/






































































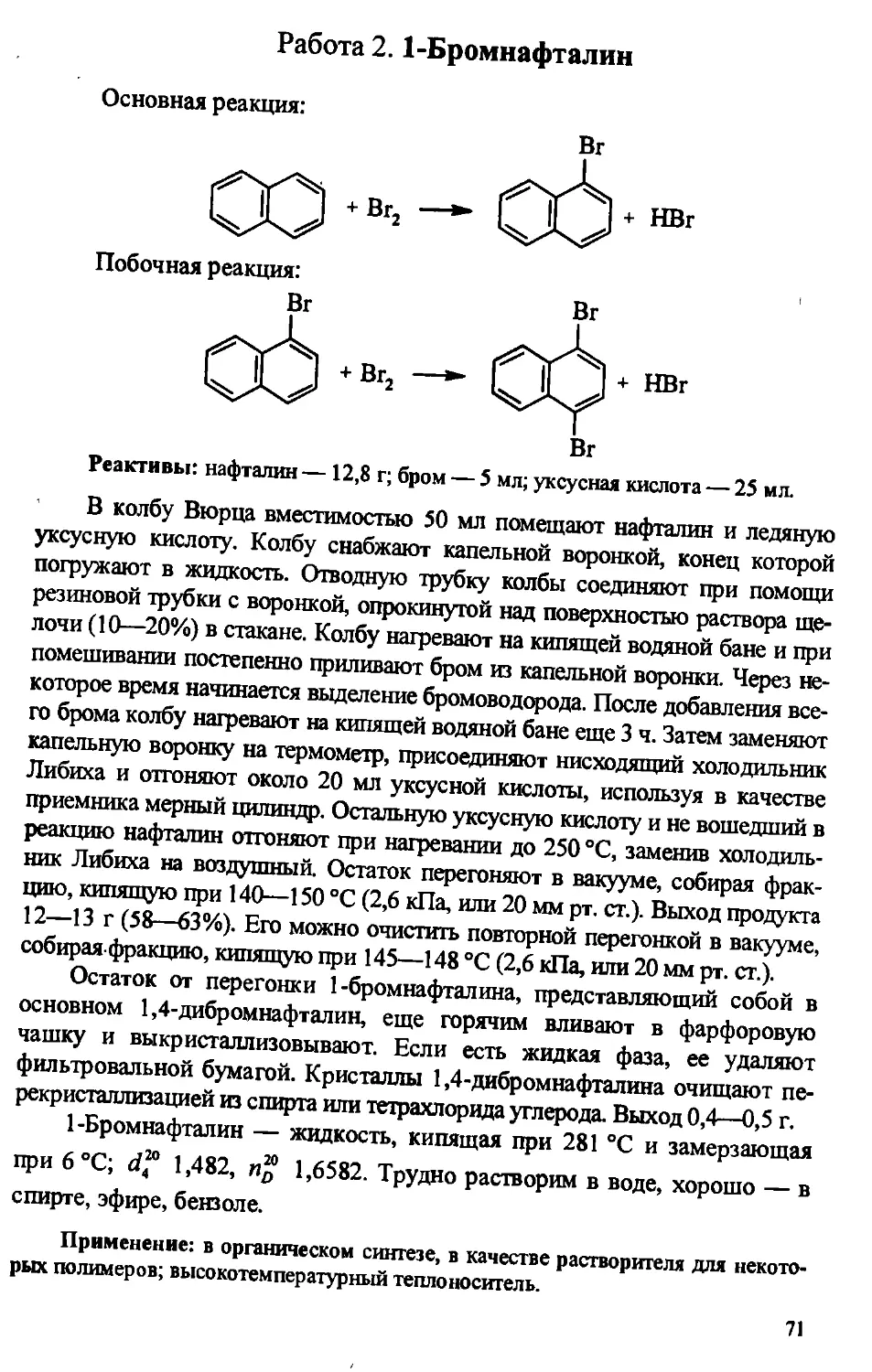






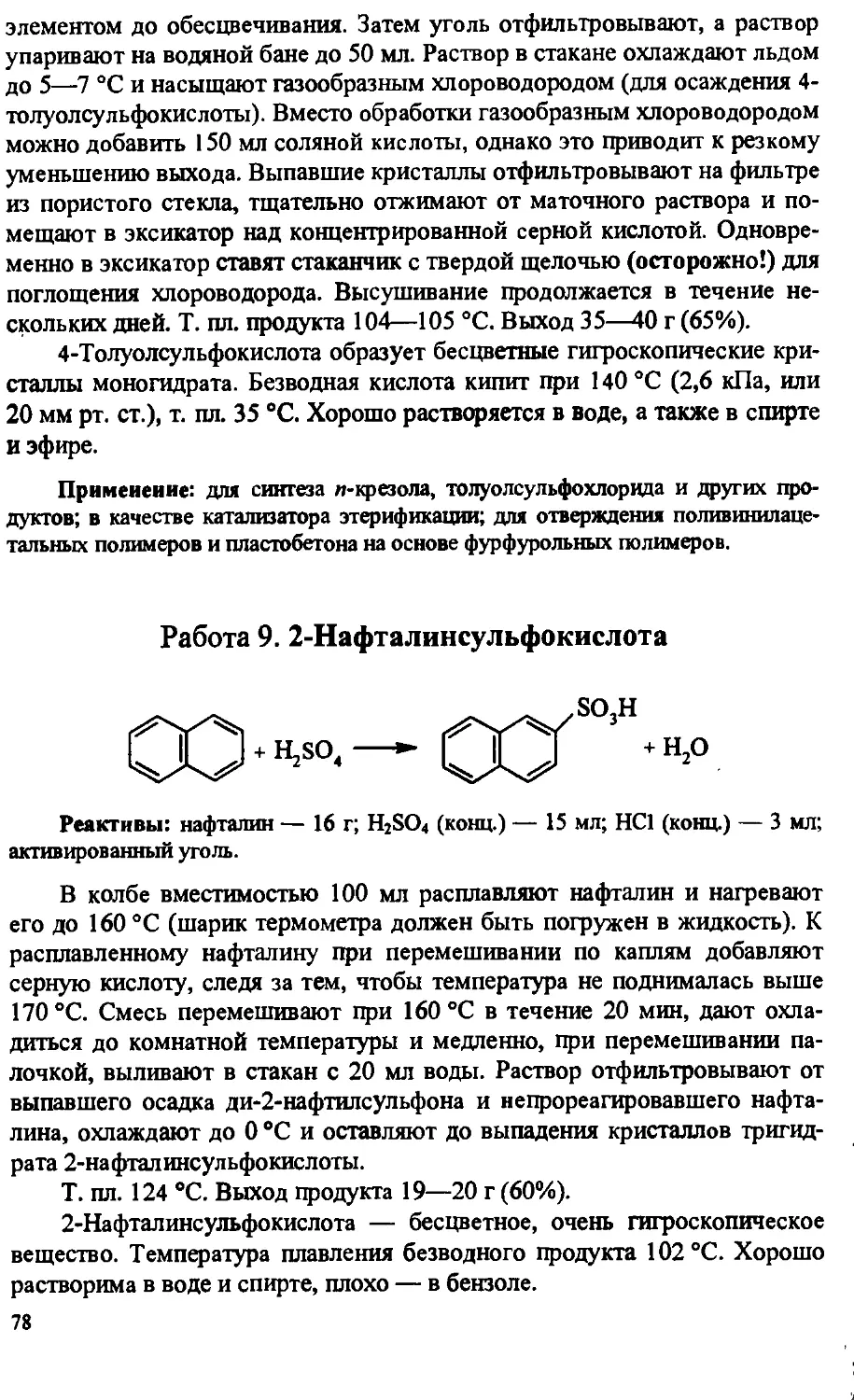







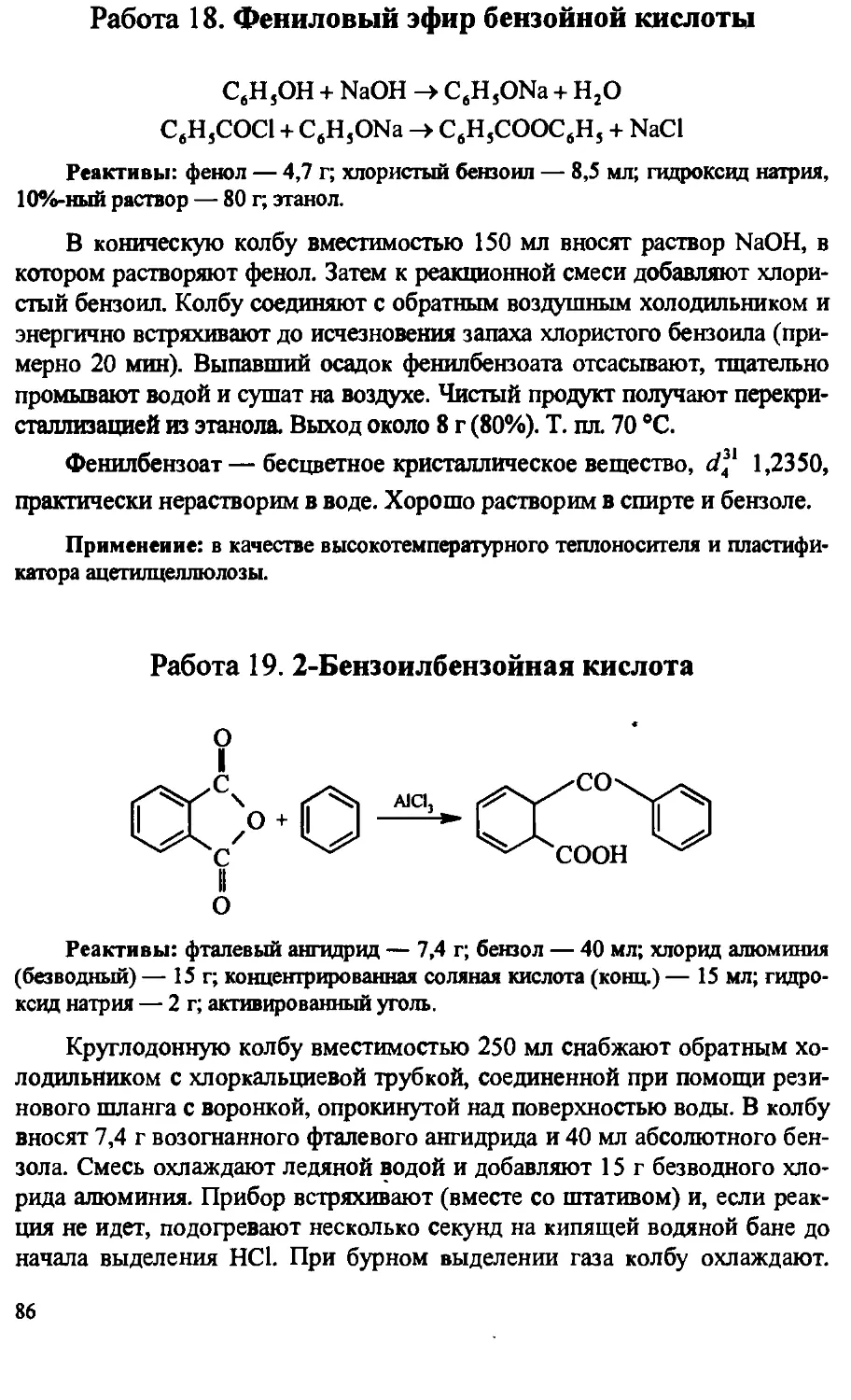









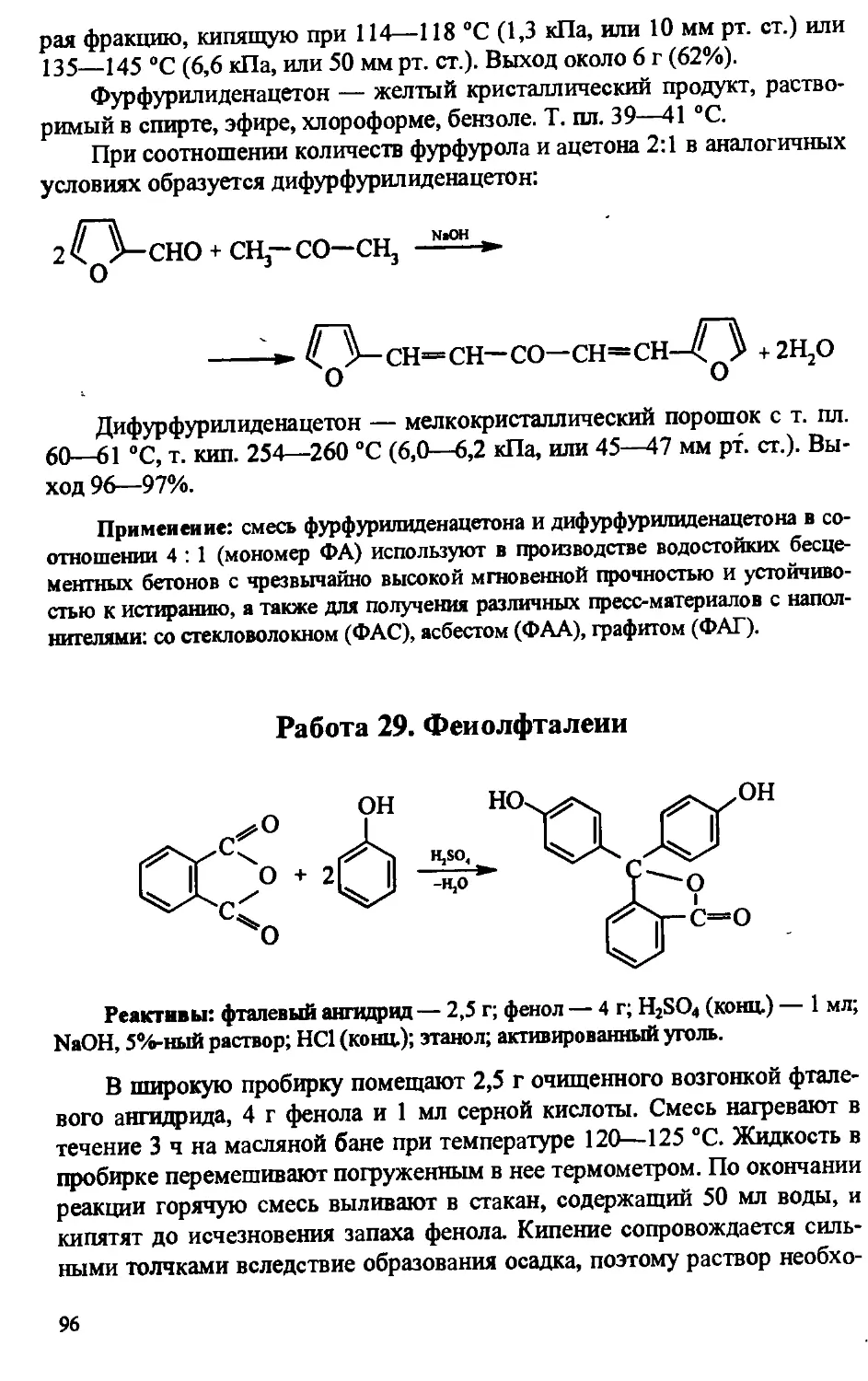























































































Author: Артеменко А.И. Тикунова И.В. Ануфриев Е.К.
Tags: органическая химия практикум
ISBN: 5-06-003987-0
Year: 2001




Text
-> 'I 7
УДК 547
ББК24.2
А 86
Рецензент — доктор химических наук, профессор И.И. Василенко
(заведующий кафедрой Белгородской ГСХА)
Артеменко А. И., Тикунова И. В., Ануфриев Е. К.
А 86 Практикум по органической химии: Учеб. пособие для студентов
строит, спец. вузов. — 3-е изд., испр. — М.: Высш. шк., 2001. —
187 с: ил.
ISBN 5-06-003987-0
Практикум представляет собой руководство к лабораторным и
семинарским занятиям. Материал подобран с учетом специализации, связанной с
химической технологией строительных материалов, производством
строительных изделий и конструкций. В практикуме рассмотрены современные
методы исследования органических соединений (спектроскопия, ЯМР,
хроматография и др.), а также основы технического анализа некоторых
полимерных соединений.
Приведены основные сведения о справочной и реферативной
литературе по органической химии.
Второе издание вышло в 1991 г.
Для студентов строительных специальностей вузов.
УДК 547
ББК 24.2
ISBN 5-06-003987-0 © ГУП «Издательство «Высшая школа», 2001
Оригинал-макет данного издания является собственностью издательства
«Высшая школа», и его репродуцирование (воспроизведение) любым способом
без согласия издательства запрещается.
Предисловие
Лабораторные работы являются важнейшим этапом учебного
процесса, совершенствующим теоретическую и практическую подготовку
будущего специалиста. Лабораторные работы по органической химии
позволяют глубже и полнее вникнуть в химические процессы, овладеть
основными законами химии. Они обычно проводятся параллельно с
изучением теоретического курса органической химии.
Настоящий практикум составлен в соответствии с программой по
органической химии для химико-технологических специальностей. Он
предназначен для студентов, специализирующихся в области химической
технологии тугоплавких неметаллических и силикатных материалов,
производства строительных изделий и конструкций, экономики и
управления в строительстве.
В первой части книги приведены правила техники безопасности при
работе в лаборатории органической химии, показаны приемы сборки
основных приборов и установок, а также перечислен необходимый
минимум лабораторного оборудования и химической посуды. Задача этой
части практикума — научить студента выполнять несложные синтезы
органических веществ, познакомить с основными методами их выделения,
очистки и идентификации, показать, как вести записи в лабораторном
журнале, дать представления о качественном и количественном анализе
органических соединений.
Вторая часть посвящена общим свойствам различных классов
органических соединений и индивидуальным особенностям их важнейших
представителей. При этом многие органические вещества, свойства
которых исследуются здесь, получают и выделяют непосредственно в
процессе опытов. Эта часть практикума (ознакомительная) рекомендована
студентам, изучающим органическую химию в небольшом объеме.
Закреплению учебного материала способствуют приводимые после каждой
темы контрольные вопросы.
Для более углубленного изучения курса органической химии в
третью часть практикума включены прописи синтезов некоторых
органических веществ. Эти синтезы сгруппированы по типам реакций. В случае
необходимости одну лабораторную работу можно заменить другой
(сходной по характеру). В конце каждой прописи синтеза приведена
краткая характеристика полученного продукта и перечислены области
его применения.
3
Авторы сочли необходимым включить в книгу синтезы и
превращения некоторых органических веществ, которые используются или могут
использоваться в строительной промышленности. Это касается прежде
всего полимерных материалов. Поэтому в практикуме рассмотрены
также методики технического анализа некоторых полимерных соединений и
других продуктов, включен раздел, посвященный физико-химическим
(инструментальным) методам исследования (УФ-, ИК- и ЯМР-спектро-
скопия, хроматография и метод электрических моментов диполей).
В приложение к книге вынесены некоторые советы по организации
самостоятельной работы студентов. Там же приведены краткие сведения
о справочной и реферативной литературе по органической химии,
рекомендации по приготовлению реактивов для лабораторных работ, правила
номенклатуры органических соединений и другой справочный материал.
В третьем издании практикума исправлены замеченные опечатки и
внесены дополнения и некоторые изменения.
Авторы выражают благодарность рецензенту — Василенко И. И.,
замечания которого способствовали улучшению книги.
Авторы
Раздел I
Общая часть
1. Основные правила и организация работы
в лаборатории органической химии
1.1. Техника безопасности в лаборатории
органической химии
Все органические вещества в той или иной степени ядовиты, а
многие из них — огнеопасны и взрывоопасны. Поэтому, работая в
лаборатории органической химии, необходимо строго соблюдать перечисленные
ниже основные правила техники безопасности,
1. Работать одному в лаборатории запрещается. Приступать к работе
можно только в присутствии преподавателя или лаборанта.
2. Во время работы в лаборатории соблюдайте тишину, чистоту и
порядок на своем рабочем месте и в лаборатории. Нельзя отвлекаться от
работы и отвлекать своих товарищей.
3. Нельзя работать в лаборатории без халата. Он должен быть сшит
только из хлопчатобумажной ткани. Запрещается держать на
лабораторном столе портфель, сумку и другие посторонние предметы. Для них
должно быть отведено специальное место.
4. В лаборатории запрещается пить воду, принимать и хранить пищу,
курить.
5. При работе следует надевать очки: а) при определении
температуры плавления в приборе с концентрированной серной кислотой; б) прн
возможном разбрызгивании и разбрасывании едких веществ
(перемешивание кислот и щелочей, дробление твердой щелочи, хлорида цинка,
сплавление в открытой чашке и т. д.); в) в случае перегонки жидкостей
при пониженном давлении и при работе с вакуум-приборами; г) при
работе с ампулами и запаянными трубками, изготовлении стеклянных
капилляров; д) при работе со щелочными металлами и плавиковой кислотой.
6. Работать с ядовитыми, раздражающими органы дыхания и сильно
пахнущими веществами необходимо только в вытяжном шкафу в
резиновых перчатках, а иногда — ив противогазе. Нельзя брать химические
вещества незащищенными руками. Сыпучие реактивы отбирайте только
сухим шпателем или специальной ложкой.
5
7. Приступая к выполнению работы, ознакомьтесь со свойствами
применяемых в синтезе веществ (огнеопасность, токсичность и т. д.).
8. Прежде чем взять необходимое количество вещества, внимательно
прочитайте надпись на этикетке лабораторной посуды, в которой
содержится это вещество.
9. Нельзя наглухо закрывать приборы для проведения реакций,
нагревания растворов и перегонки жидкостей, так как это может привести к
взрыву.
10. Нельзя держать при нагревании пробирку или колбу отверстием
к себе или в сторону стоящего рядом человека.
11. Запрещается нагревать летучие и легковоспламеняющиеся
жидкости и вещества (эфиры, петролейный эфир, бензин, спирт, ацетон,
сероуглерод и др.) на открытом пламени. Для этого пользуйтесь водяной
баней или электрической плиткой с закрытой спиралью. При перегонке
таких веществ обязательно применяйте холодильники с водяным
охлаждением. Нельзя перегонять жидкости досуха — это может привести к
взрыву или пожару.
12. Не наклоняйтесь близко к прибору, в котором идет реакция,
нагревание или перемешивание химических веществ.
13. Запрещается выливать в раковины остатки кислот и щелочей,
огнеопасных и ядовитых, плохо смывающихся и сильно пахнущих
жидкостей Для этого в вытяжном шкафу или около раковины должна стоять
специальная емкость, хорошо закрывающаяся и небьющаяся (например,
пластмассовая).
14. Не разрешается бросать в раковины бумагу, вату, стекло от
разбитой химической посуды.
15. Запрещается пробовать химические вещества на вкус, всасывать
ртом любые жидкие вещества в пипетки. При исследовании запаха
жидкости следует осторожно направлять к себе ее пары легким движением руки.
16. Категорически запрещается держать ртуть в открытой посуде.
Все приборы, содержащие ртуть, должны быть помещены на безщелевые
подносы с достаточно высокими боковыми стенками. В случае поломки
прибора, содержащего ртуть, необходимо поставить об этом в
известность преподавателя или лаборанта. Выливать ртуть в раковины
запрещается. Разлитую ртуть собирают с помощью амальгамированной
медной пластинки в специальные толстостенные банки, закрытые пробкой.
Остатки ртути, попавшие в щели пола, стола и т. д., следует обработать
20%-ным водным раствором хлорида железа (III) или порошком серы.
17. Металлический натрий следует обязательно хранить под слоем
керосина, толуола или ксилола, не содержащих следов воды. Нельзя
работать с металлическим натрием поблизости от водопроводного крана.
Приступая к работе, надо насухо вытереть стол и высушить посуду, в
которой будет проводиться реакция с металлическим натрием. После
6
окончания работы нельзя сразу мыть эту посуду водой — следует снача
ла уничтожить остатки натрия, растворяя их в спирте. Крупные остатю
натрия или его обрезки следует поместить в отдельную банку с кероси
ном (толуолом или ксилолом).
18. Концентрированные кислоты, щелочи, ядовитые и сильно
пахнущие вещества обязательно хранить в хорошо вентилируемом
вытяжном шкафу.
19. Концентрированные соляную и азотную кислоты переливать
только в вытяжном шкафу. При разбавлении кислоты необходимо
осторожно, небольшими порциями, при постоянном перемешивании
прибавлять кислоту к воде, а не наоборот! Глаза при этом должны быть
защищены очками.
20. При попадании кислот на кожу нужно быстро промыть
обожженное место струей воды, а затем — 2—3%-ным раствором соды. При
ожоге едкими щелочами надо также хорошо промыть обожженное место
водой, а затем — 2—3%-ным раствором уксусной кислоты. При
случайном попадании кислоты или щелочи в глаза тотчас промыть их большим
количеством воды, а затем обработать тампоном, смоченным в растворе
соды или борной кислоты, и вновь промыть водой.
21. Категорически запрещается хранить бром в хрупкой посуде. Для
этого применяют толстостенные склянки с притертыми пробками. Все
работы с бромом следует проводить только в хорошо вентилируемом
вытяжном шкафу, в резиновых перчатках и защитных очках. При
попадании брома на кожу необходимо немедленно протереть обожженное
место спиртом, а затем смазать глицерином.
22. Приступая к работе в лаборатории, надо ознакомиться с
имеющимися средствами противопожарной безопасности (ящик с песком,
асбестовые или войлочные одеяла, огнетушители и т. д.), аптечкой с
набором необходимых средств оказания первой помощи, а также с их
местонахождением.
23. Легковоспламеняющиеся и взрывоопасные жидкости должны
храниться в металлических шкафах в количестве, не превышающем
ежедневной потребности.
24. Запрещается перегонять эфир или диоксан без предварительной
проверки на содержание в них пероксвдов. Это может привести к взрыву!
25. Нельзя без специального разрешения преподавателя или
лаборанта переносить приборы или реактивы из одной лаборатории в другую.
26. К работе с сжатыми газами (баллонами) допускаются лица,
прошедшие специальный инструктаж по технике безопасности.
27. Если около горящей горелки чувствуется запах газа, перекройте
газ, поступающий к горелке. Затем проверьте исправность резинового
шланга и самой горелки. При обнаружении запаха газа в лаборатории
необходимо выключить газовую магистраль и тщательно проветрить ла-
7
бораторию. Категорически запрещается пользоваться спичками, а также
включать электрический свет.
28. В случае воспламенения одежды необходимо немедленно
набросить на пострадавшего халат, одеяло, пиджак и т. д. Ни в коем случае не
давать ему бежать, так как это усиливает пламя. При возникновении
пожара нужно сразу отключить вентиляцию и электроэнергию и принять меры
к ликвидации загорания. При необходимости вызвать пожарную команду.
При воспламенении эфира, бензола, бензина нельзя применять для
тушения воду. В этих случаях пламя тушат песком или асбестовым одеялом.
29. Следует бережно и аккуратно обращаться с посудой, приборами
и предметами оборудования. Старайтесь разумно экономить реактивы,
воду, газ и электроэнергию.
30. Уходя из лаборатории, проверьте, выключены ли вода, газ и
электроэнергия на рабочем месте.
1.2. Организация рабочего места в лаборатории
и порядок выполнения студентами лабораторных работ
Для проведения лабораторных работ каждому студенту отводится
определенное место в лаборатории, которое для него является
постоянным на время прохождения всего практикума. Рабочее место студента
должно быть укомплектовано необходимым набором химической посуды
и простейшими приборами.
Комплект посуды лучше помещать в верхних ящиках лабораторного
стола. Туда можно положить холодильник Либиха, набор колб
(плоскодонных и круглодонных), в том числе и колбу Вюрца для перегонки
жидкости, дефлегматор, алонж, несколько различных по вместимости
химических стаканов, капельную и делительную воронки, термометры, набор
корковых и резиновых пробок, колбу Бунзена и воронку Бюхнера,
простую воронку для фильтрования, несколько стеклянных палочек и т. д.
Штативы Бунзена с набором лапок, колец и зажимов к ним, водяные
(или другие) бани, электроплитки обычно размещают в специально
отведенном для этого месте в лаборатории.
На полках над столом можно поместить стеклянную посуду с
растворами часто применяемых химических веществ. Там же можно
расположить штатив с обычными стеклянными пробирками, промывалку с
дистиллированной водой, мерный цилиндр, градуированные пипетки,
капельницы с растворами индикаторов и индикаторную бумагу.
При проведении сложных синтезов, требующих специального
оборудования (посуда иа шлифах, различные насадки, мешалки, мотор,
реостат или автотрансформатор — ЛАТР), студент должен обратиться к
лаборанту или преподавателю для получения всего необходимого.
8
Рабочее место должно быть максимально свободным и чистым. На
столе нужно иметь только необходимую химическую посуду и реактивы.
Рядом — рабочий (лабораторный) журнал.
Студент обязан иметь при себе полотенце, мыло и тряпку для
поддержания чистоты на рабочем месте.
Реактивы, необходимые для всего лабораторного практикума,
должны храниться в специальных шкафах (лучше в отдельной комнате —
препараторской) и выдаваться лаборантом. Концентрированные кислоты
и щелочи, ядовитые и сильно пахнущие вещества обычно находятся в
вытяжном шкафу и снабжены соответствующими этикетками.
Переносить их на рабочее место строго воспрещается. Работать с ними нужно
обязательно в вытяжном шкафу.
Следует пользоваться только своей химической посудой и
приборами и ни в коем случае нельзя брать их без разрешения у своего
товарища. Тем более не следует заимствовать отдельные элементы
(колбу, термометр, холодильник и др.) из уже собранной другим
студентом установки.
Если время, отведенное на выполнение лабораторного
практикума, незначительно, то вместо синтезов органических соединений
проводится ознакомительный (малый) практикум. При этом используют
специальный штатив, содержащий набор склянок с небольшим
количеством реактивов и растворов, необходимых для выполнения работ
по определенной теме практикума. При переходе к новой теме набор
реактивов меняется.
Приступать к выполнению лабораторных работ можно только с
разрешения преподавателя (после коллоквиума). При этом выясняется
подготовленность студента к выполнению лабораторной работы. Кроме
знания методики выполнения работы студент должен ответить на ряд
контрольных вопросов, охватывающих основные теоретические разделы
органической химии (в основном, по теме выполняемой работы).
При выполнении работ III раздела студент, допущенный к работе,
составляет «Требование», в котором перечисляет все необходимое для
проведения синтеза: реактивы, посуду, оборудование и т. д. (согласно
прописи синтеза). На с. 10 приведен образец такого «Требования».
По заполненному студентом и подписанному преподавателем
«Требованию» лаборант выдает все необходимое. Затем студент
собирает установку, соблюдая при этом все правила работы со
стеклянной химической посудой и приборами. Перед началом синтеза
следует еще раз убедиться в том, что установка или прибор собраны
правильно, а взятые для работы химические реактивы строго
соответствуют прописи синтеза. Приступая к выполнению лабораторной
работы, необходимо обратить особое внимание на вопросы техники
безопасности.
Требование
Работа № Синтезируемое вещество
Фамилия, и. о. студента
Факультет, группа
Посуда
Реактивы:
Другое оборудование:
Дата Подпись студента
Подпись преподавателя
Закончив работу, необходимо разобрать установку (прибор), вымыть
и высушить посуду, убрать ее в шкаф или сдать лаборанту, а
металлические штативы, лапки, кольца, зажимы поместить в отведенные места.
1.3. Правила оформления и ведения рабочего
(лабораторного) журнала
При выполнении лабораторной работы студент обязан вести рабочий
(лабораторный) журнал, который предназначен для записи всех
наблюдений за ходом эксперимента, расчетов и полученных результатов. При
необходимости в нем зарисовывают схему установки или прибора. Делая
записи в журнале, необходимо четко излагать суть проведенного опыта
или синтеза. Такие записи проводят или в процессе выполнения работы,
или сразу же после ее окончания. При этом не следует пользоваться
черновиками — все расчеты обычно ведут на обратной стороне листа.
Переписывать полностью пропись синтеза (опыта) из руководства нет
необходимости. Отмечают лишь то, что наблюдается при выполнении
эксперимента. Желательно указывать продолжительность отдельных операций, а
также все изменения или возможные отступления от прописи.
В журнале должен быть отражен план синтеза вещества:
а) сборка установки (прибора) и подготовка исходных реактивов;
б) проведение химической реакции;
в) выделение синтезированного вещества;
г) очистка вещества;
д) определение его физико-химических констант.
Для оформления работы по синтезу органических соединений в
журнале можно использовать схему, представленную ниже.
10
Синтез
Работа №
(название синтезируемого вещества)
Литературный источник
(фамилия н инициалы автора,
название руководства, издательство, год издания, страницы)
Работа начата
1. Основная реакция:
2. Побочная реакция:
3. Расчет синтеза:
(дата, часы)
Исходные еещестеа
I
Основные
константы (из
справочника)
ИЛИ
Т
°с
г 20
Количество необходимого вещества
по теории
о
Я
в
0Э
по прописи
о
03
0Э
03
«в
г
4. Схема установки (прибора)
5. Описание хода синтеза (наблюдения, изменения прописи и т. д.)
6. Очистка сырого продукта (методика)
7. Основные константы и выход готового продукта:
Название
вещества
Выход вещества
ВГ
(или мл)
в%
от
теоретического
от выхода,
указанного
в прописи
Константы вещества
полученного при
синтезе
по
литературным данным
8. Работа окончена
9. Вещество сдано
(дата, часы)
(подпись лаборанта)
10. Работа зачтена (или не зачтена, причина)
(подпись преподавателя)
11
При выполнении ознакомительного (малого) практикума (ч. II) для
записи результатов можно предложить другую форму:
Работа №
Название темы
№ опыта
Наименование опыта
Вещества и
реактивы,
необходимые для
опыта
Условия
проведения
реакции
Уравнения
реакций
Наблюдаемые
явления
Выводы
2. Лабораторная химическая посуда и приборы
2.1. Перечень и краткое описание лабораторной
посуды и приборов
В химических лабораториях обычно используют стеклянную посуду.
Она изготавливается, как правило, из специального стекла, которое
устойчиво к кислотам, щелочам и большинству химических реагентов
(кроме фтороводорода и расплавленных щелочей), и обладает
сравнительно небольшим коэффициентом линейного расширения. Посуда из
стекла очень удобна — она прозрачна, хорошо моется и сушится и легко
поддается термической обработке. Основным ее недостатком является
хрупкость.
Стаканы обычно изготавливаются из термостойкого стекла и
бывают различной вместимости (от 50 до 1000 мл). Они служат для
вспомогательных работ — чаще с водными растворами, реже — с органическими
жидкостями.
Колбы — основная лабораторная посуда при проведении
органического синтеза. Колбы бывают круглодонные, плоскодонные,
грушевидные, конические и др. (рис. 1). Плоскодонные и конические колбы
обычно используют в качестве приемников при перегонке жидкости, для
кристаллизации, а также для приготовления растворов. Их нельзя применять
для нагревания жидких веществ до высоких температур и использовать
при вакуум-перегонке. Несоблюдение этих правил может привести к
серьезным последствиям, например взрыву. Для перегонки, в том числе и
под вакуумом, используют круглодонные колбы, которые могут быть
широкогорлыми и узкогорлыми, длинногорлыми и короткогорлыми.
Колбы могут быть двух-, трехгорлыми и т. д. Многогорлые колбы обыч-
12
Рис. 1. Колбы:
а — круглодонные; б — плоскодонные; в — двугорлая, г — трехгорлая, д — коническая,
е — грушевидные
но применяют для специальных синтезов. Круглодонные колбы,
снабженные отводной трубкой, называют колбами Вюрца (рис. 2). Они
предназначены для перегонки жидкости под атмосферным давлением.
Круглодонные колбы с боковым вертикальным ответвлением, имеющим
отводную лрубку, называются колбами Клайзена и используются для
перегонки при пониженном давлении. Применяется при перегонке и колба
Фаворского — круглодонная колба с дефлегматором и отводной трубкой.
Холодильники служат для охлаждения и конденсации паров,
образующихся при кипении органических жидкостей. Самый простой
холодильник, воздушный, представляет собой длинную стеклянную трубку.
Такие холодильники применяют при перегонке высококипящих
жидкостей. При перегонке
низкокипящих
жидкостей используют
холодильник Либиха —
такую же стеклянную
трубку, но впаянную в
другую, более
широкую. Внешняя часть
холодильника
(«рубашка») имеет два
отростка, на которые
надевают резиновые
трубки. Одну из них при- Рис 2. Колбы для перегонки:
а — колба Вюрца, б — колба Клайзена: в
соединяют к водопро- ского (с дефле;матором)
колба Фавор-
13
водному крану, а
другую отводят в раковину
(охлаждающий агент —
вода). Холодильник Ли-
биха может быть
нисходящим и обратным.
Если он используется как
обратный холодильник,
то его крепят в штативе
строго вертикально;
если же как нисходящий,
то его крепят с
небольшим наклоном
(относительно стола) в сторону
приемника. И в том и в
другом случае вода в
холодильник подается
через нижний отросток.
Для лучшей
конденсации паров кипящей
жидкости холодильник
должен быть полностью
заполнен проточной
водой. Холодильник соединяют с приемником при помощи специального
приспособления — алонжа, позволяющего направлять стекающую
жидкость. В лабораторной практике часто используют и другие типы
холодильников, например шариковые и змеевиковые, охлаждающая способность
которых лучше (рис. 3).
Капельные воронки предназначены
для медленного прибавления компонента
к реакционной смеси до или во время
проведения органического синтеза.
Воронки бывают с цилиндрическими,
шарообразными или грушевидными
емкостями для жидкости (с пробкой в верхней
части) и часто — с длинными трубками с
краном (рис. 4). Чтобы стеклянный кран
не пропускал жидкость, шлиф слегка
смазывают вазелином.
Делительные воронки применяют
для разделения несмешивающихся
жидкостей и для экстракции. Эти воронки в
отличие от капельных имеют более тол-
Рис. 3. Холодильники:
а — воздушный; б — Либиха; в — обратный шариковый;
г — обратный с охлаждаемой спиралью (змеевнковый);
д — обратный с охлаждающей спиралью (Димрота), е —
обратный с двойным охлаждением (рубашкой и
охлаждаемой спиралью)
«й
Рис. 4. Капельные воронки
14
Рис. 5. Дефлегматоры:
а — шариковый; б — елочный; в — с
насадкой
стые стеклянные стенки и меньшую
длину трубки.
Дефлегматоры бывают самой
различной конструкции и применяются
для более тщательной фракционной
перегонки (рис. 5). В верхнее
отверстие дефлегматора вставляют
термометр, а с помощью отводной трубки в
верхней части дефлегматор соединяют
с холодильником.
Хлоркалыдиевые трубки
предназначены для защиты реакционной
смеси или одного из реагирующих
веществ от влаги воздуха. В качестве
поглотителя влаги используют хорошо
прокаленный хлорид кальция.
Промывные склянки
применяются для осушки, очистки или
улавливания некоторых газов. Наиболее часто используют склянки Тищенко и
Дрекселя (рис. 6). Обычно они содержат серную кислоту или
концентрированный раствор щелочи.
Пробки служат не только для закупоривания химической посуды, но
и для соединения отдельных частей прибора. Они бывают резиновыми,
корковыми, пластмассовыми или стеклянными. Предпочтение отдают
тем или другим в зависимости от характера применяемых веществ,
условий и целей работы. Для соблюдения особой герметичности применяют
резиновые пробки. Однако резиновые, корковые и пластмассовые пробки
нестойки к действию высокой температуры и некоторых химических
реагентов. Отверстия в пробках делают специальными металлическими
сверлами, диаметр которых должен быть несколько меньше
необходимого отверстия (сверлить
начинают с узкого конца пробки).
Термометры. В
химических лабораториях чаще всего
применяют ртутные
термометры. Термометры бывают
различной конструкции.
Наиболее распространены
обычные ртутные термометры
(химические) и технические (с
а б
Рис. 6. Промывные склянки:
а — склянка Тищенко; б— склянка Дрекселя
прямой и изогнутой
трубками). Используют также и
палочковые термометры, пред-
15
Рис. 7. Газовые горелки Бунзена (а)
и Теклю (б):
1 — регулятор подачн воздуха; 2 — винт,
регулирующий подачу газа
ставляющие собой толстостенный
капилляр, на который с наружной
стороны нанесена градуировка в виде
закрашенных в черный или красный
цвет штрихов. Обычные химические
термометры позволяют измерять
температуру в пределах от -30 до 360 °С.
Наиболее распространены
термометры со шкалой в 100, 150, 200, 250,
300 и 360 °С. Иногда используют и
специальные (газонаполненные)
термометры со шкалой до 550 и даже до
750 °С. Для очень точных измерений
применяют образцовые
(«нормальные») термометры, имеющие цену
деления в 0,1 °С. Такими
термометрами обычно определяют температуры плавления и кипения веществ.
При работе с термометрами необходимо соблюдать ряд правил.
После каждого измерения температуры остывший термометр протирают
спиртом. Термометр следует хранить в специальном футляре или
отведенном для него месте в ящике лабораторного стола. Если же термометр
разбился, необходимо сразу же собрать пролившуюся ртуть и
уничтожить ее следы.
Газовые горелкн широко используют в химических лабораториях
для нагревания и прокаливания. Они просто устроены и надежны в
эксплуатации. Газовые горелки бывают, в основном, двух типов: Бунзена и
Теклю (рис. 7). Принципиальной разницы между ними нет, однако
последние более удобны в работе. Газ подводится через нижний боковой
отвод горелки. Приток воздуха регулируется специальной
регулировочной гильзой (горелка Бунзена) или нижним кольцом (горелка Теклю),
находящимися несколько выше бокового отвода. Газовые горелки в
зависимости от доступа воздуха дают как светящее («холодное») пламя (до
500 °С), так и несветящее («горячее») — с температурой до 1500 °С. При
работе с газовыми горелками необходимо строго соблюдать правила
техники безопасности.
Электрические плиты бывают с открытой и закрытой спиралью.
Первые применяют, если исключено попадание на них нагреваемого вещества.
Наиболее удобны и безопасны электрические плиты с закрытой спиралью.
Такие плиты с асбестовой нагреваемой поверхностью и выступающими
бортиками можно использовать в качестве песчаных бань. Для этого на
асбестовую поверхность достаточно насыпать ровным слоем песок.
Ни в коем случае нельзя пользоваться неисправными
электрическими плитами, например, имеющими оголенный шнур.
16
Банн для нагревания обеспечивают
равномерное и безопасное нагревание,
предохраняют реакционную смесь от перегрева,
препятствуют возникновению пожаров
(например, при повреждении стеклянной посуды).
Бани бывают водяные, песчаные, воздушные и
масляные. Теплопроводящей средой в них
служат, соответственно, вода, песок, воздух и
масла. Выбор этих бань определяется
свойствами нагреваемого вещества или реакционной
смеси, а также температурой, необходимой
для их нагревания. Так, водяные бани
применяют при нагревании веществ до 100 °С (их
желательно заполнять дистиллированной
водой); для нагрева до более высокой темпера- ?ис'8- Парообразователь:
^ , 1 — корпус; 2 — стеклян-
туры используют другие виды бань (макси- ная ^Ц 3 _ водомер.
мальная температура, достигаемая при нагре- Ное стекло
вании на электрической воздушной бане,
приблизительно 250 °С, на песчаной — не выше 400, на масляной — до 150 °С).
Следует помнить, что водяные бани нельзя применять при работе с
металлическим натрием или калием.
Парообразователь служит источником пара для перегонки с
водяным паром и для нагрева водяных бань. Он представляет собой
металлический сосуд (чаще всего — медный) со стеклянной трубкой, опущенной
почти до дна. Эта трубка предохраняет от резкого повышения давления
при сильном нагревании воды (рис. 8), т. е. играет роль
предохранительного клапана. Длина трубки должна быть не менее 70...80 см. Работать
без предохранительной трубки категорически запрещается.
Эксикаторы — емкости из толстостенного стекла — предназначены
для высушивания твердых веществ. Различают обычные и
вакуум-эксикаторы (рис. 9). В последних имеется тубус, в который на резиновой
f......»...,......^
Рис. 9. Эксикаторы:
а — обычный; б — вакуум-экснкаторы
орень
Л И С Т Г К А
302440
17
пробке или на шлифе вставляют трубку с
краном. Эту трубку через манометр и
предохранительную склянку соединяют с
водоструйным насосом и создают в
эксикаторе вакуум. Вещество, которое
подвергают сушке, на часовом стекле или в чашке
Петри помещают в эксикатор на
фарфоровую подставку. В качестве осушающего
агента применяют безводные хлорид
кальция, сульфат магния, сульфат натрня,
натронную известь, гидроксид натрня, оксид
фосфора (V) и др. Концентрированную
серную кислоту также используют для
й поглощения влаги, остатков спирта, эфира,
ацетона, анилина и пиридина, но только в
Рис. 10. Типы уплотняющих обьиныхэксика^рах.
затворов для мешалок: _ , t ч
а - с использованием ртути или Резиновые трубки (шланги) служат
глицерина; б — с использованием ДЛЯ соединения отдельных частей в прибо-
отрезка резинового шланга, 1 — pax и для подвода и отвода воды и газа.
мешалки; 2 — отрезки резинового Однако резиновые трубки легко разруша-
шланга; 3 — ртуть или глицерин, „
4 - стеклянная трубка; 5 - стек- ются ПР* Действии высокой температуры и
ляиная насадка некоторых газов (хлор, кислород, хлорово-
дород, аммиак и др.). Поэтому часто
применяют трубки из полиэтилена, которые устойчивы к действию
большинства органических веществ и агрессивных сред. Такие трубки обычно
используют только при комнатной температуре (при нагревании они
легко деформируются).
Установки для перемешивания. Часто при проведении
органических синтезов требуется перемешивание. Если оно осуществляется в
открытых сосудах, то используют стеклянные или металлические
мешалки различных конструкций. Стержень мешалки крепят встык с
валом мотора с помощью резиновой трубки, а затем его пропускают
или через отрезок стеклянной трубки, или через отверстие пробки,
прикрепленной с помощью лапки к штативу. Это необходимо для
придания мешалке нужного направления. При размешивании
реакционной смеси, изолированной от внешней среды специальными
устройствами, препятствующими действию влаги воздуха и
предотвращающими утечку летучих веществ из реактора, применяют специальные
затворы. Самым простым уплотнением может служить отрезок
резинового шланга. Полная герметизация достигается применением
ртутного или глицеринового затвора (рис. 10). Скорость перемешивания
регулируют с помощью реостата или автотрансформатора (ЛАТР).
18
Рис. 11. Форштосы
Форштосы — специальные насадки для колб. Они бывают
различных ввдов — со шлифами и без шлифов (рис. 11). Их часто используют в
качестве насадок для одногорлых колб, предназначенных для проведения
нескольких операций одновременно (например, для нагревания с
обратным холодильником при перемешивании с одновременным добавлением
вещества в реакционную смесь и т. д.).
Приборы на шлифах. В лабораторной практике широко
используется химическая стеклянная посуда со стандартными шлифами,
позволяющими быстро соединять друг с другом отдельные части химических
приборов, добиваясь при этом высокой герметичности. Одновременно
исключается контакт органических веществ с пробками, резиновыми
соединениями и т. д. Чаще используются конусообразные шлифы, которые
имеют стандартные размеры («нормальные» шлифы). Они обозначаются
номерами, соответствующими верхним диаметрам (в миллиметрах),
например 10; 10,5; 14,5; 29. Каждый шлиф состоит из двух частей: муфты и
керна. Перед сборкой приборов керн следует слегка смазать вазелином
(или вакуумной смазкой), вставить его в муфту и при легком нажатни
провернуть, добиваясь при этом прозрачности шлифа. Смазки берут
немного — она не должна выдавливаться из шлифа. Если отдельные части
прибора имеют шлифы разных размеров, то следует применять
различные переходы на шлифах.
Применение посуды со стандартными шлифами позволяет быстро и
легко собрать самую сложную установку (прибор).
2.2. Требования к лабораторной посуде и работа с ней
при проведении химического эксперимента
Приступая к работе, необходимо убедиться в чистоте химической
посуды. Даже малейшие следы загрязнений могут повлиять на ход
химического эксперимента. Храниться должна только чистая и сухая посуда.
Использованную посуду следует сразу же мыть и тщательно высушивать.
19
Загрязнения удаляют различными способами: механическими,
химическими, физическими или их комбинацией.
Посуду, не загрязненную веществами, плохо растворимыми в воде
(смола, жир и т. д.), обычно моют теплой водой с применением мыла,
синтетических моющих средств, 10%-ного раствора тринатрийфосфата и
др. При этом используют различные щетки и ерши. Вымытую посуду
несколько раз споласкивают дистиллированной водой для удаления
солей, содержащихся в обычной воде. Стеклянная посуда считается чистой,
если на ее стенках вода образует равномерную пленку.
Если посуда загрязнена органическими веществами, которые
нерастворимы в воде, то для очистки применяют органические растворители
(спирты, ацетон, тетрахлорид углерода, бензин, скипидар и др.). Однако
надо помнить, что почти все органические растворители (кроме ССЦ)
огнеопасны и работать с ними можно только вдали от огня. После
обработки посуды этими растворителями ее снова моют водой и сушат.
Иногда приходится прибегать к химическим методам очистки
посуды. Если посуду невозможно отмыть водой и органическими
растворителями, то ее моют хромовой смесью, подогретым 5%-ным раствором пер-
манганата калия, смесью соляной кислоты и пероксида водорода (смесь
Комаровского), концентрированней серной кислотой или
концентрированными (до 40%) растворами щелочей.
Особого внимания заслуживает применение хромовой смеси.
Хромовокислые соли в кислоте являются очень сильными окислителями. Поэтому хромовую
смесь часто применяют, когда никакие способы мытья не помогают. Смесь
готовят добавлением в концентрированную серную кислоту измельченного
дихромата калия (5% от массы кислоты). Перед мытьем хромовой смесью посуду вначале
ополаскивают водой, а затем заполняют подогретой смесью. Иногда, если
требуется наиболее тщательная очистка, хромовую смесь оставляют в посуде на
продолжительное время (например, на ночь). После использования хромовую смесь
сливают в специальный сосуд и хранят. Уменьшение активности смеси
контролируют визуально: свежеприготовленная хромовая смесь имеет темно-оранжевый
цвет, а в процессе использования она меняет окраску до темно-зеленого.
Вымытую хромовой смесью посуду в дальнейшем моют, как обычно.
После мытья посуду тщательно сушат (за исключением случаев,
когда предстоящая работа связана с водными растворами). Это особенно
важно, если в ней будут проводить реакции, идущие только в отсутствие
следов влаги. Применяют методы холодной сушки (без нагревания) и
методы сушки при нагревании (горячая сушка). Первый метод — самый
простой: вымытую посуду надевают на колышки специальной доски,
обычно висящей над раковиной, и оставляют до тех пор, пока посуда не
высохнет. Можно сушить посуду струей холодного или горячего воздуха.
Чтобы ускорить процесс сушки посуды, ее споласкивают вначале
небольшим количеством чистого этилового спирта, а затем — чистым эфи-
20
ром. Однако лучше сушить посуду, продувая ее горячей струей воздуха
Для этого трубку, через которую пропускают струю воздуха (от
магистрали сжатого воздуха, электровоздуходувки или меха), нагревают
пламенем газовой горелки или электроспиралью. Быстро сохнет посуда и в
специальных сушильных шкафах, но перед тем как поместить посуду в
сушильный шкаф, нужно избавиться от избытка воды в ней.
Чистую и сухую посуду хранят в закрытых шкафах или ящиках
стола. Ее можно хранить и на столе, но обязательно накрытой чистой
фильтровальной бумагой.
При работе со стеклянной химической посудой необходимо
соблюдать определенную осторожность: она часто бьется при случайных
ударах и неправильном хранении (при этом следует опасаться порезов).
Поэтому при сборке установок надо проявлять осторожность и внимание.
Так, не следует слишком туго прижимать посуду лапками для крепления
к штативу. Для этого лапки лучше оклеить мягкой тканью (фланелью)
или прокладкой, вырезанной из корковой пробки. Если лапки не
створчатые, а «пальцеобразные», то на каждый «палец» обычно надевают
небольшие отрезки резиновых трубок. Все эти простые приспособления и
правила оказываются очень полезными при работе со стеклянной посудой.
Химический эксперимент в органической химии часто
сопровождается нагреванием. Поэтому надо заранее решить вопрос, выдержит ли
данная посуда нагревание. Иногда следует заменить ее на посуду из
другого стекла — термостойкого (шоттовского или иенского). Посуда,
изготовленная из такого стекла, имеет свои фирменные знаки в виде надписей
или цветных полос (черных или красных).
При работе с посудой на шлифах часто случается, что
загрязненные в процессе эксперимента шлифы трудно разъединить простым
вращением. Для этого нужно осторожно, постукивая по муфте куском
дерева или раскачивая шлифы, попытаться их разъединить. Иногда
полезно прибегнуть к нагреванию муфты коптящим пламенем
горелки, стараясь при этом как можно меньше нагревать керн. Помогает
введение в зазор между керном и муфтой капли воды, соляной
кислоты или органических растворителей. Если не помогает и это, то
шлифы разъединяют кипячением в разбавленном растворе соляной или
уксусной кислоты.
Если требуется разрезать тонкую стеклянную трубку или палочку, то
вначале на ее поверхности делают острым ребром напильника надрез, а
затем, поворачивая ее этим надрезом вверх, растягивающим движением
обеих рук ломают. Когда такую операцию необходимо провести с
широкими и толстыми трубками, то к месту глубокого надреза (лучше по всей
окружности) подносят раскаленный докрасна конец стеклянной палочки.
Обрезанные концы трубок обязательно оплавляют в пламени газовой
горелки.
21
2.3. Правила сборки установок для выполнения
органических синтезов
Выбор установки (прибора) для синтеза определяется, в первую
очередь, задачей, стоящей перед экспериментатором, условиями проведения
реакции, а также свойствами исходных и конечных продуктов.
Сборка установки должна проводиться с большой тщательностью и
аккуратностью, так как это является непременным условием успешной и
безопасной работы. Собранные установки должны быть не только
грамотными конструкционно, но и иметь привлекательный вид.
Остановимся на общих правилах сборки приборов. Как уже
говорилось, отдельные части установки необходимо соединять друг с другом
осторожно, подбирая пробки, трубки и другие детали еще до закрепления
прибора на штативе. Если прибор собирают на шлифах, то их следует
предварительно смазать. Посуду подбирают такого размера, чтобы
реагирующие вещества занимали не более половины объема (или не более
/3 объема). Если реакционная смесь будет нагреваться, то обязательно
применяют круглодонную колбу соответствующего размера. После тоге
как собраны отдельные части установки, их закрепляют в лапках
штатива. Установку всегда собирают, начиная с ее предполагаемого «верхах
или с основного блока. Например, при сборке установки для простой
перегонки следует вначале укрепить на штативе колбу Вюрца, затем к ней
присоединить нисходящий холодильник, потом алонж и, наконец,
подвести под него приемник. Вся установка должна быть собрана в одноь
плоскости или по одной линии (за исключением некоторых случаев), без
перекосов или напряжения стеклянных частей прибора. Это особенно
важно при работе со стандартными шлифами, когда они должны
присоединяться друг к другу без особых усилий со стороны экспериментатора
В то же время нужно следить, чтобы при соединении отдельных чаете!
прибора выполнялись условия герметичности. Если стеклянные част*
установки достаточно тяжелые (например, колба с обратным
холодильником, мешалкой, капельной воронкой, термометром и т. д.), то крепии
их к штативу следует несколькими лапками. При этом дефлегматоры
мешалки, обратные холодильники крепят строго вертикально, а
нисходящие холодильники — наклонно, чтобы жидкость стекала в приемник
не попадая на пробки. Если установка предназначена для работы под ат
мосферным давлением, то необходимо, чтобы она свободно сообщалась с
атмосферой во избежание повышения давления в системе. Для защить
реагирующих веществ от действия влаги воздуха (если это нужно) ис
пользуют хлоркальциевые трубки.
Приступая к работе, следует еще раз внимательно осмотреть прибор
и убедиться в правильности его сборки.
22
3. Методы очистки и выделения
органических соединений
Полученные при синтезе вещества, как правило, содержат некоторс
количество примесей (исходные вещества, не вступившие в реакции
побочные продукты, растворители и др.)- Чтобы избавиться от них, пр*
меняют различные методы очистки и выделения органических вещесп
Эти методы довольно разнообразны и зависят, в основном, от агрегатно
го состояния соединения.
3.1. Фильтрование
Фильтрование — процесс отделения твердых компонентов смеси,
находящихся в осадке, от маточных растворов (жидких компонентов)
посредством пористой перегородки — фильтра. В качестве фильтра
обычно используют фильтровальную бумагу, которая может быть
различной пористости. Фильтрами- могут служить также различные ткани,
пористое стекло, асбест, обычная и стеклянная вата и др. При этом
необходимо помнить, что фильтрующие материалы не должны
взаимодействовать ни с растворителем, ни с отделяемым осадком.
Фильтрование можно проводить различными способами. Это
определяется как характером растворителя, так и свойствами отделяемого
вещества при фильтровании- Обычно пользуются двумя способами
фильтрования: при нормальном и пониженном давлении.
Фильтрование при нормальном давлении — наиболее простой и
часто применяемый в лабораторной практике способ, не требующий
сложных приспособлений. Для этого необходимы стеклянная воронка и
фильтр. Бумажные фильтры могут быть двух видов: простые и
складчатые. Последние применяются чаще, так как имеют большую
фильтрующую поверхность, и это намного ускоряет процесс фильтрования.
Простой фильтр можно изготовить так: квадратный кусок
фильтровальной бумаги складывают вчетверо и обрезают по окружности таким
образом, чтобы готовый фильтр имел вид конуса. При этом размер
конуса должен соответствовать размеру фильтровальной воронки.
Складчатый фильтр имеет более сложную форму. Для его
изготовления вначале поступают так же, как и для получения простого фильтра.
Затем четвертушки бумаги разгибают и на фильтре, сложенном вдвое,
делают сгибы «гармошкой», как показано на рис. 12.
Фильтр (простой или складчатый) вставляют в воронку,
укрепленную в кольце, которое присоединено к штативу зажимом. Под воронку
ставят стакан, и прибор для фильтрования холодных растворов готов
23
Рис. 12. Последовательность действий при изготовлении простого фильтра (а) и
складчатого (б)
(рис. 13). Для фильтрования горячих растворов применяют специальную
воронку, обогреваемую электрической спиралью или горячей водой (рис. 14).
При фильтровании веществ, имеющих низкую температуру
плавления, используют воронки с охлаждением.
Фильтрование при пониженном давлении (отсасывание)
применяют для ускорения процесса фильтрования. Основным прибором служат
фарфоровая воронка Бюхнера и толстостенная колба Бунзена,
соединенная через предохранительную склянку с водоструйным насосом (или
лабораторным насосом Комовского) (рис. 15). Для предотвращения
последствий возможного разрыва колбы Бунзена в ходе фильтрования ее
необходимо обвернуть полотенцем. Чтобы фильтруемая жидкость не попала
между воронкой и фильтром, на дырчатое дно воронки Бюхнера кладут
бумажный фильтр, смоченный дистиллированной водой. Диаметр фильтра
Рис. 13. Установка для
фильтрования через стеклянную воронку
24
Ша
Рис. 14. Приспособления для обогрева
фильтровальной воронки горячей
водой (а) и электрической спиралью (б):
1 — воронка для фильтрования; 2 —
специальная воронка с двойными стенками
(«рубашкой»), между которыми находится вода; 3 —
«отросток» для обогрева воды; 4 —
специальная электрическая воронка для горячего
фильтрования (электрическая печь); 5 —
электрическая спираль
должен быть немного меньше внутреннего
диаметра воронки, но таким, чтобы фильтр
полностью закрывал все отверстия.
Водоструйный насос при нормальном напоре воды
может создавать вакуум в 1—2 кПа (8—15 мм
рт. ст.). Чтобы в колбу Бунзена случайно не
попала вода от водоструйного насоса, между
ним и колбой устанавливают
предохранительную склянку (некоторые конструкции водо- Л
- т л _ ~ Рнс. 15. Воронка Бюхнера
струйных насосов позволяют обходиться без /тч v у
rJ . . (7); дырчатое дно воронки
предохранительных склянок). Фильтруемую (ввд сверху) (2); колба
жидкость необходимо доливать равномерно, Бунзена (5)
чтобы осадок все время находился под слоем
жидкости. Нужно стараться, чтобы осадок не попал между фильтром и
дном воронки Бюхнера, иначе некоторая часть осадка может попасть в
колбу Бунзена.
После окончания фильтрации осадок на фильтре отжимают
стеклянной пробкой. Если растворитель или осадок легко окисляются воздухом,
или гигроскопичны, или взаимодействуют с СО2 воздуха, то процесс
фильтрования следует проводить в атмосфере инертного газа.
3*2. Кристаллизация
Кристаллизацией называют процесс образования и роста кристаллов
из раствора, расплава или газовой среды. Это один из важнейших
методов, применяемых для очистки твердых органических веществ. Он
основан на различной растворимости органического вещества и
сопутствующих ему примесей в данном растворителе при различных температурах.
Загрязненное примесями вещество растворяют при нагревании в
подходящем растворителе, а затем горячий раствор отфильтровывают от
нерастворимых примесей и дают охладиться. Выпавший осадок
отфильтровывают и сушат.
В качестве растворителей применяют этиловый спирт, ацетон,
бензол, хлороформ, диоксан, уксусную кислоту, петролейный эфир, воду и
др. Если данные о растворимости очищаемого кристаллизацией вещества
отсутствуют, то растворитель подбирают опытным путем. Хорошо
подобранный растворитель при температуре, близкой к точке кипения,
должен растворять, по крайней мере в пять раз больше вещества, чем при
комнатной температуре. Иногда, если очищаемое вещество хорошо
растворяется в растворителе при нагревании, но плохо кристаллизуется из
него при охлаждении, кристаллизацию проводят из смеси различных
растворителей, умело подобрав их соотношение.
25
В общем случае необходимо учитывать следующие требования к
растворителям:
1) растворитель должен хорошо растворять вещество при нагревании
и плохо — при охлаждении;
2) растворитель не должен химически взаимодействовать с
очищаемым веществом;
3) растворитель желательно применять в минимальном количестве
(иначе растворенное вещество не будет полностью выделяться при
охлаждении).
Из горячего раствора сначала кристаллизуется то вещество, которое
труднее растворяется в данном растворителе или присутствует в большем
количестве.
Чтобы вызвать процесс кристаллизации в пересыщенном растворе, к
него добавляют в качестве «затравки» несколько кристаллов того
вещества, которое необходимо выкристаллизовать. Помогает и потиранш
стенок сосуда с раствором стеклянной палочкой.
Довольно часто при проведении процесса кристаллизации применя
ют активированный уголь. Из-за высокой абсорбционной активности oi
хороши удаляет из раствора окрашенные загрязнения и различные смо
лообразные продукты. Однако активированный уголь нельзя добавлять i
сильно нагретому раствору, так как это может привести к выбросу жид
кости за счет ее бурного вскипания.
При использовании легковоспламеняющихся растворителей (ди
океан, бензол, этиловый спирт и др.) растворение необходимо вести npi
осторожном нагревании в круглодонной колбе с обратным водяным хо
лодильником.
Опыт. Кристаллизация бензойной кислоты* Небольшое количе
ство (около 1 г) загрязненной бензойной кислоты растворяют в 30—50 м.
кипящей воды в открытой колбе или химическом стакане. Полученны
горячий раствор быстро фильтруют через складчатый фильтр, помещен
ный в стеклянную воронку. Собранный в стакан или коническую колб
фильтрат охлаждают ледяной водой при перемешивании. Выделяютс
белые кристаллы бензойной кислоты. Через 30 мин осадок отфильтровь
вают на воронке Бюхнера с использованием водоструйного насоса и с)
шат при 60 °С. Т. пл. 122 °С.
3.3. Перегонка
Перегонка — процесс отделения жидких веществ от нелетучих npi
месей или разделения летучих веществ с различной температурой кип<
ния. Это достигается нагреванием жидкости до кипения и последующе
конденсацией ее паров в холодильнике.
26
Рис. 16. Схема установки для простой перегонки
Существуют три способа перегонки жидкости:
а) при нормальном давлении (простая и фракционная перегонка);
б) при пониженном давлении (перегонка в вакууме);
в) с водяным паром.
Перегонка при нормальном давлении (простая перегонка). Этот
способ применяют, если разница в температурах кипения веществ,
входящих в состав разделяемой смеси, не менее 80—100 °С или если
основное вещество необходимо отделить от нелетучих примесей.
Схема установки для простой перегонки показана на рис. 16. Она
состоит из колбы Вюрца / с термометром 2, нисходящего холодильника
Либиха 3, алонжа 4 и приемника 5. Колбу Вюрца подбирают такого
размера, чтобы перегоняемая жидкость заполняла ее не более чем на 2/3
объема. При соединении колбы с холодильником необходимо, чтобы конец ее
отводной трубки выступал из пробки в холодильник не менее чем на 2—3
см. Ртутный шарик термометра должен находиться примерно на 0,5 см
ниже отверстия отводной трубки. В этом случае шарик термометра
хорошо омывается парами перегоняемой жидкости.
Если температура кипения перегоняемой жидкости не выше 120—
|30 °С, то применяют проточное водяное охлаждение. При перегонке
жидкостей с более высокой температурой кипение рекомендуется
использовать в качестве охлаждающего компонента непроточную воду (т. е. воду,
которая осталась в холодильнике после прекращения ее подачи) или
применять воздушный холодильник. Чтобы жидкость кипела
равномерно, в колбу помещают кусочки обожженного неглазурованного фарфора
или длинные стеклянные капилляры, запаянные с одного конца и погру-
27
женные в жидкость другим концом. Скорость перегонки регулируете)
скоростью поступления жидкости в приемник (не более 1—2 капель i
секунду).
При перегонке индивидуального вещества его температура кипеню
остается постоянной в течение всей перегонки. Если перегоняется смеа
двух веществ, температуры кипения которых различаются значительно
то вначале отгоняется жидкость с более низкой температурой кипения
Если температура кипения начинает возрастать, это означает, что начи
нает перегоняться другая жидкость, имеющая более высокую температу
ру кипения, чем первая. В процессе перегонки второй жидкости такж!
устанавливается постоянная температура. Таким образом, меняя прием
ники, можно собрать несколько фракций; в первых будет преобладать низ
кокипящая часть перегоняемой смеси, а в последних — высококипящая.
Если перегоняемая смесь состоит из компонентов, температуры ки
пения которых близки и которые не образуют азеотропных смесей, при
меняют дробную, или фракционную, перегонку. Для этого обычно ис
пользуют дефлегматоры и ректификационные колонки. В них часть па
ров перегоняемой смеси конденсируется (за счет охлаждения наружны!
воздухом), превращаясь в жидкость, обогащенную сравнительно высоко
кипящим компонентом. Оставшийся пар, наоборот, обогащается боле
летучей частью смеси. Таким образом, пары, проходящие через колонку
богаче летучим компонентом, чем пары, находящиеся над перегоняемо
жидкостью. Жидкость (конденсат), стекающая в колбу, наоборот, содер
жит высококипящие вещества.
Если температура кипения перегоняемых жидкостей не выше 80 °<
(спирт, ацетон, эфир, бензол и др.), то нагревание следует проводил
только на водяной бане.
Перегонка при пониженном давлении (перегонка в вакууме
Этот способ перегонки применяют, если перегоняемые вещества полис
етъю или частично разлагаются при температуре кипения (при атмосфер
ном давлении). Если перегонку проводить при пониженном давлении, т
температура кипения веществ понижается, а значит, уменьшается ил
вовсе устраняется возможность их разложения.
Схема установки для перегонки при пониженном давлении показа*
на рис. 17. Она состоит из колбы Клайзена 1 с термометром 8 и капиши
ром 2, холодильника 4, алонжа 5 и приемника 6. Колбу закрывают рез]
новой пробкой со стеклянной трубкой, оттянутой на конце в тонкий к;
пилляр. Верхнюю часть этой трубки соединяют с резиновым шланго!
имеющим зажим. При пониженном давлении в колбе через этот капшш
проходят в перегоняемую жидкость пузырьки воздуха, что способству(
равномерному перемешиванию и кипению жидкости. Скорость пропу
кания пузырьков воздуха регулируют зажимом на отрезке каучука (чтоб
зажим не полностью перекрыл резиновый шланг, в него вставляют то]
28
Рис. 17. Схема установки для перегонки при уменьшенном давлении
кую проволоку). Колбу Клайзена соединяют с нисходящим холодильни
ком Либиха Алонж должен иметь отвод для соединения системы с мае
ляным вакуумным или водоструйным насосом 7. Если нужно собрать все
фракции, входящие в состав перегоняемой смеси, применяют
специальные алонжи («пауки»), позволяющие крепить несколько приемников
(круглодонные колбочки). Давление, при котором осуществляют
перегонку, измеряют ртутным манометром 8, присоединенным к системе.
Чтобы избежать переброса продуктов в насос (при использовании
масляного насоса), между ним и прибором устанавливают поглотительную
систему (колонки с активированным углем и твердым гидроксидом
натрия). Если пользуются водоструйным насосом, то применяют
предохранительную склянку.
Работа с установкой требует особого соблюдения правил техники
безопасности. После того как прибор собран, необходимо убедиться в
ее полной герметичности и только затем заполнять колбу Клайзена
жидкостью. Колбу, если в этом есть необходимость, нагревают на
водяной бане. Перегонку проводят в защитных очках, а еще лучше — в
защитной маске. Во избежание взрыва нельзя перегонять вещества
досуха. И наконец, при выключении системы воздух не должен
быстро входить в нее.
Перегонка с водяным паром. Этим способом пользуются для
выделения из смесей веществ, которые кипят при довольно высокой
температуре, а поэтому не исключена возможность их разложения. Перегонять
с водяным паром можно только такие органические вещества, которые
нерастворимы или труднорастворимы в воде, а также не
взаимодействующие с ней. Кроме того, этот метод применяется для разделения таких
смесей веществ, из которых только одно улетучивается с паром.
29
i A
Рис. 18. Схема установки для перегонки с водяным паром
Известно, что кипение жидкости происходит, если сумма
парциальных давлений насыщенного пара воды и вещества будет равна
атмосферному давлению. Поэтому, используя перегонку с водяным паром, можно
перегонять вещества при температуре, которая значительно ниже
температуры кипения самого низкокипящего компонента. Во время перегонки
температура кипения остается постоянной, пока не перегонится одна из
жидкостей. Затем температура кипения повышается до значения,
соответствующего температуре кипения остающейся в колбе жидкости
(например, воды). Образующийся при перегонке пар содержит все
компоненты смеси в объемном соотношении, пропорциональном парциальному
давлению каждой жидкости, входящей в состав смеси.
Для перегонки с водяным паром применяют прибор, который
состоит из парообразователя 1, перегонной колбы 3, холодильника 4, алонжа 5
и приемника 6 (рис. 18). Парообразователь заполняют водой
приблизительно до половины и соединяют через тройник 2 с колбой. Колбу плотно
закрывают резиновой пробкой с двумя отверстиями. В одно из них
помещают стеклянную трубочку, нижний конец которой доходит почти до
дна колбы, а верхний с помощью короткой резиновой трубки соединяют
через тройник с парообразователем. Во второе отверстие вставляют
короткую, изогнутую под углом в 30—45° стеклянную трубку,
соединяющую колбу с холодильником. На тройник, который связывает между
собой парообразователь и колбу, надевают короткую резиновую трубку с
зажимом. Зажим остается открытым до начала перегонки для того, чтобы
в перегонной колбе не собирался конденсат в результате охлаждения
водяных паров. В колбу помещают вещество, собирают прибор и
подогревают парообразователь. Одновременно (через асбестовую сетку)
подогревают колбу. Как только начнет образовываться пар, нагревающий
жидкость в перегонной колбе, резиновую трубку, надетую на тройник,
закрывают зажимом. Спустя некоторое время в приемнике собирается
30
эмульсия (вещество в воде), расслаивающаяся при стоянии. В зависим
сти от плотности перегоняемое вещество будет образовывать верхн1
или нижний слой. Перегонку заканчивают, когда в холодильнике буд;
образовываться капли чистого дистиллята (воды). Затем открывают з
жим на тройнике (чтобы жидкость из колбы не засосалась в парообраз
ватель) и выключают парообразователь.
Опыт 1. Очистка спирта. Небольшое количество спирта (окол
30 мл), загрязненного любой растворимой примесью (водой, ацетонод
пиридином и др.), очищают методом простой перегонки. Исходя из кол*
честв взятого и очищенного спирта, рассчитайте процентный состав пер
воначальной смеси. Следует обратить внимание на показание термометр
в процессе перегонки. Сохраняется ли оно все время постоянным?
Опыт 2. Перегонка анилина с водяным паром. Смесь около 10 mj
анилина и 80—100 мл воды помещают в колбу и проводят процесс пере
гонки, как описано выше. В конце перегонки в приемнике собираете*
почти бесцветный анилин, который затем извлекают методом экстракции
(см. с. 33).
Следует обратить внимание на то, что несмотря на высокую
температуру кипения анилина (184 °С) процесс перегонки с водяным паром
был проведен при температуре около 100 °С.
3.4. Возгонка (сублимация)
Возгонка, или сублимация, связана с переходом кристаллического
вешества, нагретого ниже его температуры плавления, в парообразное
•состояние (минуя жидкую фазу), а затем при охлаждении — опять в
твердое состояние. Этим способом хорошо очищаются вещества, если
летучесть сопутствующих загрязнений отличается от летучести
основного вещества. Возгонкой можно хорошо очистить бензойную кислоту,
антрацен, нафталин, иод, серу и др.
Возгонку можно проводить при нормальном и пониженном давлении.
Для возгонки веществ при нормальном давлении вещество
помещают в фарфоровую чашку, которую накрывают перевернутой стеклянной
воронкой. Отводную трубку воронки закрывают куском ваты. Между
чашкой и воронкой помещают фильтровальную бумагу с небольшими
отверстиями во многих местах для пропускания паров. Это делают для
того, чтобы кристаллы вещества, образовавшиеся на холодной
поверхности воронки, не падали опять на возгоняемое вещество. Воронку
охлаждают, прикладывая к наружной поверхности смоченный в воде кусок
ткани. Если необходимо очистить небольшое количество вещества, то
вместо воронки можно использовать часовое стекло, которое выпуклой
31
поверхностью кладут на чашку (или стакан). Для охлаждения на часовое
стекло можно положить смоченный водой кусочек ваты.
Фарфоровую чашку с веществом медленно и осторожно нагреваки
на бане. Необходимо помнить, что даже небольшой перегрев может
привести к термическому разложению очищаемого вещества.
Опыт. Очистка нафталина возгонкой. Около 1 г технической
нафталина помещают в маленькую фарфоровую чашку и накрываю'
стеклянной воронкой. Чашку слабо нагревают, не допуская плавленю
вещества. При этом нафталин возгоняется и оседает в виде игл на стенка:
воронки. Все ли твердые вещества можно очищать подобным методом?
3.5. Экстракция
Экстракция, или извлечение, основана на различной растворимосп
веществ в двух несмешивающихся жидкостях. Чаще всего экстрагирова
нию приходится подвергать водные растворы. Для этого пользуются дели
тельной воронкой, в которую наливают раствор, содержащий экстрагируе
мое вещество и экстрагирующую жидкость, т. е. растворитель, в которо1
это вещество растворяется лучше. Растворитель для экстракции должен:
а) мало растворяться в другом растворителе, который содержит экс
трагируемое вещество;
б) заметно лучше растворять экстрагируемое вещество, чем раствс
ритель, из которого это вещество экстрагируется;
в) не должен химически взаимодействовать ни с экстрагируемы
веществом, ни с растворителем, в котором оно растворено;
г) быть сравнительно безопасным;
д) легко удаляться при выделении из него вещества.
Делительную воронку, содержащую раствор экстрагируемого вещ(
ства и растворитель (не более 2/з ее объема), закрывают пробкой и осп
рожно встряхивают, придерживая при этом пробку указательным пал]
цем. В воронке может повышаться давление за счет испарения раствор1
теля, поэтому нужно периодически открывать кран и выпускать пар
растворителя (при этом воронку держат трубкой вверх и в сторону <
стоящего рядом товарища). Кран воронки при этом также должен «смо
реть» вверх. После встряхивания делительную воронку закрепляют
штативе и оставляют в покое до полного разделения слоев. Затем откр*
вают пробку и, осторожно открывая кран, медленно сливают нижш
слой в стакан, стараясь не слить вместе с нижним слоем и верхний. О
тавшийся слой (экстракт) выливают через верхнее отверстие воронки
другую посуду и сушат подходящим осушителем. После этого раствор
тель отгоняют на бане. Обычно это не вызывает никаких затруднени
32
так как растворители, применяемые для экстракции, кипят при сравн
тельно низких температурах.
Опыт. Извлечение диэтиловым эфиром анилина из смеси аш
лии — вода. Около 10 мл 3%-ной суспензии анилина в воде помещают
делительную воронку, приливают 6 мл эфира и встряхивают. Эфирны
экстракт анилина после отстаивания будет находиться в верхнем слое,
вода — в нижнем. В дальнейшем поступают так, как описано выше. Пр
проведении опыта поблизости ие должно быть открытого огня!
3.6. Высушивание органических веществ
Вещества, полученные после очистки, содержат, как правило, следа
растворителей (воду, спирт, эфир и др.)» которые удаляют
высушиванием. Способ высушивания определяется природой вещества, его
агрегатным состоянием и характером растворителя.
Твердые вещества обычно сушат на воздухе при комнатной
температуре или в сушильном шкафу при повышенной температуре. В
последнем случае температура в шкафу должна быть значительно ниже
температуры плавления вещества. Для более эффективного высушивания
применяют эксикаторы, на дно которых помещают подходящие осушители
(Р2О5, безводные СаС12, MgSO4 и др.). Для ускорения процесса
высушивания используют вакуум-эксикатор.
Если растворитель прочно связан с веществом, его сушат в вакууме
или при повышенной температуре. Для этого применяют сушильный
«пистолет» Фишера (рис. 19). В колбу / для обогревания прибора
наливают жидкость с определенной температурой
кипения. Затем колбу соединяют с корпусом ЛПз -—
сушилки 2, в которую помещают лодочку 5 с
веществом. Сушилку закрывают ретортооб-
разной колбой 3 с осушителем 4 и создают
вакуум с помощью водоструйного насоса.
Жидкость в колбе доводят до кипения.
Жидкие вещества высушивают с
помощью твердых неорганических веществ,
способных поглощать воду и образовывать при
этом кристаллогидраты (см. приложение VII).
Эти осушители не должны взаимодействовать
с веществом и растворителем. Жидкость или
раствор встряхивают с небольшим количест-
*
к вакууму
вом (не более 1—3% от массы раствора) под- рис. 19. Сушильный «пис-
ходящего осушителя: прокаленного хлорида толет» Фишера
2-290 33
кальция, сульфата натрия, сульфата магния и др. После этого сосуд,
содержащий жидкость и осушитель, закрывают пробкой с хлоркальциевой
трубкой и оставляют стоять некоторое время. Часто осушитель
приходится менять несколько раз, добиваясь полного устранения влаги. Затем
жидкость отфильтровывают или декантируют (осторожно сливают).
4. Определение основных физических констант
органических веществ
Каждое органическое вещество характеризуется определенными
температурами плавления и кипения, плотностью, показателем
преломления и т. д. Эти величины называются физическими константами. С ю
помощью можно определить (идентифицировать) вещество, а также ус
тановить его чистоту. Для этого определяют некоторые физические кон
станты вещества и сравнивают их с литературными данными.
Рис. 20. Прибор
для определения
температуры
плавления
4.1. Определение температуры плавления
Температурой плавления вещества называете
температура, при которой это вещество из твердог
состояния переходит в жидкое.
Обычно температуру плавления определяют
приборе, состоящем из термостойкой круглодоннс
колбы и пробирки, закрепленной в этой колбе с п
мощью корковой пробки (или приваренной к гор]
колбы). В пробирку помещают термометр, на mapi
которого надевают резиновое кольцо для креплен]
капилляра (рис. 20). Колбу наполняют концентрир
ванной серной кислотой или силиконовым масло
глицерином, дибутилфталатом и т. д. (не более
объема). Если используют концентрированную с<
ную кислоту, то прибор нагревают не выше 250 с
Для работы с веществами, температура плавлеи
которых выше 250 °С, применяют простое устройст
состоящее из двух пробирок. В широкую пробит
вставляют более узкую, зазор между ними оставля
пустым. В узкую пробирку вставляют на корко!
пробке термометр.
Методика определения температуры плавле]
очень проста. Небольшое количество чистого вещ«
34
ва тщательно растирают стеклянной палочкой на часовом стекле. Зате
берут стеклянный капилляр (внутренний диаметр 0,8—1 мм, длина 50-
80 мм), запаянный с одного конца, н открытым концом опускают в веще
ство. Чтобы вещество переместилось на дно капилляра и уплотнилос!
капилляр бросают заплавленным концом вниз в стеклянную трубку дли
ной до 70 см, поставленную вертикально на стол. В капилляре долже]
быть плотный слой вещества высотой до 5 мм. Капилляр с веществол
прикрепляют резиновым кольцом к термометру (столбик вещества нахо
дится на уровне ртутного шарика) и нагревают колбу со скоростью ш
более 1 °С в 1 мнн. При определении температуры плавления
неизвестного вещества нагревание проводят быстрее (до 5—7 °С в 1 мнн), а затем
определение повторяют, но с более медленным нагреванием, особенно
вблизи точки плавления.
Определять температуру плавления необходимо в защитных очках
или защитной маске- Это особенно важно, если колба заполнена
концентрированной серной кислотой.
Началом плавления вещества считается момент размягчения
вещества и переход его в жидкое состояние, а концом — образование
прозрачной жидкости. Во время определения необходимо постоянно следить за
веществом в капилляре и одновременно — за показанием термометра.
Если вещество чистое, то оно плавится в пределах 0,5—1,0 °С. Четкая
температура плавления, как правило, является признаком его чистоты.
Многие органические вещества при плавлении разлагаются, и это
сопровождается их потемнением, а иногда и обугливанием. В этом
случае, конечно, точка плавления будет нечеткой.
В последнее время используется метод определения температуры плавления
под микроскопом — метод Кофлера. Несколько кристаллов вещества помешают
на обогревательный блок, установленный на столике микроскопа. Температуру
плавления измеряют термометром, связанным с блоком. В поле зрения
микроскопа одновременно видны кристаллы вещества и шкала термометра Этот метод
позволяет определять температуру плавления точно и быстро.
Опыт. Определение температуры плавления известного
органического вещества. Определите температуру плавления одного из
известных органических веществ (например, нафталина, бензойной
кислоты, салола и т. д.) по вышеописанной методике и сравните ее с
литературными данными.
4.2. Определение температуры кипения
Любая жидкость характеризуется определенной температурой
кипения. В отличие от температуры плавления эта константа зависит от дав-
35
ления — жидкость начинает кипеть тогда, когда давление ее паров
становится равным атмосферному давлению.
При определении температуры кипения небольшого количества
жидкости применяют метод Сиволобова. В стеклянную трубочку (внутренний
диаметр около 5 мм, а длина 2,5—3 см) помещают пипеткой несколькс
капель исследуемой жидкости. Затем в нее погружают тонкий капилляр
запаянный с верхнего конца. Трубку с жидкостью и капилляром
прикрепляют к термометру, как и в случае определения температуры плавления
При медленном нагревании из тонкого капилляра начинают выделяться
пузырьки воздуха. Температура, при которой начинается непрерывное вы
деление пузырьков, считается температурой кипения жидкости.
Опыт. Определение температуры кипения известного органи
ческого вещества по Сиволобову. Определить температуру кипенга
одной из органических жидкостей, например этилового спирта, хлоро
форма, тетрахлорида углерода, бензола и т. д., руководствуясь приведен
ной методикой.
4.3. Определение относительной плотности жидкости
Плотность вещества р равна отношению его массы т к объему V:
Относительная плотность d определяется отношением плотност
данного вещества к плотности другого при определенных условия:
Обычно относительную плотность вещества определяют по отношению
плотности дистиллированной воды при 4 °С: (i = p/pH0. Величина
зависит от температуры, при которой ее определяют. Поэтому необход]
мо указывать не только температуру, при которой проводят определени
но и температуру воды, плотность которой принята за единицу. Обычх
стандартная температура, при которой проводят определение, равна 20 °<
Например, запись d™ означает, что относительная плотность определе]
при 20 °С, а за единицу сравнения взята плотность воды при 4 °С.
Относительную плотность вещества можно выражать отношение
его массы к массе дистиллированной воды в том же объеме и при один
ковьгх условиях.
Для определения относительной плотности часто пользуются пикн
метрамщ например пикнометром Оствальда (рис. 21), Он состоит из тр
частей, которые обычно называют «носик», «баллончик» и «хвостик»
меткой. При помощи груши, присоединенной к «хвостику», через <а
сик» осторожно всасывают исследуемую жидкость несколько выше м(
ки. Затем в течение 10—15 мин пикнометр термостатируют при 20 с
36
Рис. 21. Пикнометр Оствальда:
1 — носик; 2 — проволочная петля; 3 —
метка; 4 — хвостик; 5 — баллончик
Избыток жидкости убирают при
помощи кусочка фильтровальной бумаги,
которым прикасаются к «носику»
пикнометра, добиваясь, чтобы мениск
жидкости в «хвостике» находился
точно на метке. Пикнометр тщательно
вытирают снаружи и взвешивают на
аналитических весах. Ту же операцию
проводят с дистиллированной водой.
Зная массу пустого пикнометра (с
точностью до 0,0001 г), а также
пикнометра с исследуемой жидкостью и, наконец, с водой, можно рассчитать зна
чение относительной плотности:
</ = («,-|и)/(|и2-т),
где тх — масса пикнометра с исследуемой жидкостью; т2 — масса
пикнометра с водой, т — масса пустого пикнометра.
Для быстрого, но менее точного определения относительной
плотности можно использовать ареометры. Ареометр состоит из стеклянной
трубки, нижняя часть которой несколько расширена и имеет шарик,
заполненный дробью. Верхняя, более узкая часть прибора имеет шкалу с
делениями. Чем больше относительная плотность жидкости, тем меньше
в нее погружается ареометр.
Определение относительной плотности с помощью ареометра
проводят в цилиндре, в который наливают жидкость. Ареометр осторожно
погружают в нее, стараясь, чтобы он свободно плавал, не касаясь ни
стенок, ни дна цилиндра. Значение относительной плотности жидкости
показывает деление на шкале, против которого установился уровень жидкости.
Имеются специальные ареометры: лактометры, (для определения
жирности молока), спиртомеры (для определения процентного
содержания спирта) и др.
4.4. Определение показателя преломления
Показатель преломления п — представляет собой отношение синуса
угла падения света на поверхность раздела двух сред к синусу угла
отражения света: п = sin a/sin p. Эта величина используется для
идентификации жидких веществ и характеристики их чистоты.
Показатель преломления зависит от температуры и резко меняется с
изменением длины волны света, поэтому измерения проводят при
постоянной температуре и монохроматическом свете. Обычно опыт ведут при
37
Рис. 22. Рефрактометр ИРФ-22:
а — вид справа; б — вид слева; 1 — измерительная головка; 2 — маховик для изменения
положения плоскостей призм; 3 — осветительное зеркало; 4 — окуляр; 5 — зрительная
труба; 6 — винт, 7 — зеркало для освещения шкалы; 8 — окошко; 9 — термометр; 10 —
измерительная призма
20 °С и при длине волны, соответствующей длине волны желтой линии
натрия D (к = 589,3 нм). Так, символ п% означает, что показатель
преломления был определен для линии D при 20 °С. Для большинства
жидких органических веществ показатель преломления находится в предела?
от 1,3 до 1,8.
Показатели преломления определяют с помощью рефрактометра. Н;
рефрактометре ИРФ-22 (рис. 22) исследование проводят следующим об
разом. Открыв полушарие измерительной головки 1, протирают плоско
сти призм ватой, смоченной эфиром. С помощью маховика 2 приводя
плоскости призм в горизонтальное положение. На поверхность измерь
тельной призмы 10 осторожно наносят пипеткой каплю исследуемо
жидкости и закрывают измерительную головку. Осветительное зеркало
устанавливают таким образом, чтобы свет от источника, поступая в осв<
тигельную призму через окно, равномерно освещал поле зрения. Вращг
маховик 2 и наблюдая в окуляр 4 зрительной трубы 5, находят грант
раздела света и тени. Если эта граница размыта или окрашена, то винте
6 добиваются ее четкого изображения. С помощью маховика 2 точно с
вмещают границу раздела света и тени с перекрестием сетки и снимак
отсчет по шкале показания преломления (по положению горизонтально
штриха). Зеркало для освещения шкалы 7 ориентируют так, чтобы ев
поступал в окошко 8, освещающее шкалу прибора.
38
Показатель преломления зависит от температуры, поэтому при изме
рении она должна быть постоянной (20 °С). Для наблюдения за постоян
ством температуры около измерительной головки вмонтирован термо
метр 9.
Опыт. Определение показателя преломления одной из оргаии
ческих жидкостей. На имеющемся в лаборатории рефрактометре опре
делите показатели преломления некоторых жидкостей (например,
бензола, этилового спирта, толуола, хлороформа, тетрахлорида углерода п
т. д.) и сравните их с литературными.
5. Общие представления об элементном
органическом анализе
После очистки и выделения органических веществ приступают к их
анализу. Элементный анализ органических соединений включает
качественный и количественный анализы.
5.1. Качественный элементный анализ
Качественный элементный анализ состоит в качественном
определении элементов, входящих в состав органического соединения. Для этого
сначала разрушают органическое вещество, затем превращают
определяемые элементы в простые неорганические соединения, которые могут
быть изучены известными аналитическими методами.
При сгорании органического соединения углерод, входящий в его
состав, образует оксид углерода (IV), а водород — воду. СО2 легко
обнаруживается при пропускании его через известковую воду — образуется
осадок в виде мути. Вода вызывает появление синего окрашивания
сульфата меди вследствие образования гидрата CuSO4'5H2O.
Для обнаружения азота органическое вещество сплавляют с
металлическим натрием. В результате образуется цианид натрия, который
обнаруживают по окраске берлинской лазури;
2NaCN + FeSO4 -* Fe(CN)2 + Na?SO4
4NaCN + Fe(CN)2 -> NaJFe(CN)J
3Na4 [Fe(CN)6 ] + 4FeCl3 -> Fe4 [Fe(CN)6 ]3 +12NaCl
берлинская
лазурь
39
Сера при сплавлении с металлическим натрием образует сульфид
натрия, который легко определяют в виде сульфида свинца:
S + 2Na->Na2S
Na2S + (CH3COO)2 Pb -> PbS + 2CH3COONa
Галогены легко открыть реакцией Бейльштейна. При прокаливании
в пламени горелки медной проволоки или пластинки, на которой
находится органическое вещество, содержащее галоген, пламя окрашивается
в интенсивно зеленый цвет. В отсутствие натрия или калия галоген,
содержащийся в веществе, реагирует при высокой температуре с медью:
Си(т) + -С1(г)4сиС1(г)
Летучий галогенид меди (CuCl) окрашивает пламя горелки в
зеленый цвет.
Определение кремния основано на разрушении вещества и
выделении кремния в виде SiO2 или солей кремниевой кислоты.
Опыт 1. Определение углерода н водорода в органическом
веществе (крахмале или сахаре). В сухую пробирку насыпают около 2 г
крахмала (или сахара) и 1 г оксида меди СиО. Смесь тщательно
перемешивают. В верхнюю часть пробирки помещают небольшой комочек ваты,
на который насыпают немного безводного сульфата меди. Пробирку
закрывают пробкой с газоотводной трубкой и закрепляют в штативе. Конец
газоотводной трубки вводят в заранее приготовленную пробирку с
известковой водой. Сначала прогревают всю пробирку, а затем нагревают
ту часть, где находится смесь оксида меди с веществом.
Отметьте, что произошло с известковой водой. Как изменился цвет
сульфата меди? На основании чего можно сделать вывод о наличии
углерода и водорода в исследуемом веществе?
Опыт 2. Определение галогена в галогенсодержащем оргаинче-
ском соеднненнн по Бейлыптенну. Медную проволоку, укрепленную в
корковой пробке, прокаливают в бесцветном пламени горелки до
исчезновения посторонней окраски пламени. После охлаждения кончик
проволоки смачивают исследуемым веществом (СНС13, СС14 и т. д.) и вносят в
пламя горелки. Что происходит?
Опыт 3. Определение азота в азотсодержащем органическом
соеднненнн. В сухую пробирку помещают около 0,2 г карбамида и
кусочек металлического натрия (с горошину). Пробирку закрепляют в
штативе и осторожно нагревают в пламени горелки (в вытяжном шкафу) в
течение 2—3 мин. Происходит вспышка и обугливание вещества.
Охладив пробирку со сплавом, содержащим цианид натрия, в нее
наливают смесь 2 мл этилового спирта в 10 мл дистиллированной воды.
Полученный раствор фильтруют и наносят 1—2 капли на предметное
40
стекло. Добавляют по 1 капле 0,5%-ного раствора сульфата железа (III)
хлорида железа (III). После подкисления смеси одной каплей 10%-hoi
раствора соляной кислоты выпадает осадок берлинской лазури, что ук;
зывает на присутствие азота в веществе.
Опыт 4. Определение серы в органическом соеднненнн. В сухо
пробирке смешивают несколько кристаллов органического веществ*
содержащего серу (например, сульфаниловую кислоту) с небольшим ку
сочком металлического натрия. Пробирку со смесью закрепляют в шта
тиве и нагревают на горелке в вытяжном шкафу. Раскаленную пробирку
охлаждают и добавляют 10—15 мл дистиллированной воды. После
прибавления в раствор нескольких капель раствора ацетата свинца выпадае!
черный осадок сульфида свинца.
Опыт 5. Определение кремния в кремняйорганнческом
соединении. Несколько капель исследуемого вещества, например дифенилси-
ландиола, смешивают с небольшим количеством сухого и химически
чистого карбоната натрия до состояния густого теста. Часть полученной
смеси укрепляют в ушке платиновой проволоки и вносят в
окислительную часть пламени горелки. Органическая часть смеси сгорает, а часть,
содержащая соединения кремния, сплавляется с карбонатом натрия:
(СбН5 )2Si(OH)2 +15О2 Na2C°3 > 12СО2 + 6Н2О + SiO2
SiO2 + Na2CO3 -> Na2Si03 + CO2
Полученный карбонатно-силикатный плав обрабатывают
дистиллированной водой при нагревании на часовом стекле или на крышке тигля.
1 каплю полученного водного раствора и 1 каплю раствора молибдата
аммония [5 г (NH4)2MoO4, 100 мл Н2О, 35 мл HNO3 (конц.)] помещают на
листок беззольного фильтра, который затем нагревают над пламенем
горелки или над электрической плиткой. После этого в центр пятна
добавляют 2—3 капли 0,5%-ного раствора бензидина в 10%-ной уксусной
кислоте и фильтр помещают в пары аммиака. Если присутствует кремний,
то через некоторое время появляется синее пятно, свидетельствующее об
образовании молибденовой сини и продукта окисления бензидина,
окрашенного в синий цвет.
5.2. Количественный элементный анализ
Задача количественного элементного анализа — установление
количественного (процентного) содержания элементов в анализируемом веществе.
В органическом анализе микроаналитические методы имеют большие
преимущества перед макроаналитическими. Они позволяют экономить реактивы,
требуют минимального количества анализируемого вещества. Кроме того, намно-
41
10
Рис 23. Схема установки для количественного определения углерода и водорода:
1 — трубка для очистки кислорода; 2, 5 — электрические печи; 3 — змеевик; 4 —
поглотители; 6 — кварцевая трубка для сожжения, 7 — поглотительный аппарат для воды; 8 —
поглотительный аппарат для оксидов азота; 9 — поглотительный аппарат для СО2, 10 —
аспиратор
го сокращают продолжительность выполнения анализа. Для количественного
элементного анализа созданы автоматизированные установки-анализаторы,
позволяющие быстро и с большой точностью определять из одной навески вещества
несколько элементов (например, С, Н, N, Si и др). Их применение особенно
удобно при выполнении большого количества серийных анализов (непрерывный
технологический контроль производства, клинические исследования и т. д). Здесь
рассмотрены более простые установки, широко используемые в лабораторной
практике.
Определение углерода и водорода. Определение всегда производится
совместно (из одной навески вещества) в установке, схема которой приведена на
рис. 23. В основу определения положен метод Либиха—Прегля. Он заключается в
количественном разложении органического вещества до оксида углерода (IV) и
воды, определяемых затем количественно при помощи специальных аппаратов,
содержащих вещества, химически связывающие эти оксиды. Для поглощения
оксида углерода (IV) применяют гидроксид натрия, нанесенный на асбест (аска-
рит), а для связывания воды — перхлорат магния (ангидрон).
Точно взвешенную навеску вещества (4—6 мг) помещают в платиновую
лодочку или кварцевую пробирку и сжигают в кварцевой трубке, через которую
пропускают с постоянной скоростью кислород. Трубку («зону окисления»)
нагревают электрической печью до 800—900 °С, а сожжение вещества обычно
производят газовой горелкой. За кварцевой трубкой размещают аппараты с
поглотителями для воды и оксида углерода (IV), массу которых определяют по разности
масс до сожжения и после. Зная массу поглощенных воды и оксида углерода (IV),
рассчитывают массовую долю С и Н (в %) по формулам:
b -0,2727 -100 с-0,1119.100
; (H)
8 8
где g — навеска вещества, мг, Ь — масса СО2; с — масса Н2О; 0,2727 — фактор
для С/СО2; 0,1119 — фактор для Н2/Н2О.
Определение азота. Азот определяют обычно двумя методами. Первый
метод заключается в том, что анализируемое вещество сжигают, а выделившийся
42
Рис. 24. Схема установки для количественного определения азота по Дюма:
1 — микроазотометр; 2 — груша с раствором гидроксида калия; 3 — края с нарезкой; 4 —
электрическая печь; 5 — кварцевая трубка для сожжения; 6 — Z-образная трубка; 7 —
аппарат Киппа
азот собирают в специальном приборе — азотометре, в котором измеряют его
объем (метод Дюма—Прегля). Если же анализируемое соединение разрушить
сильным окислителем, а выделяющийся азот восстановить до аммиака и
количественно определить его титрованием, то этот метод называется методом Кьельдаля.
По методу Дюма—Прегля точную навеску вещества, смешанную с оксидом
меди, сжигают в атмосфере оксида углерода (IV). Этот газ пропускают через
кварцевую трубку для сожжения перед анализом (чтобы вытеснить воздух) и
после — для вытеснения из трубки всех продуктов сгорания в азотометр. Одно из
обязательных условии проведения анализа — применение совершенно
свободного от воздуха (даже следов) СО2. Часть трубки имеет постоянное наполнение:
слой оксида меди, затем восстановленная медь (медная сетка) и опять слой
оксида меди. Анализ проводят в приборе, изображенном на рис.24. Зону постоянного
наполнения в трубке нагревают до 600—650 °С электрической печью, а навеску
сжигают на газовой горелке при температуре 700—750 °С. После сожжения
продукты сгорания медленно вытесняют током СО2 в азотометр, заполненный 50%-
ным раствором гидроксида калия. Массовую долю азота (в %) рассчитывают по
формуле
где т — масса 1 мл сухого азота при соответствующих температуре и давлении
(из таблицы); V— объем азота в азотометре; g — навеска вещества.
По методу Кьельдаля точную навеску вещества с катализатором (смесь
сульфата калия, медного купороса и глюкозы) помещают в колбу Кьельдаля,
приливают небольшое количество концентрированной серной кислоты и
нагревают до кипения. Когда содержимое колбы начинает темнеть, добавляют для
ускорения процесса разложения пероксид водорода. Смесь нагревают до полного
обесцвечивания, а затем, предварительно разбавив ее небольшим количеством
воды, переносят количественно в колбу для перегонки, в которую добавляют
43
раствор гидроксида калия. Начинают отгонять аммиак, улавливая его раствором
соляной кислоты. Добавив в эту колбу несколько капель индикатора (метиловый
красный), смесь титруют щелочью. <
Определение серы н галогенов по Шеннгеру. Точную навеску вещества,
завернутую в предварительно обеззоленный фильтр, укрепляют в платиновой
проволоке, впаянной в стеклянную палочку. Эта палочка укреплена в пробке для
колбы. Затем фильтр с навеской поджигают и сразу же вносят в колбу,
наполненную предварительно кислородом, и плотно закрывают ее пробкой. Продукты
сгорания поглощают водой с добавлением пероксида водорода. При определении
галогенов полученный раствор подкисляют азотной кислотой и титруют
раствором нитрата ртути в присутствии индикатора. При определении серы
растворенные продукты сгорания титруют раствором перхлората бария.
Определение кремния. Кремний определяют одновременно с углеродом и
водородом в быстром токе кислорода в пустой кварцевой трубке для разложения.
Аппаратура та же, что и для определения углерода и водорода. Точную навеску
вещества (А—10 мг) помещают в кварцевый стаканчик, в который добавляют
50—200 мг катализатора (оксид хрома на асбесте). Катализатор должен заполнять
стаканчик на 4/j его объема и лежать неплотными рыхлыми слоями. Стаканчик
помещают в кварцевую трубку для сожжения. Вещество подвергают быстрому
пиролитическому разложению почти без доступа кислорода при 850—900 °С.
Пары вещества попадают на раскаленный катализатор н разлагаются на нем без
образования карбида кремния. После того как навеску сожгли, взвешивают
аппараты для поглощения воды и оксида углерода(ТУ). Затем взвешивают стаканчик с
образовавшимся в нем диоксидом кремния и по привесу рассчитывают
содержание кремния.
Зная количественный (процентный) состав соединения, можно
установить его простейшую формулу — молекулярную, или брутто-формулу.
Эта формула показывает суммарный элементный состав молекулы.
Допустим, в состав органического вещества входят 53,33% углерода,
15,55% водорода и 31,11% азота. Вначале устанавливают соотношение
чисел атомов элементов в молекуле органического вещества. Для этого
процентное содержание элементов делят на их атомную массу:
53,33 : 12,01 =4,44; 15,55 : 1,008= 15,42; 31,11 : 14,007 = 2,22.
При последующем делении полученных частных на меньшее из них
(2,22) получают следующее соотношение:
Число атомов С : число атомов Н : число атомов N = 2:7:1.
Из этих расчетов следует, что брутто-формулой для данного соединения
может быть C2H7N. Но такому же соотношению элементов может
отвечать такая формула: C4H14N2. Поэтому для нахождения истинной
формулы вещества необходимо определить его молекулярную массу. Если она
окажется равной 45, то состав молекулы выразится формулой C2H7N.
Раздел II
Ознакомительный (малый) практикум
Работа 1. Предельные углеводороды (алканы)
Гомологический ряд алканов. Общая формула
гомологического ряда. Номенклатура алканов. Структурная изомерия.
Гнбриднзованное состояние атома углерода sp . Способы
получения алканов. Физические и химические свойства. Реакции
замещения: галогеннрованне, сульфохлорнрованне и сульфо-
окнсленне, нитрование. Окисление алканов. Нефть н
продукты ее переработки. Крекинг нефтепродуктов.
Опыт 1. Получение метана. В фарфоровой ступке растирают 2—3 г
безводного ацетата натрия с таким же количеством натронной извести
(смесь NaOH и СаО). Смесь помещают в пробирку I, закрытую пробкой с
газоотводной изогнутой трубкой. Пробирку укрепляют в штативе.
Свободный конец газоотводной трубки погружают в кристаллизатор с водой
2, а пробирку медленно, равномерно нагревают (рис. 25). Сначала
выделяются пузырьки воздуха, а затем метан, который собирают в пробирку
5. Для этого ее наполняют до краев водой, закрывают пальцем отверстие,
переворачивают вверх дном и вносят в кристаллизатор с водой. Подведя
под пробирку в воде газоотводную трубку, собирают метан. Закончив
наполнение пробирки метаном, сначала вынимают газоотводную трубку
из кристаллизатора, а затем отставляют горелку (электроплиту); в
противном случае вода войдет в горячую
пробирку со смесью и она лопнет.
Опыт 2. Горение метана. Метан в
пробирке поджигают (по мере горения в
пробирку приливают воду, которая вытесняет
метан). Отмечают характер пламени
(коптящее, некоптящее). По окончании горения в
пробирку быстро приливают немного
известковой воды. Что происходит?
Опыт 3. Отношение алкаиов к
бромной воде. К 1 мл гептана (или гексана)
приливают около 1 мл бромной воды и
взбалтывают, не закрывая пробирку. Происходит ли Рнс. 25. Прибор для
полуобесцвечивание бромной воды? чения метана
45
*
Опыт 4. Отношение алкаиов к окислителям. К гептану (или гек-
сану) приливают небольшое количество раствора перманганата калия и
взбалтывают. Происходит ли обесцвечивание раствора?
Опыт 5. Действие концентрированной серной кислоты на ал-
каиы. К 0,5—1 мл гептана (или гексана) добавляют 0,5 мл
концентрированной серной кислоты. Содержимое пробирки энергично встряхивают.
Не происходит никаких изменений. Почему?
Опыт 6. Действие концентрированной азотной кислоты на ал-
каиы. Опыт проводят аналогично предыдущему, но с
концентрированной азотной кислотой. Как в опыте 5, изменений нет. Какой вывод можно
сделать на основании опытов 5 и 6?
Контрольные вопросы
1. Напишите структурные формулы изомеров гептана и назовите их по
систематической номенклатуре.
2. Назовите следующие соединения по систематической номенклатуре:
<j;h3
j j
н3с—с—сн—с—сн3
С—
СН3
3. Из каких галогенопроизводных можно получить 2,4-диметилпентан по
реакции Вюрца?
4. Какой объем оксида углерода (IV) образуется при сжигании 2 моль этана?
5. При сжигании 4,4 г углеводорода образовалось 13,2 г оксида углерода(1У)
и 7,2 г воды. Плотность вещества по водороду равна 22. Найдите молекулярную
формулу этого углеводорода.
6. Как изменяется агрегатное состояние алканов в гомологическом ряду?
Работа 2. Непредельные углеводороды
(алкены и алкины)
Гомологические рады алкеиов и алкииов. Общие формулы
этих рядов. Номенклатура алкеиов и алкииов. Структурная
изомерия. Геометрическая изомерия для алкеиов. Гибридизоваииьк
состояния атома углерода spl и sp. Строение и особенности
двойной и тройной связи. Способы получения алкеиов и алкииов. Фи-
зические и химические свойства. Реакции присоединения.
Правило В. В. Марковиикова. Полимеризация алкеиов. Классификацш
алкадиеиов. Способы получения. Отдельные представители ал
кадиеиов. Эффект сопряжения. 1,2- и 1,4-присоедниеиие. Полиме
ризации диеновых углеводородов. Каучуки.
46
Опыт 1. Получение этилена.
Этилен получают в приборе, изображенном на
рис. 26. В пробирку У, укрепленную в
штативе, наливают 1 мл этилового спирта и
осторожно приливают 3 мл
концентрированной серной кислоты. Добавляют
несколько крупинок оксида алюминия
(катализатор) и закрывают пробирку пробкой, в
которую вставлен узкий конец хлоркаль-
циевой трубки 2, содержащей натронную
известь (для улавливания диоксидов серы
и углерода). Широкий конец хлоркальцие- л ^ „ „
- - « Рис. 26. Прибор для получения
вой трубки закрывают пробкой с газоот- кг j
V - S ~ этилена:
водной трубкой, на конец которой надет } __ ^„p^ co смесью спирта и
резиновый шланг. Содержимое пробирки серной кислоты; 2 — хлоркаль-
осторожно нагревают. Выждав некоторое циевая трубка; 3 — пробирка для
время, необходимое для вытеснения из бР0МН0Й воды «"" Раств0Ра пеР'
г _, манганата калия
пробирки воздуха, пропускают этилен в
бромную воду, а затем — в водный раствор перманганата калия (см.
опыты 2 и 3).
Опыт 2. Отношение этилена к бромной воде. В пробирку
наливают 2—3 мл бромной воды и пропускают в нее этилен до
обесцвечивания. Почему произошло обесцвечивание?
Опыт 3. Отношение этилена к окислителям. Этилен пропускают
в 0,01%-ный раствор перманганата калия, подкисленный серной
кислотой, до обесцвечивания раствора.
Опыт 4. Получение ацетилена. Ацетилен можно получать в
приборе для получения этилена. В пробирку наливают 3—5 мл воды и
бросают несколько кусочков карбида кальция. Выделяющийся ацетилен
используют для изучения его химических свойств.
Опыт 5. Отношение ацетилена к окислителям. Ацетилен
пропускают в пробирку, наполненную 0,01%-ным раствором перманганата
калия (подкисленного серной кислотой), до обесцвечивания раствора.
Опыт 6. Отношение ацетилена к бромной воде. В пробирку с
бромной водой пропускают ацетилен до полного обесцвечивания.
Почему происходит обесцвечивание?
Опыт 7. Получение ацетиленвда серебра. Ацетилен пропускают
в пробирку с аммиачным раствором оксида серебра Образуются бурые
хлопья ацетиленида серебра.
47
Контрольные вопросы
1. Напишите структурные формулы изомеров гексена и пеитина и назовите
их по систематической номенклатуре.
2. Назовите следующие соединения по систематической номенклатуре:
—СН CHj Н.С—С—С—CIL—СН
Н,С—CSC—СН=вСН СН3
3. Какие непредельные углеводороды образуются при действии цинковой
пыли на следующие соединения:
СНг"~"СН~"" С СН.
Вг
4. Напишите уравнения реакций взаимодействия изопрена с бромом, бромо-
водородом.
5. Напишите уравнение реакции взаимодействия ацетилена с хлороводоро-
дом. Какими свойствами обладает полученное вещество?
Работа 3. Галогеналкилы, спирты, простые эфиры
Изомерия н номенклатура галогенопронзводиых.
Способы получения. Физические н химические свойства. Реакции
нуклеофнльного замещения. Непредельные галогеналкилы.
Классификация спиртов. Изомерия и номенклатура
спиртов. Способы получения. Физические и химические свойства.
Высшие жирные спирты (ВЖС). Двух- н трехатомиые спирты.
Непредельные спирты. Правило А. П. Эльтекова.
Простые эфиры. Получение и свойства. Пероксиды.
Опыт 1. Получение бромистого изопропнла. В пробирку с
газоотводной трубкой наливают 1,5—2 мл изопропилового спирта и 2 мл
концентрированной серной кислоты. Смесь охлаждают и добавляют 1—7
мл воды. Продолжая охлаждение, всыпают в пробирку 1,5 г бромида
калия. Присоединив отводную трубку, укрепляют пробирку наклонно i
лапке штатива. Конец отводной трубки погружают в другую пробирку-
приемник, содержащую 1 мл воды и помещенную в стаканчик с водой
Реакционную смесь осторожно нагревают до кипения. Перегонку веду:
до прекращения выделения тяжелых капель бромистого изопропила. По
лученный продукт используют для следующего опыта.
Опыт 2. Отщепление галогена от галогеналкилов при действш
щелочей. Бромистый изопропил, полученный в опыте 1, промывают i
48
делительной воронке дистиллированной водой. Воду сливают, а
бромистый изопропил переливают в пробирку, в которую затем добавляю!
1—2 мл раствора гидроксида натрия. Смесь нагревают до начала
кипения, охлаждают, подкисляют азотной кислотой и добавляют несколько
капель 1 %-ного раствора нитрата серебра. Что происходит?
Опыт 3. Образование алкоголята натрия. В пробирку наливают
0,5 мл изопропилового спирта и помещают небольшой кусочек
металлического натрия. К образовавшемуся алкоголяту натрия приливают воду, а
затем добавляют несколько капель фенолфталеина. Объясните изменение
окраски.
Опыт 4. Окисление нзопропилового спирта* В пробирку
наливают 1 мл нзопропилового спирта и 2 мл хромовой смеси. Осторожно
нагревают до изменения цвета раствора. Что происходит?
Опыт 5. Образование сложного эфира. В пробирку наливают 1 мл
концентрированной уксусной кислоты и 1 мл гооамилового спирта, затем
добавляют 2 капли концентрированной серной кислоты. Смесь
осторожно нафевают и выливают в стакан с водой. Образовавшийся сложный
эфир всплывает на поверхность воды.
Опыт 6. Образование глицерата меди. В пробирку наливают 1 мл
2 н. раствора сульфата меди и 1 мл 2 н. раствора гидроксида натрия. К
выпавшему осадку гидроксида меди (II) добавляют несколько капель
глицерина. Почему происходит растворение осадка образовавшегося
гидроксида меди (II)?
Опыт 7. Получение днизопропилового эфира. Смешивают в
пробирке 1 мл концентрированного раствора серной кислоты и 1 мл
изопропилового спирта. Смесь осторожно нагревают до кипения. Что происходит?
Опыт 8. Изучение взаимной растворимости эфира н воды.
Смесь 5 мл диэтилового эфира и такого же количества воды помещают в
делительную воронку. После расслоения жидкостей эфирный слой
тщательно отделяют от водного. Добавляют к эфиру немного безводного
сульфата меди. Что наблюдается?
Контрольные вопросы
1. Напишите структурные формулы всех изомеров гексилового спирта.
2. Напишите уравнения реакций гидролиза водным раствором гидроксида
натрия следующих соединений: а) 2-хлорбутана; б) 2-хлор-2-метилпенгана; в) 1-хлор-
пронана.
3. Каким способом можно получить 2-метилпропанол-1 из ацетилена?
4. Напишите структурные формулы простых эфиров общей формулы
C$Hi2O.
49
5. Напишите уравнение реакции окисления пропилена водным раствором
перманганата калия. Назовите образовавшийся продукт.
6. Напишите уравнение реакции взаимодействия глицерина с гидроксидом
меди (П).
Работа 4. Альдегиды и кетоны
Строение карбонильной группы. Изомерия н
номенклатура альдегидов н кетой о в. Способы получения. Химические
свойства. Реакции нуклеофнльиого присоединения. Реакции
замещения и окислении. Функциональные производные оксо-
соедниеннй: ацеталн, окснмы, гндразоны, азнны. Альдольиан
н кротонов ая конденсации. Непредельные альдегиды и кетой ы.
Опыт 1. Окисление ацетальдегнда аммиачным раствором
оксида серебра (реакция «серебряиого зеркала»). В тщательно вымытую
пробирку наливают аммиачный раствор оксида серебра и добавляют
несколько капель ацетальдегида. Пробирку помещают в баню с горячей
водой. Через некоторое время на стенках образуется слой серебра
(«серебряное зеркало»). Эта реакция является качественной на альдегиды.
Попробуйте провести подобную реакцию с ацетоном. Объясните, почему
ацетон не восстанавливает серебро.
Опыт 2. Восстановление гидроксида меди ацетальдегндом. К
0,5—1 мл 1%-ного раствора медного купороса прибавляют равный объем
10%-ного раствора гидроксида натрия. К образовавшемуся голубому
осадку гидроксида меди приливают ацетальдегид и нагревают. Отмечают
изменение окраски раствора в процессе нагрева.
Опыт 3. Получение 2,4-диинтрофеиилгидразоиа ацетальдегида.
Несколько кристаллов солянокислого 2,4-динитрофенилгидразина
растворяют в 2 мл воды. Добавляют 1—2 капли 10%-ного раствора
гидроксида натрия и несколько капель ацетальдегида. Смесь встряхивают и
слегка нагревают. Что происходит?
Опыт 4, Осмолеиие ацетальдегида. К 2 мл ацетальдегида
добавляют 1 мл концентрированного раствора гидроксида калия и нагревают
смесь на водяной бане. Появляется желтая, а затем бурая окраска. При
этом выделяется полужидкий смолообразный продукт. Запах
ацетальдегида исчезает, но появляется резкий запах кротонового альдегида.
Опыт 5. Реакция с солянокислым гидроксиламииом. К 5 мл 5%-
ного раствора солянокислого гидроксиламина прибавляют 0,5 мл
ацетальдегида или ацетона. Смесь нагревают на водяной бане и добавляют
1 каплю метилового оранжевого. Отмечают изменение цвета индикатора.
50
Опыт 6. Получение гидросульфнтиого соединения ацетона. 1 м,
ацетона энергично взбалтывают с равным количеством свежеприготов
ленного концентрированного раствора гидросульфита натрия. Выпадае
кристаллический осадок гидросульфитного соединения ацетона. Эта ре
акция является качественной на карбонильную группу.
Контрольное вопросы
1. Напишите все возможные изомеры альдегидов н кетонов,
соответствующие молекулярной формуле ОНцО.
2. Напишите уравнения реакций гидролиза следующих галогенпроизводных:
а) 1,1-ДИХлорпропана; б) 1Д-дихлор-2-метилбутана; в) 2,2-днбромбутана.
3. Какие спирты образуются при восстановлении водородом следующих
соединений: а) пропаналя; б) бутаналя; в) пропанона; г) З-метилпеитанона-2?
4. Напишите уравнения реакций взаимодействия с водой (в присутствии
катализатора) следующих соединений: а) пропина; б) бутина-2.
5. Напишите уравнения реакций окисления: а) бутаналя; б) 2-метилбутаналя;
в) пропанона.
6. Объясните, почему реакцию с солянокислым гидроксиламином можно
использовать для количественного определения альдегидов.
Работа 5. Карбоновые кислоты и их производные
Строение карбоксильной группы. Одноосновные
предельные кислоты. Изомерии и номенклатура. Физические и
химические свойства. Индуктивный эффект. Функциональные
производные карбоиовых кислот: галогенангндриды,
ангидриды, сложные эфнры, амиды, гндропероксиды и пероксиды.
Высшие жирные кислоты (ВЖК). Мыла. Одноосновные
непредельные кислоты и нх свойства. Двухосновные предельные
и непредельные карбоиовые кислоты.
Сложные эфнры. Получение и свойства. Реакция этерн-
фнкацнн. Жиры. Олнфа. Воскн.
Опыт 1. Образование солей. К 0,5 мл 5%-ного раствора карбоната
натрия приливают равное количество разбавленной уксусной кислоты.
Раствор начинает пениться. Объясните это явление.
Опыт 2. Образование и гидролиз ацетата железа. В пробирку
помещают несколько капель 20%-ного раствора ацетата натрия и
добавляют столько же раствора хлорида железа (III). Появляется желтовато-
красное окрашивание. После нагревания раствора до кипения выпадают
хлопья красно-бурого цвета. Что происходит?
51
Опыт 3. Образование нерастворимых солей высших жирных
кислот. В пробирку с раствором мыла добавляют несколько капель 10%-ного
раствора хлорида кальция. Выпадает белый осадок нерастворимой соли.
Опыт 4. Гидролиз мыла. В пробирку с раствором мыла добавляют
несколько капель спиртового раствора фенолфталеина. Объясните
появление розовой окраски.
Опыт 5. Окисление муравьиной кислоты перманганатом
калия, В пробирку наливают 1 мл муравьиной кислоты и 5 мл 25%-ной
серной кислоты. К смеси добавляют 0,5 мл 1%-ного раствора перманга-
ната калия. Пробирку закрывают пробкой с газоотводной трубкой,
свободный конец которой погружают в известковую воду. Содержимое
пробирки нагревают. Через несколько секунд розовая окраска раствора
исчезает. Известковая вода во второй пробирке мутнеет. Проделайте такой же
опыт с уксусной кислотой.
Опыт 6. Окисление олеиновой кислоты перманганатом калня.
В пробирку помещают 2 капли олеиновой кислоты, 2 капли 1%-ного
раствора перманганата калия и 1 каплю 5%-ного раствора карбоната натрия.
При встряхивании розовая окраска исчезает, что указывает на окисление
олеиновой кислоты по месту двойной связи.
Опыт 7. Присоединение брома к олеиновой кислоте. В пробирку
вносят 0,5 мл бромной воды, добавляют 3—4 капли олеиновой кислоты и
энергично взбалтывают. Объясните причину обесцвечивания бромной воды.
Опыт 8. Омыление жира. К 0,5 мл подсолнечного или другого
растительного масла приливают 0,5 мл 30%-ного раствора гидроксида
натрия и смесь осторожно кипятят 5—6 мнн. Омыление считают
законченным, если взятая стеклянной палочкой капля жидкости полностью
растворится в дистиллированной воде с образованием обильной пены
при встряхивании.
Опыт 9. Изомеризация малеиновой кислоты. К 10%-ному
раствору малеиновой кислоты добавляют 0,5 г тиомочевины и нагревают на
водяной бане. Вскоре начинается обильное осаждение кристаллов фума-
ровой кислоты.
Контрольные вопросы
1. Напишите уравнения реакций получения масляной кислоты из бутана.
2. Какое вещество получится, если на йодистый этил подействовать
цианидом калия, а полученный нитрил омылить водой? Напишите уравнения реакций.
3. Как обнаружить акриловую кислоту в смеси с уксусной кислотой?
4. Какой необходимо взять кетон, чтобы при его окислении получить первые
четыре члена гомологического ряда одноосновных насыщенных карбоновых кислот?
52
5. Получите стеариновую кислоту гидролизом соответствующего жира.
6. Как изменяется рКа карбоновых кислот с введением в радикал электроно-
донорных и электроноакцепторных заместителей? Приведите примеры.
Работа 6. Азотсодержащие соединения
алифатического ряда
Амины и амиды кислот. Химические свойства. Нитрилы
и изо циан иды. Диазосоедииеиия алифатического ряда.
Органические порофоры и их использование в производстве
газонаполненных пластмасс. Органические производные угольной
кислоты. Карбамид и мел амин. Полимеры на их основе.
Опыт 1. Действие азотистой кислоты на амнны. В две пробирки
наливают по 0,5 мл 5%-ного раствора этиламина и диэтиламина.
Добавляют равные объемы насыщенного раствора нитрита натрия и по 1 мл 10%-
ноЙ соляной кислоты. В первой пробирке наблюдается выделение азота, во
второй — образуется желтая маслообразная жидкость. Объясните.
Опыт 2. Растворимость карбамида (мочевины) н его
азотнокислой солн в воде. В пробирку помещают несколько кристаллов
карбамида и 1 мл воды. Пробирку встряхивают, а затем проверяют реакцию
среды по лакмусовой бумажке или с помощью бумаги, смоченной
универсальным индикатором. К раствору осторожно приливают 1 мл
концентрированной азотной кислоты, смесь нагревают на водяной бане 5
мин, а затем охлаждают. Наблюдается выделение кристаллов
труднорастворимой соли — азотнокислого карбамида. Отметьте, что в отличие от
карбамида его соли плохо растворимы в воде.
Опыт 3. Взаимодействие карбамида с азотистой кислотой. В
пробирку наливают 1 мл водного раствора карбамида, добавляют 2 капли
концентрированного раствора соляной кислоты и 2 капли раствора
нитрита натрия. При встряхивании начинается бурное выделение пузырьков
газа — азота и диоксида углерода.
Опыт 4. Гидролиз карбамида. К 1 мл раствора карбамида
добавляют 1 мл 10%-ного раствора гидроксида натрия н нагревают до кипения.
Красная лакмусовая бумажка, поднесенная к отверстию пробирки,
синеет. Почему?
Опыт 5. Образование бнурета и его комплексной соли. 0,3—0,5 г
карбамида плавят в сухой пробирке, нагревая в пламени спиртовки или
на электроплите. Как только карбамид расплавится, подносят к пробирке
красную лакмусовую бумажку и отмечают изменение окраски.
Нагревание пробирки продолжают до тех пор, пока расплав не затвердеет. Обра-
53
зевавшийся биурет растворяют в воде и добавляют несколько капель
сульфата меди и 2 мл 10%-ного раствора гидроксида натрия. Отмечают
окраску образовавшейся комплексной медной соли биурета.
Опыт 6. Конденсация карбамида с формальдегидом. В сухую
пробирку помещают 0,5 г карбамида и добавляют 20%-ный раствор
формальдегида до получения прозрачного раствора. При осторожном
нагревании пробирки содержимое мутнеет вследствие образования карбамид-
формальдегидного полимера.
Контрольные вопросы
1. Перечислите основные свойства карбамида (мочевины). Приведите
уравнения реакций.
2. Напишите уравнения реакций взаимодействия соляной кислоты с пропи-
ламином, диэтиламином, триизопропиламином.
3. С помощью какой реакции можно различить первичные, вторичные и
третичные амины?
4. Напишите структурные формулы всех изомеров соединения с общей
формулой C$Hi3N.
5. Как меняется основность в ряду: аммиак—метиламин—ацетамид?
6. Какой диамин используется для получения полиамидного волокна «найлон»?
Работа 7. Соединения со смешанными функциями
Гндрокснкнслоты. Способы получения н химические
свойства. Оптическая изомерия. Асимметрический атом
углерода. Двухосновные четырехатомные гидрокснкнслоты.
Трехосновные четырехатомные гндрокснкнслоты.
Аминокислоты. Получение н химические свойства. Ка-
пролактам. Капрон. Общее понятие о белках. Альдегиде- н ке-
токнслоты. Ацетоуксусный эфнр. Кето-енольная таутомерия.
Опыт 1. Разложение молочной кислоты концентрированной
серной кислотой. В пробирку наливают 1 мл молочной кислоты и 1 мл
концентрированной серной кислоты. Закрыв пробирку пробкой с газоотводной
трубкой, нагревают смесь до кипения. При разложении образовавшейся
муравьиной кислоты выделяется оксид углерода, который поджигают.
Опыт 2. Качественная реакция а-гндроксикнслот с хлоридом
железа (Ш). В пробирку помещают 0,5 мл 5%-ного раствора фенола i
добавляют 1—2 капли 1%-ного раствора хлорида железа. Раствор
приобретает фиолетовую окраску. При добавлении к нему 1—2 капель молоч
ной кислоты происходит изменение окраски раствора до зеленовато
54
желтой. Это связано с тем, что а-гидроксикислоты вытесняют фенол из
комплексного фенолята с образованием лактата железа.
Опыт 3. Образование фелииговой жидкости. В пробирку
помещают 0,5 мл 2 н. раствора медного купороса и 0,5 мл 2 н. раствора гидро-
ксида натрия. К выпавшему осадку гидроксида меди (II) добавляют 3%-
ный раствор сегнетовой соли (калий-натриевой соли винной кислоты).
Образовавшийся синий раствор нагревают до кипения.
Опыт 4. Получение цитрата кальция. В пробирку вносят
несколько кристаллов лимонной кислоты и растворяют в дистиллированной
воде. Раствор нейтрализуют 10%-ным раствором аммиака и к
полученному нейтральному раствору добавляют 0,5 мл 5%-ного раствора
хлорида кальция. При кипячении раствора выпадает осадок, растворимый в
холодной воде.
Опыт 5. Реакция аминокислот с хлоридом железа (III). К 1 мл
5%-ного раствора а-аминокислоты в воде добавляют каплю 5%-ного
раствора хлорида железа (III). Объясните причину появления красного
окрашивания.
Опыт 6. Реакция аминокислот с солями меди (II) (образование
хелатов). К 1 мл 1 %-ного раствора а-аминокислоты добавляют
кристаллик медного купороса и кристаллик ацетата натрия (для создания
буферного раствора). Раствор приобретает темно-синий цвет. Объясните
причину этого. Напишите уравнение реакции.
Контрольные вопросы
1. Напишите структурные формулы а-, р-, у-гвдрокснкислот, содержащих
пять атомов углерода. Назовите эти кислоты.
2. Какие продукты образуются при отщеплении воды от а-, р- и у-
гидрокснкислот?
3. Напишите реакции взаимодействия ацетоуксусного эфира с синильной
кислотой, хлоридом фосфора (V), аммиаком.
4. Докажите с помощью соответствующих реакций наличие в гшровино-
градной кислоте двух функциональных групп.
5. Какие продукты образуются при нагревании а-, р-, у-аминокислот?
Работа 8. Углеводы (сахара)
Классификация углеводов. Моносахариды. Строение.
Глюкоза и фруктоза. Кольчато-цепная таутомерии. Стереонзомерня
моносахаридов. Химические свойства моносахаридов. Дисахарн-
ды: сахароза, лактоза, мальтоза и целлобноза. Строение. Восста-
55
навлнвающне н невосстанавлнвающне сахара. Несахароподобные
полисахариды: крахмал н целлюлоза. Строение, отлнчне в
строении. Гидролиз крахмала н целлюлозы. Простые н сложные эфнры
целлюлозы. Бумага. Сульфитно-дрожжевая бражка (СДБ). Ис-
нользованне простых эфнров н СДБ в строительстве.
Опыт 1. Окисление глюкозы аммиачным раствором оксида
серебра (реакция «серебряного зеркала»). К 2 мл аммиачного раствора
оксида серебра в тщательно вымытой пробирке приливают 1 мл 5%-ного
раствора глюкозы. Пробирку со смесью нагревают на водяной бане до
осаждения на стенках металлического серебра.
Опыт 2. Образование сахаратов. а) Получение сахарата кальция.
К 1 мл 10%-ного раствора хлорида кальция добавляют 0,5 мл 2 н.
раствора гидроксида натрия. К образовавшемуся гидроксиду кальция
приливают 5%-ный раствор глюкозы до растворения осадка.
б) Получение сахарата меди К 1 мл 10%-ного раствора медного
купороса добавляют 0,5 мл 10%-ного раствора гидроксида натрия. К
образовавшемуся осадку приливают 5%-ный раствор глюкозы до растворения осадка.
Опыт 3. Реакции дисахаридов с фелинговой жидкостью. В две
пробирки с растворами (по 0,5 мл) сахарозы и лактозы приливают по 0,5 мл
фелинговой жидкости и нагревают до кипения. Объясните причину
разного отношения этих Сахаров к жидкости Фелинга.
Опыт 4. Гидролиз крахмала. 2 мл крахмального клейстера
нагревают до кипения с равным количеством 30 %-ной серной кислоты в
течение 10 мин. Затем смесь нейтрализуют щелочью и проводят реакцию с
фелинговой жидкостью. Для сравнения подействуйте фелинговой
жидкостью на негидролизованный крахмал. Объясните разницу в поведении.
Опыт 5. Гидролиз целлюлозы. В пробирку помещают небольшой
кусочек фильтровальной бумаги и приливают 0,5 мл концентрированной
серной кислоты. Содержимое пробирки осторожно перемешивают и
нагревают на водяной бане. К раствору добавляют фелингову жидкость и
пробирку нагревают до появления желтого окрашивания.
Опыт 6. Растворение целлюлозы в реактиве Швейцера. К 5 мл
медно-аммиачного раствора добавляют кусочек ваты и перемешивают
стеклянной палочкой. Полученную вязкую жидкость разбавляют равным
количеством воды и выливают в стакан с 15 мл 2 н. соляной кислоты.
Смесь сразу же обесцвечивается и выпадает белый студенистый осадок.
Контрольные вопросы
1. Какими свойствами отличаются моносахариды от полисахаридов?
2. Приведите примеры восстанавливающих и невосстанавливающих
Сахаров. Какие реакции позволяют различить их?
56
3. Напишите уравнения реакций взаимодействия глюкозы с уксусным
ангидридом, синильной кислотой, гвдразином.
4. Напишите уравнения реакций гидролиза сахарозы, лактозы и мальтозы.
5. Изобразите схему кольчато-цепной таутомерии для глюкозы и фруктозы.
6. Как получают простые и сложные эфиры целлюлозы? Каково их
промышленное применение?
Работа 9. Кремнийорганические соединения
Элементорганнческне соеднненнн. Классификации.
Органические производные непереходных элементов. Характер
свизн углерод — элемент. Краткая характеристика элементор-
ганнческнх соединений по группам периодической системы
элементов. Магннйорганнческне соединении. Реактив Грннья-
ра. Алюмннннорганнческне соединения. Трнэтилалюмнний.
Катализаторы Цнглера — Натта. Фосфорорганнческне
соединении. Перегруппировка А. £. Арбузова. Кремннйорганиче-
скне соединении. Классификация. Получение кремннйоргаин-
ческнх мономеров. Снлоксановаи связь. Полноргаиоснлокса-
ны. Гидрофобнзаторы на основе кремннйорганнческнх
соединений. Использование в строительстве.
Опыт 1. Подвижность атома хлора в хлорсиланах. К 2 мл 2%-
ного раствора трнэтилхлорсилана в спирте добавляют 1 мл 1%-ного
раствора нитрата серебра. Выпадает белый творожистый осадок. Объясните
это явление.
Опыт 2. Расщепление алкоксисиланов иодоводородной
кислотой. В пробирку помещают 2—3 мл концентрированной иодоводородной
кислоты, добавляют 0,5 мл триэтилэтоксисилана, накрывают пробирку
фильтровальной бумагой, пропитанной раствором нитрата ртути (II), и
медленно нагревают на масляной бане до 120 °С. Бумага окрашивается в
красный цвет вследствие образования иодида ртути (II).
Опыт 3. Качественная реакции на тетраалкилсиланы. К 1 мл
тетраэтилсилана добавляют около 0,1 г безводного хлорида алюминия и
нагревают до кипения. Через 10—15 мин появляется желтое
окрашивание. После охлаждения приливают 1 мл 1 %-ного раствора кетона Михле-
ра в анилине. Через несколько минут выпадает красный осадок.
Опыт 4. Гндрофобизация строительных материалов.
Строительные материалы (керамическую неглазурованную плитку, кусок
штукатурки, бетонный кубик и т. д.) смачивают этилтрихлорсиланом или,
лучше, погружают в этилтрихлорсилан на несколько секунд. На
обработанный таким образом материал действуют водой и разбавленным
раствором соляной кислоты. Что наблюдается?
57
Опыт 5. Обнаружение кремния в ортоснлнкатах. Опыт
проводят по методике, описанной на с. 41.
Контрольные вопросы
1. Напишите формулы метилсилана, диметилсилана, диметилдихлорсилана,
диэтилхлорсилана, этилтриэтоксисилана, диметилсиландиола.
2. Приведите способы получения кремнийорганических мономеров.
3. Приведите способы получения кремнийорганических полимеров.
4. Какой продукт образуется прн гидролизе триметилхлорсилана в
присутствии аммиака?
5. Как зависит характер образующихся полиорганосилоксанов от
функциональности исходных кремнийорганических мономеров?
6. В чем сходство н отличие углерода и кремния?
Работа 10. Ароматические углеводороды
и их галогенопроизводные
Бензол — ароматическая система. Электронное строение
молекулы бензола. Понятие «ароматичности». Гомология н
изомерия ароматических углеводородов. Номенклатура.
Способы получения бензола н его гомологов. Химические
свойства. Реакции электрофнльного замещения. Механизм этих
реакций; 71- н а-комплексы. Орнентанты I н II рода. Механизм
ориентирующего влиянии заместителей.
Ароматические галогенопроизводные. Роль катализатора
в реакции галогеннровання прн замещении в ядро. Замещение
в боковой цепи. Различие условий проведения этих реакций.
Химические свойства ароматических галогенопронзводных.
Сравнение подвижности галогена в галогенопронзводных
ароматического н алифатического рядов.
Опыт 1. Действие на беизол бромной воды н перманганата ка-
лня. В две пробирки наливают по 0,5 мл бензола; в первую приливают 1
мл бромной воды, во вторую — 1 мл 1%-ного раствора перманганата
калия с водой. Содержимое пробирок энергично взбалтывают. Происходи!
ли изменение цвета в пробирках?
Опыт 2. Окисление толуола. В пробирку помещают смесь 0,5 mj
толуола и 0,5 мл 0,1%-ного раствора перманганата калия с содой. Смес]
при взбалтывании нагревают на водяной бане. Розовая окраска раствор:
постепенно исчезает, превращаясь в бурую. Какие продукты образуются
в результате этой реакции?
58
Опыт 3. Получение бромбензола (опыт проводят в вытяжном
шкафу!). В пробирку с 1 мл бензола прибавляют 1 мл 10%-ного раствора
брома в тетрахлориде углерода н вносят на кончике ножа немного
обезжиренных железных опилок. Смесь нагревают на кипящей водяной бане в
течение 5 мин, а затем содержимое пробирки сливают в стакан с водой. Бром-
бензол собирается на дне стакана в виде тяжелого маслообразного продукта.
Опыт 4. Изучение характера связи галогена с бензольным
ядром. В две пробирки помещают по 0,5 мл хлорбензола и хлористого
бензила, добавляют в каждую пробирку пятикратное количество воды и
нагревают до кипения. К горячему раствору приливают несколько капель
нитрата серебра. Отмечают появление белого осадка только в одной из
пробирок. Чем это объясняется?
Опыт 5. Горение бензола. Каплю бензола на стеклянной палочке
вносят в пламя горелки. Бензол воспламеняется и горит сильно коптящим
пламенем. Сравните с горением метана.
Контрольные вопросы
1. Напишите структурные формулы всех возможных изомеров
ароматических соединений общей формулы CgH10, CgHgCl.
2. Напишите уравнения реакций получения метилбензола, о-ксилола, изо-
пропилбензола.
3. Приведите механизм галогенирования бензола.
4. Чем различаются способы получения хлористого бензила н хлорбензола?
5. Напишите уравнения реакций следующих превращений:
СН4 "-► С2Н6 —> С2Н2 —> СбНб
6. Приведите механизм алкилирования бензола этиленом.
Работа 11. Ароматические нитросоединения
и сулъфокислоты
Ароматические ннтросоедннения. Нитрующий агент.
Химические свойства ароматических ннтросоеднненнй. Ннтросо-
едннення с интрогруппой в боковой цепн. Ацн-ннтро-
таутомерня.
Ароматические сульфокнслоты. Сульфирующие агенты.
Химические свойства ароматических сульфокнслот. Бензол-
сульфохлорнд.
Опыт 1. Получение нитробензола. В пробирку наливают 5 мл
нитрующей смеси и добавляют по каплям при постоянном взбалтывании
59
2 щ бензола. Смесь встряхивают 3 мин, а затем содержимое пробирки
выливают в стакан с холодной водой. На дне стакана собираются капли
нитробензола, которые отделяют с помощью делительной воронки.
Опыт 2. Получение днннтробензола. В пробирку помещают 5 мл
нитрующей смеси и 2 мл нитробензола, полученного в опыте 1. Смесь
нагревают на кипящей бане 5 мин при постоянном взбалтывании. Затем
реакционную смесь охлаждают и выливают в пробирку с водой, Динит-
робензол выделяется в ввде тяжелой маслянистой жидкости желтого цвета.
Опыт 3. Восстановление нитробензола. В пробирку помещают
0,5 мл концентрированной соляной кислоты, 1 каплю нитробензола и
кусочек олова. Бурно протекающая реакция ослабевает через 2—3 мин.
Смесь нагревают на водяной бане до исчезновения запаха нитробензола
(запаха горького миндаля). Одну каплю полученного раствора переносят
в другую пробирку, добавляют несколько капель бромной воды.
Появление белой мути свидетельствует об образовании анилина.
Опыт 4. Получение бензолсульфокнслоты. В пробирку
помещают 0,5 мл бензола и 0,5 мл концентрированной серной кислоты.
Содержимое пробирки нагревают на водяной бане до получения однородного
раствора, а затем выливают в пробирку с водой. Если сульфирование
закончено, образуется прозрачный раствор.
Контрольные вопросы
1. Приведите механизм реакции нитрования бензола, толуола.
2. Какие продукты получаются при нитровании толуола, изопропилбензола,
бензолсульфокислоты, фенола и хлорбензола?
3. Какие промежуточные продукты получаются при восстановлении
нитробензола в кислой, нейтральной и щелочной средах?
4. Приведите механизм реакции сульфирования бензола.
5. Какое из соединений будет легче сульфироваться и нитроваться: бензол,
толуол, нитробензол, бензолсульфокислота?
Работа 12. Кислородсодержащие соединения
ароматического ряда
Фенолы. Классификации. Одноатомные фенолы.
Номенклатура. Химические свойства. Причина кислотности фенолов.
Двух- и трехатомиые фенолы. Хииоидиая структура.
Ароматические спирты. Получение и свойства.
Ароматические альдегиды и кетоны. Получение. Свойства.
60
Ароматические карбоновые кислоты. Классификации.
Изомерии и номенклатура. Беизониаи кислота н ее
производные. Хлористый беизоил. Аитраниловая, /г-аминобеизоиная,
сульфобеизониая и салициловая кислота. Непредельные
ароматические карбоновые кислоты. Миогоосиовиые
ароматические кислоты. Лавсан.
Опыт 1. Растворимость фенола в воде, В пробирку с 10 мл
дистиллированной воды помещают около 1 г фенола. Пробирку энергично
встряхивают, а затем смеси дают отстояться. Смесь расслаивается: верхний слой —
раствор фенола в воде, а нижний — раствор воды в феноле. Пробирку снова
встряхивают, и эмульсию фенола разливают поровну в три пробирки.
Опыт 2. Цветная реакция на фенол. В пробирку с раствором
фенола добавляют 2—3 капли 1 %-ного раствора хлорида железа (III).
Появляется фиолетовое окрашивание.
Опыт 3. Получение фенолята натрия. К 1 мл водной эмульсии
фенола прибавляют 2 н. раствор гидроксида натрия до полного
исчезновения эмульсии. Если к полученному прозрачному раствору прибавить
по каплям раствор серной кислоты (до кислой реакции), то образуется
эмульсия- Почему?
Опыт 4. Получение трнбромфенола. В пробирку с водной
эмульсией фенола добавляют при постоянном взбалтывании бромную воду.
Образуется осадок трибромфенола.
Опыт 5. Получение бензоата натрия. В пробирку помещают 0,5 г
бензойной кислоты и прибавляют 2 н. раствор гидроксида натрия до
растворения кристаллов бензойной кислоты. Если к прозрачному раствору
прилить 2 н. раствор соляной кислоты, то снова выпадает осадок
бензойной кислоты.
Опыт 6. Окисление бензальдегнда кислородом воздуха. На
часовое стекло наносят 2 капли бензальдегида. Через некоторое время
появляются кристаллы бензойной кислоты.
Опыт 7. Реакция салициловой кислоты с хлоридом железа (III).
В пробирку наливают 2 мл воды и добавляют несколько кристаллов
салициловой кислоты. К полученному раствору приливают 2—3 капли 1%-
ного раствора хлорида железа (III). Объясните изменение окраски.
Опыт 8. Гидролиз ацетилсалициловой кислоты (аспирина), В
пробирку помещают 0,5 г ацетилсалициловой кислоты и приливают 1 мл
воды. Содержимое пробирки кипятят 2—3 мин, а затем добавляют
несколько капель раствора хлорида железа (Ш). Объясните изменение окраски.
61
Контрольные вопросы
1. Напишите уравнения реакций окисления толуола, ксилола, изопропилбен-
зола. Что общего в этих реакциях?
2. Какие соединения образуются из бензойной кислоты гфи действии на нее
гидроксидом магния, хлористым тионилом, этиловым спиртом в присутствии
концентрированной серной кислоты?
3. Каковы условия реакции получения фенола из хлорбензола?
4. Из салициловой кислоты получите ацетилсалициловую кислоту и фенило-
вый эфир салициловой кислоты.
5. Напишите уравнение реакции кротоновой конденсации бензальдегида с
ацетоном.
6. Какая реакция является качественной для фенола и салициловой кислоты?
Работа 13. Ароматические амины, диазосоединения,
азокрасители
Классификация. Анилин. Получение и химические
свойства. Ароматические ямины с аминогруппой в боковой цепи.
Реакция диазотироваиия. Свойства ароматических диазо-
еоедииеиий. Реакции, идущие е выделением и без выделения
азота. Реакция азосочетаиия. Азокрасители. Связь между
строением и окраской органических соединений.
Опыт 1. Основные свойства анилина. В пробирку наливают 1 мл
водной эмульсии анилина и опускают в нее красную лакмусовую
бумажку. Лакмусовая бумажка не меняет своей окраски. Почему?
Опыт 2. Получение солей анилина н разложение их щелочью. К
0,5 мл анилина прибавляют 1 каплю 50%-ной серной кислоты. Выпадает
белый кристаллический осадок сернокислой соли анилина, трудно
растворимой в воде. Аналогично получают солянокислую соль анилина (с
концентрированной соляной кислотой), которая легко растворяется в воде.
В пробирку с солью анилина приливают 1 мл 10%-ного раствора
гидроксида натрия. Происходит выделение анилина.
Опыт 3. Получение ацетаннлида (опыт проводят в вытяжном
шкафу). В пробирку помещают 0,5 мл анилина и по каплям прибавляют
равное количество уксусного ангидрида. Смесь встряхивают и
охлаждают. Через несколько минут выпадает белый кристаллический осадок аце-
танилида.
Опыт 4. Окисление анилина. В пробирку с 1 мл водного раствора
анилина прибавляют 1 мл хромовой смеси. Содержимое пробирки
быстро темнеет. Образование «черного анилина» является результатом
глубокого окисления анилина.
62
Опыт 5. Получение хлористого феннлдназоння. В пробирку
наливают 5 мл раствора солянокислого анилина и охлаждают, помещая
пробирку в стакан со льдом. Затем по каплям добавляют 20%-ный
раствор нитрита натрия до появления на иодкрахмальной бумажке синего'
пятна, которое свидетельствует об избытке нитрита натрия и,
следовательно, о конце диазотирования. Образовавшийся водный раствор
хлористого фенилдиазония используют для получения азокрасителей.
Опыт 6. Получение кислотного азокраснтеля. К 2 мл водного
раствора хлористого фенилдиазония, полученного в опыте 5, прибавляют
равное количество раствора фенола в щелочи. Появляется интенсивное
красное окрашивание (раствор должен быть все время щелочным).
Опыт 7. Разложение солей дназоння. Водный раствор хлористого
фенилдиазония нагревают 10 мин на водяной бане при 50—60 °С.
Образовавшийся фенол обнаруживают по реакции с FeCl3.
Контрольные вопросы
1. Напишите структурные формулы N, N-диметиланилина, о-толуцдина, п-
фенилецдиамина.
2. Как меняются основные свойства в ряду: анилин, дифениламин, трифени-
ламин? Почему?
3. Дайте обоснованный ответ, почему химические свойства ароматических
аминов отличаются от свойств алифатических аминов.
4. Приведите механизм реакции азосочетания.
5. Какие функциональные группы могут служить в качестве ауксохромов н
хромофоров?
6. Составьте схему синтеза азокрасителя на основе хлористого фениддназо-
ния и N, N-диметиланилина.
Работа 14. Многоядерные ароматические соединения
Дифеинл и дифеннлметай. Трифенилметан. Подвижность
атомов и групп, связанных с трифеиилметнльнон
группировкой. Устойчивость трифенилметильного радикала н нона.
Трифеиилметаиовые красители. Фталеииы. Фенолфталеин в
качестве индикатора.
Нафталин. Строение и химические свойства.
Производные нафталина. Антрацен. Антрахинои и его производные. Не-
беизондиые ароматические системы.
Опыт 1. Возгонка нафталина. В сухую пробирку помещают 0,5—
1 г нафталина и осторожно нагревают нижнюю часть пробирки, доводя
63
нафталин до плавления. На холодных стенках верхней части пробирки
наблюдается образование кристаллов нафталина.
Опыт 2. Нитрование нафталина. К 0,3—0,5 г нафталина,
помещенного в пробирку, добавляют 2 мл концентрированной азотной кислоты.
Закрыв пробирку пробкой со вставленной в нее стеклянной трубкой
длиной 20—25 см (в качестве воздушного холодильника), содержимое
пробирки встряхивают и нагревают на кипящей водяной бане 5 мин. Затем
смесь выливают в стакан с холодной водой. Нитронафталин выделяется в
виде желтого маслянистого вещества, затвердевающего при охлаждении.
Опыт 3. Бромироваиие нафталина. В сухую пробирку помещают
0,1—0,2 г нафталина, приливают 2 мл раствора брома в тетрахлориде
углерода и энергично встряхивают содержимое пробирки. Нафталин
постепенно растворяется в тетрахлориде углерода и бромируется на холоду.
Наблюдается изменение окраски раствора.
Аналогичную реакцию проводят во второй пробирке, однако
содержимое пробирки нагревают до кипения. Бромирование нафталина идет
значительно быстрее при нагревании.
Опыт 4. Получение фенолфталеина. В сухую пробирку
помещают около 0,1 г фталевого ангидрвда и примерно вдвое большее
количество фенола. К содержимому пробирки при встряхивании осторожно
прибавляют 3—5 капель концентрированной серной кислоты. Пробирку
нагревают над пламенем горелки до появления темно-красного
окрашивания. Смесь охлаждают и добавляют 0,5 мл воды. Если к водному
раствору фенолфталеина прибавить каплю раствора щелочи, то раствор
окрашивается в малиновый цвет.
Опыт 5. Получение флуоресценна. Около 0,1 г фталевого
ангидрвда и примерно в два раза большее количество резорцина помещают в
сухую пробирку и к смеси осторожно добавляют 3—5 капель
концентрированной серной кислоты. Содержимое пробирки нагревают до
появления темно-красного окрашивания, охлаждают и добавляют 1 мл воды для
растворения образовавшегося флуоресцеина.
2—3 капли полученного раствора флуоресцеина переносят в колб>
на 100 мл, добавляют 1 мл 2 н. раствора гидроксвда натрия и разбавляю!
большим количеством воды. Появляется красивая желто-зеленая
флуоресценция, исчезающая при подкислении раствора.
Контрольные вопросы
1. Напишите уравнения следующих реакции нафталина: а) окисления
кислородом воздуха в присутствии V2O5; б) восстановления водородом (в момент
выделения); в) восстановления водородом в присутствии катализатора.
64
2. Напишите схему синтеза одного из красителей трифеиилметанового ряда
3. В чем причина устойчивости трифенилметильного радикала?
4. Объясните предпочтительность образования а-производных прн электро-
фильном замещении нафталина.
5. Напишите структурные формулы следующих соединений: а) 4,4-дибром-
2,2-динигродифенила; б) 4,4-диаминодифенила; в) 4,4-дихлорднфенилметана;
г) 1-этилнафталина; д) 2-нафталинсульфокислоты; е) 2-метиланТрахинона; ж) 2-
гидрокси-3,6-нафталиндисульфокислоты.
6. Почему бесцветный раствор фенолфталеина приобретает окраску в
щелочной среде?
Работа 15. Гетероциклические соединения
Классификация гетероциклов. Пяти член иые гетер оцик-
лы с одним гетероатомом. Ароматические свойства фур аи а,
тиофеиа и пиррола. Фурфурол. Продукты конденсации
фурфурола с ацетоном. Мономер ФА. Взаимные переходы между пя-
тичлеииыми гетероциклами (реакция Ю. К. Юрьева). Иидол и
индиго. Иидигоидиые красители, Азолы. Шестичлеииые гете-
роциклы с одним гетероатомом. Пиридин и его свойства.
Пиридиновые основания. Азииы.
Опыт 1. Качественная реакция на фурфурол. На часовом стекле
смешивают 1 каплю анилина с одной каплей уксусной кислоты.
Полученным раствором смачивают полоску фильтровальной бумаги и наносят
на нее каплю фурфурола. Появляется розовое окрашивание.
Опыт 2. Окисление фурфурола. На часовом стекле смешивают
1 каплю свежеперегнанного фурфурола с 1 каплей аммиачного раствора
оксида серебра. Появляется черное пятно выделившегося свободного
серебра. О чем это говорит?
Опыт 3. Получение 2,4-днннтрофенилгидразона фурфурола.
3—4 капли свежеперегнанноп* фурфурола помещают в пробирку и
приливают равное количество раствора солянокислого 2,4-динитрофенил-
гидразина. Смесь нагревают до кипения. При охлаждении выпадают
красно-оранжевые кристаллы 2,4-динитрофенилгидразона фурфурола.
Опыт 4. Конденсация фурфурола с ацетоном. В пробирку
наливают смесь 2 мл фурфурола, 1 мл ацетона и 5 капель 10%-ного раствора
гидроксида натрия. Появляется черное окрашивание.
Опыт 5. Оснбвный характер пиридина. В пробирку помещают 1
каплю пиридина и 5 капель воды. Образуется прозрачный раствор, в
который опускают полоску красной лакмусовой бумаги. Смоченный конец
бумаги синеет.
3-290 65
помещают по 1 капле раствора пиридина, 1%-ного раствора перманганата
калия и 1%-ного раствора соды. Содержимое пробирки встряхивают и
нагревают 1—2 мин. Цвет раствора не изменяется.
Опыт 7. Качественная реакция на тиофеи. Одну каплю тиофена
помещают в пробирку, приливают 2 капли концентрированной серной
кислоты и 1 каплю изатина. Появляется интенсивное синее окрашивание.
Опыт 8. Осибвиые свойства хииолииа. В пробирку помещают
2 капли хинолина, 3—4 капли воды и по каплям, до полного растворения
хинолина, приливают 3—4 капли концентрированной соляной кислоты.
К солянокислому хинолину добавляют 3—4 капли 10%-ного раствора
гидроксида натрия. Выделяется тяжелая капля свободного хинолина.
Контрольные вопросы
1. Напишите схему взаимных превращений фурана, тиофена и пиррола по
Ю. К. Юрьеву.
2. Напишете структурные формулы следующих соединений: 2,5-диметил-
фурана, 2-хлорфурана, 2,5-диметилпиррола, 2-аминопиридина.
3. Напишите уравнения следующих реакций: а) полного и неполного
гидрирования фурана и пиррола; б) сульфирования и нитрования фурана, тиофена и пиррола.
4. Объясните причину ароматического характера фурана, тиофена и
пиррола. Какой из этих гетероциклов наиболее склонен к реакциям электрофильного
замещения?
5. Перечислите области применения фурфурола. Что такое «мономер ФА»?
Какой полимерный продукт используется на основе этого мономера в строительстве?
Работа 16. Высокомолекулярные соединения
Общее представление о полимерах. Элементарное звено.
Степень полимеризации. Период идентичности. Линейные,
разветвленные и пространственные полимеры. Химическая
классификация полимеров. Карбоцепиые и гетероцепиые
полимеры. Общие свойства высокомолекулярных соединений.
Понятие о средней массе полимеров. Гибкость макромолекул.
Различие понятий «полимер» и «пластмасса». Физические
состояния полимеров. Понятие о кристалличности полимеров.
Методы синтеза высокомолекулярных соединений.
Мономер — структурная единица полимера. Функциональность
мономеров. Свизь между строением мономера и его
способностью к образованию полимеров. Реакция полимеризации.
Элементарные акты радикальной полимеризации: ииициирова-
66
иие, рост цепи и ее обрыв. Ионная полимеризации (катиоииая
и анионная). Аниоиио-коордииациоииая полимеризация.
Катализаторы Цнглера — Натта и стереорегуляриые полимеры.
Полимераиалогичиые превращении и макромолекулир-
иые реакции. Деструкция полимеров. Стабилизация полимеров.
Опыт 1. Полимеризация стирола. В пробирку помещают 2 мл
стирола и несколько крупинок пероксида бензоила (инициатор). При
отсутствии пероксида бензоила можно воспользоваться пероксидом
водорода (2—3 капли). Закрывают пробирку трубкой, вставленной в пробку
(холодильник), и нагревают в течение 15 мин. Загустевшую жидкость
охлаждают.
Опыт 2. Получение полиметилметакрилата. В пробирку
наливают 2 мл метилметакрилата, добавляют несколько крупинок пероксида
бензоила и нагревают на кипящей водяной бане до получения
стекловидной массы.
Опыт 3. Получение полимера «капрон». В пробирку помещают
1—2 г капролактама и небольшой кусочек твердого гидроксида натрия или
калия. Содержимое осторожно нагревают до получения вязкой жидкости.
Опыт 4. Получение феиолформальдегидиого полимера. В
пробирку помещают 1 г фенола и добавляют 1 мл формалина (40%-ный
раствор формальдегида в воде). Смесь нагревают 2—3 мин, приливают 2—3
капли концентрированной соляной кислоты. Нагревание прекращают
после расслоения смеси. Воду сливают, а остаток выливают в
фарфоровую чашку или на железный лист. Образуется твердый продукт —
термопластичный полимер (новолак), растворимый в ацетоне. Чтобы
превратить новолачный полимер в резольный, к нему добавляют 0,5 мл
насыщенного раствора уротропина и осторожно нагревают, не доводя до
осмоления. Через несколько минут в пробирке получается продукт ярко-
желтого цвета — термореактивный полимер (это соединение можно
также получить, взяв в избытке формалин).
Опыт 5. Обнаружение кремнии в силиконовом каучуке.
Кусочек силиконового каучука помещают в пробирку и нагревают в пламени.
На стенках пробирки осаждается белый налет оксида кремния.
Опыт 6. Отношение каучука и пластмасс к растворителям.
Помещают в пять пробирок по кусочку натурального каучука,
полистирола, поливинилхлорида, полиэтилена и аминопласта. В каждую
пробирку приливают по 1—2 мл ацетона и выдерживают 30 мин. По истечении
указанного времени проверяют состояние образцов и делают вывод о
растворимости каучука и пластмасс.
Опыт 7. Отношение пластмасс к нагреванию и горению. В
фарфоровую чашку помещают поочередно по кусочку полиэтилена, по-
67
листирола, поливинилхлорида, полиметилметакрилата, аминопласта и
фенопласта и нагревают на электроплитке. Через несколько минут
образцы проверяют, прикасаясь к ним стеклянной палочкой. Отмечают
скорость размягчения образцов и характер этого размягчения в зависимости
от степени нагревания. Кусочки этих же пластмасс закрепляют в
проволоке (продетой через корковую пробку, чтобы было удобно держать в
руке) и вносят в пламя спиртовки. Отмечают характер горения.
Опыт 8. Характер продуктов, образующихся при разложении
пластмасс. Образцы пластмасс, используемые в опыте 7, помещают в
отдельные пробирки, закрывают их пробкой с газоотводной трубкой и
поочередно нагревают в пламени спиртовки. Выделяющиеся при
разложении газообразные продукты пропускают через раствор перманганата
калия или бромную воду, а также испытывают на лакмусовую бумажку.
Что происходит в каждом отдельном случае?
Опыт 9. Отношение пластмасс к щелочам н кислотам. В четыре
пробирки помещают поочередно по кусочку полистирола, полиэтилена,
фенопласта и аминопласта и приливают 1—2 мл концентрированной
серной кислоты. Содержимое пробирок осторожно встряхивают. Через
несколько минут сливают кислоту, промывают пластмассу водой и
определяют стойкость ее к действию кислоты.
Опыт повторяют с теми же образцами пластмасс, заменив кислоту на
20%-ный раствор гидроксида натрия.
Контрольные вопросы
1. Напишите уравнения реакций получения стирола и его полимеризации.
2. Приведите механизм радикальной полимеризации пропилена, стирола.
3. Приведите схему получения капрона из циклогексанона.
4. Приведите схему взаимодействия натурального каучука с серой
(вулканизация).
5. В чем различие между реакциями полимеризации и поликонденсации?
Приведите примеры этих реакций.
6 Какой путь получения синтетического каучука разработал С. В. Лебедев?
Приведите уравнения соответствующих реакций.
Раздел
Синтезы органических соединений
1. Галогенирование
Радикальный и ионный механизмы реакции галогеиироваиия.
Нуклеофильиое замещение при насыщенном атоме углерода.
Механизмы 5дг1 и 5дг2. Электрофильиое замещение в ароматическом ядре
(SE). Галогеиироваиие ароматических соединений. Механизм
реакции; л и а-комплексы.
Работа 1.1,2-Дибромэтан
Основная реакция:
СН3СН2ОН н^°4 > СН2=СН2 —^-» СН2Вг—СН2Вг
Побочные реакции:
2СН3СН2ОН3223 2
СН3СН2ОН + 2H2SO4 -> СН3СНО + ЗН2О + 2SO2
Реактивы: этанол — 15 мл; бром — 5 мл; H2SO4 (кона) — 25 мл; NaOH, 2 н.;
песок.
Получение этилена (рис. 27, а). В колбу Вюрца / вносят 5 г сухого
мелкого песка, 5 мл этанола и 15 мл концентрированной серной кислоты.
В капельную воронку 2 вливают приготовленную в стакане смесь 10 мл
спирта и 10 мл концентрированной серной кислоты. В это же время
готовят к работе газометр 5, полностью заполняя его водой.
^Смесь нагревают на песчаной бане 3. Через несколько Минут после
начала выделения газа его собирают в маленькую пробирку под водой и
После синтеза и тщательной очистки некоторые органические соединения
(по указанию преподавателя) подвергают физико-химическим исследованиям,
например записывают их спектры поглощения в ИК- и УФ-областях. На основании
УФ-спектров делают вывод о характере возможных электронных переходов и
наличии хромофорных групп (см. с. 121). Пользуясь табличными данными, по
положению и интенсивности характеристических полос в ИК-спектре (см. приложение
V) определяют наличие всех имеющихся функциональных групп в веществе.
69
Рис. 27. Приборы для получения этилена (а) и 1 Д-дибромэтана (б):
/ — колба Вюрца; 2 — капельная воронка; Д — песчаная баня; 4 — склянка Тищенко; 5 —
газометр; 6 — пробирка с бромом; 7 — колба с раствором гидроксида натрия
делают пробу на отсутствие воздуха (сгорание должно происходить
спокойно, без хлопка и свиста). Если выделяется чистый этилен, газометр
подсоединяют к склянке Тищенко 4. Начинают по каплям прибавлять
жидкость из капельной воронки, регулируя равномерность выделения
этилена. В газометре обычно собирают 4—5 л газа.
Бромироваиие этилена. (Работать с бромом только в вытяжном
шкафу, в защитных очках и резиновых перчатках.) Прибор (рис. 27, б) для бро-
мирования этилена состоит из газометра, склянки Тищенко, наполненной на
V3 водой (она служит для кошролирования скорости подачи этилена),
пробирки 6, в которой происходит реакция, и колбы с 2 н. раствором гидроксида
натрия 7, в которой поглощается унесенный током этилена бром. В пробирку
6 помещают кусочки стекла (для увеличения поверхности, на которой
происходит взаимодействие газа с бромом) и наливают 5 мл брома. Добавляют 1
мл воды для уменьшения потерь брома на испарение. В эту пробирку,
которую охлаждают водой и постоянно взбалтывают, пропускают из газометра
равномерный ток этилена до тех пор, пока не исчезнет окраска брома.
Сырой дибромэтан промывают в делительной воронке водой»
раствором гидроксида натрия и еще несколько раз водой. После
высушивания хлоридом кальция продукт перегоняют в маленькой колбе Вюрца,
собирая фракцию при 130—132 °С. Выход продукта 12—15 г (65—80%).
1,2-Дибромэтан — бесцветная маслянистая жидкость, застывающая
при 10 °С, т. кип. 131,4 °С, d]b 2,1785, и" U5379. Мало растворим в
воде, смешивается с обычными органическими растворителями.
Применение: для синтеза бромистого винила, этилендиамина (отвердитель
эпоксидных полимеров).
70
Работа 2.1-Бромнафталин
Основная реакция:
Вг
НВг
Побочная реакция:
Вг
НВг
Реактивы: нафталин — 12,8 г; бром — 5 мл; уксусная кислота — 25 мл.
В колбу Вюрца вместимостью 50 мл помещают нафталин и ледяную
уксусную кислоту. Колбу снабжают капельной воронкой, конец которой
погружают в жидкость. Отводную трубку колбы соединяют при помощи
резиновой трубки с воронкой, опрокинутой над поверхностью раствора
щелочи (10—20%) в стакане. Колбу нагревают на кипящей водяной бане и при
помешивании постепенно приливают бром из капельной воронки. Через
некоторое время начинается выделение бромоводорода. После добавления
всего брома колбу нагревают на кипящей водяной бане еще 3 ч. Затем заменяют
капельную воронку на термометр, присоединяют нисходящий холодильник
Либиха и отгоняют около 20 мл уксусной кислоты, используя в качестве
приемника мерный цилиндр. Остальную уксусную кислоту и не вошедший в
реакцию нафталин отгоняют при нагревании до 250 °С, заменив
холодильник Либиха на воздушный. Остаток перегоняют в вакууме, собирая
фракцию, кипящую при 140—150 °С (2,6 кПа, или 20 мм рт. ст.). Выход продукта
12—13 г (58—63%). Его можно очистить повторной перегонкой в вакууме,
собирая-фракцию, кипящую при 145—148 °С (2,6 кПа, или 20 мм рт. ст.).
Остаток от перегонки 1 -бромнафталина, представляющий собой в
основном 1,4-дибромнафталин, еще горячим вливают в фарфоровую
чашку и выкристаллизовывают. Если есть жидкая фаза, ее удаляют
фильтровальной бумагой. Кристаллы 1,4-дибромнафталина очищают
перекристаллизацией из спирта или тетрахлорвда углерода. Выход 0,4—0,5 г.
1-Бромнафталин — жидкость, кипящая при 281 °С и замерзающая
при 6 °С; df 1,482, п™ 1,6582. Трудно растворим в воде, хорошо — в
спирте, эфире, бензоле.
Применение: в органическом синтезе, в качестве растворителя для
некоторых полимеров; высокотемпературный теплоноситель.
71
Работа 3. Бромистый изопропил
Основные реакции:
KBr+H2SO4 -> KHSO4 + HBr
(CH3)2CHOH + HBri»(CH3)2CHBr+H2O
Побочные реакции:
(СН3)2СНОН—2£^СН3—СН==СН2
2(СН3)2СНОН "™ )(СН3)2СН —О —СН(СН3)2
H2SO4 + 2НВг -> 2Н2О + S02 + Вг2
Реактивы: шопропиловый спирт — 15 мл; КВг — 15 г, H2SO4 (конц) — 19 мл.
В круглодонную колбу вместимостью 250 мл приливают спирт, 8 мл
воды и при перемешивании 19 мл концентрированной серной кислоты, охлаждая
при этом колбу под струей воды. Продолжая перемешивание, к охлажденной
до комнатной температуры смеси добавляют бромид калия. Затем колбу через
дефлегматор соединяют с нисходящим холодильником Либиха и
реакционную смесь перегоняют до тех пор, пока в приемник не перестанут поступать
маслянистые капли, опускающиеся на дно. Если реакционная смесь слишком
сильно пенится, нагревание на короткое время прекращают.
Бромистый изопропил отделяют в делительной воронке от воды и
добавляют к нему осторожно, небольшими порциями
концентрированную серную кислоту до тех пор, пока она не соберется в виде отдельного
слоя. Бромистый изопропил, очищенный таким образом от изопропило-
вого спирта и диизопропилового эфира, перегоняют из маленькой колбы
Вюрца, собирая фракцию с температурой кипения 57—61 °С. Выход
продукта 8—10 г (50—65%).
Бромистый изопропил кипит при 59,4 °С, df 1,310, и£° 1,4251,
мало растворим в воде, смешивается со спиртом и эфиром.
Применение: в качестве исходного вещества в некоторых органических
синтезах.
Работа 4.4-Бромтолуол
НВг
72
Реактивы: толуол — 10 мл; обезжиренные железные опилки — ОД г; бром —
5 мл; иод.
В тщательно высушенную двугорлую колбу вместимостью 100 мл,
снабженную обратным холодильником и капельной воронкой, вносят
кристаллик иода, железные опилки, толуол и по каплям бром. Реакция
начинается через несколько минут. Ее течение регулируют, погружая
колбу в горячую или холодную воду. Бромоводород отводят из
холодильника через трубку, соединенную с опрокинутой воронкой,
накрывающей стакан с водой.
По окончании реакции смесь перегоняют с водяным паром до тех
пор, пока не перестанет собираться на дне приемника тяжелая
маслянистая жидкость. Ее отделяют с помощью делительной воронки и сушат
хлоридом кальция, нагревая на водяной бане. По охлаждении
маслянистую жидкость отделяют от затвердевшего кристаллогвдрата хлорвда
кальция и перегоняют, собирая фракцию, кипящую около 180 °С. Эту
фракцию охлаждают в смеси льда с солью, слегка потирая стенки колбы
стеклянной палочкой для ускорения кристаллизации. Образовавшиеся
кристаллы 4-бромтолуола отфильтровывают от маслянистой жидкости на
воронке Бюхнера. Т. пл. 25—26 °С. Выход, продукта 5—6 г (33—40%).
Продукт очищают перекристаллизацией из спирта.
4-Бромтолуол — бесцветное кристаллическое вещество с т. пл.
28,5 °С, т. кип. 184,5 °С, df 1,3898, п% 1,5490. Мало растворим в воде,
хорошо — в спирте и эфире.
Применение: используется для синтезов многих органических препаратов.
2. Нитрование
Получение иитроалкаиов. Реакция Коновалова (механизм,
условия, побочные продукты). Химические свойства иитроалкаиов. Аци-
иитро-форма (нитроновая кислота).
Получение ароматических иитросоедииеиий. Механизм реакции инт-
роваиия. Нитрующие агенты. Правила ориентации заместителей. Связь
ориентирующего действия с электронным строением заместителей.
Работа 5. Нитрометан
С1СН2СООН—^-> CICH2COONa ™°2 ) NO2CH2COONa
2NO2CH2COONa + H2O -> 2CH3NO2 + Na2CO3 + CO2
73
Реактивы: хлоруксусная кислота — 9,4 г; NaNCb — 7 г; NaOH, 40%-ный
раствор; НС1 (1:1); СаС12.
К смеси хлоруксусной кислоты с 20 г толченого льда прибавляют при
помешивании охлажденный льдом 40%-ный раствор гидроксида натрия до
щелочной реакции по фенолфталеину (около 7 мл). Температура при
нейтрализации не должна превышать 20 °С. Полученную смесь приливают к
раствору нитрита натрия в 10 мл воды, находящемуся в колбе Вюрца
вместимостью 100 мл. Колбу соединяют с нисходящим холодильником и
закрывают пробкой с термометром, шарик которого должен быть погружен в
жидкость. В приемник наливают 2—3 мл разбавленной (1:1) соляной кислоты.
Смесь медленно нагревают до начала выделения пузырьков СО2 (около 60—
70 °С), после чего нагревание прекращают, так как реакция экзотермична.
Если температура смеси начинает понижаться, снова осторожно нагревают
до 85 °С. Когда реакция в основном закончится, реакционную смесь
подогревают, доводя температуру к концу реакции до 110 °С. Нитрометан
начинает перегоняться при температуре около 90 °С.
Нитрометан отделяют от водной фазы в делительной воронке и удаляют
следы воды кусочками фильтровальной бумаги. Для более тщательной
очистки нитрометан сушат хлорвдом кальция и перегоняют, собирая фракцию,
кипящую при 98—102 °С. Выход продукта около 2 г (около 33%).
Нитрометан — светло-желтая или бесцветная жвдкость с запахом
горького миндаля. Т. кип. 101,2 °С, df 1,1382, и£° 1,3819. С водой
образует азеотропную смесь, кипящую при 83,6 °С. Смешивается с эфиром,
спиртом, ацетоном. Хуже растворяется в воде.
Применение: для получения хлорпикрина, нитроспиртов, взрывчатых веществ;
в качестве добавки к дизельным и реактивным тошшвам; экстрагент ароматических
углеводородов из нефти и нефтепродуктов; растворитель эфиров целлюлозы, каучу-
ков, природных смол; для электрофоретического покрытия керамики.
Работа 6. Нитробензол
Основная реакция:
С6Н6 + HNO3 "** >C6H5NO2 + Н2О
Побочная реакция:
NO.
HNO3
Реактивы: бензол — 9 мл; HNO3 (1,4 г/см3) — 10 мл; H2SO4 (1,84 г/см3)
12,5 мл; NaOH, 5%-ный раствор; СаС12.
74
В двугорлую колбу вмесгамосгао 100 мл помещают азотную кислоту и
добавляют к ней небольшими порциями при перемешивании серную
кислоту. Смесь охлаждают до комнатной температуры. В колбу вставляют
обратный воздушный холодильник и термометр, доходящий почти до дна колбы,
и через холодильник добавляют небольшими порциями бензол, встряхивая
при этом всякий раз содержимое колбы (для этого закрепление лапки в
муфту немного ослабляют). Температура смеси не должна подниматься выше
50 °С; при необходимости колбу охлаждают, погружая ее в баню с холодной
водой. После окончания прибавления бензола колбу нагревают на водяной
бане при 60 °С в течение 1 ч. Затем реакционную смесь охлаждают, помещая
колбу под водопроводный кран, переливают в делительную воронку и дают
хорошо отстояться. Верхний слой (нитробензол) отделяют, промывают его
водой, дважды — раствором гидроксида натрия (порциями по 10—20 мл) и
снова водой (40 мл). После этой промывки нитробензол будет находиться в
нижнем слое. При промывке щелочью возможно образование стойкой
эмульсии, которую разрушают, добавляя 1 каплю спирта.
Промытый нитробензол помещают в сухую колбу, снабженную
обратным холодильником, прибавляют безводный хлорид кальция и нагревают на
водяной бане до осветления жидкости. При охлаждении смеси нижний слой
(раствор хлорида кальция в воде) застывает вследствие образования
СаС12-6Н2О. Если этого не происходит, вносят еще осушитель и снова
нагревают. Сухой нитробензол переливают в колбу Вюрца и перегоняют, собирая
фракцию при 204—210 °С. Содержимое колбы ни в коем случае нельзя
перегонять досуха — это может привести к взрыву. Выход продукта 10—11 г
(78—85%).
Нитробензол — желтоватая маслянистая жидкость с запахом горько-
► миндаля. Т. кип. 210,8 °С, т. пл. 5,8 °С, d]° 1,2034, п% 1,5526. Мало
ютворим в воде, смешивается с эфиром, спиртом, ацетоном и бензолом.
Применение: для получения анилина, бензидина, некоторых красителей;
я очистки смазочных масел; в качестве мягкого окислителя в некоторых орга-
ческих синтезах (фуксина, хинальдина и др.), растворителя полиэтилентереф-
тта, растворителя в реактиве Уайта, который используется для обнаружения
>бодного оксида кальция в цементе.
Работа 7.2- и 4-Нитрофенолы
он он он
NO
2HNO3
75
Реактивы: фенол — 15 г; HNO3 (1,37 г/см3) — 28 мл; СН3СООН — 55 мл;
NaOH, 40%-ный раствор; НС1, 10 %-ный раствор.
В стакане вместимостью 250 мл растворяют 15 г фенола в 40 мл
уксусной кислоты. Смесь охлаждают льдом до 0 °С и при помешивании
стеклянной палочкой прибавляют из капельной воронки смесь 28 мл
азотной и 15 мл уксусной кислот. Температура при этом не должна
превышать 25—30 °С. Смесь выдерживают при комнатной температуре в
течение 1 ч, затем переносят в круглодонную колбу вместимостью 1 л,
куда добавляют 40%-ный раствор гидроксида натрия (до рН 6), и
перегоняют с водяным паром. При этом в холодильник следует подавать воду
из термостата с температурой около 50 °С; если же охлаждение
производят водопроводной водой, то ее периодически отключают, чтобы пар
расплавил оседающие в алонже кристаллы 2-нитрофенола. Раствор в
приемнике охлаждают, а выпавшие кристаллы отсасывают и сушат на
воздухе. Выход продукта 6—7 г (27—32%). Для очистки 2-нитрофенола
может быть использована перекристаллизация из разбавленного спирта.
Т. пл. 45 °С.
К жидкости, оставшейся от перегонки с паром, добавляют 40 мл
40%-ного раствора гидроксида натрия (для получения 4-нитрофенолята
натрия, который кристаллизуется более чистым, чем свободный 4-нитро-
фенол) и упаривают ее на водяной бане до объема 150 мл. Горячий
раствор фильтруют, а затем охлаждают для кристаллизации
4-нитрофенолята натрия. Если кристаллизация не происходит, раствор еще
упаривают. Выпавшую соль отсасывают и промывают на фильтре 10%-ным
раствором щелочи. Соль переносят в небольшой стакан, добавляют 10%-
ный раствор соляной кислоты (до явно кислой реакции) и 1 г
активированного угля, кипятят 10 мин и фильтруют через складчатый фильтр.
Выпавшие бесцветные кристаллы отсасывают, промывают 2%-ной
соляной кислотой и сушат на воздухе. Продукт очищают
перекристаллизацией из 2%-ной соляной кислоты. Выход 4-нитрофенола 6—7 г (27—32% от
теоретического). Т. пл. 114 °С. Суммарный выход нитрофенолов 12—14 г
(54—63% от теоретического).
2-Нитрофенол — бледно-желтые кристаллы с т. пл. 45,3—45,7 °С,
т. кип. 214,5 °С, d*° 1,2495. Плохо растворим в воде, хорошо — в спирте,
эфире, ацетоне, бензоле. Летуч с водяным паром.
4-Нитрофенол — желтые кристаллы с т. пл. 114,9—115,6°С, df
1,4790. Плохо растворим в холодной воде, хорошо — в горячей, очень
хорошо — в спирте и эфире.
Применение: 2-нитрофенол — в качестве промежуточного продукта в синтезе
фотоматериалов, сернистых и азокрасителей. 4-Нтрофенол используется как
фунгицид и антисептик. Оба изомера являются кислотно-основными индикаторами.
76
3. Сульфирование
Получение ароматических сульфокнслот. Сульфирующие
агенты: сериаи кислота, олеум, хлорсульфоиоваи кислота. Механизм
реакции сульфировании. Влиииие температуры и заместителей иа ход
реакции сульфировании. Сульфирование в ряду нафталина. Правила
ориентации заместителей. Химические свойства сульфокислот.
Замещение сульфогруппы иа другие атомы и группы (Н, ОН, CN,
СООН). Щелочное плавление. Восстановление сульфокислот.
Производные сульфокислот: сульфохлориды, сульфамиды, алкилсульфоиа-
ты, алкнларилсульфоиаты, алкнлсульфаты. Поиитие о поверхностно-
активных иеществах (ПАВ). Детергенты. Применение ПАВ в
строительстве.
Работа 8. 4-Толуолсульфокислота
Основная реакция:
H.SO,
SO3H +
Побочные реакции:
Н,С -V V + Н
SO3H
2SO4
Н,С
SO3H + U2SOt
-ГУ
к,о
SO3H
SO3H
Реактивы: толуол — 32 мл; H2SO4 (конц.) — 19 мл.
В круглодонную колбу вместимостью 200 мл, снабженную
обратным холодильником, помещают толуол и серную кислоту и нагревают до
кипения (если прибор собран на резиновых пробках, то колба должна
быть длинногорлой). Слабое кипение толуола поддерживают в течение
1 ч, хорошо перемешивая смесь встряхиванием колбы каждые 2—3 мин.
Через 1 ч слой толуола почти исчезает, что служит признаком конца
реакции. Теплую реакционную смесь выливают в стакан вместимостью 500
мл, в котором находится 100 мл воды, а колбу споласкивают небольшим
количеством воды. В раствор добавляют 1 г активированного угля и
кипятят под тягой на электрической плитке с закрытым нагревательным
77
элементом до обесцвечивания. Затем уголь отфильтровывают, а раствор
упаривают на водяной бане до 50 мл. Раствор в стакане охлаждают льдом
до 5—7 °С и насыщают газообразным хлороводородом (для осаждения 4-
толуолсульфокислоты). Вместо обработки газообразным хлороводородом
можно добавить 150 мл соляной кислоты, однако это приводит к резкому
уменьшению выхода. Выпавшие кристаллы отфильтровывают на фильтре
из пористого стекла, тщательно отжимают от маточного раствора и
помещают в эксикатор над концентрированной серной кислотой.
Одновременно в эксикатор ставят стаканчик с твердой щелочью (осторожно!) для
поглощения хлороводорода. Высушивание продолжается в течение
нескольких дней. Т. пл. продукта 104—105 °С. Выход 35—40 г (65%).
4-Толуолсульфокислота образует бесцветные гигроскопические
кристаллы моногидрата. Безводная кислота кипит при 140 °С (2,6 кПа, или
20 мм рт. ст.), т. пл. 35 °С. Хорошо растворяется в воде, а также в спирте
и эфире.
Применение: для синтеза /i-крезола, толуолсульфохлорида и других
продуктов; в качестве катализатора этерификации; для отверждения поливинилаце-
тальных полимеров и пластобетона на основе фурфурольных полимеров.
Работа 9. 2-Нафталинсульфокислота
H2SO4
Реактивы: нафталин — 16 г; H2SO4 (конц.) — 15 мл; НС1 (конц.) — 3 мл;
активированный уголь.
В колбе вместимостью 100 мл расплавляют нафталин и нагревают
его до 160 °С (шарик термометра должен быть погружен в жидкость). К
расплавленному нафталину при перемешивании по каплям добавляют
серную кислоту, следя за тем, чтобы температура не поднималась выше
170 °С. Смесь перемешивают при 160 °С в течение 20 мин, дают
охладиться до комнатной температуры и медленно, при перемешивании
палочкой, выливают в стакан с 20 мл воды. Раствор отфильтровывают от
выпавшего осадка ди-2-нафтилсульфона и непрореагировавшего
нафталина, охлаждают до 0 °С и оставляют до выпадения кристаллов тригид-
рата 2-нафталинсульфокислоты.
Т. пл. 124 °С. Выход продукта 19—20 г (60%).
2-Нафталинсульфокислота — бесцветное, очень гигроскопическое
вещество. Температура плавления безводного продукта 102 °С. Хорошо
растворима в воде и спирте, плохо — в бензоле.
78
Применение: для синтеза р-нафтола и р-нафтиламина, используемых в
производстве красителей; для получения синтетических дубителей,
пластификаторов бетонных смесей.
Работа 10. Сульфаниловая кислота
A +H2S0,—»> (Л~т3-то~4
Реактивы: анилин — 9,3 г; H2SO4 (конц.) — 5,5 мл; NaOH — 4 г; НО
(конц); активированный уголь.
В фарфоровой ступке небольшими порциями смешивают и
тщательно растирают анилин с серной кислотой. Полученную соль помещают в
фарфоровую чашку и нагревают на электрической плитке с закрытым
нагревательным элементом (на высоте 2—3 см над плиткой) в течение
1—2 ч. Температура реакционной массы не должна превышать 170—
180 °С. Твердая масса становится серо-фиолетовой, проба ее при
растворении в щелочи не должна выделять анилин (при наличии
непрореагировавшего анилина он выделяется в виде капель).
Еще горячую сульфаниловую кислоту измельчают и растворяют в
растворе гидроксида натрия (4 г NaOH в 36 мл воды). Раствор кипятят 5
мин с активированным углем, фильтруют и фильтрат подкисляют
соляной кислотой до кислой реакции по конго красному (рН 3). При
охлаждении выпадают кристаллы, которые отсасывают и перекристаллизовы-
вают из воды. Продукт сушат между листами фильтровальной бумаги.
Т. пл. 286—288 °С (с разложением). Выход 10—12 г (60—70%).
Сульфаниловая кислота — бесцветное твердое вещество, из воды
при 0—21 °С кристаллизуется ее дигвдрат, при 21—40 °С — моногидрат,
выше 40 °С — безводная кислота. В органических средах растворяется
плохо.
Применение: для синтеза некоторых красителей и сульфаниламидных
лекарственных препаратов, обнаружения некоторых элементов, пластификации
бетонных смесей, предотвращения коррозии металлов тампонажными растворами.
4* Алкилирование
Алкилирующие средства: галогеналкилы, спирты, диалкил-
сульфаты, диазометаи, олефииы, оксиды олефииов, О-, N- и С-алки-
лироваиие. Алкилироваиие аммиака и аминов. Действие оксида
этилена иа амины, спирты, карбоновые кислоты, фенолы.
79
Образование сложных эфиров минеральных кислот.
Способы алкилирования ароматических соединений. Алкнлнро-
ваиие по Фрнделю — Крафтсу. Хлорид алюминия в качестве
катализатора.
Работа 11. Диэтиланилин
C6H5NH2 + С2Н5Вг -> C6H5NHC2H5 • HBf
C6H5NHC2H5 • НВг + NaOH -> C6H5NHC2H5 + NaBr + H2O
C6H5NHC2H5 + C2H5Br -> C6H5N(C2H5)2 • HBr
C6H5N(C2H5)2 • HBr + NaOH -> C6H5N(C2H5)2 + NaBr + H2O
Реактивы: анилин — 10 г, бромистый этил — 30 г, гидроксид натрия —
13,2 г.
В круглодонной колбе вместимостью 100 мл, снабженной
эффективным обратным холодильником, кипятят смесь, состоящую из 10 г свеже-
перегнанного анилина и 15 г бромистого этила, до затвердевания
реакционной массы (около 2 ч). Колбу охлаждают и приливают небольшими
порциями раствор 6,6 г гидроксида натрия в 20 мл воды. Колбу при этом
охлаждают, так как возможно сильное разогревание.
Выделившийся продукт отделяют с помощью делительной
воронки и снова кипятят с большим обратным холодильником с
добавлением 15 г бромистого этила до затвердевания реакционной массы. После
охлаждения эту массу растворяют в воде, переливают в стакан и
кипятят (под тягой!) несколько минут для удаления избыточного
бромистого этила. Раствор охлаждают до комнатной температуры и к нему
добавляют небольшими порциями (размешивая) раствор 6,6 г
гидроксида натрия в 20 мл воды.
Органический слой (верхний) отделяют в делительной воронке,
сушат плавленым гидроксидом натрия, фильтруют в небольшую колбу
Вюрца и перегоняют с воздушным холодильником, собирая фракцию с т.
кип. 214—216 °С, представляющую собой почти чистый диэтиланилин.
Выход 10 г (62%).
Диэтиланилин — желтоватая маслянистая жидкость с т. кип.
216,3 °С, df 0,9351, г$ 1,5409. Мало растворим в воде, хорошо — в
спирте, эфире, бензоле, ацетоне. Является слабым основанием.
Применение: для синтеза трнфенилметановых и азокрасителей; получения
диэтиламина (используется в производстве ускорителей вулканизации); в
качестве отвердителя полиэфирных полимеров.
80
Работа 12. Ди-«-бутиловый эфир
Н2О
Основные реакции:
\ С4Н9ОН + H2SO4 -> C4H9OSO2OH
C4H9OSO2OH + С4Н9ОН -> С4Н9ОС4Н9 + H2SO4
Побочная реакция:
CH3CH2CH2CH2OSO2OH -> СН3СН2СН=СН2 + H2SO4
Реактивы: бутянол-1 — 25 мл; H2SO4 (конц.) — 2,8 мл; NaOH, 2 н. раствор.
В круглодонную колбу 1 вместимостью 100 мл с
насадкой Дина—Старка 2 (рис. 28) и обратным
холодильником вносят бутанол-1 и при размешивании,
покачивая колбу, добавляют серную кислоту. После
осторожного кипячения около 3—3,5 ч выделяется
вода в количестве, близком к расчетному. Нагревание
прекращают, реакционную смесь охлаждают и при
перемешивании вносят в делительную воронку, в
которую налито 18 мл 2 н. раствора гидроксида натрия.
Реакционную смесь отмывают от серной кислоты
раствором щелочи до щелочной реакции промывных
вод, затем — водой (15 мл).
Полученный продукт сушат хлоридом кальция, а
потом переносят в колбу вместимостью 50—100 мл,
фильтруя через складчатый фильтр. Перегоняют с
высоким дефлегматором до температуры 135 °С.
Смесь охлаждают, добавляют кусочек
металлического натрия. Заменяют дефлегматор на более короткий
и перегоняют дибутиловый эфир, собирая фракцию с
т. кип. 140—145 °С. Ни в коем случае нельзя
перегонять досуха: простые эфиры могут содержать
взрывчатые пероксиды. Выход около 10 г (56%).
Дибутиловый эфир — бесцветная жидкость с характерным запахом;
т. кип. 142,4 °С, df 0,7688, п™ 1,3992. Практически нерастворим в воде,
смешивается с эфиром, спиртом, бензолом. Кислородом воздуха
окисляется (быстрее на свету) с образованием взрывчатых пероксидов.
Применение: используется как растворитель, обладающий свойствами ди-
этилового эфира, но менее летучий; входит в состав жидкостей, способствующих
лучшему воспламенению горючего в двигателях на сильном морозе.
Рис. 28. Прибор
для определения
содержания воды
81
Работа 13. Диоксан-1,4
Основная реакция:
СН-ОН
21 ^
С
СН.ОН
2
Побочные реакции:
носн2—сн2он -> сн3сно+н2о
СН3—СНО + H2SO4 -> CH3COOH + Н2О + SO2
СН3—СНО н*<он~> > Продукты уплотнения
СН.СНО + НОСН.—СН.ОН —*» | * "СН— СН.+Н.0
3 2 2 сио^ 3 2
сн2— о.
о
Реактивы: этиленгликоль — 25 г; H2SO4 (конц.) — 2Д мл; К2СО3; КОН; Na
(металлич.).
В кругло донную колбу вместимостью 100 мл, соединенную через
дефлегматор с холодильником, помещают сухой этиленгликоль и серную
кислоту. Смесь осторожно нагревают и медленно перегоняют при
температуре 84—88 °С (к концу перегонки температура повышается до
102 °С). При этом содержимое колбы чернеет и вспенивается, выделяя
SO2. После прекращения нагревания дистиллят насыщают карбонатом
калия. Смесь переносят в делительную воронку и отделяют верхний слой,
который сушат сначала безводным карбонатом калия, а затем — гидро-
ксидом калия. Щелочь вызывает потемнение жидкости вследствие осмо-
ления уксусного альдегида.
Жидкость отделяют от щелочи, добавляют к ней несколько кусочков
металлического натрия и кипятят 30 мин с обратным холодильником.
Затем колбу охлаждают, добавляют еще немного металлического натрия
и перегоняют с дефлегматором, отбирая фракцию с т. кип. 100—103 °С.
Выход диоксана 9,5 г (54%).
Диоксан — бесцветная жидкость с т. кип. 101,3 °С, т. нл. 11,8°С,
df 1,0338, п™ 1,4224. Смешивается с водой и обычными
органическими растворителями. Образует комплексы с различными неорганическими
веществами. На воздухе медленно окисляется, образуя взрывоопасные
пероксиды.
Применение: используется для проведения реакции сульфирования и бро-
мирования в мягких условиях; очистки нефтяных масел; в качестве растворителя
растительных и минеральных масел, ацетилцеллюлозы, каучуков, полистирола,
природных смол.
82
Работа 14. Дифенилметан
С6Н6 + СбН5СН2С1—^^->СбН5СН2СбН5 + НС1
Реактивы: бензол — 25 мл; хлористый бензил — 6 мл; А1С13 (безводн.) —
2,5 г;.НС1 (конц,); NaOH, 10 %-ный раствор; Na (металлич.).
В двугорлую круглодонную колбу вместимостью 100 мл,
снабженную капельной воронкой и обратным холодильником с хлоркальциевой
трубкой, вносят 25 мл безводного бензола и 2,5 г тщательно
измельченного безводного хлорида алюминия. (Все операции с хлоридом
алюминия на открытом воздухе проводят как можно быстрее из-за его большой
гигроскопичности.) Хлоркальциевую трубку соединяют резиновым
шлангом с воронкой, опрокинутой над водой. В колбу по каплям
добавляют б мл хлористого бензила (под тягой!). После прекращения
выделения хлороводорода (обнаруживается по лакмусовой бумаге) в колбу
вносят 40 г толченого льда и 5 мл концентрированной соляной кислоты,
которая добавляется для предотвращения гидролиза хлорида алюминия.
Затем реакционную смесь переносят в делительную воронку, отделяют
бензольный слой, который промывают водой, раствором гидроксида
натрия и снова водой. Промытый бензольный раствор дифенилметана
помещают в колбу Вюрца (на 50 мл) и отгоняют бензол, а затем, убрав
холодильник, перегоняют дифенилметан при 255—265 °С. При этом
отводная трубка колбы Вюрца вставлена в приемник, который погружен в
холодную воду. Если дифенилметан окрашен и не застывает при
охлаждении, его перегоняют еще раз с 2—3 кусочками металлического натрия.
Выход около 5 г (60 %).
Дифенилметан — бесцветное твердое вещество с запахом
апельсиновой кожуры. Т. пл. 27 °С, т. кип. 264,3 °С. Нерастворим в воде, хорошо
растворим в эфире, спирте, бензоле; df 1,0059, я£° 1,5753.
Применение: для получения азо- и трифенилметановых красителей;
проведения различных органических синтезов; в качестве стабилизатора
пироксилиновых порохов, пластификатора нитроцеллюлозы и сложных эфиров целлюлозы.
5. Ацилирование
Актирующие средства: галогеиаигидриды, ангидриды,
сложные эфиры. Ацилироваиие спиртов, фенолов, аминов, аммиака. Аци-
лироваиие по Фриделю — Крафтсу. Реакция этерификации, ее
механизм. Кислотный и щелочной гидролиз. Омыление жиров.
83
Работа 15. Бутил ацетат
С4Н9ОН + СН3СООН H*so* > СН3СООС4Н9 + Н2О
Реактивы: бутанол-1 — 15 г; ледяная уксусная кислота — 15 мл; H2SO4
(конц.) — 1 мл; Na2CO3, 5%-ный раствор.
В круглодонную колбу вместимостью 50—100 мл помещают
уксусную кислоту, добавляют при перемешивании серную кислоту, а затем
вносят бутанол. Колбу присоединяют к насадке Дина—Старка и кипятят
до прекращения отделения капель воды. После охлаждения реакционную
смесь из колбы и дистиллят из насадки переносят в делительную
воронку, отделяют воду, а органический слой промывают последовательно
водой (50 мл), 5%-ным раствором соды до нейтральной реакции и снова
водой. Эфир сушат безводным сульфатом натрия, фильтруют и
перегоняют из колбы Клайзена, собирая фракцию с т. кип. 124—126 °С. Выход
16 г (70%).
Бутилацетат — бесцветная жидкость с приятным (в небольших
концентрациях) запахом, т. кип. 126,5 °С, df 0,8825, п% 1,3941. Мало
растворим в воде, смешивается с эф??ром, спиртом, ацетоном.
Применение: в качестве растворителя глифталевых, полиэфирных
полимеров, перхлорвиниловых и полиакриловых полимеров, эфиров целлюлозы, каучу-
ков, растительных масел; в качестве компонента растворителя для нитроцеллю-
лозных лаков и красок; для декалькомании керамики, металлизации керамики.
Работа 16. Ацетанилид
Основные реакции:
C6H5NH2 + НС1 -> C6H5NH2 • НС1
C6H5NH2 • HC1 + (CH3CO)2O -> C6H5NHCOCH3 + CH3COOH + HC1
Побочные реакции:
HC1 + CH3COONa -> CH3COOH + NaCl
(CH3CO)2O + H2O -> 2CH3COOH
Реактивы: анилин — 9,3 г; соляная кислота (конц.) — 8,5 мл; уксусный-
ангидрид — 12,5 мл; ацетат натрия (тв.) — 15г.
В стакане вместимостью 0,5 л к 250 мл воды приливают 8,5 мл
концентрированной соляной кислоты и при помешивании стеклянной
палочкой добавляют свежеперегнанный анилин. Раствор нагревают до 50 °С,
приливают уксусный ангидрид и перемешивают до полного растворения.
84
Затем сразу добавляют ацетат натрия и раствор снова интенсивно
размешивают. При охлаждении раствора начинает выпадать обильный осадок.
Кристаллы ацетанилида отсасывают и промывают на фильтре небольшим
количеством воды. Выход 10—11 г (74—82%). Продукт перекристалли-
зовывают из воды или (лучше) из спирта. Т. пл. 114 °С.
Ацетанилид — бесцветные кристаллы, df 1,0261, плохо растворим
в воде н бензоле, хорошо — в спирте.
Применение: в качестве жаропонижающего препарата; для синтеза 4-нитро-
анилина, используемого для получения красителей; в качестве пластификатора
нитроцеллюлозы и ацетилцеллюлозы.
Работа 17. Ацетилсалициловая кислота
(аспирин)
соон соон
ОН ^ Jn^OCOCHj
+ (СН3СО)2О *■ Г JJ + СН3СООН
Г JJ
Реактивы: салициловая кислота — 1,3 г; уксусный ангидрид — 1,2 г; H2SO4
(конц.).
В короткой пробирке, снабженной обратным воздушным
холодильником, растворяют при слабом нагревании 1,3 г салициловой
кислоты в 1,2 г уксусного ангидрида и затем добавляют каплю
концентрированной серной кислоты. Реакционную смесь нагревают 1,5 ч на
кипящей водяной бане. После этого смесь охлаждают, перемешивают
стеклянной палочкой, добавляют немного холодной воды и
отсасывают твердый продукт, который промывают сначала ледяной водой, а
затем небольшим количеством толуола. Выход 1,5 г (88%). Из
маточного раствора выпариванием можно получить еще некоторое
количество продукта.
Ацетилсалициловую кислоту перекристаллизовывают из бензола
или хлороформа. Т. пл. 134—135 °С.
Ацетилсалициловая кислота — бесцветное кристаллическое
вещество, трудно растворимое в холодной воде, лучше — в горячей.
Применение: в качестве жаропонижающего и болеутоляющего средства.
85
Работа 18. Фениловый эфир бензойной кислоты
СбН5ОН + NaOH -> C6H5ONa + Н2О
СбН5СОС1 + C6H5ONa -> СбН5СООСбН5 + NaCl
Реактивы: фенол — 4,7 г; хлористый бензоил — 8,5 мл; гидроксид натрия,
10%-ный раствор — 80 г; этанол.
В коническую колбу вместимостью 150 мл вносят раствор NaOH, в
котором растворяют фенол. Затем к реакционной смеси добавляют
хлористый бензоил. Колбу соединяют с обратным воздушным холодильником и
энергично встряхивают до исчезновения запаха хлористого бензоила
(примерно 20 мин). Выпавпгай осадок фенилбензоата отсасывают, тщательно
промывают водой и сушат на воздухе. Чистый продукт получают
перекристаллизацией из этанола. Выход около 8 г (80%). Т. пл. 70 °С.
Фенилбензоат — бесцветное кристаллическое вещество, d]x 1,2350,
практически нерастворим в воде. Хорошо растворим в спирте и бензоле.
Применение: в качестве высокотемпературного теплоносителя и
пластификатора ацетилцеллюлозы.
Работа 19. 2-Бензоилбензойная кислота
Реактивы: фталевый ангидрид — 7,4 г; бензол — 40 мл; хлорид алюминия
(безводный) — 15 г; концентрированная соляная кислота (конц.) — 15 мл;
гидроксид натрия — 2 г; активированный уголь.
Круглодонную колбу вместимостью 250 мл снабжают обратным
холодильником с хлоркальциевой трубкой, соединенной при помощи
резинового шланга с воронкой, опрокинутой над поверхностью воды. В колбу
вносят 7,4 г возогнанного фталевого ангидрида и 40 мл абсолютного
бензола. Смесь охлаждают ледяной водой и добавляют 15 г безводного
хлорида алюминия. Прибор встряхивают (вместе со штативом) и, если
реакция не идет, подогревают несколько секунд на кипящей водяной бане до
начала выделения НС1. При бурном выделении газа колбу охлаждают.
86
Для полного выделения хлороводорода колбу нагревают на водяной бане.
Затем колбу охлаждают и для разложения комплекса (хлорида алюминия
с 2-бензоилбензойной кислотой) добавляют по каплям через холодильник
100 мл воды, а потом 10 мл концентрированной соляной кислохы для
перевода в раствор основной соли алюминия. Бензол отгоняют, нагревая
колбу на электроплитке с закрытым нагревательным элементом. После
охлаждения льдом оставшийся в колбе маслообразный продукт
затвердевает. Его отфильтровывают, промывают на фильтре небольшим
количеством воды, затем растворяют в горячем растворе 2 г гидроксида натрия
в 20 мл воды и кипятят с активированным углем в течение нескольких
минут. Продукт фильтруют через складчатый фильтр. Охлажденный
фильтрат подкисляют 5 мл концентрированной соляной кислоты (до
кислой реакции по конго красному). Выделившийся маслообразный
продукт, который затвердевает при охлаждении и перемешивании,
фильтруют и промывают водой, а затем сушат на воздухе. Выход 9 г (75%).
Перекристаллизованная из бензола 2-бензоилбензойная кислота
плавится при 127 °С.
2-Бензоилбензойная кислота — бесцветное вещество, мало
растворимое в воде, хорошо — в спирте и бензоле.
Применение: используется для синтеза антрахинона (красители на его
основе широко применяются для окрашивания стеклопластиков).
б. Диазотирование и реакции диазосоединений
Действие азотистой кислоты на ароматические амины. Реакция
диазотироваиии, ее механизм. Диазотирующие агенты. Условия диа-
зотирования. Соли диазоиия, диазогидраты и диазотаты. Изомерия и
взаимные переходы этих продуктов. Реакции диазосоедииений с
выделением и без выделении азота. Реакции Гаттермаиа — Зандмайера.
Реакция азосочетаиии. Азокрясители. Основные поиития теории
цветности.
Работа 20. Метиловый оранжевый
HO3S
NaOH
NaNO2+2HCl
N+2NaCl
87
N(CH3)
V2
Реактивы: диметиланилин — 0,6 г; сульфаниловая кислота (двухводная) —
1 г; NaNO2 — 0,4 г; НС1,2 н.; NaOH, 2 а
В стакане вместимостью 50 мл растворяют 0,6 г диметиланилина в 5 мл
2 н. соляной кислоты. Диметиланилин должен раствориться полностью, о
чем судят по отсутствию маслянистого слоя. В другом стакане растворяют
сульфаниловую кислоту в 2,5 мл 2 н. раствора гидроксида натрия и
смешивают с раствором нитрита натрия в 5 мл воды. Полученную смесь
охлаждают в ледяной бане и приливают, тщательно перемешивая, к 2,5 мл
охлажденной до 1—2 °С 2 н. соляной кислоты. Образовавшаяся соль диазония
может выпасть в осадок, однако это не влияет на ход реакции азосочетания.
Солянокислый раствор диметиланилина также охлаждают ледяной
водой и при перемешивании вливают в раствор диазосоединения. Через
5—10 мин образуется густой пастообразный продукт красного цвета. К
нему добавляют 2 н. раствор гидроксида натрия (до щелочной реакции) и
нагревают до кипения (для ускорения кристаллизации). Затем смесь
охлаждают до 5 °С. Через некоторое время начинают выпадать оранжево-
коричневые листочки натриевой соли азокрасителя. Через 1,5 ч осадок
отсасывают, перекристаллизовывают из воды и сушат при 30—40 °С.
Выход почти количественный.
Метиловый оранжевый при сильном нагревании разлагается, не
плавясь. В кислой среде окрашен в розовый цвет, а щелочной — в желтый.
Применение: в качестве кислотно-основного индикатора, необратимого
окислительно-восстановительного индикатора.
Работа 21. Фенол
Основные реакции:
С 14 ТЧП-Г _l 14 Qf\ _*. Г* 14 Т&14 14QfV
О Э £. 4. Ч О 3 J Щ
Г4 +NaNO2 +H2SO4 ->[C6H5N^N]+HSO; +NaHSO4 +2H2O
H2O ->C6H5OH + N2+H2SO4
88
Побочные реакции:
[C6H5NsN]+HSO4~ + C6H5NH2 -> С6Н3— NH—N=N—C6H5
C6H5NH—N=N—C6H5 -» C6H5—N=N—C6H4—NH2
Реактивы: анилин — 9,3 г; H2SO4 (кона) — 10 мл; NaNO2 — 7,5 г; NaCl;
CaCl2; эфир; бромная вода.
В стакан вместимостью 300—500 мл наливают 50 мл воды и при
помешивании серную кислоту. В горячий раствор медленно, при
перемешивании, вносят анилин, который должен полностью раствориться.
Раствор охлаждают до комнатной температуры и постепенно прибавляют 75 г
тонкоизмельченного льда, доводя температуру раствора до 0 °С.
Содержимое стакана энергично перемешивают стеклянной палочкой. К смеси
из капельной воронки медленно прибавляют охлажденный до 0—5 °С
раствор нитрита натрия (7,5 г в 30 мл воды). Во время диазотирования
стакан должен находиться в бане с ледяной водой (температура
реакционной смеси не должна превышать 5 °С).
После того как большая часть раствора нитрита натрия прибавлена,
делают пробу на иодкрахмальную бумагу. Если через 5 мин после
прибавления порции нитрита натрия нодкрахмальная бумага синеет при
нанесении на нее капли испытуемого раствора, реакцию считают
законченной. В противном случае приливают еще раствор нитрита натрия и пробу
повторяют. Реакционная смесь должна быть кислой по конго красному. В
случае необходимости следует добавить несколько капель серной кислоты.
Полученный прозрачный раствор соли диазония переносят в кругло-
донную колбу вместимостью 500 мл и нагревают при 40— 50 °С. Колбу
периодически взбалтывают. Нагревание продолжают, пока не
прекратится выделение пузырьков азота. После этого фенол перегоняют с водяным
паром. Перегонку заканчивают, когда проба дистиллята будет давать с
бромной водой лишь слабую муть.
Дистиллят насыщают растертым в порошок хлоридом натрия (7—8 г)
для уменьшения растворимости фенола, переносят в делительную
воронку и экстрагируют эфиром (три раза по 20 мл). Эфирный раствор фенола
сушат хлоридом кальция и отгоняют эфир на водяной бане. Затем пере-
. гонку продолжают на воздушной бане (заменив водяной холодильник на
воздушный), собирая фракцию с температурой кипения 179—183 °С. В
приемнике фенол быстро кристаллизуется. Выход продукта 6 г (64%).
Фенол — бесцветное кристаллическое вещество с сильным
характерным запахом. На воздухе быстро окисляется, приобретая вначале ро-
зовый^ а затем — темно-красный цвет. Т. пл. 40,9 °С, т. кип. 181,7 °С, df
1,071, п£ 1,5403. Смешивается с водой при 66 °С, при комнатной
температуре — со спиртом и бензолом. Ядовит, обжигает кожу.
89
Применение: для многочисленных синтезов, получения красителей, ПАВ,
присадок для топлив, дубителей. Является антиокислителем, антисептиком (в том
числе и для древесины), используется для получения фенолальдегидных
полимеров; качественного и количественного определения свободного оксида кальция в
цементе.
Работа 22. 2-Хлорбензойная кислота
соон
соон
Cl
Cl
Реактивы: антраниловая кислота — 10 г; NaNC>2 — 6,5 г; Си2С1г — 3,5 г;
НС1 (конц.).
В фарфоровом стакане вместимостью 300 мл смешивают 10 г антра-
ниловой кислоты, 25 г НС1, 30 мл Н2О и 20 г льда. При охлаждении и
непрерывном помешивании в стакан постепенно прибавляют раствор
нитрита натрия в 30 мл воды (конец капельной воронки должен находиться в
жидкости). Окончание диазотирования определяют по иодкрахмальной
бумаге (синее окрашивание).
Полученный прозрачный раствор соли диазония медленно при
перемешивании вливают в раствор 3,5 г Си2С12 в 8 мл концентрированной
соляной кислоты. При этом происходит сильное вспенивание (выделение
азота) и одновременно осаждается 2-хлорбензойная кислота. По
окончании реакции осадок отсасывают, промывают небольшим количеством
холодной воды и хорошо отжимают на фильтре стеклянной пробкой.
Продукт перекристаллизовывают из воды. Т. пл. 142 °С. Выход 7—8 г
(65—70%).
2-Хлорбензойная кислота — бесцветное кристаллическое вещество,
растворимое в горячей воде, спирте, эфире, d™ 1,5440.
Применение: широко используется в органическом синтезе (например, для
получения акридиновых препаратов).
7. Окисление и восстановление
Окисление алкаиов и алкеиов. Окисление по месту двойной сви-
зи; метод Вагнера, метод Прилежаева, озонирование. Оксиды олефи-
нов. Установление строении алкенов по продуктам окислительной
деструкции. Окисление ароматических углеводородов с разрывом
90
кольца. Окисление алкилароматических углеводородов. Окисление
функциональных производных бензола (фонолы, амины, альдегиды).
Защита амиио- и гадроксигрупп. Окисление спиртов, альдегидов и
кетоиов. Правило Попова. Гидропероксиды и пероксиды алкнлов и
ацнлов. Окисляющие агенты: кислород, озои, перманганат калии,
хромовый ангидрид, гипохлориты, иодияя кислота, оксид серебра,
азотиаи кислота, азотистая кислота.
Гидрирование алкеиов и алкииов. 1,4-Присоединение.
Гидрирование ароматических углеводородов. Восстановление карбонильных
соединений, карбоиовых кислот и их производных. Восстановление
ароматических нитросоедииеиий. Бензидииовая перегруппировка.
Восстановление алифатических нитросоедииеиий.
Восстанавливающие агенты: натрий, водород, цинк, амальгамы металлов, алкоголи-
ты алюминии, алюмнннйгидриды, иодоводороднаи кислота.
Работа 23. Бензойная кислота
С6Н5—СН3 + 2КМпО4 -> С6Н5СООК + 2МпО2 + 2КОН + Н2О
С6Н5СООК + НС1 -> С6Н5СООН + КС1
Реактивы: толуол — 1 г; КМпО4 — 3,4 г; этиловый спирт; НС1 (кона).
Смесь 1 г толуола, 15 мл воды и 3,4 перманганата калия кипятят в
круглодонной колбе вместимостью 200 мл с обратным шариковым
холодильником в течение 2 ч на песчаной бане. К еще горячей (но не
кипящей) реакционной смеси через холодильник осторожно добавляют
этиловый или изопропиловый спирт до исчезновения малиновой окраски
перманганата калия. Выпавший диоксид марганца отфильтровывают и
промывают на фильтре кипящей водой (два раза по 15 мл). Промывные
воды соединяют с фильтратом и упаривают до объема 10—15 мл. Если
вновь выпадает диоксид марганца, его отфильтровывают.
К упаренному раствору бензоата калия прибавляют по каплям
концентрированную соляную кислоту до кислой реакции по конго красному.
Выпавшую бензойную кислоту отсасывают, промывают на фильтре неболышш
количеством ледяной воды и сушат на воздухе. Т. пл. 121—122°С. Выход
продукта 0,8 г (60%).
Бензойная кислота —бесцветное кристаллическое вещество со
слабым запахом, d\b 1,2659, п% 1,5397. Плохо растворима в холодной воде
и эфире, хорошо — в спирте, уксусной кислоте, пиридине.
Применение: в качестве консерванта; для производства пластификаторов,
пероксида бензоила (инициатор полимеризации); вулканизаторы силиконовых
каучуков. Используется с целью защиты арматуры в железобетоне от коррозии;
для определения свободного оксида кальция в цементе.
91
Работа 24. 4-Нитробензойная кислота
COOH +Cr2(SO4)3+ Na2SO4 + 5H2O
Реактивы: и-нитротолуол — 13,7 г; дихромат натрия — 42 г; H2SO4 (конц.) —
55 мл; NaOH, 5%-ный раствор — 100 мл.
В трехгорлую колбу вместимостью 250 мл, снабженную мешалкой,
обратным холодильником и капельной воронкой, помещают 90 мл воды,
дихромат натрия и л-нитротолуол. При перемешивании из капельной
воронки прибавляют в течение 20 мин концентрированную серную
кислоту. Происходит разогревание, при котором л-нитротолуол плавится и
начинается энергичная реакция. Ход реакции окисления регулируют
скоростью прибавления серной кислоты (избегают бурного кипения). После
прибавления всей серной кислоты и прекращения саморазогревания
реакционной смеси содержимое колбы нагревают при слабом кипении в
течение 30 мин. Охладив реакционную смесь, в колбу вливают 120 мл
воды и снова охлаждают. Выделившуюся 4-нитробензойную кислоту
отфильтровывают и промывают водой (50 мл).
Для удаления солей хрома кислоту растворяют в 100 мл 5%-ного
раствора гидроксида натрия. Осадок гидроксида хрома удаляют
фильтрованием, а к фильтрату при перемешивании добавляют
концентрированную серную кислоту до кислой реакции (по конго красному). Выпавшие
желтые кристаллы 4-нитробензойной кислоты отфильтровывают,
тщательно промывают водой и сушат на воздухе. Продукт очищают
перекристаллизацией из водного спирта. Т. пл. 242 °С. Выход около 10 г (66%).
4-Нитробензойная кислота — желтое кристаллическое вещество,
нерастворимое в воде. Хорошо растворимо в органических растворителях;
df 1,6100.
Применение: для синтеза некоторых медицинских препаратов и красителей.
Работа 25. Анилин
C6H5NO2 + 3Fe + 7НС1 -> C6H5NH2 • НС1 + 3FeCl2 + 2H2O
C6H5NH2 HC1 + NaOH -> C6H5NH2 + NaCl + H2O
Реактивы: нитробензол — 10,3 мл; железные опилки — 20 г; НС1 (конц.) —
90 мл; NaOH — 30 г; NaCl; КОН; эфир.
92
В круглодонную колбу вместимостью 500 мл вносят нитробензол и
железные опилки, затем небольшими порциями (по 1—2 мл) добавляют
соляную кислоту. После каждой порции колбу закрывают пробкой с
воздушным холодильником и хорошенько взбалтывают. После введения 20 мл
соляной кислоты оставшееся ее количество приливают порциями по
8—10 мл. Если восстановление идет слишком бурно, колбу охлаждают
водой. После прибавления всей кислоты колбу нагревают 30 мин на
кипящей водяной бане, периодически перемешивая реакционную массу.
Конец реакции определяют по исчезновению запаха нитробензола.
К горячей смеси осторожно приливают раствор 30 г гидроксида
натрия в 40 мл воды (до сильнощелочной реакции). Затем анилин отгоняют
с водяным паром. После того как из холодильника начнет стекать
прозрачный дистиллят, отгоняют еще около 40 мл жидкости.
Анилин высаливают, добавляя хорошо измельченный хлорид натрия
(20 г на 100 мл дистиллята). Соль растворяют при перемешивании, а
анилин экстрагируют эфиром, последовательно обрабатывая раствор тремя
порциями эфира (по 20 мл). Объединенные эфирные вытяжки сушат
несколькими кусочками гидроксида калия и отгоняют эфир на водяной
бане. Анилин перегоняют из той же колбы с воздушным холодильником,
добавив немного цинковой пыли. Собирают фракцию, кипящую при
184 °С. Выход около 7 г (80%).
Анилин — бесцветная маслянистая жидкость со слабым запахом, на
воздухе сравнительно быстро желтеет, приобретая темно-коричневый
цвет; df 1,0217, п™ 1,5683. Частично растворяется в воде (при 20 °С 3,4 г
в 100 мл). Смешивается с эфиром, спиртом, бензолом.
Применение: для синтеза красителей, взрывчатых веществ, антиоксццантов,
ускорителей вулканизации, пластификаторов и т. д.; для определения
ароматических углеводородов в нефти и нефтепродуктах; для получения анилинфенолфор-
мальдегидных и фенолформальдегидных полимеров, а также в качестве
ингибитора коррозии.
8. Реакции конденсации
карбонильных соединений
Альдольиаи и кретоиоваи конденсации. Особенности
конденсации кетой о в. Реакция Перкииа. Конденсация альдегидов и кетой ов с
ароматическими аминами и фенолами. Трифеиилметановые
красители и их синтез. Лейкосоедииеиия. Карбииольиые основании. Фталеи-
иы. Образование оснований Шиффа. Сложиоэфириая
конденсация.
93
Работа 26. Ацетоуксусный эфир
2СН3СООС2Н5 с*н*°"' )СН3СОСН2СООС2Н5 + С2Н5ОН
Реактивы: этилацетат — 25 г; Na (металлич.) — 2,5 г; СН3СООН, 50%-ный
раствор — 15 мл; NaCl, насыщенный раствор; Na2CO3, насыщенный раствор;
этанол.
В сухую круглодонную колбу вместимостью 100 мл, снабженную
обратным холодильником с хлоркальциевой трубкой, помещают 25 мл
сухого этилацетата и добавляют 2,5 г мелко нарезанного металлического
натрия (или натриевую проволоку). Для начала реакции добавляют
несколько капель этилового спирта. По окончании бурной стадии реакции
смесь продолжают осторожно нагревать (легкое кипение). Растворение
металлического натрия заканчивается обычно в течение часа. Непрореа-
гировавший натрий растворяют, добавляя 1—2 мл спирта. Реакционную
смесь охлаждают до 40—50 °С и прибавляют 50%-ную уксусную кислоту
до кислой реакции по лакмусу (15 мл). К полученному раствору
приливают равный объем насыщенного раствора хлорида натрия. Если при
этом выпадает осадок, его растворяют в небольшом количестве воды.
В делительной воронке отделяют верхний слой, который промывают
насыщенным раствором гидрокарбоната натрия и водой. Промытый
раствор ацетоуксусного эфира переносят в колбу Клайзена и отгоняют этил-
ацетат при температуре 95 °С. Ацетоуксусный эфир перегоняют в
вакууме, собирая фракцию с т. кип. 86—90 °С (при 3999,66 Па, или 30 мм рт. ст.).
Выход 5—6 г (40—48%).
Ацетоуксусный эфир — жидкость с характерным запахом. Обычно
представляет собой равновесную смесь двух таутомеров. Т. кип. 180,4 °С
(с разложением), т. пл. -45 °С, d]° 1,0282, п™ 1,4198. Ограниченно
растворим в воде, смешивается с эфиром, спиртом, бензолом.
Применение: для синтеза большого числа органических соединений.
Работа 27. Дибензилиденацетон
Основная реакция
Н5СНО + СН3СОСН3-^о->СбН5—СН=СН—СО—СН=СН—
Побочные реакции:
2С6Н5СНО + NaOH -> C6H5COONa + С6Н5СН2ОН
94
I I
сн3 сн3
Реактивы: свежеперегнанныЙ бензальдегид — 2,1 г; ацетон — 0,3 г; гидро-
ксид натрия — 2 г; этанол — 16 мл; этилацетат.
В колбу вместимостью 50 мл, содержащую раствор 2 г гидроксида
натрия в 20 мл воды и 16 мл этанола, при тщательном перемешивании
приливают половину заранее приготовленной смеси бензальдегида и
ацетона, поддерживая температуру 20—25 °С. Через 2—3 мин начинается
выделение осадка. Спустя 15 мин при перемешивании в колбу приливают
оставшуюся смесь бензальдегида и ацетона, смывают остатки этих
веществ из колбы, в которой они находились, спиртом (2—3 мл). Массу
перемешивают еще 20 мин, отсасывают, тщательно промывают осадок
водой и сушат на воздухе. После перекристаллизации из этилацетата
получают чистый дибензилиденацетон. Т. пл. 112 °С. Выход 1,8 г (77%).
Дибензилиденацетон — желтое кристаллическое вещество,
разлагающееся при перегонке. Нерастворим в воде, растворяется в ацетоне и
хлороформе.
Применение: реакция получения дибензилиденацетона используется для
определения небольших количеств ацетона.
Работа 28. Фурфурилиденацетон
и дифурфурилиденацетон
I ^-сно + снг-со-сн, Na0H» ( >-c№=ch-co-civ
о ^ 3 о ^
Реактивы: свежеперегнанныЙ фурфурол — 7,7 г; ацетон — 101; гидроксид
натрия, 35%-ный раствор — 15 мл; H2SO4 — 10%-ный раствор.
В колбе вместимостью 200 мл, снабженной механической мешалкой,
смешивают 7,7 г фурфурола, 60 мл воды и 10 г ацетона. Смесь при
перемешивании охлаждают до 10 °С и к ней приливают 15 мл 33%-ного
раствора гидроксида натрия. Реакционную смесь перемешивают около 2 ч и
добавляют серную кислоту (до кислой реакции на лакмус). При этом
смесь становится прозрачной и быстро расслаивается. Нижний слой
(масло) отделяют в делительной воронке и перегоняют в вакууме, соби-
95
рая фракцию, кипящую при 114—118 °С (1,3 кПа, или 10 мм рт. ст.) или
135—145 °С (6,6 кПа, или 50 мм рт. ст.). Выход около 6 г (62%).
Фурфурилиденацетон — желтый кристаллический продукт,
растворимый в спирте, эфире, хлороформе, бензоле. Т. пл. 39—41 °С
При соотношении количеств фурфурола и ацетона 2:1 в аналогичных
условиях образуется дифурфурилиденацетон:
СНО + СНз—СО—СН3
NtOH
о
сн=сн— со—сн—сн-< >+
о
Дифурфурилиденацетон — мелкокристаллический порошок с т. пл.
60—61 °С, т. кип. 254—260 °С (6,0—6,2 кПа, или 45—47 мм рт. ст.).
Выход 96—97%.
Применение: смесь фурфурилцденацетона и дифурфурилиденацетона в
соотношении 4 : 1 (мономер ФА) используют в производстве водостойких
бесцементных бетонов с чрезвычайно высокой мгновенной прочностью и
устойчивостью к истиранию, а также для получения различных пресс-материалов с
наполнителями: со стекловолокном (ФАС), асбестом (ФАА), графитом (ФАГ).
Работа 29. Фенолфталеин
О+2
Реактивы: фталевый ангидрид— 2,5 г; фенол — 4 г; H2SO4 (конц.) — 1 мл;
NaOH, 5%-ный раствор; НС1 (конц.); этанол; активированный уголь.
В широкую пробирку помещают 2,5 г очищенного возгонкой фтале-
вого ангидрида, 4 г фенола и 1 мл серной кислоты. Смесь нагревают в
течение 3 ч на масляной бане при температуре 120—125 °С. Жидкость в
пробирке перемешивают погруженным в нее термометром. По окончании
реакции горячую смесь выливают в стакан, содержащий 50 мл воды, и
кипятят до исчезновения запаха фенола. Кипение сопровождается
сильными толчками вследствие образования осадка, поэтому раствор необхо-
96
димо постоянно перемешивать. После охлаждения раствор фильтруют на
воронке Бюхнера, стараясь по возможности не переносить осадок на
фильтр. Осадок, находящийся в стакане и частично на фильтре,
промывают два раза небольшими порциями холодной воды, затем растворяют в
небольшом количестве 5%-ного раствора гидроксида натрия и
фильтруют. Темно-красный фильтрат подкисляют соляной кислотой (до рН 7).
Выпавший осадок фенолфталеина через полчаса отфильтровывают и
сушат. Продукт очищают перекристаллизацией из водного спирта с
использованием активированного угля. Выход около 2 г (38%). Т. пл. 261 °С.
Фенолфталеин — бесцветное кристаллическое вещество,
возгоняющееся выше температуры плавления. Мало растворимо в воде, хорошо —
в спирте и эфире.
Применение: одноцветный кислотно-основной индикатор. Служит также
для получения фенолфталеинформальдегндных полимеров (связующие для
стеклопластиков) и полиарилатов (электроизоляционные материалы и радиационно-
стойкие фильтрующие материалы).
9. Элементорганические соединения
Общие представлении об элемеиторганическнх соединениях.
Характер связи углерод — элемент. Органические производные
непереходных элементов. Магиийоргаиические соединения. Взаимодействие
реактива Грииьяра с соединениями, содержащими подвижный атом
водорода. Синтезы с помощью реактивов Гриньяра (реакции с
альдегидами, кетойами, производными карбоиовых кислот, оксидами оле-
фииов, диоксидом углерода, кислородом). Алюмииийорганические
соединения. Триэтилалюминий н его использование для получения
спиртов и кислот. Катализаторы Циглера — Натта.
Кремнийоргаиические соединения. Номенклатура. Получение
кремиийоргаиических мономеров. Алкилхлорсилаиы. Силаиолы, сн-
лаидиолы и силаитриолы. Сил азаны. Полиорганосилоксаиы.
Применение кремиийоргаиических соединений в народном хозяйстве. Гнд-
рофобизации строительных материалов.
Органические производные переходных металлов. Ферроцен.
Работа 30. Бензгидрол
Основные реакции:
С6Н5Вг + Mg -» C6H5MgBr
С6Н6СНО + C6H5MgBr -> (С6Н5 )2 CHOMgBr НС| )(С6Н5 )2 СНОН + MgBrCl
4-290 97
Побочные реакции:
C6H3MgBr + С6Н3Вг -> С6Н5— С6Н3 + MgBr2
C6H3MgBr + О2 -> C6H3OMgBr—^->C6H5OH + MgBrCl
Реактивы: бромбегоол — 9,5 г; магний (мет.) — 1,5 г; бензальдегид — 5,3 г;
абсолютный эфир; НС1 (1:1); Na2SO4, 40%-ный раствор; СаС12 (плавленый); иод
кристаллический.
В сухую трехгорлую колбу вместимостью 200 мл, снабженную
капельной воронкой, мешалкой и обратным холодильником с хлор-
кальциевой трубкой, помещают магниевые стружки и несколько
кристалликов иода. Колбу нагревают до тех пор, пока весь ее объем не заполнят
фиолетовые пары иода. Затем колбу охлаждают и приливают 5—7 мл
раствора сухого бромбензола в абсолютном эфире. Обычно реакция
начинается почти сразу; если же ее начало задерживается, колбу нагревают
непродолжительное время на теплой водяной бане. Остаток эфирного
раствора бромбензола приливают по каплям с такой скоростью, чтобы
наблюдалось слабое кипение эфира.
Для завершения реакции колбу нагревают на водяной бане почти до
полного растворения магния. Реакционную смесь охлаждают, погружая
колбу в ледяную баню, и по каплям, при непрерывном помешивании,
приливают раствор свежеперегнанного бензальдегида в 10 мл
абсолютного эфира. Чтобы довести реакцию до конца, реакционную смесь
нагревают 15 мин на водяной бане.
После охлаждения раствора добавляют 10—15 г льда и
выделившуюся основную соль магния растворяют в 10 мл соляной кислоты
(1 : 1). Эфирный слой отделяют, а водный раствор экстрагируют эфиром
(два раза по 10 мл). Соединенные эфирные вытяжки промывают 40%-
ным раствором гидросульфита натрия (5 мл) и сушат хлоридом кальция.
Эфир отгоняют на водяной бане. Оставшийся в колбе бензгидрол
застывает при охлаждении. Если этого не происходит, бензгидрол перегоняют
с водяным паром. Для лучшей очистки можно перекристаллизовать его
из смеси бензол — гептан (1:1). Выход продукта 5 г (45%). Т. пл. 69 °С.
Бензгидрол — бесцветное твердое вещество, плохо растворимое в
воде, хорошо — в спирте и эфире.
Применение: в качестве пластификатора полиамидных полимеров; для
синтеза бензофенона (пластификатор простых и сложных эфиров целлюлозы); для
получения некоторых красителей.
10. Реакции полимеризации
и поликонденсации
Мономеры; бутадиен, изопрен, хлоропрен, хлористый вннил, акри-
лонитрил, метнлметякрилат, стирол. Функциональность мономеров.
Реакции полимеризации. Механизмы радикальной н ионной
полимеризации. Инициаторы н регуляторы. Стереорегулирные
полимеры. Применение катализаторов Цнглеря—Натта. Сополнмеризацни.
Блок-сополимеры и привитые сополимеры. Реакции поликонденса-
цин. Феиолальдегидиые н карбамидальдегидные полимеры. Сложные
полиэфиры. Полимеры на основе фурфурола. Мономер ФА.
Эпоксидные и кремнийорганические полимеры. Тноколы. Полиуретаны.
Полиамиды. Альтины. Каучукн. Полистирол и полиакрнлаты. Особые
свойства высокомолекулярных соединений. Химические реакции
высокомолекулярных соединений: полимераналогичные превращении и
макромолекулирные реакции. Деструкции полимеров. Ингибиторы
деструкции. Полимерные материалы н их применение в
строительстве. Полимерцементы и полнмербетоны, газонаполненные пластмассы
и стеклопласты.
Работа 31. Полистирол
п
7-м способ
Реактивы: стирол — 5 г; поливиниловый спирт — 0,2 г; пероксид бен-
зоила — 0,1 г.
В двугорлую колбу вместимостью 100 мл, снабженную
механической мешалкой и обратным холодильником, вносят раствор 0,2 г
поливинилового спирта в 30 мл воды и раствор 0,1 г пероксида бензоила в 5 г
свежеперегнанного стирола. Колбу помещают в кипящую баню
(предварительно отключив нагревание бани) и включают мешалку. Поддерживая
температуру бани 80 °С, реакционную смесь перемешивают 6 ч.
Полученные гранулы отфильтровывают, промывают водой (четыре раза по 25
мл), отжимают между листами фильтровальной бумаги и сушат в вакуум-
эксикаторе до постоянной массы.
2-й способ
Реактивы: стирол — 10 г; бензол — 10 г; пероксид бензоила — 0,2 г.
99
В круглодонную колбу вместимостью 50 мл, снабженную обратным
холодильником, помещают свежеперегнанный стирол, бензол и пероксид
бензоила и нагревают 4 ч на кипящей водяной бане. Охлажденную массу
выливают в фарфоровую чашку и добавляют 10—20 мл петролейного
эфира или гексана. Растворитель сливают, а выпавший осадок
полистирола сушат в вакуум-сушильном шкафу при 80 °С (67—80 кПа, или
500—600 мм рт. ст.).
Применение: в качестве облицовочного, тепло- и звукоизоляционного
материала в жилищном строительстве, для изготовления труб, сантехнического
оборудования, латексных красок и эмали для внутренней отделки, пропитки
бетона и керамики мономером с последующей полимеризацией.
Работа 32. Полиметилметакрилат
2
P.. - ГППГТТ
Ан3
/-M
CH2
способ
T
COOCH
п
Реактивы: метилметакрилат — 5 г; персульфат аммония — 1 г.
В трехгорлой колбе вместимостью 250 мл, снабженной мешалкой,
обратным холодильником и капельной воронкой, растворяют 1 г персульфата
аммония в 100 мл воды. Колбу помещают на водяную баню, нагретую до
80 °С. По каплям, при энергичном перемешивании, добавляют 5 г свежепе-
регнанного метилметакрилата. Нагревание и перемешивание продолжают
3 ч. Затем через эмульсию пропускают водяной пар до коагуляции полимера.
Осадок отфильтровывают, промывают водой и сушат на воздухе. Если
полимер не осаждается при пропускании пара, то добавляют небольшое
количество 10%-ного раствора хлорида натрия. В этом случае полимер
промывают водой до отрицательной реакции на хлорид-ион.
2-й способ
Реактивы: метилметакрилат — 5 г; пероксид бензоила — 0,01 г.
Метилметакрилат помещают в широкую пробирку, добавляют
пероксид бензоила, неплотно закрывают пробирку пробкой и нагревают на
водяной бане. Через 20—25 мин жидкость загустевает и вскоре
превращается в твердую бесцветную массу.
100
Применение: для изготовления листового конструкционного стекла
(бесцветного и окрашенного), рассеивающих экранов для светильников,
декоративных ограждений, моющихся обоев, краски и грунтовок; для пропитки пористых
материалов с целью увеличения прочности, для придания водонепроницаемости
бетону, пропитки мономером бетона и керамики с последующей
полимеризацией; для получения керамических волокон.
Работа 33. Поливинилацетат
лСН—СН— ОСОСН3 *■
— PUL.
ОСОСН.
п
Реактивы: винилацетат — 5 г; пероксид бензоила — 0,1 г; бензол — 5 г;
ксилол — 60 мл.
В круглодонную колбу вместимостью 100 мл, снабженную
обратным холодильником, помещают 5 г свежеперегнанного винилацетата, 5
мл бензола и 0,1 г пероксида бензоила. Смесь нагревают 3 ч на водяной
бане при 80 °С. Бензольный раствор выливают в 60 мл ксилола,
отфильтровывают выпавший полимер, который сушат при 60° в течение 3 ч.
Применение: в качестве связующего и пленкообразующего компонента в
малярных красках по штукатурке и дереву для внутренней и внешней отделки;
для получения полимерцементов и полимербетонов с повышенной прочностью на
удар, изгиб и растяжение; для склеивания древесины, изготовления бесшовных
полов, кирпичных блоков с повышенной прочностью швов; для укрепления
поверхностных слоев сырых керамических изделий, а также в качестве связки и
пластификатора керамических пресс-порошков.
Работа 34. Поливиниловый спирт
2-сн-
ососн.
[ОН
п
—сн—сн-
он
лСН3СООН
п
Реактивы: поливинилацетат, 30%-ный спиртовой раствор — 5 г; этанол —
50 мл; щелочь, спиртовой раствор (1,12 г КОН или 0,9 г NaOH в 10 мл этанола).
В стакан или широкогорлую колбу вместимостью 100 мл помещают 10
мл спиртового раствора щелочи. Затем устанавливают мешалку и при ком-
101
натной температуре и интенсивном перемешивании приливают по каплям из
капельной воронки 5 г спиртового раствора поливинилацетата. По мере
омыления поливинилацетата происходит осаждение из раствора поливинилового
спирта. Продолжительность реакции 2—3 ч. Порошкообразный осадок
поливинилового спирта отфильтровывают на воронке Бюхнера и промывают
несколько раз спиртом до нейтральной реакции. Продукт сушат в вакуум-
сушильном шкафу при 30—40 °С (67—80 кПа, или 500—600 мм рт. ст.).
Применение: в качестве связующего и пластификатора при изготовлении
тонких формовочных порошков для керамики; клеев для склеивания бумаги и
дерева; для получения полимерцемекгных растворов и бетонов, для укрепления
поверхностных слоев сырых керамических изделий.
Работа 35. Фенолформальдегидный полимер (новолак)
ОН
+ «CH.0
сн
2
Реактивы: фенол — 9,4 г; формалин — 4 мл (4 г); НС1 — 0,1 мл.
В круглодонной колбе вместимостью 100 мл взбалтывают смесь
фенола и формалина до полного растворения, затем добавляют 0,1 мл
соляной кислоты, присоединяют к колбе обратный холодильник и нагревают
на водяной бане при 90—100 °С в течение 20—40 мин. Реакция
протекает с выделением теплоты. При закипании смеси нагревание прекращают.
Затем нагревание продолжают на кипящей водяной бане до разделения
реакционной массы на два слоя: верхний — водный (обычно мутный) и
нижний — густой, светло-желтый или светло-коричневый (в зависимости
от качества фенола), который представляет собой продукт поликонденсации.
Содержимое колбы сливают в фарфоровую чашку. После
охлаждения верхний слой отделяют. Оставшийся в чашке полимерный продукт
промывают теплой водой до нейтральной реакции по метиловому
оранжевому и высушивают, постепенно нагревая до 200°С.
Применение: для производства стеклопластиков; в качестве связующего
при получении минеральной ваты, стекловаты, древесностружечных и
древесноволокнистых плит, древесиослоистых пластиков; для керамических пресс-
порошков; для покрытия металлов, керамики и бетона; для производства
полимер цементных растворов, устойчивых к агрессивным средам, химически стойких
мастик и замазок, суперпластификаторов для бетонных смесей, полимеркерамзи-
тобетона, перлитовых теплоизоляционных изделий; для склеивания металла с
керамикой.
102
Работа 36. Эпоксидный полимер (ЭД-20)
i)OH^~CH"~*C/rjLCl **" WHO'
СН3
-*-СН—CH-CHj -О-/
Реактивы: 4,4'-дигвдроксвдифенилпропан (диан) — 20 г; эпихлорпщрин —
16,2r;NaOH(TB.)—7,73 г.
В трехгорлой колбе вместимостью 250 мл, снабженной обратным
холодильником, мешалкой и термометром, растворяют 5 г гидроксида
натрия в 43 мл воды и при перемешивании добавляют 4,4'-дигидроксиди-
фенилпропан. Когда он полностью растворится, вводят небольшими
порциями при перемешивании эпихлоргидрин. Смесь постепенно (в течение
45 мин) нагревают на водяной бане до 80°С. Затем добавляют раствор 2 г
гидроксида натрия в 6 мл воды и нагревают 30 мин до 80 °С. Наконец,
приливают третью порцию гидроксида натрия — раствор 0,73 г в 25 мл
воды, повышают температуру до 95 °С и нагревают еще 1 ч.
Смолообразный продукт переносят в фарфоровую чашку,
промывают несколько раз водой (50 °С) до нейтральной реакции по
фенолфталеину. Продукт высушивают в вакуум-сушильном шкафу при температуре
90 °С (66,6 кПа, или 500 мм рт. ст.). Получают светло-желтую жидкую
смесь олигомеров.
Применение: в качестве связующих для получения конструкционных
стеклопластиков, высококачественных клеев для ответственных конструкций; полн-
мерцементных растворов и полимербетонов; пеиопластов, антикоррозионных
покрытий; для реставрации природных и искусственных камней; изготовления
клеев для керамики; создания водонепроницаемых слоев на бетоне и т. д.
Работа 37. Полидиметил сил океан
+ 2(CH3)3SiCl + (w + 2)H2O
*- (CHASi—O™
—Si—О
n
. Реактивы: диметилдихлорсилаи — 10,3 г; триметилхлорсилан
H2SO4 (конц.) — 0,3 мл; ВаС12,10%-ный раствор.
2,2 г;
103
(Реакцию проводят в вытяжном шкафу!)
В колбе вместимостью 100 мл, снабженной мешалкой, нагревают
смесь диметилдихлорсилана и триметилхлорсилана с 25 мл воды в
течение 1 ч при 40—60 °С. Затем температуру повышают до 90—100 °С и
смесь выдерживают еще 1 ч. Содержимое колбы охлаждают, прибавляют
при перемешивании 0,3 мл серной кислоты и вновь в течение 1 ч греют
на кипящей водяной бане. Горячий продукт обрабатывают 10%-ным
раствором хлорида бария в количестве, эквивалентном количеству взятой
серной кислоты. Осадок сульфата бария отфильтровывают, а фильтрат
упаривают на водяной бане до получения вязкого раствора.
Применение: в качестве жаростойких и атмосферостойких покрытий; для
производства стеклотекстолита большой прочности и теплостойкости; пенопластов,
выдерживающих температуру до 400 °С; полимерцементных растворов и полимербето-
нов; для гидрофобизации строительных материалов и изделий, пластификации
бетонных смесей; в качестве антивспенивателей; для интенсификации помола клинкера и
сырья для керамических изделий; в качестве добавок к бетонным смесям (для
увеличения морозостойкости, сцепления со старым бетоном и т. д.).
Работа 38. Поликонденсация фталевого ангидрида
с глицерином
о +
снон—снрн
1
о
?
о—с—...
С—О—СЩ- СН—CHj- О—С
о
о
Реактивы: фталевый ангидрид — 11 г; глицерин (безв.) — 8,5 г.
11 г фталевого ангидрида и 8,5 г безводного глицерина (если
глицерин водный, навеску соответственно увеличивают) помещают в
фарфоровый стакан, который плотно прикрывают опрокинутой стеклянной
воронкой. Смесь быстро нагревают на воздушной бане до 180 °С и эту
температуру поддерживают в течение 2 ч. Затем температуру повышают до
200—220 °С и нагревание продолжают до образования стеклообразной
смолы, трудно растворимой в ацетоне.
Применение: для производства лаков и красок.
104
Раздел IV
Основы технического анализа
органических веществ
Технический анализ представляет собой совокупность химических,
физико-химических и физических способов исследования органического
сырья и материалов.
Основными методами технического анализа являются химические
методы.
Работа 1. Определение кислотного числа,
числа омыления и эфирного числа
Опыт 1. Определение кислотного числа. Кислотное число (К Ч.) —
масса гидроксида калия (в мг), которая необходима для нейтрализации
свободных кислот, содержащихся в 1 г анализируемого вещества.
Навеску исследуемого продукта (2—5 г), взятую с точностью до 0,02 г,
растворяют в 30—50 мл растворителя [вода, спирт, смесь эфира со
спиртом (1 : 1)], добавляют 3—5 капель фенолфталеина и титруют 0,1—0,5 н.
раствором гидроксида калия до появления слабо-розовой окраски,
устойчивой в течение 20—30 с.
Растворитель предварительно нейтрализуют 0,1 н. раствором
гидроксида калия по фенолфталеину. Если навеска не растворяется на холоду,
ее нагревают на водяной бане с обратным холодильником до полного
растворения; титрование проводят после охлаждения.
Кислотное число (в мг КОН на 1 г вещества) вычисляют по формуле
где V— объем раствора гидроксида калия, израсходованного на
титрование навески, мл; Т — титр раствора гидроксида калия (г КОН/мл),
равный NK 56,1/1000 (N— нормальность раствора; К — поправочный
коэффициент; 56,1 — эквивалент гидроксида калия); g — навеска
исследуемого продукта, г.
Опыт 2. Определение числа омыления и эфирного числа.
Количественное определение сложных эфиров основано на реакции
гидролитического расщепления (омыления).
105
Эфирное число (Э. Ч.) — масса гидроксида калия (в мг), которая
необходима для омыления сложных эфиров, содержащихся в I г
анализируемого вещества.
Число омыления (Ч. О.) соответствует массе гидроксида калия (в мг),
необходимой для нейтрализации свободных кислот и омыления сложных
эфиров, содержащихся в\ г анализируемого вещества, т. е.
Ч.О. = Э.Ч. + К.Ч.
Точную навеску вещества (1—2 г) помещают в колбу, добавляют 25 мл
0,5 н. спиртового раствора гидроксида калия. Содержимое колбы
нагревают с обратным холодильником в течение 1—1,5 ч. Затем колбу
охлаждают и через холодильник вносят 50 мл дистиллированной воды. К пробе
добавляют 2—3 капли раствора фенолфталеина и оттитровывают 0,5 н.
раствором соляной кислоты до обесцвечивания.
Параллельно в тех же условиях нагревают 25 мл 0,5 н. спиртового
раствора гидроксида калия и титруют 0,5 н. раствором соляной кислоты
(контрольная проба).
Число омыления (в мг КОН на 1 г вещества) вычисляют по формуле
где Vx — объем 0,5 н. раствора соляной кислоты, пошедший на
титрование контрольной пробы, мл; У2 — объем 0,5 н. раствора соляной кислоты,
пошедший на титрование навески исследуемого продукта, мл; Т — титр
0,5 н. раствора гидроксида калия; g — навеска исследуемого продукта, г.
Эфирное число определяют как разность между числом омыления и
кислотным числом (Э.Ч. = Ч.О. - К.Ч.).
Работа 2. Определение бромного числа
Для обнаружения двойных связей в непредельных соединениях
используют реакцию присоединения брома.
Бромное число (Б. Ч.) показывает, сколько граммов брома может
присоединиться к 100 г вещества.
Опыт 1. Определение бромного числа масел и олиф. Навеску
исследуемого вещества (около 0,2 г), взятую с точностью до 0,02 г,
помещают в коническую колбу с притертой пробкой и приливают 25 мл 0,1
н. раствора брома. Колбу плотно закрывают пробкой и оставляют в
темном месте на 30 мин. Затем добавляют 10 мл 10%-ного раствора иодида
калия, перемешивают и титруют выделившийся иод 0,1 н. раствором
тиосульфата натрия. Перед концом титрования прибавляют раствор
крахмала и титруют до исчезновения синей окраски раствора. Параллельно
ставят контрольный опыт.
106
Бромное число (в г брома на 100 г вещества) вычисляют по формуле
Б.Ч.= ft-
где V\ — объем 0,1 н. раствора тиосульфата натрия, пошедший на
титрование контрольного опыта, мл; V2 — объем 0,1 н. раствора тиосульфата
натрия, пошедший на титрование навески исследуемого продукта, мл;
0,008 — масса брома, соответствующая 1 мл точно 0,1 н. раствора
тиосульфата натрия, г; К — поправочный коэффициент к 0,1 н. раствору
тиосульфата натрия; g— навеска исследуемого вещества, г.
Опыт 2. Количественное определение эфиров акриловой
кислоты в техническом этилакрилате методом бромироваиия. Точную
навеску технического этилакрилата (0,1—0,2 г) кипятят с 25 мл 0,5 н.
спиртового раствора гидроксида калия в колбе с обратным
холодильником в течение 1—1,5 ч. После охлаждения колбы избыток щелочи
нейтрализуют 0,5 н. раствором соляной кислоты по фенолфталеину до
обесцвечивания.
К нейтрализованному раствору прибавляют 50 мл 0,1 и. раствора
бромид-броматиой смеси и 5 мл 0,1 и. раствора соляной кислоты.
Содержимое колбы оставляют на 1 ч в темном месте. Затем приливают 15 мл
10%-ного раствора KI и через 10 мин оттитровывают выделившийся иод
0,1 н. раствором тиосульфата натрия. В конце титрования добавляют
раствор крахмала. В аналогичных условиях титруют контрольную пробу.
Протекающие реакции описываются следующими уравнениями:
СН2=СН—СООС2Н5 + 2NaOH -> СН2=СН—COONa + С2Н5ОН
СН^СН—COONa + НС1 -> СН2=СН—СООН+NaCl
5КВг + КВгО3 + 6НС1 -> 6КС1 + ЗН2О + ЗВг2
СН2=СН—СООН + Вг2 ->СН2Вг—СНВг—СООН
2К1+Вг2->2КВг + 12
I2 + 2Na2S2O3 -> 2NaI + Na2S<06
Содержание этилакрилата х (в %, мае.) вычисляют по формуле
■*
g
где Vx — объем 0,1 н. раствора тиосульфата натрия, пошедший на
титрование контрольного опыта, мл; V2 — объем 0,1 н. раствора тиосульфата
натрия, пошедший на титрование навески исследуемого продукта, мл; К —
поправочный коэффициент к 0,1 н. раствору тиосульфата натрия; 0,0050 —
масса этилакрилата, соответствующая 1 мл точно 0,1 н. раствора
тиосульфата натрия, г; g — иавеска анализируемого этилакрилата, г.
107
Работа 3. Определение влаги
в органических веществах
Опыт 1. Визуальное определение влаги в уксусной кислоте
реактивом Фишера. Визуальное определение влаги реактивом Фишера
основано на зрительном восприятии изменения окраски раствора в точке
эквивалентности.
В коническую колбу вместимостью 100 мл, снабженную
автоматической бюреткой и клапаном Бунзена, помещают 20 мл абсолютного
метилового спирта. Осторожно встряхивая колбу, оттитровывают реактивом
Фишера влагу, адсорбированную на стенках колбы. Титрование ведут до перехода
красно-коричневой окраски в желтую. Затем быстро вносят навеску
уксусной кислоты, взвешенной с точностью до 0,0002 г, из капельницы и титруют
реактивом Фишера до красно-коричневой окраски раствора.
Содержание влаги х (в %, мае.) вычисляют по формуле
x = VK\00/g,
где V — объем реактива Фишера, пошедший на титрование пробы, мл; К —
водный эквивалент реактива, г/см; g — навеска пробы, г.
Дня установления водного эквивалента реактива Фишера в колбу для
титрования вносят навеску воды, взятую с точностью до 0,0002 г, и отгитро-
вывают влагу реактивом Фишера. Титр раствора вычисляют по формуле
где V — объем реактива Фишера, пошедший на титрование, мл; g —
навеска воды, г.
Опыт 2. Определение содержания воды в нефтн и
нефтепродуктах по методу Дина н Старка. Количественное определение воды по
методу Дина и Старка основано на отгонке воды из смеси
анализируемого продукта с безводным растворителем. В качестве растворителя
используют бензол, толуол, бензин. Растворитель предварительно
обезвоживают. Определение содержания воды проводят на установке,
изображенной на рис. 28. В колбу помещают 100 г нефтепродукта, взвешенного
с точностью до 0,1 г, и прибавляют 100 мл обезвоженного растворителя.
Колбу нагревают на песчаной бане. Пары, конденсирующиеся в
холодильнике, стекают в пробирку, где происходит расслаивание продуктов
отгона. Вода собирается в нижней части пробирки. Перегонку
продолжают до тех пор, пока уровень воды в пробирке не перестанет
изменяться. Содержание воды W(b %, мае.) вычисляют по формуле
где V— объем воды в приемнике-ловушке, мл; g— навеска продукта, г.
108
Работа 4. Определение окисляемостн сточных вод
Термин «окисляемость» характеризует общее содержание в воде
восстановителей (органических и неорганических), реагирующих с
сильными окислителями. Общее содержание в воде восстанавливающих
веществ определяют иодатным методом. В основе его лежит окисление
органических веществ иодатом калия в кислой среде. Органические
соединения в этих условиях окисляются до СО2, SO2) N2. Кислород,
входящий в состав соединений, расходуется на их окисление, а недостающее
для окисления количество кислорода выделяется из иодата калия:
Перманганатный метод дает менее точные результаты, так как пер-
манганат калия окисляет не все органические вещества и не всегда
полностью. Однако для сравнительной оценки загрязненности сточных вод
этим методом пользоваться можно. В кислой среде восстановление
КМпО4 происходит по реакции
МпО; + 8Н+ + 5е~ -» Мп2+ + 4Н2О
После кипячения сточной воды избыток КМпО4 восстанавливается
щавелевой кислотой по уравнению реакции
МпО; +5С2О42" + 16Н+ -> 2Мп2+ +8Н2О + 10СО2
Избыток щавелевой кислоты оттитровывают раствором пер-
манганата калия.
Опыт 1. Иодатный метод определения окисляемостн сточных
вод. В круглодонную колбу (можно использовать колбу Кьельдаля),
снабженную обратным холодильником, вносят 5 мл 10%-ного раствора
иодата калия и 25 мл концентрированной серной кислоты. Смесь
осторожно перемешивают н к ней добавляют пипеткой 10 мл анализируемой
воды. Колбу нагревают на песчаной бане (температура бани 200—250 °С)
в течение 2 ч до полного прекращения выделения паров иода. Затем в
колбу приливают 50 мл дистиллированной воды (при этом раствор
желтеет) н кипятят без обратного холодильника до исчезновения окраски и
запаха иода. Колбу охлаждают, содержимое количественно переносят в
мерную колбу на 200 мл и доводят до метки (при низкой окисляемости —
менее 100 мг кислорода на 1 л воды, разбавление излишне).
В коническую колбу с притертой пробкой вносят 1 г
кристаллического иодида калия, прибавляют 50 мл дистиллированной воды и
пипеткой добавляют около 20 мл раствора из мерной колбы. При низкой
окисляемости вносят в коническую колбу все содержимое круглодонной
колбы. Коническую колбу, закрыв пробкой, выдерживают в темном месте
109
10—15 мин. Выделившийся иод оттитровывают 0,1 н. раствором
тиосульфата натрия. В конце титрования прибавляют крахмал. В
аналогичных условиях проводят контрольный опыт с дистиллированной водой.
Окисляемость X равна массе иодата калия (мг), необходимой для
полного окисления всех восстанавливающих веществ в 1 л воды, и
определяется по формуле
v {Ух-У2)КУъЛ000Ь
ч л. — ,
W4
где У\ — объем 0,1 н. раствора тиосульфата натрия, пошедший на
титрование контрольного опыта, мл; У2 — объем 0,1 н. раствора тиосульфата
натрия, пошедший на титрование анализируемой воды, мл; К —
поправочный коэффициент к 0,1 н. раствору тиосульфата натрия; У3 —
вместимость мерной колбы, мл; К — объем воды, взятый для анализа, мл; F4 —
объем раствора, пошедший на титрование, мл; Ъ — масса иодата калия,
соответствующая 1 мл 0,1 н. раствора тиосульфата натрия, мг.
Окисляемость можно выражать и как массу кислорода (мг),
необходимую для полного окисления всех восстанавливающих веществ в 1 л
воды. В этом случае приведенная выше формула имеет вид
y_(*/1-
л. —
где 0,6667 — масса кислорода, мг, соответствующая 1 мл 0,1 н. раствора
тиосульфата натрия.
Опыт 2. Определение окисляемости сточной воды псрмаига-
иатиым методом. В коническую колбу вместимостью 250 мл наливают
75 мл дистиллированной* воды и 5 мл окисленного перманганатом калия
раствора серной кислоты*. Затем добавляют 10 мл 0,01 н. раствора пер-
манганата калия и 10 мл аналюируемой сточной воды (сильно
загрязненную воду предварительно разбавляют в мерной колбе дистиллированной
водой). В коническую колбу вставляют воронку и смесь кипятят 10—15
мин. В горячую жидкость вливают 10 мл 0,01 н. раствора щавелевой
кислоты и титруют 0,01 н. раствором перманганата калия. Аналогичным
образом проводят контрольный опыт с дистиллированной водой.
Окисляемость X (в миллиграммах КМпО4, затраченного на
окисление восстанавливающих веществ в 1 л воды) вычисляют по формуле
Получают путем прибавления нескольких капель перманганата калия к
серной кислоте до появления слабо-розового окрашивания.
110
где V— общий объем 0,01 н. раствора перманганата калия,
прибавленный в начале определения и израсходованный на титрование избытка
щавелевой кислоты, мл; Vx — объем 0,01 н. раствора перманганата калия,
пошедший на титрование 10 мл щавелевой кислоты, мл; V2 — объем 0,01 н.
раствора перманганата калия, пошедший на окисление всего количества
дистиллированной воды, мл; К3 — объем анализируемой воды, взятый
для определения с учетом предварительного разбавления, мл; 0,32 —
масса перманганата калия, мг, соответствующая 1 мл 0,01 н. раствора.
Работа 5. Определение молекулярной массы тиокола
(полисульфидного каучука)
Метод основан на взаимодействии концевых тиольных групп со
слабым раствором иода, избыток которого определяют титрованием
тиосульфатом натрия.
Точную навеску тиокола (0,2—0,3 г) помещают в коническую колбу,
растворяют в 75 мл бензола и добавляют 30—35 мл 0,01 н. раствора иода.
Затем содержимое колбы сильно встряхивают и оттитровывают избыток
иода 0,01 н. раствором тиосульфата натрия в присутствии крахмала.
Одновременно проводят контрольное титрование того же количества иода
при добавлении 75 мл бензола.
Молекулярную массу тиокола (М) рассчитывают по формуле
{VX-V2)K- 0,00127*
где g — навеска тиокола, г; 254 — молекулярная масса иода; V\ — объем
0,01 н. раствора тиосульфата натрия, пошедший на контрольное
титрование, мл; V2 — объем 0,01 н. раствора тиосульфата натрия, пошедший на
титрование навески тиокола, мл; К — поправочный коэффициент к 0,01 н.
раствору тиосульфата натрия; 0,00127 — масса иода, соответствующая 1 мл
точно 0,01 н. раствора тиосульфата натрия, г.
Работа 6. Количественное определение гидроксильных
групп в олиго- и полиэпоксидах
Для установления количества гидроксильных групп в олиго- и
полиэпоксидах проводят реакцию ацетилирования. Количество выделившейся
при этом воды определяют реактивом Фишера.
Навеску 0,5—1 г олиго- или полиэпоксида (влажность которого
предварительно найдена с помощью реактива Фишера) помещают в ко-
111
ническую колбу вместимостью 150 мл, приливают 5 мл ацетилирующего
раствора, закрывают пробкой и выдерживают в термостате 1,5—2 ч при
80—90 °С. Параллельно ставят контрольный опыт. После охлаждения
колбы вводят 5 мл пиридина, перемешивают раствор и титруют
реактивом Фишера.
Ацетилирующий раствор готовят следующим образом: смесь 200 мл
ледяной уксусной кислоты, 35 мл бензола и 5 мл .концентрированной
серной кислоты нагревают в колбе, снабженной термометром,
дефлегматором и ловушкой Дина — Старка. Смесь кипятят 4—4,5 ч, затем
отгоняют бензол до температуры 115 °С.
Содержание гвдроксильных групп х (в %, мае.) находят по формуле
х y д
£•1000 g
где V\ и V2 — объем реактива Фишера, пошедший на титрование
соответственно испытуемого раствора и контрольной пробы, мл; К — водный
эквивалент реактива Фишера, г/мл; g — навеска олиго- или полиэпоксида, г;
0,944 — отношение молекулярных масс гидроксильной группы и воды; W—
влажность полимера в пересчете на гидроксильные группы (в %),
рассчитанная по формуле W = 0,944^ (где .у — массовая доля воды, %).
Работа 7. Определение содержания эпоксигрупп
в эпоксидных полимерах
Количественное определение эпоксигрупп в полимерах основано на
реакции эпоксигрупп с соляной кислотой в среде диоксана или ацетона:
R—CH—СН2+НС1 *~ R— СН— CHj
0 ОН С1
В колбу с притертой пробкой помещают точно взвешенную навеску
полимера (0,2—0,3 г), приливают 30 мл ацетона, закрывают пробкой и
выдерживают (периодически встряхивая колбу) при комнатной
температуре до полного растворения полимера. Затем добавляют 10 мл раствора
соляной кислоты в ацетоне (раствор готовят смешением 5 мл
концентрированной соляной кислоты в 200 мл ацетона) и, закрыв пробкой,
выдерживают 30 мин. Избыток соляной кислоты отгитровывают 0,1 н.
раствором гидроксида натрия в присутствии фенолфталеина. Параллельно
проводят контрольный опыт без навески полимера по той же методике.
Массовую долю эпоксигрупп х (в %) определяют по формуле
112
_ (Vx - У?)К • 0,0043 ■ 100 _ (Vx - V2 )K • 0,43
A'— — ,
g g
где Vx — объем 0,1 н. раствора NaOH, пошедший на титрование в
контрольном опыте, a V2 — на титрование полимера, мл; К — поправочный
коэффициент к 0,1 н. раствору NaOH; 0,0043 — содержание эпоксигрупп,
соответствующее 1 мл точно 0,1 н. раствора NaOH, г; g — навеска
вещества (полимера), г.
Раздел V
Физико-химические (инструментальные)
методы исследования
органических соединений
№ физико-химических (инструментальных) методов исследования,
применяемых для установления молекулярной структуры органических
веществ, наиболее часто используются оптическая спектроскопия (в
ультрафиолетовой, видимой и инфракрасных областях спектра),
спектроскопия ядерного магнитного резонанса (ЯМР), хроматография, метод
дипольных моментов молекул, рентгеноструктурный анализ,
молекулярная масс-спектроскопия и др. С помощью этих методов получают ценную
информацию о взаимном расположении атомов в молекуле, их
взаимовлиянии, внутримолекулярных расстояниях, поляризуемости связей,
валентных углах и распределении электронной плотности и т. д.
1. Оптическая спектроскопия
Оптическая спектроскопия с успехом используется при решении
вопросов количественного и качественного анализа, структурно-группового
анализа, изучения внутри- и межмолекулярных взаимодействий,
конфигурации молекул, а также исследования различных видов изомерии. Она
применяется, в частности, при изучении кинетики химических реакций,
определении коистант диссоциации кислот и оснований и т. д.
Общие сведения. При прохождении света (ультрафиолетового, видимого или
инфракрасного, т. е. электромагнитных волн с определенной энергией) через
вещество происходит частичное или полное поглощение излучения определенных длин
волн (определенных частот). Осиовной задачей оптической спектроскопии является
исследование зависимости интенсивности поглощения света веществом от длины
волны или частоты колебания (или, что то же самое, энергии).
Энергия излучения и длина волны связаны соотношениями:
Е = Av; v = cfk; E = hcfk9
где Е — энергия; А — постоянная Планка; с — скорость света; v — частота
колебания, с"1; X — длина волны, см. Часто вместо длины волны применяют ее
обратное значение — волновое число v = 1/А,, которое выражают в обратных
сантиметрах (см"1), тогда Е = Acv.
114
Полную энергию молекулы можно рассматривать как сумму трех
составляющих — трех энергетических состояний:
Поэтому поглощенная молекулой лучистая эиергия может вызвать или переход
электрона с одного энергетического уровня на другой, эиергия которого выше,
или привести к колебанию и вращению атомов в молекуле. Другими словами,
поглощенная молекулой эиергия (в виде излучения), вызывая изменение этих
энергетических состояний, приводит к возникновению электронных,
колебательных или вращательных спектров. Таким образом, спектр — это количественное
распределение электромагнитного излучения по длинам волн или частотам
колебания (т. е. по энергиям).
Эиергия движения электронов значительно больше энергии колебания, а тем
более энергии вращения атомов в молекуле (£м > Ет > Е^), поэтому для
изменения электронной энергии, т. е. возбуждения внешних электронов (поглощение
в видимой и УФ-областях), требуется гораздо больше энергии, чем для изменения
колебательной энергии (поглощение в ИК-области). Поэтому при воздействии
ультрафиолетового излучения меняются все три вида энергии молекулы. Однако
электронные переходы происходят настолько быстро (10~14—10 15 с) по
сравнению с колебательными (период колебания ядер составляет 1<Г12—10~13 с), что за
это время ядра остаются фиксированными в пространстве (принцип Франка—
Кондона).
Весь спектр электромагнитного излучения охватывает широкий диапазон
частот — от длинных радиоволн до жесткого у-излучения. Однако
спектроскопия, изучающая спектры поглощения (молекулярная, или абсорбционная,
спектроскопия), использует лишь сравнительно небольшую его часть. Различают
ультрафиолетовую (УФ), видимую и инфракрасную (ИК) области спектра.
Оеиовиые законы ноглощення еветя. Для оптических спектров
(электронных, колебательных и вращательных) соблюдаются общие законы поглощения
электромагнитного излучения. Они определяют связь между поглощением и
количеством поглощающего вещества.
Закон Бугера — Ламберта (1729) выражает зависимость между
поглощающей способностью и толщиной слоя вещества: каждый тонкий слой
внутри гомогенной среды поглощает одинаковую долю входящего в него
светового потока монохроматического света. Этот закон в математической форме
может быть записан в виде
/ = /0 е"А|/ (при с = const и X = const),
где ki — коэффициент, зависящий от длины волны света и являющийся
индивидуальной характеристикой вещества; /0 — интенсивность входящего потока
излучения; / — интенсивность ослабленного поглощением потока излучения; / —
толщина поглощающего слоя.
Чаще пользуются логарифмической формой записи: 1п(/0//)=&,/. Следует
отметить, что в этом законе ие учитывается концентрация вещества с. Этому
закону подчиняются все вещества.
Закон Бера (1852) учитывает связь между поглощающей способностью
вещества и его концентрацией в растворе: поглощение данным тонким слоем
115
внутри гомогенной среды пропорционально числу поглощоющих частиц в единице
объем о, т. е. концентрации с.
Математически этот закон выражается формулой
/ = /0 е~*2С (при / = const и X - const),
где с — концентрация поглощающего вещества (раствора); к2 — постоянная
величина. После логарифмирования этого уравнения получим
ln(/0//)=V-
Эти два закона можно объединить одним выражением:
которое после логарифмирования принимает вид
где к — коэффициент поглощения, зависящий только от природы поглощающего
вещества, длины волны и температуры.
Заменив в последнем уравнении натуральный логарифм на десятичный,
получим для объединенного закона Бугера — Ламберта — Бера выражение
где е — молярный коэффициент п^лпиения (поглощения), или молярный
коэффициент экстинкции.
Величину lg(/0//) называют оптической плотностью D. Отсюда
D = ecl, e = D/(c/).
Если концентрацию раствора измерять в молях на литр (моль/л), а толщину
поглощающего слоя в сантиметрах, то молярный коэффициент экстинкции £
выражается в литрах на моль-сантиметр [л/(моль-см)]. Если с = 1 моль/л, а / = 1 см,
то е = D.
Закон Бера соблюдается не всегда. Отклонения от него свидетельствуют о
возможных межмолекулярных взаимодействиях (ассоциация, сольватация,
диссоциация и таутомерия молекул, комплексообразование и т. д.), протекающих в
данной среде при изменении концентрации поглощающего вещества. Поэтому
при измерении спектров поглощения в растворах различной концентрации
предварительно проверяют выполнение закона Бера. Для этого исследуют
зависимость оптической плотности D от концентрации с (при постоянных X и /)• При
соблюдении закона эта зависимость выражается прямой линией в координатах Due.
Следует отметить, что в ИК-области спектра отклонение от закона Бера
встречается чаще, так как в концентрированных растворах, которые
используются при записи этих спектров, межмолекулярные взаимодействия более выражены.
Способ изображении спектров поглощения. Если зафиксировать с
помощью специального прибора изменение интенсивности поглощения пропущенного
через вещество светового потока в зависимости от длины волны, то можно
получить спектральную кривую поглощения (спектр поглощения). Спектр поглощения
в УФ- или видимой областях выражается в виде графика, где на оси ординат
116
tgc
S
800
ZOO 220 2Ь0 260 290Х,нм ** WOO 5000 2OQO№O1W 1100 1000
а б
Рис. 29. Ввд спектров поглощения фенола в УФ- (о) и ИК-области (б)
обычно откладывают значение молярного коэффициента поглощения (экстинк-
ция) е или оптическую плотность раствора Д а на оси абсцисс — длины волн, нм
(1 нм = 1(Г7 см). Обычно вместо е используют lg е*. При построении
спектральных кривых поглощения в ИК-области по оси ординат откладывают поглощение
(или пропускание (в %), обозначаемое Г, а по оси абсцисс — частоты обратных
сантиметрах (см"1).
Внд спектров поглощения в УФ- и ИК-областях приведен на рис. 29.
Обычно спектр поглощения характеризуется длинами волн или частотами колебаний
максимумов поглощения и интенсивностью в этих максимумах.
Кроме определения Т и D спектрофотометр позволяет проводить
определения концентрации (с) и скорости изменения оптической плотности
что
очень удобно при изучении кинетики реакции. Порядок работы в этом случае
подробно изложен в «Техническом описании и инструкции по эксплуатации
спектрофотометра», которое прилагается к каждому прибору.
Задание: Если имеется возможность, запишите УФ-спектр предложенного
преподавателем органического вещества. Определите максимум поглощения и
коэффициент экстинкции. Сделайте вывод о характере возможных электронных
переходов в данном соединении.
1.1. Ультрафиолетовая (УФ) спектроскопия
и спектроскопия в видимой области
Возникновение электронных спектров. Электронные переходы.
Поглощение света в видимой и ультрафиолетовой областях спектра
связано с возбуждением молекулы вещества квантом света н последующими
электронньши переходами со связывающей (а, тс) или несвязывающеи
(п) орбитали на разрыхляющую орбиталь (а или тс*). Последняя имеет
более высокую энергию и в основном (невозбужденном) состоянии
свободна (вакантна). Энергия электронных переходов составляет 1,77—6,7 эВ,
что соответствует X = 700+200 нм, или 50 000—16 000 см'1.
* От способа выражения интенсивности зависит внешний вид спектра (рис. 30).
117
2500С
2ooot
мое
woe
soot
230 2kO 750 260 770Хш*м
Рис 30. Изменение внешнего
вида спектра от способа выражения
интенсивности. УФ-спеюр
пиридина в шкале lg е (1); г (2); D (3)
Спектры поглощения в видимой и
ультрафиолетовой областях, связанные
с электронными переходами, получили
название электронных спектров.
Рассмотрим возможные
электронные переходы, которые
классифицируют в соответствии с характером
валентных электронов.
1. N-^V-Переходы. В этом случае
имеют место переходы электронов со
связывающих орбиталей на
соответствующие разрыхляющие <т->а* и
л->тс*. Наибольшая энергия
необходима для перехода g-+g* , так как а-
связь — более прочная. Поэтому
соответствующие ей полосы поглощения находятся в УФ-области при X > 200
нм. Они характерны для насыщенных органических соединений.
Переходы тс —> тс* связаны с меньшей затратой энергии и поэтому
находятся в более длинноволновой части спектра (в видимой или
ближней УФ-области). Эти переходы наблюдаются в ненасыщенных
соединениях (наличие связей О=С или О=С). В спектроскопии полосы,
соответствующие этим переходам, называют К-полосами.
2. #-*(>-Переходы, Такие переходы совершаются с несвязывающих
орбиталей на соответствующие разрыхляющие: w->a* и w->tc\
Переходы п -> а* наблюдаются в далекой или ближней УФ-области, т. е.
требуют достаточной энергии и встречаются в насыщенных соединениях,
содержащих атомы со свободной парой электронов («-электроны).
Переходу л —> тс* соответствует наименьшая энергия, и соответствующие ей
полосы поглощения расположены в ближней ультрафиолетовой или
видимой областях спектра. Такой переход обнаруживается в соединениях, в
молекулах которых атом с неподеленной электронной парой связан с
другим атомом кратной связью (С=О, C—N, O=S и др.); полосы,
соответствующие п -> тс* переходам, называют R-полосами.
Интенсивность А>полос намного больше интенсивности Д-полос.
Типы переходов и соответствующие энергетические состояния w-, тс-
и a-электронов схематически изображены на рис. 31. Как видно из
рисунка, энергии этих орбиталей возрастают в ряду a < тс < w и тс <а.
Область электронных переходов (электронных спектров поглощения)
охватывает интервал спектра электромагнитных волн от 100 до 1000 нм
(~106—104 см"1). Эта область подразделяется на две: видимую — с интер-
118
, б • -разрыхляющая
орбитапь
ж ♦ -разрыхляющая
орбитапь
_ п-несОязы0ающая
п—я9 л+6* орбатапь
(Шлоса)
(К-полоса)
• ж- сдязыбающая
орбитапь
,6- обязывающая
орбитапь
Рис. 31. Типы электронных переходов и
соответствующие им энергетические
состояния
валом длин волн от 400 до 1000 нм
И ультрафиолетовую — с
диапазоном от 100 до 400 нм.
Последняя также делится на две части:
ближнюю — от 200 до 400 нм и
дальнюю (вакуумную) — от 100
до 200 нм.
Характер полос поглощения
определяется величиной
поглощенной энергии. Например,
полосы поглощения в УФ-области
являются довольно широкими и
плавными (размытыми). Это
можно объяснить шириной кривых распределения молекул по уровням
энергии в исходном и конечном состояниях.
Регистрация электронных спектров поглощения. Поглощение в
областях различных длин волн определяют обычно спектрофотометри-
ческим методом на приборе — спектрофотометре, принципиальная
Схема которого изображена на рис. 32.
Излучение от источника света /, проходя через линзу 2, фокусируется
на входной щели 3. Расходящийся световой пучок с помощью коллимирую-
щего зеркала 4 превращается в параллельный и попадает на
диспергирующий элемент (призма) 5. Здесь происходит разложение падающего света на
отдельные монохроматические участки (участки спектра с определенной
длиной волны). Объектив 6 превращает системы параллельных световых
пучков с разными дайнами волн в системы монохроматических
изображений. Вращением призмы 5 проецируют различные монохроматические лучи
на щель 7. При этом получают полную развертку спектра, так как щель
пропускает поочередно участок спектра с нужной длиной волны.
Монохроматическое излучение с интенсивностью /0, проходя через кювету 8 с веществом,
ослабляется до интенсивности /. Величину lg(/0//), т. е. оптическую
плотность раствора Д регистрируют системой Р, которая преобразует световую
энергию в электрические сигналы (фотоэлемент, фотоумножитель).
/ 2 J k
7 8 9
Рис. 32. Принципиальная оптическая схема спектрофотометра
119
При измерении спектров поглощения в ультрафиолетовой области в
качестве источника света используется водородная (дейтериевая) лампа
(200—350 нм), а кюветы для раствора вещества, призма и вся оптика в
приборе должны быть изготовлены из кварца (обычное стекло
непрозрачно для коротковолнового излучения). При работе в видимой области
используют тот же прибор, но в качестве источника излучения
применяют лампу накаливания (от 350 нм и далее), а кюветы могут быть
изготовлены из обычного стекла. В качестве растворителей в УФ-спектроскопии
применяют вещества, не имеющие поглощения в исследуемой области
спектра и не вступающие в химическое взаимодействие с растворенным
веществом (см. табл. 1). Для измерения электронных спектров поглощения
обычно используют сильно разбавленные растворы (10~2—10~5 моль/л).
Таблица 1. Граница пропускания спектрально чистых растворителей,
наиболее часто применяемых в УФ-спектроскопнн,
при толщине слоя 1 см (точность ±5 нм)
Растворитель
Ацетонитрил
Пентан
Гексан
Гептан
Изооктан
Циклогексан
Циклопенган
Вода
Метиловый спирт
Изопропиловый спирт
Этиловый спирт
Диэтиловый эфир
Диоксан
Дихлорметан
А., нм
190
190
195
197
197
198
198
200
202
203
205
210
211
230
Растворитель
Тетрагидрофуран
Хлороформ
Уксусная кислота
Этилацетат
Диметилсульфоксид
Тетрахлорид углерода
Диметилформамид
Бензол
Толуол
Тетрахлорэтилен
Пиридин
Сероуглерод
Нитрометан
А., нм
235
245
251
254
265
266
270
278
285
290
305
380
380
Пропуская поочередно через кювету с раствором вещества
монохроматические лучи света с различной длиной волны и определив
соответствующие значения оптической плотности, вычерчивают
спектральную кривую — зависимость интенсивности (в единицах е, lg б или D) от
длины волны (в нм).
В современной спектроскопии используют в основном двухлучевые
спектрофотометры, в которых излучение от источника света поступает
одновременно через кювету с чистым растворителем (эталон) и кювету с
раствором вещества. Такое устройство прибора избавляет от необходимости
делать два последовательных измерения величин пропускания: вначале
чистого растворителя, а затем—раствора вещества в этом же растворителе.
120
1.2. Электронные спектры поглощения
и строение органических соединений
Электроны, входящие в состав атомов и молекул, различаются по
своему энергетическому состоянию (\s-3 2s-, 2р-электроны и т. д). Для их
возбуждения требуется излучение с различной длиной волны (энергией).
Наибольшая энергия необходима для возбуждения электронов простой
С—С-связи (а-электроны), и поэтому предельные углеводороды
поглощают в области < 200 нм (а->а -переходы). Несколько меньшая
энергия требуется для возбуждения электронов других простых связей,
например атома углерода с атомом, содержащим неподеленную
пару электронов (я-электроны) (табл. 2). Такие полосы поглощения
относятся к переходу п->а*.
Таблица 2. Максимумы полос поглощения, соответствующие а~>а* н
/|->а*-переходам, для некоторых простых органических соединении
Соединение
СН3С1
СН31
СНзОН
а->ст*-переход
173
258
183
п->ст*-переход
160
200
150
Соединение
CH3NH2
СН3ОСН3
C2H5SH
*пих»НМ
а->а*-переход
213
184
225
л->а -переход
173
150
192
Этиленовые углеводороды с изолированными двойными связями
имеют интенсивную полосу поглощения, обусловленную переходом
я-»я , в области 165—200 нм (этилен поглощает при 165 нм).
Группы атомов, которые содержат одну или несколько кратных
связей, вызывающие избирательное поглощение электромагнитного
колебания в УФ-области, называются хромофорами. Хромофоры приводят
к появлению в спектрах К- или /?-полос (я-»я и п->п -электронные
переходы) и поглощают в области от 200 до 1000 нм.
Примеры типичных хромофорных систем и их максимумы
поглощения приведены в табл. 2 и 3.
Таблица .
Хромофор
С=С
С=С=€
СяС
O=N—
—NO2
с=о
3. Максимумы поглощении некоторых хромофоров
Ащах, НМ
165
225
173
240-250
271
280
Хромофор
—N=N—
—N=O
Бензол
Нафталин
340
620
665
203,254
275, 314
121
В табл. 3 приведены максимумы поглощения для хромофорных
групп, не связанных с я-электронными системами, или ауксохромами
(ОН, NH2, Alk и т. д.). Если же имеется связь (сопряжение) друг с другом
двух хромофорных групп и более или хромофора с ауксохромом, то
максимум поглощения смещается в длинноволновую область (батохромный
сдвиг). Наибольшее смещение достигается в случае веществ, в
молекулах которых хромофоры соединены непосредственно друг с другом
(например, сопряженные двойные связи). При этом по мере увеличения
числа сопряженных двойных связей в молекуле полнена поглощение
сдвигается в сторону длинных волн примерно на 30—40 нм на каждую
вводимую в цепь сопряжения двойную связь. Так, этилен поглощает при 165 нм,
бутадиен-1,3 — при 217 нм, а гексатриен-1,3,5 и декатетраен-2,4,6,8 —
при 256 и 310 нм соответственно. Каротин, содержащий в молекуле
одиннадцать сопряженных двойных связей, поглощает в видимой
области [окрашен в желтый цвет (504 нм)]. Одновременно с накоплением
групп СН=СН происходит увеличение интенсивности полос поглощения
(гиперхромный сдвиг).
Ацетилен имеет полосу поглощения при 173 нм (п—>п -переход).
Замена атомов водорода в этилене и ацетилене на алкильные группы
приводит к длинноволновому смещению максимума поглощения. Если в
цепи сопряженных связей двойную связь заменить на тройную, то это
практически не влияет на положение максимума поглощения, но
вызывает уменьшение интенсивности полосы поглощения.
В альдегидах и кетоиах, а также карбоновых кислотах и их
производных (ангидридах, галогенангидридах, амидах и др.) возможны три
типа электронных переходов: я->л: , л->л и я->а . Однако наиболее
характерным является поглощение, отвечающее переходу я—>я . Обычно
эта полоса поглощения находится в наиболее длинноволновой части
спектра, так как переходу п-+% соответствует наименьшая энергия.
Например, для альдегидов и кетонов она лежит в области 270—300 нм, для
кислот, галогенангидридов, сложных эфиров и амидов — в области 200—
230 нм. Характерной особенностью полос поглощения, вызванных л->л; -
переходами, является их низкая интенсивность (е = 10-5-50) и способность
смещаться в коротковолновую область при увеличении полярности
растворителя. Эту полосу легко идентифицировать: при добавлении кислоты
к раствору она исчезает, так как происходит связывание неподеленной
пары электронов гетероатома («-электронов) протоном.
Батохромиып сдвиг — смещение максимума поглощения в
длинноволновую область; гипсохромнып сдвиг — смещение в коротковолновую область.
Гиперхромный и гипохромный сдвиги — увеличение и уменьшение интенсивностей
соответственно.
122
Если в соединении содержатся две карбонильные группы, то их
влияние на характер спектра может проявляться по-разному, в
зависимости от их взаимного расположения. При значительном удалении групп
друг от друга каждая из них обнаруживается в спектре автономно. В случае
ос,Р-дикарбонильных соединений (например, диацетила СН3СОСОСН3)
имеются также две полосы поглощения, однако низкой интенсивности. В
спектре р-дикарбонильных соединений, в которых возможен процесс
енолизации
о о
отражается устанавливающееся равновесие между кетонной и енольной
формами.
Особый интерес представляют спектральные свойства <х,Р-нена-
сыщенных карбонильных соединений. Сопряжение двойной связи с
карбонильной группой вызывает смещение полос поглощения,
обусловленных переходами я->я и п—>я , в более длинноволновую область:
интенсивная (б = 10 000) я->я -полоса проявляется в области 220—260 нм, а
малоинтенсивная (б = 100) п->п -полоса — около 320 нм.
Азотсодержащие соединения — амины, нитросоединения, нитрилы
и азосоединения имеют в основном по две полосы поглощения. Амины
характеризуются двумя полосами, вызванными п->а -переходами, в
области 173—199 и 213—227 нм. При этом смещение в длинноволновую
область происходит при переходе от первичных к третичным аминам. В
спектре нитроалканов имеются также две полосы поглощения. Одна
относится к я->я -переходам (200 нм, б = 50 000), а вторая — л->я -переходам
(270 нм, б = 20^-40). В нитроалкенах полоса я->я*-перехода смещается в
более длинноволновую область (220—250 нм, б = 10 000).
Азосоедииеиия (—N=N—) имеют малоинтенсивные полосы
поглощения в областях 330—350 нм (е = 20*400) и 200 нм (б = 1000),
которые вызваны п-+п -переходами в азогруппе.
Спектры поглощения ароматических соединений существенно
отличаются от спектров соответствующих алифатических соединений.
УФ-Спектр бензола имеет три полосы поглощения, связанные с
переходами я-электронов. Одна из них с максимумом поглощения в
области 180 нм (б = 60 000), соответствует переходу я-»я*. Вторая имеет
максимум в области 203 нм и интенсивность б = 7400. Третья, с самым
длинноволновым максимумом и низкой интенсивностью (X = 256 нм, б = 200),
проявляет тонкую колебательную структуру в области 230—260 нм. Эта
полоса часто называется полосой бензольного поглощения и представляет
собой наиболее характерное поглощение для бензола.
123
Введение в молекулу бензола различных заместителей вызывает
изменение максимума поглощения. Это связано с электронным влиянием
заместителей на и-электронную систему бензольного кольца за счет
эффекта сопряжения и индуктивного эффекта. При этом может
наблюдаться смещение главного максимума бензольной полосы в сторону больших
длин волн с увеличением интенсивности поглощения (табл. 4).
Таблица 4. Влияние заместителей на положение полос поглощения моно-
замещенных производных бензола СбН5—R (в спирте)
р
14.
н
СН3
F
С1
Вг
ОН
SH
NH2
СН=СН2
NO2
ОСН3
СООН
Вторая полоса
поглощения, Х^
203
206
204
210
210
211
236
230
244
259
217
230
Интенсивность,
Б
7400
7000
8000
7400
7900
6200
8000
8600
12000
8000
6400
11600
Третья полоса
поглощения, Х^х
256
261
248
264
261
270
271
280
282
—
269
273
Интенсивность,
Е
200
225
500
190
192
1450
630
1430
750
—
1480
970
Увеличение числа бензольных колец в конденсированных
ароматических углеводородах вызывает смещение всех полос поглощения в
длинноволновую область. Например, если нафталин и антрацен
бесцветны СКгак = 275, 314 и 250, 380 нм соответственно), то нафтацен и пентацен
окрашены в желтый и голубой цвета соответственно (A^ = 480 и 580 нм).
Пятичлеииые гетероциклические соединения (фуран, тиофен и
пиррол) имеют по две полосы поглощения—интенсивную
коротковолновую полосу в области 200—210 нм и малоинтенсивную— в области
длинноволновой части спектра (252—350 нм) (табл. 5).
Таблица 5. Спектры поглощения пяти членных гетероциклических
соединений (в гексане)
Соединение
Фуран
Тнофен
Пиррол
Полоса I
Дим, НМ
200
—
210
Б
10000
—
15000
Полоса П
"tnix* НМ
252
235
350
Б
1
4500
300
124
Введение в пятичленные ненасыщенные гетероциклы ауксохромных
или хромофорных групп смещает полосы поглощения в длинноволновую
область и одновременно усиливает их интенсивность. Например,
фурфурол поглощает в водном растворе при Хт^ = 278 нм (б = 15 000).
Спектры шестичлеииых азотсодержащих гетероциклических
соединений (пиридин, хинолин) мало отличаются от спектров
соответствующих углеводородов. Однако в их спектрах наблюдается повышение
интенсивности длинноволновой полосы поглощения и сглаживание ее
колебательной структуры. Например, пиридин имеет два максимума
поглощения: при А^ах = 195 нм (б = 7500) и A^ = 250 нм (б = 2000). Замена
в ароматическом цикле группы СН на N приводит к появлению
электронного перехода п->п , который, однако, обнаруживается в виде
«плеча» только в парообразном состоянии, так как в растворах эта полоса
перекрывается более интенсивной полосой я->я -перехода.
1.3. Инфракрасная (ИК) спектроскопия
Возникновение ИК-спектров. Эти спектры связаны с переходами
между колебательными уровнями атомов в молекулах. Энергия таких
переходов составляет 0,008—1,987-10"19 Дж (ИК-область в общем
электромагнитном спектре занимает диапазон 10 000—400 см"1). Поэтому
ИК-излучение при взаимодействии с молекулой вызывает изменение не
электронных, а вращательных и колебательных состояний.
Молекулой вещества поглощаются только те частоты ИК-излу-
чения, энергия которых точно соответствует разностям между двумя
уровнями энергии связи. Это приводит к изменению (растяжению или
изгибу) соответствующих связей, т. е. колебанию или вращению ядер
атомов. Частоты этих колебаний зависят не только от характера
отдельных связей, но и от их окружения. Поэтому ИК-спектры по сравнению с
электронными более сложные.
Положение полосы поглощения ИК-спектров определяется
прочностью химической связи и массой связываемых атомов. Чем прочнее связь
и чем меньше масса атомов, тем выше частота поглощения данной связи,
т. е. тем большую энергию необходимо затратить на колебание связи.
Например, прочность связи возрастает при переходе от одинарной к
Практически имеют дело с колебательно-вращательными спектрами. Прн
поглощении молекулой излучения с v < 300 см"' возникает вращательный спектр,
а прн воздействии излучения с v = 300—4000 см"' происходят изменения как во
вращательных, так н в колебательных состояниях молекулы — возникает
инфракрасный вращательно-колебательный спектр.
125
н н ни н н й н й н н
V М w ч/ \/
с с с с с
/\ /\ /'. /\ /\ /\
•симметричное симметричное ножничное «серное крутильное ммтникоюе
v v» vt /Ч /
валентные колебания (v) деформационные колебания (Б)
Рнс. 33. Валентные (v) н деформационные (5) колебания в метиленовой группе
(знаки + н - означают колебания в направлении, перпендикулярном плоскости
бумаги)
двойной и тройной связям. Соответственно возрастают и их частоты
валентных колебаний— 1500—700,1800—1600 и 2500—2000 см"1.
Все колебания в молекуле можно разделить на два тнпа —
валентные и деформационные. Колебания, которые происходят вдоль оси связи
двух атомов без изменения угла между ними, называются валентными (v, у).
Колебания, связанные с изменением валентных углов (при этом длины
связей практически не меняются), называются деформационными (5).
Валентные колебания бывают симметричными (v,) и асимметричными
(уш), а деформационные — ножничными, веерными, крутильными и
маятниковыми (рис. 33). Однако разделение на валентные и
деформационные колебания условно; оно возможно только для линейных молекул
(например, ацетилена Н—О=С—Н).
Энергия валентных колебаний больше, чем энергия деформационных.
Исследование ИК-спектров большого числа органических
соединений показало, что одни и те же функциональные группы, входящие в их
состав, имеют практически одни и те же частоты колебаний. Такие
группы отличаются определенной автономностью и ведут себя независимо от
остальной части молекулы. Соответствующие им частоты колебания
называют характеристическими (см. Приложение I) и используют для
идентификации функциональных групп. К таким колебаниям относятся,
например, валентные колебания связей С=О (1740—1720 см"1), С=С
(1680—1620 см"1), С—Н (3100—2850 см"1), О—Н (3600—3200 см'1) и др.
Сравнение полос поглощения (частот колебаний) исследуемого вещества
с полосами поглощения соединений, строение которых установлено
ранее, позволяет определить структуру нового вещества. Особый интерес
представляет область 1500—700 см"1, в которой содержится большое
число полос, отвечающих, в основном, деформационным и некоторым
валентным колебаниям. Характер спектра в этом интервале частот
существенно изменяется даже при небольших изменениях в структуре
соединений. Эта область называется областью «отпечатков пальцев».
Кроме частот колебания очень важным показателем ИК-спектров
является также интенсивность полос поглощения. Этот параметр в коле-
126
/л
Рнс. 34. Принципиальная оптическая схема инфракрасного спектрофотометра:
1 — источник излучения («глобар»), 2, 3, б, 8t 9, 11 — зеркала; 4 — кювета с веществом; 5 —
входная щель; 7 — призма; 10 — выходная щель; 12 — термоэлемент ( болометр)
бательных спектрах зависит главным образом от дипольного момента
связи. Чем сильнее изменяется дипольный момент связи в процессе
колебаний, тем больше интенсивность ИК-поглощения.
Регистрация ИК-спектров. Для записи ИК-спектров поглощения
используют спектрофотометры, работающие в области 5000—400 см"1.
Принципиальная схема этих приборов изображена на рис. 34.
При измерении ИК-спектров в качестве источника света используют
штифт Глобара («глобар»), изготовленный из карбида кремния (силнта) и
нагретый до 1200—1400 °С. В качестве приемника ИК-излучения
применяется термоэлемент (болометр). Принцип его работы основан на
изменении электрического сопротивления тонкой пленки висмута при
тепловом воздействии ИК-излучения. Возникающий термоток усиливается и
регистрируется записывающим устройством. Измеряя изменение
интенсивности проходящего через вещество потока ИК-излучения, получают
спектральную кривую зависимости интенсивности (в процентах
поглощения или пропускания) от частоты (в см"1).
Особенностью ИК-спектроскопии является то, что стекло и кварц в
средней ИК-области спектра непрозрачны для ИК-излучения. Поэтому
оптические детали спектрофотометра (призмы, линзы, кюветы для
вещества) изготавливают из солей щелочных и щелочно-земельных металлов
(NaCl, KBr, Csl, LiF, CaF2 и др.). Поскольку эти соли отличаются друг от
друга границей пропускания в ИК-области, призмы, изготовленные из
них, необходимо менять при переходе от одной области спектра к другой.
Диапазон работы, см"1 Призма
13300—3800 Стекло
5000—1800 „ LiF
2000—650 NaCI
700-400 KBr
127
ИК-Спектры поглощения могут быть измерены для веществ,
находящихся в любом агрегатном состоянии. Если вещество необходимо
исследовать в твердом состоянии, то его тщательно измельчают,
перемешивают со спектрально чистым бромидом калия (-1 мг исследуемого
вещества на 250—300 мг КВг) и затем подвергают прессованию в
металлической форме в специальных прессах. В результате получают тонкие (менее
1 мм) таблетки. Вместо КВг можно использовать чистое вазелиновое
масло (нуйол), в капле которого растирают несколько миллиграммов
твердого вещества. Однако в этих случаях приходится учитывать
некоторое искажение спектра вещества за счет его взаимодействия с бромидом
калия (влияние его кристаллической решетки), а также за счет
собственного поглощения вазелинового масла в области валентных и
деформационных колебаний С—Н (например, в области 2919—2861 см"1). При
исследовании растворов веществ необходимо подобрать такой
растворитель, который бы отвечал ряду требований: не взаимодействовал с
материалом кювет (например, вода растворяет кюветы, изготовленные из
солей натрия, калия и лития), не имел собственных интенсивных полос
поглощения в исследуемой области, достаточно хорошо растворял
исследуемое вещество. Растворители, наиболее часто применяемые в ИК-
спектроскопии, приведены в табл. 6.
Следует отметить, что по характеру спектры одного и того же
вещества могут несколько отличаться между собой, если их запись
производилась в разных растворителях.
Таблица 6.
Растворитель
Ацетон
Ацетонитрил
Бензол
Бромоформ
Сероуглерод
Тетрахлорид
углерода
Хлороформ
Циклогексан
1,3-Дибромпропан
Диоксан
Дибромэтан
Растворители, применяемые в ИК-спектроскопнн
Толщина
кюветы, мм
0,1
0,1
0,1
0,2
0,1
0,1
0,1
0,1
0,1
0,1
0,1
Собственное поглощение растворителя, см*1
(в области 4000—650 см"')
3100—2900; 1800—1170; 1100—1080; 910—
830
3700—3500; 2350—2250; 1500—1350; 1060-
1030; 930—910
3100—3000; 1820—1800; 1490—1450; 1050—
1020; 680—650
3100—3000; 1190—1100; 710—650
2200—2140; 1595—1460
820—720
3020-3000; 1240—1200; 805—650
3000—2850; 1480—1430; 910—850
1450—1410; 1280—1180; 840—825
3700—2600; 1750—1700; 1480—1030; 910—830
3000—2900; 1460—1020; 950—930; 860—830;
780—750
128
Продолжение табл 6
Растворитель
Дихлорэтан
Метил формиат
Тетрахлорэтилен
Вода
Толщина
кюветы, мм
0,1
0,1
0,1
0,01
Собственное поглощение растворителя, см f
(в области 4000—650 см"')
3000—2900; 1460—1440; 1330—1230; 960—
930; 890—870; 730—650
3100—2800; 1800—1680; 1240-1140
935—875; 820-745
3650—2930; 1750—1580; 930—650
Задание. Запишите ИК-спектр вещества, предложенного преподавателем.
Отметьте частоты поглощения наблюдаемых спектральных полос и их
интенсивность. Пользуясь табличными данными (см. Приложение I), расшифруйте ИК-
спектр. Сравните выводы о строении вещества, сделанные на основании данных
ИК-спектра, с действительной структурой взятого соединения.
2. Спектроскопия ядерного магнитного
резонанса (ЯМР)
Рассмотренные методы УФ- и ИК-спектроскопии относятся к
оптической области спектра. Однако и в радиоволновой области можно
получить ценную информацию о тонкой структуре органических соединений.
Одним из основных методов радиоспектроскопии, который также
нашел широкое применение для исследования органических веществ,
является метод ядерного магнитного резонанса (ЯМР). Особенно
распространен ядерный магнитный резонанс на протонах — протонный
магнитный резонанс (ПМР).
Метод ЯМР заключается в следующем. Ядра некоторых атомов, в
том числе и водорода (протона), обладают собственным моментом
количества движения —ядерным спином, который характеризуется спиновым
квантовым числом I. При вращении заряженного ядра возникает
магнитное поле, направленное по оси вращения. Другими словами, ядро ведет
себя подобно маленькому магниту с магнитным моментом />м. Ядро с
ядерным спиновым числом / может ориентироваться во внешнем
однородном магнитном поле Но различными способами, число которых
определяется магнитным квантовым числом mh Каждой такой ориентации
ядра соответствует определенное значение энергии. Ядра некоторых
элементов, имеющих спиновое квантовое число / = ^ (!Н, 13C, 15N, 19F, 29Si,
31Р) , во внешнем магнитном поле могут находиться в двух энергетиче-
* Ядра с четными числами протонов ('2С, '60,32S) не имеют спина (/ = 0); их
магнитный момент равен нулю. Они не обладают магнитными свойствами и не
дают сигналов ЯМР.
5-290 129
ских состояниях, ориентируясь в направлении этого поля
(т, = + %) или против него (т7 = ->0- Поскольку ориентация в
направлении поля более выгодна, переменить ее на противоположную
можно только при сообщении протону дополнительной энергии (Л£) в
виде кванта электромагнитного излучения hv.
y#0,
2я
где у — константа пропорциональности, характеризующая данный тип
ядра; h — постоянная Планка; Яо — напряженность внешнего
магнитного поля.
Из этого уравнения следует, что у = (у/2я)Я0. В такой
переориентации спина и заключается основная идея, положенная в основу ЯМР.
Если образец вещества облучать радиоволнами с переменной частотой,
то можно подобрать такое значение Av, которое для конкретного протона
будет равно А£, т. е. при этом возможна переориентация спина (спин -12
меняется на спин +V2). При этом будет наблюдаться поглощение
излучения веществом, о чем свидетельствует появление соответствующего пика
поглощения. Меняя частоту в области всего спектра, можно получить
резонансные сигналы всех протонов, содержащихся в молекуле вещества.
Основным параметром ЯМР является величина химического сдвига б.
Химический сдвиг — это расстояние между резонансными сигналами
различных протонов. Химический сдвиг прямо пропорционален
магнитному полю Яо и выражается в относительных единицах, измеряемых в
миллионных долях (м. д.) приложенного поля или резонансной частоты
от сигнала эталона по формулам:
5=Я°6р"Яэт10< или 5=V°6p"V?T10<>
где Я^ - Яэт и v^ - у„ — разности резонансных значений
напряженности поля и частоты для исследуемого и эталонного образцов; Яо —
напряженность внешнего магнитного поля; v — частота генератора, Гц.
Химический сдвиг измеряют относительно протонов некоторых
эталонных веществ. В качестве такого эталона обычно применяется тетра-
метилсилан (ТМС), который дает только одну резонансную линию.
Кроме того, он химически инертен и положение его сигнала мало зависит от
растворителя. В шкале химических сдвигов протонов положение сигнала
ТМС принимается за нуль.
Число сигналов в спектре ЯМР показывает, сколько типов протонов
содержится в молекуле вещества, а химический сдвиг (положение
сигналов) определяет вид протонов (алифатические, ароматические и т. д.).
130
Интегральная интенсивность сигналов (площадь резонансных линий)
прямо пропорциональна числу протонов в атомной группировке,
входящей в состав молекулы. Химические сдвиги протонов большинства
органических соединений находятся в области 5 = Ог-10 м.д. (см. приложение VI).
Спектрометр ЯМР состоит из следующих основных частей: магнита,
радиочастотного генератора и радиочастотного детектора. Принцип
работы прибора состоит в том, что на образец вещества, помещенного
между полюсами магнита, действует радиочастотное поле генератора. При
определенной его частоте и напряженности магнитного поля происходит
резонансное поглощение радиочастотной энергии. При этом детектор
обнаруживает радиочастотные сигналы.
3. Хроматографический метод
Хроматографический метод, открытый в 1903 г. русским ученым-
ботаником М. С. Цветом, позволяет решать следующие задачи:
1) качественный и количественный анализ сложных органических
смесей;
2) очистку веществ от примесей;
3) установление индивидуальности вещества;
4) идентификацию веществ;
5) концентрирование вещества и выделение его из разбавленных
растворов или смесей.
Существует несколько вариантов хроматографического метода.
Однако все они основаны на различной подвижности растворенных веществ
при прохождении их через двухфазную систему, одна из которых
является подвижной, а вторая — неподвижной. Поэтому основной закон
хроматографии можно сформулировать следующим образом:
любая жидкая или газообразная смесь веществ разделяется в
процессе движения ее через слой сорбента, если существуют различия в
сорбционном взаимодействии между компонентами смеси и сорбентом.
В зависимости от типа физико-химического взаимодействия между
сорбентом и находящимся в растворе веществом различают три вида
хроматографии: адсорбционную, распределительную и ионообменную.
3.1. Адсорбционная хроматография
Адсорбционная хроматография основана на количественном
различии в адсорбционных свойствах компонентов разделяемой смеси. В
качестве сорбента используется твердый материал (твердая фаза), на
поверхности которого вдет процесс сорбции.
131
п На поверхности любого твердого тела
2 • ^^_ (адсорбента) имеются активные участки,
обладающие свободным силовым полем.
Попадая в это поле, молекулы другого
вещества могут удерживаться им. При этом каж-
ill'* дый элементарный участок поверхности ад-
' ' сорбента способен удерживать только одну
Рис. 35. Изотерма адсорбции молекулу. Но молекула может преодолеть
силовое поле и оторваться от поверхности
адсорбента. Это происходит в том случае, если энергия молекулы значительна.
Таким образом, при хроматографировании на поверхности адсорбента
одновременно протекают два процесса — сорбция и десорбция. Если число молекул,
закрепленных на поверхности адсорбента, больше числа молекул, покидающих его
поверхность, то адсорбция возрастает. Если же скорости сорбции и десорбции
одинаковы, то наступает состояние адсорбционного равновесия. При этом
каждой концентрации с адсорбируемого вещества отвечает определенное количество
его на адсорбенте (и). Кривая, показывающая зависимость количества
адсорбированного вещества от его концентрации в растворе при постоянной температуре
(после установления адсорбционного равновесия), называется изотермой
адсорбции (сорбции) (рис. 35). Эта изотерма описывается уравнением Лэнгмюра:
где а — количество адсорбционного вещества, соответствующее равновесному
состоянию при заданной концентрации; b — константа, характеризующая
поверхностную активность вещества; с — концентрация вещества в растворе; яю —
максимальное число мест на адсорбенте, которое может быть занято молекулами
адсорбируемого вещества.
Для каждого адсорбента характерна своя изотерма адсорбции. Эта величина
является основной характеристикой адсорбционной способности поглотителей.
Характер кривой изотермы адсорбции показывает, что увеличение концентрации
раствора выше определенного значения не приводит к дальнейшему увеличению
количества адсорбционного вещества, т. е. существует так называемый предел
адсорбции.
Хроматографическое разделение сложных смесей будет
эффективным в том случае, если правильно подобраны адсорбент и растворитель.
Адсорбент, выбранный для хроматографии, не должен химически
взаимодействовать с разделяемыми веществами, оказывать на них и на
растворитель каталитическое воздействие. Кроме того, необходимо,
чтобы адсорбент обладал как можно большим различием в адсор-
бируемости веществ разделяемой смеси, т. е. был избирательным.
Правильно подобранный адсорбент должен также обладать максимальной
поглотительной способностью.
Адсорбенты обычно делят на две основные группы: полярные
(гидрофильные) и неполярные (гидрофобные). Следует помнить, что адсорб-
132
ционное сродство полярных веществ к полярным адсорбентам
значительно выше, чем у неполярных.
В качестве адсорбентов широко применяются оксид алюминия,
активированные угли, силикагель, цеолиты, целлюлоза и некоторые минералы.
Оксид алюминия А12О3 — адсорбент амфотерного характера. На нем можно
разделять довольно широкий круг смесей веществ как в полярных, так и в
неполярных растворителях. Нейтральный оксид алюминия используют обычно для
хроматографирования из неводных растворов предельных углеводородов,
альдегидов н кетонов, спиртов, фенолов и эфиров. Активность оксида алюминия
зависит от содержания в нем влаги. Не содержащий влаги А12О3 имеет самую
высокую активность и условно принимается за единицу. Оксид алюминия с
необходимой активностью можно приготовить, смешивая свежепрокаленный А12О3 с
соответствующим количеством воды (по шкале Брокмана):
Активность А12О3
Количество Н2О, %
I
0
П
3
Ш
6
IV
10
V
15
Активность адсорбента определяется характером разделяемых веществ.
Например, для разделения углеводородов применяют А12О3 с активностью 1,5—2; для
разделения спиртов н кетонов — 2—3,5; лактонов — 3 — 4 и т. д.
Удельная поверхность оксида алюминия достигает 230—380 м2/г.
Активированные угли получают из каменных углей (например, антрацита).
Адсорбционный материал должен обладать хорошо развитой поверхностью н
достаточной прочностью. Адсорбционная способность активированных углей в 50—60 раз
выше, чем обычных (их удельная поверхность достигает 1300— 1700 м2/^).
Силикагель — высушенный желатинообразный диоксид кремния, который
получают из силиката натрия. Силикагели очень широко используются в
хроматографии для разделения смесей нефтепродуктов, высших жирных кислот (ВЖК)
н их сложных эфиров, ароматических аминов, нитро- н нитрозопроизводньгх
органических соединений и др. В отличне от активированных углей силикагель —
гидрофильный сорбент и поэтому мало пригоден для сорбции из водных
растворов (легко смачивается водой). Силикагели используют для осушения воздуха,
обезвоживания неводных растворов — бензина, керосина, масел н т. д.
Активность силикагеля зависит от содержания в нем воды — чем меньше воды, тем
выше его активность (по Брокману):
Активность силикагеля
Количество Н2О, %
I
0
П
10
Ш
12
rv
15
V
20
Силикагели обладают удельной поверхностью, равной 500—600 м2/г.
Цеолиты (молекулярные сита) — пористые кристаллические
алюмосиликаты щелочных и щелочно-земельных металлов (как природные, так и
синтетические). Известны четыре типа цеолитов (А, X, Y, М), имеющие различное
кристаллическое строение. В зависимости от характера катиона цеолиты обозначаются:
КА, NaA, CaM, NaX, KY, CaY.
Особенностью этих соединений является то, что поры их кристаллов имеют
размеры, соизмеримые с размерами молекул многих жидких или газообразных
веществ (0,4—1 им). Если молекулы веществ способны проникать в эти поры, то
133
происходит адсорбция в кристаллах цеолитов (наоборот, вещества, имеющие
более крупные молекулы, ие адсорбируются). Подбирая цеолиты с различными
размерами пор, можно проводить довольно четкое разделение смесей различных
веществ. Удельная поверхность цеолитов 750—800 м2/г.
При выборе адсорбента учитывается химическое строение веществ и
их растворимость. Например, предельные углеводороды адсорбируются
плохо, а непредельные—лучше. Функциональные группы усиливают
способность вещества к адсорбции. Большое влияние оказывает и кон-
формационное состояние молекулы вещества.
При выборе растворителя необходимо учитывать природу
адсорбента и свойства веществ в разделяемой смеси. Растворители должны
хорошо растворять все компоненты хроматографируемой смеси, минимально
адсорбироваться на выбранном адсорбенте, не вступать в химическое
взаимодействие ни с адсорбентом, ни с анализируемыми веществами.
Таблица 7. Растворители н адсорбенты, наиболее часто применяемые
в жидкостной адсорбционной хроматографии
Разделяемые смеси
Углеводороды
Галогенопроизвод-
ные углеводородов
Спирты
^
Фенолы
Альдегиды и кетоны
Карбоновые кислоты
Сложные эфиры
Растворители
Пентан, петролейный эфир,
бензин, изооктан, хлороформ,
тетрахлорид углерода,
хлорбензол, этиловый спирт,
ацетон
Пентан, изоокган,
петролейный эфир, тетрахлорид
углерода
Иэопропиловый спирт, н-бу-
тиловый спирт, диэтиловый
эфир, хлороформ, двоксан,
бензол, петролейный эфир
Петролейный эфир, бензол,
диэтиловый эфир, этиловый
спирт
Петролейный эфир, бензол,
диэтиловый эфир,
тетрахлорид углерода, сероуглерод
Бензол, петролейный эфир,
этиловый спирт, и-гепган,
нитропропан, вода (для
низших кислот)
Петролейный эфир, бензол,
диэтиловый эфир,
тетрахлорид углерода, хлороформ, н-
гексан
Адсорбенты
Активированный оксид
алюминия, силикагели,
алюмосиликатный
катализатор
Силикагели,
активированный оксид алюминия
Активированный уголь,
оксид алюминия,
силикагели
Оксид алюминия, оксид
кальция
Оксид алюминия, оксид
магния, талые,
силикагели
Талые, активированный
уголь, оксид алюминия,
силикагели
Оксид алюминия,
силикагели, активированный
уголь
134
Продолжение табл. 7
Разделяемые смеси
Амины, амиды
Нитро- и нитрозо-
соединения
Сульфокислоты
Сахара
Аминокислоты
Сероорганические
соединения
Гетероциклические
соединения
Растворители
Петролейный эфир, бензол,
тетрахлорид углерода, диэти-
ловыйэфир
Бензол» петролейный эфир,
хлористый метилен
Вода
Вода, изопропиловый спирт,
этиловый спирт, н-бугиловый
спирт, диоксан, петролейный
эфир, бензол, хлороформ
Вода, метиловый спирт,
водный раствор формальдегида,
диэтиловый эфир, раствор
фенола, ju-крезол, хлороформ
Петролейный эфир, изооктан,
спиртобензольная смесь,
этиловый спирт, ацетон
Бутиловый спирт,
диэтиловый эфир, хлороформ,
петролейный эфир, бензол,
этиловый спирт, ацетон, 0,004 а
раствор соляной кислоты,
вода, уксусная кислота
Адсорбенты
Силикагели, оксид
алюминия
Тальк, гидроксид
кальция, карбонат кальция,
силикагели, оксид
алюминия
Оксид алюминия
Боксит,
активированный уголь, силикагели,
оксид алюминия
Активированный уголь,
оксид алюминия, оксид
титана (IV), крахмал
Силикагели, оксид
алюминия
Оксид алюминия,
гидроксид кальция,
крахмал, силикагели,
кизельгур, карбонат
кальция, сахар, тальк
Растворители и адсорбенты, наиболее часто применяемые для
разделения растворов смесей веществ методом жидкостной адсорбционной
хроматографии, приведены в табл. 7.
Для выделения отдельных компонентов смеси, разделившихся при
хроматографировании, часто проводят их последовательное вымывание
(элюирование). С этой целью применяют растворители с различной де-
сорбционной способностью. Растворители можно расположить в ряд в
порядке убывания десорбирующей способности в полярных адсорбентах
(элюотропный ряд Траппе) (табл. 8).
Десорбирующая способность этих растворителей для неполярных
адсорбентов изменяется в обратном порядке.
В зависимости от способа проведения эксперимента адсорбционная
хроматография разделяется на колоночную и тонкослойную.
135
Таблица 8. Элюотропный ряд растворителей
Растворитель
Вода
Метиловый спирт
Этиловый спирт
Ацетон
Пропиловый спирт
1,2-Дихлорэтан
Этилацетат
Амилацетат
Диэлектрическая
проницаемость^
78,5
32,6
24,3
20,7
20,1
10,4
6,0
5,1
Растворитель
Диэтиловый эфир
Хлороформ
Бензол
Толуол
1,4-Диоксан
Тетрахлорид углерода
Циклогексан
Петролейный эфир
Диэлектрическая
проницаемость^
4,4
4,7
2,3
23
2,2
2,2
2,0
1,9
3.2. Распределительная хроматография
Распределительная хроматография основана на количественном
различии в коэффициентах распределения компонентов разделяемой смеси
между неподвижной и подвижной несмешивающимися жидкими фазами.
Неподвижная фаза удерживается сорбционными силами на поверхности
твердого носителя, не вступающего с ней во взаимодействие. В качестве
неподвижной фазы чаще всего используется вода (реже — другие
растворители).
В зависимости от природы твердого носителя и свойств жидкой
неподвижной фазы, а также способа получения хроматограмм различают
три варианта распределительной хроматографии: колоночная, бумажная
и тонкослойная.
Распределительная хроматография на колонках
аналогична адсорбционной колоночной хроматографии. Однако в данном
случае роль сорбента играет неподвижный растворитель. Распределение
веществ между двумя фазами в распределительной колоночной
хроматографии обычно определяют отношением количества вещества в
неподвижном растворителе к количеству вещества в подвижном. Такое
распределение концентраций называется коэффициентом распределения
данного вещества:
где Кр — коэффициент распределения; сн и сп — полярные концентрации
вещества в неподвижной и подвижной фазах.
Коэффициент распределения зависит от природы вещества,
растворителя, температуры и техники проведения эксперимента. Но он не зави-
136
сит от концентрации хроматографируемого вещества. Поэтому
коэффициент распределения для определенного соединения и определенной
системы фаз является величиной постоянной.
В методе бумажной распределительной
хроматографии в качестве носителя используется специальная бумага,
приготовленная из высокосортного хлопка. Она способна удерживать в своих
порах до 22% воды (сорбированной из воздуха), которая играет роль
неподвижного растворителя.
Для количественной оценки способности разделения веществ на
бумаге используется коэффициент R/ — отношение расстояния от линии
старта до центра пятна на бумаге (X) к расстоянию, пройденному
растворителем (X/) (от линии старта до фронта растворителя): Rf = X/Xf. Чем
больше различие в значениях Я/разделяемых веществ, тем лучше
разделение веществ. Коэффициент Rf зависит от многих факторов — природы
носителя, хроматографируемых веществ, растворителей и условий
проведения эксперимента. Однако для однотипных веществ при постоянных
условиях величина остается относительно постоянной и может служить
показателем при идентификации веществ.
Между коэффициентами Луи Кр существует такая зависимость
где Sn/SH — отношение поперечных сечений подвижной и неподвижной
фаз.
В распределительной хроматографии большое значение имеет правильный
подбор носителя и растворителя. В качестве носителя выбирают вещество,
которое хорошо удерживает неподвижный растворитель, а также инертно к
подвижному растворителю и разделяемым веществам. Такими носителями могут быть
силикагель, оксид алюминия, кизельгур, крахмал, целлюлоза и бумага. Характер
растворителя также зависит от нескольких факторов и прежде всего от природы
носителя и разделяемых химических веществ.
Если носитель обладает гидрофильными свойствами (например, силикагель,
целлюлоза), то роль неподвижного растворителя играет вода, а подвижного —
органический растворитель. Прн гидрофобном носителе (например, оксид
алюминия) в качестве неподвижного растворителя служит неполярное вещество
(масла, керосин, бензол и др.), а подвижного — полярные органические вещества
и вода
Если разделяемые вещества растворимы в воде, то в качестве подвижных
растворителей используют органические* растворители или их смесн,
насыщенные водой. В той случае, если вещества растворимы в органических
растворителях и нерастворимы в воде, применяют насыщенные растворы органических
растворителей. При этом необходимо, чтобы растворимость в подвижном
растворителе была больше, чем в неподвижном.
137
Кроме того, растворитель должен полностью разделять вещества близкого
строения (R/ должно быть не менее 0,05). Очень важно, чтобы растворитель не
вступал в химическое взаимодействие с хроматографируемымн веществами, а
также с реактивом, применяемым для проявления.
3.3. Ионообменная хроматография
Ионообменная хроматография основана иа обратимом стехио-
метрическом обмене иоиов, содержащихся в хроматографируемом
растворе, на подвижные ионы, входящие в состав ионитов (ионообменников).
В зависимости от знака заряда ионизирующих групп иониты делят
на катиониты (катиоиообменники) и аниониты (анионообменники).
Существуют также амфотерные иониты — амфолиты, которые
одновременно обменивают катионы и анионы.
Ионит — сложная система, состоящая из основной части молекул
(матрицы) и связанных с ией активных групп, способных к обмену ионов.
Если это катионит, то ои обладает кислыми или слабокислыми
свойствами, так как в его состав входят активные кислые группы: —SO3H, —
CH2SO3H, —СООН, —ОН, —РО3Н2 и др., в которых подвижными
являются водородные ионы или замещающие их катионы. Аииониты
обладают основными или слабоосиовными свойствами и содержат
группы: —N(OH)(CH3)3, =NH, —NH2, —ОН, —NHR, —i5r3 •
Разделение смеси иоиов, находящихся в растворе, происходит
вследствие неодинаковой способности их к обмену с ионами ионита.
Процесс обмена ионами можно представить следующим образом. Если
ионит с группировками, способными отщепить иои А, поместить в
раствор, в котором находятся ионы В того же заряда, то ионы А будут
покидать ионит и переходить в раствор, а ионы В в строго эквивалентном
количестве будут связываться с ионитом:
R—H + Na++Cr -> R—Na + H+ + Cl" (катионный обмен)
R— ОН + Ыа+ + СГ -> R—Cl + Na+ + OH" (анионный обмен)
Ионообменное равновесие может быть описано законом действия
масс. Из уравнения реакции обмена двух одновалентных иоиов А+ и В+
+ BZ + A+
согласно закону действия масс следует:
[BZ][A+]
Г ЛДП, ИЛИ
[AZ][B+] ** [AZ]
Если концентрации иоиов в твердой фазе обозначить через [А+] и
[В+], то уравнение будет иметь вид
138
где Л^в — коэффициент избирательности, шш константа ионного
обмена.
Иониты должны удовлетворять следующим требованиям: быть
химически устойчивыми в различных средах, механически прочными в
сухом и особенно в набухшем состоянии, обладать большой
поглотительной способностью и способностью хорошо регенерироваться.
3.4. Основные методы хроматографнческого анализа
Колоночная хроматография. Для колоночной хроматографии
обычно применяют колонки, изготовленные из стекла, у которых
отношение длины к диаметру находится в пределах 40—100. В нижнюю часть
колонки помещают стеклянную вату в ввде тампона» а затем загружают
адсорбент, суспендированный в растворителе. При
этом адсорбент должен заполнять колонку с
равномерной плотностью (рис. 36).
При получении распределительных хромато-
грамм на колонке твердый носитель вначале
растирают с растворителем, который будет служил»
неподвижной фазой. Полученную густую кашицу
суспендируют во втором растворителе (подвижная
фаза) и смесью равномерно заполняют колонку. В
случае ионообменной хроматографии иониты
предварительно подвергают специальной
обработке: например, катеонит очищают от ионов железа
и доводят до набухания.
После заполнения колонки в нее осторожно
приливают раствор анализируемого вещества (или
смеси веществ) в подобранном растворителе. При
адсорбционной и распределительной
хроматографии исследуемый раствор должен занимать в
колонке небольшой объем, покрывая поверхность
носителя или адсорбента. При ионообменной
хроматографии можно добавлять растворителя
больше. После внесения хроматографируемой смеси
приступают к проявлению хроматограммы, про- Рве. 36. Хроыато-
пуская через слой адсорбента (или носителя) чне- графическая колонка:
тый растворитель, играющий роль вымывающего 1 ~~ 1*сгаоригепь; 2 —
АДСООО&П" 3 •"■" ЗОНЫ
вещества. Если растворитель проходит через ко- £££
лонку очень медленно, процесс можно ускорить, ш вага
_ ткляя.
139
создавая в колонке небольшое разряжение (в нижней части), или,
наоборот, повышенное давление (в ее верхней части). В том случае, когда один
из растворителей перестает заметно распределять зоны отдельных
веществ по всей длине колонки, обычно переходят к другому
растворителю. На выходе из колонки раствор {элюат) собирают и отмечают
разделившиеся зоны. Очень удобен автоматический прибор (коллектор),
позволяющий отбирать равные объемы фракций элюата (по 0,5—10 мл)
через равные промежутки времени.
Контроль за хроматографическим разделением анализируемых
смесей можно осуществлять различными способами. Если разделяемые
вещества окрашены, то анализ веществ можно проводить непосредственно
(визуально) на колонке: в слое адсорбента появляются окрашенные зоны
(слои). Если же вещества неокрашены, но люминесцируют (при
освещении их УФ-излучением) или вызывают флюоресценцию некоторых
индикаторов, вводимых предварительно в адсорбент, то идентификация таких
веществ также не представляет особого затруднения.
Можно регистрировать разделяемые вещества непосредственно
после выхода из колонки. Например, для веществ,
обладающих кислыми свойствами, можно
использовать цветную реакцию с индикатором.
Иногда проводят анализ каждой порции элюата с
помощью различных физико-химических
методов (спектрофотометрического, потенциометри-
ческого, рефрактометрического и др.).
Хроматография иа бумаге. Известны
следующие разновидности хроматографии на
бумаге: одномерная, двумерная, круговая и электро-
форетическая. Одномерная и двумерная
бумажная хроматография может быть восходящей и
нисходящей.
Роль носителя выполняет бумага, которая
должна быть химически чистой, химически и ад-
сорбционно нейтральной, однородной по
плотности, обеспечивать определенную скорость движе-
_ „ п , ния растворителя и содержать достаточное количе-
Рис 37. Прибор для r r w х
получения одномерной ство неподвижной фазы, где происходит разделе-
восходящей хромато- иие смеси веществ. Специально для
хроматографии выпускают четыре сорта отечественной
бумаги (№1, 2, 3, 4), отличающиеся друг от друга по
плотности. Плотность бумаги определяет скорость
продвижения по ней растворителя. Бумага № 1 и
№ 2 — менее плотная и называется «быстрой», а
№ 3 и № 4 — более плотная, «медленная».
граммы на бумаге:
1 — зажим для бумаги; 2 —
полоска бумаги; 3 —
фронт растворителя; 4 —
стартовая линия с
нанесенными веществами; 5 —
подвижный растворитель
140
Получение одномерной восходящей хроматограммы осуществляют
следующим образом. На нижнюю часть полоски бумаги на некотором
расстоянии (~2 см) от нижнего края — линия старта — микропипеткой
наносят каплю раствора исследуемого вещества и рядом каплю красителя
(например, нейтральный красный) в растворителе (для установления
окончания процесса разделения). Подготовленную таким образом
бумажную полосу помещают в хроматографическую камеру. Роль
последней может выполнять обычный стеклянный цилиндр, на дно которого
наливают растворитель (подвижная фаза), насыщенный неподвижной
фазой (чаще — водой). Бумагу в камере закрепляют так, чтобы она
свободно спадала вниз, не касаясь стенок камеры, а нижний конец был
опущен в жидкость (рис. 37), однако линия старта с нанесенной каплей
исследуемого вещества не должна касаться растворителя. В процессе хро-
матографирования подвижная жидкость поднимается вверх по бумаге
(под действием капиллярных сил) и разделяет компоненты смеси на
зоны, которые при различных /?/движутся по бумаге с неодинаковой
скоростью. Когда фронт растворителя (с красителем) поднимется до
верхнего края полоски, хроматограмму извлекают из камеры, отмечают линию
фронта растворителя, сушат и проявляют веществами, которые при
взаимодействии с компонентами анализируемой смеси образуют окрашенные
соединения. Например, при разделении
аминокислот проявление осуществляют
0,2%-ным ацетоновым раствором нингид-
рина.
Качественно обнаружить вещества на
хроматограмме можно и по люминесценции в
УФ-свеге.
Одномерную нисходящую
хроматограмму можно получить, несколько изменив
методику выполнения опыта. Подвижный
растворитель, насыщенный неподвижным,
наливают в небольшую кювету,
закрепленную в верхней части камеры. На дно этой
камеры помещают бюкс с неподвижным
растворителем, насыщенным подвижным,
для создания в камере атмосферы
насыщенных паров, предотвращающей испарение
растворителя с бумаги (рис. 38). На полоску
Рис. 38. Прибор для
получения одномерной нисходя-
бумаги на расстоянии 5 см от верхнего края щей хроматограммы на бу-
наносят каплю исследуемого раствора. По- маге:
еле высушивания пятна этот край полоски
погружают в кювету с растворителем
(подвижная фаза), который под действием ка-
1 — стеклянная повета с
подвижным растворителем; 2 —
полоска бумаги; 3 — штатив; 4 —
стеклянная палочка
141
Рис 39. Схема получеши двумерной хроматограммы:
а — смесь, состоящая из шеста компонентов, в первом растворителе не разделилась; 6 —
та же хроматограмма перед опусканием бумаги во второй растворитель (поворот на 90°);
в — во втором растворителе смесь разделилась (А — место нанесения капли исследуемого
раствора; стрелкой показано направление движения фронта подвижной фазы)
пиллярных сил и сил тяжести перемещается вниз по бумаге. Хромато-
графирование заканчивается, когда фронт растворителя почти достигает
нижнего края бумаги. Проявление хроматограммы осуществляют, как
описано выше.
В случае, если, сложную смесь нельзя разделить с помощью одного
растворителя, применяют последовательно два растворителя с
различными коэффициентами распределения. Для этого используют квадратный
лист бумаги (20x20, 30x30,40x40 см), в левом углу которого (на
расстоянии 5 см от края) наносят капли исследуемых веществ. После их
высушивания бумагу помещают в камеру и опускают нижний край в
растворитель, проводя хроматографирование по восходящему методу. Как
только фронт растворителя достигнет верхнего края бумаги,
хроматографирование заканчивают и бумагу
высушивают. Затем, повернув ее на 90° против часовой
стрелки, помещают во вторую камеру с
другим растворителем и продолжают процесс
хроматографирования по восходящему
методу. После окончания хроматографирования
хроматограмму высушивают вторично и
проявляют. Так получают двумерную
хроматограмму (рис. 39).
Вместо обычных пятен на бумаге можно
получить концентрически расположенные
Рис 40. Получение кру- кольца. Это достигается с помощью круговой
говон бумажной хромато- бумажной хроматографии. Для ее получения
граммы: ю бумаги вырезают круг, в центр которого
I — ЭКСНКЭТОр. £ ~~ IfCCiv -—
нанесения капли раствора; наносят каплю исследуемого раствора. В ка-
3 _ круг бумаги; 4 — честве камеры может служить небольшой эк-
фитиль для подачи раствори- сикатор. Когда пятно высохнет, на круге вы-
теля;5 —-растворитель резают небольшую полоску (фитиль), кото-
142
рую отгибают, а круг укладывают на выступ в эксикаторе (рис. 40).
Полоска при этом должна быть опущена в растворитель (насыщенный
неподвижной фазой), налитый на дно эксикатора. Скорость поступления
растворителя к центру круга можно регулировать изменением ширины
фитиля.
Наиболее часто применяемые в распределительной бумажной
хроматографии системы растворителей (подвижные фазы) приведены в табл. 9.
Таблица 9. Подвижные фазы, наиболее часто применяемые
в бумажной хроматографии для разделения смесей
(неподвижная фаза — вода)
Растворители
«-Бутиловый спирт,
уксусная кислота, вода
н-Амиловый спирт,
пиридин
w-Бугиловый спирт,
муравьиная кислота
(20%-ный раствор),
вода
«-Бутиловый спирт, м-
пропиловый спирт,
вода
Этилацетат, пиридин,
вода
Этилацетат, уксусная
кислота, вода
«-Бутиловый спирт,
насыщенный аммиаком
(1,5 н.)
Тетрахлорид углерода,
уксусная кислота, вода
Соотношение
растворителей и способ
их приготовления
4 : 1 : 5 (по объему)
(после расслаивания смеси
применяется ее верхний
слой)
9 : 1 (по массе)
5 : 1 : 1 (по объему)
12:5 :3 (по объему)
2 :1 :2 (по объему)
3 : 1; 3 (по объему)
1:1 (по объему)
5:1:1 (по объему)
Разделяемые смеси
веществ
Аминокислоты,
углеводы и другие
органические вещества
а-Аминокислоты
Аминокислоты
»
Сахара
»
Алифатические кислоты
Алифатические кислоты
и их натриевые соли
Следует упомянуть также несколько других универсальных систем,
наиболее подходящих для разделения самых разнообразных
органических соединений:
фенол, насыщенный водой (5 :1).
бутилацетат, насыщенный водой;
бумага, пропитанная формамидом (подвижная фаза — хлороформ,
камера насыщена хлороформом);
143
бумага, пропитанная формамидом (подвижная фаза — бензол;
камера насыщена бензолом);
бумага, пропитанная формамидом [подвижная фаза — бензол —
циклогексан (3 : 7); камера насыщена той же смесью];
петролейный эфир — метанол — вода (2:1:1);
бумага, пропитанная керосином (подвижная фаза — 80%-ный
водный метиловый спирт + 5%-ный н-бутиловый спирт).
Тонкослойная хроматография — один из эффективных методов
шализа сложных смесей различных органических соединений. Техника
этого метода исключительно проста и доступна для любой лаборатории.
Вначале выбирают стеклянную пластинку подходящего размера
^5x5, 10x20, 20x20 и др.), которая служит носителем адсорбента. Кроме
стеклянных пластинок можно применять также пластинки из алюминие-
зой фольги, пластика и других материалов. Пластинки предварительно
обезжиривают, тщательно моют и высушивают. На подготовленную
таким образом пластинку наносят слой адсорбента, который является
неподвижной фазой. В качестве адсорбентов для тонкослойной
хроматографии используют силикагель, оксид алюминия, кизельгур,
целлюлозные порошки, а также полиамидные сорбенты (типа «капрон»). Адсор-
эент наносится на горизонтальную поверхность пластинки ровным слоем
при помощи специального прибора или ручным способом.
В тонкослойной хроматографии слой адсорбента может быть
закрещенным и незакрепленным. В первом случае в сорбируемую массу до-
эавляют связующие вещества (медицинский гипс или очищенный крах-
иал), во втором — сорбент находится на пластинке в свободном
состоянии. Пластинки с закрепленным слоем сорбента вначале сушат на
воздухе, а затем в сушильном шкафу в течение 3—4 ч при 150 °С. Для
получения пластинки с незакрепленным слоем сорбент (например, А12О3)
насыпают на стекло и
выравнивают стальным
валиком с
утолщением на концах (0,5—1
мм) или стеклянной
палочкой, на концы
которой надеты
кольца из резиновой
трубки (рис. 41).
На
подготовленную пластинку с по-
Рнс. 41. Станок для нанесения незакрепленного слоя M0U*bI° микропипетки
адсорбента (А12О3) на пластинку: наносят пробу иссле-
1 — валик; 2 — адсорбент (Al2Oj), 3 — тонкий слой адсор- , дуемого вещества (на
бента;^ —стеклянная пластинка расстоянии 1,5 см от
144
нижнего края). Если таких проб
несколько, то расстояние между
ними должно быть не менее 2 см.
Затем пластинку помещают в
камеру для хроматографирования, на Рнс. 42. Хроматографическая кювета
дно которой предварительно нали- с пластинкой:
вают небольшое количество раство- ; ~ кювега'2 ~ ««»«"»;3 - Расга0Р
рителя (подвижная фаза) (рис. 42).
Неподвижной фазой служат сорбционный слой и адсорбированный им
растворитель.
Если растворитель поступает на пластинку снизу вверх под
действием капиллярных сил, то такая хроматография будет восходящей. По мере
продвижения растворителя происходит разделение анализируемой смеси
веществ в зависимости от их сродства к адсорбенту. После окончания
разделения пластинку с адсорбентом вынимают из камеры, отмечают
фронт растворителя и обрабатывают специальными реагентами для
обнаружения пятен (например, парами иода, серной кислотой и т. д.).
В тонкослойной хроматографии, как и бумажной, важной
характеристикой степени разделения веществ является коэффициент Rf.
Работа 1. Разделение и идентификация смеси
2,4-динитрофенилгидразонов
(ДНФГ) в тонком незакрепленном слое
оксида алюминия
2,4-Динитрофенилгидразоны альдегидов и кетонов могут быть
получены при взаимодействии карбонильных соединений с 2,4-динитро-
фенилгидразином в кислой среде.
На стеклянную пластинку наносят тонким слоем оксид алюминия
(II—III степени активности) и равномерным движением валика вдоль
пластинки снимают его избыток. С помощью отдельных капилляров
наносят на стартовую линию по капле растворы стандартных ДНФГ
(«свидетели») и каплю исследуемой смеси ДНФГ (выдается преподавателем).
Площадь пятен, нанесенных на стеклянную пластинку, должна быть
минимальной (~2 мм2).
Растворы ДНФГ карбонильных соединений (бензальдегида, ацетона, аце-
тофенона, циклогексанона и др.) в хлороформе готовят заранее. Концентрация
каждого ДНФГ — ОД—0,3 г на 50 мл хлороформа. Приготовленные растворы,
которые служат «свидетелями» для определения состава контрольной задачи,
хранят в склянках с хорошо притертыми пробками.
145
В кювету наливают тройную смесь растворителей: циклогексанона,
хлороформа, нитробензола в соотношении 12:3: 1 (по объему). Высота
слоя смеси в кювете не должна превышать 1,5 см. Подготовленную
пластинку ставят в наклонном положении в кювету (нижний конец
пластинки должен быть погружен в смесь растворителей на 1 см). После этого
кювету помещают в эксикатор, который закрывают крышкой.
После того как растворитель поднимется почти до верха пластинки,
ее вынимают и измеряют расстояние от линии старта до центра
переместившегося пятна и расстояние, пройденное растворителем от линии
старта до фронта растворителя. Положение пятен на пластинке отмечают,
обводя их иглой. Из полученных данных определяют значения Rf для
стандартных ДНФГ и каждого компонента контрольной смеси.
Примерные значения Rf ДНФГ при использовании оксида алюминия III
степени активности, а также температуры плавления ДНФГ приведены в
табл. 10.
Таблица 10. Значения R/ некоторых ДНФГ в смеси цнклогексан —
хлороформ — нитробензол (12 : 3 :1) при применении А12О3
Карбонильные соединения
Бензальдегид
Циклогексанон
Ацетон
Ацетофенон
*/
0,20
0,36
0,40
0,42
Т. пл., °С
237
162
126
249
4. Метод электрических моментов диполей
Этот метод широко используется для изучения природы химической связи,
изомерии н конформации молекул, строения комплексов, взаимного влияния
атомов н атомных группировок в молекуле н т. д.
Если в молекуле центр тяжести положительных зарядов (ядер),
составляющих в сумме величину +е, не совпадает с центром тяжести отрицательных
зарядов (электронов), в сумме равных величине -е, н эти центры находятся на
некотором расстоянии / друг от друга, то молекула будет полярной н в ней возникает
электрический момент диполя р. Он равен произведению элементарного заряда
на межатомное расстояние: р = QL
Электрический момент диполя, измеряемый в кулон-метрах (Кл-м), является
векторной величиной. Принято считать, что этот вектор направлен в сторону
более электроотрицательного атома.
Состояние электрической асимметрии в молекуле, т. е. появление
электрического момента диполя, может быть вызвано несколькими причинами. Во-
первых, различие в электроотрицательности атомов, образующих связь в
молекуле, приводит к смещению электронной плотности в сторону более электроотрн-
146
цательного атома. В результате такая связь становится полярной, а молекула,
содержащая эту связь, приобретает электрический момент диполя. Вторая
причина — неодинаковые размеры атомных орбиталей, образующих связь. Так, если в
молекуле химическая связь образована за счет перекрывания орбиталей
различной формы, то в этом случае центр плотности отрицательных зарядов также
смещен в сторону от центра положительных зарядов.
Когда полярная молекула, имеющая постоянный электрический момент
диполя рлост, находится в электрическом поле (между пластинками конденсатора),
то в ней возникает индуцированный (наведенный) электрический момент диполя
рИНп. Таким образом, суммарный (результирующий) электрический момент
диполя будет составлять сумму:
При снятии Электрического поляр^д исчезает.
Направления векторов электрических моментов диполей молекул в отсутст-
вие электрического поля имеют хаотическое расположение, т. е. £/>/ш 0. При
наложении внешнего поля молекулы вещества ориентируются таким образом, что
их электрические моменты диполей будут параллельны напряженности
электрического поля (этому препятствует вращательное тепловое движение молекул).
Сумма проекций всех электрических моментов диполей на направление
напряженности внешнего поля представляет собой ориентационную поляризацию Р^
при которой У\ pi * 0. Учитывая появление индуцированного электрического
момента диполя и связанную с этим индукционную поляризацию Рят (под
влиянием электрического поля), общая (молекулярная) поляризация Р слагается из двух
составляющих: Р = Р^ + РНИЛ.
Индукционную поляризацию можно рассматривать как сумму поляризаций
электронной (Рм) и атомной (Р„):
р = р + р
Тогда
Связь между молекулярной поляризацией Р и электрическим моментом
диполя р дает формула Дебая:
е + 2 р
где е — диэлектрическая проницаемость; А/ — молекулярная масса; р
—плотность; jVa — постоянная Авогадро; а — поляризуемость; k — постоянная Больц-
мана; Т— абсолютная температура; р — электрический момент диполя.
4 р2
Выражение — nNA представляет собой ориентационную поляризацию.
Электронную поляризацию (Рм) принимают равной молекулярной рефракции
147
ft2
Рис. 43. Графический метод определения коэффициентов а и Р
вещества при D-линнн натрия (RD), а Р„ обычно не учитывают, так как Р„ « Рм.
Следовательно, Рор = Р- RD.
Подставляя вместо Р™ ее значение, получим:
D
(2)
3 п ЗкТ
Молекулярную поляризацию Р можно определить по формуле Хальверштадта—
Камлера:
ЗссК,
(3)
где £i — диэлектрическая проницаемость растворителя; \\ — удельный объем
растворителя (V = \fd );М— молекулярная масса вещества.
Значения е12 и Vn (для раствора) являются линейной функцией молярной
доли растворенного вещества W2\
Коэффициенты аир можно найти графическим методом (рис. 43):
а
Подставив значения Р и констант в уравнение (2), получим р =
Наиболее распространенным методом определения дипольных моментов
является метод, основанный на измерении диэлектрической проницаемости в
парах или разбавленных растворах полярных веществ в неполярных
растворителях. Обычно используют метод разбавленных растворов. Для этого
экспериментально находят две величины: плотность разбавленного раствора dl2 (в
неполярном растворителе, например в бензоле, дноксане и др.) и диэлектрическую
проницаемость е12. Последнюю величину определяют на приборе, который состоит
из термостатированной измерительной ячейки, двух генераторов (500 кГц),
измерительного конденсатора и осциллографа. Измерительную ячейку заполняют
раствором вещества и находят ее электрическую емкость. Изменение этой
величины компенсируют емкостью калиброванного прецизионного конденсатора.
Разностный сигнал после усиления фиксируется индикатором (осциллографом).
148
Чтобы вычислить общий электрический момент диполя молекулы исходя из
моментов отдельных связей, необходимо суммировать векторы дипольных
моментов связей по правилам векторной алгебры. Например, если число групповых
моментов равно двум (р\ и р2) и они расположены под углом <р, то суммарный
электрический момент диполя равен
Р = yiPi + Pi ±
Если же две связи с одинаковыми моментами р образуют угол ф, равный 180°, то
суммарный электрический момент диполя равен нулю.
Приложение
L Некоторые советы по организации
самостоятельной работы студентов (СРС)
Уменьшение лекционного курса по органической химии предъявляет
повышенные требованш к самостоятельной работе студентов. При подготовке к
лабораторным и практическим занятиям студент должен прежде всего изучить
программный теоретический материал. Для студентов, специализирующихся в
области химической технологии тугоплавких неметаллических и силикатных
материалов (специальность 25.08), рекомендуются следующие учебники и учетные
пособия:
1. Артемешсо А. И. Органическая химия. 4-е изд. —М.: Высшая школа, 2000.
2. Петров А. А,, Бальяи X. В., Трощенко А. Т. Органическая химия. 4-е
изд.: —М: Высшая школа, 1981.
3. Стрепнхеев А. А., Деревицкяя В. А. Основы химии
высокомолекулярных соединений. 3-е год. —М.: Химия, 1976.
4. Воробьев В. А. Основы технологии строительных материалов из
пластических масс. 2-е изд. —М.: Высшая школа, 1975.
Для закрепления теоретического материала можно использовать задачи и
упражнения из книги:
5. Альбицкая В. М., Серкова В. И. Задачи и упражнения по органической
химии. 3-е изд. —М.: Высшая школа, 1983.
Студентам специальностей 29.06 (производство строительных изделий и
конструкций) и 07.08 (экономика н управление в строительстве) рекомендуется
изучение теоретического материала по учебнику (1).
Для облегчения самостоятельной работы с учебником в лабораторном
практикуме приведены соответствующие разделы программы. При подготовке к
коллоквиуму студенты должны также ответить на контрольные вопросы,
предлагаемые для ознакомительного (малого) практикума (раздел 2) в каждой работе.
П. О номенклатуре органических соединений
Для названия органических соединений применяются, в основном, две
номенклатуры — рациональная и систематическая (ИЮПАК) . Однако широко
По первым буквам английского названия Международного союза чистой и
прикладной химии — International Union of the Pure and Applied Chemistry.
150
распространены и тривиальные названия органических соединений, которые,
однако, не отражают их строения (например, муравьиная, виноградная, молочная
кислоты, древесный спирт и т. д.).
В рациональной номенклатуре за основу названия
органического вещества принимают название простейшего члена гомологического ряда
конкретного класса углеводородов, а остальные рассматривают как его производные.
С ростом числе органических соединений возникла острая необходимость в
создании универсальной номенклатуры. Так появилась официальная, или
систематическая, номенклатура — номенклатура ИЮПАК.
Она предусматривает возможность по сравнительно простым правилам назвать
любое химическое соединение.
Предельные углеводороды (алканы). Первые четыре члена
гомологического ряда имеют тривиальные названия (метан, этан, пропан, бутан). Названия
последующих алканов производят от греческих числительных с добавлением
суффикса -ан: пентан, гексан, октан, ионан, декан.
При отрыве от молекулы алкана одного или нескольких атомов водорода
образуются радикалы — алкилы, названия которых строятся из наименований
соответствующих углеводородов с заменой суффикса -ан на -нл: СИ, — метан,
СН3 — метил; CjHe — этаи, С2Н5 — этил; С3Н8 — пропан, С3Н7 — пропил и т. д.
Ниже приведены наиболее распространенные радикалы (в скобках даны
сокращенные обозначения, широко используемые в химической литературе):
Радикал
СН2<
сн<
сн3—сн<
сн, сн* сн ^
(СН3)2СН-
(СН3)2СН-СН2-
СН2=СН-
Название
Метилен
Метин
Этилиден
Пропилид ей
Изопропил (/-Рг)
w-Бутил
Изобутил
(первичный изобутил)
вто^-Бутил (v-Bu)
трет-Бутип (Г-Ви)
Винил (Vy)
Радикал
СН3(СН2)зСН2-
CtHr
г* тт ГЧЛ^
ОС
н-со-
СН3СО
Название
Амил (Am), пентил
Фенил (Ph)
Фенилен
Бензил
Бензилидеи
(бензаль )
Бензенил
(бензилидин*)
2-Нафтил
Форм ил
Ацетил (Ас)
Бензоил (Bz)
Это название не допускается правилами ИЮПАК, но часто используется.
151
Продолжение табл
Радикал
сн-с<
СН2=СН-СНг-
Название
Винилиден
Аллил
Радикал .
снз-ед-со-
о-
Название
Толуил (о-, м-у w-)
2-Фурил
Для метила часто используется обозначение Me, для этила — Et.
Для названия разветвленных алканов по номенклатуре ИЮПАК вначале
выбирают самую длинную (главную) цепь углеродных атомов. Затем ее нумеруют,
начиная с того конца, к которому ближе расположен заместитель (радикал).
Указав положение каждого радикала цифрами и назвав их, добавляют название
главной цепи. Например:
сн3
1 21 3 4 5
сн3— с—сн2-сн2— сн
с
сн3
2,2-диметилпентан
Количество однородных радикалов обозначается приставками ди-, три-, тет-
ра- и т. д. Атомы углерода в главной цепи нумеруются так, чтобы цифры,
указывающие положение заместителей, были наименьшими:
2,2,4-триметилпентан
(но не 2,4,4-триметилпентан)
Если в разветвленном алкане имеются цепи равной длины, то в качестве
главной цепи выбирают ту, у которой имеется наибольшее число разветвлений:
сн—сн—сн—сн—сн—сн-
СН3 v-H- СН3 СН,
сн3
I
сн
■CR
2,3,5-триметил-4-пропилгептан
Следует различать разветвление цепи от ее изгибов при графическом
изображении:
CH2
СН3
H
CHj—
СН2
СН,
пропан гептан
Заместители перечисляются в алфавитном порядке или в порядке
возрастающей сложности. Более простыми считаются радикалы с меньшим числом уг-
152
леродных атомов, а при равном их числе — менее разветвленные, ненасыщенные
радикалы считаются более сложными, чем насыщенные. Например:
1 234 567 89 10
CHCH """ " "
:Hj сн(сНз)2
CHj" Сгц
3-метил-5-пропил-7-изопропиццекан
По рациональной номенклатуре все алканы рассматриваются как
производные метана, в котором один или несколько атомов водорода замещены на ал-
кильные радикалы:
CHj CHjCH2"—CHj"~CHjCHj CHjCHj
СН.СН,
метилметан дизтилметан J '
метилдиэтилизо-
пропилметан
Названия симметричных углеводородов часто строят исходя из названия
радикала с приставкой ди- (это распространяется и на другие классы углеводородов):
СН3—СО—CO—CHj СД—С6Н, СН2=СН—СН—СН2
диацетил дифенил дивинил -
Непредельные углеводороды. Ненасыщенные углеводороды с двойной
связью называют алкенами или олефинами, а с тройной — алкинами. Названия их
по номенклатуре ИЮПАК образуют от названий соответствующих алканов с
заменой суффикса -ян на -ен или -нн. Кратная связь обязательно включается в
главную цепь. Нумерацию цепи начинают с того конца, к которому ближе
двойная или тройная связь. В этом случае атомам углерода у кратной связи отдается
предпочтение по сравнению с насыщенными атомами углерода, даже имеющими
заместители:
CHj-CH— CH2
пропен
CHf- CHj— С— CHj— CHj— CHj
CH-
• 2-этилпентен-1 5-метилгексин-2
CHj—C»C—CHj—CH—
■сн—сн
СН3 CH—CH2
4-метил-З-этилпентен-1
153
Число кратных связей одного рода обозначают с помощью суффиксов -днен,
-трнен, -дннн и т. д. Двойная связь имеет преимущество перед тройной при
нумерации
СН2—СН—СН
СН—СН—СН.
—СН—О
гексатриен-1,3,5
С— С
гептен-1 -диин-4,6
СН
Правилами ИЮПАК допускается применение названий этилен, аллен,
изопрен, ацетилен только для незамещенных соединений.
Этиленовые и ацетиленовые углеводороды по рациональной номенклатуре
рассматриваются как производные этилена и ацетилена. При этом производные
этилена с двумя заместителями у разных углеродных атомов называются
симметричными, а у одного — несимметричными (независимо от истинной симметрии):
СН* Спт~СН ^ СН.
этилэтилен
,—СН2—СввСН
этилацетилен
сн3—с=
•СН,
дим етилацетилен
СН—СН—СН—CHj—СН3
смлш-метилэтилэтилен
СН—С
ч.,
СН(СН3)2
несимм -диизопропилэтилен
Ароматические углеводороды (арены) с одним бензольным ядром обычно
рассматривают как производные бензола. Положение заместителей можно
указывать цифрами или, если заместителей только два, буквами: о- (орто-), м- (мета-),
п- (пара-), которые соответствуют положениям 1,2,1,3 и 1,4:
1 Д-диметилбензол
(о-диметилбеизол,
о-ксилол)
1,3-диметилбешол
(м-диметилбензол,
Л1-КСИЛОЛ)
СН2—СН
1,4-диметилбензол
(л-диметилбензол,
w-ксилол)
1,2,3-триметилбешол
этилбешол
1-этил-З-е/псуьбугилбегаоЛ
(л1-этил-втор-бугилбензол)
Правилами ИЮПАК допускается употребление тривиальных названий —
толуол, ксилол, стирол — и их производных:
154
СН
с2н,
4-этилтолуол (w-этилтолуол)
3-этилстирол (л!-этилстирол)
Для полициклических углеводородов нумерация производится следующим
образом:
8 9 1
9 10
нафталин
6 5 4 3
фенантрен
Кислородсодержащие соединения. По систематической номенклатуре
спирты имеют суффикс -ол, альдегиды -ал (-яль), кетоны — -он, карбоновые
кислоты -овая (кислота). Количество одинаковых функциональных групп
указывается приставками дн-, три- н т. д. Кроме того, в рамках правил ИЮПАК
имеется так называемая радикально-функциональная номенклатура, в
соответствии с которой названия спиртов производят от названий соответствующих
радикалов с добавлением суффикса -овый (спирт) (названия по этой номенклатуре
приведены в круглых скобках).
По рациональной номенклатуре одноатомные спирты считаются
производными метилового спирта — карбинола СН3ОН; альдегиды — уксусного
альдегида СН3СНО; карбоновые кислоты — уксусной кислоты СН3СООН. Названия ке-
тонов образуют из названий радикалов, связанных с углеродом карбонильной
группы, н окончания -кетон. Названия по рациональной номенклатуре приведены
в квадратных скобках:
СНгтСН, СНг ОН
пропанол-1
(пропиловый спирт)
[этилкарбннол]
CHj—СН—CHj-OH
CHj—СН—СН-
ОН
2-метилпропанол-1
(изобутиловый спирт)
[изопропилкарбинол]
З-метилбутанол-2
[метилизопропил-
карбинол]
—CHj-CHj- СН—СНО
?
СН3-СН-С-СН3
C
CHj
З-метилбутанон-2 [метилизопропилке-
тон]
5-хлор-2-метилпенталь
155
сн3—сн—CHj— сн —соон
[Г-С-СНО СбН5 NH,
СНз
2,2-диметилпропаналь 2-амино-4-фенилпентановая кислота
[триметилуксусный альдегид]
CHj—9Н—CHjOH
соон
2-фенил-2-метилпро- пропандиол-1,2
пановая кислота
[диметилфенилуксусная кислота]
сн3— со—сн2— со —сн2— со—сн3 онс— сн2— сно
гептантрнон-2,4,6 пропандиаль-1,3
[диформилметан,
формилуксусный альдегид]
НООС—СН—СН—СООН
бутандиовая кислота
Положение функциональной группы, как видно из приведенных примеров,
обязательно должно указываться для спиртов н кетонов, но не для альдегидов и кислот, в
молекулах которых функциональная группа всегда находится в начале цепи.
Если в молекуле имеются две функциональные группы — карбоксильная
(или гидроксильная) н карбонильная, то приоритет в нумерации отдается
карбоксильной. По правилам ИЮПАК гидроксильная группа обозначается приставкой
гидроксн-, н карбонильная — оксо-:
hochj- сн^- соон сн— £о—соон
3-гидроксипропановая кислота 2-оксопропановая кислота
Для указания положения гидроксн- н оксогрупп иногда применяют
нумерацию греческими буквами, начиная с атома углерода, связанного с
функциональной группой:
з—СН—СООН СН—СО— £н—СН2— СООН
Z
а-гидроксипропионовая кислота у-оксовалериановая кислота
(у-кетовалериановая кислота)
Для кислородсодержащих соединений правилами ИЮПАК допускается
применение большого числа тривиальных названий.
156
Простые эфиры обычно называют по радикально-функциональной
номенклатуре:
СН3— О—СЩ
диметиловый эфир(метоксиметан)
j— CHj- О—СН(СН3)2
этилизопропиловый эфир
(2-этоксипропан)
Производные карбоновых кислот могут быть названы двумя способами
(названия, связанные с использованием тривиальных названий, приведены в
квадратных скобках):
СН— CHj— COONa
натриевая соль пропановой кислоты
(пропаноат натрия) [пропионат натрия]
сн—со—о—со—сн3
ангидрид этановой кислоты (этановый
ангидрид) [уксусный ангидрид, ацетан-
гидрид]
CH3CONH2
амид этановой[уксусной] кислоты
(этанамид) [ацетамид]
СН—СН—СН^-СООС2Н5
этиловый эфир бутановой кислоты
(этилбутаноат) [этилбутират]
CH3COCI
хлорангидрид этановой [уксусной]
кислоты (этаноилхлорид) [ацетилхло-
рид]
сн/ауз—cn
нитрил пентановой [валериановой]
кислоты (пентанонитрил) [валеронитрил]
Названия нитрилов по снстеме (ПОПАК образуют из наименований
углеводородов с добавлением слова -нитрил в качестве окончания или из названия
радикала и окончания -цианид:
С2Н—CN
пропаннитрил, этилцианид
3— CN
этаннитрил, метилцианид
Изоцианиды обычно называют, исходя из названия радикала. По
систематической номенклатуре к названию радикала добавляют слово нзоцнанид:
NC
метилизоцианид
С2Н—NC
этилизоцианид
Гетероцнклнческне соединения. По номенклатуре (ПОПАК названия гете-
роциклов производят, связывая названия приставок, обозначающих природу ге-
тероатома [окса- (0), тна- (S), аза- (N)] с корнем, обозначающим размер цикла
[-ир- (3), -ет- (4), -ол- (5), -нн- (6)], н суффиксами, указывающими на предельный
или непредельный гетероцикл: для предельных неазотистых -ан, предельных
азотистых -иднн, а для непредельных трехчленных циклов — суффикс -нн (с
азотом) или -ен (без азота). Например:
157
о
оксаиран, оксиран (оксид этилена) азаин, азин (пиридин)
По рациональной номенклатуре за основу берут название определенного ге-
тероцикла (фурана, тиофена, пиррола, пиридина и т. д.), а положение
заместителей в них обозначают цифрами или буквами греческого алфавита. Так, в пяти-
членных гетероциклах положение 2 (5) обозначают буквой а (а'), а положение
3 (4) — (3 ф'). В шестичленных гетероциклах положения 2 (6), 3 (5) и 4 обозив-
чают соответствеиио буквами а (а'),р ф') и у. Нумерацию начинают с гетероа-
тома (если их несколько, то нумеруют в порядке О, S, NH, N):
Р_Р
2-метилфуран 2-метилпиридин
(а-метилфуран) (а-метилпиридин, а-пиколин)
Для некоторых гетероциклов применяют тривиальные названия (пиррол,
пиридин, индол, хинолин, пурин и т. д.).
Полный свод правил номенклатуры ИЮПАК (по состоянию на 1965 г.)
приведен в книге: Справочник химика (дополнительный том).—Л.: Химия, 1968.
См. также Дж. Бенкс Названия органических соединений. —М.: Химия,
1980.
HL Некоторые сведения о справочной литературе
ио органической химии
А. Пособия по пользованию химической литературой
Терентьев А. П., Яновская JL А. Химическая литература и пользование
ею. —М.: Химия, 1967. Содержит описание главных химических (и некоторых
других) справочников, энциклопедий и реферативных журналов, краткую
характеристику обзорных и специальных журналов, а также перечень монографий по
всем разделам химии и химической технологии. Имеются следующие разделы:
«(Органическая химия», «(Прикладная химия», «Химия высокомолекулярных
соединений», «(Фармацевтическая химия», «Биологическая химия». Особый раздел
посвящен технике н методике работы с литературой. В приложении приведена
синоптическая таблица химических журналов (72 наименования), дающая
сведения, какие (или какой) тома вышли в том или ином году (по 1966 г.).
Потапов В. IVL, Кочетова Э. К. Химическая информация. Что, где и как искать
химику в литературе. —М: Химия, 1978. Подробно описана техника работы с
литературой с привлечением последних достижений информатики. Приводятся сведения
158
о библиотеках и библиографиях, об автоматизированных системах информации.
Отдельная глава посвящена патентной литературе и патентному делу в разных странах.
Подробно описаны основные справочники и реферативные издания.
Темннкова Т. И., Черкасова В. А. Справочная литература по органической
химии. —Л.: Изд-во ЛГУ, 1961.
Юрьев Ю. К. Практические работы по органической химии. —Изд-во МГУ,
Вып. I и П. С. 348— 398.
Грандберг И. И. Практические работы и семинарские занятия по
органической химии. — М.: Высшая школа, 1987. С. 255—268.
Лабораторные работы по органической химии/Под ред. О. Ф. Гинзбурга,
А. А. Петрова. — М.: Высшая школа, 1989.
Указатель литературы по органической химии/Сост. Г. И. Гладкова,
Г. Л. Мищенко. — М.: ВИНИТИ, 1976. Охвачен период с 1970 по 1975 г.
Химия и химическая промышленность. Указатель периодических и
продолжающихся изданий/Сост. И. Ф. Кузнецова н др. —М.: Международный центр
научной и технической информации, 1972. Включает около 4500 изданий, в
которых доля статей по химии составляет ие менее 30%.
Б. Справочная литература по химии общего характера
Справочник химика/Под ред. Б. П. Никольского н др. — Л.: Химия, 1965—
1968. Т. 1—6 и доп. Во втором томе приводятся константы важнейших
органических соединений (показатели преломления — в четвертом томе). Шестой том
посвящен технологии органических веществ. Дополнительный том содержит
полный свод правил систематической номенклатуры органических соединений
(по состоянию на 1965 г.), а также сведения по технике безопасности и указатель
ко всему справочнику.
Перельман В. И. Краткий справочник химика. — М.: Химия, 1964.
Гороновскнн И. Т., Назаренко Ю. П., Некряч Е. Ф. Краткий справочник
по химии. — Киев: Наукова думка, 1974.
Рабинович В. А., Хавнн 3. Я. Краткий химический справочник. —Л.:
Химия, 1977.
Гордон Л., Форд Р. Спутник химика. —М.: Мир, 1976. Оригинальный
справочник для химиков. Кроме табличных данных содержит обширные
теоретические сведения и сведения по лабораторной практике. Значительная часть
материала в других известных справочниках ие встречается. Около трети объема
уделено спектроскопии. Хорошо освещен вопрос, связанный с очисткой
растворителей. Имеются главы «Программы для расчетов на ЭВМ», «Статистическая обработка
результатов эксперимента». Каждый раздел снабжен обширной библиографией.
Краткий справочник физико-химических величин/Под ред. К. М. Мищенко
н А. А. Рявделя. — Л.: Химия, 1974. Содержит более 100 таблиц.
Перрн Дж. Г. Справочник инженера-химика. — Л.: Химия, 1969. Т. I.
Особый интерес представляет гл. I «Физико-химические свойства веществ», в
которой приведены подробные таблицы плотиостн растворов некоторых
органических веществ в воде.
Химические товары. Справочник/Сост. Т. П. Унанннц, Г. Я. Бахаревскнн,
А. И. Шерешевскнй. —М.: Химия, 1967—1974. Т. 1—5. В т. 1 описаны различ-
159
ные неорганические продукты, регуляторы роста растений, средства защиты
растений, антисептики; в т. 2 — красители, растворители, пластификаторы, крем-
нийорганические соединения, ПАВ и другие продукты органического синтеза; в
т. 3 — лаки, краски, грунтовки, шпаклевки, подмазки, пластмассы, герметики; в
т. 4 — различные полимеры и изделия из них (каучуки, клеи, крепители,
резинотехнические, асбестовые изделия, целлюлоза и ее производные, искусственные и
синтетические волокна). Всего в справочнике описаио около 3000 видов
химических товаров и изделий.
Краткая химическая энциклопедия. —М.: Советская энциклопедия, 1961—
1967. Т. 1—5. Содержит статьи, посвященные как целым разделам химии и
химической технологии, так и частным вопросам (реакциям, процессам, явлениям,
веществам, классам веществ). Крупные статьи снабжены библиографией.
Химический энциклопедический словарь/Под ред. И. Л. Кнунянца. —М.:
Советская энциклопедия, 1983. Многоплановое химическое справочное издание,
выпущенное впервые в нашей стране. Содержит около 9000 статей.
Значительный объем занимают статьи об индивидуальных химических соединениях.
Значительное место отведено проблемам общей химической технологии.
Оганесян Э. Т. Важнейшие понятия и термины в химии (краткий
справочник). —М.: Высшая школа, 1993. Содержит более 2000 терминов.
Артеменко А. И., Малеванный В. А., Тнкунова И. В. Справочное
руководство по химии. —М.: Высшая школа, 2001.
Вредные вещества в промышленности. Органические вещества/Под ред.
Н. В. Лазарева н Э. Н. Левиной. —М.: Химия, 1976. Т. 1,2. Токсикология сырья
и продуктов технологии органического синтеза и меры по технике безопасности
при работе с ними.
В. Специальная справочная литература по органической химии
Beilstein's Handbuch der organischen Chemie — Berlin: Springer—Verlag,
1918—1940. Bd. I—XXXI. — Крупнейший справочник по органической химии
(справочник Бейлыптейна). Четвертое издание содержит данные,
опубликованные в литературе до 1910 г., о 144000 соединений. Далее выпускались
дополнения: 1-е — охватывает литературу с 1910 по 1919 г., 2-е — с 1920 по 1929 г., 3-е — с
1930 по 1949 г., 4-е — с 1950 по 1959 г. Начат выпуск и пятого дополнения. „
Тома сгруппированы в четыре раздела: алифатические соединения (тома I—IV),
изоциклические (т. е. алициклические и ароматические) (тома V—XV),
гетероциклические (тома XVII—XXVII), природные соединения, структура которых
неизвестна или установлеиа не полностью (тома XXX—XXXI). По мере
выяснения структуры эти соединения описываются в дополнениях в соответствующих
томах. Том XXVIII представляет собой предметный указатель, XXIV —
формульный указатель.
В справочнике соединения, содержащие серу (за исключением сульфиновых
и сульфоновых кислот), селен и теллур, ие выделяются в отдельные классы, а
рассматриваются как аналоги соответствующих кислородсодержащих
соединений. Так, этилмеркаптан C2H5SH помещен после этанола С2Н5ОН, тиофен —
после фурана и т. д. Вещества с нефункциональными заместителями (галогены, NO,
NO2, NH3) в отдельные классы не выделяются. Последовательно выдерживается
принцип «от простого к сложному» — рассмотрение начинается в порядке ус-
160
ложнения, как это принято в номенклатуре ИЮПАК; последовательно
располагаются производные с одним, двумя и т. д. заместителями — сначала
однородными, затем разнородными, с распределением по старшинству. При каждом
основном члене ряда описываются сначала молекулярные соединения, потом
функциональные производные, затем — нефункциональные замещенные и их
производные и, наконец, сернистые, селенистые и теллуристые аналоги. Для каждого
вещества даны названия, приведены сведения о нахождении в природе,
образовании в различных случаях (В. — Bildung), способы получения (Darst. — Darstel-
lung), физические и химические свойства, физиологические свойства,
аналитическая характеристика. Далее рассматриваются производные, как указано выше.
Все сведения сопровождаются ссылками на оригинальную литературу. Названия
журналов сокращены иногда до одной буквы. В первом томе приведена сводная
таблица использованных журналов.
Предметный указатель составлен на основе вышедших из употребления
правил, поэтому удобнее пользоваться формульным указателем. В формульном
указателе к основным томам и первому дополнению использована система
Рихтера: соединения располагаются в порядке увеличения числа атомов углерода.
Соединения с одинаковым числом атомов углерода подразделяются на группы в
зависимости от числа присутствующих в молекуле элементов. Так, ацетон
(молекулярная формула С3Нб0) относится к группе 3 II (три атома углерода и атомы
двух других элементов). Внутри каждой группы формулы располагаются в
порддке увеличения числа атомов водорода, а затем по мере появления других
элементов (в следующем порядке О, N, Cl, Br, I, F, S, Р; далее — остальные в
алфавитном порядке). Под каждой молекулярной формулой приводятся названия
веществ и страница основного издания. В дополнительных томах применяется
двойная нумерация страниц: в верхнем правом углу указаны страницы данного
тома, наверху в середине — соответствующие страницы основного тома.
В новом формульном указателе использована система Хнлла. Молекулярные
формулы располагаются по числу атомов углерода, при определенном числе
атомов углерода — по числу атомов водорода, далее — других элементов в
алфавитном порядке их символов.
Словарь органических соединений/Под ред. И. Хейльброна и X. М. Бэнбе-
рн. —М.: Издатинлит, 1949. Т. 1—3. Представляет собой воспроизведение (без
перевода) английского издания 1946 г. Соединения расположены в алфавитном
порядке, нефункциональные производные рассматриваются как самостоятельные
соединения. Приводятся названия, структурная формула, молекулярная формула,
молекулярная масса, краткие сведения о нахождении в природе, физические и
химические свойства, методы определения, важнейшие производные (приведены
точки плавления и кипения). Литературные ссылки ие нумерованы. Всего
рассмотрено около 80 000 соединений.
Нефтепродукты. Свойства, качества, применение: Справочник/Под ред.
Б. В. Лоснковя. —М.: Химия, 1966. Рассмотрены топлива, масла, присадки,
смазки.
Поверхностно-активные вещества. Справочник/Под ред. А. А. Абрамзоиа и
Г. М. Гаевого. —Л.: Химия, 1979. Включает свойства индивидуальных ПАВ и
методы их оценки, а также состав, свойства, основные области применения.
Приведена обширная библиография.
6-290 161
Энциклопедия полимеров/Под ред. В. А. Каргнна. —М: Советская
энциклопедия, 1972—1977. Т. 1—3. Содержит обширный материал по химии, физико-
химии, механике и технологии полимеров и полимерных материалов.
Справочник по химии полимеров. — Киев: Наукова думка, 1971.
Справочник по пластическим массам/Под ред. В. М. Катаева, В. А. Попова,
Б. И. Сажнна. — М.: Химия, 1975. Т. 1, 2. В томе 1 рассмотрены полимеризаци-
онные полимеры. Специальные главы посвящены клеям, пластификаторам,
антистатикам и стабилизаторам. Том 2-й содержит описание пластмасс из гетероцеп-
ных полимеров, эфиров, целлюлозы, пенопластов, стеклопластиков. Описаны
токсикология пластмасс, методы испытаний, а также имеется предметный указатель.
Бондарь К. Е., Ершов Б. Л., Соломенно М. Г. Полимерные строительные
материалы. Справочное пособие. — М.: СтроЙиздат, 1974. Рассмотрены общие
сведения о полимерах строительных материалов, материалы для полов;
полимерные отделочные материалы; тепло- и звукоизоляционные полимерные
материалы; кровельные, гидроизоляционные и герметизирующие материалы;
конструкционные полимерные материалы; антикоррозионные полимерные материалы;
синтетические клеи и мастики.
Смыслова Р. А., Котлнрова С. В. Справочное пособие по
герметизирующим материалам на основе каучуков. —М: Химия, 1976. Описано около 60
товарных герметиков (в том числе строительного назначения).
Леснер И. М., Бермнн А. И., Словачевская Н. М. Указатель
препаративных синтезов органических соединении. —Л.: Химия, 1973. Составлен на основе
обработки более 200 книг, содержащих описание препаративных синтезов,
вышедших к 1970 г. (и частично в 1971 г.) на русском языке. Содержит ссылки по
методам синтеза более 17 000 соединений.
Г. Реферативные журналы
Реферативные журналы в виде кратких аннотаций (рефератов) отражают
содержание вСех (или большей части) публикаций в данной отрасли знаний.
В настоящее время в мире издаются два основных реферативных журнала по
химии: реферативный журнал «Химия» (Россия) и «Chemical Abstracts» (США).
Старейший реферативный журнал «Chemisches Zentralblatt», издававшийся с 1830 г.,
в 1970 г. прекратил существование. Другие реферативные издания иосят частный
характер.
Реферативный журнал «Химии» (РЖХим) представляет собой один из
крупнейших реферативных журналов, издаваемых Всероссийским институтом
научной и технической информации (ВИНИТИ). РЖХим издается с 1953 г.
Ежегодно вьтускаются 24 сводных тома (в 1953 г. — шесть). В состав сводного тома
входят 14 выпусков, которые издаются также отдельными тетрадями. Крупные
разделы сводного тома обозначены буквами русского алфавита: А. Общие
вопросы химии. Б. Физическая химия. В. Неорганическая химия. Комплексные
соединения. Г. Аналитическая химия. Д. Оборудование лабораторий. Е. Природные
органические соединения и их синтетические аналоги. Ж. Органическая химия.
И. Общие вопросы химической технологии. Л. Технология неорганических
веществ. М. Силикатные материалы. Н. Технология органических веществ. О.
Технология органических лекарственных веществ, ветеринарных препаратов и пес-
162
тицидов. П. Химия н технология пищевых продуктов, поверхностно-активных
материалов и душистых веществ. С. Химия высокомолекулярных соединений.
Т. Технология полимерных материалов.
Отдельный выпуск «К. Коррозия и защита от коррозии» в сводный том ие
входит.
Эти разделы разбиты на более мелкие, причем внутри них материал также
располагается в строго определенном порядке. Подробная рубрикация
приводится в первом номере РЖХим за каждый год.
В реферате вслед за его порядковым номером приводятся название работы
на русском языке, фамилии авторов, название работы на языке оригинала, в
скобках — язык, на котором опубликованы работа и резюме (для патентов не
приводятся), сокращенное название журнала, год, том, номер и страницы. Далее
следуют текст реферата (аннотация) и фамилия референта. (Перед рефератом
приводится его индексация по УДК, во многих случаях индексы УДК даны для всего
раздела.) До 1961 г. применялась сквозная нумерация рефератов на протяжении
всего года, сейчас каждый буквенный раздел имеет свою нумерацию. Ссылка
«РЖХим, 1980, 5Ж128» означает, что реферат опубликован в РЖХим за 1980 г., в
№ 5, в разделе «Органическая химия», порядковый номер реферата 128.
Поиск литературы осуществляется систематическим просмотром
интересующих исследователя разделов в новых номерах РЖХим, а за прошлые годы с
помощью годовых указателей. Имеются авторский, патентный, предметный и
формульный указатели. В третьей части патентного указателя приводятся
библиографические описания патентов по определенным темам (и номера
рефератов). В предметном указателе приводятся различные термины (методы
исследования, классы веществ по определенным признакам, процессы и т. д.). Для поиска
химических веществ удобно пользоваться формульным указателем. Он построен
по системе Хилла. После молекулярной формулы даются названия веществ,
соответствующих этой формуле, по номенклатуре ИЮПАК, суть информации (1—3
слова) и ссылка на реферат. Сведения о солях органических оснований и
производных, применяемых для идентификации (гидразоны, пикраты и т. д.),
приводятся при молекулярной формуле основного соединения. Соли органической
кислоты приведены и при формуле кислоты, и самостоятельно (кроме солей
щелочных и щелочно-земельных металлов, а также солей аммония).
К каждому номеру прилагаются свои авторский и предметный указатели (с
1972 г.).
Chemical Abstracts (Chem. Abstr., Ch. А., С. А.) выпускается
Американским химическим обществом с 1907 г. Журнал издается еженедельно. В нечетных
номерах публикуются рефераты по биологической и органической химии, в четных
— по высокомолекулярным соединениям, прикладной химии, физической и
аналитической химии. Весь материал разделен на 80 рубрик.
Реферат выглядит следующим образом: номер реферата (нумерация
сквозная), название публикации, авторы, место выполнения работы, название журнала,
год, том, номер выпуска (в скобках), язык публикации, текст реферата.
В С. А. очень развита система указателей. Указатели имеются к каждому
выпуску журнала; полугодовые (к каждому тому) и сводные — до 1965 г.,
десятилетние (Decennial Index), с 1966 г. — за пять лет. Имеется также формульный
указатель за 25 лет — с 1922 по 1946 г. Классические указатели — авторский,
163
патентный, предметный и формульный — составляются как для каждого тома,
так и для каждого выпуска. С 1972 г. (т. 76) предметный указатель разделен на
два — общий предметный н указатель химических соединений. Формульный
указатель имеет меньшее значение, чем в РЖХим. В нем никаких сведений,
кроме молекулярной формулы, не приводится. В случае очень распространенных
соединений дается название и отсылка к указателю химических соединений.
Имеется также ряд других указателей.
IV. Методики приготовления реактивов и препаратов
для лабораторных работ по органической химии
(в помощь лаборанту)
Аммиачный раствор оксида медн (I). К раствору 1 г медного купороса в
50 мл воды прибавляют 4 мл концентрированного раствора аммиака, затем
раствор 3 г солянокислого гидроксиламина в 50 мл воды н хорошо перемешивают.
Полученный раствор можно хранить несколько дней в хорошо закрытой склянке
(в темноте). Срок хранения можно увеличить, добавив в раствор медную стружку.
Раствор можно приготовить другим способом. Для этого предварительно
готовят два раствора: а) 1,5 г хлорида меди (П) и 3 г хлорида аммония растворяют в
20 мл концентрированного раствора аммиака и добавляют воды до общего
объема 50 мл; б) 5 г солянокислого гидроксиламина растворяют в 50 мл воды. Перед
употреблением растворы (а) и (б) смешивают в объемном соотношении 1 : 2.
Аммиачный раствор оксида серебра. К 100 мл 0,1 н. раствора нитрата
серебра добавляют 0,5 мл 25%-ного раствора аммиака. Полученный раствор хранят
в темной склянке не более 1 месяца. По истечении этого срока реактив, если он
полностью не израсходован, осторожно нейтрализуют азотной кислотой и
готовят новую порцию. Нельзя допускать испарения реактива и готовить его впрок в
больших количествах.
Аннлнн. Для очистки перегоняют (т. кип. 184 °С) с воздушным
холодильником из колбы Вюрца, в которую добавляют 0,5—1 г цинковой пыли.
Полученную бесцветную жидкость со слабым запахом хранят в плотно закрытой склянке
(желательно заполненной почти до пробки). Добавка небольшого количества
цинковой пыли стабилизирует анилин.
Анилиновая вода. Для проведения опытов по экстракции анилина его
перемешивают с водой в объемном соотношении 1 : 20 и оставляют, не разделяя
слон. Для исследования химических свойств анилина достаточно его растворить в
воде в объемном соотношении 1 : 100.
Бензол. Бензол марки ч. д. а. достаточно чист для большинства работ. Для
удаления следов воды бензол перегоняют, отбрасывая первые 10—20%
дистиллята. Т. кип. 80,2 °С.
Бром, раствор в хлороформе нлн тетрахлорнде углерода. В склянку с
притертой пробкой вносят 5 мл брома (под тягой!) и осторожно добавляют 95 мл
растворителя. Склянку хранят в эксикаторе. Работать с бромом нужно в
резиновых перчатках и защитных очках. Случайно разлившийся бром засыпают
сульфитом или тиосульфатом натрия.
164
Бромнд-броматнан смесь. 2,8 г бромата калия в 12 г бромида калия
растворяют в воде и доводят объем до 1 л (в мерной колбе).
Бромоводородная кислота. Раствор, образующийся при поглощении бро-
моводорода, перегоняют с дефлегматором, собирая фракцию, кипящую при 122—
126 °С. Эта фракция (р 1,47—1,49) представляет собой примерно 47%-ную бро-
моводородную кислоту. При перегонке сильно разбавленных растворов может
получаться кислота меньшей плотности, которую при необходимости повышают
повторной перегонкой. Кислоту хранят в хорошо закупоренной склянке в темноте.
Бромная вода. Для получения насыщенного раствора брома в воде в
склянку с притертой пробкой вместимостью 1 л вносят 6 мл брома, добавляют 500 мл
воды и энергично перемешивают. Небольшое количество брома при этом
остается нерастворенным (при длительном хранении он исчезает). Для опытов
используется раствор, разбавленный водой (1 : 2). Необходимо соблюдать меры
предосторожности.
Глюкоза, 5%-ный раствор. Готовят не менее чем за 12 ч до начала работы
с раствором. Раствор необходимо прокипятить.
2,4-Днинтрофеннлгндразнн солянокислый. 0,2 г 2,4-динитрофенилгидра-
зина растворяют при нагревании на водяной бане в 100 мл 2 н. раствора соляной
кислоты.
Железа (III) хлорид, 0,1 н. раствор. 9 г FeCl3-6H2O растворяют в 1 л воды.
При помутнении добавляют несколько капель концентрированной соляной
кислоты. Банку с кристаллическим хлоридом железа необходимо снова залить
парафином.
Известь натронная. Насыщенный раствор гидроксида натрия
перемешивают с негашеной известью (массовое отношение 2:1), выпаривают досуха в
железном сосуде, затем прокаливают при температуре около 500 °С и измельчают.
Препарат хранят в плотно закупоренных банках. Готовую натронную известь
перед использованием высушивают при ПО—115 °С (при значительном
содержании карбонатов прокаливают в муфельной печи).
Иода раствор в ноднде калня. 6 г иодида калия растворяют в 6 мл воды,
размешивают в этом растворе 2 г иода и разбавляют водой до 100 мл.
Известковая вода. Гашеную известь (гидроксид кальция) перемешивают с
водой. Хранят вместе с нерастворенным остатком, чтобы раствор был насыщен
гидроксидом кальция. Перед использованием прозрачный раствор сливают с
осадка в небольшие склянки.
Иодкрахмальная бумага. 0,5 г крахмала тщательно перемешивают с 10 мл
холодной воды. Полученную массу вливают в 100 мл кипящей воды и
продолжают кипячение до образования однородного клейстера. После охлаждения
добавляют 0,5 г иодида калия и 0,5 г кристаллического карбоната натрия,
растворенных в небольшом количестве воды. Полученным раствором пропитывают
фильтровальную бумагу и сушат ее на воздухе, избегая попадания солнечных лучей.
Высушенную бумагу режут на полоски и хранят в плотно закрытых банках.
Калия гидрокснд, 0,5 н. спиртовой раствор. Этиловый спирт кипятят 30 мин с
гидроксидом калия (10 г КОН в 1 л спирта) в колбе с обратным холодильником, а
потом спирт перегоняют (над той же щелочью). Затем в этот спирт при
перемешивании добавляют 30—35 г гидроксида калия и через сутки быстро сливают
прозрачную жидкость с осадка. Раствор хранят в темной склянке с хлоркальцие-
вой трубкой, заполненной натронной известью.
165
К алия карбонат (поташ). Перед использованием прокаливают при 200 °С.
Калня перманганат, кислый раствор. К 100 мл 1%-иого раствора перман-
ганата калия осторожно при перемешивании добавляют 5 мл концентрированной
серной кислоты.
Кальция карбид* Технический продукт разбивают молотком на кусочки
размером около 3—5 мм. Продукт обычно хранят в банке, залитой парафином.
Кальция хлорид. Отработанный хлорид кальция регенерируют, расплавляя
его на железном противие. Слой должен быть не толще 2 см (пары воды,
испаряясь, вызывают разбрызгивание, особенно большое, если слой соли толстый).
После испарения всей воды нагревание продолжают еще некоторое время (при
температуре 250—300 °С). Спекшуюся соль разбивают на куски и еще теплой кладут
в сухую банку. Для заполнения хлоркальциевых трубок используют кусочки
размером 2—5 мм.
Конго красный. Готовят 0,1%-ный раствор в воде.
Крахмальный клейстер. 2 г растворимого крахмала смешивают с 10 мл
холодной воды и вливают эту кашицу в 100 мл кипящей воды. Кипятят 5—10 мин,
охлаждают, дают отстояться и, если образуется хлопьевидный осадок, сливают с
него раствор (или фильтруют). Для стабилизации добавляют 0,1 г КОН или
салициловой кислоты (можно также 0,01 г иодида ртути).
Лактоза, 1%-ный раствор. После приготовления кипятят несколько минут.
Медн сульфат (безводный). Медный купорос нагревают в фарфоровой или
никелевой чаше на песчаной бане при постоянном перемешивании. Если
температура не превышает 220 °С, получается белый или слегка желтоватый продукт
(при нагревании свыше 220 °С сульфат меди частично разлагается, приобретая
серый цает; при этом осушающая способность понижается). Высушенный
сульфат меди растирают в ступке и хранят в сухой, плотно закрытой банке.
Медн (I) хлорид. Реактив можно приготовить двумя способами: а) 25 г
медного купороса и 65 г хлорида натрия растворяют при нагревании в 80 мл воды,
фильтруют, а затем к фильтрату добавляют раствор 7 г кристаллического
сульфита натрия (или 3,5 г безводного) в 40 мл воды. С выпавшего осадка сливают воду
и несколько раз промывают (декантацией) 2 н. раствором соляной кислоты до
исчезновения сульфат-иона (проба с хлоридом бария), а затем спиртом. Соль
сушат при 100—ПО °С и хранят в плотно закрытых банках. Препарат очень
чувствителен к окислению. Позеленевший препарат, содержащий основные соли
меди (П), вновь светлеет при промывке раствором 0,2 н. соляной кислоты;
б) к насыщенному раствору хлорида меди (П) добавляют кусочки меди. В
склянке, которую оставляют плотно закрытой на несколько дней, постепенно
выпадает белый осадок. Металлическую медь выбирают пластмассовым
пинцетом, а осадок обрабатывают, как описано выше.
Метиловый оранжевый. 0,1 г индикатора растворяют в 100 мл воды.
Натрий металлический. Банку с натрием распаивают с помощью
паяльника, натрий режут на куски (работать в резиновых перчатках!) и помещают их в
банку с керосином или вазелиновым маслом (можно также использовать ксилол,
декалин, тетралин). Для работы куски натрия извлекают из-под керосина
пинцетом, быстро осушают фильтровальной бумагой и сухим ножом отрезают кусочек
нужной величины. Остальную часть снова помещают в банку. С отрезанного
кусочка тщательно удаляют фильтровальной бумагой следы керосина, срезают тон-
166
кие корочки, а обрезки помещают в банку с натрием. Брать натрий нужно только
пинцетом. Ни в коем случае нельзя работать с натрием поблизости от
водопроводного крана.
Натрия ацетат (безводный). Кристаллическую соль CH3COONa-3H2O
нагревают в фарфоровой или никелевой чашке. Соль плавится в
кристаллизационной воде, а по испарении большей части воды застывает. Усиливая нагревание,
снова доводят соль до плавления (т. пл. 324 °С). При этом следует избегать
сильного перегрева (может начаться разложение). Расплавленную соль выливают на
металлическую пластинку с загнутыми краями и после остывания (теплую)
измельчают в фарфоровой ступке. Хранят в плотно закрытой банке.
Натрия гидросульфит (натрия бисульфит) NaHSO3. Гидрокарбонат или
кристаллический карбонат натрия заливают водой так, чтобы она чуть покрывала
кристаллы, и пропускают диоксид серы до почти полного растворения
кристаллов. Полученный зеленовато-желтый раствор хранят в склянке с притертой
пробкой. Диоксид серы получают, добавляя по каплям серную кислоту к сульфиту
натрия в колбе Вюрца с высоким отводом. В отвод желательно вложить
стекловату для задержки уносимых током газа следов жидкости. В лабораториях бывает
гидросульфит натрия технический — белый слеживающийся порошок со слабым
запахом сернистого ангидрида. Он обладает сильными восстанавливающими
свойствами даже после длительного хранения, но истинным гидросульфитом не
является. Его формула Na2S2O4-2H2O, а правильное название дитионит натрия. С
ацетоном и альдегидами осадков ие образует.
Натрии гндрокснд, 2 и. раствор. 84 г едкого натра (из расчета, что в нем
95% основного вещества) растворяют при перемешивании в 500 мл воды и
доводят объем до 1 л.
Натрия сульфат (безводный). Реактив высушивают при 200—300 °С.
Возможна регенерация отработанного осушителя.
Нитрующая смесь. Смешивают 2 объема концентрированной азотной
кислоты (р 1,42 г/см3) с 3 объемами концентрированной серной кислоты
(р 1,84 г/см3), медленно добавляя серную кислоту к азотной при охлаждении и
перемешивании.
Сахароза, 1%-ный раствор. Растворяют 10 г сахарозы (или пищевого
сахара) в 1 л воды, фильтруют и кипятят несколько минут.
Серебра нитрат, 0,2 и. раствор. 3,4, г нитрата серебра растворяют в 20 мл
воды и доводят объем до 100 мл. Для приготовления раствора используется биди-
стиллят. Посуда должна быть очень тщательно отмыта от возможных
органических загрязнений. Хранят раствор в темной склянке с притертой пробкой.
Серебра остатки, регенерация. Все растворы и осадки, содержащие
серебро, подлежат сбору и переработке. Ацетиленид серебра и «серебряное зеркало»
растворяют в разбавленной азотной кислоте.
К остаткам серебра прибавляют раствор гидроксида натрия до щелочной
реакции по фенолфталеину, нагревают на водяной бане и добавляют формалин.
Через 5—15 мин выделяется темно-серый рыхлый порошок металлического
серебра. Его отсасывают, промывают водой до удаления щелочи и хлорид-ионов и
сушат при 100 °С. К навеске высушенного осадка прибавляют избыток азотной кислоты
(1 мл азотной кислоты плотностью 1,42 г/см3 или 1,5 мл плотностью 1,31 г/см3 на 1 г
серебра) и нагревают при 50 °С до растворения. Жидкость фильтруют и
упаривают на водяной бане до появления кристаллической пленки. Если за это время
167
оксвды азота не удалились полностью, добавляют воду и упаривают вновь.
Упаренный раствор охлаждают, кристаллы нитрата серебра отсасывают, промывают
небольшим количеством ледяной воды и сушат при 110 °С. Маточный раствор и
промывные воды можно упарить и выделить дополнительное количество нитрата
серебра или же слить в склянку с остатками серебра.
Серная кислота 2 н. 56 мл концентрированной серной кислоты (р 1,84 г/см3)
медленно прибавляют к 944 мл воды.
Соляная кислота 2 н. 167 мл концентрированной соляной кислоты (р 1,19 г/см3)
разбавляют водой до 1 л.
Уксусная кислота 2 н. 116 мл ледяной уксусной кислоты разбавляют водой
до 1 л.
Фелннгова жидкость. Готовят два раствора. Раствор I. 34,6 г чистого
перекристаллизованного сульфата меди растворяют в воде, содержащей
несколько капель серной кислоты, и разбавляют до 500 мл.
Раствор II. 173 г сегиетовой соли (тартрат натрия-калия) растворяют в
200 мл воды, добавляют раствор 70 г гидроксида натрия (или 85 г гидроксида
калия) в 100 мл воды и разбавляют до 500 мл.
Фелингову жидкость готовят непосредственно перед каждой лабораторной
работой, смешивая равные объемы растворов I и II (исходные растворы могут
храниться неограниченно долго).
Если отсутствует сегиетова соль, то для приготовления раствора II
растворяют 121 г гидроксида натрия и 93,1 г чистой винной кислоты в 400 мл воды и
доводят объем до 500 мл.
Фенолфталеин. 0,2 г фенолфталеина растворяют в 165 мл спирта и
добавляют 135 мл воды.
Фншера реактив. В сухую склянку из темного стекла вносят 200 мл
пиридина, растворяют в нем 100 г иода и добавляют 600 мл метанола. Склянку плотно
закрывают и оставляют на двое суток. Затем склянку взвешивают и помещают в
баню со льдом, а через раствор пропускают диоксид серы из баллона до
увеличения массы на 45—50 г. Приготовленный таким образом реактив выдерживают
перед использованием не менее недели. Хранят, хорошо защищая от влаги.
Фруктоза, 0,5%-ный раствор. После приготовления раствор кипятят.
Фурфурол. Соломенно-желтая жидкость, быстро темнеющая на воздухе.
Очищают перегонкой, собирая фракцию, кипящую при 159—163 °С (т. кип.
чистого фурфурола 162 °С). Окисление (потемнение) фурфурола замедляют
добавкой 0,1% гидрохинона или пирогаллола.
Хромовая смесь (смесь Бекмана). В 100 мл воды растворяют 20 г
дихромата калия и добавляют 10 мл концентрированиой серной кислоты. Такая
хромовая смесь (смесь Бекмана) предназначена для окисления спиртов и анилина и не
годится для мытья посуды.
Щавелевая кислота, 0,01 и. В мерной колбе вместимостью 1 л растворяют
в 100 мл воды 0,63 г свежеперекрнсталлизованной щавелевой кислоты. Затем
добавляют 1 мл 30%-ной H2SO4 и разбавляют водой до метки.
Швейцера реактив. К раствору 10 г медного купороса в 100—200 мл воды
добавляют 100 мл 2 н. раствора гидроксида натрия. Образовавшийся осадок
отсасывают, промывают несколько раз водой до отрицательной реакции на сульфат-
168
ион и растворяют в минимальном количестве 25%-но го раствора аммиака.
Часть гидр о кс ид а меди при этом должна остаться нерастворенной. Раствору
дают отстояться и сливают декантацией. Эмпирическая формула реактива
Эфир абсолютный. Эфир проверяют на наличие пероксидов, встряхивая
его с равным объемом 2%-ного раствора иодида калия, подкисленного
разбавленной соляной кислотой. Присутствие пероксидов определяют по синей окраске
водного слоя при добавлении раствора крахмала. (Подкисленный серной
кислотой раствор ванадата аммония с эфиром, содержащим пероксиды, окрашивается в
красный цает, а такой же раствор дихромата калия — в синий.)
Если пероксиды отсутствуют, приступают к осушке, если они есть, от них
избавляются встряхиванием раствора с порошкообразным гидроксидом калия
(70 г на 1 л).
После отстаивания эфир сливают, добавляют 100 г хлорида кальция и через
сутки фильтруют. Затем в эфир вносят около 5 г металлического натрия в виде
тонко нарезанных листочков или проволоки, выдавливаемой из пресса. Если
через 24 ч не наблюдается выделения пузырьков водорода, то осушка считается
законченной, если же водород выделяется, добавляют еще 2—3 г натрия.
Эфир можно перегнать на водяной бане над натрием, предохраняя его от
атмосферной влаги, но можно обойтись и без перегонки, лишь слив его в сухую
склянку. Склянку с эфиром закрывают корковой пробкой с хлоркальциевой
трубкой. Для предотвращения окисления можно внести несколько крупинок
дифениламина или фосфорного ангидрида, или, еще лучше, несколько гранул гидрокси-
да калия, который действует одновременно и как осушитель.
V. Характеристические частоты колебаний в ИК-спектрах
основных классов органических соединений
Соединение
Алкай ы
Частота
V, СМ"'
2975—2950
2885—2860
1470—1435
1385—1370
2940—2915
2870—2845
1480—1440
Интенсивность
с.
с.
ср.
с.
с.
с.
ср.
Отнесение и характер колебаний
Va»CH3
VjCHj
2830—2815вОСН3
2820—2730bNCH3
6**си2 расщепляется в дублет для
гемдимстильной группы
v,ch2
5СН2
169
Продолжение прилож. V
Соединение
Алкены
R—СН=СН2
RR'C=CH2
RCH=CHR' (цис)
RCH=CHR' (транс)
RR'C=CHR"
RR'C=CR"R"
Днены
С=С—Ar
С=С—С=О
Алкнны
RC^CH
RO=CR'
Ароматические
соединения
Монозамещенные
Частота
V, СМ"'
750—720
2900—2880
1340
3095—3010
2975
1645—1640
995—985
915—905
3095—3075
1660—1640
895—885
3040—3010
1665—1635
730—665
3040—3010
1675—1665
980—960
3040—3010
1675—1665
850—790
1690—1670
1650
1600
-1625
1660—1580
3310—3300
2140—2100
2260—2190
3080—3030
1625—1575
1525—1475
1465—1440
1590—1575
770—730
710—690
Интенсивность
с.
ел.
ел.
ср.
ср.
ср.
с.
с.
ср.
ср.
с.
ср.
ср.
ср.
ср.
ср.
с.
ср.
п. и.
с.
ел.
с.
ел.
с.
с.
с.
ел.
ел.
ср.
п. и."]
п. и. >
п. и. J
п. н.
1}
Отнесение и характер колебаний
Маятниковое -СН2-
VCH
$сн
Va»CH2
ViCH2
vc-c
бсн неплоские
6СН неплоские
5сн2
vc-c
6СН неплоские
VCH
vc-c
бСн неплоские
VCH
vc-c
бСн неплоские
VCH
vc-c
бСн неплоские
vc-c
vc-c
vc-c
vc-c
vc-c
VCH,
vc-c
vc-c
VCH
Колебания кольца
Появляется в сопряженных
соединениях
5сн неплоские
170
Продолжение прилож. V
Соединение
1,2-Замещенные
1,3-Замещенные
1,4- н 1,2,3,4-
Замещенные
1,2,3-Замещенные
1,2,4-Замещенные
1,3,5-Замещенные
Спирты н фенолы
Альдегиды
R—СНО
С=С—СНО
Аг—СНО
Кетоны
R—CO—R'
С=С—СО—R
С=С—СО—С=С
Цнклопентаноны
Аг—СО—R
Аг—СО—Аг
CHal—CO—R
—СО—СНг—СО—
енольная форма
кетонная форма
Кислоты
R—СООН
Частота
V, СМ"'
770—735
900—860
810—750
725—680
860—800
800—770
720—685
860—800
900—860
900—860
865—810
730-^75
3670—3580
3550—3450
3400—3200
3590—3420
3200—2500
1740—1720
1705—1685
1715—1695
2880—2650
1725—1700
1695—1660
1670—1660
1750—1740
1700—1680
1670—1650
1745—1725
1640—1535
3200—2700
-1720
1725—1700
Интенсивность
с.
ср. "1
с.
ср. J
с.
ср. J
с. "1
ср. J
ср. 1
::;
п. и.
п. н.
с.
п. и.
ел.
с
с.
с.
ср.
с.
с
с.
с.
с.
с.
с.
с.
п. и.
с.
с.
Отнесение и характер колебаний
6Сн неплоские
бен неплоские
бен неплоские
бен неплоские
бен неплоские
5Сн неплоские
Voh» свободная группа ОН, узкая
полоса
Voh, межмолекулярная водородная
связь димеров, узкая полоса
Voh* межмолекулярная водородная
связь полиассоциатов, широкая
полоса
voh* внутримолекулярная
водородная связь, узкая полоса
voh» хелаты, очень широкая полоса
vc-o
vc-o
vc-o
vch. во всех альдегидах; могут
быть две полосы
vc-o
vc-o
vc-o
vc-o
vc-o
vc-o
vc-o
vc-o
voh
vc-o
vc-o
171
Продолжение прилож V
Соединение
С=С—СООН
Ar—COOH
CHal—COOH
Солн кислот
Сложные эфнры
R—COOR
С=С—COOR н
Ar—COOR
—СООС=Си
—СООАг
Ангидриды:
ациклические
насыщенные
ациклические
сопряженные
ангидриды с пяти-
членным циклом:
предельные
непредельные
Галогенангндрнды
R—CO—Hal
С=С—СО—Hal
Амиды кислот
R—CONH2
R—CONHR'
r—conr;
—NH—CO—NH—
—СО—NH—СО-
Частота
V, CM"'
1715—1680
1700—1680
1740—1715
3300—2500
1610—1550
1420—1300
1750-1735
1730—1715
1800—1770
1840—1800
1780—1740
1820—1780
1760—1720
1870—1830
1800—1760
1850—1810
1795—1740
1815—1785
1800—1770
3540—3200
1690—1650
1650—1590
3460—3100
1680—1630
1570—1510
1670—1630
1660
1790—1720
1720—1670
Интенсивность
с.
с.
с.
п. и.
с.
ср.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
п. и.
с.
с.
с.
с.
с.
с.
Отнесение и характер колебаний
vc-o
Vc-o
Vc-o
Voh> широкая, для всех кислот
votCOO-
v*coo-
vc-o
Vc-o
vc-o
VorC-O
v,c-o
VorOO
v,c-o
Vc-o
Vc-o
Vc-o
Vc-o
vc-o
Vc-o
vnh2 » Д^ полосы в разбавленных
растворах
Полоса «Амид I»
Полоса «Амид II»
Vnh, одна полоса в разбавленных
растворах
Полоса «Амид I»
Полоса «Амид II»
Полоса «Амид I»
Полоса «Амид I»
172
Продолжение прилож V
Соединение
Амнны
R—NH2
R2NH
R—NH3
r2i5h2
R3NH
Непредельные
азотсодержащие
соединения
R2C=N
C=C—C=N
C=N в цикле
R—C-N
C==C—CsN
Ar— CsN
Ar—N=N
Ннтросоедн нения
R—NO2
C=C—NO2
Ar—NO2
Частота
v, см"1
3500—3300
1650—1580
900—650
3600—3300
1650—1550
3350—3150
-1600
-1300
2700—2250
1620—1560
2700—2250
1690—1635
1665—1630
1660—1480
2260—2240
2235—2215
2240—2220
2300—2230
1565—1545
1385—1360
1530—1510
1360—1335
1550^1510
1365—1335
860—840
-750
Интенсивность
п. и.
п. и.
ср.
п. и.
ел.
ср.
ср.
ср.
с.
ср.
ср.
п. и.
п. и.
п. и.
п. н.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
с.
Отнесение и характер колебаний
vnh2 B разбавленных растворах
две полосы
o*nh2 , иеплоские
Ьт , плоские
vnh ' в разбавленных растворах
одна полоса
&NH
vNH}, широкая полоса или группа
полос
6*NH3
VNH2
5NH2
Vnh
Von
Von
Von
Von
Von
Von
VCN2
Vo,N02
j NO2
us NO2
V,NO2
VoiN02
VjNO2
t
173
Продолжение прилож V
Соединение
R—О—NO2
Серосодержащие
соединения
R—SH
R2SO
R2SO2
R—SO3H
■p ел "MU.
л—О ч^2^-^2
Гетероциклические
соединения
фуран
тиофен
пиррол, индол
пиридин
Кремннйорганнче-
скне соединения
Si—Н
Частота
V, СМ"1
1655—1610
1300—1255
2590—2550
1070—1030
1350—1300
1160—1120
1260—1150
1080—1010
700—600
1370—1300
1180—1140
3175—3137
3137—3112
1615—1639
1500
1274—1253
1109—1092
880—859
842—812
3125—3050
1520
1040
750—640
3440—3400
1565
1500
3070—3020
900—670
1650—1580
1580—1550
1570—1480
-2100
Интенсивность*
с.
с.
ел.
с.
04. С.
04. С.
С.
с.
с.
с.
с.
с. "
с.
с. =
С. 1
<■}
ml*
04. С.
ср.
IL И. "|
п. и. Г
J
ft
ср. 1
ел. 1
ср. J
ft
Отнесение и характер колебаний
аз NO 2
S NOj
VSH
vso
ViSO2
V«rSO2
V,SO2
V«SO2
vjS02
>
VCH
Колебания кольца
vc-o
Наиболее характерны для
фуранов
VCH
Колебания кольца
Самая сильная из полос тиофена
Vnh в растворах
Колебания кольца
vch и 5сн
Колебания кольца
VSi—H
174
Продолжение прилож. V
Соединение
Si—CH3
Si(CH3)2
Si(CH3)3
Si-СбН,
Si—О—Si
Si—0—C—
Si—Cl
Si—OH
Частота
Y, см'1
1260
800
1260
815—800
1250
840
755
1430—1425
1135—1090
1090—1020
810
3690
Интенсивность*
с.
с.
с.
с.
с.
с.
с.
с. \
с. J
с.
с.
Отнесение и характер колебаний
5сн,
5сн3
5сн2
vSi—О
VOH
* с.—сильная; ср.—средняя; ел.—слабая; п. и.—переменной интенсивности.
VI. Химические сдвиги протонов различных типов
Тип протона
—С^СН
—С=СН2
—С=СН—R
—С=СН—COR
(альдегиды, кетоны, кислоты, эфиры)
—СН=С—OR
—С=СН—OR
Ar—C=CHR
Ar—CH=C—COR
(альдегиды, кетоны, кислоты, эфиры)
Ar—C=CH—COR
(альдегиды, кетоны, кислоты, эфиры)
CeHjHal
C6H5NR2
QHsOR
сдатоз
слет
R—СНО
Ar—CHO
R—СООН
Химический сдвиг
5, м. д.
2,3-2,9
4,6
5,1
5,7—6,0
4,5-5,5
6,2
5,3-5,4
7,4-7,7
6,3—7,0
7,1-7,3
6,5
6,6—7,1
6,8
7,7
7,5
9,6—9,8
9,6—10,1
8,0—11
175
Продолжение прилож VI
Тип протона
Ar—CH2—Hal
Hal—CH2—COR
(альдегиды, кетоны, кислоты, эфиры)
Ar—CH2—NR2
Ar—CH2—OR
Ar—CH2—OCOR
Ar—CH2—COR
(альдегиды, кетоны, кислоты, эфиры)
R—CO—CH2—COR
(альдегиды, кетоны, кислоты, эфиры)
Химический сдвиг
5, м. д.
4,4—4,5
4,1—4,5
3,1
4,5
5,3
3,6—3,8
3,3—3,6
VII. Осушители и их применение
Осушитель
Окснд фосфора (V)
Серная кислота
Гидроксид калия,
гидроксид натрия
Карбонат калия
Металлический
натрий
Хлорид кальция
Сульфат магния,
сульфат натрия
Можно сушить
Нейтральные и кислые
газы, углеводороды и их гало-
генопроизводные, растворы
кислот
Нейтральные и кислые газы
Аммиак, амины, простые
эфиры, углеводороды
Амины, кетоны
Углеводороды, третичные
амины, простые эфиры
Углеводороды, простые
эфиры, нейтральные газы,
галогенопроизводные
углеводородов, нитросоедине-
ния
Углеводороды и их галоге-
иопроизводные, альдегиды,
кетоны, кислоты, сложные
эфиры, простые эфиры,
нитросоединения
Нельзя сушить
Основания, спирты,
простые эфиры, хлоро-
водород, фтороводород
Ненасыщенные
углеводороды, спирты, кетоны,
основания
Альдегиды, кетоны
Вещества с кислотными
свойствами
Хлорпроизводные
углеводородов (возможен
взрыв!), спирты, кислоты
Спирты, аммиак,
альдегиды, кетоны
Спирты, аммиак,
альдегиды, кетоны
176
УШ. Плотность и концентрация водных растворов серной,
азотной н соляной кислот
d20
1,000
1,005
1,010
1,015
1,020
1,025
1,030
1,035
1,040
1,045
1,050
1,055
1,060
1,065
1,070
1,075
1,080
1,085
1,090
1,095
1,100
1,105
1,110
1,115
1,120
1,125
1,130
1,135
1,140
1,145
1,150
]
:
1,155
1,160
1,165
1,170
1,175
1,180
1,185
1,190
1,195
Концентрация, %
d20
Серная
0,2609
0,9855
1,731
2,485
3,242
4,000
4,746
5,493
6,237
6,956
7,704
8,415
9,129
9,843
10,56
11,26
11,96
12,66
13,36
14,04
14,73
15,41
16,08
16,76
17,43
18,09
18,76
19,42
20,08
20,73
21,38
22,03
22,67
23,31
23,95
24,58
25,21
25,84
26,47
27,10
1,245
1,250
' 1,255
1,260
1,265
1,270
1,275
1,280
1,285
1,290
1,295
1,300
1,305
1,310
1,315
]
]
]
]
]
1,320
1,325
1,330
1,335
1,340
1,345
1,350
1,355
1,360
1,365
1,370
1,375
1,380
1,385
1,390
1,395
1,400
1,405
1,410
1,415
1,420
1,425
1,430
1,435
1,440
Концентрация, %
d20
кислота
33,22
33,82
34,42
35,01
35,60
36,19
36,78
37,36
37,95
38,53
39,10
39,68
40,25
40,82
41,39
41,95
42,51
43,07
43,62
44,17
44,72
45,26
45,80
46,33
46,86
47,39
47,92
48,45
48,97
49,48
49,99
50,50
51,01
51,52
52,02
52,51
53,01
53,50
54,00
54,49
1,490
1,495
1,500
1,505
1,510
1,515
1,520
1,525
1,530
1,535
1,540
1,545
1,550
1,555
1,560
1,565
1,570
1,575
1,580
]
]
]
]
]
1,585
1,590
1,595
1,600
1,605
1,610
1,615
1,620
1,625
1,630
1,635
1,640
1,645
1,650
1,655
1,660
1,665
1,670
1,675
1,680
1,685
Концентрация^
59,24
59,70
60,17
60,62
61,08
61,54
62,00
62,45
62,91
63,36
63,81
64,26
64,71
65,15
65,59
66,03
66,47
66,91
67,35
67,79
68,23
68,66
69,09
69,53
69,96
70,39
70,82
71,25
71,67
72,09
72,52
72,95
73,37
73,80
74,22
74,64
75,07
v 75,49
75,92
76,34
177
Продолжение прилож. VIII
Л 20
4
1,200
1,205
1,210
U15
U20
1,225
U30
U35
1,240
1,735
]
]
]
]
]
1,740
1,745
1,750
1,755
1,760
1,765
1,770
1,775
1,780
1,785
1,000
1,005
1,010
1,015
1,020
1,025
1,030
1,035
1,040
1,045
1,050
1,055
1,060
1,065
1,070
1,075
1,080
1,085
1,090
1,095
1,100
1,105
Концентрация, %
27,72
28,33
28,95
29,57
30,18
30,79
31,40
32,01
32,61
80,70
81,16
81,62
82,09
82,57
83,06
83,57
84,08
84,61
85,16
85,74
0,3333
U55
2,164
3,073
3,982
4,883
5,784
6,661
7,530
8,398
9,259
10,12
10,97
11,81
12,65
13,48
14,31
15,13
15,95
16,76
17,58
18,39
di
4
1,445
1,450
1,455
1,460
1,465
1,470
1,475
1,480
1,485
1,790
1,795
1,800
1,805
1,810
1,815
1,820
1,821
1,822
1,823
1,824
Азотная
1,190
1,195
uoo
1,205
1,210
U15
1,220
1,225
1,230
1,235
1,240
U45
1,250
1,255
U60
1,265
1,270
U75
1,280
U85
1,290
U95
Концентрация, %
54,97
55,45
55,93
56,41
56,89
57,36
57,84
58,31
58,78
86,35
86,99
87,69
88,43
89,23
90,12
91,11
91,33
91,56
91,78
92,00
Л 20
«Л
4
1,690
1,695
]
]
]
]
]
]
]
кислота
31,47
32,21
32,94
33,68
34,41
35,16
35,93
36,70
37,48
38,25
39,02
39,80
40,58
41,36
42,14
42,92
43,70
44,48
45,27
46,06
46,85
47,63
]
1,700
1,705
1,710
1,715
1,720
1,725
1,730
1,825
1,826
1,827
1,828
1,829
1,830
1,831
1,832
,833
1,834
1,835
1,380
1,385
1,390
1,395
1,400
1,405
1,410
1,415
1,420
1,425
1,430
1,435
1,440
1,445
1,450
1,455
1,460
1,465
1,470
1,475
1,480
1,485
Концентрация, %
76,77
77,20
77,63
78,06
78,49
78,93
79,37
79,81
80,25
92,25
92,51
92,77
93,03
93,33
93,64
93,94
94,32
94,72
95,12
95,72
62,70
, 63,72
64,74
65,84
66,97
68,10
69,23
70,39
71,63
72,86
74,09
75,35
76,71
78,07
79,43
80,88
82,39
83,91
85,50
87,29
89,07
91,13
178
Л2О
«4
1,110
1,115
1,120
1,125
1,130
1,135
1,140
1,145
1,150
1,155
1,160
1,165
1,170
1,175
1,180
1,185
1,000
1,005
1,010
1,015
1,020
1,025
1,030
1,035
1,040
1,045
1,050
1,055
1,060
1,065
Концентрация, %
19,19
20,00
20,79
21,59
22,38
23,16
23,94
24,71
25,48
26,24
27,00
27,76
28,51
29,25
30,00
30,74
d»
1,300
1,305
1,310
]
]
]
]
]
]
]
0,3600
1,360
2,364
3,374
4,388
5,408
6,433
7,464
8,490
9,510
10,52
11,52
12,51
13,50
]
1,315
1,320
1,325
1,330
1,335
1,340
1,345
1,350
1,355
1,360
1,365
1,370
1,375
Соляная
1,070
1,075
1,080
1,085
1,090
1,095
1,100
1,105
1,110
1,115
1,120
1,125
1,130
1,135
Концентрация^
48,42
49,21
50,00
50,85
51,71
52,56
53,41
54,27
55,13
56,04
56,95
57,87
58,78
59,69
60,67
61,69
кислота
14,49
15,48
16,47
17,45
18,43
19,41
20,39
21,36
22,33
23,29
24,25
25,22
26,20
27,18
Продоллсение
"4
1,490
1,495
1,500
1,501
1,502
1,503
1,504
1,505
1,506
1,507
1,508
1,509
1,510
1,511
1,512
1,513
1,140
1,145
1,150
]
]
]
]
]
]
]
1,155
1,160
1,165
1,170
1,175
1,180
1,185
1,190
1,195
1,198
прилож. VIII
Концентрация,"^
93,49
95,46
96,73
96,98
97,23
97,49
97,74
97,99
98,25
98,50
98,76
99,01
99,26
99,52
99,77
100,00
28,18
29,17
30,14
31,14
32,14
33,16
34,18
35,20
36,23
37,27
38,32
39,37
40,00
IX. Физические константы некоторых растворителей
Растворитель
Ацетонитрил
Ацетон
Т.пл.,°С
^43,8
-94,7
Т. кип. (при
101 кПа, или
760ммрт.
ст.)
81,6
, 56,3
е при 25 °С
37,5
20,7
л20
«4
0,7822
0,7900
179
Продолжение прилож IX
Растворитель
Анизол
Бензиловый спирт
Буганол
Бромбензол
Бензол
Вода
Гексан
Диэтиленгликоль
Диметилсульфоксид
Диметилформамид
N.N-Диметилацетамид
1,2- Дихлорэтан
1,4-Диоксан
Дифениловый эфир
Диэтиловый эфир
Диизопропиловый эфир
Дибутиловый эфир
Изоамиловый спирт
Иодбензол
Метанол
N-Метилформамид
Метилцеллозольв
N-Метилацетамид
Нитрометан
Нитробензол
Пропанол-1
Пропанол-2
Пиридин
Сероуглерод
Тетраметиленсульфон
(сульфолан)
Тетрагидрофуран
Толуол
Триэтиламин
Уксусная кислота
Формамид
Фторбензол
Хлористый метилен
Хлороформ
Хинолин
Хлорбензол
Т. пл., °С
-37.3
-15,5
-89,5
-30.6
5,5
0,0
-95.3
-6,5
18.54
-60,4
-20
-35,7
11,8
28
-116,3
-85,5
-98
-117,2
-31,4
-97,7
-3,8
-85
40
-28,6
5,8
-127
-88
-42
-112
28,45
-68
95
-115,3
16,66
2,55
-40
-96,8
-63,6
-15
-45,6
Т. кип. (при
101 кПа, или
760 мм рт.
ст.)
155
205
117,2
156,2
80,1
100
68,7
244,8
189
153
165
83,5
101,3
259
34,6
68,3
142,4
132
188,7
64,7
183
124
202
101,2
210,8
97,2
82,3
115,6
46,2
287,3
(разл.)
65,8
110,6
89,7
117,9
210,5
85
40
61,2
237,7
131,7
е при 25 °С
4,3
13,1
17,1
5,4
2,3
78,5
1,9
29,4
48,9
36,7
3,8
10,4
2,2
3,7
4,2
3,9
3,1
14,7
4,6
32,6
182,4
15,9
175,7
38,6
34,8
20,1
18,3
12,3
2,6
44
7,4
2,4
0,8
6,2
109,5
5,4
8,9
4,7
9,0
5,6
«4
0,9950
1,0450
0,8098
1,4950
0,8790
1,0000
0,6594
1,1164
1,0958(25)*
0,9487
0,9360(25)
1,2531
1,0338
1,0730
0,7134
0,7235
0,7688
0,8120
1,8154
0,7913
0,9961 (25)
0,9650
0,9420(40)
1,1382
1,2034
0,8040
0,7855
0,9820
1,2632
1,2614(30)
0,9892
0,8670
0,7293
1,0493
1,1334
1,0225
1,3360
1,4832
1,0950
1,1063
180
Продолжение прилож IX
Растворитель
Циклогексанои
Циклогексан
Тетрахлорид углерода
Этиленгликоль
Этанол (абсол.)
Этилацетат
Т. пл., °С
-16,4
-6,5
-23
-13
-114,1
-84
Т. кип. (при
101 кПа, или
760 мм рт.
ст.)
155,6
80,7
76,8
197,3
78,3
77,1
е при 25 °С
18,3
2,0
2,2
37,3
24,3
6,0
«4
0,9455
0,7786
1,5940
1,1135
0,7894
0,9006
* В скобках указана температура, при которой определялась относительная
плотность для некоторых растворителей.
X. Растворители, рекомеидуемые в качестве среды
для иекоторых химических реакций
Растворитель
Реакция
С4
3
Е
I
0Q
а
зз
•е-
А
и
0Q
а
о
S3
а.
С I
0Q
С4
а
Лнизол
Ацетон
Адетонитрил
Бензол
Вода
Дибутиловый
эфир
Диметил-
сульфоксид
Диметилфор-
мамид
Диоксан
Дихлорбензол
,2-Дихло-
»этан
181
Продолжение прилож. X
Растворитель
Циэтиловый
эфир
Метанол
Нитробензол
Нитрометан
ПетролеЙный
эфир
Пиридин
Серная
кислота
Сероуглерод
Сульфолан
Гетрагидро-
фуран
Тетрахлорид
углерода
Голуол
Грихлорэти-
аен
Уксусная
кислота
Хлористый
метилен
Хлороформ
Этанол
Этилацетат
3
со
Реакция
1
■е-
i
и
9>
S
а.
С
S о.
Я и
+
+
+
Оглавление
Предисловие 3
Раздел I. Общаи часть 5
1. Основные правила и организация работы в лаборатории
органической химии 5
1.1. Техника безопасности в лаборатории органической химии 5
1.2. Организация рабочего места в лаборатории и порядок
выполнения студентами лабораторных работ 8
1.3. Правила оформления и ведения рабочего (лабораторного)
журнала 10
2. Лабораторная химическая посуда и приборы 12
2.1. Перечень и краткое описание лабораторной посуды и
приборов 12
2.2. Требования к лабораторной посуде и работа с ней при
проведении химического эксперимента 19
2.3. Правила сборки установок для выполнения органических
синтезов 22
3. Методы очистки и выделения органических соединений 23
3.1. Фильтрование 23
3.2. Кристаллизация 25
3.3. Перегонка 26
3.4. Возгонка (сублимация) 31
3.5. Экстракция 32
3.6. Высушивание органических веществ 33
4. Определение основных физических констант органических
веществ 34
4.1. Определение температуры плавления 34
4.2. Определение температуры кипения 35
4.3. Определение относительной плотности жидкости 36
4.4. Определение показателя преломления 37
5. Общие представления об элементном органическом анализе 39
5.1. Качественный элементный анализ 39
5.2. Количественный элементный анализ 41
183
Раздел П. Ознакомительный (малый) практикум 45
Работа 1. Предельные углеводороды (алканы) 45
Контрольные вопросы 46
Работа 2. Непредельные углеводороды (алкены и алкины) 46
Контрольные вопросы 48
Работа 3. Галогеналкилы, спирты, простые эфиры 48
Контрольные вопросы 49
Работа 4. Альдегиды и кетоны 50
Контрольные вопросы 51
Работа 5. Карбоновые кислоты и их производные 51
Контрольные вопросы 52
Работа 6. Азотсодержащие соединения алифатического ряда 53
Контрольные вопросы 54
Работа 7. Соединения со смешанными функциями 54
Контрольные вопросы 55
Работа 8. Углеводы (сахара) 55
Контрольные вопросы 56
Работа 9. Кремнийорганические соединения 57
Контрольные вопросы 58
Работа 10. Ароматические углеводороды них
галогенопроизводные 58
Контрольные вопросы 59
Работа 11. Ароматические нитросоединения и сульфокислоты 59
Контрольные вопросы 60
Работа 12. Кислородсодержащие соединения ароматического ряда ...60
Контрольные вопросы 62
Работа 13. Ароматические амины, диазосоединения, азокрасители..„62
Контрольные вопросы 63
Работа 14. Многоядерные ароматические соединения 63
Контрольные вопросы 64
Работа 15. Гетероциклические соединения 65
Контрольные вопросы 66
Работа 16. Высокомолекулярные соединения 66
Контрольные вопросы 68
184
Раздел Ш. Синтезы органических соедииеиии 69
1.Галогенирование 69
Работа 1. 1,2-Дибромэтан „ 69
Работа 2.1-Бромнафталин 71
Работа 3. Бромистый изопропил 72
Работа 4. 4-Бромтолуол 72
2. Нитрование 73
Работа 5. Нитрометан 73
Работа 6. Нитробензол 74
Работа 7. 2- и 4-Нитрофенолы 75
3. Сульфирование 77
Работа 8. 4-Толуолсульфокислота , 77
Работа 9. 2-Нафталинсульфокислота 78
Работа 10. Сульфаниловая кислота 79
4. Алкилирование 79
Работа 11. Диэтиланилин - 80
Работа 12. Ди-и-бутиловый эфир 81
Работа 13. Диоксан-1,4 82
Работа 14. Дифенилметан 83
5. Ацилирование 83
Работа 15. Бутилацетат 84
Работа 16. Ацетанилид 84
Работа 17. Ацетилсалициловая кислота (аспирин) 85
Работа 18. Фениловый эфир бензойной кислоты 86
Работа 19. 2-Бензоилбензойная кислота 86
6. Диазотирование и реакции диазосоединений 87
Работа 20. Метиловый оранжевый 87
Работа 21. Фенол 88
Работа 22. 2-Хлорбензойная кислота 90
7. Окисление и восстановление 90
Работа 23. Бензойная кислота 91
Работа 24. 4-Нитробензойная кислота 92
185
Работа 25. Анилин 92
8. Реакции конденсации карбонильных соединений 93
Работа 26. Ацетоуксусныи эфир 94
Работа 27. Дибензилиденацетон „ 94
Работа 28. Фурфурилиденацетон и дифурфурилиденацетон 95
Работа 29. Фенолфталеин 96
9. Элементорганические соединения 97
Работа 30. Бензгидрол 97
10. Реакции полимеризации и поликоиденсации 99
Работа 31. Полистирол 99
Работа 32. Полиметилметакрилат 100
Работа 33. Поливинилацетат 101
Работа 34. Поливиниловый спирт 101
Работа 35. Фенолформальдегидный полимер (новолак) 102
Работа 36. Эпоксидный полимер (ЭД-20) 103
Работа 37. Полидиметилсилоксан 103
Работа 38. Поликонденсация фталевого ангидрида с глицерином 104
Раздел IV. Основы технического анализа органических веществ ...105
Работа 1. Определение кислотного числа, числа омыления и
эфирного числа 105
Работа 2. Определение бромного числа 106
Работа 3. Определение влаги в органических веществах 108
Работа 4. Определение окисляемости сточных вод 109
Работа 5. Определение молекулярной массы тиокола
(полисульфидного каучука) 111
Работа 6. Количественное определение гидроксильных групп в
олиго- и полиэпоксидах 111
Работа 7. Определение содержания эпоксигрупп в эпоксидных
полимерах 112
Раздел V. Физико-химические (инструментальные) методы
исследования органических соединений 114
1. Оптическая спектроскопия 114
186
1.1. Ультрафиолетовая (УФ) спектроскопия и спектроскопия в
видимой области 117
1.2. Электронные спектры поглощения и строение
органических соединений 121
1.3. Инфракрасная (ИК) спектроскопия 125
2. Спектроскопия ядерного магнитного резонанса (ЯМР) 129
3. Хроматографический метод 131
3.1. Адсорбционная хроматография 131
3.2. Распределительная хроматография 136
3.3. Ионообменная хроматография 138
3.4. Основные методы хроматографического анализа 139
Работа 1. Разделение и идентификация смеси 2, 4-динитрофенил-
гидразонов (ДНФГ) в тонком незакрепленном слое оксида
алюминия 145
4. Метод электрических моментов диполей 146
Приложение 150
I. Некоторые советы по организации самостоятельной работы
студентов (СРС) 150
П. О номенклатуре органических соединений 150
Ш. Некоторые сведения о справочной литературе по
органической химии 158
IV. Методики приготовления реактивов и препаратов для
лабораторных работ по органической химии (в помощь
лаборанту) 164
V. Характеристические частоты колебаний в ИК-спектрах
основных классов органических соединений 169
VI. Химические сдвиги протонов различных типов 175
VII. Осушители и их применение 176
VIII. Плотность и концентрация водных растворов серной,
азотной и соляной кислот * 177
К. Физические константы некоторых растворителей 179
X. Растворители, рекомендуемые в качестве среды для
некоторых химических реакций 181