)'.
Рис. 6.4. Изображение силикатных наночастиц с иммобилизованным
катализатором после реакции фторалкилирования, полученное
на сканирующем электронном микроскопе (SEM)
Рис. 6.5. Молекулярная структура димерного продукта: изомеры R, S и R, R/S, S
Устойчивость иммобилизованого катализатора была оценена
комплексом физико-химических методов. Вымывание комплекса
с подложки не было зафиксировано при анализе промывных
растворов после иммобилизации катализатора методами электрохимии
(низший предел обнаружения — Ю-4 М). Элементный анализ
показал, что количество катализатора после реакции не уменьшается2.
1 Dudkina Yu.B., Gryaznova T.V., Osin Yu.N., Satnikov V.V., Davydov AL4.t Fe-
dorenko S. V., Mustafma A.R., Vicic D.A., Sinyashin O.C, Budnikova Yu.H. Op. cit.
2 Ibid.
229
Установлена возможность повторного использования
иммобилизованного катализатора в серии реакций фторалкилмрования а —
метилстирола (табл. 6.13).
Таблица 6.13
Рециклизация гетерогенного катализатора в реакции
перфторалкилрования сс-метилстирола
(У
(bpy)NiBr2
naSiOrAEPTS \%
2F/RFi
ДМФА : Н:0 j.
№ опыта
1
2
3
<bpy>NiBr2 на SiOj-AEPTS \%
2 F/Rpl
^ + H(CF2)6I ДМФЛ|Н3о
I(CF2)6 X ^^ +f T ^
Днмер/мономер
1,0:1,4
1,0:1,1
1,0:0,8
Выход, %
45
42
42
После каждой реакции катализатор отделялся
центрифугированием, промывался водно-органической смесью (ДМФА : вода —
4 :1), диспергировался в небольшом количестве воды и в таком виде
вносился в следующую реакцию. Трехкратное использование такого
катализатора не ведет к снижению выхода продуктов фторалкили-
рования, гетерогенный катализатор сохраняет свою активность1.
Также следует отметить, что количество пропущенного
электричества не влияет на соотношение мономерного и димерного продуктов,
а их общий выход не увеличивается при пропускании через
реакционную смесь больше 2 Ф электричества на I моль перфторалкштга-
логеница.
Таким образом, удалось успешно реализовать реакцию перфтор-
алкилирования С<5р2)-Н-связей олефинов, катализируемую
комплексом никеля, иммобилизованным на силикатных наночастицах
с закрепленными аминогруппами. Обнаружено, что соотношение
мономерных и димерных продуктов зависит от реакционной среды.
Иммобилизация катализатора позволяет осуществлять его рецикли-
зацию без потерь активности. Этот каталитический метод является
Dudkina Yu.B., Gryaznova Т. У., Osin Yu.N., Salnikov V. У.. Dav)-dov N.A., Fe-
dorenko S. У., MusiafinaA.fi., Vicic D.A., Sinyashin O.G., Budnikova Yu.tJ. Op. cit.
230
экологически приемлемым, поскольку отвечает принципам зеленой
химии и атом-экономным, а также объединяет в себе преимущества
наногетерогенного катализа и электрохимического катализа.
6.12. НЕКОТОРЫЕ ДРУГИЕ СЛУЧАИ ЭЛЕКТРОКАТАЛИЗА
С УЧАСТИЕМ МЕТАЛЛОКОМПЛЕКСОВ
Селективное восстановление нитросоединений в аминопро-
изводные осуществлено с медиатором — комплексом СргТл* в 1 Н
водной серной кислоте1. Непрямой электролиз различных
нитробензолов, орто-замешенных эфирной (OCOR), карбонатной (OC02R),
амидной <N HC02R) группами, проводился в дихлорометане:
6 Ср2ТЮН+ + 6с + 6Н+ > 6 Cp2Ti+ + 6Н20
6 Ср2ТГ + ArN02 + 4H2G > 6 Cp2TiOH+ + ArNH2
Перегруппировка анилинов, орто-замещенных эфирной или
карбонатной группой, в N-ацилированный о-аминофенол протекает in
situ. N-аиилированные о-фенилендиамины превращаются в 2-заме-
щенные бензимидазолы при кипячении в уксусной кислоте:
О
II
s*OC —СХ
О-С—R ^^0-С—R ^^)Н
II II
О О
I II I II
но но
В последние годы была разработан принцип непрямой
анаэробной электрохимической генерации галактозы оксидазы
(GOase)3, GOase окисляет множество первичных спиртов в
соответствующие альдегиды. Синтетическое применение GOase интересно
из-за ее высокой субстратной чувствительности, способности кхе-
моселективному окислению первичных гидрокси-групп в полиолы
1 Jan Т., Лопег £>., Moinel С. // Electrochimica Acta, 1997.42, 2073.
2 Petersen A., Stekhan E. // Bioorhank&Medicinal Chemistry. 1999. 7. 2203.
231
и его стереоселективности. Электрохимическая регенерация GOase
позволяет предотвратить нежелательное образование ингибитора
энзима — пероксида водорода, который образуется при аэробной
генерации в синтезе с GOase. Схема непрямой электрохимической
генерации галактозы оксидазы приведена ниже:
Srcd-
Pltt-
■ч. s~ G0aseo4 *v ^*> Mreti -^
^^*GOase,>,-^^- J
-*-> Fe" -^
Vlc>^_
-^->
Анод
0
+
S — субстрат, Р — продукт,
M — медиатор (производное ферроцена).
В качестве медиатора использовались замешенные ферроцены,
которые проявили высокую каталитическую активность. Другие
комплексы железа(II) и рутения(П) с фенантролиновыми лигандами
оказались менее эффективны. Оптимальными условиями процесса
являются фосфотные буферы с рН 10>8.
6.13. ЗАКОНОМЕРНОСТИ И МЕХАНИЗМЫ РЕАКЦИЙ,
КАТАЛИЗИРУЕМЫХ КОМПЛЕКСАМИ НИКЕЛЯ И ПАЛЛАДИЯ
6.13.1. Кинетические закономерности
Использование электрохимического подхода для генерации
комплексов Ni(0) и других металлов в низких степенях окисления
открывает некоторые перспективы. Во-первых, это возможность
использования каталитического количества комплекса, который будет
регененироваться на электроде и таким образом перерабатывать
значительные количества различных субстратов. Во-вторых, с помощью
электрохимического подхода можно провести количественную
оценку скорости каталитической реакции, а в ряде случаев удается
расчленить суммарный процесс на отдельные стадии и оценить
реакционную способность интермедиатов, устойчивость различных
форм металлокомплекса. По внешним морфологическим признакам
можно выделить три качественно различных случая каталитических
232
волн в системе Ni2+/Ni°, L (L — PPh3, 2,2-bipy) (рис. 6.6)'. Это, во-
первых, прирост тока восстановления исходного комплекса Ni2+L„
(рис. 6.6, а), во-вторых, волна, отвечающая восстановлению более
сложного комплекса, включающего субстрат как лжанд (рис. 6.6, б),
и, в-трстьих, волна каталитического восстановления мсталлооргани-
ческого соединения — продукта взаимодействия восстановленной
формы исходного комплекса с субстратом (рис. 6.6, в). Наблюдаемую
картину с некоторыми допущениями во всех этих случаях можно
считать классической и использовать прирост тока (/*//<*) для
расчета ктф — константы скорости регенерации катализатора, которая
описывает суммарный процесс, включающий ряд последовательных
стадий2;
Ni(O)(bipy)2^=±Ni(0)(bipy)+bipy
Ni(0)(bipy)+ RHal^==i продукты
, _кхк2 [RHall
Рис. 6.6. Морфология каталитических волн в системе NP+L/Ni°L:
1 — комплекс NI"L, 2 — комплекс Ni"L в присутствии увеличивающихся количеств
органических галогенидов
Отношение угловых коэффициентов а в уравнениях к^ = оСта + Ь
в различных растворителях совпадает с обратным отношением их до-
1 Budnikova Yu.H.t Yakhvarov D.G., Margin Yu.M. Op. cit.; Будпикоеа Ю.Г.,
Юсупов A.M., Каргин Ю.М. Указ. соч.; Troupet М., Ratlin К, SockO., Meyer С,
Perichon J. // Nov J. Chim. 1986. LO. 593; Бутин К.П., Магдесиева ТВ.,
Реутов ОА. if Металлоорг. Хим 1990. 3. 534; ПоздеееаАЛ., Поподько /АЛ, 7а;-
стиков ГЛ., Жданов СМ., Игошкина Г.С., Джемилев У.М. // Изв. АН СССР.
Сер. хим. 1980. 1547.
2 Будпикоеа Ю.Г., Юсупов A.M., Каргин Ю.М. Указ. соч.
233
норных чисел. Чем ниже донорное число, тем круче наклон графика
к оси абсцисс и тем эффективнее каталитический процесс1.
Комплекс никеля с bipy оказался наиболее эффективным и универ-
сальным катштизатором — значительный каталитический эффект
наблюдался с разными субстратами — со связями C-Hal, P-CI и Si-CR
6.13.2. Связь электрохимических редокс-свойств комплексов никеля
с их реакционной способностью в реакциях дегалогенирования
Редокс-потенциалы комплексов использованы в качестве
исходной характеристики для прогнозирования реакционной
способности и механизма реакций металлоорганических соединений3.
Экспериментально оценены значения электрохимической щели
G = £о* — ^Кс<ь электрохимической отрицательности % = (£"0* +
+ £rc<j)/2 и степени переноса заряда AN= &%/TJG для ряда изучаемых
комплексов Ni(II) и Ni(0)4.
Параметры С, % и Д^ характеризующие комплекс Ni<0) с
различными лигандами, можно использовать для оценки реакционной
способности этого комплекса в реакциях окислительного
присоединения к органическим галогенидам (субстратам):
Ni(0)L„ + RHal -> R-Ni(II)HalL„
Эти параметры пригодны также для оценки взаимодействий
комплекса и различных лигандов и субстратов для получения
информации о комплексообразующих свойствах, донорно-акцеп-
торных взаимодействиях комплекс — субстрат. Это особенно важно
по отношению к фоефорорганическим субстратам, проявляющим
о-донорные и л-акцепторные свойства при взаимодействии с
переходными металлами. Наибольшее значение электрохимической
щели, характеризующей его степень «жесткости», имеет комплекс
Ni(II) с Ыру\ Для фосфиновых и фосфитных комплексов Ni(ll)C
1 Будникова Ю.Г., Юсупов A.M., Каргин Ю. М. Указ. соч.
2 Budnikova Yu.H., Yakhvarov D.G., Kargin Yu.M. // J. Organomet. Chem. 1997.
536—537. 265; Будникова Ю.Г., Юсупов A.M., Карги» Ю.М. Указ. соч.
J Budnikova Yu.H., Yakhvarov D.G., Kargin Yu.M. // J. Organomet. Chem. 1997.
536—537. 265; Будникова Ю.Г., Петрухина О.Е., Гудина Н.И.. Каргин Ю.М.
Указ. соч.; Будникова ЮГ.. Каргин ЮМ. // 3. Organomet. Chem. 1997.
536—537. 265; Будникова ЮГ., Юсупов A.M., Каргин A.M. Указ. соч.;
Budnikova Уи.Н.Жагцт Yu.M. //Жури. обш. Химии. 1995. 65. 1655.
4 Budnikova Yu.H., Yakhvarov D.G., Kargin Yu.M. // J. Organomet. Chem. 1997.
536—537. 265; Будникова ЮГ, Петрухина О.Е.. Гудина Н.Н., Каргин ЮМ.
Указ. соч.; Будникова Ю.Г., Каргин Ю.М. //J. Organomel. Chem. 1997. 536—
537. 265; Будникова Ю.Г., Юсупов А.М.+ Каргин А.М. Указ. соч.
- Budnikova Yu.H.. Yakhvarov D.G., Kargin Yu.M. // J. Organomet. Chem. 1997.
536—537. 265; Будникова Ю.Г., Петрухина О.Е., Каргин Ю.М. // Журн. обш.
234
заметно меньше. Наоборот, наименьшее значение
электрохимической щели, а следовательно, наибольшая донорная способность
и поляризуемость характерны для комплексов Ni(0) с bipy (0,60 В),
затем следует Ni(0) с phen (0,72 В) и с Ph3P (1,04 В). На основании
этих характеристик были рассчитаны значения степени переноса
заряда между комплексами и субстратами, исходя из предположения,
что комплекс Ni(H) или Ni(0) — донор, а субстрат — акцептор
электронов1. Степень переноса заряда с комплексами Ni(II) — субстрат
невелика, а с комплексами Ni(0) — больше на порядок, что
предполагает протекание в последнем случае реакций окислительного
присоединения. Знак, степени переноса заряда показывает, что комплекс
Ni(0) со всеми субстратами проявляет ярко выраженные донорные
свойства {AN >> 0).
Данные вольтамперометрических измерений, квантово-хими-
ческие расчеты и оценочные значения С, % и ДМ использованы для
объяснения наблюдаемой реакционной способности комплексов
никеля в каталитических процессах2. Реакционная способность
комплексов N1(0) в реакциях восстановления галогенорганических
соединений связана с рядом факторов — величиной
электрохимической щели G, степенью переноса заряда AN, прочностью связи Ni—P,
электронными и стерическими эффектами лигандов, наличием или
отсутствием последующих редокс-реакций. В ряду медиаторон-
восстановителей Ni(0)L, Ni(0)L~", L~" (L = bipy, phen) наблюдается
уменьшение энергии стабилизации переходного состояния при
переходе от внутри- к внсшнссфсрному процессу восстановления,
сопровождающееся соответствующим уменьшением скорости реакции
по сравнению с внутрисферным медиатором с той же движущей
силой процесса1.
Детальные представления о механизме двухэлектронного
восстановления комплекса с учетом конкурирующих гомогенных редокс-
реакций, доказательство тех или иных стадий, а также определение
лимитирующих процессов полезны для оценки вероятных
каталитических свойств интермедиатов по отношению к тем или иным
субстратам, поэтому им было уделено особое внимание4.
Химии. 1996. 66. 610; Будникова Ю.Г.. Каргин Ю.М. // J. Organomet. Chem.
1997, 536-537. 265; Будникова Ю.Г., Юсупов A.M., Каргин A.M. Ука*. соч.
1 Там же.
1 Там же.
3 Будникова Ю.Г., Петрухина О.Е., Каргин Ю.М. f/ Журн. общ. химии. 1996. 66.
1876.
4 Будникова Ю.Г., Петрухина О.Е.. Гудина Н.Н., Каргин Ю.М. Указ. соч.;
Daniels S„ BontempeUi С, Magna F., Fiorani M. // Ann. chim. 1988. 78.363; Daniele$.,
Ugo A, BontempellG.L Magna F. // Ann. chim. 1988. 78. 555; BontempeUi G,
Magno /'., Schiavon G., Corain B. // Inorg. Chem. 1981. 20. 2579.
235
При восстановлении комплекса никеля(Н) с Ыру лимитирующей
стадией является перенос второго электрона (Д£° = Е\ - Е$, где £|°
и t2 — стандартные потенциалы переноса первого и второго
электрона, соответственно) порядка —60 -s- —70 мВ, а конкурирующие
редокс-реакиии отсутствуют. При восстановлении комплекса
никеля(И) с phen лимитирующей стадией является перенос первого
электрона (Д£° = -30 +- -60 мВ), а конкурирующие редокс-реакиии
также не наблюдались. Восстановление комплекса Ni(ll) с PPh*
и фосфитами (i-PrO^P, PhP(OBu)2, (PhO)^P лимитируется стадией
переноса первого электрона и сопровождается химическими
реакциями компропорционирования (PPhj, А£° - 90 мВ) и диспропор-
ционирования (фосфиты. Д£° < 0).
Редокс-ре акции диспропорционирования и
компропорционирования характерны для мсталлокомплексов Ni(II) с большей
поляризуемостью, мягкостью, связанными с небольшими значениями
электрохимической щели (фосфиновые и фосфитные комплексы).
Протекание конкурирующих реакций должно неблагоприятно
сказываться на каталитической эффективности этих комплексов в
реакциях дегалогенирования, поскольку концентрация активной формы
катализатора в реакционной зоне падает, причем тем быстрее, чем
выше скорость этих редокс-реакций. Действительно, было показано,
что наибольшей каталитической эффективностью среди изучаемых
комплексов обладает комплекс с Ыру, при восстановлении которого
конкурирующие редокс-реакции отсутствуют. Несомненно, этот
фактор повышает реакционную способность комплекса с phen,
однако она ниже, чем с Ыру, т.е. необходимо учесть и другие факторы.
В ряду фосфиновых и фосфитных комплексов никеля
каталитическая эффективность будет определяться соотношением скоростей
отдельных стадий общего процесса — целевых, идущих в
направлении образования конечного желаемого продукта, и
конкурирующих1.
6.13.3. Механизмы реакций, катализируемых комплексами
никеля и палладия
Синтетическая значимость процессов, катализируемых
электрохимически генерируемыми комплексами металлов в низких
степенях окисления, требует развития теоретических основ этого
метода, понимания их механизмов для целенаправленного выбора
катализаторов и условий, прогнозирования результатов и управления
реакциями.
Синтез органических соединений, катализируемый в гомогенной
фазе комплексами переходных металлов, протекает по крайней мере
1 Будиикова Ю.Г., Петрухина О.Е., Гудшш Н.Н., Каргин Ю. М. Указ. соч.
236
через один окислительно-восстановительный процесс, при
котором происходит активация одного из реагентов1. Окислительное
присоединение играет ключевую роль в конверсии органического
субстрата в высокореакционноспособный интермедиат во многих
процессах, катализируемых мсталлокомплсксами2. Его механизм
описан в литературе3. При функционализации органилгалогенидов
(RX) в процессе окислительного присоединения к нульвалентному
(d10) металлокомплексу (М) происходит образование металлоорга-
нического производного:
R
M(d10) + RX > M(du')
X
Что касается механизма данного процесса, то для него были
предложены два его основных типа. Первый включает как трехцентровое
присоединение4, так и SN2-3aMeiueHHe5, второй — многостадийный
процесс, протекающей с образованием парамагнитных интермеди-
атов6. Так, наблюдаемая селективность в реакциях полигалогенидов7
свидетельствует, что механизм процесса в данном случае аналогичен
хорошо известному нуклеофильному ароматическому замещению8,
а образование парамагнитных комплексов никеля(Г) и продуктов,
образованных радикальным путем в реакциях некоторых арилгало-
генидов с комплексами Ni°, допускает возможность реализации этого
процесса по второму типу.
Цау и Кочи4 предложили радикально-цепной механизм,
включающий промежуточные парамагнитные никелевые комплексы,
чтобы объяснить образование частиц диарилникеля(Ш) в
каталитическом цикле при стехиометрических условиях по
нульвалентному никелю. В своих работах они показали, что окислительное
1 Jolly P. W., Wilke G. The organic chemistry of nickel, Part 2. Organic sin thesis. N. Y:
Acad. Press, 1975. \o\. 2; J.K. KochiJ.K. Organometallic mechanisms and catalysis.
N.Y.: Acad, Press, 1978.
2 KochiJ.K. Op. с it.
3 KochiJ.K. Op. cit.; Fahey D.R.f Mahan J.E. //J. Am. Chem. Soc. [977. 99,. 2501;
Foa M„ Cassar L. // J. Chem. Soc. Dalton. Trans. 1975. 2572; Hidai M., Kashi-
wagi 7"., Ikeuchi Т., Uchida К //J. Organomet. Chem. 1971. 30. 279; Fahey D.R.,
Mahan J. E. 11 J. Am. Chem. Soc. 1976.98. 4499.
4 HarrodJ.F.. Smith C.A., Than KA. (j J. Am. Chem. Soc. 1972. 94. 8321.
5 Cullman J.P., MacLaury M.R. //J. Am. Chem. Soc. 1974. 96. 3019.
* Kramer A. V., Osborn J.A. // J. Am. Chem. Soc. 1974. 96. 7832.
7 Fahey D R. // J. Am. Chem. Soc. 1970. 92.402.
B Fitton P., Rick E.A. // J. Organomet. Chem. 1991. 28. 287; Semmelhack M.F.,
Ryono L. // Tetrahedron Lett. 1993. 34. 2967,
9 Tsou Т. Т.. KochiJ.K. //J. Am. Chem. Soc. 1979. 101. 6319.
237
присоединение арилгалогенидоп к комплексу трифенилфосфинони-
келя(О) приводит к галогс1Гидутранс-арилттокеля( 111) (А) совместно
с парамагнитным галогенидом никеля(1) (В) как побочным
продуктом:
Ni(0)L4 + АгХ -» ArNi(II)XL2 + Ni(I)XL3
А В
nieL = PEt3,X = Cl, Br, I.
Причем соотношение выходов А/В сильно зависит от природы
галогснида (I < Br < CI), нуклсофильности заместителей и
полярности растворителя. Общая схема окислительного присоединения
представлена следующим образом:
Ni(0)L4^^Ni(0)L3+L
M(0)L, + АгХ—^->[Ni(I)LjArX£]
^ArNi(II)XL2+L
|Ni([)L3ArX"] *Q_
dilTus>Ni([)L3 + X-+Ar'...
Однако предложенный механизм не дает точного
представления о последовательности стадий, приводящих к комплексу
диарилникеля(Ш) из исходного ArNi(II)XL„ в условиях
каталитического цикла, т.е. в присутствии восстановленного металла.
Использование электроаналитических методов исследования
совместно с препаративным электросинтезом позволило Аматоре1,
Бонтемпелли2, Перишотгу3 и Каргииу4 во многих случаях определить
ключевые стадии элсктрокаталитических реакций.
Одним из первых был исследован каталитический процесс
сочетания аллильных галогенидов, который протекаете весьма высокой
скоростью и осуществляется по следующей схеме5:
Amatore С» Carre Е., Juland А., Тапака И., Torii S., Chiarotto Г, СагеШ I. Op.
cit.; Amatore С, Juland A.. Motrier L. // J. Elect roanal. Chem. 1991. 306. 141;
Amatore C, Juland A. // Acta Chem. Scand. 1990. 44. 755; Amatore C, Azzabi A/.,
Juland A. Op. cit.
Schiavon G., BontempettiG., De Nobiti M., Corain B. // Inoig. Chim. Acta. 1980.42.
211; Bontempelii G.. Danieie S., Fiarani M. // J. Electroanal. Chem. 1984. 160. 249;
Bontempelii G. Danieie J., Fforani M. // Алп. Chim. 1985, 75. 19; Bontempelii G.,
Magna F., Corain B„ Sdiiavon G. Op. cit.
Nedelec J. Y., Perichon J., Troupe! M. Op. cit.; Durandetti M., Devaud Д/., Periehon J.
Op. cit.; CannesC, Labbe£., DurandettiM., Devaud A/., Nedelec J.-Y. Op. cit.
Будникоеа ЮГ., Кешнер ТД„ Кареин ЮМ. Указ. соч.; Budnikova УЛ.,
Perichon J., Yakhvarov D.G., Kargin УМ., Sinyashin O.G. Op. cit.; Еудникова Ю.Г.,
Каргин ЮМ. // Журн. общ. химии. 2001. 71. 140.
Bontempelii G., Danieie S., Fiorani M. // Ann. Chim. 1985. 75. 19.
238
[NiHL2S4)
-3L
В данном случае катализатор — высокореакционноспособный
комплекс никеля(О) с трифенилфосфином генерируется in situ при
электрохимическом восстановлении соответствующего комплекса
Ni11, который стабилен и может быть приготовлен анодным
окислением никеля в присутствии соответствующего количества PPh3'.
Комплекс Ni°(PPh3)4 вступает в реакцию окислительного
присоединения с аллилгалогснидом, присутствующим в растворе. Это
присоединение приводит к функтпгонализации связи С-На! с
образованием производного а-аллил-никель(1),, в котором аллилъная группа
прямо связана с атомом никеля.
Этот аддукт может быть восстановлен при потенциале (н схеме Е2)
более отрицательном, чем потенциал восстановления самого
комплекса (£|), но более положительном, чем потенциал восстановления
самого субстрата (аллилгалогенида) (порядка -1,8 В отн. НКЭ). При
этом электрохимическом восстановлении происходит разрыв связи
никель-углерод и отделение аллильной группы, дальнейшее
превращение которой приводит к образованию продукта сочетания
(1,5-гексадиену). В то же время степень окисления металла
становится равной нулю, что позволяет ему вновь вступать в
каталитический цикл.
Из приведенного механизма следует, что 1,5-гексадиен может
быть синтезирован из адлилгалогенидов при электрохимическом
процессе при использовании каталитических количеств трифенил-
фосфино-никелевого комплекса. Потенциал рабочего электрода
должен быть равен или чуть меньше чем £2 (менее —1,25 В отн.
НКЭ). Эксперименты, проведенные при контролируемом
потенциале, и хроматографический анализ подтвердили эти
механистические аспекты.
Shiavon G.t Bontempelli G., Corain В. Op. cit.; Bontempelli G., Daniele S.t Fio-
rani M. //J. Electroanal, Chem, 1984. 160. 249; Bontempelli G., Daniele 5,T Па-
rani M. //Ann. Chim. 1985. 75. 19; BontempelliG., Magno F., Corain В., Schi-
avon G.//1. Electroanal. Chem. 1979. 103. 243.
239
Подобный электрокаталитический цикл был аналогично
получен для синтеза 1,5-гексадиена из аллилбромида в присутствии
комплексов никеля с 2,2-бипиридилом1. а также из алл ил хлорида
под действием электрохимически генерируемого комплекса кобальта
с 2,2-бипиридилом2. Однако в этих случаях за счет быстрого
разложения катализатора не удалось получить большого числа
эффективных циклов.
В первых работах, проведенных в 1980-е гг., была сделана
попытка обобщить представления о механизмах реагирования
различных органических галогенидов в условиях гомогенного катализа
комплексами Ni(0) с фосфиновыми лигандами^ В этих системах
возможность эффективного электрохимического процесса
обусловлена следующими факторами — понижением требуемой
энергии активации восстановления RX и циклической
регенерацией катализатора. Снижение энергии активации и восстановление
RX при менее отрицательных потенциалах (|£"их|> -Ящшт.])
изменяет путь реакции по сравнению с некаталитическим. Гетерогенное
восстановление RX протекает при значительном перенапряжении
и высоких катодных потенциалах, так что радикал, образующийся
при переносе первого электрона, восстана&тивается до карбаниона
и соответствующего углеводорода RH и не дает продуктов
сочетания.
Было показано, что во всех случаях, несмотря на различное
вольтамттерометрическое поведение комплекса NKPPlvjhXi в
присутствии RX, восстановление может протекать через ряд простых
стадий, среди которых ключевыми являются:
— электрохимическая стадия восстановления комплекса никеля
доМ(О);
— стадия активации органического галогенида через его
окислительное присоединение к комплексу Ni(0);
— стадия восстановительного элиминирования с растеплением
связи металл—углерод (либо термическое разложение, либо
дальнейшее катодное восстановление).
Следует отметить, что зафиксировать или выделить интерме-
диаты цикла авторам не удалось, поэтому, несмотря на общие верные
предположения, описание отдельных стадий не было обосновано.
Однако это единственная работа, рассматривающая и обобщающая
поведение всех классов органических галогенидов. Все возможные
химические и электрохимические стадии можно представить
следующим образом:
1 BontempeUi G.. Daniele S., Fiorani M. Op. cit.
2 Margel S., Anson F. С // J. Elec trochem. Soc. 1978. 125. 123 2.
J Shiavon G., BontempeUiG., Coram £. Op. cit.
240
[Ni"L2S4l2+
j+2e-
-* [NiuL,] *-
[Ni'LaXJ
алкан
олефин
IH2
I
RX
-X
A T В
[NinL2HX] + олефин + [Ni^RX] —* [Ni'L3X] + R'
J
Б е
■v^
X"+ [Ni0L|] + 1/2R-R — [Ni^RX]
I e
2e
*[Ni°L3R]
RX
I
fNiXl + R-R + X"«— [Ni,lL2S2] + X
R-R
R(-H)
RH
L = PPh3, S = CH3CN, X = I, BrT CI; R = Alk или Аг
Путь Б реализуется, когда а-органоникелевый комплекс
[NiL3R(X)J достаточно стабилен, как в случае PhBr и BzCI, при
низких температурах. Совершенно верно отмечено, что для
осуществления каталитического процесса в этих случаях необходимо
восстановление этих интермедиатов, однако авторы ошибочно
предполагали, что восстановительное элиминирование можно
индуцировать только на катоде при более отрицательных
потенциалах по сравнению с потенциалом пары Ni(II)/Ni(0). В качестве
восстановителя, как было показано позднее1, легко выступает
комплекс Ni(0), а каталитический процесс реализуется при iT\ii<ii)/Ni<o>-
Путь С характерен для неопентил бромида и бензил иодида при
температуре 25"С. Продукт сочетания образуется путем димери-
зации радикалов, получаемых при гемолитическом расщеплении
связи никель—утлерод в [NiL2R(X)J, Радикальный распад
определяет распределение продуктов. Путь А рализуется для бутил
бромида. В этом случае термическое разложение иикельорганического
интермедиата осуществляется по реакции р-элиминирования,
включающей в качестве промежуточного продукта гидридный
комплекс никеля. Для гомосочетания необходимо направить
реакцию по пути Б, выбрав более отрицательный рабочий
потенциал, а распределение продуктов будет определяться
конкуренцией путей А и Б.
Наиболее медленной стадией электрокаталитических циклов,
рассмотренных ранее, является окислительное присоединение
органического галогенида к комплексу N1(0). Следовательно,
приросты каталитического тока позволяют сравнить скорости этих
реакций. Предложена следующая последовательность для RX:
Bui > (CH3)jCCH2l > PhCH2Cl > PhBr, которая совпадаете их рядом
дипольных моментов.
Особое внимание в изучении механизмов каталитических
процессов, катализируемых металлокомттлексами, было уделено
реакциям сочетания ароматических галогенидов.
Электроаналитические методы позволяют получить некоторую
информацию о механизмах реакции. В общем случае на катоде
образуется комплекс нульвалентного никеля или палладия, когда
в качестве лиганда выступают PPh3, dppe или bipy. Первой стадией
каталитического цикла является окислительное присоединение
органического галогенида к нульвалентному комплексу, приводящее
к а-арилникелевому или а-арилпалладиевому интермедиату.
Оказывается, что координационно ненасыщенные соединения типа
1 Gosmini С, Lasry S„ Nedelec J.- Y.. Perkhon J. // Tetrahedron. 1998. 54. I2S9;
Durandetti Д/„ Devaud M., Perichon J. // New. J. Chem. 1996. 20. 659; Cannes C,
Labbe £., Durandetti Д/., DevaudM., Nedelec J.-Y. // J. Elcciroanal. Chem. 1996.
412, 85; Fahey D.R., Baldwin B.A. ff Inorganica Chim. Acta. 1979. 36. 269.
242
Ni°bipy', Ni°dppe2 или Pd°{PPh^)i3 намного более реакционноспо-
собны, чем соответствующие полностью координированные
комплексы. Реакционная способность частиц Ni° или Pd^ также зависит
от природы и концентрации галогенид-ионов, связанных с исходным
комплексом или добавленных в реакционную среду4. Были
предложены некоторые пути реакций, начиная с а-металлооргатшческих
интермедиатов. Детальное исследование образования бифенила
из бромбензола, катализируемого Ni-dppe, позволило предложить
каталитический цикл, в котором о-арилникелсвый интермедиат
сначала восстанавливается в соответствующий ArNi1, а затем
трансформируется в комплекс диарил-никель(П1):
АгХ е
l05M-'-c-<V^ Аг№»Х^|,8В
.J+e, ™ T ArNi'
103М
АгХ
Этот комплекс затем подвергается восстановительному
элиминированию. В результате он образует соответствующий продукт и Nil
с последующей регенерацией системы Ni05. Таким обраюм, реакция
гомосочетания протекает через промежуточное образование
парамагнитных (Ni1 и Ni"1) и диамагнитных (Ni° и Ni") частиц. Несмотря
на существование концептуальных аналогий между
электрохимическим подходом и классическим восстановлением пол действием
металлов, электрохимия демонстрирует здесь преимущества с точки
зрения возможности более глубокого понимания механизма. Так,
было ясно продемонстрировано существование порогового
потенциала (—2 В отн. НКЭ), необходимого для образования бифенила.
При менее катодном потенциале продуктом является фенилтгике-
левое производное Ph—Nill(dppe)Br. Для реализации циклического
1 Troupe! M„ Roiiin К, Sock О., Meyer <#., Perichon J. // Nov. J. Chim. 1986.10.593.
2 Атшоге С, Jutand A. // Organomelallics. 1988. 7. 2203.
J AmatoreC, Azzabi M., Jutand A. //J. Amer.Cliem. Soc. 1991. 113. 8375.
4 Hoherg //.. SchaeferD., BurkhartC, KriigerC, Roman M. //J. Oiganomet. Chem.
1984.266, 203; Oca/rain Af. DevaudA/., NedekcJ, Y., Troupe!M, //J. Oisanomet,
Chem. 1998.560. 103.
* С Amatore. A. Jutand. // Organometallics. 1988. 7. 2203; Amatore C, Jutand A.,
MottkrL. // J. Electroanal. Chem. 1991. 306. 125.
243
процесса необходимо далее восстановить его. Следует отметить, что
схема, предложенная на основании вольтамперограмм, не была
подтверждена экспериментально — ни один из постулируемых интер-
медиатов не был выделен или зафиксирован. Поскольку диффузия
молекул к поверхности электрода легко моделируется, были
выведены теоретические уравнения, описывающие кинетические
закономерности предложенной последовательности реакций и рассчитаны
значения констант скоростей различных стадий. Последняя стадия
восстановительного элиминирования является лимитирующей.
Интересный факт наблюдается при введении в реакционную
систему СОэ1. Присутствие С02 полностью подавляет реакцию
образования бифенила и порождает новую каталитическую цепь,
приводящую к продуктам карбоксилирования.
Ar-COiNi1^ ArNi(TI)XL2
Ar-NimU ArNi(l)Li
JtcoI=l-210"5M-1*c-1
C02
Выводы о механизме превращений были сделаны не только на
основе изучения продуктов реакции и вольтамперометрического
поведения исходных комплексов в присутствии субстрата, но и путем
исследования поведения промежуточных соединении и кинетических
расчетов отдельных стадий процесса. То, что присоединение СОг
к ArNi(I)L происходит примерно в 100—200 раз быстрее, чем
присоединение к АгХ. объясняет, почему направление реакции с
образованием биарилов в присутствии стехиометрических количеств С02
полностью исключается2.
По аналогии с каталитическими реакциями Ni-dppe были
предложены два гипотетических цикла для комплекса Ni-bipy,
включающие образование ст-арилникелевого интермедиата. В первом
Amatore С, Jutand A., Mottier L. // J. Eleetroanal. Chem. 1991. 306. 141; Ллш-
tore C, JutantlA, //Acta Chem. Scand 1990.44. 755.
Amatore С Jutand A,, Mottier I, // J, Electroanal. Chem. 1991. 306. 141;
Amatore C, Jutand A. // Acta Chem. Scand. 1990. 44. 755; Nedeiec J.-Y., Perichon J.,
Troupe! A/. //J. Ekctrochem. Soc. 1990. 137. 150c.
244
цикле (с левой стороны) Ni'X, который образуется в стадии
восстановительного элиминирования, диспропорционирует на Ni° и Nin
аналогично циклу с dppe-лигандом. За этим процессом следует
дальнейшее восстановление Ni"1. Также был предложен
альтернативный механизм, в котором биарил образуется в ходе мстатезиса
о-арилникелевого(И) комплекса с регенерацией Ni" (правая часть
схемы — см. ниже)2:
Ni"«Ni°
АгХ
Ar2Ni
Ar—Ar
Аг-Аг
АгХ
Ni°—Ni"
Электроаналитические методы позволили изучить реакционную
способность комплекса никеля(О) с 2,2-bipy по отношению к
монооксиду углерода и органических галогенидов в конкурентных
условиях и в результате предложить новый метод синтеза кетонов3.
На основе кинетических данных были предложены два пути
образования ацилникелевого комплекса. Восстановление Nibipy2+ в
присутствии избытка СО приводит к быстрому образованию
нереакционного комплекса Ni°(CO)2bipy:
Ni°bipy+2СО —^-> Ni°(CO)2 bipy
В соответствии с этим наблюдением отмечено, что RX не
реагирует, когда электролиз проводится в условиях пробулькивания СО,
т.е. в насыщенном по СО растворе. Таким образом, конверсия
органических галогенидов в кетоны эффективна, когда концентрация
СО ограничена, что позволяет генерировать смесь Ni°(CO)2bipy
MargelS., Anson F.C. Op, с it.
Inokuchi T., Tsuji Af, Kawajuch //., Torii S. // J. Org. Chem. 1991.56. 5945; Yama-
moto Г„ Wakobayashi S.. Osakada K. // J. Otganomet Chem. 1992,428, 228.
Ocafirain M., Dothem E., NedelecJ.Y., Troupe! M> //J. Organomet. Chem. 1998.
571. 37; Ocafrain M„ Devaud M., NedelecJ.Y.. Troupe! M. //J. Organomet. Chem.
1998.560. 103.
245
и Nl°bipy В этих условиях рассматриваются два пути образования
требуемого ацилникелевого интермедиата:
_r0., Ni°{CO):bipy м-Оогли-
Nrbipy 7-^ > NrCObipy
к,
RX кг RX
"СО! ^ dnijII
RNi"Xbipy ^—> RNi"(CO)Xbipv
i\
RCONiMXbipy
Первый путь — окислительное присоединение RX к Ni°CObipy
(с к\). Второй путь — окислительное присоединение RX к Ni°bipy (с £?),
затем присоединение СО из смешанных комплексов Nift(CO)2bipy или
Ni°CObipy или из растворенного СО (с £4)- Очень реакционные
органические галогениды, например арил иодиды, для которых к2 > к^, будут
реагировать по второму пути. Наоборот, ряд органических галогенидов
(бензил хлориды, арил бромиды, алкил галогениды) имеет среднюю
реакционную способность по отношению к Ni°bipy? и кг« А> Кроме
того, препаративный синтез кетонов проводился в растворах с
каталитическим количеством Nibipy2+ (5% от RX)1. В этих условиях скорости
реакций 2 и 3 примерно близки, а два пути образования
ацилникелевого комплекса конкурируют2. Таким образом, электрохимический
метод позволяет обосновать правильный выбор условий синтеза и
получить количественную информацию о процессе.
Система Pd—PPh3 характеризуется двухэлектронным
восстановлением а-арил-палладиевого шлермедиата3, аналогичного
соответствующему интермедиату в ранее изученной системе Ni-PPhj".
Образование биарила, вероятно, протекает по схеме восстановительного
элиминирования из диарилпалладия с последующей регенерацией Pd°:
АгХ
Аг-Аг
ArPdnX^r-2c
ArPd°
"АгХ
Ar2Pd11
Oca/rain M., Devaud M., Troupe! A/., Perichon J. Op. cit,;
Dunach E. // J. FVrichon. Synlett. 1990. 143.
Amatore C. // Novel irends in ekciroorganic synihesis / cd. S. Torii. Tokyo:
Kodansha, 1995.
Shiavon G., Bonrempelfi G., Corain B. Op. cit.; Nedefec J.-Y., Perichon J.. Troupel M.
Op. cit.
246
Таким образом, возможны различные механизмы димеризации
арилгалогенидов, катализируемой Ni или Pd. Предполагается, что,
варьируя экспериментальные условия, особенно катодный
потенциал, можно направлять реакцию по тому или иному пути.
Многие ключевые стадии механизма реакций, катализируемых
комплексами Ni(0), описывались интуитивно, без достаточных
доказательств. Для некоторых статей характерна неверная интерпретация
каталитических волн на вольтамперограммах, соответственно
ошибочные расчеты скоростей отдельных стадий1. Однако в последние
годы появились работы, предложившие подходы к изучению
механизма реакций сочетания (гомо- и кросс-) и других с участием
органических галогенидов (ароматических и гетероароматических)
под действием электрохимически генерируемых комплексов Ni(0)
bipy Здесь важным условием успеха является правильный выбор
модельных соединений, позволяющих разделить суммарный процесс
на отдельные стадии и подтвердить или опровергнуть возможные
пути реакции. Впервые успех был достигнут в исследовании2, где в
качестве модельного арилгалогенида был выбран о-Вг-толуол, который
при взаимодействии с Ni(0)bipy образует устойчивый о-комплекс
ToiNi"Brbipy в результате окислительного присоединения:
NiBbbipy + 2е -» Ni°bipy + 2Br -1,2 В отн. НКЭ
Ni°bipy + TolBr —> ToINiBrbipy, стабил.
Для выяснения деталей механизма было рассмотрено
восстановление комплекса TolNi"Brbipy в отсутствие и в присутствии других
ароматических и гетероароматических соединений. При
восстановлении при —1,35 В после пропускания Ie/моль никеля
электричества на вольтамперограмме раствора наблюдается одна обратимая
волна при —1,55 В (ранее ошибочно приписываемая другой форме
TolNinBrbipy301), соответствующая новому комплексу Tol2NiBrbipy,
который впервые был выделен и охарактеризован:
TolNinBrbipy + е -> TolNi'bipy + Вг~ -1т35 В
TolNi'bipy -» TobNiBrbipy
Устойчивость арил-а-комплексов, имеющих CHj-грушту в орто-
положении ароматического ядра, объясняется стерическими
факторами, препятствующими вращению вокруг с-связи Аг—Ni и
аксиальной атаке реагентов на атом никеля3.
1 Durandetti A/.. Devaud M., Perichon M.//i. Perichon. New. J. Chem. 1996. 20.659.
3 Budnikovn Y H,, Perichon /, Yakhvarov D.G., К<щт KA/„ Smyashin O.G. //), Or-
ganomet. Chem. 2001. 630. 185.
* Meyer G., Roltin Y., Perichon J. // J. Organomctal. Chem. 1987. 333. 263;
Fahey DR., Baldwin В.Л. Op. cil.
247
Этот комплекс обратимо восстанавливается:
TobNiBibipy+e-+[Tol2NiBibipyn -1,55 В
Как внешнесферный переносчик электронов, образуюшися
анион-радикальный комплекс может восстанавливать 2-бромтолуол
до толуола, что подтверждается хроматографическим анализом.
Конкурентной реакцией является медленное разложение [Tol2NiBrbipy]*~
в растворе с образованием То12:
[Tol2NiBrbipy]"" -» Tol2 + Ni°bipy + Вг"
Это реакция восстановительного элиминирования,
индуцированного переносом электрона. Полученные представдения по
механизму гомосочетания были использованы при рассмотрении
механизма кросс-сочетания различных арил- и гстероарилгало-
генидов, катализируемых Ni°bipy, и выяснении, на каких стадиях
и при каких потенциалах образуются целевые продукты АГ|Аг2. Так,
обнаружено, что активной формой комплекса выступает только
комплекс Ni(T), т.е.:
TolNinBrbipy + Ar-X-> Tol-Ar + NiBrXbipy
TolNi'bipy + Аг-Х -> TolArNiBrbipy -> Tol-Py + NiBrbipy
На основании достигнутых результатов сделаны некоторые обо-
шения. Каталитическая эффективность комплекса в различных
реакциях отличается. То есть один и тот же комплекс может быть
эффективен в реакции гомосочетания органических галогеншюв
и мало или совсем неэффективен в реакции кросс-сочетания двух
различных RX. Следующие факторы определяют эффективность
катализатора в общем случае.
1. Возможность быстрой регенерации катализатора, т.е.
циклизации процесса. Если металлокомллекс связывается интермедиатами
реакции, то каталитическая реакция угасает и блокируется, т.е. к-^
должна быть велика.
2. Скорость реакции восстановительного присоединения
Ar'Ni"r+) + АНХ -»Ar'Ai^Ni12"1""' X должна быть больше скорости
конкурентных реакций: распада, протонирования ArlNi<"+) и т.п.
3. Скорость реакции восстановительного элиминирования
Ar]Ar2NiX должна быть достаточно большой для успешной
регенерации катализатора (воможно, промотируемой переносом
электрона).
Электрохимически синтезированный устойчивый ст-комплекс
TolNi"Brbipy оказался удачной моделью при изучении механизма
248
образования третичных фосфинов1. Так, было показано, что продукт
кросс-сочетания со связью Р—С образуется по следующей реакции
замещения: ArNiBrbipy + Ph„PClj_„ -»Ar,_„Ph„P + NiBrClbipy, а
конкурентный путь через фосфиды никеля не реализуется. На начальных
стадиях электролиз идет при потенциале системы Ni(ll)/Ni(0).
После того как через электролит пропущена примерно половина
расчетного количества электричества, в растворе уже нет исходной
формы катализатора NiBr2bipy и процесс осуществляется при более
катодных потенциалах (от —1,5 до —1,7 В в зависимости от
природы Аг), однако продукт кросс-сочетания образуется даже более
энергично, чем на начальных стадиях электролиза. Основное
количество фосфинов образуется именно на этом этапе в результате
следующей последовательности реакций:
ArNiX + e -> ArNibipy +X" (-1.35 В)
ArNibipy + Ph2PCI -> ArPh2PNibipy
ArPh2PNiClbipy+e->[ArPh2PNiClbipyr->ArPh2P + Ni° + Cr
(от -1,5 до -1,7 В)
Важной реакцией, приводящей к целевым соединениям,
является восстановительное элиминирование из никельарилфосфидного
комплекса ArPh2PNiXbipy. Общую схему циклической регенерации
катализатора можно представить в следующем виде:
Ph2PX
Ni(0) илие
ArNi(I)
Ph2PX Ph2PAr
X
ArPh2PNiCl B ArNi(H)Cl a Ni(ll)X2
e
Ph2PAr ArCl ArC1
На начальных этапах электролиза продукты образуются по
реакциям, изображенным в правом цикле, а по мере расходования
NiBr2bipy начинают преобладать реакции левого цикла.
Использование TolNi"Brbipy в качестве модельного соединения
позволило проследить путь образования вторичных спиртов в реак-
1 Будникова Ю.Г., Карат Ю.М. // Журн. общ. химии. 2001. 71, 140; Margel S.,
Anson f.C. Op. cit.; Будникова Ю.Г., Каргип Ю.М., Япилкии В.В. Указ. соч.
249
ииях электровосстановительного сочетания органических галоге-
нидов и альдегидов, катализируемого комплексом никеля с 2,2-би-
пиридилом1. Обнаружено, что выход продукта пропорционален
концентрации NiBr2bipy и растворе. Было сделано предположение,
что ионы никеля связываются в соответствующий алкоголят
никеля и выводятся из цикла. Кроме того, было показано, что при
потенциале восстановления Ni(II)/Ni(0) —1,2 В отн. НКЭ спирт
не образуется, а целевой процесс сочетания идет только при более
катодных потенциалах, соответствующих дальнейшему
восстановлению а-комплекса TolNiBr (—1,35 В) по следующей схеме реакции:
NiBr3(bipy)+ 2e ^ Ni<0)(bipy)+2ВГ
Ni(0)(bipy) + 2-МеС6Н4Вг->2-МеС6Н4™<П)Вг(Ыру)
2-MeQR,Ni(ll)Br(bipy) + e -> 2-MeCuR,Ni(l) + Br
PhCHCf,H4Me-2
2-MeCbH4Ni(I)(bipy) + PhCHO > | + bipy
ONi
Таким образом, сочетание волътамперометрии и препаративного
электролиза является эффективным методом, демонстрирующим
возможности гомогенных электрокаталитических процессов в
исследовании механизмов реакций, а знание его механизма — основа
возможности управления и достижения высокой селективности.
6.13.4. Механизмы и кинетика лалладиевых каталитических
систем
Среди катализаторов органических реакций палладий является
наиболее универсальным, так как он катализирует образование
связей С-С, С-Н, C-N, С-О, С-Р, C-S и С-СО-С по реакциям
арил и винил галогенидов (трифлатов), аллильных производных
с нуклеофилами (реакции кросс-сочетания, реакции Стилла, Суд-
зукл, Хека, Цужи-Троста)2. В присутствии источника электронов
реакционная способность меняется на противоположную и реакции
с электрофилами позволяют образовывать связи С—С, С—Н и С—
С02\ Эффективность палладия проистекает из его способности
в нульвалентном состоянии активировать связи С—X (X = Г\ Вг5, С16,
1 Будникова Ю.Г., Кешнер Т.Д.Т Каргин Ю.М. Указ. соч.
2 Amatore С, JuiandA. // J. Organomet. Chem. ] 999. 576. 254.
3 JuiandA., Mosleh A. // J. Org. Chem. 1997. 62. 261; Jutand A.t Negri S. // Eur.
J. Org. Chem. 1998. 1811.
4 Fition P., Johnson MP., Mc Keon J.E. jj J. Chem. Soc. Chem Coramun. 1968. 6;
Fition A, Rick E.A. //J. OrganomeL Chem. 1971. 28. 287.
5 Fition P., Rick E,A. Op. cit.
6 Ibid.
250
О1) путем окислительного присоединения, которое приводит к
образованию комплекса органопалладия(П), способного реагировать
с нуклеофилами2. В результате нуклеофильной реакции получается
новый комплекс органопалладия(П), который приводит к конечному
соединению через одну или несколько стадий3. Недавно появились
работы, которые позволили пересмотреть устоявшиеся
представления о механизмах реакций, катализируемых палладием4. Показано,
что кроме ожидаемой зависимости эффективности палладиевого
катализатора от лиганда при атоме Pd возник неожиданный факт:
для данного лиганда реакционная способность также зависит
от предшественника комплекса Pd(0), поскольку существует общее
согласие с тем, что низкокоординированный 14-электронный
комплекс Pd(0)L2 всегда выступает активной частицей, инициирующей
каталитический цикл через его окислительное присоединение5.
Чтобы понять, почему различные источники Pd(0) столь
специфичны, были исследованы структура и реакционная способность
различных результирующих комплексов Pd(0) в окислительных
присоединениях. Кроме того, поскольку окислительное присоединение
может не быть определяющей скорость стадией каталитического
цикла, также были исследованы структура и реакционная
способность комплексов органопалладия(Н), полученных в результате
окислительного присоединения в последующей стадии реального
каталитического цикла, т.е. в реакции с нуклеофилами. Так,
обнаружено, что окислительное присоединение к арилгалогенидам
приводит к большому разнообразию комплексов арилпалладия(П):
нейтральным, анионным или катионным6. Например, окислительное
присоединение арилтрифлатов к Pd°(PPhj>4 даст катионные
комплексы ArPd(PPh3)2, нейтральные комплексы ArPdC1(PPh.;)2
образуются в присутствии хлорид-ионов. Кроме того, эти комплексы
могут быть связаны динамическим равновесием. Структура
комплексов арилпалладия(И) сильно зависит от структуры
эффективного комплекса Pd(0), участвующего в окислительном
присоединении, и от анионов, связанных с Pd(0). Ранее предполагалось, что
двухэлектронное восстановление PdC^PPh^h приводит к низколи-
гандированному комплексу Pd^PPhjh- Однако 3|Р-спектр этого ком-
1 JutandA., Mosleh A. // OnsanomcutUics. 1995. 14. 1810.
2 Fauvarque J.F., Jutand A. If Bull. Soc. Chim. Fr. 1976. 765; Fauvarque J.F.,
JutandA, H J. Organomet. Chetn. 1977. 132. C17: Fauvarque J.F., JutandA. //
J. OiBanomel. Chem. 1979. 177. 273; Gillie A., Stifle J.K. // J. Am. Chem. Soc.
L9KQ. 102. 4933.
■ AmatoreC, Jutand A. Op. cit,
* Ibid,
5 Ibid.
* Ibid.
251
плекса после исчерпывающего электролиза характеризуется тремя
сигналами1, относящимся к трем различным анионным комплексам,
находящимся в динамическом равновесии:
PdCl2(PPhih
2е
Ч /с\ /Г к. Ч - -,_ +
Pd(0> Pd(0)v - Pd(0)-C1 ^
Khc
L- *al
■ ■ n~
+ci KBc \ . л .
Pd(0)v
CI
Димер является наиболее реакционноспособной частицей.
Таким образом, комплекс Pdfl(PPh3)2 не существует в растворе,
когда генерирование происходит в присутствии галогенид-ионов,
а образуются анионные частицы Pd{0). Другой неожиданный
результат — это то, что окислительное присоединение происходит
быстрее в присутствии добавленных катионов (Li+, Zn2^)2.
Взаимодействие катиона с хлоридом, связанным с центром Pd(0),
приводит к образованию ионной пары и повышает реакционную
способность комплекса:
ч ч
Pd(0) «-СГ Li+ Pd(0) СГ Zn+Cl
l/ l/
Таким образом, результаты показывают, что структура и
реакционная способность Pd(0) зависят не только от предшественника
Pd<II> (влияние анионов), но и от пути генерации (влияние
катионов). В каталитическом цикле эти ионы могут влиять на природу
и кинетику окислительного присоединения.
Механизм реакций арилпалладия(Н) с нуклеофилами и кросс-
сочетания, катализируемых палладием, можно представить схемами,
избраженными ниже:
АгХ+NiT |Pd| ) ArNu + X"
Pd = Pd(0HPPh3)4,{PdC12(PPh3)2 + NuorH-}
Для низко реакционноспособных арилбромидов определяющей
скорость процесса стадией является, вероятно, окислительное при-
1 AmatoreC, A&flbiM.,Jutand A. Op. cit.
2 Amatore С, JutandA. Op. cit.
252
соединение (рис. 6.7, а)\ тогда как для более реакиионноспособньтх
трансмсталлирование (нуклсофильная атака на комплексы транс-
ArPdXL2, рис. 6.7, 6) рассматривается в качестве определяющей
скорость процесса стадии2. Восстановительное элиминирование ш
комплексов транс-ArPdNuLi также может быть определяющей скорость
процесса стадией (с + d на рис. 6.7)\ Однако все эти механистические
исследования были сделаны для различных выделенных систем,
каждая из которых изображает одну из стадий постулированного
цикла не в контексте всего каталитического цикла. Удалось
убедительно показать, что комплекс Pd(0), генерируемый при
восстановлении PdC12(PPti3)2. является анионным комплексом Pd(0)(PPh?hCl",
так что окислительное присоединение приводит к анионному пепта-
координированному комплексу арилпалладия(1Т) — ArPdXCl(PPh3)T,
который через длительный период времени дает ожидаемый транс-
ArPdX(PPh-t)34. Поскольку в этих условиях С1~ был наилучшим нук-
леофилом и в то же время наилучшей уходящей группой, это быстрое
равновесие представляет вырождающееся нуклеофилъное замещение,
протекающее по механизму SN1. На основании этих результатов был
предложен новый каталитический цикл (рис. 6.7, б). Нуклеофил
атакует промежуточный пентакоординированный нейтральный
комплекс арилпалладия(П) (Ь'на рис. 6.7), порождая анионный
пентакоординированный комплекс ArPdXNuL^, в котором группы Аг и Nu
присоединены и находятся в благоприятном пшюжении, что
обеспечивает быстрое восстановительное элиминирование (г/'на рис. 6.7).
Таким образом, можно исключить комплекс транс-ArPdXL» как ин-
термедиат в реакции кросс-сочетания в условиях катализа
комплексом PdC^PPh^h- В огсутствие любых нуклеофилов образование
TpaHC-PhPdI{PPh3)2 ускоряется катионами5. Это объясняется сдвигом
равновесия а'(Ь на рис. 6.7) в левую сторону. Если нуклеофил
достаточно сильный, то реакция будет протекать по схеме (Ь на рис. 6.7).
Очевидно, что механизм чрезвычайно запутан и может меняться в
каталитическом цикле при изменении условий реакции. Аналогично
изменения в среде реакции (например в растворителе) также могут
влиять на тонкие взаимодействия между двумя крайними циклами
в схеме» поскольку эерфект спаривания ионов будет контролировать
1 Fauvarque J.F,, Jut and A. if Bull. Soc. Chim. Fr. 1976. 765; Fauvarque J.F.,
JutandA. H J. Organomet. Chem. 1977. 132, C17: Fauvarque 1.F., Jutand A. //
J.Organomet, Chem, 1979. 177. 273,
1 Gillie At, Stille J.K. Op. cit; Neghishi £.-/., Takagashi Т., Baba S.M van Horn £>.£.,
Okukado ,V, //J. Am. Chem. Soc. 1987.109. 2393.
i Gillie A., Stille J. K. Op. cit.; Loar M., Stille JX 11 J. Am, Chem. Soc. 1981. 103, 4174;
Tatsumi K., Nakamura K, Kamiya S., Yamamaio Y, //J. Am. Chem. Soc. 1984. 106. 81R1.
4 Amatore C, Jutand A., SuarezA. // J. Am. Chem. Soc. 1993. 115. 9531.
5 Ibid.
253
доступность галогенид-ионов (высвобождающихся из арил галоге-
нилов) и катионов (связанных с нуклеофилами). При обсуждении
эффективности реакций кросс-сочетания, катализируемых
палладием, необходимо учитывать все эти факторы.
Pd(0)L, или {PdCI2L2 + Red}
I
Pd(0)L2
Аг-Nu
PdCl2(PPh3)2
I Восстановление
L
Ar-X
Рис. 6.7. Схема механизма реакций арилпалладия(Н) с нуклеофилами и кросс-
сочетания, катализируемых палладием (а) постулированного механизма (б)
нового каталитического цикла
254
Механизм реакции Хека выглядит следующим образом:
=\ + ArX + NEt3 —U \=д + ЕЫЧН+Х"
R R
[Pd] = {Pd(QAc)3 + nL}
Эта реакция, открытая в 1968 г. Хеком, в последние 15 лет
была значительно расширена и развита с новыми субстратами
и катализаторами. В присутствии хиральных лигандов могут быть
проведены энантиоселективные реакции1. Эта реакция требует
основания и обычно проводится в ДМФА. Постулируемый
механизм приведен на рис. 6.8, а. Среди каталитических систем,
эмпирически подобранных для реакции Хека, наиболее
эффективны смеси Pd(OAc)2 и фосфинов. Установлено, что фосфин
восстанавливает Pd(Il) в Pd(0)2. Эта очень быстрая стадия
реакции учтена в новом каталитическом цикле (рис. 6.8, б). Кроме
того, было показано, что Pd(0), генерируемый in situ, выступает
в виде анионных частиц Pd(0)(PPh3)2(OAc) , которые
подвергаются окислительному присоединению к арилгалогенидам через
включение короткоживущего комплекса пентакоординирован-
ного комплекса арилпалладия(П). По ревизованному механизму
реакции Хека нуклеофильная атака олефина протекает по
действием ArPd(OAc)(PPh3)2, а не ожидаемого транс-АгРДИРРпзЬ,
который никогда не образуется, если окислительное
присоединение происходит через Pd(0){PPh-,)2(OAc)~. Эта работа
подчеркивает ключевую роль анионов, рождаемых предшественниками
комплексов Pd(0) и объясняет эмпирические находки,
разбросанные в литературе, касающиеся специфики каталитических
палладиевых систем3.
1 De Meijere A., Meyer F.E. // Angcw. Chcm. Im. Ed. Engl. 1994. 33. 2379;
Ozawa F., Kubo A., Matsumoto K. Hayashi T. // Organomciallics 1993. 12.
4188.
2 Amatore C, JutandA., M'Barki M. // Organometallics 1992. 11. 3009; Amatore C,
Carre E., JutandA., M'Barki M., MeyerG. // Organometallics. 1995. 14. 5605
(1995); Amatore C, Carre E., JutandA., M'Barki M. 11 Organometallics. 1995. 14.
ISIS.
3 Amatore C, JutandA., Medeiros M.J. // New J. Chem. 1996. 20. 1143; Amatore C,
Btart E., Genet J.P., JutandA., Lemoire-Audoire S., Sa\>ignac M. // J. Org. Chem.
1995. 60.6829; Amatore C, JutandA., Meyer G // Inorg. Chim. Acta 1998. 273. 76;
AmatoreC, JutandA., MeyerGtA/mani H., Khali! F.,ChahdiO. //Organometallics.
1998. 17. 2958; Amatore C, BroekerG^ JutandA., Khafil F. // J. Am. Chem. Soc.
1997. 119. 5176; Amatore C, Carre E., JutandA. // Acta Chem. Scand 1998. 52.
100.
255
Pd(Q\c)2 + rtPPh2
HNEt/
Pd(0)L2
NEt,—-/(d) (a)>
HPdXL2 ArPdXL2
Ar-X
Pd(OAc)2 + wPPh2
i
Pd(OAcb<PPh3b
Pd(0)(PPh3)3(OAc) |
NEt3
HPd(OAc)(PPh3)2
Ar
Ar-X
NEt,
(a'l)
ArPdX(Q\c)(PPhj)2~l
ArPd((Mc)(PPh3)2 — ArPd(PPh3)2+
Ar Pd(QAc)L2
H ,b)
H R
AcO"
+ H """NEtj
HOAc
Рис. 6.8. Реакции Хека постулированного механизма (а) и нового
каталитического цикла (б)
Таким образом, в последние годы появилось много интересных
синтетических приложений, комбинирующих
электровосстановление и катализ переходными металлами. Не все, но многие из них
реально конкурируют с обычными химическими метолами как новые
256
эффективные синтетические процессы. В то же время подробно
исследовалось прямое электровосстановителъное сочетание, особенно
в связи с перспективным использованием растворимых анодов. Мы
упомянули в обзоре, что некоторые реакции, ранее описанные как
каталитические процессы, например, карбоксилирование
органических галогенидов или гомосочетание ал кил гало ген идо в, сейчас
могут осуществляться путем прямого электровосстановительного
сочетания в присутствии растворимого анода. Однако
электрохимический катализ переходными металлами остается очень полезным
и удобным методом, особенно в отношении селективности.
Последние исследования выдвинули на первый план прогресс в
изучении синергизма между переходным металлом катачизатора и
металлическими ионами, полученными при растворении анода. Так
как во многих случаях электрохимическое оборудование становится
все более простым, без сомнения, прямые и непрямые
электрохимические методы становятся все более привлекательными.
Глава 7. ЛИГАНД-НАПРАВЛЕННАЯ
ЭЛЕКТРОХИМИЧЕСКАЯ ФУНКЦИОНАЛИЗАЦИЯ
С(5Р2)-Н-СВЯЗЕЙ В ПРИСУТСТВИИ
СОЕДИНЕНИЙ ПАЛЛАДИЯ И НИКЕЛЯ
Можно считать, что органическая химия в настоящее время
нацелена на разработку идеальных превращений, направленных
на синтез сложных молекул, которые являются эффективными,
экономичными и экологически чистыми1. Реакции функционали-
зации связей углерод — водород, катализируемые переходными
металлами, которые преобразуют широко распространенные, но, как
правило, инертные С— Н-связи непосредственно в связи углерод-
углерод (С—С) и углерод-гетероагом (например. С—О, С—N или
С—Hal), являются весьма перспективными в этом отношении. В
отличие от традиционных методов, применяемых для образования
этих связей, прямое С-Н-замещение исключает необходимость
предварительной функционализации (например, галогенирование
или борилирование) исходного субстрата, что снижает число стадий
и уменьшает количество нежелательных отходов в многостадийных
процессах. Таким образом, можно достичь атом-экономии и
кратчайших путей синтеза с меньшим числом стадий1. По этой причине
функлионалшация С-Н-связей, катализируемая переходными
металлами, в металлоорганической химии часто рассматривается как
«Святой Грааль» и в последние десятилетия привлекает огромное
внимание3. Происходит лавинополобный рост количества
публикаций и интереса к этой области. Вследствие толерантности
палладия ко многим функциональным группам и возможности
образования различных типов связей (C-Ot C-Hal, C-N, C-S, С-С)
палладиевые комплексы являются особенно подходящими
катализаторами для таких преобразований4, причем ацетат палладия
1 Constable D.J.C., Dunn P.J., Hayler J.D., Humphrey G.R., LeazerJ.L., Lin-
dcrman Jr. R.J., Lorenz K., Mtmley J., РеаНтап В.А., Welts A.. Zaks A.,
Zhang T. Y. // Green Chcm. 2007. 9. 411; Sheldon R.A. // Chcm. Sot. Rev. 2012.
41. 1437.
2 Gutekunst W.R., Baran PS. //Chem. Sac. Rev. 2011.40.1976; YamaguchiJ., Yama-
guchiA.D., ItamiK. //Angcw. Chem., Int. Ed. 2012. 51. 8960.
1 Zhang A/., Zhang K, Jie X„ Zhao //.. Weipittg Su.G.Li. // Org. Chem. Front. 2014.1.
843: Lyons T. W., SanfordM.S. // Chem. Rev. 2010.110. 1147; Negishi E. //
Handbook of Organupalladium Chemistry for Organic Synthesis. John Wiley & Sons,
Hoboken, NJ. 2002. P. 229—247; HartwigJ.F. GrBanotransilion Metal Chemistry:
From Bonding to Catalysis. University Science Books, Sausalito. 2010.
4 Ibid.
2SS
используется чаще всего. Химически подобный трифторацетат
палладия (TFA) используется реже, хотя известно, что он также
может выступать эффективным катализатором и с окатал и затором
в перегруппировках, основанных на активации С-Н-связей1.
Кроме того, в некоторых случаях было обнаружено, что добавка
трифторуксусной кислоты может иметь решающее значение для
достижения высокой эффективности С—Н-функционализации2,
а трифторацетатный лиганд часто используется для улучшения
растворимости и каталитической активности комплексов переходных
металлов3.
Для решения проблемы селективной С—Н-функционализации
используют различные стратегии, наиболее распространенные
из них предполагают использование субстратов, которые содержат
заместители, способные к координации. Эти лиганды (их часто
называют «направляющей группой») связываются с металл о-центром
и селективно направляют катализатор на ближайшую С-Н-связь.
Многие переходные металлы, в том числе Pd, вступают в лиганд-
направленные реакции активации С—Н-связи (также известные как
реакции циклометаллирования).
Несмотря на то что палладий имеет пять доступных степеней
окисления (0, +1, +2, +3, и +4)4, большинство Pd-катализируемых
превращений связаны с Pd(0) и Pd(Il). Эти реакции с участием
палладия в низших валентных степенях окисления активно изучались
в последние десятилетия5, причем электрохимия внесла свой
немалый вклад в исследование их механизмов6. Однако различные Pd-
катализируемые лиганд-направленные реакции СН-окислительной
функционализации включают палладий(Ш)-7 и палладий(1У)-ин-
термедиаты*, что делает химию палладия в высших степенях окис-
1 WangX, Truesdale L.. Yu J.-Q. // J. Am. Chem. Soc. 2010. 132. 3648; Sakai N.,
RidderA., HariwixJ.F. //J. Am. Chem. Soc. 2006. 128. 8134.
1 ShanG., YangX., Ma L., Rao Г J/ Angew. Chem. Int. Ed. 2012. 51. 13070.
J Yuan D.. Huynh H. V. // OrBanometallics. 2012. 3 L 405.
4 Cotton F.A., Wilkinson G,, Murilh C.A., Bochmann M. Advanced Inorganic
Chemistry. N. Y.: John Wiley & Sons, 1999.
? Negishi E. Op. cit.; Mirica L.M., Khusnutdinova JR. // Coord. Chem, Rev. 2013.
257. 299; Miyaura A'., Suzuki A. //Chem. Rev 1995. 95. 2457; StahlS.S. // Angew.
Chem. Int. Ed. 2004. 43. 3400; Stoltz BM. // Chem. Lett. 2004. 33. 362; Gli-
gorich KM., Sigma» MS, // Chem. Commun. 2009. 3854.
ь JutandA. H Chem. Rev. 2008. 108. 2300.
7 Powers B.C.. Geibet M.A.L.. Klein./.E.M.N,, Ritler T.//1. Am. Chem. Soc. 2009.
131. 17050; Powers C, Ritter T. // Nat. Chem. 2009. 1. 302; Khusnutdinova JR.,
Rath N.P., Mirica L.M, //}. Am. Chem. Soc. 2010, 132.7303.
* Sehnal P., Taylor RJ.K., Fairlamb f.J.S. // Chem. Rev. 2010. 110. 824;
Cantv U.S. II Dalton Trans. 2009. 10409; Deprez N.R.. Sanford M.S. /I Inorg.
Chem. 2007. 46.1924.
2S9
ления важной и перспективной областью. Несмотря на эти успехи,
известно всего несколько охарактеризованных Pd<III)-KOMTUTCKCOB,
что препятствует детальному изучению металлоорганической химии
палладия(Ш). Биядерные комплексы Pd(III) недавно были
предложены в качестве активных каталитических интермедиатов в
окислительной функционализации С—Н-связей1 и перегруппировке
аза-Кляйзена2, в то время как моноядерные комплексы Pd(III)
выступают интермедиатами окислительно индуцированного
восстановительного элиминирования этана из PdflOA^-KOMiuieKcoB3,
введения молекулярного кислорода в связь Pd-Me4, сочетания Ку-
мады5. В этом контексте более детальное понимание электронных
свойств комплексов Pd(IH) должно способствовать разработке новых
катализаторов для различных металлоорганических превращений,
а также других многоэлектронных окислительно-восстановительных
реакций. Понимание свойств Pd"-, Pdm- и Pdlv-KOMmieKcoB является
необходимым для развития новых катализаторов, используемых для
этого типа превращений.
Одна из основных проблем новых способов синтеза, включающих
палладиевые катализаторы в высокой степени окисления,
заключается в том, что требуются со-окислители, в основном в стехиометри-
ческом и большем количестве, например, соли переходных металлов
или органические окислители, такие как соли серебра,
органические пероксиды и электрофильные фторные реагенты, которые часто
дороги или трудно отделимы от реакционных смесей6. Кроме того,
каждый раз, когда используются новый лиганд, или субстрат, или
условия реакции, необходим широкий скрининг для выбора
оптимального окислителя (рис. 7.1).
1 С—Н and С—X Bond Functionalization: Transition Metal Mediation / ed.
X. Rib-as, RSC Publishing, Cambridge, UK, 2013; Deprez N.R., Sanford M.S. //
J. Am, Chcm. Soc. 2009. 131,11234; Powers D.C, Benitez D„ Tkatchouk £., God-
dard III W.A., Ritter Т. Ц J. Am. Chcm. Soc. 2010. 132. 14092; Powers DC,
Ritler T. //Top. Organomet. Chem. 2011. 35. 129.
2 Eitel S. H., Bauer M.. Schwcinfutih D., Deibef N., Sarkar B., Kelm H., Kniger H. -J.,
Frey W., Peters R. // J. Am. Chem. Soc. 2012. 134.4683.
* Khumuidmova J.R., Rath N.P., Mirica L.M. Op. cit.; Land MP., Remy M.S.,
Kamimky W., Mayer J. M.. Sanford M.S. //J. Am. Chcm. Soc. 2009. 131. 15618;
KhusnutdinovaJ.R., Rath N.P.. Mirica L.M. // J. Am. Chem. Soc. 2012. 134. 2414;
BoisvertL., Denney Л/.С, KJoek H.S., Goldberg KJ. // J. Am. Chem. Soc. 2009.
131. 15802; ManolikakesG., Knachel P. //Angew. Chem. Int. Ed. 2009. 48. 205;
KhusnutdinovaJ.R., Rath N.P., Mirica L.M. // Angew. Chem. Int. Ed. 2011. 50.
5532.
4 Boisvert L., Denney M.C., Kloek H.S., Goldberg K. I. Op. cit.
1 Manolikakes G., Knochel A Op. ciL; Khusnutdinova J.R., Rath N.P., Mirica L.M.
Op. cit.
* Lyons T. IV., Sanford M.S. Op. cit.; Staht S.S. Op. cit.: Canty U.S. Op. ciu
260
(Pd11 перекурсор кат.]
Н-С N \У >R-C N
W -HY ^y
Окислитель!!!:
РЫС12 IPh2{OT0 XeF2
Phl(OAc)2 IPh(CsCSiMe3)(OT0 (OTf = трифлат)
соли серебра: AgPFfi и др.
Вг2
Th" (тиантренил катион-радикал)
Рис 7.1. Сокатализаторы реакции лиганд-направленного окислительного С-Н-
замещения
Электрачиз при контролируемом потенциале в мягких условиях
является реальной альтернативой классическим методам, что было
продемонстрировано, например, в работе Какиучи1, описывающей
эффективное электрохимическое орто-галогенированис
производных арилпиридина в присутствии солей палладия.
Было предложено развить более дешевую, «зеленую» и более
управляемую электрохимическую альтернативу этим реакциям С—
Н-замещения и использовать мягкие условия электросинтеза при
контролируемом потенциале анода для достижения поставленных
целей.
Всевозможные предлагаемые в литературе пути окислительной
С—Н-функционализации обязательно включают стадию
окисления — одну или несколько. Сделано предположение, что
окисление Рс1(1[)-комплексов можно провести эффективно на электроде,
заменив химический окислитель на электрохимическое окисление.
Это позволило бы получить предполагаемый биметаллический
комплекс Pd(III) и/или Pd(IV) препаративно в условиях электросинтеза
и использовать его в качестве модельного интермедиата для изучения
путей каталитического С—Н-замещения. Одной из задач было
осуществление всего процесса С—Н-функционализации
электрохимически, в условиях генерации окисленной формы палладиевого
катализатора на электроде, что ранее никем не реализовывалось. Также
осуществлена попытка разработать новые примеры реакций
каталитической С-Н-функиионализации на никеле, поскольку такие
примеры встречаются намного реже, чем с палладием2.
1 Kakiuchi F,t Kochl Т.. Mutsutuni //., Kobuyashl N,. Uram S., Sato Af„ Nishivamu $.,
Tanabe T. // J. Am. Chem, Soc. 209. 131. 11310.
3 Hachiya H., Himno K., Satoh Т.. Miura M. // Oig. Lett. 20O9.11. 1737; Hachiya H.,
Hirano A., Satoh Т., M. Miura M. // ChemCatChem. 2010. 2. 1403; Hachiya //.,
261
Ниже в данном разделе проанализированы результаты
исследований электрохимической окислительной функционализаиии 2-фс-
нилпиридина, выступающего и субстратом, и лигандом,
направляющим процесс в орто-положение за счет комплексообразования
с металлом по атому азота. Рассмотрены процессы аиетоксилиро-
вания, перфторацетоксилирования, перфторалкилирования, фосфо-
рилирования с участием комплексов никеля или палладия в высоких
степенях окисления, получаемых электроокислением стабильных
М( 11)-прекурсоров.
7.1. ЭЛЕКТРОХИМИЧЕСКОЕ ОРТО-АЦЕТОКСИЛИРОВАНИЕ
И ФТОРОАЦЕТОКСИЛИРОВАНИЕ 2ФЕНИЛПИРИДИНА,
КАТАЛИЗИРУЕМОЕ ПАЛЛАДИЕМ В ВЫСШИХ СОСТОЯНИЯХ
ОКИСЛЕНИЯ
Уксусная кислота, трифторуксусная кислота и карбоновые
кислоты с более длинной перфторалкильной цепью можно использовать
в качестве субстратов для замещения и функционал и зации связи
С— Н1. Важной задачей представляется установление различий
в электрохимических свойствах палладиевых циклов с ацетат-
и перфтороацетатными мостиками, которые, как было установлено,
выступают промежуточными продуктами реакции функционали-
зации С—Н-связи в фенилпиридине с использованием СН3СООН
и CF3COOH. Димерный ацетат палладия 1 известен — он был
синтезирован с использованием метода, описанного Риттером, его
структура подтверждена методом рентгеноструктурного анализа (рис. 7.2)2.
Hirano К.. Satoh Т., Miura М. // Angew. Chem.t Int. Ed. 2010. 49. 2202;
Hachiya //., Hirano К., Satoh Т., Miura M. // Angew Chem Int Ed Engl. 2010. 49.
2202; Kanyiva K.S., Kashihara N.t Nakao K. Hiyama Т.. Ohashi M, Qgoshi S. //
Dalton Transactions. 2010. 39. 10483; Kanyiva K.S.. Loebermaim F.. Nakaa Y.,
Hiyama T.//Tetrahedron Lett. 2009. 50. 3463; Nakao Y. //Chem. Rec. 2011. 11.
242; Nakao K, Idei H., Kanyiva K.S., Hiyama T. // J. Am. Chem. Soc. 2009. 131.
15996; Nakao K. Kanyiva K.S., Hiyama T. //J. Am. Chem. Soc. 2008. 130. 2448;
Nakao Г., Kashihara N.. Kanyiva K.S., Hiyama J. // J. Am. Chem. Soc. 2008. 130.
16170; Nakao Y. Morita E., Idei //., Hiyama T. // J. Am. Chem. Soc. 2011. 133.
3264; Nakao Y.. Yamada Y., Kashihara N.. Hivama T. //J. Am. Chem. Soc. 2010.
132. 13666.
1 Lyons T. W., Sanford M.S. Op. cit.; Powers DC, Geibel M.A.L., Klein J.E.M.N.,
Ritler T. Op. cil.; Dudkina Y.B., Mikhayiov [>. Y., Gryaznova T. V., Sinyashin O.G.,
Vicic D.A., Budnikova YH. // Eur. J. Org. Chem. 2012. 2114; Budnikova Y.H.,
Dudkina Y.B., Khrizanfarov M.N., Gryaznova TV. //J. Organomet. Chem. 2014. 751.
301; Mikhayiov D. Ytt., Budnikova Yu.H. // Russ. Chem. Rev. 2013.82(9), 835;
Dudkina Y.B., Mikhayiov D.Y.. Gryagiova 1\V„ TufatuUin A.L. KataevaO.N., Vkk DA.,
Budnikova Y.H. ff Organometallics. 2013. 32. 4785.
2 Powers B.C., Geibel M.A.L., Klein J.E.M.N., Ritter T. Op. cil.
262
В результате реакции циклопалладирования образуется
биметаллический комплекс Pd(ll):
РсЦОАс)г
CH2Clj, 40'С
(7.1)
Обнаружено, что синтез производных трифторацетата паллади>1
(TFA) был не так прост, как синтез нефторированных аналогов1.
Обработка трифторацетата палладия 2- фенилпиридином (PhPy)
в зависимости от используемого растворителя дает либо димерный,
либо мономерный палладиевый циклы. При использовании ди-
хлорметана в качестве растворителя биядерный комплекс 22
получается с выходом 64%. Однако, если ацетонитрил или ацетон
используется в качестве реакционной среды, образование димера
подавляется, а моноядерные комплексы 3 или 4 могут быть
получены с выходами 85 и 86% соответственно. Комплекс 5, который
имеет структуру, аналогичную структуре 4, но с более длинной фто-
ралкильной цепью, может быть получен при взаимодействии 2-фе-
нилпиридина с перфторгептановой кислотой (PFH) в ацетонитриле
(выход 88%)3.
Нефгорированные аналоги комплексов 3 и 4 не удалось выделить
при использовании тех же процедур. Однако нефторированные
моноядерные палладиевые циклы, аналогичные 3 и 4, были недавно
предложены в качестве промежуточных соединений в реакциях
активации С-Н связей, катализируемой Pd(OAc)24.
Результаты рентгеновских дифракционных исследований
кристаллов комплексов 3—5 приведены на рис. 7.3—7.5s. Палладиевые
Dudkina Y.B., Mikhayhv D. Г., Gryaznova Т. V., Tufatullin А.1., Kataeva O.N.,
Vide D,A„ Budnikova Y.H, Op. cit.
Bercaw JtE., Durrell A.C., Gray H.B., Green J.C., Hazari N., A. LabingerJ.. Win-
Ww/./J.//lnoiE-Chem,20l0.49. 1801.
Dudkina Y.B., Mikhayhv D. Y., Gryaznova T. V., Tufatultin A.I., Kataeva O.N.,
Vicic DA., Budnikova Y. H. Op. cit.
CantyAJ.,AhafardA., SanfordM.S., Yates B.F. Ц Oi^anometallies. 2013. 32. 544.
Dudkina Y.B., Mikhayhv D. Y., Gryaznova T. V., Tufatultin А.1., Kataeva O.N.,
VicicDA., Budnikova Y.H. Op, cil.
263
центры в 3,4 и 5 имеют плоско-квадратную геометрию, при этом два
атома азота лигандов связаны в от/к/нс-положении друг к другу. Триф-
торацетат всегда находятся в /ироис-положении к углероду арильного
лиганда.
Pd(TFA)2 + 2
О CF,
4 (86%)
Pd(QAc)2 + 2
=J C„FnCOOH
CHjCN, rt
5(88%)
Рис. 7.2. Реакционная способность трифторацетата палладия (II) по отношению
к 2-фенилпиридину, зависимая от растворителя
264
Рис. 7.3. ORTEP-представление молекулярного комплекса [{PhPy)Pd(TFA)]
(CHjCN)3. Терминальные эллипсоиды приведены с вероятностью 50%.
Выбранные длины связи (А):
Pd — С= 1,928; Pd — N1 = 1,995; Pd —О = 2,147; Pd — N2 = 2,015
Рис. 7.4. ORTEP-представление молекулярного комплекса [{PhPy)Pd(TFA)]
(PhPy)4. Терминальные эллипсоиды приведены с вероятностью 50%.
Выбранные длины связи (А):
Pd — С = 1,970; Pd — N1 = 2,055; Pd — О = 2,141;Pd — N2 = 2,012
Рис. 7.5.ORTEP-представление молекулярного комплекса [(PhPy)Pd(PFH)]
(PhPy)5. Терминальные эллипсоиды приведены с вероятностью 50%.
Выбранные длины связи (А):
Pd — С = 1,973; Pd — N1 = 2,017; Pd — О = 2,141; Pd — N2 = 2,041
265
Биметаллические комплексы палладия, как известно, находятся
и зависимом от температуры равновесии с моноядерными
частицами в присутствии азотистых лигандов. Равновесие было впервые
обнаружено в 1987 г. Рябовым1. В своей работе он описал
равновесие между хлор- или ацетатными мостиковыми циклопаллади-
рованными комплексами и свободными N-донорными лигандами.
Впоследствии было показано, что биядерный комплекс палладия
обнаруживает редокс-синергию в окислительном катализе2.
Обнаружено, что переход мономер—димер для трифторацетатных
производных сильно зависит от растворителя1- Так, растворитель
регулирует формирование различных комплексов палладия, а
замена реакционной среды влечет за собой изменение в его окружении
(рис. 7.6). Доказательство для этих взаимных переходов получено
методом 'Н ЯМР-спектроскопии выделенных кристаллических
комплексов 2—5 в различных растворителях. Для комплексов 2
и 3 наблюдаются быстрые взаимопревращения. Когда комплекс 2
растворяется в CDiCN, наблюдается его количественное
превращение в 3. Однако в спектре раствора 3 в CD2Cli проявляются две
группы сигналов как для биядерного 2, так и моноядерного 3
комплексов в соотношении 1 : 0,15 для 2 ; 3. Палладиевые циклы 4 и 5
сохраняются в некоординирующемся растворителе — хлороформе
или ацетоне, проявляя незначительную димеризацию. Однако в 1Н
ЯМР-спектре комплекса 4, растворенного в CD3CN, присутствуют
уширенные сигналы, которые относятся к исходному комплексу 4,
комплексу 3, а также свободному 2-фснилпиридину, возникающему
в результате быстрых процессов обмена в растворе. Такое поведение
наблюдается и для соединения 5.
Исследование комплексов с помощью ЦВА выполнено в
растворе дихлорметана или ацетонитрила, содержащем в качестве
фонового электролита B114NBF4, с электродом сравнения Ag/AgCl
(в насыщенном растворе КС1, отделенном от основного раствора
мембраной). Обнаружено, что природа растворителя сильно влияет
на окислительно-восстановительные свойства комплексов палладия.
На рис. 7.7 приведены циклические вольтамперограммы димеров 1
с ацетатным мостиком и 2 с трифторацетатным мостиком в СН2С12.
Циклические вольтамперограммы ацетатного димера 1 и трифтора-
цетатного мономера 3 в ацетонитриле представлены на рис. 7.8.
Потенциалы окисления комплексов 1—3 приведены в табл. 7.1. Следует
1 RyubovA.D. // Inorg. Chem. 1987. 26.1252.
3 Ajkws AC, Xiao D,Y, Geibel M.A.L., ЯШег 71 // J, Am, Chem, Soc, 2010. 132.
14530: Powers D.C., Ritter T. // Accounts of Chemical Research, 2012. 45. 840.
J Dudkina Y.B., Mikhaybv D. Y., Gryaznova T. V., Tufatullin A.I., Kataeva O.N.,
VickD.A.t Budnikova У.И. Op. cit.
266
отметить окислительный потенциал свободного PhPy 2J5 В в СН2С12
или 2,08 В в CH3CN, причем заметных пиков для Pd(TFA)2 в этих
условиях не наблюдалось.
Рис 7.6. Равновесие мономер-димер 2-фенилпиридил трифторацетатв
палладия (II) в зависимости от растворителя
Электрохимичесхие свойства [(PhPy)Pd(p.-OAc)]2 и [(PhPy)
Pd(p>TFA)]2 в СН2С12 и CH,CN с 0,1 М Bu«NPF6 качестве фонового
электролита были описаны ранее1. Отчетливо видно, что 1 с аце-
Bercaw J.Е., DurrellA.C, Gray H.B., Green J.С, Hazari N., A. LabingerJ,,
Winkler J.R. Op. cit.
267
татным мостиком окисляется более легко, чем 2 с TFA-мостиком (см.
рис. 7.7, табл. 7.1). Однако форма и количество наблюдаемых пиков
на циклических волыамперограммах несколько отличаются от тех,
которые наблюдались в обсуждаемых выше условиях. В проведенном
исследовании не только логически обоснован эффект растворителя,
но и получена информация о различном количестве переносимых
электронов на стадиях окисления (см. ниже). Количество электронов
определялось с использованием метода1, включавшего хроноампе-
рометрию и стационарную вольтамперометрию на микроэлектроде2.
г 150
2,00 1,60 1,20 0,80 ' 0,40 0,00
Potential, V
Рис. 7.7. ЦВА [(PhPy)Pd(p-OAc)]21 (красный) и ({PhPy)Pd(u-TFA)]2 2 (зеленый)
в СН2С12.5 ■ 103 М, 0,1 М BU4NBF4, стеклоуглеродный электрод
0,50 2,00 ' U50 ' 1,00 ' 0^50 ' 0,00
Potential, V
Рис. 7.8. ЦВА [(PhPy)Pd(M-OAc)b 1 (красный) и [(PhPy)Pd(TFA)](CH3CN) 3 (синий)
в CH3CN. 10 3 М, 0,1 М Bu„NBF4, стеклоуглеродный электрод
1 Amatore С, АшЫ М„ Сака P., Jutand A„ Lefrou С, Ratlin Y. // J. EleclroanaL
Chera. 1990. 288.45.
1 Dudkina Y.B., Mikhaytov D. Y, Gryavtova T. V., Tufatullin A.I., Kataeva O.N.,
VickD.A., Budmkova Y.H. Op. ей.
268
Таблица 7.1
Потенциалы Е1У2 окисления (В) для палладиевых циклов 1—3
Комплекс
1
2
3
Растворитель
CHiCh
Е'
0,89
1,18
Е2
1,27
1,67
Е1
1,46
—
Превращается и 2
CH,CN
Е1
0,88
Е2
1,82'
Превращается в 3
1,72*
1,90'
* Для необратимых пиков дан Е,
В растворе СН3С12 ацетатное производное 1 (рис. 7.7, сплошная
кривая) подвергалось трем последовательным обратимым
окислениям, которые можно представить так: Pd11—Pd'VPd111—Pd111-, PdIU-
Pdm/Pdl,l-Pdlv- и Pd,|,-Pdlv/Pdlv-Pdlv-napbi (было обнаружено,
что абсолютное значение количества электронов, потребляемых
на первой стадии окисления, составляет 2,2),
Предложенное обоснование окислительно-восстановительного
поведения 1 приведено на рис. 7.9. В CHjCN 1 окисляется
последовательно по обратимым и необратимым стадиям, соответствующим
переходам Pd]|-Pdl7Pd"I-Pd,M и Pdm-Pdin/Pd,v-Pdlv (рИс. 7.8,
сплошная кривая). В обоих растворителях 1 существует в виде ди-
мера.
Е1
Pdf(PhPy)2(OAc)2-2e^^^Pd3+(PhPy)Pd3+(PhPy)(OAc)2
Pdv(PnPy)Pd3+(PhPy)(OAc)2 +Pd^(PhPy)2(OAc)2 ^==^
^=^ 2Pd2+(PhPy)Pd3+(PhPy)(OAc)2
Pd^(PhPy)2(OAc)2^^Pd^(PhPy)Pd^(PhPy)(OAc)2:?==i
^|=±Pd^(PhPy)2(OAc)2
Рис. 7.9. Предполагаемый механизм электроокисления 1 в СН2С12
Данные циклической вольтамперометрии подтверждают
трансформацию нуклеарности трифторацетат-производных в
зависимости от растворителя. Вольтамперограмма димера 2 включает две
квазиобратимые двухэлектронные волны (измеренное количество
п = 2,1), отнесенных к PdI,-PdII/PdI,,-Pd111 и Pd,H-Pdm/Pd|V-Pd,v-
парам (рис. 7.7, пунктирная кривая). Моноядерный комплекс 3
269
обнаруживает два очень близко расположенных одноэлектронных
необратимых процесса окисления, соответствующих парам Pd'yPd1"
и Pdm/Pd,v (рис. 7.8, пунктирная кривая). Таким образом,
преобразование димерной формы в мономерную приводит к необратимости
электрохимического окисления'.
В спектрохимических условиях имелась возможность наблюдать
спектры ЭПР-интермедиата при потенциале первого пика окисления
димера 1 в СН2С1Э (рис. 7.10). Так как спектр ЭПР для Pdm-Pdm-
димера нельзя получить из-за наличия двух Pd,ll-ueHTpoB, сильно
антиферромагнитно спаренных2, наблюдаемый сигнал (g = 2,14
и АН = 40 Гц) согласуется с парамагнитным состоянием комплекса
со смешанной валентностью Pd"-Pd1M, образованным в реакции
компропорционирования между исходными Pd"—Pd" и конечными
Pdm—Pdm частицами (см. рис. 7.9). Аналогичная реакция
наблюдалась ранее в ЭПР-злектрохимических условиях для двухэлектронных
процессов восстановления Ni"bpyX2 (bpy = 2,2-бипиридин)3. Усилия
получить низкотемпературный спектр ЭПР 1 успехом не
увенчались, потому что сигнал исчез при замораживании раствора (150 К).
Уменьшение интенсивности спектра ЭПР с понижением
температуры в отличие от закона Кюри свойственно для димеризации и
полимеризации в растворе4. Таким образом, эта особенность, вероятно,
указывает на образование одномерной цепочки металлоциклов Pd"1
при окислении комплекса 1, что известно в литературе5.
2750 3000 3250 3500 3750
Magnetic field /G
Рис 7.10.ЭПР-спектр, полученный в ходе окисления [(РпРу)Рс1(ц-ОАс)Ь
1 (раствор CHjCIj, комнатная температура, 0,9 В отн. Ад/АдС1)
1 Dudkina Y.B., Mikhaylov D. Y., Gryaznova Т. V., TufamUin A.I., Katoeva O.N.,
VlcicD.A., Budnikova Y.H. Op. cit.
г Mirica L.M., Khusnutdinava J.R. Op. cit.
i Budnikova Yu.G, Yakhvarov D.G., Morozpv V.f., Kargin Yu.M., fl'yasovА.У., Vyakh-
ireva Yu.N., Sinyashin O.G. // Russ. J. Gen. Chem. 2002. 72. 168; Budnikova Y.H.,
Mikhaylov D. Y, Gryazflovo T. K, Sinymhin Q,G. // ECS Trans. 2010. 25. 66.
4 Khusnutdinova J. Л., Rath N.P., Mirica L.M. Op. cit.
s Morton J.R., Preston K.F. // J. Am. Chem. Soc. 1992. 114. 5454.
270
Уникальная особенность трифторацетатных производных
образовывать моно- или биядерные па-^адиевые циклы позволила
провести исследование активности различных каталитических форм.
Были исследованы некоторые электрохимические окислительные
реакций С-Н-функционатизации 2-фснилпиридина (табл. 7.2).
Таблица 7.2
Pd-каталиэируемая функционализация 2-фенилпиридина
перфторалкилкарбоновыми кислотами
\=/ RpCOOH, I0%|Pd| ^ ( О \—(
/=( Элскгрохимическос ' /^=\ О—к + /=( о
й N окисление ь, N d i N
a b
Ms
Г
Г
у.
4'
5-
б"
Г
8'
9'
Катализатор
Pd(OAc)3
PdfTFAh
Pd(OAch
Pd(OAc)2
Pd(OAe)2
PdfOAch
Pd(TFAh
Pd(OAc)3
Pd(TFA)j
Растворитель
CHjCN
CHjCN
CHjCN
CHjCN
СИгС\2
CH3CI2
CHjCN
CH,CN
CHjCN
Кислота
Q,FuCOOH
C6F,3COOH
C6FuCOOH
HC4FBCOOH
HC4FSCOOH
CH3COOH
HC4FKCOOH
CFiCOOH
CFqCOOH
Продукт (выделенный выход
по исходному PhPy)
6а (65%)
6а (62%)
ба(15%) + 6Ь(37%)
7а (73%)
7а (14%)
9а (Следы)
7а (68%)
8а (Следы)
—
* Плотность тока 10 тА/смг. 2Ф электричества на PhPy пропущено.
Остальное — димер фенилпиридина и потери при выделении.
" Плотность тока 25 тА/см\ ЗФ электричества на PhPy пропущено.
Были использованы в качестве катализаторов Pd(OAc)2
и Pd(TFA>2, а в качестве реакционной среды дихлорметан или аце-
тонитрил. Эксперименты с использованием Pd(TFA)2 в СН2С12
проведены не были вследствие его низкой растворимости. Как видно
из табл. 7.2, продукты С—Н-функционалиэации были получены с
хорошими или умеренными выходами, причем наблюдалась полная
конверсия PhPy. Отрадно, однако, что электрохимическое окисление
комплексов палладия действительно приводит к перфторкарбок-
силатным или перфторалкильной производным, как представлено
в табл. 7.2. В случае использования перфторкарбоновых кислот
с длинной цепью можно получить перфторалкильные продукты
271
за счет увеличения силы тока и количества пропущенного
электричества.
Следует отметить, что оба катализатора — Pd(OAc)2, Pd(TFA)2
одинаково эффективны в CH^CN. Можно предположить, что функ-
ционализашш PhPy перфторкарбоновыми кислотами в CH3CN
проходит через образование моноядерного комплекса палладия(П)
с фторсодержащими заместителями катализаторов как для Pd(OAc)2,
так и для Pd(TFA)2. В подтверждение этого был выделен и
охарактеризован моноядерный комплекс 3, полученный в результате обмена
карбоновых кислот и полной конверсии 1 под действием трифторук-
сусной кислоты в ацетонитриле1;
Чтобы получить более глубокое представление о
превращениях мономер — димер в ходе реакции С—Н-функционализации,
были выполнены последовательное электрохимическое окисление
и дальнейшие стехиометрические реакции с 2-фенилпиридином для
I(PhPy)Pd(u-OAc)h 1.
Димер 1 электрохимически окисляется в присутствии уксусной
кислоты в дихлорметане (рис. 7.11, путь А). Обработка этого
окисленного продукта 2-фениллиридином дает 61 % выхода продукта 9а.
Во время этой последовательности операций при электроокислении
цвет реакционной смеси меняется от желтого до
коричневато-красного, что предполагает образование биядерного
Pdin-PdI[I-комплекса, а затем, после завершения реакции, снова получается
раствор желтого цвета. Эти изменения цвета соответствуют предыдущим
исследованиям2.
В растворе СНЭСЬ полное превращение двухъядерного
комплекса 1 с ацетатным мостиком в перфторкарбоксилатномостиковый
димер происходит при его обработке избытком перфторкарбоновой
кислоты, как можно видеть из эксперимента ЦВА (рис. 7.12).
1 Dudkina Y.B., Mikhaylov D. Г., Gryaznava Т. К, Tufatullin A J., Kataeva O.N.,
VicicD.A., Budnikova Y.H. Op. cii.
2 PowersD.C, Xiao D. Г.. Geibet M.A.L, Ritter T. Op. cit,
272
0)АсОН(4экв.),
электрохимическое
окисление в CH2G2',
<ii) PhPy.
40°С,4ч
C6FI3COOH
<4 3KD.)
CHjCN
,CCH.i
CftF
ОЛс
<i) электрохимическое
окисление;
<ii) PhPy,
4 ч
>
6Г13
Рис 7.11. Превращения 1 в 9а в СН3С1а (путь А) и 1 в ба в CH3CN (путь Б)
г150
i—i—i—i—i—i—i 1—i—i—I—350
2,00 1,60 1,20 0,80 0,40 0400
Potential. V
Рис. 7.12. ЦВА [(PhPy)Pd(|i-OAc)]2 в отсутствие (красный) и в присутствие
(бэкв.) H(CF2)4COOH (фиолетовый) в СН2С12 в сравнении с [(PhPy)Pd(|jL-TFA}]2
(синий}
Добавление перфторгептановой кислоты к 1 в аиетонитриле
приводит к растворимому продукту, что предполагает фрагментацию 1
до моноядерного Рйи-комплекса (рис. 7.11, путь Б).
Электрохимическое окисление этого раствора, видимо, приводит к образованию
мононоядерного комплекса PdIV, дальнейшая обработка которого
фенилпиридином дает 6а (67%). Таким образом,
электрохимическое окисление арилпиридинов под действием комплексов
палладия в высоких степенях окисления позволяет осуществлять
направленную функционализацию субстратов перфторкарбоновыми
кислотами, а также получать полиядерные структуры из палладиевых
циклов со связями Pd—Pd1.
7Л, КАТАЛИТИЧЕСКОЕ С-Н-ЗАМЕЩЕНИЕ С УЧАСТИЕМ
ЭЛЕКТРОХИМИЧЕСКИ ГЕНЕРИРОВАННОГО N1(111) КОМПЛЕКСА
Использование никеля в реакциях окислительного С—Н-за-
мещения в литературе встречается сравнительно редко, хотя,
безусловно, поиск таких процессов перспективен с точки зрения
замены драгоценных металлов-катализаторов на более доступные
и дешевые2. После успешных результатов, описанных выше, также
1 Dudkina К.Я., Mikhaylov D.Y.. Gryaznova T.V., Sinyashin O.G., Vide D.A.,
Budnikova Y,H. Op. cit.; Budnikova Y.H„ Dudkina Y.B., Khrizanforov M.N.,
Gryaznova T.V. Op. cit.; Dudkina К A, Mikhaylov D. Y, Gryaznova T. V., Tufat-
uBlnA.L, Kataeva ОМ, Иск D.A., Budnikova Y.H. // Oiganometallics. 2013. 32.
4785.
J Hachiya H„ Hirano AT., Satoh Г., Miura M. // Org. Lett. 2009. ill 737; I idem. //
ChemCalChem. 2010. 2. 1403; lidem. // Angew, Chem. Int. Ed. 2010.49. 2202:
lidem. //Angew Chem Int Ed Engl. 2010. 49. 2202; Kanyiva K.S., Kashihara JV„
274
изучалась реакционная способность [Ni(bpy)3]2+ в
электрокаталитической реакции перфторалкилирования (рис. 7.13, А)'.
CF7H(CFi),Br, [Ni(bpy),l
-2^/RFX
Б
QFuCOjH
|Ni(bpy)3]2+ или Pd(OVc)2
или Pd2(PhPy)2(OAc)2
CF,H
I '
.(CF2)4
62-85%
-«e9
02CC6Fn ^cof
.QFl3
Рис. 7.13. Схема фторалкилирования 2-фенил пиридина
Совместное окисление 2-фенилпнридина и бН-перфторгек-
силбромида в присутствии [Ni(bpy)3]2+ при потенциале 1,3 В отн.
Ag/AgCl приводит к образованию 2-(6Н-2-перфторогексил)пири-
дина 10 с более высоким выходом (62%). чем в присутствии
палладия. На основании ЦВА [Ni(bpy)3J2+ можно сделать вывод, что
процесс протекал при потенциале, необходимом для окисления
Ni(H)BNi(IU) (рис. 7.14).
Перфторалкил-карбоновые кислоты также были
протестированы на способность вступать в реакцию с никелем и палладием,
поскольку данные кислоты считаются очень полезным источником
перфторалкил-функциональной группы2. Совместное
электрохимическое окисление 2-фенилпиридина и перфторгептановой кислоты
Nakao Y.,Hiyama Т., OhashiM., OgashiS. // DaltonTransactions. 2010.39.10483;
Kanyiva K.S., F. Loebermann, Nakao Y„ Hiyama T. // Tetrahedron Lett. 2009. 50.
3463; Nakao Y. Ц Chem, Rec. 2011.11, 242; Nakao Y., fdei //.. Kanyiva K.S.,
Hiyama T. //J. Am. Chem. Soc. 2009, 131. 15996; Nakao Y. Kanyiva K.S.,
Hiyama T. // J. Am. Chem. Soc. 2008. 130. 2448; Nakao Y, Kashihara N.,
Kanyiva K.S., Hiyama T. // J. Am. Chem. Soc. 2008. 130. 16170; Nakao Y,
Morila E., Idei H., Hiyama T. // J. Am. Chem. Soc. 2011. 133.3264; Nakao Y.,
Yamada У.; Kashihara N.. Hiyama T. //J.Am. Chem. Soc. 2010. 132. 13666.
1 Dudkina Y.B., Mikhayhv D. Y, Grvaznova T. K, Sinymhin O.G., Vkk D.A., Bud-
nikova Y.H. // Eur. J. Org. Chem. 2012. 2114.
3 McReynolds KA„ Lewis R.S.,Ackerman L.K.G.. Dubinina G.G., Brennessel W. W.,
Vide DA. // J. Fluorine Chem. 2010. 131.1108.
275
(соотношение 1:1) в присутствии различных катализаторов привело
к прямым продуктам перфторалкилирования путем декарбоксили-
рования. как видно на рис. 7.13, Б. Были получены хорошие выходы
(81% для Pd2(OAc)2(PhPy)2 и 85% для [Ni(bpy)3]2+). Это значит, что
перфторалкил-карбоновые кислоты на самом деле являются
лучшими субстратами, чем перфторалкил-бромиды в реакциях прямой
фун кционал изаии и.
1,100-
-0,138-
<
л.
* -1,875-
-2,612-
-3.850- ,
2,250 1,688 1,1250,563 0,000
Потенциал, В
Рис. 7.14. ЦВА [Ni(BF4)2(bpy)3] 10 2 М, [NBuJtPFe] 10 ' М; в MeCN. СУ электрод,
скорость развертки 100 мВ/с; э. с. FcVFc
Предполагается, что на пути к образованию продукта
генерируется иптермедиат 6а. Наличие 6а в реакциошгой смеси было
подтверждено хромато-масс-спектрометрией. Когда в качестве катализатора
используется Pd(OAc)2, в реакционной смеси наблюдались три
продукта: промежуточный комплекс палладия (пики фрагментов в масс-
спектре 106 [Pd]+, 155 |QH4-C5H4N]+, 319 [QH4-C5H4NPd-OAcl+,
378 [С6Н4—C5H4NPd—(ОАс)2]+), орто-карбоксилированные частицы
6а (1М|+ = 517) и продукт окисления 6b ([М]+ = 473), Увеличение
времени электролиза приводит к появлению окончательного продукта
6Ь. Это уменьшает количество ба, и он, в конечном счете, пропадает.
Обобщенные результаты М(1Г)/М(Ш)-катализируемого орто-фгоро-
алкилирования 2-фенилпиридина приведены в табл. 7.31.
Электрокаталитический синтез фторалкилированного
2-фенилпиридина протекает в мягких условиях при потенциалах, которые
позволяют генеририровать комплексы никеля и палладия в высокой
степени окисления (см. рис. 7.13). Для осуществления реакции
необходимы только электрокатализатор и субстраты.
Dudkina Y.B., Mikhcytov D. У.. Gryaznova Т. V., Sinyashin O.G., Viae D.A., Bud-
nikova У.И. Op. cil.
276
Таблица 73
Продукты of орто-С-Н-замещения Ph-Py с различными катализаторами
Прекурсор/
катализатор
C,,F„COOH
CF3H(CF2)5Br
Pd2(PhPy)2(AcO)2
и^ч.
т
^Xt^J^tFu
KJ
81 &
iT^4!
L.N
IiCF2)5CF2H
30%
Pd(OAc>2
if*4!
L^N
Js^^QjF,;»
IT (>18%)
+
ln2CC6F13
lsT (0-15%)
ff^4*!
L^N
Jycf2)5cf2h
4^
10%
[Ni(bipy)3p
ll^4!
L.N
^L^QF13
KJ
85%
А^Ч.
kJ*
JL.(CF2)5CF2H
62%
7.3. ЭЛЕКТРОХИМИЧЕСКОЕ С-Н-ФОСФОРИЛИРОВАНИЕ
Хотя к настоящему времени удалось осуществить разнообразные
реакции прямой функционал изаци и связей С—Н, в том числе
катализируемые переходными металлами, примеров образования связей
углерод-фосфор немного, видимо, из-за сильных координирующих
свойств фосфорных реагентов1. Электрохимические методы
замещения атома водорода фосфорсодержащими группами весьма
малочисленны, но в основном последние удачные примеры описаны
Kagayama Т., NakanoA., SakaguchiS., Ishii Y. // Org. Lcll. 2006. 8. 407; Gau Y,
WangG., Chen L., Xu P., Zhao K., Zhou Y„ Han L.-B. //J. Am. Chem. Soc. 2009.
131. 7956; Kuainobtt Y., Yoshida Т., Takai K. // J. Org. Chem. 2011. 76. 7370;
HouC, Ren K, Lang &, #«X, Xia C.t Li F. // Chem. Commun. 2012. 48. 5181;
Xiang С-В., Bian KJ., Mao X.-ft., Huang Z.-Z. // J. Org. Chem. 2012. 77. 7706;
BergerO., Petit С, Deal E.L., Montchamp J.-L. // Adv. Synth. Catal. 2013. 355.
1361; Sokobv VJ., Troitskaya L.L., Reutov O.A. // J. Organomet. Chem. 1980.
202. C58; Bolm C, Wenz K., Raabe G. // J. Organomet, Chem. 2002. 662. 23;
Stepanova Y.A., Dunina, Smoliakova LP. // Organometallics. 2009. 28. 6546.
277
только для функционализашш нитроароматических субстратов1.
Образование связи углерод—фосфор в условиях каталитического
действия переходных металлов рассматривается как важная
методология получения различных видов фосфорных соединений, таких
как фосфонаты, фосфинаты, фосфиноксиды, фосфины и тл. Однако
первые каталитические методы введения фосфорных групп в
различные структуры, важность которых значительна в связи с
требованиями зеленой химии, были разработаны совсем недавно2.
Общие проблемы осуществления реакции С—Н-замещения,
описанные выше, распространяются и на процессы с участием Р-нукле-
офилов. В последнем случае также необходимо участие окислителя,
причем для каждой пары субстратов приходится проводить
предварительные эксперименты, учитывая способность реагентов к
окислению, прежде всего нуклеофила, что не может не влиять на результат
реакции. Известные используемые соокислители являются дорогими
(например, соли металлов группы платины и серебра), а органические
окислители с высоким молекулярным весом трудно отделяются от
реакционной смеси. Причем все они часто недостаточно селективны3.
Несмотря на успех некоторых исследователей, прежде всего Му-
раками4 и Ю5 в лиганд-напраплетгном С-Н-фосфонировании
ароматических субстратов, многие осложняющие факторы, а именно
высокие температуры процесса, длительность реакции, необходимость
использования избытка дорогостоящего серебряного окислителя
AgOAc, дополнительного основания, а также реагентов для
облегчения стадии восстановительного элиминирования, не всегда
удовлетворительные выходы, свидетельствуют о том, что поиск новых,
более простых и эффективных решений, важен и перспективен.
Использовался накопленный опыт электрохимического С—Н-
ацетоксилирования и перфторалкилирования6 для разработки метода
С-Н-фосфонирования 2-фенилпиридина в условиях эдектрохимиче-
1 Cruz H., Gallardo /., Guirado G. // Еит. J. Org. chem. 2011. 7378; ЩепочкинА.В.,
Чупахин O.H., Чарушин В. //., Петросян В. А. // Успехи химии. 2013. 82(8). 747;
Каргин Ю. М, Буд'никова Ю. Г. // Ж-л общ. химии. 2001.71. № 9. 1472.
2 FengCh.-G., Ye M.,Xiao K.J., US., YuJ.-Q. //J. Am. Chem. Soc. 2013. 135.9322;
LiOu, Yam Т., Ishida N., Murakami M. // Angew. Chem. Int. Ed. 2013. 52. 9801.
1 Lyons T. W., Sanford M.S. Op. cit.; Slahl S.S. Op. cit.; Canty A.J. Op. cit.
4 LiCh., Yano Т., Ishida N.. Murakami M. Op. cit.
? Feng Ch.-G., Ye M., Xiao K.-J., Li S., Yu J.-Q. Op. cil.
* Lyons T. W„ Sanford M.S. Op. cit.; Powers DC, Geibel M.A.L., Klein J.E.M.S.,
Hitter T. Op. cit.; Dudkina Y.B,f Mikhaylov D.Y., Gryaznova T. V., Sinyashin ОС,
Vicic D.A., Budnikova Y.H. Op. Cil.; Budnikova Y.H., Dudkina Y.B., Khri-
zanforov M.N., Gryaznova T.V. // J. Organomet. Chem. 20I4. 751. 301;
Mikhaylov D.Yu.. Budnikova Yu.H. // Russ. Chem. Rev. 2013. 82(9). «35;
Dudkina Y.B., Mikhaylov D. Y., Gryaznova T.V., TufatullmA.L. KataevaO.N., Vicic D.A.,
Budnikova Y.M.Qp. Cit.
278
ского окисления с участием катализатора Pd(Q\c)2 и диэтилфосфита
в качестве доступного фосфорного прекурсора в мягких условиях'.
Применение электрохимического метода позволило провести реакцию
без специально добавленного окислителя (AgQAc или другого) при
контролируемом потенциале в более мягких условиях при комнатной
температуре, а также проанализировать отдельные стадии реакции*
Так, совместное электрохимическое окисление 2-фенилпи-
ридина, диэтилфосфита (соотношение I : 1,1, прикапывание
ДЭФ в течение всего электролиза) и ацетата палладия Pd(OAc)>
(10 мольных %) дает продукт орто-фосфонирования 2-фенилпири-
дина с выходом 35—68% в зависимости от условий процесса2:
+ (ЕЮ)2Р(0)Н
(7.3)
11
P(0)(OEt)2
Наилучший результат (68%) был получен при проведении
электролиза в присутствии ацетата натрия в качестве основания и бен-
зохинона — реагента, облегчающего стадию восстановительного
злим книро ва ния,
Для установления механизма процесса был синтезирован
промежуточный шестичленный биядерный палладиевый комплекс 12,
аналогичный [(PhPy)Pd(BuOhP(0)h, описанному Мураками\ С этой
целью было осуществлено взаимодействие ацетатного димера фе-
нилпиридилпалладия 1, полученного по ранее описанной мотодикс4,
с диэтилфосфитом (ДЭФ) (1 : 2) в ацегонитриле:
+ 2(EtO),PIIO
-2ЗДСООН
Дудкина Ю.Б., Грязнова Т.В., Катаева О.Н., Будникова Ю.Г., Синяшин О.Г, //
Изв. РАН. Сер. химия. 2014. 2641.
Дудкина Ю. Б., Грязнова Т. В., Катаева О. И., Будникова Ю. Г., Синяшин О. Г.
Указ. сом.; Grayaznova T.V.^ Dudkina Y.B., fsfamov D.R., Kataeva O.N., Sin-
yaxhirt O.G., Vide DA., Budnikova Yu.ff, //J. Organomet. Chem. 2015. 785. 68.
Li CA., Yano 7*., Ishida N., Murakami M. Op. cii.
Sakai N.t AidderA,, HarmigJ.F. Op. cil.
279
+2<EtQ);PHQ
-2CH3COOH
Et04 yOEt
0-R
Pd Pd
P-& Ч
El(/ 4OEl
12
(7.4)
Реакция протекает легко при 20'С, через сутки из
реакционной смеси выпадает комплекс 12 с выходом 90%'. Ранее2, при
получении дибутилфосфона го-производного комплекса палладия
[(PhPy)Pd(BuO)2P(0)h из а-гидроксибутилфосфоната и [(PhPy)
Pd(OAc)]2, смесь нагревали в диокеане при 120°С в присутствии
2,2 экв. К2НРО<. Очевидно, при использовании ДЭФ лигандный
обмен протекает заметно легче.
Комплекс 12 затем был окислен на Pt электроде в CH,CN при
20Т в бескислородных условиях при потенциале его первого пика
окисления с полной конверсией в единственный продукт орто-фос-
фонирования 2-фенилпиридина 11с практически количественным
выходом (в спектре Я MPil P реакционной смеси присутствует
единственный сигнал с 5/. 18 мл.)3:
(7.5)
P(0)(OR)2
Диэтиларилфосфонат 11 был охарактеризован комплексом
физико-химических методов.
Электрохимические характеристики комплекса 12 были изучены
методом циклической вольтамперометрии. Кривая ЦВА комплекса
12 характеризуется двумя необратимыми пиками окисления, причем
их потенциалы находятся в более положительной области
(Ер = 1,18 В) по сравнению с потенциалами комплекса [(PhPy)
Pd(OAc)]2l (E]p =0,62 В).
1 Дудки па Ю.Е., Гряшова Т. В., Катаева О.Н., Будникова Ю.Г., Сипя шин О. Г.
Указ. соч.; Grayaznova T.V.. Dudkina Y.B., Istamov D.R., Kataeva O.N., Sin-
yashin O.G., Vicic D.A., Budnikova Yu.H. Op. cit.
2 LfCh., Yuno Т., Jshida N., Murakami M. Op. cit.
3 Дудкина Ю.Б.. Грязнова Т. В., Катаева 0.ff„ Будникова Ю.Г., Синяшин О, Г.
Указ. соч.; Grayaznova T.V.. Dudkina Y.B., Islamov D.R., Kalae\>a O.N., Sin-
y ash in O.G., Vicic DA., Budnikova Yu.H. Op. cit.
280
Ранее было показано1, что комплекс 1 окисляется лвухста-
дийно с переносом двух электронов на каждой стадии. Первый
пик окисления является обратимым и характеризуется металло-
центрированным переносом электрона и превращением каждого
Pd(Il) в Pd(lll). На второй стадии также происходит перенос двух
электронов, однако процесс окисления Pd(Hl) в Pd(lV) не обратим
в CH3CN (см. рис. 7.8). Если при окислении 1 удалось зафиксировать
образование промежуточного Pd(lll), то в случае комплекса 12
парамагнитных интермедиатов не обнаружено. Однако, скорее всего, для
этого комплекса характерны такие же две стадии процесса окисления
(также два пика, но при более положительных потенциалах)2.
Таким образом, реакция замещения водорода в орто-положении
2-фенилпиридина на фосфонатную группу протекает в
электроокислительных условиях селективно и не требует дополнительных
реагентов, промотирующих стадию восстановительного
элиминирования, которые обязательно присутствуют во всех ранее
опубликованных работах3 (NM MI, BQ, PPfy и тд.). Воспроизведена стадия
восстановительного элиминирования4 посредством нагрева комплекса
12 в CH3CN (при 120Т) в присутствии NMMI (2 экв.) в течение 4 ч.
Однако только половина исходного комплекса распалась до продукта
орто-фосфонирования 2-фенилпиридина 11. Таким образом,
электрохимическое окисление способствует протеканию стадии
восстановительного элиминирования интермедиата 12 намного эффективнее.
Согласно потенциалам окисления ключевых интермедиатов
[(PhPy)Pd(u-OAc)b 1 и [<PhPy)Pd(Et0bP(O)h I2( фосфорный
комплекс 12 окисляется труднее, чем ацетатный 1. При сравнении
химического окисления (окислитель AgOAc)5 и электрохимического
окисления видно, что в первую очередь окисляется ацетатный
комплекс. Последняя побочная нежелательная реакция приводит к орто-
ацетоксилированию 2-фенилпиридина6. Для получения целевого
продукта 11 с хорошим выходом необходимо обеспечить условия для
преимущественного окисления фосфорного комплекса, т.е. вести
электролиз при потенциале окисления комплекса 12.
Предположительную схему электрохимического С—Н- фосфорилирования
2-фенилпиридина можно представить следующим образом:
1 Dudkina Y.B., Mikhaylov D. Y., Gryaznova Т. V., TufatuUin А.Г, Kataeva O.N.,
Vicic D.A., Budnikova Y. H. Op. cit.
2 Ibid.
3 FengCh.-G., Ye A/., Xiao K.-J.r LiS.f YuJ.-Q. Op. cit.: LiCh., Yam Т.. Ishida Л'.,
Murakami M. Op. cit.
4 Li Ch„ Yano Т., Ishida Л'., Murakami Л/. Op. cil.
5 Yamaguchi J,, Yamaguchi A,D,. Itemi K. Op, cit,; Zhang A/,, Zhang Y„ Jk X.
Zhao H., WeipingSu.G.Li. Op. cil.
* Dudkina Y.B., Mikhayhv D. Y., Gryaznova T. V„ TufatuUin A.I., Kataeva O.N.,
Vicic D.A., Budnikova Y.H. Op. cil.
281
Со
К»
Pd<OAc)2
P(0)(OEt)
1/2
(ЕЮ)2Р(0)Н
EtO OEt
Pd Pd
EtO/ XOEt
EtO OEt
*\ /°-p,
Pd Pd
EtO OEt
-2e
Проведение электролиза при потенциале окисления фосфорного
комплекса 12 благоприятствует каталитическому процессу и
увеличивает выход целевого продукта.
Таким образом, разработан новый подход к введению фосфо-
натной группы в арилпиридины на примере 2-фенилпиридина,
основанный на электрохимическом окислении смеси 2-фепилпи-
ридина и диэтилфосфита при комнатной температуре в
присутствии ацетата палладия1. Выделен промежуточный шестичленный
биядерный палладисвый цикл, связанный с фосфонатным ли-
гандом через атомы кислорода и фосфора [<PhPy)Pd(ElO)2P(0)l2,
в качестве интермедиата. Препаративное окисление последнего дает
селективно целевой продукт фосфонирования С—Н-связи с
количественным выходом2.
В настоящей главе приведены результаты публикаций последних
лет, в которых предложен новый удобный подход к продуктам С—Н-
замещения 2-фенилпиридина, основанный на электрохимической
генерации комплексов палладия или никеля в высоких степенях
окисления. Изменяя количество пропущенного электричества,
можно получать продукты либо орто-аиетоксилирования и фтораце-
токсилирования, либо производные орто-фторалкилиропания.
Фторированные заместители кислоты затрудняют окисление димерных
циклов палладия по сравнению с нефторированными соединениями.
Удалось зарегистрировать сигнал ЭПР во время электроокисления
димера [Р(з\(РпРу)г(ОАс)г] и предварительно приписать его к сигналу
димера Pd<II)—Pd(III) со смешанной валентностью, Обнаружены
существенные различия в редокс-свойствах перфторзамещенных
палладиевых комплексов и нефторированньгх аналогов, а также
моно- и биядерных комплексов. Разработанная методика позволяет
также вводить фосфонатную группу в 2-фенилпиридин.
Предлагается более дешевая, «зеленая» и более управляемая
электрохимическая альтернатива реакциям окислительной С-Н-активации. С-Н-
активация с участием более доступного никеля в высоких степенях
окисления ранее не описывалась. Преимущество подхода
заключается в исключении окислителей, традиционно применяемых для
получения каталитически активного палладия (реже никеля) в пысоких
степенях окисления для С— Н-функционализации, которые очень
дороги, трудно отделимы от реакционной смеси и не всегда
совместимы с другими функциональными группами молекулы, а также
мягких условиях и хороших выходах продуктов. Обсужденные
реакции можно представить следующим образом:
1 Дудкима Ю.В,. Грязноеа ТВ., Катаева ОМ., Будникова Ю.Г., Синяшин О.Г,
Указ. соч.; Grayazflova Т.У.. Dudkina У.В., htamov D.R., Kataeva O.M., Stn-
yashin Q.G., Vide D.A., Budnikova Yu.H. Op. ciL
2 Ibid.
283
(7.7)
/~К P<0)(OEt)2
Глава 8. ОЦЕНКА ЭФФЕКТИВНОСТИ
ГОМОГЕННЫХ ЭЛЕКТРОКАТАЛИЗАТОРОВ
С ПРИМЕНЕНИЕМ МЕТОДА ЦИКЛИЧЕСКОЙ
ВОЛЬТАМПЕРОМЕТРИИ
Повышенный интерес к «солнечным» тошшвам стимулировало
интенсивные исследования молекулярных электрокатализаторов,
способных выделять водород из протонсодержащих растворов
(рис. 8.1), восстанавливать ССЬ и окислять воду. Определение точных
характеристик этих катализаторов требует тщательного и
правильного применения электроаналитических методов. Ниже изложены
основы циклической вольтамперометрии и выделены практические
экспериментальные моменты, которые предшествуют рассмотрению
применения циклической вольтамперометрии для определения ха-
рактериегик электтюкатализаторов. Также обсуждаются основные
показатели, необходимые для сравнения катализаторов, в том числе
перенапряжение (г|), потенциал катализа {Еа1), наблюдаемая константа
скорости (AobJ, частота оборотов, зависимая от потенциала.
Циклические вольтамперные отклики обшего электрокаталитического
одноэлектронного восстановления субстрата представлены наряду
с методами извлечения количественной информации из этих данных.
Затем обсуждаегся расширение лого анализа на более сложные
электрокаталитические схемы — выделение Н2 и восстановление С02.
Рис. 8.1. Схема электрокаталитического цикла выделения водорода
8.1. ВВЕДЕНИЕ
Изучение возможностей пересмотра, как говорят,
энергетического портфеля человечества побудило исследователей к поиску
вдохновения в схемах превращения энергии в природе, таких как
285
элегантный процесс фотосинтеза преобразования солнечного света
в топливо. Гидрогеназные ферменты с их способностью
индуцировать окисление водорода или восстанавливать протоны до водорода
являются прекрасным примером катализа образования топлива
в природе, потому что водород является чистым, желательным
топливом1. Сложные структуры ферментов гидрогеназ содержат
неорганические активные сайты. Это способствовало желанию химиков-
неоргаников подражать структуре и активности гидрогеназ с целью
создания синтетических катализаторов, способных производить
водород из электронов и протонсодержащего сырья'.
Исследования этих малых молекул также помогают в
фундаментальном понимании механизма фермента и обеспечивают
платформу, на которой исследуются структурно-функциональные
взаимосвязи. Параллельно интенсивная работа проводилась в
направлении развития молекулярных катализаторов восстановления
С02 с получением жидкого углеродного топлива или прекурсоров
топлива3, а также окисления воды и необходимой полуреакции,
сопряженной с этим восстановительным процессом4. Хотя до сих
пор не ясно, возможно ли получение молекулярных неорганических
катализаторов в масштабах, необходимых для глобальных
энергетических решений5, эти комплексы могут стать неотъемлемой частью
устойчивого в энергетическом плане подхода к химическому
производству топлива в сочетании с возобновляемыми источниками
энергии, например, солнечным светом иди ветром*.
Отчасти из-за этой недавней эскалации
научно-исследовательских усилий, направленных на развитие устойчивой энергетики,
1 Vignais Р.М., Bifloud В. Ц Chem. Rev. 2007. 107.4206-4272.
2 WangM., Chen L., Sun L. // Energy Environ. Sci, 2012. 5. 6763-6778; Thoi VS.,
Sun У., Long J.R., Chang CJ. // Chem. Soc. Rev. 2013. 42. 2388-2400;
Felton G.A.N., Mebi C.A., Petro В J., VannucciA.K., Evans D.H., Glass U.S., Li-
chtenberger D.L.J. jf Organomet. Chem. 2009. 694. 2681-2699; Dempsey J.L.,
WwklerJ.R., Gray ff.B. // Comprehensive Inorganic Chemistry II /eds. J. Reedijk,
K. Poeppelmeier. Oxford U.K.: Elsevier, 2013. \bl. 8. P. 553-565.
* Benson ЕЕ.. КиЫак С.Р., Sathrum A.J., Smieja J.M. It Chem. Soc. Rev 2009.
38. 89-99; Costentin C, Robert A/., Saveant J.-M. // Chem. Sue. Rev. 2013. 42.
2423-2436.
4 Wasyienko D.J., Palmer R.D., Berlinguelie C.P, // Chem. Commun. 2013,49. 218-227;
Conception JJ., JurxsJ.W., Brennaman M.K., Hoertz P.G.. PatrocmoA,O.T.,
Murakami lha N. Y., Templeton J, L., Meyer TJ/i. Лес. Chem. Res, 2009.42.1954-1965.
* Pinaud B,A., BenckJ.D.. Seitz L.C., Forman A.J., Chen Z., Deutsch T.G., James B.D.,
Baum K.N., Baum G.N..Ardo S„ Wang //., Miller E, Jaramillo Т.Е. // Energy
Environ. Sci. 2013. 6. 1983-2002; SalhrumA.J„ Kubiak C.P J. // Phys. Chem. Lett.
2011.2372-2379.
* Lewis N.S., Nocera D.G. // Proc. Nail. Acad. Sci. U. S. A. 2006.103.15729-15735;
Gray H.B. // Nat. Chem. 2009.1. 7.
286
в последнее десятилетие наблюдается резкий всплеск использования
электроаналитических методов химиками, причем как
неорганиками, так и органиками1. Хотя протоны и электроны для
каталитического производства водорода и восстановления С02 в конечном
счете нужно брать из воды, катализаторы, как правило, оцениваются
в схемах полуреакций, в которых электроны поставляются
электродом, а протоны — кислотой. Поэтому исследователи обратились
к электрохимии, чтобы оценить катализаторы для производства
топлива. В идеале электрохимические методы могут быть
использованы для определения каталитической константы скорости,
получения информации о природе активного катализатора и помочь
исследовать механизм. Однако общий консенсус в сообществе, как
точно и правильно извлекать данные характеристики из
электрохимически* данных, отсутствует. Некоторые методы основаны на
предположениях, которые не всегда тщательно проработаны, а
следовательно, могут привести к неправильной интерпретации полученных
данных.
Настоящая глава касается исследователя, который стремится
использовать циклическую вольтамперометрию для оценки
молекулярных катализаторов. Прежде всего из литературы были выбраны
примеры по электрокаталитическому выделению водорода в
контексте нашега обсуждения, но представленные инструменты могут
быть непосредственно распространены на восстановление С02,
окисление водорода, окисление воды и восстановление кислорода,
а также другие электрокаталитические медиаторные реакции.
Основные понятия ЦВА вводятся наряду с общими
экспериментальными методами, прежде чем будут описаны конкретные уравнения
и понятия, относящиеся к молекулярному электрокатализу. Упор
делается на определении и обсуждении показателей качества
(характеристик) в рамках гомогенного катализа.
8.2. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
ЦВА является электроаналитическим методом, наиболее часто
используемым для изучения молекулярных электрокатализаторов.
В этом разделе мы кратко обсудим технику ЦВА, прежде чем
представить практические экспериментальные соображения. Чтобы
тщательно обучиться основам ЦВА, можно обратиться к
многочисленным прекрасным источникам, имеющимся в литературе2.
1 Wang М.,Chen L, Sun L, Op. cit.; ТЫ VS.. Sun Y., Long JR., ChangC. Op. cit.;
Felton GA.N.,MebiC.A., PetroBJ., VnnnuaiA.K., Evans DM. Op, cit,;Glass R.S.,
LichtenbergerD.LJ. Op. cil.
3 Evans D.H., OVonneil K.M.. Petersen R.A., Kelly MJ.J. // Chcm. Educ. 1983.
60. 290-293; Mabbott G.A.J. // Chem. Educ. 1983. 60. 697-702; Compton R.G.,
287
8.2.1. Циклическая вольтамперометрия
На рис. 8.2 показана типичная циклическая вольтамперограмма
в растворе частиц Р, подвергающихся обратимому одноэлектрон-
ному восстановлению в частицы Q. От начального потенциала, при
котором комплекс существует номинально в единственном редокс-
состоянии Р, потенциал сканируется в отрицательном направлении.
Когда потенциал достаточно отрицателен, чтобы восстановить Р,
начинает протекать катодный ток, так как Р восстанавливается до Q.
Рис В.2. Изменение концентрации ([С], тМ) в зависимости от расстояния
от электрода (х, см • 1(H) в различных точках во время записи обратимой
волны ЦВА: СР (сплошная линия); С0 (пунктирная линия). Скорость
сканирования = 100 мВ • с~\ А, В, С, D, E, F — разные профили
(цит. по: Compton R.G., Banks СЕ Understanding Voltammetry. 2nd ed. L: Imperial
College Press, 2011)
Banks C.E. Understanding \foltammetry. 2nd ed. London: Imperial College Press,
2011; Kissinger P.T., Heineman W.R.J. // Chem. Educ. 1983. 60. 702-706;
Izutsu K. Electrochemistry in Nonaqueous Solutions. Wfeinheim: Wiley-VCH Verlag
GmbH & Co. KGaA, 2003; Sawyer D. Т., SobkowiakA, Roberts J.L.
Electrochemistry tor Chemists. 2nd ed.: N. Y: John Wiley &. Sons, Ine., 1995.
268
Отклик тока определяется соотношением двух параметров:
константы скорости переноса электрона между электродом и
анализируемым веществом {кс пропорциональна к\ стандартной
гетерогенной электрохимической константе скорости присущее свойство
изучаемой пары электрод — аналит, обычно указывается в см • с ')
и транспорта (путем диффузии) электроактивных материалов к
поверхности электрода (описывается коэффициентом массопереноса,
/л Г)1. Когда перенос электрона на аналит является быстрым по
отношению к массопереносу анализируемого вещества (к" » тТ —
электрохимическая обратимость), а электрогенерированный
продукт является стабильным в масштабе времени эксперимента
(химическая обратимость)2, то наблюдаемый возникающий катодный
ток можно объяснить с помощью уравнения Нернста для реакции
Р ■+- е ^ Q, где СР и CQ — концентрации Р и Q на поверхности
электрода, а £"°'является формальным потенциалом процесса
восстановления3:
Е = Е°'+—1пЯ (8.1)
nF CQ
Когда приложенный потенциал изменяется, отношение СР и С0
регулируется в соответствии с уравнением Нернста. В пределах
быстрой электродной кинетики резкое увеличение катодного тока
наблюдается при потенциалах в области £"", так как Р
восстанавливается.
Можно предположить, что ток должен экспоненциально расти
даже при более отрицательных потенциалах, как предсказано
теорией Батлера-Фольмера4. На самом деле при достаточно
отрицательных приложенных потенциалах любые окисленные частицы
на поверхности электрода восстанавливаются, в результате чего
концентрация окисленных частиц (Ср) вблизи электрода (в пределах
диффузионного слоя) должна уменьшиться, как проиллюстрировано
профилями концентрация — расстояние, показанными на рис. 8.1.
Этот эффект проявляется на вольтамперограмме в виде пика тока
(|р) с последующим уменьшением тока по мере того, как потенциал
сканируется далее в отрицательную область. Образуется градиент
концентраций, а окисленные частицы должны транспортироваться
из массы раствора к поверхности электрода для того, чтобы протекал
1 Mabhott G.A. Op. cit; Compton R.G., Banks С. E. Op. cit.
1 Fetton G.A.N., MebiC.A., Petro B.J., VannucciA.K., Evans D.H., GlassR.S., Uchten-
hergerD.LJ. Op. cit.
} Evans DM., О 'Connetf K. M., Petersen R.A., Kelly M.J. Op. cil.
4 Bard A.J., Faulkner L.R. Electrochemical Methods: Fundamentals and
Applications. 2nd ed. Hobo ken, NJ: John Wiley & Sons, Inc., 2001.
289
дополнительный ток. Когда в ЦВА-экспериментах используются
высокие концентрации электролитов и растворы не перемешиваются,
единственным способом массонереноса анализируемого вещества
можно считать диффузию с хорошим приближением, а скорость
диффузии через обедненную диффузионную зону определяет
наблюдаемый ток.
Толщина диффузионного слоя (8) зависит от шкалы времени (/)
сканирования вольтамперограммы (6 - (/)/),/2, где D — коэффициент
диффузии Р, см2 • с1), а /обратно пропорционально скорости
сканирования v (r ~ RT/Fv)*. Таким образом, диффузионный слой
утолщается в течение одного цикла, а относительная толщина уменьшается
с увеличением скорости сканирования. При потенциале
переключения направление сканирования изменяется на противоположное,
но ток продолжает течь, так как приложенный потенциал еще
достаточно отрицательный, чтобы восстановить Р, хотя концентрация этих
частил на электроде значительно уменьшается начиная с этой точки,
а толщина диффузионного слоя увеличивается.
Важно отметить, что расходование Р соответствует
накоплению Q. По мерс того как приложенный потенциал становится
все более и более положительным, Q окисляется (так что отношение
Cp/Cq на поверхности электрода удовлетворяет уравнению Нернста),
а анодный ток протекает по принципам, описанным для катодного
тока.
8.2.2. Экспериментальные соображения
Переходя от теории к практике, рассмотрим сначала
экспериментальные условия, в которых выполняются электрокаталитические
измерения. Ниже мы обсудим каждую деталь электрохимического
эксперимента и общую практику, которой следует придерживаться.
Выбор растворителя. Хотя вода является целевым желательным
сырьем и растворителем для производства энергии с участием
солнечного света, многие молекулярные электрокатализаторы
изучаются в органических растворителях. Чаще всего выбор растворителя
диктуется растворимостью и стабильностью катализатора2.
Растворитель влияет на несколько экспериментальных факторов, включая
электролит, электрод сравнения и источник протонов (для
каталитической генерации Н2 и восстановления С02).
Электрохимические окна определяются потенциалами, при
которых данная система начинает проявлять отклик тока, не связанный
с катализатором. Растворитель, выбранный электролит, рабочий
1 Bard A. J,, Faulkner L.Л. Electrochemical Methods: Fundamentals and
Applications. 2nd ed. Hoboken, NJ: John Wiley & Sons, Inc., 2001.
2 kytsu K. Op. ch.
290
электрод, субстрат влияют на пределы, в которых могут быть
выполнены эксперименты. Во-первых, окиатителъно-восстановительныс
свойства растворителя диктуют окно потенциалов. В воде, например,
отрицательный предел окна потенциалов определяется
восстановлением воды до водорода на поверхности электрода, в то время как
положительный предел определяется окислением воды на большинстве
электродов. В неводных системах реакции, протекающие при
предельных значениях окна потенциалов, варьируются от растворителя
к растворителю, но окно потенциалов можно определить по
потенциалу, при котором ток протекает за пределами тока, наблюдаемого
для заряжения двойного слоя1. Во-вторых, электролит также может
участвовать в окислительно-восстановительном процессе, который
ограничивает окно, хотя это встречается редко, потому что общие
электролиты выбираются с учетом их инертной природы. В-третьих,
выбор рабочего электрода будет диктовать ширину потенциального
окна. Наконец, в-четвертых, прямое восстановление протонов (см.
раздел «Источники протонов») на электроде также ограничивает
полезный рабочий диапазон, когда проводятся определенные типы
катализа.
Применение растворителей с низкой проводимостью
(например, органических растворителей ацетонитрила и дихлор-
метана) может привести к большим ошибкам в измеряемом
потенциале в результате нескомпенсированного сопротивления,
который называют омическим падением. Каждая система
растворитель/электролит имеет уникальное сопротивление
раствора /?„, который вызывает падение потенциала (,Е0()).
Следовательно, потенциал на поверхности электрода задастся по
формуле £"ас,иа| = fmcHsurud - Eqd2- Величина EOD может динамически
изменяться во время эксперимента в зависимости от
измеряемого тока, а эта ошибка может быть оценена с помощью закона
Ома: E0D = im„aun:d^u- Катодные (отрицательные) токи приводят
к ошибочно слишком отрицательным измеренным потенциалам,
в то время как анодные (положительные) токи приводят к
ошибочно слишком положительным измеренным потенциалам.
Омическое падение можно снизить тремя способами: 1)
размещением кончика электрода сравнения в непосредственной близости
от поверхности рабочего электрода с использованием Луггмн-ка-
пилляра; 2) уменьшением размера рабочего электрода так, чтобы
протекат меньший ток; 3) увеличением проводимости растворителя
за счет более высокой концентрации электролита. Как средство
оценки сопротивления раствора ацетонитрил/электролит, можно
1 Izutsu К. О p. cit.
2 MyiandJ.C, Oldham K.B. // Anal. Chem. 2000. 72. 3972-3980.
291
использовать хроноамперометрию'. После того как омическое
падение уменьшаю! при настройке эксперимента, оставшаяся ошибка
омического паления может быть частично исправлена путем
использования активной положительной обратной связи, доступной в
некоторых коммерческих потенциостатах. В качестве альтернативы
омическое падение может быть приблизительно скорректировано
после сбора данных, если известно сопротивление раствора, с
помощью закона Ома2.
Электроды сравнения. Несколько электродов сравнения обычно
используется в водных электрохимических измерениях, например,
насыщенный каломельный электрод (SCE), стандартный
водородный электрод (SHE)3 и электрод серебро/хлорид серебра
(Ag/AgCl). Следует отметить, что электрод Ag/AgCl может быть
получен с различным составом электролитов и концентраций,
которые определяют электродный потенциал, хотя насыщенный
КС1 является наиболее распространенным. Однако для неводных
измерений первичный выбор электрода сравнения не
стандартизирован отчасти потому, что нет универсально надежного электрода
сравнения4. Использование водного электрода для измерений в
неводных средах вводит неопределеную переменную и неизмеря-
емый жидкостной скачок потенциала в ячейке, на который влияют
как растворитель, так и ионная сила5. Загрязнение неводного
раствора растворителя водой является другой проблемой. Кроме
того, потенциалы, измеренные в различных растворителях,
непосредственно не сопоставимы, так что неинформативно ссылаться
на водный электрод при выполнении измерений в органическом
растворителе.
Для ацетонитрила и других неводных растворителей наиболее
стабильным выбором представляется неводный электрод Ag/Ag+
(рис. 8.3)*'. Однако потенциал неводных электродов может
варьироваться в разных экспериментах благодаря изменению
концентрации Ag+, электролита или растворителя. Кроме того, следует
принимать во внимание то, что в литературе при сравнении данных
по электроду Ag+/Ag используются различные составы электрода
сравнения (природа электролита, концентрации и т.д.), что приводит
к несоответствию сообщаемых редокс-потенциалов.
1 McCarthy В. D., Martin D.J., Rountree E.S., Uiiman A.C., DempseyJ.L. // Inorg.
Chem. 2014. 53. 8350-8361.
2 Ibid.
J Ramette R. WJ. 11 Chem. Educ. 1987. 64. 885.
4 I-,utsu K. Op. cit.
5 Jzutsu K. Op. cit.; Pavlishchuk V. V., Addison A. W. 11 Inoi^. Chim. Acta. 2000. 298.
97-102.
6 Compton R.G., Banks C.E. Op. cil.; Izufsu K. Op. cit.
292
.'-
,/
1 —
- Ag проволока
-Стеклянная трубка
гСжатая насадка
трубки
Пористый стеклянный
кончик
Рис. 8.3. Конструкция электродов сравнения
Таким образом, в соответствии с рекомендациями ИЮПАК
(IUPAC — Международный союз теоретической и прикладной
химии), окислительно-восстановительные потенциалы,
измеренные с использованием неводного электрода, должны
соотноситься с внутренним соединением сравнения, в частности
ферроценом (Fc), бис (бифенил) хромом(О) (ВСг) или аналогичной
окислительно-восстановительной парой, которая родственна Fc1. Fc+/Fc
и BCr+/BCr — окислительно-восстановительные пары, имеющие
вполне определенные потенциалы и совместимые с неводными
условиями, они могут быть добавлены непосредственно к
анализируемому раствору до или после анализа интересующих комплексов
или других субстратов. Шкапы для пересчетов доступны, но ссылки
на потенциал пары Fc+/Fc (В отн, Fc+/Fc) являются
предпочтительными2.
Электрод Ag/AgNOj является обычно используемым нсводным
электродом Ag/Ag+.
Подготовка к выбору электродов, включая электрод Ag/AgN03,
показанного на рис. 8.3, следующая. Чистая поверхность серебра
может быть получена путем легкой полировки наждачной бумагой
шероховатостью 600 или погружения на 2 с в 0,1 М HN0.1. а затем
ополаскивания деионизированной водой и ацетоном. Серебряный
1 Gritzner С. Кша J. // Pure Appl. Chem. 1984. 56. 461—466; Gagne R.R.,
KovaiCA., Lisensky G.C. // Inoig. Chem. 1980. 19. 2854-2855,
1 Pavlishchuk V. V., Addison A. W. Op. cit.; Connelly N.G., Geiger W.E. // Chem. Rev.
1996.96.877-910.
293
провод вставляется в тело электрода сравнения, которое
конструируется из стеклянной трубки с викором Vycor (Coming), CoralPor
(Schott Glass) или пористым тефлоновым кончиком. Следует
проявлять особую осторожность, чтобы избежать загрязнения
исследуемого раствора ионами Ag+ из-за утечки из электрода сравнения
AgNO> Вымываемый Ag+ может усилить активность нативного
электрода к выделению водорода Н> (Ag(0), электроосажденный
на стеклоуглеродном электроде, может быть обнаружен по
необратимому восстановлению Ag(I) при примерно —0,53 0,77 В,
а также необратимому окислению Ag(0) при —0,14 В отн. Fc/Fc+
в ацетонитриле)1.
В качестве электрода сравнения также может быть использовано
псевдосравнение. Электрод псевдосравнения является неточным,
состоит из металлической проволоки (платиновой, серебряной или
золотой). Хотя псевдосравнение позволяет избежать проблемы утечки,
наблюдаемой для электродов Ag/AgNO:„ потенциал электрода
псевдосравнения плохо определяется и зависит от состава раствора
электролита. Редокс- потенциалы, измеренные при использовании
электрода псевдосравнения, можно соотносить с помощью
внутреннего соединения сравнения (Fc). В то время как электрод
псевдосравнения может быть погружен прямо в анализируемый раствор,
крайне важно изолирован электрод псевдосравнения в отсеке,
отделенном фриттой во время измерений, в котором состав образца
меняется в процессе эксперимента, например, в каталитических
экспериментах с выделением Нэ с участием кислоты, титруемой между
измерениями. Если электрод не изолирован, внутреннее сравнение
(например Fc) должно присутствовать при каждом отдельном
измерении (не добавляться после измерений). Так что любое изменение
потенциала может быть исправлено, а экспериментатор должен
знать о любой реакционной способности электрода псевдосравнения
по отношению к веществам, присутствующим в растворе или
пассивации электрода псевдосравнения.
В частности, должен быть получен фон внутреннего стандарта
только с субстратом и электролитом (без кагал и затора). И последнее:
экспериментатор также должен проверить, что внутренний стандарт
(сравнение) никоим образом не влияет на измерения. Это может
быть подтверждено путем выполнения сканирования перед
добавлением стандарта и после, чтобы проиллюстрировать
воспроизводимость данных, представляющих интерес.
Источники протонов. Выбор источника протонов для
каталитического выделения Н2 и восстановления СО? начнется важным
фактором. Следует рассмотреть и несколько других факторов: стабиль-
McCarthy BM., Martin D.J., Rountree E.S., VllmanA.C, DempseyJ.L. Op. tit.
294
ность и реакционную способность катализатора, прямое
восстановление кислоты на поверхности электрода, влияние воды на источник
прогонов, а в противном случае — неводные условия.
Реакционная способность катализатора по отношению к
источникам протонов диктуется как термодинамическими, так и
кинетическими параметрами. Важно отметить, что катализатор не может
восстановить кислоту до водорода при потенциалах, положительнее
термодинамического потенциала восстановления. В водном растворе
термодинамический потенциал выделения Н2 определяется как О В
отн. НКЭ и варьируется на —59 мВ на единицу рН с увеличением рН
в соответствии с уравнением Нернста, В безводном растворе
термодинамический потенциал выделения Н2 зависит от природы
кислоты и растворителя, гомосопряжения и концентрации кислоты.
Термодинамический потенциал выделения Н3 изданного источника
протонов (кислоты) должен быть определен для точных
экспериментальных условий, как более подробно обсуждается в разделе
«Перенапряжение (л)»1. Кроме того, наблюдаемая кислотность, а
следовательно, термодинамический потенциал восстановления молекулы
зависит от процессов гомосопряжения, гетеросопряжения и
избирательной сольватации2. Степень этого влияния диктуется природой
и концентрацией кислоты и используемым растворителем —
факторами, которые все должны быть рассмотрены при выборе
экспериментальных параметров3.
Возможность прямого восстановления протонов на рабочем
электроде следует рассматривать во всех экспериментах (рис. 8.4)4.
Потенциал, при котором рабочий электрод восстанавливает
протоны, варьируется широко и зависит от источника протонов и
электродного материала. Платина обычно используется как рабочий
электрод в электроаналитической химии в целом, но из-за своего
исключительного свойства способствовать выделению Нг при
относительно положительных потенциалах в протонодонорных растворах
она не считается хорошим выбором при изучении элсктрокатали-
тических реакций5. Ртутные электроды дают самое большое окно
1 Felton CAN., Glass R.S., Lichtenberger D.L., Evans D.H. // Inorg. Chem. 2006.45.
9181-9184: Roberts J.AS.. Bullock R.M. /j Inorg. Chem. 2013.52., 3823-3835.
1 Fourmond V., Jacques P.-A.. Fontecave M.. Artero V. // Inorg. Chem. 2010. 49.
10338-10347; Kaupmees A., Kaljurand /.. Leito I.J. // Phys. Chem. A 2010. 114.
11788-11793; CoeneeJ.F. // In Progress in Physical Organic Chemistry / eds. A.
Slreitwie&er, Jr., R.W. Tail. Hobokcn, NJ: John Wiley & Sons, Inc., 1967. Vbl. 4.
R 45—92.
3 Izutsu K. Op. cit.; Kaupmees A'., Kaljurand I., Leito I.J. Op. cit.
4 McCarthy B.D., Martin D.J., Rountree £.£„ Uliman A.C., Dempsey J.L. Op. cit.;
Felton G.A.N., Glass R.S., Lichtenberger D.L.. Evans D.H. Op. cit.;
* Felton G.A.N., Glass R.S., Lichtenberger D.L., Evans D.H. Op. cit.
295
для электрокаталитических экспериментов с добавлением кислоты,
но требования безопасности, стоимости и простоты использования
привели большинство исследователей к использованию стеклоугле-
родных (СУ) электродов. Следует отметить, что очень важно
полировать рабочий электрод перед каждым сканированием (а не только
один раз до первого сканирования), потому что загрязнение
поверхности электрода может привести к ошибочным значениям и нево-
спроизводимости'. Кроме того, следует отметить, что качество СУ
может варьироваться в зависимости от источника и производителя.
Когда эти первые два фактора рассматриваются вместе, диапазон
потенциалов для определенной кислоты может лежать между
термодинамическим потенциалом восстановления протонов и потенциалом,
при котором электрод непосредственно восстанавливает кислоту2.
i i i i i i i i i i i i i i i i
-0,5 -1,0 -1,5 -2,0
EJVoth. FcVFc
МММ
-2,5 -3,0
Рис. 8,4. Вольтамперограммы с линейной разверткой потенциала
10 мМ уксусной кислоты в ацетонитриле с 0,10 М тетрабутиламмония
гексафторфосфатом при 1,0 В/с. Электроды как указано:
GC — стеклоуглерод, Au(Hg}— амальгама золота. Электродные площади
отличаются, поэтому приведена плотность тока. Обратимый потенциал
восстановления уксусной кислоты показан для сравнения (цит. по:Felton G.A N.f Glass R.S.,
LichtenbergerD.L, Evans D.H. // Inorg. Chem. 2006.45.9181 —9184)
Интересующий нас электрокатализ должен протекать в пределах
этого интервала. Недавно были опубликованы примерные окна по-
1 McCarthy B.D., Martin D.J., Rountree ES„ Oilman AC, DempseyJ.L. Op. cit
2 Ibid.
296
тенциалов в ацетонитриле для 20 кислот'. С практической точки
зрения следует определить окно потенциалов достоверности для
экспериментальных условий путем независимого измерения
прямого восстановления субстрата (например, кислоты, С02) на рабочем
электроде. Это значение должно быть сообщено вместе со всеми
данными, чтобы избежать неправильной или вводящей в заблуждение
производительности индивидуального катализатора3.
Недавно было показано, что многие катализаторы выделения
Н2, первоначально изученные в неводных условиях, показывают
значительное улучшение каталитических токов при добавлении
воды3. Вода может а>шять на каталитические реакции несколькими
способами. Например, следы воды, как было показано и
экспериментально, и с помощью расчетов, существенно влияют на
относительную кислотность многих кислот в неводных растворителях.
Заряженные частицы, как правило, селективно сольватируются
водой. Это процесс, который особенно эффективен для анионных
сопряженных оснований с высоко локализованным отрицательным
зарядом; кислотность в органических растворителях, следовательно,
увеличивается при добавлении воды.
Кроме того, сольватация анионов через водородные связи с водой
может скрыть нюансы различий между кислотами, которые влияют
на рКа подобно эффектам заместителей4. Причем восстановление
НС104 на ртутном электроде в ацетонитриле, как было показано,
сдвигается к более отрицательным потенциалам при добавлении
воды5. Это наблюдение было связано с большой разницей в
энергиях сольватации протона между ацетонитрилом и водой (для
первого намного ниже), в результате дающей кислотный CH3CNH+,
который легче восстанавливается по сравнению с более слабой
кислотой Н30\
Вода также может влиять на механизм катализа: она может
действовать как реле протонов, отдавая протон на активный сайт и
принимая протон с внешней сферы кислоты, а малый размер воды,
1 McCarthy B.D., Martin D.J., Rountree E.S., Ullman A.C., Dempsey J.L. Op. cit.
1 McCarthy B.D., Martin D.J., Rountree E.S., Uliman A.C., Dempsey J.L. Op. cil.;
Feltou G.A.N., Glass R.S., Lichtenberger D.L.. Evans D.H. Op. cit.
} Helm M.L., Stewart MP., Bullock R.M., DuBois M.R., DuBois D.L. //Science. 2011.
333. 863-866; KHgore V.J., Roberts J.A.S., Pool D.H., Appel A.M., Stewart M.P.,
DuBois M.R., Dougherty W.G., Kassei W.S., Bullock R.M., DuBois D.L.J, //Am.
Chem. Soc. 2011, 133, 5861-5872; Kiigore U.J., Stewart MP.. Helm ML.,
Dougherty W.G., Kasset W.S., Dubois M.R., Dubois D.L., Bullock R.M. // lnorg. Chem.
201 L 50. I090S-1091S; Wiese S.. Kiigore U.J., Dubois D.L., Bullock R.M. //ACS
Catal. 2012. 2. 720—727.
4 Izutsu K. Op. cit.
* CoetzeeJ.E, Kolthoffl.M. //J. Am. Chem. Soc. 1957. 79. 6110-6115.
297
таким образом, разрешает наиболее широкий доступ к
пространственно загруженным активным центрам1. Сдвиг рКа при добаштении
воды может снизить термодинамический потенциал прямого восста-
новления кислоты на электроде, что может привести к
значительному вкладу тока этого нежелательного процесса в этом же
интервале потенциалов в качестве каталитического тока восстановления.
И последнее: концентрация случайной воды может варьироваться
от образца к образцу создавая проблемы для экспериментальной
воспроизводимости. Влияние воды, добавленной или намеренно,
или случайной, на электрокатализ подчеркивает необходимость
проведения экспериментов в строго сухих органических
растворителях, выявления возможных источников загрязнения водой и/или
количественной оценки концентрации намеренно добавленной воды
в неводных экспериментах.
8.3. ОЦЕНКА ГОМОГЕНННЫХ ЭЛЕКТРОКАТАЛИЗАТОРОВ
В последнее десятилетие появилось множество работ в области
молекулярного электрокатализа. Сообщения о электрокатализаторах
обычно описывают каталитическую активность по различным
параметрам, включая перенапряжение, наблюдаемые константы
скорости, частоту оборотов (TOF) и фарадеевскую эффективность.
Непосредственное сравнение систем затруднено, потому что источник
протонов, электролит, растворитель, рабочий электрод и электрод
сравнения почти всегда в сообщениях описываются различно. Кроме
того, методы определения параметров в лабораториях значительно
отличаются. Ниже сначала приведены краткие определения этих
показателей и ориентиры (разд. 8.3.1), затем кратко описаны
теоретические отклики ЦВА для каталитической идеализированной реакции
ЕТС\ (разд. 8.3.2) и подробно изложены методы, с помощью которых
эти параметры получены (разд. 8.3.3), а в заключение даны
иллюстративные примеры (разд. 8.3.4). Здесь мы сосредоточимся на
определении параметров непосредственно ЦВА, подчеркивая условия,
которые должны быть соблюдены, чтобы извлечь значимые
показатели. Параметры, получаемые с использованием метода
вращающегося дискового электрода, хотя и мощного, здесь не
обсуждаются в интересах краткости, а заинтересованный читатель может
обратиться к литературе2.
1 KHgore U.J., Roberts J. AS., Pool D.H., AppelA.M., Stewart MP., DuBois M.R.,
Dougherty W.a.Kasset W,S., Bullock R.M., DuBois D.L.J, Op. cit.
1 TJtoi V.S., Karunadasa H.I.,Surendranoth Y., Long J. R., Chang С J. // Energy
Environ. Sci. 2012. 5. 7762-7770; McCrory CC.L., Uyeda C, Peters J.CJ. // Am.
Chem. Soc. 2012. 134. 3164—3170.
298
8.3.1. Основные определения и показатели
1. Перенапряжение (л.) является термодинамическим
параметром, описывающим дополнительный потенциал, превышающий
термодинамическое требование, необходимый для проведения
реакции со специфической скоростью1. Оно определяется как разность
между приложенным потенциалом и стандартным потенциалом для
образования продукта В из субстрата А (ЕА/В). Подробнее
перенапряжение описано в разделе «Перенапряжение (т|)».
2. Стандартный потенциал для катализ-инишшруюшей редокс-
пары (£retini() определяет потенциал редокс-пары, при котором
происходит каталитический никл.
3. Потенциал полуволны (E^i/i) обычно определяется как точка,
в которой гомогенная каталитическая волна достигает половины
максимального тока. Из-за сложностей с этим понятием при
рассмотрении гемолитических процессов мы будем считать, что это
потенциал, при котором половина катализатора в непосредственной
близости от элемрода существует в активированном состоянии.
4. Наблюдаемая (или кажущаяся) константа скорости (&ohs)
описывает общую скорость гомогенного катализа и является полезной
для выяснения механизма реакции. В некоторых работах этот
параметр был назван TOFm„x2. В зависимости от сложности реакции
и конкретных условий каь* можно интерпретировать как состоящую
из константы одной лимитирующей стадии (нротонирования или
восстановительного элиминирования продукта) или как выражение
скорости, состоящее из констант скоростей отдельных стадий
катализа. Методы оценки к^ обсуждаются в разделах «Определение
константы скорости для ЕГС' реакции из ЦВА», «Константы скорости
для многостадийной каталитической реакции из ЦВА»,
«Определение константы скорости для многостадийной каталитической
реакции из ЦВА: Конкуренция побочных явлений», «Оценка кинетики
через численное моделирование».
5. TOF является кинетическим параметром, который определяет
каталитическую активность (раздел «Определение TOF из £<,ь&»)-
В частности, TOF является количеством молей продукта (например,
Н2. 02, СО, СН20), выделяемым в единицу времени на моль
катализатора. Важно отмстить, что для гомогенного молекулярного
катализа TOF описывает активность молекул катализатора,
содержащегося в реакционно-диффузионном слое на электроде, а не молекул
катализатора, содержащегося во всем объеме раствора. Поскольку
TOF зависит от процента молекул активированного катализатора
(потенциал-зависимый параметр), TOF равна к^ (TOFmiu), только
1 BardAJ., FaulknerL>R. Op. cit.
2 Cosrentin C, Savtant J.-M, // ChemElectroChem. 2014. 1. 1226-1236.
299
когда приложенный потенциал достаточно отрицателен (или
положителен) к редокс-паре так, что катализатор эффективно на 100%
находится в активной форме.
6. TOF0 — это экстраполированное TOF при ЕА/В°'.
Теоретическое TOF0 предлагается в других работах2 как «внутреннее* TOF
для катализатора (раздел «Определение TOF из к^»).
8.3.2. £гСгКаталитический механизм и идеализированные
отклики ЦВА
Электрокаталитические процессы, например, выделение Н2
и восстановление С02, могут протекать по многим механизмам
с многочисленными возможными ЦВА-откликами. Эти отклики
могут зависеть от нескольких переменных, включая скорость
сканирования, концентрацию катализатора и субстрата и константы
скорости каталитического механизма.
Правильное моделирование ЦВА-отклика для конкретного
катализатора, а следовательно, точное определение константы скорости
необходимы для установления механизма. Савьян с соавторами
строго определили различные ЦВА-оклики для одноэлектронного
восстановления субстрата А в В с помощью редокс-катализатора Р,
в котором лимитирующей стадией является гомогенный перенос
электрона (механизм ЕТС\ )\
Р+е "^0
Q+A—^-»Р + В
ЦВА-отклики для этого процесса зависят от параметров4
\ = \-^г
RT V ксСр
-о\
F A v J
7 сУ
где X — кинетический параметр, у — коэффициент избытка, £t. —
константа скорости внешнесферного переноса электрона от
восстановленного катализатора Q на субстрат А, СР — объемная
концентрация редокс-катализатора Р, Сд — объемная концентрация
субстрата А.
Costentin С, DroueiS., RobertM., Sttwant J.-Af. //J. Am. Chcm. Soe. 2012. 134.
11235-11242.
Ibid.
SaviontJ.-M,.Su KB.J.// Electroanal, Chem. 1984,171, 341-349;SaveanlJ.-M,
Elements of Molecular and Biomolecular Electrochemistry. Hoboken, NJ: John
Wiley & Sons, Inc., 2006.
SeveantJ.-M., Su K.BJ. Op. cil.
300
Может быть создан двумерный график зависимости X от у, а
различные предельные ЦВА-отклики могут быть разделены на зоны
(рис. 8.5)'. Хотя эта зонная диафамма специфически моделирует
одноэлектронный окислительно-восстановительный катализ с
гомогенным переносом электрона как скоростьопрсдсляюшсй стадией,
различные области и ЦВА-отклики стали общими ориентирами для
более сложных механизмов2. Кроме того, широко используемые для
определения кажущихся констант скорости уравнения (см. раздел
«Кинетические параметры: наблюдаемые константы скорости
и TOF») для электрокатализаторов исходят из этого анализа.
Рис. 8.5. Кинетическая эонная диаграмма и симулированные ЦВА-сигналы
для одноэлектронного восстановления субстрата А с помощью редокс-
катализатора медиатора Р, где X — кинетический параметр, а у— коэффициент
избытка. Компасная роза визуально показывает, как катализ может
перемещаться между зонами
(Ср —начальная концентрация катализатора, СА —начальная концентрация
субстрата, v —скорость сканирования, kt константа скорости гомогенного
переноса электрона от восстановленного катализатора на субстрат}. ЦВА-форма волны
следует соглашению, по которому отрицательные потенциалы отложены вправо,
а катодный ток—вверх. Сканирование начато с положительных потенциалов.
Формы волны адаптированы из работы Saveant J.-M. ft Chem. Rev. 2008.108.
2348—2378, а зонная диафамма перепечатана (с незначительными изменениями)
из работы Saveant J.-M., Su К.8. tl J. Electroanal. Chem. 19B4.171.341—349
1 Saveant J. -M. // Chem. Rev. 2008. 108. 2348-2378; Saveant J.-M., Su KB J. //
Electroanal. Chem. 1984. 171. 341—349.
2 Lexa D., Saveant J.-M.. Schdfer H.J., Su K.-B.r Vering В., Wang D.L.J. // Am.
Chem. Soc. 1990.112.6162-6177.
301
Следовательно, здесь обсуждаются различные зоны и ЦВА-от-
кликм, предстанленные на рис. 8.5, а также то, как разные зоны могут
быть доступны путем изменения X и у, как это было предложено
в виде компасной розы на рис. 8.5.
Зона D (нет катализа). Наблюдаемая ЦВА соответствует
обратимой окислительно-восстановительной паре P/Q.
Зона KS (S-образная, чистые кинетические условия*, субстрат
не расходуется). В зоне KS наблюдается S-образный отклик, т.е.
прямой и обратный след точно повторяют друг друга. Эта область
характеризуется ситуацией, когда концентрация субстрата на
поверхности электрода равна объемной концентрации. Следовательно,
доступ в эту область из зоны К (см. ниже) зависит от избегания
расходования субстрата на электроде и, как предложено на компасе
на рис. 8.5, может быть достигнуто за счет увеличения скорости
сканирования (следовательно, уменьшения X) так, что общее время,
требуемое на запись ЦВА-времени, когда субстрат расходуется,
является коротким2.
Зона К (чистые кинетические условия, потребление субстрата).
В зоне К конкуренция между потреблением субстрата в
лимитирующей стадии и диффузией нового субстрата к электроду приводит
к пиковым ЦВА и обратному сканированию, которое не ложится
на верхнюю часть прямого сканирования. Несмотря на расход
субстрата, обратной редокс-волны не видно, поскольку любой
восстановленный катализатор окисляется в каталитическом обороте.
Зона КТ2 (общий катализ, чистые кинетические условия,
потребление субстрата). Общий катализ наблюдается, когда катализатор
непосредственно потребляет весь доступный субстрат в масштабе
времени прямого сканирования, в результате чего наблюдается ЦВА
с двумя пиками. Каталитический ток наблюдается первоначально,
но субстрат быстро полностью расходуется, в результате чего
образуется пик. По мерс того как потенциал сканируется к более
отрицательным значениям, обратимая редокс-волна катализатора
наблюдается в том же положении без присутствия субстрата.
ЗонаКТ\ {чистые кинетические условия, потребление субстрата).
Катализатор может перейти от зоны КТ2 к зоне КТ\ при увеличении
коэффициента избытка у. С увеличением у обратимая волна
перекрывается каталитической волной, стирая различие между двумя
отдельными пиками. Повышение у далее сдвигает ЦВА-отклик
в зону К. Зоны ATI и КТ1 — специальные случаи чистой
кинетической зоны А?.
1 Saveant J.-M., Su К. В.J. Op. cit.
2 Stewart M.P., Ho M.-K., Wiese S,, Lindstrom M.L., Thogersoti C.E., Raugei S.,
Bullock R.M.. Helm M.L.J. // Am. Chem. Soc. 2013. 135. 6033-6046.
J Saveant J.-M., Su K. B. J. Op. cit.
302
Зона KD (пет потребления субстрата). Сигнал, изображенный
в зоне KD, описывает катализаторы, которые работают в условиях
отсутствия потребления субстрата (большой у) аналогично тому, что
происходит в зоне KS. Но в этом случае сигнал имеет не идеальную
S-образную форму из-за снижения величины кинетического
параметра X. Катализатор может потенциально перейти от зоны KS
к зоне KD путем увеличения скорости сканирования в ЦВА-экс-
перименте, эффективно отодвигая на второй план скорость, с
которой катализатор регенерируется. Катализаторы, которые имеют
более медленные константы скорости, чем те, которые характерны
для зоны KS, также дадут сигналы, аналогичные сигналам в зоне
KD. Используя плато тока сигнала в зоне ЛТД каталитическую
константу скорости можно определить с помощью уравнения (8.10)
(см. ниже)1.
Зоны KG и KG* (потребление субстрата). Формы волны,
показанные в зонах KG и KG*, возникают из условий, описываемых
небольшими у и АЯ Подобно зоне КГ2, в которой происходит
обший катализ, зоны KG и KG* ограничены диффузией субстрата
к электроду из объема раствора. В этом случае безразмерный
кинетический параметр для сигналов в этих зонах также мал, а это
означает, что можно перейти от зоны KTL к зоне KG/KG*, используя
катализатор с более медленной кинетикой или увеличивая скорость
сканирования, применяемую в анализе. Можно также
перемещаться из области KD в зону KG/KG*, снижая концентрацию
субстрата или увеличивая концентрацию катализатора, чтобы
уменьшить у.
Распространение на более сложные механизмы. Это обсуждение
формы волны было сосредоточено на конкретном примере од-
ноэлектронного каталитического процесса, в котором электрон
переносится на субстрат А по внешнесферному механизму. Хотя
интересующие нас электрокаталитические реакции происходят
по более сложным механизмам (см. ниже), формы волн,
представленные на рис. 8.3, являются относительно общими, потому что
понятия каталитической скорости и диффузии субстрата, как правило,
применяются в различных молекулярных электрокаталитических
системах. Например, было продемонстрировано для
электрохимических катализаторов выделения Н2, что различные зоны могут быть
доступны путем изменения скорости сканирования?. Обсуждение
этих каталитических режимов в количественном виде представлено
ниже.
1 5а want J.-M. Op, с it
2 Saveant У.-А/., Su К. В J. Op. cit.
J Stewart M.P., Ho M.-K., Wiese S.. Lindstrom M.L., Thogerson C.E., Raugci S„
Bullock R.M., Helm M.L.J. // Op. cit,
303
8.3.3. Определение критериев из ЦВА
Термодинамические параметры: перенапряжение и потенциал для
катализа
Перенапряжение (ц). Перенапряжение (т|) представляет собой
движущую силу реакции и определяется как дополнительный
потенциал, превышающий термодинамическое требование и необходимый
для проведения реакции с определенной скоростью'. Этот
термодинамический параметр вычисляется как разность между
приложенным потенциалом и стандартным потенциалом для образования
продукта В из субстрата А (£>ув )•
ц = Е-Е°т. (8.2)
(£д/в) должен быть оценен независимо, чтобы определить т\.
Возвращаясь к примеру катализаторов выделения Н2, мы подчеркиваем,
что термодинамический потенциал для выделения Н2 (^на/н,
и ^н2о/н3 является специфичным для данного источника кислоты,
ее концентрации и используемого растворителя. В водном растворе
стандартный потенциал (Я^о/н,) определяется отн. НКЭ. При рН О
водород выделяется при О В отн. НКЭ, а термодинамический
потенциал смещается на 59 мВ на каждую единицу рН, Определение
термодинамического потенциала выделения Н2 в неводных
растворителях (£на/н2) является существенно более сложной задачей, ^на/н,
пропорционален силе кислоты (определяемой константой кислотной
диссоциации кислоты в определенном кеводном растворителе S,
pKBi(A.s), а для кислот с рКи НА s > 0 Эванс определил соотношение
по формуле (8.3), которая основана на уравнении Нсриста2:
^НЛДЬ = V^ -у J JP*a,HA^ (8.3)
где ^^ — стандартный потенциал для пары сольватированный
протон/диводород. Существует несоответствие сообщенных ранее
значений ^vh, 3- Последнее измеренное значение для ацетони-
BardA.J., Faulkner L.R. Op.cit.
1 Felmi G.A. N., Glass R. S., Liehtenherger D. L.. Evans D. H. Op. с it.
i
Fclton G.A.N.. Glass R.S., Lichtenberger D.L., Evans D.H. Op. cit.; Roberts J.AS.,
Bullock R.M. Op. cit.; Fourmond V., Jacques P.-A., Fontecave M., Artero V, Op. cit.;
Daniele S.t Ugo P., Mazzocchin G.-A., BontetnpelliG. //Anal. Chim. Acta. 1985. 173.
141-148; Kolthqffl.M., IbomasF.GJ. //Phys. Chem. 1965. 69. 3049-3058.
304
трила (-0,028 В отн. Fc+/Fc) В является рекомендуемым на
основании надежности метода потенциала при разомкнутой цепи (ОСР)
(обсуждается ниже), используемого для определения этой
величины1.
В то время как уравнение (8.3) дает оценку E°nA/Hl, Роберте
и Баллок недавно сообщили о возможности прямого определения
£на/н2 путем ОСР-измеренийЛ Такой подход идеален для случаев,
где рКа данной кислоты или £н+/н неизвестны для конкретного
растворителя или когда должны быть приняты во внимание
эффекты гомосопряжения3. В этом измерении ЦВА записывается для
анализируемого раствора, состоящего из смеси
кислота/щелочь/водород и внутреннего эталона (Fc) с использованием СУ
рабочего электрода, Ag/AgC1 неводного электрода сравнения и про-
тивоэлектрода. Потенциометр используется для измерения ОСР
между электродом сравнения и электродом из платиновой
проволоки, в сущности он определяет £на/н3 . Как отмечалось выше, этот
подход не требует независимых значений pKa.HA,s> констант гомо-
сопряжения или Еи^/Ц и работает в широком диапазоне систем
кислота/основание и растворитель. Было также отмечено, что
найдено хорошее согласие между измеренными значениями и
рассчитанными по известным значениям рКа Ha.s с использованием
уравнение Нернста.
Артсро и другие также повергали сомнению применение формулы
(8.3), отмечая, что метод не учитывает концентрацию кислоты4.
Вместо этого они выступили за метод расчета теоретического
потенциала полуволны (Е[/2), который соответствует потенциалу, при
котором половина максимального тока восстановления кислоты
получается с идеальным электродом который может быть использован
вместо £"на/н25:
£1/2,НА/Н2 -EH'/Hi ~{ J |РКа.НА:>+Е0—2f ~C°~ ' ^A}
где Со — общая концентрация кислоты, £д — мера относительной
диффузии продуктов по сравнению с реагентами, и CHj — концен-
1 Roberts J.A.S.; Bullock R.M. Op. cit.
2 Ibid.
J AppeM.M,, Helm M, L // ACS Catal, 2014. 4. 630-633.
4 Fourmond K. Jacques P.-A., Fonlecave Л/., Artero V. Op. cit.
s Ibid.
305
трация растворенного водорода, соответствующая парциальному
давлению 1 бар1. Дополнительно доступны связанные выражения2
для условий, при которых кислоты проявляют гомосопряжение при
использовании констант равновесия для реакций агрегации, а также
выражения для Ещ, когда А" или Н2 изначально присутствуют в
растворе. Примечание: коррекция «=» — опечатка из уравнения,
приведенного в оригинальной статье.
Подобно случаю с Н2 стандартные потенциалы для
восстановления 02 (и окисления воды) и восстановления С02 определяются
спецификой условий. Стандартный потенциал для четырехэлек-
тронного восстановления 02 до воды (£о2/н2о) в стандартных
условиях (I М водная кислота, рН 0) составляет 1,229 В отн. НВЭ,
и, как и для Н2, термодинамический потенциал смещается
на —59 мВ на единицу рН при увеличении рН. Насколько нам
известно, стандартный потенциал этой полуреакции не был
определен в неводных средах, но в отсутствие расчетов следует отметить,
что £о2/н:о будет зависеть от источника протонов и растворителя.
Стандартный потенциал для двухэлектронного восстановления ССь
до СО в стандартных водных условиях (1 М водная кислота, рН 0)
составляет —0,106 В отн. НВЭ (и имеет ту же нернстовскую
зависимость, как Нги 02). Другие многоэлектронные, связанные с
протонами, реакции восстановления С02 и их соответствующие
термодинамические потенциалы приведены в табл. 8.151.
Термодинамический потенциал двухэлектроного восстановления С02
до СО в неводных растворителях (диметилформамид (ДМФА),
уравнение (8.5); ацетонитрил, уравнение (8.6)) в присутствии
кислот НА был оценен Саньяном, который использовал подробный
термодинамический цикл, учитывающий изменения свободной
энергии, связанные с перемещением компонентов из воды в не-
водную среду3. Насколько нам известно, такие расчеты не были
сделаны для других многоэлектронных процессов восстановления,
описанных в табл. 8.1.
{ 0 303/Г7Л
£со2/со,dmf,ha= "0.259-1 —— JpK^hadmf В отн. НВЭ; (8.5)
(О ЗОЗЙТ^
-t—p JpK..ha.ch,cn В отн. НВЭ. (8.6)
1 Fourmond К. Jacques P.-A., Fontecave M., Ariera V. Op. с it.
2 Ibid.
3 Costentin С, Drouet S., Robert M., Saveant J.-M. Op. eit.; Costentin C, Drouet S.,
Robert M., Saveant J.-At. // Science. 2012. 338. 90-94.
306
Таблица 8.1
Потенциалы восстановления С02 при рН 7,25°С, давлении
газа1 атм и 1 М — для других растворенных веществ1
Реакция
С02 + 2Н- + 2е- -»СО + Н20
COj + 2Н- + 2е- -> НС02Н + Н,0
С02 + 4Н- + 4е- -» НСНО + НЮ
С02 + 6Н- + бе- -»СНзОН + Н20
С02 + 8Н~ -1- 8е" -» СН4 + 2Н20
СО,+е"^СОГ
£"'(Вотн. НВЭ)
-0,53
-0,61
-0,48
-0,38
-0,24
-1,90
Как подчеркивается в определении, представленном выше,
перенапряжение является ключевым параметром, связанным с
кинетическим параметром. С какой же скоростью протекает катализ при
приложенном потенциале £7 К сожалению, эта характеристика часто
используется для оценки молекулярных катализаторов без ссылки
на какой-либо кинетический параметр, создавая неоднозначность
в смысловом значении. Какой лучше всего потенциал-зависимый
кинетический параметр использовать при оценке перенапряжения?
В гетерогенной электрохимии обычно сообщают плотность тока,
а взаимосвязь между плотностью тока и перенапряжением
обеспечивает классический тафелевский график.
В соответствии с уравнением Тафеля каталитический ток
увеличивается экспоненциально с увеличением перенапряжения2.
Молекулярные системы, однако, ведут себя совершенно иначе1. Катализ
«запускается* редокс-процессом (восстановлением каталитизатора Р
в частицы Q), который имеет стандартный потенциал (мы называем
его EKdox, см. разделы 8.3.2 и «Кинетические параметры:
наблюдаемые константы скорости и TOF»). Этот редокс-процесс выступает
в качестве переключателя включения-выключения катализа.
Катализатор имеет связанную гомогенную кажущуюся константу скорости
(/cobs) оборота каталитического цикла, которая является свойством
используемого механизма и, как правило, не зависит от
приложенного потенциала (хотя она может зависеть от растворителя,
концентрации кислоты, температуры; в сценарии с двумя конкурирующими
механизмами, активированными при различных потенциалах, коЫ
тогда будет потенциал-зависимой).
1 Benson Е.Е., Kubiak C.P.t Sathrum A.J., Smieja J. Op. cil.; Sutin Л'., Creutz C,
Fujita E. f( Comments Inorg, Chem. 1997. 19. 67—92.
2 Bard A J., Faulkner L.R. Op. cit.
J Appel A. M., Hetm ML. Op. cil.
307
Как описано в разделах «Определение константы скорости для
ЕГС- реакшти из ЦВА»; «Константы скорости для многостадийной
каталитической реакции из ЦВА*; «Определение константы
скорости для многостадийной каталитической реакции из ЦВА:
Конкуренция побочных явлений»; «Оценка кинетики через численное
моделирование», во многих, но не во всех случаях, коЫ может быть
определена на основе тока плато, достигаемого при потенциалах, при
которых доля активного катализатора (Q) близка к единице, как
описано равнением Нернста. В отличие от этого TOF является
потенциал-зависимым кинетическим параметром, который имеет
определенную связь с /robth (которую можно рассматривать как TOFltlU4).
Взаимоотношение TOF — п, как оно определено Савьяном, подробно
обсуждается в разделе «Определение TOF из А^»1. В анализе TOF —
Т| зависимость TOF от потенциала в области каталитической воль-
тамперограммы до токового плато отражает часть активированного
катализатора (Q). Тафелеподобные графики, полученные из этого
отношения, позволяют тестировать производительность
молекулярных катализаторов (в определенных пределах, как обсуждается
ниже).
Редокс-потенциал, инициирующий катализ (ЕпЛт), потенциал
полуволны (Есм/г) и потенциал, необходимый для катализа (£слд- Из-за
трудности корреляции приложенного потенциала с кинетическими
данными для молекулярных катализаторов параметр для описания
«перенапряжения, необходимого для катализа» — разницы между
потенциалом, необходимым для катализа {Еш) и EN^, — широко
использовался в литературе. Для данного обсуждения мы определяем этот
термин, как показано в уравнении (8.7), и дифференцируем его от т),
который был определен в предыдущем разделе (см. уравнение (8.2)):
Перенапряжение, необходимое для
реализации катализа, = £са, — Е/цв- (8.7)
В зависимости оттого, как Еса| определяется (какописано ниже),
он часто является просто грубой оценкой приложенного потенциала,
необходимого для катализа, хотя в идеальных случаях Есл[ может
содержать кинетическую информацию2. Как таковое значение,
определенное по формуле (8.7), может быть неточным параметром. По этой
причине мы отличаем этот общий термин от специфического
определения, представленного в уравнении (8,2), и резервируем термин
«перенапряжение» для параметризации катализатора, когда зна-
1 Costentin С, SaveantJ.-M. Op. cit.; Costentin C.t DrouetS., RobertM, Saveant J.-Af.
Op. cit.
2 Costentin С, Saveant J.-M. Op. cit.
308
чимые кинетические данные коррелируют с приложенным
потенциалом, который необходим для сравнения.
В последние годы определение Е^ было очень субъективным,
а различные методы определения Ет были описаны, в том числе
как: 1) потенциал, при котором появляется «начало»
каталитического пика; 2) потенциал тока в пике; 3) потенциал, при котором
электрокатализатор подвергается механистически соответствующему
окислительно-восстановительному процессу в отсутствие субстрата
(Екйах)\ 4) потенциал, при котором получают половину
максимального тока {ECul/i, следует отметить, что при £Cat/2 половина
катализатора в реакционно-диффузионном слое существует в
активированном состоянии; это широкое определение учитывает то, что для
гомолитического механизма реакции 50% катализатора не
активировано, когда достигается половина максимального каталитического
тока)'. Отсутствие стандартного метода означает, что Есп[ может
отличаться почти на 200 мВ2, давая неопределенность в оценках,
использующих различные определения. Метод 1 по сути субъективен
(какую величину тока следует рассчитать в качестве
каталитического тока?) и не точно описывает термодинамическую энергию,
связанную с каталитическим событием. Метод 2 не применяется,
потому что хорошо определенные пики, когда наблюдаются, не дают
информации о самой реакции, потому что потенциал пика зависит
от степени выработки субстрата3.
Для ЕГС- -механизма, работающего в идеальных условиях, в
которых наблюдаются канонические S-образные волны, Е^а = Ек<1ох-
Таким образом, методы 3 и 4 дадут то же значение4. Для различных
двухэлектротшх, двухступенчатых каталитических механизмов
взаимосвязь £cat/2 и Ек4т была определена в виде соответствующих
TOF — п, выражений (см. раздел «Кинетические параметры:
наблюдаемые константы скорости и TOF»)5.
Однако, когда побочные явления исключают возможность S-образ-
ного отклика (и получается отклик в форме пика, зависимый от
скорости сканирования), определение точного и достоверного Е^щ может
быть згпруднено или невозможно. Пертурбации концентрации
субстрата и скорости сканирования приведут к изменению Е^п. Аппель
и Хелм6 предположили, что потенциал Е^/2-, определенный для
неидеальной каталитической волны, будет иметь лишь небольшое
отклонение от истинного £иа|/2, но мы предупреждаем, что кинетическая ин-
1 Fourmond У.. Jacques P.~A., t'onlecave M., Artero V. Op. cit.
2 Ibid.
J Costentin C, Drouet S., Roben M., SaveantJ.-M. Op, cit.
4 Castentin C, Saveant J.-M. Op. cit.
5 Ibid.
6 AppetA.M., Helm M, L. Op. cit.
309
формация может быть только приблизительно оценена исходя из этого
подхода, а сравнительный анализ будет некорректен.
Таким образом, мы предупреждаем, что перенапряжение,
необходимое для катализа, как сообщалось в последние годы, не следует
рассматривать как общий параметр для прямого, количественного
сравнения катализаторов, описанных в независимых работах,
вследствие неодинакового использования параметра Еса1. Когда EICiio\
и fcai/j могут быть непосредственно измерены, они обеспечивают
полезную информацию, как для механистического анализа (см.
раздел «Константы скорости для многостадийной каталитической
реакции из ЦВА»), так и для сравнения катализаторов, £red(W даст
информацию о термодинамике, связанной с реакцией, в то время
как £Lai/2 помогает осуществить кинетическое сравнение одного
катализатора с другим через его часть во взаимосвязи TOF — г| (см.
раздел «Кинетические параметры: наблюдаемые константы скорости
и TOF»). Е^в является специфичным для каждого растворителя
и используемого источника протонов. На примере генерации Н2
перенапряжение, необходимое для катализа, часто может быть
уменьшено путем использования кислот с более отрицательным значением
Awh2 (слабые кислоты), если ЕкЛок и/или Eal/2 остается
постоянным. Но когда ряд катализаторов измеряют в одних и тех же
условиях и используемое определение £са1 четко коррелирует с
кинетическим параметром, перенапряжение для катализа может быть
использовано для сравнения катализаторов. Однако полная
взаимосвязь TOF — Т| будет более полезной.
Кинетические параметры: наблюдаемые константы скорости и TOF.
Определение кинетических и механистических закономерностей для
молекулярных катализаторов из ЦВА может быть сложной задачей,
особенно для многоэлектронных, многоступенчатых реакций. Ниже
подробно обсуждаются методы извлечения общей скорости катализа,
Определение константы скорости для ЕГС\ реакции из ЦВА.
Основные параметры реакции, например, кажущаяся скорость, могут
быть определены непосредственно из вольтамперограмм активных
катализаторов. В данном разделе описан процесс определения
кажущейся константы скорости (fr0bs) для простого механизма ЕГС\ с
последующим обсуждением приближений, которые позволяют оценить
параметры для более сложных реакций — электрокаталитического
выделения Н2 и восстановления С02.
Теория, лежащая в основе ЦВА каталитических реакций ЕТС\,
была сформулирована Дслахссм1, Николсоном и Шсйном2и Са-
1 Delahay P., Stiehl G.L. // J. Am. Chem. Soc. 1952. 74. 3500-3505.
3 Schwarz W.M., Sham /. // J. Phys. Chem. 1965, 69. 30-40: Nicholson R.S.,
Sham 1. //Anal. Chem. 1964. 36. 706-723,
310
вьяном с сотрудниками1. Интересующая нас реакция, приведенная
в разд. 8.3.2, повторяется здесь для ясности:
Р+е ^Q;
Q+A—^-»Р+В.
Сделаны некоторые допущения: перенос электронов между
электродом и редокс-активной парой P/Q быстрый и нернстовский;
вследствие этого гомогенный перенос электрона — (химическая)
стадия (Q + А), которая регенерирует Р, яштяется скоростьлимитиру-
ющей; Сд » Ср (большое у); коэффициенты диффузии частиц Р и Q
примерно равны; £, (исходный потенциал сканирования) достаточно
положительный, так что полученные в результате измерений
значения не зависят от этой величины; субстрат А редокс-неактивен
на электроде в интересующем окне потенциалов. Наблюдаемая (или
кажущаяся) константа скорости псевдопервого порядка каЫ
определяется формулой (8.8) (для ErCj -механизма) и характеризует
гомогенную химическую реакцию Q + А, а следовательно, не зависит
от потенциала. Проще говоря, kobs представляет собой скорость, при
которой Р образуется из Q в гомогенном процессе:
В предельном случае больших Х/Ср = k^RT/Fv (что вместе
с ранее высказанным предположением о большом y указывает на зону
KS) результирующая волна может быть описана уравнением (8.9):
nFACUDk^
' Г nF . Т (89)
1+expl—(£*-£j/0)
где п — число электронов, переданных в
окислительно-восстановительном процессе; А — площадь поверхности электрода, см2 (обычно
берется как геометрическая площадь поверхности, хотя
электрохимическая площадь поверхности является более предпочтительной);
D — коэффициент диффузии редокс-частиц Р, см2 - с '; Ср —
объемная концентрация редокс-частиц Р, моль • см-3.
Saveant J,-M., Su К.В. Op. cit.; SaveantJ.-M., VianeUo Е. (j Electrochim. Acta.
1965.10. 905-920; SaveantJ.-M., VianeUo E. // Electrochim. Acta. 1967. 12.629-
646; Andrieux C. P.. Bbcman C., Dumas-Bouchiat J.M., M'Haifa F., SaveantJ.-M. ff
J. Electroanal. Chem. 1980. 113. 19-40; SmeanrJ.-M.,Su K.B.J. If Electroanal.
Chem. 1985. 196. 1-22.
311
Важно отметить, что уравнение (8.9) показывает, что никакого
пика не наблюдается. При потенциалах значительно более
отрицательных .fp/Q (=100 мВ) уравнение (8.9) упрощается и ток /с
каталитического плато может быть описан уравнением (8.10). /с не зависит
от скорости сканирования, потому что ток регулируется только
регенерацией Р по химической реакции Q + А':
1с = пГАС$у[Щ^. (8.10)
kobs может быть выведена из /с. Часто отношение /с — &ob5
выводится по уравнению Рэндлса-Шевчика (уравнение (8Л1)), которое
описывает зависимость пикового тока /р (А) от скорости
сканирования v (В • с-1) для обратимого окислительно-восстановительного
процесса:
/P=M463*F*?(^) . (8.11)
Это дает соотношение
Уравнение (8.12) часто является желательным, потому что оно
не требует независимого измерения D, Л, или Ср для определения
katK. Следует отметить, что и в уравнении (8.10) представляет собой
число перенесенных электронов от электрода на Р, а значит,
отличается от я в уравнении (8.11), только когда /р определяется из
отдельного редокс-события (это может быть иногда необходимо из-за
некоторой помехи для интересующего процесса2) или когда
последовательные переносы электрона происходят на электроде с
последующими химическими стадиями. Это обсуждается далее в разделе
«Определение константы скорости для многостадийной
каталитической реакции из ЦВА: конкуренция побочных явлений». Но следует
отметить, что, как и указано, уравнение (8.12) предполагает, что
значение я в уравнении (8.10) равно 1. Следует еще раз подчеркнуть, что
использование уравнений (8.10) и (8.12) ограничивается кривыми
S-образной формы, полученными в зонах KSuKD (где применяется
ток плато)3. Доступ к зоне KS (и KD) требует, чтобы /с не ограничи-
1 Andrieux С. Р.. Bhcman С., Dumas-Bouchiat J.M., М'Hulta F.. Saveant J.-M. Op.
ciL
2 Stewart MP., ho At.-K., Wiese S.. Lmdstrom M.L., Thogerson C£, Raugei S.,
Bullock R.M., Helm M.L.J. Op. cit.
J Saveant J.-M. Op. cil.
312
вался диффузией субстрата; расход субстрата всегда незначителен,
так что предположение СА = СА справедливо. Экспериментально
S-образный ЦВА-отклик в зоне KS может быть получен за счет
увеличения концентрации субстрата (увеличения у) или увеличения
скорости сканирования, чтобы ограничить потребление субстрата.
Кроме того, доступ к чистым кинетическим зонам требует, чтобы
химическая реакция была быстрой по сравнению с временной
шкалой вольтамперограммы. Таким образом, в случае медленного
катализа (маленькая кс) v может быть уменьшена для увеличения
значения X. Однако необходимо проявлять осторожность, поскольку
большая скорость сканирования (и очень большая X) может привести
к потреблению субстрата внутри диффузионного слоя. Чтобы
убедиться, что используется подходящая скорость сканирования,
необходимо подтвердить независимость ic от скорости сканирования1.
Кроме того, при очень медленных скоростях сканирования
«фальшивая» форма плато может быть достигнута в результате
естественной конвекции2. И последнее: во многих случаях S-образный
волновой отклик не может быть достигнут. Побочные явления —
дезактивация катализатора, ингибирование тока продуктом адсорбции
на электроде и фоновое выделение Н2, как известно, возмущают
каталитические отклики1.
С этой точки зрения уравнения (8.10) и (8.12) обсуждались
в случае, когда наблюдаемая скорость &obs равна кеСА. Однако эти
уравнения широко используются для оценки каталитических
скоростей более сложных систем, в частности электрокаталитической
генерации Н2. В этих системах £obs — общая константа скорости
описывает общую наблюдаемую скорость оборота катализатора (см.
ниже). Правильное расширение теории ЕГС- на эти более сложные
каталитические системы требует рассмотренных здесь приближений.
Константы скорости для многостадийной каталитической реакции
из ДВА. Многостащ1йный каталитический цикл, как правило, не может
быть описан одиночной стадией или константой скорости, а скорее,
общей константой скорости &obs (TOFmax, раздел «Определение TOF
из A^bs»). которая может либо быть композицией констант скоростей
элементарных стадий в рамках цикла, либо отражать лимитирующую
стадию. Во многих случаях уравнения ЕrQ (формулы (8.10) и (8.12)),
представленные выше, могут быть непосредственно распространены
1 Helm M.L., Stewart MP., Buttock R.M., DuBois M. R., DuBois D.L. Op. cit.
2 Costentin C, RuberM., Saveant J.-M, Op. ci(.; Antirieux СР., Bhtman C, Dumas-
BouehiatJ.M., M'Halta F., SaveantJ.-M. //J. Eleclroanal. Chem. 1980. 113. 19—
40.
J Costentin С, Drouet S., Rober M., Saveant J.-At. Op. cit.
313
на многоэлектронные, многостадийные пути реакций. Однако это
не всегда так. Недавно Савьян опубликовал отклики ток-потенциала
для некоторых двухэлектронных, двухступенчатых гомогенных
молекулярных каталитических реакций1, хотя мы ограничим дискуссию
здесь лишь двухэлсктронными, двухступенчатыми процессами
(сначала представив гетеролитический путь, а закончив кратким
обсуждением гемолитических реакций). Обработка данных аналогична
приведенной в указанной работе2 и может быть распространена на другие
многоэлектронные, многостадийные пути реакции.
Различные рассмотренные здесь возможные реакции, в том числе
гетеролитические и гомолитические пути, представлены в табл. 8.2.
Каждый из этих сценариев имеет как стадии переноса электрона, так
и так называемые химические стадии с соответствующими
константами скорости. Следует отметить, что эти химические стадии не
обязательно должны соответствовать одиночной элементарной стадии,
они могут состоять из любого числа последовательных элементарных
стадий, композиция которых может быть охарактеризована с
помощью кажущейся константы скорости. Как подробно описано
ниже, производные ЕГС' -уравнений можно использовать для
извлечения kobs из тока плато для гетеролитических реакций в предельных
случаях, для которых существует скоростьопределяюшая химическая
стадия. Для промежуточных случаев вышеуказанная работа3
предоставляет детальные выражения, связывающие к0^с элементарными
константами скоростей к\ и к2. В некоторых из этих промежуточных
случаев к^ не может быть получена непосредственно от тока плато.
Гомолитический случай рассматривается отдельно, как описано
в конце этого раздела.
Гетеролитические пути реакции делятся на две категории.
Первая категория включает все электронные переносы, которые
осуществляются на электроде, а во второй категории один перенос
электрона происходит в растворе: 1 эквивалент О восстанавливает
интермедиат реакции (О, Q/или Q"). Последний путь иногда
называют переносом электрона в растворе (SET)4. Когда второй
перенос электронов является более трудным, чем первый, гомогенный
путь переноса электрона является неблагоприятным. В этих случаях
необратимая одноэлектронная ЕС волна предшествует
каталитической волне; как правило, к{ может быть определена из
положительного сдвига пика при добавлении субстрата5. В тех случаях, когда
1 Costentin С., Saveant J.-M. Op. cit.
2 Ibid
J Ibid.
4 Saveant J.-M. Su K.BJ. II Eleciroanal. Chem, 1985. 196. 1-22.
* Costen tin C., Saveant J. -M. Op. cit.
314
Таблица 8.2
Выражения для fcab< и Е1/2 для некоторых двухэлектронных, двухстадийных путей реакций
(адаптировано из работы Costentin С, SaveantJ.-M. II ChemElectroChem. 2014.1.1226—1236)
Гетеролитические реакции
Вес переносы электрона происходят на электроде*
Один гомогенный перенос электрона
ic = FACuPjD2k^
ЕЕСС
0 + с-*Р Е%/{
Р + е —Q Е°?
Q+A^B к{
B + Z—>0 + C k2
о/Р
р/о
It -It Г9
Еап/2 = ^P/Q
For k2C^k}Cl
каы — kiC7_
►Р + с — Q Е"т
Q + A—>B Aj
B + Z-^O + C k2
0 + 0-
2Р
Для *2С£ »*:,<;£
ДляА3С|<к^С
RT
-сМ/2
P/Q
,>/Йа" j
ECEC, J£p/Q *"■ ^07 В
■P + e
— Q 4
P/0
Q'+e—В i^/u
B + Z^P + CAi
For k,Cl» A,C)I
к -кГ°
Еш/i = ^p/o
*-P + e —
Q + A=>
Q'+Q->
B + Z^
Q ^P/Q
0' kt
P + B ke
P + C k2
Для k2Cl» &,CA
Продолжение табл. 8.2
Гетсролитические реакции
Все переносы электрона происходят на электроде*
k=2FAC«4DkZ
Один гомогенный перенос электрона
/С = Д4С^7Щ
obs
For *iCz«*iCa
Для k2Cl« ktCl
Чтобы определить к^, считают
что ЕТ происходит на электроде
^wit/2 - ^1
Р/0
+ —-In
\^2^А
ЕСЕС, Ep/Q < Eq'/p
£°
P/Q
Q + A—»B A,
В + Z —> Q" A2
Q/'+e — P + C £°P/0-
For*2C2»*,C£
Еал/1 = ^P/Q
Aal/2 _ ^P/Q
In
^kiCz j
E°-
Q + A—»B A,
B + Z—»Q" кг
Q"+Q^2P + C kt
p/Q
Для *2С£»А,С£
*obs = *Г~А
^cat/2 = ^V/Q
Для ^«A,C°
^obs = k2Ci
Лсв1/2 - J-P/Q"' p~
ln
ffik~A'
Окончание табл. 8J
Гетеролитические реакции
Вес переносы электрона происходят на электроде*
iK=2FAC°PjDk0
ьоЬ*
Один гомогенный перенос электрона
SAs
ЕССЕ, £рд) < £q-/p перенос электрона томно на электроде, неблагоприятный путь для гомогенного ПЭ
Заметка: процесс переноса второго электрона является диссоциативной реакцией ПЭ, и он не подчиняется закону
Нернста41. По этому сценарию надо использовать закон Батлера-Волмера, при этом константа скорости к, стадии
гетерогенного диссоциативного ПЭ и коэффициент переноса а включаются в уравнение ниже
Р + е —Q £°Р/0
Q + A^B ki
B + Z-ХУ' к2
Q"+c-~P + C £0о7н+с
Для кгС1» kxCl
E^n =[£0VP+c+|^lnA5J-^lnVMlC^
ЦдякгС1^к,С1
Етп =(^/^с+-^1п^]-^1п7аЙ"
ЕС ЕС, Ep/Q < Eq>/B перенос электрона ттько на электроде, путь неблагоприятный &w гомогенного ПЭ
* уравнение /с, выведенное в шапке, не применимо к этому сценарию
*р + е — Q
0+А^О'
Q'+e -*B
B + Z—>Р + С
£p/Q
^ОУВ
L = 2FACl4D-
Е..мп — Eci'm +^^ln
'wu/2
Q7B
RT. [uJk~&
k V*I*~A j
электродный и гомогенный пути переноса электрона являются
термодинамически приемлемыми, предпочтение одного пути перед
другим, как правило, контролируется относительными скоростями
последующих гомогенных реакций. Например, в реакции ЕСЕС Q
реагирует с субстратом А с образованием Q'c константой скорости
А"|, а далее Q'восстанавливается с образованием промежуточного В.
Когда к\ быстрая, Q'образуется рядом с электродом и
восстанавливается в гетерогенном процессе переноса электрона на электроде.
Когда к\ медленная, Q диффундирует от электрода до реакции с А,
а образованный Q'реагирует со вторым 1 эквивалентом Q в
гомогенном процессе переноса электронов с константой скорости ке.
Относительные скорости к] и ке могут быть измерены с помощью
различных способов1 или, чтобы отличить эти механизмы, вольтамле-
рограммы можно измерить как функцию СР.
Вообще говоря, /cabs можно непосредственно определить из плато
для гетерогенных сценариев, в которых скорость регенерации Р из Q
контролируется скорость лимитирующей химической стадией
{например, к2Су »&,Сд и кгСу «A,C°; см. табл. 8.2). В этих условиях
можно применить полученные уравнения для ЕГС\2, но уравнения
каталитического тока плато различаются для систем, в которых все
электронные переносы осуществляются на электроде, по сравнению
с теми, в которых доминируют гомогенные процессы переноса
электронов. Для общей многоэлектронной каталитической системы
число уникальных процессов переноса электронов, происходящих
на электроде, по отношению к катализатору (л) и эквивалентов
катализатора к числу оборотов (и ) включено в уравнения (8.10) и (8.12)
(раздел «Определение константы скорости для £,С/ -реакции
из ЦВА»), дающие уравнения (8.13) и (8.14) (исключение выделено
в табл. 8.2; учитывая данное исключение, можно применить это
более общее правило к любой многоэлектронной каталитической
системе, в которой второй перенос электрона следует за
лимитирующей стадией)3. Например, в реакции ЕСЕС с одним гомогенным
переносом электронов только Р восстанавливается на электроде
(п = 1) и для оборота необходимо 2 экв катализатора Р(п'= 2). В
реакции ЕСЕС только с электродными электронными переносами Р
и Q'восстанавливаются на электроде (л = 2), но только 1 экв
катализатора Р необходим для оборота (л '= 1):
1 Dempsev J.L., Winkler J.R., Gray H.B.J. // Am. Chcm. Sac. 2010. 132. 16774—
16776.
2 Cosrenlin C, Savvanf J,-M. Op, cit.
i Costemin C„ SaveantJ.-M. Op. cit.; Cosremin C, D/vuetS.. RoberM., SaveantJ.-M.
Op. cil.; Costentin C, Drouet S.. Passard G., Robert Л/., Saveant J.-M. // J. Am.
Chem. Sot. 2013. 135.9023-9031.
318
ic=nFAC^Dn'k^; (8.13)
I^ISK- (8л4)
Может показаться заманчивым в уравнении (8.10) для простоты
сделать п = 2 для обеих двухэлектронных, двухстадийных схем
реакции. Однако напомним из более ранних обсуждений, что к^
представляет скорость, при которой Q становится Р. В примере ЕСЕС-
реакции с одним гомогенным переносом электрона оборот в
определенном смысле является бимолекулярной реакцией, для которой
образование 1 экв продукта требуег, чтобы 2 экв Q были превращены
в Р. Поскольку обработка электрокаталитических систем только
недавно подробно обсуждалась в литературе1, ниже приведен рис. 8.6,
чтобы продемонстрировать использование уравнения (8.13) на
симулированных данных.
На рис. 8.6 представлен один механизм, в котором значения ли/Г
могут быть определены с помощью простых изменений; результат
моделирования демонстрирует разницу в ЦВА-откликах и пиковых
токах между двумя сценариями. А именно: как уравнение и
предполагает, ток плато увеличивается линейно с я и увеличивается с чп'.
Если механизм неизвестен, и выбирается как число общих процессов
переноса электрона, как это обычно делалось в литературе, Ао1м будет
недооценено с коэффициентом л, если процесс фактически
включает гомогенные электронные переносы. Если взять другую
крайность: п' выбирается в качестве общего числа процессов переноса
электронов, /fubs будет завышена с коэффициентом и, если процесс
фактически включает только электродные переносы электронов.
Осторожные экспериментаторы, не способные различить перенос
электрона на электроде и гомогенные пути переноса электронов,
могут выбрать для расчета и, равное общей сумме процессов
переноса электрона, с целью избежать завышения Aobs, но следует
проявлять осторожность, потому что в механистической информации,
почерпнутой из константы скорости, определяемой по этой
оценочной формуле, нет смысла.
В обсуждении до сих пор подчеркивалось, что для двухэлек-
тронной, двухстадийной гетеролитической реакции в
лимитирующем сценарии, включающем скорость определяющюю химическую
стадию, fc0bs может быть определена из тока плато и
соответствующего приложения из уравнений (8.13) и (8.14). При более сложных
сценариях, когда две химические стадии имеют сравнимые
константы скорости, Aob5 не может быть получена непосредственно из то-
1 Costentin С,. Saveanr J.-St. Op. cil.
319
кового плато1. В этих условиях £сп„ Ем/Ъ ток плато и информация,
полученная из результатов анализа основания волны foot-of-the wave
(раздел «Определение константы скорости для многостадийной
каталитической реакции из ЦВА: Конкуренция побочных явлений*),
6
5
4
3
2
1
О
10-
Ъ 8-
g6H
й 4Н
U
2-
0
12
10
8
6
4
2
0
= *£ЛС^От'кш
l1Kr=3,i = i:ir '
К— I, л"— ',
i=*Jlr'
п • L.n'-l,
#i = 3.n*=l,f=4.l5ir
л- Э,л'- 1,J- b.JUc '
л = I. «'= (,J= 5,A5l- '
,j-]n,f.r >
15
г -
■ I. n' =
■ H fiV"
J.fl
ft.l'lc
j
1.
P + HC ~^0
^Q\
Q + X
(QX+Q^!2_>p + QX "'(л' 11}„._|
Ек [)л= l, л'= 2
Q-hX-
QX
Li1
QX+Q - ■ >P-hQX
QX '^Up+Y
Ь\2)п-3,п'- ]
P + 3e
Q + X—'•
-0
->QX
^P-Y
P + 2e"
Q + X—=-^QX
QX i Q Jft" , V \ V\~2
QX' + Q ^Tf >P + QX^
QX "*■
^1Й
4Y
0,2 0,1 0,0 -0,1 -0,2-0,3
Potential (V)
Рис. 8.6. Моделирование механизма, приведенного в верхней правой части
рисунка, показано с соответствующими расчетными значениями тока,
полученными с использованием уравнения (8.13) для л и л'. Три примера
механизмов приведены для дополнительной иллюстрации того, как рисунки
были смоделированы. Ср = 1 10 6 моль - см 3, F = 96485 С < моль \ А = 1 см2,
С£ =1М,А=100М-1-г1
Costentin С, SaveantУ.-Л/. Op. cit.
320
должны использоваться в сочетании, чтобы получить информацию
0 скорости, соответствующую механизму. Эта тема изучена очень
подробно1. Гомолитичсский путь реакции также рассматривается
в качестве возможного механизма катализа выделения Н2, в
частности с участием кобалоксимов2. Хотя численное моделирование
использовалось для оценки этих путей на основе ЦВА-откликов,
уравнение, описывающее отклик ток-потенциал, было получено лишь
недавно\ Важно, что из-за второго порядка химической стадии
токовый отклик масштабируется с концентрацией катализатора в
степени 3/2 (табл. 8.2). Эта концентрационная зависимость, как также
показано4, влияет в целом на форму каталитического сигмоида.
Определение константы скорости для многостадийной
каталитической реакции из ЦВА: Конкуренция побочных явлений. Часто трудно
достичь S-образного каталитического токового отклика в зоне KS.
Конкурирующие побочные явления —потребление субстрата,
дезактивация катализатора и ингибирование продуктом не всегда можно
устранить с помощью экспериментальных параметров. Недавно,
однако, новый метод, названный анализом foot-of-the-wave основания
волны (FOWA), был представлен и применен Савьяном для оценки
кинетической информации для электрохимических каталитических
процессов, волътамперограммы которых не соответствуют жестким
требованиям характеристик зоны KS, но имеют доступное
«подножие» каталитической волны (т.е. где нет
окислительно-восстановительных процессов, искажающих подножие волны)5. FOWA можно
выполнить двумя способами в зависимости от сценария реакции.
Здесь FOWA сначала выведен для случая, в котором первая
химическая стадия является скоростьопределяющей. Затем
описывается стандартный FOWA, который можно применить в более обших
случаях.
FOWA начинается с уравнения (8.15), которое описывает
каталитический токовый отклик для многоэлектронной, многостадийной
реакции как функцию потенциала, полученного в условиях зоны KS
и в случае, когда первая химическая стадия является скоростьлими-
тирующей (для гетеролитических реакций, представленных
в табл. 8.2, k2Cz » fc,CA) и EM/2 = ЕЫа1Х:
1 Costentifi С, Saveanl J.-Л/. Op. cit.
1 Ни X., Brunschwig B,S,. Peters J.CJ. // Am. Chem. Soc. 2007. 129. 8988-8998;
DempseyJ.L, Winkler J.R,Gray HB. II J. Am. Chem. Soc. 2010.132.1060-1065;
Solis B.H., Hammes-SchifferS. // Inorg. Chem. 2011. 50. 11252-11262.
3 Costentifi C, Saviunl J.-Л/. Op. cit.
4 Ibid.
ь Costentin C, Robert M., Saveant J.-M. Op. cit.; Costenrin C, SaveantJ.-M, Op. cit.;
Costentin C„ Drouet S., Robert Л/., Saveant J.-M. Op. cit.; Costentin C, Drouet S.,
Passard G., Robert A/., Saveant J.-M. Op. cit.
321
1 + expl—(£-£red(M)
Когда при таких условиях достигается плато каталитического
тока, более простое уравнение (8.13) можно использовать для
извлечения Aot1s из /с. Однако возмущение тока отклика побочными
явлениями — потреблением субстрата и дезактивацией
катализатора становится все более вероятным как раз в ходе измерений,
например, при сканировании потенциала в катодном направлении.
FOWA анализирует идеализированную реакционную способность
катализатора, рассматривая только начальную часть каталитической
волны, в которой ожидается, что субстрат мало или вообще не
потребляется, а деградация катализатора или другие побочные явления
не происходят'.
При нормализации уравнения (8.15) по /р пиковый ток в
отсутствие субстрата, как определяется уравнением (8.11), получаем
^nFv (8.16)
На рис. 8.7, а представлены данные в формате i/ip от Е — £redox-
В чисто кинетических условиях без потребления субстрата
получается классический сигмовидный отклик тока, как предсказано
уравнением (8.15). Однако побочные явления вызывают отклонение
отклика тока от прогнозируемой S-формы. В примере, показанном
на рис. 8.7, а, влияние расхода субстрата увеличивается по мере того,
как объемная концентрация субстрата уменьшается. FOWA
предполагает, что катализ происходит в чисто кинетических условиях
у основания волны, и это может быть использовано для анализа
вольтамперограмм, которые отличаются от S-образной формы волны
в зоне KS в результате этих нежелательных побочных явлений.
Перестройка графика ///р по 1/{1 + екр[(пЕ/НТ)(Е — EKdox)\]
приводит к прямой линии для токовых откликов, полученных в условиях
зоны KS (рис. 8.7, б). Побочные явления приводят к отклонению
от прогнозируемой линейной зависимости, но у подножия волны
участок соответствует линейному ожиданию. Таким образом, можно
сделать линейную экстраполяцию для получения ожидаемой
линейной взаимосвязи, не имеющей побочных явлений. Из уравнения
(8.16) (уравнение (8.14) с поправкой на зависимость потенциала) мы
Costentin С, DrouetS., Robert Л/., SaveantJ.-M. Op. cit.
322
видим, что коЬ& может быть определена из наклона этой линии [т =
= 2,mRT/nPv)i/2{n'kdbb)i/1)i. Это является ключом к сведению, что
метод FOW\ может быть применен только к сценариям, описанным
выше: первая химическая стадия является лимитирующей
и Еш/г = Ejcook- Значение к^, извлеченное из этого анализа, в
соответствии с табл. 8.2 равно к£А и описывает реакционную способность
катализатора при гипотетическом отсутствии побочных явлений.
Е-Е.
redox
(V)
1+expl—(£-£rei]m)l
Рис. 8.7. Симулированные ЦВА для каталитического превращения субстрата
в продукте различными концентрациями субстрата (показано уменьшение
при переходе с синего цвета на желтый) (v = 0,1 В ■ с1, DP = 10т5 см2 • s_1 CP = 0
1 мМ, 7= 298 К, л' = 2, и &са = 50 с'1) (a). FOWA линейные графики для тех же
ЦВА. показывающие полученные линейные подгонки (б)
(адаптировано из работы Costentin С, DrouetS., Robert /И., SaviantJ.-M, //J. Am. Chem.
Soc 2012.134.11235—11242)
Уравнения FOWA также относятся к более общим сценариям,
которые не удовлетворяют условиям, выдвинутым выше, в том числе
к гетеролитическим путям, в которых вторая химическая стадия
лимитирует скорость, промежуточным случаям, в которых первая
и вторая химические стации имеют сопоставимые скорости
гемолитических реакций2. В отличие от вывода, представленного выше,
эти выражения не требуют, чтобы Ешп = ЕцАсы что важно, поскольку
Еш/2 не всегда может быть точным либо непосредственно
определенным из ЦВА, если побочные явления активны.
1 Costentin С, Drouet S., Robert М., Saveant J.-M. Op. cit.
1 Costentin С, Saveant J.-M. Op. cit.
323
Хотя подробные выводы этих FOWA-выражений выходят
за рамки этой монографии и представлены в другой работе',
их вывод можно суммировать следующим образом. Отклики ток —
потенциал для большинства сценариев в табл. 8.2 имеют форму
уравнения (8.17), в котором выражения для /с и Е^^ являются
уникальными для каждого конкретного сценария:
i=-
1+ехр
[£л*-ы]
(8.17)
У подножия волны Е » Еся1/2, а значит, c\p[F/RT\E— £cai/2)l >> 1-
Это приближение обеспечивает приближение ток — потенциал:
/сехр
["лг^^^Т
(8.18)
Замена выражений дня /t и £са,/2 обеспечивает общие выражения
для анализа основания волны (foot-of-the-wave), представленного
в табл. 8.3. Из графика тока от expf(F/RT)(E — ECju/2)\ информацию
о скорости можно почерпнуть из наклона полученной линейной
области у подножия волны. Нужно принять к сведению, что
применение FOWA может дать только информацию о скорости химической
стадии, непосредственно следующей после £"wl]OK, Это
продемонстрировано для случая ЕЕСС, где все электронные переносы происходят
на электроде (рис. 8.8).
10-г
50-
s40-
ё30-
3
и20-
ю-
п-
р+
0'
в->-
с^=Р
А-»В
z-»o +
*.
С*;
*|"
*2 =
1е4
1е2,1е6
о
Е-Е„
(У)
0,00 0,05 0.10
F
RT
б
еЧ_,
0,15 0,20
Рис. 8.8. ЦВА-отклик для случая ЕЕСС беэ потребления субстрата в случаях,
когда к2 (зеленый) и *, (синий) являются лимитирующими (о). FOWA,
показывающий, что линейная область соответствует кх независимо
от скоростьопределяющей стадии [6)
Cosfentin С, SaveantJ.-M. Op. cit.
324
Оценка кинетики через численное моделирование.
Электрохимическое программное обеспечение для моделирования (например,
DigiElch и DigiSim) позволяет осуществить ввод исходных условий —
концентрации, стандартных потенциалов и параметров ячейки,
а также механизм, по которому реакция, как предполагается, будет
протекать. Константы скорости должны сопровождать каждый шаг,
но большинство компьютерных программ для моделирования
способны выполнить «оптимальное приближение», чтобы определить
константы скорости. Важно помнить, что прямое соответствие
вольтамперограмм не гарантирует прямого соответствия механизма,
также как для любой другого вида механистического анализа. Чтобы
надлежащим образом использовать электрохимическое программное
обеспечение для моделирования, необходим строгий анализ
катализатора и его реакционной способности, так как это позволит
предложить механизм. Все константы скорости отдельных стадий
в предлагаемом механизме, которые можно определить
экспериментально, должны быть определены с помощью моделирования, чтобы
повысить точность других констант скоростей. Этот подход может
включать в себя использование знакомых методов механистического
анализа — определения констант равновесия и скоростей путем
ЯМР-исследований1, стехиометрического электролиза, измерений
в остановленном потоке или исследовании с лазерной вспышкой-
гашением2 в сочетании с нестационарной спектроскопией
поглощения. Кроме того, вольтамперограммы должны быть получены при
нескольких скоростях сканирования, концентрациях катализатора
и субстрата и подогнаны в целом по этим параметрам.
ЦВА является чрезвычайно чувствительной к окружающей среде
реакции, а следовательно, форма волны может зависеть от мелких
явлений. Поэтому моделирования каждой стадии основного
механизма, как правило, недостаточно, чтобы должным образом
смоделировать экспериментальную вольтамперограмму. Побочные
процессы — расход субстрата, гомо- и гстеросопряженис кислоты
или любой альтернативный путь (например, процессы деградации
катализатора, образование наблюдаемых частиц и т.д.) могут
повлиять на ток, а значит, должны быть учтены при моделировании.
Несколько программ позволяют моделировать эти побочные
явления3. Предлагаем читателю ознакомиться с элегантными приме-
1 Marinescu S.C., Winkler J.R., Cray Н.В. Ц Ргос. Natl, Acad. Sci. U, S. A. 2012.
109. 15127-15131.
1 Dempsey Л L., Winkler J. R., Cray H. B. Op. cit.
я Fehon G.A.N., Vannucci A.K., Chen J.t Lockett L.T., Okumura N.; Petro В J.,
Zakai U.I., Evans D.H„ Glass R.S„ Lkhtenberger DXJ. // Am. Chem. Soc. 2007.
129.12521 -12530; Bhugun /.. Lexa D„ Saveant J.-M. // J. Am. Chem. Soc. 1996.
118.3982-3983.
325
рами кинетического анализа с помощью цифрового моделирования
ЦВА для получения дополнительной информации1.
Определение TOF из Аы„. В молекулярном электрокатализе TOF
является кинетическим параметром, который отражает
каталитическую активность и параметризован по приложенному потенциалу.
Самое общее определение TOF — это количество молей
произведенного продукта в единицу времени на моль катализатора,
содержащегося в реакционном диффузионном слое (не в объеме раствора). Как
отметил Савьян, получение TOF только из реакций, протекающих
на поверхности электрода, обеспечивает полную картину
каталитических свойств-. Во многих сообщениях TOF была приравнена
непосредственно к наблюдаемой константе скорости (коЫ), которая
часто может быть получена из тока плато циклической вольтамперо-
граммы, измеренного в условиях, изложенных в разделах
«Определение константы скорости для ЕГС- реакции из ЦВА», «Константы
скорости для многостадийной каталитической реакции из ЦВА»,
«Определение константы скорости для многостадийной
каталитической реакции из ЦВА: Конкуренция побочных явлений», а именно
в чистых кинетических условиях и в отсутствие потребления
субстрата. Это предельное значение, где TOF ограничивается не
зависимой от потенциала константой скорости химической
регенерации Р (kobs) и лучше определяется как TOFm)WJ.
В области ЦВА, где каталитический ток еще не достиг своего
плато, TOF зависит от приложенного потенциала Ей отражает
процент активированного катализатора (О). Когда вес электронные
переносы нернстовские, отношения описываются уравнением (8J9)4.
Когда приложенный потенциал значительно отрицательнее Еми-2
(потенциала полуволны стационарной каталитической волны
и точки, при которой 50% катализатора активировано), уравнение
(8.19) показывает, что TOF становится независимой от
перенапряжения и достигает значения TOFmj„:
TOF
TOF = ■.'"'" -i; (8.19)
1 + ехр
_F_ V
Felion G.A.N., Vannucci A.K., Chen J., Locked L.T., Okumura N.; Petro B.J.,
Zakai {/./., Evans D.H., Glass R,S., Lichtenberger D.LJ. Op. cit.; Bhugun A,
Lexa 0.. SaveantJ.-M. Op. cit.; Roubelaltis M.M., Bediako D.K., Dogutan D.K.,
Nocera D.G. // Energy Environ. Sci. 2012. 5.7737-7740.
Costentin C, Robert M., Saveant J.-M. Op. cit.
Costentin C, SaveantJ.-M. Op. cit.
Costentin C, Robert M„ SaveautJ.-M. Op. cit.; Costentin C, SaveantJ.-M. Op. cit.;
Costentin C, DrouetS., Robert A/., SaveantJ.-M. Op. cit.
326
Для всех случаев, представленных в табл. 8.2 и 8.3, за
исключением реакции ЕЕСС с гомогенным переносом электрона и
гемолитическим процессом, TOFmiLX определяется формулой (8.20).
Когда £|СЛ или M^z являются скоростытимитируюшими, TOFmax
может быть упрошена до выражений &ohs, представленных
в табл. 8.2.
Уравнение (8.19) можно переформулировать путем включения
параметра т| (раздел «Перенапряжение (п.)»), чтобы получить значимую
взаимосвязь ТОF — ц (уравнение (8.21))':
TOF
TOF = lwri«B
1 + expl —(E-E^z)
TOF
1'-" max
(8.21)
1 + е'Ч яг( Е'«в ~ Ecat/2 тЧг дг V
ЕССЕ-мсханизм, в котором второй перенос электрона более
трудный, чем первый, является единственным исключением из
общего применения уравнений (8.19) и (8.21). Взаимосвязь TOF — т|
для этого сценария описывается уравнением (8.22), в котором а
является коэффициентом переноса, отражающим то, что второй
перенос электрона является реакцией диссоциативного переноса
электрона и не подчиняется закону Нернста2:
TOF
TOF = г „ "" =г. (8.22)
>[а—(£-,
Для другого исключения, определенного в литературе как гомо-
литический процесс, отношение TOF — т) описывается уравнением
(8.23) и считается химической стадией второго порядка (по
катализатору):
TOF
TOF = - ^-= -^ (8.23)
(£-£си1/2)
|l + exp
1 Costentin С., Saveant J.-M. Op, cit.
2 Ibid.
327
Отношение TOF — Г| (уравнение (8.21)), введенное Савьяном,
обеспечивает средство для сравнения катализаторов при различных
перенапряжениях. Уравнение (8.21), которое включает стандартный
потенциал превращения субстрата £а/в, подчеркивает, что, когда
приложенное перенапряжение составляет £|/2, TOF равна '/2TOFmi,4
(для гемолитического процесса, 0,35 TOF^)'. Как отмечалось выше,
потенциал-зависимая область отражает процент активного
катализатора. Для общих случаев наклон этой области равен F/(RT\n 10).
Для реакционной схемы ЕССЕ, в которой второй перенос электрона
является более трудным, чем первый, наклон равен a[F/(RT\n 10)],
в то время как для гомолитической реакции наклон равен
3F/{2RT\n 10).
Рисунок 8.9 иллюстрирует результат применения уравнения (8.21)
для двух катализаторов с различными значениями EPfQ и коЫ
(TOFmjJ, восстановаюшими один и тот же субстрат. Эти графики
TOF — п могут обеспечить средство оценки того, при каких
потенциалах для конкретного каталитического превращения различные
катализаторы будут наиболее полезными. Например, для
электролиза может быть оптимизировано т| или может быть
идентифицирована желаемая TOF.
1000
100
г- Ю
I
-л
(Г 1
р
Н 0,1
0,01
0,001
0,0 0,1 0,2 0,3 0,4 0,5 0,6
Перенапряжение, 6
Рис. 8.9. ГрафикTOF от приложенного потенциала для двух катализаторов
с различными величинами frobs и fp/Q, демонстрирующий взаимосвязь между
TOF и приложенным потенциалом, Пересечение с нулевым перенапряжением
даетТОРо, а на плато TOF равно fcote. Области сплошных линий указывают, какой
катализатор имеет большее TOF при определенном перенапряжении,
показывая, что, хотя синий катализатор (Т) имеет большее TOFq, черный
катализатор (2) имеет более высокое TOFmax
1 Costentin С, Saveant J.-Af. Op. cit.; Costentin C, DrouetS., Robert M., Servian/J.-M.
Op. cit.
328
Также была предложена характеристика TOF(): экстраполяция
TOF при £= £"1/в'. Идя общих случаев эта взаимосвязь описывается
следующим уравнением:
TOF0 = TOF^ exp[--^{£^B - ^1/2)]. (8.24)
Хотя TOF0 была описана как метод определения того, какой
катализатор по сути является «хорошим» и какой «плохим»2, было
отмечено, что, поскольку TOF0 зависит от расположения нуля
перенапряжения, используя различные субстраты с различными
значениями Е^ь , можно перевернуть результат определения — какой
катализатор «лучше»3.
Кроме того, как отмечалось ранее, на наклон зависимости TOF —
Л может влиять механизм процесса, что будет серьезно искажать
оценки, почерпнутые mTOF0. TOF0 не включают возможности того,
что субстраты с различными значениями £а/в могут ускорять или
замедлять гомогенную с ко ростьл имитирующую стадию, изменяя
скорость катализа. Кроме того, как отмечалось во введении TOF^,
значение TOF0 зависит от концентрации субстрата, а значения TOF0,
охватывающие три порядка величины, были определены в изучаемой
системе, в которой концентрация субстрата изменялась на два
порядка. Эта неотъемлемая зависимость TOFo, как от природы
субстрата, так и концентрации, подчеркивает необходимость сравнения
катализаторов с одними и теми же концентрацией катализатора,
субстратом и концентрацией субстрата. Маловероятно, что
катализаторы, описанные в литературе, будут изучаться с одинаковыми
по природе субстратами и концентрациями, т.е. этот показатель
полезен только для тестов в строго определенных случаях, например,
в специальном сравнительном исследовании катализаторов.
В то время как кинетические параметры (AotJS, TOFmax, и
отношения TOF — т|) являются ценными параметрами, на наш взгляд,
их абсолютная величина не является наиболее значимым
показателем для сравнения, потому что она часто зависит от ситуации
(например, концентрации субстрата). Наиболее значимой является
механистическая информация, которую можно почерпнуть из
анализа кинетики реакции. Эта информация может дать закон скорости
или константы скорости, параметры, которые обеспечивают более
значимое понимание производительности катализатора.
1 Cosfmtin С, SaveantJ.-M. Op. cit.; Castentin С. DmuerS.. Robert M., SaveantJ.-M. Op. cit.
1 Castetttin C., Dtvuet S., Rohan M>, Saveonl J. - M. Op, с it.
1 Quentel F.t Gloaguen F. // ElecLrochim. Acta 2013. 110. 641—645.
4 Costentin C, Drouet S., Robert M., Saveant J.-Al. Op. cit.
329
8.3.4. Примеры
Применение методов анализа, представленных в разд. S.3.3 для
молекулярных электрокатализаторов для генерации Н3 и других
многоэлектронных, многостадийных схем реакций, может предоставить
важную кинетическую и механистическую информацию.
Проблемы этого процесса включают выявление, минимизацию
и учет конкурирующих побочных явлений в анализе. Ниже мы
сначала представим серию симулированных волътамперограмм,
которые четко иллюстрируют влияние вредных побочных явлений
на катализ. Затем мы предлагаем пример, который иллюстрирует,
как конкурирующие побочные явления могут быть учтены, как
общая каталитическая константа скорости может быть получена
для сложной много электронной, многостадийной схемы реакции
и насколько ценная механистическая информация может быть
почерпнута из легко получаемого множества циклических
волътамперограмм.
Показательные примеры нендеализированных ДВА.
Каталитические S-образные вольтамперограммы в зоне KShu рис. 8.5
описаны уравнением (8.10). причем каталитический ток плато зависит
от площади поверхности электрода, коэффициента диффузии
катализатора, количества участвующих электронов и наблюдаемой
скорости. Хотя этот предсказанный каталитический токовый
отклик концептуально легко интерпретировать, на самом деле
эксперимент очень редко дает отклик ЦВА, близкий к идеальному
«S». Потребление субстрата в реакционном слое может привести
к нарушению предположения псевдо-первого порядка в
уравнении (8.10). Кроме того, могут произойти различные побочные
явления — распад катализатора или ингибирование продуктом,
которые помешают достижению S-образной вольтамперограммы.
Как будет рассмотрено ниже, анализ их возмущенных
каталитических токов непосредственно из уравнения (8.10) приводит к
неточным показателям-характеристикам и неправильно
интерпретированным механизмам.
Согласно уравнению (8.10) каталитический ток /с не должен
зависеть от скорости сканирования. Рисунок 8.10 показывает
смоделированные циклические вольтамперограммы для механизма ЕГС-
при различных скоростях сканирования. При 100 В • с ' отклик
представляет предсказанную S-образную вольтамперограмму с обратной
трассировкой, почти наложенной на прямое сканирование. Когда
скорость сканирования замедляется, очевидны три различия: вольт-
амперограмма имеет форму пика, указывающего на зону К,
максимум тока уменьшается с уменьшением скорости сканирования
и обратное сканирование не перекрывается с прямым сканирова-
ззо
нием. Когда зависимость тока в пике от скорости сканирования
строится, как показано для симулированных вольтампсрограмм
(рис. 8.10, вставка) и [Ni(7PPh2NC(.H4Br)2l2+ катализатора (рис. 8.11),
наблюдаются области, зависимые от скорости сканирования, и
области, не зависимые от скорости сканирования. При более низких
скоростях сканирования измерение занимает больше времени, а
субстрат будет исчерпан в реакционном слое, исключая выполнение
условий псевдо-первого порядка, необходимых для уравнения (8.12).
Наоборот, при более быстрых скоростях сканирования обшее время
измерения короче, субстрат существенно не расходуется в течение
эксперимента, а условия псевдо-первого порядка также
выполняются. Чтобы правильно определить наблюдаемую константу
скорости из циклической вольтамперограммы, каталитический токовый
отклик должен быть независимым от скорости сканирования. ЦВА
для каталитических систем всегда должны быть проанализированы
при различных скоростях сканирования, чтобы определить скорость-
независимую область сканирования и подходящую скорость
сканирования, при которой проводить анализ.
d
я
-4-*
С
и
и.
о
Potential (а. и.)
Рис. 8.10. ЦВА для механизма Е,С{, показывающая переход от зоны К к зоне KS
с увеличением скорости сканировании, При еще более высоких скоростях
сканирования (не показаны), зона КО будет достигнута. Вставка-график: ток
в пике как функция скорости сканирования. Симулируется с DigiElch: a = 0.5,
Ks = 10 000 см - с ', кЕ = 100 000 М ' - с ', Гкатализатор] = 0.002 М,
[субстрат]0 =1 М
В дополнение к причинам неточной оценки каталитической
скорости расход субстрата может также привести к неправильному
определению порядка реакции по субстрату и катализатору. Рисунок 8.12
показывает симулированные циклические вольтамперограммы для
механизма £Ю"для трех различных концентраций катализатора,
с (рис. 8.12, в) и без (рис. 8.12, а) расхода субстрата.
I
1
1
л ■ * '
• 1 VA
•
•0,IV/s
0 50 100
Scan Rate (V/s)
100 V/l
331
independent
О
1111111111111111111111111111111
5 10 15 20 25 30
Scan Rate (V/s)
Рис. 8.11. Зависимость скорости сканирования каталитического тока
в пике/„, для никелевого электрокатализатора выделения Нг из кислоты
в ацетонитриле. Наблюдаются области как зависимые от скорости
сканирования, так независимые
(адаптировано из работы Stewart M.P., НоМ.-К., WieseS., Lindstrom M.L, Thogenon C.E.,
RaugeiS., BuihxkRM. Helm M.U. ft Am. Chem. Soc 2013.135.6033—6046}
Current (a.u.)
Icalatystl
i
I
i
i —.— 4mM
f/
1,0
0,6
0,4i
0,2
0,0-
Potential (a.u.)
с
3
U
■a
No subsircie depletion!
Suhftivuc depletion!
i i i i i ii i 'i i i i
|catalyst|
lUraM
4inM
IraM
0,0 4,0 8,0 12,0
[catalyst] (mM)
6
Potential (a.u.)
Рис 8.12. ЦВА как функция концентрации катализатора для механизма frQ
при отсутствии потребления субстрата (о) и расходе субстрата (в). Средний
график (б) показывает ток в пике как функцию концентрации катализатора для
сценариев о и с, демонстрируя, что менее чем первый порядок
по катализатору можно наблюдать, когда субстрат потребляется в ходе
вольтамперограммы. Симулировано с DigiElch: a = 0,5, к} = 10 000 см ■ с-1,
скорость развертки 0,2 В • с-1, *«, = 100 000 М-1 ■ с-1, [субстрат)0 = 1 М.
В сценарии а концентрация субстрата не варьируется в ходе записи
вольтамперограммы
Когда расхода субстрата нет, вольтамперометрический отклик
имеет S-образную форму волны, а пиковое значение тока имеет
зависимость первого порядка от концентрации катализатора
(рис. 8.12, 6). Однако когда субстрат расходуется во время
эксперимента, вольтамперометрический отклик является классическим
отклонением от S-образной волны: ток не имеет плато, обратный
след не совпадает со следом в прямом направлении. Кроме того,
анализ тока в пике приводит к кажущейся зависимости от
концентрации катализатора^ которая меньше первого порядка. Измерение
332
токового отклика в зависимости от скорости сканирования должно
определить, влияет или не влияет потребление субстрата на воль-
тамперограммы.
Разложение катализатора может также индуцировать
значительные отклонения от S-образных вольтамперограмм
за время записи типичной ЦВА. На рис. 8.13 показаны ЦВА-от-
клики для механизма EjCh причем восстановленный
катализатор Q может разлагаться с константой скорости kdet:ump. В
показанном примере с быстрой каталитической константой скорости
(ке = 100 000 М-1 • с-1) значения А^сощр низкие — порядка 30 с-1,
приводящие к не-S-образной форме циклических вольтамперограмм.
Влияние разложения катализатора может быть уменьшено при
быстрых скоростях сканирования.
Ё
э
Potential (a.u.)
Рис 8.13. Симулированные ЦВА для механизма £,С' как функция скорости
распада восстанавленного катализатора Q. Симулировано с OigiElch: a = 0,5,
к = 10000 см - с~\ скорость развертки 0,2 В * с1, кв = 100 000 Ми - с\
[катализатор]0 = 0,002 М, [субстрат]0 = 1 М
Механистические и кинетические исследования катализатора
восстановления СОг. Пример Савьяна и других демонстрирует
перенос понятий, представленных в разд. 8.3.3, с их теоретических
основ на применение в механистическом анализе1. Интересующая
система — это двухэлектронное восстановление СО^ в СО
катализатором тетрафениллорфирином железа (FeTPP) и сокатализа-
тором кислотой Бренстеда. Эта работа подчеркивает как
экспериментальные тонкости, необходимые для получения наблюдаемой
константы скорости, так и последующее установление
соответствующего механизма. Первый интересующий момент в данном
примере заключается в отсутствии S-образного каталитического оклика
на ЦВА. Как отмечалось выше, это обычное явление в электрока-
1 CosTentin С, Drouet S., Passard &'., Robert M., Saveant J.-M. Op. cit.
P+e—k-»Q
Q+A—^->P+B
Q_W*aL_> decomposes^
N44500 ь"'
333
тализе, а по причинам, которые уже обсуждались, исключается
использование уравнений плато. Авторы обратились к методу FOWA,
изложенному в разделе 8.3,3, и использовали этот метод для
получения наблюдаемой скорости катализа. Представленные данные
для этого случая не могут рассматриваться как «идеальный»
сценарий (рис. 8.14; катализ происходит при потенциале пары Fe'/Fe°,
а не пары Fe'yFe1) для использования метода FOWA, поэтому важно
сначала продемонстрировать, что авторы способны его точно
применять из практических соображений. Активной формой
катализатора является Fe°TPP; однако полученный катализатор содержит
состояние окисления Fe11.
■* Fe' + e
Fe°
Fen + C02 + AH *"r > Fe1 + product
Fe"
Fe"CO
Рис. 8.14. Выбранные вольтамперограммы, показывающие обратимые волны
FeVFe1 и FeVFe0 (голубая 2) и каталитическую волну после добавления С02
и источника протонов (красная 1)
(адаптировано из работы Costentin С, DrouetS., PassardC, Robert M., Saveantl-M* II
J. Am. Chem. Soe. 2013.135.9023—9031)
FenTPP претерпевает два обратимых одноэлектронных переноса
до достижения активного состояния, что представляет две проблемы
для FOWA. Во-первых, некаталитическая редокс-активность будет
мешать линейной регрессии. Авторы обходят это пугем соединения
данных, удалив область, связанную с парой Fen/Fer, так что анализ
334
проводится только для данных пары Fe'/Fe". По-прежнему
остается остаточный ток от диффузии Fe", но он незначительно влияет
на общий анализ. Вторая проблема состоит в том, что /р,
определяемый лля FOWA, используется для аннулирования концентрации
и коэффициента диффузии катализатора. Это основывается на
предположении, что пиковый ток генерируется из одноэлектронной
нсрнстовской волны без каких-либо других воздействий. В этом
примере пара Fe'/Fe" является одноэлектронной нернстовской
волной, перекрывающейся с диффузионным шлейфом Fc"/Fe[, как
отмечалось выше. Хотя диффузионный шлейф оказывает
незначительное влияние на каталитический ток, этим нельзя пренебречь
в отсутствие субстрата. Авторы решили это с помощью пикового тока
волны Fe"/Fe' ip. Исходя из этих соображений и проводился FOWA.
Результаты FOWA были интерпретированы путем применения
стационарных приближений к интермедиатам реакции с
использованием общей каталитической константы скорости (коЬ$), полученной
для предложенного ниже механизма:
Константы равновесия.
I *• ~Fe(I) + e ■ 2~Fe(0) первые константы
скорости
2-Fe(0) + CO2=^"Fe(T)CO2~ (I) Кик\
Fe(I)C02" + С02 » "Fe(II)CO + С032 к0
-Fe<I)C02'-+AH ^=^-Fe(I)C02*-... НА (2) К2,кг
-Fe{I)C02"-+ H20=^-Fe(l)C02'-~ Н20 Kik'2
= Fe(I)C02"...H20 + H20—> Fe<lI)CO+H20 + 20H~ kQ
-Fe(I)C02*" ..H20 + C02 > Fe(II)CO + HCOf + ОН" Ц
-Fe(I)C02-...H20 + AH > Fe(II)CO + H20 + OH" + А" Ц
-Fe(l)C02-...HA + AH » Fe<H)CO +H20 +2A~ (3) k3
-Fe(I)CCh"-...HA + H20 > Fe(TI)CO + H20 + OH" + A" b"
Fe(T)C02-...HA + C02 > Fe(IT)CO + HCOj" + A" k?
Fe(il)CO + 2-Fe(0) > 2~Fe(l) + CO
I
C02, H20 + A" > продукт + АН
Здесь концентрация субстрата (С02) и сокатализаторов (НА
и Н30) рассматривалась постоянной в пределах диффузионного
слоя реакции (и равной объемным концентрациям). Важно
отметить, что кой. была получена из законов диффузии Фика. В результате
уравнение скорости требует, чтобы концентрационные параметры
катализатора и субстрата были разделены (т.е. Л:^ не должны содер-
335
жать концентрацию катализатора в качестве параметра). Это было
достигнуто тщательной заменой переменных и некоторыми
приближениями, чтобы получилъкфъ.
Конкретные роли сокатализаторов были исследованы по
изменению концентрации субстрата (C02J и сокатализаторов (НА и Н20).
Также была устано&пена подробная механистическая информация
и выявлена скоростьопределяющая концертная реакция переноса
протона-электрона и разрыва связи,
В целом этот пример является яркой иллюстрацией того, как
общая каталитическая константа скорости может быть получена
для сложной реакции, а ценные кинетические и механистические
данные могут быть извлечены из неидеализированных (S-образных)
ЦВА-откликов.
^=к, к [co2i+MMMMMktM^it)+
>2(*ЛН20|Г+*ЛС021) Aj'IHjOUj
k_7 + ki\AH] к:2+кЦАН\
[АН]+ ^ [АН]2
*_2+*3[АНГ
8.4. ЗАКЛЮЧЕНИЕ
Можно надеяться, что глубокие электрохимические
исследования новых молекулярных электрокатагшзаторов помогут
реализовать крупномасштабное производство чистого топлива и помочь
в понимании механизмов действия ферментов. Электрохимические
методы, в частности ЦВА, как описано здесь, являются мощными
инструментами установления характеристик каталитических
реакций (констант скоростей, TOF и т.д.). Однако следует осознать,
что ни одного точного метода определения электрохимических
каталитических констант скорости не существует, потому что многие
молекулярные электрокатализаторы реагируют по сложным
многостадийным путям, возможно, включающим побочные реакции,
а в результате наблюдаются сложные ЦВА-отклики. Кинетические
параметры могут быть определены или оценены с помощью методов,
рассмотренных здесь. Экспериментаторы должны быть уверены, что
анализ волътамперограмм выполняется на основании данных,
полученных в соответствующих условиях. Кроме того, важно, чтобы
сообщения о катализе сопровождались подробным описанием условий
реакции и используемых методов анализа, для того чтобы можно
было более легко сравнивать катализаторы.
Глава 9. НОВАЯ МЕТОДОЛОГИЯ
ОРГАНИЧЕСКОГО ЭЛЕКТРОХИМИЧЕСКОГО
СИНТЕЗА
Как известно, сейчас разработка экологически чистых процессов
должна следовать концепции зеленой химии и устойчивого развития
(GSC). Поэтому еще в конце прошлого века уделяли внимание
органическому электросинтезу как одному из приемлемых в
отношении экологических требовании процессов. В этом столетии были
разработаны электрохимические реакции с гораздо более низкими
выбросами и электрохимические методы, основанные на новых
идеях и концепциях. Эти исследования стали областью активных
разработок1. В данной главе описаны новые подходы в рамках
методологии органического электролиза. Хотя некоторые методики
и не являются совсем новыми, они упоминаются, потому что очень
полезны с точки зрения GSC.
9.1. ЭЛЕКТРОЛИЗ С ИСПОЛЬЗОВАНИЕМ ТВЕРДЫХ
ПОЛИМЕРНЫХ ЭЛЕКТРОЛИТОВ (5РЕ) И ЕГО ПРИМЕНЕНИЕ
В отличие от обычного органического синтеза
электрохимические реакции требуют наличия фоновой соли. Сложно и
трудоемко отделить желаемый продукт от электролитического
раствора, содержащего большое количество фоновых солей. Кроме
того, фоновые соли, как правило, не извлекают и не рсииркули-
руют, в результате они становятся отходами электролиза. В общем
случае фоновые соли хорошо растворимы в полярных
растворителях, но в зависимости от исходных органических субстратов
некоторые из них растворимы не в полярных растворителях,
а только в неполярных растворителях. Как упоминалось ранее,
правильный выбор фоновой соли и электролитического
растворителя непрост. Резким контрастом является электролиз с
использованием твердых полимерных электролитов (SPE), который
не требует электролитического раствора, поэтому эти проблемы
решаются более доступно2.
1 Yoshida J.. Кашка К., Horcajada Л., Nagaki A. // Chem. Rev. 2008. 108. 2265—
2299.
1 Ogumi Z, Nishio К., Yoshizawa Z. // Electrochim. Acta, 1981. 26. 1779-1782;
Hoormann D.4 Kubon CJorissen J.. Kroner Z., Putter И. // J. Elcctroanal. Chem.
2001.507. 215-225: Jorissen J. //J. Appl. Eleclroehem. 2003. 33. 969-977.
337
9.1.1. Принцип SPE-электролиза
Путем химической металлизации мелкие металлические частицы
осаждаются на поверхности ионообменных мембран типа Nation®.
Затем электрод из проволочной сетки или пористый электрод,
изготовленный из того же металла, прессуется к противоположной
поверхности мембраны так, чтобы получить композитный электрод SPE,
имеющий двойную роль — электрода и фоновой соли. Общие
преимущества системы SPE-электролиза для органического синтеза могут
быть представлены следутошим образом: 1) экономизация выделения
и рециклизашш фоновой соли и 2) избежание какой-либо реакции
или загрязнения со стороны фонового электролита. Существуют два
типа систем SPE. В одной электрод прижимается только к одной
стороне поверхности ионообменной мембраны, а в другой два электрода
припрессованы к обеим поверхностям. Рисунок 9.1 иллюстрирует одну
из систем SPE. Субстрат АН2 окисляется на аноде и результирующие
протоны мигрируют через катионообменную мембрану к катоду. Таким
образом достигается ионная проводимость, а именно — протонная
проводимость. Поступающие ионы водорода восстанавливаются
на катоде, генерируя газообразный водород в отсутствие субстрата
(рис. 9.1, а), в то время как субстрат В восстанавливается в ВН2 в
присутствии субстрата В (рис. 9.1, 6). В последнем случае два продукта —
А и ВН2 получаются из двух исходных субстратов — АН2 и В, поэтому
этот электролиз можно рассматривать как вид парного электросинтеза.
Анод Катод
с
Анод Катод
®
Н
2Н+
Кятиин-об\пнн.1Я
н2|
®
н
®^
2Н+
ka 111«н-иГ>\п
чембран|
Н
СвГ"
v®
ИНАЯ ^ '
Пористая Пористая
Pt Pt
а
Пористая Пористая
Pt Pt
6
Рис. 9.1. Принцип электролиза с использованием системы SPE с платиновыми
электродами
В системе электролиза SPE, в которой электрод прижимается
только к одной стороне поверхности ионообменной мембраны, про-
тивоэлектродное пространство заполнено элек'гролитическим раство-
338
рителем. В SPE-системе в качестве электролитических растворителей
можно использовать даже неполярные растворители, такие как гексан.
Кроме тою, когда исходный субстрат представляет собой жидкость или
газ, электролиз может быть выполнен без каких-либо растворителей.
Более того, количество используемого редокс-медиатора может быть
уменьшено путем иммобилизации его HaSPE композитном электроде.
На сегодняшний день описаны различные примеры органических
электросинтезов, использующих системы SPE, например:
гидрирование олефинов, восстановление нитробензола, электролиз Кольбе,
диметоксилирование фурана, синтез кегона с использованием
галогена-медиатора и т.д.1 SPE-электросинтез —это энергоэффективный
электрохимический метод, так как напряжение ячейки невысокое
из-за малого расстояния между электродами. Однако прочность SPE-
композитных электродов невысока, а загрязнение мембраны SPE
уменьшает выход по току и веществу. Эти проблемы еше не решены,
и коммерциализация SPE-органического электросинтеза не
достигнута. Оригинальной концепцией для SPE-органического
электросинтеза является топливный элемент с полимерным электролитом
(PEFC). Таким образом, разработка новых материалов, таких как
мембраны, и повышение уровня знаний в исследованиях топливных
элементов должны стимулировать прогресс SPE-электросинтеза.
9.1.2. SPE-электролиз с когенерацией (химическое производство
с использованием реакций, протекающих в топливных
элементах)
Система электролиза SPE была применена к коммерческому
электролизу воды (расщеплению воды) и топливным элементам.
Топливный элемент представляет собой устройство для
преобразования свободной энергии Гиббса химической реакции в
электричество с помощью электрохимической реакции. В топливном элементе
Н2-Ог электричество получается путем образования воды из водорода
и кислорода. Этот принцип предполагает, что каталитическое
окисление и восстановление в химическом синтезе можно преобразовать
в реакции топливных элементов на аноде и катоде. Например, Вакер-
процесс окисления этилена катализируется редокс-частицами типа
Pd2+/Pdri и Cu2+/Cu", как показано на рис. 9.2. На основании потоков
протонов и электронов в реакционной схеме можно ожидать, что
окисление этилена Вейкера в ацетальдегид кислородом будет возможно
с помощью реакций топливного элемента, как показано на рис. 9.3.
1 Ogumi Z, Nishio К., Yoshizawa Z Op. cit.; Hoormann D., Kubon C. Jorlssen /.,
Kroner /.., Putter H. Op. cit.; Jorissen J. Op. cit.; Ogumi Z., Ohashi S., Take-
haraZ, // Electrochim. Acta. 1985. 30. 121 — 124; Ogumi Z, Inaba M., Ohashi S.,
Uchida M.,Takehara Z. // Electrochim. Acta. 1988. 33. 365-369: Ogumi Z. tn-
atomiK., HinatsuJ.T.. Takehara Z. // Electrochim. Acta. 1992.37. 1295-1299.
339
н,с = сн2
ню
МеСНО
2Н+
Рис. 9.2. Механизм Вакер-реакции окисления
н2о
Фактически этилен вместе с парами воды и кислород поступают
в левый и правый отсеки, как показано на рис. 9.3, затем цепь
замыкается. В этих условиях протекает ток и ацетальдегид образуется
с высокой селективностью (95%) при Ю0°С. Таким образом, в этой
системе и поток электрического тока, и автоокисление этилена
проходят спонтанно, даже если потенциал не накладывается. Эта
система топливного элемента позволяет осуществлять так
называемую когенерацию электроэнергии и химических веществ1.
Анод Катод
Л
МеСНО
"•"
Г5
СН2 = СН2. гЬО
>
Кит
м-ипм<:ннш1
-Л
н,о
03
Пористая Пористая
Pt Pi
Рис. 9.3. Схема процессов в топливном элементе (на аноде этилен
реагируете водой с образованием паров метанола; на катоде кислород
восстанавливается в пары воды)
9.2. ЭЛЕКТРОЛИТИЧЕСКИЕ СИСТЕМЫ С ИСПОЛЬЗОВАНИЕМ
ТВЕРДЫХ ОСНОВАНИЙ И КИСЛОТ
Обычные растворимые основания легко окисляются на аноде,
а нерастворимые основания на твердой подложке на аноде не
окисляются. На основе этого факта недавно были разработаны новые
1 Otsuka К., Siimuzu К. Yamanaka I. // J, Chcm. Soc, Chem. Commun. 1988.
1272-1273; lidem.//J. Eleclrochera. Soc. 1990.137.2076-2081.
340
электролитические системы с использованием утилизируемых и
повторно редиклизуемых оснований и кислот на твердой подложке
в качестве фоновых электролитов'. Например, основания на твердой
подложке (силикагель или пористый полистирол), такие как
пиперидин, способствуют диссоциации протонных органических
растворителей — метанола и уксусной кислоты до протонов и анионов,
а в отношении результирующих протонов действуют в качестве
основных переносчиков электронного заряда (рис. 9.4).
ROH+Q^NR'R2 - RCT + Q^NHR'R2
(R = Me,Ac)
Q^NHR'R2 ^ 0"~"nR'R- + H+
Рис 9.4. Диссоциация протонных растворителей под действием твердого
основания
Эта система широко применяется в различных анодных нукле-
офильных замещениях, таких как метоксилирование и ацетокси-
лирование различных соединений (схема 9.1). Кроме того, следует
отметить, что после электролиза основание на твердой подложке
легко отделяется только фильтрацией, а продукт легко выделяется
из фильтрата* Регенерированные основания на твердой подложке
можно повторно использовать много раз, как показано на рис. 9.5.
—2е —Н+
Субстрат ^ > Продукт
•ПО
МеОН
PhS^^CFj
fN—Вое
НО \ // °Ме
ОМе
.Ар,
ОМе
ГЛ|—Вое
/=\ ,ОМе
0=Л X
\=/ ОМе
(76%)
(9.1)
(94%)
(94%)
Otsuka К., Shimizu Y-, Yamanaka f. // J, Chem. Soc, Chem. Commun. 1988.
1272—1273; Iidem. //J. Electrochem. Soc. 1990.137.2076-2081; Tqjima Т., Fuch-
igami T.//L Am. Chem.Soc. 2005.127 2848—2849; (idem. //Angew: Chem. Int.
Ed. 2005.44.4670-4679; Tajima Т., Kurihara //., Fuchigami T. // J. Am. Chem.
Soc. 2007.129.6680-6681.
341
S-H
ROH
S-H: Субстрат
Base = основание
Повторное
использование
Растворитель: МеОН (R= Me)
MeCN + AcOH (R = Ac)
S-OR*
Фильтрация
Упаривание
Рис 9.5. Регенерируемая и рециклизуемая система для анодного
метоксилирования и ацетоксилирования с помощью твердого основания.
Субстрат + основание —»нуклеофильное замещение -> электролиз —>
фильтрация —> продукт + основание. Основание можно повторно
использовать много раз
9.3. МЕДИАТОРЫ НА ТВЕРДОЙ ПОДЛОЖКЕ
Хотя медиаторы имеют много преимуществ, как описано в гл. 2,
3 и 5, они должны быть удалены из раствора электролита по
завершении электролиза. Однако длительное время медиаторы не
регенерировали после электролиза. Совсем недавно утилизация медиаторов
стала проблемой с точки зрения GSC, в частности атом-экономного
аспекта. Для того чтобы преодолеть эти проблемы, были разработаны
легко отделяемые и рециклизуемые медиаторы.
На ранней стадии развития этого подхода был разработан кросс-
сшитый поли-4-винил пиридин гидробромид как мсдиаторная
система на твердой подложке1. Как видно из (9.2), Вг~, выделяемый
из твердой основы, анод но окисляется в водном растворе
электролита, генерируя гштобромид-ион ВгО~, который окисляет спирты,
давая кетоны, а затем Вг~ регенерируется. Таким образом, медиатор
можно легко регенерировать в виде НВгсоли твердой подложки.
О \ N+HBr" + Н20^*ф-/ N+HOBr" + H2
ОН
Ч
N+HOBr
+
R'
V
он <9-2>
N+HBr+Дч + Н20
R R'
Yoshida J., NakaiR.. KawabataN.ff J. Org. Chem. 1980.45. 5269-5273.
342
Так как N-оксильные радикалы (например, TEMPO) обычно
проявляют обратимые окислительно-восстановительные свойства, как
показано на (9.3), они являются полезными медиаторами для
окисления спиртов. Была разработана дисперсная медиаторная система
на основе N-оксила, адсорбированного на поверхности силикагеля
(рис. 9.6)1.
I I II
ОН О" О
Рис. 9.6. Схема непрямого окисления спиртов с помощью силикагеля
с иммобилизованным N-оксидом
В этой дисперсной системе Вг сначала анодно окисляется,
генерируя Вг+, который окисляет N-оксид на силикагеле с получением
иона оксоаммония (N=0+) с последующим окислением спиртов
с получением кетонов. В этом случае окисление может быть
достигнуто без органического растворителя. Одним из преимуществ
дисперсной системы является следующее. После электролиза
электролитический раствор фильтруют, а оставшийся силикагель
промывают подходящими растворителями (например, ацетоном),
чтобы получить продукт. Однако во время промывки медиатор также
растворяется в органическом растворителе. Чтобы избежать
растворения медиатора, был разработан иммобилизованный на силикагеле
N-оксид, зарекомендовавший себя как. легко рецнклизуе.мый меди-
Тапака Н„ Kawakami К, Goto К.. Kuroboshi M. // Tetrahedron Lett. 2001. 42.
445—448.
343
атор1. N-оксид, иммобилизованный на полимерных частицах, был
также описан. Кроме того, комбинация электродов,
модифицированных полимерами с иммобилизованным TEMPO и хиральными
основаниями, как известно, полезна для асимметрического синтеза2
(см. также разд. 5.3).
9.4. ДВУХФАЗНЫЕ ЭЛЕКТРОЛИТИЧЕСКИЕ СИСТЕМЫ
В общем случае электрохимические реакции проводят в
гомогенных растворах электролитов. Электрохимическая реакция может
быть проведена даже в гетерогенных растворах электролитов, если
и процессы переноса электрона, и массоперенос не ингибируются
реакционной системой. Ниже в этом разделе мы будем обсуждать
двухфазные электролитические системы, такие как электролиз
в эмульсии, электролиз в суспензии, электролиз с использованием
катализа фазового переноса, и термоморфную двухфазную
электролитическую реакционную систему3.
9.4.1. Электролиз в эмульсии
Как показано на рис. 9.7, для случая, когда продукт является
окисляемым и восстанавливаемым соединением, таким как
альдегид, органическая фаза в двухфазной электролитической системе
(в общем системы, состоящей из органической и водных фаз, в
которой органические капельки мелко диспергированы в воде (водном
электролите)) часто используется для экстракции продукта, с тем
чтобы предотвратить его переокисление или избыточное
восстановление на электроде.
Причем несмешиваемые с водой органические соединения
часто подвергают электролизу в их эмульсии. Однако в этих случаях
эмульсия способствует поддержанию насыщенной концентрации
несмешшающегося органического субстрата в водном электролите,
а эмульгированные капли сами непосредственно не подвергаются
электролизу на электроде. В отличие от вышеупомянутых эмуль-
1 Тапака //., Chou /., Mine Л/.. Kuroboshi К. // Bull. Chem. Soc. Jpn. 2004. 77.
1745-1755.
2 Kubota J., Ida Т., Kuroboshi Л/., Tanaka #., Uchida Т., Shimamura K.
//Tetrahedron. 2006. 62. 4769—4773.
J Asami R., Atobc M.. Fuchigami T. Electrpolymerization of an Immiscible
Monomer in Aqueous Electrolytes Using Acoustic Emulsification // J. Am. Chem. Soc.
2005. 127. 13160-13161: Atobe Л/., Chen Л-С, Nonaka T. // Ultrasonic Effects
on Electroorganic Processes. Part II. Emulsion and Suspension Electrolyses under
Ultrasonic Irradiation // Electrochemistry. 1998. 66. 556—559; Chiba AT., Kotto K,
Kim 5., Nishimoto A'., Tada M. A Liquid-phase peptide synthesis in cyclohexane-
based triphasic ihermomorphic systems // Chem. Commun. 2002. 38. 1766—1767.
344
сионных электролитических систем прямой электролиз
эмульгированных капель может также осуществиться на электроде, если
капли содержат фоновые электролиты и их присутствие позволяет
образовать электрический двойной слой внутри капель* Но в данном
случае капли должны быть уменьшены до субмикронного диапазона
с использованием специальных поверхностно-активных веществ или
ультразвука, чтобы получить практически приемлемую скорость
реакции1.
Анод
Рис 9.7. Схема электролиза эмульсии. На аноде алкиловый спирт,
находящийся в водной фазе, окисляется в альдегид, найденный
в органической фазе. Полученный альдегид окисляется в кислоту в водной
фазе
9А.2. Электролиз суспензии
Прямой электролиз суспензий неэлектропроводных твердых
материалов, таких как большинство органических соединений,
не может иметь места в принципе, поскольку электрохимический
перенос электрона между твердым электродом и непроводящими
твердыми частицами практически не происходит, даже если они
контактируют друг с другом. Причем непрямой электролиз
взвешенных твердых частиц с использованием растворимых редокс-
медиаторов может произойти, так как он включает серию
последовательностей двух возможных стадий электрохимического
и химического переноса электрона между твердой и жидкой
фазами (электрод (твердая фаза) — растворенный медиатор
(жидкость) — суспендированные частицы (твердая фаза)) (рис. 9.8)2.
Подобно этой ситуации электролиз эмульгированных капель
жидкости может быть проведен с помощью медиаторов
переноса электрона.
1 Asami R., Atobe M.t Fuchigami T. Op. с it,
2 Atobe M., Chen P.-C, Nonaka T. Op. cit.
345
Растворимый продукт
Анод
Рис 9.8. Схема электролиза суспензии
9.4.3. Пример электролиза с использованием межфазного
катализа
Анодные ароматические замещения проводили с
использованием дисперсий органического растворителя (обычно метиленхло-
рида) в водной среде. Ток протекает в основном через относительно
проводящую водную фазу (тем самым снижая затраты на
электроэнергию), тогда как синтетические реакции, как считалось,
ограничиваются органической фазой; функция катализатора фазового
переноса заключается в том, чтобы перенести анионы в органическую
фазу и придать достаточную электропроводность этой фазе, а также
обеспечить реагент сочетания (например, CN- или СН3СОО-) для
интермедиатов, полученных из ароматических субстратов (рис. 9.9).
Анод
АгН
■♦ ArlT
-т—•►ArCHCN'^^-
->ArCN
Bu4N+CN"
Органическая
BU4N+ + И4 *a3a
NaCN
Na+ H3
Водная
фаза
:
Катод
Рис. 9.9. Пример электролиза с использованием межфазного катализа.
На аноде реакция синтеза идет в органический фазе, а на катоде ток течет
в водной фазе
346
9.4.4. Термоморфная двухфазная электрохимическая
реакционная система
Чиба с сотрудниками создал термоморфные двухфазные
электрохимические реакционные системы, состоящие из органических
растворителей разной полярности (например, циклогексан-
нитроалканы)1. Как показано на рис, 9.10, в термически смешенном
гомогенном растворе электролита анодная десульфиризация
протекает гладко, давая метид о-хинона, который затем вступает в
реакцию с терпеном, давая в качестве конечного продукта циклоаддукт.
После завершения электролиза продукт в виде раствора в иикло-
гексане может быть отделен от полярного электролитического
раствора (фаза нитроалкана) простым разделением фаз при охлаждении.
Кроме того, отделенный полярный электролитический раствор
может быть повторно использован для следующего цикла реакции.
9.5. МЕТОД КАТИОННОГО ПУЛА
Карбокатионы, генерируемые путем электрохимического
окисления, могут реагировать с нуклеофилами с образованием кова-
лентных связей. Из-за короткого времени жизни карбокатионов
необходимо генерировать их в присутствии нуклеофилов с
последующей быстрой реакцией. В вышеописанной системе используемые
нуклеофилы должны иметь более высокий потенциал окисления,
чем молекулы прекурсоров карбокатионов. Поскольку углеродный
нуклеофид легко окисляется, образование углерод—углеродных
связей с использованием карбокатионов, генерируемых анодным
окислением, как правило, затруднено. Метод катионого пула может
сохранись карбокатионы, стабилизированные соседними гетероато-
мами или ароматикой, при низкой температуре <—78°С) без
разложения. Добавление нуклеофилов к катионому пулу приводит к
желаемой нуклеофильной реакции (рис. 9.11 )2.
При низкой температуре вязкость электролитического раствора
увеличивается, и это уменьшает ионную проводимость раствора.
Днхлорметан является подходящим растворителем для метода кати-
онного пула, поскольку имеет достаточную проводимость для
электролиза. В разделенном электролизере субстрат вводится в анодную
камеру, атрифтормстансульфоновая кислота (источник протонов)
вводится в катодную камеру, чтобы обеспечить протекание
расходуемой катодной реакции. Многообразные катионные пулы успешно
генерируются, как показано на рис. 9.12. Катионный пул ионов ал-
коксикарбения получают путем анодного окисления ос-еилилышх
1 Cfiiba К,. Копо Г., Kim S., Ntshimoto К., Tada A/. Op. cit.
2 Yoshida J. // Chem. Commun. 2005.4509-4516; Yoshida J., Saito K., Nokami Т.,
NagakiA. ft Synlett. 1189—1194.
347
Добавление
субстрата
25"С
COjH
Электролиз
55°С
-S'(CH2hCOOH
оЛУ
Отделение
продукта
МеО'
25°С
ОМс Yield: 76%
I гЧ^4^4-! 4r(H02ccH2CH2s)2
Me
II I
Верхний слой: цикл ore ксан
Нижний слой: нитрометан/нитроэтан (1:4)
500 mM LiC104
Рис. 9.10. Термоморфная двухфазная электрохимическая реакционная система
эфиров при низкой температуре, причем добавление аллилсиланов
качестве углеродных нуклеофилов дает эффективно желаемые
продукты. Аналогично пул иона N-ацилиминияи пул ионов диарилкар-
бения доступны для химических превращений. На рис. 9.13
показаны примеры нуклеофильных реакций накопленных частиц ионов
N-ацилкарбения.
—С—М ~е' ._ >
I
-78ЧГ
I
» —С—Nil
I
М = Н or Si Cation pool
Рис. 9.11. Схема реакции нуклеофильного замещения методом катионого пула
Аг-^^Лг
Рис 9.12. Примеры катионных пулов
R'
I
С02Ме
I КУ
С02Ме
r'v^h^R"
п.
Рис. 9.13. Различные реакции ионного пула катиона N-ацилиминия
349
Концепция метода катионного пула может быть расширена и
применена в методе катионного потока в сочетании с системой
микрореактора (см. разд. 9.8). Этот подход позволяет не только исследовать
короткоживушие интермедиаты, генерируемые электрохимически,
но и реализовать комбинаторный синтез полезных материалов.
9.6. МЕТОДЫ, УПРАВЛЯЕМЫЕ ТЕМПЛАТОМ
В общем случае региоселективная межмолекулярная реакция
сочетания протекает труднее по сравнению с внутримолекулярной.
Темплат-направленные по внутримолекулярному пути реакции
позволяют реализовать селективные реакции сочетания, которые
в противном случае труднодостижимы. Региоселективное анодное
сочетание фенолов может быть достигнуто с помощью темплата
бора'. Сначала темплат. тетрафеноксиборат получают посредством
процедуры one-pot^ а затем его анодное окисление с последующим
гидролизом дает селективно продукт орто-орто сочетания:
Анодное окисление
—»
McCN
<9.4)
ОН
85%
Кремниевый темплат также известен2.
9.7. ЭЛЕКТРОЛИЗ В СВЕРХКРИТИЧЕСКИХ ЖИДКОСТЯХ
Коммерциализация электросинтетических процессов
сдерживалась ограниченной растворимостью субстратов и продуктов
в обычных электролитических растворах, невысокими характериети-
1 Matkowsky 1.М.. Rommel С £„ Frohlkh R., Grksbach U., Putter U.. Wald-
vogeiS.R. // Chem.Eur. J. 2006. 12. 7482-7488.
2 Hudson СМ., MoelierK.D. // J. Am. Chem. Soc. 1994.116. 3347-3356.
350
ками межфазного массопереноса для двухфазной системы, в которой
происходит реакция на разделе фаз твердая фаза (электрод) —
жидкость (электролит), низкой селективностью желаемых продуктов
реакции и сложными схемами разработки, которые часто используются
для выделения продуктов.
Сверхкритические жидкие растворители могут преодолеть многие
недостатки, связанные с обычными растворителями — водой и
органическими растворителями. Сверхкритические жидкости
традиционно применяются для хроматографии, экстракции, очистки и т.д.1
Кроме того, их все более широко признают в качестве полезных сред
для органических и полимерных синтезов в процессах лабораторного
и промышленного масштаба из-за их низкой токсичности, простоты
удаления растворителя, возможности повторного использования (ре-
циклизации) и вариации скоростей реакции2.
Сверхкритическая жидкость может быть определена как любое
вещество, которое находится выше его критической температуры
(7^) и критического давления (Рс). Она существует в однофазном
виде (рис. 9.14). Физико-химические свойства сверхкритической
жидкости находятся между свойствами жидкостей и газов и могут
варьироваться между проявляемыми газами до жидкоподобных
значений при небольших изменениях давления и температуры3.
Например, сверхкритические среды проявляют большую диффуэность
и более низкую вязкость, чем обычные жидкости. Следовательно,
Тс Т
Рис. 9.14. Фазовая диаграмма давление — температура
1 Vdtfte F., Stoddart J.F. Supercritical Fluids for Organic Synthesis. Wcinhcim: Wiley-
VCH Verlag GmbH, 2000.
1 Jessop P.G., Leimer W. Chemical Synthesis Using Supercritical Fluids. W;inheim:
Wiley-VCH \fcrlag GmbH, 1999; Kendall J.L., Ctmehs D.A., YounxJ.L., DeSi-
morte J.M. Polymerizations in Supercritical Carbon Dioxide // Chem. Rev. 1999.
99- 543—564.
J Wferf, F., Stoddart J.F. Op. cit.
3S1
сопротивление к межфазному мае со переносу также ниже по
сравнению с жидким растворителем. А субстраты и продукты, которые
плохо растворимы в жидкой фазе, могут стать значительно более
растворимыми в сверхкритических средах. Такие уникальные и
полезные свойства сверхкритических жидкостей делают их
подходящими растворителями для электросинтеза.
Обычные сверхкритические растворители, такие как диоксид
углерода и этан, имеют мягкие критические условия, но они не
способны сольватировать полярные электролиты. Реакционная среда
должна, следовательно, как правило, создаваться путем смешения
с сорастворителем (метанолом1, этанолом2, ацетоном3, ацетони-
трилом4 или ДМФА5) для получения подходящего раствора
электролита.
Домбро с сотрудниками сообщил, что диметилкарбонат был
получен в результате электросинтеза из монооксида углерода и
метанола в сверхкритической двуокиси углерода в среде метанола6:
2СНдОН + СО ^^^р900 >(СН30)2СО (9.5)
scC02-CH3OH
Они добавили 0,35 мольной доли метанола в сверхкритический
диоксид углерода, чтобы получить смесь, способную растворять соль
аммония. Такая смесь обладает ионной проводимостью, а электролиз
протекает гладко с получением диметилкарбоната и отличным
выходом по току (около 100%).
Причем Токуда с сотрудниками сообщил об
электрохимическом карбоксилировании бензилхлорида, коричного хлорида
и 2-хлорнафталина в среде сверхкритической двуокиси углерода
и ДМФА7:
1 Jun J.tFedkiw P.S. Ionic conductivity of alkali-metal salts in sub- and supercritical
caibon dioxide plus methanol mixtures // J. Electroanal. Chem. 2001.515. 113—
122.
2 Wu W., Zhang J.. Han В., Chen J.. Liu Z, Jiang Т., He J., Li W. Solubility of
room-temperature ionic liquid in supercritical C02 with and without organic
compounds// Chem. Commun. 2003.39. 1412-1413.
3 Ibid.
4 Van H., Sato Т., Komago D., YamaguchiA., Oyaizu K„ Yuasa M., Otake, K.
Electrochemical Synthesis of a Pblypyrrole Thin Film Using Supercritical Carbon Dioxide
as a Solvent// Langmuir. 2005. 21. 12303—12308.
5 Sasaki A., Kudoh H., Senboku H., Tokuda M. Electrochemical Carboxylalion of
Several Organic Hatides in Supercritical Carbon Dioxide, Novel Trends in Elec-
traorganic Synthesis / ed. S. Torii). Tokyo: Springer-\ferlag, 1998, P. 245—246.
* DombroJrRA., Prentice GA., McHugh MA. Electro-Organic Synthesis in
Supercritical Organic Mixtures//J. Electrochem, Soc. 1998.135. 2219—2223.
7 Sasaki At Kudoh //., Senboku //., Tokuda A/. Op. ciL
3S2
п
+ scC03
40"С, 80 кг • см"2
I I Зфмоль-1
Pt Mg 37,5 мА-ся"г
BuiNBr/scCOi+DMF
COOH (9.6)
Выход: 81%
XI
n
+ scCOi
3 Fmol
Pt Mg 37,5 mA'cm"
ОС
46°C, 80 kg ■ cm"2
.COOH
(97)
Yield: 64%
П"
scC02
43°C,80kgcin-2
П
pt
3Fmor
Mg 37,5mA-cm-1
3Fmor'
Pt Mg 37t5mA'gm~^
BujNBr/seCOz+DMP
COOH
(9.8)
BiuNBt/scCOi+DMF
Yield: 61%
Смесь, используемая в этой работе, также обладает
способностью сольватировать полярные электролиты, например,
аммонийные соли. В этих реакциях, так как углекислый газ играет
роль реагента, С02-насышенные условия как сверхкритические
способствуют повышению выхода продукта. Как упоминалось
выше, обычные сверхкритические жидкости, такие как
сверхкритический диоксид углерода, имеют низкую диэлектрическую
проницаемость. Следовательно, многие фоновые электролиты,
как правило, трудно растворить в этой жидкости. Поэтому, чтобы
получить подходящий электролитический раствор, к жидкости
следует добавить полярный органический растворитель. Однако
наличие сорастворителя ограничивает доступный диапазон
потенциалов и приводит к осложнениям. Использование
сверхкритических фторированных углеводородов является привлекательной
альтернативой, которая позволяет избежать этих проблем.
Например, сверхкритический фтороформ проявляет относительно
высокую растворимость, а его диэлектрической постоянной
можно управлять от 1 до 7, манипулируя либо температурой, либо
давлением scCHF, без каких-либо добавок, например, полярных
353
растворителей1. Поэтому сверхкритический фтороформ
использовали в качестве реакционной среды в электрохимических
синтезах.
Атобе с сотрудниками сообщил об электрохимическом синтезе
полипиррола и политиофсна в свсрхкритичсском фтороформе2. Оба
мономера (пиррол и тиофен) можно электрололимеризовать в
сверхкритическом фтороформе быстрее, чем в обычных органических
средах (растворах ацетонитрила), а полученные пленки имеют очень
однородную структуру (рис. 9.15).
i—i
10 цт
а
Рис. 9.15. SEM-фотографии пленок полипиррола,
полимеризованного в растворахацетонитрила (а) и сверхкритического
фтороформа (б)
Кроме того, Атобс успешно получил политиофеновые нано-
кисти с помощью темплатной электрохимической полимеризации
в сверхкритическом фтороформе (рис. 9.16)3. В этой работе на-
нопористые мембраны из оксида алюминия (толщина — 60 мкм,
размер пор — 200 нм), покрытые с одной стороны испаренной Pt
(около 500 нм),были использовали в качестве электродной
матрицы для электроосаждения политиофена в порах.
Использование специальных свойств жидкостей, таких как более высокая
диффузность и низкая вязкость, обеспечивает эффективный
транспорт мономера в пористую темплат-матрицу и наноточное
заполнение полимерами. В результате твердая политиофеновая
наношетка была получена после удаления мембраны из оксида
алюминия.
Mori Т., Li M., Kobayashi A.. Okahata К Reversible Control of Enzymatic Transgly-
cosylations in Supercritical Fluoroform using a Lipid-Coated b-D-Galactosidase //
J. Am. Chem. Soc. 2002.124. 1188-1189.
Atobe M., Ohsuka //., Fuchigami T. Electrochemical Synthesis of Polypyjrole and
Polytihiophene in Supercritical Trifluoromethane // Chem. Lett. 2004.33.618—619.
Atobe Л/.. Hzuka J.. Fuchigami 7".. Yamamoto H, Preparation of Nanostnictured
Conjugated Polymers Using Template Electrochemical Polymerization in
Supercritical Fluids // Chem. Lett. 2007. 36.1448-1449.
10 pm
354
Эффективный транспорт мономера
Нано-
пористый I
темплат %i
Р1аж>д
X,
i
Электрохимическая
полимеризация
*■
200 nm pores
/
Растпорение
темплата
*■
В суперкритической
жидкости
Политиофсн
Полн-
тиофеновая
наношеткн
Рис. 9.16. Получение политиофен-нанощетки с использованием шаблона
электрохимической полимеризации в сверхкритическом фтороформе
9.8. ЭЛЕКТРОЛИЗ В ИОННЫХ ЖИДКОСТЯХ
Ионные жидкости состоят из катионов и анионов без
растворителя, они находятся в жидком состоянии приблизительно при
комнатной температуре. Так как ионные жидкости имеют уникальные
свойства (негорючесть, термическую стабильность, нелетучесть
и возможность повторного использования), они интенсивно
изучались в качестве «зеленого» растворителя сточки зрения
фундаментальных и прикладных аспектов1. Так как они имеют также хорошую
электропроводность, много внимания было уделено их
применению в органических электрохимических устройствах.
Действительно, с недавнего времени проводятся интенсивные исследования
по их применению в перезаряжаемых литиевых батареях,
конденсаторах с двойным апектрическим слоем, влажных солнечных батареях
и топливных элементах. Далее в этом разделе объясняются структура
и физические свойства ионных жидкостей и описывается их
применение в электролитических реакциях.
9.8.1. Структуры ионных жидкостей
Ионные жидкости приблизительно разделяют на два класса
по типу катиона. Первый включает азотсодержащие гетероаромати-
ческие катионы, а второй состоит из алифатических аммониевых
катионов. Алифатические ионные жидкости подразделяются на откры-
тоцепные и циклические. По виду аниона ионные жидкости делятся
Ohio H. (ed.) Electrochemical Aspects of Ionic Liquids. Wiley, 2005; Ohtto H. (ed.)
Electrochemical Aspects of Ionic Liquids. 2nd edn. Wiley, 2011; Wehon T. // Chem.
Rev, 1999. 99. 2071—2084; Wassershad P., Keim W. // AngewChem Int. Ed. 2000.
39.3772—3789; Rogers R. D.. Seddon K./?., Volkov S, (eds) // Green Industria]
Applications of Ionic Liquids. Kluwer Academic, 2002; HepiotP.,LagrostC. // Chem.
Rev. 2008.108.2238-2264.
355
на пять классов: хлороалюминатнъте, фторнеорганические, фторорга-
нические, поли (фтористый водород) и не фтороионные. Кроме того,
в последнее время получены фосфинсодержащие ионные жидкости,
например, соли фосфония, и разработаны различные новые типы
ионных жидкостей с другими новыми анионами (рис. 9.17).
HjC
Ч/
BFf
[emimlBF*
С2Н<
Н,С
ч/
PF6-
[cmimJPFft
С2Н5
НзС^>Н;2н5
CFjS03"
|emim]CF;,SOi
(CF3S02)2N-
[emim](CF5S02)2N
or
[emim]TFSA
/0\
HjCH2C^N\>N^CH2CH=CH2
BFf
PFft-
|BP|PF6
N -C4H*
(CHj)3N+C3H7
(CF3S02)2N
[TMPAJTFSA
(CH2CH3)2N+(CH3)CH2CH2OCH,
BF4-
|DEME|BF4
W чс3н7
<CF3S02)2N~
[PP13JTFSA
ГЧСНз
(CFjS02)2N-
|P13|TFSA
(CiHs)3P+CsHu
(CF3S02)2T>T
[Pu25]TFSA
Рис. 9.17.Типичные примеры структур ионных жидкостей и их аббревиатуры
9.8.2. Гидрофильность и гидрофобность ионных жидкостей
Ионные жидкости также могут быть классифицированы на
гидрофильные и гидрофобные, как показано в табл. 9.1. Независимо
от типа катионов ионные жидкости, имеющие BF^~ и CF3SOJ в
качестве аниона, являются гидрофильными, а с PF^ и (CF3SOj)2N~ —
являются гидрофобными. Их характерное свойство проявляется
в том. что гидрофобные ионные жидкости не смешиваются
ни с водой, ни с обычными органическими растворителями, такими
как эфир и гексан, а следовательно, происходит фазовое разделение,
образование трех фаз» Поэтому продукты можно разделить путем
экстракции жидкость — жидкость, что является одним ид больших
преимуществ ионных жидкостей.
356
Таблица 9.1
Гидрофобные и гидрофильные свойства ионных
жидкостей солей имидазолиния и фосфония
Ионные жидкости
[emimj BF^
fbmim] PFs
[bmim] BF4
[emim] (CFjSO:)2N
tbmiml (CF,S02)2N
[bmim] CFjS03
[emim) CH^OSO,
[emim] CF3SO3
[emim) NOi
tri (te/7-butyl) (dodecyl) phosphoniumtetrafluoroborate
Свойства
Гидрофильные
Гидрофобные
Гидрофильные
Гидрофобные
Гидрофобные
Гидрофильные
Гидрофильные
Гидрофильные
Гидрофильные
Гидрофобные
Взаимосвязь между молекулярными структурами широко
используемых имидазолиевых ионных жидкостей и их физическими
свойствами (температурой плавления, вязкостью и
электропроводностью) заключается в следующем. С увеличением длины ал-
кильной цепи вязкость увеличивается, а точка плавления и
электропроводность снижаются. Однако соотношение между
температурой плавления и длиной ал кил ьн ой цепи не всегда соблюдается,
как показано на рис. 9.18. Чтобы уменьшить температуру
плавления, следует использовать несимметричный или объемный
катион, что приводит к слабому кулоновскому взаимодействию
катиона и аниона.
Ме-^-й
[emim]BF4
[BF4
Me'
Ml
~СбН 13
[BFJ-
ViscosUy:3L8cP(25'C)
Melting point: 14,6*C
Conductivity: l3,6mS/cm
Viscosity: 223,8 cP<25°C)
Melting point: -82°C
Conductivity: 1,04 mS/cm
Рис. 9.18. Отношение между длиной алкильной цепи ионных жидкостей
солей имидазолиния и их физическими свойствами (вязкостью, температурой
плавления и электропроводностью)
Когда в 2-положение кольца имидазолия введена метильная
группа, водородное связывание в 2-положении невозможно. Точка
плавления не уменьшается, а неожиданно увеличивается. Таким
357
образом, соотношение между водородной связью и температурой
плавления до сих пор не выяснено. Когда в 2-положение кольца
имидазолия вводят алкильную группу, затрудняется восстановление
кольца, но окисление облегчается. Кроме того, когда ион
имидазолия как катион заменяется на алифатический ион аммония,
возрастает сопротивление к окислению, а вязкость и температура
плавления также увеличиваются. В настоящее время имидазолиевые
ионные жидкости имеют самую низкую вязкость среди различных
типов ионных жидкостей (рис. 9.19).
Me
Me^N©N^Et I(CF,S02)2N]- Me-lSf-Pr [<CF?S02)2N]-
[cmim](CFiS02)2N Me
Viscosity: 28 cP (25°C) Viscosity: 72 cP <25CC)
Melting point: - 16"C Melting point: 19°C
Conductivity: 8,4 mS/em Conductivity: 3,2 mS/cm
Рис. 9.19. Отношения между катионными фрагментами ионных жидкостей
и их физическими свойствами (вязкостью, температурой плавления
и электропроводностью)
9.8.3. Полярность ионных жидкостей
Полярность ионных жидкостей можно оценить по разности
энергий между основным и возбужденным состоянием красителя
Рейхарда, который обычно используется для оценки полярности
обычных растворителей. Полярность имидаэолииевьгх и пиридини-
евых ионных жидкостей аналогична полярности этанола и немного
ниже, чем метанола. Но их полярность гораздо выше, чем
полярность MeCN и ДМФА, следовательно, она эквивалентна полярности
протонных растворителей. Полярность ионных жидкостей и
органических растворителей можно проиллюстрировать следующим
образом:
МеОН > ионная жидкость соли имидазолия > ионная жидкость
соли пиридиния > EtOH > MeCN > ДМФА.
9.8.4. Электрохимические свойства ионных жидкостей
Несмотря на то что электропроводность неводного электролита
ниже, чем водного, как показано в табл. 9.2, неводные электролиты
имеют преимущество широкого полезного окна потенциалов.
Соответственно ионные жидкости принадлежат к неводным
электролитам.
3S8
Таблица 9.2
Классификация ионных жидкостей и электропроводность
Электролит
Водная
система
Неполная
система
Кислота
Щелочной
Нейтральный
Органический
Неорганический
Ионная жидкость
35wt% H2S04/H20
30wt%KOH/H2O
30wt%ZnCl2/H2O
1 MLiPF6/EC + EMC
2 MLiAICI4/SOCl2
[emim] BF4
Электропроводность (при 25'С)
(mS-cm4)
848
625
105
9,6
20,5
13,6
Как показано в табл. 9.3, имидазолиевые ионные жидкости,
как правило, имеют относительно хорошую электропроводность,
в то время как ионные жидкости на основе соли поли (фтористого
водорода) демонстрируют весьма хорошие значения
электропроводности независимо от катиона (табл. 9.4). Соли 1-этил-З-мети-
лимидазолия (emim) в ряду ионных жидкостей показывают самую
высокую электропроводность при комнатной температуре. Кроме
того, поскольку апектропроводностъ ионных жидкостей коррелирует
с подвижностью ионов, она в значительной степени зависит от
вязкости ионных жидкостей. Значит, молекулярный дизайн ионных
жидкостей с низкой вязкостью имеет большое значение.
Алифатические аммониевые ионные жидкости проявляют более низкую
проводимость, так как они обычно имеют более высокую вязкость
по сравнению с имидазолиевыми ионными жидкостями. В
противоположность этому ионные жидкости с поли (фтористым водородом)
как анионом имеют очень низкую вязкость и поэтому хорошие
низкотемпературные свойства.
Взаимосвязь между молекулярной структурой и проводимостью
ионной жидкости имидазолия показывает, что вязкость
увеличивается с увеличением объемности N-алкильноЙ группы, связанной
с кольцом имидазолия, в результате чего происходит резкое снижение
электропроводности. Однако, даже если N-алкильную группу
заменить атомом водорода, который меньше метилъной группы, вязкость
увеличивается за счет водородных связей с противоанионом.
Проводимость, следовательно, не улучшается. Правило Вальдена, в котором
подвижность и вязкость продукта являются постоянными, может быть
применено к ионным жидкостям, а молярная электропроводность
от вязкости ионных жидкостей подчиняется следующему уравнению:
Xr\ = constant (X = molar electroconductivity, т) = viscosity).
359
Таблица 9.3
Физические и электрохимические свойства ионных
жидкостей солей ими дазо линия при 25°С
Ионная жидкость
|emim| А1СЦ
[emim) H2 iFv-,
[emim] BF4
[emim] CF,CO:
[emim] CH3SO3
[emim| CFjSO-,
[emim] (CFjC02)2N
[emim] (C2F5S02)2N
[cmim| (CF,S03bC
Температура
плавления, °C
8
-90
11
-14
39
-10
-15
-1
39
Плотность,
г- см-1
1,29
1.14
1,24
1,29"
1,25
1,38
1.52**
Вязкость,
мПа
18
5
43
35"
160
43
28
61
181
Проводимость,
mS • см-1
22,6
100
13
9,6*
2,7
9,3
8,4
3,4
1,7
Окно
потенциалов
^nod
г
(Both.
LiVLi)'"
1,0
1,5
1.0
1,0""
1,3""
1.0
1.0
0,9
1,0
5,5
5,3
5,5
4,6""
4,9""
5,3
5,7
5,8
6,0
* 20"С.
** 22"С.
"" GC (glassy carbon), 1 mA - см 2, 20 mV ■ s ',
""Pt,50mV s-'.
Таблица 9.4
Электропроводность ионных жидкостей
солей поли (фтористого водорода}
Ионная жидкость
Me4NF-4HF
Et4NF-4HF
n-Pr,NF-4HF
Et>N-3HF
[emim] H23F3.3
Электропроводность, mS ■ см '
196,6
99,2
33,6
32,6
100,0
Соответственно проводимость ионных жидкостей возрастает
с уменьшением вязкости. Естественно, что вязкость изменяется
с температурой, а электропроводность ионных жидкостей (emim
солей) резко падает с понижением температуры, как показано
на рис. 9.20. Это связано с быстрым ростом их вязкости при
понижении температуры. Данная тенденция вызывает большую проблему.
360
когда электрохимические устройства (батареи и конденсаторы),
использующие ионные жидкости в качестве электролитов,
эксплуатируются в зимний период в холодном помещении. Разработка ионных
жидкостей с отличной проводимостью весьма востребована.
15
Е
^ 10
•о
с
(3
о
• [emim] BF4
О [emim] CF4SO3
□ [emiml <CF3S02)2N
AIemim|(C2F5S02)2N
20 30
-30 -20 -10 0 10
Temperature T ("С)
Рис. 9.20. Температурная зависимость электропроводности ионных
жидкостей соли имидазолиния
Ионные жидкости электрохимически стабильны, а их
используемая область потенциалов, тх* их потенциальное окно, в котором
сама ионная жидкость не может ни окислиться, ни восстановиться,
очень велико. Это одна из характеристик ионных жидкостей.
В целом устойчивость к окислению зависит от аниона, в то же
время восстановительная стабильность зависит от катиона.
Однако, несмотря на то что ион имидазолия имеет положительный
заряд, он легче окисляется, чем BF^, благодаря ненасыщенным
связям вимидазольном кольце. Соответственно устойчивость
к окислению ионных жидкостей имидазолия не всегда связана
с окислительным разложением анионов. Среди органических
анионов трифтораиетат-ион наиболее легко разлагается в
окислительных условиях, в то время как (CFiSCbhN" и (CFiSChbC-
имеют некоторое сопротивление к окислению и не очень легко
разлагаются окислительно. Порядок устойчивости к окислению
следующий, и этот порядок согласуется с потенциалами
окисления, измеренными в растворе:
AsF6~ > PF6~ > BF4" >(CF3S02)3C_ >(CF3S02)2N_ > CF3SO; >CF3CO;
361
Причем восстановительная стабильность обычно зависит от
восстанови [ильного разложения катиона, а алифатический ион аммония
более трудно восстановить но сравнению с ионом имидазолня. Ион
2-метилимидазолия труднее восстановить на 0,3—0,5 В по сравнению
с незамещенным ионом имидазолия. Как показано на рис. 9.21,
ионные жидкости, состоящие из алифатического иона аммония
и (CF3S02)2N_ (TFSA), имеют широкое окно потенциалов — около 6 В.
Юг
с
о
-и
с
е-5
и
-10
[PP13ITFSA
[ТМРА] TFSA
[TEA] TSAC
L
l ^ [emim] TSAC
[emim] TFSI
4,0 -3,0
-2.0 -1,0 0,0 1,0
Potential (V vs. Fc/Fc+)
2,0 3,0
4,0
;n
_-"C3H7
-N+-/iC3H7
4©p
S02CF^ SO,CFt
/ /
N "N
\ \
S02CF, COCFi
k
1TMPAI |PP13i |TEAi |emim] TFSA TSAC
Working electrode: GC, Temperature: 25°C, Scan rate: 50 mV/s
Рис. 9.21. Вол ьтамперограммы с линейной разверткой в различных ионных
жидкостях
Фосфониевые ионные жидкости превосходят ионные жидкости
алифатического аммония по стойкости к окислению и имеют
широкое окно потенциалов (около 6,5 В), а также термическую
стабильность. Ионные жидкости солей поли (фтористого водорода),
однако, имеют узкое окно в сторону отрицательных потенциалов
из-за восстановления кислого протона, как показано на рис. 9.22.
Такое быстрое восстановление протона благоприятствует
преимущественному электрохимическому фторированию (см. разд. 5.7
и 5,8.6). £tjN-3HF легко окисляется из-за загрязненности свободным
амином, в то время как EUNF-4HF и Et-?N-5HF имеют превосходную
стойкость к окислению, а их протоны легко восстанаштиваются
на катоде, генерируя водород.
362
Potential (V vs. Ag/Ag+ (0,1 m))
-10 12
Me«NF 2HF
R4NF-2HF
Me < EK n-Pr
EhNOHF
R4NF-3HF
El3N-4HF
R4NF-4HF
EhN-5HF
R4NF-5HF
Et3N-6HF
Рис, 9,22. Окна потенциалов ионных жидкостей солей поли (фтористого
водорода)
Недавно предложена новая фосфониевая ионная жидкость
три (трет-бутил) (додецил) фоефония тетрафтороборат,
используемая для изучения электрохимических свойств
нерастворимых соединений в пастовых электродах1. Преимущества
предлагаемого электрода состоят в высокой проводимости
(2,63 mS • см-1), очень широком электрохимическом окне (от 2,7
до —2,9 В; это один из самых широких диапазонов доступных
потенциалов, который когда-либо ранее сообщался для ИЖ),
стабильности во времени и воспроизводимости. Электрод RTIL—
СРЕ позволяет определить вольтамперыые характеристики
редокс-активных соединений. Так, нерастворимый поли трис-
(ц2-1,1 -ферроцендатил-фенилтидрофосфинато-фенилфосфинато)
железо(Ш), тетрагидрофурана сольват {u2-[Fell(n5-C5H4—P(PhOO)
(ns-C5H4-P(PhOOH))]3Fe,l,}*THF квазиобрагимо окисляется втрех-
элсктронном процессе при не более положительным потенциале
по отношению к ферроцену
1 Khrizanforov M.,\., Arkhipova D.M., Shekurov R.P., Gerasimova Т.P., Ermo-
laev V. У., hlamov D.R., Mituykov V.A.* Kataeva O.N., Khrizanforova V. V.. Sin-
yashin O.a, Budnikova У.П. // J. Solid. State. Eleclrothem. 2015. 19. 28K3-2890.
363
9.8.5. Вольтамперометрия в ионных жидкостях
Когда ионные жидкости используют в качестве растворителей
для измерения циклической вольтамперомстрией органических
соединений, наблюдаемые редокс-токи, как правило, крайне малы.
Это происходит из-за высокой вязкости ионных жидкостей, которая
уменьшает коэффициент диффузии. Например, коэффициент
диффузии Ni(II) salen в ионной жидкости [bmini] BF4 при комнатной
температуре 1,8 • 10_м см2 ■ с-1, что более чем в 500 раз меньше, чем
в типичной органической системе растворитель — электролит типа
0,1 М Е14г4С104/ДМФА|.
9.8.6. Органические электрохимические реакции в ионных жидкостях
Хотя ионные жидкости считаются идеальной средой для
органических электролитических реакций из-за их негорючести и
достаточной электропроводности, а также широкого окна потенциалов,
до сих пор в литературе не очень много сообщении об органических
электрохимических реакциях в ионных жидкостях.
Органический электросинтез. Катодное восстановление
карбонильных соединений, таких как бензальдегид и ацетофенон, было
исследовано в ионных жидкостях. Как было отмечено,
преимущественно протекает их димеризация2. Из ацетофенона был получен
соответствующий пинакол в виде диастереомерной смеси, адиастере-
оселективность сильно зависела от используемых ионных жидкостей3:
9 -2е.2Н+ НО ,
^\ Разделенная ячейка т». y-^N^-'Ph
1 Me i Л/илпи Pn A
Ph Me , ф/ноль
/ ОН
[emirn]CF3S03 92% выход, 6% de
|Et3BuNjCF3S03 82% выход, 58% de
[Me3BuN]CF3SOi 80% выход, 54% de
(9,9)
Алифатические ионные жидкости в качестве растворителя
приводят к более высокой диастереоселективности по сравнению с
ароматическими ионными жидкостями.
Электрокаталитическое гомосочетаниеРпВг и Р1тСН>Вг
происходит в присутствии NiCl2(bpy)-комплекса в fbmim]Tf2N4:
Sweeny В.К., Peters D.G. // Eleclrochcm. Commun. 2001. 3. 712-715.
DohertyA.P.i Brooks C.A. // ElectroehimAcu». 2004. 49. 3821-3826; LttgrostC,
Hapiot Л, Vaultier M. // Green Chem. 2005. 7. 46S-474.
Dohe DohertyA.P., Brooks С A. Op. cil.
Mellah A/., Gmouh S., Vaultie M.. Jouikov V, // Electrochem. Commun. 2003. 5.
591-593.
364
2Ph(CH2bBr kw^.. . >Ph(CH2CH2)rPh (9.10)
KNl%y) jc = 0:35%
-l,4BvsAg/Ag+
2Ф/маль
Интересно, что наночаепшы Pd, генерируемые катодно в ионной
жидкости, как было показано, являются весьма эффективным
катализатором сочетания арилгалогенидов в отсутствие лиганда1. Элек-
тровосстановительнос дегалогенирование Wc-диталогенидов с
использованием Со(П)ка1еп-комплекса в fbmim] BF4 также протекает
успешно2:
осе ^ оэ->
иг 2 Со( I )salen 2 Co(l I )salen
V^__^ 68%
2с
Выделение продукта гораздо проще по сравнению с
аналогичным дегалогенированием в обычных молекулярных
растворителях, так как комплекс Co(H)salen остается в фазе ионной
жидкости при экстракции продукта нсполярным органическим
растворителем — диэтиловым эфиром. Кроме того, была
продемонстрирована возможность рециклизации системы
катализатор/ионная жидкость.
Циклические карбонаты получают путем восстановления С02
при -2,4 В отн. Ag/AgCl в присутствии эпоксидов в различных
ионных жидкостях, таких как [emim]BF4, [bmim]PF6 и [BPy]BF4
(ВРу = /7-бутилпиридиниЙ), при использовании Си катода и Mg
или А1 анода5.
Электрохимический синтез карбаматов можно осуществить путем
электролиза раствора СОг и амина в [bmimJBF^ с последующим
добавлением алкилируюшего агента4:
Вп Вп О
N-H > N-/ (9.12)
л.м f bmim BF4 .- / \ „
Ме СО4(0т1МА),55-С Ме OEt
Разделенная ячейка 87%
-2.4Bvs.Ag/AfiCl
затем EtI
Pachon P.L., ElsevierCJ., RothenbergG. //Adv. Synth. Catal. 2006. 348. 1705-1710.
She Y.,Aiobe M„ Tajim Т., Fuchigami T. // Electrochemistry. 2004. 72, 849—85 L
YangH., Gu Y, Deng Y, Siti F. // Chem. Commun. 2002. 274-275.
Feroci M., Orsini M., Rossi L., Sotgiu G., fnesrA. // J. Org. Chem. 2007. 72. 200-
203.
365
Кроме того, в ионных жидкостях было проведено
кинетическое исследование анодного сочетания ароматических соединений.
Анодное окисление 1,2-диметоксибензола привело к появлению
соответствующего тримера1:
ОМе J ~ ОМе
-6с'-ЬН%ПГТ I (9.13)
|cmim]TFSA
ОМе Л ^ Х)Ме
В этой реакции скорость димеризации в ионной жидкости
в 5—10 раз меньше, чем в ацетонитриле из-за высокой вязкости
ионной жидкости.
N-Гетероциклические карбены можно генерировать катодным
восстановлением ионных жидкостей на основе имидазолиния2.
Полученные карбены являются стабильными основаниями, которые
способны катализировать реакцию Генри3:
BF«"
Me-NUN lbmim]BFi Me-N N +2Нз
Н
О
HOv N02 ,N02
PIAH+MeN02—^г^ Н + г* (9Л4)
™ " Me'V^Bi. Ph' Ph'
81% 17%
Так как ионные жидкости, как правило, имеют гораздо более
высокую вязкость, массоперенос является довольно медленным,
как описано выше. Это является недостатком электросинтеза
в ионных жидкостях. Однако было установлено, что катодное
восстановление N-метилфталимида, промотированное ультразвуком,
приводит к более высокой конверсии и эффективности по току,
что связано с облегчением массопереноса субстрата под действием
ультразвука4:
Mellah M., Zeitouny J., Gmouh S„ Vaulrier Д/., Jouikov V. ff Electrochem. Commun.
2005. 7. 869-874.
Chowdhury S., Mohan R.S., Scott J.L. // Tetrahedron. 2007. 63. 2363—2389;
Fernet, A/., Chiamtto A/„ OmniI, SotgiuЛ/.т faesiG.A, //Adv. Synth. Catal. 2008.350.
1355—1359; Ferwi M.. Etirtson M,N„ Rossi L„ Inesi A. // ElectrochcmCommun.
2009.11. 1523-1526.
feroci A/., L'linson A/jY., Rossi L.t faesi'A, Op. cit.
Wliagran C, Banks C.E.. Pitner W.R., HardacreС Campion R.G. // Ultrason. So-
nochem. 2005. 11423-428.
366
он
NH ,2С'?" > I II NH (9.15)
|emim]NTf2 in.
0,11 Ф/моль
Ультразвук: 89% (выход по току)
Без ультразвука: 66% (выход по току)
Электрохимическое фторирование. Соли поли (фтористого
водорода), состоящие из амина и фторида аммония и фтористого водорода
(Et3N-wHF. n = 3—5; Et^NF-wHF, n = 3—5), являются ионными
жидкостями, которые имеют хорошую электропроводность из-за их низкой
вязкости1. С увеличением содержания HF (ростом п) они становятся
более анодно устойчивыми. Как описано в гл. 5, обычное
электрохимическое фторирование проводится в органических растворителях,
содержащих соли фторидов — Et3N-3HF и Et4NF-3HF2.
Однако анодная пассивация (формирование непроводящей
полимерной пленки на поверхности электрода, которая подавляет фара-
деевский ток) происходит часто, что приводит к снижению выхода
и низкой эффективности по току. Кроме того, также происходит
преимущественное аистамидированис, когда McCN используется в
качестве электролитического растворителя. Электрохимическое
фторирование без растворителей представляет собой альтернативный
способ предотвращения анодной пассивации и ацетамидирования.
Селективное электрохимическое фторирование без растворителей
бензола, нафталина, олефинов, фурана, бензофурана и фенантро-
лина впервые достигнуто с менее чем 50%-м выходом при
использовании ионной жидкости Et3N-3HF в качестве реакционной среды,
фонового электролита и источника фтора3.
Хотя Et3N-3HF и Et4NF-3HF легко окисляются (менее или около
2 В отн. Ag/Ag+), также были разработаны анодно очень стабильные
соли поли (фторида водорода), EttNF-mHF(ro > 3,5) и Et3N-5HF (3 В
отн. Ag/Ag+)4.Используя ионную жидкость Et4NF-wHF (m > 3,5),
1 Fuchigam Т., fnagi S. // Chem. Commun. 2011. 47. 10211 -10223; Fuchigami T.
Electrochemical Partial Fluorirtation // Organic Electrochemistry. 4th edn. / eds
H. Lund and O. Hammerich. N. Y: Marcel Dekker. 20(H), Ch. 25; Мотош К. //
Yoyuen (Molten Salts). 1999. 39. 7-22; Momota K., Morita M., Matsuda Y. // Elec-
trochim. Acta. 1993, 38.1123-1130.
1 Fuchigam Г., Inagi S. Op. cit.; Fuchigami T. Op. cit.: Momota K. Op. cit.;
Мотош К., Morita Л/., Matsuda Y, Op. cit.: Fuchigami T. // J. Fluorine Chem, 2007.
128.311-316.
я Meurs H.H., Eitenberg W. // Tetrahedron. 1991. 47. 705.
4 Momota K. // Yoyuen (Molten Salts). 1999. 39. 7—22; Momota K„ Morita M.,
Matsuda, Y. И Electrochim. Acta. 1993. 38.1123-1130.
367
анодное частичное фторирование аренов, таких как различные
замещенные бензола, толуола и хинолинов, было успешно проведено при
высоких плотностях тока с хорошими выходами по току (66—90%)
без растворителей1. Монофтортолуол и дифтортолуол образуются
последовательно с увеличением пропущенного электричества, однако
если далее увеличивать количество пропущенного электричества,
трифтортолуол не образуется, а начинает фторироваться кольцо2:
СН3 CH2F
-2с, -Н (Г "*| -2с, -Н'
EUNF-4HF 5 I J Et4NF-4HF *
2.0 F/mol ^^ 2,0 F/mol
1.9Vvs.Ag/Ag" 52% 2,1 V
CHF2 CHF2
(9.16)
-2e,-H ,. f ^1 -2e,-H"
Et4NF-4HF L^> EUNF-4HF
2,0 F/mol ^rC 2,0 F/mol
2,1V 47% 2,5 V
Селективное электрохимическое фторирование алифатических
альдегидов с обменом водорода при использовании EtsN-5HF дает
ацилфториды3. Селективное анодное фторирование циклических
ненасыщенных сложных зфиров в EtjN-SHF также дает
фторированные продукты и сопровождается расширением кольца4:
F" F
C/C02Et
<
; (9.17)
2,2-2,3 Vvs.Ag/Ag+
-20"С
л=1 71%
л = 0 56%
Матош К., Horio H., Kato К., Morita М„ Matsuda Y. // Electrochim. Acta, 1995,
40, 233—240; МотоШ К., Yonezawa T.r Hayakawa Y, Kato K.. Morita M„
Matsuda Y. II J. Appl. Elcclroehem. 1995.25.651-658: Momota K., Mukai A'., Kato K..
Morita M. И Electrochim. Acta. 1998. 43. 2503-2514; Momota K, Mukai K.,
Kato K., Morita M. //J. Fluorine Chem. 1998. 87. 173-178.
Momota K., Mukai K., Kato K., Morita M. Op. cit.; Momota K.r Mukai K., Kato K.,
Mori fa, M. Op. cit.
Yotwdo #., ChenS.-Q., Hatakevama Т., Нага S.. Fukuhara T. I/ Chem. Lett, 1994,
849-850.
Chen S.-Q., Hatakeyama Т., FukuharvT., Hara S„Yoneda N.// Electrochim. Acta,
1997. 42. 1951-1960; Нага S., Chen S.-Q.. Hoshio Т., Fukuhara Г., Yoneda N. //
Tetrahedron Lett. 1996.37. 8511-8514.
368
Кроме того, анодное фторирование различных производных
фенола может быть выполнено в Et3N-5HF с использованием
углеродного волокна в качестве анода с образованием производных 4,4-диф-
торощпслогексадиенона с хоропгами выходами':
^к,-2Н"
B3N-5HF
Carbon fibre cloth
(9.18)
Электрохимическое фторирование адамантанов также возможно
в Et jN-5HE Моно-, ди-, три- и тетрафтороадамантаны были селективно
получены из адамантанов при контроле потенциала окисления, а атомы
фтора были введены селективно к третичным атомам углерода2:
-2e.-Hf Г*7 I
Et3N-5HF/CH2CI2
2,3-3,0 V (vs. Ag/Agr)
:л^-+
(9.19)
Анодное фторирование циклических простых эфиров, лактонов
и открыто цепных и циклических карбонатов может быть достиг-
»гуто путем анодного окисления смеси большого количества жидкого
субстрата и небольшого количества Et4NF-4HF (только 1,5—1,7 экв.
F- на субстрат) при высокой плотности тока (150 мА ■ см-2)3:
XT ^0"^F
X = СН2, л = 0 83%
X = О, /7=1 80%
Х = 0, я = 0 77%
1 Fukuhara Г., Akivama Y, YonedaN,,Tada Т., Ham S. // TerahedronLett. 2002.43.
6583-6585.
3 Aoyama М„ Fukuhara Т., Ham 5. //J. Org. Chem. 2008. 73. 4186—4189.
i Hasegawa M., Ishii H>, Fuchigami T. // Tetrahedron Lett. 2002. 43. 1503—1505;
Hasegawa M., Ishii H., Cao Y., Fuchigami T. //J. Electrochem. Soe. 2006.153.
D162-D166.
369
о + о
¥ -2е.-Н+ II
Х"Х)СН2СгЬ z^F/moi * ОСНСН3
F
Х=ЕЮ 97%
X = Et 44%
О О
Х_У Et,NF-5HF' ^__У (9.21)
Z = CH2 75%
Z = О 87%
По этой методике субстрат селективно окисляется с получением
соответствующего монофторированного продукта с хорошим
выходом и хорошей эффективностью по току. Обнаружено, что
использование органических растворителей или большого количества
Et4NF-4HF не приводит к образованию или низкому выходу (около
10%) требуемого фторированного продукта. Выделение
фторированных продуктов проходит легко: фторированный лактон и
карбонаты можно экстрагировать растворителем, в то время как
фторированный тетрагидрофуран можно легко выделить перегонкой
реакционной смеси после электролиза. В этих случаях субстраты
преимущественно окисляются, приводя к эффективному
фторированию, поскольку используется только небольшое количество
фоновых фторидных солей. Так как в качестве катода
используется платина с низким перенапряжением водорода, кислые
протоны ионной фторидной соли преимущественно
восстанавливаются на катоде, генерируя газообразный водород. Следовательно,
диафрагма не является необходимой для электролиза. Как уже
отмечалось, небольшое количество фторидной соли достаточно для
фторирования, поэтому этот метод фторирования желателен с атом-
экономной точки зрения.
Так как фталид окисляется с трудом (отн. НКЭ), анодное
фторирование в любом органическом растворителе или системе без
растворителей не идет. Однако фторирование проходит весьма
эффективно в системе двойной ионной жидкости, состоящей из [emimj
OTfnEtjN-SHF1:
Hasegawa А/., Ishii//., fuchigami T. // Green Chem, 2003. 5. 512—515.
370
п - 3 or 5
activated cation
В этой двойной ионной системе катионный интермедиат,
генерируемый из фталида, как ожидается, имеет ТГО~-противоанион
(активированный катион А на схеме 9.22), который легко реагирует с F~,
давая фторированный фталид с выходом от хорошего до отличного.
Вязкость ионных жидкостей фторидных солей выше, чем обычных
органических растворителей, поэтому массоперенос субстратов
к поверхности анода из объема жидкости происходит медленнее,
чем в органических растворителях, что является неблагоприятным
для электролитических реакций. Как указано в разд. 9.11, известно,
что ультразвук значительно повышает массоперенос из объема к
поверхности электрода. Этот эффект также можно применить к системе
с ионной жидкостью, гак как эффективность анодного фторирования
в ионных жидких фторидных солях заметно возрастает1:
О
О
ЕЮ
SPh
-4с,-2Н
6 F/mol EtjN-3HF
Under ultrosonication
Yield: 65%
EtO
SPh
F F
EtjN-3HF/MeCN
1.6Vvs.SSCE
2,5 F/mo!
Yield: 76%
О
ЕЮ
(9.23)
SPh
Et3N-3HF/MeCN
2,2 Vvs. SSCE
20,7 F/mol
Yield: 53%
F
Следует отметить, что анодное дифгорирование этил-ос- (фенмлтио)
ацетата происходит эффективно даже в легкоокисляюшейся ионной
жидкости Et3N-3H F, которая не подходит для дифторирования.
В чистых ионных жидких фторидных солях нуклеофильность
фторид-ионов довольно низка, что приводит к низким выходам
фторирования. Было показано, что эфирные растворители, например,
ДМЭ, повышают нуклеофильность фторид-ионов, но ДМЭ до-
Sunaga T.Atobe Af., Inagi S\, Fuchlgami T. // Chem. Commun. 2009. 956—958.
371
вольно легко разлагается1. В противоположность ему PEG и даже
его олигомер устойчивы к анодному окислению. Было обнаружено,
что добавление только 3% олигомера PEG в реакционную систему
в значительной степени улучшает выход из-за его способности
координировать противокатионы фторид-ионов2:
S
О^
-4е
(9.24)
Et,N-3HF
8 F/mol
SO mA/cm"
Without additive: 22%
WithDME:18%
With PEG (MW 200): 80%
Даже в ионных жидкостях (ИЖ) HF-солей часто происходит
сильная пассивация анода. Использование медиаторов является
эффективным для решения проблемы, но разделение продукта
усложняется. Фучигами с сотрудниками разработал новую систему
непрямого анодного фторирования, использующую специфическую для
этой задачи ионную жидкость с фрагментом иодарена как медиатора
в HF-солях3. Медиатор улучшил эффективность различных
электрохимических реаюшй фторирования (рис. 9.23) и после процесса
экстракции продукта остался нетронутым в ионных жидкостях для
повторного использования в последующих циклах.
-2et -F"
pN F,I-Ph-g HPh-Q ^ pN p
5 mA/cm
4 F/mol
(CFaSO^N-
l-ph-O = i—i )-0{CH2h ■ ~\J
EWG
EWG = COOEl: 87%
Without mediator: 31 %
EWG = CN: 72%
Рис. 9.23. Схема электрохимического фторирования, использующего
специфическую для этой задачи ионную жидкость с медиатором
Нои Кт Fuchigami Т. //J. Electrochem, Soc. 2000.147.4567-4572; Shaaban M.R.,
Ishii H., Fuchigami Г. // J. Org. Chem. 2000,65. S685-86S9; Dawood KM.,
Fuchigatni T. //J. Org. Chem. 1999.64. 138-143.
Sawamura Т., InagiS.* Fuchigami T. // J. Electrochem. Soc. 2009Л 56. E26 — E28.
Sawamura Т., KuribayashiSJnagiS.,Fuchigami T. // Oig. Lett. 2010. 12.644—646.
372
Кроме того, медиатор йодбензол на полимерной подложке
(PSIB) также эффективен для непрямого анодното фторирования
в HF-солях1. В этом случае подвеска йодбензольного фрагмента
на твердой полимерной подложке не может непосредственно
окисляться, поэтому необходима двойная медиаторная система. Как
показано на рис. 9.24, анодное окисление С1~ дает С1+. который
взаимодействует с иодбензольным фрагментом с образованием PhI+CL
Эти частицы затем захватывают фгорид-ион, давая гипервалентный
[(хлор)(фтор)йод]бензольный фрагмент. Гипервалентный Йодный
фрагмент, генерируемый таким образом, окисляет субстрат, а
следовательно, регенерируется исходный PSIB. Регенерированный
медиатор PSIB повторно используют в последующих циклах, сохраняя
хороший выход (около 90%) фторированного продукта.
1/2
1/2
гл
s s
Anode
F F
Pli^Ph PlT"^Ph
-2е
/
сг *
O-Ph-I ©—Ph-l 0—Ph-1 =
Cl'-^O-Ph-l! -^Г
CI
Рис. 9.24.Схема электрохимического фторирования с использованием
медиатора на полимерном носителе
Электрополимеризацня. Группы Матте и Фучигами независимо
провели электроокислительную полимеризацию пиррола, тиофена
и анилина в различных ионных жидкостях2. Матте использовал
1-бутил-З-метилимидазолия тетрафторборат и гексафторфосфат
([bmim]BF4 и [bmim]PF6) для электрополимеризации пиррола и
анилина. Он обнаружил, что полученные таким образом я-сопряженные
полимеры обладают высокой стабильностью и могут
электрохимически никлизоваться в ионных жидкостях до миллиона раз3.
Sawamura Т.. Kuribayashi SJnatf S. Fwhlgami T. //Adv Synth. Calal. 2010. 352.
2757-2760.
Lu W„ FadeevAC Qi В., StnefaE., Mates В.Я, DingJ.r SfrinksGM., MazyrkiewiczJ-,
Zhou Д, WallaceG.C., MacFurkmeDR.,ForsythSA.. FomthM.//Science. 2002. 297.
983-987; Sek^cfii K.. AtobeM., Fuchigumi T. // Eleclrochem. Commun.4.881-885;
Sekiguchi K.,Atobe Л/., Fuchigami Т. Ц J. Electroanal. Chem. 2003.557.1—7.
Lu W„ Fadeev A.G., Qi В., SmelaE., Mattes B.R., Ding J., Spinks G.M., Mazurkie-
wicz ■/., Zhou /)., Wallace G. 61., MacFarlane D. A., Forsyth S.A., Forsyth M. Op. cit.
373
Кроме того, поскольку полимеры имеют быстрые скорости
переключения цикла (100 мс), они могут быть использованы в
качестве электрохромных окон и цифровых дисплеев. Также было
показано, что полианилин, полученный описанным образом,
весьма полезен для использования н электрохимических
преобразователях. Волокно длиной 10 мм диаметром 59 мкм мокрого
прядения полианилиновой эмералднновой основы (ЕВ)
обрабатывают трифторметансульфоновой кислотой с образованием
волокна эмералднновой соли (ES), которое обладает высокой
проводимостью — 300 S - см-1. Волокно с ES-структурой
восстанавливается двухэлектроино при —0,4 В отн. Ag/Ag+ в ионной
жидкости |bmiml BF4, давая лейкоэмералдин (LE), a (LE)
окисляется около +0,8 В отн. Ag/Ag4 с образованием исходного ES,
как показано на рис. 9.25. Таким образом, с помощью окисления
и восстановления катион ионной жидкости [bmimj соответственно
выталкивается и вводится в состав. Натяжение, следовательно,
сокращается, когда полимер окисляется из полностью
восстановленного LE-состояния в ES, и увеличивается при восстановлении
(см. рис. 9.25). В ресурсных испытаниях волокна как
электроактивность, так и электромеханические свойства сохраняются без
значительного уменьшения (< 1%) при любом напряжении или
деформации за 10 000 редокс-циклов, еслив качестве растворителя
используется безводная ионная жидкость. Значит, я-сопряженный
полимер в системе ионного жидкого электролита является весьма
перспективным для электрохимических механических
преобразователей.
CF,SOr
«h-Q-nh-(^-nh-Q-nh|
-2bmim
-2е
IbmimJCF^SO.! [bmim]CF^SO?
Рис. 9.2S. Принцип электрохимического преобразователя
Группа Фучигами использовала femim|OTf для
электрохимической полимеризации1. Исследователи обнаружили, что полиме-
1 Sekigucfii К., Atobe М.щ Fuchigami Т. // Electrochem. Commun. 2002. 4. 881 -885;
Hdem. //J. Electroanal. Chem. 557.1—7.
374
ризация пиррола в ионной жидкости (ИЖ) протекает значительно
быстрее, чем в обычных средах (водных и аистонитрильных
растворах), содержащих 0,1 М [emimJOTf в качестве фонового
электролита. Интересно, что, как показано в табл. 9.5, поверхность поли-
пирролъной пленки, полученной в чистом [emimJOTf, такая гладкая,
что не наблюдается никакого зерна. Когда пленки полипиррола
и политиофена получают в ионной жидкости, электрохимическая
емкость и электропроводность заметно увеличиваются. Это может
быть связано с чрезвычайно высокой концентрацией анионов в
качестве дотирующих примесей, что приводит к гораздо более
высокому уровню допирования. Как описано выше, полимерные пленки,
полученные в ионной жидкости, имеют более высокую
электрохимическую плотность и высоко регулируемые морфологические
структуры, а следовательно, их можно использовать в качестве
электрохимических конденсаторов высокой производительности, ионно-
просеивающих пленок, ион-селективных электродов, матрицы для
хостинга частиц катализатора и т.д.
Таблица 9.5
Физические свойства пленок полипиррола и политиофена,
полученных электрохимически в различных средах
Полимер
Пол и пиррол
Пол и пиррол
Пол и пиррол
Политиофен
Политиофен
Среда
н2о
CH3CN
[emim]OTf
CHjCN
[emimJOTf
Фактор
шероховатости"
(безразмерный)
3,40
0,48
0,29
8,60
3,30
Электрохимическая
емкость
._<Ссм-^__
77
190
250
9
45
проводность (5,
см1)
1,4 Ю-7
iti-io-fi
7,2 • Ю-2
4,1 • 10-"
L9 10-3
Уровень
допирования,
%
22
29
42
—
—
* Стандартное отклонение толщины.
Анодная полимеризация 3-(п-фторфенил)тиофена была также
проведена в [emimJNTfi. Результирующая полимерная пленка, как
было установлено, является очень гладкой'. Кроме того, описан
электросинтез иоли(3,4-этилендиокситиофена) (PEDOT) и ноли-
фенилена в ионных жидкостях2.
Naudin Е., Но ИЛ. Bmnchaud S., B/vau £., Belanger D. // J. Phys. Chem. B. 2002.
106,10585-10593.
Randriamahazako H., Please C, Chevrot £>. // Eleetrochem. Commun. 2004. 6.
299-305; Abedin S.Z.E., BorissenkoN., Endres F. // Eleetrochem. Commun. 2004.
6.422-426.
375
Ионная жидкость является рециклизуемой средой для
органического синтеза, что является одной из характерных особенностей
ионных жидкостей* Однако, когда ионная жидкость используется
в качестве электролитической среды, разложение самой ионной
жидкости часто происходит на противоположном электроде.
Повторное использование ионной жидкости является большой
проблемой. Но в случае электрохимической полимеризации эта
проблема может быть решена с помощью окислительной
полимеризации циклическим сканированием потенциала. При использовании
этого метода полимеризации мономер окисляется с образованием
полимерной пленки, которая осаждается на электроде. Кроме того,
осажденный полимер сам окисляется и восстанавливается несколько раз
во время попеременного анодного и катодного сканирования. В ходе
восстановления на рабочем электроде анодная окислительная
полимеризация мономера и окисление самого полимера (так называемое
допирование) происходят на противоположном электроде, в то время как
при окислении на рабочем электроде восстановление полимера (так
называемое дедопирование) протекает на противоположном электроде,
а следовательно, разложения ионной жидкости не происходит. Ионная
жидкость, таким образом, легко регенерируется просто экстракцией
оставшегося мономера соответствующими растворителями, а
выделенная ионная жидкость может быть использована много раз1.
Другие примеры использования ИЖ. Хотя следующие
синтетические применения ионных жидкостей не характерны для
органических веществ, возможно электрохимическое осаждение различных
металлов в ионных жидкостях. Например, Zn, Ga и In могут быть
электрохимически осаждены из смесей [EMIMJ C1 и ZnCl2, GaCh
и InClj соответственно. Разнообразные элементы и переходные
металлы могут быть осаждены из ионных жидкостей, кроме хлоридной
ионной жидкости. Кроме того, известно, что одиоэлектроиное
электрохимическое восстановление молекул кислорода в ионных
жидкостях эффективно генерирует ионы супероксида.
9.9. ТОНКОСЛОЙНЫЕ ЭЛЕКТРОЛИЗЕРЫ
Хотя электроорганические синтезы, как установлено,
выступают в качестве мощного инструмента реализации органических
процессов, они по-прежнему находятся в стадии разработки, а свой
потенциал в качестве «зеленой» методологии еще не полностью
реализовали. В обычных электросинтетических процессах к
растворителю должно быть добавлено большое количество фонового элект-
1 Sekrguciii К.. Atobe M., Fuchigami Т. // Electrochem. Commun. 2002. 4. 881 -885;
lidem. // J. Eleelroanal. Chem. 2003.557.1-7.
376
ролита* чтобы обеспечить достаточную электропроводность, но
наличие большого количества последнего может вызвать проблемы
при выделении и разработке реакционной смеси и дополнительные
проблемы отходов.
Ячейка с капиллярным зазором, разработанная компанией BASF
для коммерческого электросинтетического процесса (парный
электросинтез фталида и трет-бутилбензальдегида), позволяет
проводить электролиз даже в разбавленном растворе электролита. Ячейка
состоит из круговых дисковых электродов с небольшим
межэлектродным зазором (1—2 мм), чтобы минимизировать омическое
падение напряжения в электролите (рис. 9.26)1.
Токособиратель
Корпус ячейки
Контактные графитовые пластины
Графитовые диски -
Капиллярный зазор (1-2 mm)—
Поток электролита
Основной электрод/контакт
Вход
Выход
Продукт + Hi
Графитовый диск
Рис. 9.26. Схематическое представление электрохимической ячейки
BASF5E с капиллярным зазором. Графитовые дисковые электроды с малым
межэлектродным зазором
При этом в лабораторных экспериментах применение
тонкослойной проточной ячейки простой геометрии с рабочим и
вспомогательным электродами, непосредственно обращенными друг
к другу, позволяет проводить электросинтетические процессы в
проточном режиме в отсутствие фонового электролита (рис. 9.27)2.
Например, метоксилирование фурана проводится в анодных условиях
с помошью этой ячейки в метанольном растворе даже в отсутствие
добавленного фонового электролита3.
Jultner К. Technical Scale of Eleclroehemisiry // Electrochemical Engineering /
eds D.D. Macdonald and P. Schmuki. Wiley-VCH \fcriiiE GmbH, 2007. Ch. 1.
Paddon C.A., PritchardGJ., Thiemann T„ Marken F. Riired electrosynthesis: micro-
llow cell processes with and without added electrolyte // Electrochem. Commun.
2002.4,825-831,
Horii A, Atobe M., Fuchigami Т., Marken F. Self-supported Methoxylation and Ace-
toxylationElectrosynthesis Using a Simple Thin-laver Row Cell // J. Electrochem.
Soc.2006.153. D143-D147.
377
Сиейсер
(несколько десятков итп)
Тефлоноьаи трубка
Шприц-помпа
Рис 9.27. Схематическое представление тонкослойной проточной ячейки
с рабочим и вспомогательным электродом, непосредственно обращенными друг
к другу. Тефлоновая трубка присоединена к выходу, а шприц-дозатор — к входу
9.10. ЭЛЕКТРОХИМИЧЕСКИЕ МИКРОПОТОЧНЫЕ СИСТЕМЫ
(MICROFLOW)
Системы MicroFlow недавно привлекли к себе значительный
исследовательский интерес как со стороны научных кругов, так и
промышленности. Основные преимущества и потенциальные выгоды от
микропоточной технологии следующие: 1) очень большое отношение
поверхности к объему; 2) точный контроль температуры; 3) точный
контроль за временем пребывания; 4) строгий контроль ламинарного
потока; 5) крайне быстрая молекулярная диффузия; 6) улучшение
безопасности реакционного процесса. Как объяснено выше, системы
MicroFlow имеют много преимуществ, которые могут быть
применены в широком диапазоне органических синтезов и органических
процессов массового производства. Перенос электрона является одной
из наиболее распространенных движущих сил органических реакций,
а органический электросинтез служит простым и мощным методом для
органических процессов переноса электронов. Комплексное
использование микропоточной технологии с органическим электросинтезом
является одним из самых сложных процессов в органической химии.
Кроме того, разработаны новые системы, которые реализуются только
с помощью электрохимических MicroFlow-реакторов.
Йошида с сотрудниками сообщил, что микропоточная
электрохимическая система служит весьма эффективным методом
окислительной генерации нестабильных органических катионов при
378
низких температурах1. Этот метод называется методом катионого
потока. Электрохимический реактор для метода катионого потока
снабжен анодом из углеродной ткани и катодом из платиновой
проволоки (рис. 9.28). Анодный и катодный отсеки разделены
диафрагмой из PTFE-мсмбраны. Раствор катионного предшественника
вводится в анодную камеру, а раствор трифторметансульфоновой
кислоты (ТГОН) в качестве источника протонов вводится в катодную
камеру. Органический катион, который немедленно генерируется,
переносится в сосуд, в котором происходит нуклеофильная реакция,
дающая желаемый продукт присоединения.
Спейсер
(пористая мембрана)
Вход
*■ Выход
Рис. 9,28, Схематическое представление микропоточного электрохимического
реактора. Анод из углеродного полотна и катод из платиновой проволоки
разделены диафрагмой из пористой мембраны, вход сверху слева на анодном
отсеке, выход снизу справа от катода
Атобе с сотрудниками сообщил, что использование
параллельного ламинарного потока в микропоточном электрохимическом
реакторе позволяет эффективно генерировать ион N-ацилиминия
с последующим захватом легко окисляющимся углеродным нук-
леофилом — аллилтриметилсиланом (рис. 9.29)2. Раствор
катионного предшественника и раствор аллилтриметилсилана вводятся
параллельно. Ламинарный поток предотвращает окисление
аллилтриметилсилана на аноде. Только прекурсор окисляется, генерируя
ион N-адилиминия. Ион ГЯ-адилиминия диффундирует и вступает
в реакцию с аллилтриметилсиланом. Хотя эффективность процесса
очень низка для системы Bu4NBF4/CH3CN, использование системы
Bu4NBF4/T03 (2,2,2-трифторэтанол) {выход 59%) или ионной
жидкости (выход 62—91 %) привело к образованию целевого продукта.
Siiga S,, Okajima Af, Fujiwara К., Yoshida J. A New Approach to Conventional and
Combinatorial Organic Syntheses Using Electrochemical Micro Flow Systems //J.
Am. Chem. Soc. 2001. 123,7941 -7942.
Hani ZX, Fuchigami Т., Atohe Л/. A New Approach to Anodic Substitution Reaction
Using Parallel Laminar Flow in a Micro-Flow Reactor // J. Am. Chem. Soc. 2007.
129. 11692-11693.
379
Ul
00
о
Прокладка
Pt катод
Тсфлоновая
трубка
Шприи-помпа
Шприц-помпа
Вход1
Приемник
для сбора
анод
ЧЩель
(катодная сторона)
Стеклянный
канал
О
I
СООМе
+ ljS^^™s^^N
I
СООМе
TMS
и
Q
Стеклянная
пластина |Щсль|
СООМе
Стеклянная
пластина
Pt катод
Pi анод
Рис. 9.29. Схематическое изображение системы MicroFlow реактора с двумя входами {а) и параллельный ламинарный
поток в реакторе (б). Показанная модельная реакция является реакцией анодного замещения 1М-(метоксикарбонил)
п иррол и д и на алл илтриметил сил аном
Использование параллельного ламинарного течения в
микропоточном электрохимическом реакторе также позволяет
проводить хемоселективное катодное восстановление, причем так, чтобы
контролировать региоселективность продукта электрохимического
карбонильного аллилирования1. Электрохимическое карбонильное
аллилирование может давать либо у-, либо сс-аддукты — в
зависимости от того, что восстанавливается на катоде — альдегид или ал-
лилгалогенид. Если альдегид имеет более высокий потенциал
восстановления, аллилгалогенид преимущественно восстанавливается
с получением у-аддукта, но если потенциал восстановления аллил-
галогенида выше, то преимущественно восстана&1ИВается альдегид
и благоприятствует генерации а-аддукта. Контроль селективности
продукта (региоселективность в этой реакции), следовательно,
необходим, чтобы либо аллилгалогениды, либо альдегиды
восстанавливались хемоселективно, независимо от их потенциалов
восстановления:
«XI 2е
' +PhCHO
2Н^, -СГ
2с ,^ X. ^Ph
2Н \ -СГ
(9.25)
ОН ОН
у-аддукт а-аддукт
Для выполнения хемоселективного катодного восстановления
авторы использовали параллельный ламинарный поток жидкость —
жидкость, образующийся в электрохимическом микропоточном
реакторе. Как показано на рис. 9.30, когда два раствора (раствор аллил-
хлорида и раствор альдегида) вводятся через впускные отверстия I
и 2 микропотчного реактора, образуется стабильная межфазная
граница жидкость — жидкость, а массоперенос между входными
потоками происходит [только пугем диффузии. Субстрат, введенный через
впускное отверстие 1, может, следовательно, восстанавливаться
преимущественно в то время, когда восстановления субстрата из входа 2
(приток 2) можно избежать. В результате протекает хемоселективное
катодное восстановление, а продукт целевого кросс-сочетания
получается региоселективно. Другими словами, контроль
селективности продукта осуществляется с помощью простого переключения
потоков реагентов.
1 Amemiya F., Fuse К., Fuchigami Т., Atobe M. Chemoselective Reaction System
Using a Two Inlets Micro-flow Reactor: Application to Reductive Carbonyl Allyla-
tion// Chem. Commun. 2010. 46. 2730-2732.
381
ш
Со
Лзмсивнс типа
СНО
^ \ \ Катод
Вход Л X^y^
Вход 2 / он
С) ff/ а-аддукт
^f"^4^ */ Анод как основной продукт и™
. Vх Катпд
^-^■И» он
ВХОД 1 OOPh
ОН
пне
?cjek пакости
при, Ж. та
Вход 2 /( ,м^"
/^ ^ у-аддукт
асно ^/ Анод как основной продукт
Рис. 9.30. Хемоселективное катодное восстановление с помощью параллельного ламинарного потока в микропоточном
реакторе с двумя входами; а— режим потока для селективного восстановления бензальдегида, б — режим потока для
селективного восстановления 1-хлор-3-метил-2-бутена
9.11. ЭЛЕКТРОЛИЗ В УЛЬТРАЗВУКЕ
Многие преимущества ультразвука в химических процессах
хорошо известны и исследованы в различных областях химии, но,
возможно, самое поразительное влияние ультразвука касается
гетерогенных реакционных систем, особенно с разделом твердой
и жидкой фаз, в которых одними из полезных процессов являются
модификация размера частиц, модификация дисперсии частиц,
повышение массопсрсноса, очистка поверхностей или образование
новых поверхностей'.Так как электрохимический процесс синтеза
является типичным гетерогенным процессом на разделе фаз твердое
вещество (электрод) — жидкость (электролитический раствор),
различные эффекты, в частности способствование массопереносу,
будуг индуцироваться ультразвуком. Основной вклад в массоперенос
заключается в микроструйном потоке, возникающем в результате
асимметричного разрушения (коллапса) кавитационного пузырька
(рис. 9.31)2.Суслик с сотрудниками сообщил, что скорость потока
достигает более 100 мс~' на разделе фаз вода — твердое тело3.
Полость Сжатие
Коллапс
• о ■=> о ^
Ммкроструйный
поток
о -> о ^> щ
Твердое
тело
Рис 9.31. Схлопывание (коллапс) кавитационных пузырьков:
а — симметричный коллапс кавитационных пузырьков в объеме раствора;
б— асимметричный коллапс кавитационного пузырька на разделе поверхности
жидкость — твердое тело
Такое способствование массопереносу ультразвуком приводит
к увеличению эффективности по току в различных
электросинтезах. Например, Атобе с сотрудниками сообщил, что значительный
Walton D.J., PhulfS.S. Sonoelectrochemistry // Advances in Sonochemtstry / ed.
T.J. Mason. L,: JAI Press, 1996. Vol. 4.
LorimerP,, Mason TJ, The application of Ultrasound in Electroplating //
Electrochemistry. 1999.67.924-929.
Suslick K.S., Gawtenowski J.J., Schubert P. F., Wang H.H. Alkanc sonochemistrv //
J. Phys. Chem. 1983. 87.2299.
383
АгСНО
Ar:/3-CH3C6H4
е.Н+
1—-—>
2с, 2Н'
ультразвуковой эффект на токовую эффективность был найден
в электрохимическом восстановлении л-метилбензальдегида1:
АгСН-СНАг
1/2 I I
ОН ОН
[D1] (9.26)
АгСН2ОН
[Ml]
Как показано в табл. 9.6, выход по току при восстановлении
л-метилбензальдегида резко возрастает под влиянием
ультразвука. Селективность образования гидродимерного продукта
(D1) также увеличивается при обработке ультразвуком, а эффект
этого можно объяснить экспериментально и теоретически как
результат облегчения массопереноса молекулы субстрата к
поверхности электрода из электролитического раствора за счет
ультразвуковой кавитации2. Селективность продукта
электрохимического восстановления диметилмалеата (9.27) и бензойной
кислоты (9.28) также в значительной степени зависит от
ультразвука (см. табл, 9л>):
СНзСООСНз
СНСООСНз
II
СНСООСНз
е,Н
2с, 2Н+
1 ——>
1/2 I
СНСООСНз
СНСООСНз
сн2соосж
ID2J
СН2СООСНз
СН2СООСН3
[М2]
(9.27)
PhCOOH
2о,2Н+
-Н20
■»PhCHO
[МЗ]
2е,2Н*
*PhCH2OH
[М4]
(9.28)
Matsuda К., Atobe Af.r Nonaka T. Ultrasonic Effects on Eleclroorganic Processes,
Part I. Product-selectivity in Elect roreduct ion of Benzaldehydes// Chem. Lett.
1994.21 1619-1622.
Atobe A/., Matsuda K„ Nonaka Г. Ultrasonic Effects on Eleclroorganic Processes.
Part 4. Theoretical and Experimental Studies on Product-selectivity in Electrore-
duction of Benzaldehyde and Benzoic Acid // Electroanalysis. 1996. 8.784—788;
Atobe M.. Nonaka T. //Chem. Leu. 1997. 26. 323-324,
384
Таблица 9.6
Электровосстановление л-метилбензальдегида,
диметилмалеата и бензойной кислоты
Исходное
соединение
п-Метилбензаль-
дегид'
п- Метил бе нзаль-
ДСГИД*
я-Метилбензаль-
дегид'
Диметилмалеат*"*
Диметилмалеат*"*
Диметилмалеат""
Бе н зо й н ая кислота5'
Бензойная кислота**
Бензойная кислота5*
Режим
перемешивания
Без
перемешивания
Механическое"
Ультразвук'"
Без
перемешивания
Механическое"
Ультразвук*"
Без
перемешивания
Механическое*"
Ультразвук*"
Эффективность
по току | D11 +
[Ml],
[D2] + |M2|nm
[М31 + [М4],%
35
74
89
66
93
96
1
17
54
Селективность
D1J/IM1],
[D2]/[M2] или
[М3]/[М4]
0,0
1,0
24,0
0,0
0,3
0,4
0,0
0,2
1,5
* При электролизе пропущено 0,5 Ф моль-1 при 20 мА см-1 на РЬ катоде
в растворе 0,25 MH2SO4/50% MeOH.
" Перемешивание магнитной мешалкой.
**" Катод располагался на расстоянии 1,7 см от источника ультразвука
(диаметр — 0,6 см, 12 W, 20 kHz).
*"" Электролиз на свинцовом катоде в растворе 0,025 MKHtPOj/
Dt025MNa2HPO4/0,5 MNaCL
5* Электролиз на свинцовом катоде в растворе 0,05 MHiSO4/0,2 M уксусной
кислоты.
Как упоминалось в разд. 5.8, проводящие полимеры обладают
не только электропроводностью, но и уникальными оптическими
и химическими свойствами1. Разнообразие свойств, проявляемых
проводящими полимерами, лежит в основе использования этих
материалов в многочисленных технологических приложениях. Обычно
свойства полимеров зависят от их химической (молекулярной) и
физической (морфологической) структур. Следовательно, структуру
проводящих полимеров нужно уметь контролировать, чтобы адап-
HeinzeJ. Electrochemistry of Concluding Polymers // Organic Electrochemistry /
eds H. Lund and O. Hammerich. N. Y,: Marcel Dekker, 2001
385
тировать их к конкретным целям использования. Химические
структуры можно регулировать пугем изменения молекулярной структуры
соответствующих мономеров и выбора условий и процедур
полимеризации1. При этом методы контроля их физических структур были
относительно ограничены, но в последнее время с этой целью многие
исследования были сосредоточены на применении ультразвука в
процессах полимеризации, в частности электрополимеризации.
Осава с сотрудниками обнаружил, что качество политиофеновых
пленок, электрополимеризованных на аноде, может быть повышено
с помощью ультразвука. По обычной методике пленки становятся
хрупкими, но при помощи ультразвуковой очистительной бани
частотой 45 кГц можно получить гибкие и жесткие пленки (модуль
упругости при растяжении 3,2 ГПа и прочность 90 МПа)2.
Работа Атобе и сотрудниками, вероятно, была первым
современным изучением электрополимеризации под действием
ультразвука при низких частотах. Они опубликовали цикл статей по этой
тематике, начиная с электроорганических реакций в ультразвуковых
полях3, полимеризации анилина, которая изучалась как
электрохимическими4, так и химическими способами5, и заканчивая синтезом
наночастиц6.
Поведение полипиррольных пленок, электрополимеризованных
под действием ультразвука, также было исследовано. В результате
выявлены уникальные свойства в процессах допирования — дедопи-
рования. Авторы объясняют свои результаты по разработке
высокоплотной пленки под действием ультразвука, но при этом отмечают
деградацию пленки из-за высокой кавитации при 20 кГц (рис. 9.32)7.
1 Heinze J. Electrochemistry of Conducting Polymers // Organic Electrochemistrv /
eds H. Lund and O. Hammerich. N. V: Marcel Dekker. 2001
2 Osawa S„ fto \f.. Tanaka K., KuwanoJ. // Synth. Met. 1987. 18. 145-150.
я Atobe A/., Kaburagi Г., Nonaka T. Ultrasonic Effects on Electroorganic Processes.
Part. 13. A Role of Ultrasonic Cavitation in Electrooxidative Polymerization of
Aniline // Electrochemistry. 1999.67. 1114—1116.
4 Ibid.
5 Atobe M., Chowdhury A.N., Fuchigami Т.. Nonaka T. Preparation of Conducting
Polyaniline Colloids under Lltrasonication // Ultrason. Sonochem. 2003. 10.77—
80; Chowdhury A.N., Atobe A/., Nonaka T. Studies on Solution and Solution-Cast
Film of Polyaniline Colloids Prepared in the Absence and Presence of Ultrasonic
Irradiation// Ultrason. Sonochem. 2004. 11.77—82.
6 Park J.E., Atobe A/., Fuchigami T. Sonochemical Synthesis of Conducting Polymer-Metal
Nanoparticles Nanocomposite // Electrochim. Acta. 2005. 51. 849—854; Park J.E.,
Atobe M., Fuchigami T. Sonochemical Synthesis of Inorganic-organic HybrideNanocom-
posite Based on Gold Nanoparticles and Polypyrrole//Chem. Lett. 2005.34.96-97.
7 Atobe M., Tkuji //., Asami R., Fuchigami T. A Study on Doping-undoping Properties
of Polypyrrale Films Electropolymerized under Ultrasonication// J. Electrochem.
Soc. 2006.153. D10-D13.
386
Группа Безансона также изучила использование высокочастотного
ультразвука (500 кГц, 25 Вт) в электрополимеризации 3,4-этилен-
диокситиофена (EDOT) или полипиррола в водной среде с целью
оценки его штияния на свойства проводящего полимера. Они
показали, что: 1) усиление массопсрсноса, индуцированное ультразвуком,
повышает электрополимеризацию и 2) эффект массопереноса — это
не единственное явление, индуцированное ультразвуком в ходе
электроосаждения '.
I,8pm 1,8цт
Рис. 9.32. СЭМ-изображения полианилиновых пленок, полученных без (а)
и с (б) ультразвуком
9.12. ЭЛЕКТРОСИНТЕЗ С ИСПОЛЬЗОВАНИЕМ СПЕЦИФИЧЕСКИХ
ЭЛЕКТРОДНЫХ МАТЕРИАЛОВ
9.12.1. Электрохимический синтез с использованием
гидрофобных электродов
ГНдрофобные электроды с композитным покрытием. Большинство
электродов является гидрофильным, но гидрофобные электроды
можно получить композитным покрытием электродов (типа Ni, Zn,
Pb и т.д.) в соответствующей дисперсии соль металла — поли (те-
трафторэтилен) (PTFE) в гальванической ванне. Фторид графита
и фторированная смола, кроме (PTFE), также используются в
качестве гидрофобных композиционных материалов. В ходе покрытия
гидрофобные тонкодисперсные частицы включаются в
гальванический слой. Полученные таким образом электроды имеют хорошую
электропроводность, а их поверхность с покрытым слоем проявляет
водоотталкивающие свойства, механическую и химическую стабилъ-
' Taouil А.Е., Lallemand F.. Hthn J. К. Blondeau- Patissier V, Electrosynthesis and
characterization of conducting polypyrrole elaborated under high frequency
ultrasound irradiation// Ultrason. Sonochem. 2011. 18. 907—910; TaouilA.E.,
Lallemand F., Hihn J. Y.,MeIotJ>M., Blondeau-Patissier V,, Lakard B. Doping properties
of PEDOT films electrosynthesized under high Frequency ultrasound irradiation //
Ultrason. Sonochem. 201L 18. 140-148.
387
ность. В общем, выделение водорода и кислорода происходит
конкурентно в электролитических реакциях органических соединений
в водном растворе. В частности, выделение водорода легко
происходит в кислом водном растворе, а выделение кислорода — в
щелочном водном растворе. Но не так легко катодно восстановить
карбонильные соединения в водном кислом растворе и анодно окислить
спирты в водном щелочном растворе. Поэтому катоды с высоким
перенапряжением водорода и аноды с высоким перенапряжением
кислорода должны использоваться для электрохимических реакций
соответственно в водных кислых и щелочных растворах. Однако Ni
электроды и никель/PTFE композитное покрытие используются
для восстановления и окисления, как упоминалось выше, чтобы
получить соответствующие спирты и карбонильные соединения
с гораздо большей эффективностью по току по сравнению с
электродами без покрытия Ni1. Это обусловлено не более высокими
перенапряжениями водорода и кислорода на электроде с композитным
покрытием, подавляющими выделение водорода и кислорода, а
эффектом «собирания» субстрата в результате сильного гидрофобного
взаимодействия между гидрофобной поверхностью электрода и
гидрофобными органическими субстратами2:
СУ0Н imkoh/^McCn' С^О (9.29)
PTFE/Ni катод: 98% эффективность по току
Ni катод: 2% эффективность по току
Электроды* покрытые PTFE-волокном. Электроды, покрытые
PTFE-волокном, которые получают путем обертывания электрода
PTFE-волокнами, являются гидрофобными. Было показано, что
анодное окисление гидрохинона на этом электроде в присутствии
диенов в NaC104/MeN02 генерирует хиноны, которые
подвергаются реакции Дильса-Альдера с диенами, давая циклические
продукты с превосходными выходами-'. Использование непокрытого
стеклоуглеродного анода также генерирует хиноны из гидрохинонов,
но присутствующие диены легко окисляются и разлагаются до
реакции с хинонами. Однако при использовании стеклоуглеродного
электрода, покрытого PTFE-волокном, легко окисляемый диен за-
1 Kunugi К, Fuchigami Т., Леп H.-J., Nonaka Т. // Chem. Lett. 1989. 757-760;
Kuttugi Г., Fuchigami Т., Nonaka Т. //J. Electroanal. Chem. 1990.287. 385-388;
Kunugi Г., Nonaka T, Chang Y.B., Watanabe N. (1992) Elecirochim. Acta 1992.
37. 353-355.
2 Ibid.
J Chlba K., Jitmo M., Kuramoto R., Tada M. // Tetrahedron Lett. 1998. 39. 5527—
5539.
388
держивается на PTFE-волокне, и только полярные гидрохиноны
могут достичь электрода сквозь гидрофобные волокна, как
показано на рис. 9.33. Далее генерируемые на аноде гидрофобные хиноны
немедленно реагируют с диенами, адсорбированными на PTFE
волокнах, и в результате гидрофобные продукты также задерживаются
на волокнах, избегая дальнейшего окисления продуктов. Этот новый
способ применим для синтеза различных терпенов1.
Анод
PTFE волокно
(Гидрофобное)
ОН
C02Et
Гидрофильный
раствор
Рис. 9.33. Схема реакции Дильса-Альдера анодно генерируемых производных
хинона с диеном на поверхности электрода, модифицированного волокнами
PTFE
9.12.2. Электролитические реакции с использованием алмазных
электродов
Алмаз — твердый кристалл, состоящий из атомов углерода sp3,
образующих ковалентные связи. Он имеет превосходную
прозрачность и теплопроводность, а также механическую и химическую
стабильность. Хотя чистый алмаз не является проводящим, он
становится электропроводным при допировании примесями — бором.
Алмазные пленки, допированные бором, могут быть использованы
в качестве электродов, которые обладают отличными и
превосходными электрохимическими свойствами по сравнению с известными
металлическими и углеродными материалами электродов. Соответ-
1 Chiba К., Fukuda Л/., Kim S., Kitarw К, Tada М. // J. Org. Chem. 1999.64.7654-
7656.
389
ственно алмазные электроды, легированные бором (BDD), —
перспективные новые электродные материалы'.
Электрохимические характеристики и применение в
высокочувствительном электроанализе
Широкое окно потенциалов в водном растворе1. В DD-электроды
имеют перенапряжение водорода около — 1 В отн. Ag/AgCl, которое
сравнимо с ртутью, и перенапряжение кислорода около 2,2 В отн.
Ag/AgCl, которое выше, чем в других распространенных анодных
материалах. Следовательно, характерное свойство ВDD-электрода —
его широкое окно потенциалов, которое достигает 3,2 В. Даже
в апротонных органических растворителях, таких как MeCN,
катодное окно на 500 мВ шире, чем у Pt электрода. А такие уникальные
свойства могут привести к замене либо дорогостоящих благородных
металлов, либо токсичных тяжелых металлов.
Малый фоновый ток (малый остаточная плотность токаУ.
Емкость поверхности BDD-электрода состаштяет несколько мкФ ■ см-2,
что на два порядка меньше по сравнению со стеклоуглеродом, а его
остаточная плотность тока мала — всего несколько сотен нА • см 2.
Это связано с тем, что ни растворенный газ, ни ионы фоновой соли
не адсорбируются на поверхности BDD, затрудняя окислительно-
восстановительные реакции.
Большое перенапряжение восстановления кислорода*. Из-за
затрудненного катодного восстановления растворенного кислорода
желаемый ток восстановления можно наблюдать даже без удаления
растворенного кислорода.
Физическая и химическая стабильность и высокая долговечность1*.
BDD-электроды в основном работают без ухудшения качества
и могут быть использованы даже в агрессивном растворе. Электролиз
можно проводить при высокой плотности тока (10 А см-2), а также
при температурах до 600аС. Поверхность электрода устойчива и
практически не загрязняется адсорбцией примесей.
1 Brillas Е., Martfnez-НиШе С A. (eds) Synthetic Diamond Films —Preparation,
Electrochemistry, Characterization and Applications. \№inhcim: Wiley-VCH \fcrlag
GmbH, 2011; Fujishima A., Einaga Y. (eds) Diamond Electrochemistry. Elsevier
and BKC, 2005.
2 Bouamrane F., Tadjeddtne A., Buffer J.E., Тепле R., Levy-Clement C. //J. Eleciro-
anal. Chem. 1996. 405. 95-99; ChenQ., GrangerЛ/.С, Lister, Т.Е., Swain G.M. //
J. Electrochem. Soc. 1997.144. 3806—3812; Martin И.В., Argoitia A., Landau U.,
Anderson А.В., Angus J.C. // J. Electrochem. Soc. 1996.143. LI33—L136.
} Ibid.
4 Yam Y., Tryk DA., Hashimoto K., Fujishima A. If J. Electrochem. Soc. 1998. 145.
1870-1876.
J Brillas E., Maninez-HuitkC.A. (eds) Op. cit; Fujishima A., Einaga Y. (eds)
//Diamond Elect roc hemistrv. Elsevier and BKC, 2005; Swain G.M. // J. Electrochem.
Soc. 1994.141. 3382-3393.
390
Высокая скорость переноса электрона между редокс-частицами
и BDD-электродом*. Внешнесферные одноэлектронные
окислительно-восстановительные системы, такие как Fe(CN)*_/* ,
1гСЦ~/ " и Ru(NHj)^+ / 4, показывают обратимые волны на
циклических вольтамперограммах, а перенос электронов на электроде
протекает быстро.
Уникальная электрохимическая селективность1. BDD-электроды
с концевым кислородом, полученные обработкой в кислородной
плазме или анодным окислением, показывают селективные отклики
на конкретные химические вешества. Так, BDD-электроды имеют
широкое окно потенциалов и чрезвычайно малый фоновый ток в
дополнение к вышеуказанным особенностям, С их использованием были
разработаны высокочувствительные сенсоры для обнаружения
биологических материалов. Например, с использованием BDD-электродов
с концевыми кислородами могут быть обнаружены и количественно
проанализированы следовые количества допамина и мочевой
кислоты, даже в присутствии большого количества аскорбиновой
кислоты3. Кроме того, поскольку поверхность BDD-электродов устойчива
к загрязнению адсорбированными примесями, они могут быть
использованы в качестве электроаналитического детектора''. Совсем
недавно были разработаны BDD-микроэлектроды, которые позволяют
проводить invivo мониторинг рН для диагноза расстройства желудка
в естественных условиях, а также оценивать раковые опухоли invivo5.
Применение в органическом электросинтезе. К настоящему
времени разработаны различные реакции анодного замещения —
фторирование, метоксилирование, ацетоксилирование и цианирование
ароматических соединений, гетероциклов и сульфидов.
Электролиз Кольбе также был исследован с помощью BDD-
анода'1. Совсем недавно было показано, что BDD-анод эффективен
для региоселективного сочетания замещенных фенолов,
приводящего к бифенолам7:
1 Bouamrane F., Tadjeddine A., Butter J.E.. Теппе R., Levy-Clement С. Op. cit.;
Chen С?.. Granger М.С., Lister. Т.Е., Swain G.M. Op. cit.; Swain G.M. Op. cit.
2 РЬра Е.. Notsu ff., Miwa T.n Tryk D.A., Fujishima A. // Electrochem. Solids. Lett.
1999.2.49-51.
3 Ibid.
4 Jolly S., Koppang M., Jackson Т., Swain G.M. // Anal. Chem. 1997. 69- 4099—4107.
J Fierro S„ Yoshikawa M., Nagano O., Yoshimi K., Saya H.t Einaga Y. // Sci. Rep.
2012.2.901.
6 Wadhawan J.D., Wadhawan J.D^ Campo F.J., Campion R.G.. Foord J.S., Marken F.,
BullS.D., DaviesS.G., WaltonDJ., RykyS.//L Electraanal.Chem.200L507.135-141
7 Kirste A., Nieger \f., Malkowsky I.M., Sleeker F.. Fischer A.. Waldvogel S.R. //
Chem. Eur. J. 2009. 15. 2273-2277; Kirste A.. Sc/makenburgG., Waldvogel S.R. //
Org. Lett. 2011. 13. 3126-3129; Kirste A.. Elsler В., Schnakenburg G.,
Waldvogel S.R. II J. Am. Chem. Soc. 2012. 134. 3571-3576.
391
он
R^
ВООанод
)-OH, Et^NMe 0,SOMe OH
ЧЬЯ
(9.30)
OH
BDD-электроды также являются превосходными анодами для
генерации алкокеираликалов из спиртов. Соответственно анодное
1,2-диметоксилирование изоэвгенола было реализовано в условиях
микропотока1. Инаги с сотрудниками добился параллельных
электрохимических реакций альтернированного сополимера 9-флюо-
ренола и 9,9-диоктилфлюоренона на биполярном BDD-электроде
в ELjNOTs/Z-PrOH, приводящих к получению разноцветных
градиентных пленок2. Это отличное применение широкого окна
потенциалов BDD-электродов.
Применение в неорганическом электросинтезе. BDD-электрод
является превосходным анодом для генерации озона и пероксида
водорода из воды по сравнению с платиной и анодом из оксида
иридия3. На BDD-электроде легко генерируется газообразный
хлор, а эффективность образования хлорноватистой кислоты
высока, поэтому можно ожидать его применения для
дезинфекционных обработок. С использованием BDD-анода можно
получить надсерную кислоту из серной кислоты с выходом 70%
по току, в то время как производство аммиака из азотной кислоты
возможно с помощью BDD-катодов4. Графит реагирует с газо-
Sumi Г., Saito Т., Natsui K„ YamamotoT. Atobe M., Einaga Г., Nishiyama S. //
Angew. Chcm. Int. Ed. 2012. 51. 5443-5446.
Inagi 5., Nagai H., Tomita /., Fuchigami T. // Angew. Chcm. Int. Ed. 2013. 52.
6616-6619.
Katsuki /V„ Takahashi E.r Toyota A/., Kurosu Т., Lida M., Wakita S., Nishiki Y.,
Shimamttne Т.//У. Electrochem. Soc. 1998,145. 2358-2362.
Ferro S., De Battisti A., ComninellisCh., Haenni W. //J. Electrochem. Soc. 2000.
147. 2614—2619; Reuben C, Galun E., Cohen //.. Tenne Л., Kalish R., Muraki Y.,
Hashimoto K., Fujishima A., ButlerJ,M., Levy-Clement C. //J. Electroanal.Chem.
1995. 396. 233-239; Tenne A, Patel K., Hashimoto K.t Fujishima A. // J. Elec-
traanal. Chem. 1993.347. 409-415.
392
образным фтором, а в объеме графита образуется фторид графита,
что приводит к подавлению фарадсевского тока. Наоборот, у BDD-
электрода фторируется только поверхность, поэтому он сохраняет
свою электропроводность1. Так как он устойчив к фтористому
водороду, этот электрод, как ожидается, будет полезен для получения
газообразного фтора.
9.13. ФОТОЭЛЕКТРОЛИЗ И ФОТОКАТАЛИЗ
9.13.1. Фотоэлектролиз
Когда полупроводниковый электрод облучают, происходит
разделение зарядов, которое генерирует пары электрон (е) — дырка (h+).
Генерируемый электрон действует как восстановитель, в то время как
дырка работает как окислитель. Хонда и Фудзисима впервые
показали возможность такого фотосенсибилизированного электролиза
с использованием л-тила полупроводникового ТЮ2 и Pt электродов,
как показано на рис. 9.342. Когда поверхность ТЮ2 электрода
облучают, используя длину волны короче 410 им, протекает ток,
генерируя кислород и водород на поверхности ТЮ2 и Pt электродов
соответственно. Ti02 возбуждается при облучении, генеририруя
пары электрон — дырка, а дырки в валентной зоне движутся к
поверхности, приводя к окислению воды, в то время как электроны
в зоне проводимости движутся в объем, а затем далее перемещаются
по внешней цепи к противоположному Pt электроду, восстанавливая
протоны. Это говорит о том, что вода разлагается видимым светом
на кислород и водород без применения какого-либо внешнего
напряжения:
2й+ + Н20-П/202 + 2Н+
2с + 2Н+->Н2 (9.31)
Total: H2O^^H2 + l/202
Это инновационный прорыв. Его называют эффектом Хонды-
Фудзисимы3.
Фотоэлектрохимические ячейки имеют преимущество в
производстве кислорода и водорода в дополнение к электроэнергии.
Однако на сегодняшний день мало примеров применения такого
фотоэлектролиза к органическим реакциям.
1 Ando Т., Yamamoto A'., Kamo M., Sato ¥., Takamatsu Y., Kawasaki $., Okino F.,
Touhara H. //J. Chem. Soc., Faraday Trans. 1995. 91. 3209—3212.
2 Fujishima A, Honda K, // Nature. 1972. 238. 37-38.
J Ibid.
393
Pt
электрод
Стеклянная мембрана
Рис. 9.34. Схема фотохимической ячейки сТЮ2 электродом
9.13.2. фотокаталиэаторы
Когда цепь между ТЮ2 и Pt электродами замыкается, Pt в
конечном счете осаждается на поверхности ТЮг. Соответственно
частицу ТЮ2 с некоторым количеством Pt, осажденной на ней
(рис. 9.35), можно рассматривать как фотоэлектрохимическую ячейку
с коротким замыканием. При таком фотокатализаторе окисление
и восстаноатение происходят одновременно в двух местах на малой
частице. Так как сайты окисления и восстановления очень близки
друг к другу, были разработаны различные уникальные органические
синтетические реакции1. Например, анодное окисление лервичного
амина, как правило, образует альдегид через промежуточный имин,
тогда как тоже окисление с помощью катализатора TiQ) с осажденной
на ней Pt дает вторичный амин (см. рис. 9.35)2. В этом случае
первичный амин окисляется дыркой, генерируемой при
фотовозбуждении с образованием альдегида, который реагирует с нспрореаги-
ровавшим исходным амином с образованием промежуточного имина.
Имин немедленно восстанавливается на участке Pt, что приводит
к образованию вторичного амина. Это связано с тем, что окисление
и восстановление происходят на активных участках, близких друг
к другу. С использованием этого принципа был реализован синтез
оптически активной пипеколиновой кислоты из оптически
активного лизина за счет фотокаталитической реакции с использованием
покрытых платиной частиц полупроводникового порошка ТЮ23:
1 Fax M.N. If Асе. Chem. Res. 1983. 16. 314-332.
2 Nishrmoto S.. Ohtani /L Yoshikawa Т.. Kagiya T. // J. Am. Chem. Soc. 1983. 105.
7180-7182.
J Ohtani В.. TsuruS., Nishimoto $., Kagiya T. //J. Org. Chem. 1990. 55.5551-5553.
394
соон
соон
H2N
NH7 -2H* NH
NH2
_HjO
-NH?
L-Lysine
H70
-Н-Ю
-NH, 0HC
COOH
!N
(9.32)
COOH
L-Pipccolicacid
hv
RH2C4
NH
RCH2NH?
-2e,-2H"
H20, -NH,
-H,0
RNC=NCH3R
Рис. 9.35. Пример фотокаталитических реакций
Таким образом, фотокаталитические реакции могут считаться
беспроводными электролитическими реакциями.
9.14. ЭЛЕКТРОХИМИЧЕСКИЕ ПОЛИМЕРНЫЕ РЕАКЦИИ
Как описано в гл. 5, проводящие полимеры являются редоксак-
тивными, особенно в состоянии пленки на поверхности электрода.
В ряде практических приложений процесс допирования и дедопиро-
вания проводящих полимеров должен быть обратимым, чтобы
избежать переокисления и перевосстановления, которые могут вызвать
нежелательные реакции в структуре полимера. Из-за этого
проводящие полимеры в легированном состоянии можно рассматривать
395
какреакционноспособные частицы, способные претерпевать
последующие реакции. Дизайн специфических элекгрогенерированных
частиц, полученных из конкретных полимеров в последующих
химических реакциях, представляет собой мощное средство введения
в полимеры универсальных функшюнальностей. Это
электрохимическая полимерная реакция является типом послеполимеризаци-
онной функционализации (или просто пост-функционализации)
(рис. 9.36)'. Реакционное отношение (или степень
функционализации) повторяющихся реакционных центров влияет на различные
свойства результирующего проводящего полимера, в том числе его
поглощение, фотолюминесценцию и электрохимию. Такие анодные
реакции, как окислительное галогенирование, цианирование и пи-
ридинирование проводящих полимеров, как известно, протекают
при использовании соответствующих нуклеофильных допирующих
компонентов. Катодная реакция — восстановительное гидрирование
фрагмента 9-флуоренона в проводящем полимере также возможна.
Поэтому можно разрабогать парные электрохимические полимерные
реакции в одной ячейке и получить хорошую эффективность по току
Сопряженный полимер
I
f • • • л
Bocltuhoh.-
лгшк
Допант
, \
? - * - * [ tot ltJVKilILlH
—,*Х?—*{jk £\— fkAklillH
4 Электросинтез '
Ойратпмый перенос ■электрона
Рис.936. Электрохимические полимерные реакции
Основным преимуществом электроорганического синтеза
является возможность быстрого переключения наложенного потенциала
на рабочий электрод — включен или выключен. Применяя это
преимущество электрохимических полимерных реакций, реакционное
соотношение можно точно настроить путем точного регулирования
количества заряда, передаваемого между анодом и катодом. В
результате физические свойства сопряженного полимера можно
подбирать, регулируя реакционное отношение. Для прогресса этой
твердофазной реакции выбор электролита имеет решающее значение.
В электрохимическом фторировании полифлюоренового
производного ионные жидкие соли фтористого водорода играют роль
фоновой соли, реакционной среды и источника фтора, предотвращая
отсоединение пленки во время электролиза2:
1 Inagi S., Fuchigami Т. // MacromoL Rapid Commun, 2014. 35, 854—867,
2 Inagi S., HayashiS.. Fuchigami '/. // Chem. Commun. 2009.1718-1720.
396
(9,33)
-2e, -ArSSAr, 2F~
Et4NF-5HF
Глава 10. ОБЛАСТИ, СВЯЗАННЫЕ
С ОРГАНИЧЕСКОЙ ЭЛЕКТРОХИМИЕЙ
Мир сталкивается с серьезными проблемами: экологическими,
истощением ресурсов и актуальными проблемами энергетики.
Органическая электрохимия, в том числе электросинтез, может
использовать свой потенциал для решения этих проблем. В данной главе
описан вклад органической электрохимии в органическую
электронику, повторное использование биомассы, С1-химию и очистку
окружающей среды.
10.1. ПРИМЕНЕНИЕ В ОРГАНИЧЕСКИХ ЭЛЕКТРОННЫХ
УСТРОЙСТВАХ
Органические молекулы и полимеры с уникальными
электронными свойствами имеют много приборных приложений
(люминесцентные, проводящие материалы), а также используются для
хранения энергии. Эти материалы на основе органических соединений
превосходят неорганические материалы с точки зрения легкости,
гибкости и стоимости. Кроме того, миллионы молекулярных
конструкций органических материалов позволяют легко адаптировать
их оптоэлектронные свойства. Основные принципы органических
электронных устройств с точки зрения органической электрохимии
описаны ниже.
10.1.1. Органическая электролюминесценция1
Органические фотолюминесцентные материалы, как правило,
излучают в процессе релаксации фотовозбужденных электронов
в основное состояние (рис. 10.1). Возбужденное состояние также
можно образовать электрохимически. Когда достаточное
напряжение приложено к аноду и катоду, как сэндвич сжимающих
органический излучающий слой (тонкую пленку), происходит ин-
жекция дырок на аноде и инжекция электронов на катоде. Заряды
мигрируют внутри слоя и рекомбинируют, излучая свет (рис. 10.2).
Чтобы способствовать мягкой инжекции электронов с поверхности
электродана LUMO эмиссионного слоя, дополнительный тонкий
слой, то есть электронно-транспортный слой, вводится между
интерфейсами. Аналогичным образом дырочно-транспортный слой
используется для инжекции дырок с электрода на HOMO эмис-
1 Tang С. W., VanShke S.A. // Appl. Phys. Lett. 1987. 51.913-915: Miachke V..
Baeuerle P. // J. Mater. Chem. 2000. 10. 1471-1507.
398
сионного слоя. Хотя эти транспортные слои имеют большое
значение для инжеклии носителей заряда в эмиссионный слой, они
не должны прерывать эмиссию эмиссионного слоя. Каждый
органический слой формируют путем химической десорбции паров
(CVD) кристаллических молекул или мокрым способом (wet-
process) из раствора полимера.
Основное
состояние
Возбужденное Основное
состояние состояние
Энергия
hv
Излучат:
i
ITITC
• Электрон
Рис 10.1. Схема механизма фотолюминесценции. Два электрона
находятся в основном состоянии, а при фотовозбуждении один переходит
в возбужденное состояние. При возвращении обратно в основное состояние
органические фотолюминесцентные материалы излучают во время процесса
релаксации
Дырочно-транспортный
Излучающий слой
Энергия
Прозрачный
анод
Ре комби наши
• ••
Катод
Электрон -транспорта ы и
слой
• Электрон
О Дырка
Рис. 10,2, Схема механизма электролюминесценции
399
10.1.2. Органические фотовольтаические ячейки1
Принцип фотоэлектрического преобразования в органическом
фотоэлементе (OPVC) является процессом, обратным органической
электролюминесценции (OEL). OPVC вырабатывает электрическую
энергию через состояние разделения зарядов, полученное фото
возбуждением, в то время как электрическая энергия вызывает ин-
жекцию электронов и дырок, а последующая рекомбинация зарядов
приводит к эмиссии в процессе OEL.
Чтобы создать состояние с разделением зарядов, необходимо
получить элсктронодонорный и элсктроноакиспторный слои в тонкой
пленке OPVC (рис. 10.3). Когда фотовозбужденное состояние
(экситон) генерируется в донорном слое солнечной энергией при
близком расположении донорного и акцепторного слоев,
возбужденный электрон может спонтанно мигрировать к LUMO
акцепторного слоя, в результате чего происходит формирование состояния
с разделенными зарядами. Носители заряда затем мигрируют,
перепрыгивая через каждый слой к электродным терминалам, причем
этот процесс приводит в действие фотоэлектрический элемент.
Прозрачный
электрод
hv
Противоэлектрод
Excition
Донор Акцептор
D* А
о
D+
•
А
-•—
D+ D D А А А
LUMO
HOMO -•<>
Возбужденное Разделение Миграция дырки
состояние зарядов и электрона
• Электрон
о Дырка
Рис 10.3. Схема процесса фотоэлектронного преобразования
Дизайн соединения слоев важен для индуцирования
эффективного разделения зарядов до дезактивации экситона. Конструкция
пути к поверхности электрода генерируемых носителей заряда также
важна для высокой эффективности преобразования фотоэлектронов.
1 TangC. W. //Appl. Phvs. Leti. 1986. 48. 183-185; Gums S., NeugebauerH.. Saric-
iftci NS. И Chem, Rev. 2007. 107.1324-1338.
400
10.1.3. Солнечные элементы, сенсибилизированные красителем1
Полупроводниковый электрод (оксид металла) может
генерировать электрическую энергию на основе принципа
фотокатализаторов. Фотовозбужденный электрон в полупроводниковом электроде
достигает электрической цепи, а оставшаяся дырка принимает
электроны от окислительно-восстановительной системы в
электролите с последующим переносом электронов от противоположного
электрода на редокс-систему. Благодаря относительно широкой
щели между полосами частот полупроводниковых электродов,
УФ-свет, как правило, используется для запуска этого типа
фотоэлемента. Сенсибилизированные красителем солнечные элементы
(DSSCs) работают в видимой области солнечного света, используя
органический краситель на полупроводниковом электроде в
качестве фотосенсибилизатора. Органический краситель должен иметь
более высокий уровень LIJM0, чем полупроводниковый электрод
и более низкий уровень HOMO, чем редокс-система в электролите.
Типичная система DSSC, известная как ячейка Грацеля, состоит
из прозрачного анода, оксида титана (ЖЬ) с покрытием из
рутениевого комплекса, 1~/13_ редокс-системы в электролите и Pt катода
(рис. 10.4). Напряжение, генерируемое в DSSC, соответствует
разнице в энергетических уровнях между уровнем Ферми Ti02 и
стандартным электродным потенциалом \ /V ~ редокс-системы.
Энергия
hv
Проз
эле
про
^\j\j
Зона
юдимости/
Валентная
зона s
рачный Т1СЬ
ктрод
-©
Кра
i
Напряжение !
( ©
ситель
Электролит
Противо
--
эле к
трод
Рис 10.4. Структура ячейки Грацеля
1 O'Regan B.,Gratzel M. // Naiure 1991. 353.737-740: HagfeldtA., Grat&l M. // Асе.
Chem. Res. 2000. 33. 269-277.
401
10.1.4. Органические транзисторы1
Органические транзисторы, в которых в качестве
полупроводникового слоя используется органический материал, тщательно
изучены. Модель транзистора органического эффекта поля (OFET)
показана на рис. 10.5.
Электрод Элекгрод
истока стока
Полупроводниковый слой
_
Изоляционный слой
Электрод затвора
Рис. 10.5. Иллюстрация топ-контактного OFET
Механизм OFET заключается в следующем: наложение
напряжения индуцирует носителей заряда на границе раздела
органического и изолирующего слоев, в результате чего снижается
сопротивление между электродом истока и электродом стока*
Полициклические ароматические углеводороды и другие
сопряженные молекулы пригодны для этой цели. Межмолекулярная
подвижность носителей через органический слой является важным
фактором, поэтому контроль ориентации молекул и дизайн стыка между
органическим слоем и электродами имеют важное значение. Было
разработано несколько материалов на основе полимеров
(сопряженных полимеров) для OFET. Влажная технология (Вет-процесс)
доступна и полезна для изготовления устройств с большой площадью
при низкой стоимости, хотя подвижность носителей, как правило,
ниже, чем для высоко ориентированных малых молекул.
10.1.5. Электрохромные устройства2
Электрохимическая реакция органических молекул иногда
сопровождается резким изменением цвета. Обратимое редокс-поведение
с изменением цвета применяется для электрохромцых устройств.
Это проявление цвета и восстановление свойств, которые
отличаются от явлений люминесценции, подходят для применения в элект-
HeilmeierCH., Zanoni L.A. // J. Phys. Chem. Solid. 1964. 25. 603-611;
Yoshida Af, Ventura &T Hoshino S,.Takada N,, Kodzasa Т., Kamata T. // Jpn. J.
Appl. Phys. 2005. 44. 3715-3720; Beaujuge P.M., Reynolds J.R. // Chem. Rev.
2010. 110. 268-320.
Beaujuge P.M.+ Reynolds J.R. Op. cil.
402
ронной бумаге. Однажды окрашенные электрохимической реакцией
данное изображение и информация остаются такими даже после
отключения питания.
На рис. 10.6 показано типичное электрохромное устройство,
состоящее из сэндвичевой структуры электролита, содержащего
хромовое соединение на анодной и катодной пластинах. Для
визуализации изменения цвета в электрохимической реакции должен
использоваться прозрачный электрод. Используемые хромовые
соединения должны иметь обратимые окислительно-восстановительные
свойства, т.е. их анион-радикальное или катион-радикальное
состояния должны быть стабильными.
Катод (просвечивающий электрод)
/ е N. ЭЛСкЧрШШ
Бесцветный Окрашенный
Потребляемый перенос электрона
—С у/
Анод
Рис. 10.6. Механизм электрохромного устройства
Они могут быть закреплены на прозрачном электроде, чтобы
избежать восстановления цвета, когда цветное состояние
генерируется на противоположном электроде. Как показано в разд. 5.8.6,
использование стабильного допированного состояния и обратимых
изменений цвета проводящих полимеров для электрохромных
приложений очень удобно. Кроме того, пленкообразующие свойства
электронолимеризованных проводящих полимеров выгодны.
Разноцветные электрохромные устройства получаются путем стекинга
каждой ячейки, показывающей свои индивидуальные цвета.
10.1.6. Конденсаторы на основе проводящих полимеров
Обычные алюминиевые электролитические конденсаторы
состоят из алюминиевого анода, оксидной пленки алюминия,
электролитического раствора и алюминиевого катода (рис. 10.7).
Алюминиевые твердые конденсаторы заменяют электролитический раствор
с твердыми проводящими материалами, такими как |3-Мп02,
органические проводники и проводящие полимеры. В отличие от ионной
проводимости электролитических растворов твердые конденсаторы
работают по электронпроводящему механизму. Таким образом,
проводимость значительно улучшается. Кроме того, термические
свойства твердых конденсаторов превосходят свойства обычных.
403
Анод
Катод
Рис. 10.7. Алюминиевый электролитический конденсатор
10.2. ЭЛЕКТРОХИМИЧЕСКАЯ КОНВЕРСИЯ БИОМАССЫ В ЦЕННЫЕ
МАТЕРИАЛЫ
Биомасса является возобновляемым органическим ресурсом,
а следовательно, ее использование не приводит к проблеме
истощения, в отличие от ископаемых ресурсов, таких как нефть и уголь.
В настоящее время социальные требования к эффективному
использованию биомассы, которую можно превращать в высокозначимые
химические вещества и топлива, быстро увеличиваются. Химическая
переработка биомассы должна быть выполнена с использованием
технологии энергосбережения и с низким уровнем выбросов,
соответственно, чтобы добиться этих целей, были проведены различные
испытания с использованием каталитических и ферментативных
реакций. Как упоминалось ранее, электрохимические реакции имеют
следующие преимущества: сохранение энергии и низкое количество
выбросов. Интенсивное изучение электрохимических процессов,
применяемых для химической переработки биомассы, началось
более 30 лет назад, а ее преобразование в высокоценные химикаты
исследовалось в рамках национального проекта в США1. Сегодня
изучение трансформации биомассы становится все более важным.
Основными компонентами растительной биомассы являются
целлюлоза и лигнин, которые представляют собой полимеры.
Поскольку ни один из них не пригоден для электролиза из-за их нераст-
Nonaka Г., BaizerM.M. Ц Elcctrochim. Acta. 1983. 28.661-665; BaizerM.M. //
Tetrahedron. 1984. 40. 935—969; Chum H,L,t BaizerM.M, The Electrochemistry
of Biomassand Derived Materials. ACS Monograph Series. ACS: Washington D.C.
1985.
404
воримости в растворителях, например, в воде, была сделана попытка
превращения биомассы в полезные химические вещества либо путем
гидролиза с последующим электролизом, либо путем непрямого
электролиза с соответствующими медиаторами.
Так как водорастворимая глюкозы образуется в результате
гидролиза целлюлозы, ее превращение в глюконовую кислоту и сорбит
было достигнутое выходами 90 и 50% по току соответственно путем
электрохимического окисления и восстановления глюкозы с
использованием неразделенной ячейки (рис. 10.8). Каждый из этих
электрохимических процессов был реализован на практике отдельно
в промышленном масштабе.
Кат«
2е
>д Анод
ОН
2Н/ 1 201-Г
/ НОСН^СНСНСНСНСНСНО n
/ "1 1 1 1 \
' ОНОНОН ОН '
, -2Н+ ,
\ ?н ?н /
НОСНзСНСНСНСНСНСЮН НОСН7СНСНСНСНСООН
III I "III I
ОНОНОН ОН ОНОНОН ОН
2е
Рис 10.8. Схема парного электросинтеза глюконовой кислоты и сорбита
из глюкозы
Байзер и его коллега реализовали парный электросинтез
глюконовой кислоты и сорбита из глюкозы, используя обе электродные
реакции1.
Синтез диальдегида крахмала был проведен путем окисления
крахмала медиатором ГО^ с использованием метода непрямого
элеюролиза в экс-ячейке (выход по току 100%). Также были
разработаны другие различные электрохимические процессы переработки
биомассы, например, парный электросинтез фурфурилового спирта
и 2-фуранкарбоновой кислоты из фурфурола, полученного из геми-
целлтолозы, и электросинтез ацетона, 2-бутена и метилэтилкетона
из 2,3-бутандиола, который легко доступен в результате
ферментации глюкозы и ксилозы2.
Кроме того, была предпринята попытка анодного
окислительного превращения лигнина в малые молекулы. В результате были
получены ароматические соединения (ванилин)3. «Валдвогель»
1 Park К., Pintauro P.N., Bai&r MM., Nobe К. // J. Electrochem. Soc. 1985. 132.
[R50-1S55.
2 Li W., Nonaka Т., Chou T.-C. // Electrochemistry 1999. 67. 4—10.
J Smith CZ, Uiley JMJ>.t Hammond J Ж. // J. AppL Electrochem. 2011.41.363-375.
405
и BASF разработали способ электрохимической деградации
лигнина на алмазном аноде, допированном бором (BDD), в водном
растворе с получением производных гидроксибензальдегида —
ванилина и/или производных фенола с выходом выше чем 5% по массе1.
Совсем недавно было исследовано окисление лигносульфокислоты
на серебряном или никелевом аноде в водном щелочном растворе
с использованием ячейки проточного типа с целью повышения
селективности и выхода полезных соединений — ванилина,
ванилиновой кислоты и гваякола (Шмитт и Валдвогель; не опубликовано).
Важность эффективного использования биомассы в общемировых
масштабах будет все более возрастать, а электролиз, как ожидается,
станет мощным инструментом этого прогрессивного способа
утилизации природных ресурсов.
103. ПРИЛОЖЕНИЕ К С1 -ХИМИИ
Концепция С1-химии включает синтез различных полезных
органических соединений путем образования углерод — углеродных
связей, используюший С1-соединения в качестве исходных
субстратов, или за счет введения различных атомов в С1-соединения.
В 1980-х гг. связанные с этой проблемой исследования
проводились в рамках национального проекта, а в последнее время С1 -химия
вновь привлекла большое внимание в связи с требованиями зеленой
химии для уетойчивот развития.
Типичный пример Cl-химии — фиксация С02, что является
весьма важной темой. Поскольку СО; — конечный продукт
процессов горения, обратное преобразование и использование СОг
представляются очень актуальными объектами исследований.
Однако потенциал восстановления С02 очень отрицателен, а
следовательно, протекает конкурентная реакция катодного выделения
водорода, которая уменьшает эффективность по току и выход продукта.
Поэтому катодное восстановление интенсивно исследовали с ис-
Griesbach U., Fischer A.* Sleeker F^ Botzem J., Pelzer R., Emmeluth M., Wald-
vogel S.R. Method for electrochemically cleaving lignin on a diamond electrode.
WO 2009138368 Al, filed May 11, 2009 and issued Nov. 19, 2009; Griesbach U.,
Fischer A., Stecker F., Botzem У., Pelzer /J., Emmeluth Af, Waldvogel S.R.
Process for the electrochemical cleavage of lignin at a diamond electrode. US Patent.
20110089046 Al, filed May 11, 2009 and issued Apr. 21, 2011; Stecker F., Fischer A.,
Kirste A,. Waldvogel S.R., fiexenbrecht C, Schmitt D. Process for the preparation
ofvancllin. US Patent 20140034508 Al, filed July 3, 2013 and issued Feb. 6, 2014;
Stecker F.< Fischer A., KirsteA., VortlA., Wong СИ., Waldvogel S.R,, RegenbrechiC,
Schmitt D., Hartmer M.F. Process for producing vanillin from vanillin-comprising
compositions. US Patent 20140046099 Al, filed Julv 3. 2013 and issued Feb. 13,
2014.
406
пользованием различных катодных материалов. При тестировании
этих материалов было обнаружено, что медь наиболее эффективна
для восстановления С02'. В целях повышения эффективности
катодное восстановление СОг проводили под высоким давлением.
Катодное восстановление С02 дает муравьиную кислоту, щавелевую
кислоту, метан и др.:
е СООН
С02 >\ +НСООН + СН4 (10.1)
СООН
Совсем недавно Наката и Еинага с сотрудниками реализовали
электрохимическое восстановление СО> в морской воде с
использованием BDD-электрода в нормальных условиях, получив
формальдегид селективно с высокой эффективностью по току (74%)2:
С°2 Морехода > А НСН0 <Ю-2>
В D D катод эффекта вносгь по току
Высокая эффективность по току связана с широким окном
потенциалов и арЗ-свяэанным утлеродом в BDD.
Катодное восстановление СО проводит к образованию
квадратичной кислоты3:
СО -£->
О РН
У
(10.3)
о он
10.4. ЭКОЛОГИЧЕСКАЯ ОЧИСТКА
Электрохимическая обработка сточных вод с применением
анодного разложения является весьма перспективным методом
уменьшения содержания токсичных загрязняющих растворенных веществ.
Для оптимизации этого метода важно выбрать анодные материалы,
поскольку электролитические продукты сильно зависят от их
природы, а также от условий процесса — плотности тока и температуры4.
На сегодняшний день для анодов разработаны различные материалы.
Hon У. Electrochemical C03 Reduction on Metal Electrodes in Modem Aspects of
Electrochemistry. \fol. 42/ed. C.G. Vayenas, R.E. White, M.E. Gamboa-Aldeco.
Springer-Nferlag. 2008. Ch. 3.
Nakata K., Ozpki 71, Terashima C.SujishimaA., Einaga Y. // Angew. Chem. Int. Ed.
2014.53.871-874.
Ercoli Д., Silvestri C, Gambino S..Guainaw M„ Filardo G. // Ger. Offen. 1973. 2.
235. 882, 2 Jan; Chem. Abstr. 1973. 78. 97190h.
Panizza M., Cerisota G, // Chem. Rev. 2009. 109. 6541-6569.
407
Их классифицируют следующим образом: углерод (аморфный
углерод, графит), благородные металлы или оксиды металлов (Pt,
1Ю2, Ru02) и оксиды неблагородных металлов (Pb02, Sn02, ТЮХ).
С давних времен электролиз использовался для очистки сточных вод
анодным окислением физиологического раствора путем генерации
раствора гипохлорита натрия, который применяется для
обесцвечивания сточной окрашенной воды.
Очистка сточной воды также была проведена с использованием
озона, генерируемого при окисления воды на РЬ02. В последние
годы были разработаны ВDD-электроды, которые оказались
эффективными дня обезвреживания сточных вод, содержащих
красители, органические кислоты, фенолы, растворимые полимеры
и т.д.1 Кроме того, было показано, что алмазоподобный углерод
эффективен для деградации стойких органических соединений
фтора3. Эффективность деградации обусловлена гидроксильными
радикалами, электрогенерированными на BDD-аноде, которые
приводят к полной деградации органики до С023. Интересно, что
этот механизм разложения очень похож на случай с
фотокатализатором ТТ02. BDD-электроды также эффективны для разложения
органических добавок в гальванических ваннах, а следовательно,
была продемонстрирована возможность повторного использования
электролитической ванны. Причем было показано, что разложения
вредных органических хлоридов (например ДДТ), можно достичь
электролизом с использованием медиатора-комплекса кобальта типа
гидрофобного витамина Вц при фотооблучении4. Кроме того, была
разработана новая поточная циркуляционная система с
использованием Pd трубки в качестве катода для электрокаталитического
гидрирования и было показано, что эта система очень эффективна для
дехлорирования полихлороароматических соединений, как показано
на рис. 10.95.
1 Fail С, Gandin D., ComnineUis С, Petrel A., Haenni W. // Electrachem. Solid-State
Lett. 1999. 2. 228—130; Gandini D., MaM £, MtchaudP.A.,Haenni W., Perrei A.,
ComnineUis Ch. // J. Applied Eleclrochem. 2000. 30. 1345-1350; Rodrigo M.A.,
Michaud P.A., Duo I. Panizza Л/., Cerisota G, ComnineUis Ch. //J. Electrochem.
Soc.200]. 148. D60-D64.
1 Ocliiai Г., lizuka K, Nakata K., Murakami Т.. Tryk D.A., Koide Y., Morito Y., Fu-
jishimaA. // Ind. Eng. Chem. Res. 2011. 50. 10943-10947: Ochiai 7"., fizuka Y..
Nakata K., Murakami Т., Tryk D.A., Fujishima A., Koide У., Morimoto Y. //
Diamond Relet. Mater. 2011. 20. 64-67; Ochiai Т., Moriyama H., Nakata A".,
Murakami T.t Koide K, Morito Y., Fujishima A.//Chem. Lett. 2011. 40.682-683.
} Panizza M., Cerisota G, Op, cit.
4 Shimakoshi //., Tokunaga A/., Hisaeda Y. // Dalton Trans. 2004. 878-882.
5 Fuchigami Т., Tajima T. // Electrochemistry. 200. 74. 585—589; Kawabata Y„
Naito Y., Saitoh Т.. Kawa K.. Fuchigami Т., Nishiyama S. // Eur. J. Org. Chem.
2014. 99-104.
408
Хлор/Бром/ Иод
ароматические
соединения
(В\пд) Г^>
. ;"Г.ДЬД|
Дегалогенированные
соединения
(Выход)
Регенерация
галогена
в виде соли
(например NaX)
(Х=Вг, I)
П ро н и к н <) не н не иолоро на
Катали шое I'd чернь
(+ углеродные волокна)
Рис. 10.9. Система электрокаталитического дегалогенирования
Была предпринята попытка конверсии хлорфторутлеродов (CFC)
в полезные вещества с использованием газодиффузных электродов —
Au, CuT Pd и In:
CCUF,
21 2"
IMno,a.NaOH
->CH4 + CH2F2
(10.4)
Глава 11. ОБЛАСТИ, СВЯЗАННЫЕ
С ОРГАНИЧЕСКОЙ ЭЛЕКТРОХИМИЕЙ
Органический электросинтез можно выполнить в мягких
условиях — при комнатной температуре и нормальном давлении.
Он не требует опасных окислителей или восстановителей, например,
тяжелых металлов. Кроме того, при его использовании получают
меньше отходов по сравнению с обычным химическим синтезом.
Несмотря на такие преимущества, количество коммерческих
органических электродных процессов ограничено в отличие от
неорганических промышленных процессов1.
11.1. ДОРОГА К ИНДУСТРИАЛИЗАЦИИ
Органический синтез, как правило, требует нагревания.
Исходные материалы и продукты легко разлагаются, что уменьшает
выход и селективность при высоких температурах реакции. В
отличие от этого органический электросинтез является
синтетическим химическим процессом, который может протекать в мягких
условиях — при комнатной температуре и нормальном давлении,
что позволяет избежать термического распада органических
соединений. Если создать оптимальные электролитические условия,
органический электросинтез может быть превосходной методикой
синтеза с отличным выходом и селективностью. Чтобы уменьшить
энергию активации, в химических реакциях обычно используют
различные катализаторы, но многие из них дороги, а следовательно,
очень важны рециклизация и повторное использование
катализаторов.
В электрохимических реакциях электроды являются
электропроводящими интерфейсами, в которых электроны переносятся
и от субстратов, а также действуют в качестве катализатора
(электрокатализатора), поэтому дорогой катализатор не нужен. Кроме
того, приложенный потенциал и ток для электрохимической
реакции можно недорого и точно контролировать. Таким образом,
органический электросинтез имеет много технических преимуществ
по сравнению с обычными органическими синтетическими
процессами.
Mac dona Id D.D., Schmuki P. (cds). Electrochemical Engineering. Encyclopedia
of Electrochemistry. Vol. 5. Wiley-VCH Vferlag GmbH, 2007; Putter H. Organic
Electrocliemistry. 4th edn / eds H. Lund and O. Hammerich. Marcel Dekker, 2001.
Ch. 31; Genders J. D.. Pletcher D. Elect rosy nthesis. From Laboratory. To Pilot. To
Production. N. Y.rThe Eleclrosynthesis Co. Inc., 1990.
410
Но органический электросинтез также имеет некоторые
недостатки. Обычные химические реакции являются гомогенными,
в то время как полем реакции электролиза является гетерогенный
раздел фаз, поэтому электросинтез имеет недостаточную
производительность. Кроме того, в отличие от неорганических
электродных процессов, например, электролиза солей, в органическом
электросинтезе рабочий электрод, генерирующий продукты, как
правило, это либо анод, либо катод. Противоположный электрод,
как правило, не используется для получения ценных продуктов,
кроме парного синтеза, разработанного BASF, как описано ниже.
Хотя основная роль противоположного электрода заключается
в обеспечении протекания тока к рабочему, содержание этого
электрода необходимо, что приводит к увеличению
эксплуатационных расходов. Когда рабочим электродом выступает катод, срок
службы анода в качестве противоположного электрода становится
препятствием для индустриализации. Обычно анод легко
корродирует, а значит, для него должны быть использованы дорогие
электродные материалы. В результате стоимость обслуживания
анода часто намного выше, чем рабочего катода. Эти проблемы
затрудняют индустриализацию органического электросинтеза.
Как уже упоминалось ранее, следует признать, что органический
электросинтез имеет серьезные ограничения. Несмотря на это,
катодное гидросочетание акрилонитрила использовалось более
40 лет во всем мире. Это объясняется тем, что продукт — адипо-
нитрил выгоден из-за своей универсальности и высокого спроса
благодаря превосходным физическим свойствам. Метод катодного
гидросочетания также превосходит другие синтетические
процедуры с точки зрения стоимости энергии. Первоначально процесс
функционировал с использованием разделенной ячейки, но позже
был разработан электролиз без диафрагмы, чтобы снизить
стоимость электроэнергии.
11.2. ПРИМЕРЫ
11.2.1. Электросинтез адипонитрила
6,6-Нейлон производится путем каталитического гидрирования
адипонитрила с образованием гексаметилендиамина и с
последующими дегидратацией-конденсацией адипиновой кислоты:
NC(CH2X,CN—^Uh2N(CH2)6NH2
/iH2N(CH2)6NH2 + яНООС(СН,)4СООН _„ню>
-^щр> -^HN(CH2)6NHCO<CH2)4CO^r {UA)
411
Так как 6,6-нейлон имеет отличную прочность и долговечность,
он широко используется для производства шинного корда,
синтетических тканей, конструкционных пластмасс и т.д. Гексаметилендии-
зоцианат в качестве исходного материала для полиуретана получают
из гсксаметилендиамина. Благодаря преимуществам нейлона и
полиуретана, катодное гидросочетание акрилонитрила является важной
технологией для повседневной жизни.
Катодное гидросочетание акрилонитрила было известно
с 1940-х гг., но выход адипонитрила первоначально не был высоким.
В 1960 г. Байзер (компания «Монсанто») улучшил как выход, так
и эффективность по току гидроеочетания с помощью соли
четвертичного аммония в качестве фонового электролита1, В 1963 и 1965 г.
«Монсанто» разработала и коммерциализировала электрогидроднме-
ризацию акрилонитрила, который был произведен по низкой цене
в крупном масштабе с использованием метода SOH10,
разработанного в то время:
2CH2=CHCN——>NC(CH2)4CN (1K2)
н20
Водный раствор, содержащий большое количество соли
четвертичного аммония (приблизительно 40%), был использован для
растворения практически нерастворимого акрилонитрила в высокой
концентрации. Таким образом, процесс «Монеанто» адоптировала
в гомогенную электролитическую систему.
При этом AsahiChemicalCompany (теперь AsahiKaseiCorporation,
Япония) разработала эмульсионное катодное гидросочетание
акрилонитрила с использованием термоморфности акрилонитрила
и воды, содержащей 10% соли четвертичного аммония, и
запустила процесс в 1971 г.2 Оба процесса на ранней стадии
функционировали в разделенной ячейке, снабженной катионообменной
мембраной типа сепаратора. Позже обе компании улучшили
свои технологии, и в середине 1980-х гг. они установили
неразделенные электролизеры, чтобы уменьшить высокое напряжение
на ячейке, вызванное сопротивлением диафрагмы. Чтобы
разработать процесс в неразделенном электролизере, они
сосредоточились: 1) на высокой селективности, а также меньшей коррозии;
2) создании электролитических условий для стабильной
долгосрочной работы и систем для рециркуляции соли четвертичного
аммония/очистки продуктов; 3) разработке оборудования, в
котором взрывоопасную газовую смесь акрилонитрила и кислорода
можно обрабатывать безопасно.
1 Putter H. Op. cit.
2 ShimizuA. // Catalysts & Catalysis. 1998. 5. 269.
412
Катоды, используемые в этом процессе — Hg, Pb или Cd, имеют
более высокое перенапряжение водорода, поскольку выделение
водорода в качестве побочной реакции уменьшает выход но току
гидросочетания. При этом Fe, Ni или Pb используются как аноды
и имеют более низкие перенапряжения кислорода, чтобы
предотвратить анодное окисление органических соединений. Фоновый
электролит представляет собой смесь четвертичных аммониевых
солей и неорганической соли.
Четвертичные аммониевые соли, например соль этилтрибути-
ламмония, эффективны не только для повышения селективности
образования адипонитрила, но и для зашиты катода от коррозии.
Неорганические соли (фосфат калия и борат щелочного металла)
увеличивают проводимость электролита и предотвращают коррозию
анода.
Подробно условия осуществления этого процесса были описаны
некоторыми исследователями1. Ниже приведен пример условий
эксплуатации:
катод: Pb:
aHOfl:Fe(Ni-9%);
фоновый электролит: (EtBu3N)2HP04 + К2НР04 + КгВ407;
плотность тока: 2 кА м-2;
температура: 554С;
выход по току: 90,7%.
В настоящее время каждый год во всем мире получают более
300000 т адипонитрила катодным гидросочетанием акрилонитрила.
В 2010 г. 24% от всей продукции адиподинитрила было изготовлено
катодным гидросочетанием.
11.2.2. Электросинтез ароматических альдегидов
В компании BASF уже в течение более чем 30 лет работает
электрохимическое производство ацеталей ароматических альдегидов
типа анисальдегида и л-толуальдегида2:
СНз С<ОМе)2 СНО
R R R
R = Me, OMe
1 Macdonald D.D.t Sehmuki /\ (eds). Op. cil.; Putter H. Op. cil.; Genders J.D.,
Fletcher Z). Op. ciL; ShimizuA, Op. cit.
2 Putter H. Op. cil.
413
Окисление метилъной группы в ароматическом кольце приводит
к получению ароматических альдегидов, но обычные химические
методы с использованием окислителей часто вызывают
переокисление, что приводит к образованию п качестве побочного продукта
карбоновых кислот. Электрохимическое окисление производных
толуола на аноде в метаноле дает стабильные диметилацетали,
которые легко гидролизуются с образованием альдегидов и метанола.
Выделенный метанол может быть повторно использован для анодной
ацетализации. Этот процесс работает в ячейке с капиллярным
зазором, разработанной компанией BASF1.
11.2.3. Парный электросинтез фталида и трет-
бутилбензальдегида
Недавно компания BASF коммерциализировала парный
электросинтез фталида итрет-бутилбензальдегидадиметилацеталя (парный
электросинтез, сочетающий анодное диметоксилирование трет-бу-
тилтолуола и катодное восстановление сложного диэфира фгалевой
кислоты)2:
Катод
а.
СООМе
+4е, +4Н+
90%
СООМе
О + 2МеОН
2МеОН
(1Ь4)
'^V-'
СООМе
СН, СН(ОМе)2
Общая схема
1 ^^ХООМе
+4с, +4Н \
J> -4e,-4H'
о +
О
СН3 " СН(ОМе)2'
Фоновая соль: MeBu3N+MeS04"
________-_-_ — — -_ — — — — — — ______-__ — ___ — _j
1 Beck F., Gitthke H. 11 Chem.-Ing.-Tech. 1969. 41. 943-950.
2 Putterh.Op.ch.
414
Продукты могут использоваться в качестве средств зашиты
растений, добавок для гальванических ванн, ультрафиолетовых
поглотителей, ароматических химических веществ, фунгицидов и т.д.
Фталид был получен классической каталитической гидрогенизацией
ангидрида фталевой кислоты, но этот метод требует наличия чистого
газообразного водорода и высокого давления.
При этом в ходе анодного диметоксилирования трет-бутилто-
луола протоны преимущественно восстанавливаются на катоде,
генерируя газообразный водород. BASF приложила большие усилия,
чтобы найти приемлемый способ восстановления, который можно
использовать вместо восстановления протонов. В конце концов
удалось подобрать сочетание анодного диметоксилирования трет-бу-
тилтолуола и катодного восстаноааения сложного диэфира фталевой
кислоты, чтобы создать такой парный электросинтез (см. (1L4)).
Результатом катодного восстановления диметилового эфира
фталевой кислоты является фтатид иметанол, тогда как результирующий
метанол используется для анодного диметоксилирования трет-бутил-
толуола и дает диметилацеталь с хорошим выходом. В этом парном
электросинтезе наблюдается замечательный баланс метанола,
протона и электрона.
Этот процесс выполняется в неразделенной капиллярной
ячейке с массой биполярных круглых графитовых электродов (см.
рис. 9.26)1. Электроды имеют центральное отверстие, отделены друг
от друга прокладками и соединены последовательно. Так как соль
четвертичного аммония используется в качестве фонового
электролита, катодное восстановление метанола подавляется, а
восстановление сложного диметилового эфира о-фталевой кислоты протекает
с высокой эффективностью без образования побочного продукта.
Так как эффекгнвность обеих электродных реакций высока, КПД
использования энергии в парном синтезе гораздо выше, чем в обычных
процессах.
11.2.4. Электрохимическое перфорирование
Электрохимическое перфторирование представляет собой
процесс, в котором все атомы водорода в исходной органической
молекуле замещаются атомами фтора без генерации в процессе
электролиза элементарного фтора. Дж. Саймоне достиг
электрохимического перфторирования органических соединений в безводном
жидком HF с использованием никелевых электродов. Он получил
перфторированные продукты впервые в 1941 г.2 и является пионером
BeckF.,Guthke Я. Op.ciL
Simons J.И. //J. Elcctrochem. Soc. 1949. 95. 47-52; Idem, // J. Fluor. Chcm.
1986. 32. 7-24.
415
в области электрохимического перфорирования, а этот метод
называется процессом Саймонса. Процесс использует неразделенную
ячейку при низкой температуре, чтобы сохранить HF в виде
жидкости (точка кипения HF 19,5°С). Электрохимическое
перфорирование органических соединений, содержащих кислород, азот или
серу, приводит к образованию соли с безводным HF, которая
обеспечивает хорошую проводимость раствора безводного жидкого HF.
А в случае углеводорода должны быть добавлены соли (KF и NaF)
для придания электропроводности и реализации
электрохимического процесса перфорирования, Перфторированные продукты
из карбоновых кислот (их хлоридов), сульфокислот (их хлоридов)
и триалкиламинов получаются с хорошим выходом1:
CH3S02CI _ ~ЯС UF ) CF3S02F (11.5)
J ^ Бе-анодная HF J * * '
С'Н"С0Х E_HF ' C'H"C0F
<X = C1,F)
(11.6)
<C^»N ь-^Lhp '<C^bN (И.7)
Однако во многих случаях выход электрохимического перфто-
рирования довольно низкий из-за расщепления в процессе
электролиза углерод — углеродной связи2. Продукты перфторалкилкар-
боксильной кислоты и сульфокислоты полезны в качестве моющих
средств и смазочных материалов.
Механизм реакции обсуждался в течение многих лет3. Один
из вариантов механизма предлагает для перфторирования в качестве
важнейшего реагента анодно генерируемый фтор-радикал, а другой
включает электрогенерированные высоко окисленные фториды
никеля {Ni2F.5, NiF3 и NiF4) на поверхности никелевого анода. Они
действуют как фторирующие реагенты и электрохимически
регенерируются на никелевом аноде. Во время электролиза фторированные
продукты, которые частично имеют полярность, остаются в
электролите, чтобы повергнуться дальнейшему электролизу. Конечные
перфторированные продукты неполярны, а их удельная плотность
очень высока, поэтому они выпадают из жидкого HF на дно
электролизера в виде жидкости.
Suriyenarayattan S„ Noel M. // J. Solid State Electrochem. 2008.12.1453-1460.
Ibid,
Suriyanarayanan N.. Noel Л/. Op. cit.; Hollitzer £., Satori P. // Chem-lng-Tcch,
1986. 58. 31—38; Dimitrov Л., RMger S., Ignatyev N. K.4 Datcenko 5. // J. Fluor.
Chcm. 1990. 50. 197-205; Sartori P.. Ignat'ev N., Junger C, Juschke C, Rie-
land P. U J- Solid Stale Electrochem. 1998. 2.110-116.
416
Электрохимический процесс перфорирования теперь реализован
в больших масштабах на заводах ЗМ в США и GlassCompany и Mrtsu
bishiMaterialsElectronicChemicalsCompany в Японии. MitsuiChemicals
(Япония) также производит NF;< электрохимическим перфториро-
ванисм NH3'. NF3 используется в качестве травителя и средства
газоочистки для устройства, используемого в методе химического
осаждения из паровой фазы (CVD).
11.2.5. Другие примеры
3,6-Дихлоропиколиновая кислота (Лонтрел). Daw Chemical
Company коммерциализировала электрохимическое восстановление
в водном растворе 3,4,5,6-тетрахлорпиколиновой кислоты в 3,4-дих-
лорпиколиновую кислоту, которая находит применение в качестве
прекурсора агрохимикатов2:
2с, 2Н+
^2СГ*
соон
С1
(П.8)
Cr^N-^cooH
Как объяснялось в гл. 4 (см. рис. 4.5), высокая региоселектив-
ность связана с контролируемой ориентацией субстрата на
поверхности катода вследствие дипольного момента молекулы.
Производное р-лактама. Otsuka Chemical Company
коммерциализировала электросинтез антибиотика — производного р-лактама
(GCLE):
R'OCHN
S—SCbAr
COOR2
aq. NaCT/CH2Cl2
Pt-Pl
aq. NaCl/CHjCl;
Pt-Pt
R'OCHN
S—SO-. At
COOR2
NH,
DMF
(119)
1 TasakaA., Kawa^oe T„ ТакитЛ., YamanakaM., Tojo T.TAriisuk(t M, //J, Elcc-
trochem. Soc. 1998. 145. 1160-1164.
2 Edamura F., Kyriyacou D., Love J. US Patent 4217185. 1980; Chcm, Abstr. 1981.
94.22193.
417
R'OCHNv с
DMF
Электролиз проходит в двухфазной системе дихлорметан — вода,
а реакция протекает через анодное окисление хлорид-ионов.
Цистенк. Электрохимическое восстановление цистина в цистеин
используется в США, Японии, Китае и некоторых европейских
странах:
HOOCCHCH2S—SCH2CHCOOH *, 2Н
NH2 NH2
' ' Кятод — углерод или Ag
Де.иГ y 2HSCH2CHCOOH (ИЛ }
Катод - углерод или Ag J, „
Так как отделение цистеина от цистина является довольно
сложным, электролиз нужно продолжать до тех пор, пока весь
исходный цистеин не будет полностью израсходован. Это означает, что
на заключительном этапе электролиза электроэнергия потребляется
в основном на восстановление протонов с выделением водорода, что
приводит к снижению выхода по току, а это является проблемой,
которую еще предстоит преодолеть.
Гидроксид тетраметиламмония. Электрохимический диализ
хлорида тетраметиламмония (Япония) использует катионную
ионообменную мембрану для получения бесхлорного тетраметиламмония
гидроксида высокой чистоты:
Me4NCl ~s'-y2a2 >Me4NOH (11.11)
4 Вода ч v '
Чтобы избежать загрязнения продукта хлором, анодно
генерируемый газообразный хлор полностью удаляется из системы диализа.
Другие примеры. Компания BASF производит 2,5-диметокси—2,5-
дигидрофуран и дигидрофталевую кислоту из фурана и о-фталевой
кислоты:
Она также производит ацетоин путем анодного окисления цикло-
гексанона с применением йодного медиатора.
418
Кроме того, компания SNPF (Франция) производит
противовоспалительное средство фенопрофен катодным восстановлением
мета-(а-хлорэтил)фенил фенилового эфира в присутствии диоксида
углерода1:
соо
*.-q--co*,r TIT cii-13)
Mg растворимый
анод
Электросинтез искусственного подсластителя мальтола и т-
гидроксибензилового спирта использовали в коммерческих целях
в Японии, но этот процесс следует остановить из-за его высокой
стоимости по сравнению с альтернативными химическими
процессами и низкой цены продукции по сравнению с производимой
в других странах. Как описано выше, органические
электрохимические процессы часто конкурируют с химическими. Поэтому
необходима разработка электрохимических процессов, которые
превосходят химические. Следует подчеркнуть, что органические
электросинтетические процессы, направленные на снижение выбросов,
яа1яются весьма перспективными2.
1 Chausaard J,, Troupel M., Robin Y. // J. Appl. Electrochem. 1984. 19. 345—348,
2 Putter iJ. Op. ciL
Приложение А
ПРИМЕРЫ КОММЕРЦИАЛИЗИРОВАННЫХ ОРГАНИЧЕСКИХ
ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
А.1. Электрохимическое фторирование
Анодное фторирование и анодное метоксилирование
этилфенилтиоацетата1
Анодное частичное фторирование органических соединений, как
правило, в качестве источника фтора и фонового электролита
использует поли (фтористоводородный) комплекс амина или
аммониевой соли. В неразделенной ячейке происходит выделение водорода
путем катодного восстановления протонов, а также целевое анодное
фторирование субстрата. В растворе ацетонитрила фторирование
протекает через анодно генерируемый катионный интермедиат,
а в растворе метанола происходит селективное метоксилирование
(рис. А.1). Оптимальные условия анодного фторирования зависят
от используемых субстратов. Необходимо оптимизировать
комбинацию реакционной среды и фоновых электролитов.
Wve
Rubber plug
Hydrogen bubbles
Pi plate (2 cm x 2 cm)
1,0 M EbN-3HF/MeCN (or MeOH) + substrate
Stirring bar
Рис. A. 1. Иллюстрация установки для анодного фторирования при
постоянном токе
Анодное фторирование этилфенилтиоацетата протекает
через окисление атома серы с последующим депротонированием
1 Fuchigami Т., Shimojo М., Кото А. //). Org. Chem. 1995.60. 3450-3464; Fuch-
igami Т., Yarn //., KormoA. // J. Org. Chem. 1991. 56. 6731-6733.
420
а-положения. Стадии депротонирования способствует эффект элек-
троноакдепторной фуппы, замещенной у а-углеродафенилтио-
грунны. Монофторированный продукт сложноэфирной группы
в се-положении должен быть относительно устойчив, поэтому его
можно использовать в качестве строительного блока для
дальнейшего превращения:
-2е,-Н+
о
EtN-3HF/McCN PhS
2 Ф/мши,
-2е.-Н+
<А1>
EtN-3HF/McOH PhS
2 Ф/моль
На рис, Л. 1 показан типичный электролизер, используемый для
анодного фторирования, оборудованный анодом из платиновой
пластины и катодом (2 см х 2 см). Ячейка заполнена
электролитическим раствором, содержащим этилфенилтиоацетат (196 мг, 1 ммоль),
Et3N-3HF (1.6 мл, 10 ммоль) и аиетонитрил (8,4 мл). Электролиз при
постоянном токе (20 мА, плотность тока 5 мА • см-2) в течение 2 ч
41 мин (2 Ф * моль-1) проводят при перемешивании. Затем раствор
нейтрализуют насыщенным раствором NaHCOi и экстрагируют эти-
лацетатом (30 мл х 3). Органическую фазу промывают насыщенным
раствором соли (100 мл) и сушат над Na2S04. Растворитель удаляют
при пониженном давлении, а сырой продукт очищают с помощью
колоночной хроматографии на силикагеле с получением желаемого
фторированного продукта с выходом 50—70%. Если используют
метанол вместо ацетонитрила, селективно получают метоксилиро-
ванный продукт.
А.2. Электросинтез с использованием гидрофобного электрода
Синтез хромана из терпенов и электрогенерированного
хинометана1
Гетерореакция Дильса-Ал ьдера хинометана и терпена
рассматривается как биогенетический путь для euglobal- производных.
Электрохимическое генерирование хинометана и последующее циклоприсо-
единение к терпену, возможно, дает хроман — основную структуру
euglobal (рис. А.2).
СЫЬа К„Аткат Т., Tada А/. // Chem. Commun. 1996. 1763-1764.
421
ISJ
Электрод
сравнения
00
PTFE-волокно
COOfl
4
OH S
■/
MeOi^^OMc
vjn a i
I
МеСГ^^ТШе
A
Met)
MeO
COOH
Pt катод
СУ анод, покрытый волокном РТЕЕ
1,0 М LiCI04/MeN02 + субстрат
Рис Л.Л. Иллюстрация электролитической установки с гидрофобным электродом и синтез euglobal
Сульфид 1 (ОЛ ммоль) и а-фелландрен 2 (0,4 ммоль)
растворяют в 1,0 М LiC104/CH3N02 (15 мл). Электролиз при постоянном
потенциале (1,2 В отн. Ag/AgCl) выполняется с использованием
стеклоуглеродного анода, покрытого поли (тетрафторэтиленом)
(PTFE), и катода в виде платиновой пластины. После прохождения
1,2 Ф ■ моль-1 заряда продукт экстрагируют гексаном (10 мл)
несколько раз. Органический слой сушат над Na2S04, а продукт 3
подтверждают с помощью ТСХ.
А.З, Синтез натуральных продуктов с использованием анодного
окисления
Синтез Anibaneolignan анодным окислением производных фенола1
Хотя анодное окисление производных фенола очень полезно для
получения алициклических соединений из ароматических
соединений, разнонаправленные пути реакции затрудняют селективное
получение желаемого продукта. Если условия реакции
оптимизировать, анодное окисление фенолов может быть мошным
инструментом получения желаемых продуктов, которые трудно получить
в обычном органическом синтезе.
Согласно рис. А.2, катион А, который генерируется путем
анодного окисления фенола 1, подвергается ииклоприсоединению к изо-
сафролу, в результате чего образуется природный продукт Aniba
neolignan.
Фенол 1 (100 мг), изосафрол 2 (500 мг) и LiClO^ (200 мг)
растворяют в смеси метанол/уксусная кислота (2 : 1, 30 мл) и используют
в качестве электролитического раствора. В стеклоуглеродном
химическом стакане в качестве анода электролиз при постоянном токе
(10 мА, плотности тока — 0,19 мА • см-2) проводят в течение 130 мин
с использованием катода из платиновой проволоки. Растворитель
удаляют, а продукт очищают с помопдао колоночной хроматографии
на силикагелс.
1 Shizuri Г.. Уататига S. (1983) TeirahedranLetl. 1983. 24. 5011-5012.
423
MeO.
MeO
-2c,-H
Ar*
-2e,-HJ
UClO^/McOH-AcOH q—4 / °
MeO
OMc
Aniba neolignan (3)
A
\ I.. OMe
^O
(A.2)
А.4. Электролиз Кольбе
Синтез диэфира из моноэфира адипиновой кислоты1
Реакция Кольбе, которая включает одноэлектронное окисление
карбоксилата и последующее декарбоксшшрование с образованием
его димера, является очень эффективной для получения высших ал-
канов. Например, моноэфир адипиновой кислоты может быть
превращен в его димерное соединение:
2CH3OOC(CHj)4COOH 2с' С°2
CH3ONa/CH3OH
-2е,-С02
<А. 3)
-> СН,ООС(СН2 )8 СООСН,
CH3ONa/CH3OH
Электролитическая ячейка, оснащенная платиновым анодом
и катодом (25 мм х 30 мм, расстояние — 5 мм), заполняется
раствором метанола {250 мл), содержащем монометиловый эфир
адипиновой кислоты (120 г, 0,75 моль), мстилат натрия (4,1 г, 0,075 моль)
и пиридин (10 мл, 0,12 моль). Электролиз при постоянном токе
(1,1 А, плотность тока — 0,13 мА ■ см-2) проходит в течение 23 ч.
В процессе электролиза в этих условиях выделяется тепло, что
вызывает кипение реакционной смеси. После охлаждения до комнатной
температуры к смеси добавляют уксусную кислоту (20 мл), чтобы
ее подкислить. Растворитель удаляют при пониженном давлении,
а остаточное твердое вещество растворяют в эфире (500 мл).
Фильтрат промывают водным раствором NaHCOj и водой и сушат над
CaS04. Продукт получают с помощью перегонки при пониженном
давлении.
А.5. Непрямой электросинтез с использованием медиатора
Синтез кетонов путем непрямого анодного окисления алкоголя
с помощью йода-медиатора2
Катионные частицы йода генерируются путем анодного
окисления йодил-ионов и используются для окисления спирта с
получением кетона и регенерацией йодид-ионов. Реакция может
медиироваться каталитическим количеством йодида без оксида
металла:
tiaufeJ., Seek F. // Chem, LnB. Tech. 1970. 42.170-175.
Shono Т., Matsumura Y.. HayashiJ., Mizoguehi M. //Tetrahedron Lett. 1979. 20.
165-168.
425
R
R
R
V
CH-OH \> \ » C=0
-2e Анод
R' = R2= /XT" 93%
R1 = C6H13, R2 = Me: 99%
R1 = R2 = Cyclohexyl: 74%
R1 = Ph. R2 = Me: 100%
В электролизере получают водный раствор (15 мл) йод ид а калия
(2,49 г, 0,015 моль) и спирта (0,06 моль). Электролиз проводится
при постоянном токе с использованием платинового или
углеродного анода (пропускается 4—15 Ф ■ моль1 электроэнергии).
Органический слой отделяют, а водный слой экстрагируют диэтиловым
эфиром.
Объединенную органическую фазу промывают водным раствором
Na2S20.v Продукт очищают перегонкой.
А.6. Электросинтез проводящих полимеров
Электрополимеризация пиррола
Электрополимеризация ароматических соединений протекает
на поверхности электрода, в результате получается тонкая пленка.
Эти проводящие полимеры используются как проводящие
материалы, элсктрохромные устройства и катализаторы. Ниже показан
типичный механизм окислительной полимеризации пиррола:
Н Н
<А.5)
В ячейку типа стакана помещают электроды из платиновых
пластин (1 см х 1 см) и насыщенный каломельный электрод (НКЭ)
в качестве сравнения. В ячейку добавляют водный раствор (50 мл)
пиррола (0,1 М) и NaC104 (0,1 М). Развертка потенциала от 0,6
до 0,8 В отн. НКЭ при скорости сканирования 100 мВ * с-1 приводит
к полимеризации на рабочем электроде, которая контролируется как
показано на рис. А.З. Рабочий электрод, покрытый политшрролом,
очищают, промывая водой.
426
I 1 1 1 1 1 1 1
-0,6 -0,4 -0,2 0 +0,2 +0,4 +0,6 +0,8
Потенциал (Both. НКЭ)
Рис. А.З. ЦВА пиррола в течение электрополимеризации
427
Приложение В
ПРИМЕРЫ ОРГАНИЧЕСКОГО ЭЛЕКТРОСИНТЕЗА
Таблица В. 7
Окно потенциалов органического раствора для
электрохимических реакций (Pt рабочий электрод)
Растворитель
АсОН
Acetone
MeCN
MeCN
DMF
DMSO
McOH
MeOH
CH2C12
THF
Sulfolane
MeN03
Propylene carbonate
Фоновый
электролит
AcONa
n-Bu4NCL04
UC104
Et4NBF4
w-BuN4C104
LiClO.,
L1C104
KOH
n-BuN4CI04
UC104
NaC104
Mg(CI04)2
Et4C104
Потенциал (В отн. НКЭ)
Катодная сторона
-1,0
-1,0
-3,0
-1,8
-2,8
-3,8
-1,0
-1,0
-1,7
-3,2
-4,0
-2,6
-1,9
Анодная сторона
+2,0
+ 1,6
+2,5
+3,2
+ 1,6
+ 1,3
+ 1т3
+0,6
+ 1,8
+ 1,6
+2,3
+2,2
+ 1,7
428
Таблица В.2
Потенциалы окисления типичных органических соединений
Соединение
Электролит/растворитель
Потенциал, В
Электрод
сравнения
Aromatic compounds
Benzene
Toluene
Anisole
Biphenyl
Fluorene
Naphthalene
Pyridine
Thiophcnc
Pyrrole
NaCIO,/MeCN
NaC104/MeCN
Pr4NCI04/MeCN
NaCI04/MeCN
NaCI04/MeCN
NaCKVMeCN
NaCKVMeCN
NaCKVMeCN
NaCI04/McCN
2,00
1,93
1,76
1,48
1,25
1,31
2,20
2,10
0,46
Ag/Ag+
Ag/Ag+
SCE
Ag/Ag+
Ag/Ag+
SCE
SCE
SCE
SCE
Olefins
Ethylene
Cyclohexene
Styrenc
Bu4NBF4/McCN
NaClCVMcCN
NaC104/McCN
2,90
1,95
1,90
Ag/Ag^
Ag/Agf
SCE
Nitrogen and sulfur compounds
Acetamide
Aniline
W-MeLhy Ian i line
Nitrobenzene
Thiophenol
Dimethyl sulfide
Dimethyl disulfide
EL,NCU/MeCN
Buffer/H20
Na2S04/H20
0,1 M HCI/50%
acetone/H20
CF3C02H/CH2C1,
0,1 M HCl/MeOH
LiC104/MeCN/CH2a2
2,00
1,04
0,70
0,58
1,65
0,86
1,75
SCE
SCE
SCE
NHE
Ag/Ag+
Ag/Ag+
Ag/Ag+
Alcohols
Methanol
Ethanol
Isopropyl alcohol
t-Buty alcohol
Ally alcohol
Benzyl alcohol
Phenol
Bu4NBF4/MeCN
Bu4NBF4/MeCN
Bu4NBF4/MeCN
Bu4NBF4/MeCN
Bu4NBF4/MeCN
NaCKVMeCN
NaCKVMeCN
2,73
2,61
2,50
2,60
2,65
>2,00
1,04
Fc/Fc +
Fc/Fc +
Fc/Fc +
Fc/Fc +
Fc/Fc +
Ag/Ag+
Ag/Ag^
429
Таблица В.З
Потенциалы восстановления типичных органических соединений
Соединение
Электролит/растворитель
Потенциал, В
Электрод
сравнения
Halogen compounds
Chloromethane
L-BuLyl chloride
t-Butyl bromide
t-Butyl iodide
EUNCICU/DMF
Et4NC104/DMF
Et4NBr/DMF
Bu<NBF4/DMF
-2,76
-2,60
-2,19
-1,91
SCE
SCE
SCE
SCE
Carbony compounds
Acetone
Acctophcnonc
Formaldehyde
Acetaldehyde
Benzaldehyde
EMNBr/DMF
LiOH/75% Dioxane
pH = 8
pH=9,l
NH4C1/40%EtOH
-2,84
-1,26
-1,22
-1,51
-1,32
SCE
SCE
NHE
NHE
SCE
Quinones
1,4-Benzoquinone
1,4-Naphtoquinone
9,10-AntJiraquinone
50% EtOH
50% EtOH
95% EtOH
0,71
0,49
0,16
NHE
NHE
NHE
Olefins
Styrene
rra/w-Styrene
c/5-Styrene
Bu4NI/DMF
Bu^NI/DMF
Bu4Nl/DMF
-2,45
-2,30
-2,07
SCE
SCE
SCE
Aromatic compounds
Benzene
Naphthalene
Anthracene
Pyrene
Biphcnly
Furfural
2,6-DImethylpyridine
Nicotinamide
Bu4NBr/Mc,NH
Bu4NBr/Me-,NH
Bu4NBr/Me2NH
Bu4NBr/Me2NH
NaBPhj/THF
Britton and Robinson/ MeOH
Bu4NBr/Me2NH
Britton and Robinson/MeOH
-3,42
-2,53
-2,04
-2,29
-2.68
-1,04
-2,85
-1,34
Ag/Ag+
Ag/Ag+
Ag/Ag*
Ag/Ag+
Ag/Ag+
SCE
Ag/Ag*
Ag/Ag+
Nitrogen and sulfur compounds
Nitromethane
Nitrobenze/e
Diphenyl disulfide
Metryl phenyl sulfone
Benzenesulfonic acid
pH = 7H20
pH = 7/80% dioxane
Bu4NI/DMF
ВщШг/DMF
Me^NCI/dioxane
-0,88
-0,62
-2,75
-2,41
-1,50
SCE
SCE
Ag/Ag+
SCE
SCE
430
Таблица В.4
Физические свойства типичных растворителей
Compound
Acetic acid
Acetone
Acetonitrile (AN)
Chloroform
/V, ЛГ-DimetheIformamide
(DMF)
Dimethyl sulfoxide
(DMSO)
Ethanol
Hexamethyl-phosphoric
triamide (HMPA)
n-Hexane
Methanol
Nitrobenzene (1МИ)
Nitromcthanc (NM)
Propylene carbonate (PC)
Pyridine (Py)
Tetrahydroftiran (THF)
Water
Molecular
wieght
60,05
58,08
41,05
119,38
73,10
78,14
46,07
179,20
86,18
32,04
123,11
61,04
102,09
79,10
72,11
18,01
Boiling
point (°C)
117,8
56,2
81,6
61,27
158
189,0
(Decomp.)
78,32
235
68,7
64,75
210,80
101,2
241
115
65,0
100,00
Melting
point
<°C)
16,64
-95,4
-45,7
-63,49
-61
18,55
-114,15
7,20
-94,3
-97,68
5,76
-28,6
-49
—4K5
-108,5
0,00
Density d
(g/cm-1)
(25'C)
1,0492'
0,7845
0,7766
1,4891*
0,9443
1,096
0,7851
1,024*'*
0,6594
0,7866
1,1986
1,1312
1,19
0,9779
0,880
0,9971
Viscosity
T| milli-
poisc
(25'C)
12,2*
3,02
3,39
5,55"
7,96
19,6
10,78
—
3,258*
5,42
18,11
6,27
25,3
8,824
4,6
8,903
Relative
permittivity £r
(25*C)
6,15
20,7
35,97
4,724
36,71
46,6
24,3
29,6
1,90
32,6
34,82
35,94
64,4
12,01
7,39
78,54
Dipole
moment
u.(xl0 »
Cm)
5,67
9,76
13,06
3,40
12,86
—
5,63
—
0,00
5,63
14,03
11,53
—
7,17
5,67
6,47
Donor
number On
—
17.0
14.1
—
26,6
29,8
20
38,8
—
19,0
4,4
2,7
15,1
33,1
20,0
18,0
Acceptor
number v4n
52,9
12,5
18,9
23,1
16,0
19,3
37,1
10,6
0,0
41,3
14,8
20,5
18,3
14,2
—
54,8
Self-ion-
ization
constant
PA'runi
14,5
—
28,5
—
—
-32
18,9
—
—
16,7
-
19,5
—
—
-14,0
* 20X.
" 22,8°C.
"* 30°C.
+0,5
-0,5
-1,0
-2,0
-3,0
N1TRO
KETONES
QUATERNARY AMMONIUM?
GEM POLYCHLORIDES RCL
OEM POLYBROM1DES RBr„
BENZYL, ALLIL 1,2-
BROM IDES di-Br
I
IODIDES
ORGAN O-SULFUR
MONO CHLORIDES
MONO-BROMIDES
ACTIVATED OLEFINS
AROMATICS
1
+0.5
-0,5
-2,0
-1.0
Potential (V vs. SCE)
Рис. В.1. Области потенциала электрода для восстановления функциональных групп
-3,0
-0,5
+0,5
+1,0 +2,0
AROMATIC HIDKOCARBONS
PHENOLS
AROMATIC AMINES
ORGAN O-M ETALLIC
R-SII R-S-R
ORGANO-
SULFUR
RCOO'
CARBOXYLATE SALTS
AROMATIC
HALFDES
R-CONHj
AMIDES
RCOR
KETONES
-0,5
0
+0,5
+2,0
+ 1,0
Potential (V vs. SCE)
Рис. В2. Области потенциала электрода для окисления функциональных групп
Электроды сравнения
+597 mV
< >
1 1 1
+241 mV
+ !97mV
R/Fc
10-
5-
а 0-
|2 ■
-5-
-10Н
1,0 0,8 0^6 1),4 0^2 ОД) -0,2-0,4
Потенциал, В
НО ■ НЮ
Ag/AgNO, Ag/AgCl
О М Щ KCI)
vs. Ag/AgCl
vs. НВЭ(Н2/2Н")
vs. HR3 (Hg2/2Hg+)
vs. Fc/Fc1
Л^" ^,в Еф, AEf E\(i
Ag/AgCl +0,43 +0,37 60 +0,40
НВЭ +0,63 +0,57 60 +0,60
НКЭ +0,39 +0,33 60 +0,36
Fc/Fu+ +0,30 -0,30 60 +0,00
Оглавление
Предисловие ..„3
ГЛАВА 1. НЕСКОЛЬКО ОБЩИХ ВОПРОСОВ ОРГАНИЧЕСКОЙ
1.1. Образование двойного электрического слоя , , 14
1.2. Окислительно-восстановительные потенциалы... 15
1.3. Энергия активации и перенапряжение „ „ 17
1.4. Токи, управляемые переносом электрона и массопереносом , „ 18
ГЛАВА 2. ПРИНЦИПЫ ОРГАНИЧЕСКОЙ ЭЛЕКТРОХИМИИ.
НЕКОТОРЫЕ ФУНДАМЕНТАЛЬНЫЕ ВОПРОСЫ ЭЛЕКТРОХИМИИ
ОРГАНИЧЕСКИХ МОЛЕКУЛ 22
2.1. Рабочие электроды........... „,..„.„..... ..„...„.„..„..„..„......„. ..... 22
22. Электроды сравнения.. „..„ 25
2.3. Вспомогательные электроды....... ,..„ „ 26
2.4. Растворители и фоновые электролиты.... „ 27
2.5. Ячейки и источники питания.. ..28
2.6. Поляризационные кривые стационарного и нестационарного состояний „ 30
2.7. Потенциалы в электрохимических измерениях 34
2.8. Использование вольтамперометрии для изучения электроорганического
синтеза „ „ „ 37
2.8.1. Метод вольтамперометрии в селективном электросинтезе 37
2.8.2. Уточнение механизма реакции 39
2.8.3. Применение вольтамперометрии при выборе медиатора 40
2.8.4. Использование вольтамперометрии при выборе материала электродов.п43
ГЛАВА 3. МЕТОДЫ ИССЛЕДОВАНИЯ В ОРГАНИЧЕСКОЙ
ЭЛЕКТРОХИМИИ: ЭЛЕКТРОХИМИЧЕСКАЯ ОЦЕНКА
РЕАКЦИОННОЙ СПОСОБНОСТИ ОРГАНИЧЕСКИХ МОЛЕКУЛ 45
3.1. Выбор электролитических ячеек , , 45
3.2. Электролиз при постоянном токе или при постоянном потенциале.,... „..„..„„ 48
33. Прямой и непрямой электролиз „..„.... ,..„ „ „„„. „,.„..„,.„„„..„„„.„,.„..„50
3.4. Электродные материалы и электроды сравнения.- _ _ _ 51
3.5. Электролитические растворители и фоновые электролиты „... , „..„......„. „..„....53
3.6. Перемешивание.............. ,.„......„ „..„..„..„... 53
3.7. Контроль реагента и продукта „ .......,...„...„.„ „..„..... „... 53
3.8. Разработка, выделение и установление природы продуктов „......„..„.. 54
3.9. Выход по току и влияние источника тока .„„ 54
ГЛАВА 4. ОРГАНИЧЕСКИЕ ЭЛЕКТРОДНЫЕ РЕАКЦИИ 56
4.1. Общая характеристика электродных реакций ,..„...„ ...ч .„..„..„..„......„......„,.„....56
42. Механизм органических электродных реакций 58
4.3. Характеристики органических электрохимических реакций , 59
4.3.1. Обращение полярности.» „. 59
4.3.2. Селективность , , . 61
4.4. Молекулярные орбитали и электроны, связанные с переносом электрона 67
4.5. Электровспомогательные группы ,..„.„..... „. 69
435
45.1. Электровспомогательные группы, основанные
на молекулярных орбитальных взаимодействиях.,,.» „..„..„ , 69
45.2. Электровспомогательные группы на основе функциональных
групп с легким переносом электронов „„....., ,.„..„.„..,„..„.„.„ , 72
4.5.3. Электровспомогательные группы, основанные
на межмолекулярных координационных эффектах . 74
4.5.4. Электровспомогательные группы, основанные
на внутримолекулярных координационных эффектах.„.„..„..„..„........ „„„....75
4.6. Структура органических электродных реакций._ _ „ , , „ 76
4.6.1. Типы превращений функциональных групп „....„.„..„.......„., ..., 76
4.6.2. Тип присоединения ,.„ „ „ „ „...„ „ , „77
4.6.3. Тип включения „ 78
4.6.4. Тип замещения 73
4.6.5. Тип заместительного обмена „......„ „ _ „ „ 79
4.6.6. Тип элиминирования „..„ „ . . 80
4.6.7. Тип димеризации .. „ . .............. „ ВО
4.6.8. Кросс-димеризация ..................... „,.„.„ ,.„..„,.„.—„ „..„..,...„.—„.„..„ВТ
4.6.9. Циклизация „ „.„ „ „ „ „ В1
4.6.10. Образование полиморфизма 91
4.6.11. Тип полимеризации 92
4.6.12. Тип расщепления ~ В2
4.6.13. Тип металлироеания „ В2
4.6.14. Тип ассиметрического синтеза...... „...„„ ш ш _ „ „ В2
4.7. Электрохимически генерируемые реакционноспособные частицы» ..„ „..„....83
4.7.1. Углеродные частицы „ . » ,..„ „....83
4.7.2. Гетероатомные частицы .....„.„...„ „..„..„..„.„„.„..„.„„.„ „..„ „.„„.„..„..„..„....86
4.7.3. Халькогенные (сера, селен, теллур) частицы. „..„ .... „... „....90
ГЛАВА 5. ОРГАНИЧЕСКИЙ ЭЛЕКТРОСИНТЕЗ 92
5.1. Электрокатализ .„ . „„..... „.... „.„..„.......„.. ,..„..„ „....93
5.1.1. Классификация и типы медиаторов ..„..„.„,..„.„...,....,..„..„...„.„..„..„,.„......„......„..„..„93
5.1.2. Органические электролитические реакции с использованием
медиаторов . ,.. , 95
5.2. Электрогенерированные кислоты и основания , 100
5.2.1. Электрогенерированные основания „ „ 100
5.2.2. Электрогенерированные кислоты .„.. „.„......„...,..„..„ ...„ . 105
53. Электрохимический асимметрический синтез....„..„ - „..„. . - 107
5.4. Модифицированные электроды„,......„..,—„.,„.., ,.„..„,„„„..„,.„...„.„..„-.„..„.„„ „..,— 109
5.4.1. Электроды, модифицированные адсорбентами ,..„„,..,„..„,.„.„ .„. 110
5.4.2. Модифицированные электроды с инородными адатомами металла,..»—110
5.4.3. Химически модифицированные электроды... „.. .„ 111
5.4.4. Электроды, модифицированные (покрытые) полимером ...„„ „ „..,. 112
55. Парный электросинтез,... „.„, „ , „,..„.. „..„...„. „..„... „ 113
5.6. Реакционноспособные электроды - 117
5.7. Электрохимическое фторирование. ...„— _..„..... „.„..„..„ „..„......„...„.^.... 118
5.7.1. Электрохимическое фторирование ароматических колец - 120
5.7.2. Электрохимическое фторирование олефинов 121
5.7.3. Бензильное электрохимическое фторирование „ 122
5.7.4. Электрохимическое фторирование сульфидов „ „.„ „ „ 122
5.7.5. Электрохимическое фторирование гетероциклических соединений......... 123
5.7.6. Электрохимическое фторирование
гетероциклических соединений с PhS-группой в качестве
электровспомогательной .............. „,.„ „ ..„..„..„ ,—125
5.7.7. Электрохимическое фторирование с использованием
неорганических фторидных солей..„ m „ „ „„„. 127
436
5.8. Электрохимическая полимеризация. -,..„.„..„ „..„., ,.^..-„^^.„..„.„..^.^.„..~.„. 128
5.8.1. Электроокислительная полимеризация ароматических мономеров 129
5.8.2. Электрохимическая полимеризация „ „ ,„ ,.„,. 130
5.8.3. Условия электрохимической полимеризации.., .„., ,..„..„......„......,,..,. 131
5.8.4. Электрохимическое допирование „...,..„.... ........... ......... 132
5.8.5. Электровосстановительная полимеризация ароматических мономеров. 132
5.8.6. Применение проводящих полимеров „.„„ „ „ „ 133
5.8.7. Электрохимический синтез полисила нов _ _ „ _. 134
5.8.8. Цепная полимеризация, инициируемая
злектрогенерированными активными частицами 135
5.9. Электрокаталитический синтез фосфорорганических соединений 135
ГЛАВА 6. МЕТАЛЛОКОМПЛЕКСНЫЙ КАТАЛИЗ
В ОРГАНИЧЕСКОМ ЭЛЕКТРОСИНТЕЗЕ ♦ 146
6.1. Преимущества металлокомплексного катализа, индуцированного
электрохимически „..,.,.„ т, —„ „ ,„„,.„ .„—„т. 146
62. Общие принципы металлокомплексного электрокатализа и методы
исследования механизма реакций - „„ „ „ „ „„ ™ 148
63. Методы исследования механизма реакций, катализируемых переходными
металлами....... „„ ,..„.„,. , .. , _ „ „ „ ....„.., ,.„..„.„. 153
6.4. Реакции гомосочетания органических галоген ид ов „ „ . 155
6.4.1. Гомосочетание органических галогенидов „ , ™ ™... 155
6.4.2, Реакции кросс-сочетания органических галогенидов .,„..,..„,.„„.,...,„.„„„.„,.„. 162
65. Синтез соединений со связью металл — углерод _ - _. 169
6.6. Присоединение органических галогенидов к ненасыщенным группам . 176
6.6.1. Реакции присоединения к двойным и тройным связям С, С 176
6.6.2. Реакции присоединения к карбонильным соединениям . , 189
6.7. Электрокаталитическое восстановление диоксида углерода „ - , 198
6.8. Синтез карбоновых кислот,, .... , „ „„.., , „..„.203
6.8.1. Карбоксилирование органических галогенидов „ , 203
6.8.2. Карбоксилирование элкенильных и алкинильных соединений „ ,-„.205
6.9. Карбонилирование органических галогенидов .. 209
6.10. Электросинтез фосфорорганических соединений из хлорофосфинов
и белого фосфора , , , „..„ , 212
6.11. Электрохимический металлокомплексный катализ в реакциях
перфторалкилирования „. „ ........... ..... ,..,..„,..,. 215
6.11Л, Электрохимическое восстановление перфторалкилгалогенидов
под действием комплексов никеля и платины в низких
степенях окисления..................... ....„ , , .,.., .„.„.„..„.„ ^...,..„,„.215
6.11.2. Реакции фторалкилирования олефинов с использованием
иммобилизованного катализатора.„. „ 223
6.12. Некоторые другие случаи электрокатализа с участием металлокомплексов... 231
6.13. Закономерности и механизмы реакций, катализируемых комплексами
никеля и палладия 232
6.13.1. Кинетические закономерности ..,„—, ..,...,...„ ,...,..,..... «..» „..„.232
6.13.2. Связь электрохимических редокс-свойств комплексов никеля
с их реакционной способностью в реакциях дегалогенирования 234
6.13.3. Механизмы реакций, катализируемых комплексами никеля и палладия.236
6.13.4. Механизмы и кинетика палладиевых каталитических систем „„.250
ГЛАВА 7. ЛИГАНД-НАПРАВЛЕННАЯ ЭЛЕКТРОХИМИЧЕСКАЯ
ФУНКЦИОНАЛИЗАЦИЯ C{SP2)-H-CBfl3EU В ПРИСУТСТВИИ
СОЕДИНЕНИЙ ПАЛЛАДИЯ И НИКЕЛЯ 258
7.1. Электрохимическое орто-ацетоксилирование и фтороацетоксилирование
2-фенилпиридина, катализируемое палладием в высших состояниях
окисления , - „ . 262
437
7.2. Каталитическое С-Н-замещение с участием электрохимически
генерированного 1Ч1(Ш)-комплекса....„ „.., .. „..„..™. „..„..„..., „......„..„. 274
7.3. Электрохимическое С-Н-фосфорилирование....„ „ ....„ 277
ГЛАВА 3. ОЦЕНКА ЭФФЕКТИВНОСТИ ГОМОГЕННЫХ
ЭЛЕКТРОКАТАЛИЗАТОРОВ С ПРИМЕНЕНИЕМ МЕТОДА
ЦИКЛИЧЕСКОЙ ВОЛЬТАМПЕРОМЕТРИИ 285
8.1, Введение , , „ , „ „ „. „ 295
82. Электрохимические методы . _ . . „ . 287
8.2.1. Циклическая вольтамперометрия . 288
8.2.2. Экспериментальные соображения „ „„ .... ,.„ ,..„ „.„. 290
83. Оценка гомогеннных электрокатализаторов ...... , „ - . 298
8.3.1. Основные определения и показатели 299
83-2. EfCJ-Кэталитический механизм и идеализированные отклики ЦВА„-.«««-300
8.3.3. Определение критериев из ЦВА ~ „ „„ „ ........... 304
8.3.4. Примеры „ 330
8.4. Заключение..... „ „..„ ............ ............ ............. ........................... 336
ГЛАВА 9. НОВАЯ МЕТОДОЛОГИЯ ОРГАНИЧЕСКОГО
ЭЛЕКТРОХИМИЧЕСКОГО СИНТЕЗА 337
9.1. Электролиз с использованием твердых полимерных электролитов (SPE)
и его применение ............. ....,..„.,....,..... „.^. ^.„„.,..м„.,..„„„...„. „..„..„...,..,....,....,....337
9.1.1. Принцип SPE-злектроли» „ „ 338
9.1.2. SPE-электролиз с когенерацией (химическое производство
с использованием реакций, протекающих в топливных элементах) , 339
92. Электролитические системы с использованием твердых оснований и кислот,.„. 340
9.3. Медиаторы на твердой подложке , 342
9.4. Двухфазные электролитические системы........ , , _ „ _. 344
9.4.1. Электролиз в эмульсии...» „ . „.. . 344
9.4.2. Электролиз суспензии —„.„...„..„..„..„.., „,.„..»„.„.„..„.„......„.....„™.345
9.4.3. Пример электролиза с использованием межфазного катализа ... 346
9.4.4. Термоморфная двухфазная электрохимическая реакционная система 347
95. Метод катионного пула , .... -..., „ , . ......347
9.6. Методы, управляемые темплатом „......„ „,... . 350
9.7. Электролиз в сверхкритических жидкостях . .. , ... „— 350
9.8. Электролиз в ионных жидкостях., „......„..„...,..., ,..„..„, .„„......„... „..„ 355
9.8.1. Структуры ионных жидкостей 355
9.8.2. Гидрофильность и гидрофобность ионных жидкостей 356
9.8.3. Полярность ионных жидкостей...» „ ............ 358
9.8.4. Электрохимические свойства ионных жидкостей 358
9.8.5. Вольтамперометрия в ионных жидкостях „ „„..., „..„ „..„ ......364
9.8.6. Органические электрохимические реакции в ионных жидкостях 364
9.9. Тонкослойные электролизеры ,..„ „ „.., ... , „ ......376
9.10. Электрохимические микропоточные системы (MicroHow) . 378
9.11. Электролиз в ультразвуке 383
9.12. Электросинтез с использованием специфических электродных материалов........ 387
9.12.1. Электрохимический синтез с использованием гидрофобных
электродов , ......387
9.12.2. Электролитические реакции с использованием алмазных электродов — ЗВ9
9.13. Фотоэлектролиз и фотокатализ... - -.„ .. „ „ „..„. 393
9.13.1, Фотоэлектролиз , 393
9.13.2. фотокатализаторы ...„ „ ...... 394
9.14. Электрохимические полимерные реакции„ „ „ „ 395
438
ГЛАВА 10. ОБЛАСТИ, СВЯЗАННЫЕ С ОРГАНИЧЕСКОЙ
ЭЛЕКТРОХИМИЕЙ 398
10.1. Применение в органических электронных устройствах „ 398
10.1.1. Органическая электролюминесценция.„....„ , ,.„ „ 398
10.1.2. Органические фотовольтаичеекие ячейки... , .„ „,.„ „ _.4О0
10.1.3. Солнечные элементы, сенсибилизированные красителем»..».»......» „.„.401
10.1.4. Органические транзисторы......... . . .... . .... ......402
10.1.5. Электрохромные устройства . , . 402
10.1.6. Конденсаторы на основе проводящих полимеров..... - , 403
10.2. Электрохимическая конверсия биомассы в ценные материалы. ,...„..,...., , 404
10.3. Приложение к С1 -химии ...„ ...„.„ „. ..... ...... „„ .............. 406
10.4. Экологическая очистка 407
ГЛАВА 11. ОБЛАСТИ, СВЯЗАННЫЕ С ОРГАНИЧЕСКОЙ
11.1. Дорога к индустриализации - „ „ _ 410
11.2. Примеры- , .. „ 411
11,2.1, Электросинтез адипонитрила .. „ „ 411
112.2. Электросинтез ароматических альдегидов „ „, 413
11,2.3, Парный электросинтез фталида и трет-бутилбензальдегида „ „..,.414
112.А. Электрохимическое перфорирование._._ _ _ 41S
11.2.5. Другие примеры - „.„ .......417
Примеры коммерциализированных органических электродных процессов 420
АЛ, Электрохимическое фторирование „...„„.... 420
А.2. Электросинтез с использованием гидрофобного электрода 421
А.З. Синтез натуральных продуктов с использованием анодного окисления 423
А.4. Электролиз Кольбе .. ......— „ „ „ ...425
A.S. Непрямой электросинтез с использованием медиатора ..„„..„.„ ....„..,..„..-.425
А.б. Электросинтез проводящих полимеров 426
Прилдоение В т.«,...,.«.„»............*„....*...,..^^
Примеры органического электросинтеза _ ...428
439
По вопросам приобретения книг обращайтесь:
Отдел продаж «ИНФРА-М» (оптовая продажа):
127282, Москпа, ул, Полярная, л. 31В, стр.1
Тел, (495) 280-15-96; факс (495) 280-36-29
E-mail: books@infra-m.ru
т
Отдел «Книга—почтой»:
тел, (495) 280-15-% (доб. 246.)
Научное издание
Будникова Юлия Германовна
СОВРЕМЕННЫЙ ОРГАНИЧЕСКИЙ
ЭЛЕКТРОСИНТЕЗ
ПРИНЦИПЫ, МЕТОДЫ ИССЛЕДОВАНИЯ И ПРАКТИЧЕСКИЕ
ПРИЛОЖЕНИЯ
МОНОГРАФИЯ
Оригинал-макет подготовлен в НИЦ ИНФРА-М
ООО «Научно-издательский центр ИНФРА-М»
127282t Москва, ул. Полярная, д. 31 В, стр. 1
Тел.: (495) 280-15-96, 280-33-86. Факс: (495) 280-36-29
b-mail: books@infra-m.ni littp://w\vwinfra-m.ru