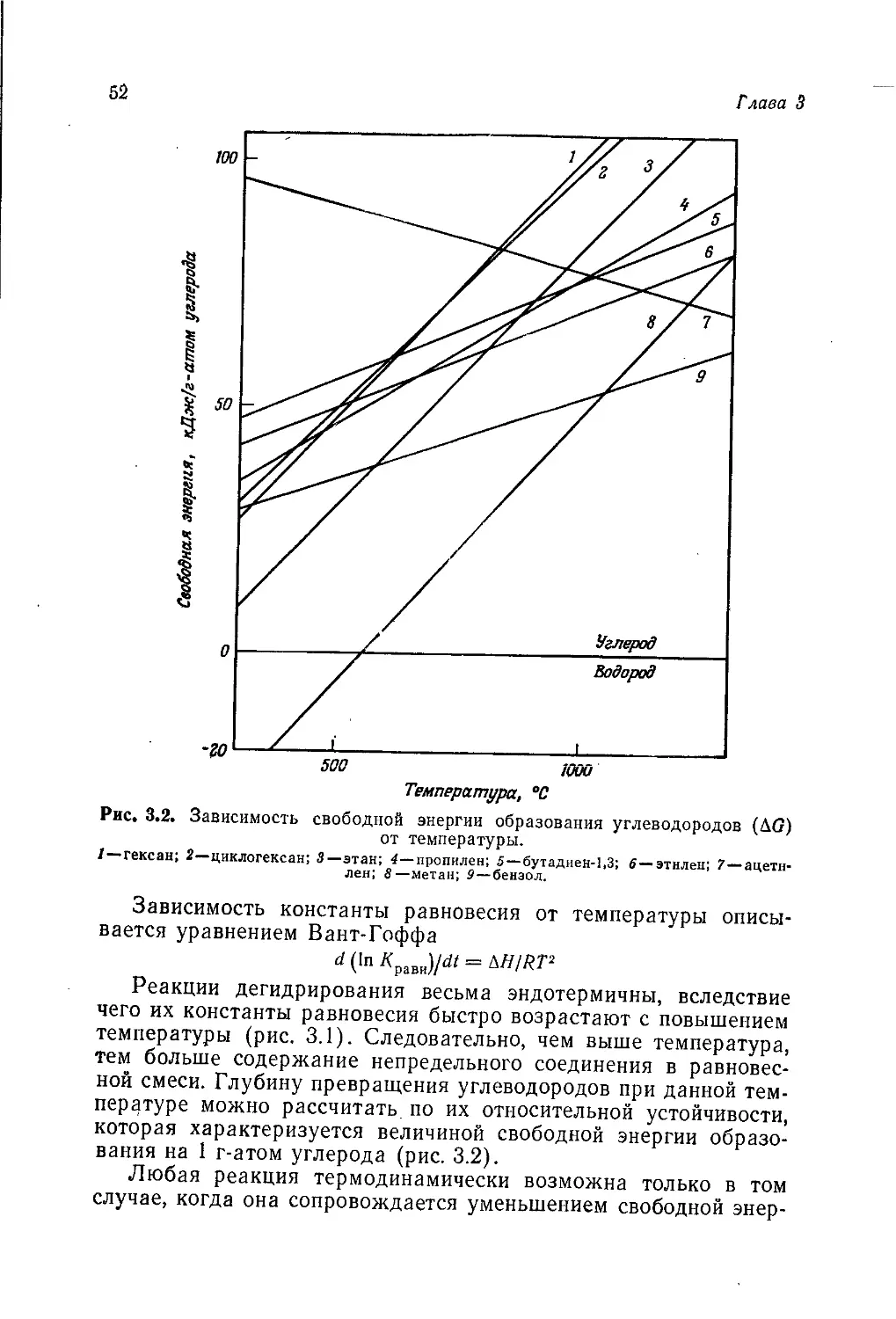
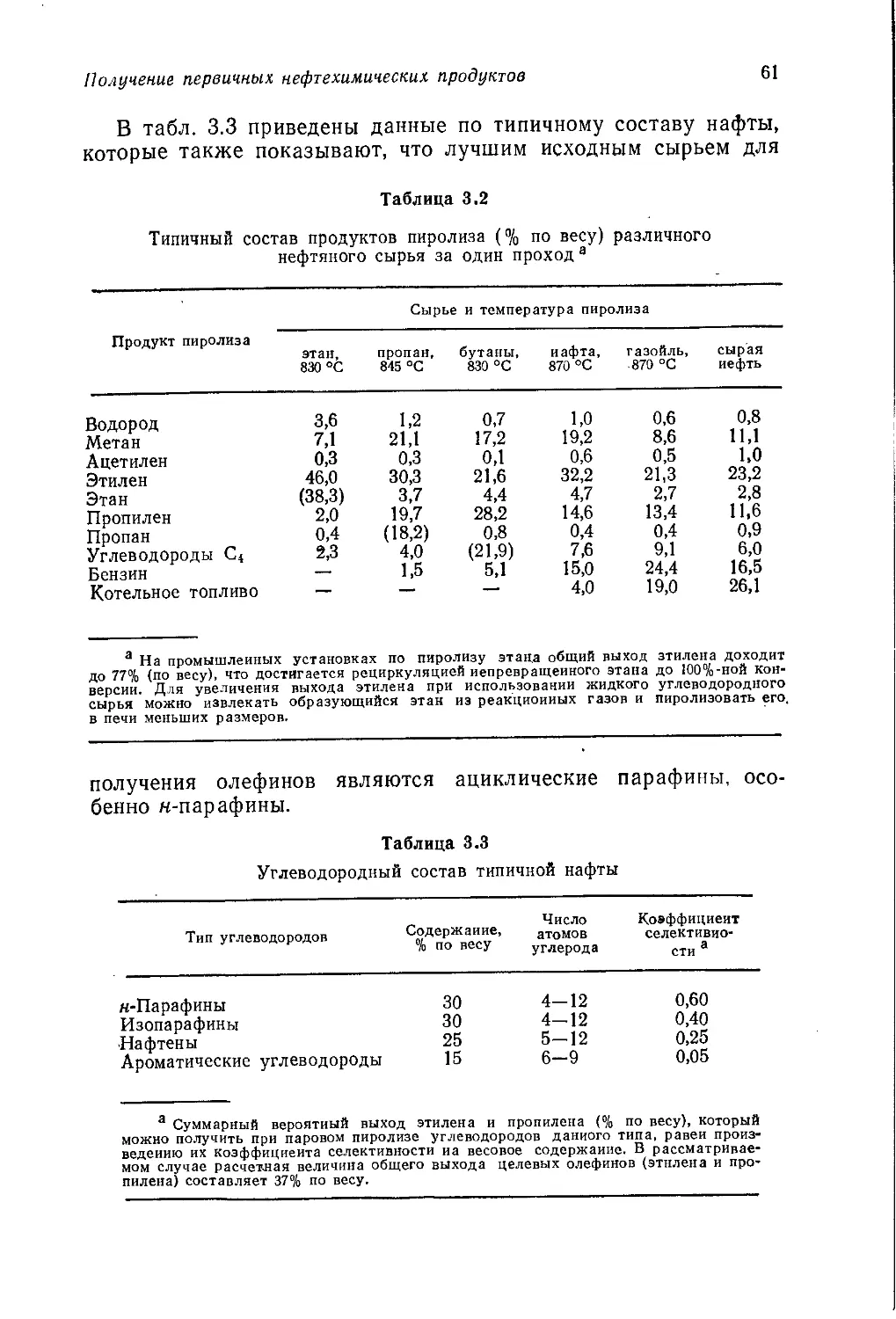
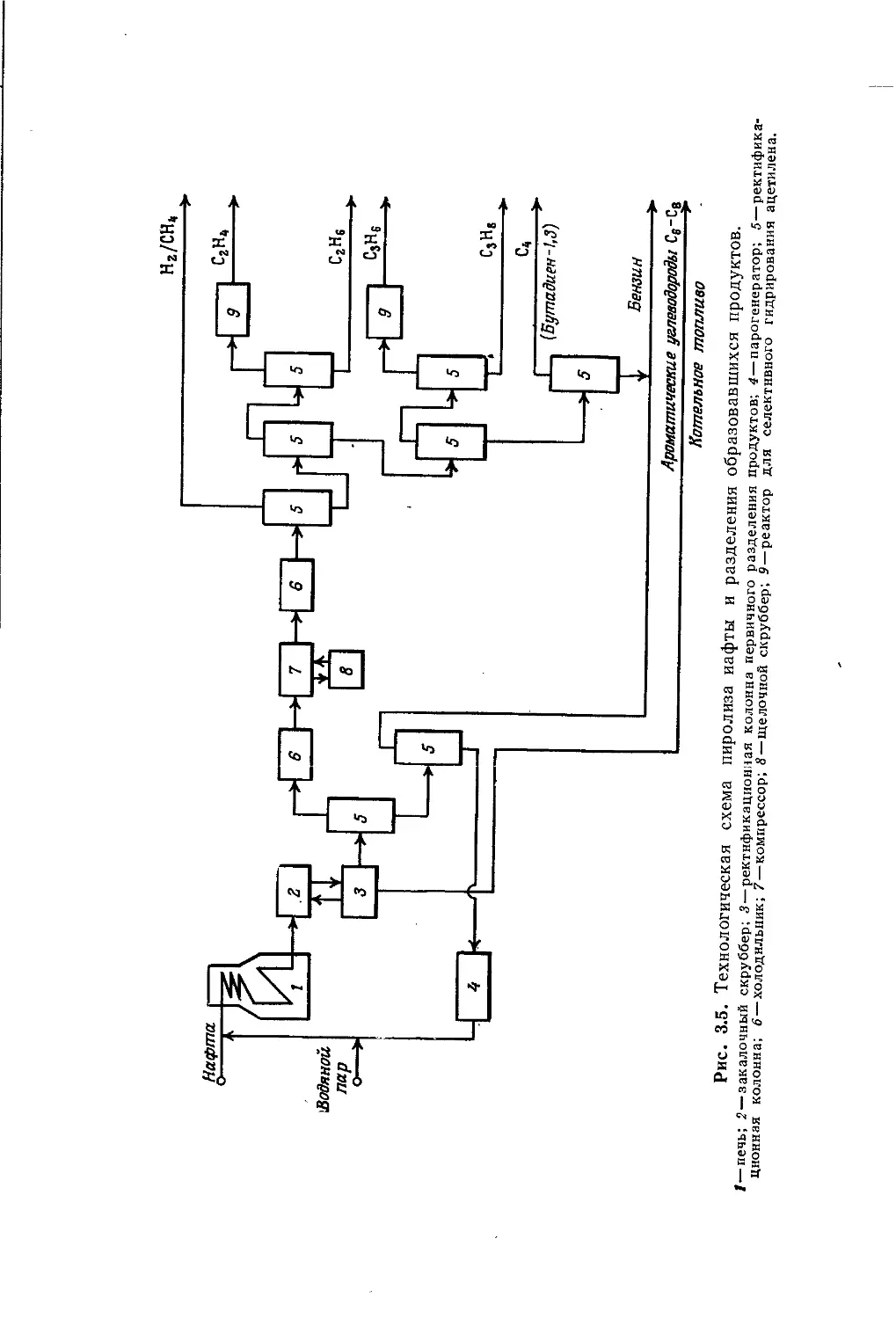
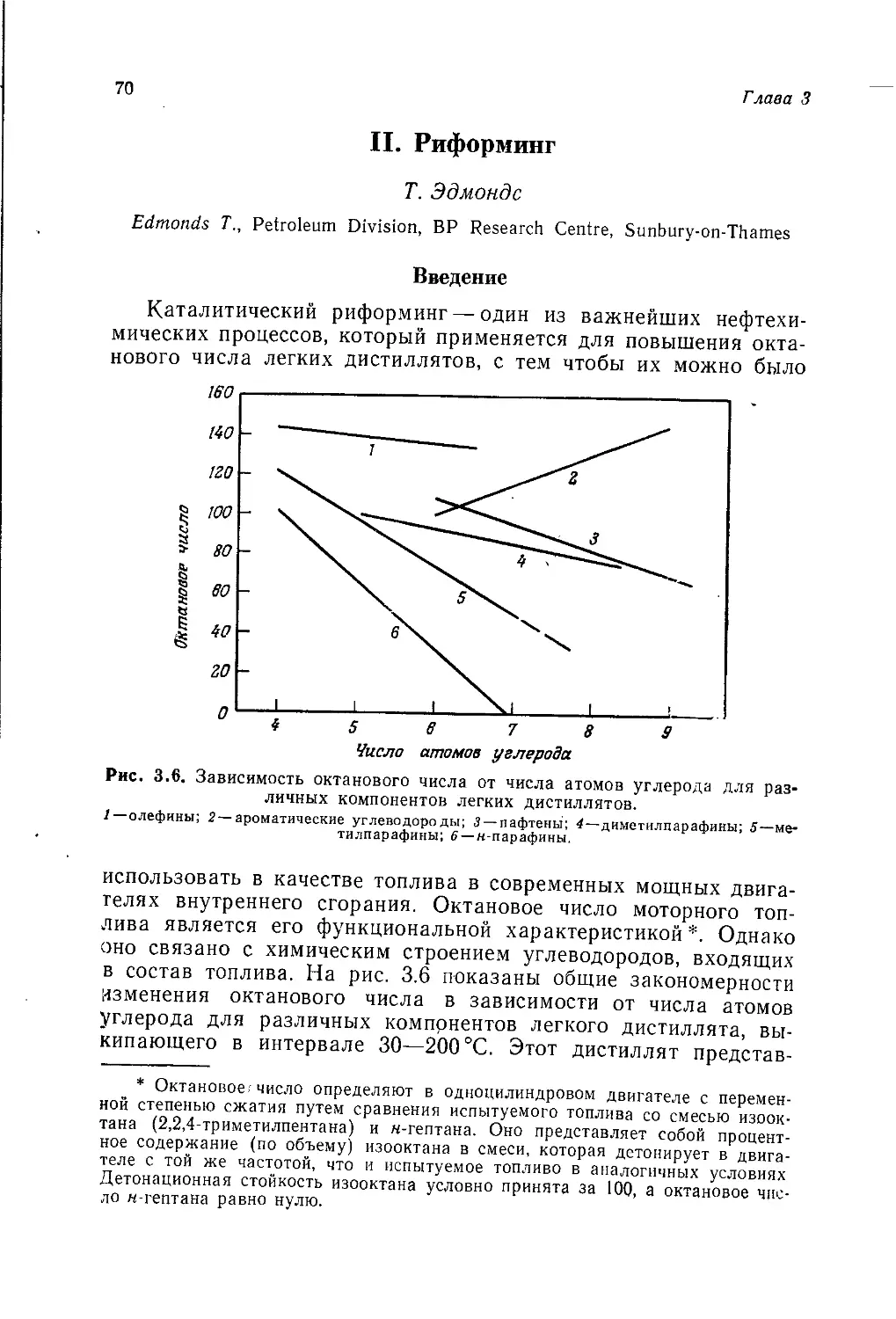
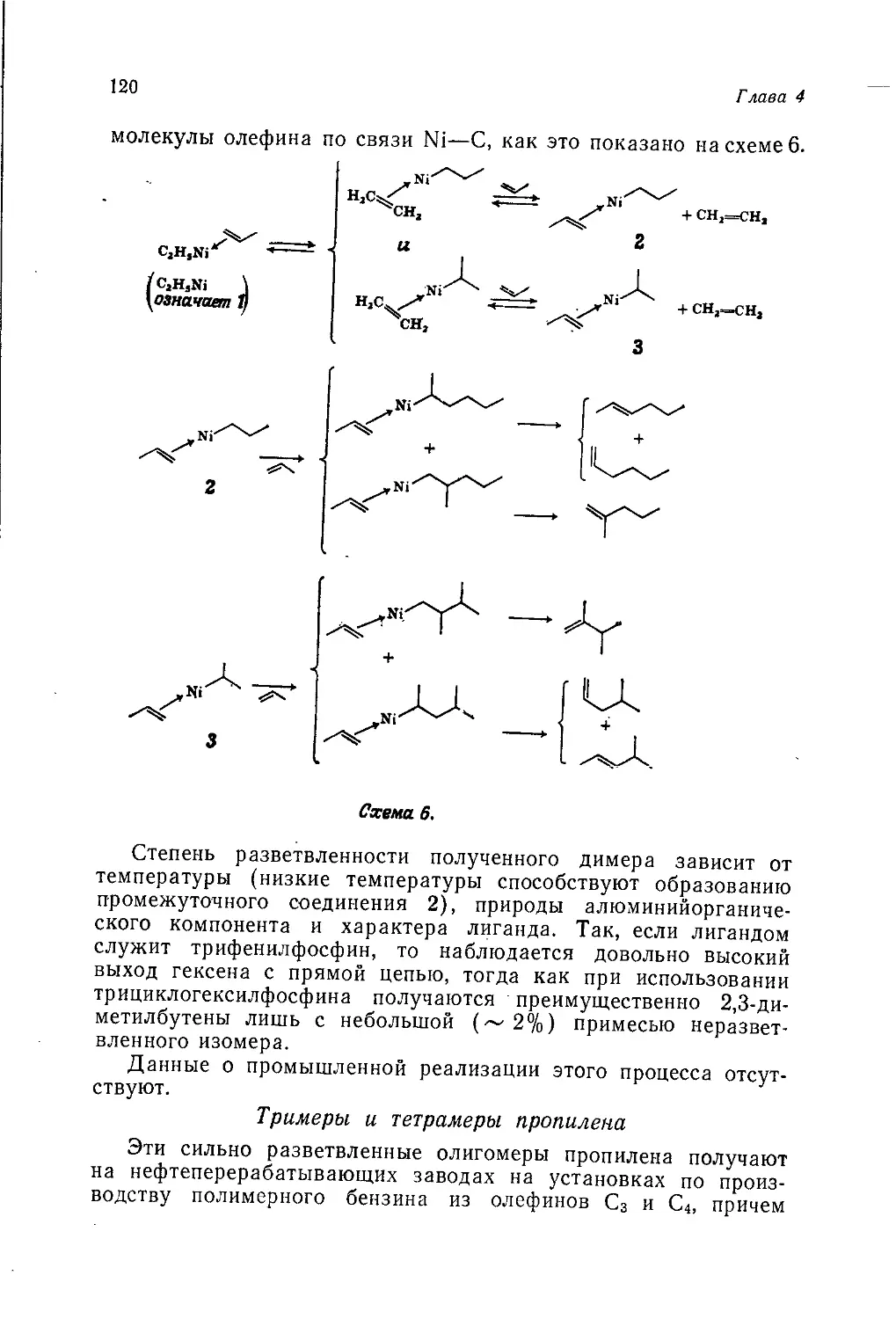
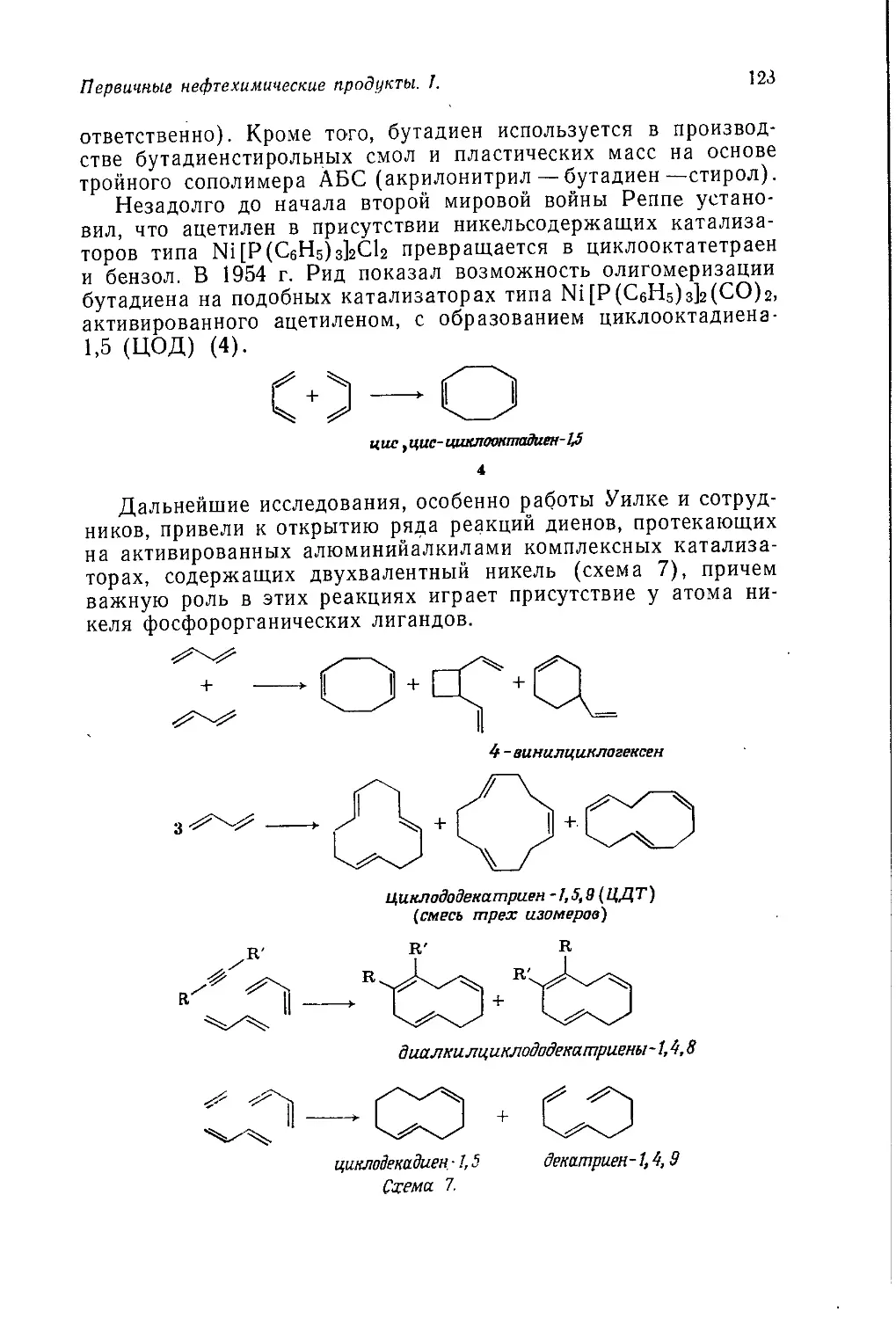
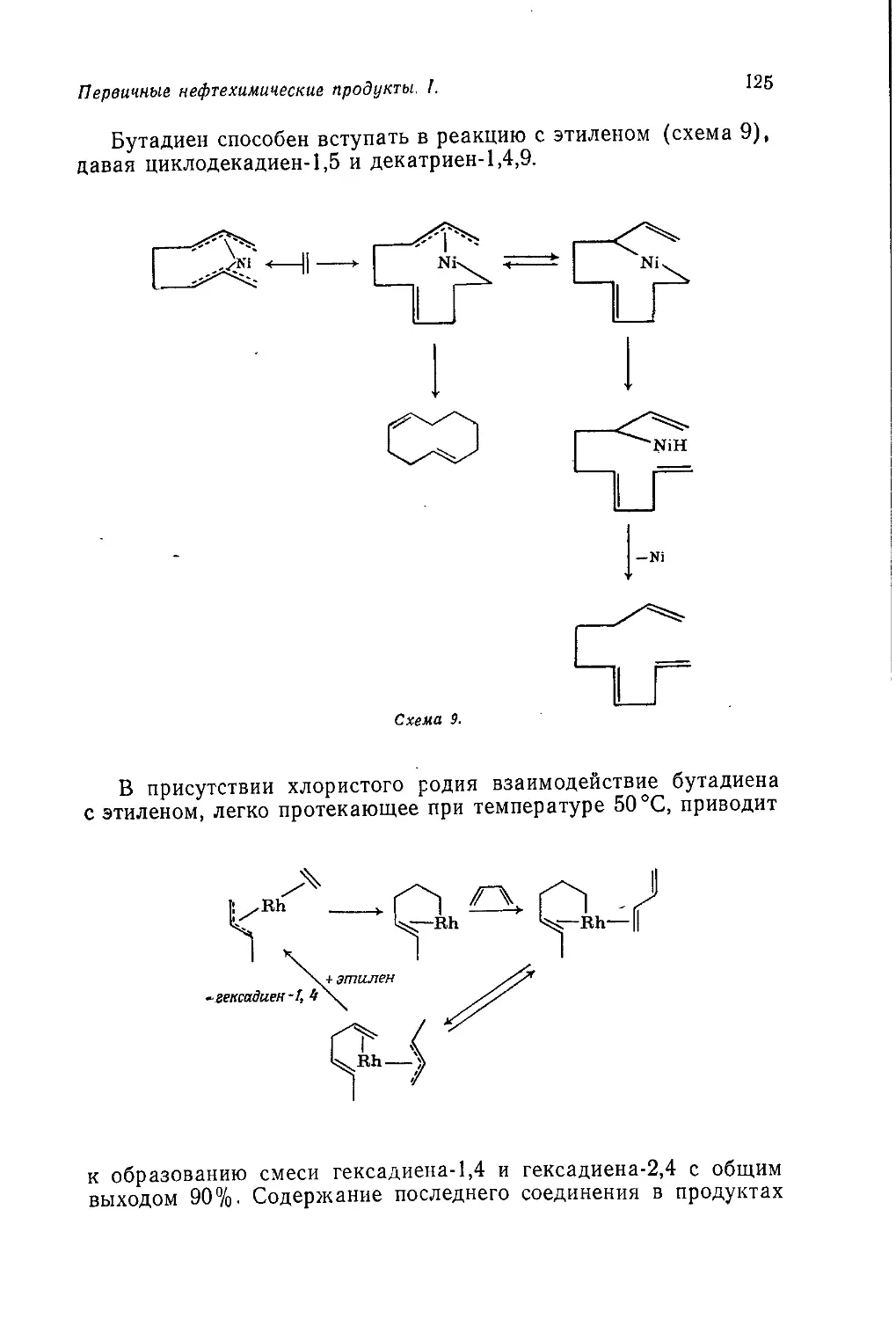
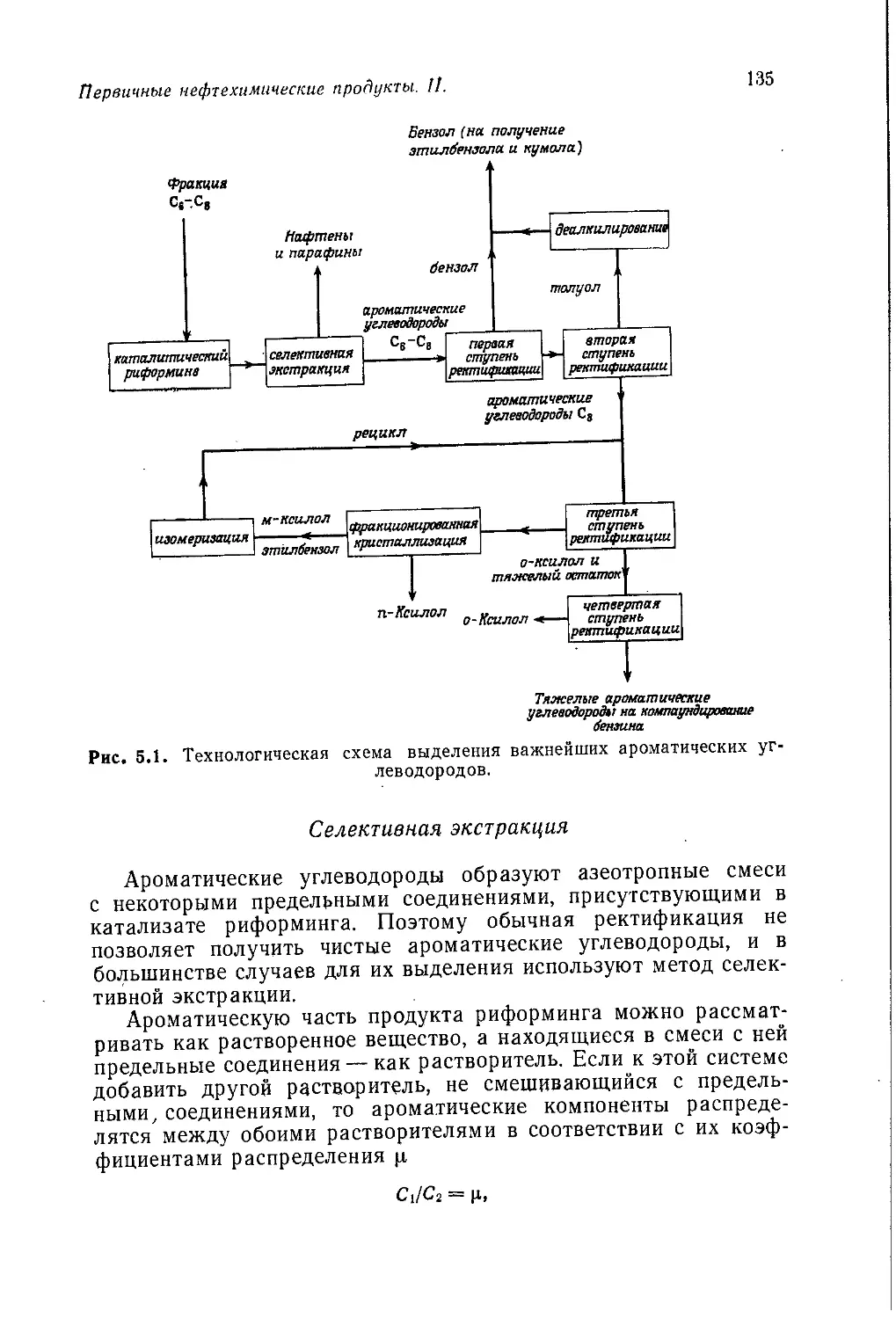
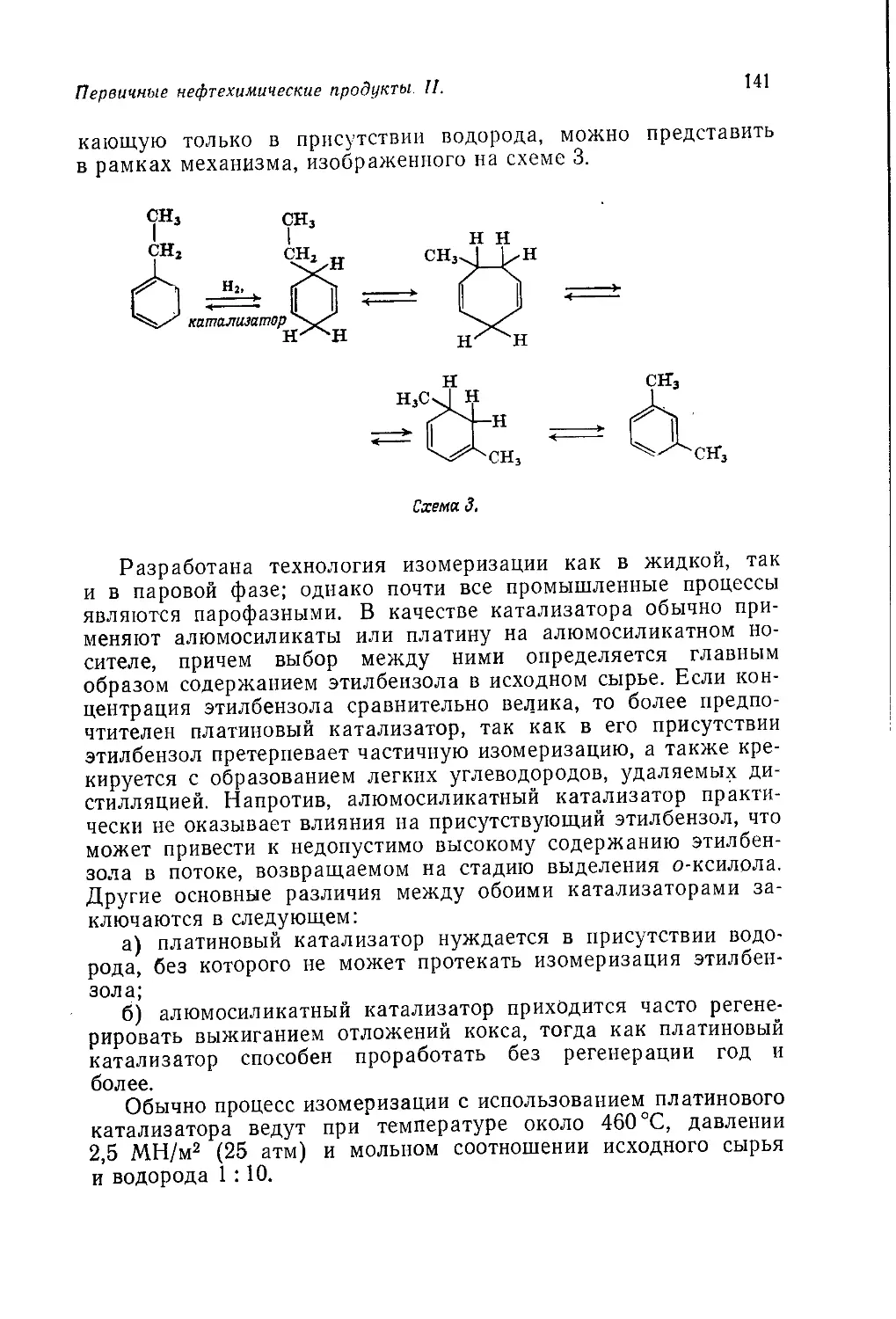
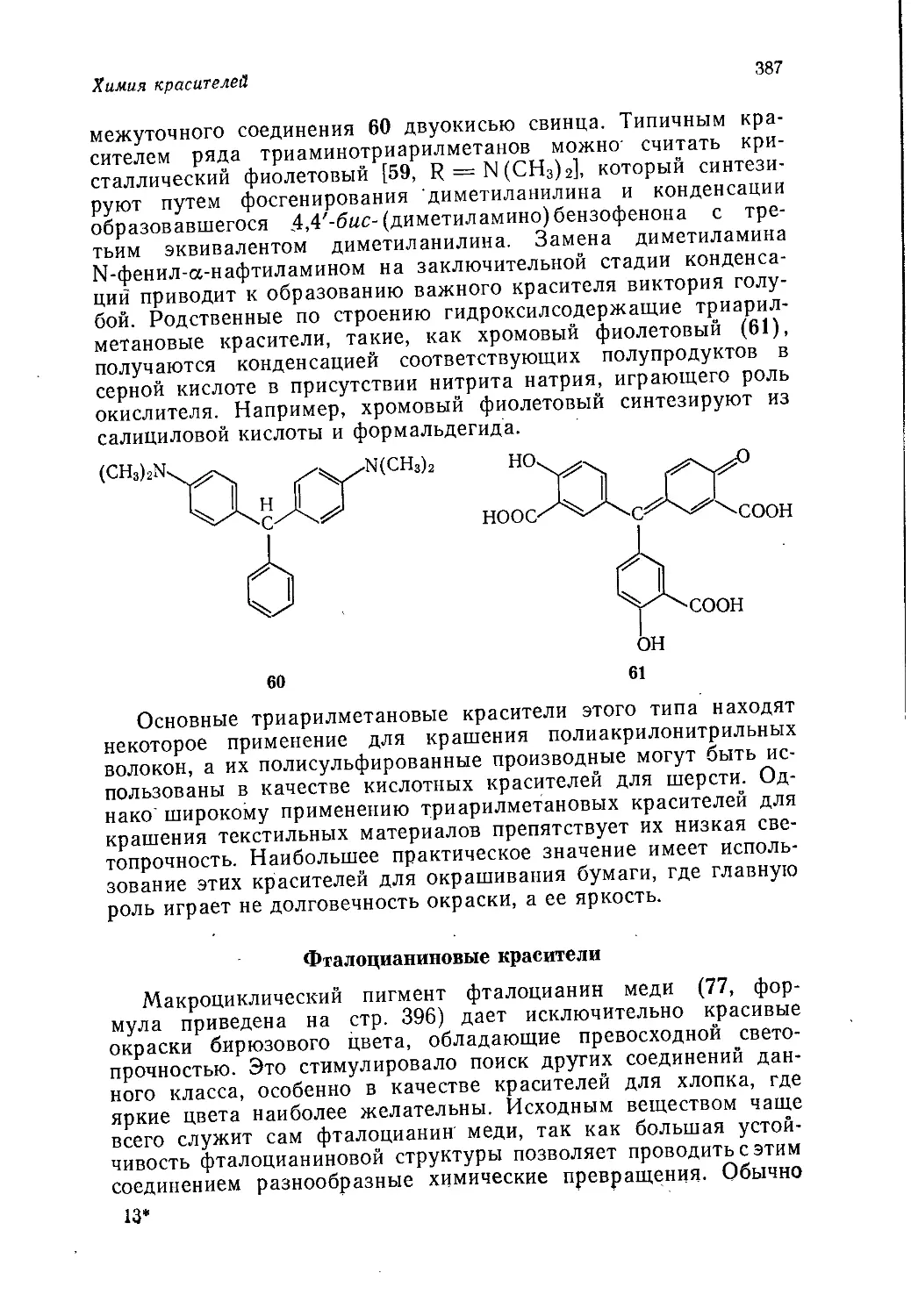
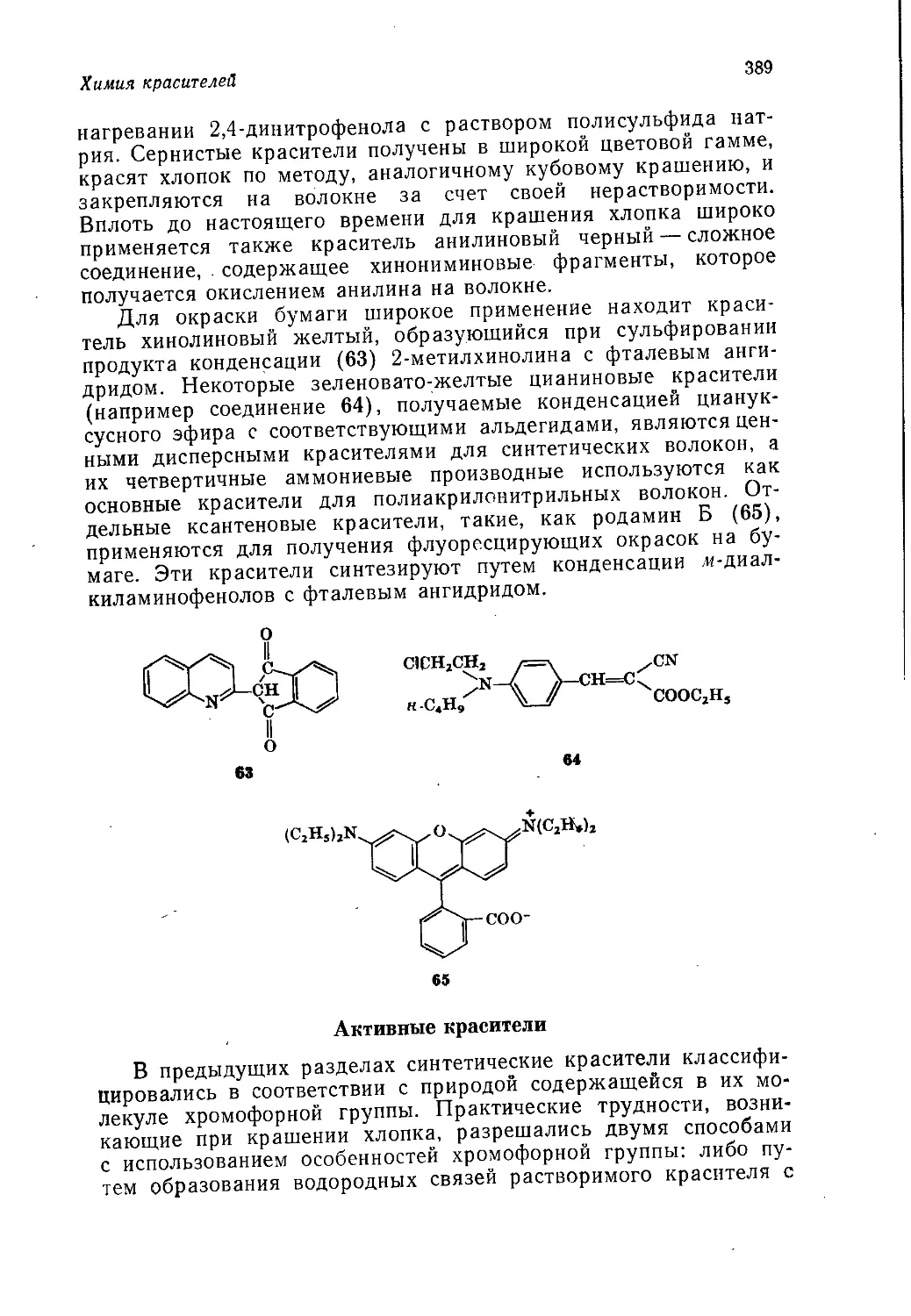
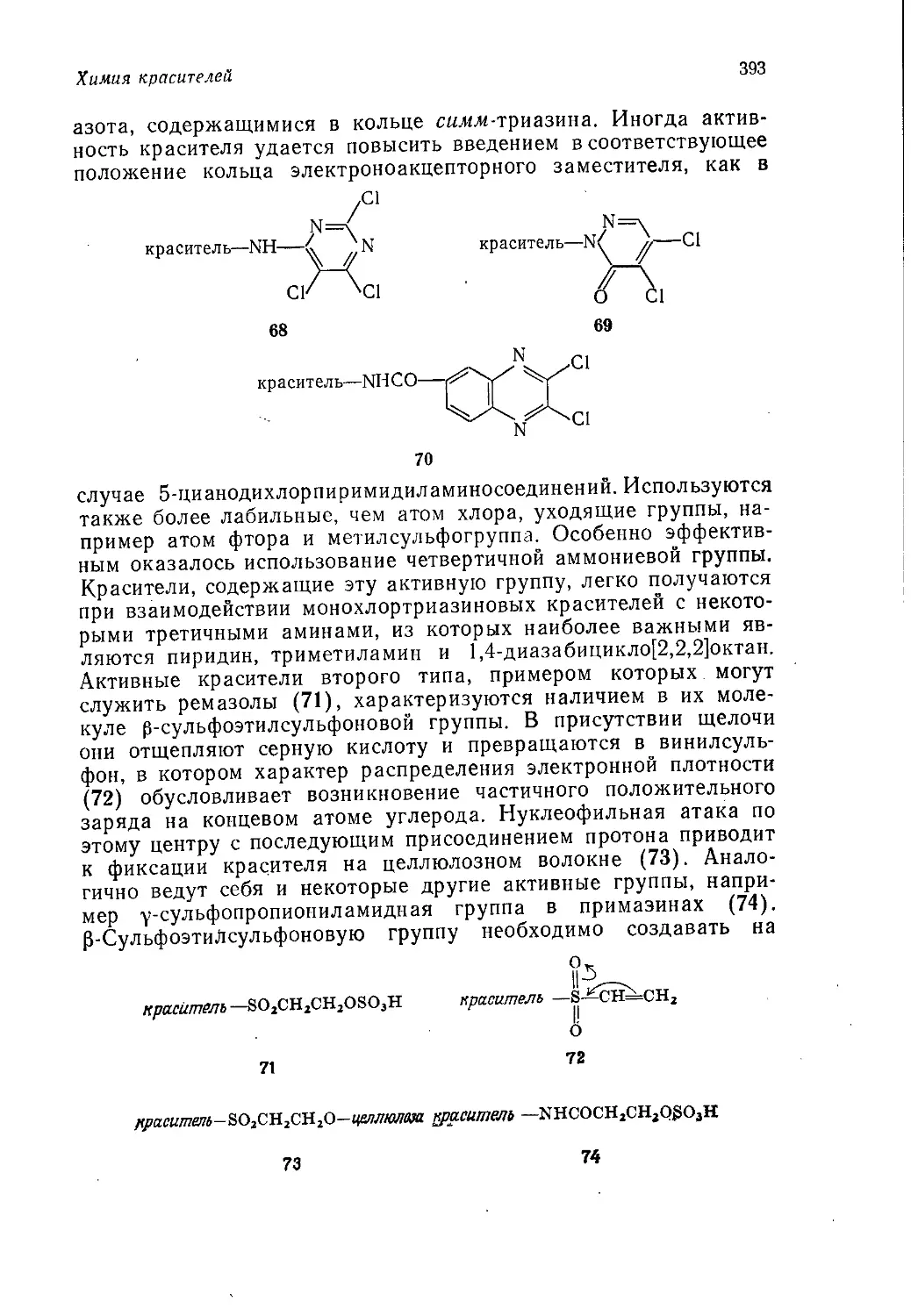
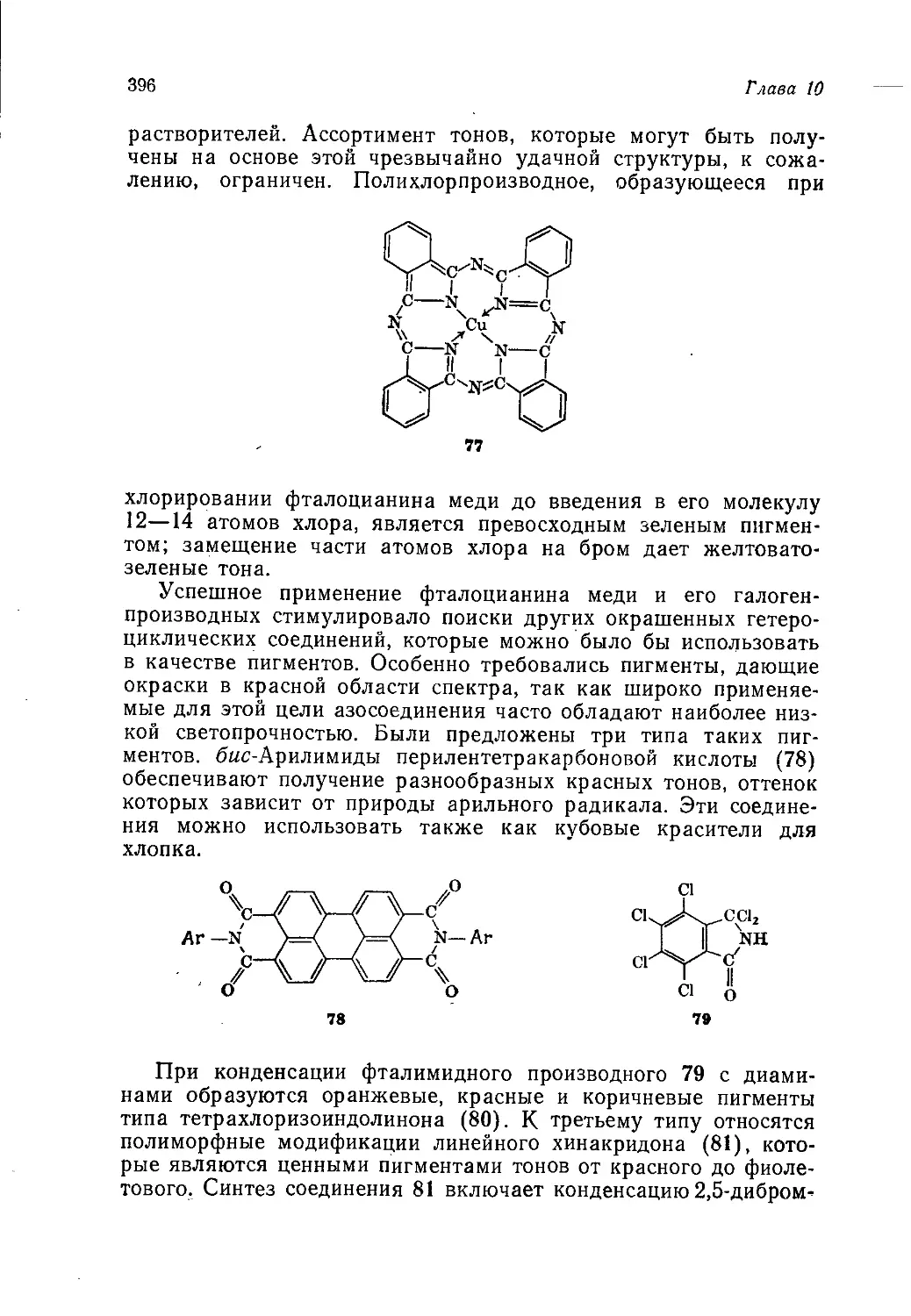
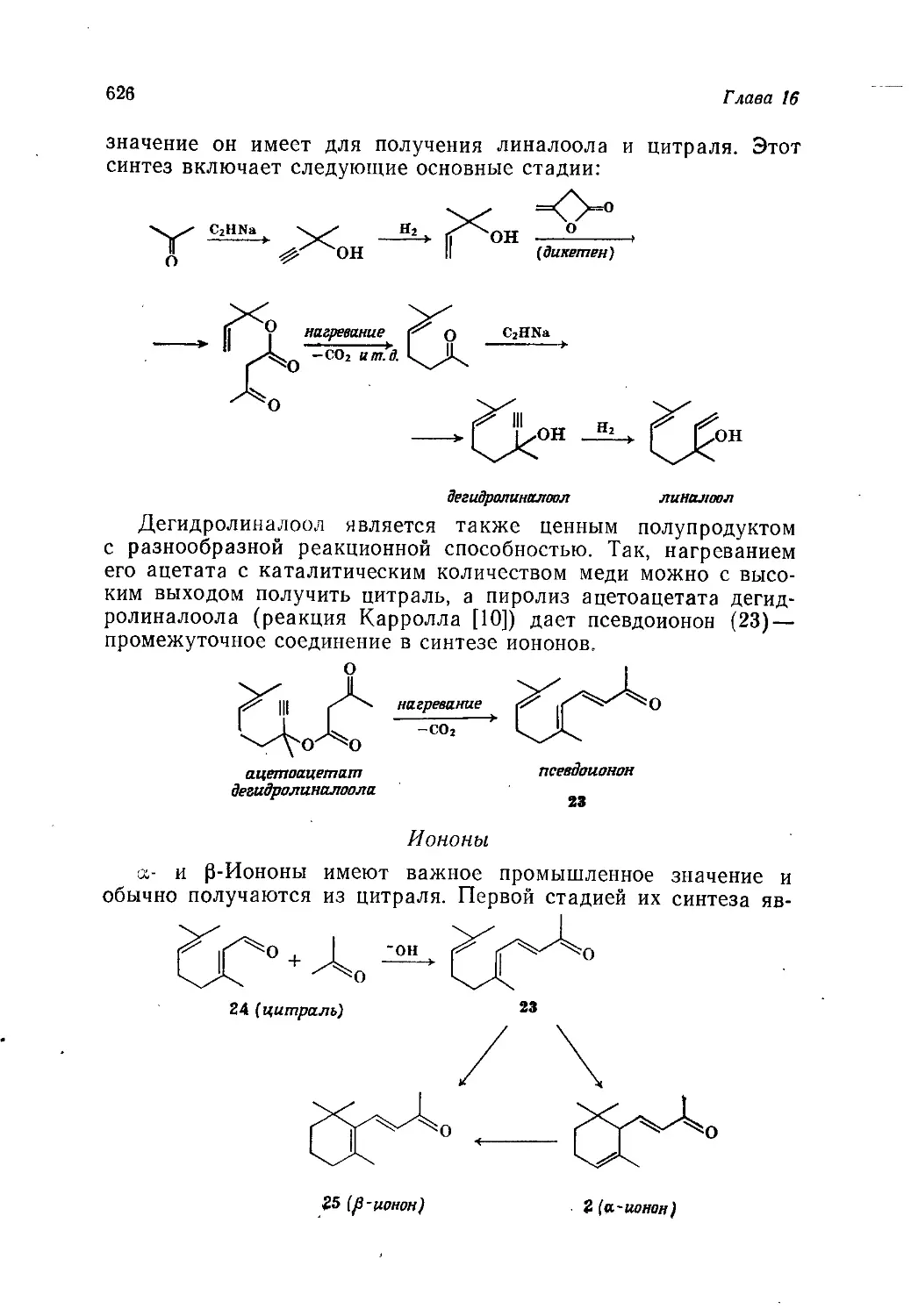
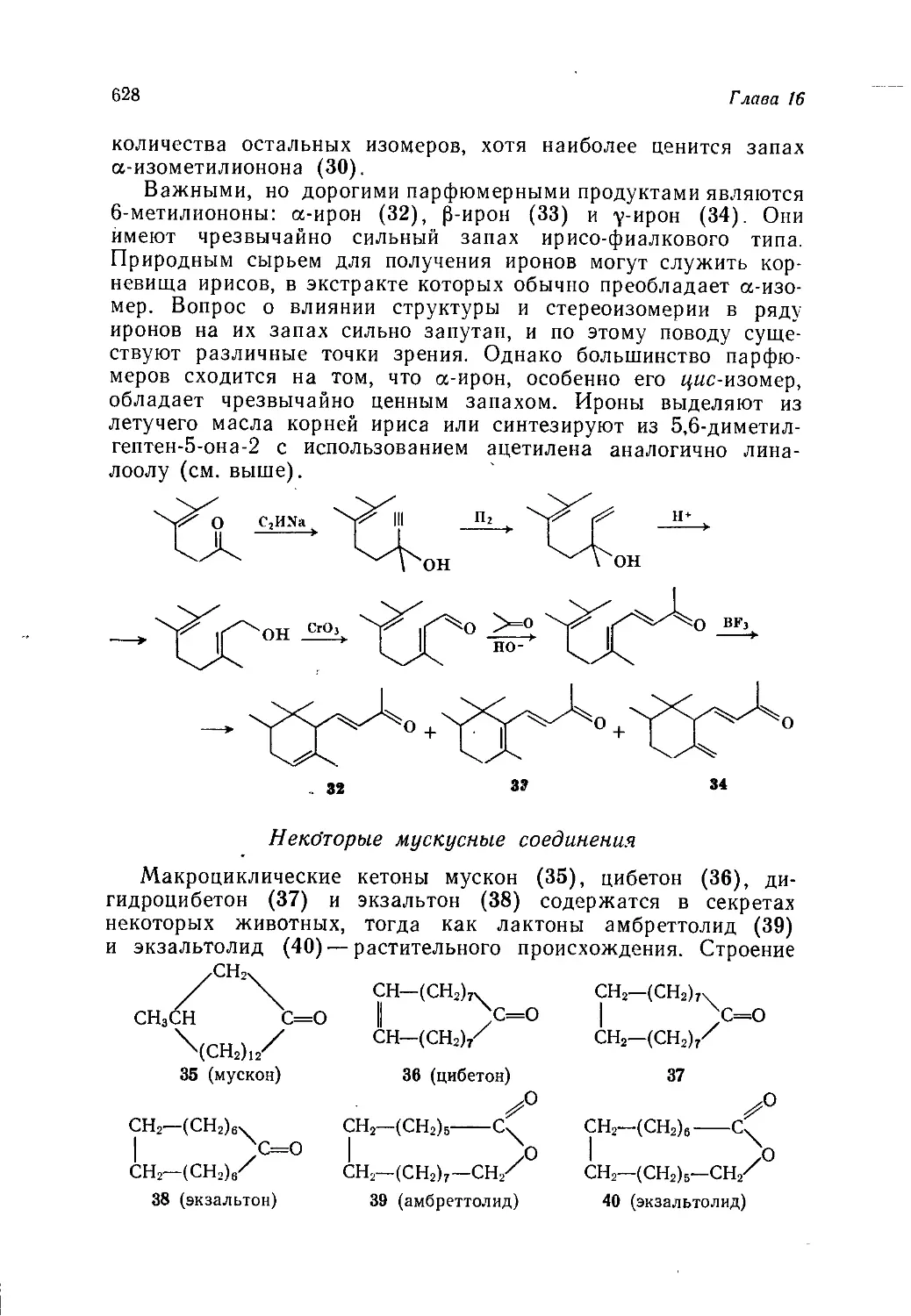
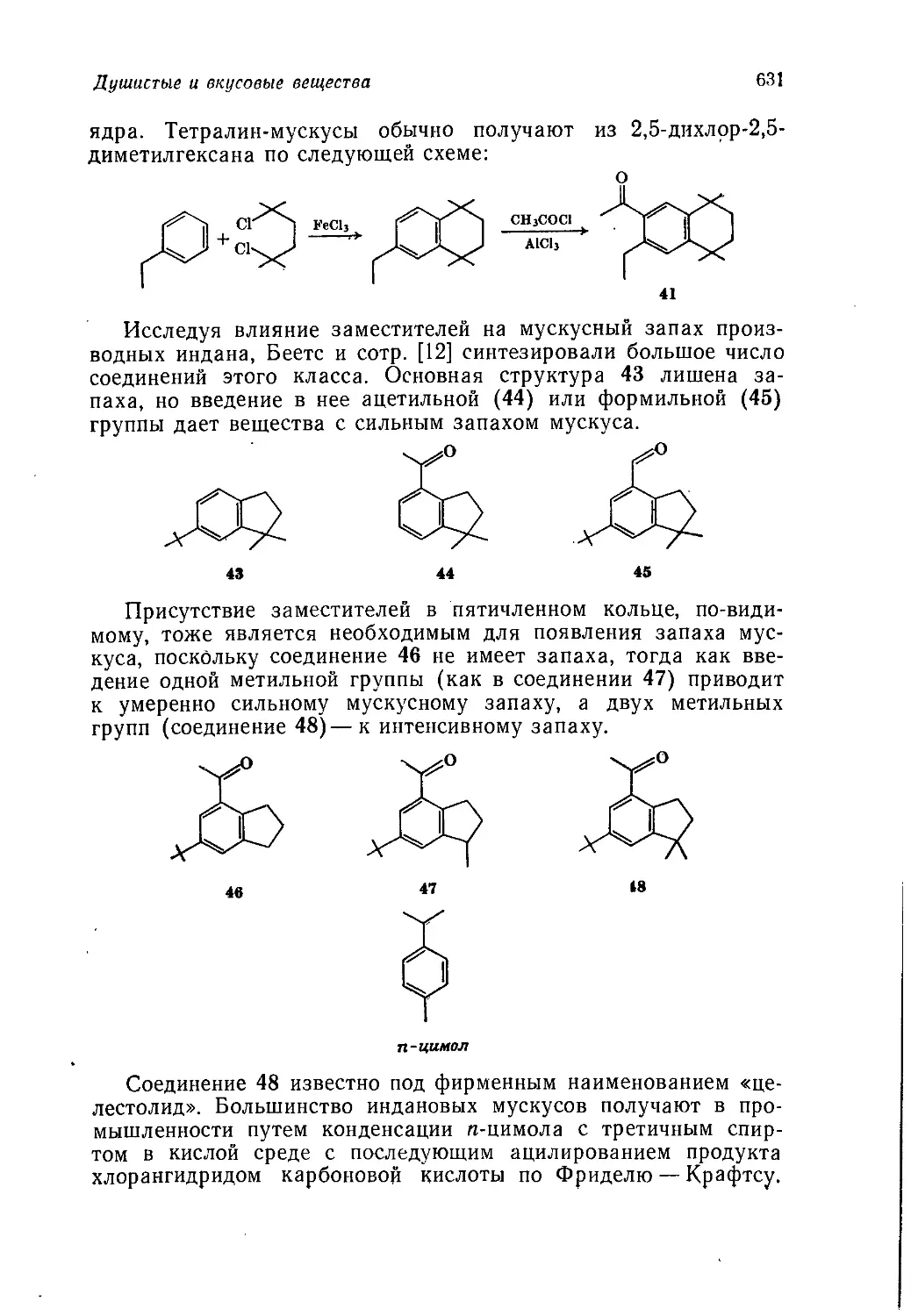
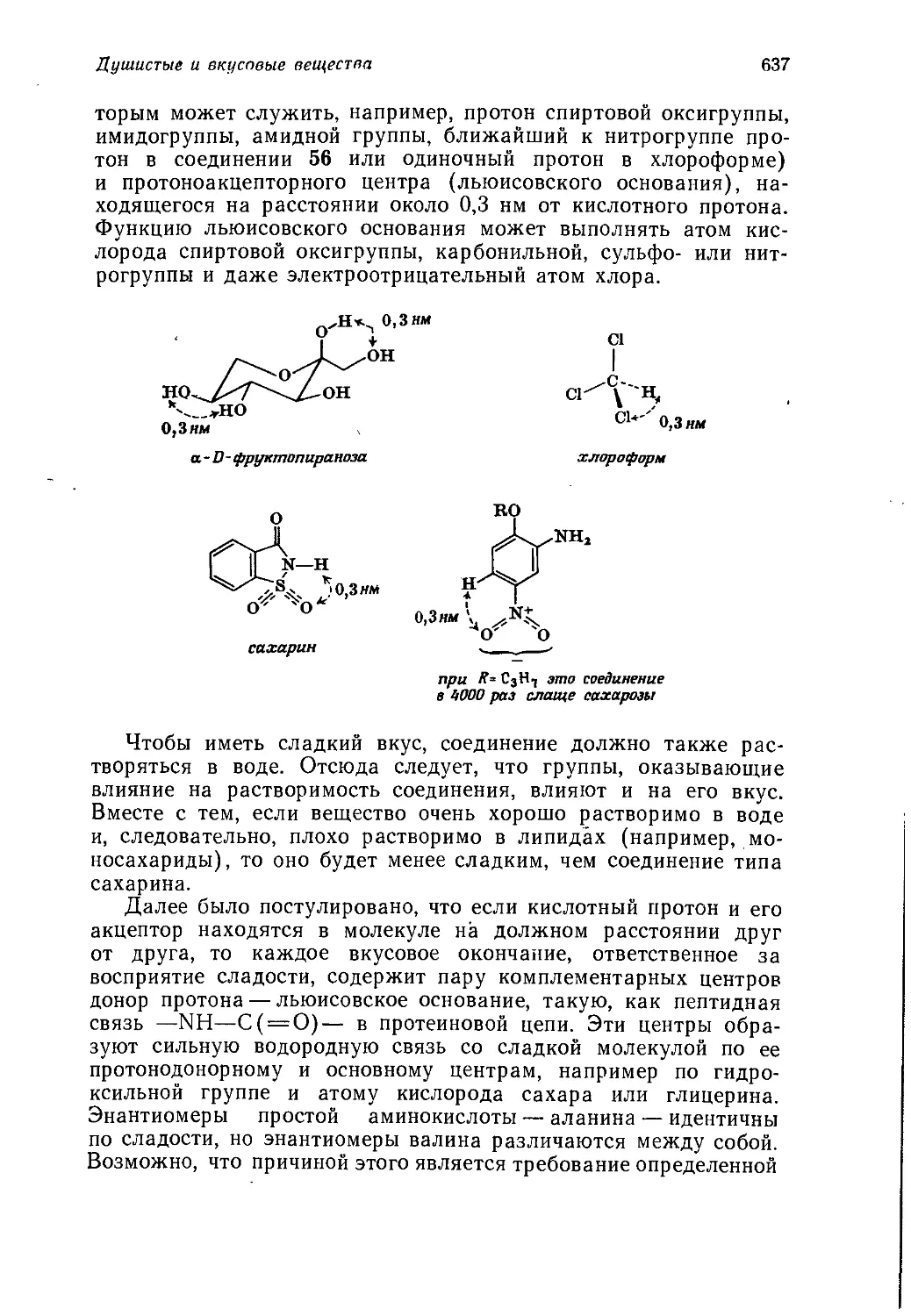
/




















































































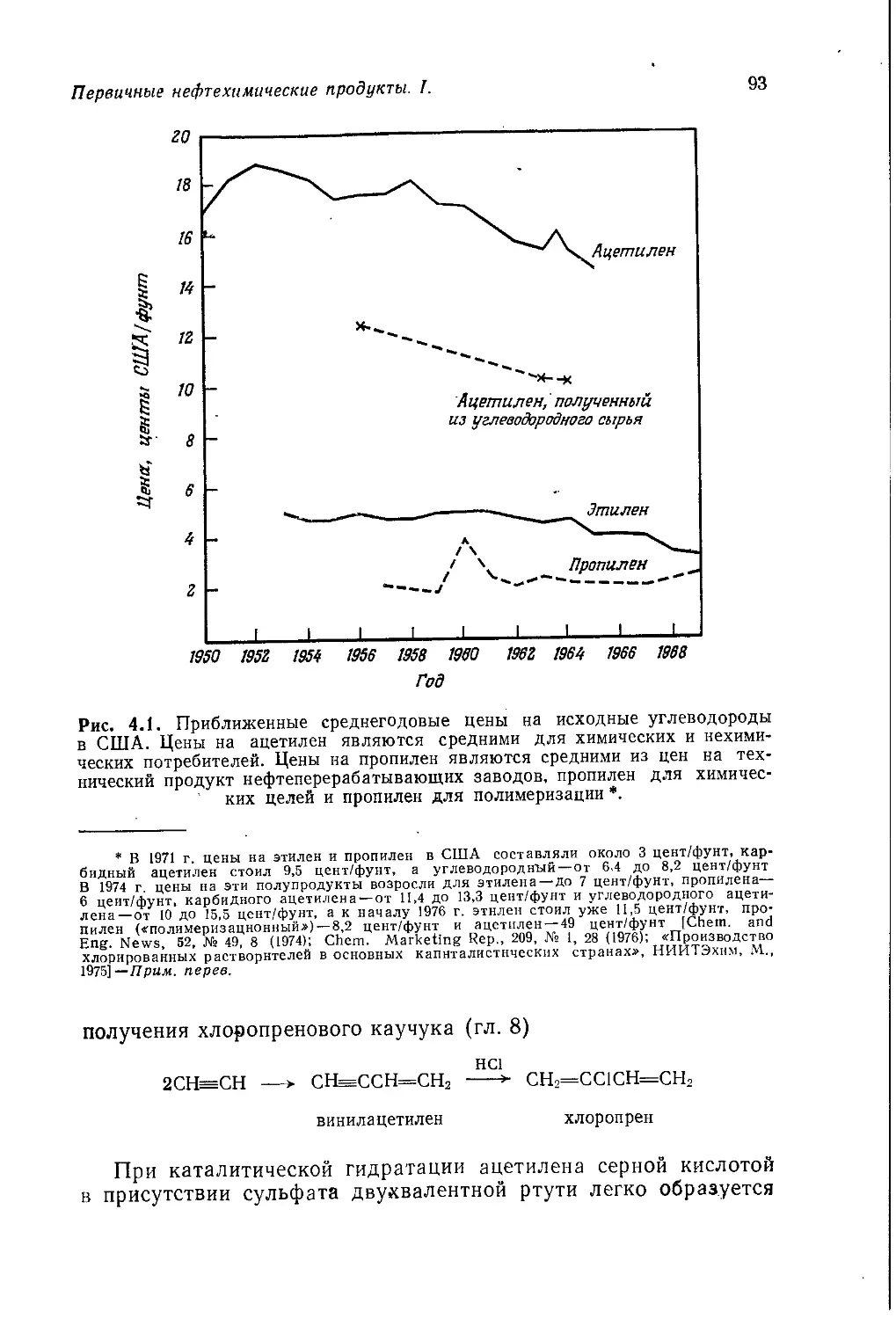
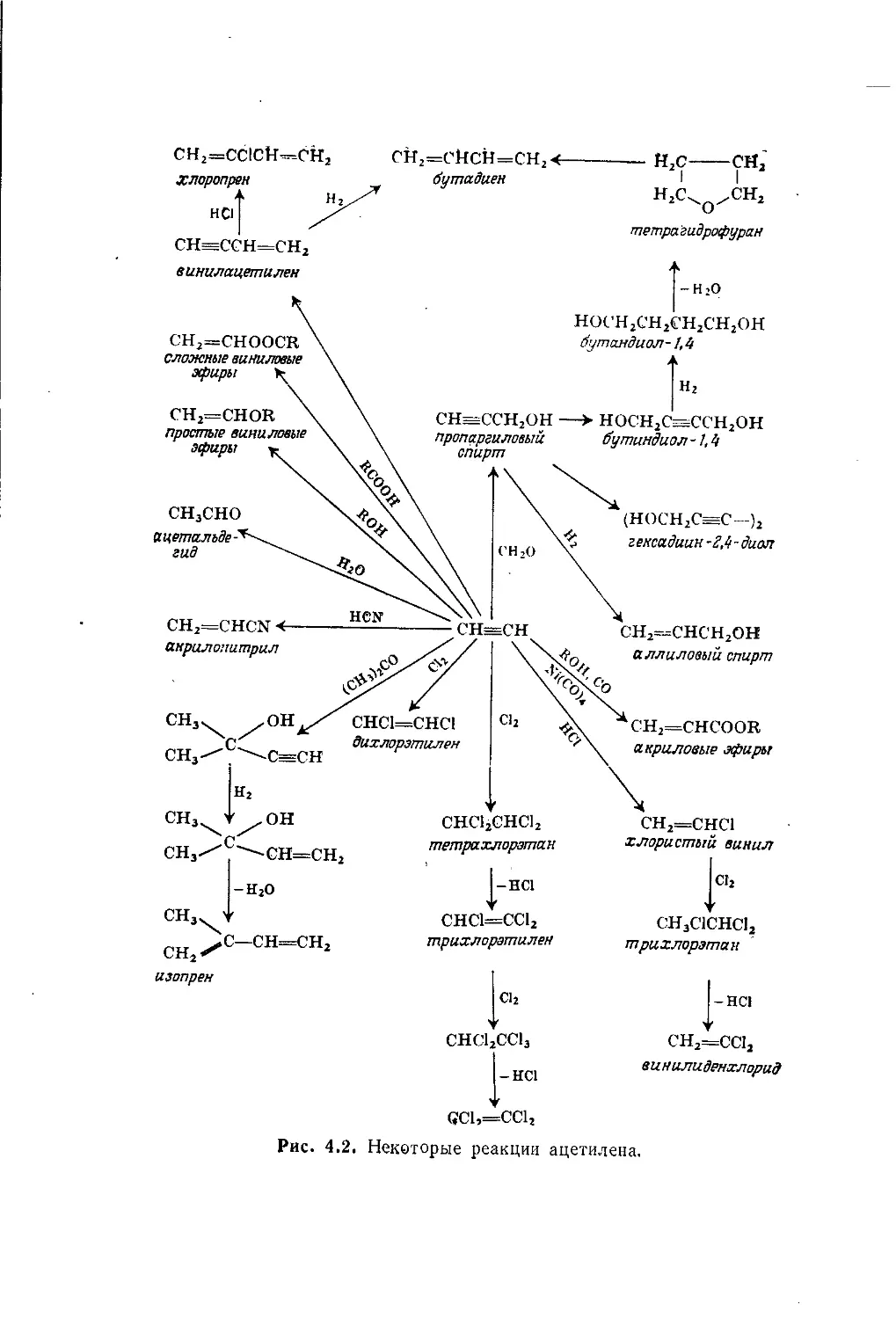



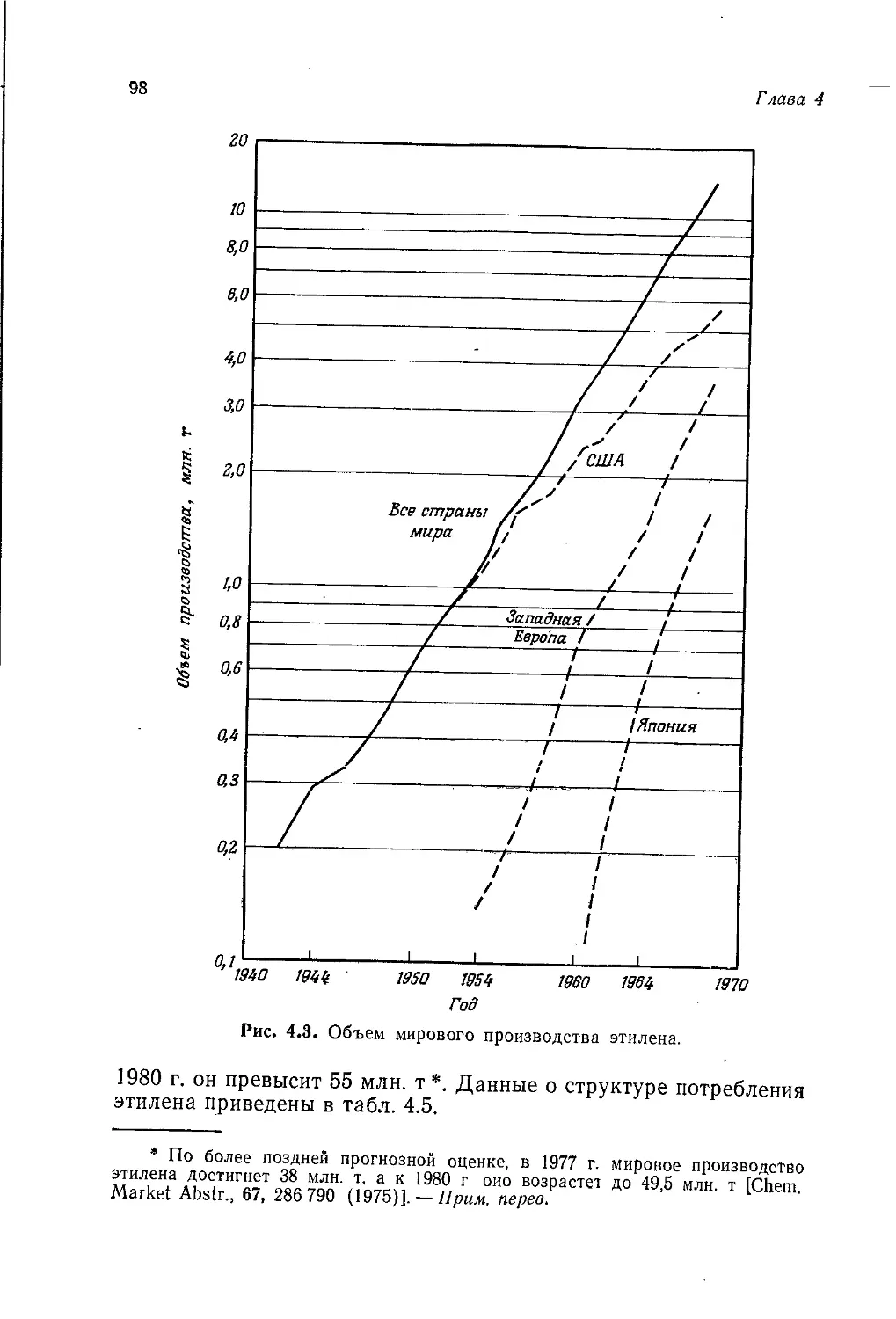
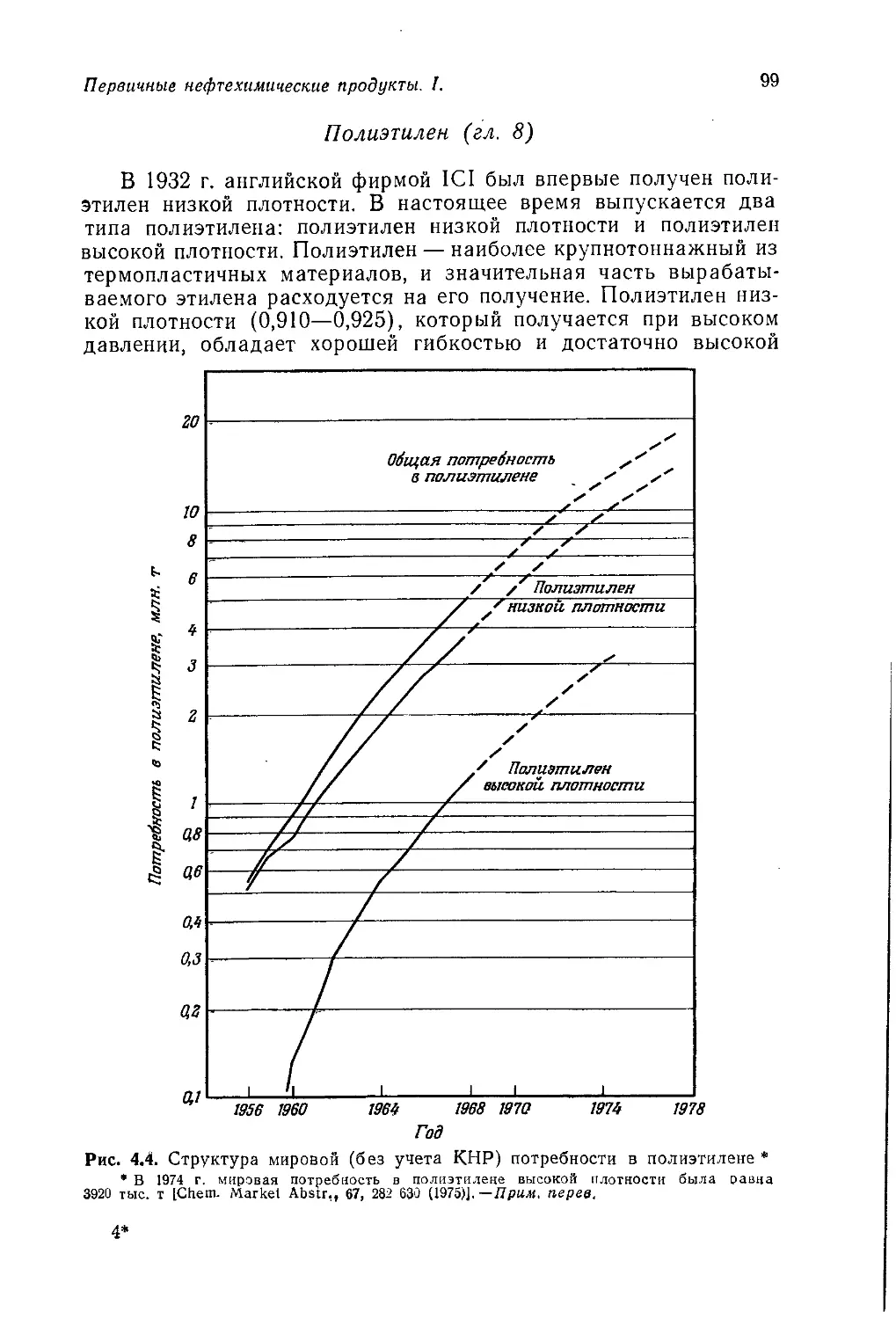





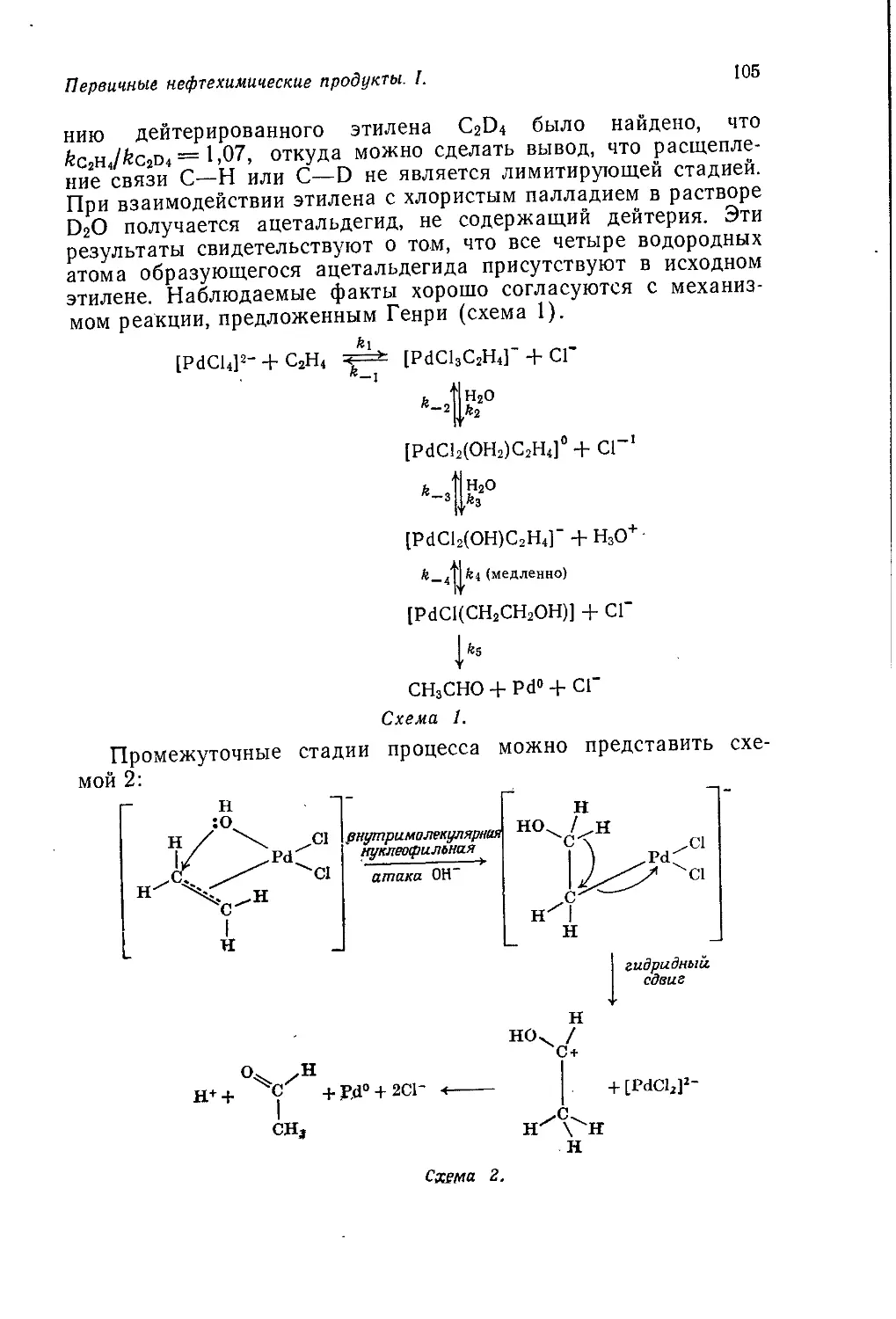

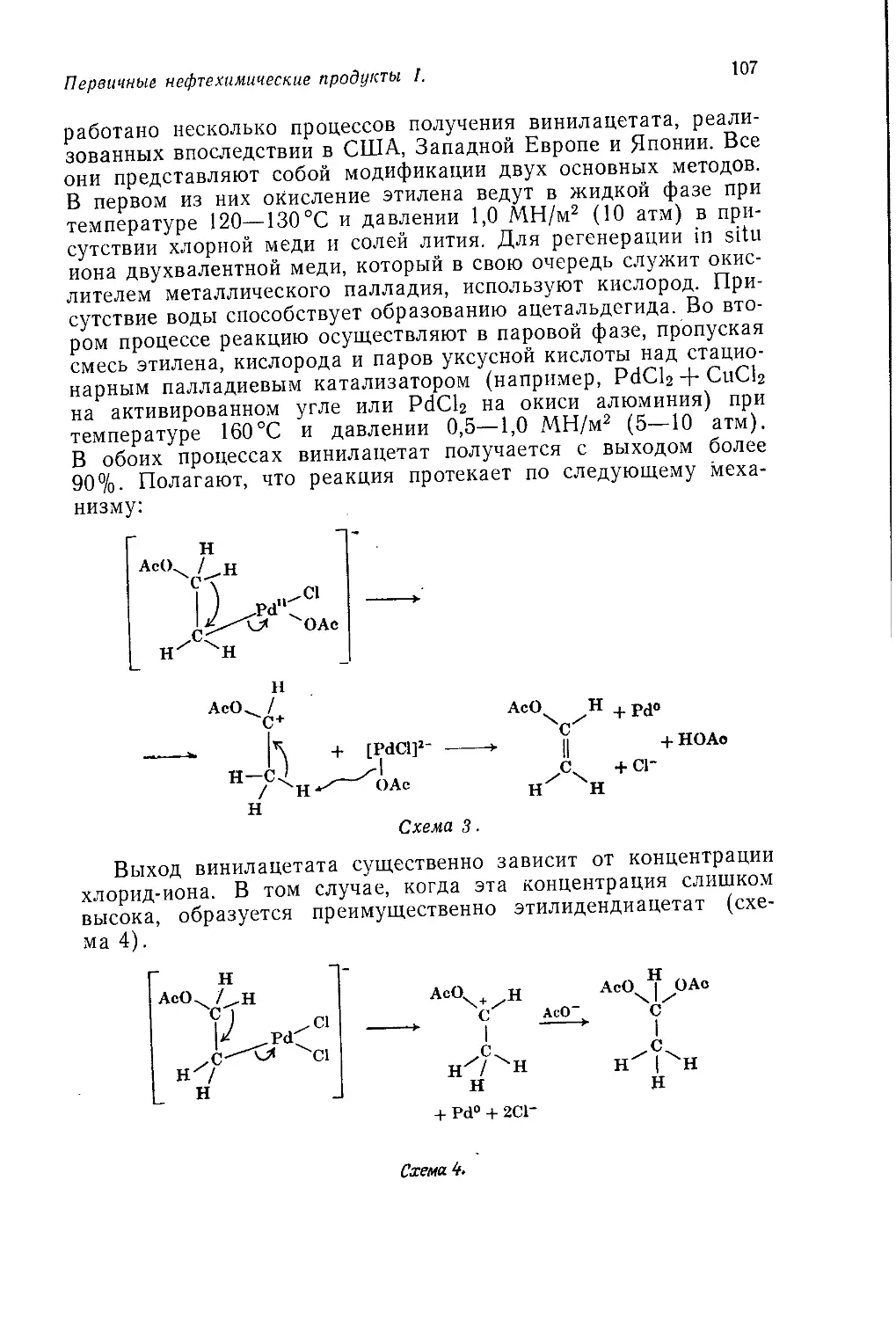








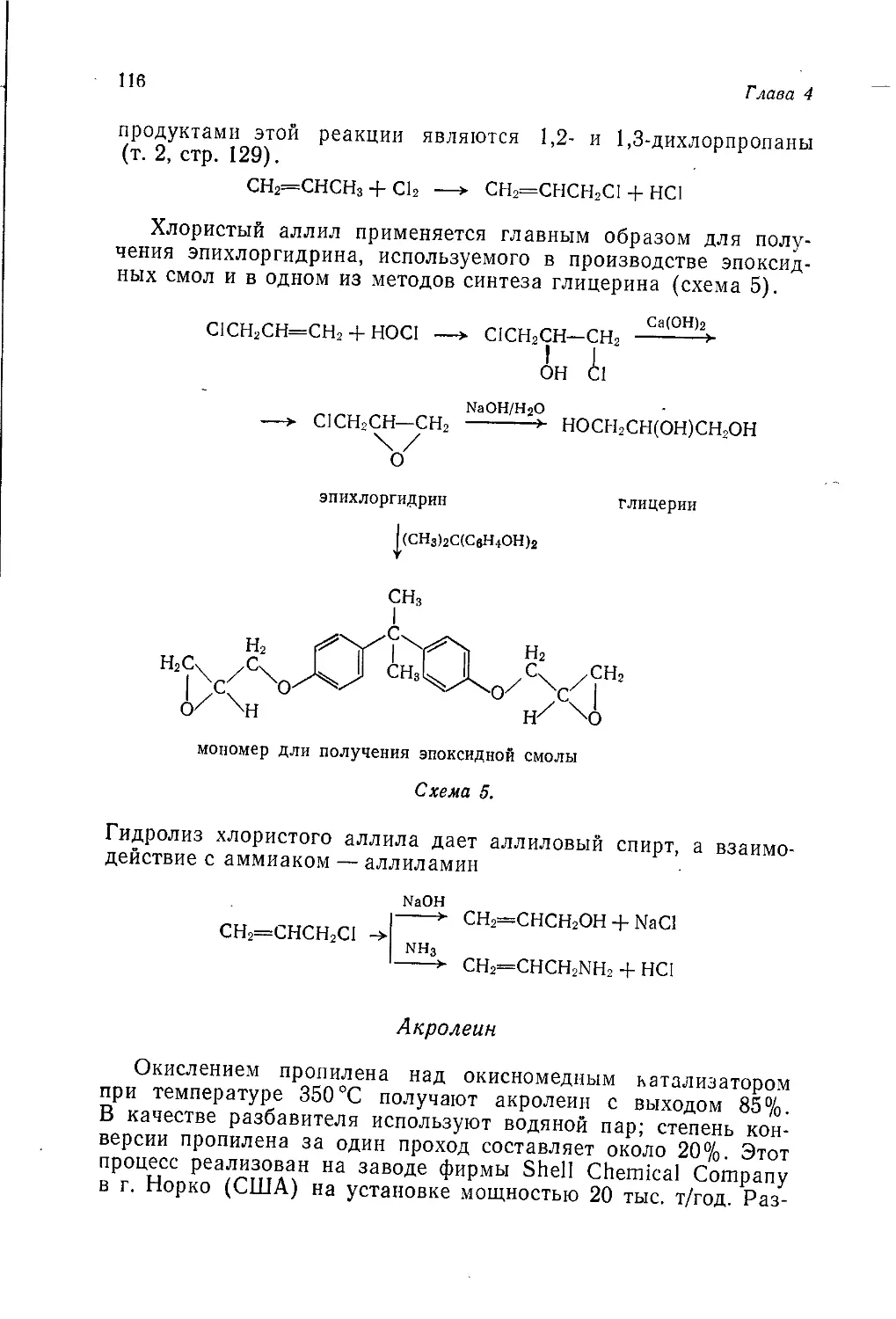

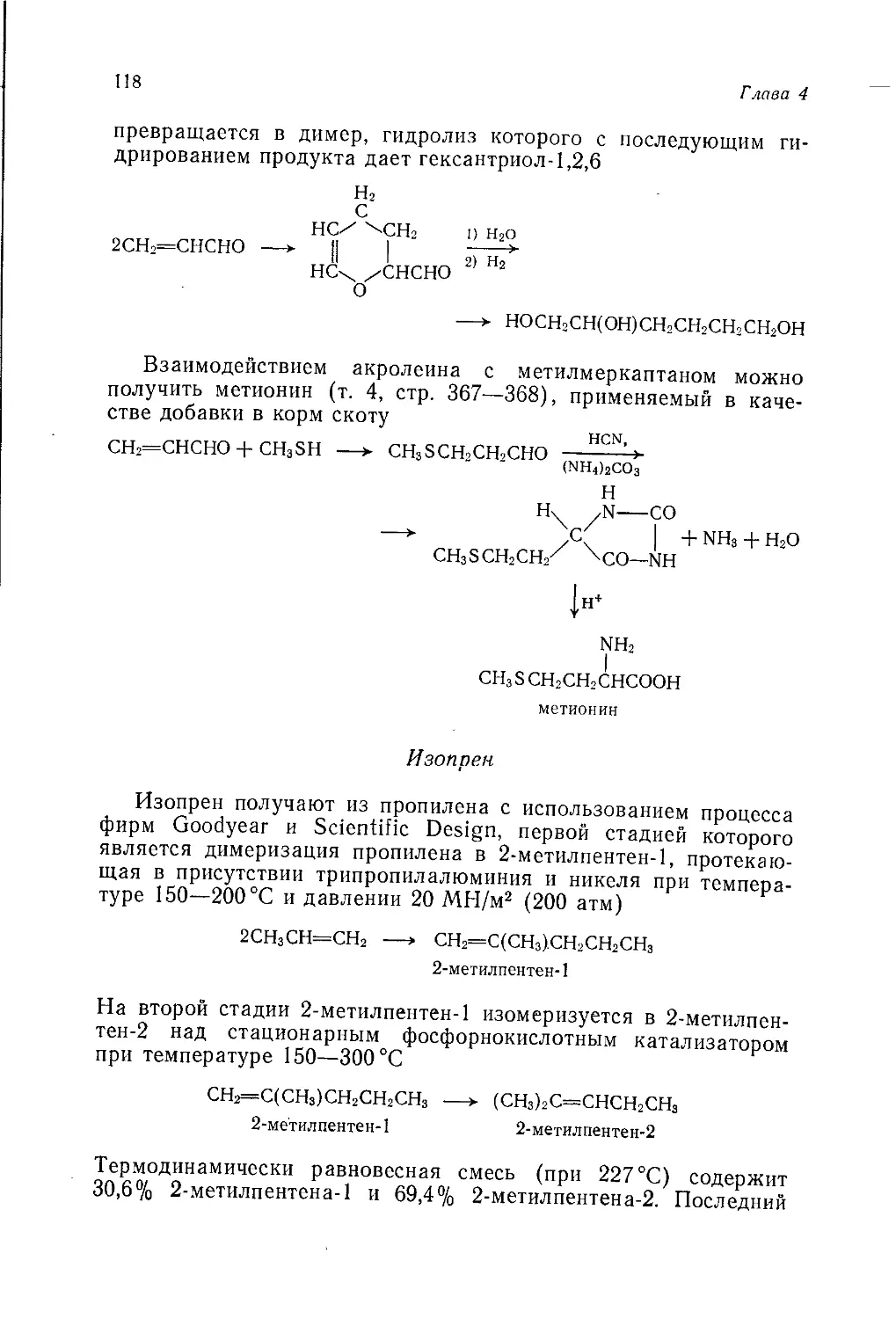

























































































































































































































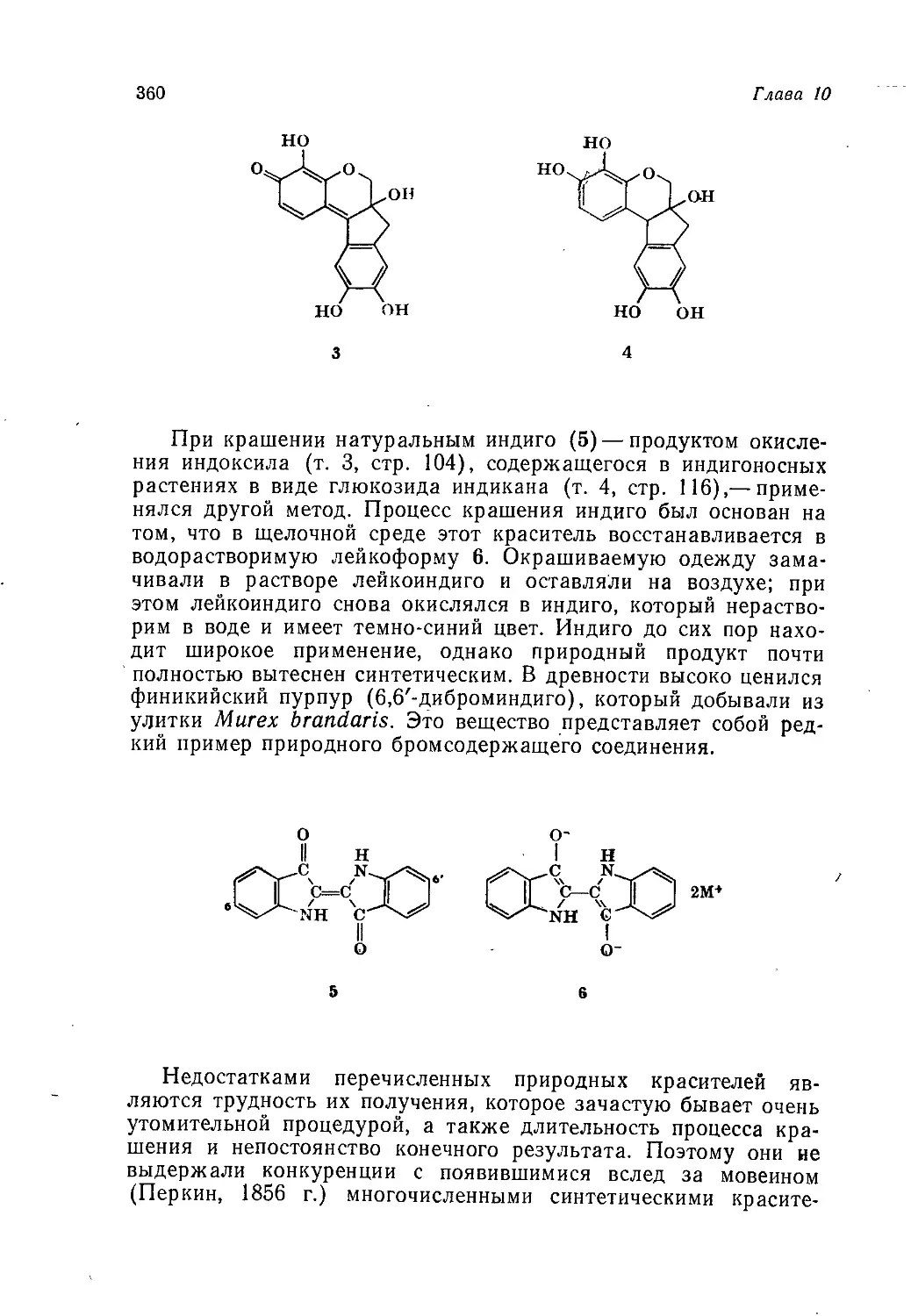
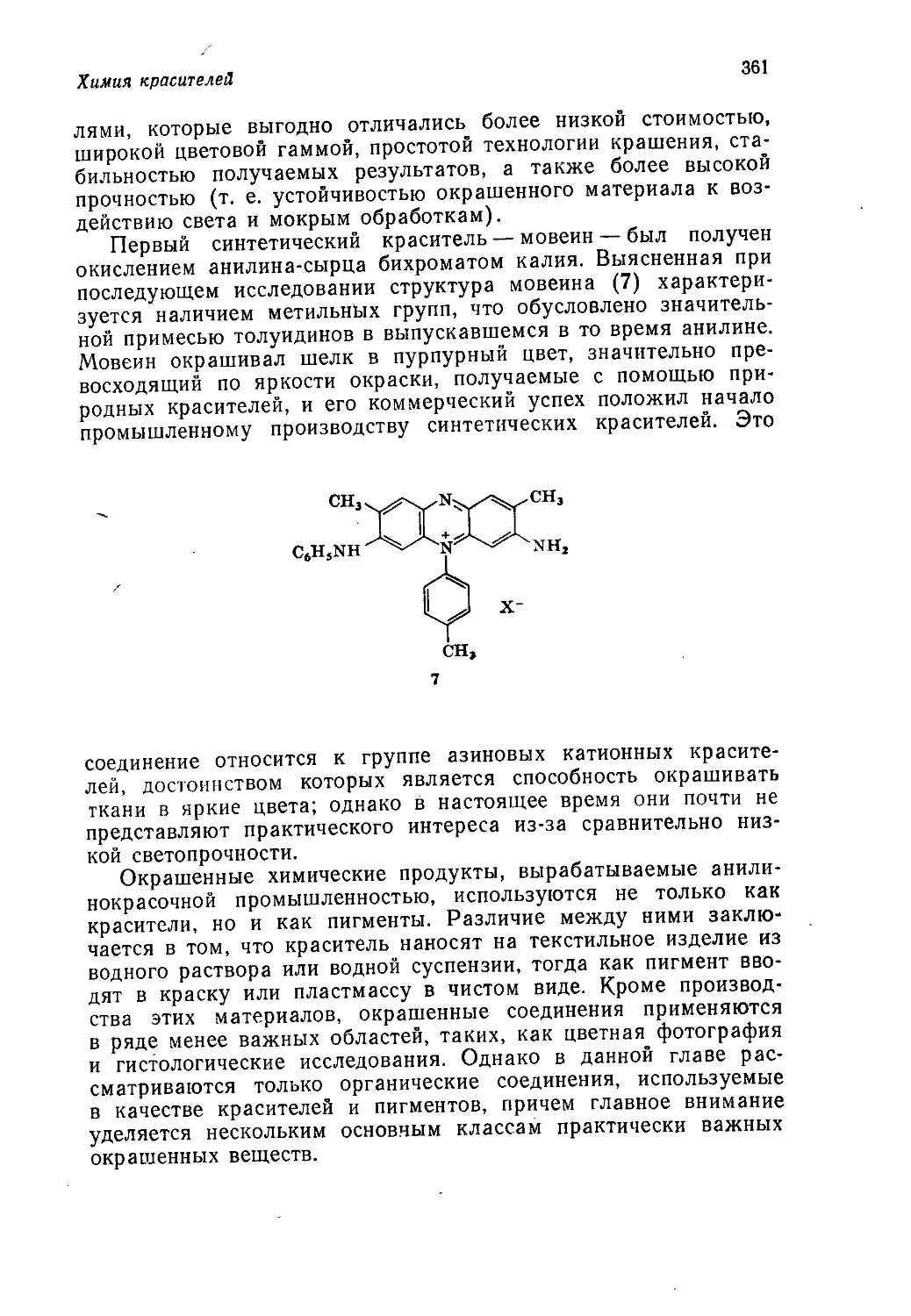
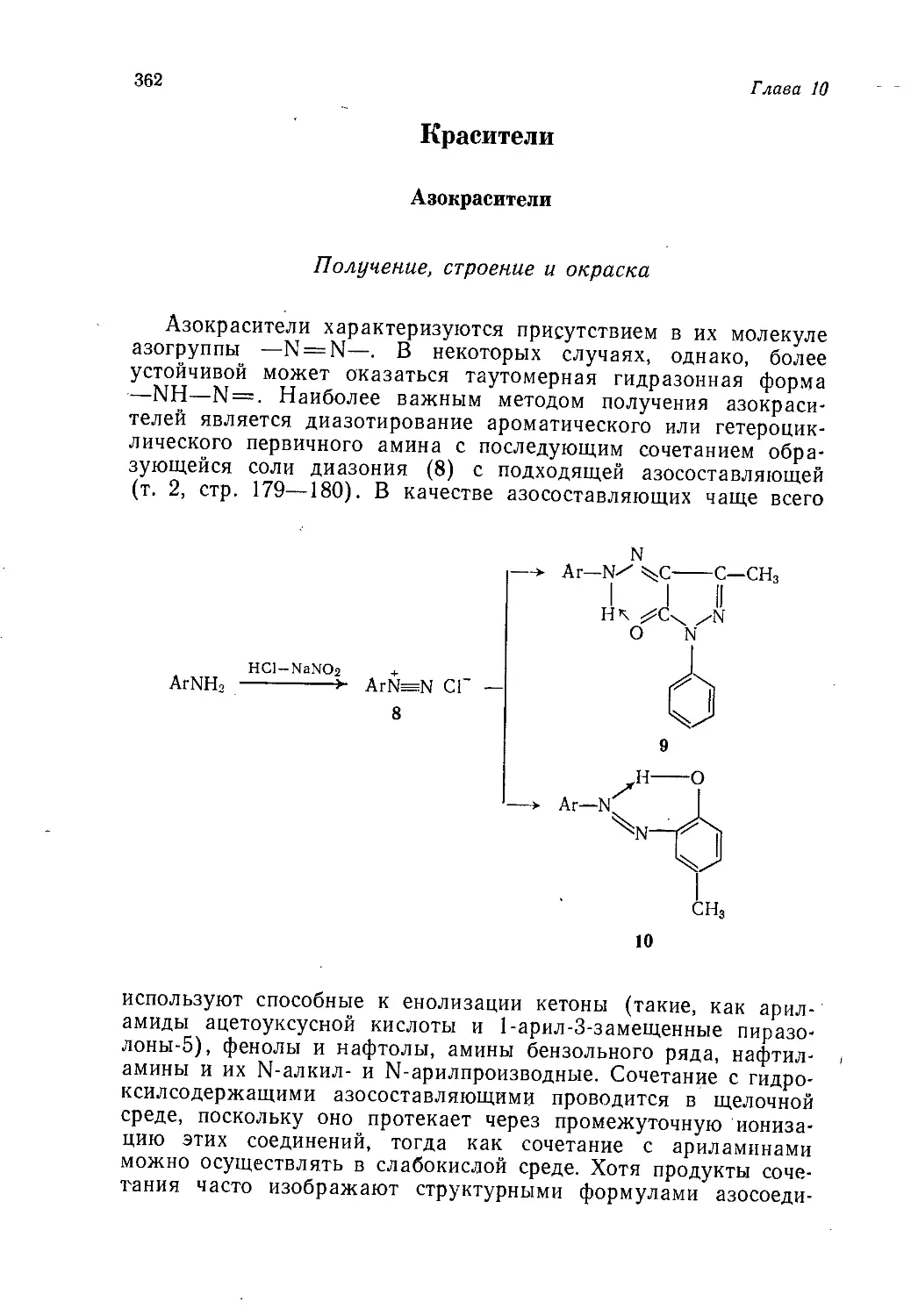

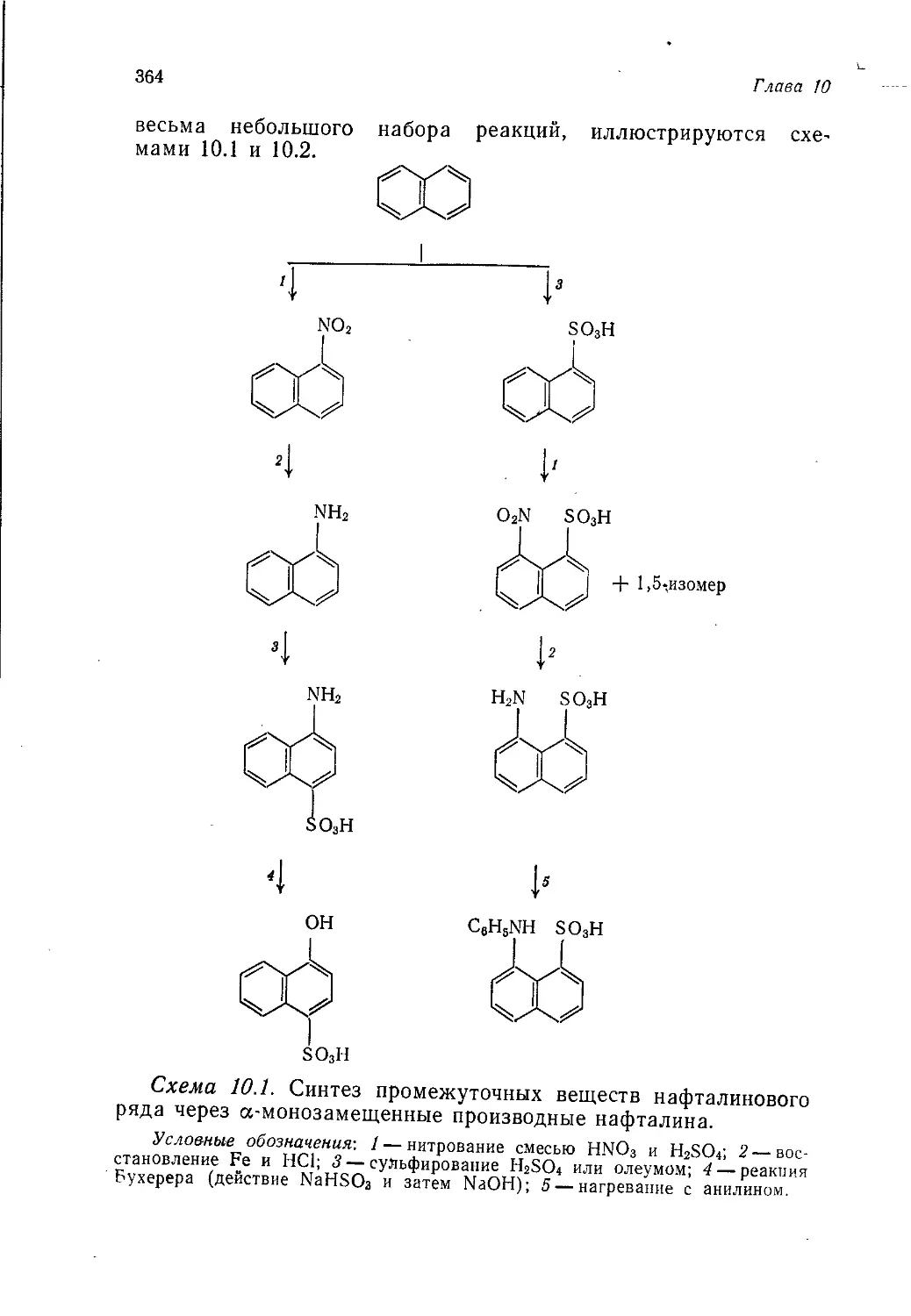






























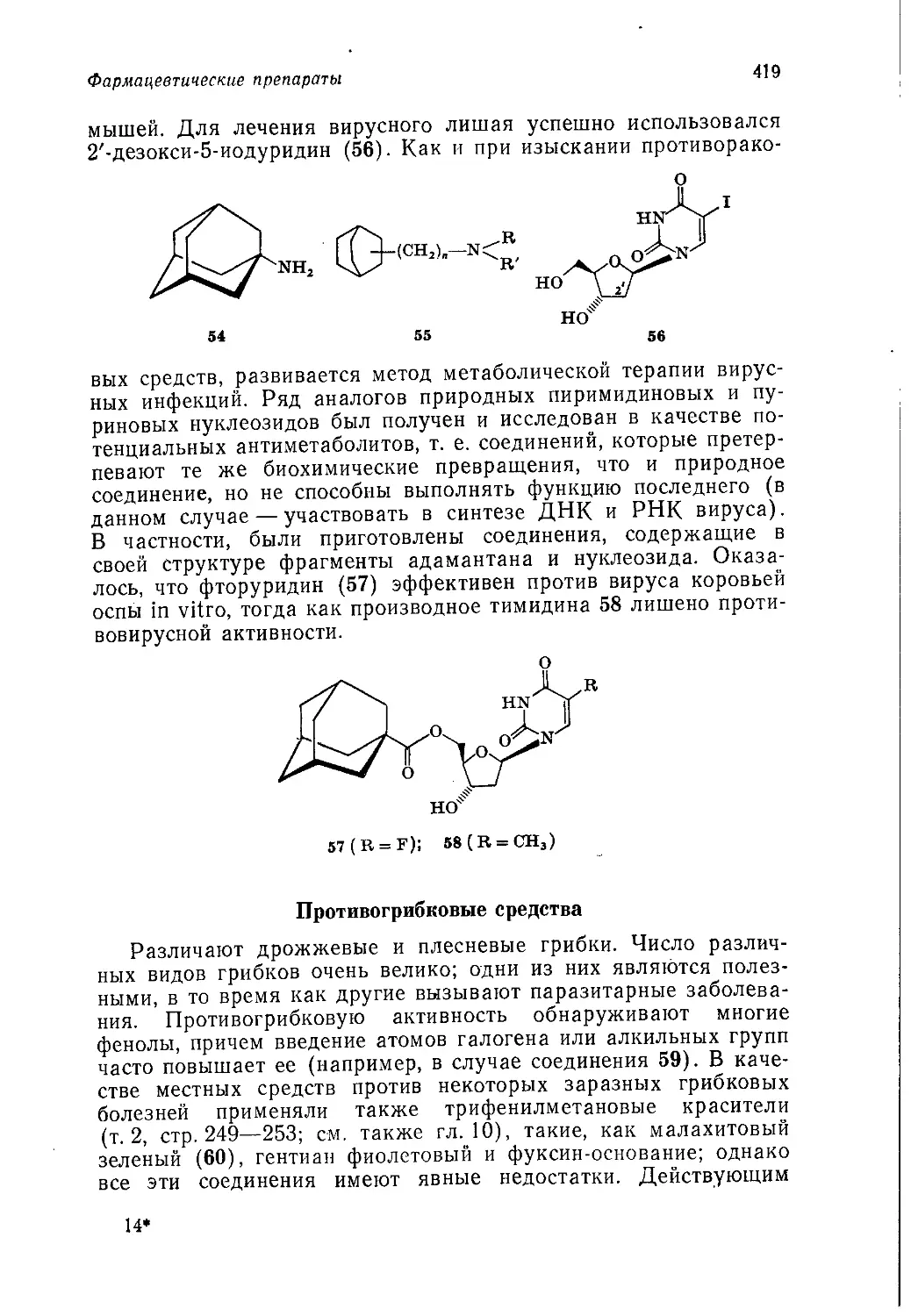
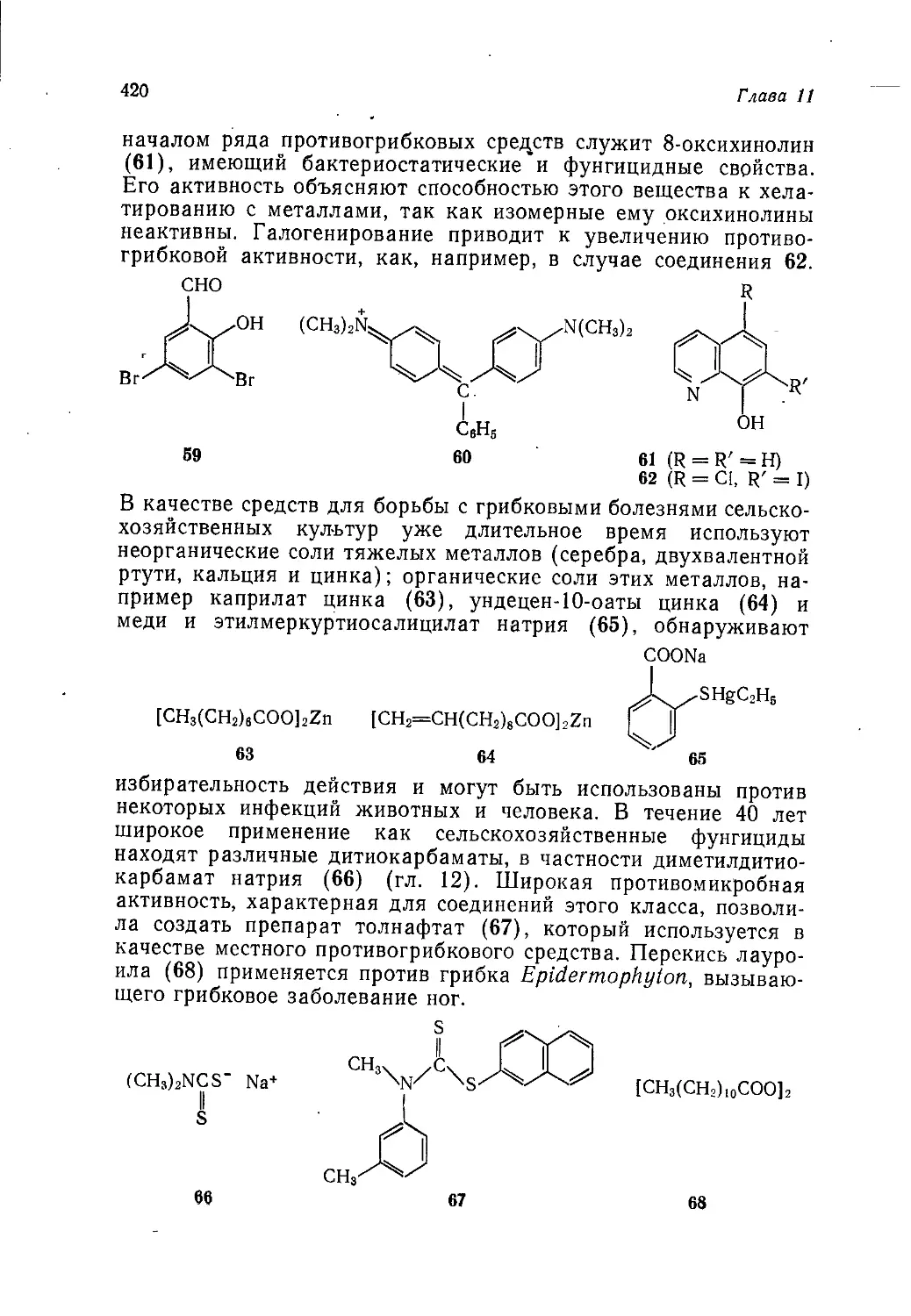
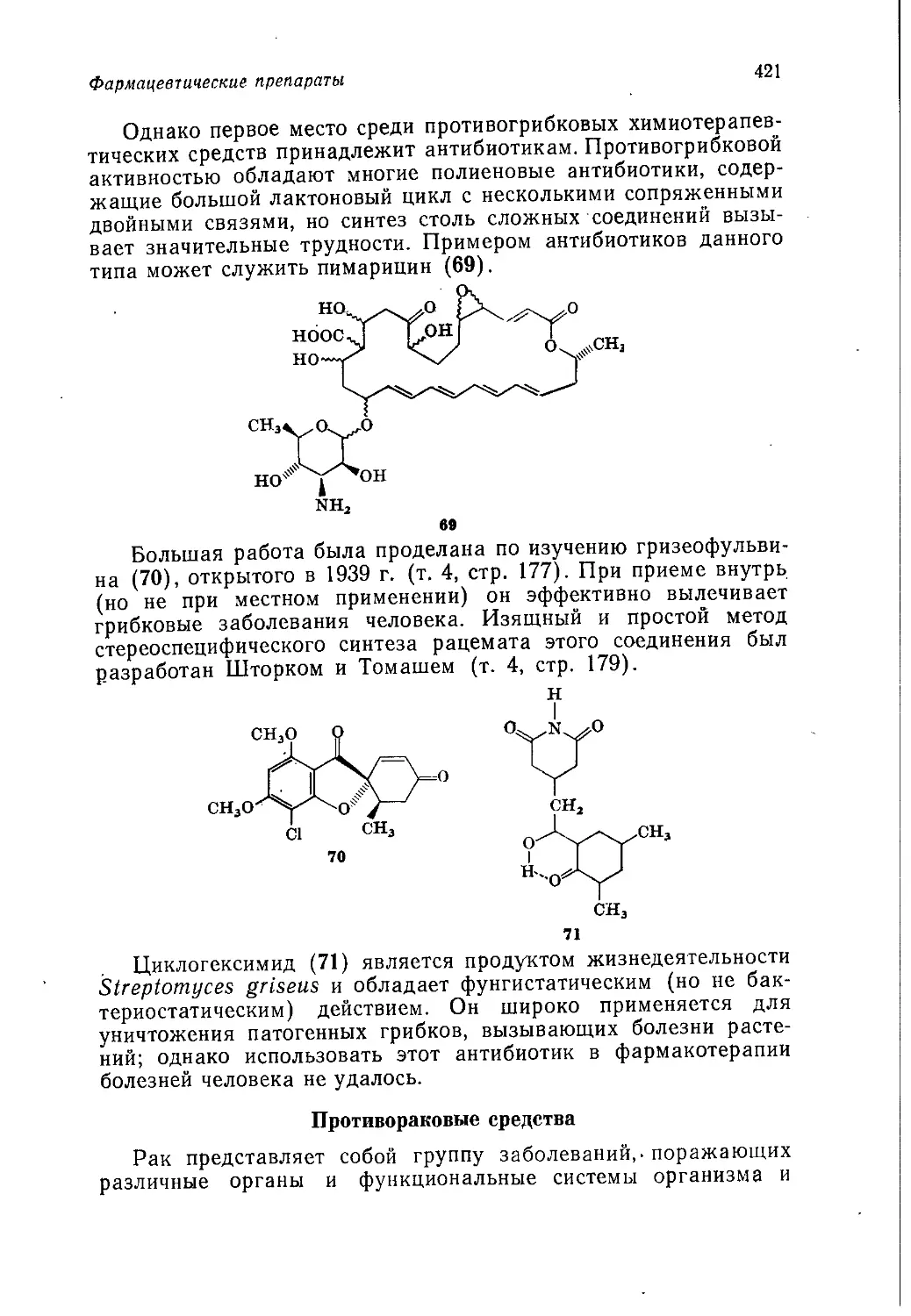
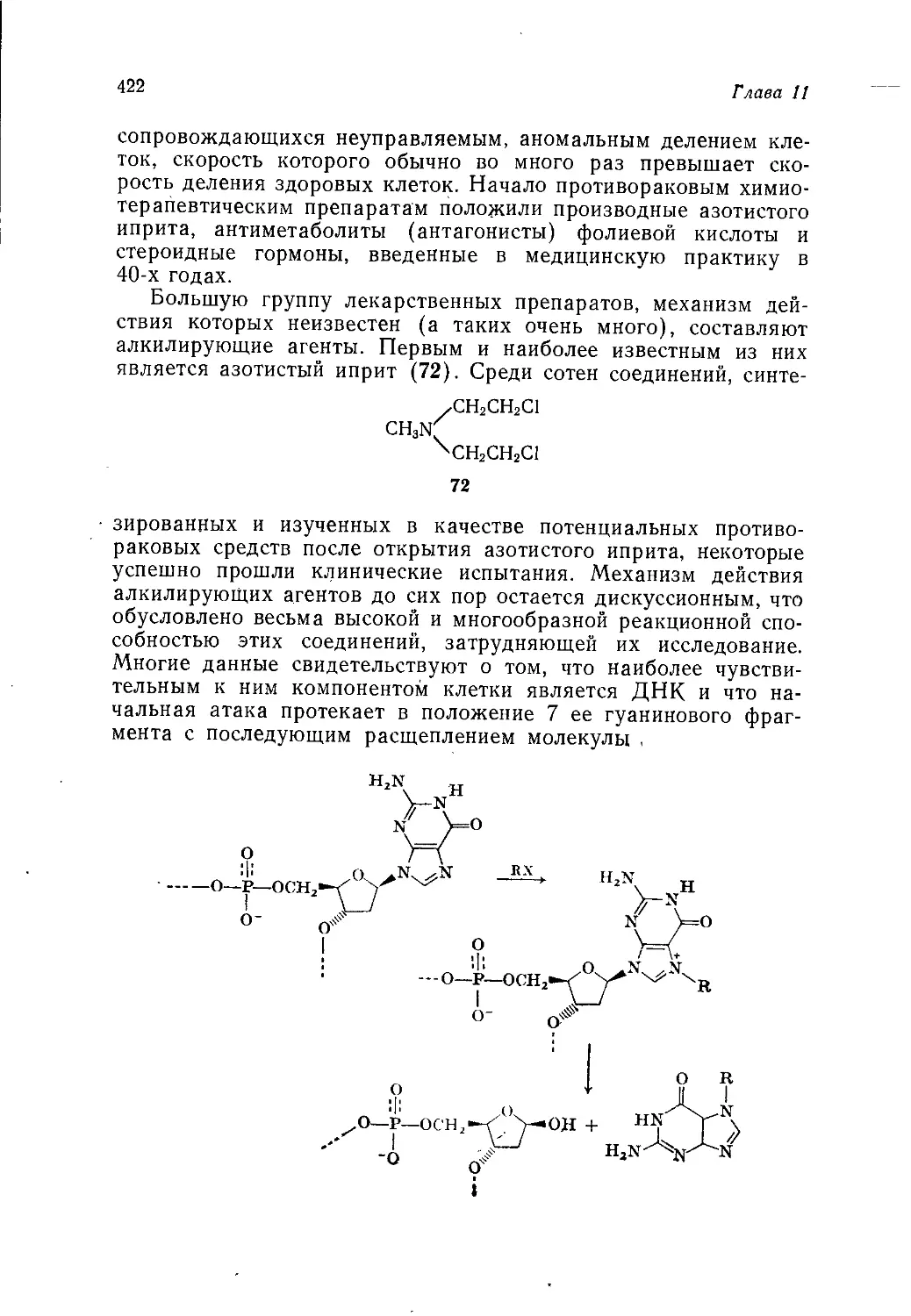


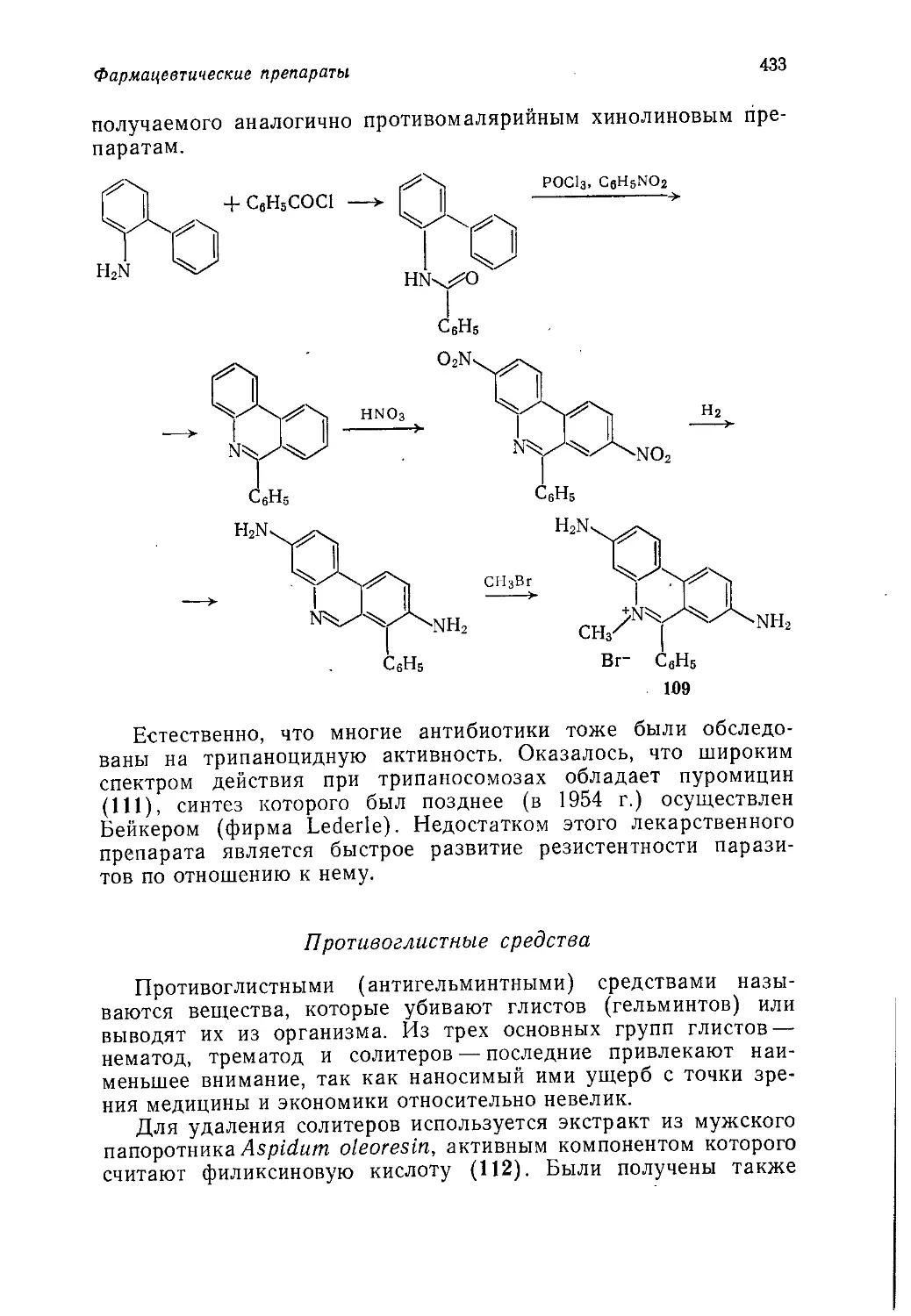
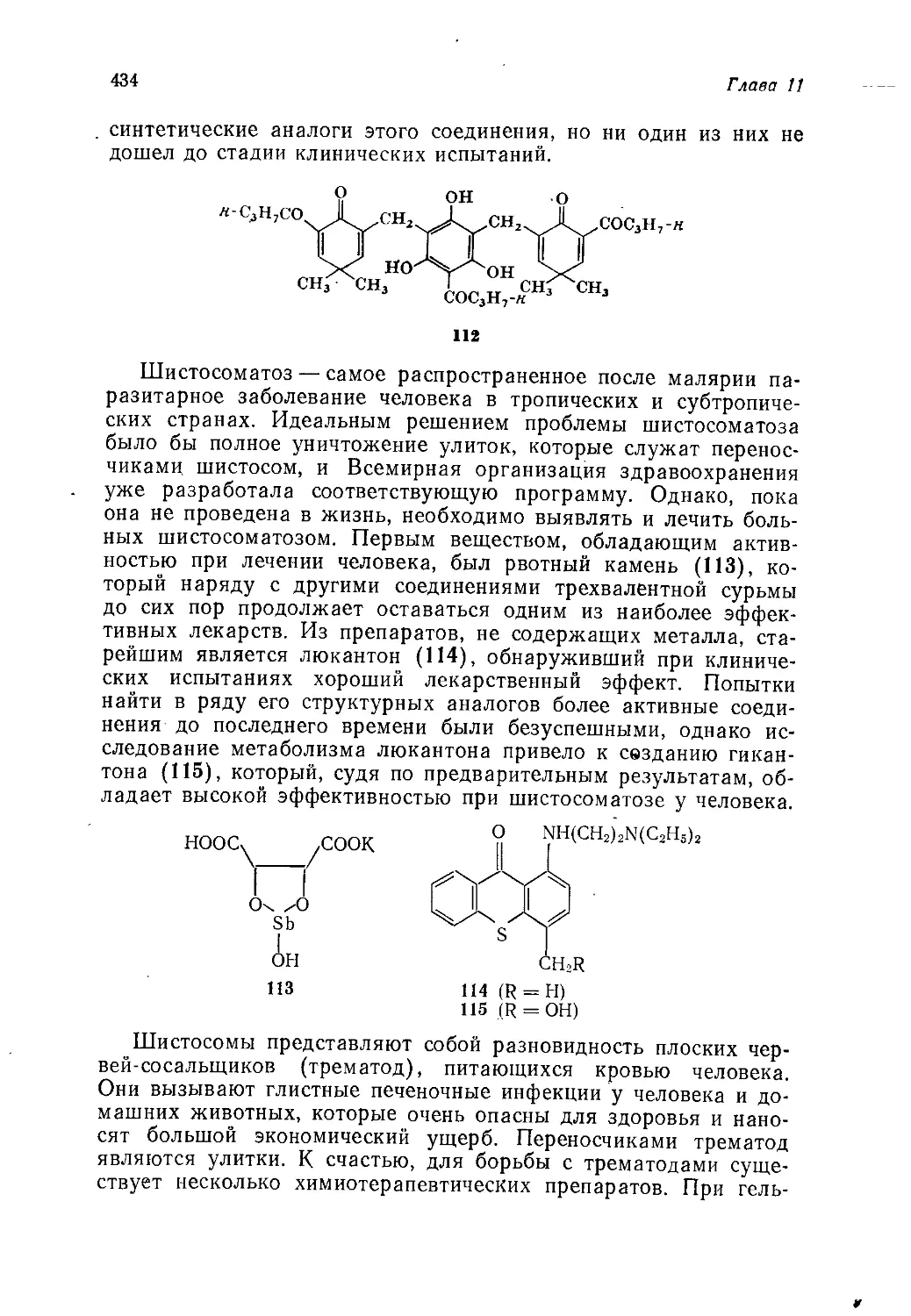
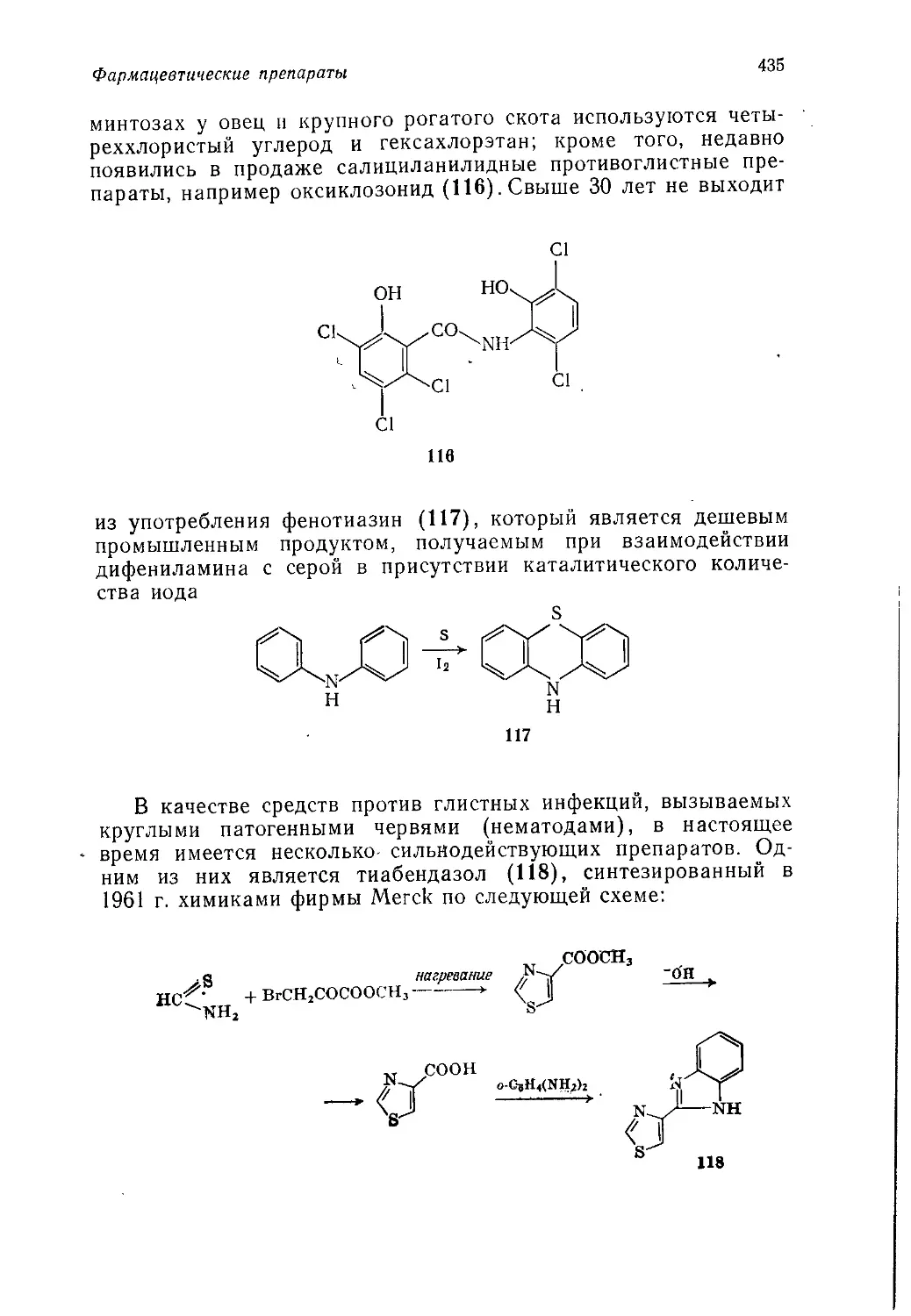
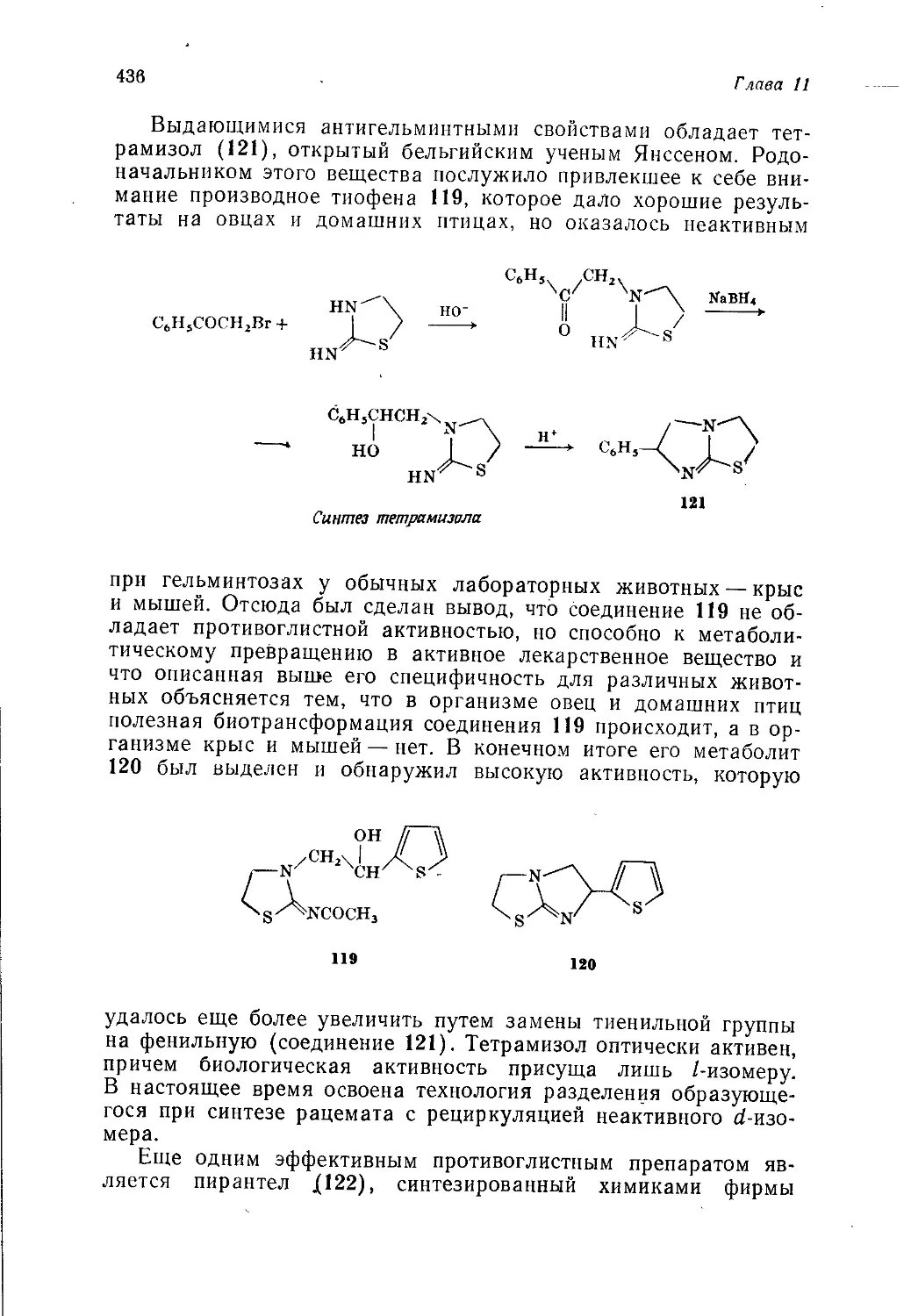
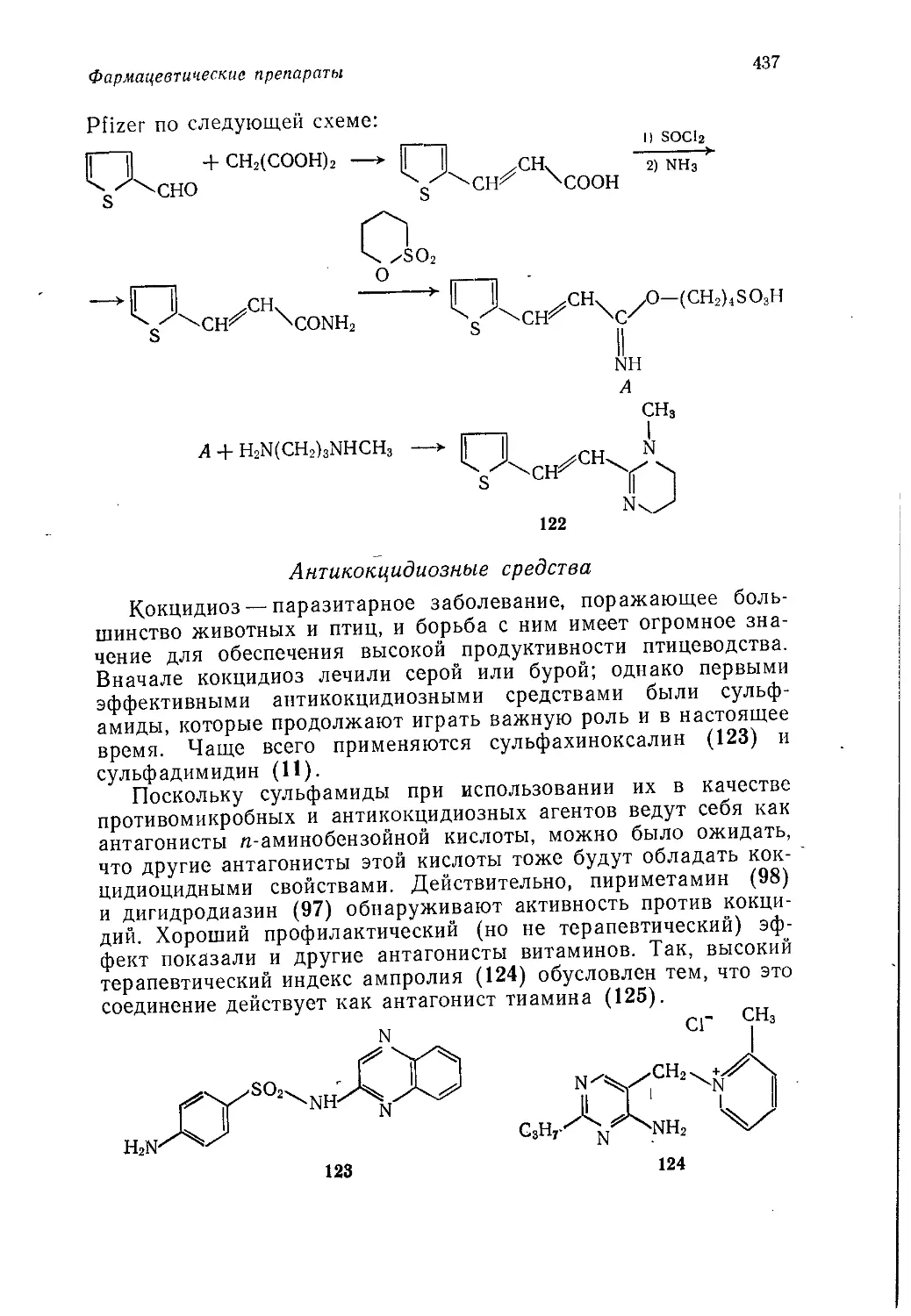


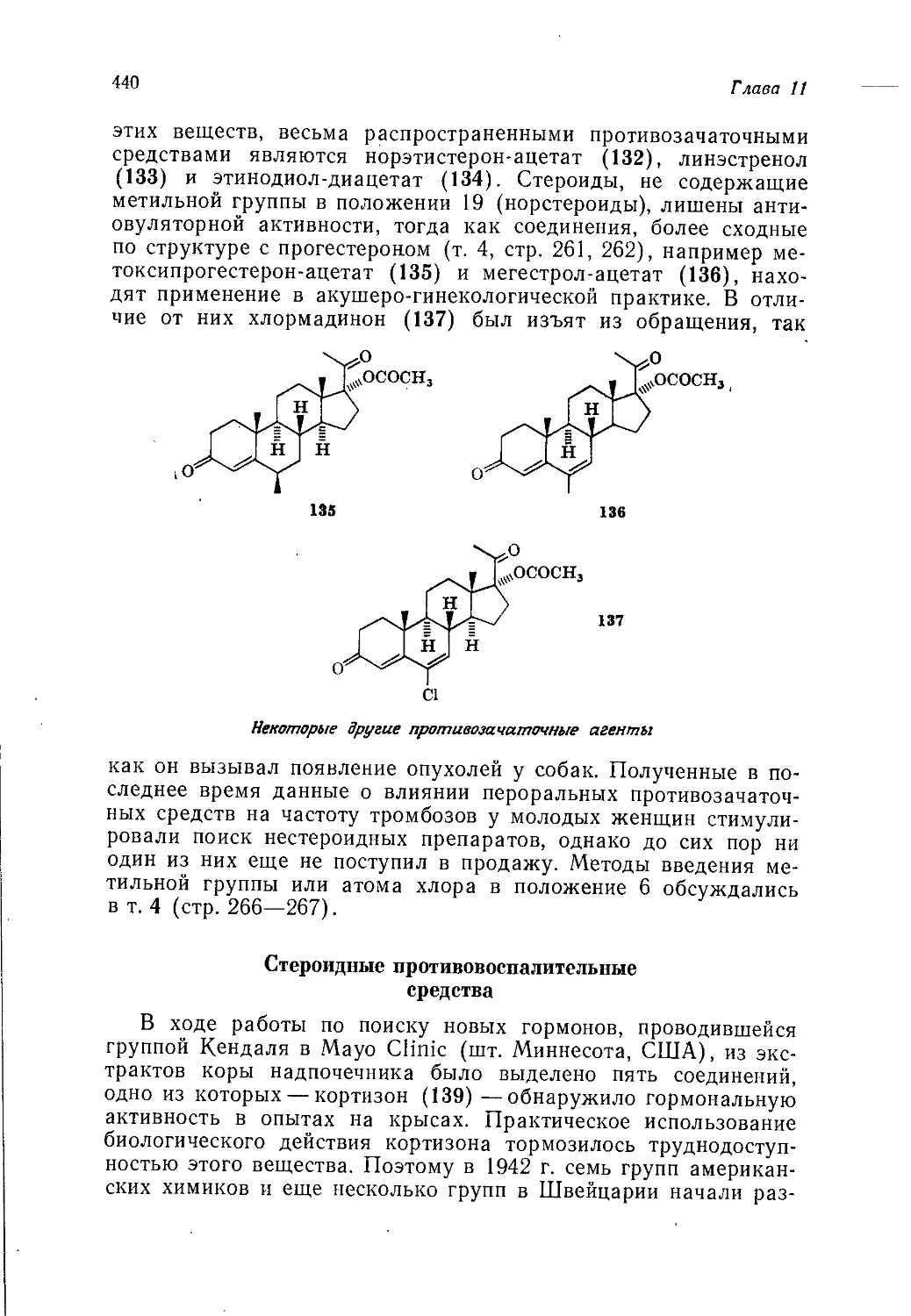

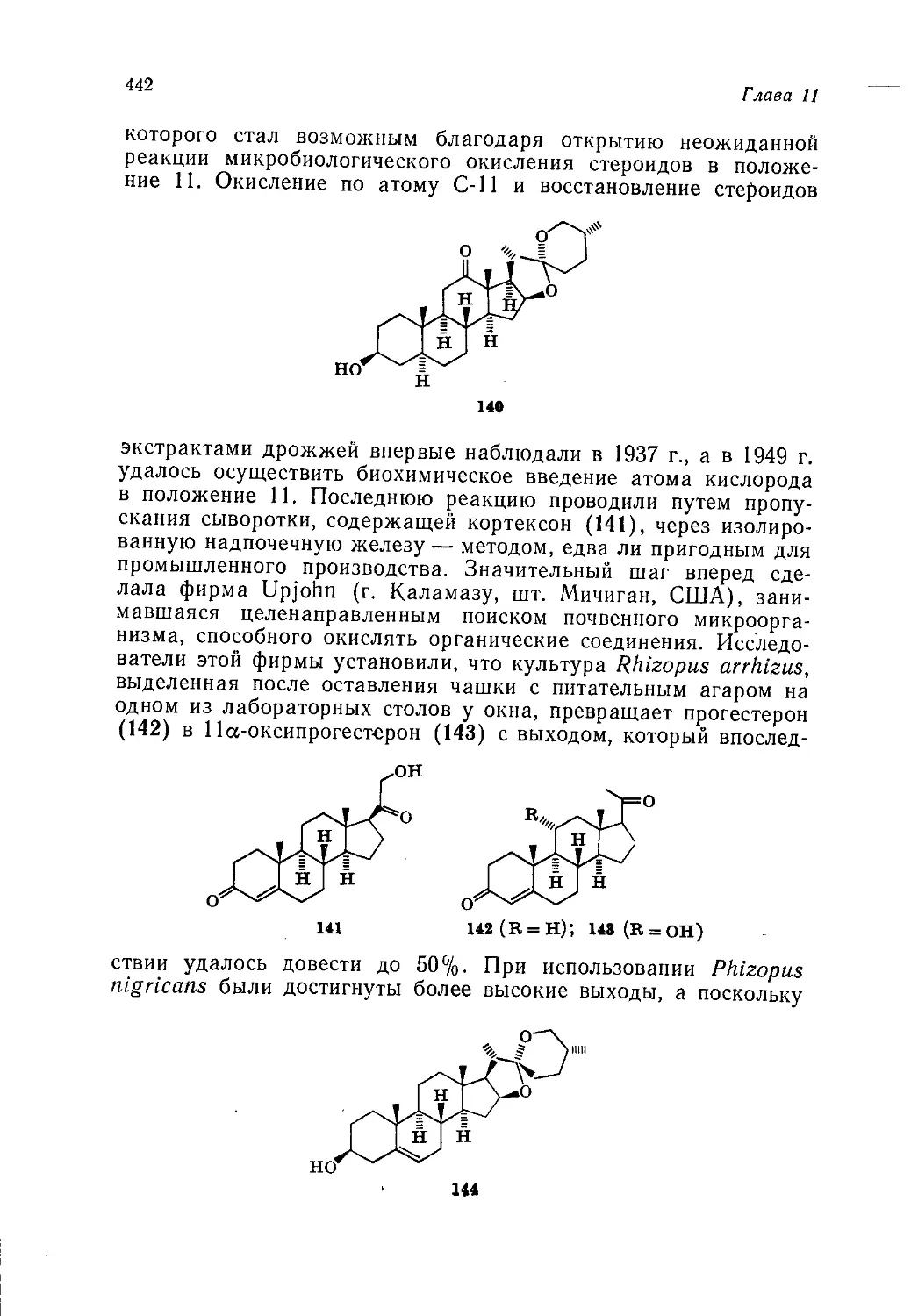
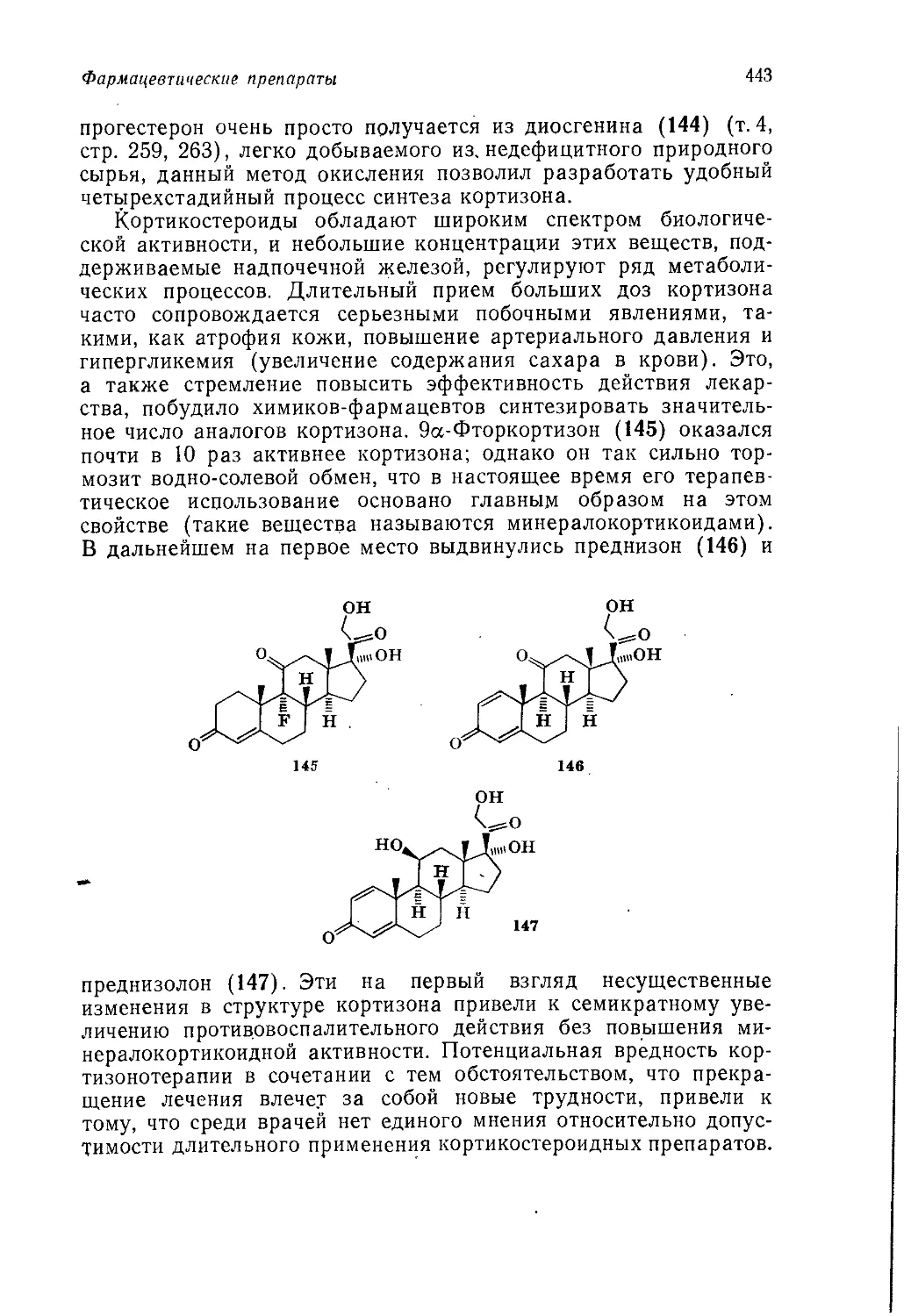

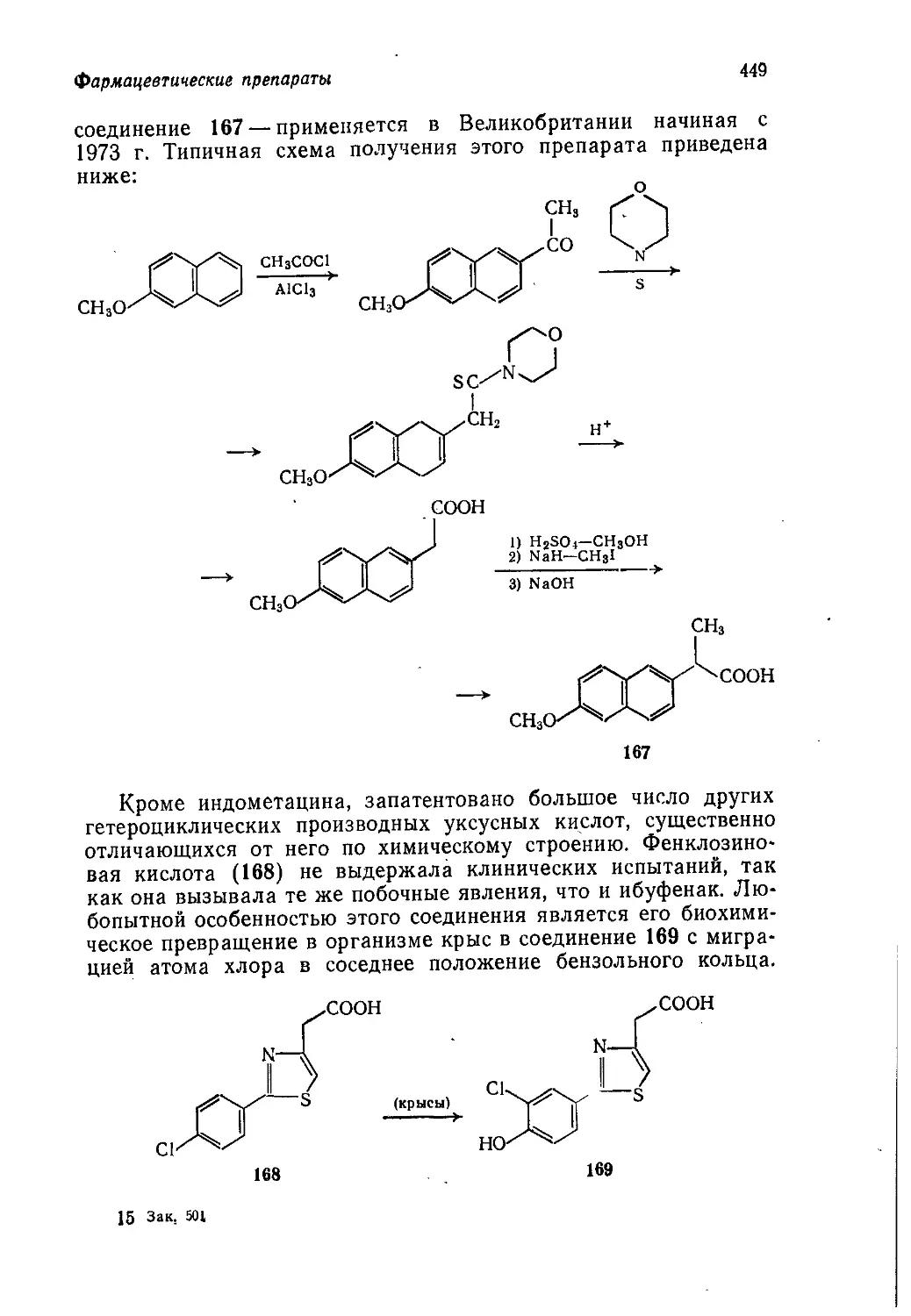


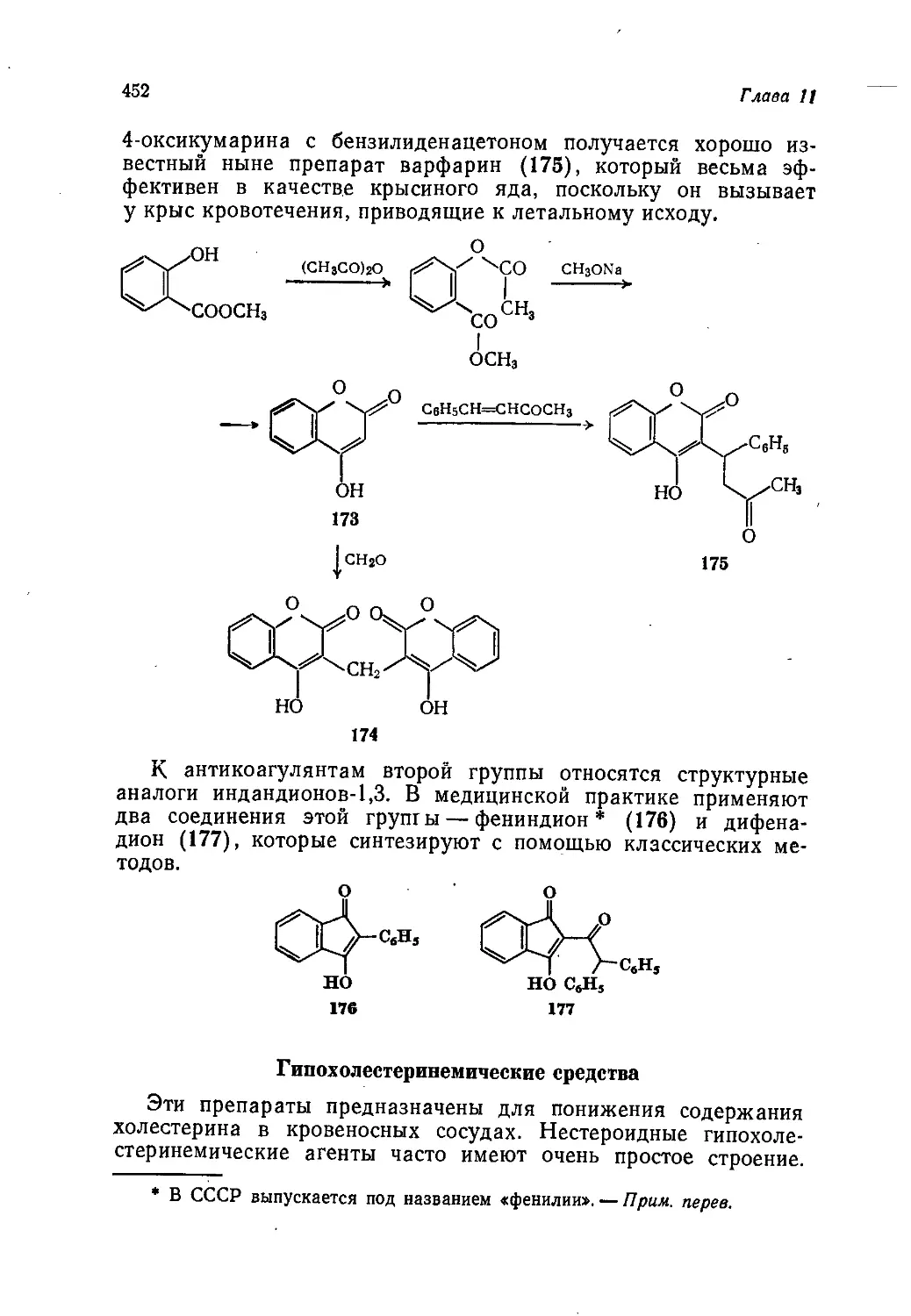
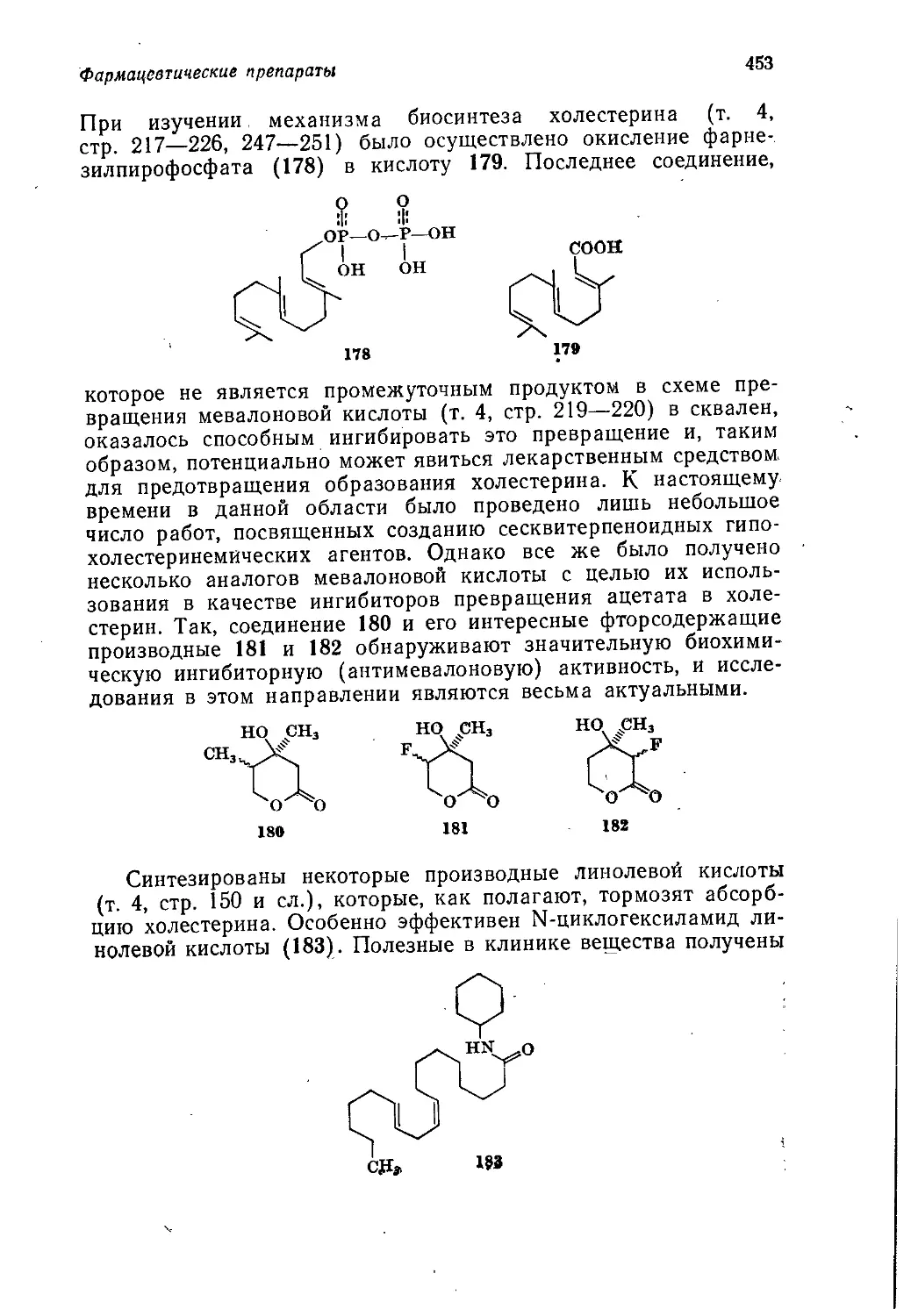
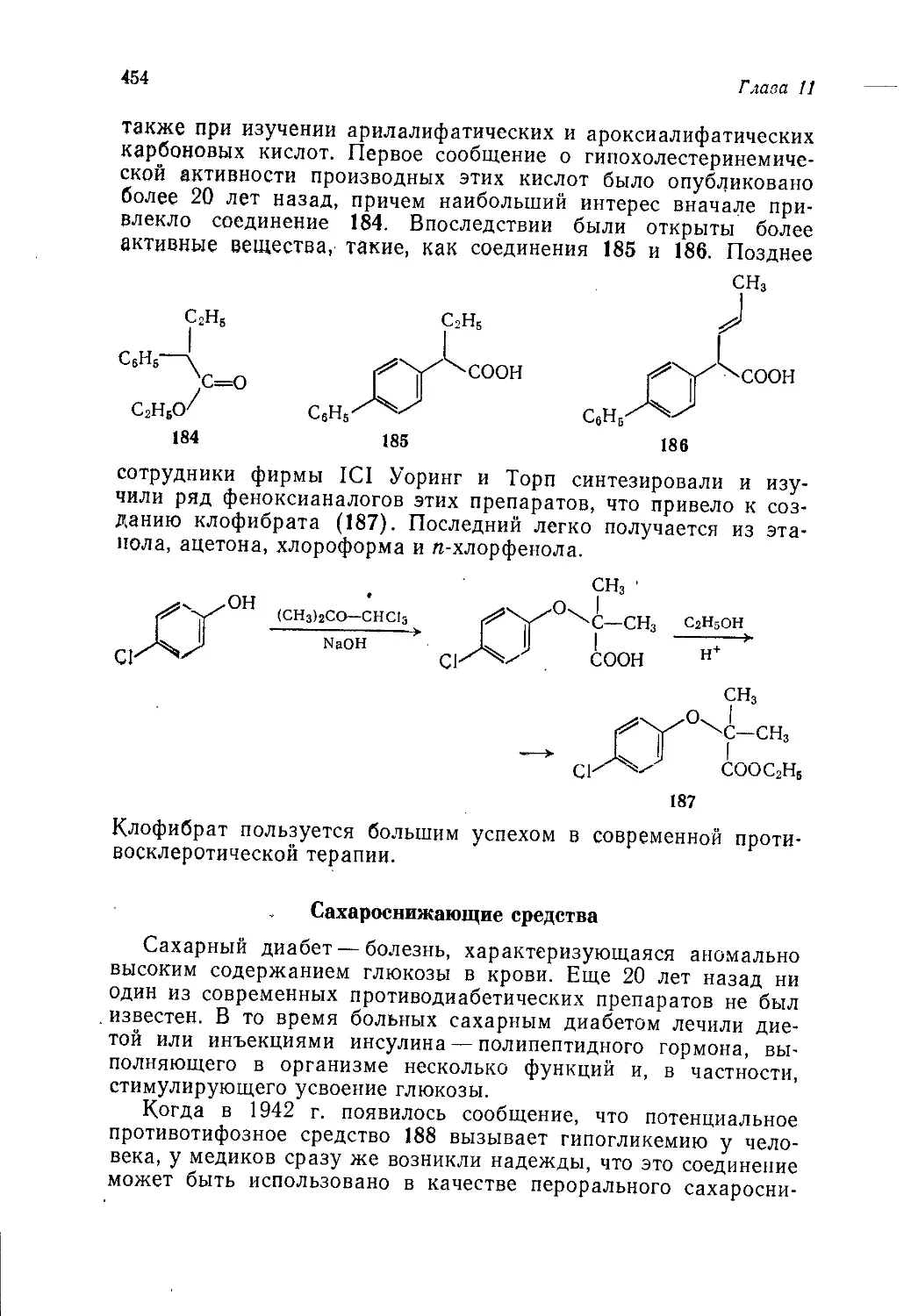
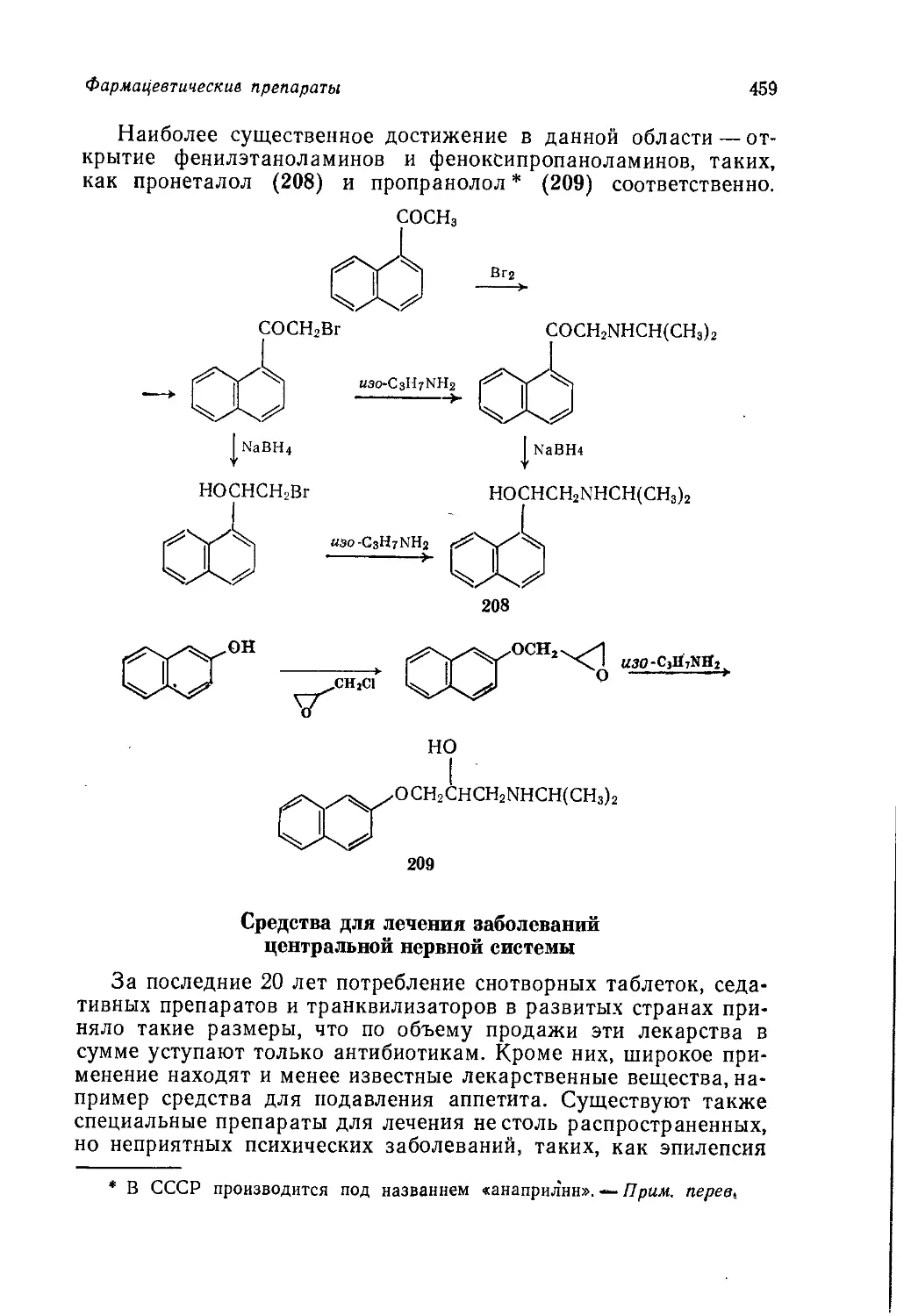

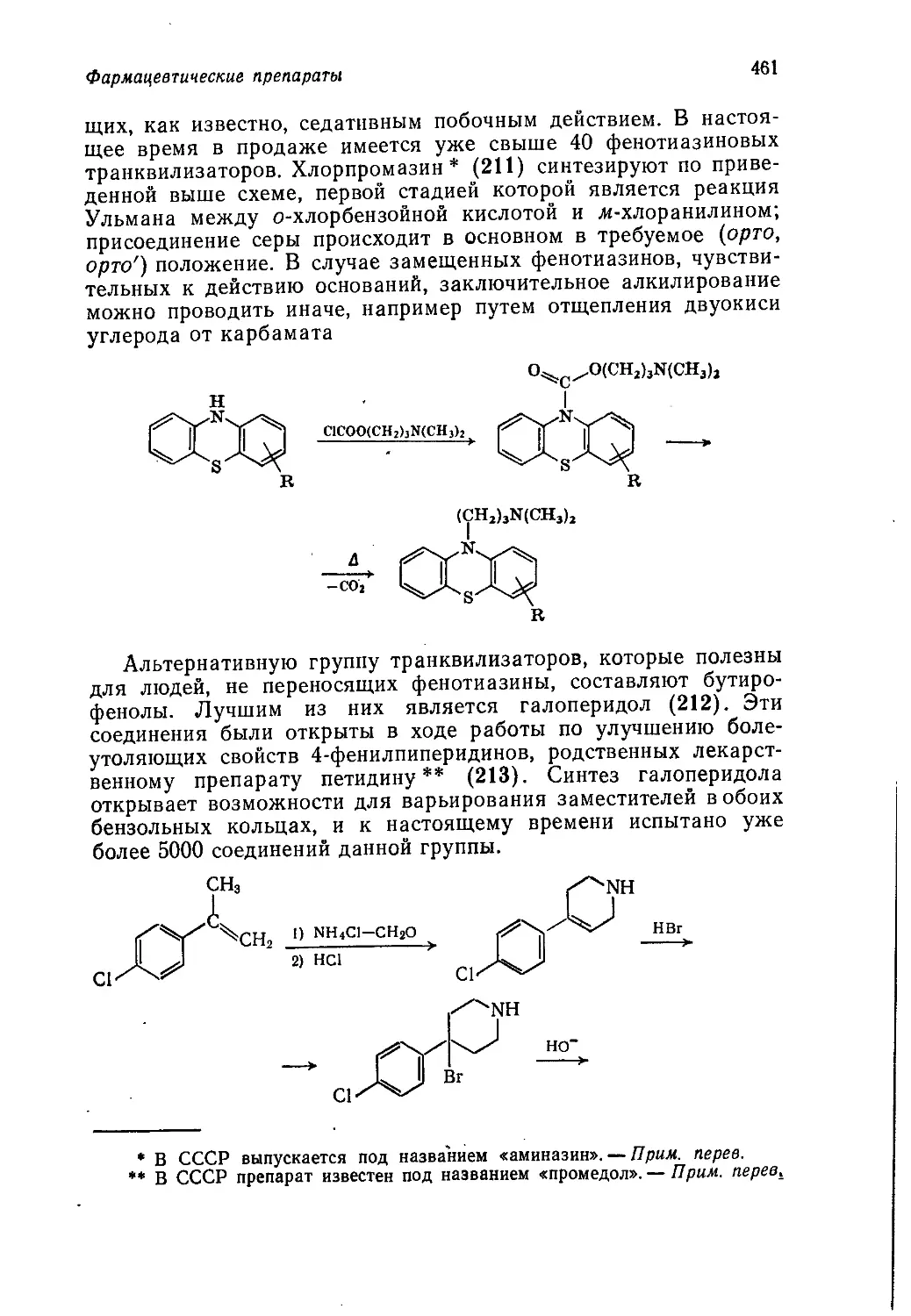
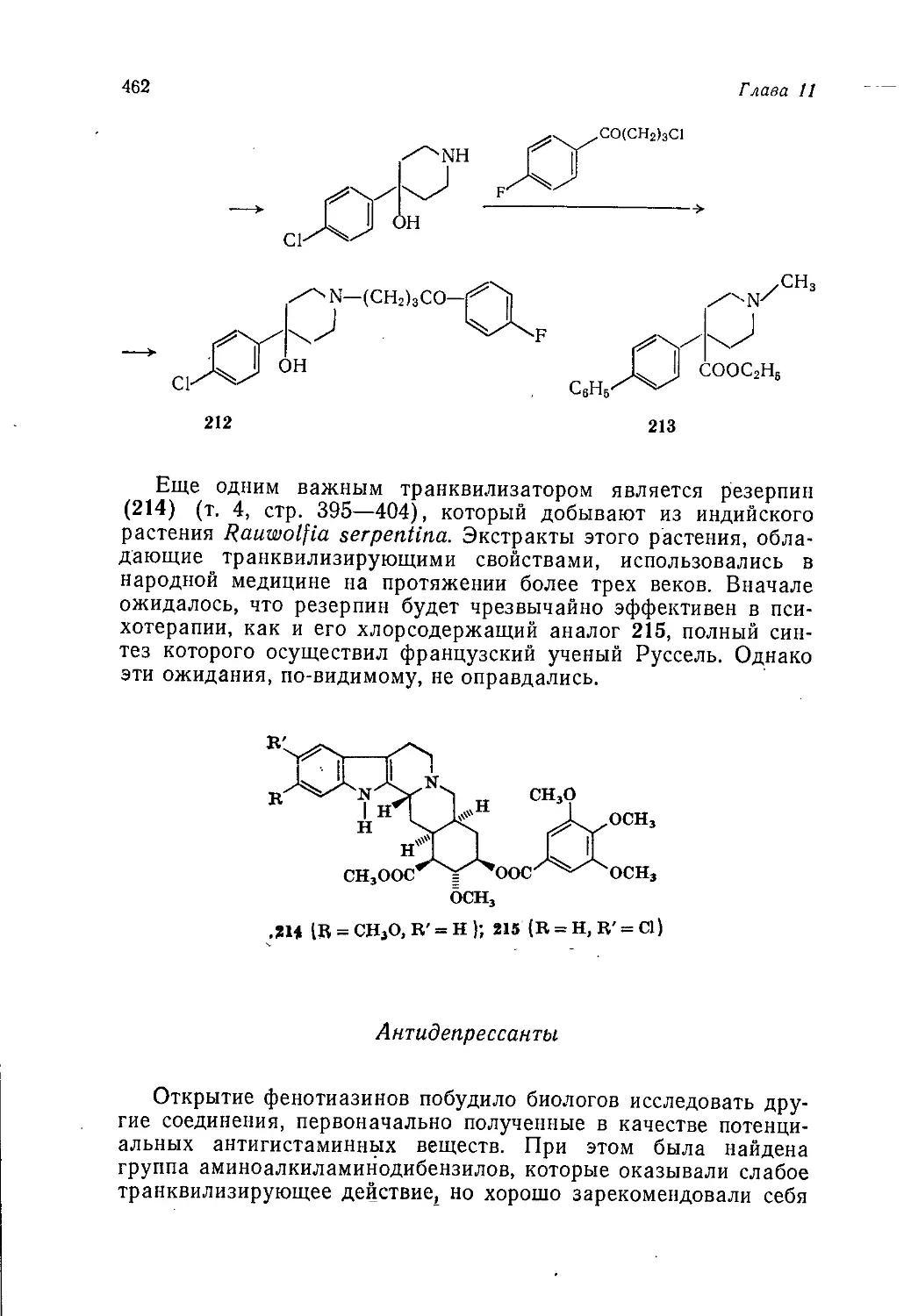
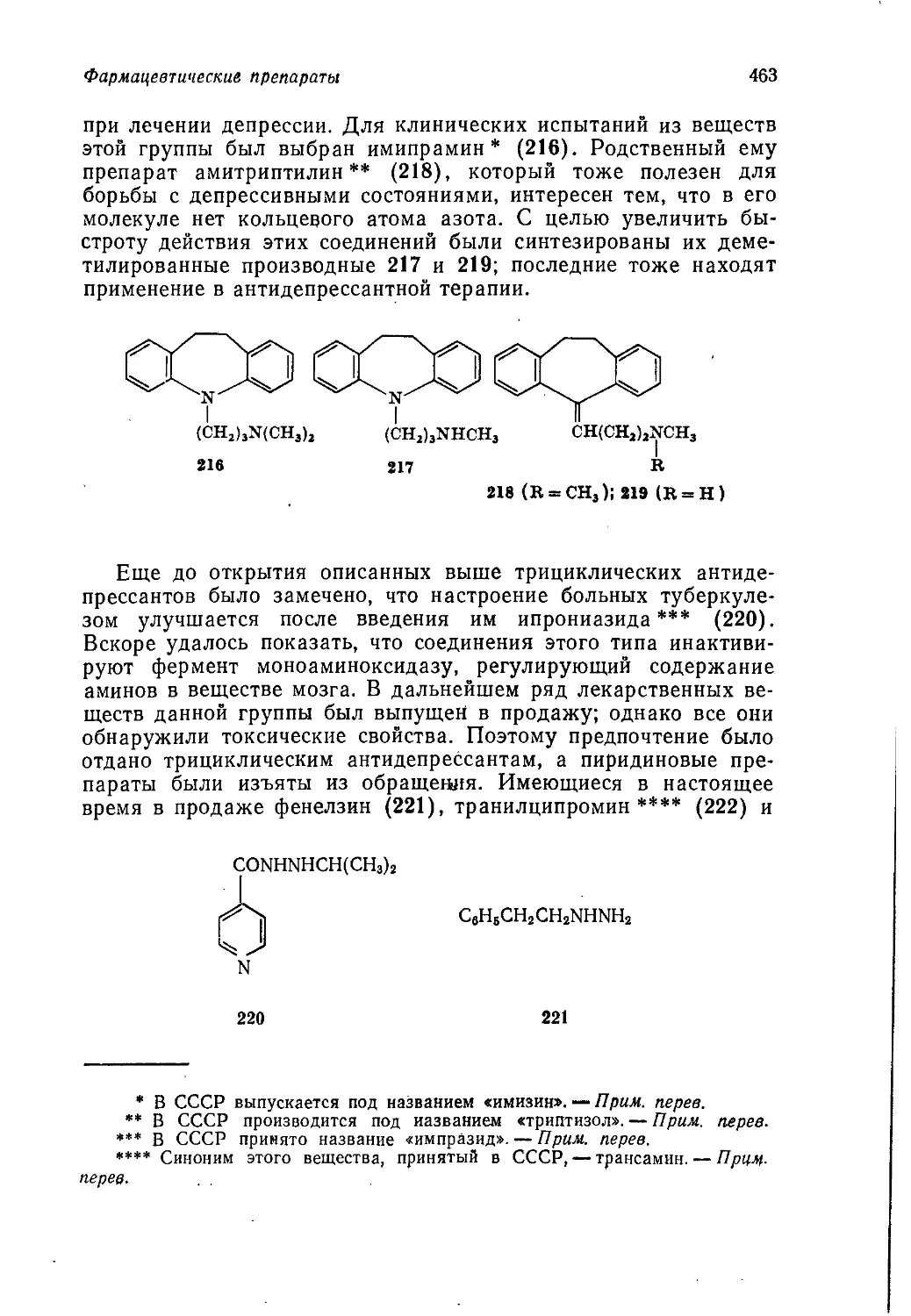







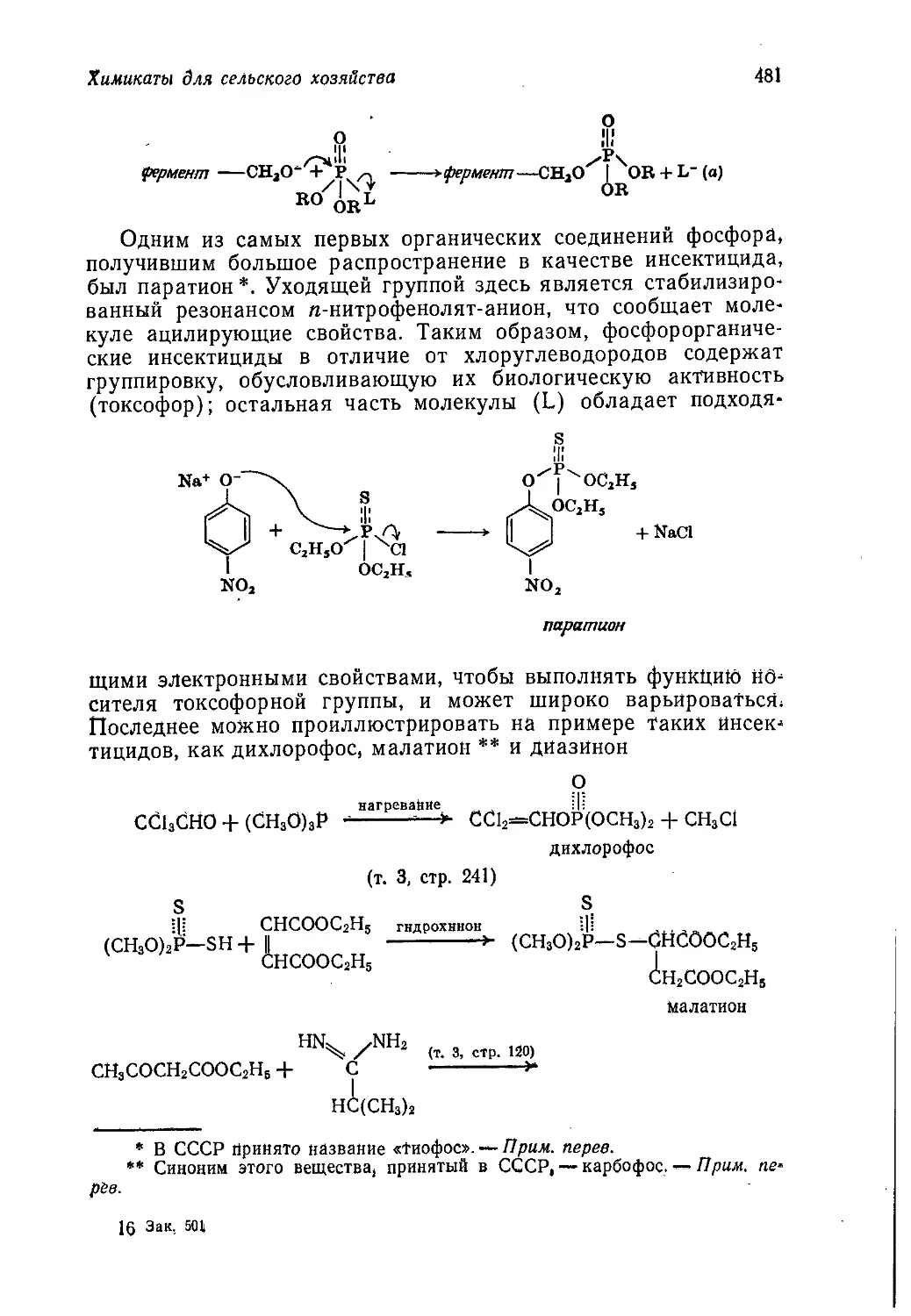
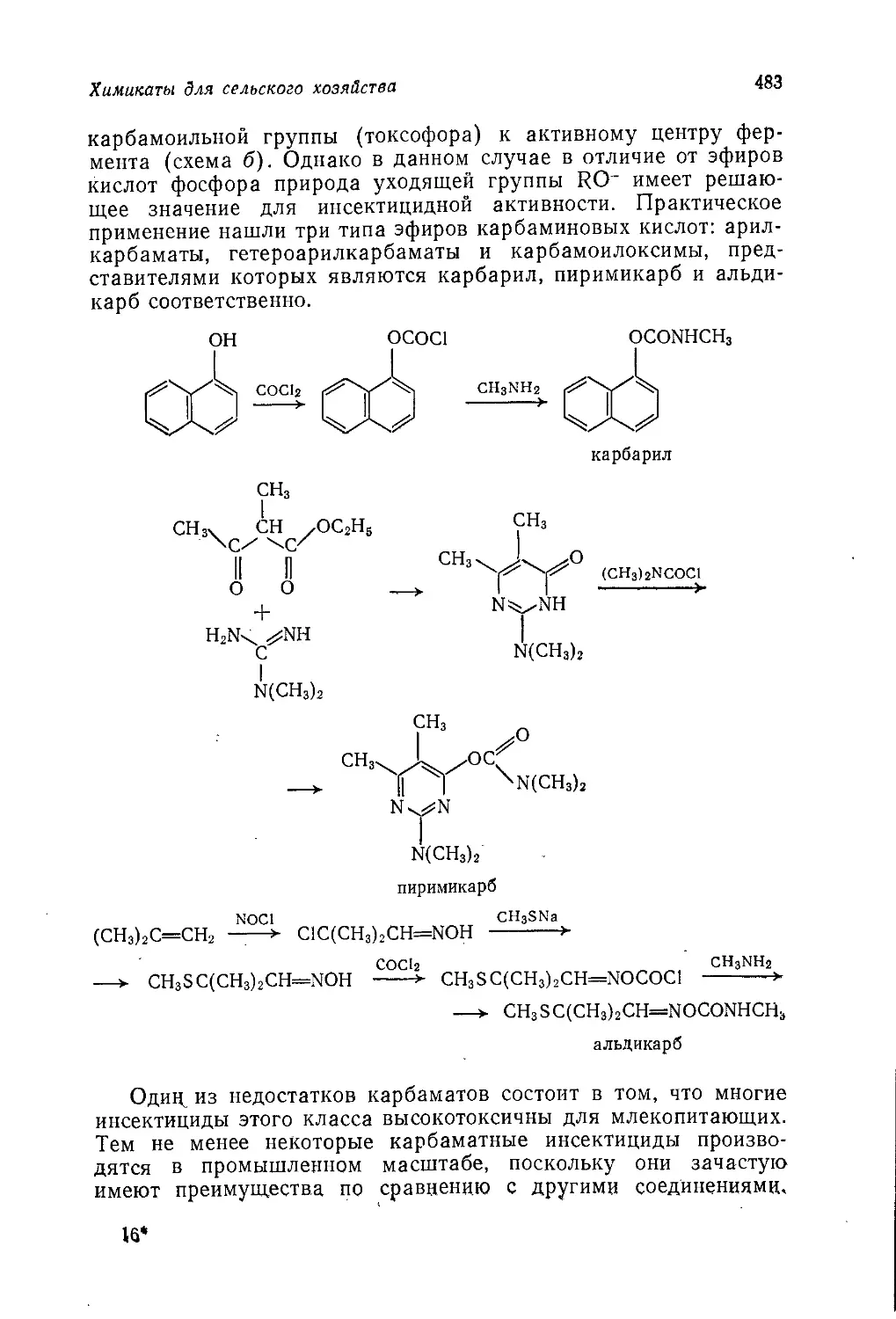
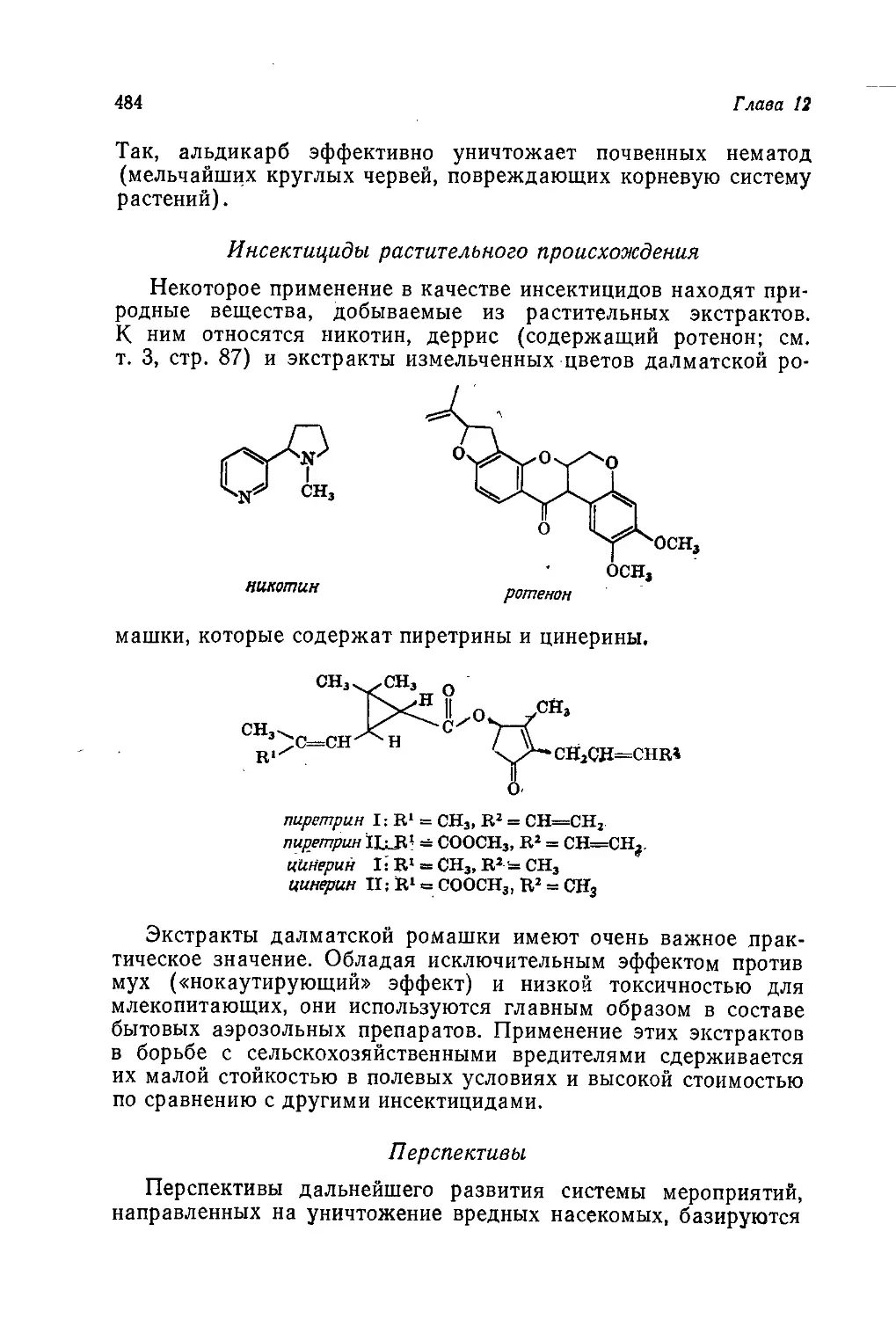




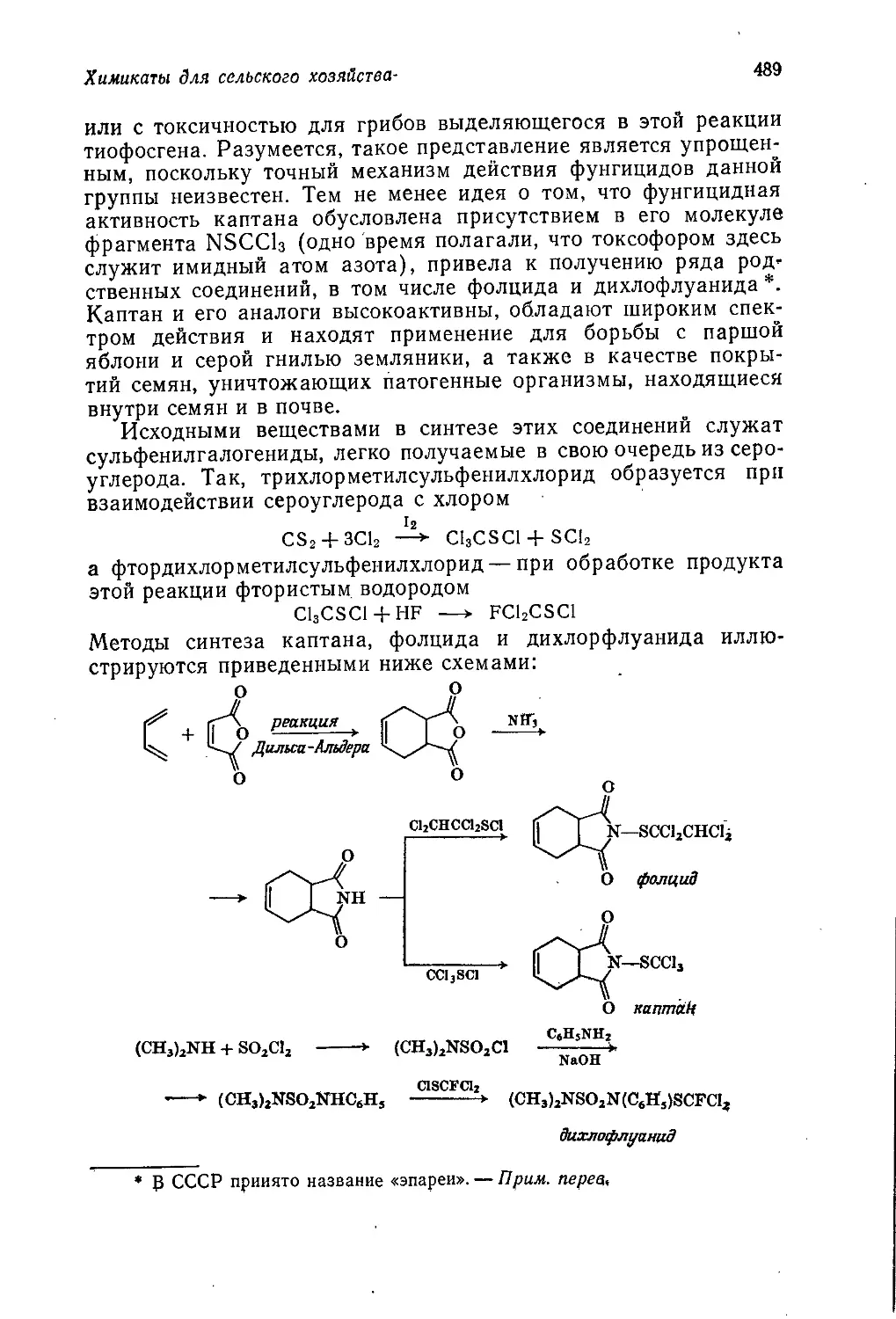

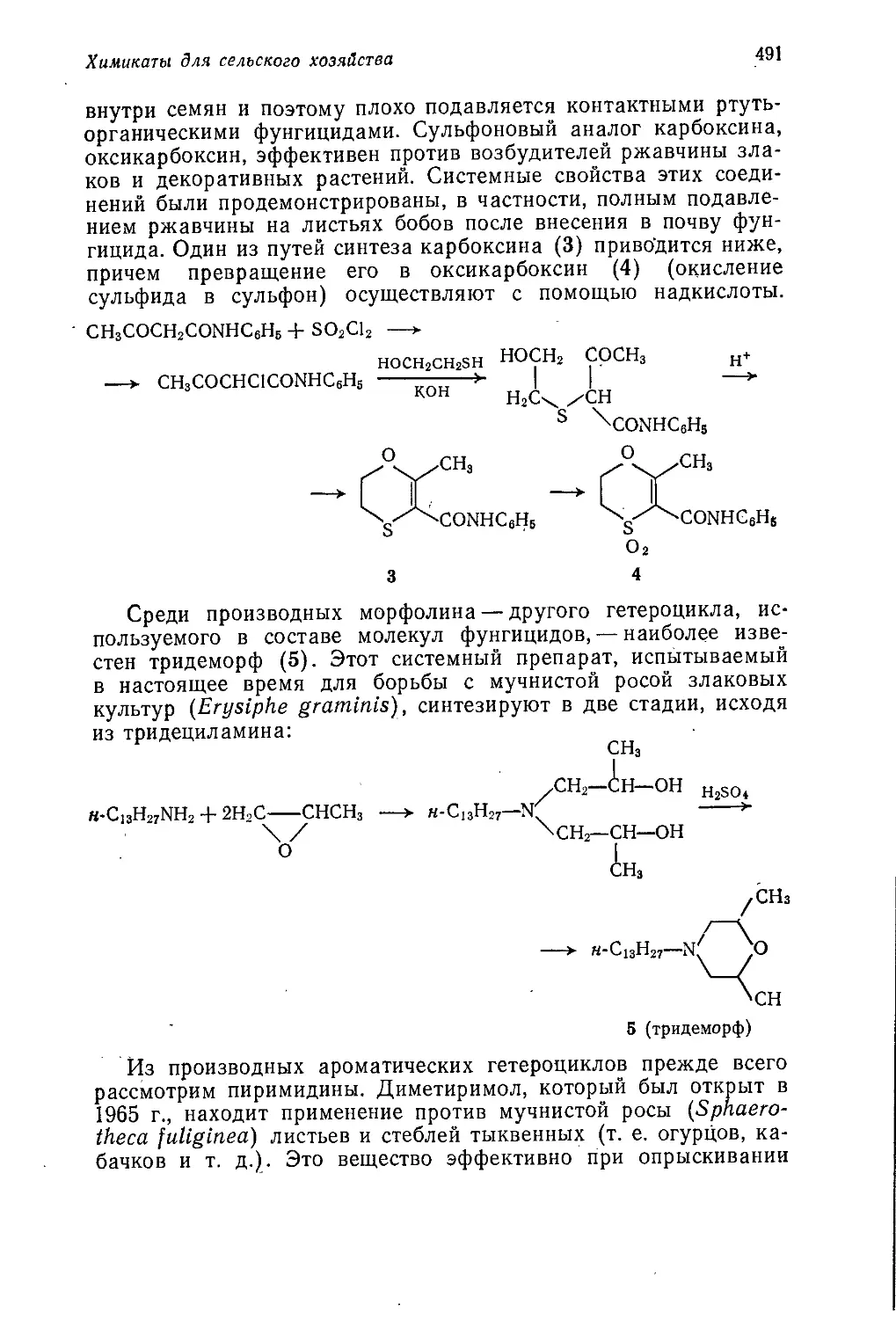
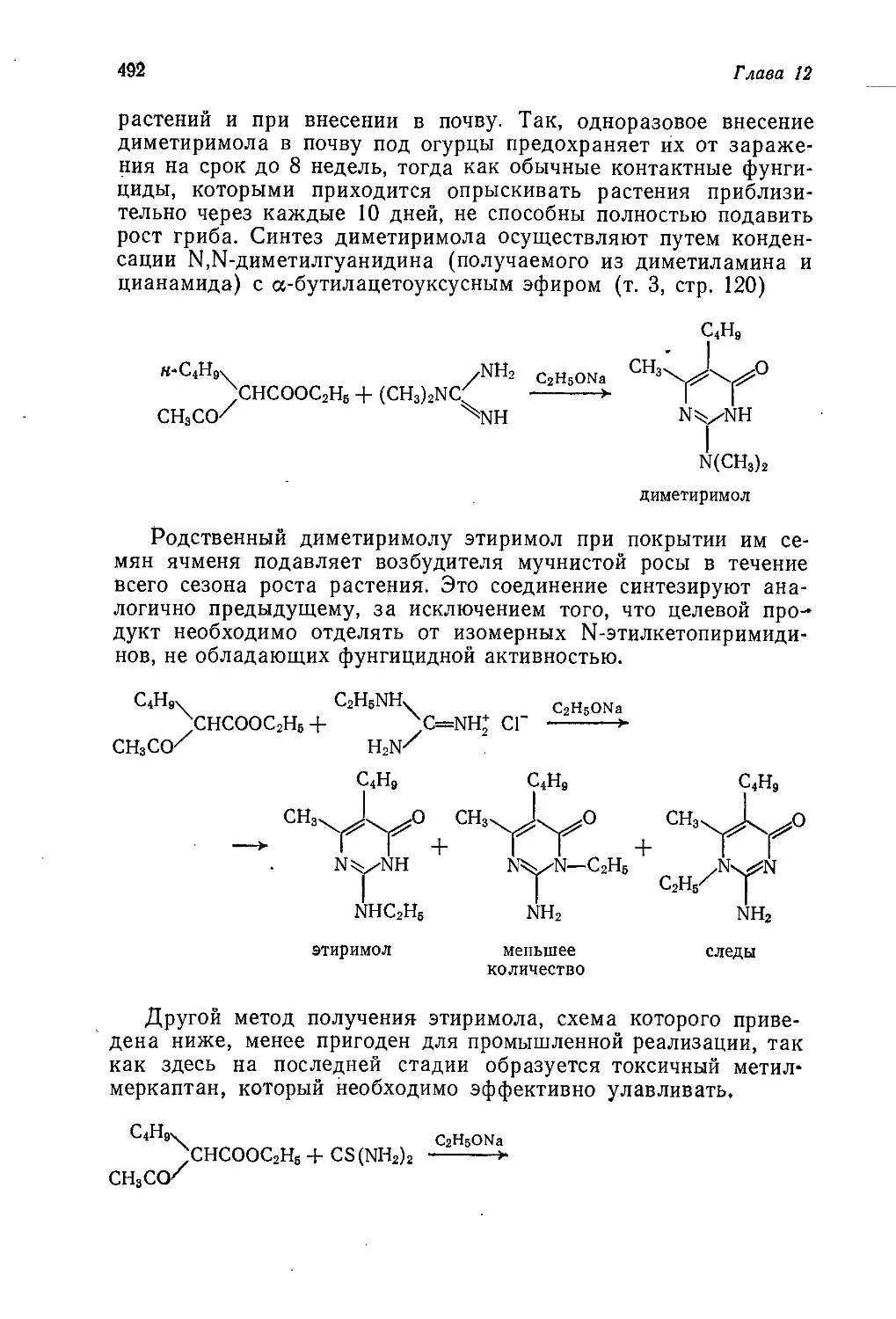
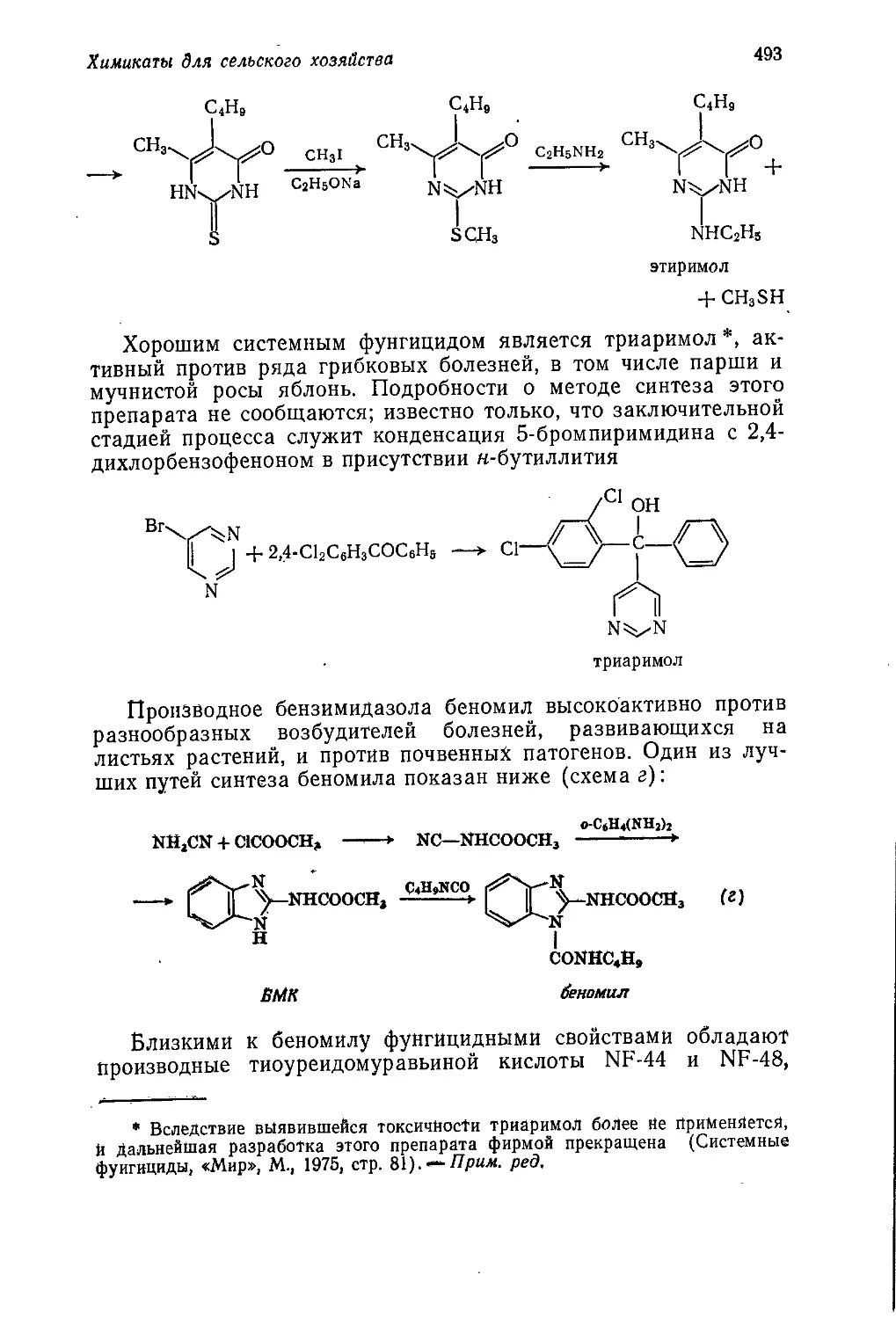
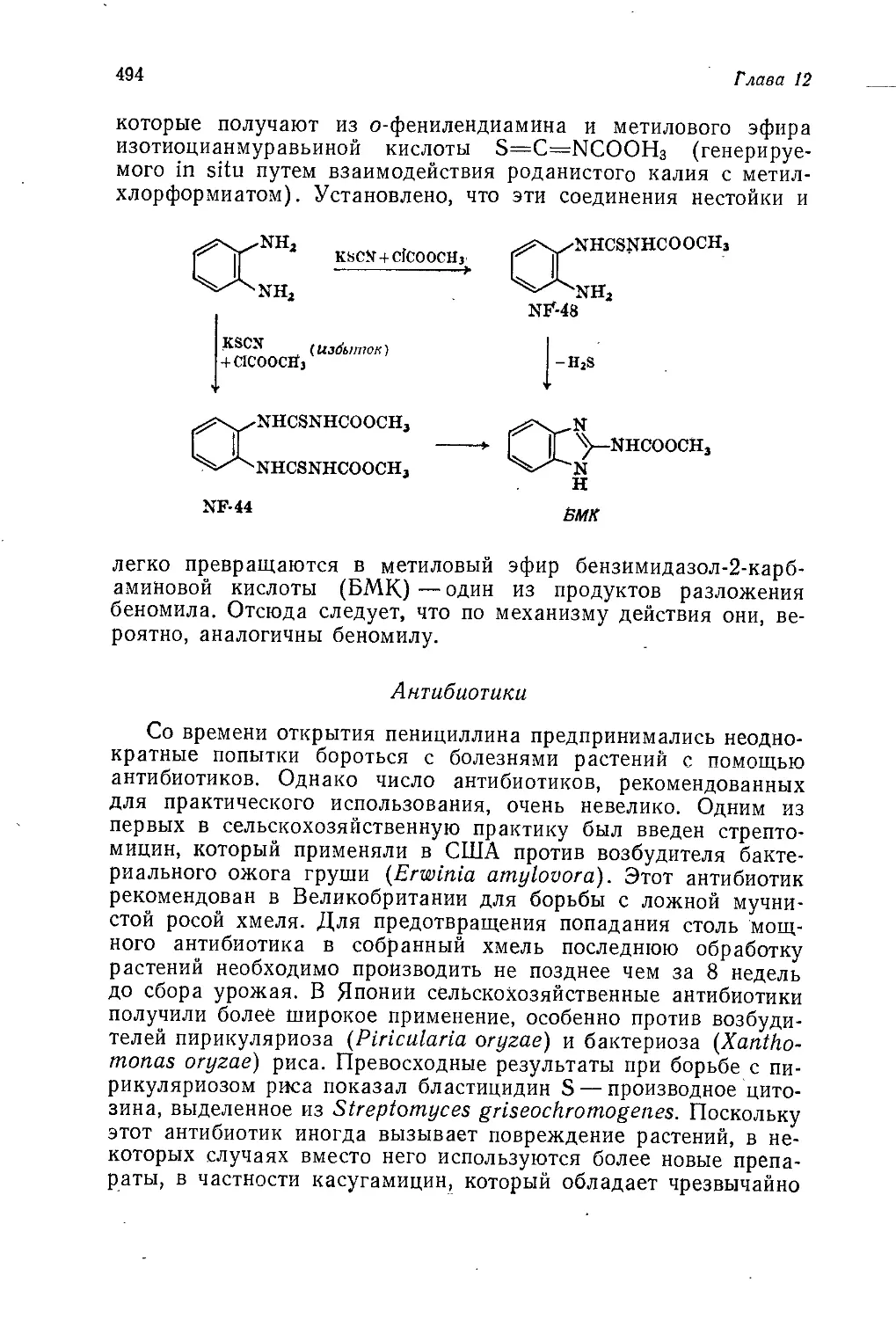


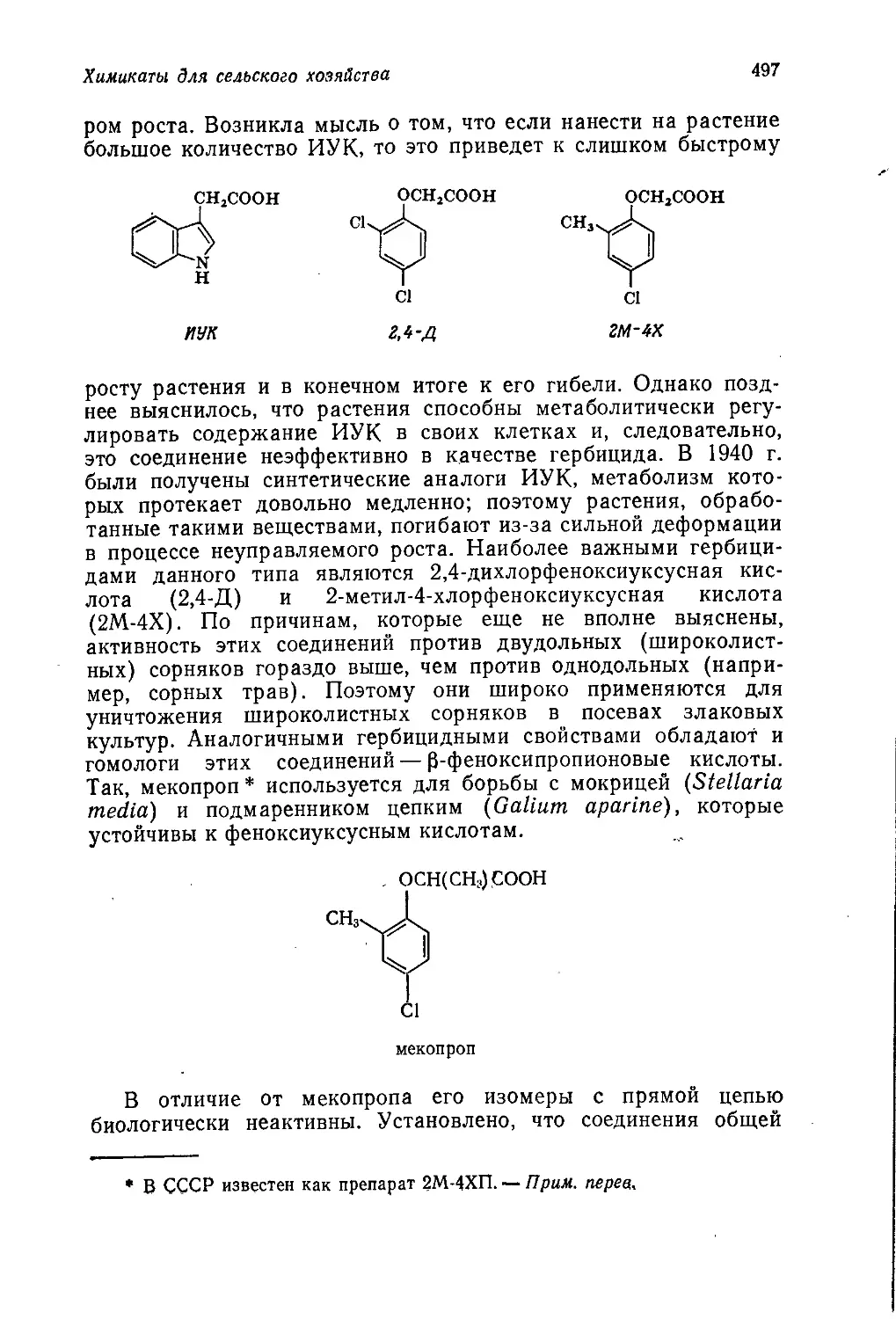
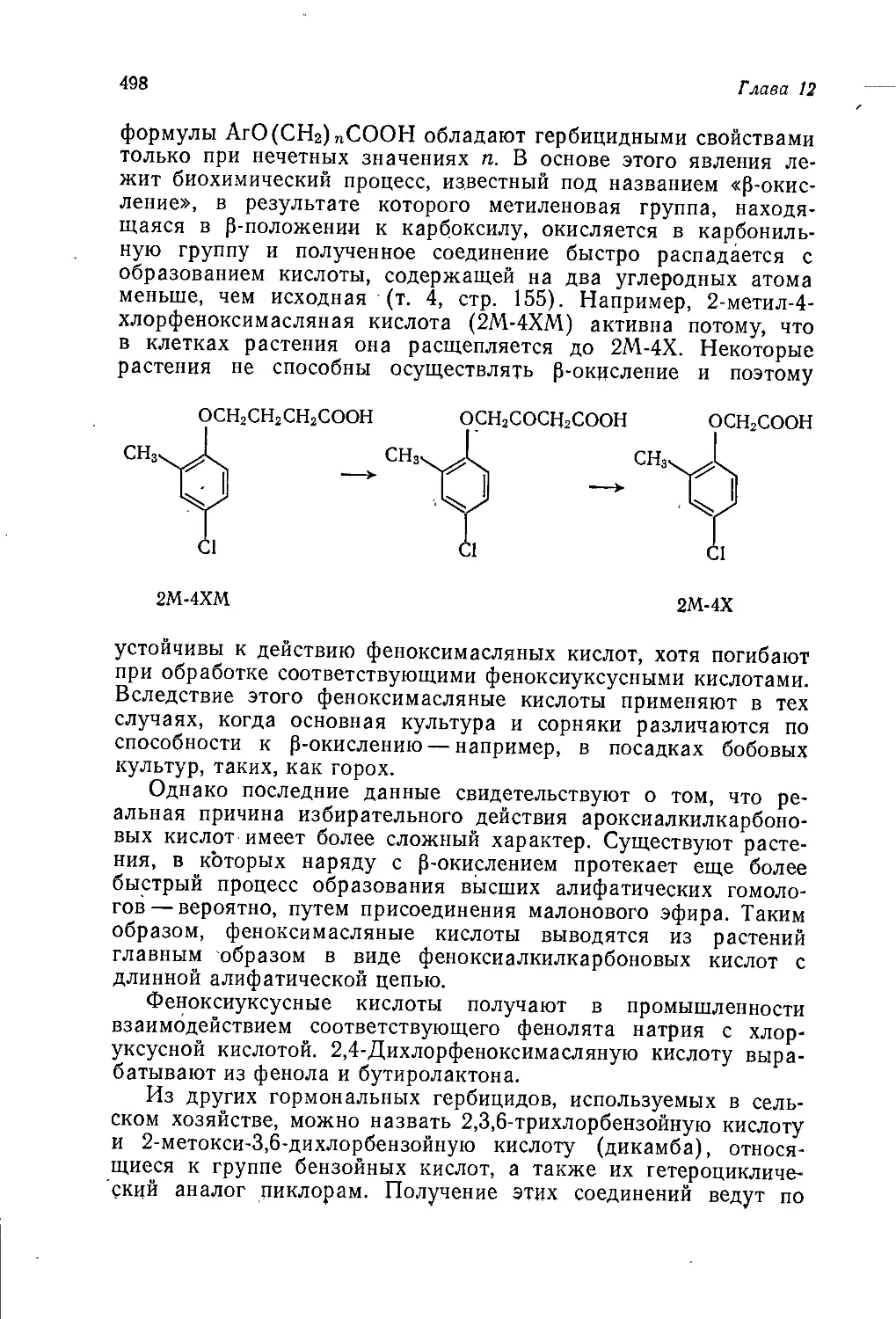
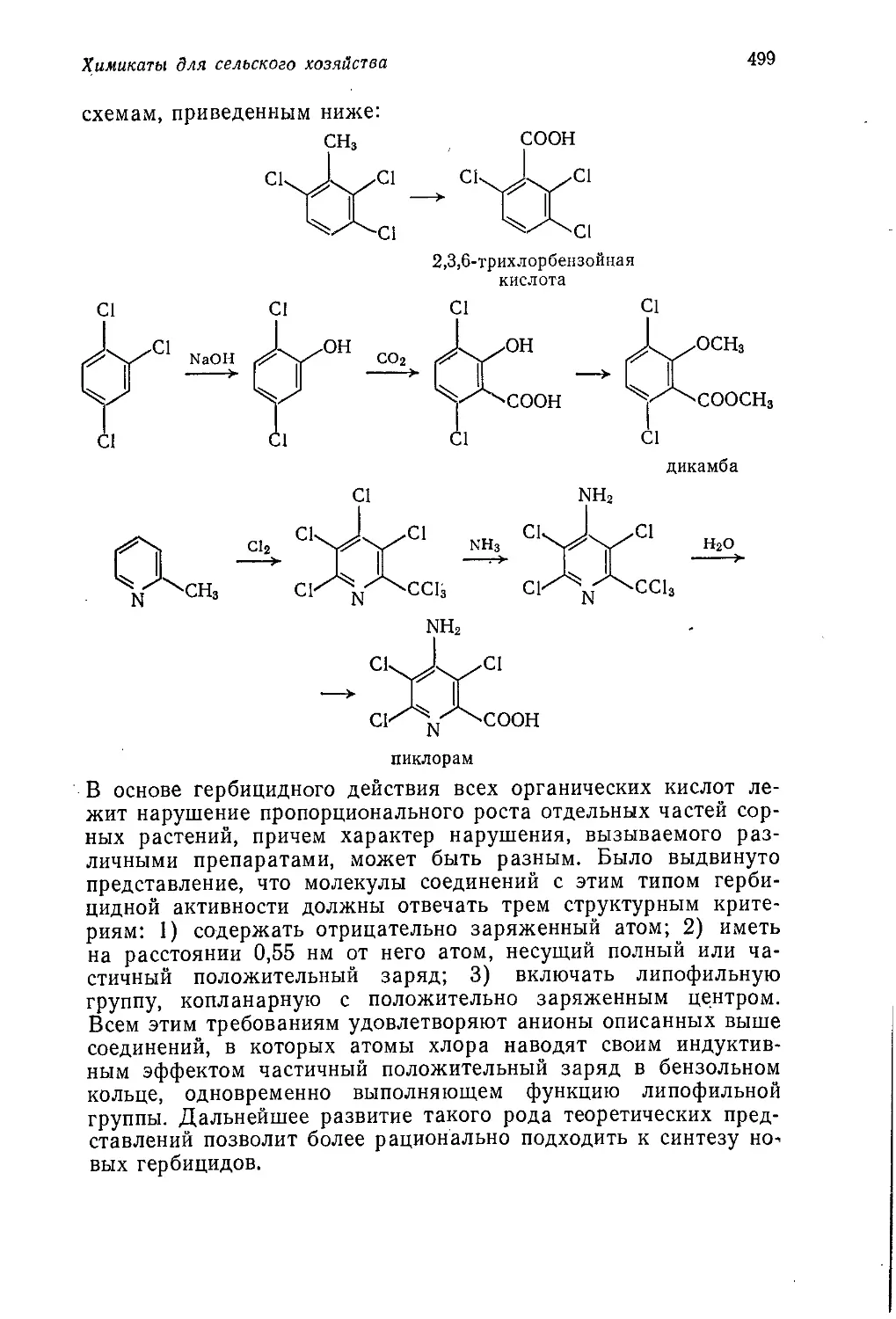

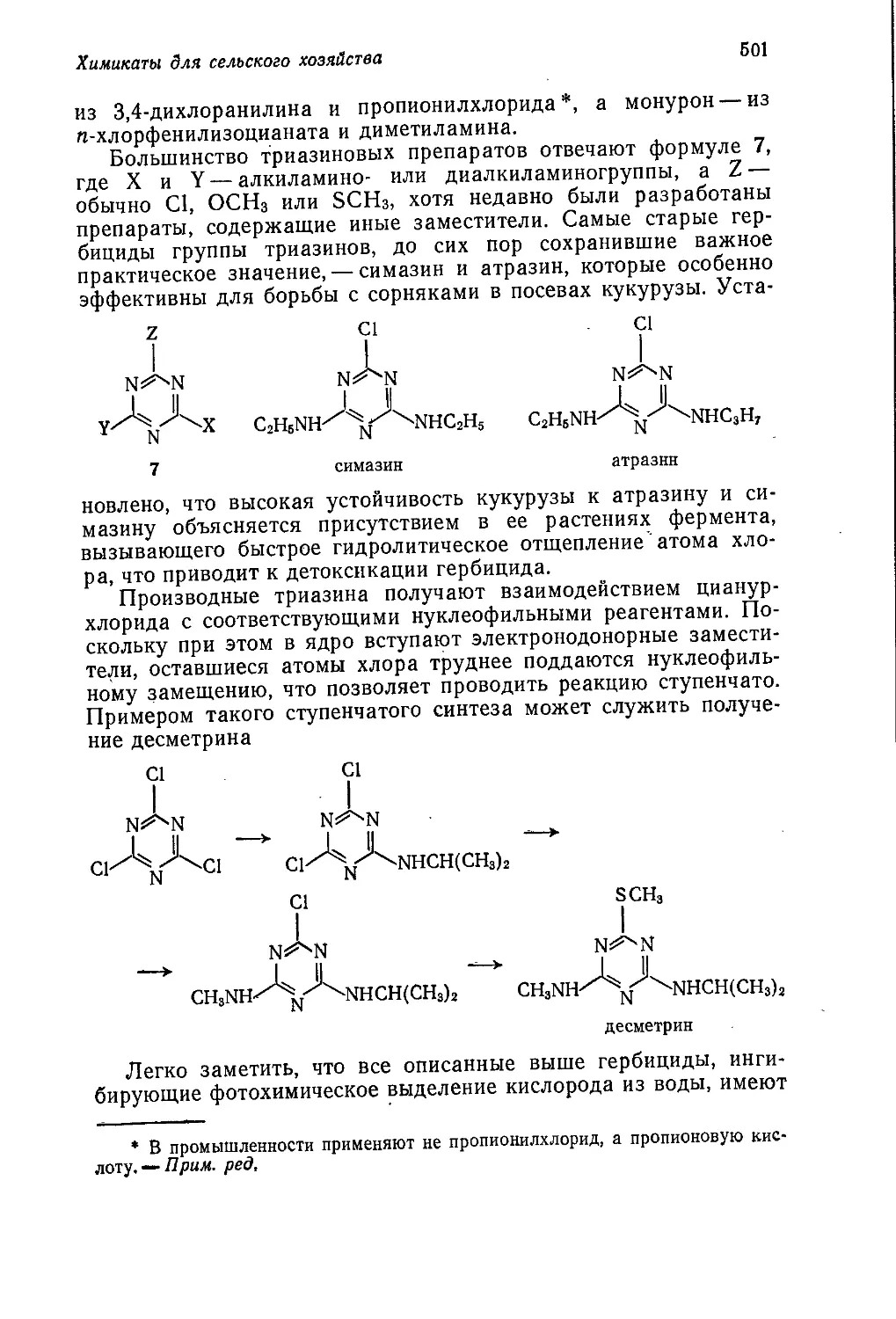
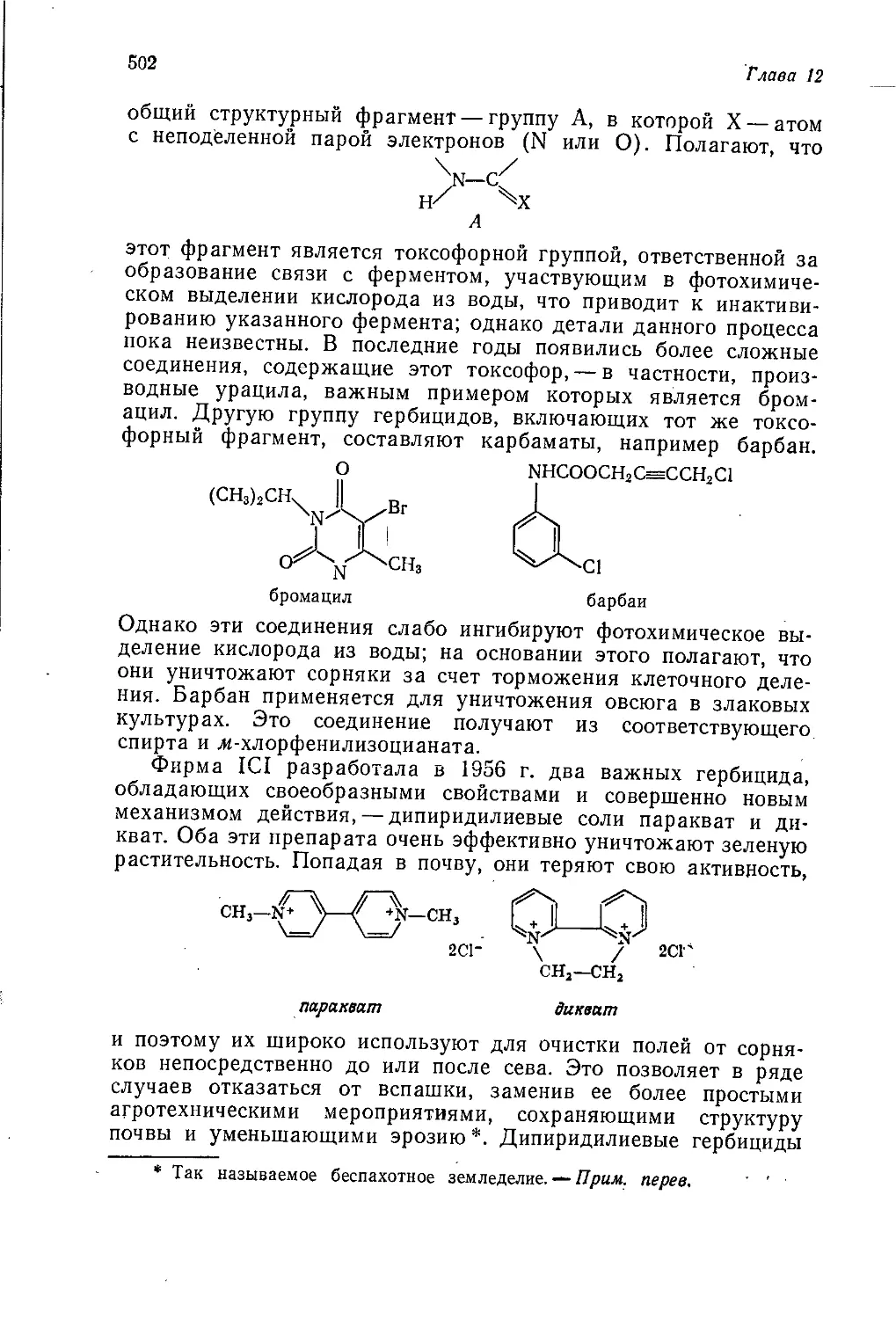
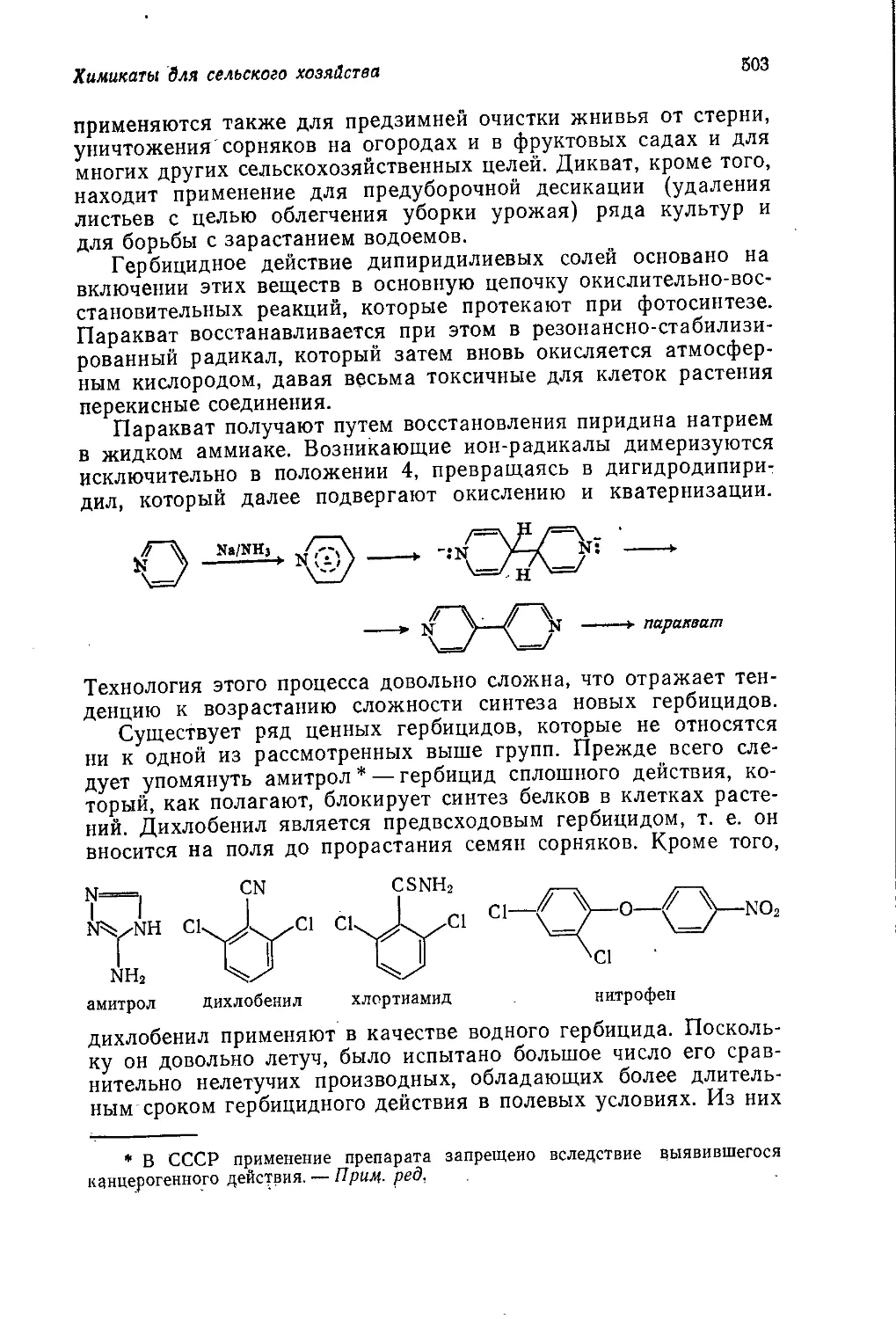
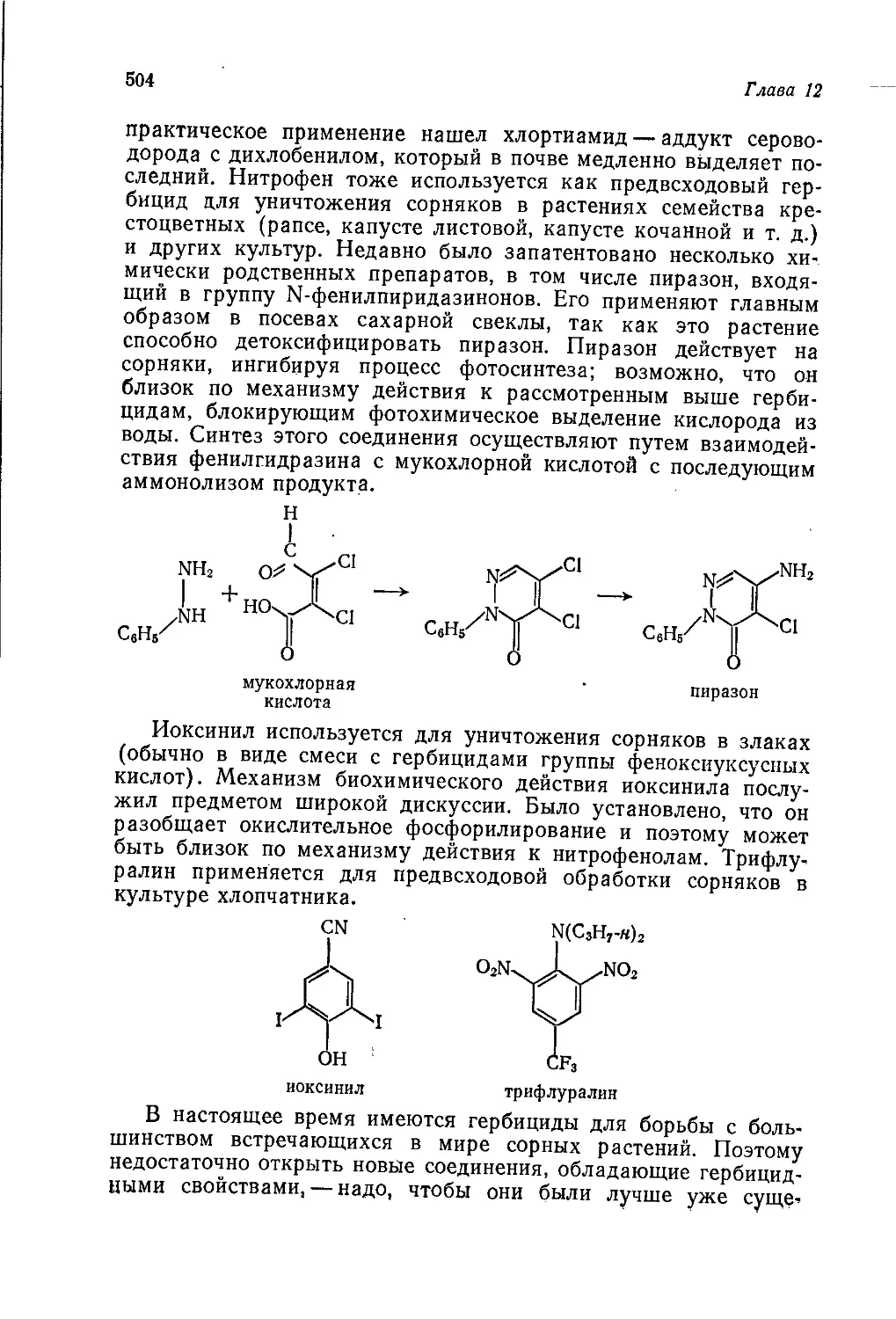


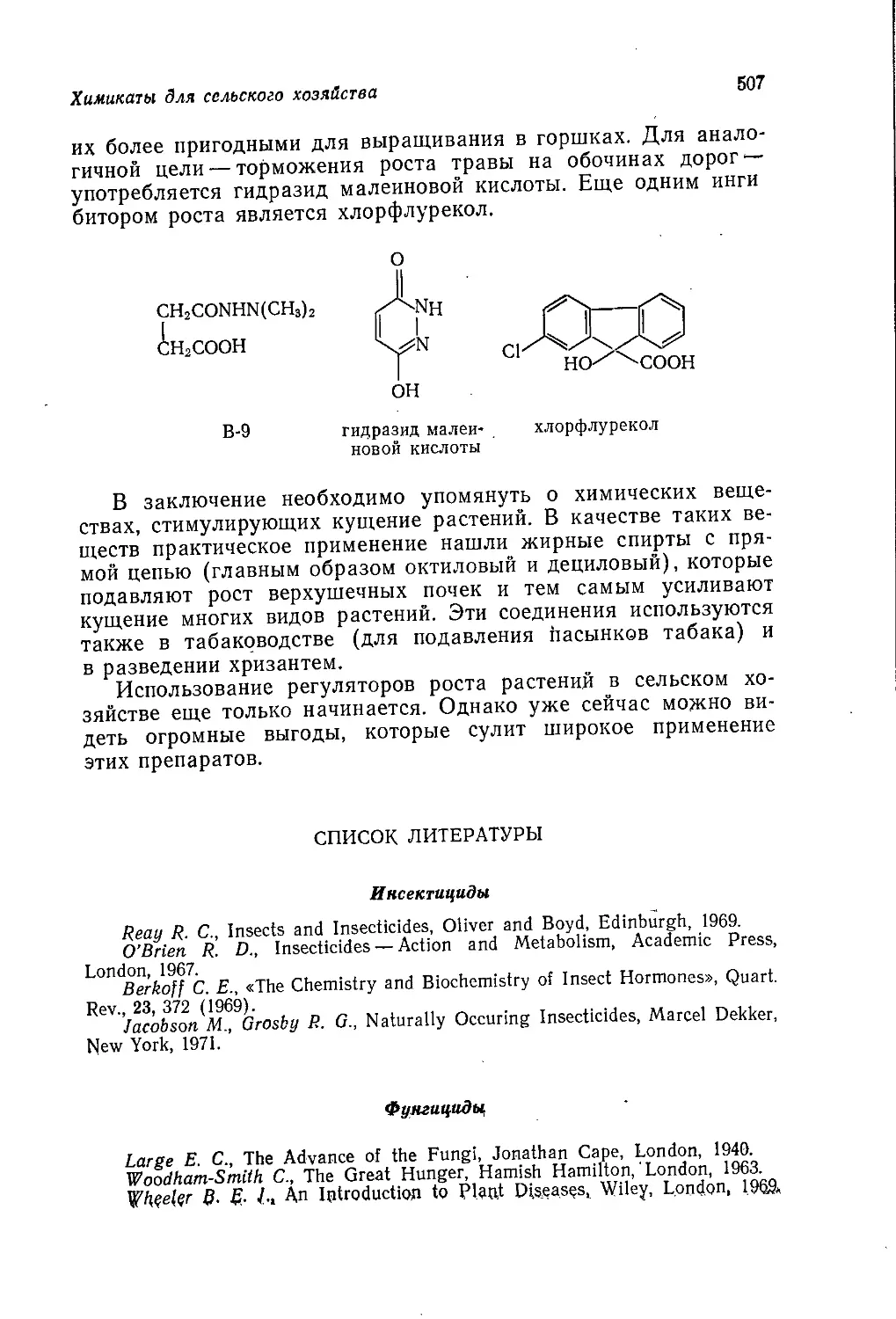




















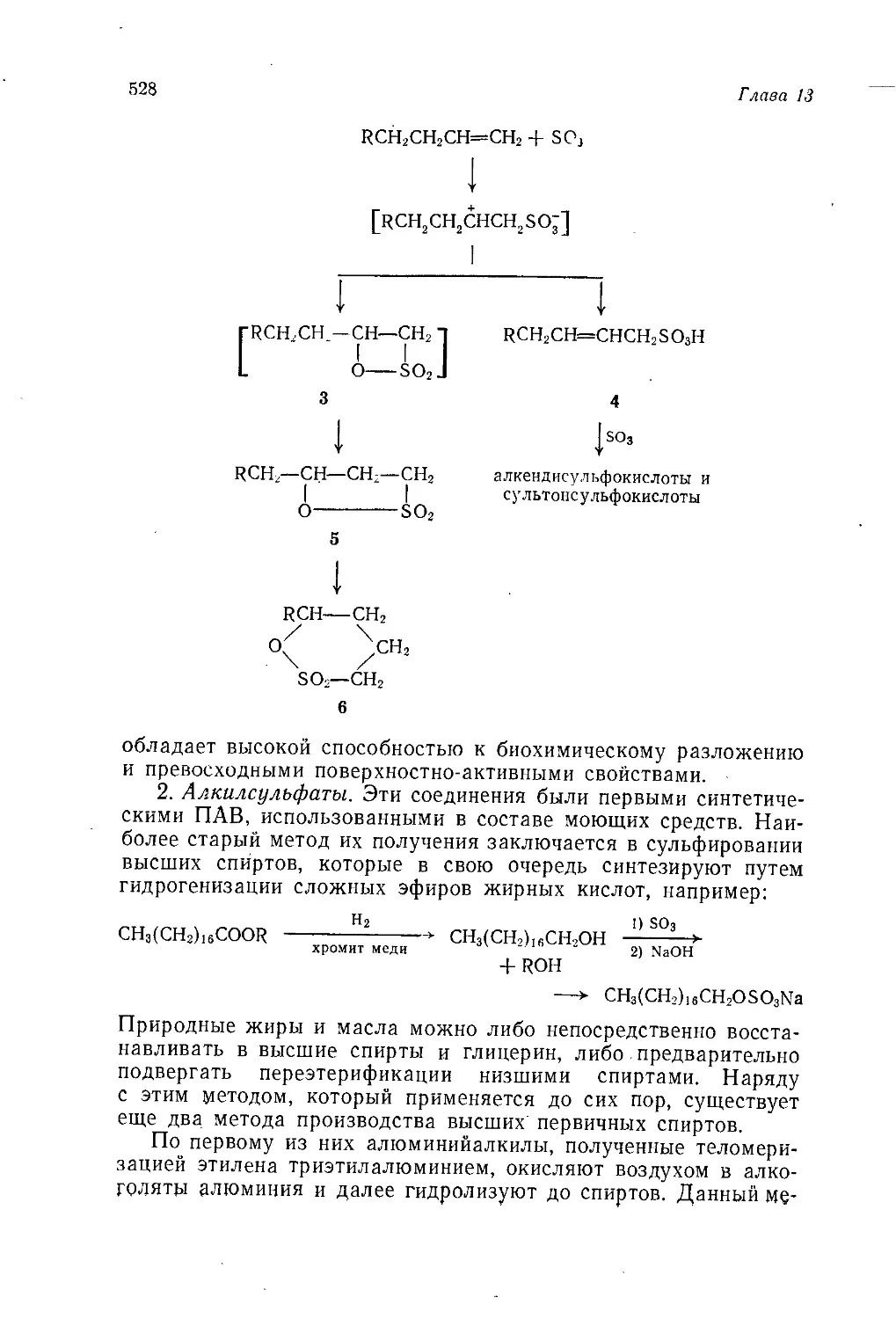







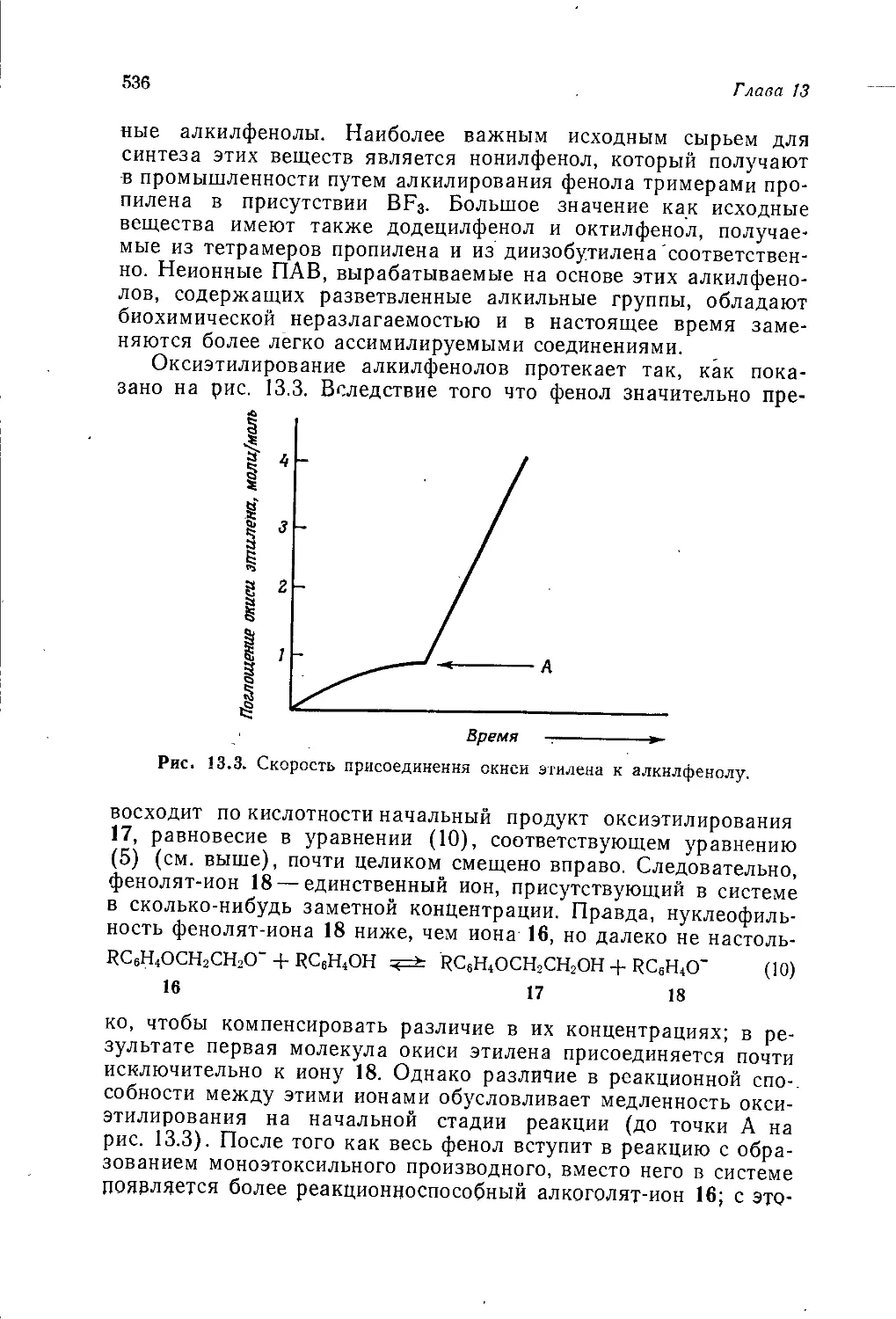












































































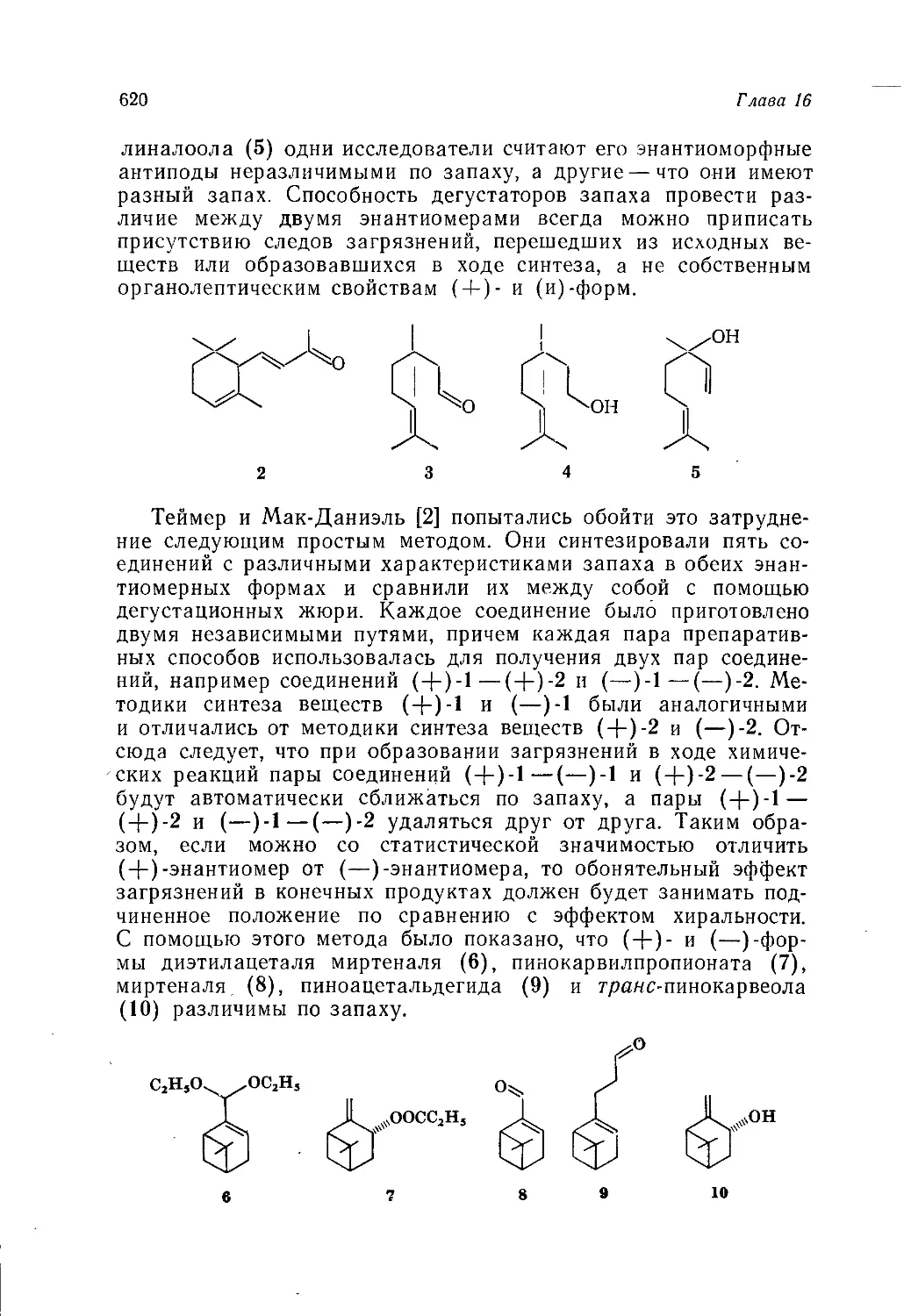
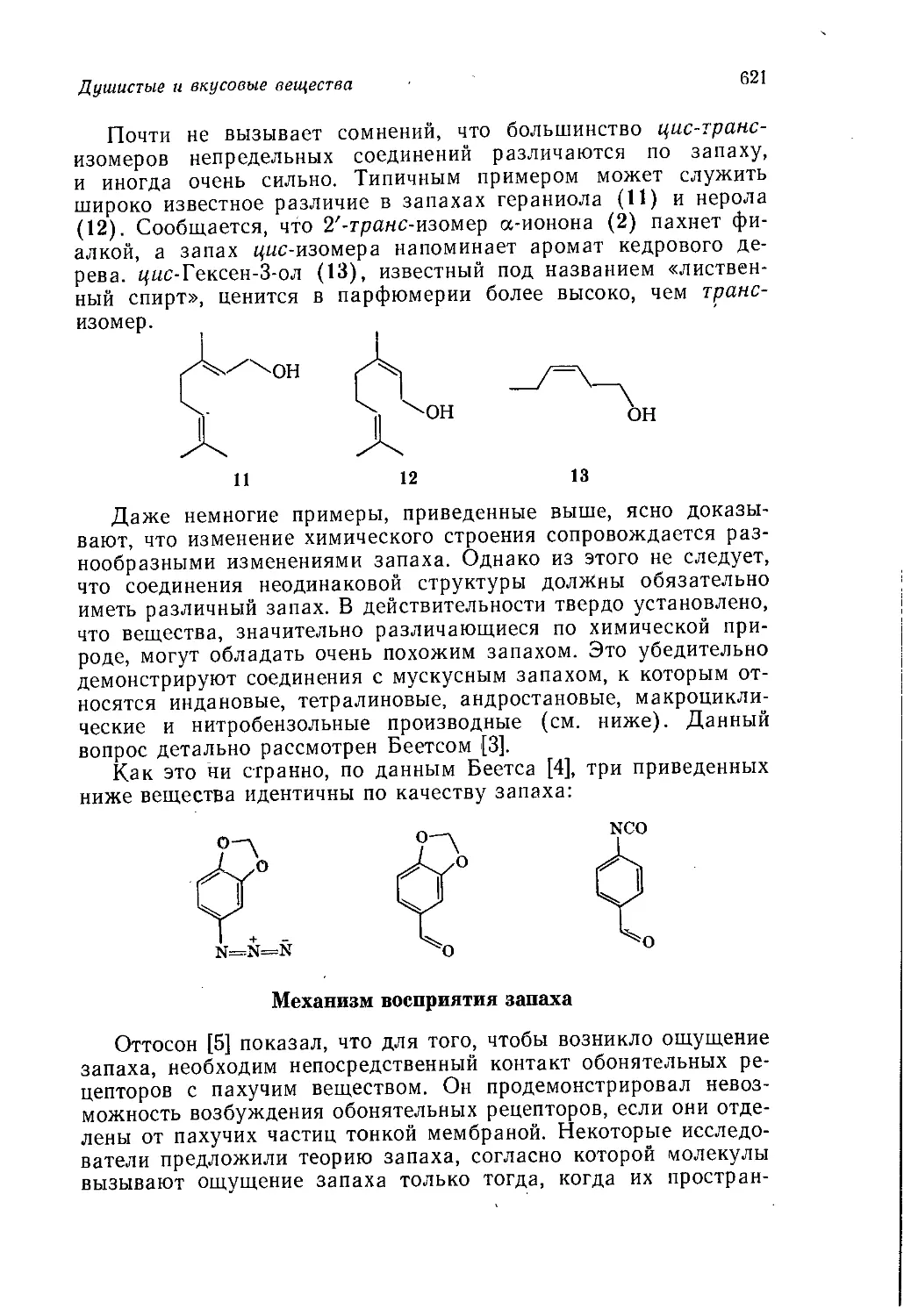


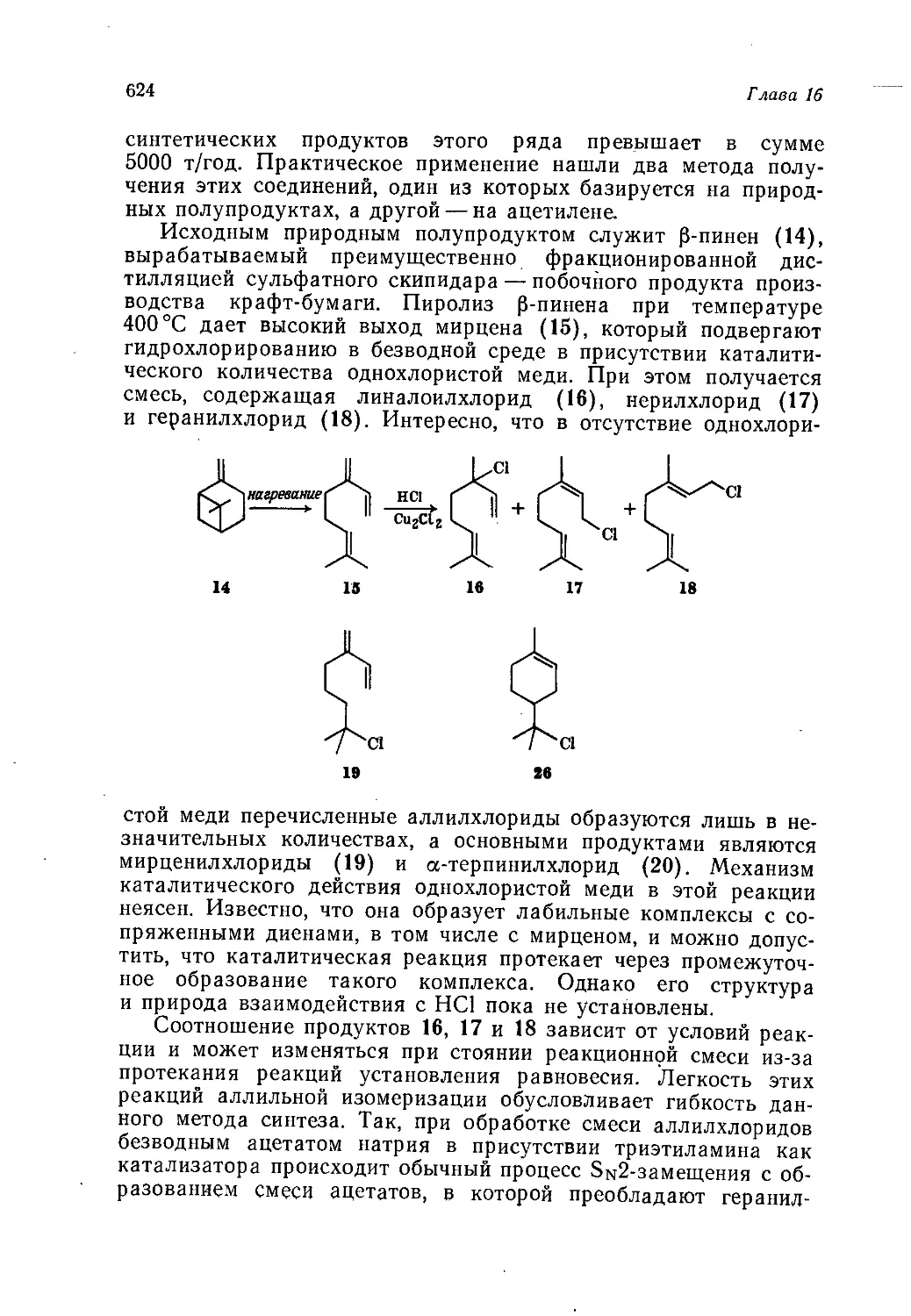
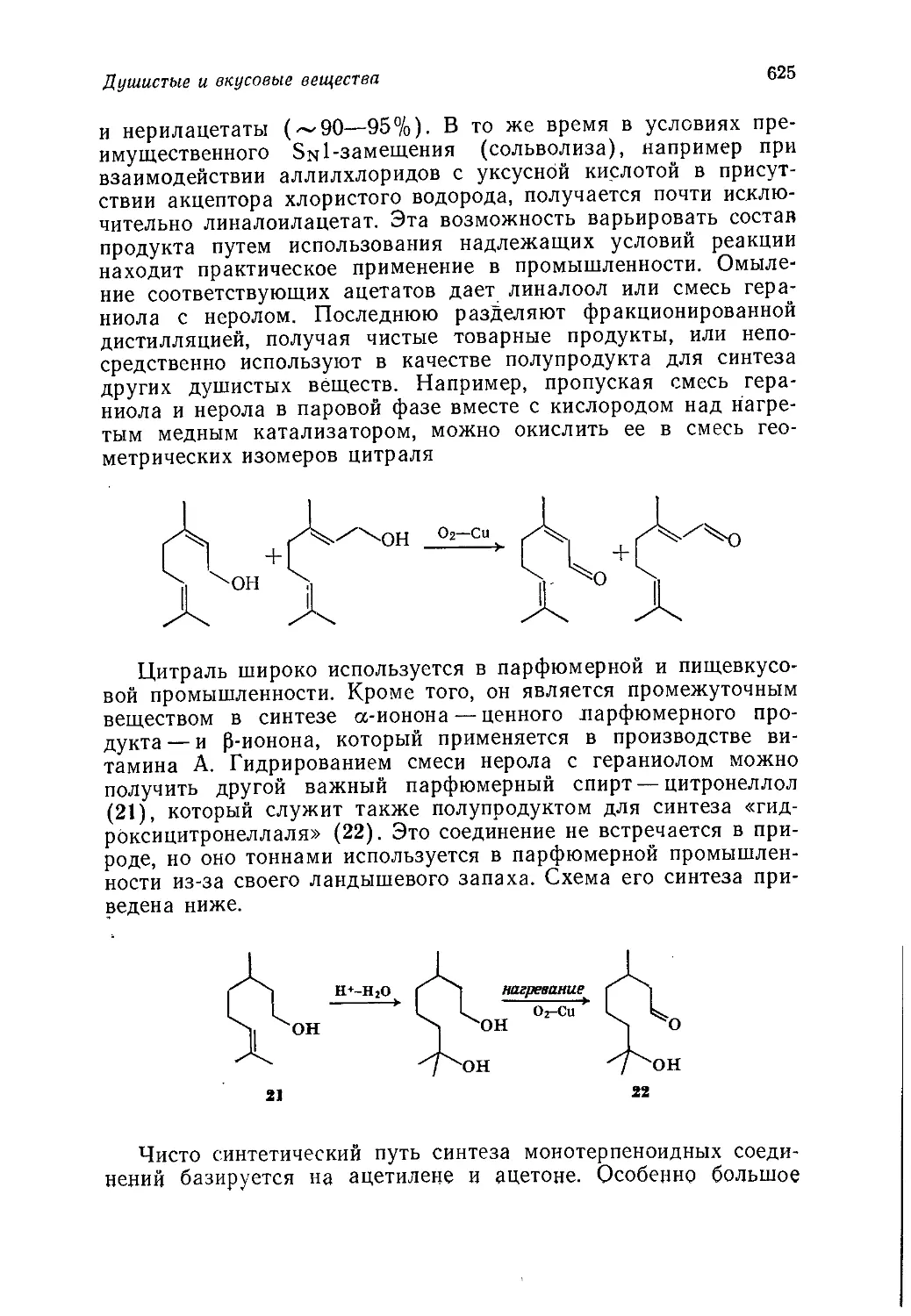








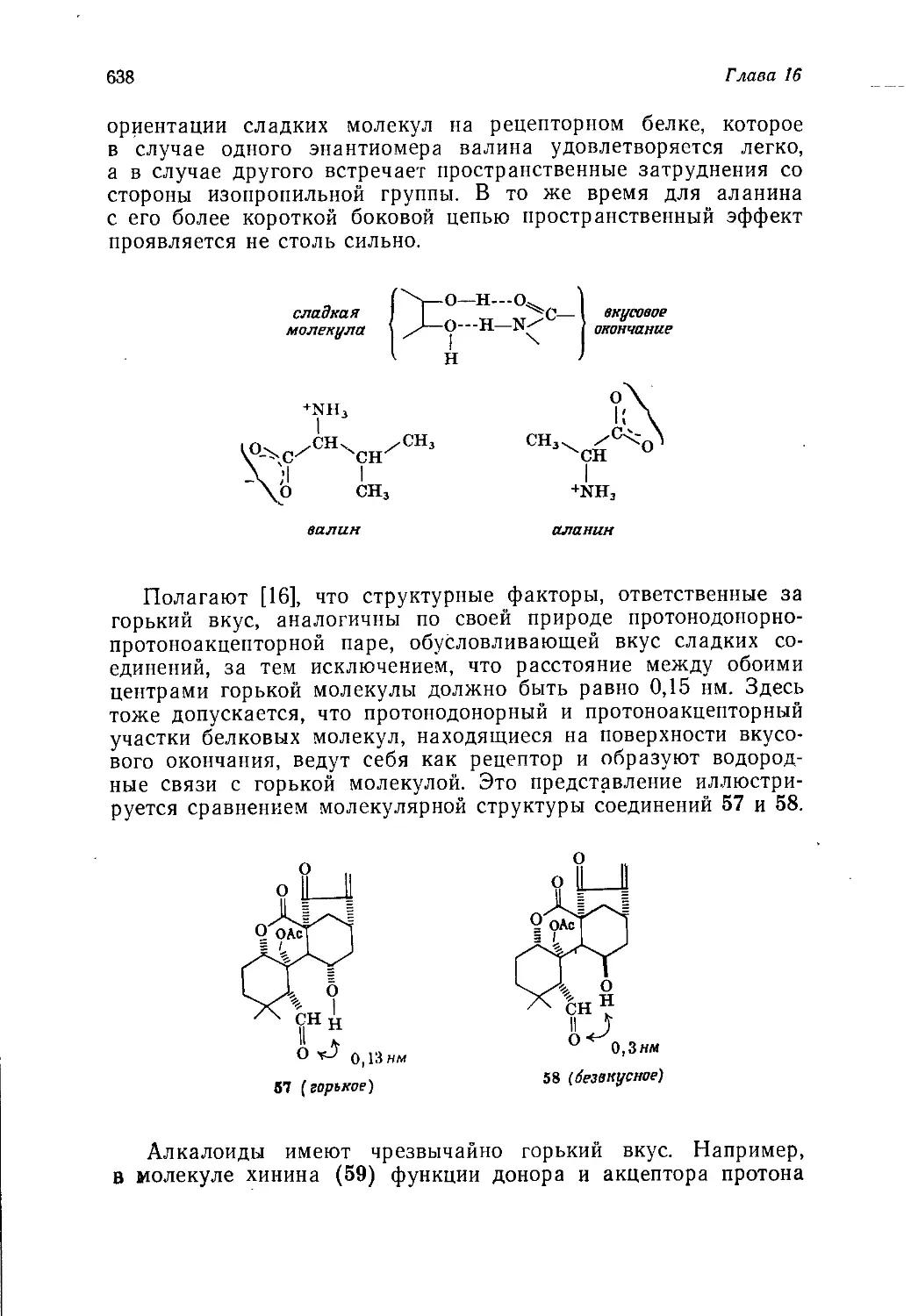
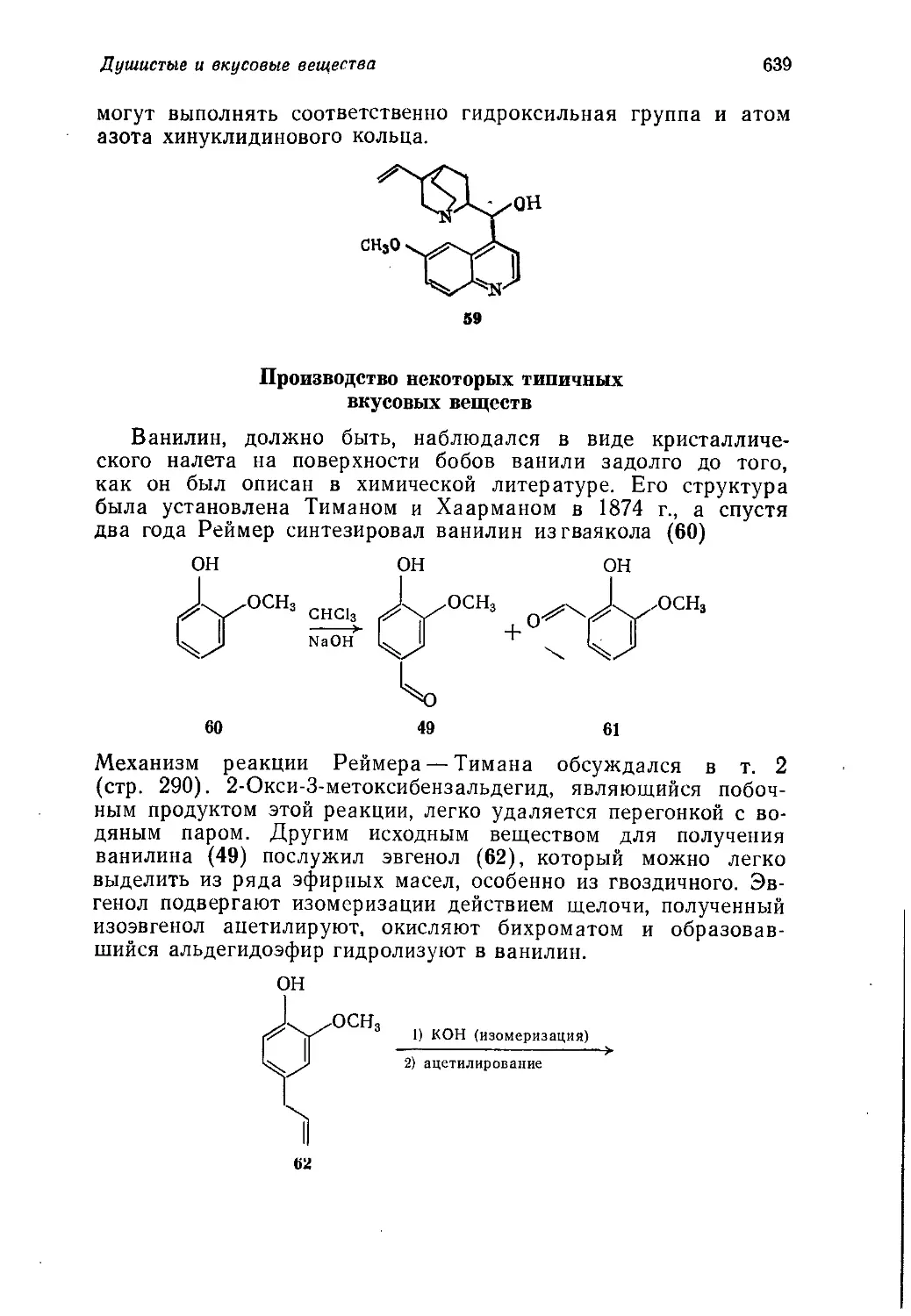

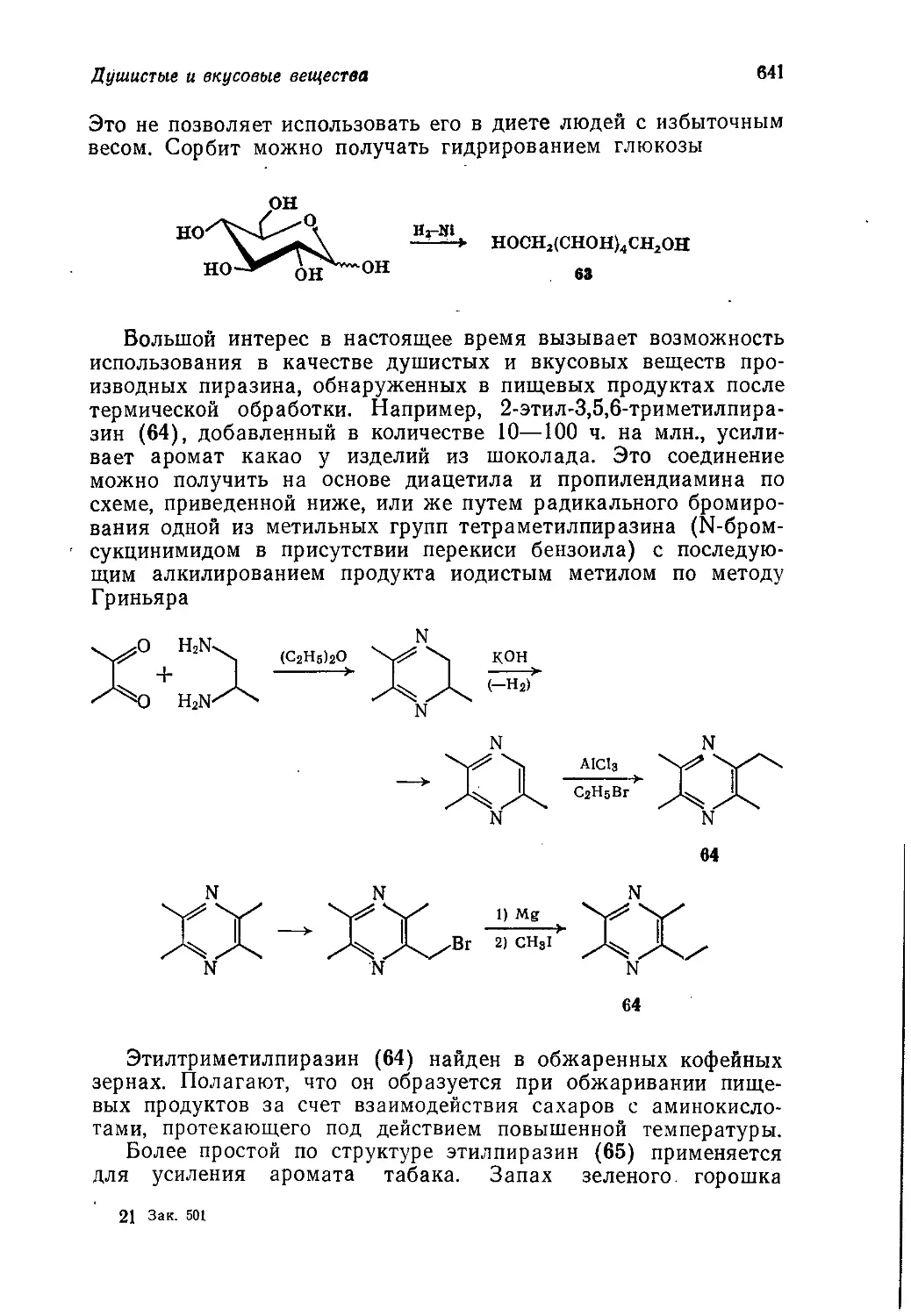
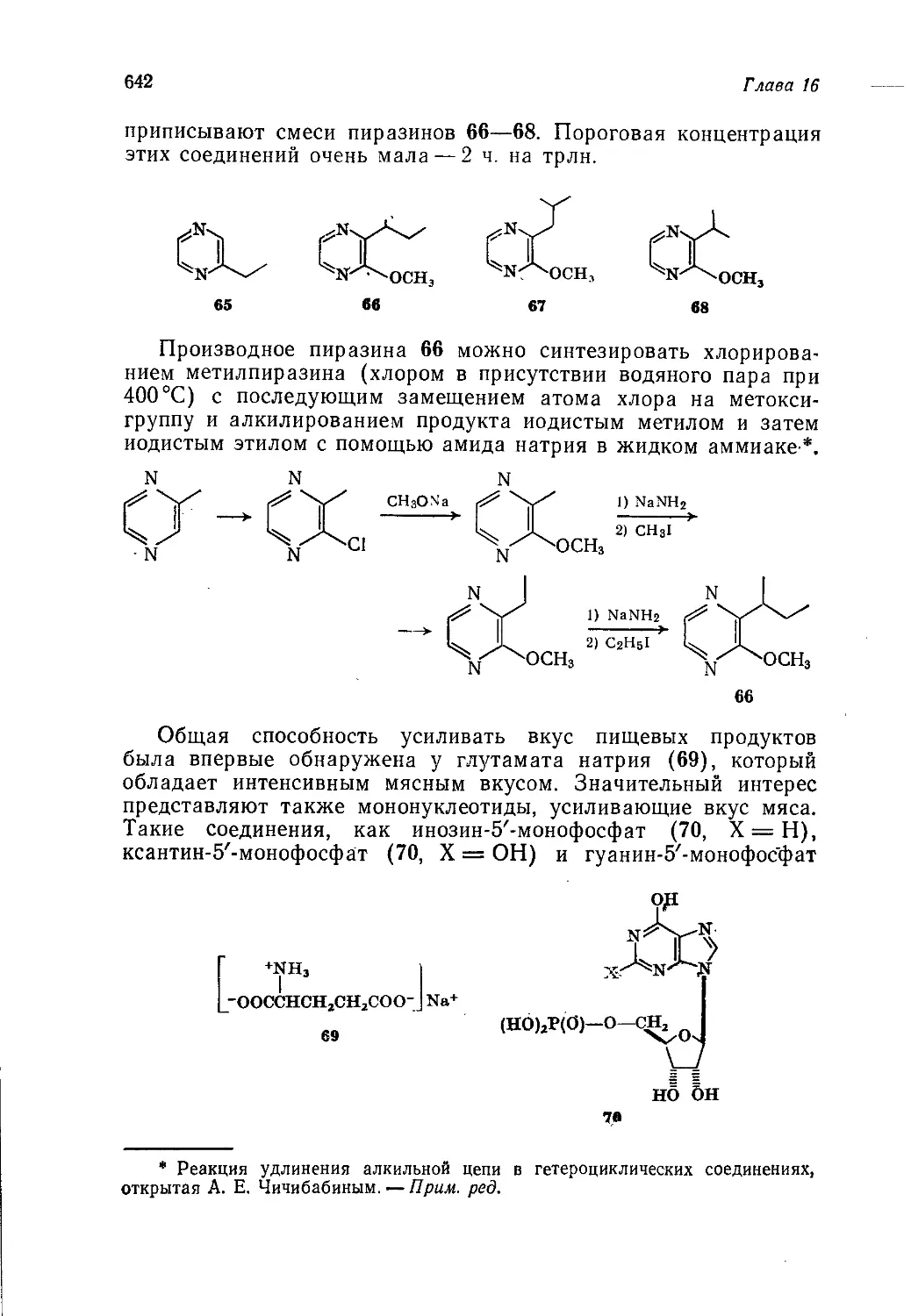





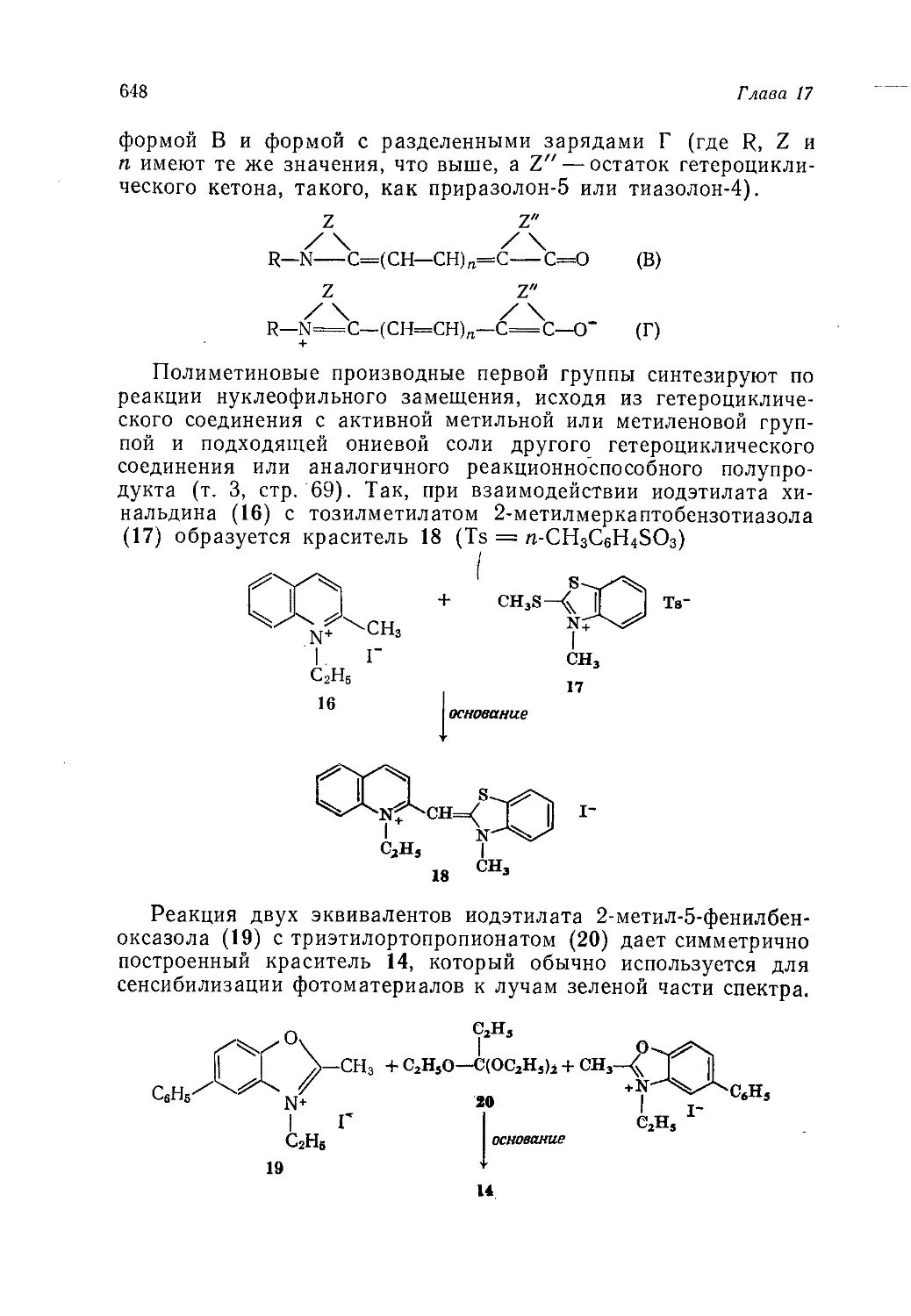
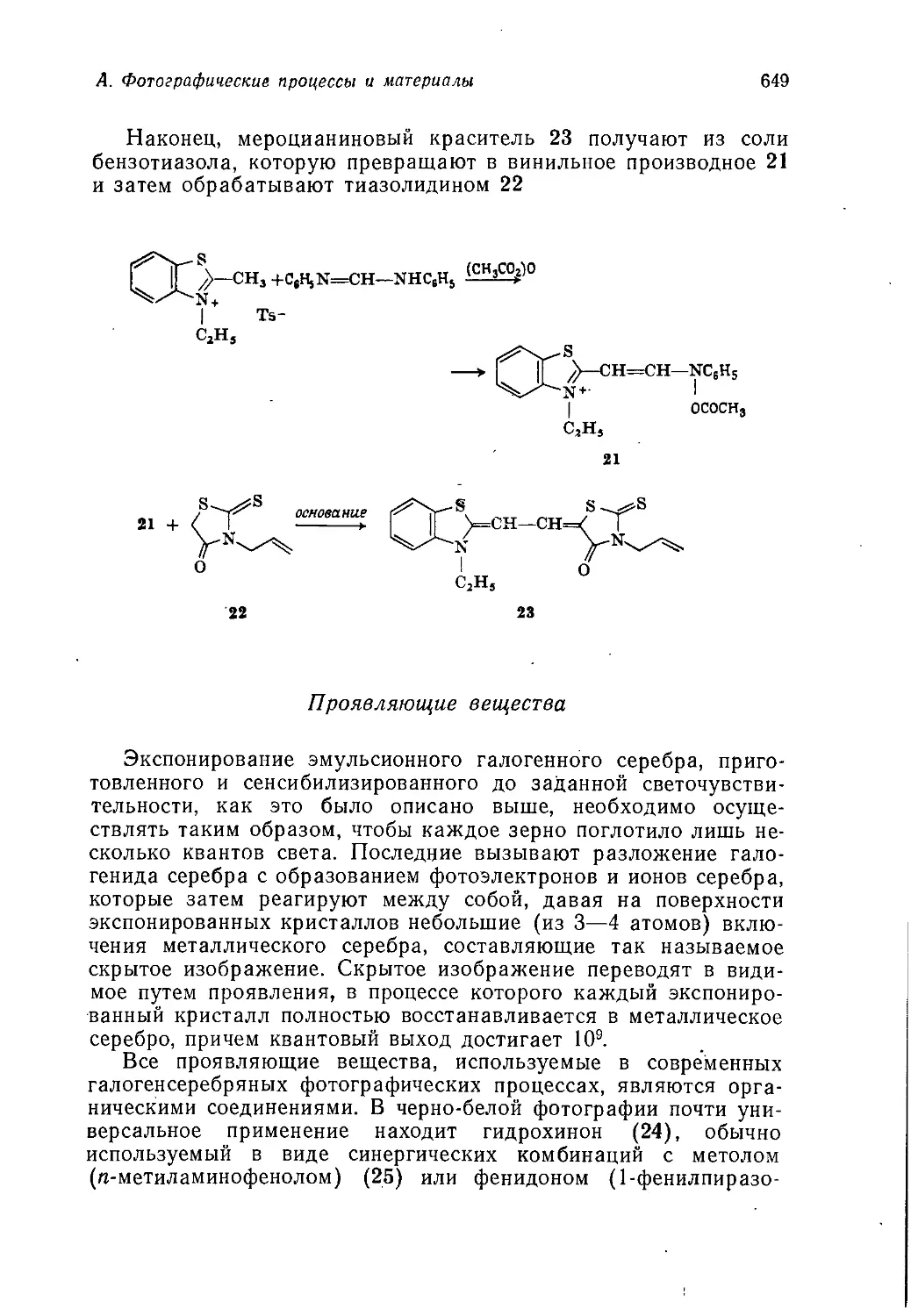
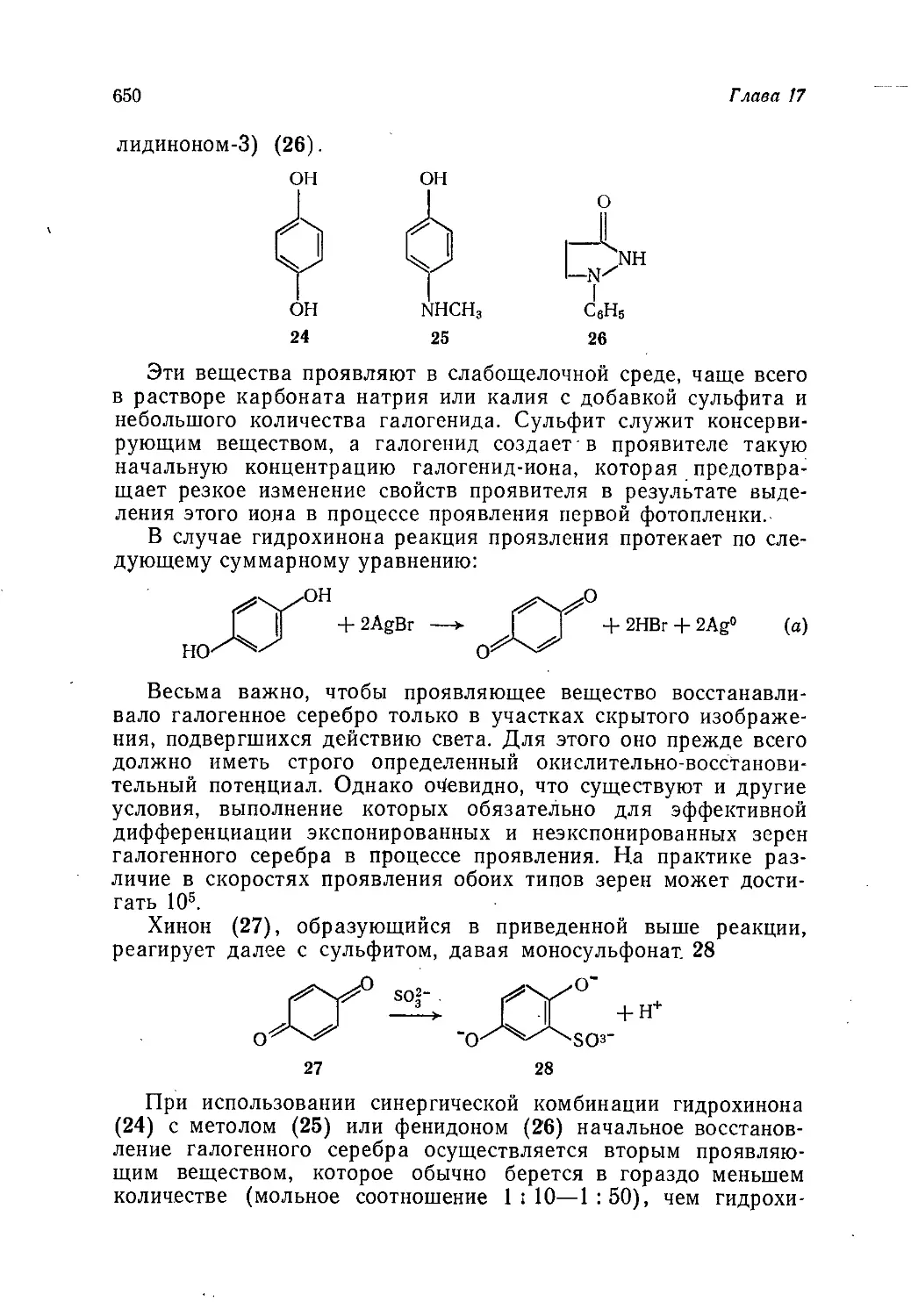
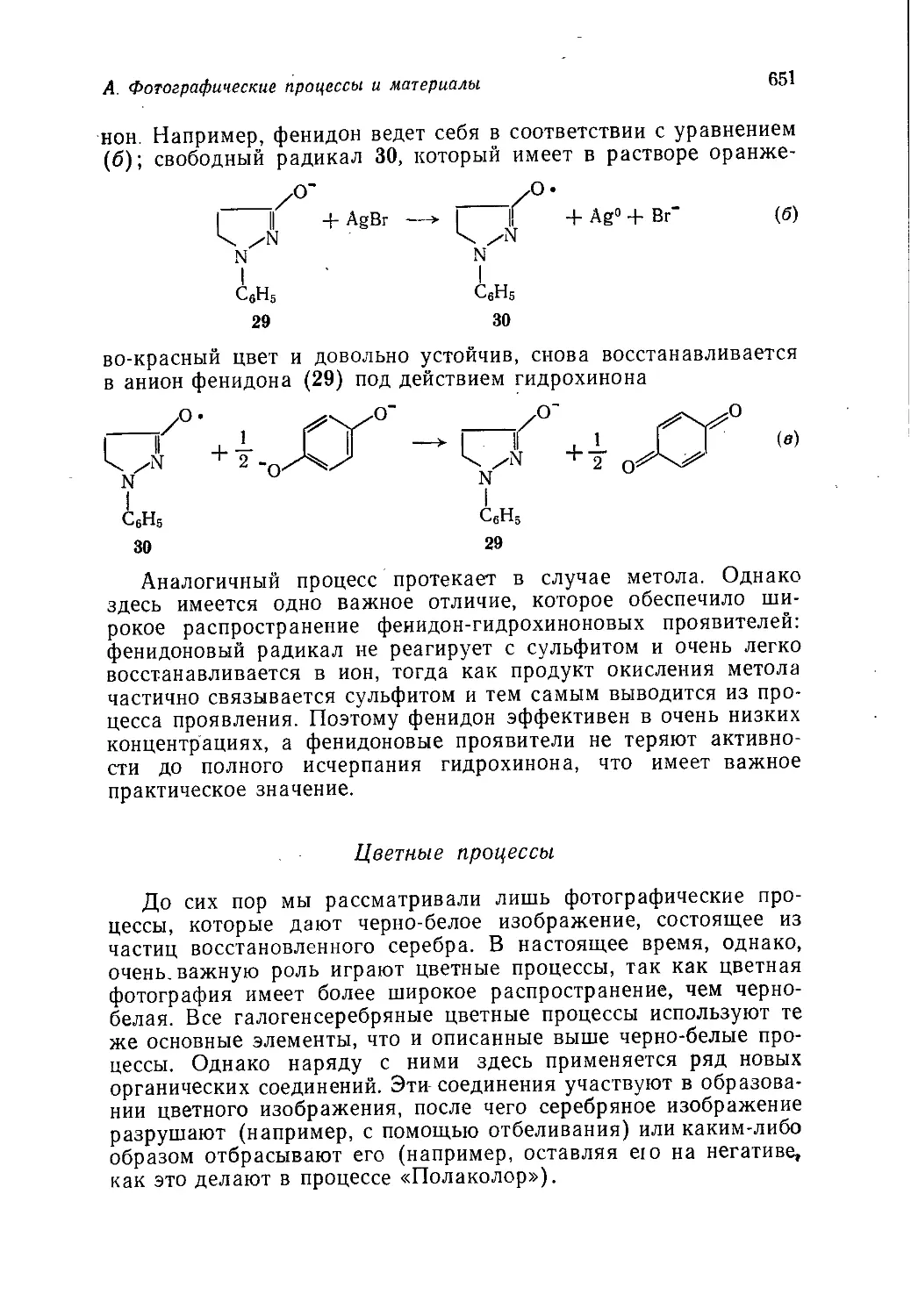
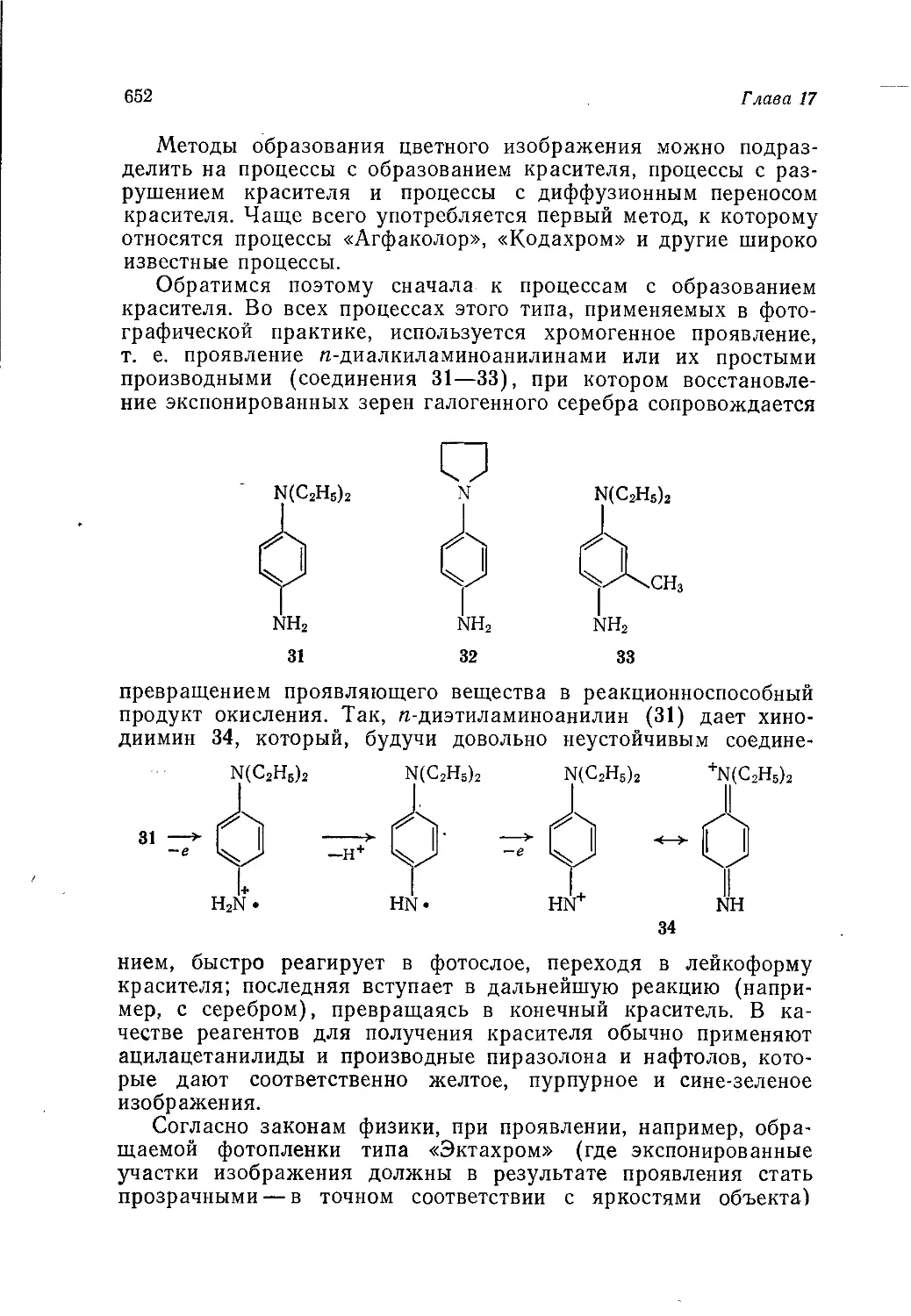
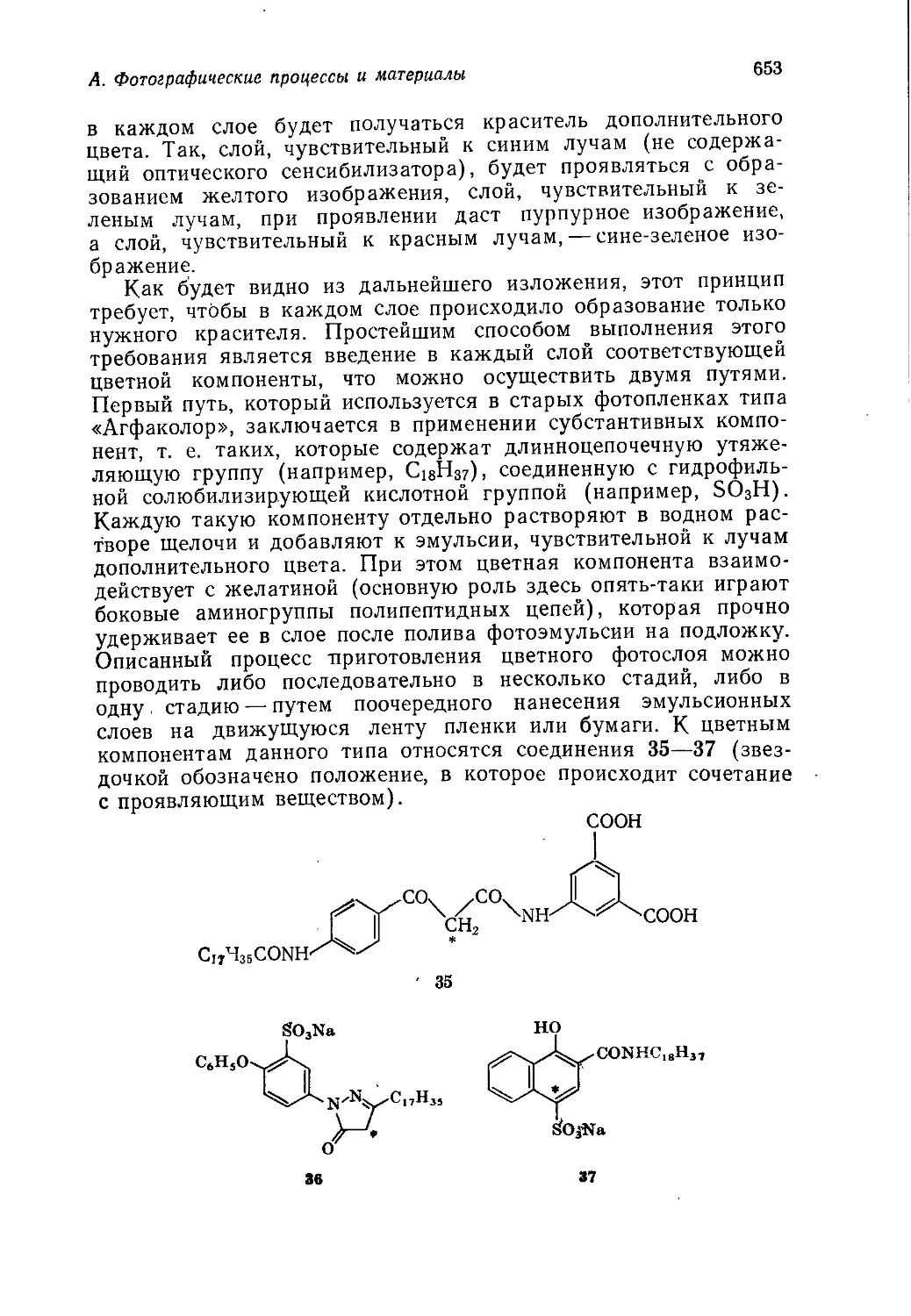
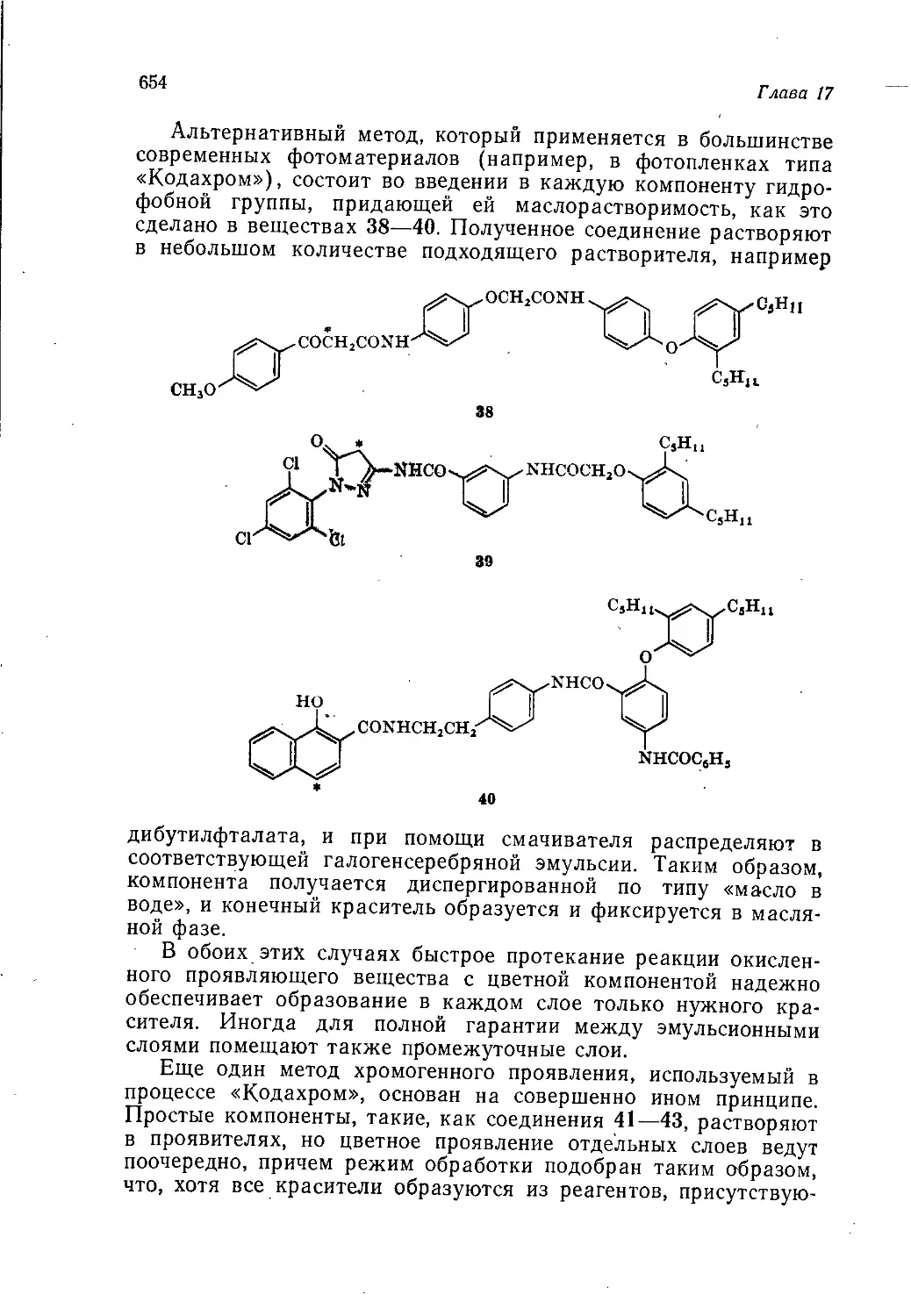
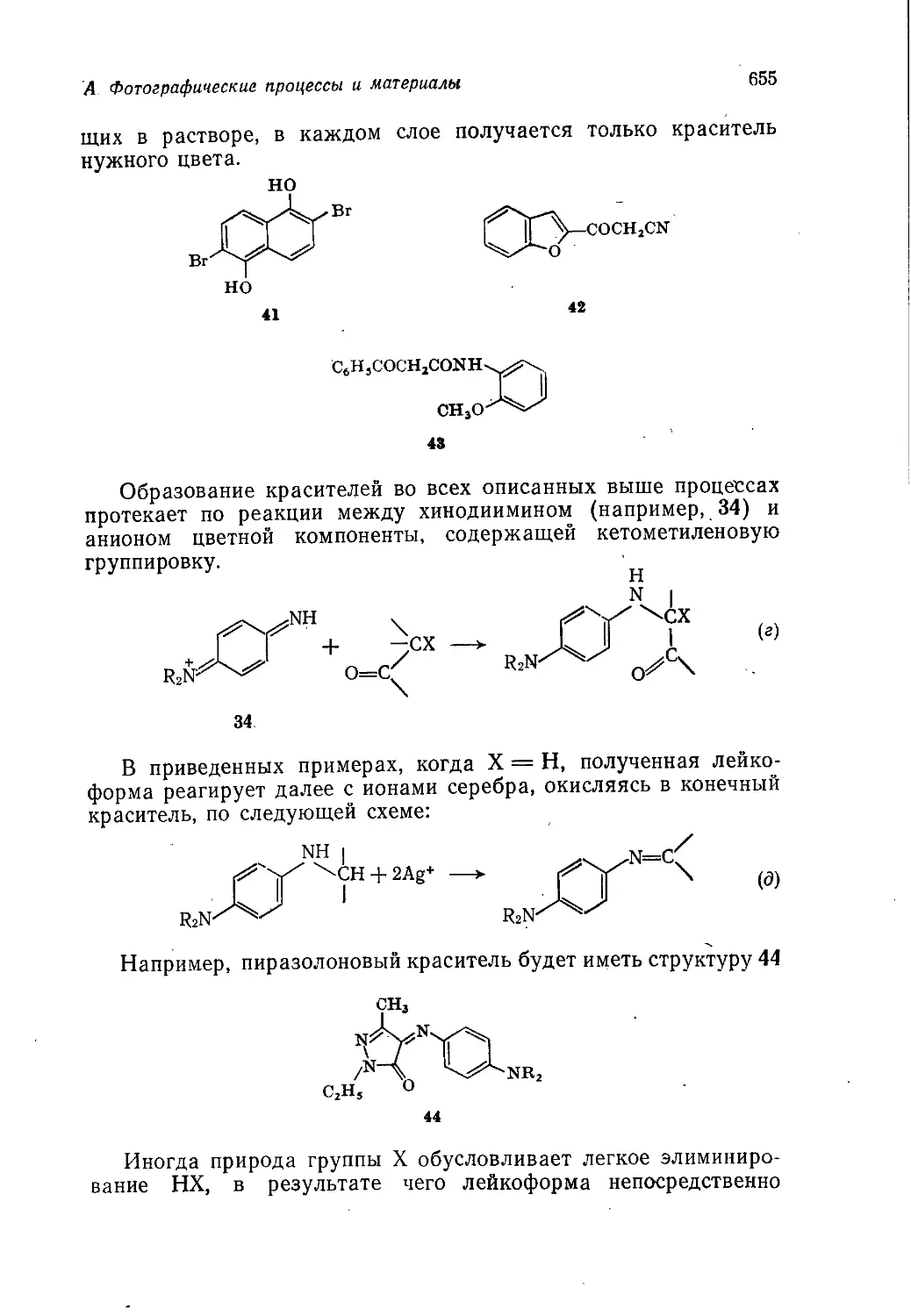

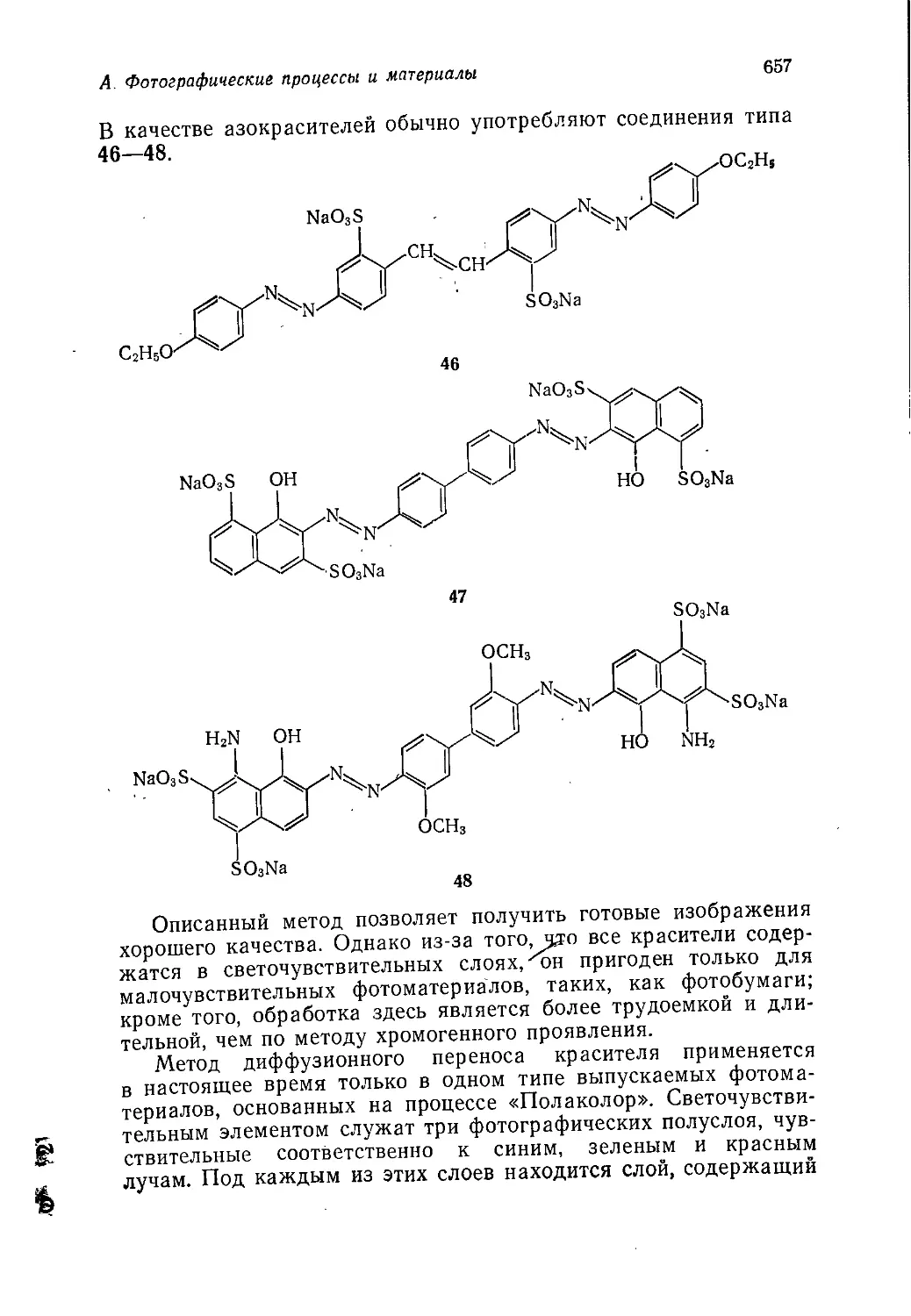
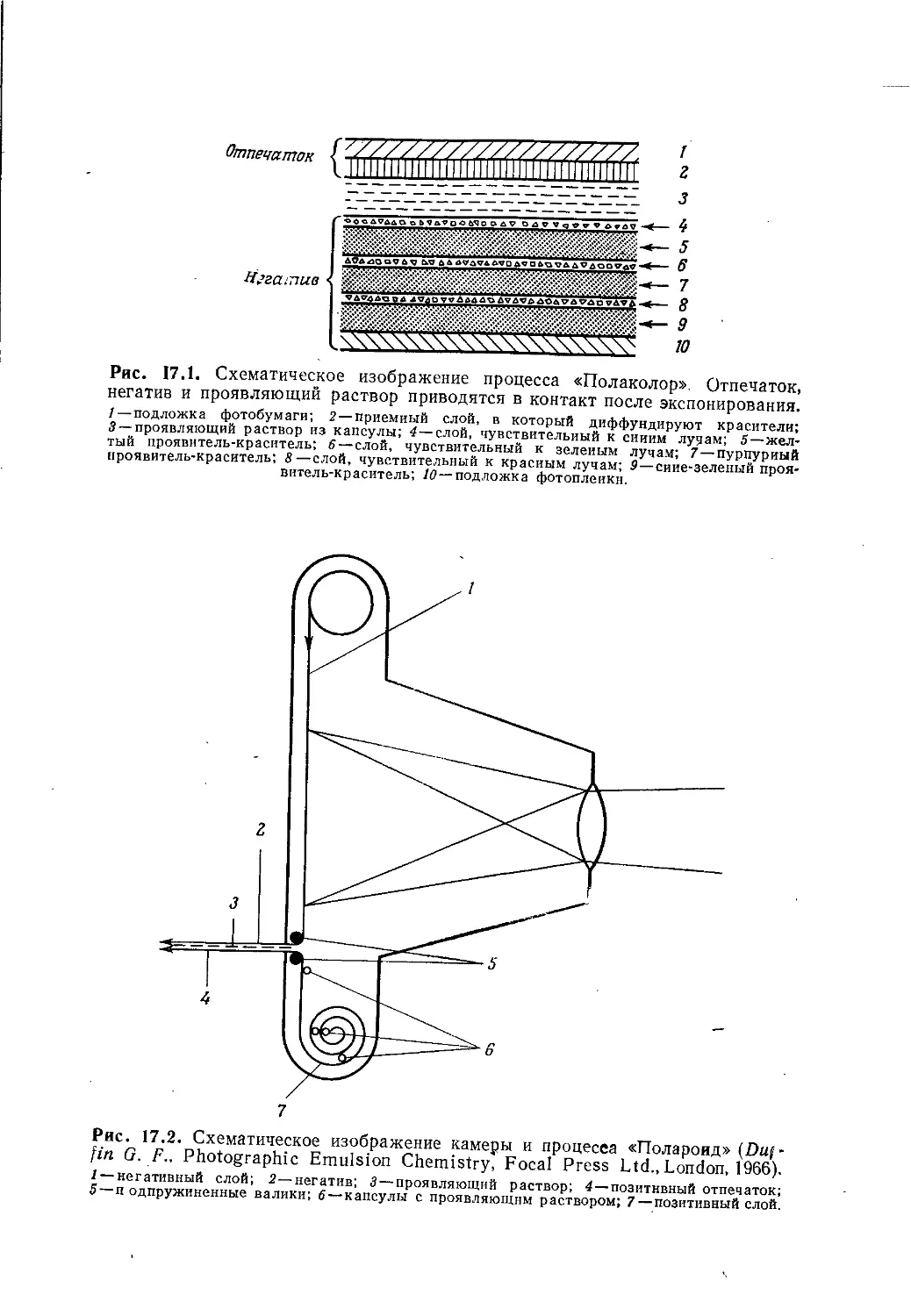



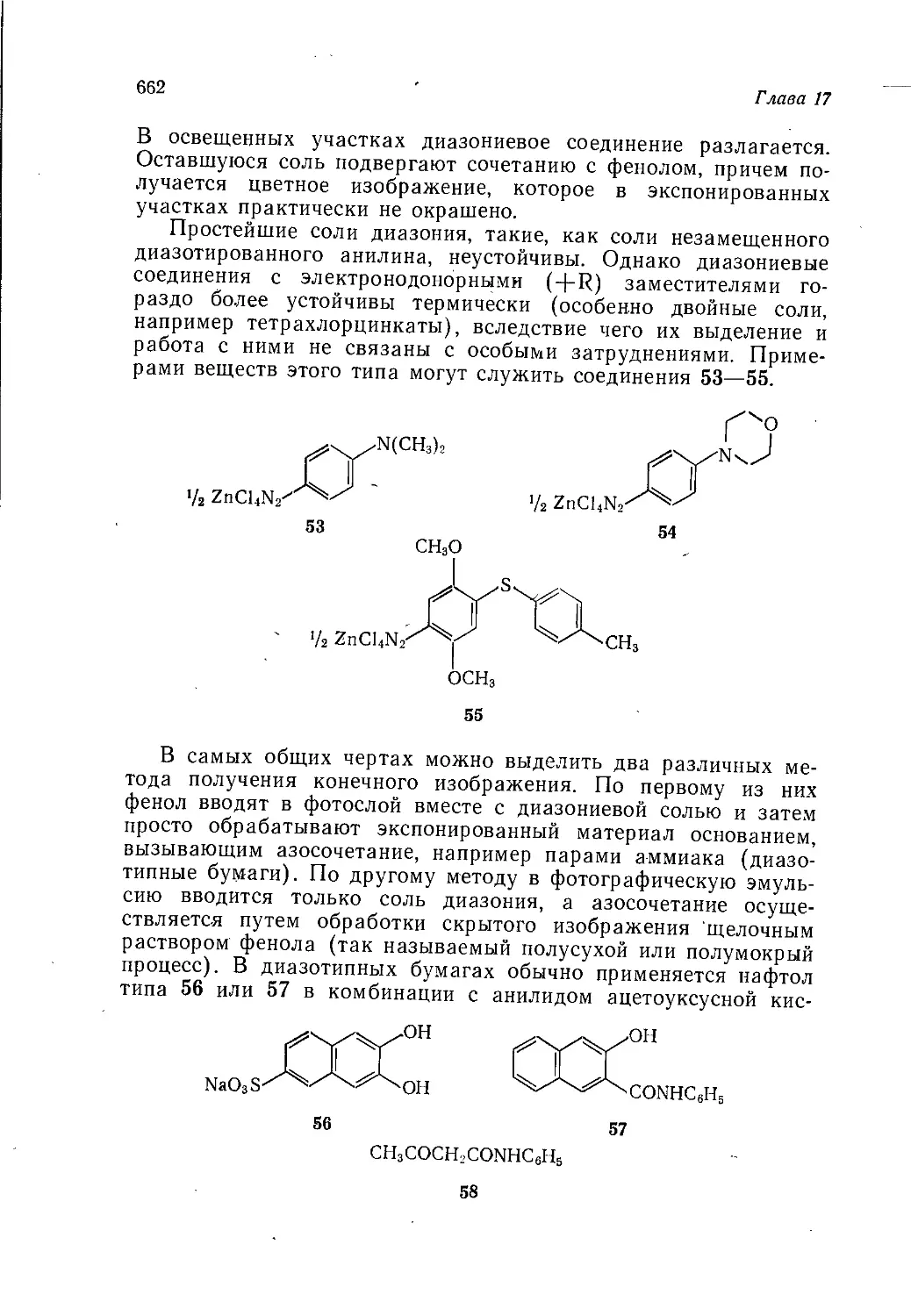

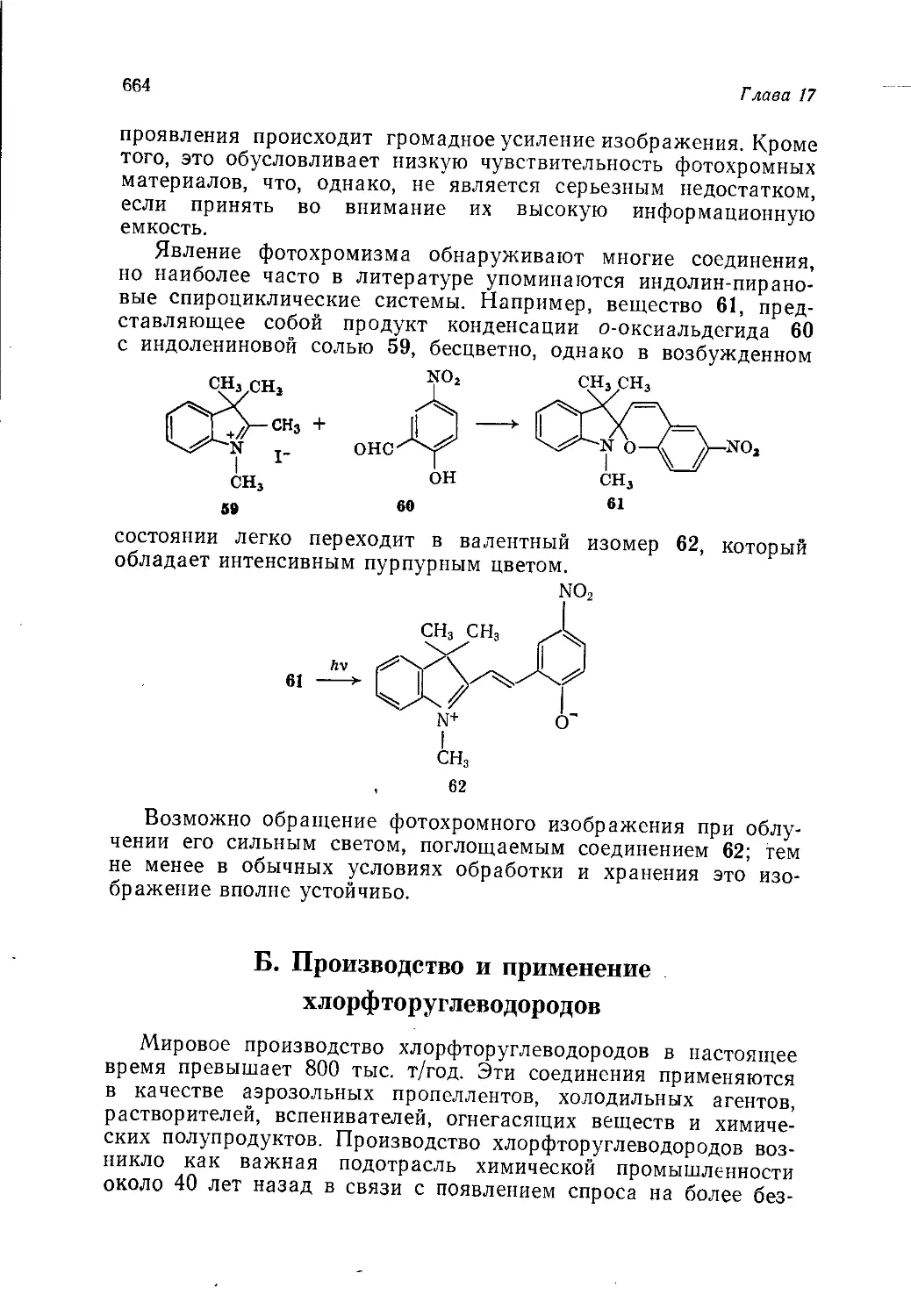







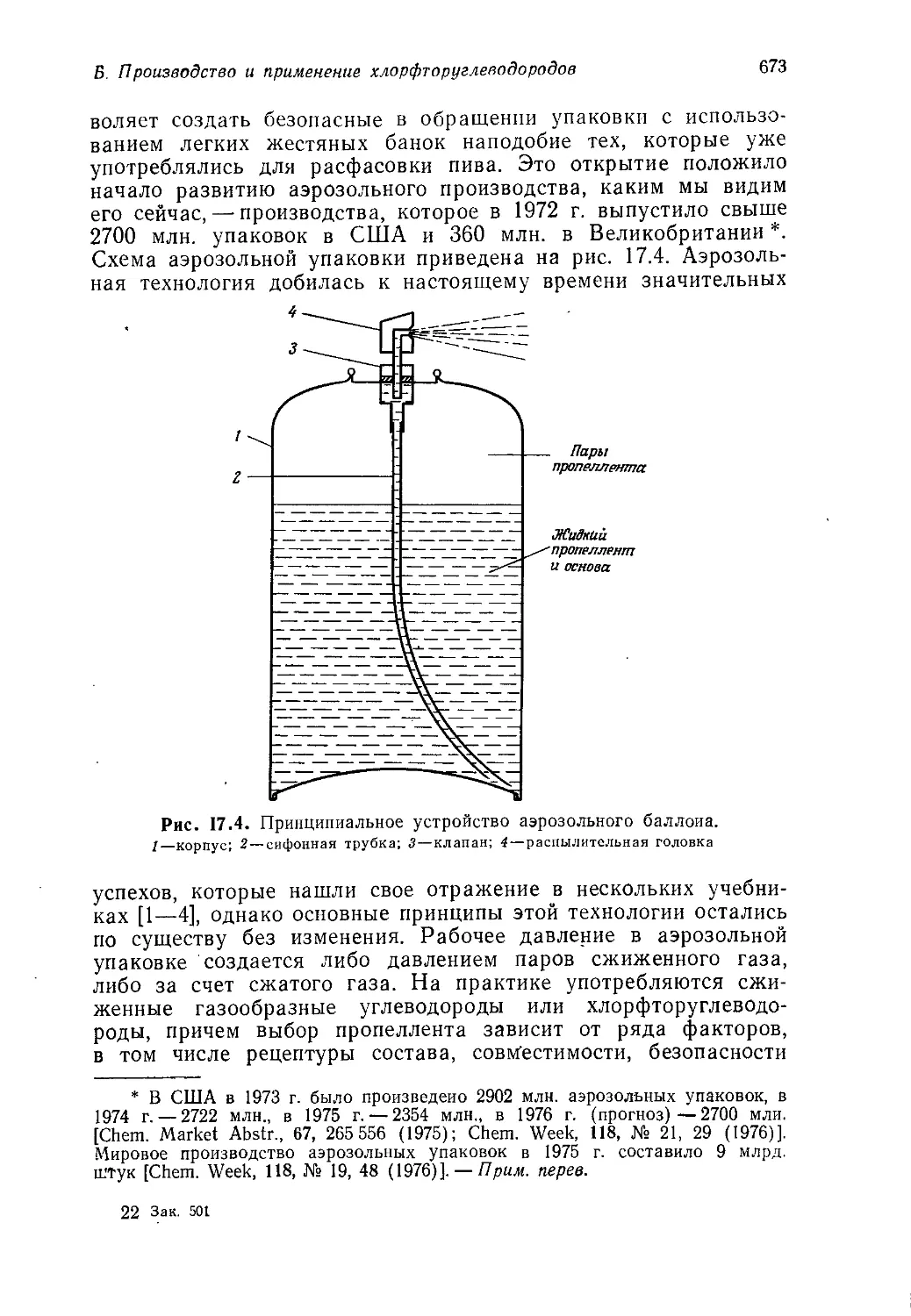































Author: Теддер Дж. Нехватал А. Джубб А.
Tags: органическая химия химия органические соединения переводная литература издательство мир
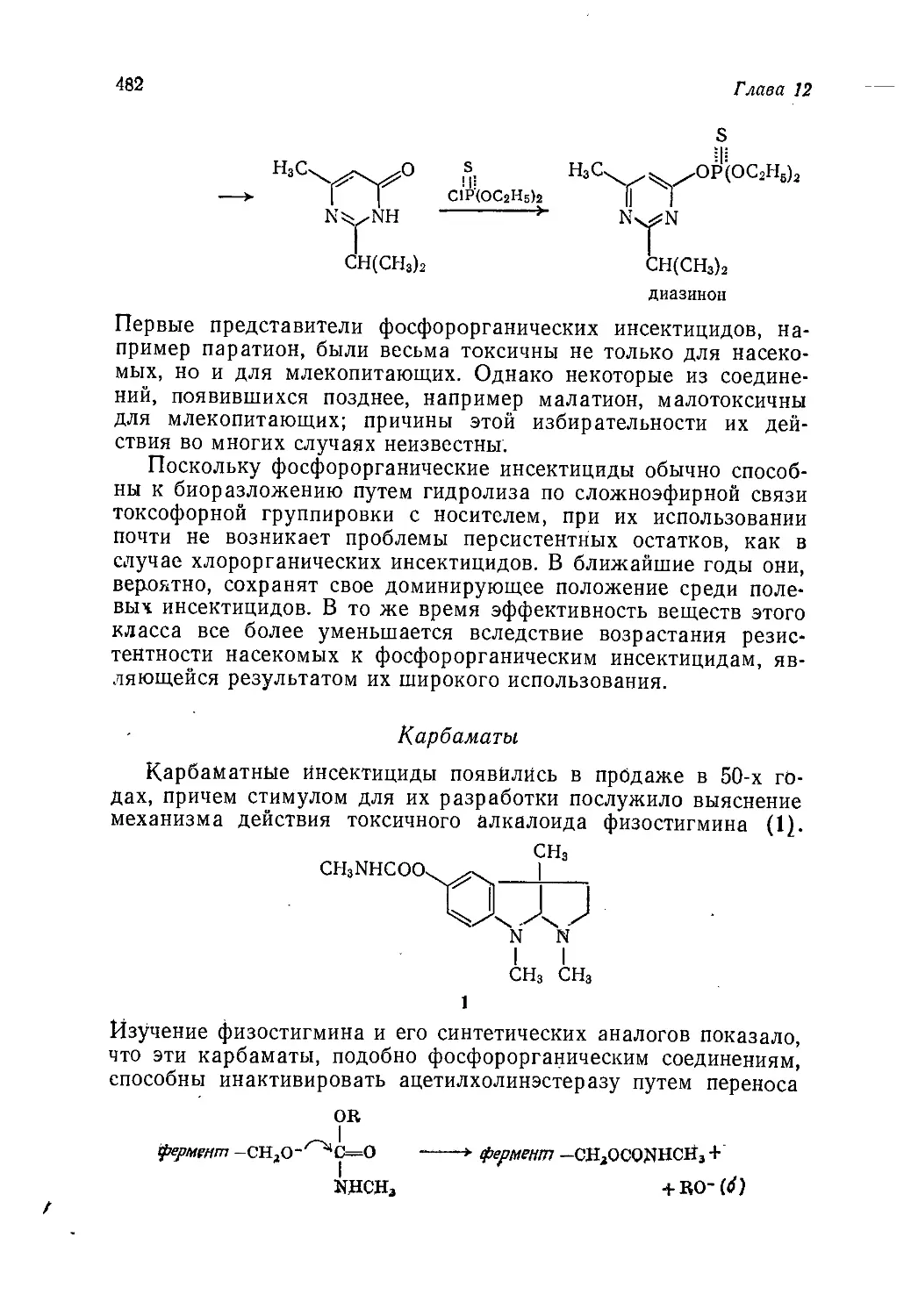
Year: 1977




Text
J. M. Tedder
University of St. Andrews
A. Nechvatal
University of Dundee
A. H. Jubb
ICI Ltd., Industrial Laboratory, Runcorn
BASIC
ORGANIC
CHEMISTRY
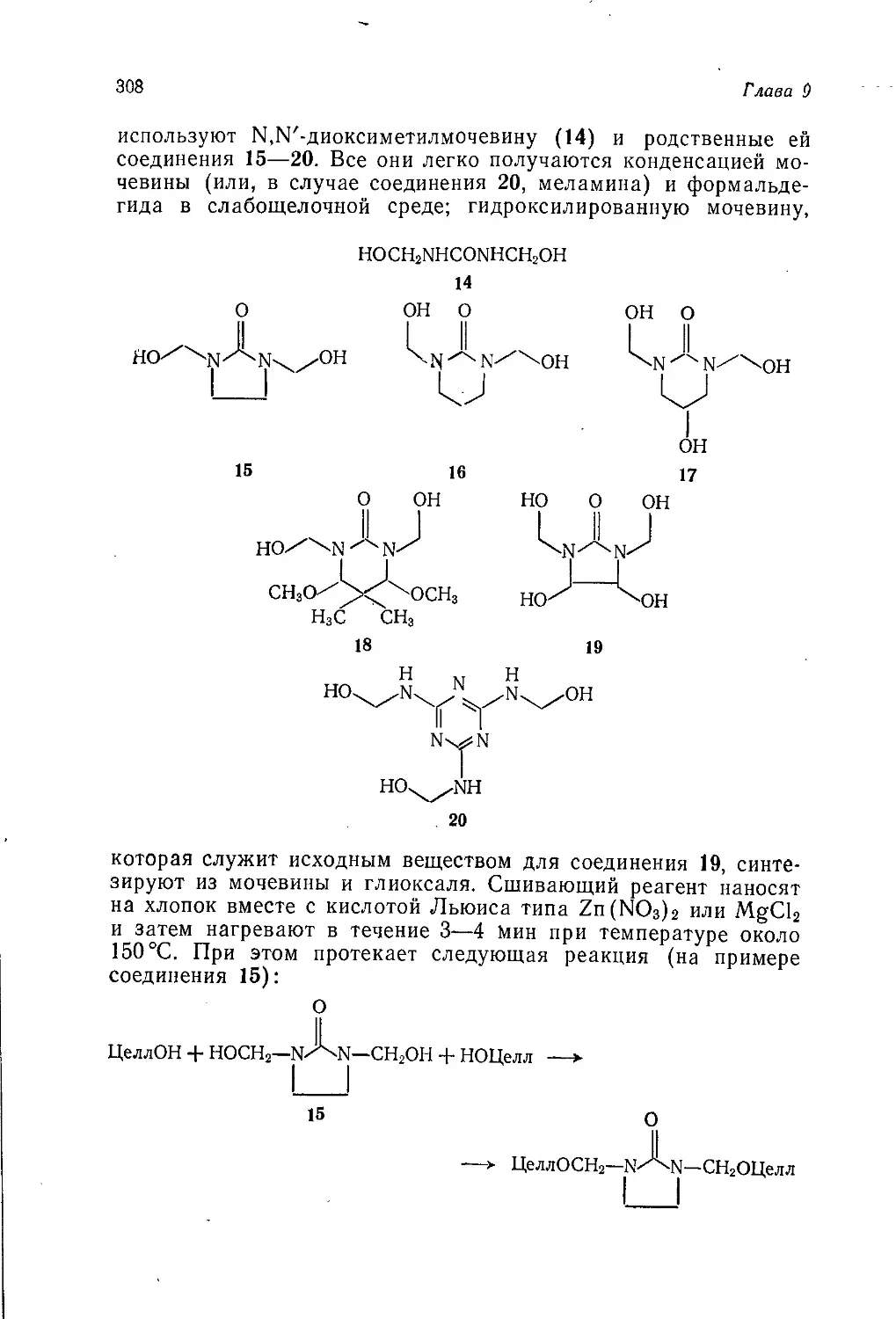
Part 5: Industrial Products
1975
JOHN WILEY & SONS
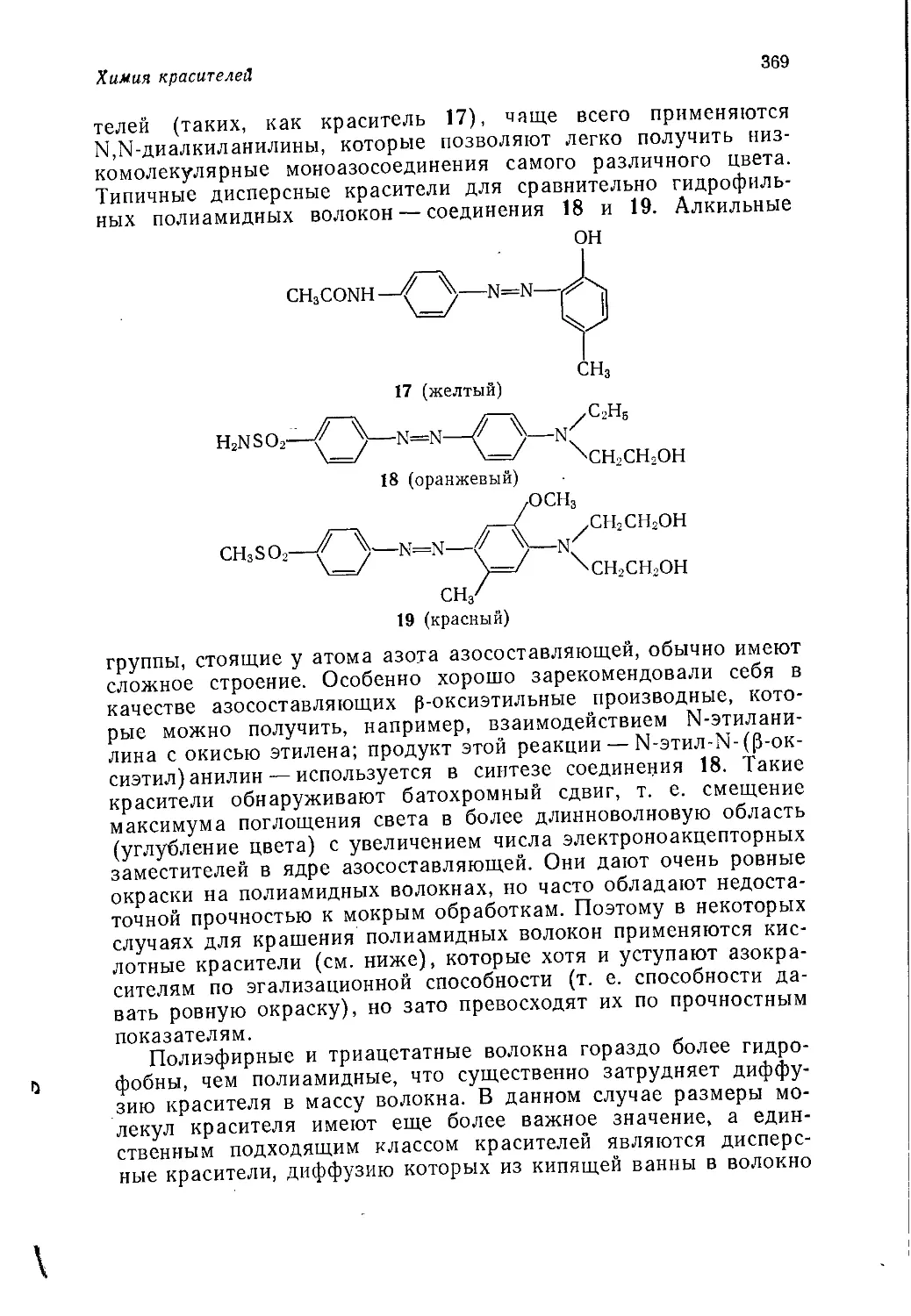
London New York Sydney Toronto
Дж. Теддер, А. Нехватал, А. Джубб
ПРОМЫШЛЕННАЯ
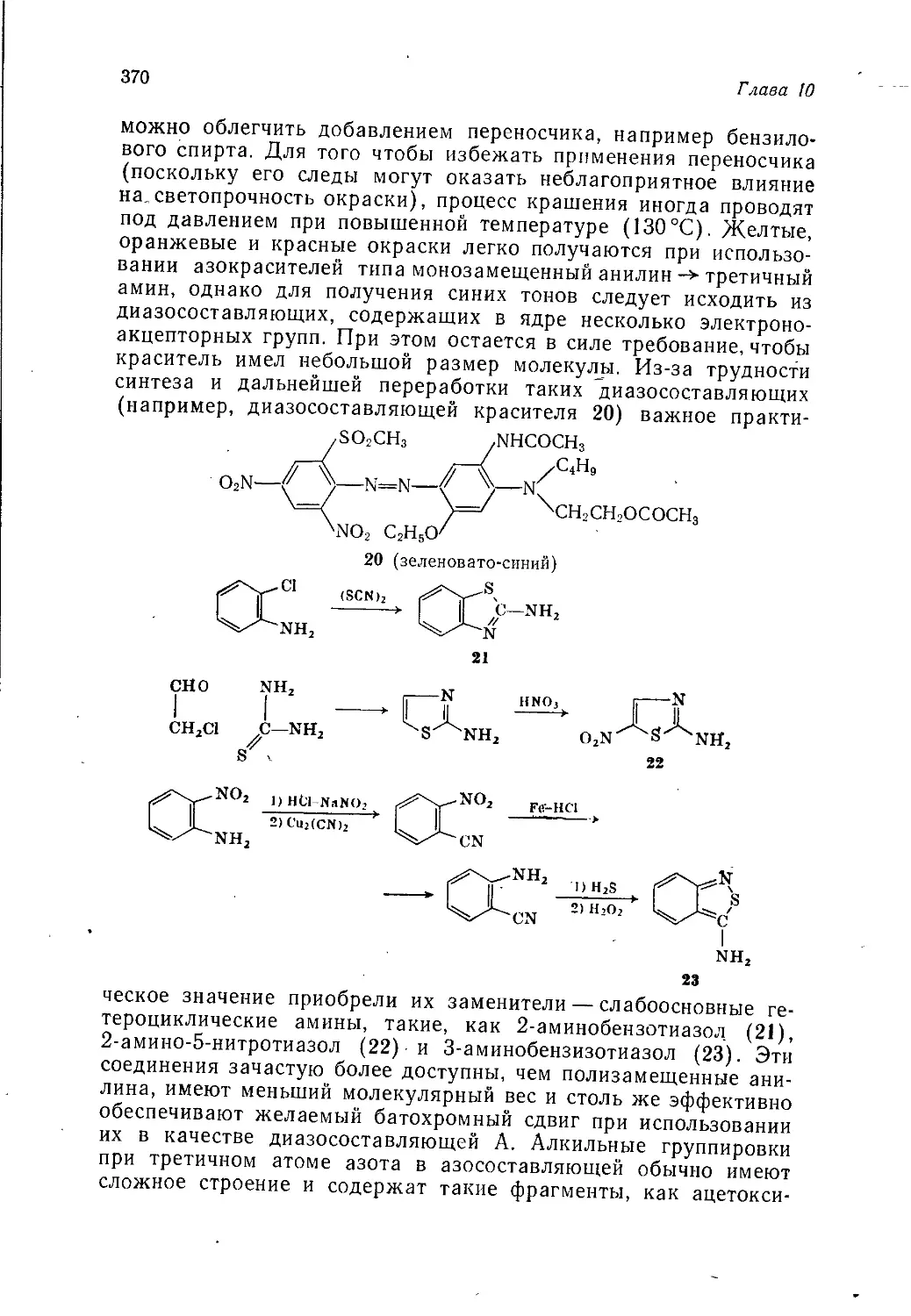
ОРГАНИЧЕСКАЯ
ХИМИЯ
Перевод с английского
канд. хим. наук Г. Я. Легина
Под редакцией
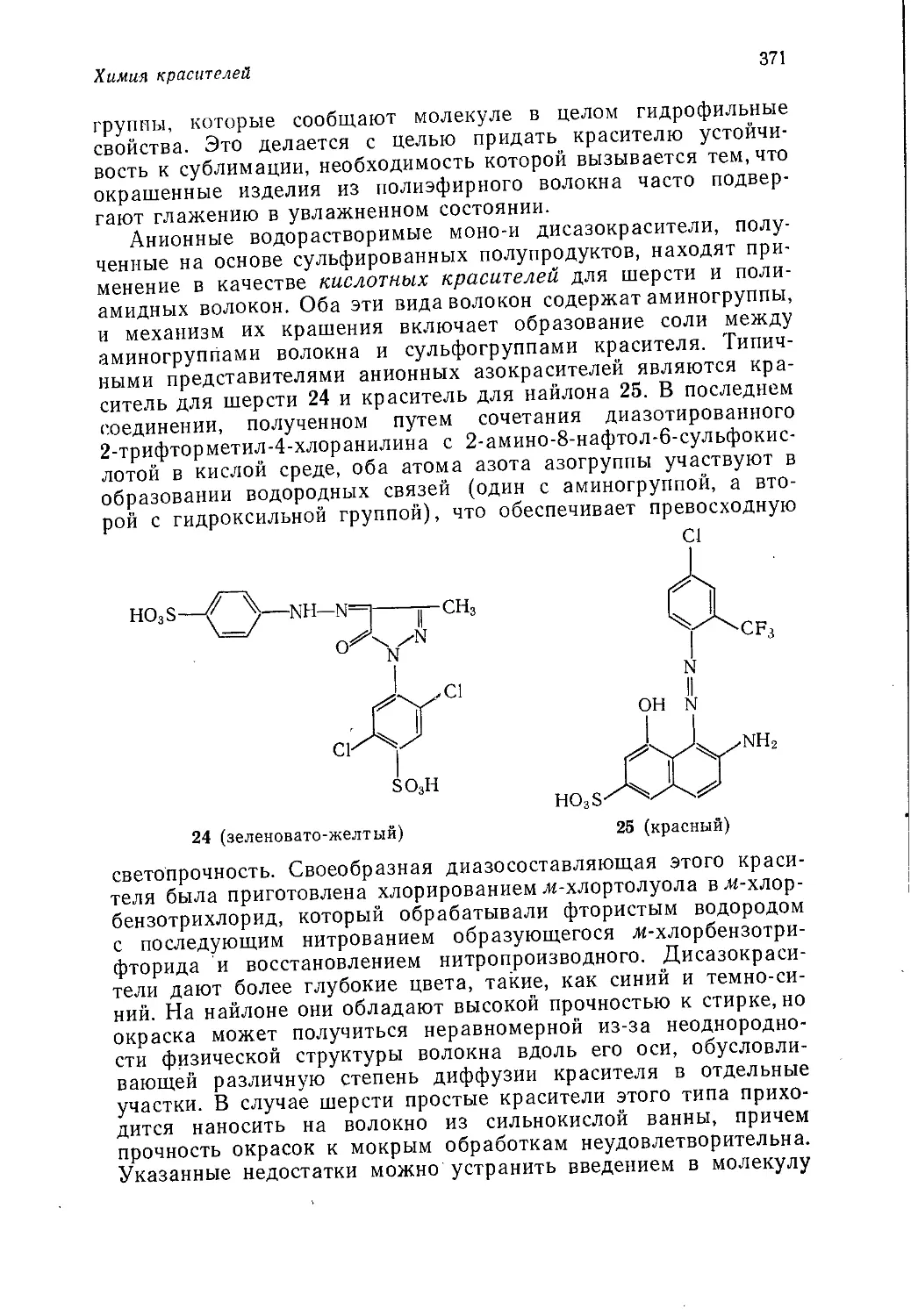
О. В. Корсунского
Издательство
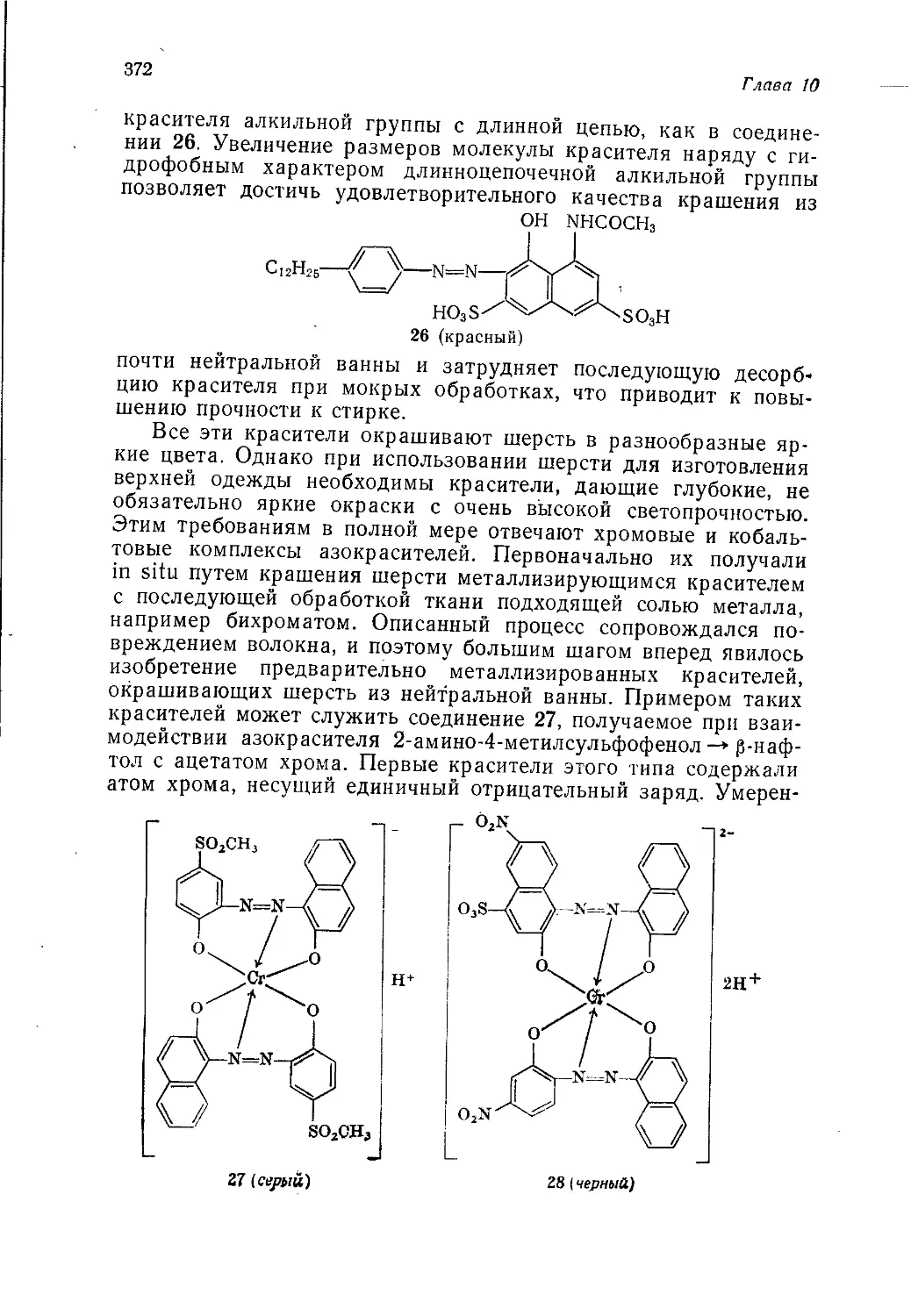
«Мир»
Москва
1977
УДК 547^.1
В книге, одним из авторов которой является крупный англий-
ский ученый Дж. Теддер, рассматриваются основы химии, на ко-
торых базируются промышленные процессы переработки органи-
ческого сырья — от переработки нефти до получения синтетиче-
ской пищи. Книга дает обобщенное представление о тенденциях
развития промышленности органической химии в странах Запад-
ной Европы, США и Японии.
Книга представляет интерес для химиков-органиков широкого
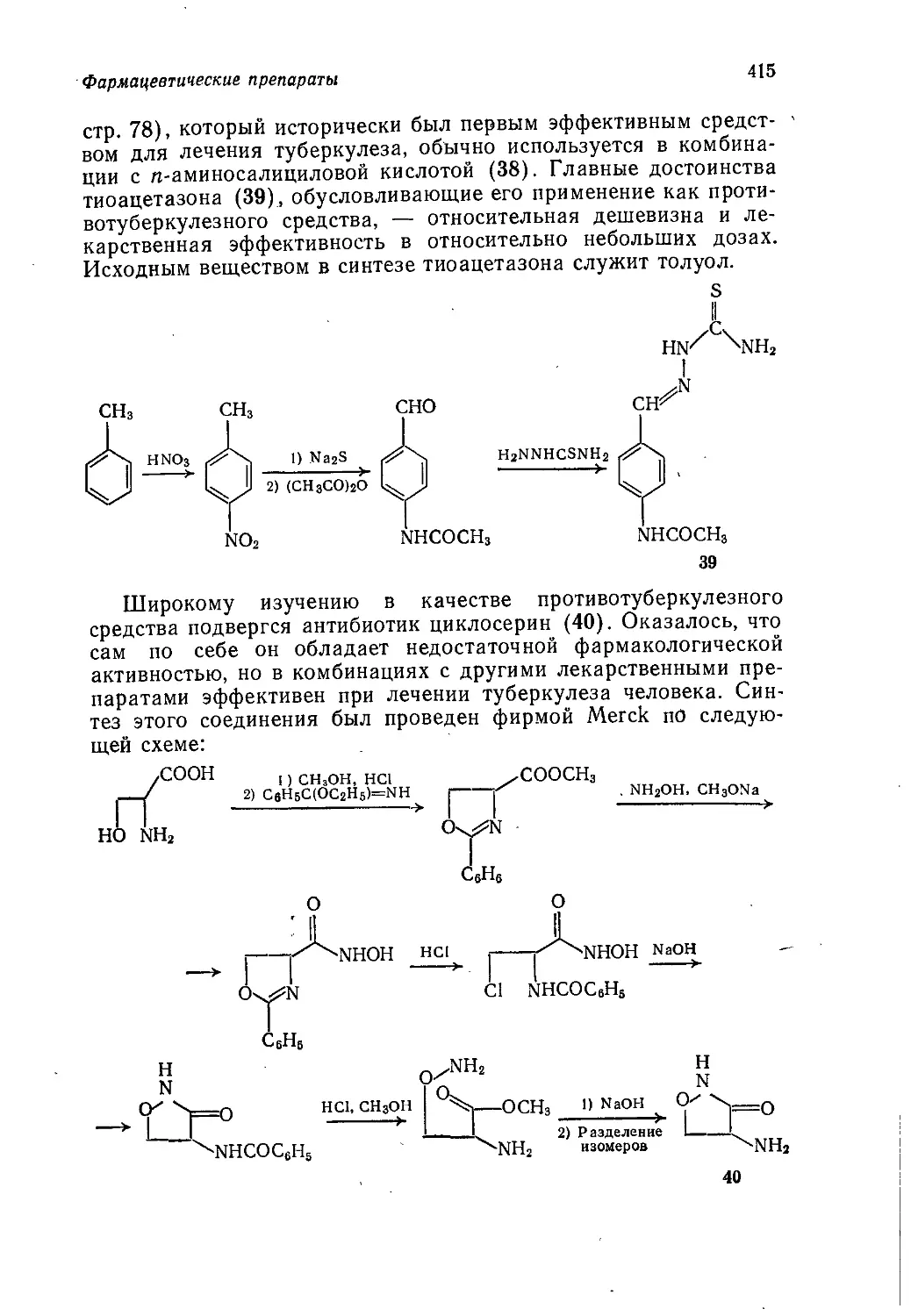
профиля — инженерно-технических и научных работников; она
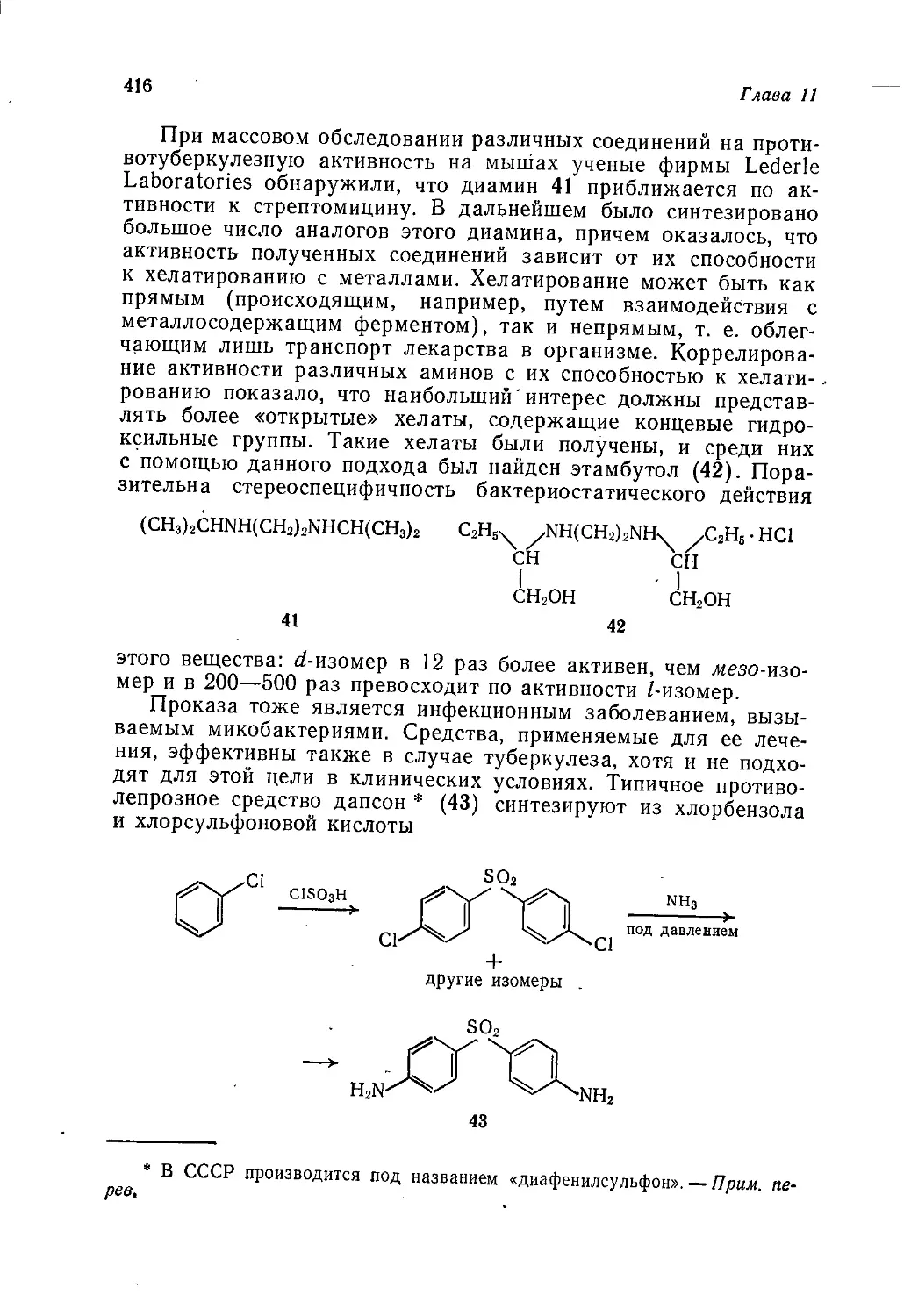
может быть рекомендована также как учебное пособие для пре-
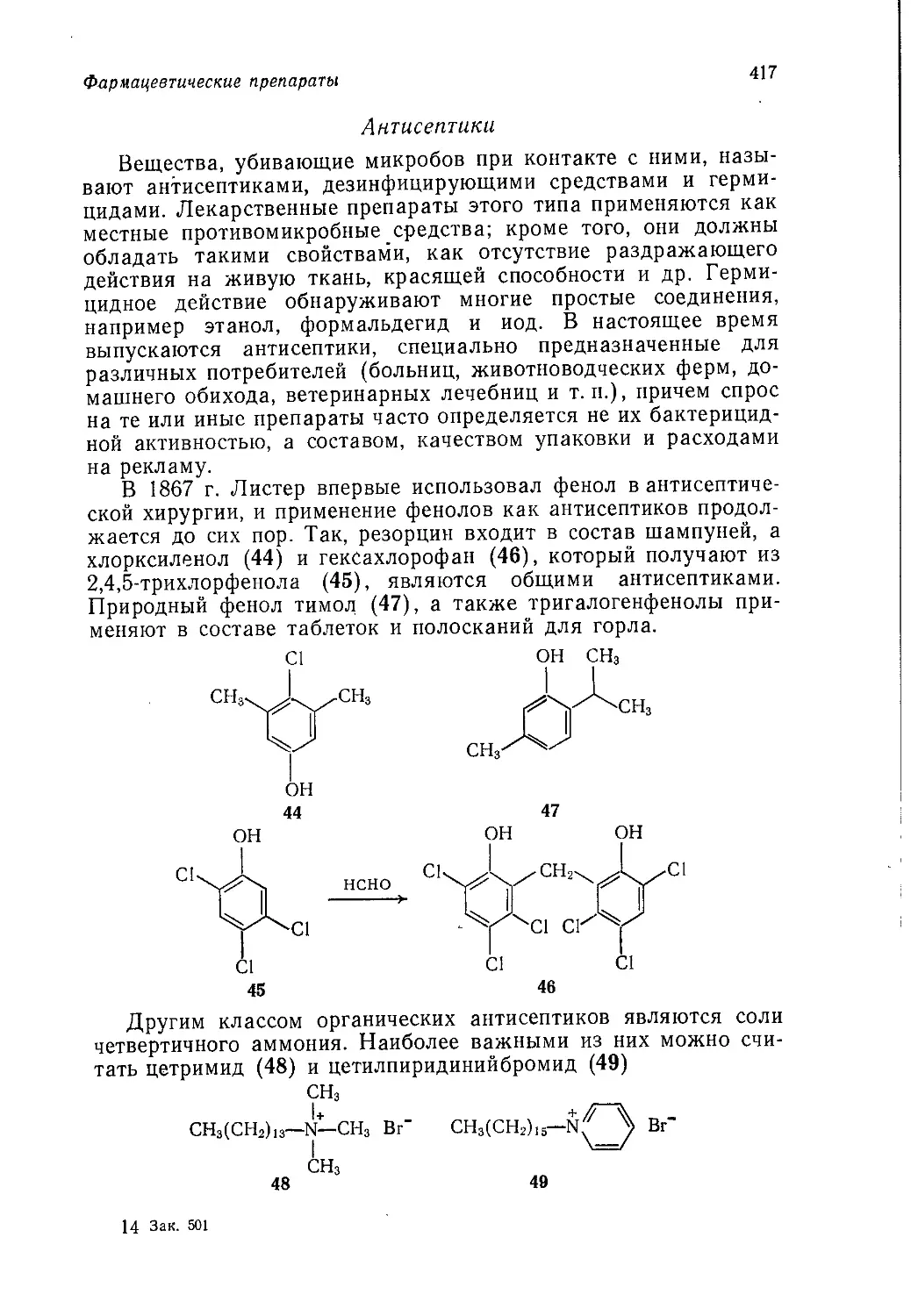
подавателей и студентов химике-технологических вузов.
48981
Редакция литературы по химии
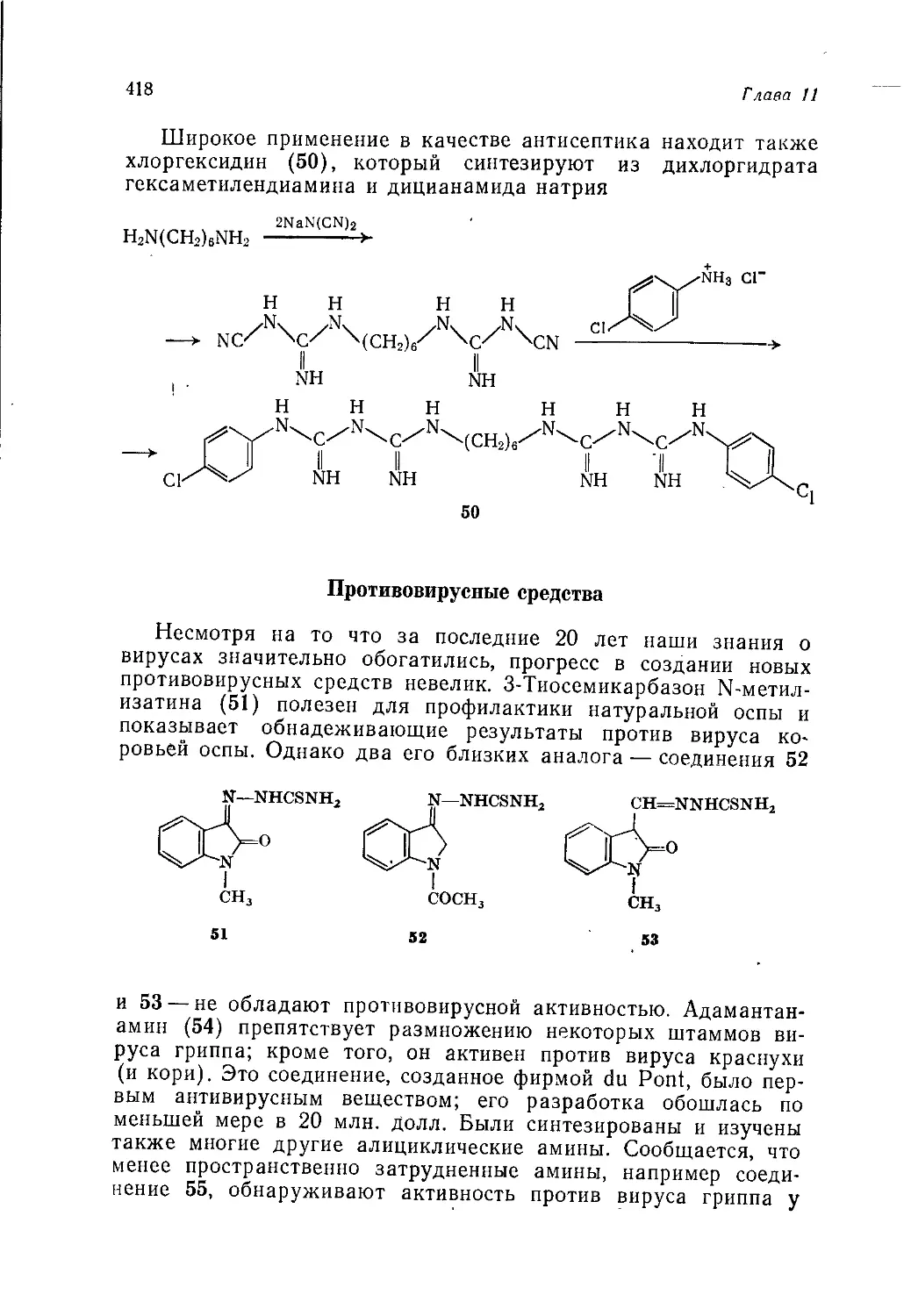
Copyright © 1975 by John Wiley and Sons Ltd.
All rights reserved. Authorized translation from English language
edition published by John Wiley & Sons, Inc.
© Перевод на русский язык, «Мир», 1977
Предисловие редактора перевода
Предлагаемая вниманию читателей книга является заклю-
чительным томом 5-томного издания, представляющего собой
учебное пособие по органической химии для университетов *.
Авторы данного тома ставили перед собой задачу ознакомить
студентов старших курсов университетов с проблемами прак-
тического использования достижений современной органиче-
ской химии, а также с работами промышленных фирм по созда-
нию новых синтетических материалов и различных продуктов,
имеющих большую промышленную ценность.
Диапазон рассматриваемых областей применения продуктов
органического синтеза необычайно широк и включает такие
редко встречающиеся в курсах общей химической технологии
разделы, как производство фреонов, химикатов для цветной и
черно-белой фотографии, душистых и вкусовых веществ, а так-
же использование продуктов и методов органического синтеза
в некоторых областях пищевой промышленности.
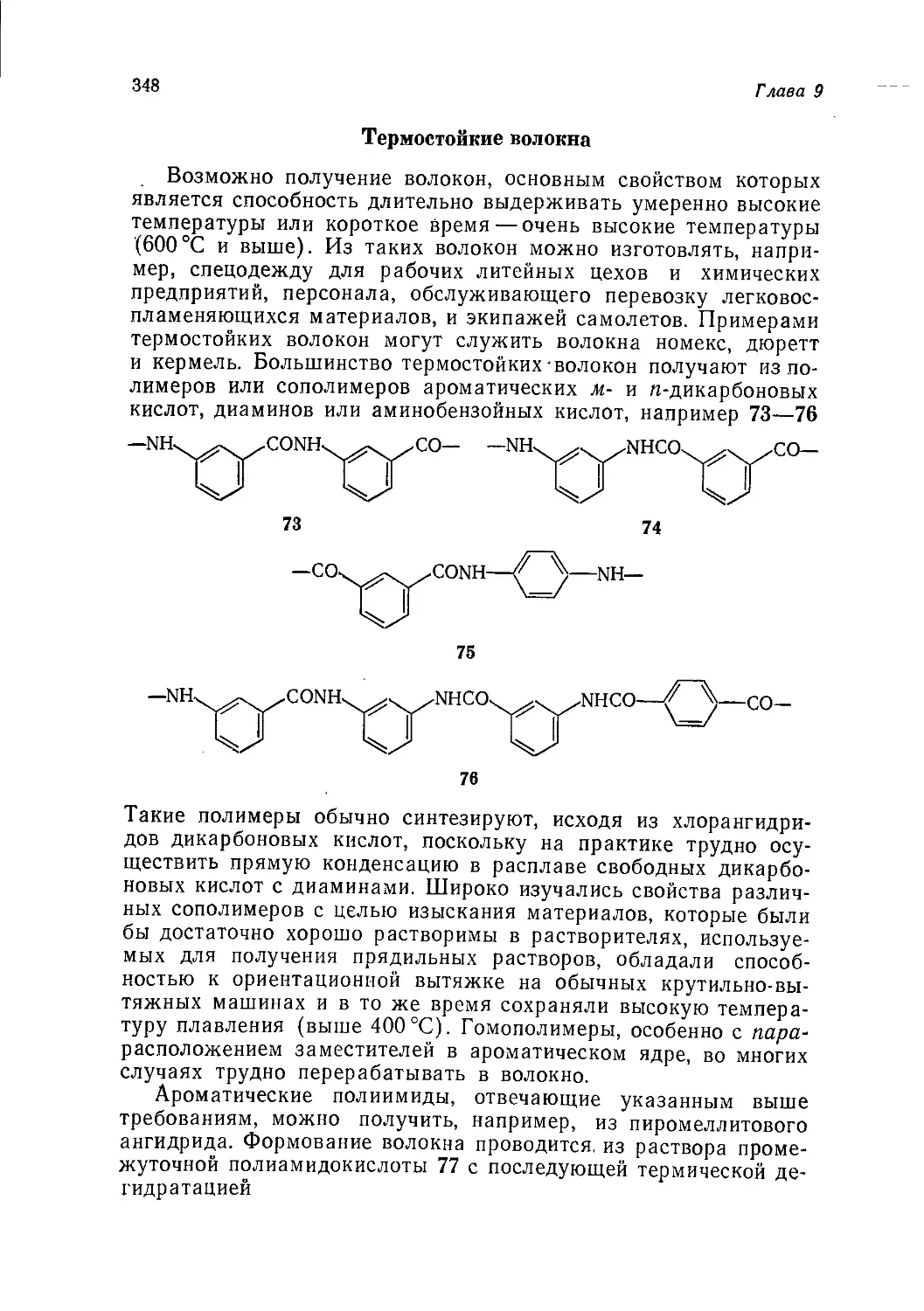
Наиболее полно разработаны четыре раздела, в которых
рассматриваются особенно быстро развивающиеся отрасли хи-
мической промышленности, являющиеся высокоприбыльными в
промышленно развитых капиталистических странах: производ-
ство продуктов тяжелого органического синтеза и пластических
масс на их основе, производство "'синтетических волокон, мою-
щих веществ и фармацевтических препаратов.
К составлению учебника были привлечены не только про-
фессора высших учебных заведений, но и ведущие специалисты
исследовательских центров некоторых основных английских
промышленных фирм, в том числе концернов ICI, British Petro-
leum, Unilever, и английского отделения американской фирмы
Minnesota Mining, которые написали для этой книги ряд спе-
циальных разделов. В результате такой совместной работы
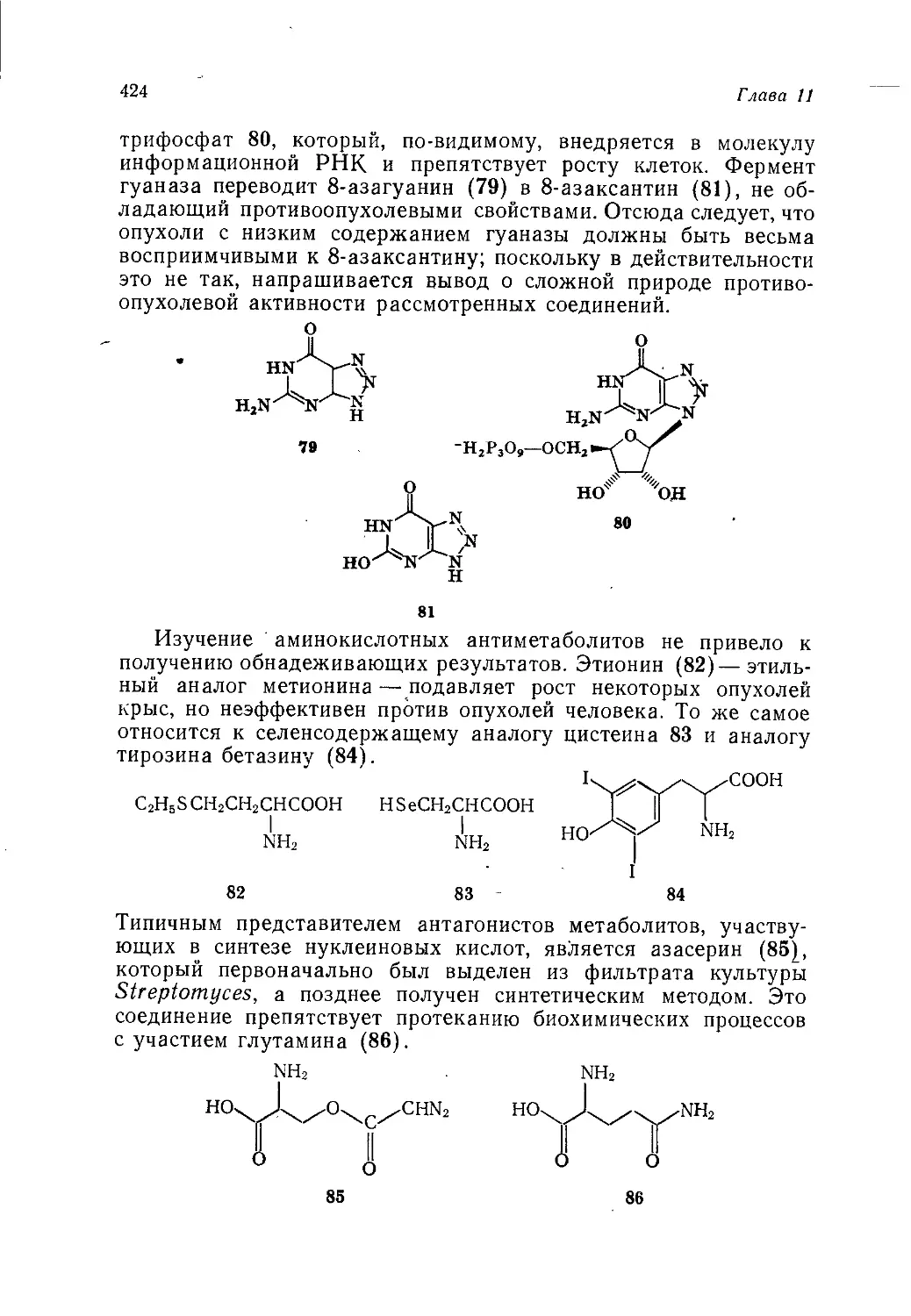
книга вышла за рамки только учебного пособия. Она в извест-
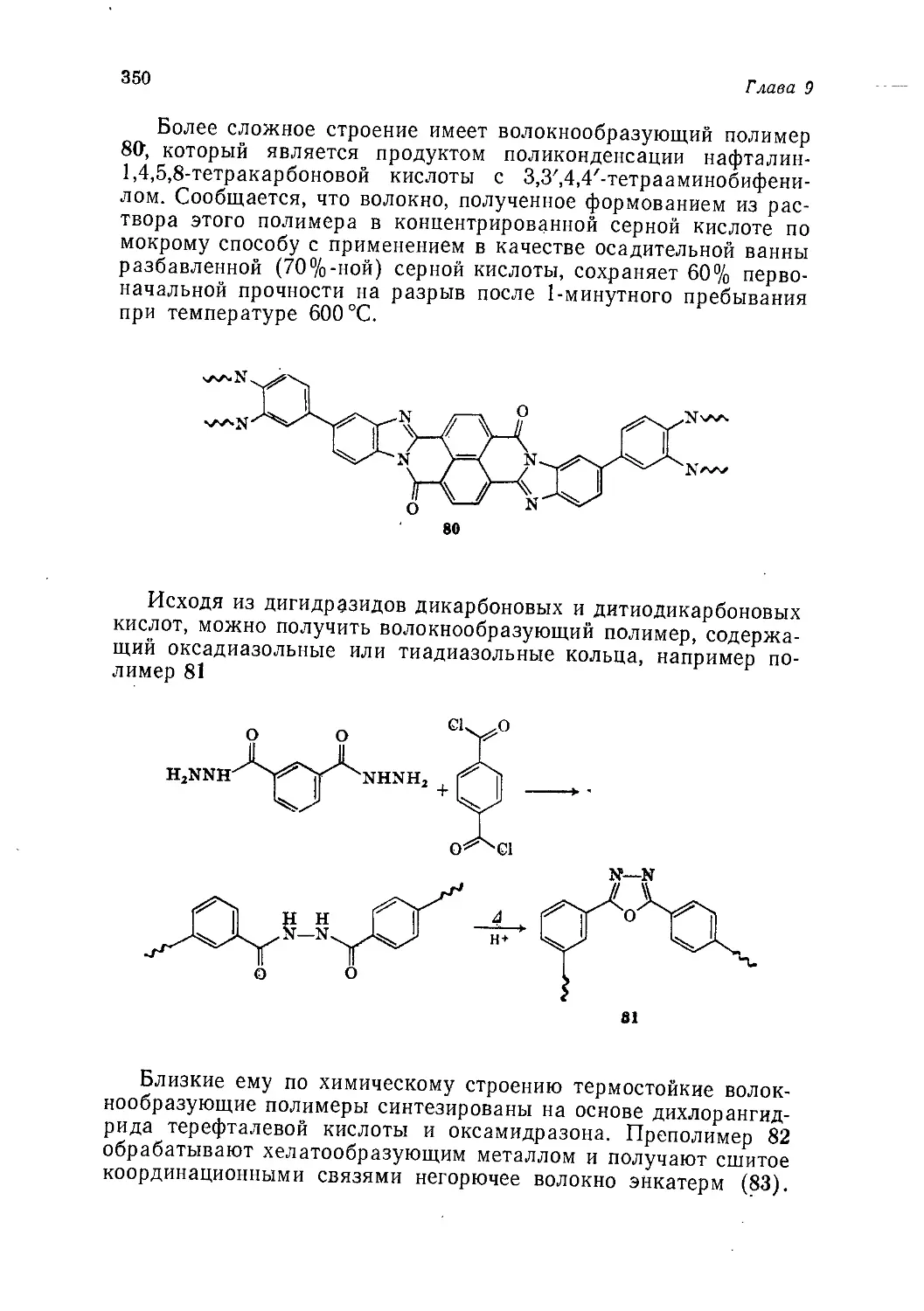
ной мере отражает концепции, установившиеся в английской
промышленности, взгляды на перспективы развития отдельных
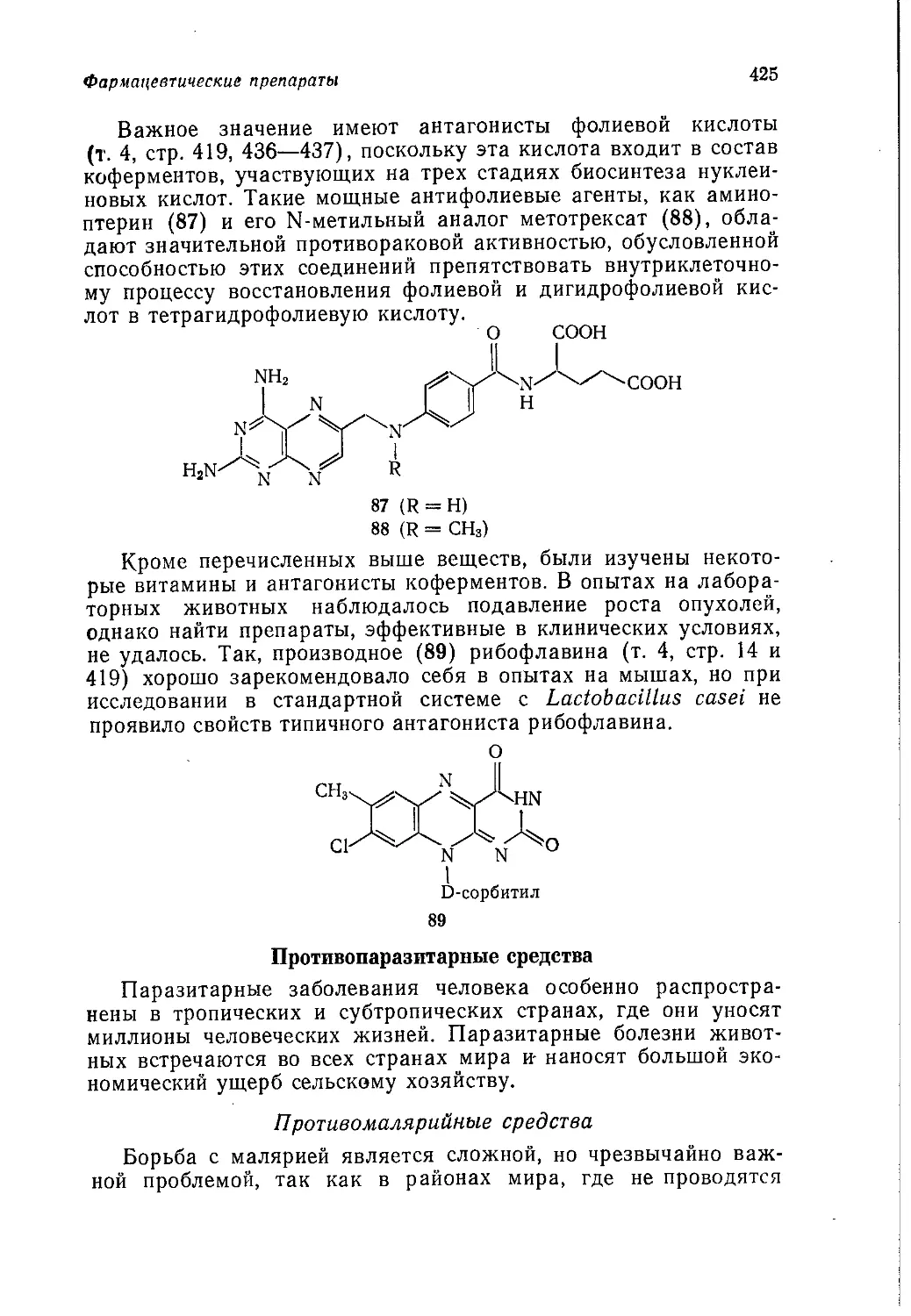
отраслей, на преимущества того или иного метода синтеза круп-
нотоннажных продуктов, желательные направления дальнейших
исследований, стоимость отдельных новых разработок. Так, по
* Первые четыре тома на русский язык не переведены.
в Предисловие редактора перевода
оценке английских специалистов, учитывая возросшие требова-
ния к защите окружающей среды, из 10 000 химических соеди-
нений, испытываемых в качестве средств химической защиты
сельскохозяйственных культур, практическое применение нахо-
дит лишь одно, а стоимость разработки такого препарата со-
ставляет в среднем 1 млн. фунтов стерлингов. Жесткие требо-
вания к тщательности и последовательности токсикологических
испытаний новых лекарственных средств увеличивают период
времени от открытия биологической активности нового соеди-
нения до выпуска его в продажу как лекарства в среднем до
7 лет.
Определенный интерес представляют оценки методов про-
мышленного производства продуктов тяжелого органического
синтеза, при наличии нескольких альтернативных путей их по-
лучения. Так, для производства фенола подтверждается об-
щепринятая точка зрения на преимущество кумольного метода,
хотя авторам известны технико-экономические подсчеты в пользу
итальянского процесса, где исходным продуктом является то-
луол, а также в пользу метода американской фирмы Scientific
Design, основанного на дегидрировании смеси циклогексанола
и циклогексанона. При рассмотрении различных методов полу-
чения капролактама выбор процесса синтеза ставится в зависи-
мость от местных условий. Оценивая различные пути получения
линейных а-олефинов, авторы указывают на такое важное пре-
имущество алюминийорганического синтеза из этилена, как воз-
можность регенерации исходного триэтилалюминия ректифика-
цией и возвращения его в цикл. Из многочисленных различных
методов получения ацетона наиболее эффективным считается
жидкофазное окисление пропилена в присутствии палладиевого
катализатора.
В любой книге отдельные разделы освещаются, как пра-
вило, не одинаково, что обусловливается научными интересами
автора и многими другими обстоятельствами. На наш взгляд,
в данной’книге недостаточно подробно освещено производство
синтетических каучуков, в результате чего не создается полного
представления о современном состоянии этой важной отрасли
нефтехимической промышленности. Весьма кратко и лишь на
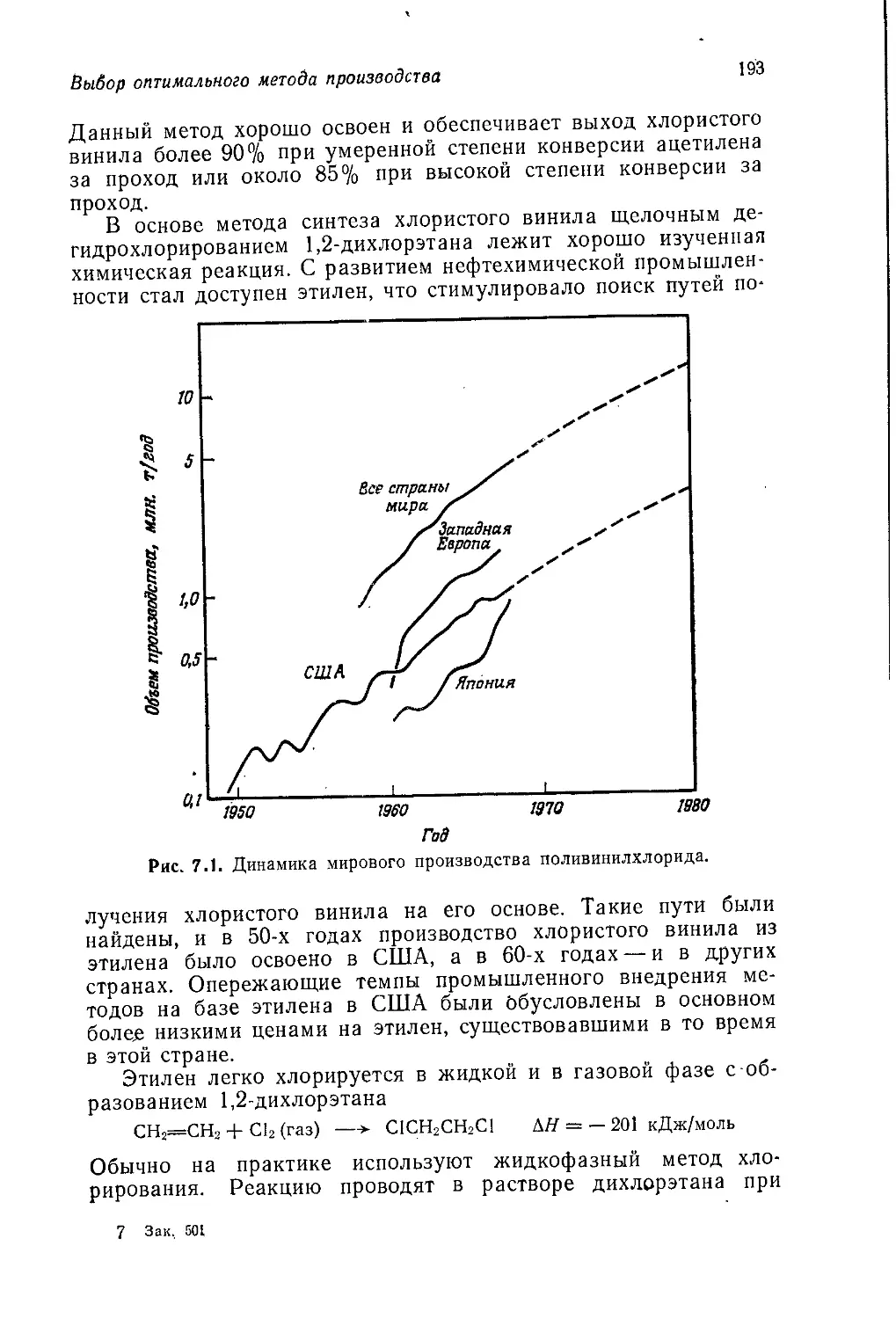
примере поливинилхлорида рассмотрена такая важная отрасль
промышленного органического синтеза, как производство и при-
менение стабилизаторов и других химикатов, необходимых в
производстве синтетических материалов на основе полимеров.
Однако эти недостатки отнюдь не умаляют достоинств книги
в целом. Она будет полезна не только студентам химических
высших учебных заведений, но и специалистам химической и
нефтехимической промышленности, работникам научно-иссле-
довательских институтов соответствующего профиля.
Предисловие редактора перевода
Следует отметить, что в ряде случаев авторы делают ссылки
на предыдущие тома этого издания. Мы сочли необходимым
оставить эти ссылки как указание на то, что в данном месте
авторы полагали целесообразным напомнить основные положе-
ния общего курса органической химии.
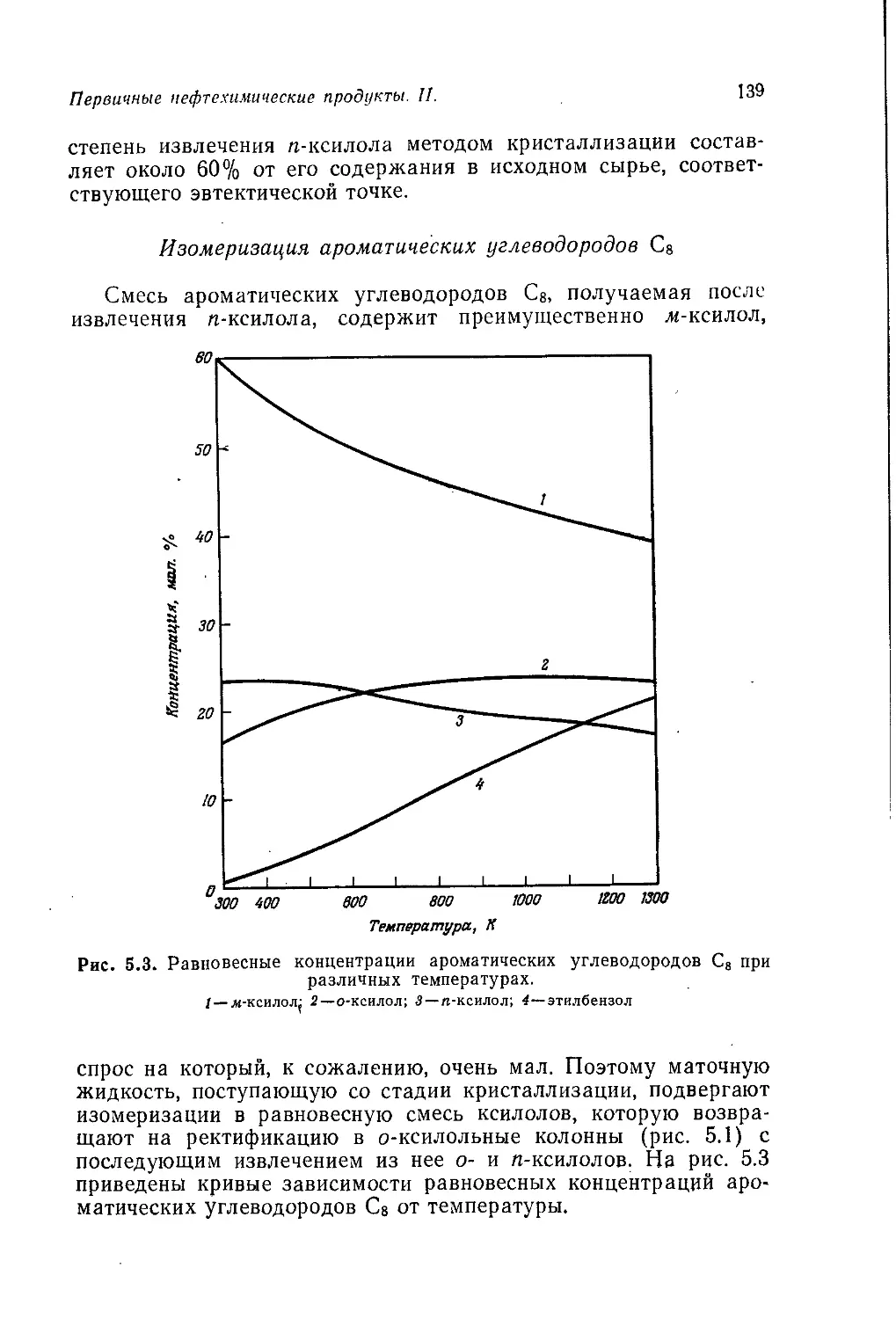
В книге приведен обширный статистический материал по
выпуску различных синтетических материалов и других химиче-
ских продуктов, главным образом в Великобритании и США.
Эти данные весьма полезны, они позволяют оценить масштабы
современной химической промышленности и сопоставить прак-
тическое значение отдельных химических продуктов. Однако
вышедшая в свет в Англии в 1975 г. книга содержит статисти-
ческие сведения, относящиеся лишь к 1972 г., которые для та-
кой высокодинамичной отрасли промышленности, как производ-
ство продуктов органического синтеза, в настоящее время уже
значительно устарели. Переводчиком книги проделана большая
работа по подбору более современного статистического мате-
риала, который, по согласованию с автором — проф. Дж. Тедде-
ром, приводится в соответствующих разделах в примечаниях
с ссылкой на источники.
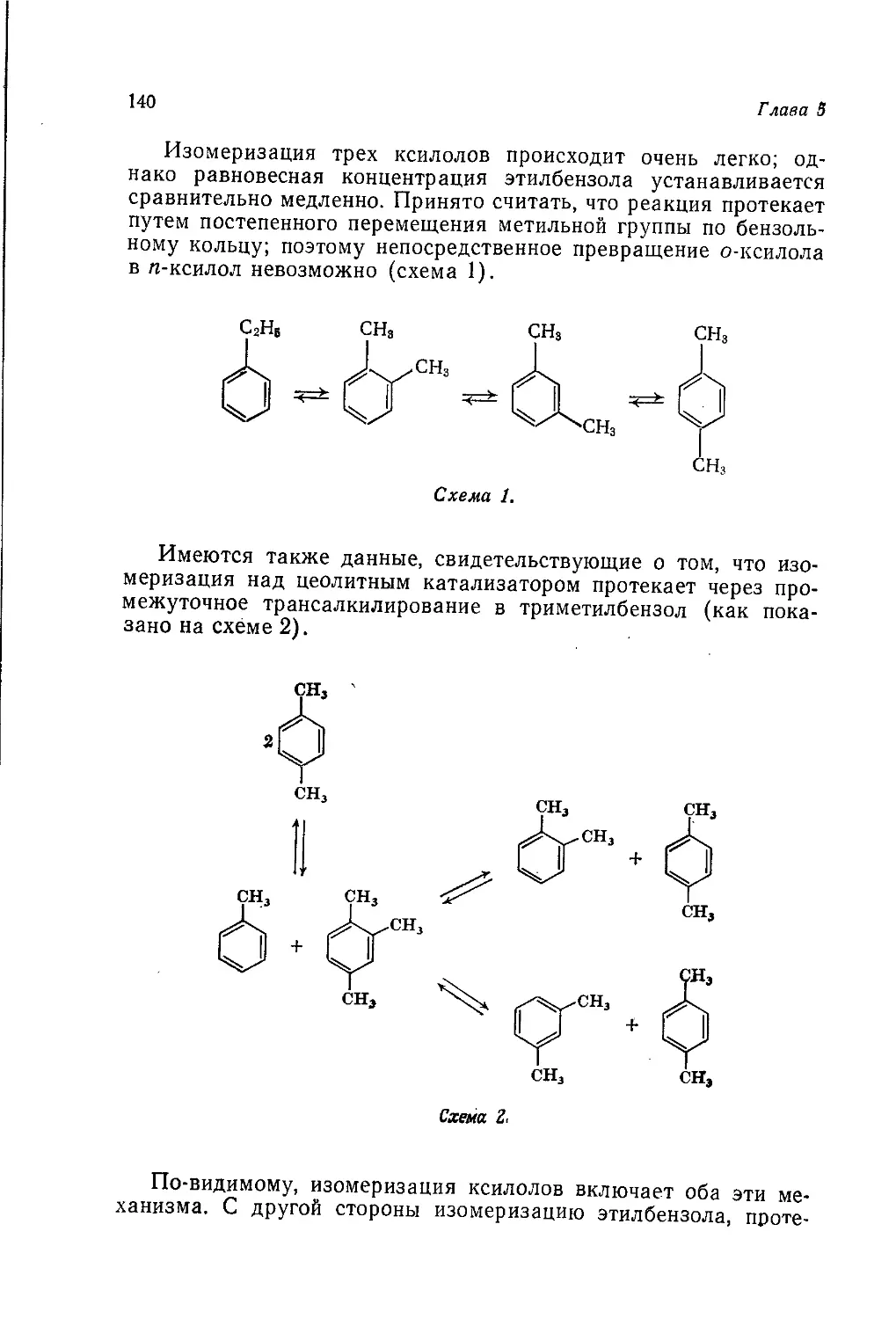
Кроме того, в дополнение к перечню литературы, рекомен-
дованной английскими авторами, приведен список работ совет-
ских исследователей по рассматриваемым в книге проблемам,
вышедших в последние годы.
О. Корсунский
Предисловие к русскому изданию
Вероятно, ни одна отрасль науки не оказывает такого боль-
шого влияния на жизнь современного человека, как химия. Мы
едим пищу, в цикле получения которой участвуют химиче-
ские удобрения и инсектициды, носим одежду, волокно для ко-
торой вырабатывают на химических предприятиях, и вообще
почти все, чем мы пользуемся в повседневной жизни, так или
иначе имеет отношение к химической промышленности. Даже
эту книгу нельзя было бы издать без помощи химии. Студенту
приходится овладевать таким огромным объемом академиче-
ских знаний, что вопросы их практического применения часто
упускаются из виду. Мы надеемся, что эта книга поможет сту-
денту лучше понять ту важную роль, которую играет химия
в повседневной жизни людей. Нам особенно приятно, что эта
книга будет издана на русском языке и ею смогут пользоваться
наши коллеги из Советского Союза.
Дж. М. Т.
Предисловие авторов
Данная книга является пятым, последним томом учебного
пособия по органической химии для химических факультетов
университетов, которое рассчитано на студентов с разной сте-
пенью подготовки — от первокурсников до выпускников. Тома.
1—3 представляли собой введение в органическую химию с точ-
ки зрения механизмов реакций. В т. 4 был изложен дополни-
тельный материал по химии природных соединений. Данный том
завершает эту серию обсуждением методов получения и свойств
промышленных продуктов органического синтеза.
Том 1 целиком воспроизводил лекционный курс. Этот курс
был подготовлен в связи с тем, что во всех существовавших
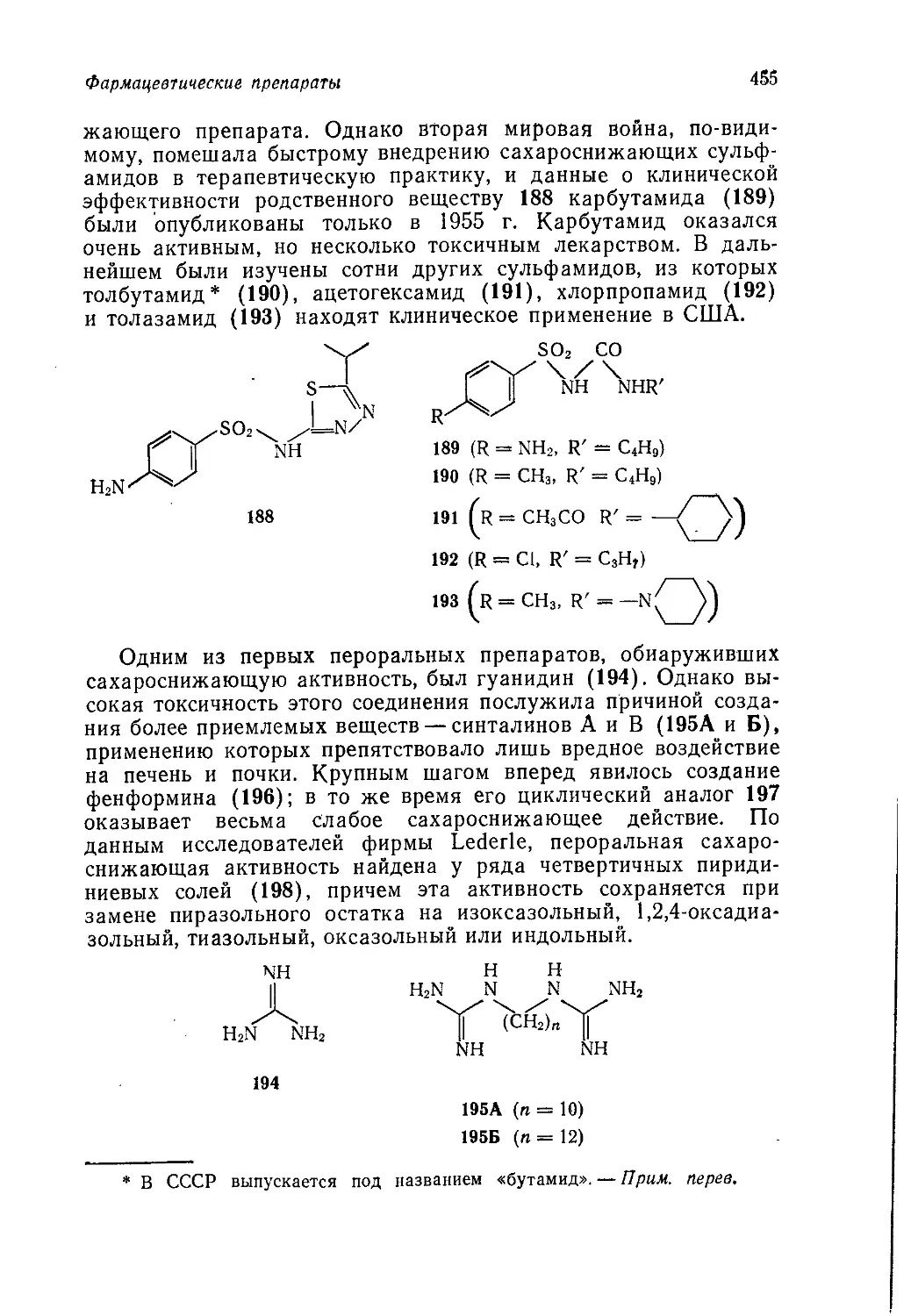
тогда учебниках органическая химия излагалась по традицион-
ной схеме «получение и свойства». Лекции и написанная по их
материалам книга имели целью облегчить понимание существа
предмета, а не просто дать студенту сумму фактов, которые он
должен выучить наизусть. За 6 лет, прошедших со времени на-
писания этой книги, произошли коренные изменения в методике
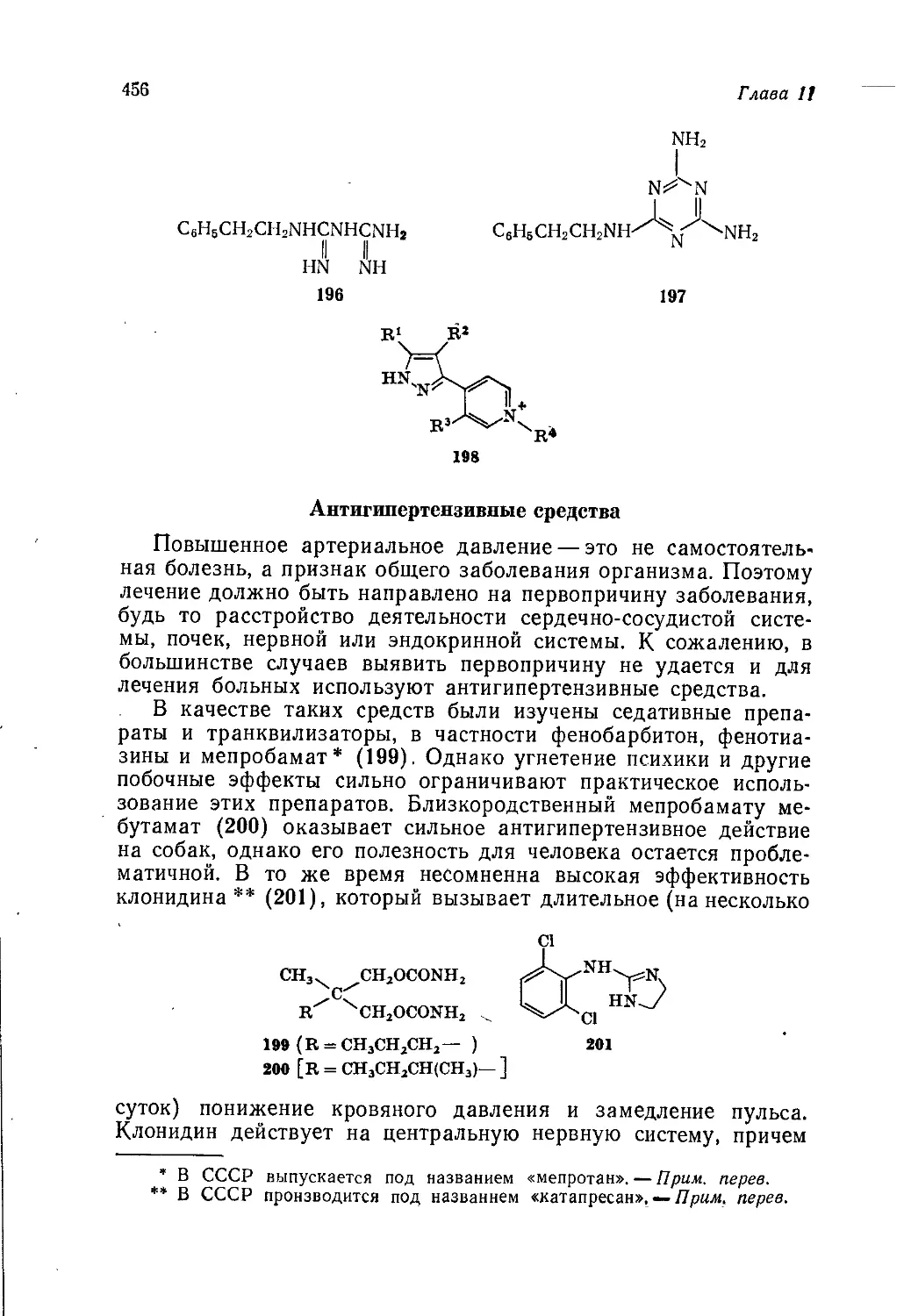
преподавания органической химии. Подход к излагаемому мате-
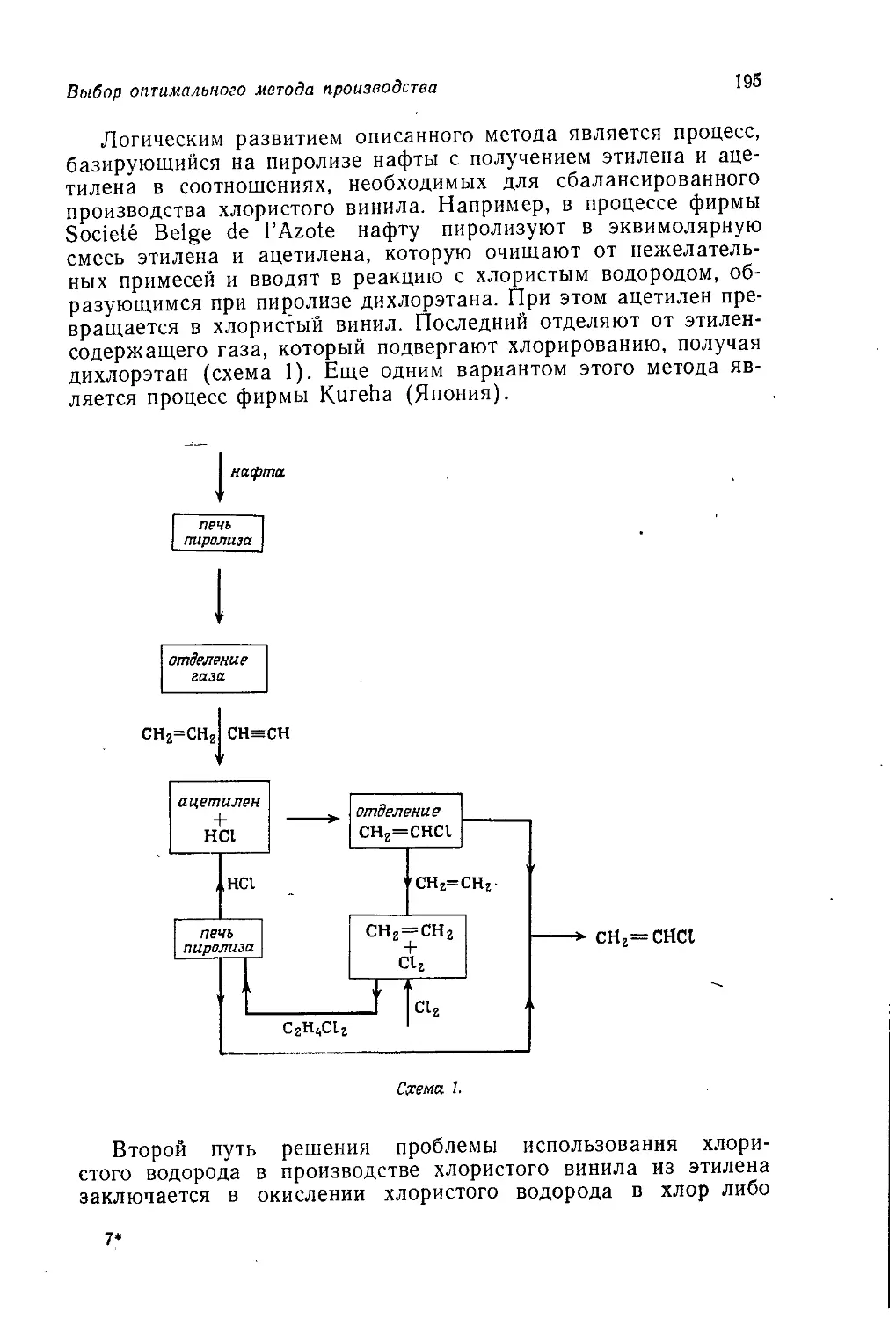
риалу с точки зрения механизмов реакций, который раньше был
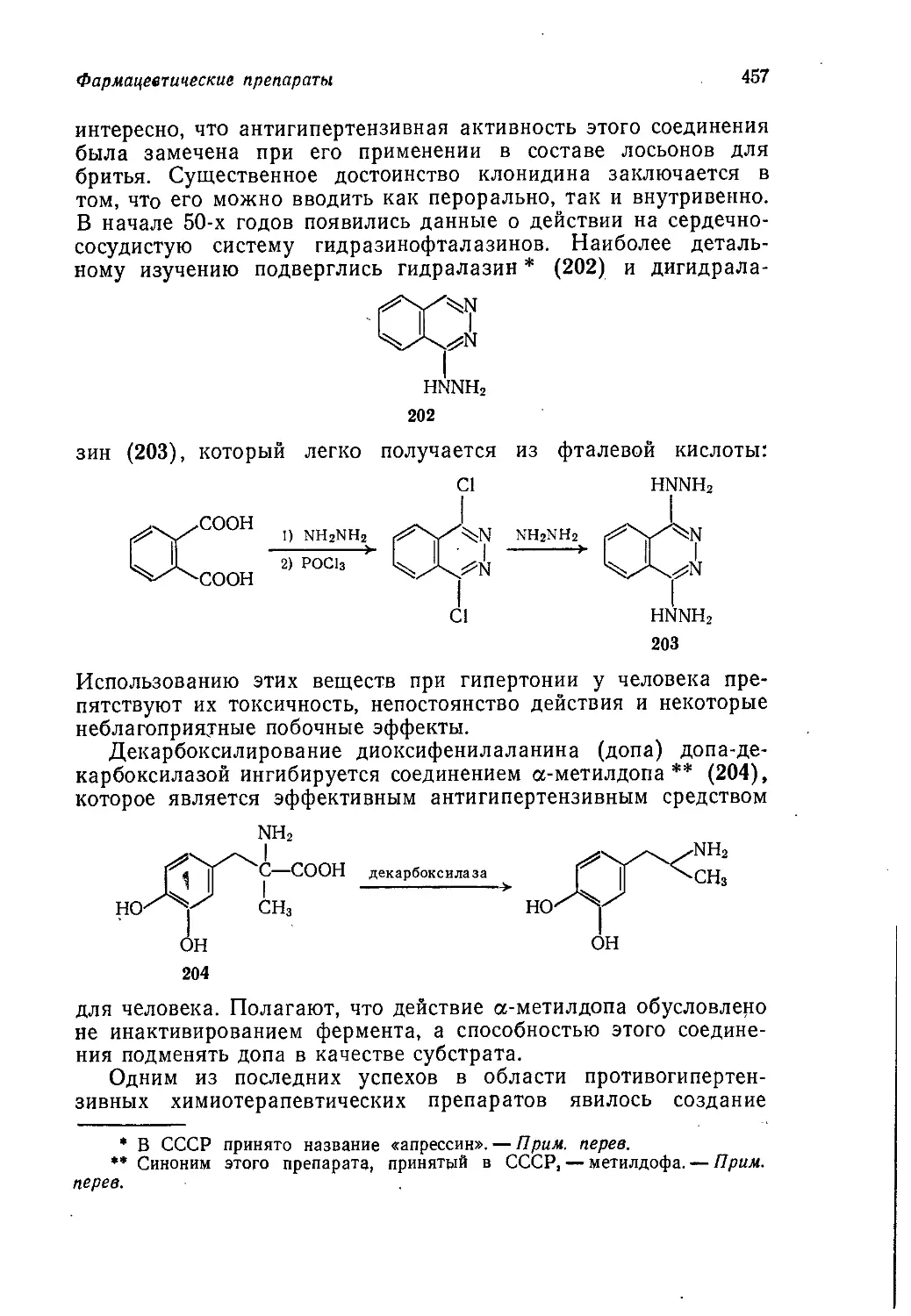
исключением, стал общепринятой нормой. Как и всякая мода,
такой подход может быть доведен до крайности; некоторые
опасные последствия чрезмерного увлечения им были отмечены
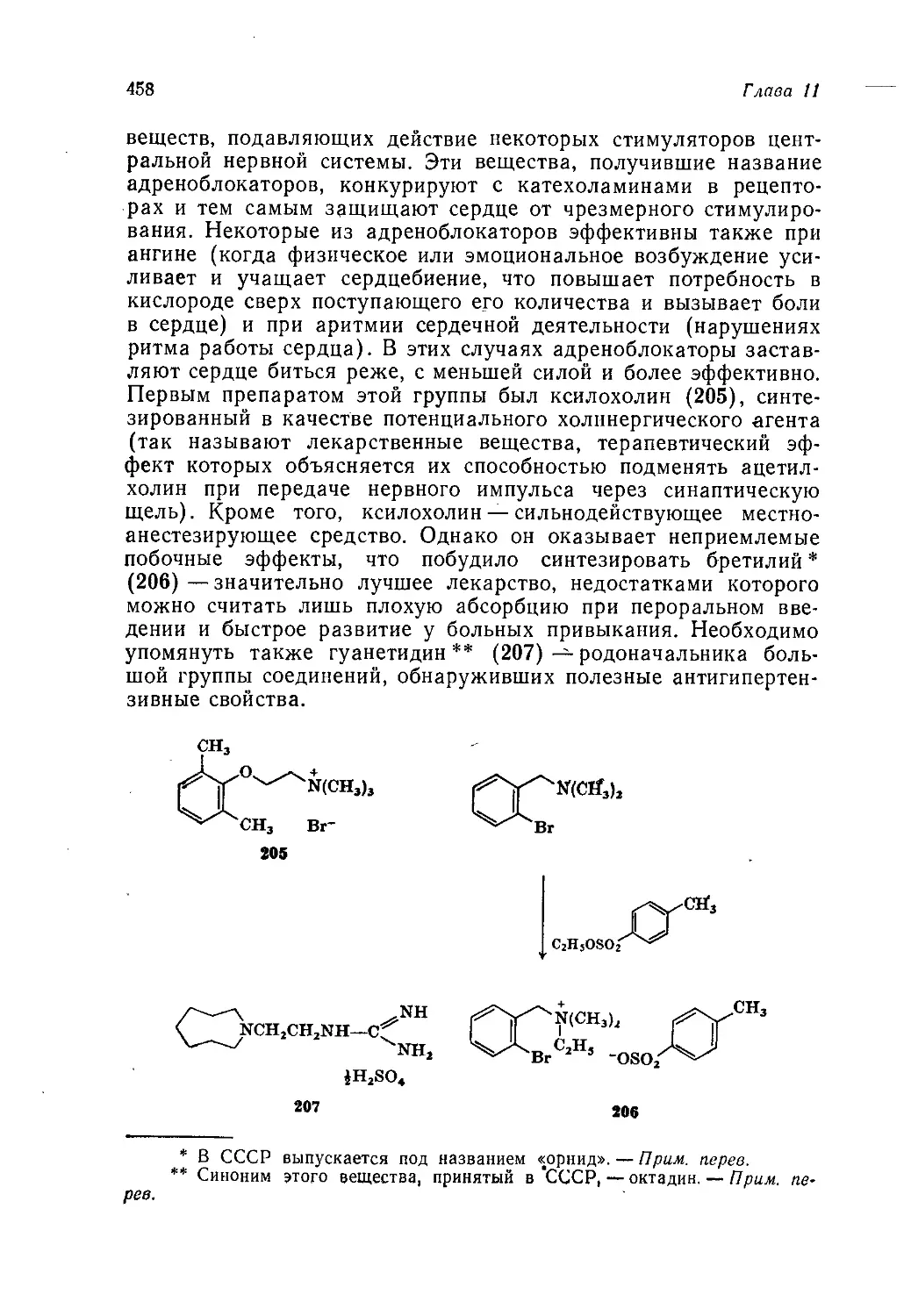
в предисловии к английскому изданию т. 2. Однако почти неиз-
бежным результатом универсального использования этого под-
хода является отказ от рассмотрения промышленных процессов,
если они не могут служить иллюстрацией какой-либо химиче-
ской реакции или химического явления. Поскольку многие про-
мышленные процессы протекают по очень сложному механизму
(иногда не вполне ясному) и часто являются гетерогенными,
нынешние выпускники университетов имеют очень смутное
представление о химической промышленности. Правда, есть
причина, по которой отсутствие промышленных процессов в лек-
ционном курсе для студентов последнего года обучения даже
желательно. Сейчас много спорят о недостатках преподавания
химии в университетах, но в целом преподаватели читают свои
лекции со знанием дела, даже если они порой неправильно трак-
туют отдельные вопросы. Однако большинство преподавателей
плохо знакомо с последними достижениями химической промыш-
ленности, и поэтому содержащиеся в их лекциях сведения по про-
мышленной органической химии, в отличие от сведений по акаде-
мическим вопросам, зачастую являются неточными и устарев-
шими. В то же время никак нельзя считать нормальным такое
положение, когда выпускники оказываются совершенно неосве-
домленными о практическом применении органической химии.
Предлагаемый читателю т. 5 «Промышленная органическая хи-
мия» представляет собой попытку исправить это положение,
10
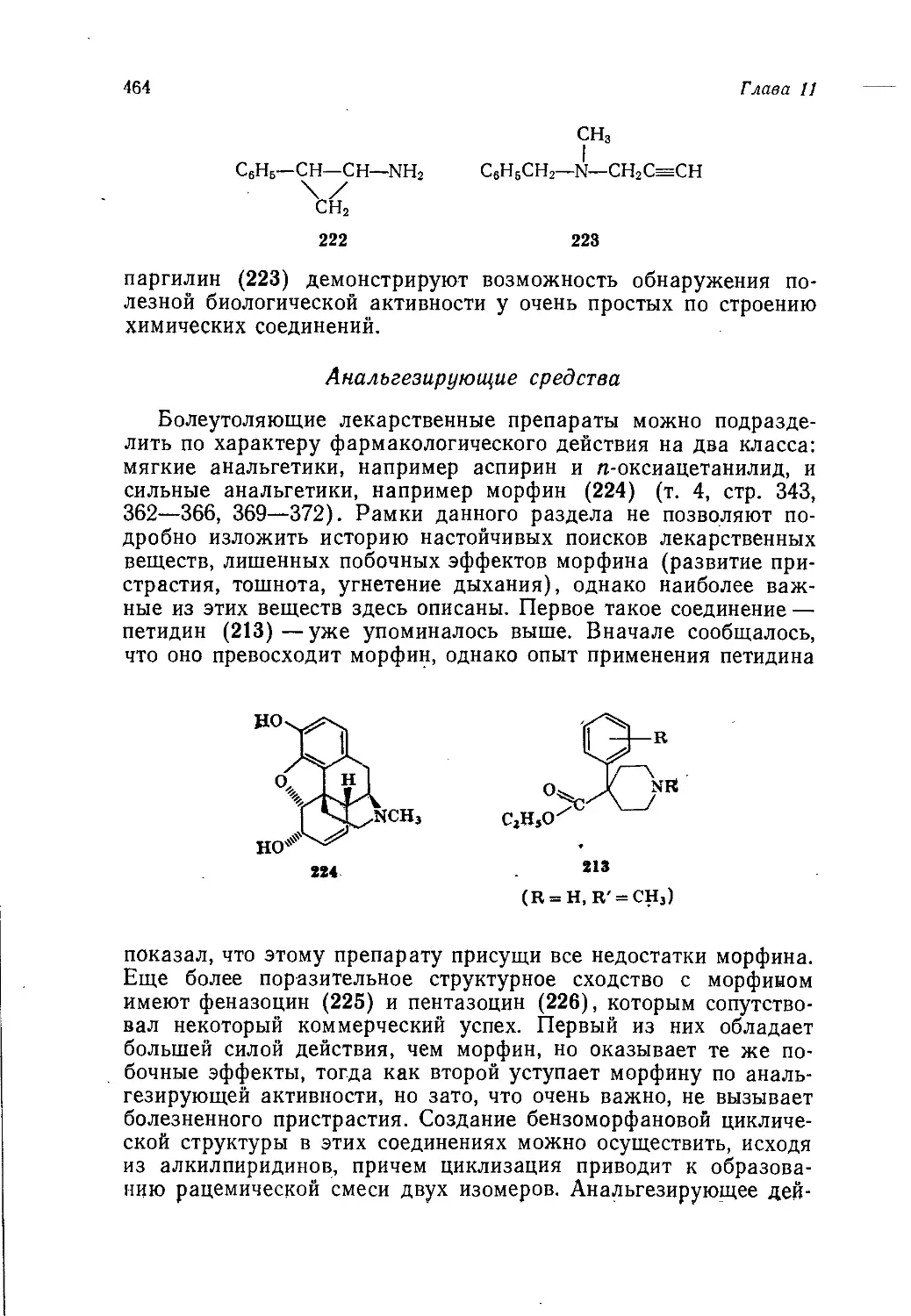
Предисловие авторов
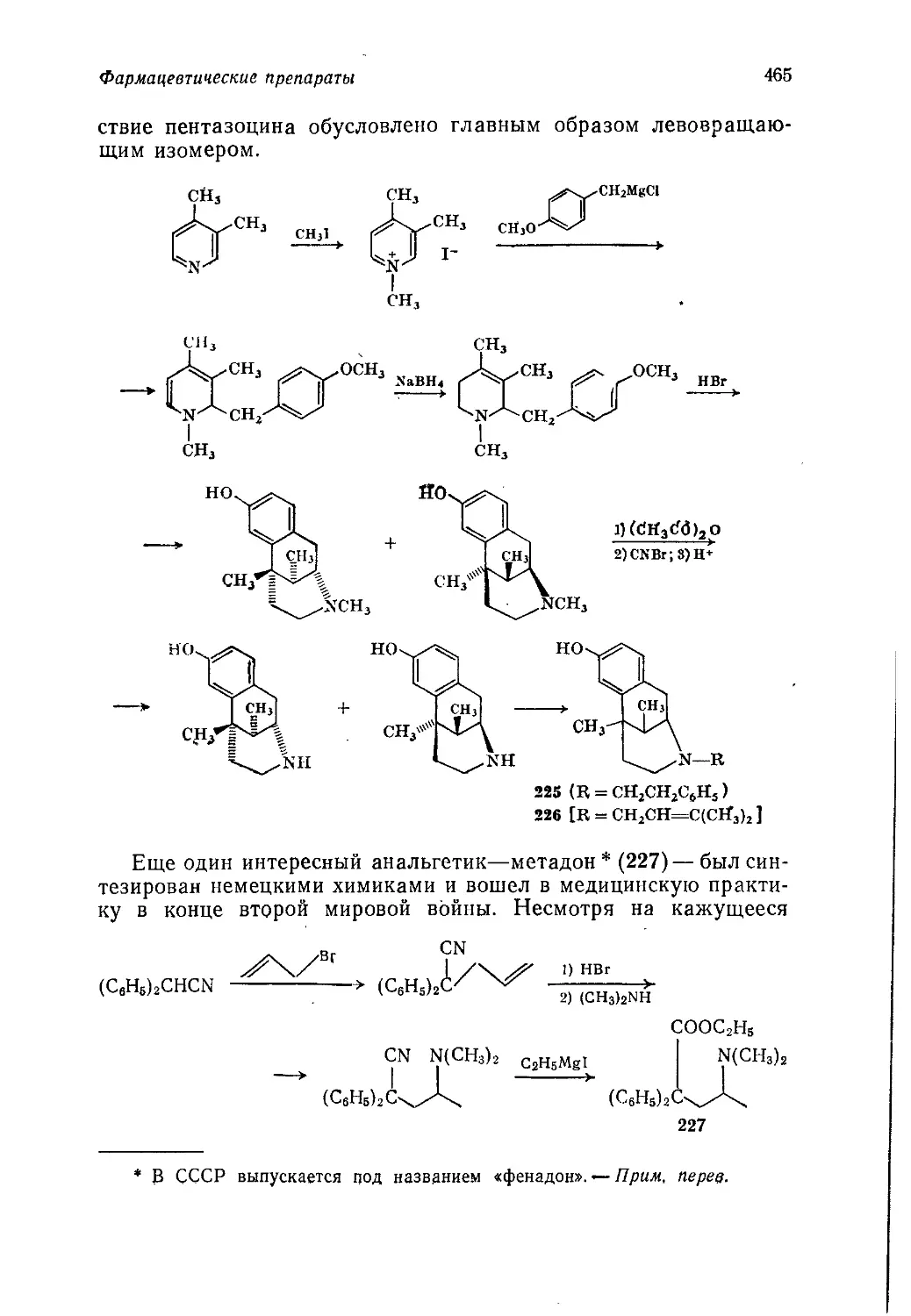
Книга начинается с введения, в котором показано, как сле-
дует увязывать технические аспекты промышленного производ-
ства химических продуктов с экономическими и социологиче-
скими аспектами. Гл. 2 посвящена видам сырья, на которых
базируется промышленность органического синтеза. В~ последу-
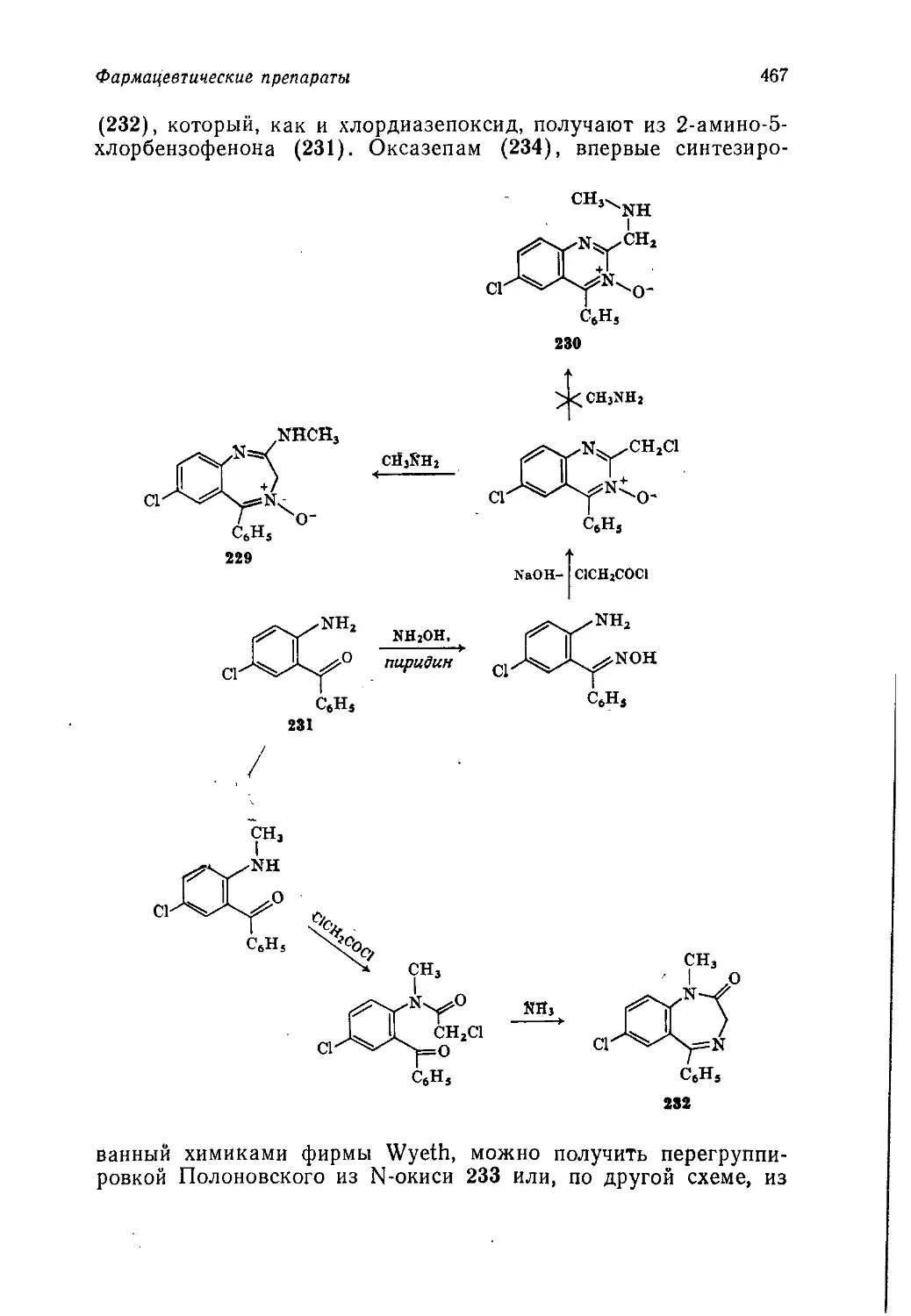
ющих четырех главах рассматриваются процессы получения
первичных органических продуктов, включая полупродукты для
производства пластмасс, волокон и др. Все эти пять глав
(гл. 2—6) относятся преимущественно к нефтехимической про-
мышленности. В гл. 7 обсуждаются различные факторы, кото-
рые принимают во внимание при выборе оптимального техно-
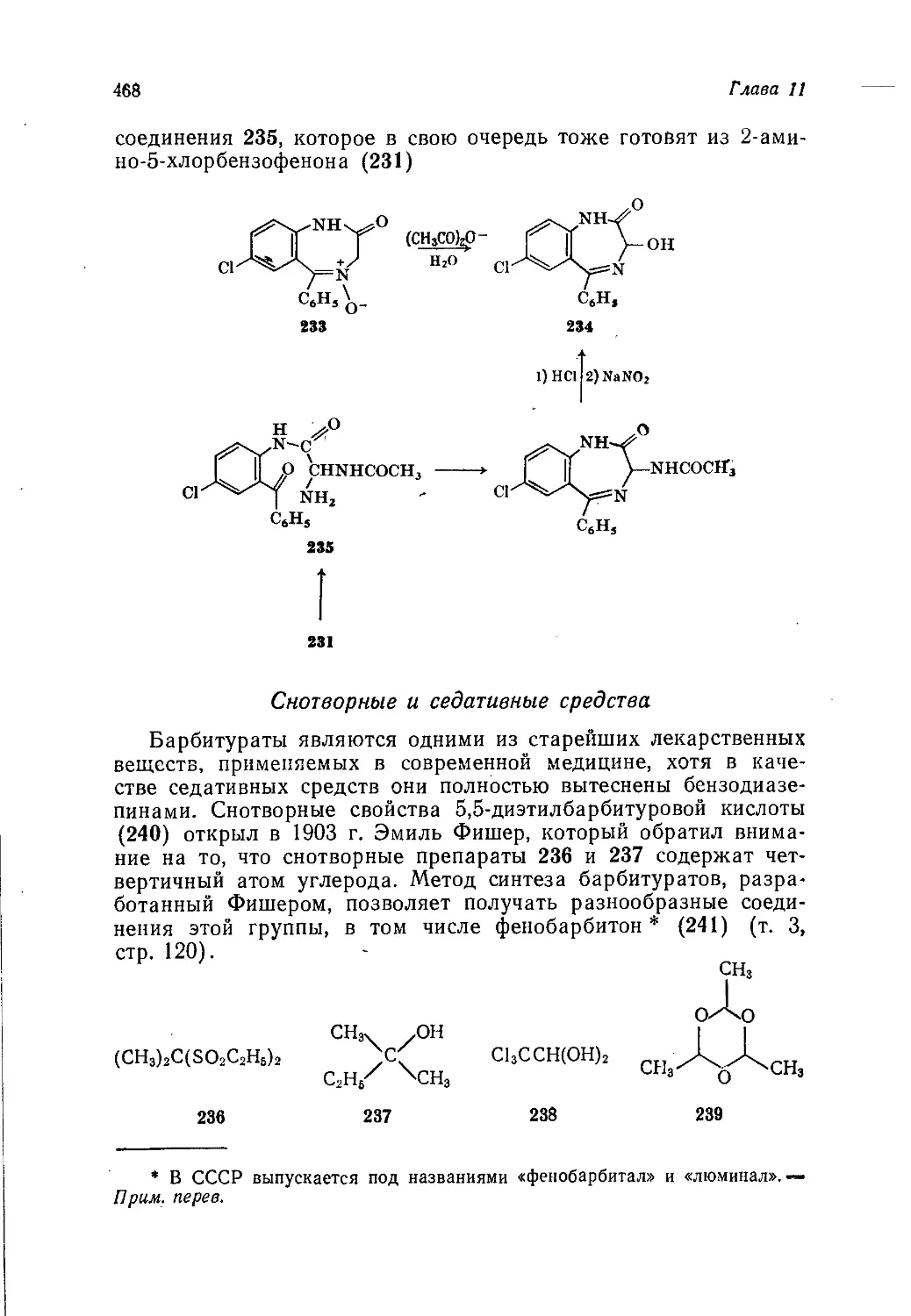
логического процесса.
Начиная с гл. 8 основное внимание переключается с процес-
сов получения тех или иных химических продуктов на сами эти
продукты. В гл. 8 и 9 рассматриваются полимерные материалы
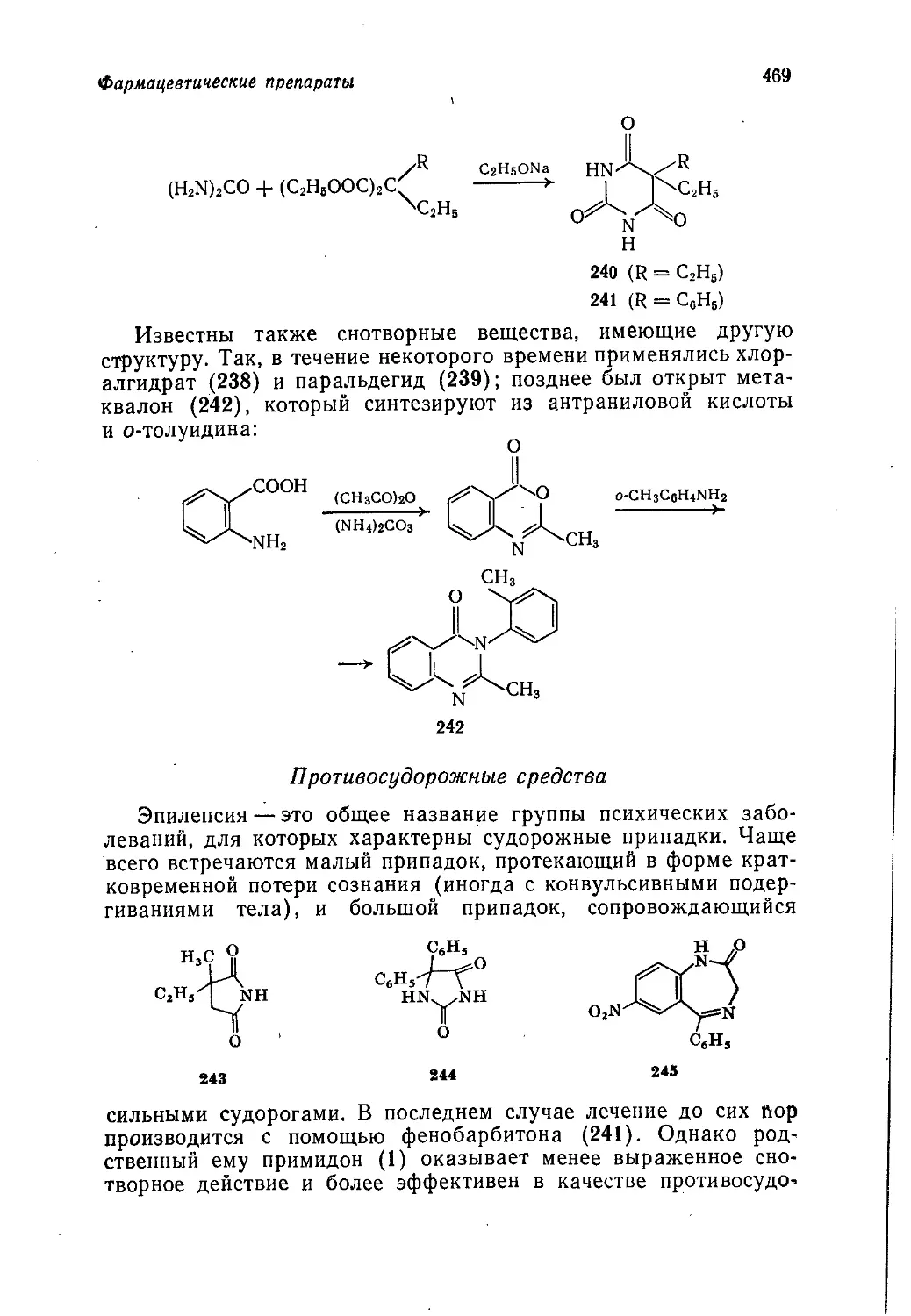
(пластмассы и химические волокна), а в следующих главах —
красители, фармацевтические препараты, органические химика-
ты для сельского хозяйства и моющие средства. Далее обсуж-
даются органические продукты, с которыми студент уже встре-
чался в тт. 1—4. В. гл. 14 описаны топлива и взрывчатые
вещества, а в гл. 15 и 16 —разнообразные химикаты, получае-
мые из природного сырья, а именно пищевые, вкусовые и души-
стые вещества. Наконец, в гл. 17 кратко рассмотрены органиче-
ские соединения, применяемые в фотографии, и фторуглеводо-
роды, которые используют как пропелленты для аэрозолей
и хладоагенты.
Многие аспекты прикладной органической химии не вошли
в эту книгу, однако- мы считаем, что в ней представлены все
наиболее важные процессы и продукты промышленности орга-
нического синтеза.
Весьма вероятно, что эта книга прежде всего привлечет вни-
мание студентов, интересующихся прикладной химией. Мы на-
деемся, что она будет полезна для них, однако должны пре-
дупредить, что эта книга рассчитана на подготовленного чита-
теля, хорошо знающего так называемую «чистую» химию. Мы
полагаем, что изложенный в ней материал будет способствовать
формированию у студента более широкого химического круго-
зора. По нашему убеждению, выпускник, не знающий промыш-
ленной химии, подобен человеку, который считает себя артистом,
но никогда не играл на сцене и не поставил ни одной пьесы.
Как и остальные тома этого учебного пособия, данная книга
предназначена для дополнительного чтения. Хочется верить, что
все изложенное в ней покажется студенту столь же интересным
и запоминающимся, как и нам.
Дж. Теддер, А. Нехватал, А. Джубб
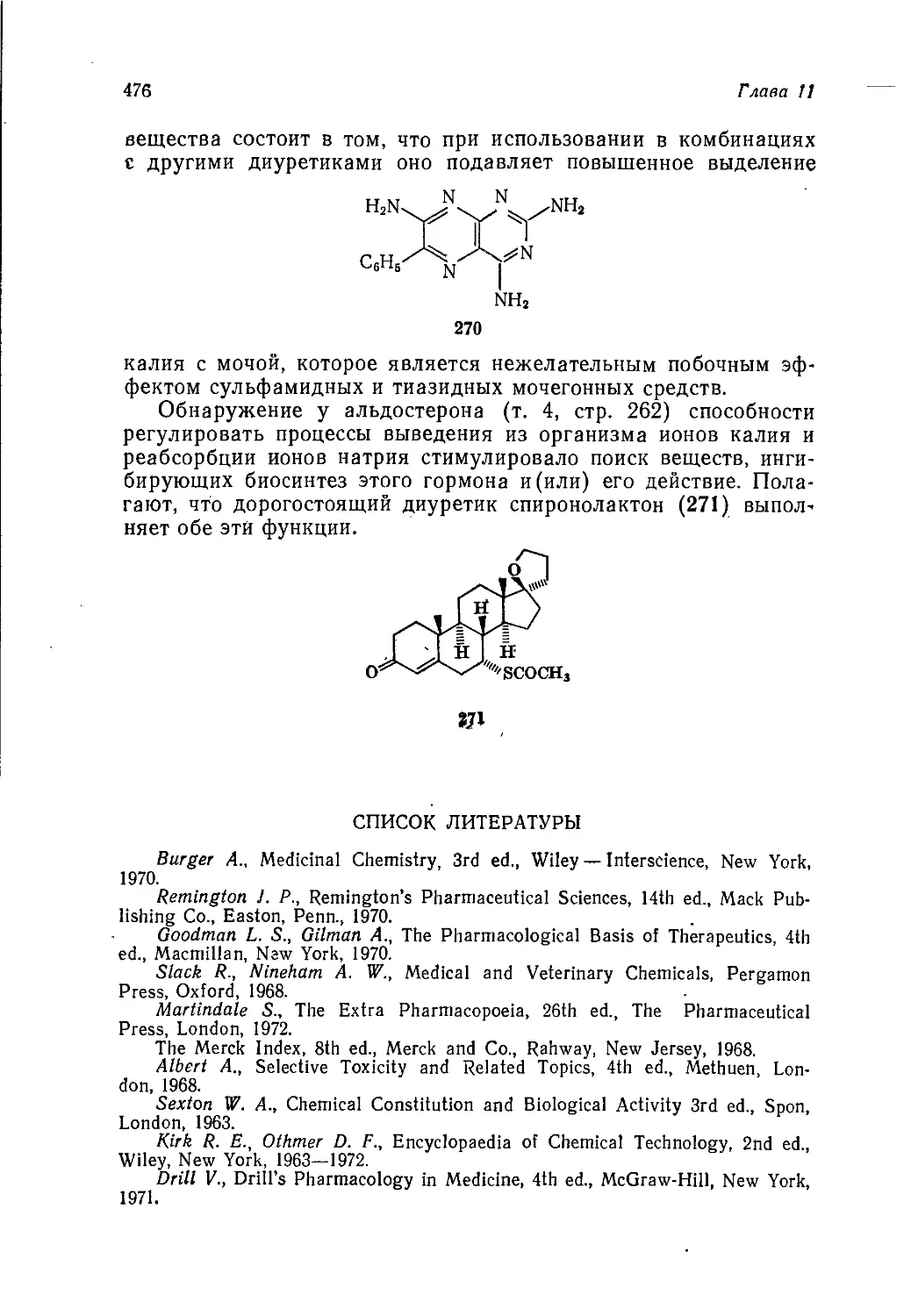
ГЛАВА 1
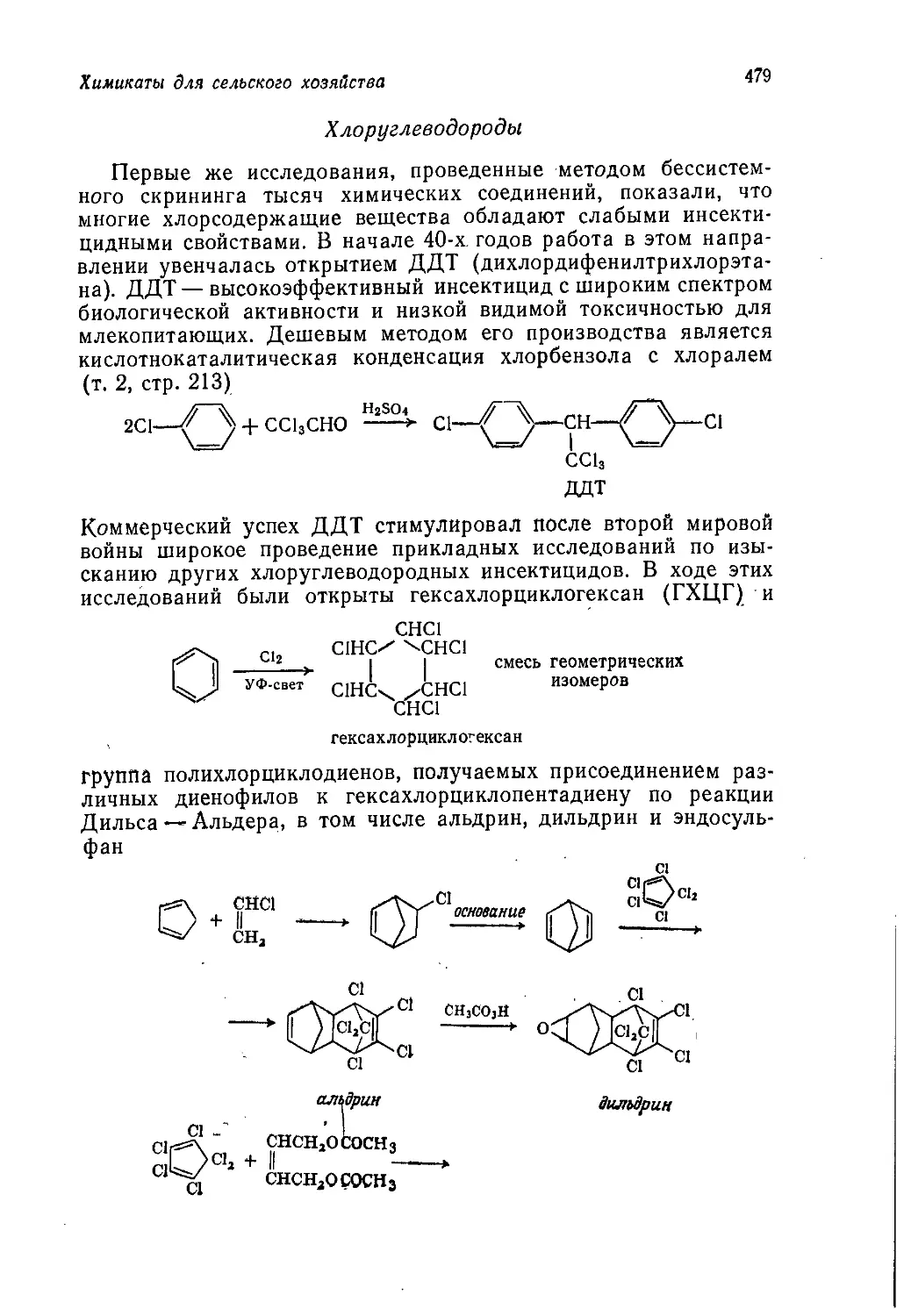
Введение
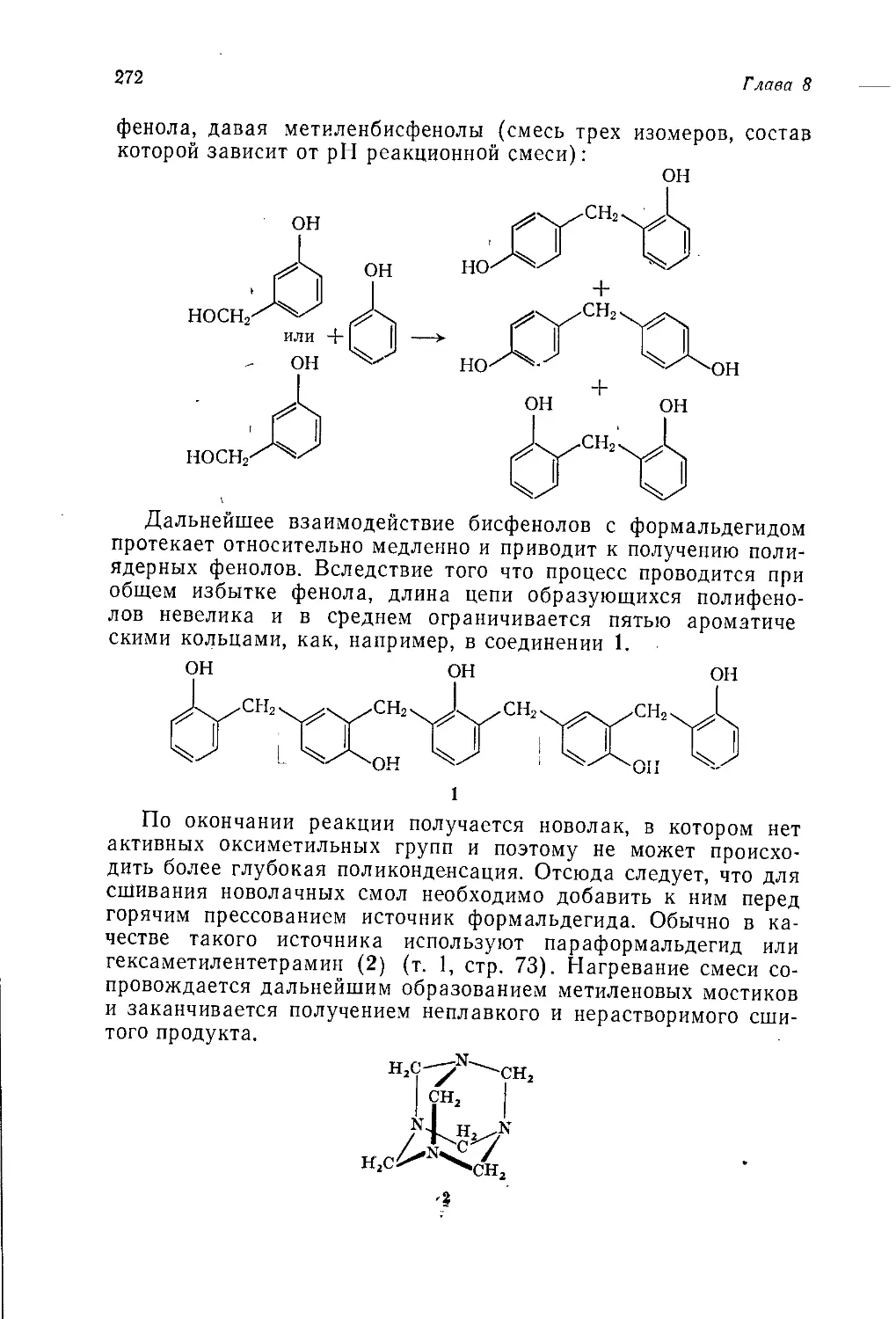
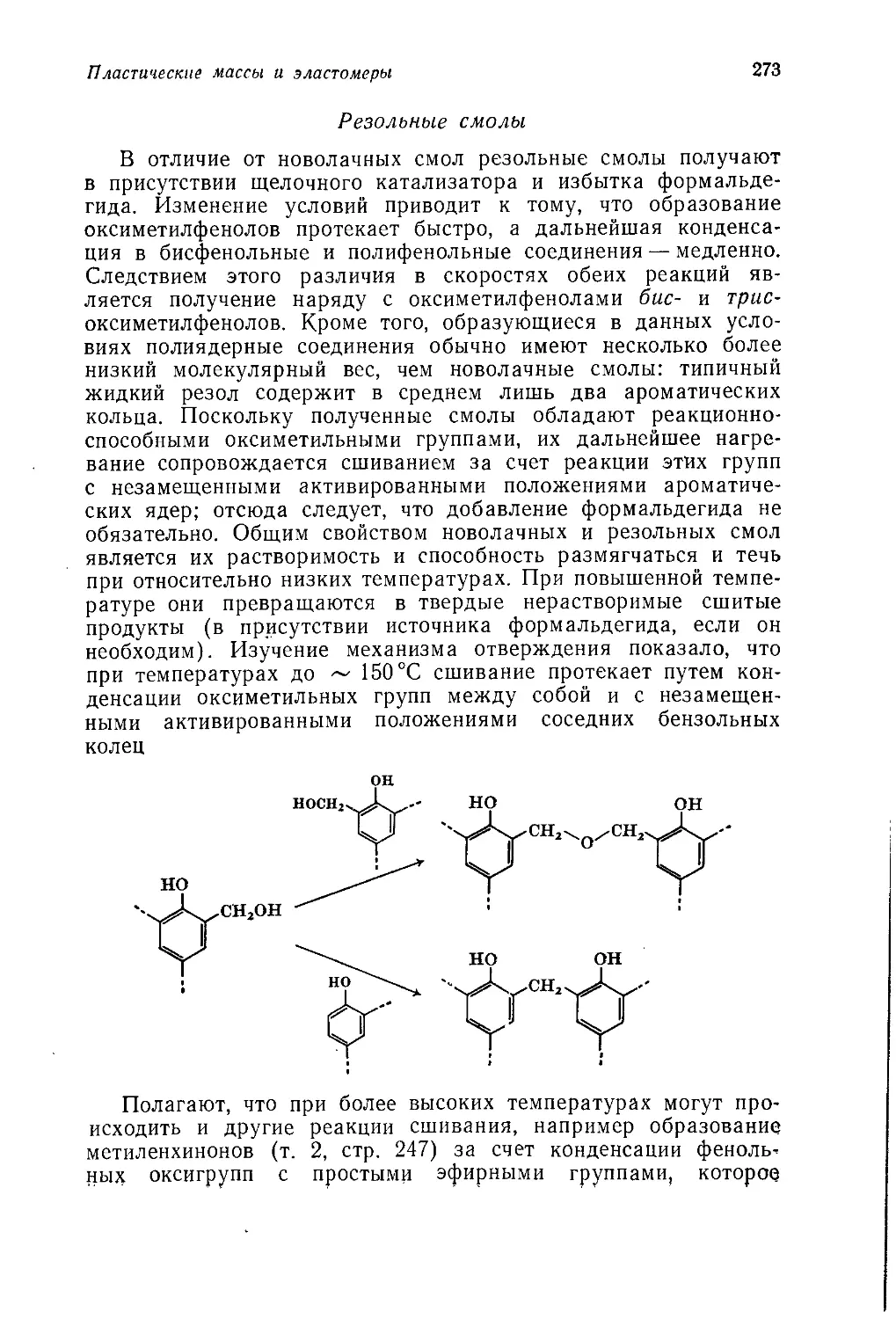
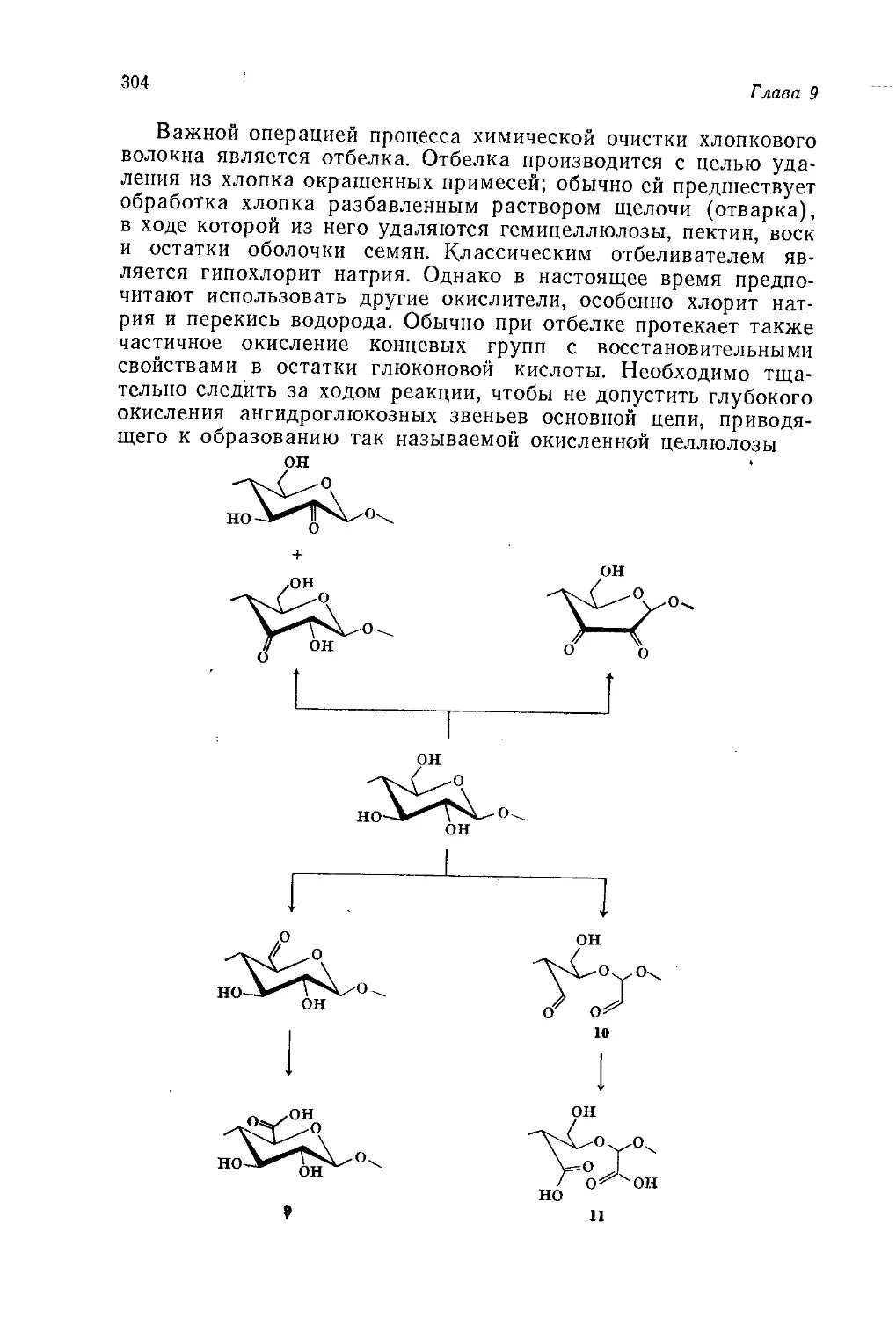
и история химической промышленности
А. Джубб
Jubb A. H.t ICI Corporate Laboratory, Runcorn
В томах 1—3 мы занимались, образно говоря, сооружением
каркаса здания органической химии. Химические реакции были
классифицированы по их механизму, а органические соедине-
ния— по их химическим свойствам и (или) строению. Том 4
был посвящен применению представлений, развитых ранее, к
природным соединениям. Последние классифицировали по ме-
тодам их биосинтеза, однако основное внимание по-прежнему
уделялось механизмам реакций, в которые вступают эти соеди-
нения. Данный том посвящен синтетическим органическим веще-
ствам, т. е. веществам, получаемым в лаборатории или на
заводе. В т. 4 упоминалось о синтезах сложных природных
соединений, преследующих не более чем дилетантскую цель со-
ревнования с природой, хотя, как было подчеркнуто, обычно при
проведении таких синтезов имеют в виду гораздо более важные
цели. Общим свойством химических веществ и методов синтеза,
описанных в данном томе, является их практическая польза,
будь то духи, которые делают человека более привлекательным,
взрывчатые, вещества для разработки залежей полезных иско-
паемых или волокно, из которого можно соткать ткань для
одежды. Важность того или иного химического вещества оцени-
вается в этой книге не с точки зрения химии, а с точки зрения
практической пользы.
Ценность любого товара целиком определяется конкретными
обстоятельствами: для путешественника в пустыне литр воды
может быть неизмеримо дороже, чем все золото Форт-Нокса *.
Особенно ярким примером в этом отношении являются лекар-
ственные препараты. Если данное лекарство — единственное
средство спасения больного от смерти, то его стоимость отно-
сительно несущественна. В то же время новый краситель, даю-
щий недостижимые ранее оттенки цвета, вряд ли найдет
применение, если он будет таким дорогим, что цена окрашенных
♦ В этом городе хранится золотой запас США. — Прим, ред.
12
Глава 1
им изделий окажется значительно выше, чем цена аналогичных
изделий, окрашенных стандартным красителем близкого, хотя
и менее привлекательного, цвета.
Поэтому химические процессы рассматриваются в этой книге
с учетом еще одного фактора — экономики. Разумеется, перво-
степенными остаются основные химические критерии осущесдви-
мости процесса с точки зрения его механизма и термодинамики.
Если процесс термодинамически неосуществим, то не может быть
и речи о его обсуждении, каким бы экономически выгодным он
ни казался. Однако в том случае, когда имеется несколько ме-
тодов получения желаемого химиката, прежде всего необходимо
принимать во внимание не техническое изящество процесса, а
его экономические показатели. Все более важным становится
и социологический фактор: экономичный и технически совершен-
ный процесс может оказаться неприемлемым из-за токсичности
побочных продуктов или вредного воздействия выделяющихся
паров на обслуживающий персонал.
Поскольку промышленность — один из основных источников
богатства страны, каждое предприятие должно стремиться к
тому, чтобы стоимость выпущенной продукции превышала за-
траты на ее производство. Последние включают такие статьи
расхода, как стоимость сырья, пара, электроэнергии и охлаж-
дающей воды (все эти затраты называются переменными, так
так они непосредственно зависят от объема производства),
а также трудовые затраты, затраты на текущий ремонт обору-
дования и зданий, накладные расходы, амортизационные отчис-
ления (средства, отчисляемые на замену износившегося обору-
дования), возмещение капитальных и пусковых затрат (эти
статьи расхода называются фиксированными затратами, так как
они не зависят от фактической загрузки предприятия и одина-
ковы, например, при коэффициенте использования мощностей
50 и 90%). Стоимость выпущенной продукции — это, как видно
из самого названия, средства, которые могут быть выручены
при ее продаже; часто она может быть определена термином
«объем реализованной продукции». Себестоимость продукции
рассчитывается, как показано в табл. 1.1.
Очень большое влияние на стоимость продукта оказывает
масштаб производства. Было найдено, что мощность промыш-
ленной установки и ее стоимость достаточно хорошо коррели-
руются логарифмическим соотношением, известным под назва-
нием «закон показателя степени 0,6»:
Стоимость проектируемой установки = г0,6 • С,
где г — отношение мощностей проектируемой и действующей
установок и С — стоимость действующей установки. Из этого
соотношения вытекает, что увеличение мощности проектируемой
Введение и история химической промышленности
13
Таблица 1.1
Структура затрат на производство этилена из нафты
на заводе мощностью 450 тыс. т/год (по данным 1973 г.)
Капитальные затраты (млн, ф. ст.)
Общая стоимость завода, включая непроизводственные ка- 35,0
пнтальные затраты “
Оборотный капитал • 2,0
Итого 37,0
Удельные затраты производства (ф. ст./т этилена)
1. Переменные затраты сырье (нафта по цене 20 ф. ст./т) катализатор н вспомогательные химические исключается стоимость побочных продуктов материалы 78,7 2,1 -53,0
чистая стоимость сырья и материалов энергетические затраты (котельное топливо, электроэнергия) газ, вода, 27,8 13,5
Итого 41,3
2. Фиксированные затраты трудовые затраты текущий ремонт оборудования и зданий налоги и страхование накладные расходы амортизационные отчисления возмещение капитальных затрат (15%) 1,3 3,1 0,8 3,1 7,8 12,3
28,4
Суммарные затраты производства, включая возмещение капи- 69,7
тальных затрат
Примечание. Приведенные цифры являются примерными и могут коле-
баться в зависимости от местонахождения предприятия.
а Под непроизводственными капитальными затратами нодразумевается стоимость со-
оруженнй, расположенных за пределами так называемой установочной лнннн, отмечающей
на генеральном плане строительства производственную зону проектируемого предприятия,
в которой размещается все технологическое оборудование. К непроизводственным капи-
тальным затратам относится стоимость складов, энергетических установок н администра-
тивных зданий, а также затраты на Подготовку участка под строительство (если не ого-
ворена иная структура капитальных затрат).
установки вдвое по сравнению с мощностью действующей будет
сопровождаться повышением ее стоимости приблизительно в
полтора раза.
Приведенное выше соотношение следует применять с осто-
рожностью. Оно справедливо лишь для сравнительно крупных
установок, не слишком сильно отличающихся по своим размерам
от аналогичных действующих установок и имеющих большую
агрегатную мощность (причем даже в этом случае показатель
14
Глава 1
степени может отличаться от 0,6). При простом же увели-
чении числа основных агрегатов стоимость установки соответ-
ственно удваивается, утраивается и т. п. В основе закона
показателя степени 0,6 лежит то обстоятельство, что стоимость,
допустим, реактора есть функция стоимости израсходованного
на его изготовление материала, которая в свою очередь является
Мощность предприятия, тыс. т/год
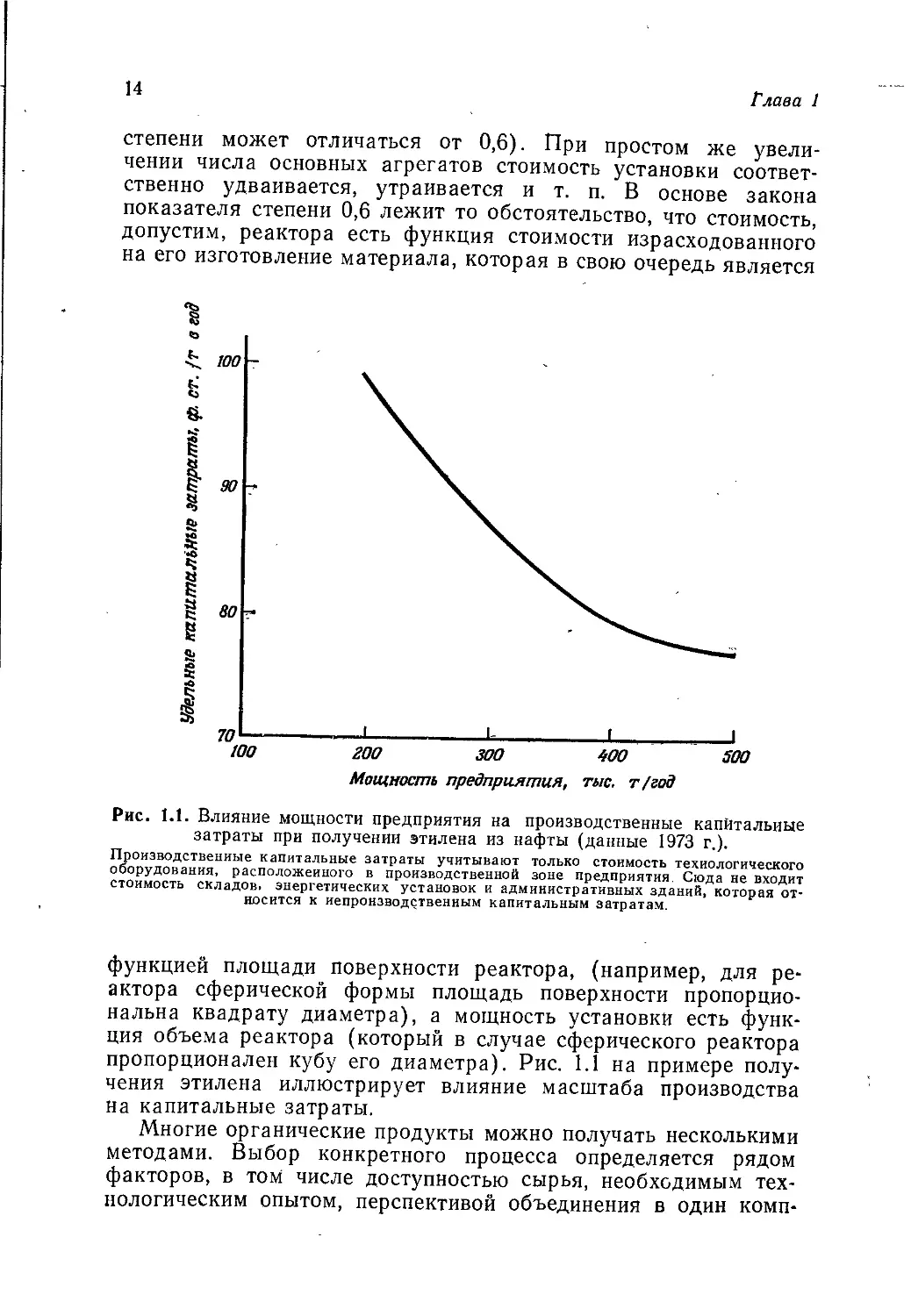
Рис. 1.1. Влияние мощности предприятия на производственные капитальные
затраты при получении этилена из нафты (данные 1973 г.).
Производственные капитальные затраты учитывают только стоимость технологического
оборудования, расположенного в производственной зоне предприятия. Сюда не входит
стоимость складов» энергетических установок и административных зданий, которая от-
носится к непроизводственным капитальным затратам.
функцией площади поверхности реактора, (например, для ре-
актора сферической формы площадь поверхности пропорцио-
нальна квадрату диаметра), а мощность установки есть функ-
ция объема реактора (который в случае сферического реактора
пропорционален кубу его диаметра). Рис. 1.1 на примере полу-
чения этилена иллюстрирует влияние масштаба производства
на капитальные затраты.
Многие органические продукты можно получать несколькими
методами. Выбор конкретного процесса определяется рядом
факторов, в том числе доступностью сырья, необходимым тех-
нологическим опытом, перспективой объединения в один комп-
Введение и история химической промышленности 15
леке нескольких процессов, мощностью установки, возможно*
стью сбыта побочных продуктов и т. д. Для разных фирм значе-
ние отдельных факторов и, следовательно, их влияние на эконо-
мику процесса может быть различным; поэтому в общем случае
нельзя сказать, какой из альтернативных процессов получения
данного продукта является лучшим.
История химической промышленности
Химическая промышленность — одна из наиболее стремитель-
но развивающихся отраслей экономики индустриально развитых
стран. Эта отрасль, которую более точно было бы рассматри-
вать как группу родственных подотраслей, вносит существенный
вклад в увеличение национального богатства *, и наличие мощ-
ной химической промышленности является для передовых стран
мира необходимым условием их дальнейшего продвижения по
пути экономического прогресса.
Наиболее старые подотрасли химической промышленности
возникли еще в XIX в. К ним относятся главным образом про-
изводство неорганических веществ на основе природного сырья,
такого, как сера и поваренная соль. Органические вещества пер-
воначально получали путем ферментации (сбраживания) сель-
скохозяйственных продуктов. До 1860 г. этим методом выраба-
тывали только пищевые вещества, такие, как уксус, и алкоголь-
ные напитки, например винный (этиловый) спирт. В конце
XIX в. было налажено промышленное производство фермента-
ционной молочной кислоты, а позднее — ферментационного
глицерина, ацетона, бутанола-1, лимонной кислоты и ряда бак-
териальных и грибковых ферментов, что было вызвано (по край-
ней мере частично) первой мировой войной. В период 1940—
1960 гг. ассортимент выпускаемых продуктов ферментации зна-
чительно расширился. В него вошли такие сложные соединения,
как антибиотики, витамины, аминокислоты и стероиды. В начале
XX в. стало быстро развиваться производство органических со-
единений из каменного угля.' На первое место здесь вышла
Германия, однако и другие угледобывающие страны — США,
Великобритания и Франция — создали в 1914—1920 гг. свою
углехимическую промышленность.
Опираясь на исследования, частично выполненные еще во
время первой мировой войны, США в 20-х годах приступили
* В 1974 г. объем потребления нефтехимических продуктов в США со-
ставлял в денежном выражении 37,5 млрд, долл., а чистый доход США от
экспорта химикатов превышал 4,8 млрд. долл. [Chem. and Ind. (London), 1975,
№ 23, 995; Chem. and Eng. News, 53, № 5, 5 (1975)]. — Прим, nepeq.
16
Глава 1
к созданию нефтехимической промышленности, сырьем для ко-
торой служат продукты переработки нефти. Вторая мировая
война способствовала быстрому развитию нефтехимии, что было
обусловлено главным образом возникновением острой потреб-
ности в стратегических материалах, таких, как каучук и бензин.
Так, если в 1925 г. США произвели из нефтяного сырья 100 т
химических продуктов, то к 1930 г. их выпуск возрос до 45 тыс. т;
затем он продолжал увеличиваться и к 1940 г. достиг 1 млн. т,
а в результате дальнейшего быстрого роста в послевоенный пе-
риод объем производства продуктов нефтехимического синтеза
в 1967 г. составил 49 млн. т.
В остальных странах мира нефтехимическая промышлен-
ность вплоть до окончания второй мировой войны практически
не существовала *. Развитие нефтяной промышленности и осо-
бенно строительство заводов по переработке нефти сделали До-
ступным сырье, на котором базируется нефтехимия. До 1955 г.
большинство стран ограничивалось производством низших оле-
финов и продуктов на их основе. Отсутствие в этих странах
природного газа заставило их ориентироваться в. выборе нефте-
химического сырья на жидкие углеводородные фракции, а до-
ступность ароматических углеводородов, получаемых из камен-
ного угля, делала их производство из нефти экономически не-
выгодным.
В странах Западной Европы нефтеперерабатывающие заво-
ды проектировались с целью удовлетворения спроса на самые
разнообразные основные нефтепродукты. Совершенно иное по-
ложение существовало в США, где громадным спросом пользо-
вался бензин. Если в США на каждую тысячу жителей в
1950 г. приходилось свыше 270 автомобилей, то для Западной
Европы эта цифра в среднем составляла 23. К тому же амери-
канские автомобили имели более мощные двигатели, что еще
более увеличивало спрос на бензин в США. Из-за невысокого
спроса на бензин нефтеперерабатывающие заводы стран Запад-
ной Европы выпускали больше тяжелых нефтепродуктов и
меньше бензина, чем заводы США (табл. 1.2).
* В СССР первое нефтехимическое предприятие было построено еще в
годы первых пятилеток. Пиролизом керосиновой фракции получался газ, со-
держащий до 20% этилена и 10—13% пропилена. Газоразделение осуществля-
лось конденсационным методом. Еще в довоенные годы из этилена, получен-
ного таким методом, вырабатывали дихлорэтан, винилхлорид, хлористый этил,
окись этилена, этиленгликоль; из пропилена — изопропанол, ацетон, ди-бром-
пропан; См. сборник «Советская химическая наука и промышленность за
50 лет», Химия, 1967, стр. 183. — Прим. ред.
Введение и история химической промышленности
17
Таблица 1.2
Структура производства основных продуктов переработки нефти
(% по весу)
Продукт США Западная Европа
1959 г 1972 Г, 1959 г. 1972 г.
Бензин 41 43 19 17
Средние дистилляты 27 29 26- 32
Котельное топливо И 6 38 37
Прочие продукты 21 22 17 14
Гог
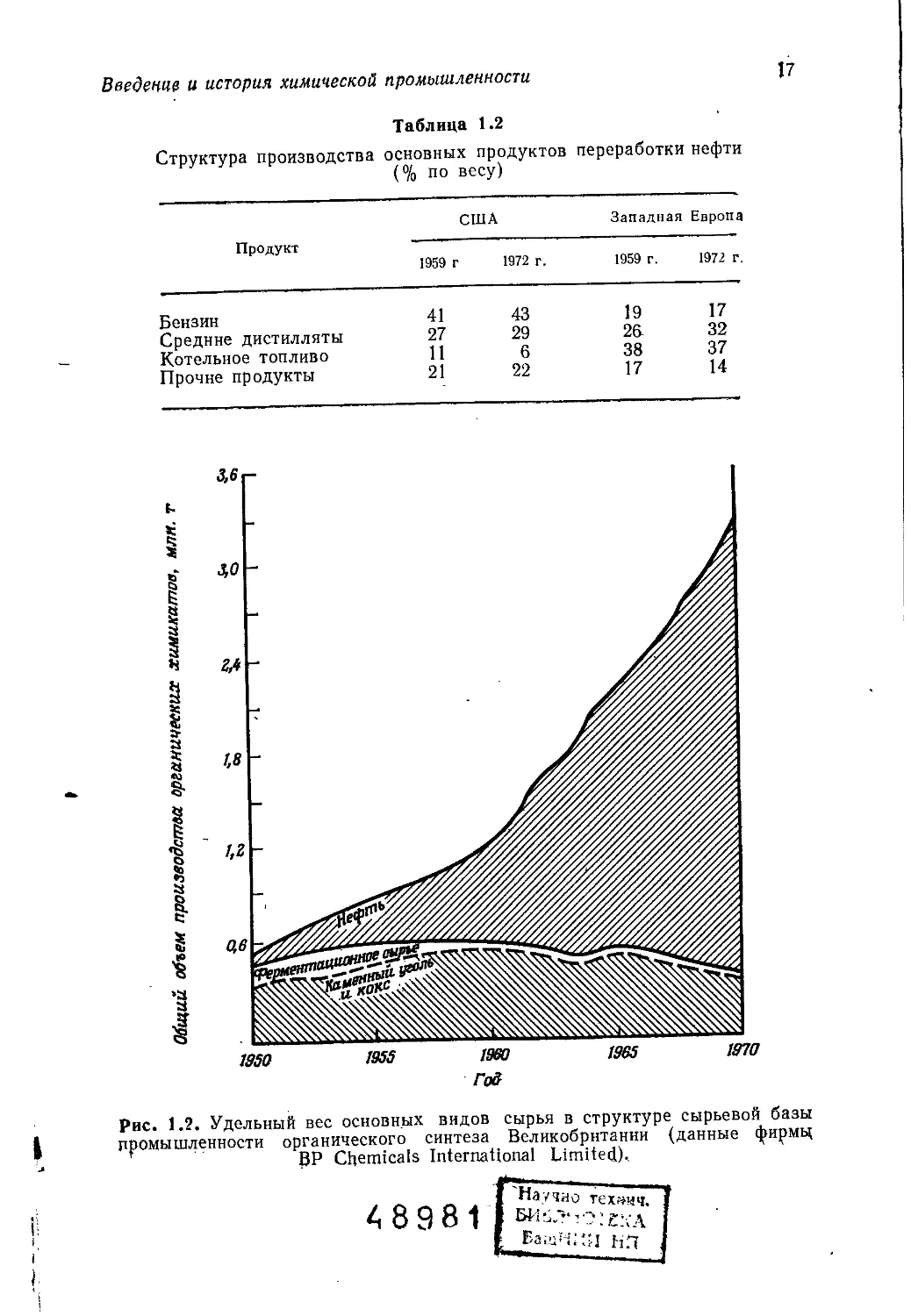
Рис. 1.2. Удельный вес основных видов сырья в структуре сырьевой базы
промышленности органического синтеза Великобритании (данные фирмц
BP Chemicals International Limited),
/ О о о а I Научно тех»яч.
4 8 9 81 | № 712: А
I нл
ЕащИ
18
Глава 1
Таблица 1.3
Динамика производства (млн. т) первичных органических химикатов
(полупродуктов органического синтеза) в капиталистических странах
[Spaght М. Е., Chem and Ind. (London), 1967, 1344]
Год США Западная Европа Прочие страны Итого В том числе нз нефтяного сырья, %
1950 2,95 0,12 0,03 3,1 40
1960 10,05 2,32 0,73 13,1 65
1965 17,0 6,9 3,1 27,0 75
1985 (прогноз) 80,0 88,0 32,0 200,0 98
2000 (прогноз) 210 240 150 600 > 99
По этим причинам нефтехимическая промышленность США
с самого начала базировалась на двух основных видах сырья:
конденсате природного газа (т. е. этане и пропане) и газообраз-
ных продуктах каталитического крекинга (т. е. этилене и про-
пилене), которые в большом количестве получались на много-
численных крекинг-установках. Напротив, в странах Западной
Европы, где нет достаточных запасов природного газа и име-
лось сравнительно мало крекинг-установок, производство низ-
ших олефинов базировалось на более тяжелых нефтепродуктах,
главным образом на нафте. В последние годы благодаря непре-
рывному увеличению числа нефтеперерабатывающих заводов и
возросшему объему перевозок сырой нефти наблюдается быст-
рый рост производства продуктов нефтехимии в различных стра-
нах мира (табл. 1.3).
На рис. 1.2 представлена динамика увеличения удельного
веса нефтехимического сырья по сравнению с углехимическим’
и ферментационным в промышленности органического синтеза
Великобритании.
Общие замечания
Теперь, рассмотрев исторические факты, мы можем задать
твопрос: почему нефтехимический синтез занял доминирующее 1
положение в промышленной органической химии, потеснив фер-
ментативный и углехимический синтез? 0
Если отбросить экономические соображения, то могущество
химии настолько велико, что практически все необходимые для
промышленности органические химикаты можно было бы син-
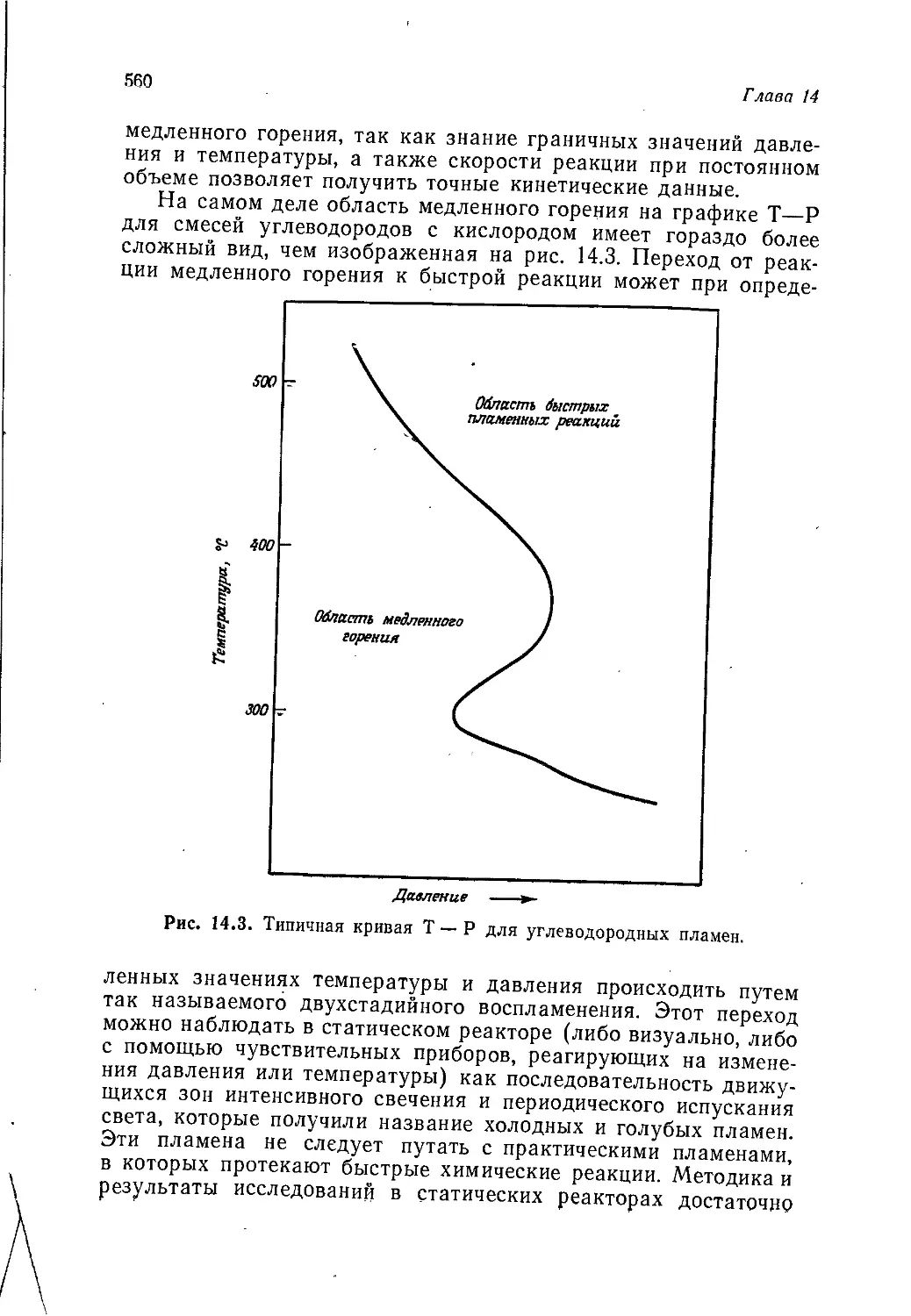
тезировать из углерода и воды. Очевидно, что в этом случае
многие методы их синтеза оказались бы довольно сложными
Введение и история химической промышленности
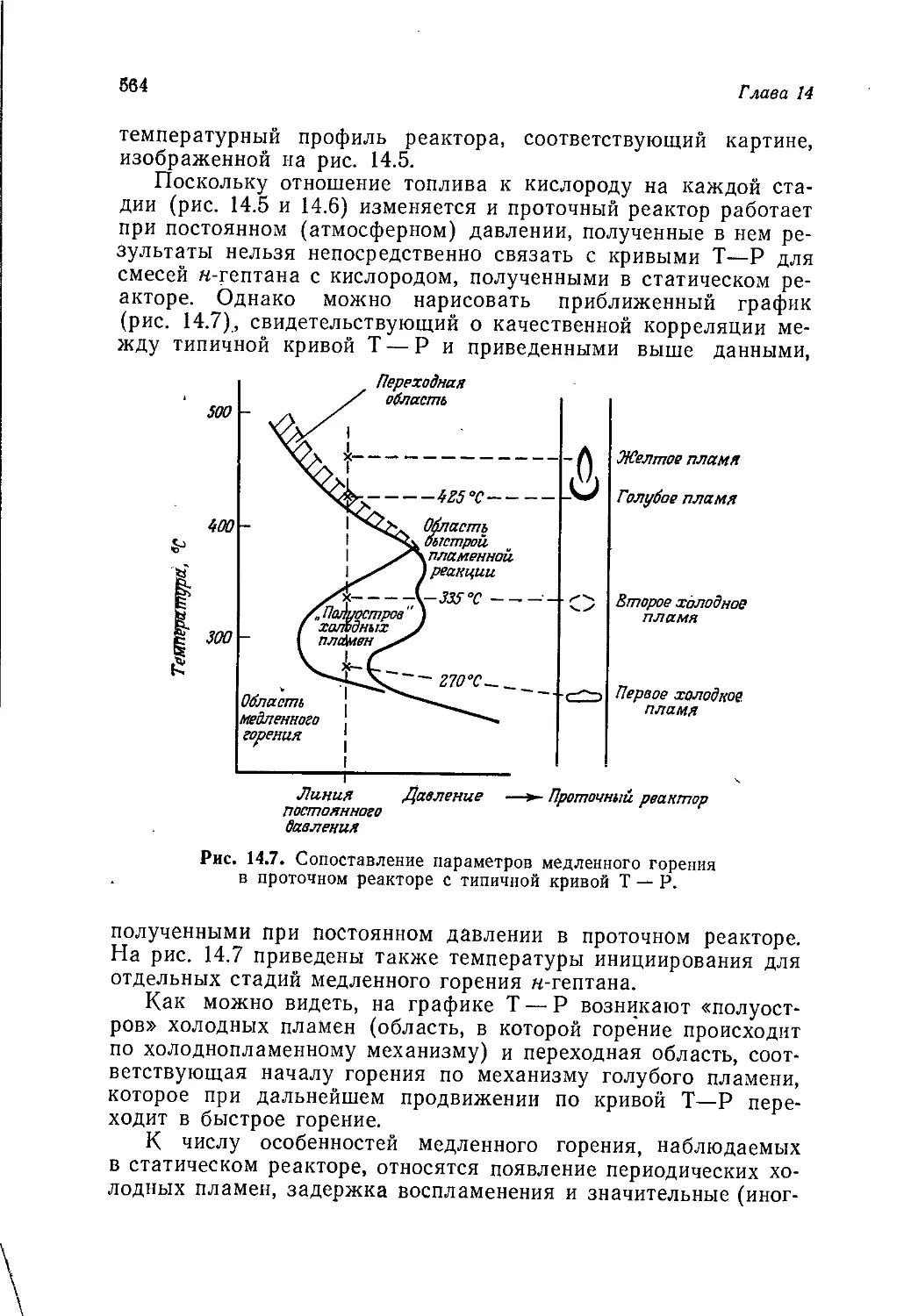
19
последовательно, дорогими. Используя более подходящее исход-
ное сырье, такое, как вещества растительного и животного про-
исхождения, каменный уголь, нефть и природный газ, мы
придем к сравнительно менее сложным и более экономичным
процессам. Выбор того или иного вида сырья будет опреде-
ляться рядом факторов, например его доступностью и стоимо-
стью по сравнению с другими видами сырья в месте расположе-
ния предприятия, характером выпускаемого продукта, наличием
подходящей технологии его производства, проектируемой мощ-
ностью предприятия, наличием необходимого капитала для его
строительства, величиной трудовых затрат, а также наличием
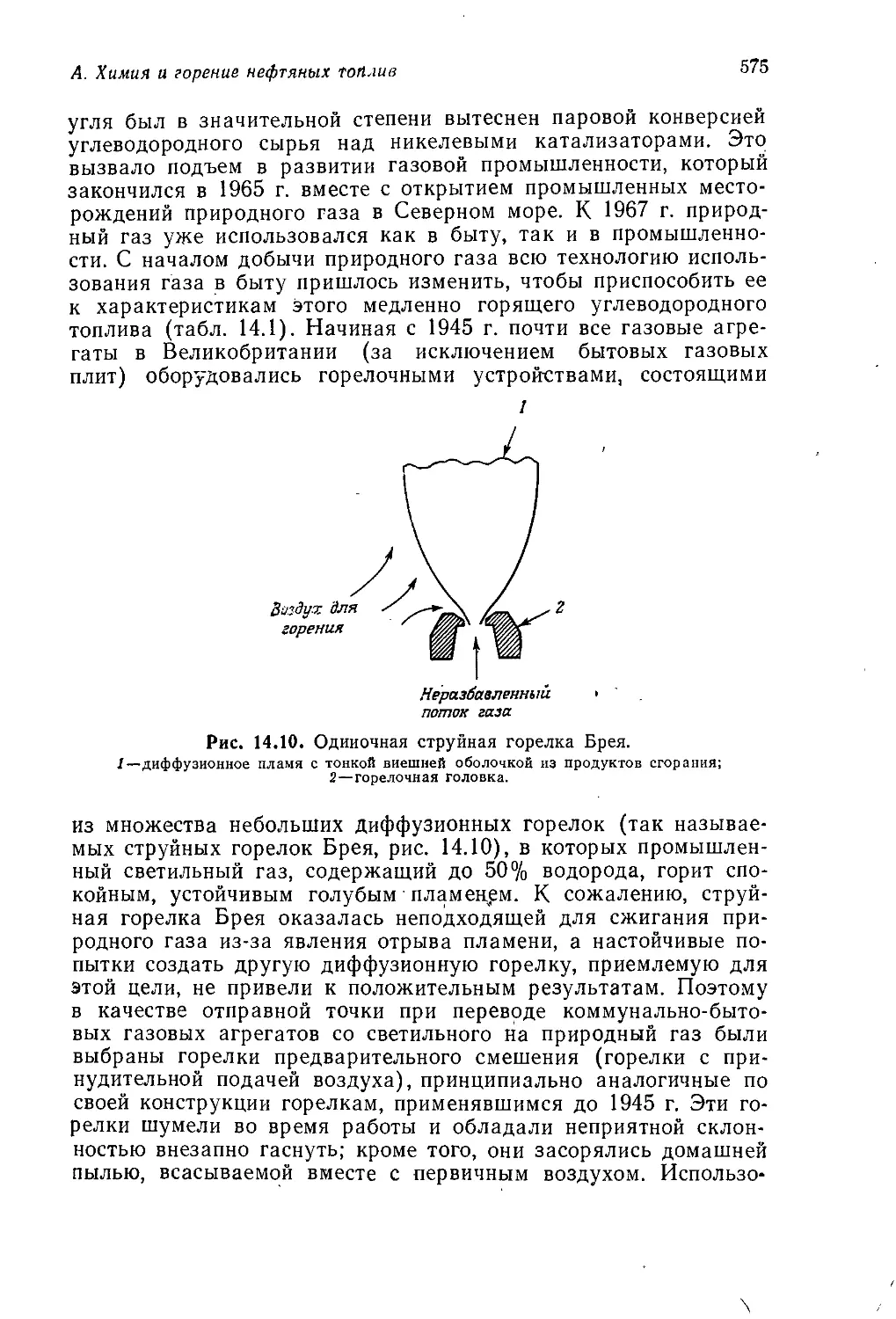
и стоимостью воды, пара и электроэнергии. Кроме того, каждый
из перечисленных выше видов сырья имеет свои преимущества
и недостатки, которые в конкретной ситуации могут стать реша-
ющими. Так, в нефти отношение водорода к углероду выше, чем
в каменном угле (1,8 по сравнению с 0,8), что делает ее более
предпочтительным сырьем для получения углеводородов; к тому
же транспортировка нефти в крупных танкерах обходится де-
шевле. Однако месторождения нефти имеются не во всех стра-
нах, и в зависимости от политической обстановки в мире
нехватка нефти в тех или иных странах мож'ет заставить их
использовать местные запасы каменного угля (как это случи-
лось в Германии во время второй мировой войны и происходит
в настоящее время в США, которые, оказавшись перед лицом
истощения своих запасов природного газа, активизировали ис-
следования в области газификации каменного угля).
Общая тенденция развития химической промышленности со-
стоит в том, чтобы избегать использования сельскохозяйствен-
ного сырья во всех случаях, когда возможно иное технологиче-
ское решение. Это обусловлено двумя основными причинами.
Во-первых, нестабильность уровня производства сельскохозяйст-
венной продукции приводят к непредсказуемым колебаниям ее
поступления. Во-вторых, повышение жизненного уровня сель-
скохозяйственных рабочих вызывает непрерывное возрастание
цен даже на побочные продукты и отходы сельскохозяйствен-
ного производства, номинальная стоимость которых ничтожна,
но затраты на сбор и транспортировку которых могут оказаться
слишком высокими. К тому же выход продукта синтеза из сель-
скохозяйственного сырья обычно ниже, чем из такого же коли-
чества нефти. Однако в некоторых случаях этот вид сырья мо-
жет быть самым лучшим или даже единственным источником
получения отдельных (обычно сложных) органических соединений.
На заре химической промышленности органические химика-
ты получали методами ферментации вследствие доступности
подходящего сельскохозяйственного сырья, наличия соответст-
вующей технологии и отсутствия более экономичных методов.
20
Глава I
Однако в настоящее время ферментативные процессы находят
ограниченное применение, поскольку обычно в них используются
водные растворы с низкой концентрацией реагентов и продуктов
реакции. Последнее затрудняет выделение и очистку образовав-
шегося продукта. Существование мощной угледобывающей про-
мышленности и многотоннажного производства кокса, необходи-
мого для получения стали и других стратегических материалов,
в которых нуждались основные страны — участники первой ми-
ровой войны, послужило базой для создания промышленности
углехимического синтеза. В то же время нефть в полтора раза
превосходит каменный уголь по теплотворной способности, не
дает при сгорании золы и обладает более высокой плотностью
и лучшими характеристиками горения. По этим причинам мно-
гие отрасли промышленности, а также транспорт позднее пере-
шли в значительной степени на использование продуктов
переработки нефти. В результате исследований, проведенных
в США в 1916—1918 гг., и развития нефтяной промышленности,
обусловленного в основном ростом числа автомобилей в этой
стране, были созданы необходимые предпосылки для возникно-
вения нефтехимической промышленности. Процесс перехода хи-
мической промышленности США на нефтяное сырье непрерывно
набирал силу, а другие страны следовали в этом отношении за
США. К настоящему времени нефть вследствие своей относи-
тельной дешевизны, которая объясняется низкой стоимостью ее
транспортировки на далекие расстояния большими танкерами
и по нефтепроводам, стала основным источником сырья для
промышленности органического синтеза. К тому же по мере
повышения жизненного уровня цены на каменный уголь, подоб-
но ценам на сельскохозяйственное сырье, увеличиваются по
сравнению с ценой на нефть, так как его добыча более трудо-
емка. Кроме того, нефтехимическая промышленность извлекает
большую выгоду из технических и научных достижений нефте-
добывающей промышленности и из повышения экономических
показателей своих собственных предприятий при переходе их
на использование непрерывных процессов и более крупных
установок.
В настоящее время масштабы производства химических про-
дуктов настолько велики, что даже если не учитывать экономи-
ческие факторы, получение их, например, из растительного
сырья вызвало бы нехватку пищевых продуктов. Более того,
выпуск синтетических волокон и синтетического каучука вы-
свобождает сельскохозяйственные площади, которые можно ис-
пользовать для удовлетворения все возрастающей потребности
человечества в продуктах питания.
Сейчас много говорят о надвигающейся угрозе энергетиче-
ского голода, обусловленного главным образом нехваткой нефти
Введение и история химической промышленности 21
и природного газа. Как будет показано в гл. 2, ресурсы этих
видов сырья действительно ограничены, однако их хватит еще
на ряд лет. Несомненно, неравномерное распределение нефтя-
ных месторождений и политическая обстановка в мире (особен-
но положение на Ближнем Востоке, где находится большая
часть мировых запасов нефти) создают определенные трудности
и неизбежно приводят к повышению цен на нефть. Это в свою
очередь заставляет многие страны шире применять другие ис-
точники энергии, такие, как горючие ископаемые, ядерную и
солнечную энергию, и искать пути более эффективного их ис-
пользования. Подобная ситуация делает возможной несколько
большую зависимость химической промышленности будущего от
коксохимического и растительного сырья.
Вместе с тем следует отметить, что в настоящее время на
получение органических химикатов (табл. 1.3) расходуется лишь
около 4% всей потребляемой нефти*. Спейт полагает, что в
1985 и 2000 гг. эта величина составит 5 и 12% соответственно.
* В 1975 г. химическая промышленность США потребляла в качестве ис-
ходного сырья 6% всей расходуемой в стране нефти и около 3% природного
газа [Chem. Market Abstr., 67, 286 887 (1975)]. — Прим, перев.
ГЛАВА 2
Источники сырья
для промышленности органического
синтеза
А. Джубб
Jubb А. Н., ICI Corporate Laboratory, Runcorn
В т. 1 (гл. 16 и 17) мы кратко рассмотрели органические
соединения, встречающиеся в природе, и уделили некоторое
внимание использованию каменного угля и нефти в качестве
исходного сырья для получения первичных химических продук-
тов. В данной главе источники сырья для промышленности ор-
ганического синтеза рассмотрены более подробно, причем осо-
бое внимание обращается на величину запасов и относительную
важность каждого вида сырья.
Сельскохозяйственное и лесохимическое сырье
Запасы минералов, каменного угля и нефти ограничены,
тогда как продукция сельского и лесного хозяйства непрерывно
возобновляется. Однако спрос на пишевые продукты и лес на
мировом рынке настолько велик, что как химическое сырье они
доступны отнюдь не в беспредельном количестве. Более того,
возрастание потребности в продуктах питания для все увеличи-
вающегося населения земного шара обусловило проведение ши-
роких исследований в области производства синтетических
белковых концентратов из нефти и природного газа. Вследствие
громадного роста выпуска химических продуктов из нефтяного
сырья объем их производства на основе сельскохозяйственного
и лесохимического сырья в настоящее время составляет лишь
небольшую часть общего производства. Тем не менее эта под-
отрасль химической промышленности продолжает играть важ-
ную, а в некоторых случаях даже незаменимую роль.
Жиры и масла
Жиры и масла получают из источников растительного и жи-
вотного происхождения. Основными компонентами жиров и
масел является смешанные триглицериды жирных кислот С16
Источники сырья для промышленности органического синтеза
23
и Cis (т. 4, стр. 150—155). Некоторые твердые растительные
жиры, например пальмоядровое и кокосовое масла, содержат
большие количества насыщенных жирных кислот от С6 (капро-
новой) до Си (миристиновой), тогда как другие содержат пре-
имущественно насыщенные Ci6 (пальмитиновую) и С!8 (стеари-
новую) и ненасыщенную Ci8 (олеиновую) кислоты. В состав
жидких растительных масел, например хлопкового и соевого,
входят главным образом ненасыщенные кислоты Cj8 (олеиновая
и линолевая). В животных жирах находятся в основном насы-
щенные С1в (пальмитиновая) и Ci8 (стеариновая) кислоты с
небольшими примесями ненасыщенных кислот Ci8. Жир рыб
содержит высоконенасыщенные кислоты. В табл. 2.1 приведены
данные о структуре мирового производства жиров и масел.
Таблица 2.1
Структура мирового производства
масел и жиров (или их эквивалентов) в 1970 г.
(прогнозная оценка) (World Agriculture, 1970)
Масло или жир Производство, млн. т
Пищевые растительные масла 18,90
Пальмовое масло 4,20
Технические масла 1,48
Животные жиры 12,51
Китовый жир 1,26
Итого 38,35
Таблица 2.2
Мировое производство глицерина (тыс. т) [Education in Chemistry, 9,
№ 1 (1972)]
США Западная Япония Мировое производ- Доля синте- тического глицерина
Год природ- сиитети- ный ческий Европа (всего) (всего) ство (всего) в мировом производстве, %
1961 63 64 95 16 255 30
1963 64 73 105 16 285 33
1965 66 91 109 18 310 33
1967 70 96 115 18 330 39
1968 73 91 125 18 335 42
1969 160 122 340 45
1970 150*
* В 1973 г. в США было произведено 163 тыс. т глицерина, в 1974 г —158 тыс, т.
в 1975 г. —119 тыс. т [Chem. Industrie, 28, 683 (1976)]. — Прим, перев.
24
Глава 2
Гидролизом различных триглицеридов получают природный
глицерин и жирные кислоты. В некоторых странах глицерин
получают также синтетическими методами (гл. 4). Данные о ми-
ровом производстве глицерина представлены в табл. 2.2. Жир-
ные кислоты используют при изготовлении самых разнообразных
продуктов, в том числе мыла, синтетических моющих веществ
(гл. 13), защитных покрытий и дезинфицирующих препаратов.
Мировое потребление мыла в период I960—1971 гг. не измени-
лось, тогда как потребление синтетических моющих средств за
это время возросло приблизительно на 160% (табл. 2.3).
Таблица 2.3
Структура и динамика мирового потребления мыла, моющих
и чистящих веществ (тыс. т) (European Chemical News,
16-th March 1973)
1963 г. 1968 г 1970 г. 1971 г. а
Мыло 6 880 6 490 6 090 6 000
Синтетические моющие ве- 3 430 7 370 8 350 8 900
щества
Чистящие вещества 460 580 6501
Прочие 140 1 010 1 280 J
Итого 10910 15 450 16 370 17 100б
Доля синтетических ве- 32 48 51 52
ществ, %
a Предварительные данные.
® В 1973 г. мировое потребление мыла и моющих веществ достигло 18 млн. т, причем
доля синтетических моющих веществ увеличилась до !/з [Chem. Market Abstr., 66, 284 230
(1973)] —Прим, перев.
Некоторые природные жирные кислоты используют в качест-
ве сырья в производстве полупродуктов для получения термо-
пластов. Например, при озонолизе олеиновой кислоты образу-
ются пеларгоновая и азелаиновая кислоты
окисление
СН3(СН2)7СН=СН(СН2)7СООН-------->
олеиновая кислота
—> СН3(СН2)7СООН + НООС(СН2)7СООН
пеларгоновая азелаиновая
кислота кислота
Пеларгоновая и азелаиновая кислоты находят применение
в производстве сложноэфирных смазочных материалов, а азе-
^аиновая кислота — при получении низкотемпературных пдастц-
Источники сырья для промышленности органического синтеза 25
фикаторов и в производстве найлона-6,9. Объем мирового про-
изводства азелаиновой кислоты, вероятно, составляет 10 тыс.
т/год.
В 1950 г. был освоен промышленный выпуск найлона-11
(рильсана), который в настоящее время вырабатывается во
Франции и Бразилии как продукт специального назначения. Его
получают из касторового масла, обработка которого метанолом
дает смесь метиловых эфиров жирных кислот. Эту смесь раз-
деляют дистилляцией и выделенный метиловый эфир рицино-
левой кислоты подвергают пиролизу, причем образуются энан-
товый альдегид и метиловый эфир ундецен-10-овой кислоты
СН3(СН2)6СН(ОН)СН2СН=СН(СН2)7СООСН3 —>
метиловый эфир рицинолевой кислоты
—> СН3(СН2)5СНО+ СН2=СН(СН2)8СООСНз
энантовый метиловый эфир унде-
альдегид цен-10-овой кислоты
Последний гидролизуют в свободную кислоту, которую обраба-
тывают НВг в присутствии перекисного катализатора, и полу-
чают с выходом около 96% соответствующую 11-бромкарбоно-
вую кислоту
ROOR —> 2RO •
RO« + HBr —> ROH + Bf
Br.-hCH2=CH(CH2)8COOH —> ВгСН2СН(СН2)8СООН
BrCH2CH(CH2)8COOH + НВг —> ВгСН2СН2(СН2)8СООН + Вг •
При взаимодействии этой кислоты с аммиаком атом брома лег-
ко замещается и образуется целевая аминокислота.
Сплавление рицинолеата натрия с 70%-ным едким натром
дает октанол-2 и себациновую кислоту. Себациновую кислоту
используют для получения найлона-6,10, потребление которого
в США в 1972 г. составляло, по оценке, 10 тыс. т.
CH3(CH2)6CH(OH)CH2CH=CH(CH2)7COONa ^25-
рицинолеат натрия
—> СНз(СН2)6СН(ОН)СН3 + NaOOC(CH2)8COONa
октанол-2 себацинат натрия
Лесохимическое сырье
Лес представляет собой самый большой по запасам и притом
непрерывно возобновляющийся сырьевой источник в мире.
Основная часть древесины распределяется между различными
потребителями, главным из которых является строительная
26
Глава 2
промышленность. Так, в 1970 г. объем мирового производства
кругляка (в пересчете на плотный объем ошкуренных пиломате-
риалов) составил 6,8 млрд, т (1,2 млрд, м3)*; кроме того, еще
~ 1 млрд, м3 лесоматериалов, как считают, был израсходован
в качестве топлива (главным образом в слаборазвитых стра-
нах) .
Рис. 2.1 показывает, как много разнообразных продуктов
можно получить из древесины. Каждый из этих продуктов об-
суждается ниже.
Бумага
Сульфитный,
щелок
Целлюлоза
Нитроцеллюлоза
Простые и сложные эфиры
целлюлозы
Вискозное волокно
Талловое масло
Скипидар
сульфитная
варка
сухая
перегонка
Древесина
Дрожжи
—► Ванилин
~ Дубильные
* вещества
сульфатная
варка
Гидролизная
глюкоза
Лигнин
Живичный
скипидар
а-Пинен
Д-Пинен
Моноцихлические
терпены
Этанол
Дрожжи
Древесная целлюлоза
Дубильные вещества
Бумага
Древесный Древесный Деготь Гад
уголь уксус
I
Метанол, ацетальдегид,
ацетон, уксусная кислота
Рис. 2.1. Химические продукты, получаемые из дрейесинЫ.
Древесная целлюлоза — один из наиболее многотоннажных
продуктов «химического использования» древесины: в 1970 г. ее
было выработано около 100 млн. т. Получение древесной цел-
люлозы не является истинным химическим процессом и в сущ-
ности представляет собой механическую переработку древесины
с целью выделения волокон целлюлозы, из которых затем де-
лают дешевые сорта бумаги. Чистую целлюлозу получают пу-
тем удаления из древесной целлюлозы таких веществ, как лиг-
нин, который в древесине связан с целлюлозными волокнами. Ее
используют, в частности, как исходное сырье в производстве
* В 1974 г. он был равен 2,5 млрд, м3 [Monthly Bull, of Statistics, 30,
№ 7, X (1976)]. — Прим, перев.
Источники сырья для промышленности органического синтеза 27
вискозных волокон (волокон из регенерированной целлюлозы)
(гл. 9). Существует несколько методов получения чистой цел-
люлозы; наиболее широко применяются сульфатный и сульфит-
ный методы. Отработанный щелок, являющийся отходом суль-
фитного процесса, содержит гексозы и может быть подвергнут
ферментации в этанол
С6Н12О6 —> 2С,Н5ОН + 2СО2
Этот метод позволяет получать в зависимости от типа вароч-
ного процесса на заводе мощностью 100 т целлюлозы в сутки
5—12,5 тыс. л спирта ежесуточно. При щелочном гидролизе
лигнинсодержащих веществ типа тех, которые присутствуют в
сульфитном щелоке, образуется ванилин (т. 4, стр. 144 и гл. 16
настоящей книги)
СНО
ОН
ванилин
Осахаривание древесины, при котором отходы лесоматериа-
лов (щепу, стружки, опилки и др.) гидролизуют концентриро-
ванной соляной или разбавленной серной кислотой, приводит
к получению остатков лигница и щелока, содержащего сахари-
стые вещества (приблизительно 4%-ного раствора). Фермен-
тацией последнего можно получить этанол и дрожжи. Широко
известный процесс Шоллера (с использованием разбавленной
серной кислоты) обеспечивает 40—50%-ный выход сахаристых
веществ и позволяет получить из каждой тонны отходов лесо-
материалов (в пересчете на сухой вес) 200—210 л 100%-ного
этанола и 40—45 кг дрожжей с содержанием белка 50%. Ана-
логичным образом можно перерабатывать в кормовые продукты
гидролизную мелассу (концентрированный раствор гидролиз-
ной глюкозы).
Ферментацией пентоз, образующихся в различных процес-
сах химической переработки древесины, можно получить в ка-
честве побочного продукта фурфурол
С5Н10О5 —> || || + ЗН2О
о^сно
фурфурол
Фурфурол является, также продуктом гидролиза некоторых рас-
тительных отходов (овсяной лузги, кукурузных початков,
28
Глава 2
стеблей сахарного тростника). Этим методом в Доминиканской
Республике, по некоторым оценкам, производится ежегодно
около 25 Тыс. т фурфурола.
В древесине содержатся различные вещества, которые не
входят в состав древесных клеток, в том числе смолы, терпены
и дубильные вещества (танниды). Основную часть вырабатывае-
мого скипидара и все талловое масло получают в качестве по-
бочных продуктов при сульфатной варке древесной целлюлозы,
причем скипидар извлекают путем ее обработки острым паром
(выход около 10 кг из 1 т древесной целлюлозы). Надрезанием
коры деревьев, подобно тому как это делают в производстве
натурального каучука, получают живицу, которую разделяют
перегонкой на канифоль и скипидар. Перегонка последнего дает
три основные фракции: а-пинен, р-пинен и моноциклические
терпены (т. 4, стр. 217). Талловое масло содержит жирные кис-
лоты типа олеиновой, а канифоль — кислоты типа абиетиновой.
В США и Великобритании все эти вещества часто называют
Naval Stores, что буквально означает «корабельные материалы»,
поскольку первоначально плохо очищенные продукты перера-
ботки живицы применяли для смоления деревянных судов с
целью придания им водоустойчивости.
Помимо дубильных веществ (т. 4, стр. 116), используемых
для выделки кож, ткань дерева, кора и листья содержат эфир-
ные масла, такие, как кедровое и сассафрасовое.
Эти эфирные масла (гл. 16) находят применение в парфкь
мерной промышленности; они представляют собой летучие ве-
щества, получаемые из растительного сырья, откуда их выде-
ляют путем экстракции или перегонки с паром. В настоящее
время из 3000 известных эфирных масел 150 вырабатываются
в промышленном масштабе. Секреты желез животных (цивет,
мускус, амбру) и млечные соки многих растений используют
в качестве фиксаторов запаха (компонентов духов, регулирую-
щих скорость испарения душистых веществ и обеспечивающих
стойкость запаха). Многие эфирные масла сейчас получают
искусственно, и в парфюмерной промышленности применяют
смеси натуральных и синтетических продуктов. В 1969 г. в США
было выпущено 24 тыс. т душистых веществ на общую сумму
22 млн. ф. ст. *
Одним из самых важных продуктов растительного проис-
хождения является натуральный каучук (гл. 8). Его получают
из млечного сока гевеи Hevea brasiliensis, родина которой —
бассейн реки Амазонки в Южной Америке. Гевея растет очень
* В 1973 г. промышленность США выработала 53,1 тыс. т душчстых ве-
ществ на сумму около - 117 млн. долл. [Chem. Market Abstr., 67, 284 70
(1975)]. — Прим, перед.
Источники сырья для промышленности органического синтеза
29
быстро и в течение 6—7 лет дает значительные количества кау-
чука, сбор которого на наиболее высокопроизводительных
участках достигает 1300—1700 кг с гектара. Несмотря на то
что все большая доля мировой потребности в каучуке удовле-
творяется за счет синтетического каучука, в настоящее время *
ежегодно вырабатывается около 2,9 млн. т натурального кау-
чука, а к 1980 г. эта цифра, по прогнозной оценке, возрастет
до 3,7 млн. т (табл. 2.4).
Таблица 2.4
Динамика мирового потребления каучука (тыс. т)
Годы Все типы каучука Натуральный каучук Доля натурального каучука, %
1950 2 302 1 722 75
1955 3 099 1 887 61
I960 4327 2 065 48
1965 6115 2 375 39
1970 8 955 2 900 32,4
1975 (прогноз) а 11 630 3 300 28,4
1980 (прогноз) 14 400 3 700 25,7
а Фактически общее мировое потребление каучука в 1975 г.
было равно 9 935 тыс. т, из которых на долю натурального каучука
приходилось 35% [Chem. Industrie, 28, 211 (1976)1.—Прим, перев.
Сухая перегонка древесины приводит к получению древес-
ного угля, выход которого составляет 20—30%. В качестве по-
бочных продуктов образуются уксусная кислота, метанол и аце-
тон, выделяемые из подсмольной воды (выход 520 л из 1 т
древесины твердых пород). В начале XX в. этот процесс был
важным источником перечисленных веществ, но затем он был
вытеснен более экономичными процессами. Так, если в 1920 г.
в США насчитывалось около 100 перегонных установок по по-
лучению уксусной кислоты, метанола и ацетона из древесины,
то в 1969 г. была закрыта последняя.
В 1967 г. лесохимическая промышленность США произвела
продукции на сумму.211 млн. долл., что составило 0,5% общей
стоимости химикатов и сопутствующих им продуктов (42,2 млрд,
долл.). В 1969 г. первая цифра возросла до 275 млн. долл., что
по-прежнему составляло менее 1% стоимости всей химической
продукции. В настоящее время США наряду с Канадой, СССР
и скандинавскими странами является главным поставщиком ле-
сохимических продуктов на мировой рынок
♦ В 1970 г. — Прим, перев.
30
Глава 2
Сахаристые вещества
В 1970 г. во всем мире было выработано около 70 млн. т
сахара, полученного из сахарного тростника и сахарной свеклы.
Маточный сироп, остающийся после удаления кристаллов са-
хара из упаренного раствора тростникового сахара (этот сироп
содержит 55% сахара, из них 35—40% сахарозы и 15—20%
инвертного сахара, т. е; глюкозы и фруктозы), известен под
названием черная патока или тростниковая меласса. Высокока-
чественной мелассой является неочищенный сок сахарного трост-
ника, в котором большая часть сахара превращена путем кис-
лотного гидролиза в инвертный сахар; она представляет собой
70—80%-ный раствор сахара, содержащий 22—27% сахарозы
и 50—55% глюкозы и фруктозы. В свекловичной мелассе при-
сутствует 48—52% сахарозы и практически нет инвертного са-
хара. Оба вида мелассы можно подвергать ферментации. По-
лученный при этом этанол, выход которого составляет 90%
(считая на сахаристые вещества), очищают дистилляцией.
Крахмал
Крахмал заключает в себе основные энергетические ресур-
сы растений (т. 4, гл. 2). Различные крахмалсодержащие про-
дукты (зерно, картофель и др.) можно использовать в качестве
исходного сырья для получения этанола, подвергая их фермен-
Таблица 2.5
Структура и динамика производства этанола в США
(по данным 1970 г.)
Исходное сырье Доля в общем производстве спиртных напитков (в пересчете на крепость выше 45°), %
1961 г. 1967 г. 1970 г. '
Синтетический 79,0 79,2 83,0
Ферментационный:
из зерна и зерновых про- 14,0 13,5 12,0
дуктов
из мелассы 3,2 2,2 0,4
из фруктовых соков 2,5 3,9 3,6
Из сульфитного щелока 1,0 1,1 1,0
Из древесной целлюлозы 0,3 0,1 —
Общий объем производства, тыс. гектолитров 12 170 13 320 14 050
Источники сырья для промышленности органического синтеза 31
тации. В случае кукурузы сначала производят обжаривание и
осахаривание зерен для перевода крахмала в поддающиеся
ферментации сахаристые вещества. Осахаривание осуществляют
обработкой разбавленной серной кислотой или ферментом диа-
стазой
диастаза
(CaHioOg)re + яНаО nCaHiaOa
крахмал
зимаза
С6Н12О6 ------> 2C2HSOH + 2СО2
Доля этанола, вырабатываемого из сельскохозяйственного и
лесохимического сырья путем ферментации, в общем объеме
выпускаемого -этанола непрерывно сокращается. Так, если в
1935 г. она составляла в США 90%, то в 1963 г. — лишь 9%
(табл. 2.5). В странах Азии (исключая КНР и Японию) и Ла-
тинской Америки в 1970 г. было получено, вероятно, около
725 тыс. т ферментационного этанола.
Путем ферментации свекловичной или тростниковой мелассы
микроорганизмом Aspergillus niger получают лимонную кислоту
CH2CQOH
воздух I
С12Н22ОП + н2о ———>- носсоон
A. niger |
СН2СООН
сахароза лимонная кислота
Молочная кислота получается методом регулируемой фер-
ментации различных углеводов, таких, как сахароза, глюкоза
или'лактоза/которые содержатся в гидролизатах кукурузного
или картофельного крахмала:
Ci2H220n Н2О —> CeHijOj -}- С6Н|2О6
сахароза глюкоза фруктоза
ферментация
С6Н12О6 -------->- 2СН3СН(ОН)СООН
молочная кислота
Ферментацией отработанных растворов производства свекло-
вичного сахара, а также растворов кукурузной клейковины и
различных углеводов можно получать глутамат натрия, лево-
вращающий изомер которого применяют в качестве усилителя
вкуса (гл. 16). При использовании растворов углеводов
32
Глава 2
протекают следующие реакции:
СОСООН
CeHijOs + 2О2 —* | + СО2 + ЗН2О
СН2СН2СООН
а-кетоглутаровая
кислота
|NH3
H2NCHCOOH NaOH H2NCHCOOH
I 4— I + H2O
CH2CH2COONa CH2CH2COOH
глутамат натрия
Углехимическое сырье
Каменный уголь образовался в результате анаэробного раз-
ложения растительных остатков под действием высоких давле-
ний и температур. Природные ископаемые позволяют просле-
дить все стадии этого процесса, начиная от образования лигни-
топодобного и лигнитового (бурого) угля и кончая битуминоз-
ным углем и антрацитом (твердым углем), который обладает
наибольшим содержанием углерода.
Помимо непосредственного использования в качестве топ-
лива, каменный уголь подвергают переработке. Существует три
основных процесса переработки каменного угля, продукты ко-
торых служат, в частности, исходным сырьем для химической
промышленности: сухая перегонка, газификация и гидрогени-
зация.
Первые два процесса различаются главным образом целью,
с которой они проводятся. При сухой перегонке стремятся полу-
чить максимальное количество кокса, а также жидкие и газо-
образные продукты, тогда как при газификации каменный уголь
превращается преимущественно в газообразные продукты. Од-
нако на практике сухую перегонку ведут таким образом, чтобы
получить либо кокс наряду с жидкими и газообразными побоч-
ными продуктами, либр светильный (коксовый) газ наряду с
коксом и жидкими продуктами.
Сухая перегонка
В процессе сухой перегонки каменный уголь нагревают без
доступа воздуха. При этом отгоняются летучие вещества и об-
разуется кокс. Природа кокса и состав летучих продуктов
зависят от типа каменного угля и температуры его нагревания.
Сухую перегонку' можно проводить при низкой (ниже 600 °C)
Источники сырья для промышленности органического синтеза
33
или высокой (выше 800°C) температуре*. Основные продукты,
образующиеся в обоих случаях, показаны на рис. 2.2.
В высокотемпературном процессе из каждой тонны камен-
ного угля получается от 34 до 47 л смолы (в зависимости от
типа коксовой печи и режима ее работы). Средний выход ка-
Каменный
уголь
сухая перегонка
Кокс <
Газ Легкое масло
(NH3, СО.СЩ.Щ) (ароматические
углеводороды)
I
Смола
(фенолы,
гетероциклические
соединения,
нафталин, антраиен)
Рис. 2.2. Продукты сухой перегонки каменного угля.
менноугольной смолы равен 38 л, что составляет приблизитель-
но 5% от веса исходного угля. Низкотемпературные процессы
дают 57—83 л смолы из каждой тонны каменного угля. На
рис. 2.3 показаны основные фракции, получаемые при перегонке
КаменноуголЬ'
ная смола
—> Легкое масло (т. кип.
до 203 °C)
—> Среднее масло (т. кип.
200-250 °C)
—> Тяжелое (креозотовое)
масло (т. Кип.
250-300 °C)
—> Антраценовое масло
(т. кип. 300—350 °C)
—> Пек
Бензол, толуол, ксилолы
пиридиновые основания,
фенолы, крезолы
Нафталин, фенолы, кре-
золы, пиридиновые осно-
вания
Нафталин, крезолы, кси-
ленолы, хинолин
Антрацен, фенантрен,
карбазол
Рис. 2.3. Продукты перегонки каменноугольной смолы.
каменноугольной смолы. Эти фракции можно подвергнуть даль-
нейшему разделению и получить индивидуальные химические
соединения желаемой степени чистоты.
Металлургический кокс получают путем высокотемператур-
ной сухой перегонки каменного угля, при которой стремятся
достичь максимального выхода кокса, а газ и смолу считают
* В советской литературе низкотемпературную сухую перегонку каменного
угля принято называть полукоксованием, а высокотемпературную — коксова-
нием. — Прим, перев.
2 Зак. 501
34
Глава ‘2
побочными продуктами. Образующаяся в этом процессе смола
содержит большое количество ароматических соединений (около
10% нафталина, 1% фенола и 0,1% пиридиновых оснований).
Последние усовершенствования в сталелитейной промышлен-
ности привели к сокращению расхода кокса (до 570 кг на 1 т
чугуна в чушках в 1969 г. по сравнению с 700 кг в 1960 г. и
1360 кг в 1917 г.), но благодаря росту выпуска стали общий
объем производства каменноугольной смолы остается примерно
на постоянном уровне.
Главной целью газовой промышленности является получение
светильного газа. Однако при этом в качестве побочного про-
дукта всегда образуется кокс. Сухая перегонка каменного угля
в металлических ретортах при температуре около 800 °C дает
низкий выход газа. Повышение температуры позволило увели-
чить выход газа, но образующийся кокс оказался непригодным
для коммунально-бытовых целей. Было найдено, что при низ-
котемпературной сухой перегонке (при 500—600 °C) получается
бездымное топливо лучшего качества. Разработано несколько
таких процессов, каждый из которых приводит к получению
каменноугольной смолы иного состава. Как правило, эти смолы
содержат меньше ароматических углеводородов, чем смола га-
зовых заводов, но зато в них присутствует около 10—12% фе-
нольной фракции с более высоким по сравнению со смолами
высокотемпературного процесса содержанием двух- и трехатом-
ных фенолов. Поэтому они до сих пор остаются единственным
источником получения пирокатехина.
Выпуск продукции газовых заводов Великобритании за по-
следние годы резко снизился, что было вызвано сначала появ-
лением процесса паровой каталитической конверсии нафты, а
затем открытием месторождений природного газа в Северном
море * (табл. 2.6).
Данные табл. 2.7 иллюстрируют все уменьшающееся значе-
ние каменноугольной смолы как источника ароматических со-
единений.
Путем нагревания кокса с негашеной известью в дуговой
электропечи его можно превратить в карбид кальция
СаО + ЗС —> СаС2+ СО
При обработке карбида кальция водой легко выделяется
ацетилен
СаС2 + Н2о —> СаО + С2Н2
* В 1965—1968 гг. на континентальном шельфе Великобритании в южном
бассейне Северного моря было открыто 5 крупных месторождений природного
газа, добыча которого к началу 1976 г. достигла 35 млрд. м3/год. За счет этих
месторождений в 1975 г. покрывалось 95% потребности Великобритании в
газе [Monthly Bull, of Statistics, 30, № 5, 38 (1976); Monthly Digest of Sta-
tistics, 1976, № 365, 61]. — Прим, перев.
Источники сырья для промышленности органического синтеза
35
Динамика Таблица 2,в и структура производства каменноугольной смолы в Великобритании (тыс. т)
Год т. _ Заводы ннзкотемпера- Коксовые Газовые турной сухой перегонки заводы заводы каменного угля
1955 1970 1100 1830 40 935 342 164
Таблица 2.7
Динамика производства продуктов переработки
каменноугольной смолы в США
Про дукт 1950 г. 1953 г. 1967 г. 1969 г. 1971 г.
Каменноугольная смола, млн. л 2835 3410 2950 2910 а, б
Бензол, тыс. т 639 700 303 339 283 в
доля в общем объеме производства бен- зола, % 91 77 9 8 8
Толуол, тыс. т 276 132 64 64,5 49
доля в общем объеме производства то- луола, % 50 26 3 1 2
Ксилолы, тыс. т 236 32,8 1,8 1,7 —
доля в общем объеме производства кси- лолов, % 14 9 1 0,8 —
Нафталин, тыс. т 131 125 236 225 159
доля в общем объеме 100 100 58 58 50
производства наф-
талина, °/о
а Знак минус означает, что данные отсутствуют.
® В 1971 г. в США было произведено 2420 млн. л каменноугольной смолы, а в 1975 г
(предварительные данные)—2820 млн. л [Chem. Marketing Rep., 209, № 19. 54 (1976)].—Прим
перев.
в В 1975 г. выпуск коксохимического бензола в США составил 216 тыс. т. [Chem
Marketing Rep., 209, № 13, 11 (1976)]. — Прим, перев.
Поскольку химия ацетилена чрезвычайно разнообразна, таким
путем можно, исходя из угля, получать многие органические
продукты. Однако, вследствие главным образом высокой энер-
гоемкости, карбидный метод производства ацетилена стал вы-
тесняться методами, основанными на использовани нефтяного
сырья. Одновременно сокращается и применение ацетилена как
полупродукта в органическом синтезе (гл. 4).
2*
36
Глава 2
Газификация
При пропускании через слой кокса или каменного угля воз-
духа, паровоздушной смеси, обогащенного кислородом воздуха
или парокислородной смеси образуются преимущественно газо-
образные продукты. В случае использования паровоздушной
смеси получается так называемый генераторный газ, процесс
образования которого включает следующие реакции, протекаю-
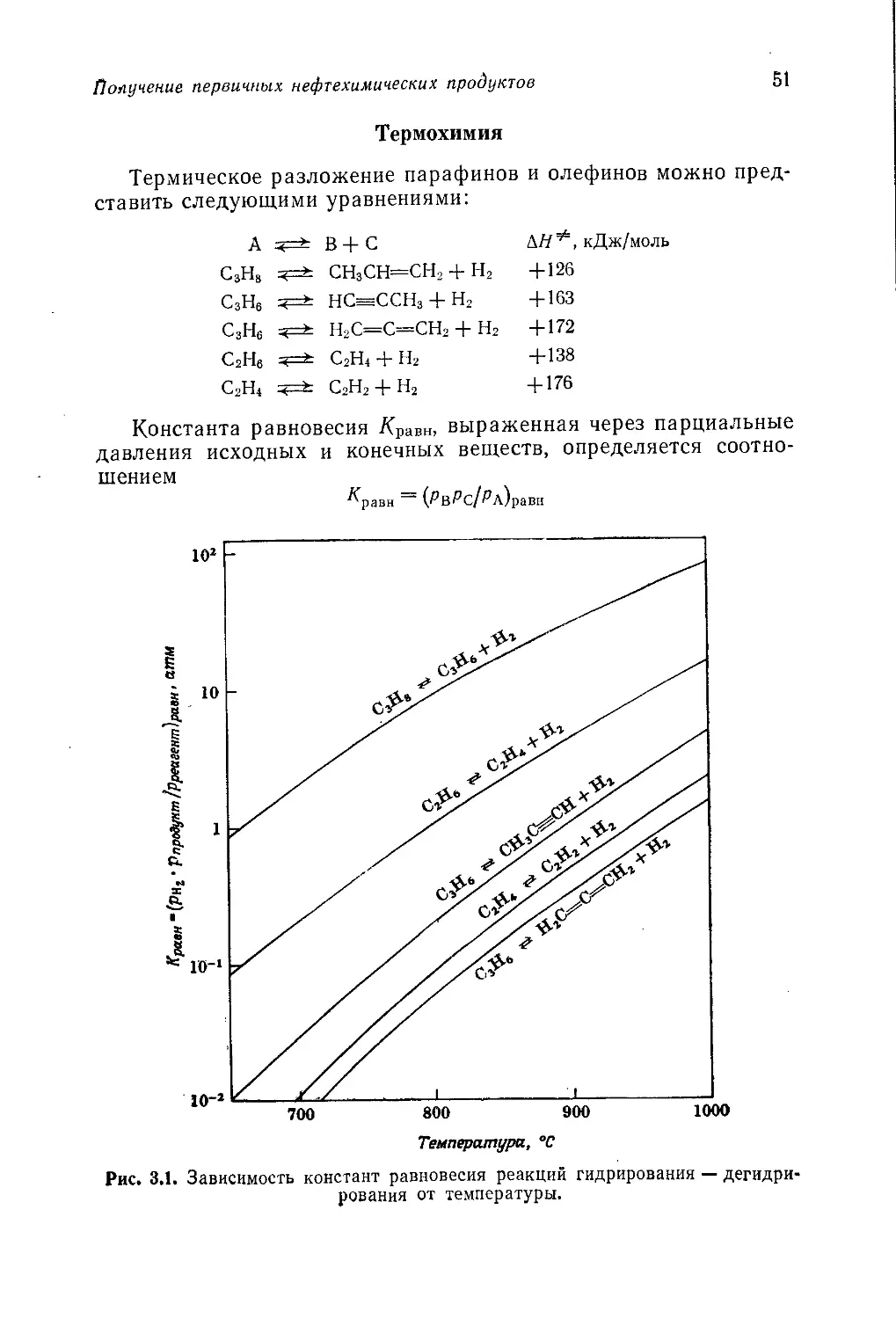
щие в различных зонах слоя:
С + ± О2 =?=* СО
неполное сгорание
С -J- О2 < СО2
полное сгорание
С + Н2О =?=* СО + Н2
С + 2Н2 =?=> СН4
С + СО2 2СО
СО + Н2О СО2+Н2
ДЯ = — 121 кДж/моль (1)
ДЯ = — 405 кДж/моль (2)
ДД = + 120 кДж/моль (3)
ДЯ = — 89 кДж/моль (4)
ДЯ = + 164 кДж/моль (5)
ДЯ = — 43 кДж/моль (6)
Реакции (3)—(5) зависят от давления, причем повышение
давления способствует образованию метана. Это обстоятель-
ство используется в процессе фирмы Lurgi, в котором газифи-
кацию каменного угля или кокса ведут под давлением в при-
сутствии кислорода и воды.
Взаимодействием каменного угля или кокса с водой [урав-
нения (3) и (7)] получают водяной газ. Так как обе эти реакции
эндотермичны, слой топлива периодически подогревают в про-
цессе пропускания через него водяного пара.
С + 2Н2О ч=* СО2 + 2Н2 ДЯ = +78 кДж/моль (7)
Повышение температуры способствует образованию окиси
углерода. Равновесный состав сухого газа при 400 °C включает
33,1% СО2 и 0,2% СО, а при 1000°С —0,3% СО2 и 49,5% СО.
До 50-х годов метод газификации каменного угля использовался
для получения синтез-газа; однако позднее он во многих слу-
чаях был вытеснен методами, основанными на использовании
природного газа и нефти.
Гидрогенизация
Впервые превращение каменного угля в нефть было осуще-
ствлено в 1869 г. Бертло, который действовал на него водородом
в момент выделения. Использование для этой цели молекуляр-
ного водорода в присутствии катализатора было предложено
Бергиусом в 1913 г. Фирма BASF разработала технологию
Источники сырья для промышленности органического синтеза
37
этого процесса, который широко применялся в Германии перед
и во время второй мировой войны. К 1939 г. в Германии насчи-
тывалось 13 действующих заводов по гидрогенизации камен-
ного угля и различных фракций каменноугольной смолы. В Ве-
ликобритании до 1939 г. действовал завод по гидрогенизации
каменного угля фирмы ICI в г. Биллингэме, но во время второй
мировой войны он был переоборудован для получения высоко-
сортного бензина путем гидрирования креозота.
При гидрогенизации пастообразную смесь тонкоизмельчен-
ного угля с нефтью обрабатывают водородом в присутствии
железного катализатора при температуре 480—500°C и давле-
нии 25 МН/м2 (250 атм). В отличие от сухой перегонки камен-
ного угля, которая дает лишь 8—10% смолы от веса взятого
угля, метод гидрогенизации обеспечивает, получение около 75%
сырой нефти. В последние годы, однако, этот метод практически
не используется.
Тем не менее ожидаемое в ближайшем будущем сокращение
запасов природного газа в США вновь пробудило значительный
интерес к газификации нефти и каменного угля. Некоторые из
изучаемых технологических процессов являются традиционны-
ми; в их числе находится процесс газификации под давлением
каменного угля фирмы Lurgi, который уже используется в про-
мышленности для получения газа с невысокой теплотворной
способностью. Еще четыре важных процесса проходят проверку
на опытно-промышленных установках: процесс «Bi-gas» фирмы
Bituminous Coal Research, процесс «COa-Acceptor» фирмы Con-
solidation Coal, процесс «Hygas» Института технологии газа
США и процесс «Synthane», разработанный в Горном бюро
США*.
Широко исследуются также процессы ожижения каменного
угля. Можно назвать четыре таких процесса, расположив их
по степени технологической проработки: процесс «Char Oil Ene-
rgy Development» (COED) фирмы FMC Corporation, заключаю-
щийся в пиролизе каменного угля в псевдоожиженном слое с
получением синтетической сырой нефти, углистого остатка и
газа; процесс «Н-Coal» фирмы Hydrocarbon Research Inc, в ос-
нове которого лежит гидрирование пастообразной смеси камен-
ного угля и нефти в псевдоожиженном слое кобальт-никель-мо-
либденового катализатора; процесс «Low Ash Coal Develop-
ment» фирмы Pittsburgh and Midway Coal Co. (филиал фирмы
Gulf Oil Co.), включающий экстракцию каменного угля при
* По прогнозной оценке, к 1985 г. в США будут ежегодно получать
~ ПО млрд, м3 синтетического газа, расходуя для этой цели 26 млн. т ка-
менного угля в год. К 2000 г. эти цифры возрастут соответственно до
1300 млрд, м3 и 300 млн. т в год [Oil and Gas J., 74, № 2, 59 (1976)]. — Прим,
перса.
38
Глава 2
высоком давлении; процесс «Consol Synthetic Fuels» (CSF)
фирмы Consolidated Coal Co. и Ведомства по исследованиям в
области каменного угля США, по которому каменный уголь
экстрагируют растворителем и экстракт гидрируют, причем по-
лученную сырую нефть можно переработатть в бензин обычными
методами крекинга и риформинга (1967 г. фирма Esso объявила
о своем намерении заняться исследованиями в этом направле-
нии, однако данные о них пока отсутствуют).
Мировая добыча каменного угля в 1971 г. составила, по
оценке, 2,1 млрд, т (из них 0,14 млрд, т было добыто в Велико-
британии, табл. 2.8). Эту цифру следует сопоставить с величи-
ной прогнозных мировых запасов извлекаемого каменного угля,
составляющей 7500 млрд, т (что вдвое больше разведанных за-
пасов, 20% которых, как считают, находится в США) *.
Таблица 2.8
Динамика добычи и потребления каменного угля в Великобритании
(млн. т)
1956 г. I960 г 1971 г.
Добыча 222,0 193,6 144,7
Потребление газовых заводов 27,8 22,3 1,8
Потребление коксовых заводов 29,3 28,5 23,2
Нефть
Сырая нефть и ее характеристики
Сырая нефть, которой практически всегда сопутствует при-
родный газ, имеет самый различный состав, меняющийся не
только от месторождения к месторождению, но и от скважины
к скважине. Главными компонентами нефти являются пара-
фины (алканы), нафтены (циклоалканы) и ароматические угле-
водороды (в основном гомологи бензола).
Первоначально сырые нефти подразделяли на парафиновые,
нафтеновые (названные так по главным компонентам их легких
фракций) и асфальтовые (названные так по главным компонен-
там их тяжелых фракций, имеющим высокий молекулярный вес
* В 1975 г. во всем мире было добыто 2,7 млрд, т каменного угля. Ми-
ровые разведанные запасы извлекаемого каменного угля в 1974 г. оценива-
лись в 4300 млрд, т, а прогнозные запасы — в 8130 млрд, т [Monthly Bull, of
Statistics, 30,. № 7, X (1876); Statistical Yearbook 1975, 27s 186 (1976)].-.
ilpUM. перев..
Источники сырья для промышленности органического синтеза
39
и низкое содержание водорода). Более правильно различать
парафиновые, ароматические и асфальтовые нефти, в каждой
из которых присутствует переменное количество нафтенов. Ве-
роятно, с химической точки зрения было бы удобнее классифи-
цировать нефти как парафиновые, нафтеновые и ароматические
в соответствии с природой преобладающих в их составе углево-
дородов. Однако следует иметь в виду, что миогие соединения,
содержащиеся в нефти, сочетают в своей молекуле сразу не-
сколько структурных фрагментов, например циклическую си-
стему и боковые алкильные группы или насыщенные кольца
вместе с ароматическими.
Кроме того, сырая нефть обычно содержит небольшие коли-
чества органических соединений серы, азота и кислорода.
Перегонка сырой нефти приводит к ее первичному разделе-
нию на жидкие фракции, кипящие в широком температурном
интервале. Приблизительный фракционный состав нефти при-
веден в табл. 2.9. Состав бензина прямой гонки в большинстве
Таблица 2.9
Фракции, получаемые при перегонке сырой нефти
Фракция Пределы выкипания, °C иыд состав
Газ
Бензин (нафта)
Газойль } сРеднйе Дистилляты
Смазочные масла
Котельное топливо
Остаток
Ниже 20
20-200
175-270
200—400
300 (в вакууме)
С4
С4—С12
Св—С|й
С is—С2
С2о—Сзо
случаев соответствует составу сырой нефти, т. е. из высоконаф-
теновой нефти получается бензин с высоким содержанием Наф-
тенов. Содержание ароматических углеводородов и нафтенов в
прямогонном бензине (бензине, полученном перегонкой нефти,
а не другими термическими или химическими методами ее пере-
работки) зависит от пределов его выкипания. Этот вид бензина
более принято называть (особенно в нефтехимической промыш-
ленности) нафтой; его конечная температура выкипания колеб-
лется в пределах от 160 до 190°С (табл. 2.10).
Следует отметить, что бензин прямой гонки, как правило,
не может непосредственно служить топливом для двигателей
внутреннего сгорания, поскольку он обладает низким октано-
вым числом (гл. 14).
Как указывалось выше, выход основных фракций при пере-
гонке сырой нефти, так же как и относительное содержание
Таблица 2.10
Состав легкой нафты, полученной из нигерийской нефти
(т. кип. 65—164 °C). Групповой состав фракции С4—С9;
парафины — 31,4%, нафтены — 48,4%, ароматические углеводороды — 6,4%
(данные фирм Phillips и Imperial Petroleum Ltd.)
Компоненты
Содержание,
% по весу
Т кип., °C
Изобутан 0,1 -11,7
н-Бутан 0,4 -0,5
Изопентан 1,6 27,8
н-Пеитаи 1,8 36,1
2,2-Диметилбутан 0,2 49,7
Циклопентан 0,7 49,3
2,3*Диметилбутан 0,5 58,0
2-МеТиЛпентан 2,0 60,3
З-Метилпентаи 1,5 63,3
н-Гексан 2,5 68,7
Метилциклопентан •4,3 71,8
2,2‘Диметилпентан 0,2 79,2
2,4*Диметилпентан 0,3 80,5
Циклогексан 3,8 80,7
3,3-Диметилпеитан 0,1 86,1
1,1 -Диметилцик лопентан 0,7 87,8
2,3-Диметилпентаи 1 1 R 89,8
2-Метилгексан । * >о 90,0
цис-Х ,3-Диметилциклопентан 1,7 91,7
З-Метилгексан 1,4 91,8
транс-Х ,3-Диметилциклопеитаи 1,5 90,7
транс-1,2-Диметилциклопентан 2,4 91,9
З-Этилпентан о 2 93,5
н-Гептан 2,2 98,4
цис-Х, 2- Диметилциклопентан 0,3 99,5
Метилциклогексан 8,9 100,9
1,1,3-Триметилциклопентан 0,9 104,9
2,2-Дйметилгексан 0,1 106,8
Этилциклопентан 0,6 103,5
2,5-Диметилгексан 0,2 109,1
2,4-Диметилгексан 0,3 109,4
1 -транс-2-цис-4-7 риметил циклопентан 0,9 109,3
1-траяс-2-гргс-3-Триметилциклопентан J 1,5 110,2
3,3-Диметилгексан 112,0
н-Октаи 1,8 125,7
н-Ноиаи 1,5 150,8
Другие парафины С8 4,4
Другие парафины С9 5,8
Другие парафины С1о 2,1
Другие нафтены С8 16,7
Другие нафтены С9 4,0
Бензол 0,3 80,1
Толуол 1,0 110,6
Этилбензол 0,5 136,2
л- и п-Ксилолы 1,9 1391; 138,3
Источники сырья для промышленности органического синтеза
41
Продолжение табл. 2.10
Компоненты Содержание, % по весу Т. кип , °C
Изопропилбензол 0,2 152,4
о-Ксилол 0,7 144,4
н-Пропилбензол 0,2 159,2
3- и 4-Этилтолуолы 0,5 161,3; 162,0
Мезитилен 0,4 164,7
2-Этил толуол 0,2 165,1
Псевдокумол 0,6 169,3
Гемимеллитол (1,2,3-триметилбензол) <0,1 176,1
Ароматические углеводороды Сю не- 0,2
установленного строения
углеводородов различного типа в нафте, зависит от места до-
бычи нефти (табл. 2.11).
Таблица 2.11
Выход продуктов (% по весу) первичной перегонки сырой нефти
различных месторождений (П — парафины, А — ароматические углеводороды,
Н — нафтены)
Место добычи Нафта Средние дистилляты Котельное топливо
Экофнск (Северное море) 19 (П -54, Н—31, А - 15) 34 47
Нигерийская светлая нефть 25 (П - 40, Н—52, А - 8) 37 38
Монагас (Венесуэла) 1 (П-8, Н — 89, А —3) 19 80
Хасси-Мессауд (Алжир) 30 (П - 65, Н—27, А-8) 40 30
Методы переработки нефти
Если бы при переработке сырой нефти ограничивались ее
перегонкой или другими физическими методами разделения,
сохраняющими неизменным химический состав нефти, то таким
образом не удалось бы удовлетворить мировую потребность в
бензине, а его октановое число было бы слишком низким. По-
этому используются химические методы переработки нефти, в
результате которых природа ее компонентов изменяется. Наи-
более важными методами нефтепереработки являются крекинг,
риформинг, алкилирование, полимеризация и изомеризация.
а) Крекинг. В процессе крекинга происходит разрыв связей
G—С и С—Н в углеводородной цепи с образованием промежу-
42
Глава 2
точных радикалов, которые затем превращаются в парафины и
олефины меньшего молекулярного веса. Три основные модифи-
кации крекинг-процесса — термический крекинг, каталитический
крекинг и гидрокрекинг — рассматриваются в гл. 3.
СН3(СН2)ХСН2СН2(СН2)//СН3 —>
—> СН3(СН2)Х_1СН=СН2+ CH3(CH2)i,CH3
б) Риформинг (гл. 3). В отличие от крекинга, при котором
тяжелые фракции превращаются в легкие путем расщепления
молекул углеводородов, риформинг основывается на изменении
молекулярной структуры углеводородов. Процесс риформинга
используют не для увеличения выхода бензина, а для улучше-
ния его качества. При каталитическом риформинге происходят
следующие основные реакции: дегидрирование нафтеновых угле-
водородов в ароматические с выделением водорода, изомериза-
ция циклических углеводородов (превращение алкилциклопен-
танов в циклогексаны) с последующей ароматизацией, дегидро-
циклизация парафинов в ароматические углеводороды и изоме-
ризация н-парафинов в изопарафины.
Кроме того, возможно протекание реакций гидрокрекинга,
таких, как деалкилирование боковых цепей циклических соеди-
нений и гидрирование олефинов.
в) Алкилирование. В нефтепереработке алкилированием на-
зывают реакцию между олефином (например, этиленом или изо-
бутиленом) и парафином (например, изобутаном), приводящую
к образованию более тяжелых соединений разветвленной струк-
туры, обладающих высоким октановым числом. Полученные
продукты часто используют в качестве компонентов наиболее
ходовых марок компаундированного бензина. На первых уста-
новках алкилирования экзотермический процесс осуществляли
в жидкой фазе при температуре 5—20 С° в присутствии серной
кислоты. В качестве катализатора можно также применять без-
водный фтористый водород (при 25—45 °C) или хлористый алю-
миний. Парафин берут в избытке, чтобы подавить реакцию
между двумя молекулами олефина (полимеризацию).
А1С13
СН2=СН, + (СН3)2СНСН3 -----> (СН3)2СНСН(СН3)2
этилен нзобутан 2,3-днметилбутан
H2so4
(СН3)2С=СН2 + (СН3)2СНСН3 -----> (СН3)2СНСН2С(СН3)з
изобутилен изобутан 2,2,4-триметилпентан
г) Полимеризация. Термин «полимеризация» в нефтеперера-
ботке относится к каталитическому превращению газообразного
олефина в жидкие непредельные олигомеры. На первых уста-
Источники сырья для промышленности органического синтеза
43
новках по полимеризации нефтяного сырья с высоким содер-
жанием олефинов использовали термический процесс (при тем-
пературе 500°C); однако позднее был разработан каталитиче-
ский процесс, что позволило повысить выходы и упростить упра-
вление реакцией. Если катализатором служит кизельгур,
пропитанный фосфорной кислотой, то при температуре 150—
220°C и давлении 3,5—5,0 МН/м2 (35—50 атм) достигается
90%-ная конверсия олефина. Запатентованы также другие ката-
лизаторы, такие, как серная кислота, хлористый алюминии и
силикат алюминия
Н+ (СНз)2С=СН2
(СН3)2С=СН2 —► (СН3)3ССН=С(СН3)2 ------------>- тример
изобутилен 2,4,4-триметилпентен-2
В последние годы метод полимеризации потерял прежнее зна-
чение, так как он дает топливо с более низким октановым чис-
лом, чем другие методы; кроме того, считают, что высокое со-
держание олефинов в моторном топливе приводит к загрязне-
нию воздуха. •
д) Изомеризация. Под изомеризацией в нефтепереработке
подразумевают перегруппировку н-парафинов в разветвленные
изомеры. Как уже отмечалось, реакции изомеризации в неболь-
шой степени протекают в некоторых из уже рассмотренных про-
цессов, например при каталитическом риформинге. В качестве
исходного сырья для изомеризации используют н-бутан, н-пен-
тан и н-гексан, а в качестве катализатора — хлористый алюми-
ний или платиновые катализаторы, которые меньше корроди-
руют аппаратуру. Так, процесс «пентафайнинг», предназначен-
ный для изомеризации смесей пентана, гексана и гептана, ведут
при температуре 300—400 °C над платиновым катализатором на
пористом носителе в присутствии водорода.
Полагают, что основная реакция протекает по приведенному
ниже механизму; однако следует отметить, что для ее начала
необходимо присутствие следов хлористого водорода и олефина.
СН3СН2СН2СН2СНСН2СН2 < -t
гептан
+ А1С13
---» СН3СН2СН2СН2СНСН2^-СН3 -f- [HAlCIj]-
CH3CH2CH2CH2CHCH3 < - СН3СЦ2СН2СН2СД—сй2
, (ИА1СЫ'
г-регпилеексан
44
Глава 2
Об относительном значении различных методов химической
переработки нефти в современной промышленности можно су-
дить по данным, приведенным в табл. 2.12.
Таблица 2.12
Мощности по переработке нефти в США
(на январь 1969 г.) а
Методы переработки
Мощность, тыс т/год
Каталитический крекинг
Каталитический риформинг
Гидрокрекинг
Гидроочистка
Алкилирование
с H2SO4
с HF
Полимеризация с Н3РО4
Изомеризация с А1С13
290
127
25
190
23
9
} 12
а На 1 января 1976 г. мощности по переработке нефти в США
имели следующую структуру (тыс. т/год): каталитический кре-
кинг—237, каталитический риформинг —180, гидрокрекинг —45, гидро-
очистка— 326, алкилирование с H2SO4 и с HF —27 и 17 соответ-
ственно и изомеризация с А1С13 —17 [Oil and Gas J., 74. № 13, 56,
57 (1976)].—Прим, перев.
Помимо рассмотренных выше, существует много других
процессов, которые играют важную роль в нефтеперерабаты-
вающей промышленности. Некоторые из них являются вспомо-
гательными при решении основной задачи — превращении сырой
нефти в товарные продукты. Сюда относятся гидрогенизацион-
ная очистка (для удаления серу-, кислород- и азотсодержащих
примесей), сольвентная очистка (экстракция селективными рас-
творителями) и дезодорирующая сероочистка. Дальнейшее рас-
ширение возможностей нефтепереработки обеспечивает исполь-
зование молекулярных сит, которые позволяют отделить «-пара-
фины от изопарафинов и циклопарафинов. Одной из главных
причин извлечения н-иарафинов из нефтяных фракций типа
газойля является большая потребность нефтехимической про-
мышленности в высокочистых «-парафинах С12 — Cis, которые
служат исходным сырьем в производстве моющих веществ и
пластификаторов.
Объем мировой добычи и потребления нефти
и ее ресурсы
Мировая добыча сырой нефти в 1972 г. составила 2,6 млрд, т,
что практически равно объему ее потребления, составившему,
Источники сырья для промышленности- органического синтеза
45
по оценке, 2,59 млрд. т. Среднегодовой прирост потребления
нефти в период 1966—1971 гг. равнялся 7,8%, а к 2000 г. уро-
вень потребления нефти, как полагают, втрое превзойдет ны-
нешний *.
В свете этих данных интересно оценить прогнозные мировые
запасы сырой нефти. Сведения о достоверных запасах место-
рождений нефти регулярно публикуются в печати, и, несмотря
на непрерывный рост ее потребления, общие разведанные за-
пасы нефтяных месторождений постоянно увеличиваются. В на-
стоящее время они оцениваются в 90,9 млрд, т (из которых 53%
на Ближнем Востоке) **.
Разведанные запасы нефти, вероятно, составляют лишь не-
большую часть действительных запасов, поскольку они не вклю-
чают дополнительные «потенциальные» резервы эксплуатируе-
мых месторождений и нефть еще не открытых месторождений.
Запасы последних в отличие от разведанных запасов невоз-
можно оценить сколько-нибудь точно, и такая оценка неизбежно
будет в значительной степени субъективной. Тем не менее в
1958 г. Геологическое управление США, детально проанализи-
ровав все данные, пришло к выводу, что мировые запасы нефти,
поддающейся извлечению если не сейчас, то в будущем, по-ви-
димому, превышают 190 млрд. т. Более поздние подсчеты увели-
чили эту цифру до 450 млрд, т, однако и последнюю величину
нельзя считать окончательной.
Помимо указанных выше дополнительных источников сырой
нефти, существуют такие ее потенциальные источники, как неф-
теносные сланцы, битуминозные пески и каменный уголь. Со-
гласно оценке, запасы обширных отложений нефтеносных слан-
цев в бассейне реки Грин-Ривер (приток реки Колорадо, США)
эквивалентны 10 млрд, т извлекаемой нефти. Огромным неф-
тяным потенциалом обладают также залежи битуминозных
.песков в районе озера Атабаска (север провинции Альберта,
Канада). Суммарные мировые запасы всех потенциальных ис-
точников нефти оцениваются Геологическим управлением США
в 1370 млрд, т, причем полагают, что около 62% этого коли-
чества поддается извлечению ***.
* В 1974 г. во всем мире было добыто 2727 млн. т нефти, а в 1975 г.—
2599 млн. т, в том числе 491 млн. т (включая газовый конденсат) в СССР,
413 млн. т в США и 352 млн. т в Саудовской Аравии [Monthly Bull, of Sta-
tistics, 30, № 5 (1976); «Правда», 1 февраля 1976 г.] — Прим, перев.
** Мировые ресурсы извлекаемой нефти возросли к концу 1975 г. до
273 млрд, т, в том числе ресурсы Саудовской Аравии — до 63 млрд, т (23%
мировых запасов нефти) [Oil and Gas J., 74, № 9, 77 (1976)]. — Прим, перев.
*** В связи с этим представляют интерес прогнозные данные о производ-
стве синтетического жидкого топлива в США в 1980—2000 гг. (в пересчете
на сырую нефть, тыс. т) (продолжение сноски см. на след, стр.);
46
Глава 2
Природный газ
Природным газом называют газ, добытый из подземных
месторождений и содержащий главным образом парафиновые
углеводороды. Чаще всего встречаются газовые месторождения
трех типов:
1. Чисто газовые месторождения, в газе которых при атмо-
сферном давлении и обычной температуре присутствуют лишь
небольшие количества жидких углеводородов. Полагают, что
месторождения этого типа, залегающие обособленно от нефтя-
ных полей, содержат около половины мировых запасов природ-
ного газа.
2. Газоконденсатные месторождения, в которых содержатся
более высокие концентрации жидких углеводородов.
3. Газонефтяные месторождения, в которых газ находится
в виде «газовой шапки» над нефтяным пластом или в виде рас-
творенного газа, выделяющегося при понижении давления в
процессе извлечения нефти из скважины.
Газообразные углеводородные компоненты природного
газа — это в основном метан и этан с переменным количеством
примесей высших парафинов. Газ, содержащий некоторое коли-
чество жидких н-парафинов (пентана, гексана и гептана), назы-
вают жирным газом. Олефины почти никогда не встречаются
в природном газе. Другими возможными компонентами природ-
ного газа являются азот, двуокись углерода, сероводород и
в ряде случаев гелий, концентрация которого может быть доста-
точной для его извлечения.
Природный газ разных месторождений сильно различается
по своему составу (табл. 2.13); более того, состав газа, добы-
ваемого из одной и той же скважины, может изменяться в про-
цессе ее эксплуатации.
Газ многих месторождений США имеет высокое содержание
этана и пропана, что в сочетании с наличием в этой стране
мощной газоперерабатывающей промышленности и развитой
сети газопроводов (общая протяженность магистральных газо-
Исходное сырье 1980 г. 1985 г. 1990 г. 1995 г. 2000 г.
Каменный уголь 0 4,6 25,6 77 205
Нефтеносные сланцы 5.1 25,6 77 103 103
Итого 5,1 30.2 102,6 180 308
[Chem. and Eng. News, 53, № 34, 7 (1975)]. — Прим, перев.
Источники сырья для промышленности органического синтеза
47
Таблица 2.13
Состав (% по объему) природного газа различных месторождений
Состав Лак (Фран- ция) Слохте- рен (Голлан- дия) Хасси- Рмель (Алжир) Сэрмаш Лос-Аид- Дашава (СССР)
(Румы- ния) желес США)
Метан 69,4 81,9 83,5 99,2 77,5 98,0
Этан 2,8 2,7 7,0 — 0,7
Пропан 1,5 0,38 2,0 — —•
Бутаи 0,7 0,13 0,8 — 16,0
Пентан 0,3 0,05 - 0,3 ——
Гексан и высшие 0,3 0,03 0,1 —
парафины Сероводород 15,2 —- 6,5 —
Двуокись углерода 9,5 0,8 0,2 0,1
Азот 0,3 14,0 6,1 0,8 — 1,2
проводов 360 тыс. км) позволяет широко использовать его в
качестве сырья для получения этилена. В Западной Европе
природный газ состоит преимущественно из метана и поэтому
непригоден для переработки в этилен. Газовые фонтаны, кото-
рые действовали в Северном море недалеко от Линкольншир-
ского побережья Великобритании, тоже давали практически
чистый метан. Однако открытые позднее нефтяные месторожде-
ния, расположенные в более северных районах моря, по-види-
мому, содержат значительные количества попутного газа, в
состав которого входят парафины Сг — С5.
Общие разведанные запасы природного газа в мире состав-
ляют, согласно подсчетам, 45,3 трлн, м3 *. В этой связи следует
отметить, что в США, чьи запасы оцениваются в 7,37 трлн, м3,
ежегодно добывается 623 млрд, м3 природного газа **. Эта
ситуация вызывает тревогу американских специалистов и яв-
ляется причиной все более широкого использования газифика-
ции каменного угля и постепенной отмены ограничений на им-
порт в США продуктов первичной перегонки нефти, особенно
нафты, которая служит сырьем для получения этилена ***. Вы-
сокий уровень потребления природного газа отчасти обусловлен
тем, что газ представляет собой один из источников первичной
энергии. Так, в 1972 г. в США сжиганием природного газа было
выработано 34% всей первичной энергии, в то время как для
* В конце 1975 г. мировые разведанные запасы природного газа оце-
нивались в 61,2 трлн, м3 [Oil and Gas J., 74, № 9, 76 (1976)]. — Прим, перев.
** Добыча природного газа (как и нефти) в США начиная с 1973 г.
непрерывно сокращается. — Прим, перев.
*** С мая 1975 г. ограничения на импорт в США нефти и нефтепродук-
тов полностью отменены. — Прим, перев.
48 Глава 2
Великобритании доля природного газа в топливно-энергетиче-
ском балансе составила 12%, а для Японии— 1,0%.
Сжиженным природным газом обычно называют метан, пере-
веденный в жидкое состояние для облегчения его транспорти-
ровки. Сжиженный нефтяной газ — это смесь углеводородов
переменного состава, содержащая преимущественно пропан и
бутан. Источниками сжиженного нефтяного газа служат природ-
ный газ и газы нефтехимических и нефтеперерабатывающих за-
водов. В последнем случае иногда пользуются термином «сжи-
женный нефтезаводской газ». Сжиженный нефтяной газ нефте-
химических и нефтеперерабатывающих заводов обычно содержит
заметные количества олефинов С3 и С4, что отличает его от при-
родного газа.
ГЛАВА 3
Получение первичных
нефтехимических продуктов
В предыдущей главе было показано важное значение нефти
как основного источника сырья для химической промышленно-
сти и приведено краткое описание характеристик сырой нефти и
методов ее переработки. Данная глава посвящена более деталь-
ному рассмотрению технологии получения различных полупро-
дуктов из нефтяного сырья.
I. Крекинг
Дж. Дагганах
Cattanach. J., Research and Development Department,
BP Chemicals International Limited, Epsom
Введение
Нефтехимические полупродукты, такие, как этилен и пропи-
лен, получают преимущественно путем пиролитического расщеп-
ления легких углеводородных фракций в присутствии водяного
пара. Поэтому промышленные установки, работающие по дан-
ному методу, обычно называют установками парового пиролиза.
В США для 90% установок используют в качестве сырья этан и
пропан, извлекаемые из природного газа, тогда как в странах
Западной Европы и в Японии почти исключительно применяются
легкие фракции нефти, известные под общим названием «нафта»
(максимальный интервал выкипаия 35—200°C). Имеются дан-
ные, что в США получил распространение пиролиз газойля (ма-
ксимальный интервал выкипания 175—360 °C) и уже разрабо-
тана технология пиролиза сырой нефти *.
* В 1975 г. в США 25% этилена производили из нафты и газойля, 39% —
из этана и 36% — из попутных газов нефтедобычи и нефтезаводских газов.
В Западной Европе из нафты в 1975 г. получали 95% этилена, из газойля —
4%, из попутных газов нефтедобычи и нефтезаводских газов—1% [Chefp.
Age, J12, № 2965, 10 (1976)]. — Прим, перев.
50
Глава 3
Термическое разложение углеводородов используется в про-
мышленном масштабе с 1912 г. Первоначально его проводили
с целью повышения выхода средних дистиллятов (с интервалом
выкипания 150—340°С). Позднее были разработаны другие ва-
рианты термического крекинга, в частности висбрекинг, при
котором происходит ограниченное расщепление углеводородных
молекул в мягких условиях, приводящее к снижению вязкости
тяжелых дистиллятов (с температурой кипения выше 250°C), и
процессы замедленного коксования и флюид-коксования нефте-
продуктов, в которых термическое расщепление ведут в жестких
условиях, вызывающих полное превращение исходного нефтя-
ного сырья в кокс, средний дистиллят, бензин (с пределами вы-
кипания 50—200 °C) и газообразные продукты. Бензин термиче-
ского крекинга непригоден для современных двигателей внут-
реннего сгорания. Поэтому процесс термического крекинга как
метод переработки нефти на моторное топливо был вытеснен
каталитическим крекингом и гидрокрекингом, при которых одно-
временно происходит глубокое расщепление молекул углеводо-
родов и их быстрая меж- и внутримолекулярная перегруппи-
ровка. Каталитические процессы не требуют применения очень
высоких температур, более селективны и обеспечивают лучшие
выходы легких дистиллятов и высококачественного бензина, чем
термический крекинг.
Каталитический крекинг служит в США главным источником
получения пропилена. Однако в производстве других нефтехими-
ческих полупродуктов процессы каталитического крекинга и
гидрокрекинга находят ограниченное применение, исключая по-
лучение исходного сырья для пиролиза. Количество олефинов,
извлекаемое из нефтезаводских газов, недостаточно для удовле-
творения нужд химической промышленности, вследствие чего
паровой пиролиз приобрел самостоятельное значение как метод
получения олефиновых углеводородов. Полагают, что в настоя-
щее время общий годовой объем мирового потребления этилена
(без социалистических стран) составляет 22 млн. т, а пропи-
лена— 11 млн. т*. Пиролизом жидкого углеводородного сырья
получают также значительные количества других полупродуктов,
таких, как бутадиен, бутилены, изопрен и ароматические углево-
дороды. Современные установки пиролиза нафты имеют годовую
мощность 250—500 тыс. т этилена и потребляют свыше 1 млн. т
сырья в год.
Печи пиролиза конструируются так, чтобы создать оптималь-
ные условия для образования требуемых олефинов. Эти условия
подбирают, исходя из общих химических и физических законо-
мерностей протекания реакций.
* Эти цифры, вероятно, относятся к 1970 г. — Прим, перев.
Получение первичных нефтехимических продуктов
51
Термохимия
Термическое разложение парафинов и олефинов можно пред-
ставить следующими уравнениями:
А В+ С
С3Н8 СН3СН=СН2 4-Н2
С3Н6 =?=> нс==ссн3 + н2
С3Н6 =₽=* Н2С=С=СН2 + н2
C2He =F=b с2н4 + н2
С2Н4 < - С2Н2 -р Н2
Д//^, кДж/моль
4-126
4-163
4-172
4-138
4-176
Константа равновесия Кравн, выраженная через парциальные
давления исходных и конечных веществ, определяется соотно-
шением
^равн ~~ (РвРс/Рл)равн
102
700 800 900 1000
Температура, °C
Рис. 3.1. Зависимость констант равновесия реакций гидрирования — дегидри-
рования от температуры.
52
Глава 3
Рис. 3.2. Зависимость свободной энергии образования углеводородов (Д(?)
от температуры.
/—гексан; 2—циклогексан; 3—этан; 4—пропилен; 5—бутадиен-1,3; этилен; 7—ацети-
лен; 8—метан; 9—бензол.
Зависимость константы равновесия от температуры описы-
вается уравнением Вант-Гоффа
Реакции дегидрирования весьма эндотермичны, вследствие
чего их константы равновесия быстро возрастают с повышением
температуры (рис. 3.1). Следовательно, чем выше температура,
тем больше содержание непредельного соединения в равновес-
ной смеси. Глубину превращения углеводородов при данной тем-
пературе можно рассчитать, по их относительной устойчивости,
которая характеризуется величиной свободной энергии образо-
вания на 1 г-атом углерода (рис. 3.2).
Любая реакция термодинамически возможна только в том
случае, когда она сопровождается уменьшением свободной энер-
Получение первичных нефтехимических продуктов 53
гии (Д6<0), причем в состоянии равновесия свободная энер-
гия системы имеет наименьшую величину. Высокие температуры
способствуют дегидрированию парафинов в олефины, тогда как
низкие облегчают протекание реакции гидрирования
С2Нб С2Н4 + Н2 (температура инверсии 720 °C)
При еще более высоких температурах этилен дегидрируется в
ацетилен
С2Н4 С2Н2 + Н2 (температура инверсии 1152°С)
Поэтому установки парового пиролиза углеводородов на этилен
работают в интервале температур 750—900 °C, тогда как процесс
получения ацетилена ведут при температурах выше 1200°C.
Важное значение при проектировании установок парового
пиролиза имеет высокая термическая устойчивость потенциаль-
ных побочных продуктов, таких, как метан, бензол и другие аро-
матические углеводороды, а также углерод.
Энергия диссоциации связей
Энергии диссоциации связей С—С и С—Н типичных углево-
дородов зависят от ближайшего окружения этих связей
(табл. 3.1). Как правило, связи С—С слабее связей С—Н, при-
Таблица 3.1
Энергии диссоциации связей (кДж/моль) в реакции Ri—R2 Ri’+R2*
R2 R1
н снэ С2н5 азо-СзН; rper-CiHe с6н5
СН3 435 368 356 351 335 389
с2н5 410 356 343 335 322 377
Я-Сзну 410 356 343 335 322 377
изо-С3Н7 397 351 335 326 305 368
трет-Ctfla 381 335 322 305 285 351
С6н5 381 389 377 368 351 418
с5н5сн2 356 301 289 285 268 331
сн2=снсн2 356 343 331 339 — 372
сн2=сн 431 385 372 364 351 414
чем их энергия определяется состоянием гибридизации электро-
нов связи и возрастает с увеличением s-характера этих электро-
нов. Кратные углерод-углеродные связи обладают гораздо более
высокой энергией, чем простые.
С—С С = С feC
D (С—С), кДж/моль 335-368 682 962
54
Глава 3
Прочность простых связей С—С, расположенных рядом с
кратными связями (т. е. a-связей), значительно повышена, что
объясняется увеличением s-характера связывающей орбитали за
счет перекрывания с $р2-орбиталью двойной связи. Последнее
приводит к уменьшению s-характера электронов соседней 0-свя-
зи С—С и ее существенному ослаблению
С=С—С—С
D (С—С), кДж/моль 682 385 343
Аналогичное влияние на энергии а- и p-связей оказывает неспа-
ренный электрон в свободном радикале. Однако учет одного
только простого расщепления наиболее слабых связей не позво-
ляет предсказать строение продуктов, образующихся при пиро-
лизе углеводородов.
Механизм процесса пиролиза
Впервые на роль свободных радикалов как фактора, опреде-
ляющего кинетику пиролиза углеводородов и строение образую-
щихся продуктов, указал более 35 лет назад Райс. Широкие
исследования, проведенные за последние 25 лет, дали большой
объем количественной информации и подтвердили основные
принципы, лежащие в основе предложенного Райсом свободно-
радикального механизма этого процесса (т. 2, гл. 11). Согласно
Райсу, процесс пиролиза углеводородов включает три основные
стадии: инициирование цепи, продолжение цепи и обрыв цепи.
1. Инициирование цепи. Первой стадией пиролиза углеводо-
родов является мономолекулярное гомолитическое расщепление
наиболее слабой связи С—С с образованием двух радикалов,
например:
С12Н26 -—>• С2Н5 • + С]оНг1 •
Вклад реакции расщепления связей С—Н с образованием ато-
марного водорода обычно ничтожен.
2. Продолжение цепи. Небольшие радикалы, образовавшиеся
на стадии инициирования цепи, способны отрывать атом водо-
рода от молекулы исходного углеводорода, причем образуются
радикал большего молекулярного веса и насыщенный углеводо-
род, например:
С2Н5 • + С12Н26 —> С2Н6 + С12Н25 •
Большие радикалы термически неустойчивы и обладают чрезвы-
чайно малым временем жизни. Поэтому они претерпевают рас-
щепление связи С—С, находящейся в 0-положении к радикаль-
ному центру, и превращаются в новый радикал
CgHuCHjCHjCHCHa —> CgH]7CHa • 4- CgHg
Получение первичных нефтехимических продуктов
55
Эта реакция p-распада повторяется до тех пор, пока не полу-
чится сравнительно устойчивый радикал
С8Н17СН2СНгСНСН3 —> СзНб + 4С2Н4 + СНз •
Описанный процесс определяет выход этилена при пиролйзе
«-парафинов. С другой стороны, изомеризация первичных ра-
дикалов высших углеводородов во вторичные, протекающая пу-
тем 1,4- и 1,5-гидридных сдвигов
сгСхс' СГ'С'ЧС с"Счс
| I I I | I
<С с. .С с
сх Сх н Сх •
с последующим p-распадом, приводит к образованию пропилена
СзН7СН2СНСНз —> СзН7. + С3Н6
Небольшие радикалы, не имеющие p-связей С—С, могут распа-
даться с образованием атомарного водорода
С2Н5 • —► С2Н4 + н •
По своей устойчивости при температурах пиролиза сравнительно
стабильные радикалы располагаются в следующий ряд:
Н«> СН3’> СгН5"> изо-С3Н7-> трет-СШэ*. Эти небольшие
радикалы способны вступать в реакцию продолжения цепи, от-
рывая от углеводорода с длинной цепью атом водорода и опять
генерируя радикал большого молекулярного веса
СНз • + CijHse —> СН, + С12Н25 •
Таким образом, имеет место цепной процесс, в котором общая
скорость расщепления исходного углеводорода может значитель-
но превышать скорость его расщепления по реакции иницииро-
вания цепи.
Вероятность образования первичных, вторичных и третичных
радикалов на стадии продолжения цепи определяется числом
первичных, вторичных и третичных атомов углерода в молекуле
исходного углеводорода и относительными константами скоро-
стей отрыва от них атома водорода (т. 3, стр. 313), которые за-
висят от разности энергий активации этих реакций
ОК = й,/й2 = (Л,/Л2)еА£/ет,
где ОК — относительная константа скорости, а А£ = £Втор—
— Дперв = 8 КДЖ/МОЛЬ ИЛИ £трет — £лерв = 17 кДж/мОЛЬ. ОтНО-
сительные константы скоростей отрыва атома Н от вторичного
и третичного углеродных атомов по сравнению с первичным при
825 °C равны 1,7 и 6,4 соответственно. Увеличение числа метиль-
ных групп в парафинах изостроения способствует образованию
третичных радикалов и затрудняет образование первичных
56
Глава 3
радикалов, распад которых приводит к получению этилена. По
этой причине выходы пропилена и высших олефинов из развет-
вленных парафинов выше, чем из неразветвленных.
3. Обрыв цепи. Обрыв цепной реакции происходит путем ги-
бели радикалов, образовавшихся на стадии продолжения цепи,
за счет рекомбинации или диспропорционирования
стенка
Н« + Н« -----► Н2 (этот процесс играет важную роль
при пиролизе этана)
СН3 • + СНз • —> С2Н6
еНд • -р С2Н9 • -> CgHg ИЛИ СН4 -р С2Н4
С2Н9 • -р С2Нд • —► С>Н1(! или С2Н9 -р С2Н4
Описанный свободнорадикальный механизм может быть ис-
пользован для предсказания строения и приблизительного соот-
ношения основных первичных продуктов пиролиза углеводоро-
дов. Обычно принимают, что после небольшого индукционного
периода концентрации радикалов в системе достигают постоян-
ного значения, соответствующего равенству скоростей их образо-
вания и дальнейшего превращения. Фактические концентрации
радикалов довольно малы и в большинстве случаев составляют
10“9—10~13 моль/л. Расщепление углеводородов протекает пре-
имущественно по реакциям продолжения цепи, причем средняя
длина кинетической цепи к, которая является мерой относитель-
ного вклада двух параллельных направлений расщепления
_ скорость расщепления по реакции продолжения цепи
скорость расщепления по реакции инициирования цепи
обычно велика (> 10). Поэтому состав и строение продуктов
пиролиза почти целиком определяются реакциями продолжения
цепи. Относительные скорости отдельных реакций продолжения
цепи зависят от природы и количества образующихся радикалов,
которые в свою очередь определяются строением исходного угле-
водорода.
Приложение рассмотренного механизма
к процессам пиролиза
различных углеводородов
Пиролиз этана и н-пентана протекает по следующим упро-
щенным схемам:
Этан
Инициирование цепи: С2Н6 —> СН3 • + СН3 •
Продолжение цепи: СН9 • + С2Н9 —> СН4 -4- С2Н5 • (передача цепи)
С2Н5 • —> с2н4 -4- Н •
Н’фС?Н6 —> н2 + с2н5.
Получение первичных нефтехимических продуктов
57
стенка
Обрыв цепи: Н • + Н • * Дй
С2Н3 • С2Н3 • > С4Н10
н-Пентан
Инициирование цепи: С3Н]2 —> СН3« + С4Н9«
С5Н12 —> с2н5. + с3н7.
Продолжение цепи: R • + C3Hi2 —► RH-j-CsHu*
где R = Н, СН3, С2Н6;
С5Нц • может быть первичным (а), вторичным (б) или
третичным (в)
а) Цеп И С—С—С—С—С-------> С2Н4 + С3Н7«
С3Н7. --->С2Н4 + СН3-
б)Цеп 2: С—С—С—С—С-------> С3Н6 + С2Н5 •
С2Н5. ---> с2н4 + н.
ЛИЛ
в) Цепь 3: С—С—С—С—С ---> CH 3СНгСН 2 = СН2 и CHS •
где С—С—С означает fl-распад
Обрыв цепи: СН3 • + СН3 • —> С2Н3
сн3 • + С2Н6 • —> С3Н8
Нафтены (циклопарафины) более устойчивы в условиях пи-
ролиза, чем соответствующие парафины. Алкилнафтены спо-
собны деалкилироваться; кроме того, в некоторой степени может
происходить их дегидрирование с образованием ароматических
углеводородов. Основными продуктами пиролиза циклогексана
являются бутадиен, этилен и водород
цнкло-С6Н12 —> С4Н3 + С2Н4 + Н2
Ароматические углеводороды термически устойчивы и, как
правило, не изменяются в условиях пиролиза. Углеводороды с
большими алкильными группами в боковой цепи распадаются
по обычному свободнорадикальному механизму. Связь С—С,
находящаяся в a-положении к ароматическому кольцу, обладает
повышенной прочностью, вследствие чего метильные группы,
присутствующие в исходном углеводороде или образовавшиеся
в процессе деалкилирования, не отщепляются. Основным резуль-
татом пиролиза ароматических углеводородов является образо-
вание продуктов уплотнения, таких, как пек и кокс
ароматические углеводороды 1—► многоядерные ароматические
углеводороды —> кокс
Изложенное выше относится только к пиролизу индивидуаль-
ных углеводородов при сравнительно малой глубине их
58
Глава 5
превращения. Если же пиролизу подвергается смесь углеводо-
родов, то радикалы, генерируемые наименее устойчивыми ком-
понентами смеси, ускоряют расщепление более устойчивых со-
единений. Вместе с тем скорость расщепления наименее устой-
чивых компонентов снижается из-за истощения запаса свобод-
ных радикалов в системе. Типичные углеводороды, присутствую-
щие в нафте, по константам скорости их расщепления в процессе
пиролиза можно расположить в следующий ряд: неразветвлен-
ные а-олефины >' сильно разветвленные парафины > «-пара-
фины изопарафины > шестичленные нафтены > пятичленные
нафтены > ароматические углеводороды. Таким образом, на-
фтены (циклопарафины), которые в индивидуальном состоянии
довольно устойчивы термически, при пиролизе нафты в значи-
тельной степени расщепляются.
Вторичные реакции
С увеличением степени конверсии парциальное давление ис-
ходных углеводородов понижается, а парциальное давление пер-
вичных продуктов их пиролиза повышается вплоть до некоторого
максимального значения, при котором скорость образования
данного продукта равна скорости его дальнейшего превращения.
При еще более высоких степенях конверсии скорости образова-
ния первичных продуктов становятся меньше скоростей их даль-
нейшего превращения, в результате чего концентрации первич-
ных продуктов уменьшаются (рис. 3.3).
Этилен обладает сравнительно большой термической устой-
чивостью; однако он может вступать во вторичные реакции типа
С2Н4 4- С2Н4 —> С2Н3 • 4- С2Н5 •
С2Н5 • -f- С2Н4 —► С2Н3 С2Нз •
С2Н3 • + С2Н3 • —> С4Нв
С+в о
Пропилен Термически менее устойчив и может претерпевать сле-
дующие основные превращения:
С3Н6 —-> С2Н3 • 4- СНз •
СН3«4-С3Н6 —> СН44-С3Н5.
С2Н3 • 4- С3Нд --> С2Н4 4~ С3Н5 •
п (С3Н5 •) —► (C3H3)rt —► ароматические углеводороды 4- Н2
Н«4-С3Н8 —> С3Н7.
С3Н7. —> С2Н44-СН3.
Получение первичных нефтехимических продуктов
59
р<— Первичные -Н
реакции
[-«--Вторичные реакции ---------Н
Время пребывания (произвольные единицы)
Рис. 3.3. Влияние жесткости условий пиролиза легкой иафты на выходы
продуктов.
Бутадиен термически устойчив, но способен вступать в реакции
типа реакции Дильса — Альдера
су-а
Реакции уплотнения олефинов, диенов и ароматических угле-
водородов приводят к образованию многоядерных ароматических
углеводородов, которые претерпевают вторичные превращения,
давая кокс. Вторичные реакции коксообразования особенно энер-
гично протекают в стационарном слое, непосредственно приле-
гающем к стенке реактора. Образующийся кокс приходится
периодически удалять путем его выжигания в токе воздуха.
60
Глава 3
Кинетика пиролиза
Высшие парафины пиролизуются легче, чем низшие, но для
парафинов с пятью и более атомами углерода энергии активации
этого процесса близки. Отсюда следует, что отношение констант
скоростей пиролитического расщепления парафинов Сп и Ст
(п > т > 5) приблизительно равно отношению соответствую-
щих предэкспоненциальных множителей А
. . -EJRT .
kn Апе м________Ап_
km Atrft~EmlR'T А”1
В начальных стадиях реакции скорость пиролиза•парафинов
практически не зависит от давления и отношения площади по-
верхности реактора к его объему. Это, по-видимому, свидетель-
ствует о том, что реакция расщепления является мономолеку-
лярной и протекает по кинетическому уравнению первого по-
рядка (т. 3, стр. 255)
Следовательно, выход продуктов пиролиза должен быть пропор-
ционален расстоянию, проходимому молекулами углеводорода
на начальных стадиях расщепления, а скорость пиролиза — воз-
растать почти вдвое при повышении температуры на каждые 20°.
В действительности скорость пиролиза все же зависит от дав-
ления, а порядок реакции больше единицы и приближается к
1,3. Как показали Райс и Герцфельд, суммарный порядок про-
цесса может изменяться в зависимости от характера реакции
обрыва цепи. Если в ней участвуют небольшие радикалы, обра-
зовавшиеся на стадии инициирования цепи, то суммарный поря-
док равен 1,5, если радикал большого молекулярного веса,
способный претерпевать р-распад, то 1,0, а если два таких
радикала, то 0,5. Промежуточные значения суммарного порядка
свидетельствуют об одновременном протекании нескольких
реакций обрыва цепи.
Исходное сырье
Потенциальный выход олефинов в значительной степени за-
висит от вида исходного сырья. Состав сырья, используемого
в мировом производстве этилена, следующий:
Этан Пропан Бутан Нафта Газойль
Доля, % 36 11 3 47 3
Лучшим сырьем являются парафины, поскольку с повыше-
нием содержания водорода в исходных углеводородах выход
олефинов возрастает (табл. 3.2).
Получение первичных нефтехимических продуктов
61
В табл. 3.3 приведены данные по типичному составу нафты,
которые также показывают, что лучшим исходным сырьем для
Таблица 3.2
Типичный состав продуктов пиролиза (% по весу) различного
нефтяного сырья за один проход а
Сырье и температура пиролиза
Продукт пиролиза этан, 830 °C пропан, 845 °C бутаны, 830 °C иафта, 870 °C газойль, •870 °C сырая иефть
Водород 3,6 1,2 0,7 1,0 0,6 0,8
Метан 7,1 21,1 17,2 19,2 8,6 11,1
Ацетилен 0,3 0,3 0,1 0,6 0,5 1,0
Этилен 46,0 30,3 21,6 32,2 21,3 23,2
Этан (38,3) 3,7 4,4 4,7 2,7 2,8
Пропилен 2,0 19,7 28,2 14,6 13,4 11,6
Пропан 0,4 (18,2) 0,8 0,4 0,4 0,9
Углеводороды С4 2,3 4,0 (21,9) 7,6 9,1 6,0
Бензин —• 1,5 5,1 15,0 24,4 16,5
Котельное топливо — — — 4,0 19,0 26,1
а На промышленных установках по пиролизу этан,а общий выход этилена доходит
до 77% (по весу), что достигается рециркуляцией иепревращеиного этана до 100%-ной кон-
версии. Для увеличения выхода этилена при использовании жидкого углеводородного
сырья можно извлекать образующийся этан из реакционных газов и пиролизовать его.
в печи меньших размеров.
получения олефинов являются ациклические парафины, осо-
бенно н-парафины.
Таблица 3.3
Углеводородный состав типичной нафты
Тип углеводородов Содержание, % по весу Число атомов углерода Коэффициент селективио- а сти
«-Парафины 30 4-12 0,60
Изопарафины 30 4-12 0,40
•Нафтены 25 5-12 0,25
Ароматические углеводороды 15 6-9 0,05
а Суммарный вероятный выход этилена и пропилена (% по весу), который
можно получить при паровом пиролизе углеводородов данного типа, равен произ-
ведению их коэффициента селективности иа весовое содержание. В рассматривае-
мом случае расчетная величина общего выхода целевых олефинов (этилена и про-
пилена) составляет 37% по весу.
62
Глава 3
Параметры процесса пиролиза
Оптимальные условия работы установок парового пиролиза,
обеспечивающие высокий выход этилена, — это короткое время
пребывания в реакционной зоне (0,1—0,4с), высокая темпера-
тура (750—900°C), низкое общее давление [ниже 0,3 МН/м2
(3 атм)], небольшое парциальное давление углеводорода (по-
дача водяного пара 25—100% от веса сырья) и быстрая «за-
калка» реакционных газов с целью подавления вторичных
реакций.
Жесткость режима пиролиза определяется главным образом
двумя независимыми друг от друга параметрами: временем
пребывания и температурой, которые для каждого типа сырья
подбирают таким образом, чтобы получить пирогаз требуемого
состава.
Время пребывания
Реакции, приводящие к образованию целевых олефинов,
можно представить уравнениями типа
С2Нб С2Н4 + Н2
1 — X X X
где х — мольная доля этана, превратившаяся за некоторое про-
межуточное время Степень достижения равновесия (СДР)
связана с константой равновесия /СраВн при времени tx отноше-
нием
СДР = "clH4wha . о < С ДР < 1,0
"с2н6
где п — мольная доля, соответствующая времени t\, и р — общее
давление углеводорода в системе. Следовательно, выход этилена
возрастает с увеличением степени достижения равновесия
(т. е. х—►Хоо при СДР-> 1,0). Однако по мере повышения кон-
центрации этилена возрастает скорость нежелательных вторич-
ных реакций. Поэтому с увеличением времени пребывания вы-
ходы целевых продуктов при данной температуре проходят
через максимум (рис. 3.3). Обычно время пребывания подби-
рают таким образом, чтобы степень достижения равновесия при
данной температуре соответствовала оптимальным выходам про-
дуктов. Как правило, степень достижения равновесия колеблет-
ся от 0,3 до 0,90, причем ее величина берется тем ниже, чем
больше степень конверсии (рис. 3.3). Соответствующие времена
пребывания составляют 0,1—1,0 с.
Получений Первичных нефтехимических продуктов
63
Температура пиролиза
Общая энергия активации для пиролиза этана приблизи-
тельно равна 222 кДж/моль, и скорость его конверсии быстро
увеличивается с температурой. Энергии активации реакций ини-
циирования, продолжения и обрыва цепи составляют приблизи-
тельно 351, 46 и 0 кДж/моль соответственно. С повышением
температуры скорость реакции инициирования цепи и, следова-
тельно, скорость образования свободных радикалов быстро воз-
растают. Напротив, скорость реакций обрыва цепи, приводящих
к гибели свободных радикалов, не зависит от температуры. По-
этому повышение температуры будет способствовать увеличе-
нию концентрации свободных радикалов в системе и возраста-
нию скорости желательных реакций, ведущих к образованию
целевых олефинов.
Поскольку реакция инициирования цепи сильно эндотермич-
на, соответствующая ей константа равновесия быстро возра-
стает с температурой, что способствует образованию свободных
радикалов. Напротив, реакции обрыва цепи сильно экзотер-
мичны, что обусловливает быстрое понижение соответствующих
им констант равновесия с температурой. Реакции продолжения
цепи почти не обладают тепловым эффектом, и соответствую-
щие им константы равновесия практически не зависят от тем-
пературы. В общем, чем выше температура, тем больше кон-
центрации свободных радикалов. Таким образом, смещение
равновесия тоже приводит к увеличению выхода этилена с по-
вышением температуры вследствие возрастания ^paвн
_ 1с2н6 „
, "С2Н4 лравн
Современные установки по получению этилена работают при
температурах от 750 °C (мягкий режим пиролиза) до 900 °C
(жесткий режим пиролиза) в зависимости от требуемого отно-
шения этилена к высшим олефинам. На рис. 3.4 показано, как
изменяется состав продуктов пиролиза с повышением темпера-
туры при одновременном уменьшении времени пребывания с
целью поддержания высокого выхода этилена.
Степень разбавления углеводорода паром
Разбавление углеводорода водяным паром понижает его
парциальное давление и тем самым способствует протеканию
реакций инициирования цепи и |3-распада за счет подавления
реакций обрыва цепи и других бимолекулярных реакций, в ко-
торых участвуют активные радикалы — переносчики цепи.
64
Глава 3
Рис. 3.4. Влияние температуры на состав продуктов пиролиза легкой нафты
/ — этилен; 2—пропилен; 3—топливный газ; 4—бензин; 5 —котельное топливо.
Степень достижения равновесия при дегидрировании выражает-
ся уравнением
Р
17
лравн
СДР = Т37
а выход этилена — соотношением
_____________________________1
1+_Л
2
х =
Типичными яв-
СДР J
Таким образом, низкое парциальное давление углеводорода
способствует увеличению выхода этилена. На практике степень
разбавления углеводорода водяным паром подбирается исходя
из типа сырья и жесткости условий пиролиза. ~
ляются следующие степени разбавления:
Сырье Этан Пропан
Подача пара, % от веса сырья 25—40 30—50
Кроме того, водяной пар понижает точку росы тяжелых про-
дуктов, предотвращая тем самым образование смолы, а также
Нафта Газойль
40-80 80-100
Получение первичных нефтехимических продуктов 65
подавляет реакции отложения кокса, пассивируя поверхность
змеевиков, и облегчает перенос тепла к реагентам. К тому же
водяной пар окисляет кокс по реакции водяного газа
С + Н2О —> СО + Н2
Устройство печи пиролиза
Печь с прямым обогревом
Наиболее распространенным современным типом печи пиро-
лиза для получения олефинов из газообразных нефтепродуктов,
нафты и газойля является трубчатая печь. Это основной агре-
гат в производстве олефинов. Технологическая схема процесса
приведена на рис. 3.5. Печь состоит из двух секций: конвекцион-
ной, в которой сырье испаряется, смешивается с водяным паром
и подогревается до температуры реакции, и радиантной, в ко-
торой подводится тепло, необходимое для прохождения реак-
ции. В каждой печи монтируется от двух до восьми змеевиков,
а сами печи устанавливаются с учетом возможности их попере-
менного выключения для удаления образовавшегося кокса, что
обеспечивает непрерывную работу всей установки. Конфигура-
ция и размеры змеевиков в радиантной секции (где протекает
пиролиз) определяются конструкцией печи и требуемым со-
ставом продуктов. Обычно они имеют длину 30—160 м и диа-
метр 50—120 мм. Температурный профиль змеевиков регули-
руется с помощью газовых горелок, расположенных у стенок
печи.
Реакционные газы быстро «закаливают», охлаждая их до
температуры 300—400 °C, чтобы подавить вторичные реакции.
«Закалка» может быть прямой (впрыскивание в газовый поток
нефтяного масла или воды) и непрямой (втеплообменниках,
включенных в технологическую линию). Отводимое тепло ре-
куперируется для получения пара высокого давления. Если
применяется непрямая «закалка», то после теплообменников
обычно дополнительно помещают скруббер, орошаемый зака-
лочным маслом.
Из куба ректификационной колонны первичного разделения
пирогаза отбирают тяжелую фракцию, смолу и закалочное
масло, а из верхней части колонны — газообразные углеводо-
роды, бензиновую фракцию и водяной пар-разбавитель. Отде-
ление бензина и водяного пара от газообразных продуктов про-
изводится в другой ректификационной колонне.
Полученный пирогаз компримируют в четыре-пять ступеней
до давления около 3,5 МН/м2 (35атм), причем на одной из про-
межуточных ступеней его промывают раствором щелочи для
3 Зак. 501
нг/сн<
---------—>
(Бутадиен-1,3)
Бензин
Ароматические углеводороды С6-С8
Котельное топливо
Рис. 3.5. Технологическая схема пиролиза иафты и разделения образовавшихся продуктов.
/—печь; 2 — закалочный скруббер; 3 — ректификационная колонна первичного разделения продуктов; 4 — парогенератор; 5 — ректифика-
цнонная колонна; 6—холодильник; 7—компрессор; 8—щелочной скруббер; 9—реактор для селективного гидрирования ацетилена.
Получение первичных нефтехимических продуктов 67
удаления кислых газов, таких, как сероводород и двуокись угле-
рода. Ацетиленовые углеводороды обычно удаляют путем селек-
тивного гидрирования пирогаза или его соответствующих фрак-
ций; можно также извлекать их селективными растворителями.
Сжатый газ, содержащий водород и углеводороды, осушают
пропусканием через окись алюминия или молекулярные сита,
охлаждают приблизительно до —70 °C и направляют в демета-
низатор. В качестве хладоагентов в различных холодильных
циклах системы разделения пирогаза используются комприми-
рованные метан, этилен и пропилен. Этилен и пропилен выде-
ляют и очищают путем низкотемпературного фракционирования
под давлением. Этан и пропан возвращают в цикл и пиролизуют
в специальных печах. Из бутан-бутиленовой фракции методом
абсорбции можно извлечь бутадиен. Фракция от С5 и выше,
выкипающая до 200°C (т. е. бензиновая фракция), содержит
значительные количества ароматических углеводородов Сб — С8,
которые можно выделить экстракцией (гл. 5). По другой схеме
присутствующие диены подвергают селективному гидрированию
и полученную фракцию используют как моторное топливо.
Другие процессы расщепления углеводородов
Получение линейных а-олефинов
Лучшим сырьем для получения а-олефинов является твер-
дая парафиновая фракция, содержащая высшие н-парафины
Си — С34. В мягких условиях крекинга (температура ~550°C,
время пребывания 3 с) целевые а-олефины С6 — Сго довольно
устойчивы. Их образование протекает по следующим уравнениям:
сн3« + С)0Н2)СН2СН2СН2С7Н15 —> СН4 + CioH21CHCH2CH2C7Hls
с10н21снсн2сн2с7н15 —> с10н21сн=сн2 +с7н!5сн2.
Таким образом, получается смесь со статистическим распреде-
лением а-олефинов, причем содержание каждого олефина тем
ниже, чем выше его молекулярный вес.
Процессы с непрямым, обогревом
Трубчатые печи обычно не позволяют использовать в каче-
стве сырья нефтепродукты с конечной температурой выкипания
выше 350 °C из-за значительного коксообразования. Однако
разработаны новые процессы пиролиза, в которых необходимое
для реакции тепло подводится с теплоносителем, предваритель-
но подогретым до температуры, значительно превышающей
температуру реакции. Теплоносителем могут служить твер-
дые вещества (кокс, огнеупорные материалы), жидкости
3*
68
Глава 3
(расплавленные металлы или соли) или водяной пар, перегре-
тый до температуры 2000 °C. Этим методом можно проводить и
пиролиз тяжелых нефтепродуктов, включая сырую нефть. Кокс
удаляется из реакционной зоны вместе с теплоносителем, а теп-
ло, полученное при его сжигании в отдельном реакторе, утили-
зируется для нагревания рециркулирующего теплоносителя.
В зависимости от того, что хотят получить — этилен или ацети-
лен, температуру пиролиза варьируют в пределах 780—1500°С, а
время пребывания — в интервале 1,0—0,1 с соответственно. Вы-
ход этилена из сырой нефти составляет 20—30% по весу.
Процесс Вульфа был разработан для получения ацетилена
и является циклическим. Сначала керамическую кладку печи,
имеющую форму пчелиных сот, подогревают горячими топоч-
ными газами. Затем в печь подают разбавленный водяным па-
ром исходный нефтепродукт, который пиролизуется на раска-
ленной керамической кладке. После этого начинается новый
цикл подогрева кладки печи.
Автотермический крекинг
При автотермическом крекинге* для получения необходи-
мого тепла сжигают часть исходного сырья, в результате чего
общий процесс становится самоподдерживающимся. Сжигание
можно проводить по так называемому методу погружного пла-
мени, но обычно его осуществляют в нижней части реактора,
используя для более полного сгорания топлива принудительную
циркуляцию через пламя твердого неорганического материала.
Температура в реакционной зоне составляет 860—970 °C, время
пребывания — около 0,2 с.
Процессы электрокрекинга
В последние годы значительный интерес привлекла идея о
непосредственном использовании электроэнергии для промыш-
ленного получения первичных химических продуктов в плазмен-
ной струе. Этим методом можно создать температуры выше
8000 °C, при которых молекулы всех углеводородов будут дис-
социировать до атомов. Строение продуктов будет зависеть от
быстроты «закалки» реакционных газов, но в любом случае эти
продукты должны иметь небольшой молекулярный вес. Такие
условия являются благоприятными для образования ацетилена,
и его прямое получение из метана, нафты, сырой нефти или
даже каменного угля технически вполне возможно.
* В советской литературе этот процесс обычно называют термоокисли-
тельным пиролизом или методом частичного окисления. — Прим. ред.
Получение первичных нефтехимических продуктов
69
Каталитический крекине
Основная часть пропилена получается в промышленности
как побочный продукт каталитического крекинга газойля на
бензин. Каталитический крекинг — низкотемпературный про-
цесс, который обычно ведут при 450—600 °C в присутствии
сильнокислотного катализатора, содержащего кристаллический
алюмосиликат (молекулярные сита). Крекинг парафинов про-
текает по карбониево-ионному механизму, включающему ста-
дию p-расщепления, аналогичного р-распаду свободных ради-
калов. Небольшой карбониевый ион реагирует с парафином,
давая карбониевый ион большего молекулярного веса, который
расщепляется на олефин и первичный карбониевый ион
С3Н3 + С12Н26 с3н6+с12н+5
с12н2+6 —> СД + С9Н*
Последний быстро изомеризуется в более устойчивый вто-
ричный карбониевый ион
С.Н,.СН„СН,СН,СН: —> С-НцСНХИХНСН,
Oil L t, i 4 О 1 1 X О
р-Расщепление этого вторичного карбониевого иона приво-
дит к образованию пропилена
С6НнСН2СН2СНСН3 ___> с6нпсн+ + с3н6
Процесс продолжается вплоть до образования наименьших кар-
бониевых ионов, содержащих три атома углерода, которые на-
чинают новую цепь. Выход пропилена составляет 2—4% веса'
исходного сырья.
Кроме каталитического крекинга, низшие олефины получают
в промышленности (правда, в меньшем количестве) в процессе
Фишера — Тропша, термическим крекингом и при газификации
каменного угля.
Гидрокрекинг
Процесс гидрокрекинга в последние годы получил широкое
распространение (в качестве альтернативы каталитическому
крекингу) как метод превращения тяжелых дистиллятов сырой
нефти в более легкие парафиновые фракции. Последние, начи-
ная от пропана и бутана и кончая нафтой, являются важным
сырьем для получения олефинов. Гидрокрекинг ведут при тем-
пературах до 450 °C и давлении 15—20 МН/м2 (150—200 атм)
в присутствии избытка водорода на бифункциональном катали-
заторе типа металлов, осажденных на кислотных цеолитах.
70
Глава 3
II. Риформинг
Т. Эдмондс
Edmonds Т., Petroleum Division, BP Research Centre, Sunbury-on-Thames
Введение
Каталитический риформинг — один из важнейших нефтехи-
мических процессов, который применяется для повышения окта-
нового числа легких дистиллятов, с тем чтобы их можно было
Рис. 3.6. Зависимость октанового числа от числа атомов углерода для раз-
личных компонентов легких дистиллятов.
/—олефины; 2 — ароматические углеводороды; 5 —нафтены; 4—диметилпарафины; 5—ме-
тилпарафины; 6 — н-парафины.
использовать в качестве топлива в современных мощных двига-
гелях внутреннего сгорания. Октановое число моторного топ-
лива является его функциональной характеристикой *. Однако
оно связано с химическим строением углеводородов, входящих
в состав топлива. На рис. 3.6 показаны общие закономерности
изменения октанового числа в зависимости от числа атомов
углерода для различных компрнентов легкого дистиллята, вы-
кипающего в интервале 30—200 °C. Этот дистиллят представ-
* Октановое-число определяют в одноцилиндровом двигателе с перемен-
ной степенью сжатия путем сравнения испытуемого топлива со смесью изоок-
тана (2,2,4-триметилпентана) и «-гептана. Оно представляет собой процент-
ное содержание (по объему) изооктана в смеси, которая детонирует в двига-
теле с той же частотой, что и испытуемое топливо в аналогичных условиях
Детонационная стойкость изооктана условно принята за 100, а октановое чис-
ло «-гептана равно нулю.
Получение первичных нефтехимических продуктов
71
ляет собой сложную смесь парафинов, циклопарафинов (наф-
тенов) и ароматических углеводородов, содержащих 5—11 ато-
мов углерода. Конкретный групповой состав дистиллята опреде-
ляется природой исходной сырой нефти. Типичные составы и
октановые числа легких дистиллятов, полученных из кувейт-
ской, иранской светлой и ливийской нефтей, приведены в
табл. 3.4. Товарный бензин должен иметь октановое число выше
Таблица 3.4
Характеристики легких дистиллятов, полученных из кувейтской (I),
иранской светлой (II) и ливийской (III) нефтей
Пределы выкипания, °C
Характеристика
30-65 65-145 145-190
I II Ш I II Ш I II III
Выход, % от веса
сырой нефти
Общее содержание
серы, ч. на млн.
(по весу)
Углеводородный со-
став, % по весу
парафины
нафтены
ароматические
углеводороды
Октановое число
2,9 2,7 2,3 10,02 12,71 10,15 6,83 7,65 6,7
200 910 17 240 880 5 960 1200 50
97,5 98 94 72 56 55 64 45,5 51
2,5 2 5,2 19 31 40,4 18 35 39,5
0 0 0,« 9 13 4,6 18 19,5 9,5
71,5 71,5 72 44 50 51,5 30 35 22
90. Таким образом, дистилляты, представленные в этой таб-
лице, обладают слишком низким октановым числом. Данные
рис. 3.6 показывают, что для повышения октанового числа не-
обходимо увеличить содержание в топливе ароматических угле-
водородов, разветвленных парафинов и олефинов при одновре-
менном уменьшении среднего молекулярного веса топлива. Эта
цель достигается при помощи каталитического риформинга, ко-
торый ведут в условиях, способствующих максимальному обра-
зованию ароматических углеводородов. Выход последних на-
столько высок, что становится экономически целесообразным
выделение из продукта риформинга индивидуальных аромати-
ческих углеводородов для химической переработки, особенно
при тщательном подборе исходного сырья по пределам выкипа-
ния и углеводородному составу.
Реакции, происходящие в процессе каталитического рифор-
минга, отражают сложность состава исходного сырья. Их можно
подразделить на три основные группы: реакции дегидрирова-
ния (а, б), изомеризации (в, г) и гидрокрекинга (д, е).
72
Глава 3
Реакции (ж) и (з) включают одновременное дегидрирование и
изомеризацию.
а) Дегидрирование циклогексанов в ароматические углеводо-
роды
цикло-СбН|2 —► С6Нв + ЗН2
б) Дегидрирование парафинов в олефины
С6Нц —> СвН12 + Н2
в) Изомеризация алкилциклопентанов в циклогексаны
г) Изомеризация н-парафинов в изопарафины
н-гексан —> 2- и 3-метилпентан, диметилбутаны
д) Дегидроциклизация парафинов в ароматические углеводороды
H-CgHu -----------------------> CgHg + 4Н2
е) Дегидроизомеризация алкилциклопентанов в ароматические
углеводороды
> CgHg + ЗН2
ж) Гидрокрекинг парафинов
С„Н14-1-Н2 —> 2С3Н8
з) Деалкилирование аел-диалкилнафтенов, образовавшихся в ре-
зультате дегидроциклизации парафинов
Большинство углеводородов, содержащихся в исходном сырье,
реагирует в процессе риформинга сразу по нескольким направ-
лениям (а — з).
Катализатором обычно служит платина, нанесенная на вы-
сокочистую окись алюминия. Такой катализатор имеет актив-
ные центры двух типов: металлические, на которых происходят
реакции гидрирования — дегидрирования, и кислотные центры
носителя, на которых осуществляется изомеризация. Реакции
гидрокрекинга могут протекать как на тех, так и на других цент-
рах. Концентрация платины в катализаторе составляет 0,3—
1,0% (по весу), причем платина должна быть нанесена таким
образом, чтобы как можно большая часть ее атомов была до-
Получение первичных нефтехимических продуктов
73
ступна для молекул исходных углеводородов. Обычно платину
наносят в форме хлороплэтиновой кислоты, которая оставляет
на поверхности окиси алюминия 0,5—1,0% (по весу) атомов
хлора, образующих кислотные активные центры.
Платиновый катализатор весьма чувствителен к отравлению
сернистыми соединениями. Поэтому сырье, поступающее на
риформинг, предварительно подвергают обессериванию до оста-
точного содержания серы ниже 3 ч. на млн. (по весу) (данные
о содержании серы в легких дистиллятах приведены в табл. 3.4).
Процесс ведут при температуре 500—525 °C и давлении
1,0—4,0 МН/м2 (10—40 атм). Для предотвращения происходя-
щей в этих жестких условиях быстрой дезактивации поверхно-
сти катализатора углеродистыми отложениями необходимо
присутствие водорода. На практике при проведении процесса
риформинга водородсодержащий газ вместе с парами углеводо-
родного сырья циркулирует через стационарный слой катали-
затора. Продукты реакции охлаждают, конденсат отделяют
и выводят из системы, а газы (содержащие главным образом
водород) возвращают в реакционную зону.
В табл. 3.5 приведены сравнительные данные по углеводо-
родному составу исходного сырья и катализата риформинга.
Эти данные показывают, что основная задача риформинга —
увеличение концентрации ароматических углеводородов при
Таблица 3.5
Углеводородный состав (% по весу) исходного сырья и катализата
риформинга, имеющих октановые числа 60 и 100 соответственно
Число атомов углерода Парафины Нафтены Ароматические углеводороды
сырье катализат сырье катализат сырье катализат
3 0 Следы
4 0,1 2,0
5 1,9 6.8 0,1 0,3
6 6,4 9,3 2,5 0,6 0,4 4,4
7 12,1 8,2 6,6 0,5 3,6 17,6
8 15,6 4,4 6,8 0,3 5,6 23,0
9 13,3 1,3 5,6 0,1 4,3 16,4
10 8,8 0,2 3,0 Следы 1,0 4,1 *
11 1,8 0 0,5 0 0 0,5
— 1
Итого 60,0 32,2 25,1 1,8 14,9 66,0
одновременном уменьшении среднего молекулярного веса (что
обеспечивает повышение октанового числа)—успешно выпол-
нена.
74
Глава 3
Термодинамика процесса
Условия, необходимые для получения максимального вы-
хода ароматических углеводородов, определяются термодина-
микой протекающих реакций. В табл. 3.6 приведены некоторые
термодинамические константы равновесия и теплоты реакций
при 500 °C для углеводородов Се-
Таблица 3.6
Термодинамические параметры реакций каталитического риформинга
Реакции
&Н, кДж/моль
Циклогексан бензол + ЗН2 6- 10
Метилциклопентан циклогексан 8,6-10
к-Гексан бензол + 4Н2 7,8-10
к-Гексан 2-метилпентан 1,1
«-Гексан 3-метилпентан 7,6-10“'
к-Гексан гексен-1 + Н2 3,7 • 10“2
+221
-15,9
+226
-5,9
-4,6
+ 130
Значения констант равновесия, представленные в табл. 3.6,
отвечают парциальным давлениям реагентов и продуктов, вы-
раженным в атмосферах. Чем выше молекулярный вес углево-
дородов, тем более благоприятна термодинамика процесса для
ароматизации нафтенов и дегидроциклизации парафинов и тем
менее благоприятна она для изомеризации парафинов и алкил-
циклопентанов.
Реакции образования бензола из циклогексана и н-гексана
сильно эндотермичны и сопровождаются выделением водорода.
Теоретически для получения максимального выхода аромати-
ческих углеводородов следует использовать возможно более
высокую температуру и возможно более низкое парциальное
давление водорода. Однако на практике оба эти параметра ли-
митируются условиями, в которых происходит быстрая дезак-
тивация катализатора из-за отложения на нем кокса. Высокая
температура и низкое парциальное давление водорода способ-
ствуют также образованию олефинов. Хотя эта реакция не
протекает в сколько-нибудь существенной степени в обычных
условиях процесса риформинга, она имеет важное значение, по-
скольку на некоторых его стадиях олефины получаются как
промежуточные соединения. Равновесие между метилциклопен-
таном и циклогексаном благоприятствует образованию первого
соединения, но вследствие быстрого дегидрирования циклогек-
сана в бензол это равновесие нарушается. В равновесной си-
стеме «-гексан— метилпентаны термодинамически наиболее вы-
годным изомером является 2-метилпентан. Кроме того, термо-
Получение первичных нефтехимических продуктов 75
динамические данные показывают, что при температуре 500°C
в этой системе теоретически должно содержаться 30—35% Ди-
метилбутанов.
Степень достижения равновесия для индивидуальных компо-
нентов исходного сырья над типичными катализаторами рифор-
минга была детально изучена, и при этом сделаны следующие
выводы:
1) циклогексаны быстро превращаются в равновесную смесь
ароматических углеводородов;
2) из метилциклопентана получается равновесное количе-
ство бензола (если не учитывать образование продуктов рас-
щепления) ;
3) концентрации диметилпарафинов, образующихся при изо-
меризации н-парафинов, отличаются от равновесных.
Однако необходимо иметь в виду, что данные, полученные
для индивидуальных соединений, не всегда позволяют точно
предсказать поведение смесей, так как компоненты смеси, на-
ходясь на поверхности катализатора, могут оказывать влияние
друг на друга.
Механизм каталитического риформинга
Детальный механизм реакций, протекающих в процессе ка-
талитического риформинга, неизвестен. Были изучены превра-
щения циклогексана, метилциклопентана, циклогексена и метил-
циклопентена над тремя различными катализаторами, один
из которых катализировал только изомеризацию, второй
н- Гексан Изогексаны
Циклогексан
Метилциклопентан < ? н-Гелеен
Изоеексены
Циклогексен Метилциклопентен
Циклогексадиен Метили иклопентадиен
Бензол
Схема реакций, протекающих при риформинге углеводородов Са
76
Глава 3
дегидрирование, а третий обладал каталитической активностью
и того, и другого типа (бифункциональный катализатор). Полу-
ченные результаты позволили предложить схему реакций ри-
форминга углеводородов С6, приведенную выше. Реакции, отме-
ченные вертикальными стрелками, протекают на металлических
активных центрах гидрирования — дегидрирования, а реакции,
отмеченные горизонтальными стрелками, — на кислотных актив-
ных центрах носителя.
Согласно этой схеме, активные центры дегидрирования необ-
ходимы только для образования бензола из циклогексана, вслед-
ствие чего данный процесс протекает как над дегидрирующим,
так и над бифункциональным катализатором. С другой сторо-
ны, дегидроизомеризация метилциклопентана в бензол проходит
через 4 стадии: дегидрирование в метилциклопентен, изомери-
зация в циклогексен и две заключительные стадии дегидрирова-
ния. Эта реакция протекает только / над бифункциональным
катализатором; более того, впоследствии было показано, что
она происходит даже при использовании в качестве бифункцио-
нального катализатора механической смеси двух компонентов,
имеющих активные центры разного типа. Полагают, что реак-
ции изомеризации на кислотных активных центрах катализатора
протекают по карбониево-ионному механизму. При дегидроизо-
меризации метилциклопентана лимитирующей стадией является
стадия изомеризации, что в рамках карбониево-ионного меха-
низма можно объяснить необходимостью образования неустой-
чивого первичного карбониевого иона. Промежуточные соедине-
ния — метилциклопентен и циклогексен — были идентифицирова-
ны в продуктах этой реакции при небольших значениях времени
контакта и глубины превращения.
Дегидроциклизация «-гексана представляет собой еще более
сложный процесс, который включает 1,5-циклизацию в метил-
циклопентан с последующей дегидроизомеризацией в бензол,
т. е. всего 6 стадий. Это привело к представлению о прямой
1,6-циклизации над активными центрами платины, которое было
подтверждено опытами с использованием монофункционального
катализатора с дезактивированными кислотными центрами. Од-
нако скорость 1,6-циклизации и, следовательно, доля этой реак-
ции в общем выходе бензола меньше, чем 1,5-циклизации.
Рассмотренная схема не учитывает реакции гидрокрекинга,
коксообразования и полимеризации, роль которых невелика.
С другой стороны, она позволяет объяснить с единой точки зре-
ния образование ароматических углеводородов как из парафи-
нов, так и из нафтенов.
Недавно были разработаны и выпущены в продажу ката-
лизаторы нового поколения для процесса риформинга. Они со-
держат два металла, осажденные на окись алюминия. При
Получение первичных нефтехимических продуктов
77
использовании этих катализаторов механизм реакции, вероятно,
остается без изменения, но процесс можно проводить в термо-
динамически более благоприятных условиях без потери актив-
ности катализатора.
III. Прямое окисление бутана и нафты
X. Грин
Green Н. S., Research and Development Department,
BP Chemicals International Limited, Hull
Введение
Самыми старыми промышленными методами получения ук-
сусной кислоты являются ферментация этанола в винный уксус
и сухая перегонка древесины (гл. 2). Во время первой мировой
войны увеличившаяся потребность в ацетоне, который выраба-
тывался из уксусной кислоты, стимулировала разработку первого
синтетического метода получения этой кислоты, основанного на
окислении ацетальдегида. Этот метод сохранил свое значение
до сих пор, хотя ацетальдегид теперь получают преимущест-
венно из этилена (процесс фирмы Wacker), а не из ацетилена
или этанола, как раньше (рис. 3.7).
1 Pd-катализатор
CA + yOj --------------СНзСНО
Другими важными методами промышленного получения ук-
сусной кислоты являются карбонилирование метанола (процес-
сы фирм BASF и Monsanto).
Rh- или Со-катализатор
сн3он + со --------------------> сн3соон
и все более широко используемое, начиная с 50-х годов, прямое
жидкофазное окисление бутана (процессы фирм Celanese и
Huis) и нафты (процесс фирмы BP Chemicals).
Что касается исходного сырья для двух последних процес-
сов, то н-бутан в больших количествах вырабатывается в США
из природного газа, тогда как в Западной Европе более до-
ступны различные марки легкой нафты, получаемые при пер-
вичной перегонке нефти. Типичная нафта, пригодная для
окисления в уксусную кислоту, имеет пределы выкипания
15—10 °C и содержит главным образом углеводороды С5—С7
(табл. 3.7).
78
Глава 3
сн=сн
СН3СНО
CHjCOOH
Рис. 3.7. Основные промышленные методы получения уксусной кислоты*.
* К концу 1974 г. мощности по производству уксусной кислоты в США составляли
около 1,22 млн. т/год. Из них 46°/о работало по методу жидкофазного окисления н бутаиа,
34°/|) —по методу окисления ацетальдегида, 16°/0 — по методу карбонилирования метанола
и 3»/с— по методу ферментации этанола; кроме того, небольшое количество уксусной ки-
слоты (до 1°/о) вырабатывали в качестве побочного продукта получения глицерина
[Hydrocarbon Process, 53, Кг 11, 103 (1974)]. — Прим, перев.
Таблица 3.7
Углеводородный состав нафты, используемой в качестве
• сырья для получения уксусной кислоты
Компоненты
Содержание,
% по весу
н-Пентан
н-Гексан
Изопентан
2- и З-Метилпентаны
Ароматические углеводороды и нафтены
Прочие парафины С4 — С8
15-25
10—20
10-20
10—20
10-20
5-10
Описание процесса
Жидкофазное окисление как бутана, так и нафты обычно
ведут при температуре 150—200 °C и давлении 3—9 МН/м2
(30—90 атм). Процесс осуществляют в автоклавах, изготов-
ленных из нержавеющей стали, так как продукты реакции обла-
дают корродирующими свойствами. Через жидкий реагент бар-
ботируют в виде мелких пузырьков воздух или кислородно-воз-
душную смесь. Реакция сопровождается значительным тепловы-
делением (1,5 МДж/кг прореагировавшего углеводорода), по-
этому реакционную смесь заставляют непрерывно циркулировать
через трубное пространство охлаждаемого водой теплообмен-
ника, который обеспечивает отвод выделяющегося тепла и под-
держание необходимой температуры. Поглощая тепло, вода пре-
Получение первичных нефтехимических продуктов 79
вращается в пар, используемый в качестве теплоносителя на
последующей стадии перегонки продуктов.
Продукты реакции либо отбирают непосредственно из реак-
тора, либо получают путем конденсации паров, выходящих из
его верхней части. В последнем случае газы, оставшиеся после
отделения жидких продуктов (в основном окислы углерода и
азот со следами кислорода), сбрасывают в атмосферу. Непре-
вращенный углеводород выделяют из конденсата и возвращают
в реактор. Если оксидат отбирают непосредственно из реактора,
то он содержит гораздо меньше углеводорода и выделение
последнего не обязательно. Кроме воды, в оксидате присутст-
вуют муравьиная, уксусная и пропионовая кислоты, промежу-
точные продукты окисления (преимущественно кетоны и слож-
ные эфиры) и немного непрореагировавшего углеводорода и
высококипящих веществ. Нейтральные компоненты, выделенные
путем перегонки, можно возвращать в реактор, чтобы повысить
выход уксусной кислоты, или же подвергать дополнительному
разделению с целью получения побочных продуктов, таких, как
ацетон и метилэтилкетон. Кислые продукты после освобожде-
ния их от воды разделяют и очищают в сложной системе рек-
тификационных колонн.
Состав продуктов окисления сильно колеблется в зависимо-
сти от состава исходного сырья и условий проведения процесса.
Обычно соотношение уксусной и муравьиной кислот находится
в пределах от 4: 1 до 8: 1, а соотношение уксусной и пропионо-
вой кислот при окислении нафты — от 5:1 до 7:1. Выход про-
пионовой кислоты из бутана незначителен. В зависимости от
типа исходного сырья из 1 т его получают 1,0—1,4 т карбоновых
кислот.
Механизмы реакций
Окисление парафиновых углеводородов в жидкой фазе со-
провождается образованием карбоновых кислот с меньшим чис-
лом атомов углерода, так как процесс протекает с расщепле-
нием углерод-углеродных связей. Таким образом, теоретически
Из молекулы «-бутана может получиться два фрагмента С2 или
один фрагмент Ci и один фрагмент Сз, что приведет к образова-
нию двух молекул уксусной кислоты или одной молекулы му-
равьиной и одной молекулы пропионовой кислот. Углеводороды
более высокого молекулярного веса, разумеется, допускают
большее число различных комбинаций.
Реакция окисления протекает по цепному свободнорадикаль-
ному механизму в две стадии. На первой стадии образуются
алкилперекисные и гидроперекисные радикалы, которые затем
распадаются по различным направлениям, превращаясь в дру-
гие радикалы или молекулы. Если скорость распада перекисных
80
Глава 3
радикалов сведена к минимуму, как, например, при низких
температурах (до 100 °C) и небольших степенях конверсии за
проход (менее 1%), то процесс окисления описывается следую-
щими реакциями:
Инициирование цепи-. инициатор — (RH + Оз — -> R- -> r. + ho2.) (1)
Продолжение цепи'. R’ + O2 - -> RO2._ (2)
ro2- + rh - -> RO2H + R. (3)
Обрыв цепи: 2R. ’(4)
r» + ro2. - —> устойчивые продукты (5)
2RO2 • (6)
Реакция отрыва атома водорода (3), которую считают ли-
митирующей, подчиняется обычной для свободнорадикальных
реакций закономерности: при переходе от третичных к вторич-
ным и далее первичным радикалам ее скорость уменьшается
(т. 1, стр. 17).
В промышленных процессах окисления, которые приходится
вести при высоких скоростях реакций (т. е. при высокой тем-
пературе), алкилперекисные, диалкилперекисные и гидропере-
кисные радикалы претерпевают побочные превращения. В ре-
зультате этого возникает очень сложное переплетение взаимо-
связанных реакций с участием углеводородов, кислых продуктов
и разнообразных промежуточных соединений.
Механизм образования карбоновых кислот может быть раз-
личным. Например, гидроперекись, получившаяся при реакции
(3), может распадаться, давая вторичный алкоксильный ра-
дикал:
СН3СН2СНСН3 —> СН3СН2СНСН3 + • ОН (7)
ООН О.
Последний в свою очередь распадается на меньший алкильный
радикал и ацетальдегид
СН3СН2СНСН3 —> СН3СН2 • + СН3СНО (8)
о.
Образовавшийся ацетальдегид окисляется далее в уксусную
кислоту.
Аналогично может протекать образование пропионовой кис-
лоты из н-пентана или из н-гексана. При этом формальдегид,
являющийся продуктом распада первичных алкоксильных ра-
дикалов, окисляется в муравьиную кислоту
СН3СН2СН2СН2О • —> СНзСН,СН2 • + НСНО (9)
Получение первичных нефтехимических продуктов
81
Кетоны могут образоваться путем диспропорционирования
[уравнение (19)] или распада [уравнение (И)] алкоксильных
радикалов, генерируемых углеводородами изостроения:
2СН3СН2СНСН3 —> СН3СН2ССН3 + СН3СН2СНСН3 (10)
о- о он
СНз
СН3СН2ССН3 —> СН3СН2. + СНзСОСНз
о.
(11)
Другие основные продукты окисления — окись и двуокись
углерода — выделяются при распаде ацильных и ацилперекис-
ных радикалов.
СН3СО. —> СН3. + СО (12)
СНзСООО • —> СН3О . + СО2 (13)
Приведенные выше реакции иллюстрируют лишь некоторые
из путей образования основных продуктов окисления. Даже при
использовании чистого бутана возможно протекание гораздо
большего числа реакций. В случае же нафты их число возрас-
тает во много раз, и, помимо целевых продуктов, могут полу-
чаться самые разнообразные кислые и нейтральные соединения
Однако большинство нежелательных продуктов способно к
дальнейшему окислению и поэтому может возвращаться в сферу
реакции.
Довольно высокие выходы уксусной кислоты при окислении
бутана и нафты, получаемые несмотря на сложный механизм
процесса, можно объяснить высокой устойчивостью этой кисло-
ты к дальнейшему окислению.
Статистические данные о производстве
и потреблении низших карбоновых кислот
Уксусная кислота
Уксусная кислота — один из важнейших нефтехимических
продуктов, о чем свидетельствуют высокие темпы наращивания
ее производства в США. Так, если в 1950—1960 гг. средний
годовой прирост производства уксусной кислоты составлял 6%,
то в 1960—1970 гг. он увеличился почти до 9% *• В 1970 г. спрос
* Рост производства уксусной кислоты в США продолжался до 1973 г.
включительно. В последующие два года ее производство резко сократилось и
в 1975 г. составило 950 тыс. т, что ниже уровня 1972 г. [Chem. and Eng. News
52, № 18, 10 (1974); 53, № 18, 30 (1975); 54, № 24, 37 (1976)]. — Прим, перев.
82
Глава 3
на уксусную кислоту в США достигал 900 тыс. т. В 1975 г.
общий объем мирового производства уксусной кислоты должен
составить, по прогнозной оценке, 2 млн. т, из которых прибли-
зительно 800 тыс. т (35—40%) намечено получить прямым
окислением парафинов.
Уксусную кислоту используют главным образом в производ-
стве ацетилцеллюлозы (через уксусный ангидрид, гл. 9), винил-
ацетата, применяемого при изготовлении водоэмульсионных кра-
сок, и ацетатных эфиров, употребляемых в качестве раствори-
телей. Из других важных, хотя и менее крупных, потребителей
уксусной кислоты можно назвать производства фармацевтиче-
ских препаратов, хлоруксусных кислот и пищевых продуктов
(маринование).
В табл. 3.8 представлены данные о структуре потребления
уксусной кислоты в США.
Таблица 3.8
Структура потребления уксусной кислоты3 в США в 1970 г.
Конечный продукт переработки или область
нрименения
Доля в общем
потреблении,
%
Ацетилцеллюлоза 44
Винилацетат (как мономер) 31
Ацетатные эфиры (как растворители) 14
Хлоруксусная кислота 3
Прочие 8
а В 1974 г доля ацетилцеллюлозы в структуре потребления уксусной
кислоты в США составляла 23%, а доля винилацетата—43%; кроме того,
10% уксусной кислоты расходовалось в производстве терефталевой кислоты
и днметилтерефталата IChem. Market Abstr,, 67, 286 106 (1975)].—Прим,
перев.
Муравьиная кислота
Оценка мировых мощностей по производству муравьиной
кислоты затруднительна, так как существует большое число
мелких производителей, вырабатывающих ее в качестве побоч-
ного продукта. В США мощности по производству муравьиной
кислоты в 1971 г. составляли 35 тыс. т/год.
Основная часть муравьиной кислоты расходуется на краше-
ние и отделку тканей, дубление кож и химический синтез. В За-
падной Европе все большее распространение получает использо-
вание муравьиной кислоты для обработки силоса с целью
предотвращения его порчи при хранении.
Получение первичных нефтехимических продуктов 83
П ропионовая кислота
Статистические данные по объему производства и потребле-
ния пропионовой кислоты отсутствуют *.
Главными потребителями ее являются производства хлорор-
ганических гербицидов, винилпропионата, используемого для
изготовления водоэмульсионных красок (только в ФРГ), и про-
пионата кальция, применяемого для сохранения свежести хлеба.
Кроме того, пропионовую кислоту используют для продления
срока хранения зерна.
* В 1973 г. в США было выработано 27,2 тыс. т пропионовой кислоты.
К 1975 г. потребность в этом продукте увеличилась до 29,5 тыс. т/год (по
прогнозной оценке, в 1976 г. потребность должна составить 33,1 тыс. т, а
в 1980 г. — 41,3 тыс. т). Структура потребления пропионовой кислоты в США
в 1975 г. выглядела следующим образом: получение пропионатов натрия и
кальция — 28%, использование в качестве консерванта зерна — 28%, производ-
ство пропионилцеллюлозных пластиков — 20%, синтез гербицидов— 17%, про-
чие цели — 7% [Chem Market Abstr., 67, 286 109 (1975); Chem. Marketing Rep.,
209, № 10, 9 (1976)]. — Прим, перев.
ГЛАВА 4
Первичные нефтехимические продукты
I. Алканы, алкены и алкины
А. Джубб
Jubb A. H.t ICI Corporate Laboratory, Runcorn
Метан и окись углерода
Синтез-газ
Метан, который используется главным образом как топливо,
добывают в виде природного газа или выделяют из газов неф-
техимических и нефтеперерабатывающих заводов. Одной из
самых важных областей применения метана является получе-
ние так называемого синтез-газа — смеси водорода с окисью
углерода в различных пропорциях. Существует два основных
метода переработки метана в синтез-газ: конверсия с водяным
паром и неполное окисление. В обоих случаях исходным сырьем
могут также служить высшие парафины.
Первая стадия процесса конверсии метана с водяным паром
включает взаимодействие обоих реагентов над катализатором,
осажденным на пористом носителе
СН4 4- Н2О -е-Д- СО -j- ЗН2 И.Н = + 205 кДж/моль
СН4 + 2Н2О СО2 + 4Н2 Д/7 = -f- 163 кДж/моль
В состав катализатора обычно входят окись никеля и различ-
ные промоторы. Процесс ведут при температуре выше 700 °C и
давлении более 2,5 МН/м2 (25 атм) в трубчатых печах, через
межтрубное пространство которых пропускают горячие топоч-
ные газы. «Паровое число» (мольное отношение водяного пара
и метана) поддерживают равным 3:1.
Обычно реакционные газы содержат непревращенный угле-
водород; поэтому применяют дополнительный реактор, в кото-
ром каталитическую конверсию с водяным паром ведут при тем-
пературе выше 1000 °C.
Окись углерода, если это необходимо, можно удалить из син-
тез-газа в «СО-конвертере», где она вступает в каталитическую
реакцию с избытком водяного пара с образованием двуокиси
углерода и водорода. Для уменьшения содержания окиси угле-
рода в конвертированном газе до значений ниже 0,5% исполь-
зуются, вероятно, цинк-медь-хромовые катализаторы при темпе-
Первичные нефтехимические продукты. I.
85
ратуре 150—300 °C. Образовавшуюся двуокись углерода уда-
ляют, если это необходимо, методами адсорбции или абсорбции.
Процесс конверсии углеводородного сырья с водяным паром
широко применяется для получения светильного газа. Так, в
1968 г. в Великобритании более 90% светильного газа было
выработано путем паровой каталитической конверсии нафты
(однако с того времени положение изменилось в связи с откры-
тием месторождений природного газа в Северном море).
В процессе неполного окисления метана, который может про-
водиться как в присутствии катализатора, так и без него, метан
окисляют недостаточным количеством кислорода.
СН4 + -i- О2 —> СО + 2Н2 ДН = —36 кДж/моль
СН4 + О2 —> СО2 + 2На
Вероятно, сначала кислород окисляет метан до двуокиси
углерода и воды, после чего протекает более медленная реакция
этих продуктов с избытком метана, приводящая к образованию
окиси углерода и водорода.
Таблица 4.1
Влияние исходного сырья на состав синтез-газа
(% по объему), полученного неполным окислением по методу
фирмы Shell (данные относятся к сухому техническому
синтез-газу, содержащему 1,4% азота)
Компонент Сырье
природный газ нафта тяжелое котельное топливо
н2 60,9 51,6 46,1
со 34,9 41,8 46,9
со2 2,8 4,8 4,3
сн4 0,4 0,4 0,4
В табл. 4.1 приведены типичные составы синтез-газа, полу-
ченного методом неполного окисления из различного углеводо-
родного сырья.
Существует несколько модификаций промышленного процес-
са получения синтез-газа неполным окислением углеводород-
ного сырья, из которых наиболее известны процессы фирм Te-
xaco, Shell и Topsoe. В этих методах тоже при необходимости
проводится конвертирование окиси углерода и удаление образо-
вавшейся двуокиси.
Если нужно получить чистую окись углерода, то синтез-газ
пропускают через аммиачный раствор соли одновалентной
86
Глава 4
меди. Поглощенную окись углерода регенерируют нагреванием
раствора, промывают водой и осушают.
Если же необходимо получить чистый водород, окись угле-
рода каталитически конвертируют водяным паром в двуокись,
которую затем удаляют методами абсорбции. В настоящее вре-
мя около 90% вырабатываемого в мире водорода получают из
нефтяного сырья.
Синтез-газ часто подвергают дополнительной переработке
с целью изменения его состава в соответствии с требованиями
потребителей. Примеры таких требований приводятся в
табл. 4.2.
Таблица 4.2
Состав синтез-газа, необходимый для проведения некоторых
синтезов на его основе
Конечный продукт синтеза илн тип реакции Необходимый состав синтез-газа, моли
Аммиак ЗН2 : ш2
Метанол 2Н2: ICO
Реакции оксосинтеза 1Н2 : ICO
Фосген, уксусная кислота, альдегиды, акрилаты СО
Перекись водорода, гидрирование бензола, восстанов- ление нитробензола и др. н2
Метанол
Получение метанола из сиптсз-газа представляет собой хо-
рошо освоенный процесс, протекающий по следующему уравне-
нию:
СО 4- 2Н2 =ё=> СН3ОН ЛЯ = - 109 кДж/моль
Реакцию между окисью углерода и водородом осуществляют
при температуре 350—450 °C и давлении 25—45 МН/м2 (250—
450 атм) над цинк-хромовыми катализаторами при неполной
(менее 20%) конверсии газа за один проход. Реакционные газы
охлаждают, отделяют метанольный конденсат, а непревращен-
ные газы возвращают в цикл. Недавно фирма IC1 предложила
новый высокоселективный медный катализатор, позволяющий
проводить процесс при температуре 250 °C и давлении порядка
7 МН/м2 (70 атм). Метанол можно получать не только из окиси,
но и из двуокиси углерода; однако эта реакция используется
в меньшей степени.
СО2 + ЗН2 —> СН3ОН -|- Н2О ДЯ = — 58,5 кДж/моль
Кроме того, некоторое количество метанола получают в ка-
честве побочного продукта при жидкофазном окислении бутана
Первичные нефтехимические продукты. I. 87
в уксусную кислоту, как это делают, например, на заводе фирмы
Celanese в г. Пампа (шт. Техас, США).
Путем парофазного окисления метанола над серебряным ка-
тализатором при температуре 500—650 °C можно получить
формальдегид
СН3ОН + уО2 —> НСНО + Н2О \Н = — 158 кДж/моль
В последние годы были разработаны железо-молибденовые ка-
тализаторы для этой реакции, которые, как сообщают, обеспе-
чивают 92%-ный выход формальдегида. Формальдегид исполь-
зуют в производстве фенолформальдегидных и мочевиноформ-
альдегидных смол (гл. 8) и в качестве полупродукта для полу-
чения пентаэритрита, гексаметилентетрамина и гликолевой кис-
лоты. Полимеры формальдегида, выпускаемые под торговыми
наименованиями «дельрин» и «селкон», являются термопластич-
ными материалами, обладающими рядом полезных свойств.
Реакция Фишера — Тропша
В 1925 г. в Институте Фишера (Германия) была открыта
реакция образования высших углеводородов из окиси углерода
и водорода, протекающая при температуре 250—300°C и дав-
лении 0,1—1 МН/м2 (1—10 атм) над железо-кобальтовым ката-
лизатором. При дальнейшем исследовании этой реакции удалось
разработать более эффективные (кобальтовые) катализаторы,
и уже в 1936 г. в Германии было налажено промышленное про-
изводство синтетических углеводородов. К 1941 г. по этому
методу работали 9 заводов, а в 1942 г. мощности по получению
синтетического топлива в Германии составляли 740 тыс. т/год.
Промышленное применение этот процесс, получивший назва-
ние «процесс Фишера — Тропша», нашел только в Германии в
период второй мировой войны.
Единственным исключением является завод фирмы Sasol
(ЮАР), на котором используется комбинация процессов фирм
Kellog (США) и Lurgi-Ruhrchemie. В первом из этих процес-
сов применяется флюидизированный железный катализатор,
движущийся в газовом потоке (соотношение Н2:СО = 6:1), и
получаются главным образом легкие углеводороды (в том числе
50—70% углеводородов нормального строения) с небольшой
примесью водорастворимых кислородсодержащих соединений, в
основном этанола. Во втором процессе, который ведут в стацио-
нарном слое железного катализатора (соотношение Н2: СО =
= 2:1), получаются преимущественно тяжелые масла и пара-
фины почти целиком (на 90—95%) нормального "строения
(табл. 4.3). Продолжающаяся эксплуатация этого завода
88
Глава 4
вызвана отсутствием в ЮАР собственных месторождений нефти,
что побуждает искать пути получения жидкого топлива из угля,
запасы которого в этой стране очень велики.
Таблица 4.3
Состав (% по весу) продуктов реакции Фишера — Тропша
Продукт Процесс с движущимся катализатором а Процесс со стационарным 8 катализатором
Углеводороды С3 — С4 7,7 5,6
Бензин (углеводороды С5—Си) 72,3 (70%) 33,4 (50%)
Среднее масло 3,4 (60%) 16,6 (40%)
Полужидкие высшие парафины 3,0 10,3
Средние парафины (т. пл. 57—60 °C) — 11,8
Твердые парафины (т. пл. 95—97 °C) — 18,0
Спирты и кетоны 12,6 4,3
Органические кислоты 1,0 Следы
3 Цифры в скобках показывают содержание олефинов в дайной фракции.
Для реакции Фишера — Тропша было предложено несколько
различных механизмов, в частности механизм, включающий
промежуточное образование карбидов. Несомненно, что реаль-
ный процесс представляет собой сложную комбинацию ряда
гетерогенных реакций; его можно описать следующими суммар-
ными уравнениями:
иСО + 2иН, —> (СН2)„ + яН2О ДЯ = —192 кДж/моль
2иСО + иН2 —>- (СН2)„ 4-«СО2
Окись углерода
Кроме синтеза метанола и реакции Фишера — Тропша, окись
углерода используют для получения фосгена и муравьиной кис-
лоты. Фосген получают путем взаимодействия избытка окиси
углерода с хлором в реакторе, заполненном активированным
углем
СО+С12 —> СОС12
Основная часть вырабатываемого фосгена расходуется на полу-
чение изоцианатов (из аминов), поликарбонатных смол (реак-
цией с диоксисоединениями) и изоциануровой кислоты (реак-
цией с аммиаком). Муравьиную кислоту получают путем абсорб-
ции окиси- углерода раствором едкого натра или метанолом при
высоком давлении
н+
CO + NaOH —> HCOONa ------► НСООН
ЫНз н+
СО + СН3ОН —> НСООСНз -------> hconh2 -----> НСООН
Н2О
Первичные нефтехимические продукты. I. 89
В настоящее время муравьиную кислоту все в большей степени
вырабатывают как побочный продукт окисления углеводород-
ного сырья в уксусную кислоту (гл. 3).
Взаимодействие окиси углерода с водой и олефинами под
давлением в кислой среде приводит к образованию трет-алкил-
карбоновых кислот. По этой реакции, известной под названием
реакции Коха, в промышленности получают из изобутилена три-
метилуксусную кислоту
н+
(СН3)2С=СН2 + СО + Н2О --> (СНз)зССООН
триметилуксусная
кислота
Реакция Коха применима также к высшим разветвленным оле-
финам, таким, как тримеры пропилена и диизобутилен. Получен-
ные карбоновые кислоты с разветвленной цепью используют в
качестве сиккативов в составе масляных лаков и красок. Ката-
литическое присоединение окиси углерода и водорода по двой-
ной связи олефинов (так называемая реакция оксосинтеза) бу-
дет рассмотрена в гл. 6.
Применение метана в различных синтезах
Метан непосредственно используется для получения синиль-
ной кислоты, сероуглерода и хлорпроизводных.
Синильную кислоту можно получить из метана несколькими
методами. В основе процесса Андруссова лежит реакция метана
с аммиаком и воздухом над платиновым катализатором при
температуре 1000 °C.
СН4 + NH3 + у О2 —> HCN + 3H2O
Модификацией этого процесса является окисление аммиака воз-
духом над сеткой из платинородиевого сплава (90% Pt %- 10%
Rh) при температуре 900 °C
4NH3 + 5О2 —4NO + 6Н2О
с последующим взаимодействием реакционных газов, охлажден-
ных до 600 °C, с метаном
2NO + 2СН4 —> 2HCN + 2Н2О + Н2
В процессе фирмы Degussa, нашедшем применение в ФРГ,
используется прямая реакция метана с аммиаком, открытая
Кюльманном в 1842 г.
ch4 + nh3 —> HCN + 3H2
Эта реакция эндотермична и проводится над платиновым ката-
лизатором в трубчатом реакторе, футерованном керамическим
90
Глава 4
материалом, при температуре около 1200—1300°С. Образую-
щийся в качестве побочного продукта водород можно утилизи-
ровать.
В процессе фирмы Shawinigan метан (или другое углеводо-
родное сырье, например пропан или более тяжелые дистилляты)
реагирует с аммиаком при температуре 1400 °C в псевдоожижен-
ном слое кокса, подогреваемом с помощью электрического тока.
По данным фирмы, выход синильной кислоты как по метану, так
и по аммиаку достигает 85%.
Кроме того, синильную кислоту можно получить из окиси
углерода через ряд промежуточных стадий:
100 °C NH3
СО + СНзОН ----—------> НСООСНз -----------------»
1 МН/м2 (10 атм) 40 °C, 1,5 МН/м2 (15 атм)
метилформиат
А12о3
-* HC0NH2^^ HCN + H2O
формамид
В последнее время синильную кислоту во все возрастающих ко-
личествах получают в качестве побочного продукта производ-
ства акрилонитрила из пропилена (гл. 6).
Сероуглерод, который традиционно получают в промышлен-
ности из древесного угля и серы, может быть также получен
взаимодействием метана с серой при температуре 700°C и дав-
лении 0,2 МН/м2 (2 атм) (процесс Thacker):
CH4 + 2S2 —CS.-l 2H2S
CH4 + S2 —> CS2 + 2H2
CH4 + 2H2S —> CS2-t-4H2
Образующийся сероводород переводят в серу по методу Клауса
H2S + j Оз —> SO2 + Н2О SO2 + 2H2S —> 3S + 2H2O
Метан менее активно реагирует с хлором, чем его гомологи.
Однако эта реакция легко протекает при температуре 400—
440°C. Варьируя соотношение хлора и метана, можно получать
различные хлорпроизводные — от хлористого метила в одном
предельном случае до четыреххлористого углерода в другом.
Так, при мольном соотношении хлора и метана 0,6: 1 образуется
смесь хлористого метила, хлористого метилена, хлороформа и
четыреххлористого углерода в мольном соотношении 6:3:1:
: 0,25. Хлористый метил можно выделить из смеси перегонкой, а
остальные продукты подвергнуть жидкофазному фотохлориро-
ванию. Из хлорпроизводных метана наибольший спрос имеет
Первичные нефтехимические продукты. 1.
91
четыреххлористый углерод.
С12 С12
СН, + С12 —> на + СН3С1 —> CH2CI2 + НС1 —>
С12
—> CHCI3 + НС1 —► ССЦ + НС1
Хлористый метил часто получают также взаимодействием
метанола с хлористым водородом.
Ацетилен
Ацетилен получают в промышленности двумя основными ме-
тодами: из карбида кальция и путем пиролиза углеводородного
сырья на этилен, при котором ацетилен образуется в качестве
побочного продукта (гл. 2). Кроме того, существует несколько
методов, в которых ацетилен получается из углеводородов как
основной продукт (гл. 3). При этом используется тот факт, что,
будучи термодинамически неустойчивым при обычной темпера-
туре, ацетилен гораздо более устойчив при повышенных темпе-
ратурах. Так, при температуре 1200 °C он является самым устой-
чивым из углеводородов (гл. 3, рис. 3.2) и поэтому может быть
получен путем их пиролитического расщепления.
Толчком к промышленному применению химии ацетилена
послужили работы немецкого ученого Реппе, которые привели
к созданию в Германии во время второй мировой войны значи-
тельных мощностей по производству различных продуктов на
основе ацетилена. Промышленное производство самого ацети-
лена в Германии достигло наивысшего уровня (440 млн. м3 в
год) в 1943—1944 гг., причем 86% ацетилена было получено кар-
бидным методом. Данные о структуре современного производства
Таблица 4.4
Производство ацетилена в 1968 г. (тыс. т)
Страна
Общий объем
производства
В том числе из нефтяного
сырья
ФРГ Австрия Испания Франция Италия США Япония 318 187 14 0 57 0 173 19 265 195 465 Данные отсутствуют а 503 46
а В 1970 г. из нефтяного сырья было выработано 210 тыс. т ацетилена.
В 1972 г. производство ацетилена составило (тыс. т): в ФРГ 352, в Испании
38, во Франции 78, в Италии 179, в США 362, в Японии 79 (из нефтяного
сырья) (Statistical Yearbook 1975, 27, 286-289 (1976)].—Прим, перев.
92
Глава 4
ацетилена отсутствуют; поэтому в табл. 4.4 представлено со-
стояние производства ацетилена на 1968 г.
В 1970 г. в США главными потребителями ацетилена, полу-
ченного из нефтяного сырья (210 тыс. т), были производства
хлористого винила (40%), винилацетата (30%), акрилатов
(22%), хлорорганических растворителей и других химикатов*.
Доля ацетилена, израсходованного на нехимические цели (такие,
как сварка и резка металлов, которая до сих пор сохраняет важ-
ное значение), была невелика.
Ацетилен приходится использовать в разбавленном виде при
давлениях не выше 0,2 МН/м2 (2 атм). Баллоны для ацетилена
рассчитаны на 12-кратное превышение рабочего давления, с тем
чтобы они могли выдержать взрывное разложение заключенного
в них ацетилена. В общем можно сказать, что, несмотря на по-
тенциальную опасность работы- с ацетиленом, обусловленную
легкостью его разложения, проведение синтезов на основе аце-
тилена и очистка полученных продуктов не представляют особых
затруднений.
Тем не менее в последние годы ацетилен как полупродукт
органического синтеза все более вытесняется этиленом, который
значительно дешевле (рис. 4.1). К настоящему времени ацетилен
сохраняет доминирующее положение лишь в небольшом числе
производств. В этой связи интересно отметить, что в 1944 г. в
Германии приблизительно половину всего этилена вырабаты-
вали селективным гидрированием ацетилена, что было вынуж-
денной необходимостью. Рис. 4.2 показывает, насколько богата
и разнообразна химия ацетилена; однако последующее обсуж-
дение затрагивает лишь те процессы, в которых он продолжает
играть важную роль.
При взаимодействии ацетилена с хлористым водородом в га-
зовой фазе над сулемой, нанесенной на активированный уголь,
легко образуется хлористый винил с выходом 85—90% (гл. 7)
СН=СН + НС1 —> СН2=СНС1 ДН = —184 кДж/моль
Парофазной реакцией ацетилена с уксусной кислотой при
температуре 170—220 °C над ацетатом цинка или кадмия, нане-
сенным на активированный уголь, получают винилацетат
СН==СН + СНзСООН —► СН,=СНООССН3
Ацетилен можно димеризовать в винилацетилен, гидрохлори-
рование которого дает хлоропрен — мономер, используемый для
* В 1975—1976 гг. потребность США в ацетилене (для химических целей)
составляла около 150 тыс. т. Основная часть ацетилена была использована на
получение хлористого винила (31%), акрилатов (26%) и винилацетата (18%)
[Chem. Marketing Rep., 210, № 19, 9 (1976)]. — Прим, перев.
Первичные нефтехимические продукты. I.
93
Год
Рис. 4.1. Приближенные среднегодовые цены на исходные углеводороды
в США. Цены на ацетилен являются средними для химических и нехими-
ческих потребителей. Цены на пропилен являются средними из цен на тех-
нический продукт нефтеперерабатывающих заводов, пропилен для химичес-
ких целей и пропилен для полимеризации *.
* В 1971 г. цены на этилен и пропилен в США составляли около 3 цент/фунт, кар-
бидный ацетилен стоил 9,5 цент/фунт, а углеводородный — от 6,4 до 8,2 цент/фунт
В 1974 г. цены на эти полупродукты возросли для этилена—до 7 цент/фунт, пропилена—
6 цеит/фунт, карбидного ацетилена — от 11,4 до 13,3 цент/фунт и углеводородного ацети-
лена—от 10 до 15,5 цент/фунт, а к началу 1976 г. этилен стоил уже 11,5 цент/фунт, про-
пилен («полимеризацнонный») —8,2 цент/фунт и ацетилен — 49 цент/фунт [Chem. and
Eng. News, 52, № 49, 8 (1974); Chem. Marketing Rep., 209, № 1, 28 (1976); «Производство
хлорированных растворителей в основных капиталистических странах», НИЙТЭхнм, М.,
1975] — Прим, перев.
получения хлоропренового каучука (гл. 8)
2СН=СН
НС1
сн=ссн=сн2 ----> СН2=СС1СН=СН2
винилацетилен
хлоропрен
При каталитической гидратации ацетилена серной кислотой
в присутствии сульфата двухвалентной ртути легко образуется
СН2=СС1СЙ=СН2
хлоропрен
HCi
СН=ССН=СН;
винилацетилен
СН2=СНСН=СН2<
бутадиен
Й2С----СИ;
н2с^оХсн2
тетра гидрофуран
-Н20
ch2=choocr
сложные виниловые
эфиры
ch2=chor
простые виниловые
эфиры
СН3
ch2=chcn <
акрилонитрил
СН3СНО
ацетальде-
гид
СН=С'Н
СНС1=СНС1
дихлорэтилен
ch2=chcoor
акриловые эфиры
НОСН2СН2СН2СН2ОН
бутандиол- /, 4
(НОСН2С=С—)2
г ексадиин -ZA- диол
СН2==СНСН2ОН
аллиловый спирт
ОН
СНз^С"^С=СН
СНз
сн3.
Н2
у ^он
.с:
сн=сн
-Н20
HCN
сн
•С—СН=СН;
изопрен
н2
СН=ССН2ОН —> НОСН2С=ССН2ОН
пропаргиловый бутиндиол -1,4
спирт
СНС12СНС12
тетрахлорзтан
СН2=СНС1
хлористый винил
-НС1
С12
СНС1=СС12
трихлорэтилен
СНзСЧСНСГ
трихлорэтан
С12
-HCI
СНС12СС13
-НС1
СН2=СС12
винилиденхлорид
(}С1,=СС12
Рис. 4.2. Некоторые реакции ацетилена.
Первичные нефтехимические продукты. I. 95
ацетальдегид* (т. 1, стр. 108)
СН=СН + Н2О —► СНзСНО
Другой метод получения ацетальдегида, разработанный Реппе,
основан на реакции ацетилена с метанолом с последующим гид-
ролизом образовавшегося метилвинилового эфира разбавленной
фосфорной кислотой. Этот метод, как и прямая гидратация аце-
тилена, в настоящее время не применяется. Однако его первая
стадия — присоединение спиртов к ацетилену — все еще находит
ограниченное применение для получения метилвинилового эфира
и его высших гомологов.
Несмотря на существование нескольких методов получения
акриловой кислоты и ее эфиров из пропилена, синтез этих соеди-
нений из ацетилена еще сохраняет важное значение.
Nl(CO)t
HteCH + CO + ROH -—„> ch2==chcoor
40 — 50 иС
В первом варианте этого процесса использовали карбонил ни-
келя в разбавленной соляной кислоте, который служил не
только катализатором, но и источником части окиси углерода.
Фирма BASF разработала процесс, в котором катализатором
является бромид никеля (0,1%) в растворе тетрагидрофурана;
образующуюся на первой стадии акриловую кислоту этерифици-
руют соответствующим спиртом.
Ацетилен может также служить полупродуктом для получе-
ния акрилонитрила
CU2CI2/NH4CI
CH=CH-+-HCN ---------->• CH,=CHCN
Однако в настоящее время этот метод практически полностью
оставлен и акрилонитрил получают главным образом из пропи-
лена (гл. 6).
Реакция ацетилена с формальдегидом приводит к образова-
нию пропаргилового спирта, который затем превращают в бу-
тиндиол-1,4
нсно нсно
ClfeCH ----> СН==ССН2ОН -----> HOCH2teCCH2OH
пропаргиловый бутиндиол-1,4
спирт
Последний продукт легко гидрируется в бутандиол-1,4, при
дегидратации которого над медным катализатором образуется
у-бутиролактон. Взаимодействием этого лактона с метиламином
при температуре 200 °C и повышенном давлении получают N-
метилпирролидон, используемый в качестве селективного
Реакция Кучерова. — Прим, перев.
96
Глава 4
растворителя для экстракции ацетилена и ароматических угле-
водородов.
Н2 —Н2
НОСН2С=ССН,ОН —> НОСН2СН2СН2СН2ОН ----------->
Н2С---СН2 CH3NH2 Н2С—сн2
ОСХ /СН2 ОСХ /СН>
О N
I
СН3
у-бутиролактон N-метилпир-
ролидон
Фирма SNAM Prpgetti разработала промышленный процесс
получения изопрена, в основе которого лежит каталитическая
реакция ацетилена с ацетоном *. Образующийся в качестве про-
межуточного продукта 2-метилбутин-3-ол-2 селективно гидри-
руют в метилбутенол, который дегидратируют в изопрен
ОН
10—40 °C | Н2
СН=СН + сн3сосн3-----------------> СНзСС=СН ---------->-
3 3 2 МН/м2 (20 атм) । Pd/Rh/BaSO4
СН3
он
I M2O3
-> СНзССН=СН2 сн2=с-сн=сн2
СНз СН3
изопрен
Хлорирование ацетилена в растворе тетрахлорэтана при тем-
пературе 80 °C в присутствии пятихлористой сурьмы или хлор-
ного железа приводит к образованию 1,1,2,2-тетрахлорэтана
(СНС1г)2. Это соединение при обработке его известью при тем-
пературе 50 °C или при пиролизе над хлоридом бария или меди,
нанесенным на активированный уголь, при температуре 220—
320 °C легко отщепляет хлористый водород, превращаясь в три-
хлорэтилен
СНС12СНС12 —> СНС1=СС12 + НС1
Трихлорэтилен применяют в качестве растворителя и для обез-
жиривания поверхности металлов. При хранении его необходимо
стабилизировать добавлением небольшого количества ацетиле-
новых спиртов. Кроме описанного метода, трихлорэтилен полу-
чают из дихлорэтана через трихлорэтан, причем благодаря раз-
* Метод получения изопрена из ацетилена и ацетона первоначально был
разработан в СССР А. Е. Фаворским, и в 1939—1941 гг. по этому методу ра-
ботала крупная опытная установка. В дальнейшем для организации крупно-
тоннажного производства изопрена были использованы более дешевые и эф-
фективные виды сырья (изобутилен, метанол, изопентан). — Прим, ред.
Первичный нефтехимические продукты I. 97
работке технологии процесса окислительного хлорирования
(гл. 6) этот метод приобретает все большее значение.
Хлорирование трихлорэтилена приводит к получению пента-
хлорэтана, который при обработке известью превращается в
перхлорэтилен:
СНС1=СС12 + С12 —> CHCI2CCI3
пентахлорэтан
Са(ОН)2
СНС12СС13 -------> СС12=СС12 + НС1
перхлорэтилен
Перхлорэтилен используют главным образом в качестве раство-
рителя для химической чистки одежды. В настоящее время это
соединение, как и другие хлорпроизводные этилена, все в возра-
стающей степени получают из этилена.
Этилен
Этилен — наиболее важный полупродукт органического син-
теза. Объем его мирового производства в период 1950—1970 гг.
возрос с 650 тыс. т до 19 млн. т (рис. 4.3), и ожидается, что к
Таблица 4.5
Структура потребления этилена в ведущих капиталистических в 1972 г. (% от общего объема потребления) странах а
R Западная Европа Продукты переработки этилена Велико- (включая бРитания Великобританию) США
Полиэтилен низкой плотности 39,5 39,5 Полиэтилен высокой плотности 6,3 13,5 Окись этилена 19,6 13,1 Хлористый винил (дихлорэтан) 16,8 16,9 Этанол 8,0 2,7 Стирол (этилбензол) 7,1 7,5 Ацетальдегид — 3,8 Прочие 2,7 3,0 28,2 11,6 20,1 15,4 6,3 9,7 2,8 5,9
Общее потребление этилена, млн. т 1,04 7,70 6 Мощности по получению этилена, 1,18 9,35 млн. т/год 9,10 9,24 в
а Объем производства этилена в Японии был равен 4 млн. т в 1970 г. и 3,5 млн. т
в 1971 г.
б В 1974 г. 10,40 млн. т [Chem. Market Abstr., 67, 286 1119 (1975)].—Прим, перев.
в На 1 января 1976 г. мощности по получению этилена в США составляли 12,2 млн.
т/год [Chem. Marketing Rep., 209, № 1, 40 (1976)].—Прим, перев.
4 Зак. 501
98
Глава 4
Рис. 4.3. Объем мирового производства этилена.
1980 г. он превысит 55 млн. т *. Данные о структуре потребления
этилена приведены в табл. 4.5.
* По более поздней прогнозной оценке, в 1977 г. мировое производство
этилена достигнет 38 млн. т. а к 1980 г оно возрастет до 49,5 млн. т [Chem.
Market Abstr., 67, 286 790 (1975)]. — Прим, перев.
Первичные нефтехимические продукты. I.
99
Полиэтилен (гл. 8)
В 1932 г. английской фирмой ICI был впервые получен поли-
этилен низкой плотности. В настоящее время выпускается два
типа полиэтилена: полиэтилен низкой плотности и полиэтилен
высокой плотности. Полиэтилен — наиболее крупнотоннажный из
термопластичных материалов, и значительная часть вырабаты-
ваемого этилена расходуется на его получение. Полиэтилен низ-
кой плотности (0,910—0,925), который получается при высоком
давлении, обладает хорошей гибкостью и достаточно высокой
Рис. 4.4. Структура мировой (без учета КНР) потребности в полиэтилене *
* В 1974 г. мировая потребность в полиэтилене высокой плотности была равна
3920 тыс. т [Chem. Market Abstr., 67, 282 630 (1975)]. —Прим, перев.
4*
100
Глава 4
разрывной прочностью, тогда как полиэтилен высокой плотности
(0,94—0,96), который был впервые получен в 1953 г. полимери-
зацией этилена при низком давлении на основе работ Циглера,
имеет более высокую степень кристалличности и характери-
зуется большими значениями предела текучести и температуры
размягчения (~ 120°C). На рис. 4.4 приведены данные о дина-
мике мировой потребности в полиэтилене.
Окись этилена
Окись этилена ранее получали взаимодействием этилена с
хлорноватистой кислотой с последующим дегидрохлорированием
образовавшегося этиленхлоргидрина известью
Са(ОН)2
СН2=СН2 + НОС1 —> С1СН2СН2ОН -------->- Н2С—сн2
'v'
Побочно получаются дихлорэтан и р.р'-дихлордиэтиловый эфир.
Преимуществами хлоргидринного метода являются высокий вы-
ход окиси этилена (85%, считая на этилен) и сравнительно
небольшие капитальные затраты на строительство установки.
Однако из-за значительного расхода хлора (2 т/т окиси этилена)
этот метод в настоящее время все более вытесняется прямым
окислением этилена, чему дополнительно способствует возмож-
ность использования установок, работающих по хлоргидринному
методу, для получения окиси пропилена. К середине 1969 г. при-
близительно 90% мощностей по производству окиси этилена в
США работало по методу прямого окисления этилена *.
Существует по крайней мере четыре промышленных процесса
прямого окисления этилена в окись этилена, причем все они пре-
дусматривают использование серебряного катализатора на но-
сителе. В процессах фирм Union Carbide и Scientific Design
окислителем служит воздух, тогда как в процессе фирмы Shell
применяется кислород. В настоящее время обычными являются
установки с единичной мощностью 100 тыс. т/год. Реакцию ведут
при температуре 200—290°С и давлении 1,0—2,5 МН/м2 (10—
25 атм), используя концентрацию этилена в сырьевом потоке
* В 1969 г. мировые мощности по производству окиси этилена составляли
3,35 млн. т/год, из которых 190 тыс. т/год (6%) приходилось на долю пред-
приятий, работающих по хлоргидринному методу, 2,12 млн. т/год (63%) —
по методу окисления этилена воздухом и 1,04 млн. т/год (31%)—по методу
окисления его кислородом. В 1975 г. общие производственные мощности воз-
росли до 4,64 млн. т/год, причем все они распределялись между установками
прямого окисления этилена воздухом (2,02 млн. т/ год, или 43%) и кислоро-
дом (2,62 млн. т/год, или 57%) [Hydrocarbon Process., 55, № 3, 69 (1976)].—
Прим, перев.
Первичные нефтехимические продукты I.
101
ниже 3% по объему. В этих условиях выход окиси этилена со-
ставляет 65—70%.
СН2=СН2 + у О2 —> Н2С-----СН2 ЛЯ = —146 кДж/моль
СН2=СН2 + ЗО2 —> 2СО2 + 2Н2О Д/7 = - 1322 кДж/моль
М
сн2=сн2 —> Н2С—сн2
\ / О
йз\ /кг
СО2
Вопрос о том, образуется ли двуокись углерода путем пря-
мого окисления этилена или же она является продуктом даль-
нейшего окисления окиси этилена, послужил предметом специ-
ального исследования. Было найдено, что kz/ki « 0,08 и k$/ki »
« 0,4, т. е. преобладает первое направление.
Основная часть окиси этилена идет на производство этилен-
гликоля
Н2С—СН2 + Н2О —> НОСН2СН2ОН
Доля этого продукта в структуре потребления окиси этилена
составляет свыше 55% для США и свыше 80% для Японии. Эти-
ленгликоль до сих пор используют главным образом в качестве
компонента антифризов. Однако все более важным потребите-
лем этиленгликоля становится производство полиэфирных воло-
кон, для которых он служит исходным сырьем. Кроме того, на
основе этиленгликоля получают поверхностно-активные веще-
ства, этаноламины и простые эфиры этиленгликоля и полиэти-
ленгликолей.
Хлористый винил
Фирма Union Carbide объявила о создании процесса прямого
хлорирования этилена в хлористый винил с выходом 87%, од-
нако никаких данных о промышленной реализации этой техно-
логии пока не имеется. Поэтому превращение этилена в хлори-
стый винил до сих пор проводят в две стадии. Первую стадию —
получение дихлорэтана — осуществляют путем прямого хлориро-
вания этилена молекулярным хлором или окислительного хло-
рирования этилена смесью хлористого водорода с воздухом
7 СН2=СН2 + Ch —> С1СН2СН2С1
СН2=СН2 + 2НС1 + у О2 —> С1СН2СН8С1 + нго
102
Глава 4
На второй стадии дихлорэтан подвергают дегидрохлорирова-
нию в хлористый винил, причем образующийся хлористый водо-
род возвращают на стадию окислительного хлорирования
этилена (если таковая имеется). Более подробно эти реакции
рассматриваются в гл. 7.
Этанол (гл. 6)
Существует два метода получения этанола из этилена. По
первому из них этилен (обычно с концентрацией 95%, но допу-
стимы и более низкие концентрации) поглощают в нескольких
последовательно расположенных абсорберах 95—98%-ной сер-
ной кислотой при температуре 55—85 °C и давлении 1,5—
4,0 МН/м2 (15—40 атм)
СН2=СН2 + H2SO4 —> CH3CH2OSO3H
моноэтилсульфат
2СН2=СН2 + H2SO4 —> (CH3CH2)2SO4
диэтилсульфат
Моноэтилсульфат и побочно образующийся диэтилсульфат под-
вергают гидролизу водяным паром
CH3CH2OSO3H + H2O —> CH3CH2OH + H2SO4
(CH8CH2)2SO4 + Н2О —> C2H5OC2H6 + H2SO4
Спирт и эфир отгоняют, а оставшуюся разбавленную серную
кислоту концентрируют и возвращают в абсорберы. Эфир и эта-
нол разделяют дистилляцией. Выход этанола составляет около
90%.
Другой метод получения этанола заключается в каталитиче-
ской гидратации этилена над фосфорной кислотой, нанесенной
на кизельгур, при температуре 300 °C и давлении 6—7 МН/м2
(60—70 атм). Степень конверсии этилена за один проход состав-
ляет лишь около 5%, однако выход этанола достигает 95%.
Этанол используют в качестве растворителя и полупродукта
для получения ацетальдегида, уксусной кислоты и ее эфиров.
Стирол
Стирол вырабатывают из этилбензола, который может быть
выделен из продуктов каталитического риформинга нефтяных
фракций или получен прямым этилированием бензола. Послед-
ний метод—алкилирование бензола этиленом в присутствии
хлористого алюминия — описан в гл. 5. Кроме того, этилбензол
получают путем парофазной реакции этилена с бензолом при
температуре 200—250°C и давлении 1,4 МН/м2 (14 атм) в ста-
ционарном слое кизельгура, пропитанного фосфорной кислотой.
В процессе „Alkar” фирмы Universal Oil Company катализато-
ром служит фтористый бор; сообщается также об использовании
в качестве катализаторов молекулярных сит.
Первичные нефтехимические продукты. I.
103
Превращение этилбензола в стирол обычно осуществляют
путем его каталитического дегидрирования. Эта реакция обра-
тима, причем образованию стирола благоприятствуют повышен-
ные температуры. Однако верхний предел температуры процесса
определяется термической нестабильностью стирола и этилбен-
зола. Пары этилбензола подогревают перегретым водяным па-
ром и пропускают при температуре 360 °C над окисноцинковым
или окиснохромовым катализатором. Степень конверсии этил-
бензола за один проход обычно достигает 40—60%, выход сти-
рола— 90—92%. Для отделения стирола от непревращенного
этилбензола используют дистилляцию в вакууме. Во избежание
полимеризации стирола при дистилляции и во время хранения
к нему добавляют ингибиторы.
+ СН2=СН2 —>
Фирма Union Carbide разработала технологию жидкофазного
окисления этилбензола в фенилметилкарбинол (а-метилбензило-
вый спирт) в присутствии ацетата марганца. Побочно обра-
зуются бензойная кислота и ацетофенон, который можно легко
превратить в карбинол. Последний при температуре 300°C легко
дегидратируется над катализатором, содержащим ТЮг, в стирол.
-що
Недавно был разработан новый метод получения стирола, на
первой стадии которого этилбензол окисляют в гидроперекись
при температуре 130°C и давлении 0,5 МН/м2 (5 атм). Степень
конверсии этилбензола составляет 13%, выход гидроперекиси —
85%. Раствор гидроперекиси этилбензола упаривают до концен-
трации 35% и обрабатывают при температуре ПО °C пропиле-
ном в присутствии нафтената вольфрама или молибдена. При
этом образуются окись пропилена (стр. ИЗ) и фенилметилкар-
бинол, который на третьей стадии подвергают дегидратации
он
ООН
над окиснотитановым катализатором, получая стирол. Сооб-
щается, что уже построено три завода (в США, Нидерландах и
Испании), работающие по этой технологии.
104
Глава 4
Ацетальдегид
До начала 60-х годов ацетальдегид в США получали преиму-
щественно окислением или дегидрированием этанола
4 275_5QQ OQ
С2Н5ОН+4о2 ---------------> сн3сно + н2о
2 Ag (катализатор)
Cu/Сг(катализатор)
с2н5он -----гй-ТооЧ"" сн3сно + н2
В Западной Европе в этот период значительную часть ацеталь-
дегида все еще вырабатывали из ацетилена.
В 1894 г. Филлипс описал реакцию этилена с хлористым пал-
ладием, в результате которой образуется ацетальдегид. Затем
эту реакцию изучали Андерсон (1934 г.) и Хараш (1938 г.).
Однако промышленное значение реакции Филлипса было оце-
нено лишь в 1956 г., когда на ее основе был разработан процесс
фирмы Wacker. Сущность этого процесса состоит в окислении
этилена в присутствии хлоридов двухвалентной меди и палладия.
Используются две модификации процесса: одностадийный ва-
риант, в котором катализатор регенерируют in situ, пропуская
через раствор кислород (фирма Farbwerke Hoechst), и двухста-
дийный вариант, в котором регенерацию катализатора ведут в
дополнительном реакторе, используя для этого воздух (фирма
Wacker-Chemie). Выход ацетальдегида, считая на этилен, равен
95%. Основные реакции, протекающие при окислении этилена в
ацетальдегид, можно представить уравнениями
СН2=СН2 + PdCl2 + Н2О —► СН3СНО + Pd + 2НС1
fu -f- 2сиы2 —► PdCl2 Ч- CU2CI2
Cu2Cl2 + j O2 + 2HC1 —> 2CuC12 + H2O
Суммарное уравнение процесса имеет следующий вид:
СН2=СН2 + у О2 —> СНзСНО
Таким образом, реакция является каталитической относи-
тельно хлористого палладия. Наряду с СиС1г предложено ис-
пользовать также другие окислители, такие, как бензохинон,
РЬОг и соли трехвалентного железа; однако в промышленности
применяют только хлорную медь.
Скорость реакции возрастает с увеличением [Pd2+] и [С2Н4] и
снижается с увеличением [С1~] и [Н+]
d[C2H4] _ 4PdCir][C2HJ
df [СГ] [Н3О+]
Это выражение справедливо также для аналогичных реакций
окисления пропилена, бутена-1 и бутена-2. В опытах по окисле-
Первичные нефтехимические продукты. 1.
105
нию дейтерированного этилена C2D4 было найдено, что
fec2H4/fec2D4 = 1,07, откуда можно сделать вывод, что расщепле-
ние связи С—Н или С—D не является лимитирующей стадией.
При взаимодействии этилена с хлористым палладием в растворе
D2O получается ацетальдегид, не содержащий дейтерия. Эти
результаты свидетельствуют о том, что все четыре водородных
атома образующегося ацетальдегида присутствуют в исходном
этилене. Наблюдаемые факты хорошо согласуются с механиз-
мом реакции, предложенным Генри (схема 1).
ki
[PdCl4p-+ С2Н4 ч==* [PdCl3C2H4]’+ СГ
*-зЬ2°
♦ 11^2
[PdCl2(OH2)C2H4]° + СГ1
[PdCl2(OH)C2H4]~ + Н3о+-
й_4||а4 (медленно)
[PdCl(CH2CH2OH)J + СГ
СНзСНО + Pd° + СГ
Схема 1.
Промежуточные стадии процесса можно представить схе-
мой 2:
О. /Н
Н++ С +F,d° + 2C1'
СИ,
н
но. /
с+
| + [PdCl2]2-
нх\хн
н
Схема 2.
106
Глава 4
В 1968 г. доля ацетальдегида, полученного на основе этилена,
в общем объеме производства ацетальдегида составляла 39%
для США, 46% для Западной Европы и почти 100% для Японии.
Свыше 80% ацетальдегида идет на получение уксусной кис-
лоты, уксусного ангидрида, бутанола-1 и 2-этилгексанола-1. Боль-
шая часть остального ацетальдегида расходуется на синтез
винилацетата, пентаэритрита, хлораля и пиридиновых основа-
ний. Бутанол-1 и 2-этилгексанол-1 получают из ацетальдегида
по следующей схеме, включающей реакции кротоновой конден-
сации и последующего гидрирования:
2СНЭСНО
|он-
СН3СН(ОН)СН2СНО
ацетальдоль
I
Н2
н2о + сн3сн=снсно --- о- > СН3СН2СН2СН2ОН
Cu/SiO2
кротоновый альдегид бутанол-1
(8О°/о)
сн3сн2сн2сно
масляный альдегид
I кротоновая
I конденсация
Н2
сн3сн2сн2сн=с—сно —СН3СН2СН2СН2СНСН2ОН
СН2СН3 ^Н2СН3
2-этилгексен-2-аль 2-этилгексанол-1
В 1972 г. мощности по производству ацетальдегида в США
оценивались в 700 тыс. т/год. Потребность в ацетальдегиде в
США в 1970 г. равнялась 620 тыс. т, а в структуре его потребле-
ния главное место занимало получение уксусной кислоты и
уксусного ангидрида (49%), бутанолов (14%) и 2-этилгекса-
нола-1 (12%).
Винилацетат
После опубликования первого сообщения о получении аце-
тальдегида из этилена появилось большое число работ, посвя-
щенных изучению реакции этилена с хлористым палладием в
уксуснокислой среде. В результате этих исследований было раз-
Первичные нефтехимические продукты !.
107
работано несколько процессов получения винилацетата, реали-
зованных впоследствии в США, Западной Европе и Японии. Все
они представляют собой модификации двух основных методов.
В первом из них окисление этилена ведут в жидкой фазе при
температуре 120—130°С и давлении 1,0 МН/м2 (10 атм) в при-
сутствии хлорной меди и солей лития. Для регенерации in situ
иона двухвалентной меди, который в свою очередь служит окис-
лителем металлического палладия, используют кислород. При-
сутствие воды способствует образованию ацетальдегида. Во вто-
ром процессе реакцию осуществляют в паровой фазе, пропуская
смесь этилена, кислорода и паров уксусной кислоты над стацио-
нарным палладиевым катализатором (например, PdCl2 + CuCl2
на активированном угле или PdCl2 на окиси алюминия) при
температуре 160°C и давлении 0,5—1,0 МН/м2 (5—10 атм).
В обоих процессах винилацетат получается с выходом более
90%. Полагают, что реакция протекает по следующему меха-
низму:
н
Ас(К /
С +
+ [PdCl]2"
/ Н '°Ас
Н
АсО Н
+ Pd°
+ НОАо
+ CI-
Схема 3
Выход винилацетата существенно зависит от концентрации
хлорид-иона. В том случае, когда эта концентрация слишком
высока, образуется преимущественно этилидендиацетат (схе-
ма 4).
АсО | ОАо
I
С
Н I н
н
+ Pd° + 2С1-
Схема 4.
108
Глава 4
Кроме этилена, винилацетат можно получить из ацетальде-
гида и уксусного ангидрида (или уксусной кислоты), причем
промежуточный этилидендиацетат подвергают термическому
расщеплению в присутствии сильной кислоты:
СНзСНО + (СН3СО)2О —> СН3СН(ОСОСН3)2 —>
—> СН3СООСН=СН2 + СНзСООН
В 1970 г. в США было выработано 360 тыс. т винилацетата *,
из которых 90% было израсходовано на получение поливинил-
ацетата, поливинилового спирта и поливинилбутираля, а осталь-
ные 10% —на сополимеризацию с другими мономерами, в част-
ности с хлористым винилом. Поливинилацетат используется
главным образом для изготовления латексов, синтетических
смол, водоэмульсионных красок и адгезивов.
Высшие олефины
Полимеризация этилена по методу Циглера может быть ис-
пользована для получения его олигомеров — главным образом
линейных а-олефинов С< — С20 (кривая распределения продук-
тов реакции соответствует функции Пуассона с максимумом при
С12). Мелкораздробленный алюминий, диспергированный в рас-
творителе, активируют размалыванием в шаровой мельнице и
обрабатывают при повышенном давлении водородом в присут-
ствии триэтилалюминия. Образовавшийся диэтилалюминийгид-
рид обрабатывают этиленом под давлением и получают триэтил-
алюминий.
2А1 ЗН2 4- 4А1(С2Нб)з —> 6А1Н(С2Н5)2
6А1Н(С2Нб)2 + 6С2Н4 —> 6А1(С2Н6)з
Часть триэтилалюминия возвращают на стадию получения гид-
рида, а остальное его количество вводят в реакцию с этиленом
при температуре ниже 130 °C и давлении 7—10 МН/м2 (70—
100 атм). При этом происходит присоединение этилена
Z(CH2CH2)XH
А1(С2Н6)з + пСН2=СН2 —> А1—(СН2СН2)9Н
\сн2сн2)2н
При более высокой температуре (130—250°С) протекает конку-
рирующая реакция
/(CH2CHS)XH С H(CH2CH2)X_,CH=CH2
Al—(СН2СН2)^Н + ЗСН2=СН2 —► (С2Н6)зА1 + j Н(СН2СН2)г-1СН=СН2
\сн2сн2)2н I H(CH2CH2)2_!CH=CH2
* В 1975 г. 515 тыс. т [Chem. and Eng. News, 54, № 24, 37 (1976)].—
Прим, перев.
Первичные нефтехимические продукты. I.
109
которая может проводиться как термически, так и каталитиче-
ски (металлический никель, температура 50°C). Обычно исполь-
зуют некаталитический процесс, с помощью которого линейные
а-олефины удается получить с выходом -~95%. Побочными реак-
циями являются миграция двойной связи и разветвление цепи.
Триэтилалюминий можно регенерировать путем дистилляции или
экстракции.
Линейные а-олефины используются в качестве полупродуктов
для получения высших спиртов, применяемых в производстве
пластификаторов (спирты С? — Сэ) и моющих веществ (спирты
С12 — С18). Этот процесс, разработанный фирмой Continental Oil
Со и впервые реализованный в США фирмой Gulf Oil, известен
под названием «альфен-процесс».
В одной из его модификаций — так называемом альфол-про-
цессе — смесь триэтилалюминиевых соединений окисляют в соот-
ветствующие триалкоксиды алюминия, которые затем гидроли-
зуют серной кислотой. В результате образуется смесь первичных
спиртов С4—Сг2-
ЗСН2=СН2 +-| Н2 + А1 —> (С2Н5)3А1
(С2Н6)зА1 + «СН2=СН2 —>
н(сн2сн2)хх О2
H(CH2CH2)y—А1 -Л
Н(СН2СН2)г//
Н(СН2СН2)хОх
\ Г12аС/4
—> Н(СН2СН2)уО—А1 ------>
Н(СН2СН2)гО//
Н(СН2СН2)ХОН
H(CH2CH2)yOH + 1 AI2(S О4)з
Н(СН2СН2)гОН
Этот процесс реализован в США и ФРГ. Полученные спирты
служат полупродуктами для производства пластификаторов
(спирты Се — Сю, которые переводят во фталаты) и моющих
веществ (спирты Сю— С]8).
Этиленпропилено^вые каучуки
В результате работ Натта по полимеризации пропилена стало
возможным промышленное производство сополимеров этилена
с пропиленом (в соотношении приблизительно 50:50) с исполь-
зованием катализаторов Циглера. Этиленпропиленовые каучуки
(СКЭП) обладают значительным сопротивлением истиранию и
высокой озоно- и кислородостойкостью; кроме того, они потенци-
ально дешевле других эластомеров. Поскольку этиленпропиле-
новые каучуки не содержат двойных связей, их можно вулкани-
зовать только перекисями, что имеет ряд серьезных недостатков.
Для преодоления этого затруднения с целью придания этилен-
пропиленовым каучукам способности вулканизоваться серой в
110
Глава 4
них вводят третий компонент (в количестве 5—10%), содержа-
щий две несопряженные двойные связи. Такие продукты назы-
вают тройными этиленпропиленовыми каучуками (СКЭПТ).
В качестве третьего сомономера используют дициклопентадиен,
гексадиен-1,4, этилиденнорборнен и другие диены. В табл. 4.6
представлены данные о потреблении этиленпропиленовых кау-
чуков.
Таблица 4.6
Динамика потребления (тыс. т) этиленпропилеиовых каучуков (СКЭПТ)
Страна или регион 1965 г. 1970 г. 1975 г. (прогноз)
США 13 75 150 а
Великобритания — 3 23
Западная Европа 1 23 85 6
Японии — 13 55 в
Мировое потребление 14 150 320 г
а В 1974 г. потребление СКЭПТ в США составило 120 тыс. т, а в 1975 г.—85 тыс. т
[Chem. Market Abstr., 68, 300 416 (1976)]. — Прим, перев.
б В 1975 г. фактическое потребление, по оценке, составило 49 тыс. т [Chem. Market
Abstr., 68 , 300 817 (1976)]. — Прим, перев.
в В 1975 г. фактическое потребление было равно 17,5 тыс. т [Chem. Market Abstr.,
68, 300 444 (1976)].—Прим, перев.
г Фактическое мировое потребление СКЭПТ в 1975 г. (без социалистических стран)
оставляло 163 тыс. т [Chem. Marketing Rep., 209, № 17, 16 (1976)]. — Прим, перев.
Пропилен
Пропилен получают из газов пиролиза углеводородного
сырья на этилен и из нефтезаводских газов. Обычно отношение
пропилена к этилену в газах пиролиза нафты составляет 0,4—0,5.
В США значительные количества пропилена расходуются неф-
теперерабатывающими заводами на получение алкилатного и
полимерного бензина.
Полипропилен
В отличие от этилена, полимеризацию которого можно прово-
дить как при низком, так и при высоком давлении, пропилен
полимеризуют только по методу Циглера. В большинстве обла-
стей своего применения полипропилен успешно конкурирует с
полиэтиленом высокой плотности. Он используется для изгото-
вления различных изделий методами литья под давлением и
экструзии; кроме того, полипропилен выпускается в виде лент,
фибриллированной пленки, непрерывной нити, моноволокна и
штапельного волокна. Более подробно технология получения
полипропилена и его стереорегулярные формы рассматриваются
в гл. 8.
Первичные нефтехимические продукты. I. 111
Акрилонитрил (гл. 6 и 7)
В 30-х годах в Германии был построен первый завод по про-
изводству акрилонитрила из ацетилена. Этот метод получения
акрилонитрила сохранял первостепенное значение до начала 60-х
годов, а затем он был в значительной степени вытеснен методом
окислительного аммонолиза пропилена (гл. 6):
СН3СН=СН2 + NH3 + О2 —> CH2=CHCN + ЗН2О
В гл. 7 сопоставляются различные способы промышленного
получения акрилонитрила и приводятся причины, по которым
окислительный аммонолиз пропилена наиболее предпочтителен.
В 1970 г. мировое производство акрилонитрила составляло,
по оценке, 1,4 млн. т *. В США мощности по получению акрило-
нитрила в 1972 г. достигали 540 тыс. т/год, а потребность в нем
в 1971 г. равнялась 460 тыс. т. В структуре потребления акрило-
нитрила в США 60% приходилось на долю производства поли-
акрилонитрильных и модакриловых волокон, 18%—на долю
производства сополимеров акрилонитрила, бутадиена и стирола
(АБС-пластиков) и сополимеров акрилонитрила и стирола (АС-
пластиков), 6% шло на синтез бутадиеннитрильных каучуков и
16% —на прочие цели, включая экспорт**.
Некоторое количество акрилонитрила перерабатывают в
акриламид, полимер которого используется, в частности, как
флоккулирующий агент.
разб. H2SO4
ch2=chcn + н2о ----------> ch2=chconh2
В дополнение к глутамату натрия (усилитель вкуса, гл. 16),
получаемому методом ферментации, японская фирма Ajinomoto
выпускает синтетический продукт, вырабатываемый из акрило-
нитрила
Со-катализатор HCN/NHa
ch2=chcn + со + н2 -------------> ohcch2ch2cn ------->
NaOH/H2O
—> NCCH(NH2)CH2CH2CN ---------> NaOOCCH(NH2)CH2CH2COOH
Ряд фирм разработали технологию получения адипонитрила
путем восстановительной димеризации акрилонитрила. Один из
этих процессов, реализованный в промышленности, основан на
электрохимическом восстановлении акрилонитрила на свинцо-
вом катоде в электролизере с диафрагмой. Полученный адипо-
* В 1974 г. во всем мире было получено -~2,3 млн. т акрилонитрила
[Hydrocarbon Process., 54, № il, 108 (1975)]. — Прим, перев.
** Мощности по производству акрилонитрила в США на 1 января 1976 г.
составляли 750 тыс. т/год [Chem. Marketing" Rep., 209, № 1, 42 (1976)], а по-
требление его в 1974 г. равнялось 536 тыс. т (из них 59% пошло на получе-
ние полиакрилонитрильных волокон, 24%—на получение АС- и АБС-сополи-
меров и 6,7%—на синтез бутадиеннитрильных каучуков) [Chem. Industrie,
28, 28 (1976)]. — Прим, перев.
112
Глава 4
нитрил используется как полупродукт в производстве найлона-
6,6 (гл. 9).
2CH2=CHCN + 2Н2О + 2е —> NC(CH2)4CN + 2ОН“ (реакция на катоде)
Н2О —> 2Н++ у О2 + 2е (реакция на аноде)
Недавно были опубликованы данные о новом методе полу-
чения найлона-6,6: присоединение к акрилонитрилу хлористого
водорода с последующей обработкой образовавшегося хлорпро-
пионитрила порошкообразным железом и получением адипони-
трила. Побочный продукт реакции — хлористое железо — восста-
навливают до металла и возвращают в процесс.
«Оксоспирты» (гл. 6)
Путем гидроформилирования пропилена с последующим вос-
становлением промежуточно образующихся альдегидов полу-
чают н-бутанол и изобутанол
СН3СН=СН2 + СО + Н2 —> СН3СН2СН2СНО + (СН3)2СНСНО
бутанол-1 изобутанол
Изопропанол (гл. 6)
Изопропанол, полученный гидратацией пропилена, был пер-
вым нефтехимическим продуктом, вырабатываемым в промыш-
ленном масштабе (его производство началось приблизительно
в 1920 г.). При этом, как и в случае этилена (см. выше), можно
использовать либо сернокислотный метод, либо метод прямой
каталитической гидратации.
н2о
СН3СН=СН2 ----> (СН3)2СНОН
Ацетон
Ацетон получают совместно с фенолом кумольным методом
(стр. 115, гл. 5 и 7), в котором пропилен является одним из ис-
ходных соединений. Кроме того, ацетон вырабатывается (все в
меньшем количестве) из изопропанола путем его дегидрирова-
ния или окисления (гл. 7).
(СН3)2СНОН ч=* (СН3)2СО + Н2
>. z-, 1 Си-катализатор
СНз)2СНОН + — О2 --------------> (СНз 2СО + Н2о (выход 85 - 90%)
2 550 — 650 °C '
Первичные нефтехимические продукты. /.
113
Фирма Shell разработала технологию жидкофазного окисления
изопропанола в ацетон и перекись водорода кислородом при
температуре 80—120°С и давлении 0,2—0,3 МН/м2 (2—3 атм)
(СН3)2СНОН + О2 —> (СН3)2СО + Н2О2
Ацетон получают также прямым окислением пропилена
(гл. 7) и как побочный продукт производства глицерина из
акролеина (см. ниже).
Окись пропилена
В настоящее время основным промышленным методом полу-
чения окиси пропилена является хлоргидринный (многие высво-
бодившиеся установки по хлоргидринному производству окиси
этилена переведены на выпуск окиси пропилена). В качестве
побочного продукта при этом образуется 1,2-дихлорпропан.
Са(ОН)2
СН2=СНСН3 + НОС1 —> СН2СНСН3 --------->• Н2С—СНСНз
I I \/
С1 он о
Изыскание технологичного метода прямого эпоксидирования
пропилена послужило предметом широкого исследования в раз-
личных странах мира. К настоящему времени в промышленности
реализован лишь один процесс этого типа, хотя в литературе
описано большое число таких процессов. Среди них заслуживает
внимания процесс фирмы Celanese, основанный на использова-
нии надуксусной кислоты и осуществляемый путем совместного
окисления пропилена и ацетальдегида (или изомасляного альде-
гида).
Фирмы Kellog и Bayer исследовали электрохимический метод
получения окиси пропилена. В разработанном ими процессе про-
пилен пропускают через анодное пространство диафрагменного
электролизера, в котором анолитом служит разбавленный рас-
твор хлористого натрия. Образующаяся на аноде хлорновати-
стая кислота реагирует с пропиленом, давая пропиленхлоргид-
рин. Последний диффундирует в катодное пространство и раз-
лагается там сильнощелочным католитом с образованием окиси
пропилена.
Реакции на аноде-.
2СГ —> С12 + 2е
С12 + Н2О —> НОС1 + Н- + СГ
НОС1 + СН3СН=СН2 —> СН3СНС1СН2ОН + СН3СН(ОН)СН2С1
114
Глава 4
Реакции на катоде-.
2е + 2Н2О —► 2НО" + Н2
СН3СНС1СН2ОН •) но-
+ } —> сн3сн—сн2 + Н2О + сг
СН3СН(ОН)СН2С1 ) \ /
о
Суммарная реакция-.
СН3СН=СН2 + Н2О —>
СН3СН—СН2 + Н2
О
Однако данные о промышленном внедрении этого процесса
пока отсутствуют. Единственным реализованным процессом пря-
мого эпоксидирования пропилена является процесс, разработан-
ный совместно фирмами Scientific Design и Atlantic Richfield и
осуществленный на трех недавно построенных установках в
США, Нидерландах и Испании. Реакция протекает в присутствии
катализатора, содержащего переходные металлы, по уравнению
СН3СН=СН2 + ROOH —► СН3СН—СН2 + ROH
х/
где ROOH = С6Н6СН(СН3)ООН или трет-С4Н9ООН
На основе данных по изучению реакции гидроперекиси трет-
бутила с циклогексеном в присутствии каталитических количеств
солей ванадия Гоулд и др. пришли к выводу, что эта реакция не
протекает ни по свободнорадикальному механизму, ни по меха-
низму окисления — восстановления и что лимитирующей стадией
является гетеролитическое расщепление связи О—О в комплексе
металла с гидроперекисью. Предложенный ими механизм можно
представить следующей схемой:
1. Быстрая необратимая активация катализатора-.
трет-СЩЮОН I..
VO(acac)2 -----------> —V^-
2. Быстрое обратимое комплексообразование-.
| | + /С4Н9-трет
—V— + трет-С4Н9ООН ч=> —V—(У
I I \он
3. Гетеролитическое расщепление связи О—О, лимитирующее суммарную
скорость процесса'.
—уХ-сК + I
I ОН
I
-I--V—OCiflg-mpeiu
Первичные нефтехимические продукты. 1.
115
4. Быстрый перенос протона'.
5. Быстрый обратимый обмен лигандами:
| + /С4Н8-трет | + хС4Н9-трет'
—V—С/ + трет-С4Нв00Н =?=t —V—с/ + трет-С4Н9ОН
I 44 I Х)Н
6. Обратимое ингибирование:
трет-С^эОН
---------->
-<--------
| + zC4H9-TpeT
—v—dz
I \н
третье 4Н9ОН
------------>
<----------
—V (трет-С4Н9ОН)2
Окись пропилена находит применение в производстве пропи-
ленгликоля (используемого для получения ненасыщенных поли-
эфиров и т. п.), пенополиуретана, поверхностно-активных ве-
ществ и др.
Кумол
Бензол алкилируется пропиленом легче, чем этиленом (гл. 5).
Эту реакцию ведут в паровой фазе над фосфорнокислотным ка-
тализатором при температуре 250 °C и давлении 2,5 МН/м2
(25 атм) или в жидкой фазе в присутствии хлористого алюми-
ния, активированного хлористым водородом, при температуре
70—100°C и давлении 0,1—0,5 МН/м2 (1—5 атм). В жидкофаз-
ном процессе катализатором может служить также серная кис-
лота.
сн3сн=сн2 + н+ ---> (СНЩСН +
+/СНз
сн
I
СН3
Кумол используют главным образом как полупродукт для
совместного получения фенола и ацетона (гл. 5). Небольшая
часть его в виде гидроперекиси применяется также в качестве
инициатора свободнорадикальной полимеризации.
Хлористый аллил
Хлорирование пропилена при температуре 450—550 °C и от-
ношении пропилена к хлору 4 : 1 приводит к образованию хло-
ристого аллила с выходом 80—85%. Основными побочными
116
Глава 4
продуктами этой реакции являются 1,2- и 1,3-дихлорпропаны
(т. 2, стр. 129).
СН2=СНСН3 + Cl2 —> СН2=СНСН2С1 + НС1
Хлористый аллил применяется главным образом для полу-
чения эпихлоргидрина, используемого в производстве эпоксид-
ных смол и в одном из методов синтеза глицерина (схема 5).
Са(ОН)2
С1СН2СН=СН2 + НОС1 —> С1СН2СН—сн2 ----------->-
ОН (L
NaOH/H2O
—> С1СН2СН—СН2 ----------> НОСН2СН(ОН)СН2ОН
V
эпихлоргидрин глицерин
|(СН3)2С(СвН4ОН)2
мономер дли получения эпоксидной смолы
Схема 5.
Гидролиз хлористого аллила дает аллиловый спирт, а взаимо-
действие с аммиаком — аллиламин
NaOH
СН2=СНСН2С1 ->
--->
NH3
--->
СН2=СНСН2ОН + NaCl
CH2=CHCH2NH2 + HCI
Акролеин
Окислением пропилена над окисномедиым катализатором
при температуре 350 °C получают акролеин с выходом 85%.
В качестве разбавителя используют водяной пар; степень кон-
версии пропилена за один проход составляет около 20%. Этот
процесс реализован на заводе фирмы Shell Chemical Company
в г. Норко (США) на установке мощностью 20 тыс. т/год. Раз-
Первичные нефтехимические продукты. /.
117
работали также катализаторы, содержащие висмут, молибден и
фосфор (аналогичные катализаторам для получения акрилони-
трила). Полагают, что реакция протекает через промежуточное
аллильное производное, образующееся на поверхности катали-
затора путем отщепления атома водорода от метильной группы
пропилена. Затем происходят быстрая изомеризация этого про-
изводного и присоединение атома кислорода. Протекание атаки
СН2=СНСНз —* СН2=СНСН2—Кат Кат— СН2СН=СН2
—-гд
СН2=СНСН2О—Кат
н
сн2=снсно
Кат—ОСН2СН=СН2
|-н
онссн=сн2
атомом кислорода по обоим концам молекулы пропилена дока-
зано с помощью меченых атомов.
Акролеин служит полупродуктом для синтеза глицерина по
методу фирмы Shell Chemical Company. Согласно этому методу,
СН2=СНСНО + (СН3)2СНОН —> СН2=СНСН2ОН + (СН3)2СО
акролеин изопропанол аллиловый спирт ацетон
СН2=СНСН2ОН —> НОСН2СН(ОН)СН2ОН
глицерин
на первой стадии акролеин обрабатывают избытком изопропа-
нола в паровой фазе при температуре 400 °C над катализатором,
содержащим магний и цинк. Выход аллилового спирта состав-
ляет 77—80%. Его гидроксилируют водной перекисью водорода
в присутствии окисновольфрамового катализатора и получают
глицерин с выходом 85—90% (считая по аллиловому спирту).
Акролеин можно окислить в акриловую кислоту с выходом
70%. Окисление ведут воздухом в присутствии водяного пара
над кобальтовым, висмутовым или молибденовым катализато-
ром при температуре 420—480 °C. Возможно и прямое окисле-
ние пропилена в акриловую кислоту над кобальтовым или мо-
либденовым катализатором (концентрация пропилена в его смеси
с воздухом и водяным паром составляет 10%). Акролеин легко
118
Глава 4
превращается в димер, гидролиз которого с последующим ги-
дрированием продукта дает гексантриол-1,2,6
Н2
С
НС/ \СН2 1) н2о
2СН2=СНСНО —> II | —г-*
НС\ /СНСНО 2) Н2
О
—> НОСН2СН(ОН)СН2СН2СН2СН2ОН
Взаимодействием акролеина с метилмеркаптаном можно
получить метионин (т. 4, стр. 367—368), применяемый в каче-
стве добавки в корм скоту
HCN,
сн2=снсно + CH3SH —► CH3SCH2CH2CHO —---------->
(NH4)2CO3
н
Нх ------СО
—> | + NH3 + Н2О
ch3sch2ch/ \со—NH
1Н+
nh2
CH3SCH2CH2CHCOOH
метионин
Изопрен
Изопрен получают из пропилена с использованием процесса
фирм Goodyear и Scientific Design, первой стадией которого
является димеризация пропилена в 2-метилпентен-1, протекаю-
щая в присутствии трипропилалюминия и никеля при темпера-
туре 150—200°С и давлении 20 МН/м2 (200 атм)
2СН3СН=СН2 —> СН2=С(СН3).СН2СН2СН3
2-метилпентен-1
На второй стадии 2-метилпентен-1 изомеризуется в 2-метилпен-
тен-2 над стационарным фосфорнокислотным катализатором
при температуре 150—300 °C
СН2=С(СН3)СН2СН2СН3 —> (СН3)2С=СНСН2СН3
2-ме'тилпентен-1 2-метилпентен-2
Термодинамически равновесная смесь (при 227°C) содержит
30,6% 2-метилпентена-1 и 69,4% 2-метилпентена-2. Последний
Первичные нефтехимические продукты. I.
119
на третьей стадии подвергают расщеплению при температуре
650—750°C в присутствии небольших количеств бромистого
водорода и водяного пара, в результате чего образуется изопрен
с выходом 65% (побочно получается также некоторое количе-
ство циклопентадиена и пиперилена, которые удаляют дистил-
ляцией)
(СН3)2С=СНСН2СН3 —> СН2=С(СН3)СН=СН2 + сн4
изопрен
В 1972 г. суммарные мировые мощности по производству
изопрена составляли ~500 тыс. т/год. Годовая мощность одного
из заводов фирмы Goodyear, работающего по описанному выше
методу, равна 50 тыс. т*. Изопрен используется в качестве мо-
номера для получения стереорегулярного полиизопрена, свой-
ства которого весьма близки к свойствам натурального каучука
(гл. 8). Объем мирового потребления полиизопрена в 1970 г.
составил 320 тыс. т.
Димеризация пропилена
В последние годы было взято значительное число патентов
на способы димеризации пропилена в присутствии каталитиче-
ской системы комплекс никеля с органическими лигандами —
алкилалюминийгалогенид. Полагают, что при повышенной тем-
пературе или в присутствии пропилена в таких системах воз-
никают каталитически активные частицы типа соединения 1,
образующегося из [(н^Н^зР^ЬИСЬ и C2H5AICI2:
(н-С4Н9)зР-/
С2Н6А1С1"
1
Согласно этому представлению, димеризация пропилена про-
текает путем быстрого замещения олефином алкильной группы,
находящейся во внутренней координационной сфере активного
комплекса, с последующим медленным внедрением второй
* В СССР крупнотоннажное производство изопрена организовано на ос-
нове двух методов: двухстадийного дегидрирования изопентана и двухстадий-
ного сйнтеза из изобутилена и формальдегида через диметилдиоксан [сб. «Син-
тетический каучук», под ред. Гармонова И. В., «Химия», Л., 1976, стр. 15].—
Прим. ред.
120
Глава 4
молекулы олефина по связи Ni—С, как это показано на схеме 6.
Степень разветвленности полученного димера зависит от
температуры (низкие температуры способствуют образованию
промежуточного соединения 2), природы алюминийорганиче-
ского компонента и характера лиганда. Так, если лигандом
служит трифенилфосфин, то наблюдается довольно высокий
выход гексена с прямой цепью, тогда как при использовании
трициклогексилфосфина получаются преимущественно 2,3-ди-
метилбутены лишь с небольшой (~2%) примесью неразвет-
вленного изомера.
Данные о промышленной реализации этого процесса отсут-
ствуют.
Тримеры и тетрамеры пропилена
Эти сильно разветвленные олигомеры пропилена получают
на нефтеперерабатывающих заводах на установках по произ-
водству полимерного бензина из олефинов С3 и С4, причем
Первичные нефтехимические продукты I.
121
использование бутиленов в последние годы существенно сокра-
тилось. Тримеры применяются главным образом для синтеза
нонилфенола и «изодеканола» *. Спрос на тетрамеры в послед-
ние годы существенно понизился в связи с сокращением вы-
пуска моющих средств на основе алкилбензолсульфонатов
(гл. 13).
Диспропорционирование пропилена
Фирма Phillips Petroleum Со разработала процесс «Triol»,
посредством которого пропилен диспропорционируется на ко-
бальтовых и молибденовых катализаторах при температуре
150—210°С и давлении 1 МН/м2 (10 атм), превращаясь в эти-
лен и н-бутилены.
Полагают, что в этой реакции быстро устанавливается обра-
тимое равновесие между пропиленом и продуктами его диспро-
порционирования, которое, по-видимому, включает образование
на поверхности металлического катализатора квазициклобута-
нового промежуточного соединения
сн3
^сн=сн2
м
сн=сн2
сн3
СН3<
CH.TTmCHj
: : м :
.СН——СН2
сн3
сн3
он
сн3^сн
сн
м II
сн2
Углеводороды С4
В отличие от этана (т. кип. —88,6°C), этилена (т. кип.
—103,6 °C), пропана (т. кип. —42°C) и пропилена (т. кип.
—47,6°C), которые можно разделить путем низкотемператур-
ной ректификации, углеводороды фракции С4 (отделяемой от
углеводородов Сг— Сз при первичной ректификации) обладают
настолько близкими температурами кипения, что не могут быть
разделены этим методом на индивидуальные компоненты
(табл. 4.7).
Бутадиен можно извлечь из бутан-бутиленовой фракции с
помощью экстрактивной дистилляции с фурфуролом или N-ме-
тилпирролидоном. Бутилены концентрируют до содержания
90—95% экстрактивной дистилляцией с ацетонитрилом, в про-
цессе которой они отделяются от бутанов. Изобутилен, обла-
дающий высокой реакционной способностью, можно выделить
* Изодеканол получают методом оксосинтеза с использованием в качестве
исходного олефина тримера пропилена. Основное применение изодеканола —
производство пластификаторов с улучшенными характеристиками.—Прим,
ред.
122
Глава 4
Таблица 4.7
Углеводородный состав бутан-бутиленовой фракции,
полученной паровым пиролизом нафты
Углеводороды С4 Т. кип. при 760 мм рт. ст., °C Типичное содержание, % по весу
н-Бутан 0,5 7
Изобутан -11,7 1
Бутен-1 -6,3 18
гщс-Бутен-2 3,7 1 7
трп«с-Бутен-2 0,9 J
Изобутилен -6,9 37
Бутадиен -4,4 30
из смеси углеводородов С4 путем абсорбции 50%-ной серной
кислотой *.
Бутадиен
Бутадиен является важным исходным продуктом в произ-
водстве синтетических каучуков (гл. 8). В табл. 4.8 приведены
Таблица 4.8
Объем производства н мощности по получению бутадиена
в ведущих капиталистических странах в 1972 г.
Страна или регион
Мощности, тыс. т/год
Производство, тыс. т
Западная Европа 1260 940
Великобритания 244 200
США 1800 553
Япония 687 579
а 563 тыс. т в 1971 г.
данные о производстве бутадиена в передовых капиталистиче-
ских странах. Большая часть вырабатываемого бутадиена (96%
в Западной Европе и 79% в США) идет на получение бутадиен-
стирольного каучука, стереорегулярного бутадиенового каучука
и бутадиеннитрильного каучука (в 1970 г. мировое потребление
этих каучуков составляло 3,5 млн. т, 730 тыс. т и 190 тыс. т со-
* В СССР крупнотоннажное производство изобутилена высокой степени
чистоты организовано более эффективным методом — путем избирательной
гидратации изобутилена, содержащегося во фракции углеводородов С4, на ка-
тионообменных смолах с последующей каталитической дегидратацией обра-
зовавшегося триметилкарбинола. Преимуществами метода являются более
простое технологическое оформление и отсутствие агрессивных корродирую-
щих сред [Чаплиц Д. И., Соболев В. М., в сб. «Синтетический каучук», под
ред. Гармоиова И. В., «Химия», Л., 1976, стр. 727—730]. — Прим, ред.
Первичные нефтехимические продукты. 1.
123
ответственно). Кроме того, бутадиен используется в производ-
стве бутадиенстирольных смол и пластических масс на основе
тройного сополимера АБС (акрилонитрил—бутадиен—стирол).
Незадолго до начала второй мировой войны Реппе устано-
вил, что ацетилен в присутствии никельсодержащих катализа-
торов типа Ni [Р(С6Н5)з]2С12 превращается в циклооктатетраен
и бензол. В 1954 г. Рид показал возможность олигомеризации
бутадиена на подобных катализаторах типа Н1[Р(СбН5)з]г(СО)2,
активированного ацетиленом, с образованием циклооктадиена-
1,5 (ЦОД) (4).
цис, цис- циклооктпадиен-1,5
4
Дальнейшие исследования, особенно работы Уилке и сотруд-
ников, привели к открытию ряда реакций диенов, протекающих
на активированных алюминийалкилами комплексных катализа-
торах, содержащих двухвалентный никель (схема 7), причем
важную роль в этих реакциях играет присутствие у атома ни-
келя фосфорорганических лигандов.
циклододекатраен-1,5,9 (ЦДТ)
(смесь трех изомеров)
диалкилциклододекатриены-1, 4, 8
циклоЗекадиен I, 5
Схема. 1.
декитриен-1,4,9
124
Глава 4
Подбирая катализатор, можно с высоким выходом получать
из бутадиена тот или иной продукт его олигомеризации. Так,
координационно ненасыщенный комплекс никеля, полученный
восстановлением диацетилацетоната никеля алюминийалкилами
в присутствии подходящих лигандов (окиси углерода, третичных
фосфинов, 1,5-ЦОД или 1,5,9-ЦДТ), чрезвычайно активно ката-
лизирует превращение бутадиена (в котором он растворен)
в ЦДТ.
В этой реакции образуется транс, транс, транс-изомер ЦДТ.
Однако при использовании в качестве катализатора системы че-
тыреххлористый титан — алкилалюминийгалогенид основным
продуктом реакции является транс, транс, цис-изомер. Послед-
ний катализатор более эффективно катализирует образование
ЦДТ, чем никельсодержащие системы.
В том случае, когда одно из координационных положений
у атома никеля блокировано сильным лигандом, становится
возможным присоединение лишь двух молекул бутадиена с об-
разованием бнс-л-аллильного комплекса (схема 8).
Схема. 8.
Применение в качестве лигандов средних фосфитов P(OR)3
способствует образованию ЦОД, тогда как при использовании
третичных фосфинов получается смесь ЦОД и винилциклогек-
сена.
Первичные нефтехимические продукты. I.
125
Бутадиен способен вступать в реакцию с этиленом (схема 9),
давая циклодекадиен-1,5 и декатриен-1,4,9.
Схема 9.
В присутствии хлористого родия взаимодействие бутадиена
с этиленом, легко протекающее при температуре 50°C, приводит
к образованию смеси гексадиена-1,4 и гексадиена-2,4 с общим
выходом 90%. Содержание последнего соединения в продуктах
126
Глава 4
реакции возрастает с повышением температуры. Аналогичным
образом эта реакция протекает в присутствии никель- и железо-
содержащих катализаторов, активированных алкилалюминие-
выми соединениями.
Гексадиен-1,4 вырабатывается в США и Японии в промыш-
ленном масштабе и используется в качестве сомономера для
получения тройных этиленпропиленовых каучуков.
Циклооктадиен-1,5 и циклододекатриен-1,5,9 производятся в
Западной Европе, США и Японии. Последнее соединение ис-
пользуется в ФРГ и Японии как полупродукт для получения
найлона-12, причем лактам со-аминолауриновой кислоты (исход-
ный мономер для найлона-12) можно синтезировать несколь-
кими способами (схема 10).
Карбонилирование циклооктадиена
С2Н5ОН дает эфир 5. Полученную из
в присутствии PdL2Cl2 и
него свободную кислоту
+ со+с2н5он
PdL2Cls
Х^СООС2Н5
б
Первичные нефтехимические продукты. I.
127
можно с высоким (более 80%) выходом превратить в азелаи-
новую кислоту путем обработки водным раствором едкого нат-
ра при высокой температуре (320°C).
(X—s^COOH
НООС(СН2)7СООН
Эти реакции пока не нашли применения в промышленности.
Гомогенное газофазное хлорирование бутадиена при темпе-
ратуре 330—400 °C приводит к образованию смеси 3,4-дихлор-
бутена-1 и 1,4-дихлорбутена-2 в мольном соотношении 3:2. По-
следний продукт при нагревании с однохлористой медью изоме-
ризуется в 3,4-дихлорбутен-1, который, будучи более низкокипя-
щим соединением, может непрерывно удаляться из реакционной
смеси отгонкой. Дегидрохлорирование 3,4-дихлорбутена-1 горя-
чим раствором едкого натра позволяет получить хлоропрен
с выходом более 90%.
СН2=СНСН=СН2 —> СН2=СНСНС1СН2С1 С1СН2СН=СНСН2С1
3,4-дихлорбутен-1 1,4-дихлорбутен-2
|-НС1
СН2=СНСС1=СН2
хлоропрен
При обработке смеси дихлорбутеиов цианистым натрием в
присутствии однохлористой меди образуется 1,4-дицианобутен-2.
Гидрирование этого продукта в паровой фазе приводит к полу-
чению адипонитрила, дальнейшее гидрирование которого дает
гексаметилендиамин.
С1СН2СН=СНСН2С1 4 NaCN
СН2=СНСНС1СН2С1 J Си2С12
Н2
ncch2ch=chch2cn —►
н2
—► ncch2ch2ch2ch2cn —> H2N(CH2)6NH2
Имеются сведения, что этот процесс до сих пор используется
на одном из заводов фирмы du Pont.
Недавно фирмы du Pont и Esso объявили о том, что ими
разработаны новые процессы синтеза адипонитрила из бута-
диена; однако сведения об этих процессах весьма скудны.
128
Глава 4
Технология фирмы du Pont включает гидроцианирование бута-
диена в присутствии катализатора, содержащего металлический
никель. На первой стадии происходит присоединение синильной
кислоты к бутадиену с образованием смеси изомерных ненасы-
щенных мононитрилов. Продукты реакции разделяют и нитрилы
изостроения подвергают изомеризации; полученную равновес-
ную смесь изомеров снова разделяют. Затем проводят присоеди-
нение второй молекулы синильной кислоты и из образовавшейся
смеси динитрилов выделяют адипонитрил. В основе процесса
фирмы Esso лежит реакция бутадиена с иодом и цианистой
медью, приводящая к получению комплекса дегидроадипонит-
рила с иодидом одновалентной меди. При гидролизе этого
комплекса водным раствором синильной кислоты с высоким вы-
ходом образуется дегидроадипонитрил и регенерируются иод и
цианистая медь, которые возвращают в реакцию.
Взаимодействие бутадиена с двуокисью серы под давлением
дает циклический сульфон, гидрированием которого получают
тетраметиленсульфон (сульфолан) — соединение с т. кип.
СНСН
СН2 СН2 + SO2 ч=Ь
S S
О2 О2
сульфолан
285 °C, смешивающееся с водой в любых соотношениях. Сульфо-
лан применяется как селективный растворитель для абсорбции
двуокиси углерода и экстракции ароматических углеводородов.
Изобутилен
Изобутилен является важным полупродуктом для получения
бутилкаучука (который содержит 95% звеньев изобутилена);
в 1970 г. потребление изобутилена в США составило 95 тыс. т,
а во всем мире — 255 тыс. т (гл. 8).
Полимеры изобутилена и его сополимеры с «-бутиленами,
представляющие собой вязкие жидкости, используются в каче-
стве «индексных» присадок к смазочным маслам (присадок, по-
нижающих температурную зависимость вязкости) и герметиков.
При абсорбции изобутилена 60 %-ной серной кислотой при
температуре 20 °C с последующим нагреванием до 90 °C легко
Первичные нефтехимические продукты. I.
129
образуется смесь димеров и тримеров (в соотношении 80: 20)
с общим выходом 90% (т. 1, стр. 65):
/СН3
СН2=С<
Х СНз
СНз СНз
I I
СНз—С—СН=С—СНз +
СНз
2,4,4-триметилпентен-2
(80% от количества
димеров)
СНз СНз
I I
+ СНз—С—СН2—С=СН> -|- тримеры
I
СНз
2,4,4-триметилпентен-1
(20% от количества
димеров)
Димеры изобутилена применяются для алкилирования фено-
лов в производстве моющих веществ и для синтеза спиртов Сэ
по реакции карбонилирования.
Взаимодействием изобутилена с пропиленом в присутствии
хлористого алюминия или фосфорной кислоты получают смесь
разветвленных гептенов, которую перерабатывают в изооктанол.
Разработано по меньшей мере четыре промышленных метода
синтеза изопрена из изобутилена. Все они базируются на реак-
ции Принса (конденсации альдегидов с олефинами). В процессе,
разработанном и осуществленном в СССР, чистый изобутилен
обрабатывают формальдегидом в 1,5%-ной серной кислоте и
полученный 4,4-диметилдиоксан-1,3 (выход 75—80%) подверга-
ют расщеплению над фосфатнокальциевым катализатором при
температуре 300—400°C с образованием изопрена (выход 85%).
Аналогичный процесс, разработанный французским Институтом
нефти (Institut Frangaise du Petrole), отличается тем, что поз-
воляет использовать в качестве исходного сырья бутан-бутиле-
новую фракцию. Это приводит (особенно при низкой концентра-
ции изобутилена) к образованию значительного количества
побочных продуктов — многоатомных спиртов.
CHav ц+
)с=СН2 + 2НСНО —>
сн3/
СН8\ /СН2 СН2\ 300—400 °C
/С\ /° -----------------**
СН3/ ХО-------СН/
4,4-диметилдиоксан-1,3
СН3
—> сн2=с—сн=сн2 + нсно + Н2О
изопрен
б Зак, 501
130
Глава 4
Разработаны различные модификации этого процесса в ряде
стран, в том числе в Японии (процесс фирмы Sumitomo) и ФРГ
(процесс фирмы Bayer). Последний процесс базируется на ис-
следованиях, проведенных в 1938—1939 гг. в Людвигсхафене
(Германия), и включает осуществление реакции Принса на
твердой ионообменной смоле и расщепление диметилдиоксана
на псевдоожиженном фосфорнокислотном катализаторе.
Более новым является процесс фирмы Marathon (США), в
котором вместо формальдегида применяется метилхлорметило-
вый эфир, причем реакцию ведут в присутствии TiCl4.
Tici4
(СН3)2С=СН2 + СН3ОСН2С1 ----> (СН3)2СС1СН2СН2ОСН3
При термическом расщеплении продукта конденсации получают
изопрен, а также метанол и хлористый водород, которые воз-
вращают на стадию синтеза метилхлорметилового эфира.
(СН3)2СС1СН2СН2ОСН3 -а---ВаНИ> СН2=С(СН3)СН=СН2 + СН3ОН + НС1
изопрен
По сообщению фирмы Marathon, общий выход изопрена, считая
на изобутилен, составляет 80%. Венесуэльская фирма Ashland
Oil уже приобрела лицензию на этот процесс.
Изобутилен гидратируется в мягких условиях (60%-ная
серная кислота, 20 °C), превращаясь в трет-бутиловый спирт
(гл. 6). При жидкофазном хлорировании изобутилена образует-
ся металлилхлорид с выходом 88%.
Фирма Escambia Chemical Corporation разработала техно-
логию получения метакриловой кислоты путем окисления изо-
бутилена азотной кислотой и двуокисью азота
[О]
СН2=С(СН3)2 —> (СН3)2С(ОН)СООН —*, СН2=С(СН3)СООН
метакриловая кислота
Кроме того, изобутилен применяют для алкилирования фено-
лов с целью получения антиоксидантов. Одним из важнейших
продуктов этого типа является бутилированный п-крезол.
н-Бутилены и н-бутан
Дегидрирование н-бутиленов и н-бутана протекает с погло-
щением тепла и приводит к получению бутадиена. Термодина-
мика этого процесса такова, что высокая температура и низкое
Первичные нефтехимические продукты. 1. 131
давление смещают равновесие в сторону образования диена.
СН2=СНСНЛ'Н3 _Н2
CH3CH2CH/.’Hj + < СН2=СНС'Н=СН2
+ lh СНзСН=СНСН3 4112 As
-н-’ -------
+ н:
В процессе фирмы Houdry дегидрирование н-бутана прово-
дят в одну стадию при температуре 590—675°C и давлении
0,5 МН/м2 (5 атм)*. н-Бутан подают в систему реакторов со ста-
ционарным слоем таблетированного алюмохромового катализа-
тора, смешанного с инертным материалом. Каждый реактор
работает 5—10 мин, после чего поток переключают на следую-
щий реактор, а в первом производят регенерацию катализатора.
Регенерацию осуществляют путем продувки водяным паром с
последующим выжиганием отложений кокса током воздуха.
Бутадиен отделяют от бутана и бутиленов с помощью селектив-
ной экстракции или экстрактивной дистилляции; бутан и бути-
лены возвращают в цикл. Общий выход бутадиена достигает
60%. Дегидрирование н-бутиленов обычно ведут в присутствии
водяного пара, чтобы понизить парциальное давление углево-
дорода и свести к минимуму коксообразование.
При гидратации бутена-1 и бутена-2 (гл. 6) получается бу-
танол-2. Из-за легкости полимеризации этих олефинов прямая
гидратация их не применяется. Обычно в качестве гидратирую;
щего агента используют 80—85%-ную серную кислоту в мягких
условиях. Дегидрированием бутанола-2 при температуре 350—
400 °C над металлическим катализатором получают метилэтил-
кетон (бутанон-2), который применяется главным образом в ка-
честве растворителя. Это соединение можно также синтезиро-
вать окислением бутена-1 и бутена-2 в присутствии хлористого
палладия (подобно получению ацетальдегида). Окисление бу-
тена-1 протекает примерно в 4 раза медленнее, чем этилена;
выход метилэтилкетона при температуре 90—120 °C и давлении
0,9—2 МН/м2 (9—20 атм) составляет 85—88%.
Чистый бутен-1 находит все более широкое применение для
- получения сополимеров с другими а-олефинами. Эпоксидирова-
ние бутена-1 дает окись бутилена, которую превращают в бутан-
диол-1,2, используемый в производстве пластификаторов и как
стабилизатор хлорорганических растворителей.
В гл. 3 было описано окисление нафты. Разработан также
ряд технологических процессов, предусматривающих использо-
вание в качестве сырья бутан-бутиленовой фракции.
* В соответствии с принципом Ле Шателье этот процесс проводят под
вакуумом при остаточном давлении 120—150 мм рт. ст. — Прим. ред.
5*
ГЛАВА 5
Первичные нефтехимические продукты.
II. Ароматические углеводороды
Введение
X. Нейлор
Naylor Н. G., Central Technical Division,
BP Chemicals International Limited, London
Большинство многотоннажных органических продуктов, со-
держащих бензольные кольца, можно синтезировать, исходя из
бензола, толуола или индивидуальных ароматических углеводо-
родов Сд. Ранее основным источником ароматических углеводо-
Таблица 3.1
Методы получения и области применения важнейших
ароматических углеводородов
Углеводород Методы получения Области применения или продукты переработки
Бензол 1. Экстракция 2. Деалкилирование толуо- ла Полистирол (через этил- бензол), фенол (через кумол), циклогексан, моющие вещества
Толуол Экстракция Повышение качества бензи- на, бензол, толуилендии- зоцианат а
Этилбензол 1. Экстракция 2. Алкилирование бензола Полистирол
п-Ксилол 1. Экстракция 2. Изомеризация аромати- ческих углеводородов С8 Полиэфирные волокна и (в незначительной сте- пени) пленки
о-Ксилол 1. Экстракция 2. Изомеризация аромати- ческих углеводородов С8 Фталатные пластификаторы, алкидные и полиэфирные смолы
.и-Ксилол Экстракция Ограниченное применение для получения изофта- левой кислоты
Кумол Алкилирование бензола Фенол (побочно получает- ся также ацетои)
а Смесь 2,4- ретанов. и 2,6-толуилендиизоцианатов, применяемая в производстве пенополиу-
Первичные нефтехимические продукты. П.
133
родов была каменноугольная смола. Однако с изобретением
двигателя внутреннего сгорания и увеличением спроса на бен-
зин стало быстро возрастать значение нефтяного сырья. Тем
не менее существенную часть бензола все еще получают из по-
бочных продуктов коксования каменного угля (гл. 2).
В табл. 5.1 приведены первичные ароматические углеводоро-
ды, которые получаются путем экстракции нефтяного сырья или
синтетическими методами, а также конечные продукты их пе-
реработки.
В табл. 5.2 представлены данные по объему производства
индивидуальных ароматических углеводородов в Западной Ев-
ропе в 1970 г. и величине производственных мощностей по каж-
дому из них.
Таблица 5.2
Объем производства и производственные мощности
по важнейшим ароматическим углеводородам в Западной Европе в 1970 г.
Ароматический углеводород
Производство, тыс. т Мощность, тыс. т/год
Бензол 2700 а 3600
Этилбензол 1400 1800
Кумол 900 1000
о-Ксилол 250 300
п-Ксилол 300 400
а В 1975 г. производство бензола в Западной Европе составляло 3310 тыс. т, а
мощности—6074 тыс. т {Chem. Industrie, 28, № 9 (1976)].— Прим перев.
Методы получения ароматических углеводородов
X. Нейлор
Naylor Н. G., Central Technical Division,
BP Chemicals International Limited, London
Содержание индивидуальных ароматических соединений в
сырой нефти очень мало. Поэтому селективной экстракции аро-
матических углеводородов предшествует получение их концен-
трата. Первой очевидной стадией концентрирования является
перегонка сырой нефти с целью выделения погона приблизитель-
ного состава С6—Се; однако и в нем содержание ароматических
углеводородов все еще невелико. Далее эту узкую фракцию
134
Глава 5
подвергают каталитическому риформингу, приводящему к увели-
чению концентрации ароматических углеводородов, что необхо-
димо не только для нефтехимических целей, но и для получения
высококачественного моторного топлива. В табл. 5.3 приведены
типичные составы исходного сырья и катализатов риформинга
Таблица 5.3
Углеводородный состав (% по весу) исходного сырья
и катализатов риформинга с различными октановыми числами
Соединения Исходное сырье Катализат с октановым числом по исследовательскому методу (без добавки тетраэтилсвинца)
90 96 100
Ароматические углеводоро- ды: бензол 0,8 4,3 5,2 6,9
толуол 3,4 20,1 23,1 26,4
этилбензол 1,1 3,2 3,7 3,8
/г-ксилол 0,9 4,0 4,5 5,1
Л/-КСИЛОЛ 1,8 8,5 8,7 10,2
о-ксилол 1,2 4,2 4,7 5,9
углеводороды С9 0 6,4 7,7 8,2
углеводороды С1о и 0 0 0 0
выше Общее содержание аромати- 9,2 50,7 57,6 66,5
ческих углеводородов Общее содержание парафи- 66,5 44,3 41,4 33,1
нов Общее содержание нафте- 24,3 5 0 1,0 0,4
ИОВ
и октановые числа последних по исследовательскому методу.
Октановое число бензина представляет собой меру его дето-
национной стойкости, причем, чем выше октановое число, тем
лучше качество бензина (гл. 3).
Все более важным источником получения ароматических
углеводородов становится также процесс парового пиролиза
нефтяного сырья (гл. 3). Полученную бензиновую фракцию гид-
рируют, чтобы перевести олефины и диены в насыщенные со-
единения, и подвергают селективной экстракции.
Методы дальнейшей переработки концентрата ароматических
углеводородов выбирают в зависимости от характера целевых
продуктов. На рис. 5.1 приведена принципиальная технологиче-
ская схема получения всех важнейших ароматических углеводо-
родов. Представленные в этой схеме процессы обсуждаются
ниже.
Первичные нефтехимические продукты. //.
135
Бензол (на получение
этилбензола и кумола)
Тяжелые ароматические
углеводороды на компаундирование
бензина
Рис. 5.1. Технологическая схема выделения важнейших ароматических уг-
леводородов.
Селективная экстракция
Ароматические углеводороды образуют азеотропные смеси
с некоторыми предельными соединениями, присутствующими в
катализате риформинга. Поэтому обычная ректификация не
позволяет получить чистые ароматические углеводороды, и в
большинстве случаев для их выделения используют метод селек-
тивной экстракции.
Ароматическую часть продукта риформинга можно рассмат-
ривать как растворенное вещество, а находящиеся в смеси с ней
предельные соединения — как растворитель. Если к этой системе
добавить другой растворитель, не смешивающийся с предель-
ными, соединениями, то ароматические компоненты распреде-
лятся между обоими растворителями в соответствии с их коэф-
фициентами распределения у,
CJC2 = у,
136
Глава 5
где С] — концентрация ароматического углеводорода в предель-
ных соединениях и С2 — их концентрация в добавленном рас-
творителе. Однако, во-первых, главную роль в этом процессе
играет селективность добавленного растворителя по отношению
к ароматической части продукта, а, во-вторых, приведенная
выше функция распределения не учитывает взаимную смеши-
ваемость обоих растворителей. Отсюда следует, что первосте-
пенное значение имеет отношение концентраций ароматических
и предельных углеводородов в обеих фазах и что более пра-
вильно будет пользоваться не коэффициентом распределения,
а коэффициентом селективности 0
₽ = [Ха/Хп]1/[Ха/Хп]ц
где хА и %п — мольные доли ароматических и предельных
соединений соответственно в фазах I и II. Если фаза I —
добавленный растворитель, то чем больше величина 0, тем
выше его селективность по отношению к ароматическому
углеводороду. Эта ситуация напоминает процесс ректификации:
эффективное разделение достигается путем многократного по-
вторения экстракции до получения продукта желаемой степени
чистоты.
В настоящее время в промышленности вместо описанного
выше периодического процесса приняты непрерывные схемы
экстракции с использованием большого числа смесителей и от-
стойников. Растворитель поступает в верхнюю часть колонны,
в низ которой противотоком подают катализат риформинга, при-
чем эффективное смешивание обоих потоков в колонне обеспе-
чивается с помощью вращающихся дисков. Вытекающий из
нижней части колонны растворитель обогащен ароматическими
углеводородами и содержит лишь небольшое количество пара-
финов и нафтенов. Последние удаляют экстрактивной отгонкой,
а ароматические углеводороды подвергают ректификации.
Остающийся в кубе ректификационной колонны растворитель
возвращают на стадию экстракции. В качестве растворителя
обычно используют сульфолан, смесь N-метилпирролидона с
этиленгликолем или диметилсульфоксид. В зависимости от при-
меняемого растворителя детали проведения процесса экстрак-
ции могут несколько варьироваться.
Получение о-ксилола и п-ксилола
В большинстве случаев потребность в индивидуальных кси-
лолах больше того их количества, которое получают путем
селективной экстракции нефтяного сырья с последующей ди-
Первичные нефтехимические продукты II.
137
стилляцией экстракта. Поэтому очень часто принципиальную
технологическую схему, приведенную на рис. 5.1, несколько
видоизменяют и выделяют о- и га-ксилолы непосредственно из
катализата риформинга. При четкой ректификации катализата
отбирают узкую фракцию, содержащую преимущественно аро-
матические углеводороды Се. Единственный изомер, который
может быть удовлетворительно выделен дистилляцией, — это
о-ксилол (табл. 5.4). Желательно, чтобы в потоке, поступающем
в о-ксилольные колонны, не было парафинов и нафтенов, так
как они способны образовывать с ароматическими углеводоро-
дами азеотропные смеси.
Таблица 5.4
Температуры кипения и плавления изомерных
ароматических углеводородов С8
Соединение
T. пл., °C
о-Ксилол
л-Ксилол
и-Ксилол
Этилбензол
144,41
139,10
138,35
136,15
-25,19
-47,88
+13,25
-95,01
Основным методом промышленного получения га-ксилола
является фракционированная кристаллизация (температуры
плавления ароматических углеводородов С» приведены в
табл. 5.4).
Количество га-ксилола, которое можно выделить из данного
количества сырья путем кристаллизации (т. е. степень извлече-
ния), определяется характером фазового равновесия для этого
сырья. В типичном случае поток, поступающий на стадию кри-
сталлизации (после предварительной отгонки из него о-ксило-
ла), содержит приблизительно 50% (по весу) л/-ксилола, 20%
(по весу) га-ксилола и 20% (по весу) этилбензола с небольшой
(2—4% по весу) примесью остатков о-ксилола. Строго говоря,
в этом случае необходимо учитывать фазовое равновесие для
четырехкомпонентной системы; однако принципиальная картина
рассматриваемого явления не изменится, если принять, что
основное значение в данном случае имеет поведение системы
л-ксилол — л-ксилол (рис. 5.2). При понижении температуры
(допустим, начиная с 25 °C, когда смесь представляет собой
однородную жидкость) кристаллизация n-ксилола не происхо-
дит до тех пор, пока не будет достигнута линия РЕ. В этой
точке начинается выпадение кристаллов чистого га-ксилола, а
138
Глава 5
состав фазы постепенно сдвигается с температурой вдоль линии
РЕ до точки Е, после чего начинает кристаллизоваться эвтек-
тическая смесь м- и n-ксилолов и весь раствор становится
твердым. Если необходимо получить чистый n-ксилол, то охла-
ждение прекращают незадолго до достижения точки эвтектиче-
ского состава Е и кристаллы n-ксилола отделяют путем филь-
трования или центрифугирования. Поведение реальной фракции
изомерных ароматических углеводородов Се очень близко к
Концентрация п-ксилола (в жидкой /разе),
МЛП. %
Рис. 5.2. Фазовая диаграмма системы .м-ксилол — n-ксилол (см. текст).
идеальному: присутствие этилбензола и о-ксилола вызывает
изменение содержания n-ксилола в эвтектической смеси лишь
приблизительно с 13 до 10 мол.%. Однако температура замер-
зания этой смеси понижается с —65 до —86 °C.
На практике смесь ксилолов охлаждают до температуры око-
ло —70 °C и затем центрифугируют, чтобы отделить выпавшие
кристаллы n-ксилола. Поскольку вместе с /г-ксилолом удаляется
некоторое количество других изомеров, удерживаемых в про-
странстве между кристаллами, полученный продукт слегка по-
догревают (при этом небольшая часть чистого n-ксилола рас-
плавляется и играет роль промывной жидкости) и снова цен-
трифугируют. Эту операцию повторяют до тех пор, пока не будет
достигнута необходимая степень чистоты n-ксилола (выше 99,2%
по весу). Промывную жидкость, полученную на всех ступенях
центрифугирования, возвращают в цикл, чтобы избежать из-
лишних потерь n-ксилола. Однако теоретически максимальная
Первичные нефтехимические продукты. И.
139
степень извлечения n-ксилола методом кристаллизации состав-
ляет около 60% от его содержания в исходном сырье, соответ-
ствующего эвтектической точке.
Изомеризация ароматических углеводородов С8
Смесь ароматических углеводородов С8, получаемая после
извлечения n-ксилола, содержит преимущественно лг-ксилол,
Рис. 5.3. Равновесные концентрации ароматических углеводородов С8 при
различных температурах.
1—л-ксилол^ 2—о-ксилол; 3—л-ксилол; 4—этилбензол
спрос на который, к сожалению, очень мал. Поэтому маточную
жидкость, поступающую со стадии кристаллизации, подвергают
изомеризации в равновесную смесь ксилолов, которую возвра-
щают на ректификацию в о-ксилольные колонны (рис. 5.1) с
последующим извлечением из нее о- и n-ксилолов. На рис. 5.3
приведены кривые зависимости равновесных концентраций аро-
матических углеводородов С8 от температуры.
140
Глава 5
Изомеризация трех ксилолов происходит очень легко; од-
нако равновесная концентрация этилбензола устанавливается
сравнительно медленно. Принято считать, что реакция протекает
путем постепенного перемещения метильной группы по бензоль-
ному кольцу; поэтому непосредственное превращение о-ксилола
в n-ксилол невозможно (схема 1).
Схема 1.
Имеются также данные, свидетельствующие о том, что изо-
меризация над цеолитным катализатором протекает через про-
межуточное трансалкилирование в триметилбензол (как пока-
зано на схеме 2).
Схема И.
По-видимому, изомеризация ксилолов включает оба эти ме-
ханизма. С другой стороны изомеризацию этилбензола, проте-
Первичные нефтехимические продукты. II.
141
кающую только в присутствии водорода, можно представить
в рамках механизма, изображенного на схеме 3.
Схема 3.
Разработана технология изомеризации как в жидкой, так
и в паровой фазе; однако почти все промышленные процессы
являются парофазными. В качестве катализатора обычно при-
меняют алюмосиликаты или платину на алюмосиликатном но-
сителе, причем выбор между ними определяется главным
образом содержанием этилбензола в исходном сырье. Если кон-
центрация этилбензола сравнительно велика, то более предпо-
чтителен платиновый катализатор, так как в его присутствии
этилбензол претерпевает частичную изомеризацию, а также кре-
кируется с образованием легких углеводородов, удаляемых ди-
стилляцией. Напротив, алюмосиликатный катализатор практи-
чески не оказывает влияния на присутствующий этилбензол, что
может привести к недопустимо высокому содержанию этилбен-
зола в потоке, возвращаемом на стадию выделения о-ксилола.
Другие основные различия между обоими катализаторами за-
ключаются в следующем:
а) платиновый катализатор нуждается в присутствии водо-
рода, без которого не может протекать изомеризация этилбен-
зола;
б) алюмосиликатный катализатор приходится часто регене-
рировать выжиганием отложений кокса, тогда как платиновый
катализатор способен проработать без регенерации год и
более.
Обычно процесс изомеризации с использованием платинового
катализатора ведут при температуре около 460 °C, давлении
2,5 МН/м2 (25 атм) и мольном соотношении исходного сырья
и водорода 1:10.
142
Глава 5
Методы селективного извлечения п-ксилола
В настоящее время в промышленности применяют еще два
метода извлечения n-ксилола из технической смеси изомеров С8.
По одному из них исходное сырье пропускают в жидком со-
стоянии через стационарный слой молекулярных сит Y-типа. При
этом п-ксилол удерживается в слое молекулярных сит, а осталь-
ные изомеры, сорбирующиеся слабее, выводятся из адсорбера.
Адсорбированный n-ксилол элюируют селективным раство-
рителем, который затем отгоняют из элюата и возвращают в
адсорбер. Поскольку товарный п-ксилол должен иметь высокую
степень чистоты (выше 99,2% по весу), элюент используется
также для удаления остатков исходной смеси из пространства
между частицами молекулярных сит. Весь процесс ведут непре-
рывно, применяя принцип движущегося слоя адсорбента (когда
исходная смесь проходит по адсорберу снизу вверх, а навстречу
ей движется адсорбент). Адсорбция n-ксилола, вымывание
остатков исходной смеси и десорбция п-ксилола происходят
в последовательных зонах слоя при непрерывном пропускании
через адсорбер потоков исходной смеси, растворителя и про-
дукта. На практике эффект движущегося слоя адсорбента соз-
дают путем последовательного изменения положения точек
ввода исходной смеси и элюента и вывода элюата, причем, чем
больше число этих точек, тем больше степень приближения
к истинно движущемуся слою. Сообщается, что описанный ме-
тод позволяет извлекать из технической смеси ароматических
углеводородов С8 свыше 95% п-ксилола со степенью чисто-
ты 99,3%.
В основе второго метода лежит реакция комплексообразова-
ния ароматических углеводородов с фтористым водородом и
фтористым бором
ArH + HF + BF3 ч=Ь AfH+BF;
Ионизированный комплекс ArHsBF^ способен экстрагировать-
ся избытком жидкого фтористого водорода. Устойчивость ком-
плексов этого типа очень сильно зависит от структуры аромати-
ческого углеводорода. Так, для м-, о- и n-ксилолов она изме-
няется в отношении 20: 2 : 1; в случае этилбензола устойчивость
комплекса значительно ниже, чем для п-ксилола. Варьируя кон-
центрацию фтористого бора в фтористом водороде, можно подо-
брать условия, при которых будет достигаться исчерпывающая
экстракция л/-ксилола. Путем сверхчеткой ректификации остатка
можно выделить чистый п-ксилол. При повышенных температу-
рах система HF—BF3 вызывает также изомеризацию ксилолов.
Первичные нефтехимические продукты. И.
143
Гидродеалкилирование толуола в бендол
Гидродеалкилирование толуола можно проводить как терми-
чески, так и каталитически, причем ни один из этих методов не
имеет особых преимуществ перед другим.
СНз
+ Н2 + СН4 ЛЯ = — 56,4 кДж/моль
В термическом варианте смесь паров толуола с водородом
в мольном соотношении ~1 :3 пропускают через реактор, в ко-
тором поддерживают температуру 650—750°C и давление 4—
6 МН/м2 (40—60 атм). Жидкие продукты подвергают дистилля-
ции, выделяя бензол. Небольшую часть рециркулирующего во-
дорода выводят из системы, чтобы уменьшить концентрацию
метана и тем самым повысить степень конверсии толуола в бен-
зол. Перед сбрасыванием в атмосферу сдувочные газы промы-
вают для извлечения содержащегося в них продукта.
Стехиометрия этой реакции показывает, что положение тер-
модинамического равновесия не зависит от общего давления
в системе. Однако повышение давления сопровождается возра-
станием скорости реакции. Кроме того, оно приводит к увеличе-
нию селективности образования бензола, подавляя побочные
процессы, такие, как синтез многоядерных углеводородов и от-
ложение кокса. Еще одной побочной реакцией является получе-
ние бифенилов, например:
2 О ОО
Поэтому для того, чтобы сместить равновесие в сторону обра-
зования бензола, в системе поддерживают высокое парциальное
давление водорода. В указанных выше условиях равновесная
смесь содержит около 3% бифенилов; эти соединения возвра-
щают в реактор с целью подавления дальнейшего их образо-
вания.
Для реакции термического гидродеалкилирования толуола
предложен цепной свободнорадикальный механизм
R • + Н2 RH + Н • (инициирование)
Н. + С6Н6СНз (С6Н6СН3)« с6н6 + сн3.
СНз• + Н2 СН4 + Н«
В каталитическом процессе обычно применяют слабокислот-
ный катализатор, позволяющий избежать протекания ионных ре-
акций, которые приводят к коксообразованию. Примерами таких
катализаторов могут служить окись хрома на окиси алюминия
144
Глава 5
и цеолиты. Процесс ведут при несколько более низких темпера-
турах, чем термическую реакцию. Механизм каталитического
гидродеалкилирования толуола мало изучен.
Получение этилбензола и кумола
алкилированием бензола
Этилбензол и кумол получают в промышленности путем ал-
килирования бензола этиленом и пропиленом соответственно.
Эта реакция, катализируемая кислотами Льюиса, известна под
названием реакции Фриделя — Крафтса, например:
+ СН2=СН2
СН2—СНз
А// = — 104 кДж/моль
Предложен ряд катализаторов алкилирования, но чаще всего
применяют хлористый алюминий в жидкофазной системе. При
температурах ниже 500 °C термодинамическое равновесие пол-
ностью смещено в сторону конечных продуктов; однако в этих
условиях протекают побочные реакции, приводящие к образо-
ванию полиалкилбензолов.
Механизм алкилирования бензола требует присутствия хло-
ристого водорода, которое обеспечивают добавлением незначи-
тельного количества воды или хлористого этила. Система хло-
ристый алюминий — хлористый водород протонирует олефин по
двойной связи. Возникающая при этом электрофильная частица
взаимодействует с л-электронагуш бензольного кольца (т. 2,
стр. 109, 212):
СН2=СН2 + НС1 + А1С13 —> [С2Н5]+[А1С14]’
[С2Н5]+[А1С14]- + С6Н6 —> [СвН6С2Нв]+[А1С14]-
[С6Н6С2Н5]+[А1С14Г —> C6HSC2H5 + НС1 + А1С13
При температуре реакции хлористый алюминий находится
в димерной форме, которая не обладает каталитическими свой-
ствами. Однако он образует комплекс с бензолом, способный
растворять дополнительное количество хлористого алюминия
в каталитически активной мономерной форме.
В промышленном процессе продукт, вытекающий из реак-
тора, направляют в отстойник, откуда хлористый алюминий воз-
вращают в сырьевой поток. Этилбензол (или кумол) выделяют
с помощью четкой ректификации, а тяжелые погоны, содержа-
щие полиалкилбензолы, вновь обрабатывают комплексом хло-
ристого алюминия с бензолом в условиях протекания транс-
Первичные нефтехимические продукты. II.
145
алкилирования, например
С2Н5 С2Н5
Продукт трансалкилирования возвращают на стадию отгонки
этилбензола.
Алкилирование бензола этиленом протекает в более жестких
условиях, чем алкилирование пропиленом. Поэтому для полу-
чения кумола можно использовать олефиновое сырье, содержа-
щее этилен, причем последний не вступает в реакцию. Напротив,
присутствие пропилена в этилене, предназначенном для получе-
ния этилбензола, нежелательно.
Бензол
А. Джубб
Jubb А. Н., ICI Corporate Laboratory, Runcorn
Наиболее крупнотоннажным продуктом дальнейшей перера-
ботки бензола является этилбензол (стр. 144), который служит
полупродуктом для получения стирола (гл. 4), в свою очередь
используемого в качестве мономера в производстве одного из
важнейших пластических материалов — полистирола (гл. 8).
Почти все методы промышленного получения фенола (стр. 157
и гл. 7) тоже основаны на использовании бензола; таким
Таблица 5.5
Структура потребления бензола в США в 1971 г.
Конечные продукты переработки бензола Потребление
тыс. т %
Этилбензол (для получения стирола) 1650 44,2
Фенол 717 19,2
Циклогексан 584 15,6
Малеиновый ангидрид 144 3,9
Алкилбензолсульфонаты 140 3,8
Анилин 132 3,5
Дихлорбензол 042 1,1
ДДТ 019 0,5
Прочие химические цели 301 8,2
Итого 3729 100,0
146
Глава 5
образом, фенол является вторым по значению продуктом пере-
работки бензола. В табл. 5.5 приведены данные о структуре по-
требления бензола в США в 1971 г., причем 92% бензола, выра-
ботанного в США в этом году, было получено из нефтяного
сырья и 8% —из коксохимического.
Циклогексан
Бензол легко гидрируется в циклогексан в жидкой или паро-
вой фазе над никелевым катализатором. Жидкофазный процесс
обычно ведут при температуре около 150—200 °C под давлением,
необходимым для поддержания жидкого состояния бензола. Су-
ществующие модификации этого процесса отличаются друг от
друга главным образом по способу отвода теплоты реакции.
С6Н6 + ЗН2 —> C6Hi2 ДЯ = — 200 кДж/моль
Циклогексан используют преимущественно как полупродукт
для синтеза адипиновой кислоты, гексаметилендиамина и капро-
лактама.
Окисление циклогексана воздухом при температуре 150 °C и
давлении 1 МН/м2 (10 атм) в присутствии нафтената кобальта
(1 ч. на млн) при степени конверсии циклогексана около 8%
приводит к образованию циклогексанола (А) и циклогексанона
(К) с общим выходом 70%, а также ряда побочных продуктов.
После удаления непревращенного циклогексана технический
продукт окисляют 50%-ной азотной кислотой при температуре
85 °C в присутствии медно-ванадиевого катализатора. При этом
получается адипиновая кислота, выход которой составляет
1 кг/кг технического продукта. Адипиновую кислоту очищают
перекристаллизацией из воды. Из маточного раствора можно
выделить смесь адипиновой, глутаровой и янтарной кислот.
гидроперекись
циклогексила
побочные продукты
(сложные эфиры,
карбоновые кислоты
и др.)
HNOg
---> НООС(СН2)4СООН
Существуют различные варианты проведения окисления цик-
логексана, которые лежат в основе ряда промышленных процес-
сов. В некоторых из них используются более низкие степени
конверсии циклогексана с целью увеличения выхода циклогек-
Первичные нефтехимические продукты. [[.
147
санола и циклогексанона. В процессе «КА» фирмы Scientific
Design окисление циклогексана ведут в присутствии борной кцс-
лоты (при степени конверсии циклогексана 10%), что обеспечи-
вает 90%-ный выход продукта. Борная кислота меняет направ-
ление разложения промежуточной гидроперекиси и, кроме того,
легко этерифицирует циклогексанол, повышая соотношение А : К
до 10:1.
В литературе имеются сообщенйя о процессах прямого окис-
ления циклогексана в адипиновую кислоту (в частности, в при-
сутствии уксусной кислоты и кобальтовых катализаторов), но
надежных данных о промышленной реализации этих процес-
сов нет.
Фирма Celanese Corporation разработала вариант процесса
«КА», в котором циклогексан окисляется воздухом до степени
конверсии около 25%. Непревращенный циклогексан отгоняют,
образовавшиеся циклогексанол и .циклогексанон выделяют ди-
стилляцией и окисляют азотной кислотой в адипиновую кислоту.
Кубовый остаток после первой стадии окисления, содержащий
сложную смесь побочных продуктов, этерифицируют пентандио-
лом-1,5 в смесь полиэфиров. Последнюю подвергают гидрогено-
лизу над хромитом меди и получают смесь диолов, из которой
отгоняют гександиол-1,6. Парофазное аминирование этого диола
аммиаком над скелетным никелевым катализатором приводит
к образованию гексаметилендиамина. Пентандиол-1,5, который
является продуктом восстановления глутаровой кислоты, обра-
зующейся при окислении циклогексана, выделяют и возвращают
на стадию этерификации. Из адипиновой кислоты в две стадии
получают гексаметилендиамин. Для этого ее переводят в ади-
понитрил путем обработки избытком аммиака в жидкой или па-
ровой фазе в присутствии дегидратирующего катализатора (фос-
фата бора), а затем полученный адипонитрил подвергают ката-
литическому гидрированию:
NH3 Н2
НООС(СН2)4СООН ——---------NC(CH2)4CN —г H2N(CH2)6NH2
' фосфат бора (350—450 °C) N1
адипиновая кислота адипонитрил гексамети-
(выход 85%) лендиамин
(выход 94%)
Малеиновый ангидрид.
Малеиновый ангидрид может быть получен парофазным окис-
лением большинства соединений, содержащих неразветвленную
цепь из четырех или пяти атомов углерода (кроме парафинов),
над окиснованадиевым катализатором. К числу таких соедине-
ний относятся также фурфурол, кротоновый альдегид и н-бутан.
Однако большинство промышленных процессов получения
148
Глава 5
малеинового ангидрида базируется на использовании бензола
(гл. 4). Пары бензола смешивают с воздухом и подают в труб-
чатый реактор, заполненный пятиокисью ванадия на инертном
носителе. Реакцию ведут при температуре 400—450 °C, атмо-
сферном давлении и времени контакта порядка 0,1 с. Поскольку
она сильно экзотермична, выделяющееся тепло снимают распла-
вом солей, циркулирующим в межтрубном пространстве реак-
тора, и используют его для генерирования водяного пара. Реак-
ционные газы последовательно охлаждают в котле-утилизаторе
и водяном холодильнике и абсорбируют водой, причем ангидрид
превращается в малеиновую кислоту. Последнюю снова дегид-
ратируют в ангидрид путем отгонки воды в вакууме или в виде
азеотропа с углеводородами. Полученный малеиновый ангидрид
очищают заключительной перегонкой в вакууме. Существует
несколько вариантов описанного процесса, различающихся глав-
ным образом оформлением стадии выделения и очистки продук-
та окисления. На некоторых заводах применяют конденсаторы
периодического действия, позволяющие извлекать свыше 85%
этого продукта в форме ангидрида. Остальную часть продукта
можно выделить в виде малеиновой кислоты путем абсорбции
водой. Выходы составляют 60—65% (считая на бензол).
v2o5 СНСОЧ
2[ | +9О2 --> 2 || /О + 4Н2О + 4СО2
СНССГ
В 1971 г. мощности по производству малеинового ангидрида
в США составляли приблизительно 140 тыс. т/год при потреб-
ности 100 тыс. т. Основными потребителями малеинового ан-
гидрида были производства полиэфирных смол (50%), фумаро-
вой кислоты (15%), пестицидов (гл. 12) (10%) и алкидных
смол (гл. 8) (5%); остальной малеиновый ангидрид расходо-
вался на получение пластификаторов, смазочных материалов и
яблочной кислоты.
Алкилбензолсулъфонаты
Синтез алкилбензолсульфонатов, которые используют в ка-
честве моющих веществ, описан в гл. 13. В том случае, когда
HF или SO3
Олефин СбНб •* R—СеН5 * RC6H4SO3H
AIGI3 ii2o(J4
R — разветвленный радикал, получается моющее вещество, не
способное к биохимическому разложению. В настоящее время
вместо таких веществ выпускаются линейные алкилбензолсуль-
фонаты, которые легко подвергаются биохимическому разло-
жению.
Первичные нефтехимические продукты. II. 149
Анилин
Анилин служит важным полупродуктом для получения краси-
телей, фармацевтических препаратов и вспомогательных хими-
катов, используемых при переработке каучуков. В промышлен-
ности анилин синтезируют так же, как в лаборатории (т. 1,
стр. 136), а именно путем нитрования бензола смесью азотной
и серной кислот с одновременной отгонкой выделяющейся воды
в виде азеотропа с избытком бензола и восстановления полу-
ченного нитробензола. Традиционный процесс восстановления
нитробензола является периодическим и осуществляется в ап-
паратах с мешалкой. Нитробензол восстанавливают чугунными
стружками в присутствии воды и 30%-ной соляной кислоты; вы-
ход анилина составляет 90—95%. Обычно такие установки
имели мощность менее 10 тыс. т/год. В настоящее время почти
повсеместно применяются более экономичные непрерывные про-
цессы каталитического парофазного гидрирования нитробензола
при температуре 270 °C на установках с мощностью до
50 тыс. т/год. Гидрирование нитробензола ведут в стационарном
слое сульфида никеля, нанесенного на окись алюминия, или
в жидкой фазе в присутствии палладиевых, никелевых или мед-
ных катализаторов на носителях. Метод получения анилина ам-
монолизом хлорбензола, применявшийся ранее, в настоящее
время уже полностью оставлен.
Основная часть анилина (свыше 50% общего объема его
потребления) идет на получение стабилизаторов и ускорителей
вулканизации каучуков. Вторым по значению потребителем ани-
лина является синтез изоцианатов для производства жесткого
пенополиуретана. Остальной анилин расходуется главным обра-
зом на получение красителей, фотоматериалов и лекарственных
препаратов*.
* Приводим данные по структуре потребления анилина в США в 1974 г.
[Chem. and Eng. News, 52, № 36, 7 (1974)]:
Конечные продукты переработки анилина Потребление
тыс. т %
Изоцианаты 97,5 40
Химикаты для переработки каучуков 86,2 35
Красители и полупродукты для их получения 13,6 6
Гидрохинон 13,6 6
Лекарственные препараты 9,1 4
Прочие 22,7 9
Итого 242,7 100
— Прим, перев.
150
Глава 5
Хлорбензолы
Хлорирование бензола при температуре 40—60 °C и мольном
отношении бензола и хлора 1 :0,6 приводит к преимуществен-
ному образованию монохлорбензола. Использование более низ-
ких отношений бензола к хлору и проведение реакции в
присутствии хлористого алюминия позволяет получать главным
образом дихлорбензолы, промышленное значение которых не-
прерывно возрастает.
С1 С1-
о-Дцхлорбензол находит применение в синтезе различных
органических полупродуктов (таких, как 3,4-дихлоранилин, ис-
пользуемый в производстве красителей) и для получения пести-
цидов. Кроме того, его употребляют в качестве растворителя.
п-Изомер применяется как дезодорант и средство для уничтоже-
ния моли.
ДДТ
Инсектицид ДДТ (дихлордифенилтрихлорэтан) вырабаты-
вают из хлорбензола (гл. 12).
Ксилолы
Дж. Пеннингтон
Pennington J., Research and Development Department,
BP Chemicals International Limited, Hedon, Hull
Получение фталевого ангидрида из о-ксилола
Еще в 1960 г. основным исходным сырьем для получения
фталевого ангидрида служил нафталин, а из о-ксилола во всем
мире вырабатывали лишь 10% этого продукта. К 1970 г. о-кси-
лол занял доминирующее положение в сырьевой базе мирового
производства фталевого ангидрида: из 1,5 млн. т произведенного
фталевого ангидрида около 55% было получено на основе
о-ксилола. Эта тенденция продолжает развиваться, и в послед-
ние годы практически все новые мощности по производству
фталевого ангидрида базируются на использовании о-ксилола *.
* В 1975 г. на нафталине базировалось лишь 22% мировых мощностей по
производству фталевого ангидрида [Chem. Age, 113, № 2981, 6 (1976)].—
Прим, перев.
Первичные нефтехимические продукты FF
151
По принятой на всех (кроме’ одного) заводах технологии
о-ксилол превращают во фталевый ангидрид путем парофазного
окисления над стационарным катализатором. Единственным ис-
ключением является завод фирмы Phone Progile (Франция)
мощностью около 15 тыс. т/год, на котором применяется метод
жидкофазного окисления о-ксилола, сходный с процессом полу-
чения терефталевой кислоты фирмы Amoco (см. ниже). Боль-
шинство новых производственных мощностей по фталевому ан-
гидриду работает по технологии фирм von Heyden или BASF.
В основных чертах оба эти процесса одинаковы, что позволяет
дать для них одно общее описание. В основе получения фтале-
вого ангидрида из о-ксилола лежит следующая реакция:
+ ЗО2
А// = — 1290 кДж/моль
В поток обеспыленного, сжатого и подогретого воздуха впры-
скивают о-ксилол, имеющий степень чистоты не ниже 95%. Для
предотвращения образования взрывоопасной смеси концентра-
цию паров о-ксилола регулируют таким образом, чтобы она
составляла менее 1 мол.%.
Реакцию ведут в трубчатых аппаратах, содержащих окисно-
ванадиевый катализатор на носителе, при температуре 350—
100 °C. Выделяющееся тепло отводится расплавом солей, цирку-
лирующим в межтрубном пространстве, и используется для
нагревания змеевиков, в которых генерируется пар высокого
давления. Недавно разработанные катализаторы обеспечивают
высокую селективность реакции (более 75% от теории) при зна-
чительной пропускной способности системы, причем благодаря
последним достижениям в области конструирования химической
аппаратуры созданы реакторы, включающие до 15 000 труб. Это
позволяет получить большую производительность в расчете на
один реактор, несмотря на низкую концентрацию о-ксилола в
сырьевом потоке.
Реакционные газы после их частичного охлаждения пропу-
скают через систему конденсаторов периодического действия,
работающих поочередно. В каждом таком конденсаторе имеются
ребристые трубы, по которым циркулирует хладоагент, вызы-
вающий вымораживание кристаллов фталевого ангидрида из
реакционных газов. Затем газовый поток переключают на дру-
гой конденсатор, а в первый подают горячий теплоноситель для
расплавления сырого продукта, стекающего в промежуточный
сборник. Выходящие из конденсаторов газы промывают водой,
чтобы извлечь из них остатки органических веществ, и сбрасы-
вают через выпускную трубу в атмосферу.
152
Глава 5
Сырой продукт, уже содержащий около 99% фталевого ан-
гидрида, подвергают термической обработке с последующей
непрерывной вакуум-дистилляцией в системе из 2—3 ректифи-
кационных колонн. Степень чистоты товарного продукта — не
ниже 99,8%. Его можно хранить, транспортировать и использо-
вать в виде плава; если, однако, необходимо получить сыпучий
продукт, то производят дополнительную операцию чешуиро-
вания.
Около половины вырабатываемого в мире фталевого ангид-
рида расходуется на нужды самого крупного потребителя этого
химиката — производства эфиров фталевой кислоты, используе-
мых в качестве пластификаторов поливинилхлорида (гл. 8).
Большая часть остального фталевого ангидрида идет на полу-
чение алкидных смол (защитные покрытия и компоненты лаков
и красок) и ненасыщенных полиэфиров (связующие для стекло-
пластиков). Алкидные смолы представляют собой группу смол
разнообразного состава, получаемых преимущественно на основе
дикарбоновых кислот, многоатомных спиртов (в частности, гли-
церина и пентаэритрита) и жирных монокарбоновых кислот;
последние ограничивают степень сшивания полимера и придают
ему растворимость в углеводородах и (в случае непредельных
кислот) способность к «высыханию» на воздухе.
Получение терефталевой кислоты
и диметилтерефталата из п-ксилола
Терефталевая кислота и диметилтерефталат расходуются в
основном на получение полиэфирных (полиэтилентерефталат-
ных) волокон, ' таких, как выпускаемый фирмой ICI терилен
(гл. 9).
До середины 60-х годов всю вырабатываемую терефталевую
кислоту переводили для окончательной очистки в диметилтере-
фталат, что связано с очень низкой летучестью и плохой раство-
римостью этой кислоты. Усовершенствование технологии произ-
водства терефталевой кислоты привело к увеличению мощностей
по получению высокочистого продукта (в 1970 г. на долю этих
мощностей приходилось около 20% общего объема мирового
производства терефталевой кислоты и диметилтерефталата, со-
ставившего приблизительно 1,75 млн. т).
Полагают, что первоначальный метод получения терефтале-
вой кислоты — окисление п-ксилола разбавленной азотной кис-
лотой — в настоящее время продолжает использоваться только
фирмой Du Pont. Тем не менее выпуск ее по этому методу пре-
вышает 200 тыс. т/год. Быстрое расширение рынка сбыта поли-
эфирных волокон и высокая рентабельность их производства
обусловили появление множества промышленных процессов по-
Первичные нефтехимические продукты. II.
153
лучения терефталевой кислоты (причем все они являются жид-
кофазными). Однако около 80% терефталевой кислоты во всем
мире вырабатывают с использованием двух процессов: процесса
фирмы Amoco (или Mid-Century) и процесса фирмы Witten.
Прямое окисление /г-ксилола в n-толуиловую кислоту в при-
сутствии небольших количеств солей кобальта или марганца
идет сравнительно легко, однако дальнейшее окисление в тере-
фталевую кислоту протекает чрезвычайно медленно. Эту про-
блему пытались решить тремя различными способами. Во-
первых, использовали промоторы, ускоряющие окисление п-то-
луиловой кислоты, такие, как бромид-ион (фирма Amoco),
некоторые кетоны и альдегиды. Во-вторых, оказалось, что
приемлемые результаты дает повышение концентрации кобаль-
тового катализатора (японская фирма Teijin). Перспективность
такого метода с точки зрения экономики будет зависеть от эф-
фективности регенерации и повторного использования кобальта.
Наконец, в-третьих, было найдено, что n-толуиловую кислоту
можно перевести в более легко окисляющиеся производные, на-
пример в ее метиловый эфир (фирма Witten). Следует также
упомянуть о процессах фирмы Henkel, нашедших применение
только в Японии, которые основаны на диспропорционировании
бензоата или изомеризации фталата калия в терефталевую кис-
лоту.
Прямое окисление:
СООН
+ ЗН2О
ДЯ = — 1365 кДж/моль
Процесс фирмы Witten:
= — 691 кДж/моль
+ СНэОН
СООН
+ Н2О
ДЯ = — 29 кДж/моль
СООСНз
154
Глава б
кН = — 674 кДж/моль
кН = — 29 кДж/моль
В процессе фирмы Amoco в систему из нескольких парал-
лельно или последовательно соединенных реакторов, через ко-
торые барботируется воздух, непрерывно подают л-ксилол (сте-
пень чистоты не менее 99%), уксусную кислоту (растворитель)
и раствор, содержащий кобальт-марганцевый катализатор и
бромид в качестве промотора. Температуру поддерживают в
пределах 175—230 °C, отводя выделяющееся тепло за счет ки-
пения реакционной смеси при соответствующем давлении, обыч-
но равном 1,5—3,0 МН/м2 (15—30 атм). В случае необходимости
содержание воды в реакционной смеси можно регулировать пу-
тем частичного отбора конденсата из обратного холодильника.
Во избежание образования в верхней части реактора взрывча-
той газовой смеси скорость воздушного потока подбирают таким
образом, чтобы концентрация кислорода в отходящих газах
была невелика.
Терефталевая кислота плохо растворима в уксусной кислоте,
и поэтому она непрерывно осаждается в процессе окисления. По
окончании реакции оксидат охлаждают и центрифугируют. Ма-
точный раствор перегоняют, чтобы отделить растворитель и не-
превращенный л-ксилол от воды и высококипящего остатка,
и возвращают в реакцию. Остаток, содержащий катализатор,
можно либо частично возвращать в цикл, либо подвергать пе-
реработке с целью извлечения компонентов катализатора, либо
считать отходом производства. Отцентрифугированный осадок
продукта промывают горячей уксусной кислотой и высушивают,
получая техническую терефталевую кислоту со степенью чи-
стоты около 99%. Выход ее превышает 90% от теории. Основ-
ной примесью является промежуточный продукт окисления —
л-карбоксибензальдегид. Техническую кислоту можно далее
очистить или перевести в диметилтерефталат (см. ниже).
Раствор бромида в уксусной кислоте очень агрессивен, вслед-
ствие чего большую часть аппаратуры приходится изготовлять
Первичные нефтехимические продукты. П.
155
из титана или дорогостоящих коррозионностойких сплавов. Од-
нако описанный процесс пригоден для окисления любых алкил-
ароматических соединений, что позволяет в случае необходимо-
сти получать на одном и том же оборудовании различные про-
дукты."
Метод прямого окисления n-ксилола в терефталевую кислоту
используется тремя фирмами. Установки, работающие по этому
методу, имеют. единичную мощность 40—70 тыс. т/год, причем
вместо бромида применяют органические промоторы, а катали-
затором служат соли кобальта. Как и в процессе фирмы Amoco,
реакцию ведут в растворе уксусной кислоты, но температура
окисления сравнительно низка (около 130°C). Фирма Mobil ис-
пользует в качестве промотора метилэтилкетон (процесс фирмы
Olin Mathieson), тогда как в процессах фирм Eastman и Тоуо
Rayon эту функцию выполняют соответственно ацетальдегид и
паральдегид. Кроме того, фирма Mobil проводит окисление с
помощью 99,5%-ного кислорода под давлением около 3,5 МН/м2
(35 атм), что обеспечивает очень высокую скорость реакции. Во
всех этих процессах часть промотора окисляется через проме-
жуточные перекисные соединения [которые способствуют под-
держанию высокого отношения Со(III)/Со(II)] в уксусную кис-
лоту, являющуюся побочным продуктом. Непревращенный про-
мотор рециркулирует вместе с растворителем. В остальном про-
цессы с использованием органических промоторов аналогичны
процессу фирмы Amoco.
Обычно техническую терефталевую кислоту этерифицируют
большим избытком метанола при высоких температурах (выше
200°С) и давлениях [около 15 МН/м2 (150 атм)]. Катализато-
рами служат амфотерные окислы металлов, такие, как SnO,
ZnO, БЬгОз и др. Полученный эфир очищают, как правило, ди-
стилляцией (иногда для этого применяют также кристаллизацию).
Альтернативный метод очистки терефталевой кислоты, раз-
работанный фирмой Amoco, заключается в гидрировании рас-
твора технического продукта (10—15% по весу) в воде или
уксусной кислоте при температуре 250—280 °C и соответствую-
щем давлении. Во избежание гидрирования бензольного кольца
используют низкое парциальное давление водорода. В качестве
катализатора применяют палладий на пористом носителе. В ре-
зультате гидрирования ц-карбоксибензальдегид и ряд окрашен-
ных примесей восстанавливаются в более растворимые соедине-
ния. При охлаждении раствора из него выпадают кристаллы
высокочистой терефталевой кислоты, которые промывают и су-
шат. В процессе фирмы Mobil техническую терефталевую кис-
лоту гидрируют и фильтруют в потоке перегретого водяного
пара; высокочистый продукт конденсируется из газового потока
и далее улавливается в циклонах.
156
Глава 5
На первой стадии непрерывного процесса фирмы Witten
n-ксилол окисляют воздухом в n-толуиловую кислоту при тем-
пературе около 150°C. Реакцию ведут без растворителя; ката-
лизатором служит растворимое в углеводородах соединение ко-
бальта (например, нафтенат или соль жирной карбоновой
кислоты), применяемое в низкой концентрации — вероятно, ме-
нее 0,1% по весу. Как всегда, необходимо контролировать со-
держание кислорода в отходящих газах. Отвод теплоты реакции
и регулирование температуры достигается за счет кипения ре-
акционной смеси [под давлением около 0,4—0,8 МН/м2 (4—
8 атм)], которое, по-видимому, дополняется ее циркуляцией
через выносные холодильники. Степень конверсии n-ксилола за
один проход, вероятно, превышает 70%. После удаления непре-
вращенного n-ксилола (с последующим возвращением его в
цикл) n-толуиловую кислоту этерифицируют метанолом при
температуре выше 200 °C и давлении более 2,5 МН/м2 (25 атм)
в отсутствие катализатора. Полученный метиловый эфир п-то-
луиловой кислоты подвергают окислению в монометилтерефта-
лат при температуре около 200 °C и давлении 1,5—2,5 МН/м2
(15—25 атм) в присутствии кобальтового катализатора, посту-
пающего с первой стадии окисления. Продукт реакции снова
этерифицируют метанолом и получают смесь, содержащую ди-
метилтерефталат. Сырую смесь разгоняют в системе вакуум-
ректификационных колонн и выделяют высокочистый диметил-
терефталат. Непревращенный метиловый эфир п-толуиловой
кислоты и другие промежуточные продукты возвращаются в
цикл; высококипящий кубовый остаток является отходом. Вы-
сокочистый диметилтерефталат может храниться и использо-
ваться в виде плавленого или чешуированного продукта.
Установлено, что окисление n-ксилола ц метилового эфира
n-Толуиловой кислоты можно совместить в одном процессе. При
этерификации полученного оксидата образуются метиловый
эфир n-толуиловой кислоты, который возвращают на окисление,
и диметилтерефталат. Данные о промышленной реализации это-
го упрощенного метода получения диметилтерефталата отсут-
ствуют.
Получение изофталевой кислоты из м-ксилола
Изофталевая кислота вырабатывается в промышленности
как в чистом виде (с чистотой выше 99%), так и с примесью
терефталевой кислоты. Объем ее мирового потребления в 1970 г.
составлял не более 50—60 тыс. т/год, однако он постоянно воз-
растает. Основная часть изофталевой кислоты получается с ис-
пользованием процесса фирмы Amoco (см. выше); главным
потребителем этой кислоты является производство ненасыщен-
ных полиэфирных смол.
Первичные нефтехимические продукты. 11. 157
Кумол
Р. Холл
Hall R. Н., Research and Development Department,
BP Chemicals International Limited, Epsom
Производство фенола и ацетона
Реакции превращения кумола в его гидроперекись и разло-
жения последней на фенол и ацетон были открыты Хоком и
Лангом при изучении аутоокисления углеводородов; результаты
этой работы были опубликованы в 1944 г. Сухой кумол (1)
встряхивали в атмосфере кислорода при температуре 85°C и
облучении ультрафиолетовым светом. При этом образовывалась
гидроперекись кумола (2), которую выделяли в виде кристалли-
ческой натриевой соли. Осторожное подкисление соли с после-
дующей экстракцией эфиром и перегонкой при пониженном дав-
лении приводило к получению чистой гидроперекиси 2. Скорость
образования гидроперекиси кумола была невелика, а выход ее
при продолжительности реакции 24 ч составлял около 7%. Об-
работка гидроперекиси кипящей 10%-ной серной кислотой в те-
чение 1,5 ч сопровождалась образованием фенола и ацетона.
Фенол выделили в форме бензоата; общий выход фенола из
гидроперекиси был близок к 75%.
СНз СНз
I о2 I н+ '
СбН6—СН —► C6HS—С—ООН —> С8Н5ОН + (СН3)2СО
СНз СНз 1
1 2 *
С6Н6ООСС6Н6
Сразу после появления работы Хока и Ланга несколько
промышленных фирм (особенно английская фирма Distillers
Company Ltd. и американская Hercules Powder Со.) заинтересо-
вались этой реакцией как потенциальным методом совместного
производства фенола и ацетона. Объединение технологии, раз-
работанной обеими фирмами, привело к созданию процесса
Distillers — Hercules, который после слияния фирмы Distillers
Со. Ltd. с фирмой BP Company Limited (в 1967 г.) стал назы-
ваться процессом «ВР — Hercules» *.
* Кумольный метод совместного получения фенола и ацетона был впер-
вые разработан в нашей стране П. С. Сергеевым, Б. Д. Кружаловым и
Р. Ю. Удрисом и реализован в промышленном масштабе в 1949 г. За рубе-
жом первая аналогичная установка вступила в строй в Канаде лишь в
1953 г. — Прим, перев.
158
Глава 5
Первой стадией процесса является синтез кумола путем ал-
килирования бензола, взятого в избытке, пропиленом (стр. 144).
Реакцию ведут либо в жидкой фазе при температуре около
100 °C с использованием хлористого алюминия в качестве ката-
лизатора, либо в паровой фазе при температуре приблизительно
250 °C и давлении 3,45 МН/м2 (34,5 атм) над фосфорной кисло-
той, нанесенной на кизельгур. В обоих вариантах процесса
алкилирования, известного в общих чертах еще до разработки
кумольного метода получения фенола, степень конверсии бен-
зола ограничивают, чтобы свести к минимуму образование про-
дуктов ди-, три- и полиалкилирования. В жидкофазном процессе
непревращенный бензол и продукты тяжелее кумола отделяют
и возвращают на стадию алкилирования.
С6Н6 + СН3СН=СН2
AICI3, жидкая фаза
Н3РО4., паровая фаза
С6Н5СН(СН3)2
Окисление кумола обычно ведут в каскаде из двух или не-
скольких последовательно соединенных реакторов. В большин-
стве случаев применяют реакторы башенного типа, хотя на не-
которых заводах используются также автоклавы с мешалкой.
Окислителем чаще всего служит воздух, однако иногда вместо
него применяется кислород. Температура реакции составляет
95—130°С. Обычно максимальную температуру поддерживают
в первом реакторе и постепенно понижают ее по мере увеличе-
ния концентрации гидроперекиси. Использование столь высоких
температур позволяет отказаться от ультрафиолетового облуче-
ния реакционной смеси, осуществляемого в работе Хока и
Ланга. Реакция протекает в безводной среде или в присутствии
небольших добавок воды. Для поддержания pH около 7 в ре-
акционную смесь обычно вводят некоторое количество щелочи.
Конечную концентрацию гидроперекиси регулируют таким об-
разом, чтобы она не превышала 40% (оптимальная концентрация
20—30%); это обеспечивает высокую селективность окисления.
Побочно образуются небольшие количества а,а-диметилбензило-
вого спирта СбН5С(СН3)2ОН и ацетофенона.
Окисление кумола в описанных выше условиях протекает по
цепному свободнорадикальному механизму:
Инициирование цепи'. RH + • X —> R« + HX [R = С6Н5С(СН3)2], гдеХ«
образуется за счет следующих реакций:
ROOH —> RO.-+-.OH
RH + RO* —> ROH + R.
RH + -OH —► R« + H2O
Первичные нефтехимические продукты. //. 159
Продолжение цепи: R • + 0_> —> R0. •
R02 • + RH —> ROOH + R •
Обрыв цепи: 2RO2 • -—► ROOR + 02 или другие стабильные продукты
2RO2. —> 2RO • + О2
RO . —> С6Н5СОСН3 + • СН3
НСНО + нсоон
Продукт, вытекающий из последнего реактора, упаривают
в вакуум-ректификационной колонне до остаточной концентра-
ции гидроперекиси около 80% и направляют на стадию разло-
жения. Отогнанный кумол очищают и возвращают на окисление.
Разложение гидроперекиси обычно ведут непрерывным ме-
тодом, раздельно подавая концентрат гидроперекиси и кислот-
ный катализатор (обычно серную кислоту) в аппарат или каскад
аппаратов с продуктами разложения гидроперекиси и собирая
вытекающий раствор в приемник. Температуру поддерживают
приблизительно при 50—90 °C, причем значительное тепловыде-
ление (226 кДж/моль) снимается с помощью охлаждающих
змеевиков или за счет испарения ацетона.
Реакция разложения имеет ионный механизм, который, по-
видимому, можно изобразить следующей схемой:
н3с
СК -ОН
сн3
СН3
-НгО
C6HSOH + (СНз)2СО
Помимо основной реакции, протекает ряд побочных процес-
сов, приводящих к образованию а-метилстирола (4) и его ди-
меров, кумилфенола (5), 2,2-б«с-(4'-оксифенил) пропана (6)
(который в промышленности обычно называют дифенилолпро-
паном) и окиси мезитила (7), а также воды, некоторого коли-
чества смолы и следов других соединений. Ацетофенон и часть
а,а-диметилбензилового спирта (3), присутствующие в сыром
концентрате гидроперекиси, проходят непревращенными через
160
Глава 5
стадию разложения.
С6Н5С(СН3)2ОН —> С8Н5С(СН3)=СН2 + Н2О
8 4 1
димеры
С6Н5ОН + С6Н5С(СН3)2ОН —> C8HSC(CH3)2— С8Н4ОН
3 5
>С6Н4ОН
2С6Н5ОН + (СН3)2СО —> (СН3)2(/ + Н2О
\с6Н4ОН
6
2(СНз)2СО —> (СНз)2С=СНСОСНз + н2о
7
Продукты расщепления нейтрализуют и разделяют ректифи-
кацией. Из верха одной или двух ректификационных колонн
отбирают ацетон, воду и почти все количество углеводородов
(кумола и а-метилстирола), которые, таким образом, отде-
ляются от фенола и высококипящих веществ. Окончательную
очистку ацетона проводят в специальной ректификационной ко-
лонне. Кумол очищают и возвращают на стадию окисления,
а а-метилстирол либо выделяют как товарный продукт, либо
гидрируют в кумол и возвращают в цикл.
Фенольную фракцию первой ректификационной колонны на-
правляют в другую колонну, где происходит отделение сырого
фенола от ацетофенона и высококипящих веществ. Кубовый
остаток либо сжигают, либо извлекают из него кумилфенол,
либо крекируют при высокой температуре (например, при
300°C), получая фенол и а-метилстирол; при этом присутствую-
щий в кубовом остатке а,а-диметилбензиловый спирт превра-
щается в а-метилстирол. Фенол и а-метилстирол возвращают
на более раннюю ступень ректификации. Иногда из дистиллята
крекинга выделяют также ацетофенон.
Сырой фенол подвергают окончательной очистке для удале-
ния следов углеводородов, окиси мезитила и других примесей.
Это достигается путем простой перегонки либо с помощью
экстрактивной дистилляции с водой или другой подходящей
жидкостью. Выход фенола и ацетона по кумолу может превы-
шать 90%, причем количество образующегося ацетона состав-
ляет около 0,6 т на 1 т фенола. Фенол, полученный кумольным
методом, имеет очень высокую степень чистоты и удовлетворяет
самым высоким требованиям.
ГЛАВА 6
Некоторые методы получения
нефтехимических полупродуктов
Промышленные процессы во многих случаях существенно
отличаются от лабораторного проведения реакций. В них ис-
пользуются значительно более высокие давления и температуры
и часто применяются гетерогенные катализаторы. В данной гла-
ве рассматриваются некоторые процессы многотоннажного неф-
техимического синтеза. Собранный в ней Материал является
далеко не исчерпывающим и имеет целью дать химику, знако-
мому только с работой в лаборатории, общее представление
о промышленном производстве органических веществ.
Окисление
В. Вуд
Wood В., Research and Development Department,
BP Chemicals International Limited, Epsom
Многие Низшие углеводороды, получаемые а промышленно-
сти нефтехимического синтеза (гл. 2), используют в качестве
исходного сырья в процессах окисления. Такие процессы охва-
тывают очень широкий диапазон реакций — от небольших из-
менений структуры углеводорода путем его неполного окисления
в эпоксиды, спирты, альдегиды, кетоны и (или) карбоновые
кислоты до полного сгорания. Продукты неполного окисления
(Например, спирты, альдегиды и кетоны) тоже могу? служить
исходным сырьем в реакциях окисления. Так, пропилен можно
сначала превратить в акролеин, а затем акролеин окислить в
акриловую кислоту
СН2=СНСН3 —► СН2==СНСНО —> СН2=СНСООН
5 Зак. 501
162
Глава 6
Перечисленные кислородсодержащие полупродукты полу-
чаются не только окислением углеводородов. Например, альде-
гиды производят методом оксосинтеза (стр. 180), а спирты —
гидратацией олефинов. Однако современная тенденция в
в промышленности направлена на использование селективных
методов прямого одностадийного окисления дешевого углеводо-
родного сырья. Данный раздел посвящен рассмотрению процес-
сов этого типа.
Наиболее распространенным окислителем для углеводородов
является кислород (часто в виде воздуха), поскольку он дешев
и доступен в неограниченном количестве. К настоящему вре-
мени методы прямого окисления кислородом (воздухом) уже
в значительной степени вытеснили методы, основанные на ис-
пользовании других окислителей (озона, хромовой и азотной
кислот, серы и ее соединений, перекиси водорода).
Реакции окисления сопровождаются выделением тепла
(табл. 6.1), и одной из важных практических проблем крупно-
Таблица 6.1
Тепловые эффекты типичных реакций окисления
углеводородов при постоянном давлении
Реакция ДН, кДж/моль
СН4 + 2- 02 —> СО + 2Н2
СН4 4* Ог —* СН2О 4- Н2О
СН4 + 2О2 —> СО2 4- 2Н2О
С2Н6 -j—— О2 —* С2Н4 4- Н2О
С2Н6 + О2 —> СНзСНО (газ) + Н2О
С2Н6 + ЗО2 —> 2СО2 + ЗН2О
-36
—282
-802
-105
—322
— 1425
тоннажного химического производства является обеспечение
эффективного теплоотвода с целью регулирования глубины
окисления и достижения оптимального выхода целевого продук-
та, поскольку нерегулируемое повышение температуры может
привести к полному сгоранию исходного углеводорода.
В последние годы в нефтехимической промышленности раз-
работаны новые окислительные методы, расширившие диапазон
получаемых продуктов за пределы кислородсодержащих соеди-
нений. Так, парофазное каталитическое окисление пропилена
в присутствии аммиака или этилена в присутствии синильной
кислоты приводит непосредственно к акрилонитрилу. Первая из
этих реакций получила название реакции окислительного ам-
монолиза, а вторая — окислительного цианирования (стр. 170
и 208).
Методы получения нефтехимических полупродуктов 163
Другими важными разновидностями реакций окисления яв-
ляются реакции окислительного хлорирования, или, более точно,
окислительного гидрохлорирования (т. е. хлорирования смесью
кислорода с хлористым водородом, стр. 168), и окислительного
дегидрирования. В последней из них алканы и (или) моноалке-
ны подвергают каталитическому дегидрированию в паровой
фазе в присутствии кислорода с образованием диенов. Напри-
мер, бутен-1 и бутен-2 превращают этим методом в бутадиен-1,3
СН3СН,СН=СН, п
О2
или —> СН2=СНСН=СН2 + Н2О
СН3СН=СНСН3
Еще две реакции сопряженного окисления — это реакции
двуокиси серы или треххлористого фосфора с н-алканами в при-
сутствии кислорода (воздуха) с образованием соответственно
алкансульфокислот (сульфоокисление) и дихлорангидридов ал-
килфосфоновых кислот
RH + SO2 + у О2 —> RSO3H
RH + РС13 + у О2 —> RP(O)C12 + НС1
Классификация методов окисления
Методы окисления, используемые в промышленности нефте-
химического синтеза, можно подразделить на жидкофазные и
парофазные и на каталитические и некаталитические. Обычно
некаталитическое окисление углеводородов в жидкой или паро-
вой фазе протекает по свободнорадикальному механизму и ча-
сто заканчивается образованием смеси продуктов. В то же вре-
мя каталитические реакции более селективны, причем роль
катализатора заключается в увеличении скорости окисления в
целевой продукт по отношению к скоростям побочных процес-
сов. Это позволяет снизить температуру реакции и тем самым
улучшить ее селективность.
Несмотря на большое число работ, посвященных выяснению
механизма катализа, в настоящее время невозможно точно
предсказать, какой катализатор необходим для данной конкрет-
ной реакции. Поэтому выбор катализатора для нового промыш-
ленного процесса и разработка технологии его изготовления до
сих пор проводятся на основе экспериментальных работ и имею-
щегося опыта.
Некаталитическое парофазное окисление. Важным процессом
этого типа является разработанный в США метод неполного
окисления пропан-бутановых смесей, называемых сжиженным
нефтяным газом (СНГ), в ряд продуктов, таких, как формаль-
6*
164
Глава 6
дегид, ацетальдегид, метанол, «-бутанол, изобутанол и уксусная
кислота. Первоначальный вариант предусматривал использова-
ние в качестве окислителя воздуха; реакцию осуществляли в
избытке СНГ при температуре 370°C и давлении 0,7—0,8 МН/м2
(7—8 атм). Позднее стали использовать кислород, что, как со-
общают, позволило увеличить производительность установки и
расширить диапазон получаемых продуктов.
Другой промышленный метод некаталитического парофаз-
ного окисления, который заслуживает упоминания, — это полу-
чение синтез-газа (смеси окиси углерода с водородом) путем
неполного окисления метана (природного газа) ограниченным
количеством кислорода при температуре 1300—1500 °C. Для об-
легчения регулирования глубины окисления кислород часто раз-
бавляют водяным паром (гл. 2). Синтез-газ используют в про-
изводстве метанола и для проведения реакций оксосинтеза
(стр. 180).
Каталитическое парофазное окисление. Очень важной реак-
цией этого типа является прямое окисление этилена в окись
этилена
С2Н4 + О2 —> Н2С—СН2 (газ) А77 = — 146 кДж/моль
Х'о/
заменившее более старый метод ее получения через этиленхлор-
гидрин
Са(ОН)2
С2Н, + НОС1 —> СН2—СН2 ------>- Н2С—СН2
II \ /
ОН С1 о
Превращение олефина в его 1,2-эпоксид называют эпоксидиро-
ванием.
Катализатором эпоксидирования этилена служит металличе-
ское серебро на инертном носителе (например, на а-окиси алю-
миния), которое промотируют соединениями щелочноземельных
металлов, например перекисью бария или окисью кальция.
В случае окисления воздухом используют газовые смеси с кон-
центрацией этилена меньше нижнего предела взрываемости
(3%) и температуру 260—290 °C; при окислении кислородом
температуру реакции можно понизить до 230°С. Давление в
реакторе обычно поддерживают равным 1—3 МН/м2 (10—
30 атм). Выход окиси этилена составляет 50—70%, причем
главным побочным продуктом является двуокись углерода.
Основную часть вырабатываемой окиси этилена подвергают
гидратации в этиленгликоль, который служит компонентом ан-
тифризов и полупродуктом для производства полиэтилентере-
фталата (гл. 9). Кроме того, окись этилена применяется как
Методы получения нефтехимических полупродуктов
165
фумигант и полупродукт для синтеза полиэтиленгликолей и не-
ионных поверхностно-активных веществ (гл. 13).
Другими каталитическими процессами парофазного окисле-
ния, используемыми в промышленном масштабе, являются окис-
ление нафталина или о-ксилола в фталевый ангидрид, бензола
или бутиленов в малеиновый ангидрид (гл. 5), пропилена в
акролеин и (или) акриловую кислоту и окислительный аммоно-
лиз пропилена в акрилонитрил (стр. 170).
Некаталитическое жидкофазное окисление. К реакциям этого
типа относится один из наиболее важных промышленных про-
цессов— окисление фракций нафты состава С4—С» в уксусную
кислоту с побочным получением муравьиной и пропионовой кис-
лот (гл. 3). Однако вследствие того, что некаталитическое жид-
кофазное окисление парафиновых углеводородов не имеет стро-
гой направленности и приводит к образованию ряда кислород-
содержащих продуктов, такие реакции часто оказываются
непригодными для промышленного производства и используются
только в случае низших парафинов, дающих относительно про-
стые смеси продуктов окисления. Тем не менее в случае высших
парафинов с прямой цепью удается повысить селективность
окисления, проводя процесс с сильно разбавленным (3—4,5%-
ным) кислородом при температуре 165—170 °C в присутствии
борной кислоты. При этом основными продуктами являются
вторичные спирты. Роль борной кислоты состоит в стабилизации
образующихся спиртов в форме боратов. Этот метод, известный
под названием реакции Башкирова, используется для жидко-
фазного окисления циклогексана в циклогексанол (см. также
гл. 4), который служит полупродуктом ^ля получения адипи-
новой кислоты, применяемой в производстве полиамидных во-
локон (гл. 9).
Еще одна реакция некаталитического жидкофазного окисле-
ния, имеющая большое промышленное значение, лежит в основе
превращения кумола в его гидроперекись в процессе производ-
ства фенола. Окисление кумола протекает путем атаки кисло-
рода по третичному атому углерода боковой цепи (гл. 5).
Каталитическое жидкофазное окисление. В последние годы
разработано большое число процессов этого типа. Один из са-
мых новых — процесс прямого окисления этилена в ацетальде-
гид кислородом (воздухом) в присутствии водной окислительно-
восстановительной системы, содержащей хлориды палладия и
двухвалентной меди. Реакция обычно осуществляется при тем-
пературе 50—140 °C и давлении 0,6—1,5 МН/м2 (6—15 атм) и
протекает путем разложения комплекса этилена с хлористым
палладием под действием воды, причем образование ацетальде-
гида сопровождается восстановлением палладиевой соли в ме-
таллический палладий. Последний вновь окисляется в присут-
'166
Глава 6
ствующей окислительно-восстановительной системе. Выход ацет-
альдегида по этилену составляет 95%; реакция сопровождается
выделением тепла (ДН = —243 кДж/моль) (гл. 4). Этим мето-
СН2=СН2 + PdCI2 + Н2О —> СНзСНО + Pd + 2НС1
2CuC12 Ц- Pd —> PdCl2 Cu2Cl2
Cu2CI2 + 2HC1 + |o2 —> 2CuC12-J-H2O
дом можно также окислять высшие олефины. Например, из про-
пилена можно получить ацетон, а из н-бутиленов — метилэтил-
кетон.
Важной модификацией описанного процесса является реак-
ция этилена с уксусной кислотой и кислородом в безводном
органическом растворителе, содержащем соли меди и палладия,
которая приводит к образованию винилацетата. Суммарное
уравнение этой реакции
С2Н4 + СН3СООН + у О, —> СН2=СНОСОСН3 + Н2О
Винилацетат применяется в производстве водоэмульсионных
красок.
Среди других реакций каталитического жидкофазного окис-
ления следует отметить получение терефталевой кислоты из
п-ксилола и изофталевой кислоты из л-ксилола (гл. 5). Ана-
логично осуществляют жидкофазное окисление толуола в бен-
зойную кислоту, которую можно использовать как полупродукт
для производства фенола (гл. 7) и капролактама (через цикло-
гексаикарбоновую кислоту, гл. 7).
Хлорирование
А. Джубб
Jubb А. И., ICI Corporate Laboratory, Runcorn
Объем мирового производства хлора в 1970 г. составлял око-
ло 20 млн. т, причем приблизительно 80% хлора было израсхо-
довано на получение хлористого винила, растворителей и пести-
цидов. Только в США потребность в хлоре в 1970 г. составляла
9,6 млн. т, из которых 59% было использовано для получения
хлорорганических растворителей.
Жидкофазное или парофазное хлорирование органических
соединений преследует две цели: получение продуктов, в кото-
рых наличие атома хлора придает им специфические свойства,
и синтез хлорсодержащих полупродуктов, которые способны
Методы Получения нефтехимических Полупродуктов
167
вступать в дальнейшие реакции замещения или элиминирования
хлора, приводящие к образованию новых функциональных
групп. Хлорирование парафинов протекает по свободноради-
кальному механизму и во многих случаях осуществляемся в
промышленности с целью получения монохлорпроизводных
(обычно смеси изомеров). При этом используют большой моль-
ный йзбыток парафина, чтобы подавить образование полихлор-
йроизводных, поскольку монохлорсоединения хлорируются при-
близительно с такой же скоростью, как и исходные парафины.
Природа ориентирующих факторов в реакциях хлорирования
обсуждалась в т. 2 (стр. 132—137).
Хлорирование олефинов при низких температурах (Ниже
250°C) идет по механизму присоединения, а при повышенных —
по механизму аллильного замещения. Присоединение хлора по
двойной связи олефинов в жидкой фазе в темноте протекает
гетеролитически
СН,С1
Х-) ^-\СН2 I
ci ' ci ’ || ----► С1? || +С1- -----» СН2С1
СН2 ''СН2
этилен
При взаимодействии хлора с олефинами в паровой фазе на
свету преобладает гомолитическое (свободнорадикальное) при-
соединение (т. 1, стр. 59). Повышение температуры способствует
аллильному замещению, так как присоединение атома хлора
к олефину обратимо, а отрыв атома водорода от аллильного ра-
дикала необратим. Например, в случае пропилена (т. 2, стр. 129)
происходят следующие реакции:
а) С1 •, + СН3СН=СН2 СН3СНСН2С1 обратимое присоединение
|+С1,
СН3СНС1СН2С1 + С1 •
1,2-дихлорпропан
б) С1 • + СН3СН=СН2 —► (СН2СНСН2) • + НС1 необратимое аллильное
аллильный замещение
радикал
| + С12
С1СИ2СН=СН2 + С1 *
хлористый аллил
Один из основных недостатков метода хлорирования с точки
зрения экономики заключается в том, что во многих случаях
16А
Глава 6
введение в молекулу органического соединения атома хлора со-
провождается выделением молекулы хлористого водорода. Это
происходит в конечном итоге даже при превращении этилена
в дихлорэтан, поскольку в дальнейшем из него получают хло-
ристый винил. В настоящее время * 1 т хлора стоит 30—40 ф. ст.,
и только утилизация хлористого водорода позволяет хлориро-
ванию успешно конкурировать с альтернативными методами.
Существует несколько способов возвращения хлористого водо-
рода в цикл, например электролиз соляной кислоты или взаи-
модействие НС1 с Fe2O3 с последующим переводом образовав-
шегося хлористого железа обратно в Ре20з, сопровождающимся
выделением хлора. Фирма Shell объявила о разработке ею тех-
нологии перспективного процесса, в основе которого лежит ме-
тод Дикона
соли Си и Fe
4НС1 + О2 —-------> 2С12 + 2Н2О
400—600 °C 2
Наибольшее внимание привлек еще один способ утилизации
хлористого водорода, который в настоящее время широко ис-
пользуется в промышленности, — окислительное хлорирование.
В этой реакции хлористый водород in situ окисляется в хлор.
Впервые метод окислительного хлорирования нашел применение
в процессе получения хлорбензола (гл. 7), разработанном при-
близительно в 1930 г. немецкой фирмой Raschig и усовершен-
ствованном затем фирмой Hooker Chemical Со. (США).
С6Н6 + НС1 + уО2 С6Н5С1 + Н2О
Позднее этот метод приобрел большое значение в производстве
хлористого винила (гл. 7)
СН2=СН2 + 2НС1 + у О2 —► Н2О + С1СН2СН2С1 —> СН2=СНС1
В табл. 6.2 приведены данные по промышленному примене-
нию хлорирования, а также (для иллюстрации относительного
значения различных реакций хлорирования) данные о произ-
водстве хлорорганических продуктов в США в 1970 г.
При высокотемпературном хлорировании в присутствии боль-
шого избытка хлора углеводороды претерпевают деструкцию с
образованием фрагментов Ci и С2, превращающихся далее
в четыреххлористый углерод и гексахлорэтан. По предложению
Хааса реакции этого типа называют реакциями хлоролиза. Аме-
риканская фирма Stauffer разработала несколько методов ути-
лизации побочных продуктов производства хлористого винила
* По-видимому, в 1972 г. В начале 1976 г. цены на хлор возросли до
43,5 ф. ст./т в Великобритании и до 125—140 долл./т в США [Chem. Age, 111,
№ 2944, 10, 23 (1975)]. — Прим, перев.
Таблица 6.2
Важнейшие промышленные процессы хлорирования
Исходное сырье Условия реакции Основные продукты а
Окись угле- рода Избыток хлора, катализа- тор — активированный уголь, давление 0,1 МН/м2 (1 атм) Температура 350—450 °C, различные соотношения СН4: С12 Фосген
Метан Хлористый метил6 (50), хлористый метилен (210), хлороформ (100) и четы- реххлористый углерод в (200)
Этан Этилен Дихлорэтан Ацетилен Трихлор- этилен Пропилен Бутадиен н-Пеитан и изопентан Парафины С9—Ci з Парафины С13—С17 Парафины Сзо—С25 Парафины С]2 — С18 Бензол Температура 300—450 °C Жидкая фаза, 40 °C, давле- ние 0,1 —1,0 МН/м2 (1 — 10 атм) Газовая фаза катализа- тор— FeCl3, давление 0,1 МН/м2 (1 атм) Жидкая фаза, катализатор— А1С13 Жидкая фаза (раствор в тет- рахлорэтане), 80 °C, ката- лизатор — SbCl3 Температура 70—80 °C, ка- тализатор — FeCl3 Соотношение С3Н6: С12 4:1, температура 500 °C Газовая фаза, 350—400 °C Температура 300 °C, давле- ние 0,5 МН/м2 (5 атм), избыток углеводорода Температура 70—120 °C, ре- акцию ведут до введения 50—70% хлора Температура 70—120 °C, ре- акцию ведут до введения 40—60% хлора Температура 70—120 °C, ре- акцию ведут до введения 40—60% хлора Монохлорирование Температура 40—60 °C, ка- тализатор — порошкооб- разное железо Хлористый этил г (280) Дихлорэтан4 (3100) 1,1,2-Трихлорэтан (100) и 1,1,2,2-тетрахлорэтан 1,1,2,2-Тетрах лорэтан Пентахлорэтан Хлористый аллил (120 в 1969 г.) 3,4-Дихлорбутен-1 и 1,4-ди- хлорбутен-2 (гл. 4) Монохлорпентаны (с после- дующим гидролизом в амиловые спирты по ме- тоду фирмы Sharpies Chemicals Inc. (США); производились до начала 60-х годов) Компоненты смазочно-ох- лаждающих эмульсий и вторичные пластифика- торы (40) е Монохлорпарафиныж для алкилирования бензола Монохлорбензол (200) и о- и п-дихлорбензолы (60) (в зависимости от соотно-
шения С6Н6 : С12)
170
Глава 6
Продолжение табл. 6.2
Исходное сырье Условия реакции Основные продукты а УФ-облучение, 40—60 °C Гексахлорциклогексан
Толуол РС13 или УФ-облучение, Хлористый бензил (40),хло- 100—150 °C, соотношение ристый бензилиден (2) и С7Н8 : С12 зависит от при- бензотрихлорид (6) роды целевого продукта
а Цифры США в 1972 г в скобках показывают объем потребления (тыс. т) данного продукта в
6 Вырабатывается также окислительным хлорированием метана , или по реакции
метанола с хлористым водородом.
в Получается также путем хлорирования сероуглерода.
г Производится также взаимодействием этанола с хлористым водородом.
Д Вырабатывается также методом окислительного хлорирования (гл. 7).
е В других странах объем потребления вторичных пластификаторов еще больше,
чем в США.
ж Эти продукты подвергают дегидрохлорированию в олефины, которые используются
для алкилирования бензола (метод применяется в ФРГ).
путем их хлоролиза в четыреххлористый углерод и перхлорэти-
лен. Хлоролиз используется в промышленности для получения
гексахлорциклопентадиена из пентана и гексахлорбутадиена из
бутанов.
Окислительный аммонолиз
Синтез акрилонитрила из пропилена
Э. Гассон
Gasson Е. J., Research and Development Department,
BP Chemicals International Limited, Grangemouth
В последнее десятилетие* мировая потребность в акрило-
нитриле (используемом главным образом в качестве полупро-
дукта для производства полиакрилонитрильных волокон и мас-
лостойких синтетических каучуков и смол) увеличивается в
среднем более чем на 10% в год. Этому в значительной степени
способствует резкое снижение затрат производства при полу-
чении акрилонитрила из пропилена и аммиака по сравнению
с другими методами его получения — из ацетилена и синильной
кислоты или из этиленциангидрина, которые в настоящее время
практически вышли из употребления (гл. 7). Наиболее быстры-
ми темпами производство акрилонитрила аммиачно-пропилено-
вым методом развивается в Западной Европе и Японии, где
* i960—1970 гг. — Прим, перев,
Методы получения нефтехимических полупродуктов 171
мощности по его получению увеличились в период 1965—1971 гг.
в 8 и 6 раз соответственно.
В 1971 г. объем мирового производства акрилонитрила этим
методом достигал, по оценке, более 1,5 млн. т, а соответствую-
щие производственные мощности в Западной Европе, Америке
(Северной и Южной) и Японии, взятые в отдельности, превы-
шали к началу 1972 г. 640 тыс. т/год.
Впервые синтез акрилонитрила взаимодействием пропилена,
аммиака и кислорода был описан в 1949 г. в патенте, выданном
на имя Косби (патентообладатель — фирма Allied Chemical}.
Однако выход акрилонитрила, приведенный в этом патенте, со-
ставлял не более 6%, вследствие чего этот новый метод вна-
чале, по-видимому, не привлек к себе внимание исследователей.
К 1950—1951 гг. Хэдли с сотрудниками (фирма Distillers Com-
pany Limited, или, сокращенно, DCL) разработал технологию
двухстадийного парофазного каталитического процесса, в кото-
ром пропилен окисляют над окисномедным катализатором, про-
мотированным селеном, в акролеин, а затем акролеин превра-
щают в акрилонитрил действием аммиака и воздуха над скис-
номолибденовым катализатором.
Несмотря на то что этот процесс не был внедрен в промыш-
ленность (так как более предпочтительным оказался односта-
дийный метод получения акрилонитрила), он обеспечивал высо-
кий общий выход конечного продукта. Кроме того, публикация
патента Хэдли и сотрудников активизировала поиск катализа-
торов для одностадийного превращения пропилена в акрило-
нитрил.
Первым успехом на пути к созданию одностадийного про-
цесса было открытие в 1957 г. Айдолем (фирма Standard Oil,
шт. Огайо, США, или, сокращенно, фирма Sohio) катализатора
на основе молибдата висмута. В 1959 г. фирма DCL разработала
катализатор, содержащий окислы олова и сурьмы. К этому вре-
мени Хэдли (фирма DCL) выдвинул общее представление, со-
гласно которому реакцию пропилена с аммиаком и кислородом
можно рассматривать как одну из многих реакций, характерных
для соединений с «активированной» метильной группой. Хэдли
назвал данную реакцию окислительным аммонолизом; в настоя-
щее время это название является общепринятым.
RCH3 + NH3 + у О2 —► RCN + 3H2O
Фирма Sohio первой (в 1960 г.) начала промышленное произ-
водство акрилонитрила окислительным аммонолизом пропилена
и продала лицензии на свой метод во многие страны. На некото-
рых заводах первоначальный катализатор реакции—фосфомо-
либдат висмута на двуокиси кремния — заменен катализатором,
172
Глава 6
состоящим из окислов сурьмы и урана, осажденных на
силикагеле, а в 1972 г. появилось сообщение о создании еще од-
ного нового катализатора. Процесс фирмы DCL (позднее пере-
именованный в процесс фирм ВР — Ugine) был впервые исполь-
зован в промышленном масштабе в 1965 г. фирмами Ugilor во
Франции и Border Chemicals в Шотландии. В 1972 г. по этому
методу работали 4 завода, тогда как процесс фирмы Sohio и его
модификации применялись более чем на 20 заводах.
Для реакции окислительного аммонолиза пропилена запа-
тентовано множество катализаторов. Однако почти все промыш-
ленные катализаторы относятся к двум основным группам: кон-
такты на основе молибдата висмута и контакты на основе окиси
сурьмы. Последние содержат по крайней мере еще один окисел
металла, такого, как олово, железо или уран. Согласно патент-
ным данным, оба типа катализаторов в последнее время были
усовершенствованы путем введения в них дополнительных про-
мотирующих добавок. Представителем третьей группы катализа-
торов, которые содержат молибдаты или вольфраматы, промоти-
рованные двуокисью теллура, может служить катализатор на
основе окислов церия, молибдена и теллура, применяемый на
заводе фирм Montecatini и Edison в Италии.
К настоящему времени накоплен обширный материал по
кинетике и механизму окислительного аммонолиза пропилена,
особенно в присутствии висмут-молибденового катализатора.
Принято считать, что реакция протекает через промежуточное
образование акролеина. Стадия превращения пропилена в акро-
леин имеет первый порядок по пропилену и нулевой по кисло-
роду (при обычных рабочих концентрациях последнего). При
низкой концентрации кислорода порядок реакции по нему при-
ближается к первому. Вторая стадия — взаимодействие акро-
леина с аммиаком и кислородом, сопровождающееся образова-
нием акрилонитрила, — имеет первый порядок по акролеину и
аммиаку и нулевой по кислороду. Поскольку вторая стадия про-
текает значительно быстрее первой, суммарная реакция обра-
зования акрилонитрила из пропилена имеет кажущийся нулевой
порядок как по кислороду, так и по аммиаку, если только они
взяты в приблизительно стехиометрических соотношениях с про-
пиленом. Энергия активации первой стадии реакции равна
79 кДж/моль, а второй (по различным данным)—от 12,5 до
29,3 кДж/моль.
На первой стадии, лимитирующей суммарную скорость реак-
ции, пропилен адсорбируется на катализаторе, отщепляя атом
водорода от метильной группы и образуя симметричный аллиль-
ный адсорбат, содержащий делокализованную л-связь:
СН2“СН—СН2
i 1
Методы получении нефтехимических полупродуктов 173
Затем отщепляется второй атом водорода от одной из метиле-
новых групп и происходит присоединение аниона кислорода,
поставляемого катализатором (но не из газовой фазы непосред-
ственно). В результате образуется акролеин, который, по край-
ней мере частично, десорбируется с катализатора в газовую
фазу. Что касается второй стадии реакции, то еще полностью не
установлено, реагирует ли адсорбированный акролеин прямо
с молекулярным аммиаком, давая альдимин, или же с имино-
(имидо)-ионом, получающимся при отщеплении атома водорода
от адсорбированного аммиака. Некоторые исследователи счи-
тают второе направление более вероятным, поскольку можно
показать, что в случае висмут-молибденового катализатора дей-
ствительно имеет место адсорбция как акролеина, так и ам-
миака. Однако превращение акролеина в акрилонитрил над
сурьмяно-оловянным катализатором, который не взаимодей-
ствует с аммиаком, тоже протекает очень быстро.
Последней стадией реакции является перенос кислорода из
газовой фазы к катализатору на место прореагировавших ато-
мов кислорода. В реакторах со стационарным слоем катализа-
тора состав сырьевого потока и условия процесса регулируют
таким образом, чтобы обратное окисление катализатора проис-
ходило одновременно и равновесно с реакцией окислительного
аммонолиза; поэтому в нормальных рабочих условиях состав
катализатора остается постоянным. В реакторах с псевдоожи-
женным катализатором может оказаться необходимым создание
особой зоны для его окисления — например, путем подачи части
воздуха ниже точки ввода остальных реагентов.
Главные реакции, протекающие при окислительном аммоно-
лизе, сильно экзотермичны
с,н6 + NH3 + -|o2 - -> ch2=chcn + ЗН2О АД = - 515 кДж/моль
СзНд + 3NH3 + 3O2 - -> 3HCN + 6Н2О ьн = — 946 кДж/моль
СзНв + 302 — -> зсо + зн2о \н = - 1075 кДж/моль
С,ч6 + |о2 - -> зсо2 + зн2о ьн = - 1925 кДж/моль
Общее количество тепла, выделяющееся в промышленном
реакторе, составляет, вероятно, около 700 кДж/моль пропилена.
Одним из главных условий поддержания оптимальной темпера-
туры в зоне реакции является отвод этого тепла. В процессе
фирмы Sohio температуру реакции регулируют путем исполь-
зования охлаждающих змеевиков, находящихся в слое псевдо-
ожиженного катализатора, а в процессе фирм ВР — Ugine эта
задача решается с помощью реакторов трубчатого типа, труб-
ки которых имеют малый внутренний диаметр и заполнены
174
Глава 6
катализатором, а межтрубное пространство — циркулирующим
теплоносителем (расплавом солей). Тепло, отводимое с охлаж-
дающей жидкостью, утилизируется в обоих процессах для полу-
чения пара высокого давления.
Реакцию проводят при температуре 400—470 °C и времени
контакта, равном 2—4 с для стационарного катализатора и
10—20 с для псевдоожиженного. Состав сырьевого потока
обычно подбирают таким образом, чтобы он находился за пре-
делами воспламенения: 5—8% пропилена, 5—9% аммиака, 0—
30% водяного пара и остальное — воздух. Отношение аммиака
к пропилену обычно колеблется от 1,0 до 1,1. При более низких
отношениях уменьшается выход акрилонитрила и возрастает
выход акролеина, что может привести к нарушению технологи-
ческого режима из-за полимеризации последнего. Непревращен-
ный аммиак необходимо удалять из реакционных газов прежде,
чем они будут охлаждены до точки росы, чтобы предотвратить
полимеризацию побочно образовавшегося акролеина. Для этого
газы обычно промывают горячим раствором сульфата аммония
в серной кислоте, из которого путем последующей кристаллиза-
ции можно получить сульфат аммония. Однако выделять эту
соль экономически невыгодно, и в настоящее время на большин-
стве заводов производственный процесс ведут таким образом,
чтобы получить максимальный выход акрилонитрила при мини-
мальном выходе сульфата аммония.
Реакционные газы после нейтрализации аммиака охлаждают
и промывают водой, которая экстрагирует из них все органиче-
ские продукты и синильную кислоту, выделяемые затем на ста-
дии дистилляции. Получают сырой акрилонитрил, содержащий
приблизительно 70—75% акрилонитрила, 12—15% синильной
кислоты, 2—4% акролеина, 1—3% ацетонитрила, 5—7% воды и
ничтожные примеси других побочных продуктов. Дальнейшая
очистка акрилонитрила в процессах фирм Sohio и ВР — Ugine
в принципе одинакова .и различается лишь порядком операций.
В последнем процессе сначала удаляют акролеин и другие аль-
дегиды. Это достигается за счет их реакции с частью синильной
кислоты, в результате которой образуются циангидрины, легко
отделяемые перегонкой. В следующей ректификационной колон-
не происходит отделение синильной кислоты в виде головной
фракции. Третьей стадией очистки является разделение акрило-
нитрила и ацетонитрила экстрактивной дистилляцией с водой,
причем в дистилляте отбирают водный акрилонитрил, а из ку-
бовой жидкости — водный ацетонитрил. Сырой акрилонитрил
сушат и подвергают окончательной очистке путем ректификации
в одной или нескольких колоннах.
В процессе фирмы Sohio сначала экстрактивной дистилля-
цией с водой отделяют ацетонитрил, после чего из головного
Методы получения нефтехимических полупродуктов 175
дистиллята отгоняют синильную кислоту. Сырой акрилонитрил
очищают от примесей ацетона и циангидринов многократной
ректификацией, при которой его попеременно выводят то с вер-
ха, то из низа ректификационной колонны.
Товарные продукты имеют очень высокую степень чистоты:
акрилонитрил — свыше 99,98% (если не считать добавленный
ингибитор), а синильная кислота (процесса ВР — Ugine) —
более 99,5%.
Надежные данные о выходе акрилонитрила в различных
промышленных процессах труднодоступны. Большинство дан-
ных, приводимых в журнальных статьях и патентах, несомненно,
получены в лабораторных реакторах (которые обычно дают зна-
чительно более высокие выходы, чем промышленные установки).
Кроме того, не всегда сообщается, как рассчитывался выход
акрилонитрила — по взятому сырью (выход за один проход)
или по превращенному сырью (окончательный выход, или се-
лективность конверсии). Часто приводится вторая величина,
которая всегда больше первой (если только конверсия не яв-
ляется количественной), хотя в случае процессов данного типа,
где отсутствует рециркуляция непревращенных реагентов, по-
казательным является только выход за один проход. К тому же
даже на лабораторных установках выход акрилонитрила изме-
Таблица 6.3
Катализаторы и выходы акрилонитрила при окислительном
аммонолизе пропилена
Первоначальный катализатор Добавки Фирма- патентообл адатель Год Выход акри- лонитрила,
Bi, Р, Mo, Si Sohio 1957 53-59
Ba, F, Si 1962 73
V SNAM 1966 67
Fe, Ni, Mn, Sumitomo 1970 84
Mo, T1
Sb. Sn — Distillers 1959 60
Cu, Fe и др. » 1963 71
Sb, Fe Sohio 1963 63
Cu, Nb » 1965 65
В, P, V Nitto 1968 73
V, Те 1969 79
Sb, U — Sohio 1962 75
Си Distillers 1963 73
— Monsanto 1969 80
Те, Mo, Ce, Si —- Montecatini — 1968 80
Edison
Те, Mo, Си, P Goodrich 1969 65
Те, Mo, Fe — SNPA 1969 78
Те, W, Fe, Si — Nitto 1970 76
176
Глава б
няется в зависимости от природы катализатора и промоторов,
состава сырьевого потока, рабочего давления и времени кон-
такта.
В табл. 6.3 приведены данные о катализаторах и выходах
акрилонитрила, взятые из патентной литературы. Эти данные
не являются полными и лишь иллюстрируют способы усовер-
шенствования различных катализаторов посредством введения
в них дополнительных промоторов и модификаторов. Выходы
акрилонитрила рассчитаны по взятому пропилену и относятся
к лабораторным установкам. Однако строгое сопоставление этих
выходов не всегда возможно из-за различий в составе исходного
сырья и времени контакта. Не исключено, что некоторые из но-
вых катализаторов уже используются в промышленности.
Приведенные в табл. 6.3 элементы, входящие в состав ката-
лизаторов, применяются в виде окислов или солей, но для крат-
кости изображения кислород здесь не показан.
Синтез пиридинов
А. Джубб
Jubb А. Н„ ICI Corporate Laboratory, Runcorn
Несмотря на то что пиридины не всегда синтезируют с ис-
пользованием реакции окислительного аммонолиза, между не-
которыми методами их синтеза и получением акрилонитрила
существует сходство, делающее возможным совместное рассмот-
рение этих процессов. Объем потребления пиридинов по нефте-
химическим масштабам невелик; тем не менее области примене-
ния отдельных пиридиновых оснований имеют большое значение.
В табл. 6.4 перечислены главные пиридиновые основания и
наиболее важные области их применения. Хотя первый синтез
пиридина был осуществлен более 100 лет назад, единственным
источником получения пиридина и его производных, за исклю-
чением 2-метил-5-этилпиридина, примерно до 1954 г. оставалась
каменноугольная смола. К настоящему времени разработано
множество методов синтеза пиридиновых оснований (часто в
виде смеси продуктов). Однако в промышленности используется
лишь небольшая часть из них, причем, кроме основного про-
дукта — пиридина, всегда получаются пиколины.
В 1970 г. мировая потребность в пиридине составляла, по
оценке, около 8500 т, из которых приблизительно 1500 т было
выработано' из природного сырья. Потребность во всех трех
пиколинах оценивалась в 1970 г. примерно в 7000 т (причем
около 800 т было получено из природного сырья), а в 2-метил-
Методы получения нефтехимических полупродуктов
177
Таблица 6.4
Пиридиновые основания и области их применения
Соединения
Пиридин
Области применения
Производство дипиридильных гербицидов и пипе-
ридина. Денатурирование спирта. Получение
фармацевтических препаратов (антисептиков
типа цетилпиридинийхлорида и антигистаминных
препаратов на основе аминопиридина)
а-Пиколин
т. кип. 129 °C
Р-Пиколин
СН3
Путем обработки а-пиколина формальдегидом
с последующей дегидратацией продукта полу-
чают 2-винилпиридин, применяемый в производ-
стве адгезивов. Получение пестицидов, кокци-
диостатическнх препаратов и дефолиантов, а
также добавок к удобрениям, пролонгирующих
сохранение азота в почве
Производство препаратов типа ниацина и ниаци-
намида, применяемых в качестве кормовых и
пищевых добавок, содержащих витамин РР
N
т. кип. 144 °C
у-Пиколин
СН3
Получение противотуберкулезных лекарственных
препаратов на основе изоникотингидразида,
поверхностно-активных веществ, растворителей
и пестицидов
N
т. кип. 145 °C
2-Метил-5-этилпиридин
т. кип. 144 °C
Производство лекарственных препаратов типа
ниацнна (пиридин-3-карбоновая кислота) и син-
тез 2-метил-5-винилпиридина, применяемого при
получении пропиточного латекса для шинного
корда и некоторых синтетических каучуков
5-винилпиридине — приблизительно в 8500 т. В начале 1972 г.
общие мировые мощности по производству синтетических и
природных пиридиновых оснований достигали, вероятно,
30 тыс. т/год.
В основе промышленного метода синтеза пиридина и 0-пико-
лина лежит реакция между ацетальдегидом, формальдегидом
и аммиаком в псевдоожиженном слое катализатора (алюмоси-
ликата). Соотношение основных продуктов реакции зависцт от
178
Глава 6
содержания формальдегида в исходном сырье, которое обычно
подбирается так, чтобы пиридин и 0-пиколин получались в ве-
совом соотношении около 1,5: 1 с общим выходом 60—70%.
4СН3СНО + ЗНСНО + 2NH3 —> + 7Н2О + Н2
N N
В отсутствие формальдегида образуются главным образом
а- и у-пиколины (суммарный выход приблизительно 68%) на-
ряду с небольшой примесью 2-метил-5-этилпиридина и 4-метил-
3-этилпиридина
СНз
6СН3СНО + 2NH3 —> +|f^ + 6H2O + H2
N ^снз N
Эти процессы, технология которых была впервые разрабо-
тана американской фирмой Reilly Tar and Chemical Corporation
и которые в настоящее время используются в промышленном
масштабе в США, Италии и Японии, базируются на работах
А. Е. Чичибабина (т. 3, стр. 125—126). Недавно фирма. British
Petroleum разработала новый вариант этих процессов, еще не
реализованный в промышленности, причем, судя по патентным
данным, добавление воздуха к исходной смеси альдегидов поз-
воляет увеличить весовое соотношение пиридина и пиколина до
5,7: 1. Та же фирма запатентовала использование кротонового
альдегида вместо ацетальдегида.
Акролеиновый метод синтеза пиридиновых оснований пред-
ставляет собой двухстадийный процесс, на первой стадии кото-
рого пропилен окисляют воздухом на стационарном висмут-мо-
либденовом катализаторе при температуре 330°C и давлении
0,3 МН/м2 (3 атм). Выходящие из реактора газы, содержащие
акролеин, смешивают с аммиаком и подают в другой реактор,
в котором реакция протекает на псевдоожиженном алюмосили-
катном катализаторе при температуре 450 °C. Описанный про-
цесс обеспечивает весовое соотношение пиридина и (3-пиколина
около 1,5:1. По некоторым сведениям, этот процесс или его
модификации применяют в Японии и Индии.
Промышленное производство 2-метил-5-этилпиридина осу-
ществляют путем конденсации паральдегида (тримера ацеталь-
дегида) с аммиаком в жидкой фазе в присутствии солей аммо-
ния как катализаторов
C2H5\f^4
4(СН3СНО)з + 3NH3 —► 3 | ] +12Н2О
N ^СНз
Методы получения нефтехимических полупродуктов
179
Раствор паральдегида в уксусной кислоте и 30—40%-ный
водный аммиак реагируют между собой при температуре 220—
260°C и давлении 5 МН/м2 (50 атм). При этом получаются ди-
алкилпиридин с выходом 60—70% и небольшие количества а-
и у-пиколинов. Этот метод реализован во Франции, Италии,
Швеции, Швейцарии, США и, предположительно, в СССР*. Ал-
килпиридины, и в частности 2-метил-5-этилпиридин, можно под-
вергнуть термическому гидродеалкилированию в присутствии
водорода при температуре 650—750 °C и давлении 0,5—5,0 МН/м2
(5—50 атм). Например, 2-метил-5-этилпиридин превращается
в смесь пиридина и а-пиколина, соотношение которых зависит
от условий реакции.
Существует еще несколько методов синтеза пиридина, ко-
торые, хотя и не были внедрены в промышленность, послужили
предметом широкого исследования и заслуживают краткого
упоминания.
1. Фирма ICI сообщила об открытии каталитической паро-
фазной реакции тетрагидрофурфурилового спирта с аммиаком,
приводящей к 60 %-ному выходу пиридина при 55 %-ной конвер-
сии исходного спирта
+ Шз ,катализато% О + 2Н2О + 2Н2
4Э0 °C \
N
X ^СИ2ОН
Аналогичный двухстадийный метод получения пиридина пу-
тем парофазного синтеза пиперидина с его последующим де-
гидрированием был предложен фирмой Quaker Oats. Выход
пиридина по этому методу составляет 80—90%.
2. Реакция Дильса — Альдера между акролеином и метил-
виниловым эфиром дает 2-метокси-5,6-дегидропиран с выходом
95%. При взаимодействии с аммиаком в паровой фазе этот де-
гидропиран превращается в пиридин, выход которого состав-
ляет 84%.
Промышленная реализация обоих методов тормозится срав-
нительно высокой стоимостью исходного сырья.
* В СССР организовано промышленное производство 2-метил-5-винилпи-
ридина (из 2-метил-5-этилпиридина) и латексов для шинной промышленности
на его основе [Кузнецов В. Л., в сб. «Синтетический каучук», под ред. Гармо-
нова И. В., «Химия», Л., 1976, стр. 606]. — Прим. ред.
180 Глава 6
Гидроформилирование (оксосинтез)
М. Лоуренсон
Lawrenson М. Research and Development Department,
BP Chemicals International Limited, Sunbury-on-Thames
Реакцию каталитического присоединения окиси углерода и
водорода по двойной связи олефинов с образованием альдеги-
дов принято называть реакцией оксосинтеза. Однако в послед-
нее время вместо этого названия чаще всего употребляют более
содержательный термин «гидроформилирование». Гидроформи-
лирование представляет собой гомогеннокаталитический жидко-
фазный процесс, приводящий к получению смеси альдегидов
с прямой и разветвленной цепью
RCH=CH2 + Н2 + СО —> RCH2CH2CHO И RCH(CH3)CHO
В том случае, когда катализатор вызывает изомеризацию двой-
ной связи в исходном олефине, возможно также образование
других изомерных продуктов. Альдегиды — довольно реакцион-
носпособные соединения, которые могут использоваться в каче-
стве полупродуктов в производстве карбоновых кислот, аминов
и многоатомных спиртов (путем их окисления, восстановитель-
ного аминирования * и взаимодействия с формальдегидом со-
ответственно). Однако основную часть вырабатываемых альде-
гидов непосредственно переводят в спирты. Наибольшим спросом
пользуются спирты с прямой цепью, так как они более устой-
чивы термически и легче образуют различные производные,
например сложные эфиры. Альдегиды изостроения, которые яв-
ляются нежелательными побочными продуктами гидроформили-
рования, применяются в качестве топлива. Однако в будущем,
как считают, их будут подвергать термическому разложению с
целью регенерации исходных веществ.
Обычно катализатором гидроформилирования служит окта-
карбонилдикобальт Со2(СО)в, который можно приготовить из
любой соли кобальта, достаточно хорошо растворимой в исход-
ном олефине. На практике для этого используют восстановление
нафтената или олеата кобальта окисью углерода. Полагают, что
активным компонентом катализатора является тетракарбонил-
гидрид кобальта НСо(СО)4 образующийся в присутствии во-
дорода, необходимого для гидроформилирования.
Реакцию обычно проводят при температуре 140—180 °C и
давлении 19—21 МН/м2 (190—210 атм). На рис. 6.1 дана прин-
ципиальная технологическая схема процесса. В реактор для
синтеза альдегидов / подают олефин, водород и окись углерода,
* Другое название этой реакции — гидроаминирование. — Прим, перев.
Методы получения нефтехимических полупродуктов
181
а также возвратные газы и рециркулирующий катализатор. Вы-
текающие из реактора продукты поступают в декобальтиза-
тор 2, где их обрабатывают кислотой. При этом карбонилы ко-
бальта превращаются в кобальтовую соль, которую можно экс-
трагировать водой и снова возвращать в цикл. Удаление ко-
бальта из продуктов реакции необходимо для предотвращения
трех нежелательных явлений: разложения катализата гидрофор-
милирования на последующих стадиях; потерь альдегидов за
счет катализа реакций конденсации; отравления катализатора,
т
Кубовый
остаток
? Изобутиловый
спирт
> Бутанол-!
Кубовый
остаток
Рис. 6.1. Технологическая схема процесса получения бутанола.
/ — реактор для гидроформилирования; 2— декобальтизатор; 3 — узел регенерации катали-
затора; 4 — ректификационная колонна для выделения альдегидов; 5 — гндрогенизатор;
6—ректификационная колонна для выделения бутанола
используемого на стадии гидрирования. Соль кобальта снова
переводят в маслорастворимую форму в узле регенерации 3 и
возвращают в реактор. Продукты реакции подвергают перегон-
ке в ректификационной колонне 4 и полученные альдегиды
подают в гидрогенизатор 5. Хотя спирты можно в значительных
количествах получать непосредственно в реакторе 1 (за счет
гидрирования образующихся альдегидов в присутствии кобаль-
тового катализатора при температуре выше 140°C), в настоящее
время принято осуществлять гидрирование в виде отдельной
стадии (например, на твердом никелевом катализаторе, который
гораздо более эффективен). Выходящие из гидрогенизатора 5
спирты разделяют ректификацией в колонне 6. Типичный ка-
тализат гидроформилирования содержит 78—82% альдегидов,
10—12% спиртов, около 2% эфиров муравьиной кислоты и 6—
182
Глава 6
8% высококипящих продуктов альдольной конденсации. Соотно-
шение линейных и разветвленных альдегидов (и спиртов) равно
3 : 1 или несколько больше.
Обычный процесс гидроформилирования приводит к превра-
щению олефина С„ в спирты C„+i. В том случае, когда высшие
олефины труднодоступны, более удобно получать высшие спирты
из низших олефинов с помощью процесса «альдокс», в котором
из олефина Сп образуются спирты C2n+2- Этот процесс отли-
чается в сущности лишь тем, что в нем используют кобальтовый
катализатор, модифицированный органической солью цинка или
другим соединением металла, катализирующим альдольную кон-
денсацию первоначально образующихся альдегидов, в резуль-
тате чего, например, пропилен превращается в 2-этилгексанол-1.
Реакции гидроформилирования и конденсации можно проводить
в общем реакторе.
СН3СН=СН2 + Н2 + СО —> СН3СН2СН2СНО + СН3СН(СН3)СНО
СН3СН2СН2СНО —> СН3СН2СН2СН(ОН)СНСНО —>
СН2СН3
Н2
—> СН3СН2СН2СН=ССНО —* СН3СН2СН2СН2СНСН2ОН
I NI I
сн2сн3 сн2сн3
Позднее появился процесс фирмы Shell, в котором исполь-
зуется карбонилкобальтовый катализатор, стабилизированный
фосфиновыми лигандами. При обработке октакарбонилдико-
бальта триалкилфосфином выделяется окись углерода и обра-
зуется ионизированный комплекс [Со(СО)з(РК3)2] [Со(СО)4],
легко переходящий в растворимую в углеводородах форму
[Со(СО)зРКз]г- Последнее соединение является непосредствен-
ным предшественником активного катализатора. Ионизирован-
ный комплекс устойчив при давлениях до 0,1 МН/м2 (1 атм),
Тогда как октакарбонилдикобальт при таком низком давлении
разлагается на окись углерода и металлический кобальт, кото-
рый не обладает каталитической активностью. Принято считать,
что активной каталитической формой является карбонилгидрид,
аналогичный по своему составу соединению, получаемому из
октакарбонилдикобальта, но содержащий вместо одной карбо-
нильной группы фосфин. В качестве катализаторов гидрофор-
милирования запатентованы также карбонилсодержащие комп-
лексы родия; однако они пока не нашли применения в промыш-
ленности.
Фосфин-кобальтовая система гораздо активнее катализирует
реакцию гидрирования альдегидов, чем незамещенный карбо-
Методы получения нефтехимических полупродуктов 183
нилгидрид кобальта, позволяя тем самым получать спирты в
одну стадию. Однако при использовании этой системы значи-
тельная часть олефина гидрируется в алкан. Соотношение ли-
нейных и разветвленных продуктов существенно выше, чем в
случае незамещенного карбонилгидрида кобальта. Типичным
является следующий состав катализата: 20% алкана, 9% спир-
тов изостроения, 64% спиртов нормального строения и 7% вы-
сококипящих соединений. Повышенное соотношение линейных и
разветвленных продуктов, как полагают, обусловлено стерео-
селективностью НСо(СО)з[Р(С4Н9)3]. Следует заметить, однако,
что вследствие протекания конкурирующей реакции гидрирова-
ния олефина общий выход особенно ценных спиртов нормаль-
ного строения возрастает ненамного. Основное преимущество
фосфинсодержащего катализатора заключается в возможности
проводить процесс в более мягких условиях —при температуре
150—195 °C и общем давлении Н2 и СО 4,0—5,5 МН/м2 (40—
55 атм), что позволяет избежать применения дорогостоящей
аппаратуры высокого давления.
Т ермодинамика
и механизм гидроформилирования
Наиболее полные термодинамические данные, имеющиеся в
литературе, относятся к реакции гидроформилирования этилена,
приводящей к образованию единственного альдегида — пропио-
нового
сн2=сн2 4- Н2 -J- СО —> СНЭСН2СНО
Теплоты образования этилена, окиси углерода и пропионового
альдегида при 25°C равны соответственно +52, —ПО и —204
кДж/моль. Отсюда \Н = — 146 кДж/моль и AG = —146 +
+ 24Т = — 61 кДж/моль (при 25°C). Следовательно, константа
равновесия этой реакции составляет 4,1 Н~2'. м4. Гидрирование
олефина является термодинамически более выгодной реакцией
(AG = —94,5 кДж/моль при 25°C).
Выделение и идентификация гидридокарбонильного или дру-
гого промежуточного соединения реакции гидроформилирования,
катализируемой октакарбонилдикобальтом, с целью выяснения
механизма этой реакции никогда не проводились. Тем не менее,
основываясь на превращениях олефинов при комнатной темпе-
ратуре, обычно принимают, что при гидроформилировании оле-
финов происходят следующие процессы: присоединение тетра-
карбонилгидрида кобальта к олефину с образованием л-комп-
лекса, перенос атома водорода, приводящий к получении)
184
Глава 6
карбонилкобальталкильного промежуточного соединения, и цис-
внедрение окисноуглеродного лиганда по связи алкил—кобальт,
в результате которого образуется карбонильная группа. Затем
происходит повторное присоединение водорода с элиминирова-
нием альдегида.
f fHR h----chr ch2r
(СО)4Со + СН2 * (СО)4Со---СН2 ч - (СО)4Со—СН2 «=£
О
II
/С»,
(СО)3Со----СН2—CH2R (CO)3Co-C-CH2CH2R —>
II
о
* (СО)4Со—С—CH2CH2R
о
н2
---» ВСН2СН2СНО + НСо(СО)4
Согласно предложенному альтернативному механизму, носи-
телем каталитической активности является НСо(СО)3, образу-
ющийся при диссоциации НСо(СО)4
НСо(СО)4 НСо(СО)3 + СО
Однако этот механизм маловероятен, поскольку гидроформили-
рование может происходить при очень высоких давлениях, ког-
да равновесие должно быть целиком смещено в левую сторону.
При использовании в качестве катализатора октакарбонил-
дикобальта и проведении реакции в условиях, когда степень
изомеризации непревращенного олефина мала, изомерные оле-
фины с концевой и внутренней двойными связями дают почти
одинаковое соотношение линейных и разветвленных продуктов.
По-видимому, это объясняется тем, что изомеризация алкиль-
ной группы, связанной с атомом кобальта, протекает с преиму-
щественным образованием линейного соединения.
В том случае, когда предшественником активного катализа-
тора служит [Со (СО)з(РНз)]г, принято считать, что природа
катализатора и промежуточных соединений аналогична посту-
лируемой для реакции с октакарбонилдикобальтом, за исклю-
чением того, что одна карбонильная группа в них замещена
на фосфин. Карбонилгидрид НСо(СО)з[Р(С4Н9)3] был синте-
зирован и введен в реакцию с этиленом в присутствии окиси
углерода при повышенном давлении. При этом удалось выде-
лить СН3СН2СОСо(СО)з[Р(С4П9)з], который реагирует с водо-
родом, давая соответствующий альдегид и {Со(СО)3[Р(С4Н9)з]}2,
185
Методы получения нефтехимических полупродуктов
что согласуется с механизмом, представленным ниже:
НСо(СО)4
4-
RCH2CH-=CH2
НСо(СО)4
+
Всн=снсн3
RCH2CH2CHj -
Со(СО)4
RCHjCHCH3
Со(СО)4
RCHCHjCH,
Со(СО)4
КСН2СН2СН2СНО
RCH2CH(CH3)CHO
RCHCHjCHj
сно
Um.i
Области применения
Размеры и структура мощностей по получению различных
продуктов гидроформилирования олефинов в Западной Европе
в 1971 г. иллюстрируют важное значение этого метода и отно-
сительные масштабы производства каждого из продуктов. Наи-
более распространенным исходным сырьем является пропилен.
Полученные из него бутанолы используются в качестве раство-
рителей (375 тыс. т/год); кроме того, бутанол-1 превращают
путем конденсации и последующего гидрирования продукта в
2-этилгексанол (585 тыс. т/год). Этерификацией этого спирта
фталевым ангидридом синтезируют диалкилфталаты, применяе-
мые в качестве пластификаторов поливинилхлорида, который
может содержать до 50% пластификатора (гл. 8). К другим
спиртам, используемым в производстве пластификаторов
(330 тыс. т/год), относятся «изооктанолы», получаемые путем
гидроформилирования содимеров пропилена и бутилена, и спир-
ты С?—Сд, которые синтезируют из узкой олефиновой фракции
Се—Се, выделяемой из продуктов крекинга твердого парафина.
Спирты, полученные гидроформилированием высших олефинов
(таких, как додецен), подвергают сульфированию с целью
приготовления полупродуктов для производства моющих ве-
ществ (в 1971 г. мощности по получению этих спиртов состав-
ляли 30 тыс. т/год, а в 1972 г. они должны были, по прогнозной
оценке, увеличиться до 36 тыс. т/год). Гидроформилирование
186
Глава 6
этилена осуществляют в гораздо меньших масштабах; получен-
ный пропионовый альдегид перерабатывают в пропанол-1 или
в пропионовую кислоту.
Гидратация олефинов
П. Лусман
Lustrum Р., Research and Development Department,
BP Chemicals International Limited, Grangemouth
Промышленное использование гидратации олефинов почти
целиком ограничивается получением этанола и изопропанола,
а также небольших количеств бутанола-2
RCH=CH2 + H2O RCH(CH3)OH
(R = Н или алкил)
Существует два метода гидратации олефинов — прямой и не-
прямой. Несмотря на значительное различие условий, химиче-
ская сущность обоих методов одинакова. Гидратация олефи-
нов— это кислотнокаталитическая реакция, которая протекает
с участием карбониевого иона, образующегося из олефина, и
протона, поставляемого кислотой (т. 1, стр. 56). При этом со-
блюдаются обычные правила ориентации (т. 2, стр. 77—81),
приводящие в случае пропилена и высших олефинов к почти
исключительному образованию вторичных спиртов.
В процессе непрямой гидратации олефинов донором прото-
нов обычно служит концентрированная серная кислота. Обра-
зующийся карбониевый ион реагирует с сульфат-ионом, давая
моно- и диэтил сульф а ты, которые на второй стадии гидролизу-
ются, выделяя этиловый спирт.
+ R\ Rx
RCHCH3 + HSO7^ /CHOSO.OH +=> /СНОБОз+Н*
СН/ СН/
R\ - ( R\ A
^CHOSOJ + RCHCHj ( /СНО J SO2
СН/ \CH3/ A
/ Rx X R4
I ^CHO I SO2 + H2O RCH(OH)CH3 + ^CHOSO2OH
\СН/ Д СН/
R\
)CHOSO2OH + H2O RCH(OH)CH3 + H2SO4
CH3/
Методы получения нефтехимических полупродуктов 187
В производстве этанола этилен абсорбируют 94—98%-ной
(по весу) серной кислотой при температуре 55—85 °C и давле-
нии 2—3 МН/м2 (20—30 атм). Абсорбцию осуществляют путем
барботирования этилена через подаваемую противотоком к нему
серную кислоту (например, в аппарате колонного типа), кото-
рая поглощает 99% этилена. Реакционную массу, содержащую
0,3—0,4 вес. ч. этилена (в форме этилсульфатов) на 1 вес. ч.
кислоты, разбавляют 1,0—1,4 вес. ч. воды и нагревают при ат-
мосферном давлении до 60—80 °C. В этих условиях гидроли-
зуется диэтилсульфат; гидролиз моноэтилсульфата осуществ-
ляют в следующем аппарате при температуре около 100 °C.
Этанол отделяют от разбавленной кислоты дистилляцией, по-
лучая водный спирт с концентрацией 50—65% (по весу). Раз-
бавленную кислоту, содержащую 35—65% (по весу) H2SO4, кон-
центрируют и возвращают на стадию абсорбции. Абсорбцию
пропилена, который химически активнее этилена, проводят в
более мягких условиях, используя 85%-ную серную кислоту
и температуру 25—30 °C. В остальном процессы непрямой гид-
ратации этилена и пропилена аналогичны друг другу.
При прямой гидратации олефинов донором протонов слу-
жит концентрированная фосфорная кислота, содержащаяся в
порах кремнеземного носителя. Последний обычно имеет фор-
му небольших шариков или цилиндров, и реакцию ведут как
типичный гетерогеннокаталитический процесс на стационарном
катализаторе. Однако гидратация в отличие от большинства
таких процессов протекает не на твердой поверхности катали-
затора, а в жидкой фазе кислоты, находящейся в порах носи-
теля.
Смесь водяного пара с этиленом в мольном отношении
0,4—0,8 пропускают через слой катализатора при температуре
250—300°C и давлении около 6 МН/м2 (60 атм). Превращение
карбониевого иона в молекулу спирта протекает по следующему
суммарному уравнению:
RCHCH3 + H2o RCH(OH)CH3 + Н+
Хотя в этой реакции, возможно, происходит промежуточное
образование этилфосфатов, следы которых содержатся в ката-
лизате, главным компонентом катализата является этанол,
вследствие чего отпадает необходимость в стадии гидролиза.
Конденсат продукта представляет собой водный раствор с кон-
центрацией этанола 10—20%. Степень конверсии этилена за
проход составляет лишь 5—6%. Большую часть неконвертиро-
ванного этилена возвращают в реактор. При этом небольшое
количество рециркулирующего газа отдувают, чтобы вывести
из системы инертные примеси (главным образом продукты по-
лимеризации этилена) и обеспечить концентрацию этилена в
188
Глава 6
рециркулирующем газе 90—95%. В том случае,когда предусмот-
рены извлечение и рециркуляция этилена, содержащегося в га-
зах отдувки, общая селективность превращения этилена в эта-
нол достигает 95%. Низкая степень конверсии за один проход
объясняется малой величиной константы равновесия газофазной
реакции при рабочей температуре. Так, при температуре 270 °C
эта величина равна 5-Ю-8 Н~<-м2. Поскольку реакция гидрата-
ции экзотермична (АЯ =—46 кДж/моль), с повышением тем-
пературы константа равновесия уменьшается, а скорость реак-
ции возрастает. Увеличение отношения водяного пара к этилену
и повышение давления сопровождаются увеличением равновес-
ной степени конверсии этилена. Однако при этом ухудшаются
экономические показатели, так как для нагревания и испарения
дополнительного количества воды необходимо затратить больше
энергии, а с повышением давления возрастают капитальные за-
траты на оборудование. Кроме того, для данной температуры
и данного соотношения реагентов существует предельное рабо-
чее давление, выше которого происходит конденсация воды на
катализаторе, приводящая к вымыванию кислоты. Поэтому ре-
альные условия проведения процесса обычно являются резуль-
татом компромисса между указанными выше противоположны-
ми техническими и экономическими требованиями и подбирают-
ся таким образом, чтобы обеспечить минимальные суммарные
затраты производства в сочетании с надежной и безостановоч-
ной работой оборудования в течение длительного времени.
Прямую гидратацию пропилена ведут в более мягких усло-
виях; однако в принципе этот процесс не отличается от опи-
санного выше. Температура реакции составляет 150—220 °C,
давление —до 3,5 МН/м2 (35 атм). При данной температуре
равновесие гидратации менее благоприятно для образования
спирта, чем в случае этилена. Однако это компенсируется тем,
что при низких температурах пропилен гидратируется быстрее,
чем этилен. При 200 °C константа равновесия приблизительно
равна 1 • 10'7 Н-1-м2. Вследствие понижения температуры для
предотвращения конденсации воды приходится применять более
низкое давление по сравнению с процессом гидратации этилена.
В итоге достигаемая степень конверсии пропилена за один про-
ход близка к степени конверсии этилена.
Преимуществами сернокислотной гидратации олефинов яв-
ляются низкие рабочие температуры и давления и высокие сте-
пени конверсии за один проход, что позволяет использовать бо-
лее разбавленное и, следовательно, более дешевое олефиновое
сырье (например, этан-этиленовую и пропан-пропиленовую
фракции). Применение разбавленного сырья в процессе прямой
гидратации неизбежно привело бы к чрезмерному увеличению
количества газов, выводимых из системы. В то же время при
Методы получения нефтехимических полупродуктов
189
прямой гидратации нет сильной коррозии оборудования, что
снижает эксплуатационные расходы, а сырой спирт получается
чище и, следовательно, легче доводится до тех жестких конди-
ций, которым в ряде случаев должен удовлетворять товарный
продукт. Легкость управления технологическим процессом в со-
четании с меньшими расходами на текущий ремонт зданий и
оборудования делает метод прямой гидратации более предпо-
чтительным для синтеза этанола и изопропанола. Однако неко-
торые фирмы продолжают считать, что для небольших устано-
вок на заводах, где нет чистого пропилена, выгоднее исполь-
зовать метод сернокислотной гидратации пропилена.
В качестве побочных продуктов при прямой гидратации эти-
лена и пропилена образуются главным образом полимеры ис-
ходного олефина, альдегиды, кетоны, простые эфиры и высшие
спирты. Эти продукты выделяют на разных стадиях очистки
целевых спиртов. Простые эфиры — диэтиловый или диизопро-
пиловый — можно подвергнуть дополнительной очистке и реа-
лизовать как товарные продукты, а полимеры использовать в
качестве топлива. В простейшей схеме выделения и очистки
продуктов гидратации олефинов сначала удаляют основную
часть воды. Это осуществляют путем двухступенчатой ректи-
фикации, причем в первой ректификационной колонне отго-
няются низкокипящие продукты, а во второй — высокойипящие
соединения и большая часть воды. Более чистый спирт полу-
чается в том случае, когда перед первичным концентрированием
производят экстрактивную отгонку спирта с водой. При этом
с головной фракцией отгоняется основная масса побочных про-
дуктов, а отбираемый из средней части колонны разбавленный
водный раствор спирта направляют на концентрирование.
Из колонн окончательного концентрирования спиртов выте-
кают водные азеотропы с весовым содержанием целевого про-
дукта 95,5% в случае этилового спирта (т. кип. 78,2 °C) или
88% в случае изопропилового спирта (т. кип. 80,1°С). Для по-
лучения безводных спиртов эти азеотропы перегоняют с органи-
ческим растворителем, извлекающим из них воду (обычно с бен-
золом).
Области применения
Первая попытка осуществить в промышленном масштабе
примитивный вариант процесса непрямой гидратации этилена
была предпринята в начале XX в. Источником этилена служил
коксовый газ. К 30-м годам этот процесс был уже хорошо
освоен, причем этилен получали из нефтяного сырья. К настоя-
щему времени гидратация этилена как метод производства тех-
нического этанола почти полностью вытеснила ферментацию
углеводного сырья. По себестоимости ферментационный этанол
190
Глава 6
может быть сравним с синтетическим только в том случае, когда
в месте расположения завода есть источник дешевого углевод-
ного сырья или когда его производство субсидируется государ-
ством с целью защиты интересов производителей сельскохозяй-
ственных продуктов. Так обстоит дело, например, в некоторых
странах Западной Европы, особенно Франции и Италии, где
до сих пор существует крупное производство ферментационного
этанола для технических целей.
Метод прямой гидратации этилена был впервые реализован
в промышленности в 1947 г. в США. По мере строительства
новых мощностей и реконструкции старых он постепенно вы-
тесняет сернокислотную гидратацию. Широкие исследования
ведутся по изысканию новых методов гидратации олефинов и
новых катализаторов, но ни одна из этих разработок еще не
нашла применения в промышленности. Поэтому в ближайшем
будущем современный процесс прямой гидратации вряд ли бу-
дет заменен каким-либо другим процессом.
Первоначально этанол использовался преимущественно как
полупродукт для окисления в ацетальдегид и уксусную кислоту.
Однако в настоящее .время потребление его для этой цели со-
кратилось, так как были разработаны альтернативные методы
синтеза указанных продуктов. С другой стороны, возросло при-
менение этанола в качестве растворителя, которое компенсиро-
вало уменьшение его расхода на химические цели. Этанол вхо-
дит в состав лаков, адгезивов, инсектицидных препаратов,
косметических средств, антисептиков и большого числа других
продуктов. В 1968 г. общие мировые (кроме СССР) мощности
по производству этанола превышали 1 млн. т/год.
Промышленное внедрение метода непрямой гидратации про-
пилена состоялось в начале 20-х годов в США. Это было одно
из первых органических производств на базе нефтяного сырья.
Процесс вытеснения непрямой гидратации пропилена прямым
методом протекал медленнее, чем в производстве этанола. В от-
личие от этанола синтез изопропанола в промышленности
проводят не только на фосфорнокислотном катализаторе, но и
на некоторых других, в том числе на восстановленной W2O5.
Согласно патентным данным, окисновольфрамовые контакты
требуют применения более высокого давления и повышенного
отношения водяного пара к пропилену; однако, будучи тверды-
ми, они устойчивы к действию водного конденсата. На боль-
шинстве недавно построенных установок прямой гидратации
пропилена используются фосфорнокислотные катализаторы.
Основная часть изопропанола подвергается дегидрированию
в ацетон. Однако этот метод синтеза ацетона все более теряет
промышленное значение из-за широкого распространения ку-
мольного процесса производства фенола, в котором ацетон полу-
Методы получения нефтехимических полупродуктов 191
чается в качестве сопутствующего продукта (гл. 7). Кроме того,
изопропанол используется как растворитель в производстве кос-
метических средств и при нанесении защитных покрытий, а
Также как антифриз. В 1968 г. суммарные мировые мощности
по получению изопропанола составляли около 1,5 млн. т/год.
Бутанол-2 вырабатывается в небольших количествах путей
непрямой гидратации бутиленов и используется главным обра-
зом для переработки в метилэтилкетон. Пропанол-1 получается
тоже в небольших количествах при сернокислотной гидратаций'
пропилена. Высшие спирты не получают в промышленности ме-
тодом гидратации олефинов. Наибольший интерес представляют
первичные спирты с прямой цепью, необходимые для производ-
ства пластификаторов; однако их нельзя получать путем гидра-
тации олефинов.
ГЛАВА 7
Выбор оптимального метода производства
Оптимальный метод получения данного химического соеди-
нения не всегда бывает технически самым простым. Выбор ме-
тода производства в значительной степени определяется такими
факторами, как цена и доступность исходного сырья, стоимость
побочных продуктов, энергоемкость технологического процесса
и т. п. Относительное значение перечисленных факторов может
изменяться в зависимости от месторасположения завода и ха-
рактера рынка сбыта выпускаемой продукции. В данной главе
рассматриваются альтернативные методы производства пяти
важных полупродуктов, на примере которых показано, как ста-
рые методы вытесняются новыми и какими соображениями
обычно руководствуются при выборе оптимального метода про-
мышленного синтеза.
Хлористый винил
А. Джубб
Jubb А. Н., ICI Corporate Laboratory, Runcorn
Хлористый винил почти целиком расходуется на получение
пластических масс, в основном поливинилхлорида (ПВХ). По-
следний является самым старым из важнейших термопластич-
ных материалов (гл. 8). Хотя полимеры хлористого винила
начали выпускать еще до второй мировой войны, многотоннаж-
ное производство ПВХ стало быстро развиваться лишь 20 лет
назад (рис. 7.1). В 1969 г. мощности по производству хлори-
стого винила в США составляли 2 млн. т/год, а к концу 1973 г.
они увеличились до ~ 3 млн. т/год. Избыток мощностей по
получению этого продукта в США, имевший место на протяже-
нии ряда лет, привел к снижению цены на него с ~ 13 цент/фунт
в начале 50-х годов до 4—5 цент/фунт в 1968 г. *
Вначале хлористый винил вырабатывали на основе ацети-
лена, причем этот метод использовался до конца 50-х годов,
особенно в Западной Европе (табл. 7.1).
HgCI2 на угле
СН=СН + НС1 > СН2=СНС1 АН ~ — 184 кДж/моль
* К концу 1975 г. цена на хлористый винил в США увеличилась до
12,6 цент/фунт [Chem, Marketing Rep., 209, Кг 3, 32 (1976)]. — Прим, перев.
Выбор оптимального метода производства
193
Данный метод хорошо освоен и обеспечивает выход хлористого
винила более 90% при умеренной степени конверсии ацетилена
за проход или около 85% при высокой степени конверсии за
проход.
В основе метода синтеза хлористого винила щелочным де-
гидрохлорированием 1,2-дихлорэтана лежит хорошо изученная
химическая реакция. С развитием нефтехимической промышлен-
ности стал доступен этилен, что стимулировало поиск путей по-
Рис. 7.1. Динамика мирового производства поливинилхлорида.
лучения хлористого винила на его основе. Такие пути были
найдены, и в 50-х годах производство хлористого винила из
этилена было освоено в США, а в 60-х годах — и в других
странах. Опережающие темпы промышленного внедрения ме-
тодов на базе этилена в США были Обусловлены в основном
более низкими ценами на этилен, существовавшими в то время
в этой стране.
Этилен легко хлорируется в жидкой и в газовой фазе с об-
разованием 1,2-дихлорэтана
СН2=СН2 + С12 (газ) — > С1СН2СН2С1 ДЯ = - 201 кДж/моль
Обычно на практике используют жидкофазный метод хло-
рирования. Реакцию проводят в растворе дихлорэтана при
7 Зак. 501
194
Глава 7
Таблица 7.1
Доля мощностей (%) по производству хлористого
.винила из ацетилена в структуре общих мощностей
Страна или регион 1955 г. 1960 г. 1965 г. 1966 г. 1967 г. 1968 г.
США 65 55 41 40 38 26 *
Западная Европа 100 96 75 73 ,62 486
а К 1972 г. эта величина уменьшилась до 10°Л.
б Общие мощности по производству хлористого винила в Западной Европе состав-
ляли в 1968 г. 2,0 млн т/год. {К началу 1976 г они возросли до 4—4,5 млн. т/год (Chem.
Market Abstr., 68, 286 113 (1976)]. — Прим, перев}.
температуре 25—50°C и давлении 0,1—2,0 МН/м2 (1—20 атм)
в присутствии хлоридов металлов. Выход дихлорэтана достигает
95% при степени конверсии этилена 98%.
Одним из первых промышленных процессов превращения ди-
хлорэтана в хлористый винил был пиролиз. Экономическое пре-
имущество этого метода состояло в отсутствии затрат щелочи
и связанного с ее использованием лишнего расхода хлора, ко-
торый составляет один атом на каждую молекулу хлористого
винила.
С12
4
НС1
t
СН2=СН2 ------> хлоратор
С1СН2СН2С1
печь
пиролиза
---> СН2=СНС1
Пиролиз проводят при температуре около 500°C над пемзой
или каолином. В этих условиях дихлорэтан дегидрохлорируется
в хлористый винил с выходом 95%, причем степень конверсии
дихлорэтана за проход поддерживают на уровне не выше 50%.
В качестве побочного продукта образуется хлористый водород,
что во многих случаях вызывает трудности, связанные с его
утилизацией. Существует три пути решения этой проблемы. Пер-
вый из них заключается в кооперировании заводов по производ-
ству хлористого винила из этилена и из ацетилена таким обра-
зом, что последний работает на хлористом водороде, поступаю-
щем с первого. Этот метод лежит в основе так называемого
«сбалансированного» процесса
СН3=СНС1
Выбор оптимального метода производства
195
Логическим развитием описанного метода является процесс,
базирующийся на пиролизе нафты с получением этилена и аце-
тилена в соотношениях, необходимых для сбалансированного
производства хлористого винила. Например, в процессе фирмы
Societe Beige de 1’Azote нафту пиролизуют в эквимолярную
смесь этилена и ацетилена, которую очищают от нежелатель-
ных примесей и вводят в реакцию с хлористым водородом, об-
разующимся при пиролизе дихлорэтана. При этом ацетилен пре-
вращается в хлористый винил. Последний отделяют от этилен-
содержащего газа, который подвергают хлорированию, получая
дихлорэтан (схема 1). Еще одним вариантом этого метода яв-
ляется процесс фирмы Kureha (Япония).
Схема I.
Второй путь решения проблемы использования хлори-
стого водорода в производстве хлористого винила из этилена
заключается в окислении хлористого водорода в хлор либо
196
Глава 7
электрохимически, либо с помощью усовершенствованного про-
цесса Дикона. Попытки улучшить метод Дикона
соли Си или Fe
4НС1 + °* 400^00^ 2CI2 + 2H2O
предпринимались неоднократно, и по этому вопросу имеется
обширная патентная литература. Однако он так и не был реа-
лизован в промышленном масштабе, хотя фирма Shell объявила
о создании перспективного технологического процесса. В при-
сутствии углеводорода реакция протекает легче и приводит к
образованию соответствующего хлорпроизвод'ного. Первым про-
мышленным процессом, в котором был использован этот прин-
цип (так называемое окислительное хлорирование), был про-
цесс производства фенола, предложенный фирмой Raschig.
Окислительное хлорирование представляет собой третий спо-
соб -утилизации хлористого водорода, выделяющегося при хло-
‘ рировании этилена. Этот метод получения хлористого винила из
этилена привлек к себе большое внимание лет 15 назад, а уже
приблизительно в 1964 г. он использовался на заводах. Прин-
ципиальная схема процесса приведена на схеме 2.
2СН2=СН2 + 4НС1 + О2 -►
СН2=СН2
| ыздух
реактор для
окислительного
хлорировали я
2СН2С1СН2С1 + 2Н2О ДН = -238 кДж/моль
C2H4CI2 [ печь
I пиролиза
CH2=CHCi
НС1
HCI
Схема 2.
Катализатор окислительного хлорирования этилена содержит
хлорную медь, осажденную на инертном носителе. Процесс про-
водят при температуре 240—320 °C; сообщается, что выход хло-
ристого винила достигает 90—95%. Реакция весьма экзотер-
мична, вследствие чего необходимо отводить выделяющееся
тепло. В условиях реакции соль меди способна улетучиваться,
что значительно сокращает срок службы катализатора. Это за-
труднение, как сообщается, можно преодолеть путем добавления
к хлорной меди солей щелочных металлов. Указанный прием
(или какие-то другие приемы) позволил нескольким фирмам
разработать катализаторы с большим сроком службы. В зави-
симости от доступности исходного сырья возможны различные
Выбор оптимального метода производства
197
варианты процесса получения хлористого винила (этиленф-хлор,
этилен + хлор + хлористый водород и т. д.).
Один из возможных путей развития описанных методов за-
ключается в создании одностадийного процесса производства
хлористого винила из этилена без промежуточного выделения
дихлорэтана. Например, можно попытаться совместить в одном
реакторе хлорирование этилена и дегидрохлорирование дихлор-
этана
2СН2=СН2 + 2НС1 + О2 —> 2СН2=СНС1 4-2Н2О
Эта идея разрабатывалась в большом числе патентов и ожив-
ленно обсуждалась в литературе. Однако она до сих пор не
реализована в промышленности, что, по-видимому, объясняется
существенным различием между оптимальными условиями обеих
реакций, следствием которого являются низкие выходы хлори-
стого винила.
Недавно фирма The Lummus Со. объявила о разработанной
ею технологии получения хлористого винила путем каталити-
ческого хлорирования этана в одном реакторе, в котором одно-
временно осуществляют стадии хлорирования, окислительного
хлорирования и дегидрохлорирования (так называемый «транс-
кат-процесс»). Подробности о составе катализатора не приводят-
ся; лишь в одном из патентов упоминается о применении в ка-
честве катализатора смеси хлоридов меди и калия. Выходы
хлористого винила превышают 80% по этану и 95% по хлору.
Удельная себестоимость продукта примерно на 8,6 ф. ст./т
ниже, чем в случае производства из этилена, что обусловлено
Таблица 7.2
Сравнительная оценка стоимости исходного сырья
(ф ст./т хлористого винила) при получении хлористого винила
разными методами (в ценах 1972 г.)
Сырье Процесс А а Процесс Б Транскат’ процесс
Ацетилен по цене 69 ф. ст/т 14,6
Этилен по цене 28 ф. ст./т 6,9 13,8 —
Этан по цене 8,6 ф. ст./т — — 5,2
Хлор по цене 23,7 ф. ст./т 14,6 15,5 13,8
Общая стоимость сырья 36,1 29,3 19,0
а Комбинация хлорирования этилена и гидрохлорирования ацетилена.
6 Комбинация хлорирования и окислительного хлорирования этилена.
198
Глава 7
главным образом меньшей стоимостью этана по сравнению с
этиленом (табл. 7.2).
С,Н6
С1г
Воздух
СН2=СНС1
рециркуляция тяжелых
хлоруглеводородов и смол
В табл. 7.3 приведены данные о структуре себестоимости и
затратах производства при получении хлористого винила раз-
личными методами исходя из ацетилена и этилена.
Кроме описанных выше, существует еще два метода синтеза
хлористого винила: окислительное хлорирование этилена хло-
ристым аммонием, предложенное в нескольких патентах, и
окисление хлористого этила над окисью хрома, осажденной на
окиси алюминия (этот метод заявлен только в одном патенте).
Однако данные о создании на базе этих реакций перспективных
промышленных процессов пока отсутствуют.
Фенол
М. Дилке
Dilke М. Н., Research and Development Department,
BP Chemicals International Limited, Epsom
Хотя небольшое количество фенола до сих пор вырабаты-
вается из каменноугольной смолы, подавляющая часть общего
объема мирового производства фенола (около 3,3 млн. т/год)
получается синтетическими методами. Синтез фенола является,
пожалуй, наиболее интересным из химических производств, так
как здесь существует наибольшее число альтернативных воз-
можностей по сравнению с любым другим многотоннажным хи-
мическим полупродуктом. В настоящее время используется
шесть, а может быть, и семь промышленных методов получения
фенола. Все эти методы рассматриваются ниже. Одни из них
названы по исходному веществу, другие — по промежуточному
продукту или какой-либо стадии процесса; часто наименование
метода связывают также с названием фирмы, которая его
Таблица 7.3
Сравнительная оценка удельных затрат (ф. ст./т) для различных процессов
получения хлористого винила (цены 1972 г., при масштабе производства 100 000 т/год)
Затраты Гндрохло- рнрование карбидного ацетилена Гидрохло- рнрование ацетилена, полученного нз нафты Сбалансиро- ванный про- цесс на базе карбидного ацетилена Окисли- тельное хлорирование этилена Комбиниро- ванный про- цесс с исполь- зованием смеси ацетиле- на и этилена
Капитальные затраты, млн. ф. ст. Стоимость основных видов исходного 2,10 6,90 2,76 3,36 5,41
сырья:
хлор по цене 24,4 ф. ст./т 15,2 15,7 15,2 15,7 15,7
карбид по цене 28 ф. ст./т 38,2 — 18,1 — —-
нафта по цене 10,1 ф. ст./т —- 9,1 — — 9,1
этилен по цене 35,5 ф. ст./т — — 8,1 17,0 —
Итого 53,4 24,8 41,4 32,7 24,8
Энергетические затраты и стоимость 3,2 7,6 4,4 3,8 7,0
вспомогательных химикатов
Трудовые затраты 1,9 1,4 1,5 0,8 1,4
Текущие расходы
ремонт оборудования и зданий 0,9 2,8 1,1 1,4 2,2
налоги и страхование 0,4 1,4 0,6 0,6 1,1
прочие непрямые затраты 0,4 1,4 0,6 0,6 1,1
амортизационные отчисления 2,4 8,0 3,1 3,9 6,2
возмещение капитальных затрат 2,2 7,1 2,8 3,4 5,6
Итого 6,3 20,7 8,2 9,9 16,2
Общие затраты на производство, нклю- 64,8 54,5 55,5 47,2 49,4
чая возмещение капитальных затрат
200
Глава 7
разработала. Применяемые в этой главе сокращенные названия
методов производства приводятся в скобках в заголовках соот-
ветствующих разделов.
Щелочное плавление бензолсульфокислого натрия
(метод сульфирования)
Этот метод синтеза фенола является самым старым (первый
завод, на котором он применялся, был построен в Германии в
1890 г.). Процесс включает четыре стадии:
СвНв 4- H2SO4 —> C6H6SO3H 4- Н2О
2СвН650зН + Na2SO3 —> 2CeH6SO3Na 4- SO2 + Н2О
CeH6SO3Na + 2NaOH —> CeH6ONa + Na2SO3 + H2O
2C6H6ONa+SO2 + H2O —> 2C6H6OH + Na2SO3
Первая реакция идет до конца только при непрерывном удале-
нии выделяющейся воды (в одном .из вариантов ее отгоняют
в виде азеотропа с бензолом). На второй стадии всегда полу-
чается также небольшое количество сульфата натрия, что обус-
ловлено применением избытка серной кислоты. Сплавление
бензолсульфокислого натрия с каустической содой проводят при
температуре около 350 °C. Превращение фенолята натрия в фе-
нол осуществляют с помощью двуокиси серы, которая обра-
зуется на второй стадии. Фенол получается в виде водного рас-
твора, из которого его выделяют дистилляцией.
Щелочной гидролиз хлорбензола
(процесс фирмы Dow)
Этот процесс, разработанный фирмой Dow Chemical Com-
pany, включает две стадии:
СбНв + СЬ —* СвНБС1 + НС1
C5H5CI + NaOH —> С6НБОН + NaCl
Хлорирование проводят при температуре ~ 40 °C в присутствии
солей трехвалентного железа, уменьшающих образование ди- и
трихлорбензолов. Монохлорбензол выделяют дистилляцией и
подвергают гидролизу при температуре около 350 °C и давлении
~ 30 МН/м2 (300 атм). Образующиеся фенолят и хлористый
натрий содержатся в нижнем водном слое; верхний слой пред-
ставляет собой дифениловый эфир, который возвращают* в цикл.
Для выделения фенола из фенолята последний подкисляют хло-
ристым водородом, образующимся на первой стадии.
Выбор оптимального метода производства
201
Парофазный гидролиз хлорбензола
(процесс фирм Raschig — Hooker)
В этом единственном парофазном процессе производства фе-
нола протекают следующие реакции:
CeH6 + HCI + 102 —> С6Н6С1 + Н2О
С6Н5С1+Н2О —> С6Н6ОН + НС1
Суммарное уравнение имеет вид
С6Нв + уО2 —> С6Н6ОН
Процесс был разработан немецкой фирмой Raschig приблизи-
тельно в 1930 г. на основе известных к тому времени реакций
и впоследствии значительно усовершенствован американской
фирмой Hooker Chemical Company.
Смесь паров бензола с хлористым водородом, воздухом и
водяным паром пропускают над катализатором (например, си-
стемой А12О3—СиС12—FeCl3) при температуре около 230 °C. При
этом ~ 98% хлористого водорода и ~ 10% бензола превра-
щается в хлорбензол (селективность реакции 90%). Остальной
превращенный бензол хлорируется до дихлорбензолов или сго-
рает. В первоначальном процессе фирмы Raschig образовав-
шийся хлорбензол отделяли от других продуктов реакции и
подвергали гидролизу в паровой фазе над фосфатнокальциевым
катализатором при температуре 420 °C. Степень конверсии хлор-
бензола за один проход составляла 10—15%, причем Около
90—95% его превращалось в фенол, а остальное количество —
в бензол. В процессе фирмы Hooker был использован усовер-
шенствованный катализатор, обеспечивающий превращение в
фенол как хлорбензола, так и дихлорбензолов, что делает не-
нужным их разделение. В ректификационной системе, состоя-
щей из четырех колонн, продукты реакции разделяют и полу-
чают фенол, соляную кислоту, бензол,, хлорбензол, дихлорбен-
золы и смолы. Эти продукты, за исключением фенола и смол,
возвращают в соответствующие аппараты установки.
Разложение гидроперекиси кумола
(кумольный метод)
СвН6СН(СН3)2 + О2 —> С6Н6С(СН3)2ООН —> С6Н6ОН + (СН3)2СО
Этот метод, который был независимо друг от друга разра-
ботан английской фирмой Distillers. Company Ltd. и американ-
202
Глава 7
ской фирмой Hercules Powder Company *, подробно описан в
гл. 5.
Окисление толуола (толуольный метод)
В основе этого метода лежат следующие реакции:
2СвН6СН3 + ЗО2 —> 2С6Н5СООН + 2Н2О
2С6Н6СООН + у ог С6Н6СООС6Н4СООН-о —>
—> С6Н5ОН + СвН5СООН + СОг
Суммарное уравнение имеет вид
С6Н6СНз + 2О2 —> С6Н5ОН + СО2 + Н2О
Впервые жидкофазное окисление толуола в бензойную кислоту
было осуществлено в промышленном масштабе в Германии во
время второй мировой войны. Толуол окисляли воздухом при
температуре 140 °C и давлении около 0,3 МН/м2 (3 атм) в при-
сутствии солей кобальта. Из сырого продукта, содержащего
около 50% бензойной кислоты, извлекали эту кислоту и не-
превращенный толуол, а бензиловый спирт или бензальдегид
возвращали на стадию окисления.
Процесс каталитического окисления бензойной кислоты в фе-
нол в присутствии солей меди, разработанный независимо друг
от друга фирмами Dow Chemical Company и California Research
Corporation, включает промежуточное образование бензоилсали-
циловой жислоты, которая далее гидролизуется и декарбоксили-
руется, образуя фенол и бензойную кислоту. По этому методу
бензойную кислоту, содержащую бензоаты меди (катализатор)
и магния (промотор), нагревают с водяным паром и цоздухом
до температуры 230—240 °C под давлением 35—70 кН/м2 (0,35—
0,7 атм). Пары фенола и бензойной кислоты непрерывно выво-
дят из реактора и подают-в ректификационную колонну, из
верхней части которой отбирают фенол и воду, а из куба —
непревращенную бензойную кислоту, возвращаемую на окисле-
ние. Одновременно из реактора выводят часть жидкого окси-
дата, который отделяют от смолистых веществ и направляют на
рециркуляцию.
Дегидрирование смеси циклогексанона
и циклогексанола (циклогексановый метод)
Данный метод был разработан фирмой Scientific Design
Company, почему его иногда называют методом этой фирмы.
>* См. прим, перев. на стр. 157,
Выбор оптимального метода производства 203
В основе этого метода лежат следующие реакции:
CqHq + ЗН2 -> С6Н,2
С6Н12 + Ог -> CjHjgO + Н2О
СвН]2 + "g-О2 —> СвНцОН
СвН10О —> С6Н5ОН + 2Н2
С6НнОН —> C6HSOH + 3H2
Гидрирование бензола в циклогексан хорошо изучено, а окис-
ление циклогексана в смесь циклогексанона и циклогексанола
является одной из стадий синтеза капролактама (стр. 223).
Фирма Scientific Design Company осуществила глубокую тех-
нологическую проработку обеих реакций вплоть до создания на
их основе промышленного процесса. В частности, этой фирмой
был разработан усовершенствованный катализатор для заклю-
чительной стадии дегидрирования.
Прямое окисление бензола
Несмотря на многочисленные попытки непосредственного
окисления бензола в фенол, преодолеть возникающие при этом
трудности, связанные с образованием бифенилов и родственных
соединений и глубоким окислением бензола, так и не удалось.
Было испытано большое число инициаторов и промоторов, вклю-
чая у-облучение; однако создание удовлетворительной техноло-
гии оказалось пока невозможным, хотя эта цель, безусловно,
весьма заманчива. В последние годы основное внимание уде-
ляется окислению бензола воздухом или кислородом (обычно
под давлением) в присутствии уксусной кислоты и катализа-
тора типа промотированной палладиевой соли с получением
смеси фенола и фенилацетата. Реакцию ведут как в жидкой,
так и в паровой фазе. Несмотря на некоторые успехи, достиг-
нутые в этом направлении, промышленный процесс прямого
окисления бензола в фенол еще не создан.
Сравнительная оценка различных методов
производства фенола
Хотя метод сульфирования и процесс фирмы Dow исполь-
зуются все в меньшей степени, до сих пор в различных странах
мира фенол получают всеми шестью описанными выше мето-
дами. Это свидетельствует о том, что выбор наилучшего метода
во многом определяется местными условиями. Среди факторов,
которые приходится принимать во внимание при сопоставлении
альтернативных методов синтеза, необходимо назвать стоимость
204
Глава 7
исходного сырья, мощность установки, капитальные затраты,
ценность побочных продуктов и степень чистоты целевого про-
дукта.
Общей чертой большинства методов производства , фенола
является то, что исходным сырьем служит бензол или продукты
его переработки. Однако толуол, применяемый в толуольном ме-
тоде, представляет собой более дешевое сырье, чем бензол.
В двух случаях используется сравнительно дорогостоящий хлор.
Если в циклогексановом методе исходят из смеси циклогекса-
нола с циклогексаноном, то себестоимость этой смеси можно
понизить, получая ее одновременно для производства фенола
и производства капролактама. Общий выход фенола по бензолу
для всех бензольных методов (исключая прямое окисление бен-
зола) составляет 80—90%, тогда как для толуольного метода
выход фенола по толуолу, вероятно, не превышает 70—75%.
Методы, включающие хлорирование, требуют применения спе-
циальных конструкционных материалов, устойчивых к коррозии,
что повышает капитальные затраты на строительство установки.
Единственный метод, в котором получается ценный побоч-
ный продукт, — это кумольный. Поскольку в нем на 1 кг фе-
нола образуется 0,6 кг ацетона, общие экономические показа-
тели данного процесса в значительной степени зависят от цены
на ацетон. Однако возможность реализации побочных продук-
тов играет важную роль и в экономике других процессов. Так,
при использовании метода сульфирования существенное значе-
ние имеет утилизация сульфита натрия (например, для нужд
целлюлозно-бумажного производства). В процессе парофазного
гидролиза хлорбензола фирмы Raschig побочными продуктами
являются полихлорбензолы. В процессе фирмы Hooker они гид-
ролизуются, что снимает проблему утилизации отходов произ-
водства. Дифениловый эфир (побочный продукт процесса щелоч-
ного гидролиза хлорбензола) и бензойную кислоту (побочный
продукт толуольного процесса) можно либо сбывать как товар-
ные продукты, либо возвращать в цикл. В том случае, когда
исходным сырьем в циклогексановом процессе служит цикло-
гексан, получающийся водород можно рассматривать как побоч-
ный продукт.
Современные методы производства фенола (такие, как ку-
мольный) обеспечивают получение товарного продукта очень
высокой степени чистоты — например, такой, какая необходима
для синтеза капролактама.
Первый завод по производству фенола кумольным методом
был построен в 1953 г.*, а вскоре после этого появилось боль-
шое число других таких же заводов. С того времени экономи-
* См. прим, перев. на стр. 157.
Выбор оптимального метода производства 205
ческие показатели процессов синтеза фенола по методу фирм
Raschig—Hooker, а также толуольному и циклогексановому ме-
тодам значительно улучшились. Однако строительство заводов,
использующих кумольный метод, продолжается, и в настоящее
время на их долю приходится 80% мирового производства син-
тетического фенола.
Основная часть фенола до сих пор расходуется на производ-
ство фенольных смол. Проведенный недавно анализ структуры
потребления фенола в Западной Европе дал следующие резуль-
таты: производство фенольных смол — 38%, синтез капролак-
тама— 25%, синтез адипиновой кислоты—18%, получение ди-
фенилолпропана— 13%, производство алкилфенолов — 4%, про-
чие цели— 2%.
Акрилонитрил
А. Джубб
Jubb А. Н., ICI Corporate Laboratory, Runcorn
Первое промышленное производство акрилонитрила было
организовано в Германии незадолго до второй мировой войны,
причем в военные годы установки по его получению на заводах
в Людвигсхафене и Леверкузене (общая мощность обеих уста-
новок составляла около 1000 т/год) обеспечивали всю потреб-
ность страны в нитрильном каучуке. Во время второй мировой
войны выпуск нитрильного каучука был налажен также в США.
С появлением на текстильном рынке в начале 50-х годов поли-
акрилонитрильных волокон спрос на акрилонитрил значительно
возрос. Так, общие мировые мощности по производству акри-
лонитрила увеличились с 20 тыс. т/год в 1950 г. до 1,5 млн. т/год
в 1970 г. (в 1972 г. они составляли, по оценке 2,3 млн. т/год*).
Первоначальный метод получения акрилонитрила, реализо-
ванный в Германии, был основан на взаимодействии ацетилена
и синильной кислоты. На первом американском заводе, постро-
енном в 1940 г. фирмой American Cyanamid в шт. Нью-Джерси,
исходным сырьем служили окись этилена и синильная кислота.
Этот метод был использован также на заводе фирмы Union
Carbide. Однако все другие заводы, построенные в различных
странах мира в течение 50-х годов, ориентировались на ацети-
леновый процесс производства акрилонитрила. В 1960 г. всту-
пил в строй завод фирмы Sohio в г. Лима (шт. Огайо, США),
на котором акрилонитрил вырабатывали путем окислительного
аммонолиза пропилена. В дальнейшем большинство новых
* В конце 1975 г. мировые мощности по производству акрилонитрила
были равны 2876 тыс. т/год [Chem. Industrie, 28, 28 (1976)].—Прим, перев.
206
Глава 7
мощностей по производству акрилонитрила во всем мире строи-
лось по этому методу, большей частью в технологическом варианте
фирмы Sohio. Вместе с тем несколько фирм разработали и реа-
лизовали свои аналогичные процессы (гл. 6). Многие заводы,
на которых акрилонитрил получали на основе ацетилена (в ча-
стности, в Западной Европе), были впоследствии закрыты.
Фирма du Pont запатентовала процесс производства акрило-
нитрила путем взаимодействия пропилена с окисью азота, ко-
торый можно рассматривать как двухстадийный вариант про-
цесса фирмы Sohio. Имеется много сообщений о том, что в
1962 г. фирма du Pont построила в г. Бьюмонте (шт. Техас,
США) завод, работающий по данному методу. Однако пола-
гают, что к настоящему времени этот завод переоборудован на
стандартный процесс окислительного аммонолиза (если на нем
вообще когда-либо использовалась другая технология). Фирма
Knapsack-Griesheim (филиал фирмы Hoechst) разработала про-
цесс получения акрилонитрила из ацетальдегида и синильной
кислоты и провела его технико-экономическое обследование на
опытно-промышленной установке мощностью 350 т/год. И в этом
случае поступившие вначале сообщения о строительстве про-
мышленной установки, работающей по новому методу, впослед-
ствии не подтвердились.
Недавно японская фирма Asahi Chemical Industry Со. сооб-
щила о создании процесса синтеза акрилонитрила из этилена и
синильной кислоты. Кроме того, фирмы ICI и Power Gas Ltd.
объявили о совместной разработке ими процесса производства
акрилонитрила окислительным аммонолизом пропана.
Ацетиленовый метод
CHsCH + HCN —> ch2=chcn
Реакцию можно проводить либо в паровой фазе при тем-
пературе 400—500 °C над катализатором, содержащим цианид
металла, либо в жидкой фазе — водном растворе с pH 1—3,
содержащем каталитическую систему Си2С1г—НС1. Парофазный
процесс дает плохие выходы акрилонитрила, тогда как в жидко-
фазной реакции выход по ацетилену достигает 80%. Ацетилен
и синильную кислоту (мольное соотношение 6: 1 —10: I) барбо-
тируют через водный раствор катализатора при температуре
70 °C и давлении, близком к атмосферному. Реакционные газы
промывают в противотоке водой, получая акрилонитрил в виде
3%-ного водного раствора. Этот раствор подвергают перегонке
с водяным паром. Дистиллят после его охлаждения разделяется
на два слоя. Акрилонитрил-сырец очищают ректификацией, а
нижний водный слой возвращают на орошение в отпарную ко-
лонну.
Выбор оптимального метода производства 207
Окислительный аммонолиз пропилена
2СН2=СНСН3 + 2NH3 + ЗО2 '—> 2CH2=CHCN + 6Н2О
Подробное описание этого процесса приведено в гл. 6.
Реакция окиси этилена с синильной кислотой
Н2С—СН2 + HCN —> HOCH2CH2CN —-* CH2=CHCN
—Н2О
о
Взаимодействие синильной кислоты и окиси этилена с обра-
зованием этиленциангидрина является экзотермической реак-
цией, протекающей в присутствии щелочного катализатора, на-
пример диэтиламина. Реакцию проводят в аппаратах периоди-
ческого или непрерывного действия в растворе этиленциангид-
рина или воды. Сообщается, что выход циангидрина составляет
95%. После очистки путем дистилляции в вакууме продукт
подвергают дегидратации в газовой фазе в реакторе непрерыв-
ного действия при температуре 250—350 °C над активной окисью
алюминия. Возможна также жидкофазная дегидратация эти-
ленциангидрина при температуре 200 °C в присутствии формиа-
та натрия или окиси кальция. Выход акрилонитрила по этому
методу, как сообщают, равен 90%.
Реакция ацетальдегида с синильной кислотой
Синильная кислота присоединяется к ацетальдегиду при тем-
пературе 10—20 °C, давая циангидрин ацетальдегида с выхо-
дом 90%. Дегидратация последнего соединения при температуре
600—700°C над фосфорно-кислотным катализатором приводит
к образованию акрилонитрила с выходом около 90%.
СН3СНО + HCN —> СН3СН(ОН)СК ——> ch2=chcn
—Н2О
Реакция пропилена с окисью азота
4СН2=СНСН3 + 6NO —> 4CH2=CHCN + N2 + 6Н2О
Этот метод описан только в патентной литературе. Реакцию
ведут при температуре 700°C над серебряным катализатором
на носителе.
Окислительный аммонолиз пропана
СН3СН2СН3 + МН3 + 2О2 —> .CH2=CHCN 4- 4Н2О
Разработка технологии процесса осуществлялась совместно
фирмами Power Gas Со и ICI Corporate Laboratory. Несмотря
208
Глава 7
на то что полностью она еще не завершена, продажа лицензий
на этот процесс уже началась. Условия проведения реакции в
общем почти такие же, как в процессе фирмы Sohio, за исклю-
чением катализатора, состав которого не сообщается. По неко-
торым данным, выход акрилонитрила близок к 65%; выходы
побочных продуктов — ацетонитрила и синильной кислоты —
примерно такие же, как при окислительном, аммонолизе пропи-
лена. Этот процесс имеет потенциальное значение для районов,
где много дешевого природного газа и отсутствует в достаточ-
ных количествах пропилен.
Реакция этилена с синильной кислотой
В 60-х годах японская фирма Asahi Chemical Industry Со.
начала патентовать различные варианты процесса получения
акрилонитрила из этилена и пропионитрила. Недавно ею был
разработан метод синтеза акрилонитрила, основанный на окис-
лительном цианировании этилена
2СН2=СН2 + 2HCN + О2 —> 2CH2=CHCN + 2Н2О
Эту реакцию проводят при температуре около 360 °C в присут-
ствии хлористого водорода в стационарном слое палладий-це-
зий-ванадиевого катализатора при низкой степени конверсии
этилена за один проход и 100%-ной степени конверсии синиль-
ной кислоты. Выход акрилонитрила составляет 74% по этилену
и 88% по синильной кислоте. Основными побочными продук-
тами являются ацетонитрил, окись углерода, двуокись углерода,
хлорпропионитрил, дихлорэтан и хлористый винил.
Сравнительная экономическая оценка
различных методов получения акрилонитрила
В конце 50-х годов средняя цена на акрилонитрил, выраба-
тываемый в США, составляла 26—29 цент/фунт при цене на
ацетилен около 11—13 цент/фунт. Пропилен же в то время стоил
около 2 цент/фунт. Несмотря на то что технология производства
акрилонитрила из пропилена сложнее (и, следовательно, стои-
мость оборудования выше), чем из ацетилена, более низкая
стоимость исходного сырья с лихвой компенсирует этот недоста-
ток.
Указанное обстоятельство нашло свое отражение в падении
цен на акрилонитрил в США с 22 цент/фунт в 1960 г. до
14 цент/фунт и ниже в 1962 г., что было связано с широким вне-
дрением окислительного аммонолиза пропилена. В настоящее
Выбор оптимального метода производства
209
время * оптовая цена на акрилонитрил, полученный этим мето-
дом, вероятно, близка к 12 цент/фунт, тогда как при получении
акрилонитрила из ацетилена, окиси этилена или ацетальдегида
только стоимость исходного сырья составила бы 8—11 цент/
/фунт**. Поэтому ни один из методов, базирующихся на трех по-
следних видах сырья, не может конкурировать с окислительным
аммонолизом пропилена, хотя в определенных условиях заводы,
использующие ацетиленовый метод, могут еще работать. В За-
падной Европе и Японии наблюдается почти такая же картина,
как в США, однако здесь значительные мощности по производ-
ству акрилонитрила появились только после 1960 г., а пропиле-
новый метод получил широкое распространение приблизительно
после 1965 г. Следствием его экономических преимуществ яви-
лось происшедшее за последние годы перераспределение доли
различных методов в структуре мощностей по получению акри-
лонитрила (табл. 7.4).
Таблица 7.4
Динамика изменения структуры мировых мощностей
по производству акрилонитрила (% акрилонитрила,
вырабатываемого различными методами)
Метод получения 193Э г. 1955 г I960 г. 1965 г 1967 г. 1971 г.
Окись этилена — HCN 95 35 12 6 M.1.W
Ацетилен — HCN 5 65 80 40 18 7
Пропилен — ЫН3 — — 8 45 75 93
Пропилен — NO — — — 9а 7 а
Ацетальдегид — HCN — — — —- —
а Данные о том, что азота, сомнительны. на этом заводе акрилонитрил получали из пропилена н окиси
Промышленное использование окислительного аммонолиза
пропана возможно лишь при значительной (не менее 10 ф. ст./т)
разнице в ценах на пропан и пропилен, что в ближайшем
будущем вряд ли будет иметь место, если учесть ожидаемое
сокращение добычи природного газа в США. Очень трудно прог-
нозировать также промышленное будущее процесса фирмы Asahi
(окислительного цианирования этилена), так как при отсутствии
подробной технологический схемы невозможно оценить капи-
тальные затраты на строительство установки. По-видимому, он
не будет обладать экономическими преимуществами перед
* В 1972 г. — Прим, перев.
** Цены иа акрилонитрил, пропилеи, ацетилен, окись этилена и ацеталь-
дегид в январе 1976 г.: 24, 7,5, 49,5, 26 и 16 цеит/фуит соответственнс* [Chem,
Marketing Rep., 209, № 3, 34 (1976)]. — Прим. перев.
210
Глава 7
пропиленовым методом, даже если в нем будет использоваться
дешевая синильная кислота, поступающая с установок окисли-
тельного аммонолиза пропилена.
Ацетон
Б. Иоумэнс
Yeomans В., Research and Development Department,
BP Chemicals International Limited, Hull
Важное значение ацетона в мировой экономике находит свое
отражение в быстром изменении характера исходного сырья для
его производства (от натурального к нефтехимическому) и в
разнообразии промышленных методов его получения, которые
разрабатывались на протяжении XX в.
Первоначально ацетон вырабатывали путем сухой перегонки
древесины. Однако этот метод не смог удовлетворить возрос-
ший во время первой мировой войны спрос на ацетон, который
тогда использовали в качестве растворителя в производстве
аэролака для крыльев самолетов и при получении бездымного
пороха (кордита). Поэтому был разработан метод синтеза аце-
тона из ферментационного этанола, который окисляли в уксус-
ную кислоту с. последующей нейтрализацией кислоты известью
и пиролизом полученной кальциевой соли
400 °C
(СН3СОО)2Са ----> (СНзЪСО + СаСОз
Как и следовало ожидать, на смену этому методу, в котором
из двух молекул уксусной кислоты образуется только одна мо-
лекула ацетона, вскоре пришел другой метод—ацетоновая
ферментация углеводного сырья, например мелассы (процесс
Вейзманна). Отличительной особенностью нового метода было
одновременное получение ацетона и бутанола-1 в равном весо-
вом соотношении. Процесс состоял из четырех стадий: выращи-
вание культуры, стерилизация мелассы, ферментация и дистил-
ляция. к стерилизованной глюкозе добавляли чистую культуру
микроорганизма, выращивали ее в течение суток при темпера-
туре 30 °C и переносили в стерилизованный раствор, содержа-
щий мелассу, сульфат аммония и буфер. В процессе фермента-
ции, продолжающемся приблизительно 2 суток, pH поддер-
живали на уровне 5—7 путем добавления водного аммиака.
Реакционную массу с концентрацией продуктов около 2%
перегоняли с водяным паром и получали дистиллят, содержа-
щий 50% продуктов. Обычно выход ацетона и бутанола-1 со-
ставлял 28—33% от веса сахара, присутствующего в мелассе.
Выбор оптимального метода производства 211
Этот метод производства ацетона доминировал до середины
1920-х гг. Однако в 1920 г. фирма Standart Oil Company of
New Jersey начала вырабатывать изопропанол путем гидрата-
ции пропилена (этот процесс считается первым нефтехимиче-
ским производством в мире). Естественно, что вскоре был раз-
работан метод окисления изопропанола в ацетон, который до
сих пор остается наиболее важным промышленным методом
получения ацетона. Синтез ацетона из изопропанола осуществ-
ляют либо прямым дегидрированием последнего, либо его окис-
лительным дегидрированием (гл. 4).
Дегидрирование изопропанола
Катализаторами этой реакции могут служить медь и никель,
а также их сплавы, окислы и соли. Однако самые лучшие ре-
зультаты дает использование окисноцинкового катализатора
(например, 7% ZnO и 2% Na2CO3 на пемзе). Основными побои
ными реакциями являются образование пропилена и диизопрс-
лилового эфира. Окисноцинковый катализатор требует приме-
нения более высокой температуры, чем медный или никелевый,
однако он обеспечивает более высокие выходы (90%-ную кон-
версию изопропанола в ацетон при селективности 98%). Роль
пемзы заключается лишь в увеличении поверхности окиси
цинка.
ZnO—пемза
(СН3),СНОН ---------> (СНз)2СО + Н2
v 400—500 °C ' V
АН = + 66,4 кДж/моль при 327 °C
Окисноцинковый катализатор имеет срок службы 1—1,5 го-
да, но нуждается в периодическом (через каждые 10—14 дней)
выжигании кокса и других органических отложений горячим
воздухом с температурой около 500 °C.
Окислительное дегидрирование изопропанола
(СН3)2СНОН + J о2 —> (СН3)2СО + Н2О
АД = — 182 кДж/моль при 295 °C
В качестве катализатора можно использовать никель или
платину. Однако обычно применяют серебряный или медный
катализатор и реакцию ведут при температуре 400—600 °C. В от-
личие от прямого дегидрирования процесс сильно экзотермичен
и селективность по ацетону несколько ниже (85—90%).
Промышленное значение метода Вейзманна еще больше
снизилось, когда был разработан еще один катализатор для
химического превращения ферментационного этанола в ацетон,
212
Глава 7
а именно система БеаОз— ZnO — СаСОз (например, в соотно-
шении 2:7:1 по весу). В присутствии этого катализатора кето-
низацию спирта ведут в газовой фазе при температуре 400—
450 °C. Данный метод использовался в основном до конца
второй мировой войны; в настоящее время он находит приме-
нение только в развивающихся странах (например, Пакистане),
где имеются местные источники дешевого ферментационного
спирта.
Вскоре после окончания второй мировой войны был разрабо-
тан кумольный метод производства фенола, в котором на одну
молекулу фенола образуется одна молекула ацетона (гл. 5).
По существу здесь ацетон получается также из пропилена.
н+ 02
СН3СН=СН2 + СаНв ----> СвН6СН(СН3)2 ~ г~~ *
—> С6Н5С(СН3)2ООН —-------—-----' > С6Н6ОН + (СНз)2СО
10%-ная (по весу) H2SO4,
~60°С
Этот метод в настоящее время является главным конкурентом
изопропанольного метода, поскольку на его долю приходится
свыше 70% объема мирового производства фенола.
В других сравнительно новых процессах ацетон вырабаты-
вают как ценный сопутствующий продукт при производстве не-
которых кислородсодержащих соединений, например уксусной
кислоты, путем окисления парафинов. Промышленное значение
приобрели два таких процесса: парофазное окисление пропан-
бутановой фракции природного газа (процесс фирмы Celanese,
который был осуществлен на заводе в американском городе Би-
шопе; в 1972 г. этот завод был закрыт) и жидкофазное окисле-
ние легкой фракции нафты, преимущественно разветвленных
парафинов С5—С?, по методу фирмы British Petroleum (значе-
ние этого процесса непрерывно возрастает). В обоих случаях
реакция протекает по сложному радикально-цепному механиз-
му, например
RH —> R.
R • + О2 —> RO2 •
RO2. + RH —> ROOH + R.
ROOH —> продукты
Конкуренция между производствами изопропанола, акри-
лонитрила, акриловой кислоты и ее эфиров за ресурсы пропи-
лена, который служит исходным сырьем для их получения, по-
вышает перспективность синтеза ацетона путем окисления па-
рафинов. Однако экономика окислительных методов, как и всех
процессов получения нескольких целевых продуктов, зависит от
спроса на все эти продукты. Поэтому доля окислительных ме-
Выбор оптимального метода производства
213
тодов в общей структуре мощностей по производству ацетона
будет определяться прежде всего увеличением потребности в
других кислородсодержащих химикатах, особенно карбоновых
кислотах.
Разработанный недавно фирмами Wacker и Hoechst метод
прямого окисления пропилена в ацетон в присутствии хлори-
стого палладия, вероятно, сможет играть важную роль только
при отсутствии более рентабельных путей использования про-
пилена. Реакцию проводят при температуре 50—120 °C и давле-
нии 5—10 МН/м2 (50—100 атм) при pH 2—3, и она протекает
через следующие стадии:
1. PdCl2 • 2Н2О [PdCl2(OH)H2O]~ + Н+
2. [PdCl2(OH)H2O]" + СН3СН=СН2 —>
—> [PdCl2(OH)(CH3CH=CH2)J" + Н2О
3. [PdCl2(OH)(CH3CH==CH2)l" + H2O —> Pd + 2Cr + H2O + (CH3)2COH
4. (СН3)2СОН —> (СН3)2СО + н+
Лимитирующей стадией является массоперенос пропилена и
кислорода, которые барботируются через реакционную смесь.
Селективность образования ацетона равна 98%. Продукт выде-
ляют дистилляцией. Регенерация палладиевого катализатора
происходит под действием хлорной меди и воздуха, поскольку
образующаяся при окислении палладия однохлористая медь
вновь легко окисляется воздухом
5. Pd + 2CuC12 —> PdCl2 + Cu2Cl2
6. Cu2Cl2 + 2HC1 + O2 —> 2CuC12-f-H2O
Области применения ацетона
Потребность в ацетоне в США увеличилась с 180 тыс. т в
1947 г. до 640 тыс. т в 1968 г. и далее до 690 тыс. т (прогноз-
ная оценка) в 1972 г. (средний годовой прирост 6%)*. В 1968г.
мощности по производству ацетона в США составляли
690 тыс. т/год, а в 1970 г. они, по прогнозной оценке, возросли
до 1 млн. т/год **. Поэтому приходится постоянно искать новые
* В 1973 г. потребность в ацетоне в США составляла 901 тыс. т., в
1974 г. — 940 тыс. т., в 1975 г. (прогнозная оценка) —895 тыс. т. [Chem. Mar-
ket Abstr., 67, 286 119, 286 1394 (1975)]. — Прим, перев.
** На 1 января 1976 г. мощности по производству ацетона в США со-
ставляли ~ 1,2 млн. т/год [Chem. Marketing Rep., 209, № 1, 40 (1976)].—
Прим, перев.
Рис. 7.2. Приблизительная структура потребления ацетона в США в 1947—
1968 гг.
Д —динамика роста потребления ацетона: 1947—1967 гг. —6% в год, 1967—1972 гг. —6°/0
в год (по оценке). Б—структура потребления: О растворитель; X синтез других раство-
рителей; □ синтез метилметакрилата; А синтез прочих химикатов (дифенилолпропана и
ДР-)
Выбор оптимального метода производства 215
области применения ацетона. Основная часть вырабатываемого
ацетона в конечном счете используется в качестве растворителя
и полупродукта для синтеза других растворителей и различных
химикатов, а также метилметакрилата (рис. 7.2 и гл. 8)*.
В конце второй мировой войны ацетон применяли главным
образом как растворитель. Однако в последующие годы этот
рынок сбыта ацетона постепенно сокращался, что, по-види-
мому, было связано с уменьшением выпуска пластических масс
на основе целлюлозы. Наиболее важными растворителями,
получаемыми из ацетона, являются метилизобутилкетон, диаце-
тоновый спирт, «гексиленгликоль» (2-метилпентандиол-2,4) и
изофорон. Дифенилолпропан [бисфенол А, 2,2-бцс-(4'-оксифе-
нил)пропан] представляет собой важное производное ацетона,
которое используют для синтеза лаковых алкидных смол, эпок-
сидных полимеров и поликарбонатов. Кроме того, из ацетона
получают лекарственные вещества, витамины, косметические
средства и вспомогательные химикаты для резин. Быстрее всего
(на 10% в год) возрастает потребление ацетона для синтеза
метилметакрилата, который необходим в производстве акрило-
вых пластиков; последние применяют для остекления самолетов
и автомобилей, изготовления дорожных знаков и как конструк-
ционные материалы.
Синтезы на основе ацетона (рис. 7.3 и 7.4)
Катализируемая основаниями альдольная конденсация
ацетона в мягких условиях (при температуре около 5 °C)
приводит к получению диацетонового спирта. Эта реак-
ция протекает очень селективно и сопровождается образо-
ванием лишь небольшого количества триацетонового спирта
(СН3)2С (ОН) СН2СОСН2С (СН3)2ОН, который является основ-
ным побочным продуктом. Главная область применения диаце-
тонового спирта — производство «гексиленгликоля», который
получают путем гидрирования спирта над скелетным никеле-
вым катализатором (никелем Ренея) при температуре 100 °C и
давлении 0,7 МН/м2 (7 атм).
* Структура потребления ацетона в США в 1973 г. (конечные продукты
его переработки): метилметакрилат — 24,6%, метилизобутилкетон—13,6%,
как растворитель для нанесения защитных покрытий— 10,36%, фармацевтиче-
ские препараты — 5,95%, как растворитель для химических процессов — 5,2%,
метакриловая кислота и высшие метакрилаты — 5,2%, дифенилолпропан —
4,65%, как растворитель при формовании ацетатных волокон — 4,1%, «гекси-
ленгликоль»— 2,6%, диацетоновый спирт — 2,5%, метилизобутилкарбинол —
2,3%, изофорон—1,8%, окись мезитила—1,4%, прочие—15,75% [Chem. Mar-
ket Abstr., 67, 286 1031 (1975)]. — Прим, перев,
216
Глава 7
СН2=СНСН2 (СН3)2СНОН
ZnO
о
сн
сн
СНз
изофорон
(СН2)2СО + Н;
но-
5°С
(СН3)2ССН2СНСН
НО ОН
A12Oj
(СН3)2ССН2СОСН3
„ гексиленгликолъ "
он
дйацетоновый
спирт
н+
СНз
СН2=ССН2СОСН
в
окись мезитила
^Побочный продукт, образующийся
наряду с ацетоном
' (СН2)2СНСН2СОСНз
метилизобутилкетон
+
(СНз)2СНСН2СНСН2
ОН
метил пентанол-2
Рис. 7.3. Схема промышленного получения различных производных ацетона
из пропилена через изопропанол.
СН2—СНСНз —> С6Н5СН(СНз)2 (куМбл)
01 Н»
С6Н50Н + (СНз)2СО i- (СНз)2СНСН2СОСНз
,________________) н
и»
нъ
С(СНз)2
ОН f дифенилолпропан')
Рис. 7.4. Схема промышленного получения различных производных ацетона
< из пропилена через кумол.
Путем дегидратации диацетонового и триацетонового спир-
тов под действием мягких кислотных агентов (например, фос-
форной кислоты) можно получить окцсь мезитила и форон
Выбор оптимального метода производства
217
(СН3)2С=СНСОСН=С(СНз)2 соответственно; однако объем
промышленного потребления этих ненасыщенных кетонов
очень мал.
Изофорон получают с помощью катализируемой основа-
ниями альдольной конденсации ацетона в жестких условиях
[при температуре около 210°C и давлении 3 МН/м2 (30 атм)].
Эта реакция протекает неселективно и даже при низкой
степени конверсии ацетона (не более 10%), способствующей
образованию изофорона, дает значительное количество других
кетосоединений. Для улучшения экономики процесса побочные
продукты вновь переводят (путем щелочного гидролиза при
температуре около 240 °C) в ацетон и далее в изофорон, что
позволяет достигнуть почти 90%-ной селективности на изофо-
рон. Основная часть вырабатываемого изофорона идет на
получение 3,5-ксиленола, который образуется при пиролизе
изофорона над окисью алюминия при температуре около
500 °C.
3,5-Ксиленол подвергают хлорированию, продукты которого
используют в производстве бактерицидных препаратов. Фирма
Veba разработала технологию получения из изофорона дикар-
боновых кислот, диолов и диаминов (схема 3) для применения
в производстве полиамидных и полиэфирных смол; однако эти
процессы пока не реализованы в промышленном масштабе.
Схема 3.
218
Глава 7
Метилизобутилкетон первоначально синтезировали из ди-
ацетонового спирта в две стадии — путем его дегидратации и
последующего гидрирования продукта. В настоящее время су-
ществуют прямые методы получения метилизобутилкетона.
Один из них заключается в реакции ацетона с водородом при
температуре 100 °C и давлении 7 МН/м2 (70 атм) над палла-
дием, осажденным на катионообменной смоле. При этом дости-
гается 30%-ная конверсия ацетона в метилизобутилкетон, со-
провождающаяся образованием небольшого количества диизо-
бутилкетона. В другом процессе смесь изопропанола с ацетоном
состава 2: 1 пропускают над катализатором, содержащим
смесь окислов редкоземельных металлов и хромит меди, при
температуре 250 °C и давлении 5 МН/м2 (50 атм). Степень пре-
вращения в метилизобутилкетон в расчете на изопропанол со-
ставляет 30% при несколько большем образовании диизобутил-
кетона.
Синтез метилметакрилата (гл. 8) представляет собой двух-
стадийный процесс. На первой стадии ацетон в присутствии
оснований в мягких условиях присоединяет синильную кислоту,
давая ацетонциангидрин (CH3)2C(OH)CN. Последний подвер-
гают гидролизу в сульфат метакриламида, который затем эте-
рифицируют метанолом.
85И-ная (по весу) H2SO4+CH3OH
(CH3)2C(OH)CN ----------------------*
—> СН2==С(СН3)СООСН3 + NH4HSO
Дифенилолпропан (бисфенол А) получают кислотнокатали-
тическим алкилированием фенола ацетоном в мягких условиях
(приблизительно при 50°C). Катализатором этой реакции слу-
жит сильная кислота (обычно хлористый водород) в присут-
ствии метилмеркаптана или тиогликолевой кислоты, выполняю-
щих функцию промотора.
При замене ацетона другими карбонильными соединениями, та-
кими, как ацетальдегид или левулиновая кислота, можно полу-
чить аналоги дифенилолпропана; однако при этом достигаются
меньшие выходы.
Выбор оптимального метода производства 219
Капролактам
А. Джубб
Jubb A. H.t ICI Corporate Laboratory, Runcorn
Синтез и полимеризация е-капролактама были впервые осу-
ществлены в конце XIX в. В 1930 г. Карозерс исследовал свой-
ства волокна, полученного из полимеров капролактама, а в
конце 40-х годов в результате работ Шлака в Германии было
налажено первое промышленное производство поли-е-капро-
амида, известного под названием найлона-6. Практически весь
вырабатываемый капролактам идет на получение найлона-6,
который наряду с его предшественником найлоном-6,6 является
одним из важнейших полиамидов, используемых в производстве
синтетических волокон (гл. 9). В 1969 г. доля полиамидных
волокон в общем объеме мирового выпуска синтетических воло-
кон (4,4 млн. т) составляла 41%, и хотя их абсолютное потреб-
ление продолжает возрастать, в ближайшем будущем они по
объему производства переместятся на второе место после поли-
эфирных волокон *. Доля найлона-6 в структуре мирового по-
требления полиамидных волокон приближается к 40%, однако
для отдельных стран и регионов соответствующие цифры сильно
различаются между собой. Так, вследствие главным образом
исторических причин на долю найлона-6 приходится около 20%
потребления полиамидных волокон в Великобритании, 50% —
в странах Европейского экономического сообщества и 25% —
в США, тогда как для Японии эта величина равна 75%.
Помимо производства синтетических волокон, найлон-6 в не-
большом количестве используется в производстве пластических
масс. Капролактам служит также полупродуктом для синтеза
ь-лизина (гл. 15).
Мировые мощности по получению капролактама составляли
в 1969 г. около 1,4 млн. т/год, из которых 17% принадлежали
США (средний годовой прирост потребности в нем в период
1962—1968 гг. был равен 17%) **. Ведущую роль в расширении
рынка сбыта капролактама играет Япония, в которой потреб-
ность в этом продукте в 1972 г. составляла, вероятно, 450 тыс. т.
* В 1975 г. во всем мире было получено 7,15 млн. т синтетических воло-
кон, из которых 44,7% составляли полиэфирные волокна и 34,6%—поли-
амидные (табл. 9.1). — Прим, перев.
** К середине 1974 г. общие мировые мощности по производству капро-
лактама достигли 2247 тыс. т/ год (в том числе 361 тыс. т/год в США), а к
концу 1975 г. они должны были составить, по прогнозной оценке, 2550 тыс.
т/год (в том числе 410 тыс. т/год в США) [Chem. Market Abstr., 66, 286 1506,
286 1507 (1974)]. — Прим, перев.
220
Глава 7
Методы получения капролактама
Вначале основным видом исходного сырья для производства
капролактама был фенол. Однако впоследствии доминирующее
положение заняли различные методы получения капролактама
из циклогексана. В настоящее время существует семь основных
промышленных методов синтеза капролактама и еще два ме-
тода, как сообщается, находятся на ранней стадии разработки.
Часть из них основана на использовании гидроксиламина в ка-
честве промежуточного продукта, причем это соединение может
быть получено несколькими путями. Таким образом, имеет.ме-
сто довольно сложный набор возможных схем проведения син-
теза (рис. 7.5 и 7.6). Ни один из методов производства капро-
лактама не имеет явных преимуществ перед остальными, и вы-
бор оптимального метода обычно зависит от местных условий.
Фенольный метод
Фирма Allied Chemical Corporation использует на своем за-
воде в Хопуэлле (шт. Виргиния, США), имеющем годовую
мощность 130 тыс.' т капролактама, модификацию немецкого
процесса времен второй мировой войны. На первой стадии фе-
нол подвергают каталитическому гидрированию в циклогекса-
нон. Непревращенный фенол (который затем возвращают в
цикл) и небольшие количества побочно образующегося цикло-
гексанола отделяют дистилляцией, а циклогексанон обрабаты-
вают аммиаком и раствором сульфата гидроксиламина с полу-
чением оксима. Последний в присутствии 20%-ного олеума пре-
терпевает бекмановскую перегруппировку (т. 2, стр. 166). Вы-
ходы на обеих стадиях составляют около 95%. Непрерывно
вытекающий из реактора сырой продукт нейтрализуют водным
аммиаком и очищают с помощью селективной экстракции.
После уд... ления растворителя производят окончательную очист-
ку капролактама путем его перекристаллизации.
Гидроксиламин синтезируют из карбоната аммония через
нитрит аммония (процесс фирмы Raschig)
no . nh3 + н2о н2о
(NH4)2CO3 2NH4NO2 ———> 2HON(SO3NH4)2 —> (NH2OH)2-H2SO4
1NC>2 oC>2
гидроксиламин- сульфат гидро-
дисульфонат аммония ксиламина
Многостадийность этого метода существенно увеличивает
себестоимость получаемого капролактама. Он является рента-
бельным только при наличии спроса на применяемый в каче-
стве удобрения сульфат аммония, который образуется на ста-
диях получения как капролактама, так и оксима (в количестве
1,8 и 2,7 т/т лактама соответственно).
Выбор оптимального метода производства
221
16
Рис. 7.5. Некоторые пути синтеза капролактама.
Вспомогательные реактивы: 1—HNO3—H2SO4; 4 — Ня; 7—HNO3; 11—НяО$; 12 — NOC1//TV;
16 —H2SO4.
Аналогичный процесс производства капролактама из фенола
разработан фирмой Dutch State Mines, которая построила в
г. Гелене завод мощностью 100 тыс. т/год. В этом процессе на
стадии оксимирования использован «гуамофосфатный» метод
синтеза гидроксиламина, что позволило снизить количество об-
разующегося сульфата аммония до 1,8 т/т лактама. По дан-
ному методу гидроксиламин получают гидрированием окислов
азота в присутствии благородного металла. Еще более новым
является бисульфат-лактамный процесс, в котором отсутствует
222
Глава 7
5 —через кумол; 6 —О2 + соль меди (процесс фирмы Dow); 7 — HNO3; 8 —кетеи; 9 —НО-!
10—отщепление Н2; 11 —Н2О2; 12 —NOC1//1V; 13 —NH2OH; 14—СН3СОООН; 15 —NH3;
16 — H2SO4; 17 —L1C1; 18 —циклизация; 19 — (NO)HSO4.
образование побочного сульфата аммония. Вместо него при ре-
гулируемой нейтрализации продукта бекмановской перегруппи-
ровки образуется бисульфат аммония, последующий пиролиз
которого дает двуокись серы, используемую для приготовления
олеума.
Еще один вариант фенольного метода был разработан в
1951 г. фирмой Emser Werke AG. В этом варианте гидроксил-
амин получают восстановлением окиси азота, что тоже снижает
Выбор оптимального метода производства 223
количество образующегося сульфата аммония на 50% по срав-
нению с процессом фирмы Raschig.
2NO + ЗН2 + H2SO< —> (NH2OH)2-H2SO4
Методы с окислением циклогексана воздухом
Жидкофазное окисление циклогексана воздухом в присут-
ствии небольших количеств солей кобальта при температуре
145—170 °C и давлении 0,8—1,2 МН/м2 (8—12 атм) приводит
к образованию циклогекс анол а, циклогексанона, а также раз-
личных карбоновых кислот и их эфиров, которые являются по-
бочными продуктами. Чтобы свести к минимуму выход послед-
них, степень конверсии циклогексана за проход поддерживают
на уровне не выше 10%. Этот метод лежит в. основе процессов,
разработанных несколькими фирмами. Среди них можно на-
звать процесс фирмы Stamicarbon, в котором степень конверсии
циклогексана за один проход составляет 4—6%, и процесс
фирмы Scientific Design, в котором для повышения выхода
циклогексанола и циклогексанона и увеличения доли первого
в смеси обоих продуктов реакцию ведут в присутствии борной
кислоты при степени конверсии циклогексана за проход около
10%. Другие существующие процессы меньше отличаются от
традиционной технологии.
Непревращенный циклогексан отделяют от сырого продукта
дистилляцией, а сложные эфиры омыляют водным раствором
щелочи. Циклогексанол и циклогексанон разделяют путем рек-
тификации, после чего циклогексанол подвергают каталитиче-
скому дегидрированию в циклогексанон. Далее кетон переводят
в капролактам, как это было описано выше.
Толуольный метод
По этому методу работает завод фирмы Snia Viscosa в
г. Тарвизио (Италия) *. Каталитическим окислением толуола
при температуре 150—170 °C и давлении около 1 МН/м2
(10 атм) получают бензойную кислоту, которую гидрируют в
жидкой фазе над катализатором из благородного металла. Об-
разовавшаяся циклогексанкарбоновая кислота при обработке
нитрозилсерной кислотой (NO)HSO4 отщепляет двуокись
углерода и превращается в капролактам. Степень конверсии
за один проход составляет около 50—60%; непревращенную
* Мощность завода 16 тыс. т/год [Europ. Chem. News, 28, № 721, 50
(1976)]. — Прим, перев.
224
Глава 7
карбоновую кислоту возвращают в цикл. Механизм этой реак-
ции окончательно не установлен.
соон
NO+ HS04-
-со2
Нитрозилсерную кислоту получают из аммиака, кислорода
и серной кислоты
2NHg -]- ЗОз —► N2O3 "Ь зн2о
N2O3 + 2H2SO4 —► 2(NO)HSO4 4-Н2О
Побочным продуктом суммарного процесса является суль-
фат аммония, выход которого равен приблизительно 4,2 т/т ка-
пролактама. Однако, как было объявлено, фирмы ANIC и Snia
Viscosa планируют совместное строительство завода мощ-
ностью 80 тыс. т/год, на котором будет применена усовершен-
ствованная технология получения капролактама, обеспечиваю-
щая снижение выхода сульфата до 2 т/т лактама *.
Фотонитрозирование циклогексана
В 1951 г. японская фирма Тогау Industries начала разраба-
тывать метод получения капролактама путем фотохимического
взаимодействия циклогексана с хлористым нитрозилом. В на-
стоящее время фирма имеет работающий по этому методу за-
вод мощностью 90 тыс. т/год и осуществляет повышение мощ-
ности этого завода до 140 тыс. т/год.
Процесс фирмы Тогау Industries является непрерывным и
обеспечивает выход капролактама по циклогексану более 80%
с одновременным получением лишь 2,2 т сульфата аммония на
1 т капролактама. Более низкий тепловой эффект перегруппи-
ровки дихлоргидрата циклогексаноноксима (107 кДж/моль) по
сравнению со свободным оксимом (187 кДж/моль) облегчает
температурный контроль. Одним из главных достижений фир-
мы Тогау было создание ртутной лампы высокого давления,
имеющей мощность 60 кВт и обеспечивающей выход капролак-
тама, равный 400 г-кВт-1-ч-1. Поскольку излучение с длиной
волны ниже 3650 А способствует образованию смол, к охлаж-
дающей воде, циркулирующей в рубашке лампы, добавляют
поглотители, а между лампой и реакционной смесью помещают
светофильтры. Однако это приводит к уменьшению светоотдачи
* В настоящее время этот завод, расположенный в г. Манфредония (Ита-
лия), уже работает [Europ. Chem. News, 28, № 721, 50 (1976)]. — Прим, пе-
рев.
Выбор оптимального метода производства 225
лампы, и оптимальное время пребывания в реакторе опреде-
ляется экономическими факторами, в том числе стоимостью
лампы, загрузкой предприятия и стоимостью электроэнергии.
Необходимый хлористый нитрозил получают из нитрозил-
серной кислоты (см. выше)
(NO)HSO4 + HC1 —> NOC1+H2SO4
Капролактоновый метод
В 1966 г. фирма Union Carbide ввела в эксплуатацию завод
мощностью 20 тыс. т/год в г. Тафте (шт. Луизиана, США), на
котором капролактам вырабатывают путем амидирования ка-
пролактона, получаемого окислением циклогексанона надуксус-
ной кислотой. Этот завод составляет единый производственный
комплекс с установкой по получению надуксусной кислоты
Преимуществом данного метода является отсутствие побочно
образующегося сульфата аммония. Однако то обстоятельство;
что наряду с капролактамом здесь получается уксусная кис-
лота (в количестве 1,1 т/т капролактама), ограничивает возмож-
ность использования этого метода районами, в которых на кис-
лоту имеется большой спрос (если только надуксусную кислоту
готовят не из перекиси водорода и уксусной кислоты, что де-
лает возможной рециркуляцию последней).
Надуксусную кислоту обычно получают по реакции
О СН3
[О] II I
2СН3СНО —> СНзСООСНОН —> СН3СОООН + СНзСНО
надацетат надуксусная I
апетальдегидгидрата кислота ]
на рецирку-
ляцию
Нитроциклогексаноновый метод
Фирма Techni-Chem предложила новый метод получения ка-
пролактама, при котором не образуются побочные продукты.
Этот метод прошел длительную проверку на опытно-промыш-
ленной установке в г. Вэллингфорде (шт. Коннектикут, США);
однако он до сих пор не реализован в промышленном масштабе.
Первой, ключевой стадией процесса является селективное
мононитрование циклогексенилацетата в среде уксусного ан-
гидрида в нитроциклогексанон. Образующая при этом уксусная
кислота используется для получения кетена, взаимодействием
которого с циклогексаноном получают циклогексенилацетат.
Нитроциклогексанон в водном растворе основания претерпевает
размыкание цикла; последующее гидрирование раствора дает
8 Зак. 501
226
Глава 7
е-аминокапроновую кислоту. Эту кислоту подвергают термиче-
ской циклизации в капролактам, который экстрагируют из вод-
ного раствора органическими растворителями и очищают пере-
кристаллизацией.
Нитроциклогексановый метод
Сообщается, что по этому методу работает установка фирмы
du Pont мощностью 25 тыс. т капролактама в год. Самой важной
стадией процесса является восстановление нитроциклогексана
в оксим. Этой реакции посвящено большое число патентов, в
которых предлагаются различные восстановители, такие, как
водород в присутствии цинк-хромового катализатора, соли
двухвалентного железа и сульфиты. В одном из патентов фир-
мы du Pont описан вариант, обеспечивающий выход оксима
80—85% при одновременном получении циклогексиламина с
выходом 8—10%. Перегруппировку оксима осуществляют обыч-
ным образом; при этом, как всегда, в качестве побочного про-
дукта образуется сульфат аммония. Кроме того, запатентован
способ прямого получения капролактама из нитроциклогексана
путем его восстановления и дегидратации.
Метод с промежуточным образованием перекиси
1-оксициклогексила
Недавно английская фирма BP Chemicals Ltd. взяла не-
сколько патентов на метод получения капролактама взаимо-
действием циклогексанола с перекисью водорода в инертном
растворителе. Образующуюся перекись 1-оксициклогексила
переводят путем обработки аммиаком в перекись 1-аминоцикло-
гексила, которую подвергают термическому разложению в при-
сутствии галогенида лития, получая расплавленный капролак-
там. Сопутствующий продукт — циклогексанон — выделяют дис-
тилляцией-и возвращают на стадию окисления.
Этот процесс находится еще на довольно ранней стадии
разработки, вследствие чего Пока Трудно оценить его промыш-
ленную перспективность. Хотя он не приводит к побочному по-
лучению сульфата аммония, одним из его недостатков, по-види-
мому, является то, что сравнительно дорогостоящая аминопере-
кись расщепляется на эквимолярные количества продукта и
циклогексанона, который приходится возвращать в цикл.
Циклогексиламиновый метод
Гидрирование анилина на никель-кобальтовом катализаторе
дает циклогексиламин, при обработке которого избытком пере-
киси водорода в присутствии молибдатов получается циклогек-
Таблица 7.5
CO Удельные затраты на производство капролактама на иболее важными методами ( в ценах 1970 г.)
Фенольный метод (процесс фирмы Allied Chem. Corp.) Процесс фирмы Dutch State Mines Процесс фирмы Snia Viscosa ' Процесс фирмы Тогау Industries
Исходное сырье годные Ьфициен- т/т ка- ]актама 1ьные • аты, т./т ка- пактама исходные >эффициен- j, т/т ка- )олактама 1ьные >аты, т./т ка- 1актама 1сходные >эффициен- I, т/т ка- эолактама «3 «5 2 «У > X « я х Л «3 ц я расходные коэффициен- ты, т/т ка- пролактама гьные )аты, т./т ка- гактама
и <п > 6 я о 3 а. о.х н е , УД^ затр । ф. с про. 0) f- о « н"'"' о (а О
Q.X Н И Sn’&ts схх н с >» т 'О' и ХП’&Е
Фенол (65 ф. ст./т) Толуол (21 ф. ст./т) Циклогексан (37 ф. ст./т) Прочие химикаты 0,89 57,9 46,9 1,06 39,2 51,1 1,11 23,3 60,7 0,93 34,4 33,2
Общая стоимость сырья За вычетом стоимости побочных продуктов сульфат аммония хлорциклогексан 4,6 104,8 -26,5 4,5 90,3 -26,0 4,1 84,0 —23,7 2,29 0,064 67,6 — 13,2 —2,5
Чистая стоимость сырья Энергетические затраты Трудовые затраты Затраты на текущий ремонт и зданий Накладные расходы оборудования 78,3 12,3 3,3 8,1 11,4 64,3 13,3 1,4 5,6 6,9 60,3 14,2 1,9 4,6 6,5 51,9 21,7 2,3 10,0 12,5
Затраты на производство, всего ф. ст. на 1 т капролактама 113,4 91,5 87,5 98,4
228
Глава 7
Таблица 7.6
Капитальные затраты для четырех важнейших
процессов производства капролактама
(по данным журнала European Chemical News)
Процесс фирмы Мощность завода, тыс. т/год Капитальные затраты а, млн. ф. ст.
Allied Chem. Corp. 25 6,7
Dutch State Mines 40 6,1
Snia Viscosa 40 6,2
Toray Industries 50 8,8
а В ценах 1969 г.
саноноксим с выходом 95%. Этот метод реализован в промыш-
ленном масштабе в СССР.
Фирма. Vickers-Zimmer разработала процесс, в котором цик-
логексиламин подвергают гидролизу при повышенной темпера-
туре в циклогексаноЛ. Последний дегидрируют в кетон и затем
обычным путем синтезируют капролактам.
Другие методы
Существует еще несколько возможных методов получения
капролактама, включающих промежуточное образование ади-
понитрила; однако ни один из них не представляет промышлен-
ного интереса.
Выводы
В табл. 7.5 представлены затраты производства для четырех
наиболее важных процессов получения капролактама, а в
табл. 7.6 — капитальные затраты для этих процессов. Приве-
денные данные наглядно показывают, какое значительное влия-
ние на экономику процесса оказывает возможность реализации
полученного сульфата аммония. Там, где такая возможность
существует, наиболее рентабельным является процесс фирмы
Snia. Напротив, в районах, где сульфат аммония находит огра-
ниченный сбыт, более предпочтителен процесс фирмы Тоуо
Rayon.
В последние годы цены на капролактам снизились до 170—
200 ф. ст./т, что обусловлено сокращением затрат на его произ-
водство и созданием избыточных мощностей при относительно
быстром росте потребления этого продукта.
ГЛАВА 8
Пластические массы и эластомеры
М. Джонс
Jones М. Е- В-, ICI Corporate Laboratory, Runcorn
Введение
Высокомолекулярные соединения обычно подразделяют для
удобства их изучения на природные и синтетические. Хотя при-
родные высокомолекулярные соединения играют чрезвычайно
важную роль в жизнедеятельности животных и растений, в этой
главе рассматриваются преимущественно синтетические поли-
меры, к числу которых относятся большинство применяемых
пластмасс и все возрастающая часть эластомерных материалов.
Современная промышленность пластических масс берет свое
начало с получения нитроцеллюлозы (1862 г.) Александром
Парксом (т. 4, стр. 93). В течение трех лет Паркс разъезжал
с выставкой изделий, изготовленных из нового материала, и ак-
тивно пропагандировал возможность его использования в про-
изводстве перочинных ножей, расчесок, обувных подметок, лент,
труб, аккумуляторных батарей и водонепроницаемых тканей.
Однако нитроцеллюлозная пластмасса за свою более чем сто-
летнюю историю никогда не была продуктом крупнотоннажного
производства.
Напротив, фенолформальдегидные смолы, полученные в на-
чале XX в. Лео Бакландом, стали одним из важнейших типов
пластических материалов. Бакланду удалось понять, что преж-
ние методы проведения реакции между фенолом и формальде-
гидом не позволяют получить продукт однородного состава.
Регулируя соотношение обоих реагентов и используя подмечен-
ное им каталитическое влияние на эту реакцию кислот и осно-
ваний, Бакланд получил вещество, которое легко формовалось
при нагревании под давлением. Открытие Бакланда имело та-
кой успех, что ко времени его смерти (1944 г.) объем мирового
производства фенольных смол достиг 200 тыс. т/год.
Промышленные методы получения полистирола и поливи-
нилхлорида, а также сополимеров типа винилхлорид — винил-
ацетат были разработаны в конце 20-х — начале 30-х годов.
Таблица 8.1
Производство пластических масс (тыс. т)
в некоторых капиталистических странах в 1971 г.
Наименование
США а
Великобри-
тания &
Япония
Термопластичные материалы
Полиэтилен низкой плотности 2010 305 951
Полиэтилен высокой плотности 848 63 394
Полипропилен 560 78 627
Полистирол и сополимеры стирола 1710 168 695
Поливинилхлорид и сополимеры ви- 1482 325 1031
нилхлорида
Прочие виниловые пластики 290 50 110
Акриловые пластики 90 15 60
Целлюлозные пластики 75 10 10
Полиамидные смолы 50 15 22
Итого 7115 1029 3890
Термореактивные материалы
Фенольные смолы 476 65 218
Мочевинные и меламиновые смолы 300 170 647
Алкидные смолы 272 63 102
Эпоксидные смолы 71 14 19
Полиэфиры 360 38 128
Полиуретаны 410 48 94
Итого 1889 398 1208
Прочие пластмассы (всех типов) 420 50 89
Итого
9424 1477 5187
Мировое производство пластических масс в 1971 г. составляло 32,1 млн. т в.
а В 1975 г. в США было получено 7890 тыс. т термопластов и 1390 тыс. т. реакто-
пластов, в том числе 1119 тыс. т полиэтилена высокой плотности и 863 тыс. т полипропи-
лена. Производство остальных полимеров не претерпело существенных изменений [Chem.
and Eng. News, 54, № 24, 37, 38 (1976)1. — Прим, перев
6 В 1974 г. производство синтетических смол и пластмасс в Великобритании соста-
вило 2467 тыс. т [Statistical Yearbook 1975 , 27, 302 (1976)], в том числе 1514 тыс. т термо-
пластов и 821 тыс. т реактопластов. Полиэтилена было получено 488 тыс. т, полипропи-
лена—222 тыс. т, полистирола и сополимеров стирола—231 тыс. т, поливинилхлорида и
сополимеров винилхлорида—367 тыс т, фенольных и крезольных смол—59 тыс. т, амино-
пластов—139 тыс. т, алкидных смол —103 тыс. т, эпоксидных смол—76 тыс. т, ненасыще-
нных полиэфирных смол—57 тыс. т и полиуретанов — 3,5 тыс. т [Annual Abstracts of
Statistics, 197$, № 112, 206].— Прим, перев.
в В 1974 г. мировое производство пластмасс составило 41,4 млн. т [Statistical Year-
book, 1975, 27, '302 0976)].— Прим, перев
Пластические массы и эластомеры
231
Однако вплоть до этого времени химизм процессов полимериза-
ции и структура полимеров практически не исследовались. Только
в результате работ Штаудингера была создана общепринятая
ныне макромолекулярная теория строения полимеров. В 20-х
годах Штаудингер предложил длинноцепочечные химические
формулы для полистирола, полиоксиметилена и натурального
каучука, а вскоре после этого его представления были подтвер-
ждены данными рентгеноструктурного исследования целлюлозы
и других кристаллических полимеров, проведенного Мейером и
Марком.
В 1929 г. появилась первая работа Карозерса, фундамен-
тальные исследования которого привели к синтезу найлона и
хлоропренового каучука. С помощью обычных реакций Каро-
зерс получил разнообразные полимерные материалы и тем са-
мым показал близкое химическое родство синтетических и при-
родных высокомолекулярных соединений.
Вторая мировая война стимулировала создание большого
числа новых синтетических полимеров. За десять лет — с сере-
дины 30-х до середины 40-х годов — был получен ряд важных
полимеров, в том числе полиэтилен, политетрафторэтилен, поли-
этилентерефталат, силиконы и новые синтетические каучуки.
В послевоенное время исследования в области химии и тех-
нологии полимеров значительно расширились, в результате чего
были синтезированы многочисленные новые типы высокомоле-
кулярных соединений и разработаны новые методы полимери-
зации. Наиболее выдающимися по свому значению из работ
этого периода, вероятно, являются работы Циглера, открыв-
шего специальные катализаторы для полимеризации этилена
При низком давлении, и Натта, разработавшего каталитические
системы для стереоспецифической полимеризации а-олефинов..
К настоящему времени химики стали глубже понимать меха-
низмы реакций полимеризации, а знание взаимосвязи между
структурой и свойствами полимеров позволяет им получать ма-
териалы с заранее заданными свойствами. Грандиозные успехи,
достигнутые в производстве полимеров, иллюстрируются дан-
ными табл. 8.1.
Терминология и классификация
Терминология
Полимерами называются соединения с высоким молекуляр-
ным весом, молекулы которых состоят из очень большого числа
фрагментов, имеющих гораздо меньший молекулярный вес.
В первом приближении большинство полимеров можно оха-
рактеризовать в рамках составляющих их структурных еди-
ниц. Некоторые полимеры, построенные из бифункциональных
232
Глава 8
(двухвалентных) структурных единиц, обладают преимуще-
ственно линейной структурой, т. е. мономерные звенья в них со-
единены в длинные цепи типа
—М—М—М—М—М—М— или —(—М—)„—
образуя линейный полимер. В данном случае группу М назы-
вают повторяющейся единицей или элементарным звеном, а ве-
личину п — степенью полимеризации; в сумме они определяют
молекулярный вес полимера. У большинства синтетических ли-
нейных полимеров, выпускаемых промышленностью, молекуляр-
ный вес составляет 104—107. Линейные макромолекулы этих
полимеров содержат концевые группы, которые в зависимости
от вида реакции полимеризации могут быть химически тожде-
ственными элементарному звену или отличными от него. Типич-
ными представителями линейных (термопластичных) полиме-
ров являются полиэтилентерефталат и полистирол. Элемен-
тарное звено полиэфира содержит остатки как терефталевой
кислоты, так и этиленгликоля
* !
—I—СО—/ }—СООСН2СН2О—I—
L \—/ _!п
тогда как в случае полистирола структурная единица тожде-
ственна по составу исходному мономеру (стиролу), при полиме-
ризации которого изменяется только расположение химических
связей
Линейные полимеры — Не единственно возможный Тип син-
тетических полимеров. Если структурные единицы являются
Три- или полифуикциоиальными, полимерные цепи разветв-
ляются или соединяются друг с другом, образуя простран-
ственную сетку. Примером полифункциональиого мономера мо-
жет служить глицерин, частичная замена которым звеньев эти-
ленгликоля в полиэтилентерефталате приводит к получению
пространственного (сшитого) полимера, имеющего приблизи-
тельно такую структуру:
—Т—Г—Т—Э—Т—Г—Т—Г—Т—
4 4
। ।
Пластические массы и эластомеры
233
где Т — остаток терефталевой кислоты, Э — остаток этиленгли-
коля и Г — остаток глицерина.
Обычно в таких случаях содержание поперечных связей
между линейными цепями близко к концентрации трифункцио-
нальных групп (Г) в полимере. Типичным представителем силь-
носшитого полимера являются фенолформальдегидные смолы
(т. 1, стр. 171 и данная глава, стр. 271), в которых остаток
формальдегида бифункционален, а остаток фенола би- или по-
лифункционален. Строение сшитых (термореактивных) полиме-
ров нельзя охарактеризовать только элементарным звеном,
вследствие чего обычно употребляют структурные формулы, по-
добные приведенной выше.
Классификация
Карозерс разделил реакции образования полимеров на два
вида: полиприсоединение и поликонденсация. Конденсационны-
ми полимерами он назвал такие полимеры, в элементарном зве-
не которых отсутствуют некоторые атомы, имевшиеся в исход-
ных веществах. Одним из полимеров этого типа является поли-
тетраметиленадипат
пНО(СН2)4ОН + «НООС(СН2)4СООН —►
—► Н— [—О—(СН2)4ООС(СН2)4СО-]„ОН + (2л - 1)Н2О
В данном случае реакция поликонденсации протекает между
гликолем и дикарбоновой кислотой, причем на каждую обра-
зующуюся сложноэфирную связь выделяется одна молекула
воды.
Полиприсоединение, согласно Карозерсу, рассматривают как
цепную реакцию, протекающую с участием агента передачи
цепи, функцию которого может выполнять ион или радикал
(,т. 1, стр. 65). При этом полимер образуется из би- или поли-
функционального исходного вещества (мономера) путем соеди-
нения его молекул без отщепления каких-либо атомов. Типич-
ными представителями таких полимеров являются продукты,
получаемые из винильных мономеров. Примером реакции поли-
присоединения может служить полимеризация метилметакрила-
та в присутствии перекиси бензоила. Первая стадия процесса —
инициирование цепи за счет термического распада перекисного
инициатора на свободные радикалы. Эти радикалы реагируют
далее с метилметакрилатом, разрывая его двойную связь и
образуя промежуточное соединение с неспаренным электроном.
Последнее присоединяется к следующей молекуле мономера,
в результате чего получается растущая цепь. Быстрый рост цепи
происходит до тех пор, пока не наступает ее обрыв, обычно
234
Глава 8
возникающий вследствие столкновения двух растущих радика-
лов. Таким образом, процесс протекает по следующей схеме:
нагревание
С6Н6СОО—ОСОС6Н6 -------->-
/СН3
CgHgCOO • т* сн2=с
^СООСНз
2С6Н5СОО •
,си3
—► С6Н6СООСН2С<
\соосн3
начальное звено
растущей цепи
СН3
С6Н5СОО—
—сн2-с
СООСНз _т
. /СНз
сн2с(
хсоосн3
растущий полимер
2С6Н6СОО—
./снз
сн2с/
^СООСНз
—> неактивный полимер
Классификация Карозерса в ряде случаев приводит к за-
труднениям, поскольку некоторые конденсационные полимеры
можно также синтезировать с помощью полиприсоединёния. На-
пример, найлон-6 может быть получен как из е-аминокапроно-
вой кислоты с отщеплением воды, так и из капролактама без
изменения химического состава мономера
* —[—NH(CH2)sCO—4----- NH2(CH2)5COOH
—Н2О
—> —[-HN-R-NHCO—]„-
Аналогично этому полимочевинные смолы можно рассматри-
вать и как продукты полиприсоединения диаминов к диизоциа-
натам (т. 2, стр. 281) и как продукты поликонденсации диами-
нов с дифенилкарбонатом
H2N—R—NH2 + OCN—R -NCO ------------
H2N-R-NH2 + C6H5OCOOC6H6 „
—JCgrlsUrl
Поэтому первоначальная классификация полимеров, предложен-
ная Карозерсом, впоследствии была усовершенствована путем
введения в нее представления о ступенчатой и цепной полимери-
зации. Согласно этому представлению, все реакции полимериза-
ции подразделяются в соответствии с их механизмом независимо
от того, сопровождаются они отщеплением каких-либо соедине-
ний или нет. Например, образование полиуретана из диола и
диизоцианата рассматривается как ступенчатая полимеризация,
Пластические массы и эластомеры
235
хотя структура полиуретанов допускает также классическую
трактовку механизма их образования как поликонденсации
с отщеплением молекулы небольшого размера. Новая классифи-
кация полимеров, учитывающая механизм их образования, поз-
воляет выделить ряд отличительных особенностей обоих видов
полимеризации. При ступенчатой полимеризации исходные ве-
щества исчезают уже на ранней стадии процесса и рост поли-
мерной цепи происходит постепенно. Для достижения высокого
молекулярного веса обычно необходима большая продолжитель-
ность реакции, причем молекулярновесовой состав продукта из-
меняется строго закономерно. Напротив, при цепной полимери-
зации концентрация мономера уменьшается постепенно, причем
уже в начале реакции начинает образовываться высокополимер.
Молекулярный вес полимера в ходе реакции остается практи-
чески постоянным, и увеличение продолжительности процесса
приводит лишь к повышению выхода полимера, но не его моле-
кулярного веса.
Связь между химической структурой
и свойствами полимера
Наиболее важными свойствами как термопластичных, так и
термореактивных синтетических полимеров являются темпера-
тура размягчения, механическая прочность, жесткость, ударная
вязкость (ударопрочность), светостойкость, устойчивость к окис-
лительным и другим химическим воздействиям. Для переработ-
ки полимеров в изделия большое значение имеет также их спо-
собность переходить в жидкое состояние.
Физико-механические свойства всех синтетических полиме-
ров можно связать с их химической структурой. Главными фак-
торами, определяющими эти свойства, являются тип и распре-
деление химических связей в молекуле полимера, ее симметрия
и пространственное строение, а также молекулярный вес поли-
мера.
Природа связи в полимерах
Полимеры, рассматриваемые в данной главе, состоят из
структурных единиц, соединенных между собой ковалентными
связями. Эти связи определяют положение отдельных атомов
в цепи полимера. Однако в рамках одних только ковалентных
связей не удается удовлетворительно объяснить все наблюдае-
мые свойства пластмасс и каучуков, вследствие чего приходится
принимать во внимание невалентные взаимодействия. Послед-
ние обычно имеют межмолекулярный характер и, несмотря на
очень небольшую по сравнению с ковалентными связями
энергию, оказывают существенное влияние на свойства поли-
мерных материалов.
236
Глава 8
Наиболее сильным из невалентных (вторичных) взаимодей-
ствий, безусловно, является образование водородных связей, ко-
торое играет важную роль, например, в полиамидах и поли-
мочевинах. Так, высокие температуры плавления полиамидов
почти целиком обусловлены существованием в них межмолеку-
лярных водородных связей. При нахождении корреляций между
структурой и свойствами полимеров необходимо также учиты-
вать дипольные и другие индукционные взаимодействия; так,
например, в полиэфирах существуют диполь-дипольные взаимо-
действия. Наконец, во всех полимерах проявляются дисперсион-
ные силы, на долю которых в неполярных молекулах, например,
углеводородных полимерах, приходится значительная часть
межмолекулярных взаимодействий.
Строение полимерных цепей
В первом исследовании строения полимерных цепей, прове-
денном Штаудингером, было установлено, что в полистироле
фенильные группы расположены вдоль макромолекулярной
цепи в правильном порядке и связаны с альтернантными ато-
мами углерода, т. е. звенья стирола соединены по типу «голова
к хвосту». Возможно также соединение мономерных звеньев по
типу «голова к голове» или неупорядоченное соединение
—СН2—СН— СН2—СН—СН2—СН—СН2— От-
соединение «голова к хвосту»
СНг—СН-СН—СН2—СН2—СН—СН—СНг—
соединение «голова к голове»
неупорядоченное соединение
Существование всех этих возможностей обусловлено тем, что
в принципе присоединение начального радикала или иона к мо-
Пластические массы и эластомеры
237
номеру винильного типа CH2=CHR может происходить по
любому из ненасыщенных атомов углерода и зависит от ста-
бильности образующегося продукта. Большинство виниловых
полимеров имеют преимущественно структуру типа «голова
к хвосту».
Другим видом изомерии полимерной цепи, который наиболее
характерен для полиэтилена, является ее ветвление. В этом
случае узловые атомы становятся трифункциональными, что
приводит к получению макромолекулы с короткими ответвле-
ниями, имеющей вид птичьего пера.
полимерная цепь с короткоцепочечными
Ответвлениями
полимерная цепь с длиннацепочечными
ответвлениями
Наконец, при обсуждении изомерии цепи полимеров, синтези-
рованных из мономеров типа CH2=CHR, следует принимать во
внимание пространственную изомерию. Штаудингер пришел к
выводу, что в таких полимерах углеродные атомы, несущие за-
меститель R, могут быть асимметрическими и иметь либо R-,
либо S-конфигурацию. С того времени, как было сделано это
теоретическое заключение, был осуществлен ряд стереоспеци-
фических синтезов полимеров, особенно из а-олефинов. Номен-
клатура полимеров данного типа была предложена Натта, ко-
торый присвоил каждому из трех возможных пространственных
изомеров свое название. Полимеры, в молекуле которых все
заместители находятся по одну сторону основной цепи, назы-
ваются изотактическими. Если заместители располагаются по-
очередно по разные стороны основной полимерной цепи, то такие
полимеры принято называть синдиотактическими. Полимеры
238
Глава 8
с неупорядоченным пространственным расположением заме-
стителей относительно полимерной цепи называют атактиче-
скими.
R R R R
\ /К А А А /
XZ ХСХ
изотактический полимер
синдиотактический полимер
R
атактический полимер
Полимеры, полученные из 1,2-дизамещенных олефинов, тоже
могут существовать в нескольких стереорегулярных формах.
Аналогично ведут себя продукты полимеризации 1,3-диенов, в
которых на каждое элементарное звено приходится одна двой-
ная связь. Однако рассмотрение многообразных структурных
возможностей, возникающих в этих случаях, не входит в задачу
данной главы.
Кристалличность
Несмотря на то что многие синтетические полимеры дают
дискретные рентгенодифрактограммы, их изучение показывает,
что ни один из полимеров не является полностью кристалличе-
ским и что в «полукристаллическом» полимере одновременно
присутствуют области с кристаллической (упорядоченной) и
аморфной (неупорядоченной) структурой. Чаще всего кристал-
личность наблюдается у симметрично построенных полимеров.
Нарушения правильной структуры, такие, как ветвление, вклю-
чения звеньев сомономера, поперечные связи, атактические уча-
стки и др., обычно вызывают уменьшение степени кристаллич-
ности. Накопление, перечисленных факторов может в конечном
итоге привести к получению совершенно аморфных материалов.
Так, атактические полимеры, смолы с большим числом попереч-
ных связей и сополимеры с высоким содержанием звеньев вто-
рого мономера, как правило, полностью аморфны.
Высококристаллические полимеры обычно имеют относи-
тельно четко выраженные температуры плавления (Гпл), ко-
Пластические массы и эластомеры
239
торне можно определять различными оптическими и термиче-
скими методами. Из-за значительной кристалличности перера-
ботку таких полимеров методами, включающими их расплавле-
ние (например, литьем под давлением и экструзией), следует
вести при температурах выше их температуры плавления.
Температура стеклования
Существует большое число некристаллических (аморфных)
полимеров, которые не способны к истинному плавлению, но
вместе с тем при достаточно низких температурах становятся
жесткими и твердыми. Температура этого превращения зависит
от природы полимера и колеблется от —70 °C для натурального
каучука до 100 °C для полиметилметакрилата. Эта температура,
при которой аморфный полимер переходит из эластичного, кау-
чукоподобного состояния в жесткое, стеклообразное состояние,
называется температурой стеклования (Тст). На молекулярном
уровне ее можно представить как температуру, при которой от-
носительно большие сегменты основной цепи полимера стано-
вятся подвижными. Ниже этой температуры значительные пере-
мещения сегментов затруднены и аморфный полимер становится
жестким и твердым, тогда как при более высокой температуре
относительная подвижность длинных участков макромолекуляр-
ной цепи обусловливает каучукоподобность полимера.
Температура стеклования определяется аморфными участ-
ками полимера. Однако, поскольку «кристаллические» органиче-
ские полимеры никогда не бывают кристаллическими на 100%,
они тоже имеют наряду с температурой плавления температуру
стеклования. При нагревании высококристаллического полимера
переход через Тст не всегда сопровождается его видимым раз-
мягчением, так как каучукоподобные аморфные сегменты мак-
ромолекул продолжают удерживаться на своих местах кристал-
лическими сегментами и размягчение в обычном смысле этого
слова наступает только при температуре плавления полимера.
Механизм цепной полимеризации
Радикальная полимеризация
Механизмы цепных свободнорадикальных реакций частично
обсуждались в предыдущих томах (т. 1, стр. 11 —12, 59, 65; т. 2,
стр. 126—129; т. 3, стр. 311—317). Процесс свободнорадикаль-
ной полимеризации этилена (т. 1, стр. 66) можно представить
следующей схемой:
Инициирование цепи: R • + С2Н4 —> RC2H4 •
Рост цепи: RC2H4 • + С2Н4 —> RCH2CH2CH2CH2 «
240
Глава 8
Передача цепи: а) внутримолекулярная
R(CH2)4CH2. —> RCH(CH2)3CH3
б) межмолекулярная
RCH2. + R'(CH2)„R" —> RCH3 4-R'CH(CH2)n-iR"
в) на третичный атом углерода
RCH2 • + (СН3)3СН —> RCH3 + (СН3)3С •
Обрыв цепи: а) диспропорционированием радикалов
RCH2CH2. + R'CH2CH2. —> RCH=CH2 + R'CH2CH3
б) рекомбинацией радикалов
RCH2- + R'CH2. —> RCH2CH2R'
Относительная скорость каждой из этих реакций зависит от
условий полимеризации. Регулируя давление, температуру, ко-
личество растворителя и другие параметры, можно получать
полимеры с различными свойствами. Например, скорость пере-
дачи цепи быстрее возрастает с температурой, чем скорость
роста цепи, вследствие чего повышение температуры приводит
к образованию более низкомолекулярного продукта.
Очень важное значение имеет природа применяемого ини-
циатора. Наиболее распространенными инициаторами полиме-
ризации являются перекись бензоила (т. 2, стр. 128), перекись
трет-бутила (т. 2, стр. 125) и азо-бис-изобутиронитрил (т. 2,
стр. 173). Все эти соединения содержат в своей молекуле отно-
сительно слабые связи, способные разрываться при повышенной
температуре с образованием свободных радикалов.
Цепная полимеризация по ионному механизму
Для получения полимеров из некоторых а-замещенных оле-
финов могут быть использованы анионные или катионные ката-
лизаторы. Типичными анионными катализаторами являются
сильные основания, такие, как натрийнафталин или амид нат-
рия, а типичными катализаторами катионной полимеризации —
кислоты Льюиса, например хлористый алюминий и фтористый
бор. Ионная полимеризация протекает очень быстро, и обычно
ее проводят при пониженной температуре в низкокипящем рас-
творителе, что облегчает отвод теплоты реакции.
Избирательную способность мономера к полимеризации по
катионному, анионному или свободнорадикальному механизму
можно связать с электронной плотностью его двойной связи.
Так, мономеры, в которых двойная связь под влиянием электро-
Пластические массы и эластомеры 241
нодонорного заместителя обогащена электронами, образуют
сравнительно устойчивые карбониевые ионы и, следовательно,
более склонны к катионной полимеризации. Типичными мономе-
рами этого типа являются простые виниловые эфиры, изобути-
лен и дигидрофуран. С другой стороны, мономеры с электроно-
акцепторными заместителями при ненасыщенном атоме угле-
рода имеют тенденцию полимеризоваться по анионному меха-
низму. Полимеризация олефинов, содержащих сильноактивиро-
ванную двойную связь(таких, как винилидендицианид), катали-
зируется даже слабыми основаниями, хотя для большинства
мономеров, полимеризуемых по анионному механизму, исполь-
зуют сильные основания. Между этими двумя предельными ти-
пами мономеров лежит большое число а-замещенных олефинов,
которые содержат в крайнем случае лишь слабоэлектроноакцеп-
торные заместители и полимеризуются по свободнорадикальному
механизму.
Анионная полимеризация. Большинство реакций анионной
полимеризации протекает при низких температурах. Однако в
некоторых случаях процесс удобнее проводить при комнатной
или даже несколько повышенной температуре. Часть реакций
этого типа, особенно если используется гетерогенный катали-
затор, обнаруживает стереоспецифичность. Данное явление на-
блюдается не только при полимеризации монозамещенных про-
изводных этилена, но и при 1,4- и 1,2-полимеризации бутадиена
и его производных. Исследование типичной реакции анионной
полимеризации (например, полимеризации стирола в жидком
аммиаке, катализируемой амидом калия) выявило следующие
закономерности: а) глубина реакции возрастает с понижением
температуры; б) молекулярный вес полимера не зависит от
концентрации амид-иона; в) при постоянной степени конверсии
молекулярный вес полимера зависит от концентрации иона ка-
лия; г) молекулярный вес полимера зависит от начальной кон-
центрации мономера; д) с понижением температуры молекуляр-
ный вес полимера увеличивается; е) образующийся полимер
содержит азот, количество которого в среднем составляет один
атом на каждую молекулу полимера; ж) полимер не содержит
остаточных двойных связей; з) амид калия не расходуется в
процессе полимеризации. Эти экспериментальные факты можно
объяснить в рамках следующего механизма реакции:
Инициирование цепи-. K+NHJ + С6Н5СН=СН2 —> NH2CH2CHC6H6 -f- К+
Рост цепи: NH2CH2CHC6H6 + СНг=СНСбН5 —>
—> NH2CH2CH(C6H5)CH2CHCoH?
242
Глава 8
Обрыв цепи-.
NH2CH2CH(C6H6)CH2CHC6H5 + NH3 —>
-* NH2-[-CH2CH(C6H5)-]—сн2сн2с6н6 + nh2
Катионная полимеризация. Катионная полимеризация про-
текает через карбониевые ионы, и при использовании кислот
Льюиса необходимо вводить сокатализатор. Последний иниции-
рует полимеризацию, причем рост цепи происходит путем при-
соединения мономера к карбониевому иону. При катализе про-
тонными кислотами, которые сами способны генерировать ка-
тион, добавление сокатализатора является излишним; в этом
случае реакция протекает по механизму, аналогичному меха-
низму свободнорадикальной полимеризации.
Примером катионной полимеризации служит полимеризация
изобутилена, катализируемая фтористым бором в присутствии
следов воды, которая протекает по следующему механизму:
Инициирование цепи: BF3 + Н2О —> ~BF3OH Н+
"BF3OH Н+ + СН2=С(СН3)2 —> (СН3)3С+ ”BF3OH
Рост цепи: (СН3)3С+ ~BF3OH + СН2=С{СН3)2 —>
СН3 СН3
—> сн3—А-сн2—А+ bf3oh
I I
сн3 сн3
Обрыв цепи: несколько возможных путей, например:
а) кинетический
СН3 - СН3 ~
I I '
сн3—С-----сн2—с------
Ан3
СНз
СН2—С+ BF3OH —>
I
сн3
б) рекомбинация
СНз -
сн3-с—
СНз “ СН3 ~
I I
сн3—С------сн2—С------
I I
СНз _ СНз
СН3- СН3
-сн2-с
С+ BF3OH
сн2—с
СНз
сн2
-|- ~BF3OH + н+
сн3 _
СНз_„ СНз
СЧз -
СН3—А—
СН3-
СНз
—сн2—с
СН2—С—ОН + BF3
СНз _
СНз _п
СНз
Пластические массы и эластомеры
243
в) передача цепи на мономер
СНз
СНз-С —
I
СНз
СНз-
СН2-С---
СНз _п
СНз СНз
СН2—С+ BF3OH + сн2=с
СНз СНз
СНз - СНз -
I I
СНз—С------сн2—с-----
I I
СНз _ СНз _
/СНз
с;
^СН2
СНз
+ СНз-С+ BF3OH
СНз
Действительный путь обрыва цепи определяется конкретной
комбинацией мономера, катализатора и условий реакции.
Координационные катализаторы
Координационные катализаторы подразделяются на две
группы: катализаторы Циглера — Натта, получаемые из металл-
алкила или гидрида металла (восстановителя) и легко восста-
навливающегося галогенида переходного металла, и катализа-
торы, .содержащие восстановленный окисел металла на носителе,
например на окиси алюминия. Типичным представителем ката-
лизаторов первой группы является система триэтилалюминий —
четыреххлористый титан, а катализаторов второй группы —
окись хрома, осажденная на алюмосиликате.
Координационные катализаторы эффективны при полимери-
зации а-олефинов и сопряженных диенов. Детальный механизм
их действия все еще является предметом дискуссии. Однако при
использовании типичных катализаторов Циглера — Натта, по-
видимому, протекают следующие реакции:
TiCl4 4- AIR3 —> TiCl3R -р A1R2C1
TiCl3R —> TiCl3+R--
TiCla + AlRs —> TiCUR + A1R2C1
или
TiCl4 + 2A1R3 —> TiCl2R2 4- 2A1R2C1
TiCl2R2 —> TiCl2R + R.
TiCl2R —> TiCl2 + R.
TiClpR + AJRj —> TiClR2 + A1R2C1
TiCl2 + A1R3 —> TiClR + AlR2Cl
Любой предлагаемый механизм полимеризации в присутст-
вии этих катализаторов должен учитывать стереорегулярность
структуры образующихся продуктов, например получение изо-
тактических поли-а-олефинов и цис-1,4-, транс-1,4- или 1,2-изо-
тактических полибутадиенов,
244
Глава 8
Скорость полимеризации с катализаторами Циглера — Натта
зависит от концентрации TiCU, природы взятого мономера, ха-
рактера алкильных групп в алюминийалкиле и мольного отно-
шения алюминийалкила к четыреххлористому титану. Истинный
носитель каталитической активности почти наверняка представ-
ляет собой сложное комплексное соединение, содержащее титан,
алюминий, атомы галогена и алкильные группы, причем поверх-
ность катализатора играет роль матрицы и независимо от гео-
метрической структуры первой присоединившейся молекулы мо-
номера оказывает влияние на стереохимию присоединения сле-
дующей молекулы.
Методы проведения полимеризации
Принято проводить различие между полимеризацией чистого
мономера или его гомогенного раствора и полимеризацией гете-
рогенной дисперсии мономера в несмешивающейся с ним жид-
кости (обычно в воде). Первые два метода называют полиме-
ризацией в массе (блочной полимеризацией) и полимеризацией
в растворе, а гетерогенные процессы подразделяют на суспен-
зионную и эмульсионную полимеризацию. Ниже каждый из этих
методов иллюстрируется примерами, взятыми из промышленной
практики.
Полимеризация в массе
По этому методу иногда проводят свободнорадикальную по-
лимеризацию хлористого винила. Процесс протекает в гетеро-
фазной системе, поскольку поливинилхлорид (ПВХ) нераство-
рим в мономерном хлористом виниле. Преимуществом метода
полимеризации в массе является отсутствие необходимости в
применении эмульгаторов, защитных коллоидов и др. Инициато-
ром обычно служит азо-бцс-изобутиронитрил. Реакцию ведут
в две стадии, повышая температуру к концу полимеризации до
60° С. Степень конверсии мономера на первой стадии состав-
ляет около 10%. На второй стадии уменьшают скорость переме-
шивания и доводят конверсию до 70%. Изменение скорости
перемешивания, как сообщается, обеспечивает благоприятное
распределение размеров частиц образующегося полимера. По-
лимеризация стирола в массе представляет собой гомогенный
процесс, так как полистирол растворим в исходном мономере.
Оба эти процесса были разработаны главным образом немец-
кими фирмами. Полимеризация стирола в массе наиболее пред-
почтительна в том случае, когда главными требованиями к по-
лимеру являются высокая оптическая прозрачность и улуч-
шенные электроизоляционные свойства. Однако она вызывает
серьезные технологические трудности, связанные с отводом
Пластические массы и эластомеры
245
значительной теплоты реакции (63 кДж/моль) и переходом по-
движной, легко перемешивающейся жидкости в высоковязкий
полимер. В типичном непрерывном процессе чистый стирол сна-
чала подвергают термически инициированной предварительной
полимеризации до степени конверсии около 30%. Полученный
сироп полимера в мономере сливают во второй реактор, где его
постепенно нагревают до температуры 200 °C. При этом поли-
меризация доходит практически до конца. Расплавленный поли-
мер освобождают от летучих и экструдируют в ленты, которые
затем измельчают в грануляторе. Высокая конечная темпера-
тура реакции необходима для того, чтобы полимер можно было
перекачивать в расплавленном состоянии по трубопроводу. Од-
нако при этом возникает опасность получения продукта с пони-
женным молекулярным весом, который образуется либо путем
равновесной деполимеризации полимера, либо непосредственно
из мономера.
Полимеризацию метилметакрилата (стр. 265) в массе ис-
пользуют в производстве листового пластика. В полимеризаци-
онную форму, состоящую из двух полированных стеклянных
пластин, плотно вставленных в наружную оболочку, заливают
сироп полимера в мономере. Этот сироп готовят путем препо-
лимеризации чистого метилметакрилата с перекисным инициато-
ром (например, перекисью бензоила) при температуре около
95 °C, причем по достижении заданной глубины превращения
смесь охлаждают, чтобы предотвратить дальнейшую полимери-
зацию. Регулирование глубины преполимеризации представляет
известную трудность, и поэтому вместо преполимера можно
-использовать сироп-раствор, полученный растворением в моно-
мере необходимого количества полимера. Обе половины формы
скрепляют струбцинами, способными поджиматься в процессе
реакции, компенсируя усадку полимеризационной массы. После
этого форму, заполненную сиропом, помещают в печь, где при
постепенном повышении температуры от 40 до 95 °C происходит
окончательная полимеризация. Время пребывания формы в печи
может достигать 1 ч; общая продолжительность процесса со-
ставляет около 15 ч. Затем форму охлаждают и удаляют из нее
готовый лист полимера.
Полимеризация в растворе
Реакции полиприсоединения обычно сопровождаются выде-
лением тепла, и в отсутствие теплоотвода они могут стать не-
управляемыми. Выше уже упоминалось о том, какое важное
значение имеет отвод тепла при блочной полимеризации сти-
рола. Проведение полимеризации в растворе снимает проблемы,
связанные с отводом выделяющегося тепла и образованием
246
Глава 8
вязкого расплава полимера. В непрерывном процессе мономер
смешивают с растворителем и подают в многосекционный реак-
тор, в первой секции которого раствор подогревается до начала
полимеризации, а в последующих — охлаждается с целью регу-
лирования скорости реакции. В последней секции раствор снова
подогревается для завершения полимеризации и облегчения ис-
парения растворителя в заключительном аппарате, где проис-
ходит удаление летучих. Этим методом вырабатывают стироль-
ный гомополимер различных марок. Например, если основные
требования к полимеру — это повышенные прочность и ударная
вязкость, то можно использовать промышленный продукт, мо-
лекулярный вес которого выше, чем у полистирола общего на-
значения. Иногда требуется также, чтобы наряду с этими свой-
ствами полистирол сохранял высокую оптическую прозрачность,
которая теряется в случае его модифицирования каучуком
(стр. 261). Понижая по сравнению с обычным уровнем содер-
жание в полистироле летучих веществ, получают полимер с по-
вышенной температурой размягчения. Для улучшения техноло-
гических свойств некоторых полимеров в них вводят внутренние
смазки типа жидких парафинов; однако обычно это приводит
к понижению температуры размягчения, вследствие чего такие
материалы в большинстве случаев используют только для фор-
мования изделий сложного профиля.
Суспензионная полимеризация
Методом суспензионной полимеризации в США производит-
ся основная часть поливинилхлорида. Реакцию проводят в вод-
ной дисперсии хлористого винила. Свойства полученного поли-
мера зависят от подбора суспендирующего агента, который
определяет размер частиц полимера, их форму и пористость.
В свою очередь от характера частиц зависят насыпной вес по-
лимера, его сыпучесть, способность абсорбировать пластифика-
тор и легкость переработки. Типичными суспендирующими
агентами являются поливиниловый спирт, водорастворимые про-
изводные целлюлозы и желатина, которые используют в концен-
трации 0,05—0,5% по весу. Иногда к полимеризационной смеси
добавляют также 0,03—0,07% (по весу) эмульгатора, например
сульфированного масла или сложного эфира, облегчающего ре-
гулирование размера и формы диспергированных частиц. Поли-
меризацию проводят в присутствии свободнорадикального ини-
циатора, растворимого в мономере (например, перекиси доде-
цила), при температуре 50—60 °C и давлении около 0,7 МН/м2
(7 атм). Продолжительность реакции в этих условиях обычно
Пластические массы и эластомеры
247
составляет 12—15 ч. Понижение температуры вызывает увели-
чение молекулярного веса и уменьшение степени разветвленно-
сти полимера. На размер частиц и, следовательно, свойства по-
лученного полимера могут оказывать влияние и механические
факторы, такие, как скорость перемешивания, конструкция и
размещение перегородок в полимеризаторе и др. По окончании
полимеризации суспензию мономера выгружают из аппарата,
отгоняют из нее остатки мономера и подвергают центрифугиро-
ванию. Порошок полимера промывают водой, сушат и упаковы-
вают в мешки.
Суспензионная полимеризация представляет собой, вероятно,
наиболее распространенный промышленный метод получения
полистирола. Периодический процесс можно проводить, диспер-
гируя стирол в воде и добавляя к смеси растворимый в моно-'
мере перекисный инициатор, например перекись бензоила. По-
скольку отводить тепло от маловязкой водной системы сравни-
тельно легко, здесь не возникает затруднений, имеющих место
при блочной полимеризации. Полимеризационную массу стаби-
лизируют добавками суспендирующих агентов, таких, как поли-
виниловый спирт; дополнительное регулирование размера час-
тиц осуществляют путем подбора скорости перемешивания, кон-
струкции полимеризатора и т. д. После окончания реакции не-
превращенный мономер и другие летучие вещества удаляют
перегонкой с паром. Недостатками этого процесса являются
низкий коэффициент использования объема реактора (так как
большую часть полимеризационной смеси составляет вода) и
получение полимера в виде, делающем необходимым его экс-
трудирование и грануляцию с целью придания ему формы,
удобной для дальнейшей переработки.
Разработана также технология суспензионной полимериза-
ции метилметакрилата. Суспензионный полиметилметакрилат
можно перерабатывать методами формования из расплава, тог-
да как блочный полимер имеет слишком высокий молекулярный
вес и непригоден для этой цели. Как и в других процессах сус-
пензионной полимеризации, проблемы, связанные с регулиро-
ванием размера частиц и их агрегацией, решаются с помощью
тщательного подбора конструкции реактора и природы суспен-
дирующих агентов, защитных коллоидов и т. д. Типичными сус-
пендирующими агентами являются окись алюминия, тальк и
карбонат магния, а типичным защитным коллоидом — натрие-
вая соль полиметакриловой кислоты. Путем суспензионной по-
лимеризации получают также сополимеры, содержащие неболь-
шое количество звеньев акрилового сомономера, например этил-
акрилата; преимущество этих сополимеров заключается в их
повышенной термостойкости.
248
Глава 8
Эмульсионная полимеризация
Эмульсионная полимеризация отличается от суспензионной
тем, что ее проводят в присутствии мыла, образующего ми-
целлы (гл. 12). Исходный мономер либо находится в виде мель-
чайших капель, стабилизированных за счет адсорбционного слоя
молекул мыла, либо солюбилизирован в мицеллах мыла. Метод
эмульсионной полимеризации широко применяется в Западной
Европе для производства поливинилхорида (стр. 258). Как и
при суспензионной полимеризации, диспергирующей средой слу-
жит вода, которая облегчает отвод выделяющегося тепла. Кис-
лород ингибирует полимеризацию, и поэтому тщательно следят
за тем, чтобы полимеризатор и вода не содержали даже следов
кислорода. В качестве инициаторов можно использовать соеди-
нения, растворимые либо в воде, либо в мономере; однако чаще
всего применяют водорастворимые инициаторы типа персуль-
фата калия. Иногда, особенно при низкотемпературной полиме-
ризации, употребляют окислительно-восстановительные системы,
в которых инициатор активируется восстановителем, например
сульфитом натрия или сернистой кислотой. Эмульсии этого типа
обычно стабилизируют такими эмульгаторами, как сульфонаты,
используемыми в сочетании с водорастворимыми полимерами,
например поливиниловым спиртом. Последний выполняет функ-
цию защитного коллоида, подавляя агрегацию частиц. Выбор
эмульгатора и защитного коллоида имеет большое значение,
так как они могут в значительной степени переходить в товар-
ный полимер.
Компаундирующие добавки
Поскольку поливинилхлорид обладает недостаточной термо-
стойкостью и склонен прилипать к металлическим поверхностям
в процессе переработки, обычно его компаундируют рядом доба-
вок. Путем компаундирования (смешения с добавками) можно
получить материалы с самыми разнообразными полезными свой-
ствами. Кроме основного полимера, компаундированный поли-
винилхлорид может содержать следующие добавки: пластифи-
катор, термостабилизатор, краситель, светостабилизатор, напол-
нитель, смазку и вторичный пластификатор. Детальное рассмо-
трение всех этих веществ не входит в задачу данной главы;
поэтому ниже обсуждаются лишь наиболее важные компаунди-
рующие ингредиенты.
Термостабилизаторы
Наглядным доказательством термической деструкции поли-
винилхлорида является постепенное изменение его цвета. При
обычной температуре переработки (150—200 °C) поливинилхло-
Пластические массы и эластомеры 249
рид из белого становится бледно-желтым, затем оранжевым,
коричневым и, наконец, черным. Термостабилизатор — это веще-
ство, замедляющее изменение окраски полимера. Другие методы
подбора термостабилизаторов, такие, как измерение скорости де-
гидрохлорирования поливинилхлорида, менее надежны, чем цве-
товой тест. К наиболее важному типу термостабилизаторов от-
носятся соединения свинца, которые реагируют с выделяющимся
при деструкции поливинилхлорида хлористым водородом, обра-
зуя хлорид свинца. Чаще всего, по-видимому, употребляется
основной карбонат свинца. Однако его недостатком является
способность выделять двуокись углерода, что может привести
в некоторых условиях к вспениванию материала. По этой при-
чине в настоящее время все более широкое применение находит
сульфид свинца (особенно в составе жестких поливинилхлорид-
ных композиций). Другие соединения свинца применяются лишь
в специальных случаях. Так, средний фталат свинца служит
стабилизатором в композициях, используемых для изготовления
термостойкой электроизоляции и патефонных пластинок типа
«хай-фи». Помимо соединений свинца, которые.-являются наи-
более распространенными термостабилизаторами поливинилхло-
рида, все шире употребляются соли других металлов, в частно-
сти стеараты, каприлаты и пальмитаты цинка и кадмия. Еще
одну группу термостабилизаторов составляют органические со-
единения олова, которые первоначально были использованы из-
за того, что они дают прозрачные композиции. Однако самые
первые представители этой группы, такие, как дикаприлат ди-
бутилолова, не придавали поливинилхлориду достаточную тер-
мостойкость; поэтому в настоящее время они постепенно вытес-
няются другими оловоорганическими стабилизаторами, напри-
мер малеатом дибутилолова.
Пластификаторы
В Великобритании потребление пластификаторов настолько
велико, что они превосходят по объему производства многие
пластмассы, уступая только полиэтилену, полистиролу и са-
мому поливинилхлориду. В сущности пластификатор — это не-
летучий растворитель поливинилхлорида, который легко смеши-
вается с ним при температуре около 150°C и повышает элас-
тичность и прочность материала. Вследствие того что молекулы
пластификатора имеют довольно большие размеры, они очень
медленно диффундируют через полимер при обычной темпера-
туре. Наиболее важным классом пластификаторов являются
эфиры фталевой кислоты, которые синтезируют из фталевой
кислоты или фталевого ангидрида и высших жирных спиртов,
содержащих около 8 атомов углерода. По экономическим
2S0
Глава 8
соображениям чаще всего применяют диизооктилфталат и фта-
латы «оксоспиртов» С7—С9. Использование фосфатных пласти-
фикаторов растет медленнее, чем фталатных, причем объем по-
требления триксиленилфосфата, имеющего более благоприятную
структуру цен, увеличивается быстрее, чем трикрезилфосфата.
Оба эти фосфата придают поливинилхлориду повышенную
огнестойкость по сравнению с аналогичными композициями,
пластифицированными фталатами, а также улучшают стойкость
к действию растворителей. Однако они токсичнее, чем фталаты,
и вызывают увеличение хрупкости полимера при низких темпе-
ратурах. В тех случаях, когда требуется высокая морозостой-
кость, обычно употребляются такие пластификаторы, как али-
фатические сложные эфиры (дибутилсебацинат или диоктил-
азелат).
Вторичные пластификаторы
Вторичные пластификаторы в отличие от первичных сами
по себе не совмещаются с поливинилхлоридом, и их распреде-
ление в нем достигается за счет первичного пластификатора.
Обычно вторичные пластификаторы дешевле первичных, и ими
можно заменять до одной трети первичного пластификатора без
существенного изменения свойств композиции.
Смазки
СмаЗки применяются для того, чтобы свести к минимуму
адгезию полимера к металлическим поверхностям оборудова-
ния при его переработке в мельницах, каландрах, экструдерах
и др. Как правило, смазки нерастворимы в полимере, и поэтому
в процессе его переработки на границе между полимером и по-
верхностью оборудования образуется тонкая смазывающая
пленка. Примерами смазок могут служить стеариновая кислота,
стеарат кальция и средний стеарат свинца. Особенно тщатель-
ного подбора смазочных материалов требует непластифициро-
ванный поливинилхлорид, промышленные марки которого мо-
гут содержать два или более смазочных материала.
Важнейшие пластические массы
Полиэтилен —[—СЩСНг—]„—
Полиэтилен был открыт в 1932 г. в ходе проводимого фир*
мой ICI изучения химических реакций при высоком давлении.
Нагревание смеси этилена с бензальдегидом при температуре
170 °C и давлении 140 МН/м2 (1400 атм) привело к образова-
Пластические массы и эластомеры 251
нию белого твердого вещества, которое оказалось полиэтиле-
ном. Этот продукт представлял собой то, что сейчас называют
полиэтиленом низкой плотности (высокого давления). Неболь-
шие значения плотности и температуры плавления полиэтилена
низкой плотности по сравнению с полиметиленом объясняются
разветвленностью его цепи. Во время второй мировой войны
объем производства полиэтилена высокого давления увели-
чился и к 1945 г. достиг 15 тыс. т/год. Усовершенствование
технологии получения и переработки полиэтилена в послевоен-
ные годы привело к очень быстрому росту производства поли-
мера низкой плотности. Однако наиболее важным достижением
этого периода явилось открытие Циглером в 1953 г. полиэти-
лена высокой плотности. Циглер установил, что соединение,
образующееся при взаимодействии четыреххлористого титана
с- алюминийалкилами, способно вызывать полимеризацию эти-
лена при умеренных температурах и атмосферном давлении.
Благодаря своей более линейной структуре полученный поли-
мер имел более высокую степень кристалличности, чем поли-
этилен низкой плотности, что обусловливало его повышенные
температуру плавления и механическую прочность. Приблизи-
тельно в одно время с открытием Циглера фирма Phillips Pe-
troleum Со. разработала процесс получения полиэтилена высо-
кой плотности при среднем давлении [4 МН/м? (40 атм)]; ката-
лизатором реакции служил СгОз на алюмосиликатном носи-
теле. Полученный этим методом продукт обычно даже более
линеен, чем полиэтилен, синтезированный по методу Циглера.
Полиэтилен низкой плотности
(высокого давления)
При довольно высоких температуре и давлении этилен пре-
вращается в полимер, имеющий большой молекулярный вес.
Полимеризация протекает по свободнорадикальному механизму
в массе (т. е. в жидком этилене), газовой фазе, эмульсии или
растворе. В соответствующих условиях реакция может иници-
ироваться большинством свободнорадикальных инициаторов,
например перекисями, гидроперекисями, персульфатами, азо-
соединениями и просто кислородом. От выбора инициатора за-
висят температура и давление, при которых осуществляют
полимеризацию (некоторые примеры, иллюстрирующие влияние
инициатора, приведены в табл. 8.2).
Несмотря на то что полимеризация этилена в присутствии
кислорода является самым старым методом получения полиэти-
лена высокого давления (процесс фирмы ICI), механизм этой
реакции окончательно не выяснен. Однако не вызывает сомне-
ния, что он отличается от механизма полимеризации, идущей
252
Глава 8
Таблица 8.2
Условия получения полиэтилена низкой плотности
Инициатор Давление, МН/мг Температура, °C
Азо-бис-изобутиронитрил 700 а 55
Перекись бензоила 75 75
Гидроперекись трет-бутила 75 100
Кислород 100-150 100-190
а Данные о промышленном производстве полиэтилена в указанных условиях отсут-
ствуют. — Прим. ред.
под действием других радикальных инициаторов. Вероятно,
этот процесс протекает через перекисное промежуточное соеди-
нение, образующееся in situ, поскольку он включает начальный
индукционный период, в течение которого происходит только
поглощение кислорода. Наблюдаемая затем полимеризация мо-
жет протекать двояким образом. Вначале происходит быстрая
реакция, а за ней в ряде случаев может следовать более мед-
ленная. Быстрая реакция возможна только в особых усло-
виях— при определенной концентрации кислорода, температуре
и давлении. Более медленная полимеризация может продол-
жаться при гораздо меньшем давлении, но обязательно после
того, как начнется более быстрая. Эти закономерности катали-
зируемой кислородом полимеризации этилена позволили про-
вести некоторую аналогию между данной реакцией и так назы-
ваемым «вырожденным взрывом» и постулировать промежуточ-
ное образование полимерной перекиси, которая далее претерпе-
вает гомолитический распад. Образование перекиси объясняет,
в частности, существование индукционного периода. Этот меха-
низм довольно хорошо согласуется с кинетическими данными.
Полимеризация этилена в присутствии других источников
свободных радикалов, например перекиси трет-бутила, обнару-
живает иные закономерности, чем реакция, инициируемая кис-
лородом. Оптимальные условия процесса зависят от природы
инициатора. Так, в случае перекиси бензоила повышение давле-
ния подавляет ветвление цепи и приводит к получению поли-
меров с т. пл. 128—133 °C.
На рис. 8.1 приведена технологическая схема типичного про-
цесса блочной полимеризации этилена при высоком давлении.
Этилен, используемый для получения полиэтилена низкой
плотности, должен иметь высокую степень чистоты. В том слу-
чае, когда инициатором полимеризации служит кислород, его
содержание в этилене должно быть точно известно и не превы-
Пластические массы и эластомеры
253
шать 0,002% по весу, а при применении других инициаторов
концентрация кислорода должна быть ниже 0,005% по весу.
Газообразный мономер компримируют и подают в реактор, где
его нагревают, поднимая давление до требуемого уровня.
В другом варианте этилен перед подачей в реактор сжижают
при меньшем давлении и более низкой температуре. Конструк-
ция реактора должна обеспечивать точное регулирование дав-
ления, эффективный отвод тепла, равномерную подачу этилена
Этилен на
Гранулированный
полиэтилен
Рис. 8.1. Принципиальная технологическая схема процесса получения
полиэтилена при высоком давлении, разработанного фирмой ICI.
1—компрессор; 2 —теплообменник; 3—реактор [190 °C, 150 МН/м2 (1500 атм)];
4—дроссельный вентиль; 5—сепаратор; 6—холодильник; 7—гранулятор.
и вывод продукта. Отсутствие надлежащего контроля за усло-
виями реакции может привести к образованию сшитого поли-
меря и даже к взрыву. По сравнению с обычными реакторами
типа автоклава с мешалкой трубчатые реакторы облегчают
управление процессом и позволяют варьировать температуру
в реакционной зоне, где она обычно сначала повышается до не-
которого максимума, а затем понижается к выходу из реактора.
При инициировании кислородом оптимальными являются тем-
пература 180—200°С и давление около 150 МН/м2 (1500 атм);
полученный полимер имеет плотность 0,92 г/см3. Этилен при
таком высоком давлении ведет себя как несжимаемая
жидкость, и данную реакцию можно рассматривать как поли-
меризацию в растворе жидкого мономера. Если используются
другие инициаторы, то они обычно находятся в растворе. В не-
которых случаях применяются также растворители, главное
назначение которых заключается в том, чтобы облегчить отвод
тепла и удаление полимера из реактора. Иногда растворитель
служит и агентом передачи цепи; однако чаще всего добавляют
254
Глава 8
специальные агенты передачи цепи, которые помогают регули-
ровать молекулярный вес и степень разветвленности полимера.
Полученный полимер легко удаляется из реактора и не нуж-
дается в дополнительной очистке. Непревращенный этилен
отделяют дросселированием, а расплавленный полиэтилен экс-
трудируют, охлаждают, гранулируют и упаковывают.
Полиэтилен высокой плотности
(низкого давления)
Производство полиэтилена высокой плотности удобно рас-
сматривать, исходя из природы применяемого катализатора.
Существует два типа катализаторов полимеризации этилена
при низком давлении: катализаторы Циглера и окиснометал-
лические катализаторы.
Полимеризация этилена в присутствии катализаторов Циг-
лера. Катализаторы Циглера представляют собой комплексные
вещества, получаемые как в растворимой, так и в нераствори-
мой форме. Нерастворимые (осажденные) катализаторы, кото-
рые чаще всего используются в промышленности, получают
путем обработки раствора соединения переходного металла
(обычно TiCU или VOC13) металлоорганическим соединением,
например триэтилалюминием, диэтилалюминийхлоридом, бутил-
литием или диэтилцинком. Первичными продуктами реакции
TiCU с диэтилалюминийхлоридом являются осадок метаста-
бильного Т1С13 и этильный радикал, который далее диспропор-
ционируется на этан и этилен:
TiCU + (С2Н5)2А1С1 —> С2Н8А1С12 + С2Н5Т1С13
C2H6TiCl3 —> С2Н5 • -|- TiCI3
2С2Н5 • —► С2Н6+С2Н4
Для образования активного катализатора необходимо, чтобы
после завершения восстановления в реакционной смеси оста-
вался избыток металлалкила. Активный катализатор, вероятно,
представляет собой восстановленный твердый хлорид титана,
на поверхности которого находится хемисорбированное алкил-
алюминиевое соединение (или соединения). Валентность пере-
ходного металла в катализаторах Циглера зависит от мольного
соотношения обоих компонентов и восстановительной активно-
сти металлоорганического соединения. Мольное соотношение
алюминия и титана оказывает прямое влияние на скорость по-
лимеризации и молекулярный вес полимера; обычно используют
соотношения от 1:1 до 2:1. Катализаторы Циглера чувстви-
тельны к методу приготовления, и их активность может изме-
няться в зависимости от порядка смешивания компонентов, кон-
центрации и температуры исходных растворов, а также от при-
Пластические массы и эластомеры
255
роды растворителя, выбранного в качестве реакционной среды,
и степени его чистоты. В большинстве случаев растворителями
служат алифатические углеводороды. Полярные растворители
(простые и сложные эфиры, кетоны и т. п.), а также следы кис-
лорода, двуокиси углерода и воды ингибируют полимеризацию.
Обычно реакцию ведут при температурах намного ниже тем-
пературы плавления полимера, который в этих условиях прак-
тически нерастворим. Полимеризация протекает в пасте или
суспензии, причем полученный после окончания реакции пасто-
образный продукт содержит не только полимер, но и катализа-
тор. Все операции с катализатором должны проводиться в
атмосфере инертного газа, такого, как азот. Активный катали-
затор можно готовить несколькими способами, например непре-
рывно синтезировать его из компонентов прямо в реакторе или
осуществлять взаимодействие компонентов в другом аппарате
и подавать в реактор уже готовый активный катализатор или
частично прореагировавшую смесь компонентов в виде густой
суспензии в разбавителе. Обычно полимеризацию проводят при
температуре 50—70°C и давлении 0,2 МН/м2 (2 атм). Продол-
жительность реакции в полимеризаторах периодического дей-
ствия составляет 1—4 л (в непрерывных процессах она прибли-
зительно такая же). Молекулярный вес продукта можно регу-
лировать различными путями, в частности варьируя состав и
концентрацию катализатора и температуру реакции или добав-
ляя в полимеризационную систему родород. По достижении за-
данной степени полимеризации реакцию прерывают добавле-
нием спирта, который солюбилизирует катализатор и облегчает
последующую очистку полимера. Продукт выделяют фильтро-
ванием или центрифугированием; для более полного удаления
остатков катализатора полимер можно промыть спиртом.
Полимеризация этилена в присутствии окиснометаллических
катализаторов. Окиснометаллические катализаторы, которые
часто называют также катализаторами Филлипса, представ-
ляют собой окись хрома, осажденную на двуокиси кремния или
алюмосиликате. Их готовят путем пропитывания носителя вод-
ным раствором СгОз или хромовой соли (нитрата или хлорида).
Обычно концентрация хрома составляет 2—3% от веса носи-
теля. Полученный катализатор активируют нагреванием в токе
сухого воздуха при температуре 400—800 °C. Перед нагрева-
нием в катализатор можно ввести промоторы, такие, как ВаО,
СиО или Fe2O3. Установлено, что с повышением температуры
активирования катализатора содержание в нем шестивалент-
ного хрома и молекулярный вес образующегося полимера
уменьшаются.
Полимеризация этилена в присутствии окиснометаллических
катализаторов представляет собой твердофазно-жидкофазный
256
Глава 8
суспензионный процесс, протекающий на поверхности катализа-
тора, диспергированного в углеводородном растворителе (изо-
октане или циклогексане). В отличие от полимеризации с ката-
лизаторами Циглера, которую можно осуществлять как по
периодическому, так и по непрерывному методу, процесс фирмы
Phillips всегда проводят непрерывно при температуре несколько
ниже или выше температуры плавления полимера (125—
160°C) и давлении, превышающем 4 МН/м2 (40 атм). Катали-
затор обычно берется в количестве 0,5% от веса растворителя,
причем он должен быть равномерно распределен в раствори-
теле по всей реакционной зоне. В нижнюю часть реактора
подают чистый мономер, а из верхней его части вытекает рас-
твор полимера, содержащий небольшое количество диспергиро-
ванного катализатора. Время от времени в реактор добавляют
свежий катализатор, чтобы обеспечить его постоянную концен-
трацию в полимеризационной смеси. Полученный раствор поли-
этилена отделяют от непревращенного мономера, фильтруют
(с целью удаления катализатора) и охлаждают. Выпавший в
осадок полимер отделяют центрифугированием.
Полипропилен —[—СН(СН3)СН2—]п—
До середины 1950-х гг. все попытки получить полиолефины
из иных мономеров, чем этилен и изобутилен, приводили к об-
разованию лишь низкомолекулярных продуктов, промышленная
ценность которых невелика. Причиной этих неудач является
протекание реакций переноса активного центра (путем отрыва
атома водорода от олефина), конкурирующих с реакциями ро-
ста цепи путем присоединения радикала. Однако в 1954 г.
Натта, продолжая исследования Циглера, обнаружил, что неко-
торые биметаллические катализаторы циглеровского типа спо-
собны превращать пропилен и многие другие а-олефины, в част-
ности 4-метилпентен-1 и бутен-1, в кристаллические полимеры.
Путем небольших изменений состава и физической природы
катализаторов этому ученому удалось получить несколько ви-
дов высокомолекулярного полипропилена, значительно разли-
чающихся по свойствам. При дальнейшем изучении было уста-
новлено, что эти свойства обусловлены различной стереорегу-
лярностью полученных продуктов (см. выше). Изотактический
полипропилен оказался похожим во многих отношениях на по-
лиэтилен высокой плотности, тогда как атактическая форма
полипропилена характеризовалась аморфной структурой и низ-
кими прочностными характеристиками. Метильные группы, свя-
занные с альтернантными атомами углерода основной цепи,
оказывают разностороннее влияние на свойства полимера. Так,
с одной стороны, они увеличивают жесткость макромолекуляр-
Пластические массы и эластомеры
257
ной цепи и обусловливают более высокую температуру плавле-
ния полипропилена по сравнению с полиэтиленом. Однако, с
другой стороны, они могут нарушать симметричность цепи и
уменьшать общую степень кристалличности полимера, наиболее
важным следствием чего является существование полипропи-
лена в нескольких стереорегулярных формах. Изотактическая
форма обладает самой высокой упорядоченностью, так как в
ней все метильные группы располагаются по одну сторону
основной цепи. Такие молекулы не могут упаковываться в кри-
сталлическую структуру полиэтилена (плоский зигзаг), по-
скольку этому мешают объемистые метильные группы. Поэтому
кристаллический изотактический полипропилен имеет структуру
спирали, на каждый виток которой приходится три мономерных
звена. Промышленный полипропилен содержит до 95% изотак-
тической формы; остальное составляет атактическая (неупоря-
доченная) форма. Последняя может присутствовать в поли-
мере как в виде отдельных молекул высокого молекулярного
веса, так и в виде сегментов различной длины, включенных в
изотактическую цепь. Общий характер влияния структуры по-
лимера на его свойства к настоящему времени уже выяснен.
Так, атактический полимер — это аморфный каучукоподобный
материал, растворимый во многих органических растворителях
и почти не представляющий интереса для промышленности.
В то же время изотактический полимер — жесткий высококри-
сталлический материал, который растворяется только при на-
гревании выше температуры плавления. Получение полипропи-
лена в присутствии катализаторов Циглера — Натта имеет не-
которое сходство с получением полиэтилена высокой плотности
в присутствии катализаторов Циглера. Исходный мономер дол-
жен быть тщательно очищен и высушен; то же самое относится
и к остальным компонентам полимеризационной смеси. Катали-
затор обычно готовят из треххлористого титана и диэтилалю-
минийхлорида. Последние реагируют между собой в инертном
углеводородном растворителе в атмосфере азота с образова-
нием суспензии активного катализатора, содержащей около
10% твердого вещества. При работе по периодическому способу
эту суспензию вместе с мономером и дополнительным количе-
ством углеводородного растворителя загружают в автоклав.
Реакция протекает при небольшом давлении и температуре
около 60 °C в течение 8 ч. Поскольку температура реакции на-
много ниже температуры плавления полимера (170°C), обра-
зующийся полипропилен не переходит в раствор и получается
в виде суспензии. По окончании реакции содержимое реактора
состоит в основном из изотактического полимера (с небольшой
примесью атактической формы), растворителя, катализатора и
непревращенного пропилена. Оставшийся мономер отдувают, а
9 Зак. 501
258
Глава 8
суспензию полимера подвергают центрифугированию. При этом
удаляется большая часть растворителя вместе с растворимым
в нем атактическим полимером. Нерастворимый изотактический
полимер обрабатывают в центрифуге метанолом, содержащим
небольшое количество хлористого водорода, который разлагает
и солюбилизирует катализатор. После дополнительной про-
мывки метанолом полипропилен сушат, смешивают с соответ-
ствующими антиоксидантами, экструдируют и таблетируют.
Изотактический полипропилен высокого молекулярного веса
находит широкое применение. Основная часть полипропилена
перерабатывается путем литья под давлением, а также исполь-
зуется для получения упаковочной пленки и полипропиленового
волокна.
Поливинилхлорид —[—СПС1СП2—]и—
По объему производства поливинилхлорид наряду с поли-
этиленом является одним-из важнейших полимеров, используе-
мых для получения пластмасс. Своему широкому применению
поливинилхлорид в значительной степени обязан успехам в со-
здании для него эффективных термостабилизаторов, поскольку
нестабилизированный гомополимер имеет низкую термостой-
кость и плохо перерабатывается в расплавленном состоянии.
Технологическое поведение поливинилхлорида можно также
улучшить путем понижения температуры формования за счет
введения в него звеньев другого мономера, например винил-
ацетата. Однако этот метод получил меньшее распространение.
Компаундирование поливинилхлорида трикрезилфосфатом и
другими нелетучими жидкостями, приводящее к получению тер-
мостабильного продукта, является одним из наиболее важных
направлений улучшения свойств этого материала.
Промышленное производство поливинилхлорида было на-
чато в Германии и США за несколько лет до второй мировой
войны. Во время войны был налажен выпуск поливинилхлорида
в Великобритании, где его применяли, в частности, как замени-
тель резины для изоляции электрических проводов и кабелей.
В первый послевоенный период основное внимание исследова-
телей было сосредоточено на поисках пластификаторов для
поливинилхлорида; однако некоторый прогресс был достигнут
также в производстве жесткого непластифицированного поли-
мера. Кроме того, были разработаны сополимеры хлористого
винила, предназначенные для покрытия полов, изготовления
патефонных пластинок и других целей. Главные достижения
последних лет связаны с усовершенствованием самого процесса
получения поливинилхлорида, причем регулирование размеров,
ситового состава, пористости и других морфологических харак-
Пластические массы и эластомеры
259
теристик частиц продукта полимеризации позволило значи-
тельно облегчить его переработку.
Полезно сопоставить свойства поливинилхлорида и полиэти-
лена в связи с различиями в их структуре. В поливинилхлориде
имеется более сильное межмолекулярное взаимодействие, обус-
ловленное присутствием в цепи атомов хлора, что приводит к
получению более твердого и жесткого материала с гораздо бо-
лее высокой температурой стеклования. Кроме того, из-за влия-
ния атомов хлора поливинилхлорид значительно полярнее по-
лиэтилена и обладает более высокой диэлектрической прони-
цаемостью. Рентгеноструктурные данные показывают, что
степень кристалличности поливинилхлорида очень мала (5%)
и что промышленный полимер имеет почти целиком атактиче-
скую структуру с лишь небольшими включениями коротких
синдиотактических сегментов. Опытами по восстановлению про-
мышленного поливинилхлорида было также установлено нали-
чие у него значительной, хотя и переменной по величине, сте-
пени разветвленности.
Низкая термостойкость поливинилхлорида несколько неожи-
данна, поскольку подобные ему по структуре хлоруглеводороды
небольшого молекулярного веса термически устойчивы. Было
высказано предположение, что она обусловлена структурными
дефектами, такими, как ветвление, изменения типа тактичности
и др. В промышленности поливинилхлорид получают путем сво-
боднорадикальной полимеризации хлористого винила, иници-
ируемой перекисями, азосоединениями, персульфатами и т. п.,
которую проводят в суспензии, эмульсии или массе.
Политетрафторэтилен —[—С F2C F2—]n—
Полимеризация тетрафторэтилена была впервые описана
в 1941 г.; вскоре после этого началось производство поли-
тетрафторэтилена в полупромышленном масштабе. Мономер
CF2=CFa полимеризуется по свободнорадикальному механизму
в присутствии небольшого количества кислорода. Реакция про-
текает при температуре 50—200°C и давлении около 7 МН/м2
(70 атм). Обычно полимер получается в виде водной суспензии,
содержащей до 60% твердого вещества; однако для переработки
методами экструзии и прессования выпускается также гранули-
рованный и порошкообразный политетрафторэтилен.
Хотя политетрафторэтилен обладает линейной структурой и,
следовательно, должен относиться к термопластичным материа-
лам, его поведение является необычным, так как он не перехо-
дит в жидкое состояние даже при 450 °C. При температуре
около 350 °C образуется гель, причем достаточно большая по-
движность молекул полимера при этой температуре вызывает
9*
260
Глава 8
слипание его частиц. Это явление лежит в основе большинства
процессов формования политетрафторэтилена, которые, таким
образом, сходны с процессами, используемыми в порошковой
металлургии и производстве керамики.
Водные суспензии политетрафторэтилена применяются глав-
ным образом для нанесения защитных покрытий, которые для
получения гладкой поверхности необходимо прогревать при
температуре выше 300 °C, как и при изготовлении прессованных
изделий из этого полимера. Политетрафторэтиленовые покры-
тия обладают превосходной химической стойкостью; кроме того,
они широко используются для получения скользких поверхно-
стей, не обладающих липкостью.
Полистирол —[—СН(С6Н5)СН2—]и—
Гомополимер стирола по своему промышленному значению
занимает третье место среди термопластичных материалов, вы-
рабатываемых в Великобритании. Его производство, как и про-
изводство полиэтилена и поливинилхлорида, было в значитель-
ной степени стимулировано второй мировой войной. Существен-
ное увеличение мощностей по получению стирола-мономера и
полистирола в послевоенное время и большие достижения в тех-
нологии пластмасс сделали возможным многотоннажное произ-
водство полистирола в качестве дешевого термопластичного
материала общего назначения. Поскольку этот полимер обла-
дает благоприятным сочетанием таких свойств, как прозрач-
ность, жесткость, легкая перерабатываемость и низкая стои-
мость, его потребление за послевоенные годы заметно возросло.
Промышленные марки полистирола обычно получают ради-
кальной полимеризацией стирола
R*
С6Н6СН=СН2 —> — [—СН(С8Н5)СН2—]„—
В результате этой реакции образуется атактический поли-
мер с неупорядоченным пространственным расположением фе-
нильных групп относительно основной цепи. Поэтому он почти
целиком аморфен и прозрачен. Под влиянием объемистых фе-
нильных групп полимерная цепь становится более жесткой, чем
в полиэтилене, что в сумме с относительно сильным межмоле-
кулярным взаимодействием вызывает повышение температуры
стеклования (до 95 °C) и делает полимер твердым и жестким
при комнатной температуре. Благодаря ряду ценных свойств
полистирол получил широкое распространение для изготовле-
ния разнообразных изделий методами литья под давлением и
вакуум-формования. Кроме того, низкая теплопроводность по-
листирола и легкость получения из него пенопласта обеспечили
Пластические массы и эластомеры
261
широкое применение этого полимера в производстве теплоизо-
ляционных материалов. Основными недостатками полистирола
являются хрупкость и низкая механическая прочность при по-
вышенных температурах. Учитывая первый из них, многие про-
изводители выпускают модифицированный полистирол, обла-
дающий более высокой ударопрочностью (см. ниже).
Полимеризация стирола представляет собой цепную реак-
цию, которая может инициироваться ионами, свободными ради-
калами или нагреванием. Процесс полимеризации проводят в
массе, суспензии, растворе или эмульсии. Последний метод при-
меняется главным образом для получения полистирольного ла-
текса, поскольку эмульгаторы ухудшают электрические и опти-
ческие свойства полимера.
Модифицирование полистирола каучуком
Применение полистирола во многом ограничивается его
хрупкостью. Однако ударопрочность полистирола можно повы-
сить путем сополимеризации стирола с другими непредельными
соединениями, например с бутадиеном. К сожалению, модифи-
цирование полистирола по этому методу обычно сопровож-
дается заметным уменьшением жесткости и понижением темпе-
ратуры размягчения, наступающими задолго до того, как до-
стигается необходимая ударная вязкость. Поэтому чаще всего
используют другой метод — введение в полистирол небольшого
числа звеньев каучука, такого, как бутадиеновый или бутадиен-
стирольный (содержащий около 30% звеньев стирола).
Модифицирование полистирола каучуком можно осуществ-
лять различными путями. В большинстве случаев каучук рас-
творяют в стироле и полученную смесь подвергают полимери-
зации в обычных условиях получения полистирола. Образую-
щийся при этом полимер содержит не только полистирол и
каучук, но и значительное количество привитого сополимера,
в котором к молекулам каучука прикреплены короткие поли-
стирольные боковые цепи. Это придает ему намного более вы-
сокую ударопрочность, чем у простых смесей полистирола с
каучуком. Для обеспечения оптимальных характеристик моди-
фицированного полистирола необходимо тщательно контроли-
ровать степень совместимости стирольного гомополимера и кау-
чука. Полная совместимость приводит лишь к незначительному
улучшению ударной вязкости, тогда как полная несовместимость
вызывает плохую адгезию между обоими компонентами и пони-
жает ударопрочность композиции. Метод прививки стирола к
каучуку позволяет регулировать степень совместимости и полу-
чать оптимальный комплекс свойств материала.
262
Глава 8
Сополимеры стирола
Несколько лет назад началось промышленное производство
сополимеров акрилонитрила и стирола (АС-пластиков). Эти
материалы существенно превосходят гомополистирол по жестко-
сти, температуре размягчения и устойчивости к действию рас-
творителей. Несмотря на то что ударная вязкость АС-пластиков
несколько выше, чем у полистирола, наличие все еще значи-
тельной хрупкости не позволяет использовать их для ряда
ответственных целей, например в качестве заменителей метал-
лов. Поэтому было налажено производство АС-пластиков
модифицированных каучуком. Такие тройные сополимеры акри-
лонитрила, бутадиена и стирола известны под названием
«АБС-пластики». Они представляют собой либо композиции
АС-пластика с бутадиеннитрильным каучуком, либо продукты
прививки акрилонитрила и стирола к полибутадиену. Первые из
них обычно получают путем предварительного легкого сшива-
ния каучука с целью уменьшения совместимости, а послед-
ние — добавлением акрилонитрила и стирола к латексу каучука
с последующим введением, инициатора (например, персуль-
фата) и полимеризацией этой смеси, причем образуется компо-
зиция, содержащая АС-пластик, полибутадиен и их привитой
сополимер.
Как можно видеть, варьируя состав полимеризационной
смеси и условия полимеризации, можно прийти к широкому
набору разнообразных АБС-пластиков. Несмотря на более вы-
сокую по сравнению с полистиролом стоимость, АБС-пластики,
удачно сочетающие в себе такие свойства, как ударопрочность,
приятный внешний вид и твердость, находят широкое примене-
ние для изготовления предметов домашнего обихода, корпусов
телефонных аппаратов, портативных чемоданов, панелей авто-
мобилей и др.
Полиоксиметилен (полиацеталь) —[—СН2О—]то—
Хотя гомополимеры формальдегида известны уже свыше 100
лет, а сам формальдегид уже более 40 лет является дешевым
крупнотоннажным химикатом, промышленный выпуск полиаце-
тального пластика начался лишь в 1959 г., когда фирма du Pont
Со. организовала его производство в США. Спустя три года
другая фирма выпустила на рынок ацетальный сополимер, по-
лученный из триоксана и циклического простого эфира. В даль-
нейшем эти два типа полиацетальных продуктов стали выраба-
тывать и другие фирмы.
Основные свойства полиацеталей — высокие деформационно-
прочностные показатели, твердость, достаточно большая удар-
П ластические массы и эластомеры
263
ная вязкость и значительная температура плавления (190 °C) —
позволяют отнести их к группе синтетических полимеров, изве-
стных под названием «конструкционные термопласты». Этим
Термином принято обозначать пластические материалы с повы-
шенной механической прочностью, такие, как поликарбонаты,
полиамиды и полисульфоны, имея в виду не только перечислен-
ные выше физико-механические свойства, но и низкую хладо-
текучесть под нагрузкой и сохранение прочности в широком
интервале температур.
Выше уже упоминалось о существовании двух основных ти-
пов ацетальных полимеров. Различия между ними обусловлены
разным синтетическим подходом к решению проблемы стабиль-
ности полимера. Чистый формальдегид легко полимеризуется по
цепному механизму как в водных, так и в неводных средах. Эта
реакция протекает очень быстро и сопровождается выделением
тепла. Она инициируется различными ионами и приводит к об-
разованию соединений, которым обычно приписывается струк-
тура полиоксиметиленов
пСН2О —> НОСН2[ОСН2]„-2ОСН2ОН
Реакция полимеризации обратима, что вполне понятно, если
учесть полуацетальный характер концевых групп. Для получе-
ния полимера с приемлемыми механическими свойствами необ-
ходимо, чтобы он имел молекулярный вес выше 30 000. Однако
Вследствие своей термической нестабильности полиоксиметилен
деполимеризуется при температуре плавления, что делает не-
возможным его формование из расплава. Селективная этерифи-
кация концевых гидроксильных групп с образованием простых
или сложных эфиров позволяет повысить термостойкость поли-
мера. Эти реакции блокирования концевых групп играют важ-
ную роль в технологии производства полиацеталей. Можно так-
же подвергать формальдегид сополимеризации, например, со
стиролом или бутадиеном. В результате этого нарушается пра-
вильное чередование атомов углерода и кислорода в полимер-
ной цепи и повышается термостойкость, поскольку возникает
препятствие ступенчатому отщеплению формальдегидных
звеньев. Сополимеры формальдегида пока еще не приобрели
промышленного значения, однако триоксановые сополимеры, в
которых используется тот же принцип блокирования концевых
групп, уже выпускаются в промышленном масштабе.
Триоксан легко полимеризуется в присутствии катионных
катализаторов по механизму размыкания цикла, образуя линей-
ный полиоксиметилен. Гомополимер, полученный из чистого
триоксана с использованием, например, каталитической си-
стемы фтористый бор — диэтиловый эфир, не отличается по
своим химическим, термическим и механическим свойствам от
264
Глава 8
других полимеров формальдегида. Он гоже нуждается в за-
щите концевых групп, и одним из наиболее удобных методов
такой защиты является сополимеризация. Однако, поскольку
многие ценные свойства полиоксиметиленов непосредственно
связаны с их высокой кристалличностью (70—75%), сущест-
венно, чтобы она в значительной своей части сохранилась и
в готовом изделии. Поэтому необходимо тщательно подбирать
тип и количество сомономера, с тем чтобы свести к минимуму
его влияние на степень кристалличности.
Как и в большинстве других процессов синтеза полимеров,
исходные вещества для получения полиоксиметилена должны
иметь высокую степень чистоты. Первой стадией процесса круп-
нотоннажного производства полиацеталя из формальдегида яв-
ляется приготовление раствора чистого безводного мономера
в инертном растворителе при минусовой температуре. В патент-
ной литературе описаны такие растворители, как пропан, цикло-
гексан и ароматические углеводороды. Запатентовано также
добавление диспергирующих агентов типа полиоксиэтилена, ко-
торые, как сообщается, способствуют образованию тонкодис-
персного полимера, что в свою очередь облегчает последующее
блокирование концевых групп. Полимеризация формальдегида
инициируется такими катализаторами, как карбонилы метал-
лов, фосфины, стибины и амины. Молекулярный вес продукта
можно регулировать добавлением агентов обрыва и передачи
цепи, например воды, этанола или муравьиной кислоты. Для
уменьшения содержания в полимере полуацетальных групп его
обрабатывают уксусным ангидридом, который ацетилирует кон-
цевые гидроксильные группы. Порошкообразный полиацеталь
с защищенными концевыми группами промывают, сушат и на-
правляют на склад.
При сополимеризации триоксана с циклическими простыми
эфирами типа 1,3-диоксолана растворителем служит хлористый
метилен или циклогексан. К нему добавляют мономеры, агенты
передачи цепи и катализаторы и проводят полимеризацию; об-
разующийся полимер осаждается в виде мелкого порошка.
В качестве катализатора обычно Используют эфират фтористого
бора или кислоты Льюиса. Полученный порошкообразный сопо-
лимер стабилизируют с помощью щелочного гидролиза в мяг-
ких условиях, который приводит к отщеплению от концов цепи
формальдегидных звеньев и обнажению звеньев сомономера,
содержащих углерод-углеродные связи. Последние обеспечи-
вают стабилизацию сополимера при его последующей термиче-
ской переработке.
В настоящее время большая часть общего объема мирового
выпуска апетальных смол приходится на долю США. Главной
областью применения этих смол является автомобилестроение
Пластические массы и эластомеры
265
и вообще производство изделий, заменяющих изделия из метал-
лов. Другие важные потребители полиацеталей — производства
средств связи, водопроводной арматуры и предметов домаш-
него обихода.
Полиметилметакрилат
СН3
СН2—С------
СООСН3_„
Полезные свойства полиметилметакрилата были оценены
еще в 20-х годах. Однако его производство стало возможным
только после нахождения удобного пути синтеза метилметакри-
лата. Листовой полимер прекрасно зарекомендовал себя во
время второй мировой войны как материал для остекления
самолетов. В послевоенные годы область применения полиме-
тилметакрилата значительно расширилась, особенно с появле-
нием различных его марок, предназначенных для переработки
методами литья под давлением и экструзии. При этом исполь-
зуется уникальное сочетание в полиметилметакрилате высокой
оптической прозрачности, твердости и светостойкости. Промыш-
ленный полимер получают путем цепной полимеризации моно-
мера в присутствии свободнорадикального инициатора
«СН2=С
I
СООСНз
В результате неупорядоченного пространственного расположе-
ния боковых метильных и сложноэфирных групп, связанных с
альтернантными углеродными атомами основной цепи, обра-
зующийся полимер имеет атактическую структуру и поэтому
аморфен и обладает оптической прозрачностью. Наличие двух
заместителей несколько ограничивает подвижность макромоле-
кулярной цепи в полиметилметакрилате. Этот фактор в соче-
тании с полярностью сложноэфирной группы обусловливает
довольно большую твердость и жесткость полимера и его высо-
кую температуру стеклования, равную 110°С. Полиметилмета-
крилат несколько превосходит по ударопрочности полистирол
общего назначения, но все же сравнительно более хрупок, чем
другие термопластичные материалы, такие, как полиэтилен и
поливинилхлорид. Наиболее важными являются оптические
свойства этого полимера, так как он поглощает очень мало
света и обладает коэффициентом светопропускания др 90% и.
более.
266
Глава 8
Метилметакрилат легко полимеризуется при хранении даже
при обычной температуре. Поэтому обычно в него добавляют
ингибитор, например, гидрохинон, который перед проведением
полимеризации необходимо удалить путем перегонки или про-
мывки водой. Инициаторами полимеризации служат перекиси
или азосоединения; однако присутствия кислорода следует избе-
гать, чтобы свести к минимуму побочные реакции. В промыш-
ленности полиметилметакрилат получают двумя методами: сус-
пензионной полимеризацией, которая дает материал для фор-
мования литьем под давлением и экструзией, или блочной
полимеризацией, при которой полимер образуется в форме
листа или стержня.
Полиэфиры
Полимеры этого класса принято подразделять на сшитые
(термореактивные) и линейные (термопластичные). Промыш-
ленное значение, полиэфиры приобрели в начале XX в., когда
для получения защитных покрытий начали применять сшитые
алкидные смолы. Линейные полиэфиры были впервые изучены
Карозерсом в 30-х годах. Однако их практическое использова-
ние началось лишь в следующем десятилетии — после открытия
волокнообразующих свойств полиэтилентерефталата. Сшитые
полиэфирные смолы, представляющие собой плавкие преполи-
меры, которые теперь в больших количествах используются для
получения стеклопластиков, стали доступными с 1946 г. В на-
стоящее время на долю этих смол приходится основная часть
вырабатываемых сшитых полиэфиров.
Сшитые полиэфиры
До проведения процесса сшивания (отверждения) поли-
эфиры обычно представляют собой вязкие жидкости или твер-
дые вещества с низкой температурой размягчения. Их получают
путем ступенчатой полимеризации гликоля, например пропилен-
гликоля НОСИ (СНз) СН2ОН, со смесью насыщенных и ненасы-
щенных дикарбоновых кислот, например фталевой и малеино-
вой. Последняя обеспечивает создание реакционноспособных
центров для последующего сшивания полиэфира. Перед отвер-
ждением ненасыщенные полиэфиры смешивают с жидким моно-
мером, таким, как стирол или метилметакрилат. Это приводит
к понижению вязкости системы, что облегчает дальнейшую ра-
боту с ней и делает возможным образование поперечных свя-
зей. Непосредственно перед нанесением ненасыщенного поли-
эфира на армирующий наполнитель (обычно стекловолокно)
к нему добавляют свободнорадикальный инициатор. Сшивание
Пластические массы и эластомеры
267
(отверждение) достигается за счет цепной реакции стирола с
ненасыщенными звеньями полиэфирной цепи. Наиболее распро-
страненным армирующим наполнителем является стеклянное
волокно, которое при одной и той же стоимости готового изде-
лия обеспечивает самую высокую механическую прочность. Сте-
кловолокно выпускается в виде ткани, плетеных матов из наре-
занных стеклонитей длиной около 5 см и заготовок, полученных
путем наложения коротких стеклянных нитей на форму и про-
питывания их связующим. С целью увеличения адгезии смолы
к волокну, которая необходима для достижения высоких проч-
ностных показателей изделия, исходное стекловолокно обычно
подвергают химической обработке, способствующей лучшему
сцеплению со смолой. Наиболее важной является обработка
стекловолокна силанами, например винилтрихлорсиланом, кото-
рый реагирует с гидроксильными группами на поверхности сте-
кла и образует с ним химические соединения, содержащие ви-
нильные группы
НО-1-Si— , ,
: I он : I
cH2=cHSici3+ : ----► сн2=сн—si -
: o-+-Si—
HO-j-Si-
Полагают, что при сшивании полиэфирной смолы эти виниль-
ные группы внедряются в ее состав путем сополимеризации и
создают химическую связь между смолой и стекловолокном.
Как уже упоминалось, чаще всего для синтеза полиэфирных
смол применяется пропиленгликоль. Этиленгликоль дает про-
дукты, обладающие худшей совместимостью со стиролом, что
обусловлено более высокой симметричностью цепи образующе-
гося полимера, тогда как другие выпускаемые в промышленно-
сти гликоли, например диэтиленгликоль и дипропиленгликоль,
дают отвержденные смолы с пониженной температурой размяг-
чения и более высоким водопоглощением. Ненасыщенным ком-
понентом в составе большинства полиэфирных смол общего
назначения служат малеиновый ангидрид и (или) фумаровая
кислота, а насыщенным кислотным компонентом — фталевая
кислота, обычно используемая в виде фталевого ангидрида. Ко-
личество малеиновой или фумаровой кислоты в полимеризаци-
онной смеси подбирают таким образом, чтобы обеспечить же-
лаемую концентрацию центров сшивания, поскольку от числа
поперечных связей в отвержденном продукте зависят многие из
его свойств. Фталевая кислота более предпочтительна, чем
288
Глава 8
другие насыщенные дикарбоновые кислоты, так как она дешева
и образует относительно негибкие химические связи, повышаю-
щие жесткость отвержденной смолы. Полиэфиры этого типа
синтезируют путем нагревания смеси дикарбоновых кислот и
(или) их ангидридов с гликолем в заданных соотношениях при
температуре 130—200 °C в атмосфере азота. Продолжитель-
ность реакции может достигать 10 ч. Обычно гликоль берут
в небольшом избытке. Процесс можно вести в присутствии вы-
сококипящего углеводорода, с которым отгоняют в виде азео-
тропа выделяющуюся воду. Ход реакции контролируют по со-
держанию свободных карбоксильных групп в полимеризацион-
ной смеси. По достижении заданной глубины реакции смесь
охлаждают и разбавляют соответствующим количеством сти-
рола (или метилметакрилата). Для предотвращения прежде-
временного гелеобразования и увеличения срока хранения полу-
ченного раствора к нему можно добавить небольшое количество
ингибитора, например гидрохинона. Перед нанесением ненасы-
щенного полиэфира на армирующий наполнитель в смесь вво-
дят перекись (обычно 0,5—2% от веса полиэфира), после чего
проводят отверждение. При изготовлении небольших изделий,
когда отвод выделяющегося тепла не вызывает затруднений,
отверждение можно осуществлять при повышенной темпера-
туре, например при 100 °C, и несколько повышенном давлении.
Однако при изготовлении крупногабаритных изделий и в дру-
гих случаях, когда применение повышенных температур затруд-
нено, отверждение ведут при комнатной температуре. Высоко-
температурное отверждение обычно проводят в присутствии
перекиси бензоила, а низкотемпературное — в присутствии пере-
киси метилэтилкетона или перекиси циклогексанона и актива-
тора (такого, как нафтенат кобальта).
Самым крупным потребителем полиэфирных стеклопласти-
ков является промышленность строительных материалов, при-
чем на производство кровельных и теплоизоляционных матери-
алов расходуется около 30% всей выпускаемой полиэфирной
смолы. Кроме того, полиэфирные стеклопластики широко ис-
пользуются в кораблестроении, химическом и транспортном
машиностроении и производстве спортивного инвентаря.
Линейные полиэфиры
Наиболее известным представителем линейных полиэфиров
является полиэтилентерефталат (т. 1, стр. 170), который нахо-
дит широкое применение в виде волокна и ориентированной
пленки. Синтез этого полимера подробно описан в гл. 9. По-
этому здесь достаточно отметить, что полиэтилентерефталат
лишь совсем недавно стал выпускаться в форме, подходящей
Пластические массы и эластомеры
269
для его переработки общими методами, типичными для термо-
пластичных материалов. Однако масштабы его использования
в производстве пластмасс пока невелики.
Поликарбонаты —[—OCOORO——
Поликарбонаты представляют собой важнейший класс тер-
мопластичных полиэфиров, применяемых для получения пласт-
масс. Впервые поликарбонат, который в сущности представляет
собой производное угольной кислоты Н2СО3, начали вырабаты-
вать в 1958 г. Синтез линейных поликарбонатов можно осуще-
ствить взаимодействием алифатического диола или бисфенола
с производным угольной кислоты. Промышленное значение
имеют лишь ароматические поликарбонаты, которые получают
из бисфенолов, например из дифенилолпропана (бисфенола А).
Полимер на основе последнего соединения является единствен-
ным поликарбонатом, выпускаемым в значительных количе-
ствах. Его производство резко возросло за последние годы, чему
способствовало уменьшение цен на исходное сырье; в настоя-
щее время * общие мировые мощности по получению поликар-
боната превышают 150 тыс. т/год.
Все промышленные марки поликарбоната синтезируют из
дифенилолпропана, который в свою очередь является продуктом
кислотнокаталитической реакции между фенолом и ацетоном
(гл. 7). Поскольку угольная кислота не существует в свободном
состоянии, вместо нее используют ее производные — фосген или
дифенилкарбонат. В основе производства поликарбонатов ле-
жат два метода осуществления ступенчатой полимеризации:
высокотемпературная переэтерификация дифенилкарбоната ди-
фенилолпропаном и низкотемпературное фосгенирование рас-
твора дифенилолпропана в присутствии акцептора хлористого
водорода.
Переэтерификацию проводят путем нагревания смеси дифе-
нилолпропана с дифенилкарбонатом в расплаве при темпера-
туре 200 °C и пониженном давлении до выделения приблизи-
тельно 85% от теоретического количества фенола
СНз
* В 1972 г. — Прим, перев.
270
Глава 8
Затем температуру постепенно повышают примерно до 300 °C
при остаточном давлении 130 Н/м2 (1 мм рт. ст.). В этих усло-
виях по мере выделения оставшегося фенола происходит нара-
стание молекулярного веса полимера. По достижении заданной
вязкости продукт экструдируют из реактора, охлаждают и гра-
нулируют.
Вариантом этого метода является процесс с использованием
двукратного избытка дифенилкарбоната в начальной смеси. Это
обеспечивает более полное вступление дифенилолпропана в ре-
акцию на первой стадии (при температуре 200°C) и сводит
к минимуму его разложение на второй стадии процесса. В при-
сутствии избытка дифенилкарбоната сначала образуется бис-
фенилкарбонат дифенилолпропана, или бис-фенилкарбонат
2,2-бис- (4'-оксифенил) пропана
после чего протекает дальнейшая полимеризация с отщепле-
нием дифенилкарбоната.
Другой метод промышленного получения поликарбоната —
взаимодействие фосгена с дифенилолпропаном либо в водной
эмульсии, либо в органическом растворителе при обычной тем-
пературе. В первом случае водный раствор бисфенолята эмуль-
гируют с органическим растворителем, не смешивающимся с во-
дой (например, с хлористым метиленом), и через эту смесь прр
перемешивании пропускают фосген. Катализаторами фосгени-
рования служат- соли четвертичного аммония. Образующийся
полимер переходит в органический слой, который перед отгон-
кой растворителя дополнительно промывают водой. Во втором
случае дифенилолпропан растворяют в пиридине, который одно-
временно играет роль акцептора хлористого водорода, выделяю-
щегося в ходе реакции. Через этот раствор при температуре
30 °C барботируют фосген, причем в течение нескольких минут
после начала реакции выпадает хлоргидрат пиридина. По мере
протекания полимеризации вязкость раствора увеличивается.
По достижении нужного молекулярного веса полимер выде-
ляют добавлением другого органического растворителя, такого,
как метанол, который растворяет пиридиниевую соль и выса-
живает поликарбонат.
Поликарбонат, синтезированный из дифенилолпропана с по-
мощью описанных методов, обладает особенно ценным сочета-
нием нужных свойств. Это прочный, жесткий и прозрачный
материал (последнее свойство обусловлено почти полной
аморфностью полимера), который благодаря высокой темпера-
Пластические массы и эластомеры
271
туре стеклования (140 °C) сохраняет высокие физико-механиче-
ские характеристики и при температурах выше 100°C. К тому
же поликарбонат — один из наиболее ударопрочных термопла-
стов, что обеспечило возможность его использования для изго-
товления самых различных изделий — от прессованных пред-
метов домашнего обихода и детских бутылочек до корпусов
электроприборов, а также в качестве конструкционного мате-
риала, заменяющего металлы.
Фенолформальдегидные смолы
Фенолформальдегидные смолы представляют собой сильно-
сшитые полимеры, получаемые ступенчатой полимеризацией
фенола с формальдегидом (т. 1, стр. 171). Они являются исто-
рически самыми первыми синтетическими материалами и с на-
чала XX в. находят очень широкое применение. В последние
50 лет объем годового производства этих термореактивных ма-
териалов непрерывно увеличивается; однако в настоящее время
они уступают по темпам прироста производства важнейшим
термопластичным материалам, таким, как полиэтилен и поли-
стирол.
Несмотря на многолетнее использование фенолформальде-
гидных смол, их точная структура до сих пор не установлена,
что объсняется трудностью их изучения, связанной с нераство-
римостью и неплавкостью отвержденных продуктов. Тем не ме-
нее известно, что структура полученной смолы очень сильно
зависит от соотношения фенола и формальдегида. Для практи-
ческих целей требуется, чтобы первичный продукт взаимодей-
ствия между фенолом и формальдегидом был плавким веще-
ством низкого молекулярного веса. Этот низкомолекулярный
преполимер превращают в конечный отформованный продукт
сшитой структуры путем нагревания под давлением. Различают
два типа плавких фенолформальдегидных преполимеров: ново-
лачные смолы и резольные смолы.
Новолачные смолы
Новолачпые смолы получаются при кислотнокаталитической
реакции формальдегида с небольшим избытком фенола (обыч-
ное мольное соотношение 0,8: 1,0). В этих условиях происходит
сравнительно медленное образование о- и п оксиметилфено-
лов *, которые затем быстро реагируют со второй молекулой
* В промышленности пластмасс группу СНзОН принято называть не ок-
симетильной, а метилольной, хотя первое название является более правиль-
ным с точки зрения номенклатуры.
272
Глава 8
фенола, давая метиленбисфенолы (смесь трех изомеров, состав
которой зависит от pH реакционной смеси):
ОН
Дальнейшее взаимодействие бисфенолов с формальдегидом
протекает относительно медленно и приводит к получению поли-
ядерных фенолов. Вследствие того что процесс проводится при
общем избытке фенола, длина цепи образующихся полифено-
лов невелика и в среднем ограничивается пятью ароматиче
скими кольцами, как, например, в соединении 1.
1
По окончании реакции получается новолак, в котором нет
активных оксиметильных групп и поэтому не может происхо-
дить более глубокая поликонденсация. Отсюда следует, что для
сшивания новолачных смол необходимо добавить к ним перед
горячим прессованием источник формальдегида. Обычно в ка-
честве такого источника используют параформальдегид или
гексаметилентетрамин (2) (т. 1, стр. 73). Нагревание смеси со-
провождается дальнейшим образованием метиленовых мостиков
и заканчивается получением неплавкого и нерастворимого сши-
того продукта.
h2c^n^ch2
I 1Н2 I
4
Пластические массы и эластомеры
273
Резальные смолы
В отличие от новолачных смол резольные смолы получают
в присутствии щелочного катализатора и избытка формальде-
гида. Изменение условий приводит к тому, что образование
оксиметилфенолов протекает быстро, а дальнейшая конденса-
ция в бисфенольные и полифенольные соединения — медленно.
Следствием этого различия в скоростях обеих реакций яв-
ляется получение наряду с оксиметилфенолами бис- и трис-
оксиметилфенолов. Кроме того, образующиеся в данных усло-
виях полиядерные соединения обычно имеют несколько более
низкий молекулярный вес, чем новолачные смолы: типичный
жидкий резол содержит в среднем лишь два ароматических
кольца. Поскольку полученные смолы обладают реакционно-
способными оксиметильными группами, их дальнейшее нагре-
вание сопровождается сшиванием за счет реакции этих групп
с незамещенными активированными положениями ароматиче-
ских ядер; отсюда следует, что добавление формальдегида не
обязательно. Общим свойством новолачных и резольных смол
является их растворимость и способность размягчаться и течь
при относительно низких температурах. При повышенной темпе-
ратуре они превращаются в твердые нерастворимые сшитые
продукты (в присутствии источника формальдегида, если он
необходим). Изучение механизма отверждения показало, что
при температурах до ~ 150 °C сшивание протекает путем кон-
денсации оксиметильных групп между собой и с незамещен-
ными активированными положениями соседних бензольных
колец
Полагают, что при более высоких температурах могут про-
исходить и другие реакции сшивания, например образование
метиленхинонов (т. 2, стр. 247) за счет конденсации феноль-
ных оксигрупп с простыми эфирными группами, которое
274
Глава 8
обусловливает обычный темный цвет пресс-изделий, получае-
мых при температуре выше 150 °C.
Важную роль играет природа катализатора, используемого
для сшивания. Например, резол способен отверждаться при
комнатной температуре под действием сильных кислот, причем
с повышением pH смеси скорость отверждения уменьшается,
проходит через минимум (при pH 7) и затем снова возрастает
в щелочной среде.
Фенолформальдегидные смолы применяются для получения
самых разнообразных прессованных изделий. Так, в электротех-
нической промышленности из них делают изоляторы и корпуса
электроприборов, в автомобилестроении — крышки распредели-
телей зажигания, корпуса плавких предохранителей и другие
детали, которые должны сочетать в себе электроизоляционные
свойства с термостойкостью. Кроме того, фенолформальде-
гидные смолы используются в производстве слоистых пласти-
ков, в которых армирующим материалом является, например,
бумага или ткань. В последние годы все больший ’ интерес
привлекает получение фенопластовых пеноматериалов, посколь-
ку они превосходят по огнестойкости пенополистирол.
Мочевиноформальдегидные смолы
Подобно фенольным смолам, мочевиноформальдегидные
смолы после отверждения представляют собой сшитый нерас-
творимый и неплавкий продукт, а до отверждения — низкомо-
лекулярное вещество, отверждаемое в процессе последующей
переработки. Промышленное производство мочевиноформальде-
гидных смол начало развиваться в 20-х годах и с тех пор не-
прерывно расширялось, причем эти смолы используются для
получения адгезивов, текстильно-вспомогательных веществ,
пресс-порошков, защитных покрытий и пеноматериалов. Меха-
низм реакции смолообразования между мочевиной и формаль-
дегидом полностью не выяснен, что, как и в случае фенолформ-
альдегидных смол, является прямым следствием трудности
исследования конечного продукта.
Начальную реакцию мочевины с формальдегидом ведут в
нейтральной или слабощелочной среде (pH 7—8). При этом
происходит образование моно- и бнс-оксиметилмочевины, на-
пример:
2H2NCONH2 + ЗСН2О —► H2NCONHCH2OH + HOCH2NHCONHCH2OH
Если отношение формальдегида к мочевине достаточно велико
для образования значительных количеств бнс-оксиметилмоче-
вины, то последующее нагревание продукта конденсации в сла-
бокцслой среде приводит к получению отвержденной смолы.
Пластические массы и эластомеры
275
Природа реакций, протекающих на этой стадии, неизвестна, од-
нако существует ряд возможных направлений: конденсация
соседних оксиметильных групп с образованием эфирных мости-
ков, конденсация оксиметильных и иминогрупп, образование
метиленмочевины, а также циклических структур.
В качестве примера рассмотрим получение мочевиноформ-
альдегидного пресс-порошка. Такие пресс-порошки нашли боль-
шое применение благодаря низкой стоимости, хорошим элек-
тротехническим свойствам, жесткости и возможности придать
изделиям из аминопластов широкую цветовую гамму. Первой
операцией является доведение pH смеси мочевины с формаль-
дегидом до 7—8 путем добавления основания (обычно едкого
натра). Смесь перемешивают при температуре 40°C в течение
1 ч, не допуская понижения pH. Полученную смолу можно скон-
центрировать, удаляя часть воды, или полностью высушить до
порошкообразного состояния. Как и в производстве фенолформ-
альдегидных пресс-порошков, перед заключительным прессо-
ванием к смоле добавляют наполнитель (древесные опилки или
древесную муку). Кроме того, если надо получить окрашенное
изделие, в состав пресс-порошка вводят различные пигменты.
Наконец, для обеспечения достаточной скорости отверждения
необходимо использовать катализатор, которым обычно яв-
ляются кислоты или соединения, способные выделять кислоту
при повышенных температурах. Примерами таких «скрытых»
катализаторов могут служить триметилфосфат, сульфамат ам-
мония и этиленсульфит. Поскольку мочевиноформальдегидные
смолы неустойчивы при хранении, к ним можно также добав-
лять стабилизатор, например гексаметилентетрамин.
Полиуретаны
Полиуретаны получают взаимодействием ди- или триизоциа-
натов с гликолями или триолами, например л-толуилендиизо-
цианата (2,4-диизоцианотолуола) с тетраметиленгликолем
СНз
J\/NC0
и
NCO
+ НО(СН2)4ОН —>-------COHN
NHCOO(CH2)4O—
Из других изоцианатов в промышленности используются
4,4/-метиленди(фенилизоцианат) CH2(C6H4NCO)2 и гексамети-
лендиизоцианат OCN(CH2)6NCO.
Реакция диизоцианата с диолом обычно приводит к образо-
ванию линейного полимера. Однако при полной или частичной
276
Глава 3
замене бифункциональных реагентов полифункциональными [на-
пример, при использовании полиизоцианата, такого, как триизо-
цианат 3, или триола типа глицерина НОСНгСЩОЬрСНгОН]
можно получить сшитые полиуретаны.
NCO
OCN—К/—СН2—/ —NCO
Линейные полиуретаны находят применение в производстве
эластомерного волокна. В этом случае их можно синтезировать
на основе полиэфиров, содержащих концевые гидроксильные
группы, например из политетраметиленгликоля
НО—[—(СН2)4—О—]„—Н
Главной областью применения сшитых полиуретанов яв-
ляется производство пенопластов. В качестве полиолов при
этом обычно используют алифатические полиэфиры с конце-
выми гидроксильными группами, полученные этерификацией
гликолей жирными дикарбоновыми кислотами. Примером таких
полиэфиров может служить полипропиленадипат (4)
НОСН(СН3)СН2О—[—СО(СН2)4СООСН2СН(СН3)О—]„—Н
4
При обработке этого соединения полиизоцианатом образуются
сшитые полимеры. Если к реакционной смеси добавить неболь-
шое количество воды, то часть уретановых групп превращается
в реакционноспособные амидогруппы, причем выделяется дву-
окись углерода. Подбирая условия проведения процесса, можно
использовать этот газ для вспенивания полимеризационной
смеси с образованием пенопласта. Свойства полученных поли-
меров можно изменять в очень широких пределах путем варьи-
рования отношения полиэфира (простого или сложного) к поли-
изоцианату и природы полиола. Кроме того, добавление раз-
личного количества воды с последующей реакцией изоцианата
с образовавшимися амидогруппами приводит к получению по-
лиуретано-полимочевинных продуктов с новым комплексом
свойств.
Эластомеры
Натуральный каучук
Многие годы натуральный каучук был единственным извест-
ным материалом, обладающим высокой эластичностью, и ши-
роко исследовался как в вулканизованном, так и в невулкани-
зованном виде. Для лучшего понимания различий между нату-
Пластические массы и эластомеры
277
ральным и синтетическими каучуками полезно кратко остано-
виться на этих работах (о биосинтезе натурального каучука
см. т. 4, стр. 221).
Латекс натурального каучука, вытекший из дерева, пред-
ставляет собой дисперсию частиц каучука в водной среде. Свой-
ства этого латекса имеют много общего со свойствами обычных
эмульсий; так, например, он способен коагулироваться. Содер-
жание сухого вещества в латексе в среднем не превышает 40%
по весу, причем приблизительно 90% этого вещества составляет
углеводородный каучук. Элементный состав последнего, как
было установлено Фарадеем еще в 1826 г., отвечает формуле
(С5Н8)П. Вскоре после этого было показано, что каучук яв-
ляется ненасыщенным углеводородом, так как он присоединяет
многие реагенты с образованием продуктов, анализ которых
свидетельствует о присутствии в каждой, группе С5Н8 одной
двойной связи. При деструктивной перегонке каучука был выде-
лен изопрен С5Н8. Поскольку полимеризация изопрена привела
к образованию вещества с каучукоподобными свойствами, было
сделано правильное заключение о том, что молекула натураль-
ного каучука построена из связанных между собой изопреновых
звеньев. Методом озонолиза было доказано, что эти звенья рас-
положены в правильном порядке, хотя роль стереоизомерии
в структуре натурального каучука была выяснена гораздо поз-
же. С помощью явления стереоизомерии можно объяснить раз-
личия в свойствах натурального каучука (гщс-1,4-полиизо-
прена) и гуттаперчи (транс-1,4-полиизопрена).
СН3х хН СН3ч ,н сн3. ,н
... /С=С\ /С=С\ xc=cz
\сн2—СИ/ \сн2—СН2/ \сн2—Сн/ \сн2-----------
натуральный каучук (^ис-полимер)
,CH2—СН2х ,Н СНзх ,СН2—СН2у .н
"Z /С==С\ xc=cz
СН3/ \сн2—СН2/ \н СН3/ \зн2------------
гуттаперча (транс-полимер)
В отличие от натурального каучука гуттаперча практически
не обладает эластичностью и поэтому имеет гораздо меньшее
промышленное значение. Хотя она, подобно каучуку, способна
вулканизоваться, ее применение обусловлено главным образом
термопластичностью и высокой химической стойкостью. Вна-
чале гуттаперчу использовали для изготовления оболочки под-
водных кабелей, а позднее из нее стали делать покрышки мячей
для игры в гольф.
Будучи ненасыщенным соединением, натуральный каучук
теоретически аналогичен по своим химическим свойствам эти-
леновым углеводородам. Однако важным дополнительным
278
Глава 8
обстоятельством является наличие в его макромолекулярной
цепи других реакционных центров — атомов углерода, находя-
щихся в a-положении к двойным связям. Атомы водорода, свя-
занные с этими углеродными атомами, весьма лабильны и легко
отщепляются с образованием реакционноспособных свободных
радикалов.
Несмотря на то что натуральный каучук вступает в реакции
присоединения, характерные для непредельных углеводородов,
эти реакции часто осложняются побочными превращениями,
включающими замещение и циклизацию. Например, реакция
с кислородом, которая имеет наиболее важное практическое
значение, обычно сопровождается разрывом полимерной цепи и
приводит к пластикации каучука и его старению. Эти явления
можно наблюдать даже при поглощении очень небольших коли-
честв кислорода: так, поглощение 1% (по весу) кислорода вы-
зывает полную деструкцию каучука и превращение его в веще-
ство, не обладающее полезными свойствами.
Детальное изучение химической сущности вулканизации
природного каучука показало, что этот процесс включает обра-
зование межмолекулярных связей (сшивание).
Вулканизация каучука представляет собой процесс его пре-
вращения из пластического в эластическое состояние, причем
происходящее при этом изменение химических и физических
свойств каучука значительно расширяет область его примене-
ния. Явление вулканизации было открыто в 1840 г. Чарльзом
Гудьиром, который заметил, что при нагревании пластициро-
ванного каучука с серой получается продукт, обладающий гиб-
костью и эластичностью. Вулканизацию осуществляют путем
добавления к пластицированному каучуку вулканизующего
агента, например серы, с последующей гомогенизацией смеси и
нагреванием ее в пресс-форме (в случае серы до температуры
выше ПО °C). Нагревание сопровождается сшиванием молекул
каучука, причем чем больше число поперечных связей, тем твер-
же полученный продукт. Вулканизованный натуральный каучук
находит большое применение; из него изготовляют шины, пори-
стую резину, подметки для обуви, изоляцию для электрических
проводов и кабелей и др.
Таким образом, вулканизованный каучук имеет структуру
пространственной сетки, в которой число поперечных связей
должно быть не слишком велико, чтобы материал не поте-
рял эластичность и гибкость. С увеличением числа поперечных
связей получается все более жесткий продукт, и в конечном
итоге образуется твердый хрупкий материал — так называемый
эбонит.
Сырой (невулканизованный) каучук — эластичное раствори-
мое вещество, чувствительное даже к небольшому нагреванию.
Пластические массы и эластомеры
279
Поэтому на практике прежде всего понижают молекулярный
вес каучука, чтобы облегчить его дальнейшую переработку, по-
сле которой каучук подвергают легкому сшиванию, чтобы вер-
нуть ему каучукоподобность и повысить эластические свойства.
Снижение молекулярного веса обычно достигается путем меха-
нического воздействия, например вальцеванием, при котором
сырой каучук испытывает деформацию сдвига. В результате
получается частично деструктированный материал, который
легче смешивается с различными добавками (антиоксидантами,
наполнителями, пигментами и др.). Эту смесь подвергают сши-
ванию (вулканизации). Наиболее употребительным вулканизую-
щим агентом является сера, которую обычно применяют в ком-
бинации с ускорителем вулканизации при температуре 150—
200 °C.
Ненаполненный вулканизат натурального каучука обладает
высоким сопротивлением разрыву в растянутом состоянии. Это
непосредственно обусловлено кристаллизацией полимера при
его растяжении. В том случае, когда от вулканизата требуется
высокое сопротивление истиранию и раздиру, в резиновую
смесь вводят тонкоизмельченный наполнитель, например сажу.
Подобным образом путем добавления антиоксидантов улуч-
шают стойкость натурального каучука к окислению. Однако
было найдено, что во многих случаях экономически более целе-
сообразно использовать синтетические каучуки.
Синтетические каучуки
Синтетические каучуки — один из наиболее важных классов
промышленных органических полимеров. Основную массу син-
тетических каучуков получают из диенов и олефинов, но суще-
ствует также большое число каучуков специального назначения,
в том числе фторкаучуки, уретановые, акрилатные, полисуЛь-
фидные и силоксановые каучуки. Хотя по объему мирового
потребления натуральный каучук превосходит любой синтетиче-
ский каучук, взятый в отдельности, по темпам прироста потре-
бления он значительно уступает, например, бутадиенстироль-
ному каучуку.
Бутадиенстирольный каучук. Этот каучук является в настоя-
щее время важнейшим синтетическим каучуком общего назна-
чения. Он нашел широкое применение еще во время второй ми-
ровой войны, что было вызвано сокращением поступления нату-
рального каучука. Обычно сополимер содержит около 75%
звеньев бутадиена и 25% звеньев стирола, причем среди пер-
вых приблизительно 80% составляют транс-\,4-звенья, а осталь-
ное— 1,2-звенья. Сополимеризацию бутадиена со стиролом
осуществляют в водной эмульсии, содержащей персульфат
280
Глава 8
'(инициатор) и меркаптан (агент передачи цепи). Продукт полу-
чается в виде латекса, подобного латексу натурального каучука,
и перерабатывается аналогичным образом, т. е. путем пласти-
кации с последующей вулканизацией приблизительно в таких
же условиях, как в случае натурального каучука.
Синтетический цис-1,4-полиизопрен. Промышленное произ-
водство этого каучука существует с середины 50-х годов. По
большинству свойств он аналогичен натуральному каучуку, но
несколько отличается от него содержанием 1(цс-звеньев, которое
обычно составляет лишь 90—95%. Практическое использование
синтетического цис- 1,4-полиизбпрена несколько сдерживается
его более низкими по сравнению с натуральным каучуком экс-
плуатационными свойствами*.
Синтетический транс-1,4-полиизопрен. Этот каучук также
выпускается в промышленном масштабе. В отличие от ^izc-изо-
мера он часто предпочитается своему природному аналогу (гут-
таперче), так как имеет более низкую стоимость и лучшие экс-
плуатационные свойства.
Этиленпропиленовые каучуки. Синтез этиленпропиленовых
каучуков был осуществлен совсем недавно и явился прямым
результатом появления металлоорганических катализаторов
типа катализаторов Циглера. Чаще всего применяется тонко-
дисперсный катализатор, получаемый из хлорокиси ванадия и
триалкилалюминия. При проведении сополимеризации в инерт-
ном углеводородном растворителе обычно образуется продукт,
содержащий 70—80% этиленовых звеньев. Поскольку реакцион-
ная способность этилена при сополимеризации гораздо выше,
чем пропилена, исходная смесь олефинов обычно содержит
большой избыток пропилена.
Для создания в молекуле этиленпропиленового сополимера
реакционноспособных центров, делающих возможным его вул-
канизацию, к исходному сырью добавляют небольшое количе-
ство третьего сомономера. Обычно им служит несопряженный
диен (например, дициклопентадиен), в котором одна двойная
связь принимает участие в сополимеризации, а вторая исполь-
зуется для образования поперечных связей.
Полиизобутилен. Полиизобутилен был впервые синтезиро-
ван путем низкотемпературной (при —80 °C) полимеризации
изобутилена в растворе под действием фтористого бора. Полу-
* Полиизопреновый каучук (СКИ), вырабатываемый в СССР в крупно-
промышленном масштабе, содержит до 98% цис-1,4-звеньев. По некоторым
физико-механическим свойствам он превосходит натуральный каучук. Лучшая
воспроизводимость свойств СКИ, получаемого по строго определенным техни-
ческим условиям, обеспечивает ему в ряде случаев преимущества перед нату-
ральным каучуком (сб. «Синтетический каучук», под ред. Гармонова И. В.,
«Химия», Л., 1976, стр. 205—207). — Прим. ред.
Пластические массы и эластомеры 281
ченный полимер обладал не только прекрасной химической
стойкостью и устойчивостью к окислению, но и некоторыми кау-
чукоподобными свойствами. Однако промышленное значение
полиизобутилен приобрел лишь после того, как были разрабо-
таны способы его сшивания. Вулканизация полиизобутилена
становится возможной при введении в полимерную цепь неболь-
шого количества звеньев изопрена или другого сопряженного
диена. Этим методом в настоящее время производятся различ-
ные марки бутилкаучука. Наиболее важные из них содержат
1—5% звеньев диена. Обычно бутилкаучук получают введением
смеси мономеров, взятых в необходимом соотношении, в раство-
ритель (например, хлористый метил), содержащий катализатор
катионной полимеризации (например, хлористый алюминий).
Молекулярный вес сополимера уменьшается с возрастанием
концентрации диена. Для получения возможно более одно-
родного продукта степень конверсии мономеров поддерживается
на уровне не выше 60%. Продукт полимеризации, имеющий вид
суспензии, энергично перемешивают с водой с целью дезакти-
вации катализатора и удаления водорастворимых примесей.
СПИСОК ЛИТЕРАТУРЫ
Stille J. К., Introduction to Polymer Chemistry, Wiley, New York, 1962.
Brydson J. A., Plastics Materials, Butterworths, London, 1969.
Бильмейер Ф., Введение в химию и технологию полимеров, ИЛ, М., 1958.
Sorenson W. R., Campbell Т. W., Preparative Methods of Polymer Chemi-
stry, Interscience, New York, 1968 (есть русский перевод издания 1961 г.:
Сёренсен У., Кемпбелл Т., Препаративные методы химии полимеров, ИЛ, М.,
1963).
Morgan Р. 117., «Condensation Polymers: By Interfacial and Solution Met-
hods», Polymer Rev., 10, 1956 (см. Морган П. У., Поликонденсационные про-
цессы синтеза полимеров, «Химия», Л., 1970).
Raff R. А. V., Allison J. В., High Polymers, Vol. XI, Polyethylene, Inter-
sciepce, New York, 1956.
Boundy R. H., Boyer R. F., Styrene, Its Polymers, Copolymers and Deriva-
tives, Hafner, New York, 1952.
Kaufman M., The History of PVC, Maclaren, London, 1969.
Kpeccep T., Полипропилен, ИЛ, M., 1963.
Boeing H. V., Unsaturated Polyesters, Elsevier, Atnstefdam, 1967.
Vale C. P-, Taylor W. G. K-, Aminoplastics, Iliffe, London, 1964.
Whitehouse A,, Pritchett E., Barnett G., Phenolic Resins, Iliffe London,
1967.
ГЛАВА 9
Волокна
Дж. Холкер
Holker J. R., Shirley Institute, Manchester
Введение
Изучение волокон сыграло важную роль в развитии химии
высокомолекулярных соединений (гл. 8). Пионерские работы
Штаудингера по выяснению структуры целлюлозы и натураль-
ного каучука (1920 г.) привели к представлению о том, что эти
вещества состоят из длинноцепочечных молекул высокого моле-
кулярного веса (т. 4, стр. 83), а не из коллоидальных ассоциа-
тов небольших молекул. Исследование Штаудингера, выводы
которого были позднее подтверждены данными по рентгено-
структурному изучению целлюлозы (Мейер и Марк, 1927 г.),
положило начало пониманию макромолекулярной природы по-
лимеров. Вскоре после этого Карозерс с сотрудниками разра-
ботали рациональные методы синтеза волокнообразующих
полимеров. Приблизительно в конце прошлого века были полу-
чены гидратцеллюлозные волокна — вискозное и медноаммиач-
ное (т. 4, стр. 93), а в 1913 г. появилось сообщение о возможно-
сти получения волокна из синтетического полимера (поливи-
нилхлорида). Однако это изобретение не было реализовано в
промышленности. Первым промышленным чисто синтетическим
волокном был, по-видимому, найлон-6,6 (т. 1, стр. 172), произ-
водство которого началось в 1938 г. Вслед за ним очень быстро
были выпущены найлон-6, волокно ПЦ (из хлорированного
поливинилхлорида), виньон (из сополимера винилхлорида с ви-
нилацетатом, 1939 г.), саран (из сополимера винилхлорида с
винилиденхлоридом, 1940 г.), полиакрилонитрильные волокна
(1945 г.) и, наконец, терилен (из полиэтилентерефталата,
1949 г.) (т. 1, стр. 170). В последующие годы не было выпущено
ни одного нового многотоннажного волокна; происходило лишь
расширение производства и улучшение свойств уже существую-
щих волокон. Вместе с тем разработаны и продолжают разра-
батываться многочисленные волокна специального назначения,
что свидетельствует о большом размахе исследований в этой
области.
Волокна
283
Несмотря на колоссальное увеличение производства хими-
ческих волокон, свыше половины общего объема выпуска воло-
кон в мире все еще приходится на долю хлопка (табл. 9.1).
Только в нескольких высокоразвитых странах хлопок уступает
по объему 'потребления искусственным и синтетическим волок-
нам.
Таблица 9.1
Производство волокон а (тыс. т) во всем мире и в Великобритании
Волокна Мировое производство Производство в Великобритании
1970 г. 1972 г. 1970 г. 1972 г.
Хлопок И 203 12 831 198 153
Шерсть 1 594 1 484 195 192
Гидратцеллюлозные 3 434 3 701 211 205
Полиамидные 1 873 2 420 143 142
Полиэфирные 1 624 2 505 79 122
Полиакрилонитрильные 1 002 1 267 113 ПО
Прочие 431 — — —
а В 1974 г. во всем мире было произведено 13 595 тыс. т хлопка, 1478 тыс. т шерсти,
3508 тыс. т гидратцеллюлозных волокон, 2593 тыс. т полиамидных волокон, 3268 тыс. т
полиэфирных, 1448 тыс. т полиакрилонитрильных и 991 тыс. т других волокон (Мировое
производство химических волокон в 1974 г., Обзорная информация, НИИТЭхим, М., 1975).
В Великобритании в том же году было выработано 169 тыс т полиамидных, 116 тыс. т
полиэфирных н 130 тыс. т полиакрилонитрильных волокон [Chem. Market Abstr., 67, 220 303
(1975)J. В 1975 г. во всем мире было получено 3,21 млн. т искусственных волокон и
7,15 млн. т синтетических, в том числе 3,22 млн. т полиэфирных, 2,36 млн. т полиамидных
н 1,36 млн. т полиакрилонитрильных [Chem. Industrie, 28, 8) (1976)]. — Прим, перев.
В 1972 г. свыше 90% мировой потребности в синтетических
волокнах удовлетворялось за счет трех наиболее важных видов
волокон, представленных в табл. 9.1. В ближайшем будущем
это положение, вероятно, сохранится, но полиэфирные волокна
выйдут на первое место, потеснив полиамидные. Все вновь раз-
работанные волокна, по-видимому, будут использоваться лишь
для специальных целей, а не т.ля изготовления товаров широ-
кого потребления.
Для химика-органика наибольший интерес в области произ-
водства химических волокон представляют разработка новых и
усовершенствование существующих методов синтеза волокно-
образующих полимеров и полупродуктов для их получения.
Химическое модифицирование готовых волокон не привлекает
особого внимания, так как волокнообразующие полимеры, как
правило, довольно инертны. С другой стороны, биохимия обра-
зования природных волокон почти не изучена, тогда как их
структура и способы улучшения их свойств путем химического
модифицирования являются предметом широкого исследова-
ния. Более подробно это различие в подходе к химическим
284
Глава 9
и природным волокнам обсуждается в приведенном ниже (по не-
обходимости сжатом) обзоре, которому предпосланы некоторые
общие замечания по поводу требований, предъявляемых к во-
локнообразующим полимерам. Все волокна принято подразде-
лять на три основные группы: природные, искусственные и син-
тетические *.
Главное требование к волокнообразующему полимеру за-
ключается в том, что длина его вытянутой молекулы должна
быть не менее 1000А (100 нм), т. е. его молекулярный вес
должен быть не ниже 10 000. Эта величина, разумеется, может
быть и выше; например, молекулярный вес необработанной (не-
деструктированной) хлопковой целлюлозы достигает 500 000.
В случае синтетических волокон молекулярный вес исходного
полимера намеренно ограничивают, поскольку прядильный рас-
твор или расплав должен иметь не слишком высокую вязкость.
У большинства волокон, сформованных из расплава, молекуляр-
ный вес составляет 10 000—20 000. Волокна, получаемые формо-
ванием из раствора, могут иметь более высокий молекулярный
вес. Для текстильных волокон характерна также определенная
степень кристалличности и (или) ориентации молекул вдоль
оси волокна. Эти свойства, присущие природным волокнам, при-
даются искусственным и синтетическим волокнам в процессе их
формования, вытягивания и термической обработки. Точность
соблюдения параметров этих процессов оказывает существенное
влияние на физико-механические и отчасти на химические свой-
ства готового волокна. В свою очередь, регулярная структура
волокна возможна лишь при определенной степени регулярно-
сти строения макромолекул, достаточной для их плотной упа-
ковки, которая необходима для возникновения сильных меж-
цепных взаимодействий (за счет водородных связей, ассоциации
диполей или сил вандерваальсова притяжения). Однако при
слишком высокой степени кристалличности волокно не только
становится очень прочным, но и делается слишком жестким и
теряет способность растягиваться в процессе его получения и
эксплуатации. Кроме того, такое волокно чрезвычайно трудно
окрасить, поскольку реакционноспособные группы почти целиком
находятся в неупорядоченных участках. Степень кристал-
личности наиболее прочных синтетических волокон, по-види-
мому, не превышает 50—60%. Исключение составляют поли-
акрилонитрильные волокна, которые обнаруживают мало при-
знаков истинной кристалличности, но вместе с тем обладают
высокой однородностью структуры по всему сечению волокна.
В неупорядоченных участках силы межцепного взаимодействия
* Последние две группы волокон носят общее название «химические во-
локна»- — Прим, перев.
Волокна
285
могут дополняться ковалентными или ионными поперечными
связями, как это имеет место в структуре шерсти и модифици-
рованного сшивающими агентами хлопка.
Большое влияние оказывает структура волокна и на его тер-
мостойкость. В отличиё от природных волокон, которые вслед-
ствие своей полярности разлагаются без плавления, синтетиче-
ские волокна в большинстве случаев термопластичны. Некоторые
из них достаточно устойчивы при нагревании выше темпера-
туры плавления, что позволяет проводить формование волокна
прямо из расплава полимера (таковы, например, найлон-6, най-
лон-6,6, полиэтилентерефталат и полипропилен). Формование
волокон из термически нестойких полимеров, особенно полиак-
рилонитрила, ацетатов целлюлозы, поливинилового спирта и
поливинилхлорида, производится более трудоемким способом:
полимер растворяют в подходящем растворителе и полученный
раствор выдавливают через отверстия фильеры в поток горя-
чего воздуха, вызывающего испарение растворителя, или в оса-
дительную ванну. Безусловно, формование из расплава (там,
где оно возможно) является наиболее предпочтительным мето-
дом получения волокна. Низкоплавкие волокна во многих слу-
чаях имеют очевидные недостатки. Например, одежда и обивка
мебели, изготовленные из таких волокон, легко прожигаются
перегретым утюгом, тлеющим табачным пеплом или горящей
сигаретой. Желательно, чтобы волокно сохраняло свою форму
при нагревании до 100 или даже 150 °C, так как от этого за-
висит максимально допустимая температура его текстильной
обработки, а также максимальная температура стирки и хи-
мической чистки полученных из него изделий. Очень важным
свойством волокна является окрашиваемость. Если природные
волокна обладают высоким сродством к водорастворимым кра-
сителям и содержат большое число реакционноспособных функ-
циональных групп, на которых сорбируется красящее вещество,
то синтетические волокна более гидрофобны, и для них при-
шлось разработать новые красители и специальные методы кра-
шения. В ряде случаев волокнообразующий полимер модифици-
руют путем введения в него звеньев второго мономера, которые
не только нарушают регулярность структуры и тем самым по-
вышают реакционную способность полимера, но и несут функ-
циональные группы, способные сорбировать красители (гл. 1Q).
Поскольку почти все синтетические волокна бесцветны, их
можно окрасить в любой желаемый цвет. Исключение состав-
ляют лишь некоторые термостойкие волокна специального на-
значения, полученные на основе полимеров с конденсирован-
ными ароматическими ядрами. Матирование синтетических
волокон производится с помощью добавки неорганического пиг-
мента, обычно двуокиси титана. Фбтоинициированное окисление
286
Глава 9
(световая деструкция) волокон сопровождается их обесцвечи-
ванием и понижением молекулярного веса, в результате чего
ухудшаются их механические свойства. Это относится как к
природным, так и к синтетическим волокнам. Среди последних
наибольшей светостойкостью обладают полиакрилонитрильные
волокна, а наименьшей — полиамидные и полипропиленовые.
Фотоинициированное окисление представляет собой свободно-
радикальную реакцию, которая ускоряется в присутствии сле-
дов некоторых металлов (например, меди и железа), а также
некоторых красителей, образующих под действием света реак-
ционноспособные свободные радикалы.
Природные волокна.
I. Волокна животного происхождения
В данном разделе рассматриваются три основных вида во-
локон животного происхождения: волокна волосяного покрова,
шелк и кожа. Все они состоят из белковых веществ (т. 4,
стр. 305—341), относящихся соответственно к кератинам, фиб-
роинам и коллагенам, и сильно различаются между собой по
химическим и физическим свойствам. Самое важное из волокон
волосяного покрова — это, конечно, шерсть. Химическое строе-
ние шерсти может существенно различаться не только у разных
пород овец, но даже и у волокон шерсти одного и того же на-
стрига. Обнаружены также различия у шелка, вырабатываемого
разными насекомыми (промышленный интерес представляет
только один тип шелка), и у коллагенов, выполняющих раз-
личные функции в организме (коллагенов кожи, мышц, костной
ткани и др.). Шерсть и шелк становятся пригодными для упо-
требления непосредственно после удаления из них веществ не-
фибриллярной структуры — шерстяного жира и серицина соот-
ветственно. С другой стороны, чтобы превратить коллагены в
кожу, необходимо подвергнуть их ряду дополнительных опера-
ций, главной из которых является дубление, придающее им
достаточную прочность и стойкость. В табл. 9.2 приведены ти-
пичные данные по аминокислотному составу шерсти, фиброина
и коллагена кожи (о последовательности соединения аминокис-
лотных остатков в белках см. т. 4, стр. 308—309).
Шерсть
Морфологическая и химическая структура шерсти чрезвы-
чайно сложна и до сих пор не выяснена полностью. Волокна
шерсти неоднородны, но в основе своей (примерно на 90%) они
состоят из внедренных в кожный покров веретенообразных кле-
Волокна
287
Таблица 9.2 Аминокислотный состав шерсти, фиброина шелка и коллагена а
Аминокислоты 'Шерсть Фиброин Коллаген
Глицин 5,6 33,3 24,5
Аланин 3,9 26,1 9,8
Валин 5,7 2,6 1,7
Лейцин 8,4 0,7 3,2
Изолейцин 3,6 0,9 1,3
Фенилаланин 4,0 1,2 2,2
Пролин 5,9 0,5 11,3
Серин 9,3 13,2 3,2
Треонин 6,3 1,3 1,7
Оксипролин -— — 9,1
Тирозин 5,4 11,5 0,5
Аспарагиновая кислота ° 6,9 2,4 5,5
Глутаминовая кислота6 14,9 2,1 13,7
Аргинин 9,2 1,0 8,9
Лизин 3,3 0,9 3,2
6-Оксилизин — —- 1,0
Г истидин 1,0 0,4 0,6
Триптофан 0,7 0,5 • ?
Цистин 11,2 Следы Следы
Цистеин 0,3 Следы Следы
Метионин 0,4 0.1, —
а Состав шерсти дан в граммах аминокислоты на 10Э г шерстя, а со с*
тав фиброина и коллагена —в граммах аминокислотных остатков на 100 г
вещества.
б Частично в форме их амидов — аспарагина и глутамина соответ*
ственно.
ток коркового слоя, под которыми иногда расположены клетки
Сердцевинного слоя. Лестничная структура клеток коркового
слоя обусловливает способность шерсти свойлачиваться. Путем
дифференциального окрашивания шерстяных волокон можно
обнаружить, что они имеют форму двойной спирали и образо-
ваны двумя полуцилиндрами (так называемыми орто- и пара-
кортексом), сплетенными друг с другом. Корковые клетки
орто- и паракортекса несколько различаются по своей
структуре.
Если рассматривать волокно шерсти в целом, то его уни-
кальная механическая прочность обусловлена наличием ряда
внутри- и межмолекулярных взаимодействий, в том числе сил
вандерваальсова притяжения, водородных связей между амид-
ными группами основной цепи и полярными боковыми груп-
пами, ионных связей (так называемых солевых мостиков)
между кислотными и основными боковыми группами и кова-
лентных цистиновых мостиков между основными цепями.
288
Глава §
Прочность мокрого шерстяного волокна при pH 1 меньше, чем
в дистиллированной воде; причиной этого, по-видимому, служит
разрыв солевых мостиков из-за протекания реакции нейтрали-
зации.
водородные связи
между основными
цепями
Солевой мостия
\__„ „„XT —/ цистиновый
1 С±12Ь—&С-Н2—< мостин
Шерсть может быть разделена химическими методами на
три основных компонента, которые сами по себе неоднородны:
фибриллярный ориентированный белок (а-кератозу), эпите-
лиальный белок (р-кератозу) и нефибриллярный матрикс
(у-кератозу), в котором диспергирована а-кератоза. Из-за
большого числа поперечных связей шерсть нерастворима во
всех недеструктирующих растворителях. Поэтому одной из наи-
более важных стадий ее разделения на компоненты является
избирательное окисление цистиновых мостиков (например, над-
уксусной кислотой) с образованием боковых групп, содержа-
щих остатки цистеиновой кислоты
СО NH СО
I I I
CHCH2SSCH2CH —> CHCH2SO3H
NH СО NH
Окисленные а- и у-кератозы высаживают из раствора раз-
бавленными кислотами или растворами сильных электролитов.
По другому методу для облегчения растворения шерсти прово-
дят восстановление ее цистиновых звеньев в цистеиновые. Для
ft-кератозы характерно высокое содержание аминокислот с не-
большими боковыми группами, делающими возможной плотную
упаковку макромолекул (ср. с фиброином шелка) и обусловли-
вающими дискретность рентгенограммы шерсти. В то же время
у-кератоза является аморфным веществом. В ней содержится
Волокна
289
основная часть связанного цистина, на химических превраще-
ниях которого в значительной степени базируется технология
переработки шерсти. При растягивании шерстяных волокон бо-
лее чем на 20% дифракционная картина начинает существенно
изменяться и при 50%-ном удлинении становится совершенно
иной. Это явление связано с постепенным изменением простран-
ственной структуры кристаллических участков волокна — пе-
реходом а-кератина (спиралевидная конформация) в р-кератин
(полностью вытянутая зигзагообразная конформация, сходная
со структурой фиброина шелка).
Значительная неоднородность структуры шерсти делает ее
полное исследование обычными методами очень трудной зада-
чей. Основную часть доступных для определения N-концевых
аминокислот, которые были идентифицированы путем динитро-
фенилнрования (т. 4, стр. 320), составляют серин, треонин, гли-
цин, аланин и валин.
Большое разнообразие реакционноспособных боковых групп
позволяет модифицировать свойства кератина шерсти химиче-
скими методами. В то же время эти группы обусловливают мно-
гие недостатки шерсти, особенно ее чувствительность к термиче-
скому обесцвечиванию, фотохимической деструкции и действию
даже слабощелочных реагентов. Большинство химических
свойств шерсти связано с наличием в ней дисульфидных цисти-
новых мостиков. При обработке шерсти водным раствором ще-
лочи или просто кипящей водой связанный цистин превра-
щается в лантибнин. Эта реакция, вероятно, протекает по сле-
дующей схеме:
но" \
:CHCH2SSCH2CH ---> )С=СН2+[S]+HSCH2CH —>
\ (CN") / \
;CHCH2SCH2CH+ Na2S(NaCNS)
Возможно также образование лизиноаланиновых мостиков
:с=сн2 + h2n(CH2)4r —► ;chch2nh(CH2)4r
Действие восстановителей, например взятого в избытке меркап-
тана, вызывает превращение цистина в цистеин, причем после-
дующая обработка цистеина мягкими окислителями или возду-
хом сопровождается обратной реакцией. Более устойчивые по-
перечные связи образуются при взаимодействии восстановлен-
ной шерсти с дигалогенпроизводными
Br(CH2)nBr
RCH2SSCH,Rz —> RCH2SH + HSCH2R' -------------->- RCH2S(CHj)„SCH2R'
щелочь
JQ Зак. 501
290
Глава &
Взаимопревращение типа цистин — цистеин лежит в основе
методов придания шерстяным изделиям несминаемости, т. е.
способности выдерживать многократное изгибание или сохра-
нять отутюженные складки в процессе ношения одежды, а также
при ее стирке и химической чистке. Все эти методы сводятся
к расщеплению жестких цистиновых мостиков с одновременным
переходом молекул шерсти под влиянием расщепляющего
агента, способного к образованию водородных связей (водяного
пара, водного раствора мочевины), в более устойчивую про-
странственную форму (переход а-кератина в р-кератин) и по-
вторному созданию цистиновых и других, более прочных попе-
речных связей.
Для инициирования разрыва цистиновых мостиков доста-
точно небольшого количества восстановителя, так как, начав-
шись, эта реакция протекает по ступенчатому механизму, напо-
минающему расстегивание застежки-молнии (тиол-дисульфид-
ный обмен)
Чаще всего в качестве восстановителя используют тиогликолят
аммония (недостатками этого реагента являются неприятный за-
пах и способность окрашивать шерсть в присутствии следов же-
леза), сульфит этаноламина и бисульфит натрия (при pH 4—6).
Последний реагент первоначально дает тиолсульфонат (соль
Бунте)
RCH2SSCH2R' RCH2SH + NaOSO2SCH2R'
На восстановительном расщеплении цистиновых мостиков и их
повторном образовании под действием окислителей, например
броматов, основаны также химические методы многомесячной
завивки волос (т. 4, стр. 316).
Другая практически важная реакция шерсти —это ее взаи-
модействие с хлорной водой, растворами гипохлоритов и орга-
ническими соединениями, способными постепенно отщеплять
хлор при определенных значениях pH и температуры, например
Волокна
291
с дихлоризоциануровой кислотой (1). Механизм данной реак-
ции не вполне ясен.
О
CIN^NH
О
О
N
Cl
1
Однако установлено, что она сопровождается глубоким окисли-
тельным превращением звеньев цистина и тирозина. Если огра-
ничить эту реакцию поверхностными участками волокна, то
происходит ослабление или разрыв поверхностных поперечных
связей. Последнее в свою очередь понижает способность шер-
стяных волокон свойлачиваться под давлением и, следова-
тельно, уменьшает склонность шерстяной ткани к усадке путем
зацепления основных цепей поперечными связями. Кроме того,
предварительная обработка хлором «активирует» поверхность
волокон шерсти и обеспечивает более прочное удерживание по-
лимерных покрытий, которые обычно наносятся на шерсть с
целью уменьшения ее усадки.
Поскольку аминокислотные звенья с реакционноспособными
боковыми группами находятся главным образом в наружных
участках шерстяных волокон, шерсть легко поддается химиче-
скому модифицированию. Помимо рассмотренных выше реакций
цистиновых звеньев, возможно, например, ацетилирование боко-
вых аминогрупп с получением волокна, обладающего значи-
тельно пониженным сродством к кислотным красителям. При
обработке реактивом Сенджера (т. 2, стр. 160) лизин дает
2,4-динитрофенильное производное, а при действии на лизин
дифтординитробензола образуются поперечные связи, как в со-
единении 2
R(CH2)4NH
NH(CH2)4R
O2N no2
2
Сшивание молекул шерсти диэпоксисоединениями (а),
гидрином (б) и димезиловыми эфирами гликолей
роятно, тоже протекает с участием аминогрупп, тогда
эпихлор-
(в), ве-
ках для
а
2R'NH2 —
•б
в
R'NHCH2CH(OH)RCH(OH)CH2NHRz
R'NHCH2CH(OH)CH2NHR'
R'NH(CH2)nNHR'
10*
292
Глава 9
прочного связывания с волокном различных активных краси-
телей (гл. 10) необходимы амино- и оксигруппы.
Шелк
Шелк — это фибриллярный (волокнистый) белок, вырабаты-
ваемый пауками и многими другими насекомыми, в особенно-
сти бабочками и мотыльками. Разные шелка сильно разли-
чаются по аминокислотному составу и структуре, так как они
предназначаются для различных целей. Например, паук прядет
неодинаковый шелк для паутины и для потомства. Чаще всего
насекомые вырабатывают шелк для создания кокона, защи-
щающего куколку. В промышленности основным источником по-
лучения шелка служит коконная пряжа личинок шелковичного
червя Bombyx mori. Некоторое количество шелка получают
также от гусениц дикого индийского и китайского тутового шел-
копряда.
Шелковое волокно В. mori представляет собой двойную нигь
из высокоориентированного фибриллярного белка фиброина,
скрепленную веществом нефибриллярной структуры серицином,
который легко удаляется кипящим мыльным раствором (так
называемое обесклеивание). Фиброин и серицин имеют различ-
ный аминокислотный состав (табл. 9.3). Для фиброина харак-
Таблица 9.3
Аминокислотный состав фиброина н серицина
(на 1000 аминокислотных остатков)
Аминокислоты Фиброин Серицин
Г лицин 445 147
Алании 293 43
Валин 22 36
Лейцин 5 14
Изолейцин 7 7
Серин 121 373
Треонин 9 87
Аспарагиновая кислота 13 148
Глутаминовая кислота 10 34
Лизин 3 24
Аргинин 5 36
Гистидин 2 12
Тирозин 52 26
Фенилаланин 6 3
Пролин 3 7
Триптофан 2 —-
Метионин 1
Цистин (половина молекулы) 2 5
(NH3 — 86)
Волокна
293
терно высокое содержание низкомолекулярных аминокислот
(глицина, аланина и серина), на долю которых в сумме прихо-
дится 86% общего аминокислотного азота. Серицин богат сери-
ном, глицином и аспарагиновой кислотой, причем часть аспара-
гиновой и глутаминовой кислот присутствует в серицине в
форме аспарагина и глутамина, гидролиз которых сопровож-
дается выделением аммиака. При диализе очень разбавленных
растворов фиброина в присутствии комплексов этилендиамина
с соединениями двухвалентной меди или, лучше, в присут-
ствии роданистого лития получаются метастабильные водные
растворы этого белка, легко коагулирующие с образованием фи-
броинового геля. Дигерирование таких растворов с химотрипси-
ном, избирательно разрывающим белковые молекулы у карбо-
ксильной группы ароматических аминокислот (т. е. в данном
случае по месту присоединения остатков тирозина), позволяет
выделить кристаллический полипептид состава Gly29Ala20SergTyr,
порошковая рентгенограмма которого аналогична рентгено-
грамме исходного фиброина. Отсюда был сделан вывод, что
кристаллические участки фиброина шелка (составляющие
около 60% его веса) построены из этого полипептида. Струк-
тура последнего (3)
GlyAlaGlyAlaGly[SerGly(AlaGly)n]i2SerGlyAlaGlyAlaGlyTyr
3 (п обычно равно 2)
была установлена в основном классическим методом неполного
гидролиза с последующим динитрофенилированием и хромато-
графическим разделением образовавшихся динитрофенилпепти-
дов (т. 4, стр. 320). C-Концевую последовательность аминокис-
лот в молекуле фиброина недавно удалось определить путем
масс-спектрометрического расщепления полностью N-метилиро-
ванных пептидов типа SerGlyAlaGlyAlaGlyTyr. При этом иден-
тифицирован гексапептид SerGlyAlaGlyAlaGly, который, таким
образом, составляет основную часть молекулы. Положение
остатков серина находят, подвергая пептид N — О-ацильной пе-
регруппировке с последующим динитрофенилированием и не-
полным гидролизом
—NHCHRCONHCHCO—
I
носн2
—NHCHRCO NH2
Н3РО4
Р2О5
2,4-(NO2)2CeH3F
снсо—
щелочь
о—сн2
2,4-(NO2)CeH3—NH
—> —NHCHRCOOH + ^СНСО-
НОСН2
Растворимые пептиды, выделенные после дигерирования с
химотрипсином, содержат большое количество (около 40%)
294
Глава 9
октапептидов Gly(AlaGly)3Туг и Gly(Gly3Ala2Val)Tyr и тетра-
пептида GlyAlaGlyTyr, на долю которых в сумме с кристалличе-
ским полипептидом 3 приходится приблизительно 75% амино-
кислотных остатков в фиброине. Остальные 25%—это преиму-
щественно остатки аминокислот с полярными или объемистыми
боковыми группами. Таким образом, молекулу фиброина можно
представить себе как цепь, включающую три сегмента: а) высо-
коупорядоченный сегмент из 84 аминокислотных остатков об-
щей длиной около 30 нм, б) сегмент с более низкой упорядо-
а б
Рис. 9.1. Складчато-цепная структура фиброина (Fox S. W„ Foster J. F.,
Introduction to Protein Chemistry, Wiley, New York, 1957).
а—-вид в направлении цепи; б—вид сбоку,
ценностью, что обусловлено присутствием в нем остатков ва-
лина и тирозина с довольно большими боковыми группами
(этот сегмент дает окта- и тетрапептидные осколки), и в) не-
упорядоченный сегмент, содержащий остатки аминокислот с
полярными и объемистыми боковыми группами.
Вся фиброиновая цепь может содержать до 2500 аминокис-
лотных остатков, а ее молекулярный вес достигать 200 000.
Структура сегмента а (рис. 9.1), построенного в основном из
глицина и аланина, настолько компактна, что растянутые мо-
лекулы фиброина (р-фиброин) способны плотно упаковываться
в виде складчатых листов, состоящих из антипараллельно изо-
гнутых цепей, соединенных многочисленными поперечными во-
дородными связями —СО---------HN—. В свою очередь листы
Волокна
295
уложены в виде лентообразных микрофибрилл, имеющих ши-
рину около 6 нм и толщину 2 нм.
Механизм образования этой высокоупорядоченной струк-
туры из первоначально вязкой нефибриллярной секреции гусе-
ницы и конформация белковых молекул до начала прядения
шелка пока не установлены.
Как текстильное волокно шелк в настоящее время является
предметом роскоши. Он существенно уступает другим волокнам
по таким показателям, как устойчивость к истиранию, несми-
наемость и стойкость к фотоокислительной деструкции. Неодно-
кратно предпринимавшиеся попытки синтезировать шелкопо-
добные полимеры а-аминокислот до сих пор не увенчались
успехом, хотя получены волокнообразующие полимеры иного
состава, которые, как сообщается, обладают некоторыми шел-
коподобными свойствами (киана и чинон, см. ниже). Из-за на-
личия сильных межмолекулярных водородных связей и значи-
тельной кристалличности шелк имеет высокую разрывную проч-
ность как в сухом, так и во влажном состоянии, а повышенная
подвижность его молекул, которая обусловлена объемистыми
аминокислотными остатками, входящими в состав неупорядо-
ченных сегментов фиброиновых цепей, придает ему высокую
эластичность. Кроме того, шелк, подобно другим белковым во-
локнам, обладает высокой гигроскопичностью (около 10%),
причем поглощение влаги происходит в основном за счет пеп-
тидных связей и других полярных участков неупорядоченных
сегментов.
2,4-Динитрофторбензол не только реагирует с концевыми
аминогруппами, но и переводит тирозин и лизин шелка в соответ-
ствующие О- и N-динитрофенилпроизводные (4 и 5). Аналогично
—СО
5
этому, используя активные дифторсоединения, можно получить
бмс-производные. Так, 3,3'-динитро-4,4'-дифтордифенилсульфон
с высоким выходом превращает тирозин в соединение 6, при-
296
Глава 9
чем кислотный гидролиз волокна, модифицированного этим
реагентом, дает свободную б«с-аминокислоту.
Кожа
Наиболее важным волокнообразующим материалом у жи-
вотных является сложный белок коллаген, присутствующий во
всех частях их тела. Особенно много его содержится в шкуре,
костной ткани и мышцах, однако он находится также во внут-
ренних органах и в глазах. Кожу получают путем специальной
обработки шкур животных, и для технологии кожевенного про-
изводства большое значение имеет понимание молекулярной и
надмолекулярной структуры коллагеновых волокон. В дермисе
шкуры эти волокна тесно и беспорядочно переплетаются, обра-
зуя плотную пространственную сетку, напоминающую войлок.
Таким образом, кожу можно рассматривать как природный не-
тканый материал.
Различные коллагены отличаются друг от друга по своему
составу. Типичный аминокислотный состав коллагена приведен
в табл. 9.2. Важной общей особенностью всех коллагенов яв-
ляется высокое содержание пролина и оксипролина, которые
оказывают существенное влияние на их молекулярную струк-
туру. Результаты, полученные классическим методом кислот-
ного гидролиза в сочетании с протеолизом трипсином, химотри-
псином и другими пептидазами (особенно высокоспецифичной
коллагеназой, выделенной из Clostridium histolyticum, кото-
рая расщепляет только фрагмент GlyProXGlyProX по связи
X—Gly), свидетельствуют о том, что аминокислотные остатки
соединяются между собой в основной цепи коллагена по типу
блок-сополимера с чередованием полярных и неполярных сег-
ментов. Своеобразие этой структуры заключается в том, что
остатки глицина занимают в цепи главным образом каждое
третье положение. Неполярные сегменты содержат преиму-
щественно пептиды типа (GlyProX)n. Многие из таких и
родственных им по строению политрипептидов, например
(GlyXPro),,, удалось синтезировать. При рентгеноструктурном
исследовании этих соединений было найдено, что особенно близ-
кими к коллагену кристаллографическими свойствами обла-
дает пептид (GlyProPro)n. Неполярные сегменты, составляю-
щие около 35% молекулы коллагена, в значительной степени
ответственны за его упорядоченную структурную компоненту и
обусловливают термостойкость коллагена. Последнее свойство
Волокна
297
связано со способностью волокон коллагена быстро сокра-
щаться приблизительно до одной трети их первоначальной дли-
ны * при нагревании до температуры сваривания Тс, которая
является различной для каждого типа коллагена и непосред-
ственно связана с температурой денатурации этого белка в рас-
творе (приблизительно на 27° выше нее). Величина Тс в общем
возрастает с увеличением суммарного содержания пролина и
4-оксипролина. На долю этих двух циклических аминокислот
приходится около 25% общего содержания аминокислот в кол-
лагене шкуры млекопитающих. Они придают белковой моле-
куле жесткую структуру, обусловленную невозможностью вра-
щения вокруг связи N—Ci пирролидинового кольца и связи
Ci—С = О и изменением пространственной направленности цепи
у каждого гетероциклического центра (7)**.
Рентгеноструктурные данные показывают, что каждый от-
дельный полипептидный участок цепи имеет форму спирали, на
один виток которой приходится три аминокислотных остатка. Мо-
лекула коллагена состоит из трех таких спиралей (так называе-
мых а-спиралей), сплетенных в сверхспираль и связанных между
собой поперечными водородными связями. Две из этих спиралей
идентичны друг другу и имеют молекулярный вес около 100 000.
Компактные остатки глицина, занимающие каждое третье поло-
жение элементарных спиралей, поочередно внедряются в «серд-
цевину» составной спирали, в то время как другие, более объ-
емистые аминокислотные остатки ориентированы наружу.
Полярные сегменты коллагеновой цепи содержат преимуще-
ственно остатки аспарагиновой кислоты, глутаминовой кислоты,
лизина и аргинина. Часть глутаминовой кислоты, по-видимому,
присоединена через у-карбоксильную группу, так как в резуль-
тате представленных ниже реакций был выделен моноальдегид
янтарной кислоты (8). Нативный коллаген не содержит доступ-
ных для определения N-концевых групп.
1) СНзОН 2,4-(NO2)2C6H3P
-NHCHCH2CH2CONH- ~ > — NHCHCH2CH2CONH-------------------------»
• 2) NH2OH J
СООН CONHOH
* Это явление известно под названием «сверхсокращение» (суперконтрак-
ция).— Прим, перев.
** Волнистыми линиями в этой глцве обозначаются цепи неопределенной
ДЛИНЫ-
298
Глава 9
► — NHCHCH2CH2CONH—
CONHO—CeH3(NO2)2
NaOH
----------------->
(реакция Лоссеня)
H2O
—> —nhchch2ch2conh- -—> — nh2 + OHCCH2CH2COOH + h2n—
nh2
8
В коллагене обнаружены также неспиральные участки — так
называемые телопептиды. Одни из них находятся на концах ос-
новных а-спиралей, тогда как другие присоединяются к «-спи-
ралям в узлах ветвления как боковые группы. Оба типа тело-
пептидов можно отщепить путем обработки нативного коллагена
протеолитическими ферментами. Телопептидные группы обра-
зуют небольшое число (1—2 на каждую тысячу аминокислот-
ных остатков) термически и химически нестабильных попереч-
ных связей. Точная природа этих связей не установлена, однако
их образование происходит, по-видимому, путем ферментатив-
ного окислительного дезаминирования остатков лизина и окси-
лизина в боковых цепях телопептидов
R(CH2)4NH2
R(CH2)2CHCH(OH)(CH2)3R
сно
R(CH2)3CH=N(CH2)4R
R(CH2)sCHO
аллизин
альдольная
конденсация
лизин
С повышением степеней окисления и сшивания раствори-
мость коллагена уменьшается. Дубление, которое является важ-
нейшей стадией превращения коллагена в кожу, заключается
в создании в его структуре прочных поперечных связей. Про-
стейшие из них, по-видимому, возникают в результате взаимо-
действия формальдегида с боковыми группами лизина, глута-
мина и аспарагина
В качестве дубящих веществ используются также природные
танниды растительного происхождения, синтетические феноль-
ное смолы (синтаны), соли хрома и ненасыщенные масда.
Волокна
299
Природные волокна.
II. Волокна растительного происхождения
Растительные волокна получают из стеблей растений (лен,
джут, конопля), из листьев растений (сизаль) или из пуха,
формирующегося на поверхности семян (хлопок, капок*). Все
они сильно различаются между собой по морфологическим свой-
ствам. Основным волокнообразующим компонентом раститель-
ных материалов является целлюлоза — линейный полисахарид,
состоящий исключительно из 1,4-связанных звеньев (З-о-глюко-
пиранозы (8А) (т. 4, стр. 83).
он
СК1
но
он
он
он
Значительные количества целлюлозного волокна получают
также из древесной целлюлозы. Однако оно слишком короткое
и не может быть непосредственно переработано в текстильное
волокно. Поэтому его используют в производстве бумаги и гид-
ратцеллюлозных волокон (волокон из регенерированной целлю-
лозы) (стр. 312). Наряду с целлюлозой в растительных материа-
лах всегда содержатся в переменных количествах полимерные
вещества нефибриллярной структуры — пектины, гемицеллю-
лозы и лигнин. Ниже рассматривается только самое важное
текстильное волокно растительного происхождения — хлопок.
Хлопок
Хлопок, который выделяют из пуха, окружающего семена
вида Gossyrium (хлопчатник), представляет собой чистую цел-
люлозу. Хлопковые волокна, слегка приплюснутые и извитые,
имеют длину до 5 см. Целлюлоза в них обладает сложной над-
молекулярной структурой, состоящей из слоев фибрилл, кото-
рые ориентированы по спирали вокруг оси волокна и образуют
полый центральный канал (люмен). Нецеллюлозных веществ
в хлопке мало, и они находятся главным образом в оболочке
(первичной клеточной стенке).
Рентгенограмма хлопка является типичной рентгенограммой
частично кристаллического полимера. Она показывает, что эле-
ментарная ячейка состоит из четырех целлобиозных звеньев,
что соседние полимерные цепи ориентированы антипараллельно
* Капок — волоски из плодов хлопчатого дерева (сейбы), культивируе-
мого в некоторых странах Азии. — Прим, перев.
300
Глава 9
друг другу и что степень кристалличности приблизительно рав-
на 70% (эта величина превышает значение, полученное из дан-
ных по реакционной способности хлопковой целлюлозы, см.
ниже). Тонкая структура элементарных фибрилл, в которые со-
единены отдельные макромолекулы целлюлозы, до сих пор
окончательно не установлена. Долгое время господствовала так
называемая мицеллярная теория, согласно которой внутри каж-
дой фибриллы макромолекулы проходят как через кристалли-
ческие, так и через некристаллические (аморфные) участки,
причем реакционноспособны только последние. Однако по
современным представлениям каждая фибрилла сама по себе
полностью кристаллична и реакционноспособные участки надмо-
лекулярной структуры представляют собой молекулы, располо-
женные на участках нарушенной кристалличности.
Реакционная способность является важной характеристикой
структуры волокна. Разработано несколько методов определе-
ния этой характеристики для хлопкового волокна, каждый из
которых, разумеется, относится только к конкретным условиям
измерения. Вероятно, самый элегантный и наиболее точный ме-
тод— это дейтерирование (в сочетании с инфракрасной спект-
роскопией), которое представляет собой парофазную реакцию
и протекает в отсутствие растворителя, способного вызывать на-
бухание волокна и, следовательно, изменять степень его упоря-
доченности. Впервые данный метод был использован при изу-
чении пленок из регенерированной и бактериальной целлюлозы.
Обработка специально приготовленных пленок из хлопковой
целлюлозы парами D2O сопровождается дейтерированием всех
реакционноспособных гидроксильных групп. В результате этого
в ИК-спектре вместо широкой полосы валентного колебания
«неупорядоченных» ОН-групп появляется новая полоса колеба-
ния групп OD при 2530 см-1, тогда как хорошо разрешенный на-
бор полос в области 3350 см-1, обусловленный колебаниями
«упорядоченных» ОН-групп, остается без изменения. Измерение
интенсивностей этих полос относительно постоянной полосы
С—Н-связи в области 2900 см-1 позволяет вычислить соотноше-
ние гидроксильных групп в упорядоченных (кристаллических)
и неупорядоченных (аморфных) участках. Для очищенного на-
тивного хлопка было найдено, что около 58% гидроксильных
групп находится в кристаллических участках волокна. Анало-
гичный результат был получен при измерении гигроскопичности
хлопка. Поэтому все волокна (за исключением наиболее гидро-
Волокна
301
фобных волокон типа полипропиленового) содержат сорбиро-
ванную воду, количество которой в общем тем больше, чем выше
влажность воздуха и чем ниже температура. Равновесное зна-
чение гигроскопичности в обычных условиях, например при от-
носительной влажности воздуха 65% и. температуре 20°С, ко-
леблется от 15% для самых гидрофильных волокон до менее
чем 1% Для полиэфирного волокна. Гигроскопичность хлопка
составляет 6—7%. Значение его реакционной способности нахо-
дят из отношения числа молекул абсорбированной воды, прихо-
дящегося на каждое звено 1,4-ангидроглюкозы, к числу моле-
кул, которое мог бы абсорбировать полностью реакционноспо-
собный хлопок (последнее число равно 1,53 моля).
В основе изящного химического метода измерения реакцион-
ной способности хлопкового волокна лежит классическая реакция
метилирования гидроксильных групп (т. 4, стр. 49 и 86). Дан-
ные этого метода количественно подтверждают представление
о полностью кристаллической структуре хлопка. Многократ-
ная обработка хлопка, замоченного в 2 М растворе едкого
натра, диметилсульфатом в диметилсульфоксиде сопровождает-
ся постепенным, но все более медленным повышением степени
метилирования волокна до тех пор, пока явно не начнется ме-
тилирование первоначально нереакционноспособных гидроксиль-
ных групп. Содержание метоксигрупп в полученном волокне
можно определить путем его полного кислотного гидролиза
в смесь глюкозы и метилглюкоз, которую подвергают триметил-
силилированию с последующим количественным разделением
образовавшихся продуктов методом газожидкостной хроматогра-
фии.
psi(CH3)3
(а+0)
Распределение гидроксильных групп, найденное этим спосо-
бом, хорошо согласуется с величиной, вычисленной для струк-
туры, в которой каждая кристаллическая элементарная фиб-
рилла построена из 80 макромолекул целлюлозы, упакованных
в блок размером 10x8, причем 48 макромолекул располагаются
302
Глава 9
внутри кристалла. Последние, таким образом, являются не-
реакционноспособными и при гидролизе должны давать толь-
ко глюкозу. Молекулярная модель показывает, что гидроксиль-
ные группы атомов С3 и С2 или Се ангидроглюкозных звеньев,
Рис. 9.2. Элементарная ячейка целлюлозы [Jeffries R„ Roberts J. G Robin-
son R. N. Textile Res. J. 38. 238 (1968)].
находящихся на широких сторонах блока, поочередно ориенти-
рованы наружу и, следовательно, обладают высокой реакцион-
ной способностью (рис. 9.2). Однако гидроксильные группы ато-
мов Сз связаны прочной водородной связью с соседними кольце-
выми атомами кислорода и поэтому малореакционноспособны.
Рис. 9.3. Конформация целлюлозы с изображением водородных связей
между гидроксильными группами атомов С3 и кольцевыми атомами кислорода.
Следовательно, при метилировании могут получиться только 2-
или 6-О-метилглюкозы. В узких сторонах блока все гидроксиль-
ные группы лежат на поверхности, но ОН-группы атомов С3
каждого второго звена ангидроглюкозы являются нереакцион-
носпособными из-за образования водородных связей с коль-
цевыми' атомами кислорода (при Cs), ориентированных внутрь
кристалла (рис. 9.3). Отсюда следует, что продуктами метилиро-
вания целлюлозы должны быть 2,6-ди-О-метилглюкоза и 2,3,6-
три-О-метилглюкоза, получающиеся в равном соотношении.
Волокна
303
Сходимость экспериментальных результатов и данных, вычис-
ленных на основе представления о фибриллярных блоках мак-
ромолекул размером 10X8 цепей, иллюстрируется приведенной
ниже таблицей:
G g2 G3 G6 бг.з Gj.e Gs,e ^2,3,6
Найдено: 56 10 1 10 2 10 1 9
Вычислено: 60 10 0 10 0 10 0 10
(G2 = 2-О-метилглюкоза и т. п.)
Хлопок легко абсорбирует воду. Однако он не растворяется
даже в растворах реагентов, энергично разрушающих водород-
ные связи, таких, как бромистый литий, хлористый цинк и моче-
вина. Вместе с тем хлопок растворим в медноаммиачном рас-
творе, в водных растворах комплексов этилендиамина с двухва-
лентной медью (куоксен) (т. 4, стр. 93) или кадмием (кадоксен)
и тому подобных реагентах. Хлопок химически устойчив к дей-
ствию водных растворов щелочей [если не считать того, что не-
большое число концевых групп с восстановительными свойст-
вами под действием щелочи превращается по довольно слож-
ному механизму в карбоксильные группы (т. 4, стр. 42)]. Однако
растворы едкого натра с концентрацией 5 М и выше вызывают
изменения в морфологической структуре хлопкового волокна
(приплюснутое и извитое волокно выпрямляется и. становится
более круглым, а полый внутренний канал почти исчезает) и
в его кристаллической структуре (превращение целлюлозы I
в целлюлозу II). Этот процесс, получивший название «мерсери-
зация», имеет важное практическое значение, так как он сопро-
вождается повышением разрывной прочности, блеска и накра-
шиваемости хлопка. Аналогичные изменения (за исключением
того, что целлюлоза I переходит не в целлюлозу II, а в другую
структурную модификацию) происходят при кратковременной
обработке хлопка безводным жидким аммиаком, в котором хло-
пок очень легко набухает («прогрейд-процесс»).
Гидролиз хлопка горячими разбавленными растворами мине-
ральных кислот приводит к образованию d-глюкозы. Реакция
протекает путем неселективного расщепления гликозидных свя-
зей, причем в качестве промежуточных продуктов можно выде-
лить различные олигосахариды, такие, как целлотетраоза, цел-
лотриоза и целлобиоза (т. 4, стр. 69). В начальной стадии
гидролитическая деструкция хлопка идет сравнительно быстро,
однако затем скорость потери в весе уменьшается и в конце кон-
цов становится постоянной. Это различие в скоростях гидро-
лиза хлопка на отдельных стадиях процесса было объяснено
наличием в структуре волокна участков с повышенной реакцион-
ной способностью («слабых мест»). При кислотнокаталитиче-
ском ацетолизе хлопка уксусным ангидридом с хорошим вы-
ходом получается октаацетат целлобиозщ. *
304
Глава 9
Важной операцией процесса химической очистки хлопкового
волокна является отбелка. Отбелка производится с целью уда-
ления из хлопка окрашенных примесей; обычно ей предшествует
обработка хлопка разбавленным раствором щелочи (отварка),
в ходе которой из него удаляются гемицеллюлозы, пектин, воск
и остатки оболочки семян. Классическим отбеливателем яв-
ляется гипохлорит натрия. Однако в настоящее время предпо-
читают использовать другие окислители, особенно хлорит нат-
рия и перекись водорода. Обычно при отбелке протекает также
частичное окисление концевых групп с восстановительными
свойствами в остатки глюконовой кислоты. Необходимо тща-
тельно следить за ходом реакции, чтобы не допустить глубокого
окисления ангидроглюкозных звеньев основной цепи, приводя-
щего к образованию так называемой окисленной целлюлозы
и
Волокна
305
Наличие окисленных звеньев делает целлюлозу весьма чув-
ствительной к обработке щелочью, так как они легко расщеп-
ляются по механизму р-элиминирования
Окисление хлопка перйодатами сопровождается почти коли-
чественным образованием диальдегида 10, который, вероятнее
всего, существует в форме мостикового полуацеталя (т. 4,
стр. 87). Этот диальдегид способен к дальнейшему окислению
хлоритом натрия в кислоту 11, восстановлению боргидридом
он
но
12
натрия в диол 12 и другим реакциям, типичным для карбониль-
ной группы. Вследствие чувствительности к щелочам диальде-
гиды этого типа и их производные не находят практического
применения. Двуокись азота окисляет хлопок, хотя и не строго
селективно, в глюкуроновую кислоту 9; модифицированная этим
способом марлевая ткань используется для изготовления кро-
веостанавливающих бинтов.
Хлопок можно модифицировать с помощью разнообразных
реакций замещения и присоединения, в том числе реакций полу-
чения простых и сложных эфиров- целлюлозы, оксиалкилирова-
ния окисями олефинов и присоединения по Михаэлю. Обычно мо-
дифицирование проводят в водной среде. Поскольку хлопок
непроницаем для большинства органических растворителей, про-
водимые в них реакции ограничиваются преимущественно по-
верхностью волокна, и для диффузии реагентов в более глубин-
ные участки приходится прибегать к специальным приемам. То
же самое относится к вискозному волокну, шерсти и другим
гидрофильным волокнам. В тех случаях, когда реагент сравни-
тельно устойчив к гидролизу, можно просто подвергнуть волок-
но набуханию в водном растворе реагента. Если же реагент
306
Г лава 9
гидролитически нестабилен, то волокну сначала дают набухнуть
в воде, а затем обезвоживают его по методу последовательного
вытеснения одного растворителя другим (например, воды диме-
тилформамидом, воды этанолом и далее бензолом и т. п.). В от-
дельных случаях можно обрабатывать волокно реагентом в па-
ровой фазе. Следует также иметь в виду, что волокна ведут
себя как полупроницаемые мембраны и при обработке их высо-
комолекулярными соединениями реагируют лишь с поверхно-
сти.
Взаимодействием хлопковой целлюлозы с алкилгалогени-
дами в присутствии щелочи получен ряд простых эфиров со-
става ЦеллОИ, где R = СНз, С2Н5, СН2СООН, CH2CONH2,
СН2Р(О) (ОН)2, СН2С6Н5, СН(С6Н5)2, CH2CH2NH2 и
CH2CH2N(C2Hs)2. Последнее соединение можно также синтези-
ровать по реакции с солью 13, образующейся in situ. В общем
виде можно считать, что даже низкая степень замещения небла-
гоприятно отражается на механических свойствах волокна.
+ но- Делл-он
C1CH2CH2NH(CZH;12 СГ-► Н2С—СН2 ---►ДеотОСН2СН2К(СгНД.
Лс1~.
сгн5 сгн5
13
Реакция хлопковой целлюлозы со смесями уксусного ангид-
рида, уксусной кислоты и хлорной кислоты протекает с образо-
ванием ацетатов целлюлозы, имеющих различные степени заме-
щения. Ацетилирование можно также осуществить путем обра-
ботки хлопка ацетилхлоридом в присутствии пиридина или ке-
теном. Описаны эфиры целлюлозы с высшими жирными кисло-
тами, мезилаты, тозилаты и т. д. Многие карбоновые кислоты,
например уксусная, бензойная, акриловая и высшие жирные
кислоты, непосредственно реагируют с хлопком в присутствии
трифторуксусного ангидрида, играющего роль «активатора» ре-
акции. Присоединение акриламида в присутствии щелочи (реак-
ция Михаэля) сопровождается введением в хлопковую целлю-
лозу групп CH2CH2CONH2 наряду с небольшим количеством
карбоксиэтильных групп, образовавшихся в результате одновре-
менно протекающего омыления амидных функций.
Детальному изучению подверглась реакция цианэтилирова-
ния целлюлозы
он~
ЦеллОН + CH2=CHCN -----> ЦеллОСН2СН2СЫ
При действии на целлюлозу изоцианатов в среде сухого пи-
ридина образуются карбаматы, а кислотнокаталитическое при-
Волокна
307
соединение простых виниловых эфиров дает ацетали
ЦеллОН+ RNCO —> ЦеллОСОМВД
ЦеллОН + CH2=CHOR —► ЦеллОСН(СН3^
Менее распространенным является замещение производных
целлюлозы. Тозилированный хлопок реагирует с аммиаком, ча-
стично обменивая тозильные группы на аминогруппы. Одним из
последних примеров реакций этого типа является синтез пред-
полагаемой хлордезоксицеллюлозы с низкой степенью замеще-
ния путем обработки хлопка хлористым тионилом в диметил-
формамиде (реактивом Вильсмайера) (т. 3, стр. 100)
, ЦеллОН
SOC12 + (CH3)2NCHO —► [(CH3)2N=CHC1] сг ----->-
—► ЦеллС] + (CH3)2NCHO + НС1
Все эти реакции не приводят к улучшению механических
свойств хлопкового волокна, и в большинстве случаев их исполь-
зование в промышленном масштабе неэкономично. Однако они
могут приводить к повышению накрашиваемости, биологической
стойкости [которое происходит, например, при регулируемом
(поверхностном) цианэтилировании или ацетилировании хлоп-
ка], термостойкости и некоторых других показателей. Ниже рас-
сматриваются другие способы модифицирования хлопка.
Очень важное практическое значение имеют реакции сшива-
ния целлюлозы, которые лежат в основе методов придания хлоп-
чатобумажным изделиям улучшенных потребительских свойств,
таких, как несминаемость, способность длительно сохранять оту-
тюженные складки и быстрое высыхание после стирки. Самым
простым способом сшивания является обработка хлопка формаль-
дегидом с образованием производных типа ЦеллОСН2ОЦелл.
Однако эта реакция плохо поддается регулированию и сопро-
вождается уменьшением разрывной прочности волокна и его
устойчивости к истиранию, которые могут стать недопустимо
низкими. Из многочисленных вариантов модифицирования фор-
мальдегидом наиболее перспективным считается обработка
хлопчатобумажных тканей с тщательно контролируемым вла-
госодержанием (около 6%) газообразным формальдегидом и
двуокисью серы. В ходе реакции in situ образуется необходи-
мый сильнокислотный катализатор, который разрушается при
последующей сушке ткани.
СН2О + SO2 + Н2О HOCH2SO3H
Хорошая несминаемость ткани как в мокром, так и в сухом со-
стоянии достигается при введении в нее менее 1% формальде-
гида.
Однако в настоящее время для придания хлопчатобумаж-
ным тканям улучшенных потребительских свойств чаще всего
308
Глава 9
используют М,М'-диоксиметилмочевину (14) и родственные ей
соединения 15—20. Все они легко получаются конденсацией мо-
чевины (или, в случае соединения 20, меламина) и формальде-
гида в слабощелочной среде; гидроксилированную мочевину,
НО CH2NHCONHCH2OH
20
которая служит исходным веществом для соединения 19, синте-
зируют из мочевины и глиоксаля. Сшивающий реагент наносят
на хлопок вместе с кислотой Льюиса типа Zn(NO3)2 или MgCl2
и затем нагревают в течение 3—4 Мин при температуре около
150°С. При этом протекает следующая реакция (на примере
соединения 15):
О
ЦеллОН + НОСН2—nAsN—СН2ОН + НОЦелл —>
15
—> ЦеллОСН2—N-
N—СН2ОЦелл
Волокна
309
Побочно может происходить полимеризация взятого диоксиме-
тиламида. Необходимое количество сшивающего реагента со-
ставляет примерно 5% от веса ткани.
Недостатком Ь^М'-диоксиметилмочевины (14) и других сши-
вающих реагентов этого типа в том случае, когда наряду со
сшиванием они претерпевают частичный гидролиз, является
чувствительность к гипохлоритам, которые иногда используются
при стирке как отбеливатели. Продукты гидролиза могут пре-
вращаться в N-хлорпроизводные; последние, разлагаясь при
нагревании (например, при глажении), выделяют хлористый во-
дород, вызывающий обесцвечивание и ослабление (гидролиз)
ткани.
Хлопок способен сшиваться эпихлоргидрином в водно-щелоч-
ной среде и диэпоксидами в той же среде или в присутствии
кислот Льюиса [чаще всего BF3, генерируемого in situ путем тер-
мического разложения Zn(BF4)2]
NaOH
ЦеллОН + Н2С—СНСН2С1 -----► [ЦеллОСН2СН(ОН)СН2С1] —>
—> ЦеллОСН2НС—СН2 —> ЦеллОСН2СН(ОН)СН2ОЦелл
^О^
2ЦеллОН + Н2С—СН—СН—СН2 —> ЦеллОСН2СН(ОН)СН(ОН)СН2ОЦелл
В качестве сшивающих реагентов были использованы также
активированные дивинильные соединения (дивинилсульфон и
легко выделяющие его вещества) в щелочной среде
но-
2ЦеллОН + CH2=CHSO2CH=CH2 -----► ЦеллОСН,СН2ЗО2СН,СН2ОЦелл
Соединение 21, полученное из тиодигликоля S (СН2СН2ОН)2,
очень быстро реагирует с хлопком через промежуточный высо-
коактивированный неустойчивый тривинилсульфониевый ион
(реактив «сульфикс А»), Дополнительная обработка сшитого
продукта сульфитом натрия лишает его нежелательных ионо-
обменных свойств вследствие образования бетаиноподобной
структуры 22.
+ НО- + 2ЦеллОН
(NaOSO2OCH2CH2)2SCH2CH2OSO; ---> [(CH2=CH)3S ОН'] ----->
21 + Na2SO3
—> ЦеллОСН2СН2ЗСН2СН2ОЦелл ОН* ------->
СН=СН2
—> ЦеллОСН2СН2ЗСН2СН2ОЦелл
ch,ch2so3
22
310
Глава 9
Сшивание набухшей (т. е. влажной) хлопчатобумажной
ткани реагентом 21 придает ей хорошую несминаемость в мок-
ром состоянии и способность к быстрому высыханию. Однако
модифицированная ткань сохраняет сминаемость в сухом со-
стоянии, и для устранения этого недостатка необходимо вто-
рично обработать ткань одним из соединений типа N.N'-диокси-
метилмочевины.
В результате модифицирования тетра (оксиметил) фосфоний-
хлоридом (23) хлопок приобретает огнестойкость, сохраняю-
щуюся и после стирки. Аналогичным действием обладает тет-
ра (оксиметил) фосфонийгидроксид, который получается при ос-
торожном взаимодействии хлорида с эквимолярным количеством
едкого натра и, по-видимоМу, существует в растворе преимуще-
ственно в форме фосфина 24.
+ ’ но-
РН3 + 4СН2О + НС1 —> Р(СН2ОН)4СГ ---> Р(СН2ОН)3
23 24
Ткань замачивают в водном растворе фосфонийхлорида и
затем обрабатывают газообразным аммиаком («аппретура
Пробан»). При этом в волокнах отлагаются сшитые полимеры
неизвестного состава, которые, вероятно, имеют структуру ти-
па 25. Разработаны также способы модифицирования хлопка
;nch2< । ,ch2n:
/PCHoNCHsP'
мен/ Vh2N'
25
смесями фосфорорганических соединений и меламиноформаль-
дегидного преполимера с последующей сушкой и отверждением
при высокой температуре (выше 140°C).
Триэтиленимид фосфорной кислоты (26) сшивает хлопковую
целлюлозу и тоже придает ей огнестойкость. Однако это соеди-
нение токсично. С другой стороны, недавно разработанный «пи-
роватекс-процесс» заключается в сшивании хлопка путем его
кислотнокаталитической конденсации с веществами типа окси-
метиламида p-диметилфосфонопропионовой кислоты (27) с по-
следующим отверждением продукта конденсации в условиях,
описанных выше для диоксиметиламидов. Исходный амид для
соединения 27 легко получается по реакции Арбузова между
/Н2СХ \
I I /N— ]—РО (CH3O)2P(O)CH2CH2CONHCH2OH
26 27
Волокна
311
амидом 3-хлорпропионовой кислоты и триметилфосфитом или
присоединением диметилфосфита к акриламиду
(СН3О)зР + C1CH2CH2CONH2 —|
—> (CH3O)2P(O)CH2CH2CONH2
(СН3О)2РОН + CH2=CHCONH2 —I
Для придания хлопчатобумажной ткани достаточной огне-
стойкости необходимо ввести в нее приблизительно 3—5% фос-
фора. Многие азотсодержащие соединения, особенно производ-
ные меламина, при использовании их в комбинации с соедине-
ниями фосфора оказывают синергический эффект.
Механизм действия огнезащитных веществ (антипиренов),
вероятно, заключается в том, что они понижают температуру,
при которой начинается пиролиз целлюлозы, и коренным обра-
зом изменяют характер процесса горения, увеличивая количе-
ство образующейся воды и коксообразных продуктов и подав-
ляя образование легковоспламеняющихся летучих соединений
и смолы [главными компонентами последней являются левоглю-
козан и 1,6-ангидро-р-О-глюкопираноза (28)]. Этот так назы-
ваемый механизм дегидратации позволяет объяснить огнеза-
он он
28
щитное действие антипиренов, содержащих фосфор и- бор. Кроме
того, выделяющиеся в процессе горения летучие соединения
фосфора могут проявлять активность в парообразном состоя-
нии, обрывая радикальные реакции продолжения цепи, проте-
кающие в пламени. Органические галогенпроизводные (исклю-
чая соединения фтора), которые служат эффективными антипи-
ренами для большого числа полимеров, по-видимому, ведут
себя именно таким образом, генерируя активные галоген-ра-
дикалы. Огнезащитные свойства антипиренов этого класса мо-
гут быть значительно повышены добавкой трехокиси сурьмы,
которая, очевидно, превращается в довольно летучие галогениды
или оксигалогениды сурьмы.
Искусственные волокна
К этому классу относятся волокна, которые получают фор-
мованием растворов природных полисахаридов или белков в
подходящую осадительную ванну. Наиболее важным видом ис-
кусственных волокон являются гидратцеллюлозные врлркна
312
Глава 9
(волокна из регенерированной целлюлозы). Меньшее значение
имеют альгинатные волокна, вырабатываемые из морских водо-
рослей, и волокна, получаемые из растительных белков, напри-
мер казеина.
Гидратцеллюлозные волокна
Существует два метода производства гидратцеллюлозных во-
локон, причем в обоих случаях используется специально очи-
щенная (облагороженная) древесная целлюлоза. Медноаммиач-
ное (бемберговское) волокно получается путем формования
раствора целлюлозы в аммиачном растворе гидроокиси меди
в кислую осадительную ванну. Вискозное волокно, которое вы-
рабатывается в .значительно большем количестве, получают пре-
вращением целлюлозы в растворимый неполный ксантогенат
(действием сероуглерода в присутствии раствора едкого натра)
ЦеллОН + СS2 + NaOH —> ЦеллОС88Ма
с последующей регенераций целлюлозы путем формования рас-
твора ксантогената в кислую осадительную ванну. Процесс про-
изводства вискозного волокна весьма сложен, однако в нем
можно выделить следующие основные операции (т. 4, стр. 93):
1. Волокнистую целлюлозу пропитывают концентрированным
раствором едкого натра и подвергают «предсозреванию» на воз-
духе. В результате протекающих при этом реакций окисления и
разрыва макромолекулярных цепей степень полимеризации цел-
люлозы понижается; кроме того, происходит вымывание остат-
ков гемицеллюлоз. Добавление небольших количеств окисли-
теля, например перекиси водорода, ускоряет предсозревание ще-
лочной целлюлозы. Разрыв цепей, обусловленный характерным
для окисленного хлопка p-элиминированием (т. 4, стр. 42), со-
провождается перегруппировкой части образовавшихся конце-
вых групп с восстановительными свойствами в более устойчи-
вые карбоксильные группы
сно сно
Н----он
НО-----н ОН
и----OGlu
Н----ОН
СН20Н
г—ОН
I—н
Н----OGlu
Н----ОН
СН20Н
СНО
СООН
Н-----н
Н-----OGlu
"ОН
н----ОН
н-----OGlu
н----ОН
сн?он
сн2он
Волокна
313
2. По достижении заданной степени деструкции щелочную
целлюлозу измельчают и смешивают с сероуглеродом, взятым
в избытке (50—60% по сравнению с теоретическим количе-
ством), учитывающем протекание побочных реакций. Послед-
ние включают образование Na2CO3, Na2CS3 и Na2S. Полученный
ксантогенат целлюлозы имеет вид сыпучего порошка оранже-
вого цвета.
3. Ксантогенат целлюлозы растворяют в разбавленном рас-
творе едкого натра и полученный вискозный раствор подвер-
гают дополнительному «созреванию». При этом происходит ча-
стичное омыление ксантогената (значение степени замещения
понижается до 0,35—0,4), и раствор становится менее вязким.
Кроме того, в процессе созревания происходит перераспределе-
ние тиокарбоксильных групп, включающее два типа реакций:
миграцию этих групп- от атомов С2 (по которым преимуществен-
но протекает начальное замещение) и С3 к атомам Се и заме-
щение гидроксильных групп, которые были нереакционноспо-
собными на стадии гетерогенного ксантогенирования.
4. Вискозный раствор выдавливают через отверстия фильеры
в кислотно-солевую осадительную ванну. Полученное волокно
промывают и подвергают ориентированию путем вытягивания.
Простая осадительная ванна содержит только серную кис-
лоту и сульфат натрия, концентрация которых зависит от усло-
вий формования и требуемых свойств волокна. В современном
вискозном производстве обычно в осадительную ванну добав-
ляют также сульфат цинка (до 6%). Стандартное вискозное во-
локно имеет неоднородную структуру, в которой степень ориен-
тации молекул целлюлозы, расположенных в наружной части
волокна (оболочке), выше, чем в сердцевине. С увеличением
концентрации сульфата цинка в осадительной ванне разница
в структурной упорядоченности оболочки и сердцевины посте-
пенно уменьшается, и в конце концов получается высокопроч-
ное волокно, целиком имеющее структуру оболочки. Влияние
сульфата цинка отчасти обусловлено тем, что в его присутствии
ксантогенат целлюлозы быстро коагулируется через сшитые
промежуточные соединения
, н+
2ЦеллОС55" —► [ЦеллОС5 5*2п2+ ‘ЗСБОЦелл] ---->- 2ЦеллОН
Дальнейшее улучшение свойств волокна достигается путем
добавления в вискозный раствор различных модификаторов.
К ним относятся амины (циклогексиламин), соли четвертичного
аммония, полиалкиленгликоли и их простые эфиры (29, R = Н
или СН3; R' = Н, алкил или арил), трпс-оксиалкиленамины (30,
R = H или СН3), оксиэтилированные алифатические амины
(31) и амиды (32) с длинными алкильными радикалами и дру-
гие соединения. Механизм действия этих модификаторов,
314
Глава 9
по-видимому, заключается в том, что они облегчают диффузию
ионов цинка в сердцевину волокна до того, как произойдет раз-
ложение ксантогената кислотой.
H(OCHRCH2)„\
H(OCHRCH2)„OR' H(OCHRCH2)m—N
H(OCHRCH2)z /
29 30
/(CH2CH2O)raH
АлкилМ^
\(CH2CH2O)mH
,(CH2CH2O)„H
Алкил CON’/
\cH2CH2O)mH
31 32
Варьирование основных параметров вискозного процесса,
таких, как степень полимеризации исходной целлюлозы, степень
ее деструкции на стадии предсозревания, степень ксантогениро-
вания и состав осадительной ванны, а также добавление моди-
фикаторов и использование различных условий формования и
вытягивания волокна позволяют получать вискозное волокно с
самыми разнообразными свойствами. Особенно важное значе-
ние имеют высокопрочная кордная нить, на долю которой при-
ходится основная часть производимого вискозного волокна, и
высокомодульные волокна, которые по своим физико-механиче-
ским свойствам и наличию фибриллярной структуры близки
к натуральному хлопку. Одним из видов высокомодульных во-
локон являются полинозные волокна, которые отличаются
устойчивостью к набуханию в концентрированных (свыше 5 М)
растворах едкого натра и поэтому могут быть использованы в
смесях с хлопком в процессе мерсеризации.
Вискозное волокно имеет кристаллическую структуру цел-
люлозы II (как и мерсеризованный хлопок, стр. 303). Его моле-
кулярный вес ниже, чем у хлопка: стандартные вискозные во-
локна содержат около 250 элементарных звеньев на одну
макромолекулу, а полинозные — около 500. Вместе с тем рент-
геноструктурные данные и результаты, полученные методом
дейтерирования, показывают, что структура вискозного волокна
значительно менее упорядочена; это обусловливает повышен-
ную гигроскопичность и реакционную способность вискозного
волокна по сравнению с хлопком. Химические свойства вискоз-
ного волокна аналогичны свойствам хлопка. Его можно окра-
шивать теми же красителями и так же улучшать его потреби-
тельские свойства путем обработки М,Г\Г-диоксиметилмочевиной
и другими сшивающими реагентами. Кроме того, вискозное во-
локно можно модифицировать веществами, добавляемыми в
прядильный раствор. Так, добавление трис-(дибромпропил) фос-
фата (ВгСН2СНВгСН2О)зРО или алкоксифосфазена 33 придает
Волокна
315
вискозному волокну огнестойкость. При введении в прядильный
раствор некоторых веществ основного характера получаются
волокна, которые окрашиваются кислотными красителями (при-
меняемыми обычно для шерсти) и одновременно обладают по-
N
(АлкилО)2Р/ :ЧР(ОАлкил)2
II I
Р
(О Алкил)2
33
вышенным сродством к прямым красителям. Однако многочис-
ленные основания и полимеры, испытанные в качестве модифи-
каторов этого типа, оказались неудовлетворительными, так как
они существенно понижают светостойкость красителей на
волокне.
Ацетатные волокна
К ацетатным волокнам, которые составляют вторую важную
группу искусственных волокон, относятся так называемые ди-
ацетатное и триацетатное волокна, имеющие степени замещения
2,4 и 2,9 — 2,95 соответственно. Ацетилирование облагороженной
древесной целлюлозы или хлопкового линтера, активированных
набуханием в горячей уксусной кислоте, проводят периодиче-
ским способом в смеси уксусного ангидрида с уксусной и серной
кислотами (вместо серной кислоты можно использовать другие
сильнокислотные катализаторы). Реакция сопровождается ча-
стичным ацетолизом целлюлозы. В ходе ацетилирования три-
ацетат целлюлозы переходит в раствор, и по окончании реакции
его высаживают разбавленной уксусной кислотой. Процесс
можно проводить в хлористом метилене, который хорошо рас-
творяет триацетат целлюлозы. Существует также метод получе-
ния триацетата целлюлозы в гетерогенной среде. В этом случае
применяемый разбавитель (например, бензол или четыреххло-
ристый углерод) не обладает растворяющей способностью, а
наилучшим катализатором является хлорная кислота; обра-
зующийся триацетат целлюлозы отделяют простым фильтрова-
нием. В прследнее время широко исследуется возможность
использования в качестве этерифицирующего агента кетена.
316
Глава 9
Поскольку в начальной стадии ацетилирование всегда про-
текает в гетерогенной среде, попытка получить диацетат пря-
мым ацетилированием целлюлозы приводит к образованию про-
дукта со слишком широким распределением по степени заме-
щения. Это затрудняет формование волокна и ухудшает его
качество. Для получения диацетата целлюлозы исходят из три-
ацетата, который подвергают регулируемому кислотнокатали-
тическому омылению путем добавления воды и серной кислоты
к его раствору в ацетилирующей смеси и последующего нагре-
вания при температуре 45—50 °C. Нагревание продолжают до
образования продукта, полностью растворимого в ацетоне, при-
меняемом в качестве растворителя при формовании диацетат-
ного волокна. Одновременно омыляются сульфаты целлюлозы,
которые, если их оставить в волокне, могут медленно гидроли-
зоваться
ЦеллОЗОзН + Н2О ЦеллОН+ H2SO4
Выделяющаяся при этом серная кислота катализирует реак-
ции гидролитической деструкции основной цепи и ацетолиза,
сопровождающиеся понижением прочности волокна, и вызывает
обесцвечивание ткани при ее термической обработке. При ис-
пользовании хлорной кислоты как катализатора перечисленные
явления не имеют места.
Термостойкость диацетата (т. пл. ~230—250 °C) и триаце-
тата (т. пл. выше 300°С) недостаточна для формования из
расплава. Поэтому получение ацетатных волокон обычно осу-
ществляют формованием из раствора по сухому способу (вы-
давливание струек раствора в поток горячего воздуха), Диаце-
татное волокно формуют из концентрированного (20—25%)
раствора в ацетоне, содержащего несколько процентов воды,
играющей роль депрессатора вязкости, а триацетатное — из
смеси хлористого метилена с метанолом или этанолом (90: 10).
Триацетатное волокно можно также формовать по мокрому
способу непосредственно из ацетилирующей смеси в осадитель-
ную ванну с разбавленной уксусной кислотой.
Разработаны способы получения частично и полностью аце-
тилированной целлюлозы путем этерификации хлопка в гетеро-
генной среде смесью уксусного ангидрида с фосфорной кисло-
той или ацетилирования вискозного волокна парами уксусного
ангидрида. Однако эти методы пока не нашли применения в
промышленности.
Разрывная прочность ацетатных волокон колеблется от низ-
кой до умеренной. В случае триацетатного волокна это можно
объяснить отсутствием выраженной надмолекулярной струк-
туры, что обусловлено большим содержанием объемистых бо-
ковых ацетильных групп. Последние мешают образованию
Волокна
317
прочных межцепных связей, хотя триацетатное волокно обла-
дает упорядоченной структурой, что установлено рентгеногра-
фически. В диацетатном волокне нерегулярность замещения
приводит к дальнейшему ослаблению сил межцепного взаимо-
действия и понижению способности к ориентации. Однако влия-
ние этого фактора отчасти компенсируется возможностью обра-
зования водородных связей с участием свободных гидроксиль-
ных групп соседних цепей. Благодаря более открытой структуре
и наличию свободных гидроксильных групп диацетатное волок-
но более гидрофильно, чем триацетатное (его гигроскопичность
составляет 7% против 3—4% у триацетатного волокна), и обла-
дает лучшей окрашиваемостью при данной температуре. Од-
нако более высокая термостойкость триацетатного волокна
позволяет проводить его крашение при высоких температурах.
Другие недостатки, такие, как легкая электризуемость, повы-
шенная загрязняемость и низкая смачиваемость (например,
красильными растворами), можно свести до минимума путем
регулируемого омыления поверхности триацетатного волокна
разбавленным раствором едкого натра (S-отделка), которое
практически не сопровождается деструкцией макромолекуляр-
ных цепей.
Синтетические волокна.
I. Полиамидные волокна
Полиамидные волокна представляют собой один из двух
важнейших видов синтетических волокон. Их подразделяют на
волокна из полимеров типа АВ (34), которые можно рассмат-
ривать как продукты полимеризации со-аминокарбоновых кис-
лот, и волокна из полимеров типа ААВВ (35), образующихся при
поликонденсации диаминов, с дикарбоновыми кислотами. Кроме
того, возможно получение сополимеров, содержащих структур-
ные фрагменты обоих типов.
~NH(CH2),CO~- ~NH(CH2)„NHCO(CH2)„CO~'-
найлон типа п+1 найлон типа n,m+Z
34 35
Несмотря на огромное число изученных полиамидов — али-
фатических, алициклических и ароматических, — в промышлен-
ном масштабе производятся главным образом два вида поли-
амидных волокон: найлон-6 (34, п = 5) и найлон-6,6 (35, п = 6,
т = 4). Последний выпускается в несколько большем количе-
стве, чем найлон-6. Широкое распространение этих волокон по
сравнению с другими полиамидами обусловлено относительной
простотой процесса их получения и удачным сочетанием
318
Глава 9
механических и химических свойств, а также наличием эконо-
мичных методов многотоннажного синтеза полупродуктов для их
производства на основе главным образом нефтехимического
сырья.
Найлон-6
Найлон-6 (36) получают термической полимеризацией капро-
лактама (лактама е-аминокапроновой кислоты) (т. 2, стр. 229)
в инертной атмосфере при температуре до 270 °C. Для иниции-
рования реакции необходим активатор (обычно вода, е-амино-
капроновая кислота или соль АГ, см. ниже) в количестве
1—2%. Экспериментальные данные свидетельствуют о том, что
процесс протекает по механизму ступенчатой полимеризации.
п Г + Н2О ---------> НО--[-—CO(CH2)5NH—Vi—CO(CH2)5NH2
\___/ 36
В качестве регуляторов роста цепи можно использовать до-
бавки монокарбоновых кислот, например уксусной. Регуляторы
предотвращают чрезмерное повышение молекулярного веса,
блокируя концевые аминогруппы. Небольшое количество исход-
ного мономера (около 10%), присутствующее в равновесной
полимеризационной смеси, перед формованием из расплава
удаляют путем экстракции водой. Кроме того, при полимериза-
ции образуется немного циклических олигомеров (37, п=1,
3—8).
NH(CH2)6CO
[CO(CH2)5NH]„
37
В процессе формования волокна в незначительной степени
происходит деструкция полимера в мономер.
Различные методы синтеза капролактама и их относительное
значение в промышленности рассмотрены в гл. 7.
Найлон-6,6
Найлон-6,6 (38) получают при термической поликонденсации
гексаметилендиамина с адипиновой кислотой (т. 1, стр. 172).
Синтез этих исходных веществ описан в предыдущих главах.
Для получения полимера с высоким молекулярным весом
реагенты необходимо брать в строго эквимолярном соотноше-
нии. Поэтому первой стадией процесса является приготовление
чистой соли гексаметилендиамина и адипиновой кислоты со-
става 1 : 1 (так называемой соли АГ) путем смешивания обоих
реагентов в растворе метанола. Концентрированный раствор
Волокна
319
этой соли нагревают под давлением в атмосфере инертного газа
до температуры около 270 °C, затем отдувают водяной пар и
завершают поликонденсацию в вакууме.
НООС(СН2)4СООН + H2N(CH2)tNH2 -►
•--»• ~C0NH(CH2)6NHC0(CH2)4~-
38
Избыток одного из реагентов вызывает преждевременное
прекращение полимеризации. Регулирование молекулярного
веса лучше всего осуществлять добавлением небольших коли-
честв уксусной кислоты; в этом случае образующийся полимер
содержит некоторое число ацетамидных концевых групп. При-
менение регуляторов необходимо для того, чтобы обеспечить
Рис. 9.4. Водородные связи в структуре найлона-6 [Holmes D.R.,Bunn С. W.,
Smith D. J., J. Polymer Sci., 17, 168 (1955)].
оптимальную вязкость расплава полимера при последующем
формовании и хорошие механические показатели волокна.
Обычно молекулярные веса найлона-6 и найлона-6,6 составляют
10000—15000.
Вытянутые волокна обоих видов найлона, судя по рентгено-
структурным данным, имеют в кристаллических участках слои-
стую структуру с высоким содержанием межмолекулярных водо-
родных связей (рис. 9.4). Однако данные ИК-спектров свидетель-
ствуют о том, что значительная часть NH-групп не участвует
320
Глава $
в образовании водородных связей и Легко дейтерируется пара-
ми D2O.
Концевые аминогруппы повышают сродство найлона к кис-
лотным и активным красителям. Концентрацию этих групп
можно вычислить из данных по титрованию раствора найлона
НС1.В феноле. Реакционноспособные концевые аминогруппы в
твердом волокне определяют путем их 2,4-динитрофенилирова-
ния. Концевые карбоксильные группы можно оттитровать рас-
твором едкого кали и этиленгликоля в горячем бензиловом
спирте.
На различии в температурах плавления найлона-6 (215—
220 °C) и найлона-6,6 (265—170 °C) основан метод получения
бикомпонентных волокон совместным формованием обоих поли-
меров через одну и ту же фильеру в условиях, исключающих
смешивание их расплавов. Образующемуся волокну можно
придать структуру типа «бок о бок» (/), «ядро — оболочка» (2)
или типа дисперсии мельчайших фибрилл одного полимера в
матрице другого (<?) *. К последнему типу относятся волокна
соре и энкатрон, в которых фибриллы полиэфира распределены
в найлоне-6 или в найлоне-6,6 соответственно.
При нагревании волокна типа 2, ядро которого содержит
найлон-6,6, а оболочка — найлон-6 (волокно гетерофил), под
давлением выше температуры размягчения найлона-6 происхо-
дит сплавление отдельных нитей в точках их перекрещивания
и получается нетканый материал, особенно пригодный для из-
готовления ковров, обивки мебели и занавесей.
Обработка найлона-6 или найлона-6,6 водным раствором
полимерной карбоновой кислоты [например, полиакриловой
кислоты (39), сополимера метилвинилового эфира с малеиновой
кислотой 1 : 1 (40) или сополимера этилена с малеиновой кис-
лотой 1:1 (41)] с последующим нагреванием приводит к обра-
~СН2—СНл ~сн,сн—сн---сн-~ ^сн2—сн2сн—сн-^
I III II
СООН OCHjCOOH СООН НООС СООН
39 40 41
* Волокна, имеющие структуру 3, называются матричными волокнами,—
Прим- перед.
Волокна
321
зованшо на поверхности волокна прочно сцепленной с ним
пленки полимера. Эта реакция может протекать путем ацили-
рования концевых аминогрупп (а), переамидирования (б) и
образования имидогрупп (в). Ее можно использовать для при-
дания волокну жесткости и меньшей загрязняемости. Данный
NH2'~^ONH~'/'^CONH~'~4CONH
NH СООН TINOCO—N'W4r-CONHrj4r-r-
со со со
а б в
процесс может происходить и при случайном пересушивании
найлоновой пряжи, прошлихтованной полиакриловой кислотой,
перед изготовлением ткани.
Другие полиамиды типа АВ
Синтезированы и изучены все алифатические полиамиды,
содержащие элементарное звено —CO(CH2)nNH— (п = 3—12).
Однако ни один из них не может сравниться по объему произ-
водства с найлоном-6 (п = 5). Влияние длины алкиленовой
цепи на температуру плавления полимеров носит характер ти-
пичного альтернирующего эффекта, причем общая тенденция
состоит в понижении температуры плавления:
п 23456789 10 11 12
Тпл, °C 330 265 260 230 223 233 200 209 188 190 179
Гомологи с п<5 представляют большой интерес из-за их
высокой гидрофильности. Так, гигроскопичность найлона-3 при
температуре 21 °C и относительной влажности воздуха 65%
составляет около 10%.
Из числа этих полиамидов в промышленном масштабе вы-
рабатываются в виде волокон найлон-11 (рильсан), найлон-7
(энант, СССР *) и совсем недавно найлон-4 (таймин). Найлон-12
не применяется самостоятельно; однако на его основе выпускают
волокна, которые приобретают адгезионные свойства при нагре-
вании. Найлон-11 и найлон-12 используются также для изготов-
ления конструкционных пластиков.
Найлон-3. При анионной полимеризации лактама р-амино-
пропионовой кислоты получается волокнообразующий полимер.
* Волокно энант в СССР не производится. — Прим, ред,
11 Зак. 501
322
Глава 9
Однако исходный мономер труднодоступен. С другой стороны,
замещенные 0-лактамы можно легко синтезировать из соответ-
ствующих производных этилена (сам этилен недостаточно
реакционноспособен) и затем подвергнуть анионной полимери-
зации в поли-р-амиды
so3 + C1CN
CISOjNCO + R‘R2C=CR3R*
----► CISOjNCO
----». R*R2C—CR3R*
CISOjN—CO
RlR2(j;—CR3R*
HN—CO
~'NH—CR‘R2—CR3R*—CO~*
Лактам а,р-диметил-р-аминопропионовой кислоты получает-
ся в виде смеси цис- и транс-изомеров, полимеры которых
NH— СН— СН3
со—^н—сн3
имеют соответственно эритро- и трео-конфигурацию. эратро-Фор-
ма обладает более высокой термостойкостью. Формование во-
локон из этих полимеров ведут мокрым способом, например
применяя раствор в метаноле, содержащем роданистый каль-
ций. Поли-р-аминомасляная кислота
~NHCH(CHs)CH2CO~
дает волокно, свойства которого во многих отношениях близки
к свойствам натурального шелка. Синтез найлона-3 можно осу-
ществить также путем полимеризации акриламида по меха-
низму последовательного соединения его молекул по реакции
Михаэля.
Найлон-4. Исходный пирролидон (лактам у-аминомасляной
кислоты) получают на основе ацетилена (гл. 4). Анионная по-
лимеризация пирролидона в присутствии двуокиси углерода,
играющей роль инициатора, сопровождается образованием по-
лимера с высоким молекулярным весом. Этот полимер можно
формовать из расплава, несмотря на то что при температуре
280 °C равновесие лактам — полимер целиком смещено в сто-
рону мономерного лактама
—> —CO(CH2)3NH—
Волокна
323
Найлон-7. Существует два пути синтеза этого полиамида
Один из них основан на использовании лактона е-оксикапроно-
вой кислоты
----* С1(СН3)3СООН > CN(CH3)3COOCzH5-^->.
—> HjN(CHj)6CO0C2Hs——► H2N(CHj)6COOH J > найлон-7
Другой метод, разработанный независимо в СССР и Израи-
ле, базируется па теломеризации этилена с четыреххлористым
углеродом
R2O2 H2SO4, н2о
СС14 + «СН2—СН2 -----> С1(СН2СН2)„СС13 -------->
42 (при П=3)
NH3 А
—> С1(СН2)6СООН [00 ос > H2N(CH2)6COOH ------► найлон-7
Найлон-9. Для получения этого полиамида можно исходить
из соответствующего гомолога 42 (л = 4), выделяемого при
теломеризации этилена в синтезе найлона-7. В более позднем
процессе исходным полупродуктом служит и-аминопеларгоно-
вая кислота, вырабатываемая из соевого масла.
Найлон-11. Найлон-11 получают полимеризацией ш-амипо-
ундекановой кислоты, синтезируемой из касторового масла
(гл. 2).
Найлон-12. Путем тримеризации бутадиена можно получить
циклододекатриен-1,5,9, который с помощью обычных методов
(гл. 4) можно превратить в лактам w-аминолауриновой кисло-
ты— исходный продукт для синтеза найлона-12.
Другие алифатические полиамиды типа ААВВ
Из других полиамидов этого типа в промышленности полу-
чают, вероятно, только найлон-6,10 и (в меньших масштабах)
найлон-10,10. Первый из них применяется главным образом для
производства жестких щеточных изделий. Исходная дикарбо-
новая кислота Сю (себациновая кислота) выделяется из про-
дуктов щелочного расщепления рицинолевой кислоты, содержа-
щейся в касторовом масле. Затем проводят поликонденсацию
СН3(СН2)6СН(ОН)СН2СН=СН(СН2)7СООН
| NaOH
I < 200 °C
СН3(СН2)6СОСН3 + НОСН2(СН2)8СООН
I NaOH, Н2О
1240 °C
СНз(СН2)5СН(ОН)СНз + НООС(СН2)8СООН
11*
324
Глава 9
себациновой кислоты с гексаметилендиамином или декамети-
лендиамином по обычному методу и получают соответствую-
щий полиамид.
Алициклические полиамиды (киана)
Синтез этих новых волокнообразующих полиамидов осу-
ществляют путем поликонденсации бис- (п-аминоциклогексил)-
метана с жирными дикарбоновыми кислотами С8, Сю и Сю или
их хлорангидридами. Исходный амин получается гидрирова-
нием продукта конденсации анилина с формальдегидом, причем
эту реакцию проводят в таких условиях, чтобы образующийся
амин содержал не менее 50% транс,транс-изомера (43), струк-
тура которого наиболее близка к плоской.
2C6H5NH2 + СН2О ---> H2N—/ V-CH2-/ y~NH2 ---------->
H2N
КЙ2
H2
Большое значение для получения полимера, способного фор-
моваться в волокно, имеет стереоизомерный состав амина, по-
скольку от содержания в нем транс,транс-, транс,цис- и
цис,цис-изомеров в существенной степени зависят физические
свойства полиамида, особенно температуры плавления (Тпл) и
стеклования (Тст) (гл. 8).
Волокно киана, сформованное из расплавленного полимера
на основе амина 43 и, по-видимому, жирной дикарбоновой кис-
лоты Сю, напоминает по внешнему виду натуральный шелк. По
прочности оно приближается к найлону-6,6, однако обладает
меньшей сминаемостью при небольших деформациях и несколь-
ко меньшей гигроскопичностью. Отличительная особенность во-
локон типа киана — более высокая по сравнению с найлоном-6
и найлоном-6,6 температура стеклования и в то же время до-
статочно низкая температура плавления, делающая возможной
переработку полимера в расплавленном состоянии. Сравнитель-
ные данные о свойствах волокон киана и найлона-6,6 приведены
в табл. 9.4.
Важным следствием высоких значений Тст волокон киана
является то, что они сохраняют стеклообразное состояние при
стирке сотканных из них изделий. Если выстиранное изделие
резко охладить от температуры выше Тст до температуры ниже
Тст (например, прополоскав его в холодной воде) раньше, чем
исчезнут отутюженные складки, то последние закрепятся на
Волокна
325
Таблица 9.4
Сравнительные данные по свойствам волокон киана и найлона-6,6
Волокно Гпл- °С т СТ’ сухое волокно °C
мокрое волокно Гигроско- пичность, %
Волокно киана на основе 240 140 3,2
кислоты С8 Волокно киана на основе 205 135 100 3,0
КИСЛОТЫ С12 Найлон-6,6 265 90 Ниже 20 4,3
изделии и останутся до тех пор, пока его снова не нагреют выше
температуры стеклования. Таким образом, изделия из волокон
типа киана обладают высокой формоустойчивостью и не нуж-
даются в глажении. Напротив, изделия из обычных (алифати-
ческих) полиамидов в увлажненном состоянии легко теряют
свою форму даже при резком охлаждении.
Разница в длине сегментов дикарбоновой кислоты и амина
в алициклических найлонах не допускает образования большого
числа межцепных водородных связей, которое происходит в
найлоне-6 и найлоне-6,6, а наличие объемистых циклогексиле-
новых групп затрудняет плотную упаковку макромолекул.
Влияние этих неблагоприятных факторов отчасти компенсирует-
ся повышенной жесткостью алициклических фрагментов по
сравнению с алифатическими, что придает волокну достаточно
большую разрывную прочность.
Сообщается, что полиамидные волокна, обладающие полез-
ными свойствами, можно получить и из других алициклических
диаминов и дикарбоновых кислот, например из циклопропилен-
или циклобутилендиаминов и циклопропан- или циклобутанди-
карбоновых кислот, а также из бис-(аминометил) циклобутанов,
транс- 1,4-циклогексилендиамина или бис- (аминометил) цикло-
гексанов и 4,4'-метилен-бнс-(циклогексанкарбоновой кислоты).
Синтетические волокна.
II. Полиэфирные волокна
Терилен
Несмотря на широкие исследования по синтезу новых поли-
эфиров, полиэтилентерефталат (терилен) до сих пор сохраняет
практически монопольное положение среди волокнообразую-
щих полимеров данного класса. Это обусловлено рядом причин
326
Глава 9
практического и экономического характера. Терефталевую кис-
лоту получают из n-ксилола путем его прямого каталитического
окисления (т. 1, стр. 170) или, в виде моноэфира, в две стадии
через n-толуиловую кислоту. Кроме того, ее можно получить
карбоксилированием бензоата калия или изомеризацией фта-
лата калия (гл. 5). Существует метод синтеза полиэтилентере-
фталата путем прямой полиэтерификации терефталевой кислоты
этиленгликолем, однако при этом необходимо использовать вы-
сокочистую кислоту. Чаще всего кислоту сначала переводят
в легко поддающийся очистке диметиловый эфир (44), который
переэтерифицируют избытком этиленгликоля, получая промежу-
точную смесь дигликольтерефталата и его олигомеров. Эту
смесь подвергают поликонденсации при повышенной темпера-
туре в вакууме с одновременной отгонкой выделяющегося эти-
ленгликоля до достижения молекулярного веса, достаточного
для формования волокна из расплава полимера. В качестве
катализатора обычно применяют окись сурьмы. Наряду с линей-
ным полиэтилентерефталатом образуется небольшое количество
циклического тримера.
сн3осо—СООСНэ-------►
44
—► НОСН2СН2ОСО—-СООСН2СН2ОН + олигомеры—8Ь^-»
—> НОСН2СН2ОСО-^^-СООСН2СН2'^ОСО-^^-СООСН2СН2ОН
Качество продукта в значительной степени определяется
степенью чистоты исходных веществ и точностью соблюдения
режима проведения процесса. На стадии формования волокна
необходимо полностью исключить присутствие влаги, так как в
противном случае протекает быстрый гидролиз полиэфира при
высокой температуре (270—280°C), сопровождающийся пони-
жением его молекулярного веса. Полученные нити подвергают
ориентированию путем их вытягивания в горячем состоянии
(при температуре выше 80°C).
Рентгеноструктурные данные свидетельствуют о том, что в
кристаллических участках ориентированного полиэтилентере-
фталата макромолекулы почти целиком вытянуты вдоль оси
волокна, причем копланарность ароматических колец (рис. 9.5)
способствует плотной упаковке молекул. Следствием такой не-
гибкой надмолекулярной структуры полиэфирных волокон
является их более высокая жесткость по сравнению с другими
синтетическими волокнами. Значительная степень ориентации и
Волокна
327
Рис. 9.5. Кристаллическая структура полиэтилентерефталата (Mark Н. F.,
Atlas S. М., Cernia Е. М., Man-made Fibres, Science and Technology, Vol.
Ill, Interscience, New York, 1968).
отсутствие полярных групп делают полиэтилентерефталат высо-
когидрофобным и обусловливают его ничтожную гигроскопич-
ность. Почти полная неспособность полиэфирных волокон набу-
хать в воде существенно затрудняет их крашение и отделку в
производстве текстильных изделий. Поэтому для повышения
скорости диффузии красителя в волокно и достижения нужной
интенсивности окраски пришлось разработать специальные
высокотемпературные способы крашения и использовать так
называемые переносчики красителя — главным образом арома-
тические соединения (например, бифенил, дифениловый эфир
и метилсалицилат), которые способны растворять краситель и
абсорбироваться полиэфиром.
328
Глава 9
Полиэтилентерефталат чувствителен к гидролизу растворами
сильных щелочей, которые вызывают его поверхностное разру-
шение, и к аминолизу (гидразингидрат быстро разъедает во-
локно). По отношению к другим реагентам терилен вполне
ОСНгСН2ОСО—/ у—СО—о—
НОСН2СН2ОН + H2NNHCO-Zy-CONHNH2
устойчив. Серьезную проблему представляет высокая загрязняе-
мость изделий из полиэфирных волокон, что объясняют их
легкой электризуемостью (заряд статического электричества
притягивает к волокну противоположно заряженные частицы
пыли и грязи) и большим сродством к маслам, жирам и смаз-
кам (которые прочно удерживают загрязнения). Этот недоста-
ток можно свести к минимуму путем прививки к терилену не-
большого числа звеньев неволокнообразующего полимера
(«пермалоза ТГ»), близкого ему по химическому строению, но
содержащего вместо части этиленгликолевых фрагментов поли-
оксиэтиленовые группы (45 с небольшим значением п). Сцепле-
~-ОСН2СН2ОСО—/ V-COO(CH2CH2O)„—со—/ 'Ч—СО—о~-
ние полимера с поверхностью волокна достигается при их крат-
ковременном совместном нагревании до 180—200°C.
С целью изыскания волокнообразующих полимеров, которые
более реакционноспособны (т. е. обладают менее упорядочен-
ной структурой), более гидрофильны, легче окрашиваются или
способны окрашиваться красителями иных типов, чем обычно
применяемые дисперсные красители, было синтезировано боль-
шое число смешанных полиэфиров, содержащих в основной
цепи полиэтилентерефталата включения звеньев других глико-
лей и дикарбоновых кислот. В качестве кислот были использо-
ваны изофталевая (46), п-оксибензойная (47) и (для улучшения
окрашиваемости волокна) сульфоизофталевая (48) кислоты.
~Ч)ОС.
СООАЛ
.соо^4 ~чоос
соолл-
АЛО
SO2H
48
Полиэфирные волокна горючи, но, поскольку они являются
термопластичными материалами и, плавясь, стекают с горящей
Волокна
329
ткани на землю, их не относят к легковоспламеняющимся ве-
ществам. Однако в комбинации с целлюлозными волокнами,
горение которых сопровождается образованием сажи, полиэфир
теряет способность стекать на землю и становится пожароопас-
ным. Точный механизм пиролиза полиэтилентерефталата на
начальных стадиях его возгорания не установлен. Вероятнее
всего, он включает образование сложноэфирных винильных
групп
соосн2сн2осо-
соон + сн2==сносо
В основе метода идентификации полиэфирных волокон лежит
их термическое разложение в отсутствие воздуха при темпера-
туре около 600 °C с выделением бензола и толуола, которые
определяют посредством газожидкостной хроматографии.
Другие полиэфирные волокна
Из большого числа полиэфиров, изученных в качестве заме-
нителей терилена, наиболее детальному исследованию подверг-
ся, вероятно, терефталат 1,4-диоксиметилциклогексана (кодель).
Селективное восстановление диметилтерефталата приводит к
образованию алициклического диэфира 49, который гидрируют
над хроматом меди в диол 50. При переэтерификации этим дио-
лом диметилтерефталата получается полимер, который можно
формовать из расплава с последующим вытягиванием и термо-
обработкой волокна обычными методами.
49 50
Для диола 50 известны как цис-, так и транс-изомеры, при-
чем по мере увеличения содержания в нем транс-формы темпе-
ратуры плавления и стеклования полученного полимера возра-
стают. На практике диол обычно содержит около 70% транс-
330
Глава 9
изомера, имеющего, вероятно, диэкваториальную конформацию
(51). Однако быстрое охлаждение расплавленного полимера,
вытекающего из отверстий фильеры, может сопровождаться
образованием метастабильных конформеров.
Изогнутые циклогексановые кольца мешают плотной упа-
ковке молекул полимера, которая наблюдается в полиэтиленте-
рефталате (о чем свидетельствуют более низкие показатели
двойного лучепреломления и плотности коделя). Это явление
должно приводить к понижению температуры плавления. Од-
нако его влияние с избытком компенсируется увеличением
жесткости макромолекулярной цепи из-за циклического строе-
ния диола, в результате чего значения температур плавления и
стеклования для коделя (на основе диола, содержащего 70%
транс-изомера) превышают соответствующие значения для те-
рилена. Кодель обладает также более высокой гидролитической
стойкостью, обусловленной пространственными затруднениями
реакции в диольных фрагментах. Недостатком коделя является
пониженная по сравнению с териленом термо- и светостойкость.
Большой интерес привлекло к себе недавно появившееся
полиэфирное волокно А-Телл на основе л-оксибензойной кис-
лоты.
—СН2СН2ООС
А-Телл
Сообщается, что оно приближается по свойствам к натураль-
ному шелку. Кроме того, в США был разработан метод полу-
чения волокна из полиэфира бутандиола-1,4 с терефталевой
кислотой. Из числа алифатических полиэфиров наиболее пер-
~4)CH2C(CH3)2CO'VV4 /^0СН2СО~'-
52 53
спективны полипивалолактон (52) и полигликолевая кислота
(53). Полипивалолактон имеет температуру плавления около
Волокна
331
240 °C и достаточно устойчив к гидролизу, что обусловлено про-
странственными затруднениями.
Волокна из полимера 53 не представляют интереса для тек-
стильной промышленности. Однако их низкая гидролитическая
стойкость превращается в достоинство при изготовлении хирур-
гических нитей (дексон), которые способны медленно рассасы-
ваться в организме, не вызывая токсических или других небла-
гоприятных побочных явлений.
Синтетические волокна.
III. Полиакрилонитрильные волокна
Полиакрилонитрильными называются волокна, содержащие
не менее 85% звеньев акрилонитрила. Если содержание звеньев
акрилонитрила составляет 35—85%, то такие волокна носят
название модакриловых (см. ниже). Способность приобретать
при текстурировании шерстеподобный внешний вид обусловли-
вает преимущественное использование полиакрилонитрильных
волокон в производстве трикотажных изделий, ковров и обив-
ки мебели. Различные методы синтеза акрилонитрила рассмот-
рены в гл. 7.
Радикальная полимеризация акрилонитрила легко протекает
в водной суспензии в присутствии стандартных окислительно-
восстановительных каталитических систем. Полимер получается
в виде порошка с молекулярным весом 75 000—150000. Его
подвергают формованию сухим способом из раствора в диме-
тилформамиде в среду горячего воздуха или мокрым способом
из раствора в диметилформамиде, диметилацетамиде или вод-
ном растворе роданистого натрия, используя подходящую вод-
ную осадительную ванну. На ряде предприятий применяется
также полимеризация акрилонитрила в растворе подходящего
растворителя с низкой константой передачи цепи, позволяющей
получать достаточно высокомолекулярный продукт (например,
в водном растворе роданистого натрия), причем образующийся
раствор полимера может непосредственно служить прядильным
раствором. Волокно из гомополимера акрилонитрила обладает
определенными недостатками, главным из которых является
плохая окрашиваемость. Поэтому почти все промышленные
полиакрилонитрильные волокна изготовляют из сополимеров
акрилонитрила. Последний легко вступает в статистическую
сополимеризацию с другими винильными и акриловыми моно-
мерами. В качестве модификаторов полиакрилонитрильного
волокна было изучено большое число таких мономеров. Трудно
установить, какие из них в настоящее время применяются в
промышленности, однако наиболее типичными сомономерами
332
Глава 9
являются. винилацетат, низшие акрилаты и акриламид. В тех
случаях, когда содержание звеньев сомономера не превышает
15%, волокно сохраняет приемлемые механические свойства.
Неупорядоченные включения звеньев сомономера эффектив-
но нарушают регулярность химической структуры и придают
волокну повышенную термопластичность и более высокое срод-
ство к красителям. Иногда в состав волокна вводят небольшое
(менее 5%) количество звеньев третьего мономера с целью
придания ему избирательного сродства к отдельным типам кра-
сителей; например, мономер с кислотными группами увеличи-
вает сродство к основным красителям, и наоборот. Среди мно-
гочисленных соединений, предложенных в качестве третьего
сомономера, можно назвать итаконовую кислоту, изобутилен-1-
сульфокислоту и сульфоалкилакриламиды (кислотные мономе-
ры) и винилпиридины (основной мономер). Другими методами
создания в полиакрилонитрильном волокне реакционноспо-
собных функциональных групп являются частичный гидролиз,
проводящий к образованию карбоксильных групп, и реакция
с гидроксиламином, в результате которых улучшается окраши-
ваемость волокна соответственно основными и кислотными кра-
сителями.
NC-Lсоон <Н)0, NC~Lon НгХ011> NC"Lc^NH
У—CLKJJH. < V—СГЧ * >—
NC—y NC—$ NC—5 NHOH
Хотя полиакрилонитрильные волокна имеют преимущественно
атактическую структуру, они характеризуются достаточно вы-
сокой прочностью. Рентгенографические данные свидетельствуют
о значительной упорядоченности надмолекулярной структуры,
но не обнаруживают никаких признаков истинной кристаллич-
ности. Последнее не удивительно, так как полимерные молекулы
лишены стереорегулярности, необходимой для плотной упаковки.
Наличие межмолекулярных связей можно объяснить сильным
диполь-дипольным взаимодействием групп CsN соседних це-
пей, энергия которого достигает или даже превышает энергию
водородных связей в целлюлозных и полиамидных волокнах. По
этой причине полиакрилонитрильные волокна растворяются
только в высокополярных растворителях. Попытки получить
стереорегулярные полиакрилонитрильные волокна были безус-
пешными; при анионной полимеризации акрилонитрила обра-
зуется окрашенный продукт.
Широко применяется в промышленности повышение извито-
сти полиакрилонитрильных волокон с целью придания им шер-
степодобного вида. В настоящее время для этого используются
бикомпонентные волокна, такие, как волокно типа «бок о бок»,
Волокна
333
сформованное из двух акрилонитрильных сополимеров с раз-
личным содержанием второго мономера (например, винилаце-
тата). Вследствие различной усадки компонентов при нагрева-
нии волокна оно становится извитым, причем достигнутая изви-
тость сохраняется вплоть до температуры, при которой она
была получена.
Модакриловые волокна. Эти сополимерные волокна, которые
согласно приведенному выше определению содержат 35—85%
звеньев акрилонитрила, были первоначально разработаны
с целью улучшения свойств поливинилхлоридных волокон (в
частности, для повышения температуры размягчения) и облегче-
ния их формования из растворов в обычных растворителях.
В настоящее время модакриловые волокна — сами по себе или
в смеси с другими волокнами — применяются в качестве огне-
стойких волокон. Они представляют собой статистические со-
полимеры акрилонитрила с винилхлоридом или винилиденхло-
ридом, полученные путем обычной радикальной сополимериза-
ции. Примерами модакриловых волокон этого типа могут
служить дайнел, теклан, верел и канекалон. Все они формуются
из раствора. Из-за нерегулярного строения макромолекул и низ-
кого содержания звеньев акрилонитрила межцепные взаимодей-
ствия в модакриловых волокнах выражены слабее, чем в поли-
акрилонитрильных. Поэтому модакриловые волокна менее проч-
ны, растворимы в менее полярных растворителях и размягчаются
при более низких температурах. Однако они сохраняют высокую
хемо- и светостойкость, присущую полиакрилонитрильным во-
локнам. В настоящее время выпускается модакриловое волокно,
приближающееся по свойствам к полиакрилонитрильным во-
локнам (акрилан модакрилик). Для получения модакриловых во-
локон предложено использовать, помимо указанных выше, боль-
шое число других галогенсодержащих сомономеров, в том числе
СН2=СНВг, СН2=С(СН2С1)СЫиСН2=СНСООСН2С(СН2Вг)3.
Однако перечисленные соединения являются дорогостоящими и
по этой причине не нашли применения в промышленном мас-
штабе.
Синтетические волокна.
IV. Полиолефиновые волокна
Полиэтиленовые волокна
Полимеризация этилена при высоких давлениях и темпера-
турах в присутствии перекисного или другого радикального ини-
циатора приводит к образованию низкоплавкого (Гпл НО °C)
продукта с низкой плотностью (0,92 г/см3), имеющего значи-
тельную степень разветвленности, что мешает плотной упаковке
334
Глава 9
макромолекул. При полимеризации этилена на окис'нометалли-
ческих или циглеровских катализаторах при низких давлениях
получается полимер с более упорядоченной (линейной) струк-
турой и молекулярным весом около 20 000. Этот полимер спосо-
бен формоваться из расплава при температуре 300 °C. Полу-
ченное волокно после его вытягивания обладает довольно низ-
кой прочностью; его Гпл равна ~130 °C, а плотность —
0,95 г/см3. Воскоподобный внешний вид, легкоплавкость, неги-
гроскопичность и высокая загрязняемость делают полиэтилено-
вые волокна непригодными для изготовления одежды (за исклю-
чением защитной спецодежды); однако они могут быть исполь-
зованы в производстве высокоусадочных волокон и как плавкие
(термосвязывающие) компоненты нетканых материалов.
Полипропиленовые волокна
В отличие от полиэтиленовых полипропиленовые волокна
имеют важное значение в промышленности. Исходным сырьем
для них служит полипропилен с преимущественно изотактиче-
ской структурой, который получается полимеризацией пропи-
лена при низких давлениях и температурах на катализаторах
ииглеровского типа в инертном углеводородном растворителе.
Атактический полипропилен не обладает волокнообразующими
Свойствами, а синдиотактический не производится в промыш-
ленности. Полимер с Тпл 165 °C и молекулярным весом до
400 000 отфильтровывают от реакционной смеси, освобождают
от остатков катализатора, добавляют антиоксидант, окрашивают
(если это нужно) и подвергают формованию из расплава с по-
следующим вытягиванием волокна. Существенно, чтобы тактич-
ность полипропилена составляла около 90%. Ориентированное
волокно может иметь высокую степень кристалличности — до
50—60%. Стремление свести к минимуму пространственное
взаимодействие между метильными группами заставляет почти
линейные молекулы полимера принимать форму спирали, в ко-
торой на каждый- виток приходится три мономерных звена,
а скелетные связи С—С поочередно находятся в транс- и гош-по-
ложениях (рис. 9.6).
Значительную часть полипропилена перерабатывают в во-
локно не формованием из расплава, а фибриллированием .пле-
ночного полимера. В одном из существующих методов экстру-
дированную пленку разрезают на узкие полоски (такой тол-
щины, чтобы из них можно было делать бечевку и грубые
ткани), которые, подвергают термическому вытягиванию. Затем
каждую полоску фибриллируют, пропуская ее через ряд близко
расположенных игл, укрепленных на вращающемся валу. При
этом происходит продольное расщепление полоски на большое
Волокна
335
число одиночных нитей, которые затем скручиваются в пряжу.
Полипропиленовые волокна используют в производстве ка-
натов, подложки ковровых изделий, мешков, пледов и одеял.
Низкая температура размягчения, гидрофобность и плохая ок-
рашиваемость препятствуют широкому применению этих воло-
кон для изготовления одежды. Улучшению окрашиваемости по-
липропилена посвящено большое число работ, так как краше-
Рис. 9.6. Спиралевидная структура изотактического полипропилена [Natta G .,
Corradini Р„ J Polymer Sci. 39, 36 (1959)].
ние исходного полимера в массе не обеспечивает того разнооб-
разия оттенков и интенсивности окраски, которые необходимы
для удовлетворения запросов покупателей. Отсутствие в поли-
пропиленовых волокнах реакционноспособных функциональных
групп, способных сорбировать краситель, не может быть исправ-
лено путем сополимеризации, как в случае полиакрилонитриль-
ных волокон: полярные мономеры дезактивируют каталитиче-
скую систему, а химическая инертность полипропилена делает
невозможным модифицирование сформованного волокна. Одним
336
Глава 9
из путей улучшения окрашиваемости полипропиленовых волокон
является добавление к исходному полимеру солей никеля, кото-
рые выступают в роли комплексообразователей по отношению
к подходящим дисперсным красителям. Кроме того, можно дис-
пергировать в исходном полипропилене какой-нибудь неволок-
нообразующий полимер, например поливинилпиридин, который
обладает сродством к кислотным красителям. Однако, хотя при
этом в волокно и вводятся реакционноспособные группы, остает-
ся нерешенной проблема, связанная с трудностью диффузии
красителя в глубь волокна, вследствие чего многие волокна, мо-
дифицированные данным методом, окрашиваются лишь с поверх-
ности. Сообщается, что создано легко окрашиваемое кислот-
ными красителями полипропиленовое волокно геркулон-404,
в котором эта проблема решена посредством диспергирования
в полипропилене микроволокон из высокополярного гидрофиль-
ного низкокристаллического полимера (состав последнего не
приводится). Микроволокна второго полимера служат путями
проникновения водного красильного раствора в массу волокна.
Полипропиленовое волокно, окрашенное этим способом, может
быть использовано в производстве ковровых ворсовых нитей и
текстильных изделий.
Другие полиолефиновые волокна
Из числа высших полиолефинов наибольший интерес при-
влекает изотактический полимер 4-метилпентена-1, получаемого
путем анионной димеризации пропилена. Этот полимер дает
чрезвычайно легкие волокна (плотность 0,83 г/см3), близкие по
прочности и эластичности к полипропиленовым волокнам. В то
же время они имеют гораздо более высокую температуру плав-
ления (240 °C) и значительно менее склонны к усадке в про-
цессе стирки и химической чистки. Кроме того, производятся
волокна из полистирола и акрилонитрил-стирольных сополиме-
ров, которые выпускаются в виде довольно толстых экструдиро-
ванных нитей, используемых для изготовления синтетической
щетины.
Эластомерные волокна
Волокна из натурального каучука
Производство волокон из каучука [^ис-полиизопрена (54)
с молекулярным весом около 50 000—100 000] осуществляют пу-
тем введения в латекс вулканизующего агента [например, серы,
Волокна
337
дитиокарбамата (55) или тиурама (56)] и последующего про-
давливания его через отверстия фильеры в осадительную ванну,
[(CH^jjNCSS]2Zn (CH3)zNCSS—SCSN(CH3)2
бб 56
содержащую разбавленную уксусную кислоту. Затем волокна
нагревают до достижения заданной степени вулканизации (сши-
вания). В нерастянутом волокне молекулы каучука преимуще-
ственно свернуты в беспорядочный клубок. Высокая эластич-
ность волокна обусловлена способностью молекул ориентиро-
ваться при растяжении вдоль его оси. Дифракционная картина
растянутого волокна имеет вид, характерный для рентгенограм-
мы кристаллического полимера. Поперечные связи выполняют
функцию «памяти», возвращая молекулы после снятия нагрузки
в первоначальную форму клубка. При наличии слишком боль-
шого числа поперечных связей (перевулканизации) волокно ста-
новится хрупким и теряет эластичность.
Полиуретановые волокна
Подавляющее большинство синтетических эластомерных во-
локон в настоящее время составляют полиуретановые волокна.
Волокнам, содержащим не менее 85% полиуретановых сегмен-
тов (т. е. полученным из блок-сополимеров, построенных из гиб-
ких и жестких-блоков), присвоено общее название «спандекс»
(US Federal Trade Commission). В качестве гибких блоков ис-
пользуют звенья простых или сложных полиэфиров с молеку-
лярным весом до 4000, а жесткие блоки создают путем реакции
гибких блоков с диизоцианатом. Типичными простыми полиэфи-
рами, применяемыми для этой цели, являются полиэтиленгли-
коль (57), полипропиленгликоль (58), блок-сополимеры этилен-
гликоля и пропиленгликоля (59) и политетраметиленгликоль
(60), получаемый полимеризацией тетрагидрофурана.
НОСН2СН2(ОСН2СН2)тОСН2СН2ОН
57
НОСН2СН(СН3)[ОСН2СН(СН3)]тОСН2СН(СН3)ОН
58
НОСН2СН2(ОСН2СН2)т[ОСН2СН(СН3)]лОСН2СН(СН3)ОН
59
О hSt НО(СН2)4О(СН2)4О~ЧСН1)4ОН-
! &
338
Глава 9
Исходные сложные полиэфиры обычно синтезируют путем
поликонденсации адипиновой кислоты с небольшим избытком
гликоля, в результате которой образуются макромолекулы с кон-
цевыми гидроксильными группами (61). Наиболее предпочти-
НОСН2СН2О[СО(СН2)4СООСН2СН2О]тСО(СН2)4СООСН2СН2ОН
61
тельны первичные концевые ОН-группы, так как они обладают
большей реакционной способностью.
Второй реагент — диизоцианат — обычно относится к арома-
тическому ряду; примером может служить техническая смесь
2,4- и 2,6-толуилендиизоцианатов (80:20 или 65:35). Диизоциа-
наты получают действием фосгена на соответствующий амин
62. В настоящее время ведутся настойчивые поиски более про-
^х^сн3 ^х^СН3 ^х^СН3
'С'ОС12
нагревание
сосц
на холоду
C1OCHN
NHCOC1
стого метода синтеза изоцианатов. Попытки получить их непо-
средственным взаимодействием нитросоединений с окисью угле-
рода не дали большого успеха.
При обработке простого или сложного полиэфира с конце-
выми гидроксильными группами НО—X—ОН избытком диизо-
цианата образуется полимер с концевыми изоцианатными груп-
пами
OCN—Y—NHCO(O—X—OCONH—Y—NHCOO—X)„OCONH—Y—NCO
Этот преполимер удлиняют, вводя его в реакцию с алифатиче-
ским диамином, гидразином или диацилгидразином, и получают
конечный продукт. Для достижения заданного молекулярного
веса можно добавить в реакционную смесь небольшое количе-
ство вторичного амина, играющего роль регулятора.
R2NH + OCN~YNCO + HjNNHj + OCN^NCO + HNR, ->
RJNCONH(/VA'NHCONHNHCONH)/v4NHCONRj
Полиуретановые волокна нельзя формовать из расплава, так
как этому препятствует термическая нестабильность исходного
полимера, обусловливающая, в частности, его деполимеризацию
с образованием диизоцианата. Поэтому их получают другими
Волокна
339
тремя способами, простейшими из которых являются сухое и
мокрое формование из раствора в диметилформамиде. В треть-
ем методе, известном под названием «реакционное формова-
ние», заключительное удлинение макромолекулярной цепи про-
водится в процессе формования. Преполимер, выходящий из от-
верстия фильеры, пропускают через водный раствор гидразина
или диамина, в котором очень быстро протекает химическая ре-
акция, сопровождающаяся образованием на поверхности во-
локна прочной оболочки полимера. Свежесформованное волокно
принимают на бобину, подвергают вытягиванию в более тонкие
нити и обрабатывают снова диамином или даже просто горячей
водой, в результате чего происходит сшивание во внутренней
части волокна. Образующиеся полимеры (63) имеют линейную
структуру и растворимы в полярных растворителях.
~SNCO + Н2О + OCN~ --> ~-NHCONH~ + СО2
63
Если проводить получение волокна в условиях недостатка
удлинителя цепи, то на концах макромолекул остаются изоциа-
натные группы, способные реагировать при повышенной темпе-
ратуре с мочевинными и уретановыми звеньями с образованием
сшитых продуктов 64. Эти реакции могут протекать при отверж-
дении внутренней части волокна или при термообработке (при
180 °C).
Ww%NHCONH'«W'NHCOO-w^
+ +
NCO NCO
NCO NCO
+ +
NHCONH'w^NHCOO'^*^
>9O“C
> 130-C
zwwNCONHzyv4^NCOO'wz‘
/СО Vo
NH NH 64
S |
NH NH
/СО /CO
ww>'NCONH'W7wNCOO,'w-
340
Глава 9
Сшивание происходит также в том случае, когда простой или
сложный полиэфир содержит небольшое число ветвлений (65),
полученных, например, путем введения в его молекулу остатков
триола типа СН3СН2С (СН2ОН)з
‘NCO
ОН
•5
NCO
Н2О
NH2NHj
NH
NH
—NHOTNH'^-NHCONH—
а или 6
—-OCNH1
NHCONH—NH—
ГН—СО—NH
или
NH—СО—NH
—OCNH
NHC0NH—NH—
Создание поперечных связей можно осуществлять только во
время формования волокна или на последующих стадиях, по-
скольку сшивание исходного полимера делает его неплавким и
нерастворимым в недеструктирующих растворителях, а следова-
тельно, и неспособным формоваться.
Для полиуретановых волокон (таких, как спанзелл и лайкра)
обнаружено два тепловых перехода второго рода. Первый из них
(при температуре ниже 0°С) связан с поведением гибких поли-
эфирных блоков, а второй (при температуре выше 100°С) —
с поведением жестких полиариленуретановых блоков. Рентге-
ноструктурные данные свидетельствуют об отсутствии кристал-
личности полиуретановых волокон при удлинениях ниже
~400%. Следовательно, в нерастянутом состоянии гибкие блоки
должны быть преимущественно разупорядочены и свернуты ана-
логично молекулам натурального каучука. Легкая растягивае-
мость гибких блоков сдерживается взаимодействием между
жесткими блоками соседних цепей, приближающимися при вы-
тягивании макромолекул друг к другу; большой объем этих бло-
ков и образование прочных межцепных водородных связей пре-
пятствуют удлинению волокна сверх определенной степени. На-
личие водородных связей обусловливает необычайно высокую
разрывную прочность полиуретановых волокон при температу-
Волокна
341
pax выше температуры стеклования. Длинноцепочечные попе-
речные связи также ограничивают удлинение волокна и способ-
ствуют его эластичному возвращению в исходное состояние.
По сравнению с волокнами из натурального каучука поли-
уретановые волокна обладают рядом недостатков. Низкая
прочность этих волокон в горячей воде затрудняет стирку изде-
лий из них. Они более чувствительны к гидролизу (особенно
волокна на основе сложных полиэфиров), а при действии щелоч-
ных реагентов и на свету они желтеют. Отбеливатели разру-
шают полиуретановые волокна; особенно неустойчивы по отно-
шению к гипохлоритам волокна из полимеров, отвержденных
гидразином.
Эластомерные волокна типа анидекс
В качестве примера волокон этой группы, получаемых на
основе акрилонитрильных сополимеров, рассмотрим волокно
аним-8. Исходными веществами служат два сополимера: сопо-
лимер 66А, содержащий звенья акрилонитрила, этилакрилата и
N-оксиметилакриламида в соотношении 70 : 20 : 10, и сополимер
66Б, синтезированный из тех же мономеров в соотношении
20:70:10. При формовании смеси обоих сополимеров из рас-
твора в диметилформамиде по сухому способу происходит ста-
тистическое сшивание их цепей с образованием высокоэластич-
СН20Н
СН2ОН
(gj COOC2HsCOOC4H5CONH cn
ного волокна, нерастворимого во всех растворителях и обладаю-
щего хорошей светостойкостью (не желтеет на свету) и гидро-
литической устойчивостью. Это уникальное сочетание свойств
объясняют оптимальным соотношением потенциальных волокно-
образующих свойств жестких блоков сополимера 66А (с высо-
ким содержанием звеньев акрилонитрила) и мягких, каучуко-
подобных блоков сополимера 66Б.
Поливинилспиртовые волокна
Поливинилспиртовые волокна вырабатываются главным об-
разом в Японии, где их объем производства достигает 150т/сут.
Исходный мономер — винилацетат — легко получается на основе
342
Глава 9
этилена или ацетилена. Радикальная полимеризация винилаце-
тата в метаноле или другом растворителе с низкой константой
передачи цепи дает виниловый полимер, линейная структура ко-
торого может быть нарушена путем переноса активного центра
от растущей полимерной цепи Р« на ацетильные группы поли-
мера (а) или мономера М(б), либо путем возникновения свобо-
днорадикальных центров в основной полимерной цепи (в).
(а)
Т' —J—> '“Т' + рн - --м-»
ООССНз OOCCHj. ООССН2СН3СН'Л~
ОАс
«о
СН2=СНОСОСН3 + Р- ---->- РН + СН2=СНОСОСН2-
СН2=СНОСОСН2. Ч-пМ ---> CH2=CHOCOCH2CH2CH^
(^Ас
Р- +СН2==СНОСОСН2СН2СН'^'
I <
I ОАс
Р—СН2СН.
21
OCOCH2CH2CH(0Ac)^ - —Р—СН2СНСН2СН(ОАс)ЛЛ-
ООССН2СН2СН(ОАс)ал
(в) СН2СН(ОАс)ал.
»м ,
ОАс ОАс ОАс
При последующем каталитическом дезацетилировании или
кислотном омылении полимера в среде метанола получается по-
ливиниловый спирт в виде нерастворимого порошка
~-СН2СН(ОАс)~- алСн2сН(ОН)^-+СН3ОАс
Одновременно отщепляются сложноэфирные боковые цепи, что
сопровождается понижением молекулярного веса полимера и
образованием карбоксильных концевых групп
ОАс ^>Ас OCOCH2w'/'p~'z| ОАс ОАс HOOCCHj'
он он он
он он
Волокно
343
Молекулярный вес поливинилового спирта может быть сни-
жен также путем его окисления перйодатом. В этой реакции,
как полагают, принимают участие 1,2-гликолевые группы (67),
образовавшиеся либо в результате полимеризации по типу «го-
лова к голове» (а), либо при рекомбинации макромолекуляр-
ных радикалов (б).
~-СН2СН. + СН=СН;
ОАс ОАс
"ЛСН—син-
^CH2CH. + -СНСН2~-
ОАс ОАс
оде ОАс
ОН ОН
67
-> ~"-СНО + онс~*
Формование волокна из полученного полимера, обладающего
преимущественно линейной и атактической структурой, ведут
мокрым способом из водного раствора, применяя в качестве оса-
дительной ванны раствор сульфата натрия. Свежесформованное
волокно вытягивают, сушат и подвергают термообработке. При
этом оно становится нерастворимым в воде, что, по-видимому,
обусловлено образованием большого числа водородных связей
(как это имеет место в целлюлозе). Однако такое волокно все
еще может давать усадку в горячей воде, и для окончательной
стабилизации его необходимо обработать формальдегидом
с целью образования циклических формальных групп и попе-
речных связей
В результате сшивания происходит блокирование части гид-
рофильных оксигрупп, находящихся исключительно в неупоря-
доченных участках волокна, т. е. модифицирование волокна.
Влияние-типа тактичности поливинилового спирта на число и
характер образующихся водородных связей находит проявление
в его растворимости в воде. Так, нерастяну’гый атактический по-
лимер с низким содержанием межмолекулярных водородных
связей растворим в холодной воде. Изотактический полимер,
344
Глава 9
в котором, вероятно, преобладают внутримолекулярные водород-
ные связи (68), растворим в кипящей воде, причем растворение
сопровождается разрывом относительно немногочисленных меж-
молекулярных водородных связей. Наконец, синдиотактический
поливиниловый спирт, содержащий главным образом межмоле-
кулярные, более прочные водородные связи, растворяется в воде
только под давлением при температуре 160 °C. К сожалению,
стереорегулярные полимеры винилового спирта можно получить
лишь специальными методами, например при катионной полиме-
ризации триметилсилилвинилового эфира с последующим омы-
лением образующегося синдиотактического полимера
CH2=CHOSi(CH3)3---»
OSi(CH3)3 И OSi(CH3)3
Нерегулярная структура поливинилспиртовых волокон и об-
условленная ею реакционная способность гидроксильных групп
являются причиной высокой гигроскопичности этих волокон
(около 6% при температуре 20°C и относительной влажности
воздуха 65%) и их легкой окрашиваемости красителями для
хлопка. Для придания поливинилспиртовым волокнам сродства
к кислотным красителям их обрабатывают аминобензальдеги-
дом, который дает с волокном циклические (69) и линейные
69 70
(70) ацетали. Модифицирование незамещенным бензальдегидом
повышает устойчивость волокна к горячей воде и улучшает его
эластические свойства.
Поливинилхлоридные (ПВХ) волокна
При эмульсионной полимеризации хлористого винила в при-
сутствии радикальных инициаторов легко образуется преимуще-
ственно атактический полимер, содержащий небольшое число
синдиотактических фрагментов. Термическая нестабильность
этого полимера не позволяет получить из него волокно формова-
нием из расплава. Однако сухое формование из растворов в сме-
Волокна
345
сях сероуглерода с другими растворителями (обычно ацетоном)
или в тетрагидрофуране дает удовлетворительные результаты.
До разработки этого метода ПВХ подвергали хлорированию до
образования растворимого в ацетоне полимера (содержащего
~ 65% хлора). Хлорирование сопровождается статистическим
введением атомов хлора в молекулы ПВХ, и образующийся по-
лимер 71 отличается по строению от сополимера винилхлорида
с винилиденхлоридом (72). Кроме того, низкопрочные волокна
(виньон, волокно МП) можно получить формованием сополи-
^сн2- СНС1—СНС1- СНС1— СН2 — СС12—СНС1—СНС1—CHjWv
71 („пиви анид")
сн2—СС12—СН2—СНС1—СН2—СНС'Ц- СН2—СНС1- -СН2—СС12'^
72
мера винилхлорида (86—90%) с винилацетатом (14—10%) из
раствора в ацетоне.
Существенным недостатком волокон из атактического ПВХ
является низкая температура размягчения, которая обусловли-
вает усадку волокна при нагревании, стирке или химической
чистке. Разработанный недавно метод получения более стерео-
регулярного и более высококристаллического ПВХ, имеющего
повышенную температуру размягчения, расширяет возможности
применения этого дешевого полимера. Полимеризация жидкого
мономера при низкой температуре (—30 °C) приводит к образо-
ванию продукта с более высоким содержанием синдиотактиче-
ских сегментов и более низкой степенью разветвленности. По-
скольку использование алкилпроизводных бора и металлалки-
лов в качестве катализаторов полимеризации сопряжено с рядом
трудностей, в одном из современных процессов применяется ка-
талитическая свободнорадикальная система, состоящая из гид-
роперекиси кумола, двуокиси серы и метилата натрия (в соот-
ношении 1:1,5: 1,6). Предполагаемый механизм реакции пред-
ставлен на приведенной ниже схеме:
NaOCH3 + SO2 —> NaOSOOCHs —> Na+ + CH3OSO?
(C6H5)(CH3)2COOH+CH3OSO2- —> CH3OH+(C6H5)(CH3)COOSOJ
(c6h5)(ch3)2cooso; —> (c6hs)(ch3)2co. + so3
nM
(C8H5)(CH3)2CO • + M —> (СбН5)(СН3)2СОМ • > полимер (M — мономер)
В отсутствие метилата полимер получается с низким выходом
и содержит 5О2-группы. Инициатор вышеприведенного состава,
взятый в количестве 0,1% от веса мономера, обеспечивает 20%-
ную конверсию мономера в течение нескольких часов. Процесс
можно вести непрерывно. Чистый твердый полимер выделяют
центрифугированием, а непревращенный мономер возвращают
346
Глава 9
на полимеризацию. Формование волокна (например, волокна
леавиль) осуществляют из раствора в горячем циклогексаноне
в осадительной ванне, содержащей циклогексанон, роду и эта-
нол (или ацетон). Свежесформованное волокно подвергают вы-
тягиванию и термообработке в обычных условиях. Степень крис-
талличности, по рентгеноструктурным данным, составляет около
35—40%.
ПВХ-волокна устойчивы к действию большинства кислот
(кроме концентрированной HNO3), щелочей, окислителей и вос-
становителей. При фотохимической аутокаталитической деструк-
ции и при пиролизе они выделяют НС1 и темнеют; возможно,
что это связано с образованием сопряженных полиеновых бло-
ков
В отличие от пленочного поливинилхлорида ПВХ-волокна не
содержат добавки пластификатора. Невоспламеняемость, при-
сущая ПВХ-волокнам, обусловливает их применение в производ-
стве огнестойких тканей, а способность усаживаться при нагре-
вании— в производстве тканей высокой плотности, используе-
мых в качестве каркаса меховых изделий с различной глубиной
утапливания меха, а также (в смесях с другими волокнами)
при изготовлении тканей с рельефным рисунком. Волокна типа
леавиль выдерживают нагревание до температуры 130 °C и по-
этому могут окрашиваться без переносчика дисперсными краси-
телями при температуре НО °C и повышенном давлении. Стирка
и химическая чистка этих волокон не вызывают особых затруд-
нений.
Другой подход к созданию хлорсодержащих волокон, кото-
рые обладают повышенной окрашиваемостью, гидрофильностью
и стойкостью к тепловой усадке, не набухают в растворителях,
применяемых при химической чистке, и вместе с тем сохраняют
негорючесть, заключается в мокром формовании волокна из
эмульсии ПВХ в растворе поливинилового спирта с соотноше-
нием полимеров 1:1. После стабилизации поливинилового
спирта (т. е. придания ему водоустойчивости обычными мето-
дами) получается волокно корделан, представляющее собой дис-
персию частиц ПВХ в поливиниловом спирте.
Поливинилиденхлоридные волокна
Хлорирование хлористого винила приводит к образованию
1,1,2-трихлорэтана, который легко дегидрохлорируется горячим
раствором щелочи или при нагревании в паровой фазе до тем-
Волокна
347
пературы 400 °C, давая 1,1-дихлорэтилен (винилиденхлорид)
с т. кип. 32 °C
СН2=СНС1 —> СН2С1СНС12 —> СН2=СС12
При хранении в отсутствие ингибитора мономер быстро окис-
ляется в весьма неустойчивую перекись. Путем обычной ради-
кальной полимеризации в растворе или в водной эмульсии он
превращается в линейный гомополимер, который легко кристал-
лизуется. Рентгенограмма кристаллического поливинилиденхло-
рида показывает, что пространственное взаимодействие между
соседними парами геминальных атомов хлора заставляет мо-
лекулы полимера принимать форму спирали (рис. 9.7).
Рис. 9.7. Спиралевидная структура кристаллического поливинилидеихлорида
[Cairo V. М., De Santis Р., Liquori А. М., Ripamonte A., J. Polymer Sci.,
Part В, 4, 821 (1966)].
Высокая температура размягчения (выше 180°C), прибли-
жающаяся к температуре разложения (210°C), и нераствори-
мость во всех обычных растворителях делают гомополимер не-
пригодным для формования волокна как из расплава, так и из
раствора. Поэтому промышленное волокно саран (содержащее
около 70% хлора) получают из статистического сополимера ви-
нилиденхлорида с ~15% винилхлорида, который можно пере-
рабатывать формованием из расплава. Это волокно обладает
высокой хемо- и светостойкостью и способностью к самозату-
ханию при поджигании на воздухе.
348
Глава 9
Термостойкие волокна
Возможно получение волокон, основным свойством которых
является способность длительно выдерживать умеренно высокие
температуры или короткое время — очень высокие температуры
(600°C и выше). Из таких волокон можно изготовлять, напри-
мер, спецодежду для рабочих литейных цехов и химических
предприятий, персонала, обслуживающего перевозку легковос-
пламеняющихся материалов, и экипажей самолетов. Примерами
термостойких волокон могут служить волокна номекс, дюретт
и кермелы Большинство термостойких волокон получают из по-
лимеров или сополимеров ароматических м- и п-дикарбоновых
кислот, диаминов или аминобензойных кислот, например 73—76
73 74
Такие полимеры обычно синтезируют, исходя из хлорангидри-
дов дикарбоновых кислот, поскольку на практике трудно осу-
ществить прямую конденсацию в расплаве свободных дикарбо-
новых кислот с диаминами. Широко изучались свойства различ-
ных сополимеров с целью изыскания материалов, которые были
бы достаточно хорошо растворимы в растворителях, используе-
мых для получения прядильных растворов, обладали способ-
ностью к ориентационной вытяжке на обычных крутильно-вы-
тяжных машинах и в то же время сохраняли высокую темпера-
туру плавления (выше 400°C). Гомополимеры, особенно с пара-
расположением заместителей в ароматическом ядре, во многих
случаях трудно перерабатывать в волокно.
Ароматические полиимиды, отвечающие указанным выше
требованиям, можно получить, например, из пиромеллитового
ангидрида. Формование волокна проводится, из раствора проме-
жуточной полиамидокислоты 77 с последующей термической де-
гидратацией
Волокна
349
Волокна типа кермель представляют собой полиимидные во-
локна (78), которые получают на основе тримеллитового ангид-
рида и ароматического диамина
Разработаны также полибензимидазольные волокна (79),
получаемые из ароматических тетрааминов
79
350
Глава 9
Более сложное строение имеет волокнообразующий полимер
80, который является продуктом поликонденсации нафталин-
1,4,5,8-тетракарбоновой кислоты с З^'ДД'-тетрааминобифени-
лом. Сообщается, что волокно, полученное формованием из рас-
твора этого полимера в концентрированной серной кислоте по
мокрому способу с применением в качестве осадительной ванны
разбавленной (70%-ной) серной кислоты, сохраняет 60% перво-
начальной прочности на разрыв после 1-минутного пребывания
при температуре 600 °C.
Исходя из дигидразидов дикарбоновых и дитиодикарбоновых
кислот, можно получить волокнообразующий полимер, содержа-
щий оксадиазольные или тиадиазольные кольца, например по-
лимер 81
H2NNH
NHNH
Близкие ему по химическому строению термостойкие волок-
нообразующие полимеры синтезированы на основе дихлорангид-
рида терефталевой кислоты и оксамидразона. Преполимер 82
обрабатывают хелатообразующим металлом и получают сшитое
координационными связями негорючее волокно энкатерм (83).
Волокна
351
Будучи подвергнутым карбонизации, оно сохраняет свою физи-
ческую структуру до температуры 1500°C.
H2NN=C(NH2)C(NH2)=NNH2 +п -С1С’0С6Н4С0С1 >
___» _ NHN=C(NH2)C(NH2)=NNHCOC6H4CO-
82
М2+
Другое новое термостойкое волокно, обладающее к тому же
превосходными электроизоляционными свойствами, было полу-
чено из циклогексанона. Путем контролируемой кротоновой кон-
денсации циклогексанон превращают в смесь 2,6-дициклогекси-
лиденциклогексанона и 2,6-дициклогексенилциклогексанона, ко-
торую подвергают дегидрированию в 2,6-дифенилфенол. При
окислительной конденсации этого двузамещенного фенола (ср.
с поведением 2,6-диметилфенола) образуется полифениленоксид
(84), из раствора которого можно по сухому способу сформо-
вать волокно. После вытягивания при высокой температуре во-
локно приобретает высококристаллическую структуру. Это во-
локно, известное под названием «тенакс», имеет 7Пл 480 °C и
Гст 235 °C.
Золотисто-коричневое термостойкое волокно кайнол, как по-
лагают, вырабатывается на основе фенольной (фенолформаль-
дегидной) смолы. Сшивание преполимера происходит в процессе
формования волокна. В отличие от большинства других волокон
352
Глава 9
кайнол имеет аморфную структуру. Ввиду низкой прочности его
нельзя использовать как текстильное волокно; однако из него
можно делать синтетический войлок и вату для подкладочных
материалов. При нагревании кайнол разлагается, выделяя глав-
ным образом воду и двуокись углерода и превращаясь в угле-
родное волокно, обладающее высокой термостойкостью.
Углеродные волокна
Углеродные волокна, используемые в композициях со смо-
лами для изготовления армированных пластиков, характери-
зуются высокой разрывной прочностью и жесткостью. Их по-
лучают из специальных' марок полиакрилонитрильных волокон
путем трехступенчатой термической обработки по строго опре-
деленному режиму во все более жестких условиях. На первой
стадии полиакрилонитрильное волокно нагревают на воздухе
при температуре 200—300 °C, одновременно вытягивая его для
поддержания высокой степени ориентации макромолекул. Окис-
ленное волокно подвергают карбонизации в атмосфере инертного
газа с повышением температуры до 1500 °C и в заключение про-
водят графитацию волокна при температуре до 2500—3000 °C.
Природа протекающих при этом химических реакций сложна и
пока еще плохо изучена. На первой стадии в полимер вводится
кислород и волокно становится устойчивым к термической де-
струкции. Для этого промежуточного материала было предло-
жено несколько структур, большинство которых основано на
представлении об образовании многоядерной системы лестнич-
ного типа с непрерывным увеличением числа сопряженных двой-
ных связей (и, следовательно, углублением окраски) в ходе
окисления. В состав этой системы входят звенья 85—87.
Структуры 85 и 86 непланарны; для них следует предполо-
жить нерегулярно чередующиеся конформации полукресла, об-
Волокна
353
разующиеся при сближении атактически расположенных нит-
рильных групп и последующей циклизации. Считается, что лест-
ничная структура окисленного волокна обеспечивает сохране-
ние его ориентации при последующей карбонизации. Поскольку
окисленное волокно все еще хорошо растворимо в некоторых
растворителях и практически не отличается по модулю Юнга
от исходного волокна, полагают, что на стадии окисления не
происходит значительного сшивания полимера.
' Карбонизация окисленного волокна сопровождается отщеп-
лением воды, аммиака и синильной кислоты. Полученный поли-
мер нерастворим во всех растворителях, что еще более затруд-
няет исследование его структуры. Полагают, что на этой стадии
происходит агрегация колец с образованием прочных низкомо-
дульных волокон, в которых углеродный скелет имеет почти
плоскую форму. Даже при температуре 1000 °C волокно сохра-
няет небольшое количество азота (~7°/о) и водорода; однако
при более высоких температурах они полностью отщепляются.
Графитация карбонизованного полимера вызывает его дальней-
шую перегруппировку, в результате которой получается кри-
сталлит, имеющий сетчатую структуру, подобную структуре гра-
фита. Углеродные волокна, близкие по свойствам к описанным
выше, можно также получить путем регулируемого пиролиза
целлюлозных волокон. Наиболее прочное сцепление со связую-
щим при изготовлении армированных пластиков достигается
в случае, когда поверхность углеродного волокна подвер-
гают предварительной химической активации, т. е. регулируе-
мому окислению воздухом или концентрированной азотной кис-
лотой для образования карбонильных или карбоксильных
групп.
Модифицирование волокон путем прививки
других полимеров
При обработке некоторыми окислителями или при действии
на них излучения высокой энергии (у-лучей, ускоренных элект-
ронов) образуются макрорадикалы, способные инициировать
привитую сополимеризацию винильных и акриловых мономеров.
Типичными окислителями, используемыми для этой цели, яв-
ляются реактив Фентона (смесь соли двухвалентного железа
с перекисью водорода, генерирующая активные гидроксильные
радикалы), персульфаты'и соли некоторых металлов, особенно
кобальта (III), марганца (IV) и церия (IV). Природные и гидрат-
целлюлозные волокна, а также волокна из поливинилового спир-
та легко модифицируются по этому методу в водных растворах,
12 Зак. 501
354
Глава 9
в которых они легко набухают (и, следовательно, обладают вы-
сокой реакционной способностью). Полиакрилонитрильные и
полиамидные волокна труднее окислить, так как они менее реак-
ционноспособны (это затруднение можно отчасти преодолеть,
применяя смеси растворителей). Полиэфирные и полиолефино-
вые волокна настолько устойчивы к окислению, что. практически
не поддаются прививке в присутствии химических инициаторов.
Однако они более склонны к прививке при облучении. Мономер
можно использовать в виде паров или раствора в простом или
смешанном растворителе, способном вызывать набухание во-
локна; так, в случае полиэфирного волокна берут смесь воды
с дихлоруксусной кислотой. Часть макрорадикалов, образую-
щихся при облучении, имеет большое время жизни и может про-
должать инициировать полимеризацию мономера в течение дли-
тельного срока после прекращения облучения волокна.
В ускорителях электронов, обеспечивающих очень высокую
энергию излучения (150 КэВ — 4 МэВ), полимеризация реак-
ционноспособных мономеров, таких, как аллилакрилат, проте-
кает за несколько секунд, тогда как в источниках у-лучей она
продолжается несколько часов. .
Выяснение механизма инициирования прививки к природным
волокнам представляет очень трудную задачу из-за наличия в
них большого числа потенциальных точек прививки. Например,
атака молекулы целлюлозы гидроксильными радикалами или
ион-радикалами «SOJ, образующимися из персульфата, может
протекать по любому из углеродных атомов ангидроглюкозных
звеньев или по всем этим атомам.
Скорость окисления солями церия (IV) в кислом растворе
зависит не только от концентрации церия, но также от концент-
рации кислоты и природы аниона. Наиболее энергичным окис-
лителем является перхлорат церия, который медленно окисляет
воду в перекись водорода. Пинакол превращается при взаимо-
действии с солями церия (IV) в 2 моля ацетона; однако в при-
сутствии способного к полимеризации мономера (акриламида)
выделяется только 1 моль ацетона. По аналогии можно предпо-
Волокна
355
дожить, что прививка к целлюлозе происходит по связи С2—С3
Сравни:
(СН3)2С(ОН)С(ОН)(СН3)2 —> (СН3)2СО + (СН3)2СОН;
(СН3)2СОН ----* (СН3)2С(ОН)СН2СНХ
В белковых волокнах потенциальными точками прививки яв-
ляются гидроксилсодержащие боковые группы, включающие
остатки серина, треонина и тирозина; в шерсти, кроме того, при
прививке могут затрагиваться цистиновые мостики.
Найлон-6 и найлон-6,6' медленно окисляются солями це-
рия (IV), претерпевая деструкцию и превращаясь в конечном
итоге в карбоновые кислоты и карбонильные соединения. В при-
сутствии подходящих мономеров, особенно акриловой кислоты
и акриламида, происходит привитая сополимеризация, которая
из-за низких скоростей диффузии окислителя и мономера в
глубь волокна может ограничиваться его поверхностью. Пола-
гают, что инициирование протекает преимущественно путем
атаки на метиленовые атомы углерода, связанные с амидными
группами:
SAZ'CONHCH2 ------>wCONHCH -—* WCONHCHW
I' I
сн2снх.
Прививку к полиэфирным и полипропиленовым волокнам не
удается инициировать химическим методом, что обусловлено
высокой хемостойкостью этих волокон. Радиационное иницииро-
вание сравнительно малоселективно, причем в присутствии воз-
духа небольшое число образовавшихся при радиолизе первич-
ных радикалов превращается в перекисные радикалы, которые
тоже способны инициировать привитую сополимеризацию
\ \ о2 \
—СН —> —С* -----► —С—О—О*
12»
356
Глава 9
Прививка к хлопку, гидратцеллюлозным волокнам и шерсти
таких полимеров, как полистирол, полиакрилонитрил, полиал-
килакрилаты, полиалкилметакрилаты и поливинилацетат, не
приводит к сколько-нибудь существенному улучшению их фи-
зико-механических свойств. Однако привитой сополимер при-
родного белка с полиакрилонитрилом используется в Японии
для производства волокна чинон, которое обладает шелкоподоб-
ными свойствами. Важной отличительной особенностью этого
волокна, которая, возможно, и послужила причиной его про-
мышленного выпуска, является способность к ориентационному
вытягиванию после проведения привитой сополимеризации.
При использовании в качестве мономеров акриламида, акри-
ловой и метакриловой кислот, винилпиридинов, оксиалкиловых
и диалкиламиноалкиловых эфиров акриловой кислоты полу-
чаются привитые сополимеры с реакционноспособными функ-
циональными группами. Обработка хлопка, модифицированного
прививкой полиакриламида, формальдегидом в присутствии кис-
лоты дает сшитое волокно с хорошими свойствами несминаемо-
сти. Аналогичное по структуре волокно 88 получается при дей-
ствии на хлопок N-оксиметилакриламида с последующей радиа-
ционной полимеризацией.
ЦетлОСЩКНСО
CONHCHjO Целл
88
Гидрофильные полимеры, привитые к полиамидным и поли-
эфирным волокнам, повышают их электропроводность и пони-
жают загрязняемость. Полимеры с гидроксильными группами
в боковой цепи улучшают окрашиваемость активными краси-
телями, а полимеры, обладающие основными или кислотными
свойствами, — окрашиваемость кислотными и основными краси-
телями соответственно. Полиамидное волокно, модифицирован-
ное путем прививки полиакриловой кислоты в форме кальцие-
вой соли, имеет повышенную устойчивость к прожиганию сига-
ретами и тлеющим пеплом.
Установление факта прохождения привитой сополимериза-
ции, а не образования механической смеси с гомополимером, яв-
ляется трудной задачей. Для ее решения применяются следую-
щие методы:
1. Дробное осаждение. Привитой сополимер найлона с акри-
ловой кислотой частично растворяется в 90%-ной муравьиной
кислоте и при ступенчатом высаживании водой дает осадки,
содержащие оба полимера. При этом удается выделить лишь
Волокна
357
незначительное количество свободной полиакриловой кислоты,
которая растворима в воде.
2. Селективная экстракция. Ацетилирование привитого со-
полимера вискозного волокна с винилпиридином сопровождается
образованием продукта, растворимого в хлористом метилене.
При экстракции этого раствора разбавленной кислотой полу-
чены лишь следы поливинилпиридина.
3. Прямая экстракция. Привитой сополимер целлюлозы с ак-
риловой кислотой обрабатывают фенилизоцианатом для количе-
ственного получения N-фенилуретановых производных по месту
свободных гидроксильных групп. При последующей экстракции
растворителем, обычно хорошо растворяющим трыс-М-фенилуре-
тан целлюлозы, удается выделить лишь небольшое количество
этого соединения.
4. Определение концевых групп. В результате отщепления
исходного волокна (например, путем гидролиза) получается
привитой виниловый полимер, содержащий определимые кон-
цевые группы
Moncrieff Р. W., Man-made Fibres, 5th ed., Heywood, London, 1970 (имеет-
ся русский перевод 3-го изд.: Монкрифф Р. У., Химические волокна, «Легкая
индустрия», М., 1964).
Mark Н. F., Atlas S. М., Cernia Е. (Eds.), Man-made Fibres: Science and
Technology, Vols. I—III, Wiley — Interscience, New York, 1968.
Cockett S. M., An Introduction to Man-made Fibres, Pitman, London, 1966.
Cook J. G., Handbook of Textile Fibres, 4th ed., Marrow, Watford, 1968.
Goodman Synthetic Fibre-forming Polymers, Roy. Inst. Chem. Lecture
Series, 1967, № 3.
Фурне Ф„ Синтетические волокна. Получение и переработка. «Химия»,
М., 1970.
Hicks Е. М., Jr., et al., «The Production of Synthetic Polymer Fibres», Tex-
tile Progress, 3, № 1 (1971).
Koch P. A., «Faserstoff-Tabellen», Textil-Industrie, 72, № 1, 21 (1970) и
ссылки, приведенные в этой работе.
Хопфф Г., Мюллер А., Венгер Ф., Полиамиды, Госхимиздат, М., 1958.
Gooaman I., Phys J. A., Polyesters, Vol. 1. Saturated Polyesters, Iliffe,
London, 1965.
McIntyre J. E., The Chemistry of Fibres, Arnold, London, 1971.
Carter M. E., Essential Fiber Chemistry, Marcel Dekker, New York, 1971.
Гетце К., Производство вискозных волокон, «Химия», М., 1972.
358
Глава 9
Ludewig Н., Polyester Fibres: Chemistry and Technology (trans. Buck B.),
Interscience — Wiley, London, 1971.
Структура волокон, под ред. Херла Д. В. С. и Петерса Р. X., «Химия»,
М., 1969.
Hearle J. W. S. Greer R., «Fibre Structure», Textile Progress, 2, № 4
(1970).
Bailey J. E., Clarke A. J., «Carbon Fibres», Chem. in Britain, 6, 484 (1970).
Meredith R., Elastomeric Fibres, Merrow, Watford, 1971.
Buist J. M., Gudgeon H. (Eds.), Advances in Polyurethane Technology,
McLaren, London, 1968.
Райт П., Камминг А., Полиуретановые эластомеры, «Химия», Л., 1973.
Ward К., Chemistry and Chemical Technology of Cotton, Interscience, New
York, 1955.
Bikales N. M., Gaylord N. G., Mark H. F. (Eds.), Encyclopedia of Polymer
Science and Technology, Interscience, New York, 1964—1972.
Alexander P., Hudson R. F., Wool. Its Chemistry and Physics, 2nd ed.,
Chapman and Hall, London, 1963 (имеется русский перевод 1-го изд.: Алек-
сандер П., Хадсон Р. Ф., Физика и химия шерсти, Гизлегпром, М., 1958).
Honeyman J. (Ed.), Recent Advances in the Chemistry of Starch and Cel-
lulose, Heywood, London, 1959.
Crewther W. G. et al., «The Chemistry of Keratin», Adv. Protein Chem. 20,
191 (1965).
Lucas F., Rudall К- M., «Extracellular Fibrous Proteins: the Silks» in Com-
prehensive Biochemistry, Vol. 26B, p. 475, Elsevier, Amsterdam, 1968.
Lucas F., Shaw J. T. B., Smith S. G., «The Silk Fibroins», Adv. Protein
Chem., 13, 108 (1958).
Traub W., Piez-K- A., «Chemistry and Structure of Collagen», Adv. Pro-
tein Chem., 25, 243 (1970).
Ramanathan N., Collagen, Wiley, New York, 1962.
Bailey A. J., The Nature of Collagen, in «Comprehensive Biochemistry»,
Vol. 26B, p. 297, Elsevier, Amsterdam, 1968.
Sorenson W. R., Campbell T. W., Preparative Methods of Polymer Che-
mistry, 2nd ed., Interscience, New York, 1968 (имеется русский перевод изд.
1961 г.: Сёренсен У., Кемпбелл Т., Препаративные методы химии полимеров,
ИЛ, М., 1963).
Roff IP. J., Scott J. R., Fibres, Films, Plastics and Rubbers, Butterworths,
London, 1971.
ГЛАВА 10
Химия красителей
К. Стид
Stead С. V., Organics Division,
Imperial Chemical Industries Limited, Blackley
С незапамятных времен человеку хотелось окрашивать тка-
ни, которыми он пользовался, в различные цвета. В поисках
подходящих красителей он прежде всего обратился к природ-
ным веществам. Некоторые из окрашенных природных соеди-
нений оказались ценными красителями, и для крашения ими
были разработаны два технологических способа. При протрав-
ном крашении исходную ткань сначала пропитывали водным
раствором соли металла (алюминия, железа, хрома или олова),
в результате чего на волокне осаждалась нерастворимая гидро-
окись металла, а затем обрабатывали раствором природного
красящего вещества, способного к образованию нерастворимого
хелатного соединения с протравой. Природные протравные кра-
сители получались из различных источников и значительно
отличались друг от друга по химическому строению; однако все
они сходны в том отношении, что содержат хелатообразующую
систему. .Наиболее важным протравным красителем природ-
ного происхождения был ализарин (1), который добывался из
корней красильной марены и окрашивал ткани в красный цвет.
Карминовая кислота (2) — красящее начало кошенили, добы-
вавшейся из самок червецов вида Coccus cacti,—давала в зави-
симости от взятой протравы окраски от красной до фиолетовой.
В настоящее время единственным природным красителем, еще
применяемым в некоторых количествах, является черный сан-
дал, получаемый из сердцевины дерева Нaetnatoxylon саопре-
chiancum. Он содержит гематоксилеин (3), который образуется
в результате окисления присутствующего в древесине гемато-
ксилина (4), и окрашивает ткани в черный цвет.
360
Глава 10
3
4
При крашении натуральным индиго (5) — продуктом окисле-
ния индоксила (т. 3, стр. 104), содержащегося в индигоносных
растениях в виде глюкозида индикана (т. 4, стр. 116),— приме-
нялся другой метод. Процесс крашения индиго был основан на
том, что в щелочной среде этот краситель восстанавливается в
водорастворимую лейкоформу 6. Окрашиваемую одежду зама-
чивали в растворе лейкоиндиго и оставляли на воздухе; при
этом лейкоиндиго снова окислялся в индиго, который нераство-
рим в воде и имеет темно-синий цвет. Индиго до сих пор нахо-
дит широкое применение, однако природный продукт почти
полностью вытеснен синтетическим. В древности высоко ценился
финикийский пурпур (6,6'-диброминдиго), который добывали из
удитки Murex brandaris. Это вещество представляет собой ред-
кий пример природного бромсодержащего соединения.
о
5
Недостатками перечисленных природных красителей яв-
ляются трудность их получения, которое зачастую бывает очень
утомительной процедурой, а также длительность процесса кра-
шения и непостоянство конечного результата. Поэтому они не
выдержали конкуренции с появившимися вслед за мовеином
(Перкин, 1856 г.) многочисленными синтетическими красите-
Химия красителей
361
лями, которые выгодно отличались более низкой стоимостью,
широкой цветовой гаммой, простотой технологии крашения, ста-
бильностью получаемых результатов, а также более высокой
прочностью (т. е. устойчивостью окрашенного материала к воз-
действию света и мокрым обработкам).
Первый синтетический краситель — мовеин — был получен
окислением анилина-сырца бихроматом калия. Выясненная при
последующем исследовании структура мовеина (7) характери-
зуется наличием метильных групп, что обусловлено значитель-
ной примесью толуидинов в выпускавшемся в то время анилине.
Мовеин окрашивал шелк в пурпурный цвет, значительно пре-
восходящий по яркости окраски, получаемые с помощью при-
родных красителей, и его коммерческий успех положил начало
промышленному производству синтетических красителей. Это
соединение относится к группе азиновых катионных красите-
лей, достоинством которых является способность окрашивать
ткани в яркие цвета; однако в настоящее время они почти не
представляют практического интереса из-за сравнительно низ-
кой светопрочности.
Окрашенные химические продукты, вырабатываемые анили-
нокрасочной промышленностью, используются не только как
красители, но и как пигменты. Различие между ними заклю-
чается в том, что краситель наносят на текстильное изделие из
водного раствора или водной суспензии, тогда как пигмент вво-
дят в краску или пластмассу в чистом виде. Кроме производ-
ства этих материалов, окрашенные соединения применяются
в ряде менее важных областей, таких, как цветная фотография
и гистологические исследования. Однако в данной главе рас-
сматриваются только органические соединения, используемые
в качестве красителей и пигментов, причем главное внимание
уделяется нескольким основным классам практически важных
окрашенных веществ.
362
Глава 10
Красители
Азокрасители
Получение, строение и окраска
Азокрасители характеризуются присутствием в их молекуле
азогруппы —N = N—. В некоторых случаях, однако, более
устойчивой может оказаться таутомерная гидразонная форма
—NH—N=. Наиболее важным методом получения азокраси-
телей является диазотирование ароматического или гетероцик-
лического первичного амина с последующим сочетанием обра-
зующейся соли диазония (8) с подходящей азосоставляющей
(т. 2, стр. 179—180). В качестве азосоставляющих чаще всего
НС1-NaNO2 +
ArNH2 ----------> ArN=N СГ
8
10
используют способные к енолизации кетоны (такие, как арил-
амиды ацетоуксусной кислоты и 1-арил-З-замещенные пиразо-
лоны-5), фенолы и нафтолы, амины бензольного ряда, нафтил-
амины и их N-алкил- и N-арилпроизводные. Сочетание с гидро-
ксилсодержащими азосоставляющими проводится в щелочной
среде, поскольку оно протекает через промежуточную иониза-
цию этих соединений, тогда как сочетание с ариламинами
можно осуществлять в слабокислой среде. Хотя продукты соче-
тания часто изображают структурными формулами азосоеди-
Химия красителей
363
нений, их действительное строение зависит от того, какая из
возможных таутомерных форм энергетически более выгодна.
Так, при использовании в качестве азосоставляющих арилами-
дов ацетоуксусной кислоты и пиразолонов-5 преобладающей яв-
ляется гидразонная форма (например, 9), а при использовании
простых фенолов — азоформа 10. Нафтолы обычно дают смеси
таутомерных форм.
Легкость, с которой можно вносить изменения в структуру
азокрасителей, обусловила их огромное промышленное зна-
чение. Для получения разнообразных азокрасителей необходи-
мо иметь широкий набор промежуточных веществ. Последние
синтезируют из сравнительно небольшого числа первичных
полупродуктов (таких, как бензол, метилбензол, фенол, наф-
талин), которые в значительных количествах вырабатыва-
лись ранее путем перегонки каменноугольной смолы. Дру-
гим источником получения этих соединений (значение кото-
рого все более возрастает) является нефть; в настоящее время
в США из нефти производится основная часть первичных полу-
продуктов тяжелого органического синтеза (гл. 2). Таким обра-
зом, необходимые промежуточные вещества получают из 10—
20 важнейших первичных полупродуктов, используя для этого
обычные химические реакции: нитрование, галогенирование,
сульфирование, восстановление, N-алкилирование, N-ацилиро-
вание, сплавление со щелочью и др. В ряду нафталина широко
используют также реакцию Бухерера (т. 2, стр. 264).
Ароматические амины бензольного ряда чаще всего получают
нитрованием бензола и его гомологов с последующим восста-
новлением нитропроизводных. Восстановителем обычно служит
чугунная стружка в ^присутствии каталитического количества
соляной кислоты; однако в последнее время все более широкое
применение находит каталитическое гидрирование. Ариламиды
ацетоуксусной кислоты синтезируют по реакции ариламинов с
дикетеном, а пиразолоны-5 — путем циклизации продуктов кон-
денсации аминов с ацетоуксусным эфиром.
В ряду нафталина направление вступления каждого после-
дующего заместителя определяется суммарным влиянием пре-
дыдущих, вследствие чего большое значение для получения со-
единений заданной химической структуры имеет порядок от-
дельных стадий синтеза. Наиболее легко протекает замещение
в a-положение, и -поэтому нитрование и низкотемпературное
сульфирование дают а-изомеры. Если же сульфирование (кото-
рое является обратимой реакцией) проводить при повышенной
температуре, то первоначально образовавшаяся а-нафталин-
сульфокислота превращается в термодинамически более устой-
чивый p-изомер. Методы синтеза важнейших промежуточных ве-
ществ для производства краситёлей, включающие использование
364
Глава 10
весьма небольшого
мами 10.1 и 10.2.
набора реакций, иллюстрируются схе-
Схема 10.1. Синтез промежуточных веществ нафталинового
ряда через а-монозамещенные производные нафталина.
Условные обозначения'. 1 — нитрование смесью HNO3 и H2SO4; 2 — вос-
становление Fe и НС1; 3 — сульфирование H2SO4 или олеумом; 4 — реакция
Бухерера (действие NaHSOs и затем NaOH); 5 — нагревание с анилином.
Химия красителей
365
Схема 10.2. Синтез 8-амино-1-нафтол-3,6-дисульфокислоты *
(условные обозначения те же, что на схеме 10.1; 6 — сплавле-
ние со щелочью).
Моноазокрасители, получаемые путем однократного диазо-
тирования и сочетания, принято обозначать как азокрасители
типа А->Е, где буква А относится к первичному амину, буква
Е — к азосоставляющей, а стрелка означает «диазотированный
и подвергнутый сочетанию с». Главным фактором, обусловлива-
ющим цвет красителя, является длина сопряженной цепи, ко-
торая в свою очередь существенно зависит от природы исполь-
зуемой азосоставляющей. Так, ариламиды ацетоуксусной кис-
лоты и пиразолоны-5 дают с простыми диазосоставляющими А
бензольного ряда зеленовато-желтые красители, фенолы и ани-
лины— красновато-желтые, нафтолы — оранжевые, а некото-
рые аминонафтолы — красные. Разумеется, большое значение
имеет и природа диазосоставляющей. Нафтиламины обеспечи-
вают более глубокие тона, чем анилины, причем, как будет по-
казано ниже, значительное влияние на цвет красителя оказы-
вают заместители в молекуле диазосоставляющей.
При многократном чередовании диазотирования и сочетания
можно получить дис- и полиазокрасители, содержащие в моле-
куле две или несколько азогрупп. Существует три метода
синтеза диазокрасителей. По первому из них ароматический ди-
амин, такой, как бензидин или 4,4'-диаминодифениламин-2-суль-
фокислота (диазосоставляющая D), подвергают тетраазотиро-
ванию, т. е. диазотированию по обеим аминогруппам, и сочета-
нию с двумя различными азосоставляющими. В результате
образуется дисазокраситель типа Ej-<-D->E2, например соеди-
нение 11, причем используется различие в реакционной способ-
ности обеих диазогрупп исходной азосоставляющей. Аналогич-
ные по строению дисазокрасители можно получить путем диазо-
тирования и сочетания нитро- и ациламинозамещенных
* Так называемая Аш-кислота. — Прим, перев.
366
Глава 10
ариламинов, после чего восстанавливают нитрогруппу или уда-
ляют ацильную защитную группу и повторно проводят диазоти-
рование и сочетание. Подобным же образом синтезируют дис-
азокрасители типа Ai->Z-<-A2 (например, краситель 12), ис-
пользуя для этого азосоставляющие Z, способные к двукратному
сочетанию, из которых наиболее важным соединением является
8-амино-1-нафтол-3,6-дисульфокислота. Третий метод заклю-
чается в сочетании диазотированного ароматического амина
с азосоставляющей, содержащей аминогруппу, чаще всего с
а-нафтиламином и его 6- или 7-сульфокислотами (такие со-
ставляющие обозначаются здесь буквой М). Диазотирование
полученного аминоазосоединения и сочетание продукта с азо-
составляющей Е приводят к образованию дисазокрасителя типа
А->М->Е (13). Глубокий цвет красителей этого типа свиде-
тельствует о наличии в их молекуле обширной сопряженной си-
стемы. С помощью описанных методов можно синтезировать
трис-, тетракис- и даже полиазокрасители (содержащие соот-
ветственно три, четыре или несколько азогрупп), которые имеют
Чрезвычайно важной характеристикой красителя является
светопрочность, т. е. устойчивость к выцветанию под действием
света. При фотохимической деструкции азосоединений разры-
вается связь N=N, причем скорость этого процесса зависит от
их структуры. Обычно заместители, оттягивающие электроны из
сопряженной системы и тем самым понижающие доступность
Химия красителей
367
электронов связи N = N, повышают светопрочность азокрасите-
лей. Такое же влияние оказывают объемистые заместители, на-
ходящиеся в орто-положении к азогруппе, которые затрудняют
подход к связи N=N. Существует еще один эффективный спо-
соб предотвращения фотохимического расщепления азогруп-
пы— связывание о,о'-дизамещенных азосоединений, способных
вести себя как тридентатные лиганды, в прочные комплексы с
металлами (в частности, с медью, хромом и кобальтом). Такие
комплексы легко дают, например, о,о'-диокси- и о-карбокси-
о'-оксиазосоединения, которые синтезируют, , применяя в каче-
стве диазосоставляющей 2-аминофенол или антраниловую кисло-
ту. Реже используются для получения комплексов аналогичные
азосоединения, в которых одна или несколько гидроксиль-
ных групп замещены аминогруппами. При обработке о.о'-диок-
сипроизводного ацетатом меди образуется прямоугольный
копланарный комплекс состава 1:1; природу связей атома меди
в этом комплексе можно изобразить формулой 14. Соли хрома
дают в нейтральной среде отрицательно заряженный октаэдри-
ческий комплекс состава 1:2 (15), а в кислой среде — положи-
тельно заряженный комплекс состава 1 : 1 (16). Последний при
взаимодействии с другим хромирующимся азокрасителем пре-
14
вращается в несимметричный смешанный комплекс состава 1 :2.
В случае солей кобальта отщепление координационно связан-
ной воды с образованием симметричного комплекса состава 1 : 2
происходит даже в кислой среде, что объясняется относительно
более высоким сродством кобальта к азотистым лигандам. Од-
нако предварительное связывание соли кобальта в комплекс,
например, с аммиаком позволяет проводить реакцию ступен-
чато. Цветовая шкала комплексных азокрасителей охватывает
всю видимую область спектра. Как правило, они имеют более
тусклые тона, чем свободные азокрасители; однако превосход-
ная светопрочность обусловливает их важное практическое
значение.
368
Глава 10
Наличие широкого ассортимента полупродуктов и хорошо
разработанных синтетических методов, обеспечивающих высо-
кие выходы, позволяет получать азокрасители самого разнооб-
разного строения и цвета, которые могут быть использованы для
крашения всех типов тканей, а также в качестве пигментов.
В настоящее время азокрасители являются наиболее важным
классом красящих веществ — на их долю приходится свыше
50% общего объема выпускаемых красителей. На примере азо-
красителей удобно рассмотреть влияние структуры красителя
на возможность его применения для окрашивания того или
иного типа волокна. Использование азосоединений в качестве
пигментов рассматривается в конце данной главы.
П рименение
Крашение волокна, как это само собой разумеется, не дол-
жно сопровождаться существенным изменением его качеств в
худшую сторону. Полученная окраска должна сохраняться в те-
чение всего срока службы волокна, откуда вытекает требование
достаточной стирко- и светопрочности красителя. Кроме того,
необходимо, чтобы краситель на волокне был устойчив и к неко-
торым другим воздействиям; однако большинство этих требова-
ний имеет специальный характер и меняется в зависимости от
типа волокна.
Для достижения высокой прочности к стирке прежде всего
необходимо знать механизм закрепления красителя на волокне.
На практике используется пять различных способов: образова-
ние твердого раствора, солеобразование, завязывание водород-
ных связей, введение в волокно нерастворимых окрашенных
соединений и образование ковалентных связей с волокном. Кон-
кретный механизм целиком определяется химическими и физи-
ческими свойствами волокна, в частности характером присут-
ствующих в нем функциональных групп и его гидрофильностью
или гидрофобностью.
Некоторые синтетические волокна, такие, как полиамидные
и полиэфирные, являются по своей природе термопластичными
материалами. При нагревании они размягчаются и затем пла-
вятся. Такие волокна легко окрашиваются дисперсными краси-
телями. Процесс крашения заключается в нагревании волокна
в водной дисперсии водонерастворимого красителя, что приво-
дит к переносу красителя в волокно с образованием твердого
раствора. В том случае, когда краситель имеет молекулы не-
больших размеров, его диффузия в массу волокна облегчается.
Идеальными с этой точки зрения являются простые водонерас-
творимые моноазокрасители. В качестве азосоставляющих, по-
мимо фенолов, используемых обычно в синтезе желтых краси-
Химия красителей
369
телей (таких, как краситель 17), чаще всего применяются
Ы,М-диалкиланилины, которые позволяют легко получить низ-
комолекулярные моноазосоединения самого различного цвета.
Типичные дисперсные красители для сравнительно гидрофиль-
ных полиамидных волокон — соединения 18 и 19. Алкильные
18 (оранжевый)
С2Н6
СН2СН2ОН
СН2СН2ОН
СН2СН2ОН
19 (красный)
группы, стоящие у атома азота азосоставляющей, обычно имеют
сложное строение. Особенно хорошо зарекомендовали себя в
качестве азосоставляющих р-оксиэтильные производные, кото-
рые можно получить, например, взаимодействием N-этилани-
лина с окисью этилена; продукт этой реакции — М-этил-Н-(|3-ок-
сиэтил) анилин — используется в синтезе соединения 18. Такие
красители обнаруживают батохромный сдвиг, т. е. смещение
максимума поглощения света в более длинноволновую область
(углубление цвета) с увеличением числа электроноакцепторных
заместителей в ядре азосоставляющей. Они дают очень ровные
окраски на полиамидных волокнах, но часто обладают недоста-
точной прочностью к мокрым обработкам. Поэтому в некоторых
случаях для крашения полиамидных волокон применяются кис-
лотные красители (см. ниже), которые хотя и уступают азокра-
сителям по эгализационной способности (т. е. способности да-
вать ровную окраску), но зато превосходят их по прочностным
показателям.
Полиэфирные и триацетатные волокна гораздо более гидро-
фобии, чем полиамидные, что существенно затрудняет диффу-
зию красителя в массу волокна. В данном случае размеры мо-
лекул красителя имеют еще более важное значение, а един-
ственным подходящим классом красителей являются дисперс-
ные красители, диффузию которых из кипящей ванны в волокно
370
Глава 10
можно облегчить добавлением переносчика, например бензило-
вого спирта. Для того чтобы избежать применения переносчика
(поскольку его следы могут оказать неблагоприятное влияние
на.светопрочность окраски), процесс крашения иногда проводят
под давлением при повышенной температуре (130°C). Желтые,
оранжевые и красные окраски легко получаются при использо-
вании азокрасителей типа монозамещенный анилин-> третичный
амин, однако для получения синих тонов следует исходить из
диазосоставляющих, содержащих в ядре несколько электроно-
акцепторных групп. При этом остается в силе требование, чтобы
краситель имел небольшой размер молекулы. Из-за трудности
синтеза и дальнейшей переработки таких диазосоставляющих
(например, диазосоставляющей красителя 20) важное практи-
NHCOCH3
/С4Н9
—N\
^СН2СН2ОСОСНз
'2 ^2‘i5'~'
(SCN);
20 (зеленовато-синий)
22
ческое значение приобрели их заменители — слабоосновные ге-
тероциклические амины, такие, как 2-аминобензотиазол (21),
2-амино-5-нитротиазол (22) и 3-аминобензизотиазол (23). Эти
соединения зачастую более доступны, чем полизамещенные ани-
лина, имеют меньший молекулярный вес и столь же эффективно
обеспечивают желаемый батохромный сдвиг при использовании
их в качестве диазосоставляющей А. Алкильные группировки
при третичном атоме азота в азосоставляющей обычно имеют
сложное строение и содержат такие фрагменты, как ацетокси-
Химия красителей
371
группы, которые сообщают молекуле в целом гидрофильные
свойства. Это делается с целью придать красителю устойчи-
вость к сублимации, необходимость которой вызывается тем, что
окрашенные изделия из полиэфирного волокна часто подвер-
гают глажению в увлажненном состоянии.
Анионные водорастворимые моно-и дисазокрасители, полу-
ченные на основе сульфированных полупродуктов, находят при-
менение в качестве кислотных красителей для шерсти и поли-
амидных волокон. Оба эти вида волокон содержат аминогруппы,
и механизм их крашения включает образование соли между
аминогруппами волокна и сульфогруппами красителя. Типич-
ными представителями анионных азокрасителей являются кра-
ситель для шерсти 24 и краситель для найлона 25. В последнем
соединении, полученном путем сочетания диазотированного
2-трифторметил-4-хлоранилина с 2-амино-8-нафтол-6-сульфокис-
лотой в кислой среде, оба атома азота азогруппы участвуют в
образовании водородных связей (один с аминогруппой, а вто-
рой с гидроксильной группой), что обеспечивает превосходную
24 (зеленовато-желтый)
25 (красный)
светопрочность. Своеобразная диазосоставляющая этого краси-
теля была приготовлена хлорированием .и-хлортолуола в л/-хлор-
бензотрихлорид, который обрабатывали фтористым водородом
с последующим нитрованием образующегося л-хлорбензотри-
фторида и восстановлением нитропроизводного. Дисазокраси-
тели дают более глубокие цвета, такие, как синий и темно-си-
ний. На найлоне они обладают высокой прочностью к стирке, но
окраска может получиться неравномерной из-за неоднородно-
сти физической структуры волокна вдоль его оси, обусловли-
вающей различную степень диффузии красителя в отдельные
участки. В случае шерсти простые красители этого типа прихо-
дится наносить на волокно из сильнокислой ванны, причем
прочность окрасок к мокрым обработкам неудовлетворительна.
Указанные недостатки можно устранить введением в молекулу
372
Глава 10
красителя алкильной группы с длинной цепью, как в соедине-
нии 26. Увеличение размеров молекулы красителя наряду с ги-
дрофобным характером длинноцепочечной алкильной группы
позволяет достичь удовлетворительного качества крашения из
ОН ЫНСОСНз
26 (красный)
почти нейтральной ванны и затрудняет последующую десорб-
цию красителя при мокрых обработках, что приводит к повы-
шению прочности к стирке.
Все эти красители окрашивают шерсть в разнообразные яр-
кие цвета. Однако при использовании шерсти для изготовления
верхней одежды необходимы красители, дающие глубокие, не
обязательно яркие окраски с очень высокой светопрочностью.
Этим требованиям в полной мере отвечают хромовые и кобаль-
товые комплексы азокрасителей. Первоначально их получали
in situ путем крашения шерсти металлизирующимся красителем
с последующей обработкой ткани подходящей солью металла,
например бихроматом. Описанный процесс сопровождался по-
вреждением волокна, и поэтому большим шагом вперед явилось
изобретение предварительно металлизированных красителей,
окрашивающих шерсть из нейтральной ванны. Примером таких
красителей может служить соединение 27, получаемое при взаи-
модействии азокрасителя 2-амино-4-метилсульфофенол —* p-наф-
тол с ацетатом хрома. Первые красители этого типа содержали
атом хрома, несущий единичный отрицательный заряд. Умерен-
Химия красителей
373
ная величина заряда обусловливала удовлетворительное кра-
шение из нейтральной ванны, а большие размеры молекулы —
превосходную прочность к мокрым обработкам. Для обеспече-
ния достаточной водорастворимости красителя необходимо при-
сутствие в нем сульфоновой или сульфамидной группы. В по-
следние годы значительный интерес привлекают смешанные
комплексы азокрасителей, такие, как соединение 28. Процесс
получения этого комплекса включает две стадии: синтез ком-
плекса азокрасителя 6-нитро-2-амино-1-нафтол-4-сульфокисло-
та-> |3-нафтол с хромом состава 1 : 1 и последующая обработка
его азокрасителем 2-амино-5-нитрофенол -> р-нафтол, в резуль-
тате чего образуется изображенное выше несимметричное со-
единение. Источником второго отрицательного заряда здесь
служит свободная сульфогруппа. Двойной отрицательный заряд
не является слишком большим для молекул такой величины, и
краситель 28 тоже обладает удовлетворительной выбирае-
мостью из нейтральной ванны и достаточной прочностью к мо-
крым обработкам.
К другому классу химических соединений принадлежат ос-
новные красители — катионные соединения, растворимые в воде
за счет присутствующего в их молекуле четвертичного атома
азота. Основные красители применяются для крашения поли-
акрилонитрильных волокон. Они образуют соли с карбоксиль-
ными и сульфогруппами, введенными в волокно с соответствую-
щими сомономерами на стадии полимеризации. Существует два
практически важных типа основных красителей, в которых чет-
вертичный атом азота либо находится в заместителе, как в кра-
сителе 29, либо входит в состав гетероцикла, составляющего
часть хромофорной группы, как в красителе 30. Оба типа кра-
сителей обычно синтезируют путем превращения соответствую-
щих окрашенных аминов, содержащих третичный атом азота,
в четвертичные аммониевые соединения. Солеобразование между
красителем и волокном обеспечивает удовлетворительную проч-
ность выкрасок. Однако слишком большая скорость адсорб-
ции красителя приводит к неравномерности окраски, и для
29 (красный)
30 (синий)
374
Глава 10
устранения этого недостатка в красильную ванну добавляют так
называемые замедлители.
Два механизма закрепления красителя на волокне, обсуж-
давшиеся выше, не подходят в случае очень гидрофильных цел-
люлозных волокон (хлопкового и вискозного), поскольку эти
волокна не обладают термопластичными свойствами и не содер-
жат кислотных или основных центров. Тем не менее некоторые
водорастворимые красители, например дисазокраситель конго
красный (31), способны непосредственно окрашивать хлопок из
водной ванны. Такие красители называются прямыми или суб-
стантивными. Большинство их относится к классу азосоедине-
ний. Субстантивность прямых красителей по отношению к
хлопку обусловлена способностью их молекул образовывать
водородные связи с волокном или друг с другом; в последнем
случае в порах волокна образуются агрегаты молекул краси-
теля, не вымывающиеся при последующих мокрых обработках.
Прямые красители должны иметь длинные плоские молекулы
и обладать минимальной растворимостью в воде, достаточной
лишь для того, чтобы можно было красить волокно из водной
ванны. Некоторые группы, такие, как остатки бензидина и мо-
чевины, а также сама азогруппа, повышают субстантивные
свойства красителя, причем для получения практически ценного
прямого красителя необходимо одновременное присутствие в мо-
лекуле нескольких таких групп. При синтезе моноазокрасителей
их следует вводить в боковую цепь, как*это сделано в соедине-
нии 32. Другой хороший метод — фосгенирование аминомоно-
азосоединения с получением бисазопроизводного мочевины. В от-
личие от рассмотренных выше технических классов красителей
важное место среди прямых красителей занимают полиазосоеди-
нения. Классическими прямыми красителями являются дважды
омедненное бисазосоединение 33 и трисазосоединение 34. Пер-
вое из них получают нагреванием дисазокрасителя 3,3'-димето-
кси-4,4'-диаминобифенил 8-амино-1-нафтол-2,4-дисульфокис-
лота с аммиачным раствором сульфата меди, причем образова-
ние медного комплекса сопровождается деметилированием
метоксигрупп. Синтез трисазокрасителя 34 представляет собой
прекрасный пример селективного сочетания. Бензидин подвер-
Химия красителей
375
гают тетраазотированию и образовавшееся соединение сочетают
в кислой среде за счет высокореакционноспособной первой ди-
азогруппы с Аш-кислотой в орто-положение к аминогруппе. За-
тем в реакционную смесь добавляют диазотированный анилин
и в щелочной среде осуществляют сочетание в орто-положение
к гидроксильной группе; при этом малореакционноспособная
вторая диазогруппа бифенильной компоненты практически не
вступает в реакцию. Наконец, добавляют м-фенилендиамин, ко-
торый медленно реагирует с образованием третьей азогруппы.
Технология крашения прямыми красителями относительно про-
ста; однако слабость и обратимый характер связей между кра-
сителем и волокном не позволяют получить окраски с высокой
устойчивостью к мокрым обработкам. Для придания прямым
красителям нерастворимости в воде и улучшения тем самым
прочности окрасок к стирке используются дополнительные опе-
рации упрочнения красителя на волокне, например диазотиро-
вание аминогрупп и последующее сочетание продукта с водо-
нерастворимой азосоставляющей или образование комплекса с
медью. Однако эти мероприятия лишь частично решают про-
блему прочности к стирке, вследствие чего прямые красители
в настоящее время применяются главным образом в тех случаях,
когда данное свойство не имеет большого значения (например,
для крашения подкладочных тканей). Главной областью при-
менения прямых красителей является окраска бумаги.
376
Г лава 10
Альтернативный подход заключается в синтезе нераствори-
мого красителя непосредственно на волокне. Этот способ полу-
чения окрасок, прочных к стирке, лежит в основе так называе-
мого ледяного крашения, базирующегося на значительной суб-
стантивности к хлопку ариламидов 2-окси-З-нафтойной кислоты.
Родоначальником последних служит нафтол АС (анилид 2-окси-
З-нафтойной кислоты) (35). Соединения этого типа получают
ОН
CONH-
35
конденсацией ароматических аминов с 2-окси-З-нафтойной кис-
лотой в присутствии треххлористого фосфора. Исходную 2-окси-
3-нафтойную кислоту синтезируют путем нагревания 2-нафто-
лята натрия в атмосфере двуокиси углерода при повышенном
давлении. При обработке ткани щелочным раствором арил-
амида 2-окси-З-нафтойной кислоты он легко сорбируется за счет
своей высокой субстантивности и удерживается на ткани до ее
пропускания через раствор диазониевой соли, приготовленной
из водонерастворимого амина. В результате реакции сочетания
на волокне образуется нерастворимое азосоединение. После-
дующая обработка окрашенной ткани горячим мыльным раство-
ром приводит к агрегации частиц красителя в порах волокна.
Нафтол АС и его аналоги дают с хлор-, нитро-, метил- и мето-
ксианилинами, которые наиболее часто используются в качестве
диазосоставляющей, преимущественно оранжевые и красные
цвета, а с дианизидином, 4-аминодифениламином и 4-(бензоил-
амино)-2,5-диметоксианилином— синие цвета. В случае приме-
нения нафтола АТ (36) удается получить желтые окраски, то-
Н3Сх /СН3
CH3COCH2CONH——NHCOCH2COCH3
36
гда как гетероциклические аналоги нафтола АС, например арил-
амиды 2-оксикарбазол-З-карбоновой кислоты, образуют окраски
коричневого цвета. Использование стойких диазосоставляющих,
выпускаемых промышленностью в форме нафталин-1,5-дисуль-
фонатов и тетрахлорцинкатов, освобождает красильные фаб-
рики от необходимости диазотировать ароматический амин. Ле-
дяные красители существенно превосходят прямые красители по
прочности окраски, но процесс крашения ими технически более
сложен. Гораздо лучшее решение проблем, возникающих при
крашении хлопка, удалось найти с помощью активных краси-
Химия красителей
377
телей. Однако, поскольку основную роль при этом играет не
характер хромофорной системы, а наличие и химическое пове-
дение активной группы, активные красители обсуждаются в спе-
циальном разделе.
Простые антрахиноновые красители
Как показывает само название, антрахиноновые красители
являются производными антрахинона (37). В молекуле послед-
него карбонильные группы центрального хиноидного цикла ве-
дут себя по отношению к боковым бензольным кольцам как
электроноакцепторные заместители. При введении в бензольные
кольца электронодонорных заместителей становится возмож-
ным образование полярных структур типа 38 и соединение
приобретает окраску. Так, если сам антрахинон имеет лишь
бледно-желтую окраску, то его 1-аминопроизводное представ-
ляет собой вещество оранжевого цвета, 1,5-диаминопроизвод-
ное — красного, а 1,4,5,8-тетрааминоантрахинон — синего.
Синтез антрахиноновых красителей базируется на антрахи-
ноне, который в свою очередь получается путем окисления ан-
трацена или ацилированием бензола фталевым ангидридом по
Фриделю — Крафтсу с последующей циклизацией образующейся
о-бензоилбензойной кислоты. Замещение антрахинона в ядро
протекает с трудом, что связано с электроноакцепторным влия-
нием карбонильных групп Галогенирование идет настолько
плохо, что практически эту реакцию можно считать неосуще-
ствимой, а нитрование, которое приходится проводить в жест-
ких условиях, дает смеси изомеров. Гораздо более важное зна-
чение имеет сульфирование, причем этот процесс является
управляемым: в присутствии солей ртути образуется а-сульфо-
кислота, а в отсутствие этого катализатора — |3-сульфокислота.
Дальнейшее сульфирование приводит к вступлению второй суль-
фогруппы в другое бензольное кольцо, в результате чего полу-
чаются смеси изомеров. При каталитической реакции вторая
сульфогруппа вводится в положение 5 или 8, а при некатали-
тической — в положение 6 или 7.
Сульфогруппы антрахинонсульфокислот обладают высокой
лабильностью и легко замещаются на другие группы. Нагрева-
ние этих соединений с известью или аммиаком под давлением
представляет собой препаративный метод получения окси- и
378
Глава 10
аминоантрахинонов, используемых в качестве промежуточных
веществ в синтезе красителей. Сплавлением с едкими щелочами
можно ввести в ядро дополнительные оксигруппы. Этот метод
находит применение при получении 1,2-диоксиантрахинона
(ализарина), которое осуществляют путем сплавления антра-
хинон- р-сульфокислоты с едким кали. Наличие электронодонор-
ного заместителя облегчает замещение в антрахиноновом ядре.
Иллюстрацией этого может служить порядок стадий при синтезе
такого ключевого полупродукта, как 1-амино-4-бромантрахинон-
2-сульфокислота (39): антрахинон переводят через «-сульфо-
кислоту в а-аминоантрахинон, который сульфируют в положе-
ние 2 и затем бромируют в положение 4. Легко протекающая
конденсация этого полупродукта с аминами в присутствии ме-
ди — наиболее общий метод получения сульфированных антра-
хиноновых красителей, таких, как ярко-синий краситель 40.
39 40 (синий)
Иногда производные антрахинона более удобно получать по
реакции Фриделя — Крафтса, используя вместо фталевого ан-
гидрида или бензола продукты их замещения. Одним из важ-
ных полупродуктов, синтезируемых этим методом, является
1,4-диоксиантрахинон (хинизарин), который образуется при за-
мене бензола на n-хлорфенол (циклизация сопровождается гид-
ролизом атома хлора). Хинизарин применяется главным обра-
зом как исходное вещество для получения простых 1,4-ди-(М-
алкиламино) антрахинонов типа соединения 41, синтез которых
осуществляют путем взаимодействия хинизарина с алкилами-
нами.
О ЫНСНз
О ШСНз
41 (синий)
Исследования в области соединений рнтрахинона давно уже
вышли за пределы их первоначальной цели — синтеза природ-
ного ализарина — и привели к созданию красителей, способных
окрашивать любые виды волокон. Хотя в ряду производных ан-
Химия красителей
379
трахинона можно получить широкую гамму цветов, наиболее
важной областью их применения является получение окрасок
от синей до зеленой, так как именно в этой области спектра
антрахиноновые красители обеспечивают непревзойденную ком-
бинацию яркости и прочности к свету. Поскольку обсуждав-
шиеся выше механизмы закрепления азокрасителей на волокне
не связаны с природой хромофорной группы, их можно в рав-
ной степени отнести и к антрахиноновым красителям. Так, про-
стые водонерастворимые „соединения типа 41 используются в
качестве дисперсных красителей для синтетических волокон, а
сульфированные водорастворимые соединения типа 40 — как
кислотные красители для шерсти и (при наличии в боковой
цепи четвертичной аммониевой группы) как катионные краси-
тели для полиакрилонитрильных волокон. Только для крашения
хлопка обычные антрахиноновые красители не находят приме-
нения, что обусловлено полным отсутствием субстантивных
свойств у антрахинона и его производных. Однако в данном
случае могут быть использованы азоантрахиноновые красители,
например соединение 42, которое является комбинацией суб-
стантивного желтого азокрасйтеля и синего антрахинонового
красителя; в результате получается прямой краситель, дающий
зеленые окраски.
42 (зеленый)
380
Г лава 10
Кубовые красители
Несмотря на то что простые сульфированные антрахиноно-
вые красители непригодны для крашения хлопка, более слож-
ные производные антрахинона являются чрезвычайно важными
красителями для этого волокна. Они получили название кубо-
вых красителей. Отличительная особенность производных ан-
трахинона, на которой основано применение кубовых красите-
лей, — способность легко восстанавливаться гидросульфитом
натрия в разбавленном растворе едкого натра с образованием
раствора динатриевой соли диоксисоединения (лейкоформы).
Последняя затем легко окисляется под действием воздуха, вновь
переходя в нерастворимую хиноидную форму. Лейкоформа са-
мого антрахинона обладает низкой субстантивностью, и поэтому
он не применяется для крашения хлопка. Однако более слож-
ные соединения достаточно субстантивны и удовлетворительно
выбираются хлопком из щелочной ванны. При последующем
выдерживании на воздухе на волокне регенерируется нераство-
римый хинон. В заключение окрашенную ткань обрабатывают
кипящим мыльным раствором для агрегации частиц красителя.
Применение продажных стойких сульфатов восстановленных
кубовых красителей устраняет необходимость получения лейко-
формы на красильных фабриках. Эти соли, которые растворимы
в воде, готовят путем восстановления кубового красителя ме-
таллом в растворе пиридина, содержащего SO3; после нанесе-
ния на волокно их можно легко окислить в кислой среде в ис-
ходный нерастворимый хинон.
Примером простых кубовых красителей, применяемых в про-
мышленности, может служить соединение 43, в котором амино-
антрахиноновые ядра связаны между собой с помощью дихлор-
ангидрида дикарбоновой кислоты в молекулу, обладающую
субстантивными свойствами. Аналогичное строение имеют антри-
миды, такие, как краситель 44, образующийся при конденсации
хлорантрахинона с а-аминоантрахиноном. Соединения послед-
него типа — не только ценные кубовые красители, но и важные
полупродукты для синтеза карбазольных кубовых красителей,
например типа 45.
43 (желтый)
Химия красителей
381
44 (оранжевый)
Карбазольное кольцо в этих соединениях образуется в резуль-
тате сплавления антримида с хлористым алюминием и хлори-
стым натрием. Бензоиламиноантримиды циклизуются еще легче;
часто для этого бывает достаточно обработать их концентриро-
ванной серной кислотой. Среди карбазольных красителей есть
и гораздо более сложные соединения. Например, практически
важный краситель индантрен хаки 2Г получается путем цикли-
зации тетраантримида, образующегося при конденсации а-ами-
ноантрахинона с 1,4,5,8-тетрахлорантрахиноном. В случае кра-
сителей столь сложного строения часто остается невыясненным
вопрос о том, действительно ли реакция циклизации прошла с
образованием всех возможных пиррольных циклов (в послед-
нем примере —четырех).
В родственной группе кубовых красителей — антрахинонази-
нах— антрахиноновые фрагменты соединены между собой
1,4-диазиновым кольцом. Родоначальником этой группы яв-
ляется важный краситель индантрон (46) *, получаемый- путем
сплавления р-аминоантрахинона с едким кали. Существуют
также кубовые красители, в молекуле которых антрахиноно-
вые ядра соединены другими гетероциклами, в том числе
* В технике чаще называется индантреном. По номенклатуре, принятой
в советской промышленности, — кубовый синий О. — Прим. ред.
382
Глава 10
акридоновыми и тиоксантоновыми кольцами. Однако они име-
ют меньшее практическое значение.
Выше рассматривались кубовые красители, в структуре ко-
торых можно легко различить остатки антрахинона. Наряду
с ними важную группу кубовых красителей составляют произ-
водные антрона (47, R = H), в молекуле которых антроновые
остатки представляют собой часть многоядерной ароматической
системы, содержащей пиренбвые и периленовые фрагменты. Ти-
пичными красителями этой группы являются дибензопиренхи-
ноны (или дибензохризены) (48) и антантроны (49). Первые
49 (оранжевый)
48 (желтый)
из них получаются путем циклизации 3-бензоилбензантрона
(51, R = COCeHs) или 1,5-дибензоилнафталина в присутствии
хлористого алюминия, причем оба исходных полупродукта син-
тезируют бензоилированием соответствующих углеводородов по
Фриделю—Крафтсу. Антантрон образуется при циклизации
1,1'-динафтил-8,8'-дикарбоновой кислоты в серной кислоте.
Само по себе это соединение не применяется в качестве краси-
теля, но его производные, например типа 49 (R = Br), имеют
практическое значение как красители. Антроновые фрагменты
присутствуют также в красителях группы пйрантрона (50), ко-
торый получается при сплавлении с едким кали 2,2'-диметил-
Химия красителей
383
1,1'-диантрахинонила, образующегося при действии меди на
1-хлор-2-метилантрахинон.
50 (оранжевый)
51
Ключевым полупродуктом в синтезе ценных кубовых краси-
телей, содержащих периленовый скелет, является бензантрон
(51, R = H). Это соединение легко получается из антрахинона
путем его обработки чугунной стружкой и глицерином в серной
кислоте. На первой стадии антрахинон восстанавливается в ан-
трон, а глицерин превращается в акролеин. Затем оба вещества
взаимодействуют между собой по механизму конденсации через
оксикетон 47 [R = СН(ОН)СН — СН2], который далее претер-
певает дегидратацию и циклизацию в бензантрон, или по меха-
низму а,р-присоединения антрона к акролеину с получением
промежуточного продукта 47 (R = СН2СН2СНО), который
циклизуется, первоначально образуя дигидробензантрон. Сплав-
ление бензантрона со щелочью дает дибензантрон (52, R = Н);
промежуточным продуктом этой реакции является 4,4'-дибенз-
антронил.
Дибензантрон * (52, R = Н) используется не только как
важный кубовый краситель синего цвета, но и как полупродукт
для синтеза многочисленных производных, имеющих большое
практическое значение. Наиболее выдающимся среди них яв-
ляется 16,17-диметоксипроизводное — каледон нефритовый зе-
леный** (52, R = OCHa). Его синтезируют окислением дибенз-
антрона или 4,4/-дибензантронила двуокисью марганца в
растворе серной кислоты с последующим восстановлением обра-
зующегося 16,17-дикетона бисульфитом натрия в диоксидибенз-
антрон и метилированием последнего.
* Другое название этого соединения — виолантрон. — Прим, перев.
** В советской литературе этот краситель известен под названием «ку-
бовый ярко-зеленый С». — Прим, перев.
384
Глава 10
Широко изучены в качестве кубовых красителей и гетеро-
циклические аналоги описанных выше соединений. Важное мес-
то в их ряду занимает флавантрон (53), получаемый сплавле-
нием р-аминоантрахинона с едким кали при высокой темпе-
ратуре.
Кубовые красители дают окраски всех цветов, обладающие
превосходной прочностью. Недостаток многих желтых красите-
лей — способность к выцветанию, т. е. фотохимическому разло-
жению при действии на' окрашенную хлопчатобумажную ткань
солнечного света. Кроме того, из-за многостадийности синтеза
кубовых красителей они дороги, и поэтому их применяют глав-
ным образом для крашения высококачественных хлопчатобу-
мажных тканей (например, драпировочных и обивочных мате-
риалов), где необходима высокая светопрочность и стоимость
красителя не имеет большого значения.
Индигоидные и тиоиндигоидные красители
После установления строения индиго — одного из важней-
ших прежде природных красителей — перед возникшей неза-
долго до того промышленностью красителей автоматически
встала задача его синтетического производства. Эта трудная
задача была настолько актуальной, что одна только немецкая
фирма .BASF истратила на поиски экономичного метода син-
теза индиго до конца прошлого века в общей сложности свыше
1 млн. ф. ст. Когда же такой метод был, наконец, найден, син-
тетический продукт быстро вытеснил природный индиго из
практики крашения.
Фирма BASF разработала два процесса промышленного
производства индиго. В первом из них фенилглицин подвер-
гают циклизации в индоксил путем сплавления с едким кали
Химия красителей
385
и полученный индоксил окисляют в щелочном растворе в ин-
диго. Во втором процессе вместо фенилглицина используется
фенилглицин-о-карбоновая кислота, что приводит к повышению
выхода красителя. Позднее было показано, что при замене ед-
кого кали амидом натрия такой же хороший выход индиго полу-
чается и на основе более дешевого фенилглицина. Этот способ
стал (и до сих пор остается) наиболее распространенным мето-
дом технического синтеза индиго.
Глубоко-синий цвет индиго обусловлен присутствием в его
молекулярной структуре ионных форм типа 54. Крашение ин-
диго осуществляется по кубовому способу, но применяемая при
этом технология отличается от технологии крашения антрахино-
новыми кубовыми красителями, поскольку почти бесцветная
лейкоформа индиго довольно устойчива. Последняя выпускается
химической промышленностью под торговым наименованием
«индиго белый», что избавляет красильные фабрики от гро-
моздкой стадии восстановления индиго. Для перерода лейко-
индиго в нерастворимую индигоидную форму часто используют
раствор азотистой кислоты. Как и в случае других красителей,
удерживающихся на волокне за счет своей нерастворимости,
важное значение имеет агрегация молекул индиго в порах во-
локна.
Синяя окраска индиго малочувствительна к влиянию заме-
стителей, поэтому лишь немногие из его производных нашли
применение в промышленности, причем все они тоже дают
окраски синих тонов. С другой стороны, тиоиндиго (55), кото-
рый получается сплавлением фенилтиогликоль-о-карбоновой
кислоты с едким кали и окислением образовавшегося продукта,
имеет красный цвет. Технология крашения хлопка тиоиндиго
не отличается от той, которая используется в случае индиго.
Простые производные тиоиндиго дают тона от оранжевого до
красного, но замещение одного или обоих бензольных ядер
остатками нафталина или антрацена позволяет получить корич-
невые, зеленые и серые тона. Кроме тиоиндиго, практическое
значение имеют также красители смешанного строения типа 56.
Другими технически важными производными индиго и
13 Зак. 501
386
Глава 10
тиоиндиго являются бромированный изомер индиго (57) и про-
дукт конденсации тиоиндоксила с аценафтенхиноном (58).
57 (гелиотроповый.)
Триарилметановые красители
Триарилметановые красители—одни из старейших синтети-
ческих красителей (т. 2, стр. 249—253). Типичная структура 59
представляет собой лишь одну из возможных формул строения
триарилметанов, причем положительный заряд распределен в
молекуле между двумя или тремя диалкиламиногруппами, на-
ходящимися в «ара-положениях к центральному атому угле-
рода. Столь значительная делокализация заряда обусловливает
яркие фиолетовые, синие и зеленые цвета триарилметановых
красителей.
(CH3)2N
СГ
R
59
N(CH3)2
Важнейшим представителем красителей диаминотриарилме-
танового ряда является малахитовый зеленый (59, R = H).Ero
получают конденсацией бензальдегида с диметиланилином в
разбавленной соляной кислоте с последующим окислением про-
Химия красителей
387
межуточного соединения 60 двуокисью свинца. Типичным кра-
сителем ряда триаминотриарилметанов можно’ считать кри-
сталлический фиолетовый [59, R = N(CH3)2], который синтези-
руют путем фосгенирования диметиланилина и конденсации
образовавшегося Д,4'-бнс-(диметиламино)бензофенона с тре-
тьим эквивалентом диметиланилина. Замена диметиламина
N-фенил-а-нафтиламином на заключительной стадии конденса-
ций приводит к образованию важного красителя виктория голу-
бой. Родственные по строению гидроксилсодержащие триарил-
метановые красители, такие, как хромовый фиолетовый (61),
получаются конденсацией соответствующих полупродуктов в
серной кислоте в присутствии нитрита натрия, играющего роль
окислителя. Например, хромовый фиолетовый синтезируют из
салициловой кислоты и формальдегида.
61
Основные триарилметановые красители этого типа находят
некоторое применение для крашения полиакрилонитрильных
волокон, а их полисульфированные производные могут быть ис-
пользованы в качестве кислотных красителей для шерсти. Од-
нако широкому применению триарилметановых красителей для
крашения текстильных материалов препятствует их низкая све-
топрочность. Наибольшее практическое значение имеет исполь-
зование этих красителей для окрашивания бумаги, где главную
роль играет не долговечность окраски, а ее яркость.
Фталоцианиновые красители
Макроциклический пигмент фталоцианин меди (77, фор-
мула приведена на стр. 396) дает исключительно красивые
окраски бирюзового цвета, обладающие превосходной свето-
прочностью. Это стимулировало поиск других соединений дан-
ного класса, особенно в качестве красителей для хлопка, где
яркие цвета наиболее желательны. Исходным веществом чаще
всего служит сам фталоцианин меди, так как большая устой-
чивость фталоцианиновой структуры позволяет проводить с этим
соединением разнообразные химические превращения. Обычно
13*
388
Глава 10
в молекулу фталоцианина меди вводят четыре заместителя —
по одному в каждое бензольное кольцо. При использовании бо-
лее мягких условий реакции образуются смеси продуктов более
низкой степени замещения. Смесь сульфокислот, которая, как
было установлено, обладает некоторой субстантивностью к
хлопку, первоначально использовалась в промышленности в ка-
честве прямого красителя. Однако она, как и все прямые кра-
сители, имела недостаточную прочность к мокрым обработкам,
вследствие чего данный подход оказался несостоятельным.
Гораздо более прочные окраски удалось получить путем по-
вышения нерастворимости фталоцианина меди. Примером мо-
жет служить краситель алциан синий 8ГН, который синтези-
руют путем тетрахлорметилирования исходного пигмента с
последующим введением продукта в реакцию с тиомочевиной,
приводящую к образованию тетраизотиурониевой соли. Ткань
после печатания подвергают прогреванию, в результате кото-
рого соль разлагается и получается нерастворимый фталоциа-
ниновый пигмент, прочно закрепленный на волокне.
Другой подход заключается в использовании так назы-
ваемых фталогенов. Исходным веществом является диимино-
изоиндолин (62), получающийся в виде умеренно растворимого
1г' 4
JL лн
нитрата при нагревании фталевого ангидрида с мочевиной, ни-
тратом аммония и молибдатом аммония. Свободное основание,
образующееся при его подщелачивании едким натром, смеши-
вают с солью меди и производят печатание на ткани; при по-
следующем нагревании получается фталоцианин меди.
Прочие красители
Помимо красителей, относящихся к описанным выше хими-
ческим классам, существует большое число других красителей,
о которых здесь уместно привести лишь самые общие сведения.
Сернистые красители — дешевые продукты, применяемые для
неответственных целей, — представляют собой сложные веще-
ства неустановленного строения, получаемые путем нагревания
различных органических полупродуктов с серой или сернистыми
соединениями. Например, сернистый черный Т образуется при
Химия красителей
389
нагревании 2,4-динитрофенола с раствором полисульфида нат-
рия. Сернистые красители получены в широкой цветовой гамме,
красят хлопок по методу, аналогичному кубовому крашению, и
закрепляются на волокне за счет своей нерастворимости.
Вплоть до настоящего времени для крашения хлопка широко
применяется также краситель анилиновый черный — сложное
соединение, . содержащее хинониминовые фрагменты, которое
получается окислением анилина на волокне.
Для окраски бумаги широкое применение находит краси-
тель хинолиновый желтый, образующийся при сульфировании
продукта конденсации (63) 2-метилхинолина с фталевым анги-
дридом. Некоторые зеленовато-желтые цианиновые красители
(например соединение 64), получаемые конденсацией цианук-
сусного эфира с соответствующими альдегидами, являются цен-
ными дисперсными красителями для синтетических волокон, а
их четвертичные аммониевые производные используются как
основные красители для полиакрилонитрильных волокон. От-
дельные ксантеновые красители, такие, как родамин Б (65),
применяются для получения флуоресцирующих окрасок на бу-
маге. Эти красители синтезируют путем конденсации лг-диал-
киламинофенолов с фталевым ангидридом.
Активные красители
В предыдущих разделах синтетические красители классифи-
цировались в соответствии с природой содержащейся в их мо-
лекуле хромофорной группы. Практические трудности, возни-
кающие при крашении хлопка, разрешались двумя способами
с использованием особенностей хромофорной группы: либо пу-
тем образования водородных связей растворимого красителя с
390
Глава 10
волокном или агрегации молекул красителя, как в случае пря-
мых красителей, либо путем получения нерастворимого краси-
теля непосредственно на волокне или агрегации его частиц при
последующей обработке окрашенных тканей, как в случае ле-
дяных, кубовых, индигоидных, тиоиндигоидных, фталоцианино-
вых и сернистых красителей. Первый способ проще по техноло-
гии, но дает недостаточно устойчивые окраски, тогда как вто-
рой, хотя он позволяет получить значительно более прочные
окраски, довольно сложен в исполнении.
Важным достижением химии красителей в послевоенный
период явилось создание активных красителей для целлюлоз-
ных волокон, впервые выпущенных в 1956 г. Эти растворимые
в воде красители позволяют получать очень прочные к мокрым
обработкам окраски по относительно простой технологии кра-
шения. Устойчивость окраски достигается за счет присутствия
в молекуле активного красителя группы, реагирующей с волок-
ном с образованием ковалентной связи, которая удерживает
краситель на волокне при последующих мокрых обработках.
Представителем самой первой группы активных красителей —
так называемой группы проциона М — может служить соедине-
ние 66, получаемое взаимодействием аминоазокрасителя с ци-
анурхлоридом. Единственное требование к исходному красителю
заключается в том, чтобы он содержал группу (обычно амино-
группу), по которой в его молекулу можно было бы ввести
активную группировку. Таким образом, исходным веществом
может быть краситель любого химического класса. Наибольшее
практическое значение имеют азокрасители, красители ряда
антрахинона и фталоцианины. Все они, как правило, содержат
сульфогруппы, придающие соединению растворимость в воде.
Возможность использовать простые исходные красители позво-
ляет легко получать чистые яркие тона.
Химическое поведение дихлор-сцлмг-триазиновых красителей
сходно с поведением обычных галогенпроизводных шестичлен-
ных азотсодержащих гетероциклов (т. 3, гл. 2). Свойства 2- и
4-галогенпиридинов (т. 3, стр. 65—66) и их аналогов в ряду пи-
римидина, пиразина и пиридазина (т. 3, стр. 77—78) позволяют
ожидать от галогентриазинов такой же, если не более высокой,
Химия красителей
391
реакционной способности (т. 3, стр. 80). На приведенной ниже
схеме представлен механизм нуклеофильной атаки аниона Х~
на молекулу красителя, содержащую остаток цианурхлорида
(67). В этом уравнении частица :Х_ представляет собой нукле-
офил типа гидроксил-иона или иона ЦеллО- Следует отметить,
- Vе' - С\/Х
N—у N—у
краситель—NH—+ S X” —> краситель—NH——►
N-\
'Cl
67
Cl
что характер распределения электронов делает активный кра-
ситель чувствительным к атаке именно анионом, а не нейтраль-
ной молекулой. Поэтому в нейтральном водном растворе такой
краситель неактивен. Можно в водный раствор активного кра-
сителя ввести ткань, добавить в красильную ванну соль, чтобы
высадить краситель на волокно, но при этом не произойдет ни-
какой химической реакции. Когда же после завершения указан-
ного физического процесса в ванну добавляют соду, поднимая
pH до ~ 10,5, происходит реакция нуклеофильного замещения,
которая идет при температуре около 20 °C. Существует два воз-
можных пути протекания реакции. Роль атакующей нуклео-
фильной частицы может играть либо ион ЦеллО-, что приводит
к фиксации красителя на волокне, либо гидроксил-ион, что вле-
чет за собой гидролиз красителя и его потери в процессе кра-
шения. Константы скорости обеих реакций приблизительно
равны. Однако тот факт, что краситель уже сорбирован волок-
ном и, следовательно, сконцентрирован в нем, а также почти
25-кратное численное преобладание ионов ЦеллО- над гидро-
ксил-ионами в порах волокна обеспечивают преимущественное
протекание фиксации красителя по сравнению с его гидролизом.
Краситель, подвергшийся гидролизу, вымывается из волокна
после окончания процесса крашения.
Несмотря на присутствие в активных красителях данного
типа двух лабильных атомов хлора, в случае гидролиза одного
из них фиксация красителя по другому маловероятна. Образую-
щееся производное оксихлортриазина ионизируется в щелочной
среде, причем заместитель О- подает электроны в гетероцикли-
ческое ядро, препятствуя образованию положительного заряда
392
Глава 10
на атомах углерода в ядре и понижая лабильность оставшегося
атома хлора Этот крайний случай наглядно демонстрирует,
какое сильное влияние может оказывать второй заместитель на
реакционную способность атома хлора в монохлортриазинах.
В других случаях в триазиновом цикле могут содержаться
электроноакцепторные заместители, которые сохраняют актив-
ность красителя и сами являются лабильными. Сюда относятся
красители на основе 2,4-динитротиофенокситриазинов, 2,4-ди-
сульфофенокситриазинов и сульфированных триазинов;. они,
однако, менее доступны, чем хлорзамещенные производные, ко-
торые по-прежнему сохраняют важное промышленное значение.
Напротив, электронодонорные заместители, такие, как
аминогруппа, замещенные аминогруппы и метоксигруппа, пони-
жают реакционную способность атома хлора. Монохлортриази-
новые красители, содержащие эти группы в качестве второго
заместителя, окрашивают только при температуре около 60 °C,
и для крашения ими часто требуются более высокие значения
pH красильной ванны. Таковы, в частности, красители групп
проциона Эйч и цибакрона, которые выбираются волокном из
горячих ванн. Эти красители хорошо подходят также для пе-
чати: краситель смешивают с щелочью, водой и загустителем и
полученную печатную пасту, которая вполне устойчива при
обычной температуре, наносят на ткань в виде рисунка, после
чего окрашенную ткань нагревают до температуры около 100 °C.
При этом краситель реагирует с волокном, а также частично
гидролизуется. Незакрепленный краситель смывается с ткани
в процессе заключительной промывки.
Как при крашении, так и при печатании желательно, есте-
ственно, чтобы гидролиз красителя происходил как можно в
меньшей степени, а его фиксация на ткани — по возможности
в большей. Этому требованию удовлетворяют появившиеся не-
давно красители групп проциона супра (для печати) и про-
циона Эйч-Е (для крашения), содержащие две активные груп-
пировки.
Многочисленные активные красители, которые были предло-
жены в качестве замены моно- и дихлортриазиновых красите-
лей, можно подразделить на два типа. К первому типу
относятся галогенгетероциклические производные, включаю-
щие, помимо хлортриазиновык красителей, трихлорпиримидил-
аминокрасители [реактоны и дркмарены (68)], а также дихлор-
пиридазоновые [примазин П (69)] и дихлорхиноксалин-6-карбо-
ниламидные [левафикс Е (70)] красители. Большинство этих
соединений уступает по активности дихлортриазинам и прибли-
жается к монохлортриазинам. Причиной этого является присут-
ствие в гетероциклическом кольце лишь двух атомов азота
по сравнению с тремя идеально расположенными атомами
Химия красителей
393
азота, содержащимися в кольце сщи.и-триазина. Иногда актив-
ность красителя удается повысить введением в соответствующее
положение кольца электроноакцепторного заместителя, как в
С1
С1
68 69
70
случае 5-цианодихлорпиримидиламиносоединений. Используются
также более лабильные, чем атом хлора, уходящие группы, на-
пример атом фтора и метилсульфогруппа. Особенно эффектив-
ным оказалось использование четвертичной аммониевой группы.
Красители, содержащие эту активную группу, легко получаются
при взаимодействии монохлортриазиновых красителей с некото-
рыми третичными аминами, из которых наиболее важными яв-
ляются пиридин, триметиламин и 1,4-диазабицикло[2,2,2]октан.
Активные красители второго типа, примером которых могут
служить ремазолы (71), характеризуются наличием в их моле-
куле р-сульфоэтилсульфоновой группы. В присутствии щелочи
они отщепляют серную кислоту и превращаются в винилсуль-
фон, в котором характер распределения электронной плотности
(72) обусловливает возникновение частичного положительного
заряда на концевом атоме углерода. Нуклеофильная атака по
этому центру с последующим присоединением протона приводит
к фиксации красителя на целлюлозном волокне (73). Анало-
гично ведут себя и некоторые другие активные группы, напри-
мер у-сульфопропиониламидная группа в примазинах (74).
Р-СульфоэтиЛсульфоновую группу необходимо создавать на
краситель— SO2CH2CH2OSO3H краситель — S-^CH=CH2
О
71
72
Краситель—SO2CH2CH2O—целлюлоза краситель —NHCOCH2CH2OgO3H
73
74
394
Глава 10
промежуточных стадиях, а не присоединять ее к окрашенному
соединению путем заключительного ацилирования. Процесс по-
лучения активного красителя может включать, например, сле-
дующие стадии: превращение 3-нитробензолсульфокислоты че-
рез ее хлорангидрид в сульфиновую кислоту, реакцию послед-
ней с этиленхлоргидрином, восстановление продукта в амин и
сульфирование. Это ограничение удалось обойти в синтезе но-
вых активных красителей ремазолов, содержащих 0-сульфо-
этилсульфамйдную группу (75), которая вводится в исходный
аминосодержащий краситель по реакции с «карбилсульфатом»
(76), полученным из трехокиси серы и этилена.
О
Н2С-Х xso2
краситель—NHSO2CH2CH2OSO3H | |
Н2С\ /О
S
О2
75 76
Хотя главной областью применения активных красителей
служит крашение хлопка, принцип закрепления красителя на
волокне за счет образования ковалентных связей можно распро-
странить и на другие волокна. В качестве подходящих мате-
риалов прежде всего следует назвать шерсть и полиамидные
волокна, которые содержат аминогруппы. Для этих видов во-
локон уже выпускаются специальные активные красители. Сюда
относятся дисперсные красители группы процинайла, предна-
значенные для окрашивания полиамидных волокон, и металл-
содержащие комплексные красители состава 1 :2 (проциланы)
для крашения шерсти. Оба ряда красителей содержат активные
группы, специально приспособленные ' для этих волокон. Так,
в проциланах активной группой является акриламидная груп-
па, причем она реагирует достаточно медленно для того, чтобы
краситель успел полностью проникнуть в массу волокна прежде,
чем он прочно закрепится на нем.
Пигменты
Второй важной областью применения окрашенных органи-
ческих соединений является производство пигментов. Органи-
ческие пигменты обладают более высокой кроющей способ-
ностью и дают более нежные тона, чем неорганические пиг-
менты, такие, как хромат свинца. Пигменты вводятся в краску
или пластмассу в чистом виде и закрепляются там в резуль-
тате отверждения материала. Таким образом, механизм погло-
щения и фиксации красящего вещества здесь не имеет суще-
ственного значения. Подобно красителям, пигменты должны
обладать светопрочностью, причем в данном случае это свой-
Химия красителей
395
ство необходимо в еще большей степени, как и вообще высокая
устойчивость к атмосферным воздействиям. В остальных отно-
шениях требования по прочности к пигментам значительно от-
личаются от требований к красителям. Так, пигменты должны
иметь достаточную термостойкость, чтобы не разрушаться в
условиях формования пластмассы или обжига эмалевого по-
крытия. Другим требованием является нерастворимость пиг-
мента в органических растворителях, с тем чтобы избежать,
например, его миграции в нанесенный на поверхность изделия
слой белой краски.
Наиболее важную группу пигментов, особенно для получе-
ния тонов от желтого до красного, составляют легкодоступные
и дешевые азосоединения. Диазосоставляющими чаще всего
служат 2,5-дихлоранилин и 2-нитро-4-метиланилин, а в качестве
азосоставляющих используют ариламиды ацетоуксусной кис-
лоты, 1-арил-3-метилпиразолоны-5, р-нафтол и ариламиды
2-окси-З-нафтойной кислоты. Лучшие из моноазопигментов
имеют удовлетворительную светопрочность, однако их устойчи-
вость к нагреванию и действию растворителей часто недоста-
точна. Основной способ улучшения этих показателей заклю-
чается в увеличении размеров молекулы путем использования
диамина, например 3,3'-дихлорбензидина, дающего бисазопиг-
мент. Стремление повысить нерастворимость пигмента повлекло
за собой тенденцию к применению все более нерастворимых со-
ставляющих, что существенно усложняет проведение заключи-
тельного диазотирования и сочетания. Чтобы обойти это за-
труднение, был разработан метод, в котором нерастворимость
придается пигменту после создания связи —N —N—. Например,
в синтезе Пигментов группы хромофтала азокарбоновую кислоту
(такую, как 2,5-дихлоранилин -> 2-окси-З-нафтойная кислота)
переводят в хлорангидрид и подвергают конденсации с диами-
ном (таким, как бензидин) в высококипящем растворителе, в
результате чего образуется весьма нерастворимый бисамид.
Помимо описанных выше чисто органических азосоединений,
широкое применение в качестве дешевых пигментов находят
нерастворимые кальциевые, бариевые и марганцевые соли про-
стых сульфированных азокрасителей. Для неответственных це-
лей используются также соли основных триарилметановых кра-
сителей с фосфорновольфрамомолибденовой кислотой, которые
значительно превосходят по светопрочное™ исходные красители.
Идеальными свойствами как пигмент обладает более слож-
ное соединение — нерастворимый и прочный фталоцианин меди
(77). Он легко получается путем нагревания фталевого анги-
дрида, мочевины, CU2CI2 и катализатора в высококипящем рас-
творителе и дает очень красивые бирюзовые окраски, имеющие
исключительную стойкость к свету и действию температуры и
396
Глава 10
растворителей. Ассортимент тонов, которые могут быть полу-
чены на основе этой чрезвычайно удачной структуры, к сожа-
лению, ограничен. Полихлорпроизводное, образующееся при
77
хлорировании фталоцианина меди до введения в его молекулу
12—14 атомов хлора, является превосходным зеленым пигмен-
том; замещение части атомов хлора на бром дает желтовато-
зеленые тона.
Успешное применение фталоцианина меди и его галоген-
производных стимулировало поиски других окрашенных гетеро-
циклических соединений, которые можно было бы использовать
в качестве пигментов. Особенно требовались пигменты, дающие
окраски в красной области спектра, так как широко применяе-
мые для этой цели азосоединения часто обладают наиболее низ-
кой светопрочностью. Были предложены три типа таких пиг-
ментов. быс-Арилимиды перилентетракарбоновой кислоты (78)
обеспечивают получение разнообразных красных тонов, оттенок
которых зависит от природы арильного радикала. Эти соедине-
ния можно использовать также как кубовые красители для
хлопка.
78
При конденсации фталимидного производного 79 с диами-
нами образуются оранжевые, красные и коричневые пигменты
типа тетрахлоризоиндолинона (80). К третьему типу относятся
полиморфные модификации линейного хинакридона (81), кото-
рые являются ценными пигментами тонов от красного до фиоле-
тового. Синтез соединения 81 включает конденсацию 2,5-дибром-
Химия красителей
397
терефталевой кислоты с анилином с последующей циклизацией
образующегося 2,5-дианилинпроизводного.
Номенклатура
Красители и пигменты продаются под фирменными наиме-
нованиями, которые обычно представляют собой зарегистриро-
ванные торговые марки фирм-производителей, указывающие,
для какой цели они предназначаются. Такими фирменными наи-
менованиями являются упоминавшиеся в этой главе названия:
алциан, каледон, процилан, процион и процинайл (фирма ICI),
цибакрон и хромофтал (фирма Ciba), левафикс и фталоген
(фирма Bayer), примазин (фирма BASF), реактон (фирма
Geigy), дримарен (фирма Sandoz) и ремазол (фирма Hoechst).
Приведенные в этой главе названия химических соединений ино-
гда отличаются от принятых в химической литературе и соот-
ветствуют тем, которые в настоящее время применяются в ани-
линокрасочной промышленности; однако в таких случаях строе-
ние соединений поясняется структурными формулами.
СПИСОК ЛИТЕРАТУРЫ
Abrahart Е. N., Dyes and Their Intermediates, Pergamon Press Ltd., Lon-
don, 1968.
Фитц-Давид Г. Э., Бланже Л., Основные процессы синтеза красителей,
ИЛ, М., 1957.
Lubs И. A., The Chemistry of Synthetic Dyes and Pigments, Reinhold Pub-
lishing Corp., New York, 1955.
Saunders К. H., The Aromatic Diazo Compounds and Their Technical Ap-
plications, 2nd ed., Edward Arnold, London, 1949 (имеется русский перевод
1-го изд.: Саундерс К., Ароматические диазосоединения и их техническое при-
менение, ГОНТИ, М„ 1938).
Венкатараман К., Химия синтетических красителей, тт. 1—4, «Химия», Л.,
1956—1975.
Виккерстафф Т., Физическая химия крашения, Гизлегпром, М., 1956.
Цоллингер Г., Химия азокрасителей, Госхимиздат, М., 1960.
ГЛАВА 11
Фармацевтические препараты
И. Элмор, Г. Браун, А. Г peep
Elmore N. F., Brown G. R., Greer A. T., Imperial Chemical Industries
Limited, Pharmaceuticals Division, Alderley Park
Введение
В течение многих столетий врачи употребляли для лечения
больных лекарственные растения. Однако лишь в XIX в. из
растений научились выделять и использовать в качестве инди-
видуальных соединений морфин и другие алкалоиды. С разви-
тием органической химии появились синтетические вещества,
обладающие фармакологическими свойствами, такие, как сер-
ный эфир (1846 г.), аспирин (1899 г.), барбитураты и мышьяко-
вистые соединения. Обнаружение в 1932 г. противомикробной
активности у красного пронтозила * (красного красителя
2',4'-диаминоазобензол-4-сульфамида) (7), с помощью которого
оказалось возможным успешно бороться с инфекционными за-
болеваниями человека и животных, вызываемыми грамположи-
тельными микробами, явилось поворотным пунктом в истории
медицины и побудило многие химические фирмы заняться поис-
ками новых лекарственных средств. Особенно мощным стимулом
для синтеза лекарственных органических соединений послужила
возникшая во время второй мировой войны потребность в про-
тивомалярийных средствах, заменяющих хинин, доставка кото-
рого из Индонезии стала невозможной. Открытие в те же годы
пенициллина было делом случая и не диктовалось настоятель-
ной необходимостью. Послевоенный период ознаменовался бур-
ным развитием химической промышленности и большими успе-
хами в создании новых фармацевтических препаратов — сначала
стероидных гормонов и антибиотиков, а затем химиотерапевти-
ческих средств для лечения заболеваний нервной и сердечно-со-
судистой системы. В настоящее время поиск новых лекарств
ведется во всех основных областях органической химии, причем
* Другое название этого соединения—нерастворимый красный стрепто-
цид; родственный ему по химическому строению сульфаниламидный препа-
рат — растворимый красный стрептоцид — одно время выпускался в СССР,—
Прим, перев.
Фармацевтические препараты 399
для проведения синтеза сложных природных соединений (пол-
ного синтеза стероидов, синтеза пептидных гормонов и проста-
гландинов) требуется большое искусство.
Фармацевтическая промышленность развивается в несколь-
ких важных направлениях. Помимо лекарственных препаратов,
она выпускает вакцины, перевязочные материалы, медицинский
гипс и другую продукцию, описание которой выходит за рамки
этой главы. В аптеках и универсальных магазинах свободно
продаются так называемые «патентованные средства», или без-
рецептурные фармацевтические препараты; обычно это хорошо
проверенные лекарства от таких недомоганий, как легкая про-
студа, кашель, расстройство желудка или головная боль. Этот
разряд лекарственных веществ, за небольшими исключениями
(антисептики и средства против кашля), не нуждается в попол-
нении новыми органическими препаратами, и для их успешной
продажи умелая реклама играет гораздо более важную роль,
чем демонстрация терапевтического эффекта. Главная сфера
деятельности фармацевтической промышленности — поиск но-
вых лекарственных средств для лечения серьезных заболеваний,
т. е. создание химиотерапевтических препаратов, которые при-
нято называть рецептурными или «этическими» *. Большое зна-
чение придается также изысканию новых химических методов
повышения продуктивности и снижения заболеваемости домаш-
него скота. Эти работы во многом развиваются параллельно с
исследованиями в области лечения болезней человека, так как
в решении обеих проблем есть немало общего.
Поиск новых «этических» лекарств проводится в основном
в двух направлениях: средства для борьбы с инфекционными
заболеваниями и средства для борьбы с неинфекционными за-
болеваниями. Рациональный химический подход к синтезу и тех
и других веществ основывается на оценке возможного механизма
их биотрансформации или на структурной аналогии с извест-
ными фармакологически активными соединениями. Все вновь
полученные лекарственные средства проходят испытания в био-
химических или биологических лабораториях, где изучается их
действие на выращиваемые in vitro или in vivo патогенные мик-
роорганизмы или на специально зараженных подопытных жи-
вотных. В том случае, когда обнаруживается терапевтиче-
ский эффект, вещество подвергают дальнейшим биологиче-
ским испытаниям более утонченными методами; в то же вре-
мя производится синтез родственных соединений (так назы-
ваемых аналогов) с целью нахождения в данном ряду наибо-
лее активного соединения. Затем одно-два самых активных ле-
* В СССР используемая авторами классификация лекарственных веществ
в зависимости от серьезности заболеваний не принята. — Прим, перев.
400
Глава 11
карственных вещества подвергают всестороннему токсикологи-
ческому обследованию: подопытным животным регулярно в те-
чение нескольких месяцев вводят определенные дозы этих со-
единений и затем тщательно отыскивают признаки нежелатель-
ного (побочного) действия препарата. Если таковые не обна-
ружены, новый лекарственный препарат проходит ограниченные
клинические испытания на людях (если он предназначается для
лечения человека), причем предварительно необходимо получить
на это разрешение соответствующего государственного органа,
который прежде, чем дать такое разрешение, внимательно
изучает накопленные данные о токсичности и других биологи-
ческих свойствах препарата.
Обычно между открытием полезной биологической активно-
сти химического соединения и началом его промышленного
производства в качестве лекарства проходит несколько лет. Так,
фармакологические свойства пропранолола * (стр. 459) были
установлены в конце 1962 г., производство его в Великобрита-
нии началось в августе 1965 г., а продажа этого соединения
в США в качестве противоаритмического средства была разре-
шена в декабре 1967 г. По последним стандартам этот срок
считается еще небольшим, так как в настоящее время большин-
ство ответственных должностных лиц требует проведения дли-
тельных токсикологических испытаний на животных, с тем чтобы
можно было лучше оценить преимущества нового лекарства и
снизить степень риска при исследовании его на людях. Напри-
мер, противоастмэтическое средство Файзона «кромоглюкат-
натрий» было открыто в 1965 г., а его продажа в США была
COONa COONa
«кромоглюкат-натрий» Файзона (интал)
разрешена только в феврале 1973 г. Недавно Федеральное
управление по контролю за качеством фармацевтических про-
дуктов США заявило, что впредь все пероральные противоза-
чаточные средства будут допускаться к продаже лишь после
семилетних испытаний. Длительные испытания будут, конечно,
значительно увеличивать промежуток времени между патенто-
ванием новых лекарственных веществ и их появлением в про-
даже.
* В СССР известен под названием «анаприлин» (здесь и далее в этой
главе в примечаниях переводчика даны названия лекарственных препаратов
по X изданию Государственной Фармакопеи СССР). — Прим, перев.
Фармацевтические препараты 401
Связь между структурой и активностью
органических лекарственных веществ
С начала возникновения фармацевтической химии предпри-
нимались попытки установить связь между химическим строе-
нием и биологической активностью. Это позволило бы осуще-
ствлять направленный синтез фармакологических средств, об-
ладающих повышенной избирательностью, эффективностью, ста-
бильностью и продолжительностью действия. К сожалению, в
большинстве случаев указанную связь удается обнаружить
лишь задним числом. '•
Существует несколько подходов к составлению программы
целенаправленного синтеза новых лекарственных препаратов.
Весьма плодотворным оказался метод модифицирования струк-
туры уже известных синтетических или природных лекарствен-
ных веществ (например, антибиотиков и стероидов), который
позволил получить ряд ценных противомикробных и противо-
воспалительных средств и пероральных противозачаточных пре-
паратов. По альтернативному методу берут небольшой фраг-
мент химической структуры известного лекарства, вводят его
в молекулы других соединений и исследуют биологическое дей-
ствие полученных веществ. При этом было найдено, в частно-
сти, что вещества, содержащие структурный фрагмент кокаина,
сохраняют анестезирующие свойства. Знание структуры извест-
ного фармацевтического препарата, обладающего потенциально
полезным побочным эффектом,- иногда позволяет усилить по-
следний до уровня, приемлемого для терапевтических целей, од-
новременно ослабив основной эффект, присущий исходному пре-
парату. Примером использования такого подхода может слу-
жить история создания сульфамидных диуретиков (мочегонных
препаратов), которые появились в результате наблюдения, что
противомикробное средство сульфаниламид * обладает моче-
гонными свойствами. Имеется много примеров создания ле-
карств, оказывающих определенное влияние на протекание био-
логических процессов. Так, ампролий вылечивает кокцидиоз
у цыплят, индюков и крупного рогатого скота за счет того, что
он блокирует метаболизм витамина Bi в организме микроскопи-
ческого паразита — кокцидия (т. е. ведет себя как «антиметабо-
лит») и поэтому токсичен для него. Менее ясна связь между
структурой и активностью в случае химических соединений, ин-
гибирующих биологический процесс. Например, алкилирующие
агенты, подавляющие рост раковых опухолей, не обязательно
должны быть родственными по химическому строению. Синте-
зированы соединения, биологическая активность 'которых
* В СССР выпускается под названием «стрептоцид». — Прим, перев.
402
Глава 11
обусловлена способностью к комплексообразованию (или хелати-
рованию) с ионами металлов. Мочегонное средство «этакрино-
вая кислота» было найдено в ряду соединений, реагирующих
с тиольными группами белков. Иногда лекарственные веще-
ства удается найти с помощью рабочей гипотезы, связывающей
фармакологическую активность со структурой. Для изучения
структуры химических веществ можно воспользоваться измере-'
нием расстояния между отдельными атомами и группами на
молекулярных моделях. Этот подход сыграл особенно большую
роль в синтезе структурных аналогов природного анальгетика —
алкалоида морфина. Попытки использовать для поиска новых
лекарственных средств идею о том, что каждому типу биологи-
ческой активности соответствуют определенные рецепторы, не
привели к положительным результатам. Однако совершенно
другой подход, базирующийся на достижениях современной
синтетической химии, которые делают возможным изучение со-
единений с ранее не встречавшимися формой и полярностью
молекулы, в ряде случаев оказался плодотворным. Примером
успешного применения этого метода может служить описан-
ный ниже синтез успокоительных средств бензодиазепинового
ряда.
Уместно остановиться на вопросе о том, что происходит с
лекарственным веществом в организме подопытного животного
или человека. Во-первых, оно может абсорбироваться в орга-
низме. В этом случае вещество, пройдя через ряд биологиче-
ских мембран и систем, достигает рецептора (вероятно, внутри
клетки) и воздействует на него. В ходе этого процесса лекар-
ственное вещество может теряться либо путем связывания с
белком, либо путем быстрого выведения из организма. Альтер-
нативной возможностью является превращение этого вещества
в другое соединение (метаболизм), которое, собственно, и об-
ладает фармакологической активностью. Длительность действия
препарата зависит также от таких его свойств, как устойчивость к
превращению в неактивные продукты (детоксикация) или раство-
римость в жировой ткани, которая может выполнять функцию
лекарственного депо. Очевидно, что большое значение имеют
полярность, форма и размеры молекул лекарственного веще-
ства. Роль полярных эффектов можно проиллюстрировать на
примере противосудорожного средства примидон* (1), который
не обладает седативными свойствами родственного ему по
структуре фенобарбитона ** (2), хотя эти соединения разли-
* В СССР принято название «гексамидин». — Прим, перев.
** В СССР выпускается под названиями «фенобарбитал» и «люминал». —
Прим, перев.
Фармацевтические препараты
403
чаются между собой лишь одной карбонильной группой. Андро-
стерон (3) является сильнодействующим андрогеном, тогда как
его эпимер 4, существенно отличающийся от него по форме (но
не по размерам) молекулы, неактивен. Изменение физических
свойств и, следовательно, биологической активности вещества
может быть достигнуто также путем введения в его молекулу
новых группировок. Так, введение или удлинение линейной ал-
кильной группы повышает растворимость в липидах (жирах),
а разветвление этой группы затрудняет биохимическое окисле-
ние вещества. Замыкание алифатической цепи в цикл делает ее
более компактной и облегчает образование связей за счет ван-
дерваальсова взаимодействия с субстратом. Атомы галогена
тоже увеличивают липофильность лекарственных веществ;
кроме того, эти атомы принимают участие в блокировании био-
химически активных центров веществ, претерпевающих метабо-
литическое превращение (инактивирование) по механизму гид-
роксилирования. Например, эстрогенный гормон хлортрианизен
(5) значительно превосходит по продолжительности действия
5
ОН
6
404
Глава 11
диэтилстильбэстрол (6). При замене карбоксильной группы на
сульфогруппу увеличивается растворимость лекарственного ве-
щества в воде. Карбоксильные группы способны декарбокси-
лироваться в биологических системах; данный процесс можно
замедлить путем этерификации этих групп. Часто неблагопри-
ятное влияние на фармакологическую активность оказывают
полярные гидроксильные группы, которые для уменьшения их
полярности можно перевести в ацилокси- или алкоксигруппы.
Если в молекуле лекарственного вещества есть атомы азота, то
их основность можно изменять, варьируя связанные с ними за-
местители. Во многих случаях хорошие результаты дает замена
бензольного кольца на фурановое или тиофеновое, хотя фураны
часто обладают недостаточной стабильностью. В то же время
при замене бензольного кольца более основными гетероциклами
часто наблюдается исчезновение биологической активности, вы-
званное изменением полярности молекулы.
Несмотря на огромное практическое значение, которое имеют
описанные выше закономерности, было бы ошибочно слепо по-
лагаться на них при получении новых фармацевтических пре-
паратов. Для каждого лекарственного препарата связь его
структуры с биологической активностью следует рассматривать
изолированно, так как наши знания в этой области все еще на-
ходятся на очень низком уровне.
Средства для лечения инфекционных
заболеваний
Противомикробные средства
Создание химиотерапевтических препаратов для лечения бо-
лезней, вызываемых микробами, представляет собой одно из
важнейших достижений фармацевтической науки и промышлен-
ности. Ассортимент синтетических лекарственных веществ и ан-
тибиотиков (продуктов жизнедеятельности микроорганизмов),
имеющихся в настоящее время в распоряжении врачей, позво-
ляет вылечивать практически любые бактериальные инфекции.
Все виды бактерий Грам произвольно разделил на два класса:
бактерии, окрашиваемые в синий цвет реактивом Грама
(смесью красителя кристаллического фиолетового с иодом), он
назвал грамположительными, а неокрашиваемые бактерии —
грамотрицательными. Для объяснения различного поведения
бактерий по отношению к реактиву Грама было выдвинуто не-
сколько гипотез. Наиболее правдоподобная из них состоит в
том, что клетки грамположительных бактерий обладают спо-
собностью поглощать кристаллический фиолетовый и иод, тогда
как у грамотрицательных бактерий клеточная оболочка содер-
жит много липидов, которые препятствуют проникновению кра-
Фармацевтические препараты
-405
сителя в клетку. Грамотрицательные бактерии менее чувстви-
тельны к существующим противомикробным средствам, чем
грамположительные, и для лечения вызываемых ими инфекций
предстоит разработать более эффективные лекарства. Общей
проблемой при лечении инфекционных заболеваний является
возникновение у бактерий резистентности (невосприимчивости)
к имеющимся препаратам; иногда эту проблему удается решить
путем использования комбинации двух лекарств, обладающих
различным механизмом действия. В последующих разделах
описаны наиболее важные противомикробные средства, приме-
няемые в современной химиотерапии.
Синтетические противомикробные средства
Несмотря на то что ряд антисептиков был известен еще до
1930 г., ни один из них не был пригоден для внутреннего упо-
требления. В 1932 г. было установлено, что красный краситель
красный пронтозил (7) излечивает стрептококковые и стафило-
кокковые заболевания. Вскоре после этого удалось выяснить,
что пронтозил разрушается в организме животных с образова-
нием сульфаниламида (8), который и является активным про-
тивомикробным агентом. Это открытие послужило толчком к
началу интенсивных работ по синтезу и изучению других сульф-
амидов во всех странах мира. В результате этих работ было
создано большое число лекарственных препаратов, часть кото-
рых используется до сих пор, несмотря на изобретение анти-
биотиков и появление резистентных к сульфамидам штаммов
микроорганизмов. Сульфамидные препараты применяются, в
частности, для лечения инфекционных заболеваний дыхатель-
ных и мочевыводящих путей, а плохо абсорбирующийся фта-
лилсульфатиазол * (9) — для лечения желудочно-кишечных ин-
фекций. Обычно сульфамиды употребляются в комбинации с
В СССР выпускается под названием «фталазол». — Прим, перев.
406
Глава It
некоторыми другими противомикробными агентами, описан-
ными ниже. На схемах 1 и 2 приведены примеры промыш-
ленных методов синтеза типичных сульфамидных препара-
тов — сульфадиазина* (10) и сульфадимидина ** (11) (т. 3,
стр. 120).
роси
он
онссщсоон
+ ------►
HN=C(NH2)2 HJN
* В СССР принято название «сульфазин». — Прим, перев.
** В СССР выпускается под названием «сульфадимезин». — Прим, пе-
рев.
Фармацевтические препараты
407
Сульфамиды уникальны в том отношении, что механизм их
действия выяснен на уровне ферментов. Эти лекарственные ве-
щества подменяют л-аминобензойную кислоту (13) в процессе
превращения пирофосфата 12 в дигидрофолиевую кислоту (14),
образуя «ложные» дигидрофолиевые кислоты; роль последних
в антимикробном действии сульфамидов пока не установлена.
Другой эффективный противомикробный агент, триметоприм
(16), ингибирует процесс превращения дигидрофолиевой
408
Глава 11
кислоты (14) в тетрагидроптеридиновое производное 15. Синтез
триметоприма осуществляют по приведенной ниже схеме:
16
Высокой антимикробной активностью по отношению к грам-
отрицательным бактериям обладают налидиксовая кислота (17)
и нитрофурантоин* (18), которые хорошо зарекомендовали
себя при лечении заболеваний мочевыводящих путей. Налидик-
совую кислоту синтезируют путем конденсации 2-амино-6-метил-
пиридина с диэтиловым эфиром этоксиметиленмалоновой кис-
лоты с последующей циклизацией, алкилированием и гидроли-
зом продукта
В СССР выпускается под названием «фурадонин».— Прим, перев.
Фармацевтические препараты
409
Нитрофурантоин получают нитрованием фурфурола (в форме
диацетата для защиты альдегидной группы) и конденсацией
продукта реакции с Ы-аминоимидазолидиндионом-2,4
о
18
Антибиотики
История о том, как Александр Флеминг открыл в 1929 г.
сильные противомикробные свойства у плесени, общеизвестна.
Однако лишь в 1940 г. Чейну и Флори удалось выделить из пле-
сени активное начало (пенициллин) и продемонстрировать его
исключительную бактерицидную активность на человеке. Все
семь природных пенициллинов представляют собой амиды, яв-
ляющиеся производными одного и того же амина (т. 4, стр. 315).
Типичным представителем этой группы лекарственных веществ
может служить бензилпенициллин (19), который до сих пор
широко применяется в терапии из-за своей дешевизны и низкой
•токсичности.
оотЬ
соон
19
Несмотря на то что пенициллины получают в промышленно-
сти >методами ферментации, проделана значительная работа
(особенно Энери-Логаном и Шиханом) по получению пеницил-
линов синтетическими методами, позволяющими прийти к но-
вым соединениям с повышенной антибактериальной актив-
ностью. В последнее время проявляется большой интерес к мо-
дифицированию химической структуры пенициллина. Огромным
успехом в этом направлении явилось открытие группой Бичема
того, что ферментация в отсутствие предшественника с боковой
410
Глава 11
(ацильной) группой приводит к образованию 6-аминопеницил-
лановой кислоты (20). Было синтезировано свыше 2000 ациль-
ных производных этого соединения, из которых наиболее цен-
ным оказался ампициллин (22), отличающийся широтой про-
тивомикробного действия, возможностью применения в виде
таблеток и относительной стойкостью к кислотам. Однако он
легко разрушается ферментом пенициллиназой и поэтому не мо-
жет быть использован для лечения заболеваний, вызываемых
резистентными стафилококками. В то же время некоторые дру-
гие полусинтетические пенициллины, например метициллин (23),
лишены указанного недостатка.
2S
Другая группа антибиотиков, химически родственная пени-
циллинам,— это цефалоспорины. Цефалоспорин С (24) яв-
ляется продуктом жизнедеятельности плесневого грибка. Он
способен разрушаться с образованием 7-аминоцефалоспорано-
Фармацевтические препараты
411
вой кислоты (25); ацилированием последней можно синтезиро-
вать ряд соединений, сходных с теми, которые получаются из
6-аминопенициллановой кислоты (20). Так, приготовленные из
кислоты 25 антибиотики цефалотин (26) и цефалоридин (27)
особенно полезны для лечения больных, страдающих инфек-
ционными заболеваниями дыхательных или мочевыводящих пу-
тей, которые обладают повышенной чувствительностью к пени-
циллинам. ' (
н
HOOCCHtCHJjCONH^
NHa N'^=^-OCOCH3
CO2H
S4
NOCI
HCOOH
coo-
Хлорамфеникол * (28), который вначале использовался очень
широко, в настоящее время находит ограниченное применение
* В СССР выпускается под названием «левомицетин». — Прим, перед.
412
Глава И
(главным образом, для лечения брюшного тифа) из-за значи-
тельных побочных эффектов.
28
Антибиотики группы тетрациклина (т. 4, стр. 198—207) об-
наруживают наиболее широкий спектр противомикробной ак-
тивности (природа их действия пока не.раскрыта) и поэтому
употребляются для лечения самых различных заболеваний, осо-
бенно пневмонии и болезней, вызываемых грамотрицательными
микробами и риккетсиями. Они являются продуктами жизне-
деятельности плесневых грибков или получаются путем модифи-
цирования химической структуры природных соединений.
В табл. 11.1 приведены важнейшие антибиотики этой группы,
выпускаемые в промышленном масштабе.
Таблица 11.1
Некоторые антибиотики группы тетрациклина
99
Заместители в структуре 29
Антибиотик R r' н R r'"
Тетрациклин Н СНз н N(CH3)2
Хлортетрациклин а С1 СНз н N(CH3)2
Окснтетрациклин н СНз он N(CH3)2
Дезметнлхлортетрациклин CI Н н N(CH3)2
Метациклин н =СН2 в н N(CH3)3
положении 6
Миноциклин н СНз он NHCH3
а Другие названия этого антибиотика—биомицин и ауреомиции.— Прим, перев.
Большую группу антибиотиков, содержащих в своей струк-
туре макроциклический лактоновый фрагмент и продуцируе-
Фармацевтические препараты
413
мых различными штаммами Streptomyces, составляют так назы-
ваемые макролиды. Наиболее известным представителем этой
группы является эритромицин (30) (т. 4, стр. 209), который по
спектру антибактериального действия близок к пенициллину и
применяется для лечения больных с повышенной чувствитель-
ностью к последнему. Особенно эффективен эритромицин при
лечении дифтерии.
30
Линкомицин (31) не относится к макролидам, но по спектру
биологической активности приближается к ним. Фузидиевая
кислота * (32), как сообщается, исключительно эффективна при
лечении стафилококковых инфекций. Имея стероидную структу-
ру, она тем не менее отличается от типичных стероидных гормо-
нов транс-син-транс-анти-транс-расиоложением колец в простран-
стве и поэтому не обладает гормональными свойствами.
Противотуберкулезные и противолепрозные
средства
Возбудителями туберкулеза и проказы являются микобак-
терии. Первое место среди противотуберкулезных лекарств
* В СССР натриевая соль фузидиевой кислоты (фузидат натрия) произ-
водится под названием «фузидин». — Прим, перев.
414
Глава И
занимает изониазид (33), сочетающий высокую активность, ма-
лую токсичность, дешевизну и возможность употребления в
форме таблеток. Это соединение легко синтезируется из у-пико-
лина. СН3 СООСНз CONHNH2 । 1 1) окисление г^\] H2NNH2 2) этерификация N N N
33
Второе по значению место принадлежит этионамиду (34) и
его гомологу протионамиду (35), которые родственны изониази-
ду по химическому строению, но не совпадают с ним по рези-
стентным штаммам (т. е. способны подавлять развитие микро-
бов, устойчивых к изониазиду).
34
Еще одно противотуберкулезное средство — пиразинамид
(36), тоже очень похожее по химической структуре на изониа-
зид, получают из хиноксалина. Туберкулезные палочки быстро
приобретают лекарственную устойчивость, и для борьбы с этим
явлением часто применяют «комбинированную терапию», т. е.
смесь лекарств. Так, антибиотик стрептомицин (37) (т. 4,
38
Фармацевтические препараты 415
стр. 78), который исторически был первым эффективным средст-
вом для лечения туберкулеза, обычно используется в комбина-
ции с n-аминосалициловой кислотой (38). Главные достоинства
тиоацетазона (39) , обусловливающие его применение как проти-
вотуберкулезного средства, — относительная дешевизна и ле-
карственная эффективность в относительно небольших дозах.
Исходным веществом в синтезе тиоацетазона служит толуол.
S
HN^ '4nh2
N
СН3 СНз СНО сн^
no2 nhcoch3 NHCOCHs
39
Широкому изучению в качестве противотуберкулезного
средства подвергся антибиотик циклосерин (40). Оказалось, что
сам по себе он обладает недостаточной фармакологической
активностью, но в комбинациях с другими лекарственными пре-
паратами эффективен при лечении туберкулеза человека. Син-
тез этого соединения был проведен фирмой Merck по следую-
щей схеме:
/СООН , J СНзОН, НС1 | 1 2) С6Н5С(ОС2Н5)=МН > |- НО Ш2 Ох о | i^^NHOH НС1' Ox^N C6HS Н XNI N ск 0х Q на, сн3он ^NHCOC6H5 /СООСНз \ . NH2OH, CH3ONa 1 > 26Н6 О । r^^NHOH NaOH Cl NHCOC6H6 42 H N 4 ОСНз 1) NaOH , O' о L 2) Разделение 1 L ^NH2 изомеров 4NHj 40
416
Глава 11
При массовом обследовании различных соединений на проти-
вотуберкулезную активность на мышах ученые фирмы Lederle
Laboratories обнаружили, что диамин 41 приближается по ак-
тивности к стрептомицину. В дальнейшем было синтезировано
большое число аналогов этого диамина, причем оказалось, что
активность полученных соединений зависит от их способности
к хелатированию с металлами. Хелатирование может быть как
прямым (происходящим, например, путем взаимодействия с
металлосодержащим ферментом), так и непрямым, т. е. облег-
чающим лишь транспорт лекарства в организме. Коррелирова-
ние активности различных аминов с их способностью к хелати-
рованию показало, что наибольший'интерес должны представ-
лять более «открытые» хелаты, содержащие концевые гидро-
ксильные группы. Такие хелаты были получены, и среди них
с помощью данного подхода был найден этамбутол (42). Пора-
зительна стереоспецифичность бактериостатического действия
(CH3)2CHNH(CH2)2NHCH(CH3)2 C2HS\ ^/NH(CH2)2NH\ /C2Hs НС1
CH CH
CH2OH CH2OH
41 42
этого вещества: d-изомер в 12 раз более активен, чем мезо-изо-
мер и в 200—500 раз превосходит по активности /-изомер.
Проказа тоже является инфекционным заболеванием, вызы-
ваемым микобактериями. Средства, применяемые для ее лече-
ния, эффективны также в случае туберкулеза, хотя и не подхо-
дят для этой цели в клинических условиях. Типичное противо-
лепрозное средство дапсон * (43) синтезируют из хлорбензола
и хлорсульфоновой кислоты
* В СССР производится под названием «диафенилсульфон». — Прим, пе-
рев.
Фармацевтические препараты
417
Антисептики
Вещества, убивающие микробов при контакте с ними, назы-
вают антисептиками, дезинфицирующими средствами и герми-
цидами. Лекарственные препараты этого типа применяются как
местные противомикробные средства; кроме того, они должны
обладать такими свойствами, как отсутствие раздражающего
действия на живую ткань, красящей способности и др. Герми-
цидное действие обнаруживают многие простые соединения,
например этанол, формальдегид и иод. В настоящее время
выпускаются антисептики, специально предназначенные для
различных потребителей (больниц, животноводческих ферм, до-
машнего обихода, ветеринарных лечебниц и т.п.), причем спрос
на те или иные препараты часто определяется не их бактерицид-
ной активностью, а составом, качеством упаковки и расходами
на рекламу.
В 1867 г. Листер впервые использовал фенол в антисептиче-
ской хирургии, и применение фенолов как антисептиков продол-
жается до сих пор. Так, резорцин входит в состав шампуней, а
хлорксиленол (44) и гексахлорофан (46), который получают из
2,4,5-трихлорфенола (45), являются общими антисептиками.
Природный фенол тимод (47), а также тригалогенфенолы при-
меняют в составе таблеток и полосканий для горла.
Другим классом органических антисептиков являются
четвертичного аммония. Наиболее важными из них можно
тать цетримид (48) и цетилпиридинийбромид (49)
СОЛИ
счи-
СНз
к
СН3(СН2)13—N—СН3 Вг“
СН3(СН2),5—N
Вг“
СН3
48
40
14 Зак. 501
418
Глава 11
Широкое применение в качестве антисептика находит также
хлоргексидин (50), который синтезируют из дихлоргидрата
гексаметилендиамина и дицианамида натрия
Противовирусные средства
Несмотря на то что за последние 20 лет наши знания о
вирусах значительно обогатились, прогресс в создании новых
противовирусных средств невелик. З-Тиосемикарбазон N-метил-
изатина (51) полезен для профилактики натуральной оспы и
показывает обнадеживающие результаты против вируса ко-
ровьей оспы. Однако два его близких аналога — соединения 52
и 53 — не обладают противовирусной активностью. Адамантан-
амин (54) препятствует размножению некоторых штаммов ви-
руса гриппа; кроме того, он активен против вируса краснухи
(и кори). Это соединение, созданное фирмой du Pont, было пер-
вым антивирусным веществом; его разработка обошлась по
меньшей мере в 20 млн. Долл. Были синтезированы и изучены
также многие другие алициклические амины. Сообщается, что
менее пространственно затрудненные амины, например соеди-
нение 55, обнаруживают активность против вируса гриппа у
Фармацевтические препараты
419
мышей. Для лечения вирусного лишая успешно использовался
2'-дезокси-5-иодуридин (56). Как и при изыскании противорако-
вых средств, развивается метод метаболической терапии вирус-
ных инфекций. Ряд аналогов природных пиримидиновых и пу-
риновых нуклеозидов был получен и исследован в качестве по-
тенциальных антиметаболитов, т. е. соединений, которые претер-
певают те же биохимические превращения, что и природное
соединение, но не способны выполнять функцию последнего (в
данном случае — участвовать в синтезе ДНК и РНК вируса).
В частности, были приготовлены соединения, содержащие в
своей структуре фрагменты адамантана и нуклеозида. Оказа-
лось, что фторуридин (57) эффективен против вируса коровьей
оспы in vitro, тогда как производное тимидина 58 лишено проти-
вовирусной активности.
Противогрибковые средства
Различают дрожжевые и плесневые грибки. Число различ-
ных видов грибков очень велико; одни из них являются полез-
ными, в то время как другие вызывают паразитарные заболева-
ния. Противогрибковую активность обнаруживают многие
фенолы, причем введение атомов галогена или алкильных групп
часто повышает ее (например, в случае соединения 59). В каче-
стве местных средств против некоторых заразных грибковых
болезней применяли также трифенилметановые красители
(т. 2, стр. 249—253; см. также гл. 10), такие, как малахитовый
зеленый (60), гентиан фиолетовый и фуксин-основание; однако
все эти соединения имеют явные недостатки. Действующим
14*
420
Глава 11
началом ряда противогрибковых средств служит 8-оксихинолин
(61), имеющий бактериостатические’и фунгицидные свойства.
Его активность объясняют способностью этого вещества к хела-
тированию с металлами, так как изомерные ему оксихинолины
неактивны. Галогенирование приводит к увеличению противо-
грибковой активности, как, например, в случае соединения 62.
R
СНО
59
N(CH3)
ОН
60 61 (R = R' = H)
62 (R = Cl, R' = I)
В качестве средств для борьбы с грибковыми болезнями сельско-
хозяйственных кулътур уже длительное время используют
неорганические соли тяжелых металлов (серебра, двухвалентной
ртути, кальция и цинка); органические соли этих металлов, на-
пример каприлат цинка (63), ундецен-10-оаты цинка (64) и
меди и этилмеркуртиосалицилат натрия (65), обнаруживают
COONa
/ S HgC2HE
[CH3(CH2)eCOO]2Zn [CH2=CH(CH2)8COO]2Zn f J
63 64 ' 65
избирательность действия и могут быть использованы против
некоторых инфекций животных и человека. В течение 40 лет
широкое применение как сельскохозяйственные фунгициды
находят различные дитиокарбаматы, в частности диметилдитио-
карбамат натрия (66) (гл. 12). Широкая противомикробная
активность, характерная для соединений этого класса, позволи-
ла создать препарат толнафтат (67), который используется в
качестве местного противогрибкового средства. Перекись лауро-
ила (68) применяется против грибка Epidermophyton, вызываю-
щего грибковое заболевание ног.
66
67
68
Фармацевтические препараты
421
Однако первое место среди противогрибковых химиотерапев-
тических средств принадлежит антибиотикам. Противогрибковой
активностью обладают многие полиеновые антибиотики, содер-
жащие большой лактоновый цикл с несколькими сопряженными
двойными связями, но синтез столь сложных соединений вызы-
вает значительные трудности. Примером антибиотиков данного
типа может служить пимарицин (69).
в»
Большая работа была проделана по изучению гризеофульви-
на (70), открытого в 1939 г. (т. 4, стр. 177). При приеме внутрь
(но не при местном применении) он эффективно вылечивает
грибковые заболевания человека. Изящный и простой метод
стереоспецифического синтеза рацемата этого соединения был
разработан Шторком и Томашем (т. 4, стр. 179).
Циклогексимид (71) является продуктом жизнедеятельности
Streptomyces griseus и обладает фунгистатическим (но не бак-
териостатическим) действием. Он широко применяется для
уничтожения патогенных грибков, вызывающих болезни расте-
ний; однако использовать этот антибиотик в фармакотерапии
болезней человека не удалось.
Противораковые средства
Рак представляет собой группу заболеваний,, поражающих
различные органы и функциональные системы организма и
422
Глава 11
сопровождающихся неуправляемым, аномальным делением кле-
ток, скорость которого обычно во много раз превышает ско-
рость деления здоровых клеток. Начало противораковым химио-
терапевтическим препаратам положили производные азотистого
иприта, антиметаболиты (антагонисты) фолиевой кислоты и
стероидные гормоны, введенные в медицинскую практику в
40-х годах.
Большую группу лекарственных препаратов, механизм дей-
ствия которых неизвестен (а таких очень много), составляют
алкилирующие агенты. Первым и наиболее известным из них
является азотистый иприт (72). Среди сотен соединений, синте-
/СНгСНгС!
CH3lZ
\ch2ch2ci
72
зированных и изученных в качестве потенциальных противо-
раковых средств после открытия азотистого иприта, некоторые
успешно прошли клинические испытания. Механизм действия
алкилирующих агентов до сих пор остается дискуссионным, что
обусловлено весьма высокой и многообразной реакционной спо-
собностью этих соединений, затрудняющей их исследование.
Многие данные свидетельствуют о том, что наиболее чувстви-
тельным к ним компонентом клетки является ДНК и что на-
чальная атака протекает в положение 7 ее гуанинового фраг-
мента с последующим расщеплением молекулы ,
Фармацевтические препараты
423
Наиболее активные алкилирующие агенты — бифункциональ-
ные соединения, что объясняется их способностью сшивать
молекулы ДНК- В числе очень эффективных веществ данного
типа следует назвать азиридины, один из которых — триэтиле-
нимино-сши.и-триазин (73) был первым алкилирующим аген-
том, пригодным для перорального введения. Были изучены
этилениминовые производные органических соединений, широко
различающихся по форме и размеру молекулы; особенно
большое значение приобрели найденные этим методом этилени-
миды кислот фосфора, например тиоТЭПА * (74).
73
Антиметаболиты препятствуют образованию или дальнейше-
му превращению нормальных клеточных метаболитов, инакти-
вируя определенный фермент (ферменты), либо подменяя один
из участков молекулы белков или нуклеиновых кислот. Весьма
подробно были исследованы, в частности, пиримидиновые и пу-
риновые антиметаболиты. Так, 5-азаурацил (75) и 5-азаорото-
вая кислота (76) способны подавлять развитие аденокарциномы
(злокачественного роста клеток кожного покрова), но неактив-
ны против лейкемии. Оба эти соединения инактивируют фер-
мент, ответственный за превращение оротовой кислоты (77) в
оротидиловую (78). 8-Азагуанин (79), представляющий собой
О
II
H№'ZXN
Н
75 (R = Н)
76 (R = СООН)
О
о N \соон
рибофосфат
78
аналог гуанина, в котором углеродный атом заменен на атом
азота, был первым (1949 г.) антиметаболитом нуклеиновых кис-
лот, обнаружившим выраженную противоопухолевую актив-
ность. Этот пурин сам по себе не является активным нача-
лом; было показано, что сначала он должен превратиться в
перев.
* В СССР употребляются названия «тиофосфамид» и «тиоТЭФ». — Прим.
424
Глава 11
трифосфат 80, который, по-видимому, внедряется в молекулу
информационной РНК и препятствует росту клеток. Фермент
гуаназа переводит 8-азагуанин (79) в 8-азаксантин (81), не об-
ладающий противоопухолевыми свойствами. Отсюда следует, что
опухоли с низким содержанием гуаназы должны быть весьма
восприимчивыми к 8-азаксантину; поскольку в действительности
это не так, напрашивается вывод о сложной природе противо-
опухолевой активности рассмотренных соединений.
81
Изучение аминокислотных антиметаболитов не привело к
получению обнадеживающих результатов. Этионин (82)—этиль-
ный аналог метионина — подавляет рост некоторых опухолей
крыс, но неэффективен против опухолей человека. То же самое
относится к селенсодержащему аналогу цистеина 83 и аналогу
тирозина бетазину (84).
C2H5SCH2CH2CHCOOH HSeCH2CHCOOH I |] [
I I nh2
NH2 NH2 mu I 2
I
82 83 - 84
Типичным представителем антагонистов метаболитов, участву-
ющих в синтезе нуклеиновых кислот, является азасерин (85),
который первоначально был выделен из фильтрата культуры
Streptomyces, а позднее получен синтетическим методом. Это
соединение препятствует протеканию биохимических процессов
с участием глутамина (86).
85
86
Фармацевтические препараты
425
Важное значение имеют антагонисты фолиевой кислоты
(т. 4, стр. 419, 436—437), поскольку эта кислота входит в состав
коферментов, участвующих на трех стадиях биосинтеза нуклеи-
новых кислот. Такие мощные антифолиевые агенты, как амино-
птерин (87) и его N-метильный аналог метотрексат (88), обла-
дают значительной противораковой активностью, обусловленной
способностью этих соединений препятствовать внутриклеточно-
му процессу восстановления фолиевой и дигидрофолиевой кис-
лот в тетрагидрофолиевую кислоту.
О СООН
87 (R = H)
88 (R = СН3)
Кроме перечисленных выше веществ, были изучены некото-
рые витамины и антагонисты коферментов. В опытах на лабора-
торных животных наблюдалось подавление роста опухолей,
однако найти препараты, эффективные в клинических условиях,
не удалось. Так, производное (89) рибофлавина (т. 4, стр. 14 и
419) хорошо зарекомендовало себя в опытах на мышах, но при
исследовании в стандартной системе с Lactobacillus easel не
проявило свойств типичного антагониста рибофлавина.
Противопаразитарные средства
Паразитарные заболевания человека особенно распростра-
нены в тропических и субтропических странах, где они уносят
миллионы человеческих жизней. Паразитарные болезни живот-
ных встречаются во всех странах мира и- наносят большой эко-
номический ущерб сельскому хозяйству.
Противомалярийные средства
Борьба с малярией является сложной, но чрезвычайно важ-
ной проблемой, так как в районах мира, где не проводятся
426
Глава 11
специальные противомалярийные мероприятия, проживает около
400 млн. человек. Первым лекарством от малярии были измель-
ченные корни китайского растения Dichroa febrifuga, действую-
щим началом которых служит алкалоид фебрифугин (90). Из
коры хинного дерева добывают другое природное противома-
лярийное средство — хинин (91) (т. 4, стр. 376—379). Захва-
90
тывающая история этого лекарственного препарата восходит к
1630-м гг., а химическое строение хинина окончательно дока-
зано в 1944 г., когда Вудворд и Дёринг провели его синтез. Во
время первой мировой войны пути подвоза хинина с Явы и Су-
матры оказались перерезанными, и немецкие химики начали
интенсивные поиски других противомалярийных средств, ис-
пользуя в качестве исходного вещества краситель метиленовый
голубой (92), который, как было обнаружено, обладает
N
92
некоторой противомалярийной активностью. Изучение структур-
ных аналогов метиленового голубого показало, что необходи-
мым условием высокой активности является наличие боковой
(диалкиламиноалкил) аминогруппы, и в 1932 г. был получен
первый синтетический противомалярийный препарат памаквин *
(93)
Другое название этого лекарства — плазмохин. — Прим, перев.
Фармацевтические препараты
427
HN—CH(CH2)3N(C,H6)2
I
СНз
93
Поскольку памаквин токсичен и неэффективен при острых про-
явлениях малярии, он был вскоре заменен мепакрином * (94),
который широко использовался вплоть до второй мировой войны.
В начале второй мировой войны большое внимание при-
влекли противомалярийные свойства 4-аминохинолинов. Среди
соединений этой группы был найден один из наиболее важных
противомалярийных препаратов — хлорохин** (95), который
можно синтезировать из л-хлоранилина
но~
* В СССР соответствующий препарат называется акрихином. — Прим,
перев.
** В СССР выпускается под названием «хингамин». — Прим, перев.
428
Глава 11
H2NCH(CH2)3N(C2H5)2
СН3
сн3
NHCH(CH2)3N(C2HS)2
Интересную группу противомалярийных средств составляют
бигуаниды, разработанные Курдом и Розе (фирма ICI) во время
второй мировой войны. Представителем этой группы является
прогуанил * (96). В организме он превращается в дигидроди-
азин 97, также обладающий антималярийной активностью. По-
следний весьма близок по химическому строению более доступ-
ным 4-амино-5-арилпиримидинам, что привело к получению пи-
риметамина ** (98)—наиболее важного лекарственного пре-
парата в ряду бигуанидов.
CN
+ СНзСН2СООС2Н5
CaHsONa
------->
CH2N2
* В СССР принято название «бигумаль». — Прим, перев.
** В СССР выпускается под названием «хлоридин». — Прим, перев.
Фармацевтические препараты
429
HN=C(NH2)2
---------->
98
В последние годы все чаще приходится сталкиваться с ре-
зистентностью плазмодиев к противомалярийным лекарствам,
что особенно ярко показала война во Вьетнаме. По этой при-
чине вновь пробудился интерес к хинину. В 1969 г. фирма Roche
объявила о разработке промышленного метода синтеза хинина.
Противоамебные средства
Амебная дизентерия (амебиаз) зачастую считается тропиче-
ской болезнью. Однако, хотя это заболевание чаще встречается
в странах с теплым климатом, оно отнюдь не ограничивается
ими. Возбудитель амебной дизентерии Entamoeba histolytica
разносится по организму человека из кишечника и внекишеч-
ных органов. Обнаружение специфических противоамебных
свойств у эметина (99) (т. 4, стр. 388—390) стимулировало
поиск других, более эффективных и менее токсичных препара-
тов родственной структуры. Из них заслуживает упоминания
только 2,3-дидегидроэметин. Описано несколько методов пол-
ного синтеза соединений этой группы.
Для лечения амебной дизентерии был использован также
конессин (100), выделенный из коры Holarrhena antidysenterica.
Бартон и Джонсон разработали два интересных метода полного
синтеза этого вещества.
Эффективность против амебиаза животных и человека про-
являют многие противомикробные антибиотики. К ним отно-
сятся хлортетрациклин (29), эритромицин (30) и окситетра-
430
Глава 11
циклин (29), а также недавно появившийся парамомицин *
(101). Кроме того, существует большое число синтетических ле-
карственных веществ, обладающих амебоцидными свойствами.
Однако сами по себе они во многих случаях оказывают побоч-
ное токсическое действие на организм. Поэтому их применяют
главным образом против резистентных штаммов амеб (иногда
в комбинации с эметином). Для лечения амебной дизентерии
используются и иодпроизводные 8-оксихинолинов. Наиболее ста-
рый из этих препаратов — хиниофон (102) —был синтезирован
в 1892 г. и долгое время применялся как антисептик и «универ-
сальное лекарство от всех болезней», пока в 1921 г. не была
обнаружена его противоамебная активность.
В 1964 г. появились первые сообщения об использовании
в качестве эффективных средств против амеб и шистосом
(стр. 434) нитросодержащих гетероциклических соединений, та-
ких, как ниридазол (103.).
102 103
Хлорохин (95), противомалярийные свойства которого были
описаны выше, находит также широкое применение для лечения
амебных инфекций.
Трипаноцидные средства
К числу трипаносомозов относятся африканская «сонная бо-
лезнь», вызываемая укусом мухи це-це, и южноамериканская
* В СССР идентичный антибиотик выпускается под названием «мономи-
цин». — Прим, перев.
Фармацевтические препараты
431
болезнь Чага, переносчиком которой является клоп триатомида.
Эти болезни очень опасны и поражают как человека, так и жи-
вотных.
Разработанный Эрлихом метод избирательного окрашива-
ния паразитических микроорганизмов различными красителями
позволил ему изучить сотни соединений и добиться замечатель-
ных успехов. Так, было установлено, что трипановый синий
(104) и афридол фиолетовый (105) обладают неплохой трипано-
цидной активностью; однако недостаток этих лекарств заклю-
чается в окрашивании тканей и жидкостей в организме «хозяи-
на». Исследователи фирмы Bayer предположили, что если дей-
ствие красителя обусловлено его фиксацией паразитом, то такой
же лекарственный эффект должны обнаруживать соединения,
обладающие высоким сродством к паразиту, но не содержащие
хромофорной азогруппы. Настойчивые восьмилетние поиски
увенчались в 1920 г. созданием препарата сурамин (106).
106
Размножаясь в животном организме, трипаносомы потреб-
ляют огромные количества глюкозы, что приводит на последних
стадиях болезни к возникновению гипогликемии. В попытке ли-
шить паразитов пищи (рациональный подход к химиотерапии
трипаносомозов) был использован сахароснижающий агент
432
Глава 11
синталин А (Ю7), который оказался эффективным препаратом.
Однако в опытах in vitro было показано, что это вещество может
действовать на трипаносомы не только через организм «хозяи-
на», но и непосредственно. В дальнейшем был синтезирован
ряд ароматических диамидинов, один из которых — пентамидин
(Ю8) — до сих пор находит клиническое применение.
Н Н
107
108
Трипаноцидные свойства обнаружены также у некоторых
акридинов, причем активность четвертичных аммониевых произ-
водных этих соединений выше. Изомерные им фенантридины
содержат хинолиновый и изохинолиновый фрагменты, обуслов-
ливающие высокую лекарственную эффективность веществ
этого класса. Один из них — димидий (109), синтезируемый на
основе 2-аминобифенила (см. схему на стр. 433).
В поисках более доступных соединений были изучены неко-
торые производные 4-аминохинолина. Наиболее значительным
результатом этой работы было создание хинапирамина (ПО),
Фармацевтические препараты
433
получаемого аналогично противомалярийным хинолиновым пре-
паратам.
POCI3. CeH6NO2
CeHs Вг_ СдНа
NH2
109
Естественно, что многие антибиотики тоже были обследо-
ваны на трипаноцидную активность. Оказалось, что широким
спектром действия при трипаносомозах обладает пуромицин
(111), синтез которого был позднее (в 1954 г.) осуществлен
Бейкером (фирма Lederle). Недостатком этого лекарственного
препарата является быстрое развитие резистентности парази-
тов по отношению к нему.
Противоглистные средства
Противоглистными (антигельминтными) средствами назы-
ваются вещества, которые убивают глистов (гельминтов) или
выводят их из организма. Из трех основных групп глистов —
нематод, трематод и солитеров — последние привлекают наи-
меньшее внимание, так как наносимый ими ущерб с точки зре-
ния медицины и экономики относительно невелик.
Для удаления солитеров используется экстракт из мужского
папоротника Aspidum oleoresin, активным компонентом которого
считают филиксиновую кислоту (112). Были получены также
434
Глава 11
синтетические аналоги этого соединения, но ни один из них не
дошел до стадии клинических испытаний.
112
Шистосоматоз — самое распространенное после малярии па-
разитарное заболевание человека в тропических и субтропиче-
ских странах. Идеальным решением проблемы шистосоматоза
было бы полное уничтожение улиток, которые служат перенос-
чиками шистосом, и Всемирная организация здравоохранения
уже разработала соответствующую программу. Однако, пока
она не проведена в жизнь, необходимо выявлять и лечить боль-
ных шистосоматозом. Первым веществом, обладающим актив-
ностью при лечении человека, был рвотный камень (ИЗ), ко-
торый наряду с другими соединениями трехвалентной сурьмы
до сих пор продолжает оставаться одним из наиболее эффек-
тивных лекарств. Из препаратов, не содержащих металла, ста-
рейшим является люкантон (114), обнаруживший при клиниче-
ских испытаниях хороший лекарственный эффект. Попытки
найти в ряду его структурных аналогов более активные соеди-
нения до последнего времени были безуспешными, однако ис-
следование метаболизма люкантона привело к созданию гикан-
тона (115), который, судя по предварительным результатам, об-
ладает высокой эффективностью при шистосоматозе у человека.
COOK
Sb
I
он
114 (R = H)
115 (R = ОН)
113
Шистосомы представляют собой разновидность плоских чер-
вей-сосальщиков (трематод), питающихся кровью человека.
Они вызывают глистные печеночные инфекции у человека и до-
машних животных, которые очень опасны для здоровья и нано-
сят большой экономический ущерб. Переносчиками трематод
являются улитки. К счастью, для борьбы с трематодами суще-
ствует несколько химиотерапевтических препаратов. При гель-
Фармацевтические препараты
435
минтозах у овец и крупного рогатого скота используются четы-
реххлористый углерод и гексахлорэтан; кроме того, недавно
появились в продаже салициланилидные противоглистные пре-
параты, например оксиклозонид (116). Свыше 30 лет не выходит
из употребления фенотиазин (117), который является дешевым
промышленным продуктом, получаемым при взаимодействии
дифениламина с серой в присутствии каталитического количе-
ства иода
117
В качестве средств против глистных инфекций, вызываемых
круглыми патогенными червями (нематодами), в настоящее
* время имеется несколько- сильнодействующих препаратов. Од-
ним из них является тиабендазол (П8), синтезированный в
1961 г. химиками фирмы Merck по следующей схеме:
СООСНэ
yg нагревание /?'-/ "ОН
нс^- + ВгСН2СОСООСН3-------------»• С I]
^НН2 S"
о-СгЩЩНЩ
436
Глава 11
Выдающимися антигельминтными свойствами обладает тет-
рамизол (121), открытый бельгийским ученым Янссеном. Родо-
начальником этого вещества послужило привлекшее к себе вни-
мание производное тиофена 119, которое дало хорошие резуль-
таты на овцах и домашних птицах, но оказалось неактивным
NaBH<
Синтез тетрамизала
при гельминтозах у обычных лабораторных животных — крыс
и мышей. Отсюда был сделан вывод, что соединение 119 не об-
ладает противоглистной активностью, но способно к метаболи-
тическому превращению в активное лекарственное вещество и
что описанная выше его специфичность для различных живот-
ных объясняется тем, что в организме овец и домашних птиц
полезная биотрансформация соединения 119 происходит, а в ор-
ганизме крыс и мышей — нет. В конечном итоге его метаболит
120 был выделен и обнаружил высокую активность, которую
NCOCH;
он
СН2\ I
ОН
120
,---N'
119
удалось еще более увеличить путем замены тиенильной группы
на фенильную (соединение 121). Тетрамизол оптически активен,
причем биологическая активность присуща лишь /-изомеру.
В настоящее время освоена технология разделения образующе-
гося при синтезе рацемата с рециркуляцией неактивного (/-изо-
мера.
Еще одним эффективным противоглистным препаратом яв-
ляется пирантел /122), синтезированный химиками фирмы
Фармацевтические препараты
437
Pfizer по следующей схеме:
+ СН2(СООН)2
с ^сно
о
S
СН,
I) SOC12
2) NH3
Антикокцидиозные средства
Кокцидиоз — паразитарное заболевание, поражающее боль-
шинство животных и птиц, и борьба с ним имеет огромное зна-
чение для обеспечения высокой продуктивности птицеводства.
Вначале кокцидиоз лечили серой или бурой; однако первыми
эффективными антикокцидиозными средствами были сульф-
амиды, которые продолжают играть важную роль и в настоящее
время. Чаще всего применяются сульфахиноксалин (123) и
сульфадимидин (11).
Поскольку сульфамиды при использовании их в качестве
противомикробных и антикокцидиозных агентов ведут себя как
антагонисты n-аминобензойной кислоты, можно было ожидать,
что другие антагонисты этой кислоты тоже будут обладать кок-
цидиоцидными свойствами. Действительно, пириметамин (98)
и дигидродиазин (97) обнаруживают активность против кокци-
дий. Хороший профилактический (но не терапевтический) эф-
фект показали и другие антагонисты витаминов. Так, высокий
терапевтический индекс ампролия (124) обусловлен тем, что это
соединение действует как антагонист тиамина (125).
123
124
438
Глава 11
125
Очень мощными антикокцидиозными агентами являются не-
которые хинолинон-4(1Н)-3-карбоновые кислоты, в частности
биквинолат (126) и метилбензокват (127). Эти вещества, не об-
ладающие противомикробными свойствами, близко родственны
127
по своей структуре антибактериальному препарату налидиксо-
вой кислоте (17), которая неактивна при кокцидиозе.
Средства для лечения неинфекционных
заболеваний
Противозачаточные средства
В 1957 г. Рок, Пинкус и Гарсиа детально исследовали
механизм задержки овуляций стероидами, угнетающими мен-
струальный цикл во время беременности (так называемыми
гестагенами, или гестагенными агентами), и установили, что
искусственное введение этих стероидов позволяет затормозить
наступление овуляции на сколь угодно большой срок. В резуль-
тате данной работы были созданы и выпущены в продажу сте-
роидные пероральные противозачаточные средства (известные
в обиходе под собирательным названием «пилюли»), которые
получили широкое распространение во всем мире и являются
одним из важнейших достижений современной медицины. Неко-
торым из первых стероидов, изученных Роком, Пинкусом и Г ар-
ена, была присуща собственная эстрогенная активность, тогда
как другие стероиды содержали примеси эстрогенных соедине-
ний. Это наблюдение привело к выводу, что присутствие эстро-
гена вносит существенный вклад в противозачаточный эффект
препарата. В настоящее время в продаже имеется ряд комбини-
рованных гестаген-эстрогенных препаратов, надежно предохра-
Фармацевтические препараты
439
няющих от беременности, что позволяет выбирать те или иные
таблетки, руководствуясь только характером их побочного дей-
ствия. В качестве эстрогенных компонентов пероральных проти-
возачаточных средств, как правило, используются этинилэстра-
диол (128) или его метиловый эфир местранол (129), которые
синтезируют из эстрона.
132 (R = COCH3)
Два из наиболее важных противозачаточных агентов — нор-
этистерон (131) и норэтинодрел (130) — можно получить из эс-
трона методами, наглядно иллюстрирующими значение защит-
ных групп и селективных реакций в химии стероидов. Кроме
440
Глава 11
этих веществ, весьма распространенными противозачаточными
средствами являются норэтистерон-ацетат (132), линэстренол
(133) и этинодиол-диацетат (134). Стероиды, не содержащие
метильной группы в положении 19 (норстероиды), лишены анти-
овуляторной активности, тогда как соединения, более сходные
по структуре с прогестероном (т. 4, стр. 261, 262), например ме-
токсипрогестерон-ацетат (135) и мегестрол-ацетат (136), нахо-
дят применение в акушеро-гинекологической практике. В отли-
чие от них хлормадинон (137) был изъят из обращения, так
Некоторые другие противозачаточные агенты
как он вызывал появление опухолей у собак. Полученные в по-
следнее время данные о влиянии пероральных противозачаточ-
ных средств на частоту тромбозов у молодых женщин стимули-
ровали поиск нестероидных препаратов, однако до сих пор ни
один из них еще не поступил в продажу. Методы введения ме-
тильной группы или атома хлора в положение 6 обсуждались
в т. 4 (стр. 266—267).
Стероидные противовоспалительные
средства
В ходе работы по поиску новых гормонов, проводившейся
группой Кендаля в Mayo Clinic (шт. Миннесота, США), из экс-
трактов коры надпочечника было выделено пять соединений,
одно из которых — кортизон (139) —обнаружило гормональную
активность в опытах на крысах. Практическое использование
биологического действия кортизона тормозилось труднодоступ-
ностью этого вещества. Поэтому в 1942 г. семь групп американ-
ских химиков и еще несколько групп в Швейцарии начали раз-
Фармацевтические препараты
441
рабатывать методы синтеза кортизона с целью его получения
в количествах, достаточных для детального исследования. Ис-
ходным соединением чаще всего служила дезоксихолевая кис-
лота (одна из желчных кислот) (138), которая в синтезе корти-
зона претерпевает ряд превращений, в том числе перемещение
атома кислорода из положения 12 в Положение 11
Синтез кортизона
В 1949 г. было описано использование кортизона для лече-
ния ревматоидного артрита — болезни, сопровождающейся вос-
палением суставов, которая причиняет больному мучительные
боли и делает его инвалидом. Естественно, что это сообщение
повлекло за собой настойчивые поиски других стероидов, обла-
дающих противовоспалительной активностью. Сразу же встал
вопрос о неудовлетворительности методов синтеза кортизона,
базирующихся на желчных кислотах, поскольку даже лучший
из них включал 30 стадий и обеспечивал общий выход целевого
продукта менее 1%. Предписываемая суточная доза для боль-
ных артритом составляла 100 мг, причем кортизон вызывал
лишь ослабление ревматоидных симптомов, а не полное излече-
ние, что обусловливало необходимость в длительном примене-
нии этого препарата. Отсюда со всей очевидностью вытекала
острая необходимость в разработке альтернативных путей син-
теза кортизона, не требующих использования желчных кислот.
Один из мыслимых подходов заключался во введении кислорода
в положение 11 сравнительно доступного стероида, не имею-
щего заместителя в кольце С (например, прогестерона). В ка-
честве другого подхода рассматривался полный синтез (вряд ли
осуществимый в больших масштабах) и последующее превра-
щение в кортизон гекогенина (140), который в то время был
труднодоступным соединением, но вскоре перестал быть дефи-
цитным. В конечном итоге удалось реализовать оба эти подхода,
но в основу промышленного производства кортизона бы.) поло-
жен метод, базирующийся на использовании прогестерона, успех
442
Глава 11
которого стал возможным благодаря открытию неожиданной
реакции микробиологического окисления стероидов в положе-
ние 11. Окисление по атому С-11 и восстановление стероидов
экстрактами дрожжей впервые наблюдали в 1937 г., а в 1949 г.
удалось осуществить биохимическое введение атома кислорода
в положение И. Последнюю реакцию проводили путем пропу-
скания сыворотки, содержащей кортексон (141), через изолиро-
ванную надпочечную железу — методом, едва ли пригодным для
промышленного производства. Значительный шаг вперед сде-
лала фирма Upjohn (г. Каламазу, шт. Мичиган, США), зани-
мавшаяся целенаправленным поиском почвенного микроорга-
низма, способного окислять органические соединения. Исследо-
ватели этой фирмы установили, что культура Rhizopus arrhizus,
выделенная после оставления чашки с питательным агаром на
одном из лабораторных столов у окна, превращает прогестерон
(142) в 11 а-оксипрогестерон (143) с выходом, который впослед-
141 142 (R = H); 143 (R = ОН)
ствии удалось довести до 50%. При использовании Phizopus
nigricans были достигнуты более высокие выходы, а поскольку
Фармацевтические препараты
443
прогестерон очень просто получается из диосгенина (144) (т. 4,
стр. 259, 263), легко добываемого из. недефицитного природного
сырья, данный метод окисления позволил разработать удобный
четырехстадийный процесс синтеза кортизона.
Кортикостероиды обладают широким спектром биологиче-
ской активности, и небольшие концентрации этих веществ, под-
держиваемые надпочечной железой, регулируют ряд метаболи-
ческих процессов. Длительный прием больших доз кортизона
часто сопровождается серьезными побочными явлениями, та-
кими, как атрофия кожи, повышение артериального давления и
гипергликемия (увеличение содержания сахара в крови). Это,
а также стремление повысить эффективность действия лекар-
ства, побудило химиков-фармацевтов синтезировать значитель-
ное число аналогов кортизона. 9а-Фторкортизон (145) оказался
почти в 10 раз активнее кортизона; однако он так сильно тор-
мозит водно-солевой обмен, что в настоящее время его терапев-
тическое использование основано главным образом на этом
свойстве (такие вещества называются минералокортикоидами).
В дальнейшем на первое место выдвинулись преднизон (146) и
преднизолон (147). Эти на первый взгляд несущественные
изменения в структуре кортизона привели к семикратному уве-
личению противовоспалительного действия без повышения ми-
нералокортикоидной активности. Потенциальная вредность кор-
тизонотерапии в сочетании с тем обстоятельством, что прекра-
щение лечения влечет за собой новые трудности, привели к
тому, что среди врачей нет единого мнения относительно допус-
тимости длительного применения кортикостероидных препаратов.
444 Глава 11
Нестероидные противовоспалительные
. средства
Лекарственные вещества этого типа недавно вновь привле-
кли к себе внимание, когда химики предприняли поиск соедине-
ний, сравнимых по терапевтической активности с препаратами
группы кортизона, но не вызывающих характерные для корти-
костероидов побочные эффекты. Первые из синтезированных
ранее соединений, обнаруживших противовоспалительные свой-
ства, были названы анальгетиками-антипиретиками (т. е. боле-
утоляющими и жаропонижающими), однако в результате
дальнейших испытаний удалось разработать методику, позво-
ляющую отличать противовоспалительную активность от других
эффектов. Нестероидные противовоспалительные средства при-
меняют при ревматоидном артрите, суставном ревматизме и
остеоартрите, которые занимают среди болезней, ограничиваю-
щих физическую активность современного человека, второе
место после сердечно-сосудистых заболеваний. Соединение,
более эффективное при ревматоидном артрите, чем аспирин,
пока не найдено. По фармакологическому действию большинство
описанных ниже препаратов является антипиретиками и аналь-
гетиками; последние особенно полезны при лечении арт-
рита.
Салицин был введен в практику химиотерапии острого (сус-
тавного) ревматизма Маклагеном в Англии в 1874 г. Однако
даже сейчас, 100 лет спустя, механизм взаимодействия этого и
других лекарственных препаратов с воспалительным центром
остается неизвестным. Для ослабления симптомов ревматоид-
ного артрита предпочитают пользоваться салицилатами, а при
лечении суставного ревматизма часто применяют аспирин в ком-
бинации с каким-нибудь антибиотиком. Аспирин (ацетилсали-
циловую кислоту) синтезируют из фенола по реакции Кольбе —
Шмидта (т. 2, стр. 218). Несмотря на эффективность и большую
длительность действия этого препарата, его прием иногда
сопровождается побочными явлениями (такими, как желудоч-
но-кишечные кровотечения и аллергия), которые нельзя игно-
рировать. Указанные явления наблюдаются и при приеме «рас-
творимого» и «нейтрализованного» аспирина, а также аспирина,
покрытого оболочкой. Недавно фирма Merck сообщила о созда-
нии салицилатного препарата флюфенизал (148), который в
Фармацевтические препараты
445
148
4 раза превосходит аспирин по антивоспалительному действию
и меньше раздражает слизистую оболочку желудочно-кишечного
тракта. Выше приведена схема синтеза флюфенизала, включаю-
щая интересную реакцию элиминирования двуокиси серы.
Примерно такую же противовоспалительную активность про-
являет азотсодержащий аналог флюфенизала 149, тогда как
изомер 150 практически неактивен.
соон
соон
149
150
Пиразолоновые противовоспалительные агенты удобно под-
разделить на две группы: пиразолоны-5, примерами которых
могут служить антипирин (151) и амидопирин (152) , и имею-
щие большее терапевтическое значение пиразблидиндионы-3,5,
С6Н5
1) Н2
2) СН2О—HCOOH
HNOs
I
с6н5
151
СвН5
152'
445
Глава 11
важнейшим представителем которых является фенилбутазон *
(153). Клиническое применение амидопирина за последние годы
упало, поскольку стало больше известно о его побочных эффек-
тах; однако этот препарат все еще часто используется в качестве
эталона при оценке активности новых противовоспалительных
средств.
Интересная биологическая активность обладающего кислот-
ными свойствами фенилбутазона (153) была открыта случайно.
Фенилбутазон был известен в качестве анальгетика и антипи-
ретика, и вначале его применяли в комбинации с амидопири-
ном (152) как веществом основного характера в надежде, что
это приведет к повышению растворимости и увеличению эффек-
тивности препарата. В 1952 г. появилось сообщение, что про-
тиворевматическая активность такой комбинации больше, чем
у содержащегося в ней амидопирина, и что фенилбутазон сам
по себе поразительно хорошо снимает симптомы артрита. Ис-
ходным веществом в синтезе фенилбутазона служит гидразо-
бензол
153 (R = Н)
154 (R =ОН)
В организме фенилбутазон превращается в оксифенилбута-
зон (154), который не уступает ему по противовоспалительному
действию, но менее токсичен. Другой аналог — сульфинпира-
зон ** (155)—является превосходным средством против подаг-
ры. Замечена тесная взаимосвязь между величиной рКа этих
соединений и их биологической активностью.
CsH5SOCH2CH2\_____
Н0 N ^C6HS
I
c6Hs
155
Еще одну группу противовоспалительных средств составляют
производные антраниловой кислоты, которые можно рйссматри-
* В СССР производится под названием «бутадиен». — Прим, перев.
** В СССР выпускается под названием «антуран». — Прим, перев.
Фармацевтические препараты
447
вать как аналоги салицилатов. Исследователи фирмы Parke-
Davis описали три интересных соединения этой группы: мефе-
намовую кислоту (156), флюфенамовую кислоту (157) и пре-
парат CI-583 * (158). Наибольшей активностью среди них обла-
0.СООН 156 [R = 2',3'-(СН3)2С6Н3]
Г 157 (R = 3'-CF3C6H4)
^NHR 158 (R = 2',3'-С12СеН3)
дает соединение 158, которое, как сообщается, в 15 раз превос-
ходит по терапевтическому индексу фенилбутазон и в 150 раз —
аспирин.
Очень большой интерес и значительные исследовательские
силы привлекают сейчас гетероарилалифатические кислоты, об-
ладающие противоартритными свойствами. В 1963 г. химиками
фирмы Merck было открыто эффективное лекарство этой груп-
пы— индометацин (159). Толчком к синтезу индольных проти-
вовоспалительных средств послужил факт повышенного выде-
ления у больных ревматизмом метаболитов триптофана (160);
кроме того, в последнее время в литературе оживленно обсу-
ждается роль серотонина (161) в воспалительном процессе.
У препаратов ряда индометацина отчетливо наблюдается
связь между рКа и биологической активностью. При их дезаци-
лировании получаются практически неактивные соединения; за-
мена ц-хлорбензоильного остатка на ц-хлорбензильный сопро-
вождается некоторым понижением активности, тогда как в
случае деметилирования метоксигруппы снижение активности
выражено более сильно. Изучение родственных индометацину
2-замещенных индолилуксусных кислот (например, соединения
162) показало, что если разделить их на два оптических изо-
мера, то противовоспалительным действием будет обладать
только (+)-энантиомер. ц-Хлорфенильная группа в этих соеди-
нениях предпочтительно занимает ц«с-положение по отношению
к метоксифенильному фрагменту не вполне копланарного ин-
дольного ядра. Близкое к ним по структуре производное индена
* Синоним этого соединения — хлофенамовая кислота. — Прим, перев.
448
Глава 11
(.163) в 2 раза уступает индометацину по биологической актив-
ности, а его транс-изомер (164) в 10 раз менее активен, чем
соединение 162. Индометацин эффективен при лечении ревма-
тоидного артрита и применяется также при подагре и остеоар-
трите. Он вызывает такие побочные явления, как головная
боль, головокружение и расстройство желудка, которые, од-
нако, можно свести к минимуму, используя умеренные дозы.
Причиной возникновения интереса к арилуксусным кислотам
послужило открытие фирмы «Boots», что ибуфенак (165) в 2—
4 раза превосходит по силе действия аспирин. Тем не менее это
лекарство было изъято из обращения после обнаружения у при-
нимавших его больных признаков повреждения печени (токси-
ческого гепатита). Вместо ибуфенака было выпущено анало-
гичное ему а-замещенное производное — ибупрофен (166), у
165 (R = H)
166 (R = СН3)
которого в отличие от других а-замещенных производных про-
пионовой кислоты, обладающих противовоспалительными свой-
ствами, активны оба энантиомера. В течение ряда лет фирма
Syntex разрабатывает лекарственные препараты на основе за-
мещенных 1- и 2-нафтилуксусных кислот, одно из которых —
Фармацевтические препараты
449
соединение 167 — применяется в Великобритании начиная с
1973 г. Типичная схема получения этого препарата приведена
ниже:
167
Кроме индометацина, запатентовано большое число других
гетероциклических производных уксусных кислот, существенно
отличающихся от него по химическому строению. Фенклозино-
вая кислота (168) не выдержала клинических испытаний, так
как она вызывала те же побочные явления, что и ибуфенак. Лю-
бопытной особенностью этого соединения является его биохими-
ческое превращение в организме крыс в соединение 169 с мигра-
цией атома хлора в соседнее положение бензольного кольца.
15 Зак. 501
450
Глава 11
Данная реакция гидроксилирования n-замещенных бензола из-
вестна под названием «NIH-сдвиг», так как она была открыта
в Национальном институте здравоохранения США (National
Institutes of Health) (г. Бетесда, шт. Мериленд) Дейли с со-
трудниками в 1966 г.
Коагулянты и антикоагулянты
В основе нарушения кровообращения может лежать либо
тромбоз (образование внутрисосудистых сгустков крови), либо
кровоточивость (аномальная тенденция к постоянным кровоте-
чениям). В последнем случае применяют природные и синтети-
ческие стимуляторы свертывания крови. Наиболее важным при-
родным стимулятором является витамин К, химия которого
была описана в т. 4 (стр. 271).
Змеиные яды представляют собой смеси веществ, оказываю-
щих специфическое действие на факторы, участвующие в свер-
тывании крови. С помощью современных методов разделения
удалось выделить ряд компонентов яда, среди которых были
идентифицированы белки и полипептиды. Коагулирующие свой-
ства ядов некоторых змей нашли применение в терапии, и не
вызывает сомнения, что возможности этих превосходных лекар-
ственных препаратов выявлены еще далеко не полностью.
Интересным коагулянтом является карбазохром-салицилат
.(ПО) — комплекс моносемикарбазона адренохрома и салици-
СН3
170
лата натрия. Адренохром представляет собой продукт окисле-
ния важного в биологическом отношении амина адреналина.
Соединение 170 нетоксично и может вводиться перорально или
внутримышечно для остановки послеоперационного кровотече-
ния (например, после удаления миндалин).
Низкомолекулярный белок протаминсульфат, выделенный из
спермы некоторых рыб, обладает слабо выраженными анти-
коагулянтными свойствами. Он хорошо переносится (т. е. не
вызывает нежелательных побочных эффектов) и поэтому при-
меняется в медицинской практике уже свыше 20 лет.
е-Аминокапроновая кислота легко получается из ее лактама
(гл. 7), который служит важным полупродуктом в производстве
найлона и в свою очередь синтезируется из циклогексанонокси-
ма путем бекмановской перегруппировки. Эта кислота полезна
Фармацевтические препараты
451
для остановки сильных кровотечений.' Недавно были описаны
родственные ей соединения 171 и 172, разработанные соответ-
ственно фирмами Kabi (Швеция) и Merck. Первое из них в
10 раз активнее е-аминокапроновой кислоты, а второе — в
60 раз. Ряд препаратов обладает способностью ускорять свер-
н
171
соон
h2n
172
соон
тывание крови при их нанесении на кровоточащий участок.
Однако такие местные коагулянты не останавливают сильные
кровотечения. К числу веществ этого типа относятся желатино-
вая пена, окисленная целлюлоза, тромбин и многие препараты,
полученные из источников животного происхождения.
Антикоагулянты замедляют процесс свертывания крови. По
механизму действия и способу применения они подразделяются
на две большие группы: вещества прямого действия (группа
гепарина) и вещества непрямого действия (пероральные сред-
ства) .
Гепарин был открыт в 1916 г. одним из студентов медицин-
ского факультета Университета Джонса Гопкинса (г. Балти-
мор, шт. Мериленд, США). Он представляет собой сернокислый
эфир полисахарида, состоящего из чередующихся звеньев d-глю-
куроновой кислоты и 2-амино-2-дезокси-в-глюкозы. Наименьшая
молекула, еще сохраняющая антикоагулянтную активность,
имеет молекулярный вес около 8000. Мощным антагонистом
гепарина, т. е. агентом, оказывающим противоположное ему
действие, является протаминсульфат. Долгое время гепарин
считался идеальным антикоагулянтом, однако основной недо-
статок гепарина состоит в том, что его приходится вводить
путем инъекций, а это зачастую неудобно при длительном-курсе
лечения. Вслед за гепарином были открыты две группы анти-
коагулянтов, которые активны при пероральном введении. В на-
чале 20-х годов в шт. Северная Дакота (США) и в Канаде
была отмечена серьезная вспышка заболевания крупного рога-
того скота, поедавшего испорченное сено из сладкого клевера.
Токсичное вещество, вызывавшее у животных сильные крово-
течения, было идентифицировано Линком в 1939 г. Оказалось,
что это дикумарол * (174), 4-оксикумариновый фрагмент кото-
рого (173) можно легко синтезировать из метилсалицилата.
Последующая конденсация 4-оксикумарина с формальдегидом
приводит к образованию дикумарола. При взаимодействии
* В СССР выпускается как антикоагулянт под названием «дикумарин». —
Прим, перев,
15*
452
Глава 11
4-оксикумарина с бензилиден ацетоном получается хорошо из-
вестный ныне препарат варфарин (175), который весьма эф-
фективен в качестве крысиного яда, поскольку он вызывает
у крыс кровотечения, приводящие к летальному исходу.
CH3ONa
174
к антикоагулянтам второй группы относятся структурные
аналоги индандионов-1,3. В медицинской практике применяют
два соединения этой групгы — фениндион * (176) и дифена-
дион (177), которые синтезируют с помощью классических ме-
тодов.
о о
но но С6Н5
176 177
Гипохолестеринемические средства
Эти препараты предназначены для понижения содержания
холестерина в кровеносных сосудах. Нестероидные гипохоле-
стеринемические агенты часто имеют очень простое строение.
* В СССР выпускается под названием «фенилии». — Прим, перев.
Фармацевтические препараты
453
При изучении, механизма биосинтеза холестерина (т. 4,
стр. 217—226, 247—251) было осуществлено окисление фарне-
зилпирофосфата (178) в кислоту 179. Последнее соединение,
СООН
179
которое не является промежуточным продуктом в схеме пре-
вращения мевалоновой кислоты (т. 4, стр. 219—220) в сквален,
оказалось способным ингибировать это превращение и, таким
образом, потенциально может явиться лекарственным средством,
для предотвращения образования холестерина. К настоящему
времени в данной области было проведено лишь небольшое
число работ, посвященных созданию сесквитерпеноидных гипо-
холестеринемических агентов. Однако все же было получено
несколько аналогов мевалоновой кислоты с целью их исполь-
зования в качестве ингибиторов превращения ацетата в холе-
стерин. Так, соединение 180 и его интересные фторсодержащие
производные 181 и 182 обнаруживают значительную биохими-
ческую ингибиторную (антимевалоновую) активность, и иссле-
дования в этом направлении являются весьма актуальными.
Синтезированы некоторые производные линолевой кислоты
(т. 4, стр. 150 и сл.), которые, как полагают, тормозят абсорб-
цию холестерина. Особенно эффективен N-циклогексиламид ли-
нолевой кислоты (183). Полезные в клинике вещества получены
ОД»
1$3
454
Глаза 11
также при изучении арилалифатических и ароксиалифатических
карбоновых кислот. Первое сообщение о гипохолестеринемиче-
ской активности производных этих кислот было опубликовано
более 20 лет назад, причем наибольший интерес вначале при-
влекло соединение 184. Впоследствии были открыты более
активные вещества, такие, как соединения 185 и 186. Позднее
сотрудники фирмы ICI Уоринг и Торп синтезировали и изу-
чили ряд феноксианалогов этих препаратов, что привело к соз-
данию клофибрата (187). Последний легко получается из эта-
нола, ацетона, хлороформа и га-хлорфенола.
(СН3)2СО—СНС1з
NaOH
СНз '
С2Н5ОН
187
Клофибрат пользуется большим успехом в современной проти-
восклеротической терапии.
Сахароснижающие средства
Сахарный диабет — болезнь, характеризующаяся аномально
высоким содержанием глюкозы в крови. Еще 20 лет назад ни
один из современных противодиабетических препаратов не был
известен. В то время больных сахарным диабетом лечили дие-
той или инъекциями инсулина — полипептидного гормона, вы-
полняющего в организме несколько функций и, в частности,
стимулирующего усвоение глюкозы.
Когда в 1942 г. появилось сообщение, что потенциальное
противотифозное средство 188 вызывает гипогликемию у чело-
века, у медиков сразу же возникли надежды, что это соединение
может быть использовано в качестве перорального сахаросни-
Фармацевтические препараты
455
жающего препарата. Однако вторая мировая война, по-види-
мому, помешала быстрому внедрению сахароснижающих сульф-
амидов в терапевтическую практику, и данные о клинической
эффективности родственного веществу 188 карбутамида (189)
были опубликованы только в 1955 г. Карбутамид оказался
очень активным, но несколько токсичным лекарством. В даль-
нейшем были изучены сотни других сульфамидов, из которых
толбутамид* (190), ацетогексамид (191), хлорпропамид (192)
и толазамид (193) находят клиническое применение в США.
189 (R = nh2, R' = С4Н9)
190 (R = СНз, R' = С4Н9)
R = СН3СО
192 (R = Cl, R' = С3Н,)
Одним из первых пероральных препаратов, обнаруживших
сахароснижающую активность, был гуанидин (194). Однако вы-
сокая токсичность этого соединения послужила причиной созда-
ния более приемлемых веществ — синталинов А и В (195А и Б),
применению которых препятствовало лишь вредное воздействие
на печень и почки. Крупным шагом вперед явилось создание
фенформина (196); в то же время его циклический аналог 197
оказывает весьма слабое сахароснижающее действие. По
данным исследователей фирмы Lederle, пероральная сахаро-
снижающая активность найдена у ряда четвертичных пириди-
ниевых солей (198), причем эта активность сохраняется при
замене пиразольного остатка на изоксазольный, 1,2,4-оксадиа-
зольный, тиазольный, оксазольный или индольный.
NH
H2N nh2
h2n n n nh2
NH NH
194
195A (n = 10)
195Б (n = 12)
В СССР выпускается под названием «бутамид». — Прим, перев.
456
Глава If
C6H5CH2CH2NHCNHCNH2
II II
HN NH
196
NH2
N^XN
I J
CeHeCH2CH2NH''^N-/\NH2
197
Антигипертензивные средства
Повышенное артериальное давление — это не самостоятель-
ная болезнь, а признак общего заболевания организма. Поэтому
лечение должно быть направлено на первопричину заболевания,
будь то расстройство деятельности сердечно-сосудистой систе-
мы, почек, нервной или эндокринной системы. К сожалению, в
большинстве случаев выявить первопричину не удается и для
лечения больных используют антигипертензивные средства.
В качестве таких средств были изучены седативные препа-
раты и транквилизаторы, в частности фенобарбитон, фенотиа-
зины и мепробамат* ** (199). Однако угнетение психики и другие
побочные эффекты сильно ограничивают практическое исполь-
зование этих препаратов. Близкородственный мепробамату ме-
бутамат (200) оказывает сильное антигипертензивное действие
на собак, однако его полезность для человека остается пробле-
матичной. В то же время несомненна высокая эффективность
клонидина * * (201), который вызывает длительное (на несколько
С1
СН3. CH2OCONH2
R xCH2OCONHj
199 (R = CH3CH3CH3— ) 201
200 [R = СН3СН3СН(СН3)— ]
суток) понижение кровяного давления и замедление пульса.
Клонидин действует на центральную нервную систему, причем
* В СССР выпускается под названием «мепротан». — Прим, перев.
** В СССР производится под названием «катапресан»,Прим, перев.
Фармацевтические препараты
457
интересно, что антигипертензивная активность этого соединения
была замечена при его применении в составе лосьонов для
бритья. Существенное достоинство клонидина заключается в
том, что его можно вводить как перорально, так и внутривенно.
В начале 50-х годов появились данные о действии на сердечно-
сосудистую систему гидразинофталазинов. Наиболее деталь-
ному изучению подверглись гидралазин * ** (202) и дигидрала-
HNNH2
202
зин (203), который легко получается из фталевой кислоты:
СООН
СООН
1) nii2nh2^
2) РОС13
NH2NH2
hnnh2
203
Использованию этих веществ при гипертонии у человека пре-
пятствуют их токсичность, непостоянство действия и некоторые
неблагоприятные побочные эффекты.
Декарбоксилирование диоксифенилаланина (допа) допа-де-
карбоксилазой ингибируется соединением а-метилдопа** (204),
которое является эффективным антигипертензивным средством
204
декарбоксилаза
---------------->
для человека. Полагают, что действие а-метилдопа обусловлено
не инактивированием фермента, а способностью этого соедине-
ния подменять допа в качестве субстрата.
Одним из последних успехов в области противогипертен-
зивных химиотерапевтических препаратов явилось создание
* В СССР принято название «апрессин». — Прим, перев.
** Синоним этого препарата, принятый в СССР, — метилдофа. — Прим,
перев.
458
Глава 11
веществ, подавляющих действие некоторых стимуляторов цент-
ральной нервной системы. Эти вещества, получившие название
адреноблокаторов, конкурируют с катехоламинами в рецепто-
рах и тем самым защищают сердце от чрезмерного стимулиро-
вания. Некоторые из адреноблокаторов эффективны также при
ангине (когда физическое или эмоциональное возбуждение уси-
ливает и учащает сердцебиение, что повышает потребность в
кислороде сверх поступающего его количества и вызывает боли
в сердце) и при аритмии сердечной деятельности (нарушениях
ритма работы сердца). В этих случаях адреноблокаторы застав-
ляют сердце биться реже, с меньшей силой и более эффективно.
Первым препаратом этой группы был ксилохолин (205), синте-
зированный в качестве потенциального холинергического агента
(так называют лекарственные вещества, терапевтический эф-
фект которых объясняется их способностью подменять ацетил-
холин при передаче нервного импульса через синаптическую
щель). Кроме того, ксилохолин — сильнодействующее местно-
анестезирующее средство. Однако он оказывает неприемлемые
побочные эффекты, что побудило синтезировать бретилий *
(206)—значительно лучшее лекарство, недостатками которого
можно считать лишь плохую абсорбцию при пероральном вве-
дении и быстрое развитие у больных привыкания. Необходимо
упомянуть также гуанетидин** (207) — родоначальника боль-
шой группы соединений, обнаруживших полезные антигипертен-
зивные свойства.
205
ч ,NH
nch2ch2nh—сГ-
' NHj
206
207
JH2SO4
* В СССР выпускается под названием «орнид». — Прим, перев.
** Синоним этого вещества, принятый в СССР, — октадин. — Прим, пе-
рев.
Фармацевтический препараты
459
Наиболее существенное достижение в данной области — от-
крытие фенилэтаноламинов и феноксипропаноламинов, таких,
как пронеталол (208) и пропранолол * (209) соответственно.
изо -GsHyNHa
----------->.
изО-СзЩИНг,
НО
OCH2CHCH2NHCH(CH3)2
209
Средства для лечения заболеваний
центральной нервной системы
За последние 20 лет потребление снотворных таблеток, седа-
тивных препаратов и транквилизаторов в развитых странах при-
няло такие размеры, что по объему продажи эти лекарства в
сумме уступают только антибиотикам. Кроме них, широкое при-
менение находят и менее известные лекарственные вещества, на-
пример средства для подавления аппетита. Существуют также
специальные препараты для лечения не столь распространенных,
но неприятных психических заболеваний, таких, как эпилепсия
* В СССР производится под названием «анаприлнн». — Прим. перев.
460
Глава II
и болезнь Паркинсона. В распоряжении врачей имеются безо-
пасные обезболивающие средства, расширившие возможности
хирургии и способствовавшие успешному исходу многих опера-
ций. Важной задачей фармацевтической промышленности в те-
чение долгого времени было создание не вызывающих пристра-
стия анальгетиков, способных заменить морфин, и к настоящему
времени эта задача отчасти уже решена. Несмотря на большой
ассортимент применяемых лекарств, не прекращается поиск но-
вых, улучшенных препаратов, особенно для борьбы с психиче-
скими заболеваниями, и исследуются возможности медицины
все в новых областях, в том числе при потере памяти. Основные
фармацевтические препараты, относящиеся к перечисленным
выше группам, рассматриваются в последующих разделах.
Транквилизаторы
До 1953 г. единственными эффективными методами лечения
психических расстройств были психотерапия (оказывающая
ограниченное воздействие на больных с возбужденной психи-
кой), электросудорожная терапия и операции на головном
мозге. Революционизирующее значение имело создание хлор-
промазина (211) и родственных ему фенотиазиновых препара-
тов, которые снимают возбуждение у пациентов и оказывают
существенный положительный эффект при шизофрении. Эти со-
единения были открыты в результате структурной модификации
дифенилметановых антигистаминных веществ (210), обладаю-
(C6H6)2CHOCH2CH2N(CH3)2
210
ckch2)3n(chs)2|ho_
(CH2)3N(CH3)2
211
Фармацевтические препараты
461
щих, как известно, седативным побочным действием. В настоя-
щее время в продаже имеется уже свыше 40 фенотиазиновых
транквилизаторов. Хлорпромазин* (211) синтезируют по приве-
денной выше схеме, первой стадией которой является реакция
Ульмана между о-хлорбензойной кислотой и л-хлоранилином;
присоединение серы происходит в основном в требуемое {орто,
орто') положение. В случае замещенных фенотиазинов, чувстви-
тельных к действию оснований, заключительное алкилирование
можно проводить иначе, например путем отщепления двуокиси
углерода от карбамата
С1СОО(СНг)зЖСН3)г
___А
-сог
Альтернативную группу транквилизаторов, которые полезны
для людей, не переносящих фенотиазины, составляют бутиро-
фенолы. Лучшим из них является галоперидол (212). Эти
соединения были открыты в ходе работы по улучшению боле-
утоляющих свойств 4-фенилпиперидинов, родственных лекарст-
венному препарату петидину** (213). Синтез галоперидола
открывает возможности для варьирования заместителей в обоих
бензольных кольцах, и к настоящему времени испытано уже
более 5000 соединений данной группы.
* В СССР выпускается под названием «аминазин». — Прим, перев.
** В СССР препарат известен под названием «промедол». — Прим, перев.
462
Глава 11
Еще одним важным транквилизатором является резерпин
(214) (т. 4, стр. 395—404), который добывают из индийского
растения Rauwolfia serpentina. Экстракты этого растения, обла-
дающие транквилизирующими свойствами, использовались в
народной медицине на протяжении более трех веков. Вначале
ожидалось, что резерпин будет чрезвычайно эффективен в пси-
хотерапии, как и его хлорсодержащий аналог 215, полный син-
тез которого осуществил французский ученый Руссель. Однако
эти ожидания, по-видимому, не оправдались.
ЛИ 1В = СН3О, R' = Н ); 215 (R = И, В' = С1)
Антидепрессанты
Открытие фенотиазинов побудило биологов исследовать дру-
гие соединения, первоначально полученные в качестве потенци-
альных антигистаминных веществ. При этом была найдена
группа аминоалкиламинодибензилов, которые оказывали слабое
транквилизирующее действие, но хорошо зарекомендовали себя
Фармацевтические препараты
463
при лечении депрессии. Для клинических испытаний из веществ
этой группы был выбран имипрамин* (216). Родственный ему
препарат амитриптилин** (218), который тоже полезен для
борьбы с депрессивными состояниями, интересен тем, что в его
молекуле нет кольцевого атома азота. С целью увеличить бы-
строту действия этих соединений были синтезированы их деме-
тилированные производные 217 и 219; последние тоже находят
применение в антидепрессантной терапии.
(CH2)3N(CH3)2
216
(CH2)3NHCHj
217
Еще до открытия описанных выше трициклических антиде-
прессантов было замечено, что настроение больных туберкуле-
зом улучшается после введения им ипрониазида *** (220).
Вскоре удалось показать, что соединения этого типа инактиви-
руют фермент моноаминоксидазу, регулирующий содержание
аминов в веществе мозга. В дальнейшем ряд лекарственных ве-
ществ данной группы был выпущен в продажу; однако все они
обнаружили токсические свойства. Поэтому предпочтение было
отдано трициклическим антидепрессантам, а пиридиновые пре-
параты были изъяты из обращения. Имеющиеся в настоящее
время в продаже фенелзин (221), транилципромин **** (222) и
CONHNHCH(CH3)2
CeH6CH2CH2NHNH2
220
221
* В СССР выпускается под названием «имизин». — Прим, перев.
** В СССР производится под названием «триптизол». — Прим, перев.
*** В СССР принято название «импразид». — Прим, перев.
**** Синоним этого вещества, принятый в СССР, — трансамин. — Прим-
перев.
464
Глава 11
СН3
С6Нб—CH—CH—NH2 С6Н5СН2—N—СН2С=СН
222 223
паргилин (223) демонстрируют возможность обнаружения по-
лезной биологической активности у очень простых по строению
химических соединений.
Анальгезирующие средства
Болеутоляющие лекарственные препараты можно подразде-
лить по характеру фармакологического действия на два класса:
мягкие анальгетики, например аспирин и n-оксиацетанилид, и
сильные анальгетики, например морфин (224) (т. 4, стр. 343,
362—366, 369—372). Рамки данного раздела не позволяют по-
дробно изложить историю настойчивых поисков лекарственных
веществ, лишенных побочных эффектов морфина (развитие при-
страстия, тошнота, угнетение дыхания), однако наиболее важ-
ные из этих веществ здесь описаны. Первое такое соединение —
петидин (213) — уже упоминалось выше. Вначале сообщалось,
что оно превосходит морфин, однако опыт применения петидина
213
(R = H, R' = CH3)
показал, что этому препарату присущи все недостатки морфина.
Еще более поразительное структурное сходство с морфином
имеют феназоцин (225) и пентазоцин (226), которым сопутство-
вал некоторый коммерческий успех. Первый из них обладает
большей силой действия, чем морфин, но оказывает те же по-
бочные эффекты, тогда как второй уступает морфину по аналь-
гезирующей активности, но зато, что очень важно, не вызывает
болезненного пристрастия. Создание бензоморфановой цикличе-
ской структуры в этих соединениях можно осуществить, исходя
из алкилпиридинов, причем циклизация приводит к образова-
нию рацемической смеси двух изомеров. Анальгезирующее деи-
Фармацевтические препараты
465
ствие пентазоцина обусловлено главным образом левовращаю-
щим изомером.
225 (R = CH2CH2C6H3)
226 [R = СН2СН=С(СЙ3)2 ]
Еще один интересный анальгетик—метадон * (227)— был син-
тезирован немецкими химиками и вошел в медицинскую практи-
ку в конце второй мировой войны. Несмотря на кажущееся
(CeH6)2CHCN
В СССР выпускается под названием «фенадон». — Прим, перед.
466
Глава 11
отсутствие структурного сходства с морфином, это вещество,
полученное из петидина, обладает идентичным с морфином спек-
тром биологической активности. Его применяют в качестве анти-
дота для снятия симптомов болезненного пристрастия к героину.
Выше приведен один из многих возможных путей синтеза мета-
дона.
Успокаивающие средства
Состояние тревоги — трудно определимое состояние, при ко-
тором человек испытывает чувства беспокойства и напряжен-
ности. Оно может длиться очень долго. Лучшими средствами
для лечения таких больных зачастую являются мягкие седатив-
ные средства. Самое старое из них — мефенезин (228), синтези-
руемый из глицидола и о-крезола, первоначально применялся
как мышечный релаксант. Попытки увеличить продолжитель-
ность его действия привели к открытию полезного влияния кар-
баматных групп. Это позволило получить мепробамат* (199),
который до сих пор применяется в качестве седативного сред-
ства.
Однако к настоящему времени мепробамат в значительной
степени вытеснен бензодиазепиновыми успокаивающими сред-
ствами, открытыми фирмой Roche с помощью стандартной про-
цедуры скрининга. Синтез этих соединений включает неожидан-
ную реакцию расширения цикла: вместо метиламинометильного
производного 230 образуется хлордиазепоксид ** (229). Другим
полезным веществом той же группы является диазепам ***
* В СССР более известен под названием «мепротан».— Прим, перев.
** Синонимы этого препарата, принятые в СССР, — либриум и эле-
ниум. — Прим, перев.
*** Синоним этого препарата — седуксен. — Прим, перев.
Фармацевтические препараты
467
(232), который, как и хлордиазепоксид, получают из 2-амино-5-
хлорбензофенона (231). Оксазепам (234), впервые синтезиро-
229
сй3Кн2
NaOH- С1СН2СОС1
232
ванный химиками фирмы Wyeth, можно получить перегруппи-
ровкой Полоновского из N-окиси 233 или, по другой схеме, из
468
Глава 11
соединения 235, которое в свою очередь тоже готовят из 2-ами-
но-5-хлорбензофенона (231)
231
Снотворные и седативные средства
Барбитураты являются одними из старейших лекарственных
веществ, применяемых в современной медицине, хотя в каче-
стве седативных средств они полностью вытеснены бензодиазе-
пинами. Снотворные свойства 5,5-диэтилбарбитуровой кислоты
(240) открыл в 1903 г. Эмиль Фишер, который обратил внима-
ние на то, что снотворные препараты 236 и 237 содержат чет-
вертичный атом углерода. Метод синтеза барбитуратов, разра-
ботанный Фишером, позволяет получать разнообразные соеди-
нения этой группы, в том числе фенобарбитон * (241) (т. 3,
стр. 120).
СНз
сн3, .он
(CH3)2C(so2c2h6)2 с13ссн(он)2
с2н/ хсн3
236 237 238 239
* В СССР выпускается под названиями «фенобарбитал» и «люминал». —
Прим, перев.
Фармацевтические препараты
469
/R
(H2N)2CO + (C2HeOOC)2C<
XC2HS
CgHsONa
------->
240 (R = C2HS)
241 (R = C6H5)
Известны также снотворные вещества, имеющие другую
структуру. Так, в течение некоторого времени применялись хлор-
алгидрат (238) и паральдегид (239); позднее был открыт мета-
квалон (242), который синтезируют из антраниловой кислоты
и о-толуидина:
Противосудорожные средства
Эпилепсия — это общее название группы психических забо-
леваний, для которых характерны судорожные припадки. Чаще
всего встречаются малый припадок, протекающий в форме крат-
ковременной потери сознания (иногда с конвульсивными подер-
гиваниями тела), и большой припадок, сопровождающийся
243
244
сильными судорогами. В последнем случае лечение до сих пор
производится с помощью фенобарбитона (241). Однако род-
ственный ему примидон (1) оказывает менее выраженное сно-
творное действие и более эффективен в качестве противосудо-
470
Глава 11
рожного средства. Еще одним стандартным лекарственным ве-
ществом, применяемым в клинике больших припадков, является
дифенилгидантоин (244).
Лучше других препаратов зарекомендовал себя при лечении
больших припадков этосуксимид (243). Внедрение новых проти-
восудорожных средств в практику психотерапии производится
с большой осторожностью, так как их приходится применять в
течение длительного, времени. Из недавно появившихся ле-
карств большую популярность приобрел нитразепам (245), осо-
бенно при лечении эпилепсии у детей.
Анестезирующие средства
В хирургической практике для общей анестезии используют-
ся газообразные вещества (такие, как закись азота) и летучие
органические соединения. Диэтиловый эфир находит примене-
ние лишь в тех случаях, когда в распоряжении врача нет совре-
менного оборудования для общего наркоза, так как это веще-
ство не требует специальной аппаратуры. Однако эфир горюч и
взрывоопасен; кроме того, для него характерна замедленность
начала и окончания действия. Галотан* (246) лишен указан-
ных недостатков и поэтому широко используется в хирургии как
общий анестетик. Это соединение получают бромированием или
хлорированием соответствующего трифторгалогенэтана
Вг2 Ch
C1CH2CF3 ---> BrClCHCF3 -<-- BrCH2CF3
246
Более удобны в применении общеанестезирующие вещества,
которые можно вводить внутривенно. Чаще всего используются
барбитураты с короткой продолжительностью действия, напри-
мер тиопентал (247) (в виде натриевой соли). Обнаружение
V С2Н5 СООСНз
11 с,н,-и
HN^—< ] 1 сн3 н 247 NHCOCH2N(C2H5)2 СН3^^\^СН3 249 NCHj^—OCOC6HS 248 ^x.COOCHjCHjNfCjH^j 250
В СССР выпускается под названием «фторотан». — Прим, перев.
Фармацевтические препараты
471
местноанестезирующих свойств у соединений группы кокаина
(248) (одного из тропановых алкалоидов; т. 4, стр. 349) от-
крыло дорогу к получению ряда синтетических эфиров бензой-
ных кислот, таких, как лидокаин* (249) и прокаин** (250).
Стимуляторы центральной нервной системы
Ксантиновые алкалоиды, содержащиеся в кофе и чае, на-
пример кофеин (251), обладают ''определенным структурным
сходством с барбитуратами, но оказывают противоположное
биологическое действие. Кофеин (т. 3, стр. 114; т. 4, стр. 418)
является слабым психостимулятором и применяется в комбина-
ции с аспирином или n-оксиацетанилидом в рецептурных сред-
ствах от головной боли. Путем модификации структуры гор-
NHCH3
СН3
СНО
С6Н/
1) н+ С6н3-
NH2
СН3 2) разделение
изомеров
СНз
мона надпочечной железы адреналина (252) (т. 4, стр. 127
и сл.) был получен более сильный психостимулятор — амфе-
тамин *** (253). Однако нежелательные побочные эффекты
препаратов группы амфетамина ограничили их использование
таблетками для подавления аппетита. Злоупотребление этими
лекарствами представляет серьезную социологическую пробле-
му, поскольку тучные пациенты считают, что они помогают им
легче переносить диету. Поэтому были предприняты попытки
снизить стимулирующее действие амфетамина, сохранив его
способность подавлять аппетит. На этом пути удалось получить
такие вещества, как фентермин (254), диэтилпропион **** (255)
* Синонимы этого соединения, принятые в СССР, — ксиканн и ксило-
каин.— Прим, перев.
** В СССР выпускается под названием «новокаин». — Прим, перев.
*** В СССР выпускается под названием «фенамин». — Прим, перев.
**** В СССР принято название «депранон». — Прим, перев.
472
Глава II
и фенметразин* (256). Широко применяемый препарат фенфлу-
рамин (257), как сообщается, не возбуждает центральную нерв-
ную систему, а повышает уровень мышечного потребления сахара.
Антипаркинсонические средства
К настоящему времени достигнут некоторый прогресс в ле-
чении таких симптомов болезни Паркинсона, как дрожание и
мышечная ригидность. В практике паркинсонотерапии приме-
няются различные лекарственные вещества; из них наиболее
эффективны тригексилфенидил ** *** (258), который можно син-
тезировать в одну стадию по реакции Гриньяра
soci2
259
* В СССР производится под названием «мефолин». — Прим, перев.
** В СССР принято название «циклодол». — Прим, перев.
*** В СССР выпускается под названием «мебедрол». — Прим, перев.
Фармацевтический препараты
473
Новым методом лечения этой болезни является попытка уве-
личить содержание диоксифенилэтиламина (допа) (261) (т. 4,
стр. 127) в мозге больного путем введения ему биосинтетиче-
ского предшественника допа — добываемого из природных ис-
точников диоксифенил-ь-аланина (ь-допа)* (260).
допа-декарбоксилаза
260 261
Последние исследования, направленные на повышение ак-
тивности антипаркинсонических препаратов и уменьшение их
побочных эффектов, включают применение допа в комбинации с
соединением, ингибирующим его декарбоксилирование в мозге,
но не в центральной нервной системе. Два таких соединения —
262 и 263 — сейчас проходят испытания. Кроме того, большой
262
263
интерес как антипаркинсоническое средство привлекает а-ме-
тилдопа (204).
Мочегонные средства
Мочегонные средства (диуретики) применяются для повы-
шения отделения мочи почками. Возникающее в результате
этого снижение содержания жидкости в организме полезно при
лечении разнообразных заболеваний, в том числе высокого кро-
вяного давления и расстройства функций почек.
Мочегонные свойства хлористой ртути (каломели) известны
с XVI в., но лишь в 1920 г. было установлено, что аналогичное
действие оказывают ртутьорганические соединения, применяе-
мые для лечения сифилиса. Однако широкое изучение ртутных
диуретиков не увенчалось созданием нетоксичных пероральных
препаратов. Большую историю имеет также использование в ка-
честве мочегонных средств ксантиновых оснований — кофеина и
теофиллина; тем не менее и в этом ряду обнаружить высокоак-
тивные диуретики не удалось.
* Синоним этого вещества, принятый в СССР, — дофа. — Прим, перев.
474
Глава 11
Исследование побочных эффектов антибактериального веще-
ства сульфаниламида (8) показало, что мочегонные свойства
этого соединения обусловлены присутствием сульфамидной
группы. Следствием этого открытия явилось создание эффек-
тивных пероральных диуретиков. Первый из них — ацетазол-
амид * (264) — был найден в ряду гетероциклических сульфами-
О N-------N
Ill
CH3/^'N/^'S^XSO2NH2
н
264
дов. Однако использованию ацетазоламида для длительного
лечения пациентов (например, при застойной сердечной недо-
статочности) мешали побочные явления и ограниченная актив-
ность этого лекарственного вещества. Дальнейшее продвижение
по «сульфамидному направлению» привело к получению хлор-
талидона (265), который применяется сейчас в клинической
26S
практике. Наблюдение Спрагью, что ациламиногруппы повы-
шают мочегонную активность, позволило ему создать диуретик
хлортиазид (266)
nh2
so2nh2
Продолжение поисков в том же направлении выявило дру-
гую группу эффективных диуретиков — производные гидротиа-
* Синоним этого вещества, принятый в СССР, — диакарб. — Прим, перев.
Фармацевтические препараты
475
зина. Одно из этих соединений — циклопентиазид * (267)—на-
шло применение в клинике. Очень большую популярность в ка-
честве сульфамидного диуретика завоевал моносульфамид
фурземид** (268).
О2
267
268
Принципиально новая группа эффективных мочегонных
средств была обнаружена в ходе работы по изысканию соеди-
нений, блокирующих тиольные группы белков, т. е. аналогичных
по механизму действия ртутьорганическим диуретикам. Так,
этакриновая кислота (269) не уступает по силе действия фур-
земиду. Синтез этой кислоты иллюстрирует применение реакции
Манниха для получения а, p-ненасыщенного кетона
ОСН2СООН
С1
с2н5сн2сос1
---------->
ОСН2СООН
CH2O-(CH3)2NH
Cl
С помощью обычной процедуры скрининга фирмы Merck,
Sharpie и Dohme открыли триамтерен *** (270). Ценность этого
* В СССР употребляется название «циклометиазид». — Прим, перев.
** В СССР выпускается под названием «фуросемид». — Прим, перев.
*** В СССР принято название «птерофен». — Прим, перев.
476
Глава 11
вещества состоит в том, что при использовании в комбинациях
с другими диуретиками оно подавляет повышенное выделение
NH2
270
калия с мочой, которое является нежелательным побочным эф-
фектом сульфамидных и тиазидных мочегонных средств.
Обнаружение у альдостерона (т. 4, стр. 262) способности
регулировать процессы выведения из организма ионов калия и
реабсорбции ионов натрия стимулировало поиск веществ, инги-
бирующих биосинтез этого гормона и (или) его действие. Пола-
гают, что дорогостоящий диуретик спиронолактон (271) выпол-
няет обе эти функции.
СПИСОК ЛИТЕРАТУРЫ
Burger A., Medicinal Chemistry, 3rd ed., Wiley — Interscience, New York,
1970.
Remington. J. P., Remington’s Pharmaceutical Sciences, 14th ed., Mack Pub-
lishing Co., Easton, Penn., 1970.
Goodman L. S., Gilman A., The Pharmacological Basis of Therapeutics, 4th
ed., Macmillan, New York, 1970.
Slack R., Nineham A. W., Medical and Veterinary Chemicals, Pergamon
Press, Oxford, 1968.
Martindale S., The Extra Pharmacopoeia, 26th ed., The Pharmaceutical
Press, London, 1972.
The Merck Index, 8th ed., Merck and Co., Rahway, New Jersey, 1968.
Albert A., Selective Toxicity and Related Topics, 4th ed., Methuen, Lon-
don, 1968.
Sexton W. A., Chemical Constitution and Biological Activity 3rd ed., Spon,
London, 1963.
Kirk R. E., Othmer D. F., Encyclopaedia of Chemical Technology, 2nd ed.,
Wiley, New York, 1963—1972.
Drill V., Drill’s Pharmacology in Medicine, 4th ed., McGraw-Hill, New York,
1971.
ГЛАВА 12
Химикаты для сельского хозяйства
И. Кей, Б. Снелл, К. Томлин
Key I, Т., Snell В. К., Tomlin С. D. S., Plant Protection Limited,
Jealott’s Hill Research Station
По некоторым данным, ежегодно около 15 000 человек на
земном шаре умирает от недоедания. Однако несмотря на это,
Население Земли возрастает и в ближайшие 40 лет, вероятно,
вдвое превысит нынешнее. В этой ситуации нельзя мириться
с тем обстоятельством, что ежегодно 10% урожая уничтожается
на полях и в хранилищах вредными насекомыми и, кроме того,
значительная часть его теряется из-за болезней сельскохозяй-
ственных культур. Люди, ослабленные хроническим недоеда-
нием, более восприимчивы к болезням, переносимым опять-таки
насекомыми (мухами, комарами и др.), что вызывает еще боль-
шую тревогу за будущее человечества. Отсюда становится ясно,
какой важный вклад вносит в повышение продуктивности сель-
ского хозяйства химическая промышленность, которая произво-
дит не только азотные и фосфорные удобрения, но и химические
средства защиты растений. В первом разделе этой главы рас-
сматриваются инсектициды — вещества, уничтожающие вред-
ных насекомых. Второй раздел посвящен фунгицидам и другим
средствам защиты растений от болезней. В третьем разделе
описаны гербициды и, наконец, в четвертом — регуляторы роста
растений. Хотя ручная или механизированная прополка всегда
входила в комплекс агротехнических мероприятий, требования
современной экономики сельскохозяйственного производства и
увеличение размеров посевных площадей делают эти традици-
онные методы борьбы с сорняками уже неприемлемыми. Присут-
ствие сорняков на возделываемых полях нежелательно не
только из-за засорения урожая основной культуры, но и по ряду
других причин. Разросшиеся сорняки конкурируют с культур-
ными растениями за питательные вещества и воду; кроме того,
они быстрее растут и поэтому лишают культурные растения
солнечного света. Желание обезопасить поля от сорняков за-
ставило фермеров повести с ними жестокую борьбу, и в настоя-
478
Глава 12
щее время стандартным является такой высокий уровень чис-
тоты зерна, который еще несколько десятков лет назад считался
недостижимым. В садоводстве указанные соображения не имеют
решающего значения. Тем не менее и садоводы охотно поль-
зуются химикатами, поскольку они во многом облегчают уход
за садом. Наконец, обработка селективными гербицидами газо-
нов позволяет поддерживать их в таком декоративном состоя-
нии, которое прежде было невозможным.
Инсектициды
Поиск синтетических инсектицидов в 30-е годы потребовал
разработки биологических тестов для оценки инсектицидной
активности тысяч испытываемых химических соединений. Это
в свою очередь вызвало необходимость создания методов выра-
щивания в лабораторных условиях насекомых различных видов
и соответствующих растений-хозяев. Обычно насекомых просто
опрыскивают испытываемыми химикатами при известной вели-
чине дозы (концентрация действующего начала в препарате,
как правило, составляет несколько сотен частей на миллион).
В случае контактных инсектицидов применяемый препарат дол-
жен обеспечивать хорошее смачивание насекомых. Остаточные
(кишечные) инсектициды следует использовать в виде препара-
тов, равномерно покрывающих воскообразную поверхность
листьев. Даже при непосредственном опрыскивании насекомых
конечный результат зависит от многих факторов, в том числе от
скорости прохождения инсектицида через наружные покровы
к центру, на который он оказывает биохимическое воздействие,
и от скорости детоксикации инсектицида ферментами насеко-
мого на пути к этому центру. Существенное значение имеет
также способность инсектицида блокировать в организме насе-
комого какой-либо жизненно важный процесс.
Возможность {фактического использования на полях инсек-
тицида, обладающего благоприятными показателями по указан-
ным выше характеристикам, определяется, в частности, тем,
насколько он устойчив к действию атмосферных осадков и сол-
нечного света (персистентность) и нетоксичен для других форм
жизни, включая человека (избирательность). Наконец, важную
роль играет стоимость инсектицида, которая во всяком случае
не должна превышать стоимости спасенной с его помощью части
урожая.
Перечисленные выше жесткие требования приводят к тому,
что из каждых 10 000 испытываемых соединений только одно
становится товарным инсектицидом. Поэтому создание нового
инсектицида обходится в настоящее время приблизительно в
1 млн. ф. ст.
Химикаты для сельского хозяйства
479
Хлоруглеводороды
Первые же исследования, проведенные методом бессистем-
ного скрининга тысяч химических соединений, показали, что
многие хлорсодержащие вещества обладают слабыми инсекти-
цидными свойствами. В начале 40-х. годов работа в этом напра-
влении увенчалась открытием ДДТ (дихлордифенилтрихлорэта-
на). ДДТ — высокоэффективный инсектицид с широким спектром
биологической активности и низкой видимой токсичностью для
млекопитающих. Дешевым методом его производства является
кислотнокаталитическая конденсация хлорбензола с хлоралем
(т. 2, стр. 213)
ДДТ
Коммерческий успех ДДТ стимулировал после второй мировой
войны широкое проведение прикладных исследований по изы-
сканию других хлоруглеводородных инсектицидов. В ходе этих
исследований были открыты гексахлорциклогексан (ГХЦГ) и
С12
УФ-свет
СНС1
С1НСХ ХСНС1
С1НСХ /СНС1
СНС1
смесь геометрических
изомеров
гексахлорциклогексан
группа полихлорциклодиенов, получаемых присоединением раз-
личных диенофилов к гексахлорциклопентадиену по реакции
Дильса — Альдера, в том числе альдрин, дильдрин и эндосуль-
фан
альдрин
С1 " *
~ СНСН2ОСОСНз
X + 11
СНСН2ОСОСН3
дильдрин
С1
С1
480
Глава 12
Ci Cl
С1/\^сн2ососн3 w H0. cirf'^'y" 2 \
—» ci2c| --------► Cl2c so
С1ЧАСН2ОСОСН3 2>S0CI> cikACH2o^
эндосульфан
О механизме действия хлорорганических инсектицидов из-
вестно только то, что они блокируют передачу нервных импуль-
сов у насекомых — вероятно, путем нарушения переноса ионов
натрия или калия через нервные мембраны. Однако очевидно,
что хлорорганические инсектициды не содержат специфической
летальной (токсофорной) группировки. Скорее всего, их ток-
сичность обусловливается структурой и конформацией молекулы
в целом — как если бы она представляла собой ключ к ящику
Пандоры, в котором заключены вредные биохимические воздей-
ствия.
Надежды на то, что эти дешевые инсектициды окажутся па-
нацеей, разрешающей проблему защиты сельскохозяйственных
культур от вредных насекомых, не оправдались. Оказалось, что
свойства, придающие хлоруглеводородам ценность в качестве
инсектицидов, в то же время делают их использование нежела-
тельным с точки зрения охраны окружающей среды. Вследствие
высокой персистентности, легкости проникновения в живые ор-
ганизмы и растворимости в жирах хлорорганические инсекти-
циды способны накапливаться в экосистеме и сохраняться в ор-
ганизме животных и птиц, попадая затем в пищу людей. Это
обстоятельство привело к запрещению (1970 г.) применения
многих соединений данной группы, особенно в тех случаях,
когда вместо них могут быть использованы другие инсектициды.
Фосфорорганические инсектициды
Почти одновременно с открытием ДДТ было найдено, что
некоторые органические эфиры фосфорной кислоты обладают
сильными инсектицидными свойствами. В определенных преде-
лах их активность может быть связана со способностью высту-
пать в качестве фосфорилирующих агентов. Инсектициды
данной группы токсичны как для насекомых, так и для млеко-
питающих, поскольку они фосфорилируют (и тем самым бло-
кируют) фермент ацетилхолинэстеразу, ответственный за воз-
никновение и передачу нервных импульсов. Инактивирующее
действие фосфорорганических ядов можно приближенно пред-
ставить как нуклеофильную атаку аниона, образовавшегося за
счет связанной с ферментом первичной гидроксильной группы,
на атом фосфора с отщеплением уходящей группы L (схема а).
Химикаты для сельского хозяйства
481
фермент —СЩО" + р ~
RO ORL
---->• фермент—СН2О | OR-t-L'(a)
OR
Одним из самых первых органических соединений фосфора,
получившим большое распространение в качестве инсектицида,
был паратион *. Уходящей группой здесь является стабилизиро-
ванный резонансом n-нитрофенолят-анион, что сообщает моле-
куле ацилирующие свойства. Таким образом, фосфорорганиче-
ские инсектициды в отличие от хлоруглеводородов содержат
группировку, обусловливающую их биологическую активность
(токсофор); остальная часть молекулы (L) обладает подходя-
паратион
щими электронными свойствами, чтобы выполнять функций но-
сителя токсофорной группы, и может широко варьироваться.
Последнее можно проиллюстрировать на примере таких инсек-
тицидов, как дихлорофос, малатион ** и диазинон
О
СС1зСНО + (СНзО)зР *--ГРеВ-^> СС12===СНОР(ОСН3)2 + СНзС1
дихлорофос
(т. 3, стр. 241)
S S
Ц: СНСООС2Н5 ГИДРОХИНОН :| =
(СНзО)2Р—SH+II ----------» (СНзО)2Р— S— СНСООС2Н5
СНСООС2Н5 1
СН2СООС2Н5
малатион
HN. zNH2 , ,
/ (т. 3, стр. 120)
CH3COCH2COOC2HS + с -------------------->*
НС(СНз)2
* В СССР Примято название «Тиофос». — Прим, перев.
** Синоним этого вещества, принятый в СССР, — карбофос. — Прим.
рёв.
16 Зак. 501
482
Глава 12
HSC-
НзС-
•ОР(ОС2Н6)2
СН(СН3)2
С1Р(ОС2Н6)2
----------->
N^N
СН(СН3)2
N^/NH
диазинон
Первые представители фосфорорганических инсектицидов, на-
пример паратион, были весьма токсичны не только для насеко-
мых, но и для млекопитающих. Однако некоторые из соедине-
ний, появившихся позднее, например малатион, малотоксичны
для млекопитающих; причины этой избирательности их дей-
ствия во многих случаях неизвестны.
Поскольку фосфорорганические инсектициды обычно способ-
ны к биоразложению путем гидролиза по сложноэфирной связи
токсофорной группировки с носителем, при их использовании
почти не возникает проблемы персистентных остатков, как в
случае хлорорганических инсектицидов. В ближайшие годы они,
вероятно, сохранят свое доминирующее положение среди поле-
вых инсектицидов. В то же время эффективность веществ этого
класса все более уменьшается вследствие возрастания резис-
тентности насекомых к фосфорорганическим инсектицидам, яв-
ляющейся результатом их широкого использования.
Карбаматы
Карбаматные Инсектициды появйлйсь в продаже в 50-х го-
дах, причем стимулом для их разработки послужило выяснение
механизма действия токсичного алкалоида физостигмина (1).
CH3NHCOO.
СНз
СНз СНз
Изучение физостигмина и его синтетических аналогов показало,
что эти карбаматы, подобно фосфорорганическим соединениям,
способны инактивировать ацетилхолинэстеразу путем переноса
OR
I
фермент - СНДО~ ЧС=О
фермент -СНзОСОМНСНз +
+ RO- «О
Химикаты для сельского хозяйства
483
карбамоильной группы (токсофора) к активному центру фер-
мента (схема б). Однако в данном случае в отличие от эфиров
кислот фосфора природа уходящей группы RO’ имеет решаю-
щее значение для инсектицидной активности. Практическое
применение нашли три типа эфиров карбаминовых кислот: арил-
карбаматы, гетероарилкарбаматы и карбамоилоксимы, пред-
ставителями которых являются карбарил, пиримикарб и альди-
карб соответственно.
ОН OCOCl OCONHCH3
карбарил
H2NX z^NH I
С N(CH3)2
N(CH3)2
ОНз
I
CH3\_X/OC\
—> f П ^N(CH3)2
N(CH3)2
пиримикарб
NOCI CH3SNa
(CH3)2C=CH2 —> C1C(CH3)2CH=NOH --------->
COC12 CH3NH2
—> CH3SC(CH3)2CH=NOH -------► CH3SC(CH3)2CH=NOCOC1 ------->
—> CH3SC(CH3)2CH=NOCONHCH3
альдикарб
Один из недостатков карбаматов состоит в том, что многие
инсектициды этого класса высокотоксичны для млекопитающих.
Тем не менее некоторые карбаматные инсектициды произво-
дятся в промышленном масштабе, поскольку они зачастую
имеют преимущества по сравнению с другими соединениями,
16*
484
Глава 12
Так, альдикарб эффективно уничтожает почвенных нематод
(мельчайших круглых червей, повреждающих корневую систему
растений).
Инсектициды растительного происхождения
Некоторое применение в качестве инсектицидов находят при-
родные вещества, добываемые из растительных экстрактов.
К ним относятся никотин, деррис (содержащий ротенон; см.
т. 3, стр. 87) и экстракты измельченных цветов далматской ро-
никотин
машки, которые содержат пиретрины и цинерины.
пиретрин I: В* = СН3, В2 = СН=СН3
пиретрин ШЛ1 = СООСН3, В2 = СН=СН?.
цИнерин I: В1 = СН3, В2 = СН3
цинерин II: В* = СООСН3, В2 = СН3
Экстракты далматской ромашки имеют очень важное прак-
тическое значение. Обладая исключительным эффектом против
мух («нокаутирующий» эффект) и низкой токсичностью для
млекопитающих, они используются главным образом в составе
бытовых аэрозольных препаратов. Применение этих экстрактов
в борьбе с сельскохозяйственными вредителями сдерживается
их малой стойкостью в полевых условиях и высокой стоимостью
по сравнению с другими инсектицидами.
Перспективы
Перспективы дальнейшего развития системы мероприятий,
направленных на уничтожение вредных насекомых, базируются
Химикаты для сельского хозяйства
485
на все более глубоком осознании того факта, что химическая
промышленность в сущности должна обеспечить защиту урожая
от этих насекомых. Отсюда следует, что, быть может, не обяза-
тельно применять химикаты, оказывающие на насекомых пря-
мое токсическое действие. Возможно, что не менее эффектив-
ными способами являются, например, отпугивание насекомых
от культурных растений с помощью так называемых репеллен-
тов, привлечение их в другие места с помощью половых или
пищевых аттрактантов и сокращение размножения с помощью
химических стерилизаторов. Действительно, есть веские основа-
ния полагать, что использование избирательных методов борьбы
с вредителями сельскохозяйственных культур позволит не толь-
ко свести к минимуму нарушение экологического равновесия, но
и даст другие, более скрытые преимущества. В частности, при
этом не будут затрагиваться хищные насекомые — естественные
враги вредных насекомых, тогда как применяемые в настоящее
время инсектициды широкого спектра действия с их тотальной
«убойной силой» уничтожают и их, и других полезных насе-
комых.
. Большое внимание в этой связи привлекло сообщение о вы-
делении и идентификации гормона насекомых, регулирующего
их метаморфоз, — так называемого ювенильного гормона. Обра-
ботка насекомых избытком ювенильного гормона на ранней
стадии развития приводит к нарушению их жизненного цикла,
в результате чего они не превращаются через стадию куколки
во взрослое насекомое, а останавливаются на первой (личиноч-
ной) стадии. Несмотря на сложность процедуры выделения
ювенильного гормона (из бабочки семейства Cecropia) и по-
путно проводимых биологических испытаний, а также на то,
что количество полученного вещества составляло лишь не-
сколько миллиграммов, удалось установить, что этот гормон
представляет собой метиловый эфир 10,11-эпокси-7-этил-3,11-ди-
метилтридекадиен-2,6-овой кислоты (2) (т. 4, стр. 243).
Интерес к ювенильному гормону был столь велик, что за
первые 4 года, прошедшие со времени выяснения его структуры,
было разработано не менее 15 синтетических методов его
получения. Многие из этих методов чрезвычайно элегантны и
включают весьма стереоселективные и стереоспецифические ре-
акции.
486
Глава 12
Судить о перспективах практического использования юве-
нильного гормона и его биологически активных аналогов пока
преждевременно. Однако уже сейчас свыше 20 ведущих хими-
ческих фирм ведут активные исследования в области синтеза и
биологических испытаний этих соединений.
Фунгициды
. Человечество с древнейших времен испытывает на себе опу-
стошительные последствия болезней растений. В XIX в. произо-
шло два крупных бедствия, которые наглядно показали необхо-
димость борьбы с массовыми заболеваниями растений: голод в
Ирландии в 1845 г., вызванный вспышкой фитофтороза карто-
феля под действием Phytophthora inj estans (картину этого го-
лода описал недавно Сесиль Вудхэм-Смит в романе «The Great
Hunger») и полное уничтожение урожая кофе на Цейлоне лис-
товой ржавчиной Hemileia vastatrix.
Начало научному изучению болезней растений положили
пионерские работы Прево, Де Бари, Пастера и др., выполнен-
ные в прошлом веке. Роль грибков, бактерий и вирусов как воз-
будителей этих болезней была выяснена в течение последних
150 лет.
Многие болезни сельскохозяйственных культур можно пред-
отвратить с помощью чисто агротехнических мероприятий, та-
ких, как севооборот, правильный выбор посевных участков, уни-
чтожение сорняков, на которых откладывают яйца вредные
насекомые, и своевременное удаление зараженных частей расте-
ний. Теоретически самым лучшим способом защиты растений
от болезней является выведение устойчивых к ним сортов. Од-
нако на практике лишь немногим сортам удается сохранить эту
устойчивость на длительное время, так как в результате эволю-
ции или селекции среди патогенных микроорганизмов непре-
рывно появляются новые вирулентные штаммы. В том случае,
когда агротехнические приемы не помогают сдержать распро-
странение болезни, используют химические препараты.
Контактные фунгициды
Согласно определению, принятому в 1943 г. Американским
фитопатологическим обществом, фунгицидом называется «хими-
ческий или физический агент, который убивает споры или мице-
лий грибков либо подавляет их рост».
Большинство фунгицидов, применяемых в сельском хозяй-
стве, относится к веществам контактного действия. Будучи дол-
ЖНЬЩ образом унесены на поверхность семдн или листьев
Химикаты для сельского хозяйства 487
растений, они образуют защитный слой, предотвращающий или
затрудняющий рост болезнетворного гриба.
Остановимся вкратце на неорганических фунгицидах, по-
скольку они имели важное историческое значение. Из медьсо-
держащих фунгицидов заслуживает упоминания бордосская
жидкость [комплекс сульфата меди CuSO4 с гашеной известью
Са(ОН)2], которая была впервые использована в 1885 г. Мил-
ларде для борьбы с ложной мучнистой росой виноградных лоз.
С того времени бордосская жидкость получила широкое распро-
странение, хотя были найдены ее заменители, обладающие
меньшими коррозионными свойствами, например хлорокись
меди 3Cu(OH)2-C11CI2. Для борьбы с настоящими мучнисторо-
сяными грибами уже свыше 100 лет пользуются элементарной
серой. Среди полисульфидных фунгицидов важное место зани-
мает известково-серный отвар (ИСО), который получают кипя-
чением извести с серным цветом. ИСО применяется до сих пор
и считается довольно эффективным фунгицидом. Однако соеди-
нения серы способны повреждать растения; особенно чувстви-
тельны к ним некоторые сорта яблонь и груш.
К числу ртутьсодержащих фунгицидов принадлежит одно-
хлористая ртуть (каломель) Hg2Cl2, которую используют в ка-
честве протравителя корней рассады против возбудителя килы
капусты (Plasmodiophora brassicae). Широкое распространение
получили также ртутьорганические соединения общей формулы
RHgX, где R — алкил или арил и X — остаток органической или
минеральной кислоты (например, CeHsHgOCOCHs), применяе-
мые для протравливания семян злаковых культур. Все эти ве-
щества дешевы и дают хороший эффект при борьбе с возбуди-
телями разных видов головни злаков, которые когда-то были
очень распространены в Великобритании. Нормы расхода ртуть-
содержащих фунгицидов обычно очень низки — около 5 г ртути
на 1 га. Большинство ртутных Препаратов токсично для млеко-
питающих и птиц. Поэтому ПоЧти не вызывает сомнений, что их
использование будет прекращено, как только удастся найти для
них эффективные и менее токсичные заменители.
Ценными органическими фунгицидами являются производ-
ные дитиокарбаминовой кислоты NH2CSSH. Их воздействие на
грибы было Открыто незадолго до второй мировой войны (пер-
воначально эти соединения были получены как ускорители вул-
канизации каучука). Фунгицидная активность свойственна ди-
тиокарбаматам различного строения. В данном разделе рассма-
триваются лишь две наиболее важные группы этих фунгицидов.
Типичным представителем соединений первой группы может слу-
жить тирам* (тетраметилтиурамдисульфид), который получают
В СССР принято название «тиурам Д». — Прим, перев.
488
Глава 12
из легкодоступных промежуточных веществ (схема в). Сре-
ди дитиокарбаматных фунгицидов второй группы следует
S
II
(CH3)2NH + CS2 + NaOH —> (CH3)2N—С—SNa
окислитель
(воздух, Н2О2 или CI2)
S S
II II
(CH3)2N—С—S—S—С—N(CH3)2 (в)
тирам
назвать набам, синтезируемый на основе этилендиамина. Так
как разбавленные растворы набама окисляются на воздухе, его
переводят в более стабильное цинковое (цинеб) или марганце-
вое (манеб) производное.
CH2NH2
I -Г 2CS2 -|- NaOH —>
CHNH2
S
ZnSO4 CH2NHC—S4
--------* I >
_ CH2NHC—sz
. f 1
CH2NHCSNa
-«-*• I — цинеб
CH2NHCSNa
11 0
S MnSO4 JI c
набам I \ „
I ' zMn
CH2—C—s/
мйнеб
ДитиокарбаМать!— дёшёвые И высокоактивные фунгициды,
которые до сих пор пользуются большим спросом. Например,
тирам применяется для борьбы с серой гнилью (Botrytis cine-
геа) земляники и салата, а манеб и цинеб подавляют рост воз-
будителя фитофтороза картофеля.
Сильные фунгицидные свойства обнаруживают многие со-
единения, содержащие трихлорметилсульфениламидную группи-
ровку N—S—CCI3. Полагают, что их фунгицидность связана
со способностью вступать в реакцию с тиольными группами в
клетке гриба
;NSCC13 + 2RSH —> >H + RSSR + CSC12 + HC1
Химикаты для сельского хозяйства-
489
или с токсичностью для грибов выделяющегося в этой реакции
тиофосгена. Разумеется, такое представление является упрощен-
ным, поскольку точный механизм действия фунгицидов данной
группы неизвестен. Тем не менее идея о том, что фунгицидная
активность каптана обусловлена присутствием в его молекуле
фрагмента NSCC13 (одно время полагали, что токсофором здесь
служит имидный атом азота), привела к получению ряда род-
ственных соединений, в том числе фолцида и дихлофлуанида *.
Каптан и его аналоги высокоактивны, обладают широким спек-
тром действия и находят применение для борьбы с паршой
яблони и серой гнилью земляники, а также в качестве покры-
тий семян, уничтожающих патогенные организмы, находящиеся
внутри семян и в почве.
Исходными веществами в синтезе этих соединений служат
сульфенилгалогениды, легко получаемые в свою очередь из серо-
углерода. Так, трихлорметилсульфенилхлорид образуется при
взаимодействии сероуглерода с хлором
12
CS2 + 3C12 —> C13CSC1 + SC12
а фтордихлорметилсульфенилхлорид — при обработке продукта
этой реакции фтористым, водородом
CI3CSCI + HF —> FC12CSC1
Методы синтеза каптана, фолцида и дихлорфлуанида иллю-
стрируются приведенными ниже схемами:
(CH3)2NH + SO2C12 -----» (CH3)2NSO2C1 * >
NaOH
’--► (CH3)2NSO2NHC6H5
CISCFCIj
-------> (CH3)2NSO2N(C6H5)SCFC1,
дихлофлуанид
* P СССР принято название «эпареи». — Прим, перев.
490
Глава 12
Важное практическое значение имеют также производные
гуанидина. Ацетат н-додецилгуанидина (додин *) выпускается в
виде катионоактивного смачивающегося порошка, предназна-
ченного для борьбы с паршой яблони. Его получают по обыч-
ной реакции синтеза гуанидинов — взаимодействием додецил-
амина с цианамидом. Кроме того, используются алкилсульфаты
н-додецилгуанидина, которые синтезируют путем обработки н-
H-Cj2H2gNH2 NH2CN СНзСООН -—
—> [h-C12H25NHC(NH2)=NH2]+CH3COO'
додин
додециламина алкилсульфатами S-алкилизотиурония
. H-C12H25NH2
NH2CSNH2+ SO2(OR)j —> [RSC(NH2)=NH2]+ROSO2O’ _--н >
—> [C12H25NHCH(NH2)=NH2]+ROSO2O’
алкилсульфат додина
Системные фунгициды
Контактные фунгициды не способны ни вылечивать заболев-
шие растения, ни проникать в сосудистую систему растений.
Поэтому они не защищают от заражения новые побеги; кроме
того, находясь на поверхности растений, они подвергаются ат-
мосферным воздействиям. Для надежного предотвращения рас-
пространения болезни приходится обрабатывать растения фун-
гицидом несколько раз за сезон, что сопряжено с большими
затратами труда и машинного времени и значительным расхо-
дом химиката. К тому же такие патогенные грибы, как возбу-
дители вилта (семейство Verticillium), которые передвигаются
по сосудистой системе растений, не достигаются и не подав-
ляются контактными фунгицидами. Перечисленные недостатки
контактных фунгицидов делают необходимым применение си-
стемных фунгицидов — соединений, проникающих в сосудистую
систему растений и свободно перемещающихся по ней, оказы-
вая при этом искореняющее действие на клетки гриба и защи-
щая от поражения патогенами все растение, в том числе и но-
вые побеги. Поиск системных фунгицидов был начат в 40-х
годах, однако лишь в последние несколько лет английские, аме-
риканские, немецкие и японские химические фирмы разработали
ряд препаратов данного типа.
В 1966 г. было найдено, что производное 1,4-оксатиина кар-
боксин** дает превосходные результаты против пыльной головни
ячменя (Ustilago nuda). Возбудитель этой болезни находится
* В СССР этот фунгицид называется «ципрекс». — Прим, перев.
** Известен также под названием «витавакс». — Прим, перев.
Химикаты для сельского хозяйства
491
внутри семян и поэтому плохо подавляется контактными ртуть-
органическими фунгицидами. Сульфоновый аналог карбоксина,
оксикарбоксин, эффективен против возбудителей ржавчины зла-
ков и декоративных растений. Системные свойства этих соеди-
нений были продемонстрированы, в частности, полным подавле-
нием ржавчины на листьях бобов после внесения в почву фун-
гицида. Один из путей синтеза карбоксина (3) приводится ниже,
причем превращение его в оксикарбоксин (4) (окисление
сульфида в сульфон) осуществляют с помощью надкислоты.
CH3COCH2CONHCeH6 + SO2C12 —>
hoch2ch2sh носн2 СОСНз н+
—> CH3COCHClCONHCeHs ———> | —>
кон Н2СХ /СН
S 'VoNHCsHs
Г YCHs
\X\CONHC6H6
cxCH‘
%/X'CONHGeH5
Среди производных морфолина — другого гетероцикла, ис-
пользуемого в составе молекул фунгицидов, — наиболее изве-
стен тридеморф (5). Этот системный препарат, испытываемый
в настоящее время для борьбы с мучнистой росой злаковых
культур (Erysiphe graminis), синтезируют в две стадии, исходя
из тридециламина:
СН3
сн2—in—он H2so4
«-Ci3H27NH2 + 2Н2С-СНСНз —> н-С13Н27—N' ---->
\ / \сн2—СН—он
0 1
сн3
/СНз
5 (тридеморф)
Из производных ароматических гетероциклов прежде всего
рассмотрим пиримидины. Диметиримол, который был открыт в
1965 г., находит применение против мучнистой росы (Sphaero-
theca fuliginea) листьев и стеблей тыквенных (т. е. огурцов, ка-
бачков и т. д.). Это вещество эффективно при опрыскивании
492
Глава 12
растений и при внесении в почву. Так, одноразовое внесение
диметиримола в почву под огурцы предохраняет их от зараже-
ния на срок до 8 недель, тогда как обычные контактные фунги-
циды, которыми приходится опрыскивать растения приблизи-
тельно через каждые 10 дней, не способны полностью подавить
рост гриба. Синтез диметиримола осуществляют путем конден-
сации КЫ-диметилгуанидина (получаемого из диметиламина и
цианамида) с а-бутилацетоуксусным эфиром (т. 3, стр. 120)
С4Н9
Н-С4Н9ч zNH2 c2H5ONa СН’\
^СНСООС2Н5 + (CH3)2N</ ------->- [ I
СН3ССи ГЧН N4/NH
N(CH3)2
диметиримол
Родственный диметиримолу этиримол при покрытии им се-
мян ячменя подавляет возбудителя мучнистой росы в течение
всего сезона роста растения. Это соединение синтезируют ана-
логично предыдущему, за исключением того, что целевой про-
дукт необходимо отделять от изомерных N-этилкетопиримиди-
нов, не обладающих фунгицидной активностью.
С4Н9>. C2H6NH, C2H5ONa
;снсоос2н5+ ;c=nh_: сг -------------->
CH3CCr H2NZ
С4Н9
N^/NH
nhc2h5
этиримол меньшее следы
количество
Другой метод получения этиримола, схема которого приве-
дена ниже, менее пригоден для промышленной реализации, так
как здесь на последней стадии образуется токсичный метил-
меркаптан, который необходимо эффективно улавливать.
СЛ\ C2H5ONa
;chcooc2h6 + cs(nh2)2 ----->
CH3CCr
Химикаты Зля сельского хозяйства
493
С4Н9
N^/NH
SQH3
N4/NH
NHC2H5
этиримол
+ CH3SH
Хорошим системным фунгицидом является триаримол *, ак-
тивный против ряда грибковых болезней, в том числе парши и
мучнистой росы яблонь. Подробности о методе синтеза этого
препарата не сообщаются; известно только, что заключительной
стадией процесса служит конденсация 5-бромпиримидина с 2,4-
дихлорбензофеноном в присутствии н-бутиллития
триаримол
Производное бензимидазола беномил высокоактивно против
разнообразных возбудителей болезней, развивающихся на
листьях растений, и против почвенный патогенов. Один из луч-
ших путей синтеза беномила показан ниже (схема г):
NH2CN + С1СООСН,
NC—NHCOOCH3
o-C6h4(NH2)2
у-NHCOOCHj -ff^C0> Н |Г y~NHCOOCH3 (г) А» 1 conhc4h,
шк беномил
Близкими к беномилу фунгицидными свойствами обладают
производные тиоуреидомуравьиной кислоты NF-44 и NF-48,
* Вследствие выявившейся токсичности триаримол более йе Применяется,
й Дальнейшая разработка этого препарата фирмой прекращена (Системные
фунгициды, «Мир», М., 1975, стр. 81). «“ Прим, ред.
494
Глава 12
которые получают из о-фенилендиамина и метилового эфира
изотиоцианмуравьиной кислоты 5=С=ЫСООНз (генерируе-
мого in situ путем взаимодействия роданистого калия с метил-
хлорформиатом). Установлено, что эти соединения нестойки и
KSCX + CiCOOCHj
-------------->
^sx^NHCSNHCO ОСН,
NEf-48
-H2S
KSCN (избыток)
+ С1СООСЙЗ
aNHCSNHCOOCH3
NHCSNHCOOCH3
NF-44
ЁМК
легко превращаются в метиловый эфир бензимидазол-2-карб-
аминовой кислоты (БМК) — один из продуктов разложения
беномила. Отсюда следует, что по механизму действия они, ве-
роятно, аналогичны беномилу.
Антибиотики
Со времени открытия пенициллина предпринимались неодно-
кратные попытки бороться с болезнями растений с помощью
антибиотиков. Однако число антибиотиков, рекомендованных
для практического использования, очень невелико. Одним из
первых в сельскохозяйственную практику был введен стрепто-
мицин, который применяли в США против возбудителя бакте-
риального ожога груши (Erwinia amylovora). Этот антибиотик
рекомендован в Великобритании для борьбы с ложной мучни-
стой росой хмеля. Для предотвращения попадания столь мощ-
ного антибиотика в собранный хмель последнюю обработку
растений необходимо производить не позднее чем за 8 недель
до сбора урожая. В Японии сельскохозяйственные антибиотики
получили более широкое применение, особенно против возбуди-
телей пирикуляриоза (Piricularia oryzae) и бактериоза (Xantho-
monas oryzae) риса. Превосходные результаты при борьбе с пи-
рикуляриозом риса показал бластицидин S — производное цито-
зина, выделенное из Streptomyces griseochromogenes. Поскольку
этот антибиотик иногда вызывает повреждение растений, в не-
которых случаях вместо него используются более новые препа-
раты, в частности касугамицин, который обладает чрезвычайно
Химикаты для сельского хозяйства s 495
высокой активностью против возбудителей грибковых заболе-
ваний и безопасен для растений и млекопитающих.
Открытие системных фунгицидов является, несомненно, круп-
ным достижением науки. До сих пор препараты системного дей-
ствия, пассивно переносимые по ксилеме (от корней к листьям),
были наиболее эффективны для защиты лишь таких растений,
с которых урожай собирается ежегодно. Фунгициды, хорошо
передвигающиеся по сосудистой системе деревьев и кустарни-
ков, пока не разработаны; попытки борьбы с болезнью голланд-
ского ильма {Ceratosystis ulmi) путем инъекции химикатов в
ствол дерева в лучшем случае привели к частичному успеху.
Кроме того, еще предстоит создать фунгициды, способные про-
никать в растения через листья и передвигаться в них по фло-
эме (от листьев к корням).
Описано несколько примеров появления у патогенных гри-
бов растений резистентности к фунгицидам. Однако при совре-
менных темпах расширения применения системных фунгицидов,
которые в ряде случаев не являются общими ингибиторами фер-
ментов, а обладают специфическим механизмом действия, при-
обретенная резистентность грибов к фунгицидам может со вре-
менем стать серьезной проблемой. Мы пока еще очень мало
знаем о факторах, влияющих на передвижение фунгицидов вну-
три растений, и о механизме избирательной фунгицидной актив-
ности. Важное значение имеет также дальнейшее накопление
знаний о физиологии и биохимии патогенных грибов и о специ-
фике взаимоотношений между растением-хозяином и паразитом.
Большинство применяемых сегодня фунгицидов было открыто
случайно или в результате широкого скрининга; однако насту-
пит время, когда стайет возможно конструировать молекулы
фунгицидов на более научной основе.
496
Глава 12
Гербициды
Первые препараты для борьбы с сорняками уничтожали всю
растительность на обработанном участке (так называемые гер-
бициды сплошного действия). Одни из этих препаратов (такие,
как соединения мышьяка) были чрезвычайно опасны, другие
(такие, как креозот) представляли собой отходы химической
промышленности и применялись в огромных дозах по сравне-
нию с современными гербицидами. Старейшим из употребляе-
мых ныне гербицидов является хлорат натрия NaC103, который
используется в качестве гербицида сплошного действия для
дорог и дорожек. Однако он легко вымывается из почвы и
поэтому имеет меньшую продолжительность действия, чем по-
явившиеся позднее гербициды длительного действия.
Среди органических соединений гербицидные свойства рань-
ше всего были обнаружены у алкилдинитрофенолов, из кото-
рых, вероятно, наиболее важным в настоящее время является
диносеб. В основе действия этих препаратов лежит торможе-
ние окислительного фосфорилирования и синтеза АТФ (т. 4,
стр. 20). Поскольку данный биохимический процесс протекает
в организме как растений, так и млекопитающих, алкилдини-
трофенолы довольно токсичны, вследствие чего их использова-
ние постепенно сокращается.
ОН
no2
диносеб
Первым избирательным гербицидом была серная кислота,
которая, несмотря на очевидные недостатки, широко применя-
лась перед второй мировой войной в посевах злаковых культур.
Разбрызгиваемая кислота стекает с узких, имеющих воскопо-
добную поверхность листьев злаков и не повреждает их, тогда
как широколистные (двудольные) сорняки не только получают
больше кислоты, но и сильнее задерживают ее. Таким образом,
в данном случае имеет место морфологическая избирательность
гербицида.
Биохимическая избирательность была впервые открыта в
ряду так называемых гербицидных гормонов. В 1928 г. был
выделен первый гормон растений — индолилуксусная кислота
(ПУК) *• Это вещество оказалось высокоактивным стимулято-
* Обычное название этого соединения — гетероауксин. — Прим, перев.
Химикаты для сельского хозяйства
497
ром роста. Возникла мысль о том, что если нанести на растение
большое количество ИУК, то это приведет к слишком быстрому
н
ДУД
сщсоон
2М-4Х
росту растения и в конечном итоге к его гибели. Однако позд-
нее выяснилось, что растения способны метаболитически регу-
лировать содержание ИУК в своих клетках и, следовательно,
это соединение неэффективно в качестве гербицида. В 1940 г.
были получены синтетические аналоги ИУК, метаболизм кото-
рых протекает довольно медленно; поэтому растения, обрабо-
танные такими веществами, погибают из-за сильной деформации
в процессе неуправляемого роста. Наиболее важными гербици-
дами данного типа являются 2,4-дихлорфеноксиуксусная кис-
лота (2,4-Д) и 2-метил-4-хлорфеноксиуксусная кислота
(2М-4Х). По причинам, которые еще не вполне выяснены,
активность этих соединений против двудольных (широколист-
ных) сорняков гораздо выше, чем против однодольных (напри-
мер, сорных трав). Поэтому они широко применяются для
уничтожения широколистных сорняков в посевах злаковых
культур. Аналогичными гербицидными свойствами обладают и
гомологи этих соединений — (З-феноксипропионовые кислоты.
Так, мекопроп * используется для борьбы с мокрицей {Stellaria
media) и подмаренником цепким {Galium aparine), которые
устойчивы к феноксиуксусным кислотам.
. ОСН(СН3)СООН
снзч^ф
С1
мекопроп
В отличие от мекопропа его изомеры с прямой цепью
биологически неактивны. Установлено, что соединения общей
♦ В СССР известен как препарат 2М-4ХП. — Прим, перев.
498
Глава 12
формулы АгО(СН2)пСООН обладают гербицидными свойствами
только при нечетных значениях п. В основе этого явления ле-
жит биохимический процесс, известный под названием «р-окис-
ление», в результате которого метиленовая группа, находя-
щаяся в p-положении к карбоксилу, окисляется в карбониль-
ную группу и полученное соединение быстро распадается с
образованием кислоты, содержащей на два углеродных атома
меньше, чем исходная (т. 4, стр. 155). Например, 2-метил-4-
хлорфеноксимасляная кислота (2М-4ХМ) активна потому, что
в клетках растения она расщепляется до 2М-4Х. Некоторые
растения не способны осуществлять р-окисление и поэтому
2М-4ХМ 2М-4Х
устойчивы к действию феноксимасляных кислот, хотя погибают
при обработке соответствующими феноксиуксусными кислотами.
Вследствие этого феноксимасляные кислоты применяют в тех
случаях, когда основная культура и сорняки различаются по
способности к р-окислению — например, в посадках бобовых
культур, таких, как горох.
Однако последние данные свидетельствуют о том, что ре-
альная причина избирательного действия ароксиалкилкарбоно-
вых кислот имеет более сложный характер. Существуют расте-
ния, в которых наряду с р-окислением протекает еще более
быстрый процесс образования высших алифатических гомоло-
гов— вероятно, путем присоединения малонового эфира. Таким
образом, феноксимасляные кислоты выводятся из растений
главным образом в виде феноксиалкилкарбоновых кислот с
длинной алифатической цепью.
Феноксиуксусные кислоты получают в промышленности
взаимодействием соответствующего фенолята натрия с хлор-
уксусной кислотой. 2,4-Дихлорфеноксимасляную кислоту выра-
батывают из фенола и бутиролактона.
Из других гормональных гербицидов, используемых в сель-
ском хозяйстве, можно назвать 2,3,6-трихлорбензойную кислоту
и 2-метокси-3,6-дихлорбензойную кислоту (дикамба), относя-
щиеся к группе бензойных кислот, а также их гетероцикличе-
ский аналог циклорам. Получение этих соединений ведут по
Химикаты для сельского хозяйства
499
схемам, приведенным ниже:
2,3,6-трихлорбензойная
кислота
пиклорам
В основе гербицидного действия всех органических кислот ле-
жит нарушение пропорционального роста отдельных частей сор-
ных растений, причем характер нарушения, вызываемого раз-
личными препаратами, может быть разным. Было выдвинуто
представление, что молекулы соединений с этим типом герби-
цидной активности должны отвечать трем структурным крите-
риям: 1) содержать отрицательно заряженный атом; 2) иметь
на расстоянии 0,55 нм от него атом, несущий полный или ча-
стичный положительный заряд; 3) включать липофильную
группу, копланарную с положительно заряженным центром.
Всем этим требованиям удовлетворяют анионы описанных выше
соединений, в которых атомы хлора наводят своим индуктив-
ным эффектом частичный положительный заряд в бензольном
кольце, одновременно выполняющем функцию липофильной
группы. Дальнейшее развитие такого рода теоретических пред-
ставлений позволит более рационально подходить к синтезу но^
вых гербицидов.
500
Глава 12
Первыми соединениями, обнаружившими избирательную гер-
бицидную активность против сорных трав, были простые гало-
генсодержащие кислоты — трихлоруксусная и 2,2-дихлорпропио-
новая (далапон) *. Они находят применение для уничтожения
многолетних сорняков, например пырея ползучего, и сорных
видов овса, однако способны повреждать и злаковые культуры.
Далапон используется также для подавления водных сорных
растений.
Биохимический процесс, который протекает в растениях и
не имеет места в организме млекопитающих, — это фотохими-
ческое выделение кислорода из воды, которое является одной
из стадий процесса фотосинтеза. Известно много соединений,
ингибирующих эту реакцию, причем, как правило, они мало-
токсичны для теплокровных. Все гербициды, действие которых,
по-видимому, обусловлено преимущественно рассматриваемым
механизмом, можно разделить на три основные группы: амиды,
производные мочевины и триазины (известны также гербициды,
относящиеся к другим группам).
Практически наиболее важными гербицидами группы ами-
дов являются анилиды. Типичным представителем последних
может служить пропанид, применяемый против такого широ-
ко распространенного сорняка, как куриное просо (Е chino
chloa crusgalli). В то же время рис устойчив к пропаниду, по-
скольку в его растениях протекает быстрый гидролиз препарата
до анилина.
NHCOC2H8
NHCON(CH3)2
пропанид
R'NHCONR2
6
монурон
Производные мочевины с гербицидной активностью обычно
имеют структуру 6, где R— алкильная группа и R'— алкильная
или метоксигруппа. Путем варьирования этих групп, а также
арильной группы в ароматических аналогах синтезировано
большое число соединений с различными физическими свой-
ствами. Одним из наиболее важных веществ данного типа яв-
ляется монурон. Получение амидов и производных мочевины,
обладающих гербицидной активностью, осуществляется с по-
мощью обычных химических реакций; так, пропанид получают
* Имеются в виду не свободные кислоты, $ их натриевые (далапои) и
другие соли. — Прим, перев.
Химикаты для сельского хозяйства
601
из 3,4-дихлоранилина и пропионилхлорида *, а монурон — из
n-хлорфенилизоцианата и диметиламина.
Большинство триазиновых препаратов отвечают формуле 7,
где X и Y — алкиламино- или диалкиламиногруппы, a Z —
обычно С1, ОСНз или БСНз, хотя недавно были разработаны
препараты, содержащие иные заместители. Самые старые гер-
бициды группы триазинов, до сих пор сохранившие важное
практическое значение, — симазин и атразин, которые особенно
эффективны для борьбы с сорняками в посевах кукурузы. Уста-
Z
N^N
Cl
I
N^N
^NHCaH
7 симазин атразнн
новлено, что высокая устойчивость кукурузы к атразину и си-
мазину объясняется присутствием в ее растениях фермента,
вызывающего быстрое гидролитическое отщепление’атома хло-
ра, что приводит к детоксикации гербицида.
Производные триазина получают взаимодействием цианур-
хлорида с соответствующими нуклеофильными реагентами. По-
скольку при этом в ядро вступают электронодонорные замести-
тели, оставшиеся атомы хлора труднее поддаются нуклеофиль-
ному замещению, что позволяет проводить реакцию ступенчато.
Примером такого ступенчатого синтеза может служить получе-
ние десметрина
С1
N^N
CH3NH'^n^\NHCH(CH3)2
SCH3
I
N^N
CH3NH^n^NHCH(CH3)2
десметрин
Легко заметить, что все описанные выше гербициды, инги-
бирующие фотохимическое выделение кислорода из воды, имеют
* В промышленности применяют не пропионилхлорид, а пропионовую кис-
лоту. — Прим. ред.
502
Глава 12
общий структурный фрагмент — группу А, в которой X — атом
с неподеленной парой электронов (N или О). Полагают, что
этот фрагмент является токсофорной группой, ответственной за
образование связи с ферментом, участвующим в фотохимиче-
ском выделении кислорода из воды, что приводит к инактиви-
рованию указанного фермента; однако детали данного процесса
пока неизвестны. В последние годы появились более сложные
соединения, содержащие этот токсофор, — в частности, произ-
водные урацила, важным примером которых является бром-
ацил. Другую группу гербицидов, включающих тот же токсо-
форный фрагмент, составляют карбаматы, например барбан.
NHCOOCH2C=CCH2C1
бромацил
барбаи
Однако эти соединения слабо ингибируют фотохимическое вы-
деление кислорода из воды; на основании этого полагают, что
они уничтожают сорняки за счет торможения клеточного деле-
ния. Барбан применяется для уничтожения овсюга в злаковых
культурах. Это соединение получают из соответствующего
спирта и лг-хлорфенилизоцианата.
Фирма ICI разработала в 1956 г. два важных гербицида,
обладающих своеобразными свойствами и совершенно новым
механизмом действия, — дипиридилиевые соли паракват и ди-
кват. Оба эти препарата очень эффективно уничтожают зеленую
растительность. Попадая в почву, они теряют свою активность,
дикват
паракват
и поэтому их широко используют для очистки полей от сорня-
ков непосредственно до или после сева. Это позволяет в ряде
случаев отказаться от вспашки, заменив ее более простыми
агротехническими мероприятиями, сохраняющими структуру
почвы и уменьшающими эрозию *. Дипиридилиевые гербициды
* Так называемое беспахотное земледелие. — Прим, перев. ’ '
Химикаты для сельского хозяйства
503
применяются также для предзимней очистки жнивья от стерни,
уничтожения сорняков на огородах и в фруктовых садах и для
многих других сельскохозяйственных целей. Дикват, кроме того,
находит применение для предуборочной десикации (удаления
листьев с целью облегчения уборки урожая) ряда культур и
для борьбы с зарастанием водоемов.
Гербицидное действие дипиридилиевых солей основано на
включении этих веществ в основную цепочку окислительно-вос-
становительных реакций, которые протекают при фотосинтезе.
Паракват восстанавливается при этом в резонансно-стабилизи-
рованный радикал, который затем вновь окисляется атмосфер-
ным кислородом, давая весьма токсичные для клеток растения
перекисные соединения.
Паракват получают путем восстановления пиридина натрием
в жидком аммиаке. Возникающие ион-радикалы димеризуются
исключительно в положении 4, превращаясь в дигидродипири-
дил, который далее подвергают окислению и кватернизации.
Na/NHj
паракват
Технология этого процесса довольно сложна, что отражает тен-
денцию к возрастанию сложности синтеза новых гербицидов.
Существует ряд ценных гербицидов, которые не относятся
ни к одной из рассмотренных выше групп. Прежде всего сле-
дует упомянуть амитрол * — гербицид сплошного действия, ко-
торый, как полагают, блокирует синтез белков в клетках расте-
ний. Дихлобенил является предвсходовым гербицидом, т. е. он
вносится на поля до прорастания семян сорняков. Кроме того,
дихлобенил применяют в качестве водного гербицида. Посколь-
ку он довольно летуч, было испытано большое число его срав-
нительно нелетучих производных, обладающих более длитель-
ным сроком гербицидного действия в полевых условиях. Из них
* В СССР применение препарата запрещено вследствие выявившегося
канцерогенного действия. — Прим. ред.
504
Глава 12
практическое применение нашел хлортиамид — аддукт серово-
дорода с дихлобенилом, который в почве медленно выделяет по-
следний. Нитрофен тоже используется как предвсходовый гер-
бицид для уничтожения сорняков в растениях семейства кре-
стоцветных (рапсе, капусте листовой, капусте кочанной и т. д.)
и других культур. Недавно было запатентовано несколько хи-
мически родственных препаратов, в том числе пиразон, входя-
щий в группу N-фенилпиридазинонов. Его применяют главным
образом в посевах сахарной свеклы, так как это растение
способно детоксифицировать пиразон. Пиразон действует на
сорняки, ингибируя процесс фотосинтеза; возможно, что он
близок по механизму действия к рассмотренным выше герби-
цидам, блокирующим фотохимическое выделение кислорода из
воды. Синтез этого соединения осуществляют путем взаимодей-
ствия фенилгидразина с мукохлорной кислотой с последующим
аммонолизом продукта.
мукохлорная
кислота
пиразон
Иоксинил используется для уничтожения сорняков в злаках
(обычно в виде смеси с гербицидами группы феноксиуксусных
кислот). Механизм биохимического действия иоксинила послу-
жил предметом широкой дискуссии. Было установлено, что он
разобщает окислительное фосфорилирование и поэтому может
быть близок по механизму действия к нитрофенолам. Трифлу-
ралин применяется для предвсходовой обработки сорняков в
культуре хлопчатника.
CN
ОН
иоксинил
трифлуралин
В настоящее время имеются гербициды для борьбы с боль-
шинством встречающихся в мире сорных растений. Поэтому
недостаточно открыть новые соединения, обладающие гербицид-
ными свойствами, — надо, чтобы они были лучше уже суще,
Химикаты для сельского хозяйства
505
ствующих препаратов, т. е. дешевле, более избирательны, без-
опаснее в обращении или более приемлемы по персистентности.
За последние 25 лет характер сорной растительности на сель-
скохозяйственных угодьях Великобритании претерпел значи-
тельные изменения. В результате главным образом применения
гормональных гербицидов широколистные сорняки, некогда
бывшие обычными спутниками злаков, почти исчезли, а их ме-
сто заняли новые виды сорных растений, более похожие на
злаковые. Основной проблемой в производстве злаковых куль-
тур сегодня является борьба с овсюгом (Avena fatua). Для ре-
шения этой и других проблем была организована широкая сеть
научно-исследовательских и прикладных институтов, занимаю-
щихся синтезом и испытаниями новых гербицидов. Прошло ме-
нее 40 лет с тех пор, как в сельскохозяйственной практике
начали применять органические гербициды, причем в течение
всего этого времени разработка новых препаратов велась пре-
имущественно эмпирическими методами. Хотя мы можем и
впредь надеяться на случайные открытия, становится все более
необходимым учитывать при создании новых гербицидов био-
химические процессы, протекающие в клетках растений, и фи-
зико-химические свойства, которыми должно обладать соедине-
ние, чтобы оно могло проникать в растение через корни и листья
и переноситься по его сосудистой системе.
Регуляторы роста растений
Все замечательные физиологические изменения, происходя-
щие с растениями в течение их жизни, протекают с участием
химических веществ. Одно из таких веществ, индолилуксусная
кислота (ИУК), упоминалось выше. Возможность использова-
ния химических соединений с целью направленного воздействия
на рост растений давно привлекает внимание химиков и биоло-
гов и является весьма актуальным вопросом в настоящее время.
В числе желательных морфологических изменений растений
сельскохозяйственных культур, особенно в наш век все более
высокой механизации уборки урожая, можно назвать повыше-
ние однородности плодов по размерам и степени зрелости,
ослабление их связи с плодоножкой к моменту сбора урожая
и изменение формы растений с целью облегчения их роста и
(Или) снятия плодов. Кроме того, было бы крайне заманчиво
найти химические препараты, которые при внесении их в почву
вместе с удобрениями повышали урожай. Другие полезные эф-
фекты регуляторов роста приводятся ниже.
2-Нафтилуксусная и 3-индолилмасляная кислоты, являю-
щиеся структурными аналогами природного регулятора роста
S06
Глава 12
ИУК, применяются в качестве стимуляторов корнеобразования
при вегетативном размножении растений черенками.
СНдСООН
гшМерелловаЯ
кислота
&нафтилуксуатя 3-индолилмасляная
кислота кислота
Вторую группу ростовых регуляторов, встречающихся в при-
роде, составляют гиббереллины. Один из них, гибберелловая
кислота, используется в различных областях, особенно в пиво-
варенном производстве (для ускорения прорастания ячменя).
Простейшим регулятором роста растений является этилен,
который оказывает на них самое различное воздействие: угне-
тает рост, ускоряет абсциссию (опадение листьев, цветов и пло-
дов) и стимулирует созревание и цветение на соответствующих
стадиях развития растений. В садоводстве «химически связан-
ный» этилен используется в форме препарата этрел * для уско-
рения созревания фруктов и облегчения их отделения. Новой
областью применения этрела, которая приобретает все болёё
важное значение, является стимулирование движения латекса
у гевеи. В растениях этрел претерпевает распад по механизму,
изображенному ниже, выделяя этилен:
о
•Н .х'-'х !IUo—н
С1-СНИСН>1<П „
»трел
С1СН2СН2Х(СН3)з ср
хлормшатхлориВ >
Существуют также другие регуляторы роста растеййй, син-
тетические аналоги которых могут найти практическое приме-
нение в будущем.
Одним из наиболее важных по своим свойствам ростовых
регуляторов является хлормекват **. Он вызывает уменьшение
длины стебля и используется для обработки злаковых культур,
чтобы предотвратить сгибание их растений под действием веса
колосьев (полегание). Одновременное увеличение нормы внесе-
ния удобрений позволяет повысить урожай.
Применяемая в садоводстве сукцинамовая кислота (препарат
В-9) задерживает рост растений, например хризантем, делая
* 2-Хлорэтилфосфоновая кислота. — Прим, перев.
** В СССР принято название «препарат ТУР», — Прим, перев.
Химикаты для сельского хозяйства
507
их более пригодными для выращивания в горшках. Для анало-
гичной цели — торможения роста травы на обочинах дорог —
употребляется гидразид малеиновой кислоты. Еще одним инги
битором роста является хлорфлурекол.
CH2CONHN(CH3)2
СН2СООН
В-9 гидразид малеи- хлорфлурекол
новой кислоты
В заключение необходимо упомянуть о химических веще-
ствах, стимулирующих кущение растений. В качестве таких ве-
ществ практическое применение нашли жирные спирты с пря-
мой цепью (главным образом октиловый и дециловый), которые
подавляют рост верхушечных почек и тем самым усиливают
кущение многих видов растений. Эти соединения используются
также в табаководстве (для подавления пасынков табака) и
в разведении хризантем.
Использование регуляторов роста растений в сельском хо-
зяйстве еще только начинается. Однако уже сейчас можно ви-
деть огромные выгоды, которые сулит широкое применение
этих препаратов.
СПИСОК ЛИТЕРАТУРЫ
Инсектициды
Reay R. С., Insects and Insecticides, Oliver and Boyd, Edinburgh, 1969.
O’Brien R. D., Insecticides — Action and Metabolism, Academic Press,
London, 1967.
Berkoff С. E., «The Chemistry and Biochemistry of Insect Hormones», Quart.
Rev., 23, 372 (1969).
Jacobson M., Grosby R. G., Naturally Occuring Insecticides, Marcel Dekker,
New York, 1971.
Фунгициды.
Large E. C., The Advance of the Fungi, Jonathan Cape, London, 1940.
Woodham-Smith C., The Great Hunger, Hamish Hamilton, London, 1963.
Wheeler В- B- l-i An Introduction to Plant Diseases, Wiley, London, 1969*
508
Глава 12
Wegler R. (Ed.), Chemie der Pflanzenschutz- und Schadlings-bekampfung-
smittel, Vol. 2, Springer-Verlag, Berlin, 1970.
Гербициды
Audus L. J. (Ed.), The Physiology and Biochemistry of Herbicides, Acade-
mic Press, London, 1964.
Fryer J. D., Makepeace R. J. (Eds.), Weed Control Handbook, 2 vols.,
Blackwell, Oxford, 1972.
Davies D. D., Giovanelli J., Ap Rees T., Plant Biochemistry, Blackwell,
Oxford, 1964.
Fogg G. E-, The Growth of Plants, Penguin, Harmondsworth, 1966.
ГЛАВА 13
Моющие вещества
К. Джонс
Jones К., Unilever Research, Port Sunlight Laboratory
Процесс мытья
Дать определение тому процессу, который мы называем
мытьем, довольно просто. Например, можно сказать, что
мытье — это очистка поверхности загрязненного материала в
ванне с жидкостью, содержащей растворенное вещество (или
вещества) — так называемое моющее вещество (детергент), —
которое способствует удалению загрязнений. Однако все осталь-
ное гораздо сложнее. Физико-химическая природа многочислен-
ных факторов, участвующих в процессе мытья, и их относи-
тельное значение существенно зависят от типа субстрата, ха-
рактера удаляемого загрязнения и условий мытья (концентрации
моющего вещества, температуры, степени перемешивания).
Стоит лишь вспомнить о том, насколько разнообразные опера-
ции мытья повседневно выполняют домохозяйки — стирка
одежды, мытье посуды и полов и, конечно, мытье рук и тела,—
чтобы увидеть, как велико число встречающихся субстратов и
как различна природа их загрязнений.
В большинстве применяемых на практике моющих систем
растворителем служит вода; поэтому в данной главе для про-
стоты рассматриваются только моющие вещества, предназна-
ченные для использования в водной среде.
Хорошая моющая система должна выполнять двоякую функ-
цию: удалять загрязнение с очищаемой поверхности и диспер-
гировать или растворять это загрязнение, не допуская его об-
ратного осаждения на отмытый субстрат. Решающее значение
чдля выполнения этих требований имеет природа сил, действую-
щих на поверхностях раздела между субстратом, загрязнением
и моющей жидкостью. Правильно составленная моющая компо-
зиция изменяет свойства этих поверхностей таким образом, что
энергия связи загрязнения с субстратом понижается. Последнее
достигается путем введения в композицию веществ, способных
адсорбироваться на пограничных поверхностях, т. е. обладающих
510
Глава 13
поверхностной активностью. Эти так называемые поверхно-
стно-активные вещества (ПАВ) можно представить как со-
единения общей формулы RX, где R — гидрофобный «хвост»
(обычно углеводородный остаток) и X — гидрофильная «голо-
ва», например группа —СОО-. В зависимости от природы R
и X такие вещества могут быть как водорастворимыми, так
и маслорастворимыми, причем соотношение гидрофильности
«головы» и гидрофобности «хвоста» в значительной степени
определяет моющие свойства соединения.
Углеводородный «хвост» водорастворимого ПАВ трудно сов-
мещается с окружающей водной средой. Однако причина это-
го— не отталкивание между ним и молекулами воды. Силы
вандерваальсова притяжения между углеводородной цепью и
молекулами воды даже несколько превосходят силы притяже-
ния между отдельными углеводородными цепями, но суще-,
ственно уступают силам притяжения между молекулами воды.
Жидкая вода имеет трехмерную структуру, в которой ее моле-
кулы соединены водородными связями, непрерывно разрываю-
щимися и образующимися вновь. Углеводородные цепи раство-
ренного ПАВ нарушают взаимодействие между ближайшими
молекулами воды, изменяя ее структуру. В большинстве случаев
раствор стремится сохранить структуру воды, вследствие чего
молекулы ПАВ вынуждены занимать такое положение, в кото-
ром их цепи были бы, по крайней мере частично, удалены из
объема раствора. Наиболее очевидный путь к достижению этой
цели состоит в накоплении молекул ПАВ на границе между
водным раствором и воздухом, а также на поверхностях раз-
дела с частицами масла и твердых веществ, если они присут-
ствуют в системе. Такая адсорбция ПАВ на пограничных по-
верхностях оказывает очень большое влияние на свойства по-
следних и особенно на межфазные натяжения, которые сильно
понижаются. Например, поверхностное натяжение 10-3 М рас-
твора ПАВ при 20 °C составляет около 0,3—0,4 мН/см, тогда
как для чистой воды эта величина равна 0,73 мН/см. При уве-
личении концентрации ПАВ в воде наступает момент, когда его
молекулы должны изыскивать другой способ удаления своих
углеводородных «хвостов» из объема раствора. Поэтому они
начинают соединяться в агрегаты более или менее правильной
сферической формы, в которых их гидрофобные участки ориен-
тированы внутрь, а полярные «головные» группы — наружу. Та-
кие агрегаты называются мицеллами, а концентрация, при
которой начинается их образование, — критической концентра-
цией мицеллообразования (ККМ). При концентрациях ниже
ККМ термодинамические свойства растворов ПАВ близки к тем,
которыми должны обладать разбавленные растворы, содержа-
щие неассоциированные молекулы. Однако при более высоких
Моющие вещества БИ
концентрациях коллигативные свойства растворов (например,
осмотическое давление) изменяются с концентрацией медлен-
нее, чем следовало бы ожидать, поскольку все молекулы ПАВ
сверх ККМ ассоциированы в мицеллы. Так как каждая ми-
целла включает большое число молекул (обычно от 20 до 200),
переход от относительно идеального к существенно неидеаль-
ному поведению происходит в очень узком интервале концен-
траций. Аналогичным образом изменяются с концентрацией и
другие, нетермодинамические свойства растворов ПАВ (прово-
димость, коэффициент диффузии и т. д.).
Роль ПАВ в удалении загрязнений
Для того чтобы объяснить, каким образом ПАВ способ-
ствуют удалению загрязнений, необходимо рассмотреть силы,
удерживающие загрязнение на субстрате, и явления, происхо-
дящие в процессе мытья. В состав загрязнения могут входить
масляные жидкости, способные течь и, следовательно, изменяю-
щие свою форму при удалении с поверхности субстрата (остат-
ки пота на одежде или остатки жирной пищи на посуде), и
частицы твердых веществ, переходящие в моющий раствор без
изменения формы (например, сажа и окислы металлов). Хотя
многие явления, участвующие в процессах удаления обоих ви-
дов загрязнений, одни и те же, каждый из них имеет и важные
отличительные особенности. Возьмем, например, сухую ткань,
содержащую твердое загрязнение (в данном случае загрязне-
ние удерживается на поверхности материала вандерваальсовы-
ми силами притяжения). Если погрузить ее в водный раствор
ПАВ и начать перемешивать, то частицы загрязнения будут
механически отрываться от поверхности ткани и переходить
в раствор. Вслед за этим происходит смачивание тех участков
поверхности ткани и частиц загрязнения, которые ранее нахо-
дились в контакте друг с другом. В результате смачивания
водным раствором ПАВ эти участки, как и остальная поверх-
ность ткани и твердых частиц, приобретают электрический за-
ряд, обусловленный адсорбцией на них ионов из раствора или
ионизацией поверхностных полярных групп. Поскольку анионы
адсорбируются легче, чем катионы, в большинстве случаев по-
верхность очищаемого материала становится отрицательно за-
ряженной. Возникающий адсорбционный заряд частично ней-
трализуется противоположным по знаку зарядом диффузного
слоя противоионов, находящихся в растворе вблизи межфазной
поверхности. Таким образом, на каждой поверхности обра-
зуется двойной электрический слой, а так как поверхности и
ткани, и частиц загрязнения обычно заряжаются отрицательно,
612 Глава 13
эти двойные слои взаимно отталкиваются, что способствует уда-
лению загрязнения.
В том случае, когда в растворе присутствует анионное ПАВ
(как в большинстве применяемых на практике моющих си-
стем), оно будет по указанным выше причинам адсорбироваться
на частицах загрязнения и на ткани. Адсорбция анионов ПАВ
происходит даже несмотря на то, что указанные поверхности
уже имеют отрицательный заряд, и сопровождается дальней-
шим увеличением этого заряда. Последнее в свою очередь при-
водит к значительному возрастанию отталкивания между двой-
ными электрическими слоями частиц загрязнения и ткани, что,
естественно, облегчает процесс мытья.
Рис. 13.1. Смачивание твердой, поверхности маслом (усв, усм
и умв — межфазные натяжения на поверхностях раздела субстрат — вода,
субстрат — масло и масло — вода соответственно).
При отделении твердого загрязнения от ткани вместо меж-
фазной поверхности частица загрязнения — ткань возникают
две новые межфазные поверхности твердое тело — жидкость.
Адсорбция моющего вещества на этих новых поверхностях су-
Щественно понижает их энергию и тем самым уменьшает ра-
боту, затрачиваемую на их образование, т. е. на удаление за-
грязнения.
Процесс удаления жидкого загрязнения может включать,
помимо рассмотренных выше, ряд специфических явлений, та-
ких, как скатывание (rolling up) капель загрязняющей жидко-
сти, ее солюбилизация и образование смешанных фаз.
Явление «скатывания» наблюдается в том случае, когда
Поверхность субстрата смочена масляным загрязнением. При
погружении такого субстрата в раствор моющего вещества бу-
дет происходить предпочтительное смачивание его поверхности
водной фазой, вызывающее удаление масла. Это можно проил-
люстрировать рис. 13.1, изображающим каплю масляного за-
грязнения на субстрате, погруженном в воду. В точке X, в ко-
торой капля соприкасается с поверхностью субстрата, действуют
Моющие вещества
513
три силы межфазного натяжения: субстрат — вода (усв), суб-
страт— масло (уСм) и масло — вода (уМв)- Краевой угол
смачивания субстрата маслом обозначим буквой 0. Все эти ве-
личины связаны между собой соотношением
. Уев = Уем + Умв COS 0 (1)
откуда
cos0 = -v—(2)
Умв
Если водная фаза содержит маслонерастворимое ПАВ, то
оно будет адсорбироваться на поверхностях раздела масло —
вода и субстрат — вода и понижать как уСв, так и умв. При
отрицательном значении cos 0 (как в случае, показанном на
рис. 13.1) оба эти изменения приведут к уменьшению cos 0, т. е.
к увеличению 0. Данная здесь трактовка «скатывания» очень
сильно упрощена, однако строгий анализ этого явления тоже
показывает, что добавление ПАВ вызывает увеличение 0 (см.
список литературы в конце главы). Другими словами, ПАВ
заставляет загрязнение как бы скатываться с субстрата, умень-
шая площадь их соприкосновения. Однако, даже если эта
площадь уменьшится почти до нуля (0 « 180°), скатившееся
загрязнение будет продолжать удерживаться на субстрате
вандерваальсовыми силами притяжения, т. е. возникает кар-
тина, описанная выше применительно к удалению твердого
загрязнения. Тем не менее очевидно, что явление «скатывания»
существенно облегчает удаление жидких загрязнений.
Многие весьма распространенные загрязнения представляют
собой полужидкие вещества, и для удаления их по механизму
«скатывания» необходимо понизить их вязкость (и, следова-
тельно, повысить текучесть), чтобы в процессе мытья они могли
достаточно быстро изменить свою форму. Отсюда следует, что
в этих случаях надо использовать повышенную температуру.
Некоторые ПАВ способны также проникать в глубь полужид-
ких загрязнений и образовывать с ними комплексы или вызы-
вать перестройку их структуры, сопровождающуюся коренными
изменениями физических свойств загрязнения. Эти изменения
тоже могут вносить существенный вклад в легкость удаления
загрязнения.
В рассмотренных выше примерах моющее действие ПАВ
обусловлено адсорбцией их молекул на межфазных поверхно-
стях. С другой стороны, солюбилизация масляного загрязнения
зависит от способности ПАВ образовывать мицеллы, поскольку
последние ведут себя в водной среде как неполярная фаза,
в которой могут растворяться маслянистые вещества. В боль-
шинстве процессов мытья солюбилизация играет относительно
17 Зак. 501
514
Г лава 13
небольшую роль, поскольку ПАВ обычно берется в низкой
концентрации (например, при стирке в домашних стиральных
машинах — в концентрации от 0,01 до 0,3%) и его хватает лишь
для частичной солюбилизации масляного загрязнения. Един-
ственным важным исключением является мытье кожи и волос
с использованием туалетного мыла. Так, при мытье рук кон-
центрация мыла в растворе достигает 10%, и в этих условиях
солюбилизация может вносить существенный вклад в процесс
удаления загрязнений.
Обратное осаждение загрязнения
Если мы хотим получить в результате мытья чистый суб-
страт, необходимо предотвратить возможность обратного оса-
ждения (ресорбции) удаленного загрязнения. В предыдущем
разделе было показано, что в большинстве процессов мытья
основная часть загрязнения не растворяется, а диспергируется
в моющем растворе. Поэтому при отсутствии некоторого барье-
ра, препятствующего ресорбции, она неизбежно будет проис-
ходить при каждом соударении частиц загрязнения с отмытым
субстратом.
Молекулы ПАВ, адсорбированные на поверхности частиц за-
грязнения и отмытого субстрата, затрудняют обратное осажде-
ние по нескольким различным механизмам. Как было описано
выше, в результате адсорбции ионизированных молекул ПАВ
усиливается взаимное отталкивание двойных электрических
слоев твердых частиц или капелек загрязнения, с одной сто-
роны, и отмытого субстрата, с другой, что не только способ-
ствует удалению загрязнения, но и создает барьер для его
повторного осаждения. Обычно адсорбция ПАВ на частицах
загрязнения и субстрате происходит таким образом, что угле-
водородные «хвосты» ориентируются внутрь, а полярные «го-
ловные» группы — наружу. Последние сильно гидратируются, и
образовавшиеся плотные гидратные оболочки играют роль до-
полнительных барьеров, препятствующих ресорбции. Кроме
того, адсорбированные молекулы ПАВ понижают поверхност-
ную энергию частиц загрязнения и субстрата, уменьшая тем
самым движущую силу их соединения.
Все эти явления имеют положительное значение для пред-
отвращения обратного осаждения. Однако ресорбция все же
продолжает оставаться серьезной практической проблемой, осо-
бенно на стадии полоскания выстиранных изделий, когда ПАВ,
адсорбированное на частицах загрязнения и субстрате, частич-
но десорбируется в промывную ванну. Для подавления про-
цесса ресорбции в товарные моющие средства вводят специаль-
ные антиресорбционные добавки. Наиболее важной из них
Моющие вещества
515
является натриевая соль карбоксиметилцеллюлозы (целлюлоза,
в которой около половины ОН-групп замещено на группы
ОСНгСООЫа), эффективно предупреждающая обратное оса-
ждение загрязнений на хлопчатобумажные ткани. Механизм
действия этой добавки окончательно не установлен. Однако,
поскольку натриевая соль карбоксиметилцеллюлозы представ-
ляет собой анионоактивный полиэлектролит, считают, что она
адсорбируется на поверхности частиц загрязнений и (или)
хлопка и тем самым усиливает отталкивание их двойных элек-
трических слоев.
Активные добавки
Проведенное выше обсуждение касалось лишь поведения
ПАВ в деионизированной воде. В растворах, содержащих дру-
гие электролиты, особенно соли с многовалентными катионами,
ПАВ ведут себя несколько иначе, и это необходимо принимать
во внимание при рассмотрении реальных моющих систем.
Возьмем в качестве примера стирку в домашних условиях —
вероятно, наиболее важный из процессов мытья. Многие дома
снабжаются жесткой водой, содержащей ионы Са2+ и Mg2+,
и хорошее моющее средство должно эффективно отмывать за-
грязнения в такой воде. Ионы жесткости оказывают неблаго-
приятное влияние на моющую способность ПАВ различными
способами. Во-первых, кальциевые соли многих ПАВ плохо
растворимы в воде, вследствие чего в жесткой воде ПАВ будет
растворяться в лучшем случае лишь частично и стирка будет
малоэффективной. Во-вторых, одежда часто бывает загрязнена
потом, содержащим жирные кислоты; в жесткой воде они дают
нерастворимые кальциевые соли, которые не удаляются с ткани.
Третьим и, пожалуй, самым неприятным результатом присут-
ствия в моющей среде ионов жесткости является повышенная
концентрация электролитов, уменьшающих взаимное отталки-
вание двойных электрических слоев. Особенно сильное влияние
оказывают многовалентные катионы, которые значительно об-
легчают ресорбцию и мешают удалению загрязнений с поверх-
ности ткани.
Поэтому хорошее моющее средство должно эффективно вы-
водить ионы Са2+ и Mg2+ из раствора. С этой целью в состав
моющих композиций включают так называемые активные до-
бавки, которые либо изолируют ионы жесткости, либо осаждают
их. Типичными примерами активных добавок могут служить
этилендиаминтетрауксусная кислота, полиакрилат натрия и наи-
более важная из применяемых в настоящее время добавок —
триполифосфат натрия (Па5РзОю) .
17*
616
Глава 13
Как указывалось в начале этой главы, процесс мытья вклю-
чает множество различных явлений, а на практике приходится
иметь дело с огромным разнообразием субстратов и загрязне-
ний. Вследствие этого создание общей теории процесса мытья
невозможно. Проведенное выше обсуждение преследовало цель
показать лишь некоторые из важнейших аспектов поведения
ПАВ в процессе мытья. Более подробные сведения о механизме
мытья читатель может найти в работах, приведенных в списке
литературы.
Мыла и мыльные моющие средства
Жировые мыла, вырабатываемые из натуральных жиров и
масел, являются классическими ПАВ. Натриевое мыло употреб-
ляется с незапамятных времен. Несмотря на то что после вто-
рой мировой войны объем потребления этого продукта сокра-
тился, он продолжает сохранять почти монопольное положение
в мировом производстве туалетных мыл, а в развивающихся
странах твердое кусковое мыло до сих пор служит наиболее
важным моющим средством для стирки. В Великобритании су-
щественная часть стиральных порошков изготовляется на основе
мыла.
Природные жиры и масла представляют собой смеси три-
глицеридов, т. е. сложных эфиров глицерина с жирными кис-
лотами (карбоновыми кислотами с длинной углеводородной
цепью; см. т. 1, стр. 154, и т. 4, стр. 150). Мыла — это соли
жирных кислот, получаемые путем омыления триглицеридов рас-
творами щелочей, обычно едкого натра.
CH2OOCR‘ R1 COONa СН2ОН
I + I
CHOOCR2 + 3NaOH —> R2 COONa + СНОН
I + I
CH2OOCR3 R’COONa CH2OH
Важнейшим сырьем для производства мыла являются твер-
дый животный жир, получаемый из коровьих и бараньих туш,
и кокосовое масло. В табл. 13.1 приведен приближенный состав
жирных кислот, присутствующих в этих смешанных триглицери-
дах. Кроме них, используются и другие виды сырья (в зависи-
мости от их доступности и цены), такие, как пальмовое масло,
содержащее главным образом жирные кислоты Ci6 и Ci8, и
пальмоядровое масло, сходное по составу с кокосовым. В ряде
случаев масла подвергаются перед омылением различным опе-
рациям, например осветлению кислой фуллеровой землей для
удаления окрашенных примесей или (при использовании масел
Моющие вещества
617
Таблица 13.1
Жирнокислотный состав твердого животного жира
и кокосового масла
Содержание, %
Кислота
Формула
Каприловая
Каприновая
Лауриновая
Миристино-
вая
Пальмити-
новая
Стеариновая
Олеиновая
Линолевая
СНз(СН2)6СООН СН3(СН2)8СООН СН3(СН2)10СООН СН3(СН2)иСООН 8-9 7-10 45-51 16-18 2—4
СН3(СН2)14СООН 7-10 29-32
СН3(СН,)16СООН 1-3 19-26
Чис-СН3(СН2)7СН=СН(СН2)7СООН 5—8 40-43
цис, цис-СН3(СН2)4СН=СНСН2СН=СН(СН2)7СООН 1—3 2
с высоким содержанием ненасыщенных глицеридов) частичной
гидрогенизации (т. 1, стр. 156) с целью улучшения их цвета
и стабильности.
Омыление
Условия процесса мыловарения должны обеспечивать не
только омыление триглицеридов, но и получение мыла с низким
содержанием воды, удаление окрашенных примесей и отделе-
ние побочно образующегося глицерина простым ,и удобным ме-
тодом.
Классический процесс, который применяется уже много лет
и до сих пор играет важную роль в мировом производстве мыла,
заключается в нагревании жира с едким натром в открытых
котлах, снабженных устройствами для подвода обогревающего
пара и для разделения фаз. В одном из вариантов этого про-
цесса жировое сырье подвергают омылению при точно регу-
лируемых значениях температуры, содержания воды и щелочи,
с тем чтобы полученное мыло содержало лишь очень небольшое
количество избыточной щелочи. Затем добавляют сухую соль,
в результате чего смесь разделяется на две фазы — мыльное
ядро и подмыльный щелок, причем последний представляет со-
бой разбавленный водный раствор соли, в котором содержится
примерно половина образовавшегося глицерина. Эти фазы раз-
деляют и к мыльной массе добавляют воду и соль, чтобы из-
влечь дополнительное количество глицерина. После удаления
518
Глава 13
второго щелока мыльное ядро обрабатывают строго дозиро-
ванным количеством воды и отделяют образовавшийся мыльный
клей (разбавленный раствор, содержащий окрашенные примеси
и немного мыла). Полученное ядровое мыло, в котором присут-
ствует около 30% воды, подвергают дальнейшей переработке.
Существует много вариантов описанного процесса, причем
все они требуют большой затраты времени из-за многочислен-
ности последовательных операций и длительности отстаивания.
Разработан ряд более быстрых непрерывных процессов, которые
зачастую более экономичны, чем периодический котловой про-
цесс. Одни из них являются модификациями котлового процесса
и включают те же операции щелочного омыления жирового
сырья и высаливания образовавшейся массы с последующим
разделением фаз. Другие базируются на совершенно иной схеме
и включают гидролиз жира водой при высоких температурах
(~ 250 °C) и давлениях [4—5 М.Н/м2 (40—50 атм)] в противо-
точных реакторах колонного типа, отделение образовавшихся
сырых жирных кислот от водного глицерина и очистку жирных
кислот путем дистилляции с последующей нейтрализацией их
едким натром или другой щелочью.
Получение моющих средств на мыльной основе
Туалетные мыла. Ядровое мыло, полученное омылением жи-
ров, содержит около 30% воды. Прежде всего содержание воды
снижают до --—12%. Затем вводят небольшое количество доба-
вок (парфюмерные отдушки, антикоагулянты типа этиленди-
аминтетрауксусной кислоты, отбеливатели типа TiO2 или краси-
тели, а в некоторых случаях также гермициды) и смесь
тщательно перемалывают в многоступенчатых вальцовых мель-
ницах, получая однородную мыльную стружку. Эту стружку
пропускают через шнек-пресс (червячный экструдер, напоми-
нающий большую мясорубку), производящий прессование
стружки и выдавливание непрерывного мыльного бруска, из
которого штампуют готовое кусковое мыло. Обычно туалетное
мыло содержит 20—50% кокосового мыла и 50—80% мыла из
твердого животного жира; иногда оно содержит также до 10%
свободных жирных кислот. Более дорогое чисто кокосовое мыло
применяется в тех случаях, когда большое значение имеют хо-
рошее пенообразование и высокая растворимость в холодной
воде. Такие мыла прекрасно моют в мягкой воде, но в жесткой
воде они дают нежелательный осадок нерастворимого кальцие-
вого мыла. Недавно разработаны туалетные мыла на основе
синтетических ПАВ, кальциевые и магниевые соли которых хо-
рошо растворимы в воде. Хотя в некоторых странах мира такое
мыло получило значительное распространение, ему не удалось
Моющие вещества
519
вытеснить жировое мыло с его доминирующего положения в
производстве туалетных мыл.
Стиральные порошки. Типичный мыльный стиральной поро-
шок содержит 50—60% натриевого мыла (главным образом из
твердого животного жира), 8—20% пербората натрия, 3—6%
силиката натрия и небольшие количества усилителей пенообра-
зования, натрийкарбоксиметилцеллюлозы, оптических отбелива-
телей (стр. 543), отдушек и воды. Возможно также присутствие
карбоната и (или) фосфатов натрия. Товарной формой яв-
ляются порошки, получаемые холодным распылением. Смесь
ингредиентов (исключая термически нестойкие вещества, такие,
как перборат натрия) подвергают перегреву при повышенном
давлении и разбрызгивают через форсунки в распылительных
башнях, где образовавшиеся капельки за время их падения
быстро высыхают. При этом получаются хорошо знакомые всем
гранулы, которые смешивают с сухими термически нестойкими
ингредиентами.
В мыльные порошки не надо вводить активные добавки,
так как в жесткой воде они образуют с ионами кальция нерас-
творимое кальциевое мыло. Это приводит к необходимости
использовать такое количество порошка, чтобы его хватило и
на осаждение ионов Са2+, и на создание в растворе достаточно
высокой концентрации ПАВ для обеспечения хорошего моющего
действия. Отсюда следует, что при стирке в жесткой воде рас-
ход мыльных порошков увеличивается.
Когда-то стиральные порошки на мыльной основе пользо-
вались большим спросом. Однако к настоящему времени в
большинстве стран они почти полностью вытеснены синтетиче-
скими стиральными порошками на основе алкилбензолсульфо-
натов (стр. 521). Исключением в этом отношении является, в
частности, Великобритания, где на долю мыльных порошков
все еще приходится значительная часть общего выпуска сти-
ральных порошков.
Другие области применения мыла. Хотя во многих случаях
вместо мыла теперь применяются синтетические ПАВ, оно до
сих пор используется, например, при отделке тканей, в процес-
сах эмульсионной полимеризации, а также в производстве кос-
метических средств, полировочных составов и водоэмульсионных
красок.
Некоторые проблемы,
связанные с производством и применением мыла
Наряду с большим числом существенных достоинств мою-
щие средства на основе мыла имеют ряд недостатков, важней-
ший из которых — образование нежелательного осадка при
стирке в жесткой воде.
520
Глава 13
Доступность натуральных масел и жиров (а следовательно,
и цены на них) колеблются непредсказуемым образом, причем
во многих случаях увеличить поступление этого сырья нелегко.
Например, поступление кокосового масла зависит от климати-
ческих условий и весьма непостоянно. К тому же его можно
увеличить лишь путем закладывания новых плантаций кокосо-
вой пальмы, что, естественно, требует гораздо большего вре-
мени, чем строительство нового завода. Твердый животный жир
является отходом мясоперерабатывающей промышленности, от-
куда следует, что его поступление непосредственно зависит от
объема производства мяса. При этом следует принять во вни-
мание, что высококачественный говяжий жир употребляется в
пищу, и хотя на получение мыла идут более низкие сорта жира,
увеличение численности населения Земли и обусловленное им
повышение потребности в продуктах питания и моющих сред-
ствах приведут к усилению конкуренции между этими потреби-
телями за высококачественные животные жиры (если только
промышленность моющих средств не перейдет на другие виды
сырья).
Кроме того, углеводородный состав цепи природных жирных
кислот, имеющихся в распоряжении производителей моющих
средств, весьма ограничен. Большинство жиров и масел содер-
жит преимущественно жирные кислоты Ci2 либо Сы и Cia, и если
для какой-то конкретной цели потребуется, например, мыло из
кислот С14—С16, его просто нельзя будет рентабельно получить
на базе природного сырья.
Эти и многие другие причины лежат в основе все большего
сокращения производства моющих средств на основе мыла. На-
чиная с конца второй мировой войны все более широкое приме-
нение находят синтетические ПАВ, которые получаются из де-
шевого и легкодоступного нефтехимического сырья и во многих
случаях обладают лучшим моющим действием, чем мыло.
Синтетические поверхностно-активные
вещества
Поверхностно-активные вещества принято классифицировать
в зависимости от знака заряда их «головной» группы. Разли-
чают анионные, неионные и катионные ПАВ, примерами кото-
рых могут служить соответственно CH3(CH2)nOSO3Na+,
СНз(СН2)п(ОСН2СН2)8ОН и [CH3(CH2)17]2N+(CH3)2C1-. Каждый
из этих классов включает очень большое число соединений, и в
данной главе будут рассмотрены только те ПАВ, которые имеют
действительное или потенциальное практическое значение. Бо-
лее подробные сведения можно найти в работах, приведенных
в списке литературы.
Моющие вещества 521
Анионные поверхностно-активные вещества
Анионные ПАВ составляют самый важный в настоящее вре-
мя класс поверхностно-активных веществ; в 1969 г. их мировое
производство приближалось к 1 млн. т. Наибольшее практиче-
ское значение имеют соединения, содержащие насыщенную уг-
леводородную цепь из 10—15 атомов углерода, так или иначе
связанную с сульфатной или сульфонатной группой. Эти соеди-
нения применяются главным образом для получения бытовых
стиральных порошков и средств для мытья посуды.
Биохимически неразлагаемые алкилбензолсульфонаты с раз-
ветвленной алкильной группой. Первыми из синтетических
анионных ПАВ многотоннажными промышленными продуктами
стали соединения на основе алкилбензолов, получаемых путем
тетрамеризации пропилена и присоединения образующейся
смеси сильно разветвленных додеценов к бензолу.
фосфорнокислотный СНз СНз СНз
катализатор | СаНа
4СНзСН=СН2 ----—----------* СНз—С=С—СН—СН—СНз —-----------Г?“
I HF или А1С1з
СН2СН2СНз
СНз СНз СНз
—> СНз—С-----СН—СН—СН— СНз
X. СН2СН2СНз
и
Тетрамеризация пропилена, так же как и последующее алкили-
рование бензола по Фриделю — Крафтсу, протекает через про-
межуточные карбониевые ионы (т. 2, стр. 111), в которых могут
происходить перегруппировки углеродного скелета и процессы
гидридного сдвига. В приведенном выше уравнении структуры
тетрамера пропилена и додецилбензола изображены лишь одной
из многочисленных возможных формул.
Из додецил бензолов можно получить с очень высоким выхо-
дом додецилбензолсульфонат натрия, 'который является хоро-
шим ПАВ, обладающим превосходной пенообразующей способ-
ностью. К тому же он вырабатывается из дешевого и легкодо-
ступного сырья, вследствие чего этот продукт полностью
I) SOg или олеум
С12Н25С6Н5 _ “ “ * п-С12Н25С6Н4 S O3Na
2) NaOn
отвечает всем техническим и экономическим требованиям, предъ-
являемым к синтетическим ПАВ. Поскольку додецилбензолсуль-
фонат кальция гораздо лучше растворим в воде, чем кальциевое
мыло, при стирке с додецилбензолсульфонатнымя моющими
средствами в жесткой воде необходимо вводить в них активные
522
Глава 13
добавки. Эффективной и дешевой активной добавкой может слу-
жить триполифосфат натрия NasPeOio- Стиральные порошки на
основе додецилбензолсульфоната и триполифосфата натрия, по-
лучившие широкое распространение после второй мировой
войны, к концу 50-х годов почти полностью вытеснили в боль-
шинстве промышленно развитых стран мыльные стиральные по-
рошки. Однако в настоящее время стало очевидным, что мою-
щие вещества на базе тетрамеров пропилена имеют один очень
серьезный недостаток — они довольно медленно подвергаются
биохимическому разложению. Это означает, что значительная
часть додецилбензолсульфоната натрия, попавшего в систему
бытовой канализации, проходит без изменения через водоочист-
ные сооружения и поступает в реки и озера, вызывая появление
неприятной пены на поверхности очистных и природных водое-
мов. Такая пена нежелательна не только с эстетической точки
зрения, но и потому, что она сильно затрудняет процесс водо-
очистки и мешает нормальному поступлению атмосферного кис-
лорода в природные воды. Отсюда следует, что применение био-
химически неразлагаемых ПАВ в районах с высокой плотностью
населения совершенно недопустимо.
Биохимически разлагаемые алкилбензолсульфонаты с нераз-
ветвленной алкильной группой. Медленность биохимического
разложения алкилбензолсульфонатов, полученных на базе тет-
рамеров пропилена, обусловлена, высокой степенью разветвлен-
ности их алкильной группы. Алкилбензолсульфонаты натрия
с прямой алкильной цепью гораздо быстрее подвергаются био-
химическому разложению. В начале 60-х годов было разрабо-
тано и реализовано в промышленности несколько методов полу-
чения линейных алкилбензолов, и в настоящее время производи-
мые на их основе алкилбензолсульфонаты уже вытеснили в
большинстве промышленно развитых стран ПАВ на основе тет-
рамеров пропилена. Эти алкилбензолсульфонаты дешевы, обла-
дают отличными моющими свойствами и поэтому считаются
самыми лучшими из существующих сейчас ПАВ. Хотя в ряде слу-
чаев с ними успешно конкурируют некоторые позднее появив-
шиеся ПАВ (стр. 525), можно полагать, что они долго будут
сохранять важное значение.
Неразветвленные длинноцепочечные боковые алкильные
группы в этих алкилбензолсульфонатах получаются из жидких
н-парафинов Сю—Cj5 или из твердого парафина (фракции
Сю—С35). Исходные жидкие парафины выделяют из керосино-
вых дистиллятов нефти путем селективной адсорбции на моле-
кулярных ситах или карбамидной депарафинизации. Оба эти
метода дают парафины с прямой цепью, имеющие степень чи-
стоты свыше 90%. Полученные н-парафины переводят в моно-
хлорпроизводные или в олефины с неконцевой двойной связьщ,
Моющие вещества
523 .
которыми алкилируют бензол по реакции Фриделя — Крафтса.
Твердый парафин подвергают крекингу в олефины с концевой
двойной связью (а-олефины), имеющие самую различную длину
цепи. Образующуюся смесь разгоняют, отбирая фракцию
Сю — Сю, которую используют для алкилирования бензола. На
денормализация
Керосин
молекулярными „
ситами н- Парафины С12 смесь
--------;---г-* (г большинстве---*• втор-монохлорпарафинов
или карбамидная случаев
депарафинизация Cw-C,5)
каталитическое
дегидрирование
каталитическое
дегидрохлорирование
Смесь олефинов
С неконцевой двойной
связью
дензол
AlClj
Твердый
парафин
Линейный
алкилбензол
Рис. 13.2. Пути получения линейных алкилбензолов.
рис. 13.2 приведена схема различных путей получения линейных
алкилбензолов; последующие стадии производства ПАВ на их
основе описаны ниже.
Хлорирование парафинов — это радикальная реакция, кото-
рая дает смесь продуктов замещения (т. 1, стр. 17; т. 2, стр. 132).
Например, при хлорировании додекана получаются почти в рав-
ных соотношениях 2-, 3-, 4-, 5- и 6-монохлордодеканы наряду
с меньшим количеством 1-изомера. В реакцию вступает лишь
20—30% парафина, так как при большей степени конверсии
образуются значительные количества полихлорпроизводных.
Последнее недопустимо по той причине, что в этом случае резко
снижается качество конечного продукта из-за присутствия в нем
524
Глава 13
большого количества примесей динатриевых солей алкилен-
ди (бензолсульфокислот), а также моносульфонатов натрия, со-
держащих индановые (I) и тетралиновые (2) циклы. Смесь
R
R R'
1 2
хлорпарафинов и парафина после стадии хлорирования направ-
ляют в алкилатор, где ее обрабатывают большим избытком бен-
зола в присутствии хлористого алюминия как катализатора. Из-
быток бензола и парафин отгоняют и возвращают на соответ-
ствующие стадии процесса, а алкилбензол очищают путем
ректификации. Описанная рециркуляционная схема проще, чем
схема, предусматривающая разделение смеси хлорпарафинов и
парафина перед алкилированием, так как различие в темпера-
турах кипения «-парафинов и их монохлорпроизводных гораздо
»меньше, чем «-парафинов и полученных на их основе алкилбен-
золов. Вследствие низкой степени конверсии парафина в хлор-
парафины концентрация олефинов с неконцевой двойной связью
в смеси с непревращенным парафином, выходящей со стадии
дегидрохлорирования, составляет 20—30%. Альтернативный ме-
тод получения олефинов — дегидрирование парафинов — тоже
требует использования низких степеней конверсии, с тем чтобы
обеспечить высокий выход моноолефинов с неконцевой двойной
связью, практически свободных от примеси диенов. Таким обра-
зом, и в этом случае получается смесь олефинов с парафином,
что позволяет использовать рециркуляционную схему, аналогич-
ную описанной выше.
Свойства линейных алкилбензолсульфонатов натрия зависят
как от длины алкильной цепи, так и от места ее соединения
с бензольным кольцом. Например, 2-(сульфофенил) алканы зна-
чительно отличаются по свойствам от своих изомеров, в кото-
рых сульфофенильная группа связана с одним из центральных
углеродных атомов алкильной цепи, и в частности уступают им
по пенообразующей способности.
Изомерный состав алкилбензола частично определяется изо-
мерным составом олефина или хлорпарафина, используемого
для алкилирования бензола, однако главную роль играют усло-
вия алкилирования и природа катализатора. Фтористый водо-
род вызывает изомеризацию олефинов, а А1С13 способен изоме-
ризовать олефины, хлорпарафины и полученные алкилбензолы.
Степень изомеризации этих соединений можно варьировать, под-
бирая условия реакции. Так, содержание 2-фенилдодекана в про-
Моющие вещества
525
дукте алкилирования бензола додеценом-1 можно изменять в
пределах от 14% (катализатор HF, температура 55 °C, раствори-
тель— гексан) до 44% (катализатор А1С13, температура 0 — 5 °C,
без растворителя). Алкилбензолы с низким содержанием 2-фе-
нилалкана обычно считаются наиболее ценными полупродук-
тами, и рядом фирм разработаны собственные методы регулиро-
вания реакций изомеризации с тем, чтобы свести к минимуму
присутствие этого изомера.
Сульфирование. Алкилбензолы в промышленности сульфи-
руют олеумом или газообразной трехокисью серы. Оба эти ре-
агента дают высокие выходы и приводят к преимущественному
образованию ге-моносульфокислоты.
Метод сульфирования олеумом, не требующий сложного обо-
рудования, был первым методом получения алкилбензолсульфо-
натов и широко применяется до сих пор. По этому методу ал-
килбензол обрабатывают избытком олеума (обычно 20%-иого),
превращая его с выходом более 95% в сульфокислоту. Затем
к реакционной смеси добавляют воду с таким расчетом, чтобы
концентрация серной кислоты снизилась до -~71%. При этом
сульфомасса разделяется на два слоя: слой разбавленной сер-
ной кислоты и слой алкилбензолсульфокислоты, содержащей
около 10% H2SO4. Алкилбензолсульфокислоту отделяют и ней-
трализуют раствором едкого натра. Полученная паста, содер-
жащая 40—55% алкилбензолсульфоната и 5—6% сульфата нат-
рия, пригодна для переработки в большинство товарных мою-
щих средств.
Метод сульфирования трехокисью серы позволяет избежать
применения избытка серной кислоты и является самым эконо-
мичным. Реакция SO3 с алкилбензолами протекает чрезвычайно
быстро и сопровождается выделением большого количества теп-
ла. Избыток трехокиси серы нежелателен, так как при этом
получается окрашенный продукт. Современный процесс сульфи-
рования заключается в пропускании газообразного SO3, раз-
бавленного воздухом (до концентрации SO3 3—10% по объему),
над пленкой алкилбензола, стекающего по поверхности тепло-
обменника. Это обеспечивает эффективную абсорбцию трех-
окиси серы из газового потока и быстрый отвод теплоты
реакции. Такая конструкция сульфуратора позволяет точно регу-
лировать температуру и соотношение SO3 и алкилбензола и по-
лучать высококачественный продукт, практически не содержащий
серной кислоты. Далее сульфокислоту, как обычно, нейтрали-
зуют раствором едкого натра, получая 40—60%-ную пасту, при-
годную для переработки в товарные моющие средства (стр. 543).
Другие синтетические анионные ПАВ. С точки зрения по-
верхностной активности присутствие бензольного кольца в ал-
килбензолсульфонатах не обязательно. Фактически оно служит
526
Глава 13
лишь удобным промежуточным звеном между гидрофильной
сульфогруппой и гидрофобной алкильной цепью. Поэтому были
разработаны и другие способы соединения длинной алкильной
цепи с анионной группой, которые описаны ниже. Большинство
анионных ПАВ, полученных этими способами, при использова-
нии их для стирки и в других областях применения моющих ве-
ществ не уступают алкилбензолсульфонатам, хотя вместе с тем
им присущи свои технические и экономические преимущества и
недостатки. Возможно, что в будущем некоторые из новых ани-
онных ПАВ составят серьезную конкуренцию алкилбензолсуль-
фонатам. Однако вряд ли какое-либо из них достигнет того по-
ложения, которое вот уже 15 лет занимают эти вещества.
1. Олефинсульфонаты. В начале 60-х годов высококачествен-
ные линейные а-олефины с длинной цепью стали доступными
продуктами промышленного производства. Эти соединения полу-
чают двумя методами. Первый из них — крекинг твердого пара-
фина, упоминавшййся выше при описании синтеза алкилбензо-
лов; он Дает дешевые олефины довольно хорошего качества,
которые содержат небольшие примеси диенов и нафтенов. В ос-
нове второго метода лежит теломеризация этилена триэтилалю-
минием или другими алюминийорганическими катализаторами,
приводящая к получению очень чистых, но несколько более до-
рогих а-олефинов. Оба метода дают смесь олефинов различного
молекулярного веса, вследствие чего цены на фракцию Сю — -Сю,
используемую в производстве моющих веществ, зависят от спро-
са на побочно образующиеся олефины, содержащие менее 10 и
более 18 атомов углерода.
Процесс теломеризации этилена включает две стадии. На
первой стадии этилен реагирует с триэтилалюминием при уме-
ренной температуре и довольно высоком давлении, например
при 100°C и 10 МН/м2 (100 атм); в этих условиях происходит
наращивание алкильной цепи (а). По достижении заданной сте-
пени теломеризации условия реакции изменяют, существенно
СН3СН2(СН2СН2)рх
(СН3СН2)3А1 4- (р + q + г)Н2С=СН2 —+ СН3СН2(СН2СН2)9-А1 (а)
СН3СН2(СН2СН2)/
повышая температуру (например, до 200—300 °C) или добавляя
реагент типа коллоидального никеля, в результате чего имеет
СН3СН2(СН2СН2)Р>Ч
СН3СН2(СН2СН2)<;—А1 4- ЗН2С=СН2 —►
CH3CH2(CH2CH2)rZ
СН3СН2(СН2СН2) р-, сн=сн2
—> CH3CH2(CH2CH2)Q_1CH=CH2 4- (СН3СН2)3А1 (б)
CH3CH2(CH2CH2)r-1CH=CHi
Моющие вещества 527
место реакция вытеснения (б). При этом образуются высшие
олефины и регенерируется триэтилалюминий, который возвра-
щают на первую стадию. Таким образом, суммарная реакция
заключается в превращении этилена в смесь высших олефинов.
В самом простом случае — при однократном проведении каждой
из стадий — полидисперсность образующейся смеси олефинов
отвечает функции распределения Пуассона. Разработано не-
сколько процессов, предусматривающих многократное чередова-
ние стадий наращивания алкильной цепи и вытеснения высших
олефинов, с тем чтобы получить продукт с более узким распре-
делением полимергомологов. Однако на практике производство
только олефиновой фракции Сю—Сю, вероятно, обходилось бы
слишком дорого, и поэтому производители а-олефинов, рабо-
тающие по методу теломеризации, должны решать задачу пол-
ного использования образующихся олефинов с различной дли-
ной цепи.
Фракцию а-олефинов Сю — Сю подвергают сульфированию
трехокисью серы в пленочных реакторах (см. выше). Реакция
протекает весьма неоднозначно и приводит к образованию ал-
кенсульфокислот и сультонов наряду с меньшими количествами
сультонсульфокислот, алкендисульфокислот и других продук-
тов. В результате нейтрализации и гидролиза этой смеси
раствором едкого натра получается смесь алкенсульфонатов
натрия (~6О°/о), оксиалкансульфонатов натрия (~22°/о) и
дисульфонатов (~18%). Механизм образования основных про-
дуктов сульфирования можно представить схемой, приведенной
на стр. 528.
На стадии нейтрализации и гидролиза сульфокислоты просто
вступают в реакцию нейтрализации, а сультоны превращаются
в смеси оксиалкансульфонатов и алкенсульфонатов; например,
из сультона 5 получаются RCH = CHCH2CH2SO3Na, RCH2CH —
= CHCH2SO3Na и RCH2CH(OH)CH2CH2SO3Na. Единственным
конкретным доказательством присутствия в продуктах сульфи-
рования р-сультона 3 (который, по-видимому, неустойчив, так как
выделить его не удалось) является идентификация в продуктах
подщелачивания смеси веществ, образующейся в мягких усло-
виях сульфирования, 2-оксиалкан-1-сульфоната натрия, полу-
чающегося, вероятно, из этого сультона. В обычных производ-
ственных условиях, когда сульфирование ведут в пленочном
реакторе, среди оксиалкильных соединений преобладают 3- и 4-
оксиалкансульфонаты — производные сультонов 5 и 6 соответ-
ственно. Ненасыщенная сульфокислота 4 частично вступает в
дальнейшую реакцию с SO3, давая аналогичный ряд продуктов
дисульфирования.
Образующаяся в итоге всех этих реакций сложная смесь
сульфонатов, известная под названием «а-олефинсульфонат»,
528
Глава 13
RCH2CH2CH=CH2 + SOj
[rch2ch2chch2so;]
RCH,CH —CH—CH2
I I
о—so2
3
RCH2—CH—CH—CH2
I I
о------so2
5
RCH2CH=CHCH2SO3H
4
|s°3
алкендисульфокислоты и
сультонсульфокислоты
RCH—CH2
°\ /CH2
SO2—CH2
6
обладает высокой способностью к биохимическому разложению
и превосходными поверхностно-активными свойствами.
2. Алкилсульфаты. Эти соединения были первыми синтетиче-
скими ПАВ, использованными в составе моющих средств. Наи-
более старый метод их получения заключается в сульфировании
высших спиртов, которые в свою очередь синтезируют путем
гидрогенизации сложных эфиров жирных кислот, например:
н2 О so3
CH3(CH2)16COOR -------------> СН3(СН,)16СН2ОН ----->
хромит меди ' 2) NaOH
+ ROH
—► CH3(CH2)i8CH2OSO3Na
Природные жиры и масла можно либо непосредственно восста-
навливать в высшие спирты и глицерин, либо предварительно
подвергать переэтерификации низшими спиртами. Наряду
с этим методом, который применяется до сих пор, существует
еще два метода производства высших первичных спиртов.
По первому из них алюминийалкилы, полученные теломери-
зацией этилена триэтилалюминием, окисляют воздухом в алко-
голяты алюминия и далее гидролизуют до спиртов. Данный ме-
Моющие вещества . 529
тод дает высококачественные спирты, но, как и при получении
высших олефинов, продукты реакции имеют широкое распреде-
ление полимергомологов. В этом процессе, в отличие от про-
цесса получения а-олефинов, алюминийорганические соединения
не могут быть возвращены в цикл (гл. 4).
Вторым методом получения высших спиртов является про-
цесс гидроформилирования (гл. 6), в котором олефин вводят
в реакцию с окисью углерода и водородом в присутствии ко-
бальтового катализатора. Первоначально в этой реакции обра-
зуются альдегиды, но условия процесса подбирают таким обра-
зом, чтобы альдегиды in situ восстанавливались в спирты. При-
веденная ниже схема реакции упрощена, так как катализаторы
гидроформилирования вызывают также и изомеризацию олефи-
нов. а-Олефины гидроформилируются быстрее, чем олефины
н2 + со
СН3(СН2)9СН=СН2 — — >
С02(СО)з
Н2
—> [СНз(СН2)вСН(СНз)СНО + СНз(СН2)9СН2СН2СНО] —>
—> СНз(СН2)9СН(СН3)СН2ОН 4- СНз(СН2)яСН2СН2СН2ОН
с неконцевой двойной связью, и поэтому, несмотря на изомери-
зацию, полученный продукт содержит главным образом спирт
с прямой цепью и 2-метилалканол-1, как показано на схеме.
Другие спирты изостроения, кроме 2-метилалканола-1 (2-этил-,
2-пропил- и т. д.), образуются в меньших количествах из оле-
финов с неконцевой двойной связью, появляющихся в результате
изомеризации исходного сырья. Состав продукта существенно
зависит от условий гидроформилирования, причем содержание
спиртов с прямой цепьк) может колебаться в пределах 45—90%,
а спиртов изостроения — от 10 до 55%.
В качестве сульфирующего агента чаще всего применяется
трехокись серы, взаимодействие которой с первичными спир-
тами, например в пленочных реакторах, дает алкилсульфаты
с выходом до 95%. Возможно также использование серной и
хлорсульфоновой кислот, однако на практике эти реагенты
употребляются реже.
Первичные алкилсульфаты несколько дороже алкилбензол-
сульфонатов и находят ограниченное применение. Родственные
им сульфаты полиоксиэтиленовых эфиров жирных спиртов, на-
пример соединение 7, которые получают путем конденсации пер-
вичных спиртов с окисью этилена (стр. 537) и сульфирования
образующихся оксиэтилированных производных, широко приме-
няются в составе жидких препаратов для мытья посуды.
СНз(СН2)н(ОСН2СН2)зО5Оз№
7 '
530
Глава 13
Вторичные алкилсульфаты получают взаимодействием длин-
ноцепочечных а-олефинов с 90—-95%-ной серной кислотой по
приведенной ниже схеме (эта схема является упрощенной, так
как на ней не показано образование изомерных втор-алкилсуль-
H2SO4
СН3(СН2)9СН=СН2 ------>
8
NaOH
—> CH3(CH2)9CH(CH3)OSO3H -------► CH3(CH2)eCH(CH3)OSO3\a
10 11
/OCH(CH3)(C-H2)SCH3
o,s^
ХОСН(СН3)(СН2)9СН3
9
фатов путем перегруппировки промежуточных карбониевых
ионов). Побочное образование диалкилсульфатов 9 является
нежелательным; однако использование избытка серной кис-
лоты позволяет лишь частично подавить эту реакцию. Суще-
ствует два способа решения данной- проблемы. Первый способ
состоит в применении растворителя и подборе условий реакции
таким образом, чтобы моноалкилсульфат 10 выпадал в осадок
и нс мог далее присоединять вторую молекулу олефина. По вто-
рому способу продукт сульфирования обрабатывают избытком
едкого натра и полученный щелочной раствор нагревают. При
этом диалкилсульфат гидролизуется в желаемый моноалкил-
сульфат натрия 11 и вторичный .спирт, который экстрагируют
углеводородным растворителем и используют в других нефте-
химических производствах.
Алкилсульфаты вырабатываются в промышленном масштабе
и входят в состав некоторых жидких моющих средств, выпу-
скаемых в Великобритании для технических и бытовых целей.
3. Алкансульфонаты. Линейные парафины, которые упоми-
нались выше в качестве исходного сырья для получения алкил-
бензолсульфонатов, можно непосредственно превратить в ПАВ
общей формулы RR'CHSO3Na по реакции с двуокисью серы и
кислородом. Эта реакция, называемая реакцией сульфоокисле-
ния, протекает по радикально-цепному механизму и инициирует-
ся УФ-светом или у-излучением (т. 1, стр. 179). Наблюдаемые
закономерности сульфоокисления индивидуальных «-парафинов
можно объяснить в рамках следующих элементарных реакций
531
Моющие вещества
(символом RH обозначена молекула парафина):
инициирование
RH ---------R-
R • + SO2 —> RSO2.
RSO2* + O2 —> RSOzOO-
RSO2OO- + RH —> RSO2OOH + R«
rso2ooh —> RSO2O« + «OH
rso2o« + rh — -> rso2oh + R-
' -OH + RH —> H2O + R"
rso2ooh + H2o + so2 —► RSO2OH + H2SO4
Одной из замечательных особенностей сульфоокисления яв-
ляется то, что после того, как поглотится определенное неболь-
шое количество лучистой энергии, дальнейшее облучение можно
прекратить, поскольку процесс становится самоподдерживаю-
щимся. Действительно, если после выхода процесса на стацио-
нарный режим и прекращения облучения остановить подачу
двуокиси серы и кислорода, то реакция может еще 2—3 суток
находиться как бы в скрытом состоянии и вновь начинает энер-
гично идти при возобновлении пропускания SO2 и О2. Это
явление обусловлено промежуточным образованием надсульфо-
кислот, которые играют роль химических инициаторов. В резуль-
тате сульфоокисления получается смесь вторичных алкансуль-
фокислот; для подавления образования дисульфокислот, которые
слишком полярны и поэтому обладают низкими поверхностно-
активными свойствами, процесс проводят с использованием
большого избытка парафина. Последний отделяют от алкан-
сульфокислот в сепараторе и возвращают на стадию сульфиро-
вания.
Если в исходном парафине присутствуют йримеси олефинов
или разветвленных парафинов, то они ингибируют реакцию
сульфоокисления. Полагают, что эти соединения улавливают
реакционноспособные радикалы, образуя неактивные аллильные
или третичные алкильные радикалы, не способные к эффектив-
ному продолжению цепного процесса. Один из путей практиче-
ского решения данной проблемы заключается в том, что реак-
цию начинают со специально очищенным парафином, а когда
процесс выйдет на режим, вместо него используют менее чистый
парафин, получаемый с помощью обычных промышленных ме-
тодов (например, путем карбамидной депарафинизации).
Вторичные алкансульфонаты легко подвергаются биохими-
ческому разложению и хорошо зарекомендовали себя как мою-
щие вещества в различных областях применения.
В32
Глава 13
Присоединение бисульфита натрия к а-олефинам приводит
к образованию первичных алкансульфонатов натрия
RCH=CH2 + NaHSO3 —► RCH2CH2SO3Na
Эта реакция тоже идет по цепному радикальному механизму
(инициаторами обычно служат перекиси, например трет-
С4Н9ООСОСбН5) и включает следующие основные стадии:
hso; + х
хн + so-
so;. + rch=ch2 —> rchch2so;
rchch2so; + hso; —> rch2ch2so:
rch2ch2so; + so;
Главной проблемой при осуществлении данного процесса яв-
ляется подбор растворителя, который был бы способен раство-
рять как неполярный олефин, так и бисульфит натрия. Чаще
всего используются смеси низших спиртов (например, изопро-
пилового) с водой, которые хотя и не дают гомогенного раствора
в начале реакции, но достаточно хорошо растворяют оба ре-
агента.
Первичные алкансульфонаты натрия, полученные этим мето-
дом, довольно плохо растворимы в воде, что ограничивает воз-
можность их применения. Однако они могут быть использованы
в комбинации с другими ПАВ.
Неионные поверхностно-активные вещества
Неполные ПАВ содержат длинную гидрофобную алкильную
группу, присоединенную к высокополярной нейтральной группе.
Полярная группа должна быть достаточно гидрофильной, чтобы
гидрофобная группа могла «войти» в водный раствор. Этому
требованию удовлетворяют остатки сахаров, в частности глю-
козы, и, например, н-додецил-р-глюкозид (12) ведет себя как
неионное ПАВ.
но"А
но
ОН «'HjlnCH;
Некоторые промышленные неионные ПАВ действительно по-
лучают на основе сахаров и родственных им полиолов. Тем не
менее наибольшее практическое значение имеют неионные ПАВ
на основе спиртов общей формулы 13, где гидрофильной груп-
пой служит полиоксиэтиленовая цепь,1? — углеводородный оста-
ток с длинной цепью, а X — промежуточное звено, такое, как
Моющие вещества 533
—О— или —СОО—. Эти спирты синтезируют путем обработки
подходящих соединений структуры RXH окисью этилена. Реак-
RXH+.nH2C----СН2 —> RX[CH2CH2O]„H
''о'7
13
ции данного типа принято называть реакциями оксиэтилирова-
ния, а их продукты представляют собой смеси веществ с раз-
личным числом оксиэтиленовых групп (—ОСН2СН2—), связан-
ных с гидрофобной группой R. Величина п в формуле 13 — это
среднее число оксиэтиленовых звеньев, присоединенных к гидро-
фобной группе. Как показано ниже, соотношение продуктов
с разным числом оксиэтиленовых звеньев определяется свойст-
вами RXH и условиями оксиэтилирования. Неионные ПАВ —
жидкости или твердые воскообразные вещества, имеющие ряд
важных отличительных особенностей по сравнению с анионными
и катионными ПАВ. Они вызывают более сильное понижение
поверхностного натяжения при эквивалентной концентрации и
обладают меньшими ККМ, чем ионные ПАВ, содержащие ту же
гидрофобную группу. Это обусловлено тем, что в них отсут-
ствует электростатическое отталкивание между полярными
группами, имеющее место при ориентации молекул ионных ПАВ
на границах раздела фаз и внутри мицелл, что облегчает ад-
сорбцию неионных ПАВ на межфазных поверхностях и их аг-
регацию в мицеллах.
Кроме того, неионные ПАВ необычно ведут себя при раство-
рении в воде. Если водный раствор полиоксиэтиленового неион-
ного ПАВ нагревать, то по достижении так называемой темпе-
ратуры помутнения (Гц) он становится мутным. При температу-
рах выше Тп образуется второй слой, содержащий основную
часть ПАВ, а при более низких>температурах ПАВ почти цели-
ком смешивается с водой в любых соотношениях. Значение Гп
данного вещества зависит от строения его гидрофобной группы
и числа оксиэтиленовых звеньев в его молекуле. Чем длиннее
углеводородная цепь, тем, естественно, больше требуется окси-
этиленовых групп, чтобы она могла «войти» в водный раствор.
Например, н-децильное производное с тремя оксиэтиленовыми
звеньями практически нерастворимо в воде, тогда как при на-
личии четырех оксиэтиленовых групп оно растворяется в воде
при комнатной температуре. В случае н-гексадецильной цепи
вещество становится растворимым при наличии 5—6 оксиэтиле-
новых звеньев. Эти факты принято объяснять тем, что раство-
рение неионных'ПАВ в воде при температурах ниже их Та об-
условлено гидратацией полиоксиэтиленовой группировки; по мере
повышения температуры довольно непрочная гидратная оболоч-
ка молекул ПАВ постепенно расшатывается и наконец (при
534
Глава 13
температуре помутнения) настолько разрушается, что вещество
становится нерастворимым.
Реакция оксиэтилирования. Окись этилена реагирует с соеди-
нениями, содержащими подвижный атом водорода, только в при-
сутствии щелочного или кислотного катализатора или если это
соединение само обладает основными свойствами (например, яв-
ляется амином).
1. Щелочное оксиэтилирование. Вследствие напряжения в
трехчленном цикле окись этилена чувствительна к нуклеофиль-
ной атаке и в присутствии щелочного катализатора вступает
в реакции конденсации с соединениями типа RXH по уравне-
ниям (3) — (9):
RXH + В' <=* RX’ + ВН ' '
RX~ + H2C--СН2 —> RXCH2CH2O" (4)
V и
К1
RXCH2CH2O +RXH RXCH2CH2OH + RX" (5)
*2
RXCH2CH2O +Н,с---СН2 —> RXCH2CH2OCH2CH2O_ (6)
V 15
К2
RXCH2CH2OCH2CH2O~ + RXH RXCH2CH2OCH2CH2OH + RX" (7)
K3
RXCH2CH2OCH2CH,O" + RXCH,CH2OH 4=4=
4=4 RXCH2CH2OCH2CH2OH + RXCH2CH2O~ (8)
&3
RXCH2CH2OCH2CH2O" + H2C--CH2 —►
v
—> RXCH2CH2OCH2CH2OCH2CH2O" и T. Д. (9)
Сначала соединение RXH взаимодействует с катализатором, да-
вая соответствующий анион RX- [уравнение (3)]. Последний реа-
гирует с окисью этилена с образованием алкоголят-иона 14
[уравнение (4)], который либо присоединяет еще одну молекулу
окиси этилена, превращаясь в алкоголят-ион 15, содержащий
две оксиэтиленовые группы [уравнение (6)], либо отрывает про-
тон от RXH, регенерируя анион RX" [уравнение (5)]. Ион 15
вступает далее в аналогичные реакции, первый набор которых
представлен уравнениями (7) — (9). Процесс сопровождается
присоединением все новых и новых оксиэтиленовых групп и за-
Моющие вещества
535
канчивается тогда, когда будет израсходована вся окись эти-
лена.
Обратимые реакции (3), (5), (7), (8) и др., в которых про-
исходит только перенос протона, протекают быстро, а реакции
размыкания цикла по механизму Sn2 [уравнения (4), (6), (9) и
др.] — гораздо медленнее. Состав продукта зависит от количе-
ства взятого катализатора, кислотности RXH и RX [СН2СН2О]ПН,
определяющей значения /Со, Ki и т. д., и нуклеофильности анио-
нов RX- и RX [СНзСНгО]^ определяющей константы скорости
k2 и т. д.
Реакция самого катализатора с окисью этилена, не показан-
ная на схеме, иногда играет очень важную роль. Чаще всего
в промышленности используются щелочные катализаторы (обыч-
но едкий натр) в количестве около 0,5 мол.%. При оксиэтилиро-
вании спиртов гидроксил-ион не может непосредственно выпол-
нять функцию катализатора, так как его основность недоста-
точна для отрыва протона от молекулы спирта. Поэтому первой
стадией процесса является взаимодействие НО- с окисью эти-
лена, приводящее к образованию аниона этиленгликоля, кото-
рый представляет собой гораздо более сильное основание и
способен катализировать оксиэтилирование спирта
НО' + Н2С--СН2 —> НОСН2СН2О"
V
НОСН2СН2О" + ROH НОСН2СН2ОН + RO"
Образующийся этиленгликоль может вступать в дальнейшие
реакции, аналогичные описанным выше, превращаясь в поли-
этиленгликоли, которые являются побочными продуктами
НОСН2СН2ОН + иН,С--СН2 —> НО[СН2СН2О]„+1Н
Тот же результат, естественно, получается в том случае, когда
исходные вещества содержат воду. Поэтому для эффективного
протекания оксиэтилирования необходимо применять очень не-
большие количества щелочи и воды.
При осуществлении оксиэтилирования на промышленных
установках смесь гидрофобного компонента RXH и катализа-
тора нагревают до 120—190 °C и затем при строгом регулирова-
нии температуры и давления впускают окись этилена со ско-
ростью, точно соответствующей скорости ее поглощения. Реак-
ция сопровождается выделением большого количества тепла, и
концентрацию окиси этилена в реакционной массе поддержи-
вают на минимальном уровне обеспечивающем достаточную
скорость оксиэтилирования.
2. Оксиэтилированные алкилфейолы. Первыми неионными
ПАВ, получившими широкое применение, были оксиэтилировац-
536
Глава 13
ные алкилфенолы. Наиболее важным исходным сырьем для
синтеза этих веществ является нонилфенол, который получают
в промышленности путем алкилирования фенола тримерами про-
пилена в присутствии BF3. Большое значение как исходные
вещества имеют также додецилфенол и октилфенол, получае-
мые из тетрамеров пропилена и из диизобутилена соответствен-
но. Неионные ПАВ, вырабатываемые на основе этих алкилфено-
лов, содержащих разветвленные алкильные группы, обладают
биохимической неразлагаемостью и в настоящее время заме-
няются более легко ассимилируемыми соединениями.
Оксиэтилирование алкилфенолов протекает так, как пока-
зано на рис. 13.3. Вследствие того что фенол значительно пре-
Рис. 13.3. Скорость присоединения окнси этилена к алкнлфенолу.
восходит по кислотности начальный продукт оксиэтилирования
17, равновесие в уравнении (10), соответствующем уравнению
(5) (см. выше), почти целиком смещено вправо. Следовательно,
фенолят-ион 18 — единственный ион, присутствующий в системе
в сколько-нибудь заметной концентрации. Правда, нуклеофиль-
ность фенолят-иона 18 ниже, чем иона 16, но далеко не настоль-
RC6H4OCH2CH2O' + RCeH4OH jfxh RC6H4OCH2CH2OH + RC6H4O' (10)
16 17 18
ко, чтобы компенсировать различие в их концентрациях; в ре-
зультате первая молекула окиси этилена присоединяется почти
исключительно к иону 18. Однако различие в реакционной спо-
собности между этими ионами обусловливает медленность окси-
этилирования на начальной стадии реакции (до точки А на
рис. 13.3). После того как весь фенол вступит в реакцию с обра-
зованием моноэтоксильного производного, вместо него в системе
появляется более реакционноспособный алкоголят-ион 16; с это-
Моющие вещества
537
го момента оксиэтилирование ускоряется и протекает нормаль-
но, приводя к полидисперсности продукта, близкой к функции
распределения Пуассона [константы равновесия в уравнении,
соответствующем уравнению (8), мало отличаются от единицы,
а константы скорости реакций, соответствующих уравнениям
(6), (9) и т. д., близки между собой].
3: Оксиэтилированные спирты. В качестве исходных веществ
при получении неионных ПАВ этого типа применяются первич-
ные спирты с длинной цепью, вырабатываемые путем теломери-
зации этилена, гидроформилирования олефинов или восстанов-
ления жиров.
Первичные спирты почти не уступают по кислотности своим
полиоксиэтилированным производным, а алкоголят-ионы, обра-
зующиеся как из спирта, так и из этих производных, очень
близки по нуклеофильности. Поэтому полидисперсный состав
продукта щелочного оксиэтилирования приблизительно отвечает
функции статистического распределения. Вместе с тем кривая
этого состава несколько более полога, чём кривая распределе-
ния Пуассона, и продукт оксиэтилирования содержит существен-
но больше непревращенного спирта, чем следовало бы ожидать
исходя из статистических представлений. Таким образом, пре-
вичные спирты, как и алкилфенолы, легко реагируют с окисью
этилена в присутствии щелочных катализаторов, давая про-
дукты с узким распределением полимергомологов, имеющим
максимум при степенях оксиЭтилирования, близких к числу
молей окиси этилена, присоединившемуся к исходному спирту.
Вторичные спирты ведут себя несколько иначе. Эти исходные
вещества имеют важное значение в производстве неионных
ПАВ. Их получают путем окисления н-парафинов Сю—Сю воз-
духом (при содержании 5% О2 в азоте) в присутствии борной
кислоты при температуре 165 °C. Реакция протекает по приве-
денному ниже уравнению, где R и R' — н-алкильные группы.
Процесс носит радикально-цепной характер, и замещение про-
ВД'НСОХ О /OCHRR'
о2 ^в/ н2о
3CH2RR< + ЗН3ВО3 —> | | ----► RR'CHOH
О\ /О
В
I
OCHRR'
исходит преимущественно по вторичным атомам углерода, в ре-
зультате чего образуется эквимолярная смесь всех возможных
изомерных вторичных спиртов.
Поскольку эти спирты существенно менее кислотны, чем их
полиоксиэтилированные производные, равновесие в уравнении
538
Глава 13
(11) сильно смещено в левую сторону.
RR'CHOH + RR'CH(OCH2CH2)„OCH2CH2O’
19
RR'CHCT + RR'CH(OCH2CH2)nOCH2CH2OH (11)
20
По этой причине в обычных условиях щелочного оксиэтили-
рования (концентрация основания ~0,5 мол.%) на долю
иона 20 приходится лишь небольшая часть общего содержания
алкоголят-ионов в системе, даже если количество добавленной
окиси этилена очень невелико. Этот факт, а также близость
нуклеофильной реакционной способности ионов 19 и 20 позво-
ляет сделать вывод, что окись этилена конденсируется преиму-
щественно не с исходным спиртом, а с последующими продукта-
ми, давая широкое распределение полимергомологов, максимум
которого приходится на значения п, существенно превы-
шающие число введенных в реакцию молей окиси этилена. Даже
при использовании очень большого избытка окиси этилена (на-
пример, 20 молей/моль спирта) значительная часть спирта
остается непрореагировавшей. Продукты со столь большой по-
лидисперсностью обладают неудовлетворительными свойствами,
что вынуждает проводить оксиэтилирование вторичных спиртов
иными методами.
Один из очевидных путей решения этой проблемы заклю-
чается в переводе всего спирта в алкоголят. Это позволяет из-
бежать непосредственного влияния равновесных реакций и
приводит к почти статистическому распределению степеней
оксиэтилирования. Однако этот метод является дорогостоящим.
Использование эквимолярного количества едкого натра дает в
конечном итоге просто раствор полиэтиленгликоля в исходном
спирте (стр. 535), а применение короткоцепочечных алкоголя-
тов натрия или прямое растворение металлического натрия во
вторичном спирте невыгодно по экономическим соображениям.
Поэтому на практике используется двухстадийная схема, опи-
санная на стр. 539.
Оксиэтилированные спирты и алкилфенолы представляют
собой наиболее важные неионные ПАВ. Они входят в состав
бытовых моющих средств, рассматриваемых ниже, и находят
широкое применение в промышленности, в том числе в произ-
водстве текстильных изделий, бумаги, каучука и водоэмульси-
онных красок.
4. Оксиэтилированные жирные кислоты. Оксиэтилирование
жирных кислот протекает во многом так же, как и оксиэтили-
рование фенолов (кислотный компонент образует анион с низ-
кой нуклеофильностью). Однако в данном случае возникают
осложнения, поскольку условия щелочного оксиэтилирования
Моющие вещества
539
весьма благоприятны для быстрой переэтерификации. Поэтому
полученные продукты содержат наряду с моноэфиром полиэти-
ленгликоля с жирной кислотой свободный полиэтиленгликоль
и его сложный диэфир.
Неионные ПАВ этого типа используются как в быту, так и
в промышленности. Однако они уступают по объему потребле-
ния оксиэтилированным спиртам и алкилфенолам.
5. Оксиэтилированные амины. Алкиламины обладают доста-
точной основностью, чтобы непосредственно реагировать с
окисью этилена. Начальными продуктами конденсации являются
диэтаноламины, образующиеся по Бм2-реакции
,СН2СН2ОН
RNHs + 2Н2С—-СН2 —> RN^
\ / \сн2сн2он
Путем обычного щелочного оксиэтилирования диэтаноламинов
можно получить высшие оксиэтилированные производные.
Эти соединения используются главным образом в тех слу-
чаях, когда они подвергаются протонированию и ведут себя как
катионные ПАВ (см. ниже).
6. Кислотное оксиэтилирование. Механизм оксиэтилирова-
ния, катализируемого кислотами Льюиса, менее изучен, чем
механизм щелочного оксиэтилирования. Было сделано предпо-
ложение, что этот процесс протекает по схеме Sn1(h), вклю-
чающей размыкание эпоксидною кольца с последующим при-
соединением гидрофобного компонента. Однако в большинстве
н2с-сн2 BF,r н2с-сн2 _____„ о—сн2сн2 кхн
О ” O-BF3 -BFj
(в)
О—CHjCHjXHR НО—CHjCHjXR
___> I ----► I —► HOCH2CH2XR
-bf3 -BF3 -BFS
случаев более вероятной представляется Бм2-атака(г) на ком-
плекс окиси этилена с катализатором. Действительный меха-
А
RX: Н2С—СН2 +
I \+/ _ ----► RHXCH2CH2OBF3----*
Н <>O-BF3 (г)
RXCHjCHj—ОН
-” -L -7SF-* RXCH2CH2OH
низм кислотного- оксиэтилирования зависит от строения компо-
нента RXH, природы катализатора и условий реакции и, по-ви-
димому, является промежуточным между SnI и Sn2. Как и сле-
довало ожидать на основании этих механизмов, при кислотном
540 л
Глава 13
катализе распределение продуктов с различной степенью окси-
этилирования в значительно меньшей степени связано со струк-
турой исходного соединения, чем при щелочном катализе.
В качестве кислотных катализаторов оксиэтилирования наи-
более часто применяются кислоты Льюиса (BF3, SnCl4, SbCl5).
Реакция протекает быстро и сопровождается выделением тепла.
Несмотря на то что ее ведут в более мягких условиях, чем
щелочное оксиэтилирование (при ~80сС против ~ 150°C), она
дает худшие выходы и больше побочных продуктов. Главными
побочными продуктами являются полиэтиленгликоль (обра-
зующийся, вероятно, за счет дегидратации исходного спирта с
последующим полиприсоединением окиси этилена) и цикличе-
ские соединения типа диоксана, которые скорее всего получают-
ся по реакциям наподобие тех, что приведены на схеме (<9).
Из-за образования побочных продуктов оксиэтилирование в
Je2
RX(CH2CH2O),CH2CH2—OXj x-CHj
НО+—BF3
Н2
Нг<? ? +HOBF3
---> ВХ(СН2СН2О)„СН2СН2—о+ сн2 (5)
сн2
н2
RX(CH2CH2O),CH2CH2OH + H1V ?
| (К ^сн2
-BF3 СН2
присутствии кислотных катализаторов применяется редко; един-
ственное важное исключение — это двухстадийное оксиэтилиро-
вание вторичных спиртов, имеющее большое практическое зна-
чение.
Как указывалось выше, при щелочном оксиэтилировании
вторичных спиртов получается слишком широкое распределение
продуктов. Образование полиоксиэтилированного спирта с узким
распределением полимергомологов, близким к распределению
Пуассона, и хорошими поверхностно-активными свойствами
может быть достигнуто следующим образом. Спирт, содержа-
щий кислотный катализатор, обрабатывают сначала небольшим
количеством окиси этилена (1 моль/моль), после чего непревра-
щенный спирт отгоняют и возвращают в цикл. К кубовому
остатку (содержащему главным образом монооксиэтилирован-
ный спирт с небольшими примесями полиоксиэтилированных
Моющие вещества
541
производных) добавляют остальное количество окиси этилена и
проводят реакцию в обычных условиях щелочного катализа,
получая продукт с заданной степенью оксиэтилирования.
Катионные поверхностно-активные вещества
В молекуле катионных ПАВ гидрофобная алкильная цепь
присоединена к положительно заряженной гидрофильной группе.
Все практически важные поверхностно-активные вещества этого
класса представляют собой соединения четвертичного аммония
или амины.
Наиболее распространенный путь их синтеза (так называе-
мый fatty route) базируется на жирных кислотах с длинной
цепью. Последние взаимодействием с аммиаком при темпера-
туре 200—300 °C переводят в нитрилы, которые далее гидрируют
либо в соответствующие первичные амины, либо во вторичные
амины, содержащие в молекуле две алкильные группы с длин-
ной цепью [схема (е)]. Первичные амины, такие, как н-окта-
дециламин (21), получают при проведении гидрирования на
скелетном никелевом катализаторе при температуре 100—150°C
и давлении 1,5—7 МН/м2 (15—70 атм) в присутствии аммиака,
Н-С17Н35СООН ——> W-ClyHggCN —
—> m-C17H35CH2NH2
21
(е)
—> (w-C18H37)2NH
22
добавляемого, чтобы подавить образование вторичного амина.
Вторичный амин, например ди-(н-октадецил)амин (22), обра-
зуется при температуре выше 200 °C и удалении из реактора
образующегося аммиака.
Из аминов получают ряд производных, используемых в ка-
честве ПАВ, причем наиболее важными являются соединения
четвертичного аммония типа 23, ацетаты аминов (24) и алифа-
тические диамины (25). Синтез этих соединений протекает по
сн3с1
• RsNH (ch3)2nr;ci-
23
СН3С1
NaOH
СН3СООН
(CH3)3NR+Cr
RNH2
NH3R 'ООССНз
24
ch2=chcn
Н2
rnhch2ch2cn —► rnhch2ch2ch2nh2
25
542
Глава 13
приведенным выше уравнениям, где символом R обозначены
наиболее часто применяемые длинноцепочечные алкильные
группы, получаемые на базе смеси карбоновых кислот твердого
животного жира.
Другую важную группу веществ данного класса составляют
соли четвертичного имидазолиния типа 26 (где R— углеводо-
родные остатки продуктов гидрирования твердого животного
жира) и полиоксиэтилированные амины (стр. 539).
ЛЧ-СН2
R—сх + I сн3о—so;
\n—сн2
н3с/ \ch2ch2nhcor
26
Существуют и другие методы получения катионных ПАВ,
но из них практическое значение имеет лишь следующий:
НВг NR3 +
RCH=CH2 ------->- RCH2CH2Br ---> RCH,CH,NR' Вг"
перекиси z z t
где RCH = CH2 — олефин с длинной цепью. Радикальная реак-
ция присоединения бромистого водорода протекает быстро и
гладко; обработка образующегося продукта, содержащего кон-
цевой атом брома, аминами с короткой цепью, например диме-
тиламином, дает целевые четвертичные аммониевые соединения.
Вследствие того что длинноцепочечные основные молекулы
катионных ПАВ несут положительный заряд, а большая часть
межфазных поверхностей в водном растворе заряжена отрица-
тельно, эти ПАВ обладают высокой субстантивностью по отно-
шению к большому ряду материалов. Адсорбция катионных
ПАВ на межфазных поверхностях уничтожает электростатиче-
ское отталкивание, способствующее протеканию процесса мытья,
и поэтому соединения этого класса нельзя использовать как
обычные моющие вещества. Однако субстантивность, приводя-
щая к образованию защитной поверхностной плёнки, а в неко-
торых случаях также бактериостатические и бактерицидные
свойства катионных ПАВ являются ценными в ряде специаль-
ных областей применения. Важнейшими из них являются сле-
дующие: смягчение текстильных тканей (четвертичные аммоние-
вые соли с двумя длинноцепочечными алкильными группами,
стр. 547) модифицирование поверхности минералов (четвертич-
ные аммониевые соли), флотация руд (ацетаты аминов) и
получение асфальтовых эмульсий (оксиэтилированные амины и
другие производные); кроме того, катионные ПАВ употребляют-
Моющий вещества
543
ся в качестве ингибиторов коррозии в нефтяной промышлен-
ности и как бактериостатические и бактерицидные средства (соли
алифатических диаминов и соединения четвертичного аммония).
Бытовые моющие средства
на основе синтетических ПАВ
Синтетические ПАВ применяются в самых различных об-
ластях, часть которых упоминалась выше. Здесь можно привести
лишь некоторые подробности о наиболее знакомой читателю
области применения этих веществ — бытовых моющих сред-
ствах.
Средства для стирки. Они подразделяются на два важных
класса: универсальные стиральные порошки (наиболее важный
вид моющих средств, содержащих одно ПАВ), предназначен-
ные главным образом для общей стирки, т. е. стирки белого
белья, сильно загрязненной одежды и большинства окрашенных
текстильных изделий, и специальные стиральные порошки и
жидкости, предназначенные для стирки шерстяных и легко ли-
няющих изделий, а также тонкого белья. Очевидно, что основ-
ным назначением стиральных средств является удаление за-
грязнений с одежды и других текстильных изделий; поэтому
они содержат ПАВ, активную добавку и антиресорбционные
агенты. Кроме того, средства для стирки должны быть удобны
и приятны в обращении и способствовать сохранению у много-
кратно эксплуатируемых и стираемых изделий свежего внеш-
него вида. Для придания моющим средствам этих дополнитель-
ных качеств в них вводится ряд специальных ингредиентов. Тех-
нология стирки в разных странах неодинакова, так как она
зависит от социальных традиций и конструкции используемых
стиральных машин. Это следует принимать во внимание при
разработке рецептуры средств для стирки.
1. Оптические отбеливатели. После нескольких циклов
стирка — эксплуатация многие белые изделия желтеют или
сереют. Для борьбы с этим явлением в состав стиральных
средств обычно включают оптические отбеливатели. Действие
оптических отбеливателей заключается в том, что они погло-
щают ультрафиолетовый свет (при ~360 нм) и вновь испус-
кают поглощенную энергию путем флуоресценции в синей части
видимого спектра (при 430—440 нм). Возникающее при этом
«посинение» изделия компенсирует пожелтение и делает изде-
лие визуально более белым, причем, поскольку поглощение
света происходит за пределами видимой области спектра, а из-
лучение—внутри нее, то цвет изделия становится ярче. Из
сказанного следует, что оптический отбеливатель должен по-
глощать преимущественно в ультрафиолетовой области
544
Глава 13
солнечного света и что интервал его поглощения не должен за-
ходить в видимую область, так как это приведет к пожелтению
изделия, особенно если падающий свет содержит мало ультра-
фиолетовых лучей. Излучение за пределами указанного выше
интервала может вызвать нежелательное появление тусклого,
блеклого тона. Кроме того, оптические отбеливатели должны
обеспечивать высокий квантовый выход, быть светопрочными и,
если они дороги, быть эффективными при очень низком содер-
жании в стиральном составе. Поскольку невозможно удовле-
творить все эти требования с помощью одного вещества, кото-
рое годилось бы для любых тканей, обычно в рецептуру сти-
ральных средств вводят комбинацию оптических отбеливателей,
подобранную таким образом, чтобы эффективно отбеливать и
натуральные, и синтетические волокна. В случае хлопчатобу-
мажных тканей используются вещества, действующие как пря-
мые красители. Чаще всего это соединения с большими плос-
кими молекулами, содержащими сопряженную систему кратных
связей (например, соединение 27) и сульфогруппы, придающие
им растворимость в воде. Концентрация их в стиральной ком-
позиции обычно составляет 0,1—0,8%. ЕГ случае полиамидных
волокон применяют оптические отбеливатели, которые ведут
себя как дисперсные красители и способны диффундировать в
волокно. Из-за вязкости полиамидов процесс диффузии проте-
кает довольно медленно, и по этой причине наилучшие резуль-
таты дают соединения с небольшими неполярными молекулами
(например, соединение 28); обычно их берут в количестве
0,02—0,1%.
'""N Nz
SO3Na SO3Na
27 [X = NHC6HB, Y = N(CH2)4O1
H,C---C—C6H4Cl-n
I U
H2CX /N
N
C6H4SO2NH2-n
28
2. Химические отбеливатели. В процессе стирки должно про-
исходить удаление не только общего загрязнения, но и отдель-
Моющие вещества
545
них пятен (например, пищевого происхождения). Обычно это
достигается либо путем использования стиральных средств, со-
держащих химический отбеливатель, либо путем добавления
в ванну самостоятельного отбеливателя — чаще всего водного
раствора гипохлорита натрия. Единственным химическим отбе-
ливателем, широко применяемым в составе стиральных порош-
ков, является перборат натрия NaBO2-H2O2-3H2O. Во время
стирки эта соль отщепляет перекись водорода, которая при тем-
пературе выше ~70°С эффективно отбеливает ткани. Можно
также включать в рецептуру соединения, реагирующие в сти-
ральной жидкости с гидроперекисным ионом, генерируемым
перборатом, с образованием надкислот. Надкислоты обладают
. n-CH3COOCeH4SO3Na + НОО- —> n-"OC6H4SO3Na + СН3СОООН
отбеливающими свойствами при температурах ниже 60°C, и,
следовательно, такие композиции более эффективны при стирке
в мягких условиях, чем чисто перборатные стиральные составы.
3. Ферменты. Хотя обычные стиральные порошки весьма эф-
фективны, они не всегда удаляют трудноотмываемые пятна
белковых веществ, например пятна крови. Для устранения
этого недостатка в последнее время стали выпускать порошки,
содержащие протеолитические ферменты. Последние лучше
всего действуют при замачивании изделий в холодной воде пе-
ред стиркой в горячей ванне. Однако они весьма эффективны
и непосредственно в процессе стирки. Ферментные добавки вы-
зывают расщепление белковых веществ во время замачивания
и обеспечивают полное удаление трудноотмываемых пятен в ре-
зультате последующей стирки.
4. Пенообразование. Традиционное представление, что для
успешного отстирывания белья необходима обильная пена, до
сих пор бытует среди домохозяек в некоторых странах, особенно
там, где все еще широко распространены ручная стирка
и стирка в вертикальных машинах. Это представление справед-
ливо лишь в случае мыльных порошков, которые до появления
синтетических ПАВ были единственным типом стиральных по-
рошков. Однако при использовании композиций на основе син-
тетических ПАВ прямой связи между отстирывающей и пено-
образующей способностью не существует, и можно создать со-
ставы, почти не дающие пены, но обладающие хорошим
моющим действием.
Первые моющие средства, включавшие синтетические ПАВ,
прекрасно пенились; такие композиции продолжают сохранять
важное значение и в настоящее время. С ростом популярности
автоматических стиральных машин, большинство которых не
приспособлено для использования сильнопенящихся моющих
средств (так как пена выбрасывается из машины) и в которых
13 Зак. 501
546
Глава 13
процесс стирки происходит в условиях полной герметичности,
исключающей возможность определения степени пенообразова-
ния, все более широкое распространение получают слабопеня-
щиеся стиральные составы.
Высокая пенообразующая способность обычно достигается
за счет применения анионного ПАВ в комбинации с так назы-
ваемым «усилителем ценообразования», например этанолами-
дом 29 или окисью амина 30. В том случае, когда необходимо
слабое ценообразование, используется тройная смесь анионного
н-Ci 1H23CONHCH2CH2OH
h-Ci2H2BN(CH3)2
О'
29
30
ПАВ, жирового мыла и неионного ПАВ. Типичные составы
сильно- и слабопенящихся универсальных стиральных порош-
ков приведены в табл. 13.2. Нормальная рабочая концентрация
этих веществ в растворе колеблется в пределах 0,1—.1%,
Таблица 13.2
Типичные составы универсальных стиральных порошков
Содержание, %
Ингредиенты
сильнопеия-
слабопенящиеся щиеся компо-
композиции ' ЗИцИИ
Алкилбензолсульфонат натрия а 3-12 14—20 е
Натриевое мыло 6 2-10 —
Неионное ПАВ в 2-5 —
Усилитель пенообразования г — 2
Фосфаты натрия д 25—60 30-50
Перборат натрия 0-30 0-20
Силикат натрия 4-8 5-10
Сульфат натрия 4-18 10-15
Натрийкарбоксиметилцеллюлоза 1-2 0,5-1,5
Оптические отбеливатели, отдушка, ферменты До 100 До 100
(если необходимо) и вода
а Обычно с линейной алкильной группой С.о —С15.
6 Обычно из твердого животного жира.
в Например, спирты Ci2—Cie или ионилфенол, к которым присоединено 9—18 окси-
этиленовых звеньев.
г Обычно RCONHCH2CH2OH, где R—алкильная группа Сц—Ct7,
Д Обычно 8О°/о‘ИЫй триполифосфат натрия Na5P3Oi0.
е В одной очень хорошей рецептуре около половины алкилбензолсульфоиата заме-
нено алкилсульф атом натрия, полученным на базе твердого животного жира.
Моющие вещества
547
Специальные стиральные средства позволяют применять бо-
лее мягкие условия стирки, необходимые для легко линяющих
изделий, шерстяных изделий и тонкого белья. В их состав вхо-
дят те же основные ингредиенты, что и в состав универсальных
порошков, но уменьшено содержание химического отбеливателя
и активной добавки и несколько ниже pH стирального раствора.
Некоторые композиции даже совсем не содержат ни активной
добавки, ни отбеливателя.
Производство стиральных порошков обычно ведется следую-
щим образом. Пасту, полученную в результате сульфирования
алкилбензола, смешивают с другими термостойкими ингредиен-
тами и распыляют через форсунки, расположенные в верхней
части распылительной сушильной башни. Образующиеся мел-
кие капли, падая в потоке горячего воздуха, высыхают и пре-
вращаются в хорошо знакомые всем сферические гранулы сти-
рального порошка. Последние смешивают с сухими термически
нестойкими ингредиентами (перборатом, ферментами и др.)
и получают готовый порошок, который должен быть сыпучим
и не слеживаться при хранении и применении; для уменьшения
слеживаемости к порошку добавляют силикат натрия.
В некоторых странах мира использование фосфатов в каче-
стве активных добавок считается нежелательным, так как было
установлено, что они способствуют эутрофикации — ускорен-
ному росту водорослей, приводящему к зарастанию водоемов.
В ряде государств эутрофикация стала серьезной проблемой,
особенно в Швеции, США и Швейцарии. Хотя фосфаты, со-
держащиеся в моющих средствах, составляют лишь часть
соединений фосфора, которыми обогащаются природные воды
по вине человека (наряду с ними важную роль играют также
фосфаты, присутствующие в удобрениях и канализационных
стоках), угроза эутрофикации может стать причиной замены
этих активных добавок другими соединениями, если только
удастся найти для них заменители, столь же эффективные, без-
опасные для людей, но более приемлемые с точки зрения за-
щиты окружающей среды.
Смягчители. Некоторые изделия, например полотенца и пе-
ленки, при обычной стирке и сушке могут стать жесткими. Если
же заключительное полоскание проводить в воде с добавкой
катионного ПАВ, то последнее адсорбируется на ткани и при-
дает ей приятную мягкость на ощупь. Добиться того же резуль-
тата путем введения катионного ПАВ в моющее средство на
основе анионного ПАВ нельзя, так как это приводит к образо-
ванию в стиральном растворе комплекса катионного и анион-
ного ПАВ, не обладающего поверхностной активностью. Не-
смотря на неудобство использования двух разных составов,
смягчители находят широкое применение, и спрос на них
1?* .
548
Глава 13
постоянно возрастает. Самыми лучшими смягчающими аген-
тами являются четвертичные аммониевые соединения типа 31
и 32, содержащие две длинноцепочечные алкильные группы.
+ ДУ—СН2
(«-C18H37)2N(CH3)2 СГ я-С18Н37—с^+ | X-
ж—сн2
н3с/ \ch2ch2nhcoc18h37-«
31 32
В товарной форме смягчители обычно представляют собой вод-
ный раствор или пасту, содержащую 5—8% четвертичной ам-
мониевой соли, 0—1 % неионного ПАВ, оптические отбеливатели
и парфюмерную отдушку.
Жидкие средства для мытья посуды. Очевидно, что посуду
приятнее всего мыть горячей водой с использованием моющих
средств, дающих устойчивую пену. Поэтому хороший состав для
мытья посуды должен не только способствовать удалению
остатков пищи и диспергированию их в моющей ванне, но и об-
разовывать обильную пену, которая не гасится жирами и дру-
Таблица 13.3
Типичные составы жидких средств для мытья посуды
Содержание, °/п
Ингредиенты
рецептура 1 рецептура 2
ПАВ
Алкилбензолсульфонат натрия а 18-26 10-15
Натрийсульфат оксиэтилированного спирта * 6 10-18 —
Оксиэтилированиый алкилфенол в — 2-10
Этаноламид я-алкилкарбоновой кислоты г 1-5 —
Солюбилизирующие агенты д Этанол 5-15 5-15
Мочевина 2-8 2-8
Ксилолсульфонат натрия 2-8 2-8
Отдушка, красители, другие добавки и вода До 100
До 100
а Обычно содержит неразветвленные алкильные группы Ci<—Сц.
б K-CnbWOCHjCHzhOSOsNa нли смесь соединений со степенью оксиэтилироваиия,
близкой к 3.
в Например» октнлфенол илн ноннлфенол, к которому присоединено 10 — 12 окснэти-
леновых звеньев.
г Например, я-СпНгзСОЫНСНаСНаОН.
Д Обычно вводят одну-две из этих добавок. Все три добавки одновременно приме-
няются редко.
Моющие вещества 549
гими пищевыми остатками. Так как мытье посуды сопряжено
с погружением рук в раствор моющего средства, последнее не
должно также раздражать кожу. В качестве средств для мытья
посуды обычно применяются водные растворы, содержащие
смесь нескольких ПАВ и добавки типа этанола, ксилолсульфо-
ната натрия и мочевины, удерживающие ПАВ в растворенном
состоянии и придающие композиции необходимую степень вяз-
кости. Кроме того, часто вводятся и другие ингредиенты — на-
пример, отдушка, красители и глицерин. В продаже имеется
ряд жидких средств для мытья посуды, в которых содержание
ПАВ колеблется от 10% (дешевые составы) до 40% (более до-
рогие составы). Типичная рецептура двух концентрированных
композиций приведена в табл. 13.3.
СПИСОК ЛИТЕРАТУРЫ
Шварц А., Перри Дж., Берч Дж., Поверхностно-активные вещества и
моющие средства, ИЛ, М., 1960.
Kirk-Othmer, Encyclopedia of Chemical Technology, Interscience, New York,
1963, разделы Detergency, Soap, Sulfonation and Sulfation and Surfactants.
Durham K. (Ed)., Surface Activity and Detergency, Macmillan, London,
1961.
Shinoda K., Solvent Properties of Surfactant Solutions, Edward Arnold,
London, 1968.
Sittig M., Practical Detergent Manufacture, Noyes Development Corpora-
tion, Park Ridge, New Jersey, U. S. A., 1968.
Schick M. J., Nonionic Surfactants, Edward Arnold, London, 1967.
Normal Paraffins, Supplement to European Chemical News, December 2nd,
1966.
Sarkar A. K-, Fluorescent Whitening Agents, Merrow Publishing Co. Ltd.,
Watford, England, 1971.
Evans W. P„ «Cationic Fabric Softeners», Chem. and Ind. (London), 1969,
893.
ГЛАВА 14
А. Химия и горение нефтяных топлив
Д. Уайтхед
Whitehead D. М., BP Research Centre, Sunbury-on-Thames
Б. Горючие и взрывчатые вещества и пороха
О. Гуртон
Gurton A., Nobel’s Explosives Company Limited
А. Химия и горение нефтяных топлив
Несомненно, что среди миллионов людей, ко-
торые в глубокой древности пользовались ог-
нем и поклонялись ему, находились такие, ко-
торые размышляли над тайной огня.
Вильям Крукс, 1861 г.
Огонь и тепло
Огонь, который получается при горении топлива, на протя-
жении более чем 100 000 лет оставался загадочным явлением,
имеющим огромное значение для жизни человека, почти пол-
ностью находящимся в его власти, и вместе с тем, до сравни-
тельно недавнего времени, совершенно непонятным.
Природа тепла, порождаемого огнем, была одним из самых
первых объектов для серьезного размышления. Древнегрече-
ские философы считали огонь (наряду с воздухом, водой и зем-
лей) одним из первичных элементов, и представление об огне
как о «материи», переходя из одной теории в другую, просуще-
ствовало до начала XVII в. В то время зарождался современ-
ный научный подход к явлениям окружающего мира, и опыты
Френсиса Бэкона по выяснению структуры пламени свечи, ве-
роятно, положили начало процессу постепенного пересмотра
представлений о природе горения и тепла. В дальнейшем в этом
процессе приняли участие многие знаменитые ученые — химики,
физики и инженеры. Основываясь на исследованиях своих пред-
шественников, Антуан Лавуазье опубликовал в 1783 г. класси-
ческий труд «Соображения о флогистоне» («Reflexions sur la
А. Химия и горение нефтяных топлив
351
Phlogistique»), который не только заложил фундамент количе-
ственной теории тепла, но и послужил основой для возникнове-
ния химической науки, за что Лавуазье заслужил звание отца
современной химии. Таким образом, уже с ранних времен изу-
чение химических явлений и горения развивалось в самой тес-
ной связи друг с другом.
Начиная с этого периода, и особенно после промышленной
революции в Англии, внимание к теоретическим и практиче-
ским аспектам процесса горения постоянно возрастает, порой
переходя в повальную увлеченность. Многие из тех, кто зани-
мался этой проблемой, были фактически учеными-любителями,
непосредственно связанными с практикой, что отчасти объяс-
няет тесное переплетение интересов науки и производства, ко-
торое так плодотворно проявило себя в конце XVIII—XIX вв.
Один из претендентов на академическую карьеру, Джеймс
Джоуль, не был принят на должность профессора в универси-
тете Сент-Эндрюса из-за небольшого физического дефекта. Его
хорошо известная, важная работа по установлению теплового
эквивалента механической энергии, приведшая к открытию пер-
вого закона термодинамики, была выполнена в то время, когда
он был владельцем большого пивоваренного завода. В этой
связи может возникнуть вопрос: смог бы Джоуль работать
столь же успешно, если бы ему удалось заняться академической
деятельностью, пусть даже в таком крупном научном центре,
как университет Сент-Эндрюса?
Со времени промышленной революции зависимость челове-
чества от энергии, получаемой путем сжигания топлив, воз-
росла во много раз, и можно лишь сожалеть о том, что в теку-
щем столетии практическое применение фундаментальных
исследований в области горения проявляется очень слабо. Все
крупные достижения в практике стационарного горения (раз-
личные пламена) и сжигания топлив в двигателях внутреннего
сгорания транспортных средств (бензиновом и дизельном) были
получены эмпирически, без использования результатов широких
изысканий, проведенных на политехнических факультетах ин-
ститутов и в университетах.
Тем не менее очевидно, что в настоящее время под давле-
нием таких факторов, как борьба с загрязнением окружающей
среды, сокращение ресурсов энергии и непрерывное повышение
цен на топливо, в технологии процессов горения начинают про-
исходить некоторые важные изменения. Это позволяет на-
деяться на то, что в самые ближайшие годы вновь восстано-
вится тесное сотрудничество науки о горении с производством,
которое имело место в XVIII—XIX вв.
Что же мешает такому сотрудничеству? Чтобы ответить на
этот вопрос, рассмотрим существующие нефтяные топлива,
552
Глава 14
которые играют столь большую роль в жизни современного чело-
века, и основные химические явления, обусловливающие их
способность гореть, выделяя тепло. Затем обсудим некоторые
важные вопросы технологии сжигания топлив, в разработку ко-
торых химики вносили и будут вносить значительный вклад по-
средством практического применения результатов фундамен-
тальных исследований процессов горения. За последние 20 лет
в технологии сжигания топлив произошли очень важные пере-
мены, причины которых в большинстве своем выходят за пре-
делы технических проблем и поэтому лежат вне компетенции
ученых. При кратком рассмотрении этих изменений будут вы-
делены те проблемы, с которыми сталкиваются ученые-исследо-
ватели, работающие в такой динамичной и приобретающей все
большее значение области, как технология сжигания топлив.
Основные нефтяные топлива
Жидкие нефтяные топлива наряду с каменным углем и при-
родным газом принято называть первичными топливами, так
как их применение почти не требует какой-либо предваритель-
ной обработки (исключая простое физическое разделение на
фракции путем перегонки). Горение каменного угля в данной
главе не рассматривается, так как эта тема заслуживает спе-
циального обсуждения (см. [1]).
Наиболее распространенные газообразные и жидкие топлива
и их типичные свойства приведены в табл. 14.1 и 14.2.
Таблица 14.1
Теплотехнические характеристики обычных газообразных топлив
Топливо Теплотворная способность Теоретически необходимое отношение воздуха к топливу (по объему) Скорость пламени (максималь- на и), см/с Пределы воспламеие- * НИЯ, °/ о по объему
кДж/м3 сухого топлива кДж/кг сухо- го топлива (при 15 °C)
Водород 12 100 141 900 2: 1 260 4-75
Светильный газ 18 400 32 700 4 :1 100 5-31
Метан 37 700 55 600 9:1 33,8 5-15
Технический 93 900 50 400 23: 1 39,6 2-11
пропан Технический бутан 121 800 49 500 30: 1 36,6 1,7-9
Светильный газ относится к вторичным топливам, поскольку
он не встречается в природе, а получается путем либо коксова-
ния или газификации каменного угля, либо паровой конверсии
А. Химия и горение нефтяных топлив
553
Таблица 14.2
Типичные свойства обычных жидких топлив
Топливо Плотность при 15,5 °C Пределы выкипания, °C Содержа- ние серы, % по весу Тепло- творная способ- ность, кДж/кг Теоретически необходимое отношение воздуха к топливу (по весу)
Бензин 0,75 30—180 Ниже 0,1 47 300 14,7: 1
Керосин 0,78 150-260 - 0,1 46 500 14,7: 1
Газойль 0,84 180-350 0,6 45 600 14,7: 1
Котельное топливо 0,96 180 — выше 370 2,5 43 900 14,0 : 1
жидких нефтяных топлив. В нем содержится значительное ко-
личество водорода.
Природный газ может залегать как самостоятельно, так
и совместно с сырой нефтью или каменным углем. Главным ком-
понентом природного газа является метан, содержание которого
в газе различных месторождений колеблется от ~70 до 95%
по объему.
Сжиженный нефтяной газ (СНГ) содержит пропан и бутан.
Он особенно удобен для портативных газонагревательных при-
боров, так как легко сжижается под давлением.
Бензин, самое летучее из жидких нефтяных топлив, исполь-
зуется почти исключительно в автомобильных двигателях с иск-
ровым зажиганием.
Керосин (англичане в обиходе называют его парафиновым
маслом) служит в Англии и некоторых других странах, осо-
бенно Австралии, важнейшим топливом для коммунально-быто-
вых отопительных устройств. Аналогичная по составу, но отве-
чающая более жестким требованиям фракция — так называе-
мый авиационный керосин (АК)—используется в двигателях
дозвуковых и сверхзвуковых реактивных самолетов.
Газойль является основным видом коммунально-бытового
топлива в континентальной Западной Европе, США и Канаде.
В Англии и за ее пределами он широко используется под назва-
нием дизельное топливо в двигателях мощных тепловозов пас-
сажирских и товарных поездов.
Котельное топливо (преимущественно недистиллятное) — это
остатки от перегонки сырой нефти и крекинга нефтепродуктов
или отходы производства смазочных масел. В продаже имеется
несколько марок котельного топлива, которые различаются
главным образом по вязкости. Присутствие серы в нефтяных
остатках, которое раньше считалось нежелательным лишь из-за
корродирующих свойств продуктов горения, в настоящее время
554
Глава 14
вновь привлекает серьезное внимание в связи с непрерывным
ужесточением норм на содержание загрязнений в дымовых
газах.
Доля различных нефтяных топлив в~ структуре переработки
сырой нефти зависит от относительного спроса на них в данной
стране. На рис. 14.1 представлены типичные структуры спроса
для Западной Европы, Японии и США (см. также гл. 1). Мало
кто знает о том, что в США до 80% сырой нефти, а в Западной
Европе и Японии — до 90% используется в качестве топлива.
Лишь небольшая (но имеющая очень важное значение) доля
Рис. 14.1. Структура спроса на нефтяные топлива в США, Западной Ев-
ропе и Японии в 1970 г.
♦ Включая нефтезаводское топливо и потери.
нефти расходуется на химические цели — производство раство-
рителей, пластмасс, каучуков, моющих веществ и др.
Таким образом, главным потребителем нефти является теп-
лоэнергетика. Теперь рассмотрим основные химические явле-
ния, происходящие при горении, которые позволяют использо-
вать топливо как источник энергии в современной промыш-
ленности.
Химия горения
Практические пламена
Горение можно в широком смысле определить как относи-
тельно быструю экзотермическую реакцию, а пламя — как реак-
цию горения, способную распространяться в пространстве с до-
звуковой скоростью (в отличие от детонации, которая распро-
страняется со сверхзвуковой скоростью).
/
А. Химия и горение нефтяных топлив
555
Способность пламени к распространению делает возможным
создание системы с непрерывным подводом топлива и воздуха,
в которой будет поддерживаться стационарное пламя. Это
пламя можно стабилизировать с помощью горелки. Скорость
подачи топлива обычно, колеблется от нескольких миллилитров
в минуту для «дежурного» пламени бытовых газогорелочных
устройств до нескольких тонн в час в случае факела промыш-
ленной мазутной форсунки.
А Спокойное
диффузионное
пламя
Шумящее
аэрированное
пламк
\ Beci. необходимый
‘ \ для горения воздух
/у УУ Вторичный
Г \ воздух
Воздушные
окна
закрыты
Воздушные
окна
открыты
\\ Первичный
| j \ воздух
Неразбавленное
топливо
а
Неразбавленное
топливо
Рис. 14.2. Пламя горелки Бунзена.
а — диффузионное пламя; б —пламя предварительно смешанных газов (аэрированное
пламя).
Различают два основных типа стационарного пламени:
1. Диффузионное пламя, где неразбавленный поток топлива
и весь воздух, необходимый для горения, смешиваются между
собой путем диффузии через поверхность пламени. В зависи-
мости от скорости подачи топлива и скорости его смешивания
с воздухом диффузионное пламя может быть ламинарным или
турбулентным. Практическими примерами диффузионного пла-
мени являются пламя горелки Бунзена при закрытых воздуш-
ных окнах (рис. 14.2,а), пламя свечи, простой факел сжигае-
мого нефтезаводского газа и пламя, получаемое при капельном
горении жидкого топлива. Длина диффузионного пламени, как
следует из этих примеров, может составлять от нескольких сан-
тиметров до многих метров.
2. Пламя предварительно смешанных газов, где топливо
смешивается перед поджиганием с частью необходимого по сте-
хиометрии воздуха (обычно состав полученной смеси ограничи-
вается пределами воспламенения). Этот прием’называется пер-
вичной аэрацией. Для более полного сжигания топлива в пламя
556
Глава 14
вводится вторичный воздух. Практическим примером пламени
предварительно смешанных газов может служить пламя бунзе-
новской горелки с открытыми воздушными окнами (рис. 14.2,6).
Несмотря на то что большинство промышленных пламен от-
носится к турбулентному диффузионному типу, химия диффу-
зионных пламен изучена еще недостаточно. Устойчивость, форма
и яркость этих пламен определяется главным образом физиче-
скими процессами турбулентной диффузии. Показано, что в
установившемся режиме горения топливо и кислород не нахо-
дятся в контакте друг с другом, а разделены пограничным слоем,
в котором концентрация каждого из них снижается до нуля.
Реакция протекает на обеих поверхностях этого горячего слоя,
и общий механизм горения углеводородных топлив, по-видимо-
му, сводится к образованию углерода (путем пиролиза) на внут-
ренней стороне пограничного слоя и образованию активных ра-
дикалов (вероятно, гидроксильных) на его наружной стороне.
Таким образом, частицы, реагирующие между собой в погра-
ничном слое, не являются исходными реагентами. Детали меха-
низма горения в диффузионном пламени пока не выяснены, хотя
число работ, посвященных изучению физической картины про-
цесса «смесеобразования», очень велико. Некоторые проблемы,
относящиеся к практическим диффузионным пламенам, обсуж-
даются ниже.
Пламена предварительно смешанных газов принято характе-
ризовать скоростью пламени, т. е. скоростью распространения
фронта пламени в негорящую смесь топлива с воздухом. Вели-
чина этой скорости зависит преимущественно от состава горю-
чей смеси, температуры и давления. В том случае, когда пламя
стабилизировано на горелке, скорость фронта пламени может
превышать скорость газовоздушного потока, и тогда возникает
опасность «проскока» пламени в подаваемую смесь. Диффу-
зионное пламя не способно к «проскоку». (Подробное обсужде-
ние структуры различных пламен можно найти в моногра-
фии [2]).
В практическом пламени предварительно смешанных газов
быстрое выделение в реакционной зоне большей части энталь-
пии реакции дает очень высокие температуры. Подсчитано, что
толщина этой зоны в типичном пламени составляет десятые доли
миллиметра, максимальный градиент температуры достигает
100 000°С/см, а ускорение газа более чем в 1000 раз превышает
ускорение под действием силы тяжести. Поэтому не удиви-
тельно, что явления молекулярного переноса — теплопроводно-
сти, конвекции и диффузии — играют важную роль в процессах
горения и обусловливают многие особенности структуры и устой-
чивости пламен предварительно смешанных газов. Перечислен-
ные физические факторы оказывают влияние и на химизм
А. Химия и горение нефтяных топлив 557
горения, так как радикалы, инициирующие пламенные реакции,
могут легко переноситься по механизму термодиффузии или
концентрационной диффузии навстречу потоку реагентов из вы-
сокотемпературных зон пламени в низкотемпературные. Это
явление вызывает определенные затруднения в установлении
точной природы химических реакций инициирования горения.
Физический перенос тепла или активных частиц придает пла-
мени способность к самораспространению, что обусловливает
возможность стабилизации стационарного пламени на горелке
и позволяет использовать его на практике для различных быто-
вых и промышленных целей.
Из всего сказанного можно сделать вывод, что пламя —
это своеобразная химическая система, свойства которой в зна-
чительной степени определяются физическими процессами. Те-
перь рассмотрим химизм реакций, протекающих в пламени.
Быстрые пламенные реакции
Большинство явлений взрыва и горения газовых смесей
можно объяснить в рамках разветвленно-цепного механизма.
Распространение цепной реакции происходит посредством актив-
ных частиц, которые при этом исчезают, генерируя новые актив-
ные промежуточные соединения. Если в реакции образуется два
или несколько видов активных промежуточных продуктов, то
цепь, как принято говорить, разветвляется, а скорость реакции
перестает быть постоянной и начинает экспоненциально возра-
стать, приводя к взрыву или воспламенению газовой смеси, при
которых типичное время реакции составляет от 1 до 10-13 с.
Описанный механизм, в котором выделение энергии происхо-
дит в форме активных промежуточных частиц (атомов и ради-
калов), не следует смешивать с механизмом теплового взрыва,
в котором энергия выделяется в виде тепла. Тепловой взрыв
имеет место в том случае, когда скорость выделения энергии
в виде тепла превышает скорость ее рассеивания в окружаю-
щую среду. Однако число процессов горения, которые можно
адекватно объяснить в рамках одной лишь тепловой теории,
весьма невелико.
Строгое количественное описание поведения любой пламен-
ной системы возможно только при детальном знании механизма
пламенных реакций. К настоящему времени надежно установлен
лишь механизм горения водорода в кислороде. Исходя из этого
механизма и соответствующих термохимических данных, были
выведены уравнения континуума для системы водород—кисло-
род, что позволило описать поведение водородного пламени си-
стемой дифференциальных уравнений и затем вычислить его
физические характеристики. Эта процедура была выполнена
558
Глава 14
применительно к «простой» системе Н2—О2, причем экспери-
ментальные данные по структуре пламени медленно горящей
смеси Н2—О2—N2, богатой водородом, очень близко совпадают
в широком интервале состава этой смеси с результатами расче-
тов на ЭВМ.
Предполагаемый механизм постулирует промежуточное об-
разование радикалов Н«, «ОН, О- и НО2« и включает стадии
(1) — (13) (М— «третье тело»)
.он + н2 - Н2О + Н. (1) продолжение цепи
Н. + О2 - HO.-J-O. (2) разветвление цепи
О. + Н2- -> НО. + Н. (3) разветвление цепи
Н. + О2 + М - —> НО2 • м (4)
Н«+НО2. - -> НО.-р.ОН (5)
Н« + Н2О. - -> О. + Н2О (6)
Н. + НО2. - -> н2 + 02 (7)
НО» + НО2. — -► н2о + 02 (8)
о- + но2« - .ОН+О2 (9)
Н.4-.Н+М — > Н2 + М (10) рекомбинация
Н.-+-НО. + М — -> Н2о + М (11) рекомбинация
Н. + О. + М — -> НО.-+-М (12)
Реакция инициирования цепи, вероятно, протекает по уравнению
Н2-|-О2 —> 2НО • (13)
Эта стадия не представлена в общей схеме, так как ее вклад
в суммарную кинетику процесса очень мал.
Ведущая кинетическая цепь реакции окисления водорода
кислородом отвечает элементарным реакциям (1) — (3), которые
протекают с участием атомов и радикалов и образуют единую
разветвленную цепь. Однако в водородно-кислородных пламе-
нах по мере прохождения этих реакций все более важную роль
начинают играть и другие стадии пламенного процесса, и когда
реакции (1) — (3) становятся существенно обратимыми или гене-
рируемые ими активные частицы гибнут в конкурирующих ре-
акциях, разветвление цепи прекращается. Таким образом, в пла-
мени (в отличие от фронта детонации) имеет место динамиче-
ское равновесие, и значительная доля энтальпий горения акку-
мулируется в радикалах и атомах, присутствующих в пламени
в высоких концентрациях. Энергия радикалов и атомов рассеи-
вается в реакциях рекомбинации, медленно протекающих по
уравнениям третьего порядка [главным образом по уравнениям
(10) и (11)]. Эти реакции идут во времени и пространстве еще
довольно долго после завершения реакций продолжения и раз-
ветвления цепи и обусловливают медленное приведение актив-
А. Химия и горение нефтяных топлив
559
ных частиц в состояние теплового равновесия со сгоревшими
газами. Твердые поверхности, окружающие пламя, могут спо-
собствовать протеканию реакций обрыва цепи, адсорбируя при-
близившиеся к ним радикалы и атомы. Затем происходит ре-
комбинация захваченных активных частиц с образованием
устойчивых молекул.
При изучении горения окиси углерода в кислороде и более
позднем исследовании химизма горения метана было показано,
что обе эти реакции во многом аналогичны по своему механизму
реакции горения водорода. Поэтому в настоящее время принято
считать, что характеристики водородного пламени можно ис-
пользовать и для процессов горения углеводородов.
Реакции, медленного горения
Помимо быстрых пламенных реакций, существуют также ре-
акции медленного горения, которые подробно изучаются с на-
чала нынешнего столетия.
Область медленного горения, в которой взаимодействие
углеводородов с кислородом протекает относительно медленно
и не сопровождается быстрыми пламенными реакциями, лежит
в интервале ~200—600 °C. Продолжительность индукцион-
ного периода, а также время реакций в этой области состав-
ляют от десятых долей секунды до десятков минут. Таким обра-
зом, реакции медленного горения легче поддаются детальному
исследованию, чем очень быстрые пламенные реакции, описан-
ные выше.
На рис. 14.3 приведено сильно упрощенное изображение так
называемой кривой Т — Р (температура — давление), которая
разделяет область реакций медленного горения углеводорода в
кислороде и область «взрывообразных» быстрых пламенных ре-
акций (называемых иногда реакциями истинного горения).
Можно видеть, что по мере повышения температуры смеси дав-
ление, необходимое для инициирования быстрых реакций, про-
ходит через минимум и максимум, обусловленные сложной за-
висимостью скоростей этих реакций от температуры. Область
вблизи минимума носит название «полуостров». Кривые Т—Р
почти всех углеводородных топлив удивительно похожи друг на
друга по форме и положению относительно шкалы температур.
Однако их положение относительно шкалы давлений сильно
различается. Например, в случае стехиометрических смесей нор-
мальных парафинов с кислородом кривая Т—Р сдвигается с
увеличением длины цепи молекулы топлива в сторону более
низких давлений.
Кривые Т—Р были получены в статических реакторах. Эти
реакторы широко используются при изучении процессор
560
Глава 14
медленного горения, так как знание граничных значений давле-
ния и температуры, а также скорости реакции при постоянном
объеме позволяет получить точные кинетические данные.
На самом деле область медленного горения на графике Т—Р
для смесей углеводородов с кислородом имеет гораздо более
сложный вид, чем изображенная на рис. 14.3. Переход от реак-
ции медленного горения к быстрой реакции может при опреде-
Рис. 14.3. Типичная кривая Т — Р для углеводородных пламен.
ленных значениях температуры и давления происходить путем
так называемого двухстадийного воспламенения. Этот переход
можно наблюдать в статическом реакторе (либо визуально, либо
с помощью чувствительных приборов, реагирующих на измене-
ния давления или температуры) как последовательность движу-
щихся зон интенсивного свечения и периодического испускания
света, которые получили название холодных и голубых пламен.
Эти пламена не следует путать с практическими пламенами,
в которых протекают быстрые химические реакции. Методика и
результаты исследований в статических реакторах достаточно
А. Химия и горение нефтяных топлив
561
полно описаны в литературе [3], и поэтому здесь их подробное
рассмотрение является излишним.
Хотя по причинам, указанным выше, при изучении реакций
горения широко применяются статические реакторы, проточные
реакторы дают более сильное зрительное впечатление при
наблюдении медленного горения, так как они позволяют стаби-
лизировать двухстадийное воспламенение, сделав его доступным
Воздух, *
. подогретый,
до torn х
S00 °C
200 ° С
Кислород
Смесь инертного *
газа, с топливом
Рис. 14.4. Вертикальный проточный реактор,
обогреваемый горячим воздухом.
для визуального исследования. Описаны эксперименты со сме-
сями н-гептана и кислорода [4], в которых был использован вер-
тикальный проточный реактор, дающий разделенные пламена.
Близкий по конструкции реактор применялся и автором данной
главы; некоторые из полученных результатов приведены ниже.
На рис. 14.4 изображена схема обогреваемого вертикального
проточного реактора диаметром около 25 мм, в котором в потоке
инертного газа, находящегося при атмосферном давлении, под-
держивается температурный градиент от 200 до 600 °C. Если
с инертным газом смешать пары углеводорода (например,
^-гептана) и затем медленно вводить в эту смесь кислород, то
562
Глава 14
Рис. 14.5. Различные пламена «-геп-
тана, стабилизированные в верти-
кальном проточном реакторе.
и — голубое пламя; б—второе холодное
пламя; а—холодное пламя.
можно проследить всю карти-
ну медленного горения, кото-
рую удобно наблюдать в за-
темненной лаборатории.
Сначала в верхней части
реактора появляется голубая
«дымка». Она медленно рас-
пространяется вниз навстречу
потоку реагентов и спустя не-
сколько минут стабилизирует-
ся у основания реактора в фор-
ме ясно различимого бледно-
голубого пламени с низкой
интенсивностью свечения. Это
так называемое холодное пла-
мя. Вопреки ожиданиям, в хо-
лодном пламени расходуется
лишь небольшая доля вводи-
мого топлива и кислорода.
Причиной этого является от-
рицательный температурный
коэффициент реакции, обус-
ловленный изменением веду-
щего механизма разветвления
цепи. Максимальная темпера-
тура пламени составляет лишь
~ 150 °C (отсюда термин «хо-
лодное» пламя).
При повышении концентра-
ции кислорода холодное пла-
мя остается, но вслед за ним
появляется гораздо более яр-
кое голубовато-синее «голу-
бое» пламя, стабилизирующее-
ся немного выше холодного
пламени. В голубом пламени
потребляется значительно
большее количество топлива и
кислорода, но еще не все.
Иногда между первым холод-
ным и голубым пламенами
можно наблюдать ряд холод-
ных пламен (многократные хо-
лодные пламена).
Дальнейшее увеличение
концентрации кислорода со-
А. Химия и горение нефтяных топлив
563
провождается появлением желтого пламени (визуально похо-
жего на пламя свечи), которое располагается следом за голу-
бым пламенем. Это пламя, в котором расходуется остальное
количество топлива и кислорода, соответствует на графике Т—Р
точке, лежащей в области быстрой пламенной реакции.
Стадии медленного горения, предшествующие образованию
желтого пламени (два холодных пламени и голубое пламя), яв-
ляются устойчивыми и хорошо воспроизводятся при обратном
Рис. 14.6. Температурный профиль разделенных пламен н-гептана.
1—градиент температуры в отсутствие пламен; 2—области горячего («видимого») пла-
мени; 3 — голубое пламя; 4—-второе холодное пламя; 5 —первое холодное пламя.
уменьшении концентрации кислорода. Изображения этих пла-
мен приведены на рис. 14.5.
При последующем увеличении подачи кислорода вся струк-
тура становится нестабильной, и в конечном итоге горение с
ускорением (иногда переходя в детонацию) распространяется
вниз, по направлению к огнепреградителю, расположенному у
входа в реактор.
В постоянных условиях каждая из стадий горения в проточ-
ном реакторе характеризуется определенными профилями тем-
пературы и состава, анализ которых позволяет еще глубже про-
никнуть в механизм химических превращений, происходящих
при двухстадийном воспламенении. На рис. 14.6 представлен
564
Глава 14
температурный профиль реактора, соответствующий картине,
изображенной на рис. 14.5.
Поскольку отношение топлива к кислороду на каждой ста-
дии (рис. 14.5 и 14.6) изменяется и проточный реактор работает
при постоянном (атмосферном) давлении, полученные в нем ре-
зультаты нельзя непосредственно связать с кривыми Т—Р для
смесей н-гептана с кислородом, полученными в статическом ре-
акторе. Однако можно нарисовать приближенный график
(рис. 14.7), свидетельствующий о качественной корреляции ме-
жду типичной кривой Т — Р и приведенными выше данными,
Линия Давление
постоянного
давления
—»- Проточный реактор
Рис. 14.7. Сопоставление параметров медленного горения
в проточном реакторе с типичной кривой Т — Р.
полученными при постоянном давлении в проточном реакторе.
На рис. 14.7 приведены также температуры инициирования для
отдельных стадий медленного горения н-гептана.
Как можно видеть, на графике Т — Р возникают «полуост-
ров» холодных пламен (область, в которой горение происходит
по холоднопламенному механизму) и переходная область, соот-
ветствующая началу горения по механизму голубого пламени,
которое при дальнейшем продвижении по кривой Т—Р пере-
ходит в быстрое горение.
К числу особенностей медленного горения, наблюдаемых
в статическом реакторе, относятся появление периодических хо-
лодных пламен, задержка воспламенения и значительные (иног-
А. Химия и горение нефтяных топлив 566
да десятки минут) колебания индукционного периода, т. е. вре-
мени от момента смешивания реагентов до начала реакций мед-
ленного горения.
Хотя процессы медленного горения исследованы еще недо-
статочно, принято считать, что двухстадийное воспламенение
возникает в результате химического и термического превраще-
ния горючей смеси под действием холодного пламени, которое
переводит систему из режима медленного горения в режим
быстрых пламенных реакций (истинного горения).
Таким образом, любая теория медленного горения углеводо-
родов должна объяснять следующие явления: а) двухстадийное
воспламенение; б) многократные холодные пламена; в) периоди-
ческие холодные пламена (которые при определенных экспери-
ментальных условиях наблюдаются как в статическом, так и
в проточном реакторах); г) задержку воспламенения (в стати-
ческом реакторе); д) существование «полуостровов» на кривых
Т — Р всех углеводородов; е) отрицательный температурный
коэффициент реакции, который вызывает угасание холоднопла-
менной вспышки по достижении температуры всего лишь
~ 150 °C; ж) непостоянство положения кривой Т — Р различных
углеводородов относительно шкалы давлений.
Значительные успехи в объяснении некоторых из этих явле-
ний были достигнуты в 1925—1935 гг., когда рядом ученых было
начато изучение реакций медленного горения, а также пламен-
ных процессов на основе теории цепных реакций (эта теория
рассмотрена выше на примере реакции между водородом и кис-
лородом).
В частности, Н. Н. Семенов установил, что феноменология
медленного горения углеводородов в газовой фазе успешно объ-
ясняется в рамках цепной реакции, протекающей с вырожден-
ным разветвлением. Этот термин применяется для обозначения
частного случая разветвленно-цепной реакции, в которой раз-
множение активных промежуточных частиц происходит очень
редко, вследствие чего суммарная скорость процесса постепенно
снижается до нуля из-за протекания побочных реакций или пол-
ного исчерпания реагентов.
Н. Н. Семенов постулировал, что при вырожденном разветв-
лении стабильная промежуточная частица, имеющая довольно
большое время жизни, превращается далее либо в свободные
радикалы (что сопровождается разветвлением цепи), либо
в другой стабильный продукт
, ---------------> радикалы
«1 разветвление цепи
Т0ПЛИ30 + О2 —► Z — йз
-> инертные продукты
566
Глава 14
где kj, k2 и k3 — относительные константы скорости. Таким об-
разом, не все частицы, обозначенные символом Z, вызывают
разветвление, что приводит к задержке воспламенения, или вы-
рождению цепи.
Однако теория Н. Н. Семенова не принимает во внимание
тепловые эффекты, которые несомненно имеют место в холодно-
пламенных реакциях. Поэтому многие исследователи, позднее
занимавшиеся этой проблемой, пытались разработать теорию,
охватывающую все наблюдаемые явления.
Недавно был предложен ряд математических моделей, в ко-
торых рассматривается не детальный химический механизм, а
общая кинетическая схема с гипотетическими активными про-
межуточными частицами.
Осцилляционно-кинетическая модель Д. А. Франк-Каменец-
кого, основанная, на представлении об осцилляции концентра-
ций двух основных промежуточных продуктов (вероятно,
перекисей и альдегидов), является несовершенной, поскольку
из нее вытекает, что характер осцилляции не зависит от тем-
пературы и давления, что противоречит экспериментальным
данным.
Термокинетическая модель, разработанная Салинковым, по-
видимому, лучше предсказывает холоднопламенные явления; од-
нако она не объясняет отрицательный температурный коэффи-
циент реакции в области холодного пламени.
Более поздняя теория Янга и Грея (1969 г.), которая яв-
ляется развитием теории Салинкова и учитывает термокинети-
ческую связь между концентрацией промежуточного продукта и
температурой, объединяет многие представления предшествую-
щих исследователей. Она позволяет предсказывать все наблю-
даемые в холодном пламени явления, используя приемлемые
данные по константам скоростей и энтальпиям'реакций, причем
результаты вычислений на ЭВМ количественно согласуются с
результатами эксперимента в статическом реакторе.
Хотя может показаться (вследствие больших успехов, до-
стигнутых за последние 70—80 лет), что фундаментальный ме-
ханизм медленного горения углеводородов в статических реак-
торах близится к количественному пониманию, упомянутые
выше математические теории мало что говорят о природе про-
межуточных частиц и не могут быть использованы для точного
объяснения феноменологии горения в проточных реакторах.
Отсюда очевидно, что предстоит проделать еще значитель-
ную работу, прежде чем будет детально установлен меха-
низм горения для всех систем углеводородное топливо
кислород.
А. Химия и горение нефтяных топлив Б67
Использование полученных данных
по химизму горения
Ниже рассматриваются некоторые области техники, в ко-
торых с успехом используются или могут быть использованы
фундаментальные химические данные о процессах горения.
Кинетика горения
Отсутствие теоретических знаний о процессах, происходящих
в применяемых и вновь разрабатываемых пламенных системах,
не помешало развитию технологии сжигания топлив. Для хи-
мика в прошлом одним из камней преткновения при исследова-
нии горения была сложность выполнения необходимых матема-
тических расчетов. Однако теперь, когда разработаны методы
решения систем нелинейных дифференциальных уравнений лю-
бой степени сложности, можно с уверенностью предсказать бы-
стрый прогресс в наших знаниях о кинетике пламенных реакций.
В качестве примера можно указать на расчетную модель выде-
ления окиси азота в двигателе с искровым зажиганием, создан-
ную на основе известных кинетических, термодинамических и
физических данных. Эта модель может быть также использована
для предсказания влияния рабочих параметров двигателя и
рециркуляции выхлопных газов на количество образующихся
окислов азота. Аналогичная работа проводится в настоящее
время по изучению промышленных диффузионных пламен с це-
лью нахождения методов предсказания концентрации окислов
азота в продуктах -горения и путей ее уменьшения. Важность
таких исследований для целей прогнозирования и для борьбы с
загрязнением окружающей среды очевидна.
Детонация топлива в двигателях
Недостаточность наших знаний о природе основных пламен-
ных реакций стала особенно остро чувствоваться в начале XX в.,
когда был изобретен двигатель с искровым зажиганием и воз-
никла проблема борьбы с неуправляемым горением топлива,
приводящим к возрастанию скорости передачи тепла от сгорев-
ших газов к стенкам камеры сгорания, снижению мощности
двигателя, опасности его разрушения и шуму. Это явление полу-
чило название «детонация» (т. 1, стр. 174). К настоящему вре-
мени накоплено много данных, которые свидетельствуют о связи
детонации с самовоспламенением топливо-воздушной смеси (че-
рез стадии образования холодного и голубого пламен), происхо-
дящим до прихода фронта пламени, возбужденного искрой.
Ключ к решению этой проблемы был найден совместными
568
Г лава 14
усилиями инженера и химика (Миджли и Бойд, 1922 г.), которые
ввели в состав топлива тетраэтилсвинец (ТЭС). Механизм- инги-
бирования холоднопламенных реакций под действием ТЭС в то
время был неизвестен, да и в настоящее время он полностью не
выяснен. Вероятно, этот механизм включает распад ТЭС в хо-
лодном пламени с образованием «тумана» из мелкодисперсных
частиц окислов свинца, на поверхности которых происходит
обрыв цепных реакций окисления углеводородов. Тем самым
предотвращается переход к реакциям голубого пламени, приво-
дящим к зажиганию топливо-воздушной смеси. Уникальные ан-
тидетонационные свойства алкильных производных свинца, по-
видимому, обусловлены тем, что в их присутствии холоднопла-
менные реакции сами поставляют энергию, необходимую для
ингибирования процесса зажигания. Описанный механизм под-
твержден опытами в проточном реакторе, упоминавшемся выше,
в которых добавление к топливу небольших количеств ТЭС не
оказывало влияния на холодное пламя, но вызывало медленное
исчезновение из реактора голубого пламени.
В Западной Европе существует тенденция к использованию
все более компактных двигателей с высокой степенью сжатия.
Однако требования охраны окружающей среды, ограничиваю-
щие применение этилированного бензина, вынуждают вновь
вернуться к двигателям с более низкими степенями сжатия.
В связи с этим детальное изучение природы реакций медлен-
ного горения, протекающих в двигателях с искровым зажига-
нием, и, возможно, регулирование этих процессов приобретают
все более важное значение.
Холодные пламена
Установлено, что положение, в котором холодное пламя
стабилизируется в проточном реакторе, можно варьировать с
помощью небольших изменений октанового числа подаваемого
топлива. На основе этого был разработан прибор непрерывного
действия для определения октанового числа бензинового потока,
работающий по принципу стабилизации холодного пламени в
одном положении путем изменения давления в реакторе. В при-
боре используется контур обратной связи, содержащий два чув-
ствительных элемента (рис. 14.8). Изменение давления может
быть прокалибровано в октановых числах, шкала которых охва-
тывает около 10 единиц в ту и другую сторону от октанового
числа стандартного топлива. Этот метод постепенно вытесняет
дорогостоящий и длительный метод определения октанового
числа в стандартном двигателе, снабженном устройством для
регистрации детонации топлива; очевидно, что он может обес-
печить значительное сокращение затрат производства на неф-
А. Химия и горение нефтяных топлив
569
теперерабатывающих заводах. Природа данного явления точно
неизвестна; возможно, что оно связано с влиянием давления и
молекулярной структуры топлива на индукционный период ре-
акций медленного горения.
Были отмечены случаи возникновения холодного пламени
в бензобаках самолетов, когда температура у поверхности топ-
лива превышала ~ 200 °C. Небольшое повышение давления в
результате прохождения холодного пламени в статическом па-
ровом пространстве над топливом может оказаться достаточным
Рис. 14.8. Холоднопламенный измеритель октанового числа.
/—контрольно-измерительное сервоустройство; 2—датчики; 3 — обогреваемый реактор;
4—индикатор давления; 5—регулятор давления.
для разрушения топливных баков, что представляет потенциаль-
ную опасность. В настоящее время проводятся широкие иссле-
дования как в статических, так и в проточных реакторах с
целью борьбы с этим явлением, которое может стать особенно
опасным для сверхзвуковых самолетов, где топливо одновре-
менно служит хладоагентом, предотвращающим перегревание
обшивки.
Многоядерные ароматические углеводороды, образующиеся
при двухстадийном воспламенении бензиновых топлив в про-
точном реакторе, были подвергнуты сравнительному исследо-
ванию вместе с отложениями продуктов неполного сгорания, из-
влеченными из выхлопной трубы бензинового двигателя. Заме-
чательное сходство тех и других по составу и последователь-
ности отложения (по высоте реактора и по длине выхлоп-
ной трубы) демонстрирует подобие механизмов реакций угле-
водородов при низких и высоких давлениях и лишний раз
570
Глава 14
подчеркивает все более важное значение лабораторных иссле-
дований в деле борьбы с загрязнением воздушной среды.
Другими областями потенциального применения данных по
изучению холоднопламенных реакций являются производство
продуктов тяжелого органического синтеза путем окисления
углеводородного сырья и разработка методов химического ини-
циирования горения дизельного топлива посредством впрыски-
вания в топливо в точке его ввода в двигатель продуктов холод-
нопламенной реакции. Ни одна из этих возможностей еще не
реализована на практике; однако они иллюстрируют направле-
ния, в которых может развиваться использование явления мед-
ленного горения, представлявшего ранее чисто теоретический
интерес.
Дистиллятные топлива
Результаты фундаментальных исследований по горению уг-
леводородов не нашли широкого применения при конструиро-
вании коммунально-бытовых горелочных устройств, работаю-
щих на дистиллятных нефтепродуктах, поскольку немногие
Ьтлоричныи
воздух
Первачньтй
воздух
Подача
топлива
a d
Рис. 14.9. Секция горелки резервуарного типа.
а —низкое пламя; б — высокое пламя. 1—отражательные плиты; 2—корпус; 3 — воздушные
окна; 4—горячая испарительная поверхность.
встретившиеся проблемы (главным образом неполное сгорание
топлива) были легко решены эмпирическими методами.
Единственное исключение составляла проблема образования
отложений продуктов предкамерного сгорания (так называемых
углеродистых отложений) у основания горелок резервуарного
типа, что обусловлено проскоком тепла от пламени к парам на-
гретого топлива, находящегося в резервуаре (рис. 14.9). Де-
А. Химия и горение нефтяных топлив
571
тальный механизм этого явления неизвестен. Было выдвинуто
предположение, что парафиновые радикалы, получающиеся в
реакциях типа тех, которые протекают в холодном пламени,
реагируют при температурах ~ 400 °C с ароматическими угле-
водородами, присутствующими в топливе, давая ароматические
радикалы; последние довольно нестабильны и полимеризуются
на металлической стенке с образованием высокомолекулярных
отложений. Для инициирования этого процесса необходимо лишь
небольшое количество кислорода. Легко доказать, что образо-
вание нагара можно подавить путем сокращения промежутка
времени между испарением и сгоранием топлива, а также путем
исключения контакта паров с холодными металлическими по-
верхностями.
Следует отметить, что употребление в обиходе таких терми-
нов, как «смолы» и «кокс», объясняется сложностью природы
углеродистых отложений. Все эти отложения состоят преиму-
щественно из углерода (содержание водорода составляет не-
сколько процентов). Поэтому они достаточно полно характери-
зуются отношением С:Н и основными кристаллографическими
параметрами.
Капельное горение
Обычно сжиганию жидкого топлива в бытовых и промыш-
ленных горелочных устройствах предшествует его распыление
до капелек размером ~ 50—500 мкм. Это приводит к значи-
тельному увеличению поверхности топлива и облегчает его вос-
пламенение и горение (протекающее преимущественно по диф-
фузионному типу).
Прежде чем закончится выгорание каждой капельки, в не-
посредственной близости от нее и внутри нее протекает большое
число взаимосвязанных процессов. Такие процессы, как испаре-
ние, дистилляция, крекинг, смешивание, образование и горение
сажи, были детально изучены на модельных системах с учетом
всех типов форсунок, в которых топливо распыляется сжатым
воздухом. Котельное топливо содержит высококипящие фракции
нефти, которые могут частично крекироваться. Это приводит к
задержке зажигания и нежелательному образованию сажи, со-
стоящей из пустотелых сферических коксоподобных частиц, на-
зываемых ценосферами. Для химика особый интерес представ-
ляет тот факт, что внутри капелек или сажевых частиц по мере
их нагревания и протекания процесса дистилляции могут про-
исходить быстрые химические реакции. В этих реакциях прини-
мают участие компоненты золы, в частности натрий и ванадий,
превращения которых имеют очень большое значение: извест-
но, что высокотемпературная коррозия горячих металлических
572
Глава 14
поверхностей в котлах и газовых турбинах связана с образова-
нием сложных соединений натрия и ванадия.
Резюмируя современное положение в области исследования
капельного горения, можно отметить, что большинство выпол-
ненных работ относится к изучению смешивания капелек топли-
ва с воздухом и горячими сгоревшими газами, испарения капе-
лек и общих проблем турбулентности. Все большее внимание
привлекает вопрос о влиянии химического состава топлива на
скорость горения, так как этот фактор может затруднять ис-
пользование очень высоких скоростей сжигания топлив.
Проблемы загрязнения окружающей среды
и коррозии
Из всех путей, которыми современная цивилизация загряз-
няет воздушный бассейн, главным является сжигание горючих
ископаемых (каменного угля, нефти и газа). Поэтому выяснение
природы нежелательных пламенных реакций, приводящих к об-
разованию вредных продуктов, имеет очень важное значение и
является первым шагом на пути к их частичному или полному
подавлению. Особенно большой интерес привлекают в настоя-
щее время реакции образования двуокиси серы (SO2), трехокиси
серы (SO3), окислов азота [(NO)J и сажи.
При горении большая часть серы, присутствующей в нефтя-
ном топливе, окисляется до двуокиси серы и лишь не более 3%
последней подвергается дальнейшему окислению в SO3. Хотя
трехокись серы неустойчива при высоких температурах, при тем-
пературах ниже ~ 325 °C она соединяется с водяным паром, со-
держащимся в продуктах горения, и образует серную кислоту.
Кислота может выбрасываться в атмосферу в виде устойчивого
тумана или конденсироваться на металлических поверхностях,
имеющих температуру ниже ~ 120 °C, вызывая их сильную кор-
розию.
Для некоторых районов мира окислы азота представляют со-
бой наиболее важную часть проблмы загрязнения воздушного
бассейна, так как они играют значительную роль в образовании
фотохимического «смога», часто наблюдающегося в ряде круп-
нейших городов США. Реакции, протекающие на солнечном
свету между МОЖ и парами углеводородов, приводят к возник-
новению слезоточивого тумана, в котором обнаружены такие
ядовитые соединения, как пероксиацетилнитрат. В настоящее
время разрабатываются нормы предельного содержания в воз-
духе NOx, SO2 и других загрязнений.
Вопросу о том, как свести к минимуму образование трех-
окйси серы и окислов азота в процессах горения, посвящено
огромное число работ. Большинство исследователей сходится
А. Химия и горение нефтяных топлив
573
на том, что эти соединения возникают в реакциях, протекающих
с участием атомарного кислорода. В настоящее время разрабо-
таны правила эксплуатации горелок при малом избытке воз-
духа, позволившие уменьшить образование обоих загрязнений.
Однако очевидно, что радикальным решением проблемы было
бы полное подавление их образования.
Механизм образования сажи (т. е. частиц нагара или дыма)
в пламенных процессах послужил предметом интенсивных лабо-
раторных исследований и опытных работ. Во многих странах
уровень ее содержания в дымовых газах ограничивается в зако-
нодательном порядке. Важным свойством пламен является све-
тимость, обусловленная испусканием света раскаленными ча-
стицами сажи. Выгорание этих частиц представляет собой нор-
мальную составную часть процессов факельного горения (осо-
бенно при форсуночном сжигании жидких топлив); однако в
некоторых случаях богатые топливо-воздушные смеси горят не-
полно и выделяют с дымовыми газами много свободной сажи.
Для борьбы с этим явлением прибегают не только к улучшению
конструкции горелок, но и к использованию металлсодержащих
присадок к топливам. Полагают, что такие присадки вызывают
каталитическую диссоциацию водяного пара с образованием
гидроксильных радикалов, которые чрезвычайно быстро реаги-
руют с частицами сажи, давая газообразные продукты, в том
числе СО (ср. с реакцией водяного газа). Однако присутствие
металла в продуктах сжигания топлив, содержащих указанные
присадки, может в свою очередь явиться экологически вредным
фактором, что заставляет искать другие пути подавления обра-
зования сажи в пламени [5].
Англия — одна из немногих стран, где содержание сажи в
нижних слоях атмосферы за последние годы (начиная с .1956 г.,
когда были приняты «Законы о чистом воздухе») существенно
понизилось — в основном за счет сокращения применения угля
для отопления домов и в промышленности. Этот факт еще раз
показывает, что многое- можно сделать путем простой замены
вида топлива, не прибегая к помощи фундаментальной науки.
Однако с ужесточением законодательства по охране окружаю-
щей среды научно обоснованная технология сжигания топлив, в
основе которой лежит глубокое знание пламенных процессов,
будет приобретать все более важное значение в решении возни-
кающих при этом проблем.
Заключение
Из сказанного выше с очевидностью следует, что изучение
химизма горения может внести значительный вклад в развитие
технологии сжигания топлив, помогая решать самые различные
прикладные проблемы.
574
Г лава 14
Хотя еще долгое время технология сжигания топлив будет
по-прежнему развиваться в основном на базе эмпирического ме-
тода проб и ошибок, инженерам придется все чаще прибегать к
использованию результатов научных исследований, чтобы быст-
рее и с наименьшей затратой сил и средств прийти к оптималь-
ному решению стоящих перед ними задач.
Для показа необходимости учета реальной действительности
в этой главе рассмотрены некоторые факторы (преимуществен-
но нетехнического характера), вызвавшие недавно существен-
ные изменения в мировой практике сжигания топлив, и приве-
дены примеры нововведений, которые были сделаны под влия-
нием этих факторов.
Технологические процессы горения
Значение вида топлива
Приблизительно до 1955 г. около 90% потребности Велико-
британии в топливе удовлетворялось за счет каменного угля.
Уголь широко использовался для отопления домов, на тепло-
электростанциях, в парогенераторных и водонагревательных
установках и на железнодорожном транспорте. Разумеется, со
времени промышленной революции XVIII в. каменноугольные
и другие топочные устройства претерпели значительные усовер-
шенствования. Однако теплотехнические принципы их работы
практически не изменились, и все более интенсивные (особенно
с конца прошлого века) фундаментальные исследования процес-
сов горения не оказали, да, вероятно, и не могли оказать, боль-
шого влияния на развитие Технологии сжигания топлив.
В 50-х годах в Великобритании произошла «топливная рево-
люция», во время которой в стране начала создаваться много-
топливная экономика. Весьма обнадеживающими оказались
первые шаги ядерной энергетики. Поиски альтернативных источ-
ников сырой нефти (частично вызванные осложнением полити-
ческой обстановки на Ближнем Востоке) привели к появлению
на английском внутреннем рынке довольно значительного коли-
чества нефтяных топлив, которые весьма успешно конкуриро-
вали по стоимости со все более возрастающим в цене каменным
углем. В результате этого доля нефти в топливно-энергетиче-
ском балансе Великобритании в период 1958—1967 гг. увеличи-
лась с 15 до 37%, и, продолжая расти несколько менее быст-
рыми темпами, к 1973 г. достигла ~ 50%. В 60-х годах «топлив-
ная революция» получила новый толчок: метод производства
«быстроходного» * светильного газа путем коксования каменного
* Т. е. быстрогорящего. — Прим, перев.
А. Химия и горение нефтяных топлив
575
угля был в значительной степени вытеснен паровой конверсией
углеводородного сырья над никелевыми катализаторами. Это
вызвало подъем в развитии газовой промышленности, который
закончился в 1965 г. вместе с открытием промышленных место-
рождений природного газа в Северном море. К 1967 г. природ-
ный газ уже использовался как в быту, так и в промышленно-
сти. С началом добычи природного газа всю технологию исполь-
зования газа в быту пришлось изменить, чтобы приспособить ее
к характеристикам этого медленно горящего углеводородного
топлива (табл. 14.1). Начиная с 1945 г. почти все газовые агре-
гаты в Великобритании (за исключением бытовых газовых
плит) оборудовались горелочными устройствами, состоящими
Рис. 14.10. Одиночная струйная горелка Брея.
1—диффузионное пламя с тонкой внешней оболочкой из продуктов сгорания;
2—горелочная головка.
из множества небольших диффузионных горелок (так называе-
мых струйных горелок Брея, рис. 14.10), в которых промышлен-
ный светильный газ, содержащий до 50% водорода, горит спо-
койным, устойчивым голубым пламенрм. К сожалению, струй-
ная горелка Брея оказалась неподходящей для сжигания при-
родного газа из-за явления отрыва пламени, а настойчивые по-
пытки создать другую диффузионную горелку, приемлемую для
этой цели, не привели к положительным результатам. Поэтому
в качестве отправной точки при переводе коммунально-быто-
вых газовых агрегатов со светильного на природный газ были
выбраны горелки предварительного смешения (горелки с при-
нудительной подачей воздуха), принципиально аналогичные по
своей конструкции горелкам, применявшимся до 1945 г. Эти го-
релки шумели во время работы и обладали неприятной склон-
ностью внезапно гаснуть; кроме того, они засорялись домашней
пылью, всасываемой вместе с первичным воздухом. Использо-
576
Глава 14
вание таких горелок для обслуживания газовых приборов, на-
ходящихся в кухне и жилых комнатах (т. е. котлов централь-
ного отопления и радиационных газовых каминов), нежела-
тельно, так как шум пламени и звук воздуха, выходящего через
сопла инжектора, беспокоят людей. Однако за пределами Вели-
кобритании, где газонагревательные приборы принято устанав-
ливать не в квартирах, а в подвальных помещениях или вне
дома, горелки предварительного смешения являются вполне
приемлемыми и находят широкое применение.
В этой связи не удивительно, что начиная с 50-х годов
наблюдается заметная активизация исследований по технологии
сжигания нефтяных топлив в различных аспектах их промыш-
ленного использования, а несколько позднее — и работ по тех-
нологии сжигания метана для коммунально-бытовых целей. Эти
исследования проводились не только фирмами; ряд очень важ-
ных работ был выполнен, причем на самом высоком уровне, в
университетах и на политехнических факультетах институтов
Великобритании. Однако следует заметить, что многие важные
достижения были сделаны под давлением факторов экономиче-
ского, экологического или коммерческого характера, которые не
всегда давали возможность проводить длительные фундамен-
тальные исследования. Тем не менее эти достижения способ-
ствовали более всестороннему пониманию работы устройств для
создания практических пламен, что имеет важное перспективное
значение на тот случай, когда вновь возникнут обстоятельства,
требующие дальнейшего теплотехнического усовершенствования
этих устройств.
Ниже рассматриваются некоторые примеры таких усовер-
шенствований, относящиеся к недавнему прошлому или ближай-
шему будущему.
Сжигание природного газа
для бытовых целей
Научно-исследовательские и опытные работы, проводимые до
и во время осуществления программы переоборудования всех
коммунально-бытовых газовых приборов в Великобритании, пре-
следовали две цели: а) улучшение надежности работы и прием-
лемости по уровню шума горелок предварительного смешения
(с принудительной подачей воздуха) и б) проектирование и раз-
работка «конверсионной» горелки диффузионного типа для сжи-
гания природного газа.
В последнем случае исходили из того, что «конверсионные»
горелки будут устанавливаться там, где все еще используется
светильный газ, с тем чтобы при переходе на природный газ
можно было бы просто модифицировать газовый агрегат с учетом
А. Химия и горение нефтяных топлив
577
изменения теплотворной способности топлива и скорости пламе-
ни. При разработке «конверсионной» горелки ориентиром служи-
ли преимущества старой диффузионной струйной горелки Брея.
Координирование работы над созданием новых горелочных
устройств было поручено исследовательскому и испытательному
центру фирмы British Gas Corporation в Уотсон-Хаузе (Лон-
дон), который провел широкие испытания многочисленных про-
тотипов, разработанных на различных экспериментальных пред-
приятиях.
Усовершенствование существующих горелок с принудитель-
ной подачей воздуха шло в двух основных направлениях: а) усо-
вершенствование головок горелок предварительного смешения
для предотвращения проскока пламени и повышение устойчиво-
сти пламени путем создания небольших «дежурных» пламен,
стабилизирующих основные пламена, за счет соответствующей
конструкции головки и б) изменение конструкции проточной ча-
сти сопел инжектора, через которые поступает первичный воз-
дух на предварительное смешение с газом, с целью понижения
уровня шума.
Несмотря на значительные организационные и технические
трудности, которые пришлось преодолеть, главная часть про-
граммы переоборудования бытовых и промышленных газовых
агрегатов была успешно выполнена. Этого удалось достигнуть
благодаря использованию лучших образцов горелок предвари-
тельного смешения.
В то же время разработка подходящей конструкции диффу-
зионной горелки для сжигания газообразных углеводородов с
низкой скоростью пламени, несмотря на проведение широких
фундаментальных и прикладных исследований, до сих пор
остается проблемой большого практического значения.
Глубокое изучение горения метана в диффузионном пламени
показало, что задача создания метановой горелки, близкой по
характеристикам к диффузионной струйной горелке Брея для
светильного газа, по-видимому, неразрешима. В качестве осно-
ваний для такого вывода было указано на то, что оба газа
сильно отличаются друг от друга по характеристикам горения
и что при сжигании светильного газа скорость воздушного по-
тока у начала пламени значительно выше, чем при сжигании
метана, а это способствует лучшему смешиванию газа с возду-
хом и тем самым повышает устойчивость пламени более быстро-
горящего топлива.
В недавно опубликованном обзоре исследовательского центра
фирмы British Gas Corp., посвященном последним упехам в раз-
работке диффузионной горелки для сжигания природного газа,
описаны три конструкции, которые после тщательных лабора-
торных испытаний были выбраны для районных испытаний [6].
19 Зак. 501
578
Глава 14
В первой горелке неразбавленный поток газа поступает
через щель в нижней части смесителя, где он смешивается с воз-
духом. Вторая горелка (так называемая голландская сетчатая
горелка) снабжена головкой с многочисленными отверстиями
для основных пламен и дополнительными прорезями для не-
больших «дежурных» пламен; такая головка принципиально не
отличается от головок обычных горелок с принудительной пода-
чей воздуха. Третья горелка, сконструированная в BP Research
Centre [7], резко отличается от обычных газовых горелок
Рис. 14.11. Панельная горелка фирмы ВР с пористым керамическим
излучателем.
1 — близко расположенные воздухопроводы; 5 —зона горения над пористой плитой;
3— поперечный поток газа; 4 — непористая нижняя плита; 5—пористая плита»
через которую проходит газ.
(рис. 14.11). Как объявила фирма British Gas Corp., на базе
этой горелки, возможно, удастся разработать котел для цент-
рального отопления жилых помещений, имеющий размер сто-
роны основания не более 20 см и снабженный терморегулятором
и дистанционно включающимся дутьевым вентилятором. Ни
одна из описанных выше горелок пока не выпущена в продажу.
В последние годы разработка газовых агрегатов для ото-
пления жилых домов привлекает очень большое внимание. Об-
щеизвестная ныне способность газогорелочных устройств значи-
тельно повышать компактность настенных отопительных котлов
была впервые использована приблизительно в 1967 г., когда
начали применяться высокопроизводительные (снабженные вен-
А. Химия и горение нефтяных топлив
579
тилятором) жаротрубные котлы, вызвавшие поток новых за-
манчивых идей. Успехи, достигнутые в этом направлении, хо-
рошо описаны в обзоре Суджа [8].
Промышленный малогабаритный котел
Одним из наиболее важных достижений 60-х годов, связан-
ных с ростом применения нефтяных топлив, было создание
малогабаритного промышленного парового котла, работающего
на жидком топливе, который заменил прежние двухтопливные
котлы, работавшие на котельном топливе и каменном угле. Этот
котел собирается на заводе-изготовителе и доставляется на ме-
сто монтажа полностью готовым к эксплуатации и требующим
Топочные
Вода.
Рис. 14.12. Схема малогабаритного дымогарного котла с тремя газоходами.
1—«проходы» для конвекционного обогрева; 2—горелка; 3—дымогарная труба.
лишь подключения к газопроводу. Горелка и все системы котла
рассчитаны на максимальный к. п. д. (до ~ 80%) в широком
интервале нагрузки и обеспечивают более полное, чем у преж-
них котлов, сгорание топлива, высокую гибкость в работе и
низкие эксплуатационные и капитальные затраты.
Важнейшей областью применения тяжелых нефтяных остат-
ков, если не считать выработку электроэнергии, является в на-
стоящее время получение водяного пара. В некоторых районах
с высокой плотностью населения действуют законы, требующие
использовать для небольших промышленных котлов только дис-
тиллятные топлива, с тем чтобы снизить содержание серы в ды-
мовых газах. Более высокая стоимость этого топлива может ча-
стично компенсироваться за счет уменьшения отложений сажи
и облегчения технического обслуживания котла.
На рис. 14.12 приведена схема малогабаритного дымогар-
ного котла, показывающая трубу радиантного обогрева и
19*
580
Глава 14
несколько пучков относительно узких труб для теплопередачи
конвекционного типа.
Научно-исследовательские и опытные работы, особенно в
области улучшения конструкции газовых горелок и создания ав-
томатических паровых и водонагревательных котлов с низким
содержанием вредных веществ в дымовых газах, продолжаются.
Для повышения температуры поверхности котла и тем самым
снижения коррозии трехокисью серы было предложено исполь-
зовать вместо воды или водяного пара высокотемпературные
органические теплоносители.
Выплавка чугуна
Более 95% мирового производства чугуна выплавляется в
домнах (рис. 14.13), и поэтому в 50-х годах были начаты иссле-
дования по повышению экономичности доменного процесса.
Рис, 14.13. Упрощенная технологическая схема доменного процесса.
1 — домна; 2—очистка доменного газа; 3—впрыскивание котельного топлива; 4—подо-
греватель воздуха.
Сокращение запасов коксующегося угля (применяемого в
доменном процессе для получения тепла, а также окиси угле-
рода, необходимой для восстановления железной руды) и свя-
занное с этим его удорожание обусловило все более широкое
А. Химия и горение нефтяных топлив
581
применение, начиная с 50-х годов, нефтепродуктов (особенно
котельного топлива), которые впрыскивают в горячее воздуш-
ное дутье с целью уменьшения расхода кокса. В настоящее
время этот метод является общепринятым; он позволяет повы-
сить производительность печи и улучшить ее работу. Данный
пример служит замечательным образцом успешного технологи-
ческого нововведения, которое довольно быстро окупило рас-
ходы на его разработку и капитальные затраты. Существует
тенденция к дальнейшему увеличению доли кокса, заменяемой
котельным топливом, которая объясняется как экономическими
соображениями, так и тем, что присутствующая в этом топливе
сера переходит в шлак и не выделяется с отходящими газами в
виде SO2. Таким образом, доменная выплавка чугуна является
экологически безвредной областью применения высокосернистых
котельных топлив.
Однако при дальнейшем повышении скорости подачи ко-
тельного топлива возникают сложные технические проблемы,
связанные с капельным горением впрыскиваемого топлива в по-
токе горячего воздуха, поступающего со скоростью, близкой к
скорости звука, и имеющего температуру до 1000 °C. Возможно,
что результаты исследований в смежных областях, например
в разработке ракетных или газотурбинных двигателей, укажут
пути улучшения качества горения жидкого топлива в горячем
воздушном дутье.
Горелки для высокоскоростного
сжигания топлива
Методы получения компактных пламен с более высокой
скоростью теплопередачи, чем от обычно применяемого в прак-
тике пламени, получили название высокоскоростного сжигания
топлив.
Применение подкотельных горелочных устройств, обеспечи-
вающих высокоскоростное сжигание топлива, дает двоякую вы-
году: а) повышение теплоотдачи вследствие увеличения скоро-
сти удара пламени о стенки, усиливаемой, вероятно, за счет
скрытой теплоты рекомбинации (обрыва цепи) активных частиц
на «холодных» теплоотводящих стенках, и б) экономические
преимущества, обусловленные уменьшением размеров топочной
камеры.
Проблемы, возникающие при высокоскоростном сжигании
нефтяных топлив, связаны с увеличением степени конверсии SO2
в SO3 и фиксацией атмосферного азота в виде окислов азота,
происходящей при высоких температурах пламени, как это ука-
зывалось выше.
582
Глава 14
Разработанный недавно метод высокоскоростного сжигания,
обеспечивающий лишь незначительное образование окислов азо-
та, основан на принципе двухступенчатого сжигания. На первой
ступени в форсунку подается недостаточное количество воздуха,
а на второй в продукты горения дополнительно вдувают воздух
и доводят сгорание топлива до конца. Этот технологический
прием не получил широкого применения в промышленности.
Сжигание топлив в псевдоожиженном слое
Другим многообещающим промышленным (а возможно, и
бытовым) «реактором» для экологически безвредного сжигания
топлива, обеспечивающим к тому же высокую скорость горения,
является камера с псевдоожиженным топливом.
Основная часть первых работ по изучению «поверхностного»
и «каталитического» горения была выполнена фирмой Bone. Это
направление начало развиваться в Великобритании приблизи-
тельно в 1965 г. в качестве наиболее перспективного подхода
к созданию нового метода сжигания каменного угля. Инертным
флюидизированным материалом служит преимущественно ка-
менноугольная зола, которую смешивают с мелкозернистым
углем, имеющим строго определенный размер частиц. Через от-
носительно невысокий слой этой смеси продувают воздух, кото-
рый флюидизирует ее; начальное зажигание можно осуществить
газовым факелом. Тепло, выделяющееся при горении угля в
псевдоожиженном слое, непосредственно нагревает змеевики,
расположенные в топочном пространстве. «Прогоревшие» части-
цы удаляют, смешивают со свежим топливом и снова возвра-
щают в зону слоя. В настоящее время изучается возможность
получения тепла путем сжигания котельного топлива, впрыски-
ваемого в псевдоожиженный слой огнеупорного материала.
В США и Великобритании уже работают экспериментальные
реакторы, в которых уголь или котельное топливо сжигается
в псевдоожиженном слое. Этому методу предсказывают боль-
шое будущее.
В литературе приводятся следующие результаты изучения
горения псевдоожиженных топлив и вероятные тенденции даль-
нейшего развития этой технологии:
1. Возможно достижение полного сгорания топлива в самом
слое. При введении в псевдоожиженный слой извести удается
связать до 100% серы, присутствующей в высокосернистых топ-
ливах.
2. При рабочей температуре слоя 800 °C поддерживается
очень высокая интенсивность теплоотдачи, а теплонапряжен-
ность топочного объема втрое превышает эту величину для
А. Химия и горение нефтяных топлив
583
обычных котлов. Образование окислов азота при столь низких
температурах почти в два раза меньше обычного.
3. Дополнительные преимущества могут дать топочные
камеры, эксплуатируемые при повышенном давлении. Расчет по-
казывает, что парогенератор на флюидизированном топливе, ра-
ботающий под давлением 2 МН/м2 (20 атм), в 25 раз компакт-
нее обычной установки такой же производительности. Это позво-
ляет получить значительную экономию капитальных затрат.
Если такой парогенератор соединить с турбиной для выработки
электроэнергии, то полученный агрегат будет иметь более высо-
кий к. п. д., чем обычные теплоэлектростанции. На рис. 14.14
приведена схема экспериментальной топочной камеры для сжи-
гания топлива в псевдоожиженном слое под давлением.
В настоящее время общепринятым является мнение, что для
практического внедрения данного метода осталось решить чисто
Рис. 14.14. Топочная камера для сжигания топлива в псевдоожиженном слое
под давлением.
/—псевдоожиженный слой; 2—трубы, отводящие 60—70°А тепла.
584
Глава 14
инженерные проблемы, так как теоретические и экологические
аспекты сжигания топлив в псевдоожиженном слое в основном
отработаны.
Заключение
Кто-то сказал, что ученого и инженера-практика, занимаю-
щихся сжиганием топлив, разделяет «река невежества и пред-
рассудков». Лет десять назад эта мысль была, безусловно, спра-
ведлива, но сейчас, когда значительно возросла роль экономи-
ческих и экологических факторов и возникла нехватка энергии
во всем мире, подул «ветер перемен». Налицо признаки того,
что проводимые работы стали более целенаправленными, укре-
пилась связь университетов и институтов с промышленностью
(по линии выполнения государственных программ и др.) и при-
кладные направления исследований встречают более широкое
применение и поддержку. Это растущее осознание необходимо-
сти практических нововведений на базе достижений фундамен-
тальной науки происходит в то время, когда достигнут реальный
прогресс в изучении природы пламен и механизма горения в
различных условиях, результаты которого описаны в предыду-
щих разделах. Поэтому осторожный оптимизм, прозвучавший
в данной главе относительно роли химиков в настоящих и буду-
щих достижениях технологии сжигания топлив, является обосно-
ванным, хотя автор отдает себе отчет в том, что химия процесса
горения чрезвычайно сложна.
В будущем химикам совместно с представителями других
наук придется изучать реакции, происходящие не только в пла-
менах, но и при значительно более высоких температурах (выше
~4000°C), когда вещество находится преимущественно в форме
ионов и изолированных атомов и можно говорить о «новой хи-
мии». Однако это уже иная тема, и если она будет когда-нибудь
описана, то, возможно, придется рассматривать вопрос о пере-
оценке методов синтеза некоторых важных химикатов, которые
трудно получать при обычных температурах. Не приведет ли это
к новой промышленной революции?
СПИСОК ЛИТЕРАТУРЫ
1. Lowry Н. Н. (Ed.), The Chemistry of Coal Utilisation, Wiley, New York,
1963.
2. Фристром P. M., Вестенберг А. А., Структура пламени, «Металлургия»,
M., 1969.
3. Льюис Б., Эльбе Г., Горение, пламя и взрывы в газах, «Мир», М.,
1968.
4. Johnson J. Е., William К. О., Carhart Н. W., The Vertical Tube Reac-
tor — A Tool for Study of Flame Processes, 7th Symposium (International) on
Combustion, Butterworth, London, 1959, p. 392.
Б. Горючие и взрывчатые вещества и пороха
585
5. Combustion Science and Technology, 5, 187—272 (1972), Special Issue,
«Soot in Flames».
6. Dutton В. C., Harris J. A., Kavarana B. J., «Non-Aerated Burners for
Natural Gas», J. Inst. Gas Engrs., 11, 476 (1971).
7. Desty D. H., Whitehead D. M., «New Burners for Old», New Scientist,
1970, 147.
8. Sugg P. C., «Opportunities in Appliance Development for Natural Gas»,
J. Inst. Gas Engrs., 9, 601 (1969).
Б. Горючие и взрывчатые вещества и пороха
Несмотря на важную роль, которую играет взрывное горе-
ние смесей паров нефтяного топлива с воздухом в двигателях
и движителях, такие смеси не находят практического примене-
ния в качестве метательных средств для стрельбы или запуска
ракет, а также в качестве боевых или промышленных бризант-
ных взрывчатых веществ. Причиной этого является слишком
низкая плотность воздуха. Чтобы устранить указанный недо-
статок, можно использовать твердый окислитель. Первым прак-
тически важным порохом и взрывчатым веществом был, ве-
роятно, черный порох — смесь древесного угля и серы (горючие)
с нитратом калия (окислитель). Черный порох устойчив при
хранении и воспламеняется при местном нагреве, причем его
горение . сопровождается выделением энергии и газообразных
продуктов. Однако ему присущи серьезные недостатки, преодо-
леть которые удалось благодаря развитию органической химии.
Фтор как окислитель слишком дорог для большинства прак-
тических целей, вследствие чего лучшим окисляющим элемен-
том является кислород. Среди продуктов химических реакций
с участием кислорода наименьшее количество энергии содержат
окислы. В случае органических горючих обычно образуются
вода, двуокись углерода и некоторое количество окислов азота.
Последние, однако, легко восстанавливаются углеродом и во-
дородом, и различие в содержании энергии между азотом и его
окислами невелико.
Отсюда можно сделать вывод, что меньше всего энергии
содержат связи С—О, Н—О и N—N. Связи С—С, С—Н и N—О
содержат большее количество энергии. Таким образом, чтобы
построить из атомов С, Н, N и О молекулу, способную пере-
группировываться с выделением энергии, необходимо исполь-
зовать кислородсодержащие группы типа —NO, —NO2 или
—ONO2 в сочетании с алкильными или арильными группами.
Например, нитрометан CH3NO2 обладает взрывчатыми свой-
ствами, но при взрыве выделяет много окиси углерода и водо-
рода, которые в присутствии внешнего источника кислорода
могут окисляться, выделяя дополнительную энергию.
586
Глава 14
Тринитрометан (нитроформ) CH(NO2)3 тоже является
взрывчатым веществом. Однако он содержит избыток кисло-
рода, и в продуктах его взрыва присутствуют окислы азота и
свободный кислород, способные в присутствии других горючих
веществ давать дополнительное количество энергии. Идеальное
взрывчатое вещество должно иметь нулевой кислородный ба-
ланс * и содержать весь кислород в форме связей N—О. Таких
соединений немного; примером их может служить нитрогликоль
(-CH2ONO2)2
CH2ON02
| —> 2СО2 + 2Н2О + N2
CH2ONO2
Нитрозосоединения нестабильны и поэтому представляют
лишь ограниченный интерес в качестве взрывчатых веществ.
Нитросоединения гораздо более стабильны. К ним относятся
такие боевые бризантные взрывчатые вещества, как 2,4,6-три-
нитротолуол (ТНТ), Ы-метил-2,4,6-тринитрофенилнитрамин (тет-
рил) и циклотриметилентринитрамин, или Ы^'Ы^-тринитрогек-
сагидро-сшюи-триазин (гексоген).
ТНТ
тетрил
Н2
o2n4 с /NOa
н2с^ /сн2
N
NO2
гексоген
Почти все без исключения боевые бризантные взрывчатые
вещества имеют отрицательный кислородный баланс. Однако
это не имеет существенного значения, так как окись углерода
и водород не уступают по работоспособности в качестве про-
дуктов взрыва двуокиси углерода и воде **, а токсичность СО
не играет в данном случае никакой роли.
В качестве порохов и промышленных взрывчатых веществ
гораздо более полезными оказались органические нитраты. Они
менее стабильны, чем нитросоединения, но при введении стаби-
* Кислородным балансом называется процент избытка или недостатка
кислорода в молекуле взрывчатого вещества по сравнению с количеством,
необходимым для полного окисления горючих элементов (углерода и водо-
рода).— Прим, перев.
** Имеется в виду равенство молярных объемов смеси СО + Н2 и смеси
СО2 + Н2О. образующейся при более полном окислении горючих элементов в
молекуле взрывчатого вещества. — Прим, ред-
Б. Горючие и взрывчатые вещества и пороха
587
лизирующих добавок могут храниться многие годы при темпе-
ратуре почти любой зоны земного шара.
Тринитрат глицерина (нитроглицерин) (т. 1, стр. 35) был
первым органическим взрывчатым веществом , получившим
широкое применение. Основным недостатком нитроглицерина
является то, что он жидкий. Однако при пластификации им
нитроцеллюлозы (т. 4, стр. 93) образуется вещество, которое
в зависимости от типа и количества исходной нитроцеллюлозы
может иметь вид от желеобразной массы до хрупкого твердого
материала. Такие вещества используются в производстве без-
дымных порохов (кордит и так называемый баллистит) и «гре-
мучего студня» (динамита)—наиболее важного взрывчатого
вещества для подземных работ.
Из нитроцеллюлозы, пластифицированной нитроглицерином,
можно прессовать пороховые шашки с очень точным соблюде-
нием заданных размеров. При поджигании они горят по поверх-
ности, причем скорость горения зависит от концентрации газо-
образных продуктов вблизи горящей поверхности и, следова-
тельно, от давления. В настоящее время можно получить порох,
который горит или разлагается на простые вещества со строго
определенной скоростью, необходимой для поддержания уско-
рения метательного снаряда по всей длине канала ствола.
Можно также получать ракетные заряды, горящие с постоянной
заданной скоростью, обеспечивая постоянную тягу двигателя.
Современные пороха уже не всегда содержат нитроглицерин.
Некоторые (так называемые одноосновные) пороха состоят
лишь из нитроцеллюлозы и модификаторов. Существуют также
пороха, представляющие собой смеси органических смол с пер-
хлоратом аммония или хорошо дегазированные смеси на основе
бризантных взрывчатых веществ.
Важнейшими областями применения нитроглицерина яв-
ляются подземная добыча полезных ископаемых, разработка
карьеров и прокладка туннелей. Для всех этих работ нитрогли-
церин обладает значительными преимуществами перед черным
порохом. Он имеет гораздо большую энергию взрыва, разру-
шает даже самые твердые породы и дает при взрыве очень сла-
бое пламя (что позволяет использовать его в угольных забоях).
Однако сам по себе нитроглицерин не обеспечивает перечислен-
ные преимущества. Нобель обнаружил, что нитроглицерин не
способен взрываться при поджигании, но если взять немного
фульмината ртути * и поджечь эту соль, то от нее произойдет
сильный взрыв нитроглицерина. Это открытие было положено
в основу разработанного Нобелем «патентованного детонато-
ра»— закрытой с одного конца медной трубки, содержащей
* В технике более принято название «гремучая ртуть». — Прим, перев.
588
Глава 14
фульминат ртути. Взрыв, вызываемый детонатором Нобеля,
представляет собой новое явление — детонацию, на котором ба-
зируется практическое использование всех современных бри-
зантных взрывчатых веществ, как боевых, так и промышлен-
ных. До изобретения этого детонатора превращение взрывчатых
веществ в заряде обычно носило характер термического разло-
жения, которое сначала распространяется с горячими газооб-
разными или твердыми продуктами по макроповерхностям, а
затем захватывает (путем теплопроводности или диффузии) все
более глубокие слои, что сопровождается ростом температуры
и заканчивается полным выгоранием заряда. Механизм реакции
термического разложения может быть различным, причем су-
щественное влияние на него оказывает кинетика процесса. Од-
нако лимитирующим фактором часто является не скорость хи-
мической реакции, а скорость диффузии образовавшихся про-
дуктов в массу взрывчатого вещества.
При детонации по взрывчатому веществу распространяется
ударная волна, инициирующая химическую реакцию разложе-
ния. Во фронте этой волны вещество подвергается сильному уда-
ру, приводящему к резкому повышению давления. Непосред-
ственно за фронтом сжатое взрывчатое вещество претерпевает
химическое превращение, оставаясь в уплотненном состоянии,
и лишь в задней части этой реакционной зоны начинается рас-
ширение конечных продуктов. Последние движутся по направ-
лению к фронту, а не от него, причем в установившемся со-
стоянии разность скорости ударной волны и массовой скорости
конечных продуктов равна местной скорости звука. Другими
словами, детонация — это ударная волна, непрерывно поддер-
живающаяся за счет химической реакции. Скорость ударной
волны определяется энергией, выделяющейся в реакции, а меха-
низм этой реакции не имеет большого значения.
Почему же не детонирует черный порох? Причина этого за-
ключается . в недостаточной скорости химической реакции его
разложения. Для заряда бесконечных размеров скорость дето-
нации не зависит от скорости и механизма реакции, но в случае
реальных (т. е. конечных) зарядов возможно расширение про-
дуктов детонации в боковых направлениях, что меняет картину
рассматриваемого явления'.
На рис. 14.15 изображен идеальный случай — бесконечный
цилиндрический заряд взрывчатого вещества, в котором реак-
ционная зона просто распространяется по веществу до тех пор,
пока оно полностью не прореагирует. На рис. 14.16 показан
реальный случай, когда длина реакционной зоны невелика и
определенно меньше диаметра заряда. Это означает, что необ-
ходимым условием Детонации является достаточно большая ско-
Б. Горючие и взрывчатые вещества и пороха
589
D
Взрывчатое вещество
Продукты
реакции
Реакционная зона
Продукты
Рис. 14.15. Детонация бесконечного заряда бризантного взрывчатого ве-
щества. Продукты движутся вслед за фронтом со скоростью W, причем
D — 1F = местная скорость звука (где D — скорость детонационной волны).
рость химической реакции, обеспечивающая выделение значи-
тельной энергии в «сверхзвуковой» реакционной зоне.
Скорость детонации нитроглицерина в трубке диаметром
10 мм составляет около 8000 м/с. Длина реакционной зоны в
этих условиях меньше диаметра заряда (она равна -~2 мм),
вследствие чего реакция должна завершаться значительно быст-
рее, чем за 1 мкс. Конечно, при этом развивается очень высокая
температура—до 6000 К, но, несмотря на это, диффузия конеч-
ных продуктов от одной частицы испарившегося вещества к
другой протекает слишком медленно по сравнению с временем
реакции. Поэтому черный порох не способен детонировать при
указанном диаметре заряда, тогда как нитроглицерин легко де-
тонирует. Основной причиной служит то, что в нитроглицерине
горючее и окислитель находятся в тесном контакте друг с дру-
гом, так как входят в состав одной и той же молекулы.
Тогда почему же большинство бризантных взрывчатых ве-
ществ не детонирует при поджигании пламенем? Профессор
Боуден с большой группой сотрудников изучил механизм воз-
буждения детонации взрывчатых веществ ударом, трением,
нагревом и т. д. Вкратце его выводы сводятся к тому, что
Рис. 14.16. Реальный случай детонации бризантного взрывчатого вещества.
590
Глава 14
возбуждение детонации носит в основном тепловой характер и
что горение, или дефлаграция, взрывчатого вещества переходит в
детонацию в том случае, когда она протекает в замкнутом объе-
ме с быстрым ускорением или сопровождается образованием
ударных волн. Существуют взрывчатые вещества, у которых
такой переход наступает очень быстро, но для большинства
органических взрывчатых веществ он происходит с трудом. Это
обусловлено тем, что на начальных стадиях тепловое воспла-
менение представляет собой эндотермический процесс, приво-
дящий к образованию молекул, которые затем реагируют экзо-
термически. Отсюда следует, что реакция на ранних стадиях
лимитируется эндотермическими превращениями взрывчатого
вещества, причем рассеивание энергии в окружающую среду за
счет теплопроводности, конвекции и др. протекает достаточно
быстро, чтобы предотвратить ускорение реакции.
Взрывчатые вещества, детонирующие от пламени, удара
или трения, называются первичными *. Отличие их от вторич-
ных, или бризантных, взрывчатых веществ является скорее не
качественным, а количественным. Почти все первичные взрыв-
чатые вещества имеют простой состав и представляют собой
соли необычных кислот, например фульминаты и азиды. Наибо-
лее ценными из них являются фульминат ртути Hg(ONC)2 и
азид свинца Pb(Ns)2, которые термически стабильны, но спо-
собны к быстрому переходу начавшегося термического разло-
жения в детонацию. Для всех этих соединений первая стадия
реакции, вероятнее всего, носит экзотермический характер; од-
нако имеются и другие теории, объясняющие поведение первич-
ных веществ.
Органические первичные взрывчатые вещества включают
азиды (такие, как циануртриазид), соли тяжелых металлов с
нитрофенолами (пикрат и тринитрорезорцинат свинца) и ряд
необычных соединений с высоким содержанием азота, например
тетразен.
Однако свинцовые соли нитрофенолов и тетразен, как пра-
вило, используются лишь в качестве сенсибилизирующих доба-
вок к азидам, поскольку они обладают недостаточной иниции-
рующей способностью и нуждаются в прочной оболочке для
перехода их горения в детонацию.
Современные взрывчатые вещества
Идея Нобеля о том, что взрывчатые вещества гораздо без-
опаснее в обращении, чем черный порох, но обладают значи-
тельно большей работоспособностью при подрыве их неболь-
* Или инициирующими. — Прим, перев.
Б. Горючие и взрывчатые вещества и пороха
591
шим количеством чувствительного взрывчатого вещества, полу-
чила дальнейшее подтверждение в свойствах большинства со-
временных взрывчатых веществ. На рис. 14.17 и 14.18 пред-
ставлена типичная цепь взрывчатых веществ, используемая при
взрывных работах. Все опасные элементы этой цепи заключены
в герметичный капсюль-детонатор. Взрыв капсюля-детонатора
инициируется электрическим током, проходящим по мостику
накаливания, который перегорает со вспышкой, поджигающей
0,1 г азида свинца. Последний настолько быстро разлагается
Проволочка, нагреваемая
электрическим током
Свинцовая соль
. нитрофенола
Смесь хлората
с животным углем
Pb(N3)s
тэн
Рис. 14.17. Схема электродетонатора. После вспышки свинцовой соли ни-
трофенола смесь хлората с углем начинает гореть горячим пламенем, затем
азид свинца разлагается с детонацией на свинец и азот и от возникшего
ударного импульса детонирует тетранитрат пентаэритрита C5Ha(NO3)4 (ТЭН),
хотя при нагревании он разлагается довольно медленно.
на свинец и азот, что возникает ударная волна, от которой де-
тонирует нитроэфир (тетранитрат пентаэритрита, сокращенно
ТЭН). Детонация распространяется в 6-миллиметровом капсю-
ле-детонаторе и затем переходит в имеющий еще меньший диа-
метр детонирующий шнур, содержащий тот же нитроэфир
(рис. 14.18). Этот гибкий шнур опущен в скважину, выбурен-
ную в горной породе. Проходя по шнуру, детонационная волна
возбуждает взрыв прикрепленных к нему патронов-боевиков,
снаряженных органическими нитросоединениями (а иногда
также и нитроэфиром), которые обладают низкой чувствитель-
ностью, но безотказно детонируют при диаметре заряда не ме-
нее 23 мм.
592
Глава 14
Рис. 14.18. Скважинный заряд в горной породе.
Д —забойка из горной породы; Б —смесь нитрата аммония с дизельным топливом; Б —ос-
новной заряд водонаполнеиного или нитроглицеринового взрывчатого вещества; Г—пат-
роны-боевики, содержащие ТНТ и ТЭН (такой состав меиее чувствителен, чем чистый
ТЭН, но легко детонирует от детонирующего шнура), /-капсюль-детонатор; 2—детони-
рующий шнур; 3—горная порода.
Диаметр скважины превышает 100 мм. В нижнюю часть ее
укладывается плотно прилегающий к стенкам заряд В, пред-
ставляющий собой либо смесь нитроглицерина, нитроцеллюлозы,
нитрата аммония и горючих добавок, либо водонаполненное
взрывчатое вещество. Нитрат аммония способен взрываться и
сам по себе, но он имеет низкую энергию взрыва и к тому же
детонирует в скважине диаметром 100 мм только в виде тонко-
дисперсного порошка с малой насыпной плотностью. При взрыве
нитрата аммония выделяется свободный кислород, использова-
ние которого путем добавления древесной муки позволяет вдвое
увеличить энергию взрыва. Если же смешать нитрат аммония
и горючее с нитроглицерино-нитроцеллюлозным студнем, то по-
Б. Горючие и взрывчатые вещества и пороха
593
лученное взрывчатое вещество будет обладать максимальной
энергией взрыва.
Водонаполненные взрывчатые вещества бывают двух видов:
сенсибилизированные и несенсибилизированные. Первые пред-
ставляют собой загущенные водные растворы нитрата аммония,
содержащие ТНТ или другое вторичное взрывчатое вещество,
которое детонирует от патрона-боевика и возбуждает экзотер-
мический распад нитрата аммония. Несенсибилизированные
водонаполненные взрывчатые вещества — это загущенные вод-
ные растворы нитрата аммония или других нитратов, содержа-
щие избыток твердого окислителя и горючие добавки, присут-
ствующие в растворе или суспензии. Если горючие компоненты
растворимы в воде, то они равномерно распределяются в рас-
творе окислителя (нитрата аммония), хотя контакт между
ними и не столь тесный, как в молекуле химического соедине-
ния. Такие растворы оказались взрывчатыми веществами с го-
раздо меньшей чувствительностью, чем органические нитросое-
динения, однако их можно сенсибилизировать путем введения
пузырьков воздуха. Включения воздушных пузырьков при воз-
действии на них скачка давления играют роль центров мест-
ного разогрева , и воспламенения взрывчатого вещества. Одним
из методов воздушной сенсибилизации является использование
обычной алюминиевой пудры. Гидрофобная поверхность частиц
алюминия удерживает мельчайшие пузырьки воздуха, которые,
разогреваясь, быстро поджигают алюминий и в результате сами
становятся еще горячее. Таким образом, данная смесь, не-
смотря на необходимость диффузии продуктов для протекания
реакции, способна к достаточно быстрому химическому превра-
щению, что обусловлено быстротой воспламенения, высокой тем-
пературой, развивающейся при реакции, и тесным контактом
реагентов.
Выше заряда водонаполненного взрывчатого вещества в
скважине располагается низкоплотная смесь Б (рис. 14.18), со-
держащая нитрат аммония, дизельное масло и воздух. И в этом
случае воздушные пузырьки облегчают детонацию нитрата ам-
мония, однако важное значение имеет также качество контакта
окислителя с углеводородом. Нитрат аммония, применяемый в
составе смеси Б, приготовляют путем сливания расплава соли,
содержащей ~5% воды, с верха грануляционной башни и
сушки полученных гранул. В процессе сушки продукт приобре-
тает абсорбирующие свойства, что позволяет маловязким жид-
костям проникать в глубь гранул, обеспечивая максимальную
степень контакта.
Взрывные работы не всегда ведут, используя полную цепь
капсюль-детонатор — детонирующий шнур — патрон-боевик —
взрывчатое вещество. При подземных работах применяются не
594
Глава 14
скважины, а шпуры небольшого диаметра, в которых взрывча-
тое вещество должно детонировать непосредственно от капсю-
ля-детонатора. Этому требованию удовлетворяют геликниты
(смеси ~30% нитроглицерина с нитрованным хлопком, нитра-
том аммония и жидким топливом) и специальные порошкооб-
разные составы (аналогичные смеси с меньшим содержанием
нитроглицерина). Некоторые из них предназначены для работ
в угольных забоях и содержат пламегасители, эффективно пре-
дотвращающие воспламенение рудничного газа даже в самых
жестких условиях испытаний.
Хотя взрывчатые вещества имеют и военное применение,
наибольшее количество их в настоящее время используется для
подземной и открытой добычи полезных ископаемых, при про-
кладке дорог, сооружении туннелей и т. п. Главным компонентом
всех промышленных взрывчатых веществ является нитрат ам-
мония, так как он дешев, легкодоступен и нечувствителен к
внешним воздействиям. Выше было показано, как высвобож-
дается энергия, заключенная в нитрате аммония, с помощью
других взрывчатых веществ. Предпринято несколько интерес-
ных попыток найти заменители нитрата аммония, которые в
перспективе могут даже привести к изменению структуры про-
изводства взрывчатых веществ. Так, азотная кислота образует
с органическими веществами очень мощные взрывчатые смеси.
Смесь жидкого кислорода с древесной мукой весьма чувстви-
тельна к внешним воздействиям и способна детонировать. Ис-
пользовались также растворы нитрата гидразина.
Для некоторых целей, например для проведения взрывных
работ при высоких температурах и давлениях, приходится раз-
рабатывать специальные взрывчатые вещества. Обычные вто-
ричные взрывчатые вещества хорошо переносят высокие давле-
ния, но их чувствительность при этом снижается из-за сжатия
газовых пузырьков. Для борьбы с этим явлением во взрывча-
тое вещество можно ввести тонкодисперсный тяжелый мате-
риал, частицы которого при детонации играют роль «горячих
точек». В то же время обычные продажные взрывчатые веще-
ства плохо переносят температуры выше 200 °C. После тщатель-
ного изучения влияния структуры на термостойкость взрывча-
тых веществ были синтезированы довольно необычные органи-
ческие соединения, часть которых нашла применение при иссле-
дованиях космического пространства. Примерами таких веществ
могут служить гексанитростильбен и тетранитробензтетразен
(дибензо-[а,е,£]-[1,2,5,6]-тетразоцин). Главными особенностями
этих соединений являются высокая температура плавления,
симметричная структура и присутствие всего кислорода в форме
групп NO2.
Б. Горючие и взрывчатые вещества и пороха
595
гексанигпросгпильбен
В табл. 14.3 приведены методы промышленного получения
и основные области применения взрывчатых веществ в настоя-
щее время.
В заключение необходимо сказать несколько слов о ядерных
взрывчатых веществах. В принципе органические взрывчатые
вещества можно заменить ядерными, однако до этого еще да-
леко. Современные ядерные устройства очень сложны, слишком
громоздки для большинства мирных целей и к тому же дают
при взрыве радиоактивные продукты. Даже применение ядер-
ных взрывчатых веществ для сооружения подземных хранилищ
является слишком опасным, так как может привести к загряз-
нению окружающей среды.
Таблица 14.3
Взрывчатые вещества и основные области их применения в настоящее время
Взрывчатые вещества
Получение
Применение
Первичные взрывчатые вещества
Фульминат ртути Hg(ONC)2
Азид свинца Pb(Ns)2
Тетразен C2H8NioO
Стифнат свинца РЬО2С6Н(ПО2)з • Н2О
Диазодинитрофенол C6H(NO2)2ON2
Ртуть растворяют в азотиой кислоте
и добавляют спирт. По окончании
выделения окислов азота выпав-
шую соль отфильтровывают и про-
мывают
Na + NH3 —> NaNH2; NaNH2+
+ N2O —> NaNs; 2NaNs (рас-
твор) + Pb(NO3)2 —► Pb(N3)2
(осадок)
Сульфат аминогуанидина вводят
в реакцию с нитритом натрия, при-
водящую к образованию 1-гуанил-
4-(нитрозоаминогуанил)тетразона
(тетразена)
Резорцин нитруют до 2,4,6-тринитро-
производного, которое растворяют
в водном растворе MgO; получен-
ный раствор магниевой соли обра-
батывают нитратом свинца
Взаимодействие азотистой и пикра-
миновой кислот
Снаряжение капсюлей-детонаторов
с медной оболочкой и ряда удар-
ных взрывателей. Применяется все
в меньших масштабах и вскоре мо-
жет выйти из употребления
Снаряжение капсюлей-детонаторов и
ударных взрывателей. Обычно ис-
пользуется в смеси с флегматиза-
торами (декстрин, желатина, эфиры
целлюлозы), понижающими его чув-
ствительность
Сенсибилизатор в ударных составах
Сенсибилизатор в капсюлях-детона-
торах, снаряжаемых азидом свинца.
Применяется также в ударных со-
ставах
Снаряжение капсюлей-детонаторов
(главным образом в США)
Вторичные взрывчатые вещества
Нитроглицерин (тринитрат глицерина)
C3H5(NO3)3
Нитрогликоль (этиленгликольдини-
трат) С2Н4(ЫО3)2
Нитроцеллюлоза СвН7О2(ЫО3)3
Нитрат аммония NH4NO3
Тетранитрат пентаэритрита (ТЭН)
C5H8O4(NO2)4
Тринитротолуол (ТНТ) C7H5(NO2)3
Циклотриметилентринитрамин (гексо-
ген) С3Н6Ы6О6
Нитрование пентаэритрита азотной
кислотой
Нитрование толуола смесью азотной
и серной кислот
Нитрование глицерина смесью азот-
ной и серной кислот
Нитрование этиленгликоля смесью
азотной и серной кислот
Нитрование целлюлозы (хлопковой
или древесной) смесью азотной и
серной кислот
Нейтрализация газообразного аммиа-
ка азотной кислотой
Нитрование уротропина
Взрывчатые смеси дробящего дей-
ствия, предназначенные для про-
мышленных взрывных работ. Двух-
основные пороха (вместе с нитро-
целлюлозой)
Используется как добавка к нитро-
глицерину для улучшения морозо-
стойкости составов
Загущение нитроглицерина для полу-
чения «гремучего студня» и произ-
водство порохов
Применяется в сочетании с нитрогли-
церином в гелигнитах и взрывча-
тых веществах, предназначенных
для работ в угольных забоях. Во-
донаполненные взрывчатые веще-
ства, содержащие ТНТ или алюми-
ний + воздух
Основной заряд в капсюлях-детона-
торах, снаряжение детонирующих
шнуров. В смеси с ТНТ применяется
для патронов-боевиков и специаль-
ных зарядов
Для водонаполненных взрывчатых
веществ, патронов-боевиков и дру-
гих целей, упомянутых выше, а так-
же для снаряжения боеприпасов
Главным образом для снаряжения
боеприпасов. В промышленности
используется там, где требуется
повышенная термостойкость, на-
пример при взрывных работах в стан
леплавильных, и чугунолитейных
печах
ГЛАВА 15
Пищевые вещества
А. Кроссли
Crossley Л., Unilever Research, Port Sunlight Laboratory
Большинство пищевых продуктов имеет природное проис-
хождение, откуда проистекает большое разнообразие их хими-
ческого строения и состава. Дело осложняется еще и тем об-
стоятельством, что мы пока очень мало знаем о различных ма-
кромолекулярных компонентах пищевых продуктов, в том числе
сложных липопротеинах, гликолипидах, мукополисахаридах и
мукопротеинах. К тому же многие из этих веществ в том виде,
в каком они присутствуют в живой клетке, весьма лабильны и
претерпевают значительные изменения в процессе переработки
пищевого сырья. Поэтому точное выяснение химической при-
роды пищевых веществ и их превращений при переработке воз-
можно лишь в немногих случаях. Обычно же приходится соби-
рать информацию из самых различных источников, вследствие
чего изучение пищевых веществ по существу находится на
стыке большого числа наук — от органической химии и биохи-
мии до физики, инженерной биохимии и биологии.
Классификация пищевых веществ на углеводы, жиры и бел-
ки является слишком упрощенной. Более правильно подразде-
лять их в зависимости от происхождения, т. е. на мясные, рыб-
ные и растительные вещества.
Переработкой пищевого сырья занимается ряд производств
пищевой промышленности, такие, как мукомольное, сахарное,
маслобойное, а также консервные производства — холодильное,
мясо-, рыбо- и овощеконсервные и сушильное. Кроме этих «пер-
вичных» производств, существуют предприятия по дальнейшей
переработке полученных на них полуфабрикатов в такие про-
дукты, как хлеб, кондитерские изделия, шоколад, маргарин и
разнообразные молочные продукты.
Некоторые подробности об удельном весе различных пище-
вых продуктов в рационе жителей Великобритании приведены
в табл. 15.1.
Пищевые вещества
599
Таблица 15.1
Потребление продуктов питания в Великобритании
на душу населения в 1969 г.
Продукты питания Количество, Калорийность, Состав, г/100 г продукта, не подвергнутого кулинарной обработке
г/неделя Дж
белки жиры углеводы
Молоко 3700 285 3,2 4,0 5
Сыр 112 1800 26 35 —
Жиры, масло и мар- 320 3460 — 89 —
гарин Яйца 250 680 12 13
Мясные продукты 1100 500-1800 18-23 2,5-43 —
Рыба 140 328 18 0,36 —
Сахар и варенье 530 1100-1700 0,36 — 72—107
Овощи 2460 104-374 0,36-2,1 — 5-16
Фрукты 840 60—149 0,36-1,1 — 2,9-20
Хлеб, мука и хлебо- 1620 1040-1800 6,4-8,2 1,4-2,1 54-82
булочные изделия Прочие 280 — — — —
Поскольку в рамках этой главы невозможно детально рас-
смотреть все пищевые производства, для примера были выбра-
ны маслобойно-жировая и хлебопекарная промышленность.
Кроме того, обсуждается два вопроса общего значения — порча
продуктов питания и ее предотвращение и новые источники пи-
щевых веществ.
Особого упоминания заслуживает проблема пищевых доба-
вок, которая наиболее интересует химиков-органиков. Возрос-
шая глубина переработки пищевого сырья повлекла за собой
повышение спроса на химические добавки типа красителей
(гл. 10), вкусовых веществ (гл. 16), консервантов, желатиниза-
торов и эмульгаторов, подсластителей, витаминов и др.
(табл. 15.2). Эти вещества часто получают путем имитирования
состава природных материалов, однако обычно конечной целью
является разработка более дешевых и многофункциональных
синтетических добавок. Независимо от того, встречаются полу-
ченные добавки в природе или нет, их метаболизм в организме
человека подвергают тщательному исследованию, так как по-
ступают все новые данные о неблагоприятном воздействии не-
которых натуральных и синтетических добавок на жизненные
процессы.
Производство масел и жиров
Ежегодно в мире производится свыше 10 млн. т пищевых
масел и жиров. Экстракционные масла и жиры подвергают
600
Глава 15
Таблица 15.2
Некоторые пищевые добавки
Класс
Антиоксиданты
Консерванты
Эмульгаторы и стабили-
заторы (структури-
рующие добавки)
Подсластители
Улучшители муки
Красители
Пищевые кислоты
Увлажнители
Глазирующие средства
Антивспениватели
Средства для предупре-
ждения комкования
Средства для повыше-
ния выхода муки из
зерна
Секвестранты
Пропелленты
Вкусовые вещества
Усилители вкуса
Примеры
Бутилированные оксианизол и окситолуол, раз-
личные эфиры галловой кислоты, аскорбиновая
кислота, токоферолы
Бензойная и сорбиновая кислоты, бифенилол-2
Стероилтартрат, моноглицериды высших жирных
кислот, ацетил-, лактил- и цитроилглицериды,
некоторые глицериды многоосновных кислот,
эфиры сорбитана с жирными кислотами, эфиры
целлюлозы, натрийкарбоксиметилцеллюлоза,
агар, альгиновая кислота, каррагенин, летицин,
казеинаты натрия и кальция, крахмал и моди-
фицированные крахмалы
Сахарин, цикламат, пептиды
Перекись бензоила
Различные азокрасители, Р-каротин
Уксусная, лимонная, молочная, яблочная, янтар-
ная кислоты
Сорбит
Пчелиный воск, спермацет
Стеарат натрия, силиконы
Стеарат магния
Спермацетовое масло, бутилстеарат, силикон
Глицин, соли этилендиаминтетрауксусной кис-
лоты (ЭДТА)
Фреоны (хлорфторуглеводороды)
Лактоны, уксусная кислота, гидролизованный про-
теин, сложные эфиры
Глутамат, инозинат и гуанилат натрия
специальной переработке (рафинированию) с целью удаления
различных примесей. При этом получаются такие важные по-
бочные продукты, как летицин (смесь фосфолипидов, извлекае-
мая в основном из соевого масла) и жирные кислоты. Рафини-
рованные масла содержат преимущественно триглицериды (т. 4,
стр. 150) наряду с меньшими количествами ди- и моноглицери-
дов. Некоторые типичные жирные кислоты приведены в
табл. 15.3. ch2oocr СН2ОН СН2ОН ch2oocr
1 CHOOCR' । CHOOCR 1 снон 1 CHOOCR'
CH2OOCR" । CH2OOCR' 1 ch2oocr CH2OP(O)OCH2CH2N(CH3)3
триглицерид диглицерид моноглицерид он летицин
Пищевые вещества
601
Таблица 15.3
Некоторые важные природные жирные кислоты
Кислота Формула
Пальмитиновая Стеариновая Олеиновая Линолевая Линоленовая СН3(СН2)|4СООН СН3(СН2)16СООН СН3(СН2)7СН=СН(СН2)7СООН СН3(СН2)4СН=СНСН2СН=СН(СН2)7СООН СН3(СН2)2СН=СНСН2СН=СНСН2СН=СН(СН2)7СООН
Глицериды природных жиров весьма специфичны по своему
составу. Так, ненасыщенные кислоты растительных масел содер-
жат двойные связи почти исключительно в ^^-конфигурации,
а остатки различных жирных кислот занимают специфические
положения в молекулах триглицеридов. Для масел раститель-
ного происхождения типично присутствие остатка ненасыщен-
ной кислоты в положении 2: например, в масле бобов какао
содержится 40% 1-пальмитоил-2-олеоил-3-стеароилглицерина и
практически нет его изомеров. Физические свойства масел и
жиров определяются не только природой жирных кислот, вхо-
дящих в их состав, но и распределением изомерных триглице-
ридов, которое характерно для каждого конкретного масла или
жира.
Увеличение спроса на различные переработанные жиры по-
влекло за собой потребность в расширении свойств природных
жиров, что породило разнообразные методы их модифицирова-
ния. Так, распределение кислотных остатков в глицеридах
можно варьировать путем переэтерификации, катализируемой
следами натрия или алкоголятов натрия. При этом достигается
статистическое распределение остатков жирных кислот. Напри-
мер, 1-пальмитоил-2-олеоил-3-стеароилглицерин (ПОС) дает
Таблица 15.4
Продукты переэтерификации 2-олеоил-1,3-дистеароилглицерина
(СОС)
Глицерид Количество, %
Тристеароилглицерин (ССС) 29,6
2-Олеоил-1 3-дистеароилглицерин (СОС) 14,8
1-Олеоил-2,3-дистеароилглицерин (ОСС) 29,7
1-Стеароил-2,3-диолеоилглицерин (СОО) 14,8
2-Стеароил-1,3-диолеоилглицерин (ОСО) 7,4
Триолеоилглицерин (ООО) 3,7
602
Глава 15
смесь 14 разных глицеридов. Простой пример реакции переэте-
рификации приведен в табл. 15.4.
На практике переэтерификации подвергают смеси различных
масел, что приводит к очень сложной композиции глицеридов и
обеспечивает значительное модифицирование свойств продукта.
Точный механизм и природа активного катализатора этой реак-
ции до сих пор остаются дискуссионными.
Неполные глицериды (моно- и диэфиры глицерина) еще бо-
лее лабильны и могут претерпевать внутримолекулярную пере-
этерификацию в очень мягких условиях по следующей схеме:
60% 40%
Наиболее интересным методом модифицирования жиров и
масел, вероятно, является гидрогенизация (т. 1, стр. 156).
О важности этого метода наглядно свидетельствует тот факт,
что ежегодно во всем мире гидрируют для пищевых целей бо-
лее 1 млн. т масел. Гидрогенизация используется для повыше-
ния температуры плавления жиров, а также для понижения
содержания в них глицеридов высоконенасыщенных кислот, на-
пример линоленовой, которые при хранении превращаются в по-
бочные продукты путем аутоокисления.
Гидрогенизацию обычно проводят в отсутствие растворителя
при температуре 100—180 °C и давлении 0,1—0,5 МН/м2 (1—
5 атм) в присутствии мелкораздробленного никелевого катали-
затора на кизельгуре или силикагеле. Насыщение двойных свя-
зей не доводят до конца, что способствует большему разнообра-
зию продуктов. В табл. 15.5 представлен состав продуктов, по-
лученных из простого по строению эфира линолевой кислоты
при двух различных степенях гидрогенизации.
Можно видеть, что в процессе гидрогенизации происходит
не только насыщение двойных связей, но и образование транс-
изомеров и различных изомеров положения. Гидрогенизация
является селективной реакцией, причем скорость гидрирования
Пищевые вещества
603
Таблица 15.5
Продукты частичной селективной гидрогенизации метилового эфира
линолевой кислоты при температуре 100 °C в присутствии никеля,
осажденного на силикагеле
Содержание диена или олефина (%)
при продолжительности гидрогенизации
Дней или олефин _____________________________________
4 мин 7 мин
цис,гщс-Диен (непревращенный эфир линолевой кислоты) цис,транс- и транс,цис-Цкены Моноолефин (цифра при Д указывает 72,5 0,5 цис транс 37,2 1,0 цис транс
положение двойной связи) Д4 Д5 д6 д7 д8 д9 ДЮ Д11 дч Д13 Д14 Насыщенный эфир (метилстеарат) 0,02 0,02 0,05 0,09 0,08 0,12 0,20 0,30 0,35 1,00 6,1 1,25 0,78 3,7 0,90 3,4 5,0 1,30 0,25 1,00 0,16 0,38 0,7 0,05 0,07 0,20 0,23 0,28 0,30 0,58 0,64 1,10 2,3 13,5 3,7 1,9 7,2 2,0 7,0 11,6 3,2 0,75 2,3 0,42 0,87 1,6
жирной кислоты тем выше, чем больше двойных связей присут-
ствует в ее цепи. В случае идеально селективной гидрогениза-
ции образование полностью насыщенных соединений должно
наблюдаться только после того, как все полиненасыщенные
эфиры восстановятся в мононенасыщенные. В действительности
же селективность сильно зависит от условий на поверхности
катализатора, и, подбирая катализатор и условия проведения
реакции, можно существенно варьировать степень гидрогениза-
ции. Аналогично можно регулировать соотношение цис- и транс-
изомеров. Последнее имеет важное значение, поскольку транс-
изомеры плавятся значительно выше ^ыс-изомеров (например,
триолеин имеет т. пл. —5 °C, а его транс-изомер, триэлаидин,—
т. пл. 40°C). Очевидно, что свойства гидрированных жиров
определяются главным образом количеством образовавшихся
насыщенных и транс-ненасыщенных глицеридов. Существующие
промышленные процессы гидрогенизации различаются в сущно-
сти условиями, обеспечивающими заданную степень содержания
этих компонентов в конечном продукте. Этим путем могут быть
получены кулинарные жиры, масло для обжаривания, салатное
масло, а также жиры, пригодные для производства маргарина,
мороженого и кондитерских изделий.
604
Глава 15
Заслуживает упоминания еще один важный аспект приме-
нения липидов как пищевых продуктов, а именно роль линоле-
вой кислоты в питании человека. Линолевая кислота не синте-
зируется в организме, и поэтому она целиком поступает вместе
с пищей. Все больше фактов свидетельствует о том, что потреб-
ление этой кислоты в довольно больших количествах является
необходимым для .предотвращения атеросклероза—главной
причины смерти. Однако высокое содержание линолевой кис-
лоты в обычных продуктах недостижимо, и в настоящее время
ведутся интенсивные поиски способов как можно более длитель-
ного сохранения кислоты в цис,цис-форме при переработке пи-
щевого сырья, например путем применения более мягких усло-
вий гидрогенизации. Кроме того, можно было бы получить
продукт с аналогичными физическими свойствами переэтерифи-
кацией глицеридов, содержащих линолевую кислоту, более на-
сыщенными соединениями.
Природные ненасыщенные жирные кислоты родственны по
структуре гормоноподобным соединениям — простагландинам
(т. 4, стр. 161), которые обладают сильной и разнообразной фи-
зиологической активностью и в настоящее время широко изу-
чаются. Это родство демонстрируется приведенными ниже фор-
мулами, которые иллюстрируют один из доказанных путей пре-
вращения линолевой кислоты in vivo. Будущее значение этого
открытия для питания человека и состава пищевых продуктов
пока трудно оценить, но, по-видимому, оно должно' сыграть
очень важную роль.
СН3(СН2)4СН=СНСН2СН=СН(СН2)7СООН
цис цис
линолевая кислота
(биотрансформация)
дегидрирование,
удлинение цепи (на 2С),
дегидрирование
СН3(СН2)4СН=СНСН2СН СНСН2СН=СН(СН2)3СООН
цис цис || || цис цис
СН СН
Н2
арахидоновая кислота
I
он
СН3(СН2)4СНСН=СН—СН—СН—СН2СН=СН(СН2)3СООН
I I
нс 54
H0/V 0
Н2
простагландин Е2
Пищевые вещества
605
Хлебопечение
Мука и мучные изделия занимают такое важное место в пи-
щевом рационе цивилизованного человека, что заслуживают
специального рассмотрения. Кроме того, обсуждение этого во-
проса позволяет наглядно показать сложные эффекты и взаи-
модействия белковых, углеводных и липидных компонентов
муки при получении и хранении хлеба.
Зерно разных сортов пшеницы сильно различается по своей
пищевой ценности. «Сильные» пшеницы содержат обычно 13—
14% белковых веществ, а у «слабых» эта величина снижается
до 6%. В Великобритании хлебопекарную муку получают путем
размалывания смеси зерен «сильных» и «слабых» пшениц с од-
новременным удалением основной части оболочек и зародышей,
причем достигается ~ 72%-ный выход муки, содержащей 11—
12% белка. Полученную муку отбеливают двуокисью хлора,
обесцвечивающей каротиноиды (можно также добавить к муке
небольшое количество бромата калия) и обогащают солями
кальция и железа и никотиновой кислотой (витамином РР).
Изготовление хлеба складывается из трех стадий: приготов-
ление теста, сбраживание теста и выпечка. Традиционный про-
цесс включает смешение муки с дрожжами, солью, водой и, если
необходимо, с некоторыми другими ингредиентами, сбражива-
ние теста в течение 1—3 ч и выпекание. По мере протекания
брожения тесто увеличивается в объеме и приобретает способ-
ность удерживать заключенный в нем газ в виде мельчайших
пузырьков.
Созревание теста и развитие у него вязкоэластических
свойств принято объяснять образованием белками клейковины
пространственной сетки путем сшивания белковых молекул, при-
сутствующих в отдельных частицах муки. Эти молекулы нахо-
дятся в исходной муке в форме плотно свернутых клубков и
удерживаются в такой конфигурации физическими силами, в
частности внутримолекулярными ковалентными дисульфидными
мостиками между остатками цистеина. Перемешивание теста
сопровождается разрывом некоторых сравнительно слабых ко-
гезионных связей (таких, как водородные связи), что делает
возможным гидратацию, набухание и развертывание молекул
белков в солевом растворе теста. Это влечет за собой ряд
внутри- и межмолекулярных химических реакций белков и за-
канчивается образованием устойчивой трехмерной структуры
созревшего теста. Согласно общепринятому представлению, важ-
нейшими из этих реакций, по-видимому, являются реакции тиол-
дисульфидного и дисульфид-дисульфидного обмена.
В современных интенсифицированных непрерывных процес-
сах хлебопечения сшивание клейковинных белков ускоряется
606
Глава 15
введением окислителей (аскорбиновой кислоты и бромата ка-
лия). Важную роль в созревании теста играет также присут-
ствие небольшого количества жира, хотя его роль пока не
выяснена.
После того как тесто созреет, его разделяют на порции и
выдерживают в условиях, способствующих жизнедеятельности
дрожжей. При этом протекают ферментативные реакции, вызы-
вающие гидролиз части мучного крахмала в более короткоце-
почечные сахариды и в конечном итоге распад глюкозы на эта-
нол и углекислый газ. Последний обеспечивает разрыхление теста
и является необходимым для получения хлеба большого объема
с хорошим пористым мякишем. При выпечке дрожжи разру-
шаются и действие ферментов прекращается. Структура хлеба
образуется в результате разрыва оболочки у части крахмальных
зерен (т. е. клейстеризации части крахмала) в процессе выпечки,
причем белки клейковины образуют сплошной остов мякиша.
Вкус хлеба имеет довольно сложную химическую природу;
однако можно считать, что он представляет собой сочетание
вкуса мякиша и корки. Вкус мякиша обусловлен побочными
продуктами действия дрожжей на крахмал, тогда как вкус
корки является результатом реакции Майяра («реакции побу-
рения»)— взаимодействия различных восстанавливающих саха-
ров с аминогруппами белков
+ H2NC—соон
N—С—СООН
гЦ) ।
Свойством хлеба, ухудшающим его качество при хранении,
является черствение. В этом процессе участвуют оба компо-
нента пшеничного крахмала — амилоза и амилопектин, хотя по-
лагают, что более важную роль играет амилопектин.
Амилоза при выпечке хлеба удаляется из крахмальных зерен
и образует мягкий гель, который очень быстро стареет, приобре-
тая линейную структуру с водородными связями между глюкоз-
ными звеньями соседних цепей. Амилопектин, оставшийся в зер-
нах, имеет более объемистые молекулы, находящиеся в «ветви-
стой» (open-tree) конфигурации. Через несколько дней моле-
кулы амилопектина переходят в форму пучка с образованием
межцепных водородных связей, однако в отличие от линейной
молекула
амилопектина
в „ветвистой"
конфигурации
I I_) I конфигурация
Ч пучка
Пищевые вещества
607
амилозы цепи амилопектина входят в состав одной и той же
молекулы. Ретроградацию амилопектина можно отчасти сделать
обратимой путем нагревания хлеба до температуры 50—60°C.
На этом явлении основано «освежение» черствого хлеба с по-
мощью повторного нагревания.
Процесс черствения хлеба протекает несколько дней и зави-
сит от температуры, причем максимальная скорость черствения
наблюдается при ~2°С. При температуре глубокого холода
(—20 °C) молекулярное движение замедляется настолько, что
черствение не происходит. Использование эмульгаторов, напри-
мер моноглицеридов, которые дают комплексы с крахмалом, по-
зволяет увеличить срок свежести хлеба за счет замедления
реакций превращения компонентов крахмала.
Порча пищевых продуктов и борьба с ней
Пищевые продукты непрерывно атакуются всеми формами
жизни. Около 30% всех выпускаемых продуктов питания унич-
тожается насекомыми; кроме того, значительная часть их пора-
жается грибами. Поэтому не удивительны те большие потери
пищевой продукции, которые имеют место уже после сбора уро-
жая или убоя скота. Помимо таких явных вредителей, как жуки
и грызуны, огромный ущерб наносят грибы и микроорганизмы.
К этому следует добавить нежелательные изменения продуктов
питания под действием содержащихся в них ферментов, а также
аутоокисление и другие спонтанные химические реакции.
Вся история цивилизации неразрывно связана с попытками
решить проблему сохранения пищевых продуктов. Сухое и бе-
режно хранимое зерно не портится в течение весьма длитель-
ного времени, и до наших дней дошли образцы зерна из глубо-
кой древности в почти неповрежденном состоянии. В то же
время влажное зерно поражается грибами, даже находясь в ко-
лосе. Головневые и ржавчинные грибы являются частыми го-
стями полей зерновых культур, причем паразитирующий на ржи
гриб Clariceps purpurea выделяет группу опасных токсинов,
лизергиновая кислота
афлатоксин
608
Глава 15
относящихся к амидам лизергиновой кислоты (т. 4, стр. 344, 389).
Болезнетворный гриб масличных культур Aspergillus flavus,
паразитирующий главным образом на арахисе, выделяет афла-
токсин Bi и другие токсичные соединения, которые обладают
сильным канцерогенным действием на млекопитающих. Этот
гриб послужил причиной вспыхнувшей- в 1960 г. в Англии
«индюшачьей болезни икс». Однако еще более опасны смер-
тельно ядовитые вещества белковой природы, вырабатываемые
анаэробной бактерией Clostridium botulinum, которые отно-
сятся к числу самых токсичных из известных ядов.
Задолго до зарождения идеи о существовании микроорга-
низмов люди придумали способы удаления их из пищевых про-
дуктов. Так, было подмечено, что некоторые продукты, подверг-
шиеся кислому брожению, все еще вполне пригодны для упо-
требления и сохраняются лучше, чем свежие. Маринование в
уксусе — один из старейших методов консервирования пищи.
Копчение, обезвоживание, засолка, консервирование с сахаром,
замораживание известны с незапамятных времен; в основе всех
этих способов лежит понижение «активности воды». Метод сте-
рилизации пищи в герметичных сосудах был разработан во
время наполеоновских войн: француз Н. Аппер (1809 г.) пред-
ложил проводить ее в стеклянных бутылях, а англичанин П. Дю-
ранк (1810 г.) —в жестяных банках. Однако лишь в 1839 г. по-
следний способ получил широкое распространение в Америке.
В 1864 г. Пастер в своем знаменитом эксперименте показал, что
мясной бульон может долго стоять на открытом воздухе, если
только исключить попадание в него микробов.
Необходимость стерилизации продуктов в жестяных банках
является большим недостатком, так как это удорожает их стои-
мость и часто ухудшает вкус или меняет структуру. Поэтому
были предприняты настойчивые поиски путей усовершенствова-
ния прежней технологии консервирования. С изобретением пли-
точного скороморозильного аппарата (Кларенс Бердсей, 1929 г.)
появилась возможность быстро замораживать пищевые продук-
ты, избегая образования крупных кристаллов льда и нарушения
структуры. В 1906 г. была описана низкотемпературная сушка
жидких веществ (М. д’Арсонваль и Ф. Бордасм), а позднее был
разработан метод обезвоживания овощей с сохранением их
структуры и вкусовых качеств путем предварительного накалы-
вания, чтобы облегчить удаление влаги из внутренних участков
(С. Гунсон и Дж. П. Сэвидж, 1957 г.).
К настоящему времени существуют надежные способы по-
давления развития микроорганизмов в пищевых продуктах, од-
нако гидролиз и аутоокисление жиров остаются серьезной проб-
лемой. Даже пастеризованное масло быстро прогоркает в ре-
зультате протекания гидролиза с образованием ряда жирных
Пищевые вещества 609
кислот с короткой цепью, в том числе масляной (пороговая кон-
центрация ощущения 25 ч. на млн), а «обезвоженные» кокосо-
вые орехи ненамного превосходят по сроку хранения свежие.
Другие животные жиры и растительные масла тоже могут про-
горкнуть, причем хотя их триглицериды не содержат короткоце-
почечных жирных кислот, последние могут образовываться за
счет реакций аутоокисления и разрыва цепи. Однако еще более
быстрым следствием аутоокисления является образование альде-
гидов, которое протекает по следующей схеме:
Цис цис цис инициирование
C2H5CH=CHCH2CH=CHCH2CH==CH(CH2)7COOR -------->
эфир линоленовой кислоты
цис цис , цис
— > C2H6CH=CHCH2CH=CHCHCH=CH(CH2)7COOR —>
свободный радикал
цис . транс цис о2
— > C2H5CH=CHCH2CHCH=CHCH=CH(CH2)7COOR —>
продолжение
цис транс цис цепи
—-----------------------------------------------> C2H5CH=CHCH2CHCH=CHCH=CH(CH2)7COOR ---------->
I (отрыв H
L „ от другой
(J—молекулы)
Цис Транс Цис разрыв цепи
—-----------------------------------------------> C2HsCH=CHCH2CHCH=CHCH=CH(CH2)7COOR ---------->
ООН
гидроперекись
цис цис
— > С2Н6СН=СНСН2СНО + • CH=CHCH==CH(CH2)7COOR + • ОН
цис-гексен-3-аль радикальный побочный
продукт
Аутоокисление эфиров линоленовой кислоты
Гексеналь представляет собой лишь один из целого ряда аль-
дегидов и других соединений, получающихся при аутоокислении
масел, содержащих эфиры линоленовой кислоты. Однако он
обладает очень сильным запахом зеленой фасоли (пороговая
концентрация ощущения 0,11 ч. на млн), который характерен
для таких масел вообще и соевого масла в особенности. В окис-
ленных маслах идентифицированы также ненасыщенные альде-
гиды, имеющие еще меньшие пороговые концентрации. Не
исключено, что эти соединения частично ответственны за харак-
терный вкус и оттенки вкуса многих пищевых продуктов, напри-
мер рыбы, цыплят, постного мяса, животного жира и гидриро-
ванного рыбьего жира.
Добавление антиоксидантов к пищевым продуктам с целью
замедления их аутоокисления в ряде случаев не приводит к осо-
бому успеху (например, в случае очищенного свиного сала).
Большинство растительных масел сами по себе содержат при-
20 Зак. 501
610
Глава 15
родные антиоксиданты (токоферолы), которые при отсутствии
инициаторов окисления предохраняют их от окислительной
порчи, превращаясь в безвредные окисленные продукты типа
хинонов. Полагают, что в некоторых условиях аутоокисление
а,-токоферол
идет по механизму свободнорадикальной атаки на метильные
группы с образованием димеров и в конечном итоге формаль-
дегида, однако точная схема протекания этого процесса при
комнатной температуре пока не установлена. Ряд тяжелых ме-
таллов, например медь или железо, являются активными ини-
циаторами окисления масел и жиров, вследствие чего их часто
удаляют или связывают в менее активные соединения хелати-
рующими агентами, такими, как лимонная или этилендиамин-
тетрауксусйая кислоты.
Новые источники пищевого сырья
Из трех основных компонентов пищи человека наименее де-
фицитными являются углеводы. В некоторых развивающихся
странах ощущается нехватка пищевых жиров. Однако самой
распространенной и важной проблемой является белковое голо-
дание. Поэтому в настоящее время основное внимание уделяется
изысканию путей и методов расширения производства пищевого
белка.
Белки
Возможно, что когда-нибудь поставщиком необходимых пи-
щевых белков станет химический синтез, но сейчас имеется еще
немало неиспользуемых природных ресурсов. Значительных
успехов предстоит достигнуть, например, на путях использова-
ния ряда новых сельскохозяйственных источников белка и мик-
робиологической переработки неорганических веществ, углевод-
ных отходов и углеводородов нефти в усвояемые белки.
Белковая «мука», получаемая из шротов масличных семян,
богата высококачественными белками. Проблемы, связанные
госсипол
Пищевые вещества 611
с ее использованием, — разрушение ростовых ингибиторов в сое-
вых бобах, удаление токсичного госсипола из ядра хлопковых
семян и уничтожение токсичных плесневых грибов в ядрах ара-
хиса — в основном уже решены. Белковую муку можно вводить
в состав различных продуктов питания и даже формовать в
нити, похожие на волокна мяса.
Большое внимание как потенциальный источник пищевого
сырья привлекает зеленая водоросль Chlorella, встречающаяся
в природе в виде тины в прудах и реках. При выращивании на
неорганических питательных веществах она дает двукратное
увеличение богатой белками биомассы всего за 2—6 час. В от-
личие от протеина высших растений протеин Chlorella не обед-
нен лизином, метионином и триптофаном.
Значительных масштабов достигло выращивание микроорга-
низмов, особенно дрожжей, на углеводном сырье. При этом в
качестве питательных веществ используются меласса (побочный
продукт производства тростникового сахара) и сульфитный ще-
лок (отходы целлюлозно-бумажной промышленности). Хотя по
своим физико-химическим свойствам искусственный белок ана-
логичен молочному белку (казеину), он целиком идет на корм
скоту. Однако можно ожидать, что проводимые в настоящее
время интенсивные исследования с культурой микроскопиче-
ского гриба, выращиваемого на гидролизате крахмала, приведут
к созданию крупнотоннажного производства белка, пригодного
для питания людей.
Некоторые почвенные бактерии и дрожжи способны усваи-
вать алифатические углеводороды в присутствии неорганических
источников азота типа солей аммония, давая белки, пригодные
на корм скоту. В одном из последних методов природный газ
химически перерабатывают в метанол, который затем исполь-
зуют как питательную среду для специально выведенных микро-
организмов. Аминокислотный состав конечного продукта ме-
няется в зависимости от вида микроорганизма, источника
углерода и условий ведения процесса, но во всех случаях полу-
чается продукт, пригодный для откорма скота.
Синтез белков включает ступенчатое присоединение амино-
кислот в заранее заданной последовательности; в настоящее
время он ограничивается лишь лабораторным приготовлением
относительно простых молекул. Процедура синтеза облегчается,
если аминокислота ковалентно связана с водонерастворимым
полимером, и этот метод может лечь в основу будущих промыш-
ленных процессов получения синтетического белка (т. 4,
стр. 338). Сообщается, что потенциально съедобное вещество
образуется в результате статистической полимеризации амино-
кислот в «протеиноиды», имеющие молекулярный вес более
8000.
20*
612
Глава 15
Любой синтетический метод должен исходить из доступных
аминокислот. Последние используются также для повышения
биологической ценности продуктов, бедных теми или иными
аминокислотами. К счастью, многие аминокислоты можно полу-
чить из природных источников. Ниже приведены схемы про-
мышленного получения лизина и метионина:
сн2==снсно + ch3sh --->
—> CH,SCH2CH2CHO
KCN
COC12
HNO3
H2SO4
CH3SCH2CH(NH2)COOH
1) разделение
изомеров .
2") гидролиз (NaOH)
3) HCl
хлоргиВрат
L -лизина
ci-
Углеводы
Углеводы в виде зерновых продуктов вырабатываются в
очень большом количестве, а извлечение сахарозы из сахарного
тростника или сахарной свеклы лежит в основе крупнотоннаж-
ного производства сахара. D-Глюкозу вырабатывают в промыш-
ленности путем кислотного или ферментативного гидролиза
крахмала (т. 1, стр. 158), как это показано на приведенной ниже
схеме:
D-глюкоза
Пищевые вещества
613
Чисто синтетические методы получения углеводов пока от-
сутствуют. Однако определенный интерес в этом отношении
представляет опубликованная несколько лет назад работа, в ко-
торой исходным веществом служит виниленкарбонат, получен-
ный из нефтехимических полупродуктов
винилен-
карсюнат
—> ВгСН2—[СН(ОН)]3—СНО
Жиры.
Первое промышленное производство синтетических жирных
кислот было организовано в Германии во время второй мировой
войны. В этом процессе алканы (продукты гидрогенизации ка-
менного угля по Фишеру — Тропшу) окисляли воздухом в при-
сутствии водного раствора перманганата калия (гл. 6). После
фракционирования и очистки получалась фракция кислородсо-
держащих производных линейных и разветвленных углеводоро-
дов Сю—С20 с четным и нечетным числом атомов углерода, ко-
торую этерифицировали глицерином. Полученный синтетический
жир очищали и использовали для приготовления маргарина.
Аналогичным методом — окислением высококипящих пара-
финов нефти — в настоящее время вырабатываются значитель-
ные количества синтетических жирных кислот в странах Восточ-
ной Европы. Сырые кислоты перед очисткой подвергают нагре-
ванию с целью дегидратации оксикислот. Разработана также
технология, включающая олигомеризацию этилена с последую-
щим окислением или карбонилированием олигомеров, однако
полученные продукты более дорогие. Хотя эти синтетические
кислоты не применяются для пищевых целей, они позволяют
существенно сократить техническое потребление натуральных
пищевых жиров и тем самым увеличить долю этих жиров, ис-
пользуемую в производстве продуктов питания.
Перспективы производства пищевых
продуктов
Полагают, что в будущем жители развитых стран будут
предъявлять более высокие требования к разнообразию ассор-
тимента продуктов питания. Повышенным спросом будут
614
Глава 15
пользоваться продукты, не требующие сложной кулинарной об-
работки и длительно сохраняющие свои вкусовые и питательные
качества. С другой стороны, быстрый рост населения Земли
значительно осложняет проблему мировых пищевых ресурсов.
Решение этих противоположных проблем необходимо искать на
путях научно-технического прогресса.
Важное значение приобретут требования, которым должны
отвечать продукты питания. Для многих стран вопросы обеспе-
чения сбалансированного и достаточного питания населения, по
всей вероятности, станут самыми главными. В то же время
успехи науки о питании приведут к пересмотру структуры пи-
щевого рациона. Уже сейчас имеются веские данные о необхо-
димости увеличения содержания в нашей диете линолевой кис-
лоты, и повышение биологической полноценности многих продук-
тов глубокой переработки пищевого сырья путем введения в них
витаминов и жизненно важных минеральных веществ должно
предусматриваться в законодательном порядке.
На базе достижений различных отраслей науки значительно
продвинется вперед технология пищевых производств. Так, от-
крытие структуры а-спирали стимулировало познание строения
белка; аналогичные успехи имеются и в химии углеводов. Раз-
виваются также методы изучения комплексов типа липопротеи-
нов. Эти и другие исследования помогут нам глубже понять фи-
зическую природу пищевых веществ и найти возможные пути их
модифицирования. Другим перспективным направлением яв-
ляется изучение ферментативных процессов. В настоящее время
разрабатываются простые синтетические системы, имитирующие
природные ферменты, и рассматриваются различные непрерыв-
ные процессы ферментативного синтеза пищевых продуктов.
Здесь следует ожидать существенного прогресса, однако прой-
дет еще немало времени, прежде чем наука полностью переве-
дет производство продуктов питания из области искусства в об-
ласть технологии.
ГЛАВА 16
Душистые и вкусовые вещества
Э. Коули, У. Фордхем
Cowley Е., Fordham ЙЛ D., Bush Boake Allen Ltd., London,
Самыми первыми вкусовыми веществами были, вероятно,
ароматические пряности. Например, бутоны гвоздичного дерева
употреблялись в Китае за тысячу лет до нашей эры. Рост тор-
говли пряностями был одним из главных показателей экономи-
ческого развития в средние века. Использование терпеноидных
соединений в медицине восходит к античным временам, однако
изготовление приятно пахнущих композиций на цветочной осно-
ве началось лишь в XV—XVI вв. на юге Франции. В XVII в.
лавандовое и розмариновое масла производились уже в боль-
ших количествах, а в 1725 г. появилась знаменитая «Кельнская
вода» (одеколон). В большинстве своем первые душистые и
вкусовые вещества природного происхождения представляли со-
бой терпены или терпеноиды (т. 4, гл. 5).
Огромное значение для химии душистых и вкусовых веществ
имела разработка современных аналитических методов, из ко-
торых наиболее важным является газовая хроматография. Боль-
шинство ароматных продуктов и многие вкусовые вещества
представляют собой чрезвычайно сложные смеси. Например,
лимонное масло, как это будет показано ниже, содержит не
менее 60 различных идентифицированных соединений. Хотя вкус
лимона определяется, вероятно, лишь небольшой частью этих
соединений, как вкус, так и запах чувствительны даже к нич-
тожным количествам того или иного компонента смеси; поэтому
специфический запах или вкус смеси далеко не всегда обуслов-
лен ее основными ингредиентами.
. Мы не будем касаться здесь физиологических (и психологи-
ческих) аспектов запаха и вкуса и целиком сосредоточим свое
внимание на вопросах, связанных с органической химией. Вкус
и запах неизбежно имеют отчасти субъективный характер; по-
этому мы обсудим только различные типы природных соединений,
616
Глава 16
используемых в качестве душистых и вкусовых веществ, а также
получение и применение некоторых синтетических и полусинте-
тических продуктов этой категории.
Душистые вещества
У. Фордхем
Fordham W. D., Bush Boake Allen Ltd., London
Эфирные масла
Большинство эфирных масел содержит разнообразные моно-
терпеноиды, такие, как (+)-лимонен, а-пинен, цитраль, гера-
ниол, карвон и борнеол. В последние годы были выделены
также различные сесквитерпены, в том числе а-цедрол, ветиве-
рол и а-санталол. Кроме того, в эфирных маслах присутствуют
многие ароматические соединения, например фенилэтиловый
спирт, эвгенол и метилантранилат. Некоторые эфирные масла,
особенно те, которые применяются в кулинарии, содержат также
алифатические соединения серы и азота. Иногда эфирное масло
состоит преимущественно из одного химического вещества, и
такие масла, разумеется, используются в качестве исходного
сырья для их получения. Например, масло Eucalyptus globulus
содержит 75% цинеола, лемонграссовое масло — от 75 до 80%
цитраля, а масло, выжатое из кожуры цитрусовых плодов,—•
90% (+)-лимонена. Однако перечисленные примеры являются
исключениями; обычно же эфирные масла (наиболее важные
из которых рассмотрены ниже) представляют собой сложные
смеси различных химических веществ.
Розовое масло производится в Болгарии из двух видов розы:
розовой розы Rosa damascena Mill и белой розы Rosa alba Linn.
Франция и Марокко культивируют преимущественно белую
розу Rosa centifolia Linn. Существует несколько методов пере-
работки цветов розы, которые дают три типа розового масла:
конкрет, абсолю и отто.
Розовый конкрет получают путем экстракции только что со-
бранных цветов летучим растворителем, таким, как бутан или
очищенный петролейный эфир. После отгонки растворителя
остается темная вязкая масса, которую называют конкретом.
Она содержит -~60% нелетучих соединений жирного ряда и
35—45% летучих соединений, в том числе фенилэтиловый спирт
(60%), (—)-цитронеллол (20%), гераниол и нерол (12%) и
следы ароматических веществ. Конкреты находят ограниченное
Пушистые и вкусовые вещества
617
применение в производстве косметических средств и расхо-
дуются главным образом на получение абсолю.
Экстракция конкрета этиловым спиртом приводит к отделе*
нию большей части пахучих веществ от нерастворимого мате-
риала. Остаток после отгонки спирта представляет собой жел-
тое вязкое масло, называемое абсолю, которое широко приме-
няется для получения парфюмерных композиций. Розовое
абсолю богато фенилэтиловым спиртом, (—)-цитронеллолом, ге-
раниолом и неролом.
При гидродистилляции (перегонке с водяным паром) цветов
розы Rosa damascena Mill получается бледно-желтая жидкость
с очень сильным цветочным, медвяным ароматом, которая из-
вестна под названием отто (розовая вода). Она содержит
(—)-цитронеллол и гераниол наряду с небольшими количе-
ствами линалоола, фенилэтилового спирта, цитраля, эвгенола,
карвона и сесквитерпенов..
Санталовое масло — один из самых старых парфюмерно-
косметических материалов. Его вырабатывают из Santalum al-
bum L., произрастающего в Восточной Индии, и Eucarya spicata
Sprag., родиной которого является Сумм (Западная Австралия).
Ост-индское санталовое масло получают путем гидродистилля-
ции древесины и корней дерева, а австралийское — комбина-
цией селективной экстракции и гидродистилляции древесины.
Оба масла используются в парфюмерии благодаря сильному и
стойкому запаху и хорошей смешиваемости с другими эфир-
ными маслами. Они содержат преимущественно сесквитерпено-
вый спирт и «-санталол.
Гераниевые масла получаются в промышленности из не-
скольких видов растений рода Pelargonium, вследствие чего
термин «гераниевое масло» в сущности неправилен. Главным
поставщиком гераниевого масла на мировой рынок является
о. Реюньон (бывший о. Бурбон), продукцию которого часто на-
зывают бурбоновским гераниевым маслом. Это масло имеет
очень сильный запах, похожий на запах розы, и благодаря своей
низкой стоимости широко используется в парфюмерно-космети-
ческих композициях, особенно в отдушках для мыл и косметиче-
ских средств. К основным компонентам гераниевого масла
относятся цитронеллол (40—50%) и гераниол (20—25%), кото-
рым сопутствуют небольшие количества линалоола, «-терпинео-
ла, фенилэтилового спирта, эвгенола, (—)-изоментона, гексен-3-
ола-1 и сесквитерпеновых углеводородов и спиртов.
Из травянистых растений рода Cymbopogon, произрастающих
только в некоторых районах Цейлона и Явы, добывают цитро-
нелловые масла. Они содержат преимущественно гераниол и
цитронеллол и применяются лишь как дешевые отдушки для
моющих и чистящих составов и других бытовых средств. Кроме
618
Глава 16
того, цитронелловые масла являются промышленными источни-
ками гераниола и (+)-цитронеллаля, которые сами по себе
имеют важное практическое значение. (-(-)-Цитронеллаль, на-
пример, используется в качестве полупродукта для синтеза
(—)-ментола. Однако масштабы производства гераниола и
цитронеллаля из этих масел в настоящее время невелики, так
как более рентабельным считается их получение синтетиче-
скими методами.
Связь между запахом и молекулярной структурой
душистых веществ
Характер и интенсивность обонятельного ощущения, возбуж-
даемого химическим соединением, определяется его молекуляр-
ной структурой. Однако выяснение связи между ними ослож-
няется следующими факторами: а) отсутствием объективных
методов оценки запаха; б) неспособностью человека количе-
ственно определять эффекты запаха; в) сомнительностью чис-
тоты многих исследованных препаратов и г) низкой чувстви-
тельностью химических и физических методов анализа по срав-
нению с органолептическим испытанием. Большинство запахов
имеет сложную природу, хотя мы воспринимаем их как прос-
тые, причем есть только один способ установления связи между
различными запахами — сравнение их между собой. Так, про-
фессиональные парфюмеры говорят между собой на «языке
ощущений», в который входят такие выражения, как «зеленая
нота», «высокая цветочная нота», «тяжелые полутона» и т. п.
Наиболее таинственное свойство запахов заключается в том, что
их комбинации не подчиняются каким-либо определенным за-
кономерностям. Многие соединения в чистом виде имеют запах,
резко отличающийся от того, который получается при подме-
шивании к ним других пахучих веществ. Знание взаимных
влияний различных запахов составляет основу парфюмерного
искусства.
Принято считать, что ощущение запаха возникает при кон-
такте химического соединения с подходящими обонятельными
рецепторами. Однако природа этого взаимодействия неизвест-
на. Эмпирическим путем установлены некоторые атомы и груп-
пы атомов (названные осмофорами), которые оказывают силь-
ное влияние на запах. Например, введение в органическое со-
единение атома серы неизменно придает ему очень сильный и,
как правило, неприятный запах. Характерным действием на за-
пах обладает карбонильная группа, тип влияния которой ме-
няется, если она находится в сопряжении с двойной углерод-
углеродной связью. Однако это лишь одна сторона дела, так
как необходимо принимать во внимание и стереохимию моле-
Душистые и вкусовые вещества 619
кулы. Влияние стереоизомерии на органолептические свойства
отчасти аналогично ее влиянию на химические свойства соеди-
нений. Особенно ярким примером в этом отношении являются
два изомера 3-(2,2-экзо-3-триметил-экзо-5-норборнил) циклогек-
санола, из которых эпимер 1, содержащий транс-аксиальную
оксигруппу, имеет очень сильный запах сандалового дерева, то-
гда как изомер с цис-экваториальной оксигруппой полностью
лишен запаха.
1
Известно много других примеров органолептических разли-
чий стереоизомеров циклических соединений, таких, как борнеол
и изоборнеол, ментолы и карвоментолы и пр. Нейве [1] собрал
убедительные доказательства в пользу того, что отчетливый,
интенсивный запах (или даже отсутствие запаха) связан с кон-
формационной жесткостью молекулы, которая способствует раз-
витию постоянных внутримолекулярных взаимодействий, осо-
бенно у соединений довольно высокого молекулярного веса.
Этот подход позволяет объяснить общеизвестный факт, что вы-
сокомолекулярные кетоны полностью проявляют присущий им
запах только при сильном разбавлении. Превосходным приме-
ром могут служить, в частности, макроциклические кетоны,
имеющие мускусный запах.
Вопрос о влиянии хиральности на запах в настоящее время
несколько запутан, и в литературе по этому поводу высказы-
ваются противоречивые суждения. Так, а-ионон (2) был раз-
делен на оптические изомеры и изучен рядом исследователей,
но полученные ими выводы не согласуются друг с другом. Одна
группа авторов утверждает, что ( + )- и (—)-формы разли-
чаются по запаху, тогда как другая придерживается точки зре-
ния, что они пахнут одинаково, но в 10 раз слабее, чем рацемат,
приготовленный смешением обоих энантиомеров. В общем
можно сказать, что запах, оптических изомеров, по-видимому,
различается, если они имеют жесткую структуру, но для алифа-
тических соединений это правило как будто не соблюдается.
Сообщается, например, что обнаружить органолептическое раз-
личие между (+)-, (—)- и рацемическими формами цитронел-
лаля (3) или цитронеллола (4) невозможно. В случае
620
Глава 16
линалоола (5) одни исследователи считают его энантиоморфные
антиподы неразличимыми по запаху, а другие — что они имеют
разный запах. Способность дегустаторов запаха провести раз-
личие между двумя энантиомерами всегда можно приписать
присутствию следов загрязнений, перешедших из исходных ве-
ществ или образовавшихся в ходе синтеза, а не собственным
органолептическим свойствам ( + )- и (и)-форм.
Теймер и Мак-Даниэль [2] попытались обойти это затрудне-
ние следующим простым методом. Они синтезировали пять со-
единений с различными характеристиками запаха в обеих энан-
тиомерных формах и сравнили их между собой с помощью
дегустационных жюри. Каждое соединение было приготовлено
двумя независимыми путями, причем каждая пара препаратив-
ных способов использовалась для получения двух пар соедине-
ний, например соединений (+)-1—(+)-2 и (—)-1—(—)-2. Ме-
тодики синтеза веществ (+)-1 и (—)-1 были аналогичными
и отличались от методики синтеза веществ (+)-2 и (—)-2. От-
сюда следует, что при образовании загрязнений в ходе химиче-
ских реакций пары соединений (+)-1—(—)-1 и (+)-2 — (—)-2
будут автоматически сближаться по запаху, а пары (+)-1 —
(+)-2 и (—)-1—(—)-2 удаляться друг от друга. Таким обра-
зом, если можно со статистической значимостью отличить
(+)-энантиомер от (—)-энантиомера, то обонятельный эффект
загрязнений в конечных продуктах должен будет занимать под-
чиненное положение по сравнению с эффектом хиральности.
С помощью этого метода было показано, что (+)- и (—)-фор-
мы диэтилацеталя миртеналя (6), пинокарвилпропионата (7),
миртеналя (8), пиноацетальдегида (9) и транс-пинокарвеола
(10) различимы по запаху.
в
8
9
10
Душистые и вкусовые вещества
621
Почти не вызывает сомнений, что большинство цис-транс-
изомеров непредельных соединений различаются по запаху,
и иногда очень сильно. Типичным примером может служить
широко известное различие в запахах гераниола (11) и нерола
(12). Сообщается, что 2'-тщащс-изомер а-ионона (2) пахнет фи-
алкой, а запах цис-изомера напоминает аромат кедрового де-
рева. цис-Гексен-З-ол (13), известный под названием «листвен-
ный спирт», ценится в парфюмерии более высоко, чем транс-
изомер.
Даже немногие примеры, приведенные выше, ясно доказы-
вают, что изменение химического строения сопровождается раз-
нообразными изменениями запаха. Однако из этого не следует,
что соединения неодинаковой структуры должны обязательно
иметь различный запах. В действительности твердо установлено,
что вещества, значительно различающиеся по химической при-
роде, могут обладать очень похожим запахом. Это убедительно
демонстрируют соединения с мускусным запахом, к которым от-
носятся индановые, тетралиновые, андростановые, макроцикли-
ческие и нитробензольные производные (см. ниже). Данный
вопрос детально рассмотрен Беетсом [3].
Как это ни странно, по данным Беетса [4], три приведенных
ниже вещества идентичны по качеству запаха:
Механизм восприятия запаха
Оттосон [5] показал, что для того, чтобы возникло ощущение
запаха, необходим непосредственный контакт обонятельных ре-
цепторов с пахучим веществом. Он продемонстрировал невоз-
можность возбуждения обонятельных рецепторов, если они отде-
лены от пахучих частиц тонкой мембраной. Некоторые исследо-
ватели предложили теорию запаха, согласно которой молекулы
вызывают ощущение запаха только тогда, когда их простран-
622
Глава 16
ственная форма позволяет им точно «садиться» на участки носо-
вых рецепторов, имеющие соответствующий профиль. Эймур [6]
развил это представление и попытался найти корреляцию между
размерами и формой молекул, с одной стороны, и их запахом,
с другой, на основе данных для ~600 химических соединений.
Он предположил, что существуют первичные типы запаха, ко-
торые были определены им как эфирный, камфарный, мускус-
ный, цветочный, мятный, острый и гнилостный, причем теория
Эймура делает попытку связать эти запахи с формой и разме-
рами молекул. Так, камфарный запах считается характерным
для сферических молекул диаметром 0,7 нм, мускусный — для
дискообразных молекул диаметром ~1,0 нм, цветочный — для
молекул, имеющих форму бумажного змея с гибким хвостом,
мятный — для клинообразных молекул, эфирный — для неболь-
ших или тонких молекул, острый — для электрофильных моле-
кул и гнилостный — для нуклеофильных молекул. Все реальные
запахи рассматриваются как комбинации перечисленных семи
основных запахов, получающиеся в результате воздействия мо-
лекул пахучего вещества одновременно на два или несколько
различных участков обонятельных рецепторов.
Некоторым биологическим подтверждением этой теории яви-
лось открытие Гэстлендом и сотр. [7] восьми разных видов обо-
нятельных рецепторов, в обонятельном органе лягушки.
Однако теория Эймура встретила серьезные возражения.
В частности, ей, вероятно, противоречит наличие запаха горь-
кого миндаля у синильной кислоты, которая совершенно
отличается по структуре своего профиля1 от других соединений,
сходных с ней по запаху, например бензальдегида. Кроме того,
эта теория критиковалась на том основании, что в литературном
обзоре Эймура, посвященном пахучим химическим веществам,
содержатся лйшь описания запахов, которые носят субъектив-
ный характер, зачастую неопределенны, иногда противоречивы
и во многих случаях неправильны (хотя бы из-за отсутствия
контроля степени чистоты испытуемых препаратов). Недоста-
точное внимание к чистоте препаратов послужило одной из
главных причин той путаницы, которая существует сейчас в во-
просе о связи структуры с запахом.
Теория профильно-функциональных групп, предложенная
Беетсом [8], базируется на предположении, что центральную
роль в процессе восприятия запаха играют профиль молекулы
пахучего вещества и положение его функциональных групп. Из
этой теории вытекают следующие следствия:
1. Отдельные детали молекулярного профиля можно менять,
не затрагивая общего характера запаха, если только такая за-
мена не сопровождается введением полярных групп, способных
оказывать влияние на ориентационную форму молекулы.
Душистые и вкусовые вещества
623
2. Функциональные группы тоже могут быть заменены дру-
гими группами, близкими к ним по полярности, без изменения
характера запаха.
3. Соединения, не обладающие полярными группами (напри-
мер, насыщенные углеводороды) или содержащие функциональ-
ные группы, доступ к которым стерически затруднен (и которые
поэтому малоактивны химически), имеют не очень отчетливый
запах или даже вовсе лишены запаха.
4. Близко расположенные полярные группы способны взаи-
модействовать с одной и той же функциональной группой в мо-
лекуле пахучего вещества, образуя единый осмофор Если же
полярные группы удалены друг от друга, то такое расположе-
ние вызывает дезориентацию молекулы и, как следствие этого,
ослабление запаха.
В 1928 г. Дайсон предположил существование связи между
запахом и частотой колебания атомов или групп атомов в мо-
лекуле пахучих веществ. Позднее Райт [9] установил связь за-
паха с положением полосы низкочастотного (ниже 800 см-1)
поглощения в колебательных спектрах душистых соединений
и привел статистически значимые корреляции в спектрах ком-
бинационного рассеяния 16 веществ, имеющих нитробензольный
запах. Аналогичные корреляции были найдены Райтом в дале-
кой инфракрасной области спектра для 10 соединений с мус-
кусным запахом, 10 соединений с немускусным запахом и 13 со-
единений, обладающих запахом свежей зелени. Кроме того,
Райт указал, что в случае оптически активных соединений нет
ни одного примера такой ситуации, когда один энантиомер
имеет запах, а другой совсем не пахнет; отсюда он заключил,
что первичный обонятельный процесс носит не химический,
а физический характер.
Таким образом, вопрос о механизме восприятия запаха до
сих пор является предметом дискуссии.
Промышленный синтез некоторых
душистых веществ
Гераниол, нерол, линалоол, цитраль
До сравнительно недавнего времени все эти важные ацикли-
ческие монотерпеноидные соединения получали исключительно
из природного сырья, такого, как цитронелловое, немецкое ро-
зовое и лемонграссовое масла. Однако за последние 5—10 лет
были разработаны методы промышленного синтеза, продукты
которых способны успешно конкурировать по качеству, цене
и объему производства с'веществами природного происхожде-
ния. Согласно оценке, в настоящее время мировое производство
624
Глава 16
синтетических продуктов этого ряда превышает в сумме
5000 т/год. Практическое применение нашли два метода полу-
чения этих соединений, один из которых базируется на природ-
ных полупродуктах, а другой — на ацетилене.
Исходным природным полупродуктом служит р-пинен (14),
вырабатываемый преимущественно фракционированной дис-
тилляцией сульфатного скипидара — побочного продукта произ-
водства крафт-бумаги. Пиролиз р-пинена при температуре
400 °C дает высокий выход мирцена (15), который подвергают
гидрохлорированию в безводной среде в присутствии каталити-
ческого количества однохлористой меди. При этом получается
смесь, содержащая линалоилхлорид (16), нерилхлорид (17)
и геранилхлорид (18). Интересно, что в отсутствие однохлори-
14 15
стой меди перечисленные аллилхлориды образуются лишь в не-
значительных количествах, а основными продуктами являются
мирценилхлориды (19) и а-терпинилхлорид (20). Механизм
каталитического действия однохлористой меди в этой реакции
неясен. Известно, что она образует лабильные комплексы с со-
пряженными диенами, в том числе с мирценом, и можно допус-
тить, что каталитическая реакция протекает через промежуточ-
ное образование такого комплекса. Однако его структура
и природа взаимодействия с НС1 пока не установлены.
Соотношение продуктов 16, 17 и 18 зависит от условий реак-
ции и может изменяться при стоянии реакционной смеси из-за
протекания реакций установления равновесия. Легкость этих
реакций аллильной изомеризации обусловливает гибкость дан-
ного метода синтеза. Так, при обработке смеси аллилхлоридов
безводным ацетатом натрия в присутствии триэтиламина как
катализатора происходит обычный процесс 8к2-замещения с об-
разованием смеси ацетатов, в которой преобладают геранил-
Душистые и вкусовые вещества
625
и нерилацетаты (~90—95%). В то же время в условиях пре-
имущественного SnI-замещения (сольволиза), например при
взаимодействии аллилхлоридов с уксусной кислотой в присут-
ствии акцептора хлористого водорода, получается почти исклю-
чительно линалоилацетат. Эта возможность варьировать состав
продукта путем использования надлежащих условий реакции
находит практическое применение в промышленности. Омыле-
ние соответствующих ацетатов дает линалоол или смесь гера-
ниола с неролом. Последнюю разделяют фракционированной
дистилляцией, получая чистые товарные продукты, или непо-
средственно используют в качестве полупродукта для синтеза
других душистых веществ. Например, пропуская смесь гера-
ниола и нерола в паровой фазе вместе с кислородом над нагре-
тым медным катализатором, можно окислить ее в смесь гео-
метрических изомеров цитраля
Цитраль широко используется в парфюмерной и пищевкусо-
вой промышленности. Кроме того, он является промежуточным
веществом в синтезе а-ионона — ценного ларфюмерного про-
дукта — и р-ионона, который применяется в производстве ви-
тамина А. Гидрированием смеси нерола с гераниолом можно
получить другой важный парфюмерный спирт — цитронеллол
(21), который служит также полупродуктом для синтеза «гид-
роксицитронеллаля» (22). Это соединение не встречается в при-
роде, но оно тоннами используется в парфюмерной промышлен-
ности из-за своего ландышевого запаха. Схема его синтеза при-
ведена ниже.
Чисто синтетический путь синтеза монотерпеноидных соеди-
нений базируется на ацетилене и ацетоне. Особенно большое
626
Глава 16
значение он имеет для получения линалоола и цитраля. Этот
синтез включает следующие основные стадии:
Зегидрапиналоол линалоол
Дегидролиналоол является также ценным полупродуктом
с разнообразной реакционной способностью. Так, нагреванием
его ацетата с каталитическим количеством меди можно с высо-
ким выходом получить цитраль, а пиролиз ацетоацетата дегид-
ролиналоола (реакция Кэрролла [10]) дает псевдоионон (23) —
промежуточное соединение в синтезе иононов.
ацетоацетат
дегидролиналоола
нагревание
-со2
Иононы
а- и р-Иононы имеют
важное
промышленное значение и
обычно получаются из цитраля. Первой стадией их синтеза яв-
25 (р-ионон)
2 (а-ионон)
24 (цитраль)
Душистые и вкусовые вещества
627
ляется альдольная конденсация цитраля (24) с ацетоном в ще-
лочной среде с образованием псевдоионона (23), который затем
подвергают циклизации кислотными реагентами в смесь изо-
мерных иононов. При использовании фосфорной кислоты или
фтористого бора получается главным образом а-ионон (2),
а при применении концентрированной серной кислоты — р-ионон
(25). Как показали советские исследователи, и в том, и в дру-
гом случае первоначально образуется а-ионон, который в при-
сутствии серной кислоты изомеризуется в полностью сопря-
женный р-ионон.
а-Ионон широко применяется в парфюмерной промышлен-
ности, так как его запах напоминает запах фиалки. В отличие
от него р-ионон находит лишь ограниченное применение как
душистое вещество и используется преимущественно в качестве
полупродукта для синтеза витамина А. Оба ионона употреб-
ляются в пищевкусовой промышленности, особенно для имита-
ции ягодных ароматов.
При конденсации цитраля с метилэтилкетоном образуются
метилпсевдоиононы (26 и 27), каждый из которых можно под-
вергнуть циклизации в а- или р-метилионон. Таким образом,
существует четыре структурных изомера метилионона — а-изо-
метилионон (30), а-н-метилионон (28), р-изометилионон (31)
и р-н-метилионон (29). С точки зрения парфюмерии такая мно-
24
гочисленность изомеров создает большие трудности, рассмотре-
ние которых выходит за рамки данной главы. Достаточно ска-
зать, что все технические метилиононы содержат переменные
628
Глава 16
количества остальных изомеров, хотя наиболее ценится запах
а-изометилионона (30).
Важными, но дорогими парфюмерными продуктами являются
6-метилиононы: а-ирон (32), р-ирон (33) и у-ирон (34). Они
имеют чрезвычайно сильный запах ирисо-фиалкового типа.
Природным сырьем для получения иронов могут служить кор-
невища ирисов, в экстракте которых обычно преобладает а-изо-
мер. Вопрос о влиянии структуры и стереоизомерии в ряду
иронов на их запах сильно запутан, и по этому поводу суще-
ствуют различные точки зрения. Однако большинство парфю-
меров сходится на том, что а-ирон, особенно его tjwc-изомер,
обладает чрезвычайно ценным запахом. Ироны выделяют из
летучего масла корней ириса или синтезируют из 5,6-диметил-
гептен-5-она-2 с использованием ацетилена аналогично лина-
Некбторые мускусные соединения
Макроциклические кетоны мускон (35), цибетон (36), ди-
гидроцибетон (37) и экзальтон (38) содержатся в секретах
некоторых животных, тогда как лактоны амбреттолид (39)
и экзальтолид (40) — растительного происхождения. Строение
\сн2)12/
35 (мускон)
СН—(СН2)7\
СН—(СН,)/7
СН2—(СН2)?Х
I \=0
СН2—(СН2)/
Зв (цибетон)
СН2—(CH2U
I /=О
СН2—(СН2)/
СН2—(СН2)5----С
I
СН2— (СН2)7— сн2
37
СН2—(СН2)в—с.
I /О
СН2—(СН2)6—сн/
38 (экзальтон)
39 (амбреттолид)
40 (экзальтолид)
Душистые и вкусовые вещества
629
ряда этих макроциклических соединений, в частности цибетона
и мускона, было установлено Ружичкой в 20-х годах.
В последующие годы были проведены значительные теоре-
тические исследования в ряду макроциклических душистых ве-
ществ, однако пока не удалось найти хорошие методы их син-
теза, представляющие промышленный интерес. Поэтому все эти
соединения очень дороги. В большинстве синтетических методов
(т. 3, гл. 3, стр. 190—194) исходят из дикарбЬновых кислот или
их простейших производных. Раньше получение макроцикличе-
ских кетонов чаще всего осуществляли путем пиролиза соли ди-
карбоновой кислоты с металлами второй или третьей группы
или циклизацией промежуточного эфира бромкарбоновой кис-
лоты. Первый метод дает очень низкие выходы, а недостаток
второго при осуществлении его в промышленном масштабе за-
ключается в необходимости очень большого разбавления реак-
' ционной смеси для получения хороших выходов целевого про-
дукта. Одним из наиболее полезных препаративных методов
является ацилоиновая конденсация, в основе которой лежит
способность а,со-дикарбоновых кислот при обработке их мелко-
дисперсным натрием превращаться в циклические ацилоины
(реакция протекает, по-видимому, на поверхности металла).
При восстановлении цинком и соляной кислотой ацилоины пе-
реходят в макроциклические кетоны.
СООС2Н.
Макроциклические кетоны можно окислить далее кислотой
Каро (H2SO5) в соответствующие лактоны. Эта реакция ис-
пользуется на практике для получения многих макроцикличе-
Как кетоны, так и лактоны макроциклического ряда обла-
дают очень интенсивным запахом мускуса и широко исполь-
зуются в парфюмерных композициях. Кроме того, они задер-
живают испарение более летучих компонентов композиций и
часто оказывают синергический и усиливающий эффекты на ду-
шистые и вкусовые вещества. Эти эффекты в некоторых случаях
ощущаются уже в концентрациях порядка 0,01 ч. на млн.
630
Глава 16
Мускусный запах обнаруживают также некоторые аромати-
ческие нитросоединения. Из них наибольшее промышленное
значение имеют мускус-ксилол, мускус-амбрет (мускус амбро-
вый) и мускус-кетон. Карпентер с сотр. [11] детально изучили
мускус-ксилол мускус-амбрет мускус-кетон
соотношение между структурой и запахом изомеров и гомоло-
гов мускус-амбрета и пришли к выводу, что мускусный запах
обусловлен наличием третичной алкильной группы в орто-поло-
жении к алкоксигруппе. Существенную роль играет, по-види-
мому, и трет-бутильная группа, так как при ее замене на втор-
бутильную группу запах мускуса исчезает.
В промышленности нитроароматические мускусные соеди-
нения получают по реакции Фриделя — Крафтса с последую-
щим нитрованием продукта. Например, синтез мускус-кетона
из л-ксилола осуществляется по следующей схеме:
Мускусным запахом обладает также ряд тетрагидронафта-
линов, из которых практическое применение в настоящее время
находят лишь версалид (41) и тоналид (42). Наличие запаха
мускуса у соединений этого ряда, вероятно, связано с присут-
ствием карбонильной группы в боковой цепи ароматического
Душистые и вкусовые вещества
631
ядра. Тетралин-мускусы обычно получают из 2,5-дихлрр-2,5-
диметилгексана по следующей схеме:
о
41
Исследуя влияние заместителей на мускусный запах произ-
водных индана, Беетс и сотр. [12] синтезировали большое число
соединений этого класса. Основная структура 43 лишена за-
паха, но введение в нее ацетильной (44) или формильной (45)
группы дает вещества с сильным запахом мускуса.
43 44
Присутствие заместителей в пятичленном кольце, по-види-
мому, тоже является необходимым для появления запаха мус-
куса, поскольку соединение 46 не имеет запаха, тогда как вве-
дение одной метильной группы (как в соединении 47) приводит
к умеренно сильному мускусному запаху, а двух метильных
групп (соединение 48)—к интенсивному запаху.
48
48
п~ цимол
Соединение 48 известно под фирменным наименованием «це-
лестолид». Большинство индановых мускусов получают в про-
мышленности путем конденсации n-цимола с третичным спир-
том в кислой среде с последующим ацилированием продукта
хлорангидридом карбоновой кислоты по Фриделю — Крафтсу.
632
Глава 16
Индановые мускусы устойчивы, не обесцвечивают мыла
и косметические изделия и сравнительно дешевы, что позволяет
предсказать им значительное применение в парфюмерно-косме-
тической промышленности.
Вкусовые вещества
Э. Коули
Cowley Е., Bush Boake Allen Ltd., London
Анализ и идентификация вкусовых
веществ
Появление инструментальных методов анализа, особенно га-
зовой хроматографии, масс-спектрометрии и инфракрасной
спектроскопии, сделало возможной идентификацию вкусовых
веществ, присутствующих в пище даже в ничтожных количе-
ствах. Зачастую такие компоненты оказывают решающее влия-
ние на органолептические свойства пищевых продуктов. Одним
Таблица 16.1
Летучие компоненты аромата малины
Свыше 1,0 ч. на млн. 0,1 -1,0 ч. на млн.
Ацетальдегид цис-Гексен-З-аль Ацетоин Г ексен-2-аль Гексаналь а-Ионон Гераниол Чис-Гексен-З-ол-1
0,01—0,1 ч. на млн. Ниже 0,01 ч. на млн.
Ацетон р, р-Диметилакролеин Пентен-2-аль Пентен-1-ол-З Гексанол-1 З-Метилбутен-З-ол-1 Бутанол-1 Этанол Диацетил Акролеин Пропаналь Пентанон-2 Р-Ионон Метанол Пентанол-1 тра«с-Бутен-2-ол-1
из первых было подвергнуто анализу «ароматическое» начало
ягод малины [13]. Экстракт ягод малины концентрировали
и обрабатывали 2,4-динитрофенилгидразицом для удаления
Таблица 16.2
Летучие компоненты груши сорта «бартлетт»
Метилдеканоат а’ 6
Этилдеканоат а' 6
Этнллауринат а
Этнлмиристат а’ б’ в
Метилпальмитат а’ б’ в
Этилпальмитат б’ в
Метилстеарат а’ б’ в
Метил-транс-октен-2-оат 6
Этил-транс-октен-2-оат а’6
Этил-транс-децен-2-оат а’ 6
Метил-гргс-децен-4-оат а’ б> е’ ж
Этил-гргс-децен-4-оат б’ в’ е
Метил-транс-додецен-2-оат а' б’
Этил-транс-додецен-2-оат а' б' в
Этил-гргс-додецен-6-оат 61 в
Метил-г<цс-тетрадецен-8-оат 6
Этил-гр/с-тетрадецен-8- оат 6
Метилолеат а’ 61 в
Этанол а'
Пропанол а
Бутанол3' 6
Пентанол а’ 6
Гексанолы а’ б' е
Гептанол а
Октанол а' 6
Метилацетат а
Этилацетат а’ 6
т-т а
Пропилацетат
Бутилацетат а’ 6
а а, б
Амилацетат ’
а б
Гексилацетат '
а б
Гептилацетат ’
Октилацетат а’ 6
цис-Хексенилацетат 6
Метилкаприлат а
Этилкаприлат а’ 6
1Лътнл-т ране,цис-декадиен-2,4-оат а’0, г
Этил-транс, гргс-декадиен-2,4-оат а’ б’ г> ж
Пропил-транс, /<ас-декадиен-2,4-оа т а’ б’ в'ж
Бутил-транс, гргс-декадиен-2,4-оат6, ж
Гексил-транс, ф/с-декадиен-2,4-оат а' в’ ж
Метил-транс, транс-декадиен-2,4-оат б’ ж
Этил-транс, транс-декадиен-2,4-оат б’ ж
Пропил-транс, транс-декадиен-2,4-оат б’ ж
Гексил-транс, транс-декадиен-2,4-оат а' б’ в’ ж
Метил-цис, транс-декадиен-2,4-оат б’ ж
Этилдодекадиеноат а> б> в
Метил-транс, цас-додекадиен-2,6-оат а’ б’ в
Этил-транс, цис-додекадиен-2,6-оат а’ 6
Метил-цыс, «{ас-тетрадекадиен-5,8-оат а’ б’ в’ 3
Этнл-цис, «{«с-тетрадекадиен-б,8-оат а’ б' в> 3
Этилгексадека диеноат а’ 6
Этил-транс. транс, цис-декатриен-2,4,7 (?)-оат а’ б’ в
Этил-транс, цис, гргс-додекатриен-2,6,9 (?)-оат а’ 6
Метил-транс, транс, цас-тетрадекатриен-2,4,8 (?)-оат б' в
Этил-транс, транс, г{ас-тетрадекатриен-2,4,8 (?)-оат 61 в
Метил-4-оксикротонат б’ г’д’ ж
634
Глава 16
Продолжение табл. 16.2
Этил-4-оксикротонат б’ г' ж
Метил-З-оксикаприлат б’ в> ж
Этил-З-оксикаприлат б’ в
Сесквитерпены, триненасыщенные соединения а’ б’ г
Примечание. Соединения идентифицированы следующими методами:
а Газовая хроматография. д Температура плавления.
® ИК-спектроскопия. е Химические производные.
в Масс-спектрометрия. ж Встречный синтез.
г УФ-спектроскопия. 3 ЯМР-спектроскопия.
карбонильных соединений. Остаток сохранял характерный запах
малины, который оказался обусловленным ~ 10 различными
спиртами и метилацетатом. Выделенные и идентифицирован-
ные фракции ароматического начала малины представлены
в табл. 16.1.
В качестве другого примера можно привести данные
табл. 16.2, в которой перечислены соединения, найденные Джен-
нингсом [14] при изучении летучих компонентов груши сорта
«бартлетт» с использованием всех современных аналитических
методов.
Связь между химической структурой
и органолептическими свойствами
Как мы уже видели при обсуждении запахов душистых со-
единений, даже незначительные изменения в их структуре, та-
кие, как переход от одного оптического изомера к другому, со-
провождаются существенным изменением запаха. Аналогичное
явление наблюдается среди вкусовых веществ, запах которых
сильно колеблется даже в ряду соединений близкой структуры.
Например, широко известен грушевый запах изоамилацетата.
В табл. 16.3 приведены данные, показывающие, как изменяется
качество запаха у сложных эфиров, гомологичных изоамил-
ацетату.
Гераниол (3,7-диметилоктадиен-2,6-ол), который является
ценным парфюмерным продуктом, имеет также важное значе-
ние как пищевкусовой ароматизатор. Органолептические харак-
теристики некоторых эфиров гераниола приведены в табл. 16.4.
Сопоставление химической структуры и органолептических
свойств вкусовых веществ не позволяет выявить какую-либо об-
щую закономерность. У сложных эфиров, представленных
в табл. 16.3 и 16.4, сила запаха практически одинакова, но ка-
чество запаха существенно меняется с изменением структуры
углеродного скелета. Совершенно иная картина наблюдается
Пушистые и вкусовые вещества
635
Таблица 16.3
Изменение запаха у родственных сложных эфиров
Соединение
Запах
Варьирование ацильной группы
Изоамиловый эфир уксусной кислоты
Изоамиловый эфир пропионовой кислоты
Изоамиловый эфир масляной кислоты
Изоамиловый эфир коричной кислоты
Изоамиловый эфир салициловой кислоты
Грушевый
Абрикосово-сливовый
Сливовый
Фруктовый, пряный
Клубничный
Варьирование алкильной группы
Метиловый эфир масляной кислоты
Этиловый эфир масляной кислоты
Бутиловый эфир масляной кислоты
Изоамиловый эфир масляной кислоты
Яблочный
Ананасовый
Фруктовый масляный
Сливовый
Таблица 16.4
Качество запаха у гераниола и его сложных эфиров
Соединение
Запах
Гераниол
Г ераниолацетат
Г ераниолбутират
Гераниолформиат
Гераниолфенилацетат
Г ераниолпропионат
Розовый
Нежно-розовый
Цветочный, но резкий
Малиновый
Медвяно-розовый
Горький, виноградно-розовый
в случае ванилина (49) — действующего начала ванили — и так
называемого этилванилина (50), которые идентичны по каче-
ству запаха и различаются лишь по его интенсивности (этил-
ванилин пахнет в 2,5 раза сильнее, чем ванилин). Аналогичное
изменение в силе запаха наблюдается при переходе от маль-
тола (3-окси-2-метилпирона-4) (51) к этилмальтолу (52). Эти
сединения обладают одинаковым качеством запаха, но у этил-
производного он в 10 раз сильнее.
49 (ванилин)
50 (этилванилин) 51 (мальтол) 52 (этилмальтол)
636
Г лава 16
Мальтол используется в пищевой промышленности для улуч-
шения вкусового аромата хлеба и кексов, куда он добавляется
в количестве 50—100 ч. на млн. Натуральный мальтол содер-
жится в жженом солоде и в карамели (жженом сахаре),
а также в корке хлеба.
Подобно душистым веществам, вкусовые вещества послу-
жили предметом широких исследований с целью установления
механизма восприятия вкуса. Органолептические свойства дан-
ного соединения зависят от субъективного ощущения, возни-
кающего при раздражении чувствительного нервного оконча-
ния, причем вкус вещества очень часто связан с его запахом.
Однако если не учитывать сложные комбинации вкусовых
и обонятельных ощущений, то принято различать четыре основ-
ных качества вкуса: кислый, сладкий, соленый и горький. Фи-
зиологическое ощущение двух последних качеств можно объяс-
нить взаимодействием ионов с вкусовыми окончаниями языка.
Так, хлорид, бромид и иодид натрия и хлориды калия и аммо-
ния имеют соленый вкус, который, по-видимому, обусловлен
взаимодействием катиона и аниона полностью диссоциирован-
ной соли с языком. В то же время бромид калия и иодид аммо-
ния обладают горько-соленым вкусом, а соли более тяжелых
металлов первой группы (хлорид цезия, бромиды цезия и руби-
дия и иодид калия) имеют преимущественно горький вкус. По
аналогии тот факт, что большинство кислот в водном растворе
обладает кислым вкусом, приводит к выводу, что ощущение
кислого вкуса обусловлено взаимодействием ионов Н3О+
с языком.
Ощущения горького и сладкого менее очевидно связаны
с определенными химическими свойствами веществ. Сладкие
соединения имеют самую разнообразную структуру — от поли-
оксипроизводных (таких, как моно- и дисахариды), хлоро-
форма и некоторых аминокислот (с радикалом R выше С2Н5)
до имидов и производных мочевины [таких, как сахарин (53)
и дульцин (54)], а также некоторых 2-амино-4-нитрофенилал-
киловых эфиров (56). Было высказано предположение [15], что
63 (сахарин)
54 (дульцин) 55 (цикламат натрия)
общей чертой всех этих столь различных соединений является
присутствие в сладкой молекуле слабокислотного протона (ко-
Душистые и вкусовые вещества
637
торым может служить, например, протон спиртовой оксигруппы,
имидогруппы, амидной группы, ближайший к нитрогруппе про-
тон в соединении 56 или одиночный протон в хлороформе)
и протоноакцепторного центра (льюисовского основания), на-
ходящегося на расстоянии около 0,3 нм от кислотного протона.
Функцию льюисовского основания может выполнять атом кис-
лорода спиртовой оксигруппы, карбонильной, сульфо- или нит-
рогруппы и даже электроотрицательный атом хлора.
а- О- фруктопираноза.
С1
С1 \ Ъ
с1*' 0,3 им
хлороформ
сахарин
при R* С3Н7 это соединение
в 4000 раз слаще сахарозы
Чтобы иметь сладкий вкус, соединение должно также рас-
творяться в воде. Отсюда следует, что группы, оказывающие
влияние на растворимость соединения, влияют и на его вкус.
Вместе с тем, если вещество очень хорошо растворимо в воде
и, следовательно, плохо растворимо в липидах (например, мо-
носахариды), то оно будет менее сладким, чем соединение типа
сахарина.
Далее было постулировано, что если кислотный протон и его
акцептор находятся в молекуле на должном расстоянии друг
от друга, то каждое вкусовое окончание, ответственное за
восприятие сладости, содержит пару комплементарных центров
донор протона — льюисовское основание, такую, как пептидная
связь —NH—С( = О)— в протеиновой цепи. Эти центры обра-
зуют сильную водородную связь со сладкой молекулой по ее
протонодонорному и основному центрам, например по гидро-
ксильной группе и атому кислорода сахара или глицерина.
Энантиомеры простой аминокислоты — аланина — идентичны
по сладости, но энантиомеры валина различаются между собой.
Возможно, что причиной этого является требование определенной
638
Глава 16
ориентации сладких молекул на рецепторном белке, которое
в случае одного энантиомера валина удовлетворяется легко,
а в случае другого встречает пространственные затруднения со
стороны изопропильной группы. В то же время для аланина
с его более короткой боковой цепью пространственный эффект
проявляется не столь сильно.
вкусовое
окончание
сладкая
молекула
+NH3
валин аланин
Полагают [16], что структурные факторы, ответственные за
горький вкус, аналогичны по своей природе протонодонорно-
протоноакцепторной паре, обусловливающей вкус сладких со-
единений, за тем исключением, что расстояние между обоими
центрами горькой молекулы должно быть равно 0,15 нм. Здесь
тоже допускается, что протонодонорный и протоноакцепторный
участки белковых молекул, находящиеся на поверхности вкусо-
вого окончания, ведут себя как рецептор и образуют водород-
ные связи с горькой молекулой. Это представление иллюстри-
руется сравнением молекулярной структуры соединений 57 и 58.
57 (горькое)
58 (безвкусное)
Алкалоиды имеют чрезвычайно горький вкус. Например,
в молекуле хинина (59) функции донора и акцептора протона
Душистые и вкусовые вещества
639
могут выполнять соответственно гидроксильная группа и атом
азота хинуклидинового кольца.
Производство некоторых типичных
вкусовых веществ
Ванилин, должно быть, наблюдался в виде кристалличе-
ского налета на поверхности бобов ванили задолго до того,
как он был описан в химической литературе. Его структура
была установлена Тиманом и Хаарманом в 1874 г., а спустя
два года Реймер синтезировал ванилин из гваякола (60)
Механизм реакции Реймера — Тимана обсуждался в т. 2
(стр. 290). 2-Окси-З-метоксибензальдегид, являющийся побоч-
ным продуктом этой реакции, легко удаляется перегонкой с во-
дяным паром. Другим исходным веществом для получения
ванилина (49) послужил эвгенол (62), который можно легко
выделить из ряда эфирных масел, особенно из гвоздичного. Эв-
генол подвергают изомеризации действием щелочи, полученный
изоэвгенол ацетилируют, окисляют бихроматом и образовав-
шийся альдегидоэфир гидролизуют в ванилин.
ОН
ОСН3
1) КОН (изомеризация)
------------------------->
2) ацетилирование
62
640
Глава 16
1) окисление
----------->
2) гидролиз
В 30-годах был разработан еще один метод получения ва-
нилина, базирующийся на химических превращениях гваяциль-
ных фрагментов лигнина (т. 4, стр. 143). Этот процесс заклю-
чается в обработке сульфитного щелока щелочью и экстракции
щелочной соли ванилина спиртом. В настоящее время мировое
производство ванилина достигает нескольких тысяч тонн в год.
До сих пор рассматривались главным образом пищевкусо-
вые ароматизаторы. Однако большим спросом пользуются
также и неусвояемые подсластители, так как многие тучные
люди или больные диабетом стремятся сократить удельный вес
углеводов в своем рационе. Важнейшим подсластителем яв-
ляется сахарин (имид о-сульфобензойной кислоты) (53).
Сахарин производится из толуола по схеме, приведенной
выше, причем побочно образующийся я-толуолсульфохлорид
отделяют от орто-изомера путем кристаллизации. Сахарин при-
мерно в 550 раз слаще сахара (сахарозы). Запрещенный не-
давно во всем мире цикламат натрия (55) в 30 раз слаще, чем
сахар, однако основная ценность этого соединения обусловлена
его синергическим эффектом, оказываемым на сахарин: смесь
цикламата натрия с сахарином не дает остаточного горького
привкуса, характерного для сахарина.
Сорбит (63) используется в качестве подсластителя продук-
тов питания для больных диабетом. Недостатком сорбита яв-
ляется почти столь же высокая калорийность, как сахарозы
Душистые и вкусовые вещества
641
Это не позволяет использовать его в диете людей с избыточным
весом. Сорбит можно получать гидрированием глюкозы
НОСН2(СНОН)4СН2ОН
63
Большой интерес в настоящее время вызывает возможность
использования в качестве душистых и вкусовых веществ про-
изводных пиразина, обнаруженных в пищевых продуктах после
термической обработки. Например, 2-этил-3,5,6-триметилпира-
зин (64), добавленный в количестве 10—100 ч. на млн., усили-
вает аромат какао у изделий из шоколада. Это соединение
можно получить на основе диацетила и пропилендиамина по
схеме, приведенной ниже, или же путем радикального бромиро-
вания одной из метильных групп тетраметилпиразина (N-бром-
сукцинимидом в присутствии перекиси бензоила) с последую-
щим алкилированием продукта иодистым метилом по методу
Гриньяра
(СгНбЬО
А1С13
С2Н5Вг
Этилтриметилпиразин (64) найден в обжаренных кофейных
зернах. Полагают, что он образуется при обжаривании пище-
вых продуктов за счет взаимодействия сахаров с аминокисло-
тами, протекающего под действием повышенной температуры.
Более простой по структуре этилпиразин (65) применяется
для усиления аромата табака. Запах зеленого, горошка
21 Зак. 501
642
Глава 16
приписывают смеси пиразинов 66—68. Пороговая концентрация
этих соединений очень мала — 2 ч. на трлн.
Производное пиразина 66 можно синтезировать хлорирова-
нием метилпиразина (хлором в присутствии водяного пара при
400°C) с последующим замещением атома хлора на метокси-
группу и алкилированием продукта иодистым метилом и затем
иодистым этилом с помощью амида натрия в жидком аммиаке *.
Общая способность усиливать вкус пищевых продуктов
была впервые обнаружена у глутамата натрия (69), который
обладает интенсивным мясным вкусом. Значительный интерес
представляют также мононуклеотиды, усиливающие вкус мяса.
Такие соединения, как инозин-5'-монофосфат (70, Х = Н),
ксантин-5'-монофосфат (70, X = ОН) и гуанин-5/-монофосфат
+NH, I
-ООССНСН2СН2СОО-J Na+
69
* Реакция удлинения алкильной цепи в гетероциклических соединениях,
открытая А. Е. Чичибабиным.— Прим. ред.
Душистые и вкусовые вещества 643
(70, X = NH2), имеют пороговую концентрацию ощущения
~ 0,1 % и дают синергические композиции с глутаматом нат-
рия. Возможно, что в случае этих нуклеотидов вкусовые клетки
человека возбуждаются лишь тогда, когда соединение точно
соответствует активной структуре или активным центрам вку-
совых рецепторов.
Продолжаются фундаментальные исследования по физио-
логии вкуса, но результаты их можно ожидать лишь в буду-
щем. Однако как интересно было бы полностью объяснить жгу-
чий вкус перца или холодок ментола!
Что касается перспектив развития промышленности вкусо-
вых веществ, то можно предсказать дальнейшее усложнение
структуры синтетических соединений по мере открытия хими-
катов с более эффективными «ключевыми» свойствами, рост
потребления синтетических вкусовых добавок параллельно
с увеличением спроса на продукты питания высокой степени
готовности и удорожанием природных вкусовых веществ, не-
которые новые успехи в областях, касающихся общего усиле-
ния вкуса и первичных вкусовых ощущений и, наконец, уста-
новление физиологической природы восприятия запаха и вкуса.
СПИСОК ЛИТЕРАТУРЫ
1. Naves У. R., «Relationship between Stereochemistry and Olfactory Pro-
perties», Vlth Mediterranean Symposium on Smell, La France et ses Parfums,
13(68), 160 (1970).
2. Theimer E. T., McDaniel M. R., J. Soc. Cosmet. Chem, 22, 15—26
(1971).
3. Beets M. J., Molecular Structure and Organoleptic Quality, Soc. Chem.
Ind. Monogr., 1957, No 1, p. 38.
4. Beets M. J., Amer. Perfumer Cosmet., 76, 54 (1961).
5. Ottoson D., Acta Physiol. Scand., 35, Suppl. 122 (1956).
6. Amoore J. E., Proc. Sci. Sec. Toilet Goods Ass., 1962, No 37, Suppl. 1,
p. 13.
7. Gesteland R. C., Lettvin J. V., Pitts W. H., Rojas A., Olfaction and
Taste (Ed. by Zotterman Y.), Macmillan, New York, 1963, p. 19.
8. Beets M. J., Perfums, Cosmet. Savons, 5(4), 167 (1962).
9. Wright R. H„ J. Appl. Chem., 4, 611, 615 (1954).
10. Carroll M. F., J. Chem. Soc., 1940, 704.
11. Carpenter M. S., Easter W. M., Wood T. F., J. Org. Chem., 16, 589
(1951).
12. Beets M. J., Van Essen H., Meerburg W., Rec. Trav. Chirn., 77, 854
(1958).
13. Moncrieff R. W., The Chemical Senses, 3rd ed., Leonard Hill, London.
1967.
14. Jennings W. G., Flavour Symposium, Paris, 1971.
151 Schallenberger R. S., Acree T. E., Nature, 216, 480 (1967).
16. Kubota T., Rubo I., Nature, 223, 97 (1969).
21*
ГЛАВА 17
А. Фотографические процессы и материалы
Дж. Даффин
Duffin G. F., Minnesota ЗМ Research Limited, Harlow
Б. Производство
и применение хлорфторуглеводородов
Б. Джойнер
Joyner В. D., I. S. С. Chemicals Limited, Avonmouth
А. Фотографические процессы и материалы
Галогенсеребряные процессы
Несмотря на большое внимание, которое уделялось и уде-
ляется химическим методам получения фотографического изо-
бражения (преимущественно с помощью органических соедине-
ний), на практике важнейшим светочувствительным элементом
до сих пор остается галогенное серебро. Последнее обычно
представляет собой бромид серебра с небольшой примесью
иодида (в более чувствительных фотоматериалах) или смесь
хлорида и бромида серебра (в менее чувствительных фотомате-
риалах). Однако существенную роль в составе светочувстви-
тельных слоев играют и органические соединения.
Желатина
Галогенное серебро используется в светочувствительных
слоях в виде мелких кристалликов (зерен) диаметром 0,1 —
1,5 мкм, диспергированных в коллоидном связующем — жела-
тине, к которой иногда добавляют другие полимеры. Эта сус-
пензия зерен галогенного серебра в желатине называется
фотографической эмульсией. Желатина как диспергатор гало-
генного серебра играет важную роль в фотографическом про-
цессе, где она оказалась поистине незаменимым материалом.
В частности, желатина обеспечивает равномерное распределе-
ние галогенного серебра в теплом водном растворе во время
А. Фотографические процессы и материалы
645
роста зерен (оствальдовского созревания), а при последующем
охлаждении превращается в студень, образуя твердый матрикс
готовой фотопленки. Желатина легко и обратимо набухает, что
обеспечивает легкое проникновение и выведение из фотослоя
водных растворов, применяемых при обработке фотоматериа-
лов; наконец, она служит носителем конечного черно-белого или
цветного изображения, так как содержит реакционноспособные
группы, делающие возможным затвердевание слоя с получением
хороших устойчивых изображений.
По химическому строению желатина представляет собой по-
липептид с молекулярным весом ~ 100 000. Ее можно получить
в почти монодисперсной форме, однако технический продукт
обычно несколько деструктирован и имеет средний молекуляр-
ный вес около 70000. Гидролиз фибриллярного белка колла-
гена, выделенного из костей или шкур крупного рогатого скота
или свиней, ведут в присутствии извести или минеральной кис-
лоты. При этом происходит не только расщепление макрострук-
туры коллагена на отдельные цепи, но и гидролиз пептидных и
других связей, вследствие чего конечное содержание концевых
и боковых реакционноспособных групп, имеющее решающее
значение для качества продукта, зависит от условий получения
желатины. В заключение желатину экстрагируют горячей водой
и выделяют из экстракта путем упаривания.
К обычным аминокислотам, остатки которых присутствуют
в желатине, относятся глицин (1), аланин (2), пролин (3) и
оксипролин (4), причем последние две аминокислоты харак-
терны и для коллагена. Гуанидиновый фрагмент аргинина (5),
H2NCH2COOH
1
CH3CH(NH2)COOH
2
,СН2—СН—СООН
н2с' I
ХСН2—NH
/СН2—снсоон
< I
'СН2—NH
как полагают, играет важную роль в процессе застудневания
желатины, который включает обратимое образование водород-
ных связей.
Н/ /ОН
СК с
NH2 СНО Хг/
HsN\ । 1 II У
VnH(CH2)3CHCOOH СН2О СНО J_____I
НЬГ Cl^
5 6 7 8
Задубливание желатины, которое обычно осуществляют, до-
бавляя к жидкой эмульсии перед поливом ее на подложку аль-
дегиды типа формальдегида (6), глиоксаля (7) или мукохлорной
646
Глава 17
кислоты (8), протекает с участием прежде всего е-амино-
групп лизина (9) и гидроксильных групп серина (10) или окси-
пролина (4).
H2N(CH2)4CH(NH2)COOH HOCH2CH(NH2)COOH
9 10
Стабилизаторы
Фотографическую эмульсию получают путем приготовления
и выращивания кристаллов галогенйого серебра, образующих-
ся в результате реакции двойного обмена между растворимой
солью серебра и смесью галогенидов щелочных металлов. Роль
органической химии в этом процессе (если не учитывать важ-
ную функцию желатины) невелика. Однако, даже достигнув
заданных размеров, кристаллы галогенного серебра (потенци-
альная скорость роста которых примерно пропорциональна их
величине) все еще имеют низкую светочувствительность. Поэтому
их приходится сенсибилизировать, создавая на поверхности
кристаллов ничтожные включения сульфида серебра. Состоя-
ние, соответствующее оптимальной степени сенсибилизации фо-
тографической эмульсии, неустойчиво. Дальнейший рост или
перераспределение включений сульфида может затормозить
рост кристаллов или привести к образованию вуали, т. е. спо-
собности проявляться у неэкспонированных участков изображе-
ния. Единственными удовлетворительными стабилизаторами
оптимально сенсибилизированной эмульсии являются гетероцик-
лические органические соединения, дающие комплексы с сереб-
ром, особенно тетраазаиндены типа 4-окси-6-метил-1,3,3а,7-тетра-
азаиндена (11). Из других гетероциклических стабилизаторов
он
11
N-N
с*н5
12
можно упомянуть 1-фенил-5-меркаптотетразол (12) и бензотри-
азол (13). Перечисленные соединения вводят в виде раствора
в фотографическую эмульсию, достигшую оптимальной степени
сенсибилизации.
Сенсибилизирующие красители
Сами по себе кристаллы галогенного серебра, будучи бес-
цветными или желтыми, чувствительны только к синему и фио-
летовому свету. Однако существуют некоторые красители, кото-
А. Фотографические процессы и материалы
647
рые при введении их в эмульсию расширяют диапазон чувстви-
тельности на всю видимую область спектра (панхроматические
фотоматериалы) или даже в близкую инфракрасную область.
Все хорошие оптические сенсибилизаторы принадлежат к классу
цианинов, которые подразделяются на две группы. Первую
группу составляют истинные цианины, представляющие собой
резонансные гибриды форм А и Б, где Z и Z' — гетероцикличе-
ские остатки, которые часто содержат в кольце не только азот,
Z Z'
R—N===C—(СН=СН)„—СН=С—-N—R' X’ (А)
+
Z Z'
R—N—-Ь=СН— (CH=CH)n—C==N—R' X- (Б)
+
но и другие гетероатомы, X- — анион, R и R'— алкильные или
подобные им группы и п — нуль или целое число; атомы угле-
рода в полиеновой цепи могут иметь заместители. Примерами
соединений этой группы могут служить красители 13, 14, 15.
s
N
S^/SCH3
Вг-
• СНз
С2Н.
13
14
15
n-n
Вторая группа оптических сенсибилизаторов — это мероци-
анины, являющиеся винилогами амидов, структура которых
включает резонансное взаимодействие между незаряженной
648
Глава 17
формой В и формой с разделенными зарядами Г (где R, Z и
п имеют те же значения, что выше, a Z" — остаток гетероцикли-
ческого кетона, такого, как приразолон-5 или тиазолон-4).
z Z"
R—N——С==(СН— СН)„=С—С=О (В)
Z Z"
R—N==t—(СН=СН)Л—С=^С—0“ (Г)
Полиметиновые производные первой группы синтезируют по
реакции нуклеофильного замещения, исходя из гетероцикличе-
ского соединения с активной метильной или метиленовой груп-
пой и подходящей ониевой соли другого гетероциклического
соединения или аналогичного реакционноспособного полупро-
дукта (т. 3, стр. 69). Так, при взаимодействии иодэтилата хи-
нальдина (16) с тозилметилатом 2-метилмеркаптобензотиазола
(17) образуется краситель 18 (Ts = ra-CH3CfiH4SO3)
основание
Реакция двух эквивалентов иодэтилата 2-метил-5-фенилбен-
оксазола (19) с триэтилортопропионатом (20) дает симметрично
построенный краситель 14, который обычно используется для
сенсибилизации фотоматериалов к лучам зеленой части спектра.
U
19
Д. Фотографические процессы и материалы
649
Наконец, мероцианиновый краситель 23 получают из соли
бензотиазола, которую превращают в винильное производное 21
и затем обрабатывают тиазолидином 22
ф /)—СН3+С6Н,К’=СН—NHC6H5 (£Цз£°г')0
^N +
I Ts-
С2Н5
ососн.
21 +
22 23
Проявляющие вещества
Экспонирование эмульсионного галогенного серебра, приго-
товленного и сенсибилизированного до заданной светочувстви-
тельности, как это было описано выше, необходимо осуще-
ствлять таким образом, чтобы каждое зерно поглотило лишь не-
сколько квантов света. Последние вызывают разложение гало-
генида серебра с образованием фотоэлектронов и ионов серебра,
которые затем реагируют между собой, давая на поверхности
экспонированных кристаллов небольшие (из 3—4 атомов) вклю-
чения металлического серебра, составляющие так называемое
скрытое изображение. Скрытое изображение переводят в види-
мое путем проявления, в процессе которого каждый экспониро-
ванный кристалл полностью восстанавливается в металлическое
серебро, причем квантовый выход достигает 109.
Все проявляющие вещества, используемые в совре)менных
галогенсеребряных фотографических процессах, являются орга-
ническими соединениями. В черно-белой фотографии почти уни-
версальное применение находит гидрохинон (24), обычно
используемый в виде синергических комбинаций с метолом
(п-метиламинофенолом) (25) или фенидоном (1-фенилпиразо-
650
Глава !7
Лидиноном-З) (26).
ОН
ОН
ОН
24
NHCH3
25
CeHg
26
Эти вещества проявляют в слабощелочной среде, чаще всего
в растворе карбоната натрия или калия с добавкой сульфита и
небольшого количества галогенида. Сульфит служит консерви-
рующим веществом, а галогенид создает'в проявителе такую
начальную концентрацию галогенид-иона, которая предотвра-
щает резкое изменение свойств проявителя в результате выде-
ления этого иона в процессе проявления первой фотопленки.
В случае гидрохинона реакция проявления протекает по сле-
дующему суммарному уравнению:
+ 2НВг + 2Ag° (а)
Весьма важно, чтобы проявляющее вещество восстанавли-
вало галогенное серебро только в участках скрытого изображе-
ния, подвергшихся действию света. Для этого оно прежде всего
должно иметь строго определенный окислительно-восстанови-
тельный потенциал. Однако очевидно, что существуют и другие
условия, выполнение которых обязательно для эффективной
дифференциации экспонированных и неэкспонированных зерен
галогенного серебра в процессе проявления. На практике раз-
личие в скоростях проявления обоих типов зерен может дости-
гать 105.
Хинон (27), образующийся в приведенной выше реакции,
реагирует далее с сульфитом, давая моносульфонат 28
При использовании синергической комбинации гидрохинона
(24) с метолом (25) или фенидоном (26) начальное восстанов-
ление галогенного серебра осуществляется вторым проявляю-
щим веществом, которое обычно берется в гораздо меньшем
количестве (мольное соотношение 1:10—1:50), чем гидрохи-
А. Фотографические процессы и материалы
651
нон. Например, фенидон ведет себя в соответствии с уравнением
(б); свободный радикал 30, который имеет в растворе оранже-
+ AgBr
+ Ag° + Вг“
30
(б)
во-красный цвет и довольно устойчив, снова восстанавливается
в анион фенидона (29) под действием гидрохинона
Аналогичный процесс протекает в случае метола. Однако
здесь имеется одно важное отличие, которое обеспечило ши-
рокое распространение фенидон-гидрохиноновых проявителей:
фенидоновый радикал не реагирует с сульфитом и очень легко
восстанавливается в ион, тогда как продукт окисления метола
частично связывается сульфитом и тем самым выводится из про-
цесса проявления. Поэтому фенидон эффективен в очень низких
концентрациях, а фенидоновые проявители не теряют активно-
сти до полного исчерпания гидрохинона, что имеет важное
практическое значение.
Цветные процессы
До сих пор мы рассматривали лишь фотографические про-
цессы, которые дают черно-белое изображение, состоящее из
частиц восстановленного серебра. В настоящее время, однако,
очень, важную роль играют цветные процессы, так как цветная
фотография имеет более широкое распространение, чем черно-
белая. Все галогенсеребряные цветные процессы используют те
же основные элементы, что и описанные выше черно-белые про-
цессы. Однако наряду с ними здесь применяется ряд новых
органических соединений. Эти соединения участвуют в образова-
нии цветного изображения, после чего серебряное изображение
разрушают (например, с помощью отбеливания) или каким-либо
образом отбрасывают его (например, оставляя ею на негативе,
как это делают в процессе «Полаколор»),
652
Глава 17
Методы образования цветного изображения можно подраз-
делить на процессы с образованием красителя, процессы с раз-
рушением красителя и процессы с диффузионным переносом
красителя. Чаще всего употребляется первый метод, к которому
относятся процессы «Агфаколор», «Кодахром» и другие широко
известные процессы.
Обратимся поэтому сначала к процессам с образованием
красителя. Во всех процессах этого типа, применяемых в фото-
графической практике, используется хромогенное проявление,
т. е. проявление n-диалкиламиноанилинами или их простыми
производными (соединения 31—33), при котором восстановле-
ние экспонированных зерен галогенного серебра сопровождается
31 32 33
превращением проявляющего вещества в реакционноспособный
продукт окисления. Так, га-диэтиламиноанилин (31) дает хино-
диимин 34, который, будучи довольно неустойчивым соедине-
нием, быстро реагирует в фотослое, переходя в лейкоформу
красителя; последняя вступает в дальнейшую реакцию (напри-
мер, с серебром), превращаясь в конечный краситель. В ка-
честве реагентов для получения красителя обычно применяют
ацилацетанилиды и производные пиразолона и нафтолов, кото-
рые дают соответственно желтое, пурпурное и сине-зеленое
изображения.
Согласно законам физики, при проявлении, например, обра-
щаемой фотопленки типа «Эктахром» (где экспонированные
участки изображения должны в результате проявления стать
прозрачными — в точном соответствии с яркостями объекта)
А. Фотографические процессы и материалы
653
в каждом слое будет получаться краситель дополнительного
цвета. Так, слой, чувствительный к синим лучам (не содержа-
щий оптического сенсибилизатора), будет проявляться с обра-
зованием желтого изображения, слой, чувствительный к зе-
леным лучам, при проявлении даст пурпурное изображение,
а слой, чувствительный к красным лучам, — сине-зеленое изо-
бражение.
Как будет видно из дальнейшего изложения, этот принцип
требует, чтобы в каждом слое происходило образование только
нужного красителя. Простейшим способом выполнения этого
требования является введение в каждый слой соответствующей
цветной компоненты, что можно осуществить двумя путями.
Первый путь, который используется в старых фотопленках типа
«Агфаколор», заключается в применении субстантивных компо-
нент, т. е. таких, которые содержат длинноцепочечную утяже-
ляющую группу (например, С18Н37), соединенную с гидрофиль-
ной солюбилизирующей кислотной группой (например, SO3H).
Каждую такую компоненту отдельно растворяют в водном рас-
творе щелочи и добавляют к эмульсии, чувствительной к лучам
дополнительного цвета. При этом цветная компонента взаимо-
действует с желатиной (основную роль здесь опять-таки играют
боковые аминогруппы полипептидных цепей), которая прочно
удерживает ее в слое после полива фотоэмульсии на подложку.
Описанный процесс приготовления цветного фотослоя можно
проводить либо последовательно в несколько стадий, либо в
одну. стадию — путем поочередного нанесения эмульсионных
слоев на движущуюся ленту пленки или бумаги. К цветным
компонентам данного типа относятся соединения 35—37 (звез-
дочкой обозначено положение, в которое происходит сочетание
с проявляющим веществом).
СООН
- 35
654
Глава 17
Альтернативный метод, который применяется в большинстве
современных фотоматериалов (например, в фотопленках типа
«Кодахром»), состоит во введении в каждую компоненту гидро-
фобной группы, придающей ей маслорастворимость, как это
сделано в веществах 38—40. Полученное соединение растворяют
в небольшом количестве подходящего растворителя, например
дибутилфталата, и при помощи смачивателя распределяют в
соответствующей галогенсеребряной эмульсии. Таким образом,
компонента получается диспергированной по типу «масло в
воде», и конечный краситель образуется и фиксируется в масля-
ной фазе.
В обоих этих случаях быстрое протекание реакции окислен-
ного проявляющего вещества с цветной компонентой надежно
обеспечивает образование в каждом слое только нужного кра-
сителя. Иногда для полной гарантии между эмульсионными
слоями помещают также промежуточные слои.
Еще один метод хромогенного проявления, используемый в
процессе «Кодахром», основан на совершенно ином принципе.
Простые компоненты, такие, как соединения 41—43, растворяют
в проявителях, но цветное проявление отдельных слоев ведут
поочередно, причем режим обработки подобран таким образом,
что, хотя все красители образуются из реагентов, присутствую-
А Фотографические процессы и материалы
655
щих в растворе, в каждом слое получается только краситель
нужного цвета.
но
Вг
42
|Р coch2cn
Вг
НО
41
Образование красителей во всех описанных выше процессах
протекает по реакции между хинодиимином (например, 34) и
анионом цветной компоненты, содержащей кетометиленовую
группировку.
В приведенных примерах, когда X = Н, полученная лейко-
форма реагирует далее с ионами серебра, окисляясь в конечный
краситель, по следующей схеме:
(.д)
Например, пиразолоновый краситель будет иметь структуру 44
44
Иногда природа группы X обусловливает легкое элиминиро-
вание НХ, в результате чего лейкоформа непосредственно
658
Глава 17
переходит в краситель без окисления серебром. На практике при-
меняются два типа цветных компонент, замещенных в активное
положение: хлорпроизводные и фенилазопроизводные (соедине-
ния последнего типа имеют собственную окраску и служат для
корректировки цвета изображения на негативных фото-
пленках) .
Важно отметить, что существует два больших класса цвет-
ных фотоматериалов: обращаемые материалы, непосредственно
дающие позитивное изображение, и материалы, дающие сначала
цветной негатив, с- которого затем производят печать на другом
цветном материале и получают конечный позитивный отпечаток.
В позитивных фотопленках можно, использовать субстантивные,
маслорастворимые или проявитель-растворимые компоненты,
тогда как в негативных — только недиффундирующие компо-
ненты.
Теперь перейдем к цветным процессам с разрушением кра-
сителя. Все выпускаемые в продажу фотоматериалы этого типа
основаны на применении метода «проявление с отбеливанием».
Каждый слой содержит тщательно подобранный азокраситель
цвета, дополнительного к цвету спектральной чувствительности
эмульсии. В результате первого, черно-белого проявления в
экспонированных участках возникает серебряное изображение.
Затем пленку помещают в сильнокислый раствор, содержащий
агент комплексообразования с серебром (например, тиомоче-
вину) и так называемый катализатор отбеливания красителя,
типичным примером которого является 2,3-диметилхинокса-
лин (45)
45
При этом протекает следующая реакция:
I A—N=N—В + 4Ag + 4НХ —> A—NH2 + BNH2 + 4AgX
где А и В — остатки молекул азокрасителей, подобранных
таким образом, чтобы образующиеся вещества легко вымыва-
лись из фотослоя. Последующая обработка приводит к полному
удалению галогенида серебра и других соединений, оставляя
первоначальный азокраситель только в участках, не подверг-
шихся при экспонировании действию света. Как и процессы с
образованием красителя, данный метод является, полностью
стехиометрическим и дает изображения с хорошей градацией.
Принцип отбеливания красителя используется в системе
«Ильфорд калор принт» и в более поздней системе «Цибахром».
А. Фотографические процессы и материалы
657
В качестве азокрасителей обычно употребляют соединения типа
46—48.
47
48
Описанный метод позволяет получить готовые изображения
хорошего качества. Однако из-за того, что все красители содер-
жатся в светочувствительных слоях,'он пригоден только для
малочувствительных фотоматериалов, таких, как фотобумаги;
кроме того, обработка здесь является более трудоемкой и дли-
тельной, чем по методу хромогенного проявления.
Метод диффузионного переноса красителя применяется
в настоящее время только в одном типе выпускаемых фотома-
териалов, основанных на процессе «Полаколор». Светочувстви-
тельным элементом служат три фотографических полуслоя, чув-
ствительные соответственно к синим, зеленым и красным
лучам. Под каждым из этих слоев находится слой, содержащий
Иггатив
Г
г
з
4
5
в
7
8
9
10
Рис. 17.1. Схематическое изображение процесса «Полаколор», Отпечаток,
негатив и проявляющий раствор приводятся в контакт после экспонирования.
1—подложка фотобумаги; 2—приемный слой, в который диффундируют красители;
3—проявляющий раствор из капсулы; 4—слой, чувствительный к синим лучам; 5—жел-
тый проявитель-краситель; 6—слой, чувствительный к зеленым лучам; 7—пурпурный
проявитель-краситель; 8—слой, чувствительный к красным лучам; 9—сиие-зеленый проя-
витель-краситель; 10—подложка фотопленки.
Рис. 17.2. Схематическое изображение камеры и процесса «Полароид» (Duf-
fin G. F.. Photographic Emulsion Chemistry, Focal Press Ltd., London, 1966).
1 — негативный слой; 2—негатив; 3— проявляющий раствор; 4—позитивный отпечаток;
5—подпружиненные валики; б—капсулы с проявляющим раствором; 7—позитивный слой.
А. Фотографические процессы и материалы
659
проявитель-краситель, что является существенно важным эле-
ментом всей системы (рис. 17.1). Фотопленка с этой многослой-
ной эмульсией, намотанная на одну катушку фотоаппарата, вы-
полняет функцию негатива. На другую катушку намотана фо-
тобумага, имеющая приемный слой, причем в нескольких
местах к ней прикреплены капсулы с проявляющим раствором.
После экспонирования пленка и бумага прижимаются друг
к другу, а между ними образуется слой проявляющего рас-
твора, вытекающего из раздавленных капсул (рис. 17.2). В за-
ключение позитив отделяется от негатива. Во время обработки
происходит восстановление галогенного серебра в освещенных
участках и окисление проявителя-красителя, переводящее его
из растворимого состояния в нерастворимое. Молекула прояви-
теля-красителя имеет структуру типа 49. В данном случае по-
лучающийся хинон уже не обладает способностью растворяться
ОН
СН2СН2—краситель
ОН
49
в щелочном растворе, присущей хинолу. Поэтому в неосвещен-
ных участках проявитель-краситель будет диффундировать в
Рис. 17.3. Схематическое изображение самого последнего (1973 г.) фотома-
териала типа «Полаколор».
/ — эмульсионные слои и слои с проявителем-красителем; 2 — прозрачный приемный слой;
3—проявляющий раствор, выдавливаемый нз капсулы после экспонирования, который
содержит белое непрозрачное вещество; 4— черный противоореольиый слой.
приемный слой, где он взаимодействует с протравным фиксато-
ром и образует конечное позитивное изображение цветного ори-
гинала.
660
Глава 17
Сравнительно недавно фирма Polaroid объявила о разра-
ботке новой системы с использованием принципиально новой
конструкции камеры и другой структуры светочувствительного
элемента и приемного слоя, в котором образуется конечное
цветное изображение. Пленка и отпечаток объединены в еди-
ном комплекте, который автоматически выталкивается из ка-
меры через прижимное устройство, раздавливающее капсулу
с проявляющим раствором. Как показано на рис. 17.3, экспони-
рование производится через приемный слой, находящийся на
обратной стороне пленки. Цветное изображение образуется
в сущности тем же методом диффузионного переноса краси-
теля, но вся система остается в форме единого комплекта. Бе-
лый фон отпечатка получается при помощи взвешенного веще-
ства, которое распределено в проявляющем растворе, выдавли-
ваемом из капсулы в пространство между приемным слоем
и экспонированным светочувствительным элементом.
Другие органические компоненты
галогенсеребряных процессов
Имеется много других аспектов галогенсеребряной фотогра-
фии, связанных с органической химией. Так, заслуживают упо-
минания смачиватели — длинноцепочечные алкилсульфонаты,
способствующие получению ровного эмульсионного слоя на
подложке фотопленки или фотобумаги. Представляет интерес
и сама эта подложка. Уже много лет после того, как отказались
от использования пожароопасной нитроцеллюлозы (целлулои-
да), фотопленки изготовляют на подложке из ацетилцеллю-
лозы, чаще всего триацетата. Последнее название, впрочем, не
совсем правильное. Для получения ацетилцеллюлозы сполна
ацетилированный продукт подвергают частичному гидролизу до
достижения степени ацетилирования 80—85%. Такой полимер
легче перерабатывается литьем из растворителей типа хлорис-
того метилена.
За последние годы все большую популярность приобретают
полиэфирные подложки, особенно для фототехнических пленок
(например, позитивных и флюорографических). По химиче-
скому составу эти подложки аналогичны волокну терилен, вы-
рабатываемому из полиэтилентерефталата. Они получаются пу-
тем горячего экструдирования полимера в виде листов, которые
затем подвергают ориентационной вытяжке в продольном и по-
перечном направлениях — точно так же, как это делают в про-
изводстве волокна.
В некоторые фотопленки вводятся полиоксиэтилированные
соединения— сами полиэтиленгликоли или их производные
А. Фотографические процессы и материалы
661
с концевой арильной группой (50), где R=H или арил и
п= 10—50.
R(OCH2CH2)„OH
50
Эти соединения оказывают влияние на ход проявления, ускоряя
обработку самых высокочувствительных фотопленок, что имеет
огромное значение, если учесть большую чувствительность со-
временных материалов. В ряде других случаев — например, при
применении пленок типа «лит» (названных так потому, что с их
помощью изготовляют полутоновые литографии)—полиокси-
этилированные соединения позволяют достигнуть очень высо-
кой контрастности, необходимой для получения качественных
растровых изображений.
Для некоторых .специальных целей применяются также ор-
ганические полимеры. Они обеспечивают повышенную гибкость
фотослоя (полиэтилакрилатные латексы) и улучшают ряд фи-
зических свойств, например матируют поверхность пленки или
предотвращают возникновение заряда статического электриче-
ства. С помощью гетероциклических меркаптосоединений типа
51 можно производить вирирование серебряных изображений
в красивые черные тона, а поглотители УФ-лучей типа соеди-
нения 52 придают хороший внешний вид цветным фотоотпе-
чаткам.
s^sx^sa
HN-N
51
Бессеребряные процессы
Диазопроцесс
Несмотря на огромное число органических процессов, опи-
санных в патентной литературе, наиболее популярными ос-
таются процессы, базирующиеся на светочувствительных солях
диазония. При экспонировании фотослоя, содержащего диазо-
ниевую соль, в ультрафиолетовом свете (обычно используется
свет ртутной лампы с длиной волны 365 нм) протекает следую-
щая реакция:
hv
ArN2X 777? Агон + N2 + HX
662
Глава 17
В освещенных участках диазониевое соединение разлагается.
Оставшуюся соль подвергают сочетанию с фенолом, причем по-
лучается цветное изображение, которое в экспонированных
участках практически не окрашено.
Простейшие соли диазония, такие, как соли незамещенного
диазотированного анилина, неустойчивы. Однако диазониевые
соединения с электронодонорными (+R) заместителями го-
раздо более устойчивы термически (особенно двойные соли,
например тетрахлорцинкаты), вследствие чего их выделение и
работа с ними не связаны с особыми затруднениями. Приме-
рами веществ этого типа могут служить соединения 53—55.
i/2 ZnChN
55
В самых общих чертах можно выделить два различных ме-
тода получения конечного изображения. По первому из них
фенол вводят в фотослой вместе с диазониевой солью и затем
просто обрабатывают экспонированный материал основанием,
вызывающим азосочетание, например парами аммиака (диазо-
типные бумаги). По другому методу в фотографическую эмуль-
сию вводится только соль диазония, а азосочетание осуще-
ствляется путем обработки скрытого изображения щелочным
раствором фенола (так называемый полусухой или полумокрый
процесс). В диазотипных бумагах обычно применяется нафтол
типа 56 или 57 в комбинации с анилидом ацетоуксусной кис-
56
57
CH3COCH2CONHC6Hs
58
А. Фотографические процессы и материалы
663
лоты (58). Нафтол дает синий краситель, а анилид — желтый,
так что в сумме получается довольно хорошее черно-белое изо-
бражение.
В полусухом процессе цветной компонентой чаще всего
служит флороглюцин (1,3,5-триоксибензол). Это соединение
реагирует с диазониевой солью с образованием моно-, дис-
и трис-азокрасителей, имеющих соответственно желтый, корич-
невый и синий цвета, комбинация которых дает желаемое хо-
рошее черное изображение.
Процесс Кальвара
Этот процесс тоже основан на использовании солей диазо-
ния, но отличается тем, что полученное изображение состоит из
пузырьков азота. Диазониевая соль типа обычного п-диметил-
аминопроизводного 53 диспергируется в слое сополимера акри-
лового эфира с акриловой кислотой. Экспонирование вызывает
выделение азота, который при тщательном подборе полимера
образует мельчайшие пузырьки; кроме того, выделяющийся in
situ 'азот инициирует дополнительную полимеризацию макро-
молекул, что повышает сохранность этих пузырьков. Таким
образом, конечное изображение выглядит как белое помутнение
на фоне прозрачного полимерного слоя; однако благодаря рас-
сеиванию проходящего света, обусловленному его отражением
на поверхностях раздела твердое вещество — газ, плотность изо-
бражения в хорошо коллимированном световом пучке (таком,
как луч диаскопа) весьма высока. Это позволяет получать на
экране качественное черно-белое изображение.
По альтернативному методу можно дать возможность обра-
зовавшемуся азоту медленно улетучиться, подвергнуть остав-
шуюся диазосоль термическому разложению и стабилизировать
вновь выделившиеся газовые пузырьки путем дополнительной
полимеризации материала фотослоя. Первый метод дает нега-
тивное изображение, тогда как второй — позитивное.
Фотохромные системы
Еще один процесс, привлекший в последнее время большое
внимание, базируется на образовании отчетливого цветного
изображения непосредственно при экспонировании, т. е. на яв-
лении фотохромизма. Этот процесс особенно пригоден для по-
лучения микроизображений, поскольку он имеет молекулярную
природу, когда каждый квант света вызывает изменение лишь
в одной молекуле светочувствительного элемента. Последнее
позволяет получать чрезвычайно тонкие изображения по срав-
нению с галогенсеребряным процессом, в котором на стадии
664
Глава 17
проявления происходит громадное усиление изображения. Кроме
того, это обусловливает низкую чувствительность фотохромных
материалов, что, однако, не является серьезным недостатком,
если принять во внимание их высокую информационную
емкость.
Явление фотохромизма обнаруживают многие соединения,
но наиболее часто в литературе упоминаются индолин-пирано-
вые спироциклические системы. Например, вещество 61, пред-
ставляющее собой продукт конденсации о-оксиальдегида 60
с индолениновой солью 59, бесцветно, однако в возбужденном
состоянии легко переходит в валентный изомер 62, который
обладает интенсивным пурпурным цветом.
NO2
СН3
62
Возможно обращение фотохромного изображения при облу-
чении его сильным светом, поглощаемым соединением 62; тем
не менее в обычных условиях обработки и хранения это изо-
бражение вполне устойчиво.
Б. Производство и применение
хлорфторуглеводородов
Мировое производство хлорфторуглеводородов в настоящее
время превышает 800 тыс. т/год. Эти соединения применяются
в качестве аэрозольных пропеллентов, холодильных агентов,
растворителей, вспенивателей, огнегасящих веществ и химиче-
ских полупродуктов. Производство хлорфторуглеводородов воз-
никло как важная подотрасль химической промышленности
около 40 лет назад в связи с появлением спроса на более без-
Б. Производство а применение хлорфторуглеводородов
665
опасные хладоагенты. Используемый в английской литературе
для их обозначения термин «chlorofluorocarbons» (хлорфтор-
углероды) является неправильным, так как подразумевает, что
эти соединения состоят только из углерода, хлора и фтора. Для
большинства крупнотоннажных материалов данного класса это
название соответствует действительности, но наряду с ними
важное значение имеют также другие соединения, содержащие
такие элементы, как водород или бром. Поэтому правильнее
было бы называть их галогенированными углеводородами. Пер-
воначально в холодильной технике использовали газообразные
хладоагенты типа аммиака, двуокиси серы и хлористого метила,
применение которых сопряжено с рядом проблем, обусловлен-
ных токсичностью и пожароопасностью этих соединений. Ам-
миак до сих пор находит очень широкое применение в крупных
промышленных холодильных установках. Однако начавшееся
в 20-х годах повсеместное употребление бытовых и торговых
холодильников вызвало острую необходимость в нетоксичных
и негорючих хладоагентах. Использование хлорфторуглеводо-
родов в качестве холодильных агентов было предложено Хенне
и Миджли, а позднее фирма Е. I. du Pont de Nemours and Co.
Inc. выпустила их в продажу под названием «фреоны». В на-
стоящее время эти соединения производятся рядом фирм в раз-
личных странах мира. Основные производители хлорфторугле-
водородов указаны в табл. 17.1, причем приведенные в ней дан-
ные являются далеко не исчерпывающими.
Таблица 17.1
Хлорфторуглеводороды, выпускаемые в промышленном масштабе
Страна Фирма Фирменное наименование
США и Канада Allied Chemical Е. I. du Pont Kaiser Chemicals Pennwalt Union Carbide Дженетрон Фреон Кайзер Изотрон Юкон
Великобритания ICI I. S. C. Chemicals Арктон Айсион
Франция Pechiney Ugine Kuhlmann Флужен Форан
ФРГ Hoechst Kali Chemie Фриген Калтрон
Италия Montecatini Edison Алгофрен
Япония Daikin Kogyo Mitsui Fluorochemicals Asahi Glass Дайфлон Асахифлон
Австралия Australian Fluorine Chemicals Айсион
Нидерланды Zinc Organon FCC
666
Глава 17
Химический состав
Все продажные хлорфторуглеводороды представляют собой
галогенированные производные обычных алканов и содержат по
крайней мере один атом фтора. Фтор — самый активный из из-
вестных элементов, и в неконтролируемых условиях он бурно
реагирует с многими веществами. Однако для фторалканов ха-
рактерна низкая химическая активность, причем их реакцион-
ная способность тем меньше, чем больше содержание фтора
в молекуле.
Для получения хлорфторуглеводородов используют три ос-
новных исходных соединения, каждое из которых при взаимо-
действии с фтористым водородом дает ряд производных:
а) Четыреххлористый углерод
СС14 —> CC13F —> CC12F2 —> CC1F3 —► CF4
б) Хлороформ
СНС13 —> CHClsF —> CHC1F2 —> CHF3
в) Гексахлорэтан
СС13—СС13 —► CC1F3—CC12F —> CC1F2—CC1F2 —> CC1F2—CF3
Другие соединения, имеющие практическое значение, содер-
жат наряду с фтором бром, например:
г) CBrF3, CBrF2—CBrF2
или являются циклическими фторуглеводородами, например:
F2C—cf2
д) I j октафторциклобутан
F2C—CF2 .
Последовательное замещение атомов хлора на фтор сопро-
вождается понижением температуры кипения, и большинство
хлорфторуглеводородов при нормальных температуре и давле-
нии представляют собой газы. Однако, за исключением не-
скольких соединений, критическая температура которых лежит
ниже обычной комнатной температуры, хлорфторуглеводороды
способны находиться при этой температуре в сжиженном со-
стоянии. Именно на этом свойстве и основаны многие области
применения хлорфторуглеводородов.
Номенклатура
Одна из особенностей хлорфторуглеводородов, которая бро-
сается в глаза даже при поверхностном ознакомлении, — это
похожие и очень длинные названия. Указанное обстоятельство
может привести к путанице, вследствие чего была разработана
единая система цифровых обозначений хлорфторуглеводородов,
которые можно использовать либо вместе с их фирменными
Б. Производство и применение хлорфторуглеводородов
667
наименованиями в деловых документах, либо с буквой R в об-
щей литературе. В основе этой системы обозначений лежит
трехзначное число XYZ, где Z — количество атомов фтора в мо-
лекуле, Y — число атомов водорода, увеличенное на единицу,
и X — число атомов углерода, уменьшенное на единицу (если
последняя величина равна нулю, то цифру X опускают).
При этом принимается, что остальные атомы, которые не-
обходимо добавить в молекулу для соблюдения четырехва-
лентности углерода, — это атомы хлора.
Описанную выше систему номенклатуры лучше всего объ-
яснить на конкретных примерах. Так, дихлордифторметан
СС12Р2 обозначается индексом R12, т. е. здесь X = О, Y=1
и Z = 2. Хлордифторметан, для которого X — О, Y = 2 и Z = 2,
обозначается индексом R22. 1,2-Дихлор-1,1,2,2-тетрафторэтан
имеет обозначение R114 (X = 1, Y = 1 и Z = 4).
Присутствие атомов брома в. молекуле указывается бук-
вой В, после которой идет цифра, показывающая число атомов
брома. Например, CBrClF2 обозначается как R12B1, a CBrF3 —
как R13B1.
Существуют и другие детали этой системы номенклатуры,
в частности касающиеся обозначения различных изомеров; од-
нако их рассмотрение выходит за рамки данной главы.
Производство
Ключевой стадией получения хлорфторуглеводородов яв-
ляется реакция замещения атомов хлора на фтор, которую ведут
в жидкой или паровой фазе.
В жидкофазном процессе смесь соответствующего хлоругле-
водорода (например, четыреххлористого углерода) и безводного
фтористого водорода нагревают в автоклавном реакторе, со-
держащем хлорфторид сурьмы (катализатор), при температуре
100—150°С и давлении 1—3 МН/м2 (10—30 атм). Парофазный
процесс осуществляют при атмосферном давлении и более вы-
сокой температуре (250—300 °C) в присутствии импрегнирован-
ного угольного катализатора. В этом случае четыреххлористый
углерод и безводный фтористый водород раздельно испаряют,
смешивают, подогревают в теплообменнике выходящими из ре-
актора газами и пропускают над катализатором.
При использовании в качестве исходного вещества четырех-
хлористого углерода оба процесса дают смесь RH (CC13F)
и R12 (CC12F2)
2СС14 + 3HF —> CCI3F + CC12F2 + ЗНС1
Соотношение продуктов можно варьировать в широких преде-
лах, изменяя условия реакции, скорость подачи реагентов и т. д.
668
Глава 17
Полученная смесь содержит также следы CCIF3, однако обычно
стремятся избежать образования этого соединения, так как по-
требность в нем весьма невелика.
Очистка продуктов заключается в удалении из них хлорис-
того водорода и следов непревращенного фтористого водорода
путем перегонки под давлением или водной промывки в скруб-
берах с последующим разделением смеси на чистые компоненты
дистилляцией.
Получение CHC12F из CHCI3 проводится аналогичным об-
разом, но в производстве хлорфторуглеводородов этанового
ряда имеются небольшие отличительные особенности. Хотя
выше в качестве исходного вещества упоминался гексахлор-
этан, в действительности это соединение является твердым ве-
ществом (т. пл. 187 °C) и поэтому обычно готовится прямо в ре-
акторе из тетрахлорэтилена и хлора. Одновременно в реактор
подают фтористый водород, причем скорости сырьевых потоков
и условия реакции регулируют таким образом, чтобы обеспе-
чить завершение присоединения хлора до начала фторирования.
Существует 6 возможных продуктов фторирования гексахлор-
этана с различным содержанием фтора — от CCI3CCI2F до
CF3CF3, но практическое значение имеют только три из них:
CC12F — CC1F2 (R113), CC1F2 —CC1F2 (R114) и CC1F2 —CF3
(RH5).
Степень чистоты
По некоторым данным, хлорфторуглеводороды — самые чис-
тые из многотоннажных химикатов, и качество ежегодно выпус-
каемых в мире свыше 800 тыс. т этих соединений подтверждает
такую оценку. Холодильная техника, за исключением одного-
двух очень специализированных случаев, предъявляет чрезвы-
чайно жесткие требования к степени чистоты хладоагентов,
а поскольку в качестве последних используются почти все
хлорфторуглеводороды, чаще всего они целиком вырабаты-
ваются с учетом этих требований.
Обычные стандартные показатели чистоты хлорфторуглево-
дородов квалификации «хладоагентный» приведены в табл. 17.2.
Эти показатели достаточно надежно гарантируют удовлетвори-
Таблица 17.2
Технические условия на фторуглеводороды квалификации «хладоагентный»
Максимальное содержание воды, ч. иа млн. 10
Интервал выкипания (95—5%), °C 0,25
Содержание высококипящего остатка, % по объему 0,01
Содержание непоглощаемых газов, % по объему 1,5
Максимальная кислотность в пересчете на хлорид-ион, ч. 0,5
на млн.
Таблица 17.3
Свойства и применения хлорфторуглеводородов
Цифровое обозначение Формула Температура кипения, °C Абсолютное давление пара прн 21 °C, атм Применения
113 cci2f—ccif2 47,6 0,4 Растворитель, хладоагент, химический продукт
114В2 CBrF2—CBrF2 47,3 0,4 Огнегасящий агент
11 CC1F3 23,8 0,9 Пропеллент, хладоагент, вспениватель, растворитель
21 CHC12F 8,9 1,6 Потенциальный хладоагент; может также использо- ваться как рабочая жидкость
114 CCIFs—ccif2 3,8 2,0 Пропеллент, хладоагент •
12В1 CBrClF2 -4,0 2,7 Огнегасящий агент
С-318 C4F8 -5,8 2,8 Пропеллент
12 CC12F2 -29,8 6,0 Пропеллент, хладоагент, вспениватель
115 CC1F2—CF3 -38,7 7,9 Хладоагент, пропеллент
22 CHClFj -40,8 9,6 Хладоагент, химический полупродукт
13В1 CBrF3 -57,8 15,0 Огнегасящий агент, хладоагент
13 CCIF3 -81,4 33,3 Хладоагент
14 CFi -128,0 38,2 а Хладоагент (применяется очень редко)
а Критическое давление.
670
Глава 17
тельные эксплуатационные качества любого конкретного
соединения при использовании его как холодильного агента.
Однако, кроме контроля указанных в таблице показателей, ис-
пользуется также газохроматографический анализ, с помощью
которого проверяют химическую чистоту индивидуальных ве-
ществ и состав смесей хлорфторуглеводородов, применяемых
для некоторых целей.
Области применения
Применение важнейших хлорфторуглеводородов, имеющих
практическое значение, иллюстрируется данными табл. 17.3.
Гла-вной областью применения этих соединений продолжает
оставаться холодильная техника. Вместе с тем R11 и R12 ис-
пользуются преимущественно как пропелленты для аэрозольных
упаковок, и общая тенденция заключается в постепенном пе-
ремещении холодильной техники на второе место в структуре
потребления хлорфторуглеводородов (табл. 17.4).
Таблица 17.4
Структура потребления (%) хлорфторуглеводородов
в США и Великобритании
Области применения США Великобритания
Ппопелленты 50 70
Хладоагенты 28 10
Вспениватели 5 10
Растворители 5 5
Химические полупродукты 10 3
Прочие 2 2
Холодильные агенты
Выбор хладоагента определяется величиной необходимого
понижения температуры. В тех случаях, когда эта величина не
слишком велика, применяется один из высококипящих хладо-
агентов, тогда как для достижения низких температур необхо-
димы соединения с низкой температурой кипения. Следует
отметить, однако, что рабочие температурные интервалы от-
дельных холодильных агентов значительно перекрываются.
Самым распространенным хладоагентом является дихлорди-
фторметан (R12), который пригоден для использования в цен-
тробежных, ротационных и поршневых компрессорах. Он при-
Б. Производство и применение хлорфторуглеводородов
671
меняется в крупных установках для создания искусственного
климата, а также в бытовых холодильниках. В компрессорных
системах центробежного типа, входящих в состав промышлен-
ных кондиционирующих установок, часто используется трихлор-
фторметан (R11). Хлордифторметан (R22) находит широкое
применение для получения низких температур в бытовых и про-
мышленных морозильных аппаратах. Важное значение имеют
также азеотропные смеси типа CCI2F2+CH3CHF2 и CCIF2CF3+
+CHC1F2. Другие хлорфторуглеводороды используются лишь
в специальных случаях.
Хотя были проведены некоторые исследования по примене-
нию хлорфторуглеводородов в абсорбционных холодильных
циклах, на практике эти соединения используются только в па-
рокомпрессорных агрегатах. Холодильный эффект в системе
с компрессионным циклом получается за счет испарения жид-
кого хладоагента в стороне низкого давления замкнутого
цикла. Далее пары механически засасываются компрессором и
возвращаются в сторону высокого давления. Этот процесс луч-
ше всего объяснить на примере работы домашнего холодиль-
ника.
1. Сжиженный хладоагент, находящийся под давлением соб-
ственного пара, соответствующим температуре жидкости, про-
ходит через регулирующий вентиль в камеру большего объема
(сторона низкого давления системы). Там жидкость испаряется,
причем переход ее в пар сопровождается расширением; при
этом происходит понижение температуры. Необходимое для ис-
парения тепло отбирается от окружающей металлической кон-
струкции — холодной плиты или морозильного отделения холо-
дильника.
2. Пары засасываются компрессором и сжимаются до высо-
кого давления, причем при компрессии происходит повышение
температуры.
3. Горячие сжатые пары при прохождении по радиатору
охлаждаются и конденсируются в жидкость. Радиатор пред-
ставляет собой решетку из тонких трубой, расположенную на
задней стенке холодильника (в промышленных холодильниках
чаще всего применяется теплообменное устройство с водяным
охлаждением).
4. Жидкий хладоагент проходит к регулирующему вентилю,
и весь цикл повторяется снова.
Приведенная схема работы парокомпрессорной системы от-
ражает основные стадии процесса. Многостадийные, или кас-
кадные, системы, в которых горячие пары хладоагента охлаж-
даются за счет испарения второго хладоагента, циркулирующего
в отдельном цикле, представляют собой лишь усовершенствова-
ние описанной выше основной схемы.
672
Глава 17
Аэрозольные пропелленты
Строго говоря, аэрозоль — это взвесь мельчайших частиц
твердого вещества или жидкости в газе или воздухе (например,
дым или туман). Однако в быту под аэрозолем подразумевают
упаковку, содержимое которой выводится наружу благодаря
избыточному давлению, существующему в этой упаковке. Сюда
относятся такие разнообразные препараты, как инсектициды,
средства по уходу за волосами, краски и кремы для бритья. Во-
преки распространенному мнению, основные принципы упа-
ковки веществ под давлением были описаны в патентной
и другой технической литературе еще в начале нашего века,
а регулируемое клапанное устройство для бутылей было запа-
тентовано в 1862 г. Тем не менее принято считать, что отцами
настоящей аэрозольной упаковки были норвежец Эрик Рот-
хейм, который в 1933 г. запатентовал упаковку, находящуюся
под давлением, а также Гудью и Салливан, которые в 1943 г.
получили патент на аэрозольный инсектицид.
В начале 40-х годов американские войска, сражавшиеся на
Тихом океане, несли от укусов тропических насекомых более
крупные потери, чем в результате боевых действий. Гудью
и Салливан — ученые Энтомологического ведомства США, ра-
ботавшие над проблемой наиболее эффективного и удобного
применения инсектицидов, попробовали растворить инсектицид-
ные вещества в небольшом количестве масла и смешать этот
раствор с дихлордифтор метаном (R12), чтобы получить упа-
ковку с самоподдерживающимся внутренним давлением. Давле-
ние паров CC12F2 при 21 °C равно 500 КН/м2 (5 атм), что обе-
спечивает мгновенное испарение сжиженного дихлорфторметана
при выбрасывании раствора в атмосферу; это вызывает рас-
пыление раствора на мельчайшие капли инсектицида, находя-
щегося в форме чрезвычайно тонкодисперсного тумана (аэро-
золя). Образующийся инсектицидный туман сохраняется в ат-
мосфере довольно значительное время и поэтому гораздо
эффективнее уничтожает летающих насекомых, чем использо-
вавшиеся ранее грубодисперсные взвеси, создаваемые механи-
ческими распылителями. За последующий период (до 1945 г.)
было изготовлено около 40 млн. «аэрозольных бомб», которые
положили начало массовому применению аэрозолей в быту.
Первые аэрозольные упаковки были непригодны для быто-
вых целей, так как применявшееся в них высокое давление
требовало использования дорогих и тяжелых стальных балло-
нов. Однако очень быстро было найдено, что если смешать ди-
хлордифторметан с трихлорфторметаном (R11), то полученный
смешанный пропеллент будет иметь более низкое давление пара
[для смеси 50:50 оно равно 250 КН/м2 (2,5 атм)], которое поз-
Б. П роизводство и применение хлорфторуглеводородов
673
воляет создать безопасные в обращении упаковки с использо-
ванием легких жестяных банок наподобие тех, которые уже
употреблялись для расфасовки пива. Это открытие положило
начало развитию аэрозольного производства, каким мы видим
его сейчас,—-производства, которое в 1972 г. выпустило свыше
2700 млн. упаковок в США и 360 млн. в Великобритании *.
Схема аэрозольной упаковки приведена на рис. 17.4. Аэрозоль-
ная технология добилась к настоящему времени значительных
Рис. 17.4. Принципиальное устройство аэрозольного баллона.
/—корпус; 2—сифонная трубка; 3—клапан; 4—распылительная головка
успехов, которые нашли свое отражение в нескольких учебни-
ках [1—4], однако основные принципы этой технологии остались
по существу без изменения. Рабочее давление в аэрозольной
упаковке создается либо давлением паров сжиженного газа,
либо за счет сжатого газа. На практике употребляются сжи-
женные газообразные углеводороды или хлорфторуглеводо-
роды, причем выбор пропеллента зависит от ряда факторов,
в том числе рецептуры состава, совместимости, безопасности
* В США в 1973 г. было произведено 2902 млн. аэрозольных упаковок, в
1974 г. — 2722 млн., в 1975 г. — 2354 млн., в 1976 г. (прогноз)—2700 мли.
[Chem. Market Abstr., 67, 265 556 (1975); Chem. Week, 118, № 21, 29 (1976)].
Мировое производство аэрозольных упаковок в 1975 г. составило 9 млрд,
штук [Chem. Week, 118, № 19, 48 (1976)]. — Прим, перев.
22 Зак. 501
674
Глава 17
и экономических соображений. Сжатые газы (азот, двуокись
углерода, закись азота) применяются гораздо реже, поскольку
их рабочее давление падает по мере расходования продукта
и увеличения объема парового пространства.
В случае пропеллентов типа сжиженных газов такое паде-
ние давления не имеет места, так как, несмотря на постепенное
испарение жидкого пропеллента для заполнения все возрас-
тающего парового пространства, давление пара над сжиженным
газом остается постоянным вплоть до тех пор, пока не испа-
рится последняя капля жидкости.
Обычно основным пропеллентом служит дихлордифторме-
тан, давление пара которого понижают до приемлемого уровня
с помощью трихлорфторметана или других летучих растворите-
лей — таких, как этиловый спирт, хлористый метилен, метил-
хлороформ и т. и. Однако некоторые составы содержат
значительное количество воды (например, кремы для бритья, оде-
колоны, духи и некоторые фармацевтические продукты). Три-
хлорфторметан и хлорорганические растворители склонны к гид-
ролизу, и поэтому вместо них в подобных случаях для понижения
давления CC12F2 применяют 1,2-дихлортетрафторэтан (R114).
Единственные хлорфторуглеводороды, используемые в каче-
стве аэрозольных пропеллентов, — это CCI2F2, CC13F и CC1F2—
CC1F2. Однако в специальных случаях употребляются и другие
соединения. В частности, CHC1F2 (R22) находит весьма огра-
ниченное применение в аэрозольных составах, требующих по-
вышенного давления, a CC1F2—CF3 (R115) и цикло-С.^ допу-
щены Федеральным управлением по контролю за качеством
фармацевтических продуктов США к использованию в аэро-
зольных продуктах питания, таких, как взбитый крем.
Некоторое представление о разнообразии ассортимента и
основных категориях аэрозольных продуктов, выпускаемых в
Великобритании (где производство аэрозолей является само-
стоятельной отраслью промышленности, объем продукции кото-
рой в настоящее время * составляет около 7 упаковок в год на
душу населения), дает табл. 17.5.
Приведенные в табл. 17.5 широкие категории аэрозольных
продуктов охватывают более 300 различных типов составов
приблизительно 2000 наименований.
Растворители
1,1,2- Трихлортрифторэтан (R113) является очень мягким
(или, как часто говорят, «селективным») растворителем, при-
годным для снятия масел, смазки и тому подобных загрязнений
* В 1972 г. — Прим, перев.
Б. Производство и применение хлорфторуглеводородов
675
Таблица 17.5
Области применения хлорфторуглеводородных аэрозолей
в Великобритании (данные 1972 г.)
Категория продуктов Производство, млн. шт./год Доля в общем выпуске, %
Средства для ухода за волосами 117 32,2
Дезодоранты и средства против пота 53 14,6
Одеколоны и духн 16 4,4
Кремы для бритья 11 3,0
Прочие средства личной гигиены 3,5 1,0
Полировочные составы 34,5 9,5
Инсектициды 26 7,8
Освежители воздуха 23 6,4
Средства для чистки духовок 7 1,9
Средства для подкрахмаливания белья 5,5 1,6
Прочие бытовые средства 13 3,6
Фармацевтические средства 11 3,0
Средства для ухода за автомобилями 9,5 2,6
Краски и другие покрытия 15,5 4,3
Средства промышленного назначения 8 2,2
Прочие 7 .1,9
360,5
с большинства пластических материалов. Поэтому он может
быть использован в электротехнической и электронной промыш-
ленности, где существуют многочисленные проблемы, связан-
ные с очисткой поверхностей контакта металлов с пластмас-
сами. Другая область применения 1,1,2-трихлортрифторэтана —
это точное машиностроение, в частности производство авиакос-
мической и военной техники, где особенно важны такие свойства
растворителей, как химическая стойкость, отсутствие корроди-
рующего действия и способность легко испаряться, не оставляя
остатка. В большинстве случаев степень чистоты трихлортри-
фторэтана, отвечающая квалификации «хладоагентный», яв-
ляется достаточной, однако в США при проведении программы
космических исследований в значительных количествах приме-
няется еще более чистый препарат (квалификации «white
room»).
Кроме того, 1,1,2-трихлортрифторэтан употребляется для
химической чистки одежды. Мягкость растворяющего действия
и низкая температура кипения делают это соединение весьма
подходящим для обработки нежных или трудно очищаемых из-
делий— таких, как изделия из меха, замши и термически не-
стойких тканей. Для этой цели находит применение также RU,
который имеет несколько более высокую растворяющую способ-
ность. Последнее позволяет использовать его вместо перхлор-
22*
676
Глава 17
этилена, который широко применяется в настоящее время при
химической чистке одежды. Трихлорфторметан служит также
обычным растворителем для промывки холодильных систем
(особенно в процессе технического обслуживания или после
ремонтных работ) перед их заполнением свежим хладоаген-
том — чаще всего CCI2F2 или CHCIF2. Преимущества CCI3F в
данном случае заключаются в том, что он хорошо растворяет
масло и смазку, не действует на пластмассы, используемые в
герметичной конструкции двигателя, и легко удаляется путем
эвакуирования системы.
Вспениватели
Реакция между изоцианатами и многоатомными спиртами,
применяемая в производстве эластичного пенополиуретана, со-
провождается выделением тепла, которое может быть использо-
вано для испарения летучей жидкости, добавленной к исходным
реагентам, с целью образования пены. В качестве летучей жид-
кости употребляется трихлорфторметан (R11), причем его
можно либо заранее смешивать с многоатомным спиртом, либо
вводить в реакционную смесь в момент ее загрузки в заливоч-
ную машину. Вспененную реакционную массу пропускают затем
через нагретый тоннель для завершения реакции. Трихлорфтор-
метан не принимает участия в химическом процессе — это чисто
физический вспенивающий агент.
В случае жестких полиуретановых пен, которые обычно ис-
пользуются для обеспечения теплоизоляции и имеют структуру
с замкнутыми порами, применение хлорфторуглеводородных
вспенивателей дает дополнительное преимущество: пары хлор-
фторуглеводородов обладают меньшей теплопроводностью, чем
другое обычное вспенивающее вещество — двуокись углерода.
Успешно выполняет функцию вспенивателя трихлорфторметан
(R11) в комбинации с вспомогательными реагентами, подобран-
ными таким образом, чтобы получилась жесткая пена с замк-
нутыми порами. Для получения вспененной массы непосредст-
венно в формах применяется так называемый метод ступенча-
того вспенивания с использованием дихлордифторметана (R12).
В этом методе в полую форму вспрыскивается предварительно
вспененная жидкая композиция, имеющая консистенцию мыль-
ной пены при бритье, которая образуется в результате очень
быстрого испарения дихлордифторметана на выходе из смеси-
теля. Такой способ обеспечивает проникновение пеномассы даже
в самые труднодоступные участки формы и позволяет понизить
давление в форме благодаря уменьшению необходимой степени
расширения массы. Дихлордифторметан применяется также в
производстве поливинилхлоридной пены по пластизольному ме-
тоду. В данном случае CCI2F2 вводится под давлением в поли-
Б. Производство и применение хлорфторуглеводородов
677
винилхлоридный пластизоль, который удерживают под давле-
нием вплоть до момента его заливки на подложку. Происходя-
щее при этом понижение давления до атмосферного вызывает
увеличение объема паров CCI2F2 и вспенивание пластизоля. При
последующем пропускании пеномассы через нагретый тоннель
протекает дальнейшее расширение и отверждение ПВХ.
Еще одним полимером, который подвергают вспениванию в
форме листов и плит с помощью CChF, является полистирол.
Этот процесс несколько напоминает процесс вспенивания ПВХ:
дихлордифторметан смешивают под давлением с жидким поли-
мером, а затем сбрасывают избыточное давление через головку
экструдера. Полученный пенополистирол может иметь самую
различную форму — от 70-миллиметровых плит, используемых
в качестве конструкционного теплоизолирующего материала, до
тонких листов толщиной несколько миллиметров, которые при-
меняются в производстве упаковочных материалов методом
термоформования.
Огнегасящие агенты
Все хлорфторуглеводороды не горят и в той или иной сте-
пени способны гасить огонь. Однако чаще всего в качестве ог-
негасящих агентов используются бромсодержащие соединения,
приведенные в табл. 17.6.
Таблица 17.6
Индекс при R Формула Т. кип., °C Торговое наименование
114В2 CBrF2—CBrF2 47,3 Хейлон 2402. флуобрен
12В1 CBrCIF, —4,0 Хейлон 1211, BCF
13В1 CBrF3 —57,8 Хейлон 1301 ВТМ
Эффективность огнегасящего агента очень сильно зависит
от устройства огнетушителыюй системы в целом и от таких
факторов, как рабочее давление, скорость выпуска л конструк-
ция сопла. Поэтому трудно сказать, какой агент вообще будет
наилучшим в данном конкретном случае. Все перечисленные
выше бромфторуглеводороды представляют собой огнегасящие
вещества типа испаряющейся жидкости, однако по способу при-
менения они несколько различаются между собой.
Благодаря низкой температуре кипения и связанному с этим
высокому давлению пара бромтрифторметан (RI3B1) достигает
пламени в форме пара. Последнее обстоятельство наряду с
практическим отсутствием токсических свойств делает это
678
Глава 17
вещество весьма подходящим для использования в системах
«полного затопления», когда средство пожаротушения фиксиро-
вано в стационарном положении и автоматически срабатывает
при повышении температуры сверх определенного уровня. Пер-
воначально такие системы устанавливались в отсеках авиацион-
ных двигателей, а в настоящее время их все шире применяют и
для пожарной защиты гражданских объектов — в художествен-
ных галереях, музеях, библиотеках и вычислительных центрах.
Наиболее важной причиной применения бромтрифторметана во
всех этих случаях является то, что тушение пожара водой может
принести не меньше убытков, чем сам пожар, тогда как газооб-
разное огнегасящее вещество не оказывает никакого вредного
воздействия. К тому же всегда существует возможность случай-
ного срабатывания стационарной системы пожаротушения или
утечки огнегасящего агента, вследствие чего нетоксичность по-
следнего имеет очень большое значение.
Самым распространенным огнегасящим веществом типа
испаряющийся жидкости в Великобритании является CBrClF2
(R12B1), который обычно обозначается символом BCF [от анг-
лийского Bromo-Chloro-di-Fluoro-methane (бромхлордифторме-
тан)]. Он не слишком быстро испаряется при выпускании в
атмосферу и достигает пламени в форме облака паров, содержа-
щего капельки жидкости. Это повышает вероятность приникно-
вения огнегасящего агента к центру пожара. Такие вещества
хорошо подходят для ручных и передвижных средств пожаро-
тушения, особенно при борьбе с загораниями бензина и других
горючих жидкостей. Кроме ручных и передвижных огнетуши-
телей, CBrC!F2 используется в стационарных системах пожаро-
тушения, которыми оборудуются самолеты, электроагрегаты,
машинные отделения и т. д. Это подтверждает сделанное выше
утверждение, что можно говорить о пригодности применения в
той или иной ситуации только всей системы пожаротушения в
целом, а не отдельно взятого огнегасящего агента.
Химические полупродукты
Хлордифторметан (R22) служит исходным веществом для
получения тетрафторэтилена
600 °C
2CHC1F2 ----> CF2=CF2 + 2НС1
Полимеризация тетрафторэтилена описана в гл. 8. Другой фтор-
полимер вырабатывается на основе хлортрифторэтилена, кото-
рый получают обработкой 1,1,2-трихлортрифторэтана (R113)
цинковой пылью в среде этанола
CC12FCC1F2 —> CC1F=CF2 + C12
В. Производство и применение хлорфторуглеводородов 679
Транспортировка и хранение
Очень низкая химическая активность хлорфторуглеводородов
и жесткие требования к ним по степени чистоты позволяют со-
вершенно безопасно хранить и перевозить эти вещества в бал-
лонах из малоуглеродистой стали.
Транспортировка хлорфторуглеводородов в больших емко-
стях ограничивается главным образом поставками для .пред-
приятий, производящих аэрозоли, куда они поступают в желез-
нодорожных и автомобильных цистернах емкостью, например,
20 т. В Великобритании хлорфторуглеводороды перевозятся
железнодорожными цистернами только при продаже их на эк-
спорт, однако в континентальной Европе и в США этот способ
транспортировки является обычным.
Хлорфторуглеводороды химически инертны, и для их транс-
портировки и хранения можно использовать почти все без ис-
ключения обычные материалы. Из металлов не рекомендуется
употреблять только цинк, оцинкованную сталь, магний и сплавы
с большим содержанием цинка или магния. Хотя некоторые
пластмассы и эластомеры подвергаются воздействию ряда хлор-
фторуглеводородов, имеется достаточный ассортимент этих ма-
териалов, пригодных для изготовления прокладок, сальников
и т. п.
В этой главе была сделана попытка дать читателю общее
представление о технических достижениях, которые явились
результатом открытия примерно 40 лет назад первых хлорфтор-
углеводородных соединений. Эти вещества не только все шире
используются в традиционных сферах своего применения, но и
находят новые, неизвестные ранее области использования. По-
мимо специфических физических свойств, которые определяют
выбор конкретных соединений для тех или иных целей, в основе
успехов промышленности хлорфторуглеводородов лежат такие
важные преимущества веществ этого класса, как негорючесть,
очень низкая токсичность, химическая устойчивость и низкая
реакционная способность. Несомненно, что перечисленные фак-
торы будут играть первостепенную роль и в дальнейшем рас-
ширении применения хлорфторуглеводородов.
СПИСОК ЛИТЕРАТУРЫ
1. Herzka A., Pickthall J., Pressurised Packaging (Aerosols), Butterworth,
London, 1961.
2. Shepherd H. R., Aerosols: Science and Technology, Interscience, New
York, 1961.
3. Herzka A., International Encyclopaedia of Pressurised Packaging (Aero-
sols), Pergamon Press, Oxford, 1966.
4. The Aerosol Handbook, Wayne and Dorland and Co., 1972.
Список дополнительной литературы*
ОБЩАЯ ЛИТЕРАТУРА
Лебедев Н. И., Химия и технология основного органического и нефтехи-
мического синтеза, «Химия», М., 1975.
Паушкин fl. М., Адельсон С. В., Вишнякова Т. П., Технология нефтехими-
ческого синтеза, ч. I—II, «Химия», М., 1973—1975.
К гл. 1
Бахирко Б. А., Экономика химической промышленности, «Высшая школа»,
М., 1975.
К гл. 2
Дубин 3. Ю., Технология лесохимических производств, «Лесная промыш-
ленность», М., 1968.
Шарков В. И.,Сапотницкий С. А., Дмитриева О. А., Туманов И. Ф., Тех-
нология гидролизных производств, «Лесная промышленность», М., 1973.
Тютюнников Б. Н., Науменко П. В., Товбин И. М„ Фаниев Г. Г., Техно-
логия переработки жиров, «Пищевая промышленность», М., 1970.
Литвиненко М. С., Химические продукты коксования (Производство и ис-
пользование), «Техника», Киев, 1974.
Альтшулер В. С., Новые процессы газификации твердого топлива, «Нед-
ра», М„ 1976.
Суханов В. П., Переработка нефти, «Высшая школа», М., 1974.
Федоров В. С., Нефтеперерабатывающая и нефтехимическая промышлен-
ность СССР в девятой пятилетке, «Химия», М., 1976.
К гл. 3
Суханов В. П., Переработка нефти, «Высшая школа», М., 1974.
Магарил Р. 3., Теоретические основы химических процессов переработки
нефти, «Химия», М., 1976.
Черный И. Р., Производство мономеров и сырья для нефтехимического
синтеза, «Химия», М., 1973.
Брунштейн Б. А., Клименко В. Л., Цыркин Е. Б., Производство синтети-
ческих кислот из нефтяного и газового сырья, «Химия», Л., 1970.
К гл. 4
Фальбе Ю., Синтезы на основе окиси углерода, «Химия», Л., 1971.
Миллер С. А., Ацетилен, его свойства, получение и применение, т. 1,
«Химия», Л., 1969.
Список дополнительной литературы составлен переводчиком книги.
Список дополнительной литературы
681
Бобков С. С., Смирнов С. К., Синильная кислота, «Химия», М„ 1970.
Черный И. Р., Производство мономеров и сырья для нефтехимического
синтеза, «Химия», М., 1973.
Мономеры для поликонденсации, под ред. Стилла Дж. К. и Кемпбел-
ла Т. В., «Мир», М., 1976.
Синтетический каучук, под ред. Гармонова И. В., «Химия», Л., 1976.
К гл. 5
Сулимов А. Д., Производство ароматических углеводородов из нефтяного
сырья, «Химия», М., 1975.
Молдавский Б. Л., Кернос Ю. Д., Малеиновый ангидрид и малеиновая
кислота, «Химия», Л., 1976.
К гл. 7
Харлампович Г. Д., Чуркин Ю. В., Фенолы, «Химия», М., 1974.
К гл. 8
Стрепихеев А. Л-, Деревицкая В. Д., Основы химии высокомолекулярных
соединений, «Химия», М., 1976.
Технология пластических масс, под ред. Коршака В. В., «Химия», М.,
1976.
Чирков Н. Матковский П. Е., Дьячковский Ф. С., Полимеризация на
комплексных металлоорганических катализаторах, «Химия», М., 1976.
Фойгт И., Стабилизация синтетических полимеров против действия света
и тепла, «Химия», Л., 1972.
Соболев В. М., Бородина И. В., Промышленные органические каучуки,
«Химия», М., 1977.
Синтетический каучук, под ред. Гармонова И. В., «Химия», Л., 1976.
К гл. 9
Павлов С. А., Шестакова И. С., Касьянова А. А., Химия и физика высо-
комолекулярных соединений в производстве искусственной кожи, кожи и
меха. «Легкая индустрия», М., 1976.
Роговин 3. А., Основы химии и технологии химических волокон, т. 1 и 2,
«Химия», М., 1974.
К гл. 10
Степанов Б. И., Введение в химию и технологию органических красителей,
«Химия», М., 1977.
Беленький Е. Ф., Раскин И. В., Химия и технология пигментов, «Химия».
Л., 1974.
К гл. 11
Мелентьева Г. А., Фармацевтическая химия, т. 1 и 2, «Медицина», М.,
1976.
Рубцов М. В., Байчиков А. Г., Синтетические химико-фармацевтические
препараты, «Медицина», М., 1971.
Машковский М. Д., Лекарственные средства, ч. 1 и 2, «Медицина», М.,
1972.
682
Список дополнительной литературы
К гл. 12
Мельников Н. Н., Химия и технология пестицидов, «Химия», М., 1974.
Системные фунгициды, «Мир», М., 1975.
Майер-Боде Г., Гербициды и их остатки, «Мир», М., 1972.
Стонов Л. Д., Дефолианты и десиканты. Химические средства предубо-
рочного удаления листьев и высушивания сельскохозяйственных растений,
«Химия», М., 1973.
Мельников Н. И., «Синтетические регуляторы роста растений и герби-
циды», Усп. химии, 45, 1473 (1976).
К гл. 13
Неволин Ф. В., Химия и технология синтетических моющих средств, «Пи-
щевая промышленность», М., 1971.
Абрамзон А. А., Поверхностно-активные вещества. Свойства и примене-
ние, «Химия», Л., 1975.
Тютюнников Б. Н., Науменко И. В., Товбин И. М., Фаниев Г. Г., Техно-
логия переработки жиров, «Пищевая промышленность», М., 1970.
К гл. 14А
Проскуряков В. А., Авербух А. Я., Химия и топливо, «Знание», М., 1973.
Соколик А. С., Самовоспламенение, пламя и детонация в газах, Изд-во
АН СССР, М„ 1960.
Михеев В. П., Медников Ю. П., Сжигание природного газа, «Недра», Л.
1975.
Мержанов А. Г., «Проблемы горения в химической технологии и метал-
лургий», Усп. химии, 45, 827 (1976).
К гл. 14Б
Горст А. Г., Пороха и взрывчатые вещества, «Машиностроение», М.,
1972.
Светлов Б. Я-, Яременко Н. Е., Теория и свойства промышленных взрыв-
чатых веществ, «Недра», М., 1973.
К гл. 15
Калмыков П. Е., Логаткин М. Н., Современные представления о роли со-
ставных частей пищи, «Медицина», Л., 1974.
Трисвятский Л. А., Лесик Б. В.. Курдина В. Н., Хранение и технология
сельскохозяйственных продуктов, «Колос», М.. 1975.
Тютюнников Б. Н., Науменко П. В., Товбин И. М., Фаниев Г. Г., Техно-
логия переработки жиров, «Пищевая промышленность», М„ 1970.
Ауэрман Л. Я-, Технология хлебопекарного производства, «Пищевая про-
мышленность», М., 1972.
К гл. 16
Райт Р. Наука о запахах, «Мир», М., 1966.
К гл. 17А
Основы технологии светочувствительных материалов, под ред. Шебер-
стова В. И., «Химия», М.. 1977.
Миз К., Джеймс Т„ Тебрия фотографического процесса. «Химия», Л., 1973.
Предметный указатель
Абсолю 616, 617
АБС-сополимеры 111, 123, 262
Адамантанамин 418
Адипиновая кислота 146—147, 318,
338
Адипонитрил 127, 147
Адреналин 471
Адреноблокаторы 458
Азелаиновая кислота 24, 25, 127
Азид свинца 590, 591, 597
Азотистый иприт 422
Акриламид 111, 311, 332, 356
Акриловая кислота 95, 117, 161, 356,
357
Акрилонитрил 111, 112, 205 и сл., 331,
341
получение 95, 111, 170 и сл., 206
и сл.
статистические данные 205, 209
экономика производства 208—
210
Акрихин см. Мепакрин
Акролеин 116 и сл., 171 и сл., 178
Ализарин 359, 378
Алкидные смолы 148, 152
Алкилбензолсульфонаты 148, 519, 521
и сл., 529
Алкилирование 42
бензола 102, 115, 144, 148, 158,
521, 523-524
фенолов 129, 130
Аллиламин 116
Аллиловый спирт 116, 117
Альдикарб 483, 484
Альдрин 479
Алюминийалкилы 108, 119, 123, 124,
526—527, 528—529
Амантадин см. Адамантанамин
Амидопирин 445, 446
Аминазин см. Хлорпромазин
е-Аминокапроновая кислота 226, 234,
450
8-Амино-1 -нафтол- 3,6-дисульфокисло-
та см. Аш-кислота
Аминоптерин 425
n-Аминосалициловая кислота 415
Амитриптилин 463
Амитрол 503
Аммиак 85, 170 и сл., 177—179, 207,
303, 307, 377
Ампициллин 410
Ампролий 401, 437
Амфетамин 471
Анаприлин см. Пропранолол
Анидекс 341
Анилин 149, 226, 361
Антипирены 311
Антипирин 445
Антрахинон 377, 378
Антуран см. Сульфинпиразои
Апрессин см. Гидралазин
Ароматические углеводороды 33—35,
39—41, 57, 71—73, 132 и сл.
статистические данные 35, 133
Аспирин 398, 444
АС-сополимеры 111, 262, 336
А-Телл 330
Атразин 501
Ауреомицин см. Хлортетрациклин
Ацетазоламид 474
Ацетальдегид 77, 80, 97, 102, 106, 155,
177—178, 207
получение 95, 104 и сл.
Ацетилен 91 и сл.
получение 34, 35, 53, 67, 68, 91
реакции 92, 93, 95, 96, 123, 169,
192, 206, 625, 628
Ацетилсалициловая кислота см. Аспи-
рин
Ацетилцеллюлоза 315 и сл., 660
Ацетогексамид 455
Ацетон 15, 29, 79, 210 и сл.
получение 112—113, 157 и сл.,
166,. 190—191, 204, 210 и сл.
реакции 96, 215 и сл.,
статистические данные 213—215
Ацетонитрил 121, 174
Аш-кислота 365, 366, 375
Барбан 502
684
Предметный указатель
Бензидин 365, 374
Бензин 16, 37, 39, 42, 71, 134—135,
553, 554
Бензойная кислота 103, 202, 223
Бензол 33, 74, 132, 135
алкилирование 102, 115, 144,148,
158, 521, 523—524
гидрирование 146,203
прочие реакции 148, 168, 200,201,
203
статистические данные 35, 133,
145, 146
Беном ил 493, 494
Бетазин 424
Бигумаль см. Прогуанил
Биквииолат 438
Биомицин см. Хлортетрациклин
Бластидин S 494—495
Бретилий 458
Бутадиен 50, 67, 121 и сл.
Бутадион см. Феиилбутазои
Бутамид см. Толбутамид
Бутан 48, 69, 77 и сл., 86, 130—131,
147, 163—164, 212
Бутандиол-1,4 95, 330
Бутанол-1 15, 106, 112, 135, 210
Бутанол-2 131, 191
трет-Бутанол 130
Бутанои-2 см. Метилэтилкетон
Бутилены 121, 122, 128, 130—131
Бутилкаучук см. Полиизобутилен
Ванилин 27, 635, 637
Варфарин 452
Верел 333
Версалид 630—631
Виктория голубой 387
Винилацетат 92, 106 и сл., 166, 332,
345
Винилацетилен 92—94
Вииилиденхлорид 94, 333, 345, 347
2-Винилпиридин 177, 357
Винилхлорид см. Хлористый винил
Виньон 282, 345
Витавакс см. Карбоксин
Водород 86, 143
Волокна 282 и сл.
альгинатные 312
ацетатные 315 и сл., 369
белковые 286, 295, 312, 355
бикомпоиентиые 320, 332—333
вискозные 312 и сл., 357
гидратцеллюлозные 27, 312, 353
из натурального каучука 336—
337
медноаммиачные 312
модакриловые 331, 333
- МП 345
полиакрилонитрильные 331 и сл.,
352, 354, 379, 387, 389
полиамидные 312 и сл., 356, 371
поливинилиденхлоридные 344 и
сл.
поливинилспиртовые 341 и сл.,
353
поливинилхлоридные 344 и сл.
полинозные 314
полиолефиновые 333 и сл., 354
полипропиленовые 334 и сл.
полиуретановые 337 и сл.
полиэтиленовые 333—334
полиэфирные 325 и сл., 354, 356,
369, 371
ПЦ 282
статистические данные 283
термостойкие 348
углеродные 352
эластомерные 336 и сл.
Вулканизация 109, 278 и сл., 337
Газойль 39, 49, 553, 554
Галоперидол 461
Галотан 470
Гекогенин 441
Гекса меТилендиамин 127, 147, 318
324, 418
Гексаметилентетрамин 87, 272, 275,
597
Гексамидии см. Примидон
Гексан 46, 80
Гексахлорциклогексан 170, 479
Гексахлорэтан 168, 435, 666, 668
Гексиленгликоль 215
Гексоген 586, 595
Гераниол 616, 617, 621, 625, 634
Гидралазин 457
Гидратация 93—94, 102, 112, 130,
131, 186 и сл.
Гидрирование 127, 147, 149, 220, 223,
226
Гидрогенизация 36, 528, 602—603
Гидродеалкилирование 143, 179
Гидрокрекинг 42, 50, 69, 72
Гидроксиламин 220—222, 332
Гидроперекись
кумола 115, 157—159, 201, 345
трет-бутила 114
циклогексила 146
этилбензола 103
Гидроформилирование 89, 112, 180
и сл., 529
Гидрохинон 649 и сл.
Предметный указатель
685
Гидрохлорирование 92, 93
Гидроцианирование 95, 128, 205 и сл.,
218
Гикантон 434
Глицерин 15, 23, 24, 116, 117, 152,
517—518, 549, 613, 637
Глутамат натрия 31—32, 111, 642—
643
Графитация 352—353
Гризеофульвин 421
Гуанетидин 458
Гуттаперча 277
Дайнел 333
Далапон 500
Дапсон 416
Дегидрирование 42, 72, 74, 76, 103,
104, 112, 130—131, 203, 211
Дегидрохлорирование 97, 102, 127
Дегидроциклизация 42, 72, 76
Декаметилендиамин 324
Дексон 331
Десметрин 501
Детонация
в двигателях 567—568
взрывчатых веществ 588 и сл.
Диазепам 466—467
Диазинон 481—482
Диакарб см. Ацетазоламид
Диафенилсульфон см. Дапсон
Дигидралазин 457
Диизобутилен 89, 129
Диизоцианаты см. Изоцианаты
Дикамба см. 2-Метокси-3,6-дихлор-
бензойная кислота
Днкват 502—503
Дикумарин см. Дикумарол
Дикумарол 451
Дильдрин 479
Диметилтерефталат 152 и сл., 326
Дйметиримол 491—492
Димидий 432
2,4-Динитрофторбензол см. Реактив
Сенджера
Диносеб 496
N, N'-Диоксиметилмочевина 308, 314
Диосгенин 443
Дифенилгидантоин 470
Дифенилолпропан 159, 205, 215, 218,
269
Дихлобенил 503, 504
Дихлордифенилтрихлорэтан (ДДТ)
150, 479
Дихлордифторметан 667, 670, 672,
674, 676—677
Дихлорофос 481
1,2-Дихлортетрафторэтан 667, 668,
674
Дихлорэтан 97, 100, 101, 169, 193 и сл.
Дихлофлуанид 489
Диэтиловый эфир 102, 189
Диэтилпропион 471
Додин 490
L-Допа 473
Дофа см. L-Допа
Древесина 26 и сл., 77
«Закалка» реакционных газов 65,68
Ибупрофен 448
Ибуфенак 448, 449
Изобутнлеи 42, 89, 121, 128—130,
242—243
Изодеканол 121
Изониазид 414
Изооктанол 129, 185
Изопрен 50, 96, 118—119, 129—130
Изопропанол 112, 113, 117, 187 н сл.,
211, 218, 532
Изопропилбензол см. Кумол
Изофталевая кислота 156, 328
Изоцианаты 88, 149, 275—276, 306,
338, 357
Имизин см.-Имипрамин
Имипрамнн 463
Индиго 360, 384—385
Индометацин 447 и сл.
Индолилуксусная кислота (ИУК)
496—497, 505
Инсулин 454
Интал 400
Иоксинил 504
Иононы 621, 625, 626 и сл.
Ипразид см. Ипрониазид
Ипрониазид 463
Ироны 628
Кадоксен 303
Канекалон 333
Капролактам 205, 219 и сл., 227—228,
318
Каптан 489
Карбарил 483
Карбид кальция 34, 91
Карбоксин 490—491
Карбонизация 351, 352, 353
Карбофос см. Малатион
Карбутамид 455
Касторовое масло 25, 323
Касугамицин 494—495
686
Предметный указатель
Катализаторы
бифункциональные 69, 76
гидроформилирования 111, 180
и сл.
дезактивация 74
координационные 243
олигомеризации бутадиена 123
и сл.
Филлипса 255
Циглера — Натта 243, 254—255,
257, 280
Катапресан см. Клонидин
Каучуки
бутадиеннитрильный 111, 122
бутадиеновый 122
бутадиенстирольный 122, 262, 279
натуральный 28, 29, 276 и сл.,
336—337
нитрильный 205
статистические данные 29
этиленпропиленовый 109, 126,280
Киана 295, 324—325
Клонидин 456—457
Клофибрат 454
Кодель 329
Компаундирующие добавки 248 и сл.,
258
Кокаин 471
Конго красный 374
Конессин 429
Конкрет 616
Корделан 346
Кортизон 440 и сл.
Красители 362 и сл.
активные 389 и сл.
анионные 371 и сл.
антрахиноновые 377 и сл., 380
и сл.
дисперсные 346, 368 и сл., 379,
389
индигоидные 384 и сл.
катионные 361, 379
кислотные 336, 356, 371 и сл.,
379, 387
кубовые 380 и сл.
ледяные 376
основные 356, 373—374, 387, 389
природные 359—360
протравные 359
прямые (субстантивные) 315,
374—375, 376, 379
сернистые 388—389
тиоиндигоидные 385 и сл.
триарилметановые 386—387, 419
фталоцианиновые 387—388
Кианиновые 389
Крашение
кубовое 360, 385
ледяное 376
протравное 359
Крекинг 41, 42, 44, 50, 68, 69
Креозот 33, 37, 496
Критическая концентрация мицелло-
образования 510—511, 533
Ксикаин см. Лидокаин
Ксилокаин см. Лидокаин
Ксилолы 33, 35, 132 и сл., 150 и сл.
Ксилохолин 458
Кумол 115, 132—133, 144, 157—158,
165
Куоксен 303
Лайкра 340
Леавиль 346
Левомицетин см. Хлорамфеникол
Либриум см. Хлордиазепоксид
Лизергиновая кислота 607—608
Лимонная кислота 15, 31
Линалоол 617, 620, 625, 626
Линкомицин 413
Линолевая кислота 23, 604, 609
Люкантон 434
Люминал см. Фенобарбитон
Макролиды 413
Макроциклические кетоны 628 и сл.
Малеиновый ангидрид 147—148
Малатион 481, 482
Малахитовый зеленый 386, 419
Манеб 488
Мебедрол см. Орфенадрин
Меласса 27, 30, 31
Мепакрин 427
Мепробамат 456, 466
Мепротан см. Мепробамат
Метадон 465—466
Метаквалон 469
Метан 46 и сл., 84 и сл., 143, 164,
169
Метанол 29, 77, 86 и сл., 91, 156,258,
342, 611
Метилбензокват 438
Метилвиниловый эфир 95, 179
2-Метил-5-винилпиридин 177, 179
а-Метилдопа 457, 473
Метилдофа см. а-Метилдопа
Метиленовый голубой 426
Метилметакрилат 215, 218, 233, 245,
247, 266
N-Метилпирролидон 95, 121, 136
Предметный указатель
087
2-Метил-4-хлорфеноксимасляная кис-
лота 498
2-Метил-4-хлорфеноксиуксусная кис-
лота 497, 498
Метилэтилкетон 79, 131, 155, 166, 191
2-Метил-5-этилпиридин 178—179
Метициллин 410
Метол 649—650
Метотрексат 425
Мефенезин 466
Мефолин см. Фенметразин
Мидантан 418
Мирцен 624
Мовеин 361
Модифицирование
вискозных волокон 314—315
коллагена 298
поливинилспиртовых волокон
343—344, 353
полистирола 261
синтетических волокон 353 и сл.
хлопка 305 и сл.
шерсти 289 и сл.
Молекулярные сита 44, 69, 102,522—
523
Молочная кислота 15, 31
Мономицин см. Парамомицин
Монурон 500—501
Морфин 402, 464
Мочевиноформ альдегидные смолы 87,
274—275
Мыло 24, 248, 516 и сл.
Набам 488
Надуксусная кислота 113, 225
Найлон-3 321—322
Найлон-4 321—322
Найлон-6 219, 282, 317, 318, 319, 320,
321 355 357
Найлон-6,6 112, 219, 282, 317, 318 и сл.,
324—325, 355
Найлон-6,10 25, 323
Найлон-7 321, 323
Найлон-9 323
Найлон-11 25, 321, 323
Найлон-12 126, 321, 323
Налидиксовая кислота 408, 438
Натрийкарбоксиметилцеллюлоза 515,
519
Нафта 18, 39, 49
гидрокрекинг 69
конверсия с водяным паром 85
окисление 77 и сл., 165, 212
пиролиз 50, 65, 66, 122, 195
Нафталин 33, 34, 35, 150, 363
Нафтол АС 376
Нафтол АТ 376
Нерол 616, 617, 621, 625
Ниридазол 430
Нитразепам 470
Нитрат аммония 593, 597
Нитроглицерин 587, 597
Нитрозилсерная кислота 223—225
Нитроцеллюлоза 229, 587, 597
Нитрофен 503, 504
Нитрофурантоин 408—409
Новокаин см. Прокаин
Новолачные смолы 271
Окисление 161 и сл., 212
акролеина 117
бензола 147—148, 203
бутана 77 и сл., 86
бутиленов 131, 166
ксилолов 151, 152 и сл., 156
кумола 157—158, 165
метана 84, 164
пропилена 104, 113, 116—117,166,
178, 213
толуола 202
циклогексана 146—147, 165, 223
этанола 104
этилбензола 103
этилена 100, 104 и сл., 107, 165—
166
Окислительное дегидрирование 163,
211
Окислительное хлорирование 97, 163,
168
бензола 168, 201
этилена 101, 168, 196—197
Окислительное цианирование 162,208
Окислительный аммонолиз 162, 171
и сл.
пропана 207
пропилена 111, 170 и сл., 205—
206
Окись
бутилена 131
мезитила 159, 216
пропилена 113 и сл.
углерода 36, 85, 86, 88, 169, 338
этилена 97, 100—101, 164, 205,
207, 534 и сл.
Оксазепам 467—468
Оксикарбоксин 491
Оксиклозонид 435
Окситетрациклин 412, 429—430
«Оксоспирты» 112
Октадин см. Гуанетидин
Октановое число 41, 42, 43, 70 и сл.,
134
688
Предметный указатель
Олеиновая кислота 23, 24, 28
а-Олефнны 58, 67, 108 и сл., 523—
524, 526—527, 529
Оптические отбеливатели 543—544
Орнид см. Бретилий
Орфенадрнн 472
Отверждение 273 и сл.
Отто 616, 617
Пальмовое масло 23, 516
Памаквин 426—427
Паракват 502—503
Паральдегид 178, 469
Парамомицнн 430
Паратион 481, 482
Паргилин 464
Пенициллины 409
Пентазоцин 464—465
Пентан 46, 57, 80, 169
Пентахлорэтан 97, 169
Пентаэритрит 87, 106, 152
Переносчики красителя 327, 370
Персистентность 478
Петидин 461
Пигменты 361
Пимарицин 421
а-Пинен 28
(З-Пинен 28, 624
Пиперидин 177, 179
Пиперилен 119
Пиразинамид 414
Пнразои 504
Пирантел 436—437
Пиретрины 484
Пиридиновые основания 33, 34, 106,
176 и сл., 414
Пириметамин 428
Пиримикарб 483
Пирокатехин 34
Пиролиз 25, 49 и сл., 108, 194, 195
Плазмохин см. Памаквин
Пластикация 279
Пластификаторы 24—25, 44 109 131,
148, 249—250
вторичные 169, 250
фталатные 152, 185, 249—250
Полиацеталь см. Полиоксиметилен
Поливинилацетат 108, 342 •
Поливинилбутираль 108
Поливинилиденхлорид 347
Поливиниловый спирт 108, 342 и сл
346
Поливинилхлорид 248 и сл„ 258—
259, 344 и сл.
Полиизобутилен 128, 280—281
Полиизопрен 119, 277, 280, 336
Поликарбонат 88, 269 и сл.
Полимеризация
акрилонитрила 331
анионная 241, 321—322
винилацетата 342
винилиденхлорида 347
изобутилена 242—243
капролактама 318
катионная 242
в массе 244
метилметакрилата 238—239, 245,
247, 266
олефинов 42, 76, 131
пропилена 257—258
радикальная 233—234, 239—240,
342, 345
в растворе 245
стирола 241—242, 246, 247, 260—
261
ступенчатая 234—235
суспензионная 246
тетрафторэтилена 259
формальдегида 262 и сл.
хлористого винила 244, 246—Z247,
248, 344—345
цепная 234—235, 239 и сл.
эмульсионная 248, 344
этилена 239—240, 250 и сл.,
333—334
Полнметилметакрилат 215, 265—266
Полиоксиметилен 262
Полипропилен НО, 256 и сл., 334 и сл.
Полистирол 260—261, 336
Политетрафторэтилен 259—260
Полиуретаны 115, 275 и сл., 337 и сл.
Полиэтилен 97, 99—100, 251 и сл.,
254 н сл., 333—334
Полиэфиры
линейные 152, 266, 268, 325 исл.
ненасыщенные 115, 148, 152, 156,
266 и сл.
Преднизолон 443
Преднизон 443
Примидон 402, 469
Природный газ 18, 46 и сл„ 553,
576 и сл.
Прогуанил 428
Прокаин 471
Промедол см. Петидин
Пронеталол 459
Пронтозил 398, 405
Пропан 18, 48, 49, 51, 67, 69, 90,207 *
Пропан ид 500
Пропилен 18, 49, 67, ПО и сл.
полимеризация НО, 257
получение 61, 69, ПО
7
Предметный указатель
689
реакции 58, 103, 111 и сл., 119
и сл., 129, 144, 166, 169, 170
и сл., 178, 187 и сл., 207, 213,
521
статистические данные 50
тетрамеры 120—121, 521
тримеры 89, 120—121, 536
Пропионовая кислота 79, 80, 83
Пропионовый альдегид 183, 18’6
Пропранолол 400, 459
Протаминсульфат 451
Протионамид 414
Пуромицин 433
Реактив
Грама 404
Сенджера 291, 293, 295, 320
Реакция оксосинтеза см. Гидрофор-
милирование
Резерпин 462
Резольные смолы 273—274
Рильсан см. Найлон-11
Риформинг 42, 44, 70 и сл., 134
Родамин Б 389
Розовая вода см. Отто
Салицин 444
Саран 282, 347
Сахарин 636—637, 640
Светильный газ 34, 85, 552—553, 574
и сл.
Себациновая кислота 25, 323—324
Седуксен см. Диазепам
Селективная экстракция 67, 131, 132,
135—136, 220
Серицин 292—293
Сернистый черный Т 388
Сероуглерод 90, 312
Сжиженный нефтяной газ 48, 163,
553
Симазин 501
Синильная кислота 89, 90, 128, 173—
175, 205 и сл.
Синталин А 432, 455
Синталин В 455
Синтез-газ 36, 84 и сл., 164
«Скатывание» 512—513
Смягчители 547—548
Соевое масло 23, 323
Спандекс 337
Спанзелл 340
Спиронолактон 476
Стеклопластики 266—268
Стирол 97, 102—103, 241, 246, 247
Стрептомицин 414—415, 494
Стрептоцид см. Сульфаниламид
Сукцинамовая кислота 506
Сульфадимезин см. Сульфадимидии
Сульфадимидин 406, 437
Сульфазин см. Сульфаназин
Сульфаназин 406
Сульфаниламид 401, 405, 474
Сульфахиноксалин 437
Сульфинпиразон 446
Сульфолан см. Тетраметиленсульфон
Сульфоокисление 163, 530—531
Сурамин 431
Сшивание (см. также Вулканизация,
Отверждение)
волокон 'типа анидекс 341, 343
коллагена 298
полиуретановых волокон 339—
340
шерсти 291
целлюлозы 307 и сл.
Таймин см. Найлон-4
Теклан 333
Температура
помутнении 533
сваривания 297
стеклования 239
Тенакс 351
Терефталевая кислота 152 и сл., 326
Терилен 152, 282, 325 и сл.
Терофен см. Триамтерен
Тетраметиленсульфон 128, 136
Тетрамизол 436
Тетранитрат пентаэритрита 591, 592,
597
Тетрафторэтилен 259, 678
1,1,2,2-Тетрахлорэтан 96, 169
Тетрациклины 412
Тетрил 586
Тиабендазол 435
Тимол 417
Тиоацетазон 415
Тиоиндиго 385
Тиопентал 470
Тиофос см. Паратион
Тиофосфамид см. ТиоТЭПА
ТиоТЭПА 423
ТиоТЭФ см. ТноТЭПА
Тирам 487
Тиурам Д см. Тирам
Токоферолы 609
Толазамид 455
Толбутамид 455
Толнафтат 420
Толуол 33 и сл., 132 н сл., 143, 223,
415
С-
690
ГГредметный указатель
Тоналид 630— 631
Транилципромин 463
Трансамин см. Транилципромин
Триамтерен 475
Триаримол 493
Тригексилфенидил 472
Триглицериды 22, 23, 516, 600 и сл.,
609
Тридеморф 491
Триметоприм 407
Тринитротолуол 586, 597
Триоксан 262—263
Триполифосфат натрия 515, 522, 547
Триптизол см. Амитриптилин
Трифлуралин 504
2,3,6-Трихлорбензойная кислота 498—
499
1,1,2-Трихлортрифторэтан 668, 674—
675, 678
Трихлорфторметан 676
Трихлорэтилен 96, 97, 169
ТУР 506
Уксусная кислота 92, 102, 106, 154,
155, 315—316, 318, 319
получение 29, 77 и сл.
статистические данные 82
Уксусный ангидрид 106, 303, 315—
316
Уротропин см. Гексаметилентетрамин
Фенадон см. Метадон
Феназоцин 464
Феиамии см. Амфетамин
Фенелзин 463
Фенидон 649 и сл.
Фенилбутазои 446
Фенилин см. Феииндион
'Фениндион 452
Фенклозииовая кислота 449
Фенметразин 472
Фенобарбитал см. Фенобарбитон
Фенобарбитон 402, 468—469
Фенол 34, 198 и сл., 417
получение 33, 157 и сл., 200 и сл.
реакции 220, 271 и сл., 274—275,
444, 498
статистические данные 205
Фенолформальдегидные смолы 87,
205, 229, 271 и сл„ 351
Фенотиазин 435
Фентермин 471
Фенфлурамин 472
Фенформин 455
Фибрилла 299 и сл.
Фиброин 287, 292 и сл.
Физостигмин 482
Филиксиновая кислота 433—434
Флюфенизал 444—445
Фолцид 489
Формальдегид 87, 95, 129, 177, 262
и сл., 271 и сл., 298, 307—308, 343,
356, 417, 451, 645
Фосген 88, 169, 270, 338
Фракционная кристаллизация 137
и сл.
Фталазол см. Фталилсульфатиазол
Фталевый ангидрид 150—152, 377,
378, 388, 389, 395
Фталилсульфатиазол 405
Фталоцианин меди 387—388, 395—
396
Фторотан см. Галотан
Фузидиевая кислота 413
Фузидин см. Фузидиевая кислота
Фульминат ртути 587, 590, 597
Фурадонин см. Нитрофурантоин
Фурземид 475
Фуросемид см. Фурземид
Фурфурол 27, 28, 121, 147
Хинапирамин 432
Хингамин см. Хлорохин
Хинин 398, 426, 429
Хиниофон 430
Хинолиновый желтый 389
Хлопок 299 и сл., 356, 379, 387
Хлоралгидрат 469
Хлорамфеникол 411
Хлорбензол 150, 168, 169, 200—201,
416, 479
Хлоргексидин 418
Хлордиазепоксид 466—467
Хлордифторметан 667, 671, 674, 678
Хлоридин см. Пириметамин
Хлорирование 166 и сл.
ацетилена 96, 169
бензола 150, 168, 169, 170, 200
201
бутадиена 127, 169
изобутилена 130
метана 90, 169
окиси углерода 88, 169
олефинов 167
парафинов 169, 523
ПВХ 345
пропилена 115, 167, 169
этана 169, 197
этилена 101, 167, 169, 193 и сл.
Хлористый аллил 115—116, 169
Предметный указатель
691
Хлористый винил 97, 169, 192 и сл.
полимеризация 244, 246—247,248,
333 344 347
получение 92, 168, 192—193, 197,
199
статистические данные 192, 194
Хлористый метил 90—91, 169
Хлористый метилен 90—91, 169
Хлоролиз 168, 170
Хлоропрен 92, 127
Хлороформ 90, 169, 666, 668
Хлорохин 427, 430
Хлорпромазин 460, 461
Хлорпропамид 455
Хлорталидои 474
Хлортетрациклин 412, 429
Хлортиазид 474
Хлортиамид 503—504
Хлортрианизен 403
Хлорфлуренол 507
Хромовый фиолетовый 387
Целестолин 631
Целлюлоза 26, 299 и сл., 354—355,
451
Цетилпиридииийбромид 417
Цетримид 417
Цефалоспорины 410—411
Циклогексан 146—147, 203, 223, 224
Циклогексанол 146—147, 203, 223,
226
Циклогексанон 146—147, 203, 223,
226, 351
Циклогексиламин 226, 313
Циклогексимид 421
Циклододекатриен-1,5,9 123, 124, 323
Циклодол см. Тригексилфеиидил
Циклометиазид см. Циклопентиазид
Циклопентадиен 119
Циклопентиазид 475
Циклосерин 415
Циклотриметилентринитрамин см.
Г ексоген
Цинеб 488
Цинерииы 484
Ципрекс см. Додин
Цитраль 625 и сл.
Цитронелла ль 618, 619
Цитронеллол 616, 617, 619
Черный порох 585, 588
Черный сандал 359
Четыреххлористый углерод 90, 169,
435, 666, 667
Чиион 295, 356
Шелк 292 и сл.
Шерсть 286 и сл., 355, 371, 372, 379,
387
Эластомеры см. Каучуки
Элениум см. Хлордиазепоксид
Эметин 429, 430
Энант см. Найлон-7
Эндосульфан 479—480
Эикатерм 350
Эпихлоргидрин 116, 309
Эпоксидирование 100, 113 и сл., 131,
164
Этакриновая кислота 402, 475
Этамбутол 416
Этаи 18, 46, 49, 56, 60, 61, 67, 169
Этанол 15, 97, 104, 210, 211—212, 417,
549
получение 27, 30, 31, 102, 187
и сл.
статистические данные 30, 102,
190
Этилбензол 97, 103, 132, 137, 139 и сл.
получение 144—145
статистические данные 133
2-Этилгексаиол-1 106, 135
Этилен 13, 18, 49, 58, 61, 67, 68, 92,
97 и сл., 121, 125, 166, 506
алкилирование бензола 102, 144
затраты производства 13
гидратация 102, 187 и сл.
гидроформилирование 183, 186
каталитическая теломеризации
108, 526—527
окисление 77, 104, 107, 166
окислительное хлорирование 101,
196—197, 198
полимеризация 239—240, 250 и сл.
получение 49, 53, 60, 62 и сл.
статистические данные 50, 97,
98
теломеризации с CCU 323
хлорирование 169, 193
эпоксидирование 100, 164
Этиленгликоль 101, 136, 164, 326
Этиленхлоргидрин 100, 164
Этионамид 414
Этионин 424
Этиримол 492—493
Этосуксимид 470
Этрел 506
Эутрофикация 547
Ювенильный гормон 485—486
Содержание
Предисловие редактора перевода ................................. 5
Предисловие к русскому изданию...................................8
Предисловие авторов..............................................9
Глава 1. Введение и история химической
промышленности....................................................11
История химической промышленности.........................15
Общие замечания...........................................18
Глава 2. Источники сырья для промышленности
органического синтеза............................................ 22
Сельскохозяйственное и лесохимическое сырье....22
Жнры и масла . .........................................22
Лесохимическое сырье....................................25
Сахаристые вещества.....................................30
Крахмал ................................................30
Углехимическое сырье......................................32
Сухая перегонка....................................... 32
Г азификация............................................36
Гидрогенизация .........................................36
Нефть ....................................................38
Сырая нефть и .ее характеристики........................38
Методы переработки нефти................................41
Объем мировой добычи и потребления нефти и ее ресурсы . . 44 •
Природный газ.............................................46
Глава 3. Получение первичных нефтехимических
продуктов....................................................... 49
I. Крекинг......................................................49
Введение ...............................................49
Термохимия ...............................................51
Энергия диссоциации связей................................53
Механизм процесса пиролиза................................54
Приложение рассмотренного механизма к процессам пиролиза
различных углеводородов.................................56
Вторичные реакции.......................................58
Кинетика пиролиза.........................................60
Исходное сырье............................................60
Параметры процесса пиролиза...............................62
Время пребывания........................................62
Температура пиролиза .................................. 63
Содержание
693
Степень разбавления углеводорода паром .................. 63
Устройство печи пиролиза ................................... 6^
Печь с прямым обогревом...................................65
Другие процессы расщепления углеводородов....................67
Получение линейных а-олефинов.............................67
Процессы с непрямым обогревом.............................67
Автотермический крекинг ................................. 68
Процессы электрокрекннга..................................68
Каталитический крекинг .................................. 69
Гидрокрекинг ............................................ 69
II. Риформинг ....................................................70
Введение ....................................................76
Термодинамика процесса.......................................74
Механизм каталитического риформинга..........................75
III. Прямое окисление бутана и нафты ......... ... 77
Введение................................................... 77
Описание процесса............................................78
Механизмы реакций............................................79
Статистические данные о производстве н потреблении низших
карбоновых кислот ...................................... 81
Уксусная кислота ........................................ 81
Муравьиная кислота........................................82
Пропионовая кислота.......................................83
Глава 4. Первичные нефтехимические продукты.
I. Алканы, алкены и алкины.................................. 84
Метан и окись углерода....................................
Синтез-газ ............................................
Метанол................................................
Реакция Фишера — Тропша................................
Окись углерода.........................................
Применение метана в различных синтезах.................
Ацетилен .............................. ..................
Этилен ...................................................
Полиэтилен.............................................
Окнсь этилена..........................................
Хлористый вннил........................................
Этанол ................................................
Стирол ................................................
Ацетальдегид ..........................................
Винилацетат............................................
Высшие олефины.........................................
Этиленпропиленовые каучуки ............................
Пропилен..................................................
Полипропилен ..........................................
Акрилонитрил ..........................................
Оксоспирты ............................................
Изопропанол ...........................................
Ацетон ................................................
Окись пропилена........................................
Кумол ...............................-.................
Хлористый аллил........................................
Акролеин ..............................................
Изопрен ...............................................
84
84
86
87
88
89
91
97
99
100
101
102
102
104
106
108
109
110
ПО
112
113
115
115
116
118
694
Содержание
Димеризация пропилена..................................119
Тримеры и тетрамеры пропилена..........................120
Диспропорционирование пропилена ...................... 121
Углеводороды С4 :........................................121
Бутадиен ..............................................122
Изобутилен ............................................128
н-Бутилены и н-бутан...................................130
Глава 5. Первичные нефтехимические продукты.
II. Ароматические углеводороды.................................. 132
Введение ................................................132
Методы получения ароматических углеводородов.............133
Селективная экстракция ............................... 135
Получение о-ксилола и н-ксилола........................136
Изомеризация ароматических углеводородов С8............139
Методы селективного извлечения и-ксилола...............142
Гидродеалкилирование толуола в бензол ................ 143
Получение этилбензола и кумола алкилированием бензола . . 144
Бензол...................................................145
Циклогексан ...........................................146
Малеиновый ангидрид ...................................147
Алкилбензолсульфонаты .................................148
Анилин . . . ..........................................149
Хлорбензолы ...........................................150
ДДТ....................................................150
Ксилолы .................................................150
Получение фталевого ангидрида из о-ксилола.............150
Получение терефталевой кислоты и диметилтерефталата из
п-ксилола ..........................................152
Получение изофталевой кислоты из з«-ксилола............156
Кумол ...................................................157
Производство фенола и ацетона..........................157
Глава 6. Некоторые методы получения
нефтехимических полупродуктов ...................................161
Окисление ...............................................161
Классификация методов окисления........................163
Хлорирование ........................................... 166
Окислительный аммонолиз..................................170
Синтез акрилонитрила из пропилена .................... 170
Синтез пиридинов.........................................176
Гидроформилирование (оксосинтез).........................180
Термодинамика и механизм гидроформилирования ......... 183
Области применения.....................................185
Гидратация олефинов .................................... 186
Области применения.....................................189
Глава 7. Выбор оптимального метода
производства.....................................................192
Хлористый винил........................................ 192
Фенол ...................................................198
Щелочное плавление бензолсульфокислого натрия (метод суль-
фирования) .........................................200
Содержание
695
Щелочной гидролиз хлорбензола (процесс фирмы Dow) . 200
Парофазный гидролиз хлорбензола (процесс фирм Raschig —
Hooker) ..............................................201
Разложение гидроперекиси кумола (кумольный метод) . . . 201
Окисление толуола (толуольный метод).....................202
Дегидрирование смеси циклогексанона и циклогексанола (цик-
логексановый метод).....................................202
Прямое окисление бензола................................203
Сравнительная оценка различных методов производства фе-
нола ................................................203
Акрилонитрил .....................'.......................205
Ацетиленовый метод......................................206
Окислительный аммонолиз пропилена.......................207
Реакция окиси этилена с синильной кислотой..............207
Реакция ацетальдегида с синильной кислотой..............207
Реакция пропилена с окисью азота........................207
Окислительный аммонолиз пропана.........................207
Реакция этилена с синильной кислотой .................. 208
Сравнительная экономическая оценка различных методов по-
лучения акрилонитрила ................................. 208
Ацетон....................................................210
Дегидрирование изопропанола ........................ 211
Окислительное дегидрирование изопропанола................211
Области применения ацетона...............................213
Синтезы на основе ацетона............................... 215
Капролактам ..............................................219
Методы получения капролактама . 220
Фенольный метод..........................................220
Методы с окислением циклогексана воздухом................223
Толуольный метод.........................................223
Фотонитрозирование циклогексана ........................ 224
Капролактоновый метод.................................. 225
Нитроциклогексаноновый-метоц.............................225
Нитроциклогексановый метод...............................226
Метод с промежуточным образованием перекиси 1-оксицикло-
гексила ............................................ 226
Циклогексиламиновый метод................................226
Другие методы............................................228
Выводы ..................................................228
Глава 8. Пластические массы и эластомеры ...........................229
Введение .................................................229
Терминология и классификация..............................231
Терминология ............................................231
Классификация............................................233
Связь между химической структурой и свойствами полимера . . 235
Природа связи в полимерах................................235
Строение полимерных цепей................................236
Кристалличность ........................................ 238
Температура стеклования ................................ 239
Механизм цепной полимеризации.............................239
Радикальная полимеризация .............................. 239
Цепная полимеризация по ионному механизму................240
Координационные катализаторы.............................243
Методы проведения полимеризации...........................244
Полимеризация в массе....................................244
696
Содержание
Полимеризация в растворе............................ 245
Суспензионная полимеризация .......................... 246
Эмульсионная полимеризация ............................248
Компаундирующие добавки................................248
Термостабилизаторы.....................................248
Пластификаторы ........................................249
Вторичные пластификаторы...............................250
Смазки ................................................250
Важнейшие пластические массы.........................................250
Полиэтилен —[—СН2СН2—1„—
Полиэтилен низкой плотности (высокого давления) .
Полиэтилен высокой плотности (низкого давления) .
Полипропилен —[—СН(СН3)СН2—]п—................
Поливинилхлорид —СНС1СН2—]п—..................
Политетрафторэтилен —[—CF2CF2—]п—.............
Полистирол —[—СН(С6Н5)СН2—]„—.................
Модифицирование полистирола каучуком........
Сополимеры стирола
Полиоксиметилен (полиацеталь) —[—СН2О—]„—
СНз
250
251
254
256
258
259
260
261
262
262
Полиметилметакрилат —
—СН2—С--------
СООСНз _п
265
Полиэфиры .........................................
Сшитые полиэфиры.................................
Линейные полиэфиры...............................
Поликарбонаты —[—OCOORO—]„—.......................
Фенолформальдегидные смолы.........................
Новолачные смолы.................................
266
266
268
269
271
271
Резольные смолы........................................273
Мочевииоформальдегидные смолы............................274
Полиуретаны .............................................275
Эластомеры ..............................................276
Натуральный каучук.....................................276
Синтетические каучуки..................................279
Список литературы .......................................281
Глава 9. Волокна............................................... 282
Введение .................................................282
Природные волокна. I. Волокна животного происхождения . . 286
Шерсть .................................................286
Шелк ...................................................292
Кожа ...................................................296
Природные волокна. II. Волокна растительного происхождения 299
Хлопок .................................................299
Искусственные волокна....................................311
Гидратцеллюлозные волокна...............................312
Ацетатные волокна..................'....................315
Синтетические волокна. I. Полиамидные волокна.............317
Найлон-6 ...............................................318
Найлон-6,6..............................................318
Другие полиамиды типа АВ...............................321
Другие алифатические полиамиды типа ААВВ................323
Содержание ^97
Алициклические полиамиды (киана).......................324
Синтетические, волокна. И. Полиэфирные волокна...........325
Терилен ...............................................325
Другие полиэфирные волокна.............................329
Синтетические волокна. III. Полиакрилонитрильные волокна . . 331
Синтетические волокна. IV. Полиолефиновые волокна .... 333
Полиэтиленовые волокна ................................333
Полипропиленовые волокна ............................. 334
Другие полиолефиновые волокна..........................336
Эластомерные волокна.....................................336
Волокна из натурального каучука .................. .... 336
Полиуретановые волокна.................................337
Эластомерные волокна типа анидекс......................341
Полнвинилспиртовые волокна............................34,1
Поливинилхлоридные (ПВХ) волокна.................• • • 344
Поливинилиденхлоридные волокна ....................... 346
Термостойкие волокна ................................... 348
Углеродные волокна ..................................... 352
Модифицирование волокон путем прививки других полимеров . 353
Список литературы .......................................357
Глава 10. Химия красителей . ......................................359
Красители ......................................................362
Азокрасители ............................................ 362
Получение, строение и окраска...........................362
Применение..............................................368
Простые антрахиноновые красители..........................377
Кубовые красители.........................................380
Индигоидные и тиоиндигоидные красители....................384
Триарилметановые красители .............................. 386
Фталоцианиновые красители ............................... 387
Прочие красители ........................................ 388
Активные красители........................................389
Пигменты .......................................................394
Номенклатура..............................................397
Список литературы.........................................397
Глава 11. Фармацевтические препараты..............................398
Введение ................................................398
Связь между структурой и активностью органических лекар-
ственных веществ ..................................401
Средства для лечения инфекционных заболеваний ........ 404
Противомикробные средства................................404
Синтетические противомикробные средства................405
Антибиотики ...........................................409
Противотуберкулезные и противолепрозные средства .... 413
Антисептики ...........................................417
Противовирусные средства.................................418
Противогрибковые средства................................419
Противораковые средства..................................421
Противог.аразитарные средства ...........................425
Противомалярийные средства.............................425
Противоамебные средства................................429
698
Содержание
Трипаноцидные средства.................................430
Противоглистные средства...............................433
Антикокцидиозные средства..............................437
Средства для лечения неинфекционных, заболеваний............438
Противозачаточные средства...............................438
Стероидные противовоспалительные средства..............440
Нестероидные противовоспалительные средства..............444
Коагулянты и антикоагулянты..............................450
Гипохолестерннемические средства.........................452
Сахароснижающие средства.................................454
Антигипертензивные средства..............................456
Средства для лечения заболеваний центральной нервной си-
стемы ...................................................459
Транквилизаторы........................................460
Антидепрессанты........................................462
Анальгезирующие средства...............................464
Успокаивающие средства.................................466
Снотворные и седативные средства....................468
Противосудорожные средства........................, . . 469
Анестезирующие средства................................470
Стимуляторы центральной нервной системы................471
Антипаркинсонические средства ........................ 472
Мочегонные средства......................................473
Список литературы........................................476
Глава 12. Химикаты для сельского хозяйства ...........................477
Инсектициды ............................................478
Хлоруглеводороды .....................................479
Фосфорорганические инсектициды........................480
Карбаматы ........................................... 482
Инсектициды растительного'кроисхождения...............484
Перспективы ..........................................484
Фунгициды ..............................................486
Контактные фунгициды..................................486
Системные фунгициды.............'.....................490
Антибиотики ..........................................494
Гербициды ..............................................496
Регуляторы роста растений...............................505
Список литературы.......................................507
Глава 13. Моющие вещества.........................................509
Процесс мытья.............................................509
Роль ПАВ в удалении загрязнений.........................511
Обратное осаждение загрязнения..........................514
Активные добавки........................................515
Мыла и мыльные моющие средства............................516
Омыление ...............................................517
Получение моющих средств на мыльной основе..............518
Некоторые проблемы, связанные с производством и приме-
нением мыла...........................................519
Синтетические поверхностно-активные вещества..............520
Анионные поверхностно-активные вещества.................521
Неионные поверхностно-активные вещества.................532
Содержание
699
Катионные поверхностно-активные вещества..................541
Бытовые моющие средства на основе синтетических ПАВ . . 543
Список литературы.........................................549
Глава 14. А. Химия и горение нефтяных топлив......................550
Б. Горючие и взрывчатые вещества и пороха................550
А. Химия и горение нефтяных топлив................................550
Огонь и тепло............................................550
Основные нефтяные топлива............................. . 552
Химия горения............................................554
Практические пламена...................................554
Быстрые пламенные реакции..............................557
Реакции медленного горения ........................... 559
Использование полученных данных по химизму горения . . . 567
Кинетика горения.......................................567
Детонация топлива в двигателях ....................... 567
Холодные пламена . ....................................568
Дистиллятные топлива...................................570
Капельное горение .................................... 571
Проблемы загрязнения окружающей среды и коррозии . . . 572
Заключение.............................................573
Технологические процессы горения . . 574
Значение вида топлива..................................574
Сжигание природного газа для бытовых целей.............576
Промышленный малогабаритный котел......................579
Выплавка чугуна .......................................580
Горелки для высокоскоростного сжигания топлива .... 581
Сжигание топлив в псевдоожиженном слое.................582
Заключение........................................... 584
Список литературы........................................584
Б. Горючие и взрывчатые вещества и пороха.........................585
Современные взрывчатые вещества........................590
Глава 15. Пищевые вещества....................................... 598
Производство масел и жиров.......................... : . 599
Хлебопечение ............................................605
Порча пищевых продуктов и борьба с ней..................607
Новые источники пищевого сырья...........................610
Белки .................................................610
Углеводы...............................................612
Жиры ..................................................613
Перспективы производства пищевых продуктов...............613
Глава 16. Душистые и .вкусовые вещества...........................615
Душистые вещества............................................... 616
Эфирные масла............................................616
Связь между запахом и молекулярной структурой душистых
веществ .............................................618
Механизм восприятия запаха...............................621
Промышленный синтез некоторых душистых веществ .... 623
700
Содержание
Гераниол, нерол, линалоол, цнтраль.....................623
Иононы ................................................626
Некоторые мускусные соединения.........................628
Вкусовые вещества................................................ 632
Анализ и идентификация вкусовых веществ..................632
Связь между химической структурой и органолептическими
свойствами ..........................................634
Производство некоторых типичных вкусовых веществ .... 639
Список литературы . . . '................................643
Глава 17. А. Фотографические процессы и материалы.................644
Б. Производство и применение хлорфтор-
углеводородов .......................................... 644
А. Фотографические процессы и материалы ..........................644
Галогенсеребряные процессы ..............................644
Желатина ........................•.....................644
Стабилизаторы .........................................646
Сенсибилизирующие красители............................646
Проявляющие вещества ..................................649
Цветные процессы ......................................651
Другие органические компоненты галогенсеребряных процес-
сов ...............................................660
Бессеребряные процессы...................................661
Диазопроцесс ..........................................661
Процесс Кальвара.......................................663
Фотохромные системы....................................663
Б. Производство и применение хлорфторуглеводородов................664
Химический состав........................................666
Номенклатура ........................................... 666
Производство ............................................667
Степень чистоты...................................... 668
Области применения ......................................670
Холодильные агенты.....................................670
Аэрозольные пропелленты................................672
Растворители ..........................................674
Вспениватели ......................................... 676
Огнегасящие агенты.....................................677
Химические полупродукты.....................’..........678
Транспортировка и хранение.............................679
Список литературы....................................... 679
Список дополнительной литературы ................................ 680
Предметный указатель..............................................683
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформлении, ка-
честве перевода и другие просим присылать по адресу:
129820, Москва, И-110, ГСП, 1-й Рижский пер., д. 2.
ИБ № 652
Дж. Теддер,
А. Нехватал,
А. Джубб
ПРОМЫШЛЕННАЯ
ОРГАНИЧЕСКАЯ
ХИМИЯ
Редактор Г. Шкляева
Художник Г. Валетов
Художественный редактор Н. Блинов
Технический редактор А. Резоухова
Сдано в набор 2/П 1977 г. Подписано к печати
28/VI 1977 г. Бумага тип. №2 60X90’/i6=22 бум. л.
печ. л. 44. Уч.-изд. л. 43,62. Изд, № 3/8916.
Цена 4 р. 20 к. Зак. 501.
ИЗДАТЕЛЬСТВО «МИР>
Москва, 1-й Рижский пер., 2
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени
Евгении Соколовой -Союзполиграфпрома при
Государственном комитете Совета Министров СССР
по делам издательств, полиграфии
и книжной торговли.
198052, Ленинград, Л-52, Измайловский проспект, 29.
ИМЕЕТСЯ В ПРОДАЖЕ
Лер Р., Марчанд А.
ОРБИТАЛЬНАЯ СИММЕТРИЯ В ВОПРОСАХ И ОТВЕТАХ
Пер. с англ., «Мир», 1976, 1 р. 06 к.
Учебное пособие по овладению практическими приемами пред-
сказания возможности протекания синхронных реакций путем
рассмотрения симметрии молекул. Вначале автор знакомит
читателя с основными выводами теории Вудворда — Хоффма-
на, далее приводит задачи, решение которых требует приме-
нения теоретических выводов к конкретным химическим си-
стемам, и в конце книги даются подробные ответы на задачи.
Книга предназначена для химиков-органиков и физикохимиков,
занимающихся вопросами химической кинетики и реакционной
способности соединений; особенно ее следует рекомендовать
студентам и преподавателям, специализирующимся в указан-
ных областях.
Уважаемый читатель!
Заказ на эту книгу направляйте по адресу:
191040. Ленинград, Пушкинская ул., 2, магазин № 5 «Техниче-
ская книга».
Книга будет выслана наложенным платежом.
ИМЕЕТСЯ В ПРОДАЖЕ
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА НУКЛЕИНОВЫХ
КИСЛОТ
Пер. с англ., «Мир», 1976, 2 р. 33 к.
В книге комплексно рассматриваются физико-химические ме-
тоды, применяемые для исследования структурных и конфигу-
рационных изменений нуклеиновых кислот и их компонентов.
Книга посвящена изучению электрических, оптических и маг-
нитных свойств исследуемых объектов.
Предназначена д^я специалистов, работающих в области фи-
зико-химической биологии, — химиков-органиков, биохимиков,
биофизиков, специалистов в области молекулярной биологии и
генетиков.
Уважаемый читатель!
Заказ на эту книгу направляйте по адресу:
191040. Ленинград, Пушкинская ул., 2, магазин № 5 «Техниче-
ская книга».
Книга будет выслана наложенным платежом.