






































































































































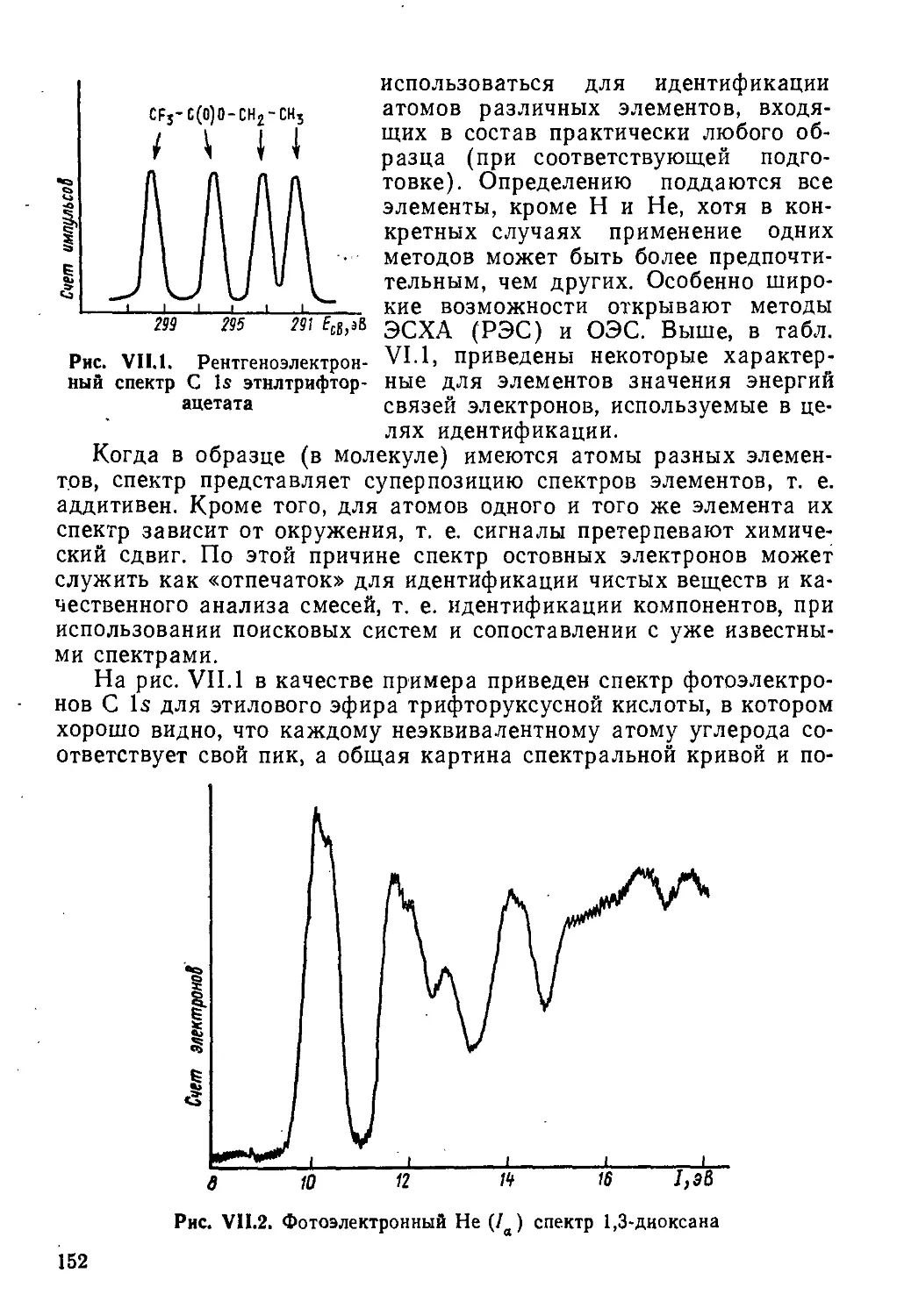





























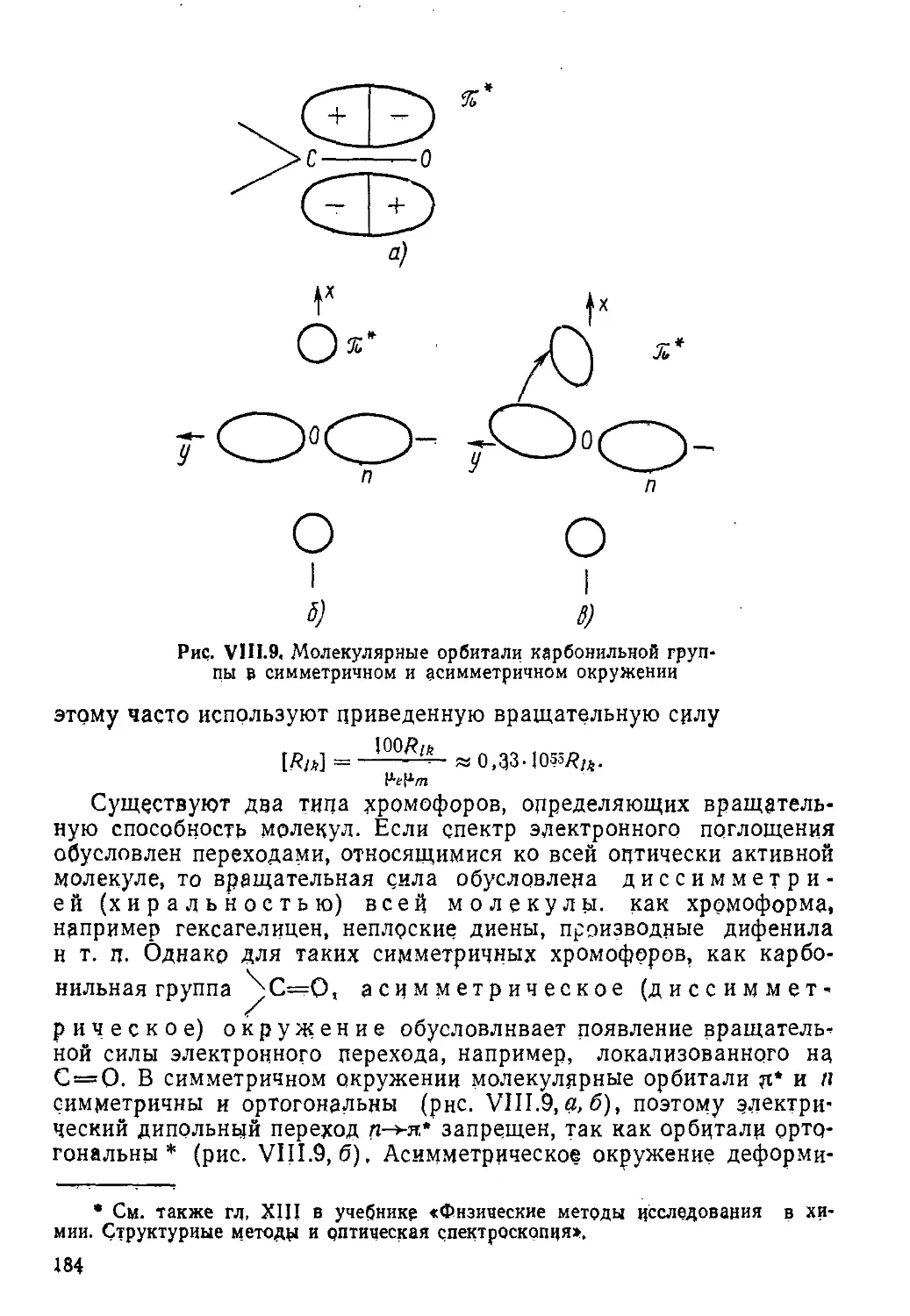


















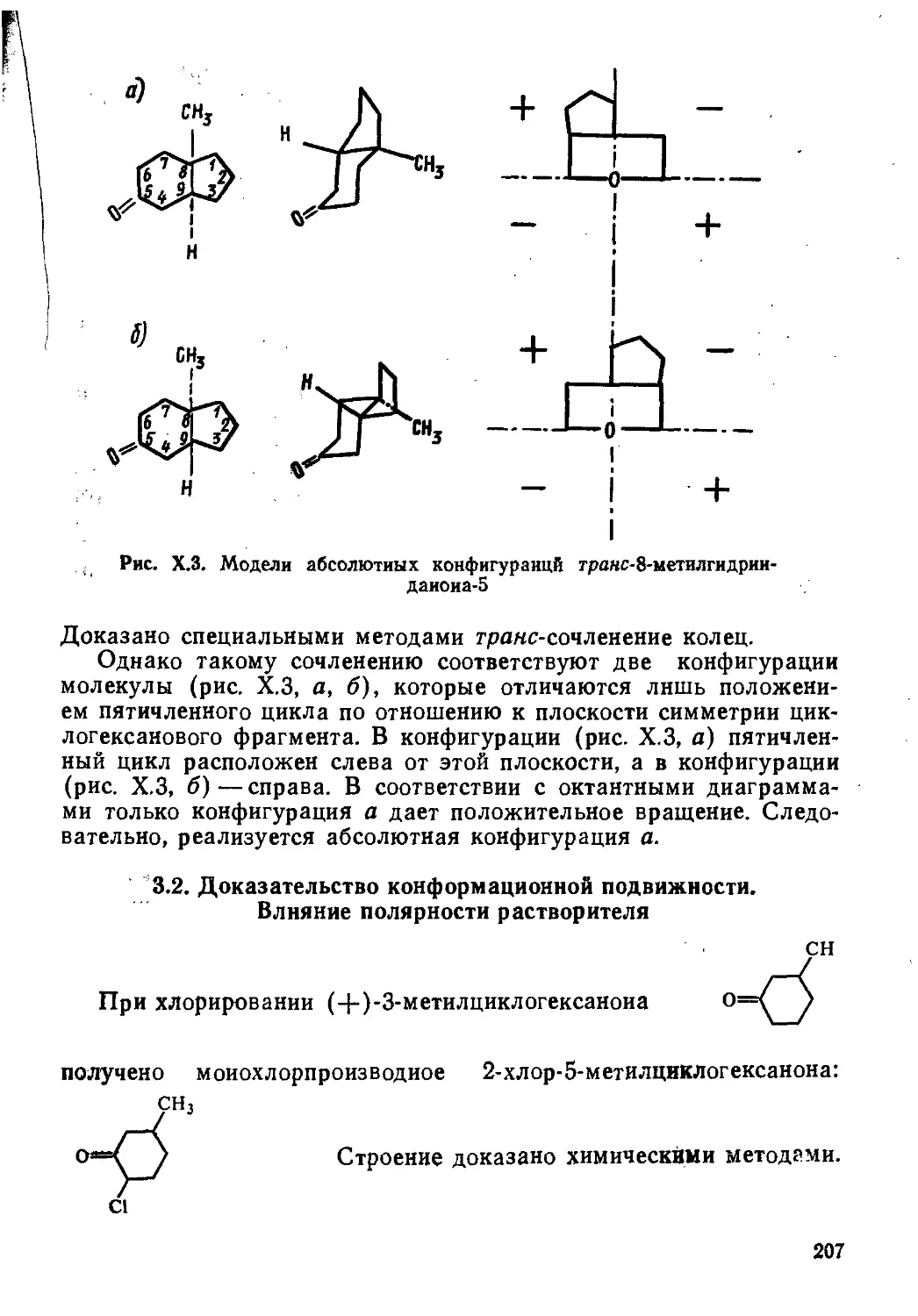
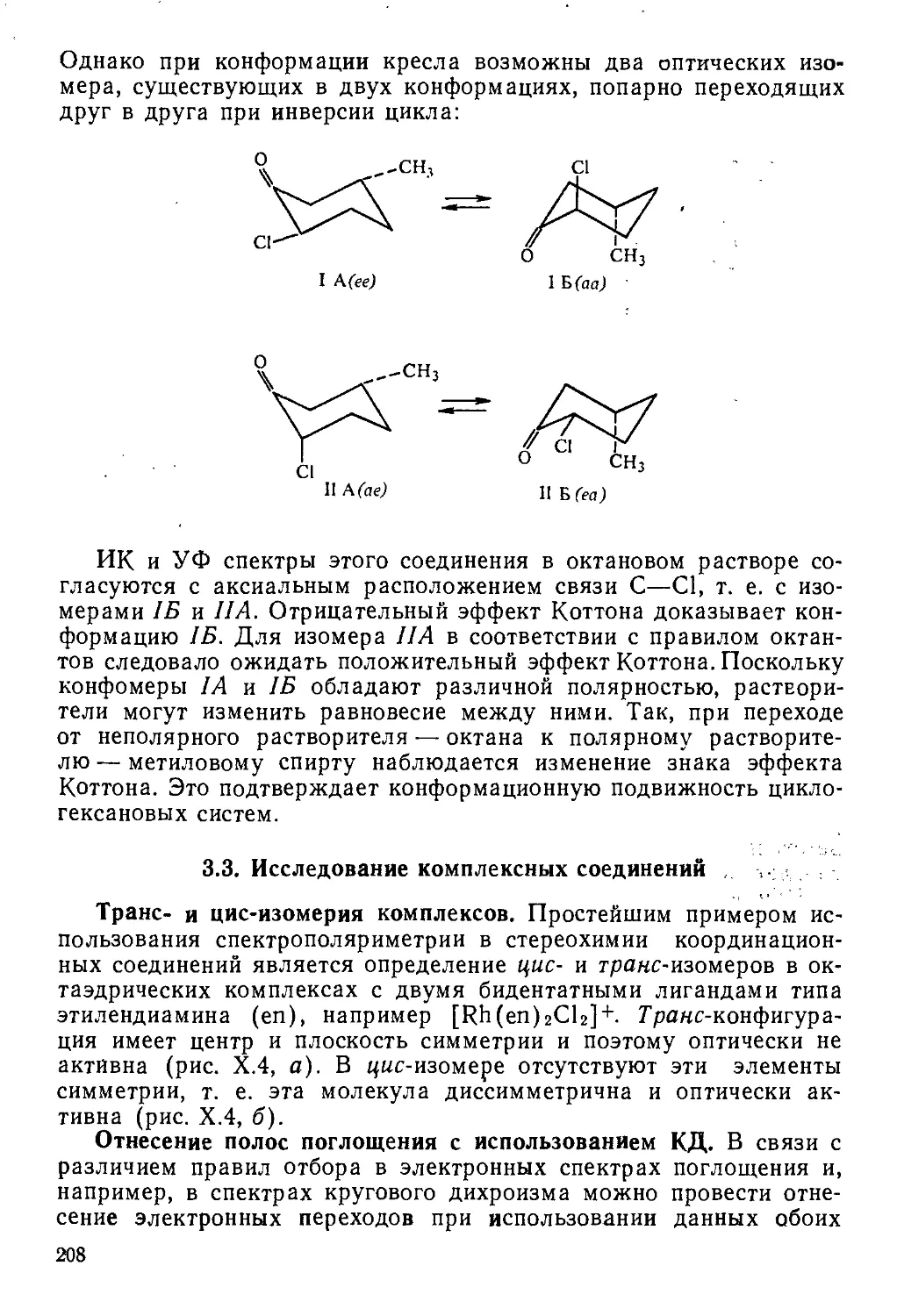





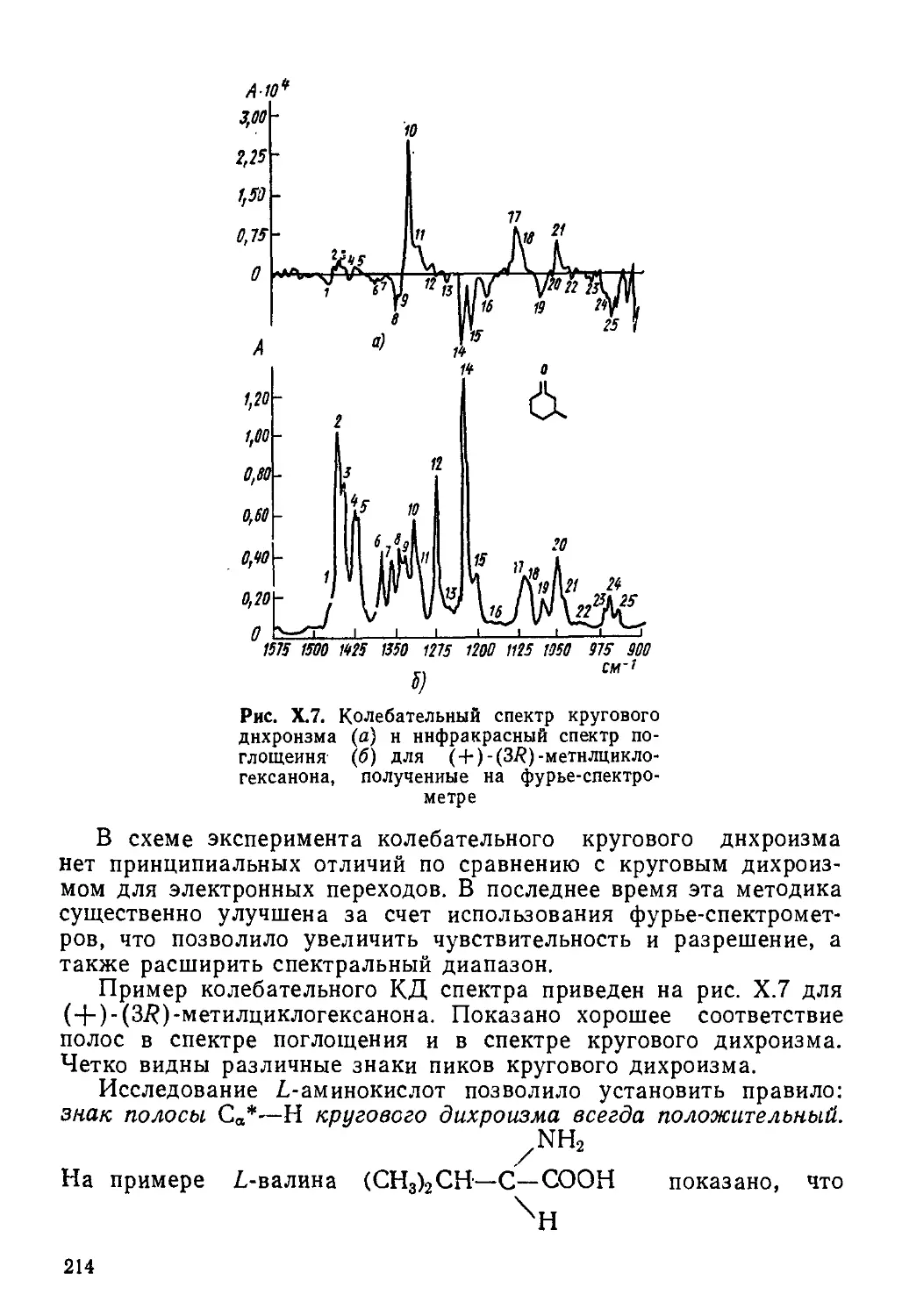
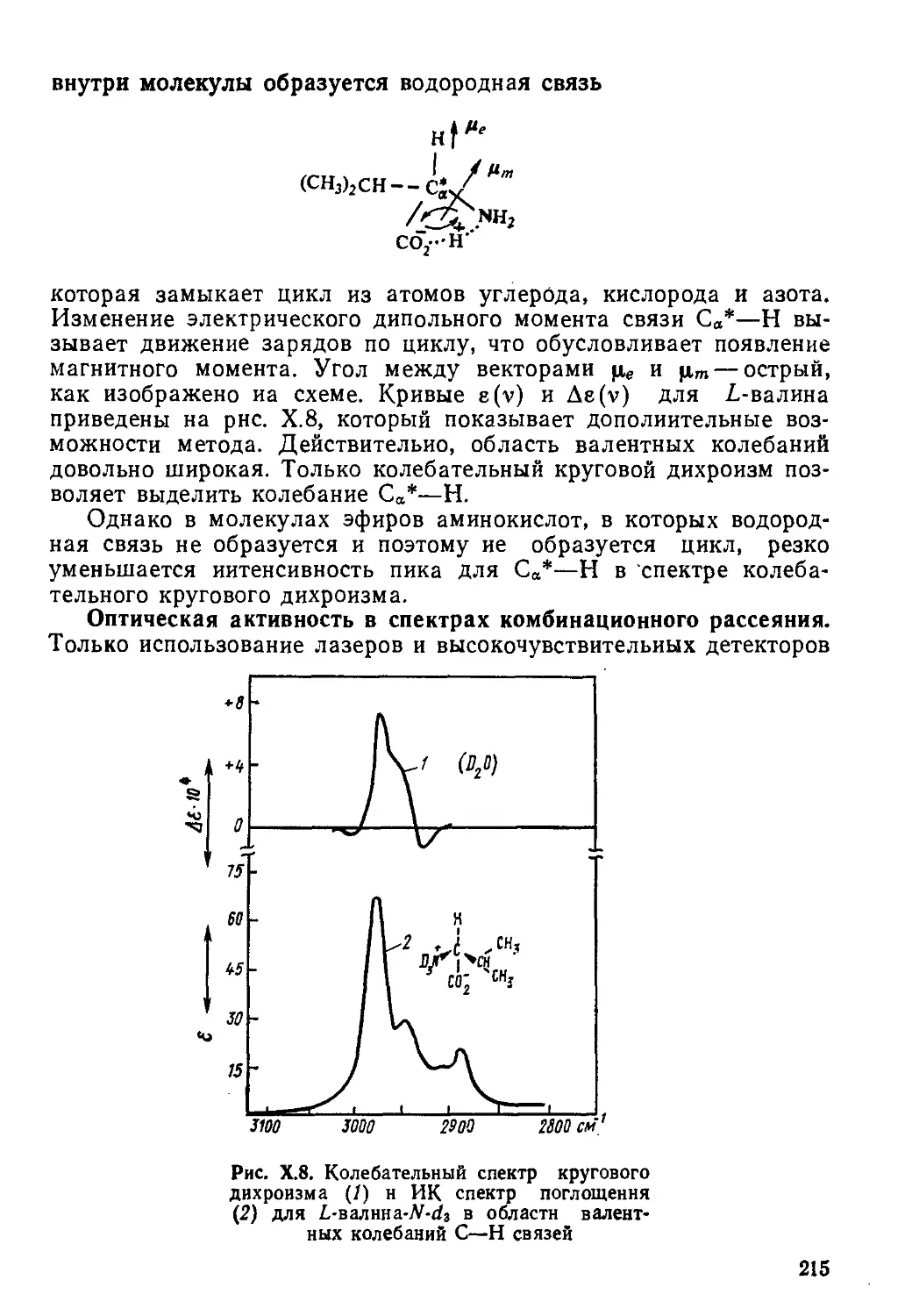









































































Author: Пентин Ю.А. Вилков Л.В.
Tags: химия физическая химия химическая физика физика переводная литература издательство высшая школа междисциплинарные методы
ISBN: 5-06-00071-0
Year: 1989




Л. В. Вилков Ю.А. Пентин
ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
В ХИМИИ
Резонансные и электрооптические методы
Допущено Государственным комитетом СССР по народному образованию в качестве учебника для студентов химических специальностей высших учебных заведений
Москва «Высшая школа» 1989
ББК 24.5 В44
УДК 541.1
Рецензенты: кафедра физической химии Казанского государственного университета (зав. кафедрой проф. Ю. М. Каргин) и проф. В. И. Мннкин (НИИ физической и органической хнмин, Ростовский государственный университет)
Вилков Л. В., Пентин Ю. А.
В44 Физические методы исследования в химии. Резонансные и электрооптические методы: Учеб, для хим. спец, вузов.— М.: Высш, шк., 1989. — 288 с.: ил.
ISBN 5-06-00071-0
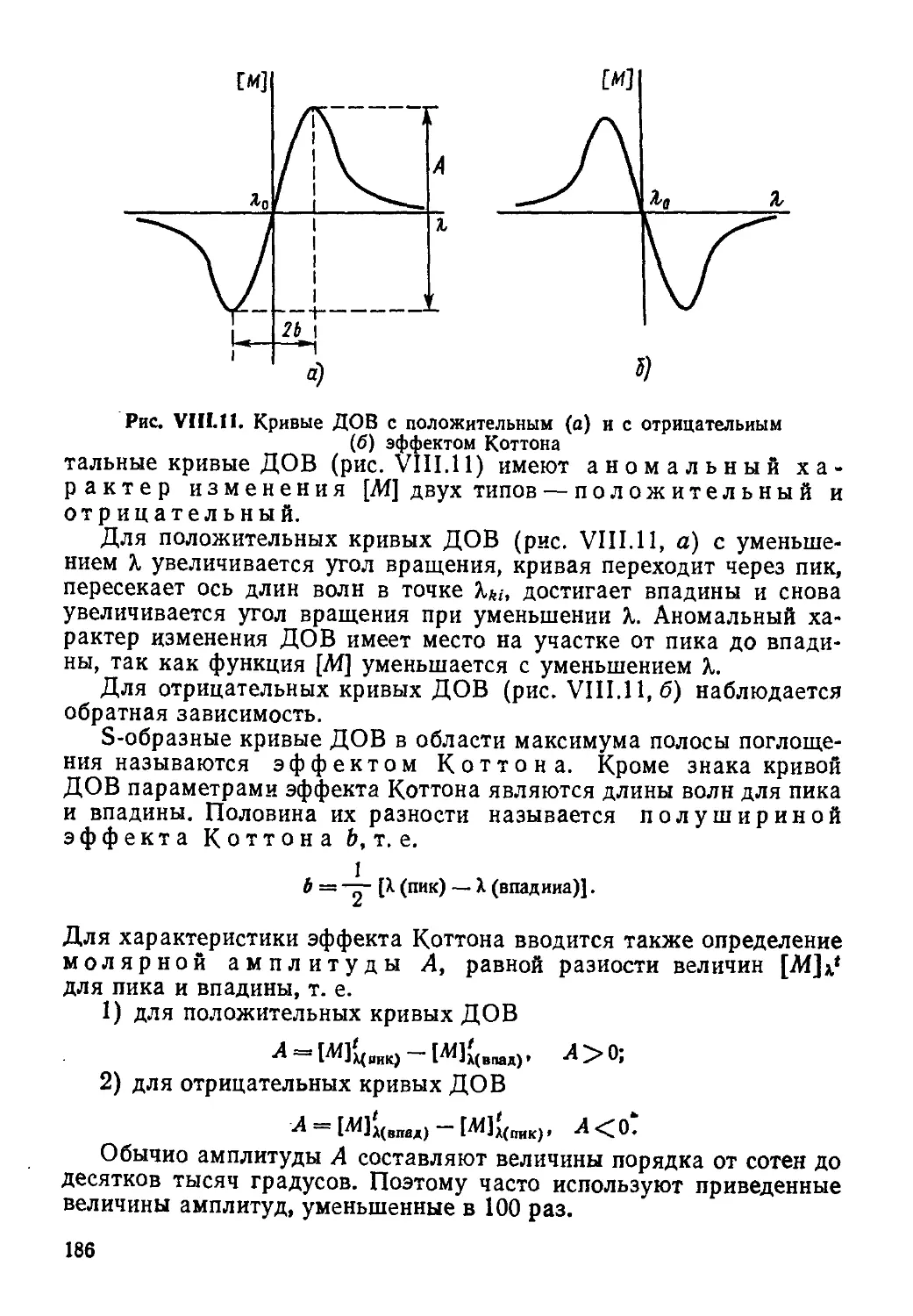
В учебнике представлены методы ядерного магнитного резонанса, электронного парамагнитного резонанса, ядерного квадрупольного резонанса, мессбауэровской, рентгеновской, фотоэлектронной и оже-спектроскопин, дисперсии оптического вращения, кругового дихроизма, аномального рассеяния рентгеновских лучей, эффекты Керра и Фарадея.
Последовательно рассматриваются физические основы методов, схемы экспериментов и условия их проведения, анализируются возможности методов, их взаимосвязь и совместное использование для определения физических параметров молекул и веществ, приводятся примеры применения методов в химических исследованиях.
1708000000(4309000000) —533 ББК 24.5
В 001(01)—89 И7—89
Учебное издание
Вилков Лев Васильевич Пентин Юрий Андреевич
ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ В ХИМИИ. РЕЗОНАНСНЫЕ И ЭЛЕКТРООПТИЧЕСКИЕ МЕТОДЫ
Зав. редакцией С. Ф. Кондрашкова. Редактор В. Н.Бораненкова. Мл. редакторы С. М. Ерохина, Л. С. Макаркина. Художественный редактор Е. Д. Косырева. Технический редактор Н. А. Битюкова. Корректор С. К. Завьялова
ИБ № 7406
Изд. № ХИМ-879. Сдано в набор 17.05.89. Подп. в печать 13.11.89. Формат 60Х88'/1«. Бум. офс. кн.-жури. Гарнитура литературная. Печать офсетная. Объем 17,64 усл. печ. л. + фора. 0,25 усл. печ. л. 17,89 усл. кр.-отт. 18,10 уч.-изд. л. + форз. 0,29 уч.-изд. л. Тираж 8700 экз. Зак. ЛЬ 1377. Цена 95 коп.
Издательство <Высшая школа», 101430, Москва, ГСП-4, Неглииная ул., д. 29/14.
Московская типография № 8 Госкомпечати СССР. 101898, Москва, Центр, Хохловский пер., 7.
ISBN 5-06-00071-0
© Л. В. Вилков, Ю. А. Пентин, 1989
ПРЕДИСЛОВИЕ
Данная книга является продолжением учебника «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия» (М., Высшая школа, 1987, в дальнейшем для краткости мы будем ссылаться на часть I), где были рассмотрены многие общие вопросы, в частности, касающиеся прямых и обратных задач, временной шкалы (характеристического времени).
Здесь изложены методы, основанные на изучении магнитных свойств и электромагнитных взаимодействий ядер и электронов, оптической активности веществ, свойств электронных оболочек (распределение электронной плотности в молекулах).
Авторы поставили перед собой задачу познакомить с основами теории, особенностями эксперимента, возможностями и ограничениями физических методов. Это позволит грамотно выбирать и использовать тот или иной метод или группу методов для решения конкретных проблем. В учебнике приведена учебная, монографическая и справочная литература.
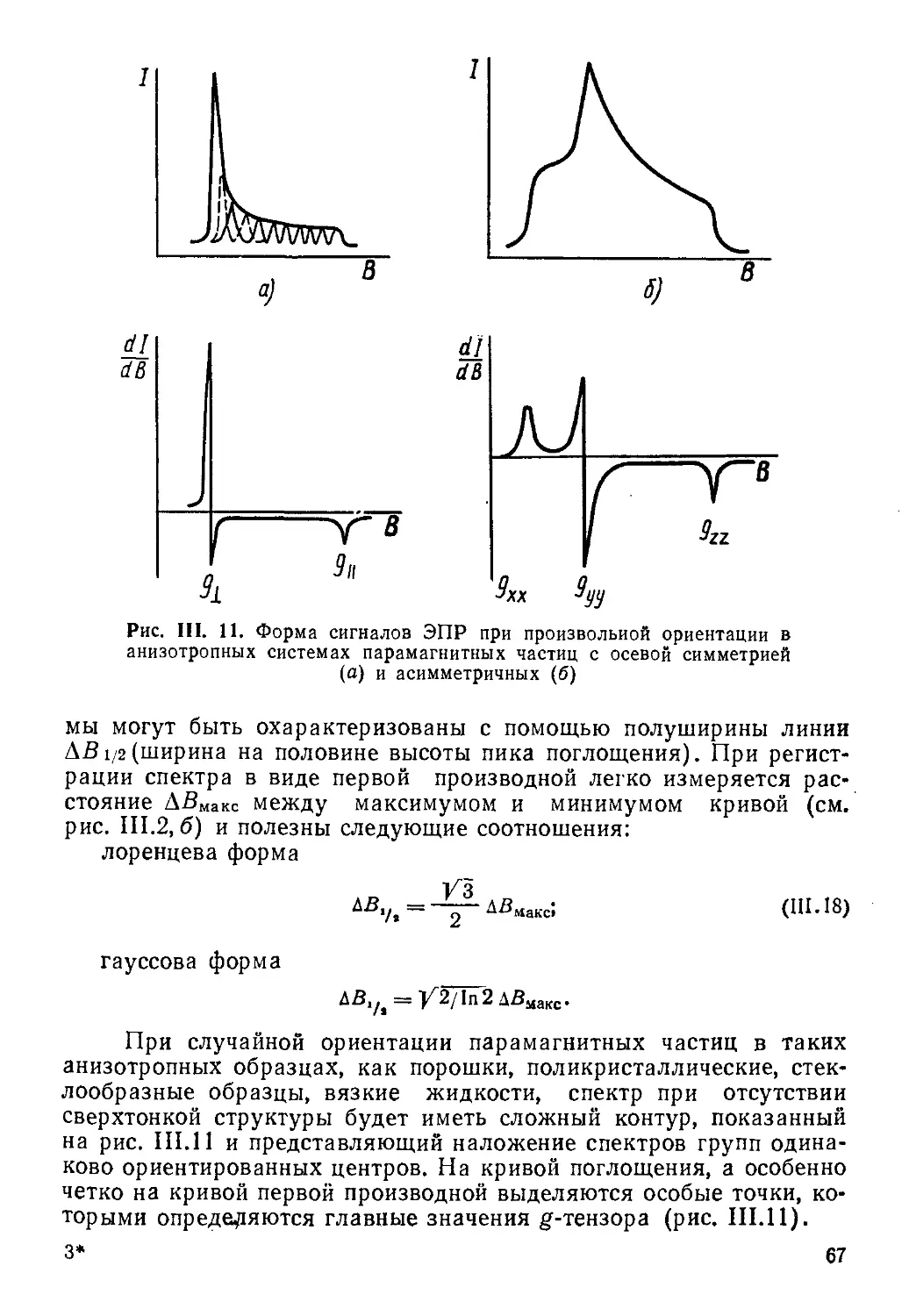
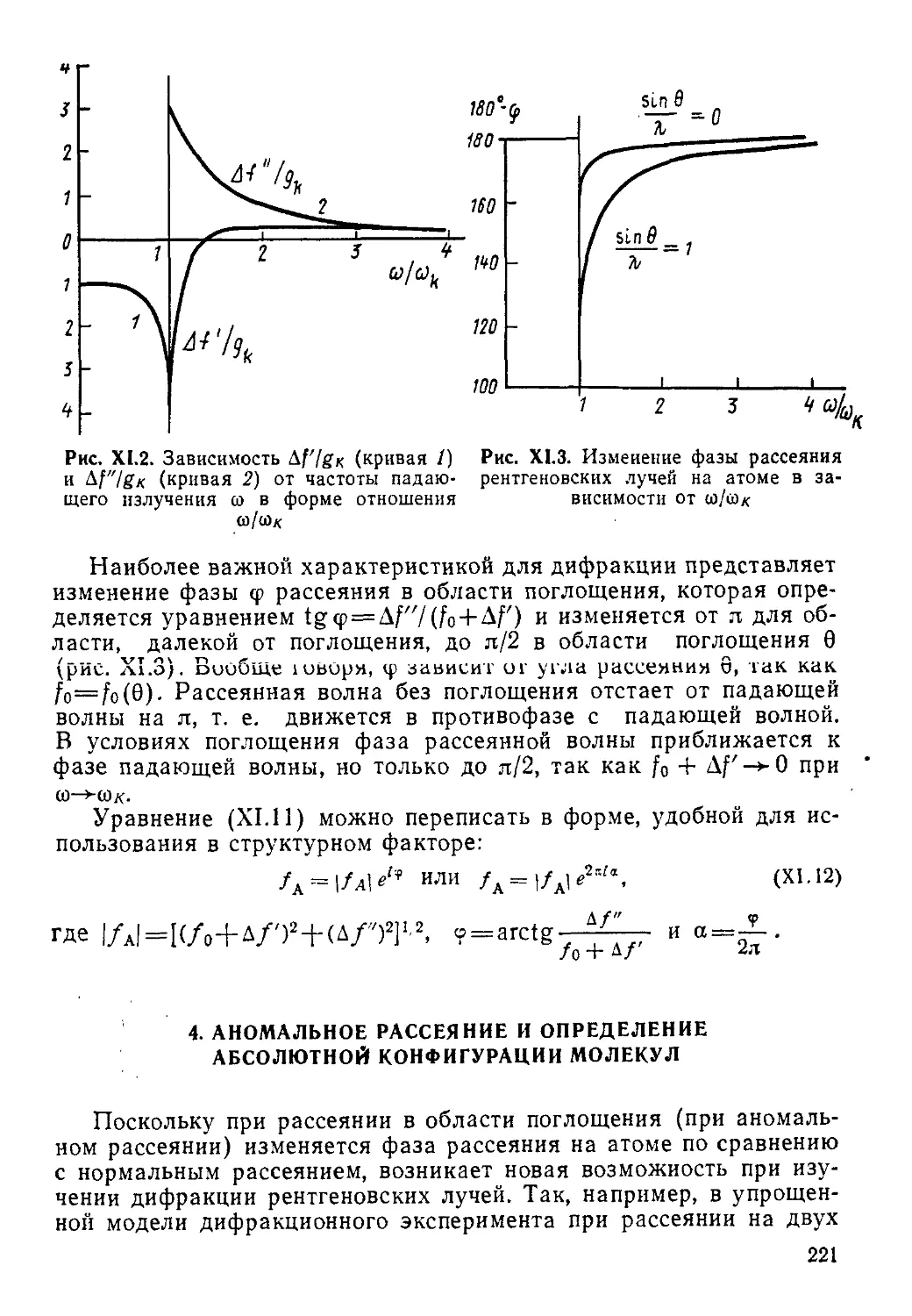
Все резонансные методы, рассмотренные в данном учебнике, относятся в сущности к спектроскопическим и основываются на изучении переходов между какими-то энергетическими состояниями системы при ее взаимодействии с каким-либо полем или излучением.
Различия между методами заключаются, во-первых, в характере состояний, а во-вторых, в диапазоне частот (разностей энергий между этими состояниями).
Так, в случае спектроскопии ЯМР и ЭПР уровни энергии, между которыми происходят переходы, появляются у системы только во внешнем магнитном поле (зеемановские уровни), т. е. не являются ее собственными. В то же время в спектроскопии ЯКР и ядерного гамма-резонанса (ЯГР или мессбауэровской спектроскопии) так же, как во всех методах оптической спектроскопии, соответствующие энергетические состояния ядер или вообще систем являются их собственными, т. е. существуют как свойство системы без наложения внешних полей.
Важно отметить, что переходы с частотами, отличающимися для разных методов иногда на много порядков, как в резонансных методах (ЯМР, ЭПР, ЯКР, ЯГР)> зависят от строения химических частиц (электронной структуры, окружения ядра, геометрии ядерного скелета, симметрии и т. д.), что больше всего и представляет интерес для химика.
3
Некоторые из физических методов особенно широко используются в химических лабораториях, например спектроскопия ЯМР и ЭПР, спектрополярнметрия (ДОВ и КД), и поэтому они рассмотрены подробнее. В то же время с помощью менее распространенных методов, таких, как рентгеновская и фотоэлектронная спектроскопия (ФЭС), ядерный квадрупольный резонанс, мессбауэровская спектроскопия, эффект Фарадея и др., получают также чрезвычайно важную информацию, поэтому некоторые из этих методов стали быстро развиваться, например ФЭС, и применение их химиками постоянно расширяется. Вообще ценность любого метода проявляется только тогда, когда он применяется для решения конкретных химических задач, и особенно возрастает при совместном использовании с другими методами.
В приложении приведены оглавление учебника «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия», принятые обозначения величин и часть основных формул и уравнений.
Предисловие и заключение написаны авторами совместно, первый — третий разделы — Ю. А. Пентиным, четвертый и пятый — Л. В. Вилковым.
Авторы выражают свою искреннюю благодарность рецензентам | А. Н. Верещагину!, А. В. Ильясову, Ю. М. Каргину, В. И. Минкину за внимательное прочтение рукописи и ряд ценных указаний, а также В. С. Гурману, Н. М. Сергееву, А. Ю. Пентину за сделанные ими по отдельным главам замечания, что способствовало улучшению книги.
Авторы
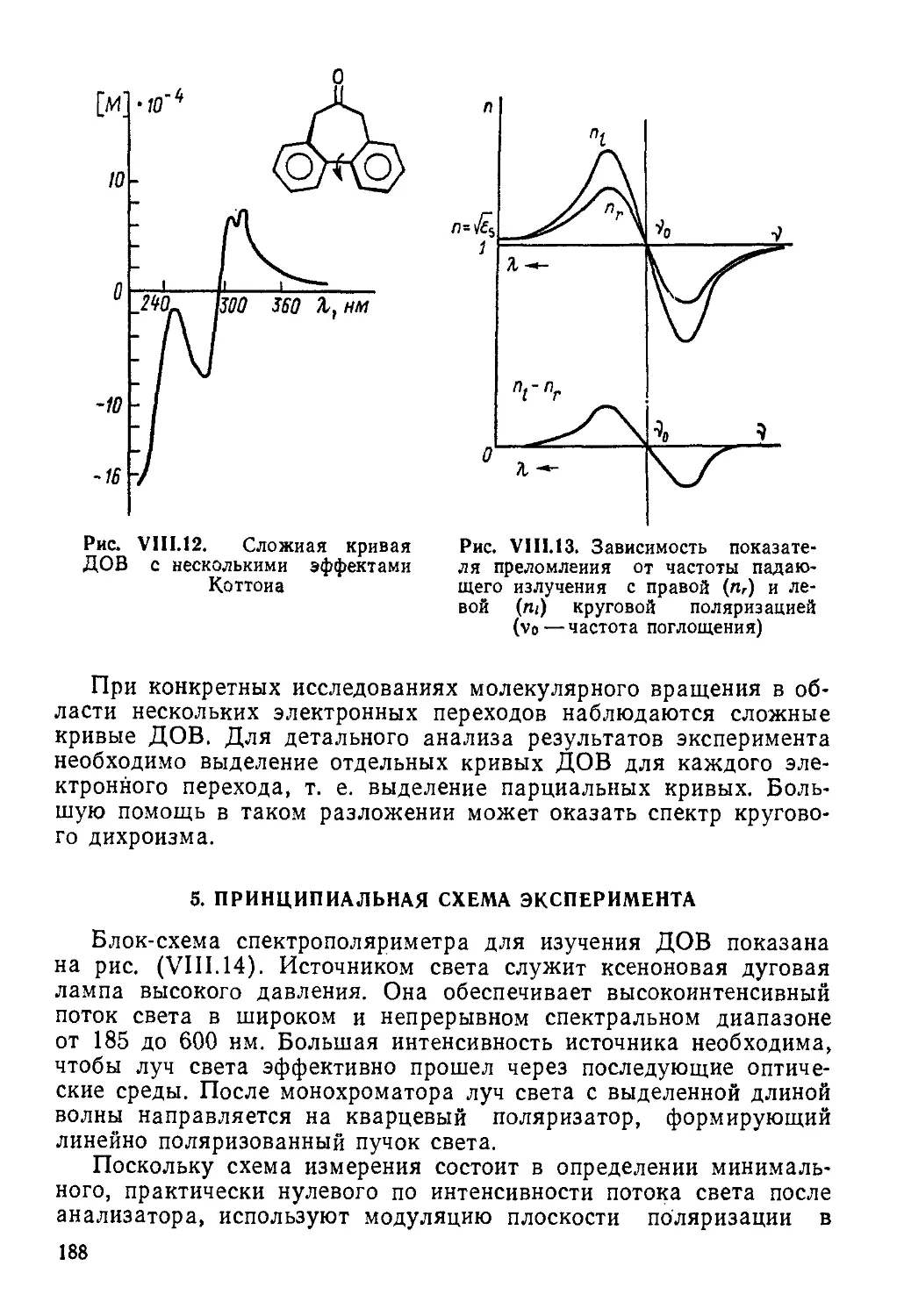
МЕТОДЫ МАГНИТНОГО РЕЗОНАНСА ЯДЕР И ЭЛЕКТРОНОВ
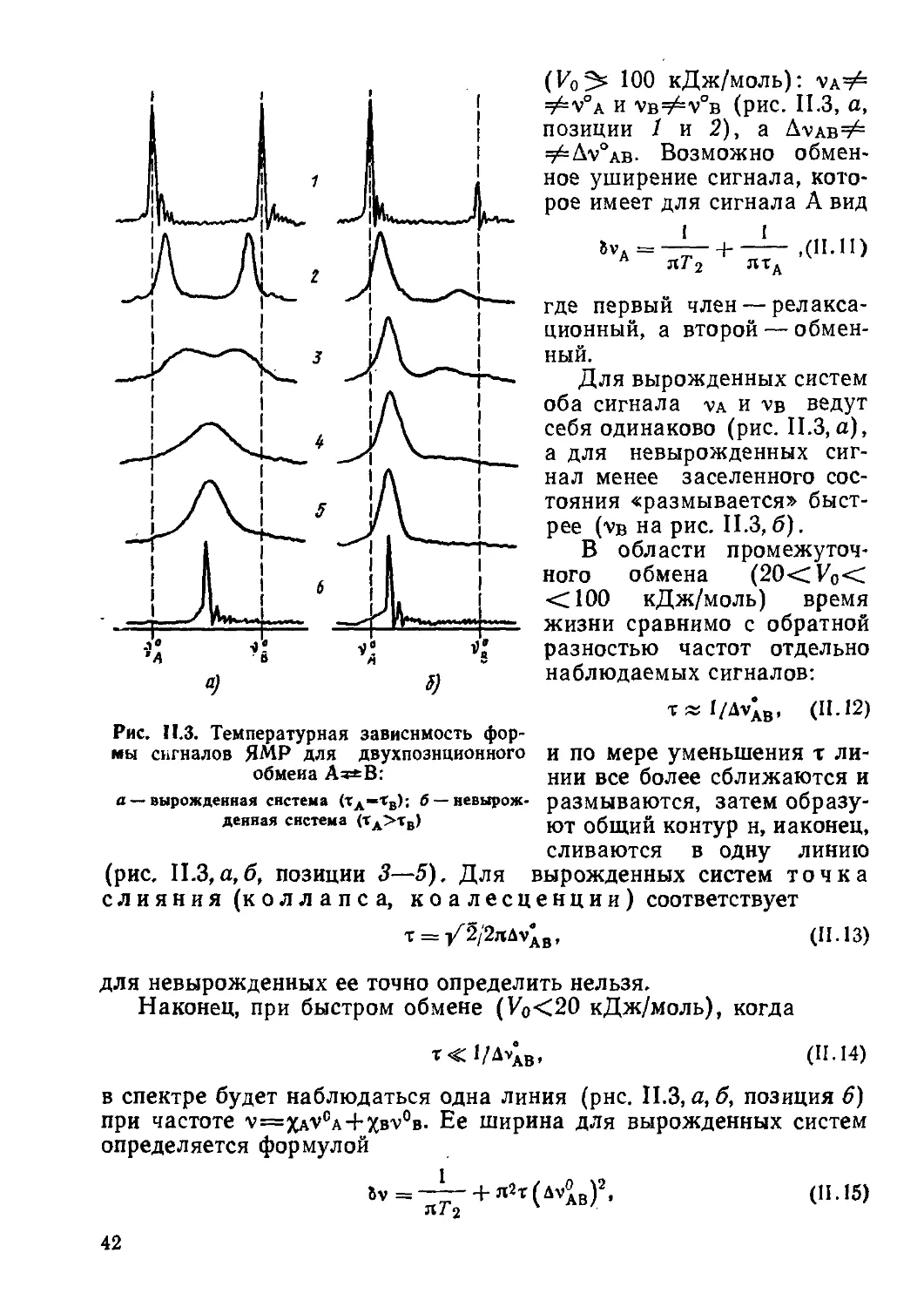
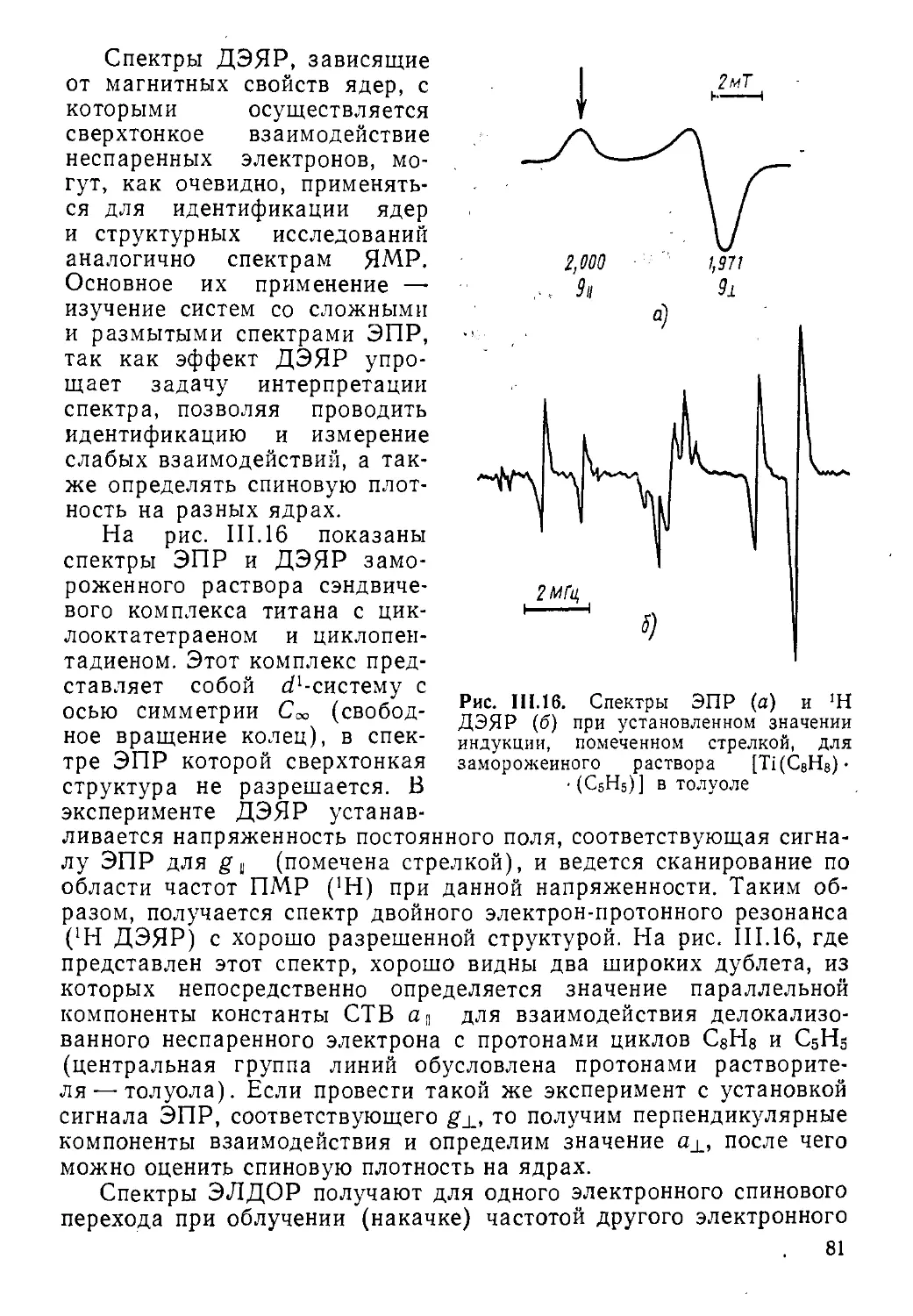
Ядерный магнитный резонанс (ЯМР) и электронный парамагнитный резонанс (ЭПР) — два метода радиоспектроскопии, позволяющие изучать структуру и динамику молекул, радикалов, ионов в конденсированных и газовой фазах вещества. Спектры ЯМР обладают высокой специфичностью и широко применяются для идентификации соединений, в структурно-аналитических целях, а также для изучения быстрых обменных процессов. Спектроскопия ЭПР — метод исследования парамагнитных частиц и центров, кинетики и механизмов процессов, происходящих с их участием. Особенно большой прогресс в развитии методов спектроскопии ЯМР и ЭПР, достигнутый в последние годы, связан с появлением импульсных фурье-спектрометров, двухмерной спектроскопии и техники множественного ядерного, электрон-ядерного и электрон-электрон-ного резонанса.
Атомные ядра и электроны, имея определенный электрический заряд, могут обладать и некоторым магнитным моментом, причем у ядра он примерно на три порядка меньше, чем у электрона. Молекула как система, состоящая из этих заряженных частиц, также может характеризоваться вектором магнитного момента, который связан главным образом с орбиталь
ным и спиновым движениями электронов. Еще одной характеристикой молекулы является тензор магнитной восприимчивости. Этими свойствами и определяются явления, происходящие при нахождении молекулы в магнитном поле. К важнейшим физическим методам исследования, связанным с изучением результатов взаимодействия молекул вещества с постоянным и переменным внешними магнитными полями, относятся методы радиоспектроскопии ЯМР и ЭПР.
Открытие эффектов магнитного резонанса произошло в середине 40-х годов. В 1944 г. советский физик Е. К. Завойский впервые наблюдал поглощение электромагнитных радиоволн парамагнитным веществом, т. е. ему принадлежит заслуга создания метода ЭПР. Большой вклад в развитие этого метода внесли в дальнейшем также Б. М. Козырев, Д. Ингрэм и многие другие советские и зарубежные ученые. Что касается изучения переходов между ядерными зеемановскими уровнями в магнитном поле и разработки метода ядерного, в частности, протонного магнитного резонанса (ПМР) в конденсированных средах, то первыми в 1946 г. это независимо сделали американские физики Ф. Блох и Э. М. Парселл со своими сотрудниками. Конструирование и серийный выпуск промышленностью ПМР-спектрометров относится к середине 50-х, а ЭПР-спектрометров — к середине 60-х годов. Для спектроскопии ЯМР на других отличных от протонов ядрах приборы высокого разрешения стали производиться в 60—70-х годах. Бурное развитие и совершенствование экспериментальных и расчетных методов ЯМР и ЭПР на базе современной техники и ЭВМ за последние десятилетия привело к широкому и плодотворному их внедрению в химические исследования.
Спектроскопию ЯМР по своему распространению можно сравнивать, пожалуй, только с ПК спектроскопией, хотя круг исследуемых методов ЯМР объектов несколько ограничен, так как не все ядра обладают магнитным моментом. Спектры ЯМР высоко характеристичны и по своей неповторимости, особенно при резонирующем ядре 13С, сравниваются, как и колебательные спектры, с отпечатками пальцев. Это связано с тем, что вид и характеристики спектра ЯМР зависят от взаимодействия ядер и электронов, т. е. от структуры всей молекулы.
6
Спектроскопия ЭПР применяется не столь широко, так как этим методом могут исследоваться лишь объекты, обладающие парамагнитным моментом, т. е. частицы (молекулы, радикалы, ионы и др.) с неравным нулю суммарным электронным спином, парамагнитные центры в кристаллах и т. д. При наличии эффекта ЭПР из спектра получают ценнейшую информацию о структуре и динамике изучаемых систем. Этим методом решают разнообразные задачи химической кинетики от выяснения механизмов простых свободно-радикальных реакций до изучения сложных биологических процессов и многие другие структурно-аналитические задачи.
Хотя методы ЯМР и ЭПР основываются, вообще говоря, на одних и тех же принципах изучения резонансных переходов между. зеемановскими уровнями спиновых систем, количественные различия в абсолютных значениях магнитных моментов и их знаках, а также различный характер изучаемых объектов и решаемых задач обусловливают то, что эти методы развивались практически независимо и имеют существенные отличия в теории и экспериментальном воплощении. В то же время есть ряд аспектов, где явления ядерного и электронного магнитного резонанса тесно переплетаются. Это прежде всего методы множественного резонанса, например двойного электрон-ядерного резонанса (ДЭЯР). Проще рассматривать совместно также химическую поляризацию ядер и электронов и т. д.
ГЛАВА I
СПЕКТРОСКОПИЯ ЯМР (ОСНОВЫ ТЕОРИИ)
1. ФИЗИЧЕСКИЕ ПРИНЦИПЫ МЕТОДА
1.1. Магнитный момент ядра и его взаимодействие с магнитным полем
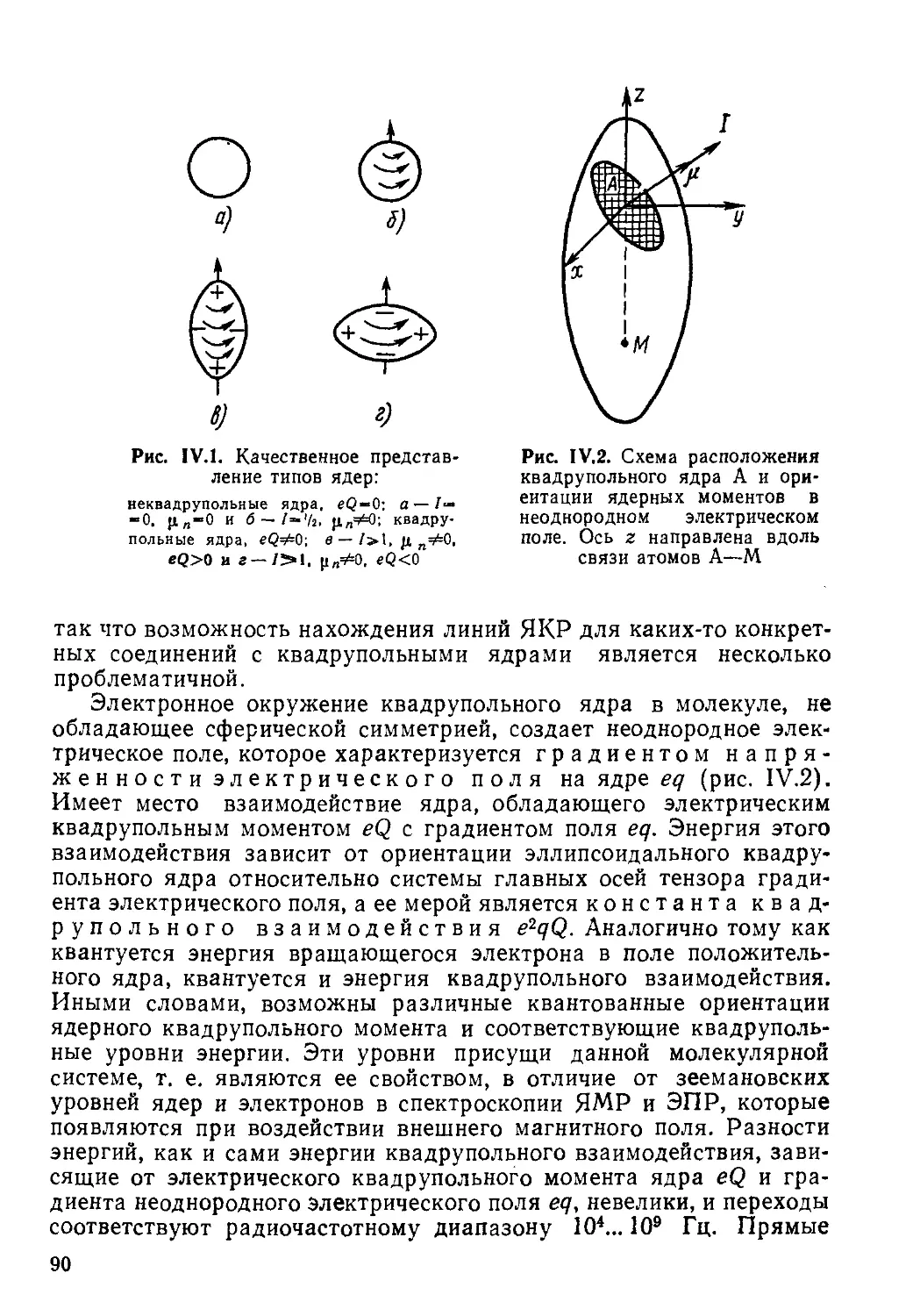
Атомное ядро состоит из протонов и нейтронов, обладающих спином 1/2, и может также иметь отличный от нуля результирующий спин I, т. е. угловой момент количества движения, характеризуемый вектором Р=Й1, где Й=й/2л, h — постоянная Планка. Отсутствие или наличие спина ядра и его значение определяются числом протонов и нейтронов, т. е. связаны с такими характеристиками ядра, как его заряд (порядковый номер элемента), равный сумме зарядов протонов, и массовое число (сумма масс протонов и нейтронов). Существуют следующие зависимости ядерного спина от этих величин.
7
1. При четных значениях заряда и массового числа, т. е. при четных числах протонов и нейтронов: ядерный спин 1 = 0, например, у таких очень распространенных изотопов, как 12С, 16О, 32S и др.
2. У всех элементов с нечетным массовым числом при любом порядковом номере, т. е., когда числа протонов и нейтронов разной четности, ядра имеют полуцелочисленный спин: 1 = Ч1> 3/г, 5/г> например, у изотопов 'Н, nB, 13С, 17О, 19F, 27А1, 31Р и др.
3. При четном массовом числе и нечетном заряде, т. е. нечетном числе как протонов, так и нейтронов, ядро обладает целочисленным спином: /=1, 2, 3....например, у изотопов 2Н, 10В, 14N,
30Р и др.
Согласно законам классической электродинамики вращение электрически заряженной частицы вокруг некоторой оси дает магнитное поле, совпадающее по направлению с осью вращения. Такая система характеризуется магнитным моментом, пропорциональным угловому моменту количества движения, и эту модель можно использовать для положительно заряженного атомного ядра.
Магнитный момент протона, называемый ядерным магнетоном
где е — заряд; тп — масса протона; с — скорость света; рп = = 5,05-10~27 А-м2. Для ядер, обладающих спином, пропорциональность магнитного момента угловому моменту количества движения выражается соотношением
(1.2)
где коэффициент пропорциональности уп называется гиромагнитным или магнитомеханическим отношением ядра (отношение магнитного момента к угловому).
Ядерный магнитный момент может быть выражен также через так называемый ядерный g-фактор, представляющий безразмерную постоянную gn, и ядерный магнетон рп:
(1.3)
Рп — gn$nl>
а в единицах ядерного магнетона имеем скалярный магнитный момент, по определению равный gn=gn/.
Значения I, уп, gn определяются природой ядра и представляют табулируемые константы. Магнитные свойства ядер некоторых изотопов приведены в табл. 1.1.
Вектор спина 1 = — р согласно квантовой механике связан со ft
спиновым квантовым числом I соотношением
|1| =//(/ + !),
(1-4)
где |1| —модуль, т. е. длина вектора I. 8
Таблица 1.1. Магнитные свойства некоторых ядер
Изотоп Естественное содержание, % Спин I V„, радиаиХ ХС-’-А-'м Sn в единицах рп
>н 99,984 '/2 336,19 5,585 2,792
2Н 0,015 1 51,61 0,857 0,857
13С 1,108 '/2 84,55 1,405 0,702
UN 99,635 1 24,30 0,403 0,404
15N 0,365 v2 —34,08 —0,56 —0,283
17О 0,037 —45,59 —0,757 , —1,893
100 '/2 316,41 5,25 2,627
29Sj 4,70 ’/2 —66,84 — 1,111 —0,555
31р 100 У2 136,22 2,26 1,131
35С1 75,4 3/2 32,87 0,54 0,822
37С1 24,6 3/2 27,44 0,45 0,683
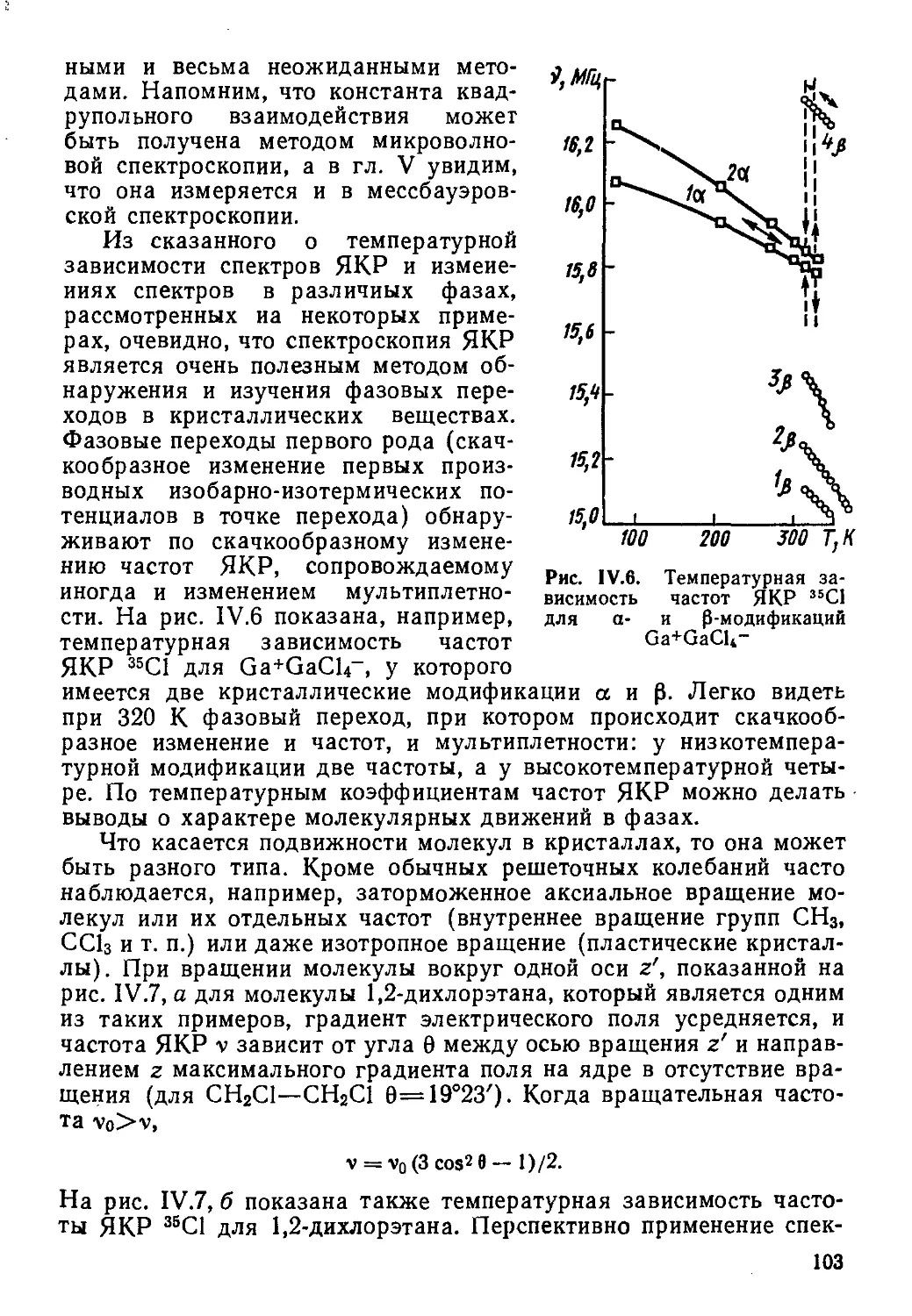
В отсутствие внешнего магнитного поля любые ориентации вектора ядерного магнитного момента в пространстве равновероятны, т. е. квантовые спиновые состояния вырождены.
При наложении постоянного магнитного поля В* возникает взаимодействие между ним и магнитным моментом ядра которое при квантово-механическом описании выражается гамильтонианом:
^ = -р„В. (1.5)
Энергия этого взаимодействия зависит от ориентации вектора магнитного момента относительно направления поля. Возможен лишь некоторый дискретный набор проекций, т. е. компонент вектора ядерного спина в любом заданном направлении, определяемых магнитным квантовым числом т/, которое принимает 27+1-значений, т. е. от +7 до —7. Если направление магнитного поля В выберем по оси z лабораторной декартовой системы координат (Вг = В), а 7г — проекция ядерного спина на эту ось, то гамильтониан взаимодействия ядра с полем (1.5) запишется в виде:
M = -ynhBIz = -g,&nBIz. (1.6)
Квантование проекции 1г приводит к тому, что возможны только 27+1 дискретных стационарных состояний с разрешенными собственными значениями энергии Ег, полностью описываемых собственными функциями состояний Т/.
* Мы будем характеризовать магнитное поле, как это принято в современной литературе, его индукцией В, измеряемой в единицах Тл (тесла — в СИ, межднародное обозначение Т): 1 Т=104 Гс (гаусс — в СГС). Вместе с тем иногда используется также напряженность магнитного поля Н, размерность которой совпадает с размерностью магнитной индукции В. Напряженность в 1 Э (эрстед— в СГС) соответствует полю с индукцией в вакууме 1 Гс. Все формулы электромагнетизма, не содержащие электрических величин, в системах СИ и СГС совпадают.
9
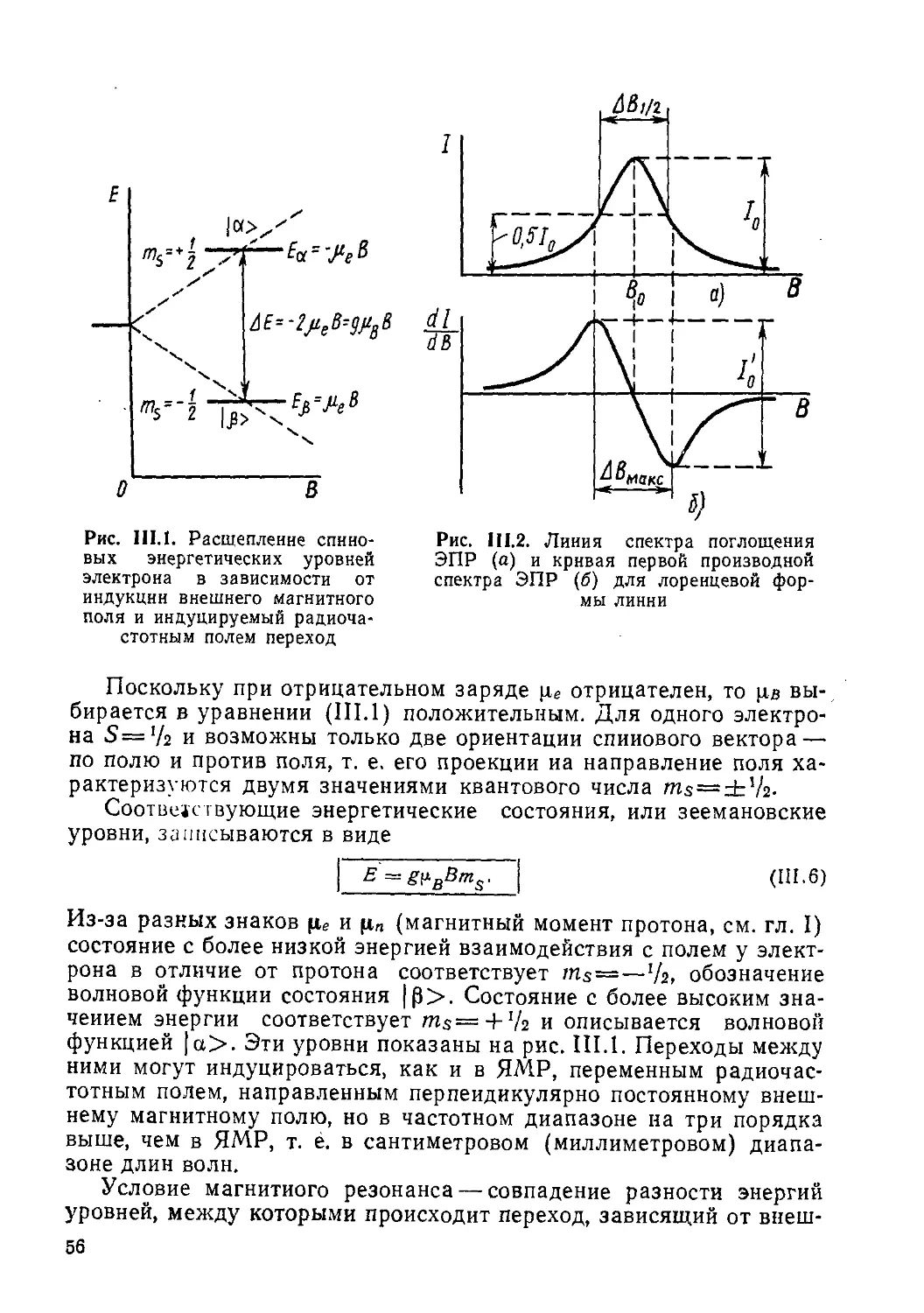
Рис. 1.1. Схема энергетических уровней протона в магнитном поле
Так, например, у протона или другой частицы со спином */г возможны только два значения квантового числа тг. +’/2 и —’/г. т. е. два спиновых состояния с энергиями:
1 1
Еь = ~РтгВ = уЛВ = - — g^nB
И (1.7)
которые описываются функциями, обозначаемыми, соответственно, символами |а> и |₽>.
В классической физике энергия взаимодействия магнитного момента с магнитным полем выражается формулой Зеемана:
В = —(р X В) = —cos (рВ),
(1.8)
т. е. зависит от абсолютных значений ц, В и угла между векторами (рВ). Е——цВ, когда вектор ц ориентирован «по полю» cos (pB) = L и такое название сохраняют за состоянием |а>; Е—цВ, когда вектор р ориентирован «против поля» cos (рВ) — — —1-состояние |р>.
Схема, показывающая расщепление энергетических уровней частицы со спином I—'h, которое прямо пропорционально напряженности постоянного магнитного поля, представлена на рис. 1.1.
В макроскопическом ансамбле частиц, помещенных в постоянное магнитное поле В, равновесная заселенность спиновых состояний при данной температуре определяется законом Больцмана:
Nt = exp (—Ei/kT),
где Ni — вероятность нахождения частицы на i-м уровне; E'i — энергия уровня; kT — тепловая энергия (k — постоянная Больцмана, Т — абсолютная температура).
В реальных условиях, т. е. при обычных в ЯМР значениях уп, В и Г, избыток заселенности ннжннх уровней очень невелик (порядка ~10-5—IO-6). Так, например, для протонов в магнитном поле ~1,25 Т (7/~ 1,25-104 Э) при комнатной температуре отношение NajNtfv 1,000007.
1.2. Условие ядерного магнитного резонанса
Как в любом другом спектроскопическом методе, переходы между энергетическими, в данном случае спиновыми, уровнями, сопровождающиеся изменением энергии системы, удовлетворяют
10
общему условию:
ДЕ = hv.
(1-9)
т. е. могут происходить с испусканием или поглощением кванта электромагнитного излучения с частотой v. При равновесной заселенности уровней избыток частиц в более низком энергетическом состоянии достаточен для того, чтобы при облучении образца экспериментально наблюдались спектры поглощения. При одинаковой заселенности уровней никаких сигналов ЯМР не будет наблюдаться, так как вероятность переходов в обоих направлениях одинакова (|а>-> |р> и ||3> -> |а>). При неравновесно высокой заселенности верхнего энергетического уровня могут фиксироваться сигналы эмиссии.
Для возбуждения переходов на образец, помещенный в постоянное однородное магнитное поле, необходимо воздействовать переменным магнитным полем Bv = B0vcos(2Ttv/ + 6), сравнимым по энергии с ДЕ зеемановских уровней системы. Резонансное поглощение электромагнитного излучения происходит при условии, что вектор осциллирующего магнитного поля перпендикулярен направлению постоянного магнитного поля BV±B и для рассматриваемой двухуровневой системы удовлетворяется равенство
hv = ynhB = ga$nB, (1.10)
представляющее так называемое условие ядерного магнитного резонанса.
Порядок значений разности энергий спиновых состояний ядер в магнитных полях порядка 1Т таков, что резонансные частоты лежат в радиодиапазоне (1—100 МГц), поэтому спектроскопия ЯМР и относится к методам радиоспектроскопии. Поскольку уп>. gn, I — это характеристики ядер, значения резонансных v и В изотопов отличаются (см. Н и N в табл. 1.2).
Квантово-механическая вероятность перехода между i-м и /-м спиновыми состояниями, характеризуемыми магнитным квантовым числом ntj, а следовательно, и интенсивность сигнала ЯМР пропорциональны квадрату модуля матричного элемента момента перехода, представляющего интеграл вида: Р°°| ,f Wt'/xWjdTl2, для которого принята также запись:
pc<|<V7|Z^|iFz>]2. (1.11)
Оператором перехода является проекция спина на ось х, откуда следует необходимость наложения высокочастотного поля, перпендикулярного направлению постоянного поля В, совпадающего с осью г.
Как для представленной на рис. 1.1 двухуровневой системы при спине /='/2, так и для более сложных систем, когда />’/2, правило отбора для разрешенных переходов: Дт/=±1.
Из уравнения (1.10) следует, что условия резонанса можно Достигать двумя путями: либо меняя частоту v переменного электромагнитного поля, например, переключая диапазоны частот
11
Таблица 1.2. Резонансные частоты некоторых ядер в магнитном поле 2,35 Т
Ядро v, МГц Ядро V, МГц Ядро V, МГц
*н 100,00 “N 7,22 19р 94,07
2Н 15,35 15N 10,13 29Si 19,86
13С 25,14 17О 13,56 31 р 40,48
источников и (приемников) при неизменной напряженности постоянного магнитного поля Н = const, либо меняя напряженность постоянного поля (полевая развертка) при неизменной частоте v = const переменного поля. На старых моделях спектрометров обычно регистрируется зависимость интенсивности поглощения электромагнитного излучения постоянной частоты от значения напряженности постоянного магнитного поля. ЯМР спектрометры с разверткой по полю имеют обычно источники электромагнитного излучения постоянной частоты порядка 1О7...1О8 Гц (60, 100, 200 ... МГц).
Частота связана с индукцией постоянного поля, как следует из (1.10), простым соотношением
и в некоторых случаях более удобно использовать угловую резонансную частоту
« ==2jtv ==уВ, (1-12)
единица измерения которой радиан-с-1. Эта частота совпадает с ларморовой частотой прецессии вектора магнитного момента вокруг направления магнитного поля В в классической модели ЯМР, предложенной Ф. Блохом, которая позволяет объяснять некоторые экспериментальные факты, например форму резонансной линии.
В названной модели рассматривается вектор макроскопического магнитного момента (намагниченности), представляющего векторную сумму отдельных ядерных моментов:
w
М = У, v-nt.
Г-1
Компонента вектора М в направлении постоянного магнитного поля
М‘| =1пВ,
где коэффициент пропорциональности %а называется ядерной магнитной восприимчивостью.
12
Рис. 1.2. Вектор суммарной ядерной намагниченности М, прецессирующий вокруг вектора В: во вращающейся системе координат (') эта прецессия отсутствует, ио осуществляется поворот М вокруг , т. е. переориен-
тация ОТ 2 К —2
Если вектор М направлен под углом а к оси z, совпадающей с направлением поля В (рис. 1.2), то при круговом движении (прецессии с ларморо-зой частотой т) компоненты Мц —Мг и остаются постоянными, причем проекции Al* и меняются по синусоидальному закону со сдвигом фаз на 90°. Такое состояние вектора М в реальных условиях не является равновесным.
Процессы продольной релаксации, т. е. стремление Af)( к равновесному значению Мп° со скоростью Ri, и поперечной релаксации— спад — происходят во времени (0 по экспоненциальному закону:
АГ, (П = АГ|
(О = Л< е-ЧТ^ (из)
где Т\ (~l/Ri)—время продольной релаксации (называемой также спин-реше-
точной); То— время поперечной релаксации (в общем случае и называется также временем спин-спиновой релаксации).
В классической модели ядерный магнитный резонанс связывают, по существу, с переориентацией вектора М из равновесного положения в направлении z в направление —г. Такая переориентация может происходить с помощью переменного магнитного поля Bv. Для этого необходимо, чтобы вектор Bv вращался в плоскости ху (см. рис. 1.2) с угловой частотой, близкой к ларморовой частоте прецессии со вектора магнитного момента М. При совпадении указанных частот переменное поле как бы «следит» за вектором М, возбуждая его прецессию вокруг вектора Bv. Это и приводит к переориентации М относительно В в системе координат, вращающейся вместе с векторами М и В, относительно неподвижной оси z, совпадающей с направлением В (см. рис. 1.2). Прецессия М относительно Bv медленная, так как это поле слабое (амплитуда B°v мала). Угол поворота вектора М во вращающейся системе координат от направления z (в плоскости уг) через промежуток времени t определяется соотношением
a = ynBj. (1.14)
Таким образом, при / = 0 а = 0, при t=a/2ynBv имеем так называемый 90°-ный импульс (а = 90°), а когда время полной ориентации t = n/(ynBv), — 180°-ный импульс (а=180°).
13
Рис. 1.3. Принципиальная блок-схема спектрометра ЯМР с полевой разверткой:
I — генератор ВЧ; 2 — радиочастотный мост; 3— усилитель; 4— детектор; 5 — регистрирующее устройство (и — сигнал дисперсии, и —сигнал поглощения); 6 — генератор развертки поля (питание электромагнита); 7 — полюса электромагнита; 8 — образец в ампуле; 9— катушка; 10— шиммирующие катушки
1.3. Реализация условий магнитного резонанса
Существуют различные способы реализации условий резонанса и получения спектров ЯМР, но принципиальные моменты следует рассмотреть сразу, перед дальнейшим изложением теории и практических применений метода.
Схема простейшего стационарного спектрометра показана на рис. 1.3. Образец в ампуле помещается в сильное однородное магнитное поле В, создаваемое постоянным электромагнитом, и одновременно находится в катушке под непрерывным воздействием высокочастотного поля небольшой мощности В,- В случае полевой развертки при постоянном значении частоты генератора v = (o/2n осуществляют медленное сканирование в резонансной области, плавно меняя В. При достижении условия резонанса, т. е. когда значение В удовлетворяет уравнению (1.10), происходит поглощение энергии излучения заданной частоты, фиксируемое по отклонению пера регистрирующего устройства.
При режиме частотной развертки катушка питается напряжением с частотой со, линейно меняющейся во времени, а постоянное поле В неизменно.
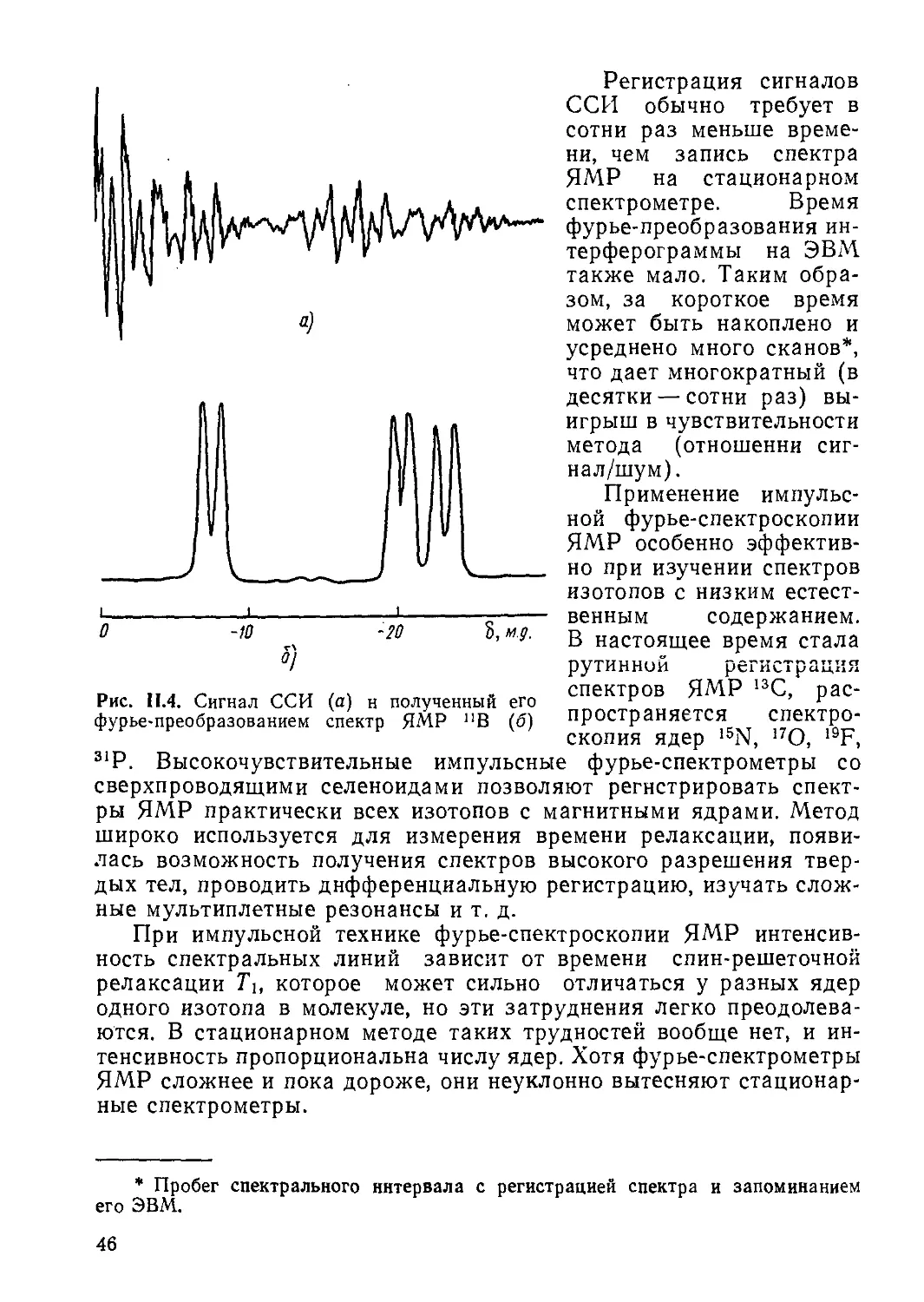
Когда вектор суммарной намагниченности М поворачивается вокруг оси х во вращающейся системе координат (см. рис. 1.2), происходит затухание индуцированных компонент Мх, Му (М±) и так называемого сигнала спада свободной индукции (ССИ), как показано на рис. 1.4. Величина М± представляет
14
Рис. 1.4. Сигнал спада свободной индукции
комбинацию двух сдвинутых по фазе на 9(jf колебаний:
= —и sin u>t 4- v cos u>t, где и называют сигналом дисперсии (проекция М на направление, перпендикулярное Bv), a v — сигналом поглощения (проекция М на направление Bv), эти сигналы могут выделяться с помощью фазочувствительного детектора (см. рис. 1.3).
Вид и интенсивность сигналов ЯМР зависят от скорости сканирования, т. е.
времени прохождения через резонансную область. Дело в том, что спиновая система обладает некоторой инерционностью, определяемой временем поперечной (спин-спнновой) релаксации Т2. При большой скорости сканирования происходит расфазировка кругового движения векторов М и В, что приводит к падению интенсивности сигнала ЯМР, но в пределах времени релаксации Т2 периодически может наступать повторная фазировка с появлением сигналов спадающей интенсивности, называемых виглями (от англ, whiggles) и имеющих вид затухающих колебаний. Наличие виглей свидетельствует о высокой однородности внешнего поля, т. е. характеризует хорошее качество спектра. При медленном сканировании, когда удовлетворяется условие
dv
— «(Av)?,
где Av — ширина резонансной области, сигналы ЯМР имеют симметричную форму (без виглей).
Для регистрируемого обычно сигнала поглощения характерна «колоколообразная» форма линии. При развертке по частоте [от развертки по полю, т. е. величины В, можно перейти к частоте, используя соотношение (1.12)] могут измеряться четыре параметра сигнала: Vo — резонансная частота (частота максимума кривой поглощения); А — интенсивность в максимуме (амплитудная); So — интегральная интенсивность (площадь регистрограммы сигнала); Avi/г — ширина линии на полувысоте: А/2.
В квантово-механическом описании при выполнении условия ЯМР осуществляется переход спиновой системы из нижнего энергетического состояния в верхнее с поглощением энергии. При этом может происходить нарушение равновесного больцмановского распределения ядерных спинов и так называемое насыщение (выравнивание заселенности состояний) с постепенным исчезновением сигнала ЯМР. Чтобы насыщения не происходило, система Должна успевать релаксировать к равновесному состоянию. Это определяется соотношением индукции накладываемого переменно-
15
го магнитного поля Bv, которая должна быть достаточно мала, и временем Т[ спин-решеточной (продольной) релаксации, возвращающей систему к равновесию за счет безызлучательных переходов. Эти переходы индуцируются локальными флуктуирующими полями, появление которых связано с молекулярным движением, а выделяющаяся при них энергия переходит в тепловую энергию (решетки). /
С увеличением Bv интенсивность сигнала (А и So) сначала растет, достигая максимума, а затем, при больших значенийх В», падает. Большая скорость развертки по полю (или частоте) позволяет использовать высокую индукцию Bv, не вызывая насыщения при значительном повышении интенсивности сигнала*. Время релаксации Т\ обычно порядка нескольких (до десятков) секунд. К его уменьшению приводят увеличение вязкости, например, при понижении температуры, парамагнитные добавки, наличие квад-рупольных ядер.
Наблюдаемая экспериментально ширина линии сигнала ЯМР Avi/г (подстрочный индекс '/г далее опускаем) включает естественную ширину Ave и аппаратурное уширение Ava, обусловленное в основном неоднородностью постоянного магнитного поля В:
Av = Ave 4- Ava.
Естественная ширина линии обратно пропорциональна времени спин-спиновой (поперечной) релаксации Т2:
Ave — \)пТ2. (1.15)
Всякое изменение напряженности постоянного поля вызовет согласно (1.12) изменение резонансной частоты Av=yAB/2n, с чем и связано уширение Ava, которое на современных спектрометрах не превышает десятых долей герца. В невязких обезгаженных жидкостях Т2аТ} (от нескольких до десятков секунд), т. е. Ave составляет сотые доли герца. Наблюдаемая ширина линий в спектрах ЯМР, вообще говоря, может меняться в очень широких пределах, но даже так называемые узкие линии (Av^l Гц) имеют ширину не меньше десятых долей герца (для ПМР — 0,3... 0,5 Гц). При использовании обезгаженных эталонов на современных спектрометрах достигается разрешение 0,2 Гц, т. е. когда однородность магнитного поля и разрешающая способность спектрометра достаточно высоки, определяющее значение для наблюдаемой ширины линии имеет характер исследуемых образцов.
В твердых телах Т2<^Т\, время релаксации Т2 мало (составляет доли секунды). То же характерно для вязких жидкостей, растворов парамагнитных веществ и некоторых других систем.
* Учитывая также сказанное о зависимости интенсивности сигнала ЯМР от скорости сканирования, можно поиить необходимость поиска компромиссного решения вопроса о ее соотиошеинн с величиной магнитной индукции (напряженности поля).
16
Так называемые широкие линии в спектрах ЯМР могут иметь ширину до 105 Гц. Возможностью регистрировать ЯМР спектры практически с любой шириной линий обладают современные импульсные спектрометры с фурье-преобразованием сигнала ССИ. Для (записи линий с шириной порядка 103 Гц используют иногда и стационарные спектрометры с регистрацией первой производной сигнала, например, при изучении спектров ЯМР твердых тел. Практически всегда запись первой производной кривой поглощения практикуется в спектроскопии ЭПР (см. гл. III).
2. ХИМИЧЕСКИЙ СДВИГ И СПИН-СПИНОВОЕ ВЗАИМОДЕЙСТВИЕ
2.1. Экранирование ядер электронами
Выше рассматривалось только взаимодействие ядер с внешним магнитным полем и полностью игнорировалось влияние электронного окружения и взаимодействие спинов ядер между собой. Для химии метод ЯМР важен прежде всего именно потому, что резонансные частоты ядер зависят от тонких магнитных взаимодействий, т. е. в конечном счете от особенностей строения и распределения электронной плотности в молекулах.
Движение электронов вокруг ядра в условиях внешнего магнитного поля В создает на ядре дополнительное магнитное поле В', которое пропорционально и направлено противоположно при-ложснному поляризующему полю:
В' (1-16)
Таким образом, реально на ядро действует некоторое эффективное поле:
В„ = В +В‘ = (1 — о)В, (1.17)
В — В„
Где а=------S— безразмерная величина, называемая кон-
стантой экранирования (обычно приводится в миллионных долях: м. д., т. е. 10-6). В изолированных атомах причиной появления поля В' (1.16) являются диамагнитные токи, и эффективное поле Вл (1.17) всегда меньше приложенного В, т. е. од>0. Такое экранирование называют диамагнитным, как и константу экранирования од. В молекулах при наложении внешнего поляризующего поля возникает слабый парамагнетизм, т. е. константу экранирования можно представить как сумму двух вкладов:
а = ая4-ап,
причем парамагнитный вклад оп<0 (направление парамагнитного тока противоположно диамагнитному). Это значит, что в молекулах о ядер может быть как положительна, так н отрицательна. Константа экранирования для ядер трех важнейших изотопов в
17
Таблица 1.3. Коистаиты экранирования ядер некоторых изотопов в атомах и простых молекулах, м. д. /
Изотоп ад (атома) Молекула а 1<WCTnl )
*н 18 На 27 6,4
13С 261 сн4 187 2,7
1вр 464 f2 — 150 0,7
изолированных атомах и простых
молекулах приведены в
табл. 1.3. При положительном значении константы экранирова-
ния, например ядер в атоме или молекуле водорода, для получения сигнала ЯМР напряженность внешнего поля должна быть
выше по сравнению с напряженностью поля для неэкранированного ядра (1Н+), так как эффективное поле (1.17) ниже.
Константы экранирования для атомов и простейших, например двухатомных молекул, рассчитываются методом квантовой химии и определяются экспериментально. Но вообще точно определить значение о не представляется возможным.
2.2. Химические сдвиги сигналов ЯМР
Из-за указанных выше трудностей на практике гораздо удобнее измерять не абсолютные значения константы экранирования, а
разности:
8 — оэт — ох,
(1.18)
где Оэт — константа экранирования ядра в каком-то эталонном веществе, а ох — константа экранирования того же ядра в исследуемом образце.
Если константа экранирования о определена для вещества (раствора), т. е. включает атомный и молекулярный вклады, а также составляющую, обусловленную межмолекулярными взаимодействиями o', то она представляет абсолютный химический сдвиг сигнала данного ядра (или группы эквивалентных ядер). Разность б таких констант для эталона и образца (1-18) или сдвиг сигнала ЯМР образца относительно выбранного эталона (в том же растворителе) называют относительным или просто химическим сдвигом.
Учитывая соотношение (1.12) при постоянном значении частоты vo (генератора), можно записать:
Ygx = у(1-8)Вэт 2л 2л ’
откуда следует выражение б через экспериментально определи-18
мЫе параметры:
\ Вэт — z?x
I 8 - B„
или^ переходя к резонансным частотам:
' V9T - VX
8 =----------
v3T
(1.19)
(1-20)
Порядок значений резонансных частот очень велик (>106 Гц) по сравнению с разностью Av = v3T—vx, а частота генератора vo мало отличается от резонансной частоты (v3t), поэтому окончательно для химического сдвига, измеряемого в миллионных долях (м.д.), имеем:
Av-106 8 =---------
Vo
(1.21)
(в зарубежной литературе единицы 10-6 обозначают ppm—parts per million).
В протонном магнитном резонансе (ПМР) в качестве эталонного вещества используют тетраметилсилан или ТМС (Si(CHs)4), относительно очень узкой и интенсивной линии которого измеряют химические сдвиги:
SH= ---------—106 (1.22)
в так называемой б-шкале ТМС (м.д.). При этом относительный сдвиг самого ТМС принимается равным нулю, а сигналы при более низкой напряженности поля согласно уравнениям (1.19)— (1.22) имеют положительный химический сдвиг, т. е. рост его соответствует смещению сигнала в сторону более слабого поля. Чем меньше экранировано ядро, тем больше в этой шкале его химический сдвиг. В ТМС экранирование протонов очень сильное, поэтому для большинства соединений химический сдвиг сигналов ПМР положительный.
Значительно реже используется также т-ш кал а (ТМС) химических сдвигов, которая связана с б-шкалой простым соотношением:
Г = 10,00-8, (1.23)
т. е. относительный сдвиг для ТМС принят за 10,00, и в сторону слабого поля убывает химический сдвиг сигналов. Совсем мало распространенной является отрицательная шкала б, хотя она по физическому смыслу и более строгая.
На рис. 1.5 в качестве примера приведен спектр ПМР низкого разрешения для этилового спирта с указанием химических сдвигов в б- и т-шкалах и соотношения экспериментально изме-
19
Рис. 1.5. Спектр ПМР С2Н5ОН низкого разрешения
ренных интегральных Интенсивностей сигналов разных групп протонов.
Вообще химические сдвиги для протонов занимают диапазон несколько более 10 м. д., а стандартная ошибка их измерения составляет ±0,001 м. д. Значение измеряемого химического сдвига зависит от внешних факторов: растворителя, концентрации и температуры
образца. Особенно сильна эта зависимость для таких функциональных групп, как ОН, NH, SH, и некоторых других. Так, например, изменение протонного химического сдвига для группы ОН при переходе от концентрированного раствора спирта в СС14 к разбавленному достигает ~5м. д., а при экстраполяции к бесконечному раз
бавлению значение напряженности поля должно быть даже выше, чем для протонов группы —СНз (рис. 1.5). Это объясняется, очевидно, различной степенью участия ОН-групп в образовании водородных связей. Отсюда понятна важность применения метода разбавления при изучении межмолекулярных взаимодействий. Специфиче-
ские влияния среды обнаруживаются также по сильной температурной зависимости химических сдвигов.
Так называемые истинные химические сдвиги соответствуют
разряженным газам или растворам в инертных неполярных растворителях при экстраполяции к бесконечному разбавлению. Обычно же используются 5—10%-ные растворы.
Химические сдвиги ядер изотопа 13С также измеряют в м.д. б-шкалы относительно ТМС:
^тмс vr
= ----— 10^, (1.24)
v0
причем нх значения лежат в диапазоне до ~230 м.д., а стандартная ошибка измерений составляет "—0,05 м.д. Влияние внешних факторов (растворитель, температура) на значение химического сдвига 13С, как правило, невелико (изменение менее —-0,5 м.д. при указанном выше диапазоне значений бс)-
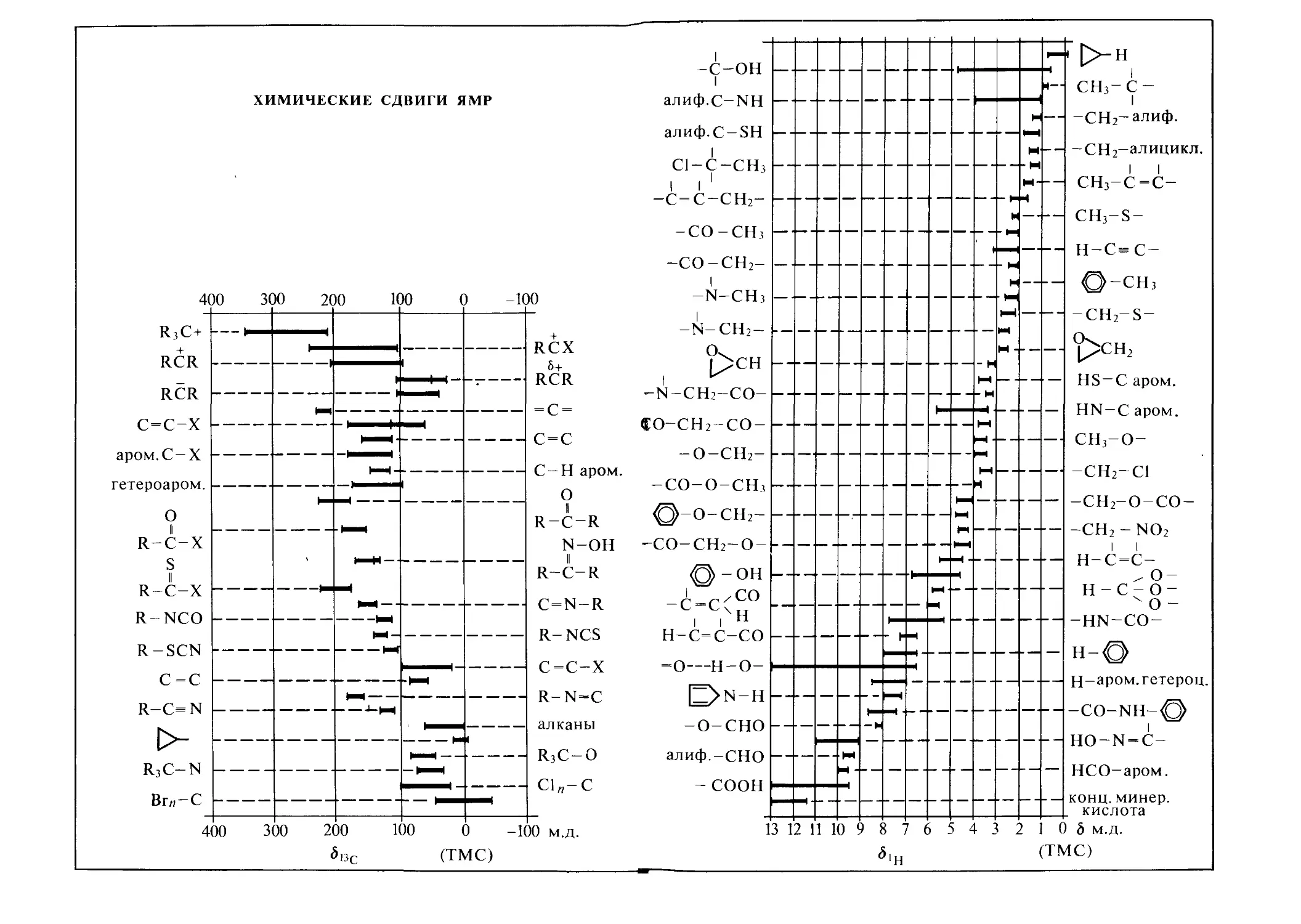
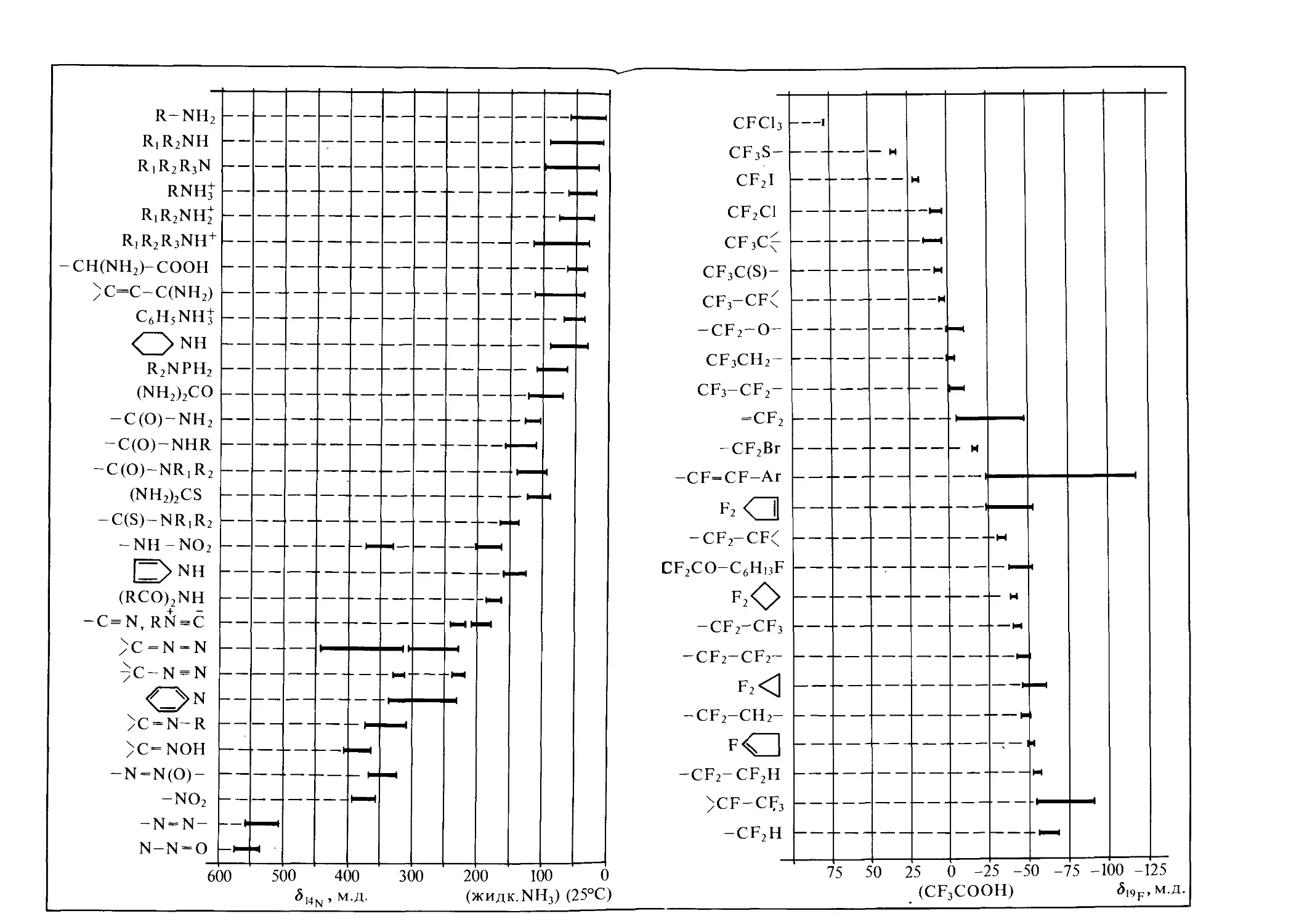
Характеристичность химических сдвигов позволяет широко ис пользовать их в исследованиях структуры соединений. Для определения различных структурных фрагментов молекул и функциональных групп составлены многочисленные таблицы и корреляционные диаграммы. На рис. 1.6 схематично показаны диапазоны наблюдаемых химических сдвигов для некоторых ядер, а кор-
20
1 н СН3СООН н’>8 C6H5CHCljCMl'H Н2СССН2 С2Н5ОН CHjCOOH С2Н5ОН тмс
"в 12 Я7 8 i & '2 0 . В(СН,)3 C6HsBCl2BCl3 8F3 В13 Д1(8НД NaBH4
”с 1 » 1 I 1 I BO W 20 0 -20 -Щ -60 (CH5)»CO KCN HCQDH CtHt ССЦ CHOI, CH5Ct CH3I CHjI, ♦ f ♦ f i 3 4 ♦ 4(
w N Л 50 0 -io -ioo -150 -200 (CNj)2IK§>NO Nuno, C6H5N--NC6Hs NH4NO, KCN CH3CN £>NH N2H„NHA 4 ♦ 4 | 4 4 ♦ 4 4 iNH3
,7о W 300 ' 200 100 , 0 -100 -200 -300 -000 NaNOj (CHj^CO S02 NaNO3 Fe(C05) CH3COOH HjQ
100, , 600 500 Ш 300 4 ’ 100 0 ГгМ CIF3 CFCt3 PF3CF, CFjCOOH HF CFH3 * ‘ 4 4 3 i| 4 4kf 4 4
З'р 500 000 300 2'00 ioo 0 -ioo -200 W1 Vе"’ V
Рис. 1.6. Примерные диапазоны химических сдвигов для некоторых ядер в различных химических соединениях
реляционные диаграммы для соединений, содержащих ядра ’Н, 13С, 15N, 19F, приведены на форзацах, где указаны также соответствующие стандарты.
Для протонов химические сдвиги относительно очень малы по сравнению с другими ядрами, и все-таки их небольшие различия дают очень важную информацию об окружении того или иного протона в молекуле.
С увеличением порядкового номера элементов наблюдается тенденция увеличения интервалов значений химических сдвигов, т. е. магнитное поле на ядре, обусловленное взаимодействием приложенного поля с электронным окружением, может меняться для тяжелых ядер в более широких пределах, а у легких ядер изменение констант экранирования много меньше.
Если диапазон химических сдвигов 13С и 19F шире, чем для протонов, примерно в 20 раз, для 31Р — в 40 раз, то для таких ядер, как 14’15N, 17О, он превышает сдвиги сигналов ПМР на два порядка и близок к 1000 м.д. Вообще же для атомов разных элементов с магнитными ядрами в неорганических и органических соединениях области сигналов ЯМР варьируются очень сильно—• от сравнительно узких (7Li, 23Na, 27А1) до чрезвычайно широких (77Se, 125Те).
Химические сдвиги сигналов ЯМР магнитных изотопов одного и того же элемента в одинаковых соединениях и относительно соответствующих изотопных эталонов практически совпадают, т. е. так называемый изотопный сдвиг, если и бывает, то весьма не
21
значителен. Так, например, для ЯМР на 2D обнаруживаются те же закономерности, какие наблюдаются в спектрах ПМР. Аналогично обстоит дело в спектрах ЯМР на 10В и UB, 14N и 15N и т. д.
Основным источником получения структурной информации и данных для идентификации соединений по спектрам ЯМР на разных ядрах следует считать эмпирические закономерности, таблицы и корреляционные диаграммы химических сдвигов.
2.3. Спин-спиновое взаимодействие и мультиплетность спектров ЯМР
Между ядерными спинами в молекулах существует взаимодействие, которое приводит к расщеплению, т. е. мультиплетности сигналов ЯМР. Химический сдвиг сигнала, представляющего мультиплет, определяется по центру мультиплета. Число компонент мультиплетов зависит от количества взаимодействующих неэквивалентных ядер.
Спиновая система, в которой все ядра характеризуются одним и тем же гиромагнитным отношением уп (фактором gn), называется гомоядерной, в противном случае — гетероядерной.
Два ядра любой спиновой системы, дающие сигналы с разными значениями химических сдвигов, называют химически неэквивалентными; при одинаковых химических сдвигах ядра называют химически эквивалентными (или изохронными). Случайное совпадение сигналов ЯМР иногда можно выявить, например, варьированием растворителя или других условий эксперимента. Истинная эквивалентность имеет место при молекулярной симметрии. В этом случае спиновую систему можно отнести к какой-то точечной группе симметрии и рассматривать, используя аппарат теории групп.
В литературе по спектроскопии ЯМР не только для гетеро-ядерных систем, но даже в случае гомоядерной спиновой системы, например образованной только протонами, химически неэквивалентные ядра или группы таких ядер принято обозначать различными буквами латинского алфавита: А, В, С, ..., X, Y, Z. При этом в зависимости от соотношения разности химических сдвигов Д6 и величины расщепления сигналов взаимодействующих ядер, т. е. силы взаимодействия, эти ядра обозначают либо буквами начальной части алфавита: АВ, АВ2, АВС и т. п. — когда величина Д6 сравнима с расщеплением сигналов, либо буквами начальной и конечной частей алфавита: АХ, АХ2, XAY и т. п.— когда Дб много больше расщепления*.
* Следует четко отличать эти обозначения ядерных спиновых систем от аналогичных по виду общих по типу формул двухатомных и многоатомных молекул (например, АВ, АХг, АХ4, АХУ2 и т. д.), широко используемых в других главах учебника, как и вообще в литературе по строению молекул и физическим методам исследования.
22
При наличии в группе п эквивалентных ядер, например Ап, для описания состояний спиновой системы, характеризуемых величиной проекции суммарного спина 1г или 2 т>> вводят мультипликативные функции, представляющие произведения
функций отдельных спинов (если 7=’/г, то а и 0). Для п спинов имеется 2П мультипликативных функций, но при этом число значений S/п/ равно п+1, т. е. некоторым значениям проекции суммарного спина Iz отвечает несколько мультипликативных функций, описывающих вырожден
Таблица 1.4. Состояния сястемы
двух эквивалентных спинов Аг (/—W
Мультипли- Smr Кратность
кативиые функции вырождения
00 —1 1
ар 0a 0 2
aa + 1 1
ные состояния.
Так, например, для системы эквивалентных спинов Л2 существуют четыре состояния, описываемые, как показано в табл. 1.4. Состояние, для которого /2 = 0, называется двукратно вы
рожденным, так как описывается двумя мультипликативными функциями. В общем случае кратность вырождения состояний
для системы из п эквивалентных спинов определяется с помощью коэффициентов биномиального разложения (а+1)", образующих при разных п так называемый треугольник Паскаля /тобгт т ш
В соответствии с квантово-механическим правилом отбора Д(2^/) = —1. которое определяется интегралами матричных элементов момента перехода вида (1.11), для системы Ач эквивалентных спинов с одинаковой валентностью возможны переходы между состояниями: 00->-ар, 00->0а, а0->-аа, 0а-»-аа.
с другими ядрами будет наблюдаться один нерасщеплеиный (синглетный) сигнал.
Два неэквивалентных магнитных ядра А и Б, для которых характерны резонансные частоты va и vb или химические сдвиги
23
6д и &б, могут взаимодействовать между собой, что приводит к расщеплению сигналов ЯМР. При этом спиновая система может относиться или к типу АВ, или к типу АХ, для которого разность химических сдвигов Д6 = 6д—6х намного больше величины расщепления сигналов от ядер А и X. Рассмотрим этот предельный случай.
Энергия взаимодействия ядер выражается через скалярное произведение векторов спинов:
Е = hj ах^а^х,
(1.25)
где Jax — константа спин-спинового взаимодействия, измеряемая обычно в герцах.
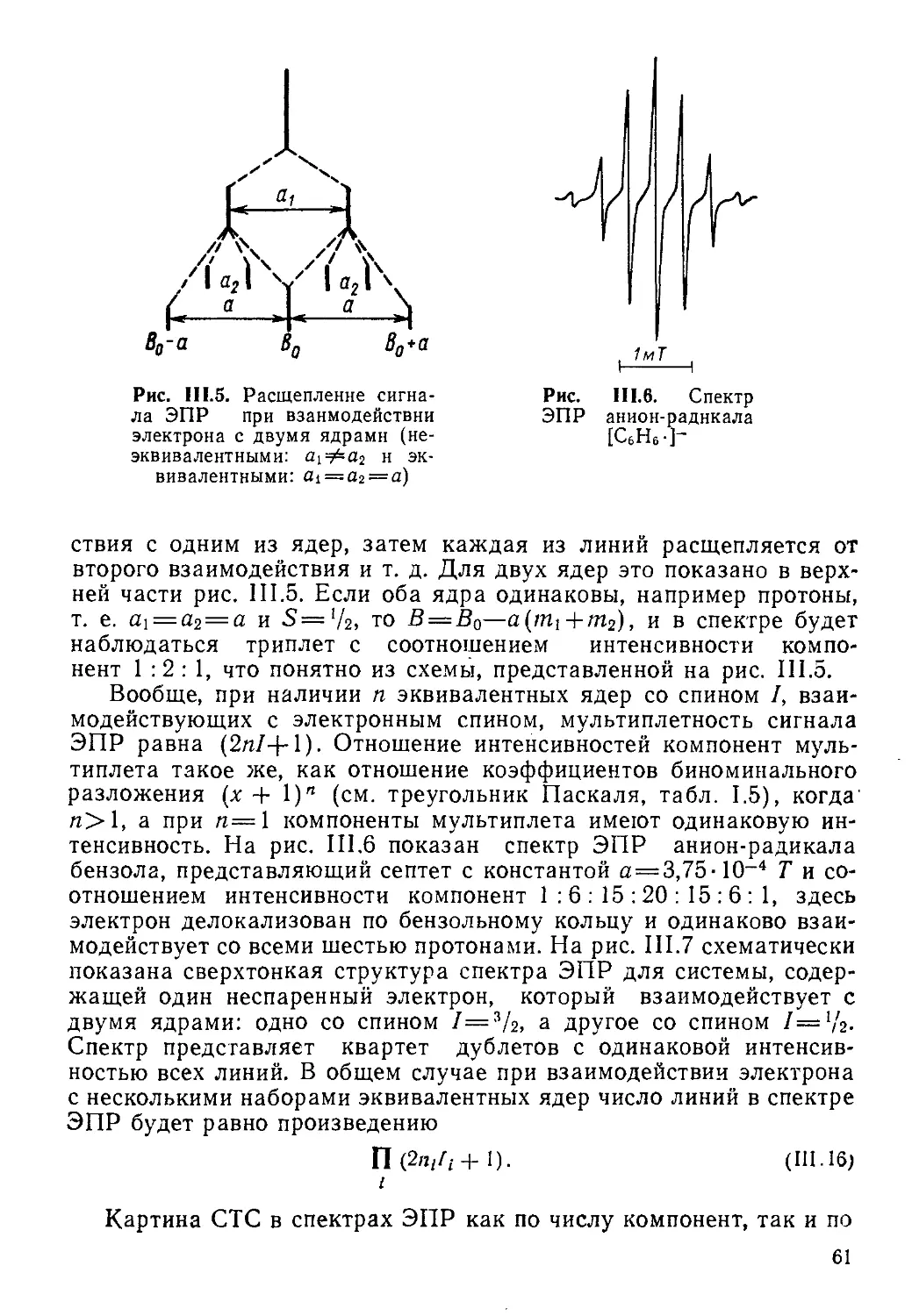
Влияние спинового состояния одного ядра на положение зеемановских уровней и резонанс другого несколько упрощенно можно описать следующим образом. Пусть в системе ядер АХ спин 1х ориентирован против поля В, что соответствует состоянию рх, тогда локальное магнитное поле на ядре А будет понижено по сравнению с тем, какое было бы в случае отсутствия ядра X. Это приведет к тому, что для достижения условия резонанса потребуется приложить поле более высокой напряженности, т. е. выше будет и резонансная частота [согласно 1.12], как это показано на схеме рис. 1.7. Если ядро X находится в состоянии ах, т. е. спии 1х ориентирован по полю, то на ядре А локальное поле повысится, т. е. для резонанса потребуется наложение поля более низкой напряженности, чем в отсутствие ядра X. Таким образом, в спектре ЯМР будет наблюдаться дублетный сигнал ядра А. Расстояние между компонентами дублета (в Гц) и будет константой спин-спинового взаимодействия:
т. е. две частоты дублета могут быть выражены соотношением v = va±TZax-
Спиновые состояния аир практически равнозаселеииы (см. выше), поэтому интенсивности линий в дублете одинаковы (1 : 1).
Совершенно аналогичным образом можно описать и результат влияния спиновых состояний ядра А (Рд и ад) на сигнал ЯМР ядра X, который также будет наблюдаться в виде дублета с расстоянием между компонентами, равным JAx. Таким образом, в целом спектр ЯМР спиновой системы АХ состоит из четырех линий или двух дублетов, по центрам которых определяются химические сдвиги бд и 6х, а по расстоянию между- компонентами дублетов — константа JAx- Последняя, являясь характеристикой
24
A
x
Рис. 1.7. Схема расщепления сигналов ЯМР в результате спин-спинового взаимодействия двух ядер А и X со спинами 1='/2
Рис. 1.8. Вид спектра ПМР этильного радикала (подкисленный раствор С2Н5ОН)
внутримолекулярного взаимодействия ядер, ие зависит от напряженности внешнего магнитного поля Н.
Для спиновой системы типа АВ в дублетных сигналах ядер А и В по мере уменьшения разности А6 и относительного увеличения Jab интенсивность внутренних компонент квадруплета 2 и 3 (рис. 1.7) будут возрастать и они будут сближаться, а интенсивность внешних компонент 1 и 4 — падать, пока, как имеет место в предельном случае системы Аз (см. выше), компоненты 2 и 3 не сольются (при Д6 = 0), а компоненты 1 и 4 не исчезнут.
При взаимодействии атомных групп, содержащих несколько ядер, спектр ЯМР, естественно, усложняется. Спектр ПМР этильного радикала, например в подкисленном спиртовом растворе (и аналогично в молекулах CH3CH2R, где R— невзаимодействующий атом), при достаточном разрешении имеет вид, представленный на рйс. 1.8. В такой системе, относящейся к типу Аз-^2> спиновые состояния группы Х2 описываются, как было показано для двухспиновой системы в табл. 1.4. Эти состояния протонов группы СН2 влияют на резонансный сигнал протонов метильной группы СН3, который и представляет поэтому триплет в соответствии с числом возможных значений суммарного спина системы Х2. Соотношение интенсивностей компонент в триплете 1:2:1, что соответствует соотношению вероятностей (кратности вырождения), влияющих состояний группы СН2 с данным суммарным спином (см. табл. 1.4).
Возможные спиновые состояния группы СН3 (трехспиновой системы Аз) показаны в табл. 1.6.
В этом случае имеется четыре возможных значения суммарного спина, и влияние спиновых состояний группы СН3 приводит соот-
25
Таблица 1.6. Состояния системы трех спинов Аз ветственно к квадруп-летному сигналу ПМР
Мультиплнкатнвн ые функции Кратность вырождения метиленовой группы СН2 с соотношением интенсивностей линий
оо 00а 0а0 0аа а0а ааа а00 аа0 -3/S -Ч2 +'/2 +’/« 1 3 3 1 1 :3:3:1. Центры триплета и квадруплета в спектре ПМР радикала СНзСН2 представляют химические сдвиги
протонов соответственно метильной и метиленовой групп, а расстояния между компонентами обоих мультиплетов дают константу про-тон-протонного спин-спинового взаимодействия 3/нн (через три связи). Соотношение общих (суммарных) интегральных интенсивностей мультиплетов СН3- и СН2-групп остается в соответствии с отношением чисел протонов в группах, т. е. 3 : 2,
В общем случае число компонент в мультиплете спиновой системы Ат, обусловленном спин-спиновым взаимодействием с системой Бп (п — число ядер Б, обладающих спином 1Б), определяется формулой (2«75+1), а при спине /б = '/2, как в ПМР, это число равно (п+1). Соотношение интенсивностей компонент всегда определяется кратностями вырождения состояний спиновой системы Бп, находимыми при данном значении п, как было показано выше (см. табл. 1.5).
При спин-спиновой связи какой-то группы магнитных ядер Ат с несколькими другими группами, например Бп, Bi и т. д., число компонент в мультиплетном сигнале группы Ат для спектров первого порядка формально равно произведению мультиплетностей, обусловленных каждой из указанных групп в отдельности: (2п1Б.+ 1)(21/в+1)...
На рис. 1.9 схематично представлено, как мог бы выглядеть спектр ЯМР системы с тремя неэквивалентными парами ядер: Х2—А2—Уг, когда их спины равны ’/2, имеет место спин-спиновое взаимодействие только соседних групп, а константы Jax=Jay (например, спектр ПМР метиленовых групп молекулы R'—СН2— —СН2—СН2—R", в которой R'#=R" и не дают дополнительных расщеплений).
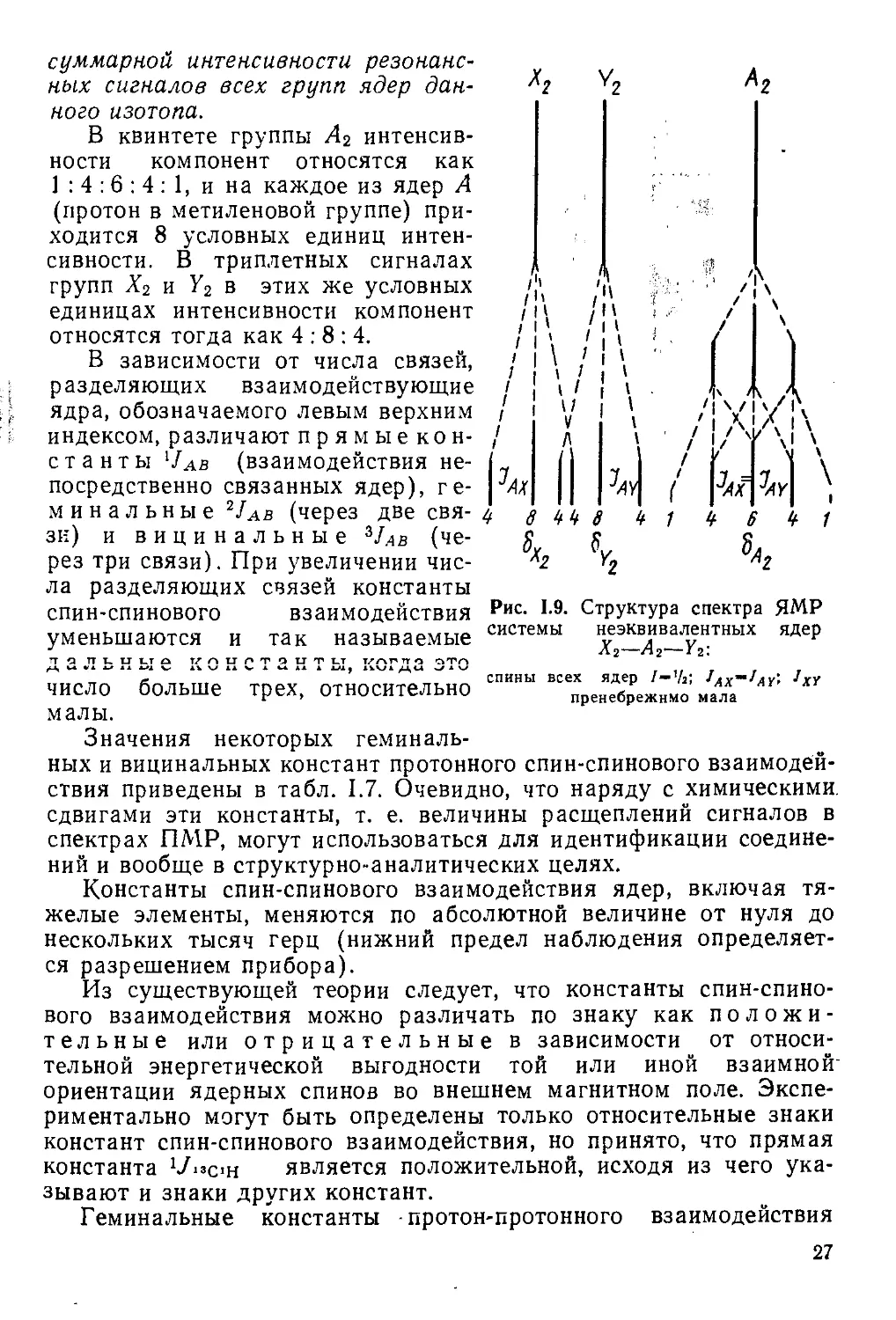
Видно, что сигнал группы А2 (средняя метиленовая группа) прн этом является квинтетным, а сигналы двух других групп представляют перекрывающиеся триплеты. В каждом из мультиплетов в отдельности соотношение интенсивностей компонент можно определить, используя треугольник Паскаля, но интересно представить соотношение интенсивностей по всему спектру в единой шкале. Это легко сделать на основании фундаментального правила ЯМР о постоянстве приходящейся на одно ядро доли
26
суммарной интенсивности резонансных сигналов всех групп ядер данного изотопа.
В квинтете группы А2 интенсивности компонент относятся как 1 : 4 :6 : 4 : 1, и на каждое из ядер А (протон в метиленовой группе) приходится 8 условных единиц интенсивности. В триплетных сигналах групп Х2 и Y2 в этих же условных единицах интенсивности компонент относятся тогда как 4:8:4.
В зависимости от числа связей, разделяющих взаимодействующие ядра, обозначаемого левым верхним индексом, различают прямыекон-станты Чав (взаимодействия непосредственно связанных ядер), геминальные 2Jab (через две связи) и вицинальные 3JAB (через три связи). При увеличении числа разделяющих связей константы спин-спинового взаимодействия уменьшаются и так называемые Д 2 Л ЬН Ы О К0НСТ2НТЫ когдз это число больше трех, относительно малы.
У2 Аг
Л2 '2 ”2
Рис. 1.9. Структура спектра ЯМР системы неэквивалентных ядер Х2-Я2-У2:
спины всех ядер I—'h\ ^xy
пренебрежимо мала
Значения некоторых геминаль-
ных и вицинальных констант протонного спин-спинового взаимодействия приведены в табл. 1.7. Очевидно, что наряду с химическими, сдвигами эти константы, т. е. величины расщеплений сигналов в спектрах ПМР, могут использоваться для идентификации соединений и вообще в структурно-аналитических целях.
Константы спин-спинового взаимодействия ядер, включая тяжелые элементы, меняются по абсолютной величине от нуля до нескольких тысяч герц (нижний предел наблюдения определяется разрешением прибора).
Из существующей теории следует, что константы спин-спинового взаимодействия можно различать по знаку как положи
тельные или отрицательные в зависимости от относительной энергетической выгодности той или иной взаимной ориентации ядерных спинов во внешнем магнитном поле. Экспериментально могут быть определены только относительные знаки констант спин-спинового взаимодействия, но принято, что прямая константа У-зщн является положительной, исходя из чего ука-
зывают и знаки других констант.
Геминальные константы протон-протонного взаимодействия
27
Таблица 1.7. Значения некоторых констант спнн-спннового взаимодействия протонов '7нн(<=2, 3, 4, 5) в различных структурных фрагментах
Фрагмент '^НН' Гн Фрагмент z/hh- Гц
R'_СН2—R" !7, —10... 15 СН2—сн2 !/, 4-6
Н2СО V, +40,2 ...42,4 3/цис, 4,4 Стране, 3,1
Н2С=СО V, —15,8
СН3ОН Ч —10,8 Hn Чаа, 7... 14
^СО 3Jae, 1...7
.сн \ II сн2 3Ле, 2... 4
сн /
^со7 н
СН3аСН2вОН !7, —21,5 3/АВ, ±6,7 ...7,2 ГОГ 3/орто, 7... 8
сн2=сн2 V, ±2,3 3/Чис, 11,5
транс, 19,0 НА нЦАх^ОСНз 3/ав, 7,98 Vbb', 7,53
Н ОСН3 Х,с=с/ !J, —2,0 3/цис, 7,0 нДО^осн3 Vab', 1,45 Vaa', 0,4
/ S н н "J Транс, 1**, а НА’
37своб.вращ, 6 .. 9 Vab, 7,75
CHR2—CHR2 37гош, 2 ... 6 Vbc, 7,22
3/транс, 9 ... 16 нв 3JCd, 4,67
СН3 СН2 z\! сн3 сн2 V, —4,5 37цис, 6... 12 3*^транс, 4 ... 7 Н Cl Vac, 0,96 VBD, 1,98 Vad, 0,75
меняются в пределах от —21,5 Гц (циклопентендион) до +42,4 Гц (формальдегид). По значениям этих констант можно судить о различного рода электронных эффектах. Электронодонорные заместители при метиловой группе приводят к увеличению константы 27нн- Влияние заместителя зависит также от его пространственного положения по отношению к паре геминальных протонов, т. е. обратно, из анализа рассматриваемых констант можно делать выводы о пространственном строении молекул.
Из теории следует, что вицинальные константы спин-спинового взаимодействия протонов 37нн, как правило, должны иметь положительный знак. Это наряду с положительным значением V;»ch (см. выше) служит отправной точкой для определения знаков в других случаях. Константы 3/Нн меняются в более узких преде-28
лах (4...20 Гц), чем геминальные, но они также, а иногда даже более чувствительны к структурным и конформационным изменениям. По значениям 3/нн легко идентифицируются цис- и транс-изомеры замещенного этилена (3/нн цис <3/нн транс)•
При исследовании конформаций алициклических соединений и поворотной изомерии производных этана можно использовать зависимость констант 37нн от двугранного угла фрагмента Н — С — С — Н, иллюстрируемую рис. 1.10 и приближенно описываемую уравнением Карп-луса:
Рнс. 1.10. Зависимость вицинальной константы спнн-спинового взаимодействия для фрагмента Н—С—С—Н от торсионного угла
Г cos2 6 + Т (в от 0 до 90°) •'НН= ^/^2cos2В+Г (в от 90 до 180°),
где /С]=8,5 Гц, /(2=9,5 Гц, Т = —0,28 Гц.
В общем случае для фрагмента X—С—С—Y зависимость константы 3JXY от двугранного угла 0 может быть записана в виде
3JXY = A cos 2В 4- В cos В 4- С, (1.27)
где А, В и С — эмпирические коэффициенты.
Константы спин-спинового взаимодействия протона с другими ядрами и тяжелых ядер между собой варьируются в очень широком диапазоне — от десятков до тысяч и более герц (в абсолютных величинах), что на некоторых примерах иллюстрирует табл. 1.8.
Спин-спиновую связь ядер рассматривают иногда как суммарный результат трех эффектов взаимодействия ядер и электронов. Во-первых, магнитный момент ядра оказывает воздействие на электрическое поле, обусловленное орбитальным движением электронов, а это поле, в свою очередь, взаимодействует с магнитным моментом другого ядра. Во-вторых, имеет место взаимодействие магнитных диполей, в котором участвуют не только ядра, но и электроны. И, наконец, учитывая симметрию атомных s-op-биталей, надо иметь в виду отличную от нуля электронную спиновую плотность на ядрах — так называемое контактное взаимодействие Ферми. При спин-спиновой связи протонов именно это взаимодействие является наиболее важным.
В случае других ядер дело обстоит много сложнее. Как и для химических сдвигов, приближенные теоретические расчеты констант спин-спинового взаимодействия обычно не приводят к хорошим результатам.
(1-26)
29
Таблица 1.8. Значения некоторых коистаит спин-снинового взаимодействия ядер разных элементов 'Jab (/=1,2)
Соединение Взаимодействующие ядра "лв- Гц
Н2ВН2ВН2 “В—>н ‘7=137 (терминальная)
» ‘7=48 (мостиковая)
СеН12 (циклогексан) *»с—>н ‘7=140
СН3ОН То же ‘7= 144
НСООН >7=218
CH2CI2 ‘7=162
СН3С! >7=193
СНВгз ‘7 = 208
СН2ВГ2 ‘7 = 185
СНзВг >7=153
Н—F >9F—‘Н ‘7 = 615
CH3F То же =7 = 44
CH2F2 27 = 53
CHF3 27=81
РН3 9‘Р—‘Н ‘7=179
О2Р—О—РОз ( ‘7=620
1 31р_1р 27=17
Н 1 О2Р—РОз ( 31 р 1р ‘7 = 444 1
1 27=94 .
н ( 3J р 1р ‘/ = 480 .
PF, 31р 19р ‘7=1405
POC!2F V =- И 70
F2PO(OH) | ‘7=980
На практике чаще используют эмпирические закономерности и корреляционные соотношения. Для сравнения спин-спиновоп связи ядер различных элементов оказались полезными так называемые приведенные константы (Н-А~2-м~3), учитывающие гиромагнитные отношения взаимодействующих ядер:
2л^ав
АВ~ ftYAYB
(1-28)
Значения приведенных постоянных не меняются для разных изотопов одного и того же элемента. Эту упрощенную константу удобнее использовать в приближенных расчетах.
Анализ структуры спектров ЯМР, рассмотренный выше, касался в основном достаточно простых спектров первого порядка, но часто наблюдаются гораздо более сложные спектры не первого порядка, которые на первый взгляд кажутся непонятными. Это случается тогда, когда разность химических сдвигов двух типов ядер не отличается в несколько раз от значений константы спин-спинового взаимодействия, как бывает при наблюдении спектров первого порядка, для которых характерно неравенство 30
kf^J. В спектрах не первого порядка изменяются положение и интенсивность сигналов ЯМР, как говорилось, например, о спектре спиновой системы АВ (с. 25), и расшифровка их требует других подходов.
Поскольку химический сдвиг б зависит от напряженности внешнего магнитного поля, а константа спин-спинового взаимодействия — нет, то регистрация спектров ЯМР при более высокой напряженности поля позволяет увеличить отношение Дб : J, т. е. приблизить картину спектра к первому порядку.
Спектры не первого порядка возникают также у систем с несколькими наборами химически эквивалентных, ио магнитно неэквивалентных ядер, что связано с симметрией системы. В этом случае никакое изменение напряженности поля не приблизит вид спектра к первому порядку.
Анализ спектров не первого порядка, если они не сводятся к первому, требует специального математического аппарата и моделей для расчетов положения и интенсивности линий, а также моделирующих и итерационных программ для использования ЭВМ. Когда в спиновой системе много взаимодействующих ядер, учитывают свойства симметрии с целью факторизации гамильтониана и сведения задачи к решению нескольких более простых. Так или иначе, в результате проводимого анализа сложных спектров не первого порядка получают значения химических сдвигов и констант спин-спинового взаимодействия, а иногда и важную ттлгтгч птттж'т'л ттт. ттчгт/ч тттт/Кгчг» w о тттлл тт пттг»тткеоп птиллито ттх. ULTO Quavu
1 j XV <1 I V 1 11W44 * W4V44XJU V
констант.
ГЛАВА II СПЕКТРОСКОПИЯ ЯМР (ПРИМЕНЕНИЕ И ТЕХНИКА ЭКСПЕРИМЕНТА)
1. ПРИМЕНЕНИЕ В СТРУКТУРНЫХ ИССЛЕДОВАНИЯХ
Спектроскопия ЯМР наряду и в сочетании с другими физическими методами является эффективным методом исследования химического строения молекул, стереохимической конфигурации и конформации. В гл. I была рассмотрена связь спектров ЯМР со структурой молекул и очевидно, что для решения обратной задачи, т. е. получения данных о структуре соединения, требуется использовать по возможности все параметры спектра ЯМР, а это по крайней мере следующие данные:
1) химический сдвиг сигнала ЯМР, определяемый по центру сигнала (мультиплета);
2) мультиплетность сигнала, связанная с числом взаимодействующих ядер и их спинами;
3) константы спин-спинового взаимодействия Чдв ядер;
31
4) отношение интенсивностей компонент мультиплета;
5) интегральная интенсивность сигналов (мультиплетов).
Как отмечалось, корреляционные таблицы и диаграммы химических сдвигов для разных изотопов (см. форзацы)—это первое, что может дать ориентировку относительно наличия тех или иных структурных элементов в молекуле. Однако все возможные группировки ядер и их сочетания, как и непрерывно растущее число данных, не могут быть охвачены никакими таблицами и диаграммами. Их можно, конечно, хранить в памяти ЭВМ, обслуживающих информационно-поисковые системы, которые уже существуют в некоторых научных центрах.
Существенный разброс значений химических сдвигов, наблюдаемый для одних и тех же структурных фрагментов молекул различных соединений, ограничивает возможности структурных исследований с применением корреляционных таблиц и диаграмм только решением некоторых простейших задач. В связи с этим практический интерес представляют расчетные оценки химических сдвигов, основывающиеся на различных теоретических концепциях. Квантово-механические методы расчетов, к сожалению, пока оказываются малоэффективными, и обычно используются чисто эмпирические подходы с привлечением разнообразных корреляционных соотношений и аддитивных схем расчета химических сдвигов.
В качестве примера можно привести корреляцию химического сдвига протонов с электроотрицательностью заместителей, позволяющую кроме оценки значений 6 по известной шкале электроотрицательностей проводить и обратную процедуру — по значениям 6 определять электроотрицательность заместителя, от которой зависит электронная плотность около протонов. Так, для фрагмента —СНХ—СН— при увеличении электроотрицательности атома X сигнал ближайшего протона смещается в сторону меньшей напряженности поля, т. е. химический сдвиг растет (a-эффект), а сигнал более удаленного протона — в сторону более сильного поля, т. е. химический сдвиг падает (р-эффект). Или, например, для протонов этильного радикала в соединениях СН3—СНг—X химические сдвиги могут быть представлены зависимостью
®сн3 — 8сна = 1 >464£х—2,61 м.д., (П.1)
где £х — электроотрицательность X. Аналогичные линейные зависимости наблюдаются и для ряда других соединений RX (R— различные радикалы), но пределы применимости уравнений вида (П.1), конечно, весьма ограничены.
Один из примеров простых аддитивных схем расчета химических сдвигов представляет следующее уравнение (Грант и Пол), по которому можно определить химический сдвиг в спектроскопии ЯМР 13С для k-ro углеродного атома 6ftc в ацикличе-32
ских углеводородах:
— ®ЭТ + 2д п^^1< (И-2)
Г-1 где бэт — эталонный сдвиг; nki —.пело заместителей в i-м положении относительно k-vo атома; Ay — инкремент химического сдвига от i-ro заместителя (равен 9,1; 9,4; — 2,5; 0,3 и 0,1 м.д. для заместителей в а-, 0-, у-, б- и е-положениях).
Предложены и более детализованные эмпирические соотношения и таблицы инкрементов, позволяющие рассчитывать химические сдвиги для некоторых рядов соединений с достаточно высокой точностью.
При анализе взаимосвязи химических сдвигов со структурой и пространственным строением молекул широко применяется также метод аналогий и модельных соединений, которые выбираются так, чтобы они имели структуру или фрагменты, близкие к возможным в рассматриваемом соединении, и химические сдвиги для них были бы известны. Например, химический сдвиг 31Р при данном валентном состоянии атома варьируется, как было видно, в очень широких пределах в зависимости от заместителей. Объяснить и тем более рассчитать химический сдвиг для каждого конкретного соединения — задача чрезвычайной сложности. Однако если известны химические сдвиги для нескольких родственных соединений ряда, например РХ3, PX2Y, PXY2, PY3 и т. п., то можно предсказать химический сдвиг для неизученного соединения путем простой интерполяции или экстраполяции. На рис. II. 1 иллюстрируется, как в ряду фосфинов с заместителями —Н, —PF2 и SiH3 предсказывается химический сдвиг для соединений PH(PF2)2 и P(PF2)2(SiH3). По таким предсказываемым значениям химических сдвигов можно затем идентифицировать еще не изученные соединения.
В спектроскопии ПМР, когда протонные химические сдвиги сравнительно невелики, из-за перекрывания мультиплетных сигналов спектры часто бывают сложными. Как уже указывалось, один из возможных путей упрощения спектров не первого порядка (приближения их к первому порядку за счет увеличения разницы химических сдвигов сигналов) является повышение напряженности постоянного поля Н (т. е. использование соответствующего спектрометра). Другим путем упрощения является применение метода двухмерной спектроскопии ЯМР. При отсутствии таких возможностей иногда используется еще один путь — добавка небольших количеств комплексных соединений парамагнитных ионов лантаноидов с дикетонами так называемых сдвигающих реагентов. Эти реагенты могут индуцировать разные по величине сдвиги резонансных сигналов у различных групп протонов, увеличивая расстояния между сигналами и устраняя их перекрывание. Это происходит в результате образования ионом лантаноида до-2—1377 33
О -100 -200 -500 -WO
Рис. ПЛ. Закономерность изменения химического сдвига
б 31Р в замещенных фосфинах с Н, PFa и SiH3 в качестве заместителей:
кружками показаны предсказываемые значения
полнительных координационных связей с какими-то определенными группами атомов исследуемого соединения. Наиболее распространенными сдвигающими реагентами являются хелатные комплексы Еи(Ш) и Рг(Ш) с дикетонами:
в частности с 2,2,6,6-тетраметил-3,5-гептандионном [Р=С(СН3)з], 1,1,1,2,2,3,3-гептафтор-7,7-диметил-4,6-октандионном (R=C3Fz)
и др.
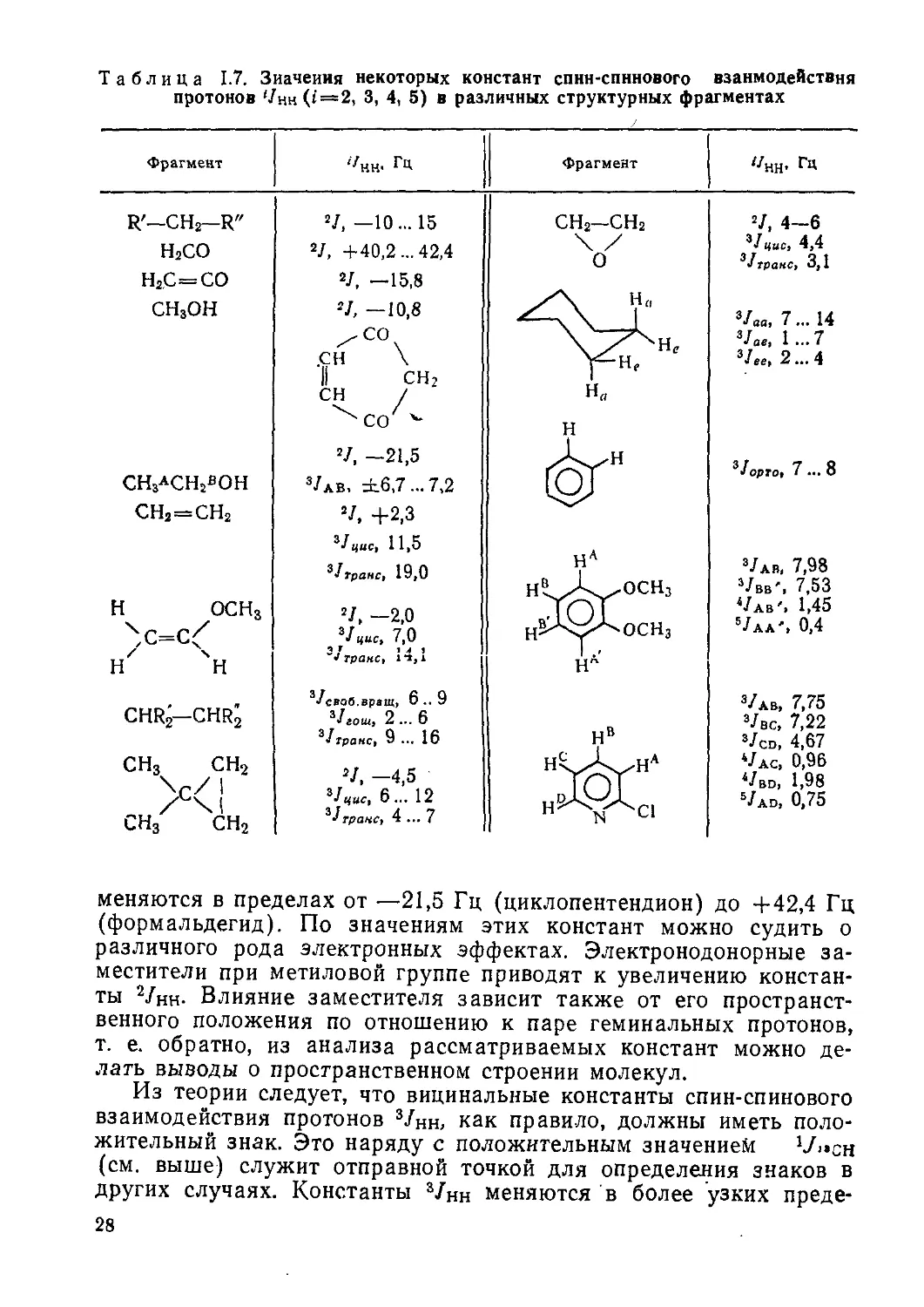
В специальных учебных пособиях и руководствах приводится много примеров и упражнений по расшифровке спектров ЯМР, особенно органических соединений. Рассмотрим здесь только один из таких примеров. На рис. II.2 представлен спектр ПМР полученного ацетилированием ароматического соединения. Брутто-формула соединения C10H13NO2, т. е. оно относится к производным ряда СпН2п-8 и кроме бензольного кольца содержит, очевидно, двойную связь С = О ацетильной группы СН3СО. В спектре ПМР видно шесть сигналов. Самый интенсивный синглетный сигнал при 2,1 м.д. относится к протонам ацетильной группы. Один из заместителей, несомненно, содержит этильную группу, дающую в спектре квартет (6=4 м.д.) и триплет (6 = 1,4 м.д.). Судя по химическому сдвигу протонов группы —СН2, она не связана непосредственно с бензольным кольцом, а связана с атомом кис-
34
ТМС
в 7 6 5*3 2 1 О В
S, м.д.
Рис. II.2. Спектр ПМР соединения C10H13NO2
лорода: —О—С2Н5 (вариант —СООС2Н5 отпадает, так как второй атом кислорода содержится в ацетильной группе). Слабый синглетный сигнал (6 = 7,9 м.д.) отвечает одному протону, и на основании значения б, а также большой ширины его можно было бы отнести к фенольной ОН-группе, что не будет согласовываться с брутто-формулой и предыдущими заключениями, или к амидной группе NH, что и соответствует действительности, т. е. имеет заместитель— NHCOCH3. Оставшиеся четыре линии спектра следует отнести к образующим спиновую систему А2В2 и находящимся в этих парах в орто-положении друг по отношению к другу протонам бензольного кольца, о чем говорят как положение сигналов по шкале химических сдвигов, так и интенсивности линий. Отсюда следует вывод о пара-замещении бензольного кольца, и окончательное заключение о строении исследуемого соединения, выражаемом формулой:
nhcocHj
Значения констант спин-спинового взаимодействия, как следует из сказанного (см. гл. I; 2.3), также могут служить для целей идентификации и вместе с мультиплетностью и соотношением интенсивности компонент сигнала несут ценную структурную информацию. Прямые и геминальные константы ('/ и 2/) характеристичны для типов связей атомов с магнитными ядрами, т. е. для валентных состояний атомов или гибридизации АО. Так, например, 2* 35
константа 'Vcm имеет в зависимости от гибридизации АО углерода примерно следующие значения (Гц): sp3~125, sp2~160, sp~250; по значениям константы VsipiH^lSO и ~400 Гц легко различить грех- и четырехкоординированный атом фосфора и т. д. Чувствительны эти константы, конечно, и к ближайшему окружению связей.
Вицинальные константы 3/ и константы более далекого взаимодействия спинов зависят от пространственного строения фрагментов (молекул). Например, по константам 3/ идентифицируются цис-, транс-, син-, анти-, поворотные н конформационные изомеры (см. табл. 1.7 и рис. 1.10) при условии статической изомерии, т. е. когда потенциальные барьеры изомеризации достаточно велики. Имеется также возможность изучения методом спектроскопии ЯМР, в частности, по константам спин-спинового взаимодействия хиральности молекул.
Хиральность — это свойство объекта быть несовместимым со своим зеркальным отображением. Так, например, молекулы, у которых нет зеркально-поворотной симметрии, являются хираль-ными. Молекула называется прохиральной, если она может быть превращена в хиральную единственным изменением какого-либо ее фрагмента. В тех и других молекулах некоторые группы ядер, казалось бы химически эквивалентные, могут быть магнитно неэквивалентными, что проявляется в спектрах ЯМР. Такое явление, называемое диастереотопией ядер, наблюдается по спектрам ЯМ1Р при совмещении в одной молекуле хиралыюго и прохирального фрагментов.
Например, в прохиральной молекуле
о
Р1Г О ^''F F
две группы — OPF2 эквивалентны, так как через фрагмент I
Ph—Р=О проходит плоскость симметрии, но в каждой из групп
PF2 два атома фтора неэквивалентны, чего и не требует симметрия. Это проявляется в константе спин-спинового взаимодействия 2/FF. Вообще, в оптически активных молекулах неэквивалентность ядер X в пирамидальных группах —МХ2 (—PF2, —NHs) или тетраэдрических группах —MX2Y (например, —CH2R, SiH2R н др.) не зависит от высоты барьера внутреннего вращения этих групп, в то же время при внутреннем вращении плоских групп —МХ2 и тетраэдрических групп —МХ3 потенциальный барьер обычно настолько низок, что ядра X становятся эквивалентными.
36
Существенное значение для структурных исследований, особенно в неорганической химии, имеет влияние изотопов низкого природного содержания («примесных спинов») на спектр ЯМР основных ядер изучаемого соединения. Когда рассматриваются спектры ЯМР на ядрах 'Н, 19F, 31Р, 103Rh и др. ('/ = 1/г), природное содержание которых 100% или близко к таковому (в обогащенных образцах), а в молекулах присутствуют только такие ядра, указанной проблемы не возникает. Но часто при исследовании спектров ЯМР приходится сталкиваться с проявлением эффекта «примесных спинов», например изотопов 13С, 29Si, 183W и др.
Так, большинство молекул WF6 ( — 86%) содержит немагнитное ядро W(7 = 0), и только 14% содержит ядро 183W(7 = 7г), которое дает септетный сигнал ЯМР, обусловленный спин-спиновым взаимодействием с шестью эквивалентными ядрами 19F, т. е. спектры ЯМР самих примесных ядер определяются обычными правилами, обсуждавшимися выше для изотопов высокого содержания. Все 100% молекул WF6 дают, однако, спектр ЯМР 19F, но для —86% молекул этот спектр представляет синглет, а для 14%—дублет с расщеплением Vwf, обусловленный спин-спиновым взаимодействием с ядром 183W. Таким образом, спектр ЯМР 19F соединения WF6 в итоге представляет как бы триплет из центральной линии и двух сателлитов с отношением интенсивностей примерно 1 : 12 : 1 [(14 : 86) ~ (1 : 6)]. Из такого рода спектров получают много полезной структурной информации.
Естественное содержание большинства примесных изотопов со спином / = !/2 составляет около или менее 15%, так что при наличии в соединении нескольких ядер данного элемента вероятность нахождения в одной и той же молекуле двух ядер примесного изотопа мала и ею можно пренебречь. Поэтому, например, в спектрах ЯМР 13С не наблюдается расщепления основных сигналов, обусловленного спин-спииовым взаимодействием ядер !3С между собой.
Исключение составляют примесные изотопы ксенона и платины. В частности, естественное содержание 195Pt около '/3 (33,8%), так что спектр ЯМР ядер со спином 1=4%, связанных в молекуле с атомом платины, будет состоять из трех линий с отношением интенсивностей 1:4:1, и по такой картине спектра можно установить наличие атомов Pt.
В случае систем (Pt2X), когда X связан с двумя ядрами Pt, картина несколько сложнее. Доля молекул, равная (2/з)2 = 4/9, не имеет примесных спинов, и наблюдаемый спектр ЯМР ядер X представляет синглет. У (1/з)2 = 1/э части молекул оба ядра 195Pt примесные, т. е. спектр X будет триплетным с соотношением интенсивностей 1:2:1. Наконец, в остальных (4/э) молекулах будет одно примесное ядро, и спектр X представляет дублет (1 : 1). Суммарный спектр ЯМР X системы (Pt2X) является та-
37
ким образом квинтетом с соотношением интенсивностей 1:8:18:8:1 или, пренебрегая крайними слабыми сателлитами, увидим приблизительно триплет 1:2:1, картина опять-таки весьма характерная для PtjX.
2. ФИЗИКО-ХИМИЧЕСКИЕ ПРИМЕНЕНИЯ
Характеристики спектров ЯМР находят применение не только в структурно-аналитических, но и в других целях. Найдено много корреляционных соотношений спектральных параметров в рядах соединений с другими физико-химическими характеристиками. Как уже указывалась, например, зависимость химического сдвига от электроотрицательности заместителей в ближайшем окружении данного атома. В физической органической химии находят применение корреляции б с индексами реакционной способности, постоянными Гаммета и Тафта заместителей в ароматических соединениях и т. п.
Такого рода корреляционные соотношения могут служить не только для оценки значений химических сдвигов, но позволяют решать и обратную задачу — определять по экспериментальным значениям б нужные физико-химические характеристики. Найдена, например, следующая зависимость a-постоянных Гаммета от химического сдвига протонов &nh« в замещенных анилинах для 5—15%-ных растворов в ацетонитриле:
О = 1,027 (3NH,-3,84). (П.З)
Успешно применяют спектроскопию ЯМР для изучения донорно-акцепторных комплексов, причем химические сдвиги 13С, как оказалось, являются, например, более информативными, чем сдвиги *Н в отношении механизма комплексообразования. Хотя в общем значения Sisc не коррелируют с зарядовыми плотностями, спектроскопия ЯМР 13С широко используется для изучения электронной структуры органических и элементорганических соединений и, несомненно, позволяет получать важные данные.
Большую роль спектроскопия ЯМР сыграла в развитии теоретических концепций органической химии, касающихся, в частности, строения и стереохимии интермедиатов и механизмов химических реакций. Получены структурные данные о таких интермедиатах многих практически важных химических реакций, какими являются карбкатионы и карбанионы. Например, в случае изопропильного катиона значения химических сдвигов 8>н и 8.зС показывают значительное дезэкранирование магнитных ядер, особенно углерода, а значение константы спин-спинового взаимодействия V13CH свидетельствует о практически плоской структуре центральной части катиона (т. е., что гибридизация центрального атома углерода близка к sp2). Исследуют как классические кар-бониевые ионы, так и неклассические а-мостиковые карбкатионы, 38
которые идентифицируются по спектрам ЯМР, в частности, по сравнительно большому значению константы Мен-
Для изучения механизмов и кинетики химических реакций применяется, как известно, метод струи и остановленной струи. В последние годы большие успехи достигнуты в этом направлении благодаря применению спектроскопии ЯМР для наблюдения за ходом реакции с одновременной идентификацией промежуточных продуктов (интермедиатов). В проточных системах происходит некоторое дополнительное уширение линии, определяемое соотношением
1 I 1
л/2(эф) лГ2(стат) f
vjifi Т2(3ф)—наблюдаемое или эффективное время спин-спиновой релаксации в потоке; Т^стат)— время релаксации для статического образца; t — время пребывания образца в измерительных катушках.
Чем больше скорость потока (меньше t), тем больше уширение, но оно предсказуемо и может быть скорректировано. Метод остановленной струи используется для измерения скорости образования и распада интермедиатов, когда после остановки потока многократно и быстро осуществляется регистрация соответствующей области спектра. Такие исследования получили развитие особенно в результате появления и внедрения в практику импульсной фурье-спектроскопии.
Эффективный метод изучения химических реакций, протекающих с промежуточным образованием радикальных пар, основывается на использовании явления так называемой химической поляризации ядер (ХПЯ), о котором вместе с явлением химической поляризации электронов еще будет сказано в главе, посвященной рассмотрению спектроскопии ЭПР.
Интенсивность линий в спектрах ЯМР может использоваться не только для расшифровки спектров и структуры молекул, но, как очевидно, и для количественного анализа. Метод спектроскопии ЯМР применяется в этих целях как в статических условиях (например, ЯМР 19F и на других ядрах) при соответствующей калибровке и принятии мер предосторожности для устранения ошибок из-за явлений насыщения переходов, так и в динамических условиях, о чем уже говорилось.
Для проведения количественного анализа смесей различного рода (растворитель и растворенное вещество, смеси изомеров, включая оптические изомеры, изотопомеров, реакционные смеси) Используются интегральные интенсивности сигналов, которые пропорциональны концентрациям компонент. При этом, конечно, важно, чтобы сигналы были достаточно смещены друг относительно друга, т. е. чтобы компоненты были спектрально «разрешены».
39
Следует оговорить, что спектры ЯМР оптических изомеров в оптически неактивной (ахиральной) среде не различаются между собой. Для индуцирования их неэквивалентности, делающей возможным исследование оптической чистоты, в раствор обычно добавляют хиральный сдвигающий реагент, например трис-(3-три-фторметилоксиметилен-о!-камфорато)-европий:
°Х/°
3. ДИНАМИЧЕСКИЙ ЯМР
Спектроскопия ЯМР широко и успешно применяется для исследования равновесных химических превращений и обменных процессов, при которых периодически меняется строение, а значит, электронное окружение магнитных ядер и спин-спиновое взаимодействие ядер, т. е. химические сдвиги б и константы /. К таким процессам относятся как внутримолекулярные превращения (заторможенное внутреннее вращение, инверсия пирамидальной системы связей у азота, инверсия циклов, таутомерия и т. д.), так и межмолекулярные обменные и другие равновесные химические реакции (протонный обмен в водных растворах карбоновых кислот, аммиака, лигандный обмен, рекомбинация ионов, биохимические взаимодействия фермент—субстрат и т. д.).
Характеристическое время метода ЯМР (временная шкала) охватывает диапазон от 10-1 до 10~6 с. В соответствии с принципом неопределенности Гейзенберга
Д£Дг?>Й, (II.5)
учитывая, что A£=/iv, а М отождествляя с временем жизни т ядра в одном из состояний, можно записать:
(П-6)
где Av — разность резонансных частот (химических сдвигов или констант спин-спинового взаимодействия).
Рассмотрим простейший случай реакции двухпозиционного обмена:
А^В
где А и В — формы молекул с разным положением обменивающих* ся ядер, время жизни которых тд и тв.
Будем считать, что ядра в состояниях А и В имеют одинаковое время спин-спиновой релаксации Т2, и введем эффективное время жизни т=хаТа=ХвТв, где молярные доли ха и хв (Ха=1— Хв) легко 40
выражаются через константу равновесия. Для вырожденных случаев, когда потенциальные ямы для молекулярных форм А и В одинаковы, например, для устойчивых шахматных конформаций этана при заторможенном внутреннем вращении:
или для конформаций кресла циклогексана:
на
Ха=Хз=1/2, а т=Та/2=Тв/2.
Время жизни связано с константами скорости реакций; для рассматриваемого случая k\=l/rk, и обе величины находятся в зависимости от потенциального барьера (энергии активации) обменной реакции и температуры. Из теории активного комплекса известна следующая зависимость константы скорости:
ьт
k — n—-—ехр(—ДС*/7?Г), (II.7)
л
где " — трансмиссионный коэффициент; k — постоянная Больцмана; ДС * — свободная энергия активации (AG* =Д/7*—TAS*). Часто принимают равенство свободной энергии активации потенциальному барьеру (AG* «Д/7* « Vo, предполагая Д5*=0 и х=1), тогда записывают:
k = Л exp (-Vo/ЛГ), (II.8)
а пренебрегая зависимостью коэффициента А от температуры,
|fe = 6,2-1012exp (—У0/ЛГ)7| (II-9)
Формулой (П.9) обычно и пользуются для оценок Vo^AG* по константам скоростей обменных процессов, найденным методом динамической спектроскопии ЯМР.
Вид сигнала ЯМР сложным образом зависит от т и резонансных частот, но, следуя (П.6), по соотношению т и Av°ab=v°a—v°b можно выделить следующие характерные области обмена. Если удовлетворяется соотношение
t»1/Av;b, (II. Ю)
то наблюдаются отдельные сигналы v°a и v°b для состояний А И В, т. е. обмена нет, или происходит медленный обмен
41
Рис. 11.3. Температурная зависимость формы сигналов ЯМР для двухпознционного обмена Аз* В:
а — вырожденная система (тА—тв); б —невырожденная система (тА>тв)
ЮО кДж/моль): va=H= #=v°a и vb#=v°b (рис. П.З, а, позиции 1 и 2), a Avab¥= #=Avoab- Возможно обменное уширение сигнала, которое имеет для сигнала А вид
где первый член — релаксационный, а второй — обменный.
Для вырожденных систем оба сигнала va и vb ведут себя одинаково (рис. П.З, а), а для невырожденных сигнал менее заселенного состояния «размывается» быстрее (vb на рис. П.З,б).
В области промежуточного обмена (20<V0< < 100 кДж/моль) время жизни сравнимо с обратной разностью частот отдельно наблюдаемых сигналов:
t»I/Avab, (11.12)
и по мере уменьшения т линии все более сближаются и размываются, затем образуют общий контур н, наконец,
сливаются в одну линию
(рис. П.З,а,б, позиции 3—5). Для вырожденных систем точка слияния (коллапса, коалесценции) соответствует
т = /2/2лДуав
(11.13)
для невырожденных ее точно определить нельзя.
Наконец, при быстром обмене (У0<20 кДж/моль), когда
Г«1/А*;в, (И. 14)
в спектре будет наблюдаться одна линия (рнс. П.З, а, б, позиция 6) при частоте v=xavca+xbv°b. Ее ширина для вырожденных систем определяется формулой
6v=-4- + n2T(Av°B)2, (11.15)
Л/2
42
а для невырожденных
bv = + 4«2x1x2b (Av1b)2(ta + тв). (П-16)
При медленном и промежуточном обмене в вырожденных системах отношение наблюдаемой разности частот Avab к разности Av°ab в отсутствие обмена
Avab / 1 \1/2
--= 1 - —---------~• (11.17) AVAB \ 2rf2t2(AVAB)2 )
При больших значениях Avab анализ формы линии существенно затруднен.
Метод динамического ядерного резонанса позволяет изучать кинетику реакций первого порядка с константами скорости от 10 до 106 с-1, что соответствует свободным энергиям активации (барьеру) от десятков до 100 кДж/моль. Быстрые процессы обнаруживаются по уширению линий дополнительно к релаксационному. Если для надежности идентификации такого дополнительного уширения принять его величину не менее 2 Гц, то, используя формулу (11.15) для вырожденных систем, можно оценить верхний предел констант скоростей, допустимых для определения при заданной разности Av°ab:
*пред " ’“’л2(Д*°Ав)2- (II. 18)
Из формулы видно, что возможности метода зависят от разности резонансных частот.
В спектроскопии ЯМР эксперимент обычно проводится при температурах в диапазоне ~ 120...470 К, но не всегда удается исследовать спектры в достаточно широком интервале температур даже этого диапазона, что ограничивает круг изучаемых процессов. Так, даже при нагревании до ~ 200 °C (верхний температурный предел, обусловленный конструкционными характеристиками спектрометров) для систем с энергией активации обменного перехода ~80...100 кДж/моль будет наблюдаться лишь начало медленного обмена.
В невырожденных системах, если содержание одной из форм менее 1% (хв<0,01), т. е. разность энергий форм AG (или, грубо, АГ — разность глубины потенциальных ям) более ~15 кДж/моль, то чувствительность метода динамического ЯМР становится недостаточной для фиксирования обменного процесса. Оптимально его применение к изучению состояний при сравнимых заселенностях.
Рассмотрим некоторые примеры. Упоминавшаяся ранее инверсия циклогексанового кольца в конформациях кресла приводит к «обмену» аксиальных и экваториальных протонов местами. При комнатной температуре в спектре ПМР наблюдается только один сигнал и лишь Цри понижении температуры (до-------70°C) видны
43
отдельные линии аксиальных и экваториальных протонов. Процесс инверсии циклогексана можно отнести к типу промежуточного обмена вырожденной двухпозиционной системы. Методом динамического ЯМР константа скорости обмена (при указанной Т) найдена как k=60 с-1, а барьер инверсии V0=42,2 кДж/моль.
Интересный пример четырехпозиционного обмена представляют конформационные переходы: кресло^кресло Л'-хлорпиперидина:
Здесь переход аксиальной конформации в экваториальную может происходить как путем инверсии шестичленного цикла, характеризуемой константой скорости klt так и путем инверсии пирамидальной системы связей при N — константа скорости k2. Методом динамического ПМР (характеристическое время ЯМР ’Н 10-4 с) были определены константа первого процесса k\ и потенциальный барьер инверсии цикла Роцикл~56,5 кДж/моль. Второй процесс, хотя относится тоже к промежуточному обмену, оказывается несколько быстрее. Он изучен методом динамического ЯМР 1гС(/~ 10-6 с), и барьер инверсии связей при N оценен как V0"HB~ «46 кДж/моль. Оба значения барьера являются эффективными величинами, так как это слабо невырожденная система, т. е. аксиальная и экваториальная конформации C5H10NCI отличаются по энергии (AV/«AG°«6 кДж/моль, конформация е обладает меньшей энергией), и энергии активации переходов а-+е и е-*-а также отличаются (на указанную величину).
Осложнения при изучении динамических процессов, возникающие из-за наличия спин-спинового взаимодействия, его температурной зависимости (в меньшей степени), одновременно протекающих обменных реакций и некоторых других причин, обходят, применяя методы множественного резонанса и другую технику спектроскопии ЯМР, рассматриваемую ниже.
4. ТЕХНИКА И МЕТОДИКА ЭКСПЕРИМЕНТА
4.1. Спектрометры ЯМР
Устройство спектрометров ЯМР имеет в своей основе возможность выполнения условий ядерного магнитного резонанса (1.10), и при рассмотрении в гл. I (1.3) принципов реализации этих усло
44
вий была показана блок-схема спектрометра стационарного типа (см, рис. 1.3). Современные спектрометры ЯМР предназначены обычно для работы на разных ядрах, что достигается изменением диапазона генерируемых частот. Это относится как к спектрометрам стационарного типа, так и к импульсным фурье-спектрометрам, которые отличаются способом генерирования высокой частоты.
После внедрения в 60-х годах электронно-вычислительной техники в физический эксперимент была реализована возможность получения спектров ЯМР высокого разрешения путем фурье-преоб-разования сигнала ССИ (см. гл. I; 1.3) после воздействия короткого (порядка 10-5—10“6 с) мощного (от 1 кВт) импульса электромагнитного поля с несущей частотой v. Действие импульса Bv продолжительностью tp состоит в повороте вектора намагниченности М на угол а, равный согласно (1.14) ynBvtp.
Частота v не обязательно должна совпадать с резонансной частотой, так как при малой продолжительности импульса его можно представить как целый интервал частот Av или сумму бесконечного числа гармоник (членов разложения в ряд Фурье). Этот интервал, обратно пропорциональный продолжительности импульса, можно представить как v±l/tp.
При этом в интервале Av найдется частота, совпадающая с резонансной vo, т. е. произойдет поворот вектора намагниченности в
плоскость хц и наведение в катушке ССИ, если tn^—-—• Так, на-
4Дм
пример, микросекундный импульс может возбудить все переходы ядер с резонансными частотами в интервале ~50 кГц. При неравенстве частоты импульса и резонансной частоты v#=vo будет наблюдаться картина, аналогичная виглям при быстром прохождении узкого сигнала в стационарном спектрометре ЯМР (см. рис. 1.4), что обусловлено затухающими колебаниями ССИ частоты vo—v.
На практике осуществляется многоимпульсная (с промежутками t между импульсами в несколько секунд) последовательность с накоплением сигнала ССИ и фурье-преобразованием полученной интерферограммы на ЭВМ. Интерферограмма, представляющая супер-позицию ССИ, является функцией времени f(t) и зависит от спектра резонансных переходов ядер (ЯМР), который обозначим как функцию F(v). Экспериментатора интересует обратная задача— получение спектра ЯМР, что достигается фурье-преобразованием временной функции в частотную:
F(>) =2л J e^fyydt. (11.19)
На рис. П.4 показан, например, вид сигнала ССИ и полученный из него после фурйе-преобразования спектр ЯМР (в шкале б ПВ).
45
Регистрация сигналов ССИ обычно требует в сотни раз меньше времени, чем запись спектра ЯМР на стационарном спектрометре. Время фурье-преобразования интерферограммы на ЭВМ также мало. Таким образом, за короткое время может быть накоплено и усреднено много сканов*, что дает многократный (в десятки — сотни раз) выигрыш в чувствительности метода (отношении сиг-нал/шум).
Применение импульсной фурье-спектроскопии ЯМР особенно эффективно при изучении спектров изотопов с низким естест-
венным содержанием.
В настоящее время стала рутинной регистрация спектров ЯМР 13С, распространяется спектроскопия ядер 15N, 17О, 19F,
31Р. Высокочувствительные импульсные фурье-спектрометры со
।_______________1_______________।______________-
о -ю -?о S, м.р.
5/
Рис. П.4. Сигнал ССИ (а) н полученный его фурье-преобразованием спектр ЯМР "В (б)
сверхпроводящими селеноидами позволяют регистрировать спектры ЯМР практически всех изотопов с магнитными ядрами. Метод
широко используется для измерения времени релаксации, появилась возможность получения спектров высокого разрешения твердых тел, проводить дифференциальную регистрацию, изучать сложные мультиплетные резонансы и т. д.
При импульсной технике фурье-спектроскопии ЯМР интенсивность спектральных линий зависит от времени спин-решеточной релаксации Т1( которое может сильно отличаться у разных ядер одного изотопа в молекуле, но эти затруднения легко преодолеваются. В стационарном методе таких трудностей вообще нет, и интенсивность пропорциональна числу ядер. Хотя фурье-спектрометры ЯМР сложнее и пока дороже, они неуклонно вытесняют стационарные спектрометры.
* Пробег спектрального интервала с регистрацией спектра и запоминанием его ЭВМ.
46
Принципиальная блок-схема импульсного спектрометра показана на рис. II.5. Основные его преимущества следующие. Прежде всего это высокая мощность генератора (до 1 кВт, тогда как в стационарном спектрометре доли ватт) импульса электромагнитного поля порядка 10-1...l Т. Контур датчика ЯМР, подводящий радиочастотное поле Bv, должен
Рис. II.5. Блок-схема импульсного фурье-спектрометра ЯМР:
/ — образец; 2 — полюса магнита; 3 — ЭВМ; 4 — генератор; 5 — усилитель мощности; 6 — приемник; 7 — аналого-цифровой преобразователь; 8 — цифро-аналоговый преобразователь; 9 — самописец; 10 — телетайп (оператор)
надежно работать в этих жестких условиях и быть чувствительным к слабым сигналам ССИ в промежутках между импульсами, поэтому его связь с генератором и приемником должна удовлетворять более
высоким конструкционным требованиям, чем в стационарном спектрометре. Это же относится ко второму контуру, подводящему вторую частоту V2 для двойного резонанса (см. ниже), и третьему контуру датчика, служащему для стабилизации условий резонанса. Для формирования всех видов импульсов имеются модулятор и программирующий блок. Фазочувствительный детектор обеспечи-
вает высокое отношение сигнала к шуму.
На совмещенную с фурье-спектрометром ЯМР электронно-вычислительную машину, являющуюся, по существу, его неотъемлемой составляющей частью, возглагаются функции управления спектрометром по заданной программе или в соответствии с командами, подаваемыми оператором. ЭВМ формирует импульсы, накапливает сигнал ССИ и преобразует его в спектр, хранит информацию в памяти и по команде выдает или в цифровом виде, или через цифроаналоговый преобразователь графически. Кроме того, ЭВМ может выполнять много других операций по обработке данных, улучшению качества спектра, упорядочению и систематизации информации.
Импульсные фурье-спектрометры выпускаются с железными и сверхпроводящими магнитами. К магнитам в любом спектрометре предъявляются особые требования, и до 70% стоимости прибора приходится на магнит и обеспечивающие его работу системы (питание, стабилизация и др.). Основное требование — достаточно высокая индукция (1... 7 Т), однородность и стабильность статического поля в той части ампулы с образцом, которая находится в резонансной катушке, помещаемой между полюсами магнита. Зазор между полюсами бывает обычно 20 ...40 мм, стандартная ампула представляет цилиндр длиной 15;.. 20 см и диаметром 5 ...
47
... 10 мм, а высота его части, находящейся в катушке, варьируется (~10 см); используют и микроампулы. Простой и эффективный способ повышения однородности поля на образце — вращение ампулы вокруг ее продольной оси со скоростью ~20...30 об/с. Однородность поля увеличивают также с помощью дополнительных тонких электрических катушек (шиммы), накладываемых на полюсные наконечники магнита (см. рис. 1.3). Используются также различные системы стабилизации ЯМР спектрометров.
4.2. Двухмерная спектроскопия ЯМР
В конце 70-х годов появилась новая перспективная методика импульсной спектроскопии — двухмерная спектроскопия ЯМР* в разных ее разновидностях в зависимости от набора формируемых импульсов. При многоимпульсной последовательности промежутки между импульсами влияют на вид получаемого спектра ЯМР. Можно получить, например, ряд спектров обычным фурье-преобразова-нием сигнала ССИ при закономерном увеличении времени задержки t (промежутков между импульсами). В каждом из этих спектров интенсивность представлена как функция частоты v. После этого при фиксированном значении частоты в этом наборе спектров можно провести еще одно преобразование сигнала эха ССИ, представив интенсивность как функцию времени t задержки импульсов.
Если это проделать для всех частот, то в результате и будет получен «двухмерный» спектр как функция двух частот: частоты импульса (поля) v и частоты следования импульсов 1/7.
Наиболее важным вариантом методики 2D ЯМР является так называемая двухмерная б—/-спектроскопия, в которой дается зависимость интенсивности от двух аргументов — химического сдвига б и константы спин-спинового взаимодействия J. Эти аргументы откладываются на взаимно перпендикулярных осях координат, а фактически трехмерное изображение спектра напоминает горный рельеф (рис. II.6). При 7=const (б-сечение) получается набор синглетных сигналов групп неэквивалентных ядер, а при каждом б = const (J-сечение) получается информация о мультиплетно-сти сигналов и константах спин-спинового взаимодействия. Метод позволяет избежать перекрывания мультиплетов и упростить расшифровку спектров. Большим преимуще-
Рис. II.6. Вид спектра 13С ЯМР А в с D
НОСН2—СН2—СН(ОН)—СНз в трехмерном пространстве, полученного методом двухмерной (2D) .
6-7-спектроскопии
* Иногда обозначают 2D ЯМР (от англ, two-dimensional — двухмерная). 48
ством является также более высокое разрешение в двухмерном спектре ЯМР.
Метод 2D ЯМР спектроскопии требует высококачественной аппаратуры и более длительного времени эксперимента, чем в обычной импульсной фурье-спектроскопии, что обусловлено необходимостью большего числа сигналов ССИ.
4.3. Двойной резонанс
Обычный эксперимент в спектроскопии ЯМР предусматривает наложение одного радиочастотного поля B = BV° cos(2nv/ + 6) перпендикулярно статическому полю BV±B (однократный резонанс, см. гл. I; 1). Однако большинство современных спектрометров ЯМР дают возможность работать в условиях двойного резонанса, когда дополнительно к полю регистрации Въ накладывается второе возмущающее радиочастотное поле BV2, причем также BV2J-B. Если наблюдают спектр ЯМР ядер А на частоте vi для системы взаимодействующих ядер [ИХ], то частота возмущающего поля v2 выбирается в резонансной области ядер X, что обозначается следующим образом: А— {X}, например 13С—{'Н} (ядра !3С наблюдаются, протоны облучаются).
Методы двойного резонанса различают по видам в зависимости от способа генерации частоты v2, способов изменения (развертки) vi и v2 и от мощности возмущающего поля (амплитуды В°Чг). В разных методах могут наблюдаться разные эффекты возмущения спектра — изменение распределения интенсивности и положения линий, упрощение и детализация спектра. По наблюдаемому спектру кроме данных о ядрах А получают и информацию и о ядрах X без прямого наблюдения спектра ЯМР этих ядер, а некоторые данные могут быть получены только методом двойного резонанса.
Рассмотрим спиновую систему [ЛВ], энергетические уровни которой во внешнем магнитном поле схематично показаны на рис. II.7. Первая буква в обозначении мультипликативной функции спинового состояния относится к ядру А, а вторая — к ядру В. При тепловом равновесии различия в заселенности уровней пропорциональны разностям их энергий, так как по сравнению с kT они малы. На рис. II.8, а показан спектр однократного ЯМР в таких условиях.
Если частота v2 совпадает с частотой некоторого перехода, например Bi (см. рис. II.7), и амплитуда у/?»2 сравнима по величине с фактором (ЛТ'г)-1''2, тс выравнивается заселенность уровней аа и а|3, т. е. происходит насыщение уровня ар по ядру В. Тогда интенсивность линии, соответствующей переходу Alf уменьшится, линия перехода А2 — увеличится, а линия перехода Bi исчезнет (рис. II.8,б). Это явление называется ядерным эффектом
49
Спиновое состояние
Изменение заселенности при Сличении с частотой перелова :
Не . меняется Увеличивается
Не меняется Уменьшается
Увеличивается Не меняется
Уменьшается Не меняется
Рис. II.7. Схема уровней энергии и переходов для спиновой системы [ДВ], 7лз=й=0
Оверхаузера (ЯЭО), нй котором и основан метод спектроскопии межъядерного двойного р е з о н а н с а (ИНДОР*).
Аналогичным образом облучение системы [АВ] частотой перехода Bz приведет к увеличению интенсивности линии перехода А] и к уменьшению интенсивности линии Аг. Один из способов представления спектров ИНДОР показан на рис. II.8, в; в такой записи спектр при насыщении частотой перехода В} является разностью спектральных кривых, показанных на рис. II.8,а и II.8,б). Легко получают такие спектры на обычных спектрометрах непрерывного типа (или стационарных), проводя сканирование по частоте поля V2. Когда V2 совпадает при сканировании с частотой перехода BJr наблюдается картина, показанная на рис. II.8, б, а когда V2 совпадает с частотой перехода В2, наблюдается обращение этих пиков (А] — положительный, А2 — отрицательный). Таким образом, спектр ИНДОР при наблюдении только линий ядер А позволяет определить и резонансные частоты ядер В.
Величина ЯЭО зависит от механизмов релаксационных процессов. Когда при двойном резонансе А — {В} доминирующее значение имеет диполь-дипольный механизм релаксации, эффект максимален и описывается величиной
ЯЭОмакс = 1 + ув/2уд. (11.20)
Например, в случае ,3С—'Н ЯЭОмакс~3,0, что дает существенное увеличение отношения сигнал/шум в спектрах ЯМР 13С двойного резонанса. Указанные механизм релаксации и значение ЯЭО обычно относятся только к непосредственно связанным атомам углерода и водорода.
* От англ, internuclear double resonance (INDOR).
50
Рис. II.8. Вид спектров ЯМР спиновой системы [ЛВ]:
а — спектр однократного резонанса; б — эффект Оверхаузера при насыщении перехода В\\ в — спектр ИНДОР, эквивалентный разности спектров а и б
При отрицательном значении какого-то из гиромагнитных отношений ув или ул ЯЭОМакс может быть отрицательным. Так, например, для 29Si—{'Ю ЯЭОмакс ~—1,5, а для 15N—{'Н} ЯЭОмакс~—4. Но ожидаемый эффект может вообще иногда не наблюдаться, так как минимальная и максимальная величины ЯЭО могут иметь разные знаки, т. е. на практике не исключается ЯЭО = 0. Очевидно, что данные о ЯЭО полезны при изучении механизмов релаксационных процессов.
Поскольку вклад диполь-дипольно-го механизма релаксации зависит от расстояния между ядрами (для ядер со спином '/2 он обратно пропорционален шестой степени расстояния), то ЯЭО может использоваться в конформационных исследованиях. Так, например, применяя гомоядерный двойной резонанс 'Н—{1Н}, регистрируют сна
чала обычный спектр однократного ПМР, а затем накладывают поле BV2 с частотой \’2 в резонансной области какой-то определенной группы протонов. В разностном спектре будут наблюдаться ПМР только от протонов, расположенных близко к облучаемым, т. е. имеющих с ними спин-спиновую связь. Последовательно проводя такой эксперимент с разными группами протонов (меняя V2), можно получить полное представление об относительном расположении протонов в молекуле.
Для регистрации спектров двойного резонанса применяются как стационарные, так и импульсные методы. В фурье-спектрометрах выравнивание заселенностей достигается применением 90°-ного импульса, и получаемый эффект в изменении интенсивностей ничем не отличается от наблюдаемого на стационарных. В то же время 180°-ный импульс обращает спины В, т. е. меняет их поляризацию, а значит, обращает и заселенность соответствующих уровней. Это приводит к более кардинальному изменению интенсивности линий, соответствующих переходам А, т. е. значительно повышает чувствительность метода.
Кроме рассмотренного метода ИНДОР, отличают другие виды двойного резонанса в зависимости ог величины уВ-^. Так называемый тиклинг линий спектра происходит при значениях у#,, ~Av,/3=(7'17'2)~'i/’ и сопровождается, как правило, ЯЭО и дублетным расщеплением линий. Картина тиклинга сложна, как и его теория, требующая привлечения громоздкого математического
51
аппарата. Селективный двойной резонанс наблюдается при увеличении амплитуды до когда происходит
упрощение спектра за счет частичного коллапса мультиплета.
Тотальный двойной резонанс, или метод спиновой развязки, требует дальнейшего повышения мощности второго поля. При можно добиться полного коллапса рас-
щеплений, связанных с облучаемььм ядром. Этот метод находит широкое применение. В спектроскопии ПМР он используется для упрощения спектров и доказательства спиновой связи групп. В спектроскопии ЯМР 13С обычно бывает необходимая полная развязка от всех протонов (|3С—{'Н}).
Для неорганических соединений, когда константы спин-спиново-го взаимодействия очень велики (1000 Гц и более), проблема спиновой развязки встречает большие трудности. Требуемая мощность второго поля становится настолько велика, что может вызывать плавление или кипение образца. Перспективна здесь импульсная методика, которую полезно применять и по другим причинам. Она позволяет, например, раздельно получать эффекты изменения интенсивности и частоты или изменения интенсивности и спиновой развязки.
В методе тройного резонанса кроме поля регистрации Bvt, на образец накладываются еще два поля: В„2 и В„3. Ничего принципиально нового по сравнению с двойным резонансом это не дает, но возможны различные сочетания рассмотрных выше видов двойного резонанса. Например, одно из полей используют для спиновой развязки с !Н, а другое поле — для создания тиклинга. При наличии спектрометров ЯМР на многие ядра метод тройного резонанса применяется редко, но при использовании только спектрометра ПМР требуется иногда его применять.
В следующей главе будет рассмотрен метод двойного электрон-ядерного резонанса, которым также получают информацию, аналогичную получаемой из спектров ЯМР.
4.4. Образцы, растворители, стандарты
Агрегатное состояние исследуемого вещества играет большую роль в спектроскопии ЯМР. Как уже упоминалось, использование специальной импульсной техники и другие специальные приемы открыли в последние годы возможность получать хорошие спектры ЯМР для образцов в любом агрегатном состоянии. Однако устранение трудностей, связанных с дипольным уширением линий ЯМР в твердых образцах и вязких жидкостях, остается предметом особого рассмотрения. В настоящее время для твердых образцов успешно исследуются спектры высокого разрешения на 13С, 14N, 29Si и других ядрах.
52
Ограничения в изучении спектров ЯМР газов, обусловленные низкой интенсивностью, также устраняются применением высокочувствительных фурье-спектрометров, но для сложных нелетучих веществ исследования паров практического значения все-таки почти не имеют. Возможно исследование токсичных и агрессивных веществ и, наоборот, объектов in vivo.
В подавляющем большинстве случаев спектры ЯМР регистрируются для невязких жидкостей и растворов. При этом подготовка образца предусматривает выбор ампулы, растворителя, концентрации раствора, стандарта для измерения химического сдвига и, если необходимо, сдвигающих реагентов, калибровочных эталонов и других добавок. Жидкость или раствор должны быть, конечно, тщательно очищены и отфильтрованы от гетерогенных частиц. Особенно важно удаление парамагнитных и ферромагнитных примесей, приводящих к чрезвычайно сильному уширению линий и даже исчезновению спектра. Хотя, как было сказано выше, добавка некоторых парамагнитных комплексов — сдвигающих реагентов, не только не портит спектр, а бывает даже полезной. Важен также контроль температуры образца.
В спектроскопии ЯМР используются такие стандартные растворители, как тетрахлорид углерода CCU, хлороформ CH3CI н дейтерохлороформ CD3CI, ацетон (СНз)2СО и дейтероацетон (CD3)2CO, бензол СбНе и дейтеробензол CgDg, диметилсульфоксид (CH3)2SO и др. Различные специальные растворители применяются для проведения исследований при высоких и низких температурах (например, дейтеродиметилсульфоксид и метанол или фреон-12), а также в случаях избирательной растворимости образцов (например, СН3СООН, C5H5N и C5D5N, Н2О и D2O).
Оптимальные концентрации выбираются с учетом допустимого уровня отношения сигнал/шум и растворимости исследуемого вещества. В спектроскопии ПМР содержание вещества в растворе выражают обычно в молярных долях (%), что удобно при оценке относительных интенсивностей сигналов. Навеска вещества т (г), требуемая для приготовления раствора с концентрацией С, определяется формулой
VfMBC т ..----------
Л1р(1 - С)
где V — объем растворителя; р—плотность растворителя; Мр и Мв — молярные массы растворителя и вещества. Минимальная концентрация вещества составляет примерно 10~2...10-3 моль/л.
О стандартах, применяемых для измерения химических сдвигов 6, уже говорилось (см. гл. I; 2.2). На практике используют методы внутреннего или внешнего с т а н д а р т а. В первом методе стандарт (например, 1—2 капли ТМС) вводится непосредственно в раствор исследуемого вещества. При этом считается, что стандарт инертен по отношению к растворителю и растворенному
53
веществу (не образует ассоциатов и т. п.). Когда это не так, применяют метод внешнего стандарта. Для этого раствором стандарта (например, ТМС) заполняют ампулу меньшего диаметра, помещаемую в основную ампулу и каким-то образом центрируемую в ней. При втором методе необходимо вводить поправку на различие в объемной диамагнитной восприимчивости растворов исследуемого вещества и стандарта.
В спектроскопии ЯМР на разных ядрах есть, конечно, свои методические особенности приготовления образцов. Например, для ЯМР 13С требуются обычно ампулы большего диаметра (8...25 мм), чем для ПМР (~5 мм), а в качестве стандартов химических сдвигов, помимо ТМС, иногда используются дейтерированные соединения и т. д. (подробно см. специальную литературу и справочники).
ГЛАВА III
СПЕКТРОСКОПИЯ ЭЛЕКТРОННОГО ПАРАМАГНИТНОГО РЕЗОНАНСА
1. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
Метод спектроскопии ЭПР, являющийся одним из довольно широко применяемых и продуктивных физических методов структурных и кинетических исследований в химии, применим только к парамагнитным образцам. К таким образцам относятся частицы, имеющие неспаренные электроны — свободные радикалы, ион-ради-калы, молекулы в триплетных состояниях, комплексы переходных металлов и др., а также фазы, содержащие свободные электроны и другие парамагнитные центры.
Как уже упоминалось, явления ЭПР и ЯМР основаны на одном и том же принципе магнитного резонанса. Различия между ними заключаются в величинах | це | 3> | Цп | и знаках магнитных моментов и взаимодействий, что приводит к серьезным отличиям в теории и эксперименте.
Наличие электронного спина и связанного с ним магнитного момента обусловливает возможность снятия вырождения спиновых состояний внешним магнитным полем и индуцирования переходов между ними. Эти переходы происходят с поглощением энергии электромагнитного излучения в микроволновой (30...2 мм) области (СВЧ диапазон 9...35 ГГц; интервал значений индукции постоянного магнитного поля 0,34—1,25 Т), что и называют электронным парамагнитным резонансом. В зарубежной литературе используется термин электронный спиновый резонанс (ESR), однако в рассматриваемом методе радиоспектроскопии состояния из-за спинорбитальной связи не являются чисто спиновыми, поэтому более адекватно название ЭПР или даже парамагнитный резонанс.
54
Именно орбитальный вклад в магнитный момент частицы меняет условия резонанса, что проявляется в значении g-фактора (Ланде), и это первая характеристика спектра ЭПР. Второй важнейшей чертой, содержащей большую информацию, является сверхтонкая структура спектра, обусловленная электрон-ядерным спин-спиновым взаимодействием. В спектрах ЭПР анизотропных образцов, содержащих парамагнитные центры с 1, может наблюдаться также тонкая структура, связанная с расщеплением спиновых уровней энергии в нулевом поле, т. е. без наложения внешнего магнитного поля. Определенную информацию несет ширина сигналов ЭПР. Сам факт наблюдения спектра говорит прежде всего о том, что хотя бы какая-то часть образца содержит парамагнитные частицы или центры, т. е. имеет неспаренные электроны.
1.1. Условие ЭПР
j Электрон, обладая собственным моментом количества движения (опином) и являясь электрически заряженной частицей, имеет магнитный момент:
= (1I1.I)
где S — вектор спинового углового момента (в единицах й=й/2л); цв — магнетон Бора (р.в=ей/(2тс)=9,27-10-24 А>м2; е — заряд электрона; т — масса покоя электрона; с — скорость света); g — безразмерная величина (g-фактор Ланде), равная для свободного электрона 2,00232.
В отсутствие внешнего поля спиновые векторы ориентированы беспорядочно, т. е. спиновые состояния вырождены. При наложении внешнего магнитного поля В гамильтониан взаимодействия с ним
= (III. 2)
запишется в виде
& = gpBBSz. (1П.З)
Ось z совпадает с направлением поля. В общем случае парамагнитной частицы (при одном или нескольких иеспаренных электронах) суммарный вектор S связан со спиновым квантовым числом S известным соотношением:
|S| = /S (S+ 1), (111.4)
а его проекция, входящая в выражение (Ш.З):
Sz=fims, (III.5)
где ms — квантовое число, которое может принимать значение от —S до +S (как и проекция Sz в единицах Й), т. е. всего 2S + 1-значений.
55
Рис. III.l. Расщепление спиновых энергетических уровней электрона в зависимости от индукции внешнего магнитного поля и индуцируемый радиочастотным полем переход
I
Рис. II 1.2. Линия спектра поглощения ЭПР (а) и кривая первой производной спектра ЭПР (б) для лоренцевой формы линии
Поскольку при отрицательном заряде це отрицателен, то цв выбирается в уравнении (Ш.1) положительным. Для одного электрона S='/2 и возможны только две ориентации спинового вектора — по полю и против поля, т. е. его проекции иа направление поля характеризуются двумя значениями квантового числа ms=±’)2.
Соответствующие энергетические состояния, или зеемановские уровни, записываются в виде
E=g\>-BBms. (Ш.6)
Из-за разных знаков и (магнитный момент протона, см. гл. I) состояние с более низкой энергией взаимодействия с полем у электрона в отличие от протона соответствует тз~—1/z, обозначение волновой функции состояния |р>. Состояние с более высоким значением энергии соответствует ms— + 'i2 и описывается волновой функцией |а>. Эти уровни показаны на рис. III.1. Переходы между ними могут индуцироваться, как и в ЯМР, переменным радиочастотным полем, направленным перпендикулярно постоянному внешнему магнитному полю, но в частотном диапазоне на три порядка выше, чем в ЯМР, т. ё. в сантиметровом (миллиметровом) диапазоне длин волн.
Условие магнитного резонанса — совпадение разности энергий уровней, между которыми происходит переход, зависящий от внеш-56
него поля В, с энергией кванта электромагнитного излучения, A£=/iv, а для одного электрона
ЬЕ = — 2jj.sS = gp-£B = Дм. (III.7)
Достижение этого условия осуществляется обычно разверткой по полю, т. е. варьированием В при постоянной частоте излучения (v=const).
Резонансный сигнал в спектре ЭПР обычно регистрируется в виде зависимости от напряженности поля первой производной интенсивности спектра поглощения, как это показано на рис. III.2, а, б, что позволяет лучше выявить особенности и разрешить структуру спектра.
1.2. Положение резонансного сигнала и g-фактор
В качестве параметра, определяющего положение линии резонансного поглощения в спектре ЭПР, можно рассматривать так называемый спектроскопический фактор расщепления Ланде или g-фактор, равный отношению электронного магнитного момента к полному угловому моменту. В теоретической спектроскопии для свободных атомов (в газовой фазе) получено следующее выражение этого фактора:
/(J + 1) + S(S+1) + I(£+1)
27(7+1)
где S — суммарный спин (спиновое число);/. — суммарный орбитальный момент; J — полный угловой момент (при Рассел — Саун-дерсской спинорбитальной (LS) связи он принимает значения от |L + S| до |L-S|).
Чисто спиновое значение g-фактора для свободного электрона (S=l/2, L = 0, /='/2) по формуле (III.8) получается равным go=2, а приведенное выше более точное значение 2,00232 содержит релятивистскую поправку. Для неспаренного электрона во многих свободных радикалах g-фактор также близок к этому значению и может отличаться от него только во втором или даже третьем знаке после запятой, но вообще, например, у соединений переходных металлов и других парамагнитных систем значения g-фактора меняются довольно в широких пределах (до нескольких единиц).
Отклонение g-фактора Ag от чисто спинового значения, обусловленное спин-орбитальной связью, может быть как отрицательным, так и положительным. Оно тем больше по абсолютной величине, чем сильнее спин-орбитальное взаимодействие: возрастает, например, с увеличением порядкового номера элемента, и чем меньше А£ уровней, между которыми происходит переход. Приложенное внешнее магнитное поле Ввнеш индуцирует дополнительный орбитальный момент количества движения, а орбитальное движение
57
со. эл.
$2&9?
+49 -Ад В6неш
Рис. Ш.З. Схема изменения g-фактора в результате спин-орбитальной связи
электрона создает в свою очередь магнитное поле Внавед, направленное противоположно приложенному полю. Электронный спин находится, таким образом, в локальном магнитном поле ВЛОк, равном сумме приложенного и наведенного полей Влок — Ввнеш" Внавед, в этом и заключает-ся спин-орбитальная связь. Чем больше наведенное поле, тем меньше локальное поле на спиновой системе и меньше g-фактор, а напряженность внешнего поля Ввнеш для достижения условия резонанса должна быть выше — это соответствует отрицательному отклонению (—Ag)
от чисто спинового значения g-фактора, как показано на схеме для gi (рис. Ш.З).
Возможна другая ситуация, например, такого распределения неспаренных электронов по разным орбиталям, что локальное поле оказывается на спиновой системе увеличенным, т. е. gi выше чисто спинового значения g0 и резонанс происходит при более низком значении Ввнеш, это соответствует положительному отклонению (+Ag), как для g2 (рис. Ш.З).
Таким образом, появление резонансных пиков при разных значениях индукции внешнего магнитного поля, когда развертка спектра проводится по полю при постоянной частоте, зависит прежде всего от g-фактора. Поскольку это так и поскольку g-фактор отражает характер спин-орбитального взаимодействия в системе, то в известном смысле чисто формально и условно этот параметр можно сравнивать с химическим сдвигом в спектрах ЯМР, хотя информативность g-фактора ниже.
До сих пор говорилось о g-факторе как о скалярной величине, но это можно делать только при рассмотрении спектров ЭПР изотропных образцов, например растворов. В общем случае g-фактор— величина тензорная, и условия резонанса зависят от ориентации парамагнитного объекта относительно поля. При свободном движении парамагнитных частиц в газе или растворе все ориентации равновероятны и происходит усреднение, так что тензор становится сферически симметричным, т. е. характеризуется единственным параметром g. То же относится к другим изотропным системам. На практике, однако, часто исследуют спектры ЭПР анизотропных систем, таких, как замороженные растворы, парамагнитные центры в монокристаллах, объекты в матрицах, различные твердые образцы и др. Во всех этих случаях g-фактор должен рассматриваться как симметричный (имеющий осевую симметрию) или асимметричный (неаксиальный) тензор. Его при соответствующем выборе системы координат всегда можно диагонализовать и получить три главных значения g-фактора: gxx, gyy и gzz. Если при 58
сферической симметрии они все равны, то для систем с осевой симметрией имеются два различных фактора: gn = gz (ось z совмещается с осью симметрии высшего порядка) и g±, а для систем низшей симметрии все три главных значения неаксиального тензора различны.
При исследовании анизотропных образцов спектр ЭПР зависит, таким образом, от их ориентации относительно поля. Измеряя, например, спектр монокристалла при различных углах, принципиально возможно определить главные значения тензора g-фактора. Если при осевой симметрии тензора 0 — угол, образуемый осью z с направлением поля, то эффективный g-фактор.
£-2 = ^2 COS2B+ Sin26, (111.5)
а условие резонанса запишется в виде
В ——— (g2 cos20 + g\ sin2б)~'''2, (III. 10)
^в
т. е. для разных 0 регистрируются сигналы ЭПР при разных значениях В.
1.3. Электрон-ядерное взаимодействие и сверхтонкая структура спектра ЭПР
Если неспаренный электрон находится вблизи ядра со спином 7=И=0, то в результате взаимодействия их магнитных моментов происходит расщепление зеемановских уровней энергии электрона в соответствии с более сложной зависимостью энергии от поля:
Е = amsmr (HI. II)
где а — константа сверхтонкого взаимодействия (СТВ); mj — квантовое число проекции ядерного спина.
При взаимодействии электрона с одним ядром, спин которого Т=1/2, каждый из уровней расщепляется на два (рис. Ш.4).
В соответствии с правилами отбора Ams=±l и Ат;=0 возможны два индуцированных перехода:
а
h^ = ^E1=gl>.BB
„а
h42 = &Е2 — g^BB + — .
Разность частот переходов (МГц или см-1) равна константе СТВ:
Дм = М2 — mj = а .
Так, например, для атомов водорода в твердой матрице в спектре ЭПР наблюдается не один пик, а дублет с центром при g=2,0023.
В большинстве случаев структура спектров ЭПР свободных ра-
59
ms
mI
/ ' 2 1
---\
\ ’2."
гл"ГЛ“-'р^\
4
l«> _ 7 ,
£w=r^BB4a
"i^Eafi=i9^B'4a
iji> i 4
---^ = -2^ВЙ + Г
'г
ja>
Рис. III.4. Схема расщепления электронных зеемановских уровней при взаимодействии с ядерным спином /=’/2 и индуцируемые переходы
дикалов в жидкой фазе может быть объяснена аналогичным образом, представляющим первый порядок теории возмущений. При взаимодействии неспаренного электрона с двумя ядрами (1) и (2) энергии уровней будут зависеть от двух ядерных квантовых чисел mIt которые обозначим т1 и т2:
ЬЕ = ms(giiBB + alml+a2m2), (III. 12)
где Ci и а2— константы СТВ с ядром (1) и (2) соответственно. При указанных выше правилах отбора энергии разрешенных переходов
Д£ = Ьч = gpBB 4- almi + a2m2. (HI. 13)
Константы СТВ можно выразить и в единицах индукции поля В (тесла), если поделить их на g'p.B. Принимая для свободного электрона резонансное значение Во при постоянной частоте v, т. е. условие резонанса 1гу=§ц,вВ0, при развертке спектра по полю (при вариации его вблизи Во) для резонансных линий взаимодействующего с ядрами электрона из (III.13) получим
В ~ Во — aimi — а2т2,
а в общем случае
В = Во — 2 а‘П1‘
(Ш.14)
(Ш.15)
Таким образом, при взаимодействии электрона с несколькими ядрами общую картину спектра ЭПР можно представить так; сначала имеем расщепление сигнала от наиболее сильного взаимодей-60
a
B0-a
В0*а
1мТ
B0
Рис. 111.5. Расщепление сигнала ЭПР при взаимодействии электрона с двумя ядрами (неэквивалентными: а,--^а2 н эквивалентными: ai = a2 = a)
Рис. III.6. Спектр ЭПР анион-раднкала [С6Н6-]-
ствия с одним из ядер, затем каждая из линий расщепляется от второго взаимодействия и т. д. Для двух ядер это показано в верхней части рис. III.5. Если оба ядра одинаковы, например протоны, т. е. aj = a2 = a и S=!/2, то В = В0—a(mi + m2), и в спектре будет наблюдаться триплет с соотношением интенсивности компонент 1:2:1, что понятно из схемы, представленной на рис. III.5.
Вообще, при наличии п эквивалентных ядер со спином I, взаимодействующих с электронным спином, мультиплетность сигнала ЭПР равна (2л/4-1)- Отношение интенсивностей компонент мультиплета такое же, как отношение коэффициентов биноминального разложения (х + l)" (см. треугольник Паскаля, табл. 1.5), когда' л>1, а при п=1 компоненты мультиплета имеют одинаковую интенсивность. На рис. III.6 показан спектр ЭПР анион-радикала бензола, представляющий септет с константой а = 3,75-10~4 Т и соотношением интенсивности компонент 1 : 6 ; 15 : 20 : 15 : 6 : 1, здесь электрон делокализован по бензольному кольцу и одинаково взаимодействует со всеми шестью протонами. На рис. III.7 схематически показана сверхтонкая структура спектра ЭПР для системы, содержащей один неспаренный электрон, который взаимодействует с двумя ядрами: одно со спином 1=3/2, а другое со спином /=’/2-Спектр представляет квартет дублетов с одинаковой интенсивностью всех линий. В общем случае при взаимодействии электрона с несколькими наборами эквивалентных ядер число линий в спектре ЭПР будет равно произведению
П (2л/7(-+ I). (Ш.16)
i
Картина СТС в спектрах ЭПР как по числу компонент, так и по
61
Рис. IH.7. Сверхтонкая структура спектра ЭПР при взаимодействии неспаренного электрона с двумя ядрами (/д = 3/2 и /в=’/2)
соотношению их интенсивностей определяется практически по тем же правилам, что и в спектрах ЯМР.
В теории рассматриваются разные механизмы взаимодействия электрона и ядра в магнитном поле. Важнейший из них, так называемое контактное взаимодействие Ферми, связано с наличием на ядре электронной плотности неспаренного электрона. Такое взаимодействие тем больше, чем больше s-характер орбитали, на которой находится электрон.
Величина константы СТВ, т. е.
расстояние между линиями в мультиплетах, характеризует степень делокализацйи неспаренного электрона и зависит от спиновой плотности на ядрах. Спиновая плотность — это не то же, что плотность неспаренного электрона. Дело в том, что его орбиталь может поляризовать спины спаренных электронов на прилежащей о-связи, т. е. каждый из них будет несколько больше относиться к одному из связанных атомов, чем к другому. Поэтому на каждом из ядер будет некоторая спиновая плотность, даже на том, на котором нет плотности неспаренного
электрона.
Существует также прямое взаимодействие векторов моментов магнитных диполей электрона и ядра, которое зависит от величины момента ядра и от угла, образуемого вектором ядро — электрон, с направлением магнитного поля. В изотропных системах при хаотическом движении частиц это взаимодействие усредняется. В общем случае, как и g-фактор, константа СТВ а — величина тензорная. Только для изотропных систем этот тензор характеризуется одним параметром (сферическая симметрия), а для анизотропных систем имеет два (симметричный волчок — эллипсоид вращения) или три (асимметричный волчок) независимых параметра. Удобно разделить тензор СТВ на изотропную и анизотропную части. Анизотропная составляющая связана как раз с прямым дипольным взаимодействием и обратно пропорциональна кубу расстояния между ядром и электроном, усредненного по волновой функции электрона. При значительной анизотропии тензора СТВ спектры ЭПР сильно усложняются и для их анализа требуется компьютерная обработка с соответствующими программами, составленными по алгоритмам решения задач с разной записью гамильтонианов взаимодействия сложных систем с полем.
Как уже упоминалось, рассмотрение спектров ЭПР ведется нами
62
с учетом первого порядка теории возмущений. Отметим здесь только одно интересное отличие, к которому приводит учет более высоких порядков теории возмущений. При введении соответствующих поправок могут в определенных условиях измениться правила отбора переходов. Например, становится отличной от нуля вероятность переходов ae0n-<->-Pean(Affis = ±l, Am;=±l), запрещенных в первом порядке теории возмущений (см. рис. Ш.4). При этом интенсивность линий, соответствующих разрешенным по первому порядку переходам, несколько уменьшится.
1.4. Электрон-электронное взаимодействие и тонкая структура спектров ЭПР анизотропных систем
Выше рассматривались в основном системы с одним неспаренным электроном, но существует много парамагнитных систем, изучаемых методом спектроскопии ЭПР, в которых имеется несколько неспаренных электронов. Это, например, комплексы переходных металлов, молекулы в триплетных состояниях и др.
Если молекула диамагнитного вещества (молекулы в основном состоянии, которое синглетно, не имеют неспаренных электронов 5=0) может иметь возбужденное триплетное состояние (два неспа-
ренных электрона приводят к суммарному электронному спину 5=1), время жизни которого больше характеристического времени метода, то можно регистрировать спектр ЭПР молекул в этом состоянии, как для обычных парамагнитных частиц. В магнитном
поле происходит зеемановское расщепление триплетного состояния на три подуровня, как показано на рис. Ш.8, а. Два возможных по правилу отбора \tns = ±l перехода, также указанных на рисунке, происходят с одинаковым изменением энергии (т. е. частотой v или значением индукции В постоянного поля), и в спектре ЭПР будет наблюдаться один сигнал.
В поликристаллическом или замороженном стеклообразном образце из-за анизотропии спин-орбитального и диполь-дипольного
взаимодействий даже в отсутствие внешнего магнитного поля (в ну-
левом поле) происходит снятие спинового вырождения, т. е. расщепление на три подуровня: ms= + l, ms== =—1 и ms=0 или, в частном случае, на два подуровня: ms=±l и ms=0 (рис. Ш.8, б). После зеемановского расщепления подуровня ms==hl во внешнем магнитном поле энер-
ms=+/
ms=0
ms-1
ms=tf /
ms=° Г\ч ms=0 । б-*1
Рис. III.8. Расщепление триплетного состояния (S=l) в магнитном поле в отсутствие (а) и при наличии (б) крамерсовского расщепления в нулевом поле
63
'^-rr<Z w
mS~ + 2
'ть~~'г
i
ms~ " 2
гии переходов Ams— +1 и Ams=—1 теперь различны (hv\^hvi.) и сигнал в спектре ЭПР будет дублетный.
Существует теорема Крамере а, согласно которой у систем с четным числом неспаренных электронов низшее по энергии состояние в нулевом поле соответствует ms=0, как и показано на рис. III.8, б
для триплетного состояния молекул. Более высокие по энергии состояния
Рис. III.9. Квартет уровней системы со спином S = 3/2 прн большом крамерсовском расщеплении в нулевом поле
из-за электростатического и спин-орбитального взаимодействия могут быть в отличие от случая, представленного на на рис. III.8, б, и не вырождены в отсутствие внешнего магнитного поля. Для анизотропных систем с нечетным числом неспаренных электронов при расщеплении в нулевом поле произвольной симметрии всегда существуют по крайней мере дважды вырожденные состояния. Это вырождение, называемое крамерсовским, снимается внешним магнитным полем, как показано на рис. III.8, б для системы с электронным спином 5=1 и на рис. III.9 для системы со спином 5=3/2.
Когда расшепление в нулевом поле мало по сравнению с gpBp0, наблюдаются два сигнала вблизи g~2, как было сказано выше о переходах в триплетном состоянии. Но если крамерсовское расщепление велико, как часто бывает, например, у ионов переходных металлов, то ^-фактор сильно отличается от значения g=2. На рис. III.9 показано очень большое расщепление, когда переход между низшим и высшим уровнями в обычном диапазоне радиочастот и напряженностей поля не происходит. Возможен единственный переход, показанный на рис. III.9, — между уровнями ms — — */2 и ms— + {k, а переходы между ms = — '/2 и ms=—3/2; ms— + l/2 и ms=+3/2 лежат за пределами наблюдаемого спектрального диапазона. При большом расщеплении в нулевом поле иногда вводят так называемое эффективное спиновое квантовое число | ms | для самого низкого уровня энергии. Оно равно нулю — при четном числе неспаренных электронов и ’/2 — при нечетном их числе. Система в таком состоянии или не имеет спектра ЭПР — или дает простой спектр | ms | = ’/2 в обычной области частот (напряженностей поля).
Следствием расщепления в нулевом поле является также возможность нарушения правила отбора Дт$ = ±1, приводящего к ус-
64
ложнению спектров из-за большого числа наблюдаемых частот переходов. Расщепление в нулевом поле зависит также от анизотропии объекта, и при более низкой симметрии, чем кубическая, картина становится сложнее.
1.5. Интенсивность, ширина и форма линии
Интенсивность сигнала ЭПР определяется, как и в ЯМР спектрах, вероятностью переходов между спиновыми состояниями (1) и (2), индуцируемых радиочастотным полем, поляризованным перпендикулярно внешнему магнитному полю В, которая равна
Л112 = (В74Я2) I < Tilp-J т2 > I2 г (v), (III. 17)
где — перпендикулярная составляющая оператора магнитного дипольного момента; Г (v)—нормализованная функция формы линии'
Для получения оптимального сигнала желательны достаточно
высокая напряженность поля и радиочастота, малая ширина линии и, конечно, достаточная концентрация парамагнитных частиц. При тепловом равновесии заселенность |р> спинового состояния электрона несколько выше и преобладает поглощение энергии радиочастотного поля с переходом электронов в верхнее |а> состояние. Заселенность уровней может меняться
в процессе эксперимента, но выравнивание заселенности и исчезновение сигнала поглощения не происходит из-за существования механизмов безызлучательного перехода электронов на нижний уровень, называемых релаксационными процессами. Энергия, полученная от радиоизлучения, может передаваться спиновой системой окружения, например, в виде фононов решетки, и такой процесс называется, как уже говорилось в гл. I, спин-решеточной релаксацией (Т[). Время жизни т верхнего состояния уменьшается также из-за индуцированного испускания и при этом, как следует из принципа неопределенности бДАт—й, возрастает неопределенность энергии состояния и происходит уширение линии (рис. III.10, а, б). Существует, кроме того, механизм спин-спиновой релаксации (Т^), определяемый беспорядочным распределением полей ядерных и электрон
Рис. III.10. Зеемановские уровни энергии (а) и линия поглощения (б) с учетом принципа неопределенности
3—1377
65
ных спинов вокруг неспаренного электрона, которое «размывает» его уровни энергии и также приводит к уширению линии. Таким образом, ширина линии определяется величиной дЕ, которая тем больше, чем меньше Ат, завсящее от времен релаксации Т\ и TV Если время релаксации велико, то заселенность верхнего уровня будет возрастать, а интенсивность сигнала ЭПР падать из-за насыщения. При малом времени релаксации линия будет широкой из-за принципа неопределенности. Уширяют сигнал и нерелаксационные процессы, в частности тонкое и сверхтонкое спин-спйновое взаимодействие (см. выше), обменные процессы и др. Что касается обменных процессов, то принципы эффекта являются общими для спектроскопии ЭПР и ЯМР и обсуждались в гл. I, однако при рассмотрении спектров ЭПР должен учитываться не только обмен ядер, но и обмен электронов.
Релаксационные процессы в спектроскопии ЭПР жидкой фазы играют большую роль, чем в ЯМР, так как типичное время релаксации для электрона в этих условиях составляет 10-6 с, а для ядер ~10 с.
Характеристическое время метода ЭПР много меньше, чем ЯМР, и для изучения методом ЭПР доступны процессы с относительно низкими энергиями активации.
Для сужения сигналов ЭПР на практике часто приходится прибегать к сильному охлаждению образцов жидким азотом или даже гелием, или водородом, что прежде всего позволяет увеличить время спин-решеточной релаксации. Это особенно бывает необходимо при изучении солей переходных металлов н редкоземельных элементов. Для снижения эффектов, вызываемых спин-спиновой релаксацией и обменными процессами, прибегают также к разбавлению образцов диамагнитными веществами и изоляции парамагнитных центров друг от друга в матрицах и при замораживании растворов.
Абсолютная интенсивность сигнала ЭПР надежно не измеряется и является величиной неопределенной, относительные же интенсивности сигналов в принципе пропорциональны полному числу несла-ренных электронов системы. При количественных измерениях используются интегральные интенсивности, получаемые двойным интегрированием спектральной кривой зависимости первой производной от напряженности поля:
ат (В) dB
dB2.
Приближенно иногда измеряют и пиковые интенсивности.
В спектрах ЭПР разбавленных растворов линия почти всегда имеет лоренцову форму (см. рис. III.2,а), а для твердых образцов в некоторых случаях наблюдается гауссова форма линии. Эти фор-
66
Рис. III. 11. Форма сигналов ЭПР при произвольной ориентации в анизотропных системах парамагнитных частиц с осевой симметрией (а) и асимметричных (б)
мы могут быть охарактеризованы с помощью полуширины линии АВ 1/2(ширина на половине высоты пика поглощения). При регистрации спектра в виде первой производной легко измеряется расстояние АВмакс между максимумом и минимумом кривой (см. рис. III.2, б) и полезны следующие соотношения:
лоренцева форма
Уз
^1/а = -^-ДВМакс; (Ш.18)
гауссова форма
ДВ,д ~ У2/1п2 ДВмакс.
При случайной ориентации парамагнитных частиц в таких анизотропных образцах, как порошки, поликристаллические, стеклообразные образцы, вязкие жидкости, спектр при отсутствии сверхтонкой структуры будет иметь сложный контур, показанный на рис. III.11 и представляющий наложение спектров групп одинаково ориентированных центров. На кривой поглощения, а особенно четко на кривой первой производной выделяются особые точки, которыми определяются главные значения g-тензора (рис. Ш.П).
3* 67
2. ПРИЛОЖЕНИЯ СПЕКТРОСКОПИИ ЭПР
Применение методов спектроскопии ЭПР в химических исследованиях весьма разнообразно. Но грубо можно говорить о двух направлениях — одном, касающемся в основном структурных аспектов, и другом — динамики процессов. К первому относится изучение структуры органических, неорганических и комплексных радикалов и ион-радикалов, парамагнитных центров в твердых телах и т. д., а ко второму — изучение механизмов и кинетики химических реакций, обменных процессов и т. д.
2.1. Структурные исследования
Извлечение структурной информации из экспериментальных данных по спектрам ЭПР, т. е. решение соответствующей обратной задачи, основывается на рассмотрении связи спектра со структурой, которая проводится обычно в рамках метода МО ЛКАО. Как уже говорилось, по величине и знаку g-фактора (изотропные системы) или компонентам g-тензора судят о характере парамагнитной частицы, ее заряде и распределении электронной плотности. Даже в органических (углеводородных) радикалах, у которых g-фактор близок к спиновому значению, по нему все-таки можно различать, например, положительные и отрицательные ион-радикалы: он больше у отрицательных ионов.
Поскольку сверхтонкое взаимодействие связано с делокализацией электронов, то по экспериментальным значениям констант СТВ можно, как уже отмечалось, проводить количественную оценку электронной и спиновой плотности иа разных атомах изучаемых радикалов и судить об их строении.
Рассмотрим в порядке возрастания числа атомов в парамагнитной частице с одним неспаренным электроном некоторые достаточно простые радикальные системы. Ряд интересных проблем возникает при использовании спектров ЭПР в исследованиях двухатомных радикалов типа АН и АВ, позволяющих проверить современные представления об их электронном строении. Определены компоненты тензоров g-фактора и сверхтонкого взаимодействия гидроксильного радикала ОН и ион-радикала NH в разных средах, характеризующие распределение электронной и спиновой плотности. К так называемым л-радикалам типа АВ относят, например, N2- О2-, NO, СЮ и др., а к о-радикалам — F2~, Cl2~, FC1~, XeF, KrF и др. Из данных спектроскопии ЭПР по этим радикалам сделан, в частности, вывод об убывании относительной электроотрицательности атомов в ряду Kr>F>Xe>Cl.
Трехатомные радикалы типов АВ2 или ХАВ могут быть линейного или углового строения, что зависит от распределения плотности неспаренного электрона, т. е. относительной заселенности (что иногда представляют в терминах электроотрицательности) у ато-68
мов А и В. При увеличении заселенности у атома А валентный угол должен увеличиваться, но при относительно большой электроотрицательности атома В это влияние на геометрию радикала может быть пренебрежимо мало. При угловой структуре радикала спектр ЭПР в зависимости от общего числа электронов в системе и анизотропии образца имеет характерный вид, позволяя определять спиновую плотность
Рис. III.12. Спектр ЭПР иои-радика. ла “B'H-j:
на центральном атоме и оценивать валентный угол. Так, например, для радикала NO2 было по-
лучено значение валентного угла слабые пики 133°, совпадающее в пределах
ошибок с установленным прямыми экспериментальными методами (134°). В идентифицированном методом спектроскопии
обусловлены присутствием изотопа 10В
ион-радикале СО2~
ЭПР в облученном
формиате натрия, локализация неспаренного электрона на 13С оказалась больше сравнительно с 14N в NO2~, что соответствует большему значению валентного угла (ближе к 180°). Эти выводы
согласуются с особенностями реакционной способности этих частиц: большая склонность ион-радикала СОг~ к димеризации, при-
соединение водорода к атому углерода, а не к кислороду, как в радикале NO2, и т. д.
В четырехатомном ион-радикале ВН3", достаточно сложный спектр которого, состоящий из 16 сигналов, показан на рис. III.12, по константам СТВ при взаимодействии неспаренного электрона с ядром ”В(/=3/2) и тремя протонами рассчитана спиновая плотность на атоме бора и водорода и установлена плоская структура
радикала.
Методом спектроскопии ЭПР изучен довольно большой ряд четырехатомных неорганических радикалов АВ3, которые, вообще говоря, могут иметь плоское или пирамидальное строение. Для радикалов С1О3~, СО3~ и других найдена осевая симметрия g- и
а-тензоров и определены их параллельные и перпендикулярные компоненты, а для радикала NO3 — существенная асимметрия тензоров. По данным спектроскопии ЭПР, как и других методов, например колебательной спектроскопии, конфигурация радикалов СН3 и NH3+ близка к плоской.
В радикале СН3 протоны лежат в плоскости с атомом С в центре, а на перпендикулярной этой плоскости 2рг-орбитали находится неспаренный электрон (л-радикал), т. е. его плотность в указанной плоскости равна нулю. Однако спиновая плотность на протонах не
69
равна нулю и в спектре ЭПР проявляется изотропность СТВ — четыре равноотстоящие линии с отношением интенсивностей 1 : 3 : 3 : 1, а = 22,5-10~4 Т. Это объясняют контактным взаимодействием и поляризацией а-орбиталей связей С—Н.
Для пятиатомных радикалов ДВ4 можно было бы предполагать тетраэдрическую конфигурацию, но согласно теореме Яна —Теллера для трижды вырожденных дублетных электронных состояний 2Fi и 2Рг (при одном неспаренном электроне S=1/2, а мультиплет-ность равна 2) правильные тетраэдрические конфигурации внутренне нестабильны. Изменение симметрии радикала от Та до С2а может быть обусловлено как возмущением Яна — Теллера, так, например, и несимметричным внешним окружением, когда соседние катионы располагаются относительно атомов В радикала таким образом, что эффективная симметрия понижается. Если радикал имеет конфигурацию искаженного тетраэдра, то интерес представляют степень искажения и выяснение вклада разрыхляющих, связывающих и несвязывающих молекулярных орбиталей (например, симметрии «1 и /г) в конечное состояние. Эта задача в принципе решается с привлечением спектроскопии ЭПР.
Проведен анализ спектров ЭПР и сделаны заключения о строении целого ряда радикалов типа ДВ4. В частности, показано, что радикал PF4 имеет слегка искаженную тетраэдрическую конфигурацию, а у анион-радикала PF4~ она сильно отклоняется от симметрии Та. Примерами других изученных пятиатомных радикалов являются РО42-, С1О42-, SO4~, SeO4~ и др.
Большинство органических радикалов принадлежит к типу, уже упоминавшемуся ранее и называемому л-радикалами, у которых неспаренный электрон локализован в основном на рг-орбитали или находится на молекулярной л-орбитали. Для таких радикалов предложена (Мак Коннел) линейная зависимость константы ан сверхтонкого взаимодействия с протоном от л-электронной плотности ря на соседнем с ним атоме углерода: ан = фря, где величина Q — эффективная константа для однотипных радикалов почти не
R \
меняется (для алкильных радикалов 1 Н Q^2,4...2,5-10~3 Т).
R2z
Величина ря вычисляется по формуле ря=П (1—Дх,), значения i
\Xi для разных заместителей табулированы в специальной литературе.
Спектры ЭПР ароматических радикалов и ион-радикалов показывают, как уже видели на примере анион-радикала бензола (см. гл. III; 1.3), что неспаренный электрон делокализован по углеродной циклической системе и одинаково взаимодействует со всеми протонами, спиновая плотность на которых отлична от нуля. В качестве еще одного примера можно привести анион-радикал п-бензо-семихинона (четыре протона)
70
спектр которого представляет квинтет с константой СТВ а= = 2,37 • 10-4 Т и соотношением интенсивности компонент 1 : 4 :6 :4 :1.
В органических о-радикалах неспаренный электрон локализован на a-орбитали. Примером является формильный радикал Н—С = О, для которого характерна большая константа СТВ (а=1,32-10~2 Т), обусловленная вкладом s-орбитали от атома водорода и не наблюдаемая в л-радикалах. Радикал винил также относится к a-радикалам, найденные для него константы СТВ различны для взаимодействия с каждым из трех протонов (указаны при формуле в 10-3 Т);
Если использовать значение константы СТВ для свободного атома водорода а = 50,68-10~3 Т (эталонная ls-орбиталь), то можно по отношению к ней константы ан оценивать спиновую плотность на ls-орбиталях атомов водорода в таких комплексах, обозначаемую рн, т. е. существует прямая пропорциональность между спиновой плотностью и константой СТВ. Так, на транс-протоне винилового радикала относительная спиновая плотность рнтракс=6,86/50,68= = 0,135, а на tftzc-протоне рнч“с=3,42/50,68=0,067, т. е. в два раза меньше, и еще в два раза она понижается на гем-протоне.
Спектроскопия ЭПР служит мощным методом изучения молекул в триплетных состояниях, причем это могут быть не обязательно возбужденные состояния. Так, например, у молекулы Os или дифе-нилметилена, получаемого при облучении дифенилдиазометана:
IN
II
N"
триплетным является основное состояние. Возбужденные, долгоживущие триплетные состояния существуют, как известно, например, у ароматических углеводородов типа нафталина. При изучении органических радикалов, захваченных в твердых телах, получают компоненты ^-тензоров и тензоров сверхтонкого взаимодействия с разными ядрами (*Н, 13С, 14N, 19F и др.).
Несколько особый интерес представляет исследование спектров
71
ЭПР комплексов переходных металлов. Важность их изучения обусловлена использованием для идентификации соединений по специфической картине СТС, получаемой информацией о распределении электронной плотности, спиновой плотности на разных ядрах, о том, какие заняты d-орбитали, т. е. о направлении ян-теллеров-ского возмущения и т. д. При этом следует, конечно, заметить, что интерпретация спектров указанных комплексов встречает немалые трудности. Дело в том, что переходные металлы могут иметь несколько приближенно вырожденных орбиталей и несколько неспаренных электронов. В свободном ионе Sd-орбиталей вырождену, но в комплексе взаимодействие их с лигандами различно и происходит разделение на две или более групп орбиталей. Например, в октаэдрическом комплексе имеется трижды вырожденный нижний уровень и дважды вырожденный верхний (у других типов комплексов орбитали группируются по-другому).
Если даже ограничиться рассмотрением только октаэдрических комплексов переходных металлов первого ряда, то и для них имеется большое разнообразие, так как у них может быть до 10 d-электронов и до 5 неспаренных электронов. При анализе спектров ЭПР этих систем необходимо знать число указанных электронов, рассматривать возможные эффекты Яна — Теллера и крамерсов-ское вырождение (см. выше). Для переходных металлов второго и третьего рядов спин-орбитальное взаимодействие возрастает, так что наблюдение и интерпретация спектров ЭПР их комплексов становится еще гораздо труднее.
Однако спектры парамагнитных комплексов редкоземельных элементов, у которых /-оболочка заполнена не более чем наполовину (4/7) и спин-орбитальное взаимодействие мало, бывают обычно очень четкими и информативными, например, для Gd(III). Для других элементов наблюдение спектров ЭПР требует гелиевых температур. Если возбужденные электронные состояния лежат близко к основному состоянию, то время спин-решеточной релаксации Т\ мало, и линии уширяются, т. е. для наблюдения спектров требуется понижать температуру.
Следует обратить внимание на то, что парамагнитные соединения обычно не дают хороших спектров ЯМР: в них резонансные полосы очень широки, так что при изучении этих соединений большое преимущество имеет спектроскопия ЭПР, позволяющая получать структурную информацию, в том числе аналогичную даваемой методом спектроскопии ЯМР.
Так, например, из значения g-фактора и сверхтонкой структуры спектра ЭПР получают прямые данные о числе и типах ядер, обладающих спином, с которыми взаимодействует электрон. Иллюстрацией может служить определение по виду расщепления и константе СТВ степени делокализации неспаренного электрона, а по g-факто-ру — основного места его нахождения в продукте окисления хром (III)порфиринового комплекса:
72
СбН5
СбН5
На рис. III.13 показан спектр ЭПР этого продукта. Центральный мультиплет обусловлен взаимодействием неспаренного электрона с четырьмя эквивалентными ядрами I4N(Z= 1), а слабые сателлиты (мультиплета) указывают на взаимодействие с 53Сг(7 = 3/2, природное содержание 9,55%). По центру мульти-
Рис. III.13. Спектр ЭПР продукта окисления хром (III) порфиринового комплекса
плета определено значение
g=l,982((—\g), указывающее на достаточно сильную спин-орби-тальную связь. Опыт с меченым 17О окислителем показал что О связан с Сг, который находится в центре комплекса. На основа
нии всех этих данных сделан вывод, что в результате окисления образуется сР-оксохром(V)порфирин (отрыв двух электронов), Альтернативная структура оксида хром(У)порфирина с неспа-
ренным электроном на лиганде, а не на атоме металла должна была бы давать сверхтонкую структуру спектра ЭПР, обусловленную взаимодействием его с протонами, и значение g-фактора, близкое к go = 2,002, что не согласуется с экспериментом.
2.2. Кинетические и другие исследования
Плодотворное использование спектроскопии ЭПР для изучения механизмов и кинетики химических реакций основывается на следующем. Во-первых, уже сама возможность обнаружения в реагирующей системе парамагнитных центров — атомов, радикалов, являющихся промежуточными продуктами сложных процессов, позволяет предполагать какой-то, например, свободнорадикальный механизм их протекания. Если же, во-вторых, по структуре спектра ЭПР удается идентифицировать парамагнитные частицы, то предполагаемые механизмы получают дополнительное обоснование. Нако-
73
нец, наблюдение за изменением концентрации отдельных парамагнитных центров во времени дает информацию о кинетике процессов.
При помощи так называемой струевой методики, когда реакционная смесь с большой скоростью пропускается через ячейку, находящуюся в резонаторе, удается поддерживать в нем достаточно высокую концентрацию образующихся короткоживущих радикальных продуктов в течение времени, необходимого для регистрации спектра. Это позволяет не только наблюдать промежуточные продукты, но и получать константы скорости отдельных элементарных стадий процесса. Таким методом исследованы, например, цепные реакции, протекающие в смесях Н2 + О2, F2 + CCI4, Р2 + СНзВг (в газовой фазе). Метод ЭПР широко используется для изучения цепного окисления углеводородов в жидкой фазе, реакций радикала ОН с различными спиртами, механизма и кинетики радикальной полимеризации и т. д.
Получили большое распространение методы так называемых спиновых меток и спиновых ловушек. В первом из них к диамагнитной молекуле присоединяют стабильный радикал —
«метку», например, типа —О так, чтобы его свободная ва-
R2z
лентность была незатронутой. В зависимости от характера связи метки и ее окружения она может быть либо закреплена жестко, тогда возникает анизотропия, либо иметь какие-то степени свободы вращения. Вид спектра ЭПР в любом случае служит источником информации об исходной молекуле. Могут использоваться и сразу две метки, присоединяемые к разным частям молекулы.
В методе «спиновых ловушек» в исследуемую систему вводится непарамагнитная молекула «ловушка», которая реагирует с короткоживущим радикалом, образуя стабильный радикал. С помощью этого радикала получают данные о кинетике и механизме процессов в изучаемой системе. В качестве ловушки в реакциях окисления органических соединений пероксидом никеля использовался, например, нитробензол, который фиксировал образующиеся при окислении радикалы R по схеме:
N-R I
о
По СТС спектра ЭПР получающегося стабильного радикала судят об исходном радикале R.
В условиях матричной изоляции при понижении температуры до 77 К (жидкий азот) или до 4,2 К (жидкий гелий) удается стабилизировать на продолжительное время даже чрезвычайно реакционноспособные частицы (атом Н, радикалы СН3, С2Н5 и др.). При этом оказывается возможным не только изучать спектры ЭПР 74
различных парамагнитных центров, но и получать информацию о кинетике радиационно- и фотохимических процессов, подвижности реакционных центров в твердой фазе и ее связи с рекомбинацией радикалов, явлений адсорбции. По изменению формы линий изучается кинетика быстрых обменных процессов (спиновый обмен между радикалами, перенос электрона, заторможенное внутреннее вращение и т. д.).
Так, например, при повторной изомеризации анион-радикала терефталевого альдегида в результате заторможенного внутреннего вращения альдегидных групп
магнитное окружение неспаренного электрона меняется и в условиях медленного обмена наблюдаются отдельные спектры цис- и транс-форм. Действительно, в цис-изомере две пары эквивалентных протонов — это а и Ь, с и d, а в транс-изомере — а и с, b и d. В обоих случаях спектр будет представлять триплет триплетов, но с разным соотношением интенсивностей, и при медленном обмене они накладываются друг на друга. При изомеризации g-фактор не меняется, а сумма констант СТВ в спкеграх изомеров одна и та же, т. е. крайние линии спектров изомеров совпадают. Как оказалось, при изучении интенсивностей компонент отношение концентраций поворотных изомеров транс!цис= 1,4. Большая устойчивость транс-изомера была установлена избирательным дейтерированием. По ширине линий оценено минимальное время жизни т« 4,7-10~7 с.
Важная область применения спектроскопии ЭПР — изучение химических процессов в биологических объектах, в частности метаболизма, при использовании парамагнитных зондов (меток). Это основывается на исключительно высокой чувствительности метода к крайне малому содержанию парамагнитного вещества. Так, например, используемый иногда в калибровочных целях для определения числа неспаренных электронов радикал 1,1-дифенил-2-пикрилгидра-зил (ДФПГ) обнаруживается по сигналу ЭПР при нахождении в резонаторе в количествах 10~12 г. Как один из примеров можно привести изучение кинетических закономерностей взаимодействия Дифениламина с ДФПГ. Лимитирующей стадией процесса является
75
реакция
(С6Н5)2 N - NC6H2 (NO2)3 + (С6н5)2 NH ->
-> (С6Н5)2 N - NHC6H2 (NO2)3 + (C6H5)2 N
По спектру ЭПР можно следить за скоростью уменьшения концентрации ДФПГ и определить по этой кинетической зависимости порядок реакции.
Методом ЭПР определены степени окисления и координационные числа металлов, а также получены данные о лигандах во многих металлопротеинах в замороженных растворах. Один из примеров— оксид хромпорфиринового комплекса.
В спектроскопии ЭПР имеется также круг объектов, которые представляют собой простейшие парамагнитные центры — электроны или «дырки» в твердых телах или растворах. Это могут быть, например, захваченные электроны в кристаллах, в частности различных галогенидов щелочных металлов, называемые F-центрами. При нагревании кристалла, например LiF, в присутствии паров металла и последующего быстрого охлаждения образуется вакансия аниона, занимаемая электроном, т. е. F-центр. Система имеет характерную окраску, обусловленную F-полосой поглощения в видимой области оптического спектра, а в спектре ЭПР появляется широкая полоса F-центров в области чисто спинового значения S’-фактора. Ширина сигнала связана с перекрыванием линий сверхтонкой структуры, обусловленных взаимодействием с ядром окружающих катионов и в меньшей степени с ядрами анионов. Плотность захваченного электрона в основном локализуется на вакансии и мало размывается на окружение, хотя между вакансией и шестью окружающими ее катионами решетки идет «конкуренция» за электрон. Так, при увеличении размеров катиона и постоянном анионе (вакансии) s-характер электронной плотности на шести ближайших катионах возрастает, а при одном и том же катионе и увеличении размеров аниона (от F- к С1~) s-характер электронной плотности на катионах убывает. Существуют и некоторые другие электронно-избыточные центры и предложены различные теоретические модели их описания.
Когда на кристаллы галогенидов щелочных металлов воздействуют излучением высоких энергий, кроме F-центров могут образоваться также электронно-дефицитные И-центры (дырки).
Предложена модель возникновения F-центра, согласно которой происходит потеря электрона анионом, например С1~->-С1, а образующийся атом галогена стабилизируется при сближении с сосед* ним анионом и образовании ион-радикала: С12_. При этом о*-ор-биталь частицы, занятая неспаренным электроном, представляет комбинацию рх-орбиталей атомов галогена. Такая модель нашла подтверждение в наблюдении V-центров как двухатомных молекул, ориентированных в направлении (НО) кристалла.
76
Наконец, так называемые сольватированные электроны возникают, например, в металлоаммиачных растворах. Приближенная модель таких парамагнитных центров предполагает нахождение электрона в полости, окруженной шестью молекулами NH3. Возможна также ассоциация одного или двух таких сольватированных электронов с сольватированным же катионом (катионами) щелочного металла, т. е. образование одно- или двухэлектронных частиц— ассоциатов.
При комнатной температуре ^-фактор для узкой полосы такого электрона равен 2,0008 и не зависит от иона металла. Наблюдается сильное сверхтонкое взаимодействие с ядрами 14N и очень слабое взаимодействие с протонами и ядрами металла.
Получают и исследуют методом спектроскопии ЭПР также гидратированные и другие сольватированные и захваченные электроны, например, в застеклованных спиртах, расплавах солей и т. д.
3. ТЕХНИКА И ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДИКИ СПЕКТРОСКОПИИ ЭПР
3.1. Общие сведения
Принципы устройства и действия спектрометров ЭПР сходны со спектрометрами ЯМР, а отличия связаны в основном с различиями области частот и диапазона напряженности магнитного поля. Обычно стандартные приборы рассчитаны на получение спектров ЭПР при частотах 9,5 ГГц (Х-полоса), 25 ГГц (/С-полоса) и 35 ГГц (Q-полоса), а индукция магнитного поля меняется в диапазоне
0,34... 1,25 Т. Работа на X полосе ведется обычно с растворами и при изучении систем, не требующих очень высокого разрешения спектров, а при высоких частотах и напряженности поля чаще исследуются твердые образцы малого размера и при низких температурах.
На рис. III.14 показана блок-схема спектрометра ЭПР. От микроволнового источника, в качестве которого используется клистрон, поток излучения направляется по волноводу. Пройдя через ферритовый изолятор,
Рис. 111.14. Принципиальная блок-схема спектрометра ЭПР:
1 — клистрон; 2 — ферритовый изолятор; 3 — аттенюатор (ослабитель); 4 — радиочастотный мост;
5 —детектор; 5 —усилитель; 7 — регистрирующее устройство; 8 — модулятор; 9 — полость резонатора; /0 —образец; 11 — электромагнит
77
предотвращающий возврат волн, и ослабитель, он попадает в радиочастотный мост; на одном плече этого моста находится ячейка с образцом, помещаемая между полюсами электромагнита. Ячейка расположена в полости резонатора, ее размер выбирается таким, чтобы образовалась стоячая волна. Исследуемый образец помещается в область наивысшей плотности энергии, а полость с образцом устанавливается так, что его положение соответствует также области однородного внешнего магнитного поля. При выполнении условия резонанса и поглощении энергии излучения возникает разбалансировка моста, и прошедшее напрямую радиоизлучение попадает на кристаллический детектор. Полученный сигнал усиливается и регистрируется на самописце. Применяют обычно модуляцию магнитного поля малой частоты и фазочувствительный детектор, так что сигнал регистрируется, как уже отмечалось, в виде первой производной кривой поглощения. При стабилизированной частоте сканирование для достижений условий резонанса ведется по полю.
В спектроскопии ЭПР очень важен характер исследуемых образцов. Этим методом можно изучать газы, растворы, замороженные растворы, монокристаллы, порошки. Имея в виду релаксационные процессы, обычно выбираются условия, при которых парамагнитные частицы или центры рассредоточены в диамагнитной матрице. Этим условиям удовлетворяют, например, такие парамагнитные образцы, как растворы или твердые тела, в которых парамагнитные центры генерируются при облучении. При изучении ионов переходных металлов часто используют технику выращивания монокристалла с изоморфно замещенной парамагнитным ионом решеткой диамагнитного вещества. Когда монокристалл вырастить не удается, исследуют порошки, содержащие парамагнитные ионы и получаемые соосаждением.
Замороженные растворы обычно получают в виде стекол, для чего подбирают соответствующие растворители или смеси растворителей. Некоторые из них указаны в табл. III.1, где приведены только двухкомпонентные смеси с рекомендуемым соотношением компонентов 1 :1 или 2: 1. Часто используют другие двух-, трех- и даже четырехкомпонентные смеси с самыми различными соотношениями компонентов.
Указанные в таблице спирты, как и воду, можно использовать только когда образец дает очень сильный сигнал и раствор помещается в ампулу с маленьким диаметром, так как эти растворители, имеющие высокую диэлектрическую проницаемость, сильно поглощают энергию микроволнового излучения. Пирекс, из которого изготавливают ампулы для образцов, в какой-то степени также поглощает микроволновое излучение и может сам давать сигнал ЭПР, поэтому если отношение сигнала к шуму мало, то предпочтительно брать ампулу из кварца.
78
Таблица Ш.1. Некоторые стеклообразующие растворители и смеси для спектроскопии ЭПР
Чистые вещества
Смеси
З-Метилпеитаи Изопеитаи
Метилциклопеитаи
Метилциклогексан Изооктаи
Толуол
Триэтаноламин 2-Метилтетрагидрофу-раи
Ди-н-пропиловый эфир Дис-транс-декалии Борная кислота Фосфорная кислота Сериая кислота Глицерин Триацетат глицерина
Сахар Этанол
Пропанол-1
Бутанол-1
Изопропаиол
(1:1)
З-Метилпеитаи/изопеитаи Метил циклопеитаи/метил-циклогексаи
Пропаи/пропилен Пропилеигликоль/вода Диэтиловый эфир/изопеи-таи
Толуол*/метилеихлорид Толуол/ацетои Толуол/метаиол Толуол/этаиол Т олуол/ацетонитрил Толуол/хлороформ
(2:1) Этилеигликоль/вода Диэтиловый эфир/пеи-тен-2(цис- или транс-)
Изопентан/З-метилпеитан 2-Метилтетрагидрофу-раи/метанол
или 2-Метилтетрагидро-фураи/пропноиитрил
или 2-Метилтетрагидро-фуран/метиленхлорид
Толуол можно брать и больше половины.
3.2. Методы двойного резонанса
Существует несколько методов множественного резонанса в спектроскопии ЭПР, из которых основными являются рассматриваемые ниже двойной электрон-ядерный резонанс (ДЭЯР) и электрон-электронный двойной резонанс (ЭЛДОР или ЭДР) *. Как правило, хорошо разрешенные спектры ЭПР регистрируются для невязких жидкостей и кристаллов при низких температурах, а для многих структурно неупорядоченных сред характерны неразрешенные или плохо разрешенные спектры. Основной задачей развития указанных специальных методов явилось повышение спектрального разрешения. В методе ДЭЯР оказывается одновременное воздействие на систему при неразрешенной сверхтонкой структуре в спектре ЭПР двух переменных электромагнитных полей, одно из которых вызывает электронные, а второе — ядерные зеемановские переходы.
* В зарубежной литературе используют сокращения ENDOR и ELDOR для электрои-ядериого и электрои-электроиного двойных резонансов.
79
Рис. III.15. Схема энергетических уровней и релаксационных переходов в спиновой системе с одним иеспаренным электроном (S = ‘/2) и ядром (/=’/2)
В методе ЭДР на спиновую систему также воздействуют двумя переменными полями, но оба поля вызывают электронные зеемановские переходы, а эффект состоит в изменении относительной интенсивности линий многокомпонентного спектра ЭПР.
На рис. III.15 повторена схема энергетических уровней при сверхтонком взаимодействии в системе с одним неспаренным электроном и ядром со спином !/2, которая была уже показана в правой части рис. III.4, но теперь для наглядности две пары уровней (средних и крайних) сдвинуты по горизонтали в разные стороны. По правилам отбора разрешены два электронных спиновых перехода е(1) и е(2), показанные на обоих этих ри
сунках, и два ядерных спиновых перехода п(1) и п(2) (см. рис.
III.15). Необходимо отметить, что константа сверхтонкого элек-
трон-ядерного взаимодействия может быть сравнима и даже больше резонансной частоты перехода в спектре ЯМР (в отсутствие взаимодействия). Переходы типов п(1) и п(2) в ЯМР не могли бы наблюдаться из-за очень широкой резонансной полосы.
В многоуровневых системах, подобных показанной (рис. III.15) при воздействии достаточно мощных радиочастотных полей, может
происходить спиновая поляризация, т. е. возникать неравновесная заселенность уровней с выравниванием заселенности и насыщением каких-то из них. Эта спиновая поляризация и лежит в основе уже рассмотренных в гл. II методов множественного резонанса в спектроскопии ЯМР, а также явлений ДЭЯР и ЭЛДОР, в которых при изучении спектра ЭПР под действием сильного поля (накачки) насыщаются, соответственно, ядерный или электронный зеемановский переход. Измененный спектр ЭПР регистрируется при этом с помощью второго СВЧ-поля (наблюдения).
На рис. III.15 показаны также запрещенные перекрестные электронно-ядерные переходы X и X' с изменением суммарного магнитного квантового числа, равным 0 и ±2. Вероятности этих переходов Wx и Wx> могут быть отличны от нуля, а, например, при низких температурах, когда основной релаксационный процесс обусловлен взаимодействием электронного спина с колебаниями решетки, Wx, Wx' >Wn. Этот случай особенно важен для метода ДЭЯР. Вероятности всех типов релаксационных процессов зависят от анизотропных свойств образца (g-тензора и а-тензора) и различного рода подвижности парамагнитных частиц (центров).
80
Рис. III.16. Спектры ЭПР (а) и 'Н ДЭЯР (б) при установленном значении индукции, помеченном стрелкой, для замороженного раствора (Ti(C8H8)- (С5Н5) ] в толуоле
Спектры ДЭЯР, зависящие от магнитных свойств ядер, с которыми осуществляется сверхтонкое взаимодействие неспаренных электронов, могут, как очевидно, применяться для идентификации ядер и структурных исследований аналогично спектрам ЯМР. Основное их применение — изучение систем со сложными и размытыми спектрами ЭПР, так как эффект ДЭЯР упрощает задачу интерпретации спектра, позволяя проводить идентификацию и измерение слабых взаимодействий, а также определять спиновую плотность на разных ядрах.
На рис. III.16 показаны спектры ЭПР и ДЭЯР замороженного раствора сэндвиче-вого комплекса титана с цик-лооктатетраеном и циклопентадиеном. Этот комплекс представляет собой (/'-систему с осью симметрии бД, (свободное вращение колец), в спектре ЭПР которой сверхтонкая структура не разрешается. В эксперименте ДЭЯР устанав
ливается напряженность постоянного поля, соответствующая сигналу ЭПР для g I (помечена стрелкой), и ведется сканирование по области частот ПМР ('Н) при данной напряженности. Таким образом, получается спектр двойного электрон-протонного резонанса (*Н ДЭЯР) с хорошо разрешенной структурой. На рис. III.16, где представлен этот спектр, хорошо видны два широких дублета, из которых непосредственно определяется значение параллельной компоненты константы СТВ afl для взаимодействия делокализованного неспаренного электрона с протонами циклов С8Н8 и С5Н5 (центральная группа линий обусловлена протонами растворителя— толуола). Если провести такой же эксперимент с установкой сигнала ЭПР, соответствующего g±, то получим перпендикулярные компоненты взаимодействия и определим значение ад_, после чего можно оценить спиновую плотность на ядрах.
Спектры ЭЛДОР получают для одного электронного спинового перехода при облучении (накачке) частотой другого электронного
81
спинового перехода. Изменение интенсивности линии ЭПР на частоте vo при насыщении другого перехода с частотой vp выражается как эффект ЭЛДОР отношением
где I и /о — интенсивности линий наблюдения при наличии и в отсутствие накачки. Величину R регистрируют в зависимости от разности частот vo—vp, а для представленной на рис. III.15 системы уровней пик в спектре ЭЛДОР будет наблюдаться при частоте v=a. И в более сложных системах из нескольких линий спектра ЭЛДОР можно прямо определять константы СТВ. Это особенно ценно при больших расщеплениях или когда спектр усложнен многими взаимодействиями.
В спектрах ЭЛДОР могут наблюдаться также линии, соответствующие накачке запрещенных переходов (Wx, WX'), из которых может быть получена информация как о константах СТВ, так и о средних частотах ЯМР. Вообще методы ЭЛДОР и ДЭЯР являются взаимно дополняющими, причем первый более информативен в случае систем с сильными, а ДЭЯР — со слабыми сверхтонкими взаимодействиями. Преимущества методов двойного резонанса перед обычной спектроскопией ЭПР в достижении не только более высокого спектрального, но и временного разрешения. Этими методами плодотворно исследуются различные релаксационные процессы. Методом ЭЛДОР, например, можно наблюдать более медленные, чем в спектроскопии ЭПР, процессы, время протекания которых сравнимо с временем электронной спиновой релаксации Т\е. Методами спектроскопии двойного резонанса достигается также высокое пространственное разрешение при необходимости изучения рассредоточенных парамагнитных центров в образце. Именно методом ДЭЯР, например, изучались F-центры в кристаллах галогенидов металлов и устанавливалась протяженность размытия плотности захваченного анионной вакансией электрона.
В соответствии с принципами методов двойного резонанса техника этих методов, как видно из сказанного, имеет свои особенности: в спектрометрах имеются два источника радиочастотного излучения (накачки и наблюдения) и две регистрирующие системы. Для проведения эксперимента необходима возможность перестройки частоты источников в широком диапазоне, т. е. сканирования по частоте, в отличие от обычных спектрометров, где осуществляется сканирование по полю. Существуют также приборы с импульсными источниками и с регистрацией методом электронного спинового эха,
3.3. Химическая поляризация ядер и электронов
Обычные спектры поглощения ЯМР и ЭПР молекул, ионов, радикалов регистрируются, как отмечалось, в условиях равновесной заселенности магнитных зеемановских ядерных и электронных 82
уровней, подчиняющейся больцмановскому тепловому распределению. Такая равновесная заселенность может, однако, по разным причинам нарушаться, например, в методах множественного резонанса при воздействии мощных радиочастотных полей.
Может происходить и так называемая химическая поляризация ядер и электронов. Дело в том, что неравновесная заселенность зеемановских уровней может создаваться и при элементарных химических актах в образующихся частицах или в тех состояниях, из которых эти частицы возникают (триплетные состояния, радикальные пары и т. п.). В таких ситуациях будут наблюдаться не обычные спектры магнитного резонанса (спектры поглощения), а либо с аномальной интенсивностью поглощения, либо даже спектры испускания.
Аномально высокая интенсивность сигналов поглощения соответствует повышенной заселенности более низкого зеемановского уровня, т. е. созданию так называемой положительной химической поляризации ядер или электронов: 1; jVpg/yVOe^> 1.
С другой стороны, спектры эмиссии соответствуют повышенной заселенности более высокого зеемановского уровня, т. е. отрицательной химической поляризации ядер или электронов: 1;
А7ре/А7я < 1. Те и другие спектры содержат уникальную информацию об элементарных актах рождения частиц, о самих этих частицах и характере состояний — предшественников этих частиц.
Наблюдение эффектов химической поляризации ядер и электронов в спектрах ЯМР и ЭПР возможно лишь в процессе протекания элементарных актов, так как сверхравновесная заселенность зеемановских уровней каждой частицы сохраняется только в течение времени ядерной (1...30 с) или электронной (10-1...10~5 с) релаксации. Поэтому при изучении этих явлений проводят реакции непосредственно в.магнитном поле спектрометров и непрерывно ведут регистрацию спектров.
В настоящее время химическая поляризация ядер (ХПЯ) исследована во многих реакциях термического распада перекисей и азосоединений, процессах изомеризации с миграцией молекулярных фрагментов и др. На рис. III.17 в качестве примера показано, как выглядит спектр ПМР продуктов, получающихся при термическом распаде перекиси ацетилбензоила в тетрахлорэтилене при 100 °C; видны несколько сигналов эмиссии (направлены вниз), обусловленных отрицательной сверхравновесной ХПЯ. Для каждого пика на рисунке указано его отнесение. Обнаружение эффектов химической поляризации является надежным критерием существования радикальных стадий реакции.
Как ХПЯ, так и химическая поляризация электронов (ХПЭ) бывает двух типов — интегральная (для сигнала в целом) и мультиплетная, когда наблюдаются разные знаки поляризации компонентов в сторону более высокой и низкой от центра мультиплета
83
5
Рис. III.17. Вид спектра ПМР, полученного в процессе реакции термического разложения перекиси ацетилбеизоила
_|__i'll
4 3 2/0 8,мд.
Рис. 111.18. Мультиплетная химическая поляризация в спектре ПМР этилиодида (реакция ал-килиодидов с литийэти-лом)
/°-°\ Н5С6—С. ^с —сн3
(100°С, в C2CI4), с наблюдаемой химической поляризацией ядер образующихся продуктов:
1 — СеНв; 2 — С6Н5СНЭ; 3 — СвН5СООСН3; 4 —СНзС1; 5 — С2Н,;
« — СИ,
напряженности поля. На рис. Ш.18 показан вид мультиплетной ХПЯ в спектре ПМР этилиодида, полученного в реакции литийэти-ла с алкилиодидами.
Теория химической поляризации ядер и электронов сложна и продолжает разрабатываться, но уже в настоящее время открываются большие возможности анализа механизмов и кинетики химических реакций с использованием указанных эффектов.
Методы, представленные в разделе, особенно спектроскопия ЯМР, относятся к наиболее широко применяемым в химических исследованиях. Хотя и не все ядра, в том числе и очень распространенных изотопов важнейших элементов, дают спектры ЯМР, этот метод настолько информативен и продуктивен, что практически ни одна химическая группа исследователей не может без него обойтись. Оба метода являются высоко чувствительными (в большей степени спектроскопия ЭПР) и обладают большой специфичностью.
Современное техническое воплощение методов позволяет исследовать вещества в различных агрегатных состояниях и решать множество структурно-аналитических задач, изучать кинетику и механизмы химических процессов, получать данные, стимулирующие развитие теоретических представлений. Очень важно, что спектро-84
скопия ЯМР, ЭПР и методы множественного, в том числе смешанного, резонанса взаимно дополняют друг друга.
Для успешного использования методов магнитного резонанса как в химии, так и в смежных областях науки и техники необходимо понимание основ их теории, кратко изложенных в гл. I, III, знание практических возможностей и особенностей эксперимента — гл. II, III.
Постоянное совершенствование и появление принципиально новой техники эксперимента, автоматизация и сочетание с ЭВМ открывают все новые возможности и перспективы применения методов. В качестве примеров достижений бурно развивающегося приборостроения в рассматриваемой области можно указать на современные импульсные фурье-спектрометры, появление техники двухмерной спектроскопии ЯМР и уже упоминавшегося множественного резонанса. Повышение чувствительности, спектрального, временного и пространственного разрешения, которое дает эта новая техника, приводит к дальнейшему расширению получаемой информации и поднятию ее на другой, более высокий уровень. Понятно поэтому, что интерес к развитию теории методов спектроскопии ЯМР и ЭПР и практическому их применению не только не ослабевает, но продолжает неуклонно расти.
Контрольные вопросы
Г л а в а I
1. Как выражается энергия взаимодействия ядра, обладающего ненулевым спииом, с внешним магнитным полем?
2. Почему для индуцирования переходов между зеемановскими энергетическими уровнями осциллирующий вектор радиочастотного поля должен быть перпендикулярен направлению постоянного магнитного поля?
3. Что такое спин-решеточная н спии-спиновая релаксация? Как соотносятся их времена в разных агрегатных состояниях вещества?
4. Запишите, исходя из условия ЯМР для двухуровневой системы, выражение частоты через индукцию поля.
5. Перечислите и запишите выражения и шкалы химических сдвигов в ЯМР.
6. Что влияет на величину химических сдвигов в ПМР? Почему химические сдвиги в ЯМР I9F и 13С меняются в значительно большем диапазоне, чем в ПМР?
7. Что такое константа экранирования ядра? В виде каких составляющих ее можно представить?
8. Как связаны обычные и приведенные константы спин-спинового взаимодействия? В чем преимущества приведенных констант?
9. Как соотносятся интегральные интенсивности сигналов ЯМР для групп ядер одного и того же изотопа н интенсивности компонентов мультиплетов?
10. Укажите основные параметры и характерные черты спектров ЯМР первого порядка. В чем отлнчие спектров первого и более высоких порядков?
Глава II
11. С какими физико-химическими характеристиками установлены корреляции химических сдвигов в ЯМР?
85
12. Показанный на рис. II.4 спектр ПВ ЯМР получен для аииоиа BgHu-. О каком строении аииоиа говорит этот спектр?
13. Если известно, что в CH2F2 2ZFH = 50,2 Гц, то какой вид спектра 19F ЯМР будет у соединения CDHF2?
14. В спектре 31Р ЯМР для соединения P4S3 наблюдаются дублет (1:1) н квартет (1:3: 3:1) с соотношением интегральных интенсивностей дублет: квар-тет=3 : 1. Какова структура соединения?
15. Что такое сдвигающие реагенты в ЯМР и что дает их применение в структурных исследованиях?
16. В чем заключается эффект «примесных спинов» и как его можно использовать для определения некоторых ядер?
17. Каковы критерии медленного, быстрого и промежуточного обмена ядер в спектрах ЯМР?
18. При отсутствии обмена протонами между А—Н и В—Н линии ПМР отстоят для иих иа 250 Гц. При комнатной температуре происходит обмен, и линия отстоят иа 25 Гц. Концентрации частиц одинаковы (0,2 моль/л), а время спии-решеточиой релаксации велико. Рассчитайте время жизни протона у А—Н и найдите константу скорости обмена.
19. В чем суть методов множественного резонанса?
20. Что такое двухмерная спектроскопия (2D) ЯМР?
Глава III
21. Объясните условие ЭПР и его реализацию в экспериментальном воплощении метода.
22. Что такое g-фактор Лайде и как ои влияет иа положение сигнала ЭПР?
23. Каковы правила отбора для переходов между зеемановскими уровнями по электронному и ядерному спиновым квантовым числам в системах с электрон-ядериым сверхтонким взаимодействием?
24. Константа СТВ с 13С в метиловом радикале а1ЯС = 41 • 10—«Т, а константа протонного СТВ я,н = 23.10-4Т. Попытайтесь схематично изобразить спектр ЭПР радикала 13СНз[Со1е Т. et al. Mol. Phys., 1, 406 (1958)].
25. Как возникает тонкая структура спектров ЭПР анизотропных систем? Что такое крамерсовское расщепление?
26. В чем суть метода «спиновых меток»? Какие данные ои позволяет получать?
27. Какие методы миожествеииого резонанса применяют в спектроскопии ЭПР (совместно с ЯМР)?
28. В каких случаях возникает неравновесная заселенность зеемановских спиновых состояний?
МЕТОДЫ КВАДРУПОЛЬНОГО И ГАММА-РЕЗОНАНСА ЯДЕР
Спектроскопия ядерного квадрупольного резонанса (ЯКР), относящаяся к радиоспектроскопическим методам, и метод мессбауэровской спектроскопии, называемый также методом ядерного гамма-резонанса (ЯГР), используются в структурных исследованиях и позволяют получать уникальную информацию о распределении электронной плотности и характере химических связей по сдвигам резонансных сигналов ядер и параметров градиента неоднородного электрического поля на ядрах, создаваемого электронным окружением. Эти данные важны как опорные для теоретической и квантовой химии. Оба метода применимы для исследования только твердых образцов. Исключительно высокая чувствительность обоих методов к малейшим изменениям электрических полей открывает возможность исследования широкого круга проблем, связанных с внутри- и межмолекулярными взаимодействиями.
Два рассматриваемых в разделе метода на первый взгляд, казалось бы, имеют мало общего. Действительно, если иметь в виду частотные (Гц) диапазоны, в которых происходят соответствующие квантовые переходы, то они отличаются на десять-пятнадцать порядков (в спектроскопии ЯКР 104... 109 Гц, а в мессбауэровской спектроскопии 10!s... 10i9 Гц). Это ведет к тому, что
техническое воплощение методов не имеет ничего общего. Есть, конечно, у каждого из них и не только аппаратурная специфика. Но при всем том многие данные, получаемые химиками при использовании этих методов, по своему характеру весьма близки и касаются электронной структуры, а шире — химического строения веществ.
Метод ядерного квадрупольного резонанса стал применяться в химических исследованиях в конце 40-х — начале 50-х годов. Ценность и перспективность метода заключаются в возможности получать с его помощью количественные данные об изменении электрических полей на ядрах атомов в молекулярных кристаллах, т. е. о распределении электронной плотности и характере химических связей в основном состоянии молекулярных систем. Это один из немногих методов прямого определения градиентов электрических полей, дающий, таким образом, возможность экспериментальной проверки квантово-химических приближений и расчетов. В отличие от методов магнитного резонанса в спектроскопии ЯКР сдвиги резонансных частот зависят только от внутримолекулярных электрических воздействий. Внешнее магнитное поле иногда требуется налагать при изучении асимметрии неоднородного электрического поля в некоторых системах.
Гамма-резонансная ядерная флуоресценция, т. е. испускание и поглощение у-квантов при ядерных переходах без затраты энергии на отдачу ядра, была открыта Р. Л. Мессбауэром в 1958 г. Эффект назван поэтому его именем, как и разработанный метод спектроскопии. Источником излучения и объектом, поглощающим его, являются ядра одного и того же изотопа, соответственно, в возбужденном и основном состояниях. В ядерной физике ядра с одинаковыми зарядами и массовыми числами, но разными энергиями и временами жизни (полураспада) называют изомерами. Время жизни изомеров играет огромную роль в гамма-резонансной спектроскопии, определяя ширину линий. Большим достоинством метода является высокая монохроматичность у-излучения (узость линии) и высокое спектральное разрешение. Положение резонансного сигнала или так называемый изомерный сдвиг зависит от электронного окружения ядер. Метод мессбауэровской спектроскопии позволяет получить такие же данные о градиенте электрического поля иа ядрах, как и метод спектроскопии ЯКР, 88
а некоторые данные аналогичны получаемым в спектроскопии
ямр.
Для химиков чрезвычайно важна та эмпирическая основа для изучения закономерностей распределения электронной плотности в молекулярных системах, которую представляют данные мессбауэровской спектроскопии, как и спектроскопии ЯКР. На них опираются также квантово-механические расчеты многоэлектронных систем. Оба рассматриваемых метода имеют также прикладное значение в химических исследованиях, как структурно-аналитические.
ГЛАВА IV
ЯДЕРНЫЙ КВАДРУПОЛЬНЫЙ РЕЗОНАНС
1. ОСНОВЫ ТЕОРИИ
1.1. Общие сведения
Ядерный электрический квадрупольный момент eQ является мерой отклонения распределения электрического заряда в ядре от сферической симметрии. Качественно можно представить четыре возможных типа ядра (рис. IV.1). Если суммарный спин ядра /А и, следовательно, его магнитный момент Цп равны нулю (рис. IV. 1, а), то распределение заряда в ядре характеризуется сферической симметрией, и квадрупольный момент eQ отсутствует. Распределение заряда остается сферическим, т. е. eQ = 0, и при спине ядра /д = '/2, когда цл=Н=0 (рис. IV. 1, б). Если (цл#=0), то сферическая симметрия распределения заряда нарушается, и появляется электрический квадрупольный момент eQ#=0. На рис. IV.l,s и IV.1, г показаны, соответственно, два случая: eQ>0, когда распределение заряда вытянуто вдоль спиновой оси, например у 14N (eQ = 0,019 барн), и eQ<0, если оно ориентировано перпендикулярно этой оси, например у 35С1 (eQ=—0,082 барн).
Размерность квадрупольного момента, вообще говоря, определяется произведением заряда на квадрат расстояния, но обычно в качестве единицы измерения используется барн=10-28 м2. Все известные величины ядерных квадрупольных моментов невелики и лежат в пределах —2 < eQ < +10 барн.
Эффект ЯКР может наблюдаться на ядрах ограниченного числа элементов (несколько десятков ядер, включая разные изотопы). Среди них такие распространенные элементы, как В, N, Cl, Br, I, А1, Си, As, Sb и др. В то же время ядра Н, С, F, Р и ряда других важнейших элементов не имеют квадрупольного момента и не дают спектров ЯКР. Наиболее часто методом ЯКР исследуются галогеносодержащие соединения и несколько реже соединения азота. При этом примерно для половины изучавшихся соединений с азотом и ~20% соединений с хлором эффекта ЯКР наблюдать не удалось,
89
Рис. IV.2. Схема расположения квадрупольного ядра А и ориентации ядерных моментов в неоднородном электрическом поле. Ось z направлена вдоль связи атомов А—М
Рис. IV.1. Качественное представление типов ядер:
неквадрупольные ядра, eQ-0: а — I— -0. цл-0 и б —7-72. цл=#0; квадру-вольные ядра, е<?¥=0; в — 7>1, g п¥=0, eQ>0 и г — />1, рл¥=0, eQ<0
так что возможность нахождения линий ЯКР для каких-то конкретных соединений с квадрупольными ядрами является несколько проблематичной.
Электронное окружение квадрупольного ядра в молекуле, не обладающее сферической симметрией, создает неоднородное электрическое поле, которое характеризуется градиентом напряженности электрического поля на ядре eq (рис. IV.2). Имеет место взаимодействие ядра, обладающего электрическим квадрупольным моментом eQ с градиентом поля eq. Энергия этого взаимодействия зависит от ориентации эллипсоидального квадрупольного ядра относительно системы главных осей тензора градиента электрического поля, а ее мерой является константа квадрупольного взаимодействия e2qQ. Аналогично тому как квантуется энергия вращающегося электрона в поле положительного ядра, квантуется и энергия квадрупольного взаимодействия. Иными словами, возможны различные квантованные ориентации ядерного квадрупольного момента и соответствующие квадруполь-ные уровни энергии. Эти уровни присущи данной молекулярной системе, т. е. являются ее свойством, в отличие от зеемановских уровней ядер и электронов в спектроскопии ЯМР и ЭПР, которые появляются при воздействии внешнего магнитного поля. Разности энергий, как и сами энергии квадрупольного взаимодействия, зависящие от электрического квадрупольного момента ядра eQ и градиента неоднородного электрического поля eq, невелики, и переходы соответствуют радиочастотному диапазону 104... 109 Гц. Прямые 90
методы наблюдения ЯКР используются для частот выше 2 МГц. Поглощение излучения, связанное с квадрупольными переходами, можно наблюдать только в твердой фазе, так как в газе и жидкости хаотическое движение и неупорядоченность молекул усредняют градиент электрического поля на ядре и расщепления квадрупольных уровней энергии не происходит.
Значения квадрупольных моментов ядер обычно известны, и экспериментальные исследования спектров ЯКР проводятся для получения частот переходов, констант квадрупольного взаимодействия, а значит, е^ипараметров асимметрии градиента электрического поля г] (см. ниже), т. е. структурных данных, информации о распределении зарядов и характере химических связей. Например, чем больше ионный характер связи с данным атомом, тем меньше величина градиента поля и e2^Q. Обратно, чем более ковалентной является химическая связь, тем выше соответствующая константа квадрупольного взаимодействия. Данные ЯКР предоставляют возможность экспериментальной проверки результатов квантово-механических расчетов и приближенного рассмотрения ряда проблем, связанных с внутри- и межмолекулярными взаимодействиями. Метод спектроскопии ЯКР важен как аналитический при работе с твердыми веществами, для которых не представляет трудности выращивание больших монокристаллов.
1.2. Электростатическое взаимодействие квадрупольного ядра с электрическим полем
Для наглядности рассмотрим классическую теорию взаимодействия ядра с внешним электрическим полем. Квадрупольный момент eQ связан с функцией распределения по объему заряда ядра Ze. Фиксируем систему координат с началом в центре ядра. Пусть р(хь Х2, Хз) =р (г)—плотность ядерного заряда в точке с координатами Х\, х2, х3 (г — радиус-вектор), a U(xi, х2, Хз) — U(г) — потенциал, создаваемый в этой точке всеми окружающими ядро зарядами. Тогда энергию электростатического взаимодействия заряда Ze с внешним электрическим полем можно записать в виде
Е — f р (Х1, х2, х3) U (*1. Х2, x3)dv ==J Р (г) и (г) do. (IV. 1)
Интегрирование ведется по объему ядра, вне которого р(г)==О. На эту величину Е отличается полная энергия ядра в веществе от энергии «обнаженного» ядра. Поскольку потенциал U(г) меняется по объему ядра мало в связи с малостью размеров по сравнению с обычными расстояниями между ядром и окружающими его зарядами, можно представить потенциал в виде разложения в степен
91
ной ряд:
3 3
£/(г) = 6Z(0) + V (-—-) + У ("5"^—)•*/•*/+••.• (IV.2)
XjV dxt Jo 2 dxidxj Jo
Можно заранее выбрать направление осей координат, так чтобы перекрестные члены (вторые производные потенциала при i=£j) равнялись нулю (система главных осей), а если учесть также, что интеграл по всему объему / p(r)du=Ze, то подстановка (IV.2) в (IV.1) приводит к следующему разложению энергии по мультиполям:
3 3
Е = eZU (0) + У ) f (г) dv + V 2 (“гН (г)dv + • • • •
(IV.3)
Первый член («монопольный») в этом выражении представляет энергию взаимодействия кулоновского точечного заряда ядра Ze с окружающими зарядами, т. е. не зависит от ориентации ядра. Можно отметить, что он не представляет интереса также и при сравнении энергии основного и возбужденного состояний ядра (гл. V), так как Ze и U(0) для них не различаются. Второй член («дипольный») в выражении (IV.3) также исчезает, так как р(г)==р(—г)> т. е. центры массы и распределения плотности заряда ядра совпадают, ядро не обладает электрическим дипольным моментом, и интегралы типа J x,p(r)du равны нулю. По тем же причинам инвариантности по отношению к изменению знака координат исчезают все члены с нечетными степенями х,. Таким образом, интерес представляет лишь третий, квадрупольный, член , 3 , „,гг ч „
(IV. 4)
1-1 \ '"'I /о
Обозначим Uа — ( градиент электрического поля в направ-
\ дх* /о
лении оси Xt в точке Xi—О. Вводя r2~x12+x22+xz2, выражение (IV.4) приводят к виду
r2p(r)dv. (IV.5)
2 Г2 \ 14
i~— ₽(r)dv + -^
Второй член инвариантен по отношению к изменению ориентации ядра (равен 0 : У (7z/=V2(7=0 — уравнение Лапласа, см.
i
ниже) и при рассмотрении энергии квадрупольного взаимодействия не нужен. Этот нулевой член введен, чтобы придать Е в (IV.5) нужные свойства симметрии. Он обусловливает сдвиг энергетического уровня ядра и важен в мессбауэровской спектроскопии (гл. V).
92
Первый член в выражении (IV.5) описывает взаимодействие квадрупольного момента ядра с неоднородным электрическим полем в точке Xi=л‘2=Хз=0 и обусловливает квадрупольное расщепление энергетического уровня ядра на два или несколько подуровней. Введя
Q/7=if(3x2-r2)p(r)dvF (IV.6)
можно преобразовать выражение этого члена к виду
1 °
(IV.7)
Энергия квадрупольного взаимодействия Eq отлична от нуля только в том случае, когда не равен нулю интеграл (IV.6), т. е. распределение заряда ядра не имеет сферической симметрии. Наличие спина ядра 1^1 придает распределению заряда ядра эффективно цилиндрическую симметрию. Если принять за главную ось эллипсоида вращения, представляющего тензор квадрупольного момента, ось i—z, то, учитывая, что Qxx + Qyy + Qzz=O, имеем Qxx=Qpy=—Qzz/2. Таким образом, для определения квадрупольного момента ядра нужен, как уже говорилось, всего один параметр Q = QZ2, а выражение энергии квадрупольного взаимодействия (IV.7) в координатах i=x, у, z можно переписать в виде
Eq = v \uzz - 4- {Uxx + £7^)1. (Iv.8)
0 L 2 J
Градиент неоднородного электрического поля, создаваемого на ядре окружающими зарядами, также представляет собой симметричный тензор, след которого Uxx+ Uyy + Uzz=0 *, а в системе главных осей тензор диагоналей. Введем новые обозначения элементов этого тензора: qxx—Uxx, qyy = Uyy, qzz—Uzz. При сферической симметрии поля qxx—qyy = qZZt т. е. Eq—Q. При осевой симметрии поля, что часто встречается на практике, qxx=qyy=^qzz> т. е. для характеристики градиента электрического поля также достаточен один параметр: q—qzz или eq (е— единичный заряд). Ось z может совпадать, например, с направлением химической связи атома А, имеющего квадрупольное ядро, с другим атомом М (см. рис. IV. 2).
Для асимметричного поля принято выбирать главные оси так, что \qzz\ > \qyv\ > |<7ххj - Кроме наибольшей по модулю компоненты qzz, т. е. градиента поля eq, в этом случае вводится параметр
* Это известное из фнзикн дифференциальное уравнение Лапласа, описывающее потенциал электростатического поля в области, не содержащей заряда:
Э2Ц dW oW
vj2(J == ” -4-----4- -------- 0.
V дХ2 ду2 дг2
93
а с и м м е т р и и г]—|^хх—quv\lqzz, являющийся мерой отклонения поля от осевой симметрии и лежащий в пределах 1 ^т]^0. Эти две величины позволяют полностью охарактеризовать тензор градиента электрического поля в системе главных осей.
1.3. Квадрупольные уровни энергии и переходы
Чтобы перейти от классического выражения энергии (IV.7) к квантово-механическому гамильтониану, достаточно заменить Qn (1V.6) соответствующим квантово-механическим оператором:
(IV.9)
Собственные значения энергии можно получить, оценив соответствующие матричные элементы перехода между ядерными состояниями \1т>\
(IV.10)
где I — полный угловой момент или спин ядра; т— его проекция на ось г или ядерное магнитное квантовое число, принимающее (2/+1) значений (от +1 до—1). Поскольку разность энергий между основным и первым возбужденным состояниями ядра очень велика (~1021 Гц) по сравнению с величиной Eq (ниже 109 Гц), то всеми ядерными состояниями, кроме основного, можно пренебречь.
Симметрия квантово-механического оператора совпадает с рассмотренной симметрией тензора Qu, а в зависимости от симметрии тензора градиента электрического поля Un получаются выражения энергии квадрупольного взаимодействия как функции q или q и г].
В общем случае квантово-механическую задачу на собственные значения энергии квадрупольного взаимодействия строго решить трудно, особенно при нецелочисленных спинах ядер и симметричном градиенте поля. Однако для некоторых конкретных систем эта задача решена точно.
Так, например, квадрупольные уровни энергии ядра с квадру-польным моментом eQ в неоднородном электрическом поле с осевой симметрией и градиентом eq определяются формулой
Из этого выражения видно, что уровни энергии дважды вырождены по mi, так как не зависят от знака mi. Так, например, при 7=3/г ("В, 35>37С1, 79>81Вг и др.) возможно появление двух дважды вырож-
94
денных уровней при т1 = ±^2 и Ш/ = ±3/2 (рис. IV.3). Энергии этих уровней в соответствии с уравнением (IV.11) равны:
Р _ е2^
± Va 4
а их разность
ДЕ = Е± 8/> — E±1/t = e2qQ
2 Рис. IV.3. Квадрупольные уровни энергии ядер с
полуцелочисленным спнном при осевой симметрии Введем В дальнейшем поля, создаваемого окружением, и возможные обозначение e2qQI^ = K- переходы
Осевая симметрия по-
ля возникает, например, в молекулах СН3С1, СН3Вг, причем наибольший градиент поля совпадает с направлением связи углерод— галоген, принимаемым за ось z. Для указанного случая
можно ожидать один переход между уровнями при выполнении условия резонанса hv = AE, где v — частота радиочастотного диапазона, поглощаемая образцом. Из частоты поглощения можно непосредственно найти константу квадрупольного взаимодействия e2qQ = 2hv. Обычно ее выражают в единицах частоты (МГц), т. е. в рассмотренном случае она равна просто удвоенной частоте ЯКР.
Используя уравнение (IV.11), можно найти квадрупольные уровни и соотношения между частотами переходов и e2qQ для ядер с другими значениями I в аксиально-симметричных полях. Так, для 7=5/2 (27А1, I21Sb, 1271 и др.), также показанном на рис. IV.3, возникают три уровня энергии: Е±1р, Е±3,з и Ei5/i , и при обычных условиях все они заселены. Правило отбора квадрупольных переходов Д|т/| = 1, так что возможны два перехода:. E±i 2-^-Е±3,-2 и Е±у2~*~ -+Е+5/2, причем частота второго перехода в два раза больше первого: v2=2vi.
Получены точные решения на собственные значения энергии для спинов 7=1 и 7=3/2 в асимметричном поле, когда уравнение (IV.11) уже непригодно. При 7=1 (14N, 33S и др.) получаются три показанных на рис. IV.4, б квадрупольных уровня энергии, которые описываются формулами:
Е± =
e2gQ (1 ± 1) 4/ (2/ - 1)
= К(1 ±1),
(IV.12)
95
m=0
m-±1
Eq-2K
„ e^qQ
0 = ~ 2/(2/ + 1) = ~ К'
Рис. IV.4. Квадрупольные уровни энергии н частоты переходов для ядер со спином 1—1 в аксиальносимметричном (а) н асимметричном (5) полях
Возможны переходы (рис. IV. 4, б) с частотами v+ = K(3 + t])//j и v- = K(3—г])/7г, из которых непосредственно определяется как константа квадрупольного взаимодействия е27<2 = 4/<, так и параметр асимметрии т]. Если параметр т] достаточно велик, то правило отбора Дт = ±1 нарушается и возможен также переход с частотой vd = 27<T]//i. В аксиально-симметричном поле (т] = = 0) уровни Е+ и Е- вырождены (Е+ = К), и возможен только
один переход с частотой ч = ЪК11г (рис. IVA,a). Асимметрия градиента электрического поля на ядре 33S имеет место, например, в молекуле CH3SH, где валентный угол ZCSH — 920.
Число квадрупольных уровней при нецелочисленных спинах, как видно на рис. IV.3, равно (/Ч-’/э) и все они дважды вырождены. Для ядер с 1=г1ъ в несимметричном поле получаются следующие
выражения квадрупольных уровней энергии:
E^=K(t^)
{ ч2 V/«
(IV.13)
и возможен только один переход с частотой
= / h.
\ з У /
(см. рис. IV.3). Таким образом, по единственной частоте ЯКР нельзя определить одновременно e2qQ и параметр асимметрии т]. Для органических соединений в большинстве случаев и функция
, -п2 у/* 3 )
мало чувствительна к изменению ц. Поэтому частота
ЯКР определяется в основном величиной e2qQ (т. е. градиентом поля eq). В слабых магнитных полях (0,5... 2-10~2 Т) можно наблю
дать смещение невырожденных и расщепление дважды вырожденных (по тт) квадрупольных уровней (эффект Зеемаиа). В результате увеличивается число возможных переходов и наблюдаемых частот ЯКР, что позволяет находить e2qQ и г].
Для других значений спинов точных решений задачи нет, но выведены уравнения (в виде полиномов) и составлены таблицы для вычисления e2qQ и г] из частот ЯКР-спектров. Схема относительного расположения квадрупольных уровней энергии для других ядер с
96
полуцелочисленными спинами в аксиально-симметричном поле и возможные переходы также показаны на рис. IV.3. При т) = 0 частоты переходов при заданном I относятся как простые целые числа: 1: 2:3:4.
1.4. Интенсивность, ширина и мультиплетность сигнала
Следует заметить, что хотя сами квадрупольные энергетические уровни обусловлены взаимодействием электрического квадрупольного момента ядра с неоднородным электрическим полем, индуцированные переходы между ними связаны с взаимодействием магнитного момента ядра с переменным (радиочастотным) магнитным полем, так как энергия взаимодействия квадрупольного момента с электрическим полем в 1014 раз меньше энергии магнитного дипольного взаимодействия.
Вероятность переходов, а следовательно, интенсивность в спектрах ЯКР зависит от направления вектора переменного магнитного поля В по отношению к главным осям тензора градиента неоднородного электрического поля на ядре. При осевой симметрии градиента электрического поля интенсивность ЯКР максимальна при В±г, а при В||г вероятность перехода (интенсивность) равна нулю. Поэтому для монокристаллов по зависимости интенсивности ЯКР от угла поворота можно в принципе локализовать систему главных осей. В случае порошков интенсивность ЯКР составляет ~50% максимальной интенсивности, которую можно получить для соответствующего монокристалла.
Кроме измеряемых частот ЯКР, из которых определяют e2qQ и т), характеристическим параметром этих спектров является также ширина линий Av. Она меняется от ~700 Гц (для 14N) или ~1 кГц (для 35С1) до ~50 кГц (93Nb), что составляет максимум. Ширина линии связана с временами релаксации 7\— спин-решеточ-ной и Т2— спин-спиновой, представляющими во многих случаях специальный интерес. Кроме того, Av сильно зависит от дефектов и напряжений, а в неидеальных кристаллах и от содержания примесей, так как все это приводит к статистическому разбросу значений градиента поля.
Небольшой вклад в ширину линии вносят магнитные взаимодействия, в частности и магнитное поле земли, равное 5-10~5Т (для 35С1 и 14N его вклад в ширину линии будет около 150 Гц).
Наконец, немаловажной характеристикой является мультиплетность сигналов ЯКР, которая обусловлена тем, что химически эквивалентные атомы могут находиться в кристаллографически неэквивалентных позициях При этом возникают различия кристаллического поля в местах резонирующих квадрупольных ядер, приводящие к «кристаллическим сдвигам» частот ЯКР. Число компонент мультиплетов т зависит от числа сортов кристаллографически неэквивалентных мояекул в элементарной ячейке N и числа наборов 4—1377 97
кристаллографически неэквивалентных атомов в каждой молекуле л/:
х
(IV.14) /-1
Отношение интенсивностей линий в мультиплете равно отношению чисел резонансных атомов в каждом неэквивалентном положении.
2. ПРИЛОЖЕНИЯ И ИНТЕРПРЕТАЦИЯ СПЕКТРОВ ЯКР
2.1. Частоты ЯКР
Как следует из теории (§ 1), частоты ЯКР зависят от квадрупольного момента ядра и градиента электрического поля на ядре. Квадрупольные моменты ядер eQ меняются для элементов довольно закономерно по периодической системе, увеличиваясь сверху вниз по группам и справа налево по периодам, но с некоторыми исключениями. Изменение значений констант e2qQ для атомов не симбат-но eQ, так как зависит также от электронной конфигурации атома, т. е. eq^T Например, eQ для элементов левых подгрупп больше, чем для правых, a e2qQ, наоборот, для элементов правых подгрупп больше, чем для левых. У большинства квадрупольных ядер eQ>0.
В зависимости от окружения ядра в молекулярной системе (в кристалле) частота ЯКР может меняться по сравнению с частотой для атома в очень широких пределах. Например, для одновалентного атома А можно указать следующие закономерности изменения или сдвига частоты. Наибольшей является зависимость ее от атома партнера М, с которым связан атом А; изменение частоты ЯКР атома А может достигать 1500%.
При одном и том же атоме М в зависимости от его валентного состояния (гибридизации АО) частота может меняться в пределах до 50%. В зависимости от окружения, т. е. заместителей при М в данном валентном состоянии, изменения частоты могут происходить в пределах до 20%. Расщепления линий ЯКР могут превышать 2% частоты также при разных конформациях молекул, что позволяет иногда определять наличие конформеров и соотношение между ними. Такого же порядка сдвиги частоты ЯКР возможны за счет эффектов кристаллического поля в молекулярных кристаллах. Что касается изменений частот, вызываемых наличием меж- и внутримолекулярных водородных и других ассоциативных связей, то они могут составлять от 3 до 40% частоты.
Больше всего изучались спектры ЯКР соединений изотопов 35С1 и 37С1. Природное содержание 35С1 много выше (75,53%), и будем рассматривать здесь данные только по этому изотопу. Линии ЯКР 37С1 в эквивалентном окружении ядра просто лежат ниже по часто-98
Рис. IV.5. Интервалы частот ЯКР 35С1 для некоторых типов связей и классов соедине-ний L
те (в соответствии с отношением квадрупольных моментов 37Q/35Q— 0,785) и примерно в 3 раза менее интенсивны. На рис. IV.5 представлены некоторые характерные интервалы частот ЯКР 35С1 для различных типов связей и соединений. Для ковалентных связей хлора частоты попадают в основном в интервал ~30...40 МГц, хотя, как видно на схеме, бывают и отклонения. У многих неорганических соединений хлора частоты лежат ниже указанного интервала, что свидетельствует о большем ионном характере связей. Вообще же диапазон частот ЯКР для 3SC1 составляет от ~70 (FC1 70,7) до ~2 (FeCl2 2,37) МГц. Поскольку оба изотопа хлора имеют спин /=3/2, то, как было сказано выше, обычно по спектру ЯКР нельзя определять параметр асимметрии поля т). Этот недостаток в какой-то степени компенсируется свойствами хлора, образующего в органических соединениях практически только a-связи, когда в хорошем приближении можно принимать т) = 0. Но во всяком случае, согласно уравнениям (IV. 13), имеется слабая зависимость v от г].
У соединений liN (аминов, гетероциклических соединений, нитрилов, цианидов, нитритов) частоты ЯКР лежат в диапазоне от 0,8 до 6 МГц, но ниже 1,5 МГц их наблюдать очень трудно, если вообще возможно. А, например, для приведенных в табл. IV.1 других элементов интервалы частот ЯКР составляют: 75As~30... 120 МГц, 122> 123Sb~ 10... 180 МГц (2 перехода у 121Sb, 3 перехода у 123Sb, 1= ==7/2), 209 Bi~15... 115 МГц (4 перехода). Найдены многие эмпирические закономерности изменения частот ЯКР в рядах и различных классах неорганических, органических и высокомолекулярных соединений.
Частоты линий ЯКР, т. е. «константы» e2qQ, имеют явно выраженную температурную зависимость. Так, например, для 14N частота монотонно уменьшается примерно на 0,1 МГц при повышении температуры от 77 до 300 К, что характерно для низкочастотных линий. У 35С1 изменения могут составлять 0,5... 1 МГц, но известны 4* 99
факты отсутствия или даже противоположного хода изменения (увеличения частоты с температурой). При фазовых переходах наблюдается скачкообразное изменение v(e2^Q).
При изучении влияния давления на e2gQ обнаружен рост частоты с увеличением давления, что объясняется возрастанием ковалентности связей и увеличением плотности (сближением частиц). Влияние давления всегда должно рассматриваться с учетом температурного эффекта и изменения объема.
Точные значения частот, полученные при известных условиях и приводимые в справочниках, могут служить для целей идентификации. Данные о мультиплетности сигналов, получаемые по частотам характеристики градиента электрического поля (e2^Q и р), и другие данные содержат, как отмечалось в § 1, большую информацию, интересующую химика. Извлечение ее, т. е. решение обратной задачи метода, основывается на модельных представлениях и приближенных расчетах, некоторые из них рассматриваются ниже.
2.2. Структурные приложения
В связи с тем, что спектры ЯКР получают для кристаллов, решающее значение для их интерпретации и извлечения структурной информации Имеет знание основ кристаллохимии и кристаллографии, а прежде всего симметрии молекул и кристаллических структур. Как уже указывалось, квадрупольное ядро каждого не только химически, но и кристаллографически неэквивалентного резонирующего атома характеризуется своим сигналом ЯКР, т. е. значениями e2qQ и г]. Этим обусловлена мультиплетность т, т. е. число линий ЯКР, соответствующее числу неэквивалентных позиций резонирующих атомов одного и того же изотопа (IV.14). Соотношение интенсивностей линий мультиплета записывается в виде
i 1: i-2 : Z3 :... : im = q\: qt: qj :... : qm,
где (k от 1 до tn) — кратность кристаллографически неэквивалентных атомов в данной пространственной группе.
Если известна структура молекулярного кристалла или по крайней мере число молекул в элементарной ячейке I и пространственная группа, то общее число резонирующих атомов Г=п1, где п —
т число резонирующих атомов в молекуле (я^я/), или Г—l^ q^.
ы
Используя эти соотношения, можно объяснить наблюдаемый спектр/' ЯКР, а при неизвестной структуре кристалла, но при заданной фор-»; муле и симметрии молекулы представить возможные способы раз-У мещения молекул в решетке, т. е. получить по спектру ЯКР важную, ‘ структурную информацию.
Рассмотрим некоторые примеры. Молекула тетрахлор-п-фени-лендиамина.
100
NH,
относится к точечной группе симметрии D2h, и все четыре атома хлора в ней химически эквивалентны. Структура же кристалла такова, что две молекулы (/=2) в элементарной ячейке кристаллографически эквивалентны (?/=!), но симметрия каждой молекулы понижена (С,- или 1). Это значит, что в каждой молекуле есть два набора (п; = 2) кристаллографически неэквивалентных атомов С1, кратность каждого из которых ?й=2. Тогда мультиплетность сигнала 3SC1 будет равна 2, а отношение интенсивностей в мультиплете 1:1; интегральная интенсивность мультиплета пропорциональна общему числу резонирующих ядер.
Спектр ЯКР 35С1 низкотемпературной кристаллической фазы пентахлорида фосфора PCU состоит из десяти линий, которые образуют две группы: четыре линии при более высоких, а шесть линий при более низких частотах. Этот спектр согласуется с другими известными данными о кристаллизации пентахлорида фосфора в виде РС14+РС16-, т. е. четыре высокочастотные линии относятся к катиону, а шесть низкочастотных — к аниону. Интересно, что здесь все десять атомов С1 оказываются кристаллографически неэквивалентными. При быстром охлаждении паров пентахлорида фосфора образуется другая метастабильная фаза, дающая в спектре ЯКР три линии. Две из них относят к кристаллографически неэквивалентным атомам хлора в аксиальных (а) положениях тригонально-бипирамидальной конфигурации PCI5, а третью — к экваториальным (е) атомам хлора, которые, очевидно, кристаллографически эквивалентны:
а
В случае монофтортетрахлорида фосфора PFCh при аксиальном (а) замещении фтором также есть данные о двух кристаллических модификациях. В спектре ЯКР 35С1 одной из них наблюдается четыре линии равной интенсивности, что свидетельствует о кристаллографической неэквивалентности всех четырех атомов С1 (симметрия молекулы понижена до Ci или 1). В другой модификации наблюдается две линии с соотношением интенсивности 1 :3, т. е. существует’два набора кристаллографически неэквивалентных
101
атомов (аие), а симметрия молекулы в кристалле, очевидно, сохраняется.
В рассмотренных примерах число наборов кристаллографически эквивалентных молекул в ячейке было равно единице Но,
например, в кристалле Cs+GaCU- число линий ЯКР 35С1 равно удвоенному числу атомов хлора, т. е. имеется уже два набора кристаллографически неэквивалентных анионов в ячейке.
По спектру ЯКР было идентифицировано соединение Na+[Ga2C17]-. В спектре наблюдалось две линии 69Ga и семь 35С1, один из сигналов хлора при более низкой частоте, чем шесть остальных. Сравнение с другими спектрами привело к заключению о следующей структуре аниона:
все атомы которого (Ga и С1) кристаллографически неэквивалентны, а в ячейке один набор кристаллографически эквивалентных анионов.
Структурные данные получают также находя параметр асимметрии поля т). Так, например, обнаружение достаточно большого т| по спектру ЯКР 1271 для НЮ3 показало отсутствие осевой симметрии этой молекулы и привело к предположению структуры Ю2(ОН).
Иногда спектры ЯКР используют и для получения данных о таких геометрических параметрах частиц, как валентные углы и межъядерные расстояния. Конечно, эти данные не обладают высокой точностью, но могут служить прикидочными при изучении сложных структур кристаллов. Например, при изучении РВгзО в виде монокристалла были измерены зеемановские расщепления каждой линии ЯКР Вг в зависимости от ориентации кристалла, удалось определить его пространственную группу (РПта)-
В предположении о том, что главные оси градиента электрического поля на атомах Вг совпадают с направлением связей Р—Вг, были рассчитаны валентные углы, оказавшиеся равными в пределах 107,6... 107,7е. Эти значения примерно на 2° завышены по сравнению с данными рентгеноструктурного анализа.
Методом спинового эха в двойном резонансе были измерены константы квадрупольного взаимодействия 55Мп и 2D в M(CO)sD и изучено прямое диполь-дипольное взаимодействие ядерных спинов. Из этих данных было рассчитано межъядерное расстояние Мп—D (1,61 ±0,01) • 10—1 нм, прекрасно согласующееся с найденным методом нейтронографии (1,601±0,016) • 10-1 нм. Для Мп(СО)5Н позднее было определено, что расстояние Мп—Н равно (1,59±0,02)Х Х10-1 нм. Такие исследования пока очень редки, но являются примером того, что сходные данные могут иногда быть получены раз
102
ными и весьма неожиданными методами. Напомним, что константа квадрупольного взаимодействия может быть получена методом микроволновой спектроскопии, а в гл. V увидим, что она измеряется и в мессбауэровской спектроскопии.
Из сказанного о температурной зависимости спектров ЯКР и изменениях спектров в различных фазах, рассмотренных иа некоторых примерах, очевидно, что спектроскопия ЯКР является очень полезным методом обнаружения и изучения фазовых переходов в кристаллических веществах. Фазовые переходы первого рода (скачкообразное изменение первых производных изобарно-изотермических потенциалов в точке перехода) обнаруживают по скачкообразному изменению частот ЯКР, сопровождаемому иногда и изменением мультиплетно-сти. На рис. IV.6 показана, например, температурная зависимость частот ЯКР 35С1 для Ga+GaCLr, у которого
Рис. IV.6. Температурная зависимость частот ЯКР 35С1 для а- и р-модификаций Ga+GaCl4-
имеется две кристаллические модификации аир. Легко видеть при 320 К фазовый переход, при котором происходит скачкооб-
разное изменение и частот, и мультиплетности: у низкотемпературной модификации две частоты, а у высокотемпературной четыре. По температурным коэффициентам частот ЯКР можно делать выводы о характере молекулярных движений в фазах.
Что касается подвижности молекул в кристаллах, то она может быть разного типа. Кроме обычных решеточных колебаний часто наблюдается, например, заторможенное аксиальное вращение молекул или их отдельных частот (внутреннее вращение групп СН3, СС13 и т. п.) или даже изотропное вращение (пластические кристаллы). При вращении молекулы вокруг одной оси г', показанной на рис. IV.7, а для молекулы 1,2-дихлорэтана, который является одним из таких примеров, градиент электрического поля усредняется, и частота ЯКР v зависит от угла 0 между осью вращения z' и направлением z максимального градиента поля на ядре в отсутствие вращения (для СН2С1—СН2С1 0=19°23/). Когда вращательная частота Vfl>V,
v = v0 (3 cos2 в — 1)/2.
На рис. IV.7, б показана также температурная зависимость частоты ЯКР 35С1 для 1,2-дихлорэтана. Перспективно применение спек-
103
Рис. IV.7. Пространственное изображение молекулы СНгС!—СН2С1, вращающейся вокруг оси г' (а), и температурная зависимость частоты
ЯКР 3SC1 (б)
троскопии ЯКР также для изучения фазовых переходов, связанных с конформерией молекул, т. е. появлением степени свободы заторможенного внутреннего вращения и конформационных переходов, например транс-гош или аксиально-экваториальных. Полиморфизм, обусловленный конформерией молекул, хорошо известен, например, для некоторых фреонов, замещенных циклогексанов и др.
При фазовых переходах второго рода нет скачкообразного изменения спектральных характеристик, но изменение симметрии кристалла может приводить к плавному изменению мультиплетности. При переходах типа «порядок—беспорядок», кроме того, наблюдается резкое уширение линий ЯКР из-за неупорядоченности системы.
Применение импульсных спектрометров ЯКР позволяет обнаруживать сигналы большой ширины (~2% от значения частоты против ~0,02% при стационарных методах). Это сделало возможным исследование структур с неустранимыми элементами беспорядка. К таким системам относятся, в частности, кристаллические полимеры. Данные спектроскопии ЯКР позволяют судить о структуре, характере расположения и подвижности полимерных молекул в кристалле. Изучены спектры ряда хлорсодержащих полимеров. У поливинилхлорида, например, в спектре найдено восемь компонентов сигнала, которым должно соответствовать восемь типов кристаллографически неэквивалентных атомов хлора. Частотный диапазон сигнала от 36,56 до 38,18 МГц свидетельствует о наличии химической неэквивалентности (различном химическом окружении) атомов С1 в полимере. Изучались и неорганические полимеры с малой степенью беспорядка и достаточно узкими линиями, например, на основе (МГал2)п и (МГал3)п, где М — металл, а Гал — галоген. 104
2.3. Интерпретация градиента неоднородного электрического поля на ядре
Происхождение и величину градиентов электрических полей на ядрах атомов в молекулах приближенно объясняют с точки зрения характера химических связей и распределения электронной плотности в рамках теории МО ЛКАО. В молекулярных кристаллах основной вклад в градиент поля на ядре дают валентные электроны рассматриваемого атома, а в простейшем подходе Таунса и Дейли для таких атомов, как 14N и галогены, показывается, что градиент создают главным образом р-электроны валентной оболочки. Исходное положение этого подхода состоит в том, что градиент электрического поля в направлении z (например, совпадающем с направлением связи, см. рис. IV.2) в молекуле eqMOa можно выразить через градиент электрического поля в свободном атоме eq^ в виде линейного соотношения:
<^0.,=-(IV.15)
где Up — величина, зависящая от строения молекулы и числа несбалансированных р-электронов в данной структуре.
Если для простоты рассмотреть сначала атом с одним валентным электроном, характеризуемым квантовыми числами п, I, mi, то градиент поля в свободном атоме, создаваемый этим электроном в направлении углового момента для состояния mi — l, равен:
с, , /3 cos2 9 - 1 \ л 2/е / 1 \
= eqnlmi = е J | itnimi (2 ---] dv = - \ •
(IV.16)
Для водородоподобных функций средняя величина
/ 1 \__________2Z3______
\ г3 / п?а301(1 + 1)(2/ + 1) ’
где Z — эффективный заряд ядра; а0——5---боровский радиус
те2
атома водорода.
Показано, что в интересующем нас случае при участии одного валентного р-электрона в химической связи, когда компонента градиента создаваемого им поля в направлении z соответствует т=0 (при 1=1),
eqP = eqnVi=-^- е <г~з> . (IV.17)
О
Эти величины можно получить из данных по тонкой структуре оптических спектров атомов.Для некоторых изотопов онн приведены в табл. IV.1.
105
Таблица IV.1. Атомные градиенты р-электрониого поля и константы квадру-польиого взаимодействия для некоторых элементов
Изотоп Спин I eQ, барн Электрон <r—3>-10-21, CM-S <7p10-=\ CM*"3 e2<7pQ, МГц
“N 1 0,019 2p 22,5 — 18,0 —8—14
35C1 3/« —0,082 3p 48 —38,4 109,7
75As 0,29 4p 51 —40,8 —412
121Sb 5/° —0,33 5p 88 —70,4 954
209Bi ’/e —0.46 6p 166 — 132,8 1500
Из таблицы видно, что величина qp с ростом квантового числа п уменьшается, хотя и не в строгом согласии с теорией. При расчетах <г-3> и qP по спектральным данным вводят ряд поправок, в частности, на конфигурационное взаимодействие, поляризацию внутренних оболочек и эффект экранирования. Последние два эффекта учитывают с помощью так называемого фактора Штернхаймера для свободного атома — уоо, путем умножения e^q^Q на (1—у»). Этот учет особенно важен для ионных кристаллов, так как в них градиент электрического поля на ядре сильно зависит от соседних ионов, а для р-электронов атома и внешних зарядов факторы Штернхаймера различны. Известные значения уоо для атомов и ионов табулированы в специальной литературе.
Полный градиент электрического поля на ядре А в молекуле eq^ox=----- может быть представлен в виде суммы одноэлектрон-
dz^
ных вкладов от всех молекулярных орбиталей (МО):
= (IV.18)
i
где Ni — заселенность i-й МО, а
(IV.19)
где игг= [ (3 cos2 0—1)/г3а]; г к — расстояние «элементарного» заряда ejT)2 до ядра А, а 0 — угол, образуемый вектором г с осью z.
Представляя МО Т,- в виде ЛКАО
Vl ~ '%ci'V (IV-20)
j
и подставляя уравнение (IV.20) в (IV. 19), получим выражение од-ноэлектроиного вклада:
= (™.21)
J
где
qib = J <f* (J = k или j k). (IV.22)
106
Дальнейшее упрощение состоит в пренебрежении всеми интегралами вида (IV.22), кроме q'i (j — относится к атому А). Вклады электронов, находящихся на АО всех других атомов, кроме А, как и зарядов ядер в qiA, т. е. интегралы qhh (k не относится к атому А), пренебрежимо малы, как и интегралы перекрывания qlh с атомом А, а тем более qM (I тоже не относится к атому А), и могут не рассматриваться. Тогда вместо (IV.21) запишем
. (IV.23)
i
Орбитали атомного остова можно не учитывать, так как они имеют сферическую симметрию и не дают вклада в qtA. На практике поляризация внутренних электронных оболочек все-таки имеет место и, как указывалось, может учитываться фактором Штернхай-мера. Но поправка пренебрежимо мала и при рассмотрении причин возникновения градиента поля на ядре сумма в уравнении (IV.23) берется только по валентным орбиталям атома А. Более того, возможны следующие дальнейшие упрощения. Сферически симметричные s-орбитали не дают вклада в р(А, а вклады р-, d- и f-орбиталей относятся (если использовать водородоподобные функции) как 21 :3: 1. Тогда можно ограничиться рассмотрением только р-орби-талей, а сумму (IV.23) представить в виде
<7* = CUX + <fyy + c\qz, (IV.24)
где
q1 = ^pjU^pj&v (J = x,y,z) (IV.25)
показывает вклад электрона на р;-орбитали в z-компоненту градиента поля на ядре, а величины с2/ в сущности представляют статистические веса или заселенности соответствующих р;-орбиталей в в МО данной связи.
Вводят параметр qp = qzz( = qyy = qxx) как «градиент поля, создаваемого одним несбалансированным р-электроном», величина которого определяется экспериментально. Известно также, что qzx= = qzv{ = qxz=qyz = qxv = qyx') = ilzqp. Используя этот параметр и обозначив cj'2=nj (заселенность р;-орбитали), можно переписать (IV.24) в виде
\пг- — (пх+пд) qp.
(IV.26)
Согласно (IV.18) z-компонента полного градиента поля на ядре А ермол получается суммированием одноэлектронных вкладов вида (1V.26) по всем МО Тг.
Теперь можно записать в рассмотренном приближении выражения измеряемых экспериментально в спектроскопии ЯКР величин,
107
т. е. константы квадрупольного взаимодействия e2qQ и параметра асимметрии тр
е2?С?мол = 2 N‘ ["*' — у ("х/ + лгд)1 ^qpQat, (IV.27)
где сумма по i представляет множитель Up в (IV. 15) и
а1 = -у 2 Ni (Пх‘ ~ Пуй • (IV.28)
I
где
д = е2?9мол Чр ~ ^qpQar
(IV.29)
Значения e2qpQ для некоторых элементов приведены в табл. IV.1.
Несмотря на существенные упрощения, эта теория до настоящего времени составляет основу для интерпретации данных ЯКР и во. многих случаях дает удовлетворительные результаты. Она же служит для решения таких обратных задач, как суждения об электронной структуре и характере химических связей в молекулах, степени их ионности, степени двоесвязности, s-характере или степени гибридизации АО. Конечно, попытки разделить разные факторы, влияющие на градиент электрического поля на ядре в молекуле, создает известную неопределенность в интерпретации интегрального эффекта квадрупольного взаимодействия с помощью теории Таунса и Дейлн. Но строгой теории градиента неоднородного электрического поля на ядре в настоящее время нет, хотя попытки более строгого рассмотрения задачи делались.
Сами понятия об указанных выше свойствах химических связей тоже определяются весьма условно. Ионность I связывают с разностью электроотрицательностей атомов, причем разные авторы по-разному (как по-разному определяется и величина электроотрицательности х)'.1= (ха—%в) (Горди),7 = 1—exp^ Ха~Хв j (Полинг) и т. д. По Таунсу и Дейли, например, в случае связей хлора ионность входит в линейное соотношение е<7Мол~ (1—I)eqPr (см. ниже). Степень двоесвязности л также выражают различным образом, например через компоненты градиента поля: л= ~ ^хх—^!1У , или свя-
3 Цат зывают ее с параметром асимметрии и константами квадруполь-ного взаимодействия: п =—л—и т< п. Что касается s-xa-
2
рактера связи, то при sp-гибридизованных орбиталях его оценива-ют по формуле s= —— , где р— валентные углы (109°28' cos 3 — 1
или 120° и т. д.). Таунс и Дейли предложили для связей атомов галогена (Гал — М) правило: если электроотрицательность Хм на
108
0,25 отличается от электроотрицательности %Гал, связанного с М галогена, то s-характер гибридизации равен 15°/0, в противном случае он равен нулю. Предполагается, что d-гибридизация всегда меньше 5%.
Предложена следующая формула для градиента электрического поля на ядре атома галогена в молекуле:
= (!-*+<* ±/-л)е?агтал( (IV.30)
где знак плюс перед I берется, если хГал <хм , а знак минус, если ХГал >zM Формулу используют только для ковалентных связей.
2.4. Корреляции спектральных параметров ЯКР с другими физико-химическими характеристиками
В методе спектроскопии ЯКР, как и в других физических методах исследования, химики всегда стараются провести корреляцию получаемых данных с химической информацией и данными других методов. Данные ЯКР сопоставляются, в частности, с данными ЯМР, мессбауэровскими и ПК спектрами и т. д. Найдены полезные корреляции частот ЯКР некоторых изотопов с константами ионизации рКа карбоновых кислот, о-параметрами Гаммета и Тафта, индексами реакционной способности и др.
Так, например, найден ряд зависимостей вида v=vo + аХст,, где ст,— постоянная Гаммета заместителей для различных замещенных бензолов и гетероциклов. В частности, для хлорзамещен-ных бензола получено корреляционное соотношение:
v35cl =(34,826 + 1,024 2°/) ±°.3б МГц (IV.31)
(коэффициент корреляции г=0,96). Точность определения ст-посто-янных заместителей по уравнению (IV.31) для производных хлорбензола оценивается как ±0,50 при изменении их значений в пределах нескольких единиц. Соотношение (IV.31) может использоваться для орто-, мета- и пара-ззмешенных хлорбензола, но, как было показано, для жета-замещенных оно выполняется несколько строже, чем для пара-замещенных.
Получены аналогичные корреляционные соотношения, связывающие частоты ЯКР галогенов (Cl, Br, I) с ст*и-постоянными Тафта для разных заместителей R, например, в соединениях типа R1R2R3MCI при М = С, Si, Ge, Sn. Предложены также следующие корреляции величин рКа карбоновых кислот для замещенных уксусной кислоты ХСН2СООН с частотой ЯКР 35С1 в XCH2CI:
рКа = (17,47 -0.3930VC1) ±0,11;
для кислот RCOOH с частотой в RC1:
рКа = (6,82 - 0,0592vcl) ±0,04,
правда, эти соотношения дают заметные отклонения ДНЯ ТМШх X и R, как F, СН3О— и CH3S—.
109
Найдены корреляции частот ЯКР с потенциалами ионизации, например, положительная линейная зависимость va„ci от первого потенциала ионизации четырех хлорметанов. Линейные зависимости наблюдаются также между частотами ЯКР и полярографическими потенциалами полуволн (при восстановлении, например, иодбензо-лов, хлорнитроалканов).
Сопоставление с ИК. спектрами показывает хорошую корреляцию частот некоторых валентных колебаний, например, v(Si—Н) в соединениях типа RiRzHSiCl или v(C—Cl) в алкилхлоридах RC1 с частотами ЯКР 35С1. Наблюдается линейная зависимость между числом несбалансированных р-электронов, которое определяется отношением е2р^2мол/(e2qQat), в галогензамещенных (Cl, Br, I) метанах и константами спин-спинового взаимодействия Ч'зс-н в молекулах этих соединений.
3. АППАРАТУРА И МЕТОДИЧЕСКИЕ ОСОБЕННОСТИ
Существуют стационарные и импульсные методы наблюдения сигналов ЯКР в области от ~2 до 1000 МГц. Основные блоки про-
стого стационарного спектрометра регенеративного типа показаны на схеме, рис. IV.8. Исследуемый образец помещают в катушку колебательного контура LC с обратной связью. Частота колебаний в контуре v может плавно меняться изменением емкости С. При выполнении условия резонанса A£=/iv (Д£— разность энергий квадрупольных уровней) происходит поглощение образцом радиочастотной энергии, что меняет активную составляющую проводимости контура LC, т. е. его добротность. Изменение напряжения на контуре детектируется и усиливается. В стационарных методах для наблюдения сигналов ЯКР применяется частотная или магнитная (зеема-
новская) модуляция. Последняя существенно увеличивает отношение сигнала к шуму (приблизительно в 100 раз).
В сверхрегенеративных схемах задается прерывистый режим ко-
Рис. IV.8. Упрощенная блок-схема регенеративного спектрометра ЯКР:
1 — образец в индукционной катушке колебательного контура LC; 2 — генератор-детектор; 3 — блоки усилителя; 4 — фазочувствнтельный детектор; 5 — самописец;
6 — фазовращатель опорного напряжения; 7 — блоки модулятора
лебаний генератора-детектора, так что спектр частот сверхрегенеративного спектрометра содержит, помимо основной частоты vo ряд сателлитов vo± ±/iv (/г=1, 2, 3, ..., v — частота гашения). Поэтому вместо одной линии ЯКР на таком спектрометре регистрируется 2/г+1 сигналов, из которых только
110
центральный соответствует частоте ЯКР, что представляет определенные неудобства, особенно в случае богатых линиями спектров ЯКР. Но в то же время эти спектрометры дают возможность сканирования в широком частотном диапазоне, т. е. находить и детектировать широкие полосы. Простой регенеративный спектрометр полезен при работе в области низких частот (до 100 МГц). На выходе стационарных спектрометров может применяться многоканальное накопление, повышающее чувствительность этих приборов.
Недостатками стационарных методов является малая вероятность обнаружения сигнала: для многих образцов спектры ЯКР не обнаруживаются, а иногда и в принципе не могут быть обнаружены. Нередко возникают трудности с детектированием, большой длительностью эксперимента и низкой чувствительностью.
Аналогично тому как это делается в ЯМР фурье-спектроскопии, спектры ЯКР получают также, регистрируя кривую спада свободной индукции после наложения мощных радиочастотных импульсов прямоугольной формы. Реализуемый на спектрометрах метод импульсного квадрупольного спинового эха обеспечивает большой выигрыш в чувствительности и разрешении, которое в этом случае практически определяется естественной шириной линии и не зависит от аппаратурных факторов.
Существует также возможность использования метода двойного резонанса, в котором участвует ядро с легко наблюдаемым ЯМР, связанное с квадрупольный ядром или близко к нему расположенное. В результате двойного резонанса может происходить передача энергии от системы уровней ЯМР к системе ЯКР, в итоге частоты ЯКР находят, наблюдая изменения в спектре ЯМР, что дает огромный выигрыш в чувствительности.
Метод спектроскопии ЯКР, конечно, менее широко распространен в химических лабораториях, чем многие другие физические методы. Это отчасти связано со сложностью и малой доступностью аппаратуры и жесткими условиями проведения эксперимента (низкие температуры, термостатирование и т. д.), а также с ограниченностью объектов: определенный круг ядер, кристаллические образцы, причем лучше монокристаллы, чем порошки. Масса образцов, необходимая для исследования, сравнительно велика и составляет от десятых до нескольких граммов и даже десятков граммов. Но хотя и круг решаемых этим методом проблем тоже сравнительно не так широк, многие получаемые с его помощью данные бывают уникальны, и спектроскопия ЯКР в целом является очень ценным методом в химических исследованиях.
ГЛАВА V
МЕССБАУЭРОВСКАЯ СПЕКТРОСКОПИЯ
1. ОБЩАЯ ХАРАКТЕРИСТИКА И ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
Метод мессбауэровской спектроскопии, называемой иногда спектроскопией ядерного гамма-резонанса (ЯГР), основан на изучении поглощения у-излучения какого-то ядра-источника ядром того же изотопа, находящимся в исследуемом образце. Возможность такого поглощения, т. е. у-резонанса, зависит не только от разности энергий возбужденного и основного состояний ядер. Условия резонанса соблюдаются только тогда, когда устранен также эффект отдачи ядер при испускании и поглощении у-квантов, а также скомпенсирован каким-то образом эффект Допплера. Метод получил свое развитие именно с того момента, когда это было понято, а еще раньше экспериментально был найден простой и едва ли не единственно возможный путь ликвидации потерь на отдачу.
Применение метода в химии базируется на зависнмостн энергии ядерных переходов или разности энергий ядерных состояний, между которыми происходит переход, от химического окружения ядра, т. е. взаимодействий ядра с этим окружением. Связанные с этим различия в энергиях переходов чрезвычайно малы (на десять порядков меньше) по сравнению с энергиями самих у-квантов (переходов), имеющих порядок ~ 104... 105 эВ, что соответствует области частот 1012... 1013 МГц (107... 109 см-1).
Однако разрешающая способность метода достаточно велика, чтобы его можно было успешно использовать в структурных химических исследованиях. Этому способствует высокая монохроматичность излучения: естественная ширина линии rT==Avi/2 (ширина на полувысоте пика) обычно лежит в пределах 10~10... 10-1® от значения энергии у-лучей.
Эффект Мессбауэра получен для нескольких десятков элементов, но далеко не для всех из них эффект находит применение в химии. Данные о наиболее широко используемых изотопах приведены в табл. V.I.
Имеется ряд определенных условий, которые должны выполняться, чтобы наблюдался эффект Мессбауэра, и не у всех элементов они выполняются, хотя возбужденные состояния ядра элемента могут быть известны. Прежде чем перейти к этим условиям, рассмотрим само явление ЯГР.
Для простоты возьмем сначала систему из свободных атомов в газовой фазе. Ядра этих атомов могут быть в основном и возбужденном состояниях, т. е. на соответствующих уровнях энергии Eg и Ее. Разность этих энергий равна энергии перехода ядра АЕ=Ее — —Eg—Er, который может происходить с испусканием или поглощением у-кванта, причем в первом случае ядро перейдет в основное,
112
а во втором — в возбужденное состояние. Энергия у-кванта приблизительно равная энергии перехода, настолько велика, что в отличие от более длинноволнового излучения, например УФ, видимого или ИК, при обычных атомных и молекулярных массах существенное значение приобретает отдача, т. е. ее скоростью v и энергией ER пренебречь уже нельзя.
Без потери общности можно рассмотреть одномерную полукласси-ческую задачу, поскольку, как показано на схеме (рис. V.1), излучение у-кванта ядром источника (радиоактивного изотопа) и отдача
Ее~
Ет
Е9 "
=М
Ее
-hi'
Е9.
Рис. V.I. Схема взаимодействия ядер в возбужденном и основном состоянии с у-квантом (свободные
* атомы)
этого ядра происходят в противоположных направлениях, а направления движения у-кванта и отдачи ядра, способного его поглотить, совпадают. В момент испускания у-кванта энергия ядра радиоактивного изотопа сверх энергии покоя в основном состоянии составляет Ет+ 4iMVx2, где М — масса ядра, Vx — скорость его теплового движения. После испускания имеем систему из у-кван-та и ядра в основном состоянии с добавкой к его скорости движения скорости отдачи v, так что энергия этой системы равна E1+i/2M(Vx + v)2. По закону сохранения энергии
Ет + у MV2 = + у- М (Vx + v)2 = + -j- MV2 + MvVx + у MV2,
(V.1)
отсюда видно, что энергия перехода и энергия у-кванта разнятся на величину
Ет-Е^ = 1/2Mv2 + MvVx = Er + Ed. (V.2)
Первое слагаемое и есть э н ер г и я отдачи Er, а второе, связанное со скоростью поступательного движения Vx,— энергия эффекта Допплера ED, за счет которого происходит уширение полосы у-излучения. Вообще говоря, Ev зависит от направления движения ядра и того, как оно соотносится с направлением движения у-кванта. Кривая распределения испускаемых у-квантов по энергиям
Ef = Ет — ER — Ed
(V.3)
показана на рис. V.2, а слева.
113
Рис. V.2. Распределение испускаемых и поглощаемых у-квантов по энергиям:
а — для свободных атомов; б — для ядер в кристаллической решетке при низких температурах
Удобно далее перейти к обычным сопоставимым характеристикам излучения и ядер. Энергию у-кванта можно выразить, как Ev— —PfC, где ру — импульс кванта, с — скорость света. Импульс ядра р при отдаче равен, но противоположен по знаку импульсу у-кванта, т. е. р=— Рт=-------.Энергию отдачи ядра из уравнения (V.2)
с
можно выразить через импульс ядра следующим образом: Ец= = xl2Mv2= (Mv)2/2M=p2/2M, а следовательно,
ER = E*/(2Mc2)- (V.4)
Учитывая приближенное равенство для энергии теплового движения: 1/2MV2xcakT (й— постоянная Больцмана), можно записать Vx— (2kT/M)'\ а подставляя эту величину в выражение для энергии Допплера, получим: ED=MvVx= (2Mv2kT)'h = 2(ERkT)'lK, используя (V.4), окончательно имеем
Ed = Ел (2kT/M&)i/2. (V.5)
В у-резонансной спектроскопии ядра источника и образца одинаковы и отличаются только энергетическим состоянием. Чтобы ядро, совершающее тепловое движение и находящееся в основном состоянии, могло поглотить у-квант, испущенный источником, и перейти в возбужденное состояние, претерпев отдачу, энергия кванта должна быть равна
E^Er+ER+ED. (V.6)
Кривая распределения таких квантов, показанная на рис. V.2, а справа, симметрична по отношению к кривой распределения испущенных у-квантов с энергиями (V.3), так как энергии отдачи и эф-114
фекта Допплера одинаковы по абсолютной величине. Как видно, область перекрывания площадей, ограниченных двумя кривыми (заштрихованная часть на рис. V.2), очень мала. Это значит, что мала вероятность у-резонанса для свободно двигающихся в газовой или жидкой фазах атомов или молекул, т. е. мала вероятность того, что испущенный возбужденным ядром у-квант будет поглощен ядром, находящимся в основном состоянии. Главной причиной несовпадения энергии испущенного у-кванта и энергии, которая необходима, для того чтобы у-квант мог быть поглощен, является несовпадение по знаку больших энергий отдачи ER.
Открытие Мессбауэра заключалось именно в решении проблемы, связанной с энергией отдачи. Как следует из уравнения (V.4), Er можно уменьшить, сильно увеличив массу М. Если, например, ядро-излучатель и ядро-поглотитель фиксированы в жесткой кристаллической решетке, энергия отдачи может передаваться решетке только в определенных количествах, так как колебания решетки квантованы. Если ER меньше колебательных квантов, то эффективной массой М в выражении (V.4) становится вся масса кристалла, т. е. энергия отдачи пренебрежимо мала. Правда, на практике оказывается, что возбуждение колебаний решетки кристалла при поглощении у-квантов все-таки возможно, так что совсем пренебречь величиной Ея нельзя.
Допплеровское уширение уменьшается, как следует из уравнения (V.5), и в результате увеличения эффективности массы, и за счет понижения температуры и уменьшения кинетической энергии излучателя и поглотителя. Происходит сближение и сужение кривых распределения у-квантов по энергиям, как показано на рис. V.2, б, и перекрывание их увеличивается. Часть всего излучения при Е^Ет, которая поглощается и определяет интенсивность мессбауэровского спектра, зависит от температуры и подвижности частиц в твердом теле.
Теперь можно перечислить условия, необходимые для наблюдения эффекта Мессбауэра.
Во-первых, должен происходить процесс релаксации возбужденных ядер с испусканием у-излучения и в нем должна участвовать достаточная часть ядер, так как возможны и другие релаксационные процессы, например с эмиссией электронов.
Во-вторых, энергия у-квантов должна лежать в пределах 10< <EV< 150 кэВ, т. е. и энергия ядерного перехода должна быть соответственно велика, но энергия отдачи не должна превышать колебательных квантов решетки.
В-третьих, период полураспада мессбауэровского возбужденного ядра должен лежать в пределах 1 </>/,,< 100 нс, т. е. время жизни состояния (мессбауэровского уровня) должно быть достаточно большим, чтобы принцип неопределенности не мог сильно сказываться на измерении Ет, но и достаточно малым, чтобы получались достаточно интенсивные и широкие линии,
115
Рис. V.3. Схемы радиоактивного распада с образованием мессбауэровских атомов 57Fe (а) и 119Sn (б)
так как очень узкие линии трудно или даже невозможно наблюдать.
В-четвертых, у излучателя (мессбауэровского возбужденного ядра) должен быть долгоживущий предшественник—материнский радиоактивный изотоп, достаточно удобный в обращении. Распад этого изотопа должен проходить через стадию образования мессбауэровского уровня.
Наконец, основное состояние изотопа должно быть устойчиво, а сечение поглощения должно быть достаточно велико. Необходимо или достаточное природное содержание этого изотопа, или возможность легко проводить обогащение.
На рис. V.3, а в качестве примера представлена схема широко используемого в мессбауэровской спектроскопии распада радиоактивного материнского изотопа 57Со с образованием при захвате электронов возбужденных состояний изотопа 57F* и переходом ядер в основное состояние 57Fe. Изотоп 57Со доступен (получают в циклотроне) и удовлетворяет как материнский изотоп четвертому условию. Из верхнего возбужденного состояния 57Fe* меньшая часть ядер (9%) непосредственно переходит в основное состояние 57Fe с испусканием у-квантов высокой энергии, а большая часть (91%) — в более низкое возбужденное состояние (мессбауэровский уровень)» удовлетворяющее третьему условию, из этого состояния и осуществляется мессбауэровский переход. Изотоп 57Fe в основном состоянии удовлетворяет последнему условию, и хотя его природное содержание всего около 2%, этого достаточно. Именно такое ядро и является партнером мессбауэровского возбужденного ядра, т. е. поглощает испущенный им у-квант, переходя при акте ЯГР в возбужденное состояние.
116
На рис. V.3, б в качестве другого примера представлен распад радиоактивного олова с образованием промежуточного мессбауэровского уровня, удовлетворяющего необходимым требованиям. Аналогичные явления наблюдаются и для многих других элементов с порядковыми номерами от 25 до 96 (см., например, табл. V. 1), имеющих подходящие изотопы в соответствии с указанными условиями. Энергия ядерных переходов более легких элементов слишком высоки.
Рис. V.4. Вид кривой зависимости поглощения у-квантов от скорости движения источника относительно поглощающего вещества
Для химии эффект Мессбауэра, как уже отмечалось, важен тем, что энергия ядерного перехода Ет, а значит, и энергия испускаемого или поглощаемого у-кванта Е-; зависят не только от самого ядра (изотопа элемента), но и от других факторов. Это прежде всего электронное окружение ядра, а также внутренние и внешние электрические и магнитные поля. В качестве источника у-излучения и его поглотителя в мессбауэровской спектроскопии используются разные вещества. Таким образом, ядра одного и того же изотопа в источнике и поглощающем веществе находятся, вообще говоря, в разном окружении, т. е. Дущст)1# Дгщогл), а энергия испускаемого у-кванта Дущст) такова, что он не может быть поглощен ядром поглотителя, Ецае-г^Е^пот^ и явление ЯГР не происходит.
Настройка источника монохроматического у-излучения для получения мессбауэровских спектров может достигаться за счет эффекта Допплера. Дело в том, что £\,(ИСт) включает как составляющую энергию этого эффекта (см. выше зависимость Ev от ED и от скорости движения ядра), и ее можно в некотором интервале варьировать, двигая с какой-то скоростью v источник относительного поглощающего вещества. Это движение модулирует частоту у-кван-тов, и, когда энергия фотона Ey=hv' становится равной £Т(погл), он поглощается ядром поглотителя, т. е. происходит ЯГР. Чем больше скорость движения источника в направлении поглотителя ( + и), тем больше Е-;. Наблюдаемые в мессбауэровской спектроскопии разности энергии Aft= | ДТ(ист>—Дущогл) | соответствуют относительным скоростям движения порядка миллиметра в секунду, которые легко осуществляются и точно измеряются.
Таким образом, мессбауэровский спектр регистрируется, как показанная на рис. V.4 кривая зависимости интенсивности поглощения
у-излучения от скорости движения источника относительно поглощающего вещества, которая фактически эквивалентна зависимости от энергии или частоты у-квантов. Значение скорости движения источника, соответствующее максимуму поглощения у-квантов неподвижным поглотителем (минимум прохождения), обозначают v0.
117
Таблица V.1. Характеристики* некоторых ядер, используемых в мессбауэровской спектроскопии
Изотоп a, % <«/, нс E 7, кэВ гт. мм-с-1 Go, смМО-18 Знак Д8/8
5 Те Ч2 72 2,17 99,3 14,412 0,192 2,57 . .
119Sn V2 7, 8,58 18,3 23,875 0,626 1,40 +
121Sb 7s 72 57,25 3,5 37,15 2,1 0,21 +
125Te V, 7, 6,99 1,535 35,48 5,02 0,28 +
129 J 7S 7? — 16,8 27,72 0,59 0,38 +
129Xe Ч2 72 26,44 1,01 39,58 6,85 0,24 +
197 Au Ч2 ’/2 100 1,892 77,34 1,87 0,041 +
e — ядерные спины основного (g) и возбужденного (e) состояний; a — природ
иое содержание; Ъд—период полураспада; Е-^ — энергия у-излучения; Г 7 — естественная ширина линии; Со — сечение поглощения; AR и R — объясняются в тексте.
Необычные для других спектроскопических методов (УФ, ИК, ЯМР и др.) единицы измерения можно всегда сопоставить с привычной энергетической или частотной шкалой. Так, например, при £\,(ист) = = 14,41 кэВ (57Fe) изменение скорости движения источника уИст на 1 мм-с-1 изменяет Еу на 4,8-10~8 эВ (или частоту vT на ~20 МГц).
Чаще всего в мессбауэровской спектроскопии в качестве источника у-излучения (излучатель) используется какое-либо стандартное вещество, а поглощающим у-кванты веществом является исследуемый образец. Иногда, наоборот, изучаемое вещество содержит возбужденные ядра, а стандартом является поглотитель у-квантов, а не излучатель (например, в некоторых системах галоген — ксенон), но если не делается специальных оговорок, то имеется в виду первый вариант.
2. ПАРАМЕТРЫ МЕССБАУЭРОВСКИХ СПЕКТРОВ
Наибольшее влияние на изменение энергии £\,(Обр) по сравнению с £т(ист) оказывают три главных типа взаимодействия ядра с его химическим окружением в образце:
1) кулоновское взаимодействие ядра с электронами;
2) квадрупольное взаимодействие с градиентом электрического поля на ядре (для ядер со спином />*/2);
3) магнитные взаимодействия.
2.1. Изомерный (химический) сдвиг
Изменение энергии ядерного перехода, т. е. энергии поглощаемого образцом у-кванта по сравнению с испускаемым:
® ^'((обр) '^7 (ист),
(V.7)
118
Рис. V.5. Схема энергетических уровней и переходов для ядра s7Fe при отсутствии н наличии градиента электрического поля на ядре (а), инд спектра Fe(CO)6 с квадрупольным расщеплением (б)
связанное с различием электронного окружения ядер в образце и источнике, называется изомерным или химическим сдвигом и измеряется как значение скорости (мм-с-1) движения источника, при которой наблюдается максимум поглощения у-квантов (если сигнал синглетный). При экспериментальном определении б по центру сигнала используется термин центровой сдвиг, а изомерный (химический) сдвиг отличается от него поправкой на небольшой допплеровский вклад от теплового движения мессбауэровских ядер (рис. V.5).
Электроны, взаимодействуя с ядром, влияют на относительное расположение основного и возбужденного состояний ядра. Энергия этого электростатического взаимодействия зависит как от электронной плотности на ядре, характеризуемой квадратом модуля электронной волновой функции l'F(O) |2, так и плотности ядерного заряда р(г). Используя выражение этой энергии при перекрывании объема ядра электронным облаком:
Е = -^-ле I Чг (0) [ 2 Jp(r)r2di>
для возбужденного и основного состояний ядра (и умножая на eZ, где е — единичный заряд; Z — порядковый номер элемента), можно получить следующее выражение для измеренного сдвига:
2
» = £Г(обр)-£Г(ист)=-7Яе2'г[<Г2>г“ <Г2>*][ 1*(°)/обр-1*(0) |®Ст] .
(V.8)
где <72>== ( J p(r)r2cto) / jp(r)cb — квадрат эффективного радиуса ядерного заряда.
119
В более приближенном рассмотрении это выражение упрощают, считая, что играют роль прежде всего s-электроны (только s-электронная функция на ядре 4rs(0)=/=0, а другие орбитали лишь несколько влияют на s-электронную плотность на ядре за счет эффектов экранирования). Принимая, что ядра сферические, для эффективного радиуса ядра в основном состоянии вводят обозначение R, а разность радиусов ядер в возбужденном и основном состояниях обозначают Д/?. Тогда получают:
8 = (4Л/5) («2Z/?2) (д/?//?) [ j (0) |2бр - | (0) 12ист] (V.9)
(формулу (V.9) можно перевести в СИ, используя электрическую постоянную ео).
Изомерный сдвиг 6 может быть как положительным, так и отрицательным в зависимости от знака AR/R. Если эффективные размеры (радиус) возбужденного ядра больше, чем ядра в основном состоянии, т. е. плотность ядерного заряда pe<pg, a AR/R>Q, то при увеличении s-электронной плотности на ядре в образце по сравнению с источником, согласно уравнению (V.9), будет наблюдаться положительный сдвиг ( + 6). Обратно, при ре>рё и AR/R<0 изомерный сдвиг отрицателен (—б) при увеличении s-электронной плотности на ядре в образце. Так, для изотопа 57Fe \R/R<Q и изомерный сдвиг при увеличении s-электронной плотности отрицателен, а для других, приведенных в табл. V.1 элементов, AR/R>0 и при росте s-электронной плотности на ядре изомерный сдвиг будет положительным.
При экспериментальном измерении изомерных химических сдвигов и их применениях всегда важно, какой используется стандарт. Так, например, для исследования мессбауэровских спектров на 57Fe официальным стандартом является соединение этого изотопа Na2[Fe(CN)5NO], а теперь обычно используют металлическое железо. Для 119Sn общепринятым стандартом является обычно SnOj и т. д.
Некоторые применения изомерных сдвигов в химических исследованиях будут рассмотрены ниже.
2.2. Квадрупольное расщепление
В результате электрического квадрупольного взаимодействий (см. гл. IV) в мессбауэровском спектре возникает тонкая мультик плетная структура сигналов, которая зависит от спинов ядер в об* новном и возбужденном состояниях и от градиента электрического поля на ядрах. Ядро, совершающее мессбауэровский переход, мо-жет в одном из состояний или часто в обоих состояниях обладать спином />’/2, а значит, и квадрупольный моментом eQ. Если при этом имеется также градиент электрического поля, создаваемого окружением на квадрупольном ядре (eq=/=0), то ядерные уровни
120
энергии расщепляются на несколько подуровней. Характер и величина расщепления зависят от константы квадрупольного взаимодействия e2qQ, а при отсутствии осевой симметрии электрического
поля также от параметра асимметрии гр
На рис. V.5, а показана схема энергетических уровней для ядра 57Fe, имеющего в основном состоянии спин 7g = 1/2, eQ = 0, а в возбужденном состоянии спин /е=3/2, eQ=#0. При сферической симметрии электрического поля е<?=0 и никакого расщепления верхнего уровня не будет, например, в правильных тетраэдрических или октаэдрических структурах. При наличии градиента поля верхний уровень расщепляется на два подуровня в зависимости от квантового числа | nii|, например, при осевой симметрии поля в триго-нальнобипирамидальной структуре соединения Fe(CO)s. Правило
отбора для мессбауэровских переходов Дт; = 0, ±1, и синглетная при е<7=0 линия расщепляется на дублет (рис. V.5, б), при этом центровой сдвиг б определяют по центру дублета (у0). Расстояние между компонентами А (мм-с-1) называют квадрупольным расщеплением. При осевой симметрии поля A = e2<yQ/2 (см. гл. IV). В общем случае из картины квадрупольного расщепления получают информацию, аналогичную извлекаемой из спектров ЯКР, т. е. определяют e2qQ (МГц) и ц, хотя, конечно, по одному только Д все данные о тензоре градиента электрического поля получить нельзя. Необходимо бывает проводить исследования во внешнем магнитном поле и с монокристаллическими образцами. Само значение Д часто, однако, также бывает полезным при решении некоторых химических проблем (см. ниже).
Когда ядро в обоих состояниях имеет квадрупольный момент eQ#=0, картина расщепления в мессбауэровском спектре становится сложнее, но и более информативна, как
Рис. V.6. Схема энергетических уровней и переходов для изотопа 1291 прн наличии градиента электрического поля на ядре (а) и качественный вид спектра (б)
121
a)
Рис. V.7. Схема энергетических уровней и переходов для ядра 119Sn в магнитном поле (о) и вид сверхтонкой магнитной структуры мессбауэровского спектра (б)
это иллюстрирует, например, рис. V.6, на котором показана схема переходов и качественный вид мессбауэровского спектра для изотопа 1291.
2.3. Сверхтонкая структура магнитных взаимодействий
Если на ядро со спином /#=0 действует магнитное поле, то происходит зеемановское расщепление ядерных уровней энергии на 2Z-4-1 компонентов, что подробно рассмотрено в гл. I. В мессбауэровском переходе участвуют два состояния ядра, характеризуемые спинами Ig и 1е, от которых зависит расщепление уровней энергии Eg и Ее на соответствующее число компонентов. Ядер-ные переходы между зеемановскими подуровнями основного и возбужденного состояний подчиняются правилам отбора Д/П/=0, ±1, и в результате этих переходов наблюдается сверхтонкая структура (СТС) мессбауэровского спектра. Для четкого ее наблюдения требуются достаточно сильные поля с индукцией В> >1 Т.
Магнитное поле на ядре может создаваться как внешними источниками, так и магнитными моментами атомов в самом образце. Последнее относится прежде всего к ферромагнитным и антиферромагнитным веществам. Для парамагнитных веществ из-за быстрой релаксации электронных спинов СТС мессбауэровских спектров наблюдать труднее, обычно это оказывается возможным только при очень низких температурах.
122
Соотношение интенсивностей линий зеемановского спектра зависит от угла 0 между направлением пучка у-излучения и направлением магнитного поля. На рис. V.7, а представлена схема зеемановских подуровней двух состояний ядра 119Sn и переходов между ними, а на рис. V.7, б — вид спектра в отсутствие градиента электрического поля на ядре (eq=0) и квадрупольного расщепления Д, когда направления магнитного поля и пучка у-излучения совпадают. Компоненты наблюдаемого секстета расположены симметрично относительно центрового сдвига 6, т. е. скорости v0 соответствующей синглетной линии в отсутствие магнитного поля. Интенсивность компонентов секстета в случае магнитного поликристаллического образца соотносятся как 3:2:1:1:2:3.
Интерпретация мессбауэровских спектров при одновременном квадрупольном и магнитном сверхтонких взаимодействиях весьма сложна. Как и в спектроскопии ЯКР (см. гл. IV), для облегчения интерпретации иногда используют внешнее магнитное поле, с помощью которого можно бывает определить как направление градиента электрического поля на ядре eq, так и параметр асимметрии гр
3. ПРИМЕНЕНИЕ В ХИМИИ
Мессбауэровская спектроскопия находит применение в различных областях науки и техники, в частности, кроме химии она широко используется в физике твердого тела, геологии и биологии. К обратным задачам данного метода, которые решаются химиками, относятся определение степени окисления атомов, участвующих в мессбауэровских переходах, их валентных состояний, структурные определения, заключения о распределении электронной плотности, характере химических связей и т. п.
3.1. Эмпирические корреляции
Из сказанного об изомерном сдвиге с очевидностью следует, что возможна его корреляция со степенью окисления атома элемента в молекуле исследуемого вещества. Рассмотрим это иа примере изотопа 119Sn. Электронная конфигурация нейтрального атома (валентной оболочки) 5s25p2. Ион Sn2+ формально имеет конфигурацию 5s2, и изомерный сдвиг для него (Д/?//?>0) будет положительным ( + и) относительно Sn (и0)> так как s-электронная плотность на ядре увеличилась из-за отсутствия эффекта экранирования 5р-электронов. Однако у иона Sn4+ удаляются и бз-электроны, что приводит к уменьшению s-электронной плотности на ядре и отрицательному (—у) изомерному сдвигу. Некоторые данные о изомерных сдвигах 119Sn приведены в табл. V.2.
Считается, что в оловоорганических соединениях изомерный сдвиг относительно SnOj меньше 2 мм-с-1 указывает на четырех-
123
Fe (мет)
Fe______
0), $-т W.S-f (!),s*2 m,s^i (s),s-0 (Ш), s-i m 5=f m, 54 (D7),S’7 W, *=' CW, $ = ' диамагн коммент.
Рис. V.8. Корреляционная диаграмма интервалов изомерных сдвигов 57Fe для соединений железа и различных валентных состояниях (в скобках) относительно металлического железа:
S — суммарный электронный спин. Для наиболее типичных состояний железа дана штриховка
меньшей степени, чем заполнение
валентное состояние олова, а б>2,5 мм-с-1 также четко свидетельствует о двухвалентном состоянии. Эмпирически оценены так называемые парциальные изомерные (центровые) сдвиги 6/ для различных заместителей в соединениях четырехвалентного олова, так что полный сдвиг 6 для SnWXYZ можно оценивать как сумму парциальных сдвигов 6w, 6х, 6y, 6z-
На рис. V.8 приведена корреляционная схема, которая дает представление об интервалах изомерных сдвигов 57Fe для соединений железа. Степень окисления железа может меняться от 0 до 6, и охарактеризовать ее по изомерному сдвигу 57Fe не так просто. Изменение заселенности электронами d-орбиталей влияет на s-электронную плотность в р-орбиталей. Но все-таки и по
известным сдвигам для железа, хотя интервалы их, как видно из рис. V.8, перекрываются, делаются важные заключения.
Таблица V.2. Изомерные сдвиги 119Sn в некоторых соединениях олова относительно SnO2 как стандарта
Соедниение d, MM'C-1 Соединение 6, MM’C’1 Соединение 6, MM'C—1
SnFt —0,47 Sn(CH3)4 1.21 SnFa 3,20
SnCl4 0,85 Sn(C6H5)4 1,22 SnCh 4,07
SnBr4 1.15 SnO 2,71 SnBra 3,93
Snl4 1,55 SnS 3,16 Snla 3,85
SnH4 1,27 SnSO4 3,90
Железо входит составной частью во многие биосистемы, в частности гемопротеины и системы небелковой природы (например, содержащиеся в микроорганизмах). В химии жизненных процессов существенную роль играют окислительно-восстановительные реакции порфириновых комплексов железа, которое может в них находиться в состояниях Fe(II) и Fe(III). В Этих реакциях участвуют как электроны лигандов (их л-орбиталей), так и желе-124
за, а его степень окисления можно определять, применяя мессбауэровскую спектроскопию.
Для некоторых элементов эмпирические корреляции сдвигов представляют проблему и весьма условны. Так, например, у 197Аи интервалы сдвигов настолько перекрываются, что степени окисления элемента определить не представляется возможным.
В более благоприятных случаях экспериментальные данные по изомерным сдвигам могут служить основой для квантово-химических расчетов распределения электронной плотности, заполнения орбиталей и т. п.
Если в мессбауэровском спектре наблюдается квадрупольное расщепление, что говорит о наличии градиента электрического поля на квадрупольном ядре, то это исключает высокую (тетраэдрическую, октаэдрическую) симметрию окружения ядра. В частности, по квадрупольному расщеплению было установлено, что соединение (SnF.4)n в твердом состоянии не имеет тетраэдрической симметрии, а полимерно:
F F F
Интересным является вопрос о так называемой стереохимической активности неподеленных электронных пар, который помогает выяснять мессбауэровская спектроскопия. Если в случае легких элементов свободная пара всегда играет роль в определении стереохимической конфигурации, то у тяжелых элементов это не очевидно. В мессбауэровском спектре ионов ТеХб2- (X — С1, Вг, I) не наблюдается, например, квадрупольного расщепления, т. е. они имеют строение правильного октаэдра. Такой же вывод следует из данных рентгеноструктурного анализа и колебательной спектроскопии. Видимо, электронная пара занимает 5х-орбиталь и поэтому не является стереохнмически активной. С другой стороны, у иона IF6~ и изоэлектронной молекулы ХеЕб неподеленная пара стереохимически активна, так что они имеют структуру искаженного октаэдра, и наблюдается квадрупольное расщепление.
Иногда асимметрия октаэдрических и других комплексов, например, при замещении не приводит к достаточно сильному градиенту электрического поля на ядре центрального атома, чтобы могло наблюдаться заметное квадрупольное расщепление. Поэтому интерпретация мессбауэровских спектров должна проводиться с осторожностью. Как уже отмечалось, помочь в интерпретации может применение внешнего магнитного поля, например, при изу
125
чении комплексов железа со смешанной валентностью (Fe(II) и Fe(III)].
Рассмотрим пример определения структуры соединения 12Вг2С14 по мессбауэровскому спектру 1291 при известном спектре (табл. V.3) и строении 12С16 как плоской симметричной молекулы с двумя мостиковыми и четырьмя концевыми атомами С1:
ci ci С1
ci ci С1
По аналогии можно структур:
предположить следующие пять плоских
Br CI
Cl С1 Вг
Br CI Вг
Cl CI
В отличие от одного мультиплета в спектре 12С1б (см. табл. V.3) наблюдаются два мультиплета одинаковой интенсивности, т. е. атомы I в молекуле неэквивалентны. Это сразу же исключает структуры I, II и III, в которых атомы I находятся в одинаковом окружении (в соответствии с симметрией структур), т. е. эквивалентны. Параметры одного из наблюдаемых мультиплетов (2) близки к параметрам мультиплета для 12С1б, т. е. один из атомов I в 12Вг2С14 имеет аналогичное с атомами I в 12С1б окружение: два концевых и два мостиковых атома С1. Отсюда следует однозначный вывод, что молекула 12Вг2С14 имеет структуру IV.
Таблица V.3. Параметры мессбауэровских спектров галогенидов 1291
Молекула д, мм-с—1 e^qQ, МГц
12С1в laBrsCli 3,50±0,10 (1) 2,82±0,02 (2) 3,48±0,02 +3060±Ю 4-2916± 10 -4-3040=4=10
126
3.2. Динамические эффекты
Как всякий физический метод, мессбауэровская спектроскопия имеет свое характеристическое время. Если изучаемое соединение само меняется во времени, т. е. происходят, например, обменные процессы или переходы одной формы соединения в другую, то при этом могут, естественно, меняться как изомерный сдвиг, так и градиент электрического поля на ядре (квадрупольное расщепление) и внутренние магнитные поля. Поэтому важно, как соотносятся времена жизни разных форм образца (частота нх перехода) н характеристическое время метода. Разность частот мессбауэровских переходов источника и образца имеет порядок 108 с-1 (порядок величины частотной характеристики изомерного сдвига см. выше). Для того чтобы можно было наблюдать отдельные сиг
налы и измерять изомерные сдвиги для двух переходящих друг в друга форм образца, частота их перехода не должна превышать 107 с-1. Таким образом, порядок характеристического времени метода мессбауэровской спектроскопии можно оценить как 10-7 с.
В комплексе железа со смешанной валентностью, т. е. атомами Fe(II) и Fe(III), в спектре будут наблюдаться два сигнала, только если любой переход электрона между этими атомами будет происходить достаточно медленно. Например, в комплексном соединении [Ее11Ее2шО(СНзСОО)б(Н2О)з] при температуре 290 К в мессбауэровском спектре наблюдается один усредненный синглетный сигнал (рис. V.9), указывающий на протекание быстрого обменного процесса (высокочастотный переход электрона). При по-
нижении температуры этот сигнал постепенно расщепляется, причем наиболее четко структура сигналов, указывающих на наличие двух неэквивалентных атомов Fe, проявляется лишь при 17 К. Сделан вывод о переходе электрона в пределах фрагмента Fe3O, а энергия активации оценена в 470 см-1.
Методом низкотемпературной мессбауэровской спектроскопии удалось зафиксировать и изучить неустойчивый при комнатной температуре тетрахлорид ксенона. При температуре 4,2 К время жизни ХеС14 по крайней мере сравнимо с характеристическим временем метода. В ионах 1291С14 иод дает 0-излучение и осуществляется переход в 129ХеС14. За время мессбауэровского перехода сохраняется плоская квадратная конфигурация и в спектре наблюдается квадрупольное расщепление. Изомерный сдвиг близок по величине к сдвигу хорошо изученного XeF4, устойчивого при обычных температурах, а квадрупольное рас-
-4-2 0 ♦ 2 *4 и^мм с'’
Рис. V.9. Температурная зависимость мессбауэровского спектра 57Fe в соединении [FeHFe21!1O-
• (СН3СОО)6(Н2О)3]
127
Рис. V.10. Блок-схема мессбауэровского спектрометра:
/ — двигающийся источник; 2 — коллиматоры н экраны (РЬ); <3неподвижный образец; 4 — детектор н счетчик импульсов; 5 — неподвижный цилиндрический рассеиватель (образец); 6 — двигающийся конический рассеиватель (образец); 7 — неподвижный источник
щепление несколько меньше.
Зависимость квадрупольного расщепления от температуры свя-. зана с различной подвижностью частиц в решетке, т. е. полем сил и соотношением подвижности с характеристическим временем метода. Изменения сверхтонкой магнитной структуры обусловлены релаксационными процессами, т. е. изменением заселенности зеемановских магнитных подуровней в зависимости от температуры.
4. ТЕХНИКА И ОСОБЕННОСТИ ЭКСПЕРИМЕНТА
’ Принципиальная блок-схема мессбауэровского спектрометра представлена на рис. V.10. О некоторых требованиях к источнику— стандартному веществу — уже говорилось выше. Следует добавить, что это должно быть доступное, хорошо воспроизводимое, чистое вещество, которое дает узкую линию, по возможности без структуры. Некоторые из стандартных веществ были указаны в § 2.1. Следует отметить, что для 129Хе часто используется как источник 1291 в тетраэдрическом анионе IO4_. Хотя это искусственный изотоп иода, но он является долгоживущим и накоплен в достаточно больших количествах. Источник (или поглотитель) дви-128
гается мессбауэровским вибратором с точно контролируемой и измеряемой с помощью специальных механизмов скоростью в разных направлениях (к образцу и от него). Поглощающее вещество, как показано на рис. V. 10, в одном из вариантов (рис. V. 10, а) помещается на пути у-излучения, которое может просто проходить через толщу образца и поглощаться им. В другом варианте (рис.
V.10, б) у-излучение подает на образец под каким-то углом, так что ядра в образце, поглотив у-кванты и возбудившись, затем их рассеивают.
Пропущенные или рассеянные после поглощения (в зависимо- ’ сти от варианта)у-кванты затем коллимируются и попадают на детектор, за которым следует счетчик импульсов. Результирующие кривые зависимости числа импульсов от скорости движения источника также показаны для обоих вариантов на рис. V.10. Скорость движения, соответствующую максимуму поглощения (минимуму пропускания) или рассеяния, т. е. изомерному (центровому) сдвигу, обозначают v0. Счет у-квантов и точная корреляция его со скоростью движения источника представляют некоторые трудности.
Положение сигнала (линии) измеряется в лучшем случае с точностью +0,01 мм-с-1. Различия изомерных сдвигов А6 и квадрупольные расщепления А могут не очень сильно отличаться по величине от обычной ширины сигналов. Поэтому возможны перекрывания сигналов, и в сложном контуре разбираться бывает нелегко. Иногда в этом помогают модельные расчеты спектров и сравнение с экспериментальными кривыми, но всегда остается неопределенность, связанная с выбором модели и предположениями о ширине и форме линий.
Необходимость работы в широком интервале температур и при очень низких температурах (до 1 К и ниже), что бывает связано также с необходимостью работы в сильных магнитных полях, получаемых на магнитах в условиях сверхпроводимости, обусловливает большую сложность и дороговизну не только основного, но и необходимого для мессбауэровской, спектроскопии дополнительного оборудования. Недавнее открытие «высокотемпературной» сверхпроводимости, достигаемой на некоторых керамиках уже при температурах жидкого азота (а не гелиевых, как раньше), приведет, возможно, к существенному упрощению и удешевлению аппаратуры.
В мессбауэровской спектроскопии применяется также метод матричной изоляции при низких температурах; исследовались, в частности, атомы железа и молекулы Fe2 в различных матрицах (в Хе, N2, СН4 и др.), а также и другие объекты.
Что касается аналитических возможностей метода и такой характеристики, как чувствительность, то, когда образец является поглотителем у-квантов, они сравнительно с многими другими методами невысоки/ В этом случае для исследования необходимы
5-1377
129
большие массы (г) вещества, а содержание мессбауэровского элемента должно быть не менее 0,5... 1%; если же образец является излучателем у-квантов, то достаточно содержание порядка тысячных процента. К особенностям метода относятся также возможность работы только с твердыми образцами' и ограниченность круга объектов.
В последние годы получили развитие исследования мессбауэровских спектров при высоких давлениях (до мегабар). Интервал достигаемых давлений определяется наличием сверхпрочных материалов, например, таких, как алмаз, и соответствующей конструкцией камеры с образцом (аналогично кюветам высокого давления в ИК спектроскопии и рентгеноструктурном анализе). Хотя высокие давления сравнительно слабо влияют на электронные оболочки атомов, измеряемые в зависимости от давления параметры мессбауэровских спектров несут новую информацию о взаимодействии ядра с электронным окружением. По сравнению с другими методами мессбауэровская спектроскопия в исследованиях при высоких давлениях отличается даже большей чувствительностью к изменениям энергии взаимодействия.
Эксперимент под высоким давлением существенно усложняется и удлиняется время его проведения. Во-первых, более интенсивен фон из-за рассеяния от деталей камеры и большой проникающей способности жесткого у-излучения, что приводит к ухудшению отношения сигнала к шуму. Во-вторых, малость образца в камере высокого давления требует для нормальной скорости счета использования источников с высокой удельной активностью. Длительность эксперимента предъявляет повышенные требования к стабильности всей экспериментальной установки.
Существует метод, называемый мессбауэровской спектроскопией электронов конверсии (МСЭК.) Этот метод основывается иа регистрации возникающих при конверсии у-квантов электронов или рентгеновских лучей. Эмиссия электронов конверсии с различных оболочек атомов обусловлена рассеянием энергии при возбуждении атомных ядер, чем эти электроны отличаются от фотоэлектронов, испускаемых при облучении атомов или молекул УФ- или рентгеновским излучением (см. разд. 3), когда атомные ядра не возбуждаются.
Доля конвертированных у-переходов может быть достаточно большой, чтобы иметь аналитические применения. Измеряемой величиной, характерной для изотопа элемента, является коэффициент конверсии
а=а'/(1 — а'),
где а'—отношение числа конвертированных переходов к общему числу переходов. Измерительная техника МСЭК обычно представляет устройства с обратным рассеянием.
Метод МСЭК эффективен и используется преимущественно для
130
исследования поверхностей и слоев толщиной 5—300 нм, что, как и в случае методов рентгено- и фотоэлектронной спектроскопии, рассмотренных ниже, связано с глубиной выхода электронов, обладающих определенной кинетической энергией. Метод может применяться также для изучения in situ взаимодействий между поверхностью твердых тел и жидкой фазы. Таким образом, МСЭК имеет значение прежде всего, говоря о химии, для изучения явлений адсорбции, коррозии, образования пассивированных слоев, образовав ния фаз, а также в катализе. Все эти применения МСЭК аналогичны применениям методов, имеющих в настоящее время более широкое распространение и представленных в следующем разделе.
* * *
Рассмотренные в разделе методы исследования дают ценнейшую информацию о строении, электронных эффектах и передаче взаимного влияния групп в органических, элементорганических, неорганических и координационных соединениях. Как спектроскопия ЯКР, так и мессбауэровская спектроскопия оказались весьма полезными при изучении некоторых биохимических объектов и проблем, показана перспективность их применения в макромолекулярной химии. Получено много интересных эмпирических корреляций параметров, определяемых из спектров ЯКР и ЯГР, с другими физико-химическими характеристиками веществ. Оба метода позволяют исследовать структуру и динамику твердых фаз, фазовые переходы, подвижность молекул в кристаллах и многие другие проблемы.
Как уже неоднократно подчеркивалось, ценность получаемых обоими методами данных о распределении электронной плотности, градиентах электрического поля на ядрах трудно переоценить. Они необходимы для более глубокого понимания характера химических связей и являются тем материалом, на котором проверяются используемые химиками теоретические построения, а также базируются как приближенные, так и более строгие квантово-химические расчеты.
Конечно, методы спектроскопии ЯКР и мессбауэровской спектроскопии не столь широко распространены и применяются в химических исследованиях, как ЯМР, ЙК или масс-спектроскопия и некоторые другие. Это связано как с малой доступностью и сложностью приборного оборудования, так и с ограниченностью круга объектов и решаемых проблем. В обоих методах эффекты, на которых они основаны, наблюдаются на ядрах далеко не любых элементов и изотопов, а, кроме того, исследоваться могут только твердые образцы, количества которых, необходимые для работы, довольно велики.
С развитием экспериментальной техники некоторые ограничения будут, возможно, устраняться, но и в настоящее время хими-5* 131
ки, зная некоторые уникальные возможности рассмотренных методов, не могут их не применять в своей исследовательской работе.
Контрольные вопросы
Глава IV
1. Почему для описания квадрупольного момента ядра, величины вообще тензорной, достаточно одного параметра Q?
2. Сколько квадрупольных уровней энергии имеют ядра с полуцелочнслен-ным спнном в поле с осевой симметрией? Как соотносятся частоты переходов между ними (при »] = 0)?
3. Чем определяется мультнплетность сигналов ЯКР квадрупольных ядер данного изотопа?
4. В спектре ЯКР на 14N в твердом пиридине C5H5N при 77 К наблюдаются линии при 3,90 н 2,95 МГц. Найдйте значения e2qQ и т).
5. Природное содержание 59Со, имеющего спин /=7/2, равно 100%. Сколько переходов будет наблюдаться в спектрах ЯКР K’Co(CN)e н КзСо(СЬ1)3Вг? Выразите энергию переходов через e2qQ.
6. Сколько всего частот будет наблюдаться в спектре ЯКР К+(А12Вг?)“ (учесть, что существуют изотопы 79Вг н 81Вг), если структура вещества такая же, как у Na+(Ga2Cl7)~, которая показана и обсуждена на с. 102?
7. Предполагается, что РС13О реагирует с ТеС14, образуя аддукт РС13О-•ТеС14, а с А1С13 — ионное соединение РС12О+А1С14_. Как это можно доказать методом спектроскопии ЯКР?
8. Электроны каких атомных орбиталей вносят больший вклад в градиент неоднородного электрического поля на ядре в молекулярном кристалле согласно теории Таунса и Дейли?
9. Какие факторы оказывают наибольшее влияние на сдвиг частоты ЯКР атома данного элемента в молекулярном кристалле?
Глава V
10. Какие условия должны выполняться, чтобы для ядер какого-то элемента наблюдался эффект Мессбауэра?
11. Как связана скорость движения источника у-нзлучення (или образца, поглощающего у-квант) с энергией испускаемых (поглощаемых) квантов?
12. Какими главными факторами определяется изомерный (химический) сдвиг в мессбауэровской спектроскопии?
13. В днфеннлолове изомерный сдвиг относительно SnO2 найден равным +6=1,56 мм-с-1, а цнклопентадненнлолове +6 = 3,74 мм-с-1. В каком валентном состоянии находится олово в этих соединениях? Какое из этих соединений является, таким образом, полимерным, т. е. может быть представлено формулой: [Sn(R)+, а какое— мономерным: Sn(R)2 (R—С6Н3 нлн С3Н5)?
14. Изомерный сдвиг 57Fe в комплексном анноне: [(C6H5S)2FeS2MoS2]2- относительно металлического железа равен 0,33 мм-с-1. Что можно сказать о степени окисления Fe и Мо?
15. С чем связана тонкая и сверхтонкая структура мессбауэровских спектров, какие данные из нее получают?
16. В каком из соединении: SnCl2Br2, SnCl3Br илн SnF3I—квадрупольное расщепление &Eq больше н почему?
17. Перечислите основные химические применения мессбауэровской спектроскопии.
МЕТОДЫ РЕНТГЕНОВСКОЙ
И ФОТОЭЛЕКТРОННОЙ СПЕКТРОСКОПИИ
Группа методов рентгено- и фотоэлектронной спектроскопии, включая оже-спектроскопию, позволяет получать данные об энергиях отрыва электронов от атомов и молекул как с внешних — валентных оболочек, так и с внутренних оболочек атомного остова. Это эффективные методы структурных исследований и высокочувствительные неразрушающие аналитические методы изучения молекул в газовой фазе, поверхности твердых тел, биологических объектов и полимеров. Особенно широко и продуктивно они применяются в катализе, адсорбции, электронике, а также как методы прямого измерения энергетических характеристик электронных состояний атомов и молекул. Эти характеристики являются уникальными в отношении возможности сопоставления их с теоретическими представлениями и модельными расчетами.
Если рентгеновские спектры испускания, поглощения и флуоресценции были известны и стали применяться еще в первой половине нашего века, то новые методы анализа и исследования веществ, которые можно условно объединить под общим названием — методы фотоэлектронной спектроскопии, разрабатывались лишь в 50-х и 60-х годах параллельно в СССР, Швеции,
Англии и США, Их применение в химии началось в конце 60-х, а соответствующие серийные приборы появились лишь в 70-х годах и постоянно совершенствуются.
Здесь имеются в виду методы, которые основываются на явлениях фотоэффекта, получаемого при использовании монохроматического электромагнитного излучения, и вторичной электронной эмиссии. Собственно фотоэлектронной спектроскопией (ФЭС) называют метод, в котором вещество облучают в вакуумной УФ области электромагнитного спектра. Приоритет открытия явления эмиссии фотоэлектронов в газах под действием УФ облучения, положившего начало развитию метода ФЭС, принадлежит Ф. И. Вилесову (СССР). В рентгеноэлектронной спектроскопии (РЭС, или ЭСХА*, что означает электронная спектроскопия для химического анализа) используют монохроматическое рентгеновское излучение. Создателем этого метода применительно к изучению поверхности твердых тел является шведский ученый К- Зигбан. Для возбуждения эмисии электронов может использоваться также электронный пучок, тогда говорят о методе индуцированной электронной эмиссии спектроскопии**.
Релаксационный процесс вторичной электронной эмиссии дает начало методу оже-электронной спектроскопии ОЭС ***, разработанному Д. Харрисом (США).
Вся эта группа методов вместе с абсорбционной и эмиссионной спектроскопией в УФ и видимой областях, включая спектры люминесценции и в меньшей степени по распространенности рентгеновские спектры, используется в структурных исследованиях, в частности, позволяет получать информацию об электронной структуре вещества, а также в аналитических целях.
Главным практическим применением методов фотоэлектронной спектроскопии с уникальными возможностями и эффективностью является исследование поверхности твердых тел, тонких пленок и
* В зарубежной литературе применяют аббревиатуру ESCA.
** 1ЕЕ.
*** В латинской транскрипции AES, по имени французского физика П. Оже (Р. Auger).
134
слоев вплоть до мономолекулярных. Наконец, неоценимо значение полученных данных об энергиях связи электронов для теоретической и квантовой химии.
ГЛАВА VI
ФИЗИЧЕСКИЕ ОСНОВЫ МЕТОДОВ И ЭКСПЕРИМЕНТАЛЬНАЯ ТЕХНИКА
1. ОБЩИЕ ПРИНЦИПЫ
Методами ФЭС, РЭС и ОЭС измеряют кинетическую энергию £Кин испукаемых фото- и оже-электронов, что позволяет определять значения энергии связи электронов Eni (п и I — квантовые числа уровня) в атомах на всей совокупности уровней. Эти величины, как очевидно, характерны для атомов каждого элемента, но зависят и от электронного окружения атома в исследуемом образце. Для твердых образцов определяют именно энергию связи электрона, обозначаемую £Св (или Ent), а для газов всегда приводят энергию ионизации £Нон-
Энергия коротковолнового УФ излучения (/ivClO2 эВ) достаточна для того, чтобы выбить электрон из валентной оболочки атомов или с молекулярных орбиталей, а рентгеновские лучи (и
СВоВоВн.
г
лйже-ё
ШУ////'/////
------ Зона проВодимости ______1------УроВень Ферми Валентная зона
К
Рис. VI.1. Схема возбуждения и релаксации электронов при ионизирующем облучении:
а — фотоэлектронная эмиссия: б — рентгеновское поглощение; в — рентгеновская флуоресценция; г — оже-процесс; зачерненными кружками обозначены электроны, иезачериениымн — образующиеся вакансии
Lpi Li
L1
135
электронный пучок) обладают достаточной энергией (/гу>103эВ) для возбуждения эмиссии электронов с внутренних оболочек.
Рассмотрим процесс фотоэлектронной эмиссии в РЭС вместе с рентгеновским поглощением и релаксационными процессами рентгеновской флуоресценции и эмиссии оже-электронов в одноэлектронном приближении по схеме, представленной на рис. VI. 1 для диэлектрика.
При фотоэлектронной эмиссии (рис. VI. 1, а) измеряемая кинетическая энергия £Кии свободного электрона, выбитого квантом излучения hv с атомного уровня, характеризуемого квантовыми числами п и I, по закону сохранения энергии равна:
£KHH = Av — Eni — <f,
(Vl.l)
где Eni — энергия связи электрона на данном уровне [для показанного случая Eni=ECB(K) *]; <р — работа выхода (для твердого тела) или энергия отдачи (для газа).
Не рассматривая подробно, следует указать только, что величина <р или пренебрежимо мала (энергия отдачи атома или молекулы при испускании фотоэлектрона, за исключением фотоионизации водорода), или может быть учтена как постоянная для данного прибора (работа выхода материала спектрометра). Работу выхода каждого образца обычно нет необходимости знать, поскольку образец находится в электрическом контакте со спектрометром. Таким образом, при измеренной Ект1 и известной частоте монохроматического излучения v непосредственно определяется энергия связи электрона
ЕСВ -- ^КИИ’
(V1.2)
В энергетическом спектре фотоэлектронов, представляющем кривую зависимости числа фотоэлектронов от кинетической энергии (или энергии связи электронов в атомах), наблюдаются четкие узкие полосы, каждая из которых соответствует определенному электронному уровню; например, уровням показанным на рис. VI.1, соответствует спектр вида, приведенного на рис. VI.2, а. Интенсивность полос пропорциональна содержанию эквивалентных (с учетом окружения, т. е. химического строения) атомов данного элемента. Информация, извлекаемая из фотоэлектронного спектра, прежде всего из положения пиков, сходна с той, которая может быть получена из рентгеновских спектров поглощения (или, соответственно, из УФ спектров). При прохождении через слой образца непрерывного спектра рентгеновского из
* Напомним, что буквами К, L, М и т. д. принято обозначать электронные оболочки (уровни), характеризуемые квантовым числом п, равным, соответственно, 1, 2, 3 и т. д.
136
лучения, как и для оптических спектров поглощения (в УФ, видимой или ПК областях), происходит ослабление рентгеновских лучей по экспоненциальному закону:
Л = 'о/‘М, (VT.3)
где 7ov — интенсивность входящего в образец излучения; Iv — интенсивность после прохождения слоя толщиной I; k — коэффициент ослабления.
Вообще говоря, ослабление может происходить не только за счет поглощения рентгеновских лучей, по и за счет когерентного и пекогерентного (эффект Комптона) рассеяния. Однако рассеяние играет существенную роль только для легких элементов и высокочастотного (коротковолнового) рентгеновского излучения. Уже при порядковом номере элемента Z = 20 вклад рассеяния составляет доли процента, т. е. kv можно принять за коэффициент поглощения. Он се
Рис. VI.2. Общий вид спектров:
лективно ЗаВИСИТ ОТ частоты, а — фотоэлектронов; б — рентгеновского погло-так как при достаточной энер- щения: 8 - реит"не°э"°®р*НоУаоресценцни: г — гни фотона hv' происходит выбивание электронов с какой-
либо оболочки атома ( см. рис. VI. 1, б) и наблюдается резкий скачок поглощения. Низкочастотная граница такого скачка называется краем поглощения. В рентгеновском спектре поглощения каждому электронному уровню соответствует свой край поглощения (рис. VI.2, б). Энергия кванта рентгеновского излучения hv, необходимого для выбивания электрона с данного уровня без эмиссии (например, в зону проводимости, как на схеме рис. VI.1, а), может быть сопоставлена с величинами, входящими в уравнения (VI.1) и (VI.2):
hv’ = hv — Ект
(VI.4)
т. е.
hv'=Ent = ECB(J<)
137
(если электрон выбивается с /(-уровня). Таким образом, частота края поглощения, как и положение соответствующего пика в фотоэлектронном спектре, непосредственно определяет энергию связи электрона на данном уровне.
Положение края поглощения и значение коэффициента поглощения также зависят от порядкового номера элемента и окружения атома (в молекуле, кристалле, вообще среде). В отличие от УФ, видимого и ПК излучения коэффициент поглощения рентгеновских лучей сравнительно обычно мал, чем объясняется их легкая проницаемость через различные вещества.
Методы фотоэлектронной спектроскопии, в частности ЭСХА, имеют большое преимущество перед рентгеновским поглощением в силу гораздо более высокой чувствительности и разрешающей способности. Пики в фотоэлектронных спектрах относительно более узкие, четкие и интенсивные, их положение, т. е. и сдвиги могут быть измерены с высокой точностью.
При фотоионизации или возбуждении электрона с какой-то оболочки (например, К или L) каким-либо облучением в этой оболочке образуется вакансия («дырка»). Различные по характеру релаксационные процессы, схематично показанные на рис. VI.1,6, могут привести к заполнению данной вакансии.
Если осуществляется переход электрона с какой-то внешней электронной оболочки на вакансию внутренней оболочки, то при этом может происходить испускание кванта рентгеновского излучения. Это так называемая рентгеновская флуоресценция. Так, например, при /(-захвате по схеме, показанной на рис. VI. 1, в, энергия кванта /(а1-излучения равна разности соответствующих энергий связи электронов:
Av4Kai) = £cB(K)-ScB(W-
При переходе электрона с уровня Ln возникнет другая линия /(-серии характеристического спектра (рис. VI.1, в) рентгеновского излучения:
fiv'"(Kai) = ECB(K)-ECB(Lu).
(VI.5)
Данное явление лежит в основе метода рентгенофлуоресцентного анализа (РФА). Если заполняются вакансии в L оболочке в результате перехода электронов с М оболочки, то получается L серия линий рентгеновского спектра и т. д. Частота характеристического излучения зависит от атомного номера электрона (по закону Мозли квадрат частоты определенной серии и атомный номер связаны линейной зависимостью), на чем основывается качественный РФА. В основе количественного анализа
138
лежит, как во многих других методах, пропорциональность интенсивности излучения числу излучающих атомов.
Относительные интенсивности линий в сериях характеристического рентгеновского спектра определяются соответствующими правилами отбора, т. е. вероятностями квантовых переходов, а частоты, как уже было сказано (см. равенство VI.5), дают разности энергии квантовых уровней
Рис. VI.3. Зависимость выходов флуоресценции (1) и оже-электронов (2) при вакансии в К-оболочке от атомного номера Z элемента
электронов.
Другой возможный релаксационный процесс — это безызлучательный переход электрона из внешней валентной оболочки на вакансию во внутренней оболочке атома; освобождающаяся при этом энергия, равная разности соответствующих энергий связи электрона, 'например ЕСЪ(К)—Есъ(Цп) или ЕСЪ(К)—Есъ(Цг), может привести к эмиссии электрона с одного из уровней (рис. VI.1, г) внешней оболочки, например Ьщ, как показано на схеме рис. VI.1, г, поскольку £Св(Ьш)<£'Св(Х)—£Св(Ьц). Это так называемый KLL — оже-процесс. Измеряемая в RLL ОЭС кинетическая энергия выбрасываемых оже-электронов определяется равенствами типа
£кин 11) — £св (Ю ^св (^11) — ^-св (^ш)-
(VI.6)
Из равенства (VI.6) видно, что при известной входящей в него первой разности, например, из данных о характеристическом рентгеновском спектре и по измеренной кинетической энергии оже-электронов можно определять энергию связи электрона на уровнях внешней оболочки. Спектр регистрируется как функция распределения оже-электронов N(£КИн) (рис. VI.2, г) или производной dN(EКИИ )/d£КИИ ПО £"кин-
Возможны также LMM — оже-переходы, когда заполнение вакансии, образовавшейся после облучения в L оболочке, в результате безызлучательного перехода электрона с М оболочки приводит к выбросу оже-электрона с другого уровня той же оболочки и др.
Вероятность оже-процесса уменьшается с увеличением энергии первичного ионизирующего излучения или пучка электронов (ионов) и атомного номера Z элемента. Для выхода рентгеновской флуоресценции имеет место обратная зависимость. При атомных номерах 3^Z<14 для химического анализа используют обычно KL1---оже-переходы, а при 14^Z^38 LMM — переходы. В слу-
чае энергий переходов >10 кэВ преобладает рентгеновская флуо-
139
ресценция. Зависимость выходов флуоресценции WK и оже-элект-ронов от атомного номера для заполнения вакансий в /(-оболочке иллюстрируется рис. VI.3, где видно, что сумма этих величин равна единице, а превышение выхода флуоресценции над, выходом оже-электронов начинается с Z~34.
Рентгеновское излучение может, конечно, выбивать фотоэлектроны и с внешних электронных уровней но энергии ионизации при эмиссии таких «внешних» фотоэлектронов обычно измеряются методом ФЭС при возбуждении УФ излучением.
Аналогично тому как РЭС связана с рентгеновскими спектрами поглощения и рентгеновской флуоресценцией, метод ФЭС связан с электронными УФ спектрами поглощения и релаксационными процессами фотолюминесценции (флуоресценции и фосфоресценции) в УФ и видимой областях спектра (см. учебник «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия»),
2. ПАРАМЕТРЫ И СТРУКТУРА ФОТОЭЛЕКТРОННЫХ СПЕКТРОВ
2.1. Химический сдвиг
Положение сигнала, или энергия связи электрона Есв, измеряемая в спектре, определяется, как уже указывалось, прежде всего электронной конфигурацией атома. Таким образом, полный фотоэлектронный спектр атома представляет собой набор сигналов, соответствующих s-, р-> d-, f-,... электронам оболочек атомного остова, как показано, например, для металлического кобальта на рис. VI.4. Атом в молекуле какого-то вещества характеризуется спектром, близким по виду к его спектру в веществе сравнения, хотя сигналы могут быть несколько сдвинуты. Атомы всех элементов, исключая водород и гелий, могут идентифицироваться и определяться по фотоэлектронному спектру (методами РЭС, ОЭС и др.). Некоторые удобные для идентификации линии ряда элементов приведены в табл. IV. 1.
В химических исследованиях с применением методов рентге-но- и фотоэлектронной спектроскопии чаще всего интерес представляет не абсолютное значение энергии связи Есв (эВ), а ее изменение для данного электронного уровня атома одного и того же элемента в разных соединениях:
Д£св = £°«р-£*7л. (VI.7)
Разность эн гргии связи некоторого электронного уровня (атома) в исследуемом соединении (образце) и веществе сравнения (эталоне) называют химическим сдвигом. В качестве эталона выбирают или простое вещество данного элемента, или какое-то дру-140
гое специально оговоренное вещество сравнения, содержащее атомы данного элемента (например, NaCl — для определения химических сдвигов уровней атома С1 в его соединениях). О выборе стандартов и уровней сравнения как начале отсчета энергий связи будет сказано в следующем
? 35 И
,______II 1ч*
О 500 1000
Рис. VI.4. Рентгеноэлектронный спектр металлического кобальта
параграфе.
На рис. VI.4 для 2р-электронов Со, как и в табл. VI. 1 для
ряда элементов, можно видеть расщепление сигналов переходов с 2р- и Зр-уровней. Это расщепление, наблюдаемое также для сигналов фотоэлектронов с d- и /-уровней, обусловлено квантованием полного момента количества движения J. Для неспаренного р-эле-ктрона (как й р-электронной вакансии) квантовое число орбитального момента 1=1, а спиновое $ = ±7г, отсюда возможны два р-уровня, обусловленные спин-орбитальной связью и характеризуемые квантовыми числами полного момента ]=3/2 и J = = 7г- Аналогично, для d-уровней имеем /=5Д и / = 3/2, а для /-уровней — /=7/2 и / = 5Д. Так что энергии связи (химические сдвиги) обозначают указанием символов элемента и соответствующего уровня, например С Is, S 2р3/2, Pt 4f7/2 и т. д. Если нижний индекс опускается, то имеют в виду наиболее интенсивный пик или усредненный по мультиплету сигнал.
Химические сдвиги уровней атомного остова позволяют разли-
чать атомы одного и того же элемента в разном окружении в молекуле или каком-то образце. Эти сдвиги невелики (не превышают нескольких электрон-вольт) и перекрывание линий разных элементов мало вероятно, учитывая, что для большинства из них. наблюдается несколько линий. В то же время возможны, однако, случайные совпадения пиков химически неэквивалентных атомов одного элемента, так как интервал значений химических сдвигов не столь велик (~10 эВ), даже имея в виду минимальную ширину линии (0,2 эВ).
Если, рассматривая молекулу, можно считать, что основные электронные уровни образующих ее атомов сохраняются и смещаются мало, то валентные электронные оболочки меняются при образовании химических связей весьма существенно. Иными словами, если электроны внутренних оболочек относятся в сущности к атомам молекулы и могут быть описаны с помощью аппарата АО, то валентные электроны должны рассматриваться в терминах МО, которые строятся обычно в приближении ЛКАО. Об этом приближении уже неоднократно говорилось выше, а все, что касается важнейших свойств МО, таких, как симметрия, локализация на атомах, связях или фрагментах и локальная симметрия и т. д., одинаково важно при рассмотрении электронных УФ спектров (см. учеб-
141
— Таблица VI.1. Энергия связи (эВ) электронов на уровнях атомного остова некоторых рядов элементов при возбуж-
ЬО денин линией К? Al (1487 эВ)
Li Be в с N О F Ne
15 55 111 188 284 399 532 686 867
Na Mg А1 Si н s Cl Ar
2р 31 52 2р 73 99 135 164 200 247
2s 63 89 74 100 136 165 202 247
15 1072 1305 25 118 149 189 229 270 320
К Ca Sc Ti V Cr Мп Fe Со Ni Си Zn Ga Ge As Se Br Kr
Зр 18 26 32 34 38 . 43 49 56 60 68 74 87 103 122 141 162 182 214
107 129 147 168 189 223
35 34 44 54 59 66 74 84 95 101 112 120 137 158 181 204 232 257 289
2р 294 347 402 455 513 575 641 710 779 855 931 1021 1116 1217 1323 1436
297 350 407 461 520 584 652 723 794 872 951 1044 1143 1249 1359 1476
25 377 438 500 564 628 695 769 846 926 1008 1096 1194 1298 1413
ник «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия») и фотоэлектронных спектров.
Измеряемые методом ФЭС энергетические характеристики электронов валентных оболочек в молекулах могут интерпретироваться по-разному в зависимости от выбираемого теоретического подхода и приближения. При испускании фотоэлектрона происходит переход из основного электронного состояния исходной молекулы в основное (или иногда в возбужденное) электронное состояние ионизованной молекулы. Наиболее строго было бы рассматривать, как в любой другой спектроскопии, энергию ионизации как энергию перехода между этими двумя состояниями, характеризуемыми соответствующими ВОЛНОВЫМИ функциями VMon и Vhoh- Но обычно Используют более простые и доступные подходы, например метод МО в одноэлектронном приближении.
2.2. Спин-орбитальная связь в молекулах и некоторые другие эффекты
Как и в атомах (см. выше), спин-орбитальная связь может приводить к расщеплению электронных уровней энергии молекул. Например, у парамагнитных молекул (NO, О2) при высоком разрешении наблюдается слабое расщепление линий фотоэлектронного спектра (~1 эВ), обусловленное спин-орбитальной связью неспаренного электрона. Вообще эта связь для двухатомных и линейных многоатомных молекул описывается векторной схемой электронных моментов — орбитального, спинового и полного. Последний выражается через проекции двух первых на ось молекулы формулой
Й = Д + 2Л, (VI.8)
где Л=|Л1£|—абсолютное значение проекции суммарного орбитального момента, а — проекция суммарного спинового момента (подробнее см. учебник «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия»).
У линейных молекул при Л=1 и 2т=1/2 имеем два состояния: лз/2 и т/г, т. е. происходит расщепление сигнала в результате спин-орбитальной связи на 2 компонента (рис. VI.5). Для нелинейных молекул типа симметричного волчка (при наличии поворотной оси симметрии Сп порядка nj>3) сигналы могут расщепляться только при ионизации удалением электрона с вырожденных МО (двукратно — е и трехкратно f). Так, например, в ряду молекул RI при R = H, СНз, С(СН3)з, SiH3 наблюдается расщепление сигнала, соответственно, на 0,66; 0,63; 0,56 и 0,55 эВ, причем в случае групп R^H сигнал относится к делокализованной несвязывающей орбитали неподеленной пары иода, частично являющейся также дважды вырожденной е-орбиталыо групп R^H.
При ионизации молекулы, находящейся в вырожденном электронном состоянии, ион также остается в вырожденном состоянии.
143
Рис. VI.5. Фотоэлектронный спектр НВг (ионизационный потенциал) при возбуждении Не (/ )
Спонтанное снятие этого вырождения происходит в результате эффекта Яна — Теллера. Так, например, при образовании иона СН4+ удалением электрона в СН4 с одной из трижды вырожденных орбиталей /2 вместо одного пика наблюдается три максимума, а при плохом разрешении — широкая полоса. Ян-тел-леровское возмущение дважды вырожденного состояния приводит к появлению двух максиму-^ мов, частью не разрешенных!
т. е. также к уширению полосы. Аналогичное возмущение длй двухатомных молекул называют эффектом Реннера-Теллера.
В связи с крайней неустойчивостью атомов в фотоионизирован-ном состоянии возникает сателлитная структура рентгеноэлектронных спектров. Когда удаляется электрон внутренней оболочки, происходит сильное и внезапное возмущение всей системы с возбуждением и даже эмиссией вторичных электронов. При этом возможны два случая. В первом (shake up) процесс возбуждения приводит к появлению в спектрах основных уровней сателлитных пиков с меньшей Екип (большей ЕСв) по сравнению с основными. Второй процесс (shake off) приводит к возбуждению электронов в континуум и не дает дискретной сателлитной структуры. Кроме сателлитов shake up в спектрах могут наблюдаться дополнительные линии, обусловленные оже-переходами и потерями энергии на неупругие соударения. Интерпретация сателлитной структуры рентгеноэлектронных спектров представляет нелегкую проблему, так как могут быть самые разные механизмы возникновения мультиплет-ности сигналов.
2.3. Колебательная структура фотоэлектронных спектров
Для не очень сложных молекул так же, как в абсорбционных УФ спектрах и спектрах люминесценции, при достаточном разрешении может наблюдаться колебательная структура фотоэлектронных спектров. Имея в виду наличие у молекул (и многоатомных ионов) различных электронно-колебательно-вращательных состояний, соотношение (VI.2) для энергии связи электрона в молекуле (на какой-то молекулярной орбитали) можно переписать так:
Eqb = h'i £кин — I ^Екол -]- Д£вр, (VI.9)
где / — энергия адиабатической ионизации, представляющая энергию перехода между основными (нулевыми колебатель
144
ными) состояниями исходной и ионизованной молекулы, а Д£кол и Д£Вр — возможные при фотоионизации изменения колебательной и вращательной энергии.
Обычно составляющую Д£Вр не рассматривают, так как вращательная структура фотоэлектронных спектров не наблюдается даже в газе (недостаточное разрешение, большие молекулярные массы).
Кроме понятия"адиабатической энергии удаления электрона вводится также понятие энергии вертикальной ионизации, соответствующей электронному переходу, при котором геометрическая конфигурация (межъядерные расстояния и валентные углы) молекулярного iioiki неизменна по сравнению с исходной молекулой. Очевидно, что если исходная молекула находится в основном электронно-колебательном состоянии, то энергия вертикальной ионизации может быть равна или превышать энергию адиабатической ионизации, т. е. Д^кол равна нулю или положительна. Принципиально возможно и отрицательное значение Д£Кол, если молекула ионизуется из колебательно-возбужденного состояния.
Строгого правила отбора для Дц колебательных переходов, как и в оптической электронной спектроскопии, в фотоэлектронных спектрах нет, и часто наблюдается хорошо развитая колебательная структура полос. Она видна, например, на рис. VI.5, где приведен фотоэлектронный спектр бромоводорода. Соответствующий более низкому значению энергии / дублет интенсивных узких пиков без колебательной структуры относится к ионизации с несвя-зывающей орбитали Вг и обусловлен спин-орбитальной связью (см. гл. VI; 2.2). Полоса при более высоких энергиях I относится к ионизации со связывающей орбитали и расстояния между пиками ее структуры соответствуют частоте валентного колебания v(H—Вг) ионизованной молекулы. В ФЭС также справедлив принцип Франка —Кондона, т. е. наиболее вероятны вертикальные переходы.
У многоатомных молекул, имеющих ЗУ—6 (ЗУ—5) колебательных степеней свободы (У — число атомов), если даже колебательная структура разрешена в фотоэлектронном спектре, интерпретация ее представляет нелегкую задачу. Однако из нее также получают информацию как о колебательных частотах и структуре ионизованной молекулы, так и характере МО исходной молекулы.
2.4. Интенсивность фотоэлектронных пиков
Строгой теории абсолютных интенсивностей фотоэлектронных спектров нет. Характерной чертой является пропорциональность интегральной интенсивности числу атомов соответствующего элемента (РЭС) и степени вырожденности орбитали, с которой выбивается электрон. Вообще, наблюдаемая интенсивность является
145
сложной функцией (/) многих параметров:
Л = / (5, Ni, 'i, 0, Xz, Ki, С,), (VI.10)
где F — аппаратурный фактор, учитывающий чувствительность анализатора энергий электронов и детектора, интенсивность потока фотонов и другие особенности конструкции прибора; Nt — число атомов, испускающих электроны с i-го уровня атома элемента; Oj — сечение фотоионизации данного электронного уровня; р,- — параметр асимметрии; 0 — угол между направлениями вылета фотоэлектрона и ионизирующего излучения; X, — эффективная глубина выхода электронов; Ki — параметр, учитывающий наличие сателлитов; С,- — фактор, учитывающий ослабление потока фотоэлектронов в загрязняющем поверхностном слое (углеводородном или другом) толщины d.
Обычно известны не все параметры, входящие в уравнение (VI. 10), и на практике при количественном анализе используют относительные интенсивности. Параметрами Ki и Ct, а точнее их отношением для двух линий (1=1 и 2), когда X<>d, можно пренебречь, как и постоянным значением F. Таким образом, отношение интенсивности двух линий можно записать в виде
Параметры асимметрии р,, входящие в Л,г, для разных уровней и элементов табулированы в специальной литературе (как и значения щ); угол 0 бывает известен (обычно равен 90°); Х< можно оценить (см. ниже), т. е. может быть рассчитано.
2.5. Глубина выхода фотоэлектронов
При оценке длины свободного пробега X фотоэлектрона (глубины выхода) на практике обычно предполагают, хотя это и не совсем строго, экспоненциальную зависимость интенсивности /=f(X). Величину X экспериментально оценивают тогда по ослаблению интенсивности сигнала подложки при напылении на нее пленки известной толщины:
Z = Zo exp (— <f/X), (VI.12)
где I — измеряемая интенсивность; /о — интенсивность пика для чистой подложки; d — толщина пленки.
Другой способ заключается в измерении увеличения интенсивности выбранного пика при наращивании толщины пленки образца:
/ = /„[!— ехр(—rf/X)], (VI.13)
где /<» — интенсивность данного пика для массивного образца (d-> —>-оо).
146
Глубина выхода Л. зависит, конечно, от характера образца (плотности и упаковки), энергии связи электронов, а значит, и Ент, угла эмиссии фотоэлектронов. При фиксированных углах Л. является приближенной функцией £'кин(Х«£'кнн-1/2).
При малых энергиях связи электронов, т. е. больших кинетических энергиях (валентные оболочки), фотоэлектроны могут преодолевать толщину до ~ 10 нм, а при энергиях связи порядка сотен и 1000 эВ глубина выхода фотоэлектронов не превышает обычно 2... 3 нм, а иногда и десятых долей нанометра, т. е. толщины атомных и молекулярных слоев.
3. ТЕХНИКА И МЕТОДИКА ЭКСПЕРИМЕНТА
3.1. Аппаратура
Спектрометры, применяемые для получения фотоэлектронных
спектров, имеют существенные конструкционные отличия в зависимости от их конкретного предназначения. Однако принцип действия, схема возбуждения и анализа энергий электронов имеют много общего. На рис. VI.6 показана принципиальная блок-схема фотоэлектронного спектрометра, на которой это можно проиллюстри-
ровать.
Источники. В ЭСХА для возбуждения электронов внутренних оболочек источником излучения служит рентгеновская трубка. Обычно используется монохроматическое излучение /faMg с энергией 1253,6 эВ или 7<аА1— 1486,6 эВ. Ширина возбуждающей линии порядка 1 эВ. Если необходимо получить высокое разрешение, используют дополнительную монохроматизацию (кристаллами), что приводит к сужению возбуждающей линии и увеличению разрешающей способности прибора.
В ФЭС внешних электронных оболочек возбуждающим источником УФ излучения является, как правило, резонансная гелиевая лампа (разрядная трубка) с капиллярным коллиматором, направляющим на образец узкий
луч света. Используются линии Не I — 21,2 эВ (собственная ширина линии менее 10~1 2 эВ) и Не II—40,8 эВ.
В ОЭС для возбуждения оже-электронов служит электронная
Рис. VI.6. Принципиальная блок-схема фото-(рентгено-) электронного спектрометра:
1 — источник; 2 — образец; 3 — электрля-
ный анализатор; 4 — приемник; 5 — регистрирующее устройство; 6 — защита от внешних полей (магнитного поля Земли)
147
пушка, дающая узкий (менее 10~4 м) первичный пучок электронов с энергиями до 5 кэВ. оже-процесс происходит наряду с эмиссией фотоэлектронов также при использовании рентгеновского излучения, например КаА1 нлн KaMg в ЭСХА, но спектры оже-электро-нов можно отличить, так как в отличие от фотоэлектронов их Ект не зависит от hv возбуждающего излучения. Поток возбуждающих фотонов hv под определенным углом (обычно ~45°) падает на образец, помещаемый вблизи входной щелн монохроматора. Электронный пучок в ОЭС обычно падает вертикально.
Анализаторы. Выбитые электроны поступают через входную щель в электронный анализатор-монохроматор, в котором происходит разделение электронов по скоростям (моментам). Электроны с соответствующей кинетической энергией попадают через выходную щель на детектор.
Анализаторы могут быть с магнитной или электростатической фокусировкой, но последние имеют преимущество в защите от внешних электромагнитных помех, и в современных спектрометрах применяются анализаторы типа электростатического конденсатора. Геометрическая форма анализатора и режим пропускания через него электронов могут быть различны. Но обычно проводится предварительное торможение электронов на входе, а между образцом и анализатором создается некоторый потенциал. Этим добиваются лучшего разрешения, хотя н за счет некоторой потери чувствительности.
Разрешающая способность электростатического анализатора определяется отношением АЕ/ЕК1т- На практике современные спектрометры позволяют получить разрешение сигналов фотоэлектронов с внутренних уровней до 0,2 эВ, а с внешних уровней до 0,02 эВ, которое бывает достаточно для наблюдения колебательной структуры спектра, т. е. разрешающая способность приборов с источником, дающим узкую линию возбуждающего излучения, имеет порядок ~ 10~3.
Приемники. Обычно в качестве детекторов используют фотоэлектронные умножители. Усиленный после детектора сигнал поступает на самописец или через интерфейс на совмещенную с прибором ЭВМ.
Спектр фотоэлектронов получают, сканируя или поле анализатора, или замедляющее поле. Регистрация может проводиться непрерывно или ступенчато (по точкам). Для улучшения отношения сигнала к шуму необходимо усреднение по многократным сканам или увеличение времени счета импульсов в каждой точке. Имеющиеся в современных спектрометрах микропроцессоры и мини-ЭВМ управляют работой системы и обеспечивают накопление сигналов, усреднение, сглаживание, разложение сложных контуров на отдельные компоненты, вычитание фона, дифференцирование, интегрирование и другую обработку спектров.
Образцы. Можно получать фотоэлектронные спектры твердых
148
или газообразных (при низких давлениях) образцов, жидкости для исследования замораживают или испаряют. При изучении твердых образцов особенно необходим высокий вакуум для предохранения от загрязнений поверхности адсорбируемыми частицами, иногда необходимо охлаждение.
Даже свежеприготовленные образцы часто оказываются для ФЭС сильно загрязненными. Например, металлические поверхности на воздухе сразу покрываются оксидными пленками. Даже в вакууме почти всегда по сигналу С 1s обнаруживается пленка масла (от вакуумного насоса). Правда, этот сигнал бывает полезен и часто используется для калибровочных целей. Специальные камеры для подготовки образцов при спектрометрах позволяют без вынесения на воздух обрабатывать образцы, чистить поверхности ионной или электронной бомбардировкой, менять и т. д.
к 3.2. Стандарты для учета зарядки образцов
0 и калибровки спектрометров
. Поверхностный слой образца может, в принципе, заряжаться как положительно, так и отрицательно по отношению к спектрометру. Отсюда следует необходимость учета разности потенциалов поверхностного слоя образца и материала спектрометра в виде поправки ±£зар к измеряемой величине ЕСъ- Для оценки этой поправки используют различные внешние и внутренние стандарты.
Самым распространенным, так называемым внешним стандартом отповерхности, является уже упоминавшийся выше сигнал С 1s (285,0 эВ) от углеводородной пленки, образующейся на образце в результате проникновения в спектрометр паров масла от вакуумного насоса, дегазации прокладок и т. д. Иногда в качестве внешнего стандарта на поверхности используют линии от напыленных пленок химически инертных металлов, например Au 4/7/2, Pd 3ds/2, или адсорбированных ионов аргона.
Применяют также внешний стандарт от добавки, в частности, использовалась линия F 1s от добавляемого к веществу LiF. Выбор вещества стандарта и способ подготовки пробы требуют особого внимания, так как вообще этот метод менее надежен.
В качестве внутреннего стандарта может быть принят сигнал от какого-то атома в соединениях исследуемого ряда, если измеряемая величина ЕСв для этого атома в данном ряду соединений практически не меняется. В этом случае все другие измеряемые значения Есз нормируются к значению Есзсг выбранного атома-стандарта. Например, в ряду соединений [Р(СбН5)з]2Р1Х2, где X — различные ацидолиганды, за стандартное значение EC3CT принимают энергию связи электрона С 1s в фенильных группах. Если измеренное значение £'Свст=284,0 эВ, а значение Есз С 1s при отсутствии заряда считается равным 285,0 эВ, то для получения правиль
на
них значений Есв, например Pt 4/, нужно просто прибавить к измеренным значениям 1,0 эВ.
Вообще учет зарядки образцов является одной из центральных методических проблем в фотоэлектронной спектроскопии, которая далеко еще не решена. В любом случае при публикации спектров должны указываться использованный стандарт и значение энергии, принятое за начало отсчета.
3.3. Комплексные установки и методики
Рис. VI.7. Схема одновременного ионного травления и получения оже-спекТров при послойном анализе состава поверхности образца:
1 — образец; 2 — кратер травления; 3 — ионный пучок; 4 — возбуждающий электронный пучок; 5 — анализируемая точка (перемещается по поверхности)
Перспективны экспериментальные установки, объединяющие несколько методов в одном приборе и реализующие, таким образом, комплексный подход к изучению образцов. Это может быть ЭСХА и ОЭС, а дополнительно также ФЭС и некоторые другие методы, например масс-спектрометрия. Сопоставление данных двух-трех разных методов для одного и того же образца (участка поверхности) без его перемещения в камере прибора, в условиях высокого вакуума позволяет получить более обширную и разностороннюю информацию, облегчая интерпретацию результатов.
При необходимости исследовать изменение состава образца в зависимости от глубины проводится послойный анализ, который выполняется при совместном использовании ЕСХА и ОЭС или ОЭС с ионным травлением. Послойные спектры ЕСХА получают при последовательном чередовании травления и регистрации спектра. Большое значение имеет при этом равномерность травления по всему анализируемому участку поверхности, который задается диаметром пучка ионной пушки (0~2... 3 мм).
Конструкция ионной пушки, в которой разгоняется поток ионов, например Аг+, Кг+, обеспечивает высокую скорость травления практически без загрязнения поверхности. В сочетании с высокой чувствительностью детектора электронов быстрое травление позволяет профилировать по глубине слой толщиной до 1000 нм в течение нескольких минут.
При использовании ОЭС травление и регистрация спектра проводятся одновременно (рис. VI.7). Образец бомбардируется сфокусированным электронным пучком и анализируется энергия вторичных электронов (оже), а при одновременном ионном травлении получают послойные профили.
150
При сканировании электронного пучка, которое также осуществляется достаточно быстро (метод сканирующей оже-ми-кроскопии СОМ), получают данные о двухмерном распределении элементов по площади (растр), а сочетание с ионным травлением позволяет проводить трехмерный анализ поверхностного слоя образца.
3.4. Рентгеио-флуоресцентные спектрометры
В спектрометрах, предназначенных для качественного и количественного РФА, анализируемые на содержание элементов пробы облучаются рентгеновским излучением от трубок, питаемых от генератора высокого и стабильного напряжения и тока. Материалами анодов рентгеновских трубок для определения различных элементов могут служить W, Мо, Ст, Rh н другие металлы. Измеряемые и контрольные пробы (твердые и жидкие) в кюветах, изготовляемых из разных материалов (С, Al, Ni и др.), помещаются в специальное устройство, обеспечивающее их быструю смену и установку в нужное положение, а иногда и вращение вокруг своей оси.
Характеристическое флуоресцентное излучение, даваемое пробой, коллимируется, и параллельный пучок лучей после прохождения через абсорбер (ослабитель) падает на плоский кристалл анализатора. Возможно использование нескольких сменных коллиматоров и ослабителей, а также кристаллов, служащих для спектрального разложения рентгеновского излучения. В ассортимент кристаллов-анализаторов входят LiF, Ge, Si, кварц, графит и ряд других. Диспергирование излучения кристаллической решеткой с заданной постоянной происходит вследствие селективного отражения под углом, зависящим от длины волны.
Отраженное от кристалла излучение попадает иа детектор. Для коротковолнового излучения используют сцинтилляционные детекторы, например Nal, активированный таллием, а в длинноволновом диапазоне—счетчики Гейгера. Для управления системой, регистрации спектров, выполнения измерений и обработки данных в современных приборах используется микропроцессорная техника и ЭВМ.
ГЛАВА VII
ПРИМЕНЕНИЕ МЕТОДОВ
ФОТОЭЛЕКТРОННОЙ СПЕКТРОСКОПИИ В ХИМИИ
1. СТРУКТУРНО-АНАЛИТИЧЕСКИЕ ПРИМЕНЕНИЯ
1.1. Элементный анализ и идентификация соединений
Вся группа методов фотоэлектронной спектроскопии, как и методы рентгеновской спектроскопии (РФА и абсорбционной), могут
151
СР5- с(о)о-сн2-сн3
_____I____I____I_____I________
299 295 291 Ес$,з&
Рис. VII.1. Рентгеноэлектронный спектр С 1s этнлтрифтор-ацетата
использоваться для идентификации атомов различных элементов, входящих в состав практически любого образца (при соответствующей подготовке). Определению поддаются все элементы, кроме Н и Не, хотя в конкретных случаях применение одних методов может быть более предпочтительным, чем других. Особенно широкие возможности открывают методы ЭСХА (РЭС) и ОЭС. Выше, в табл. VI.1, приведены некоторые характерные для элементов значения энергий связей электронов, используемые в целях идентификации.
Когда в образце (в молекуле) имеются атомы разных элементов, спектр представляет суперпозицию спектров элементов, т. е. аддитивен. Кроме того, для атомов одного и того же элемента их спектр зависит от окружения, т. е. сигналы претерпевают химический сдвиг. По этой причине спектр остовных электронов может служить как «отпечаток» для идентификации чистых веществ и качественного анализа смесей, т. е. идентификации компонентов, при использовании поисковых систем и сопоставлении с уже известными спектрами.
На рис. VII. 1 в качестве примера приведен спектр фотоэлектронов С 1s для этилового эфира трифторуксусной кислоты, в котором хорошо видно, что каждому неэквивалентному атому углерода соответствует свой пик, а общая картина спектральной кривой и по
Рис. VII.2. Фотоэлектронный Не (/а) спектр 1,3-диоксана
152
ложение пиков характерны для данного соединения. Фотоэлектронные спектры внешних, валентных электронов, т. е. получаемые при ионизации с МО, также специфичны, как это показывает, например, спектр 1,3-диоксана (рис. VII.2), причем их общий вид зависит от строения молекулы обычно даже в большей степени, чем спектр остовных электронов.
1.2. Структурная информация
Применение методов ФЭС для структурно-группового анализа основывается на том, что значения Есв электронов в функциональных группах и вообще в некоторых структурных фрагментах мало зависят от строения молекулы (образца) в целом. Иными словами, химический сдвиг \ЕСВ определяется в основном ближайшим окружением данного атома А, т. е. достаточно характеристичен для функциональной группы (структурного фрагмента). В табл. VII.1 такие данные приведены для некоторых групп. В структурном анализе важно также, что относительная интенсивность максимумов, соответствующих разным группам, пропорциональна их числу в данном соединении.
Таким образом, уже по химическим сдвигам и распределению интенсивности сигналов для остовных электронов можно делать некоторые выводы о строении соединений. Помогают при этом и некоторые закономерности, рассматриваемые ниже.
Уже говорилось о связи колебательной структуры полос фотоэлектронных спектров со строением молекул и распределением электронной плотности (см. гл. VI; 2.3). По форме и колебательной структуре полос можно делать выводы о характере орбитали, с которой удаляется электрон, не только для двухатомных, но и для некоторых многоатомных молекул. Когда электрон удаляется со связывающей орбитали, то из-за ослабления связи частота соответствующего валентного колебания в ионе будет ниже, чем в исход-
Таблица VII.1. Энергии связи электронов £св (эВ) в некоторых функциональных группах (относительно стандарта С 1s £св=285 эВ)
Группа £св. С 1s Группа £CB, N Is Группа Есв< А 2Р
—соон —соом R3OOH r2co СОз2- Карбиды 289,5 289,0 286,7 288,0 289—290 282—283 RNO2 no2-NO3-r4n+ NH3 NCS-CN- Нитриды 406,0 403,6... 404,6 407... 408 401 ... 402 400,4 398,5 398,2... 399 397... 398 Силициды РО*2-Фосфиды SO32-so?-Сульфиды СЮ3-сю4-Хлориды 99... 100 133 128... 130 167... 168 169... 170 161... 162 206... 207 208... 209 198... 200
» А — Si, Р, S, С1.
153
ной молекуле, что и имеет место в примере с бромоводородом (см. гл. VI; 2.3). В случае несвязывающих орбиталей ионизация также приводит иногда к появлению колебательной структуры.
Так, например, в фотоэлектронном спектре аммиака полоса, обусловленная ионизацией с несвязывающей орбитали неподеленной электронной пары, имеет развитую колебательную структуру с расщеплениями, соответствующими уменьшенной частоте симметричного деформационного (зонтикового) колебания NH3. Это подтверждает зависимость валентных углов молекулы от наличия не-поделенных пар на атоме азота, а также указывает на то, что ион NH3+ в основном состоянии является практически плоским (валентные углы сильно увеличены по сравнению с нейтральной молекулой).
Если в какой-то вибронной полосе фотоэлектронного спектра обнаруживается увеличение колебательной частоты (расщепление) по сравнению с исходной молекулой, то можно объяснить это ионизацией с антисвязывающей (разрыхляющей) орбитали, локализованной на фрагменте, к которому относится колебание, и судить о структуре данного фрагмента.
Сложную картину представляют фотоэлектронные спектры комплексов переходных металлов. В связи с наличием d-электронов в них значительно сильнее, чем у молекул с замкнутыми оболочками, релаксационные эффекты, а порядок расположения уровней у иона и молекулы может быть разным. Для разумной интерпретации спектров этих комплексов необходимо сопоставление их в рядах родственных соединений. Важным моментом при изучении фотоэлектронных спектров комплексов является также то, что d-электроны сильнее возбуждаются линией Не (II), чем Не (1а), в отличие от s- и р-электронов. Поэтому в спектре, возбуждаемом линией Не (II), полосы, относящиеся к ионизации с d-орбиталей, интенсивнее, чем в спектре того же образца, возбуждаемом линией Не(1«).
1.3. Количественный анализ
Рассматриваемые методы обладают чрезвычайно высокой чувствительностью, но как аналитические методы используются в основном для количественного определения состава поверхностных слоев. Относительные и абсолютные пределы обнаружения элементов в оже-спектроскопии, например, составляют, соответственно, 10-1... 10~3% и 10~12... 10~16 г. Метод ОЭС имеет также преимущества в высоком пространственном разрешении, обеспечивающем возможность проведения тонкого локального анализа.
Хотя ЭСХА несколько уступает ОЭС, но также характеризуется относительным пределом обнаружения 0,1... 1% при абсолютном пределе порядка ~10~8 г. Это позволяет анализировать атомные и молекулярные слои и при этом определять элемент, один атом 154
которого в соединении приходится на 100...200 и более атомов других элементов, например Со в витамине Bi2 (в молекуле один атом Со и 180 атомов других элементов).
При определении концентрации элементов по глубине образца последовательно удаляют тонкие верхние слои образца ионным травлением. Проводя ре-
гистрацию спектров, получают зависимость интенсивности от времени травления, а при известной скорости травления — от глубины образца. На рис. VII.3 показано, например, изменение интенсивности линий рентгеноэлектронного
Рис. VII.3. Зависимость интенсивности I рентгеноэлектронных линий:
1 — С 1S; 2 — о 1S; 3 — S1 2р; < — А1 2р — от времени ионного травления образца пленки иа алюминиевой подложке
спектра в зависимости от времени ион-
ного травления полимерной пленки, содержащей Si, С, О, нанесенной иа алюминиевую подложку. Первоначально наблюдается падение интенсивности линий С 1$и О 1s, связанное с удалением слоя загрязнений. Для пленки характерно постоянное содержание компонентов. После стравливания большой толщины пленки наблюдается падение интенсивности линий С 1s и Si 2р. Изменение интенсивности линий О 1s характеризует стравливание оксида AI2O3 на подложке, когда линия А1 2р растет.
Количественный анализ состава поверхности изучаемого образца основывается на использовании уравнения (VI.11) для определения отношения концентраций Vi/Vs. Относительная погрешность обычно не превышает 20%, но иногда из-за неточности используемых значений щ, а также в связи с пренебрежением фактором Ci может быть значительно больше (до 100%). Для более точного нахождения N1/N2 используют метод градуировочных кривых, представляющих зависимости Л/Л от известных отношений N1/N2. Для построения таких кривых берутся относительные интенсивности, полученные на приборе с тем же аппаратурным фактором что
и у спектрометра, используемого в анализе.
Ионное травление изменяет относительное содержание элементов на поверхности образца. Наибольшее влияние на получаемые при анализе профили оказывают шероховатость поверхности после травления и эффекты выбивания и распыления. Определение истинного распределения концентраций по глубине — задача трудно решаемая. Как и большинство обратных задач физических методов, она относится к некорректно поставленным задачам и требует привлечения некоторой априорной информации о зависимости концентраций от глубины, а также повышения устойчивости решения по отношению к экспериментальным ошибкам с помощью ре-
гуляризующих алгоритмов.
155
2. ТЕОРЕТИЧЕСКОЕ МОДЕЛИРОВАНИЕ И ОБЪЯСНЕНИЕ ХИМИЧЕСКИХ СДВИГОВ
Существует три основных модели, или подхода, к расчетам энергии связи электронов и объяснению химических сдвигов. Первый подход основывается на оценке эффективных зарядов атомов и сопоставлении их разности с химическими сдвигами для разных образцов. В самой грубой модели для оценки зарядов используется просто шкала электроотрицательностей. Например, заряд на атоме Ni(II) в комплексах оценивают как разность электроотрицательностей атома и присоединенных к нему лигандов. Даже такая оценка удовлетворительно отражает разности зарядов и ход изменения химических сдвигов, а полуэмпирические методы квантовой химии (ППДП, ЧПДП и др.) позволяют получить существенно лучшие результаты.
Согласно упрощенной параметрической модели сферических зарядов энергию связи внутреннего электрона, зависящую от эффективного заряда данного атома и взаимодействий с зарядами других атомов, представляют при выбранном уровне отсчета энергии в виде
£св = *<7+2 (<?//%), (VII.I)
где k = <Z.e1lr'> — константа кулоновского взаимодействия*; г — средний радиус валентной оболочки; q — эффективный заряд данного атома; qi— эффективные заряды других (г-х) атомов; Rt— расстояния между данным и i-м атомами.
Записав эти выражения для энергии связи электрона в атоме исследуемого вещества и эталона и взяв разность, получим для химического сдвига формулу
Д£св = Д£ (q) + ДК, (VII.2)
где слагаемые представляют собой разность первых и вторых членов выражений (VII.1), причем AV называют энергией маде-лунговского взаимодействия.
Для ионной связи химический сдвиг сигнала по отношению к свободному атому выражают как
ДЕСВ = g (1/г —а/7?), (VII.3)
где а — постоянная Маделунга; R — расстояние между ядрами противоположно заряженных ионов.
* В атомных единицах т=е=Ь —е0=1.
156
Рассчитанные по формуле (VII.2) значения химических сдвигов могут сильно отличаться (до десятков электрон-вольт) от наблюдаемых экспериментально. Существенное улучшение сходимости дает учет изменения энергии релаксации, т. е. дополнение уравнения (VII.2) членом \Ерел:
д£св = Д£(?)+дк + д£рел. (VII.4)
Согласно предложенным моделям энергия релаксации складывается из двух составляющих — внутриатомной, которая определяется взаимодействием «дырки» во внутренней оболочке с электронами данного атома, н межатомной, определяемой взаимодействием «дырки» с электронами окружающих атомов. Вторая составляющая важна только для конденсированных фаз. Релаксационный потенциал оценивают по разности энергий молекулы и иона, рассчитываемой с помощью полуэмпирических методов квантовой химии.
Второй подход использует теорему Купменса, утверждающую примерное равенство орбитальной энергии и энергии связи Есв электрона. Сравнение экспериментальных данных ФЭС по химическим сдвигам с полученными в результате квантово-механических расчетов орбитальными энергиями позволяет более обоснованно интерпретировать спектр, т. е. проводить отнесение пиков, а также оценивать делаемые в расчетах допущения. В то же время рассчитанные значения энергии обычно плохо согласуются с большими абсолютными значениями Есв- Можно лишь надеяться, что относительные значения, т. е. разности рассчитанных энергий, правильно отражают различия энергий связи, т. е. химические сдвиги EEct для изучаемых объектов. Полуэмпирические методы квантовой химии даже для молекул, образованных атомами элементов первого ряда, не только не дают количественного соответствия рассчитанных энергий МО и энергий связи электронов, но иногда приводят к неправильному порядку относительного расположения уровней энергии.
В третьем, наиболее строгом подходе энергию связи электрона представляют в отличие от теоремы Купменса как разность полных энергий молекулы и иона, получающегося при удалении электрона. Этот подход следует применять только при условии проведения полных неэмпирических квантово-механических расчетов (ab initio) с учетом эффектов корреляции электронов при разном их числе в молекуле и ионе, а также релаксационных эффектов в ионе из-за наличия электронной «дырки». Столь сложные расчеты практически возможны лишь для очень небольших молекул.
Наиболее широко и успешно применяется пока первый подход, так как существуют достаточно простые и доступные методы оценки эффективных зарядов, а также учета поправок на релаксационные эффекты, позволяющие объяснять наблюдаемые химические сдвиги и даже удовлетворительно предсказывать их.
157
3. НЕКОТОРЫЕ ЗАКОНОМЕРНОСТИ И КОРРЕЛЯЦИИ ХИМИЧЕСКИХ СДВИГОВ
3.1. Связь с эффективным зарядом и степенью окисления
Как следует из уравнения (VI 1.1) и показано экспериментально, ДЕСв коррелирует с эффективным зарядом атома q. В качестве примера на рис. VII.4 показана зависимость ДЕСв 2рз/2 от заряда атома Fe. Заряды атомов или рассчитываются (по электроотрицательностям или полуэмпирическими и неэмпирическими методами квантовой химии), или находятся из сдвигов линий эмиссионных рентгеновских спектров.
Возможны, однако, отступления даже от симбатности хода изменения значений q и &ЕСВ, так как существенное влияние на химический сдвиг оказывает величина ДУ (VII.2), поэтому правильнее последовательно проводить корреляцию заряда с величиной ЕЕСВ—ДУ. Разделение суммарного химического сдвига на вклады от эффективного заряда и от потенциала Маделунга имеет смысл прежде всего для ионных соединений. В молекулах сдвиги коррелируют с молекулярным электростатическим потенциалом, который в отличие от эффективного заряда является не условной, а измеряемой физической величиной.
На основании корреляционных соотношений типа (VII.2) с эмпирически найденными параметрами (k и другие) проводят и расчеты зарядов qi на атомах по измеренным химическим сдвигам ДЕсв. Получаемые в рамках метода наименьших квадратов значения зарядов удовлетворительно согласуются с величинами, рассчитываемыми полуэмпирическими методами квантовой химии.
Поскольку химический сдвиг линии для внутренних уровней опорционален эффективному заряду А, а заряд растет с увеличением степени окисления, то очевидно, что химический сдвиг коррелирует и с последней. Чем выше положительная степень окисления, тем больше сдвиг в сторону более высоких значений Есв (положительный сдвиг). При отрицательных степенях окисления наблюдается сдвиг в сторону меньших значений Есв (отрицательный сдвиг). Если характер окружения данного атома в соединениях, где он имеет разную степень окисления, меняется не сильно, т. е. соседние группы или лиганды одинаковы, то с ростом степени окисления на единицу химический сдвиг увеличивается примерно на 1 эВ, во всяком
атома л в соединении
Рис. VII.4. Зависимость энергии СВЯЗИ Есв Fe 2р3/2 от формального заряда q на атоме железа:
1~ Fe(C,H5)2; 2- K<Fe(CN)s; 3-KcFe(CN)«; 4 — Naa[Fe(CN)sNO]
случае наблюдается примерно линейная зависимость Есв от степени окисления.
Такую зависимость можно проследить для соединений на рис. VII.4, где точкам 2 и 3, например, соответствуют степени окисления Fe(II) и Fe(III). В комплексе Na2[Fe(CN)5NO], представленном на рисунке точкой 4, в зависимости от того, какой заряд приписать нитрозогруппе NO: положительный, нулевой (группа нейтральна) или отрицательный, атом железа мог бы иметь степень окисления (II), (III) или (IV). Судя по химическому сдвигу Ре2рз/2 (711,0 эВ в отличие от 710,0 эВ для Кз[Ре(С\)е] и 708,7 для K4[Fe(CN)e]), степень окисления железа в комплексе равна (IV). Это указывает и на то, что заряд нитрозогруппы отрицательный.
В положительных ионах металлов электроны остова сильнее притягиваются ядром, так что энергии связи электронов в оксидах, солях и т. п. выше, чем в нейтральных атомах металлов. В соединениях углерода также наблюдаются, как видно на рис. VII. 1, разные линии С 1s в зависимости от окружения атомов углерода (электроотрицательности заместителей).
Хотя химический сдвиг коррелирует с состоянием окисления атома элемента в образце, однако иногда из-за малых значений сдвигов бывает трудно определить их характерные неперекрываю-щиеся интервалы для каждой степени окисления того или иного элемента в разных образцах. Например, в молекулах Mo(CO)4(PPh3)2, МоС12(СО)з(РРЬз)2, MoCl4(PPh3)2 и МоС13О(РРЬз)2, в которых формально степени окисления Мо равны, соответственно, (0), (II), (IV) и (V), энергии связи Мо З^-эле-ктронов найдены равными 230,9; 232,7; 235,3 и 235,5 эВ, т. е. по крайней мере степени окисления (IV) и (V) надежно по химическим сдвигам определить нельзя.
3.2. Аддитивность химических сдвигов
Химический сдвиг для атома данного элемента А при данной степени окисления зависит, как уже отмечалось, главным образом от его ближайшего окружения. Положительный сдвиг Д£Св увеличивается с ростом электроотрицательности заместителей В; при атоме А, поэтому при интерпретации фотоэлектронных спектров полезно использовать известные ряды, характеризующие относительную электроотрицательность атомов и групп.
Кроме того, общий химический сдвиг Д£АСВ приближенно можно считать аддитивной величиной, т. е. представить в виде суммы:
A5cab = 2^(A-Bz), (VII.5)
I
где Д£(А—В,) — вклад, обусловленный связью А—В,-. При всей ограниченности таких схем применение их в РЭС оправдывается возможностью экспериментальной оценки сдвига внутреннего уровня
159
Таблица VII.2. Инкременты ДЕ(С—Вг) для ДЕсв С 1s *
в. ДЕ, эВ вг ДЕ, эВ
-—CHnRm —0,1 —О- или ОМ 0,6
-CH„Fm 0,3m -OR 1,6
-CH„Clm 0,2m = 0 3,0
—CH„(OR)m 0,2m = S 1,1
—CH„(NH„)m 0,0 —nh2 0,8
—CH2(COOX) 0,6m —F 2,7
—cox 0,3 —Cl 1,5
—coox 2,3 —Br 1,1
* £свст=285 эВ и по определению при Bj—Н инкремент ДЕ—0,0; R — углеводородный радикал или Н; М — металл; X—CHS, CF3, OR, Cl.
атома в различных окружениях и анализа ряда находимых экспериментально закономерностей.
В табл. VII.2 приведены, например, инкременты АД для расчета AfcB С Is в алифатических соединениях относительно стандарта £,Свст = 285 эВ. Рассчитанные по схеме с этими инкрементами значения сдвигов удовлетворительно согласуются с экспериментальными. Так, для трифторацетона СР3СОСН3 рассчитанные сдвиги энергий связи С 1s атомов углерода (слева направо) равны 8,4; 3,8; 0,3, а экспериментально найденные — 7,8; 3,7; 0,4 эВ.
Аддитивные схемы с найденными экспериментально инкрементами предложены для расчета химических сдвигов линий в соединениях многих элементов, и надо сказать, что величины АД (А—В,) для разных А, но одинаковых В, коррелируют между собой (коэффициенты корреляции ~0,95).
3.3. Корреляция химических сдвигов с данными других методов
Поскольку химический сдвиг в РЭС или энергия связи внутренних электронов зависит от всего распределения электронной плотности в окружении данного атома, то естественно ожидать, что эти характеристики будут связаны с данными других методов, также характеризующими электронную структуру, в частности, с параметрами рассмотренных в предыдущих разделах спектров ЯМР, ЯКР и Мессбауэра (ЯГР).
Действительно, существует корреляция между химическими сдвигами 6 в ЯМР спектроскопии и АДСВ в РЭС, что можно пояснить следующим образом. Для ядра А бд определяется разностью констант Лод в стандарте и образце. Поскольку внутренние электронные оболочки практически не меняются от соединения к соединению, при расчете диамагнитного экранирования Абд их можно не учитывать. Влияние электронов атомов X,-, связанных с А, можно 160
(VII.6)
учесть введением создаваемого ими потенциала, обратно пропорционального расстоянию Я, между X/ и А. Тогда с учетом (VII.1) и (VII.2) можно прийти к выражению
дяА = Г — д£св + ^2д (Qi/Ri) L i
Это соотношение полезно для расчета А о а по экспериментальным значениям АЕСВ и отражает связь АЕСВ с од, хотя она может быть более сложной.
Найдены также корреляции, например, между энергиями связи Есв электронов и частотами спектров ЯКР, отражающими градиент электрического поля на ядре С1, в соединениях хлора. Хотя и не с очень высокими коэффициентами корреляции, обнаружены также линейные зависимости между изомерными сдвигами 6Е в мессбауэровских спектрах соединений Fe и Sn и химическими сдвигами АЕСв в РЭС.
По спектрам ЯКР и ЯГР (см. гл. IV и V) можно определять константу квадрупольного взаимодействия e2qQ и параметр асимметрии т] градиента электрического поля на ядре.
Поскольку симметрия градиента поля влияет на ширину и расщепление, то возможна корреляция между указанными параметрами и шириной рентгеноэлектронных линий, хотя эффект их уширения сравнительно невелик.
Любопытно, что химические сдвиги ДЕСВ в РЭС сравнимы с теп-лотами химических реакций и может быть установлена корреляция между ними. Так, например, энергия процесса типа
М ->М+ (Is) + е- . - (VII.7)
где М — молекула; М+ — ион, получающийся при удалении внутреннего ls-электрона атома (указано в скобках), равна величине Есв (ион).
Можно сделать предположение, что вследствие релаксации электронов в M+(ls) атом с удаленным электроном после заполнения вакансии 1s как бы становится атомом элемента с порядковым номером на единицу больше. Например, после процесса типа (VII.7)
(!$)+<?-
имеем переход
NH+ (1з)->ОН+
Заселенность орбиталей в NH3+ после релаксации электронов близка к заселенности орбиталей в ОНз+. Поэтому можно ожидать, что энергия связи электронов Есв N 1s в молекуле NH3 приблизительно равна энергии перехода NH3+->OH3+. С использованием такого подхода, известного как метод эквивалентных остовов, установлена связь между термодинамическими величинами (теплотами сооТвет-6—1377 161
ствующих реакций) и химическими сдвигами линий С 1s, В Is, N Is, О Is, Р 2р, S 2р.
Найдены также другие корреляции химических сдвигов Д£св, например, со сродством к протону и т. д.
4. АДСОРБЦИЯ, КАТАЛИЗ И ДРУГИЕ ОБЛАСТИ ПРИМЕНЕНИЯ
Методы рентгено- и фотоэлектронной спектроскопии в применении к явлениям адсорбции позволяют изучать и решать ряд проблем. С одной стороны, это идентификация продуктов на адсорбенте, исследование электронной структуры адсорбатов в зависимости от строения адсорбента и нахождение энергетических характеристик взаимодействия адсорбат — адсорбент. С другой стороны, это определение мест локализации адсорбированных молекул, поверхностной концентрации, степени покрытия поверхности, изучения кинетики адсорбции или каталитической реакции, выяснение механизмов адсорбции и каталитического действия металлов и сплавов и т. д.
При физической адсорбции или образовании хемосорбционной связи происходит сдвиг в спектрах фотоэлектронов. Изменение энергии связи электрона на i-м уровне можно представить в виде
Д£св I ~ £цгаз) ~ Е1 (аде), (VII.8)
где £/(газ) — энергия связи i-ro уровня молекулы в газе; £/<адс) — энергия того же уровня в адсорбированной молекуле. К величине £»(адс) нужно прибавить еще работу выхода электрона для образца, покрытого адсорбатом (<рд), которой раньше пренебрегали, но здесь £>(газ) определяется по отношению к уровню вакуума и этого делать нельзя. При физической адсорбции сдвиги обусловлены главным образом изменением межатомной энергии релаксации £рел при переходе молекулы из газообразного в конденсированное состояние. Таким образом, адсорбционный химический сдвиг, т. е. общее изменение энергии уровня при адсорбции, можно определить по формуле
Д£/ = Д£Свt "г Д£рел + Та- (VII.9)
Первое слагаемое обусловлено хемосорбционной связью. Энергии релаксации валентных и внутренних уровней адсорбата находят с помощью модельных представлений и расчетов. Чтобы избежать сложностей, связанных с определением величины <рд, обычно анализируют разностные спектры, вычитая из спектров адсорбат+адсорбент спектры адсорбента.
При адсорбции молекулы определенным образом ориентируются и фиксируются на поверхности адсорбента. Это приводит к тому, что интенсивность I фотоэлектронных спектров валентных оболочек зависит от ориентации МО относительно угла падения и вектора поляризации ионизирующего излучения, а также от направления вы-
162
хода фотоэлектрона. Угловую зависимость 1 в ФЭС используют как для идентификации валентных орбиталей, так и для определения способа координации и геометрии расположения молекул на субстрате. Показано, например, что молекулы СО при адсорбции на Ni координируются перпендикулярно поверхности металла, а при адсорбции на Pt угол зависит от кристаллографической грани: для плоскости (111) он прямой, а для (НО) почти на 30° меньше.
В катализе очень важно знание поверхностного состава катализатора и установление зависимостей его активности от состава, а состава — от температуры и других условий реакции. Эти проблемы успешно решаются рассматриваемыми здесь методами. Обычно поверхностный состав катализаторов существенно отличается от объемного даже до проведения каталитической реакции. Обогащение поверхности какими-то элементами, например переходными металлами, может сильно влиять на каталитическую активность. Так, при исследовании катализатора Ni—Zr—Н, в котором содержание Ni на поверхности зависит от времени термической обработки, найдена прямая зависимость его активности от роста поверхностного содержания Ni. Иногда, правда, максимум каталитической активности соответствует сравнительно узкому интервалу изменения концентрации какого-то компонента.
Большое значение для понимания механизмов катализа, эффектов промотирования и отравления катализаторов имеет сопоставление поверхностного состава до и после каталитической реакции. В последнее время исследования модельных и реальных катализаторов показали особенно высокую эффективность комплексного применения методов РЭС и ОЭС. Хотя в ОЭС при двукратной (и более) ионизации возрастает роль трудно учитываемых многоэлектронных процессов, этот метод даже при простых подходах к интерпретации результатов позволяет получить очень важные и обширные данные при выяснении причин промотирования и отравления катализаторов.
Так, например, в реакциях гидрирования — дегидрирования углеводородов на платиновых катализаторах, по данным исследования, отношения интенсивностей оже-пиков О (515 эВ) и Pt (237 эВ) обнаружен неожиданный промотирующий эффект хемосорбированного кислорода, тогда как окисление Pt приводило к дезактивации катализатора.
В качестве другого примера приведем результаты исследования с применением РЭС, ОЭС и сканирующей оже-микроскопии (СОМ) причин потерь активности катализатора — триоксида молибдена на подложке из оксида алюминия, который применяется для удаления серы из газов, выделяющихся в процессе дегазации каменного угля. Методами ЭСХА и ОЭС сначала получают обзорные прямые спектры фото- и оже-электронов образцов свежеприготовленного и отработанного катализатора. Приведенные на рис. VII.5, а полные спектры ЭСХА' показывают наличие в обоих образцах алюминия, 6* 163
Рис. VII.5. Спектры фотоэлектронов свежеприготовленного (/) и отработанного (II) катализатора (Мо03 на подложке А12О3):
а — полный спектр ЭСХА+оже; б — участок спектра ЭСХА высокого разрешения
кислорода, молибдена и углерода. Сигнал углерода сильнее в спектре отработанного катализатора, а сигналы молибдена — в спектре свежеприготовленного. На отработанном катализаторе имеется также сера.
На рис. VII.5, б показаны спектры высокого разрешения пиков Мо 3d. В спектре свежеприготовленного образца энергии связи электронов характерны для триоксида Мо03, а спектр отработанного катализатора указывает на наличие двух соединений молибдена Мо03 и MoS3. На основании этих данных можно было предположить два механизма отравления катализатора: либо образование по всей поверхности слоя углерода, либо формирование на активной поверхности МоО3 слоя MoS3.
С целью выявления, какой из механизмов имеет место, применили СОМ. Оказалось, что молибден распределен по подложке сравнительно равномерно. Сера локализована на небольших участках главным образом в местах повышенного содержания молибдена (рис. VII.6). Углерод распределен на поверхности равномерно (на рисунке не показано). Таким образом, отравление катализатора происходит в результате покрытия его углеродной пленкой, а не химической дезактивации.
Вполне очевидно, что методы рентгене- и фотоэлектронной спектроскопии с успехом могут применяться для изучения явлений коррозии, отжига и т. п., связанных с окислением металлов и сплавов, а также диффузии, сегрегации элементов, например, на изломах при термической обработке и т. д. Широко используются рассматриваемые методы в современной микроэлектронике, открывая уникальные возможности контроля качества н обнаружения неисправностей уст-164
роиств с атомно-молекулярными проводящими и непроводящими слоями, в физике твердого тела, изучении зонной структуры вещества, в изучении биологических объектов и других областях науки и техники.
0)
Рис. VII.6. Оже-изображения локализации серы (а) и молибдена (б) на поверхности отработанного катализатора
Рассмотренные в разделе методы характеризуются прежде всего высокой чувствительностью, специфичностью и большой широтой возможных применений, хотя и предназначены главным образом для исследования поверхности твердых тел и молекул в газовой фазе. В некоторых аспектах их можно сопоставлять с какими-либо другими физическими методами исследования, а в некоторых отношениях они обладают совершенно уникальными возможностями. Например, эмиссионный спектральный анализ может найти себе конкурента в методе РЭС при определении химических элементов. Фотоэлектронные спектры более специфичны, чем абсорбционные рентгеновские спектры и
УФ спектры, характеризуясь более узкими линиями и достаточно высоким разрешением. Многие данные, получаемые из фотоэлектронных спектров, хорошо коррелируют с данными других методов.
Уникальность методов рентгено- и фотоэлектронной спектроскопии — в возможности детального изучения тонких поверхностных слоев. При совместном использовании нескольких методов, включая применение оже-микрозонда, открывается возможность исключительно тонкого локального, а с ионным травлением — и профилированного послойного анализа твердых образцов с разрешением по поверхности 50—200 нм, а по глубине от 1 до нескольких нанометров. Уникальны также количественные энергетические характеристики, получаемые из фотоэлектронных спектров, и представляющие опорные данные для развития квантовой теории строения молекул и веществ.
Не менее важным является выход рассматриваемых неразрушающих методов анализа и контроля в различные отрасли народного хозяйства. В настоящее время они имеют огромное значение для таких технологических направлений, как катализ, металлургия,
165
борьба с коррозией, изготовление и эксплуатация электронных схем и т. д. Совершенствование приборной техники методов фотоэлектронной спектроскопии, развитие их теории и широкое внедрение в практику научных исследований и для решения технических проблем является одной из важнейших задач научно-технического прогресса.
Контрольные вопросы
Г л а в а VI
1. В чем основные отлнчня рентгеноэлектронных (ЭСХА) н фотоэлектронных (УФЭС) спектров?
2. Какова кинетическая энергия фотоэлектрона, испускаемого с С ls-орбн-талн (ЕСв=294 эВ) под действием излучения Ка А1 с энергией кванта Е1 == = 1487 эВ? Какая будет скорость у этого фотоэлектрона?
3. Какую энергию связи имеет электрон, если под действием излучения с длиной волны Л=58,4 нм Не (/ ) он испускается с кинетической энергией £кив= 11,75 эВ?
4. В чем суть релаксационного оже-процесса и рентгеновской флуоресценции?
5. Чем характеризуются рентгеновские спектры поглощения, каковы нх параметры?
6. Дайте определение химического сдвига в рентгено- и фотоэлектронной спектроскопии.
7. Какими параметрами характеризуются фотоэлектронные спектры молекул? Что такое потенциалы или энергии адиабатической и вертикальной ионизации?
8. От чего зависит интенсивность фотоэлектронных пиков?
9. Как определяется глубина выхода фотоэлектронов?
10. Каковы отличительные особенности аппаратуры для различных методов рентгеновской н фотоэлектронной (включая оже-) спектроскопии?
Глава VII
11. Охарактеризуйте аналитические возможности методов рентгено- и фотоэлектронной спектроскопии.
12. Как энергия связи электрона £Св на некотором уровне зависит от эффективных зарядов данного атома и окружающих атомов?
13. В чем суть основных подходов к объяснению химических сдвигов в фотоэлектронных спектрах?
14. Как коррелирует химический сдвиг в ЭСХА со степенью окисления атома элемента?
15. В чем состоит аддитивность химических сдвигов?
16. Что такое адсорбционный химический сдвиг, как он выражается?
17. Какие преимущества дает комплексное применение методов ЭСХА, ОЭС и СОМ при изучении поверхностей (в катализе, микроэлектронике и т. д.)?
МЕТОДЫ ИССЛЕДОВАНИЯ ОПТИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
Оптическая активность веществ является важной стереохимической характеристикой. Данные о вращении плоскости поляризации оптически активных веществ послужили основой для создания Вант-Гоффом и Ле-Белем в 1874 г. теории о пространственном тетраэдрическом строении метана и его производных. Позднее эти представления нашли применение для многих классов веществ, включая комплексные соединения, а также биологически активные вещества.
Простую поляриметрию заменили методы дисперсии оптического вращения (ДОВ) и кругового дихроизма (КД), которые позволили изучать более полно оптические характеристики оптически активных веществ как функции длины волны излучения. Современные методики ДОВ и КД позволяют определять абсолютную конфигурацию молекул (правда, на полуэмпирической основе), химическое строение, конформации и некоторые спектральные характеристики молекул.
Полное решение задачи об абсолютной конфигурации молекул дает метод аномального рассеяния рентгеновских лучей, который используется для этих целей с 1949 г.
Одним из важнейших свойств Молекул, особенно^природных соединении, является свойство хиральности, или оптической активности. Оно обусловлено существованием зеркальноподобных H3Oi меров — энантиомеров.
В отличие от геометрических изомеров энантиомеры эквивалентны по своим физическим и химическим свойствам. У них одинаковые температуры
плавления и кипения, давление пара, плотность, показатель преломления, для неполяризованного света — колебательный и электронный спектры, одинаковая реакционная способность к ахиральным реагентам.
Однако энантиомеры по-разному вращают плоскость поляризации линейно поляризованного света. Существенно отличаются также реакции энантиомеров с хиральными реагентами или реакции, катализируемые хиральными реагентами.
Основным условием хиральности молекул является отсутствие центра симметрии, плоскости симметрии, зеркально-поворотной оси симметрии Sn в молекуле.
Наглядной моделью хиральных молекул может служить правая или левая спирали.
Спиральная модель молекул позволяет качественно объяснить многие свойства хиральности и определить номенклатуру хиральных молекул.
Для исследования оптически активных веществ широко используются три метода:
1) дисперсия оптического вращения (ДОВ);
2) круговой дихроизм (КД);
3) аномальное рассеяние рентгеновских лучей.
Первые два метода позволяют лишь относительно устанавливать Конфигурацию энантиомеров. Только рентгеноструктурный анализ с использованием аномального рассеяния рентгеновских лучей решает вопрос об абсолютной конфигурации хиральных молекул.
Учитывая то, что метод кругового дихроизма является одним из спектральных методов, он дает возможность изучать также электронное строение молекул.
Рассматривая более широко исследования оптически активных веществ, следует указать на хроматографический метод и метод ЯМР, которые здесь не излагаются. В первом методе используют хиральные неподвижные фазы в качестве адсорбента. Во втором методе создают условия для различий в химических сдвигах и интенсивностях отдельных сигналов энантиотропных групп за счет их взаимодействий с хиралЬным растворителем или хиральным сдвигающим реагентом (см. гл. II).
168
ГЛАВА VIII
ДИСПЕРСИЯ ОПТИЧЕСКОГО ВРАЩЕНИЯ
1. ЛИНЕЙНО ПОЛЯРИЗОВАННОЕ ИЗЛУЧЕНИЕ.
КРУГОВАЯ ПОЛЯРИЗАЦИЯ СВЕТА
Классические представления об электромагнитном излучении в форме монохроматической волны основаны на том, что электрическое поле и магнитная индукция В волны перпендикулярны друг другу и перпендикулярны направлению распространения излучения (рис. VIII.1). Если проекция осциллирующего вектора электрического поля на плоскость, перпендикулярную направлению распространения луча, представляет линию, то такой луч называют линейно поляризованным (иногда называют плоскополяризованным). В том случае, когда такие проекции ориентированы по всем направлениям, луч света неполяризован.
Электромагнитное излучение распространяется в форме волны и характеризуется амплитудами |<Уо| или |Во|, круговой частотой w и скоростью v\ например, для волны в положительном направлении оси г поле имеет вид (рис. VIII.1):
g,x=»ecos<»^ - , (VIII. 1)
= ^°cos _Л.) =-L^oCOS , где с —скорость света в вакууме; е —диэлектрическая проницаемость; цмагнитная проницаемость среды; со —2nv и v —частота волны.
Скорость распространения электромагнитной волны в среде можно выразить через скорость излучения в вакууме с и показатель преломления П‘,
v = c/n. (VIII,2)
Бодну электрического поля можно записать в виде
cos <0 - -у-) . (VII 1.3)
Величину nz называют оптической длиной пути луча. Уравнение для волны иногда выражают через волновой вектор к, длина которого равна 2л/Х. Так как c=Xv и co==2nv, то аргумент косинуса
/ пг \ „ ( nz \
ш < — J = 2л, t — -— I =5= — k'z,
\ с ) \ Av )
где й'==2лД' иД'=Х//г.
169
X
Рис. VII 1.2. Сложение и разложение линейно поляризованных лучей
Рис. VIII.1. Схема распространения линейно поляризованного света вдоль осн г.
Показано периодическое изменение в одной фазе электрических и магнитных полей с одинаковой амплитудой
Тогда имеем:
$х = %0 cos (at - k’z). (VI11.4)
Уравнения (VIII.3) и (VIII.4) описывают волну, движущуюся вправо, так как через промежуток времени Д/>0 все точки косинусоиды <?’x=const передвинуты вправо на Az>0. Электрическая и магнитная волны распространяются с одинаковыми фазой, периодом и скоростью. Линейно поляризованный луч можно представить в виде суммы двух линейно поляризованных волн в перпендикулярных плоскостях, которые отличаются только амплитудами (рис. VIII.2). Иными словами, сложение двух линейно поляризованных волн, отличающихся только амплитудами, дает линейно поляризованный луч.
Рассмотрим другой пример. Распространяются два луча <SX и (S’у с одинаковой амплитудой, но один луч опережает другой на четверть волны, т. е. оптическая длина пути увеличена Х/4. Тогда если
I nz
<£х = $0 cos <«> / —-
\ с
то, используя соотношения (o=2jtv и с—Xv, получим I nz + Х/4 \ Г /
= $0 cos о>11 —-------1 = й0 cos ш 11
= $0 sin <о 11 — —). (VIII.5)
\ c j
Два перпендикулярных линейно поляризованных луча Sx и Sy с опережающей разницей в фазе л/2 для Sy образуют луч с круговой поляризацией по правой спирали (рис. VIII.3). Если линейно поляризованный луч Sx опережает Sy на четверть волны, то образуется луч с круговой поляризацией по левой спирали (рис. VIII.3).
170
Рис. VIII.3. Образование луча с круговой поляризацией влево (а) и вправо (б) путем сложения двух линейно поляризованных лучей с разницей фаз ±л/2
Таким образом, сложение двух перпендикулярных линейно поляризованных лучей с разностью фаз в л/2 приводит к лучам в форме левой или правой спирали соответственно. Если смотреть навстречу направлению распространения луча, то в левой спирали вектор электрического поля вращается по кругу по часовой стрелке и называется правым лучом 8Г, а для правой спирали — по кругу против часовой стрелки (рис. VIII.4) и называется левым лучом St.
В векторной форме, используя единичные векторы i вдоль х и j вдоль у, можно записать напряженность электрического поля для пр авого луча-
(nz О ( . ( пг \)
t —--1} 1 — sin ш / —-)} j
с } ( \ с }\
(VIII.6)
171
и для левого луча:
Рис. VIH.4. Схема движения векторов ST (а) и 8/ (б):
Г ( nz %i — “ 11 — -~
! пг go sin <» / — — И.
, \ C ))
(VIII.7)
Сложение уравнений Sr и Si дает линейно поляризованный луч, так как этот луч имеет только одну проекцию на ось х:
% = %r+8i=2
I / пг
0 (cos <о / —----
I \ с .
(VIII.8)
проекция со стороны наблюдателя, смотрящего на источник излучения
лучу, конец вектора
Если амплитуды лучей с правой и левой круговой поляризацией различны, то на проекции в плоскости, перпендикулярной огибающей опишет эллипс. Такой луч имеет
эллиптическую поляризацию. Наибольшая ось эллипса будет суммой амплитуд лучей с круговой поляризацией, а мень-
шая ось — разностью этих амплитуд.
Идея о разложении линейно-поляризованного света на два луча с левой и правой круговой поляризацией была предложена О. Френелем для феноменологического объяснения явления оптической активности веществ. Лучи с разной круговой поляризацией имеют раз-
ную скорость распространения в оптически активных веществах, следовательно, разные показатели преломления (двулучепреломление), т. е. vr^=vi и nr^=ni.
Если луч линейно поляризованного света вошел в оптически активное вещество в точке z и прошел в этой среде расстояние d см вдоль z, то соответствующие ему две компоненты с левой и правой круговой поляризацией будут иметь следующий вид (предполагается, что поглощения нет):
nz + nid с
nz 4- n\d \*|
~7~ ’
и
(VIII.9)
’ nz + nrd / nz + nrd
t---------l — ( Sin a 11 —
k Cl) I \
с
(VIII.10)
После выхода из оптически активного вещества в результате сложения двух лучей с левой и правой круговой поляризацией получаем снова линейно поляризованный луч. Действительно сложение дает
g = + g; = 2$0 COS и
Пг + П1
> 2с
Пг~п
a cos ш------
/К 2с
d 1 +
+1 sin a
Пг—Щ
d I j ? = 4-
(VIII.11)
172
Рис. VIH.5. Схема изменения плоскости поляризации луча при прохождении через слой толщиной d оптически активного вещества
Поскольку за пределами оптически активного вещества показатели преломления двух лучей равны, т. е. nr=ni=n, то в результате имеем
/ n(z ±d) \
g = 2g0 cos <о И - - i. (VIII.12)
\ c /
Однако положение плоскости поляризации линейно поляризованного луча изменилось, поскольку перед выходом из вещества вектор напряженности электрического поля 8 имел компоненты 8Х по оси х и 8У— по оси у (рис. VIII.5). Их отношение определяет угол поворота плоскости поляризации. Принято считать за положительное направление вращение по часовой стрелке, если смотреть вдоль оси z на источник излучения (рис. VIII.5). Поэтому необходимо ввести знак минус для тангенса или изменить знак разности пг — п(:
П! — П-
tg а = _ = tg ш> г„ d (VI 11.13)
©х
Наконец, можно выделить выражение для угла поворота плоскости поляризации:
(VIII.14)
Это уравнение Френеля, которое показывает, что положительное вращение наблюдается в случае ni>nr.
Следовательно, угол поворота плоскости поляризации света определяется разностью значений показателей преломления щ—пг.
173
Поскольку а зависит от длины пути луча и концентрации вещества в исследуемом растворе, то принято рассчитывать удельное вращение в градусах для толщины слоя 10 см или 1 дм при заданных температуре t (°C) и длине волны % (нм):
(VIII.I5)-* а С/
где d — толщина слоя, дм; С — концентрация, выраженная в граммах оптически активного вещества, содержащегося в 100 мл раствора.
На основе удельного вращения вычисляют молекулярное (молярное) вращение [град/(дм-моль-мл)]:
t И
(vin.ie)
где М — молярная масса вещества.
Здесь важно отметить, что вращение плоскости поляризации оптически активного вещества очень чувствительно к разности ni—пг, которая имеет порядок ~ 10-6. Столь небольшие различия приводят к вращению на угол более 10° для .D-линии натрия. Следует отметить, что в обычных рефрактометрических исследованиях точность определения показателя преломления не превышает 10-4. Удельное вращение [а] Л естественно, различно для разных веществ и составляет десятки и даже сотни градусов. Оно зависит от %, а также от применяемого растворителя. Молекулярное вращение достигает величин порядка десятков тысяч градусов, что, конечно, не имеет определенного тригонометрического смысла, но важно как физико-химическая характеристика оптически активного вещества в данных условиях.
2. КВАНТОВО-МЕХАНИЧЕСКОЕ РАССМОТРЕНИЕ ОПТИЧЕСКОЙ АКТИВНОСТИ
И СПИРАЛЬНАЯ МОДЕЛЬ МОЛЕКУЛЫ
Феноменологические представления о различии показателей преломления для лучей с правой и левой круговой поляризацией не дают возможности установления более глубоких связей явления оптического вращения и молекулярных свойств. К сожалению, в теории оптической активности, как и в теориях ряда других методов, не достаточно полно решена прямая задача и поэтому ограничено решение обратной задачи метода. Прямая задача состоит в определении экспериментально измеряемого угла вращения а на основе молекулярных свойств. Взаимодействие света с веществом связано с характером волновых функций электронного состояния и их изменениями в электромагнитном поле волны. Однако волновые функции для электронных состояний многоатомной молекулы из-за 174
трудностей задачи не известны с достаточной точностью. Поэтому так же, как и для электронных спектров, можно получить лишь частичное решение, вывести общие условия наблюдения явления вращения плоскости поляризации и выявить параметры, определяющие вращение.
Ввиду крайней громоздкости вывода основного уравнения, которое дано в специальной литературе (например, см. У. Козман: «Введение в квантовую химию». М., ИЛ, 1960), проследим лишь главные этапы вывода и физический смысл получающихся величин.
Распространение света в веществе с точки зрения классической теории связано с осцилляцией электронов в атомах и молекулах, которую вызывает падающий свет. Электромагнитная волна света, как указывалось, представляет систему двух взаимно перпендикулярных полей: электрического и магнитного. Обычно для задачи распространения света в веществе рассматривают только электрическую компоненту электромагнитной волны, так как сила Лоренца, действующая на электрон со стороны магнитного поля, равна е [vxB], где v — скорость электрона, В — магнитная индукция. Эта сила мала из-за малой величины v/c (<gQ=cB0).
Однако при анализе задачи распространения света в оптически активном веществе необходимо учитывать и влияние магнитного поля (рис. VIII.6). Используя теорию возмущений, можно получить выражение для электрического дипольного момента це1-, индуцированного полями <£ и В электромагнитной волны в молекуле, которая находилась в состоянии Ф,-, в виде суммы двух слагаемых:
(VIII.17)
где цег(ё’) и gei(B) —электрические дипольные моменты, иидуци-рованные полями 8 и В падающей волны (рис. VIII.6, а) ;в = —.
Компонента це>(В) обусловлена изменением во времени вектора В, которое вызывает возникновение электрического поля 8 (В), параллельного или антипараллельного В в зависимости от свойств молекул (рис. VIII.6, б и рис. VIII.6, в). Это означает, что це,-(В) перпендикулярно Hei(g’).
Возникновение переменного электрического дипольного момента в молекуле рег(В) под влиянием переменного магнитного поля может быть качественно объяснено на основе спиральной модели молекулы, которая наиболее удобна для описания оптической активности. Такая модель подсказана экспериментами по распространению линейно поляризованного излучения в микроволновом диапазоне (Х~3 см) на отрезках левых и правых спиралей из медной проволоки диаметром 6...7 мм и длиной ~10 мм. В этих экспериментах доказано вращение плоскости поляризации совокупностями произвольно ориентированных спиралей одного типа.
175
С помощью классической электронной теории получены полуко-личественные результаты оптического вращения для спиральных моделей молекул. Качественно появление электрического дипольного момента це1(В) в молекуле видно из рис. (VIII.6, б) (VIII.6, в). Молекула показана в виде фрагмента спирали, ось которой направлена вдоль вектора В.
Через виток правой спирали (рис. VIII. 6,6) проходят силовые линии магнитного поля В световой волны, перпендикулярные линиям электрического поля %. Положительное изменение во времени В, т. е. В<0, вызывает индуцированное поле $инд такого направления, что его магнитное поле ВИНд противоположно вызвавшему его появление полю В. Это изменение ВИНд вызывает движение положительного заряда вдоль спирали, которое приводит к появлению электрического дипольного момента це»(В). В случае правой спирали це/(В) направлен влево, а для левой спирали — вправо (рис. VIII.6, в). Направления дипольных моментов даны согласно физическому определению вектора электрического дипольного момента рт отрицательного полюса к положительному.
Индуцированные дипольные моменты цеДй’) и це/(^) в молекуле изменяются во времени с такой же частотой, как и падающая волна. Это приводит к вынужденному излучению, т. е. к распространению света в среде. Однако имеются следующие особенности такого излучения, Векторы $ и В изменяются в одной фазе. Но вектор В отстает по фазе на 90° от падающей волны (при дифференцировании косинус переходит в синус со знаком минус). Колеблющиеся диполи Це/($) и (В) в веществе, представляющем слои молекул, создают плоский фронт рассеянного излучения, фаза которого отстает, в свою очередь, от фазы колеблющихся диполей также на 90°. Это означает, что волна, рассеянная диполем це/(В), отстает от фазы подающей волны на 180° и от Це/(8’) на 90°. Но интенсивность волны, рассеянной диполем це/(#), значительно меньше, чем интенсивность падающей волны, которая и определяет фазу волны в направлении поля 8. Модуляция волны в направлении 8 волной, рассеянной диполем рои(8), влияет на изменение скорости прохождения суммарной волны — уменьшает ее в соответствии с уравнением v = cln. Таким образом, для волны электрического поля падающего линейно поляризованного света при прохождении в оптически активном веществе должно наблюдаться вращение плоскости поляризации, так как кроме компоненты электрического поля в плоскости падающей волны появляется перпендикулярная компонента.
Квантово-механическое рассмотрение возникновения индуцированных моментов це/($) и це/(В), как уже говорилось, основано на теории возмущений. Величина це/($) молекулы в состоянии i определяется поляризуемостью молекулы а/ для vt^vs/:
176
V el (Ю = a(g.
Величина at определяется по уравнению
_ 2 V Чк! । Рг» । 2
~ 3/1 Т ^1 ~ '-2 ’
(VIII.18)
(VIII.19)
где = J 4fft*|ie4fidT — электрический дипольный момент перехода; Ч’ь, 4f>— волновые функции состояний k н I; це= — оператор электрического дипольного момента; Г; — радиус-
Еъ Е i *
вектор заряда в/; мЛ/ =----------частота перехода; у — частота
. h.
падающего света.
Электрический дипольный момент, индуцированный магнитным полем, можно получить из общего выражения
Ре, (В) = У т; (B)peV; (В) dT, (VIII.20)
где гГ,(В) является волновой функцией состояния i молекулы, которая находится в магнитном поле В и имеет дополнительную энер. гию —цтВ (цт индуцированный магнитный момент молекулы). Проведение преобразований уравнения для це>(В) дает в хорошем приближении выражение
Ре,(В) = -3,В. (VIII.21)
Для произвольно ориентированных молекул в жидкости или газе величина р, (Кл'М3-В-1) определяется по уравнению
В, = — У —5-^------' • (VIII.22)
В уравнение (VIII.22) входит новая физическая величина Rih, называемая вращательной силой перехода i-^k и выражаемая уравнением
Rik = Im(<Wi\MWk> <^ft|rf/>), (VIII.23)
где Im —мнимая часть скобки < ... > означают интегрирование [см. уравнение (VIII.20]; це —оператор электрического дипольного момента, как в уравнении (VIII.20) (представляет действительную величину); —оператор магнитного дипольного момента (представляет мнимую величину):
ей V , , eh V Г. ( д
2т 2т V7 ду}
/ д й \ , I д д
+ j х» -—- — z -— +k m ~Z—г —*1~л— \ 1 dzj dxj j \ 7 dxj 7 dyj
177
6)
Леба я спирмь
Рис. VIH.6. Схема возникновения индуцированного электрического дипольного момента РеЛ’
а — под влиянием & и В; б—-для фрагмента правой спирали; в — для фрагмента левой спирали; стрелкой отмечено движение заряда по спирали
Величина Rik определяет правила отбора и непосредственно связана с электронными переходами. Ее единица измерения соответствует произведению единиц измерений моментов, т. е. [це] = Кл-М И [рт]=А-М2 = =Кл-м2-с-1, поэтому [/?;*]= = Кл2.м3-с-‘.
Пропорциональность ре,(В) и В можно рассматривать как эмпирическую зависимость в соответствии с рис. VIII.6, а величину Р—как вращательную поляризуемость.
В то же время магнитный дипольный момент Hmz(S’) индуцируется переменным электрическим полем в направлении S. Но электрическое поле излучения этого магнитного диполя <8s(pm/(8’)) направлено параллельно це(В), совпадает по фазе и эквивалентно по амплитуде. В результате эти поля суммируются, так что имеем уравнение
S s = £,(МВ)) +
~ s (f*« (в)).
При этом напомним, что МВ) И,; pm(g)d.gs; ^(1М^))Н £,;ЗММВ))И W, и 8SA_ g.
Рассеянная волна gs увеличивается в —- -
раз за счет поля
Лоренца (и — показатель преломления). Учитывая, что BQ=golc, амплитуда рассеянной волны равна
dN п2 +2 { ш \2
& os — ; И I о
*о \ с J
178
где ео — электрическая постоянная; d — толщина слоя; N — число молекул в единице объема; $о — амплитуда падающей волны.
Вывод этого уравнения дается, например, в кн. У. Козмана «Введение в квантовую химию» М., ИЛ, 1960.
Угол вращения плоскости поляризации а определяется отношением ffasl&a. По величине он мал и выражается следующим уравнением (в градусах):
180 % Os 180 dN п2д-2 ( ш
а =---- arctg----- =-------------;— Р I —
Л Stj Л е0 3 \ С
Величина р в уравнении (VIII.22) выражена в виде функции от частоты падающего излучения v. Поэтому сразу можем получить зависимость а от v:
_ 240 dN n2 + 2 ( ч \2 у Rlk ~ h *о 3 \ с / к Al -
(VIII.24)
Однако в экспериментах по вращению плоскости поляризации света часто используют зависимость угла вращения а от длины волны света %. Используя соотношение Xv = c, имеем для а:
а =
240
dN
*0
и2 4-2 У AjRik Ч 19 I2
" к ----------к!
(VIII.25)
Чтобы перейти к выражению молекулярного вращения в соответствии с уравнениями (VIII.15) и (VIII.16), необходимо в уравнениях (VIII.24) и (VIII.25) выразить N (число молекул в 1 см3) через концентрацию С' (число граммов оптически активного вещества в 100 см3), подставляя
N = Nk ,
А ЛН00
где Na — постоянная Авогадро; М — молярная масса.
Тогда, представляя а в виде произведения a=a°dN, находим
[al a°dN а°
Ш] = Л4 -Н- = 100Л1 ——--------= N, ——
1 J 100 /С'100 А 10
где 1=0,Id, так как I выражено в дециметрах.
Окончательно имеем для зависимости молекулярного вращения от v:
24 ЛГА п2 + 2 / \ \2 у Rih h 3 к с ) ’
(VIII.26a)
и для зависимости от X:
[Л1 ](= -Д- — • (VIII.256)
1 n he1 гд 3 X2-xL-
Таким образом, параметрами молекулярного вращения являются две молекулярные характеристики: Л/»- — длйна волны (или vs; частота) электронного перехода и — вращательная сила этого перехода.
Уравнение для молекулярного вращения объясняет основные условия и характер явления оптической активности.
Первый основной вывод из уравнений (VIII.26a) и (VIII.266) состоит в том, что вращение связано с электронным переходом. Вращательная сила не равна нулю, когда электронный переход возможен, как говорят, по механизму электрического дипольного перехода и по механизму магнитного дипольного перехода. Первый переход можно представить как линейное перемещение электрического заряда, а второй — как движение заряда по окружности, т. е. полное движение заряда совершается по спирали.
Некоторые электронные переходы в молекулах имеют нулевое значение магнитного дипольного момента перехода. Поэтому даже интенсивные электронные переходы могут не проявляться в оптической активности. Но даже слабые по интенсивности электронные переходы, но имеющие значительный магнитный дипольный момент перехода, обладают оптической активностью. Роль вращательной силы в оптической активности аналогична вероятности или величине квадрата модуля момента электрического дипольного перехода, называемого иногда также силой диполя, которому пропорционален коэффициент поглощения в электронном спектре. Вращательная сила определяется скалярным произведением
Rki = (Veiklbnlil) = IM*1 |tW/| cos fl, (VIII.27)
где juieifc и pmki — векторы электрического и магнитного дипольных моментов перехода; 0 — угол между ними. Поэтому для оптического вращения важно соотношение направлений и pmki.
Электронные переходы обладают особенным свойством, которое выражается так называемым правилом сумм. Это правило состоит в том,что
2 = (VIII.28)
» I * >
Оно доказывается с использованием свойства полноты ортонор-мированной системы волновых функций V. Через эти функции может быть выражена б-функция Дирака
8(г-г') = 2 ^(г')^А(г). »-о
где г и г' — обобщенные переменные функций.
180
Суммирование в уравнении (VIII.28) можно поменять местами с интегрированием. Имеем
<Wml^>} = S Г <(r)^A(«-)drJ ^(r')^Z(r')dr' =
= П ^(г)|?г|Гт [2 (г) ^(г')1 (г') dr dr'
I k J
-П(r>i*«£n8 <r' ~ r> (r')dr df' = J ^z(ОмЛ (Г) dr.
Учитывая, что це и цт— эрмитовы операторы, последний интеграл есть действительная величина. Это и доказывает равенство (VIII.28).
Физический смысл уравнения (VIII.28) состоит в том, что в молекуле электронные переходы имеют разные знаки Rik. Следовательно, для определенной геометрической конфигурации знак вращательной силы Rik будет зависеть от типа электронного перехода.
3. СИММЕТРИЯ МОЛЕКУЛ И ОПТИЧЕСКАЯ АКТИВНОСТЬ
Качественное решение вопроса, будет ли Rik равна нулю или нет, получается на основе симметрии молекулы. Сначала чисто эмпирически Л. Пастером было установлено, что молекулы, являющиеся зеркальными отображениями друг друга, оптически активны
и вращают плоскость поляризации одинаково, но в противоположных направлениях. В 1905 г. В. Фойгт сформулировал более общее правило: оптически активная молекула не должна иметь зеркальноповоротную ось Sn и, в частности, плоскость симметрии и центр симметрии i = S-,. Оптически активные молекулы могут иметь, симметрию Сп и Dn. Это правило полностью подтверждается рас
смотрением свойств симметрии операторов и Цт.
Требования симметрии на основе теории представлений групп
формулируются следующим образом:
вращательная сила электронного перехода Rik как скалярное
произведение векторов электрического щчд и ментов переходов не равна нулю только в том случае, когда векторы ^eik и цты принадлежат полносимметричному представлению, т. е. A, A', At, Ag или Alg.
В качестве примеров оптически активных молекул могут служить молекулы с асимметрическим атомом углерода, произ- . водные метана CXZYW, например СНзСН(ОН)СгН5. Для таких молекул можно представить спиральную модель, если рассматривать положение заместителей на
магнитного pcmki мо-
модель асимметрической молекулы
CH3CH(OH)C2HS
181
поверхности цилиндра (рис. VIII.7). Производные дифенила
имеющие неплоское строение, т. е. <р^=0, являют
ся примером оптически активных молекул без асимметрического атома углерода. Частным случаем этих производных является клас-
no2 соон
сический объект 6,6'-динитродифеновая кислота ,
ноос o2n
в которой фенильные кольца свернуты на угол <р—70° и отсутствует центр инверсии. Однако ось Съ сохраняется. В молекуле гекса-
гелицена ) \ \ отсутствуют какие-либо заместители, но
\ / ( >
V / \__f
из-за стерических препятствий молекула неплоская и имеет форму фрагмента спирали. Среди комплексных соединений, обладающих оптической активностью, выделяются симметричные (симметрия £>з) трис-диаминовые (трнс-хелатные) комплексы переходных металлов типа [Co-en3]3+, [Re-en3]3+, [Со (С2О4) 3]3— и т. п. [еп этилендиамин— C2H4(NH2)2]. имеющие октаэдрическую Конфигурацию связей лигандов и центрального атома (рис. VIII.8, а).
Каждую из этих моделей молекул можно рассматривать с точки зрения принадлежности к спирали, например правой — энантиомера I и левой — энантиомера II по отношению к оси С2. Эти комплексы имеют также ось третьего порядка. Проекция вдоль оси С3 имеет иной вид для энантиомеров I и II и характер спирали изменяется на левый для I и правый для II (рис. VIII.8, б).
Комплексы переходных металлов могут быть оптически активными, если диссимметрию (отсутствие симметрии) молекул создают следующие факторы:
1) расположение хелатных колец относительно центрального атома;
2) расположение монодентатных лигандов относительно центрального атома;
3) конформация хелатных колец;
4) координация оптически активных лигандов;
5) координация асимметрического донорного атома.
182
I
Правая спираль
I левая спираль
Рис. VIII.8. Схема октаэдрических трис-хелатиых комплексов симметрии £>з: дугой показан бидентатный лиганд (например, еп— -NHjCHjCHjNHj); представление о виде спирали дает движение вдоль дуги лиганда в направлении от ближайшего атома 1 к дальнему атому 2
Важно отметить, что неплоское строение хелатного кольца может вызвать диссимметрию. Донорный атом в свободном лиганде обычно имеет симметричное окружение, но его присоединение к атому металла может создать асимметрию.
Величину вращательной силы 7?,^ приближенно можно оценить исходя из того, что дипольный электрический момент перехода =3,3-10-30 Кл-м (1£>), а магнитный дипольный момент равен одному магнетону Бора, т. е. |цт(л| =0,9- 1О-23 А-м2. Это означает, что
Ящх 3,0-10-53 Кл2.Мз.с-». (VIII.29)
Это значение получено для гексагелицена и является, по-видимому, максимальным, поскольку в других молекулах сомножители fieik и Umih могут принимать меньшие значения, чем в молекуле гексагелицена. В более общем случае вращательная сила составляет лишь малую часть (несколько процентов) от указанного значения. По-
183
Рис. V1II.9. Молекулярные орбитали карбонильной группы в симметричном и асимметричном окружении
этому часто используют приведенную вращательную силу (/?/*] = - « 0,33- |055/?м.
Существуют два типа хромофоров, определяющих вращательную способность молекул. Если спектр электронного поглощения обусловлен переходами, относящимися ко всей оптически активной молекуле, то вращательная сила обусловлена диссимметрией (хиральностью) всей молекулы, как хромоформа, например гексагелицен, неплрские диены, производные дифенила н т. п. Однако для таких симметричных хромофоров, как карбонильная группа ^С=О, асимметрическое (диссимметрическое) окружение обусловливает появление вращательной силы электронного перехода, например, локализованного нд С=О. В симметричном окружении молекулярные орбитали ft* и /I симметричны и ортогональны (рнс. VIII.9, а, б), поэтому электрический дипольный переход запрещен, так как орбцталц ортогональны * (рис. VIII.9, б). Асимметрическое окружение деформи
* См. также гд, ХШ в учебнике «Физические методы исследования в химии. Структурные методы и оптическая епектроскопчя».
184
рует п- и л-орбитали. Появляется возможность спирального «движения» электрона при переходе п-+-л* (рис. VIII.9, в), т. е. появляется вращательная сила, которая, правда, значительно меньше, чем тогда, когда молекула является сама диссимметричным хромофором, например гексагелицен.
В общем случае возмущения симметричного хромофора внешним полем в соответствии с теорией групп существует правило: статическое возмущение, которое индуцирует хиральную активность в симметричном хромофоре, должно быть антисимметричным при инверсии или отражении относительно какой-либо векторной координаты в симметричном хромофоре.
4. Кривые дов. эффект коттона
Рис. VIII.10. Гладкие положительная (?) и отрицательная (2) кривые дисперсии оптического вращения (ДОВ)
Уравнение (VIII.24) показывает, что при изменении длины волны будет наблюдаться изменение угла вращения плоскости поляризации: дисперсия оптического вращения (ДОВ). Если измерения проводят при длине волны то наблюдают плавные кривые ДОВ: положительные для правого вращения (по часовой стрелке), отрицательные — для левого вращения. Знак вращения определяется знаком вращательной силы Rik (рис. VIII.10).
Поскольку в общем случае не известны волновые функции молекул, отсутствует возможность точного расчета оптического вращения. Это обстоятельство объясняет то, что в методе ДОВ широко распространены и используются полуэмпирические и эмпирические приемы анализа экспериментального материала.
Плавные кривые приближенно описываются эмпирическим уравнением Друде для одной или двух составляющих:
= <VIII-30) где X/— длина волны ближайшего максимума поглощения, нм; коэффициент Kj пропорционален вращательной силе /-го перехода.
Уравнение Друде и более точные уравнения (VIII.26, а) и (VIII.26, б) не описывают вращение плоскости поляризации света в области поглощения, так как согласно этим уравнениям при v-+Vkt или А-*-?.*, должно наблюдаться бесконечное увеличение [М]. Для невырожденной изолированной полосы поглощения эксперимен-
185
Рис. VIII.11. Кривые ДОВ с положительным (а) и с отрицательным (б) эффектом Коттона
тальные кривые ДОВ (рис. VIII.И) имеют аномальный характер изменения [Л1] двух типов — положительный и отрицательный.
Для положительных кривых ДОВ (рис. VIII.11, а) с уменьшением 1 увеличивается угол вращения, кривая переходит через пик, пересекает ось длин волн в точке ХЛ(-, достигает впадины и снова увеличивается угол вращения при уменьшении X. Аномальный характер изменения ДОВ имеет место на участке от пика до впадины, так как функция [Л1] уменьшается с уменьшением X.
Для отрицательных кривых ДОВ (рис. VIII.11, б) наблюдается обратная зависимость.
S-образные кривые ДОВ в области максимума полосы поглощения называются эффектом Коттона. Кроме знака кривой ДОВ параметрами эффекта Коттона являются длины волн для пика и впадины. Половина их разности называется полушириной эффекта Коттона Ь, т. е.
b = —- [X. (пик) — X (впадииа)].
Для характеристики эффекта Коттона вводится также определение молярной амплитуды А, равной разности величин [Л4]х‘ для пика и впадины, т. е.
1) для положительных кривых ДОВ
^ = [Л1]1(Вик)-[Л1К(вПад). л>°;
2) для отрицательных кривых ДОВ
Л = [Л4]х(впад) ~ [ЛЛл(пик)> А <0.
Обычно амплитуды А составляют величины порядка от сотен до десятков тысяч градусов. Поэтому часто используют приведенные величины амплитуд, уменьшенные в 100 раз.
186
Более полное квантово-механическое рассмотрение процесса взаимодействия излучения с веществом в области поглощения приводит к качественному согласованию экспериментальных и теоретических кривых ДОВ. При этом учитываются процессы поглощения, вынужденного испускания и спонтанного излучения. В результате в уравнении (VIII.22) для вращательной поляризуемости 0 в знаменателе появляется комплексное число lysiv, где yw — положительная постоянная (2у*,= 1/т=ЛА(-, т — время жизни возбужденного состояния, Aki — коэффициенты Эйнштейна спонтанного испускания *). Предполагается, что y*,<Cv*(.
Таким образом, более точное выражение для р имеет следующий вид:
р = —(VI1I.3I) ЗлЛ
Подстановка действительной части величины р в уравнение для молекулярного вращения [Л1] как функции v дает
. 24 «2+2/ v \2 у
h »о 3 (с) й ( 2ki—«2)2 + Yi?2
(VIII.32)
Для v—vki это уравнение приводит к [Л1] =0. При положительных Hik знак [Л1] определяется разностью (v2*z—
—v2).
На основе эффекта Коттона можно оценить вращательную силу электронного перехода Rik по параметру К/ уравнения (VIII.30) или приближенно из произведения:
0,36-Ю-56
Л(й = -Aiiibik •
^z
Знак Atk определяет знак Rik-
При наложении нескольких эффектов Коттона получают более сложный характер кривых ДОВ (рис. VIII.12).
Необходимо отметить, что характер кривых ДОВ подобен зависимости показателя преломления от длины волны (рис. VIII.13). Для лучей с правой и левой круговой поляризацией показатели преломления пг и щ изменяются таким образом, что их разность «г—пг воспроизводит эффект Коттона. Кривые n;(v) и пг(у) пересекаются при v=vftz- Аномальный характер зависимости показателей преломления пАу), пг(у) и угла вращения a(v), пропорционального пАу)—пг(у), также обусловлен спонтанным излучением при поглощении.
* См. также гл. XIII в учебнике сФизические методы исследования в химии. Структурные методы и оптическая спектроскопия:».
187
о
При конкретных исследованиях молекулярного вращения в области нескольких электронных переходов наблюдаются сложные кривые ДОВ. Для детального анализа результатов эксперимента необходимо выделение отдельных кривых ДОВ для каждого электронного перехода, т. е. выделение парциальных кривых. Большую помощь в таком разложении может оказать спектр кругового дихроизма.
5. ПРИНЦИПИАЛЬНАЯ СХЕМА ЭКСПЕРИМЕНТА
Блок-схема спектрополяриметра для изучения ДОВ показана на рис. (VIII.14). Источником света служит ксеноновая дуговая лампа высокого давления. Она обеспечивает высокоинтенсивный поток света в широком и непрерывном спектральном диапазоне от 185 до 600 нм. Большая интенсивность источника необходима, чтобы луч света эффективно прошел через последующие оптические среды. После монохроматора луч света с выделенной длиной волны направляется на кварцевый поляризатор, формирующий линейно поляризованный пучок света.
Поскольку схема измерения состоит в определении минимального, практически нулевого по интенсивности потока света после анализатора, используют модуляцию плоскости поляризации в 188
Рис. VIII.14. Принципиальная схема эксперимента в методе ДОВ:
1 — ксеноновая лампа; 2 — монохроматор; 3— поляризатор; 4 —кювета; 5 — анализатор; 6 — детектор
ячейке Фарадея или простое качание поляризатора на несколько градусов около среднего значения. Ячейка Фарадея представляет кварцевый стержень внутри соленоида (см. гл. X). Переменное магнитное поле соленоида приводит- к колебаниям плоскости поляризации света.
После модуляции плоскости поляризации луч света проходит через кювету с образцом, длина которой обычно составляет от 1 до 20 мм. Далее луч света попадает на анализатор и детектор-фотоумножитель. Электронно-механическая система обеспечивает автоматический поворот анализатора в скрещенное с поляризатором положение.
Точность измерения угла поворота плоскости поляризации оптически активным веществом составляет ~ 0,003°. Особенность эксперимента по измерению ДОВ состоит в том, что при прохождении линейно поляризованного света через оптически активное вещество имеет место не только поворот плоскости поляризации. Вышедший из кюветы луч приобретает эллиптичность вследствие различного поглощения лучей с круговой поляризацией вправо и влево. Модуляция плоскости поляризации необходима также, чтобы учесть этот эффект.
Для проведения эксперимента по ДОВ необходимо использовать стандарты с известными молекулярными вращениями и исключить или учесть оптическую активность материала кюветы.
Метод ДОВ обладает высокой чувствительностью, поскольку для получения кривых ДОВ достаточно около 10~5... 10-6 г вещества.
ГЛАВА IX
КРУГОВОЙ ДИХРОИЗМ
1. ПОГЛОЩЕНИЕ ЛУЧЕЙ С РАЗЛИЧНОЙ КРУГОВОЙ ПОЛЯРИЗАЦИЕЙ
Круговой дихроизм — явление различного поглощения двух лучей с правой и левой круговой поляризацией.
Феноменологическая теория распространения света через ве-
189
щество с поглощением основана на комплексном представлении показателя преломления п:
n = n — ix, (IX.1)
где п — действительный показатель преломления; х — коэффициент поглощения.
Уравнение для волны в направлении z будет иметь вид
Л zn. 1 az* , (. z \
g = goa V с > с е >. (IX.2)
Из уравнения (IX.2) следует, что амплитуда прошедшего через вещество луча уменьшается пропорционально ехр^—— zzj.Поскольку интенсивность луча пропорциональна квадрату амплитуды, получаем закон Бугера — Ламберта — Бера для z—d,-.
г г Г 2WX 1
Z = /Oexp--------. (IX.3)
L с J
Выразим теперь поглощение или оптическую плотность А:
. . /о 4 л
А = Ig~ = ~xd Ige. (IX.4)
1 к
Если введем молярный десятичный коэффициент экстинкции [л/(моль-см)]
A 4nxlge
где С — концентрация (моль/л), то получаем уравнение для I в виде
I = /о-1О-,м. (IX.6)
Для лучей с правой и левой круговой поляризацией молярные коэффициенты экстинкции обозначаются 8г и ei соответственно. Тогда имеем
/Г = /О.Ю“‘^ и Zz = /O.io-,'C</. (IX.7)
В оптически активной среде Zr=£zi. Поэтому при прохождении линейно поляризованного света через оптически активное вещество имеет место не только поворот плоскости поляризации света, но и круговой дихроизм. Поэтому прошедший свет обладает эллиптической поляризацией. Это новая важная характеристика луча света. Ее физический смысл состоит в следующем.
Амплитуды правого и левого лучей, прошедших через оптически активное вещество, будут различны и выражаться уравне-190
ниями, сходными с (VIII.9) и (VIII.10):
%г = '$ог Г-fcos <0 ft — j — (sin o> ft — — — I j j I; (IX. 8)
= g0Z[(cos o> ft ——Пг nid •)] i 4- (sin aft — Пг + П;^—jl . (IX.9) Ц \ C /) t \ C J) J
В уравнениях (IX.8) и (IX.9) амплитуды So, и Soi включают коэффициенты экстинкции соответственно:
Г a I Г 2л 7
= $0 ехР----dy’r == $0 ехР —d*r
|_ С J 1*1
и
Г 2л 1
®ol ~ ехР —-i dKl I •
После выхода лучей из вещества их сумма дает луч с эллиптической поляризацией. Если Soi>^or, то вектор суммарной волны вращается влево (рис. IX.1). Поскольку частота вращения векторов Sr и Si одинакова, но направление различно, максимальная амплитуда суммарного луча будет равна Sor-\-<£oi, а минимальная амплитуда Sor—Sr>i-
Эллиптичностью называют величину 0, равную отношению малой амплитуды к большой-.
%ог — &ы
{£ 0 =-------------- .
®ог + ®01
(IX. 10)
Угол 0 очень мал по величине. Поэтому tg0«0 и ( 2л .. ехр^—— dr.' ( 2л ехр
— %01 %ог +
+ ехр
. (IX.11)
Сокращая на ехр
,получаем
Г 2л 1
1 — ехр—-~d(*i — xr) я
------—------------ («-«,).
1 + ехр ———d (xz — хг) • A i
(IX. 12)
Так как xj—xr«t;l (редко больше 10-6... 10“7), то, разлагая в ряд, упростим выражение для 0:
9 = -—(xz-«r). (IX. 13)
Это уравнение очень похоже на уравнение Френеля (VIII.14). Молярная величина эллиптичности имеет следующее
191
Рис. IX. 1. Схема возникновения эллиптичности:
б —линейно поляризованный луч вдоль оси х; б —линейно поляризованный луч после прохождения через оптически активное вещество (без кругового дихроизма); в — эллиптически поляризованный луч вдоль оси а — эллиптически поляризованный луч после прохождения через оптически активное вещество (с круговым дихроизмом)
выражение:
[8] = у-
(IX. 14)
где I — длина кюветы, дм (/=0,ld); С' — концентрация, выраженная числом граммов вещества в 100 мл раствора.
Существует связь между молярной эллиптичностью и молярным коэффициентом экстинкции. Используя равенство С—10 C'jM, моль/л, можно получить:
18 000
4л
Де = 3300 Де.
(IX. 15)
Для измерения эллиптичности необходимо определять отношения интенсивности луча в двух перпендикулярных направлениях. 192
При этом различие в интенсивности очень большое, ч~о вызывает практические трудности измерений. Величина Де непосредственно связана с основным определением кругового дихроизма и общими характеристиками спектров поглощения.
Коэффициенты е/ и ег так же, как и средний молярный коэффициент по-
глощения е= , имеют сходную по форме зависимость от частоты. В разумном приближении зависимость Ae(v) описывается гауссовой формой:
Рис. IX.2. Гауссова зависимость (в определенном приближении) молярного коэффициента экстинкции для лучей с круговой поляризацией е/ и ег и их разности Де от частоты поглощаемого света (е/> >6г)
Де = Де0 ехр
(IX-16)
где А характеризует полуширину полосы; Аео — дихроичное поглощение в максимуме полосы.
В связи с нелинейностью связи v
и А, зависимость Ае(^) будет несколь-
ко отличаться от Ae(v). Некоторые исследователи предпочитают гауссову форму для Де (А,).
Если в молекуле имеется несколько оптически активных переходов, то соблюдается правило аддитивности
[*)]=£[()]; И ДЕ = £Д£/,
(IX.17)
где [0], и Ае, — парциальные молекулярные величины для /-го перехода.
Наглядную зависимость Ae(v) = ez(v)—e,(v) показывает рис. IX.2. Если каждая из кривых er(v) и ez(v) имеет гауссову форму и отличается существенно только высотой пика, то их разность будет также иметь гауссову форму.
Общая картина прохождения линейно поляризованного света через вещество может быть представлена как средняя для правого и левого лучей в соответствии с уравнением
Z = Zoe
4к .
~~Mld е А
4к
(IX. 18)
= ——
2
Преобразуем уравнение (IX. 18) с использованием среднего коэффициента Хср= (хН-хг)/2:
7—1377 193
Д» / *>•
I _ V V (г>“ Г 4л 1 2л
= Z0 exp ——— rfxcp ch——rf(xz— xf).
I A J A
(IX. 19)
В большинстве
ch — d(xz —xf)pt; I A J
случаев круговой дихроизм мал, т. е.
1. Поэтому в обычных спектрофотометриче
ских исследованиях коэффициент экстинкции равен среднему коэффициенту Хер. Для изучения кругового дихроизма необходимы специальные исследования поглощения лучей с определенной круговой поляризацией.
2. СВЯЗЬ КРУГОВОГО ДИХРОИЗМА И ВРАЩАТЕЛЬНОЙ СИЛЫ ПЕРЕХОДА
Ранее было показано, что феноменологическая теория распространения света позволяет при введении комплексного показателя преломления рассматривать два явления: вращение плоскости поляризации и кругового дихроизма. Такой же подход возможен и на более глубокой физической основе. Комплексная величина показателя преломления означает, что поляризуемость молекулы является также комплексной величиной.
Поскольку вращение плоскости поляризации обусловлено на молекулярном уровне вращательной поляризуемостью р, также можно подойти к обобщению этого явления, представляя величину р как комплексную:
р = р'+/р".
Тогда и угол вращения а является комплексной величиной:
а = а' + 1а".
Проекции вектора электрического поля $ на оси х и у для угла поворота плоскости поляризации а (см. рис. VIII.6) будут выражены общими соотношениями (фазовые множители zn/c опущены с целью упрощения записи уравнений):
= g0 exp (i<at) cos a и
%y = —$0expsin a.
Теперь перейдем к новой системе координат g и т], которые являются главными осями для эллипса, описываемого электрическим вектором (см. рис. IX. 1). Так как угол поворота плоскости поляризации есть действительная величина, то имеем для поворота почасовой стрелке
cos a' — %у sin a'
194
и
~ % х sin а' + g’ycos а'.
Подставим <$х и ёу, выраженные в комплексной форме, = ^oexPVwO [cos a cos а' + sin а sin а'] = = <£0 exp (iat) cos (а — а') = exp (iat) cos (la")
И
= goexp (Zwi) [cos a sin а' — sin a cos а'] — <£0 exp (Zwi) sin (a' — a) = = $0 exP (*“O sin (—ia") = —$o exp (W) sin (ia").
Используем известные соотношения
cos (ia") = ch а" И sin (ia") — i sh a".
Тогда получаем
= $o exP (^“0 ch a" и
— — z$0 exp (icoi) sh a".
Действительная часть этих выражений для проекций электрического поля выражается уравнениями
7?е {^} = $о cos at ch a" = (1£й ch a") cos at
И
He {$>;} = 8*п “i sh a" = (<£0 sh a") sin at.
Поскольку оси g и г) являются главными осями эллипса, можно выразить эллиптичность 0 в виде
Re {$>;} sh a"
tg fl =------------= —--------
He{^} ch a"
-th a".
Для малых значений 0 имеем 0=—а". Отрицательный знак соответствует вращению вектора $ по часовой стрелке тогда, когда поглощение луча с левой круговой поляризацией больше, чем с правой, т. е. ez>er, но &i<.8r.
Таким образом, мнимая часть а дает эллиптичность
„ „ 180 dN nt + 2 (а \2
а" = —0 =------------------- —— 0" —
л «о 3 \ с j
(IX.20)
При этом учтено, что вклады от изменений магнитной индукции и электрического поля во времени равны и суммируются. Величину р" можно получить иа основе квантовой теории (уравнение VIII.31), что дает:
Г Т + Yjwv2]. (IX.21)
ft
7*
195
Из уравнений (IX.20) и (IX.21) получаем выражение для зависимости эллиптичности от вращательной силы
' “ ттг т? J!Lr~ ? -,!)2 + т5?!1 (1Х -22)
или для молярной эллиптичности, используя уравнение (VIII.26, а),
f9]*= ПГ 2 -v2)2 + viz-2] • (IX.23)
Чтобы найти связь вращательной силы /?,* с интегральными характеристиками экспериментально получаемых данных Ае как функции v, проведем интегрирование дроби [0] k/v:
°° 8
f 24 Na г v2
\ [в]1Л<Ь=-----— 7 RikVki 1 -т-5--й----i----dv. (IX.24)
JLJV ЛС2 е0 J С*/"-2) +V»/-2
При интегрировании уравнения (IX.23) обычно пренебрегают зависимостью п от V, так как в основном этот член обусловлен растворителем, который не поглощает в области поглощения исследуемого вещества.
Интеграл в уравнении (IX.24) преобразуется следующим образом прн замене v2=/:
00 00
/• у2 1 f* _
J (^_V2)2 + Y2;v2 dV = Т j y7/(t2 ~ 2T*‘7 + + =
— C 1/7 I Г« L ° f ^к1 2 \ f . 4 1
“ 2 \ 2 -^ + v*zjd<- 2yw •
При этом можно использовать таблицы интегралов, в которых найдем, что . '''. г
Г V х dx __________л______ " '-!
J ах2 + 2Ьх~с ~ У2а(уас~Ь) ' ' ?
Таким образом, имеем
f j 24 Na л 12л Na vi г,
I ---d'' = 7T-----X Rm = —r-0------7, Rik-
J i he2 en 2 he2 en T-
0 k s < . >.
Выразим теперь вращательную силу для одного перехода:
he2 8о (* . zfY 941
^ = "7^7 I ~(IX.25) 12л Na .) v
о
196
Учитывая равенство (IX. 15), получим
„ „ № (
Rlk = 265 —— e0 I лМа J о
Дг(ч) ------dv.
(IX.26)
Если используем гауссову форму для Ae(v) согласно уравнению (IX.16), то найдем очень простую зависимость от параметров дихроичной кривой Ae(v):
Л«2
Rik = 265 —— е0 лЛГд
л_= , >25_ (0_51 2 v*/ ’
(IX.27)
Таким образом, из экспериментальных измерений полосы дихроичного поглощения можно найти вращательную силу электронного перехода, являющуюся характеристикой электронного перехода молекулы.
3. СХЕМА ЭКСПЕРИМЕНТА. ФОРМИРОВАНИЕ ЛУЧЕЙ С КРУГОВОЙ ПОЛЯРИЗАЦИЕЙ
Проблема измерения кругового дихроизма, т. е. измерение Ае = е/—бг, состоит в том, что эксперимент проводится в области полосы поглощения, а отношение Де/е (е — коэффициент экстинкции)— очень малая величина. Для выполнения эксперимента необходимо формирование лучей с круговой поляризацией для заданной длины волны.
В настоящее время эти проблемы решены различными способами. Повышены интенсивность источников излучения и чувствительность детекторов. По существу, эти части установок для кругового дихроизма могут быть одинаковыми с таковыми в спектрополяриметрах для измерений дисперсии оптического вращения. В связи с тем, что неизвестно такое дихроичное вещество, для которого один из коэффициентов поглощения ez или ег был бы очень мал, принципиальным является узел прибора для формирования лучей с круговой поляризацией. Для этого используется так называемая четвертьволновая пластинка.
Если линейно поляризованный луч попадает на двулучепре-ломляющую четвертьволновую пластинку, то он разлагается на два луча во взаимно перпендикулярных направлениях (рис. IX.3). Оптические пути лучей с перпендикулярной поляризацией, определяемые произведением толщины пластинки d на показатель преломления для каждого из лучей, например, параллельного оси п и, соответственно, перпендикулярного п± этой оси пластинки, должны отличаться на величину ±Х/4, т. е.
л К d — n^d — ±Х/4 = dbn. (IX.28)
197
X
Рис. IX.4. Проекция траектории луча в направлении распространения z после прохождения через четвертьволновую пластинку:
а — круговая поляризация вправо (т./'о V2 \
y®0<:os“/; % у уЙоз1Пш/):
б — круговая поляризация влево (i/o lz2 \
Sx=7®0cos“/:
Рис. IX.3. Разложение падающего линейно поляризованного луча а с помощью четвертьволновой пластинки:
ось х параллельна оптической оси кристалла. Электрическое поле волны расположено под углом g к оси х; выбрано J5—п/4
В этом случае два выходящих линейно поляризованных луча имеют разность фаз л/2, что и обусловливает формирование луча с круговой поляризацией.
В качестве четвертьволновой пластинки служит пластинка кристалла, вырезанная параллельно оптической оси (рис. IX.3). Если луч света распространяется вдоль оси z и падает на пластинку под углом р к оси х, то можно принять, что линейно поляризованный луч представляет результат сложения линейно поляризованных лучей по осям х и у (левая система координат). Амплитуды лучей вдоль х и у будут равны соответственно
й’ох = $оcos ₽ к sin ₽•
(IX. 29)
Тогда волны вдоль х и у, прошедшие сквозь пластинку с показателями преломления пх и пу, соответственно, будут иметь следующий вид:
{ fijcd \
= g0 cos р cos <л / —---] (IX.30)
\ с /
и
/ пud \ 7 fi^d \ "|
Ъу = %о sin ₽ COS <0 Н - -у- 1 = sin ₽ cos М------------— 1 _ <р j .
198
Разность фаз двух лучей определяется величиной
“ (пу — пх) d 2nv 2л
ф =-------------- =----knd = —— knd .
с с \
(IX.31)
Если And=X/4, то ф=л/2. Для р=л/4 амплитуды 80х и Soy становятся равными друг другу. При этих условиях уравнения для лучей существенно упрощаются и, если время отсчитывать с момента выхода лучей из пластинки, получаем два луча:
/2
= —— й’о cos
и
/2 (IX. 32)
^у = -^- go sin at.
Как видели ранее, сумма двух плоскополяризованных лучей с одинаковыми амплитудами и разностью фаз в л/2 дает луч с круговой поляризацией. Изменение знака р на —л/4 изменяет знак у амплитуды Sy и, следовательно, направление круговой поляризации (рис. IX.4). Измерение поглощения при различных ориентациях падающего линейно поляризованного луча позволяет определять Д$, т. е. круговой дихроизм.
Теперь необходимо получить лучи с круговой поляризацией при различных длинах волн. Это означает, что для заданных величин X должна быть соответствующая толщина d с учетом зависимости Дп(Х):
<*(*) = (IX. ЭЗ)
4Дп (X)
Чтобы сохранить разность фаз, можно увеличить толщину в 2k-}-+ 1 раз (k— целое число).
Возникающие технические проблемы решаются несколькими способами. Так, используется система тонких призматических пластин из кварца, которые могут скользить друг относительно друга, чтобы изменялась толщина d в соответствии с уравнением (IX.33). Другой способ получения четвертьволновой пластинки с t/(X) состоит в использовании электрооптического эффекта Покельса.
Если из одноосного кристалла вырезана пластинка перпендикулярно его оси (рис. IX.5), то электрическое поле, приложенное к плоскостям пластинки, вызывает появление двулучепреломления— различия показателей преломления вдоль осей х и у (пх и Пу). Это различие не зависит от толщины пластинки, а определяется лишь длиной волны света и величиной приложенного напряжения. Обычно используется кристалл дигидрофосфата аммония (ДФА). В качестве прозрачного электрода применяют тонкий слой глицерина, прикрываемого пластинкой из плавленого кварца.
199
Рис. IX.5. Принципиальная схема измерения КД:
/ — ксеноновая лампа; 2 — монохроматор; 3 — поляризатор; 4 — четвертьволновая пластинка; 5 — кювета; 6 — детектор
В отсутствие электрического поля пластинка ДФА изотропна. Синусоидальное поле изменяет поляризацию падающего линейно поляризованного света, например, от круговой вправо до круговой влево.
Схема прибора для измерения кругового дихроизма показана на рис. IX.5. Источником света служит ксеноновая лампа высокого давления. Далее луч света проходит монохроматор и поляризатор. После формирования луча с круговой поляризацией в блоке четвертьволновой пластинки он пропускается через кювету с веществом. Поглощение регистрируется фотоумножителем и далее записывающей системой. Для каждого значения v (или X) в интервале X от 185 до 600 нм получают Л, Л, /0 и, соответственно, е;, бг и е или Ae(v) = e;(v)—er(v) и е. Обычно измеряют Ae(v). При этом
Де=ДЛ/Cd, (IX.34)
где АА=А[—Аг — разность оптических плотностей для лучей I и г.
Чувствительность прибора для измерения кругового дихроизма должна быть значительно выше, чем обычного спектрофотометра, так как разности оптических плотностей очень малы (АА<^/1ср) • Также высокие требования предъявляются к материалам кювет. Широко используется кварц, обработанный специальным термическим способом. Недопустимы механические напряжения кварцевых стекол.
В настоящее время существуют комбинированные приборы, предназначенные для съемки кривых ДОВ и КД. При соответствующей настройке можно получать спектры УФ.
Экспериментально наблюдаемые кривые КД для отдельной полосы имеют действительно гауссову форму (см. рис. VIII.2), положение максимума которых соответствует электронному переходу.
200
4. ВЗАИМОСВЯЗЬ ДИСПЕРСИИ ОПТИЧЕСКОГО ВРАЩЕНИЯ И КРУГОВОГО ДИХРОИЗМА
Уравнения (VIII.24) и (IX.27), например, показывают, что как явление ДОВ, так и КД зависят от вращательной силы электронного перехода, которая определяет знаки и величину обоих эффектов. Это означает, что между этими явлениями имеется определенная связь. Ее можно установить, если использовать аналогию явлений дисперсии оптического вращения и дисперсии света, а также кругового дихроизма и поглощения, о которых говорилось ранее.
В свою очередь доказано, что явления дисперсии и поглощения света взаимообусловлены. Физический смысл этой связи состоит в том, что поглощение света в области электронных переходов, а затем передача поглощенной энергии лучу в периодическом процессе вызывает изменение скорости прохождения света через вещество, следовательно, изменение показателя преломления. Частично поглощенная энергия претерпевает диссипацию, что приводит к обычному поглощению. При этом интенсивность луча уменьшается.
Количественно взаимосвязь явлений ДОВ и КД выражена соотношениями типа уравнений Крамерса — Кронига:
оо
2 С* X'
[/И,Н^)1=— [6пСЛ1., dX' (IX.35)
Л J А2 — (А У
О
И
оо
[0« (X)] = f [Mik (X-)] (V)2 d\', (IX.36)
ЛА J A2 — (A у
0
где [Mi*]—молярное вращение (см. VIII.16); [9xft]—молярная эллиптичность (см. VIII.15) и (VIII.16).
Первоначально подобные уравнения были введены для действительной и мнимой частей показателя преломления или диэлектрической проницаемости. У. Моффит и А. Московиц показали, что эти соотношения, называемые дисперсионным и, могут быть применены для ДОВ и КД в форме уравнений (IX.35) и (IX.36).
В соответствии с этими уравнениями функция [MXft(X)] является нечетной функцией X, а функция [0»*(Х)] — четной. Зная одну из функций, можно вычислить другую. Эти уравнения полезны для разложения спектров ДОВ при использовании данных КД. Из этих уравнений, в частности, получено, что
A-fe = 4O28Aeo;ft, (IX. 37)
где Aik — амплитуда эффекта Коттона (см. гл. XIV).
Кроме того, дисперсионные соотношения позволяют приближенно оценить вращательную силу из параметров кривой ДОВ:
201
Rlk = 0,36-10-5» , (IX.38)
где Aik— амплитуда; bit — полуширина эффекта Коттона; v,* — частота электронного перехода.
ГЛАВА X
ПРИМЕНЕНИЕ СПЕКТРОПОЛЯРИМЕТРИИ В ХИМИИ
1. ОБЩИЕ ВОПРОСЫ ИСПОЛЬЗОВАНИЯ МЕТОДОВ дов и кд
Методы ДОВ и КД практически используются совместно. Задачи, решаемые обоими методами, являются в основном общими и состоят в определении:
1) абсолютных конфигураций органических и комплексных соединений;
2) химического строения, т. е. взаимного расположения атомов и групп атомов в молекуле;
3) конформации молекулы или ее фрагментов, например конформации металлоцикла в комплексном соединении;
4) характеристик электронных переходов;
5) характеристик взаимодействия хиральных молекул с молекулами растворителя, специфическими реагентами и т. п.;
6) качественного и количественного состава веществ (аналитические приложения).
Сравнения возможностей ДОВ и КД показывает, что принципиально они одинаковые. Однако имеются и некоторые отличия.
Для ДОВ характерно проявление вклада вращения вдали от электронных переходов. Поэтому ДОВ представляет более эффективный метод обнаружения слабой оптической активности химически чистого вещества по сравнению с КД- Это обусловливает предпочтительное использование ДОВ в аналитических целях. Из-за «фоновых» эффектов кривые ДОВ приобретают «индивидуальность» и служат удобным инструментом для идентификации. Спектры ДОВ находят применение при изучении кинетики процессов. По аналогии с изобестическими точками в спектрах поглощения наблюдают изовращательиые точки на нулевой линии ДОВ в процессе, например, рацемизации (общие точки пересечения кривых удельного вращения [а]?; рис. Х.1).
Круговой дихроизм близок по природе к электронным спектрам поглощения. В отличие от ДОВ кривая КД имеет относительно более простую, гауссову форму максимума (положительного или отрицательного). Это облегчает определение длины волны поглощения и при перекрывании нескольких максимумов облегчает разделение сложной кривой на отдельные составляющие. Поэтому для спектроскопических целей предпочитают использовать метод КД, особенно для комплексных соединений. Полосы
202
кривых КД обладают меньшей шириной, чем полосы электронных спектров, в связи с тем, что некоторые колебания, вносящие значительный вклад в электронно-колебательные спектры, не эффективны для явления оптической активности.
Разрешение спектров УФ, КД и ДОВ можно расположить в таком порядке: КД>ДОВ>УФ. Спектры УФ обладают меньшим разрешением из-за значительного числа перекрывающихся полос электронноколебательных переходов.
Кривые КД более удобны для расчета силы вращения электронного перехода, которая количественно характеризует оптическую активность.
Образно сравнивают ДОВ с инфракрасной спектроскопией, а КД с электронными спектрами поглощения.
2. ЭМПИРИЧЕСКИЕ ЗАКОНОМЕРНОСТИ.
ПРАВИЛА БРЮСТЕРА И ОКТАНТОВ
Поскольку в настоящее время отсутствуют возможности строгого расчета волно-
[а] -ю'3
_______।______। ।
600 500 000 300
Л, нм
Рис. Х.1. Кривые ДОВ для (—)soo-fFe рЬепзр+, наблюдаемые в процессе рацемизации:
— начальная кривая;
--------кривая, полученная через 20 мин; —--------
кривая, полученная через 50 мни. Видны точки нзо-вращення
вых функций молекул, использование мето-
дов ДОВ и КД основано в основном на эмпирических закономер-
ностях и правилах.
Наиболее общими правилами являются следующие.
1. Правило смещения Фрейдеиберга. Если два аналогично построенных диссимметричных соединения претерпевают одинаковые химические изменения, вызывающие сдвиг оптического вращения в одном и том же направлении, то оба соединения, по всей вероятности, имеют одну и ту же конфигурацию.
2. Правило оптической суперпозиции Ваит-Гоффа. Асимметрические атомы углерода вносят независимые вклады в общее молекулярное вращение, равное сумме этих вкладов.
3. Вицинальное правило Чугаева — Куиа — Фрейдеиберга Знак оптического вращения определяется ближайшим окружением асимметрического центра. Заместители, находящиеся на значительном расстоянии, могут изменить величину, но не знак вращения. В гомологическом ряду молекулярное вращение стремится к предельному значению. Так, для карбинолов
сн3
он (X-H,Br,coNH2 и др.) наблюдается правое вращение.
сн2сн2х
203
Но введение заместителя с новой полосой поглощения, расположенного вблизи асимметрического центра, может изменить знак вращения, как, например, в карбинолах, показывающих левое вращение:
СН3 CHj
Н—------ОН и н------j-ОН
I I
CH-CH-CN CHjCN
4. Правила Брюстера. На основе спиральной модели Дж. Брюстером предложено правило, связывающее относительную величину поляризуемости и знак вращения при 7?-линии натрия для определенной абсолютной конфигурации. Это правило формулируется следующим образом:
Асимметрический атом X в абсолютной конфигурации, изображенной схематически с разными заместителями А, В, С и D, яв-в I
А------с
I I D
ляется правовращающим, если поляризуемости заместителей уменьшаются в порядке A>B>C>D. Например, в молекуле иодопропионовой кислоты имеет место уменьшение рефракций, соон
I------сн3
। । н
пропорциональных величинам поляризуемостей, в следующем порядке: I (13,954) >СООН (7,226)>С//3 (5,719) >Н (1,028), который определяет правое вращение.
Используя таблицы рефракций атомов и групп атомов и экспериментальные данные о знаке вращения для 77-линии натрия, возможно предсказать абсолютную конфигурацию молекул.
Для сложных молекул, в которых оптическая активность возникает вследствие определенных конформационных особенностей, изменяющих симметрию молекулы, необходимо учитывать форму звеньев цепи, соответствующих правой и левой спирали. Простейшим фрагментом такой цепи являются три звена, представляющих связи между атомами и расположенных в гош-конформации. Если движение вдоль звеньев от его ближайшего конца к дальнему совершается по часовой стрелке, то такая цепь будет правоспиральной и правовращающей. Движению против часовой стрелки соответствуют левая спираль и левое вращение. Для трех звеньев 4 1 правая спираль (правое вращение) и левая
204
спираль (левое вращение) показаны проекциями Ньюмена I и II соответственно:
1
I
Для молекулы с тетраэдрическими центральными атомами 2 и 3 следует учитывать шесть гош-конфигураций, создающих относительные гош-положения заместителей:
1
Молярное вращение представляет сумму вкладов от всех шести конформаций, пропорциональных их рефракциям. Брюстером также используется уравнение для количественной оценки молярного вращения гош-конформаций на основе модели спирали.
Следует еще раз отметить, что указанные правила относятся к оптической активности вдали от полосы поглощения.
5. Правило октантов. Одно из наиболее важных проявлений оптической активности связано с внутренне симметричным хромофором, например С = О, который находится в асимметричном окружении. Большой экспериментальный материал для производных циклогексанона позволил сформулировать правило октантов, нашедшее очень широкое применение и развитие для других классов соединений. Оно связывает знак эффекта Коттона с положениями замещающих групп по отношению к карбонильной группе. На рис. Х.2 показано расположение четырех октантов, задаваемых плоскостями А, В и С, пересекающихся в точке на связи С=О. Плоскость А является плоскостью симметрии цикла. В плоскости В находится карбонильная группа с двумя атомами углерода цикла С2 и С6. Плоскость С перпендикулярна плоскостям А и В, пересекает связь С = О и выделяет четыре октанта, называемых задними. Проекция со стороны карбонильной группы на задние октанты позволяет удобно представить влияние заместителей на знак вращения. Так, аксиальные и экваториальные заместители у атома 3 приводят к отрицательному эффекту Коттона, а у атома 5— к положительному. Экваториальные заместители у атомов 2 и 6 не влияют на оптическую активность. Аксиальные заместители у атома 2 вызывают вращение вправо (положительное), а у атома 6 — влево (отрицательное). Заместители у атома 4 не влияют на оптическую активность.
205
Рис. Х.2. Правило октантов для производных циклогексанона:
а — стереопроекции плоскостей А, В, С выделяют октанты относительно связи С—О; б —• вид спереди на задние октанты (акс — аксиальный; экв —экваториальный)
Если предположить аддитивность влияния заместителей, то вращательная сила может быть представлена алгебраической суммой вкладов заместителей.
Правила октантов применяют также к циклобутанонам, циклопентанонам и циклогептанонам.
Для ряда классов соединений найдены специальные правила, позволяющие связать стереохимию и знак эффекта Коттона (ок-тантное правило для хиральных моноолефинов, секторное правило для бензоатов и лактонов, правило квадрантов для ароматического хромофора с асимметрическим центром в бензильном положении и т. п.).
3. ПРИМЕРЫ ИСПОЛЬЗОВАНИЯ
ДОВ и кд
3.1. Определение абсолютной конфигурации
Проблема биосинтеза непосредственно связана с оптической активностью соедине-
ний, обусловленной хиральностью соединений. Поэтому определение абсолютной конфигурации представляет очень важную задачу. Для решения такой задачи неоценимую роль играет правило октантов, которое позволяет обойтись без экспериментального изучения родственных соединений (сравнительный метод).
Так, в молекуле (+)-8-метилгидринданона-5, вращающей вправо, имеются два асимметрических центра С (8) и С (9) (рис. Х.З).
206
Рис. Х.З. Модели абсолютных конфигуранцй грйнс-8-метилгидрии-даиоиа-5
Доказано специальными методами траяс-сочленение колец.
Однако такому сочленению соответствуют две конфигурации молекулы (рис. Х.З, а, б), которые отличаются лишь положением пятичленного цикла по отношению к плоскости симметрии циклогексанового фрагмента. В конфигурации (рис. Х.З, а) пятичленный цикл расположен слева от этой плоскости, а в конфигурации (рис. Х.З, б) — справа. В соответствии с октантными диаграммами только конфигурация а дает положительное вращение. Следовательно, реализуется абсолютная конфигурация а.
3.2. Доказательство конформационной подвижности.
Влияние полярности растворителя
При хлорировании (-(-)-З-метилциклогексаноиа
сн
получено моиохлорпроизводиое 2-хлор-5-метйлцнКЛогексанона:
С1
Строение доказано химическими методами.
207
Однако при конформации кресла возможны два оптических изомера, существующих в двух конформациях, попарно переходящих друг в друга при инверсии цикла:
ИК и УФ спектры этого соединения в октановом растворе согласуются с аксиальным расположением связи С—С1, т. е. с изомерами 1Б и ПА. Отрицательный эффект Коттона доказывает конформацию 1Б. Для изомера ПА в соответствии с правилом октантов следовало ожидать положительный эффект Коттона. Поскольку конфомеры IA и 1Б обладают различной полярностью, растворители могут изменить равновесие между ними. Так, при переходе от неполярного растворителя — октана к полярному растворителю — метиловому спирту наблюдается изменение знака эффекта Коттона. Это подтверждает конформационную подвижность циклогексановых систем.
3.3. Исследование комплексных соединений
Транс- и цис-изомерия комплексов. Простейшим примером использования спектрополяриметрии в стереохимии координационных соединений является определение цис- и транс-изомеров в октаэдрических комплексах с двумя бидентатными лигандами типа этилендиамина (еп), например [Rh(en)2Cl2]+. Транс-конфигурация имеет центр и плоскость симметрии и поэтому оптически не активна (рис. Х.4, а). В цис-изомере отсутствуют эти элементы симметрии, т. е. эта молекула диссимметрична и оптически активна (рис. Х.4, б).
Отнесение полос поглощения с использованием КД. В связи с различием правил отбора в электронных спектрах поглощения и, например, в спектрах кругового дихроизма можно провести отнесение электронных переходов при использовании данных обоих 208
транс
Рис. Х.4. Изомеры иона [Rh еп2С12]+
методов. Так, октаэдрические комплексы Со3+ типа [Соеп3]3+ имеют две характерные полосы поглощения: ~21400 см-1 и ~ 29 600 см-1 не очень высокой интенсивности (lge~2).
Правила отбора (см. ч. I, гл. XIII) запрещают электронные переходы между (/-уровнями свободного атома или иона, т. е. (/-<-► (/-переходы, поскольку (/-АО имеют центр симметрии. В электронных спектрах центросимметричных ионов и молекул запрещены переходы типа g+->-g и и-^и, разрешены только переходы
Однако несимметричные колебания лигандов или несимметричное окружение центрального иона металла приводят к нарушению этих правил и появлению линий поглощения. Так, для конфигурации d6, которую имеет ион Со3+, в сильном октаэдрическом поле лигандов все электроны спарены и находятся на трижды вырожденном * уровне /2г. Электронная конфигурация t62g в соответствии с правилами теории групп образует состояние 'Aig, т. е. синглетное полносимметричное состояние.
При поглощении кванта излучения возможен переход одного электрона на более высокий уровень, которым является дважды вырожденный уровень симметрии eg в соответствии с расщеплением в октаэдрическом поле пятикратно вырожденных (/-уровней свободного иона (рис. Х.5). При электронной конфигурации возбужденного иона (/52g£g) реализуется четыре состояния: два типа симметрии 'Tig и 'T^g для синглетных состояний и два 3Tis и 3T2g для триплетных состояний. Хотя триплетные состояния ниже по энергии, но вероятность переходов с сохранением спина электрона
* Часто трижды вырожденные состояния (уровни), особенно в колебательной спектроскопии, но иногда н в электронной (см. учебник <Физическне методы исследования в химии. Структурные методы н оптическая электроскопия». Раздел четвертый), обозначают буквами f(F), а не t(T).
209
Рис. Х.5. Схема уровней электронной энергии иона Со3+ и октаэдрическом поле Oh и в поле симметрии D3\
стрелками показаны переходы в спектрах поглощения (СП) и в спектрах кругового дихроизма (КД)
выше, так что следует рассматривать только переходы 'Ajg-^-'Tjg и 'Aig-^Tig. Именно эти переходы объясняют появление полос поглощения в видимой и ближней ультрафиолетовой областях спектров комплексов переходных металлов при условии, как об этом говорилось, некоторого нарушения центросимметричного состояния.
Для установления взаимного расположения уровней 'Tig и *T2g требуется знание энергии электронного отталкивания, которая может быть оценена лишь в определенном приближении. Октаэдрические комплексы типа [Со епз]3+ имеют D3 симметрию. Понижение симметрии от Oh к Ь3 должно привести к расщеплению уровней энергии + М2 и + Mi (рис. Х.5) и, следо-
вательно, к увеличению числа переходов. Но такое расщепление мало и не проявляется в спектрах поглощения, что свидетельствует о превалирующем влиянии только ближайшего откаэдрического окружения Со(III).
Исследование кругового дихроизма показывает, что только один из двух переходов и обладает значи-
тельной вращательной силой (рис. Х.6). Поскольку оператор магнитного дипольного момента принадлежит к типу симметриии Tig группы Он как оператор вращения RxRy или Rz, то только переход имеющий прямое произведение A\g'XT\g=Tlg, бу-
дет происходить по механизму магнитного дипольного перехода. Симметрия уровня Т2е не допускает такого перехода.
В строго октаэдрическом поле лигандов магнитные и электрические дипольные переходы исключают друг друга, поскольку при-210
Рис. Х.6. Кривые спектра поглощения (СП) и кругового дихроизма (КД)
надлежит к разным типам симметрии точечной группы Он. Понижение симметрии до Dz в [Со епз]3+ создает условия для проявления оптической активности перехода, соответствующего переходу М1г->-~^T\S группы Oh. В связи со сложностью задачи определения вида волновых функций различных состояний молекулярных ионов не имеется строго теоретического анализа вращательных сил перехода. Однако накоплен значительный экспериментальный материал, который позволяет вывести эмпирические закономерности.
Обнаружилось, что правила отбора для Oh-симметрии достаточно надежны
для диссимметричных молекул и вращательные силы переходов, соответствующих верхнему хТ\г в группе Oh, значительно больше, чем для xTzg, поскольку для последнего уровня необходимо создавать условия как для электрического, так и магнитного дипольных моментов перехода.
В молекулярном ионе симметрии D3 уровни T\s расщепляются на 'Е и lAi. Согласно таблице характеров для этих уровней возможна оптическая активность обоих переходов 'А<-+'Е и “Л^'Лг, поскольку z и Rz, рассматриваемые как операторы электрического и магнитного дипольных моментов, принадлежат к неприводимому представлению Аз, а х и Rx и у и Ry принадлежат к неприводимому представлению Е.
Для комплексов кубической симметрии с ионами М(аа)зп+ и цис-М.(аЬ)3п+, где аа — симметричный и ab — несимметричный бидентатный лиганд, для d3, ds и спин-спаренной d6 конфигураций ионов экспериментально на основе спектров КД установлено, что наиболее низким по энергии является состояние Де-группы симметрии Oh- Полоса перехода с участием этого уровня имеет две компоненты КД — Е и Л. Большой экспериментальный материал свидетельствует о том, что в октаэдрических комплексах Д-компонента превалирует и лежит ниже по энергии, чем Л-ком-понента. Для идентификации Д-компоненты используется ряд приемов, например изучение спектра КД кристалла, ориентированного так, что луч проходит вдоль оси Сз и дает только £-компо-ненту.
Корреляция спектрополяриметрических данных с диссиметри-ей комплексов. Ранее указывалось на то, что диссиметрия комплексных соединений может быть обусловлена асимметрией ближайшего к центральному иону окружения, асимметрией лигандов и их конформаций и т. п. Рассмотрим только конфигурационный эффект, а именно возможность определения хиральности в трис-бидентатных комплексах типа [Со еп]3+.
211
к сожалению, до настоящего времени не используется единая номенклатура для описания хиральности (абсолютной'конфигурации) комплексных соединений. Поэтому можно привести ряд обозначений. Так, для проекции, перпендикулярной оси Сз (см. рис. VIII.8), при движении по цепи бидентатного мостика от ближайших трех атомов лигандов к дальним влево (левый винт) получаем хиральность L(C3) = A. Однако относительно оси Сз (см. рис. VIII.8) эта конфигурация будет иметь правый винт или правую хиральность, т. е. D(C2) = D. Все обозначения эквивалентны, т. е. D == = L(C3) = Л. Комплексы противоположной
хиральности (см. рис. VIII.8) имеют правый винт относительно оси Сз, поскольку движение от ближайших атомов к дальним вдоль мостиков совершается вправо, но имеют левый винт относительно оси С2. Эквивалентными обозначениями будут L=s= = L(C2)^D(C3)=A.
Специальными исследованиями было установлено что положительный дихроичный максимум С-компоненты первой полосы поглощения соответствует Л-конфигурации, а отрицательный — Д-конфигурации. Соответственно A-компонента имеет отрицательный максимум КД для Л-конфигурации и положительный для Д-конфигурации.
Такие исследования основаны на сопоставлении знака вращения Е и А — компонент первой полосы поглощения комплексов типа [М(аа)з] и [М(аЬ)3] с абсолютными конфигурациями, которые определены методами рентгеноструктурного анализа, КД в ориентированном кристалле или какими-либо другими.
Такая закономерность широко используется для установления абсолютной конфигурации трис-бидентатных комплексов на основе только спектров КД.
Предпринимались попытки распространить это правило на менее симметричные комплексы, в частности молекулы симметрии Сз. Соответствующее правило сформулировано следующим образом: комплексы симметрии Сз с положительной вращательной силой перехода А имеют конфигурацию D == Л. Трудности состоят в идентификации перехода А.
4. КОЛЕБАТЕЛЬНАЯ ОПТИЧЕСКАЯ АКТИВНОСТЬ
В последние пятнадцать лет развивается новое направление в исследовании естественной оптической активности молекул на основе колебательных инфракрасных спектров и спектров комбинационного рассеяния.
Источником стереохимической информации о хиральных молекулах являются не только электронные переходы, но и колебательные переходы. Для ряда классов соединений электронные переходы лежат в коротковолновой УФ области, которая не доступна 212
для исследования. Это существенно ограничивает возможности изучения кругового дихроизма этих соединений.
Колебательная оптическая активность может проявляться в большем числе переходов, соответственно числу колебаний в молекуле, в которых участвуют все части молекулы. Получаемая информация позволяет идентифицировать абсолютную конфигурацию молекул по знаку, например, колебательного кругового дихроизма. Обычные колебательные спектры различных конформеров довольно похожи друг на друга. Но в колебательных спектрах кругового дихроизма существенно увеличиваются различия экспериментальных данных, поэтому идентификацию конформера можно сделать с большей уверенностью.
Известно, что для определения силовых полей молекул практически недостаточно одних спектроскопических данных, так как число колебательных частот молекулы всегда меньше числа силовых постоянных. Кроме того, часто из-за перекрывания полос в спектре возникают трудности с выделением полос отдельных колебаний. Использование колебательного кругового дихроизма помогает в решении этого вопроса, поскольку правила отбора могут существенно различаться для отдельных полос в области их перекрывания, например, v(C*—Н) в Л-валине
Г /н
H3N—С*—СН(СН3)г .
^сог
Данные колебательной оптической активности также могут существенно помочь в установлении электронного строения молекул.
Колебательный круговой дихроизм. Вращательная сила колебательных переходов так же, как и электронных, определяется скалярным произведением электрического и магнитного моментов перехода. Так, для s-ro нормального колебания при переходе из основного состояния в первое возбужденное имеем выражение
^oi = hn I (Фо +1) W W +о) }•
Поскольку цт=-^- V _£С(Г. хр(), где qt — заряд i-й частицы, 2 mi
г, и р/ — ее радиус-вектор и импульс соответственно, то из-за больших величин масс ядер существенно уменьшаются магнитные моменты и, следовательно, вращательные силы перехода. По сравне-нию с электронными переходами это уменьшение составляет несколько порядков (до 10~3... 10~5).
Существует несколько моделей для расчета R. Теория метода продолжает совершенствоваться. Но даже упрощенные модели молекул с фиксированным распределением зарядов на атомах дают возможность интерпретации эксперимента.
213
Рис. Х.7. Колебательный спектр кругового днхронзма (а) н инфракрасный спектр поглощения (б) для (+)-(3/?)-метнлцикло-гексанона, полученные на фурье-спектро-метре
В схеме эксперимента колебательного кругового дихроизма нет принципиальных отличий по сравнению с круговым дихроизмом для электронных переходов. В последнее время эта методика существенно улучшена за счет использования фурье-спектромет-ров, что позволило увеличить чувствительность и разрешение, а также расширить спектральный диапазон.
Пример колебательного КД спектра приведен на рис. Х.7 для (+)-(37?)-метилциклогексанона. Показано хорошее соответствие полос в спектре поглощения и в спектре кругового дихроизма. Четко видны различные знаки пиков кругового дихроизма.
Исследование L-аминокислот позволило установить правило: знак полосы Са*—Н кругового дихроизма всегда положительный.
znh2
На примере L-валина (СН3)2СН—С— СООН показано, что
214
внутри молекулы образуется водородная связь
(СН3)2СН—
COj-H'
которая замыкает цикл из атомов углерода, кислорода и азота. Изменение электрического дипольного момента связи Са*—Н вызывает движение зарядов по циклу, что обусловливает появление магнитного момента. Угол между векторами и цт — острый, как изображено иа схеме. Кривые e(v) и Ae(v) для Л-валина приведены на рнс. Х.8, который показывает дополнительные возможности метода. Действительно, область валентных колебаний довольно широкая. Только колебательный круговой дихроизм позволяет выделить колебание Са*—Н.
Однако в молекулах эфиров аминокислот, в которых водородная связь не образуется и поэтому не образуется цикл, резко уменьшается интенсивность пика для Са*—Н в спектре колебательного кругового дихроизма.
Оптическая активность в спектрах комбинационного рассеяния.
Только использование лазеров и высокочувствительных детекторов
75-
Рис. Х.8. Колебательный спектр кругового дихроизма (/) и ИК спектр поглощения (2) для /.-валнна-Л^-^з в области валентных колебаний С—Н связей
215
позволило наблюдать спектры комбинационного рассеяния с круговой поляризацией.
Те же взаимодействия, которые определяют дисперсию оптического вращения и кругового дихроизма, определяют спектры комбинационного рассеяния с круговой поляризацией. Поскольку индуцированный электрический дипольный момент пропорционален тензору электрической поляризуемости атп и вращательной поляризуемости (индексы тип относятся к электронным состояниям), разность в интенсивности рассеяния лучей с левой и правой круговой поляризацией \I-Ii—Ir = M(v) будет определяться произведением
<Ф? Ф/> <Фу |РтЯ| ’ ( <+/ 1<?^1 ФУ > )2 ’
^7 \ t'v.s / \ Q'xs /
где ф,-, -ф/ — волновые функции колебательных состояний i и / соответственно; Qs — нормальная координата.
В хиральных молекулах для всех или почти всех колебатель-ных координат величины —— и —не равны нулю.
dQs &Qs
В настоящее время уже возможно сравнение колебательных спектров кругового дихроизма в инфракрасной области и соответствующих спектров комбинационного рассеяния для широкого диапазона частот.
ГЛАВА XI
АНОМАЛЬНОЕ РАССЕЯНИЕ РЕНТГЕНОВСКИХ ЛУЧЕЙ-МЕТОД ОПРЕДЕЛЕНИЯ
АБСОЛЮТНОЙ КОНФИГУРАЦИИ
1. АБСОЛЮТНАЯ КОНФИГУРАЦИЯ МОЛЕКУЛ В ДЕКАРТОВОЙ СИСТЕМЕ КООРДИНАТ
В общем курсе кристаллохимии рассматриваются методы исследования структуры кристаллов — рентгеноструктурный анализ, нейтронография и, частично, электронография. Однако не дается изложение специального метода рентгеноструктурного анализа, который используется для определения абсолютной конфигурации молекул. Такая задача возникает при изучении оптически активных веществ. В гл. VIII, IX и X представлены оптические методы исследования оптически активных веществ. Особенность этих методов состоит в том, что легко определить с их помощью различие в абсолютной конфигурации молекул, но нет возможности прямого отнесения экспериментальных данных по ДОВ или КД к определенному энантиомеру. Именно эту проблему и решает метод аномального рассеяния рентгеновских лучей.
216
Рис. Xl.i. Энантиомеры («) и (S) в правой системе координат (х, у, z) молекулы PFCIBr
Абсолютная конфигурация молекулы задается, например, в какой-либо системе координат — правой или левой. Обычно принята правая система. В этой системе есть однозначное соответствие конфигурации молекулы и координат атомов. Энантиомеры отличаются друг от друга знаком координат какой-либо оси как следствие отражения в плоскости или знаком всех координат как следствие отражения в начале координат. Так, на рис. XI.1 показано отражение в плоскости ху, т. е. координаты энантиомеров асимметричной молекулы PFCIBr отличаются только знаком z. Для энантиомеров R и S, связанных центром инверсии i, имеем соотношение
г (/?) = — г (S)
или (XI. 1)
!•(/?)= rR(x, у, Z) и r(S) = rs(-x, -у, —Z), где R— правая, S— левая конфигурации по системе Кана, Инголь-да и Прелога*.
* Если расположить молекулу, например PFCIBr, таким образом, что атом фосфора будет находиться за условной плоскостью по отношению к атомам Вг, С1 и F, то их расположение по часовой стрелке от «старшего» атома Вг к С1 н далее к F определяет /^-конфигурацию, а расположение «по старшинству» в противоположную сторону— S-конфигурацию.
217
Переход от rs(x, у, z) к rs(—х, —у, —z) осуществлялся или поворотом вокруг оси z, или двумя отражениями oXz и oyz, которые не изменяют форму энантиомера.
2. НОРМАЛЬНОЕ РАССЕЯНИЕ И ЗАКОН ФРИДЕЛЯ
В условиях нормальной дифракции рентгеновских лучей длина волны падающего излучения X меньше длины волны собственных электронных переходов в атоме (а частота v, соответственно, больше vk), т. е. Х<Хк и v>vk. Это позволяет использовать приближение рассеяния рентгеновских лучей свободным электроном. Такой электрон становится источником сферической волны с амплитудой f1. Атомная амплитуда рассеяния А (6) является результатом сложения волн, рассеянных всеми электронами атома, пропорциональна f1 и зависит от угла рассеяния 0 и плотности распределения электронов в атоме. Обычно атомной амплитудой рассеяния называют безразмерную величину f(0) =А (0)//1. С увеличением угла рассеяния 0 функция f(0) резко уменьшается от величины Z (порядковый номер) до нуля. В принятом приближении функция f(0) является действительной.
Дифракционная картина в рентгеноструктурном анализе описывается с помощью обратной решетки, которую образуют дифрагированные монокристаллом лучи. Для любого кристалла прямая и обратная решетки имеют одинаковую симметрию. Векторы обратной решетки а*, Ь* и с* связаны с векторами кристаллической решетки а, b и с следующим образом. Вектор а* перпендикулярен плоскости, в которой находятся векторы b и с прямой решетки. Соответственно векторы Ь* и с* перпендикулярны плоскостям на векторах а и с, а и Ь. Между векторами прямой и обратной решеток установлены следующие соотношения:
* Ьхс и* сХа а* ==----- Ь* ~------
vc VC
где Vc — abXc=l/V*c — объем элементарной ячейки монокристал- • ла; V*c — объем ячейки в обратной решетке. ;
Векторы кристаллической и обратной решеток образуют сопря- j женный набор. Это означает, что скалярное произведение любых j двух одноименных векторов равно единице, т. е. J
аа* =Ы>* =сс* = 1,
или в более общем виде I
_ а X Ь с* =-------
Vc
« ( = 1 i =-j .
u.u, = { „ , . ' ‘ ' I
1 > 1 I =0 I
где u=a, b или с и u* = a*, b* или c*.
Если структура размещена на какой-либо решетке, то ее дифракционная картина размещена на решетке, обратной к исходной’. 218
Узлы обратной решетки задаются векторами Н. Эти векторы перпендикулярны плоскостям кристалла с индексами hkl и направлены из начала обратной решетки, совпадающей с началом кристаллической решетки, в точку обратной решетки с теми же индексами hkl.
Измеряемая интенсивность /(Н) рассеяния рентгеновских лучей монокристаллом пропорциональна квадрату модуля структурного фактора F(H):
I (Н) ~ (Н)|2 = F (Н) 5* (Н), (XI.2)
где H = /ia* + &b* + Zc*; Н — радиус-вектор узла обратной решетки; h, k, I — Миллеровские индексы; а*, Ь*, с* — векторы элементарной ячейки в обратном пространстве; | Н | =2sin Q/K— \/dhki', dhkt— межплоскостное расстояние.
Структурный фактор является суммой атомных амплитуд, умноженных на соответствующий фазовый множитель:
5(H) = 2 /;(|Н|)Л'НгЛ (XI.3)
J
где г, — радиус-вектор атома или узла кристаллической решетки /; ri — X/a-1-yib + ZjC; а, Ь, с — векторы элементарной ячейки; Xj, У1, г, — координаты в относительных единицах — в долях а, b и с.
По определению Н и Г/ имеем
H.rj — hxj + kyj + lzj. (XI.4)
Для действительных /7( | Н |) получаем, используя равенство— Н = Н,
5* (Н) = 2 fj (|Н|) е-2"'Нг/ = 2 fj (|Н| Л'НГ> = 5 (Н)
и _ * (XI.5)
5* (Н) = 2 fj (|Н|) e“2,t'Hr/ s 2 fj (I U|) = F (H).
Как следствие имеем
I (H) ~ F (H) 5* (H) = F (H) F (H) = F (fij F (H) ~ / (H). (XI. 6)
Уравнение (X.6) является выражением закона Фриде л я, в соответствии с которым
/(Н) = /(Н). (XI. 7)
Это означает, что дифракционная картина имеет центр симметрии. Поэтому из такого эксперимента нельзя установить, имеет ли реальный кристалл центр симметрии. Поскольку для левого и правого энантиомеров Г/(7?)=—r/(S) [см. уравнение (XI. 1)], то Fr (Н) = 2 /у (|Н |) еЫНг/Л) = 2 fj (|Н|) e-2"'Hr/5> = (Н) = Fs (Й)
и _ (XI. 8)
5* (H) = 5S(H) = 5’(H).
219
Следовательно,
/j?(H) = /s(H) = /s(H) = /j?(H).
(XI. 9)
Закон Фриделя не позволяет, в частности, определить абсолютную конфигурацию молекул.
3. РАССЕЯНИЕ РЕНТГЕНОВСКИХ ЛУЧЕЙ В ОБЛАСТИ ПОГЛОЩЕНИЯ АТОМА
Согласно классической электронной теории уравнение движения связанного в атоме электрона вдоль х под действием падающего излучения имеет следующий вид:
х 4-ух + <*>1х = ez“z, (XI. 10)
т
где со/с — собственная круговая частота колебаний электрона; со — частота падающего излучения; у — коэффициент затухания.
Из решения этого уравнения можно найти для одного электрона амплитуду рассеяния ft: и затем атомную амплитуду аномального рассеяния /л в комплексной форме:
при X > fry|
= /о(8) + V' ) + /ДГ" ( -JL-, (XI.11)
J \“к )’
при X « [ГД
где gK — сила осциллятора, пропорциональная вероятности /С-пере-хода; fo(0)—атомная амплитуда рассеяния свободными электро-
/ (О \ / (О \
нами (когда у=(Ок=0); / И ’ ч ~~ дисперси-
онные поправки, зависящие от со. Имеются таблицы величин fo(0), Af и Af".
Уравнение (XI.11) получают в два этапа. Сначала рассматривают рассеяние при |г/|, т. е. при больших X. Это позволяет не учитывать зависимость рассеяния от угла 0. Затем делается переход к |г;-| и находят f0(9)- Величины Af' и Af" не зависят от угла 0, так как обусловлены рассеянием самой внутренней электронной оболочки, имеющей малые размеры.
Зависимости Af' и Af" от со показаны на рис. XI.2. Поправка Af' в основном отрицательна, что соответствует уменьшению амплитуды рассеяния в резонансной области. Поправка Д/">0, т. е. всегда положительна.
220
Рис, Х1.2. Зависимость t^f'IgK (кривая /) Рис. Х1.3. Изменение фазы рассеяния и bf"lgK (кривая 2) от частоты падаю- рентгеновских лучей на атоме в за-щего излучения со в форме отношения висимости от со/со/с
со/сок
Наиболее важной характеристикой для дифракции представляет изменение фазы ср рассеяния в области поглощения, которая определяется уравнением tg<p=Af"/ (f0 + Af) и изменяется от л для области, далекой от поглощения, до л/2 в области поглощения 0 (рис. XI.3). Вообще ювиря, ср зависит от угла рассеяния 6, гак как fo=fo(6)- Рассеянная волна без поглощения отстает от падающей волны на л, т. е. движется в противофазе с падающей волной. В условиях поглощения фаза рассеянной волны приближается к фазе падающей волны, но только до л/2, так как f0 + Af'->0 при * GJ—>"С0к.
Уравнение (XI.11) можно переписать в форме, удобной для использования в структурном факторе:
/А = |/л1^’ или /А = |/А] еы*, (XI. 12)
где |/а|=1(/о+А/')2+(Д///)2]1-2, ? = arctg У' ; и
/о + д/
4. АНОМАЛЬНОЕ РАССЕЯНИЕ И ОПРЕДЕЛЕНИЕ АБСОЛЮТНОЙ КОНФИГУРАЦИИ МОЛЕКУЛ
Поскольку при рассеянии в области поглощения (при аномальном рассеянии) изменяется фаза рассеяния на атоме по сравнению с нормальным рассеянием, возникает новая возможность при изучении дифракции рентгеновских лучей. Так, например, в упрощенной модели дифракционного эксперимента при рассеянии на двух
221
Рис. Х1.4. Упрощенная модель рассеяния на двух атомах: а— рассеивает нормально; б — аномально
атомах (рис. XI.4) условия аномального рассеяния позволяют отличить левое расположение этих атомов от правого, поскольку разность фаз лучей 1 и 2 будет определяться разностью хода Д=АС = ВС, одинаковой в обоих случаях, и разностью фаз лучей, рассеянных нормальным атомом А и аномальным В, т. е.
Д<рв==?(/?) —?(А)—д (рис. х.4,а), Д?Й=«(А) ——^-Д
Л Л
(рис. Х.4,б). Как уже говорилось, <р(А)=л, а <р(В)=л/2. Следо-, л 2л , А л 2л . „
вательно. лля Дф_ =------------—Д. а для Дсрл=------------Д. Раз-
X ‘“ 2 X
2
2
личным фазам Дсра и Дфе соответствуют различные интенсивности рассеяния. При нормальном рассеянии <р(В)=<р(А) и Д<ра = Дфб.
Теперь можно показать, что в общем случае аномального рассеяния для структур, не имеющих центра симметрии, не выполняется закон Фриделя. Действительно,
₽ /пч Vi/ , 2кГНг, 2к/(а +Нг.)
ЛНН)~2,|/Лу|е 1 е у = 2|Лур J 1 •
Для F*a(H) имеем
^a(H) = 2I fAj |e-21t(a;+Hr/ .
Тогда
/д(Н) ~ FAF*A = (2 I fA] I е^(я/+НгР)(2 I fA] I г-2^‘ЛНг/).
Для центросимметричных отражений получаем
Ia (Н) ~ Fa (Н) F\ (Н) = /2 I fA} I e2’t,<*/-Hr? W2 । ?А} I e-2«'(«/-Hr/>
222
Это означает, что
/А(Н)^/А(Н). (XI.13)
При изучении энантиоморфных структур соотношение г;(/?) = = —r/(S) [(см. уравнение (Х.1)] приводит к следующему уравнению:
F ar = 2 1 fA) I = 2 I fA] I = FAS (H).
Далее получаем
'ar (H) ~ FAr (H)/^ (H) = FAS(H)F*as (H) ~ IAs(a),
t. e.
'ar (H) = /AS (H) I as (H) = I ar (H). (XI.14)
Следовательно, в случае аномального рассеяния энантиоморфными структурами рефлексы, связанные центром симметрии, имеют различную интенсивность и их отнесение позволяет определить абсолютную конфигурацию.
Один из путей определения энантиомерной структуры состоит в том, что сначала проводят обычное рентгеноструктурное исследование в области нормального рассеяния, из которого находят структуру вещества, но с точностью до знака всех координат атомов. Затем проводят эксперимент в условиях аномального рассеяния хотя бы одного атома. Измеренные величины /(Н) и 7(H) сравнивают с рассчитанными на основе структурных амплитуд, т. е. сопоставляют уравнения для разностей этих величин:
/эксп (Н) - /Эксп (Н) = k [ I Far (Н) Р - | F AR (Н) 12] (XI.15)
или
/эксп (Н) - /эксп (й) = k [ I Fas I 2 _ | Fas (H) 12] . (XI.16)
На практике реализуется или одно, или другое уравнение.
Впервые этот метод был использован в 1951 г. Бийво для определения абсолютной конфигурации аниона винной кислоты в двойной натрийрубидиевой соли Na, ЕЬ-СДПОбРНгО. В качестве источника использовалось /Са-излучение циркония с Х=0,0784 нм. При этом атом Rb давал аномальное рассеяние, так как поглоще
* Проекционная формула Фишера для иатрийрубидиевой соли (+)-виииой кислоты означает, что атомы главной цепи, расположенной вертикально, находятся за плоскостью соответствующих асимметричных атомов углерода, а атомы и группы атомов по линии, перпендикулярной главной цепи, находятся перед плоскостью, в которой расположены рассматриваемые асимметричные атомы углерода.
223
ние атома Rb происходит при Л=0,0814 нм. Найденная абсолютная конфигурация аниона совпала с относительно произвольно принятой ранее как R.R-конфигурация*:
COORb+
I
н—с*—он
I
но—с*—н
I _
COONa+
натрийрубидиевая соль ( + )-винной кислоты
Метод аномального рассеяния широко используется также для установления абсолютных конфигураций комплексных соединений и биоорганических соединений___________________________________
Рассмотренные методы исследования оптически активных веществ разделяются на два типа. Методы ДОВ и КД относятся к оптическим методам, аномальное расстояние рентгеновских лучей — к дифракционным.
Используемые длины волн в ДОВ и КД имеют порядок нанометров, т. е. существенно превышают размеры молекул. Поэтому в этих экспериментах возможно определять лишь оптические характеристики излучения, такие, как состояние поляризации в ДОВ или коэффициент поглощения при прохождении через вещество. Они значительны по величине лишь тогда, когда в молекуле имеется хромофор, т. е. группа атомов или атом со значительным коэффициентом экстинкции.
В методах ДОВ и КД вопрос об абсолютной конфигурации решается лишь приближенно или качественно, так как в строгой теории методов, связывающей оптические свойства и молекулярную конфигурацию для расчета вращательной силы перехода, необходимо задавать электронные функции молекулы для основного и возбужденных состояний. К сожалению, такая информация достаточной точности отсутствует, что и приводит к установлению различных правил и закономерностей, к развитию полуэмпирических методов при использовании ДОВ и КД.
Одним из важнейших применений ДОВ и особенно КД наряду со стереохимическими являются спектроскопические исследования, определение типов электронных переходов и электронного строения.
Новое направление развития этих методов представлено колебательным круговым дихроизмом в ИК спектроскопии и определением разности в интенсивности рассеяния лучей с правой и левой круговой поляризациями в спектрах комбинационного рассеяния. В этой области следует ожидать новых важных результатов.
Практически единственным методом, позволяющим определять абсолютную конфигурацию, является метод аномального рассеяния рентгеновских лучей. Благодаря автоматизации эксперимента существенно расширяется база структурных данных.
224
Контрольные вопросы
Глава VIII
1. Что такое линейная н круговая поляризация излучения? Какая связь между ними?
2. Какой параметр оптически активной среды определяет вращение плоскости поляризации линейно поляризованного света? Как графически на основе фаз распространяющихся лучей пояснить явление вращения плоскости поляризации?
3. Выведите формулу Френеля и поясните ее физическое содержание,
4. Определите понятия «удельное» и «молярное вращение».
5. В чем состоит недостаток феноменологической теории вращения плоскости поляризации линейно поляризованного света?
6. Как спиральная модель молекулы объясняет вращение плоскости поляризации? Почему возникает составляющая электрического поля, перпендикулярная падающему полю?
7. Что такое вращательная сила электронного перехода и каков ее физический смысл? Как формулируется и доказывается правило сумм?
8. Каковы требования к симметрии молекул, обладающих оптической активностью?
9. Каковы факторы, определяющие диссимметрию комплексов переходных металлов?
10. В чем состоит принципиальная схема эксперимента в методе ДОВ? Каковы требования к условиям эксперимента?
11. Что такое аномальный характер кривых ДОВ? Каковы причины его возникновения?
Глава IX
1. Сформулируйте закон Бугера — Ламберта — Бера для лучей с круговой поляризацией.
2. Определите понятие «круговой дихроизм».
3. Какова связь кругового дихроизма с эллиптичностью луча?
4. В чем состоит связь кругового дихроизма и вращательной силы электронного перехода?
5. Как экспериментально определить вращательную силу перехода?
6. Объясните схему эксперимента в методе КД. Как формируется луч с круговой поляризацией?
7. Как устанавливается связь между молярной дисперсией оптического вращения и функцией эллиптичности луча, прошедшего через оптически активную среду?
Глава X
1. Перечислите задачи, решаемые методами ДОВ и КД.
2. Сравните возможности ДОВ и КД.
3. Какие закономерности, помогающие в решении стереохимических задач методами ДОВ и КД, вы можете назвать? Что такое правило Брюстера? В чем состоит правило октантов?
4. Как решаются задачи установления абсолютной конфигурации методами ДОВ и КД в комплексах переходных металлов?
5. Как решаются задачи отнесения полос поглощения с использованием КД?
6. В чем состоит колебательная оптическая активность?
8—1377 225
Глава XI
1. Как в декартовой системе координат можно выразить соотношение правой (Я) и левой (S) коифвгураций?
2. Сформулируйте условия нормального и аномального рассеянии рентгеновских лучей атомами.
3. В чем состоит закон Фриделя?
4. Почему из закона Фриделя следует невозможность различить дифракционную картину энантиомеров?
5. В чем состоит особенность выражения для амплитуды атомного аномального рассеяния?
6. Покажите графически, как изменяются дисперсионные попраики в зависимости от частоты падающего излучения.
7. Какова зависимость фазы аномального рассеяния атома от частоты падающего излучения?
8. Докажите, что в случае аномального рассеяния рентгеновских лучей имеется возможность определить абсолютную конфигурацию энантиомера.
МЕТОДЫ ИЗУЧЕНИЯ ПОЛЯРИЗУЕМОСТИ И МАГНИТНОЙ ОПТИЧЕСКОЙ АКТИВНОСТИ
Поляризуемость молекул является одним из важнейших физических (электрических) свойств молекул, определяющих межмолекулярные взаимодействия, а также взаимодействия с внешним электрическим полем. Обычно средние значения поляризуемости, определяемые на основе измеряемых величин рефракции, могут быть дополнены данными релеевского рассеяния и эффект-' Керра. В результате получают главные значения эллипсоида поляризуемости молекулы, используемые для оценки главных значений эллипсоидов поляризуемости химических связей, конформаций молекул в растворах, изучения проблемы взаимного влияния атомов в молекуле.
Внешнее магнитное поле вызывает деформацию электронной оболочки, которая обусловливает оптическую активность ахиральных молекул (эффект Фарадея).
8
В химических исследованиях широкое распространение иашло использование экспериментальных молярных рефракций, которые пропорциональны поляризуемости, для изучения химического строения соединений, специфических взаимных влияний атомов в молекуле, эффектов сопряжения и т. д.
Однако в рефрактометрических измерениях определяют лишь среднее
значение электронной поляризуемости молекулы. В самом общем случае электронная поляризуемость молекулы различна в различных направлениях, что выражается в тензорных свойствах поляризуемости. Количественное описание поляризуемости дается через три главных значения те и з о р а электронной поляризуемости* (bi, bi и Ьз), которые соответствуют определенным направлениям главных осей. Только вдоль этих осей совпадают направления векторов напряженности электрического поля & и индуцированного дипольного момента ц. На этих трех значениях можно построить эллипсоид, который называется эллипсоидом поляризуемости. Прн этом величины bi и &з составляют половины главных осей эллипсоида поляризуемости.
Экспериментальные и расчетные методы рефрактометрии излагаются довольно подробно в химических практикумах и специальных руководствах. Поэтому здесь эти методы, основанные на измерениях показателя преломления, не рассматриваются.
В настоящем разделе будут рассмотрены методы определения степени деполяризации релеевского рассеяния и двойного лучепреломления в явлении Керра, вызванного приложенным электрическим полем к исследуемому веществу в жидкой фазе.
Релеевское рассеяние света в газах и растворах, а также явление Керра в газах и растворах позволяют измерять независимо анизотропию оптической (электронной) поляризуемости молекул:
Y2 = ~ l(ii - fc)2 + (fc - *з)2 + («з - ii)2] •
Располагая данными о молярной рефракции, можно рассчитать среднюю оптическую поляризуемость молекулы:
Ь — — (bl + Ъг + Ьз).
* В этом разделе приняты другие обозначения для электронной поляризуемости и степени деполяризации по сравнению с гл. VI—X учебника «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия». Это обусловлено тем, что в литературе по этим методам используются именно эти обозначения.
228
На основе экспериментальных данных, полученных из релеев-ского рассеяния света, эффекта Керра и молярной рефракции, возможно н определение молекулярных характеристик (Ь\, Ьъ, д3).
Основные задачи, которые решают с использованием указанных методов, следующие:
1) определение главных значений эллипсоида электронной поляризуемости молекул химических связей и групп атомов;
2) изучение эффектов взаимного влияния;
3) определение пространственного строения молекул, включая конформацию;
4) изучение внутреннего вращения и определение конформационного состава.
При решении этих задач предполагают, что в соответствии со свойством аддитивности значения поляризуемости молекулы (молярной рефракции) при заданном пространственном строении могут быть вычислены по данным поляризуемости связей (так называемая валентно-оптическая схема).
Важно то, что рассматриваемыми здесь методами определяют конформации молекул в растворах. Такие структурные методы, как рентгеноструктурный анализ или газовая электронография, практически неприменимы для этих условий.
Явление Фарадея выявляет индуцированную анизотропию вещества в магнитном поле для лучей с правой и левой круговой поляризацией. Получаемые данные в виде численных значений угла поворота плоскости поляризации линейно поляризованного света или кривых дисперсии магнитного оптического вращения или магнитного кругового дихроизма используются для изучения электронного строения молекул.
ГЛАВА XII
РЕЛЕЕВСКОЕ РАССЕЯНИЕ СВЕТА
1. РЕЛЕЕВСКОЕ РАССЕЯНИЕ СВЕТА В ГАЗАХ И РАСТВОРАХ
В приближении упругого рассеяния электрическое поле излучения &= й’о cos at, падающего на изотропную молекулу, индуцирует электрический дипольный момент р, меняющийся с частотой падающей волны со:
p = i?ocos“^> (ХП.1)
где&—электронная поляризуемость молекулы.
В свою очередь колеблющийся диполь является источником рассеянной сферичес кой волны электрического поля:
U. (t — Г/с) . ш2 Ь'$л [ Г \
---^sin0 =—— —^-со8<ф- — sine, (XII.2) 4леос2г С2Г 4л»0 \ с )
229
Рис. Xll.l. Система координат для наблюдения релеевского рассеяния света: а —рассеяние изотропными молекулами: б —рассеяние анизотропными молекулами;
—индуцированный электрический дипольный момеит с компонентами uz и и — интенсивности лучей, рассеянных вдоль х и поляризованных вдоль z и у (нижние индексы). Верхний индекс указывает поляризацию падающего света
где <?<;= | g’s|—напряженность электрического поля рассеянного излучения; г — расстояние от центра сферы; 0 — угол между ц и направлением рассеянного луча.
Максимальное рассеяние будет наблюдаться при 0 = л/2. Поэтому эксперимент проводят при наблюдении рассеяния под углом 90° к падающему лучу (рис. XII.1).
Поскольку рассматриваемая молекула изотропна, то поляризованное вдоль z падающее излучение вызовет рассеянное излучение при 0 = л/2, поляризованное также вдоль z, поскольку So и ц коллинеарны.
Анализ картины рассеяния показывает, что в идеально однородной среде не должно быть явления рассеяния, так как вторичные волны гасят друг друга. М. Смолуховский и А. Эйнштейн разработали теорию рассеяния света на флуктуационных неоднородностях, возникающих из-за теплового движения молекул.
Для интенсивности рассеянного света изотропными молекулами получено уравнение
Л2
I = 10 —-----n^Vb2 sin2 9,
(XII.3)
где Iq — интенсивность падающего света; п\ — число молекул в единице объема; V — объем, занимаемый рассеивающими молекулами.
Физический смысл этого уравнения состоит в том, что доказано независимое рассеяние отдельными молекулами [сравните с уравнением (ХП.2)].
В случае анизотропных молекул индуцированный дипольный момент ц не совпадает по направлению с Sz [(см. рис. XII.1)]. Доста
230
точно рассмотреть проекцию р, на плоскость гОу, поскольку компонента цх не дает излучения в направлении х. Таким образом, только компоненты pz и Цу являются источником рассеянного излучения 1гг и Izy (верхний индекс указывает поляризацию падающего света, нижний — рассеянного) с поляризацией вдоль гну.
Так же, как и при рассмотрении рассеяния изотропными молекулами, нужно ожидать эффекта рассеяния на флуктуациях плотности для средней поляризуемости молекулы и на флуктуациях ориентации анизотропно поляризующихся молекул, т. е. на флуктуациях анизотропии молекул. Однако вывод соответствующих уравнений может быть упрощен в связи с тем, что достаточно рассмотреть интенсивность поляризованного рассеяния света отдельной анизотропной молекулой, ориентация которой усреднена.
Поскольку интенсивность поляризованного света пропорциональна квадрату индуцированного дипольного момента, имеем, например,
/ / 3 '2\
Izz~ i?cos2^ J>’ <хпл)
где bq 2, 3) —главные значения электронной поляризуемости; 8г — поле падающего излучения; bzz— диагональный элемент матрицы b в лабораторной системе координат; cos zq — направляющий косинус.
Направляющие косинусы могут быть выражены через эйлеровы углы и с их помощью может быть проведено изотропное усреднение по ориентациям.
При рассеянии света, поляризованного вдоль z, получены следующие уравнения для Izz и Izy:
л2 /
= Л)------Г Л1И62 +
Х4г2е20 к
и
Л2 3
/’ = /о------
у X4r2j2 45
(XII.5)
где b — средняя поляризуемость молекулы; у — анизотропия поляризуемости. Величина Izy составляет деполяризованную часть интенсивности. Более удобной величиной является коэффициент деполяризации, который рассчитывается из экспериментальных величин Izy и Izz. Этот коэффициент выражается также через молекулярные характеристики у и b соотношением
_ 3?2
I* 4562 + 4у2
(XII.6)
231
Если падающий луч поляризован по осн х, то
ly = lz = /о—п1у~7Г"'<2-у X4r2s2 45
(XII.7)
Прн этом коэффициент деполяризации Au=/Xj<//Xz= 1- Компоненты 1ху и Ixz необходимы прн рассмотрении рассеяния естественного света, поскольку прн нспользованнн естественного света можно считать, что /го=/"'о=^о/2. Каждая из компонент падающего света /го и /хо будет давать вклад в суммарную интенсивность рассеянного света с поляризацией нлн вдоль г, или вдоль у, т. е. для рассеяния с поляризацией вдоль z получаем /z нз уравнений (XII.5) и (XII.7): д2 / 7 \
/г = + Iх = /0 — пу *2 + — Y2J (XII.8)
и 1У для рассеяния с поляризацией вдоль у:
„ г л2 6
(XII.9)
Коэффициент деполяризации для падающего естественного света равен
/ у 6у2
77 = 45Й2 + 7у2 •
(ХП.Ю)
Уравнения (XII.6) и (ХП.Ю) дают оценку максимальной деполяризации в случае, если в молекуле, например, &i#=0 и 62=Ьз=0. Тогда А„=1/з и Аи=’/2. По мере уменьшения различий в величинах bg (д= 1,2,3) Ав и Аи приближаются к нулю.
Из уравнений (XII.6) и (ХП.Ю) можно выразить у2. Например, из уравнения (ХП.Ю) имеем
Y2=45ft2. -ц
6 — 7ДЦ
(XII.il)
Таким образом, нз эксперимента по релеевскому рассеянию света газов получаем уравнения, связывающее трн главных значения: Ь\, Ь2, Ь3.
Теория рассеяния в жидкостях н растворах сталкивается со значительными трудностями в связи с необходимостью учета изменений внутреннего поля и флуктуации плотности н ориентации. Нанлучшнм приближением к условиям газовой фазы являются разбавленные растворы в инертных, неполярных н не образующих комплексов растворителях.
Поскольку рассеяние света является суммой двух слагаемых — рассеяние изотропное и анизотропное [см. уравнения (ХП.5) н 232
(XII.8)], можно рассматривать суммарное рассеяние в следующем виде, например, для естественного света:
л2 (
h + ly = h---7*1^ +
2X4f2s2 к
Y2 I — ^изотр 4“ ^анизотр-
(XII.12)
Для сравнения рассеяния различных веществ вводят величину R — коэффициент р а с с е я н и я, или п о с т о я н н а я Р е л е я.
R— г —$изогр 4" ^анизотр» (XII.13)
‘tjV
где
л2 л2 13
^изотр = 7 и ^анизотр о” Л1 зс V2- (XII.14)
2к4£2 2к4£2 45
Такое преобразование полезно для того, чтобы отдельно рассматривать величину 7?анизотр, непосредственно связанную с у2.
Для растворов (/?аиизотр) 12 представляют как сумму слагаемых для растворителя (/?анизотр)1 и растворенного вещества а внутреннее поле молекул учитывают коэффициентом т. е.
(^?анизотр)12 = (^?анизотр)1 4“ (^?анизотр)2 =
(XII.15)
Определение (/?анизОтр)2 проводят при последовательном разбавлении раствора. Измерения у2 для паров и разбавленных растворов доказывают удовлетворительное согласование данных.
(Ланизотр) 2, ' П2 + 2 \2 к з I ’
л2____13 / п2 + 2 \2
2Х4£2 45 к 3 J 1
2. СХЕМА И УСЛОВИЯ ЭКСПЕРИМЕНТА
Для определения молекулярной анизотропии определяют интенсивность света, рассеянного под углом 90° к падающему и поляризованного вдоль z и у (рис. XII.2). В последнее время источником света в основном служат газовые лазеры непрерывного действия. При использовании ламп необходимы монохроматоры. Если изучают рассеяние поляризованного света, то перед ячейкой с веществом устанавливают поляризатор. Конструкция ячейки предусматривает поглощение отраженных внутрь ячейки лучей. Постоянство температуры обеспечивается термостатированием. На пути рассеянного луча устанавливают анализатор и четвертьволновую пластинку, которая превращает линейно поляризованный луч в луч с
233
Рис. XII.2. Схема эксперимента для исследования релеевского рассеяния:
I — лазер; 2 — поляризатор; 3 — ячейка; 4 — анализатор; 5 — четвертьволновая пла* стинка; 6 — детектор
круговой поляризацией. Это необходимо для того, чтобы работа фотоумножителя не зависела от поляризации рассеянного излучения.
Чтобы исключить систематические ошибки, измеряют относительные интенсивности фототока t:
* ис __ /?ис
*ст Ret
где числители относятся к исследуемому веществу, а знаменатели —- к стандартному.
В качестве стандарта используют, например, бензол.
ГЛАВА XIII ЭФФЕКТ КЕРРА
1. ЗАКОН КЕРРА
Изотропные вещества в однородном электрическом поле большой напряженности обладают способностью к двулучепреломлению монохроматического линейно поляризованного луча света, распространяющегося перпендикулярно приложенному полю. Это явление было открыто в 1875 г. Керром в экспериментах со стеклом (прозрачное изотропное вещество), а также с жидкостями. Лишь в 1930 г. наблюдали эффект Керра в газах и парах. Таким образом, эффект Керра представляет электрооптическое явление, которое состоит в том, что изотропное вещество, помещенное в электрическое поле, приобретает свойство оптически одноосного кристалла с оптической осью, направленной вдоль приложенного поля, т. е. внешнее электрическое поле вызывает искусственную анизотропию вещества. Такое воздействие поля обусловлено тем, что анизотропные молекулы изотропного вещества под влиянием поля преимущественно ориентируются вдоль поля (рис. XIII.1). Наличие постоянного электрического дипольного момента молекул усиливает этот эффект.
Если плоскость падающего линейно поляризованного луча S составляет некоторый угол (обычно для максимального эффекта выбирают 45°) с направлением внешнего поля <£г, то такой луч может быть разложен на две составляющие параллельную <£г, и #х, перпендикулярную <8г (рис. XIII.2). В связи с анизотропией 234
Рис. XIII.1. Искусственная анизотропия вещества в однородном электрическом поле &. Показана ориентация молекулы
Рис. XIII.2. Представление падающего линейно поляризованного луча в виде суммы лучей $ ц (вдоль
поля <8г) и <8 । пер-
пендикулярно полю S г); <хо задают равным 45°
вещества в электрическом поле ёг для лучей <FN и будут различаться показатели преломления п№ и га±. На выходе из ячейки Керра
лучи £Гц И можно представить следующими уравнениями: - Д coso,^- lc n.) (XIII.1)
и £х = “Йгсозоф - — nJ , (X1II.2) х У 2 \ с )
где <Г0— амплитуда; co=2nv; I — длина ячейки Керра; с — скорость света.
Разность фаз двух лучей имеет вид
I 2л
? = _„х)= _—^{Лл _„х). (ХШ.З)
Сложение лучей &и и <% ± дает эллиптический луч. В гл. VIII было показано, что сложение двух компонент с разностью фаз, равной нулю, дает линейно поляризованный луч, а сложение двух линейно поляризованных лучей в перпендикулярных плоскостях, имеющих одинаковые амплитуды и разность фаз л/2, приводит к лучу с круговой поляризацией. Промежуточный более общий случай соответствует эллиптической поляризации. Эллиптичность луча определяется разностью фаз ф. Доказательство этого дано ниже.
Количественно явление Керра выражается законом Керра
— I (Лл - пх)= 2лВД^
(XIII.4)
или
Дп -- п j — пх = М3$2г,
(X11I.5)
235
1
где В — постоянная Керра, характерная для каждого вещества при заданной температуре и длине волны.
Квадратичная зависимость Ал от напряженности поля &г определяет независимость Ал от его направления. В оптическом эксперименте определяют разность Лц—лх.
Величины В могут быть положительными и отрицательными. С увеличением температуры константа Керра уменьшается. Для жидкостей В значительно больше, чем для газов.
Константа Керра В зависит от длины волны. Поэтому для удобства вводят величину К.=В'к1п. Поскольку В = —, то
(XIII.6)
Величины В и К зависят от таких важных свойств молекул, как электрическая поляризуемость и ее анизотропия, а также дипольный момент молекулы.
2. МЕТОДИКА ЭКСПЕРИМЕНТА
Общая схема эксперимента приведена на рис. ХШ.З. :
Источником излучения может служить натриевая (589 нм) или ртутная (546 нм) лампы, а также лазеры.
Монохроматическое излучение, полученное от гелий-неонового (632,8 нм) лазера или с помощью монохроматора от указанных ламп, проходит через поляризатор, направляющий плоскость поляризации света под углом 45° к электрическому полю ячейки Керра. Ячейка Керра представляет сосуд, в который вмонтированы электроды конденсатора, создающего поле 15...50 кВ/см. При ширине зазора между обкладками 2...3 мм подводится напряжение 10...15 кВ. Длина ячейки для жидкостей и растворов составляет 10...25 см, для газов и паров — до 1...1.5 м. При работе с газами с целью повышения чувствительности увеличивают давление.
После прохождения через ячейку Керра луч имеет эллиптическую поляризацию, так как несмотря на то, что амплитуды лучей
Рис. ХШ.З. Схема эксперимента для изучения эффекта Керра:
/ — лазер; 2 — поляризатор; 3 —ячейка Керра; 4 — четвертьволновая пластинка; 5 — анализатор; $ —детектор
236
и й’х одинаковы, их фазы различны. В аналитической форме уравнение эллипса можно получить следующим образом. Перепишем уравнения (XIII.1) и (XIII.2) в более общей форме, принимая за t величину t—1—пхъ исходя из произвольного угла между
8 г И 8,
$! = a cos wt,
И (XIII.7)
х — Ь COS (<о/ + у) ,
где а и Ь — амплитуды лучей и cfx, равные, соответственно, cos (О') и ffosin
Исключим из уравнения (XIII.7) время t следующими подстановками:
cos u>t =--- и % , = Ь (cos wt cos у — sm u>i sin y) =
л -*•
= ЬI-------cos у — sin wt sin у
Затем преобразуем второй член в скобках:
Я, ---cosy а
sin at sin у =
b
возведем в квадрат:
Я2Х sin2 <at sin2 cp = - 4-
2£,$х
COS2 у —-------cos у
ab
а2
сложим с выражением:
COS2 a>f sin2 у =
sin2 у,
получаем:
«2 гл 2 ср
+ ------— cosy = sin2 у.
Это есть уравнение эллипса, т. е. вектор электрического поля суммарного луча после выхода из ячейки Керра описывает эллипс.
Затем можно найти ориентацию эллипса и величины его полуосей для условий эксперимента по изучению явления Керра.
Упрощенное преобразование системы координат дает уравнение для угла поворота 0 от направления поля 8г‘
2а Ь ^20= а2_ 62 cos?«
237
Следовательно, при <р=л/2 0=0 ,т. е. оси эллипса совпадают с направлениями и ёх. Если падающий луч S направлен под углом 45°к<!Гг, то а=-^-=й = -^-. В этом случае угол 0 также равен 45°, т. е. главная ось эллипса а совпадает с вектором S (рис, XIII.4), В этой повернутой системе координат уравнение эллипса приобретает каноническую форму и его полуоси равны:
a sin <f a sin <р
а' =--- — и Ь' =---------------г ,.... =— .
У 1 — cos <р У ' + cos ср
Полученные выражения могут быть использованы для определения различия фаз q> следующим образом.
Эллиптический луч света выходит из ячейки Керра и направляется на четвертьволновую пластинку, ось которой совпадает с осью эллипса. Тогда эта пластинка становится компенсатором и превращает луч с эллиптической поляризацией в линейно поляризованный луч, который претерпевает поворот на угол а от направления S, определяемый анализатором (см. рис. ХШ.З). Измерения светового потока регистрируются детектором.
Найдем связь между а и ф. Эллиптический луч, падающий на четвертьволновую пластинку, может быть представлен системой уравнений для двух лучей в перпендикулярных плоскостях, разность фаз между которыми л/2, г. е.
$а, = a' cos u>r;
Рис. XI 11.4. Эллиптически поляризованный луч после прохождения ячейки Керра, представленный в системе координат z и у в виде суммы двух лучей с амплитудами а и Ь соответственно, а также в системе главных осей с амплитудами а' и Ь'
— Ь' sin u>t = b' cos tot
л \
2/
(знак вращения результирующего луча зависит от знака Ь'). Действие четвертьволновой пластинки состоит в том, что она снимает разность фаз двух перпендикулярных лучей и превращает их в лучи, распространяющиеся в одной фазе, т. е.
^ь, = a' cos tot;
'6Ь, = Ь’ cos u>t.
Но эти два луча в результате сложения дают линейно поляризованный луч, плоскость поляризации которого повернута к оси а' на угол а, выражаемый через отношение полуосей:
238
, b' V 1 + COS <P ‘g « = —~ = - •
а у 1 — cos <p
Тригонометрические преобразования дают
tga=tg?/2.
Таким образом, имеем удивительно простой результат:
a = T/2. (XIII.8)
Измеряя с помощью анализатора угол поворота плоскости поляризации света, прошедшего ячейку Керра и четвертьволновую пластинку, получаем возможность определить <р и, наконец, «ц— что и требуется от эксперимента. Возможно также прямое измерение разницы «ц—с использованием компенсаторов.
Измерения проводят при определенной температуре или при нескольких температурах. Термостатирование ячейки Керра осуществляется с точностью до ~0,2°. Точность измерения углов поворота составляет 10-5... 10~3 и достигает в ряде установок 10-7 рад.
3. ТЕОРИЯ ЭФФЕКТА КЕРРА
Задача теории состоит в установлении связи разности Пц— измеряемой экспериментально, с электронной поляризуемостью молекул в направлении поля н перпендикулярно полю.
Введем систему координат такую, что ось х направлена вдоль ячейки Керра (вдоль падающего луча), ось у перпендикулярно полю и ось z вдоль поля (см. рис. XIII.4).
Показатель преломления вещества, прн отсутствии межмолекулярного взаимодействия обусловлен средней электронной поляризуемостью молекул Ь, а их связь устанавливается уравнением молярной рефракции Лоренца — Лоренца:
«2 _ 1 м 1
<хш'9>
где М — молярная масса; р — плотность; N\ — постоянная Аво-гадро.
Внешнее электрическое поле создает внутреннее поле * + 2gr, действующее на молекулу в направлении г. Сред-3
нее значение поляризуемости молекул вдоль z обозначим через Б2* и в перпендикулярном направлении — Буу. Введем молярную константу Керра тК. следующим образом:
239
Используя уравнение (XIII.9), получаем
т
З^о
bzz---by и
3 \2
* 4- 2 /
(XIII.11)
Уравнение (XIII.11) дает возможность выразить тК через молекулярные параметры поляризуемости, т. е. найти теоретическое выражение для тК.
Однако величину т/( можно также представить в виде функции чисто экспериментально наблюдаемых величин, в том числе постоянной Керра В. Для этого в уравнении (XIII.11) заменим разность Б22—Ъуу=\Ь на выражение, которое найдем следующим образом. Поскольку величина АБ мала, будем искать ее дифференцированием по b уравнения (XIII.9). Используя правило дифференцирова-, и \' и’и — v'u ния дроби — =----------------, получим
\ V / V2
bn\n М 1 —
—— =—ЛГАД^ (XIII.12)
(п2 -Г Д2 р Зе0
При этом предполагалось, что эффект электрострикции равен нулю, т. е. изменение АУ=0 при $/=#0. Таким образом, имеем
QnXn М Зел
Дй = -------ГТ- • (XIII. 13)
(п2 4-2)2 р Л\ k '
Подставим это выражение в уравнение (XIII.11) и найдем тК. При этом выделим в качестве множителя Ап:
М 6п / 3 \2 1
р («2^2)2 \ е 4-2 ) $2
(XIII.I4)
Учитывая закон Керра в форме (XIII.5) и принимая, что Ап= =Пц—пх, получаем (8Z2 сокращается)
М Ж
6п
(п2 4- 2)2
(XIII.15)
Уравнение (XIII.15) дает экспериментально определяемую величину тК, выраженную через макроскопические свойства исследуемого вещества. Теперь задача состоит в том, чтобы выразить величину Б22—Ъуу—АЪ через молекулярные параметры поляризуемости, п £ +2 £
ь присутствии поля Fz —------молекулы будут ориентироваться
вдоль поля. В хорошем приближении, которое дает согласие с экспериментом, предполагается, что поле Fz не изменяет поляризуемость молекул. Поэтому усредненные по ориентациям молекулы компоненты тензора поляризуемости Ъц и Б1;- в лабораторной системе х, у, г будут иметь вид: 240
3
Ьц = 2 ьч <С°*ЦУ1)> q-l
И
(XIII.16)
з
Ь:1 = 5 < C0S (?') C0S (?/) > ’
<7 = 1
где i(j)=x, у, z\ bq—b\b2bz — главные значения эллипсоида поляризуемости молекулы; cos(gi) и т. д. — направляющие косинусы; < > — знак усреднения по ориентации молекулы.
При проведении вычислений удобно ввести углы Эйлера ф, гр, 0 для перехода от системы координат в молекуле (главные оси 1,2,3) к системе х, у, z, связанной с ячейкой Керра. Принимая достаточным классическое распределение Больцмана по углам, можно записать в общем виде выражение для усреднения, например
__и
ПГcos • cos (qj)e кг sin fldBdipd?
<cos (qi) cos(<?J) > = —---------------------------- , (XIII.17)
JjJ e liT sin BdBd^d?
где U=U(8, ф) —потенциальная энергия молекулы в поле Sz.
Ввиду осевой симметрии поля функция U не зависит от ф. Направляющие косинусы явно выражаются через 0, ф и ф. Функция U может быть выражена через параметры молекулы и напряженность внутреннего поля Fz:
U = (XIII.18)
3
где pz= cos (<?г); — компонента электрического дипольного
г- 1
момента молекулы, произвольно ориентированной в системе координат х, у, z; gg (9= 1,2,3) —компонента постоянного дипольного момента молекулы в системе главных осей эллипсоида поляризуемости; azz — компонента статической поляризуемости молекулы (azz= — baTzz+b3nzz — сумма атомной и электронной поляризуемости).
п - и
Для экспериментально используемых полей-------< I, что позво-
ляет ограничиться первыми членами разложения в ряд:
ехр kT + kT + 2 kT + 2W2
(XIII.19)
В результате перехода к системе х, у, z и усреднения получается диагональная матрица
241
~bxx О О
о ЪуУ о
О О b2z
byy о о
О ~Ьуу о
О о bzz
(XIII.20)
В КОТОрОИ 5хх — Вуу. В явном виде
*«==
ftj 4~ ft2 4- bj
3
_3_
45kT
[(ct! — a2) (bt — Ы) 4- (a2 — a ) (ft2 — *з) +
F2
+ («з - ai) (b3 - *01 + 45~fe2Zr2 [(fx? - p!) (bl ~ bi) -b (p! - nl) (bi - Ы) +
4* (рз — Pi) (*з—fti)] = b + F2 (9j + 02)
И
(XIII.21)
byy = b- F^+Qi)^,
(Х1П.22)
где
0i = (45ЛГ)-12 («/ - «/) (bl - bj)
(X111.23)
02 = (45Л2Г2)-! 2 (p? - Py) (b[ - bj)
Следовательно,
з , Aft = — F": (Aj + 02).
(Х1П.24)
Обычно член 0i называют анизотропным, а 02 — дипольным. Подставим разность b2Z—Ъуу из уравнений (XIII.24), в уравнение » 4- 2 (ХШ.Н), учитывая, что Fz =-------------'$z. Тогда получим
3
тК = (01 4- 02) • (XII 1.25)
2г0
Величина тК в системе СИ измеряется в м5-Кл2-Дж-2-моль_1 или м5 • В-2 • моль-1.
Изложенная классическая теория эффекта Керра обоснована для излучения, длина волны которого далека от длины волны поглощения вещества.
Постоянная Керра В или молярная константа Керра тК могут иметь как положительные, так и отрицательные значения. Знак этих величин обусловлен только дипольным членом 02. Анизотропный член 01 всегда больше нуля, так как последовательность уменьшения или увеличения главных значений статических щ, аг, аз и электронных bit Ьг, Ь3 поляризуемостей одинакова. Однако в 242
молекулах возможно несовпадение последовательностей щ, |12, Цз и Ь\, b2, b3. Тогда член @2 становится отрицательным, как, например, в молекулах SO2, СН3ОСН3, СНС13 и т. п. В этих молекулах Ь±>Ьп, т. е. электрический дипольный момент молекулы перпендикулярен наибольшему значению поляризуемости.
Выражение для @1 может быть упрощено в связи с тем, что в соответствии с экспериментальными и теоретическими данными принимают следующее соотношение величин:
1,1 (или 1,05). (XIII.26)
Тогда
е1= 1,1 (45*г)~12 <bi - W-
Молярная константа Керра многокомпонентной системы аддитивна и может быть записана в следующей форме, если Nt — молярная доля:
mK=^NimKi.
(XIII.27)
Это уравнение используется при исследовании растворов. Так, для двухкомпонентной системы уравнение (XIII.15) записывается в виде
где индексы (1) и (2) относятся к растворителю и растворенному веществу, соответственно, а (12) —к раствору.
Величину т/С2 находят экстраполяцией к бесконечному разбавлению из уравнения
(Л)и l‘m [лЛ12 — тК1 (1 — л2)]/л2.
(XIII.29)
Более точная теория эффекта Керра для конденсированных сред еще не завершена.
Таким образом, эффект Керра позволяет определить уравнение, связывающее макроскопические свойства вещества в соответствии с уравнением (XIII.15) с главными значениями поляризуемости молекулы и проекциями дипольного момента молекулы на главные оси эллипсоида поляризуемости.
243
4. ПРИМЕНЕНИЕ МЕТОДА РЕЛЕЕВСКОГО РАССЕЯНИЯ СВЕТА И ЭФФЕКТА КЕРРА
4.1. Определение главных значений эллипсоида поляризуемости молекул
В результате измерений молярных рефракций
+ +
3 анизотропии поляризуемости на данных по релеевскому рассеянию
Y2 = У [«1 - «2)2 + («2 - М2 + (iS - it)2]
и анизотропного ©t и дипольного @2 членов по данным эффекта Керра (определяемых раздельно при исследованиях в довольно широких интервалах температур в газах или из тК и у2 в случае жидких систем)
45Й7'01 (Р'/Ра) = (it - i2)2 4- (ij - is)2 + (i3 - it)2
и
45*27'202 = P2 (2*t - b2 - is) + p! (2i2 - i3 - ij) + (2i3 - it - i2)
в принципе возможно определение величин главных значений поля-ризуемости (bit b2, Ь3).
Так, в простейшем случае анизотропии, когда Z>t=/=Z>2=Z»3, например, для линейных молекул и молекул типа симметричного волчка, имеющих оси Сп при п^З, достаточно двух уравнений
ЗЬ = Ь[ + 2*2
и
Y2=(*1-*2)2,
которые дают решение при привлечении дополнительных данных, так как
Y = ± (it — Ъ2).
В более общем случае также необходимо использовать теоретические соображения или проводить анализ ряда замещенных для од* нозначного отнесения величин Ь\, Ь2, Ь3.
Наиболее обоснованной системой уравнений для определения Ьи b2, Ь3 является система, полученная из экспериментов для газовой фазы. Использование данных для растворов и тем более жидкостей предусматривает учет эффективного поля молекул в конденсированной фазе.
Для полярных молекул можно пренебречь членов 0j. Эффект Керра дает практически величину 02.
244
Для аксиально симметричных молекул, не имеющих дипольного момента, например этилен С2Н4, п-дизамещенные бензола, нафталин и т. п., явление Керра дает ту же информацию, что и релеев-ское рассеяние света. Поэтому в этих случаях принципиально возможно определить только две величины из трех.
4.2. Определение главных значений эллипсоида поляризуемости химической связи и группы атомов
В основе использования поляризуемостей молекул лежит свойство аддитивности этих величин по связям и группам атомов как экспериментальная закономерность. Каждой связи может быть сопоставлен эллипсоид поляризуемости. Ориентация его главных осей задается симметрией связи. Одна из осей совпадает с направлением связи, а главное значение этой продольной поляризуемости обозначается bL (или bt ). Для одинарных связей предполагают эллипсоид вращения и поперечные поляризуемости обозначают Ьт (или bj_). Для кратных связей типа С=С, С = О, C = N и т. п. перпендикулярно плоскости атомов, присоединенных к кратным связям, вводится вертикальная поляризуемость bv (или Z>±) (рис. XIII.5).
Аддитивность поляризуемости молекул подчиняется правилам тензорного суммирования. Предполагается, что конфигурация молекулы известна. Для произвольных осей эллипсоида поляризуемости молекулы используется обычное преобразование осей эллипсоида поляризуемости связи к заданной системе х, у, г:
*хх=2 2 bq^(qx); *ху=2 2 cos(?x)cos(^),
р q-L,T,V р q—L,T,V
(XIII.30)
где р — номер связи; q=L, Т, V — индексы главных значений поляризуемости связей; cos (qx) и cos (qy) — направляющие косинусы
Затем осуществляется переход к главным осям поляризуемости молекулы. Экспериментальные значения bi, b-?, Ьз для молекулы позволяют в соответствующей группе молекул определить Ьь, Ьт, bv связей. Если отсутствуют, например, данные по светорассеянию, то возможно использование констант Керра двух соединений известной структуры, содержащих исследуемую связь или группу. При этом необходимо, чтобы направление электрического дипольного момента по отношению к главным осям было различно.
Рис. ХП1.5. Главные полуоси эллипсоида поляризуемости:
а — одинарной и б — двойной связи углерод-углерод (6Ь, ьт, м
245
Таблица XIII.1. Величины bLt Ьт и bv (10~40 Кл-м^В-1) для ряда связей углеводородов
Связь Модельное соединение bL ьг
С-Н Алканы 0,72 (0,72) (0,72)
с—с Алканы 1,10 0,30 (0,30)
С—Сар* Алкилбензолы 1,37 ... 1,46 0,27- -0,41
Сар—Сар Бензол 2,49 0,23 0,66
С—С1 Хлористый метил 4,07 2,18 (2,18)
Сар—CI Хлорбензол 4,68 2,15 1,65
* Сар — атом углерода в арильной группе.
На основе данных по светорассеянию и эффекту Керра были определены bL, bT, bv многих связей * (табл. XIII.1).
Нарушение аддитивности позволяет выявлять эффекты взаимного влияния. Так, показано, что в молекулах СН2Х2 по сравнению с СНзХ происходит резкая перестройка эллипсоидов поляризуемости связей С—X (табл. XIII.2).
Таблица XIII.2. Разности поляризуемостей связей С—X _______в молекулах СН2Х2 и СН3Х (10-441 Кл-м2-В->)
X дьг ДЬу
С1 0,09 —0,41 0,01
Вг —0,01 0,59 0,13
I 1,69 1,38 —0,04
Изменения полностью локализованы в плоскости X—С—X (&т), а перпендикулярная составляющая by практически сохраняет свое значение [Д&( = &,с~х (СН2Х2) — &,(.СН3Х)].
Таблица ХШ.З. Отклонения от аддитивных величин поляризуемостей в пара-замещенных бензолов X —С6Н4—Y (10“40 Кл-м!В~') *
X Y ДЬ1 Дй2 ДЬз
no2 С1 0,82 0,33 —0,39
no2 (CH3)2N 14,22 13,14 — 13,60
no2 СНаО 3,00 —0,32 —0,32
* — вдоль X—Y, Д&2 — в плоскости бензольного кольца перпендикулярно X—Y, ДЬз — перпендикулярно плоскости.
Как проявление эффекта сопряжения трактуются отклонения от аддитивных величин (табл. ХШ.З) в системах пара-замещенных бензолов X—СбН4—Y.
* Аддитивная схема по Ле Февру предполагает 6L(C—•Н)=6т(С — Н) = = МС —Н).
246
Можно отметить увеличение A&t и уменьшение Д&з. Более детальная аддитивная схема, предложенная В. М. Татевским, практически включает эффекты взаимного влияния в систему расчета.
4.3. Изучение конформаций и внутреннего вращения молекул
Расчет по аддитивной схеме электронных поляризуемостей молекул, а именно величин Ь\, &2, &з для различных конформаций молекул позволяет после сравнения с экспериментальной постоянной тК решить вопрос о конформации. При этом необходимо знать значения валентных углов в молекуле. Например, для молекулы ди-метилхлорметилфосфинксида С1СН2Р(О) (СНз)2 возможны две конформации: транс и гош
о
С1
транс
гош
Рис. ХШ.в. Рассчитанная кривая тК(ф) для CgF«—CeFs
Сопоставление рассчитанных (при заданных валентных углах) и экспериментальных значений тК показывает, что равновесие этих форм существенно сдвинуто в сторону тракс-формы. Действительно, „Л (траке) =61-10~27; тК(гош) =— 64-10~27; тКэксп=
= 49-10-27 м5-В-2-моль-1 (раствор в СС1<).
Таким методом можно определить конформацию циклических систем, положение заместителя, например аксиальное или экваториальное, угол поворота групп, соединенных одинарной связью. Так, в молекуле СвНв—СвН5 константа Керра сильно зависит от угла вращения ф около связи С—С. Эту зависимость можно вычислить в форме т *(ф) (рис. XIIL6). Экспериментальное значение для раствора в циклогексане тК(эксп) = =43,2-10~27 м5-В~2-моль~1 соответствует Ф==62°, что хорошо согласуется с ф=70° для газовой фазы, полученной методом газовой электронографии, и совпадает с данными рентгеноструктурного исследования.
При исследовании равновесий с участием нескольких компонентов полезно комплексное использование методов: эффекта Керра, электрических дипольных моментов н релеевского рассеяния света.
Существенным ограничением рассмотренных методов является аддитивная схема,
247
необходимость априорного знания величин валентных углов, использование экспериментальных данных для растворов, где теория метода недостаточно строга.
ГЛАВА XIV •
ЭФФЕКТ ФАРАДЕЯ
1. ЯВЛЕНИЕ ФАРАДЕЯ. СХЕМА ЭКСПЕРИМЕНТА
В 1845 г. М. Фарадей открыл действие магнитного поля на прохождение света в веществе. Сущность явления состоит в том, что линейно поляризованный луч света, прошедший через оптически неактивное прозрачное вещество, изменяет плоскость поляризации, если вещество помещено в магнитное поле, направленное вдоль луча.
Схема эксперимента показана на рис. XIV.1. Источником света может служить ртутная лампа. Монохроматор выделяет излучение с определенной длиной волны X (частот v или <d=2jiv). Далее поляризатор формирует линейно поляризованный луч, который направляется в отверстие в магните (электромагните), ось которого совпадает с направлением магнитного поля В. При использовании электромагнитов значения индукции достигают 1 Т с однородностью 10-9 Т/см в зазоре см. Поляриметрическая кювета для жидкостей длиной ~3 см и объемом ~2 см3 термостатируется и фиксируется в зазоре латунными держателями. Естественно, что технические данные установок могут несколько отличаться. Анализатор позволяет определять угол поворота плоскости поляризации с высокой точностью (до ~ 10~3 град). Так же могут исследоваться газы и твердые вещества, а в частности молекулы, изолированные в матрице. Регистрация прошедшего излучения производится фотоэлектрическим методом. Поскольку измерение угла поворота осуществляется методом компенсации, т. е. до полного исчезновения прохождения света, вводится компенсатор (рис. XIV.1).
В 1854 г. Верде установил количественное соотношение для вращения плоскости поляризации в магнитном поле а:
a=V(\,T)dB, (XIV.1)
Рис. XIV.1. Схема прибора для наблюдения эффекта Фарадея:
1 — источник излучения; 2 — монохроматор; 3 — поляризатор: 4 — электромагнит; 5 — кювета; 6 — анализатор; 7 — фотоумножитель
248
где V(X, Т) — постоянная Верде, зависящая от длины волны X и температуры Т, является характеристикой вещества; d — длина кюветы, см; В — модуль индукции магнитного поля. Строго говоря, а пропорционально cos 0, где 0 — угол между направлением распространения света н магнитного поля. Обычно 0 равно нулю.
Для растворов в случае невзаимодействующих веществ соблюдается аддитивность, т. е.
а араст-ль "4* ®раств. в-во’ (XIV.2)
где для концентрации С растворенного вещества имеем
^раств. в-во [Траста, в-во] CdB. (XIV.3)
Знак угла вращения а принимается положительным для вращения плоскости поляризации по часовой стрелке, если наблюдатель смотрит на источник света и распространение света совпадает с направлением магнитного поля, т. е. условие знаков вращения совпадает с таковым для спектрополяриметрии (см. гл. VIII, рис. VIII.5). Для моля вещества вводится молярное вращение чистого вещества:
или для растворов:
V а
= — . (XIV.4)
С С ио
где р — плотность вещества.
Эффект Фарадея является общим для любых прозрачных веществ. Для оптически активных веществ угол поворота представляет сумму углов: 1) угол вращения плоскости поляризации оптически активного вещества и.2) угол вращения линейно поляризованного света вследствие эффекта Фарадея.
Постоянные Верде очень малы по численному значению (сотые доли угловых секунд). Лишь для ферромагнитных металлов углы поворота достигают значений до градуса и более. В основном многие вещества вращают плоскость поляризации излучения влево при .D-линии натрия. Некоторые парамагнитные вещества имеют положительное вращение. Уравнение Верде [см. уравнение (XIV.1)] выражает зависимость знака а от направления В. Поскольку а не зависит от направления луча света по отношению к В, возможно использование многоходовых кювет (рис. XIV.2). Этот эффект можно объяснить тем, что cos0 = cos(—0). Физически это означает, что при обратном направлении прохождения луча света его плоскость поляризации вращается в противоположную сторону по отношению к этому лучу, а по отношению к полю В — в том же направлении,
249
Рис. XIV.2. Использование многоходовой кюветы в эксперименте по эффекту Фарадея
т. е. получается двойное вращение (2 а). Многократное отражение позволяет увеличить а до десятков градусов.
Знак вращения зависит от длины волны используемого излучения. Более полная информация о веществе может быть получена при определении дисперсии магитного оптического вращения (ДМОВ), т. е. при изучении функции а=а(2и), или a=a(v). Однако можно изучать поглощение света луча с правой и левой круговой поляризацией или зависимость Ae(v)=ez(v) — —Br(v) от v, называемую магнитным круговым дихроизмом (МВД).
2. ТЕОРИЯ ЭФФЕКТА. СВЯЗЬ С ЭФФЕКТОМ ЗЕЕМАНА
С точки зрения классической электронной теории прохождение света через прозрачное вещество обусловлено осцилляцией электронов в молекулах (атомах), вызванных внешними полями световой волны и магнитного поля в случае эффекта Фарадея. Колеблющийся электрон является источником вторичной волны, которая представляет проходящий через вещество свет.
Поскольку индукция магнитного поля световой волны значительно меньше внешнего поля, в уравнениях учитывают только последнюю, т. е. | Ввиеш| = | В| =Вг. Направление распространения света выбирают за ось z. Вектор напряженности электрического поля световой волны 8 находится в перпендикулярной плоскости ху (рис. XIV.3). Если не учитывать затухания, то дифференциальное уравнение колеблющегося электрона можно записать в следующем виде с использованием выражения для силы Лоренца:
г+ «>ог = —(£ + [гХВ]),
т
(XIV.5)
где г—радиус-вектор; — собственная частота колебаний электрона; т — масса и ё— заряд электрона. Для проекций вектора г имеем, учитывая, что ВХ=ВУ = О и [гХВ]= (уВг)х— (хВг)у:
* ~ ~ + “с* = ~ ^х
tn. tn
и (xiv.6)
• • е « 2 е „
У 4- т 4- == —
т т
250
Рис. XIV.3. Схема для векторов 8г и 8i в декартовой системе координат, в которой ось z направлена вдоль поля В
Линейно поляризованный луч S «на входе» (г=0) может быть представлен суммой двух лучей с круговой поляризацией ёг и St (см. гл. VIII), компонентами которых являются функции Sx и Sy. Для правой системы координат, (рис. XIV.3) лучи с правой и левой круговой поляризацией выражаются уравнениями:
<£*r = $ocos“^ ^'yr = — <fo sin
и (XIV.7)
%Х1 = $о cos %yl = +^’osinwi'.
Представление в комплексной форме двухмерного вектора Sr или Si позволяет одним уравнением задать каждый из лучей
== *&хг 4* ^уг 2=1 exp ( - iwt)
и (XIV.8)
%i = %xt + »8’уг = 8’оехР( + со-
множитель ехр(—lent) вращает вектор So с постоянной угловой скоростью ю в правой системе координат по часовой стрелке, т. е. вправо от наблюдателя, а ехр(йо/) вращает влево.
Тогда систему уравнений (XIV.8) можно объединить, доумно-жив второе на г = / — 1. Тогда получим:
“ . (х + ‘ у) — z Т7" (х + + ш0 Iх + 1у) — ($х + *$!/)•
d/2 т dt и т
(XIV.9)
Решение уравнения (XIV.9) представим в видех + н/=Го ехр (±цо/). После подстановки этого выражения и дифференцирования найдем,
251
что
_______(e/m)%o______ (wq — м2) j. (ешВг)/т
(XIV.10)
Поскольку не учитывалось затухание колебаний в уравнении (XIV.5), в уравнении (XIV.10) получена действительная функция. Теперь можно вывести уравнение для показателя преломления, так как
Л2=е= 1+р/^£о, (XIV.11)
где поляризация вещества Р=УУёг или P=Ner. Таким образом, запишем с учетом уравнений (XIV.8) и (XIV.I0) и ё——е
„2 _ 1 + -У [е2/(те0.)
Г (“о — “2) — KeBz)/m] О)
и
(XIV.12)
(XIV.13)
2 < , N
— 1 ”Т” п - в
(в* — аг) + (еВ£/т.)ш
Из уравнения (XIV.I2) найдем —«г2 и затем щ—пг. Имеем:
п2_п2= _дг._£!___________2(е/т) Вги>
mzo (о)2 — ш2)2—(еВго)/т)2
Учитывая, что рассматривается случай, когда £о<юо, т. е. рассеяние в области, далекой от поглощения, можно пренебречь членом “j в знаменателе уравнения (XIV. 13). Перепишем это урав-
2 2
nl ~nr nt + nr „
нение для nt — пг=— -------, где п= . Получаем
2п 2
т2е0 п(о)0 —о)2)2
(XIV.H)
В гл. XIII было показано, что вращение плоскости поляризации определяется уравнением
« = Y" (ni - nr) d = (nz - nr) d.
Следовательно, в случае эффекта Фарадея
УеЗ 0)2rfB, а= “ 2пт^с ’ (XIV.15)
Если ш<(во, то а<0, т. е. вращение происходит влево [см. уравне-252
Рис. XIV. 4. Схема действия силы Лоренца:
для левого круга (а) при движении электрона против часовой стрелки частота вращения увеличивается, для правого круга (о) — уменьшается
Рис. XIV.5. Кривые дисперсии Пг(со) и «/(со) и разность «/(со)—«г(со) =Д«(со) в магнитном поле Bz
ние (XIV.13)]. Постоянная Верде в указанном приближении имеет вид
о)2
v=—(XIV.16)
В ряде случаев удобно выразить постоянную Верде через производную дп/да>. Так как в уравнении (XIV.12) то
. т
е дп
-----о»—. (XIV.17).
2тс--'
Для этого уравнения не нужна, оценка соо- Оно полезно также при рассмотрении свободных электронов, когда <»о=О.
При переходе к области поглощения в эксперименте по эффекту Фарадея необходимо учитывать эффект Зеемана — расщепление спектральных линий испускания и поглощения в магнитном поле. Согласно упрощенной схеме эффекта Зеемана влияние магнитного п'оля в направлении z состоит в том, что колеблющиеся в плоскости ху электроны можно рассматривать как вращающиеся по и против часовой стрелки (рис. XIV.4). Однако сила Лоренца Гл = = —e[vXB] = —е[гХВ] будет изменять частоту вращения электронов. Для левого круга частота увеличивается, поскольку сила Fa направлена в центр (правило правой руки) и
е
= coq 4" До) == o)q -f" т» (XIV. 18)
Для правого круга сила F к направлена от центра и частота уменьшается:
(XIV.19)
253
Из рис. XIV.4, а видно, что движение электрона по левому кругу может находиться в резонансе с лучом St, а движение по правому кругу (рис. XIV.4, б)—с Sr- Уравнения (XIV.18) и (XIV.19) можно получить из (XIV.12) при Тогда для слабых магнитных полей Bz величина будет близка к нулю. Уравнения
(XIV.12) можно преобразовать в новую систему:
е2 N л, = 1 4- и ( еВ* Y 2 (“° 2т ) “ е2 N И/ — 1 4- • ' / еВ, \2 2от )
(XIV.20)
Таким образом, дисперсионные кривые пг(ю) и щ(ю) будут иметь одинаковый характер, но смещены по оси абсцисс. На рис. XIV.5 представлены кривые п/(ю), пг(а) и щ(ю)—пг(а) в широкой области со, включающей поглощение. Эти кривые могут быть получены из уравнений с учетом поглощения. В связи со сдвигом вправо по оси ю для кривой п/(ю) для области ю<С<оо имеем nt—пг<0, т. е. вращение влево. Однако в области поглощения nt>nr, т. е. вращение положительное. Затем снова вращение становится отрицательным.
Эффект Зеемана для нескольких электронов с учетом спина электрона имеет более сложный характер («аномальный» или сложный эффект Зеемана). Его рассмотрение следует проводить на основе квантовой механики.
В магнитном поле В вырожденные электронные энергетические уровни Е атомов и молекул для полного момента импульса /=#0 расщепляются на ряд уровней, расстояние между которыми пропорционально | В| — В. Частоты перехода между уровнями Е" и Е', например, в спектрах поглощения будут определяться уравнением
u = uo + “SjMj.-),
(XIV.21)
где g — фактор Ланде; Mj—магнитное квантовое число, которое изменяется от —J до +J.
Спектры поглощения подчиняются следующим правилам отбора: ДА4=0, ±1. При наблюдении вдоль поля ДЛ4=±1. Переход для ДЛ1= + 1 обладает круговой поляризацией влево, а для ДЛ4=—1—вправо (рис. XIV.6). Нижний уровень соответствует М<0, так как в поле В разность энергий AE=gpBEM (рБ — магнетон Бора — единица магнитного момента). Из-за расщепления основного состояния изменяется его заселенность и, следовательно, интенсивность поглощения для компонент различна.
254
Е"
у=о
Е1
И'=0
Е"
J =1
О
-1
О
&М=-1 ДМ= + 1
3''= 1
М
О
Ej____
1'=1/2
+ 1/2
1/2
м
+ 1/2
-1/2
-1/2 £М=-1*М^1
S) М +2
-2
О
О -1
Т^2
Е
Г*
О -1 ди=-1 дм=+1 5)
Рис. XIV.6. Эффект Зеемана — расщепление вырожденных уровнен в поле:
показаны переходы с правилами отбора АЛТ—±1 для испускания нли поглоще-ння вдоль поля В и определенной поляризацией г или I
-1
Дм=-1 ДМ=*1
Е
и
Таким образом, квантово-механическое объяснение эффекта Фарадея состоит в том, что в магнитном поле В имеет место расщепление вырожденных электронных уровней атомов и молекул, переходы между которыми происходят с поляризацией излучения вправо (ЛМ=—I) и влево (ДАГ= + 1).
Если в молекуле нет вырождения электронных уровней, то эффект Фарадея возникает в связи с возмущением собственных волновых функций в поле фо и потерей ими центра и плоскости симметрии. Так, возмущенная волновая функция ф может быть выражена через функцию в отсутствие поля фо и волновые функции возбужденных состояний молекулы фй уравнением
Ф = Фо + (XIV.22)
ь
255
В первом порядке теории возмущения
- в 4г №
J on
Ck ~ -------, (XIV.23)
E0 — Ek
где 0 — угол, отсчитываемый от В; Ео и Eh— энергии состояний фо и фй.
Анализ уравнения (XIV.23) показывает, что если фо=2рг, то дает чувствительный вклад в сл, т. е. в деформацию фо-
Для орбитали фо=2з в возмущении участвует 4/-орбиталь. Эффект Фарадея, обусловленный л-связью, будет больше, чем обусловленный о-связью, поскольку разница в энергии Е2р—Ем меньше, чем E2s—E4f. К сожалению, как это видно из уравнения (XIV.22), точное решение прямой задачи затруднено в связи с тем, что волновые функции молекул неизвестны.
3. МАГНИТНЫЙ КРУГОВОЙ ДИХРОИЗМ (МКД) И ДИСПЕРСИЯ МАГНИТНОГО ОПТИЧЕСКОГО ВРАЩЕНИЯ (ДМОВ)
Эффект Фарадея зависит от частоты используемого света. Изменяя частоту падающего света в значительном интервале, можно получить зависимость угла вращения от частоты а (со), т. е. кривую ДМОВ. Для приведенных примеров снятия вырождения уровней в поле (рис. XIV.6) кривые ДМОВ будут существенно различны. Для переходов (рис. XIV.6, а) дисперсионные кривые показателей преломления п;(ю) и пг(а>) сдвинуты относительно друг друга, и кривая ДМОВ показана на рис. XIII.5. Для вырожденного основного электронного состояния (рис. XIV.6, б) заселенности расщепленных подуровней в магнитном поле различны. Это существенно изменяет форму кривой а (со) даже больше, чем различие в собственных частотах соо(г) и соо(/). Поэтому кривая разности щ(со)—пг(<о) практически по форме повторяет кривую п(со) или кривую ДОВ (см. гл. VIII).
При вырождении основного и возбужденного электронных состояний (рис. XIV, виг) электронным переходам будут соответствовать более сложные кривые ДМОВ.
Эффекты Фарадея в виде ДМОВ очень чувствительны к наличию в молекуле расщепления электронных уровней энергии, хотя само расщепление невелико, особенно по сравнению с шириной полосы поглощения. Сдвиг же кривых пг(ю) и пДю) и разность этих кривых простираются на значительный интервал частот. Так, расщепление линий в электронных спектрах испускания или поглощения молекул в полях ~ 1 Т составляет ~ I см-1. В то же время ширина полос в конденсированной фазе достигает Ю3 см-1.
Однако практически более удобно изучение МКД (магнитный круговой дихроизм), которое заключается в измерении кривых 256
Дев (со) = е/(co)—ег(со) для различных со (е/ (со) и ег(со) —молярные коэффициенты поглощения). Кривые МКД для данного электронного перехода в меньшей степени зависят от наличия других переходов, т. е. от эффектов фона, чем кривые ДМОВ.
Схема эксперимента МКД аналогична КД (см. гл. IX). После четвертьволновой пластинки лучи с круговой поляризацией г и I проходят через кювету, находящуюся в поле В. Обычно исследуют растворы, жидкости илн твердые тела.
Методами теории возмущений получено следующее выражение для Дев (со) при переходе из состояния а в состояние / изотропного вещества
f! С \ 1
A f (<>) + I В -I g (ш) ,
\ kT j J
(XIV.24)
где величины А, В, С выражаются через электрические и магнитные дипольные моменты перехода
А = —у" У[<Ф/ I V-mz 1 Фр — <Фа I Pros 1фа>] I™ ( <Фа 1‘Ргх I Фр <Ф/ I V-ej I Фа > ) I «а
(XIV.25)
о 3 (Ф> I Р'пг I Фа ) . , . — . , , , I I
В = ~ 2л1т Zi --------1--------(<Фа 1 Ргх I Фр <Фр Ргу I Ф*>~
da j h“ka
—-<фа1ргрфр <Ф; I Йх I Р>) + 2 <Ф/1^г|1ра> (<фа| £ел| фр X
X <Фа | Реу I Фа> — <Фа I Ргу I Ф/> < Фа I Ргх I Фа >)] I (XIV.26)
С = ^у- V <фа I bnz | Фа> ^(<Фа I Ргх I Фр <ФрРгрФа>), (XIV.27)
где da — степень вырождения основного уровня а; |imz — оператор магнитного дипольного момента вдоль оси z.
Функция g (со) представляет форму линии поглощения прн частоте со/g с полушириной у;а:
“3У;а
£ = / 2 2V_L 2v2 ’
(шуа-“2) +“2Y;a
а функция /(ю) —ее производную:
- и2) Y/g
Ч(Ра-“2)2 + “^а]2'
Обе функции представлены на рис. XIV.7.
(XIV.28)
(XIV.29)
9—1377
257
Величина А определяется вырождением основного или возбужденного электронного состояния, т. е. связана с эффектом Зеемана первого порядка. Коэффициент В существует для любого перехода и не зависит от вырождения, так как определяется смешением электронных состояний в магнитном поле. Эта величина включает только недиагональные элементы матрицы оператора магнитного дипольного момента. Коэффициент С не равен нулю
Рис. XIV.7. Зависимость функ- только при вырождении основного ций — g(co) и — f(<o) электронного состояния, особенно для
нечетного числа электронов в молекуле. Этот терм определяет зависимость МКД от температуры, поскольку заселенность расщепленных в магнитном поле уровней будет различной.
Суммарная кривая Дев(ю) зависит от соотношения А : В : С. Если превалирует коэффициент А, то Дев(и) изменяется как дЦды. При С»А кривая Дев(ю) имеет симметричный вид функции Я(<»)-Коэффициент В обычно меньше А и С; которые определяют форму кривой МКД. Задача эксперимента состоит в том, чтобы определить А, В, С и на их основе оценить знаки и численные значения магнитных и электрических дипольных моментов переходов.
Величины А, В и С определяются из экспериментальных кривых МКД методами моментов. Поскольку экспериментально удобнее измерять молярную эллиптичность [0]м=ЗЗОО Де, для коэффициентов А, В, С соответственно используют следующие выражения (в СИ):
А = 3,093-10-81J dw (» — <ор) [6]м/«>
(XIV.30)
1и
В + —— = -3,093-10-85 d»[6]M/», (XIV.31)
J
где (Ufa — центр полосы.
Чтобы найти величины В и С, изучают температурную зависимость МКД. Для двух и большего числа переходов картина МКД усложняется в связи с перекрыванием кривых МКД. Важно то, что положительный пик МКД ([6]м>0 и ez—ег=Де>0) соответствует отрицательной величине коэффициента В и наоборот.
Поскольку А и С обусловлены вырождением, оии не равны нулю для молекул, обладающих осью симметрии Сп при п^З. Необходимо отметить, что энантиомеры имеют одинаковые кривые МКД. При изучении энантиомера наблюдается аддитивность МКД и КД. 258
4. ПРИМЕНЕНИЕ ЭФФЕКТА ФАРАДЕЯ В ХИМИИ
4.1. Аддитивные свойства постоянной Верде
В связи с трудностями расчета эффекта Фарадея, возникающих из-за отсутствия точных выражений для электронных волновых функций молекул, в органической химии используются эмпирические закономерности, связывающие константу Верде с химическим строением молекул. Так, для гомологических рядо& производных ХСН2(СН2)пСНз, где X — СН3, ОН, СНО т. д., имеет место аддитивность, например, величин VM(CH2). Соединения с кратной связью имеют величину VM больше, чем для насыщенных соединений.
На основе данных при исследовании большого числа органических и элементорганических соединений установлено, что каждой связи между атомами А и В можно отнести парциальную величину
А В \
ЛА ЛВ /
где А и В — элементы, пА и «в — число валентных электронов.
Так, для ряда связей углерод — углерод получено (в мкрад) в С2Н6^(-^-_-^-)=18,5; в СН2=СН2 = =135;
в СН=СН Ум(—~~ = —С 'j =173. При переходе от связен \ 4 4 /а+п
углерод — углерод к связям углерод — элемент (IV группы) Vm (у—увеличивается от 74 мкрад для С — Si до 278 мкрад для С — РЬ. В одном периоде имеет место увеличение /С Е \
Ум---------) при переходе от IV группы к VII.
\ 4 пЕ J
Отклонения от аддитивности могут быть интерпретированы как проявление специфических эффектов взаимного влияния атомов и групп атомов. Так, в молекулах с сопряженными связями наблюдаются значительно большие величины Ум, чем полученные в расчете по аддитивной схеме. В производных бутадиена это превышение (экзальтация) составляет ~ 120 мкрад. В системах С^С—С==С отклонение меньше (около 63 мкрад). Данные подобного типа очень полезны для выявления характера взаимного влияния. На основе данных по эффекту Фарадея было высказано предположение о резком уменьшении ароматичности в молекулах фторбензола СбН5Р, фурана, пиррола, увеличение, ароматичности в N-окиси пиридина
по сравнению с самим пиридином.
9
259
4.2. Изучение электронных переходов в комплексных соединениях
с помощью МКД
Прямые наблюдения эффекта Зеемана сложных молекул из-за большой ширины полос в электронном спектое практически невозможны. Метод МКД позволяет определить электронные переходы в магнитном поле, поскольку они имеют различную круговую поляризацию (см. рис. XIII.6). При использовании МКД изучается сумма полос поглощения, сдвинутых на небольшую величину, но имеющих разный знак Дев=ег+(—ег). Информация об электронных переходах в эффекте Фарадея в форме МКД и в эффекте Зеемана в принципе одинакова.
Важно отметить, что по сравнению с электронной спектроскопией метод МКД потенциально позволяет обнаружить слабый переход вблизи сильной полосы, так как Де/е такого перехода может быть больше, чем для сильной полосы, или
Рис. XIV.8. Качественное гопоставленне электронного гпектра поглощения (а) и МКД (б) для металлопор-фнринов
имеет противоположный знак. Здесь ситуация такая же, как и в случае КД оптически активных молекул (см. гл. IX).
Таким образом, метод МКД является одним из вариантов метода электронной спектроскопии.
В подавляющем большинстве молекул органических соединений основной электронный уровень не вырожден. Это означает, что коэффициент С равен нулю [см. уравнение (XIV.24)].
Наиболее широко МКД используется для исследования неорганических и комплексных соединений, включающих основания нуклеиновых кислот и полинуклеотиды. Так, в электронном спектре металлопорфиринов, имеющих симметрию Dih, наблюдаются две полосы (рис. XIV.8). Оба возбужденных состояния дважды вырождены. Из кривой МКД видно, что коэффициент А первого перехода во много раз больше второго. Для различных металлов это соотношение составляет в среднем 9:1. Значение В коэффициента существенно зависит от заместителей в кольцах. МКД очень чувствителен к степени окисления железосодержащих порфиринов.
На примере молекулы T1CI4, основное состояние которой спин-спарено (а6), можно показать эффективность применения метода МКД. Для этой молекулы был решен вопрос о том, на какой возбужденный уровень совершается электронный переход. Верхние занятые уровни Ц и tz принадлежат несвязывающим молекулярным орбиталям лигандов (рис. XIV.9). Из МКД эксперимента по знаку
260
Рис. XIV.9. Возможные схемы электронных переходов для TiCh
и величине Цт следует, что электронный переход (рис. XIV.9, а) должен быть записан как Ai Т% (t\ -► е*). Расчеты на основе ППДП МО оказались ненадежными, так как привели к выводу о переходе (рис. XIV.9, б). Благодаря высокой чувствитель-
ности метод МКД позволяет изучать запрещенные переходы в тетраэдрических соединениях, ионов редкоземельных элементов и ряда других ионов.
4.3. Аналитические применения эффекта Фарадея
Созданы установки промышленного типа, работающие в переменных магнитных полях. Измерения вращения плоскости поляризации производятся методом компенсации при сравнении магнитооптических параметров изучаемого и стандартного веществ. Точность измерения углов поворота составляет -~0,05'.
В связи с высокой чувствительностью и возможностями автоматизации метод на основе эффекта Фарадея используется для качественного и количественного анализа жидкостей определения элементного и молекулярного состава вещества с точностью до тысячных долей процента (мае.) в любом диапазоне изменения концентраций. В настоящее время можно проводить анализ редкоземельных элементов, хлорсодержащих органических соединений, соединений с двойными, тройными и сопряженными кратными связями, неорганических и органических кислот и т. п.
261
Оптические методы нашли широкое применение в решении задач химического строения и физических свойств молекул различных классов. Важно отметить, что для определения главных значений тензора электронной поляризуемости используются данные нескольких методов, например данные по молекулярной рефракции, степени деполяризации релеевского рассеяния, двулучепреломления (электрического эффекта Керра) и электрических дипольных моментов. Такая интеграция методов требует более строгого подхода в интерпретации определяемых физических величин. Особенно этот вопрос остро стоит в связи с использованием теории взаимодействия излучения с изолированными молекулами. Учет влияния молекул жидкой среды требует дальнейшей разработки теории.
В плане развития экспериментальных исследований необходимы данные по поляризуемости ионов свободных радикалов молекул в возбужденных состояниях, которые могут быть использованы для изучения механизмов химических реакций.
В этом разделе методика эффекта Фарадея стоит несколько особо, так как данные этого метода находят основное использование в области спектроскопии. Вместе с тем аддитивность константы Верде в определенном смысле соответствует аддитивности поляри-зуемостей. В настоящее время требуется более глубокая теория, связывающая химическое строение и величины постоянных Верде.
Наиболее важно применение эффекта Фарадея, а именно магнитного кругового дихроизма, в относительно высокосимметричных системах, таких, как координационные соединения, ароматические соединения и биологически активные соединения. Этот метод имеет значительные преимущества перед методом электронных спектров поглощения. Однако слишком еще преобладает эмпирический подход в анализе экспериментальных данных. Необходимо дальнейшее развитие теории метода.
Контрольные вопросы
Глава ХП
1. Почему наиболее эффективно изучать рассеяние света под углом 90° к падающему?
2. Какая роль флуктуационных неоднородностей в теории релеевского рассеяния?
3. Почему анизотропия молекул вызывает деполяризацию рассеянного излучения?
4. Что такое степень деполяризации рассеяния и как она зависит от характера поляризации падающего излучения и величины анизотропии поляризуемости?
5. Какие условия необходимы, чтобы теория деполяризации рассеяния в газах могла быть использована для рассеяния в жидкостях и растворах?
6. Как применяется принцип аддитивности в теории деполяризации рассеяния?
7. Опишите схему эксперимента по измерению степени деполяризации рассеянного света.
262
ГлаваХ!!!
1. Как формулируется закон Керра? В чем состоит искусственная анизотропия веществ в электрическом поле?
2. Опншнте схему н условия эксперимента для наблюдения электрического явления Керра.
3. Почему при прохождении ячейки Керра линейно поляризованный луч становится эллиптически поляризованным?
4. Сформулируйте определение молярной константы Керра.
5. Какие экспериментальные данные необходимы для расчета молярной константы Керра индивидуального вещества?
6. Выведите уравнение молярной константы Керра, выраженной через главные значения тензора электронной поляризуемости.
7. Объясните качественно температурную зависимость константы Керра.
8. Чем определяется знак молярной константы Керра?
9. Как выражается аддитивность в уравнении для молярной константы Керра?
10. Как решается задача определения главных значений тензора электронной поляризуемости молекул?
11. Как решается задача расчета главных значений тензора электронной поляризуемости химических связей в молекулах?
12. Как решается задача о конформации молекул на основе экспериментальных величин молярных констант Керра?
Глава XIV •
1. В чем состоит явление Фарадея?
2. Сформулируйте уравнение Верде.
3. Опишите схему и условия эксперимента для изучения явления Фарадея.
4. Как классическая электронная теория объясняет явление Фарадея? Выведите уравнение для угла вращения линейно поляризованного света при прохождении через вещество параллельно приложенному магнитному полю.
5. В чем состоит связь явлений Фарадея н Зеемана?
6. Почему в явлении Фарадея наблюдается искусственная анизотропия для лучей с правой и левой круговыми поляризациями?
7. Какие величины входят в уравнение для магнитного кругового дихроизма молекул? Каков их физический смысл?
8. Какие применения в химии имеют явление Фарадея и магнитный круговой дихроизм?
ЗАКЛЮЧЕНИЕ
В учебнике изложены теоретические основы, схемы и условия эксперимента, а также наиболее важные применения в химии более тринадцати физических методов исследования. Некоторые методы исследования развиваются в нескольких направлениях и поэтому нельзя назвать точно их число.
Естественно, что материал книги является продолжением уже вышедшей книги «Физические методы исследования в химии. Структурные методы и оптическая спектроскопия» (см. список литературы).
В целом в двух книгах представлено более двадцати физических методов. Не все методы одинаково широко используются. Различна значимость результатов исследования. Тем не менее можно сказать,
263
что данные каждого метода являются уникальными. Несмотря на достигнутую разработанность ряда методов, процесс развития, углубления и расширения применений практически всех методов продолжается. Особенно следует отметить методы масс-спектрометрии, ИК фурье-спектроскопии, лазерной КР, ЯМР, рентгеновской и фотоэлектронной спектроскопии, а также магнитного кругового дихроизма.
К большому сожалению, следует указать на то, что некоторые методы практически не используются в нашей стране из-за отсутствия соответствующей аппаратуры, что существенно снижает уровень исследований. Это касается частично новинок спектроскопии ЯМР, а также фотоэлектронной спектроскопии, колебательного кругового дихроизма, магнитного кругового дихроизма. Можно надеяться на то, что дальнейшее развитие научного приборостроения ликвидирует этот пробел.
Вне зависимости от практических возможностей использования того или другого метода постоянную ценность представляют принципиальные возможности методов. Освоение этого материала должно способствовать более глубокому пониманию и решению химических проблем.
Важным вопросом является развитие практических навыков в использовании физических методов. Реально, по-видимому, это осуществить на ограниченном числе методов типа масс-спектрометрии, ИК, УФ и ЯМР спектроскопии и некоторых других методах.
Обе книги могут быть полезными для преподавания предметов «Математика» и «Физика», так как выделяют те разделы этих предметов, которые важны для химиков. Так, кроме дифференциального и интегрального исчисления химику, активно использующему физические методы в своей работе, необходимы разделы линейной алгебры, теории групп и интегральных преобразований. Для решения обратных задач методов особое значение имеют вычислительные методы. С точки зрения преподавания физики важно уделить внимание вращательному движению, магнитным явлениям и, конечно, квантовой механике, ее приближенным методам решения уравнения Шредингера, особенно методу теории возмущений. Некоторые задачи физического практикума также могут ориентироваться на дальнейшее использование в практике физических методов исследования в химии.
С целью взаимного согласования курсов лекций материал данного учебника также может быть использован в курсах «Строение молекул», «Физическая химия», «Органическая химия», «Неорганическая химия», «Аналитическая химия» и др.
ПРИЛОЖЕНИЕ
ОГЛАВЛЕНИЕ К ЧАСТИ I
Предисловие............................................................. 3
Введение. Общая характеристика физических методов....................... 5
1. Прямая и обратная задачи методов ................................ 5
2. Спектроскопические методы исследования........................... 7
3. Дифракционные методы............................................. 9
4. Оптические и другие методы..................................... 11
5. Характеристическое время метода................................. 11
6. Значение физических методов для теоретической химии............. 13
7. Современный уровень и перспективы развития физических методов 14
Контрольные вопросы ............................................... 16
Первый раздел. Методы масс-спектрометрии............................... 17
Глава I. Процессы ионизации и принципиальные схемы масс-спектрометров ............................................................... 19
1. Ионизация атомов и молекул...................................... 19
2. Процесс ионизации и типы ионов ................................. 19
3. Методы ионизации................................................ 24
4. Принципиальные схемы масс-спектрометров ........................ 28
4.1. Магнитный масс-спектрометр ................................... 28
4.2. Динамические масс-спектрометры................................ 33
4.3. Спектрометр нон-цнклотронного резонанса ...................... 35
Глава II. Применение масс-спектрометрии ............................... 36
1. Идентификация и установление строения веществ................... 36
2. Определение потенциалов ионизации молекул н появления нонов . 40
3. Масс-спектральные термодинамические исследования................ 43
4. Масс-спектрометрня в химической кинетике......................... 53
Контрольные вопросы ............................................... 56
Второй раздел. Методы определения электрических дипольных моментов молекул....................................................... 57
Глава III. Теоретические основы методов................................ 59
1. Электрический дипольный момент молекулы......................... 59
2. Энергия молекулы во внешнем электрическом поле.................. 60
3. Ориентационная поляризация молекул.............................. 62
4. Эффект Штарка н квантово-мехаинческнй подход к выводу ориентационной поляризации молекул ..................................... 64
5. Диэлектрик в электрическом поле................................ 68
Глава IV. Экспериментальные методики и применение данных по электрическим дипольным моментам молекул в химии . . 71
1. Первый метод Дебая — определение электрического дипольного момента молекул паров веществ........................................ 71
2. Второй метод Дебая — определение электрических дипольных моментов молекул веществ в разбавленных растворах.................. 72
3. Отклонение молекулярного пучка в неоднородном электрическом поле .............................................................. 74
4. Метод электрического резонанса.................................. 76
5. Использование данных по дипольным моментам в химии .... 78
Контрольные вопросы ............................................... 82
Третий раздел. Методы определения геометрического строения молекул................................................................ 83
265
Глава V. Микроволновой метод, исследовании вращательных спектров молекул...................................................... 85
1. Вращательные спектры поглощения молекул................... 85
2. Методика эксперимента в микроволновой вращательной спектроскопии ........................................................... 94
3. Методы расчета геометрических параметров молекул.......... 97
4. Определение электрических дипольных моментов молекул .... Ю5
5. Исследование внутреннего вращения и инверсии молекул .... Ю7
6. Некоторые результаты микроволновых исследований ...... 1 Ю
Глава VI. Чисто вращательные спектры комбинационного рассеянии 113
1. Теоретические основы метода....................................113
2. Методика эксперимента вращательной спектроскопии КР .... 120
3. Определение геометрии молекул..................................121
Глава VII. Метод газовой электронографии..............................122
1. Основные этапы развития газовой электронографии................122
2. Рассеяние электронов атомами...................................123
2.1. Упругое рассеяние электронов атомами..........................124
2.2. Неупругое рассеяние электронов атомами .......................130
2.3. Полная интенсивность атомного рассеяния.......................131
3. Рассеяние электронов молекулами............................ . 132
3.1. Молекулярная составляющая интенсивиости рассеяния ..... 132
3.2. Преобразование Фурье в газовой электронографии ...... 134
3.3. Двухатомные молекулы...................................... . 136
3.4. Кривые радиального распределения..............................142
3.5. Многоатомные молекулы.........................................143
4. Методика эксперимента в газовой электронографии............... 146
4.1. Принципиальная схема электронографа ......................... 146
4.2. Микрофотометрирование.........................................149
4.3. Выделение молекулярной составляющей интенсивности рассеяния 150
5. Расшифровка электронограмм.................................... 151
6. Влияние внутримолекулярных колебаний на конфигурацию молекул, определяемую методом газовой электронографии.................155
7. Возможности метода газовой электронографии.....................160
8. Определение геометрии молекул при совместном использовании электроиографнческих и спектроскопических данных..................162
9. Некоторые стереохимические результаты электронографических исследований ...................................................... 163
Контрольные вопросы ...............................................167
Четвертый раздел. Методы колебательной И К и КР спектроскопии ............................................................ 169
Глава VIII. Теоретические основы колебательной спектроскопии ... 171
1. Квантово-механическое представление колебательных спектров . . 171
2. Основы классической теории колебательных спектров .............181
3. Практический расчет колебательных спектров.....................185
Глава IX. Симметрия молекул н нормальных колебаний....................191
1. Общие представления о симметрии молекул.....................!. 191
2. Качественные представления о симметрии колебаний............. 194
3. Результаты теоретико-группового анализа колебаний..............199
4. Резонанс Ферми.................................................204
5. Эффекты кристалличности........................................205
Глава X. Анализ и интерпретация спектров. Определение симметрии н структуры молекул...............................................206
1. Выводы из сопоставления ИК и КР спектров...................... 206
2. Поляризация полос в спектрах КР................................215
3. Контуры вращательной структуры полос ...................217
266
4. Групповые или характеристические частоты.......................220
5. Изотопные эффекты .............................................227
Глава XI. Другие применения колебательных спектров....................230
1. Определение силовых полей молекул..............................230
2. Корреляции силовых постоянных молекул с другими свойствами . 235
3. Крутильные колебания и потенциальные барьеры внутреннего вращения ............................................................238
4. Использование фундаментальных частот для расчета колебательных вкладов в термодинамические функции...........................242
5. Идентификация соединений и качественный анализ смесей........ 244
6. Количественный анализ .........................................247
7. Исследование равновесий .......................................252
8. Комплексы с водородными связями ...............................255
9. Кинетические исследования......................................257
10. Колебательная спектроскопия высокомолекулярных соединений . 258
Глава XII. Приборы и экспериментальная техника........................264
1. Техника н методики ИК спектроскопии........................... 264
1.1. Принципы устройства и действия ИК спектрометров............. 265
1.2. Характер и подготовка образцов...............................271
1.3. Дополнительные приспособления. Исследования специфических образцов ..........................................................276
2. Нарушенное полное внутреннее отражение (НПВО)..................278
3. Техника спектроскопии КР.......................................283
3.1. Спектральная аппаратура и образцы ...........................283
3.2. Резонансное и инверсное КР ..................................286
3.3. Методы нелинейной спектроскопии КР...........................287
Контрольные вопросы ............................................. 290
Пятый раздел. Методы электронной УФ спектроскопии.................... 293
Глава XIII. Основы теории электронных спектров молекул ...............295
1. Общая характеристика свойств электронных состояний.............295
2. Номенклатура н символика электронных состояний.................299
3. Классификация электронных переходов, их относительное положение 306
4. Правила отбора и интенсивность переходов.......................313
Глава XIV. Применение электронных спектров ...........................320
1. Структурно-спектральные корреляции.............................320
1.1. Органические соединения......................................320
1.2. Неорганические и комплексные соединения......................327
2. Аналитические применения.......................................329
2.1. Качественный анализ и идентификация веществ..................329
2.2. Количественный анализ........................................330
Глава XV. Техника и методики электронной спектроскопии .... 333
1. Аппаратура абсорбционной спектроскопии ........................333
2. Подготовка образцов............................................337
3. Спектроскопия с дифференцированием, разностная спектроскопия и двухволновая спектроскопия ...................................... 339
4. Спектры люминесценции.................т........................342
4.1. Теоретические основы ........................................342
4.2. Практическое применение н техника люминесцентной спектроскопии .............................................................347
Контрольные вопросы ..............................................352
Заключение ...........................................................354
Приложение............................................................355
Литература ...........................................................356
Предметный указатель..................................................359
ПРИНЯТЫЕ ОБОЗНАЧЕНИЯ ВЕЛИЧИН
а0 — Боровский радиус
с — скорость света е — заряд электрона h — постоянная Планка k —постоянная Больцмана Т — абсолютная температура те — масса покоя электрона тр — масса покоя протона
А — длина волны излучения или потока частиц v — частота излучения <о—круговая частота ге — равновесное межъядерное расстояние
• tfi теег
п — показатель преломления
<...> — знак усреднения (интегрирования с нормировкой)
8о — электрическая постоянная Na — постоянная Авогадро
М — молекулярная масса
В— универсальная газовая постоянная |Хв — магнетон Бора р — плотность вещества АВС — вращательные постоянные
la, 1 ь, 1с — моменты инерции в главных осях инерции а, Ь, с е—диэлектрическая проницаемость
Цо — собственный электрический дипольный момент Mq — проекция момента перехода (Q=X, У, Z)
Е — напряженность электрического поля
Те, 4fB, 4fr, Ее, Ev, Ег — волновые функции и энергии электронного, колебательного и вращательного состояний соответственна
ОСНОВНЫЕ ФОРМУЛЫ И УРАВНЕНИЯ
I. Общая характеристика физических методов
1. Прямая задача метода: Ах = и, где А — оператор; х— совокупность характеристик вещества; и — результат измерений. Обратная задача метода: х = =В(и). Возможности методов определяются решением обратной задачи.
2. Спектроскопические методы исследования основаны на уравнениях
м = А-1 (£'-£") и Mq= <Ф' | | Ф") (Q= X, У, Z).
3. Дифракционные методы: рентгенография, электронография и нейтронография. Уравнение де Бройля А = й/(тпо) (т, v — масса и скорость частицы). Условие дифракции: Kcjr (г — межъядерное расстояние). Соотношение интенсивностей рассеяния: 7р : Ia : 1В = 1 : 106: 10-2.
4. Оценка характеристического времени метода А/=т из принципа) неопределенности Гейзенберга (А£А/^ ft); I
К 1 т -------—------•
Д£
268
II. Методы масс-спектрометрии
1. Ионный ток Ij иоиов типа /, регистрируемый детектором в масс-спектрометре: \
где 1е — электронный ток ионизирующей пушки; nj — число молекул / в единице объема; pj — сечеиие ионизации; I — длина пути электронов в ионном источнике.
2. Основное уравиеие для расчета регистрируемых масс иоиов т в масс-спектрометре с магнитным анализатором:
т г^-В^
~ = IV ’
где г — радиус траектории; В—магнитная индукция; V— ускоряющее напряжение.
3. Применения масс-спектрометрии:
a) /W(M) = BmIn>
где ПИ (М) — потенциал ионизации молекулы М; Bmin — минимальная энергия поиизируощих электронов;
б) b(R1-R2)«Z7Z7(R+)-Z7«(Ri),
где D(Rl—Rt) — энергия разрыва химической связи Rt—Rt', ПП (Ri+) — потенциал иоявления Ri+; ПИ (/?]) —потенциал ионизации радикала Rt;
К
в) ll==KjI}T=------ IjT,
а]
где pj — парциальное давление газа /; К;—коэффициент чувствительности для иоиов Tina j; К—константа чувствительности прибора. Определяют ДЯ, (суб-лимации;, Ал г (реакции), и др.
т —•
III. Методы определения электрических дипольных моментов
1. Уравнение Дебая для молярной поляризации Рт вещества в газовой фазе: е - 1 М 1 ( Но \
в + 2 р Зе0 Nk \ aD + 3kT ' ’
где цо-(дипольный момент; aD — деформационная поляризуемость.
2. сравнение молярной рефракции Лоренца — Лореица: . «2_ 1 М 1 „ ,
~ П2 + 2 ‘ р “ 3s0 А ’
где 6 — электронная поляризуемость в переменном электрическом поле высокой частоть.
3. Смещение молекулярного пучка в
иеодиородиом электрическом поле
<2 Р-о$ г
2 = 3kT '
ati Fz
« = “у = •— 2 tn
где а--ускорение молекул в поле т — их масса; t—время пролета.
4. Летод электрического резонанса (область вынужденного испускания) ^7рез ~ И (ро, — Е (р-р, z* 4ь» t > М — 1),
где момент инерции линейной молекулы; J, М — квантовые числа.
269
IV. Микроволновой метод исследования вращательных спектров молекул
1. Частоты переходов в спектрах линейных молекул н молекул типа симметричного волчка |
4 = 2S(/ + 1). |
2. Молекулы типа асимметричного волчка: 1а<1ъ<1с (А>В>С). Параметр аснммегрнн
2В - А - С
А —С
Частоты переходов являются сложной функцией v=v (Д, В, С, 1Х, Ь). Величина т изменяется от — J до +J. В новых обозначениях JT=JK_ +2; т= = Л-1—ТСн, где К-i — квантовые числа К вытянутого волчка; К+1 — сплюснутого волчка.
3. Эффект Штарка. Вращательная энергия для двухатомных н линейных молекул в поле <?Гг:
ЗМ2-/(/ + 1) г( й’г)-* 1(1 + )- 2Лб/(/ + 1) ’ (2/ —1)(2/ +3) ’
Вращательная энергия для симметричного волчка
КМ
Обобщенное уравнение для асимметричного волчка
g — a,b, с
где p,g—составляющие дипольного момента по осям g=a, b, с; Ае —(коэффициенты как функции параметров х, Jz и Mj2.
4. Определение барьера внутреннего вращения Vo. При
Vo V0n2?2
V (?) = -y-d —cos пТ) ® °4—
8«4p
Vo =------
n*
I np>
где /пр = ~~r------J A — моменты ннерцни относительно связи как »сн вра-
щення.
Частоты крутильного колебания vKP определяют из эксперимента.
V. Чисто вращательные спектры комбинационного рассеяния
pg1 — оператор электрического дипольного момента, индуцированного полем световой волны Sq :
= aQ<g’l?,
где aQ = Ba5B-1—тензор поляризуемости молекулы в лабораторной снетке ко-эрдннат Q—X, У, Z; а4—тензор поляризуемости в системе главных ос:й эл
270
липсоида поляризуемости молекулы q=x, у, г и В — ортогональная матрица направляющих косинусов cos Qt.
Уравнение для частоты перехода в спектрах линейных молекул при Д/= = ±2 с учетом центробежного растяжения D„(DV<^.BV}
i | Д* | = (4BV-6DO)(J +3/2)-8Z?i,(Z 4-3/2)з.
Для (линейных молекул симметрии D«,h имеет место чередование в интенсивности линий.
VI. Метод газовой электронографии
1. Потная интенсивность атомного рассеяния
/ат(з)=-^-{ 1/(3) P + ~Y
где /0— интенсивность падающего излучения; f (s) — |f (s) |e*n(s) — амплитуда
атомного
рассеянна
рассеяния; |/(s) | и т) (s)—табулированы; S(s)—функция иеупругого 4л
; R — расстояние от центра рассеяния; s= -у— sin0/2; 0 — угол рас-
сеяния.
2. Мслекулярная составляющая интенсивности рассеяния многоатомных молекул
sM (s) = V gn (s) —-— exp
2
где
gi](s) = | fi (s) | | fj(s) | cos (ng,-(s) - ig; (s))
/ *
I Л(з)2 I
4Sk(s) -
la — ам1литуды колебаний пар атомов; гац = reij —------
reij межъяд риое расстояние в молекуле с малыми колебаниями.
3. Юивая радиального распределения
эффективное
2 С
f (г) = — I sMc (з) ехР ( — is2) sin sr ds,
О
где Мс
s) —кривая sAf(s) с поправками на неядерное рассеяние. Пр1 ближеиио
/(и «2
________ci}_______ г {r~rn{j)2 -relJ У 2Я (2b Л-ll) еХР “ 2 (2b + I*,)
Кривая дает спектр межъядериых расстояний.
VII. Методы колебательной и ИК и КР спектроскопии
1. Теоретические основы:
а) Колебательное уравнение Шредингера
= Ev^v.
271
При использовании нормальных координат Qk и в приближении Борна — Оппенгеймера
= П (Qft) и ~ 2 *
k
в гармоническом приближении
2/
h'iei I Vb + ~
где t)b=l, 2, 3 — колебательное квантовое число; ves—• колебательна^ постоянная; dh—степень вырождения колебательного состояния (<Д = 1, 2, 3].
б) Интенсивность поглощения в ИК-спектрах пропорциональна ква грату момента перехода: Afx = <4^ |[хх|Чг' > и т. д.; цх, pv, — проекции ц; — верхнее, Чг” — ннжнее состояния.
в) КР-спектры определяет индуцированный полем излучения S электрический дипольный момент
Ми = а&,
где а — включает элементы amn (т, п=х, у, г) тензора поляризуемо:ти.
Для всех точечных групп симметрии указаны отличные от нуля компоненты Мг (1=х, у, z) и am„ (m, п = х, у, г) в таблицах характеров точечных групп.
г) Классическая теория колебательных спектров. Уравнения для кинетической Т и потенциальной энергии V:
т и v =
ИЛИ
27' = {?}ТИ?11 и 2r={9}Fl|9||, где tij — коэффициенты кинетической энергии; ftj— силовые постоянше; q — обобщенные естественные координаты и q — скорости.
Для гармонических колебаний, используя уравнение Лагранжа, юлучаем
(F — XT) || /|| =0,
где IIZII — матрица-столбец амплитуд изменения координат q (qs = l} со; (УХ/ + + d); yx=2nv, v — частота; t — время, б — фаза); F и Т — матрицы f * j и I j.
Уравнение в форме детерминанта (вековое уравнение)
|F-XT |=0
определяет п корней Xk=4jA!s (й = 1, 2, ...,п). Для каждого Хь (или vft) находится k-й набор амплитуд ||/||к или lhj (j=l, 2, 3, ...,n).
Полное матричное уравнение для амплитуд и частот
fl = tla,
где А — диагональная матрица квадратов частот; L — матрица линейисо преобразования нормальных координат Q.
l/?ll=L||Q||, в которых п
2T = ^Q2k и 27 = 2^.
Прямая колебательная задача — расчет всех Хь и на основе мгриц Т и F. На практике вводят G матрицу (G=T-1)> тогда вековое уравнение принимает форму
272
|GF — ХЕ I =0, где E у- единичная матрица, и полное колебательное уравнение преобразуется к виду!
\ GFL = LA.
Обратная колебательная задача — расчет элементов матрицы F (силовых постоянных) — относится к классу некорректных задач.
2. Симметрия молекул н нормальных колебаний. Число колебаний типа симметрии & которое включает трансляции и вращения, дается уравнением
л’= Т 2 (/?)!?(«).
где /г — 1 исло операций симметрии в группе; ht — число операций в i-м классе (при отсутствии в молекуле осей симметрии выше второго порядка все Лс = 1); Х<(7?)— характер приводимого представления для операции R (в i-м классе); Xs;(R) —| характер неприводимого представления для операции 7? (в i-м классе), 3. Поляризация полос в КР-спектрах. pn=/±//j— степень деполяризации, измеряемая экспериментально, где /п >—интенсивность рассеянного света, поляризованного параллельно поляризованному падающему свету; — интенсивность рассеянного света, поляризованного в перпендикулярном направлении.
В случае КР-спектров
I 33'2
! Рп 45а'2+43'2 ’
где а' и Р' — производные поляризуемости и анизотропии поляризуемости по Qk.
Для всех неполносимметричных колебаний а\ = 0 и рПк = 3/4, т. е. колебания полностью деполяризованы (dp).
Степень деполяризации полносимметричных колебаний (A, Ab А', Ае) находится в интервале 0^рпк<3/4, так как а'ьУ=0. Это означает, что соответствующие полосы КР полностью или частично поляризованы (р).
4. Изотопные эффекты
а) — = ]/ — ,
СО; У (Л
где со и со,, |х и ре — частоты и приведенные массы исходной и изотопзвмещенной двухатомной молекулы соответственно.
б) Правило произведений
—отношение зависит только от масс атомов и геометрической структуры, в) Правило сумм
п т
2 2^ = 22Ь
I 1-1 U /-1
— если совокупность изотопных разновидностей молекул разбивается на группы I и II так, что в группах имеется по одинаковому числу эквивалентных атомов каждого изотопного вида, то сумма квадратов фундаментальных частот всех молекул одной группы (I) равна соответствующей сумме для другой группы (II).
273
5. Правило Бэджера для двухатомных молекул а1) ke = (Гг-^/У ’ где — силовая постоянная; ге—равновесное межъядериое расстояние; ац, I'tj — эмпирические постоянные, находимые для элементов периода, ато^ы которого образуют молекулу. I
6. Количественный анализ. Объединенный закон Бугера — Ламберта — Бера
I —Т I Р А 'х 'ог где—иитеисивиость выходящего из вещества излучения; /ох— иитеисивиость входящего света; ах—коэффициент поглощения, л-моль_,-см“’; с — концентрация, моль-л-1; I — толщина поглощающего слоя, см.
7. Нарушенное полное внутреннее отражение:
а) Закон Сиеллнуса „ . л2 sin 6
n2 sin 0 = n2 sin <₽ н - = л21 — —:--->
Л! sm <р
где Л! и п2 — показатели преломления двух сред; 0 и <р — углы падения и отражения. 1
б) Глубина проникновения света при полном внутреннем отражении! в оптически менее плотную среду
.____________*1_________
п 2л (sin2 0 —л^)1/2 ’ где Xi -— длина волны света в оптически более плотной среде; л21 — отношение показателей преломления.
VIII. Методы электронной УФ-спектроскопии
1. Основы теории электронных спектров молекул:
а) Электронное уравнение Шредингера
HeWe = Et^e,
где — оператор Гамильтона и 4%—электронная волновая функция прн закрепленных ядрах, например, в равновесной конфигурации; Ee=Ee(R)—собственное зиачеине электронной энергии, зависящее от ядерных координат Re.
Для последовательности состояний с энергией EeW^EgW^Ee<2>... приняты обозначения соответственно X, А, В, С и т. д.
б) Двухатомные и линейные молекулы. В основе классификации состояний — модуль проекции суммарного орбитального момента количества движения на ось молекулы |ML| = А и проекция суммы электронных спинов иа ось молекулы St с обозначением Sm. Величина А принимает значения О (S), 1 (П), 2(A), 3(Ф)...
Полный момент количества движения молекулы
2 = А + .
Общее обозначение состояния 25+1А2, напРнмеР, 2^з/2 и 2П|/2.
в) Многоатомные молекулы. Электронные состояния X, А, В, С... классифицируют по типам симметрии данной точечной группы с указанием мультиплет-иости (тильда относится к символам, определяющим порядок возбуждения состояний): XlAg, А2В2, А'А2, В2В\ и т. п.
г) Правила отбора и иитеисивиость переходов. Коэффициент Эйнштейна для поглощения
274
8лз
Втл= —|ММ|2. on!
где Мпт— момент перехода.
Интенсивность поглощения для единичной плотности излучения
СГЛ = B,nnNmhvmn = 8jt3\mn Nm |Л4т"|2 = f е (v) <b,
где e(v) — экспериментальная функция молярного коэффициента экстинкции. Сила осциллятора
теНчтп
Т _ _ — — —— л
Момент перехода
М = f <Х’ J dTs-
где 4fe, Y,,, 4fs — волновые функции электронных, колебательных и спиновых состояний.
Правило отбора по спину AS = 0.
Правило отбора по симметрии: тройное прямое произведение типов симметрии
х ;е х
должно относиться к полиосимметричиому типу точечной группы, которой принадлежит молекула.
Квадрат первого интеграла — фактор Франка — Кондона; в виброииых спектрах правило отбора связано с неравенством нулю интеграла J ф'*ф'*р.еЧг'еЧГ v, если ои относится к полиосимметричиому типу.
2. Применения электронных спектров. Для многокомпонентных систем закон Бугера — Ламберта—Бера подчиняется аддитивности
А = Z 2 *‘с‘-i
IX. Спектроскопия ЯМР
1. Ядериый магнитный момент цп определяет энергию идра в магнитном поле В
Е — — цпВ = — рпВ cos (р.л,В), ЦпУ=0 дли ядер с разной четностью чисел протонов, нейтронов, а также при четном массовом числе и нечетном заряде, т. е. при /У=0, где I — спии ядра.
2. Условие резонанса для двухуровневой системы при /=*/г
h-ч = 'irfi-B = gn$nB , где уп—гиромагнитное отношение ядра; рп — ядериый магнетон; gn—ядериый g-фактор.
3. На ядро атома в молекуле действует эффективное поле
Вл = В + В- = (1 - а) В, где а — константа экранирования.
4. Химический сдвиг сигнала ЯМР исследуемого вещества X. бх рассчитывают по формулам:
275
В миллионных долях
Av
8 =---10S,
где v0 — частота генератора и v0«vaT.
5. Энергия спин-спииового взаимодействия магнитных ядер
£ = A/aj/a/x ,
где/ах—константа спии-спннового взаимодействия (Гп); /А и /х — спины ядер А и X. Величины Iах зависят от величины поля В.
6. Динамический ЯМР.
При равновесии двух форм А В с равными концентрациями по измеренному времени жизни этих форм т определяют константу скорости реакции обмена
6,2-1012exp (—70/7?Г)
и V0=AG* —потенциальный барьер.
X. Спектроскопия электронного парамагнитного резонанса (ЭПР)
1. Условие ЭПР
Д£ = — 2(леВ = gpBB = h-ч,
где ре — магнитный момент электрона (ре^>рп); рв — магнетон Бора; g— фактор Лайде (для свободного электрона 2,00232).
При исследовании анизотропных образцов
g2 = g\ cos2 0 + g\ sin2 9,
где 0 — угол оси г с направлением поля В.
2. Электрои-ядерное взаимодействие и сверхтонкая структура ЭПР.
Вблизи ядра с /У=0 энергия электрона задается уравнением
Е = gpBBms + amsml,
где ms — спиновое квантовое число электрона (проекция спииа иа направление В); т, — то же, для ядерного спина: а — коистаита сверхтонкого взаимодействия (СТВ).
Число линий в спектре ЭПР при взаимодействии электрона с несколькими наборами nt эквивалентных ядер равно
П (2Я;Л + 1-) i
XI. Ядериый квадрупольный резонанс
1. Ядериый электрический квадрупольный момент eQ — мера отклонения распределения электрического заряда в ядре от сферической симметрии; eQ=/=0, если /а^1 (/а — спин ядра). Все известные величины eQ лежат в пределах — 2<eQ< + 10 бари (барн=10~28 м2).
2. В системе координат, связанной с аксиально симметричным электрическим полем, главные значения тензора градиента поля q^x—q^y^qa. ось г направлена вдоль максимального градиента eqIt-, полная энергия взаимодействия ядра и поля в квантово-механическом приближении равна
e2Qq[3mj -/(/ + !)]
= 4/ (21 — 1) ’
276
где eq—qtI. Уровни энергии дважды вырождены. Для /=3/2, например, имеются ^±1/2 и £±з/2 и т- Д-
3. Асимметрическое поле характеризуется параметром асимметрии т] = = lqxx — <hv\lqzz и, например, для /=3/2 выражение для частоты содержит т]
> XII. Мессбауэровская спектроскопия
1. Согласно закону сохранения энергии при испускании у-кванта ядром с массой М имеем
Ет + -j~MV2x = + у М (V х + v)2 ,
где Ет — Ее—Eg — разность в энергии возбужденного Ее н основного Eg состояния ядра; Vx — скорость его теплового движения; Е^ — энергия у-кваита; v — скорость отдачи
Ет — Mv2 + MvVх— Er -г Ed,
где Er—энергия отдачи; Ео — энергия эффекта Доплера.
Так как импульсы ядер и у-кванта равны по абсолютной величине, то рп = =р1=Е1[с и ER = Ey2/(2 Мс2). Для теплового движения ~ QkT/М)1!2, сле-довательио,
ED — Е, (2kT/Mcrf/2.
Уменьшение Er достигается исследованием кристалла, когда М можно заменить иа Л1кр, т. е. £,|<Pji=£,2v/(2AfKpC2).
2. Условия поглощения у-кваитов при Ет (источник) У=£т (поглотитель) (при £п«0)
5Т (ист) = Ет (ист) + Eq (v) = Ет (погл), где v — скорость источника.
XIII. Методы рентгеновской и фотоэлектронной спектроскопия
1. При фотоэлектронной эмиссии под действием кванта излучения можно определить энергию связи электрона
^СВ = “ 7?кин>
где £кин—измеряемая кинетическая энергия удаленного электрона.
В рентгеновском спектре поглощения каждому электронному уровню (К, L\, Е\\, Lui и т. д.) соответствует свой край поглощения (низкочастотная граница полосы).
При К захвате электрона образующейся вакансией наблюдается рентгеновская флуоресценция и частота излучения v" соответствует разности энергий, например:
h”" (^в,) = ^св (К) — Есз (£щ).
Возможен также переход электрона с высшего уровня иа вакансию (например, £ц->-/С), при котором высвобождаемая энергия идет иа удаление электрона с другого высокого уровня (например, £ni).
277
В оже-процессе:
£кин (Kinlin) = £св (К) — £св (£ц) — £св(£ш)-
Зная из рентгеновских спектров разность £св(К) —£св(£ц) н измеряя Екяв оже-эле’ктронов, можно определить £св(£ш).
2. Химический сдвиг — разность энергий связи электрона для одних и тех же уровней энергии в образце и эталоне
др р°бр р эт “ссв — с п1 с-,ч1 •
XIV. Дисперсия оптического вращения
1. При прохождении через оптически активную среду показатели преломления для лучей с левой круговой поляризацией щ и правой пг ие равны, что определяет поворот плоскости поляризации линейного поляризованного света а (уравнение Френеля):
где d — толщина слоя, дм.
, Afa
2. Молекулярное вращение [А1]х = —-— ,
где М — молярная масса вещества. Функция [Af (X)] * называется крибой дисперсии оптического вращения (ДОВ).
3. Кваитово-мехаиическая форма зависимости:
NА п2 + 2 ( ' (''hi у2)___
' Х= А ’ е0 3 ( С ) 2» ( 2._,2)2+у2?2 ’
где коэффициент Vm — частота перехода v — частота падающе-
его света; Rik— вращательная сила перехода
Rik=Im | цг | | 1 ,
где Im {...}—мнимая часть {...}; Y, и — волновые функции молекулы; ре оператор электрического дипольного момента; цт — оператор магнитного дипольного момента.
В форме скалярного произведения = | Цг II цл I cos 9,
где 0 — угол между це и цт.
4. если векторы це и ц™ принадлежат полиосимметричиому пред-
ставлению, т. е. А, А', Аь Ае или Aig.
Симметрия молекул и оптическая активность: а) молекулы оптически активны, если принадлежат к группам симметрии Сп или Dn; б) молекулы оптически неактивны, если имеют плоскость симметрии, центр симметрии или ось Sn.
5. В области v«Vki отрезок экспериментальных и теоретических кривых ДОВ имеет аномальный, например S-образный, характер для Rik>0 и называется эффектом Коттона.
XV. Круговой дихроизм
1. Для лучей с правой и левой круговой поляризацией молярные коэффициенты экстинкции ег и е; соответственно в оптически активной среде не равны друг другу, т. е. егУ=р.;. Это явление называется круговым дихроизмом.
2. Экспериментально измеряют зависимость Ae = ei — er = Ae(v). Для гауссовой формы
[/ v — \2 j
— ----л---
\ / J
278
где Де;* — дихроичное поглощение в максимуме полосы ik-, Де;*>0 для положительного эффекта Коттона и Де;*<0 для отрицательного эффекта Коттона; Д;* — полуширина полосы.
3. Расчет из экспериментальных данных КД
Rik = 26>5 е0 |/4 • = 1 >25..
яЛГА V 2 ч/ti 'М
XVI. Аномальное рассеяние рентгеновских лучей — метод определения абсолютной конфигурации
1. В правой системе координат х, у, г по системе Каиа, Иигольда и Прелога обозначают правую конфигурацию молекулы г(7?)=гв (х, у, г), а левую — / s (—х, —у, —г).
2. Закон Фриделя: при условии (X* — длина волны собственных переходов) дифракционная картина имеет центр симметрии (нормальное рассеяние): /(Н)=/(Н), где / — интенсивность рассеянных рентгеновских лучей от монокристалла; Н — радиус-вектор узла обратной решетки; Н = —Н.
Закон Фриделя ие позволяет_ определить абсолютную конфигурацию молекул, так как /л(Н)=/8(Н)=/8(Н)=/н(Н). _
3. При условии рассеяние аномально и /д(Н)^=/л(Н).
Абсолютную конфигурацию определиют из сопоставления /лэксп(Н) — _/Аэксп(Н) и /Антеор(Н) _ /Адте0Р(Н) или /л8те°Р(Н) —/латеор(Н).
XVII. Методы изучения электронной поляризуемости молекул
1. Рэлеевское рассеяние света. Из эксперимента определяют отношение До = = /v1 2//zz, где 1гг и 7VZ— иитеисивиости рассеянного излучения, поляризованного по осям гну соответственно, а падающее излучение поляризовано по оси г. Тогда имеет место уравнение
1гг + 4у2 ’
(6| + 62 4“ 63 \
Ь —-------------1 ; i>i, bi, b$ —
О /
главные значения эллипсоида поляризуемости молекулы; у — анизотропия по-ляриуземости
Y2 = [(61 — 6г)2 + (6г — 63)2 + (63 — 6j)2].
2. Эффект Керра, а) Закон Керра
Дл = ля — nL=\B S2Z,
где n я — показатели преломления для лучей с длиной волны К параллельных и перпендикулярных напряженности электрического поля <3^; В — постоянная Керра. Экспериментально определяют Ап.
б) Экспериментальная молярная константа Керра
тК =
*1
/ 3 у
U + 2/
м_ м 6л ( 3 Y р (п2 4- г)2 4-2/
где R । и R± — молярные рефракции для лучей, параллельных и перпендикулярных полю %г.
279
в) Теоретическое выражение
m^ = V-^A(01+02), /ео
где в1=(45йТ)-1 У (а; — а;) (bi — Ь/)(а.щ-) — статическая поляризуемость) и е2 = (45А2Г2)-1 2 (р. - Ру) (bl - bj)
(Mi(J) — компоненты электрического дипольного момента).
г) Из измерений R, у2, тК и ц определяют b2 и Ьз-
XVIII. Эффект Фарадея
1. Уравнение Верде
a=V(\, T)dB,
где а — поворот плоскости поляризации света после прохождения через слой d вещества в магнитном поле В; V (Д Т)—постоянная Верде. Кривая а (X) — дисперсия магнитного оптического вращения (ДМОВ).
2. Для молярного вращения
V"
VM = — М 2 р I
где Vi — постоянные для химических связей.
3. Магнитный круговой дихроизм (МКД)
Д8В (со) = tlB (со) - ггВ (со) = -6,4723.10-7 |д/ (со) + [в + g (co)J ,
где величины А, В и С выражаются через электрические и магнитные дипольные моменты переходов, функция g(co) представляет лоренцеву форму линии поглощения, а /(со) —ее производную.
ЛИТЕРАТУРА
Общая
Вилков Л. В., Пеитин Ю. А. Физические методы исследования в химии. Структурные методы и оптическая спектроскопия. — М.: Высшая школа, 1987.
Данцер К., Тан Э., Мольх Д. Аналитика: Пер. с ием. — М.: Химия, 1981.
Драго Р. Физические методы в химии. — М.: Мир, 1981. Т. 1, 2.
Зоркий П. М. Симметрия молекул и кристаллических структур. — М.: Изд-во МГУ, 1986.
Макаров Е. Ф., Озеров Р. П., Усенбаев К. У. Физические основы резонансных методов исследования в химии. — Фрунзе: Изд-во IФрунзенского политехнического института, 1978.
Миикин В. И., Симкии Б. Я., Миняев Р. М. Теория строения молекул. — М.: Высшая школа, 1979.
Татевский В. М. Строение молекул, —М.: Химия, 1977.
Экспериментальные методы химической кинетики/Под ред. Н. М. Эмануэля и Г. Б. Сергеева — М.: Наука, 1980.
Ebsworth Е. А. V., Rankin D. W. Н., Cradok S. Structural Methods in Inorganic Chemistry. — London: Blakwell Scientific Publications, 1987.
Physical Chemistry an Advanced Treatise, v. IV Molecular Properties/Ed. by D. Hederson. — N. Y. — L.: Academic Press, 1970.
Кпервому разделу
Бучачеико А. Л. Химическая поляризация ядер и электронов. — М.: Наука, 1974.
Вертц Дж., Болтон Дж. Теория и практические приложения метода ЭПР. — М.: Мир, 1975.
Гереон Ф. Спектроскопия ЭПР высокого разрешения. — М.: Мир, 1973.
Гюнтер X. Введение в курс спектроскопии ЯМР. —М.: Мир, 1984.
Жуике А. Ядерный магнитный резонанс в органической химии. — М.: Мир, 1974.
Ионин Б. И., Ершов Б. А., Кольцов А. М. ЯМР-спектроскопия в органической химии. —Л.: Химия, 1983.
Иоффе Б. В., Костиков Р. Р., Разин В. В. Физические методы определения строения органических соединений. — М.: Высшая школа, 1984.
Казицина Л. А., Куплетская Н. Б. Применение УФ-, ИК- и ЯМР-спектроско-пии в органической химии. — М.: Высшая школа, 1971.
Керриигтон А., Мак-Леглан Э. Магнитный резонанс и его применение в химии.— М.: Мир, 1970.
Лундин А. Г., Федин Э. И. ЯМР-спектроскопия. — М.: Наука, 1986.
Попл Дж., Шнейдер В., Бернстейн Г. Спектры ядерного магнитного резонанса высокого разрешения. — М.: ИЛ, 1962.
Сильверстейн Р., Бассиер Г., Моррил Т. Спектрометрическая идентификация органических соединений. — М.: Мир, 1977.
Сергеев Н. М. Спектроскопия ЯМР- — М.: Изд-во Мир, 1981.
Эткинс П., Саймонс М. Спектры ЭПР и строение неорганических радикалов.— М.: Мир, 1970.
Ко второму разделу
Вертхейм Г. Эффект Мессбауэра. — М.: Мир, 1966.
Гречишкин В. С. Ядерные квадрупольные взаимодействия в твердых телах.— М.: Наука, 1973.
Иркаев С. М., Кузьмин Р. Н., Опаленко А. А. Ядерный гамма-резонанс. — М.: Изд-во МГУ, 1970.
Мессбауэровская спектроскопия/Под ред. У. Гонзера. — М.: Мир, 1983.
281
Семин Г. К., Бабушкина Т. А., Якобсон Г. Г. Применение ядерного квадрупольного резонанса в химии.— Л.: Хнмня, 1972.
Фрауэнфельдер Г. Эффект Мессбауэра. — М.: Атомиздат, 1964.
Химические применения мессбауэровской спектроскопии / Под ред. В. И. Гольданского и Р. 3. Хербера. — М.: Мир, 1970.
Шпинель В. С. Резонанс гамма-лучей в кристаллах. — М.: Наука, 1979.
К третьему разделу
Зигбан К-, Норминг К., Фальман А. и др. Электронная спектроскопия: Пер. с англ. — М.: Мир, 1971.
Карлсон Т. А. Фотоэлектронная и оже-спектроскопия: Пер. с англ. — Л.: Машиностроение, 1981.
Лосев Н. Ф., Самгунова А. Н. Основы рентгеиоспектрального флуоресцентного анализа. — М.: Химия, 1982.
Миначев X. М., Антошин Г. В., Шапиро Е. С. Фотоэлектронная спектроскопия и ее применение в катализе. — М.: Наука, 1981.
Нефедов В. И., Черепин В. Т. Физические методы исследования поверхности твердых тел. — М.: Наука, 1983.
Нефедов В. И. Рентгеиоэлектрониая спектроскопия химических соединений. — М.: Химия, 1984.
Нефедов В. И. Рентгеноэлектронная спектроскопия химических соединений и материалов//В сб.: Физическая химия. Современные проблемы. — М.: Хнмня, 1986. С. 230.
Пентин Ю. А., Тарасевич Б. Н. Новые методы спектроскопии в хнмнн. — М.: Знание, 1975.
Шульман А. Р., Фридриков С. А. Вторично-эмиссионные методы исследования твердого тела. — М.: Наука, 1977.
К четвертому разделу
Вайнштейн Б. К. Современная кристаллография. — М.: Наука, 1979. Т. I.
Джеймс Р. Оптические принципы дифракции рентгеновских лучей. — М.: ИЛ, 1950.
Джерасси К. Дисперсия оптического вращения. — М.: ИЛ, 1962.
Дисперсия оптического вращения н круговой дихроизм в органической хн-мнн/Под ред. Г. С н а т ц к е. — М.: Мнр, 1970.
Дунина В. В., Рухадзе Е. Г., Потапов В. М. Получение н исследование оптически активных веществ. — М.: Изд-во МГУ, 1979.
Козман У. Введение в квантовую хнмню. — М.: ИЛ, 1960.
Хокинс Е. Абсолютная конфигурация комплексов металлов. — М.: Мнр, 1974.
К пятому разделу
Верещагин А. Н. Поляризуемость молекул. — М.: Наука, 1980.
Вуке М. Ф. Расстояние света в газах, жидкостях н растворах. — Л.: Изд-во ЛГУ, 1977.
Вуке М. Ф. Электрические н оптические свойства молекул и конденсированных сред. — Л.: Изд-во ЛГУ, 1984.
Волькенштейн М. В. Молекулярная оптика. — М., Л.: Гостехнздат, 1951
Жевандров Н. Д. Применения поляризованного света. — М.: Наука, 1978.
Калитеевский Н. И. Волновая оптика.—М.: Высшая школа, 1978.
Свердлова О. В. Электронные спектры в органической химии. — Л.: Хнмня, 1985.
Фабелинский И. Д. Молекулярное рассеяние света. — М.: Наука, 1965.
предметный указатель
Абсолютная конфигурация молекул 204, 206, 217
Аддитивные схемы расчетов в ЯМР 31
Адсорбционный химический сдвиг 162
Анизотропия электронной поляризуемости 228
Аномальное рассеяние рентгеновских лучей 168, 221
Асимметрии параметр 91, 94
Бари 89
Брюстера правило 204
Бугера—Ламберта — Бера закон 190
Ваит-Гоффа правило оптической суперпозиции 203
Верде постоянная 248, 253, 259
Вероятность перехода ЯМР 11
Вигли 15
Волна излучения
— — без поглощения 169
----с поглощением 190
Вращательная сила перехода 177
Вращение
— молекулярное 174
— удельное 174
Гамма-резоиаисиая ядериая флуоресценция 88
Гиромагнитное отношение 8
Главные значения эллипсоида поляризуемости ----------молекул 244 ----------химических связей 245
Глубина выхода электронов 146
Градиент электрического поля иа ядре 92, 93
Двойной резоиаис 49
Двойной электрон-ядериый резонанс (ДЭЯР) 7, 79
Двухмерная спектроскопия ЯМР 33, 48
Диамагнитная константа экранирования 17
Диастереотопия ядер 36
Дипольный момент
----индуцированный электрический 177, 229
---магнитный 177
Дисперсии сигнал 14
Дисперсия оптического вращения (ДОВ) 168
Дисперсия магнитного оптического вращения (ДМОВ) 250, 256
Дисимметрия комплексов переходных металлов 211
Доплера эффекта энергия 113, 114
Зеемана эффект 255
Излучение колеблющегося диполя 229
Изомерный сдвиг 88, 119
Изохронные ядра 22
ИНДОР 50
Индукция магнитного поля 9
Индуцированной электронной эмиссии спектроскопия 134
Интенсивность излучения 190
— амплитудная (ЯМР) 15
— интегральная (ЯМР) 15
— ФЭС 145
— ЭПР 66
— ЯКР 97
Ионизация
— адиабатическая 144
— вертикальная 145
Ионное травление 150
Кана, Иигольда и Прелога система 217
Карплуса уравнение 29
Квадрупольное расщепление 121.
Квадрупольного взаимодействия константа 90, 95
Квадрупольиый электрический момент 89, 93
Керра константа молярная 239
Керра постоянная 236
Керра эффект (явление) 234
Коалесценция (коллапс) 42
Колебательный круговой дихроизм 213
Комптона эффект 137
Комплексы трис-хелатиые переходных металлов 182
Контактное взаимодействие Ферми 29, 62
Конформация хелатных колец 182
Коэффициент деполяризации 231
283
Край поглощения 137
Крамерса — Кронига типа уравнения 202
Крамерса теорема (вырождение, расщепление) 64
Кривые ДОВ 186
«Кристаллический сдвиг» в ЯКР 97
Круговой дихроизм (КД) 168, 189
Купменса теорема 157
Ланде g-фактор 55, 57, 59
Ларморова частота 12
Лоренца-Лоренца уравнение 239
Магнетон
— Бора 55
— ядерный 8
Магнитная восприимчивость ядерная 12
Магнитное квантовое число 9
Магнитный круговой дихроизм 250
— момент электрона 54
--- ядра 8
Магнитомехаиическое отношение 8
Маделунга взаимодействие - (константа) 156
Межъядерный двойной резонанс 50
Мессбауэра уровень 115
— эффект 112
Множественный резонанс 44, 49
Мозли закон 138
Молярная константа Керра 239
— рефракция 244
Мультиплетиость ЯМР 22, 26
— ЭПР 61
— ЯКР 97
Мультипликативные функции 23
Намагниченность 12
Напряженность магнитного поля 9
Оверхаузера ядерный эффект (ЯЭО) 50
Оже-процесс 139
Оже-спектроскопия (ОЭС) 134
Оператор дипольного момента
— магнитного 177
— электрического 177, 180
Оптическая активность 168
— длина пути луча 169
— плотность 190
Остовов эквивалентных метод 161
Отдачи энергия (скорость) 113, 114
Парамагнитный зонд (метка) 74, 75
Парциальные изомерные сдвиги 124
Паскаля треугольник 23
Поглощения сигнал 14
284
Показатель преломления 169, 190, 252
Пою^ельса электрооптический эффект
Поляризация излучения (света)
----- круговая 169
----- линейная 169
----- эллиптическая 172
Поляризуемость молекулы
— вращательная 178
----- статическая 241
-----электронная 177, 228
Поперечная релаксация 13, 15, 16
Правило(а)
— аддитивности 193
— октантов 205
— оптической суперпозиции Вант-Гоффа
(см. Вант-Гоффа правило опти-
ческой суперпозиции)
— отбора в спектрах КД 209
-----в электронных спектрах 209
-----ЭПР 59, 63
-----ЯМР11
— сумм 180
— Фойгта 181
Приведенные константы спин-спино-вой связи 30
Примесные спины 37
Продольная релаксация 13, 16
Протонный магнитный резонанс (ПМР) 6, 19
Релаксация 13
Релеевское рассеяние света 229
Релея коэффициент (постоянная) 233
Реииера — Теллера эффект 144
Рентгеновская флуоресценция 138
Рентгеновское поглощение 136
Рентгеноэлектрониая спектроскопия 139
Рефракция молярная 244
Решетки прямая и обратная 218
Саттелитная структура РЭС 144
Сверхтонкого взаимодействия константа ЭПР (СТВ) 59
Сдвигающие реагенты 33
Система координат правая 170, 217
Сканирующая оже-микроскопия (СОМ) 151
Спада свободной индукции сигнал (ССИ) 14
Спин ядра 7
Спиновая плотность 62
Спиновой развязки метод 52
Спиновых меток и ловушек методы 74
Спин-решеточная релаксация 13, 16
Спии-спиновая релаксация 13, 15, 16, 65
Спии-спинового взаимодействия константа 24
Спиральная модель молекулы 168, 176, 178, 182, 204
Стандарты ЯМР 53
— ФЭС 149
Стационарный спектрометр ЯМР 14
Струи метод 39
Структурный фактор 219
Тауиса — Дейли теория 105
Тиклииг 51
Тонкая структура ЭПР 63
Тройной резонанс 52
Условие ЭПР 57
— ЯМР 11
Фарадея эффект (явление) 248
Ферми-коитактиое взаимодействие 29
Фишера формула 223
Фотоэлектронная спектроскопия 134
Франка — Кондона принцип 145
Фрейдеиберга правило смещения 203
Френеля уравнение 173
Фриделя закон 218
Характеристический спектр рентгеновского излучения 138
Химическая поляризация электронов 83
----- ядер 39
Химический сдвиг ФЭС 140
-----ЯГР 119
-----ЯМР 18
Хиральность 36, 168, 212
Хромофор 184, 224
Центровой сдвиг 119
Четвертьволновая пластинка 197
Чугаева — Куна — Фрейдеиберга вицинальное правило 203
Ширина линии ЯМР 15, 16
Штерихаймера фактор 106
Эйнштейна коэффициент спонтанного испускания 187
Экранирования коистаита 17
Экстинкции коэффициент
-----малярный 190
Электронная спектроскопия для химического анализа (ЭСХА) 134
Электрои-электроииый двойной резонанс (ЭЛДОР или ЭДР) 79
Электрон-ядериый двойной резонанс (ДЭЯР) 79
Эллиптичность луча 191
Энантиомеры 168, 217
Эталоны ЯМР 19
Эффективное спиновое квантовое число 64
Эффект Коттона 186
Ядериый g-фактор 8
Яна — Теллера теорема 70
-----эффект 144
Ячейка Фарадея 189
Ячейка Керра 236
ОГЛАВЛЕНИЕ
Предисловие............................................................. 3
Первый раздел. Методы магнитного резонанса ядер и электронов .... 5
Г лава I. Спектроскопия ЯМР (основы теории)............................. 7
1. Физические принципы метода....................................... 7
1.1. Магнитный момент ядра и его взаимодействие с магнитным полем 7
1.2. Условие ядерного магнитного резонанса......................... 10
1.3. Реализация условий магнитного резонанса....................... 14
2. Химический сдвиг и спин-спиновое взаимодействие................ 17
2.1. Экранирование ядер электронами................................ 17
2.2. Химические сдвиги сигналов ЯМР................................ 18
2.3. Спин-спиновое взаимодействие и мультиплетность спектров ЯМР 22
Глава II. Спектроскопия ЯМР (применение и техника эксперимента) . . 31
1. Применение в структурных исследованиях.......................... 31
2. Физико-химические применения................................... 38
3. Динамический ЯМР............................................... 40
4. Техника и методика эксперимента................................ 44
4.1. Спектрометры ЯМР.............................................. 44
4.2. Двухмерная спектроскопия ЯМР................................. 48
4.3. Двойной резонанс.............................................. 49
4.4. Образцы, растворители, стандарты.............................. 52
Глава III. Спектроскопия электронного парамагнитного резонанса .... 54
1. Теоретические основы метода..................................... 54
1.1. Условие ЭПР................................................... 55
1.2. Положение резонансного сигнала и g-фактор..................... 57
1.3. Электрон-ядерное взаимодействие и сверхтонкая структура спектра ЭПР............................................................. 59
1.4. Электрон-электронное взаимодействие и тонкая структура спектров ЭПР анизотропных систем........................................ 63
1.5. Интенсивность, ширина и форма линии........................... 65
2. Приложения спектроскопии ЭПР................................... 68
2.1. Структурные исследования...................................... 68
2.2. Кинетические и другие исследования............................ 73
3. Техника и экспериментальные методики спектроскопии ЭПР ... 77
3.1. Общие сведения................................................ 77
3.2. Методы двойного резонанса..................................... 79
3.3. Химическая поляризация ядер и электронов.................... 82
Контрольные вопросы........................................... . 85
Второй раздел. Методы квадрупольного и гамма-резонанса ядер.......... 87
Г лава IV. Ядерный квадрупольный резонанс.............................. 89
1. Основы теории................................................... 89
1.1. Общие сведения................................................ 89
1.2. Электростатическое взаимодействие квадрупольного ядра с электрическим полем.................................................... 91
1.3. Квадрупольные уровни энергии и переходы....................... 94
1.4. Интенсивность, ширина и мультиплетность сигнала............... 97
2. Приложения и интерпретация спектров ЯКР........................ 98
2.1. Частоты ЯКР................................................... 98
2.2. Структурные приложения......................................... 100
286
2.3. Интерпретация градиента неоднородного электрического поля иа
ядре......................................................... 105
2.4. Корреляции спектральных параметров ЯКР с другими физикохимическими характеристиками..................................... 109
3. Аппаратура и методические особенности.......................... ПО
Глава V. Мессбауэровская спектроскопия............................... 112
1. Общая характеристика и теоретические основы метода............ 112
2. Параметры мессбауэровских спектров............................ 118
2.1. Изомерный (химический) сдвиг................................ 118
2.2. Квадрупольиое расщепление................................... 120
2.3. Сверхтонкая структура магнитных взаимодействий.............. 122
3. Применение в химии............................................ 123
3.1. Эмпирические корреляции..................................... 123
3.2. Динамические эффекты....................................... 127
4. Техника и особенности эксперимента............................ 128
Контрольные вопросы ............................................. 131
Третий раздел. Методы рентгеновской и фотоэлектронной спектроскопии 133
Глава VI. Физические основы методов и экспериментальная техника . . . 135
1. Общие принципы................................................ 135
2. Параметры и структура фотоэлектронных спектров................ 140
2.1. Химический сдвиг............................................ 140
2.2. Спин-орбитальная связь в молекулах и некоторые другие эффекты ........................................................... 143
2.3. Колебательная структура фотоэлектронных спектров............ 144
2.4. Интенсивность фотоэлектронных пиков......................... 145
2.5. Глубина выхода фотоэлектронов............................... 146
3. Техника и методика эксперимента .............................. 147
3.1. Аппаратура.................................................. 147
3.2. Стандарты для учета зарядки образцов и калибровки спектрометров .......................................................... 149
3.3. Комплексные установки и методики............................ 150
3.4. Рентгено-флуоресцентные спектрометры........................ 151
Глава VII. Применение методов фотоэлектронной спектроскопии в химии 151
1. Структурно-аналитические применения........................... 151
1.1. Элементный анализ и идентификация соединений................ 151
1.2. Структурная информация...................................... 153
1.3. Количественный анализ....................................... 154
2. Теоретическое моделирование и объяснение химических сдвигов . . 156
3. Некоторые закономерности и корреляции химических сдвигов ... 158
3.1. Связь с эффективным зарядом и степенью окисления............ 158
3.2. Аддитивность химических сдвигов............................. 159
3.3. Корреляция химических сдвигов с данными других методов . . 160
4. Адсорбации, катализ и другие области применения............... 162
Контрольные вопросы.............................................. 166
Четвертый раздел. Методы исследования оптически активных веществ . . 167
Глава VIII. Дисперсия оптического вращения........................... 169
1. Линейно поляризованное излучение. Круговая поляризация света . 169
2. Квантово-механическое рассмотрение оптической активности и спиральная модель молекулы.......................................... 174
3. Симметрия молекул н оптическая активность..................... 181
4. Кривые ДОВ. Эффект ........................................... 185
5. Принципиальная схема эксперимента............................. 188
287
Глава IX. Круговой дихроизм........................................ 189
1. Поглощение лучей с различной круговой поляризацией.......... 189
2. Связь кругового дихроизма и вращательной силы перехода .... 194
3. Схема эксперимента. Формирование лучей с круговой поляризацией 197
4. Взаимосвязь дисперсии оптического вращения и кругового дихроизма ....................................................... 201
Глава X. Применение спектрополяриметрии в химии.................... 202
1. Общие вопросы использования методов ДОВ и КД................ 202
2. Эмпирические закономерности. Правила Брюстера и октантов . . 203
3. Примеры использования ДОВ и КД.............................. 206
3.1. Определение абсолютной конфигурации....................... 206
3.2. Доказательство конформационной подвижности. Влияние полярности растворителя ............................................ 207
3.3. Исследование комплексных соединений....................... 208
4. Колебательная оптическая активность......................... 212
Глава XI. Аномальное рассеяние рентгеновских лучей — метод определения абсолютной конфигурации..................................... 216
1. Абсолютная конфигурация молекул в декартовой системе координат 216
2. Нормальное рассеяние и закон Фриделя........................ 218
3. Рассеяние рентгеновских лучей в области поглощения атома . . . 220
4. Аномальное рассеяние и определение абсолютной конфигурации молекул........................................................ 221
Контрольные вопросы............................................ 225
Пятый раздел. Методы изучения поляризуемости и магнитной оптической активности...................................................... 227
Глава XII. Релеевское рассеяние света.............................. 229
1. Релеевское рассеяние света в газах и растворах............. 229
2. Схема и условия эксперимента................................ 233
Глава XIII. Эффект Керра........................................... 234
1. Закон Керра................................................. 234
2. Методика эксперимента....................................... 236
3. Теория эффекта Керра........................................ 239
4. Применение метода релеевского рассеяния света и эффекта Керра 244
4.1. Определение главных значений эллипсоида поляризуемости молекул ......................................................... 244
4.2. Определение главных значений эллипсоида поляризуемости химической связи и группы атомов................................... 245
4.3. Изучение конформаций и внутреннего вращения молекул .... 247
Глава XIV. Эффект Фарадея.......................................... 248
1. Явление Фарадея. Схема эксперимента......................... 248
2. Теория эффекта. Связь с эффектом Зеемана.................... 250
3. Магнитный круговой дихроизм (МКД) и дисперсия магнитного оптического вращения (ДМОВ).................................... 256
4. Применение эффекта Фарадея в химии.......................... 259
4.1. Аддитивные свойства постоянной Верде...................... 259
4.2. Изучение электронных переходов в комплексных соединениях с помощью МКД.................................................... 260
4.3. Аналитические применения эффекта Фарадея.................. 261
Контрольные вопросы............................................ 262
Заключение......................................................... 263
Приложение..........................................................265
Литература......................................................... 281
Предметный указатель............................................... 283
Отсканировал Семенкяенко Владимир
chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
ХИМИЧЕСКИЕ СДВИГИ ЯМР
400 300 200 100 0 -100
R3C+ RCR RCR с=с-х аром.С-Х гетероаром. О II R-C-X S II R-C-X R-NCO R-SCN с=с R-ON R3C—N Вгл-С RCX 5+ RCR = С = С = С С-Н аром О 1 R-C-R N—ОН II R-C-R C = N —R R-NCS с=с-х R-N-C алканы R3C-0 С1„-С
—
н- 4'1,! 1 |i| 1 1 1 1 1 1 I 4 I 1 L—J 1
н и—ч 1
—
4 D0 3 D0 2( 30 1 >13С D0 (ТЬ 3 -К 4С) 30 м.д.
-с-он 1 алиф.С-NH алиф.С-SH Cl-C-СНз 1 1 1 - с=с-сн2- - СО-СНз -СО-СН2-1 -N—СНз -N-CH2- ( 1>сн -N-CH2-CO- WCH2-CO- -о-сн2- -СО-О-СНз 0-О-СН2- -СО-СН2-О- 0-ОН _ ' /СО 1 |хн н-с=с-со =о—н-о- ON-H -о-сно алиф.-СНО - соон • [>-h СНз-C -1 -CH2- алиф. -СН2-алицикл. 1 1 СНз-C=C- CH3-S-н-с^с-0-СНз -CH2-S- <|>СН2 HS — С аром. HN —С аром. СН3-О- -CH2-CI -сн2-о-со--сн2 - no2 1 1 н-с=с-о-н-с-о- х о - -HN-СО- Н-0 Н-аром. гетероц -CO-NH-0 HO-N-C-НСО-аром. конц. минер. кислота
4—
— - — — — — 1 1 1 I 1 1 1 1 1 1 1 । । । । । 1 I ! г! ! ! । L i 1 i । i 1! ' - м - н l нт , ; i 1 H
—• ; Uni , I i i 1 ! I1!1!!' — i ! : 1 1 i i i i 1 1 1 1 1 । ! ! ।
— —
— — — - — — —
— -и
м м - — —
н—
1 3 1 2 1 1 10 8 ; 6 5 4 3 2 j С 5 м.д.
(ТМС)
r-nh2 r,r2nh R1R2R3N RNHj r,r2nh2 R!R2R3NH+ -CH(NH2)-COOH )C=C-C(NH2) C6H5NH3 <22> nh r2nph2 (NH2)2CO -C(O)-NH2 -C(O)-NHR -C(O)-NRiR2 (nh2)2cs -C(S)-NR,R2 -nh-no2 E> nh (RCO)2NH -ON, RN=C >C =N = N 7C-№N )ON-R )C=NOH -N-N(O)- -no2 -№N- N-N-O — — — — —
— — — — H
— ^1 -4
— —-4
ч — —
— * - HH-
—-4
600 500 400 300 200 100 0
5]4 , м.д. (жидк.МНз) (25°С)
CFC13 CF3S-cf2i cf2ci CF3C^ CF3C(S) — CF3— CF< -cf2-o-cf3ch2-cf3-cf2-=cf2 CF2Br —CF=CF—Ar H2O -cf2-cf< cf2co-c6h13f F2<0> -cf2-cf3 - cf2—cf2-f2<| -cf2-ch2- F^ 1 -cf2-cf2h )CF-Cf3 -cf2h 1 - — — I I T 1 У I 1 X 1 1 1 1 J 1 1 1
— M
-M 4
— — 1 1 1 П 1 1 1 1 III 1 1 ! 1 1 — — M h
75 50 25 6 -25 -50 -75 -100 -125 (CF3COOH) <519р,м.д
Л. В. Вилков Ю.А. Пентин
ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ В ХИМИИ