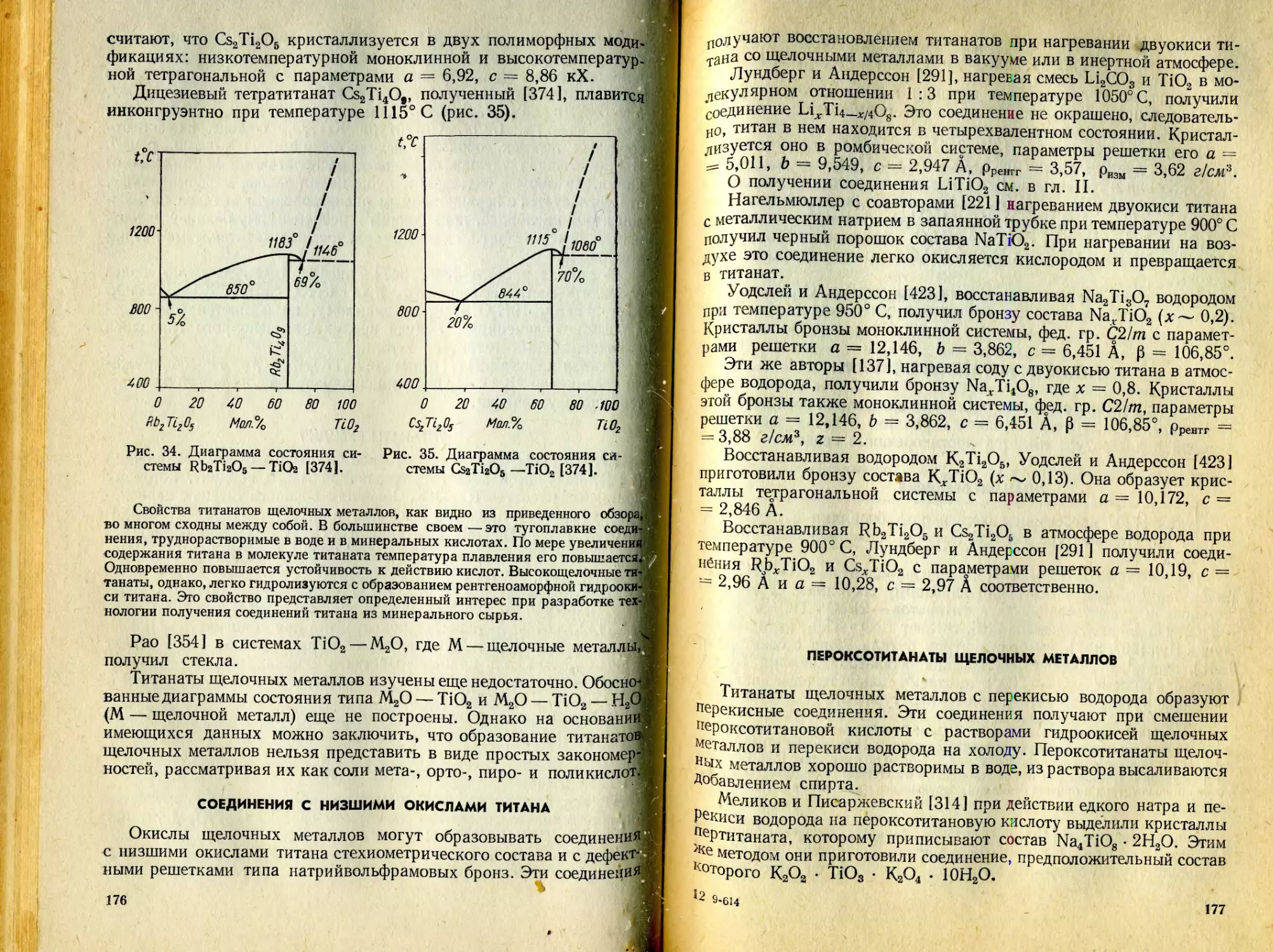
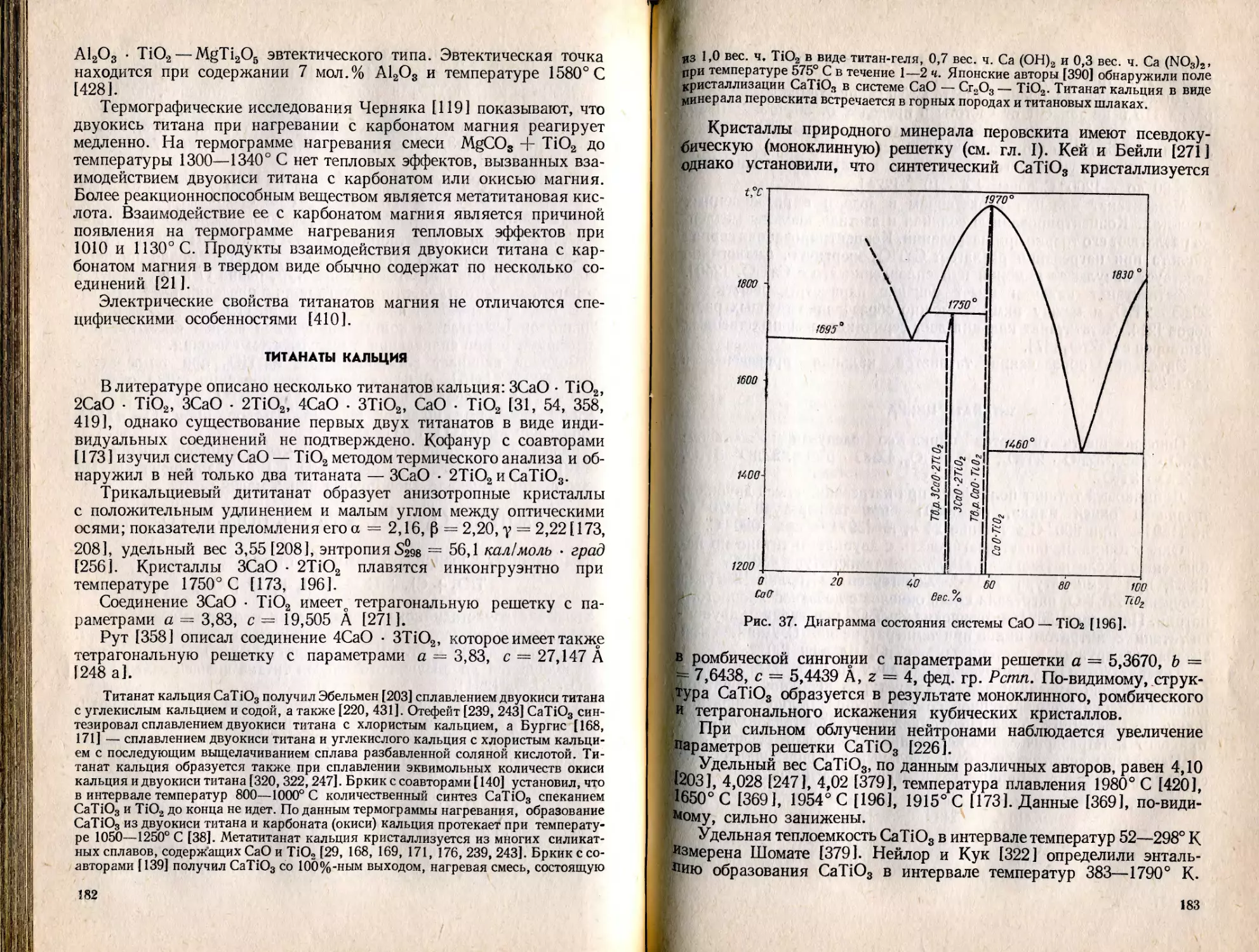
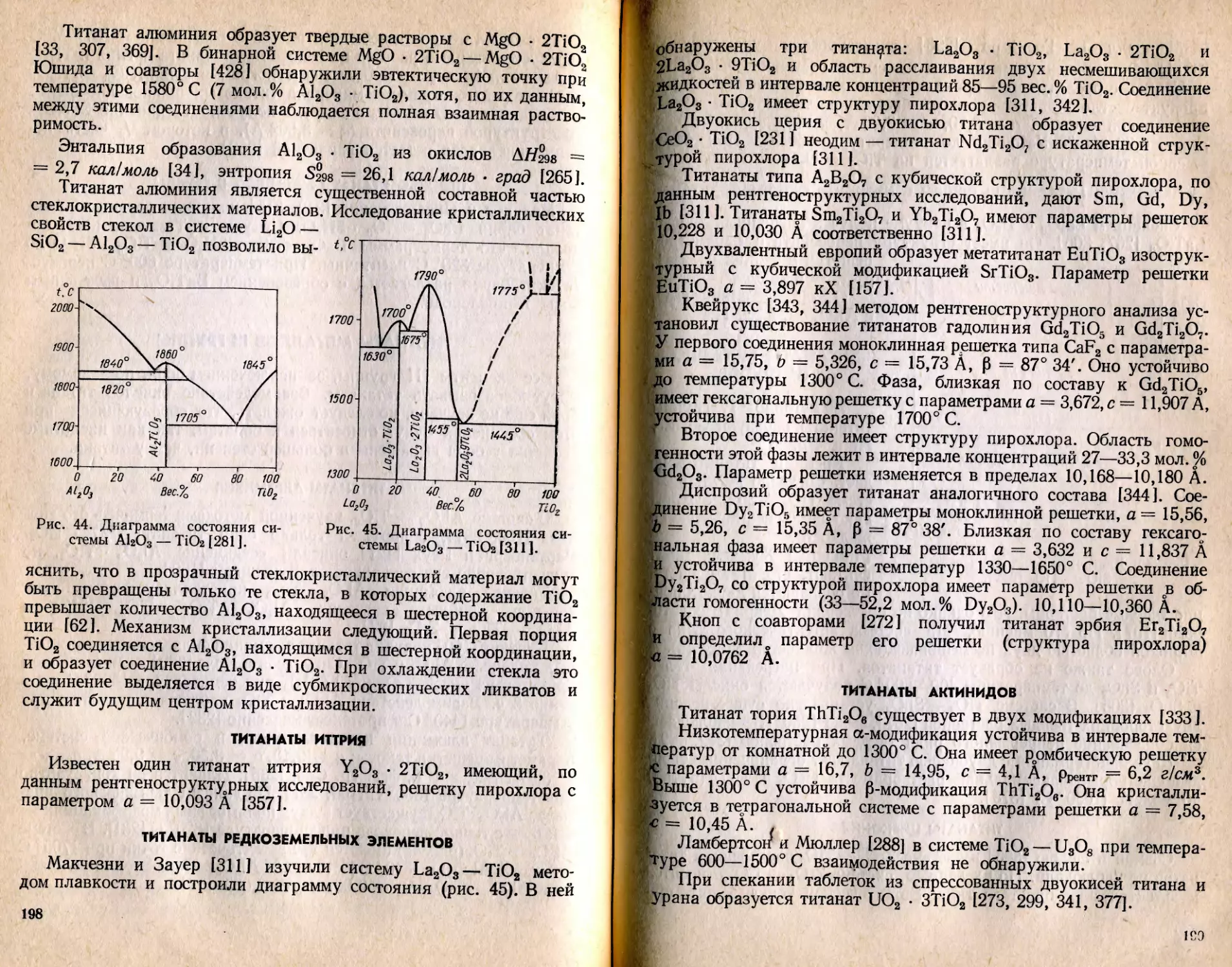
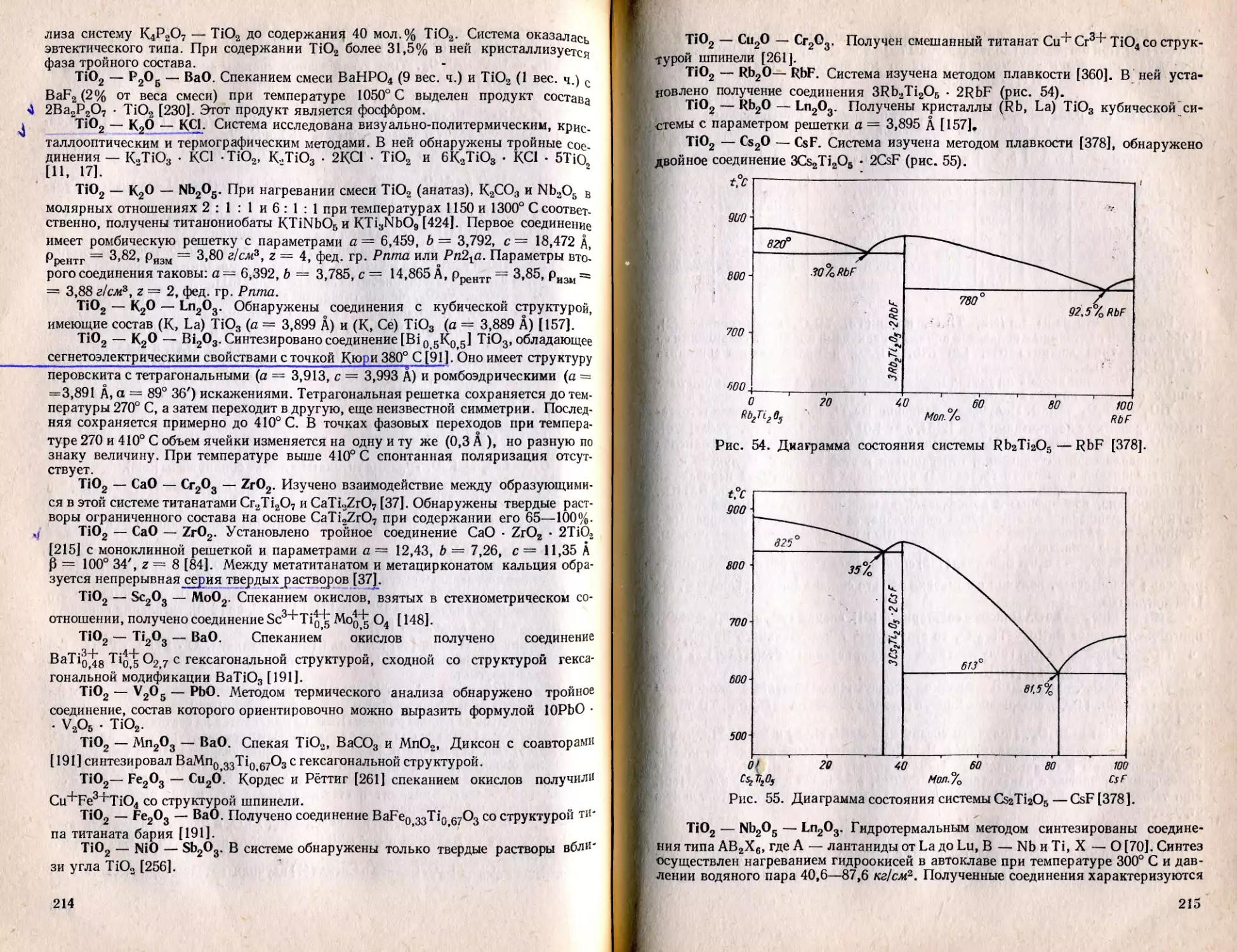
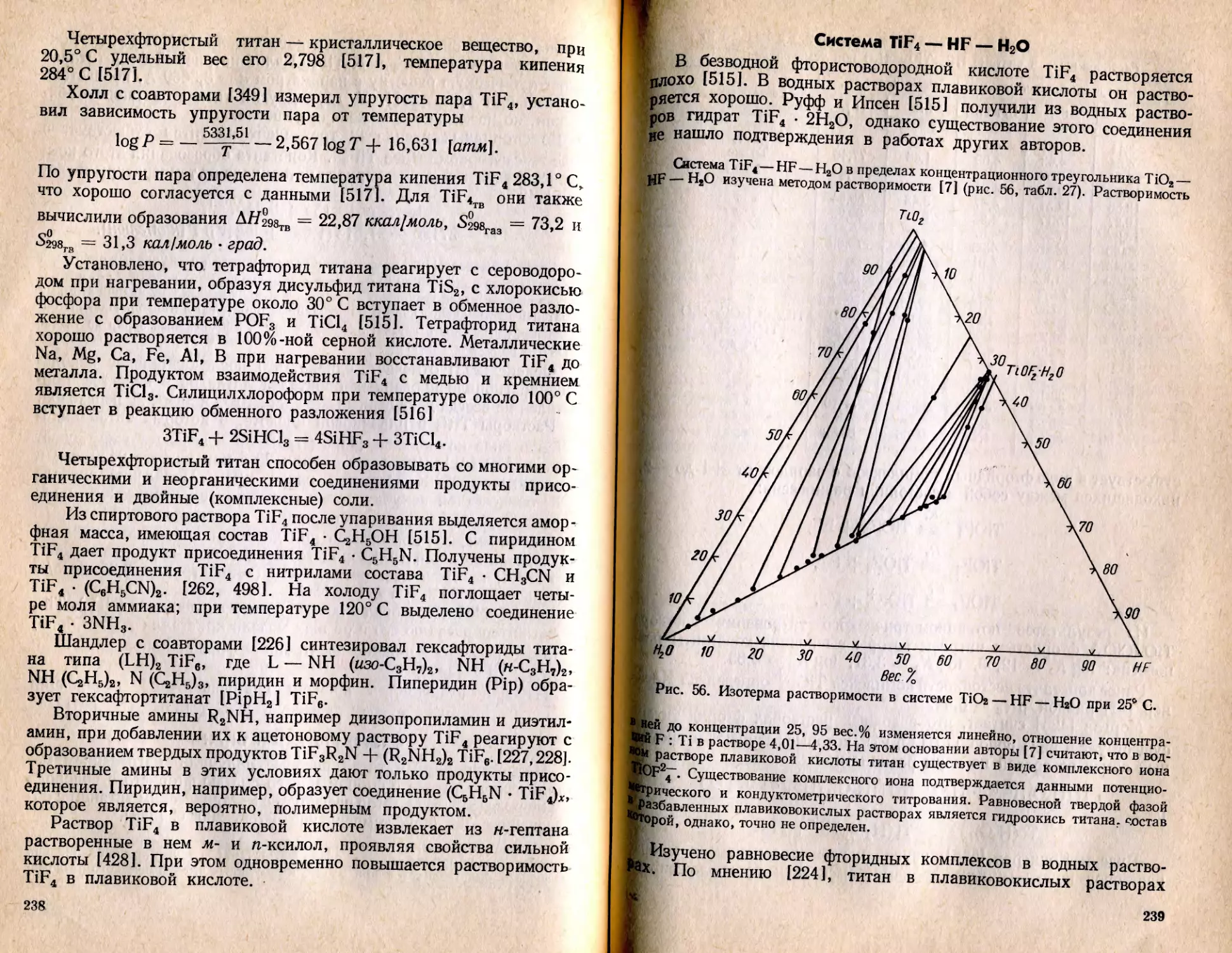
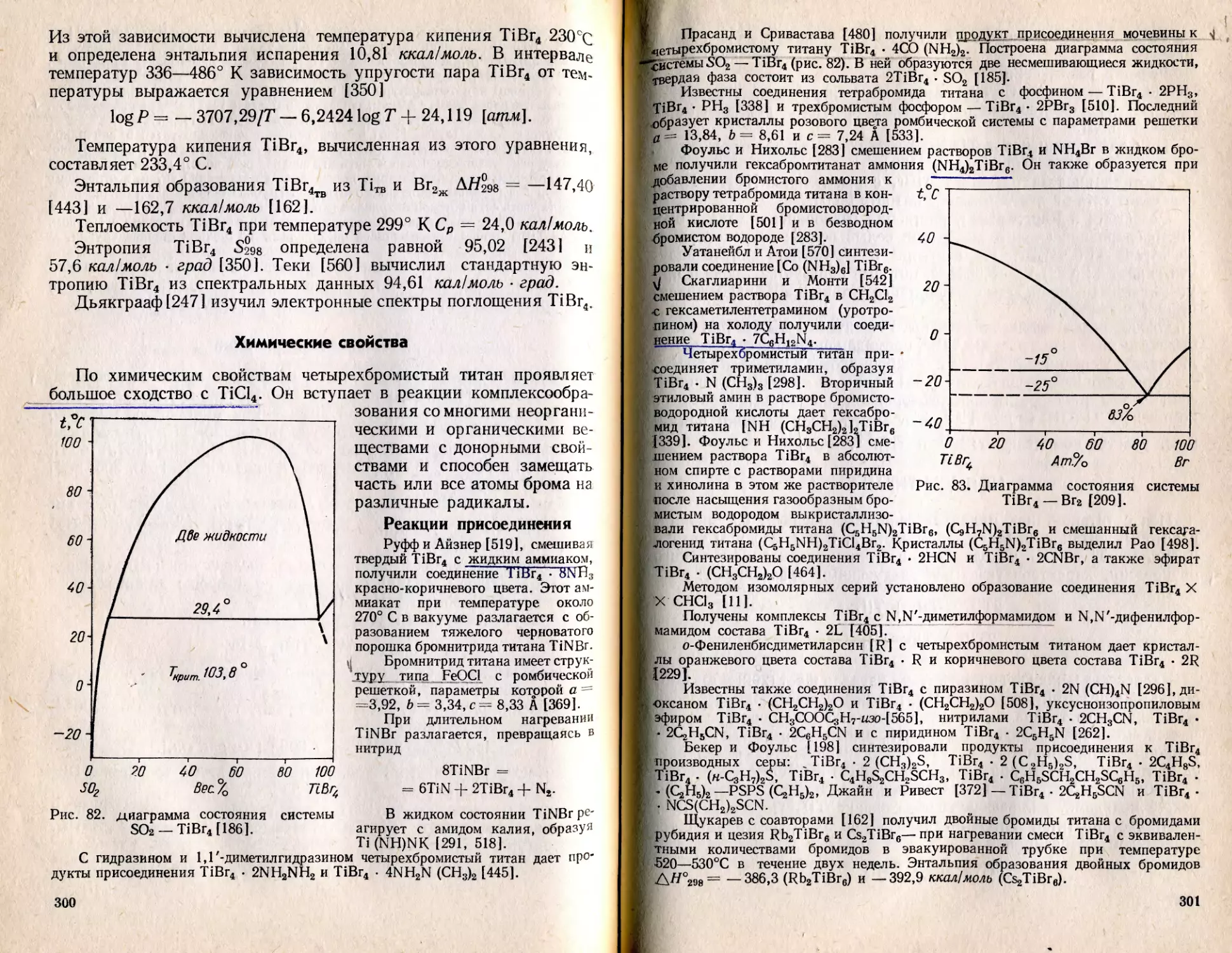
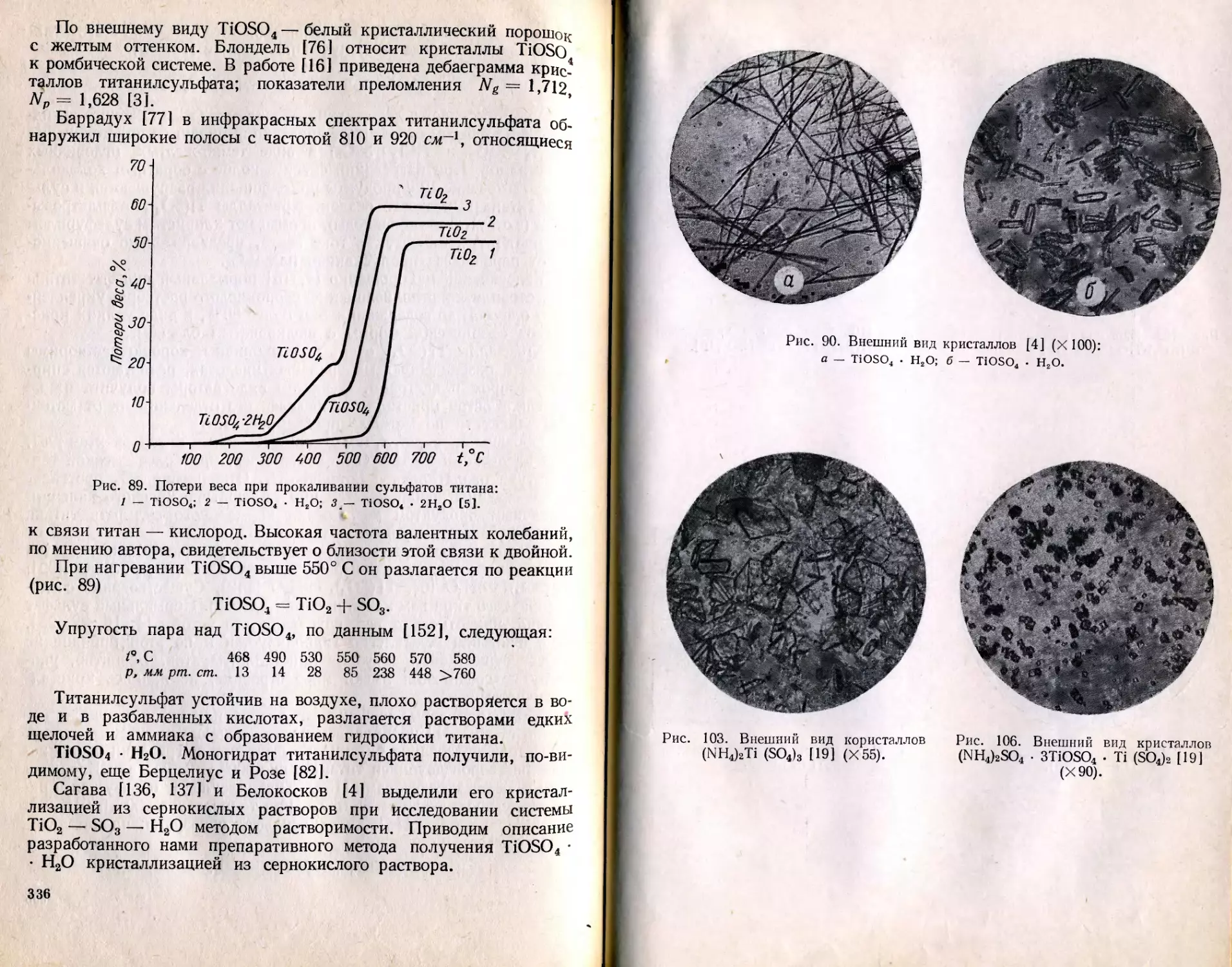
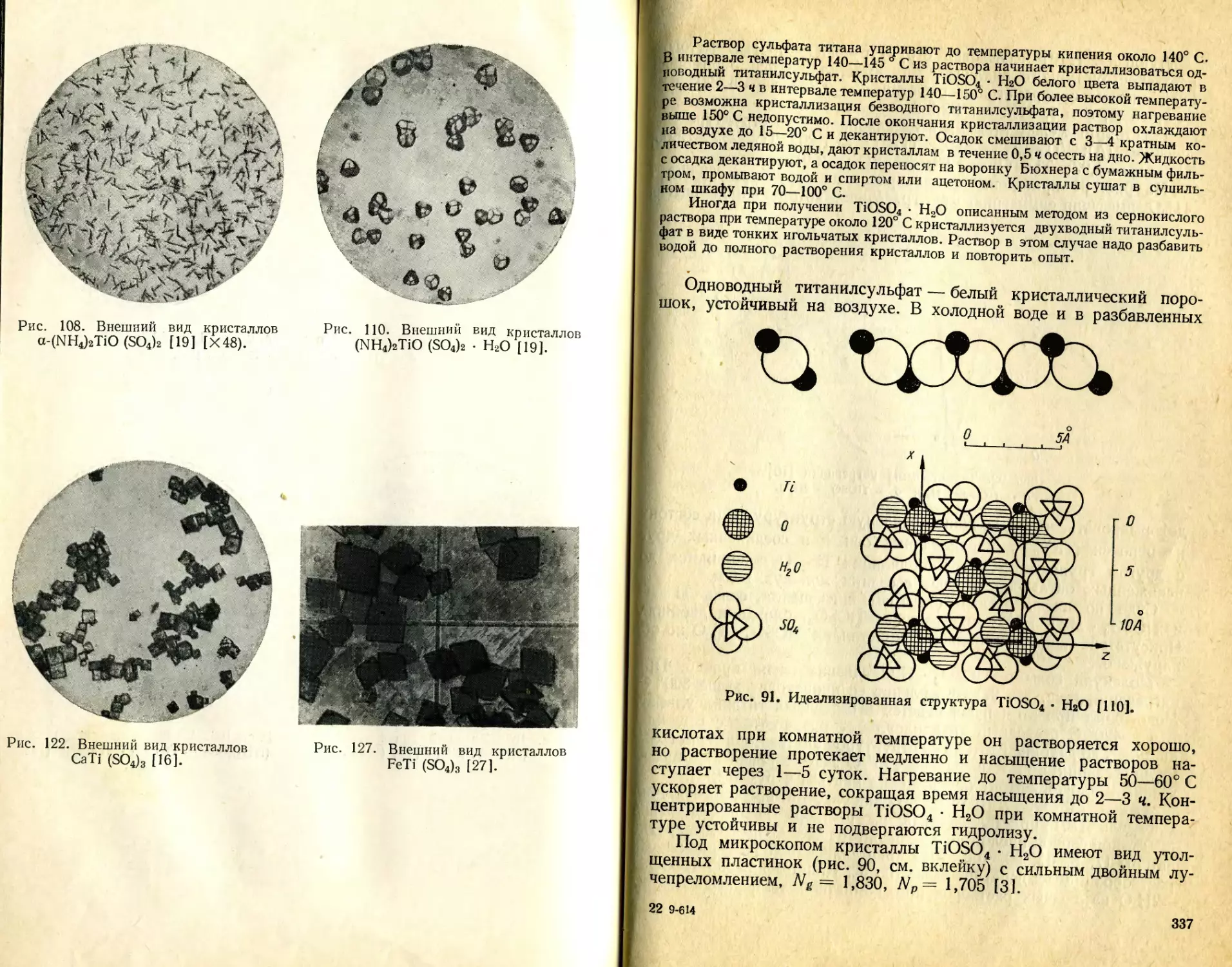
/









































































































































































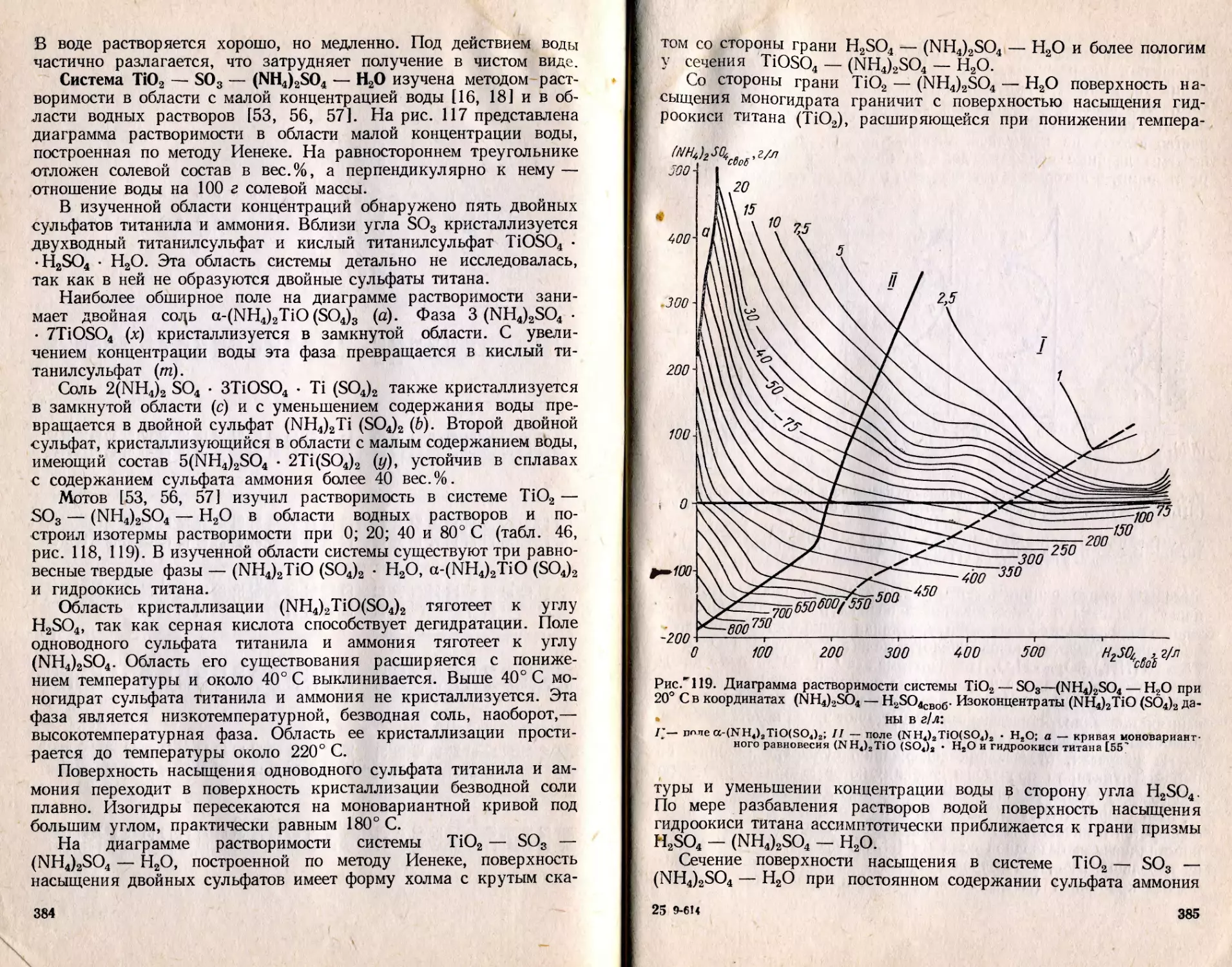




















Text
АКАДЕМИЯ НАУК УКРАИНСКОЙ ССР
ИНСТИТУТ ОБЩЕЙ И НЕОРГАНИЧЕСКОЙ ХИМИИ
Я. Г. ГОРОЩЕНКО
ХИМИЯ
ТИТАНА
«НАУКОВА ДУМКА» КИЕВ —1970
УДК 546-—821
540
ПО
В монографии освещается современное состояние
химии титана на основе обзора мировой литературы и
исследований автора. Книга состоит из восьми глав, в
которых рассматриваются основные классы соединений
титана: окислы, гидроокиси, титанаты, галогениды,
сульфаты, металлические соединения и др. Приводятся
краткие сведения о физических свойствах титана, на-
хождении его в природе, описаны важнейшие виды ти-
тановых руд.
Предназначается для научных работников, препо-
давателей вузов и инженеров. Может быть реко-
мендована для студентов и аспирантов, занимающихся
углубленным изучением химии титана.
Ответственный редактор
чл.-корр. АН УССР
И. А. ШЕКА
101—69 М
КИЕВСКИЙ ПОЛИГРАФИЧЕСКИЙ КОМБИНАТ
2—Б—2
ПРЕДИСЛОВИЕ
Химия титана за послевоенные годы стала быстро развиваться в связи с рас-
ширением производства и применения различных титансодержащих материалов в
технике.
Титан и его сплавы, обладающие высокими коррозионными свойствами и бла-
гоприятным сочетанием прочностных свойств с малым удельным весом, находят все
более широкое применение в морском судостроении, самолетостроении, химическом
машиностроении, при изготовлении ракет и космических аппаратов. Двуокись ти-
тана характеризуется превосходными пигментными свойствами и в больших мас-
штабах используется в качестве белого пигмента в лакокрасочной, бумажной, тек-
стильной и других отраслях промышленности. Четыреххлористый титан стал ос-
новным исходным материалом для производства металлического титана, двуокиси
титана, титанорганических полимеров и низших хлоридов титана, служащих ката-
лизаторами в органическом синтезе. Большой интерес представляют титанаты
бария, свинца, стронция при изготовлении сегнетоэлектриков, изделий для инфра-
красной техники, термоизоляционных материалов (гексатитанат натрия с волок-
нистой структурой).
Общее число работ по химии титана, опубликованных за последние 20 лет,
превысило уже три тысячи. Большинство из них посвящено синтезу и изучению
свойств различных соединений титана. Наибольшее внимание исследователей при-
влекают те классы соединений титана, которые перспективны для создания новых
неорганических материалов. К ним относятся титанаты, хлориды, сульфаты, метал-
лические, титанорганические и комплексные соединения. Некоторые работы по
химии титана приобрели важное общенаучное значение. Например, гексакомплек-
сы трехвалентного титана часто приводятся в учебных пособиях в качестве класси-
ческих образцов для иллюстрации закономерностей расщепления электронных
уровней центрального иона под действием кристаллического поля лигандов.
В мировой литературе известно несколько книг и значительное число обзорных
статей по химии титана и отдельных классов его соединений. Однако в настоящее
время еще нет обобщающей монографии, освещающей современное состояние хи-
мии титана и доступной широкому кругу научных работников и специалистов,
занятых в титановой промышленности. Это затрудняет ориентирование среди боль-
шого количества публикаций в периодической печати и препятствует использова-
нию достижений науки в химической технологии и металлургии титана.
Предлагаемая читателям книга обобщает работы по химии титана, опублико-
ванные до середины 1965г. Литература, появившаяся в печати завремя подготовки
рукописи к изданию, приложена в виде дополнительных списков к соответствую-
щим главам книги. В главе о сульфатах титана освещены результаты собственных
3
исследований автора и его сотрудников. В качестве основных библиографических
источников использованы справочники Гмелина1, Меллора2, Паскаля3 и рефера-
тивные журналы.
Автор выражает благодарность ответственному редактору чл.-корр. АН УССР
И. А. Шеке и рецензентам докт. техн, наук А. К* Шаровой и канд. хим. наук
И. С. Чаусу за их ценные замечания.
1 G m е 1 i п.— Handbuch anorg. Chem., 42, Titan, 195L
2 Mel lor J. W.— A comprehensive Treatise on inorg. and theoret. Chem.,
7 1930
3 A 1 b e г t Ph., Chretien A., Fl ah a u t J., Freundlich W.,
Langeron J.-P., Lehr P.— Titano — zirconium — hafnium — thorium.
Paris, Masson et Cie, 1963.
ГЛАВА I
ОБЩИЕ СВЕДЕНИЯ О ТИТАНЕ
ИСТОРИЯ ОТКРЫТИЯ ТИТАНА
Титан был открыт в результате исследований, проведенных в
конце XVIII в. английским священником Грегором, увлекавшимся
геологией и минералогией, и немецким минералогом и химиком Клап-
ротом. В 1790 г. Грегор проанализировал черный песок, собранный
в Менаккате (провинция Корнуэлл, Англия), и обнаружил в нем
46,6% окиси железа и марганца, 3,5 кремнекислоты и 45% белого
окисла неизвестного металла, который он назвал менакканумом [119,
120]. Черный песок — менакканит, судя по его химическому со-
ставу, был минералом ильменитом.
В 1795 г. Клапрот [141] проанализировал красный песок из Венгрии и обнару
жил в нем неизвестный металл, который назвал титаном. Песок этот оказался ми-
нералом, известным теперь как рутил. Затем титан был обнаружен Клапротом в
коричневом минерале, полученном из Испании и названном им титанитам (минерал
сфен). В 1797 г. Клапрот [142] проанализировал песок из Менаккана, подтвердил
результаты Грегора и пришел к выводу, что титан и менакканум Грегора — одно
и то же вещество. Так было завершено открытие нового химического элемента, на-
звание которого — титан — стало общепризнанным в научной литературе.
В течение последующих 20—25 лет после работ Грегора и Клапрота система-
тическое изучение титана не проводилось. Правда, еще в 1796 г. Лампадиус [153]
пытался получить металлический титан нагреванием его двуокиси с углем, но из-за
большого сродства титана к кислороду, углероду и азоту ему это не удалось. Ме-
таллический титан, загрязненный значительным количеством примесей, выделил
Берцелиус в 1825 г. действием металлического натрия на фтортитанат калия K2TiF(j
[90], а также Веллер и Девилль в 1850 г. [204]. Чистую двуокись титана впервые по-
лучил Розе в 1823 г. [ 173].
В 1826 г. Берцелиус и Розе [172] определили атомный вес титана, оказавшийся
равным 48,6. Эта величина близка к принятой в настоящее время.
На протяжении XIX в. титан не привлекал к себе особого вни-
мания химиков, так как долгое время не находил практического
применения. Первым толчком к расширению исследований по химии
титана послужило создание в 1916 г. промышленного производ-
ства двуокиси титана, которая оказалась прекрасным белым пигмен-
том, превосходящим по своим качествам цинковые и даже свинцовые
белила. Бурное развитие химии титана началось после разработки
Кролем в 1940 г. [151] метода производства металлического титана,
обладающего ценными свойствами и представляющего большой
интерес для многих отраслей новейшей техники.
В последние 15—20 лет получили широкое развитие исследова-
ния металлических соединений титана, а также галогенидов, которые
5
стали применяться в производстве металлического титана, титан-
органических соединений, представляющих интерес для создания
термостойких пластмасс, комплексных соединений и др.
ИЗОТОПНЫЙ СОСТАВ и атомный вес
Титан относится к IV группе периодической системы химиче-
ских элементов Д. И. Менделеева. Порядковый номер его 22. В при-
роде известно пять стабильных изотопов титана [80, 81, 126] с мас-
совыми числами 46, 47, 48, 49 и 50. Искусственно получено четыре
радиоактивных изотопа титана с массовыми числами 43, 44, 45 и 51.
Распространенность изотопов титана в природе характеризуе-
тся следующими данными (%): 46Ti —7,95; 47Ti — 7,55; 48Ti —
73,45; 49ti — 5,51; 50Ti — 5,34 [125, 126]. Хогг [121 ], учитывая боль-
шую разницу в массах изотопов титана, предполагал возможность
разделения их в природных процессах. Для проверки этого пред-
положения был изучен изотопный состав минералов ильменита и
рутила из различных месторождений и продажной двуокиси титана.
Изотопный состав титана в исследованных образцах изменяется ме-
нее 1%. Средние значения распространенности изотопов титана в
изученных образцах равны (%): 46Ti — 7,99; 47Ti — 7,32; 48Ti —
73,99; 49Ti — 5,46; 50Ti —5,25. Это находится в хорошем согласии с
данными предыдущих исследований [125, 126], и, таким образом,
заметного разделения изотопов титана в природных процессах, свя-
занных с минералообразованием, не наблюдается.
Атомные массы изотопов титана, вследствие дефекта массы при
образовании ядер из элементарных частиц, как известно, отличаю-
тся от целых чисел. По данным масс-спектрографических исследо-
ваний Гизе и Бенсона [114], стабильные изотопы титана имеют сле-
дующие массы:
Изотопы Массы изотопов по
водородной шкале
4вТ1 45,9672420 ±1,8
47Ti 46,9666862 ±3,2
48Ti 47,9631912± 1,6
49Ti 48,9634294 ±2,1
50Ti 49,9606687 ±2,3
что хорошо согласуется с данными микроволновой спектроскопии
и ядерных исследований.
Изотоп 43Ti образуется по реакции 40Са (а, п) 43Ti [177]. Он имеет
период полураспада 0,58 сек.
Шарп и Даймонд [185] получили облучением окиси скандия про-
тонами с энергией 30—45 Мэе изотоп 44Ti. По их данным, период по-
лураспада 44Ti (^Ti -> 44Ge) 23 года. Хейзенг и Цинг [129] 44Ti полу-
чили облучением скандия дейтерием 45Sc (а, Зп) и нашли, что период
полураспада его равен около 1000 лет. Хонда с соавторами 1127, 128]
6
определил период полураспада 44Ti 200 лет. Как видим, в данных
различных авторов имеются значительные разногласия. Недавно
Уинд и соавторы [203] произвели тщательную очистку 44Ti экстрак-
цией 20%-ным раствором я-гептанола в петролейном эфире из со-
лянокислых растворов. Они нашли период полураспада 44Ti равным
46,4 года. Близкая величина — 48,2 года — установлена Мерилен-
дом и Хейманом [166].
Изотоп 46Ti можно получить по нескольким ядерным реакциям,
например: 42Са (a, n)45Ti, 45Sc (р, n)45Ti и др. [78]. Люис [159] опре-
делил период полураспада 46Ti 3,5 ч, Андерсон [79] — 3,0 ч.
Изотоп 51Ti также может быть получен по нескольким реак-
циям, например: ^Ti (п, у) 51Ti [123, 184], 48Са (a, ri) 51Ti [123],
б4Сг (п, а) 61Ti [1231. Период полураспада его, по определению
разных авторов, равен 5,79 [183], 5,8 [194] и 5,89 мин [123].
Пуларикас и Финк [171] измерили абсолютные сечения акти-
вации ядер стабильных изотопов титана при облучении их ней-
тронами с энергией 14,8 (табл. 1).
Таблица 1
Активация ядер титана
Изотоп Абсолютное сече- ние активации нейтронами с энергией 14,8 Мэв, барн Период по- лураспада Реакция
50Ti 27,0 1,8 мин (п, р) —> 50Sc
50Ti 48,0 22 мин (п, р) —> б05с
50Ti 9,0 — (л, у) —51Ti
49Ti 29,0 — (л, р) —49Sc
48Ti 58,0 44 ч (п, р) —*• 48Sc
47Ti 230,0 3,45 дня (л, р) —* 47Sc
46Ti 50,4 3,06 ч (л, 2 л) —► 45Ti
46Ti 520,0 85 дней (л, р) —> 46Sc
Флюгге и Маттаух [111] вычислили средний атомный вес титана
исходя из изотопного состава и получили величину 47,8891 (по фи-
зической шкале). В 1961 г. Бакстер с соавторами [87, 88, 89] в по-
следний раз определял атомный вес титана по соотношению
TiCl4 : 4Ag и TiBr4 : 4Ag и нашел его равным в среднем 47,90. Атом-
ная комиссия утвердила атомный вес титана по углеродной шкале
47,90 [97].
Радиус свободного атома титана (2,32 А) вычислен Слейте-
ром [186].
Джефриз [137] определил ядерные спины и магнитные моменты
изотопов 47Ti и 49Ti, равные соответственно б/2, 7/2 и —0,78706,
—1,1022.
7
ФИЗИЧЕСКИЕ СВОЙСТВА
По внешнему виду титан в компактном состоянии — металл се-
ребристого цвета. Он существует в двух полиморфных модификаци-
ях: низкотемпературной с гексагональной плотно упакованной ре-
шеткой (а) и высокотемпературной с кубической объемноцентриро-
ванной решеткой (р).
Полиморфизм титана установлен по данным рентгеноструктур-
ных исследований [96, 182]. Подтверждено существование поли-
морфных модификаций титана дилатометрическим методом [38, 150]
и исследованием теплоемкости и термоэлектродвижущей силы [134—
136].
Температура превращения гексагональной модификации ти-
тана в кубическую зависит от чистоты исследуемых образцов. При-
меси многих веществ, например азота, марганца, железа, кисло-
рода и др., заметно изменяют температуру а-> (3-превращения ти-
тана как в сторону ее повышения, так и в сторону понижения [67].
Между а -> p-превращением титана и изменением упругих кон-
стант (£ — модуль нормальной упругости и G — модуль сдвига)
установлена определенная связь.
Температура полиморфного превращения технического титана,
по данным [54], лежит в пределах 850—880° С, по данным [163] —
в пределах 860—960° С. В наиболее чистом, иодидном, титане, согла-
сно измерениям термоэлектрических свойств, а -> ^-превращение
происходит при температуре 883—887° С [206], а по изменению рав-
новесного давления водорода — при 882,5° С [163]. Последняя ве-
личина считается наиболее достоверной.
Балихин [3] при электролизе титана выше 883° С получал крис-
таллы P-Ti, а ниже — a-Ti.
Прецезионные измерения параметров решетки а-титана высокой
чистоты дали следующие результаты:
а, А
2,9504
2,9506
2,95111
с'у
4,6833
4,6788
4,68433
Литера-
тура
[100]
[196
[205
Параметр решетки кубической модификации титанаопри 900° С —-
3,3065 А [109], при комнатной температуре — 3,282 А [155].
Плотность а-титана при исследовании наиболее чистых образ-
цов равна (г/сл8) 4,507 [ПО], 4,489 [208], 4,505 [100], плотность
Р-титана при 900° С — 4,319 г/см? [96].
Измерение температуры плавления титана сопряжено с боль-
шими трудностями из-за активного поглощения металлом в нагре-
том состоянии газов, в частности кислорода и азота. В старых ра-
ботах температуры плавления титана приводятся в пределах от 1750
до 1850° С. Эти данные следует считать завышенными из-за недо-
8
статочной чистоты образцов металла. Небольшие количества при-
месей кислорода и азота в титане заметно повышают его температуру
плавления. В работах последних 10—15 лет исследователи пользо-
вались более чистыми образцами титана, полученными методом иодид-
ного рафинирования и нашли температуру плавления для него рав-
ной (° С) 1725 [48]; 1696 [76]; 1680 [92]; 1668 [104]; 1716 [124];
1707 [162]; 1690 [168]; 1672 [169]; 1650 [178]; 1665 [179]. Наиболее
достоверную величину из этого перечня определить трудно; сред-
нее значение будет 1684° С.
Температура кипения титана, вычисленная Несмеяновым [44]
из упругости пара над металлом, составляет 3442° К.
Берри и Рейнор [86] измерили коэффициенты линейного рас-
ширения а-титана. Средний коэффициент линейного расширения
вдоль оси а при температуре от комнатной до 700° С равен 11,033 X
X 10~6, а вдоль оси с—13,37 • 10“6. Шпридборо и Христиан [189]
для а-титана определили средний коэффициент расширения в ин-
тервале температур от 0 до 600° С вдоль оси а — 9,55 • 10~6, вдоль
оси с— 10,6 - 10~6. Средний коэффициент линейного расширения
Р-титана при 900—1070° С равен примерно 12,0 • 10~6. Авторы для
а-титана вычислили дебаевскую температуру 0 = 270 ± 30° К.
Давление пара титана в твердом состоянии измерено методом
Лэнгмюра (испарение на нити) [91, 98, 99, 107]. Несмеянов [44]
считает, что наиболее надежными являются результаты, приведен-
ные в работах [91, 107 ] и по ним с помощью электронной машины вы-
числил вероятные коэффициенты, входящие в уравнение зависимос-
ти упругости пара титана от температуры, и давление пара через
определенные интервалы температуры. Зависимость давления пара
титана от температуры, по Несмеянову, выражается уравнениями
для твердого титана
lg Р = 32,51129 —27017,08т-1 + 0,61128 • 103Т —
— 6,768421g Т [мм pm. cm.]
(Т = 1300— 1980° К);
для жидкого титана
lg Р - 296,01329 — 65632,18т-"1 +
+ 7,87692 . 103Т — 85,14729 lg Т [мм pm. cm.]
(Т = 1980 —3000° К).
Вычисленная по данным упругости пара энтальпия сублимации
ана /7о = Н2,5, А= 113,2 ккал/г-am [44].
Найп с соавторами [143] измерил теплоемкость титана Се при
1,2—4,5° К. Зависимость ее от температуры выражается уравнением
CQ = уТ + ₽Т3,
где у = 3,346—3,351 мкдж!молъ • град2, (3 = 427—430. Дебаев-
ская температура
0д - 12л4ВД°К.
Эйвен с соавторами [82] нашел, что теплоемкость очень чистого
титана в интервале температур 4—15° К выражается уравнением
С - 3,88 - 10~37 + 2,60 • 10~573 [дж1г-ат-град],
где у = 3,88 • 10~3 дж!г-ат • град2, что близко к данным [143 L
Дебаевская температура определена 0д = 421° К.
Данные разных авторов [95, 101, 148, 1901 по теплоемкости ти-
тана в интервале температур 13—360° С согласуются. Ниже при-
водятся результаты измерения [148]:
Т, °К 15,44; 49,04; 104,49; 148,7; 198,46; 248,05; 305,51
Се, кал!г-апг-град 0,046; 1,085; 3,583; 4,654; 5,305; 5,682; 6,005
Измерена теплоемкость титана при температуре 600—1345° К
[176]. При температуре 600°К С = 0,72 дж!г-ат • град. Вблизи точ-
ки полиморфного превращения (882° С) С = 0,93 дж!г-ат • град.
За точкой перехода теплоемкость титана уменьшается до
0,75 дж/г-ат • град и далее остается постоянной.
Козен и Джонстон [148] вычислили AS^s = 7,33 кал!молъ • град.
Энтальпия титана определена экспериментально [20].
Для а-титана
АЯГэв = 8,6417 + 0,7025 • 10^4Т2 + 0,3467 • 10Т-1 —
— 3745 [кал [г-ат]
(7- 298 — 1155° К);
для (3-титана
Д//^8 = 7,9687— 1753 \кал1г-ат}
= 1155— 1400° К).
Эти данные были подтверждены Серебряковым и Гельд [54].
Голутвин [20] нашел, что теплота а -> [3-превращения титана
составляет 820 кал!г-ат.
Гиллес с соавторами [115, 147] вычислил термодинамические
функции титана при температуре 0—8000° К.
Электрическое сопротивление металлического титана в сильной
степени зависит от его чистоты, в частности от содержания примеси
кислорода [207]. С повышением температуры электросопротивление
титана возрастает, и при температуре а -> (3-перехода на кривой
температурной зависимости электросопротивления появляется рез-
кий излом.
При низкой температуре титан переходит в сверхпроводящее
состояние. Температура перехода (°К), по данным разных авторов,
равна 0,53 [103]; 0,558 [187]; 0,387 [188]; 0,37, 0,49 [193]. Штиль
ю
и Гейн 1193] показали, что титан относится к числу «жестких» сверх-
проводников, на свойства которых существенно влияют внутренние
напряжения кристаллической решетки, обусловленные механичес-
кой деформацией, вызванной наличием в испытуемых образцах при-
месей. Заметное влияние на переход титана в сверхпроводящее
состояние оказывает даже малая примесь кислорода (около
0,005%).
Определено поверхностное натяжение о — 1588 дин!см металли-
ческого титана при температуре плавления [198].
Удельная магнитная восприимчивость титана при температуре
20° С равна 3,36 • 10~6 [146].
Измерена постоянная Холла R низкотемпературной гексаго-
нальной решетки а-титана 99,99%-ной чистоты в интервале темпера-
тур 350—1100° К [180, 181 ]. При комнатной температуре R = — 2х
X 10“п м31к. При температуре 675° К величина R меняет знак и при
1100° К возрастает до + 3,5 • 10~н мЧк.
Кришнан и Джайн [149] определили, что константы в уравнении
термоионной эмиссии титана i = AT2 exp (—qlkT) составляют
<р — 3,95 и А = 44 а/см2 • град2.
НАХОЖДЕНИЕ В ПРИРОДЕ
Титан относится к числу наиболее распространенных в природе
элементов. В земной коре только девять элементов превышают по
количеству титан (О, Si, Al, Fe, Са, Mg, К, Na, Н), среднее содержа-
ние которого равно 0,61 вес.% [12, 13]. По распространенности в
земной коре титан среди металлов стоит на седьмом месте после А1,
Fe, Са, Mg, К и Na.
Титан входит в состав почти всех магматических и происходящих
из них пород. В морской воде его больше, чем железа, в 1,73 раза
[133]. В 1 м3 воды океана содержится 1 мг титана [116]. В морских
донных отложениях он находится в виде самостоятельных минера-
лов и обломков горных пород. Максимальное содержание титана
в отложениях морского дна Охотского моря равно 2,89% [47].
Содержание титана в почвах из различных частей света колеб-
лется в значительных пределах (0,5—1,5% ТЮ2 [14]). В пахотных
и пустынных почвах его меньше, в глинистых — больше. Глинистые
вещества серо-бурых подзолистых почв Пакистана содержат 0,94—
1,39% ТЮ2 [138].
Титан входит в состав бокситов большинства месторождений.
На основании статистических данных [83] установлено следующее
содержание ТЮ2 в бокситах разных стран (%): Венгрия — 2,
Италия — 1,2—3,0, Франция — 0,6—5,8, Индия — 6—8, Джор-
джия и Калифорния — 2—4, Австралия — 5, Золотой берег — 2—4.
В бокситовых шламах, получающихся при производстве глинозема
по способу Байера, накапливается до 30% TiO2 [106].
п
Титан обнаружен в золе торфа, каменного угля, нефти [156, 108],
а также во всех живых организмах [199, 200]. Подобно Na, Mg, Al,
Са, К, V, Сг, Мп, Со и Ni он входит в состав живого вещества в зна-
чительно меньших количествах, чем в состав почв [93]. Содержание
титана в органических тканях рыб составляет в среднем 10“4 вес. %
[199]. Организм человека содержит титан в таких же количествах*
как As, В, F, РЬ и V [199]. В ядрах клеток коры головного мозга
у человека, коровы и собаки установлено постоянное наличие титана
[16]. В среднем содержание титана в организмах животных, обитаю-
щих на суше, равно 8 • 10~4вес.% [11].
Присутствие титана установлено на Солнце и звездах. Относи-
тельное содержание титана на Солнце равно 4,96, в метеоритах —
4,76 (десятичный логарифм относительной распространенности ато-
мов водорода равен + 12) [21 ]. На Солнце и звездах титан находится
в ионизированном состоянии и в виде молекул ТЮ. Полосы погло-
щения ТЮ обнаружены в спектрах большинства наиболее холодных
звезд: гигантов и сверхгигантов от Л4Х до Л44, например а-Геркулес,
р-Пегас.
Содержание титана в метеоритах зависит от их природы. В же-
лезных метеоритах оно равно 6 • 10“5 — 3 • 10-4 вес. % [2011; ка-
менные метеориты в среднем содержат 6 - 10~2 вес.% титана [165].
В хондритах титана меньше, чем в земной коре. Явнель и Дьяконова
[75] определили следующее содержание титана в метеоритах
(вес.%): хондриты — 0,072, углеродистые хондриты — 0,04, ахон-
дриты, бедные кальцием,— 0,06, ахондриты, богатые кальцием,—
0,24. Отмечены метеориты с очень высоким содержанием титана, на-
пример, в троилите метеорита Norton County найдено 6,4 вес. % ти-
тана [164].
КОРРОЗИОННЫЕ СВОЙСТВА ТЕХНИЧЕСКОГО ТИТАНА
И ЕГО СПЛАВОВ
Современная промышленность производит технический титан
и его сплавы с А1, содержащие присадки Сг, Mo, V, Мп, Sn и Nb
(табл. 2). По микроструктуре титановые сплавы разделяются на од-
нофазные (а- и Р-сплавы) и двухфазные, содержащие одновременно
а- и р-модификации титана.
Коррозионные свойства титана и его сплавов рассмотрены в ра-
ботах [70, 117, 132, 140, 197].
Технический титан (ВТ1 и ВТ2) и многие его сплавы обладают
высокой коррозионной устойчивостью по отношению к агрессивным
средам. Высокая коррозионная стойкость титана и его сплавов объяс-
няется образованием на их поверхности окисной пленки, облаго-
раживающей электрохимические потенциалы [29, 192].
При комнатной и повышенной температурах технический титан
на воздухе и в атмосфере чистого кислорода не окисляется, сохра-
12
13
няя блестящую поверхность. Детали, изготовленные из технического
титана, устойчивы на воздухе до температуры 350° С.
До температуры 800° С окисление титана происходит медленно.
Как показали электронографические исследования [102], при тем-
пературе 300° С на поверхности титана возникает пленка кубиче-
ского окисла TiO. Выше 400° С пленка моноокиси исчезает и обра-
зуется пленка анатаза и рутила, а при температуре выше 500° С —
только рутила.
По наблюдениям [23], в результате окисления титана на воздухе
при температурах 350—450 и 700—900° С на его поверхности обра-
зуется рутил, а при температуре 500—600° С — смесь рутила с
небольшим количеством низшего окисла Ti3O5. Азот с титаном не
дает особых нитридных фаз, а, видимо, входит в решетку окислов.
В результате исследования кинетики поглощения техническим
титаном кислорода установлено, что процесс окисления протекает
по двум механизмам [77]. При низкой температуре поверхность ти-
тана покрывается окисной пленкой. Процесс окисления описывае-
тся следующим уравнением:
In (/ + 3) = W + Т<2,
где t — время, V — объем адсорбированного кислорода, и
— константы.
При высокой температуре кислород диффундирует внутрь ме-
талла. Процесс окисления титана в этих условиях описывается
уравнением
lg [(С - Cf)/(CZ - Q)] = - К - 2,3//т,
где С — концентрация кислорода, вес.%; Cz— первоначальная
концентрация кислорода в металле (0,12%); Cf — конечная концен-
трация кислорода, соответствующая образованию ТЮ2; /С — кон-
станта; t — время; т = аЧпЧЭ (а — размер эквивалента куба, см;
D — коэффициент диффузии).
Показано, что диффузия кислорода в а-титане при температуре
800—900° С подчиняется параболическому закону, за исключением
начального периода, в который количество растворяющегося кис-
лорода пропорционально разности максимально возможной и су-
ществующей концентраций [51, 52]. Параболический характер диф-
фузии кислорода свидетельствует о наличии на поверхности титана
при температуре 800° С защитной пленки из кислорода. Выше 800° С
на поверхности титана образуются TiO и TiN, ускоряющие процесс
окисления металла.
Сплавы титана с железом (до 30 вес. % Fe) обладают меньшим со-
противлением окислению, чем чистый титан. Сплавы железа с тита-
ном (до 50 вес. % Ti) характеризуются повышенной жаропрочнос-
тью по сравнению с чистым железом [17].
Титан и его сплавы в присутствии сильных окислителей (дымя-
щей азогной кислоты, хлора и кислорода) способны возгораться не
14
только при температурах, характерных для автоклавных процес-
сов, но и при комнатной температуре [5,27,43 ]. Монолитные образцы
титана, с поверхности которых снята защитная окисная пленка,
возгораются в атмосфере кислорода при достижении определенного
давления [157]. Это давление Борисова и Варданов [5] называют
критическим. Критическое давление кислорода, при котором проис-
ходит загорание титана и его сплавов при комнатной температуре,
таково:
Марка сплава Критическое дав- ление кислорода, атм Марка сплава Критическое дав- ление кислорода, атм
ВТ1-1 20—25 ВТ6 8
ВТЗ-1 8 ВТ8 13
ВТ5 9—12 ВТ10 11
ВТ5-1 9—12 ВТ14 13
ОТ4 10 ВТ15 12
ОТ4-2 7
Водяной пар и азот повышают критическое давление.
Титан, покрытый окисной (защитной) пленкой, инертен [43].
Самовозгорание его наступает только после удаления с поверхности
металла этой пленки.
Взаимодействие технического титана с водяным паром начинае-
тся при температуре 350—370° С [39]. Выше 400° С кинетика вза-
имодействия титана с водяным паром подчиняется параболическому
закону.'
При температуре 800—1000° С титан окисляется водяным паром
до рутила [4] по закону Am ~ aft*, где Ат — привес образца,
t — время, а — константа [158]. Водород, поглощающийся тита-
ном при разложении воды, распределяется между твердым раство-
ром и газовой фазой. Из-за растворимости водорода объем металла
увеличивается, что приводит к непрерывному разрушению возни-
кающей окисной пленки на его поверхности.
Окисляемость титана водяным паром при температуре 700—
1200° С выше окисляемости его на воздухе [34], что обусловлено
более рыхлой структурой окалины.
Титан весьма стоек к морской воде и морской атмосфере [113,
131, 161], к влажному хлору [161] и раствору FeCl3 [197].
Галоиды в отсутствие влаги окисляют титан [69]. Листовой ти-
тан в атмосфере сухого хлора саморазогревается и воспламеняется
через 14 ч, в жидком броме при комнатной температуре — через
10 мин, в сухом кристаллическом иоде — после нагревания до тем-
пературы 100° С — через 15 мин.
Титан обладает удовлетворительной стойкостью к 20%-ным со-
ляной и серной кислотам при температуре до 10° С, а при нагрева-
нии выше 20° С значительно корродирует [1, 58].
Титан заметно корродирует в 20%-ной фосфорной кислоте при
температуре 20° С [18].
Поверхность титана легко пассивируется. Добавка к соляной
и серной кислотам нитрата натрия сообщает поверхности титана
15
90S
положительное значение электродного потенциала и уменьшает
его коррозию. Для торможения коррозии титана в 20%-ных соляной
и серной кислотах при температуре 20° С достаточно добавить к
ним 0,01 % нитрата натрия [58].
Эффективными ингибиторами коррозии титана в соляной и серной
кислотах служат нитроанилины, нитрофенолы и другие окислители
[8]. Коррозию титана в соляной и серной кислотах тормозят хлор,
бром и йод [69, 191]. Сохранению пассивной поверхности титана
способствует накопление в растворе ионов Ti4+ [2].
Исследование пассивации титана в 1-н. серной кислоте показало,
что в активной области он переходит в раствор в виде ионов Ti3+,
а в пассивной — Ti4+ [68]. Ионы Ti3+ оказывают сильное депасси-
вирующее действие на титан. Потенциал пассивации титана мало
зависит от температуры. Стационарный потенциал титана с пониже-
нием температуры смещается в положительную сторону, при 25° С
он близок к потенциалу пассивации, а при 15° С лежит в пассивной
области. Пассивация титана, по предположениям [68], обусловлена
адсорбцией атомных ионов-радикалов кислорода и образованием
в пленке и у ее поверхности энергетического барьера, препятствую-
щего процессу растворения.
Титан устойчив к действию 10%-ного раствора едкого натра.
Азотная кислота всех концентраций действует на титан при ком-
натной температуре незначительно [113]. К кипящей азотной и
соляной кислотам при комнатной температуре титан более стоек,
чем нержавеющая сталь типа 1Х18Н9Т [139]. К органическим кис-
лотам стойкость титана и нержавеющей стали примерно одинакова.
Титан устойчив до температуры 100° С к антрониловой, азелаиновой,
адипиновой, бензойной, лимонной, монохлоруксусной, параамино-
бензойной, салициловой, тетрахлоруксусной, фумаровой, фталевой,
феноксиуксусной, яблочной и янтарной кислотам [10]. Смесь ледя-
ной уксусной кислоты с уксусным ангидридом, винная, муравьиная
и щавелевая кислоты сильно его корродируют.
Титан быстро растворяется в плавиковой кислоте всех концен-
траций [191], потенциал растворения его равен приблизительно
0,77 в. Растворение титана в кислотах сопровождается образованием
ионов Ti3+ [191].
Высокой коррозионной устойчивостью отличаются и многие
сплавы титана.
Сплавы АТЗ, АТ4, АТ6, АТ8 и АТ 10 стойки к дистиллированной
воде при комнатной температуре и водопроводной при температуре
до 100—170° С [61 ]. Эти сплавы после пребывания в течение 5000 ч
в атмосфере Тбилиси, Рустави и Батуми, где они подвергались воз-
действию дымовых газов—копоти, сероводорода, окислов азота,
сернистого газа, аммиака, углекислоты и др., не теряют декоратив-
ных свойств и отражательной способности полированной поверхно-
сти. Лучше всех в этом отношении оказались сплавы АТЗ и АТ4. Тер-
мическая стабильность их сохраняется до температуры 450—550° С.
16
Как и технический титан, сплавы АТ2, АТЗ, АТ4, АТ6, АТ8,
АТ9 и АТ 10 показали чрезвычайно высокую стойкость к морской
воде [29]. Механические свойства этих сплавов после длительного
нахождения в морской воде не ухудшились.
Сплавы ВТ15, ВТ14, АТ21, АТ22, АТ23 показали хорошую кор-
розионную стойкость к серной кислоте с концентрацией 83 г!л в при-
сутствии сульфата никеля при комнатной температуре [46]. Ионы
никеля активируют коррозию титановых сплавов.
Титановые сплавы АТЗ, АТ4, АТ6, АТ8, АТ9 и АТ 10 при ком-
натной температуре абсолютно стойки к соляной и азотной кисло-
там всех концентраций, а также к серной кислоте с концентрацией
до 15% [60] (коррозия менее 0,1 мм/год). При комнатной темпера-
туре они стойки к царской водке и 30%-ной фосфорной кислоте.
В кипящих азотной, соляной и серной кислотах растворяются.
Нестойки также к плавиковой кислоте.
Тавадзе с соавторами [59, 62, 64] изучил стойкость титановых
сплавов в технологических растворах пищевой промышленности.
Сплавы АТЗ, АТ4, АТ6 и АТ8 обладают хорошей коррозионной стой-
костью к уксусной и муравьиной кислотам до температуры кипения.
Они оказались стойкими также к разным сокам и маринадам при
комнатной температуре и не изменяют их органолептических свойств.
Сплавы АТЗ и АТ8 отличаются высокой стойкостью в условиях про-
изводства кофеина, винного уксуса; к охлаждающим растворам пи-
воваренного производства [62]; к винодельческим растворам при
производстве сухих, крепких и сладких вин; к консервным раство-
рам с поваренной солью и без нее; к чайным растворам с таннином
и без него [59]. В местах сварных соединений эти сплавы стойки к дей-
ствию олеиновой, щавелевой, винной, уксусной и молочной кислот
и технологических растворов консервных и винодельческих произ-
водств [64].
Стойкость титановых сплавов ATI, АТ2, АТЗ, АТ4, АТ6 и АТ8
к настойкам иода, нашатырного аниса, грудного эликсира и жень-
шеня в 10 раз превышает стойкость луженой меди [63], а к экстрак-
там диголен-нео, водяного перца и чебреца — в 15 раз.
Стойкость сплава АТ к производственным растворам таннина
и галловой кислоты превышает стойкость технического титана в 90,
луженой меди в 300 и нержавеющей стали 1Х18Н9Т в 220 раз [63].
Присадка к титану циркония повышает его коррозионные свой-
ства, так как цирконий более стоек к действию минеральных кис-
лот, чем титан [1, 18].
ФИЗИОЛОГИЧЕСКОЕ ДЕЙСТВИЕ ТИТАНА И ЕГО СОЕДИНЕНИЙ
Металлический титан физиологически безвреден [130]. Могилев-
ская [41 ] вводила взвесь пыли металлического титана и его двуокиси
белым крысам интрахеально. В течение 11 месяцев существенных
2 Э-614
17
изменений в организме животных не замечено. Действие пыли ме-
таллического титана, его двуокиси и карбида на белых крыс ана-
логично действию окиси железа [42].
Четыреххлористый титан более токсичен, чем хлористый водо-
род. Работа с TiCl4 противопоказана людям с заболеваниями орга-
нов дыхания и сердечно-сосудистой системы [42].
ГЕОХИМИЯ И ГЕОЛОГИЯ
Геохимические свойства титана изучены Щербиной [73]. Явля-
ясь лиофильным элементом с большим ионным радиусом и высокой
валентностью, титан входит в состав природных минералов в анион-
ной форме. Обычно в минералах титан присутствует в высшей ва-
лентной форме. Только в редких случаях встречаются минералы,
содержащие трехвалентный титан, например лиловые авгиты. _
Высокая валентность и большой ионный радиус титана способ-
ствуют проявлению этим элементом амфотерных свойств в природ-
ных минералах.
Характерными комплексными соединениями титана среди при-
родных минералов являются изополикислоты. Известны, однако, и
гетерополисоединения, например титаносиликаты.
Из-за большого ионного радиуса титан обладает склонностью к
образованию труднолетучих природных соединений с ионной связью.
Летучесть поэтому имеет подчиненное значение в переносе тита-
на в геохимических процессах.
В условиях щелочной среды дериватов нефелин-сиенитовой агпа-
итовой магмы перенос титана происходит, вероятно, в форме от-
носительно легкорастворимых щелочных. титано-силикатов. ..
Главными минералами титана являются соединения с железом
и кальцием.
Закономерности образования месторождений титановых руд
сформулированы Рехенбергом [175] и Малышевым [35, 36, 37]. При-
водим описание их, следуя работам последнего.
Все известные месторождения титановых руд могут быть отнесены к трем груп-
пам: магматогенным, экзогенным (осадочным) и метаморфическим. Они приурочены
к определенным эпохам рудообразования. Месторождения титановых руд группи-
руются в титанорудные районы и провинции, причем в пределах одного района мо-
гут встречаться месторождения разного генетического происхождения. Каждой из
трех групп титанового рудопроявления характерны пространственная и генети-
ческая связи с определенными для них комплексами горных пород.
Магматические месторождения титановых руд связаны с интрузивными комп-
лексами основных и щелочных пород. Месторождения титановых руд в гранито-
идах и типичных ультраосновных массивах неизвестны. Рудные тела месторождений
этого типа обычно залегают в самих интрузивах и не выходят за их пределы. Они
образуются из остаточных обогащенных рудными компонентами растворов габбро-
идной и щелочной магмы.
Форма рудопроявления зависит от интенсивности тектонических подвижек и
степени раскристаллизованности интрузий во время этих подвижек.
18
Месторождения титана, связанные с основными породами, в зависимости от
комплекса вмещающих пород, бывают четырех типов.
С анортозитовыми и габброанортозитовыми массивами связаны ильменитовые,
гематито-ильменитовые и магнезитовые, а иногда рутило-ильменитовые руды.
К массивам габбро, габбро-норитов и габбро-амфиболитов приурочены в ос-
новном ильменито-магнетитовые руды, а в ряде случаев и титаномагнетитовые.
С малыми интрузиями габброидной магмы, представленными дайкообразными,
а иногда и пластообразными телами габбро-диабазов, связаны месторождения бо-
гатых титаном титаномагнетитов, преимущественно в виде зон вкрапленных руд.
В ультраосновных массивах габбро-пироксенитово-перидотитовых формаций
встречаются месторождения бедных титаном титаномагнетитовых руд. Они обычно
как титановые месторождения не имеют практического значения.
Титановые месторождения, связанные с интрузивными комплексами щелочных
пород, менее распространены. Известные рудопроявления, связанные со ще-
лочными породами, распределяются на три генетических типа.
К ультраосновным щелочным массивам приурочены в основном кнопитотитано-
магнетитовые месторождения, к сложному комплексу луяврит-фаяит-уртитов —
месторождения лопарита.
В нефелиновых и щелочных сиенитах известны месторождения сфена, ильмено-
рутила, реже — ильменита.
Магматические месторождения титановых руд пространственно и генетически
связаны с интрузивными комплексами габброидной магмы и приурочены к масси-
вам докембрийского и реже нижнепалеозойского возраста. В более молодых по воз-
расту габброидных массивах богатые титаном крупные месторождения неизвестны.
Экзогенные месторождения представляют наибольший практический интерес,
так как они характеризуются крупными запасами легкообогатимых руд. Осадоч-
ные месторождения образуются за счет разрушения коренных месторождений ти-
тана и габброидных и щелочных пород, всегда содержащих повышенное количество
титановых минералов.
Наиболее благоприятны для образования богатых титаном крупных экзоген-
ных месторождений эпохи развития коры выветривания и последующего ее раз-
мыва, сопровождающегося сортировкой и переотложением продуктов выветри-
вания.
Кора выветривания габброидных пород может сама представлять непосред-
ственный интерес для добычи руд с целью извлечения ильменита с попутным полу-
чением каолинов. Она также является лучшим источником для образования бога-
тых россыпей.
Наиболее крупные и богатые по содержанию титановых минералов прибрежно-
морские россыпи как современные, так и древние. Наряду с титановыми (ильменит,
лейкосен, аризонт, рутил) в них нередко содержатся монацит, циркон и другие
минералы.
Титановые россыпи образовались в эпоху верхнего протерозоя, девона, кар-
бона, мезокайнозоя.
Метаморфические месторождения титановых руд приурочены к древним мета-
морфическим толщам докембрия и реже — нижнего палеозоя. Собственно мета-
морфические месторождения титана представлены зонами вкрапленных руд рутила
или рутила и ильменита. Они образуются в результате метаморфизма титансодер-
жащих габброидных пород и глинистых титансодержащих осадочных пород. Руды
этих месторождений бедны титаном.
Другой тип метаморфических месторождений — метаморфизованные комплек-
сы из ранее образовавшихся месторождений титановых руд магматических и древ-
них россыпей. В результате метаморфизма улучшается качество руд, так как тита-
номагнетит распадается на ильменит и магнетит, а за счет ильменита образуется
рутил. Метаморфические комплексы — основные источники образования рутило-
вых россыпей.
2*
19
ТИТАНОВЫЕ МИНЕРАЛЫ
В земной коре обнаружено около ПО титановых минералов,
которые могут быть разделены на несколько типов (табл. 3). Однако
важное промышленное значение имеют только ильменит, аризонит,
рутил, перовскит (кнопит), сфен и титаномагнетит. Остальные мине-
ралы относятся к числу редких, не образующих значительных
месторождений (табл. 3). Приводим описание важнейших титановых
минералов.
Ильменит (Fe, Mg, Мп) TiOs [15, 22], или титанистый железняк
и менакканит. Название получил от Ильменских гор на Урале, где
был найден [152]. Ильменит имеет изменчивый состав — от FeTiO3
(кричтонит) до MgTiO3 (гейкилит) и MnTiO3 (пирофанит). В неиз-
мененном ильмените может содержаться лишь незначительное ко-
личество Fe2O3. Обычно состав ильменита близок к кричтониту.
Кристаллизуется он в тригональной сингонии, пространствен-
ная группа R3. Размеры элементарной ячейки следующие: а =
= 5,52 А, а = 54,84° (ромбоэдрическая установка); а = 5,126,
с = 14,333 А (гексагональная установка) [84]. По внешнему виду
обычно толстотаблитчатые кристаллы, также острые ромбоэдры, час-
то в тонких пластинках и листочках. Излом роговистый. Твердость
5—6, уд. вес 4,5—5,0. Цвет в массе черный или коричнево-красный.
Непрозрачен. Блеск полуметаллический. Слабо магнитен.
Ильменит является второстепенным минералом в изверженных
породах, замещает магнетит, особенно в габбро и диоритах. В ги-
пергенных условиях превращается в лейкоксен (TiO2 • /гН2О) и
рутил [30]. Коренные месторождения ильменита есть в США (штаты
Виргиния, Нью-Йорк, Миннесота, Калифорния и др.), Норвегии
(юго-западная часть близ Эгерзунда), Канаде (районе Квебека) [145,
167], Южно-Африканской Республике, Мозамбике, Танганьике,
Китае, Корее, Индии. Промышленные запасы ильменитовых пес-
ков встречаются в Индии (штат Траванкор) [195], Австралии (Но-
вый Южный Уэллс), Японии (профектура Хонду), США (Флорида,
Лос-Анжелос) и Бразилии. В СССР крупные месторождения иль-
менитовых руд находятся на Урале (Кусинское, Медведевское),
россыпные месторождения — на Украине (Житомирская обл.).
В табл. 4 приведен химический состав ильменита из крупных мес-
торождений.
Аризонит Fe2O3 • TiO2 [15, 22]. Минерал с моноклинной ре-
шеткой. Излом раковистый. Твердость 5,5, уд. вес 4,25. Цвет темный,
стально-серый. Np < Ng, Nm < 2,62. Оверхольт с соавторами [170]
и Михеев [40] рассматривают аризонит как смесь гематита, иль-
менита, анатаза и рутила. Руднева с соавторами [53] считает ари-
зонит самостоятельным минералом, хотя рентгеновская структура
его выражена не отчетливо.
Встречаются рассыпные месторождения аризонита. Крупные за-
пасы аризонита имеются в Индии (Квилон), Сенегале, СССР
20
Таблица 3
Минерал
Формула
Литература
Титаномагнетит
Ильменит
Кричтонит
Пикроильменит
Манганоильменит
Магнетоильменит
Хёгбомит
Гейкелит
Пирофанит
Сенаит
Таозит
Перовскит
Кнопит
Лопарит
Кеннедит
Ниоболопарит
Улигит
Оливейраит
Делоренцит
Иттрокразит
Аризонит
Браннерит
Металопарит
Пирохлор
Пиррит
Мауцелиит
Левизит
Дербилит
Коппит
Гатчеттолит
Ниоботанталотитанит
Ромеит
Атопит
Шнербертит
Веслинит
Приазовит
Титановые минералы
Окислы
| Fe3O4, содержащий Ti
Группа ильменита
(Fe, Mg, Мп), TiO3
FeTiO3
(Fe, Mg), TiO3
(Fe, Mn)TiO3
FeTiO3 + FegO4
(Al2Fe2[Mg, Ti])O3
MgTiO3
MnTiO3
(Mn, Fe, Pb)TiO3
(Al, Ti)2O3
Группа перовскита
CaTiO3
(Ca, Ce)TiO3
(Ca, Ce, Na) (Nb, Ti)O3
(Fe2, Mg)Ti3O9
Лопарит с повышенным содержанием
ниобия
Ca[Zr, Ti]2O5 c Al2TiO5
3ZrO2 2TiO2 • 2H2O
Титанат Fe, U, Y
Водный титанат иттровых земель
Fe2O3 • 3TiO2
(UO, TiO2, UO2)TiO3
Измененный лопарит
Группа пирохлора
(Na, Ca)2 (Nb, Ti)2 (O, F)7
(Ca, Na, Fe)2 (Nb, Ti, Ta)2O6 (F, OH, O)
(Ca, Pb, Na) (Sb, Fe, Ti)O6
(Ca, Na, Fe)2 (Sb, Ti)2O6 (Fe, OH, O)
6FeO • 5TiO2 • Sb2O5 (?)
Пирохлор, содержащий К и TR
Пирохлор, содержащий U и Н2О
Содержит Nb, Та, Ti, Са, Мп, Се, Al, Th
Сурьмяный пирохлор
» »
» »
» »
Титанотанталониобат Al, Ca, U и др.
[22]
[22, 94]
[22]
22:
22
[22]
22,105
[144]
66]
[22]
22
22
22
22
22
145]
[22]
22
22
16
22
22
22
22
22
[22]
[22]
22
174
21
Продолжение табл. 3
Минерал
Формула
Литература
Фергусонит
Ризёрит
Тиррит
Брагит
Форманит
Кохемит
Группа фергусонита
(Y, Ег, Се, U, Fe) (Na, Та, Ti)O4
Титансодержащий фергусонит
Разновидность фергусонита
» »
Фергусонит, содержащий Та
Фергусонит, содержащий Y, Fr, Th, Zr
[221
22
22
22
45
[45]
Ампангабеит
Полимигнит
Хлопинит
Рутил
Нигрин
Ильменорутил
Хромрутил
Анатаз
Псевдобрукит
Брукит
Стрюверит
Группа самарскита
(Y, Ег, U, Са, Th)2 (Nb, Та, Fe, Ti)3O18
(Са, Fe, Се, Y, Na)2 (Ti, Zr, Nb)3O9
Самарскит с титаном
Группа рутила
TiO2
(Ti, Fe)O2
5TiO2[(Fe, Mn) (Ta, Nb)2O6]
(Ti, Cr)O2
TiO2
Fe2TiO5
TiO2
Ильменорутил c Ta
[22]
22
[22]
•
[20, 202]
[22]
[22]
[22]
22
22
22
Эвксенит
Тантэвксенит
Линдокит
Поликроз
Бломстрандин
Приорит
Эшвегеит
Бетафит
Писекит
Менделеевит
Лоранскит
Виикит
Нуолаит
Эпистолит
Ненадкевичит
Иринит
Мурманит
Ионструпит
Мозандрит
Ринкит
Ринколит
Чевкинит
Баотит
Бафертисит
Титанониобаты
(Y, Ca, Ce, U, Th) (Nb, Ta, Ti,)2O6
Эвксенит c Ta
Ториево-кальциевый эвксенит
(Y, Се, Ca, W, Th) (Ti, Nb, Ta)2O6
(Y, Er, Ca, Th, U) (Ti, Nb, Ta)2O6
To же
Измененный эвксенит
(U, Ca, Th, Y, Ce) (Nb, Ta, Ti)3O9 nH2O
Титано-тантало-ниобат U, TR
(U, Ca, Fe) (Nb, Ta, Ti)2O6OH
(U, Ce, Ca) (Ta, Nb, Zr)2O6
Разновидность эвксенита
» »
Ниобат, содержащий SiO2, TiO2, Na2O,
(Na, Ca) (Nb, Ti) [Si2O7] • 2H2O
(Th, Ce, Ca, Sr, Na, K)4 [Ti, Nb, Ta,
Fe]6 [O, OH]l7
NaTi2 [SiO4] [OH] • H2O
Силикат Ce, Ca, Na, Ti, F
Близок к ионструпиту
» » »
10CaSiO3(Ce)2(TiO3)3 • 3CaFe2
(Ca, Fe)Ce2(Si, Tij3O, О
Ba(Ti, Nb)oSio07
BaFe2TiSi2O9 “
[22]
22
22
22
22
22
22
22
22
22
22
22
22
22]
[32]
[7]
[22]
22
22
22
22
[22
[56]
[57]
22
Продолжение табл. 3
Минерал
Формула
Литература
Дабунлевит
ферсманит
Шетелигит
Цирконолит
Белянкинит
Герасимовскит
Танталополикраз
Титанобетафит
Бломстрандит
Ломоносовит
Ловчоррит
Щербаковит
Самиресит
Эшинит
Эльсвортит
Циркелит
Кальковскит
Ловенит
Розенбушит
Лампрофиллит
Рамзаит
Лоренцит
Молентраафит
Юкспорит
Кильгауит
Титанит (сфен)
Греенволит
Чевкинит
Астрофиллит
Бенитоит
Т итаноэльпидит
Ловозерит
Нептунит
Джоакинит
Лейкосфенит
Чинлузуит
Нарсарсукит
Шёрломит
Линозит
(К, Na, Ba, Са, Мп) (Ti, Nb, Fe, Mg)
(Si, Al)2 (O, OH)7 . nH2O -
(Ca, Na)2 (Ti, Nb) (O, Fe)3 . (Ca,
Na)2Si (O, F)3
(Ca, Y, Sb, Mn)2 (Ti, Nb, Ta)2 (O, OH)7
Цирконотитанат U, Th, Nb и др.
TiO2 - Nb2O5 • ZrO2 • CaO -H2O
Ниобобелянкит
(Y, Ce, Ca, U, Th) (Ti, Nb, Ta)2O6
Батафит c Ti
(U, Ca, Fe, Ce) (Nb, Ta, Ti)3O9 • nH2O
Na2Ti [(Si, P)O4]
Разновидность ринколита
Na, К, Ba2 (Ti, Nb)2 (Si2O7)2
Бетафит c Pb и U
(Ce, Ca, Th, U) (Ti, Nb)2O6
(U, Ca, Fe) (Nb, Ta, Ti)2O6, OH
(Ca, Zr, Fe2+)2 (Ti, Nb, Zr)2O7
T итаносиликаты
(Fe, Ce)2O3 . 4(Ti, Si)O2
(Ca, Na) (Zr, Nb, Ti, Fe, Mn)FSiO4 (?)
6(Ca, Na) (O, F) . 2(Zr, Ti)O2 • 4SiO2
Титаносиликат Sr и Na
Разновидность лоренцита
Na2(TiO)2Si2O7
Титаносиликат Ca и Na
Титаносиликат Na, К, Ca, Sr, Ba
(Ca, Y, Ce) (Ti, Al, Fe3+)SiO5
CaTiSiO5
(Ca, Mn)TiSiO5
(Ca, Ce, Y, Th) (Ti, Fe3+)SiO6
(K2, Na2, Ca) (Fe2+, Mn)4 (Ti, Zr) [Si2O„
OH]
BaTi[Si3O9]
NaiTi, Zr)He[Si3O9]2
(H, Na, K) (Ca, Mn, Mg) (Zr, Ti)H6
[Si3O9]2
Na2FeTi[Si4O12]
NaBa[Ti, Fe]3 fSi4O12]
Na4Ba(TiO)2 (Si2O5)5
2(Na, K)2 • l/2(Mn, Ca) . 0,3(Ti,Zr)O2 •
14SiO2 • 9H2O
Na(Ti, Fe) [(O, OH, F)Si4O10]
Ca3(Fe, Ti)2 [(Si, Ti)4]3
Mg2SiTiO6 • CaMgSi2Oe
[55]
[22]
[45]
[45]
[45]
[45]
[45]
[45]
[22]
[45]
[22]
[45]
[22]
[22]
[28]
[6]
[22]
[15]
[22]
22
22
22]
22
[22:
[22]
[22]
[22, 50]
[22]
[22:
[15]
22
22
[22, 24]
22]
22
221
23
Продолжение табл. 3
Минерал
Формула
Литература
Варвикит
Кальциркит
Фрайденбергит
Льюисит
Тамеллит
Прочие минералы
(Mg, Fe)3 TiB2O8
Титаноцирконат
Na2O • Fe2O3 • 7TiO2
5СаО • 2ТЮ2 3Sb2O5
Ba2(Fe3+ Ti, Mg)2 [O(Si4OI2)] • H2O
[22]
[26]
[1' 12]
[22]
[160]
Украина, Центральная часть РСФСР). Химический состав аризо-
нита приведен в табл. 5.
Рутил TiO2 [15, 22,401. Кристаллизуется в тетрагональной син-
гонии, фед. гр. Р4!тпт. Параметры решетки рутила см. гл. IV.
Кристаллы обычно призматические, как правило, вытянутые, ино-
гда тонковолокнистые, часто тонкоигольчатые. Излом неяснорако-
вистый. Хрупок. Твердость 6,0—6,5, уд. вес 4,18—4,25. Блеск ме-
таллическо-алмазный. Цвет красновато-коричневый, переходящий
в красный, иногда желтоватый, синеватый, фиолетовый, черный; в
проходящем свете ярко-красный. Черта светло-бурая.
Таблица 4
Химический состав ильменита в %
Компо- нент Вирги- ния (США) [85] Нью-Йорк (США) [85] Траван- кор (Ин- дия) [85] Иври (Ка- нада) [85] Норвегия [85] Кусин- ское мес- торожде- ние (Урал) [85] р. Соби (УССР) [23]
тю2 51,30 44,30 54,30 42,50 42,30 44,00 52,77—62,25
FeO 37,90 36,70 26,00 31,10 33,90 31,40 29,45—36,68
Fe2O3 1,60 4,40 15,50 20,70 12,90 16,90 1,41—12,73
SiO2 4,60 3,20 1,40 0,88 3,50 1,84 0,52—1,50
A12O3 0,55 0,19 1,10 1,05 1,80 - — 1,25—1,30
P2O5 0,17 0,07 0,26 Следы 0,01 0,15 •“ 1,1 -•
ZrO2 — 0,006 2,18 — 0,03 —•
MgO 2,35 0,80 0,85 2,00 1,60 2,76 0,20—0,28
MnO . 0,70 0,35 0,40 0,04 0,35 0,72 0,40—0,95
CaO 0,59 1,00 0,08 0,10 0,20 0,10—0,14
v2o5 0,07 0,24 0,20 0,36 0,40 — 0,12—0,14
Cr2O3 Нет 0,001 0,07 0,15 0,07 0,05 —
SnO2 0,02 0,001 0,06 0,001 0,001 — —--
CuO 0,0005 0,004 0,01 0,08 0,60 — IM
CoO - - —• - • 0,02 * .
Ce2O 0,01 0,002 0,08 — — — - —
PbO 0,02 - ' 0,01 >» —
wos —• 0,005 Д* 0,001 * >' — -
NiO - — 0,04 — 0,02 — —- ——
Au — 0,006 1 •1 “ — —
Pt — 0,02 — 1- - — ।
Nb 7" - 0,002 0,10 • 0,005
24
Таблица 5
Химический состав аризонитовых руд и концентратов в %
Компонент Квилон (Ин- дия) [85] Синегал [85] УССР (образцы)
1 2 3
TiO2 60,30 54,70 61,35 62,36 62,40
FeO 9,70 7,30 — 0,57
Fe2O3 24,80 30,30 26,69 26,60 27,16
Si02 1,40 1,30 1,56 1,80 0,68
AlgO3 1,00 0,50 2,63 2,63 0,68
PA 0,17 0,17 i — 0,10
ZrOg 0,60 2,37 »
Mg<5 0,65 1,90 2,50
MnO 0,40 1,32 — -— 0,89
CaO 0,15 0,10 — — 0,66
v2o6 0,26 0,27 — *—- 0,14
CrgOg 0,14 0,23 2,03 1,68 1,53
SnOo 0,01 — — •
CoO 0,01 — " - -—— - —
wo3 0,004 — “ - 1
Au 0,004 — —-—
Pt 0,02 - - “ -
Nb 0,10 —- - — —
s — — - - — — - 1,41
Одноосный, положительный, наиболее светопреломляющий из
всех породообразующих минералов и обладает чрезвычайно высоким
двупреломлением. Показатели светопреломления рутила приведены
в гл. IV.
з, Nb2O5, Та2О5, WO3
Рутил является второстепенным минералом в таких извержен-
ных породах, как рогообманковый диорит, сиенит, гранит, амфи-
болит. Часто образуется в кварце или в полевом шпате в виде иголь-
чатых кристаллов, пронизывающих кварц.
Коренные месторождения рутила известны в США (Арканзас),
на Мадагаскаре, в Бразилии, Канаде (Квебек) и Норвегии, россып-
ные месторождения — в Австралии, США (Флорида) и СССР (Укра-
ина [25]).
Содержание TiO2 в рутиле меняется в пределах 94—98%. Рутил
содержит в виде примесей FeO, V2O5, Сг2О
и SnO2.
Перовскит (кнопит) CaTiO3 [15, 22, 40]. Псевдокубический ми-
нерал моноклинной сингонии [210], фед. гр. Р211т, а, Ь, с»7,60 А,
Р ~ 90°, у псевдокубической ячейки а = 7,59—7,65 А [211], об-
наружен Креммером на Урале [174].
Кристаллы обычно имеют кубический облик. Грани куба со
ш фиховкой параллельно ребрам. Спайность по кубу весьма со-
вершенная. Излом неровный до неяснораковистого. Хрупкий. Твер-
дость 5,5, уд. вес 4,0. Блеск алмазный до металловидно-алмазного.
Цвет светло-желтый, медово-желтый, оранжево-желтый, красно-
бурый, сиреневато-черный. Черта бесцветная, черно-желтая.
25
Показатели преломления изменяются в пределах 2,34—3,37 [71 ].
Двойное лучепреломление слабое.
Перовскит может образовывать изоморфные смеси с NaCeTi2O6,
Na2Nb2O6 и Ca (Fe3+)Nb2O6, в результате чего получаются минера-
лы: кнопит, дизаналит, лопарит. Химический состав перовскита
приведен в табл. 6.
Таблица 6 Таблица?
Химический состав перовскита Химический состав сфена в %
(кнопита) в % .
Образец
Компонент Образец 1 [19] 2 [118]
1 [9] 2 [72]
SiO2 TiO2 ThO2 TR2C3 Nb2O5 Ta2O5 0,12 56,36 2,20 0,64 0,09 54,88 0,22 7,99 1,71 0,16 SiO2 TiO2 ThO2 tr2o8 Nb2O5 Ta2O5 A12O3 Fe.,O3 29,96 39,12 0,41 0,56 30,85 35,74 0,10 1,33
AI2O3 Fe2O3 FeO 0,80 1,60 Нет 0,09 0 74 0,55 0,84 1,98 1,36
МпО CaO SrO MgO V2O5 K2o Na2O 0,06 37,20 Следы 0,10 0,42 Следы 30,87 1,56 0,03 0,06 0,13 1,77 FeO MnO CaO SrO MgO KoO Na2O 0,31 0,04 27,51 0,08 0,13 0,39 0,02 0,07 27,78 0,05
H2O“ - 0,12 H2O“ 0,14 —
H2O+ 0,32 0,25 H2o+ 0,12 WWW
Сумма 99,82 100,67
Перовскит мало распространенный минерал. Известно только
одно крупное месторождение перовскита [31 ].
Титанит (сфен) CaTiSiO5 [15, 22, 40]. Минерал моноклинной сис-
темы, фед. гр. С2/с. Параметры решетки следующие: а = 6,55,
в = 8,70, с = 7,43 А, Р = 11,9°43', z = 4 [209]. Кристаллы сфе-
на имеют разнообразный облик (призматические, часто клинообраз-
ные). Твердость 5,0—5,5, уд. вес 3,40—3,56. Блеск от алмазного до
смолистого. Цвет бурый, серый, желтый, зеленый, розово-красный,
черный. Черта белая, слегка розоватая. Показатели преломления
Ng = 2,092, Nm = 1,970, Np = 1,950.
Титанит содержит в различных количествах примеси Fe, Мп,
TR, Nb, F и др. Химический состав его приведен в табл. 7.
Титанит широко распространен как второстепенный минерал в
изверженных породах. Встречается в основных рогообманковых
гранитах, сиенитах, диоритах и особенно характерен для нефели-
новых сиенитов. Крупные месторождения титанита известны в Ти-
роле, Италии и Франции. В СССР месторождения титанита имеются
на Кольском полуострове (Хибины) и Урале (Вишневая гора).
26
ОСОБЕННОСТИ ХИМИЧЕСКИХ СВОЙСТВ ТИТАНА
И КЛАССИФИКАЦИЯ ЕГО ХИМИЧЕСКИХ СОЕДИНЕНИЙ
Химические свойства титана, как и других элементов, опреде-
ляются строением внешней электронной оболочки атома. Исходя из
электронной структуры атома титана, можно объяснить особен-
ности его химического поведения и выяснить наиболее характерные
классы соединений, образуемые этим элементом.
Титан относится к IVB группе периодической системы элементов
Д. И. Менделеева. Электронная оболочка атома титана имеет строе-
Рис. 1. Зависимости числа внешних электронов от
условной идеальной плотности металлов [49].
ние ls22s22p63s23p63d24s2. Атом титана парамагнитен, у него два
неспаренных электрона и три незаполненных d-орбитали.
Электроны 3d- и 45-орбиталей титана имеют примерно одинаковые
энергии связи с ядром, равные 6,9 и 6,8 эв соответственно. Электроны
нижележащей Зр-орбитали имеют высокую энергию связи, равную
35 эв, и поэтому они практически не могут принимать участия в хи-
мических реакциях. Вследствие этого высшая окислительная валент-
ность титана равна 4. Титан может также проявлять окислительную
валентность, равную 3. В таких соединениях, как TiO, TiS, TiSe,
TiTe, имеющих соотношения между атомами, соответствующие двух-
валентному состоянию, он проявляет металлическую природу связи.
В элементарном состоянии титан обладает металлическими свой-
ствами. При образовании металлических соединений принимают
участие все четыре электрона атома титана, находящиеся за преде-
лами аргоновой оболочки. Это вытекает из характера изменения
идеальной плотности металлов первого большого периода (рис. 1)
НУ]. Таким образом, металлическая валентность титана равна его
окислительной валентности, т. е. 4.
27
Титан образует металлические соединения со многими металлами
и неметаллами. В соединениях с последними металлическая природа
связи титана проявляется обычно, если на один его атом приходится
менее четырех валентных единиц неметалла. В противном случае
валентные электроны титана утрачивают свободу и образуются со-
единения с ковалентной связью.
Будучи переходным металлом с незаполненными Sd-орбиталями^
титан проявляет склонность к образованию многочисленных ком-
плексных соединений. В образовании связи в комплексных соеди-
нениях принимают участие незаполненные 3d, 4р-орбитали.
В водных растворах соли титана мало устойчивы и частично или
полностью гидролизуются. Устойчивыми являются только соли ти-
тана с анионами, обладающими сильными комплексообразующими
свойствами (F~, CNS-, SOl”, С2О1“ и др.).
Для титана характерно образование основных (окси) солей.
По-видимому, большая устойчивость оксисолей объясняется энерге-
тической выгодностью связи 2р-электронов с 3d-электронами титана.
В соответствии с положением в периодической системе химиче-
ских элементов титан проявляет амфотерные свойства. С основания-
ми он образует многочисленные титанаты, которые можно рассмат-
ривать как соли гипотетических титановых кислот.
Для титана характерно также образование титаноорганических
соединений. В этих соединениях связь между металлом и углеродом
осуществляется чаще всего через атомы кислорода и других эле-
ментов. Известны, однако, соединения и с непосредственной связью
титана с углеродом, которые отличаются малой устойчивостью.
Все известные соединения титана можно разделить на следующие
классы: металлические соединения, окислы, гидроокиси, титанаты,
галогениды, сульфаты, фосфаты, комплексные соединения с орга-
ническими лигандами, титанорганические соединения и др. Эта клас-
сификация положена в основу систематизации экспериментального
материала данной монографии.
Литература
1. Андреева В. В., Глухова А. И.— Ж- прикл. хим., 1962, 35, 1771.
2. Андреева В. В., Казарин В. И.— ДАН СССР, 1958, 125, 1048.
3. Балихин С. В.— Изв. АН СССР. Металлы, 1965, 2, 77.
4. Бардин И. П., Ревякин А. В. — В кн.: Титан и его сплавы, 2. Изд-во
АН СССР, М., 1958, 119.
5. Борисова Е. А., Барданов К- В.— Бюлл. цветной металлургии,
1963, 2.
6. Бородин Л. С., Быкова А. В., Капитонова Т. А., Пятен-
к о Ю. А. — ДАН СССР, 1960, 134, 1188.
7. Борнеман-Старинкевич И. Д.— Зап. Всесоюзн. минер, об-ва,
1959, 88, 207.
8. Б р ы н з а А. П., Герасвотина Л. И., Хмелевская С. А.—
Изв. высш. уч. зав. Химия и хим. техн., 1964, 3, 450.
9. Бурова Т. А.— В кн.: Минералы Хибинских и Ловозерских тундр. Изд-во
АН СССР, М., 1937.
28
in Бубницкая C.M., Струн кин В. А., Зальцман Т. Д., Со-
рокин Ю. И. Металловедение титана. «Наука», М., 1964, 144.
1 Виноградов А. П.— Труды биохим. лабор. АН СССР, 1935, 3, 5.
2* Виноградов А. П.— Геохим., 1962, 7, 291.
3 Виноградов А. П.— Геохим., 1956, 1, 6.
/Виноградов А. П. Геохимия редких и рассеянных химических эле-
ментов в почвах. Изд-во АН СССР, М., 1950.
5 В и н ч ел л А. Н., Винчелл Г. Оптическая минералогия. ИЛ, М., 1953.
16* В о й н а р А. О.— Биохим., 1953, 18, 29.
17* Войтович Р. Ф.— Ж- физ. хим., 1965, 39, 458.
18* Г л у х о в а А. И., Андреева В. В.— Ж. прикл. хим., 1962, 35, 1778.
19^ ГорощенкоЯ. Г. Физико-химические исследования переработки редкозе-
’ мельных титанониобатов сернокислотным методом. Изд-во АН СССР, М.-Л.,
I960.
20. Голутвин Ю. М.— Ж- физ. хим., 1959, 33, 1798.
21. Г р и ш т е й н Д. Л. Ядерные процессы в звездах. ИЛ, М., 1957.
22. Д а н а Э. С. Описательная минералогия. ОНТИ, Л.-М., 1937.
23. Д я д ч е н к о М. Г.— ДАН УРСР, 1958, 4, 445.
24. Ефимова А. Ф., Катаева 3. Т.— ДАН СССР, 1959, 129, 896.
25. Жердева А. Н., Абу л ев и ч В. К-— В кн.: Минеральное сырье, 1.
Госгеолтехиздат, М., 1960, 26.
26. 3 д о р и к Т. Б., С и д о р е н к о Г. А., Б ы к о в а А. В.— ДАН СССР,
1961, 137, 681.
27. И о ф ф е В. Г.— Изв. высш. уч. зав. Цветная металлургия, 1964, 6, 125.
28. Калита А. П. и др.— Труды ИМГРЭ, 1962, 8, 201.
29. К о н о п к и н а 3. И.— В кн.: Титан и его сплавы, 7. Изд-во АН СССР, М.,
1962, 274.
30. К у к о в с к и й Е. Г., К о н о н о в Ю. В.— Геолог, ж., 1961, 21, 97.
31. Кухаренко А. А., Багдасаров Э. А.— Материалы Всесоюзн.
н.-и. геол, ин-та, 45. 1961, 37.
32. Кузьменко М. В., Казакова М. Е.— ДАН СССР, 1955, 100, 1159.
33. Лайнер Д. И., Цыпин М. И.— Изв. АН СССР. ОТН. Металлургия и
топливо, 1959, 5, 131.
34. Л у ч к и н Г. П., И л ь и н Г. Г.— Физика металлов и металловедение,
1956 2 521
35. Мал ыш е в И. И.— ДАН СССР, 1957, 112, 311.
36. М а л ыш е в И. И.— Сб. научи.-техн, информации. Мин-во геол, и охраны
недр, 1955, 11,35.
37. Малышев И. И. Закономерности образования и размещения месторож-
дений титановых руд. Госгеолтехиздат, М., 1957.
38. Минц Р. С., Шелест А. Е., Малков Ю. С.— В кн.: Титан и его
сплавы, 10. Изд-во АН СССР, М., 1963, 95.
•39. Мирочников В. С., Резниченко В. А.— В кн.: Титан и его
-лавы, 9. Изд-во АН СССР, М., 1963, 186.
40. Михеев В. И. Рентгенометрический определитель минералов. Госгеол-
техиздат, М., 1957.
49 ^0Гилевская Я-— Гигиена и санитария, 1956, 3, 20.
^.Могилевская О. Я., Мельникова Е. А., Мезенце-
43 Ff3 — Цветные металлы, 1957, 4, 51.
• п е л е н ь И. М. Металлургия цветных металлов и методы анализа. Метал-
44 £УРГизДат> М., 1965, 323.
м е я н о в Ан. Н. Давление пара химических элементов. Изд-во АН
СССР, М., 1961.
46* НИ°бИЙ И тантал- Справочник для геологов. Госгеолтехиздат, М., 1959.
пи колаева С. А., Зиновьев В. А. Металловедение титана.
«Наука», М., 1964, 139.
• Остроумов Э. М.— Геохим., 1956, 1, 90.
6 Ivr? Р н ° В- Н.— Изв. АН СССР. ОТН. Металлургия и топливо, 1962,
29
49. Полинг Л. Общая химия. «Мир», М., 1964, 402,
50. Пэн Ужи-чжун, Ма Чже-шэ н.— Кэсюэ Тунбао, Кехие тунбао,
Кехие tongbao, 1963, 5, 67.
51. Ревякин А. В.— Изв. АН СССР. ОТН. Металлургия и топливо, 1961, 5,
113.
52. Ревякин А. В.— В кн.: Титан и его сплавы; 8. Изд-во АН СССР, М., 1962,
175.
53. Р у д н е в а А. В., М о д е л ь М. С., Соловьев В. И.— В кн.: Титан
и его сплавы, 9. Изд-во АН СССР, М., 1963, 10.
54. Серебренников Н. Н., Г е л ь д П. В.— Изв. высш. уч. зав. Цветная
металлургия, 1961, 4, 80.
55. С е м е н о в Е. И., Бурова Т. А.— ДАН СССР, 1955, 101, 1113.
56. Семенов Е. И., X у н Вен-Син, К а л итон ов а Т. А.—
ДАН СССР, 1961, 136, 915.
57. С е м е н о в Е. И., У ж а н Пэй-шань. Sci. Record, New Series, 1959*.
3, 12.
58. Сорокин Ю. И., Цейтлин X. Л. Металловедение титана. «Наука»,
М., 1964, 160.
59. Т а в а д з е Ф. Н., Манджгаладзе С. Н., Даш ни ан и Т. С.,
Лордкипанидзе И. Н.— В кн.: Титан и его сплавы, 7, Изд-во
АН СССР, М., 1962, 246.
60. Тавадзе Ф. Н., Манджгаладзе С. Н., Лордкипани-
дзе И. Н., Д а ш н и а н и Т. С.— В кн.: Титан и его сплавы, 7. Изд-во АН
СССР,М., 1962,253.
61, Тавадзе Ф. Н., П а н д ж г о л а д з е С, М., Дашниани Т. С.,
Лордкипанидзе И. Н.— В кн.: Титан и его сплавы, 7. Изд-во
АН СССР, М., 1962, 263.
62. Тавадзе Ф. Н., Л о ш х и Т. А.— В кн.: Титан и его сплавы, 10. Изд-во
АН СССР, М., 1963, 154.
63. Т а в а д з е Ф. Н., Манджгаладзе С. Н., Лордкипани-
дзе И. Н., Дашниани Т. С. — В кн.: Титан и его сплавы, 10. Изд-во
АН СССР, М., 1963, 151.
64. Тавадзе Ф. Н., Л а ш х и Т. А. Металловедение титана. «Наука», М.,
1964, 166.
65. Титановые сплавы. Всесоюзный ордена Ленина научно-исследовательский ин-т
авиационных материалов. Отд. научн.-техн. информации. М., 1959.
66. Тихоненков И. П., Казакова М. Е.— Зап. Всесоюзн. минер,
о-ва, 1957, 86, 641.
67. Ф е д о т о в С. Г. —Изв. АН СССР. Неорг. материалы, 1965, 1, 1737.
68. Цветова Р. В., Красильников А. И.— Ж- физ. хим., 1965, 39,
207.
69. Ц е й т л и н X. Л., Файнгольд Л. Л., Стрункин В. А. Метал-
ловедение титана. «Наука», М., 1964, 150.
70. Ш в а р ц Г. Л., Макарова Л. С., А к и м ц е в а А. П. Таблицы кор-
розионной стойкости титана и его сплавов в различных агрессивных средах.
М., 1961.
71. Ш и л и н Л. Л.— ДАН СССР, 1940, 28, 347.
72. Шилин Л. Л., Янченко М. Т.— ДАН СССР, 1962, 144, 639.
73. Щ е р б и н а В.— ДАН СССР, 1952, 85, 839.
74. Юрк Ю. Ю. Редкие минералы пегматитов Приазовья. Изд-во АН УССР, К-»
1956.
75. Я в н е л ь А. А., Дьяконова М; И.— В кн.: Метеоритика, 15. 1958,
136.
76. Andenstedt Н. К-, Pequignot J. R., R ay m er J. M.— Trans.
Am. Soc. Metals, 1952, 44, 990.
77. Alexander W. A., P d g e о n L. M.— Canad. J. Research., 1950, 28B,
60.
78. Allen J. S. V., P о о 1 M. L., К u r b a t о v J. D., Quill L. L.— Phys,
Rev., 1941, 60, 155, 425.
30
70 Andersson G.— Phil. Mag., 1954, 45, 621.
In A st о n F. W.~ Proc. Roy. Soc., 1935, A149, 396.
я? A s t о n F. W.~ Nature, 1934, 133, 684.
яо A v e п M. H., С r a i g R. S., W a i t e T. R., W a 1 1 a c e W. E.— Phys.
Rev., 1956, 102, 1263.
яч Bardossy G., Bardossyne L. Z.— Fold, kosl., 1953, 83, 230.
84* В a r t h T. F. W., P о s n j а к E.— Z. Kristallogr., 1934, 88, 265.
Я5* Barksdale J. Titanium. The Ronald press Co, N. Y., 1949.
»fi’ Berry R. L. P., R а у n о r G. W.— Research., 1*953, 6, 215.
Я7* В axter G. P., F e r t i g G. J.— J. Am. Chem. Soc., 1923, 45, 1228.
88* В a x t e r G. P., В u t 1 e г A. Q.— J. Am. Chem. Soc., 1926, 48, 3121.
89* Baxter G. P., Butler A. Q.— J. Am. Chem. Soc., 1928, 50, 408.
90. В e r z e 1 i u s J. J.— Pogg. Ann., 1825, 4, 1.
91. В 1 о c h e r J. M., Campbell J. E.— J. Am. Chem. Soc., 1949, 71, 4040.
92* В 1 au dr у В. J., D a a h e A. H.— Trans. AIME, 1962, 224, 770.
93*. Bobko E. W.— Boden kunde Aflanzenernahr, 1937, 4, 328.
94. * Bourn. Catalogue de la Collection mineralogi ique particuliere du Roi, 430,
Paris, 1813. *
95 Burk D. L., Estermann Friedberg S. A.— Z. Phys. Chem.*
(BRD), 1958, 16, 183.
96. Burgers W. G., Jacobs F. M.— Z. Kristallogr., 1936, 94, 299.
97. C a m e г о n A. E., W i c h e г s E.— J. Am. Chem. Soc., 1962, 84, 4175.
98. С a r p e n t e r L. G., R e о v e 1 1 F. R.— Nature, 1949, 163, 527.
99. C a r p e n t e r L. G., Mair W. N.— Proc. Phys. Soc., 1951, 64, 57.
100. Clark T. C.— Trans. AIME, 1949, 185, 588.
101. Cl usi us K-, Franzosini P.— Z. Phys. Chem. (BRD), 1958, 16, 194;
Helv. chim. acta, 1958, 41, 1342.
102. С о n j e a u d P.— J. Rech. Centre natl. research. Sci., 1955, 32, 273.
103. Daunt J. G., H e e г С. V.— Phys. Rev., 1949, 76, 715.
104. Deardorff D. K-, Hayes E. T.— J. Metals, 1956, 8, 509.
105. Dick. Min. Soc., London, 1893, 10, 145.
106. Demodaran V., Gupta J.— Scient. and Industr. Res., 1955, 14,
B292.
107. Edwards J.W., Johnston H. L., Ditmars W. E.— J. Am.
Chem. Soc., 1953, 75, 2467.
108. E n d e 1 1 J.— Braunkohle, 1952, 4, 446.
109. Eppelsheimer D. S., Penman R.R.— Nature, 1950, 116, 960.
110. F a s t J. D.— Z. anorg. Chem., 1939, 241, 42.
111. Flugge S., Mattauch J. — Phys. Z., 1943, 44, 181.
112. Frenzel G.— Neues Jahrb. Mineral. Monatsh., 1961, 1, 12.
113. Gee E. A., Golden L. B., Lusby W. E.— Ind. Eng. Chem., 1949,
41, 1668.
114. Gi'ese C.F., Benson J. L.— Phys. Rev., 1958, 110, 712.
115. G i 1 1 e s P. W., Wheatley Q. L.— J. Chem. Phys., 1951, 19, 129.
116. ° Id b e r g D. E.— Annual Rev. Phys. Chem., 12, Palo Alto, Calif., 1961,
117.
118.
119.
120.
121.
122.
, Acherman W. L.— Ind. Eng. Chem
• j
1952, 44, 1930.
Gordillo С. E.— Bull. Acad. nac. cient., 1958, 40, 135.
Gregor W— J. Phys., 1791, 72, 152.
Gregor W— Crell s. Ann., 1791, 40, 55, 103.
« ° ё g J- E— Canad. J. Chem., 1954, 32, 1039.
".a m b e r g. Geologiska Foreningens i Stockholm Forhand lingar 1890, 12,
098 (Stokkholm).
Ida TJP 0 n d w-R-> Kundu D. N., Pool M. L.— Phys. Rev., 1933,
VU, 157.
24‘ ?au^r M-> Kamen E. L„ Kessler H. D., McPherson D. J—
125 Metals, 1951, 3, 881.
n 1 b b s R. F.— Nucl. Data, Nat. Bur. Stand. Cirk., 1950, 449, 43.
31
126. H i е г А. О.— Phys. Rev., 1938, 53, 282.
127. Honda М., Lal D.— Nucl. Phys., 1964, 51, 363.
128. Honda M., Arnold J. R.— Geochim. cosmochim. acta, 1961, 23, 219.
129. Huizenga J. R., Wing J.— Phys. Rev., 1957, 106, 90.
130. Hudson H. E.— J. Roy. Inst. Chem., 1956, 80, 187.
131. Hutchinson G. E., Permar P. H.— Corrosion, 1949, 5, 319.
132. Ind. Eng. Chem., 1954, 1, 103A.
133. I s h i b a s h i M., Higashi S.— Bull. Soc. Solt. Sci., Japan, 1958, 12,
322.
134. Jaeger F. M., Rosenbohm E., Fonteyne R.— Proc. Acad.
Amsterdam, 1936, 39, 442.
135. Jaeger F. M., Rosenbohm E., Fonteyne R.— Proc. Acad. Am-
sterdam, 1936, 39, 462.
136. J a e t e r F. M., Rosenbohm E., Fonteyne R.— Rec. Trav.
Chim., 1936, 55, 615.
137. Jeffries C. D.— Phys. Rev., 1953, 92, 1262.
138. Karim A., Kham D. H.— Soil. Sci., 1955, 80, 227.
139. Kiefer G. C.-L Iron Age, 1952, 170, 170.
140. Kiefer G.C., Harple W. W.— Metal Progress, 1953, 63, 74.
141. Klaproth M. H. Beitrage zur chemischen Kentnis der Mineral korper, 1.
Posen—Berlin, 1795.
142. Klaproth M. H. Beitrage zur chemischen Kentnis der Mineralkorper,
2. Posen —Berlin, 1797.
143. Kneip G. D., Betterton J. O., Scarbrough J. O.— Phys.
Rev., 1963, 130, 1687.
144. К nor r i ng О., Cox K- G.— Mineral Mag., 1961, 32, 676.
145. Knoerr A. W.— Eng. Mining. J., 1952, 153, 72, 76, 79.
146. К о j i m a H., T abble R. S., Williams D. E.— Proc. Roy. Soc.,
1961, A260, 130, 237.
147. К о 1 s к у H., Gilles P. W.— J. Chem. Phys., 1954, 22, 232.
148. Kothen C. W., Jonston H. L.— J. Am. Chem. Soc., 1953, 75, 3101.
149. Krishnan K. S., Jain S. C.— Nature, 1952,170, 759.
150. Kroll W.— Metallwirtsch., 1939, 18, 77.
151. Kroll W.— Trans. Electrochem. Soc., 1940, 78, 35.
152. Kupffer A. T.— Kast. Arch. Nat., 1827, 10, 1.
153. Lampadius W. A.— Crells Ann., 1796, 1, 259.
154. de Lapparent J.— Tschermak, 1937, 49, 11.
155. Levinger B. W.— J. Metals, 1953,5, 195.
156. Leutwein F.— Freiberger Forschungsh., 1956, C, 28.
157. Littman F. E., Church F. M., Kinderman E. M.— J. Less-
Common Metal, 1961, 3, 367.
158. Lohberg K-, Schiliecher H.-W.— Z. Phys. Chem. (BRD), 1958,
15, 223.
159. Luis M.— Phys. Rev., 1952,88, 225.
160. Mazzi F., Rossi G.—• Z. Kristallogr., 1965, 121, 243.
161. Maschinenmarket, 1959, 65, 17, 15.
162. Maykuth D. J., Ogden H. R., Jaffee R. J.— Trans. AIMEt
1953 197 225.
163. McQui’llan A. D.— J. Inst. Metals, 1950, 78, 249.
164. Moore С. B., Brown H.— J. Geophys. Res., 1963, 68, 4293.
165. Moore С. B., Brown H.— Geochim. cosmochim. acta, 1962, 26, 495.
166. Moreland P. E., H e у m a n n D.— J. Inorg. Nucl. Chem., 1965, 27,
493.
167. M о r e s s g W. B.— Canad. Paint and Varnish Mag., 1951, 25, 22, 24, 65.
168. Ogden H.R., Maykuth D. J., Finaly W. L., Jaffee R. I. —
J. Metals, 1951, 3, 1150.
169. Or i an i R.A., Jones T. S.— Rev. Sci. Instrum., 1954, 25, 248.
170. Overholt J. L., V a u x G., Rodda J. L.— Amer. Min., 1950, 35,;
117.
32
i7i Poul ari kas A., Fi nk R. W.— Phys. Rev., 1959, 115, 989.
179’ Rose H.— Pogg. Ann., 1829, 15, 149.
.7q Rose H.— Gilb. Ann., 1823, 73, 74, 76.
174 Rose H.— Ann. Phys. Chem., 1839, 48, 558.
175 Rechenberg H. P.— Neues Jahrbuch Minerallogie, Monatsh., 1955, 4,
87
176 Rozier H. L.— J. Appl. Phys., 1963, 34, 2350.
177 S c h e 1 b e r g A. D., S a m p s о n M. B., Mitchell A. C. G.— Phys.
’ Rev., 1948, 74, 1239.
178 Schofield T. H., Bacon A. E.— J. Inst. Metals, 1953, 82, 167.
79* Schofield T. H.— Proc. Phys. Soc., 1954, B67, 845.
180 S с о v i 1 G. W.— J. Appl. Phys., 1953, 24, 226.
81’ Scovil G.W.-J. Appl. Phys., 1956,27, 1196.
182. Schulze A.— Z. Metalkunde, 1930, 23, 261.
183. Sergent B. W., Y a f f e L., Gray A. P.— Canad. J. Phys., 1955, 31
235.
184 Seren L., Friederlander H. N., Turkel S. H.—Phys. Rev.,
1947,72, 888.
185. Shorp R.A., Diamond R. M.— Phys. Rev., 1954, 93, 358.
186. Slater J. C.— Phys. Rev., 1930, 36, 57.
187. S m i t h T. S., D a u n t J. G.— Phys. Rev., 1952, 88, 1172.
188. Smith T. S., Gager W. B., Daunt J. G.— Phys. Rev., 1953, 89,
654.
189. Spreadborough J., Christian J. W.— Proc. Phys. Soc., 1959,
74, 609.
190. Stalinski B., Bieganski Z.— Roczn. Chem., 1961, 35, 273.
191. S t r a u m a n i s M. E., Chen P. C.— Metall, 1953, 7, 85.
192. S t r a u m a n i s M. E., В a 1 1 a s s J. J.— Z. anorg. Chem., 1955, 278, 33.
193. S t e e I e M. S., Hein R. A.— Phys. Rev., 1953, 92, 243.
194. Sterk M. J., Nussbaum R. H., Wepstra A. H.— Physica, 1955,
21, 441.
195. S w a r u p D.— Eastern Metals Rev., 1953, 6, 157.
196. Szanto J.— Madyar tud. akad. musz. tud. oszt. kosl., 1955, 16, 233.
197. Taylor D. F.— Ind. Eng. Chem., 1950, 42, 639.
198. T i 1 1 e J., Kelly J. C.— Brit. J. Appl. Phys., 1963,14, 717.
199. Vernadskij V. I.— Rev. gen. Sci. pures appl., 1923, 34, 42.
200. Vernadskij V. I.— Rev. gen. Sci. pures appl., 1924, 35, 9.
201. Wenke H.— Z. Naturforsch., 1960,15a, 953.
202. Werner A. G., Handbuch dieMineralogie, 1. 1803, 55.
203. Wing J.„ W a h 1 g r e n M. A., Stevens С. M., О r 1 a n d i -
n i K. A.— J. Inorg. Nucl. Chem., 1965, 27, 487.
204. Wohler F., Deville H.-S. C.— Gilb. Ann., 1850, 73, 34.
205. Wood R. M.— Proc. Phys. Soc., 1962, 80, 783.
~06. W о r n e r H. W.— Australian J. Sci. Research., 1951, 4A, 62.
207. Wyatt J. L.— J. Metals, 1953, 5, 903.
олп 4 0 n s e г B. W.— Metal Progress, 1949, 55, 346.
209. Zachariasen W.— Z. Kristallogr., 1930, 73, 7.
2 0. Z e d 1 i t z O.— N. J. Min. Bull., 1939, 75, 245.
21 b Z e d 1 1 t z O.— Naturwis., 1943, 31, 369.
Дополнительная литература
Белов Б .IL, Богомолова Н. Б., Цветкова В. И., Чирков Н. М.
^месь (С5Н5)2 TiCl2 — Al (С2Н5)2 — катализатор полимеризации этилена.—
в и Кинетика и катализ, 1967, 8, 265.
иоградов Ю. М. Перспективы применения новых сплавов титана в хи-
ическом машиностроении.— В кн.: Титановые сплавы для новой техники.
«Наука», М., 1968, 74.
3 9 611
33
Глазунов С.Г. Современные титановые сплавы.— В кн.: Титановые сплавы
для новой техники. «Наука», М., 1968, 13.
Гопиенко Вал. Г., Г о п и е н к о В. Г. Взаимодействие хлора с титаном при
повышенной температуре.— Ж. прикл. хим., 1968, 41, 940.
Е л ю т и н В. П., М а у р а х М. А., Миш у к В. Я., Свердлов Г. М. Тепло-
та плавления титана.— Изв. высш. уч. зав. Цветная металлургия, 1967, 1,
60.
Китайгородский И. И., Ходаковская Р. Я., Б е у с М. М. Ис-
следование влияния двуокиси титана на ситализацию стекла.— Труды ИХТИ,
1966, 50, 10.
Куш ни р Н. А., Лунев В. М., Романов А. А., Соболев Л. И.,
Середа Н. И. Взаимодействие аммиака, водорода и азота с титаном.—
Укр. физ. ж., 1968,13, 87.
Лайнер Д. И., Бай А. С., Слесарева Е. Н., Цыпин М. И. Неко-
торые характеристики процесса окисления титана.— Физ. металлов и матери-
аловедение, 1965, 20, 864.
Павлушкин Н. М., ЖиткевичЗ.В., Колесов Ю. И., Пет-
ров С. В. Титансодержащие стекла.— Изв. АН СССР. Неорган. материалы,
1967, 3, 733.
Рубцов А. Н., Гопиенко В. Г. Гидрирование металлического титана
пропаном.— Ж. прикл. хим., 1968, 41, 418.
Расцветаева Р. К-, Симонов В. И., Белов Н. В. Кристалли-
ческая структура виноград овита.— ДАН СССР, 1967, 177, 832.
Тихомиров В. И., Дьячков В. И. Кинетика окисления титана кисло-
родом при высоких температурах.— Ж- прикл. хим. 1967, 40, 2405.
Шестопал В. О. Теплоемкость и образование вакансий в титане при высоких
температурах.— Физ. тв. тела, 1965, 7, 3461.
Aldridge А. М., Р 1 е n d 1 Н. S., А 1 d г i d g е J. Р. Время жизни и масса
Ti43.—Nucl. Phys., 1967, А98, 323.
Connillo E., Mazzi F., Rossi G. Кристаллическая структура не-
птунита.— Acta crystallogr., 1966, 21, 200.
Deschuyter M., Hoste J. Среднее сечение реакции (и, р) на титане в
реакторе.— Radiochim. acta, 1967, 7, 198.
Facetti J., Flegenheimer J., Trabal E. Новый изотоп титана
52Ti.— Radio chim. acta, 1966, 5, 143.
Gupta R. P., Daual В. Решеточная теплоемкость титана.— Phys, status
solidi, 1966, 13, 257.
Kofstad P. Высокотемпературное окисление титана.— J. Less-Common Me-
tals, 1967, 12, 449.
Kur era V., Novak P., Franz F., Koritta J. Коррозия анодно-по-
ляризованного титана в соляной кислоте.— Collect. Czechosl. Chem. Com-
muns, 1967, 32, 2049.
Ko ver F., Musselin M. J. О природе и свойствах тонких диэлектрических
пленок, полученных анодным окислением титана.— Onde electr., 1967, 47,
375.
Kohlhaas R., Braun M., Vollmer О. Атомные теплоемкости титана
в области высоких температур.— Naturforsch., 1965, 20а, 1077.
Moore Р. В., Louisnathan J. Фресноит — необычная координация ти-
тана.— Sci., 1967, 156, 1361
ГЛАВА II
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ ТИТАНА
к металлическим соединениям титана мы относим соединения
его с металлами и неметаллами, обладающие определенной совокуп-
ностью физических свойств. Признаки, по которым соединения от-
носятся к металлическим, рассмотрены нами ранее [34]. Следует,
однако, отметить, что до настоящего времени понятие «металличе-
ское соединение» точно не сформулировано. Некоторые авторы пы-
таются определить его, исходя только из природы химической связи
[80, 137, 139], но это односторонний подход. Примером может слу-
жить попытка Корнилова [80] провести классификацию металли-
ческих соединений только на основе природы химической связи, в
результате которой в число «металлидов» попали и неметаллические
вещества, как, например, соединения металлов I и II групп с неме-
таллами VI и VII групп.
Титан образует металлические соединения со многими металлами и неметал-
лами. Металлохимические свойства титана рассмотрены в работах [61, 62, 65,
68, 69, 73], закономерности образования титановых сплавов — в работах [3, 39,
134, 390]. Сплавам титана с металлами и неметаллами посвящены монографии
Еременко [50, 51] и Маквилл ан [396].
Химические элементы по характеру взаимодействия с титаном можно разде-
лить на пять групп (табл. 8).
К группе элементов, не вступающих в химическое взаимодействие с титаном и
не образующих с ним твердых растворов, относятся щелочные и щелочноземельные
металлы, исключая бериллий [69], и инертные газы. Отсутствие взаимодействия ти-
тана со щелочными и щелочноземельными металлами объясняется большим разли-
чием атомных радиусов и сравнительной близостью их металлических свойств.
К элементам, образующим с титаном твердые растворы ограниченного соста-
°тносятся иттрий, скандий, редкоземельные элементы [146], а также вольфрам
[387] и торий [209].
Небольшое количество элементов (Zr, Hf, V, Та, Мо) образует с ним непрерыв-
ные твер дые р астворы.
с Галогены, отличаясь от титана большой электроотрицательностью, образуют
с ним соединения с ковалентной и ионной связями. Подобно галогенам ведут себя
ояв*40"0? ^сеРа» если титан реагирует с ними в состоянии высшей валентности,
£«~НОИ СоеДинения титана с кислородом и серой низшей валентности обладают
металлическими свойствами.
талл амУю Мирную группу представляют элементы, образующие с титаном ме-
вичнГеСКИе соеДинения. Их насчитывается более пятидесяти. К ним относятся ти-
перехо неметаллы с малыми атомными радиусами (Н, В, С, N, Р, S), большинство
металлатп металлов с недостроенными d-и f-электронными оболочками, бериллий,
и лантЫ 1 РУппы периодической системы, за исключением скандия, иттрия
кремнийНИД°В’ металлы подгрупп А IV, V и VI групп периодической системы и
3*
35
Таблица 8
Взаимодействие титана с химическими элементами
Характер взаимодействия титана с
элементами
Элементы
Группа 1. Элементы, не образующие
химических соединений и твердых
растворов
Группа 2. Элементы, образующие твер-
дые растворы ограниченного состава
Группа 3. Элементы, образующие твер-
дые растворы неограниченного состава
Группа 4. Элементы, образующие хи-
мические соединения валентного типа
Группа 5. Элементы, образующие ме-
таллические соединения
Не, Li (?), Ne, Na, Аг, К, Ca, Kr, Rb (?),
Sr (?), Xe, Cs (?), Ba (?), Rn, Fr (?), Ra(?)
Mg, Sc, Y, La, Ce, Pr(?), Nd, Pn (?),
Sm (?)? Eu (?), Gd (?), Tb(?), Dy (?),
Ho (?), Er (?), Tu (?), Yt(?),Lu(?), W,
Th
V, Zr, Mo, Hf, Ta
F, Cl, Br, J, At (?), O, S
H, Be, В, C, N, O, Al, Si, P, S, Cr, Mn, Fe,
Co, Ni, Cu, Zn, Ga, Ge, As, Se, Nb, Tc (?),
Ru, Rh (?), Pd, Ag, Cd (?), In, Sn, Sb, Те,
Re, Os, Ir, Pt, Au, Hg, T1 (?), Pb, Bi,
Po (?), Ac (?), Pa (?), U, Np (?), Am (?),
Cm (?), Bk (?), Cf (?), Es (?), Fr (?), Md (?),
No(?),Lr (?)
В настоящей главе рассматриваются только системы, в которых
образуются металлические соединения титана. Описание остальных
систем, имеющих меньший интерес для химии титана, см., например,
в [50, 396].
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ ТИТАНА
БИНАРНОГО СОСТАВА
Титан образует металлиды бинарного состава с характерными
для этого класса соединений структурами типа чистых металлов,
с высоким координационным числом (в том числе фазы Лавеса), низ-
ким координационным числом (фазы типа никельарсенидных), фазы
внедрения. Среди металлических соединений титана распростране-
ны фазы переменного состава бертоллидного типа, поэтому для
определения их индивидуальности следует принимать во внимание
не только брутто-формулы, но и кристаллические структуры.
ТИТАН — ВОДОРОД
Как и большинство переходных металлов, титан обнаруживает
значительное сродство к водороду. На холоду титан с водородом
не взаимодействует, однако при нагревании поглощает его весьма
энергично. Взаимодействие титана с водородом наблюдали многие
исследователи еще до того, как появилась возможность получения
36
алла высокой чистоты [200, 340, 445, 467, 505]. Титан поглощает
ме1ог)одпри травлении в кислотах [17, 316, 342, 535] и при элект-
лизе водных растворов с использованием его в качестве катода
roll 341 ]• Заметное поглощение газообразного водорода металличе-
ким титаном начинается при температуре 375°С [159, 311 ] и являе-
С я обратимым процессом [317]. Образовавшийся продукт взаимо-
действия титана с водородом — гидрид — при нагревании под ва-
У ZMOM диссоциирует. Диссоциация начинается при температуре
100° С, но до 350° С протекает медленно; энергичное выделение во-
дорода происходит после нагревания гидрида титана до 650° С [88],
а при 1000° С водород выделяется полностью за короткое время [111,
159,160,200].
Продукты взаимодействия титана с водородом — твердые и,
как правило, нелетучие вещества, которые Сивертц и Готта [466]
азвали полуметаллами. По мнению [307, 308], гидриды титана, как
и других переходных металлов, относятся к металлическим соеди-
нениям.
Гвоздов и Журенкова [40] считают, что растворенный в титане
водород находится в атомарном состоянии.
В старых работах высказывались предположения, что наряду
с нелетучими гидридами титан подобно кремнию образует также
и летучий гидрид состава TiH4 [338, 340, 445, 467]. В последующем
это предположение не подтвердилось [15, 158, 292, 420]. Однако
недавно в иных условиях доказано существование TiH4 [206]. Он
образуется наряду с хлоргидридами TiH3Cl и Т1Н2С12 при облу-
чении смеси газообразного водорода и четыреххлористого титана,
насыщенной парами ртути, ультрафиолетовым светом с длиной вол-
ны 2537 А в вакууме при давлении 4—8 мм рт. ст. Присутствие в
продуктах реакции TiH4 и хлоргидридов титана доказано масс-
спектроскопическим методом.
Максимальное количество водорода, растворяющегося в ме-
таллическом титане, соответствует формуле Т1Н2 [41, 104, 108, 222,
264, 265, 270]. Гидриды с большим содержанием водорода при насы-
щении им титана не получены.
Макквиллан [391, 392] изучил изотермы поглощения водорода
металлическим титаном и построил диаграмму состояния системы
Ti —Н. Согласно его данным, в этой системе существуют три фазы:
«-твердый раствор водорода в гексагональной модификации титана,
₽-твердый раствор водорода в кубической модификации титана и
-фаза, являющаяся гидридом состава TiH2. Водород стабилизи-
рует фазу на основе (3-титана и понижает температуру а -> (3-пе-
рехода. Эта диаграмма подтверждена работами других авторов.
Леннинг с соавторами [272, 357] методом микроструктуры уста-
новил, что при температуре 320° С (3-фаза претерпевает эвтектоид-
ный распад на а- и у-фазы. Хэгг и Шипко [293] на основании изу-
чения равновесной упругости водорода в системе Ti —Н установили
существование четвертой 6-фазы, на что указывали ранее Гибб и
37
Крушвиц [264]. На рис. 2 приведена диаграмма состояния системы
Ti—н, построенная Еременко [50] по данным работ [391, 392] с уче-
том результатов исследований [264, 265, 272, 293, 357].
а-Фаза является твердым раствором водорода в гексагональной
модификации титана. Согласно [272],
водорода в а-титане составляет около
максимальная растворимость
8 ат.%, по [339] — 5,8 ат.%,
по [527] — 6 ат.%. По-видимому,
наиболее достоверными следует
считать данные Макквиллана
[391], установившего, что ма-
ксимальная растворимость во-
дорода в а-титане при эвтек-
тоидной температуре 7,9 ат. %.
При комнатной температу-
ре в а-титане растворяется
0,1 ат.% водорода.
Параметр решетки а-тита-
на от внедрения водорода
не изменяется [391 ], хотя ра-
нее [309] указывалось на из-
менение параметров решетки
а-титана при поглощении во-
дорода.
[3-Твердый раствор водо-
рода, по Хэггу [309], про-
Рис. 2. Диаграмма состояния системы стирается ДО содержания в
Ti —Н [50]. сплавах 50 ат.% Н, по Шреть-
ену с соавторами [222],— до
31 ат.% Н. Более достоверны, по-видимому, данные [391], по
которым растворимость водорода в [3-титане при эвтектоидной тем-
пературе 33,6—38 ат.%.
Ямагуши и Отсука [533] считают, что при действии на титан
48%-ной фтористоводородной кислоты ими получен в качестве [3-фа-
зы моногидрид титана TiH с параметром решетки а — 4,37 А. Это
утверждение, однако, противоречит данным [357, 391], из которых
видно, что [3-фаза при комнатной температуре не может быть сохра-
нена даже после ее закалки.
у-Фаза имеет область гомогенности в пределах между составами
сплавов TiH и TiH2 [309]. Предельное содержание водорода в об-
ласти гомогенности у-фазы, по данным [267] и других авторов, от-
вечает формуле Tibli,99. Структура у-фазы окончательно еще не
выяснена. По мнению некоторых авторов, у-фаза является соедине-
нием TiH2 с кубической гранецентрированной решеткой, параметр
которой с увеличением содержания водорода изменяется от 4,396 до
4,455 А 1222, 264, 267, 309, 357, 391, 476]. Согласно [264, 267, 311 ],
в у-фазе атомы водорода находятся в тетраэдрических пустотах
кубической гранецентрированной решетки титана. Сталинский с
соавторами [481 ], основываясь на данных исследования гидрида
38
яна состава TiHi.eoz — TiHi>969 методом протонного магнитно-
ТИТпезонанса, полагает, что атомы водорода распределяются хао-
° ски в местах кристаллической решетки, расположенных в
тйЧраЭдрической конфигурации относительно атомов титана. В рабо-
Т6 [149, 267, 476] отмечается, что кубическая гранецентрирован-
ная у-фаза при содержании водорода более 60. ат. % имеет тетраго-
нальные искажения.
6-Фаза изучена еще недостаточно, состав и структура ее неиз-
вестны.
Кинетика гидрирования металлического титана газообразным
водородом описана в работах [43, 111, 125, 271, 465]. Скорость по-
глощения водорода титаном существенно зависит от состояния по-
верхности металла и сильно тормозится окисной пленкой [125].
Компактный технический металл гидрируется водородом с наиболь-
шей скоростью при температуре 650—700° С [20].
Природа химической связи в гидридах титана обсуждается в не-
скольких работах. Малючков и Финкельштейн [97] на основании
исследований протонного магнитного резонанса считают, что в а-
гидриде титана связь атомов водорода с атомами титана носит метал-
лический характер. В [3-гидриде титана атомы водорода менее по-
движны и характер связи водорода с металлом несколько меняется.
Вайнштейн и Жураковский [17] изучили тонкую структуру
К-спектров гидрида титана с содержанием 1 и 2 вес. % Н. С повышени-
ем содержания водорода в гидриде титана наблюдается постепенное
ослабление длинноволновой линии поглощения (переход ls-электро-
на в Зй-полосу). Эти данные подтверждают гипотезу о металлопо-
добном состоянии водорода в гидриде титана вследствие передачи
электрона водорода в Зй-полосу атомов титана.
Антонова [5] проанализировала результаты исследования фи-
зических свойств гидридов переходных металлов различными ме-
тодами, в том числе методами парамагнитного резонанса и рентгено-
спектрального анализа, и пришла к заключению, что связь в этих
соединениях должна быть ионно-ковалентной, нелокализованной.
По внешнему виду гидрид титана с малым содержанием водо-
рода мало отличается от металла, из которого он получен. По мере
увеличения содержания водорода различие между гидридом и ме-
таллом становится более явственным. Продукт с большим содержа-
нием водорода никакими внешними признаками металлического
титана не обладает. Он легко рассыпается в порошок серого цвета,
переходящего в черный, в зависимости от степени его измельчения.
Плотность гидрида титана меньше плотности чистого металла,
ивертц и Готта [466] установили, что гидрирование титана до со-
става Г1Н1>64 понижает плотность с 4,52 до 3,91 г/см3.
Сталинский и Биганский [482] изучили термодинамические свой-
а нестехиометрических гидридов титана. Теплоемкость Ср, теп-
ппи°Д9о^а^Ие И ЭНТР°ПИЯ гидридов титана состава TiHi,607 и TiHi,7i8
н Уо ]\ равны соответственно 7,28 и 7,30 кал/молъ • град.
39
1208,2 и 1195,6 кал!моль, 7,29 и 7,24 кал!моль • град. Для гидридов
состава TiHi^oz, TiHi ,718 и TiHi,974 ЛН298 составляет соответ-
ственно — 27,3;—28,0; —29,5 ккал! мольТЛ¥1х, S^8 —25,1; 26,9 и
30,0 кал!молъ • град.
Теплота растворения водорода в а-титане составляет
21,6 ккал!моль Н2 [392].
При комнатной температуре гидрид титана — устойчивое на воз-
духе вещество, не реагирующее с разбавленными кислотами. При
нагревании на воздухе он воспламеняется и сгорает. Вначале при
температуре около 400° С выгорает водород, а при 800—900° С сго-
рает титан.
Гидрид титана применяется при получении металлических соеди-
нений титана с бором, кремнием и другими металлами [15]; восста-
новлении металлов из окислов; в качестве катализатора при гидри-
ровании органических веществ; для получения чистого водорода. Из
1 кг гидрида титана можно получить 300 л чистейшего водорода [20].
Исследование дейтеридов титана показало, что они по своим свой-
ствам аналогичны гидридам титана [267, 292, 385, 468].
Виберг и Узон [506] получили из TiCl4 и LiAlH4 в эфирном рас-
творе при температуре —110° С двойной гидрид титана и алюминия
TiH4 • 4А1Н3 — вещество светло-желтого цвета, очень неустойчи-
вое и распадающееся уже при температуре —85° С.
Барбарас с соавторами [207] в результате взаимодействия жидко-
го четыреххлористого титана с твердым борогидридом лития выде-
лил борогидрид трехвалентного титана Ti (ВН4)3. Это твердое ве-
щество зеленого цвета, очень неустойчивое и при комнатной тем-
пературе разлагающееся с выделением водорода и диборана.
ТИТАН — БЕРИЛЛИИ
Сплавы титана с бериллием получают нагреванием порошкооб-
разной смеси металлов до температуры 900—1600° С.
В системе Ti — Be обнаружены твердые растворы на основе
а-и |3-титана и несколько металлических соединений. Растворимость
бериллия в а-титане не превышает 9,1 ат.% [249, 266]. Раствори-
мость бериллия в (3-титане при 955° С находится в пределах 5,1 —
9,8 ат.% [221].
Сплавы титана с бериллием, содержащие 9,8—42,9 ат.% Be,
двухфазные. Одна из твердых фаз образована на основе (3-титана.
Природа второй фазы точно еще не установлена. Вероятно, это фаза
на основе TiBe или Ti4Be3 [266]. При большем содержании бериллия
сплавы состоят также из двух фаз, одна из которых имеет состав
TiBe? и является фазой Лавеса типа MgCu2 с параметром решетки
а = 6,42 А [266].
В богатой бериллием части системы найдены соединения Ti2Be17
в виде двух модификаций (a-форма, изоморфная с Nb2Be17 и (3-форма,
изоморфная с Th2Ni17 [536]) и неупорядоченная фаза на основе TiBe12.
40
И средняя кристаллизуется, по-видимому, в гексагональной сис-
1 ме с параметрами решетки а =- 29,44, с = 7,33 А и z = 48 ато-
Lob 1443, 444].
м Гиллам и Руксби [263] в системе, богатой бериллием, обнаружь
пи две Фазы — тетрагональную TiBe12, изоструктурную с VBe12,
Л гексагональную Ti2Be17. Параметры решетки TiBe12 изменяются
Рис. 3. Диаграмма состояния системы Ti — В [421].
в зависимости от содержания титана в сплаве от а — 7,27 и с ~
= 4,195 А (1,5% Ti) до а - 7,35 и с - 4,24 А (31 % Ti). Фаза Ti2Be17,
по данным рентгеноструктурных исследований, идентична с фа-
зой TiBe12, описанной Раухле и Рундле [443]. Она имеет параметры
решетки а = 7,35 и с = 7,26 А.
ТИТАН — БОР
Образование боридов титана при нагревании металлов впервые
наблюдал Муассан [397, 399], но химических соединений определен-
ного состава он не получил.
По имеющимся в литературе сведениям, титан с бором образует
твердый раствор ограниченного состава и металлические соединения
^В [415], Ti2B5 [430], TiB [249], TiB2 [249] и TiB12 [275]. Бориды
титана получают кратковременным нагреванием металлов в высоком
вакууме до температуры 1900—.2000° С [249]; технические сплавы —
[406?Р°ЛИЗОМ Расплава> состоящего из буры, TiO2 и CaF2 (флюс)
алюмотермическим восстановлением смеси окислов ТЮ2 +
41
b
h
+ B2O3 [219] и восстановлением в вакууме при нагревании смеси
TiO2 и угля с В2О3 или В4С [107, 344].
Полти с соавторами [421] изучил систему Ti—В методом метал-
лографии и рентгеноструктурного анализа и построил диаграмму
состояния (рис. 3), используя при этом также и данные других ав-
торов.
Растворимость бора в а- и р-титане небольшая, менее 0,1%
1266, 275, 415, 421].
Соединение Ti2B существует при температуре выше 1800° С. Оно
имеет простую тетрагональную решетку с параметрами а = 6,11
и с = 4,56 кХ [430]. Полти и соавторы [421] высказали предполо-
жение, что эта структура может отвечать также и «другой тетраго-
нальной решетке с параметрами а = 5,24 и с = 7,602 А.
Моноборид титана TiB имеет узкую область гомогенности вблизи
стехиометрического состава. Он образуется по перитектической
реакции
при температуре 2200° С и имеет кубическую гранецентрирован-
ную решетку. Параметр решетки,опо данным [273, 319, 430], а =
= 4,24 А, по [421], а = 4,26 А, ризм = Ррентг = 5,09 г/см3. Дек-
кер и Каспер [235] нашли, однако, что моноборид титана кристал-
лизуется в ромбической системе с параметрами а = 6,12, b = 3,06
и с = 4,56 А.
Диборид титана TiB2 характеризуется значительной областью
гомогенности вблизи стехиометрического состава. Он кристалли-
зуется в гексагональной системе. По Эрлиху [249], в согласии с
[406, 421] параметры решетки а = 3,026 и с = 3,213 А, плотность
TiB2 ризм = 4,40 г!см3 [344]. Температуру плавления TiB2 различ-
ные авторы определили равной (%): 2900 [344], 2920 [431 ], 2620 [132],
2300 [133]. Последние два значения Еременко [50] считает зани-
женными.
Энтальпию образования TiB2 определяли несколько авторов,
но в их результатах нет необходимого согласия. По Самсонову [141 ],
А//298 = —70,05 ккал/молъ. Крестовников и Вендрих [861, проана-
лизировав литературные данные, приняли А7/298 = —70,0 ккал!моль.
Уильямс [507] на основании критического анализа литературных
данных и прямых калориметрических опытов приводит значение
Д//298 = —50,0 ккал/молъ. Эту величину он подтвердил масс-спек-
троскопическими измерениями. Шиссель и Трулсон [462] с помо-
щью масс-спектроскопических измерений определили AZ/298 (TiB2) =
— —52,1 ккал/молъ.
Энтропия TiB2S298 = 7,52 кал/моль • град [86], энтальпия ис-
парения TiB2, по данным масс-спектроскопических измерений, рав-
на —424 ккал/молъ (462).
42
С является псевдобинарной с эвтекти-
пдойная система TiB2 _
Г1ЮЙ точкой при температуре 2560 К [93].
Либорид титана очень твердое вещество. Твердость его по шкале
выше 9, микротвердость, по данным Киффера [344], равна
ЖХи2, Самсонова [132],- 3370 кг/мм*.
6 Диборид титана обладает высокой окалиностойкостью и жаро-
очностью. В соответствии со своими физическими свойствами он
применяется для изготовления твердых сплавов, идущих для про-
изводства режущего инструмента, штампов, фильер, а также тиглей
для плавки металлов, чехлов термопар и пр.
Бориды Ti2B5 и TiB12 изучены еще недостаточно.
Рис. 4.
ТИТАН — УГЛЕРОД
Впервые карбид титана получен Шимером [464] в виде остатка
при растворении титанистого чугуна в соляной кислоте. Муассан
[397, 399] и Шнайдер [467] наблюдали образование карбида при на-
гревании металлического ти-
тана с углем.
Титан с углеродом образу-
ет твердый раствор на основе
а- и р-титана и два карби-
да — TiC [4641 и TiC2 [446].
Предположение о существо-
вании карбида титана состава
Ti2C [320] не подтвердилось
в последующих работах.
Диаграмма состояния си-
стемы Ti—С во всей области
концентраций еще не построе-
на. На рис. 4 приведена диа-
грамма состояния части систе-
мы Ti—С до содержания
50 ат. % С, построенная по
данным металлографических
и рентгеноструктурных исследований
ления образцов определяли методом
расплавления.
1 1 Уст0»ов^но, что ва-титане при температуре 900° С растворяется
’ ат- С [318]. Температура а -> ^-превращения титана под вли-
решеХи^ИМеСИ уГЛеРода П0ВЬ1Шается с 882 до 920° С. Параметры
U тапа gt пнсдреш
Еременко и Третьяченко [55] вы
УглеР°да в сс-титане 0,64
вами^9^1Р^ТуРе РаствоРяется ок
в обнаружил несколько
Диаграмма состояния системы
Ti — С [213].
[213]. Температуру плав-
начинающегося и полного
~ J---'"АА1-1 J.1WUU1LUUV.1 V/l v. X ICipdlVlCipDl
LpwnLa'TTTaHa от внедрения углерода изменяются незначительно.
. 1 выяснили, что максимальная раствори-
ат. %. В [3-титане при перитектиче-
около 3,3 ат. % С. Кадофф с соавто-
вВтит Jнесколько меньшую растворимость углерода
। итане, не превышающую 0,8 ат. %.
43
Система Ti^—C до содержания 5 вес. % С была повторно изучена
Курнаковым и Троневой [91], подтвердившими, в основном, дан*
ные [2131.
Карбид титана TiC имеет широкую область гомогенности, гра-
ницы которой еще окончательно не установлены. Эрлих [246] счи-
тает, что со стороны бедных углеродом сплавов граница 6-фазы
(TiC) проходит при содержании 16,5 ат.% С, а в более поздней ра-
боте [249] — при 14 ат.% С. Нортон с соавторами [405] границу го-
могенности со стороны богатых углеродом сплавов проводит при
содержании 22—26 ат.% С, а Уманский и Хидекель [150] — при
25—30 ат.% С. Агте и Моерс [162], а также Эрлих [249] нашли, что
TiC растворяет значительное количество углерода при плавлении,
однако в твердом состоянии растворы с высоким содержанием уг-
лерода метастабильны.
Кристаллизуется TiC в кубической системе [167]. Параметры
его решетки с увеличением содержания углеродаос 13,3 до 20,0 вес. %,
по данным [214], изменяются от 4,312 до 4,330 А. Рентгенографиче-
ским методом определено, что TiC с содержанием около 25 ат.% С
имеет параметр решетки а = 4,262 А, а с содержанием около
50 ат.% С — а = 4,318 А [38]. Параметры решетки TiC стехиомет-
рического состава определяли многие авторы. По наиболее новым
данным, полученным при исследовании образца, приготовленного
из иодидного титана и углерода высокой чистоты, а = 4,3281 А [1 ].
Образцы карбида титана с примесью кислорода имеют меньший па-
раметр решетки, так как внедрение кислорода в решетку вызывает
ее сжатие [214].
Карбид титана — тугоплавкое вещество. Различные авторы при-
водят для точки плавления TiC следующие данные (° К): 3400—
3500 [251 ], 3410 [162], 3420 [140] 3530 [345]. Твердость карбида ти-
тана по шкале Мооса равна 9,5, микротвердость — 2850 кг!мм2,
по [89], и 3000 кг/мм2, по [140]. Плотность TiC, вычисленная по пара-
метру решетки и измеренная, равна 4,915 г! см3. Карбид титана имеет
высокую электропроводность, понижающуюся с повышением тем-
пературы, как у металлов. Отрицательные значения коэффициента
Холла и термо-э. д. с. указывают на преимущественно электронный
характер электропроводности карбида титана [96]. Теплопроводность
карбида титана приведена в [290, 498]. Средний коэффициент ли-
нейного расширения в интервале температур 25—2040° С выражае-
тся уравнением [295]
агп = 6,96 • 10’6 + 0,93 (Т — 25) • 10“6.
Данные о магнитной восприимчивости и электросопротивлении
монокарбида титана имеются в работах [206, 274, 346], термодина-
мические— в [336, 366, 408]. Согласно Хемпфри [300], энтальпия
образования TiC A/7s98 = —43,8 ккал!моль. Морозова и соавторы
[115] установили, что энтальпия образования TiC зависит от тем-
пературы, при которой он получен. По их данным, образец TiC,
44
лученный выше 2000° С, имеет А/7°98 = —53,3, а при 1900° С,—
лс 0 ккал!'моль. Вероятно, величина энтальпии образования зависит
’ гранулометрического состава порошков TiC.
Водные растворы кислот, не обладающих окислительными свой-
вами, и щелочей на карбид титана не действуют. При сплавлении
с едкими щелочами и фторидами щелочных металлов карбид ти-
тана разлагается.
В компактном состоянии до температуры 800 С карбид титана
кислородом воздуха не окисляется; порошкообразный продукт за-
горается на воздухе при температуре 500—600° С.
С азотом карбид титана вступает в реакцию по уравнению
[53, 123].
TiC + -|-N2<>TiN +С.
В атмосфере гелия карбид титана реагирует с окисью магния
при нагревании до температуры 1600—1900° С с образованием ТЮ,
MgoTi04, Mg и СО [496]. Окисел TiO с TiC дает твердый раствор
Ti (О, С).
Спеканием TiC с карбидом бора В4С при температуре выше
1650°С получен борид титана TiB2 [404].
Для получения карбида титана (под карбидом титана обычно
понимают монокарбид, так как он является единственной, легко об-
разующейся в системе Ti—С карбидной фазой) предложено много
различных методов: выделение из ферросплавов, содержащих ти-
тан и углерод [378, 464]; сплавление двуокиси титана с углем в элек-
тропечи [397, 399, 467]; восстановление тетрахлорида титана нагре-
ванием с окисью углерода, углеводородами или углем в атмосфере
водорода [48, 103, 167, 203, 405]; взаимодействие металлического ти-
тана с сажей при взрыве тетрила [301 ] и т. д. Промышленное зна-
чение для получения карбида титана имеет метод, основанный на
науглероживании металлического титана или двуокиси титана при
нагревании их смеси с сажей в атмосфере водорода ниже точки плав-
ления [106, 162, 237, 251, 452]. Полученный по этому методу кар-
бид титана содержит углерода меньше, чем это соответствует фор-
муле, так как процесс восстановления двуокиси титана протекает
по схеме
TiO2 Ti3O5 Ti2O3 TiO Ti(O, C),
11 ОЛ 1 wv/pciJuuuruibM v paviovpa iiv п iivj lav,
р0, 162, 103, 105, 136, 150, 321], содержащего до 18,5% С [103].
аман и Рамахандран [446] из четыреххлористого титана и эти-
би а ПРИ высокой температуре получили новую кубическую кар-
сост ФазУ’ Она имеет параметр решетки а = 3,13 А, а возможный
ав ее выражается формулой TiC2. Детально эта фаза еще не
Ti екаа^анчивзется образованием твердого раствора TiO и TiC [10,
р0, 162, 103, 105, 136, 150, 321], содержащего до 18,5% С [103].
^аман и Рамахандран [446] из четыреххлористого титана и эти-
бил дГ" *'4<vunu» icimiepaiype получили попу to куиическую кар-
спе^10,/33^’ Она имеет параметр решетки а = ~ ~ ‘
изучена. - -
45
ТИТАН — АЗОТ
Титан с азотом образует твердые растворы на основе а- и (3-мо-
дификаций и два нитрида — Ti2N и TiN.
Впервые нитриды титана получили Розе [458] и Либих [369]
нагреванием паров TiCl4 с аммиаком, однако природу этих соедине-
ний установил Веллер [519—521 ]. В литературе описано много раз-
личных методов получения нитридов титана: непосредственное со-
единение металлического титана при температуре выше 800° С с га-
Рис. 5. Диаграмма состояния системы Ti — N [212,
213, 421].
зообразным азотом [160, 162, 169, 219, 397, 399, 408, 486, 508] или
с аммиаком [169, 252, 253, 520]; нагревание двуокиси титана в токе
аммиака в течение 4 ч при температуре 1500° С [110, 118, 254, 451 ];
нагревание гидрида титана с азотом или аммиаком [228, 255]; на-
гревание двуокиси титана с углем в токе азота [162, 251, 382, 450,
523, 524]; пропускание паров TiCl4 в смеси с азотом над раскален-
ным углем [167, 168]; термическое разложение при пропускании
смеси паров TiCl4 с азотом и водородом над вольфрамовой прово-
локой, нагретой до температуры 1450° С [167, 168, 223, 388, 389,
432]; нагревание смеси паров TiCl4 с N2 и Аг в плазменной горел-
ке [418].
Диаграмму состояния системы Ti—N впервые построил Нильсен
[403], используя данные [246, 249, 318], затем уточнили [212, 213,
421] (рис. 5).
46
ратуру
сплавах при температуре ниже 1050° С, имеет узкую область го-
гональной системе. Хольмберг [291 ] тетрагональной фазе приписы-
[291, 535]. Изменение параметров решетки а-титана
На диаграмме состояния Ti — N есть область твердого раствора
азота в а- и (3-титане, фаза на основе мононитрида титана TiN (6-фаза)
и фаза на основе нитрида титана предположительного состава Ti3N
(е-фаза).
Примесь азота стабилизирует a-фазу титана и повышает темпе-
ратуру а ₽-превращения. Мононитрид титана обладает обширной
областью гомогенности. Второй нитрид титана Ti3N, обнаруженный
в сплавах при температуре ниже 1050° С, имеет узкую область го-
могенности в пределах 6,8—8,9 вес. % N и кристаллизуется в тетра-
гональной системе. Хольмберг [291 ] тетрагональной фазе приписы-
вает состав Ti2N.
Твердый раствор на основе а-титана простирается до состава
TiNo,20-0,22 [291, 535]. Изменение параметров решетки а-титана
при растворении в нем азота изучалось в [212, 217, 291, 318, 421].
Согласно [291], параметры решетки а-титана в результате внедрения
азота изменяются в пределах «=2,9506—2,9711, с=4,6798—4,7843 А.
Нитрид титана Ti2N имеет тетрагональную решетку типа анти-
рутила, фед. гр. Р^21тпт с параметрами а = 4,9452, с = 3,0342 А,
z = 2, Ризм 4,86, Ррентг 4,19 а/см .
Мононитрид титана TiN (6-фаза) обладает обширной областью
гомогенности. Нижняя граница ее простирается, по Эрлиху [2491,
до состава TiN0,42, по данным Хольмберга [291], для отожженных
образцов — доТ{Хто,б; неотожженные образцы имеют нижнюю границу
6-фазы при еще меньшем содержании азота [291]. Верхняя граница
гомогенности 6-фазы простирается до состава TiNj,i6 [12].
6-Фаза имеет кубическую гранецентрированную решетку, па-
раметр которой меняется в зависимости от содержанияо азота. Эрлих
[249] нашел для состава TiN0,42 б-фазы а = 4,213 А, Хольмберг
[291 ] для состава TiN0,6 — а = 4,2238 А, ризм = 4,95, ррентг =
= 4,97 г/см?. Параметр решетки 6-фазы стехиометрического состава,
по данным различных авторов, таков: 4,32 [167], 4,23 [233],
4,225 Д302] 4,234 [249], 4,40 [201, 202], 4,235 [12], 4,2346 [384] и
4,210 А [38]. Еременко [50] считает наиболее достоверной величи-
ну а = 4,235 А [12].
Самсонов и Верхоглядова [138] изучили микротвердость и элек-
тросопротивление сплавов титана с азотом в области гомогенности
6-фазы. В отличие от карбида титана, изменение микротвердости
и электросопротивления TiJST является нелинейным, что свидетель-
ствует о наличии ионной связи наряду с металлической.
Цвет TiN зависит от степени дисперсности и изменяется от
светло-коричневого до бронзово-желтого.
Мононитрид титана плавится при температуре 2930° С [250, 251 ]
с частичным разложением и выделением азота. Он, подобно карбиду,
очень твердое (микротвердость его 2160 кг!мм2 [132]) и электропро-
°Дное вещество. При низких температурах TiN переходит в сверх-
4^8?оОДЯ1цее состояние, температура перехода, согласно [383],
К, а по данным [299], 5,6° К.
47
Определена средняя теплоемкость TiN в интервале температур
298—1003° К, которая зависит от содержания в нем азота [116].
Хемпфри [300] определил энтальпию образования TiN A/Z^s^
= —80,4 ккал!моль, что находится в согласии с данными [289]
(—79,4 ккал/молъ). Термодинамические свойства TiN описаны в
[50, 135, 409, 487]. ’
Мононитрид титана устойчив к действию кислот. При нагревании
с горячими водными растворами едких щелочей разлагается с выде-
лением аммиака. Аммиак также выделяется при анодном растворе-
нии TiN. С моноокисью титана TiN образует односторонние твердые
растворы, причем до состава TiO0,7N0,3 образуется твердый раствор,
а до состава TiO0,4N0,3 заполняются дефектные места решетки TiO
[475].
С окисью углерода TiN не взаимодействует. Кислород и двуокись
углерода при температуре выше 1200° С окисляют TiN до TiO2 и N2.
Установлено, что при температуре около 1300° С быстро и обра-
тимо протекают следующие реакции [488]: ’
TiN + 4НС1Г ТС14 + 2Н2 + 0,5N2,
TiN + 3HCL TiCL + 1,5Н» + 0,5N2,
TiN 4- 2HClr TiCL + H, + 0,5N2.
* 1 i a I 7 A
Первая реакция протекает слева направо преимущественно при
температуре ниже 1500° С, а вторая и третья — выше 1500° С. По-
этому возможен перенос TiN в результате пропускания над ним па-
ров НС1 в трубке, нагреваемой до 1600° С с одного конца и охлаж-
даемой до 1300° С с другого.
При анодном растворении TiN выделяется азот, что, по мнению
[428], указывает на металлическую природу связи. Согласно [58],
в TiN существует смешанный тип связи — ковалентной и металли-
ческой. В области гомогенности наблюдается увеличение металли-
ческой компоненты связи по мере приближения к нитриду стехио-
метрического состава. d-Электроны титана в химическую связи во-
влекаются в малой степени.
ТИТАН — КИСЛОРОД
В этом разделе рассматриваются только низшие окислы титана,
обладающие металлическими свойствами.
В системе Ti — O получено 13 окислов: Ti6O, Ti3O, TiO, Ti2O3,
Ti3O5, Ti4O7, Ti5O9, TieOu, Ti7O13, Ti8O15, Ti9O17, Ti10O19 и TiO2.
Окислы Ti6O и Ti3O недавно обнаружены в сплавах системы Ti—О
[29, 72, 75, 76, 82] при исследовании ее методом физико-химического
анализа. На диаграмме состав — свойство в системе Ti —О составу
этих окислов отвечают особые точки на кривых коэффициента тер-
48
1еского расширения, модуля упругости Е и характеристической
^мпературы 0. Соединение Ti6O устойчиво до температуры 820—
С, а 1— Д° 1400° С. О существовании окисла Ti3O, однако,
°оедполагали ранее [178, 414] на основании рентгеноструктурных
П следований сплавов и на основании исследования сплавов ме-
тодом электросопротивления [510]. При исследовании структуры
окалины на металлическом титане [306], а также литых сплавов
методом электросопротивления [511] было высказано предположе-
ние о существовании соединения Ti6O.
Окисел состава TiO получен в недостаточно чистом виде еще в
1814 г. нагреванием промасленной нефтью двуокиси титана [368].
Моноокись титана готовят нагреванием металлического титана с его
двуокисью при температуре 1550° С в вакууме в течение 2 ч. [4, 4G7,
486]; нагреванием двуокиси титана с металлическим магнием [2001
или металлическим кальцием [5093 при температуре 1400—1500° С;
нагреванием стехиометрических количеств TiC и Ti2O3 при темпера-
туре 1500—1750° С в вакууме или в инертной атмосфере [120].
Окисел Ti2O3 впервые выделен в 1847 г. нагреванием двуоки-
си титана в токе водорода в виде продукта недостаточной чисто-
ты [238]. Для его приготовления предложено нагревание двуокиси
титана с магнием при температуре 1100—1200° С в течение 3 ч в ат-
мосфере водорода [200]; нагревание смеси двуокиси титана с углем
в вакууме при 1450° С в течение 20 ч [407, 486]; нагревание смеси
TiO2 + TiO при 1000—1200° С в инертной атмосфере [119]; нагре-
вание металлического титана с двуокисью титана при 1550° С в те-
чение 5—6 ч [4, 433].
Окисел Ti3O5 впервые получен Пфордтеном [429] восстановлением
двуокиси титана металлическим магнием в атмосфере водорода при
температуре 700—800° С [200]; газообразным водородом при 900—
1300° С [126, 5091; углеродом в вакууме при 1350° С [407, 486]; уг-
леродом и металлическим титаном при 1200—1600° С с выдержкой
4—8 ч [4]; а также при взаимодействии смесей окислов TiO + 2TiO2
и Ti2O3 + TiO2 при 1400° С [4].
Окислы Ti4O7, Ti5O9 и другие, представляющие гомологический
ряд TinO2n__i, где п = 4, 5, 10, открыты Андерссоном с соав-
торами [178] в результате рентгеноструктурного исследования
сплавов в системе Ti —О при комнатной температуре. Сплавы гото-
вили из двуокиси титана и металлического титана тремя различными
способами: сплавлением в дуговой печи в атмосфере аргона, нагре-
ванием в электрической печи сопротивления в атмосфере аргона
при температуре 1200° С и нагреванием образцов в эвакуированной
кварцевой трубке.
До Андерссона образование в системе Ti —О самостоятельных
Ж-ж В °бласти составов Ti02,o —TiOi,9o (a-фаза), TiOi,8 —TiOi,7
ф-фаза ) и TiO]t56—TiOiт46 (у-фаза) было отмечено Эрлихом [244];
существование фазы с областью гомогенности в пределах состава
1.80—ТЮ1>8з указывал Филоненко с соавторами [151].
4 9-614
49
Изучено равновесие окислов титана состава TiOi>75 — ТЮцэо
со смесями Н2О/Н2 в газообразной фазе при температуре 1030° С
и зависимость удельной электропроводности от содержания в спла-
вах кислорода [13]. Из кривой зависимости парциальных давле-
Рис. 6. Зависимость состава окислов титана от отношения парциальных давлений
На и Н2О в сосуществующей с ними газовой фазе:
а — 1030° С [13], б — 986 и 1038° С [117].
ний Н2 и Н2О над окислами титана (рис. 6, а) в интервале состава
TiOi.75 — ТЮ1.90 видно, что существует шесть фаз переменного со-
става, непосредственно смыкающихся друг с другом. Следовательно,
при температуре около 1000° С в системе Ti — О нет дискретных
соединений гомологического ряда, существование которых установ-
лено Андерссоном при комнатной температуре. Данные по электро-
50
проводности, полученные при температуре 20 и 300° С, не противо-
речат результатам Андерссона о существовании соединений состава
TisO5, Ti4O7, Ti6Ou, Ti9O17 и Ti10O19. Однако Хойд и Айринг [156]
экспериментальные данные [13] истолковали как совпадение с ре-
зультатами Андерссона.
Морозова и Конопелько [117] повторили опыты [13], в которых
для повышения точности образцы окислов титана выдерживали в
токе Н2 — Н2О более длитель-
ное время (рис. 6, б). Авторы
подтвердили существование'
гомологического ряда соеди-.
нений Андерссона. Однако,
по их предположениям, пере-,
ход от одного соединения к
другому в некоторых случаях
является непрерывным.
Картину фазовых превра-'
щений, предложенную Андерс-
соном, подтверждает измене-
ние магнитной восприимчи-
вости в области состава
TiOi.5-TiOi.87 (рис. 7) [18].
Соединение Ti5O9 получи-
ли восстановлением TiO2 во-
дородом, Халимов и Резни-
ченко [155], фазу Ti6On —
Филоненко с соавторами [151]
и Модель с Уколовой [113].
Высшим окислом титана в системе Ti - О является ТЮ2. Методы
Рис. 7. Изотерма магнитной восприимчи-
вости при температуре 80° К (на абс-
циссе отложено значение х в формуле
ТЮ,) [18].
ее получения и свойства описаны в гл. IV. Кроме перечисленных
окислов, в системе Ti—О отмечено существование так называемой
6-фазы [177, 178, 198, 225, 474], образующейся в сплавах при тем-
пературе ниже 925° С и содержании 15—23 вес.% О. Состав 6-
фазы окончательно еще не установлен. Вероятно, она образуется
на основе окисла Ti3O2 или Ti4Og.
Диаграмма состояния системы Ti—О построена Бумпсом с со-
авторами [198] и затем уточнена [72, 75, 82, 234, 474] (рис. 8, 9).
Примесь кислорода стабилизирует tx-фазу титана и повышает
температуру а-> p-превращения до 1740° С [123, 198, 318, 322,
390].
Обширное поле на диаграмме Ti —О занимает твердый раствор
кислорода в а-титане. Область a-твердого раствора простирается,
По [244, 245] (рентгеноструктурные данные), до состава ТЮо42>
по [178] ,— до ТЮ0,5 (изменение параметров решетки), по [19],—
ДО TiO0,5—TiOo.85, ПО [24],—до TiO0.89 и по [478],—до ТЮ0,84-
Согласно [178], a-фаза образуется в результате внедрения ато-
°в кислорода в октаэдрические пустоты гексагональной решетки.
4*
51
Параметры решетки при этом возрастают от 2,95 до 2,96 для а йог
4,68 до 4,85 А — для с, что не противоречит данным Ростокера [448],
проследившим за изменением параметров решетки до содержания
34 вес. % О. Макаров и Кузнецов [98] также показали, что общее
число атомов в элементарной решетке a-фазы увеличивается с по-
вышением содержания кислорода и достигает 2,92 при составе
ТЮо451, тогда как число атомов титана в ячейке считается неиз-
Рис. 8. Диаграмма состояния системы Ti—О [474].
менным и равным 2. Поэтому a-фаза относится к соединениям
переменного состава и построена по типу внедрения атомов кисло-
рода в октаэдрические пустоты титановой части решетки.
Страуманис с соавторами [477] высказал другое представление
о структуре a-фазы. Изучив растворение окислов титана ТЮХ во
фтористоводородной кислоте, они пришли к заключению, что эти
сплавы представляют твердые растворы титана и его окислов. Их
мнение, однако, противоречит данным Макарова и Кузнецова [98],
установивших, что ячейка a-фазы содержит два атома титана неза-
висимо от содержания кислорода (в пределах гомогенности фазы).
В области a-фазы Корнилов и Глазова [72, 75, 84] обнаружили
существование TieO и Ti3O (см. рис. 9).
Включение кислорода в решетку а-титана искажает ее и ока-
зывает сильное влияние на физические свойства сплавов [50].
52
Корнилов и Глазова [74, 77] полагают, что кислород при рас-
ворении в а-титане образует связь металлического типа. При со-
ставе Ti3O частично появляется ковалентная связь [77].
С а-Титан имеет дырчатую проводимость. Вхождение кислорода
в решетку а-титана приводит к уменьшению термо-э. д. с. и констан-
ты Холла; в интервале содержания кислорода 4,2—5,7 ат.% про-
водимость а-титана меняется с дыр-
чатой на электронную.
фазе TiO на диаграмме состояния
Ti — О отвечает обширная область го-
могенности [244]. Поле TiO, по мне-
нию различных авторов, имеет сле-
дующие границы гомогенности:
ТК^-ТЮнзз [244, 245, 246, 249],
TiOo’92-TiOi,26 (800° С) и TiOo,78-
-ТЮ1,26 (1400° С) [198], ТЮо,89-
-TiOi,20(1300° С) [24], TiOo.82-TiOi.i7
[448], TiO0.64 — TiOi.26 [178]. Эрлих
[244] и Андерссон с соавторами [178]
установили, что фаза TiO, не подвер-
гавшаяся длительному отжигу, имеет
кубическую гранецентрированную ре-
шетку типа NaCl с хаотическим рас-
пределением вакансий титановой и
кислородной решеток. Все позиции
титановой решетки заняты при ниж-
ней границе, а кислородные места при
верхней границе гомогенности. При
составе TiOi.oo 15% обеих атомных
позиций остаются вакантными.
Рис. 9. Диаграмма состояния
системы Ti — О до содержания
40 ат.% О [72, 82].
Денкер [236] полагает, что высокая равновесная концентрация
вакансий, наблюдаемая в фазе TiO, является энергетически выгод-
ной. Образование вакансий уменьшает среднее число валентных
электронов в ячейке и понижает уровень Ферми, который в безде-
фектном кристалле TiO лежит выше разрыхляющих состояний.
Пара вакансий понижает энергию кристалла TiO на 1,1 э&. При этом
разрыхляющие орбитали освобождаются от электронов, а связу-
ющие остаются заполненными. По оценке Ямашито [535], ширина
Зб/-зон в TiO равна около 6 эв.
Ария и Попов [8] предложили модель строения TiO со структу-
Рои типа NaCl, основанную на субмикронеоднородной структуре.
По их представлениям, во всей области гомогенности в решетке су-
ществуют одно- и трехвалентные катионы, которые образуют само-
стоятельные области состава TiOo.s (содержащего Ti+) с вакансией
половины кислородных позиций и ТЮ1.5 (содержащего Ti3+) с ва-
кансией т/3 всех позиций атомов титана. Оба сорта имеют структуру
53
NaCl и распределяются по объему статистически, вследствие чего
нет сверхструктурных линий. С представлением о том, что в решетке
моноокиси титана существуют катионы Ti3+, согласуются инфра-
красные спектры [9] и изменение средней теплоемкости [56].
Андерссон с соавторами [178] установил, что фаза типа NaCl
при составе ТЮО,637 имеет параметры решетки а ~ 4,201 А, ризм
= 5,00, ррентг — 5,20 г/см3, а при составе TiOi,275 — а — 4,168 А,
Ризм = 4,80, Ррентг = 6,26 г!см3. По Эрлиху [244], параметр решет-
ки ТЮ типа NaCl с увеличением содержания кислорода уменьшается
с 4,184 до 4,152 А; Ростокер [448] установил, что при составе
TiOc#82 ci = 4,181, а при составе TiOi,i7 — я = 4,162 А. В пределах
области гомогенности параметр решетки ТЮ не изменяется прямо
пропорционально содержанию в сплавах кислорода.
Параметр кубической фазы при стехиометрическом составе TiOi.oo,
по данным разных авторов, таков: 4,235 [182], 4,17 [113], 4,1807
[433], 4,182 А [177]. Наиболее вероятны значения, приведенные
в последних двух работах. s
При стехиометрическом составе TiOi.oo на кривых магнитной
восприимчивости [19], энтальпии образования [7] и граммформуль-
ного состава [114] имеются особые точки, наличие которых подтвер-
ждает существование в системе Ti — О химического соединения со-
става TiO, на основе которого образована фаза моноокиси титана.
Найлор [407] в результате исследования теплоемкости фазы на
основе ТЮ установил наличие фазового перехода при температуре
991° С. Существование фазового перехода подтверждено в работах
[178, 234, 323, 347, 474, 512]. Андерссон и соавторы [178] считают,
что низкотемпературная фаза TiO существует при 900° С и ниже.
Плотность низкотемпературной фазы ТЮ стехиометрического со-
става, равная 4,91 г!см3, близка к плотности фазы типа NaCl —
4,95 г/см3. Авторы поэтому полагают, что структуры этих фаз близ-
ки. В [177] приведена рентгенограмма низкотемпературной моди-
фикации TiO.
Бумпс с соавторами [198] в образцах сплавов, содержащих 15—
23 вес.% О, отжигавшихся в течение длительного времени, обнару-
жил фазу, обозначенную им 6. Существование этой фазы подтвер-
ждено другими авторами [177, 178, 474]. Андерссон с соавторами [178]
показал, что 6-фаза устойчива только ниже 900° С. По данным [198],
6-фаза имеет тетрагональную решетку с параметрами а — 5,333,
с = 6,645 А. Андерссон [179] считает, что 6-фаза имеет гексагональ-
ную решетку с параметрами а = 4,991, с = 2,879 A, z = ЗТЮХ,
фед. гр. РЫтпгт. Структура 6-фазы аналогична структуре дефек-
тной фазы типа 8-TaN.
Образцы фазы состава TiO>i>0 в интервале температур от точки
кипения азота до 370° С обладают свойствами полупроводников;
образцы состава TiO<i>0 имеют металлическую проводимость [374].
Согласно [77], связь кислорода с титаном в фазе TiO — ковалент-
ного типа.
54
Карлсон и Мозер [210] методом МО ЛКАО с учетом самосогла-
сОвания рассчитали молекулу TiO. Они показали, что состояние
гзд является основным. Состояние 32~ более высокое по энергии,
qeM ^Д-, d1/^ и d1^. Карлсон и Несбет [211 ] рассчитали прибли-
женные волновые функции -состояния молекулы TiO.
Моноокись титана — вещество золотисто-желтого цвета. Энталь-
пия образования его Д//298=—125,4 ккал!моль [7]. Гровс и соавторы
[287] измерили упругость пара TiO методом Кнудсена. Зависимость
упругости пара от температуры выражается уравнением
lg Р (TiO) = — 29421/Т — 0,584 • 10~3 . Т + 10,43 [мм pm. cm.].
Вычисленная по упругости пара энтальпия испарения ТЮтв->
, г ТЮгаз Д/7о — — 134,55 ккал/молъ.
Поланд и соавторы [422] изучили скорость окисления TiO при
температуре 802—924° С кислородом воздуха. Реакция подчиняется
параболическому закону с энергией активации 45 ккал/молъ.
При растворении TiO в серной кислоте, разбавленной водой 1:1,
образуется осадок основного сульфата трехвалентного титана [127].
Энергия активации растворения TiO в серной кислоте равна
15 ккал/моль.
Зеликман с соавторами [60] установил, что TiO начинает хло-
рироваться газообразным хлором в присутствии угля при темпера-
туре 300° С. Хлорирование протекает с образованием TiCl4 и TiO2.
Моноокись титана при взаимодействии с хлором и серой не обра-
зует оксихлорида и оксисульфида. По мнению Лецерфа [359], это
служит признаком того, что она не является истинным окислом.
К такому же выводу пришел и Штрауманис с соавторами [480], изу-
чивший растворение окислов титана ТЮХ в плавиковой кислоте.
По его данным, взаимодействие ТЮХ в щавелевой кислоте сопрово-
ждается электрохимическим процессом (Ti + ЗН+-> Ti3++ 1,5Н2)
и химическим растворением (Ti2O3 + 6HF — 2TiF3 + ЗН2О).
Показано, что при уменьшении концентрации кислорода в ти-
тане сродство его к металлу увеличивается [337].
Изучено изменение сродства кислорода к титану при образова-
нии твердого раствора Ti (О, С) [109]. По мере уменьшения содер-
жания кислорода в твердом растворе энергия связи его с титаном
увеличивается. При температуре 1300° С и содержании кислорода
0,05 вес. % Д Z (связь Ti — О) = AZ (связь Са — О) = 113 050 кал.
Выше 1500° С энергия связи титана с кислородом больше энергии
связи кальция с кислородом.
Фаза Ti2O3, по Эрлиху [244], характеризуется узкой областью
гомогенности в пределах состава TiOi,46 — TiOi,56- Андерссон с со-
авторами [178] верхний предел гомогенности этой фазы относит
к составу TiOl.si.
Нейлор [407] на основании исследования теплоемкости пришел
к заключению, что фаза Ti2O3 имеет полиморфное превращение при
55
температуре 200° С. Существование фазового перехода в Ti2O3
подтвердили Фукс и Вухер [256] по изменению магнитной воспри-
имчивости, но они приводят более высокую температуру полиморф-
ного превращения, равную 473° С.
Магнитная восприимчивость х соединения Ti2O3 ниже точки
фазового перехода практически не зависит от температуры. Вблизи
точки перехода наблюдается резкое возрастание х. Выше точки пе-
рехода х увеличивается с повышением температуры, но медленно.
Фазовый переход по Вухеру [513] является переходом от анти-
ферромагнетизма кристаллической решетки к антиферромагне-
тизму молекул, т. е. к спариванию спинов двух соседних магнитных
ионов.
Абрахамс [171 ] фазовый переход в Ti2O3 наблюдал при темпера-
туре 660° К. Исходя из анализа рентгенограмм, автор считает, что
фазовый переход в Ti2O3 связан с переходом парамагнитного со-
стояния выше точки превращения в антиферромагнитное. Пирсон
[433] также наблюдал фазовый переход в Ti2O3, но при температуре
350° С.
Высокотемпературная модификация Ti2O3 имеет ромбоэдрическую
структуру типа корунда [244]. По данным различных авторов,
ячейка Ti2O3 при стехиометрическом составе имеет следующие
параметры решетки: а = 5,155/с = 13,607 А [177]; а = 5,149, с —
= 13,642 А, фед. гр. 7?3с, z = 6 [4023; а = 5,148, с = 13,636 А, ризм ==
— 4,5868 г/см3, z = 6 [479] (гексагональная установка); а = 5,433,
а — 56° 34', ризм = 4,61, ррентг = 4,57 г/см3, фед.огр. z —
= 2 [1713; а = 5,414 А, а - 56°44'[198]; /7-5,248 А, а - 56°39'
[433] (ромбоэдрическая установка). Атомы, находящиеся в решетке
Ti2O3 в избытке, частично располагаются в пустотах, а частично —
в правильных положениях, что вызывает появление вакансий на
местах ионов противоположного знака [4793.
Окисел Ti2O3 — вещество фиолетового цвета с температурой
плавления 1820° С [185], Д#298 =—362,8 ккал/моль [7]. Гровс
с соавторами [282] определил давление упругости пара Ti2O3 по
методу Кнудсена. Зависимость упругости пара от температуры вы-
ражается уравнением
IgP (Ti2O3) - —64700/Т— 1,26> IO”3? + 21,65 [мм рт. ст.].
При испарении Ti2O3 разлагается на TiO и TiO2. Энтальпия ис-
парения 296,0 ккал/моль.
В условиях комнатной температуры Ti2O3 является полупро-
водником р-типа [433, 534]. Выше 450° К Ti2O3 ведет себя как ме-
таллический проводник.
Соединение Ti2O3 — антиферримагнетик с 7\ф — 248° К, х^. —
-0,874 • 10~6 при 298° К [170]. Выше Таф магнитная восприим-
чивость приблизительно следует закону Кюри — Вейса со значени-
56
м 0 2000° С. Магнитный момент иона Ti3+ в Ti2O3 равен 1,2 рв-
Пирсон [433] определил магнитную восприимчивость 1,09 • 10~&
при 20° С. Природа связи кислорода с титаном в Ti2O3 ковалентная
и частично ионная [77].
В отличие от TiO окисел Ti2Os является истинным. Он способен
образовывать различные химические соединения. С окислами ме-
таллов Ti2O3 дает соединения типа сложных окислов. Лецерф и Гар-
ду [358, 359], нагревая смесь Ti2O3 с окислами Li2O, MgO и МпО,
получил соединения LiTiO2 (кубическая решетка типа NaCl, фед.
гр. FmStn, а = 4,140 A, z = 2, ризм = 3,80 г/см3), MgTi2O4 (решетка
шпинели, фед. гр. FdStn, а = 8,474 А, z = 8, ризм = 4,05 г/см3),
MnTi2O4 (решетка шпинели, фед. гр. Fd3m, а = 8,600 A, z — 8,
Ризм = 4,37 2/СЛ13).
Соединение MgTi2O4, по [128], с титанатом магния Mg2TiO4 об-
разует твердые растворы состава «Mg2TiO4 • mMgTi2O4, в которых
ионы Mg2+ и Ti3+ могут изоморфно замещаться ионами Fe2+, Мп2‘~,
Ti2+, Ni2+, Со2+, Ti3+, А13+ и др.
В системе Ti2Os — А12О3 в пределах 12,9—79,1 мол. % Ti2Oa
имеется одна фаза [196]. Предельная растворимость Ti2O3 в A12OS
при температуре 1700° С равна 12,5 мол. %. Японские авторы нашли
меньшую растворимость Ti2O3 в А12О3 [532].
В серной кислоте Ti2O3 растворяется медленнее, чем TiO. Энер-
гия активации при растворении Ti2O3 в серной кислоте, разбавлен-
ной водой 1:1, равна 21 ккал/моль [127].
Окисел Ti3O5, по мнению Хемпфри [300], является высокотем-
пературной фазой, а найденный Нейлором [407] при температуре
177° С фазовый переход — метастабильным. Однако Азбрик и Маг-
нели [175] подтвердили существование двух модификаций Ti3O5—
высокотемпературной и низкотемпературной. Фазовый переход вы-
сокотемпературной модификации Ti3O5, по их данным, лежит при
температуре около 120° С.
Жданов и Русаков [57, 131] установили, что на основе окисла
Ti3O5 (высокотемпературная модификация) образуется минерал
аносовит. Аносовит, выделенный из титанового шлака, кристалли-
зуется в ромбической системе с параметрами решетки а = 3,74, b =
= 9,466, с = 9,715 А. Магнели с соавторами [174, 177, 178] изу-
чил рентгеноструктуру монокристалла Ti3O5 по методу Вейссен-
берга и установил, что он кристаллизуется в моноклинной системе
с параметрами решетки а = 9,757, b = 3,802, с = 9,452 А, а =
= 93,11°, Ризм = 4,20, ррентг = 4,24 г/см\ z = 4. Авторы пола-
гают, что кристаллы Ti3O5, выделенные Ждановым и Русаковым из
титанового шлака, содержали примесь MgO, которая стабилизи-
ровала структуру псевдобрукита.
Халимов и Резниченко [153], восстанавливая титанаты магния,
получили твердый раствор т (MgO • 2TiO2) . nTi3O5, имеющий рент-
генограмму, сходную с аносовитом.
57
Окисел Ti3O5 — вещество синего цвета. Энтальпия образования
его—587,0, по данным [7], и—558,16 ккал!моль, по [128]. Последние
нашли выражение для изобарного потенциала
&L (Ti3O5) = —579068 + 100,687 [ккал/моль]
(7 = 328 — 1553° К).
При нагревании Ti3O5 с серной кислотой, разбавленной водой
1 : 1, он растворяется легче, чем Ti2O3. Энергия активации при раст-
ворении равна 18 ккал!моль [127]. 1
Диаграмма состояния системы Ti — О (см. рис. 8) не отражает
истинных фазовых равновесий при составе Ti3O5 — TiO2. Структура
этой части диаграммы состояния нуждается еще в уточнении. I
Эрлих [244] в области концентраций TiOijo—TiOi.so обнару-
жил обширную область гомогенности. Филоненко с соавторами [151 ]
получил окисел состава Ti2O3 • (3—4)TiO2. Существование этого
окисла подтверждено в работе [113]. Андерссон с соавторами [176,
178], как уже сообщалось выше, в интервале TiOi>7 — TiOi>9
установил существование серии окислов, представляющих гомологи-
ческий ряд TinO2n-i, где п = 4—10. Структура второго члена
гомологического ряда Ti5O9 изучена Андерссоном [180] методом
Гинье и Вейсенберга. Окисел Ti5O9 имеет триклинную решетку с
параметрами а — 5,569, b = 7,120, с = 8,865 А, а = 97,55°, [3 = \
= 112,34°, у = 108,50°, ризм = 4,29, рреНтг = 4,31 г!см\ z = 2 J
фед. гр. 71. Структура образована из ТЮ6-октаэдров, соединенных
в цепочки по пять октаэдров в каждой. Цепочки соединены по вер-
шинам и ребрам октаэдров в бесконечные слои рутилового типа тол-
щиной, равной длине цепочки. Структура участков, непосредственно
примыкающих к плоскости раздела двух рутильных слоев, близка
к структуре типа корунда. На её основе выведены общие выражения
для параметров решетки, ее объема и координат атомов всех
членов серии [181]. Члены гомологического ряда TirtO2rt-i обладают
близкой структурой, в основе которой лежит структурный тип
рутила. Каждый член серии построен из горизонтальных блоков
рутильного типа, бесконечных в двух измерениях, имеющих тол-
щину /г7Ю6-октаэдров (п— число гомологического ряда).
Халимов и Резниченко [154] получили окисел Ti5O9 в виде тем-
но-синего вещества восстановлением TiO2 водородом. По их рас-
четам, энтальпия образования Ti5O9 Д//?4оо = — 1044,10 ккал!моль.
В области состава системы TiOi,9—TiO2>0, по данным [178],
существуют двухфазные сплавы. Одной из фаз является рутил,
линии которого появляются на рентгенограммах при отношении
0 : Ti > 1,95. Природа второй фазы еще не установлена. i
Руднева с соавторами [129] фазу состава TiOi,95-2,00 называет
непрозрачным рутилом. Я
Наличие двухфазной области в интервале составов TiOita68 —
TiOi.992, по данным измерения э. д. с., отмечено в работе [193].
Установлено, что в результате диссоциации TiO2 образуется продукт
стехиометрического соединения TiO2_x [185]. Величина х за-
висит от давления кислорода в газовой фазе.
При плавлении рутила в дуговой печи состав его изменяется до
содержания TiOi,90 в результате потери кислорода. Полученные
при этом образцы на рентгенограммах не дают линий рутила [178].
Крайней фазой в изученной части системы Ti —О является дву-
окись титана со структурой рутила [178]. Две другие модификации
двуокиси титана — анатаз и брукит — в изученных условиях яв-
ляются метастабильными фазами.
Брауэр и Литке [185] считают, что рутил плавится при темпера-
туре 1870° С и образует эвтектику с Ti2O3 при 1665° С. Состав эв-
тектики TiOi ,72-
Соединения с содержанием О : Ti > 2 в двухкомпонентной си-
стеме Ti — О не обнаружены.
Исследована система TiOx (х = 1,5—2) методом электронного
парамагнитного резонанса [6]. В сплавах изученного состава отме-
чен сигнал, соответствующий Ti3+, наблюдающийся до температу-
ры — 100° С. §--Фактор изменяется от 1,963 при х = 1,760 до 1,955
при х — 1,998.
ТИТАН — АЛЮМИНИЙ
Впервые сплавы титана с алюминием получил Веллер [525], на-
гревая смесь двуокиси титана, криолита и металлического алюминия.
Рис. 10. Сводная диаграмма состояния системы Ti—Al [50].
59
о I
Часть диаграммы состояния си-
Ti — А1, богатой титаном [70].
Рис. 11.
стемы
Опыты по получению сплавов титана с алюминием описаны в рабо-
тах [278, 360, 361, 377], однако индивидуальность полученных
алюминидов титана убедительно не доказана. В настоящее время
сплавы титана с алюминием получают нагреванием чистых метал-
лов или алюмотермическим
восстановлением TiO2.
В системе Ti — Al образу-
ются взаимные твердые раст-
воры ограниченного состава и
металлические соединени я
Ti6Al, Ti3Al, TiAl и TiAl3.
Соединение Ti6Al обнару-
жили в сплавах титана с алю-
минием по излому на кривой
зависимости константы Холла
[44] и на диаграммах состав —
свойство [70]. Соединение
Ti3Al установили по сверх-
структурным линиям, указы-
вающим на существование
упорядоченной фазы типа
Ti3Sn [218]. Образование
TisAl подтверждено в [67,
70, 79, 163].
Огден с соавторами [416]
обнаружил соединение TiAl.
Оно имеет обширную область
гомогенности — от 33 до 46
вес. % А1. Фаза на основе TiAl кристаллизуется в тетрагональной
системе и имеет решетку типа упорядоченной CuAu. С увеличением
содержания алюминия от 33 до 48 вес. % параметр решетки а
уменьшается от 4,00 до 3,98 кХ, а параметр с увеличивается от
4,06 до 4,07 кХ [230].
Манхот и Рихтер [377] получили TiAl3, нагревая K2TiF6 с ме-
таллическим алюминием. Затем TiAl3 обнаружил Ван-Эркеленс
[243] в системе Ti —Al методами термического анализа и микрострук-
туры, но приписал ему неправильный состав TiAl4. Манхот и Лебер
[380] уточнили состав этого соединения и выделили его в чистом
виде. Возможно, что и другие авторы получили TiAl3, но также при-
писывали ему неправильный состав TiAl4.
Соединение TiAl3 кристаллизуется в тетрагональной сингонии
с параметрами решетки а = 5,421, с = 8,574 кХ, z = 4 [257].
Плавится TiAl3 конгруэнтно при температуре 1355° С [380].
Диаграмма состояния системы Ti—Al впервые построена Ван-
Эркеленсом [243], а затем уточнена [27, 63, 70, 197, 224, 240, 416,
473]. Наиболее сложной оказалась часть системы с высоким содер-
жанием титана. До настоящего времени строение ее в различных
60
работах трактуется по-разному. Мы приводим сводную диаграмму,
составленную Еременко (рис. 10), и часть диаграммы для области
с высоким содержанием титана в трактовке Корнилова с соавторами
(рис. И), которая наиболее правильно отражает состояние равно-
весия в высокотитанистых сплавах.
ТИТАН — КРЕМНИЙ
Взаимодействие титана с кремнием и силиконитридом впервые
наблюдали Шутценбергер и Колсон [472]. Муассан [398, 400, 401]
получил силициды титана неопределенного состава нагреванием
рутила с кремнием в дуговой электропечи.
Рис. 12. Диаграмма состояния системы Ti — Si [297].
Силициды титана могут быть получены сплавлением металлов
в вакууме или в атмосфере инертных газов; алюмотермическим вос-
становлением смеси двуокисей титана и кремния [220, 303, 304,
305]; действием газообразного Ti С14 на металлический кремний при
высокой температуре [362]; восстановлением двуокиси титана си-
лицидом кальция [258]; электролизом расплавленной смеси ТЮ2 —
^SiFg [413] и сплавов силикатов щелочных и щелочноземельных
металлов с двуокисью титана [232].
В системе Ti — Si обнаружены силициды титана следующего
состава: Ti5Si3, TiSi и TiSi2; построена диаграмма состояния системы
*1 — Si (рис. 12).
61
Соединение Ti5Si3 имеет переменный состав и кристаллизуется
в гексагональной сингонии. Параметры решетки, по данным Пиетри-
ковского и Дувеца [435], равны а — 7,465, с = 5,162 А. Фреундлих
с соавторами [258] нашел их равными а = 7,41, с~ 5,15 А. Михеев
с соавторами [112] обнаружил силицид Ti5Ss в системе Ti—Al —
Cr — Fe — Si — В. Он относится к числу тугоплавких соединений.
Температура его плавления 2120° С [435].
Голутвин [36, 37] определил для Ti5Si3A//298 = —147 ккал/моль.
По данным этого автора, температурная зависимость энтальпии
образования Ti5Si3 выражается уравнением
АЯГ (Ti5Si3) = 58,227 + 2,871 • 10~3Т2 + 2,646 • Ю6?-1 —
— 2649 [кал/моль]
(7 = 298—1170° К).
Моносилицид титана кристаллизуется в ромбической системе.
По Агееву и Самсонову [2], параметры решетки TiSi а ~ 3,611,
b ~ 4,960, с = 6,479 кХ, z = 8, рреНтг = 4,34, рИЗм — 4,21 г/см\
а по Бруклю с соавторами [199], а ~ 6,53, b = 3,63, с = 4,98 кХ.
Голутвин [36] измерил теплоту сгорания TiSi и по ней вычислил
энтальпию образования AZ/fgs = —39,2 ккал/моль. Им же [371
найдена температурная зависимость энтальпии образования
&НТ (TiSi) = 15,437+ 0,8832 • 106 • 7-1 —7563 [кал/моль]
(7 = 298- 1350° С).
Дисилицид титана TiSi2 получен нагреванием металлического
титана и кремния [397, 399]. Ди силицид титана плавится конгруэнт-
но при температуре 1520° С [297]. Кристаллизуется он в ромбиче-
ской системе с параметрами решетки а = 8,252, b — 4,742, с =
= 8,540 А [373]. Это соединение наиболее легко получается в чи-
стом виде по сравнению с другими силицидами титана [303, 304,
305, 258, 413].
Энтальпия образования TiSi2 = —42,9 ккал/моль [36].
Зависимость энтальпии образования от температуры выражается
уравнением [37]
\НТ (TiSi2) = 14,947 + 4,16 • 10~3 • 72 + 0,455 • 106 • 7”1 —
— 6350 [кал/моль]
(7 = 298 — 1180° К).
Дисилицид титана дает ограниченные твердые растворы с диси-
лицидом ниобия [25 ].
ТИТАН — ФОСФОР
Фосфиды титана получены нагреванием TiCl4 с РН3 [279, 459,
522], нагреванием металлического титана и фосфора [397, 399].
62
в системе Ti — Р установлено существование нескольких фосфи-
нов, однако диаграмма состояния этой системы еще не построена.
Шенберг [4831 получил фосфид титана Ti3P, который имеет струк-
туру типа Fe3P с параметрами решетки а = 10,000 и с = 5,017 А.
Это же соединение получено нагреванием металлического титана
п фосфора в запаянной кварцевой трубке при температуре 800° С
в течение 48 ч [363]. По Лундстрему [363], параметры решетки сле-
дующие: а = 9,956, с = 4,988 А, фед. гр. Р^21п. Фазу Ti3P полу-
чил Кнаузенберг с соавторами [343].
Брауэр с соавторами [186] синтезировал фосфид титана Ti5P3,
обладающий узкой областью гомогенности. Кристаллы Ti5P3 имеют
гексагональную решетку, параметры которой, по [186], а = 7,234,
с = 5,090 А, Ризм = 4,85 г!см3, z = 2, фед. гр. Р63 (тст), по [204],
d = 7,2381, с = 5,088 А, Ризм ~ 4,85, Ррентг~ 4,856 с!см?. Атом
фосфора находится в окружении девяти атомов титана, образующих
искаженный четырнадцатигранник.
Установлено, что фаза Ti6P3 существует в области концентраций
TiPo 6, — TiP0 68 и имеет параметры решетки а = 7,219 и с = 5,096 А
[343].
Бильтц с соавторами [192] получил фосфид титана Ti2P. Это же
соединение синтезировано Верейкиной и Самсоновым [22] пропу-
сканием фосфина над металлическим титаном при температуре
800° С.
Бильтц с соавторами [192] получил также фосфид титана TiP
с областью гомогенности TiP0,92 — TiP0i94. По [262], область
гомогенности при температуре 1700° К находится в пределах
TiPo,945—TiP0,96. Бахмейер и соавторы [194] определили параметры
решетки этой фазы а = 3,506 и с = 11,73 кХ (гексагональная син-
гония). Фаза TiP, по данным [343], существует в области концент-
раций TiPo.ss — TiPi.oo и имеет параметры решетки а — 3,488 и с —
= 11,191 А. Шёнберг [484] нашел, что фаза TiP имеет гексагональ-
ную решетку, которую можно рассматривать какосверхструктуру
типа NiAs с параметрами а = 3,487, с = 11,65 А, рреНтг = 4,27,
Ризм = 4,08 г!см3, z = 4, фед. гр Р^/ттс.
В области концентраций TiP0,35 — TiP0,6i и TiP0,68 — TiP0,96 су-
ществует еще по одной фазе, состав которых не установлен. ’
Пропуская пары фосфина над металлическим титаном при тем-
пературе 850° С, Верейкина и Самсонов [22] получили фосфид ти-
тана TiP.
Хуллигер [294] получил дифосфид титана TiP2, имеющий струк-
туру типа ZrAs2 с параметрами а — 6,181, b — 8,256 и с = 3,346 А.
ТИТАН — СЕРА
Впервые сульфид титана состава TiS2 выделил Розе [456, 457],
Нагревая двуокись титана с серой в парах сероуглерода. Леви [362]
Наблюдал взаимодействие между металлическим титаном и серой
пРи нагревании до красного каления.
63
3,37 г! см3.
В настоящее время сульфиды титана получают при нагревании
металлического титана с серой в запаянной кварцевой трубке при 1
температуре 750° С около двух недель и пропускании сероводорода
над металлическим титаном при температуре 550—900° С [47, 310].
В литературе описано много сульфидов титана, индивидуальность
которых нередко требует еще дополнительного подтверждения [424—
426, 491 ]. Диаграмма состояния системы Ti — S еще не построена.
Сульфид титана Ti2S описан в работах [152, 191, 310]. Авторы
[191 ] считают, что на основе Ti2S существует фаза с областью гомо-
генности TiSo,25 — TiSi,o- Кристаллы Ti2S имеют тетрагональную
симметрию с параметрами решетки а — 4,375, с = 8,370 А, р =
= 4,68 г/см3[ 152]. Микротвердость их равна 620 кг/мм2 [152]. Они не
растворяются в разбавленных соляной и серной кислотах, а при на-
гревании до 700—900° С сгорают с образованием ТЮ2 и SO2 [152].
Уадслей [514] исследовал сплавы состава Ti2-f-xS (где 0,2 <х <
< 1) методом рентгеноструктурного анализа и нашел фазу, являю-
щуюся промежуточной между TiS2 (фед. гр. С6) и TiS (фед. гр. В8).
Кристаллы состава Ti^22 S4 имеют гексагональную решетку с па-
раметрами а = 3,41, с = 11,46 А, рИзм = 3,35, ррентг =
Голубцова [32] при анодном растворении сплава металлического
титана с серой выделила порошкообразный продукт, которому при-
писывает, по данным химического анализа, состав Ti5S3.
Хан и Несс [284, 298] получили сульфид титана Ti3S2 со струк-
турой типа карбида вольфрама с параметрами решетки а = 3,28 —
3,27, с = 3,20—3,21 А.
Моносульфид титана TiS получен несколькими авторами [191,
283, 298, 310, 325]. По мнению [191], TiS является основой фазы
с областью гомогенности в пределах состава TiSi.oo — TiSi ,04-
Жаквин и Жаннэ [325] в сплавах титана с серой состава TiSog7 —
TiS,, ,38 при температуре 1000° С обнаружили высокотемпера-
турную модификацию TiS с гексагональной решеткой типа
NiAs (TiS0,97 — TiSi.oe), фазу с ромбической решеткой (TiSi,25) и
фазу с моноклинной решеткой (TiSi,285 — TiSi.sos)- I
Установлено, что высокотемпературная модификация TiS являе-
тся сверхструктурой низкотемпературной модификации этого со-
единения [283]. Параметры решетки высокотемпературной модифи-
кации TiS таковы: а = 3,41, с = 3,26 кХ, z = 9, Ррентг = 4,0 г! см?,
фед. гр. R3m. Низкотемпературная модификация TiS переходит в
высокотемпературную при температуре 1000° С необратимо.
Жаннэ [314] в системе Ti — S обнаружил соединение состава
Ti3S5. Он получил также сульфид титана, которому приписывается
состав Ti3S4 [314, 315]. Дебаеграмма Ti3S4проиндицирована в монок-
линной системе с параметрами а = 5,98, b — 3,440, с = 10,14 А, Р»
= 95°, 44', ррентг = 3,798 г/см3 [315]. Хан и Хардер [283] предло-
жили через Ti3S4 обозначать фазу, которая до них обозначалась
как Ti2S3. Кожина с соавторами [85] считает, что Ti3S4 явля-
ется одним из промежуточных соединений на основе фазы TiS3. В
64
результате рентгеноструктурных исследований системы Ti — S
установлено образование сульфида Ti2S3 [173, 191]. По мнению
1191], это соединение — основа фазы с областью гомогенности в пре-
делах состава TiSiдз — TiSi,5. Сульфид Ti2S3 имеет гексагональную
решетку с параметрами а = 3,42, с = .1,4 А, рреНтг = 3,75 г!см\
2=1, фед. гр. C6/mmc [283]. Дисульфид титана TiS2 описан в
работах [173, 191, 249, 310, 314, 419]. Эрлих [249] показал, что TiS2
имеет гексагональную решетку типа Cdl2, параметры которой, по
[249], а = 3,405, с = 5,685 Л; по [310],— а = 3,39, с = 5,70 А,
2=1, ррентг = 3,15 г/сл3; по [314], — а = 3,397, с =5,691 А.
Кожина с соавторами [85] считает, что фаза TiS2, по-видимому,
имеет структуру типа Cdl2, фед. гр. РЪ^тс, но параметр с удвоен-
ный по сравнению с данными предыдущих авторов.
По мнению Жаннэ [314], в дисульфиде титана связь Ti — S кова-
лентная с небольшой долей ионной. Таким образом, присоединение
двух атомов серы к одному атому титана, подобно присоединению
двух атомов кислорода, приводит к исчезновению металлической
связи и металлических свойств у дисульфида титана, характерных
для сульфидов низшей степени окисления. Этому выводу противо-
речит указание Мактаггарта [379] на то, что дисульфид титана об-
ладает металлической проводимостью и выпрямительными свой-
ствами.
Хан и Хардер [283], нагревая металлический титан с серой в от-
ношении 1 : 4 в вакууме при температуре 600° С, получили трисуль-
фид титана TiS3, кристаллы которого имеют моноклинную решетку
с параметрами а = 4,98, Ь — 3,37, с = 17,6 кХ, р = 97,5°, р =
= 3,21 г/см3, 2 = 4, фед. гр. Р2Х или Р2г/т. Данные Жаннэ и
и Бенара [313, 314] о параметрах решетки TiS3 (а = 4,97, b = 3,42,
с = 8,78 А, Р = 97°10', ризм = 3,20 г!см?) согласуются с данными
[283], за исключением параметра с, которому приписывается в два
раза меньше значение. Херальдсен и соавторы [296] подтвердили
данные Жаннэ, найдя следующие параметры решетки TiS3: а =
= 4,973, Ь = 3,433, с = 8,714 А, Р = 97,47°.
Кожина и соавторы [85] на основании рентгеноструктурного
исследования считают, что в системе Ti — S существуют только две
гомогенные области в пределах составов TiS0,77 — TiSi,17 на основе
девятислойной плотнейшей упаковки атомов серы, свойственной
высокотемпературной модификации TiS, и TiSi.si — TiSj,97 на ос-
нове четырехслойной плотнейшей упаковки, свойственной фазе
TiS2.
Измерена упругость пара над сплавами титана и серы в пределах
состава TiS2 — TiS3 и вычислена теплота диссоциации TiS3, равная
-— 36,6 ккал!моль [296].
Логинов и Дали [95] изучили магнитную восприимчивость сулы*
фидов титана в интервале состава TiS — Ti2S3 при темпера-
туре 91—973° К. Температурная зависимость магнитной воспри-
имчивости оказалась сложной. Это свидетельствует об отсутствии
/ \ ' 'Д‘
5 9-614 65
локализованных d-электронов у ионов титана. Авторы
роятным, что d-электроны различных атомов титана
с образованием атомных связей.
считают
ве~
спариваются
ТИТАН — ХРОМ
В системе титан — хром обнаружено одно металлическое соеди-
нение состава TiCr2 [31, 71, 147, 226, 231, 280, 365, 502]. Высказы-
вавшиеся предположения о существовании других хромидов тита-
на, например TisCr2 [501] и Ti2Crs [54, 64, 393], не нашли подтверж-
дения.
Дувец и Тейлор [231 ] установили, что TiCr2 имеет кубическую
а = 6,929 кХ. Левинжер [365]
решетку типа MgCu2 с параметром
Рис. 13. Диаграмма состояния системы
Ti—Сг [502].
показал, что TiCr2 существу-
ет в двух полиморфных моди-
фикациях. Кубическая моди-
фикация устойчива при темпе-
ратуре ниже 1000° С. Выше
1300° С она превращается в
гексагональную со структу-
рой типа MgZn2 с параметра-
ми решетки а — 4,922 и
с = 7,945 А. В других рабо-
тах указываются следующие
температуры полиморфного
превращения TiCr2 (°C): 1235
[501], 1220 [71], 1150—1165
[280].
Хромид титана TiCr2 —
соединение переменного со-
става. Область гомогенности его, по данным различных авторов,
приходится на сплавы со следующим содеражанием хрома: 59 —
70 ат.% [54], 60—65 ат.% [226], 60—62 ат.% [64]; 61—63 вес.%
[147].
Голубцова [33] разработала метод получения порошка TiCr2
анодным растворением титанохромового сплава в электролите, со-
стоящем из 5 мл соляной кислоты (уд. вес 1,19), 3 г янтарной кислоты
и 1000 мл метанола.
Температура плавления TiCr2 равна 1585° С [71].
Диаграмма состояния системы титан —хром построена Макквил-
ланом [393] и затем уточнена в работах [54, 64, 502] (рис. 13).
Корнилов и Борискина [78] изучили систему TiCr2 — TiFe^
Железо способствует стабилизации твердого раствора на основе
гексагональной модификации TiCr2. При содержании Fe > 8,5 вес.%
полиморфное превращение в твердых сплавах (Ti, Fe) Сг2 не наблю-
дается, если Fe < 8,5 вес.%, полиморфное превращение происходит
при температуре ниже 1200° С. В системе TiCr2 — ZrCr2 существу-
66
ет непрерывная серия твердых растворов между изоморфными
модификациями исходных й- и у-фаз, разделенных узкой
(б + у)-областью [81]. Свечников с соавторами [148] изучил систе-
мы Т1Сг2—NbCr2 и Ti—NbCr2.
ТИТАН — МАРГАНЕЦ
Вальбаум [528] в результате рентгенографических исследований
сплавов титана с марганцем установил существование соединения
TiMn2 с гексагональной структурой типа MgZn2 и параметрами ре-
Рис. 14. Диаграмма состояния системы Ti—Мп [145].
тетки а ~ 4,81, с — 7,88 кХ. Существование TiMn2 подтверждено
в работах [145, 281, 287, 381, 515]. Савицкий и Копецкий [1451
нашли параметры решетки TiMr^ равными а = 4,812, с = 7,817 кХ.
128 Соединение TiMn2 плавится конгруэнтно при температуре 1325° С
В системе Ti — Мп установлено существование фазы, близкой
по составу к TiMn [386], и показано, что эта фаза имеет структуру
типа о-фазы в системах Fe — Сг, Со — Сг и других [242]. Парамет-
ры решетки о-TiMn а = 8,862, с = 4,533 кХ.
Уатерстрат с соавторами [515] в системе Ti — Мп установил су-
ществование двух фаз риф, имеющих состав, также близкий к-
5*
67
TiMn. Фаза Ф устойчива при температуре ниже 900° С, имеет тет-
рагональную решетку с параметрами а — 8,19, с — 12,81 А и
содержит 50,5 вес. % Мп. Выше 900° С устойчива р-фаза (52 вес. %
Мп), кристаллическая структура которой точно еще не определена.
В части системы Ti — Мп, богатой марганцем, предполагается
существование соединений TiMn3 [287] и TiMn4 [145].
Мейкут с соавторами [386] построил диаграмму состояния си-
стемы Ti —Мп, которая была затем уточнена [145, 390] (рис. 14).
ТИТАН — ЖЕЛЕЗО
Сплавы титана с железом впервые получены Вёлером и Девил-
лем [524]. К настоящему времени в системе Ti — Fe описаны три
металлических соединения: Ti2 Fe, TiFe и TiFe^
Рис. 15. Диаграмма состояния системы Ti—Fe [50].
Существование соединения Ti2Fe установили Полонис и Парр
[427]. До них о существовании соединения этого состава с кубиче-
ской решеткой сообщили Лавес и Вальбаум [372]. Однако Ростокер
[449 ] не подтвердил данные этих авторов, высказав предположение,
что они, по-видимому, за Ti2Fe приняли тройные фазы Ti3Fe3O и
Ti4Fe2O.
О существовании соединения TiFe предполагал Вальбаум [526].
Дувец и Тейлор [227] получили его и определили, что оно имеет ку-
бическую решетку типа CsCl с параметром а = 2,696 А и z = 2.
68
Уитт и Вальбаум [504] установили существование соединения
TiFe-2 со структурой типа MgZn2, а Спич [469] подтвердил. Ранее
этому соединению приписывался неверный состав TiFe3 [328, 367,
492]. Параметры решетки TiFe2 таковы: а = 4,769, с = 7,745 А,
2 = 12. Голубцова [30] TiFe2 выделила в чистом виде анодным ра-
створением сплавов.
Диаграмма состояния системы Ti — Fe построена в нескольких
вариантах. Еременко [50] обобщил данные работ [66, 239, 399,
427, 455, 502, 516] и построил сводную диаграмму состояния
(рис. 15).
ТИТАН — КОБАЛЬТ
Сплавы титана с кобальтом впервые получили Стевенхаген и
Шухардт [470, 471 ]. В системе Ti — Со, подобно Ti — Fe, обнару-
жено три соединения: Ti2Co, TiCo и TiCoa- В работах [94, 356, 417]
, Рис. 16. Диаграмма состояния системы Ti—Со [50].
сделаны попытки построить диаграмму состояния системы Ti — Со.
Еременко [50] обобщил результаты предыдущих авторов (главным
образом [94, 417]) и построил сводную диаграмму состояния
(Рис. 16).
Лавес и Вальбаум [372] установили, что Ti2Co имеет кубическую
гранецентрированную решетку с параметрами а = 11,283 А и
Ррентг == 5,64 г/см3, по [227], и а = 11,30 А, по [417].
У соединения TiCo кубическая объемноцентрированная решетка
типа CsCl, изоморфная с TiFe и TiNi; параметр решетки а —
69
= 2,988 кХ [227]. Фаза TiCo характеризуется узкой областью
гомогенности вблизи теоретического состава.
По данным [355], соединение TiCo2 существует в двух полиморф-
ных модификациях. Ранее Кестер [354] этому соединению приписьь
вал неправильный состав TiCo3. Обе модификации обладают сход-
ной структурой типа MgNi2, но отличаются по химическому составу.
Модификация, содержащая больше кобальта, чем TiCo2, имеет па-
раметры решетки а = 4,715 и с = 15,37 кХ, а более бедная кобаль-
том — а = 4,720 и с = 15,392 кХ [227].
ТИТАН — НИКЕЛЬ
В системе титан — никель известны три интерметаллических со-
единения: Ti2Ni, TiNi и TiNi3. Построить диаграмму состояния
системы Ti — Ni пытались многие авторы [370, 371, 375, 423, 490,
Рис. 17. Диаграмма состояния системы Ti—Ni [375].
499, 526]. На рис. 17 представлена диаграмма состояния, построен-
ная Марголиным с соавторами [375].
Соединение Ti2Ni получил Вальбаум [5261. В элементарной ячей-
ке этой фазы содержится 96 атомов, параметр ячейки, по данным
[16], а = 11,310 кХ, по данным [449], а = 11,29 кХ.
Вальбаум [526] получил также TiNi в виде кубической фазы с
объемноцентрированной решеткой типа CsCl. Эта фаза относится
к бертоллидному типу с областью гомогенности в пределах 54—
58 вес.% Ni. Ячейка TiNi3 имеет параметр а = 2,980 кХ и г ==
= 2 [16].
70
Соединение TiNi3, полученное Фогелем и Вальбаумом [499],
имеет плотноупакованную гексагональную решетку с параметрами
а = 5,086, с = 8,304 кХ, по [373], и а = 5,093, с = 8,276 кХ, по
[227]. Тейлор и Флойд [489], Багряницкий и Тяпкин [16] нашли,
что параметр с имеет величину в два раза меньше. По их данным,
параметры решетки таковы: а = 2,5454, с = 8,2900 кХ [489] и а =
5,370, с-8,306 кХ [16].
ТИТАН — МЕДЬ
Образование сплавов титана с медью наблюдал Шнайдер [467]
при нагревании K2TiF6 с металлическими натрием и медью в атмо-
сфере водорода.
Рис. 18. Диаграмма состояния системы Ti—Си [324].
В системе Ti — Си обнаружены соединения Ti2Cu, TiCu, Ti2Cu3,
TiCu2, TiCu3. Исследованию диаграммы состояния системы Ti—Си
посвящено много работ [23, 324, 348, 447, 463, 493], однако обоснован-
ная диаграмма состояния еще не построена. На рис. 18 приведена диа-
грамма состояния по Джокайнеру с соавторами [324], которая
нуждается в уточнении с учетом результатов работ [23, 493].
Соединение Ti2Cu получили Лавес и Вальбаум [372]. По их дан-
ным, оно имеет кубическую гранецентрированную решетку с 96
итомами. Ростокер [449] определил параметр решетки а — 11,24 А.
71
В отличие от [372], Энс и Марголин [241 ] считают, что фаза Ti2Cu
имеет гексагональную решетку с параметрами а — 4,164, с ~
= 3,611 А. Величины этих параметров близки к найденным Карл-
соном [348], который соединению Ti2Cu приписывает состав Ti3Cu.
Таким образом, структура фазы на основе Ti2Cu окончательно еще
не выяснена.
Фазы, в основе которых предполагается соединение TiCu, по-
лучены Карлсоном [348]. Одна из них (у) имеет тетрагональную ре-
шетку, а вторая (6) — гексагональную (структура типа AuCu).
Существование TiCu подтверждено [23, 324, 447, 493], однако струк-
тура этого интерметаллида еще окончательно не установлена.
Соединение Ti2Cu3 получил Джокайнен с соавторами [324],
а соединение TiCu2 — Тжебиатовский с соавторами [493], но струк-
тура их также еще не выяснена.
Соединение TiCu3, по мнению [373], имеет деформированную гек-
сагональную плотноупакованную решетку. Рауб с соавторами [447],
однако, нашел, что решетка ромбическая. Параметры решетки в
области гомогенности изменяются в пределах а = 5,518—5,152,
b = 4,526—4,522 и с = 4,335—4,336 А.
ТИТАН — ЦИНК
В литературе известно шесть соединений титана с цинком, но диа-
грамма состояния системы Ti — Zn еще не построена.
Лавес и Вальбаум [372] сообщили о получении соединения TiZn
с объемноцентрированной решеткой типа CsCl, а также TiZn2 и
TiZn3. Интерметаллид TiZn2, описанный в работах [285, 434, 437],
имеет кубическую решетку типа MgZn2 с параметром а ~ 5,064 А
и z = 12о. У TiZn3 также кубическая решетка с параметром а —
= 3,029 А. Пиетраковский [437] параметр TiZn3 нашел равным а ~
= 3,9322 А и z = 4.
Гейне и Цвиккер [285] получили TiZn5, плавящийся конгру-
энтно при температуре 1445 °C.
Растворяя цинк в титане диффузионным методом, Пиотровский
[434] приготовил титанид TiZn10, который идентифицировал хими-
ческим, металлографическим и рентгеноструктурным методами.
Гейне и Цвиккер [285] установили, а Пиотровский [434] под-
твердил существование TiZn15, образующегося по перитектической
реакции
Ж + TiZn5 TiZn15.
ТИТАН — ГАЛЛИЙ
В системе Ti — Ga обнаружено три соединения: Ti3Ga, Ti2Ga
и TiGa3 (рис. 19).
Соединение TiGa имеет гексагональную решетку типа Ni2Zn с
параметрами а = 4,514 и с = 4,50 А [164, 165, 166].
72
у TiGa3, описанного Вальбаумом [529], тетрагональная струк-
тура типа TiAl3 с параметрами решетки а = 5,559 и с = 8,109 А.
В области концентраций от 23 до 26 ат. % Ga Андерко обна-
4
ружил фазу а2, которая,
по-видимому, основана на
соединении Ti3Ga [164 —
166].
ТИТАН — ГЕРМАНИЙ
13001
нос-
до о-
10 15 20 25
30 35
Ga
В системе Ti — Ge ус-
тановлено три соединения:
Ti6Ge3, TiGe и TiGe2. Мак-
квиллан [395] сделала на-
бросок диаграммы состоя-
ния Ti — Ge.
Пиетраковский и Дувец
[435] установили, что соеди-
нение Ti5Ge3 имеет гексаго-
нальную решетку с пара-
метрами а = 7,537 и с —
= 5,223 А. Моногерманид титана
шеткой с параметрами а = 3,809, b = 5,235 и с = 6,834 А [2].
Дигерманид титана также имеет ромбическую решетку с парамет-
рами а = 8,594, Ъ = 5,030 и с = 8,964 А [531].
700
0
Ti
Рис.
19. Диаграмма состояния
Ti— Ga [166].
системы
TiGe обладает ромбической ре-
ТИТАН — МЫШЬЯК
Получены два арсенида титана — TiAs и TiAs2. У моноарсени-
да титана TiAs гексагональная решетка с параметрами а = 3,635,
с = 12,040 кХ, z = 4 TiAs, по [364, 494], и а — 3,64 и с =
12,28 кХ, по [194].
При длительном нагревании металлического титана и мышьяка
в запаянной кварцевой трубке выше 820° С образуется диарсенйд
титана TiAs2. Он имеет ромбическую решетку с параметрами а =
== 13,27, Ь = 6,96, с = 3,50 А, ризи — 6,30, рРентг = 6,31 г/см\
фед- ГР- Рппт, 2=8.
ТИТАН — СЕЛЕН
В системе Ti —Se обнаружены пять селенидов: Ti3Se2, TiSe,
А155е8, Ti3Se4 и TiSe2. Диаграмма состояния системы Ti — Se еще
построена.
Соединение Ti3Se2 получено нагреванием порошкообразных ти-
Тана и селена в атмосфере водорода [419]. Оно имеет область гомо-
генности в пределах состава TiSeo,5 — TiSeo.7 и кристаллизуется
73
в гексагональной системе со структурой типа NiAs [249 ]. Параметры
решетки а = 3,57 и с = 6,32 — 6,30 А, при составе TiSe0,66 следую-
щие: а = 3,56 и с = 6,30 кХ [284].
Эрлих [247, 2491 синтезировал моноселенид TiSe с гексагональ-
ной структурой типа NiAs и параметрами решетки а — 3,55, с =3
= 6,22 А. Гронволд и Лэнгмюр [277] для состава TiSei.os опреде-
лили параметры решетки а = 3,571 и с = 6,297 А. Хан и Несс
[284] нашли, что фаза TiSe простирается до состава TiSeo,75. В более
поздней работе [298] эти авторы установили, что фаза TiSe в области j
гомогенности (TiSeo.75 — TiSe^o) кристаллизуется в виде двух мо-
дификаций. Одна из них является гексагональной с параметрами ре-
шетки а — 7,14 и с = 11,9 А, а вторая — триклинной. 1
Диселенид титана получил Эрлих [247, 249о]. Он имеет решетку?
типа Cdl2 с параметрами а = 3,54 и с — 5,98 А. Между структурой
TiSe (типа NiAs) и TiSe2 (типа Cdl2) наблюдается гомогенный пе-|
реход.
В области сплавов с составом TiSei>22 — TiSei,36 существует фаза
на основе Ti3Se4 [188, 208]. Она кристаллизуется в моноклинной
системе. Параметры решетки ее а — 6,40 — 6,35, b = 3,57, с =
- 12,05 — 12,01 А, р = 90° 30' — 90° 5Г. Чевретон и Бертаут
[215] в согласии с [188] нашли, что Ti3Se4 имеет структуру типа
NiAs с моноклинной решеткой, параметры которой а = 6,40, b =
= 3,57, с - 12,04 А, р = 90° 42', фед. гр. 2/m. 1
Установлено, что структура сплавов состава TiSei>2 — TiSe2,oo
резко меняется с изменением содержания селена [277]. Сплавы со-
става TiSei.2 имеют гексагональную, TiSei,3—моноклинную, а
TiSei>4 — TiSe2,o — гексагональную структуру. Бернуссети Джаннин
[187], изучая изменение параметров решетки фазы на основе TiSe/
показали ее существование в области состава TiSeit42 — TiSeit96.
В сплавах состава TiSei,6 Бернуссет [189, 208] обнаружил су-
ществование моноклинной модификации с параметрами ячейки а =4
- 3,608, b = 6,19, с = 2x5,97 А, Р = 90° 22', ризм = 5,35 г/см* п
гексагональной, имеющей параметры а = 3,5939 и с = 5,9827 А.
Переход гексагональной модификации в моноклинную сопровож-
дается удвоением параметра с.
Чевретон и Брунье [216] нагреванием титана и селена в кварце-
вой трубке при температуре 800° С в течение двух дней получили
соединение Ti5Se8 с моноклинной решеткой типа NiAs. Это соедине-
ние, вероятно, идентично с моноклинной фазой Бернуссета. I
Фаза TiSe2 является металлическим проводником с выпрямитель-
ными свойствами [379].
ТИТАН — НИОБИЙ
При высоких температурах взаимодействие титана с ниобием^
не наблюдается. Елютин с соавторами [49] установил, что ниже
1600° С в сплавах титана с ниобием образуется соединение TiNbi
74
область существования которого расширяется с понижением тем-
пературы. Образование соединения TiNb подтверждено [45] по
резкому изменению коэффициента диффузии скандия в сплавах с
содержанием 10 вес. % Ti. Однако Шахова и Будберг [157] соеди-
нения TiNb в сплавах титана с ниобием не обнаружили. Они счи-
тают, что предыдущие авторы за TiNb приняли твердый раствор
карбида титана в ниобии.
Кузьмин и Моисеева [92] измерили упругость пара титана над
его сплавами с ниобием при температуре 1600—1800° К. Упругость
пара выражается уравнением IgP = А — BIT.
ТИТАН — РУТЕНИЙ, ТИТАН — ПАЛЛАДИЙ
1600Л
1400
1000]
600 -I
1720°
1160
(P+d-Ti
РОгП
104 О"
#+р-Т1
882°
В системе Ti — Ru Лавес и Вальбаум [372] обнаружили интер-
металлид TiRu с кубической гранецентрированной решеткой типа
CsCl. Иордан [326] определил
параметр решетки а = 3,06 А.
В системе Ti — Pd Лавес
и Вальбаум [372] установили
существование трех соедине-
ний: Ti2Pd, Ti2Pd3 и TiPd3.
Соединение Ti2Pd имеет куби-
ческую гранецентрированную
решетку с 96 атомами в ячей-
ке. У TiPd3 гексагональная
решетка с параметрами а =
= 5,486 и с = 8,976 А [530].
Рудницкий и Бирун [130]
изучили систему Ti — Pd ме-
тодами термического анализа,
ми кр остр у кту р ы, ми кр отвер -
дости и другими и построи-
ли диаграмму состояния
(рис. 20). В отличие от [372],
они установили существова-
ние в системе Ti — Pd одного
соединения TiPd2, образующегося при температуре 1040° С из
Двух твердых растворов: Р и у. Система Ti -— Rh не изучена.
о-*
20 40 60 80 100
P(f Ат.% TL
Рис. 20. Диаграмма состояния системы
Ti —Pd [130].
ТИТАН — СЕРЕБРО
Получение сплавов титана с серебром при нагревании смеси се-
ребра, двуокиси титана, окиси серебра и алюминия или угля было
описано в патентах Росси [453]. Однако в нескольких старых ра-
ботах существование химических соединений в системе Ti — Ag
отрицалось.
75
При сплавлении металлического титана с серебром в дуговой
печи образуется TiAg, который имеет упорядоченную гранецентри-
рованную тетрагональную структуру типа CuAu с параметрами ре-
шетки а = 4,096 и с = 4,069 кХ [503]. J
Шуберт [463] определил параметры решетки TiAg, в отличие
от [503], равными а ~ 11,8 и с — 8,14 А. Он также получил соеди-
нение Ti2Ag с решеткой типа Zr2Cu и параметрами а — 2,95 и
с = 2,90 А.
Уорнер 1518] обнаружил третье соединение титана с серебром
состава TisAg. Оно имеет тетрагональную решетку с упорядоченной
структурой типа Cu3Au, изоморфной с Ti3Cu, Zr3Cu и Zr3Ag, пара-
метры решетки а = 4,187, с = 3,950 Л, рнзм = 6,30, рреНтг =
= 6,24 г/см3, z = 4. Обоснованная диаграмма системы Ti — Ag
еще не построена.
ТИТАН — ИНДИЙ
Андерко с соавторами [163, 165, 166] в системе Ti — In обнару-
жил соединение Ti3In с упорядоченной гексагональной структурой
типа Mg3Cd. Оно образуется в результате эвтектоидного распада
твердого раствора индия в р-титане при температуре 840—850° С.
Джонсон и Прозен [327] нашли в сплавах титана с индием инкон-
груэнтно плавящееся соединение Ti3In4, которое имеет тетрагональ-
ную решетку с параметрами а = 10,094, с = 3,052 A, z = 2, ризм ~
— 6,42, ррентг = 6,44 г!см?.
ТИТАН — ОЛОВО
Растворение титана в расплавленном олове наблюдал Муассан
[397, 399]. !
' В сплавах Ti — Sn установлено существование соединений Ti3Sn,
Ti2Sn, Ti5Sn3 и Ti6Sn5.
Соединение Ti3Sn, полученное Пиетраковским [436], имеет гек-
сагональную решетку с параметрами а = 5,916 и с — 4,764 А. Су-
ществование его подтверждено в работах [26, 517]. Параметры
решетки, по данным [517], таковы: а = 4,766, с = 5,920 A.- Ti3Sn
плавится конгруэнтно при температуре 1663° С [439].
Соединение Ti2Sn, имеющее гексагональную решетку с парамет-
рами а = 4,653, с = 5,700 А, рРентг “ 6,57 г/см3, получено Пиетра-
ковским и Фринком [439]. I
Пиетраковский и Дувец [435] получили металлид Ti5Sn3, харак-
теризующийся гексагональной решеткой с параметрами а = 8,049
и с = 5,454 А. :
Пиетраковский и Фринк [439] обнаружили также TieSn5, который
кристаллизуется в гексагональной системе с параметрами а — 9,22,
с = 5,69 А, ррентг = 6,85 г/см3. TLSm плавится конгруэнтно при
температуре 1494°С [439]. I
76
Рис. 21. Диаграмма состояния системы Ti — Sn [50].
Еременко обобщил данные о системе Ti — Sn и построил ее
диаграмму состояния (рис. 21k
ТИТАН — СУРЬМА
В системе Ti — Sb обнаружены следующие соединения: Ti4Sb,
Tiir7Sb, TisSb, Tii.2Sb, TiSb и TiSb2.
Соединение Ti4Sb получили Новотный и Функ [4121. Оно имеет
упорядоченную гексагональную решетку с параметрами а = 2, а'=
5,946, с = 4,798 А, РреНТГ = 5,66 г!см?, фед. гр. £>О19.
Кьексхус с соавторами [349] получил металлиды Ti3Sb, Tii./Sb
и Tii>2Sb. Фаза Ti3Sb существует в двух полиморфных модифика-
циях — кубической, типа ^-вольфрама с параметром решетки а =
= 5,2186 А, и тетрагональной — с параметрами решетки а —
::: 10,465, с = 5,2639 A, z = 8, фед. гр. 14/mCm. Фазы Tii>7Sb
и Ti] 2Sb кристаллизуются в ромбической системе с параметрами а =
2= 10,172, b = 8,348, с = 7,135 А и а = 14,55, b = 16,34, с =
5,31 А соответственно.
Соединения TiSb и TiSb2 получили Новотный и Песл [410].
Первое из них кристаллизуется в гексагональной системе с решет-
кой типа NiAs и параметрами а = 4,070 и с = 6,305 А. У второго
^единения тетрагональная решетка типа СиА12 с параметрами а=
6,666, с = 5,817° А. Къексхус с соавторами [349] нашел пара-
метры решетки TiSb а = 4,115, с = 6,264 А.
ТИТАН — ТЕЛЛУР
В системе Ti—Те обнаружены следующие соединения: Ti2Te>
Ti5Te4,TiTe, TisTe4 и TiTe2. Теллуриды титана образуются при нагре-
вании металлического титана и теллура в кварцевой запаянной трубке.
Соединение Ti2Te получили Хан и Несс [284, 298]. Оно имеет
область гомогенности в пределах TiTeo,25 — TiTeo,7 и кристалли-
зуется в тетрагональной системе с параметрами решетки а = 14,3^
с = 3,58 A, z - 25 TiTeo.s [298]. 1
Фаза Ti5Te4 описана Гронволдом с соавторами [276, 454]. При
стехиометрическом составе она имеет тетрагональную решетку с
параметрами а — 10,164, с = 3,7720, А 2 = 2, фед. гр. 14/m I
Монотеллурид титана TiTe описал Эрлих [247, 245]. Он имеет!
структуру типа гексагонального NiAs с параметрами ячейки а = ,
— 3,83, с — 6,39 А [249]. Хан и Несс [284] установили, что TiTe
имеет область гомогенности в пределах TiTeo,75 — TiTei.oo, в кото- ;
рой кристаллизуется фаза типа NiAs. Параметры решетки, по их
данным, таковы: а = 7,71, с = 12,60 А [298], т. е. удвоенные по
сравнению со значениями, полученными Эрлихом.
Чевретон и Бертаут [215] описали теллурид состава Ti3Te4 со
структурой типа моноклинного NiAs и параметрами решетки а =
= 6,82, b = 3,85, с = 12,66 А, ₽ = 90° 28'. I
Эрлих [247, 249] получил дителлурид титана TiTe2 со структурой ]
типа Cdl2. Параметры решетки его а = 3,75, с — 6,51 А. Дител- |
лурид титана обладает металлической проводимостью. Эрлих [2491.1
считает, что между моно- и дителлуридами титана существует не-1
прерывный переход от гексагональной структуры типа NiAs (TiTe)
к тетрагональной типа CdT2 (TiTe2). Рааум с соавторами [454] сооб-
щил, что в системе Ti — Те существует фаза Ti2-%Te2, гомоген-1
ная от 55,4 до 66,67 ат. % Те. При содержании 60 ат. % Те она j
имеет структуру типа моноклинного NiAs, а в интервале 60 —
66,67 ат. % Те — гексагональную типа Cd (ОН)2. Фаза Ti2—xTeJ
построена по типу вычитания. |
ТИТАН — РЕНИЙ |
Тжебиатовский и Немец [495] в системе Ti — Re установили су-4
ществование только одного соединения состава T^Re^. Оно имеет I
решетку типа а-марганца с параметром а = 9,54 кХ и числом ато-1
мов в ячейке г = 58. Савицкий и Тылкина [143] подтвердили I
существование этого соединения, обладающего большой микротвер-1
достью, равной 2000 лса/лии2. I
ТИТАН — ОСМИЙ, ТИТАН — ИРИДИЙ, ТИТАН — ПЛАТИНА |
Лавес и Вальбаум [372] в системе Ti — Os обнаружили соедине,- j
ние TiOs с кубической решеткой типа CsCl и параметром а — 3,07 A j
[326].
78
Еременко с соавторами [52] в системе Ti — Ir в области сплавов,
содержащих 1—55 ат. % 1г, обнаружил соединение Ti3Ir (у-фаза)
с решеткой типа Сг8О, имеющее параметр а = 5,00 кХ, и соедине-
ние Tilr. Последнее существует в виде двух модификаций — высо-
котемпературной с решеткой типа CsCl, а = 3,10 кХ и низко-
температурной 6, структура которой еще не определена.
Кёнихсбергер и Шиллинг [352] наблюдали сплавление титана
с платиной при температуре выше 400° С. В системе Ti — Pt обна-
ружено пять соединений: Ti3Pt [229], Ti2Pt, Ti2Pt3, TiPt3 [372]
и TiPt8 [440].
Соединение TiPt оимеет структуру типа 0-вольфрама, параметр
решетки а = 5,033 А.
Фаза Ti2Pt кристаллизуется в кубической системе, образуя гра-
нецентрированную решетку типа Ti2Ni. Структура Ti2Pt3 еще не
установлена.
Триплатинид титана Ti2Pt3 имеет структуру, подобную TiZns
[3721. По мнению [530], структура TiPt3 подобна AuCu3, параметр
решетки а = 3,90 А. Дувец и Джордан [229] нашли, что TiPt3
имеет фед. гр. РтЗп, а = 5,033 A, z = 2.
У TiPts тетрагональная решетка с параметрами а = 8,312, с =
= 3,897 A, z = 2, фед. гр. [440].
ТИТАН — ЗОЛОТО
В системе Ti — Au найдено пять соединений: Ti3Au, TiAu,
TiAu2, TiAu4 и TiAu6. Еременко [50], используя данные [438,-447],
построил диаграмму состояния системы Ti — Au (рис. 22).
ч Соединение Ti3Au впервые обнаружили Лавес и Вальбаум [372].
Оно имеет структуру типа (3 -вольфрама с параметром решетки а~
~ 5,096 А, фед. гр. РтЗп, z — 2Au. Данные [372] подтверждены
Маквилланом [394]. Однако по Шуберту [463], yoTi3Au тетрагональ-
ная решетка с параметрами а — 3,13, с = 19,94 А, фед. гр. 14 ттт.
Ti3Au плавится конгруэнтно при температуре 1932° С [438].
Пиетраковский с соавторами установил существование моноаури-
Да титана [438]. Он имеет значительную область гомогенности, пла-
вится конгруэнтно при температуре 1490° С.
Диаурид титана TiAu2, полученный Рауби с соавторами [447],
плавится конгруэнтно при температуре 1485° С. Он имеет гексагоо-
нальную плотно упакованную решетку с параметром а = 2,79 А
и отношением с!а ~ 1,71. Диаурид титана плавится конгруэнтно
пРи температуре 1455° С.
Пиетраковский [441] открыл соединение TiAu4, образующееся
п° перитектической реакции при температуре 1123° С.
Рауб и соавторы [447] установили существование соединения
^AUg с тетрагональной гранецентрированной решеткой, имеющей
Параметр а = 4,073 А и отношение da = 0,974.
79
ТИТАН — РТУТЬ
Интерметаллиды титана со ртутью получаются нагреванием ме-
таллов в эвакуированных трубках.
Интермета л л ид Ti3Hg существует в двух полиморфных модифи-
кациях [437]. Низкотемпературная модификация устойчива в ин-
Рис. 22. Диаграмма состояния системы Ti —Au [50].
I
тервале температур 540—760° С. Она имеет о кубическую решетку
типа 0-вольфрама с параметрами а = 5,1888 А и z = 8. Высокотем-
пературная модификация устойчива выше 815° С. Она имеет так-
же кубическую решетку, однако типа AuCu3 с параметрами а =
= 4,1654 А и г = 4.
У TiHg тетрагональная решетка типа AuCu с параметрами а =
= 3,009, с = 4,041 A, z = 2.
ТИТАН — СВИНЕЦ
Растворение титана в расплавленном свинце наблюдал еще Муас-
сан [397, 399]. Взаимодействие между расплавленным свинцом и
порошкообразным титаном начинается при температуре выше 500” С
[124].
80
Новотный и Песл [411]
получили металлид Ti4Pb,
имеющий гексагональную
решетку с параметрами
а = 5,985 и с = 4,845 А.
Феррар и Марголин [260]
подтвердили их данные,
найдя параметры решетки
равными а = 5,962, с =
== 4,814 А.
Феррар и Марголин
[260] в системе Ti — Pb
обнаружили существование
фазы (у), основанной на
соединениях Ti3Pb или
Ti2Pb (рис. 23).
ТИТАН — ВИСМУТ
В системе Ti — Bi уста-
новлено существование од-
ного соединения состава
Ti4Bi [410, 411]. Кристал-
Рис. 23. Диаграмма состояния системы
Ti — Pb [260].
।
лическая структура его
еще не определена.
ТИТАН — УРАН
В системе Ti — U обна-
ружено одно металлическое
соединение состава TiU2. Ди-
аграмма состояния системы
Ti — U построена Уди и Бун-
жером [497], а затем повтор-
но исследована Наптоном
[350] (рис. 24).
Соединение TiU2 имеет
гексагональную решетку [183]
с параметрами, по данным
1497], а = 4,817, с = 2,844 А,
поданным [351 ],— а = 4,828,
с = 2,847 А. Гомозов и соав-
торы [35] подтвердили, что
фаза TiU2 образуется при
закаливании сплавов, со-
держащих 18,3 ат.% Ti
(о-фазы).
® 9-614
81
РАСТВОРИМОСТЬ ЭЛЕМЕНТОВ
В МЕТАЛЛИЧЕСКОМ ТИТАНЕ
В ТВЕРДОМ СОСТОЯНИИ
В литературе имеются данные о растворимости в ос- и р-титане многих эле-
ментов и некоторых соединений (табл. 9).
Галоиды с титаном образуют соединения с ионной связью, поэтому раствори-
мость их в а- и Р-титане как физический процесс не имеет реального смысла.
Анализ данных табл. 9 показывает, что а-титан является лучшим растворите-
лем для многих элементов, чем Р-титан. Если все элементы по растворимости в ти-
тане условно разделить на труднорастворимые (растворимость менее 1 ат.%) и
легкорастворимые (растворимость более 1 ат. %), то окажется, что из числа изу-
ченных, к труднорастворимым в а-титане относится 24 элемента, а в р-титане —
только 4. Неограниченной растворимостью в а-титане обладает только его ближай-
ший аналог — цирконий и, по-видимому, гафний, растворимость которого еще не
изучена, а также уран при температуре выше 900° С. В р-титане растворимость всех
элементов, как правило, выше, чем в а-титане. Семь элементов из числа изученных
(V, Сг, Zr, Nb, Мо, Та, U) образуют твердые растворы неограниченного состава.
Труднорастворимыми в а- и Р-титане являются металлы I и II групп и инерт-
ные газы, за исключением водорода, бериллия и магния.
С понижением температуры растворимость всех элементов в а-титане уменьша-
ется. При комнатной температуре растворимость большинства элементов в а-тита-
не ничтожно мала.
О растворимости металлов и неметаллов в а- и р-титане см. [46].
Растворимость титана изучена еще в немногих металлах (около 20) (табл. 10).
Кроме металлов, с которыми титан образует твердые растворы неограниченного со-
става, растворимость его в марганце, железе, кобальте, никеле, меди и золоте ве-
лика, а в магнии, алюминии, цинке и олове мала. Растворимость титана в щелочных
и щелочноземельных металлах, за исключением магния, точно еще не определена,
но она, по-видимому, не превышает 0,01 ат.%.
Таблица 9
Растворимость металлов и неметаллов в металлическом титане
(в твердом состоянии)
Эле- мент a-Ti P-Ti
t, °C Раствори- мость, ат.% Литера- тура f, °C Раствори- мость, ат.% Литера- тура
н 335 7,9 [391 335 33,6—38,0 [391]
Be — 9,1 [249; — 5,1—9,8 [221]
В — 0,5 [221] — 0,5 [221]
В 750—1300 0,25 [421] 750—1300 0,25 [421]
С 920 0,64 [55] 1750 3,3 [551
N 1000 20,4 [213] 2000 6,5 [213]
О 800—1700 33,7 [198] 1740 5,5 [198]
Mg — До 3,0 [261] — До 3,0 [261]
Al 0—1100 38,3 [416] 1600 30,8 г416]
Si 850 0,67 П21] 1320 5,0 г2971
Si 600 0,52 [121] — — -
P —— 0,1 [266] — —II»
S - ' 0,03 [266] — — — •
Ca -—• 0,06—0,08 [312] т—" — 1
Sc 600 2,0 [146] 1430 7,5 [146]
82
Продолжение табл. 9
3-Ti
cx-Ti
Эле- мент /, °C Раствори- мость, ат.% Литера- тура f, °C Раствори- мость, ат.% Литера- тура
V 785 1,5 [221] Вьше 900 Неограничен- [172]
ная раство-
римость
Сг 450 0,34 111] Выше 1380 То же [502]
Сг 800 0,2 [111] — — — - 11 '
Мп 550 0,4 [15] иоо 30,0 [390]
Fe 585—600 0,45 [15] 1(80 22,2 [502]
Со 685 0.5—0,7 [417[ 1(20 14,3 [417]
Ni 750 0,2 [375] S65 10,9 [375]
Си 798 1,2 [15] 1000 14,1 [324]
Zn 400 0,02 [268] --
Ga 700 12 [166] 1530 29 [166]
Ge 500 1,8 [395] 1410 7,3 [395]
Ge 897 4,0 [395] —
As 0,17 [70] — — — —
Se - - - 0,16 [266] — — —-
Y 600 0,5—0,6 [146] 1400 0,81 [146]
Zr — ' 11 Неограничен- [259] — Неограничен- [259]
ная раство- ная раство-
римость римость
Nb 600 1,57 288 Выше 1600 То же 288]
Mo 600—885 0,5 [288] Выше 885 » » [288]
Ag 850—860 7,0 [518] 11(0 25 [518]
In — до 10,5 [163, 165, Более 10,5 [163, 165,
166] 166]
Sn 865 8,5 [439] 1605 16 [439]
Sb — Малая рас- [4Ю] — Малая рас- [410]
творимость творимость
Те - — 0,18 266] — — —
La 900 1,77 [142] 1520 1,50 [259]
La 20 0,72 [142]
Ce 600 0,42 [142] 1450 0,9 [142]
Nd 600 0,63 [144] 1550 1,0 [144]
Ta 550 3,7 [387] 80( Неограничен- [387]
ная раство-
римость
w 715 0,4 [387] 188С 20 387
Re 1,0 [143] 40 [143]
Au 832 4,4 [438] 1367 15 [438]
Au 400 0,5 [432]
Pb 752 4,3 [260] 1305 15,9 [260]
Pb 600 0,71 [260] — — ” ~ •
и 900 Неограничен- [350] Выше (00 Неограничен- [350]
ная раство- ная раство-
римость римость
и 655 1 [350] — —
Al.0. о 600 1,5 [29, 83] 1300 3,5 [28]
6*
83
Таблица 10
Растворимость титана в металлах
^Металл 6 °C Растворимость, ат.% Литерату- ра
Mg 651 0,0049
Mg 850 0,030
Al 400 0,12
Al 640 0,25
V Выше 1000 Неограниченная раствори- мость
Мп 1204 (6-Mn) 7,5
Fe 1300 (a-Fe) 6,0
Fe Комнатная температура (a-Fe) 2,0
Fe Максимальная растворимость 0,64
Co 800—1200 20
Ni 1150 13,0
Ni 750 9,4
Cu 870 (a-Cu) 5,69
Cu 300 (a-Cu) 0,76
Zn 0,0003
Zr —— " Неограниченная раствори- мость
Nb Выше 1600 То же
Mo Выше 1600 » »
Sn Близ точки плавления 0,002—0,003
Ta Выше 830 Неограниченная раствори- мость
W 1880 2,2
Au 1115 10,5
U Выше 900 Неограниченная раствори- мость
u 723 (6-U) 4,0
[161]
[161]
[190]
[190]
[172]
[231]
{500]
[500]
[94]
[490]
[490]
[23]
[23]
[285]
[259]
[288]
[53]
[387]
[387]
[447]
[350]
[350]
МНОГОКОМПОНЕНТНЫЕ МЕТАЛЛИЧЕСКИЕ
СОЕДИНЕНИЯ ТИТАНА
Многокомпонентные металлические соединения титана исследо-
вали, главным образом, методом рентгеноструктурного анализа.
Число изученных систем и полученных соединений еще недостаточ-
но для выяснения закономерностей их образования. Поэтому мы
ограничимся перечнем исследованных систем и известных высоко-
компонентных соединений титана (табл. 11).
84
Гм>|Г"| ч !!> П Г«"П | II If" ТГ'ЧГ*1
СГ> CM TH ,—1 о —1 со О СО Ю to оэ Tf
О СО СО СОСОСОСОСОСОСОСОСЧСО
О] СО СО СО СО СО СО СО СО СО СО СО СО
1—'- t • " ...•
«—1 СО ’ СО О’
со со со со о
СО СО СО СО xf
Ь——11—— Ь—i L_—J 1——I
СО СО СО COCO СО
оо Ю LQ Ю Ю LO
СО СО со СО СО
t—n ilh— >| । |U., »U—U Ь-м
«—н CO
1 I
(N
i -Q
CO CD
85
Продолжение табл. 11
CU
то
н
86
Литература
1. А в г у с т и н и к А. И., Климашин Г. М., Козловский Л. В.-—
Изв. АН СССР. Неорг. материалы, 1965, 1, 830.
2. А г е е в Н. В., Самсонов Г. В.— ДАН СССР, 1957, 112, 853.
3, Агеев Н.В. Химия, металловедение и обработка титана. Итоги науки.
Технические науки, II. Изд-во АН СССР, М., 1959, 5.
4. А г е е в Н. В., Р е з н и ч е н к о В. А., У к о л о в а Т. П., Мо-
дель М. С.— В кн.: Титан и его сплавы, 2. Изд-во АН СССР, М., 1959, 64.
5. Антонова М. М.— Изв. АН СССР. Неорг. материалы, 1965, 1, 1758.
6. А н т у ф ь е в В. В., Васильев Я. В., Вотинов М. П., Хари-
тонова О. К., Харитонов Е. В.— Физ. тв. тела, 1962, 4, 1496.
7. А р и я С. М., Морозова М. П., Вольф Э.— Ж- неорг. хим., 1957,
2 13.
8. Ария С. М., Попов Ю. Г. — Ж. общ. хим., 1962, 32, 2077.
9. А р и я С. М., Голомолзина М. В.—-Ж- общ. хим., 1963, 33, 1389;
Физ. тв. тела, 1962, 4, 292.
10. Б ел я к о в а Е., Ко м ар А., Ми х ай л ов В.—Металлург, 1939, 14, 28.
11. Белякова Е. П., Комар А., Михайлов В. В.— Металлург,
1940, 4, 5.
12. Бергер А. X.— Ж- физ. хим., 1939, 13, 1483; 1939, 13, 372; Acta Physico-
chim. URSS, 1939, 11, 617.
13. Богданова H. И., Пироговская Г. IL, Ария С. М.— Ж- не-
орг. хим., 1963, 8, 785.
14. Борискина Н. Г., Корнилов И. И.— Ж. неорг. хим., 1964, 9,
1163.
15. Борискина Н. Г., Мясникова К- П.— В кн.: Титан и его сплавы,
7. Изд-во АН СССР, М., 1962, 61.
16. Багряницкий Ю. А., Тяпкин Ю. Д.— В кн.: Доклады на совеща-
нии по исследованию диаграмм состояния металлических систем (краткое со-
держание). Изд-во АН СССР, М., 1956, 111.
17. Вайнштейн Э. Е., Жураковский Е. А.— Изв. АН СССР. ОХН,
1959, 8, 1493.
18. В а с и л ь е в Я. В., Ари я С. М. — Изв. АН СССР. Неорг. материалы,
1965, 1, 347.
19. В а с и л ь е в Я. В., X р ы ч е в а Д. Д., Ария С. М.— Ж. неорг. хим.,
1963, 8, 788.
20. В а ш к о в О. И., Гаврилова В. К. —В кн.: Титан и его сплавы, 2.
Изд-во АН СССР, М., 1959, 145.
21. В е н г л о с к и С., Бокий Г. Б., Победи нс ка я Е. А.— Ж*
структ. хим., 1964, 5, 64.
22. В е р е й к и н а Л. Л., Самсонов Г. В.— Ж. неорг. хим., 1960,5, 1888.
23. Вигдорович В. Н., Крестовников А. И., Мальцев М. В. —
Изв. АН СССР. ОТН, 1958, 2, 149.
24. В о л ь ф А., Толкачев С. С., Кожина И. И.— Вести. ЛГУ, 1959,
10, 87.
25. Гл адышевский Е. И., Л я х В. И., Сколоздра Р. В.,
СтадныкБ. И.— Порошковая металлургия, 1964, 2, 15.
26. Г л азов а В. В., К у р н а к о в Н. Н.— ДАН СССР, 1961, 138, 835.
27. Гл аз ов а В. В.— ДАН СССР, 1965, 160, 109.
28. Глазова В. В.— ДАН СССР, 1965, 164, 567.
29. Глазова В. В., Корнилов И. И.— Изв. АН СССР. Неорг. материа-
_ лы, 1965, 1, 1834.
30. Г О Л у б ц о в а Р. Б.— ДАН СССР, 1958, 118, 89.
31. Голубцова Р. Б.— ДАН СССР, 1961, 137, 593
32. Голубцова Р. Б.~ ДАН СССР, 1961, 137, 859.
33. Голубцова Р. Б.— В кн.: Титан и его сплавы, 7. Изд-во АН СССР, М.,
1962, НО.
Г о р о щ е н к о Я. Г. Химия ниобия и тантала. сНаукова думка», К-, 1965.
87
35. Г о м о з о в Л. И., Рубцова Л. А., Иванов О. С.— В кн.: Строение
и свойства сплавов урана, тория и циркония. Госатомиздат, М., 1963, 92.
36. Голутвин Ю. М.— Ж. физ. хим., 1956, 30, 2251.
37. Голутвин Ю. М.— Ж. физ. хим., 1959, 33, 1798.
38. Г о р б у н о в Н. С., Шишаков Н. А., Садыков Г. Г., Бабад-
Захряпин А. А,—Изв. АН СССР. ОХН, 1961, 11, 2093.
39. Глазунов С. Г., Молчанова С. Г. Диаграммы состояния сплавов
титана. Оборонгиз, М., 1954-
40. Г в о з д о в С. П., Ж У Р е н к о в а А. А.— Изв. высш. уч. зав. Черная
металлургия, 1960, 9, 8.
41. Гвоздов С. П., Журенкова А. А.— Научн. докл. высш, школы.
Металлургия, 1958, 32.
42. Г л а д ы ш е в с к и й Е. И., М а р к и в В. Я-> Кузьма Ю. Б., Чер-
кашин Е. Е.— В кн.: Титан и его сплавы, 10. Изд-во АН СССР, М., 1963,
71.
43. Г р а д ш т е й н А. Е.— Ж. прикл. хим., 1961, 34, 2784.
44. Г р у м - Г р ж и м а й л о Н. В., Корнилов|И. И., Пылае-
ва Е. Н., В о л к о в а М. А.-— ДАН СССР, 1961, 137, 599.
45. Грум-Гржимайло Н. В.— Изв. АН СССР. ОТН, 1957, 7, 24.
46. Гуляев Б. Б.— ДАН СССР, 1965, 164, 574.
47. Дубровская Г. Н., Оганесян В. X.— Изв. АН Арм. ССР.
Хим. наука, 1964, 17, 387.
48. Е л ю т и н В. П., Пенекин Г. И., Лысов Б. С.— Изв. высш. уч.
зав. Черная металлургия, 1963, 11, 5.
49. Елютин В. П., Бернштейн М. Л., Павлов Ю. А.— ДАН СССР,
1955, 104, 546.
50. Е р е м е н к о В. Н. Титан и его сплавы. Изд-во АН УССР, К-, I960.
51. Еременко В.Н. Многокомпонентные сплавы титана. Изд-во АН УССР,
К 1962
52. Еременко В. Н., Штепа Т. Д.> Чураков М.М. — ДАН УССР,
1964, 6, 763.
53. Еременко В. Н., Великанова Т. Я-— Ж- неорг. хим., 1962, 7,
1750.
54. Еременко В. Н.— В кн.: Вопросы металловедения и термообработки.
Труды Ин-та черной металлургии АН УССР, М., 1954, 8, 10.
55. Е р е м е н к о В. Н., Третьяченко Л. А.— Порошковая металлур-
гия, 1964, 6, 27.
56. Е р о ф е е в а М. С., Лукиных Н. Л., Ария С. М.— Ж- физ. хим.
1961, 35, 772.
57. Ж Д а н о в Г. С., Русо нов А. А.— Труды Ин-та кристаллографии
АН СССР.М., 1954, 9, 165.
58. Жураковский Е.А., Дзелановский В. П.— Порошковая ме-
таллургия, 1964, 5, 57.
59. 3 е л и к м а н А. Н., Горовиц Н. Н.— Ж. прикл. хим. 1952,25, 134.
60. 3 е л и к м а н А. Н., Сегерчану П. Ж-— Ж- общ. хим.1956, 26, 3.
61. Корнилов И.И.—Изв. АН СССР. ОХН, 1954,3,392.
62. Корнилов И. И., Будберг П. Б.— Усп. хим., 1956, 25, 1474.
63. К о р н и л о в И. И., Пылаева Е. Н., Волкова М. А.— Изв.
АН СССР. ОТН, 1956, 7, 771.
64. К о р н и л о в И. И., Михеев В. С., Чернова Т. С.— Труды
Ин-та металлургии им. А. А. Байкова, 1957, 2, 126.
65. Корнилов И. И.— Ж. неорг. хим., 1958, 3, 360.
66. К о р н и л оjb И.|И., Б о р и'с к!и н aj^ Н.| Г.— ДАН СССР, 1956, 108,
1083.
67. Корнилов И. И., Нартова Т. Т.— ДАН СССР, 1961, 140, 829.
68. К о р н и л о в И. И., Будберг П. Б. Диаграммы состояния двойных
и тройных систем титана. Изд. ВИНИТИ АН СССР, 1961.
69, Корнилов И. И.— В кн.: Титан и его сплавы. 7. Изд-во АН СССР, ЛЕ,
1962, 5.
70. Корнилов И. И., Пылаева Е. Н., Волкова М. А.— В кн.г
Титан и его сплавы, 10. Изд-во АН СССР, М., 1963, 74.
71 Корнилов И. И., Шахова К. И., Будберг П. Б,—ДАН СССР.
1963, 149, 1340.
72. Корнилов И. И., Глазова В. В.— ДАН СССР, 1963, 150, 313.
73 Корнилов И. И. — В кн.: Титан и егс сплавы, 10. Изд-во АН СССР, М.»
1963, 5.
74. Корнилов И. И., Глазова В. В. — Физ. металлов и металловедение»
1964 18 457.
75. Корнилов И. И., Глазова В. В.— ДАН СССР, 1964, 154, 638.
76. К о р н и л о в И. И., Глазова В. В.— В кн.: Металловедение титана.
«Наука». М., 1964, 15.
77. Корнилов И. И.— Изв. АН СССР. Металлургия и топливо, 1964, 6, 19.
78. К о р н и л о в И. И., Борискина Н. Г.— Ж- неорг. хим., 1964, 9Г
702.
79. К о р н и л о в И. И., Пылаева Е. Н., Волкова М. А., Кри-
пякевич П. И., М а р к и в В. Я.— ДАН СССР, 1965, 161, 843.
80. Корнилов И. И.— Изв. АН СССР. Неорг. материалы, 1965, 1, 1635.
81. К о р н и л о в И. И., БудбергН. Б., Ш а х о в а К. И., Алисо-
ва С. П.— ДАН СССР, 1965, 161, 1378.
82. Корнилов И. И., Глазова В. В.— Изв. АН СССР. Неорг. материа-
лы, 1965,1, 1778.
83. Корнилов И. И., Глазова В. В.— Изв. АН СССР. Металлы, 1965,
1, 189.
84. Корнилов И. И., Глазова В. В.— Изв. АН СССР. Сер. хим., 1966,
1, 16.
85. К о ж и н а И. И., Попов Ю. Г., Толкачев С. С.— Вести. ЛГУ,
4. Физика и химия, 1964, 22, 115.
86. Крестовников А. Н., В е н д р и х 24. С.— Изв. высш. уч. зав. Цвет-
ная металлургия, 1959, 2, 54.
87. К р и п’ я к е в и ч П. I., М а р к i в В. Я.— ДАН УРСР, 1963, 12, 1606.
88. Крылов Б. С.— В кн.: Титан и его сплавы, 10. Изд-во АН СССР, М., 1963,
159.
89. К о в а л е в с к и й А. Е., Петрова Л. А. Микротвердость. Труды
совещания по микротвердости, 1951, 170.
90. Куцев В. С., О р м о н т Б. Ф.— Ж- физ. хим., 1955, 29, 597.
91. К у р н а к о в Н. Н., Тронева М. Я-— Ж. неорг. хим.. 1961, 6, 1347.
92. Кузьмин А. А., Моисеева Т. Г.— Физ. металлов и металловедение»
1964, 18, 925.
93. Левинский Ю. В., МалибековС. Е.— Ж- неорг. хим., 1965, 10,
588.
94. Л и в ш и ц Б. Я-, X а р и н Я- Д.— Ж- неорг. хим., 1958, 3, 685.
95. Логинов Г. М., Далин X.—Ж. неорг. хим., 1962, 7, 682.
96. Львов С. И., Немченко В. Ф., Косолапова Т. Я., Самсо-
н о в Г. В.— ДАН СССР, 1964, 157, 408.
97. Малючков О. Т,, Финкельштейн Б. Н.—ДАН СССР, 1959,
127, 822.
Макаров Е. С., Кузнецов Л. М.— Ж. структ. хим., 1960, 1, 170.
М а р к i в В. Я., Т е с л ю к М. Ю.— ДАН УРСР, 1962, 12, 1607.
Д., Ворошилов Ю. В., Г л а дыш евски й Е. И.—
Изв. АН СССР. Неорг. материалы, 1965, 1, 890.
98.
99.
100. Маркив В. Я
чг * ________
ini’ ^aP кив В^я.— Изв. АН СССР. Металлы, 1966, 1, 156.
сон
сон
сон
1770.
сон
J02. Меер
103. М е е р
104. Меер
1П~ 1940, 13,
Ю5. Меер
,6> 197.
106. Меер
сон
Г. А.— Редкие металлы, 1934, 5 6.
Г. А., Л и п к е с Я. М.— Ж- прикл. хим., 1939, 12, 1759.
Г. А., Кац Г. А., Хохлова А. В.— Ж- прикл. хим.,
Г. А.— Изв. сектора физ. хим. анализа ИОНХ АН СССР, 1943,
Г. А., Крейн О. Е.— Ж- прикл. хим., 1952, 25, 134.
89
T>'I
107. Меерсон Г. А., Самсонов Г. В. — Ж. прикл. хим., 1954, 27, 1115.
108. Меерсон Г. А., Колчин О. П.— Труды Минцветзолото, 25. Метал-
лургиздат, М., 1955, 195.
109. Меерсон Г. А.— Изв. АН СССР, ОТН. Металлургия и топливо, 1962,
3, 33.
110. М е е р с о н Г. А., Р а к и т с к а я Е. М.— Изв. АН СССР. Неорг. матери-
алы, 1965, 1, 80.
111. М и х е е в В. С., Чернова Т. С.— В кн.: Титан и его сплавы, 7. Изд-во
АН СССР, М., 1962, 68.
112. Михеев В. С., Чернов Т. С., Мясникова К. П., Марко-
вич К- П.— Изв. АН СССР. Металлургия и горное дело, 1964, 2, 156.
113. М о д е л ь М. С., Уколова Т. П.— Ж. неорг. хим., 1957, 2, 2274.
114. М о р о з о в а М. П., Вольф Э., Балова Т.— Вести. ЛГУ, 1959,
1,4, 78.
115. М о р о з о в а М. П., Хрипун М. К-, Ария С. М.— Ж. общ. хим.,
1962, 32, 2072.
116. Морозова М. П., Хернбург М. М., Черезова Л. А.— Вести.
ЛГУ. Физика и химия, 4, 22, 169, 1965.
117. М о р о з о в а М. П., Конопельке М. В.— Вести. ЛГУ, 1965, 4, 148.
118, О с т р о у м о в Е. А.— Зав. лаб., 1935, 506.
119. Пат. США № 2 681851 от 21.06.1954 г.
120. Пат. ФРГ № 924927 от 14.04. 1955; РЖХим., 1956, 26184П.
121. Пылаева Е. Н., Волкова М. А.—В кн.: Титан и его сплавы, 7. Изд-
во АН СССР, М., 1962, 74.
122. Попова И. А., Рабезова В. И. — В кн.: Титан и его сплавы, 7.
Изд-во АН СССР, М., 1962, 105.
123. Портной К- И., Левинский Ю. В.— Ж- физ. хим., 1963, 37, 2627.
124. Рабинович Б. Н., Чижиков Д. М.— Изв. АН СССР. ОТН, 1956,
7, 114.
125. Ревякин А. В., Резниченко В. А.— В кн.: Титан и его сплавы,
2. Изд-во АН СССР, М., 1959, 126.
126. Резниченко В. А., Халимов Ф. Б.— В кн.: Титан и его сплавы,
2. Изд-во АН СССР, М., 1959, 11.
127. Резниченко В. А., Хромова Н. В.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 89.
128. Резниченко В. А., Халимов Ф. Б.— В кн.: Титан и его сплавы,
9. Изд-во АН СССР, М., 1963, 42.
129. Руднева А. В., Модель М. С.— Изв. АН СССР. Металлургия и
горное дело, 1963, 1, 59.
130. Рудницкий А. А., Б и р у н Н. А.— Ж- неорг. хим., 1960, 5, 2414.
131. Русаков А. А., Жданов Г. С.—ДАН СССР, 1951,77,411.
132. Самсонов Г. В.— ДАН СССР, 1952, 86, 329.
133. Самсонов Г. В., Марковский Л. Я.—Усп. хим., 1956, 25, 190.
134. Самсонов Г. В., Н е ш п о р В. С., Л а н г е Л. В.— Металловедение
и обработка металлов, 1956, 1, 35.
135. С а м с о н о в Г. В., У м а н с к и й Я- С. Твердые соединения тугоплав-
ких металлов. Металлургиздат, М., 1957.
136. Самсонов Г. В.— Укр. хим. ж., 1957, 23, 287.
137. Самсонов Г. В.— Порошковая металлургия, 1959, 2, 3.
138. Самсонов Г. В., Верхоглядова Т. С.— ДАН СССР, 1961, 138,
342.
139. Самсонов
140. Самсонов
1, 180.
Г. В.— Изв. АН СССР. Неорган. материалы, 1965, 1, 1803.
Г. В., П а д е р н о В. Н.— Изв. АН СССР. Металлы, 1965,
141. С а м с о н о в Г. В.— Ж- прикл. хим., 1965, 28, 919.
142. Савицкий Е. М., Бурханов Г. С.— Ж. неорг. хим., 1957, 3, 2609. I
143. Савицкий Е. М., Т ы л к и н а М. А. Ж. неорг. хим., 1958, 3, 820.
144. Савицкий Е. М., Бурханов Г. С.— Ж. неорг. хим., 1960, 7, 1751.
145. Савицкий Е. М., Копецкий Ч. В.— Ж- неорг. хим., 1960, 5, 2422.
90
146 Савицкий Е. М., Бурханов Г. С.— В кн.: Титан и его сплавы,
7. Изд-во АН СССР, М., 1962, 51.
147, Свечни ков В. Н., Кочержинскнй Ю. А., Латыше-
ва В. И.— В кн.: Научн. труды Ин-та металлофизики АН УССР, 1962, 6,
132.
148. С в е ч н и к о в В. Н. Кочержинскнй Ю. А., Латыше-
ва В. И.— Научн. труды Ин-та металлофизики АН УССР, 1964, 19, 192.
149. Софьина В. В., Павловская Н. Г.— Ж. физ. хим., 1960,34, 1104.
150. Уманский Я. С., Хидекель С. С.— Ж- физ. хим., 1941, 15, 983.
151. Филоненко Н. Е., Кудрявцев В. И., Лавров И. В.—ДАН
СССР, 1952, 86, 561.
152. Филоненко Н. Е„ Кудрявцев В. И.— ДАН СССР, 1953, 88,
891.
153. Халимов Ф.Б., Резниченко В. А.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 14.
154. Халимов Ф. Б., Резниченко В. А.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 21.
155. Халимов Ф. Б., Резниченко В. А.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 22.
156. Хойд Б. Г., Айринг Л.— Ж. неорг. хим., 1965, 10, 2837.
157. Ш а х о в а К- И., БубдергП. Б.—В кн.: Титан и его сплавы, 7. Изд-во
АН СССР, М., 1962,78.
158. Alexander Р. Р.— Metals and Alloys, 1937, 8, 263.
159. Alexander P. P. — Metals and Alloys, 1938, 9, 45.
160. Alexander P. P.— Metals and Alloys, 1938, 9, 179.
161. A u s t К- T., P i d g e о n L. M.— J. Metals, 1949, 1, 585.
162. A g t e C., Moers K.— Z. Anorg. Chem., 1931, 198, 233.
163. Anderko K-, S a g e 1 K-, Zwicker U.—Z. Metallkiinde, 1957, 48, 57.
164. Anderko K-, Zwicker U.— Naturwis., 1957, 44, 510.
165. Anderko K-— Naturwis., 1957, 44, 88.
166. Anderko K-— Z. Metallkiinde, 1958, 49, 165.
167. van Arkel A. E.— Physika, 1924, 4, 286.
168. van Arkel A. E., de В о e r J. H.— Z. anorg Chem., 1925, 148, 845.
169. A p а и Д., Хаяси X., Накамура С., Адзума Н.~-Rept.
Govt Industr. Res. Inst. Nagoya, 1962, 11, 119.
170. Abler S. F., S e 1 w о о d P. W.— J. Am. Chem. Soc., 1954, 76, 346.
171. A d r a h a m s S. C.— Phys. Rev., 1963, 130, 2230.
172. Adenstedt H. K-, RequignotJ. R., Raymer J. M.— Trans.
Am. Soc. Metals, 1952 , 44, 990.
173. Abendroth R.P., Schlechten A. W.— Trans. AIME, 1959, 215,
145.
174. Asbrink S., Magneli A.— Acta chem. Scand., 1957, 11, 1606.
175. Asbrink S., M a g ne 1 i A.— ActaCrystallogr., 1959, 12, 575.
176. Andersson S., Magneli A.— Naturwis., 1956,43, 495.
177. Andersson S., et a 1.— Acta chem. Scand., 1957, 11, 1653.
178. Andersson S. et a 1.— Acta chem. Scand., 1957, 11, 1641.
179. Andersson S.— Acta chem. Scand., 1959, 13, 415.
1^0. Andersson S.— Acta chem. Scand., 1960, 14, 1161.
181. Andersson S., Jahnberg L.— Arkiv kerr.i, 1964, 21, 413.
182. В r a k k e г H.— Z. Kristallogr., 1928, 67, 547.
183. Bloch J.— Compt. rend., 1964, 259, 1343.
184. В о k L. D. С., О d end a al P. E.— J. Afric. Chem. Inst., 1957, 10, 40.
185. Brauer G., Littke W.— J. Inorg. Chem., 1950, 16, 67.
186. Brauer G., Gingerich K-, Knausenberger M.— Angew.
1O Chem. 1964, 76, 187.
187. Bernusset P., Jeannin Y.— Compt. rend., 1962, 255, 934.
!~8. Bernusset P., Jeannin Y.— Compt. rend., 1962, 255, 2973.
J89. Bernusset P.— Compt. rend., 1963, 257, 2840.
iy0. Buckle H.— Metal If orsch., 1946,1, 437.
91
191. В i 1 t z W., Erlich P., Meisel K.— Z. anorg. Chem., 1937, 234, 97.
192. В i 11 z W., Rink A., Wiechmann F.— Z. anorg. Chem., 1938,
238 395
193. Blumenthal R.N., Whitmore D. H.— J. Electrochem. Soc., 1963,
110, 92.
194. Bachmayer K-, Nowotny H., Kohl A.—Monatsh. Chem., 1955,
86, 39.
195.
196.
197.
Boiler H., Nowotny H.— Monatsh. Chem., 1964, 95, 1272.
Belon L., Borestier H.— Compt. rend., 1964, 258, 4282.
Bumps E. S., Kessler H. D., Hansen M.— J. Metals, 1952, 4
609.
198. Bumps E. S., Kessler H. D., Hansen M.— Trans. Am. Soc. Me-
tals, 1953, 45, 1008.
199. В r u к 1 C., Nowotny H., Schob O.— Monatsh. Chem., 1961, 92,
781.
200. Billy M.—Ann. chim., 1921, 16, 5.
201. Becker K., Ebert F.— Z. Phys., 1925, 31, 268.
202. Becker K-, Ebert F.— Z. techn. Phys., 1930, 11, 148.
203. Burgers W. G., Basart J. C.— Z. anorg. Chem., 1934, 216, 209.
204. Barnighausen H., Knausenberger M., Brauer G.— Acta
Crystallogr., 1965, 19, 1.
205. Breisacher P., Siegel B.— J. Am. Chem. Soc., 1963, 85, 1705.
206. В i 11 h e r H., G о r e t z к i H.— Monatsh. Chem., 1960, 91, 616.
207. Barbaras G. B. et al.— J. Am. Chem. Soc., 1951, 73, 4583.
208. Benard J., Bernusset P.— Z. anorg. Chem., 1964, 328, 231.
209. Carlson O. N. et al.— J. Metals, 1956, 8, 132.
210. Carlson K. D., Moser C.— J. Phys. Chem., 1963, 67, 2644.
211. Carlson K. D. Nesbet R. K-—J. Chem. Phys., 1964,41, 1051.
212. C a d о f f J., P о 1 t у A. E., Margolin H., Nielsen J. P.— New
York University, Final, Rept. on Contract № DA-30-069-ORD to Watertawn
Arsenal 1952
213. C a doff J., Nielsen J. P.— J. Metals, 1953, 5,248.
214. C a d о f f J., Nielsen J. P., Miller E. Warmfeste und Karrosion
bestandige Sinterwerkstoffe, 2. Plansee Siminar «De re metallica», 1956, 216.
215. Chevreton M., Bertaut F.— Compt. rend., 1962, 255, 1275.
216. Chevreton M., В г u n i e S.— Bull. Soc. mineral, et cristallogr., 1964,
87,277.
217. Clark H. T.— J. Metals, 1949, 1, 588.
218. Clark D., Terry J. C.— Bull. Inst.Metals, 1956,3, 13, 116.
219. C h i о 11 i P.— J. Am. Ceram. Soc., 1952, 35, 123.
220. Cotter P. G., Kohn J. A. Potter R. A.— J. Am. Ceram. Soc.,
1956, 39, 11.
221. С r a i g h e a d С. M., Simmons O. W., Eastwood Z. W.— 1
J. Metals, 1950, 2, 485.
В i c h о r a
222. C h r e t i e n A., Freundlich W-,
1954, 238, 1423.
M.— Compt. rend.
223. Campbell I. E. et al.— J. Electrochem. Soc., 1949, 96, 318.
224. Сато, Хаун, Я н ь-цин, К о н д о,— J. Japan Inst. Met., 1959, 23,
456.
225. Chih-Chung Wang, Grant N. J.— J. Metals, 1956, 8, 18.
226. Cuff F. B., Grant N. J., Floe C. F.— J. Metals, 1952, 4, 848.
227. Duwez P., Taylor J. L.— J. Metals, 1950, 2, 1173.
228. Duwez P., Odell F.— J. Electrochem. Soc., 1950, 97, 299.
229. Duwez P., Jordan С. B.— Acta Crystallogr., 1952, 5, 213.
230. Duwez P., Taylor J. L.— J. Metals, 1952, 4, 70.
231. Duwez P., Taylor J. L.— Trans. Am. Soc. Metals, 1952, 44, 495.
232. D Oder о M.—J. Chim. Phys., 1952, 49, СП.
233. D a w i h 1 W., R i x W.— Z. anorg. Chem., 1944, 244, 191.
234. DeVries R.C., Roy R.— Am. Ceram. Soc., Bull., 1954, 33, 370.
92
935. Decker B.F., Kasper J. S.— Acta Crystallogr., 1954, 7, 77.
936. Denker S. P.— J. Phys. Chem. Solids, 1964,25, 1397.
937. Dranteley, Beckman n.— J. Am. Chem. Soc., 1930, 52, 3956.
938. Ebelmen J.J.— Ann. Chim. Phis., 1847, 20, 392.
239. E и ce E., Margolin H.— J. Metals, 1956, 8, 572.
240. E n с e E., Margolin H.— Trans. AIME, 1961, 221, 15L
241. E n с e E., Margolin H.— Trans. AIME, 1961., 221, 320.
242 Elliott R.P., Levi ng er B.W., Rostoker W.— Trans. AIME,
1954, 200, 228.
243. van Erckelen E.— Metall und Erzt, 1923, 20, 206.
244. Ehrlich P.— Z. Elektrochem., 1939, 45, 362.
245. Ehrlich P.— Z. anorg. Chem., 1941, 247, 53.
246. Ehrlich P.— Angew. Chem., 1947, A59, 163.
247. Ehrlich P.— Angew. Chem., 1948, A60, 68.
248. Ehrlich P.— Z. angew. Chem., 1949, 260, 3.
249. Ehrlich P.— Z. anorg. Chem., 1949, 260, 1.
250. Friedrich E.— Z. Phys., 1925, 31, 813.
251. F r i e d r i c h E., Sit tig L.— Z. anorg Chem., 1925, 144, 169; 1925,
143, 293.
252. Friedel C., Guerin J.— Ann. chim. Phys., 1876, 8, 24.
253. Friedel C., Guerin J.— Bull. Soc. chim., 1875, 24, 530.
254. Friedel C., Guerin J.— Compt. rend., 1876, 82, 972.
255. Foster L. S.— Metall Progress, 1952, 62, 150.
256. F о e x M., W u c h e r J.— Compt. rend., 1955, 241, 184.
257. Fink W. L., van Horn K. R., Budge P. M.— Trans. AIME, 1931,
3, 421.
258. Freudlich W., Chretien A., Bichara M. — Compt. rend.
1954 239 1141.
259. Fast J. D.— Rec. trav. chim., 1939, 58, 973.
260. Ferrat P., Margolin H.— J. Metals, 1955, 7, 101.
261. Fredricson J. W.— J. Metals, 1955, 7, 368.
262. Gingerich K. A.— Nature, 1965, 200, 877.
263. Gillam E., Rooksby H. P.— Acta Crystallogr., 1964, 17, 762.
264. Gibb T. R. P., Kruschwitz H. W.— J. Am. Chem. Soc., 1950, 72,
5365.
265. Gibb T. R.P., McScharry J., Bragdon R. W.— J. Am. Chem.
Soc 1951 73 1751.
266. Goldhoff R.M., Shaw H.L., Graighead С. M., J a f f e e R. I.—
Trans. Am. Soc. Metals, 1953,45, 941.
267. G a к e 1 H. L.— Acta Crystallogr., 1958, 11, 46.
268. Gebhardt E.— Z. Metallkunde, 1941, 33, 355.
269. G о г e t z к i H., Bittner H., N о w c t n у H.— Monatsh. Chem.,
1964, 95, 1521.
270. Guldbransen E.A., Andrew K. F.—- J. Metals, 1949, 187, 741.
271. Guldbransen E. A., Andrew K. F.— J. Electrochem. Soc., 1949,
96, 364.
272. Graighead С. M., Lenning G.A., Jaffee R. J.— Trans. AIME,
1952, 194, 1317.
273. Glaser J. W.— Trans. AIME, 1952, 194, 391.
274. Qlasser F. W., I v a n i с к W.— J. Metals, 1952, 4, 387.
275. Greenhouse H. M., Accountius О. E., Sisler H. H.—
J. Am. Chem. Soc., 1951, 73, 5086.
276. Gronvold R., Kjekshus A., Raaum F.— Acta Crystallogr.,
n 1961, 14, 930.
277. Gronvold F., Langmyhr F. J.— Acta chem. Scand., 1961, 15, 1949.
278. G u i 1 1 e t L.— Bull. Soc. Nat. Ind., 1902, 101, 244.
279. G e w e с к e J.— Lieb. Ann., 1908, 361, 79.
280. Gross K. A., L a m b о r t I. R.— J. Inst. Metals, 1960, 88, 416.
281. Goldschmidt H. J.— Metallurgia, 1957, 56, 17.
93
282. Groves W. О., Hoch M., Johnston H. L. — J. Phys. Chem.
1955 59 127.
283. Hahn H., Harder B.— Z. anorg. Chem., 1956, 288, 241.
284. Hahn H., Ness P.— Naturwis., 1957, 44, 581<
285. Heine W., Z w i с к e r U.— Z. Metallkunde, 1962, 53, 380.
286. Heine W., Z w i с к e r U.— Z. Metallkunde, 1962, 53, 386.
287. Hellawell A., Hume-Rothery W.— Phys. Trans. Roy. Soc.
1957, 249, 417.
288. Hansen M., Kamen E.L., Kessler H.D., McPher-
son D. J.— J. Memals, 1951, 3, 881.
289. Hoch M., D i n g 1 e d у D. P., J о n s t о n H. L. — J. Am. Chem. Soc.,
1955 77 304.
290. Hoch M., V a i d i J.— J. Am. Ceram. Soc., 1963, 46, 245.
291. Holmberg B.— Acta chem. scand., 1962, 16, 1255.
292. Hagen H., Sieverts A.— Z. anorg. Chem., 1930, 185, 225.
293. Haag R. M., S h i p к о F. J.— J. Am. Chem. Soc.* 1956, 78, 5155.
294. H u 1 1 i g e r F.— Nature, 1964, 204, 775.
295. H о u s к a C. R.— J. Am. Ceram. Soc., 1964,47, 310.
296. Haraldsen H., Kjekshus A., Rost E., Steffensen A.—
Acta Chem. Scand., 1963, 17, 1283.
297. Hansen M., Kesler H. D., McPherson D.J. — Trans. Am. Soc.
Metals, 1952, 44, 518.
298. Hahn H., Ness P.— Z. anorg. Chem., 1959, 302, 17.
299. Hardy G. F., Hulm J. K-— Phys. Rev., 1954, 93, 1004.
300. Humphry G. L.— J. Am. Ceram. Soc., 1951, 73, 2261.
301. H о r i g u c h i Yoshikazu, Nomura Yokan — Bull. Chem.
Soc. Japan, 1963, 36, 486.
302. Hoffmann W., S c r a d e r A.— Arch. Eisenhiittenwesen, 1936, 10, 65.
303. Honigschm id O.— Compt. rend., 1906, 143, 249.
304. Honigschmid O.— Monatsh. Chem., 1906, 27, 1067.
305. Honigschmid O.— Monatsh. Chem., 1907, 28, 1017.
306. H u r 1 e n T.— J. Inst. Metals, 1960, 49, 128.
307. Hagg G.— Z. Phys., 1931, B12, 33.
308. Hagg G.—Z. Phys., 1929, B6, 221.
309. Hagg G.— Z. phys. Chem., 1931, Bll, 433.
310. Hagg G., Schonberg N.— Arkiv kemi, 1954, 7, 371.
311. Huber H., Kirschfeld L., Sieverts A.— Ber., 1926, 59, 2891.
312. Inst. Min. met. Bull., 1956, 66, 1.
313. J e a n n i n G., Benard J.— Compt. rend., 1958, 246, 614.
314. J e a n n i n G.— Ann. Chim., 1962, 7, 57.
315. J e a n n i n G.— Compt. rend., 1963, 256, 3111.
316. Jame W.J., Strumanis M. E.— J. Electrochem. Soc., 1959, 106, 631.
317. Jaffee R. I., Campbell I. E.— J. Metals, 1949, 185, 646.
318. J a f f e e R. I., Og den H. R., Maykuth D. J.— Trans. AIME, 1950,
188, 1261.
319. Juze R., Hahn H.— Z. anorg. Chem., 1940, 244, 133.
320. Jacobson B. Westgren. — Z. phys. Chem., 1933, B20, 362.
321. J u n к e r E.— Z. anorg. Chem., 1936, 228, 97.
322. Jenkins A. E., Worner H. W.— J. Inst. Metals, 1951, 80, 157.
323. Jenkins A. E.— Trans. Am. Soc. Metals, 1953, 45, 1025.
324. J о u к a i n e n A., Grant N. J., F 1 о e C. F.— J. Metals, 1952, 4, 766.
325. Jacquin Y., Jeannin Y.— Compt. rend., 1963, 256, 5362.
326. Jordan С. B.— Trans. AIME, 1955, 203, 832.
327. Johnson R. G., P г о s e n R. J.— Trans. AIME, 1962, 224, 397.
328. J e 1 1 i n h a u s W.— Z. anorg. Chem., 1936, 227, 33.
329. J e i t s c h к о W., Nowotny H., Benesovsky F.— Monatsh.
Chem., 1963, 94, 1198.
330. J e i t s c h к о W., Nowotny H., Benesovsky F.— Monatsh.
Chem. 1963, 94, 1201.
94
q31 Jeitschko W., Nowotny H., Benesovsky F.— Monatsh.
Chem., 1963/64, 95, 178.
332 Jeitschko W., Nowotny H., Benesovsky F.— Monatsh.
Chem., 1963/64, 95, 319.
333. J e i t s c h к о W., Nowotny H., Benesovsky F.— Monatsh.
Chem., 1964, 95, 436.
334. Jeitschko W. et al.— Monatsh. Chem., 1964, 95, 1004.
335. J e i t s c h к о W., Nowotny H., Benesovsky F.— Monatsh.
Chem., 1964, 95, 431.
336. Kelley К- K.— Ind. Eng. Chem., 1944, 36,865.
337. Kubaschewsky O., Dench W.— J. Inst. Metals, 1953, 82, 87.
338. К 1 a u b er A.— Z. anorg. Chem., 1930, 185, 225.
339. Koester W., Banquertm L., Evert M.— Z. Metallkunde, 1956,
47, 564.
340. Klauler A.— Z. anorg. Cehm., 1921, 117, 243.
341. Kirschfeld L., Sieverts A.— Z. phys. Chem., 1929, A145, 227.
342. К i к u г о O.— J. Scient. Res. Inst., 1955, 49, 313.
343. К n a u s e n b e r g e г M., Brauer G., Gingerich K. A.—
J. Less-Common Metals, 1965, 8, 136.
344. Kieffer R., Benesovsky F., Honak E. R.-— Z. anorg, Chem.,
1952, 268, 191.
345. Kieffer R., Kolbe F.— Powder Metallurgy, 1949, 4, 4.
346. Klemm W., Schuth W.— Z. anorg. Chem., 1931, 201, 24.
347. Kuylenstierna U., Magneli A.— Acta chem. scand., 1956, 10,
1195.
348. Karlson N.— J. Inst, Metals, 1951, 79, 391.
349. KjekshusA., GronvoldF., TholjornsenJ.— Actachem.
Scand., 1962, 16, 1493.
350. Knapton A. G.— J. Inst. Metals, 1955, 83, 497.
351. К n a p t о n A. G.— Acta Crystallogr., 1954, 7, 457.
352. Konigsberger J., Schilling K-—Ann. Phys., 1910, 32, 181.
353. Karlson N.— Nature, 1951, 168, 558.
354. Koster W.— Arch. Eisenhiittenwesen, 1934, 8, 471.
355. Koster W., W agner E.— Z. Metallkunde, 1937, 29,230.
356. Koster .— Z. Metallkunde, 1950, 41, 63.
357. Lening G. A., Craighead С. M., J a i f ee K- F.— J. Metals, 1954,
6, 367.
358. Lecerf A., Hardy A.— Compt. rend., 1961, 252, 131.
359. Lecerf A.— Ann. chim., 1962, 7, 513.
360. Levy L.— Compt. rend., 1888, 106, 66.
361. Levy L.— Ann. chim. Phys., 1892, 25, 449.
362. Levy L.— Compt. rend., 1896, 121, 1148.
363. Lundstr от T.— Acta cem. Skand., 1963, 17, 1166.
364. Lukaszewicz K-, Trzebiatowski W.— Bull. Acad. Pol. Sci.,
1954 2 277.
365. Levinge’r B. W.— J. Metals, 1953, 5, 196.
366. Levinson L. S.— J. Chem. Phys., 1965, 42, 2891.
367. Lamort J.— Ferrum, 1914, 11, 225.
^08. L a u g i e r A.— Ann. chim. et phys., 1814, 89,306,317.
L i e b i c h J.— Pogg. Ann., 1831, 21, 159.
^0. Long J. R.— Met. Progress, 1949, 55, 364.
^°ng J. R.— Met. Progress., 1949, 55, 191.
^aves W al lb aum H.J.— Naturwis., 1939, 27, 674.
47л Laves F., Walbaum H. J.— Z. Kristallogr., 1939, 101, 78.
77k’ c L ar e n M. G. Sci. Ceram., 2. London — N. Y., Acad, press, 1965, 407.
Margolin H., Ence E., Nielsen J. P.— J. Metals, 1953, 5, 243.
M e 1 1 e r K-, Arndt H. H. — Naturwis., 1950, 47, 10, 224.
M a n c h о t W., Richter P.— Lieb. Ann., 1907, 375, 140.
°- McKenna P. M.— Powder Metall. Bull., 1950, 5, 88.
95
379. McT a g g а г t F. К.— Aust r. J. Chem., 1958, 11, 471.
380. Manchot W., Leber A.— Z. anorg. Chem., 1926, 150, 26.
381. Margolin H., E n с e E.— J. Metals, 1954, 6, 1267.
382. Moers K-— Z. anorg. Chem., 1926, 153, 7.
383. Matthias В. T., H u 1 m J. K-— Phys. Rev., 1952, 87, 799.
384. Munster A., Sagel K-— Z. Elektrochem., 1953, 57, 571.
385. Morton J. R., Stark D. S.— Trans. Farad. Soc., 1960, 56, 351.
386. M а у к u t h D. J., Ogden H. R., Jaffee R. I.— Trans. AIME, 1953,
197 225
387. M а у к u t h D. J., Ogden H. R., Jaffee R. I— J. Metals, 1953, 5,
231.
388. Munster A., Ruppert W.— Naturwis., 1952, 39, 349.
389. Munster A., Ruppert W.— Z. Elektrochem., 1953, 57, 564.
390. McPherson J., Hansen M.— Z. Metallkunde, 1954, 45, 76.
391. McQuillan A. D.— Proc. Roy. Soc., 1950, A204, 309.
392. McQuillan A. D.— J. Inst. Metals, 1951, 79, 371.
393. McQuillan M. K-— J. Inst. Metals, 1951, 79, 379.
394. McQuillan M. K-— J. Inst. Metals, 1954, 82, 511.
395. McQuillan M. K-— J. Inst. Metals, 1955, 83, 485.
396. McQuillan A. D.,McQuillan M. K. Titanum. Baterworths scien-
tific publication, London, 1956.
397. Moissan H.— Compt. rend., 1895, 120, 290.
398. Moissan H.— Compt. rend., 1896, 123, 13.
399. Moissan H.— Ann. chim., 1896, 9, 229.
400. Moissan H.— Compt. rend., 1896, 122, 1297.
401. Moissan H.— Compt. rend., 1898, 125, 839.
402. N e w n h a m R. E., Haan J. M.— Z. Kristallogr., 1962,117, 235.
403. Nielsen J. P. Titanium Simposium U. S. office of Naval Research, 1949,
153.
404. Nelson J. A., Willmore T. A., Womeldorph R. C.—J. Elec-
trochem. Soc., 1951, 98, 465.
405* Norton J. L, Blumenthal H., Sinderband S. J.— Trans.
AIME 1949 185, 149.
406. Norton J.T., Blumenthal H., Sinderband S. J.— J. Metals,
1949 1, 749.
407. N a у fo r B. F.— J. Am. Chem. Soc., 1946, 68, 1077.
408. Naylor B. F.— J. Am. Chem. Soc., 1946, 68, 370.
409. Naylor B. F.— J. Am. Chem. Soc., 1946, 68, 964.
410. Nowotny H., Pesl J.— Monatsh. Chem., 1951, 82, 336.
411. Nowotny H., Pesl J.— Monatsh. Chem., 1951, 82, 344.
412. Nowotny H., Funk R., Pesl J.— Monatsh. Chem., 1951, 82, 513.
413. Nowotny H.# Schachner H., Kieffer R., Benesovs-
ky F.— Monatsh. Chem., 1953, 84, 1.
414. Nowotny H., Dimakapoulu E. — Monatsh. Chem. 1959, 90, 620.
415. Ogden H.D., Jaffee R. L— J. Metals, 1951,3,335.
416. Ogden H. R., M а у к u t h D. J., Finlay W. Z., Jaffee R. I.—
T Mpfak 1Q51 Я
417. 6 r r e 1 F. L, Eon t ana M. G.— Trans. Am. Soc. Metals, 1955, 47, 554.
418. Opfermann W.— Monatsber. Dtsch. Akad. Wiss., Berlin, 1964, 6, 92.
419. О f t e d о 1 I. — Z. Phys. Chem., 1928, 134, 301.
420. Paneth F., Mathies M.— Ber., 1922, 55, 775.
421. Platy A. E., Margolin H., Nielsen J. P.— Trans. Am. Soc.
Mpfak 1Q^4 46 319
422. Poland D. E., Kuriakose A. K., Margrove J. L.— J. Phys.
Chem., 1965, 69, 158.
423. Poole D. M., Hume-Rothery W.— J. Inst. Metals, 1955, 83, 473.
424. Pfordten O.— Lieb. Ann., 1886, 234, 290, 294.
425. Pico n.— Bull. Soc. chim., 1934, 1, 919.
426. Pico n.— Compt. rend., 1934, 197, 1415.
96
лот. Р о 1 о n i s D. H., Parr J. G.— J. Metals, 1954, 6, 1148,
428. Philipp W.— Acta Metallurg., 1962, 10, 583.
429. Pfordten O.— Lieb. Ann., 1887, 237, 201.
430. Post B., Glaser J. W.— J - Chem. Phys., 1952, 20, 1050.
431. Post B., Glaser F. W., Moskowitz D.— Acta Metallurg., 1954,
2, 20.
432. Pollard F. H., W oodwards P.— J. Chem. Soc., 1948, 1709.
433. Pearson A. D.— Phys. Chem. Sol'ds, 1958, 5, 316.
434. Piotrowski W.— Zesz. nauk. Politechn. lodzk., 1963, 51, 33.
435. Pietrakowsky P., Duwez P.— J. Metals, 1951, 3, 772.
436. Pietrakowsky P.— J. Metals, 1952, 4, 211.
437. Pietrakowsky P.— Trans. AIME, 1954, 200, 219.
438. Pietrakowsky P., Frink £. P., Duwez P.— J. Metals, 1956,
8, 930.
439. Pietrakowsky P., Frink E. P.~ Trans. Am. Soc. Metals, 1957,
49 339.
440. Pietrakowsky P.— Nature, 1965, 206, 291.
441. Pietrakowsky P.— J. Inst. Metals, 1961—1962, 90, 434.
442. Rieger W., Nowotny H., Eenesovsky F.— Monatsh. Chem.,
1964, 95, 1573.
443. Rauechle R. F., Rundle R. E.— Acta Cristallogr., 1952, 5, 85.
444. Rauechle R., Rundle R. E.— Acta Cristallogr., 1953, 6, 107.
445. Renz C.— Ber., 1906, 39, 249.
446. Raman S., Ramachandrar G. N.— Current Sci., 1962, 31, 321.
447. Raub E., Walter P., Engel M.— Z. Metallkunde, 1952, 43, 112.
448. Rostoker W.— J. Metals, 1952, 4, 981.
449. Rostoker W.— J. Metals, 1952, 4, 2096.
450. Ruff O., Eisner F.— Ber., 1908, 41, 2250.
451. Ruff O.— Ber., 1909, 42, 900.
452. Ruff O., Brintzinger H.—Z. anorg. Chem., 1923, 129, 267.
453. Rossi A G.— Пат. США № 802941, 812, 764, 1902 г.; № 1024474, 1912 г.
454. R a a uni F., GronvoldF., KjekshusA., Haraldsen. H.—
Z. anorg. Chem., 1962, 317, 91.
455. Roe W.P., Fisher W. P.— Trans. Am. Soc. Metals, 1952, 44, 1030.
456. Rose H.— Gilb. Ann., 1823, 73, 129.
457. Rose H.— Svenska Acad. Handl., 1821, 247.
458. Rose H.— Pogg. Ann., 1829, 16, 58.
459. Rose H.— Pogg. Ann., 1832, 24, 141.*
460. S c h m i d H.— Acta Crystallogr., 1964, 17, 1080.
461. Shoemaker С. B., Shoemaker D. P.— Acta Crystallogr., 1965, 18,
900.
462. Schissel P. O., Trulson О. C.— J. Phys. Chem., 1962, 66, 1492.
463. Schubert K.— Z. Metallkunde, 1965, 56, 197.
464. S h i m e r P. W.— Proc. Roy. Soc., 1837, 42, 89.
465. Sieverts A., Roell E.— Z. ancgr.Chem., 1925, 146, 149.
466. Sieverts A., Gotta A.— Z. anorg. Chem., 1928, 172, 1.
467. Schneider E. A.— Z. anorg. Chem., 1895, 8, 94.
468. S i d h e r S. S., Heaton L. R., Z a u b e r i s D. D.— Acta Crystallogr.,
1956 9 607.
469. S p e i c h G. R. — Trans. AIME, 1962 224, 850.
470. Stev enhagen A., Schuchardt E.— Ber., 1899, 32, 1513.
471. Stevenhagen A., Schuchardt E.— Ber., 1902, 35, 909.
472. Schutzenberger P., Colson A. — Compt. rend., 1882, 94, 1710.
473. S a g e 1 K., Schulz E., Z w i с < e r U — Z. Metalkunde, 1956, 47,
529.
474. S c h о f i 1 d T. H., Bacon A. E.—J. Inst. Metals, 1955, 84, 47.
Schmitz-Dumont O., Steiberg K-— Naturwis., 1954, 41, 117.
^76. Sidhu S., Heaton L., Zauberis D. D.— Acta Crystallogr., 1956,
9, 607.
97
477. S t r aum a n i s M. E., Cheng С. Н.» SchlechtenA. W.— J. Elek.
trochem. Soc., 1955, 103, 439.
478. Str aumanis M. E. Li H. W.— Z. anorg. Chem., 1960, 305, 143.
479. S t r a um a n i s M. E. E j i m a T.— Acta Crystallogr., 1962, 15, 404.
480. Straumanis M. E. et al.— J. Electrochem. Soc., 1965, 112, 262.
481. Stalin ski B., Coogan С. K-, Gutowsk у H. S.— J. Chem. Phys.
1961, 34, 1191.
482. Stalins к i B., Bieganski Z. —Bull. Acad. Polon. Sci. Ser. sci.
chim., 1962, 10, 247.
483. Schonberg N.— Acta chem. Scand., 1954, 8, 1460.
484. Schonberg N.— Acta chem. Scand., 1954, 8, 226.
485. Snell P.-O.— Acta Chem. Scand., 1967, 21, 1773.
48 5. S h о m a t e С. H.— J. Am. Chem. Soc., 1946, 68, 310.
487. Shomate С. H.— J. Am. Chem. Soc., 1946, 68, 964.
488. Schafer H., Fuhr W.— Z. anorg. Chem., 1962, 319, 52.
489. Taylor A., Floyd R. W.— Acta Crystallogr., 1950, 3, 285.
490. Taylor A., Floyd R. W.— J. Inst. Metals, 1952, 80, 577.
491. Thorpe E.— J. Chem. Soc., 1885, 47, 493.
492. Tofaute W., Buttinghaus A.— Arch. Eisenhiittenwesen, 1938^
12, 33.
493. Trzebiatowski W., В er a к G., Romotowski T.— Roszn.
chem., 1953, 27, 426.
494. Trzebiatowski W., Lukaszewicz K.— Roszn. chem., 1954r
28, 150.
495. Trzebiatowski W., N i e m i c J.— Roszn. che m., 1955, 29.
496. U e 1 z H. F. Y.— J. Am. Ceram. Soc., 1950, 33, 340.
497. U d у С. M., Bounger F. M.— Trans. AIME, 1954, 200, 207.
498. Vasilos T., Kingery W. D.— J. Am. Ceram. Soc., 1954, 37, 409.
499. Vogel R., Wallbaum H.J.— Arch. Eisenhiittenwesen, 1938, 12, 299.
500. Vogel R., Ergang R.— Arch. Eisenhiittenwesen, 1938, 12, 149.
501. Vogel I.R., Wanderott B.— Arch. Eisenhiittenwesen, 1940, 14, 279.
502. van Thyne R. J., Kessler H. D., Hansen M.— Trans. Am.
Soc. Metals, 1952, 44, 974.
503. van Thyn R. J., Rostoker W., Kessler H. D.— J. Metals,
1953, 5, 670.
504. Witte H., Wallbaum H. J.— Z. Metallkunde, 1938, 30, 100.
505. Winkler C.— Ber., 1891, 23, 2662.
506. W i b e r g E., U s о n B.— Z. Naturforsch., 1951, 6B, 392.
507. Williams W. S.— J. Phys. Chem., 1961, 65, 2213.
508. Weiss L., Kiser H.— Z. anorg. Chem., 1910, 65, 345.
509. Wyss R.— Ann. chim., 1948, 123, 215.
510. Wasilewski R.I.— Programm AIME, 1957, 3.
511. Wasilewski R. I.— Trans. AIME, 1962, 1, 224.
512. Wang С. C., Grant N.J.— J. Metals, 1956, 8, 184.
513. W ucher J.— Compt. rend., 1955, 241, 288.
514. W a d s 1 у A. D.— Acta Crystallogr., 1957, 10, 715.
515. Waterstrat R. M., Das B. D., Beck P. A. — Trans. AIME, 1962,
224 512.
516. Worner H. W.— J. Inst. Metals, 1951, 79, 173.
517. Worner H.W.—J.Metals, 1953,81,521.
518. Worner H. W. — J. Inst. Metals, 1954, 82, 222.
519. W 6 h 1 1 e r F.— Pogg. Ann., 1849 78, 401.
520. W 6 h 1 1 e r F.— Lieb. Ann., 1850, 73, 34, 47.
521. W 6 h 1 1 e r F.— Ann. chim. et phys., 1850, 29, 175.
522. W 6 h 11 e r F.— Lieb. Ann., 1853, 87, 375.
523. Wohl 1 er F., Deville H.-C. S.— Lieb. Ann., 1857, 103, 230.
524. Wo h 1 1 er F., Deville H.-S. C.— Ann. chim. phys., 1858, 52, 92.
525. W 6 h 1 1 e r F.— Lieb. Ann., 1860, 113, 248.
526. Wallbaum H.J.— Arch. Esenhiittenwesen, 1940, 14, 521.
98
527. Wedler G., Strothenk H.— Z. phys. Chem., 1966, 48, 86.
528. Wallbaum H.J.— Z. Kristallogr., 1941, 103, 391.
529. Wallbaum H. J.— Z. Metallkunde, 1942, 34, 118.
530. Wallbaum H.J.— Naturwis., 1943, 31, 91.
531. W a 11 b a u m H. J.— Naturwis., 1944, 32, 76.
532. Хорибэ Тосиясу, Кувабара, Сэндзо — J. Chem. Soc. Ja-
pan, Ind. Chem., Sec., 1964, 67, 276.
533. Yamaguchi S., Otsuka R. — Z. anorg. Chem., 1957, 291, 131.
534. Yahia J., Frederikse H. P. R.— Phys. Rev., 1961, 123, 1257.
535. Yamashito J.— Techn. Ret. ISSP, 1963, A, 76; РЖХим., 1965, 14Б,
48.
536. Z a 1 k i n A. et al.— Acta Crystallogr., 1961, 14, 63.
Дополнительная литература
Августиник А. И., Дроздецкая Г. В., Орданьян Л. В. Взаи-
модействие карбида титана с водой.— Порошковая металлургия, 1967, 6, 53.
Августиник А. И., Голикова О. А., Климашин Г. М., Коз-
ловский Л. В. Влияние кислорода на некоторые свойства карбида тита-
на.— В кн.: Исследования в области химки силикатов и окислов. «Наука»,
М. — Л., 1965, 244.
Августиник А. И., Козловский Л. В., Климашин Г. М.
К вопросу о высокотемпературном взаимодействии карбида титана с некото-
рыми окислами.— Изв. высш. уч. зав. Хтмия и хим. технология, 1966, 4,
528.
Арбузов М. П., X а е н к о Б. В. Распределение электронной плотности
в карбиде титана.— Порошковая металлургия, 1966, 4, 74.
Айвазов М.И., Мелехин В.Ф. Кристаллизация нитрида титана из га-
зовой фазы.— Изв. АН СССР. Неорган. материалы, 1967, 3,2109.
Буланов Ю. П., Кузнецова Г. В Исследование условий анодного
выделения фазы Tin Nim С.— Зав. лабор., 1967, 33, 407.
Балакир Э. А., Крылов Ю. И., Кузьминская Л. Н., М о и -
сейцева 3. К- Совместимость окисления магния с моноокисью титана.—
Изв. АН СССР. Неорган. материалы, 1967, 3, 685.
Б р ы т о в И. А., Р у м ш М. А., П а р о б ец А. С. Рентгеноспектральное ис-
следование моноокиси титана в области гомогенности и нитрида титана.— Физ.
тв. тела, 1968, 10, 794.
Борискина Н. Г., Корнилов И. И. К вопросу о строении систем
Ti — Fe и Ti — Сг — Fe.— В кн.: Новые исследования титановых сплавов.
«Наука», М., 1965, 61.
Будберг П. Б., Шахова К. И., Алисова С. П. Исследование систе-
мы TiCr2 — ZrCr2.— В кн.: Новые исследования титановых сплавов. «Наука»,
М., 1965, 37.
Будберг П. Б., Шахова К. И. Диаграмма состояния системы титан — тан-
тал.— Изв. АН СССР. Неорган. материалы, 1967, 3, 656.
Богомолов В. Н., С о ч а в а Л. С. ЭП? в частично восстановленном ру-
тиле.— Физ. тв. тела, 1967, 9, 3355.
Вайнштейн Э. Е., Чирков В. И., Блохин С. М. Рентгеноспект-
ральное исследование химической связи в окислах титана.— Изв. Сибирского
отд. АН СССР. Сер. хим., 1967, 11, 12.
Ворошилов Ю. В. Горшкова Л. В., Попова Н. М., Федо-
ров Т. Ф. Тройные системы Ti — Zr — С и Ti — Hf — С.— Порошковая
металлургия, 1967, 5, 81.
* е л ь д П. В., Швейкин Г. П., А л я м о в с к и й С. И., Ц а а й В. А.
Электронная структура и взаимная растворимость моноокисей титана, ванадия
г и ниобия.— Ж. неорг. хим., 1967,12, 200.
*лазоваВ. В., Кен и на Е. М. Исследование взаимодействия в систе-
ме Ti3O — Ti3Sn.— Докл. АН СССР, 1968,180, 106.
7*
99
Глазова В. В., Корнилов И. И. Взаимодействие между субоксидами
титана и циркония.— Ж. неорг. хим., 1967, 12, 3159.
Глазова В. В. Легирование титана. «Наука», М., 1966.
Горяйнов В. М., Юдин Б. Ф., Ав густи ни к А. И. Исследование
высокотемпературного взаимодействия в системе TiC — ZrO2.— Огнеупоры,
1966, 10, 50.
Г опиенко Вал. Г., Г о п и е н к о В. Г. Взаимодействие хлора с титаном.
Ж. прикл. хим., 1968, 41, 940.
Гузей Л. С., Соколовская Е. М., Григорьева А. Т. К вопро-
су о диаграмме состояния ниобий—титан.— Вести. МГУ. Химия, 1966,5,
79.
Грушина В. В., Родин А. М. Взаимодействие водорода со скандием и
сплавами Ti — Sc.— Ж- физ. хим., 1968, 42, 466.
Голубцова Р. Б. Исследование интерметаллидной фазы системы
Ti — Al — Fe — Si — В — Сг.— Зав. лабор., 1965, 31, 534.
Дубровская Г. Н. Исследование взаимодействия порошка титана с серово-
дородом.— Порошковая металлургия, 1965, 9, 6.
Дубровская Г. Н. Термическая устойчивость сульфидов переходных ме-
таллов IV группы.— Укр. хим. ж., 1967, 33, 997.
Дудкина Л. П., Корнилов И. И. Диаграмма равновесия системы
TiNi — TiFe. —Изв. АН СССР. Металлы, 1967, 4, 184.
Дьячков В. И., Тихомиров В. И. Влияние давления кислорода на
кинетику высокотемпературного окисления титана.— Вести. ЛГУ, 1968, 4,
158.
Еременко В. Н., Листовничий В. Е. Диаграмма состояния систе-
мы Ti — Р.— В кн.: Новые исследования титановых сплавов. «Наука».
М., 1965, 20.
Еременко В. Н., Штепа Т. Д., Г р и ц е н к о Э. Е. О промежуточных
фазах в сплавах титана с иридием и родием.— В кн.: Новые исследования ти-
тановых сплавов. «Наука», М., 1965, 25.
Еременко В. Н., Буянов Ю. И., Прима С. Б. Строение диаграммы
состояния системы Ti — Си.— Порошковая металлургия, 1966, 6, 77.
Еременко В. Н., Ш т е п а Т. Д., Сиротенко В. Г. О промежуточных
фазах в сплавах титана с иридием, родием и осмием.— Порошковая метал-
лургия, 1966, 6, 68.
Еременко В. Н., Листовничий В .Е. Способ синтеза халькоге-
нидов титана.— В кн.: Халькогениды. «Наукова думка», К- 1967, 66.
Еременко В. Н., Листовничий В.Е. Структура и свойства сплавов
титана с серой.— В кн.: Халькогениды. «Наукова думка», К. 1967, 69.
Жураковский Е. О. Об электронном строении фосфидов титана. —
ДАН УССР, 1967, А, 1127.
Жураковский Е.А. Рентгеновские К-спектры поглощения титана, вана-
дия и германия в германидах.— Порошковая металлургия, 1966, 8, 70.
Захаров А. М., Савицкий Е. М. Исследование диаграммы состояния
тройной системы W — Zr — Ti.— Изв. АН СССР. Металлы, 1966, 5, 159.
Захаров А. М., Савицкий Е. М. Исследование диаграммы состояния
системы Мо — Ti — С.— Изв. АН СССР. Металлы, 1968, 1, 162.
Ивановский Г. Ф., Ширяев А. Т. Сорбция конденсированной тонкой
пленкой титана водорода при низких давлениях.— Ж. физ. хим., 1965, 39,
2464.
Крылов Б. С. Растворимость водорода в титане.— Изв. АН СССР. Металлы,
1966, 2, 144.
Корнилов И. И., Нартова Т. Т. Фазовое равновесие и свойства сплавов
квазибинарной системы Ti3Al — Ti3Sn.— В кн.: Новые исследования титано-
вых сплавов. «Наука», М., 1965, 30.
Корнилов И. И., Волкова М. А., Пылаева Е. Н., Крипяке-
в и ч П. И., М а р к и в В. Я. Исследования диаграммы равновесия богатых
титановых сплавов системы Ti —Al.— В кн.: Новые исследования титано-
вых сплавов. «Наука», М., 1965, 48.
100
Корнилов И. И., Матвеева Н. М. Термохимическое исследование
сплавов системы Ti — Al в области сс-твердого раствора.— В кн.: Новые ис-
следования титановых сплавов. «Наука», М., 1965, 56.
Корнилов И. И., Глазова В. В. О термической устойчивости соедине-
ний в системе Ti — О.— Ж- неэрг. хим., 1966, 10, 1660.
Корнилов И. И., Глазова В. В. Квазибинарная система Ti — А12О3.
— В кн.: Новые исследования титановых сплавов. «Наука», М., 1965, 3.
Корнилов И. И. Состояние и перспективы исследований в области
металлохимии титана.— В кн.: Титановые сплавы для новой техники.
«Наука», М., 1968, 24.
Корнилов И. И., Дудкина Л. П. Пластичность TiNi.— Цветная ме-
таллургия. Бюлл. МЦМСССР, М., 1967, 11, 38.
Корнилов И. И., Волкова М. И. Фазовые равновесия в системе
Ti _ Al —V.— Изв. АН СССР. Металлы, 1968, 1, 176.
Корнилов И. И., Михеева В. С., Маркович К. П. Исследование
фазового равновесия части системы Ti — Al — Сг — Fe — Si — В.— Изв.
АН СССР. Металлы, 1967, 3,211.
Котляр Е. Е., Назарчук Т. Н. О некоторых химических свойствах
карбидов переходных металлов [V—V групп периодической системы.— Изв.
АН СССР. Неорган. материалы, 1966, 2, 1778.
К а л и н ю к Н. Н. Определение растворимости водорода з жидких металлах
(Ni и Ti) методом безтигельной плавки.— В кн.: Методы определения и ис-
следования газов в металлах. «Наука», М., 1968, 238.
Колачев Б. А. О природе гидридов переходных металлов.— Ж. физ. хим.,
1967, 41, 670.
Колобова К. М., Немнонсв С. А. Перераспределение З^-электронов
в эквиатомных сплавах Ti —Fe.— Физ. металлов и металловедение, 1968, 25,
267.
Лакомский В. И., Кал инк к Н. Н. Растворимость водорода в жидком
титане при 2103—2580° К.— Изв АН СССР. Металлы, 1966, 2, 149.
Минц Р. С., Беляева Т. Ф , Малков Ю. С. Изучение системы
Ni3Al — NiTi.— Изв. АН СССР. Металлы, 1967, 2, 172.
М а р к и в В. Я., Г л а д ыш е в с к и й Е. И., Федорук Т. И. Фазовые
равновесия в системе Ti — Со — Si.— Изв. АН СССР. Металлы, 1966, 3, 179.
М а р к и в В. Я., Г л а дышевский Е. И., Крипякевич П. И.,
Федорук Т. И. Система Ti — Ni — Si.— Изв. АН СССР. Неорган. ма-
териалы, 1966, 2, 1317.
М а р к и в В. Я., Лысенко Л. А., Гладышевский Е. И. Система
Ti — Fe — Si.— Изв. АН СССР. Неорг. материалы, 1966, 2, 1980.
Маркив В.Я-, Крипякевич П. И. Тройные металлические соединения
титана с медью и галлием.— Кристаллография, 1966, 11, 859.
Маркив В. Я., Г л а д ы щ е в с х и й Е. И., С к о л о з д р а Р. В., К р и’-
пякевич П. И. Тройные соединения титана.— ДАН УССР, 1967, 3, 266.
Михеев В. С., Чернова Т. С. Исследование части системы Ti — Al —
— Сг — Fe — Si — В.— Изв. АН СССР. Металлы, 1967, 2, 206.
Морозова М. П., Хернбург М. М. Энтальпия образования нитритов
титана.— Ж- физ. хим., 40, 1966, 1125.
Нартова Т. Т., Андреев О Н. Исследование некоторых физических
свойств и жаропрочности непрерывных твердых растворов Ti — Zr и Ti — Hf.
Изв. АН СССР. Металлы, 1967, 6,180.
Немнонов С. А., Колобова К- М. Рентгеновские спектры, электронная
структура и свойства металлических соединений титана.— Физ. металлов и
металловедение, 1966, 22, 680.
Нешпор В. С., Климашин Г. М. Теплопроводность карбида титана в
зависимости от содержания углерода в области гомогенности.—Изв. АН СССР.
Неорг. материалы. 1965, 1, 1545.
Ор д а н ь я н С. С., Круглова Г. В., Ав густи н и к А. И. Ди-
аграмма состояния системы Ti — С — Re.— Изв. АН СССР. Неорг. мате*
риалы, 1968, 4, 1086.
101
Оганесян В. X. Физические свойства сульфидов титана.— В кн.: Халько-
гениды. «Наукова думка», К., 1967, 160.
Петрусевич И. В., Нисельсон Л. А., Беляев А. И. О получении
силицидов титана совместным восстановлением тетрахлоридов титана и крем-
ния водородом.— Изв. АН СССР. Металлы, 1965, 6, 52.
П е ш е в П. Получение кристаллического диборида титана из газовой фазы.—
Изв. Института общ. и неорг. хим., 1966, 4, 53.
Переляев В. А., Швейкин Г. П., Алямовский С. И. О взаимной
растворимости Ti2O3 и V3O3. — Изв. АН СССР. Неорг. материалы, 1968, 4,
1372.
Пауков Е. П., Хриплович Л. М., Филаткина В. С. Теплоем-
кость диборида титана при низких температурах.— Ж- физ. хим., 1967, 41,
1621.
Родякин В. В., Выхорев А. Ф., Ткалич В. С., Понома-
рев В. Д. Растворимость титана в жидком магнии.— Изв. высш. уч. зав.
Цветная металлургия, 1967, 6, 55.
Самсонов Г. В., Синельникова В. С., Копылова В.П. Алю-
мотермическое восстановление двуокиси титана.— Ж- прикл. хим., 1966, 39,
729.
Самсонов Г. В. Система Ti — Be.— Усп. хим., 1966, 35, 779.
Самсонов Г. В., Антонова М. М. Гидридные фазы титана.— Укр.
хим. ж., 1966, 32, 555.
Самсонов Г. В., Оганесян В. X. Исследование природы химической
связи полупроводниковых сульфидных фаз.— Изв. АН СССР. Неорган. ма-
териалы, 1966, 2, 1757.
Самсонова Н. Н., Будберг П. Б. Влияние ванадия и молибдена на
свойства и фазовые превращения металлида TiCr2.—Порошковая металлур-
гия, 1966, 8, 49.
Самсонова Н. Н., Будберг П. Б., Корнилов И. И., Аса-
но в У. А. Взаимодействие металлида TiCr2 с молибденом.— Изв. АН СССР.
Неорг. материалы, 1966, 2, 1878.
Самсонова Н. Н., Будберг П. Б., Корнилов И. И.. Аса-
но в У. А. Взаимодействие металлида TiCr2 с ванадием.— Изв. АН СССР.
Неорг. материалы, 1966, 2, 1882.
Старостина Т. С., Сидоров Л. Н., Акишин П. А., Кара-
сев Н. М. Масс-спектрометрическое исследование состава пара над система-
ми TiC — Си ZrC — С.— Изв. АН СССР. Неорг. материалы, 1967, 3, 727.
Страшинская Л. В., Миронова Н. В. Контактное взаимодействие
нитрида титана с титаном, цирконием и ванадием в вакууме.— Изв. АН СССР.
Металлы, 1966, 4, 143.
Третьяченко Л. А., Еременко В.Н. Строение тройной системы
Ti — у — С.— Изв. АН СССР. Неорг. материалы, 1967, 3, 2009.
Федоров Т. Ф„ К у з ь м а Ю. Б. Система титан — железо — бор.— Изв.
АН СССР. Неорг. материалы, 1967, 3, 1498.
Чирков В. И., Вайнштейн Э. Е. О некоторых результатах рентгено-
графического исследования высших окислов титана (TiOj 75 _ 2 00).— Изв.
АН СССР. Неорг. материалы, 1967, 3, 1022.
Чирков В. И., Вайнштейн Э. Е., Васильев Я-В. Исследование
рентгеновских эмиссионных спектров титана в окислах Ti2O3 и Ti3O5.— Изв.
АН СССР. Неорг. материалы, 1967, 3, 1017.
Чирков В. И., Вайнштейн Э. Е. Природа химической связи в окислах
титана переменного состава ТЮ0>85_ 1>2о-“ ДАН СССР, 1965, 165, 1354.
Alcock С. В., Zador S. Термодинамические исследования разбавленных
растворов дефектов в структуре рутила.— Proc. Brit. Ceram. Soc., 1967, 8,
231.
Adelberg L. M., Cadoff L. H., Tobin J.M. Температура эвтектик,
образованных углеродом и карбидами металлов IV и V групп.— J. Am. Сегат.
Soc., 1966, 49, 573.
102
gerodias G., Chevreton M. Структурное исследование тройных селени-
дов.— Compt. rend., 1965, 261, 2202.
poller H., N о w о t n у H. Кристаллическая структура Ti5AS3.— Monatsh.
Chem., 1965, 96, 565.
g lumen t h a 1 R. N., Baukus J., Hirthe W. M. Исследование де-
фектной структуры нестехиометрического рутила ТЮ2_Л.— J. Electrochem.
Soc., 1967, 114, 1, 172.
gr un ie S., Chevreton M. Структурное изучение нового селенида
титана Ti9Se4.— Compt. rend., 1967, C264, 449.
gukowiecki J. Механизм синтеза карбида титана с участием углеводоро-
дов.— Prace Inst, hutn., 1966, 18, 35.
Roller Н., Nowotny N. Кристаллохимическое исследование монофосфи-
дов и моноарсенидов в системах Ti — (Cr, MoW) — (Р, As).— Monatsh. Chem.,
1965, 96, 852.
Bernsset P. Изучение системы титан—селен.— Rev. chim. miner., 1966,
3, 135.
Beaudouin J. Приготовление дисилицида титана путем электролиза при
высокой температуре.— Compt. rend., 1966, С263, 993.
Beaudry В.J. Влияние титана на температуру плавления и превращения ит-
трия.— J. Less-Common Metals, 1968, 14, 370.
Bruckner W., KI ei ns t ii k K.,Schul ze G. E. Расположение атомов
в гомогенной области фаз Лавеса ZrFe2 и TiFe2.— Phys. Status Solidi, 1967,
23, 475.
Bruckner W. Нейтронографическое исследование TiFe.—Krista’l und Technik,
1967, 2, K5—K7.
Carlson K. D., Claydon Ch. P., Moser С. Электронное строение и
свойства основного состояния мононитрида титана.— J. Chem. Phys., 1967,
46, 4963.
Chang L. L. Y., Scroger M. G., P h i 1 l]i p s В. Фазовые равновесия
в системе Ti — W — О.— J. Less-Common Metals, 1967, 12, 51.
Conroy E. L., Park К- С. Электрические свойства TiS2.— Inorg. Chem.,
1968, 7, 459.
Gherman t J.-L., Deschanveres А. Наблюдение полос выделения ти-
тана в карбиде титана нестехиометэического состава.— Compt. rend., 1967,
С265, 382.
Dou Йосихико. Получение диборида титана.— Нихон киндзоку гаккайси,
1966, 30, 106.
Denker S. Р. Электронные свойства моноокиси титана.— J. Appl. Phys., 1966,
37, 142.
Dubertet A., Lehr Р. Изучение твердости сплавов титан — кислород.—
Compt. rend., 1966, С263, 591.
Freundlich W., Nematolah F. M. Две тройные интерметаллические
фазы Zr Fe Si и Ti Fe Si. — Compt. rend., 1966, C262, 1000.
Frisch C. R., Forman A. R. Ядерный магнитный резонанс Ti в сплавах
Ti — Н.— J. Chem. Phys., 1968, 48, 5187.
F 1 i n k E., W i e g e г s G. A., J e 1 1 i n e k F. Система титан — сера.— Rec.
trav. chim., 1966, 85, 869.
Giorgi T. A., Ricca F. Термодинамические свойства водорода и дейтерия
в а-титане. Niuovo cimento Suppl., 1967, 5, 472.
Grievenson P. Исследование системы Ti — С — N.— Proc. Brit. Ceram.
Soc., 167, 8, 137.
Gerrish M. E., Stobbs J.J. Окисление уран-титановых сплавов в двуо-
киси углерода при 450° С.— J. Inst. Metals, 1967, 95, 284.
Ganglberger Е., Nowotny FL, Benesovsky F. Соединение
. Ti6Cu8Be]5.— Monatsh., 1965, 96, 12C6.
Gilles J. С. Сплавы TiO2 — TiN. — Rev. Hautes temperat. et refract., 1965,
2, 237.
103
Greenaway D. L., Nitsche R. Приготовление и оптические свойства
халькогенидов IV—VI групп со структурой Cdl2.— J. Phys. Chem. Solids,
1965, 26, 1445.
H a v a 1 d а А. Исследование равновесия в системе Ni — Сг — Ti — Al — W.
— Kovove mater., 1967, 5, 3.
Hauptman Z., Schmidt D. Banerjee S. К. Транспортная система
Ti2O3/HCl.— Collect. Czechosl. Chem. Communs, 1967, 32, 2421.
Holliday J. E. О природе химической связи в TiC и TiC0,8.— J. Appl. Phys.,
1967, 38, 4720.
Н i 1 t i E. Новые фазы в системе титан — кислород.— Naturwis., 1968, 55, 130.
Higashi Iwami, Atoda Tetsuzo. Параметры решетки диборита ти-
тана.— Sci. Papers Inst. Phys, and Chem. Res., 1966, 60, 32.
Huber J. E. Теплота образования диборида титана.— J. Chem. Eng. Data,
1966, 11, 430.
H о 1 1 о x G. E., S m a 1 1 m a n R. E. Продукты окисления диборида титана.—
Philos. Mag. 1966, 13, 1.
H i 1 t i E., Laves F. Рентгеноструктурное исследование TiO.— Naturwis.,
1968, 55, 131.
Jacquin Y., Huber M.— Изучение фазовой диаграммы сера — селен —
титан. — Compt. rend., 1966, С262, 1059.
Jones Р. M.S., Е 1 1 i s Р., Smith J. R., A s 1 e t t T., Hutche-
son C. G. Рентгенографическое исследование гидрида титана.— Atomic
' Weapons Res. Estebl. U. K. Energy Author., 1966, 60/66.
Jeitschko W., Nowotny H. Кристаллическая структура Ti3SiC2.—
Monatsh., 1967, 98. 329.
J о u b e r t C.-J. Соединения титана с вакансиями.— Bull. Soc. fcanc. mineral,
et cristallogr., 1967, 90, 598.
Киносита M., Хамано Й. Синтез диборида титана методом восстанов-
ления углеродом.— J. Ceram. Assoc. Japan, 1966, 74, 244.
Keys L. К., M u 1 a у L. К. Изучение магнитных свойств фаз. Магнели систе-
мы титан — кислород.— J. Appl. Phys., 1967, 38, 1466.
Keys L. К. Магнитные свойства Ti3O5.— Phys. Letters, 1967, A24, 628.
Lun ds tr dm T., Snell P. О. Исследование кристаллических структур и
фазовых превращений в системе титан — фосфор.— Acta Chem. Scand., 1967,
21, 1343.
Lynch С. T., М е г s о 1 S. А., V a h 1 d i с k F. W. Микроструктура TiB2.—
J. Less-Common Metals, 1966, 10, 206.
Meissner H.-G., Schubert К- Структура фазы Ti6Sn5.—Z . Metallkun-
de, 1965, 56, 475.
M e u 1 1 e r M. H., Knott H. W. Кристаллические структуры Ti2Cu, Ti2Ni,
Ti4Ni2O, Ti4Cu2O.— Trans. Metallurg. Soc. AIME, 1963, 227.
M i 1 e t С. Исследование сплавов системы уран — плутоний — титан — угле-
род.— Mem. Sci. Rev. Metallurgie, 1967, 64, 261.
Nickl J., Sprenger H. О монокристаллах тройного соединения TiCuSi.—
Naturwis., 1967, 54, 181.
Nickl J., Sprenger H. О монокристаллах тройного соединения TiCuGe.—
Naturwis., 1967, 54, 515.
Owens J. P., Conard B. R., Franzen H. E. Кристаллическая структу-
ра Ti2S.— Acta crystallogr., 23, 77, 1967.
О k а А. Синтез TiHx.— Bull Soc. Japan, 1967, 40, 2284.
Pfeifer H. U., Bhan S., Schubert К. О строении системы Ti—Ni—Cu
и некоторых квазигомологических сплавах.— J. Less-Common Metals,—
1968, 14, 291.
Pearce M. L., Marek R. W. Образование карбидов кремния и титана мето-
дом химического осаждения из пара.— J- Am. Ceram. Soc., 1968, 51, 84.
Р I о v n i с k R. И., V 1 a s s e M., Wold А. Синтез и структурные свойства
некоторых тройных халькогенидов титана.— Inorg. Chem., 1968, 7, 127.
Peshev Р., Bliznakov G. Боротермическое приготовление диборидов
титана.— J. Less-Common Metals, 1968, 14, 23.
104
peshev Р. Получение карбида титана осаждением из газовой фазы. — Док л.
Болг. АН, 1966, 19, 141.
peshev Р., Niemyski Т. Получение кристаллического диборида титана
по реакции в газовой фазе.— J. Less-Common Metals, 1966, 10, 133.
pundqvist S., Tansuriwong S. P. Тройные арсениды титана с ко-
бальтом и никелем.— Acta Chem. Scar.d.* 1967, 21, 813.
paub E., Raub C. J., Roschel E. Compton V. B., rG e b a 1 -
1 e T. H., M a t t h i a s В. T. Твердые растворы Fe в a-Ti.— J. Less—Com-
mon Metals, 1967, 12, 36.
penter B., Weber R. Система TiO — LiTiO2.— Naturwis., 1966, 53, 251.
peid A.F., Sienko M. J. Некоторые характеристики титано-натриевых бронз.—
Inorg. Chem., 1967, 6, 321.
Raman A., Schubert К- Исследование систем Ti — Ni — Al и
Ti — Cu,— AL— Z. Metallkunde, 196E, 56, 99.
possteutscher W., Schubert К. Структурные исследования в неко-
торых системах Ti4-”5 — В4*”5.— Z. Metallkunde, 1965, 56, 813.
possteutscher W-, Schubert К- О некоторых сплавах Ti — Zn и
Ti — Cd — Z. Metallkunde. 1965 , 56, 730.
Staub F., Josz К. Исследование системы Cu — Ni3Ti. Zesz. nauk Politechn.
slaskiej, 1967, 189.
Stewart R.W., Cutler LB. Влияние температуры и парциального дав-
ления кислорода на окисление карбида титана.— J. Am. Ceram. Soc., 1967,
50, 176.
Stadelmaier Н. Н., S с h б b е 1 J.-D., Jones R. А. Фаза т в тройной
системе Со — Ti — В.—Metalls, 1967, 21, 171.
Suzuki A., Wahlbeck Р. G. Исследование парообразования системы ти-
тан—теллур.— J. Phys. Chem., 1966, 70, 1914.
Sprenger Н., Nickl J. Новый силицид титана Ti5SL.— Naturwis., 1967,
54, 645.
Snell P.-О. Кристаллическая структура TiP.— Acta Chem. Scand., 1967, 21,
1773.
Такэути С., Сундзуки К- Дефектная структура нестехиометрических
фаз TiO и VO.— J. Japan inst. Metals, 1967, 31, 611.
Toesca S., Colson J.-C., Barret P. Изучение кинетики сульфирова-
ния титановой пыли сероводородом.— Compt. rend., 1967, С266, 266.
Watanable D., C a s t 1 e r J. R., Jos t sons A., Malin A. S. Упо-
рядоченная структура окиси титана.— Nature, 1966, 210, 5039, 934.
Wahlbeck P. G., Gilles P. W. Уточнение фазового состава системы
титан — кислород.— J. Am. Ceram. Soc., 1966, 49, 180.
Watanable D., Castle г, Jostsons A., Malin A. S. Упорядо-
ченная структура TiO.— Acta crystallogr., 1967, 23, 307.
Wolfgruber H., Nowotny H., Benesovsky F. Кристаллическая
структура TigGeCg.— Monatsh., 1967, 98, 2403.
Wahlbeck P. G. Gilles P. W. Энергия диссоциации TiOre3 и высокотем-
пературное испарение и термодинамика окислов титана.— J. Chem. Phys.,
1967, 46, 2465.
W a n g F. Е., Ernst D. W. Фазовый переход в TiNi.— J. Appl. Phys., 1968,
v 39, 2192.
v a h 1 d i e k F. W. Микроструктура монокристаллов карбида титана. — J. Less-
Common Metals, 1967, 12, 429.
a h 1 d i e k F. W. Свойства диборида титана.— J. Less-Common Metals, 1967,
v 12, 202.
v 0 n K., Nowotny H., Kieffer R. О кристаллической структуре кар-
богидридов переходных металлов.— Monatsh., 1968, 98, 2164.
ГЛАВА III
ГИДРООКИСИ ТИТАНА
Как класс химических соединений гидроокиси — это продукты
взаимодействия металлов с гидроксильными группами. Титан, бу-
дучи переходным металлом, образует гидроокиси в различном ва-
лентном состоянии. Известны гидроокиси титана с валентностью
2, 3 и 4. Одновалентный титан устойчивой гидроокиси не образует.
Металлическая связь титана с кислородом и водородом в этом слу-
чае энергетически более выгодна, чем ковалентная связь с гидро-
ксогруппой. i
Из растворов солей титана гидроокиси Ti-З и Ti-4 осаждаются
основаниями при pH 4 и 1,5 соответственно в виде хлопьевидных
аморфных малорастворимых в воде продуктов [13]. Однако титан,
как, впрочем, и другие многовалентные металлы, не может прочно
удерживать гидроксогруппы. Его гидроокиси после образования
претерпевают различные изменения и имеют поэтому сложное строе-
ние. Индивидуальные свойства их маскируются протеканием про-
цессов полимеризации, гидратации, дегидратации, адсорбции и
образования водородных связей.
Гидроокиси Ti-2 и Ti-З в присутствии воды медленно разлагают-
ся с выделением водорода [56, 140] и легко окисляются кислородом
воздуха. В соответствии с общими закономерностями основность
гидроокисей титана повышается с уменьшением валентности.
роокиси дву- и трехвалентного титана — слабые основания. Гидро-
окиси титана-4 обладают амфотерными свойствами,
они
способны
образовывать соли с кислотами и основаниями.
ГИДРООКИСИ ТИТАНА НИЗШЕМ ВАЛЕНТНОСТИ
Велер [1401 и Пфордтен [НО] получили гидроокись двухвалент-
ного титана Ti (ОН)2 осаждением из раствора TiCl2 аммиаком, гид-
роокисями щелочных металлов и сернистым аммонием. По их описа-
ниям, Ti (ОН)2—хлопьевидный осадок темно-черного цвета,
который растворяется в разбавленной соляной кислоте с окрашива-
нием раствора в желтый цвет. Растворы гидроокиси титана-2 в
концентрированной соляной кислоте коричневые [НО]. Кислород
воздуха окисляет Ti (ОН)2 до Ti (ОН)3. Цвет при этом изменяется
106
через синий до белого. При хранении Ti (ОН)2 разлагается с выде-
лением водорода [140].
фукс [57] и Эбельмен [56] осаждением из раствора солей Ti-З
аммиаком и гидроокисями щелочных металлов получили гидро-
окись трехвалентного титана Ti (ОН)3. Осадок гидроокиси темно-
коричневого цвета, при хранении разлагается с выделением водо-
рода. Окраска его при этом становится черной, затем голубой и,
наконец, белой. Гидроокись титана-3 описана в работах [61, 115].
Гидроокись титана-3 образуется также при гидролизе водных
растворов солей титана, например TiCl3, при нагревании [66]. Ке-
ниг и Пфордтен [86] получили гидроокись титана из титаната
j\ja3TiO3.
Пексок и Флетчер [116] установили, что гидролиз солей трехва-
лентного титана при pH 3—-4 ограничивается образованием моно-
мерных ионов Ti (ОН)2”- Полимерные ионы в этом интервале pH
не образуются. В более щелочных растворах, с pH > 4, образуются
двуядерные гидроксокомплексы по реакции
Ti (ОН)2+ -> TiOTi (ОН)3+ + Н+.
Раствор гидроокиси титана при этом окрашивается в черный цвет
(^шах — 700 нм). Двуядерные комплексы не дают типичной для ио-
нов Тг3 реакции с винной кислотой.
При быстром добавлении щелочи к водному раствору солей ти-
тана-3 получается мономерная гидроокись титана Ti (ОН)3.
В растворах с pH > 5 ионы Ti3+ быстро окисляются протонами.
Гидроокись титана-3 относится к числу сильных восстановителей.
Она восстанавливает нитраты до аммиака, азобензол до гидразобен-
зола и пр.
ГИДРООКИСЬ ТИТАНА-4 *
В ранних работах [50, 53, 85, 88, 112, 119, 121, 124] гидроокиси
титана подразделялись на ортотитановую и метатитановую кислоты.
Гидроокись титана, полученная осаждением на холоду из водных
растворов солей титана действием щелочей,” представляет собой
аморфный хлопьевидный осадок, растворимый без остатка в раз-
бавленных минеральных кислотах [112, 118, 120]. В продуктах,
промытых водой и высушенных при комнатной температуре, содер-
жится боле^дауад^одей 6°^ J» I г-жа титана. Фрике [58] предло-
жил называть эти продукты ортотитановой или а (а) -титановой
Кислотой.
Гидроокись титана, полученная гидролизом растворимых солей и
осаждением основаниями при нагревании, представляет собой бе-
лый кристаллоподобный осадок", нерастворимый в разбавленной
серной, азотной и соляной кислотах [88, 121, 124]. В ней содержится
после промывания водой и сушки при комнатной температуре около
107
I ®
Хяпля воды титана, Фонке Г581 назвал эту гидроокись ме^
татитановой илир (Ъ)-титановой кислотой. 11
Бертон [43] установил, что орто- и метатитановая кислоты имеют
различные полосы поглощения в ультрафиолетовой области света^
Граница полосы поглощения ортотитановой кислоты, полученной
из водных растворов TiCl4 действием щелочей, лежит при длине
волны 350 нм, а у метатитановой кислоты, полученной из водных 2
растворов TiCl4, спектр поглощения такой же, как у рутила. , 1
В патентах [23] предложено различать еще одну разновидность 1
1 гидроокиси титана — у-титановую кислоту, которую получают из. |
водных растворов солей титана осаждением щелочами и гидролизом- |
Она превращается в рутил при нагревании до температуры ниже
800° С. Я
Термины орто- и метатитановая кислота широко распространены я
в литературе, особенно довоенной, хотя и нет строго определенных!
признаков, по которым их можно было бы различать. В настоящей’!
работе мы не будем проводить различия между отдельными видами!
гидроокиси титана вследствие неопределенности их как химических!
индивидуумов. Гидроокись титана будет рассматриваться как одна, а
фаза, состав и свойства которой изменяются в широких пределах. <
Я
ПОЛУЧЕНИЕ ГИДРООКИСИ ТИТАНА Л
Гидроокись титана может быть получена тремя различными ме-1
тодами: осаждением основаниями из растворимых солей титана,!
разложением титанатов щелочных металлов разбавленными кисло-Я
тами и гидролизом солей титана и высокощелочных титанатов. Я
Получение гидроокиси титана осаждением основаниями из растворов солей tnu - j
тана. Осаждение гидроокиси титана основаниями можно проводить из растворов^
сульфатов, хлоридов, нитратов, хлоратов, фторидов и некоторых других раствори- 1
мых солей. Величина pH, при которой начинается образование нерастворимого 1
осадка гидроокиси титана, зависит от концентрации раствора и природы содержа-1
щихся в растворе анионов. Комплексообразующие ионы, например F~, повышают!
pH осаждения гидроокиси титана. В присутствии некоторых комплексообразую!
щих агентов, например винной кислоты, гидроокись титана из водных растворов!
слабыми основаниями вообще не осаждается.
Из сернокислых растворов гидроокись титана начинает осаждаться при pH]
1—1,5 [13, 20]. Окончание осаждения наступает при рН^^ДО (рис. 25).
Из разбавленных фторидных растворов титана едкие щелочи (едкий натри
вначале осаждают основные соли, которые устойчивы до pH 5^2—7,0 [14]. При бо4|
лее высоких значениях pH они разлагаются с образованием гидроокиси титана!
В качестве оснований — осадителей гидроокиси титана — используются ед!
кие натр и кали [11, 96], водные растворы аммиака [1, 85, 96, 128, 137, 146], раст-?|
воры поташа и соды [96]. Природа осадителя, по-видимому, оказывает существен-1
ное влияние на состав и свойства образующейся гидроокиси титана. Например, pH I
раствора, находящегося в контакте с осадком, из которого осаждена гидроокись !
титана едким натром, не изменяется со временем. Если же гидроокись титана oca-d
ждена аммиаком, то pH раствора со временем понижается (рис. 25). Вероятно, при!
осаждении гидроокиси титана аммиаком наряду с гидроокисью титана образуется |
небольшое количество нестойких основных солей, разлагающихся при стоянии.
108
I
Получение гидроокиси титана разложением титанатов кислотами. При дей-
ствии на титанаты некоторых щелочных металлов разбавленными кислотами они
разлагаются с образованием гидроокиси титана. Этим методом можно получать
гидроокись титана только из высокощелочных титанатов калия и натрия, например
К TiO3 [42, 107], Na2TiO3 [95]. Он нашел практическое применение для получе-
й^'ТИДроикиСИТИТйИ^,” использующейся в ус-
оряющей превращение анатаза в рутил в производстве титановых пигментов.
Получение гидроокиси титана методом гидролиза. Водные растворы многих
,,оЛей титана неустойчивы. При значительном разбавлении или нагревании они
Рис. 25. График потенциометрического титрования
сульфата титана едким натром и аммиаком: О —
осаждение NaOH, — осаждение NH4OH.
подвергаются гидролизу с образованием гидроокиси титана Сернокислые растворы
титанилсульфата гидролизуются в широкой области концентраций. Как следует из
диаграммы состояния системы ГЮ2 — SO3 — Н2О, область существования гидро-
окиси титана простирается от 0 до 40% H2SO4 [8]. Область существования гидро-
окиси титана в системе TiO2 — ЙС1 — HQC) при температуре 0° С простирается от
05о 36>74% НС1 [28]. Система ТЮ2 — HNO3 — Н2О методом растворимости еще не
изучена. Судя по неустойчивости нитрата титана, в этой системе также имеется об-
ширное поле гидроокиси титана.
Легко гидролизуются многие титанорганические соединения, например тетра-
эфиры Ti (OR)4, где R = —С2Н-3, —С3Н7, —С4Н9 и др. [70, 72, 87].
- Фториды титана относятся к числу более стойких соединений. В водных раст-'^
иорах оннГне гидролизуются в заметной степени даже при большом разбавлении и
Длительном кипячении.
Гидролизу в водных растворах с образованием гидроокиси титана подвергают-
ся титанаты щелочных металлов. Под действием горячей воды гидролизуются с об-
разованием гидроокиси титана гетранатриевый титанат Na1TiO4 [104], динатрие-
вый титанат Na2TiO3 [33, 69, 118], восьминатриевый пентатитанат Na8Ti5O14. По-
видимому, гидролизуются и вызокощелочные титанаты калия, рубидия и цезия,
110 свойства этих соединений изучены еще недостаточно.
Гидролиз растворов титанилсульфата нашел применение для получения гид- ,
Р°окиси титана в крупных промышленных масштабах. Растворы титанилсульфата
в в°де и разбавленной серной кислоте, хотя и находятся в метастабильном состоя-
ть гидролизуются обычно с малой скоростью. Исследование кинетики гидроли-
а титанилсульфата показывает, что скорость гидролиза зависит от многих факто-
Р°в» главными из которцх являются концентрация в растворе двуокиси титана,!
тнощение H2SO4 : TiO2, температура [6, 25, 32, 46, 73, 99, 111, 149]. Особен-/
109
но существенно влияют на скорость гидролиза, состав гидроокиси титана ц
ее физические свойства добавки так называемых зародышей. Зародыши добавляю,
тся в раствор сульфата титана перед гидролизом для его ускорения и получения
гидроркиси с определенными физико-техническими свойствами. Методы приготов.
ления зародышей описаны во многих патентах и руководствах по технологии про.
изводства титановых пигментов [35]. Физико-химическая природа зародышей рас-
смотрена ниже.
О СОСТАВЕ ГИДРООКИСИ ТИТАНА
Изучению гидроокиси титана посвящено значительное количе-
ство работ, однако наши представления о ее составе еще неполные.
Это объясняется тем, что гидроокись титана — фаза, которая обра-
зуется во многих физико-химических системах, существует в ши-
роких интервалах концентраций и температур и медленно прихо-/
дит в состояние равновесия. Состав гидроокиси титана из-за боль-
шой ее удельной поверхности зависит также от адсорбции ею из,
растворов различных примесей. Полное представление о составе-
гидроокиси титана можно получить только, изучив ее методом фи-
V зико-химического анализа и проследив за изменениями в процессе^
прихождения системы в состояние равновесия. Препаративные ме-
тоды не позволяют определить истинный состав гидроокиси титана
вследствие изменения его при отмывании осадка от маточной\
жидкости. I
В литературе существуют две точки зрения на состав гидроокиси
титана.
Одни исследователи полагают, что гидроокись титана состоит из соединений
определенного состава. Розе][ 121 ] орто- и метатитановые кислоты рассматривал как
соединения, имеющие состав Н4ТЮ4 и Н2ТЮ3. Симон [133] полагал, что гидроокиси/
всех многовалентных металлов при старении превращаются в гидраты определен-?,
ного состава или разлагаются на окислы металлов и воду. Рейндерс и Киз [123],
высушивая гидроокись титана над Р2Об, получили продукт состава TiO2 -1
• 2,13Н2О. Бильтц с соавторами [42], высушивая гидроокись титана в вакууме
прй^те’мпёфатуре 35° С, выделил продукт состава^. Соединение тако-1
го же состава, в котором вода частично замещена на аммиак, получено обезво-
живанием гидроокиси титана в жидком аммиаке при температуре —79° С. Основы-
ваясь на этих данных, авторы допускают существование метатитановой кислоты 1
как соединения определенного состава. По данным [147], гели гидроокиси титана, j
осажденные из растворов основаниями при тащдй^уу^еОу -С, после обезвоживания
vhx при температуре 12° С имеют состав TiO2 • ЗН2О, а полученные осаждением при]
кипячении — TiO2 • 2Н2О. Миронов [20] методом потенциометрического титрова-
ния и растворимости установил, что осаждение гидроокиси титана едким натром /
при комнатной температуре сопровождается связыванием с одним атомом осажден-
ного титана двух гидроксильных групп. На этом основании автор считает, что оса-
док гидроокиси титана имеет состав ТЮ (ОН)2. Ульянов [29] исследовал систему j
"Ti (SO4)2 — H2SO4 — NaOH — H2O при температуре 20 и 25° С методами раствори-
мости и кажущегося объема осадка. Он обнаружил в этой системе образование
гидроокиси титана состава Н2ТЮ3 с некоторым количеством соосажденной серной j
кислоты. Касимов [11] изучил состав осадков гидроокиси титана, полученных из
сернокислых и хлоридных растворов, методом третьего компонента. По его дан-
ным, гидроокись титана имеет состав Н2ТЮ3 и H2Ti2O6. Автор, однако, при опре- I
делении состава твердых фаз пользовался необоснованным графическим методом. I
Другие исследователи считают, что гидроокись титана — соединение перемен-
ного состава. К этому выводу пришли Корнелли и Уольтер [50] в результате ис-
110
к <
спедования гидроокиси титана методом термического анализа и методом дегидрата-
ций Ван-Блеимена.
рискин [4] приводит следующий сосзав гидроокиси титана, полученной гид-
ролизом татанилсульфата, промытой водой и высушенной при комнатной темпе-
ратуре (вес. %):
TiO2 so3 h2d Влага Сумма
81,5 4,4 7,1 6,7 99,7
76,7 7,6 7J6 7,9 99,8
- 72,8 7,3 13J0 6,7 99,8
74,8 7,0 9J6 8,5 99,8
Из данных анализов следует, что так называемая метатитановая кислота является
продуктом переменного состава, содержащим кроме двуокиси титана и воды зна-
чительное количество SO3. Тройной состав метатитановой кислоты установлен так
же в работе Леках [17], изучившей гидролиз титанилсульфата при различной тем-
пературе. Состав образцов гидроокиси титана, полученных Леках, таков (вес. %):
°C TiO2 so3 H2o
100 80,0 8,0 12,0
150 88,7 5,7 5,6
200 93,9 4,0 2,1
300 98,0 1,3 0,7
С повышением температуры содержание SO3 и Н2О в гидроокиси титана зако-
номерно уменьшается. Гидроокись титана, полученная при температуре 300° С,
состоит практически из чистой двуокиси тирана с очень небольшой примесью SO3 и
воды.
О СТРОЕНИИ ГИДРООКИСИ ТИТАНА
Исследование строения гидроокиси титана различными мето-
дами должно дать представление с характере связей титана с гид-
роксо-группами, молекулами воды к другими обособленными груп-
пами атомов, входящими в состав этого продукта (SO4, Cl, F и др.)
Пассарини [109] изучил строение гидрэокиси титана методом инфракрасной '
спектроскопии. Исследуемые образцы гидрсокиси титана, полученные осаждением
из кислого раствора солей титана карбонатом калия, содержали 56 и 81% воды. /
Спектры поглощения их снимали в интервате длин волн 1300—2000 нм. Максиму-
мы в спектрах поглощения приходились на длины волн 1443 и 1930 нм и совпадали
с максимумами спектров поглощения чистой воды. Отсюда автор сделал вывод, что
гидроокись титана состоит из двуокиси, с которой молекулы воды связаны адсорб-
ционными силами, подобными капиллярным.
Недавно изучен состав гидроокиси титана как фазы в системах
TiOa — SO3 — Na2O — Н2О и TiQj — SO3 — Н2О. Для опреде-
ления общего содержания компонентов использованы метод индиф-
ферентной добавки, для определения содержания гидроксильных
гРУпп — метод ацидиметрического титрования после обмена их на
ионы фтора в концентрированном растворе фтористого калия [8а].
Из сернокислых растворов гидроокись титана осаждается едким на-
тРом при комнатной температуре в виде твердой фазы четверного
ш
состава, изменяющегося со временем. Свежеосажденный продукт
содержит в расчете на 1 моль TiO2 2—3 моля Н2О (общей), 0,01—
—0,1 моль Na2SO4 и 0,01 — 0,08 моля SO3. В этом продукте, кото*
рый по классификации Фрике [58] является ортотитановой кисло*
той и должен содержать четыре гидроксильных группы, связанных
. с одним атомом титана, на самом деле обнаружено наличие только
I двух связанных с титаном гидроксильных групп. По химическому
составу осадки так называемой ортотитановой кислоты являются
гидратированной метатитановой кислотой TiO (ОН)2 • пН2О с при-
месью кислотных остатков SO4 и ионов Na+. Кислотные остатки SO.
с гидроокисью титана связаны очень прочно, не отмываются водой
и разбавленными кислотами. Они, по-видимому, структурно свя:
v заны с гидроокисью титана, имеющей полимерное строение. Навер-
ное, ионы Na+ адсорбированы на поверхности осадка, так как легко
отмываются от него водой.
При длительном хранении при комнатной температуре под ма-
точной жидкостью гидроокись титана, осажденная из сернокислых
растворов едким натром, постепенно отщепляет кристаллизацион-
ную воду и гидроксо-группы, превращаясь в практически безводный
продукт. Примеси SO3 и Na2SO4 при старении гидроокиси титана не
отщепляются от нее и полностью удерживаются осадком. R
Из кипящих сернокислых растворов сульфата. титана образуется
осадок гидроокиси титана с первоначальным содержанием двух
структурно-связанных гидроксильных групп с одним атомом титана.
Однако при температуре кипения растворов гидроксильные группы
легко отщепляются от осадка (в течение нескольких часов) и по-
следний превращается в практически безводную двуокись титана,
содержащую в виде примеси 0,01—0,08 молей SO3 на один моль
TiO2. В гидролизных осадках сульфата титана, выпадающих из
разбавленных сернокислых растворов при комнатной температуре,
содержится 0,04—025, молей SO3 в расчете на один моль ТЮ2.
Из приведенного краткого обзора следует, что гидроокись тита-
на — продукт сложного состава. Она может содержать переменное
количество связанных с титаном гидроксильных групп, струк-
турную воду, кислотные остатки и адсорбированные катионы. В ре-
зультате старения происходит отщепление от металла гидроксиль-
ных групп и воды и превращение гидроокиси в безводную двуокись
титана, удерживающую в связанном состоянии кислотные остаткй
и адсорбированные на поверхности катионы. По своему химическо-
му составу свежеосажденная гидроокись титана может рассматри^
ваться как гидратированная метатитановая кислота, а подвергшая-
ся старению — безводная двуокись титана.
у Джере и Папиль [82] в инфракрасном спектре поглощения гидроокиси титана
установили существование частот 3125, 1613 и 1149 см—1, ответственных за валент-
ные колебания ОН-групп, деформационные колебания Н2О и валентные колебания
связи TiO. Авторы полагают, что гидроокись титана является полцмердым продуй^
том.
112
Вейзер и Миллиген [144] изучили гидроокись титана методами рентгеногра-
ьии» фазового анализа и потенциометрии и пришли к заключению, что она пред-
ъявляет собой гидратированную двуокись титана. Аналогичный взгляд на строе-
ние гидроокиси титана высказал Дерибере [52].
По данным рентгеноструктурных исследований свежеосажденная
гЯДроокись титана, полученная из хлоридных, сульфатных и дру-
гих растворов при комнатной температуре, имеет аморфное строение
[68, 139, 145, 146, 148]. Берестнева с соавторами 13] показала, что
титановый гель, полученный гидролизом TiCl4 на холоду, имеет
аморфное строение также и по данным электронографических иссле-
дований. Судя по магнитной восприимчивости, рентгеноаморфная /
гидроокись титана имеет структуру анатаза [155, 156]. На кривой
^магнитной восприимчивости ее в зависимости от содержания воды
асе точки ложатся на линии магнитной восприимчивостью
чистой воды и безводного анатаза. ~~
Аморфная гидроокись титана при стоянии подвергается старе-
нию, которое заключается в частичном обезвоживании гидроокиси
л титана и образовании мелкокристаллического осадка. Вейзер и со-
авторы [143, 147] сообщают,что ортотитановая кислота стареет под
водой, превращаясь в продукт со структурой анатаза. Состарив-
шаяся гидроокись титана, поглощающая мало воды и не проявляю-
’ щая при сорбции и десорбции гистерезиса, на рентгенограммах дает
слабые линии анатаза [147]. Старение ускоряется кипячением су-
спензии гидроокиси титана с водой.
Воду гидроокись титана удерживает непрочно. В вакууме она^
полностью обезвоживается уже при 7?° С [42]. При нагревании на
воздухе гидроокись титана полность.о разрушается при темпера-
туре около 500° С. Превращение гидроокиси титана в двуокись со-
провождается экзотермическим тепловым эффектом, равным 10 —
20 кал!г TiO2, вызванным уменьшением поверхности образца [141].
Уилка [148] сообщил, что при нагревании до 300° С гидроокись
титана превращается в анатаз, рутил или смесь этих модификаций
Двуокиси титана в зависимости от способа ее получения. При тем-
пературе 700—900° С все осадки гидроокиси титана превращаются
в рутил. Тонкие пленки гидроокиси титана не образуют кристал-
лической структуры даже при длительном нагревании до темпера-
туры 550° С [49].
Джерти Патель [82] методом термографии и рентгенографии
Установили, что гидроокись титана состава TiO (ОН)2 теряет струк-
турную воду при температуре около 160° С. Переход в кристалли-
ческую форму происходит около 250° С. Кристаллический анатаз *
образуется при температуре около 350° С, а превращение его в ру-
тил происходит при температуре около 810° С.
Гидролиз растворов солей титана при нагревании сопровождае-
тся образованием кристаллических осадков со структурой рутила,
анатаза или брукита. Берестнева с соавторами [5? в продуктах гид-
ролиза горячих растворов четыреххлористого титана с помощью
S Э-614 из
"я rW
электронографических исследований обнаружила кристаллики всех Ж
трех модификаций двуокиси титана. Инуцука [81 ] электронографии
чески нашел в тонких пленках на стекле, образовавшихся при Я
гидролизе TiCl3 и TiCl4 и высушенных при температуре ЮО^ С, > I
кристаллики волокнистого брукита. Однако в продуктах ^п^кали^ р
вания гидроокиси титана, как отмечают Памфилов и Иванчева [24/
27], присутствие брукита достоверно никем не доказано. . д
Структура двуокиси титана, в которую превращается гидроокись Я
при прокаливании, зависит от условий ее получения. ЛН
Из сернокислых растворов осаждается гидроокись титана, ко-
торая при прокаливании превращается сначала в анатаз, а затем
в интервале температур 800—1000° С — в рутил [26, 135, 146];Я
Из хлоридных растворов образуется гидроокись титана со струкЛк
турой рутила [26, 135, 146]. При парофазном гидролизе четырех-Я
хлористого титана в интервале температур 300—750° С получаетсяу!
анатаз [31 ]. При более низкой температуре (20—200° С) отмечена9
еще^одна фаза, структура которой не выяснена. Я
В результате гидролиза азотнокислых растворов, по [26], по-1
лучается гидроокись титана в форме анатаза, а по данным [146],—
рутила; в форме анатаза получается также гидроокись титана из 1
растворов, содержащих избыток ионов МОГ и С1“. Я
Примесь сульфатов способствует образованию гидроокиси тичН
тана со структурой анатаза. Как показал Шоссбергер [135], из хлоЯ
ридных и нитридных растворов, содержащих 5% примеси H2SOJH
или Na2SO4, образуется анатаз. Я
Факторы, определяющие структуру гидроокиси титана, полу- К
чаемую при гидролизе растворимых солей, окончательно еще не вы- Л
яснены. Памфилов и Иванчева [26] считают, что образованию гид-Я
роокиси титана со структурой анатаза способствует присутствие вЯ
растворе сильных комплексообразующих агентов, например ионов !
sol~. Я
Роден [125] исследовал гранулометрический состав гидроокиси Я
титана. Продукт гидролиза сульфата титана, по данным электрон-
номикроскопических исследований, состоит из геля с р азмер амиЯ
частиц 100—200 мкм. Эти частицы в свою очередь состоят из агре-1
гатов размером по 0,6—0,7 мкм, образовавшихся из слипшихся зе- ’
рен размером по 60—75 нм, примерно по 1000 штук в одной частице,?*
связанных адсорбированными ионами SO1~ В каждом зерне содер-
жится около 20 микрокристаллов, размером приблизительно 20 АйЯ
Электронномикроскопические исследования гидроокиси титана,$
полученной гидролизом четыреххлористого титана, показали, чт(ЖВ
она состоит из шаровидных частиц размером около 100 А [15, 16]Я
Промежутки между шаровидными частицами обусловливают пори-q
стость геля. Длительное нагревание гелей в маточном растворе
уменьшает пористость гелей вследствие более плотной упаковки
частиц. Замена воды на жидкость с меньшим поверхностным натя-
114
жением способствует образованию гелей с более крупными и менее
плотно упакованными частицами. Поверхность гидроокиси титана,
полученной гидролизом, в 10—20 раз превышает поверхность ча-
стиц двуокиси титана, полученной из этой гидроокиси [67].
Гидролизная двуокись титана, не прошедшая термообработку,
имеет ярко выраженную бидисперсную структуру. Термопаровая
обработка резко влияет на тонкопористую часть структуры образца
и практически не влияет на широкопористую [10].
Гидроокись титана легко подвергается фотохимическому окис-
лению под действием ультрафиолетовых лучей. Органические веще-
ства способствуют образованию гидроперекиси, окрашивающей гид-
роокись титана в желто-оранжевый цвет [117]. Чистая гидроокись
титана, полученная из тетраэтилтитана, приобретает желтый цвет
под действием длинноволнового ультрафиолетового облучения. При
нагревании окрашенных в результате фотохимического окисления
образцов гидроокиси титана они обесцвечиваются при температуре
600° С. Желтая окраска гидроокиси титана обусловлена содержа-
нием гидроперекиси состава TiO3 • 2Н2О. Присутствие гидропере-
киси доказывается исследованием спектров поглощения и выделе-
нием свободного иода по реакции КГ
О СОСТОЯНИИ ГИДРООКИСИ ТИТАНА В ВОДНЫХ РАСТВОРАХ
Состояние гидроокиси титана исследовали преимущественно в
слабокислых растворах. Осаждение гидроокиси титана из водных
растворов указывает на существование в них гидрокси-ионов тита-
на, находящихся в равновесии с ионами гидроксила. Состав гидрок-
си-ионов титана и равновесия их в водных растворах солей титана
изучалось в нескольких работах [21, 22, 34, 54, 100] методами по-
лярографии, потенциометрического титрования, ионного обмена и
распределения между водными растворами и несмешивающимися
органическими жидкостями. Эти методы позволяют установить на-
личие взаимодействия в водных растворах между ионами титана
и гидроксильными ионами, определить заряд ионов и концентра-
цию их. Однако они не позволяют определить состав ионов, так как
одинаковый заряд могут иметь ионы различного состава. Например,
ионы титана с зарядом 1+ могут иметь состав TiOOH+ и Ti (ОН)з’
и при исследовании их методом потенциометрического титрования
или ионного обмена неразличимы. Вследствие этого схемы ионных
равновесий в растворах солей титана, описанные в работах различ-
ных авторов, являются условными. Они составлены исходя из пред-
положений о существовании в растворах ионов титана определен-
ного состава.
Набиванец [21], предположив, что в растворах солей титана су-
ществует однозарядный ион TiOOH+, определил константу его дис-
социации Д’ = 1,5 • 10~13 (18° С).
ч.
Либерти [100] полагает, что в растворах с pH 0,5 — 3,0 суще-
ствует ион Ti (ОН)з~, подвергающийся гидролизу по реакции
Ti (ОН)^ + Ш2О = Ti (0Н)ж!Ж) + гН+.
По распределению титана между водными растворами и растворами
хлороформа, содержащими 8-ортооксихинолин, и бензола, содер-
жащими теоноилтрифторацетон, автор рассчитал константы гидро-
лиза: pKi=i = 1,8, pKi=2~ 4,20 и рК?'=з = 6,31 (рис. 26). Автор
также вычислил произведение растворимости [ТЮН3+] • [ОН“]3 =
= 1СГ39'34. В другой работе Набиванец и Лукачина [22] методами
Рис. 26. Распределение титана между различными
формами гидроокиси [100].
растворимости и ионного обмена определили общие (Д’) и частные
(k) константы нестойкости гидроокиси титана, оказавшиеся равны-
ми: Ki = 1,1 • Ю“8, = 6,7 • КГ36, К3 = 2,1 • 10“48, = 2,1 •
. ю~59, kx = k2 = 6,1 • 10“18, k3 = 3,2 • 10"13, Аг4 = 1,0 • 10““.
Близость значений kr и k2, по мнению авторов, указывает на то,
что присоединение первых двух гидроксильных ионов к Т14+ проис-
ходит почти с одинаковой энергией и приводит к образованию ста-
бильных ионов титана по схемам
Ti4+ + 2Н2О = 2Н+ + Ti (ОН)г+,
Ti4* + Н,0 = 2Н+ + ТЮ2+.
АДСОРБЦИОННЫЕ СВОЙСТВА ГИДРООКИСИ ТИТАНА
Гидроокись титана — пористое вещество, обладающее адсорб-
ционными свойствами [78]. Она адсорбирует газы, органические
вещества, соли и другие, даже если получена непосредственным
осаждением из раствора хлорида титана аммиаком [137]. Высоко-
активные адсорбенты из гидроокиси титана получаются после спе-
циальной обработки.
Описано много методов получения сорбентов из гидроокиси титана, облада-
ющих разнообразными свойствами.
116
При гидролизе титанэфиров типа Ti (OR)4, где R — С2Н5, — СдН7 и — С4Н9,
пропУсканием паР0В воды через бензольный или спиртовый раствор получены гели
с удельной поверхностью около 300 мЧг и радиусами пор 10 — 100 А [70, 72]. Вер-
ховский с соавторами [7] гидролизом TiCl4 получил титановый гель, на котором хо-
рошо адсорбируется аммиак, сернистый газ и щелочи. В работах некоторых ав-
торов [1, 95, 107, 128] описано получение сорбентов осаждением гидроокиси титана
из солянокислых растворов аммиаком и другими щелочами.
Существенное влияние на адсорбционные свойства гидроокиси
титана оказывает термическая обработка. Максимальную адсорб-
ционную емкость имеют образцы гидроокиси титана, высушенные
при температуре 97—214° С [126].
Авалиани и соавторы [1 ] изучили влияние термической обработ-
ки на адсорбционные свойства гидроокиси титана в интервале тем-
ператур 20—450° С. Судя по поглощению азота, гель гидроокиси
титана, подвергшийся термической обработке в вакууме в интервале
температур 20—300° С, представляет собой смешанно-пористый ад-
сорбент с размером пор 40 zA и удельной поверхностью скелета
400 мЧг. После термической обработки выше 300° С свойства адсор-
бента резко изменяются, удельная поверхность его уменьшается.
При нагревании в вакууме до температуры 450° С гель гидроокиси
титана частично теряет кислород, окрашиваясь в цвет от серо-ко-
ричневого до черного.
Гели гидроокиси титана адсорбируют разнообразные вещества.
На 1 г геля, высушенного при 100° С, адсорбируется 8,5 г аммиака
(—16° С) и 28 см3 СО2 [107]. Магнус и Киффер [101] полагают, что
гидроокись титана с двуокисью углерода реагирует и химически,
так как кривые адсорбции точно не воспроизводятся. Адсорбцию
аммиака на гидроокиси титана изучил Хигути [77], а двуокиси уг-
лерода — Бишоф и Адкинс [45]. На гелях гидроокиси титана адсор-
бируется сернистый газ [76, 79], бензол, хлороформ [2, 74, 80, 122,
138], вода [75, 128]. С осадком гидроокиси титана соосаждаются
радиоактивные изотопы: 45Zr, 95Nb, 32Р, 60Со, 59Fe, 89Sr, 144Се,
125Sb, 106Ru, 131I, 137Cs[98].C гелем метатитановой кислоты при гид-
ролизе сернокислых растворов титана соосаждается частично V5+
1138]. Гидроокись титана предложена в качестве адсорбента для
извлечения из растворов Ри4+ и Ри6+. Из растворов карбоната нат-
рия на гидроокиси титана адсорбируется цирконий, ниобий и ру-
тений [89].
Бернс и Уолдрон [40] описали получение гидроокиси титана,
обладающей свойствами неорганического катионита.
КОЛЛОИДНО-ХИМИЧЕСКИЕ СВОЙСТВА ГИДРООКИСИ ТИТАНА
Гидроокись титана образует коллоидно-дисперсные растворы в
ВиДе положительно и отрицательно заряженных золей, которые под
Действием электролитов могут образовывать гели и желатинизиро-
ваться.
117
Впервые коллоидные растворы гидроокиси титана получили
Розе [63, 119] и Грахам [64]. Розе получил золь гидроокиси титана,
подвергая гидролизу TiCl4, Грахам — из желатинизированной гид-
роокиси титана методом диализа. Оба исследователя наблюдали
желатинизирование концентрированных золей титана.
Положительные золи гидроокиси титана образуются в результате
гидролиза хлорида титана [30 ,36, 102, 119], пептизации [105, 108,
132, 136, 152] и диализа водного раствора TiCl4 [ 152]. Гидролиз соб-
разованием золей идет при медленном приливании четыреххлори-
стого титана в холодную или горячую воду. При этом образуются
прозрачные гидрофильные или молочно-белые гидрофобные золи с
ультрамикроскопическими частицами [30]. Гидроокись титана пеп- |
тизируется в положительный золь действием кислот со слабыми ком-
плексообразующими свойствами (азотная, уксусная, пропионовая,
муравьиная) [136]. я
Отрицательные золи гидроокиси титана получают пептизацией 1
[9, 51, 60, 136, 152], перезарядкой положительных золей [12, 90, I
113], растиранием гидроокиси титана в фарфоровой ступке [113,
151], диализом положительных золей до наступления коагуляции I
с последующей пептизацией винной кислотой и очисткой повторным -в
диализом в течение 10—12 дней [60] и др. Пептизируют гидроокись
титана с образованием отрицательных золей кислоты, имеющих ком-
плексообразующие свойства (винная, щавелевая, молочная, яблоч- I
ная, галловая, серная) [136], едкий натр [152], тринатрийфосфат I
[113]. Перезарядка положительных золей гидроокиси титана про- |
исходит под действием едкого натра [12, 59,90] и тринатрийфосфата
[113]. Растиранием гидроокиси титана, полученной осаждением из 1
сернокислого раствора аммиаком, удалось получить очень концент-, Я
рированные золи, содержащие до 414 г!л ТЮ2 [ИЗ]. V
Золи гидроокиси титана, вследствие ее амфотерности и высокой j
адсорбционной способности, имеют сложное строение. Томас и Сте- Я
вард[136] считают, что коллоидные частицы гидроокиси титана
могут содержать многоядерные оловые или оксометаллические ком- А
плексы, гидроксильные и акво-группы, некоторые ионы противо- 4
положного знака, координационно связанные с центральным ато- S
мом. Изучив спектры поглощения эритрозина, адсорбированного I!
на положительном золе гидроокиси титана, Киселева и Каргин ]
[94] пришли к выводу, что потенциалопределяющим ионом поло- я
жительных коллоидных частиц служат ионы водорода. Отрицатель- |
ные золи, полученные пептизацией винной кислотой, имеют состав а
частиц (ТО2)105 • С4Н6Об [9, 51]. Форма частиц отрицательных зо-
лей, по наблюдениям в ультрамикроскопе, шарообразная [59]. Зо- 1
ли, полученные гидролизом четыреххлористого титана, дают рент- |
генограммы рутила [103]. Тонкая структура коллоидных частиц I
золей гидроокиси титана, сольватация, строение двойного электри- |
ческого слоя на поверхности раздела с дисперсионной средой прак- I
тически еще не изучены.
118
Гидрозоли титана замерзают при более низкой температуре, чем
чистая вода. Понижение температуры замерзания Медведев и Га-
линкер [19] использовали для определения размера коллоидных
частиц.
Отрицательные золи гидроокиси титана, по измерениям Пр ин-
цена и Пеплинского [114], имеют изоэлектрическую точку при
pH 6,7.
Гхош и Ракшид [65] измерили £ потенциал золя гидроокиси ти-
тана в присутствии различных электролитов (NaCl, Na2SO4,
K^Fe (CN)e, лимоннокислый натрий). В среднем при эквикоагу-
ляционных концентрациях £ = +27,8 мв.
Положительные золи гидроокиси титана при старении становят-
ся очень устойчивыми к коагуляции электролитами [41, 102]. Раз-
бавленные положительные и отрицательные золи с концентрацией
0,5% TiO2 не коагулируются при вымораживании их жидким воз-
духом [55].
В области коагуляции титановых золей изменение прохождения
света в зависимости от активности преобладающего иона идет по
S-образной кривой [150].
Александрова и соавторы [3] изучили кинетику медленной коа-
гуляции адсорбционно насыщенного и ненасыщенного золей гидро-
окиси титана фотометрическим методом. Коагуляция первого про-
текает в одну стадию с предшествующим индукционным периодом.
Второй золь коагулирует в две стадии без индукционного перио-
да. Авторы полагают, что механизм медленной коагуляции золей
гидроокиси титана в определенный период, независимо от хими-
ческой природы дисперсной фазы и стабилизатора, носит общий
характер и обусловлен степенью адсорбционной насыщенности
системы.
Этанол промотирует сенсибилизацию золя гидроокиси титана
[47].
К ВОПРОСУ О ПРОМЫШЛЕННЫХ МЕТОДАХ ПОЛУЧЕНИЯ
ГИДРООКИСИ ТИТАНА ГИДРОЛИЗОМ СУЛЬФАТОВ ТИТАНА
В ВОДНЫХ РАСТВОРАХ
Гидроокись титана получается в крупных промышленных масштабах гидроли-
зом растворов сульфата титана в виде так называемой метатитановой кислоты [4,
35]. Последняя перерабатывается в двуокись титана, использующуюся в качестве
белых титановых пигментов. Гидролиз растворов сульфата титана, как уже отме-
чалось, проводится в присутствии титановых зародышей, ускоряющих процесс.
Зародышами служат золи гидроокиси титана с концентрацией TiO2 20—50 г/л.
Высокой активностью обладают только золи, зерна которых состоят из частиц раз-
мером около 20 А [125].
Титановые зародыши получаются осаждением гидроокиси титана из сернокис-
лых растворов едким натром. Методы приготовления зародышей разработаны
эмпирически. Механизм действия их еще неизучен. Сопоставление данных по ки-
нетике гидролиза растворов сульфата титана показывает, что зародыши ускоряют
первую стадию процесса.
119
Константа скорости реакции гидролиза сульфата титана в присутствии заро-
дышей увеличивается в десятки раз. Вероятно, зародыши снижают энергию,
активации реакции гидролиза, являясь центрами мицеллообразования.
ГИДРОПЕРЕКИСЬ ТИТАНА
Впервые гидроокись титана упоминается в работе Шенка [127].
Веллер [142, 153] получил гидроперекись титана осаждением аммиа-
ком и едкими щелочами из растворов сульфата и хлорида, содержа-
щих перекись водорода. Гидроперекись титана образуется также
при обработке свежеосажденной гидроокиси титана перекисью во-
дорода.
Классен [48] установил, что из разбавленного спиртового раство-
ра сульфата и хлорида титана получается осадок, в
котором
содер-
жание активного кислорода даже при значительном избытке пере-
киси водорода равно 1 г-ат на 1 моль ТЮ2. Исходя из этого, автор
приписал гидроперекиси титана состав, выражаемый формулой
ТЮ3 • aq. Меликов и Писаржевский [106] состав гидроперекиси ти-
тана выразили формулой ТЮ3 • 2Н2О.
Джере и Патель [84] описали следующую методику получения
чистой гидроперекиси титана. Свежеосажденную гидроокись титана
суспензируют в воде при температуре 0° С и к суспензии добавляют
по каплям при перемешивании избыток 20%-ного раствора пере-
киси водорода. Через 4—5 ч осаждается гидроперекись титана.
Осадок отфильтровывают, промывают водой и высушивают 4—6
дней над Р2О5. Полученный продукт имеет вид желтого порошка.
Состав гидроперекиси титана Джере и Патель [83] выражают фор-
мулой ТЮ2 (ОН)2.
Билли [37, 391 при гидролизе перекисного сульфата титана и
калия К2 [TiO2 (SO4)2] получил продукт, состав которого изменяе-
тся в пределах Ti2O5 • хН2О— Ti2O5,27-xH2O. Осаждением из серно-
кислых и солянокислых растворов автор [38] получил гидропере-
кись титана, состав которой колеблется в пределах Ti2O5 • aq —
— TiO4 • aq.
Шварц с соавторами [129, 130], гидролизуя соль К2 [TiO2 (SO4)21
при температуре 0°С из разбавленного водного раствора, полу-
чил осадок, который после промывания ацетоном имеет состав
TiO3 • 2Н2О.
Макаров и Ладейнова [18] изучили состав гидроперекиси тита-
на, образующейся при температуре 0—20° С, в широком интервале
концентраций перекиси водорода. Состав осадка оказался постоян-
ным и выражается формулой TiO3 • 2Н2О. Таким образом, гидро-
окись титана образует, вероятно, только одно перекисное соедине-
ние. Описанные Билли препараты, по-видимому, состояли из смеси
гидроперекиси и гидроокиси титана.
Строение гидроперекиси титана Шварц с соавторами [129, 1301
г . /О
выражает формулой Ti[(OOH)OH3, или OTi^f | • 2Н2О. Шенк
120
[1341 приписывает гидроперекиси титана строение OTiX | • 2Н2О.
Макаров и Ладейникова [181 изучили дегидратацию гидропере-
киси титана и сняли термограмму нагревания. Судя по полученным
кривым, строение гидроперекиси титана выражается формулой
Ti (ОН) (ОН)3, т. е. она относится к соединениям гидроперекисно-
го типа.
Гидроперекись титана — желтый рентгеноаморфный порошок
[84], диамагнитен {84, 154], на воздухе медленно разлагается с вы-
делением кислорода [129, 130]. Через восемь суток после получения
состав ее выражается формулой Ti2O5, через три месяца — TiO2 [93].
При нагревании гидроперекись титана вспыхивает [129, 130]. Не-
которые авторы [62, 134] наблюдали при встряхивании раствора
гидроперекиси титана и К2Сг2О7 с эфирсм извлечение в органичес-
кую фазу синего комплекса пироксихрома. Образование синего
комплекса служит признаком отщепления от гидроперекиси титана
перекиси водорода. Шварц с соавторами [129, 130], однако, при
встряхивании суспензии гидроперекиси титана с водой образование
перекиси водорода не обнаружил.
Свежеосажденная гидроперекись титана растворяется в присут-
ствии перекиси водорода в водных растворах аммиака и гидроокисей
щелочных растворов. Щелочные растворы гидроперекиси титана
бесцветны. При стоянии они разлагаются, выделяя кислород [48].
Суспензия гидроперекиси титана с раствором йодистого калия
при pH 7,5—8,0 и температуре 0° С выделяет свободный иод [131].
Гидроперекись титана растворяется в фосфорной и щавелевой
кислотах. В присутствии винной кислоты аммиак не осаждает из
водных растворов осадка гидроперекиси титана [97].
Свежеосажденная гидроперекись титана с раствором перекиси
водорода образует коллоидный раствор желтого цвета, который при
испарении превращается в тиксотропный гель [91, 92]. Небольшие
Добавки электролитов вызывают коагуляцию золя гидроперекиси
титана.
Литература
ГАвалиани К- Е-, Цицишвили Г. В., Окропиридзе Ц. К.»
Адолашвили М. Г.— Труды Ин-та химии АН Груз. ССР., 1964, 17, 65.
2. Алексеевский Ж- В., Белоцерковская Г. М.— Ж. общ.
хим., 1936, 6, 370.
3- Александрова Е. М., Ш и ц Л. А., Тюрикова О. Г.— Коллоид,
ж., 1964, 26, 645.
4- Беленький Е. Ф., Р и с к и н И. В. Хкмия и технология пигментов.
Госхимиздат, Л., 1960.
5> Берестнева 3. Я., Карецкая Г. А., Каргин В. А.— Кол-
t ЛОЙД, ж., 1950, 12, 338.^
6. Брусиловская Б. Е., Оттомановская О. М.— Ж. общ. хим.,
1940, 10, 677.
121
7. Верховский С. Е., Верховская А. К., Карсу цель М. В—
Ж- прикл. хим., 1938, 11,4.
8. Горощенко Я. Г. Физико-химические исследования сернокислотного
метода переработки редкоземельных титанониобатов. Изд-во АН СССР, М.,
1960.
8а. Гор ощенко Я. Г., Бирюк Л. И.— Укр. хим. ж., 1968, 34, 54.
9. Думанский А. В., Пусковский Б. С.— ЖРФХО, 1930, 62, 469.
10. Исирикян А. А., Казьменко Н. А., Кисилев А. В.—Кол-
лоид. ж., 1964, 26, 675.
4 11. Касимов А. К.— Изв. АН Каз. ССР. Сер. хим., 1956, 10, 10.
12. Каргин В. А.— Ж- физ. хим., 1933, 4, 597.
13. К л и м е н к о Н. Г., С ы р о к о м с к и й В. С.— Зав. лабор., 1947, 13,
1029.
14. Ласточкина А. А., Шека И. А.— Укр. хим. ж., 1964, 30, 898.
< 15. Леонтьев Е.А., Л укьянович В. М.— В кн.: Методы исследования
структуры высокодисперсных и пористых тел. Изд-во АН СССР, М., 1959,
19.
« 16. Леонтьев Е. А., Лукьянович В. М., Неймарк И. Е.,’П и*
онтковская М. А.— Изв. АН СССР. ОХН, 1958, 9, 1037.
17. Л е к а х Н. Б. Автореф. канд. дисс., Харьков, 1964.
18. М а к а р о в С. 3., Л а д е й н о в а Л. В.— Изв. АН СССР. ОХН, 1961,
6, 958.
19. М е д в е д е в П. И., Г а л и н к е р И. С.— Коллоид, ж., 1962, 24, 717.
20. М и р о н о в Н. Н. Труды по химии и химической технологии, 1. Горький,
1963, 85.
21. Н а б и в а н е ц Б. И.— Ж. неорг. хим., 1962, 7, 417.
22. Н а б и в а н е ц Б. И., Лукачина В. В.— Укр. хим. ж., 1964, 30, 1123.
23. Пат. США № 2326156, 1943; № 2403228, 1946.
,24. Памфилов А. В., Иванчева Е. Г.— Ж- общ. хим., 1937, 7, 2774.
• *25. Памфилов А. В., Иванчева Е. Г., Соболева И. М.—
Ж- прикл. хим., 1939, 12, 226.
26. Памфилов А. В., Иванчева Е. Г.— Ж- общ. хим., 1939, 9, 1739.1
' 27. Памфилов А. В., И в а н ч е в а Е. Г.— Ж. общ. хим., 1940, 10, 154.
28. Топтыгина Г. М., Морозов И. С.— Ж. прикл. хим., 1961, 6, 1685.
29. Ульянов А. И.— Изв. АН СССР. ОТН, 1960, 4, 580.
30. Ф о д и м а н Е. В., Каргин В. А.— Ж. физ. хим., 1934, 5, 716.
31. Щ е г р о в Л. Н.— Ж. физ. хим., 1963, 37, 912.
’32 . Askenasy Р., Heise К-— Z. anorg. Chem., 1931, 196, 256.
33. Auger V.— Compt. rend., 1923, 117, 1302.
34. Beukenkamp J., Herrington K. D.— J. Am. Chem. Soc., 1960,
82 3025
35. Barksdale J. Titanium N. Y., 1949.
36. Brintzinger H., Brintzinger W.— Z. anorg. Chem., 1931, 196,
44.
37. Billy M.— Ann. chim., 1921, 16, 5.
38. Billy M., San-Galli J.— Compt. rend., 1932, 194, 1126.
39. Billy M.—Compt. rend., 1921, 172, 1411.
40. В a r n e s К. C., Waldron J. C. Res. Group. U. K. Atomik Energy Author.
№ AFRF-R 1965 4894
41. Bhatia L. S., Ghosh S.—J. Ind. Chem. Soc., 1930, 7, 687. JI
42. В i 11 z W., Leher G. A., Rahlfs O.— Z. anorg. Chem., 1940, 244,
281
. 43. В e r t о n A.— Compt. rend., 1938, 207, 625. В
44. Becker H., Klein E., Rechman H.— Farbe und Lack, 1964, 10#
779.
45. В i s h о f f F., Adkins H.— J. Am. Chem., Soc., 1925, 47, 807.
• 46. Сакаи H., Йосикава К., Судзуки M., К а б а с и С.—J. Chem-1
Soc. Japan. Ind. Chem. Sec., 1961, 64 , 613, A36. •
47. Chakravarti K-, Ghosh B. N.— J. Ind. Chem. Soc., 1964, 41, 559. J
122
48. Classen A.— Ber., 1888, 21, 370.
49, Con j e a u d P.— Compt. rend., 1954, 238, 2075.
50. С a r n e 1 1 у T., Walter J.— J. Chem. Soc., 1884, 53, 59. ►
51. Dumanski A., В u n t i n A., К n ig a A.— Roll. Z., 1927, 41, 108.
52. Deri here M.— Ann. Hyg. Publ., 1941, 18, 133.
>53, Demoly A.— Compt. rend. Traw. Chim. 1842, 5, 325.
54. Delafosse D.— Compt. rend., 1955, 249, 1991.
55. Djatschkowsky S. J.— Koll. Z., 1931, 54, 287.
»56. E be Imen J. J.— Ann. chim. et phys., 1847, 20, 385. -
57 F u c s J. N.— J. prakt. Chem., 1939, 18, 495.
58. Fricke F.— Roll. Z., 1934, 69, 312.
59. Freundlich H., Kross W.— Roll.Z., 1930, 52, 37.
60. F о d i m a n E. V., R a r g i и V. A.— Acta physicochim. URSS, 1934/35,
1, 220.
51. Friedel C., Guerin J.— Compt. renl4 1875,81, 889.
62. F i c h t e r F., G о 1 d о c h A.— Helv. chim. acta, 1930, 13, 1200.
53. G r a c h a m T.— Pogg. Ann., 1864, 123, 529.
64. G г a c h a m T.— Proc. Roy. Soc., 1863, 18, 335.
65. Ghosh B. N., R a к s h i t S. С,— Sci. and Culture, 1953, 18, 498.
66. Gutbier A., Ottenstein B., Leutheuser E., Lassen R.,
A 1 1 a m F.— Z. anorg. Chem., 1927, 162,67.
67. H a r v e у E. N.— J. Am. Chem. Soc., 1943, 65, 2343.
68. FI e d v a 1 1 J. A.— Ark. kem. min., 1921, 8, 1.
69. Holmquist P. J.— Bull. geol. inst. Upsala, 1896/97, 3, 211.
.70. Harris M. R., Whitaker G.— J. Aopl. Chem., 1962, 12, 490.
71. Harris M. R., Whitaker G.— J. Appl. Chem., 1963, 13, 198.
72. H a r r i s M. R., Whitaker G.— J Appl. Chem., 1963, 13, 348.
•73. Hixon A. W., Plechner W. W.— Ind. Eng. Chem., 1933, 25, 262.
74. H i g u t i J.— Sci. Pap. Inst. Tokyo, 37, 980—981; Chem. Zentr., 1940, 11,
3313.
75. H i g u t i J.— Sci. Pap. Inst. Tokyo, 32, 718—723; Chem. Zentr., 1938, 1,
3314.
76. H i g u t i J.— Sci. Pap. Inst. Tokyo, 29, 529—631; Chem. Zentr., 1936, II,
1506.
77. H i g u t i J.— Sci. Pap. Inst. Tokyo, 31, 577—682; Chem. Zentr., 1937, II,
1331.
78. H i g u t i J.— Sci. Pap. Inst. Tokyo, 35, 872—874; Chem. Zentr., 1939,1,4020.
79. Higuti J.— Sci. Pap. Inst. Tokyo, 27/28, 585—600; Chem. Zentr., 1936, I,
3806.
B0. Higuti J.—Sci. Pap. Inst. Tokyo, 36, 915—919; Chem. Zentr., 1939, II, 3679.
Bl. Inuzuka H.— J. Japan Ceram. Assoc., 1941, 49, 416.
*82. J e r e G. V., Patel С. C— J. Sci. Ind. Res., 1961,’B20, 292
83. J e r e G. V., Patel С. C.— J. Inorg. a. NuciTCtem., 1961, 20, 343.
84. J e r e G. V., Patel С. C.— Z. anorg. Chem., 1962, 319, 175;
’85. К п о p.— Pogg. Ann., 1862, 123, 351.
86 .,К 6 n i g T., P f о r d t e n O. D.— Ber., 1889, 22, 2070.
87. Knoll H., Frydrych R., Kiihnhold U.— Naturwis., 1958, 45,
262.
88. К о w a 1 e v s к у W.— Z. anorg. Chem., 1900, 25, 189.
89. К e n n e d у J., P e с к e 11 J. W., Perkins R.— Res. Group. U. K.
Atomic Ener., Author., 1964, AERE-R4516.
90. Kargin V. A.— Acta phisicochim. URSS, 1934/35, 1,64.
91. Katzoff S., Roseman R.— Compt. rend., 1939, 208, 1733.
92. Katzoff S., Roseman R.— J. Am. Chem. Soc., 1935, 57, 1384.
93. К h а г к a r D. P., Patel С. C.— Current Sci., 1955, 24, 413.
94. К i s s e 1 e v a W. W., Kargin V.— Acta physicochim. URSS, 1940, 12,
589«
£>. К 1 о s к у S.— J. Phys. Chem., 1930, 34, 2621. .
y8. К 1 о s к у S., M a t z a n о C.— J. Phys. Chem., 1925, 29, 1125.
123
97. Levy L.— Ann. Chim. Phys., 1892, 25, 453.
98. Levi H. W. Treatment and Storage High-Level Radioactive Wastes. ^Vien
na, 1963, 587; РЖХим, 1964, 11, Б, 398.
e 99. L i g о r i о C., Work L. T.— Ind. Eng. Chem., 1937, 29, 213.
100. Liberti A., Chiantella V., Corigliano F.—J. Inorg.
a. Nucl. Chem., 1963, 25,415.
4 101. Magnus A., Kieffer R.— Z. anorg. Chem., 1929, 179, 215.
102. M a j u m d a r S. K-— J. Ind. Chem. Soc., 1929, 6, 357.
103. Milligan W.O. Beiser H. B.— J. Phys. Chem., 1936, 40, 1095.
104. M о 1 1 a r d E.— Compt. rend., 1872, 75, 472.
105. Mehta S. M., Joseph O.— Ind. Chem. Soc., 1933, 10, 177.
106. M e 1 i к о f f P., Pissarjewsky L.— Ber., 1898, 31, 678, 935.
107. Nikitin N. J., J u r j e w W. J.— Z. anorg. Chem., 1928, 171, 28L
108. Owens H.S., Morris R.— J. Phys. Chem., 1938, 42, 563.
109. P a s s e r i n i L.— Gazz., 1935, 65, 529.
ПО. P f о r d t e n O. D.— Lieb. Ann., 1887, 237, 201.
*111. Parravano N., Caglioti V.— Gazz., 1934, 64, 429.
Д12. P f о r d t e n O.— Ber., 1884, 17, 727.
113. Parravano N., Caglioti V.— Gazz., 1934, 64, 450.
П4. Princen L. H. de Vena-Peplinski M.— J. Coll. Sci., 1964, 19
786.
115. P о 1 i d о г i E.— Z. anorg. Chem., 1899, 19, 306.
**116. Pecsok R.L., Fletcher A. N.— Inorg. Chem., 1962, 1, 155л*
^117. Petit J., Poisson R.— Compt. rend., 1955, 240, 312.
.118. Rose H.— Gilb. Ann., 1823,73,67.
.119. Rose H.— Pogg. Ann., 1844, 1, 507.
\120. Rose H.— Pogg. Ann., 1845, 53, 267.
121. Rose H.— Pogg. Ann., 1863, 61, 507.
122. R о h 1 a n d P.— Z. anorg. Chem., 1909, 65, 108.
123. R e i n d e r s W., Kies H. L.— Rec. traw. chim., 1940, 59, 785.
. 124. Rammelsberg C.— Ber. Berl. Akad., 1874, 490.
125. R о h d e n C.— Chim. et Ind., 1956, 75, 287.
126. Subramanaya R.S., Subbarao K-, Sahjivarao B.— Proc
Ind. Acad. Sci., 1947, A25, 186.
127. Schon n.— Z. anorg. Chem., 1870, 9, 41.
128. Subbarao K-— J. Phys. Chem., 1941, 45, 500.
129. Schwarz R., S e x a u e r W.— Ber., 1927, 60, 5C0.
130. Schwarz R., Giese H.— Z. anorg. Chem., 1928, 176,209.
131. Schwarz R., Heinrich F.— Z. anord. Chem., 1935, 223, 387.
132. Schneider E. A.— Z. anorg. Chem., 1895, 8, 81.
133. Simon A.— Kolloid. Z., 1928, 46, 161.
134. Schenk M.— Helv. chim. acta, 1936, 19, 625.
135. Schossberg F.— Z. Kristallogr., 1942, 104, 358.
136. Thomas A. W., Stewart W. G.— Kolloid. Z., 1939, 86, 279.v
137. Unger er E.— Z. Pflanzenernahrung Diingung, 1931, A21, 129.
138. Uno S., Inokuma T.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1951, 54
785
139. V e v i G. R.— Aiorn. Chim. Ind. applicata, 1925, 7, 410; Ch. A., 1926, 2947
140. W о h 1 1 e r F.— Lieb. Ann., 1850, 73, 34.
141. W о h 1 1 e r L.— Kolloid. Z., 1926, 38, 97.
142. Weller A.— Ber., 1882, 15, 2599.
*143. Weiser H. В., M i 1 1 i d a n W. O.— J. Phys. Chem., 1934, 38, 513.
144. Weiser H. B., Milligan W- O.— Chem. rev., 1939, 25, 1.
145. Weiser H. B., Mi 1 1 igan W. O.— J. Phys. Chem., 1940, 44, 108L
146. Weiser H. B., Milligan W. O., Cook E. J.— J. Phys. Chem-
1941, 45, 1227.
147. Weiser H. B., Milligan W. O., Simpson W. S.— J. Phys
Chem., 1942, 46, 1051.
148. W i 1 к a S.— Acta chem. Scand., 1954, 8, 1796.
124
Lins К.— Angew. Chem., 1936, 49, 489.
, Hausknech P.— Ber., 1913, 46, 3763.
i49. Work L. T., T u w i n e г S. B., Gloster A. I.— Ind. Eng. Chem.,
! 1934, 26, 1263.
< го W a n n о w H. A.— KolL Bein., 1939, 50, 367.
51. Wegelin G.— Kolloid. Z., 1914. 14, 65.
i52. W i n t g e n K-, Lins K.— Angew. Chem., 1936, 49, 489.
153- Weber A.— Z. anal. Chem., 1884, 23, 410.
154. Wedekind E., Hausknech P.— Ber., 1913, 46, 3763.
155. Zimens К. E., H e d v a 1 1 J. A.— Trans. Cahlmers. Univ. Technol.,
1 1942,9,14.
156. Z i m e n s К. E., H e d v a 1 1 J A.— Charmas tekn. Hogskalas Handl.,
1942, 9, 26.
Дополнительная литература
Беляев И. H., Артамонова G А. Исследование гидроокисей титана,
циркония и совместно осажденных гидроокисей титана, свинца, циркония и
свинца.— Ж- неорг. хим., 1966, 11, 464.
бобыренко Ю. А., Ш е й к м а н А. И., Долматов Ю. Д. О размерах
кристаллов зародышевых золей ТЮ2 в зависимости от условий синтеза.—
Ж. прикл. хим., 1967, 49, 716.
Долматов Ю. Д., Бобыренко Ю. Я., Шейкман А. И. О росте
частиц ТЮ2 в процессе термического гидролиза.— Ж. прикл. хим., 1967, 49,
911.
Долматов Ю. Д., Бобыренко Ю. Я., Шейкман А. И. О кинетике
термического гидролиза Ti-4 и росте частиц ТЮ2.— ЖВХО, 1966, 11, 35L
Комиссарова Л. Н., Шацкий В. М. Сорбция скандия при гид-
ролитическом осаждении титана.— Ж. прикл. хим. 1966, 39, 2211.
Колосницина И. В., Петрова Р. С., Изучение адсорбционных
свойств двуокиси титана газохрома~о графическим методом.— Коллоид, ж.
1967, 29, 815.
Heither-Wirguin С., Albu У. А. Структура гидроокисей и равнове- ✓
сие обмена на них.— J. Inorg. a NucL Chem., 1966, 28, 2379.
Levi Н. W., Schiewer E. Обменная адсорбция катионов на TiO2 • aq.— *
Radio chim. acta, 1966, 5, 126.
ГЛАВА IV
ДВУОКИСЬ ТИТАНА
Высший окисел титана — двуокись — в природе встречается
в форме трех минералов: рутила, анатаза и брукита, представляю-
щих из себя полиморфные модификации, отличающиеся различным
кристаллическим строением. Искусственные методы разработаны
только для получения двуокиси титана со структурой рутила и ана-
таза. Двуокись титана производится в больших количествах в виде
мелкокристаллических продуктов, используемых преимущественно
в качестве пигментов. Разработаны однако и методы выращивания
монокристаллов рутила и анатаза, отличающихся высокими коэф-
фициентами преломления (большими, чем у алмаза) и представ-
ляющими интерес для изготовления полудрагоценных камней. !
Образование искусственных кристаллов брукита наблюдали лишь немногш
исследователи в продуктах гидролиза солей титана и при физико-химическом ис
следовании некоторых систем. Брукит обнаружен, например, при петрографи-
ческом изучении системы FeO — ТЮ2 (гл. V). Бах [86] установил, что брукит об-
разуется при нагревании тонкой пленки гидроокиси титана (800 А), полученной
гидролизом тетраэтил- и тетрабутилтитана, при температуре 250°—400° С в присут-
ствии ионов щелочных металлов.
*
КРИСТАЛЛИЧЕСКОЕ СТРОЕНИЕ
Полиморфные модификации двуокиси титана имеют следующие
пространственные группы: рутил — — Р4/тпт, анатаз —
Dih — IMamd [НО, 188, 219], брукит — — Pabc [134, 2401
Рутил и анатаз кристаллизуются в тетрагональной системе, брукит-
в ромбической. В ряде работ определены параметры решеток син
тетических рутила и анатаза (табл. 12). 0
Эрлих [148] определил параметры решетки искусственного ру
тила: а = 4,592, с — 2,960 A, d%5 = 4,197; молекулярный объев
измеренный 19,04 см3! моль, вычисленный 18,80 см2/моль. Шоссбер
гер [257] для рутила, полученного из четыреххлористого титана
нашел а = 4,598, с — 2,960 А.оПараметры решетки синтетической
анатаза а = 3,784, с — 9,505 A, z — 4 [257]. И
Атомы титана в ячейке рутила расположены по вершинам и 1
центре элементарной ячейки, атомы кислорода — по диагонали!
базисных плоскостей и по перпендикулярным к ним диагоналям
126
проходящим через центр ячейки. Титан в решетке рутила окружен
шестью атомами кислорода в виде слегка деформированного октаэд-
ра 1168, 219, 284] (рис. 27). Два атома кислорода в ячейке рутила
Таблица 12
Параметры решеток природных минералов двуокиси
титана в А
Кристалличес- кая модифика- ция а ъ с Литература
Рутил 4,529 2,926 ’284]
9,504 — 2,940 Г290]
4,589 —— 2,926 [159]
4,591 — 2,984 [144]
4,659 • 2,996 [285]
4,589 — 2,956 [184]
4,5014 —- 2,8989 [282]
4,584 2,953 [205]
4,586 2,956 [38]
4,601 - 2,97 [33]
Анатаз 3,738 —* 9,389 [284]
3,805 —— 9,56 [285]
3,788 — 9,519 [184]
3,777 —- 9,501 205]
3,775 — 9,490 [38,151]
Брукит 9,166 5,436 5,135 [39,151]
9,155 5,450 5,163 [274]
9,185 5,447 5,145 [240]
• 9,219 5,451 5,15 107
9,12 5,41 5,10 220]
располагаются в той же плоскости, что и титан, а по два атома кис-
лорода находятся на поверхностях, расположенных на расстоянии
с/2 выше и ниже указанной плоскости. Заселенные октаэдры имеют
по два общих ребра с соседними октаэдрами и образуют цепи в на-
правлении оси с. В ячейке находятся по две молекулы Коор^.
Динаты атомов титана: 0, 0, 0; х/2» 1/2, х/2; координаты атомов кисло-
рода: х,х, о; х, х, о\ г/2 + х, 1/2 — х, 1/2t 1l2 — x, V2 + x, х/2[270].
Параметр х определяет расстояния между атомами кислорода и
титана. По данным Ленарда-Джонса и Дента [203], х = 0,302.
Более новое значение х = 0,306 получил Баур [105]. Приняв по-
следнее значение х, Богородицкий с соавторами [9] вычислил £] =
1,944±0,004 A, S2 = 1,9887 ± 0,006 А.
Анатаз также построен из кислородных октаэдров с атомами ти-
тана в центре октаэдров. Октаэдры анатаза отличаются от рутило-
ВЬ1Х только расстоянием между атомами титанГТГкислорода, лежа-
н£ими в одной плоскости. Атомы кислорода в решетке анатаза обра-
зуют плотнейшую кубическую упаковку. Зигзагообразные цепочки
113 заселенных ТЮ6-октаэдров в структуре анатаза чередуются с
127
у незаселенными. Каждый октаэдр имеет четыре общих ребра с со
, сёдним (рис. 28).
В ячейке анатаза содержится 4 молекулы ТЮ2. Координаты
2?
О-О
рутила
• Тс
О' 0
Рис. 28. Строение анатаза [126].
Рис. 27. Структура
а — элементарная ячейка; б — сочле-
нение октаэдров в решетке рутила.
4 — х [270]. Вегард[284] нашел,
2»
V, /2 ’ ’ ' 2>
что х = 0,208.
Паркер [236] определил расстояния между атомами титана и кис-
лорода в решетке анатаза: Oi 2 — Ti = 1,95 Л (|| с) и Оз 4 5 6 Ti =|
= 1,90 А. ’ ...........
I Энергия решетки рутила отличается от энергии решетки анатаза
только на 0,5% [109].
Атомы кислорода в бруките образуют искаженную четырехслой*
ную (топазовую) плотнейшую упаковку, слои параллельны (100)®
[106, 240, 293]. Атомы титана находятся в октаэдрических пустотах^
Каждый октаэдр TiOe имеет общие ребра с двумя соседними. Эти
128
обшие ребра несколько укорочены по сравнению с другими ребрами
[106]. Атомы титана несколько смещены от центра октаэдров (на
q 2 0,1 А). Два октаэдра из каждой группы трех октаэдров, свя-
занных общими ребрами, слагают в направлении оси с зигзагообраз-
ные цепи, которые связаны тройными октаэдрами, и двойные сетки,
параллельные (100) (рис. 29).
Исследованию структуры кристаллических модификаций дву-
окиси титана посвящены работы [218, 237, 239].
0 0-0
Рис. 29. Строение брукита [219].
Шредер [258] считает, что анатаз диморфен с точкой перехода
\ 642°С. Диморфность анатаза доказывается наблюдениями за из-
мененйем угла призмы с ростом температуры
Рутил в системе Ti — О имеет узкую область гомогенности. Она
простирается, по данным Магнели с" соавторами [76, 77, 80], в пре-
делах TiOi,96 — Ti02,oo, а по Штрауманису с совторами [266],—
— ТЮг.оо (гл. II).
По мнению [266], решетка рутила в области гомогенности имеет
Дефектную структуру из-за недостатка в ней ионов кислорода.
Вакансии кислорода составляют до 0,034 иона на решетку, или до
0,85% всех кислородных позиций в решетке. Для компенсации по-
терь отрицательных зарядов часть атомов титана находится в виде
ионов Ti3+, чем и объясняется темная окраска рутила с дефектной
структурой. Верхняя граница фазы рутила по сравнению со стехио-
метрическим составом TiO2 не содержит избытка кислорода [290].
Кислород может адсорбироваться в интервале 500—900° С только
кристаллами рутила. Адсорбция кислорода обратима и имеет место
Ка активных центрах, образованных загрязнениями на поверхности
кристаллов.
Установлено, что в вакууме 10~5 — 10—6 мм рт. ст. при температуре выше
г .рС рутил теряет кислсрод [115, 111, 198]. Вес образца рутила, по Цофстаду
1 уо], в зависимости от давления кислорода в интервале температур 1200—1500° С
^меняется пропорционально р~'1'/б,где р —давление кислорода, В нестехиомет- 9
9'614 129
рическом рутиле имеются кислородные вакансии, концентрация которых бо-
лее 10‘19 см3.
Форланд [152] изучил дефекты в решетке рутила, измеряя потери веса при тем-
пературе 934—1194° С и парциальном давлении кислорода 10"7 —ХОГ^атм. Выше-
1000° С, по его данным, потери веса рутила также пропорциональны р”1/б. Дефек-
ты решетки состоят из вакансий кислорода и электронов, не связанных с этими
вакансиями. Наибольшее, полученное автором, содержание вакансий кислорода
равно 0,384% (температура 1194° С и р = 10~13,5 атм.). В интервале температур
t 1099—1194° С средняя энтальпия образования вакансий кислорода в рутиле рав^
на —130 ккал/молъ.
Наличие вакансий кислорода в рутиле подтвердили Хауль и Дюмген [181],
исследуя диффузию О18.
Параметры решетки рутила стехиометрического состава ТЮ2,
по Штрауманису с соавторами [266J, равны (25° С): а = 4,5937,
с = 2,9581 А, объем ячейки 62,423 А3, ризм = 4,2493 г!см3. I
Недавно Бенделиани с соавторами [12а] нагреванием рутила
при температуре 400—1500° С и давлении 40—120 кбар получили
четвертую модификацию двуокиси титана, метастабильную в нор-
мальных условиях. Она имеет структуру типа а-РЬО2 ромбиче-*
ской системы, фед. гр. РЬсп, параметры решетки tz = 4,531» Ь =
= 5,498, с = 4,900 А, РРентг “ 4,35 г!см3. Обратный переход этой
модификации двуокиси титана в рутил происходит при нагрева-
нии ее в течении 4 ч до температуры 600° С. Непременным условием
образования четвертой модификации двуокиси титана является при-
сутствие в качестве минерализатора воды-
< Хиллераус [1831 считает, что решетки анатаза и рутила практи-
чески не поляризуются благодаря потери атомами титана четырех
электронов. Энергия решетки рутила 0,3218 ккал)моль. Малое зна-
чение её свидетельствует о молекулярной природе кристалла рутила.
Природа связи титана с кислородом в двуокиси титана оконча-
тельно еще не определена. Аркель [82], а также Кардона и Харбек
[130] считают, что в рутиле связь титана с кислородом носит ион-
ный характер. В другой работе Аркель [81 ] на основании изучения
поляризации ионов пришел к выводу, что решетка рутила является
промежуточной между ионной и молекулярной. Это мнение разде-
ляют Андерсен [79] и Грант [167].
Матосси [213] на основе анализа спектров комбинационного рас-
сеивания и инфракрасных спектров пришел к заключению, что ру-
тил — молекулярный кристалл. Титан прочно связан только с
двумя атомами кислорода из шести его окружающих. Однако спектр
рутила отличается от изолированной молекулы двуокиси титана.
Богородицкий с соавторами [9] считает, что в пользу наличия
ковалентной связи между атомами титана и кислорода в рутиле сви-
детельствует неравенство расстояний Si и S2. Он предполагает су-
ществование плоских ковалентных связей по линии Slf однако
доля ковалентной связи пока еще не может быть вычислена.
130
МЕТОДЫ ПОЛУЧЕНИЯ ДВУОКИСИ ТИТАНА
В старых работах описываются методы получения всех трех мо-
дификаций двуокиси титана, однако до появления рентгенографии
не было надежных методов идентификации структуры мелкокристал-
лических продуктов и потому приводимые в этих работах данные о
природе полиморфных модификаций двуокиси титана нередко бы-
вают сомнительными.
7 Рутил получен прокаливанием гидроокиси титана [251, 252, 136, 173—176]
и гидролизом паров TiCl4 при температуре 100—1200° С [177, 171]. Отефейт [169,
170] получил рутил из расплавов двуокиси титана в KF, CaF2, K2SiF6, СаС12 +
+ SiO2; при обработке титаната калия хлористым калием; фтортитаната калия фто-
ристым калием при действии на смесь СаГ2 + CaTiO3 хлористым и фтористым во-
дородом. Грив и Увейт [165] кристаллы рутила получили из расплава двуокиси
титана. Михель [216, 217] наблюдал образование мелких игольчатых кристаллов
рутила при нагревании смеси ильменита с пиритом в Графитовом "тигле при
температуре 1200° С.
j Анатаз получили Розе [251] и Отефейт [173, 176] при^ощбом калении гидро-
окисититана, осажденной аммиаком из сернокислого раствора. В более поздних ра-
ботах получение анатаза прокаливанием гидроокиси титана описано многими ав-
торами.
Розе [251] сообщил о получении брукита в результате длительного нагревания
гидроокиси титана до красного каления. Получение бгукита действием паров во-
ды на TiCl4 при красном калении описано Даубри *131, 133]. Хольгерссон и
ХерлинД177] получили брукит гидролизом Т1С14 при температуре 800° С. Бруси-
ловский и Брусиловская [11] сообщили, что при нагревании гидроокиси титана до
белого <к а ления образуется смесь анатаза и брукита, а выше 1000° С — рутил.
Памфилов и Иванчева [52, 54] считают, что сообщения о получении брукита
искусственными методами не нашли подтверждения в последующих работах и
являются ошибочными.
Блюменталь [103] для^препаративного получения рутила рекомендует прово-
дить гидролиз четыреххлористого титана в присутствии сульфата аммония, улучша-
ющего фильтруемость осадков. Отфильтрованная и промытая водой гидроокись
титана прокаливается в течение 1 ч при температуре 955° С.
Мойнов и Резниченко [39] получили рутил сжиганием четыреххлористого ти-
тана кислородом.
Блюменталь [103] рекомендует получать анатаз гидролизом сульфата титана и
прокаливанием полученной гидроокиси титана при температуре 540° С.
Высокочистая двуокись титана со структурой анатаза образуется при прока-
ливании гидроокиси титана при температуре 700—800° С, полученной гидролизом
двойного сульфата титанила и аммония (NH4)2TiO (SO4)2 • Н2О (гл. VII).
Описанными методами двуокись титана получается в виде белого
микрокристаллического порошка с размерами частиц 0,1—Q5 ммк
и с удельной поверхностью 3—14 м2/г [113].
Сандерна с соавторами [129] разработал метод получения моно-
кристаллов спектрально чистого анатаза крупных размеров. Ме-
таллический титан, не содержащий кремния, растворяется в аммиач-
ном растворе 90 %-ной перекиси водорода. Содержащиеся в металле
примеси Fe, Мп, Mg, Sn, Ni, Al и Ag в аммиачном растворе пере-
киси водорода не растворяются и отделяются от раствора фильт-
рованием. После удаления перекиси водорода образуется гель
гидроокиси титана, из которого нагреванием до 200° С получаются
кристаллы анатаза размером до 3 мм.
Методы получения монокристаллов рутила из расплавов солей
описаны Аникиным с соавторами [1]. Крупные кристаллы рутила
образуются при охлаждении сплава Na2B4O7— TiO2+ 5 — 6% LiF
от 1200 до 800° С со скоростью 0,5—1 град!ч, а также разложением
TiF4 парами воды при температуре 1050—1120° С над сплавом
KF — A1F3 — TiO2.
Гидротермальный синтез рутила проводится перекристаллиза-
цией двуокиси титана в растворах хлоридов (15—20%) тетрабора-
тов] (5—20%) и фтористого калия (1—6%) при температуре 500° С
и давлении 900—1000 атм [1]. В гидротермальных условиях рутил
кристаллизуется из растворов в NH4HCO3, (NH4)2 СО3, NH4F и
NaF [34]. Перекристаллизацию двуокиси титана в рутил на за-
травку удалось осуществить только в растворе NaF и KF.
О ПРЕВРАЩЕНИИ АНАТАЗА И БРУКИТА В РУТИЛ
Диаграмма состояния системы Ti — О изучена только при дав-
лении, не превышающем одной атмосферы. Вследствие этого харак-
тер полиморфных превращений окислов титана, в том числе и его
двуокиси, еще не изучен. В интервале температур от 100 до 1850° С
при нормальном давлении наблюдается только одностороннее прев-
ращение анатаза и брукита в рутил, который в этих условиях яв-
ляется стабильной модификацией [161]. Превращения анатаза в
брукит и наоборот экспериментально не осуществили. В горных
породах анатаз, брукит и рутил нередко встречаются совместно,
однако анатаз и рутил обычно являются минералами вторичного
происхождения [20], а брукит — гидротермальным минералом [40].
Некоторые авторы [153, 230] считают, что превращение анатаза
и брукита в рутил носит монотропный характер. Это значит, что на
диаграмме состояния двуокиси титана (/? — t - диаграмма) точки
равновесного превращения лежат выше точки плавления и являют-
ся, таким образом, гипотетическими. О монотропном характере
превращения анатаза и брукита в рутил судят по необратимости
этого процесса [1121. Барблан с соавторами [88, 89] пришел к вы-
воду о монотропности превращения полиморфных модификаций
двуокиси титана в результате исследования энергий кристалличе-
ских решеток.
ПРЕВРАЩЕНИЕ АНАТАЗА В РУТИЛ
Превращение анатаза в рутил наблюдается в интервале темпера-
тур от 300 до 1000° С [121, 257]. Обычно оно происходит при тем-
пературе 800—850° С [123, 185, 186].
На скорость превращения анатаза в рутил оказывают влияние
многие факторы: способ получения анатаза, содержание в нем npif-
132
месей, размер частиц, температура и продолжительность нагревания,
а также примесь минерализатороз и затравки из рутилизирую-
щих зародышей [112, 121, 150, 264, 289, 305]. Характерно, что мо-
нокристаллические образцы анатаза превращаются в поликристал-
лический продукт, хотя переход сводится к незначительной пере-
стройке кристаллической решетки [265].
Рис. 30. Схема получения кристаллических модификаций двуокиси
титана [231].
Анатаз, полученный гидролизом сульфата титана, превращается в рутил при
более высокой температуре, чем полученный из хлоридных растворов [121]. Из раз-
бавленных растворов получается анатаз, который легко превращается в рутил
[230]. Хорошо отмытая от примеси SO3 ггдроокись титана превращается в рутил
при температуре 300° С [257]. Превращение анатаза,шолученного осаждением ам-
миаком из водных растворов, в рутил ниже 655° С очень замедляется [309].
Парравано [2311 обобщил закономерности превращения различ-
ных форм двуокиси титана и представил их схемой (рис. 30). Эта
схема не является исчерпывающей, так как не учитывает влияния
на структурные превращения двуокиси титана различных факторов.
Никем не доказано также существование аморфной двуокиси тита-
на. По-видимому, можно говорить только о рентгеноаморфном про-
дукте, который из-за мелкокристаллического строения не дает чет-
ких линий на рентгенограммах, но обнаруживает кристаллическую
структуру при исследовании его электронографическим методом.
Тем не менее схема Парравано отражает в известной мере последова- \
дельность превращения различных форм двуокиси титана в зависи-
мости от метода получения и основные ее положения не вызывают
133
возражений. В частности, аморфная (рентгеноаморфная) двуокись
титана, как правило, вначале превращается в анатаз и только затем
в рутил. Из сернокислых растворов двуокись титана получают' в
форме анатаза. Из хлоридных и нитратных растворов можно полу*
чить рентгеноаморфную двуокись титана непосредственно в форме
рутила или анатаза. Брукит превращается только в рутил. |
Не менее существенное влияние на превращение анатаза в рутил
оказывают примеси. Одни из них замедляют превращение, а другие
(минерализаторы и затравки) ускоряют его. Влияние примесей на
превращение анатаза в рутил было известно еще Эбельмену [150,
136, 172].
Замедляют переход анатаза в рутил примеси ионов РО4“ [230], SO4—[53,
88, 89,’121], хлориды и нитраты щелочных металлов [124, 289]. Температура пере-
хода анатаза в рутил повышается при старении гидроокиси титана и при содер-
жании примесей, отщепляющих серную кислоту [257]. Образованию структуры
анатаза при нагревании тонких пленок гидроокиси титана способствует примесь
кварца [86].
Рао с соавторами [254] с помощью рентгеновского метода изу-
чил влияние примесей ионов Al3+, Zn2-H, РО3-, SOl^, С1“ на переход
анатаза в рутил при температуре 708—870° С. Все примеси в коли-
честве 5 ат. % стабилизируют анатаз. По величине уменьшения ста-
билизации они располагаются в ряд: РО^-, SO|~, А13+, СГ-, Zn2+.
Хлор-ионы в количестве 1—5% ат. не влияют на переход анатаза Bi
рутил.
Памфилов и Иванчева [53] считают, что замедление превращения анатаза в
рутил происходит вследствие комплексообразования. Шоссбергер [257] объясняет
стабилизацию анатаза вхождением SO^” в решетку в виде твердого раствора. Бар-
блан и соавторы [88, 89] полагают, что стабилизацию решетки анатаза примесью
ионов 5О4~можно объяснить особенностями ее строения. Решетку анатаза можно
рассматривать как деформированную решетку типа NaCl с наполовину заполнен-
ными местами катионов. Включение ионов SO4~ в эту решетку затрудняет ее пере-
стройку.
Иного взгляда придерживаются Сулливан и Колеман [263], изучившие вли-
яние ионов SO4~ на превращение анатаза в рутил с помощью меченых атомов се-
ры. Они отвергают гипотезу, что SO4 оказывает влияние на превращение анатаза
в рутил, и считают, что превращение анатаза в рутил зависит от дефектов структур
ры решетки TiO2 и величины удельной поверхности препаратов. Нагревая раствор
сульфата титана, они установили по изменению активности 35S, что ионы SO4“ свя-
заны с двуокисью титана двумя способами. В первом слое расположены компактно-
о 2
связанные ионы SO4 . В остальных слоях ионы SO4 находятся в капиллярно-
конденсированном состоянии. При нагревании анатаза выше 300° С вначале ос-
вобождаются капиллярно-конденсированные ионы SO" по уравнению реакции
первого порядка. Удаление компактно-связанных ионов SO4““
зависит от степени
покрытия поверхности. Температурная зависимость
распределения
ионов
so|-
описывается модифицированной экспоненциальной функцией Больцмана, а констан-;
та скорости реакции определяется уравнением Аррениуса. Концентрации ацата-
за в начале и конце процесса связаны между собой уравнением первого порядка.
134
Ускоряют превращение анатаза в рутил примеси НС1 [13, 169, 173, 176, 289],
P4NO3 [289], В2О3 [150], Na2WO4 [172, 276], ванадаты щелочных металлов [112],
КоСг207 [112], Ва (ВО3)2 и Ва3 (PO4)t [230], NaF, LiCl (?) [121, 230], ZnCl2 [121],
>F2, CaCl2, пары воды [169], соли фосфорной кислоты [150], ZnO [121, 206], MgO,
Sb3Q? [206], KOH, NH3 [230] и др. Бунтики [112] наблюдал полное превращение
янатаза в рутил под действием минерализаторов при температуре 575 и частич-
ное - при 400° С.
Судзуки и Котера [264] отмечают влияние размера частиц двуокиси титана на
скорость превращения анатаза в рутил.
На скорость превращения анатаза в рутил существенно влияет окружающая
атмосфера [264, 273]. Газообразный водород ускоряет превращение анатаза в ру-
тил; в вакууме превращение замедляется [273].Положительное влияние на скорость
перехода анатаза в рутил атмосферы водорода объясняется образованием анионных
вакансий в решетке TiO2, что способствует диффузии ионов О2“ на поверхность
частиц, где превращение протекает активнее. В вакууме в решетку внедряются
ионы Ti3-^, замедляющие ее перестройку.
Переход анатаза в рутил ускоряется с повышением давления [277]. Гидроокись
титана, нагревшаяся при температуре 850° С под давлением 1000 кг!'см2 в течение
18 ч, превратилась в рутил только частично, а при давлении 7000 кг!см2 — пол-
ностью.
По данным Рао с соавторами [ 114, 254], кинетика превращения чистого анатаза
в рутил и с примесями РО4“, SO4““, А13"^", С1“ и Zn2-*" описывается уравнением ре-
акции второго порядка.
При температуре 610° С скорость превращения спектрально чистого анатаза в
рутил очень мала [114, 255], а выше 730° С становигся большой [ 114]. Энергия ак-
тивации равна около 100 ккал/молъ.
В более поздней работе Рао [255] считает, что кинетика превращения анатаза в
рутил в интервале температур 625—698° С согласуется с уравнением реакции пер-
вого порядка. Энергия активации около 8 ккал/молъ. Энтальпия превращения
оценена по величине площади максимума на дифференциальной кривой термограм-
мы нагревания и равна около — 100 ккал/молъ.
В присутствии примеси SO^ энергия активации превращения анатаза в рутил
равна 120 ккал/молъ и возрастает с увеличением содержания SO2"- [310].
Судзуки с соавторами [124, 264] изучил превращение анатаза в рутил методом
измерения диэлектрической проницаемости. В интервале температур 750—800° С
кинетика превращения описывается уравнением реакции первого порядка. Энер-
гия активации реакции на воздухе НО —116 в атмосфере кислорода 92—
96 ккал/молъ. Добавка к двуокиси титана 1 % LiCl повышает энергию активации до
150 ккал/молъ.
Иоганарасимхан [309] исследовал скорость гревращения анатаза в рутил,
полученного аммиачным гидролизом из четыреххлсристого титана. Кинетика про-
цесса описывается уравнением реакции первого порядка, энергия активации равна
110 ккал/молъ.
Превращение анатаза в рутил при температуре 460—600° С сопровождается
резким изменением удельной поверхности образца [17, 61] и его плотности [61].
Изменение этих свойств сказывается на каталитической активности двуокиси ти-
тана, о чем можно судить по глубине фазовых превращений [61].
Кубо и соавторы [196] при длительном натирании анатаза (в течение несколь-
ких дней), полученного гидролизом сульфата титана, наблюдали превращение его
в аморфный продукт. Анатаз, полученный из четыреххлористого титана, в резуль-
тате натирания превращается в рутил.
Содержание анатазной и рутильной форм двуокиси титана в порошкообразных
образцах определяется методом рентгенофазового анализа [75]. Оеме [229] предло-
жил определять содержание анатаза и рутила по измерению диэлектрической про-
ницаемости. Измерения производятся в иммерсионной жидкости, погрешность оп-
ределения равна ±2% .
135
ПРЕВРАЩЕНИЕ БРУКИТА В РУТИЛ
Превращение брукита в рутил изучалось на образцах природных
минералов. Бунтини [112] установил, что брукит начинает превра-
щаться в рутил при температуре 525° С. Рао и Иоганарасимхан
[256, 308] установили, что скорость превращения брукита в рутил
при температуре 715° С еще ничтожно мала. Примеси минерализа-
торов и тонкое измельчение кристаллов ускоряет превращение.
Пик на термограмме нагревания брукита, отвечающий превращении:
его в рутил, лежит при температуре около 750° С. Кинетика пре-
вращения описывается уравнением реакции первого порядка. Энер-
гия активации равна 54—67 ккал!молъ.
При облучении брукита нейтронами наблюдается изотропное
увеличение параметров решетки [182].
ФИЗИЧЕСКИЕ СВОЙСТВА
Чистая двуокись титана — вещество белого цвета [55]. При
нагревании она окрашивается в желтый цвет, исчезающий после
охлаждения.
В некоторых работах [192, 293, 306] сообщалось об окрашивании
двуокиси титана при нагревании в желто-коричневый, коричнево-
зеленый, темно-коричневый и серо-коричневый цвета. Появление
темных оттенков и необратимого окрашивания двуокиси титана при
нагревании объясняется, по-видимому, недостаточной чистотой ис-
пытуемых образцов.
Расплавленные перлы двуокиси титана светло-желтые [300].
Кристаллы технических продуктов со структурой анатаза, по-
лучаемых на лакокрасочных заводах, имеют округлую форму, а
рутила — вытянутую [104, 108]. Углы у кристаллов двуокиси ти-
тана, как показали наблюдения под электронным микроскопом,
исчезают задолго до плавления при температуре 800—1400° С, что
, является следствием высокой подвижности атомов в поверхностных
слоях граней [22]. Частицы двуокиси титана при нагревании их до
температуры 1000—1300° С спекаются. Кинетика спекания опре-
деляется диффузией ионов кислорода [125].
ВЛИЯНИЕ ПРИМЕСЕЙ НА ЦВЕТ ДВУОКИСИ ТИТАНА
Цвет двуокиси титана зависит от совершенства кристаллической
решетки. Деформация решетки под влиянием примесей служит
причиной окрашивания.
Как показали Джонсон и Уейл [191], включение бесцветных
катионов с валентностью более четырех (Sb5+, Та5+, W6+, Мо°+) в
решетку рутила вызывает появление синего оттенка. Окраска йо-
136
нов Fe3+ и Сг3+ усиливается решеткой рутила. Катионы с валент-
ностью менее четырех, например щелочноземельных металлов, ок-
рашивают рутил в желтый цвет. Голубоватый цвет рутила вызван \
частичной потерей его решеткой атомов кислорода и образованием v-'
ионов Ti3+.
Окраска двуокиси титана очень чувствительна к содержанию
даже малых следов некоторых примесей (табл. 13).
Таблица 13
Минимальное количество примесей, вызывающих
окрашивание двуокиси титана (13)
Примесь
Содержание
примеси, %
Окраска двуокиси титана,
вызванная примесями
Сг2О3 0,00015 Коричнево-желтоватая
СоО 0,0007 Серо- желтоватая
СеОо 0,0015 Желтая
СиО“ 0,003 Се ро- желтоватая
Ре2^3 0,003 Желтоватая
МпО 0,003 Серая
v205 0,007 Серо-голубая
РЬО 0.01 Серая
Наиболее чувствительна к окрашиванию под влиянием при-
месей рутильная форма двуокиси титана, вероятно, вследствие того,
что рутил имеет более плотно упакованную решетку, чем анатаз.
Парравано и Каглиоти [23Э ] наблюдали пожелтение рутила при со-
держании 0,003% Fe3+, тогда как анатаз, содержащий 0,009 %Fe3+,
остается еще бесцветным.
Двуокись титана обладает свойством фототропии, заключаю-
щемся в окрашивании, под действием света и нагревания, которое в
темноте исчезает [233, 303—305]. Фототропию проявляют образцы
анатаза, обработанные растворами солей Fe3+, Ni24", Сг34~, Мп24-,
Си2+ и прокаленные при температуре около 1000°'С до частичного
превращения анатаза в рутил [214, 306]. Под действием света фото-
тропные образцы окрашиваются в коричневый цвет, который исче-
зает в темноте. При нагревании фототропных образцов двуокиси
титана от 250 до 600° С появляется окраска от желто-коричневого
До коричнево-зеленого оттенка, исчезающая после охлаждения. При
более высокой температуре фототропные образцы двуокиси титана
окрашиваются в серо-желтый цвет необратимо.
По мнению Мактаггарта и Бира [214], механизм фототропии сво-
дится к окислению примесей, концентрирующихся на поверхности
кристаллов анатаза или на границе раздела анатаз-рутил, под дей-
ствием кислорода, выделяемого ТЮ2 при освещении или нагревании
\Например, Fe24" Fe34~). В темноте и при охлаждении идет обрат-
ный процесс.
137
ЛЮМИНЕСЦЕНЦИЯ
Катодные лучи, как установил еще Крукс [128], вызывают
желтую люминесценцию двуокиси титана, которая усиливается
примесью Nd2O3 [222]. Причиной появления люминесцентного све-
чения служит протекание обратимых восстановительно-окислитель-
ных процессов под влиянием облучения [223].
Техническая двуокись титана флуоресциирует под действием
фиолетового света [92, 179].
Природный рутил проявляет в ультрафиолетовом свете светло-
синюю, направленную наклонно к оси с, поляризованную люминес-
ценцию [235]; природный анатаз флуоресцирует зеленым поляризо-
ванным светом параллельно оси с.
При нагревании двуокиси титана в водородном пламени с из-
бытком водорода возникает красное люминесцентное свечение [224].
В избытке кислорода двуокись титана светится синим светом до
температуры 1200° С. Расплавленная двуокись титана также све-
тится синим светом (три полосы с длиной волн 625, 550 и 425 нм).
ПОГЛОЩЕНИЕ И ОТРАЖЕНИЕ СВЕТА
Границы поглощения длинных волн ультрафиолетового света
субмикроскопическими порошками рутила и анатаза лежат соответ-
ственно при длине волны 397 и 377 нм [93, 84]. Интенсивная по-*
лоса поглощения рутила и анатаза около 350 нм относится к типу
полос поглощения решетки [275]. v - .;Д
Инфракрасные спектры поглощения рутила, анатаза и брукита
сходны между собой и содержат полосы с максимумами частот около
700—650 и 550 см~х [307]. В спектре рутила имеется очень слабая
полоса поглощения около 420 см~х. Рутил резко поглощает свет,
при 2530—2800 см~х [278]. Некоторые авторы наблюдали полосы
поглощения рутила с частотой 1852 и 1530 слг4, которые не были
обнаружены Иоганарасимханом [307]. |
Окислы титана обладают различной способностью к отражению
световых волн. Максимумы поглощения света окислами титана в'
интервале длин волн 200 — 800 нм равны (в эв): TiO — 3,5, Ti2O
2,25 и 3,4 TiO2 (рутил) — 3,00, TiO2 (анатаз) — 3,23, TiO2 (бру-
кит) — 3,26 [120]. |
В отражательном спектре рутила в области длин волн 390 —
420 нм наблюдается резкий обрыв [288]. Падение коэффициента от-
ражения рутила в этой области спектра, по мнению [278], вызваны
недостатком кислорода в рутиле и содержанием примесей металлов.
Основываясь на различной способности к отражению короткий
волн (фиолетовая и ультрафиолетовая области спектра), Дантума
[135] разработал фотографический метод идентификации анатаза и
рутила.
з
\
138
СВЕТОПРЕЛОМЛЕНИЕ
Все три кристаллические модификации двуокиси титана име-^
высокие коэфридиенты лучепреломления и отличаются большим
дойным-”лучепрёломлением. Причиной последнего служит зна-
чительная поляризуемость (а0) кислорода в решетках кристаллов
двуокиси титана [87]. Показатели преломления можно объяснить,
приняв во внимание анизотропию а0. Для рутила принято, что ион
кислорода О2~ имеет две поляризуемости: параллельную а0 | и
перпендикулярную сс0 | плоскости ионов титана. Для X = 5893 ;
д — осо || = 2,58, а0 | = 1,73; для % = со — а0 || = 2,43, сс0 | =
= 1,63.
Экспериментально коэффициенты преломления двуокиси титана измерялись
многими авторами. В среднем коэффициенты преломления рутила — 2,72 и аната-
за — 2,55 [242]. Дисперсия коэффициентов рутила и анатаза в зависимости от дли-
ны волны приведена з табл. 14.
Таблица 14
Коэффициенты преломления анатаза и рутила [258]
при 25° С (со — обыкновенный луч, & —
необыкновенный луч)
X, А Анатаз Рутил
«со «е «со «8
4358,3 2,7688 2,6576
5460,7 2,5948 2,5161 2,6505 2,9467
5893 2,5612 2,4880 2,61124 2,8993
6234,3 2,5890 2,8712
6907,5 2,5097 2,4445 2,5555 2,8294
Коэффициенты пэеломления брукита при температуре 25° С, по измерения
Шредера [258], таковы:
М А па «V
4916,1 2,6717 2,6770 2,809
5893 2,5831 2,5843 2,7004
Кристаллы синтетического рутила имеют показатели преломления в натр не -
вом пламени [211] Псо = 2, 605 и иг = 2,901.
Т аблица 15
Значения коэффициентов в уравнении Раджакришнана
коэф- фи- циент Анатаз Рутил Брукит
(0 е (0 е а V
а 2,4485 2,8325 2,2868 2,6034 2,9858 2,6957 2,4285
b 2,3998 1,8492 2,7275 3,7199 2,1036 2,3130 2,9641
с 0,10 0,18 0,10 0,20 0,18 0,16 0,05
139
Раджикришнан [243] выразил зависимость показателей преломления всех трех. ’
модификаций двуокиси титана от длины волны следующим уравнением:
В
п2-1=а+-^=хг-сЯ’
где 10 = 0,2870 мкм\ значения коэффициентов а, b и с приведены в табл. 15.
Из-за большого лучепреломления двуокись титана как пигмент з
обладает высокой укрывитостью, т. е. способностью окрашивать а
поверхности в белый цвет с малым удельным расходом вещества. Я
Этим объясняется широкое применение двуокиси титана в качестве 1
белого пигмента в лакокрасочной и других отраслях промышлен- I
ности. Наибольшей укрывитостью обладает рутил. Я
Характерно, что показатели преломления у рутила и анатаза 1
больше, чем у алмаза («Na = 2,4172). Поэтому синтетические кри-1
сталлы рутила и анатаза применяются для украшений как полу- 1
драгоценные камни. 'I
МАГНИТНЫЕ СВОЙСТВА
Двуокись титана парамагнитна [37]. Удельная магнитная вое-1,
приимчивость рутила при комнатной температуре, по Эрлиху [148],|
X • 10е = 0,08. В интервале температур от +20 до —180° С удель-!
ная магнитная восприимчивость рутила практически не изменяется..
Зименс и Хедвалл [312] определили для рутила х • Ю6 — 0,13 и]
анатаза х • 10е = 0,09. Хил и Селвуд [180] нашли для двуокиси;
титана, модификация которой неизвестна, х • Ю6 = —0,30, что!
свидетельствует о диам'тнитности образца. Мазуркевич с соавтора-'1
ми [37] также для образца неизвестной кристаллической структуры!
определил х • 10е = —0,170. .1
По характеру изменения магнитной восприимчивости двуокись,
титана ведет себя как антиферромагнетик [262]. 1
Частичное восстановление двуокиси титана увеличивает ее пан
рамагнитность [37]. Я
ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА I
Диэлектрические свойства двуокиси титана зависят от кристал-1
лической структуры и проявляют анизотропию. а
Либих и Рубенс [201] измерили диэлектрическую проницаемость^
природного рутила в направлениях, параллельном и перпендикуляр- ,
ном оси с: 8ц = 167, е± = 83; по данным Шмидта [269], ец +1
= 173 и ех = 89. Средняя диэлектрическая проницаемость рутила]
е = 117 [260]. 'Я
Температурные коэффициенты диэлектрической проницаемости
рутила в интервале от —200 до +100° С отрицательны и равны у,, = '
= —8 • 10~4, Yx = —5 • 10“4 [145]. • ]
140
Л
Диэлектрическая проницаемость синтетических образцов рутила
определена Церфоссом с соавторами [311] — ей = 180, =
=== 89—92, Ксендзовым [30] — е = 95 (20° С, 105 — 106 гц), Ер-
оера и Кетелааром [146] — 8 = 114, Берберихом и Беллом [91]
£ = 104 (30° С, 60 гц).
Синтетический анатаз имеет среднюю диэлектрическую прони-
цаемость 8 = 48 [146], а природный рутил в направлении оси а
= 78 [260]. Данные по измерениям диэлектрической проницае-
мости синтетических образцов двуокиси титана имеются в работах
[5, 268].
ЭЛЕКТРОПРОВОДНОСТЬ
При комнатной температуре двуокись титана — диэлектрик, а
при высокой температуре — проводник.
Удельная электропроводность двуокиси титана Ери комнатной
температуре, по [210], Ю-25^-1- сж-^по [294], 10~16— lO^cw-1 X
X см~х (прокаленный образец чистой двуокиси титана), по [91],
10“13 — 10“14 ом-1 • см-1 (технический продукт).
Носителями тока в рутиле являются поляроны большого радиу-
са [147]
Потеря кислорода двуокисью титана сопровождается увеличе-
нием электропроводности и превращением ее из диэлектрика в про-
водник. Зависимость уделыюй электропроводности двуокиси титана
от содержания кислорода, по измерениям Вервега [287], характе-
ризуется следующими данными:
Состав образца ТЮ2,0000 ^^1,9995 ^^1,995
Удельная электропровод-
ность, 1О“~10 10—1 0,8
ПОЛУПРОВОДНИКОВЫЕ СВОЙСТВА
Честер и Брадхурст [119] обнаружили появление электропро-
водности в образце рутила, помещенного в раствор сульфата натрия,
через который пропускался водород. Авторы считают, что проводи-
мость рутила возникла в результате внедрения водорода в решетку
Рутила.
Полупроводниковые образцы двуокиси титана получили и япон-
ские исследователи [194]. Удельное сопротивление этих образ-
цов в интервале температур 10 — 400° С изменялось от 0,05 до
30 000 ом • см. а термо-ЭДС — от 180 до 950 лш/°С.
РАБОТА ВЫХОДА ЭЛЕКТРОНА
Работа выхода электрона с поверхности двуокиси титана и её <
Твердых растворов с WO3 и Fe2O3 измерена Бондарем [7], а также
^зоновой и Кейер [63]. По данным Бондаря, она равна 98 мв.
141
Адсорбция кислорода, воды и перекиси водорода увеличиваю?
работу выхода электрона из TiO2 [59]. При совместном присутствие
кислорода и воды контактная разность потенциалов двуокисц
титана (работа выхода электрона) с ростом температуры проходи?
через максимум, что авторы объясняют образованием перекиси водо-
рода в результате протекания каталитической реакции,
примеси железа, молибдена, меди и серебра к рутилу
работу выхода электрона, а большие — увеличивают
Примесь окиси алюминия увеличивает работу выхода
Небольшие
уменьшают
(табл. 16)
электрона
Таблица 16
Влияние примесей на контактную разность
потенциала рутила относительно золотого электрода
сравнения [7]
Содержа- Контактная разность потенциалов, мв
ние при- месей, ат. % Железо Молибден Медь Серебро
0,0003 —53 —125 90 104
0,003 —51 —122 —65 —54
0,006 82 —94 - —165
0,03 211 16 0 60
0,3 340 180 380 68
Бондарь [7] изучил потенциал эмиссии электронов с поверхности двуокиси ти
тана. При низких температурах в вакууме (10“3 —10“4мм рт. ст.) эмиссия не на
блюдается даже при напряжении 3000—4000 в. Кислород снижает потенциал по
явления эмиссии до 1200 в. Адсорбция двуокисью титана полярных молекул (вода
ацетон) сопровождается резким падением потенциала появления эмиссии вслед
ствие концентрирования электронов у поверхности под влиянием поля, созда
ваемого этими молекулами. Между потенциалом появления эмиссии и работой вы
хода электрона с поверхности двуокиси титана существует определенная завися
мость. Увеличение работы выхода при добавлении к двуокиси титана молибдена
приводит к росту потенциала появления эмиссии.
ПОЛЯРИЗАЦИЯ ТИТАНА В РУТИЛЕ
Руфф [241], решив уравнение локализованного поля, наше-
поляризуемость титана в рутиле стехиометрического состава рав
ной 2,2 А3. Эта величина в несколько раз превышает значение поля
ризуемости иона Ti4+, которая составляет
0,272 А3 [24]. Повы
шенное значение поляризуемости титана в рутиле согласуется
результатами изучения распределения электронной плотности
объясняется влиянием кристаллического поля.
ТОЧКА ПЛАВЛЕНИЯ
Результаты определения точки плавления двуокиси титана еле
дующие (°C): 1850 [302], 1825 [300], 1855 [301], 1847 [280J, 184с
142
[232], 1830 [143], 1840 [127]. Точки плавления двуокиси титана,
по данным различных авторов, колеблятся в пределах 30° С.
Наиболее вероятной поэтому будет средняя величина, равная
1841°С.
ЛИНЕЙНОЕ РАСШИРЕНИЕ И ТЕПЛОПРОВОДНОСТЬ
Штрауф [261] измерил коэффициенты линейного расширения ру-
тила, анатаза и брукита в интервале температур 0—30° С вдоль
кристаллографических осей, получив при этом следующие значения:
Модификация аа - 106 ab • 106 ас • 106
Рутил 7,192 — 9,743
Анатаз —2,8801 — 6,6424
Брукит 14,4938 19,2029 22,0489
Штрауманис с соавторами [2661 определил коэффициенты линей-
ного расширения рутила в интервале 10—60° С: аа = 6,9 • 10-6,
ас — 9,9 • 10“6. При температуре 40° С для рутила, по данным
[154], аа = 7,14 • 10“6, ас ~ 9,19 • 10“G, объемный коэффициент
расширения у = 2,347 • 10“5.
Данные о теплопроводности двуокиси титана приведены в ра-
ботах [101, 189, 195].
УПРУГОСТЬ ПАРА
Давление пара двуокиси титана, рассчитанное по прибли-
женной формуле Нернста, при 2000° К равно 3 мм рт. ст., при
3000° К —боле? 1 атм [102].
Гровс с соавторами [158] определил упругость пара двуокиси
титана методом Кнудсена, выразив зависимость ее от температуры
уравнением
lg Р = __ 0,492 • 10~3Т 4-11,19.
Энтальпия превращения ТЮ2тв -> ТЮ2 , вычисленная по уп-
ругости пара, ДЯо == 138,9 ккал!моль.
ТЕПЛОЕМКОСТЬ
Теплоемкость рутила в интервале 69—175° К Макдональд и
Зейтц [209] выразили уравнением
Cv^D{~ + 2E~y
Шомате [267] измерил теплоемкость синтетических анатаза и
Рутила (табл. 17).
143
Таблица 17
Молярная теплоемкость синтетических рутила
и анатаза [267]
Рутил Анатаз
Г, С Ср, кал/моль. • • град. Т, ° К кал/моль. • • град.
52,5 1,572 52,5 1,345
61,9 2,073 60,1 1,870
70,8 2,594 68,7 2,416
79,5 3,127 77,2 2,995
100,1 4,438 95,2 4,259
120,2 5,740 115,2 5,587
140,4 7,004 134,9 6,775
160,7 8,165 155,5 7,914
180,7 9,190 175,6 8,942
201,1 10,09 195,9 9,827
221,2 10,91 216,3 10,68
240,9 11,56 235,8 11,35
261,3 12,22 256,1 11,97
281,0 12,76 276,5 12,62
297,7 13,14 295,8 13,17
298,16 13,16 298,16 13,22
интервале
По данным Артура
удельная теплоемкость
[83], в
двуокиси титана
температур 200—800° С
(структура не указана)
А
При температуре 350° С Сто2 = 0,210 кал/г • град.
По Нейлору [221], теплоемкость рутила и анатаза
(в кал/моль град)
Срутил = 17,14 + 0,000987 — 350 000/72
(298 — 1800° К),
Санатаз = 17,21 + 0,001087 — 359 000/72
(400— 1300° К).
СТАНДАРТНАЯ ЭНТРОПИЯ И ЭНТАЛЬПИЯ ОБРАЗОВАНИЯ
Макдональд [209] определил S°98 = 12,45 кал/моль • град. По |
данным Шамоте [267], для синтетического рутила S°98 — 12,01,
анатаза — S298 =11,93 кал/моль • град.
Энтальпию образования двуокиси титана измеряли многие ав-
торы (табл. 18). Температурная зависимость энтальпии образования
рутила и анатаза (ккал/моль), по Нейлору [221 ], выражается урав
нениями:
ДЯрутил = 17,147 + 0.0004972 + 350 000/7 — 6328
(298 — 1800° К),
144
ЛНтт = 17,21т + 0,00054Т2 + 359 000/7 - 6383
(400— 1300° К).
Таблица 18
Энтальпия образования TiO2 по реакции
Т>тв + = Т'Огг
—А//,
ккал/моль
Кристалли-
ческая моди-
фикация
ТЮ2
Литера-
тура
97,7
218,4
220,1
218,1
218,7
225,3
219,0
224,9
Комнатная
»
»
20
20
19
20
Рутил
Рутил
»
»
»
»
296
207
259
245
246
178
247
[4]
СВОБОДНАЯ ЭНЕРГИЯ
Свободная энергия образования рутила по реакции Ti + О2 =
— TiO2 AF298 = —212,430 ккал!моль [209].
Температурная зависимость свободной энергии {ккал/моль) вы-
ражается уравнениями, по Ричардсону и Зефферсу [244],
= — 217,500 + 0,04147
(25— 1800° С),
по Макдональду и Зельтцу [209],
223,80+ 0,04127
(1500 — 1800°С).
ПРОЧИЕ СВОЙСТВА
Энергия смачивания порошка анатаза в воде и органических
Жидкостях равна около 1 кал!г, или нескольким эргам на 1 см2 по-
верхности [84, 85].
При нагревании с водяным паром до температуры 500—1000° С
и Давлении 1500—7000 апгм рутил становится заметно летуч [212].
ХИМИЧЕСКИЕ СВОЙСТВА
Двуокись титана при комнатной температуре отличается малой
Реакционной способностью. При нагревании активность двуокиси ти-
Тана повышается, она вступает в реакцию с кислотами, основаниями,
10^-614 145
реагирует с галогенидами щелочных металлов, восстанавливается
металлами, водородом, углеродом и т. д. В
Лукс и Прошель [202] попытались охарактеризовать кислот-
но-основные свойства двуокиси титана в расплаве NaCl + KF с
двуокисью марганца в качестве индикатора. При атмосферном дав-
лении кислорода двуокись титана по отношению к двуокиси мар-
ганца ведет себя как основание, а при давлении 0,005 атм — как
слабая кислота. Окислы Na2O, ВеО и А12О3 с двуокисью титана
реагируют как основания, а В2О8 и SiO2 — как кислоты. Двуокись
титана является, таким образом, несколько более слабой кислотой, I
чем двуокись кремния и окись бора. Н
Характерная особенность некоторых образцов двуокиси тита-
на — высокая фотохимическая активность, проявляющаяся при 1
действии видимого и ультрафиолетового света уже при комнатной;!
температуре. |
Из полиморфных модификаций двуокиси титана наиболее актив- ]
ной в химическом отношении является анатаз, а более инертной — '
рутил. I
Измерение ^-потенциала и спектрофотометрические исследования , ;
поверхности частиц двуокиси титана, на которых адсорбирован мен -
тиловый красный, указывают на изменение заряда их в зависимое-1
ти от температуры прокаливания, т. е. от кристаллической струк-1
туры [208]. Двуокись титана, прокаленная ниже 800° С и имек|1
щая структуру анатаза, подвергается гидролизу по схеме';
ТЮН Ti+ + ОН и заряжается положительно, а со структур I
рой рутила —по схеме ТЮН ТЮ““ + Н+ и заряжается отрица-
тельно. 1
ТЕРМИЧЕСКАЯ УСТОЙЧИВОСТЬ |
При температуре 1100—1200° С в вакууме или в атмосфере 'I
азота двуокись титана отщепляет кислород, окрашиваясь в синий
цвет [297]. Ш
По данным масс-спектроскопических исследований. [68], Подя
вакуумом (10~6 мм рт. ст.) и температуре 1600° К двуокись^
титана образует ионы Ti"*", а при более высокой температуре —М
TiO. С повышением температуры до 2000° К количество ионов Ti j]
возрастает, а затем уменьшается, и наряду с заряженными образуются
нейтральные частицы Ti, TiO и ТЮ2. При температуре 2000° К |
в вакууме около 0,1% паров двуокиси титана находится в форме.
ионов. Двуокись титана испускает ионы также при нагревании И
ее до белого каления на вольфрамовой проволоке [100].
ж
ДЕЙСТВИЕ КИСЛОТ И ОСНОВАНИЙ |
Будучи амфотерным окислом, двуокись титана образует соли Е
с кислотами и основаниями.
146
С двуокисью титана из минеральных кислот образуют соли только
плавиковая и серная [118]. Остальные кислоты реагируют очень
слабо, и соли их получаются только косвенными путями.
Растворы гидроокисей щелочных металлов на прокаленную дву-
окись титана (рутил и анатаз) действуют при нагревании под высо-
ким давлением. В результате спекания двуокиси титана с безводными
окислами и карбонатами оснований получаются соли титановых
кислот — титанаты, о чем более подробно сказано в гл. V. Сплавы
двуокиси титана с окислами щелочных металлов — стекловидные
продукты, раскристаллизовывающиеся только при специальной тер-
мической обработке.
РАСТВОРИМОСТЬ В СОЛЕВЫХ РАСПЛАВАХ
Растворимость двуокиси титана в расплавах различных солей
изучалась, главным образом, в связи с проблемой электролити-
ческого получения титана. Исследователи при этом обычно инте-
ресовались величиной раст-
воримости двуокиси титана,
которую определяли препа-
ративным методом, и не об-
Таблица 19
Растворимость двуокиси титана в
расплавах фторидов щелочных
металлов [21], вес. %
ращали внимания на состоя- ние фазовых равновесий. Фториды щелочных метал- Растьори- тель 1000° c 1100°C 1200° C
лов растворяют значитель- 1
ное количество двуокиси ти- LiF NaF 0,39 0,71 2.5 04 1,67 50 11
тана [2, 21, 46]. Раствори- KF 33,09 46,04 UVj 1 1 58,39
мость двуокиси титана во фто-
ридах щелочных металлов повышается с увеличением атомного
веса щелочного металла (табл. 19). Растворы двуокиси титана во
фтористом натрии подчиняются уравнению Шредера [21].
В хлоридах щелочных металлов двуокись титана растворяется
в незначительных количествах (0,07—0,2%) [2, 26].
Хлористый кальций в чистом виде двуокись титана практически
не растворяет [18]. Если в расплаве хлористого кальция содержит-
ся примесь окиси кальция, то последняя реагирует с двуокисью
титана, образуя титанаты, в виде которых и переходит в раствор.
По измерениям некоторых авторов, в хлористом кальции растворяе-
тся 0,5—0,7% ТЮ2 [26, 31, 67].
Растворимость двуокиси титана в расплавленном криолите, по
Хасанову [71 ], составляет 3,0%, по данным Ануфриевой и Ивано-
ва [2] — 3—7%; в эвтектической смеси Na3AlF6 — А12О3 при тем-
пературе 1000° С растворяется 5% ТЮ2 [6].
Дармоис и Ролин [142] считают, что в расплаве криолита дву-
окись титана не подвергается диссоциации.
Во фтортитанате калия K2TiF6, по Коломицкому [28], растворяет-
Ся До 15% TiO2 (900° С), по данным Ануфриевой и Иванова [2],—
10*
147
14% TiO2; в более поздней работе Коломицкий и соавторы [29]
установили, что максимальная растворимость двуокиси титана
в K2TiF6 равна 7%.
Двуокись титана растворяется в сплавах фтортитаната калия
с галогенидами щелочных металлов. Оно [228] нашел, что в сплаве
K2TiF6 — NaCl растворимость TiO2 равна 3%, в сплаве K2TiF6—
KF — 5%. По Коломицкому [28], в эвтектическом сплаве K2TiF6—
NaCl растворяется до 5% ТЮ2 (650—700° С), а вообще растворимость
двуокиси титана в этой системе зависит от содержания K2TiF6 и
составляет 5—10% [31 ]. Кристаллооптическим методом установлено,
что со сплавами системы K2TiF6 — NaCl двуокись титана не
вступает в химические соединения [48].
Меерсон и Смирнов [43] установили, что эвтектика в квазибинар-
ной системе (2В2О3 — СаО — CaF2) при температуре 925° С содержит
8,5% TiO2.
В метафосфате натрия растворяется 0,42% ТЮ2 (720° С), в пи-
рофосфате — 13,7% (1000° С) [3].
Двуокись титана хорошо растворяется в расплавленной буре
[74]. Растворимость ее сильно увеличивается с повышением темпе-
ратуры.
ДЕЙСТВИЕ СУЛЬФАТОВ И СУЛЬФИТОВ
Двуокись титана реагирует с бисульфатами и сульфатами
щелочных металлов. Сплавление двуокиси титана с бисульфатами
щелочных металлов и аммония при не очень высокой температуре со-
провождается образованием двойных сульфатов титана. Сульфаты
и сульфиты щелочных металлов реагируют с двуокисью титана при
температуре около 1000° С, образуя титанаты [70]. С сульфатом
аммония двуокись титана реагирует при температуре 400—500° С
[47]. Фактически реакция протекает между двуокисью титана и би-
сульфатом или пиросульфатом аммония, на которые сульфат аммо-
ния разлагается выше 300° С. ]
ДЕЙСТВИЕ ВОССТАНОВИТЕЛЕЙ
Двуокись титана восстанавливается до низших окислов или
металлического титана водородом, окисью углерода и многими
металлами. I
Восстановление водородом
Атомарный водород при комнатной температуре с двуокисью
титана не реагирует [187]. При температуре 2000°С и давлений
130 атм двуокись титана восстанавливается водородом до монооки-
си [225].
Назу [226] сообщил, что в интервале температур 750—1000° С двуокись тита-
на восстанавливается водородом до Ti2O3 без образования промежуточных продук-
тов. Константа равновесия реакции Л^р Рн2о/Рн2, по его данным, выражается
148
I уравнением lg Kp = 1,903 — 2754 Г”1. Снопова i Ротков [66], а также Вейс [297] в
интервале температур 700—1200° С наблюдали при восстановлении двуокиси тита-
на водородом образование только одного окисла Ti2O3. Однако утверждение этих
I авторов об отсутствии в продуктах восстановления других промежуточных окис-
лов не нашло подтверждения в последующих работах. Как показали Богданова и
Морозова с соавторами, работы которых рассмотрены в главе II, при восстановле-
: нии двуокиси титана водородом в зависимости от соотношения о в газовой
। фазе образуется серия окислов.
I Крестовников и Ломов [32] на основании литературных данных определили
энтальпию и изобарно-изотермический потенциал: реакции 2TiO2 + Н2 = Ti2O3 +
I *4" В2О.
Д#т = 4095 + 0,8747 + 3,856 • 10~372 [кал/моль],
&ZT = 4095 —2,012571g 7 —3,856 • 1СГ372 — 10,17 [кал/моль].
I Продукты восстановления двуокиси титана водородом обладают
I полупроводниковыми свойствами [95]. Мактаггарт [215] при восста-
I новлении двуокиси титана водородом е электрическом разряде по-
I лучил соединения типа вольфрамовых бронз. В этих бронзах, по
I мнению автора, содержатся ионы водсрода Н^, заменяющие ще-
I л очной металл в бронзах обычного типа.
Водород в смеси с азотом при температуре 1750° С восстанавли-
вает двуокись титана до нитрида TiN [299].
Восстановление окисью углерода
Снопова и Ротков [66], восстанавливая двуокись титана окисью
углерода при температуре 700—1100° С, получили продукт, состав
которого выражают формулой Ti3O5. Азсайг и соавторы [78] пола-
гают, что восстановление двуокиси титана окисью углерода проте-
кает по реакции
l/xTiO2 + СО = lfxTiO2_x + СО2.
Вначале образуется фаза со структурой рутила (х< 0,06), а затем
другая, состав которой авторы еще не усгановили, но она, по-видимо-
му, относится к типу соединений Андерссона (см. гл. II).
Действие углерода
Брентли и Бекман [90] взаимодействие двуокиси титана с углеро-
дом выражают уравнением
TiO2 + ЗС = TiC +• 2СО.
Мейерсом и Крейн [44], как уже отмечалось в гл. II, показали,
что восстановление двуокиси титана углеродом протекает ступен-
' чато с образованием промежуточных соединений и заканчивается
получением твердого раствора Ti (О, С). Эту точку зрения разделяют
Куцев и Ормонт [27]. В числе промежуточных продуктов восстанов-
ления они обнаружили фазы TiOj.z (1600—2000° С), Ti3O5 (1700—
149
1900° К), Ti2O3 (2000° К), карбидную фазу, графит. Фаза TiO1>7
при избытке углерода переходит в оксикарбидную состава TiC/^.
Аналогичная схема восстановления двуокиси титана углеродом
описана Кубо с соавторами [197]. I
Крестовников и Ломов [32] по литературным данным вычис-
лили энтальпию и изобарно-изотермический потенциал реакции
TiO2 + ЗС = TiC + 2СО
\НТ = Ю9 400 — 47 [кал!моль},
\ZT = 109 400 — 9,27 lg Т— 110,867 }кал!моль}.
Смесь углерода и карбида бора реагирует с двуокисью титана
по уравнению [65].
2ТЮ2 + В4С + ЗС = 3TiB2 + 4СО.
Действие аммиака
Дуглас с соавторами [137] установил, что в интервале температур
900—1700° С аммиак восстанавливает двуокись титана до низшего
окисла TiOi^s. Затем процесс восстановления идет через ряд про-
межуточных окислов вплоть до TiO. Моноокись титана с аммиа-
ком образует нитрид титана.
♦
Восстановление гидридом титана
и металлическим титаном
При спекании смеси двуокиси титана с металлическим титаном об-
разуются окисли, существующие в системе титан-кислород (гл. II).
Если двуокись титана и металлический титан нагреваются в эвакуи-
рованной трубке, не соприкасаясь друг с другом, протекает реакция
TiO2 TiO2_x + х/2О2.
Анатаз при температуре 800—850° С темнеет из-за частичной
потери кислорода и при 900—1000° С превращается в рутил несте-
хиометрического состава. Затем рутил восстанавливается до TiOi 956
(1000° С) и Ti3O5 (1050° С) [23].
Богоявленская и соавторы [8] произвели термодинамическую
оценку возможных реакций восстановления двуокиси титана гид-
ридом титана в расплаве едкого натра. Возможной реакцией являе-
тся образование моноокиси титана
TiO2 + NaH = TiO + NaOH.
Изобарно-изотермический потенциал этой реакции Az64s ==
— —88090 кал!моль.
Частично может протекать также реакция
TiO + 2NaOH = Na2TiO3 + 2Н.
150
Восстановление натрием
Чистый натрий при температуре 950° С не восстанавливает
а3уокись титана [250]. Пары натрия восстанавливают двуокись
гИтана до смеси TiO + 3Na2O • Ti2O3 [199].
Восстановление магнием
При медленном нагревании двуокиси титана с магнием обра-
зуется титанат магния MgTiO3 [116, 295, 297]. Если магний
взят с избытком, то образуются смешанные окислыМ§ТЮ2 и MgTi2O4
[295]. Шретьен и Вейс [116] в более поздней работе также отмечают,
что до температуры 1200° С магний восстанавливает двуокись ти-
тана только до моноокиси. Однако Рутковский [249] полагает, что ,
магний восстанавливает двуокись титана по реакции
TiO2 + 2Mg = Ti + 2MgO + 74120 кал.
Но нагревая двуокись титана с магнием в атмосфере водорода при
температуре 930° С, автор получил продукт с содержанием 82,6%
Ti, т. е. близкий к составу TiO.
Восстановление кальцием и другими металлами
Металлический кальций восстанавливает двуокись титана при
температуре 950° С до металлического титана [250]. Шретьен и
Вейс [116] показали, что восстановление двуокиси титана кальцием
при температуре до 1200° С протекает без образования промежуточ-
ных соединений.
Гидрид титана начинает реагировать с двуокисью титана при
температуре 500° С. При температуре 900° С реакция идет очень
быстро, заканчиваясь за 5 мин [156]. Водород не принимает учас-
тия в восстановлении двуокиси титана гидридом кальция. Восста-
новителем служит только кальций [42].
, Карбид и цианамид кальция при температуре около 1000° С реа-
I гйруёт "с П^вуокись^^ нитрид и карбонитрид титана
! [41 ]. В атмосфере азота восстановление двуокиси титана цианамидом 't-
кальция приводит к получению почти чистого нитрида титана.
। Силициды кальция [CaSi2, CaSi и Ca2Si) с двуокисью титана
образуют металлический титан или силициды титана [1551.
Металлические цинк, молибден и медь при температуре 1100° С s
восстанавливает двуокись титана [297].
ДЕЙСТВИЕ ГАЛОГЕНОВ И ИХ СОЕДИНЕНИЙ
Фторирование двуокиси титана элементарным фтором не изу-
чено.
, Двуокись титана реагирует с трехфтористым бромом, образуя
Фторид титана [149].
151
При температуре 500 С двуокись титана фторируется трехфто*
ристым бором BF3. Продуктами реакции являются TiF4 и (BOF)
[27].
Двуокись титана реагирует с фтористым калием [15]. Реакция
начинается при температуре 400° Сив интервале 650—700° С идет
очень быстро. В продуктах реакции содержатся фтортитанаты
калия K2TiF6 и K2TiOF4.
Новоселова и Бацанова [45] установили, что двуокись титана
реагирует с кремнефтористым натрием. Бамбуров и Фотиев [14]
термодинамическими расчетами показали, что взаимодействие дву-
окиси титана с кремнефтористым натрием проходит по реакции
взаимного обмена
TiO2 + Na2SiF6 = Na2TiF6 + SiO2.
Бамбуров [16] изучил взаимодействие двуокиси титана с фтор-
силикатом калия. По литературным данным автор рассчитал изо-
барно-изотермический потенциал реакции |
TiO2 + 2KF + SiF4 - K2TiF6 + SiO2,
AZr = — 43 880 — 28,147" [кал [моль]. I
В интервале температур 298—1200° К AZ имеет отрицательное
значение, что указывает на возможность протекания реакции слева
направо. 1
Газообразный хлор начинает реагировать с двуокисью титана
при температуре 775—825° С [25, 160, 200, 227]. Пользуясь при!
ближенной формулой Нернста, Памфилов и Шихер [51 ] вычислили
константу равновесия реакции
х 1V^2 т ^^12 — х "Г ^2
Ig/G- 9367/7+ 0,43.
При температуре 800° С хлорируется около 1% ТЮ2 [200]?
По другим данным [160], 1 л хлора при температуре 800° С хлори-
рует 0,1 мг ТЮ2, а при 1000° С — 10 мг TiO2. Продуктом взаимодей-
ствия хлора с двуокисью титана является TiCl4 [200]. Оксихлориды
титана при пропускании над двуокисью газообразного хлора и че-
тыреххлористого титана до температуры 1122° К не образуются
[ 157].
В присутствии угля двуокись титана начинает хлорироваться
газообразным хлором при температуре 300—400° С, а при 410—
500° С реакция идет уже весьма энергично [49, 60]. Основными про-
дуктами хлорирования двуокиси титана в присутствии угля являю-
тся TiCl4, СО, СО2, СОС12 и С12. Протекающие процессы описываю-
тся уравнениями:
2»
СО2 + С12 = СОС12.
152
Двуокись марганца катализирует хлорирование двуокиси титана
[491.
Годнев и Памфилов [19] провели термодинамический расчет
равновесных продуктов хлорирования двуокиси титана в присут-
ствии угля и общем давлении 1 атм (табл. 20). Основные продукты
Таблица 20
Равновесные парциальные давления газообразных продуктов хлорирования
двуокиси титана газообразным хлором в присутствии угля (общее
давление 1 атм)
t, °C Рсо РСО2 PTiCl4 PCOCL л РС12
400 0,0054 0,4346 0,500 2,3-10-8 1,38-10~7
600 0,1750 0,3700 0,455 5,7-10-7 1,46-10-5
800 0,6000 0,0470 0,353 4,9-10-7 7,4.10“5
1000 0,6623 0,0027 0,335 9,9.10-8 1,12-10~4
хлорирования двуокиси титана — TiCl4, СО и СО2. Фосген и хлор
в газовой фазе в равновесных условиях находятся в небольших
концентрациях. При температуре ниже 600° С хлорирование про-
текает с преимущественным образованием четыреххлористого титана
и двуокиси углерода. Выше 600° С основными продуктами реакции
Таблица 22
Константа равновесия и равновесные
парциальные давления при хлорировании
двуокиси титана смесью хлора и окисью
углерода (общее давление 1 атм)
Таблица 21
Состав газообразных продук-
тов хлорирования двуокиси
титана газообразным хлором
в присутствии угля
4 °C СО2, об. % СО, об. % t °с К РТ1СЦ Рсо2 Pel =РСО At
430 84,7 15,3 227 1,059-10-31 0,33 0,66 1,1-10-8
480 530 81,0 74 5 19,0 25,5 427 4,932-10—20 0,33 0,66 9,0- Ю-6
650 54,8 45,2 627 1,552-10— 3 0,33 0,66 3,7-10”4
720 12,4 87,6 827 2,014-10-9 0,33 0,66 4,0-16“3
920 2,6 97,4 1027 1,377-10-6 0,32 0,64 1,7-10“2
1227 1,603-10—4 0,29 0,58 6,3-10~2
являются четыреххлористый титан и окись углерода. Расчетные
Данные согласуются с экспериментальными (табл. 21), полученными
Памфиловым и Штанделем [50].
Памфилов и Шихер [51 ] вычислили константу равновесия ре-
. акции
TiO2 + 2С12 + 2СО = TiCl4 + СО2
и рассчитали равновесные концентрации продуктов реакции
(табл. 22). До температуры 1000° С равновесие при хлорировании
153
двуокиси титана смесью хлора и окиси углерода сдвинуто в сто-
рону образования четыреххлористого титана и двуокиси углеро-
да. Следовательно, в термодинамическом отношении смесь газообраз-
ного хлора и окиси углерода до температуры 1000° С может эффек-
тивно хлорировать двуокись титана до четыреххлористого титана.
Газообразный хлористый водород реагирует с двуокисью титана
выше 800° С по реакции [69]
TiO2 + 4НС1 = TiCl4 4- 2Н2О — 182,9 ккал.
Уоррен и Шимицу [298] вычислили свободную энергию ре-
акции
TiO2 + 2С + 4НС1 = TiCl4 + 2СО + 2Н2.
При температуре выше 1500° К свободная энергия имеет отри-
цательную величину и, следовательно, реакция протекает самопро-
извольно с образованием TiCl4.
Труст и Отефейт [279] при температуре красного каления не
наблюдали взаимодействия между двуокисью титана и четырех-
хлористым кремнием. Раутер [248], однако, установил частичное
хлорирование свежеосажденной двуокиси титана четыреххлористым
кремнием при температуре 360° С. * ]
Двуокись титана хлорируется с образованием четыреххлористоп)
титана при нагревании ее с хлоропроизводными углеводородов,
например СС14, СНС13, С2С16 [117, 140, 141, 291 ], фосгеном [72],хло-
ридами серы [36, 98, 138] и др. Четыреххлористый углерод хлори-
рует двуокись титана, будучи в жидком состоянии [96]. При ки-
пячении смеси двуокиси титана с четыреххлористым углеродом в
колбе с обратным холодильником за 30 ч хлорируется до 70% TiO2.
Газообразный треххлористый алюминий при температуре 220—
360° С реагирует с двуокисью титана по реакции . I
A1CL + TiO2 = TiCl4 + 2А1ОС1.
3 ’ 2 I
Аналогично на двуокись титана действует и жидкий хлористый алю-
миний [21].
Действие брома, иода и их соединений на двуокись титана изу-
чено еще мало.
При нагревании двуокиси титана в смеси с углем до красного
каления в токе газообразного брома образуется TiBr4 [10, 139].
i
ФОТОХИМИЧЕСКАЯ АКТИВНОСТЬ
Фотохимическая активность — свойство вещества стимулиро-
вать протекание химических процессов под действием света.
Высокая фотоактивность свойственна только анатазу. Рутил
имеет пониженную фотоактивность, причем она меньшая у рутила,
полученного прокаливанием анатаза, чем гидролизом четыреххло-
ристого титана [561?
154
Бондарь [7] показал, что анатаз, полученный из сернокислых
растворов и содержащий 1 % K2SO4, активен в отношении фотосорб-
ции кислорода и воды при облучении светом с длиной волны 180 —
940 нм. По мнению автора, активными центрами сорбции кислорода
являются сульфогруппы, содержащиеся в двуокиси титана в виде
рримесей. На рутиле и двуокиси титана, полученной из хлоридных
растворов, при длине волны 180—360 нм наблюдается фотодесорбция
кислорода.
Двуокись титана сенсибилизирует фотохимическое окисление
коасителей [164,166], водного раствора аммиака [167, 163]. Наиболее
активно действие света в области собственного поглощения двуокиси
титана при длине волны менее 400 нм. На солнечном свету в присут-
ствии органических веществ (глицерин, миндальная кислота, трост-
ииковый сахар) и восстановителей (SnCl2) двуокись титана вследствие
восстановления становится бледно-серой [99]. Скорость фотовосста-
новления зависит от предварительной обработки двуокиси титана
и от среды, поглощающей кислород [193]. Фотовосстановление сво-
дится к отщеплению с поверхности двуокиси тигана кислорода,
который окисляет соприкасающиеся с ней органические вещества
[253, 292]. Фотоактивность двуокиси титана в красочной пленке
вызывает ее разрушение. Явление это получило название «меления»,
так как окрашенная поверхность разрушенной пленки пачкается,
как поверхность, покрытая мелом.
Тодиско [281] и Лунд [204] механизм меления двуокиси титана
объясняют действием фотона света на валентные электроны иона ки-
слорода, что приводит к реакции
2TiO2 4* hy == Ti2O3 + О.
Образующийся по этой реакции кислород реагирует с органически-
ми веществами красочной пленки, разрушая ее.
Малые примеси тяжелых металлов усиливают фотоактивность
Двуокиси титана, а большие (> 0,1%) снижают ее [56L
Фотоактивность двуокиси титана и атмосферостойкость красоч-
Bbix покрытий, определяемая ею, как показал Памфилов с соавто-
. Р^ми [58], находятся в прямой связи. Модифицирование двуокиси
Титана гидратом окиси алюминия повышает атмосферостойкость
Лакокрасочных покрытий на основе двуокиси титана.
Центрами фотоактивности двуокиси титана в красочной пленке
служат ионы Ti3+- Увеличение парамагнитной восприимчивости
двуокиси титана при ее частичном восстановлении влечет за собой
Увеличение и фотоактивности [37].'
I Различия в фотоактивности анатаза и рутила, по мнению Лунда
I 04], нельзя объяснить различием в энергиях решеток или погло-
Т'Внии света. Автор высказывает предположение, что малая фотоак-
ьН8ность рутила является следствием более плотной упаковки ре-
^етки *'
Фотоактивность двуокиси титана может проявляться и в проте-
кании окислительных процессов. Рутил, окрашенный в серый цвег
ионами Ti3+, под действием ультрафиолетового облучения обес-
цвечивается. Механизм обесцвечивания состоит во взаимодействий
атомарного кислорода, образующегося под действием ультрафиоле-
тового цвета, с квазибинарными электронами, локализованными hJ
’поверхности пигмента, содержащей ионы Ti3+ [286]. |
В работе [272] описан способ исследования фотоактивности об-
разцов двуокиси титана, основанный на окислении ионов __Ag+-
Кларк и Вондьюдис [122] изучили изменения, вызываемый
ультрафиолетовым облучением смеси двуокиси титана и нитрат^
серебра. Двуокись титана, смоченная раствором нитрата серебрЗ
и высушенная в темноте, после облучения в отсутствие влаги при!
обретает розовую окраску, исчезающую со временем. В контакте
с влагой образец медленно темнеет. Потемнение необратимо и вы
звано диффузией коллоидного серебра на поверхность образца. I
Двуокись титана обладает фотокаталитической активностью!
Молекулы воды, адсорбированные на ее поверхности, способны окис!
ляться до перекиси водорода [7, 190]. Памфилов и МазуркевиЛ
[57] показали, что фотокаталитическая активность двуокиси ти!
тана зависит от обработки ее поверхности.
ХЕМОСОРБЦИЯ Я
Двуокись титана сорбирует кислород. Раппопорт с соавтора*]
ми [62] показал, что сорбция кислорода на двуокиси титана имфЛ
две формы. Первая порция кислорода адсорбируется необратима
при температуре 90° К, последующие адсорбируются физически
и при нагревании до температуры 500° К, необратимо переход»
в более заряженную форму. I
Исследования работы выхода электрона и электропроводности
в процессе сорбции кислорода на двуокиси титана позволили усТЯ
новить две формы хемосорбции: заряжено необратимую и нейтраль*:
но необратимую [69]. 8
Примесь WO3 к двуокиси титана в виде твердого раствора су*
щественно влияет на хемосорбцию кислорода [53]. Предполагается
ся, что активными центрами хемосорбции кислорода являются элеК*
троны, захваченные поверхностными акцепторными уровнями, КО*'
торые могут быть образованы пустыми кислородными узлами.^
Исирикян и соавторы [73] получили модифицированные гексаЦ
нолом и диметилдихлорсиланом образцы двуокиси титана. В ре<г
зультате хемосорбции гексанола образуется плотный слой привить<
к поверхности пигмента гексанольных радикалов. ХемосорбиИ^
диметилдихлорсилана сопровождается параллельно идущей пол I
меризацией и образованием рыхлой полисилоксановой пленки. |
156
При адсорбции на двуокиси титана лауриновой кислоты обра-
зуется мономолекулярный слой 1234]. На 1 г ТЮ2 адсорбируется
4^-5 мг лауриновой кислоты.
Сахарова и Шутова [64] изучили адсорбцию олеата натрия на
пигментной двуокиси титана. По их данным, характер адсорбции
зависит не от свойств пигмента, а от особенностей поверхностно-
активного вещества, что объясняется специфическими свойствами
растворов коллоидного вещества с дифильным характером.
Литература
К Аникин И. Н., Наумова И. И., Румянцева Г. В.— Кристал-
лография, 1965, 10, 230.
2. Ануфриева Н. И., Иванов А. И.— Изв. АН СССР. ОТН. Метал-
лургия и топливо, 1960, 4, 9.
3. Андреева В. Н.— Укр. хим. ж., 1958, 24, 23.
4. А р и я С. М., Морозова М. П., Вольф Э.— Ж- неорг. хим., 1957,
2, 13.
5 Беляев И. Н., Новосильцев Н. С., X о д а к о в А. Л., Шу-
льман М. С.—ЖТФ, 1951, 21, 547.
6. Беляев А. И. Физико-химические процессы при электролизе алюминия.
Металлургиздат, М., 1947.
7. Бондарь П. Б. Автореф. канд. дисс. Черновицкий гос. ун-т, Черновицы,
1964.
8. Богоявленская Н. В., Черненко В. И., Бабченко В. А.,
Выдра Э. И.— Укр. хим. ж., 1965, 31, 793.
9. Богородский Н. П., Митюрева И. А., Фридберг И. Д.—
Физ. тв. тела, 1962, 4, 2393.
10. Б е р д о н о с о в С. С., Л а п и ц к и й А. В., Власов Л. Г.— Вести»
МГУ. Сер. II, хим., 1963, 1, 38.
11. Б р у с и л о в с к и й А. М., Брусиловская Б. Е., Носо-
в и ч Е Б.— Труды Ин-та лаков и красок, 1939, 2, 23.
12. Б е р е с т н е в а 3. Я., К а р е ц к а я Г. А., Каргин В. А.— Коллоид,
ж., 1950, 12, 338.
12а. Бенделиани Н. А., Попова С. В., Верещагин Л. В.— Гео-
хим., 1966, 5, 499.
13. Б е л е н ь к и й Е. Ф., Р и с к и н И. В. Химия и технология пигментов.
Госхимиздат, Л., 1960.
14. Б а м б у р о в В. Г., Фотиев А. А.— Труды Ин-та химии. Уральский
филиал АЙ СССР, Свердловск, 1963, 7, 195.
15. Б а м б у р о в>В. Г., Д е м е н е в Н. В.— Труды Ин-та химии. Уральский
филиал АН СССР, Свердловск, 1963, 7, 19.
16. Бамбуров В. Г.— Труды ин-та химия. Уральский филиал АН СССР,
1963, 7, 27.
17. Ганическо Л. Г., Киселев В. Ф.— ДАН СССР, 1961, 138, 608.
13. Гриневич В. В., К а з а й н А. А.— Вкн.: Титан и его сплавы, 4. Изд-во
1п АН СССР, М., 1960, 147.
1^' Г с д н е в И. Н., Памфилов А. В.— Ж. общ. хим., 1937, 7, 1264.
Д ана Э. С. Описательная минералогия. ОНТИ, М.— Л., 1937.
^1-Делимарский Ю. К., Будераская Г. Г.— Укр. хим. ж., 1962,
99 2г?’ 565-
Дорин В. А.— ЖТФ, 1955, 25, 577. v
Дорин В. А., Тартаковская Ф. М.— Ж. неорг. хим., 1959, 4,
?635-
^•Жданов Г. С. Физика тв. тела. Изд-во МГУ, М., 1962.
3 ел и к м а н А. Н., Сегорчиану Т.— Ж- общ. хим., 1956, 26, 625.
157
26. Иванов А. И., Гоп и ен ко В. Г.— Труды ВАМИ, 1957, 40, 365.
27. Куцев В. С., О р м о н т Б. Ф.— Ж- физ. хим., 1955, 29, 597.
28. Коломицкий Ф. М.— Изв. АН Каз. ССР. Сер. металлургии, обогаще
ния и огнеупоров, 1958, 2, 7.
29. Коломицкий Ф. М., Милов А. И., Пономаренко В. Д.^
Изв. АН Каз. ССР. Сер. металлургии, обогащения и огнеупоров, 1961, 1, 26
30. К с е н д з о в Я. М.— ЖТФ, 1950, 20, 117.
31. Коломицкий Ф. М., Пономарев В. Д.— Изв. высш. уч. зав;
Цветная металлургия, 1959, 5, 106.
32. Крестовников А. Н., Ломов А. Л.— Труды Ин-та цветных метали
лов им. М. И. Калинина, 1960, 33, 8.
33. Ковалев Г. А.— В кн.: Материалы Центр, н.-и. геол.-разв. ин-та, 2. Гео-
химия, 1937.
34. Кузнецов В. А., Пантелеев В. В.— Кристаллография, 1965, Ю
445.
35. Кейер Н. П., Михайлова И. Л., Сазонова И. С.— Кинет. J
кат., 1964, 5, 1086.
36. Л у г и н с к и й Г. П.— Ж. общ. хим., 1936, 6, 1108.
'3 7. Мазур кевич Я. С., Новальковский Н. П., Памфи-
лов А. В., Савицкий А. В.— Укр. хим. ж., 1965, 31, 252.
38. М и х е е в М. И. Рентгенометрический определитель минералов. Госгеол-
техиздат, М., 1957.
39. М о й н о в С. Г., Резниченко В. А.— В кн.: Титан и его сплавы,
9. Изд-во АН СССР, М., 1963, 166.
40. Минералы, 2. Изд-во АН СССР, М., 1965, 9.
41. Меер сон Г. А., Якимова Л. М., Шведова Т. А.— Изв.!
АН СССР. ОТН. Металлургия и горное дело, 1963, 1, 69.
42. М е е р с о н Г. А., К о л г и н О. П.— Атомн. энергия, 1957, 2, 253.
43. М е е р с о н Г. А., Смирнов М. П. Химия редких элементов, 2. Изд-во
АН СССР, М., 1955, 130.
44. М е е р с о н Г. А., Крейн О. Е.— Ж- прикл. хим., 1952, 25, 134.
45. Новоселова А. В., Бацанова Л. Р.— Изв. Сиб. отд. АН СССР1
1960, 8, 421.
46. Пономарев В. Д., Коломицкий Ф. М., Путилин Ю. М,—I
Изв. высш. уч. зав. Цветная металлургия, 1958, 6, 78.
47. Полякова В. М., Чернявская Е.И. Труды Ин-та химии. Ураль-
ский филиал АН СССР, Свердловск, 1963, 7, 33.
48. Путилин Ю.М., Галузо В. Н., Пономарев В.Д. Труды Ин-та>
металлургии и обогащения АН Каз. ССР, 1962, 5, 95.
49. Памфилов А. В., Худякова А. С., Штандель Е. Г.— Жя
общ. хим., 1935, 5, 605.
50. Памфилов А. В., Штандель Е. Г.— Ж. общ. хим., 1937, 7, 258.
51. Памфилов А. В., Ших ер М. Г.— Ж. общ. хим., 1937, 7, 2760.
52. Памфилов А. В., Иванчева Е. Г.— Ж. общ. хим., 1937, 7, 2774J
’153. Памфилов А. В., Иванчева Е. Г.— Ж. общ. хим., 1939, 9, 1739J
54. Памфилов А. В., Иванчева Е. Г.-— Ж. общ. хим., 1940, 10, 1541
55. Памфилов А. В., Иванчева Е. Г., Трехлетов К. Ф- —|
Ж- прикл. хим., 1940, 13, 1310.
^56 . Памфилов А. В., М у ш и й Р. Я., Мазуркевич Я. С.— Укр*“
хим. ж., 1962, 28, 589.
57. П а м ф и л о в А. В., Мазуркевич Я. С.—Укр. хим. ж., 1964, 30, 431
58. Памфилов А. В., Бондарь П. Г., Мазуркевич Я. С.— Укр*-
хим. ж., 1965, 31, 768.
59. Памфилов А. В., Бондарь П. Г., Мазуркевич Я. С.— Укр4
хим. ж., 1965, 31, 48.
60. Резниченко В. А., Соломаха В. П.— В кн.: Титан и его сплавы.
Изд-во АН СССР, М., 1960, 39.
’61. Рубинштейн А. М., Куликове. Г.— Изв. АН СССР. ОХН, 195k;
21, 133. ' .
158
62. Раппопорт В. А.— ДАН СССР, 1963, 153, 871.
63. С аз он о в а И.С., К е й е р Н. П.— Кинет, и кат., 1965, 6, 448.
64. С а х а р о в а М. Г., Шу то в а А. И.— Лакокрасочные материалы и их
применение, 1964, 3, 23.
65. С а м с о н о в Г. В.— Ж- прикл. хим., 1955, 28, 1018.
66. С н о и о в а Е. В., Ротков Н. И.— Советская металлургия, 1936, 12,
11.
67. С к л я р е н к о С. И., 71 и п к е с Я. М.— Ж- прикл. хим., 1940, 13, 51.
68. Стародубцев С. В, Тимошкина Ю. И.— ЖТФ, 1949, 19, 606.
.69. Ф игу р овская Е. Н., Киселев В. Ф., Волькен-
штейн Ф. Ф.— ДАН СССР, 1965, 161, 1142.
70. Ф о т и е в А. А., Ефремова 3. Д.— Тр. Ин-та химии. Уральский фи-
лиал АН СССР, Свердловск, 1963, 7, 45.
71. Хасанов Е. И.— Легкие металлы, 1936, 12, 16.
72. Черепнев А. А. Проблемы хлорирования в области редких и рассеян-
ных элементов. Металлургиздат, М., 1947.
,73 . Ширикян А. А., Киселев А. В., Ушакова Е. В.— Коллоид,
ж., 1964,26,45.
74. Шень Цинь-нан, Д е л и м арский Ю. К.— Укр. хим. ж., 1961,
27, 454.
75. Ш у г а м Е. М.— Хим. пэом., 1956, 7, 426.
76. AnderssonS., С о 1 1 е п В., Kuylenstierna U.,Magne-
1 i А.— Acta chem. Scand., 1957, 11, 1641.
77. A n d e r s s о n S., Collen B., Kruuse G., Kuylenstier-
na U., Magneli A., Pestmalis H., A shrink S.— Acta chem.
Scand., 1957, 11, 1653.
78. A s s a у a g P., D о d e M., F a i v r e R.— Compt. rend., 1955, 240, 1212.
79. A n d e r s e n H. G.— Phys. Rev., 1960, 120, 1606.
80. A s b г i n k S., Magneli A.— Acta chem. Scand., 1957, 11, 1606.
81. A r k e 1 A. S. — Rec. traw. chim., 1926, 45, 437.
82. A г k e 1 A. E.— Phys., 1924, 4, 286.
83. Arthur J. S.— J. Appl. Phys., 1950, 21, 732.
84. В о у d G. E., Harkins W. D.— Phys. Rev., 1939, 55, 237.
85. В о у d G. E., H a r k i n s W. D.— J. Am. Chem. Soc., 1942, 64, 1190.
86. Bach H.— Naturwis., 1964, 51, 10.
87. В о 11 о n H. C., Fawcett W., Gurney I. D. C.— Proc. Phys. Soc.,
1962, 80, 199.
88. Barblan F., В a n d er b erger E., Niggli P.— Helv. chim. acta,
1944, 27,88.
89. Barblan F. F.— Schweiz, mineralog. petrogr. Mitt., 1943, 23, 295.
90. Br anHey L. R., Beckman A. O.— J. Am. Chem. Soc., 1930, 52,
3956.
91. В e r b e r i c h L. J., Bell M. E.— J. Appl. Phys., 1940, 11, 681.
92. В e u t e 1 E., Ku t zer In i gg A.— Monatsh. Chem., 1931, 57, 9.
93. В e r t о n A.— Compt. renc., 1939, 208, 1898.
94. Billy M., В e г t о n A.— Compt. rend., 1938, 206, 1631.
95. Breckenridge R. G., H о s b e r W. R.— Phys. Rev., 1953, 91, 793.
96. В a r d a w i 1 A. B., Collier F. N., T i r 1 e S. Y. — Less-Common Me-
tals, 1965, Of 20.
97. В a u m g a r t e r P., Bruns W.— Ber., 1941, 74, 1232.
96. В о u г i о n F.— Ann. chim. et phys., 1910, 20, 547.
99. В e и г о t h A.— Z. Wiss. Phot., 1915, 14, 217.
°0. Blewett J. P., Jones E. J.— Phys. Rev., 1936, 50,(464.
91. В о t e z N., H e r t e n s t e i n H.— Phys. Z., 1913, 14 ,332.
J92. Born F.— Z. Elektrochem., 1925, 31, 309.
}03. Blumenthal W. B.— Ceramic Age, 1948, 51, 320.
94. Bowman A., Oil J.— Colour chemist’s Assoc., 1952, 35, 314.
J95. Baur W. H.— Acta Crystallogr., 1956, 9, 515.
106. Baur W.— Acta Crystallogr., 1961, 14, 214.
159
107. Bernal J. D. (см. статью F. C. Phillips) — Mineral Mag., 1932/34,
23, 126.
108. Becker H., Klein E., Rechman FI.— Farbe und Lack, 1964, 10,
779.
109. В о 1 1 и о w F.— Z. phys., 1925, 33, 741.
ПО. В u er g er M. J., Butler R. D.— Am. Mineralogist, 1938, 23, 417.
111. Brauer G., L i t t к e W.— J. Inorg. a. Nucl. Chem., 1960, 16, 67.
112. В u n t i к у E.N.— J. Res. Natl. Bur. Standards, 1933, 11, 719.
113. В u b 1 e R. А., В e к w i t h J. В., FI о n i g M.— J. Am. Chem. Soc., 1945,
67, 1554.
114. Czanderna A. W., Rao R. N. С., H о n i g J. M.— Trans. Farad.
Soc., 1958, 54, 1069.
115. Czanderna A. W., H о n i g J. M.— J. Phys. Chem., 1959, 63, 620.
116. Chretien A., Wyss R.— Compt. rend., 1947, 224, 1642.
117. Camboulives P.— Bull. Soc. Chim., 1910, 7, 616.
118. Chen e, Deissenberg, Martin-Borre t.— Ann. Chim. Ana-
lyt., 1946, 28, 197.
119. C h e s t e г P. F., В r a d h u r s t D. H.— Nature, 1963, 199, 1056. ’
120. Companion L. A., W у a 11 R. E.—J. Phys. Chem. Solids, 1963, 24,
1025.
121. С и н p и к и, К у б о, Като. — J. Chem. Soc. J арап. Ind. Chem.
Sec., 1953, 56, 149, 832; 1954, 57, 259.
122. Clark W. С., V о n d j i d i s A.— Nature, 1964, 203, 635.
123. С о n j e a u d P.— Compt. rend., 1954, 238, 2075.
124. Судзуки А., Котэра Й.— Tokyo kogyo shikensho hokoku, Repts Govt
Chem. Ind. Res. Inst., Tokyo, 1964, 59, 116.
125. Chaklader A. C. D., T h i r i a r J.— Compt. rend., 1964, 259, 1969.
126. Cromer D.T., Herrington K-— J - Am. Chem. Soc., 1955, 77, 4708.
127. С о u g h a n о u г L. W., Roth R. S., Prosse V. A.— J. Res. Nat>
Bur. Stand., 1954, 52, 1.
128. Crookes W.— Proc. Roy. Soc., 1881, 32, 206.
129. Czanderna A. W., C e i f f о r d A. F., H о n i g J. M.— J. Amer.
Chem. Soc., 1957, 79, 5407.
130. Cardona M., H a r b e k e G.— Phys. Rev., 1965. 137, 1467.
131. D a u b r e e A.— Compt. rend., 1849, 29, 227.
132. D a u b r e e A.— Compt. rend., 1850, 30, 383.
133. D a u b r e e A.— Compt. rend., 1854, 39, 135.
134. Donnay J.D. H.— Am. Mineralogist, 1941, 26, 195.
135. Dantuma R. S.— Verfkroniek, 1954, 27, 64, 126.
136. Deville H. - S. C. — Compt. rend., 1861, 53, 161.
137. Douglass D. U., Pierre G. R. S., S p 1 i s e r R.— Metallurg. Soc,
Conf., 1961, 8, 705.
138. Darzens G., Bourion F.— Compt. rend., 1911, 153, 270.
139. D u p p a F. B.— Proc. Roy. Soc., 1856, 8, 42.
140. Demarcay E.— Ber., 1887, 20, 96.
141. Demarcay E.— Compt. rend., 1887, 104,111.
142. Darmois E., Rolin M.— Compt. rend., 1949, 229, 933.
143. D г у s M., Trzebiatowski W.— Roszn. chem., 1957, 31, 489.
144. Dewey W. P.— Phys. Rev., 1924, 23, 763.
145. Eucken A., Buchner A.— Z. phys. Chem., 1935, B27, 321.
146. Errera J., Ketelaar H.— J. Phys. Rael., 3, 239, 1932.
147. E a g 1 e r D. M.— J. Phys. Chem. Solids, 1934, 25, 1243.
148. Ehrlich P.— Z. Elektrochem., 1939, 45, 362.
149. E m e 1 e u s H. J., Woolf A. A.— J. Chem. Soc., 1950, 164.
150. E b e 1 m e J. J.— Ann. chim. et phys., 1851, 33, 34; Compt. rend., 1851r
32, 330; Lieb. Ann., 1851, 79, 205; 1851, 80, 212.
151. First Supplementary Cards of x-ray diffraction data, compiled and publisched by
joint commitee of A. S. T. M. Amer. Soc. for x-ray and Electron Diffraction, and
Inst, of Phys, of London, 1944.
160
«52. F 0 г 1 a п К-— Acta chem. Scand., 1964, 18, 1267.
153. В i г к s L. S., Friedman H.— Rev. Sci. Instrum., 1947, 18, 576.
154. F i s e a u H.— Compt. rend., 1866, 82, 1133.
155. Freundlich W., Cl. retienA., Bichara M.— Compt. rend.,
1954, 239, 1141.
156. Freundlich W., Bichara M.— Compt. rend., 1954, 238, 1324.
157. Farber M., Darnell A. J.— J. Chem. Phys., 1955, 23, 1460.
158. G г о v e s W. O., Hoel. M., Johnston H. L. — J. Phys. Chem.,
J 955, 59, 127.
159. Greenwood G.— Phil Mag., 1924, 48, 654.
160. G a 1 m i c h e P.— Ann. chim., 1948, 3, 243.
161. Goldschmidt V.M.— Ser. Acad. Oslo, 1927, 8.
162. Gopalarao G.— Z. phys. Chem., 1939, 184, 377.
163. Gopalarao G., Dhar N. R.— Soil Sci., 1931, 31, 379.
164. GoodeveC. F., Kitchener J. A.— Trans. Farad. Soc., 1938, 34, 570.
165. Grieve J., White J.— J. Roy. techn. Coll., 1939,4, 441.
166. Goodeve C. F.— Trans. Farad. Soc., 1937, 33, 340.
167. Grant F. A.— Ref. Mod. Phys., 1959, 31, 646.
168. Goldschmidt V. M.— Scr. Vidensk. Selsk., Math.-nat. KI., Oslo, 1926,
1, 17.
169. Hautefeuille P.— Ann. chim. et phys., 1865, 4, 129.
170. Hautefeuille P.— Compt.rend., 1863,57, 148.
171. Hautefeuille P.— Compt. rend., 1864, 59, 188.
172. Hautefeuille P.— Compt. rend., 1880, 90, 668.
173. Hautefeuille P.— Ann. chim. et phys., 1890, 21, 419.
174. Hautefeuille P.— Lieb. Ann., 1865, 133, 194.
175. Hautefeuille P.— Bril. Soc. Chim., 1864, 2, 194.
176. Hautefeuille P.— Compt.rend., 1890,110, 1038.
177. Holgersson S., Herrlin A.— Z. anorg. Chem., 1931, 198, 69.
178. Heumann B., Kroger C., Kunz H.— Z. anorg. Chem., 1934, 218,
379.
179. Haitinger M., Feigel F., Simon A.— Mikrochem., 1931/32,
10, 117.
180. Hill F. N., S e 1 w о о d P. W.— J. Am. Chem. Soc., 1949, 71, 2522.
181. Haul R., Dumbgen G — J. Phys. Chem. Solids, 1965, 26, 1.
182. H a u s e г О., Schenk M.— Phys. Status Solidi, 1964, 6, 83.
183. Hylleraus E. A.— Z. Kristallogr., 1927, 65, 469.
184. Huggins M. L.— Phys. Rev., 1926, 27, 638.
185z. Huttig G. F.— Angew. Chem., 1940, 53, 35.
186. Huttig G. F., К os t er ion K.— Kollod. Z., 1939, 89, 202.
187. I w a s e K., N a s u N.— Kinzoku nokenkyu, 1937, 14, 89.
188. J о h n s e n A.— C. Min., 1919, 97.
189. J annetar E.— Compt. rend., 1892, 114, 1352.
190У J о n a s K.— Veszpremi vegyipari eguet. Kozl., 1964, 8, 17.
191. Johnson G., Wey 1 W. A.— J. Am. Cer. Soc., 1949, 32, 398.
192. J u n к e r E.— Z. anorg. Chen., 1936, 228, 97.
193. Jacobsen A. E.— Ind. Eng. Chem., 1949, 41, 523.
194. К а т о а к а, Судзуки.— Bull. Electrochem. Lab., 1954, 18, 12.
195. Kinger у W. D., Francl J., Coble R. L., Vasil os T.—
J. Am. Cer. Soc., 1954, 37, 107.
196. К у б о T. и др.— Kagyokagaku zasshi, J. Chem. Soc. Japan Ind. Chem. Sec.,
1963, 66, 318.
197. Кубо T., Синрики К., Ханадзава T.— Kogyo kagaku zasshi,
J. Chem. Soc. Japan. Ind. Chem. Sec., 1961, 64, 619.
198. К о f s t a d P.— J. Phys. Chem. Solids., 1962, 23, 1579.
199. Konig T., Pfordten O.— Ber., 1889, 22,2070.
~00. К a n g г о W., Jahn R.— Z. anorg. Chem., 1933, 210, 325.
*91. Liebich T., Rubens H.— Ber. Berl. Akad., 1921, 211.
^92. Lux H., Pr o^s he 1 E.— Z. anorg. Chem., 1948, 257, 59.
И 9-614
161
203. Lenard-Jons J. E., Dent В. M.— Phil. Mag., 1927, 3, 1204.
204. Lund E.— Skand. tidskr. farg. och lack, 1957, 3, 57.
205. Legrand C., Delville J.— Compt. rend., 1953, 236, 944*
206. Mochulsky M.— Ind. Quim (Buenos Aires), 1947, 9, 213.
207. M i x t e r W. G.— Am. J. Sci., 1912, 33, 45.
208. Morimoto T., Sakamoto M.— Bull. Chem. Soc. Japan, 1963, 36fc
1369.
209. McDonald H. J., Seitz H.— J. Am. Chem. Soc., 1939, 61, 2405.
210. Meyer W.— Z. techn. Phys., 1935, 16, 355.
211. Moore С. H.— Trans. AIME, 1949, 184, 194.
212. Morey G. W., I n g e r s о n E.— Am. Mineralogist, 1942, 27, 227.
213. M a t о s s i F.-~ Z. Phys., 1963, 173, 1.
214. McTaggart F.K., Bear J.— J. Appl.Chem., 1955, 5, 643.
215. McTaggart F. K-— Nature, 1963, 199, 339.
216. Michel L.— Compt. rend., 1892, 115, 1020.
217. Michel L.— Bull. Soc. Min., 1893, 16, 37.
218. N i g g 1 i P.— Schweiz, mineralog. petrogr. Mitt., 1942, 22, 305.
219. N i g g 1 i P.— Z. Kristallogr., 1921, 56, 119.
220. Niggli P. Lehrbuch derMineralogie, 2 AufL, 2. Berlin, 1926, 606.
221. Naylor B.F.— J. Am. Chem. Soc., 1946, 68, 1077.
222. Nicholas E.L., Boardman L. J.— J. Opt. Soc. Am., 1930, 20, 1151
223. Nicholas E. L.— J. Opt. Soc. Am., 1930, 20, 106.
224. Nicholas E. L.— Phys. Rev., 1923, 22, 240.
225. Newbery E., Prury J. N.— Proc. Roy. Soc., 1916, A92, 276.
226. N a s u N.— Sci. Rep. Tohoku, 1936, I, 25, 510.
227. Okazaki K., Moriyama J., Kush i m a J.— Mem. Fac. Engng^.
Kyoto Univ., 1957, 19, 291.
228. О n о K-, Matsushima T., Hata K-, Kurigama T.— ScL
Rep. Res. Inst. Tohoku Univ., 1957, A9, 227; Ch. A., 1957, 17 522.
229. О e h m e F.— Farbe und Lack, 1958, 64, 319.
230. Parravano N., C a g 1 i о t i V.— Gazz., 1934, 64, 703.
231. Parravano N.— Chim. Industria, 1938, 20, 1.
232. Pierre P. D. C. St.— J. Am. Ceram. Soc., 1952, 35, 188.
233. Parmelee C. W., Badger A. E.— J. Am. Cer. Soc., 1934, 17, 11
234. Petit J., Jacovic M., Helm a J.-H., Bosshard G.— Compt.
rend., 1963, 257, 3878.
235. Pochettino A.— Z. Kristallogr., 1931, 51, 113.
236. Parker R. L.— Z. Kristallogr., 1924, 59, 1.
237. Pauling L.— Z. Kristallogr., 1918, 67, 377.
238. Pauling L.— J. Am. Chem. Soc., 1926, 51, 1010.
239. Pauling L.— J. Am. Chem. Soc., 1927, 49, 765.
240. Pauling L., Sturdivant J. H.— Z. Kristallogr.,
1929, 69, 557.
1928,
68, 239;
241. R u f f a A. R.— Phys. Rev., 1964, 133, 1418.
242. Rosengarten F.— Cornell Eng., 1953, 19, 16.
243. R a d h а к r i s h n a n T.— Proc. Ind. Acad. Sci., 1952, 35A, 117.
244. Richardson F. D., Jeffes J.H. E.— J. Iron Inst., 1948, 160, 20!
245. Roth W. A., Becker G.— Z. Phys. Chem. Bodenstein-Fertbd., 1931
246.
247.
248.
249.
250.
251.
252.
253.
254.
Roth W. A., Becker G.— Z. phys. Chem., 1932, 159, I.
Roth W. A., Wolf U.— Rec. trav. chim., 1940, 59, 511.
R a u t e r G.— Lieb. Ann., 1892, 270, 235.
Rutkowski W.— Prace Inst. Min-wa hutn., 1956, 8, 85.
Ruff O., Brintzinger H.— Z. anorg. Chem., 1923, 129, 267.
Rose H.— Pogg. Ann., 1844, 61, 507.
Rose H.— Gilb. Ann., 1823, 13, 67.
Renz C.— Helv. chim. acta, 1921, 4, 961.
Rao C. N. R., Turner A., H о n i g J. M.— J. Phys. Chem.
1959, 11, 173.
Solids,
162
255. Rao R. N. C.— Canad. J. Chem.,. 1961, 39, 498.
256. Rao C? N. R., Yoganarasimhan S. R., Fa er h P. A. — Trans.
Farad. Soc., 1961, 57, 504.
257. Schoss b[er.g,er F.— Z. Kristallogr., 1942, 104, 358.
258. Schroder A.— C. Min. A, 1927, 379; Z. Kristallogr., 1928, 66, 493; 1928,
67, 485.
259. Sieverts A., Gotta A.— Z. anorg. Chem., 1931, 199, 384.
260. S m i d t W.— Ann. Phys., 1903, 11, 114.
261. S c h г a u f Q.— Z. Kristallogr., 1884, 9, 433.
262. S a n t e n J. H.— Chem. Weekblad , 1949, 45, 745.
263. Sul 1 i van W.F., Coleman J. R.— J. Inorg a. Nucl. Chem., 1962, 24,
645.
264. Suzuki А., К о t e r a Y.— Bud. Chem. Soc. Japan, 1962, 35, 1353.
265. Shannon R. D., P a s к J. A.— Am. Mineralogist, 1964, 49, 1707.
266. StraumanisM. E., EjimaF., James W. J.— Acta Crystallogr.,
1961 14, 493.
267. Shomate С. H.— J. Am. Chem. Soc., 1947, 69, 218.
268. S|t e v e 1 s J. M.— Rec. trav. chim., 1947, 66, 71.
269. Schmidt W.— Ann. Phys., 1902, 9, 919.
270. Strukturber., 1. 1913/28.
271. Schafer H., Goser С., В a у er L.— Z. anorg. Chem., 1950, 263, 87.
272. S b г о 1 1 i W., В e r 1 о t t i E.— Ann. chim. 1959, 49, 1143.
273. Shannon R. D.— J. Appl. Phys., 1964, 35, 3414.
274. Schroder A.— Z. Kristallogr., 1928, 66, 493.
275. Schultz G. V.— Z. Phys., 1964, 179, 473.
276. T r a u b e H.— N. Tb.Min. Beilagebd., 1895/96, 10, 470.
277. T h i e n c h i N.— Compt. rend., 1946, 222, 1178.
278. Trieber E., Stenius A., Lang W., Berndt W.—Acta chem,
Scand., 1954, 8, 1865.
279. Troost L., Hautefeuille P.— Compt. rend., 1872, 75, 1819.
280. Trombe F., Foex M., Henry C.— Compt. rend., 1946, 223, 317.
281. Todisco S.— Ind.Vernice, 1954,8, 17.
282. T о к a d у L.— Mat. termeszettudomany Ertesito, 1927, 44, 247.
283. T i e e d § E., Birnbrauer E.— Z. anorg. Chem., 1914, 87, 129.
284. V e g a r d L.— Phil. Mag., 1916, 32, 505.
285/ V e g a r d L.— Skr. Acad. Oslo, 1925, 11,1.
286. V о n d j i d i s A. G., С 1 а г к W. C.— Nature, 1963, 198,278.
287. V e r w e g G. J. W.— Philips techn. Resch., 1947, 9, 47.
288. V r a t и у F., M i с a 1 e F.— Trans. Farad. Soc., 1963, 59, 2739.
289. Weiser H. В., M i 1 1 i g a n W.O., Cook E. J.— J. Phys. Chem.,
1941, 45, 1227.
290. Williams C. W.— Proc. Roy. Soc., 1917, A93, 418.
291. Watts C. W., Bell C. A.— J. Chem. Soc., 1878, 442.
292. Wey 1 W.A., Fori and T.— Ind. Eng. Chem., 1950,42,257. '
293. W e у 1 R.— Z. Kristallogr., 1959, 111, 401.
294. W a i n e r E.— Trans. Electrochem. Soc., 1946, 89, 331.
295. Winkler C.— Ber., 1890, 23, 2642.
296. Weiss L., Kaiser H.— Z. anorg. Chem., 1910, 65, 345.
297. Wyss R.— Ann. chim., 1948, 3, 2:5.
298. Warren I. H., S h i m a z u H.— Can. Min. and Metallurg. Bull., 1965,
58, 551; repts.— trans. Can. Inst. Min. and Metallurg., 1965, 68, 169—178.
299. W a r t e n b e r g H., Broy J., Reinicke R.— Z. Elektrochem.,
1923 29, 214.
300. W artenberg H., Prophet E.— Z. anorg. Chem., 1932, 208, 369.
301. W artenberg H., Busch H. J., S a r a n E.— Z. anorg. Chem., 1937,
230, 257.
302. W artenberg H., G u r r W. —Z. anorg. Chem., 1931, 196, 374.
303. Williamson W. O.— Nature, 1939, 143, 279.
304. Williamson W. О,— Nature, 1937, 140, 238.
163
305. Williamson W. О.— Trans. Brit. Ceram. Soc., 1940, 39, 345.
306. W i 1 1 i a m s о n W. O.— Min. Mag., 1940, 25, 513.
307. Y oganar as i mh an R. C.— Ind. J. Chem., 1963, 1, 360.
308. Yoganarasimhan S. R.— J. Scient. and. Ind. Res., 1962, B21, 88.
309. Yoganarasimhan S. R.— Ind. J. Chem., 1963, 1, 358.
310. Yoganarasimhan S. R., Rao C. N. R.— Trans. Farad. Soc., 1962,
58 1579
311. Z e r f о s s S., Stokes R. G., Moore С. H.— J. Chem. Phys., 1948,
16, 1166.
312. Z i mensK. E., Hed val 1 J. A.— Svensk. Kem. Tidskr., 1941, 53, 1.
Дополнительная литература
Ария С. М., Балашова Е. П., Владыкина И. Е. Магнитная вое-
приимчивость твердых растворов двуокиси молибдена в двуокиси титана.—
Вести. ЛГУ, 1967, 10, 129.
Б у р ы л е в Б. П., Боровик Г. Р., Ушаков Н. В. Влияние двуокиси
титана на восстановление пятиокиси фосфора из апатита углеродом.— Изв.
высш. уч. зав. Химия и химическая технология, 1967, 10, 1136.
Бенделиани Н. А., Попова С. В., Верещагин Л. Ф. Синтез но-
вой модификации двуокиси титана, устойчивой при высоких давлениях.—
Геохимия, 1966, 5, 499.
Бамбуров В. Г., Фотиев А.А. О взаимодействии двуокиси титана с ге-
ксафторсиликатом калия.— Изв. Сиб. отд. АН СССР, 1963, 3, 42.
Богомолов В. Н. Влияние гидростатического сжатия на электропровод-
ность и эффект Холла в рутиле.— Физ. тв. тела, 1966, 8, 3659.
Богомолов В. Н., М и р л и н Д. Н. Инфракрасное поглощение в прово-
дящих кристаллах рутила.— ЖЭТФ, 1967, 5, 293.
Голубенко А. Н., Резухина Т. Н. Свойства высших окислов титана.
Изв. АН СССР, Неорг. материалы, 1967, 3, 101.
Иида Такэаки, Надзуки Хироси. Влияние легирующих добавок
на фотопроводимость TiCX (рутил).— J. Chem. Soc. Japan. Indastr. Chem.
Sec., 1967, 70, 1285, A82. -
Исирикян А. А., Козьменко И. А. Зависимость адсорбции азота,
аргона и кислорода на рутиле от температуры прокаливания образца в ваку-
уме.— Коллоид, ж., 1968, 30, 220.
Коган Н. Д., Ниуберг Л. В., Базилевич 3. А. Электрофорез су-
спензий двуокиси титана в водных средах.— Лакокрасочные материалы и их
применение, 1966, 5, 21.
Кето в А. Н., Колесов И. М. Термографическое исследование хлориро-
вания двуокиси титана в пятиокиси ниобия.— В кн.: Сборник научных тру-
дов. Пермский политехнический институт, 1965, 18, 42.
Лу Тун-син, Раппопорт В. Л. Изучение адсорбции кислорода на
двуокиси титана методом ЭПР.— Вестник ЛГУ, 1966, 10, 45.
Машенко Л. И., Казанский В. Б., Парийский Г. Б., Шара-
пов В. М. Адсорбция кислорода на восстановленной двуокиси титана.—
Кинет, и кат., 1967, 8, 853.
Мастихин В. М., К е й е р Н. П., Кефели Л. М. О природе возникно-
вения сигнала ЭПР при адсорбции кислорода на рутиле.— В кн.: Методы ис-
следования катализаторов и каталитических реакций. Новосибирск, 1965,
3, 77.
Памфилов А. В., Мазуркевич Я- С., Бондарь П. Г., Л у к и-
н ю к Р. А. Низкотемпературная эмиссия из двуокиси титана.— Укр. хим.
ж., 1966, 32, 144.
Памфилов А. В., Мазуркевич Я- С., Бондарь П. Г. Влияние
освещения на работу выхода электрона из двуокиси титана.— Укр. хим. ж.,
1965, 31, 918.
164
Резниченко В. А., Соломаха В. П. Изучение хлорирования окислов
титана.—В кн.: Проблемы металлургии титана. «Наука», М., 1967, 79.
Рапопорт В. Л. Измерение электропроводности порошкообразной TiO2.—
В кн.: Методы исследования катализаторов и каталитических реакций.
Новосибирск, 1965, 1, 381.
Самсонов Г. В. Синельникова В. С. Алюмотермическое восстанов-
ление TiO2 в вакууме.— Изв. высш. уч. зав. Цветная металлургия, 1966, 4,
65.
/Толстая С. Н., Михайлова С. С., Таубман А. Б., Ува-
ров А. В. Адсорбция поверхностноактивных веществ на двуокиси титана.—
ДАН СССР, 1968, 178, 148.
У мэя К., И с с д а Т., С а в а м у р а К. Реологические свойства суспензий
двуокиси титана.— J. Soc. Mater, Sci., Japan, 1968, 17, 282.
Фигуровская E.H. Влияние форм хемосорбции кислорода на работу выхо-
да электрона и электропроводности двуокиси титана. — В кн.: Проблемы ки-
нетики и катализа, «Наука», М., 1968, 12, 149.
Фотиев А. А., Шейкман Ф. П. Взаимодействие двуокиси титана с фто-
ристым калием.— Ж- неорг. хим., 1967, 12, 2563.
Шейкман Ф. П., Фотиев А. А. Термодинамический расчет реакций,
возможных в системе KF — TiO2 — SiO2.— Труды Ин-та химии, 14. Ураль-
ский филиал АН СССР, Свердловск, 1967; ЮЗ.
Boutin Н., Р г a s k Н., lyenger R. D. Изучение состояния воды в
рутиле.— J. Catolysis, 1967, 9, 309.
Boehm Н. Р., ЬГе г m a n n М. Определение активного водорода, термическое
обезвоживание и регидроксилирование.— Z. anorg. Chem., 1967, 352.
С о г n a z Р. F., Н о о f f J. Н. С., Р 1 u i у m F. J., Schmidt G. С. Изуче-
ние с помощью ЭПР координации кислорода на поверхности TiO2, лишенной
части кислорода.— Disc. Farad. Soc, 1966, 41, 290.
Сирасаки Т., Окада М., Морикава К. Химическая структура и
каталитическая активность TiO2 — NiO и TiO2 — Fe2O3 катализаторов, при-
готовленных методом сверхгомогенного осаждения.— J. Chem. Soc. Japan.
Ind. Chem. Sec., 1965, 68, 593. 365.
^Carnahan R. D., Brittain J. О. Изучение кинетики восстановления
кристаллов рутила спектроскопическим методом.— J. Am. Ceram. Soc., 1965,
, 48.
• С h i d 1 е у В. Е.} Р а г к е г F. L., Т а 1 b о t Е. А. Свойства гидратированной
двуокиси титана как высокотемпературного ионообменного материала.—
Res. Group. U. К. Atomic Energy Author., 1966, AERE-R 5220.
Che M., N accache Cl., Imelik B., PrettreM. Изучение ЭПР час-
тично восстановленной двуокиси титана.— Compt. rend., 1967, 24, 1901.
- Саге аг о G. Адсорбция катионных поверхнсстно активных веществ на TiO2
из водных растворов.— Ohio J. Sci.,4967, 67,237.
D а у R. Е., Р а г t i t t G. D. Толщина адсорбционного слоя спирт + углево-
дород на рутиле.— J. Phys. Chem., 1967, 71,3073.
Day D.E., Lewis К. E., Parfitt G.D. Адсорбция спиртов на поверхно-
сти рутила. ШРАС, 1965, М., Abstr. A. and В, 1965, 51.
Delmaire J.-P., Wallet N., DuquesnoyA. Об изменении удель-
ной электропроводности окислов титана при высокой температуре.— Compt.
rend., 1967, С264, 1290.
* Doucet S. Синтез x. . . анатаза при низкой температуре.— Bull. Soc. fcanc.
mineral et crktallogr., 1967, 90, 111.
Gruenwold T. B., Gordon G. Самодиффузия кислорода в нестехиомет-
рическом рутиле TiO2_x.— Israel J. Chem., 1967, 5, 12. -
Gonzaler F., Madinabeitia G., Trillo J. M., Munuera G.
Поверхностные свойства двуокиси титана.— Colog. quim. fis. proc, supecfic.
solid, Madrid, 1964; Madrid, 1965, 243.
v G u у P., Remy L. Спектры поглощения TiO,.— J. chim. phys. et phys.-chim.
biol., 1966, 63, 827.
165
H i 1 1 е г t L. Фазовая диаграмма TiO2 — CaFo.— Acta Chem. Scand., 1965, 19,
1516.
Iyengar R. D., С о d e 1 1 M., Karra J.S., Turkevich J. Исследо-
вание поверхностной химии рутила методом спин-электронного резонанса.—
J. Am. Chem. Soc., 1961, 88, 5055.
Iwaki T., MiuraM. Исследование поверхности двуокиси титана. — J. Sci.
Hiroshima Univ., 1967, Ser. A, Div. 2, 31, 209.
Juillet F., Long J.,Teichner S.-J. Поверхностные свойства тонкоиз-
мельченной двуокиси титана.—Compt rend., 90-е Congr. nat. Soc. Savantes,
Nice, 1965, Sec. Sci. TI, Paris, 1966, 155.
Kralky O., Leppold H. Исследование рассеяния рентгеновских лучей
под малыми углами на эмульсии двуокиси титана в льняном масле.— J. Col-
loid and interface Sci., 1966, 22, 197.
Keesmann I. Гидротермальный синтез брукита.— Z. anorg. Chem., 1966,
346, 30.
ч/Lewis К. E., Parfitt G. D. Изучение поверхности рутила с помощью ИК-
спектров.— Trans. Farad. Soc., 1966, 62, 204.
Lavelle J. A., Zet t lemoyer А. С. Теплота погружения рутила в ор-
ганические жидкости.— J. Phys. Chem., 1967, 71, 414.
Long J., Juillet F., Teichner S.-J. Свойства двуокиси титана (аната-
за), полученной в пламенной печи.— Rev. hautes temperat. et refract., 1965,
2, 163.
ZL ong J., Teichner S.J. Каталитическая активность двуокиси титана (ана-
таза) в реакции окисления окиси углерода.— Bull. Soc. chim. France, 1965, 9,
2625.
Moser J. В., Blumenthal R. N., W h i f more D.H. Термодинами-
ческое изучение нестехиометрического рутила TiO2-x.— J. Am. Ceram. Soc.,
1965, 48, 384.
♦ McLean W.R., Ritchie M. Реакции на двуокиси титана: фотоокисление
аммиака.— J. Appl. Chem., 1965, 15, 452.
MiuraM., Naono H., Iwaki T. Адсорбция трифосфата на поверхности
двуокиси титана.— J. Sci. Hiroshima Univ., 1966, Ser. A, Div, 2, 30, 57.
M c G о w n D. N. L., Parfitt G. D. Скорость коагуляции рутила в раство-
рах аэрозоля ОТ в п-ксилоле.— Disc. Farad. Soc., 1967, 42, 225.
McDaniel С. L., Schneider S. J. Фазовые соотношения в системе
TiO2 — IrO2 — J. Res. Nat. Bur. Standards, 1967, A71, 119.
«Munuera G., Trillo J. M. Механизм пожелтения пигмента TiO2. — Bol.
Soc. esp. ceram., 1967, 6, 477.
M i с a 1 e F. J., Z e t t 1 e m а у e г А. С. Кинетика десорбции воды с поверхно-
сти рутила.— J. Colloid and Interface Sci., 1967, 24, 468.
Navrotsky A., KI ep p a O. J. Энтальпия перехода анатаз — рутил.—
J. Am. Ceram. Soc., 1967, 50, 626.
Niemyski T., Piekaczyk W. Выращивание монокристаллов рутила
(TiO2) методом химического транспорта с TiCl4.— J. Krystal Growth, 1967, 1, 177.
Piekarczyk W. Получение чистой двуокиси титана.— Internat. Symp.,
Berlin, 1966, 213.
Pandey H. N. Теоретический расчет упругих постоянных и теплоемкости ру*-
। тила.— Phys. Status Solidi, 1965, 11, 743.
Primet M., Pichat P., Mathieu М.-V., P r e 11 г e M. Исследование ад-
сорбции окиси углерода двуокисью титана методом ИК-спектроскопии.—
Compt. rend., 1967, 265, В681.
<Perny G., Lorang R. Схема уровней энергии двуокиси титана.— J. chim.
phys. et phys.-chim. biol., 1966, 63, 833.
«Ropoport V. L. Адсорбция кислорода на двуокиси титана.— ШРАС 1965,
М., 1965, Abstr. A and В, 43.
Roy R., Greer R. Т. Захват протонов в TiO2.— Solid-State Communs,
1967, 5, 109.
Roy P. К., Coble R. L. Растворимость магния, двуокиси титана и метати-
таната магния в окиси алюминия.— J. Am. Ceram. Soc., 1968, 51, 1.
166
- 0 ] у m о s i F. Инициирование взрывов перхлората аммония катализаторами
Сг2О3 — TiO2.— Magyar kern, folyoirat, 1965, 71, 346.
Smith W. R., Ford D. G. Адсорбция на гетерогенных поверхностях двуоки-
си титана и гомогенных поверхностях угля.— J. Phys. Chem., 1965, 69, 3587.
Sastry R. L. Влияние некоторых примесей на превращение анатаз — рутил.—
• Indian J. Chem., 1965, 3, 414.
S р i v а с k М. Хемосорбция кислорода на двуокиси титана, индуцированная
у-излучением.— Internal. J. Appl. Radiat. anc Isotopes, 1967,18, 145.
S t a u f f J., H us ter H. J. Исследование ЭПР хемилюминесценции соедине-
ний четырехвалентного титана с ОН-и О2Н-радикалами.— Z. Phys. Chem.,
1967, 55, 39.
.Sakata К i m i k □, Sakata Tamio. Получение и структурные данные
для фазы (Tii—х Vx) О2.— Japan J. Арр. Phys., 1967, 6, 112.
Simons P. Y., D a c h i 1 1 e F. Структура TiO2 II (фазы TiO2 при высоком
давлении).— ActaCrystallogr., 1967, 23, 334.
^Shannon R. D., PaskJ. А. Кинетика превращения анатаза в рутил.— J. Am.
Ceram. Soc., 1965, 48, 391.
„ T а к э а к а И., Хироси Н. Энергетические уровни ионов Fe2 *" и Со2 *" в
TiO2 — J. Chem. Soc. Japan. Ind. Chem. Sec., 1967, 70, 1624.
Д e k i z Y., Legrand С. Влияние старения на полиморфный переход в оки-
си титана.— Compt. rend., 1965, 261, 3619.
а с k m a n Р. Н., HirtheW.M., Frounfelker R.E. Энергия связи
в рутиле.— J. Phys. Chem. Solids, 1967, 28, 1525.
W е 1 t п е г W., McLeod D. Спектроскопия окисей титана в неоновой и ар-
гоновой матрицах при 4 и 20° К.— J. Phys. Chem., 1965, 69, 3488.
fW i t t k е J. Диффузия ионов переходных металлов в рутил (TiO2).— J. Elec-
trochem. Soc., 1966, 193, ИЗ.
White W. В. Применение инфракрасной спектроскопии к проблемам упорядо-
чения — разупорядочения в простых ионных твердых телах.— Mater. Res.
Bull., 1967, 2, 381.
Vergnoux A.M., Giordano J., M. FcexM. Изучение роста кристал-
лов рутила с использованием солнечной печи.— J. Phys. Chem. Solids, 1967,
1, 67.
V a h 1 d i с k F. W. Фазовый переход TiO2 при различных давлениях.— J. Lees-
Common Metals, 1966, 11, 99.
V ergnoux A.-M., Giordano, Foex M. Получение монокристаллов
рутила в солнечной печи.— Compt. rend., 261, 1965, 3343.
* 2 е t 11 е m о у е г А. С., Mi call F. J., Naraga К-S., Iyengar R. D.
Природа воды, адсорбированной на рутильной форме двуокиси титана.—
' IUPAC 1965, М, Abstr. A and В, 1965, 55.
ТИТАНАТЫ
»
| .fXt
Двуокись титана с окислами металлов образует титанаты. Извест*
ны титанаты бинарного состава, содержащие кроме двуокиси тиг |
тана окислы одного металла, и более сложного состава, содержащий : ,
окислы двух или нескольких элементов. По-видимому, титанаты
могут давать окислы следующих металлов: Li*, Be, Na*, Mg*,1
Al *, К *, Ca *, Sc, V, Cr *, Mn *, Fe *, Co *, Ni *, Cu, Zn *, GaS
Ca *, Sc, V, Cr *, Mn *, Fe *, Co *, Ni *, Cu, Zn *, GaS
Rb *, Sr *, Y *, Zr *, Nb *, Mo, Tc, Ru, Rh, Pd, Ag, Cd *J|
In, Sn, Sb, Cs *, Ba *, La *, Ce *, Pr, Nd *, Pm, Sm *, Eu *, Gd *S
Tb, Dy*, Ho, Er, Tu, Yb, Lu, Hf*, Ta, W, Re, Os, Ir, Pt, Au, Hg,|
TI, Pb *, Bi *, Po, Al, Fr, Ra, Ac, Th *, Pa, U *, Np, Pu, Am, СтД
Bk, Cf, E, Fm, Md, No, Lw. у
Общим методом получения титанатов служит спекание окисловЯ
карбонатов и некоторых других соединений металлов с двуокисью |
титана при температуре выше 1000° С. В качестве минерализующих
добавок, облегчающих кристаллизацию, служит фтористый натрийД
вольфрамат натрия, бура и другие плавни. Титанаты щелочных I
и щелочноземельных металлов получаются также методами гидро- S
термального синтеза: нагреванием двуокиси или гидроокиси ти-1
тана с гидроокисями металлов и водой в автоклавах под давлением I
[143]. Я
Все известные титанаты — вещества, нерастворимые в воде, с
высокими точками плавления. Только некоторые титанаты щелочно- |
земельных металлов реагируют с водой, подвергаясь гидролизу.Я
• В кристаллических структурах титанатов отмечено присутствие I
октаэдрических комплексов TiO6. В основу, например, кристалле- л
химической систематики титанатов стронция положены октаэдры I
TiO6, степень полимеризации которых зависит от стехиометрическо- 1
го состава [52]. В метатитанате стронция октаэдры ТЮ6 сочленяю-
тся между собой вершинами, образуя трехмерную псевдокубиче- I
скую связь с кубооктаэдрическими пустотами, которые заполняют-
ся катионами металлов для компенсации избыточного заряда.
Некоторые титанаты обладают сегнетоэлектрическими свойсг- я
вами, т. е. способностью спонтанно изменять поляризацию при на-
гревании. Классическим представителем сегнетоэлектриков являе-
* Титанаты уже синтезированы.
168
Тся титанат бария ВаТЮ3. Его сегнетоэлектрические свойства
были открыты одновременно Вулом и Гольдманом в СССР, Вайне-
ром и Соломоном в США и Огава в Японии [64].
ТИТАНАТЫ МЕТАЛЛОВ I ГРУППЫ
Титанаты, по-видимому, могут давать все металлы первой груп-
пы периодической системы элементов. Однако к настоящему времени
изучены только титанаты щелочных металлов. Титанаты меди,
серебра и золота еще не получены.
ТИТАНАТЫ ЛИТИЯ
Пукал [330] нагреванием смеси карбонатов лития с двуокисью
титана при температуре 950° С получил два титаната лития —
Li4TiO4 и Li2TiO3. Однако Кордес [262, 2631 не подтвердил индиви-
дуальность первого соединения. Дилитиевый титанат получили
также Барблан [141 ] и Кутолин [63], изучившие его инфракрасный
спектр поглощения. Образование Li2TiO3 наблюдалось в системах
Li, Na|Fe, TiO3 [7, 95], Li, K|F, TiO3 [7, 96], Li, K|TiO3, SiO3
[10, 97], Li, K|TiO3, P2O7 [7, 13] при изучении их методом терми-
ческого анализа. По внешнему виду Li2TiO3 — белый порошок.
Кристаллизуется он в кубической системе типа NaCl, фед. гр.
FdSm [262, 263], параметр решетки а = 4,1355 А [141]. При нагре-
вании до температуры 800—1000° С параметр решетки удваивается
и на рентгенограмме появляются сверхструктурные линии.
Сегнетоэлектрические свойства у дилитиевого титана, на воз-
можность существования которых указывалось в работах [63, 89],
до настоящего времени не обнаружены.
Показатель преломления Li2TiO3 2,087, энтальпия образо-
вания Д/Лэв = —398,9 ккал/моль [81 ], энтропия S298 =
— 21,9 кал/моль • град.
Дилитиевый титанат в воде не растворяется. Разлагается
серной и соляной кислотами и их смесями только при нагревании.
В системах Li2TiO3 с Li2SO4, Li2MoO4, Li2WO4, LiPO3, LiCl
и LiF, как показали исследования их методом плавкости, до тем-
пературы 1100° С наблюдается полная взаимная нерастворимость
[7, 124]. Дилитиевый титанате солями Na2TiO3, Na2SiO3, K2TiO3,
NaF, KF, RbF образует сплавы эвтектического типа. Эвтектики ле-
>нат при содержании LiTiO3 (экв. %) и температуре (°C) соответ-
ственно: 18,5 и 992; 26 и 788; 11,5 и 750; 24 и 844; 27,5 и 654; 25,5
и 651 [7].
Барблан [141; спеканием карбоната лития с двуокисью титана
в соотношении 1 : 2 получил дилитиевый дититанат Li2Ti2O5. Спе-
канием рассчитанных количеств смеси Li2CO3 и ТЮ2 приготовлен
титанат Li4Ti5O12 со структурой шпинели [283].
169
Лецерф [295], нагревая смесь окиси лития и двуокиси титана
при температуре 1000° С в атмосфере аргона, получил сложный
окисел LiTiO2, в котором титан находится в трехвалентном состо-
янии. Восстановление титана произошло за счет металлического
лития, образующегося в результате термической диссоциации окиси
лития. Сложный окисел имеет кубическую решетку, фед. гр. FtnStn,
а = 4,140 А, ризм = 3,80 г/см3, z = 2. 1
Хуммель и Тин [238] синтезировали дилитиевый трититанат
Li2O • ЗТЮо.
ТИТАНАТЫ НАТРИЯ
В литературе описано значительное число титанатов натрия.
Маллард [316, 317], нагревая двуокись титана с едким натром
в отношении 1 : 8 при температуре 800° С в течение 5 ч, получил
тетранатриевый титанат Na4TiO4. Как полагает Конжюд [178],
это соединение образуется при конденсации паров TiO2 на поверх-
ности кристаллов хлористого натрия при температуре выше 450° С.
Тетранатриевый титанат образуется при сплавлении двуокиси
титана или природных минералов рутила, анатаза и брукита с
перекисью натрия [408]. Реакция начинается при температуре
250° С и сопровождается выделением кислорода. I
Розе [347], нагревая двуокись титана с 5—12-кратным коли-
чеством соды, наблюдал выделение из расплава двуокиси углерода
в количестве около 0,9 моля на 1 моль ТЮ2. На основании этого он
заключил, что в расплаве двуокиси титана с содой образуется ди-
натриевый титанат Na2TiO3. Динатриевый титанат получен нагре-
ванием смеси двуокиси титана с содой многими авторами [14, 130,
194, 321, 326, 330, 412]. Он кристаллизуется в системах Li, Na|F,
TiO3 [95], Li, Na|F, SiO3 [79], образуя короткие призматиче-
ские кристаллы, температура плавления которых, по данным [42],
равна 1030° С, по [95] — 1020° С, удельный вес 3,19 [326, 380].
Энтальпия образования метатитаната натрия из элементов
А//298 = —379,5 ккал!моль [80], энтропия образования S298
= 29,1 кал!моль • град, энтальпия плавления — 16,81 ккал!моль,
энтропия плавления 12,90 кал/моль • град [326]. Шомате [380]
определил теплоемкость кристаллов Na2TiO3 в интервале темпе-
ратур от 52 до 298° К и энтропию в интервале 298—1600° К.
Кей и Майлс [269] провели рентгеноструктурное исследование
кристаллов Na2TiO3, полученных из растворов. Они имеют иска-
женную структуру перовскита с ромбической решеткой. Параметры
решетки а = 5,4941, b — 7,7508, с ~ 5,5130 A, z = 4, фед. гр-
РС2гп.
Динатриевый титанат при сплавлении его с солями NaCl, Na2CrO4>
Na2SO4, Na2WO4 и Na2MoO4 образует несмешивающиеся жидкости
[123]. С метаванадатом натрия при сплавлении он дает двуокись
титана. Кристаллизация из расплава двуокиси титана служит ука*
170
Занием на то, что система Na2TiO3 — NaVO3 является нестабиль-
ном сечением системы Na2O — TiO2 — V2O5.
Под действием горячей воды Na2TiO£ гидролизуется [130, 220,
3471- Нагревание ускоряет гидролиз. Динатриевый титанат легко
аСтворяется в разбавленной соляной кислоте.
Нигли [320], сплавляя двуокись титана с содой в атмосфере
0Э2, получил восьминатриевый пентатитанат. Это соединение син-
те3ировал Д'Анс [131], нагревая двуокись титана с едким натром,
рудников и Тресвятский [24] при исследовании системы Na2O —
TiO2 методом термического анализа установили, что Na8Ti5O14 *
плавится конгруэнтно при температуре 1027° С. Образование
Ма8Т15О14 в системе Na2O — TiO2 отмечает также Беляев с соавтора-
ми [14, 18].
Восьминатриевый пентатитанат легко гидролизуется под дей-
ствием воды и разлагается разбавленными кислотами. В результате
образуется рентгеноаморфная гидроокись титана.
Кормимболуф [172], нагревая при температуре 800° С двуокись
титана (1 вес. ч.) с содой (2 вес. ч) и вольфраматом натрия (5 вес. ч.),
получил тетранатриевый трититанат Na4Ti3O8. Кристаллы этого со-
единения имеют моноклинную решетку и нерастворимы в воде.
Нагревая смесь двуокиси титана с содой в присутствии вольфра-
мата натрия, этот же автор [172] синтезировал динатриевый дити-
* танат Na2Ti2O5. Его получили также и другие авторы [320, 412,
141, 380]. Эта соль кристаллизуется в ромбической, системе, пара-
метры решетки ее а = 13,49, Ь= 16,66, с = 3,80 A, z = 8 [141].
Удельный вес, по данным Барблана [141], = 3,422, по [321,
380] — 3,40.
Динатриевый дититанат плавится при температуре 985° С [412].
Энтальпия плавления — 26,230 ккал!моль, энтропия плавления
20,85 кал!моль • град [321 ], энтропия образования S298 =
= 41,5 кал!моль • град [380]. Шомате [380] определил теплоем-
кость Na2Ti2O5 в интервале температур от 52 до 298° К и энталь-
пию в интервале 298—1600° К-
В отличие от более щелочных титанатов натрия динатриевый
Дититанат трудно растворим в воде и в холодной соляной кислоте.
Водой он заметно не гидролизуется. Однако в горячей соляной кис-
лоте растворяется легко.
Сплавляя двуокись титана с содой, вольфраматом натрия и
в°льфрамовой кислотой, Кормимболуф [172] получил динатрие-
вый трититанат Na2Ti3O7. Этот титанат был затем синтезирован
Холмквистом [220] сплавлением TiO2 и Na2CO3 в отношении 2 : 1
в примесью NaF в качестве плавня, а также и другими авторами
24, 133, 141, 250, 320, 321, 380, 403, 412]. Динатриевый трити-
^нат, по данным [412], плавится конгруэнтно при температуре
]128°С, а по другим данным [369],— при 1125°С. Жункер [2501
вРи изучении системы Na2O — TiO2 методом термического анализа
ь области высоких концентраций двуокиси титана не наблюдал
171
образования конгруэнтно плавящихся соединений. Он считает
что Na2Ti3O7 плавится инконгруэнтно при температуре 1120°(у
Будников и Тресвятский [24] также полагают, что Na2Ti3O7 пла1
вится инконгруэнтно при температуре 1130° С. Я
Беляев и Беляева [18] это соединение обнаружили в системе
NasTiOs — TiO2. I
По внешнему виду Na2Ti3O7 представляет собой белые кристалл^/
моноклинной системы. Андерссон и Уодсли [133] определили параме-
системы
Рис. 31. Диаграмма состояния
Na2O — TiO2 [24].
тры решетки а = 8,751, Ь =
= 3,804, с =9,135 А,
= 101,57°, ризм = 3,40, рРеитг
= 3,43 г/см3, z = 2, фед. гр,
P21/m. Поданным Холмквист^
[220], чистые иглы Na2Ti3(Y
имеют удельный вес а4 =41
= 3,507, удельный вес поЯ
р ошкообр азного продукта
d|8= 3,4925, по Нейлору [321J
и Шомате [380], удельный
вес равен 3,44.
Шомате [380] определил
удельную теплоемкости
Na2Ti3O7 в интервале 52 -4
298° К и энтальпию в интерК
вале 298—1700° К. Энтропи^
образования Na2Ti3O7 S®98 ==
= 55,9 кал/моль • град, эн!
тальпия плавления -Ц
37,100 кал/моль, энтропия
плавления 26,48 кал/моль «
• град. Я,
Динатриевый трититанат в соляной кислоте почти не растворяе-
тся даже при нагревании. В горячей серной кислоте он растворяе-
тся также плохо, но лучше, чем кристаллическая двуокись!
титана.
При нагревании двуокиси титана с едким натром и содой в гиДЯ
ротермальных условиях под давлением образуется динатриевый,
тетратитанат Na2Ti4O9 в виде волокнистых кристаллов [403]. |
Д'Анс и Лефлер [131] нагреванием двуокиси титана с содойТ
получили динатриевый пентатитанат Na2Ti5On. Эта соль получения
также Люксом [284] из расплава двуокиси титана в смеси Na2SO4^!
K2SO4 и японскими авторами [403] при гидротермальном синтезе^
Беляев и Голованова [14] наблюдали образование Na2Ti5Ou прд
исследовании тройной системы Na2O — V2O5 — TiO2.
Андерссон [136] получил динатриевый гексатитанат нагреванием^
соды с двуокисью титана. Эта соль в виде волокнистых кристалл;
172
была приготовлена нагреванием двуокиси титана с едким натром
0 содой в гидротермальных условиях [403].
Динатриевый гексатитанат имеет моноклинную решетку с пара-
метрами а = 15,131, b = 3,745, с = 9,159 А, 0 = 99,30°, Ррентг -
3,51, ризм = 3,48 г!см3, z = 2, фед. гр. С2/т.
При обработке двуокиси титана едким натром под давлени-
ем в условиях Байеровского процесса производства алюминия
образуются гидраты титанатов натрия Na2O • 6TiO2 • 7Н2О и
gj\ja2O • TiO2 • 7Н2О [376].
Из приведенного обзора видно, что двуокись титана с окисью
натрия дает титанаты разнообразного состава. Правда, большинство
титанатов натрия получено препаративными методами и поэтому
йндивидуальность их нуждается в подтверждении методами физико-
химического анализа.
Будников и Тресвятский построили диаграмму состояния систе-
мы Na2O — TiO2 в области высокой концентрации TiO2 (рис. 31)»
ТИТАНАТЫ КАЛИЯ
Нагреванием смеси стехиометрических количеств К2О и TiO2
(анатаза) в серебряной лодочке в атмосфере аргона получен орто-
титанат калия К4ТЮ4 [400]. Соединение это представляет собой
белый кристаллический порошок, очень гигроскопичный, водой гид- ч
ролизуется, образуя гидроокись титана. При нагревании выше 650° С
разлагается по реакции
к4тю4 к2тю3 + к20. s
Дебаеграмма его указывает на изогипность структуры с K^SnCV
Демоли [37] получил из водного раствора четырехводный дика- г
лиевый титанат К2ТЮ3 • 4Н2О. Ниггли [320] и Нисиока [326]
приготовили дикалиевый титанат К2ТЮ3 нагреванием двуокиси
титана с карбонатом калия, Пукал 1330] — спеканием двуокиси ти-
тана с бикарбонатом калия.
Дикалиевый титанат кристаллизуется из расплавов в системах
Li, K||F, SiO3 [19], Li, K||F, TiO, [96], Li, K||TiO3, SiO3 [97],
K||TiO3, Cl [17]. При сплавлении с KC1, K2CrO4, K2SO4, K2WO4
11 K2MoO4 он образует несмешиваюгциеся жидкости.
Кристаллы K2TiO3 имеют ромбическую симметрию, параметры
рещетки его а == 10,04, b — 6,95, с = 5,46 А, фед. гр. D^.
7 Температура плавления К2ТЮ3 равна 828° С, при температуре
°4° С он претерпевает полиморфное превращение [71. Энтальпия
энтропия плавления ^TiOg, по данным Нисиока [3261, равны
Ответственно —10,6 ккал/молъ и 9,645 кал/молъ • град, по Беля-
и Голованову [16], —15,65 ккал/молъ и 14,24 кал/молъ • град,
^тальпия образования K2TiOa из элементов Д/7298 =
384,6 ккал/молъ [83].
173
При нагревании смеси двуокиси титана и углекислого кал^;
в атмосфере СО2 образуется дикалиевый дититанат K2Ti2O5 [320J
Барблан [141] получил его непоср
титана с карбонатом калия в соотно
ствия углекислого калия с анатаз
рутилом — выше 800° С. Беляев
и Беляева [17] наблюдали обра-
зование K2Ti2O5, при исследовании
системы К|| TiO3, Cl.
Дикалиевый дититанат пла-
вится при температуре около 980° С
цственным спеканием двуокиси
гении 2:1. Реакция взаимодеэд
м начинается выше 680° С, а с
Рис. 32. Структура K2TiO5 [135].
Рис. 33. Диаграмма состояния
стемы КгТЪОй —ТЮ2 [374].
[320], удельный вес его d\5 — 3,485 [141], показатель преломления
больше 1,74 [141]. Он кристаллизуется в моноклинной системе!
Параметры решетки, по [135], а = 11,374, b = 3,799, с — 6,616 А/
Р = 100,10°, фед. гр. С2/т; по [374], а = 11,38, b = 3,85, с
= 13,16 кХ, р = 99°, рРентг = 2,949, ризм = 2,923 г!см3, вероятч
ная фед. гр. Ст. Структура K2Ti2O5 состоит из тригональный
бипирамид, в которых каждый атом титана окружен пятью атомамЯ
кислорода (рис. 32). Две бипирамиды имеют одну общую грань/
и эти сдвоенные полиэдры образуют бесконечную цепь вдоль оси Я
Цепи связаны общими атомами кислорода в двумерный слой состав
ва Ti2O5~> между слоями находятся атомы калия, каждый из который
необычным способом несимметрично окружен восьмью атомами киед
лорода (на рис. 32 изображена проекция, связь одного из атомов
калия с атомами кислорода изображена пунктирными линиямйИ
Шмитц-Думонт и Рекхард [374] получили дикалиевые титанаЯ
состава K2Ti3O7 и K2Ti5On, плавящиеся инконгруэнтно при темпов
ратуре 1050 и 1300° С соответственно. Они построили диаграмм
состояния системы K2Ti2O5 — ТЮ2 (рис. 33). Я
По [134], кристаллы K2Ti3O7 относятся кофед. гр.С2/т; параме^
ры решетки а = 11,37, b = 3,80, с — 6,62 А, £ = 101,1°, г ==’И
174
Цид-Дрезднер и Биргер [175] сплавлением двуокиси титана с кар-
бонатом калия получили дикалиевый гексатитанат в виде тонких
призматических кристаллов, вытянутых вдоль оси Ь. У кристаллов
K2Ti6Oi3 моноклинная симметрия, параметры решетки а = 15,582,
b 3,82, с = 9,112, р = 99,7°, рРентг = 3,581, ризм = 3,58 г/см3,
z = 2, фед. гр. С2/т. Структура K2Ti6O13 состоит из зигзаго-
образных цепочек, образованных октаэдрами TiO6 вдоль оси Ь.
В промежутках титановых цепочек располагаются крупные ионы
калия. Титановые октаэдры и кубы калия слегка деформированы,
что согласуется с неравномерным распределением зарядов на каж-
дом атоме кислорода, приводящим к необходимому балансу валент-
ностей. Гексатитанат калия изоструктурен с гексатитанатами натрия
и рубидия.
В старых работах [346, 347] описаны титанаты калия более
сложного состава, например К2О • 6Т1О2 • ЗН2О, К2О • 12ТЮ2,
К3О • 12TiO2 • 9Н2О. Они, по-видимому, не являются индивиду-
альными соединениями, будучи продуктами неполного гидролиза
высокощелочных титанатов калия.
Беляев и Беляева [ 171 в системе K2TiOs — КС1 — TiO2 обнаружи-
ли соединение 2К2О • 13ГЮ2, индивидуальность которого нуждае-
тся еще в уточнении,
ч
ТИТАНАТЫ РУБИДИЯ
Шульц [372], сплавляя двуокись титана с карбонатом рубидия
(1:1), получил в виде гигроскопичной массы продукт, которому
приписывает состав Rb2TiO3. Сплавляя двуокись титана с карбона-
том рубидия в отношении 2 : 1 при температуре 1200° С, он при-
готовил дирубидиевый дититанат Rb2Ti2O5. Эта соль образуется
в виде ромбических кристаллов с большим коэффициентом свето-
преломления, удельный вес d\6 = 3,888 [141].
Шмитц-Думонт и Рекхард [374] сплавлением двуокиси тита-
на с карбонатом рубидия получили дирубидиевый тетратитанат
Rb2Ti4O9, который плавится конгруэнтно при температуре 1183° С.
Андерссон [136] синтезировал дирубидиевый гексатитанат
Rb2Ti6O13. Эта соль кристаллизуется в моноклинной, системе с па-
раметрами решетки а = 15,89, b = 3,82, с = 9,11 А, Р = 100,4°,
Ррентг = 4,07 г!см3, фед. гр. С2/т.
Шмитц-Думонт и Рекхард построили диаграмму состояния
системы Rb2Ti2O5— TiO2 (рис. 34).
ТИТАНАТЫ ЦЕЗИЯ
При сплавлении двуокиси титана с карбонатом цезия получается
Титанат в виде гигроскопической массы состава Cs2TiO3 и титанат
состава Cs2Ti2O5 [372, 373]. Дицезиевый дититанат образует боль-
шие кристаллы с температурой плавления 892° С. По мнению [141],
кристаллы Cs2Ti2O5 ромбической системы, другие авторы [374]
175
считают, что Cs2Ti2O5 кристаллизуется в двух полиморфных моди-
фикациях: низкотемпературной моноклинной и высокотемператур.
ной тетрагональной с параметрами а = 6,92, с ~ 8,86 кХ.
Дицезиевый тетратитанат Cs2Ti4O9, полученный [374], плавится
инконгруэнтно при температуре 1115° С (рис. 35). Л
Рис. 34. Диаграмма состояния си-
стемы Rb2Ti2O5 — TiOa [374].
Рис. 35. Диаграмма состояния си-
стемы Cs2Ti2O5 —TiO2 [374].
Свойства титанатов щелочных металлов, как видно из приведенного обзора,
во многом сходны между собой. В большинстве своем —это тугоплавкие соеди-
нения, труднорастворимые в воде и в минеральных кислотах. По мере увеличения
содержания титана в молекуле титаната температура плавления его повышается.
Одновременно повышается устойчивость к действию кислот. Высокощелочные ти-
танаты, однако, легко гидролизуются с образованием рентгеноаморфной гидрооки-
си титана. Это свойство представляет определенный интерес при разработке тех-/
нологии получения соединений титана из минерального сырья.
Рао [354] в системах TiO2 — М2О, где М — щелочные металлы,
получил стекла. |
Титанаты щелочных металлов изучены еще недостаточно. Обосно*
ванные диаграммы состояния типа М2О — TiO2 и М2О — TiO2 — H2Oj
(М — щелочной металл) еще не построены. Однако на основании
имеющихся данных можно заключить, что образование титанато^
щелочных металлов нельзя представить в виде простых закономер-]
ностей, рассматривая их как соли мета-, орто-, пиро- и поликислот.
СОЕДИНЕНИЯ С НИЗШИМИ ОКИСЛАМИ ТИТАНА
Окислы щелочных металлов могут образовывать соединения
с низшими окислами титана стехиометрического состава и с дефекту
ными решетками типа натрийвольфрамовых бронз. Эти соединения
улучают восстановлением титанатов при нагревании двуокиси ти-
,аца со щелочными металлами в вакууме или в инертной атмосфере.
Лундберг и Андерссон [291], нагрегая смесь Ы2СО3 и ТЮ2 в мо-
лекулярном отношении 1:3 при температуре 1050°С, получили
"соединение LixTi4_z/4O8. Это соединение не окрашено, следователь-
но, титан в нем находится в четырехвалентном состоянии. Кристал-
лизуется оно в ромбической системе, параметры решетки его а =
'= 5,011, Ъ = 9,549, с = 2,947 А, рреНгг = 3,57, ризм = 3,62 г/смл.
О получении соединения LiTiO2 см. в гл. II.
Нагельмюллер с соавторами [221 ] нагреванием двуокиси титана
с металлическим натрием в запаянной трубке при температуре 900° С
получил черный порошок состава NaTiO2. При нагревании на воз-
духе это соединение легко окисляется кислородом и превращается
в титанат.
Уодслей и Андерссон [423], восстанавливая Na2Ti3O7 водородом
при температуре 950° С, получил бронзу состава Na/l'iO, (х~ 0,2).
Кристаллы бронзы моноклинной системы, фед. гр. С21т с парамет-
рами решетки а = 12,146, b = 3,862, с — 6,451 А, 0 = 106,85°.
Эти же авторы [137], нагревая соду с двуокисью титана в атмос-
фере водорода, получили бронзу NaxTi4O8, где х — 0,8. Кристаллы
этой бронзы также моноклинной системы, фед. гр. СИт, параметры
решетки а = 12,146, Ь = 3,862, с = 6,451 А, 0 = 106,85°, рреНтг =
= 3,88 г!'см3, z — 2.
Восстанавливая водородом K2Ti2O5, Уодслей и Андерссон [423]
приготовили бронзу состава KxTiO2 (х ~ 0,13). Она образует крис-
таллы тетрагональной системы с параметрами а = 10,172, с —
= 2,846 А.
Восстанавливая Rb2Ti2O5 и Cs2Ti2O;, в атмосфере водорода при
температуре 900° С, Лундберг и Андерссон [291 ] получили соеди-
нения RbzTiO2 и CsxTiO2 с параметрами решеток а — 10,19, с =
— 2,96 А и а = 10,28, с = 2,97 А соответственно.
ПЕРОКСОТИТАНАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Титанаты щелочных металлов с перекисью водорода образуют
перекисные соединения. Эти соединения получают при смешении
пероксотитановой кислоты с растворами гидроокисей щелочных
Металлов и перекиси водорода на холоду. Пероксотитанаты щелоч-
ных металлов хорошо растворимы в воде, из раствора высаливаются
Добавлением спирта.
Меликов и Писаржевский [314] при действии едкого натра и пе-
рекиси водорода на пероксотитановую кислоту выделили кристаллы
пертитаната, которому приписывают состав Na4TiO8 • 2Н2О. Этим
же методом они приготовили соединение, предположительный состав
Которого К2О2 • ТЮ3 • Као4 • ЮН2О.
52 9-614 177
I
4*
Уолтон [413] сплавлением двуокиси титана с перекисью натри^
получил пертитанат натрия Na2TiO4, а Шварц и Хенрих [361 ] __
пертитанат калия K4TiQ
Перекисные титанаты дают аммоний и амины, хотя, будуЧи
слабыми основаниями, они не образуют нормальных титанатов,
устойчивых в водных растворах. И
Меликов и Писаржевский [313, 315] получили пертитанат, имею-
щий, по их данным, состав (NH4)2O2 • ТЮ3 • Н2О2. ;Я
Куровский и Ниссельман [264] описали пероксотитанаты следу,
ющего состава: (СН3 • NH3O)2 • TiO4 • ЗН2О (желто-зеленый по-Ц
рошок),
2 [(C2H5)2NH2 ’• HTiO4] • Н2О2 14Н2О
2 [(С3Н7 • NH3O) • HTiOJ • Н2О2 • 2Н2О
рошок).
Пертитанаты изучены еще весьма недостаточно. Строение при-
веденных выше соединений нуждается в уточнении.
(С2Н5 • NH3O) • НТЮ4 • 1,5Н2О (желтый порошок).
( (желтый порошок) Л
(желто-зеленый по-
•; 1 'i 1 а
ТИТАНАТЫ МЕТАЛЛОВ II ГРУППЫ
Двуокись титана образует соединения с окислами всех металлов
II группы, за исключением окиси бериллия, о взаимодействии с Я
которой в литературе имеются противоречивые сведения. И
*
ТИТАНАТЫ БЕРИЛЛИЯ
Пукал [330] нагреванием оксигидрата бериллия с двуокисью
титана при температуре 1270° С получил дибериллиевый титанат
Be2Ti2O6.
Вартенберг и соавторы [419] исследовали систему ВеО—ТЮ2 ме-
тодом термографии и построили диаграмму состояния. В ней они
установили существование двух титанатов — 2ВеО • ТЮ2 и ВеО •
• ТЮ2. По данным Блюмена [20], титанат ВеО • TiO2, который по-
лучил Вул [407, 384], имеет структуру рутила. Лэнд с соавторами И
[281 ], однако, отрицает существование титанатов бериллия. По его I
данным, в системе ВеО — TiO2 существуют только твердые растворы
на основе рутила и эвтектика при температуре 1670° С и 85% TiO2-
Кокко [183], изучив систему ВеО — ТЮ2 от 0 до 50 вес. % при температуре
1600° С методами микроструктуры и рентгенографии, не обнаружил в ней ни хи-
мических соединений, ни твердых растворов, что было подтверждено [68]. Глушко-
ва и Исупова [49] высказали предположение об отсутствии взаимодействия меж,-
ду двуокисью титана и окисью бериллия, основываясь на изучения энтальпии об-
разования,
Вул и соавторы [45] исследовали диэлектрические свойства
титанатов металов II группы периодической системы. Однако по
их работе трудно судить, к титанатам какого состава относятся ЦХ
478 * j
1
экспериментальные данные, так как в ней не приведены химические
анализы образцов и результаты рентгеноструктурных исследований.
ТИТАНАТЫ МАГНИЯ
В системе MgO— ТЮ2 обнаружены три титаната магния —
2 MgO • TiO2, MgO - ТЮ2 и MgO - 2TiO2 [ 72, 184, 336, 420].
Димагниевый титанат получил Отефейт [239, 244] нагреванием
2 вес. ч. TiO2, 1 вес. ч. MgO и 10 вес. 4.MgCl2. Кристаллы титаната
выделяются в чистом виде при выщелачивании сплава горячей
водой и уксусной кислотой. О получении его сообщается в работах
[21, 184, 297, 420].
Димагниевый титанат плавится, по данным [184], при тем-
пературе 1732° С, по [420],— при 1840е С. Массажа [297]
считает, что это соединение плавится инконгруэнтно. Показатель
преломления его 1,972 [72].
Кристаллы Mg2TiO4 имеют кубическую решетку типа шпинели
[216].о Параметр решетки, по мнению различных авторов, равен
8,44 А [323], 8,412 А [155], 8,41 А [158, 159], удельный вес — 3,52
[239, 244, 345] и 3,53 [232]. Бережной [34] вычислил энтальпию обра-
зования Mg2TiO4 из окислов Н298 = —4,4 ккал/молъ (см. также
табл. 23).
Таблица 23
Энтальпия образования (— Д#298» ккал/молъ) титанатов щелочноземель-
ных металлов [52]
Формула Из окис- лов Из элементов Формула Из окис- лов Из элементов
Mg,TiO4 4,1 517,2 Sr3Ti2O7 72,0 946,2
MgTiO3 6,3 375,6 (374,7) SrTiO8 32,4 399,0 (398,9)
' MgTiao5 4,4 599,2 SrTi3O5 36,3 628,4
Ca2TiO4 19,0 526,7 Ba2TiO4 45,7 538,0
Ca3Ti2O7 37,7 944,1 BaTiO3 38,7 397,6 (394,8)
CaTiO3 18,2 395,5 (397,4) BaTi2O5 45,3 629,7
CaTi2O5 19,5 622,3 BaTi3O7 46,4 856,3
Sr2TjO4 37,9 545,6 BaTj4O9 46,0 1081,4
Примечание. В скобках приведены калориметрические данные [80,81]-
В закаленных образцах Mg2TiO4 происходит замещение Mg2+ на
Ni24* # в медленно охлажденных образцах это замещение не обнару-
жено.
Димагниевый титанат образует твердые растворы со структурой
Шпинели с MgAl2O4 [33] и MgCr2O4 [37].
В воде и в разбавленных кислотах на холоду Mg2TiO4 не
растворяется. При длительном нагревании растворяется в разбавлен-
ной соляной и азотной кислотах.
12*
179
Отефейт [240], нагревая смесь двуокисей титана и кремния с
хлористым магнием, наряду с силикатом магния MgSiO3 получил
титанат магния MgTiO3.
Титанат магния образуется при нагревании двуокиси титана
с металлическим магнием при температуре около 1200° С по реакции
[414]:
Рис. 36. Диаграмма состояния системы MgO — ТЮг [184].
В чистом виде MgTiO3 получен нагреванием двуокиси титана
с 10-кратным количеством хлористого магния и небольшим коли-
чеством хлористого аммония. Избыток плавней удаляют выщелачи-
ванием сплава водой и уксусной кислотой [239, 244].
Для получения титаната магния одни авторы [401, 253, 369] спе-
кали смесь окислов при температуре около 1200° С, другие [379, 322,
330] — ПрИ 1300—1350° С (И </); Ивасе и Нисиока [246] вначале
нагревали смесь окислов 3—4 ч при температуре 1420° С, а затем
полученный сплав измельчали и повторно нагревали в пламени
водородной атомарной горелки.
Кофанур и Депрозе [174] и Блюмен [21] установили образова-
ние MgTiO3 в системе MgO — ТЮ2 при исследовании ее методом
термического анализа (рис. 36). Бережной и Гулько [33] показали,
что поле MgTiO3 имеется в тройной системе MgO — А12О3 — TiO2-
180
Титанат магния кристаллизуется в форме гексагональных таб-
литчатых кристаллов [239, 240], фед. гр. 7?3, z = 2 [217]. Пара-
метры решетки MgTiO3 в ромбоэдрической установке, по [217J,
таковы: а = 5,40 А, а = 55°Г, по данным [332], а = 5,54 А,
а = 54° 39'. Плотность кристаллов, поОтефейту [239, 240], 3,91 г/см3,
по Шомате [379], 3,84 г/см3, показатели преломления N = 1,95,
No = 2,28 [184].
Титанат магния плавится конгруэнтно [297], температура плав-
ления его 1583° С, по [184], и 1565° С, по [369].
Энтальпия образования MgTiO3 из окислов АН298 —
= —2,4 кал/моль [34], энтропия S298 = 17,8 кал/моль • град [379].
Вул и Гольдман [41 ] установили, что диэлектрическая проница-
емость MgTiO3 при комнатной температуре равна 17 и не меняется
от перемены частоты электрического поля в пределах 5—4 • 106 гц.
В воде и в разбавленных кислотах MgTiO3 не растворяется.
Однако он разлагается концентрированной серной кислотой при
нагревании и при сплавлении с бисульфатом аммония.
Водород начинает восстанавливать MgTiO3 при температуре
около 800° С [415]. В результате восстановления смеси окислов
MgO + ТЮ2 водородом при температуре 1200° С получаются ми-
нералы титановых шлаков: твердый раствор mMgTiO • nTi2O3
и аносовит mMgTi2O5 • nTi3O5.
Газообразный хлор в присутствии угля начинает реагировать
с MgTiO3 при температуре около 500° С [87]. Хлорирование идет
в две стадии. Вначале хлорируется окись магния
MgTiO3 + С12 4- 4- С = MgCl2 + TiO2 + 4- СО2,
затем начинает хлорировать двуокись титана
TiO2 + С12 + С TiCl4 + СО + СО2.
Вследствие двухстадийного хлорирования титаната магния непро-
хлорировавшийся остаток может обогащаться двуокисью титана.
При нагревании с углем до температуры 2400° С MgTiO3 вос-
станавливается до смешанного окисла титана низшей валентности
MgO • Ti2O3 [294].
Танака [401 ] спеканием окиси магния с двуокисью титана полу-
чил магниевый дититанат MgTi2O5. Существование этого титаната
в системе MgO — TiO2 подтверждено и другими исследователями
1184, 21]. Он кристаллизуется в ромбической системе, параметры
решетки его а = 3,7, b = 9,8, с — 10,0 А [85].
Магниевый дититанат плавится при температуре 1645° С, по [369],
при 1652° С, по [184]. Энтальпия образования MgTi2O5 из окислов
АН298 = —5,6 кал/моль [34].
С титанатом алюминия А12О3 • TiO2 магниевый дититанат обра-
зует взаимные твердые растворы [33, 298, 428]. Бинарная система
181
А12О3 • TiO2— MgTi2O5 эвтектического типа. Эвтектическая точка
находится при содержании 7 мол.% А12О3 и температуре 1580° С
[428].
Термографические исследования Черняка [119] показывают, что
двуокись титана при нагревании с карбонатом магния реагирует
медленно. На термограмме нагревания смеси MgCOs + TiO2 до
температуры 1300—1340° С нет тепловых эффектов, вызванных вза-
имодействием двуокиси титана с карбонатом или окисью магния.
Более реакционноспособным веществом является метатитановая кис-
лота. Взаимодействие ее с карбонатом магния является причиной
появления на термограмме нагревания тепловых эффектов при
1010 и 1130° С. Продукты взаимодействия двуокиси титана с кар-
бонатом магния в твердом виде обычно содержат по несколько со-
единений [21].
Электрические свойства титанатов магния не отличаются спе-
цифическими особенностями [410].
ТИТАНАТЫ КАЛЬЦИЯ
В литературе описано несколько титанатов кальция: ЗСаО • ТЮ2,
2СаО • ТЮ2, ЗСаО • 2ТЮ2, 4СаО . 3TiO2, CaO • ТЮ2 [31, 54, 358,
419], однако существование первых двух титанатов в виде инди-
видуальных соединений не подтверждено. Кофанур с соавторами
[173] изучил систему СаО — ТЮ2 методом термического анализа и об-
наружил в ней только два титаната — ЗСаО • 2ТЮ2 и СаТЮ3.
Трикальциевый дититанат образует анизотропные кристаллы
с положительным удлинением и малым углом между оптическими
осями; показатели преломления его а = 2,16, (3 = 2,20, у = 2,22 [173,
208], удельный вес 3,55 [208], энтропия S298 = 56,1 кал!моль • град
[256]. Кристаллы ЗСаО • 2ТЮ2 плавятся инконгруэнтно при
температуре 1750° С [173, 196].
Соединение ЗСаО • ТЮ2 имеето тетрагональную решетку с па-
раметрами а = 3,83, с — 19,505 А [271].
Рут [358] описал соединение 4СаО • ЗТЮ2, которое имеет также
тетрагональную решетку с параметрами а — 3,83, с ~ 27,147 А
[248 а].
Титанат кальция CaTiO3 получил Эбельмен [203] сплавлением двуокиси титана
с углекислым кальцием и содой, а также [220, 431]. Отефейт [239, 243] CaTiO3 син-
тезировал сплавлением двуокиси титана с хлористым кальцием, а Бургис [168,
171] — сплавлением двуокиси титана и углекислого кальция с хлористым кальци-
ем с последующим выщелачиванием сплава разбавленной соляной кислотой. Ти-
танат кальция образуется также при сплавлении эквимольных количеств окиси
кальция и двуокиси титана [320, 322,247]. Бркик с соавторами [140] установил, что
в интервале температур 800—1000° С количественный синтез CaTiO3 спеканием
CaTiO3 и ТЮ2 до конца не идет. По данным термограммы нагревания, образование
СаТ1О3 из двуокиси титана и карбоната (окиси) кальция протекает при температу-
ре 1050—1250° С [38]. Метатитанат кальция кристаллизуется из многих силикат-
ных сплавов, содержащих СаО и TiO2 [29, 168, 169, 171, 176, 239, 243]. Бркик с со-
авторами [139] получил CaTiO3 со 100%-ным выходом, нагревая смесь, состоящую
182
/
яз 1,0 вес. ч. TiO2 в виде титан-геля, 0,7 вес. ч. Са (ОН)2 и 0,3 вес. ч. Ca (NO3)2,
при температуре 575° С в течение 1—2 ч. Японские авторы [390] обнаружили поле
кристаллизации CaTiO3 в системе СаО — Сг2О3 — TiO2. Титанат кальция в виде
минерала перовскита встречается в горных породах и титановых шлаках.
Кристаллы природного минерала перовскита имеют псевдоку-
бическую (моноклинную) решетку (см. гл. I). Кей и Бейли [271]
однако установили, что синтетический СаТЮ3 кристаллизуется
Рис. 37. Диаграмма состояния системы СаО — ТЮ2 [196].
в ромбической сингонии с параметрами решетки а = 5,3670, b =
= 7,6438, с = 5,4439 А, z = 4, фед. гр. Рстп. По-видимому, струк-
тура СаТЮ3 образуется в результате моноклинного, ромбического
и тетрагонального искажения кубических кристаллов.
При сильном облучении нейтронами наблюдается увеличение
параметров решетки СаТЮ3 [226].
Удельный вес СаТЮ3, по данным различных авторов, равен 4,10
1203], 4,028 [247], 4,02 [379], температура плавления 1980° С [420],
1650° С [369], 1954° С [196], 1915° С [173]. Данные [369], по-види-
Мому, сильно занижены.
Удельная теплоемкость СаТЮ3 в интервале температур 52—298° К
измерена Шомате [379]. Нейлор и Кук [322] определили энталь-
пию образования СаТЮ3 в интервале температур 383—1790° К.
183
Девриз с соавторами построил диаграмму состояния системы
СаО — TiO2 (рис. 37).
Диэлектрическая проницаемость CaTiO3 при температуре 25° с
равна 115 и не зависит от частоты в пределах от 50 до 4 • 106 гц.
По данным [75], диэлектрическая проницаемость CaTiO3 опреде-
ляется колебаниями кристаллической решетки.
Коэффициент линейного расширения в интервале температур
от —80 до —1200° С равен 1,4 • Ю"5 [224].
Метатитанат кальция нерастворим в воде и в разбавленных
кислотах. Концентрированные соляная и азотная кислоты медлен-
но разлагают его только при нагревании. Концентрированная серная
кислота при нагревании разлагает CaTiO3 энергично. Аналогично
действует бисульфат аммония при сплавлении его с CaTiO3 [240].
Метатитанат кальция имеет близкие параметры решетки с
ЗСаО • 2ТЮ2 и между ними возможно образование твердых раст-
воров [21 ]. Метатитанат кальция дает серию непрерывных твердых
растворов с CaZrO3 [37].
Энтальпии образования титанатов кальция приведены в
табл. 23.
ТИТАНАТЫ ЦИНКА
Описано шесть титанатов цинка со следующим составом:
2ZnO • TiO2, 3ZnO • 2ТЮ2, ZnO • TiO2, 4ZnO • 5TiO2, 2ZnO * 3TiO.
и ZnO • 3TiO2.
Дицинковый титанат получается при нагревании смеси двуокиси
титана и окиси цинка, по [330] — при температуре 1270° С,
по [180] — при 900° С в течение 24 ч, по [397] — при 1050° С.
Окись цинка начинает реагировать с двуокисью титана по на-
блюдению Коле и Нельсона [180], при температуре 430° С, а по
[395], при температуре 700° С. Хольгерсон и Херлин [232] для
получения Zn4TiO4 нагревали смесь окислов с добавкой хлористого
натрия. Пассерини [339] синтезировал Zn2TiO4 нагреванием двуоки-
си титана с нитратом цинка при температуре 900° С. Дицинковый
титанат обнаружил в системе ZnO — TiO2 Блюмен [20].
Титанат Zn2TiO4 существует в виде двух полиморфных модифи-
каций — высокотемпературной со структурой неполной обратной
шпинели и низкотемпературной со структурой полной обратной
шпинели [166, 165]. Медленное охлаждение кристаллов Zn2TiO4
приводит к тетраэдрическому искажению кубической решетки.
Параметры решетки Zn2TiO4 и удельный вес определялись не-
сколькими авторами. Данные их измерений следующие:
а, А° Удельный вес Литература
8,410 5,43 [339]
8,44 5,33 232
8,460 5,295 [397]
8,46 5,28 [180
8,41 5,30 [146]
184
0 20 40 60 60 100
ZnO Мол70 TlG;.
Рис. 38. Диаграмма состояния
системы ZnO—ТЮг [187].
В Zn2TiO4 атомы цинка частично могут замещаться атомами
никеля. Структура шпинели при этом сохраняется [167].
Титанат Zn2TiO4 диамагнитен, молекулярная магнитная воспри-
имчивость его 63 • 10~6 [338], энтальпия образования A F&8 =
= —391,6 ккал/молъ (из элементов) и 0,3 ккал/молъ (из окислов) [82],
энтропия S298 == 32,8 кал/молъ • град, [265].
Леви [286] получил титанат цинка 3ZnO • 2TiO2. О существо-
вании этого соединения в системе ZnO — TiO2 заключили Сигиура
и Икеда [383] по скорости испарения
цинка.
I Метатитанат цинка ZnTiO3 приго-
товил Леви [285, 286] нагреванием
2 вес. ч. ТЮ2,8 вес. ч. ZnSO4 и 3 вес. ч.
,K2SO4. Пукал [330] получил ZnTiOs,
нагревая смесь окислов при темпера-
туре 1270° С, Вул [407] и Смит [384]—
нагревая смесь окислов при темпера-
туре 1500° С под давлением.
I Японские авторы [402] синтезиро-
вали ZnTiO3 при нагревании двуоки-
си титана с плавленным хлористым
цинком при температуре 500—600° С
в течение 48 ч,
I Кофанур с соавторами [173] уста-
новил в системе ZnO — TiO2 сущест-
вование только ограниченных твердых
растворов (до 10—15 вес.% ТЮ2) и
ZnTiO3.
Г Структура ZnTiO3 окончательно
еще не определена, а данные различ-
ных авторов противоречивы. Кофа-
нур с соавторами [173], определил,
что ZnTiO3 имеет ромбическую решетку
с параметрами а — 4,806, b = 5,032
и с = 5,477 А. Показатели преломления а — 2,33, р = 2,38,
• V = 2,41. Японские авторы [402] установили, что у ZnTiO3 струк-
тура ильменита с параметрами решетки а = 5,076, с = 13,920 А.
Сканави и Маслов [99] считают, что ZnTiO3 имеет структуру шпи-
нели. Другие авторы [146] приписывают ZnTiO3 гексагональную
структуру с параметрами решетки а — 5,077, с — 13,92 А.
Диэлектрическая проницаемость ZnTiO3 — 30 [41], энталь-
Ийя образования АН298 = —309,1 ккал/молъ (из элементов) и
*^0,4 ккал/молъ (из окислов) [82].
Титанат цинка 4ZnO • 5TiO2 получил Леви [286].
Бартрам и Слипитс [1461 в системе ZnO — TiO2 установили
существование титаната цинка состава 2ZnO • TiO2. Он имеет
185
дефектную структуру типа шпинели, параметр решетки а =
8,395 А, ррентг = 4,517 г/см*.
Леви [286] выделил титанат цинка ZnO • 3TiO2 в виде серо-
стальных игл с удельным весом 4,92. Существование титанатов
4ZnO • 5TiO2 и ZnO • 3TiO2 в системе ZnO — TiO2 подтверждено
[383].
Дулин и Рейз [187] построили диаграмму состояния системы
ZnO — TiO2, в которой установили существование только двух сое-
динений (рис. 38).
ТИТАНАТЫ СТРОНЦИЯ
Известно шесть титанатов стронция, имеющих состав
3SrO • TiO2, 2SrO • TiO2, 3SrO • 2TiO2, SrO • TiO2, 2SrO • TiO2
и SrO • 2TiO2.
Тристронциевый титанат 3SrO • TiO2, по данным [185], об-
разуется в системе SrO — TiO2 в интервале 1560—1760° С. Выше и
ниже этой температуры он неустойчив и распадается на окись
стронция и дистронциевый титанат.
При спекании двуокиси титана с карбонатом стронция обра-
зуется дистронциевый титанат Sr2TiO4 [330, 394]. Пукал [330], од-
нако, в своей работе не привел данных, подтверждающих индивиду-
альность этого соединения. Войцеховская [416] показала, что SrTiO4
образуется при спекании двуокиси титана и карбоната стронция
при температуре около 950° С. Выше 1000° С он разлагается по ре-
акции
SrTiO4 + TiO2 = 2SrTiO3.
Дистронциевый титанат существует в двух полиморфных моди-
фикациях— а и Р — с температурой перехода около 1525° С и
плавится инконгруэнтно при I8600 С [185].
Тарте [391] с помощью ИК-спектров показал, что в структуре
Sr2TiO4 присутствуют октаэдры TiOe. Структура Sr2TiO4 рассмотрена
и в работе [52].
Тристронциевый дититанат Sr3Ti2O7 устойчив ниже 1580° С [112].
Выше этой температуры он разлагается с образованием SrTiO3
и высокотемпературной модификации Sr2TiO4. Титанат Sr3Ti2O7 об-
разует двухсторонние твердые растворы со SrTiO3 [185].
Метатитанат стронция SrTiO3 получили спеканием TiO2 и SrO
при температуре 1270° С Пукал [330], при 1000° С Гофманн [2231
и при 1500° С Вул [407]. Судя по термограмме нагревания, SrTiO3
образуется при спекании окислов при температуре около 1300° С.
Савагути [182] вырастил монокристаллы SrTiO3. Они имеют
структуру перовскита. Параметр решетки, по [217], равен 3,92 А,
по [223], 3,899 А; плотность кристаллов 5,11 г!см3 [223].
Японские авторы установили [201], что при охлаждении кристал-
лов SrTiO3 до температуры 100° К обнаруживается ясно выраженное
186
^вупреломление в направлении погасания (100) и появляются двой-
никовые пластины тетрагонального габитуса. Это служит подтвер-
ждением фазового превращения кубической решетки в тетрагональ-
ВуЮ-
Литл [282] измерил параметры решетки SrTiO3 в интервале
температур 4,2—300° К и установил, что кроме псевдокубической,
существуют еще три полиморфные модификации SrTiO3. При тем-
пературе НО—300° К существует кубическая модификация с коэф-
фициентом линейного расширения 9,4 • 10~б град~1 и параметром
решетки 3,915 А (290° К).
При температуре 65—110° К существует тетрагональная фаза
с отношением параметров da
35—65° К —ромбическая мо-
дификация. Ниже 10° К ав-
торы встретили еще одну ку-
бическую форму с ромбоэд-
рической решеткой. Фазовые
превращения SrTiO3 происхо-
дят в результате деформации
кубической решетки по всем
трем направлениям.
Броус с соавторами [157]
определил для кубической мо-
дификации SrTiO3 параметр
решетки 3,893 кХ.
При понижении темпера-
туры поляризуемость титана
в SrTiO3 падает, а кислоро-
да — растет. Поведение элек-
тронной поляризации SrTiO3
= 1,00056, а в интервале температур
гооо-
1800-
1600-
1400-
2040°
1600°
1830°
1140
1830
1640
20 40 60 80 100
Мол°/0 Т10г
Диаграмма состояния системы
SrO— TiO2 [188].
0
SrO
Рис. 39.
согласуется с отрицательным температурным коэффициентом пока-
зателя преломления параэлектрической SrTiO3 [213].
Матосси [303] по данным частот = 5464 и v2 = 178 см-1,
наблюдающихся в ИК-спектрах SrTiO3, вычислил приближенные
значения силовых постоянных (мкдин/А): к (Ti — О) = 1,06 и
к (Sr — О) = 0,11. Полученные данные согласуются с данными для
квазимолекулы TiO2. Атор показал, что предположение о конфигу-
рации типа TiO6 в случае титанатов не соответствует эксперименту.
Метатитанат стронция плавится конгруэнтно при температуре
020° С [394]. Плотность его равна 5,11 г!см3 [223], диэлектричес-
ки проницаемость при комнатной температуре 155 [41].
Дриз и Тжебиатовский [188] построили диаграмму состояния
Лстемы SrO — TiO2, в которой обнаружили только три соединения:
*TiO4, Sr3Ti2O7 и SrTiO3 (рис. 39).
Cfg Смоленский [88] обнаружил, что SrTiOg обладает сегнетоэлектрическими свой-
Ми с точкой Кюри около 10° К. По данным [51], диэлектрическая проница-
187
емость SrTiO3 в интервале температур от точки кипения жидкого гелия до комнат-
ной температуры изменяется в пределах 315—320 и не зависит от частоты электри-
ческого поля. При температуре ниже 77° К наблюдается резкое возрастание ди-
электрической проницаемости. В интервале температур 3—4° К выше точки кипе-
ния гелия диэлектрическая проницаемость SrTiO3 остается постоянной, достигая
величины примерно 15 000. Авторы изучили гистерезис диэлектрической проница-
емости при 293,77 и 4,2° К, но петли гистерезиса не обнаружили.
Богданов с соавторами [25 ] исследовали двойное лучепреломление
и диэлектрические свойства SrTiO3 в интервале 4,2—300° К. Они
установили фазовые переходы при температурах ПО и 60° К и пред-
полагают, что в области температур 4,2—20° К существует третий
переход. Методом рентгеноструктурного анализа подтверждено, что
при температуре 100° К наблюдается превращение псевдокубиче-
ской структуры SrTiO3 в псевдомоноклинную.
Метатитанат стронция при температуре 1/4° К является сверх-
проводником [78].
В спектре комбинационного рассеяния SrTiO3 найдено 7 линий
[323].
Кан и Легендеккер [255, 274] показали, что заполненные электронами валент-
ные полосы SrTiO3 образованы в основном из 2р-орбиталей кислорода, а пустые —
из З^-орбиталей титана. В кубических кристаллах SrTiO3 и BaTiO3 имеется шесть
низших эллипсоидальных зон проводимости, вытянутых в направлении (100) в
^-пространстве (пространство квазиимпульсов). Спинорбитальное расщепление
снимает вырождение при k = 0 и приводит к появлению дополнительных зон про-
водимости, расположенных несколько выше самой нижней зоны проводимости.
Симонек и Сроубек [371] критикуют расчеты запрещенных зон проводимости
в титанате стронция, проведенные авторами [255, 274]. Они считают, что упомяну-
тые авторы не учли влияния на энергию ионизации ионов смещения ковалентного
заряда и ионов кислорода на ионы титана.
Метатитанат стронция образует непрерывную серию твердых
растворов с цирконатом стронция SrZrO3 [55, 56], ферратом висмута
BiFeO3 [115] и титанатом бария [229].
Клевенжер [177] синтезировал серию соединений перовскитового
типа SrFeO3 — SrTiO3. Железо в этих соединениях присутствует
в виде Fe4+ и Fe3+. Электрический баланс при замещении Fe°+
на Fe4+ обеспечивается за счет потери кристаллами части атомов кис-
лорода.
Дистронциевый трититанат описан в старых работах [168, 171] и
его индивидуальность нуждается в подтверждении. Энтальпии об-
разования титанатов стронция см. в табл. 23.
ТИТАНАТЫ КАДМИЯ
Известен только один титанат кадмия CdTiO3. Он впервые был
получен Пукалом [330] нагреванием двуокиси титана и окиси каД'
мия при температуре 1270° С. Христенсен и Рассмуссен
получили его нагреванием гидроокисей титана и кадмия с раствор0^1
188
н-н. NaOH в автоклаве при температуре 380—450° С под давлением
300—500 атм.
Г Титанат кадмия существует в виде двух полиморфных модифи-
каций с точкой перехода 1050° С. Низкотемпературная модификация
CdTiO3 имеет структуру ильменита, а высокотемпературная — пе-
ровскита [290, 331 ]. У ромбической модификации со структурой
ильменита параметры решетки а = 10,594, b = 7,600, с = 10,812 кХ
(300, 301 ]. Псевдокубическая модификация со структурой перовски-
та, по измерениям Захариазена [430], имеет параметр решет-
ки а = 3,75 А. Мегоу [300, 301] для моноклинной (псевдокубиче-
ской решетки) CdTiO3 определила параметры: а = с = 3,784, b =
= 3,800 кХ, ₽ = 91° 10'.
к Кей и Мейле [269] исследовали кристаллы CdTiO3, выращенные
из раствора. Они имеют искаженную структуру перовскита с пара-
метрами решетки а = 5,348, b = 7,615, с = 5,417 А.
Е. Титанат кадмия обладает сегнетоэлектрическими свойствами,
точка Кюри лежит около 50° К [88], диэлектрическая проницае-
мость равна 62 [41].
ТИТАНАТЫ БАРИЯ
В литературе описаны шесть титанатов бария: 2ВаО • TiO2,
ВаО • ТЮ2, 2ВаО - 3TiO2, ВаО • 2ТЮ2, ВаО • 3TiO2 и
ВаО • 4TiO2.
Е Дибариевый титанат Ba2TiO4 получил Пукал [330] нагреванием
смеси двуокиси титана и углекислого бария в отношении 2 : 1
(мол.) при температуре 1270°С. Существование этого титаната бария
было отмечено многими авторами [364, 392,348,336], изучавшими
систему ВаО — TiO2.
* Кубо и Синрики [277—279] получили Ba2TiO4 спеканием дву-
окиси титана с окисью или карбонатом бария при температуре 1300°C
® течение 1 ч. Эти же авторы [280] получили Ва2ТЮ4 нагреванием
брикетов двуокиси титана с азотнокислым барием до температуры
И)00° С. Карбонат бария начинает реагировать с двуокисью титана
fc образованием Ва2ТЮ4 при температуре 860—900° С [209, 279].
Дибариевый титанат имеет моноклинную решетку с параметра-
ми а = 6,12, b = 7,70, с = 10,50 А, ₽ - 93° 8', г = 4, фед. гр.
^/т [144].
К Тарте [391] по данным ИК-спектров в области 700—800 см~~{
# твердых растворах (Ba, Sr)2TiO4 и (Ba, Sr)2 (Si,Ti)O4 индентифи-
ДИровал присутствие тетраэдров TiO4. Тетраэдры слабо искажены.
® твердых растворах (Ba, Sr) — (Si, Ti)O4 титан замещает кремний
Р тетраэдрах SiO4. В отличие от Sr2TiO4, в котором титан присутствует
? виде октаэдров, в титанате бария Ва2ТЮ4и в твердых растворах
*®а, Sr)2TiO4 титан находится в форме тетраэдров TiO4.
Температура плавления Ba2TiO4 по [392], равна 1820° С, а по
1348], I8600 С.
189
Кристаллы Ba2TiO4 растворяются в 4-н. уксусной и соляной
кислотах.
Титанат бария BaTiO3 образуется при нагревании смеси двуокиси титана ц
карбоната бария [142, 209, 277, 279, 330, 350* 393, 407]. Карбонат бария начинает
реагировать с двуокисью титана, по [363], при температуре выше 600° С, по [209],__
выше 740° С; по [279],— выше 700° С. Однако, как установили Тжебиатовский и
Войцеховская [393], при взаимодействии карбоната бария с двуокисью титана
ниже 1000° С вначале образуется в качестве промежуточных соединений более
щелочные титанаты, чем метатитанат. Эти промежуточные титанаты при темпера-
туре около 1300° С полностью разлагаются и получается BaTiO3 со 100%-ным вы-
ходом.
Кубо и Синрики [280] синтезировали BaTiO3 нагреванием брикетов двуокиси
титана с нитратом^бария при температуре 600—800° С. По данным [127], метати-
танат бария получается спеканием смеси гидроокиси титана, осажденной из серно-
кислого раствора, и нитрата бария при температуре 600° С.
Метатитанат бария выделяется в виде мелкокристаллического осадка при на-
гревании смеси свежеосажденных гидрата гидроокиси бария Ва (ОН)2 • 8Н2О и
гидроокиси титана с 1-н. раствором едкого натра под давлением 300—500 атм и
температуре 380—450° С [179]. 4 лашен [206] получил мелкокристаллический
осадок с размером частиц 1—5 жа добавлением диспергированной в воде гид-
роокиси бария к раствору тетрапропоксититана в пропиловом спирте.
Дербушир с соавторами [190] получил ВаТЮ3 прокаливанием двойного ок-
салата бария BaTiO (С2О4)2, а Тамман и Калсинг [396] — нагреванием гидрооки-
сей бария и титана до температуры 800° С.
Кристаллизация BaTiO3 наблюдается при охлаждении барий- и титансодер-
жащих стекол [222].
Несколько авторов наблюдали образование ВаТЮ3 в системе ВаО — TiO2
при изучении ее методами физико-химического анализа [21, 336, 348, 364, 392].
Бурсиан и Смирнова [23] охлаждением жидкой фазы получили монокрис-
таллы BaTiO3 размером в несколько сантиметров. Крупные монокристаллы BaTiO3
со структурой перовскита получили нагреванием смеси TiO2, ВаСО3 и ВаС12
с молекулярным отношением 1:1, 4 : 3,3 в синтеркорундовом тигле при темпе-
ратуре 1250° С в течение 12 ч [156, 163, 318, 319].
Маттиас [318], Бурбанк и Эванс [156] получили гексагональные кристаллы
BaTiO3 из расплава TiO2 и ВаСО3 в смеси углекислых калия и натрия, а Кей
[270] — из мелкокристаллического титаната бария нагреванием с расплавом сме-
си карбонатов калия и натрия при температуре 900° С в течение 20 ч с последую-
щим медленным охлаждением.
Маттиас [318] и Кей [270], наряду с гексагональными кристаллами, наблю-
дали образование из щелочного расплава моноклинных кристаллов BaTiO3.
Сасаки [94] вырастил монокристаллы BaTiO3.
Беляев и Соколович [5] установили, что в системе Na, К, Ba || TiO3,
СО3 метатитанат бария занимает обширное поле.
Метатитанат бария плавится конгруэнтно при температуре
1610° С, по данным [392], а по [348], — при температуре 1680° С.
Известно несколько полиморфных модификаций, в которых кри-
сталлизуется BaTiO3.
Кубическая модификация BaTiO3 со структурой перовскита
устойчива в интервале температур 120—1460° С [348]. Выше 1460°С
она превращается в гексагональную модификацию. Эрн [202] на
дифференциальной кривой нагревания BaTiO3 обнаружил двойной
эндотермический эффект при температуре 800° С, который, по его
мнению, обусловлен превращением кубической модификации в иную-
Однако других доказательств в пользу этого предположения автор
190
не приводит. По данным [156], гексагональные кристаллы BaTiO3,
полученные из расплава, переходят в кубические при охлаждении
их до температуры 1050° С.
Кубическая модификация ВаТЮ3 при охлаждении сохраняет
структуру перовскита, но превращается последовательно в тетра-
Рис. 40. Температурная зависимость спонтанной поляризации
BaTiO3, измеренная в направлении псевдокубической оси (001) [309].
тональную модификацию при температуре 120° С, в орторомбиче-
скую около 0° С и в ромбоэдрическую при —80° С [210 ]. Все три псев-
докубические модификации BaTiO3 обладают сегнетоэлектриче-
скими свойствами. Акулов и Мельгуй [3] показали, что все типы
псевдокубических решеток BaTiO3 можно получить из теории ди-
польных решеток и закона анизотропии, как простое следствие изме-
нения ориентации вектора спонтанной поляризации относительно
осей кристалла.
I Переход кубической решетки BaTiO3 в тетрагональную и ром-
боэдрическую наблюдается по спектру электронного парамагнит-
ного резонанса ионов Fe3+ в кристалле титаната бария [199].
I Фазовые переходы BaTiO3 происходят и под действием облуче-
ния нейтронами. Шнек [378] наблюдал превращение тетрагональной
модификации BaTiO3 в кубическую под действием быстрых нейтро-
нов в реакторе мощностью 4,2 • 108 п/см2 при температуре 80° С.
Кубическая фаза ВаТЮ3, по мнению Шенка, метастабильна, так
как при температуре 1100° С в результате облучения нейтрона-
ми мощностью 6,2 • 1018 п/см2 превращается в тетрагональную
фазу.
$ Кубическая (идеальная) модификация BaTiO3 относится к фед.гр.
РщЗт, а = 4,0011 кХ, (120° С) и 4,0040 кХ (200° С) [302].
В Тетрагональная модификация имеет деформированную решетку
, тнпа перовскита. Параметры ее решетки при температуре 20° С
Равны а = 3,9865, с ~ 4,025 кХ [302]. Блатхер с соавторами [163],
191
изучая пироэлектрические свойства, заключил, что тетрагональная
модификация имеет фед. гр. Р4тт. У орторомбической модификации
BaTiO3 фед. гр. Стт, ромбоэдрической модификации — R3m [210].
Рис. 41. Размеры псевдокубической элементарной ячейки BaTiO3 [226]. (V —
объем ячейки).
Рис. 42. Температурная зависимость диэлектрической проницае-
мости BaTiOo, измеренной по направлению псевдокубических осей
(010) и (001) [309].
с
При температуре —90° С параметры решетки последней а = 3,999 А,
а = 90° 14' [349]. . г
Гексагональная модификация BaTiO3 имеет фед. гр. ССй1тть,
z = 6, а = 5,735, с = 14,05 А [156, 200], кристаллы ее оптически
положительные, одноосные, п0 = 2,28, пе = 2,42 [104].
192
Диксон с соавторами [191] предполагает, что образование гек-
сагональной модификации BaTiO3 связано с частичной потерей
кислорода и образованием ионов Ti3+.
При охлаждении кубической модификации BaTiO3 происходят
последовательные фазовые превращения, сопровождающиеся изме-
нениями параметра псевдокубической решетки, поляризации и ди-
электрической проницаемости (рис. 40, 41, 42). Приблизительные
температуры сегнетоэлектрических переходов по данным, приведен-
ным на рис. 40, 41, 42, равны 120, 5 и —90° С. Сегнетоэлектрические
переходы в кристаллах ВаТЮ3 одновременно являются фазовыми
переходами, энтальпию и энтропию которых определяли несколько
авторов (табл. 24).
Таблица 24
Энтальпия (кН, кал/моль) и энтропия кал/моль град)
фазовых переходов BaTiO3
Из ромбоэдрической в орторомбическую Из орторомбической в тетрагональную Из тетрагональной в кубическую Литера- тура
—ан AS —АН AS —АН AS
8 14,3 12 0,04 0,07 0,06 16 22 15,5 26 0,058 0,076 0,054 0,091 10 47 50 47 0,12 0,125 0,12 [225] [163 367 [411 [399а]
Сегнетоэлектрические свойства ВаТ1О3 открыли Вул и Гольдман [41—43].
Вначале авторы установили, что на кривой зависимости диэлектрической проница-
емости титаната бария от температуры имеется максимум. Затем они получили
образец титаната бария, который не обладал сегнетоэлектрическими свойствами
[44]. Рентгеноструктурные исследования позволили установить, что несегнето-
электрическая модификация имеет кубическую структуру типа перовскита с па-
^►аметрами а — b = с = 4,04 А, 0 = 90°. Эту модификацию ранее получила Мегоу
301]. Сегнетоэлектрической является тетрагональная модификация с парамет-
рами решетки д==3,98 и с=4,04 А. Иого [251] установил, что параметры тетраго-
нальной решетки в интервале температур 17—85° С изменяются по закону
'1 а = 3,98429 + 5,4216 • 10~5/ + 2,3562 • К)-7/2,
I с = 4,022 698 — 6,36 • 10 ~7t — 6,6081 • 10~7/2.
Превращение тетрагональной модификации в кубическую происходит при
В неполяризованном состоянии кристаллы BaTiO3 имеют ку-
бическую решетку и не обладают пьезоэлектрическими свойствами.
® силу кубической структуры кристаллов BaTiO3 в них имеется
система из трех осей (100), система из шести осей (ПО) и система
Из четырех осей (111), по направлению которых может происходить
спонтанная поляризация. При понижении температуры кристал-
ЛЬ1 BaTiO3 последовательно поляризуются по направлениям (001),
о 10) и (111).
«-S14 193
Кристаллохимические данные свидетельствуют в пользу обра-
зования структуры BaTiO3 из октаэдров ТЮб“. Косвенное под-
тверждение существования октаэдров вытекает из результатов изу-
чения ИК-спектров. Исследуя ИК-спектры отражения BaTiO3, Де-
мешина и Мурзин [53] обнаружили частоту v3 ~ 185 см~\ соответ-
ствующую колебаниям ионов Ва2+ относительно октаэдра TiOl".
Показано путем расчета колебательных частот в ячейке BaTiO3
и сопоставления полученных данных с результатами экспери-
ментальных измерений, что связь в титанате бария ковалентная
[189]. Ковалентный характер связи подтверждается исследованиями
эмиссионных рантгеновских спектров [120].
Появление сегнетоэлектрических свойств в титанате бария Шу-
ваев [120] объясняет большим зарядом и малым ионным радиусом
титана. Это создает условия для перемещения атомов титана в пре-
делах TiOe~-октаэдров.
Нельсон [325] показал, что сегнето- и антисегнетоэлектриче-
ские соединения типа перовскита могут образовывать только ионы
с незаполненными d-орбиталями.
j Климовский [275] изучил влияние давления (о) на свойства
монокристаллов BaTiO3. При давлении менее 3500 кг/см2 точка
Кюри (Tk) кристалла понижается линейно со скоростью dTk/da =
= —4,8 • 10~3 град • см2/кг. Скрытая теплота перехода сегнето-
электрической модификации в параэлектрическую при давлении
менее 3500 кг/см2 равна около 37 кал/моль, а выше 3500 кг/см2 —
около 12 кал/моль.
Бобович и Бурсиан [26] обнаружили в монокристаллах BaTiO3,
выращенных из расплава ВаС12, спектр комбинационного рассе-
яния, состоящий из трех полос — 500, 550 и 695 см~1.
Метатитанат бария образует твердые растворы с титанатами стронция, свинца
и другими, в которых также проявляются сегнетоэлектрические свойства BaTiO3.
Исследованию твердых растворов BaTiO3 посвящено много работ в связи с большим
их значением для производства сегнетоэлектрических материалов [1, 39, 106, 118,
151, 162, 248а, 308, 337, 351, 365, 366, 381, 417]. Шолохович и Ходаков [121] пока-
зали, что в монокристаллах твердого раствора BaTiO3 с BaSnO3 сегнетоэлектри-
ческие свойства выражены более ярко, чем в поликристаллических образцах. При
вхождении в решетку метатитаната бария Со, Ni, Nb и Та наблюдается закономер-
ное увеличение параметра решетки [66]. Вблизи точки Кюри параметр решетки
BaTiO3, содержащего примеси, равен 4,008 А .
В метатитанате свинца растворяется до 32 мол. % BaTiO3 [65]. Малые количе-
ства Sb3+ (до 0,1 вес. %) дают смешанные кристаллы с BaTiO3 типа (Ba, Sb3+)
(TH", Ti3-!-) О3. В решетке этих кристаллов некоторые места титана остаются пусты-
ми. При содержании Sb3-^ более 0,25 вес. % образуются твердые растворы в резуль-
тате замещения двух ионов Ti4-^" на ионы Sb3+ и Sb5+ [204].
Подобно Ba2TiO4 метатитанат бария растворяется в уксусной
и 4-н. соляной кислотах [278].
При нагревании двуокиси титана и карбоната бария с хлористым
барием после выщелачивания спека разбавленной соляной кислотой
194
a
5
Рис. 43. Диаграмма состояния системы ВаО — TiOa^
а — по Резу и Рою [348]; б — по Щепочкиной [126].
получается дибариевый трититанат 2ВаО • ЗТЮ2 [168, 170, 171,
279]. Кристаллы этого титаната имеют удельный вес 5,91 и отли-
j чаются большим двойным лучепреломлением. Этот же титанат мож-
но получить спеканием двуокиси титана с окисью бария при тем-
пературе 1300° С в течение 1 ч [278] и нагреванием брикетов из дву-
окиси титана и нитрата бария до 1000° С [280].
В результате оптических и рентгеноструктурных исследований
установлено, что кристаллы с большим содержанием титана, чем в
BaTiO3, имеют тетрагональную (псевдокубическую) структуру [164].
Кристаллы 2ВаО • 3TiO2 нерастворимы в разбавленных кисло-
тах, но растворяются в концентрированной соляной кислоте.
При исследовании системы ВаО— TiO2 был обнаружен барие-
вый дититанат ВаО • 2TiO2 [348, 364, 392]. Это соединение устой-
чиво в интервале температур 1210—1322° С [348], плавится инкон-
груэнтно при 1322° С, по [348], и при 1315° С, по [392].
Демоли [193] сообщил о получении им титаната бария состава
ВаО • 3TiO2. Существование его в системе ВаО — TiO2 подтвер-
дили и другие авторы [67, 336, 348]. По мнению Реза и Роя [348],
он плавится при температуре 1357° С. Келер и Карпенко [67]
сообщили, что ВаО • 3TiO2 образует кристаллы ромбической сис-
темы в виде табличек с ярко выраженной спайностью в двух направ-
лениях под углом 65°, показатели преломления Ng = 2,43, Np =
= 2,39. Однако Вайнштейн с соавторами [40], основываясь на ре-
зультатах исследования тонкой структуры К-спектров поглощения,
высказал сомнение в существовании этого титаната в качестве инди-
видуального химического соединения.
Сканави [98] получил бариевый тетратитанат ВаО • 4TiO2,
существование которого было подтверждено [67, 161, 348, 336,
369, 364]. По [392], ВаО • 4TiO2 плавится инконгруэнтно при тем-
пературе 1465° С, по [348], — при 1428° С. Кристаллы ромбической
системы с показателями преломления Ng = 2,42 и Np — 2,35 [67].
Диаграмма состояния системы ВаО — ТЮ2 построена многими авторами [67,
126, 348, 392] (рис. 43а, б). Келер и Карпенко [67] на построенной ими диаграмме
состояния уточнили превращения ниже кривой солидуса. Они подтвердили сущест-
вование соединения ВаО • 3TiO2 и твердого раствора TiO2 с метатитанатом бария’
Энтальпии образования титанатов бария приведены в табл. 23.
СОЕДИНЕНИЯ С НИЗШИМИ ОКИСЛАМИ ТИТАНА
Эти соединения изучены еще недостаточно.
Окись магния с Ti2O3 образует соединение MgTi2O4 (см. гл. П)>
которое с титанатом магния Mg2TiO4 может давать твердый раствор
состава nMg2TiO4 • mMgTi2O4 [86].
При восстановлении титанатов магния с содержанием 5—10%
MgO водородом при температуре 1200° С получается окисел Ti3Qy
образующий твердый раствор состава mMgTi2O5 • nTi3O5 [254J-
1%
В результате восстановления смесей с содержанием 15% MgO
образуется твердый раствор на основе метатитаната магния и Ti2O3
состава /nMgTiO3 • nTi2O3. Рентгенограмма mMgTiO3 • nTi2O3
сходна с рентгенограммой минерала титановых шлаков аносовита.
Спеканием SrO и Ti2O3 в вакууме получено соединение SrTiO2,5
со структурой перовскита (а = 3,902 А), в котором V6 часть по-
зиций кислорода остается вакантной [2681.
Г ПЕРОКСОТИТАНАТЫ МЕТАЛЛОВ II ГРУППЫ
Меликов и Писаржевский [312, 315] получили перекисный ти-
танат бария ВаО2 • ТЮ3 • 5Н2О. Суцуки и Тамизава [382] по-
казали, что термограммы ВаО2 • TiO3 • ЗН2О и TiO3 • ЗН2О до
температуры 320° С идентичны. При температуре 600° С пероксо-
титанат бария разлагается с образованием BaTiO3 и выделением
воды и кислорода.
I ТИТАНАТЫ МЕТАЛЛОВ III ГРУППЫ
|< Все элементы III группы, за исключением бора, по-видимому,
могут образовывать титанаты. Взаимодействие окислов титана и
бора еще не изучено, но следует ожидать, что образующиеся при
этом соединения будут относиться к боратам, так как кислотные
свойства у бора выражены в большей степени, чем у титана.
ТИТАНАТЫ АЛЮМИНИЯ
В системе А12О3 — ТЮ2, изученной методами термического ана-
лиза [160, 281], обнаружен только один титанат А12О3 • ТЮ2.
Вартенберг и Реух [421] описали соединение А12О3 • TiO2, но
существование его оспаривается Бунтингом [160].
К Титанат алюминия А12О3 • ТЮ2 кристаллизуется из спла-
' вов в тройных системах А12О3 — TiO2 — SiO2 [217] и
А12О3 — TiO2 — ZrO2 [32].
!Бунтинг [ 160 ] получил титанат алюминия сплавлением эквимоль-
ных количеств А12О3 и ТЮ2, Сигурдсон и Коле [369] — нагреванием
брикетов из смеси TiO2 и А1 (ОН)3 при температуре 1500° С в те-
чение 3 ч. Взаимодействие А12О3 с ТЮ2 наблюдается только при
температуре 1440° С и протекает медленно [233].
К Титанат алюминия кристаллизуется в ромбической^ системе,
параметры решетки таковы: а = 9,40, b = 3,36, с = 9,95 А, ррентг =
= 3,77 г!см3, z = 8, фед. гр. С2221 [235]. Плавится он конгруэнтно
при температуре I8600 С [160, 281]. Поданным термического ана-
- лиза, Al2O3-TiO2 существует в двух полиморфных модификациях —
аир — с точкой перехода при температуре 1820° С [281]. В системе
А12О3 — ТЮ2 (рис. 44) имеются две эвтектические точки при 1705° С
(23 вес.% А12О3) и 1840° С (61,5 вес.% А12О3). Показатели преломле-
ния ALO3 • TiO2 = 2,06, Np = 2,035 [22].
197
Титанат алюминия образует твердые растворы с MgO • 2TiO,
[33, 307, 369]. В бинарной системе MgO • 2TiO2 — MgO • 2TiO2
Юшида и соавторы [428] обнаружили эвтектическую точку при
температуре 1580° С (7 мол. % А12О3 • TiO2), хотя, по их данным,
между этими соединениями наблюдается полная взаимная раство-
римость.
Энтальпия образования А12О3 • ТЮ2 из окислов ДЯгэв =
= 2,7 кал/моль [34], энтропия 3°98 = 26,1 кал/моль • град [265].
Титанат алюминия является существенной составной частью
стеклокристаллических материалов. Исследование кристаллических
Рис. 44. Диаграмма состояния си-
стемы AI2O3 — TiOa [281 ].
Рис. 45. Диаграмма состояния си-
стемы LaaO3 — ТЮг [311].
яснить, что в прозрачный стеклокристаллический материал могут
быть превращены только те стекла, в которых содержание TiO2
превышает количество А12О3, находящееся в шестерной координа-
ции [62]. Механизм кристаллизации следующий. Первая порция
TiO2 соединяется с А12О3, находящимся в шестерной координации,
и образует соединение А12О3 • TiO2. При охлаждении стекла это
соединение выделяется в виде субмикроскопических ликватов и
служит будущим центром кристаллизации.
ТИТАНАТЫ ИТТРИЯ
Известен один титанат иттрия Y2O3 • 2TiO2, имеющий, по
данным рентгеноструктуорных исследований, решетку пирохлора с
параметром а = 10,093 А [357].
ТИТАНАТЫ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
Макчезни и Зауер [311] изучили систему La2O3 — TiO2 мето-
дом плавкости и построили диаграмму состояния (рис. 45). В ней
198
обнаружены три титаната: La2O3 • TiO2, La2O3 • 2TiO2 и
2La2O3 • 9TiO2 и область расслаивания двух несмешивающихся
жидкостей в интервале концентраций 85—95 вес. % TiO2. Соединение
Ьа20з ’ ТЮ2 имеет структуру пирохлора [311, 342].
К Двуокись церия с двуокисью титана образует соединение
СеО2 • TiO2 [231 ] неодим — титанат Nd2Ti2O7 с искаженной струк-
турой пирохлора [311].
Ж Титанаты типа А2В2О7 с кубической структурой пирохлора, по
данным рентгеноструктурных исследований, дают Sm, Gd, Dy,
Jb [311 ]. Титанаты Sm2Ti2O7 и Yb2Ti2O7 имеют параметры решеток
10,228 и 10,030 А соответственно [311].
L Двухвалентный европий образует метатитанат EuTiO3 изострук-
турный с кубической модификацией SrTiO3. Параметр решетки
EuTiO3 а = 3,897 кХ [157].
9 Квейрукс [343, 344] методом рентгеноструктурного анализа ус-
тановил существование титанатов гадолиния Gd2TiO5 и Gd2Ti2O7.
У первого соединения моноклинная решетка типа CaF2 с параметра-
ми а = 15,75, о = 5,326, с — 15,73 А, 0 = 87° 34'. Оно устойчиво
до температуры 1300° С. Фаза, близкая по составу к Gd2TiO5,
имеет гексагональную решетку с параметрами а = 3,672, с = 11,907 А,
/устойчива при температуре 1700° С.
J Второе соединение имеет структуру пирохлора. Область гомо-
генности этой фазы лежит в интервале концентраций 27—33,3 мол. %
$л}б2О3. Параметр решетки изменяется в пределах 10,168—10,180 А.
Н Диспрозий образует титанат аналогичного состава [344]. Сое-
динение Dy2TiO5 имеет параметры моноклинной решетки, а = 15,56,
b = 5,26, с = 15,35 А, 0 = 87° 38'. Близкая по составу гексаго-
нальная фаза имеет параметры решетки а — 3,632 и с — 11,837 А
и устойчива в интервале температур 1330—1650° С. Соединение
Dy2Ti2O7 со структурой пирохлора имеет параметр решетки в об-
Iласти гомогенности (33—52,2 мол. % Dy2O3). 10,110—10,360 А.
I Кноп с соавторами [272] получил титанат эрбия Er2Ti2O7
и определил параметр его решетки (структура пирохлора)
л = 10,0762 А.
I
I ТИТАНАТЫ АКТИНИДОВ
Титанат тория ThTi2Oe существует в двух модификациях [333].
К Низкотемпературная a-модификация устойчива в интервале тем-
ператур от комнатной до 1300° С. Она имеет ромбическую решетку
Эс параметрами а = 16,7, b — 14,95, с = 4,1 А, Ррентг — 6,2 г/см3.
Выше 1300° С устойчива P-модификация ThTi2Oe. Она кристалли-
.-зуется в тетрагональной системе с параметрами решетки а = 7,58,
с = 10,45 А. .
я Ламбертсон и Мюллер [288] в системе ТЮ2 — U3O8 при темпера-
туре 600—1500° С взаимодействия не обнаружили.
I При спекании таблеток из спрессованных двуокисей титана и
Урана образуется титанат UO2 • 3TiO2 [273, 299, 341, 377].
Маршалл и Госкатра [296] синтезировали уранат титана
Т^+ц^Об осаждениемам миаком из смеси растворов TiOSO4 ~l
+ UO2SO4 с последующим прокаливанием осадка выше 700° С. Это
продукт светло-оливково-зеленого цвета, «кристаллизуется в гек-
сагональной системе, а = 3,70, с — 3,90 A, z = 1/2, Ррентг = 6,58,
ризм = 6,72 г/см9. Устойчив до температуры 950° С. При более
высокой температуре разлагается на TiO2 и U8O8.
СОЕДИНЕНИЯ МЕТАЛЛОВ III ГРУППЫ С НИЗШИМИ ОКИСЛАМИ ТИТАНА
Спеканием La2O3 с Ti2O3 в вакууме получен смешанный окисел
LaTiO8 [267, 306]. Он имеет структуру перовскита с параметром
. о
1800 -
1700А
1600 4
1713°
Рутил+ж
1500-
1400._
0
SCO.
Две жидкости
1780°
1550°
Кристобалит+рутил
1830
О’
решетки а = 3,92 А, z=l,
ррентг = 6,26 г!см?.
О взаимодействии Ti2OE
и А12О3 см. в гл. II.
ТИТАНАТЫ МЕТАЛЛОВ
IV ГРУППЫ
ZZ7 4Р 60 80 100
Вес°/ ТЮг
Из металлов IV группы
титанаты образуют Ge, Zr,
Hf и Pb.
Кремний титанатов не
дает. Согласно [407], в си-
стеме ТЮ2 — SiO2 сущест-
вуют две несмешивающие ся
жидкости.
Рис. 46. Диаграмма состояния системы
SiO2— TiO2 [352].
Риккер и Хуммель [352]
считают, что в этой систе-
ме образуются двухсторон-
ние твердые растворы, что однако не подтвердил Девриз с соавто-
рами [197]. По его мнению, эта система эвтектического типа с
широкой областью двух несмешивающихся жидкостей (рис. 46).
Олово также не образует титанатов. При нагревании гелей
TiO2 и SnO2 до температуры 400—600° С получаются окислы TiO2
и SnO2 [305]. В системе TiO2 — SnO2 установлено существование
двухсторонних твердых растворов [287], выше 1350° С — наблюдает-
ся образование непрерывного ряда твердых растворов [342, 429].
Двуокись германия с двуокисью титана дает ограниченные твер-
дые растворы. Максимальная растворимость GeO2 в TiO2 (рутил)
40 мол. % (1050° С) [375].
Г температуре 1600° С и содержании около 50—55 мол % ZrO ДВу-
I малма™Тп”ствошЕН“Я7Д??’Т вми™е твеР™ Растворы. Матен-
Я%, раХортЙГт^ ворутиле "Р" температуре 1600"С
ZrO2 (тетрагональная модифи-
кация) 37%.
Однако Кофанур и соав-
торы [173] обнаружили в си- 1Япп_
к стеме ZrO2 TiO2 образование
титаната состава ZrO2 • TiO2 и
и построили диаграмму состоя-
1'ния системы (рис. 47). Этот ти-
танат встречается также и в
1 ?,и^мах мё° — Zr°2 — TiO2
I84 3 и А12°з - ZrO2 - TiO2
llozj. Существование его в си-
стеме ZrOa — ТЮ2 подтверди-
Или Броун и Дувец [154]. Это
соединение при температуре
320 С вступает в эвтектоидное
взаимодействие с моноклин-
Вной модификацией ZrO2.
«Максимальная растворимость
|ZrO2 в Т1О2 при температуре
1760° г ____________
примесь TiO2 понижает температуру превращения тверд
гвора ZrO2 с тетрагональной решеткой в твердый раствор с
линнои решеткой с 980° С до 340° С. Растворимость TiO.
линнои модификации ZrO2 незначительна.
окко [183] обнаружил, что твердые растворы на основе TiO,
уществуют до содержания 23 вес. % ZrO2, на основе ZrO2 • TiO,—
«оразуются в интервале концентраций от 56 до 64% ZrO,. Предел
186?°РИМОСТИ Т*°2 В тетРагональной модификации ZrO2 15 мол. %
Энтальпия образования ZrO2 • TiO2 из окислов Д^98 =
== — 3 ккал!моль, S298 = 22,2 кал/моль град [35]. Фогель [4101
изучил электрические свойства ZrO, • TiO,
а А*
1820
T8p.ZrOz TCOZ
1840°
1760
ТИТАНАТЫ ЦИРКОНИЯ
Совман и Эндрю [385] в системе ZrO2 — TiO2 образования тита-
натов не наблюдали. По их данным, система ZrO2 — TiO2 относится
к эвтектической простого типа. Эвтектическая точка лежит прй
1700-
1600
Тетраг.
тб.р. ZrO2
।
-Ц 1---------1—-----
40 60 80 100
Мол°/0 Т10,
Диаграмма состояния системы
ZrO2 — TiOa [173].
С данными (385] и равна около 18 мол.%,
твердого рас-
। с моно-
'2 в мо-
о
Zr0z
Рис. 47.
?0
ТИТАНАТЫ ГАФНИЯ
I Соединение НЮ2 • TiO2 кристаллизуется в ромбической сис-
— hiO2 образуются также твердые растворы наоснове
₽есистемеРТЮТрЫ^П^11е^-И ° = 4’77, Ь = 5’°5, с = 5,58 А 1473
р iO2 и НЮ2.2
200
201
ТИТАНАТЫ СВИНЦА
Спеканием окиси свинца и двуокиси титана получено два ти-
таната — 2РЬО - TiO2 и РЬО • TiO2. Красная окись свинца начи-
нает взаимодействовать с анатазом при температуре 350° С, с ру-
тилом при 390° С [153]. В интервале температур 400—500° С про-
исходит преимущественно окисление РЬОдоРЬ3О4, а выше 500° С —
образование титанатов свинца. Кристаллы PbTiO3 получены также
методом гидротермального синтеза при нагревании гидроокисей
титана и свинца с 1-н. раствором NaOH в автоклаве [179].
Систему РЬО — TiO2 изучили Беляев и Нестерова [4 ] методом
термического анализа. В этой системе установлено два инконгруэнт-
но плавящихся соединения — Pb2TiO4 (т. пл. 1024° С) и PbTiO3
(т. пл. 818° С).
Комори [259] разработал метод выращивания монокристаллов
PbTiO3.
Матсуо и Сасаки [304] обнаружили образование твердых раство-
ров PbTiO3 с РЬО. Они отрицают существование титаната 2РЬО- TiO2.
Титанат свинца обладает сегнетоэлектрическими свойствами с
точкой Кюри 800° К [88]. По диэлектрическим свойствам РЬТЮ3
сходен с титанатом бария BaTiO3 [410].
По данным термографических исследований, PbTiO3 имеет эндо-
термический эффект фазового превращения при температуре 490° С
[207]. Превращение относится к типу переходов сегнетоэлектрик —
параэлектрик и связано с превращением тетрагональной решетки
в кубическую.
Титанат свинца дает твердые растворы с титанатом кальция
[90] и стронция [61]; с цирконатами стронция [109], бария [108]
и свинца [105, 117, 249]; с метаниобатомсвинца [117]; со станнатом
кальция CaSnO3 [113]; с ферратами лантана LaFeO3 [1101 и висму-
та BiFeO3 [112, 114], с алюминатом лантана LaAlO3 [111 ] и с дру-
гими соединениями. Твердые растворы PbTiO3 с метатитанатами
кальция, стронция и бария обладают сегнетоэлектрическими свой-
ствами.
Изучены диэлектрические свойства и структура твердых раст-
воров в системе PbTiO3 — SrTiO3 — Bi3/2 TiO3 [107].
Исследованы ИК-спектры титанатов CaTiO3, BaTiO3 и PbTiO3
в области 2—300 мкм [93]. В спектре обнаружены две интенсивные
полосы с триплетной структурой около 550 и 360 см~л. В области
малых частот отчетливо проявилась полоса поглощения около 180-—
190 см"1-
ТИТАНАТЫ МЕТАЛЛОВ V ГРУППЫ
К настоящему времени изучены только титанаты ниобия и
висмута. Ванадий титанатов не образует. В системе V2O5— TiO3,
которая исследована методом термического анализа, обнаружена
небольшая взаимная растворимость окислов [4].
202
ТИТАНАТЫ НИОБИЯ
$ По данным [356], в системе Nb2O5— TiO2, изученной методом
«термического анализа, образуется два конгруэнтно плавящихся
соединения (рис. 48), имеющие состав 3Nb2O5 • TiO2 (т. пл. 1480° С)
й Nb2O5 • TiO2 (т. пл. 1490° С). На диаграмме состояния имеются
«три эвтектические точки при содержании TiO2 21 мол. % (1465° С),
около 38 мол. % (1465° С) и окло 58 мол. % (1475° С). По-видимому,
соединения двуокиси титана с пятиокисью ниобия следует рассмат-
ривать не как титанаты, а как ниобаты, так как кислотная функция
1500-
1480-
1450-
T6.p.TiO2
Ж
60
100
NtbOc
48. Диаграмма состояния си-
стемы Nb2O5—TiO2 [356].
20 40
о
TiOz
Рис.
ячейкой p-Nb2O5 и
у ниобия более сильно выражена,
чем у титана [360].
И Гольдшмидт [218] изучил систе-
му Nb2O5 — TiO2 методом рентгено-
]структурного анализа. Он отметил,
что в р> Nb2O5 (высокотемператур-
ная модификация) растворяется
по 30 мол. % TiO2 и подтвердил
существование в этой системе фазы
Nb2O5 • TiO2. Последняя кристал-
зуется в моноклинной системе,
(вдементарная ячейка ее сходна с
оноклинной : *
меет следующие параметры: а =
b = 3,76, с = 20,2 кХ, р = 120°. Автор отрицает
существование в системе самостоятельной фазы 3Nb2O5 • Т1О2.
Io его наблюдениям, между p-Nb2O5 и Nb2O5 • TiO2 нет двух-
зной области. При высокой температуре в TiO2 (рутил) растворяе-
я до 20% Nb2O5, но при охлаждении этот твердый раствор рас-
адается на рутил и фазу Nb2O5 • TiO2.
I Рутил и двуокись ниобия образуют серию непрерывных твердых
створов.
| Уодслей [426] установил в системе Nb2O5— TiO2 существование
динения Ti2Nb10O29.
У ниобата титана TiNb2O7 моноклинная решетка с параметрами
= 11,93, b = 3,81, с = 20,44 А,
тг = 4,29 г/см3, z = 6, фед. гр. А2/т [421]. Он кристаллизуется
Двух полиморфных модификациях — ромбической и моноклинной.
араметрыо ромбической модификации равны: а = 28,50, b = 3,805,
= 20,51 А, фед. гр. Атта, моноклинной — а = 15,57, b = 3,814,
20,54 А, ₽= 113° 41'[427].
Соединение TiO2 • 12Nb2O5 кристаллизуется в моноклинной сис-
Ме с параметрами решетки а — 29,78, b == 3,821, с = 21,12 А,
94,9°, рИзМ = 4Ррентг = 4,53 г/см3, 2 = 2, фед. гр. С2
120° 10'; ризм = 4,25
203
ТИТАНАТЫ ВИСМУТА
4
Титанаты висмута привлекли внимание исследователей в связи
с проявлением сегнетоэлектрических свойств [100—102]. Титанат
висмута Bi4Ti3O12, полученный [59, 103, 387, 406], кристаллизуется
в тетрагональной системе с параметрами решетки а = 5,44, с =
= 32,83 А [387]. Он имеет сегнетоэлектрический фазовый переход,
температура которого, по данным
различных авторов, 730° С [59],
675° С [387], 643° С [103, 406],
670° С [92].
Субборао [388] сообщил о полу-
чении титаната висмута Bi2Ti4O11}
имеющего два фазовых перехода
при температурах 250 и 1200° С.
Выше 1285° С этот титанат разла-
гается с образованием Bi2Ti2O7 и
Bi4Ti3O12.
Беляев с соавторами [15] сделал
первую попытку построить диаграм-
му состояния системы Bi2O3 — TiO2.
В ней он обнаружил соединения
Bi24TiO38, Bi4Ti3O12 и Bi2Ti3O9. Ти-
танат Bi24TiO38 имеет кубическую
объемноцентрированную решетку с
параметром а = 9,05 кХ и пла-
вится при температуре 844° С. Тита-
нат Bi2Ti3O9 имеет фазовый пере-
Рис. 49. Диаграмма состояния си- температуре 180—260° С.
стемы Bi2O3 — Т1О2 [92]. гд r г jr
3 При повторном изучении систе-
мы Bi2O3— TiO2 (рис. 49) Сперан-
ская с соавторами [92] обнаружила титанаты — 4Bi2O3 • TiO2,
2Bi2O3 • 3TiO2 и Bi2O3 • 4TiO2, — плавящиеся инконгруэнтно при
температуре 865, 1210 и 1275° С соответственно.
СОЕДИНЕНИЯ МЕТАЛЛОВ V ГРУППЫ
С НИЗШИМИ ОКИСЛАМИ ТИТАНА
Система NbO2 — TiO2 характеризуется непрерывной серией твер-
дых растворов [219].
Келер [257] получил ортониобат и ортотанталат трехвалент-
ного титана и измерил параметры их кристаллических решеток.
Оба соединения имеют структуру типа рутила. Параметры решеток
таковы: TiNbO4 — а = 4,712, с = 2,996 A, z = 1,98, роРеНтг = 5,1^
ризм = 5,04 г/W; TiTaO4 — а = 4,689, с = 3,07 A, z = 1,97>
Ррентг “ 7,204, ризм “ 7,10 г/см3.
204
ТИТАНАТЫ МЕТАЛЛОВ VI ГРУППЫ
Металлы VI группы в состоянии высшей валентности являются
кислотообразующими окислами и с титаном должны давать соли
соответствующих кислот. В состоянии низшей валентности они прояв-
ляют основные свойства и с двуокисью титана образуют титанаты.
К настоящему времени изучены только титанаты трехвалентного
хрома.
При спекании Сг2О3 и TiO2 при температуре 1440° С образуется
гитанат хрома Сг2О3 • 2TiO2 [233, 234 R Взаимодействие между
полуторной окисью хрома и двуокисью титана начинается при тем-
пературе около 1170° С. В системе Сг2О3 — TiO2 — А12О3 был обна-
ружен титанат хрома, вероятный состав которого выражается фор-
мулой Сг2О3 • TiO2 [236, 237].
d| При нагревании анатаза и брукита с окисью хрома взаимодей-
ствие между ними не наблюдалось [205]. Рутил при температуре
1200° С медленно реагирует с окисью хрома. До 3,5 мол. % Сг2О3
растворяется в рутиле, образуя твердый раствор. При содержании
около 12 мол. % Сг2О3 дает сложный окисел со структурой нестехио-
метрического рутила.
V В системе СаО — Сг2О3 — TiO2 при температуре 1000° С японские
авторы [390] не обнаружили титанатов хрома.
К Азо с соавторами [138] спеканием двуокиси титана с окисью
хрома в отношении 1 : 3 (мол.) при температуре 1300° С получил
термистер.
№ ТИТАНАТЫ МЕТАЛЛОВ VII ГРУППЫ
Металлы VII группы так же, как
и металлы VI группы, образуют ти-
танаты, находясь в состоянии низ-
шей валентности. Известны пока
только титанаты марганца.
К. В системе МпО — TiO2 получены
^соединения Mn2TiO4 иМпТЮ3 [214,
418]. Диаграмму состояния этой
системы построили Грив и Уайт
'{214] (рис. 50).
При спекании аморфнрй двуоки-
си титана с карбонатом, марганца в
Соотношении Мп : Ti = 2 : 1 при тем-
пературе 500 и 1000° С в атмосфере
‘ водорода, насыщенной парами во-
•$ЙЫ, образуется титанат Mn2TiO4
Рис. 50. Диаграмма состояния си-
стемы МпО — ТЮг [214].
1298]. Это соединение можно получить также нагреванием
смеси TiO2 + МпСО3 в атмосфере азота. При медленном охлаж-
дении спека образуется модификация (а-1) с тетрагональной
205
решеткой, параметры которой равны а = 8,7261, с = 8,567 А. При
быстром охлаждении получается кубическая модификация (Р-1)
с гранецентрированной решеткой. Она имеет пространственную
группу Fd3m и структуру типа шпинели, а = 8,679 А.
Переход а -> £ происходит при температуре 770° С. Обе модифи-
кации Mn9TiO4 обладают ферромагнитными свойствами. Метатита-
нат марганца MnTiOg (368 ] имеет структуру ильменита и встреча-
ется в природе в виде минерала пирофанита._
Виллере и соавторы Т409] определили температуру Кюри Mn2TiO4
78° К. Эти же авторы получили твердые растворы Мп^х) V^i-x) •
• Tix+ О4 и изучили их магнитные свойства.
Лецерф и Гарди [292, 293] получили MnTi2O4 (см. гл. II).
ТИТАНАТЫ МЕТАЛЛОВ VIII ГРУППЫ
ТИТАНАТЫ ЖЕЛЕЗА
К настоящему времени исследованы только титанаты подгруп-
пы железа.
Титанаты железа представляют большой практический интерес,
так как они находятся в основе минералов титановых руд (кричто-
нита, аризонита, псевдобрукита и др.). Железо, по-видимому, мо-
жет образовывать титанаты, будучи в двух- и трехвалентном состо-
янии. Взаимодействие окиси железа с двуокисью титана, однако,
изучено еще недостаточно.
В системе FeO — TiO2 обнаружены титанаты 2FeO • TiO2, FeO •
• TiO2 [334] и FeO • 2TiO2 [74, 76, 85, 125]. Дититанат железа
впервые получен Щепочкиной [125] при исследовании системы
FeO — TiO2. Его можно синтезировать нагреванием вюстита и TiO2
X (анатаз) при температуре 1200° С в течение 21 ч в атмосфере СО +
+ СО2 [76]. Дититанат железа образуется обычно наряду с другими
твердыми фазами. Выход его зависит от соотношения между содер-
жанием окиси и двуокиси углерода в газовой фазе. Иногда полу-
чается чистый дититанат железа без примеси других твердых фаз.
Наиболее часто он получается в чистом виде при содержании в
__ газовой фазе 50% СО,
Титанаты Fe2TiO4 и FeTiO3, по данным [334], образуют между
собой твердые растворы, что отрицает Новохатский с соавторами
[76].
Поулард [334] сообщил о получении титаната 2Fe2O3 • 3TiO2
с параметрами решетки а = 9,30, с = 9,50 А. Однако Бертаут
и Дуриф [152] отрицают существование этого соединения. Они
показали, что в результате нагревания смеси окислов Fe2O3 и TiO2
в соотношении 2 : 3 (мол.) при температуре 1200° С образуется псев-
Добрукит Fe,O3 • TiO2. ~ " 7
По мнению [191, 2581, в системе Fe2O3 — TiO2 существует только
одно соединение Fe2O3 • TiO2 (в «сухом виде»). В гидротермаль-
206
FeO 2FeOTtO? FeO-TiO, FeO-2TiO? TIO
2 Bee.% 2
5
Рис. 51. Диаграмма состояния системы FeO — TiOa:
a — по Щепочкиной; б — по Макчезни и Муану [310].
ных условиях (в «мокром виде») кристаллизуется второе соединение
Fe2O3 • 3TiO2, сходное с аризонитом.
Титанаты Fe2TiO4, FeTiO3 и FeTi2O5 образуются в тройной
системе FeO — Fe2O3 — TiO2 [386, 398].
Диаграмму состояния системы FeO — ТЮ2 построили авторы
£28, 76, 125, 310], однако, между их данными нет еще полного со-
гласия.
По [28], в этой системе существует два титаната железа --
Fe2TiO4 и FeTiO3, образующих между собой твердые растворы.
На диаграмме состояния [125] при-
<$ ведено также соединение FeTi2Or
(рис. 51, а). Макчезни и Муан [310]
построили диаграмму состояния,
сходную с диаграммой [125]
(рис. 51, б).
Недавно система FeO — TiO2 была
повторно исследована Новохатским с со-
авторами [76]. Он с помощью термодина-
мических расчетов установил, что дитита-
нат железа FeTi2O5 образуется в термоди-
намически неустойчивой системе FeO —
TiO2 (брукит) (рис. 52, а). В термодина-
мически устойчивой системе FeO — TiO2
(рутил) FeTi2O5 не существует (рис. 52, б).
Поэтому диаграмма состояния [28], на
которой отсутствует FeTi2O5, относится к
равновесной системе FeO — TiO2 (рутил),
а диаграмма состояния [125] — к нерав-
новесной системе FeO — TiO2 (брукит).
Дититанат Fe2TiO4 имеет ку-
бическую решетку с параметром
а = 8,51 А [74]. Метатитанат
FeTiO3 кристаллизуется в тетраго-
нальной системе. Он лежит в ос-
нове природного минерала ильме-
у нита, описанного в главе I, где
приведены также и параметры ре-
Рис. 52. Диаграмма состояния си-
стемы FeO—Т1О2 [76]:
а — FeO — TiO2 (рутил); б — FeO — ТЮ2
(брукит).
шетки. У дититаната FeTi2O3 ромбическая решетка с параметрами
а = 3,77, b = 9,68, с = 9,84 А [125], ризм = 4,46 г/см3 [76].
Тодд и Кинг [399] измерили теплоемкость Fe2TiO4 и Fe2TiO5
в интервале температур 51—298° К. На кривой температурной
зависимости теплоемкости Fe2TiO4 есть максимумы при температу-
ре 56,0 и 99,1° К.
Энтальпия образования FeTiO3 ЛН298 = —299,7 (из элементов)
и —10,4 ккал/моль (из окислов) [73], изобарный потенциал AZ°98
= —280,4 (из элементов) и —9,7 ккал/моль (из окислов).
Энтропия Fe2TiO4 S298 = 39,0, энтропия Fe2TiO5 —
= 37,4 кал/моль • град [399].
208
Зависимость изобарного потенциала образования (кал/моль)
FeTi2O8, по [86], выражается уравнением
AZ = —507 050 4- 94,27 (1000 — 2000° К).
Однако Новохатский с соавторами [76 ] считает, что фактически
изучено равновесие реакции
FeTiOg -J- Н2 Fe 4- TiO2 (рутил) + Н2О
и найдена для нее зависимость AZ.
, Шмаль и соавторы [386] определили изобарные потенциалы
восстановления Fe2TiO4 и FeTiOg (ккал/моль) по реакциям
Fe2TiO4 + СО = FeO 4- FeTiO3 4- СО2, (1)
AZX = —2905 + 5,637
FeTiO3 + СО = Fe 4- TiO2 4- CO2, (2)
AZ2 = 5081 4- 1,477(800— 1000° C).
Белякова [30] вычислила свободную энергию разложения иль-
менита хлористым водородом по реакции
FeTiO3 4- V4O2 4- ЗНС1 = FeCl2 4- TiO2 + 172Н2О
в интервале температур 400—1200° С.
Термодинамические характеристики образования и восстанов-
ления дититаната железа приведены в табл. 25 [76].
Таблица 25
Изобарный потенциал и энтальпия образования дититаната железа
Реакция
AZy’ ккал/моль
127!»
ккал/молъ
FeO + 2TiO2 (рутил) == FeTi2O5
FeO 4- 2TiO2 (брукит) = FeTi2O5
TiO2 (брукит) TiO2 (рутил)
FeTi2O6 -|- CO — FeO 4- TiO2 (рутил) 4-
+ CO2
FeTi2O5 4~ CO = Fe 4~ TiO2 (брукит) 4~
4-CO2
—9560 4- 4,02 T
-11030 4-4,32 T
—730 4- 0,15 T
5222 4-1,196 T
6687 4- 0,898 T
—9,6
—11,0
-0,7
5,2
6,7
Характерной особенностью системы FeO — Fe9O3 — Ti09 является
существование твердых растворов между Ре2О3 (гематит) и титана-
тами железа. Природу этих твердых растворов исследовали несколько
авторов, но результаты их противоречивы.
Поулард [334] установил наличие в системе FeO — Fe2O3 — TiO2
двух твердых растворов между FeTiO3 и Fe2O3, имеющих состав
(Tit±^, Fel+, Fe1±s) О где 0 < у < V3 и (Fe^, Ti*+) О3, где
0 < X < Vg.
Шевалье с соавторами [181] показал, что твердые растворы
FeTiOg — Fe2O3, по данным рентгеноструктурных исследований,
14 9-614
209
можно отнести к шпинелям трех типов: х Fe3O4 • (1—x)Fe2TiO4,
#Fe3O4 • (1 — у) FeTiO3 и z Fe2O3 (1 — z) FeTiO3. Природные ми-
нералы магматического происхождения (титаномагнетиты и ильме-
ниты) представляют собой твердые растворы этих типов, в которых
Fe2+ частично замещается на Mg2+ и Mn2+ , a Fe3+ — на А13+.
Вебстер и Брайт [422] изучили тройную систему FeO—
—Fe2O3 — TiO2 при температуре 1200° С и установили образо-
вание трех твердых растворов: Fe2O3— FeTiO3, состав которого вы-
ражается формулой FemTi2_mO3; Fe3O4 — Fe2TiO4 состава FenTi3_nQ^
со структурой шпинели; Fe2TiO5 — FeTi2O5 с ромбической структу-
рой состава FepTi3 О5. Поля первичной кристаллизации всех этих
твердых растворов в системе FeO — Fe2O3 — TiO2 обнаружил Тей"
лор [398].
Тейлор [389] исследовал фазовые отношения в системе
FeO — Fe2O3 — TiO2 в зависимости от парциального давления кисло-
рода при температуре 1300° С. Автор установил существование
непрерывных твердых растворов между гематитом и ильменитом,
а также между титанатами Fe2O3 • TiO2 и FeO • 2TiO2.
Сплавы состава xFe2O3 (1 — х) FeTiO3, полученные нагревани-
ем смеси ильменита и гематита, при х < 0,17 парамагнитны, а
при 1 > х > 0,17 ферромагнитны [353].
Изучена также система FeO — Fe2O3 — TiO2 [429].
ТИТАНАТЫ КОБАЛЬТА И НИКЕЛЯ
trc
1800
1700
1600
1500
1400
1300
О 20 40 60 80 100
СоО Мол°/0 Т102
Рис. 53. Диаграмма состояния си-
стемы СоО— Т10г [289].
Левин и Мак Мурдье [289] по-
строили диаграмму состояния си-
стемы СоО — TiO2 и отметили в ней
существование трех титанатов ко-
бальта: 2СоО • ТЮ2, СоО • ТЮ2
и СоО • 2TiO2 (рис. 53). Согласно
[276], титанат кобальта Co2TiO3
получен спеканием двуокиси тита-
на с окисью кобальта при высо-
кой температуре. Об этом соедине-
нии ранее упоминалось в работе
[192], где ему приписывали струк-
туру шпинели.
Метатитанат кобальта CoTiO3,
синтезированный [276, 324], име-
ет ромбоэдрическую решетку с па-
раметрами а = 5,4846 А,а=55°01',
фед. гр. R3 [324].
4 . При низких температурах СоТЮ8 ащ^ферромагнешкUQ51. На
кривой его магнитной восприимчивости при температуре 38 К
имеется максимум (температура Нееля).
210
По [48], термодинамические константы CoTiO3 следующие:
Д//298 = —288,1 ккал!моль, = —269,0 ккал/моль, So98 =
23,8 кал/моль • град.
Метатитанат кобальта восстанавливается при температуре 1230—
1356° С окисью углерода с образованием дефектной структуры.
Более глубокое восстановление приводит к образованию металли-
ческого кобальта и двуокиси титана [48].
При спекании смеси окислов получается титанат трехвалентного
кобальта Co2Ti2O5 [276].
Финк [212] наблюдал образование титанатов кобальта — Co2TiO4,
CoTiO3 и CoTi2O5 — в качестве продуктов твердофазных реакций,
протекающих между спрессованными окислами СоО и TiO2 при
температуре 1370° С.
Никель дает метатитанат NiTiO3 со структурой ильменита [368].
Диникелевый титанат Ni2TiO3 со структурой шпинели упоминает-
ся в работе [192].
Г СМЕШАННЫЕ ТИТАНАТЫ
Под смешанными (сложными) титанатами понимаются соедине-
ния, образованные двуокисью титана с окислами нескольких ме-
таллов и неметаллов. В основу классификации смешанных титанатов
следовало бы положить химический состав и кристаллическую струк-
туру. Однако смешанные титанаты и их структура изучены еще
недостаточно. Наиболее достоверным и надежным признаком сме-
шанных титанатов остается пока только химический состав и со-
став систем, в которых они образуются.
г Поскольку все титанаты состоят из двуокиси титана и окислов
металлов и неметаллов, всеобъемлющей будет классификация
их на основе физико-химических систем, которая, однако, имеет
существенный недостаток. Она не учитывает химических свойств
соединений, и на ее основе невозможно сгруппировать титанаты
со сходными свойствами. Но она и не исключает дополнительной
классификации смешанных титанатов по химическим и структур-
ным признакам. Однако, несмотря на указанные недостатки,
мы придерживались классификации смешанных титанатов на ос-
нове физико-химических систем. Физико-химические системы нами
располагаются в порядке возрастания порядковых чисел элементов,
входящих в состав системы.
TiO2 — Li2O — Na2O — NaF. В этой системе методом термического анализа
обнаружено соединение 5Li2TiO3 • 13Na2TiO3 • 4NaF [95].
TiO2 — Li2O — L ’ — KF. Методом термического анализа установлено су-
ществование соединения lOLiF • Li2TiO3 • 28KF [96].
Ti02 — Li2O — A12O3. Получен смешанный окисел LiAlTiO4 со структурой
Шпинели [261].
TiO2 — Li2O — V2O3. Получено соединение LiVTiO4 со структурой шпинели,
параметр кубической решетки а = 8.35 А [150].
Ц*
211
TiO2 — Li2O — Cr2O3 . Выделено соединение LiCr3"^TiO4 co структурой шпи-
нели, параметр решетки а — 8,32 А [150, 261].
TiO2 — Li2O — Fe2O3. Синтезировано соединение LiFe3^“TiO4 со структурой
шпинели, а = 8,35 А [150].
TiO2 — Li2O — СоО. Получен смешанный титанат лития и кобальта
LiCoTi3O8 [252].
TiO2 — Li2O — ZnO. Получен титанат LiZnTi3Og [252].
TiO2 — Li2O — Rh2O3. Синтезировано соединение LiRhTi3 O8 co структурой
шпинели, параметр кубической решетки а = 8,36 А [150].
Ti09 — Li90 — CdO. Получен титанат LiCdTi3O8 [252].
TiO2 — Li2O — La2O3 . Установлено существование титаната (Li, La) TiO3 co
структурой перовскита, a = 3,861 A [157].
TiO2 — Na2O — NaF. В этой системе обнаружено соединение 3Na2TiO3 .
• 2NaF с температурой плавления 899° С [122], что не подтвердили авторы [95],
исследовавшие систему методом термического анализа. Они, однако, установили’
что в двойной системе образуется соединение 4Na2TiO3 • NaF, плавящееся инкон-
груэнтно при температуре 918° С.
TiO2 — Na2O • NaCl. Обнаружено тройное соединение 4Na2TiO3 • NaCl, крис-
таллизующееся в виде пластинок с показателями преломления 1,682 [18].
TiO2 — Na2O —1А12О3 — SiO2 — СаО. Принц [340] изучил систему альбит —
титанит (NaAlSi3O8 — CaSiTiO5 )и альбит — анортит — титанит (NaAlSi3O8 —
Ca2Al2Si3O8 — CaSiTiO5), но, кроме титанита, не обнаружил в них смешанных
кристаллов.
ТЮ2 — Na2O — SiO2 . При исследовании системы методом термического ана-
лиза установлено существование соединений Na2O • TiO2 • SiO2, 13Na.,0 .
. 13TiO2 • SiO2 и 13Na О . 13SiO2 • TiO2 [8].
TiO2 — Na2O — P®05. Получено соединение 6Na2O • 3TiO2 • 4P2O5 [329].
( TiO2 — Na2O — K2O. Изучено взаимодействие метатитанатов калия и натрия
и установлено, что они образуют между собой эвтектику, лежащую вблизи состава
K2TiO3 [326]. В средней части бинарной системы Na2TiO3 — K3TiO3 существуют
две несмешивающиеся жидкости.
В отличие от [326], в результате термографического и рентгеноструктурного
изучения системы K2TiO3 — Na2TiO3 авторы [16] обнаружили соединения K2TiO3-
. Na2TiO3 (устойчиво в интервале температур 800—900° С) и 2K2TiO3 • Na2TiO3
(существует ниже 800° С).
ТЮ2 — Na2O — Fe2O3 . В этой системе известен минерал фреденбергит [211].
По мнению [211], кристаллы его имеют гексагональную решетку с параметрами
а = 9,62, с = 22,4 А г = 6, рреНт = 4,38, рИЗм = 4,3а/сл«3. Уодслей [425] считает,
что фреденбергит имеет моноклинную решетку с параметрами а — 12,305, b =
= 3,822, с = 6,500 А, Р = 107° 18х, ризм = 4,30, рреНтг = 4,00 г/см3. Параметры
решетки фреденбергита близки к параметрам решетки бронзы Na0f2TiO2. На этом
основании Уодслей предлагает состав фреденбергита выражать формулой
Na0>22 (Ti, Fe) О2.
Байер и Гоффманн [147], спекая окись железа и двуокись титана с нитратом
натрия при температуре 100Э — 1200° С в течение 15 ч, получили соединения
Na2O • Fe2O3 • 6TiO2 и Na2O • Fe2O3 • 7TiO2. Оба соединения кристаллизуются в
моноклинной системе с близкими параметрами решетки (а = 12,25, b — 3,81,
с = 6,49 А, р = Ю7°8', ризм = 3,95 г/см3).
Ti0o — Nao0 — La9OQ. Получен титанат (Na, La) TiO3 co структурой перов-
скита, a — 3,865 A [157]. .
TiO2 — Na2O — Bi2O3. Синтезирован титанат (Na0,5 Bi05) TiO3, обладающий
сегнетоэлектрическими свойствами [91], точка Кюри лежит около 320° С. Это со-
единение имеет структуру перовскита с тетрагональными и ромбическими искажс-
212
(ГяИЯМИ, параметры решетки а = 3,891 А, а = 89° 36' [57]. Изучены твердые раство-
ры Nao,5Bio,5Ti03 с РЬТЮ3 [58].
TiO2 — NaF— А1РО4. При исследовании системы смешанных кристаллов не
обнаружено [260].
TiO2 — NaF — РЬО. Изучил Беляев [6].
ТЮ2 — NaCl. Беляев и Беляева [18] при температуре до 1100° С в этой си-
стеме наблюдали полную нерастворимость TiO2 в NaCl.
TiO^ — MgO — AI2O3 — СаО. Смешанные титанаты не обнаружены [132,.
369].
TiO2 — MgO — А12О3 — SiO2 — ZrO2. Система изучалась Бережным и Гуль-
ко (36].
TiO2 — MgO — Si02. В системе возможно образование соединения типа тита-
ната, но Бережной [31] не обнаружил его существования.
TiO2 — MgO — СаО. Установлено существование только простых титана-
тов — перовскита (CaTiO3) и гейкелита (MgTiO3). Из сплавов с избытком СаО крис-
таллизуется перовскит, а при избытке MgO гейкелит [31].
ТЮ2 — MgO — Сг2О3. Между титанатом магния Mg2TiO4 и хромитом магния
MgCr2O4 образуются неограниченные твердые растворы [37].
TiO2 — SiO2— А12О3. Титанатов тройного состава в системе не обнаружено
[50, 128, 149]. Спеканием порошкообразной смеси TiOs, А12О3 и ВаСО3 получен
смешанный титанат СаО • А12О3 • 2TiO2c удельным весом 4,06 [27].
TiO2 — SiO2 • AI2O3 — СаО. Изучены частные разрезы анортит — перовскит
£аО • А12О3 • 2SiO2 — CaTiO3) [328], анортит — титанит (СаО • А12О3 • 2SiO2 —
SiTiO5) [248, 340], геленит — перовскит (2СаО • А12О3 — SiO2 — CaTiO3)
[327], анортит — титанит — воллостанит (СаО • А12О3 • 2SiO2 — CaTiSiO5 —
CaSiO3) [245]. Однако, кроме титанита, других сложных титанатов в этой системе
не обнаружено. Система исследована в работе [129].
ТЮ2 — SiO2 — К2О. Тройные титанаты в ней не обнаружены [355].
ТЮ2 — SiO2 — СаО. Описаны два тройных соединения — CaTiSiO5 и
СаО • 2TiO2 • 2SiO2 [355]. Между f-CaSiO3 и CaTiO3 существует твердый
раствор [195], образующийся в результате частичного замещения Si4~^ на Ti4+ в
p-CaSiO3.
Титаносиликат CaTiSiO5, известный как минерал титанит или сфен, получен
•Нагреванием двуокиси титана и двуокиси кремния с хлористым кальцием [241,
242], а также нагреванием смеси СаСО3, TiO2 и SiO2 [145, 169, 247, 362].
Сфен кристаллизуется в моноклинной системе (см. гл. I). Удельный вес его
Д497, температура плавления 1382, по [247] 1386° С, по [340]; энтальпия образова-
ния АН<298-= 54,2 ккал/моль [35].
Гинсберг и Нигосьян [46] получили титаносиликат СаО • 2TiO2 • 2SiO2. Бе-
режной [31] в системе СаО — TiO2 — SiO2, кроме сфена, других смешанных тита-
Шатов не обнаружил.
TiO2 — SiO2 — СаО — CaF2. Исследованы псевдобинарные разрезы между эвтек-
тиками двойных систем TiO2 — СаО и TiO2 — SiO2 с CaF2 [227]. Самая низкая темпе-
ратура ликвидуса на первом разрезе (1320° С) приходится на состав 60% TiO2, 14%
раО и 26% CaF2. Образцы сплавов, охлажденные до температуры примерно 20° С,
^Одержат рутил, перовскит и флюорит. На втором разрезе низшая температура
^Давления сплавов (1220° С) приходится на состав 4,5% TiO2, 40,5% SiO2 и 55%
bF2- Сплавы, содержание большее количество TiO2 и SiO2, после охлаждения со-
держат кристобалит, 'Л ид и мит и стекловидную фазу, а при избытке CaF2 —
Ьекловидную фазу и флюорит.
ТЮ2 — SiO2 — ВаО. Спеканием смеси TiO2, А12О3 и ВаСО3 получены титано-
кликаты бария — BaTiSiO5 и BaTi2SiO7 с удельным весом 4,25 и 4,85 соответ-
Вгвенно [27].
тю2 - Р2О5 - К2О. Беляев и Сигида [9] изучили методом термического ана-
213
лиза систему К4Р2О7 — TiO2 до содержания 40 мол.% TiO2. Система оказалась
эвтектического типа. При содержании TiO2 более 31,5% в ней кристаллизуется
фаза тройного состава.
ТЮ2 — Р2О5 — ВаО. Спеканием смеси ВаНРО4 (9 вес. ч.) и TiO2 (1 вес. ч.) с
BaF2(2% от веса смеси) при температуре 1050° С выделен продукт состава
4 2Ва2Р2О7 • TiO2 [230]. Этот продукт является фосфором.
ТЮ2 — К2О — КО. Система исследована визуально-политермическим, крис-
таллооптическим и термографическим методами. В ней обнаружены тройные сое
динения — K,TiO3 • КС1 • TiOo, K.,TiO3 • 2КС1 • TiO2 и 6KoTi03 . KC1 - 5TiQ
[П, 17]. - - - - 2
TiO2 — K2O — Nb2O5. При нагревании смеси TiO2 (анатаз), K2CO3 и Nb2O5 в
молярных отношениях 2 : 1 : 1 и 6 : 1 : 1 при температурах 1150 и 1300° С соответ-
ственно, получены титанониобаты KTiNbO5H KTi3NbO9 [424]. Первое соединение
имеет ромбическую решетку с параметрами а — 6,459, b = 3,792, с = 18,472 А,
Ррентг = Ризм = г!см\ 2=4, фед. гр. Рпта или Рп2га. Параметры вто-
рого соединения таковы: а = 6,392, b = 3,785, с = 14,865 А, Ррентг = 3,85, ризм =
= 3,88 г!см3, z = 2, фед. гр. Рпта.
ТЮ2 — К2О — tn2O3. Обнаружены соединения с кубической структурой,
имеющие состав (К, La) TiO3 (а = 3,899 А) и (К, Се) TiO3 (а = 3,889 А) [157].
TiO2 — К2О — Bi2O3. Синтезировано соединение [Bi 0 5К0 5] TiO3, обладающее
___сегнетоэлектрическими свойствами сточкой Кюри 380° С [91]. Оно имеет структуру
перовскита с тетрагональными (а = 3,913, с = 3,993 А) и ромбоэдрическими (а =
=3,891 А, а = 89° 36') искажениями. Тетрагональная решетка сохраняется до тем-
пературы 270° С, а затем переходит в другую, еще неизвестной симметрии. Послед-
няя сохраняется примерно до 410° С. В точках фазовых переходов при темпера-
туре 270 и 410° С объем ячейки изменяется на одну и ту же (0,3 А ), но разную по
знаку величину. При температуре выше 410° С спонтанная поляризация отсут-
ствует.
ТЮ2 — СаО — Сг2О3 — ZrO2. Изучено взаимодействие между образующими-
ся в этой системе титанатами Cr2Ti2O7 и CaTi2ZrO7 [37]. Обнаружены твердые раст-
воры ограниченного состава на основе CaTi2ZrO7 при содержании его 65—100%.
TiO2 — СаО — ZrO2. Установлено тройное соединение СаО • ZrO2 • 2TiO2
215] с моноклинной решеткой и параметрами а — 12,43, b — 7,26, с — 11,35 А
3 = 100° 34', 2=8 [84]. Между метатитанатом и метацирконатом кальция обра-
зуется непрерывная серия твердых растворов [37].
TiO2 — Sc2O3 — MoO2. Спеканием окислов, взятых в стехиометрическом со-
отношении, получено соединение Sc3+Ti40+ Мо4+ О4 [148].
ТЮ2 — Ti2O3 — ВаО. Спеканием окислов получено соединение
с гексагональной структурой, сходной со структурой гекса-
гональной модификации BaTiO3 [191].
TiO2 — V2O5 — РЬО. Методом термического анализа обнаружено тройное
соединение, состав которого ориентировочно можно выразить формулой ЮРЬО •
• V2O5 . TiO2.
TiO2 — Mn2O3 — ВаО. Спекая TiO2, ВаСО3 и МпО2, Диксон с соавторами
[191] синтезировал ВаМп0 33Ti0 67О3 с гексагональной структурой.
TiO2— Fe2O3 — Cu2O. Кордес и Рёттиг [261] спеканием окислов получили
Cu+Fe3+TiO4 со структурой шпинели.
TiO2 — Fe2O3 — ВаО. Получено соединение BaFe0 33Т£0 б7О3 со структурой ти-
па титаната бария [191].
ТЮ2 — NiO — Sb2O3. В системе обнаружены только твердые растворы вблИ'
зи угла TiO3 [256].
214
TiO2 — Cu2O — Сг2О3. Получен смешанный титанат Cu‘“Cr3-^ TiO4co струк-
турой шпинели [261].
TiO2 — Rb2O— RbF. Система изучена методом плавкости [360]. В ней уста-
новлено получение соединения 3Rb2Ti2O5 • 2RbF (рис. 54).
TiO2 — Rb2O — Ln2O3. Получены кристаллы (Rb, La) TiO3 кубической си-
стемы с параметром решетки а = 3,895 А [157],
TiO2 — Cs2O — CsF. Система изучена методом плавкости [378], обнаружено
двойное соединение 3Cs2Ti2O5 • 2CsF (рис. 55).
Рис. 54. Диаграмма состояния системы Rb2Ti2O5 — RbF [378].
Рис. 55. Диаграмма состояния системы Cs2Ti2O5 — CsF [378].
TiO2 — Nb2O5 — Ln2O3. Гидротермальным методом синтезированы соедине-
ния типа АВ2Х6, где А — лантаниды от La до Lu, В — Nb и Ti, X — О [70]. Синтез
осуществлен нагреванием гидроокисей в автоклаве при температуре 300° С и дав-
лении водяного пара 40,6—87,6 кг!см2. Полученные соединения характеризуются
215
фед. гр. Pbnm и ромбической структурой природного минерального ряда эшинит —
приорит [69].При нагревании до температуры 750—950° С структура их изменяется,
превращаясь в структуру типа природного минерального ряда эвксенит —
поликраз, фед. гр. Рсап [71] (Табл. 26).
Таблица 26
Параметры решеток редкоземельных титанониобатов р-модификации
со структурой эшинит-приорита [70]
Формула
Формула
LaTiNbO6 5,45 11,01 7,60 TbTiNbO6 5,23 10,98 7,44
CeTiNbO6 5,43 10,99 7,56 DyTiNbO6 5,19 10,97 7,43
PrTiNbO6 5,38 10,99 7,54 ErTiNbO6 5,17 10,97 7,39
NdTiNbO6 5,33 10,98 7,51 TuNbTiO6 5,15 10,98 7,37
SmTiNbO6 5,29 10,98 7,48 LbTiNbO6 5,13 10,97 7,36
EuTiNbO6 5,28 10,98 7,47 YuTiNbO6 5,12 10,96 7,35
GdTiNbO6 5,27 10,98 7,45
Показано, что ряды Ln (Nb, Ti)2O6 и Ln (Та, Ti)2O6 морфотропные [2].
В обоих рядах лантаниды с большими ионными радиусами образуют соединения со
структурой типа эшинита (Ge, Ln) (Nb, Ti)2Oe, а малые — типа эвксенита
(Y, Ln) (Nb, Ti)2O6.
При замещении ниобия на тантал морфотропный переход происходит при лан-
танидах с малыми ионными радиусами.
ТЮ2 — Nb2O5 — ВаО. Получено соединение Ba6Ti2Nb8O30, имеющее тетраго-
нальную решетку с параметрами а — 12,54, с = 4,01 A, z = 1, фед. гр. Р4Ьт [370].
TiO2 — Nb2O5 — ВаО — РЬО. Сриканта и соавторы [366] сплавлением окислов
синтезировали серию соединений, относящихся к этой системе. Получено семь
групп соединений четверного состава: 1) кубические со структурой перовскита,
2) тетрагональные, изоморфные с высокотемпературной модификацией ВаТЮ3,
3) ромбические, изоморфные с ромбической низкотемпературной модификацией
BaTiO3, 4) кубические со структурой пирохлора, 5) ромбические изоморфные с
PbNb2O6, 6) тетрагональные со структурой типа натрий-вольфрамовой бронзы,
7) ромбические типа BaNb2O6.
TiO2 — МоО2 — Ln2O3. Брикснер [148] спеканием окислов, взятых в стехиомет-
рических количествах, получил соединения общего состава Ln3"^Ti^MoQ^O4, где
Ln—лантаниды.
TiO2 — RhO2 — ВаО. Диксон с соавторами [191], спекая TiO2 и ВаСО3 с RhO2,
приготовил титанат BaRu0 33Ti0 67О3 со структурой гексагонального ВаТЮ3.
4 TiO2 — Ag2O — La2O3. Получены кубические кристаллы состава (Ag, La) ТЮз
с параметром а = 3,874 А [157].
TiO2 — SnO2 — La2O3. В системе образуются смешанные кристаллы
состава Lao (Sn, Ti)2O7, содержание La2O3 в которых доходит до 27% (Sn, Ti) 09 —
до 13,3% [342].
TiO2 — ВаО — Bi2O3. Получены соединения BaBi4Ti4O15 и Ba2Bi4Ti5Ol8, ко-
торые являются сегнетоэлектриками [59J. В них наблюдается переход от ромби-
ческбьГструктуры к тетрагональной. При температуре 20° С Ba2Bi4Ti5018oHMeeT
ромбическую решетку с параметрами а — 5,514, b = 5,526 и с = 50,370 А [60].
При температуре около 310° С наблюдается фазовый переход в тетрагональную
структуру:
ромб. (С$ <г=тетраг. О($.
ТЮ9— La2O3— РЬО. Титанат лантана с титанатом бария дает твердые раст-
воры типа La^Baj—ХТЮ3 [254]. Кристаллы этих твердых растворов при х < 0,85—
216
0 9 имеют кубическую решетку типа перовскита, а при х > 0,9 — ромбическую
решетку.
TiO2 — Ln2O3 — WO2. Брикснер [148] спеканием окислов синтезировал сое-
/деления общего состава Ln3+Ti0i5W40+o4, где Ln — лантаниды.
ТЮ2 — La2O3 — Т12О3. Получены кристаллы (Ti, La) TiO3 кубической син-
ении с параметром решетки а — 3,875 А [ 157].
TiO2 — WO3 — РЬО. Титанат и вольфрамат свинца образуют соединение
предположительного состава 3PbTiO3 • PbWO4 [12].
TiO2 — IrO2 — ВаО. Спеканием окислов TiO2, ВаСО3 с 1гО2 приготовлено сое-
динение BaIr0,33Ti0,67O3 со структурой гексагонального BaTiO3.
ТЮ2 — РЬО — В120з. Беляев с соавторами [15] получил соединения
BiaPbTi4Ols и Bi2PbTi4O12.
Гидротермальный синтез. При гидротермальном синтезе в системе Na2O —
ZnO — SiO2 — Н2О в результате растворения титановых вкладышей автоклава
образовались кристаллы титаносиликата натрия Na2TiOSiO4 [77]. Фаза имеет те-
трагональную решетку с параметрами а = 6,48, с = 5,08 А, г — 2, фед. гр. PMntntn.
Литература
,1 Авер ья нов а Л. Н., Беляев И. Н.— Ж. неорг. хим., 1961, 6, 501.
2. Александров В. Б.— В кн.: Четвертая конференция молодых научных
сотрудников ИМГРЭ. Изд-во АН СССР, М., 1962, 81.
3. Акулов Н. С., Мельгуй М. А.— ДАН БССР, 1963, 7, 661.
4. Беляев
5. Беляев
6. Беляев
7. Беляев
8. Беляев
9. Беляев
10. Беляев
И. Н., Нестерова А. К-— Ж- общ. хим., 1952, 22, 396.
И. Н., Соколович М. Л.— Ж. прикл. хим., 1952, 25, 657.
И. Н.—ДАН СССР, 1954, 95, 535.
И. Н., С и г и д а Н. П.— Ж. общ. хим., 1956, 26, 1553.
И. Н., С и г и д а Н. П.— Ж. неорг. хим., 1958, 3, 433.
И. Н., С и г и д а Н. П.— Ж. неорг. хим., 1958, 3, 425.
И. Н., С и г и д а Н. П.— В кн.: Физико-химический анализ
солевых систем. Ростов-на-Дону, Ростовский ун-т, 1962, 51.
11. Беляева А. Г.— В кн.: Материалы 4-й научной конференции аспирантов.
Ростов-на-Дону, Ростовский ун-т, 1962, 97.
12. Б е л я е в И. Н., Аверьянова Л. Н.— В кн.: Физико-химический
анализ солевых систем. Ростов-на-Дону, Ростовский ун-т, 1962, 65.
13. Беляев И. Н., С и г и д а Н. П.— Ж. неорг. хим., 1958, 3, 440.
14. Б е л я е в И. Н., Голованова Т. Г.— Ж- неорг. хим., 1962, 7, 2760.
15. Беляев И. Н., Смолянинов Н. П., Кальницкий Н. Р.—
Ж. неорг. хим., 1963, 8, 384.
©.Беляев И. Н., Голованова Т. Г.— Ж- неорг. хим., 1965, 10, 1877.
7. Б е л я е в И. Н., Беляева А. Г.— Ж- прикл. хим., 1965, 38, 1280.
Й. Б е л я е в И. Н., Беляева А. Г.— Ж- неорг. хим., 1965, 10, 467.
.Бычкова Н. А. Автореф. канд. дисс. Ростовский ун-т, 1953.
20. Б л ю м е н Л. М.— Изв. АН Туркм. ССР, 1951, 5, 70.
Б л юм е н Л. М./ - Изв. АН Туркм. ССР, 1953, 6, 49.
•2, Б р о н В. А., П о д н о г и н А. К-— ДАН СССР, 1953, 93, 93.
№ Б у р с и а н Э. В., Смирнова Н. П. — Кристаллография, 1963, 8,
799.
^•Будников П. П., ТресвятскийС. Г.— ДАН УРСР, 1954, 5, 371.
I Богданов С. В., Каштанова А. М., Киселева К. В. — Изв.
, АН СССР. Сер. физ., 1965, 29, 896.
^•Бобович Я. С., Б у р с и а н Э. В.— Оптика и спектр., 1961, 11, 131.
!?. Богородицкий Н. П., Зеленкова И. Е., Прохватило-
в а В. Г., Ф р е д б е р г И. Д.— ДАН СССР, 1955, 104, 543.
Белянкин Д. С., Торопов Н. А., Лапин В. В. Физико-хими-
ческие системы силикатной технологии. Промстройиздат, М., 1954.
I 217
29. Белякова Е., Комар А., Михайлов В.— Металлург, 1940
15 3. *
30. Белякова Е. П.— Укр. хим. ж., 1963, 29, 778.
31. Бережной А. С.— Огнеупоры, 1950, 15, 350, 446.
32. Бережной А. С., Гулько Н. В.— ДАН УРСР, 1955, 1, 77.
33. Б е р е ж н о й А. С., Гулько Н. В.— Укр. хим. ж., 1955, 21, 158.
34. Бережной А. С.— ДАН УРСР, 1962, 1, 65.
35. Б е р е ж н о й А. С.— ДАН УРСР, 1962, 3, 387.
36. Б е р е ж н о й А. С., Гулько Н. В.— В кн.: Научные труды Украин-
ского н.-и. ин-та огнеупоров, 6. 1962, 53, 92.
37. Бережной А. С., Гулько Н. В., Гавриш А. М.— ДАН УРСР
1964, 12, 1614.
38. В а л я е в X. С., Гай И. А.— Труды Гос. исслед. электрокерам, ин-та,
1957, 2, 39.
39. Веневцев Ю. Н., Бондаренко В. С., Жданов Г. С., Чка-
лова В. В., Стемберг Н. Г.— Кристаллография, 1961, 6, 375.
40. Вайнштейн Э. Е., Бриль М. Н., Копелев Ю. Ф.— ДАН СССР
1959, 126, 744.
41. В у л Б. М., Гольдман И. М.— ДАН СССР, 1945, 46, 139; 1945, 49, 79.
42. В у л Б. М., Гольдман И. М.— ДАН СССР, 1945, 46, 154.
43. В у л Б. М., Гольдман И. М.— ДАН СССР, 1946, 51, 21.
44. В у л Б. М., Гольдман И. М.— ДАН СССР, 1948, 60, 41.
45. В у л Б. М., Гольдман И. М., Разбаш Р. Я-— ЖЭТФ, 1950, 20,
465.
46. Гинсберг А., Нигосьян С.— Изв. геологии, комиссии, Л., 1924,
43, 397.
47. Година Н. А., Келер Э. К., Руденко В. С.— Ж- неорг. хим.,
1960, 5, 2794.
48. Голубенко А. Н., Устинов О. А., Резухина Т. Н.— Ж- физ.
хим., 1965, 39, 1164.
49. Г л у ш к о в а В. Б., Исупова Е. Н.— Изв. АН СССР. Неорг. материа-
лы, 1965, 1, 1143.
50. Г л е б о в С. В., К а р к л и т А. К-, Савкевич И. А., Миль-
ш е н к о Р. С. — Огнеупоры, 1947, 12, 152.
51. Горина Ю. И., Каштанова А. М., Максимова Г. В., Ска-
на в и Г. И. — Кристаллография, 1961, 6, 473.
52. Гребенщиков Р. Г., Торопов Н. А.— ДАН СССР, 1964, 158, 710.
53. Демешина А. И., Мурзин В. Н.— Физ. тв. тела, 1962, 4, 2980.
54. Ершов Л. Д.— Труды Гипроцемента, 1940, 1, 5.
55. И с х а к о в X. Ш., Келер Э. К-— Ж- неорг. хим., 1962, 7, 1946.
56. Исхаков X. Ш., Келер Э. К.— Ж- неорг. хим., 1962, 7, 1958.
57. Иванова В. В. К а п ы ш е в А. Г., Веневцев Ю. Н., Жда'
н о в Г. С.— Изв. АН СССР. Сер. физ., 1962, 26, 354.
58. И с у п о в В. А., С т р е л е ц П. Л., Серова И. А., Яценко Н.Д-
Широбоких Т. М.— Физ. тв. тела, 1964, 6, 790.
59. И с м а и л з а д е И. Г.— Азерб. хим. ж., 1961, 5, 91.
60. И с м а и л з а д е И. Г.— Кристаллография, 1963, 8, 852.
61. Крамаров О. П., Ход а ков А. Л., Шолохович М. Л., Фе-
сенко Е. Г.— В кн.: Сегнетоэлектрики. Ростов-на-Дону, Ростовский ун-т,
1961, 5.
62. Кондратьев Ю. Н.— ДАН СССР, 1963, 153, 1370.
63. Кутолин С. А.— Изв. АН СССР. Неорг. материалы, 1965, 1, 928.
64. К е н ц и г В. Сегнетоэлектрики и антисегнетоэлектрики. ИЛ., М., 1960.
65. Круглы Я. Я., Максимова О. С.— Изв. АН Латв. ССР. Сер. хим-
1963, 6, 705.
66. К у д з и н А. Ю.— Кристаллография, 1964, 9, 295.
67. Келер Э. К-, Карпенко Н. Б.— Ж. неорг. хим., 1959, 6, 1125.
68. Келер Э. К-, Исупова Е. Н.— Ж. неорг. хим., 1960, 5, 433.
69. Комков А. И,— ДАН СССР, 1959, 126, 641.
218
70. Комков А. И., Белопольский М. П., Ч е р н о р у к С. Г.,
Колпаков Д. А.— ДАН СССР, 1962, 147, 687.
71. Комков А. И,— ДАН СССР, 1963, 148, 1182.
72. Лапин В. В.— Труды совещ. по керамическому сырью. Изд-во АН СССР,
М., 1948, 126.
73. Л е н е в Л. М., Новохатский И. А.— Изв. АН СССР. Металлургия
и горное дело, 1964, 4, 87.
74. Модель М. С.— ДАН СССР, 1962, 146, 871.
75. М у р з и н В. Н., Демешина А. И., Богданов С. В.— Изв.
АН СССР. Сер. физ., 1965, 29, 920.
76. Новохатский И. А., Лене в Л. М., Савинская А. А.— Изв.
АН СССР. Металлы, 1965, 2, 65.
77. Н и к и т и н а А. В., Илюхин В. В., Литвин Б.Н., Me л ьн и-
к о в О. К., Белов Н. В.— ДАН СССР, 1964, 157, 1355.
78. Наука и жизнь, 1965, 1, 84.
79. Нестерова А. К- Автореф. канд. дисс. Ростовский ун-т, 1953.
80. П а н ф и л о в Б. И., Феодосьев Н. EL— Ж. неорг. хим., 1964, 9,
2685.
81. Панфилов Б. И., Феодосьев Н. Н.— Ж- неорг. хим., 1964, 9,
2693.
82. Панфилов Б. И., Феодосьев Н. Н,— Ж. неорг. хим., 1965, 10,
298.
83. Панфилов Б. И., Феодосьев Н. Н.— Ж- неорг. хим., 1965, 10,
1844.
84. ПятенкоЮ. А.,Пудовкина 3.
85. Руднева А. В., М а з е л ь М. С.—
ное дело, 1963, 1, 59.
В.— Кристаллография, 1964, 9, 98.
Изв. АН СССР. Металлургия и гор-
86. Резниченко В. А.,Халимов Ф. Б.-— В кн.: Титан и его сплавы,
9. Изд-во АН СССР, М., 1963, 42.
87. Резниченко В. А., СоломахаВ. П.— В кн.: Титан и его сплавы,
9. Изд-во АН СССР, М., 1963, 136.
88. Смол енски й Г. А.— ДАН СССР, 1950, 70, 405.
89. Смоленский Г. А., Кожевникова Н. В.— ДАН СССР, 1951,
76, 519.
90. Смоленский Г. А.— Ж* техн, физ., 1950, 20, 137.
91. Смоленский Г. А., Исупов В. А., Аграновская А. И.,
Крайник Н. Н.— Физ. тв. тела, 1960, 2, 2982.
92. С п е р а н с к а я Е. И., Р е з И. С., К о з л о в а Л. В., Скори-
ков В. М., С л а в о в В. И.— Изв. АН СССР. Неорг. материалы, 1965,
1, 232.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
Стеханов А. И., Карамян А. А., Асгафьев Н. И.— Физ. тв.
тела, 1965, 7, 157.
Сасаки X.— Нихон буцури гаккайси, Buturi, 1965, 20, 44.
Сигида Н. П., Беляев И. Н.— Ж- неорг. хим., 1957, 2, 1119.
Сигида Н.П., Беляев И. Н.— Ж- неорг. хим., 1957, 2, 1128.
Сигида Н. П., Беляев И. Н.— В кн.: Физико-химический анализ со-
левых систем.\ остов-на-Дону, Ростовский ун-т, 1962, 43.
Сканави Г. И.— ДАН СССР, 1948, 59, 41.
Сканави Г. И., Маслов В. В.—ЖЭТФ, 1954, 27, 735.
Сканави Г. И., Демешина А. Н.— ЖЭТФ, 1956, 31, 565.
Сканави Г. И., Матвеева Е. Н.— ЖЭТФ, 1956, 30, 1049.
Сканави Г. И., Ксендзов В. М., Тригубенко Б. А., П р о -
хватилов В. Г.— ЖЭТФ, 1957, 33, 320.
Тамбовцев Д. А., Скориков В. М., Же л у де в И. С.— Крис-
таллография, 1963, 8, 889.
Трифанов И. А.— Кристаллография, 1964, 9, 923.
Ф у к у м и С.— Нихон буцури гаккайси, Buturi, 1965,20,55.
Фесенко Е. Г., Прокопало О. И.— Кристаллография, 1961.
6, 469.
t
219
107. Фридберг В. Я., ФрейнденфельдЭ. Ж-, К р у ч а н Я. Я.-—
Изв. АН СССР. Сер. физ., 1960, 24, 1387.
108. Федулов С. А., Жданов Г. С., Рез И. С.—Кристаллография,
1961, 6, 681.
109. Федулов С. А., Веневцев Ю. Н.— Физ. тв. тела, 1961, 3, 3371.
110. Ф е д у л о в С. А., В е н е в ц е в Ю. Н., Жданов Г. С., Д ж му-
ха д з е Д. Ф.— Изв. АН СССР. Сер. физ., 1962, 26, 357.
111. Федулов С. А., Веневцев Ю. Н., ДжмухадзеД. Ф.— Крис-
таллография, 1962, 7, 408.
112. Федулов С. А., Веневцев Ю. Н., Жданов Г. С., Смажев-
с к а я Е. Г., Рез И. С.— Кристаллография, 1962, 7, 77.
113. Федулов С. А., Веневцев Ю. Н.— Кристаллография, 1964, 9,
358.
114. Федулов С. А., Ладыжинский П. Б., Пятигор-
ская Л. И., Веневцев Ю. Н.— Физ. тв. тела, 1964, 6, 475.
115. Федулов С. А., Пятигорская Л. И., Веневцев Ю. Н.—-
Кристаллография, 1965, 10, 291.
116. Халимов Ф. Б., Резниченко В. А.— В кн.: Титан и его сплавы,
4. Изд-во АН СССР, М., 1960, 14.
117. X о д а к о в А. Л., Шолохович М. Л.— Изв. высш. уч. зав. Сер. физ.,
1961,2,85.
118. Черепанов А. М.— В кн.: Рост кристаллов, 3. Изд-во АН СССР, М.,
1961, 457.
119. Черняк Я. Н.— Огнеупоры, 1953, 7, 313.
120. Шуваев А. Т.— Изв. АН СССР. Сер. физ., 1959, 23, 569.
121. Шолохович М. Л., Ходаков А. Л.— В кн.: Рост кристаллов, 3.
Изд-во АН СССР, М., 1961, 463.
122. Шолохович М. Л.— Ж. общ. хим., 1955, 25, 1900.
123. Шолохович М. Л., Баркова Г. В.— Ж. общ. хим., 1956, 26, 1266.
124. Шолохович М. Л., Баркова Г. В.— Ж. общ. хим., 1954, 24, 218.
125. Щепочкина Н. И.— Труды пятого совещания по экспериментальной и
технической минералогии и петрографии. Изд-во АН СССР, М., 1958, 493.
126. Щ е п о ч к и н а Н. И. Автореф. канд. дисс. Ин-т геологии рудных место-
рождений, петрографии, минералогии и геохимии АН СССР, М., 1956.
127. Японский пат. № 1232 от 24.02. 1955 г.; РЖХим, 1957, 41678П.
128. Ag am a wi V. М., White J.— Trans. Brit. Ceram. Soc., 1952, 51, 293
129. Agamawi V. M., White J.— Trans. Brit. Ceram. Soc., 1953, 52, 271.
130. Auger V.— Compt. rend., 1923, 117, 1302.
131. D’ A n s J., L о f f e r J.— Ber., 1930, 63, 1446.
132. Armand D. L., Cole S. S.— J. Metals, 1949, 1, 909.
133. Andersson S., Wadsley A. D.— Acta Crystallogr., 1961, 14, 1245.
134. Andersson S., Wadsley A. D.— Acta chem. Scand., 1961, 15, 663.
135. Andersson S., Wadsley A. D.— Nature, 1960, 187, 499.
136. Andersson S.— Acta Crystallogr., 1962, 15, 194.
137. Andersson S., Wadsley A. D.— Acta Crystallogr., 1962, 15, 201.
138. A s о S., О n u k i A., Nischizawa H.— J. Electrochem. Soc. Japan,
1951 19 145.
139. В г c i с B. S., M i 1 i c e v S., S i f t a г J.— Croat. Chem. Acta, 1961, 33,
169.
140. Brcic B.S., Brencic J., Siftar J.— Croat. Chem. Acta, 1963, 35,
135.
141. В a r b 1 a n F. F.— Schwez. mineralog. petrogr. Mitt., 1943, 23, 295.
142. В о b г о w E. N.— Пат. США, №2421984; Ch. A., 1947, 7286.
143. Balduzzi F., Stenemann S.— Швейцарский пат., №287553, 1953;
Chpm 1(ЖЧ 7 944
144. Bland J.’A.— Acta Crystallogr., 1961, 14, 875.
145. Bellen E., Sustschinsky P.— Z. Kristallogr., 1904, 38, 266.
146. Bartram S. F., SI epet у s R. A.— J. Am. Ceram. Soc., 1961, 44, 493-
147. Вауг G.,[Hof f mann W.— Z. Kristallogr., 1965, 121, 9.
220
148. В г i x n er L. Н.— Inorg. Chem., 1964, 3, 600.
149. В о dger А. Е., D о п е у L. М.— Glass Ind., 1940, 21, 309.
150. В 1 a s s е G.— J. Inorg. a. Nucl. Chem., 1963, 25, 230.
151. В о с h у n s к i Z., Rzeszowski R., Szurkowski B.— Prace
komis. mat.-przyrodn. Poznan towarz. pryiaciol nauk, 1964, 11, 179: РЖХим.,
1965, 15Б, 489.
152. Bertaut E. F., Durif A.— Bull. Soc. franc, mineral, et cristallogr.,
1956, 79, 176.
153. Bergstein A.— Chem. listy, 1956, 50, 3.
154. Brown F., Duwez P.— J. Am. Ceram. Soc., 1954, 37, 129.
155. В u s s e m W.. Schusterius C., Ungewiss A.— Ber. Deutsch,
keram. Ges., 1937, 18, 434.
156. Burbank R. D., Evans H. T.— Acta Crystallogr., 1948, 1, 330.
157. Brous J., Fankuchen I., Banks E.— Acta Crystallogr., 1953,
6, 67.
158. Barth T. F. W., P о s n j а к E.— J. Washington Acad., 1931, 21, 255.
159. Barth T. F. W., P о s n j а к E.— Z. Kristallogr., 1932, 82, 328, 333.
160. Bunting E. N.— J. Res. Nat. Bur. Stand., 1933, 11, 725.
161. Bunting E. N., Shelton G. R., Cremer A. S.— J. Am. Ceram.
Soc., 1947, 30, 114.
162. Bunting E. N., Shelton G. R., Creamer A. S.— J. Res. Nat.
Bur. Stand., 1949, 43, 237.
163. В 1 a t t n e г H., К б n z i g W., Merz W.— Helv. phys. acta., 1949,
22, 35.
164. В 1 a t t и e r H., Matthias B., Merz W.— Helv. phys. acta, 1948.
20, 225.
165. В i 1 1 i e t Y., P о i x P.— Bull. Soc. Chim., France, 1963, 3, 477.
166. В i 1 1 i e t Y., Poix P., Michel A.— Compt. rend., 1963, 256,
4217.
167. В i 1 1 i e t Y., Poix P., Michel A.— Bull. Soc. Chim. France, 1963,
10, 2196.
168. Bourgeois L.— Z. Kristallogr., 1888, 14, 281 .
169. Bourgeois L.— Ann. chim. et phys., 1883, 29, 479.
170. Bourgeois L.— Bull. Soc. Chim., 1886, 46, 263.
171. Bourgeois L.— Compt. rend., 1886, 103, 143.
172. Cormimboluf H.—Compt. rend., 1892, 115, 824.
173. С о u g h а п о u r L. W., Roth R. S. De Prosse V. A.—J. Res.
Nat. Bur. Stand., 1954, 52, 37.
,174 . Coughanour L. W., De Prosse V. A.— J. Res. Nat. Bur. Stand.,
1953, 51, 85.
175. C i d - D г e s d n e r H., Buerger M. J.— Z. Kristallogr., 1962, 117,
411.
• 176. Carstens C. W-, Kristof f ersen K-— N. Jb. Min. Beilagebd.,
1931, 62, 163.
1177. Clevenger T. R.— J. Am. Ceram. Soc., 1963, 46, 207.
1178. С о n j e u d P.— Q mpt. rend., 1954, 239, 1210.
/179. Christensen A. N., Rasmussen S. E.— Acta chem. Scand., 1963,
17, 845.
|180. Cole S. S., Nelson W. K-— J. Phys. Chem., 1938, 42, 245.
481. C h e v a 1 1 i e r R., В о 1 f a J., Mathieu S.— Bull. Soc. frac, miner,
et cristallogr., 1955, 78, 307, 365.
j 182. Cabar ути Э.— Buturi, 1965, 20, 49.
, 183. Cocco A. — Ann. chim., 1958, 48, 587.
1184. Cocco A., Scromek N.— Ann. chim., 1961, 51, 1194.
[185. Cocco A., Massazza F.— Ann. chim., 1963, 53, 883.
1186. Cocco A.— Ann. chim., 1965, 65, 153.
187. Dulin F. H., Rose D. E.— J. Am. Ceram. Soc., 1960, 43, 125.
H88. D г у s M., Trzebiatowski W.— Roszn. chem., 1957, 31, 489.
1189. D vorak V., Janovec V.— Чехосл. физ. ж., 1962, B12, 461.
221
190. Derbyshire S. W., Fraker A. C., St a de 1 m a i er H. H.—
Acta Crystallogr., 1961, 14, 1293.
191. Dickson J. G., Katz L., Ward R.— J. Am. Chem. Soc., 1961, 83,
3026.
192. D a t t a R. K., Roy R.— Nature, 1961, 191, 169.
193. D e m о 1 у A.— Compt. rend. Trav. Chim., 1849, 5, 334.
194. D e m о 1 у A.— Compt. rend. Trav. Chim., 1849, 5, 331.
195. DeVries R. C., Roy R., Osborn E. F.— J. Am. Ceram. Soc., 1955,
38, 158.
196. De Vries R. C., Osborn E. F., Roy R.—J. Phys. Chem., 1954,
58, 1069.
197. DeVries R. C., Roy R., Osborn E. F.— Trans. Brit. Ceram. Soc.,
1954 53 525.
198. Ernst T.— Z. angew. Mineral., 1943, 4, 394.
199. Elliott W. R., Rjorkstam J. L.— J. Phys. Chem. Solids, 1964, 25,
1273.
200. Evans H. T., Burbank R. D.— J. Chem. Phys., 1948, 16, 634.
201. Etsuro S., Atsushi K., Yoichi K.— J. Phys. Soc. Japan, 1963^
18, 459.
202. E r n V.— Am. Ceram. Soc., 1963, 46, 295.
203. E b e 1 m e n J. J.— Compt. rend., 1851, 32, 711.
204. Eberspacher O.— Naturwis., 1962, 49, 155.
205. F 1 6 г к e О. W.— Ber. Deutsch. Keram. Ges., 1961, 38, 133.
206. Flaschen S. S.— J. Am. Chem. Soc., 1955, 77, 6194.
207. F e t i v e a u M. et al.— Bull. Soc. Chim. France, 1965, 2, 450.
208. Fisk H. G.— J. Am. Ceram. Soc., 1951, 34, 9.
209. Freundlich W.— Compt. rend., 1953, 236, 1895.
210. Forsbergh P. W.— Phys. Rev., 1949, 76,1187.
211. F r e n z e 1 G.— N. Jr. Mineral. Monatsch., 1961, 1, 12.
212. Fink G.— Z. Phys. Chem., 1965, 44, 122.
213. Granicher H., Lawless W. N.— Helv. phys. acta, 1965, 38, 362.
214. Grieve J., White J.— J. Roy. Techn. Coll. (Glasgow), 1940, 4, 660.
215. Goughanour L. M.— J. Res. Nat. Bur. Stand., 1955, 54, 191.
216. Goldschmidt V. M.— Naturwis., 1926, 14, 481.
217. Goldschmidt V. M.— Scr. Akad. Oslo, 1926, 8, 1.
218. Goldschmidt H. J.— Metallurgia, 1960, 62, 211.
219. Goldschmidt H. J.— Metallurgia, 1960, 62, 246.
220. Holmquist P.J.— Bull. geol. Inst. Upsala, 1896/97,3,211.
221. H agenmiil ler P., Lezerf A., Onill on M.-~ Compt. rend.,.
1962 255 928
222. H e r c z о g A.— J. Am. Ceram. Soc., 1964, 47, 107.
223. Hofmann A.— Z. phys. Chem., 1935, Б28, 65.
224. Hippel A. et a 1.— Ind. Eng. Chem., 1946, 38, 1097.
225. Harwood M. G., Popper P., Rushman D. F.— Nature, 1947,
160, 58.
226. Hauser O., Schenk M.— Phys. Status Solidi, 1964, 6, 83.
227. H i 1 1 e r t L.— Acta chem. Scand., 1965, 19, 1774, 1986.
228. Hardy A. et al.— Compt. rend., 1964, 259, 3462.
229. Hegenbarth E.— Phys. Status Solidi, 1965, 9, 191.
230. H e n d e r s о n S. T., Rauby P.W.— J. Electrochem. Soc., 1951, 98,
479.
231. Ha ay man P. W.~ Chem. Weekblad., 1948, 44, 603.
232. Holgersson S., Herrlin A.— Z. anorg. Chem., 1931, 198, 69.
233. Hamelin M.— Rev. Met., 1950, 47, 324.
234. Hamelin M.— Bull. Soc. Ceram., 1951, 11, 13.
235. Hamelin M.— Compt. rend., 1954, 238, 1896.
236. Hamelin M.— Compt. rend., 1955, 240, 79.
237. Hamelin M.— Bull. Soc. Chim. France, 1957, 11 —12, 1421.
238. Hummel F. A. Tien T. Y.— J. Am. Ceram. Soc., 1959, 42, 206.
239. H a u t e f e u i 1 1 e P.— Compt. rend., 1864, 59, 733.
240. H a u t e f e u i 1 1 e P.— Ann. chim. et phys., 1865, 4, 161.
241- H a u t e f e u i 1 1 e P.— Ann. chim. et phys., 1865, 4, 155.
242. H a u t e f e u i 1 1 e P.— Lieb. Ann., 1865, 134, 24.
243. H a u t e f e u i 1 1 e P.— Lieb. Ann., 1865, 134, 166.
244. H a u t e f e u i 1 1 e P.— Ann. chim. et phys., 1865, 4, 167.
245. Iwase K. , N i s i oka U.— Sci. Rep. Tohoku. K- Honda anniversary vol
1936, 444.
246. Iwase К., N i s i о к a U.— Sci. Rep. Tohoku, 1937, 1, 26, 592.
247. I w a s e K-, F u к u s i m a M.— Sci. Rep. Tohoku, 1932, 1, 21, 114.
248. Iwase K-, Saito Y.— Kinzoku no Kenkyu, 1934, 11, 539.
248a. Ikeda T.,— J. Phys. Soc. Japan, 1959, 14, 1286.
249. Ikeda T., Okano T.— Japan J. Appl. Phys., 1964, 3, 63.
250. Junker E. —Z. anorg. Chem., 1936, 79.
251. J о h о P.— Z. Kristallogr., 1964, 120, 329.
252. J о u b e r t J.-C., D u r i f A., V a r a m b о n.— Bull. Soc. franc, mineral,
et cristallogr., 1963, 86, 92.
253. J a n d e r W., Bunge K-— Z. anorg. Chem., 1938, 239, 418.
254. Johnston W. D., S e s t r i c h D.— J. Inorg. a Nucl. Chem., 1961, 20,
32.
255. Kahn A. H., Legendecker A. J.— Phys. Rev., 1964, 135, 1321.
256. Кино А., Судзуки M.— Когё кагаку дзассы, 1964, 67, 432.
257. Keller С.— Z. anorg. Chem., 1962, 318, 89.
258. Karkhanavala M. D., Mom i n A. C.— Am. Ceram. Soc., 1954, 42,
399.
259. Комори C.— Buturi, 1965, 20, 203.
260. Kuan-Han Sun, Eastman Kodak C o.— Пат. США № 2430539,
1947.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.
274.
275.
276.
277.
278.
279.
280.
281.
282.
К о r des Е., Rottig Е.— Z. anorg. Chem., 1951, 264, 34.
К о г d е s Е.— Z. Kristallogr., 1935, 92, 139.
К о г des Е.— Fortschr. Mineralog., 1934, 18, 27.
Kurowski Е., Nissenmann L.— Вег., 1911, 44, 224.
King E. G.— J. Am. Chem. Soc., 1955, 77, 2150.
Kay H.F., Vousden P.— Phil. Mag., 1949, 40, 1019.
Kestigian M., Ward R.— J. Am. Chem. Soc., 1954, 76, 6027.
К e s t i g i a n M., Dickinson J.G., Roland W.— J. Am. Chem.
Soc., 1957, 79, 5598.
Kay H. F., Miles J. L.— Acta Crystallogr., 1957, 10, 213.
К а у H. F.— Acta Crystallogr., 1948, 1, 229.
Kay H. F., Bailey P. C.— Acta Crystallogr., 1957, 10, 219.
Knop O., Brisse F., Castell iz L. S u t a r n o.— Canad. J.
Chem., 1965, 43, 2812.
К a i m a n S.— Canad. Mineral., 1959, 6, 389.
Kahn A. H., Leyendecker A. J.— Phys. Semiconduct., Paris, 1964.
33.
Klimowski J.— Bull Soc. amis Sci. et lettres Poznan, 1964, B, 17, 59.
К у б о T. и др.— Kogyo kagaku zasshi, J. Chem. Soc. Japan Ind. Chem.
Sec., 1964, 67, 1502; РЖХим 1965, 18Б465.
Kubo T., Shinriki K-— J. Chem. Soc. Japan, Ind. Chem. Sec. 1952,
55, 137.
Kubo T., Shinriki K-— J. Chem. Soc. Japan, Ind. Chem. Sec., 1951,
54, 268.
Kubo T., Shinriki K.— J. Chem. Soc. Japan, Ind. Chem. Sec., 1952,
55, 49.
Kubo T., Shinriki K-— J. Chem. Soc. Japan, Ind. Chem. Sec., 1953,
56, 335.
Lang S. M., Fillmore C. L., Maxwell L. H.— J. Res. Natl. Bur.
Stand., 1952, 48, 298.
Lytle F. W.— J. Appl. Phys., 1964, 35, 2212.
I
223
283. L e n d I e t M., L e n s e n M.— Compt. rend., 1961, 253, 2982.
284. Lux H.— Z. Elektrochem., 1949, 53, 45.
284a. Linz A., Herrington K.— J. Chem. Phys., 1958, 28, 824.
285. Levy L.— Compt. rend., 1898, 107, 421.
286. Levy L.— Ann. chim. et phys., 1892, 25, 433.
287. Lietz J., Nadop G.— Naturwis., 1955, 42, 389.
288. Lambertson W. A., Mueller M. H.— ANL — 5312 (sept. 14, 1954)e
289. Levin E. M., McMurdie H. F. — Phase diagrams for Ceramists*
Am. Ceram. Soc. Columbus, 1959, II, 1027.
290. Liebertz J., Rooymans C. J. M.— Z. phys. Chem., 1965, 44, 242.
291. Lundberg M., Andersson S.— Acta chem. Scand., 1964, 18, 817.
292. Lecerf A.— Ann. chim. 1962, 7, 513.
293. Lecerf A., Hardy A.— Compt. rend., 1961, 252, 131.
294. Lecerf A.— Compt. rend., 1961, 252, 3253.
295. Lecerf A.— Compt. rend., 1962, 254, 2003.
296. Marshall R. H., Hoekstra H. R.— J. Inorg a Nucl. Chem., 1965
27, 1947.
297. Massazza F.— Ann. chim., 1961, 51, 78.
298. Moore С. H., Sigurdson H.— J. Metals, 1949, 1, 914.
299. M a n о j 1 о v i с M., R i s t i с M., Maleic S. S.— New Nucl. Materi-
als ind. Nun-Metals Fuels. I. Vienna, 1963, 245.
300. M e g a w H. D.— Trans. Farad. Soc., 1946, 42A, 224.
301. M e g a w H. D.— Proc. Phys. Soc., 1946, 58B, 133.
302. M e g a w H. D.— Proc. Roy. Soc., 1947, A189, 264.
303. Mat ossi F.— Ann. Phys., 1963, 11, 22.
304. Matsuo Y., Sasaki H.— J. Am. Cer. Soc., 1963, 46, 409.
305. Milligan W. O., Holmes B. G. — J. Phys. Chem., 1953, 57, 11.
306. Moeller C. W.— J. Chem. Phys., 1957, 27, 983.
307. Moore С. H., Sigurdson H.— J. Metals, 1949, 1, 917.
308. Мори С., и др.— Nat. Techn. Rept., 1964, 10, 32.
309. Merz W. J.— Phys. Rev., 1949, 76, 1221.
310. McChesney B., Muan A.— Am. Mineralogist, 1961, 45, 572.
311. McChesney J. B., Sauer H. A.— J. Am. Ceram. Soc., 1962, 45, 416.
312. Melikoff P., Pissarjewsky L.— Z. anorg. Chem., 1898, 18, 58.
313. Melikoff P., Pissarjewsky L.— Z. anorg. Chem., 1898, 18, 89.
314. Melikoff P., Pissarjewsky L.— Ber., 1898, 31, 678.
315. Melikoff P., Pissarjewsky L.— Ber., 1898, 31, 952.
316. Mallard E.— Compt. rend., 1872, 75, 472.
317. Mallard E.— Ann. chim. et phys., 1873, 28, 86.
318. Matthias B.— Phys. Rev., 1948, 73, 808.
319. Matthias В. T., Breckenridge R. G., Beaumont D. W.~
Phys. Rev., 1947, 72, 532.
320. N i g g 1 i P.— Z. anorg. Chem., 1916, 98, 241.
321. Naylor B. F.— J. Am. Ceram. Soc., 1945, 67, 212.
322. Naylor B. F., Cook O. A.— J. Am. Chem. Soc., 1946, 68, 1003.
323. Naraganan P. S., Vedam K.— Z. Phys., 1961, 163, 158.
324. Newnham R. E., Fang J. H., Santoro R. P. — Acta Cristal-
logr., 1964, 17, 240.
325. Nelson C. W.— Mass. Inst. Technolog. Lab. Insulat. Res. Cambridge, 1963-
326. N i s i о к a U.— Sci. Rep. Tohoku, 1934, 1, 23, 259.
327. N i s i о к a U.— Sci. Rep. Tohoku, 1935, 24, 710.
328. N i s i о к a U.— Sci. Rep. Tohoku, 1935, 24, 707.
329. О u v г a г d L.— Compt. rend., 1890, III, 147.
330. P u к a 1 1 W-— Silikat.-Z. 1914, 2, 109.
331. P о s n j а к E., Barth T. F. W.— Z. Rristallogr., 1934, 88, 271.
332. Posnjak E., Barth T. F. W.— Z. Kristallogr., 1934, 88, 276.
333. Perez J. M., Mondange H., Col longues R.— Bull. Soc. Chi171’
France 1961 1 79
334. P о u i 1 1 a r d E.— Ann. chim. et phys., 1950, 12, 164.
224
1335. P arg a P on d al I., В ergt K.— Tonind-Z., 1933, 57, 1000.
336. Pfisterer H., H ey w ang W., R am isch E.— Ber. Deutsch.
Keram. Ges., 1956, 33, 178.
337. Partington I. R., Planer G. V., Boswell I. I.— Phil. Mag.,
1949, 40, 157.
338. P о i x P., Michel A.— Bull. Soc. Chim. France, 1961,2, 352.
339. P a s s e r i n i L.— Gazz., 1930, 69, 957.
340. Prince A. T.— J. Geol., 1943, 51, 1.
341. Patchett J. E., Nuffield E. W.— Canad. Mineral., 1960, 6, 483.
342. Padurow N. N., Schusterius C.— Ber. Deutsch, keram. Ges., 1954,
31, 391.
343. Queyroux F.— Bull. Soc. franc, mineral, et cristallogr., 1963, 86, 295.
344. Queyroux F.— Compt. rend., 1964, 259, 1527.
345. Rieke R., U n g e w i s s A.— Ber. Deutsch, keram. Ges., 1936, 17, 237.
346. Rose H.— Pogg. Ann., 1849, 74, 563.
347. Rose H.— Gilb. Ann., 1823, 73, 83.
348. Rase D. E., Roy R.— J. Am. Ceram. Soc., 1955, 38, 102.
349. Rhodes R. G.— Acta Crystallogr., 1949, 2, 417.
350. Rushman D. F., Strivens M. A.— Proc. Phis. Soc., 1947, 59, 1011.
351. R u s h m a n D. F., Strivens M. A.— Trans. Farad. Soc., 1946, 42A,
231.
352. Ricker R. W., Hummel F. A.— J. Am. Ceram. Soc., 1951, 34, 271.
L353. R e i t h 1 e г J. С., В о 1 t a J.— Bull. Soc. franc, mineral, et cristallogr.,
1963, 86, 68.
354. R а о В. V.— J. Am. Ceram. Soc., 1964, 47, 455.
355. Rao B., J a n a к i r am a V. — Phys. Chem. Glasses, 1963, 4, 22.
356. Roth R. S., Coughanour L. M.— J. Res. Nat. Bur. Stand.,1955,
55, 209.
357. Roth R. S.— J. Res. Nat. Bur. Stand., 1956, 56, 17. |
358. Roth R. S.— J. Res. Nat. Bur. Stand., 1958, 61, 437.
359. Roth R. S., W a d s 1 ey A. D.— Acta Crystallogr., 1965, 18, 724.
360. Roth R. S., W a d s 1 e у A. D., Gotehouse В. M. — Naturwis.,
1964, 51, 262.
361. Schwarz R., Heinrich F.— Z. anorg. Chem., 1935, 223, 387.
362. Smolensky S.— Z. anorg. Chem., 1912, 73, 293.
363. S p i e s s G.— Ber. Deutsch, keram. Ges., 1961, 38, 495.
364. S t a t t о n W. O.— J. Chem. Phys., 1951, 19, 33.
365. Sawada S., Nomura S.— Rept. Inst. Sci. Technol., 1950, 4, 217.
366. Srikanta S. et al.— Acta Crystallogr., 1962, 15, 255.
367. S h i r a n e G., Takeda A.— J. Phys. Soc. Japan, 1952, 7, 1.
368. Shiran G., Pickart S. J., Ishikawa Y.— J. Phys. Soc. Japan,
1959 14 1352.
369. Sigurdson H., Cole S. S.— J. Metals, 1949, 1, 905.
370. Stephenson N. C.— Acta Crystallogr., 1965, 18, 496.
371. S i m a n e к E., S г о u b e к Z.—• Phys. Status Solids, 1965, 8, K47.
372. Schulz A. H.— Dissq '., Bonn, 1947.
373. Schmitz-DuMont O., Schultz A. H.— Monatsh. Chem., 1952,
83, 648.
1 374. Schmitz-DuMont O., Reckhard H.— Monatsh. Chem., 1959,
90, 134.
I 375. Sarver J. F.— Am. J. Sci., 1961, 259, 701.
j’ 376. S u g i у a m a N.— Light Metals, 1957, 24, 26.
J-377. Schrocke H.— N. Jr. Mineral. Monatsch., 1958, 70.
.378. Schenk M.— Naturwis., 1965, 52, 537.
! 379. Shomate С. H.— J. Am. Chem. Soc., 1946, 68, 964.
380. Shomate С. H.— J. Am. Chem. Soc., 1946, 68, 1634.
381. S о к u d о T.— J. Phys. Soc. Japan, 1960, 15, 2112.
382. Suzuki T., Tamizawa S.— J. Chem. Soc. Japan, Ind. Chem. Sec.,
1951, 54, 443.
15 9-614 225
383. Sugi ига M., Ikeda К. — J. Japan Ceram. Assoc., 1947, 55, 16, 30, 45
62, 97, 113, 121, 144; Ch. A., 1951, 7763i.
384. Smith G. S.—Chem. Age, 1945, 53, 429.
385. S о w m a n H. G., Andrew A. I.— J. Am. Ceram. Soc., 1951, 34, 298.
386. S c h m a h 1 N. G., Frisch B., Hargarter E.— Z. anorg. Chem.,
1960, 305, 40.
387. Subb ar ao E. C — Phys. Rev., 1961, 122, 804.
388. Subbarao E. C.— J. Am. Ceram. Soc., 1962, 45, 564.
389. Taylor R.W.— Am. Mineralogist, 1964, 49, 1016.
390. Тад а с и H., Кэсакими M.— Kogyo kagaki zasshi, J. Chem. Soc.
Japan, Ind. Chem. Sec., 1963, 5, 673.
391. T a r t e P.— Nature, 1961, 191, 1002.
392. Trzebiatowski W., D г у s M.— J. Berak. Roczn. Chem., 1954, 28,
21.
393. Trzebiatowski W., Wojciechowska J.— J. Damm. Expert-
entia, 1950, 6, 138.
394. Trzebiatowski W., D г у s M.— Roszn. Chem., 1955, 29, 964.
395. Tammann G., Raising H.— Z. anorg. Chem., 1925, 149, 21.
396. Tammann G., Raising H.— Z. anorg. Chem., 1925, 149, 68.
397. Taylor N. W.— Z. Phys. Chem., 1930, 139, 241.
398. Taylor R.W.— J. Am. Ceram. Soc., 1963, 46, 276.
399. Todd S. S., King E. G.— J. Am. Chem. Soc., 1953, 75, 4547.
399a. Todd S. S., Lorenson R. E.— J. Am. Chem. Soc., 1952, 74, 2043.
400. Tournoux M., Devalette M.— Bull. Soc. Chim. France, 1965, 8,
2337.
401. Tanaka Y.— J. Soc. Chem. Ind. Japan Suppl., 1939, 42, 202B.
402. Тэруитиро К., Масанори K-, Ю к у а к и М.— J. Chem. Soc.
Japan, Ind. Chem. Sec., 1963, 66, 403.
403. Таки С., Танака К-— J - Chem. Soc. Japan, Ind. Chem. Sec., 1963, 66,
417.
404. Umezu S., К ak i uch i F.— Nipp. kogyo kwaisi, 1930, 46, 866.
405. Uitert L. G. et a 1.— J. Phys. Chem. Solids, 1964, 25, 1447.
406. Uitert L. G., Egerton L.— J. Appl. Phys., 1961, 32, 959.
407. V u 1 В. M.— Nature, 1945, 156, 480.
408. Viltange M.— Compt. rend., 1954, 239, 61.
409. Villers G., L e с e r f A., R a u 1 t M.— Compt. rend., 1965, 260, 3017.
410. V о 1 g e r J.— Research., 1954, 7, 230.
411. V о 1 g e r J.— Res. Rep., 1952, 7,21.
412. W aschbur n E., Bunting E. N.— J. Nat. Bur. Stand. 1934, 12, 239.
413. Walton J.H.— J. Am. Chem. Soc., 1907, 29, 481.
414. Winkler C.— Ber., 1890, 23, 2658.
415. Wyss R.— Ann. chim. a phys., 1948, 3, 215.
416. Wojciechowska J.— Roczn. chem., 1953, 27, 216.
417. W a k u S.— Rev. Electr. Commun. Lab., 1964, 12, 33; 1964, 12, 123.
418. W h i t e J., H о w a t D. D., Hay R. — J. Roy. Techn. Coll. (Glasgow).
1934, 3, 231.
419. W artenberg H., Reuch H. J., Saran E.— Z. anorg. Chem.,
1937 230 257
J29. W artenberg H., Prophet E.— Z. anorg. Chem., 1932, 208,
373.
421. W artenberg H., Reuch H. J.— Z. anorg. Chem., 1932, 207, 1.
422. Webster A. H., В r i g h t N. F. H.— J. Am. Ceram. Soc., 1961, 44, 110»
423. Wadsley A. D., Andersson S.— Nature, 1961, 192, 551.
424. Wadsley A. D.— Acta Crystallogr., 1964, 17, 623.
425. Wadsley A. D.— Z. Kristallogr., 1964, 120, 396.
426. Wadsley A. D.— Acta Crystallogr., 1961, 14, 660.
427. Wadsley A. D.— Acta Crystallogr., 1961, 14, 664.
428. Yoshida T., Ueoka H., T a k e i T.— J. Electrochem. Soc. Japan,
1960, 28, 612.
226
f429. Yoshida Т., Takei Т.—J. Electrochem. Soc. Japan, Overseas Ed.,
1960, 28, 195.
430. Zachariasen W. H.— Skr. Akad. Oslo, 1928, 4, 111.
431. Zedlitz O.~ N. Jb. Min. Beilgebd., 1940, 75,245.
Дополнительная литература
дграновская А. И., Саксонов Ю. Г. Кристаллическая структура
твердых растворов в системах Mg2TiO4— MgFe2O4 и Mg2TiO4— MgCr2O4. —
Кристаллография, 1966, 11, 200.
Александров Л. А., Лискер К. Е., Фрейдберг И. Д. Электри-
ческие свойства твердых растворов двойных сплавов на основе титаната каль- <,
ция.— Изв. АН СССР. Сер. физ., 1967, 31, 1898.
Аренд Г., Ц оу ф о в а П., Новак И. В. О кислородной нестехиометрии
титаната бария.— В кн.: Рост кристаллов. «Наука», М., 1965, 81.
Беляев Э. К., Сафиуллин Н. Ш., Панасенко В. М. К вопросу «
взаимодействия двуокиси титана с углекислым натрием.—Изв. АН СССР. Не-
орг. материалы, 1968, 4, 97.
Беляев И. Н., Аверьянова Л. Н., Беляева И. И. Исследование
системы РЬО — PbTiO3 — PbWO4.— В кн.: Исследования в области химии
и технологии минеральных солей и окислов. «Наука», М.— Л., 1965, 151.
Беляев И. Н., _ Г о л о в ан о в а Т. Г. Система K2TiO3 — Na2TiO3. — j
Ж- неорг. хим., 1966, 10, (877.
Беляев И. Н., Аверьянова Л. Н., Беляева И. И. Твердофазные4
реакции титанатов двухвалентных металлов.— Ж- неорг. хим., 1966, 11, 1183.
Биндар Е.И. Фрейденфельд Э. Ж- Кинетика термофазных реакций
в системе PbNb2O6 — Bp, ТЮ3.— Учен. зап. Рижского политехи, ин-та,
1965/1966, 16, 259. *
Бородин В. 3.» Кузнецов В. Г., Лезгинцева Т. Н. Диэлектри-
ческие и оптические исследования монокристаллов ВаТЮ3 в диапазоне инфра-
красных частот.— Изв. АН СССР. Сер. физ., 1965, 29, 1982.
Ба т ы г и н В. Г. Об образовании и некоторых свойствах титанатов натрия. — *
|Ж. неорг. хим., 1967, 12,1442.
бович я. С., Петровский Г. Т. О структуре стекол бинарных си-
стем РЬО2 — TiO2 и Cs2O — ТЮ2.— Ж. прикл. спектроскопии. 1966, 4, 353.
брова А. М., Жигун И. Г., Брагина М. И., Фотиев А. А.
ИК-спектры поглощения некоторых соединений титана.— Ж- прикл. спектро-
скопии, 1968, 6, 96.
брова А.М., Фотиев А. А. Поведение титанатов натрия при гидрохи-
мической переработке. — Труды ин-та химии, 14. Уральский филиал
АН СССР, 1967, 65.
брова А. М., Фотиев А. А., Карапетьянц М. X. Титанаты
натрия.— Ж- физ. хим., 1966, 49, 1835.
р е ж н о й А. С., Гулько Н. В. О субсолидусном строении четырех-
компонентной системы MgO — Сго03 — А12О3— ТЮ2.— ДАН СССР, 1965,
164, 384.
режной А. С., Гулько Н. В. Субсолидусное строение шестиком-
понентной системы Fe2O3—Сг2О3—А12О3 — ZrO2 — TiO2 — SiO2.—ДАН УССР,
1966, 2, 200.
режной А. С., Гулько Н. В. Диаграммы состояния систем Сг2О3 —
TiO2 и Cr2O3 — ZrO2.— ДАН УССР, 1968, 3, 250.
режной А. С., Гулько Н.В. Субсолидусное строение шестикомпо-
нентной системы MgO — Сг2О3— А12О3— ZrOo — TiO2 — SiO2.— Укр. хим.
Ж., 1965, 31, 881.
ргин В.В., Милюков Е.М. Расслаивание и кристаллизация стекол в
системе ZnO — А12О3 — SiO2 — TiO2.— Ж- прикл. хим., 1968, 41, 1941.
227
Вербицкая Т. Н., Буракова Т. Н. Кристаллооптическое изучение
системы PbTiO3 — PbZrO3 — PbSnO3.— Изв. АН СССР. Сер. физ., 1965
29, 2059.
Вербицкая Т. Н., Буракова Т. Н. Оптическое исследование фазово-
го состава титаната бария с малыми добавками окиси хрома.— Кристаллогра-
фия, 1967,12, 436.
В е л и к о д н ы й Ю. А., К о в б а Л. М., Трунов В. К- Твердые растворы
в системе TiO2 — CrNbO4.— Ж. неорг. хим., 1967, 12, 3212.
Волков В. Л., Фотиев А. А. О взаимодействии титанатов и силикатов
натрия с окислами кремния и титана.— Труды Ин-та химии, 14. Уральский
филиал АН СССР, Свердловск, 1967, 79.
Галахов Ф. Я-, Коновалова С. Ф. О ликвационных явлениях в систе-
ме Li2O — TiO2 — SiO2.— Изв. АН СССР. Неорг. материалы, 1965, 1, 1399.
Глазирин З.М., Фотиев А.А. Оптическое исследование титанатов нат-
рия.— Кристаллография, 1965, 10, 2.
Голубенко А. Н., Резухина Т. Н. Термодинамические свойства ти-
таната никеля.— Ж- физ. хим., 1965, 39, 1519.
Дидковская О. С., Климов В. В., Веневцев Ю. Н. Влияние
германия на структуру и свойства сегнетоэлектрика PbTiO3.— Изв. АН СССР.
Неорг. материалы, 1968,4, 157.
Зорина М. Л., Зорин А. П. Изучение структуры стекол системы SrO —
SiO2 — TiO2 с помощью ИК-спектров поглощения.— Изв. АН СССР. Неорг.
материалы, 1967, 3, 2276.
Зорин А. П., Зорина М. Л. Некоторые свойства и структура стекол си-
стемы ВаО—SiO2 — TiO2.— Изв. АН СССР. Неорг. материалы, 1966, 2, 18, 6.
Иванова Г. В., Дяткина М. Е. Молекулярные орбитали ортотитанат-
ного иона.— В кн.: Радиоспектроскопические и квантовохимические методы в
структурных исследованиях. «Наука», М., 1967, 26.
Исмаилзаде И. Г., Нестеренко В. И. О температурах сегнетоэлект-
рических переходов в BiBi2TiNbO9 и BiBi2TiTaO9.— Изв. АН СССР. Неорг.
материалы, 1968, 4, 760.
Исмаилзаде И. Г., Нестеренко В. И., М и р и ш л и Ф. А., Рус-
тамов П. Г. Рентгенографическое и электрическое исследование системы
Bi4Ti3O12 — BiFeO3.— Кристаллография, 1967, 12, 468.
Кашлинский А. И., Чечерников В. И. Магнитные свойства твердых
растворов РЬТЮ3 — LaFeO3.— Изв. АН СССР. Сер. физ., 1967, 31, 1839.
Каретников Г. С., Кудряв це в Н. Т., Гол овч а некая Р. Т.
Изучение щелочных растворов метатитаната натрия в присутствии глицери-
на.— Ж- физ. хим., 1965, 39, 2298.
V Кейер Н. П., Сазонова И. С., Михайлова И. Л., Белослуд-
ц е в а Т. Б. Каталитическая активность титаната никеля.— Кинетика и ка-
тализ, 1966, 7, 920.
Кириллов В. В., Исупов В. А. Исследование реверсивной диэлектри-
ческой проницаемости от напряженности поля в поликристаллическом ВаТЮз
и твердых растворах (Ba, Sr) TiO3.— Изв. АН СССР. Сер. физ., 1967 , 31, 1835.
Комиссарова Л. Н., Покровский Б. И., Нечаева В. В. О взаи-
модействии окиси скандия с двуокисью титана.— ДАН СССР, 1966, 168, 1076.
Комиссарова Л. Н., Покровский Б. И., Спиридонов Ф. М-
Взаимодействие окиси скандия с окислами элементов подгруппы титана."
В кн.: Химия высокотемпературных материалов. «Наука», М.-Л., 1967, 96-
____Ку т олин С.А., В у л их А.И. Синтез_метатитанатов щелочных металлов®
вакууме.— Ж- неорг. хим., 1965, 10, 140.
Кутолин С.А., Вулих А.И. Метатитанаты щелочных металлов.— В кН-'
Методы получения химических реактивов и препаратов, 16. М., 1967, 14.
Леонов А. Й., Пирютко М. М., Келер Э. К- Влияние газовой сред®*
и температуры на реакции в системе Се — Ti — О и сравнение свойств титан®'
тов редкоземельных элементов.— Изв. АН СССР. Сер. хим., 1966, 5, 787.
Леонидова Г. Г., Волк Т. Р. Исследование фазового перехода титана^
бария.— Физ. тв. тела, 1965, 7, 3344.
228
Дима р ь Т. Ф., Кривобок В. И., Находкова А. П., Б а-
т у р 3. Е. О синтезе твердого раствора системы BaTiO3 — PbTiO3 — CaTiO3
методом совместного осаждения. — Труды Всесоюзн. н.-и. ин-та хим. реакти-
вов и особо чистых хим. веществ, 2. 1966, 342.
Д и с ке р К- Е. Электрические свойства твердых растворов системы (Sr, Bi2/ )
TiO3 — CaTiO3.— Изв. АН СССР. Сер. физ., 1967, 31, 1832J
Лошкарев Б. А., Семириков И. С. Некоторые свойства и структура
керамики системы ZnO — TiO2-— Изв. АН СССР. Неорг. материалы, 1967,
3, 1467.
Лошкарев Б. А., Сычева Н. А., Устьянцева Т. А. Спекание и
некоторые свойства материалов в системе NiO — TiO2.— Изв. АН СССР. Не-
орг. материалы, 1967, 3,2040.
Лошкарев Б. А.. Семириков И. С. Условия образования и диэлект- V
рические свойства в системе Zn2TiO4—CaTiO3.— Ж- прикл. хим., 1966, 39,
Мартынов С. В., Петров В.М. Сверхвысокочастотные диэлектрические
свойства титаната бария с добавкой NiNb2O7 и СоТа2О7.— Изв. АН СССР.
Сер. физ., 1967, 31, 1888.
Медведков Б. Е., Н и Л. П., Пономарев В. Д. Взаимодействие
двуокиси титана со щелочными и высокомодульными (а = 30) алюминатными
растворами при 280° С.— Изв. АН Каз. ССР. Сер. хим., 1967, 3, 5.
Находнова А. П., Савенкова Г. Е., Климов В.В. Об электро-
проводности твердых растворов системы ВаТЮ3 — CaTiO3 — PbTiO3.— Изв. V
АН СССР. Неорг. материалы, 1968, 4, 244.
'Наумова И. И., Аникин И. Н. Растворимость двуокиси титана в расплав-
ленных боратах щелочных металлов.— Ж- неорг. хим., 1966, 11, 1746.
| Н овохатский И. А., Л е н ев Л. М., Савинская А. А. Термодина-
мические функции образования соединений в системе FeO — TiO2.— Изв.
высш. уч. зав. Черная металлургия, 1966, 2, 5.
Пол аидо в И. Н., М ы л о в В. П. Исследование сегнетоэлектрической кера-
мики Ba(Ti, Sn)O3 при высоких давлениях.— Физ. тв. тела, 1967 9,
2319.
( П ролесковский Ю. А., Акулович В.М. Почернение спектральных
линий, вызванных присутствием примесей в TiO2 — А12О3 и TiO2 — MgO.—
Becui АН БССР. Сер. х!м., н., 1967, 1, 51.
П я т е н к о Ю. А. О кристаллохимической трактовке некоторых промежуточных
фаз в системах СоО — HfO2 (ZrO2) и TR2O3 — TiO2(ZrO„).—ДАН СССР,
173, 1967, 634.
,<Рождественска М. В., Кунин В.Я. Электрические свойства моно-»
кристаллов SrTiO3.— Изв. АН СССР. Сер. физ., 1967, 31, 1915.
Сафиуллин Н. Ш.,Беляев Э. К- Исследование механизма образования
титанатов натрия в смесях углекислого натрия с двуокисью титана.— Изв.
АН СССР. Неорг. материалы/1968, 4, 557.
чСафиуллин Н. Ш., Беляев Э. К- Щелочное разложение измененных
хромсодержащих ильменитов.— Укр. хим. ж., 1968, 34, 217.
С аркисов Э. С., Бердников В. Р., Хожаинов Ю. М., Голо-
вина Г. П. Получение титаната самария Sm2Ti2O7. — Изв. АН СССР.
Неорг. материалы, 1968,4, 1371.
Саркисов Э. С., Бердников В. Р., Хожаинов Ю. М., Голо-
вина Г. П. Исследование взаимодействия гидроокисей неодима и титана.—
Изв. АН СССР. Неорг. материалы, 1968, 4, 787.
Саркисов Э. С., Бердников В. Р., Головина Г. П. О хими-
ческом взаимодействии гидроокисей титана и лантана. — Изв. АН СССР.
Неорг. материалы, 1967, 3, 1627.
Синяков Е.В., Дудник Е.Ф. Электрические свойства монокристаллов
твердых растворов BaTiO3 — CdTiO3.~ Изв. АН СССР. Сер. физ., 1967, 31,
1786.
229
Синяков Е. В., Флёрова С. А., К у быш к и н О. А. Влияние одно-
стороннего механического давления на фазовые переходы в монокристаллах
BaTiO3. —Изв. АН СССР. Сер. физ., 1967, 31, 1768.
Смажевская Е. Г., Федулов С. А., Ривкин В. И., Родиче-
в а В. Н., Подол ьнер Н. А., Белая В. С., Альбац В. И. Ис-
следование систем PbTiO3 — (Ko,5Bio,5) TiO3 и PbTiO3 — (Na0,5Bi0',5) TiO3__
Изв. АН СССР. Неорг. материалы, 1966, 2, 1850.
Сперанская Е. И., Скориков В. М. О титанатах висмута.— Изв.
АН СССР. Неорг. материалы, 1967, 3, 241.
СиняковЕ. В., ДудникЕ. Ф., Флёрова С. А. Влияние механи-
ческого давления и процессы поляризации в монокристаллах BaTiO3 и твердых
растворах BaTiO3 — ZnO2.— Физ. тв. тела, 1966, 8, 2848.
Соловьев С. П., Кузьмин И. И., Харченко В. А. Влияние облу-
чения на электрофизические свойства титаната бария и материалов на его
основе.— Изв. АН СССР. Сер. физ., 31, 1967, 1757.
Столыпин Ю. Е., Исупов В. А. Пьезоэлектрические свойства системы
твердых растворов PbTiO3 — PbZrO3 — PbMg0,5Wo,503.— Изв. АН СССР.
Сер. физ., 1967, 31, 1814.
Томашпольский Ю. Я-, Веневцев Ю. Н. Сегнетомагнетизм в си-
стеме Pb2CoWOe — BaTiO3.— Физ. тв. тела, 1965, 7, 3126.
Т у р и к А. В. К теории дисперсии диэлектрической проницаемости поликри-
сталлического BaTiO3 в области сверхвысоких частот.— Изв. АН СССР. Сер.
физ., 1967, 31, 1861.
Фотиев А. А., Боброва А. М., Глазырин М. П. Исследование фа-
зового состава системы ТЮ2 — Na2O — Na2CO3.— Труды Ин-та химии, 9.
Уральский филиал АН СССР, Свердловск, 1966.
Фотиев А. А., Волков Л. В. Условия разделения двуокисей кремния и
титана при переработке титано-кремниевого сырья с использованием соды.—
Труды Ин-та химии, 14. Уральский филиал АН СССР, Свердловск, 1967, 87.
Шапиро 3. И. Исследование структуры и диэлектрических свойств твердых
растворов в системах LiTiO3 — CdTiO3 и LiNbO3 — CgTiO3.— Изв. АН СССР.
Неорг. материалы, 1967, 3, 2245.
Шефтель И. Т., Заславский А. И., Лейкина Б.Б., Текстер-
Проскурова Г. Н. Полупроводниковые твердые растворы системы
BaTiO3 — SrTiO3 — BaSnO3.— Изв. АН СССР. Сер. физ., 1967, 31, 1828.
Штерберг А. А., Кузнецов В. А. Получение кристаллов РЬТ1О3 в гид
ротермальных условиях.— Кристаллография, 1968, 13, 745.
Янсон Г. Д., Фейденфельд Э. Ж-, Скомороха И. М., Макси-
мова О. С. Кинетика образования титаната свинца. Учен. зап. Рижского
политехнического ин-та, 1965/1966, 16, 387.
Akimoto S., Sy о п о Y. Разложение некоторых титанатов со структурой
шпинели при высоких давлениях.— J. Chem. Phys., 1967, 47, 1813.
Angelin L., Pinton M., C e v 1 e s G. Системы на основе TiO2 и их по-
ведение в плазмотроне.— Dinamica reasioni chim., Roma 1967, 247.
Arend H., Coufova P., Novak J. Химические процессы, протекающие
при восстановлении кристаллов титаната бария водородом.— J. Am. Сегат.
Soc., 1967, 50, 22.
Arend Н., Novak J.— Влияние расплава флюорита на химический состав
монокристаллов BaTiO3. — Kristall und Technik, 1966, 1, 93.
Ault J.D., Welch A. J. E.— Система окись иттрия — двуокись титана.—
Acta Crystallogr., 1966, 20, 410.
Asbrink G., Magneli A.— Рентгеноструктурное исследование ряда сме
шанных окислов систем со структурой псевдобрукита.— Acta chem. Scand.,
1967, 21, 1977.
Bayer G., Florke O. W., Hoffmann W., Schell H.-J. Расслаи-
вание и кристаллизация в стеклах системы Na2O — TiO2 — SiO2. Glastechn.
Ber., 1966, 39, 242.
Bayer G., Hoffman W. Сложные щелочные окислы титана Ах ==
“ г/)16 со структурой типа МпО2.— Am. Mineralogist, 1966, 51, 511.
230
gayer G., Hoffmann W. Кристаллизация стекол в системе Na2 — TiO2 —
SiO2.— Glass Technol., 1966, 7, 94.
ger t au t E.-F., P a t г a t G. Структура LiGaTiO4.— Bull. Soc. France mi-
ner. et crystallogr., 1965, 88, 586.
gemst A., Delaunois С. Изучение роли TiO2 в системе SiO2 — TiO2 —
K2O.— Verres et refract, 1967, 21, 11.
g i 1 1 iet Y., Morge ns t er n - В a d ar au I., Poix P., Michel A.
Структура упорядоченного Ln2TiO4.— Compt. rend., 1965, 260, 4780.
grandt R. C., Ansell G. S. Выращивание монокристаллов титаната ба-
рия.— Mater. Res. Bull., 1967, 2, 585.
Bratton R. J., Tien T. Y. Фазовые переходы в системе BaTiO3 —
KNbO3.— J. Am. Ceram. Soc., 1967, 50, 90.
В h i c 1 e V. G., В h a s i п H. С. Изучение системы SrTiO^7 — Co с помощью
эффектаМассбауэрa.— Phys. Rev., 1967, 159, 586.
В 1 asse G. Новые титанаты типа АВ2Об.— J. Inorg. a. Nucl. Chem., 1966, 28,
1122.
В 1 a s s e G. Кристаллическая структура ATiNbO6, ATiTaO6 и ASbO6 (A —
— TR) Mater. Res. Bull., 1967, 2, 497.
Carlsson L. Изменение кристаллической структуры BaTiO3.— Acta Crysta-
llogr., 1966, 20, 459.
Chandrasekaran K. S., Mohanlal S. К. Явление аномальной дис-
персии в структуре титаната бария.— Acta Crystallogr., 1965, 19, 853.
Cocco А.; С h i а с i d h I. Исследование системы ZrO2 — TiO2 — SrTiO3.—
Ann. chim., 1965, 55, 1350.
Cooke F. W., Brandt R. C., DeVries R. C., Ansell G. S. Диэлек-
трические характеристики старения и микроструктура в системе BaTiO3 —
CaTiO3.— J. Am. Ceram. Soc., 1966, 49, 648.
Collongues R., etall. Структура и свойства соединений, образованных
окислами редкоземельных элементов с TiO2.— Bull. Soc. chim. France, 1965,
4, 1141.
,'Couf ova P., Novak J., H 1 a s i v с о v a N. Гидроксил, как дефект ре-
шетки перовскита BaTiO3.— J. Chem. Phis., 1966, 45, 3171.
Colwell J. H. Теплоемкость сверхпроводящего смешанного титаната.— Phys.
Letters, 1967, A25, 623.
К haussy M. C., Vincent Н., Joubert J.-С. Область существования
фазы шпинели в системе TiO2 — ZnO — Li2O.— Bull. Soc. chim. France, 1966,
1, 198.
К ummins S. E., Cross L. E. Электрические и оптические свойства крис-
таллов Bi4Ti3O12.— J. Appl. Phys., 1968, 39, 2268.
gD a 11 a R. K-, Rustam R. Устойчивость Ni2TiO4.— Z. Kristallogr., 1965,
121, 410.
iDeschanvres A., Reveau B., Tollemer F. Замещение двух-
валентного металла в титанатах типа перовскита на медь.— Bull. Soc. chim.
Trance, 1967, 11,4077. ‘
К) e shpandeV. V., Rama R.K. Фазовые исследования в системе ZrOo —
TiO2.—Current Sci., 1967, 36, 374.
' F orster R.H., Hall E. О. Нейтронографическое изучение Fe2TiO4.— Acta
Crystallogr., 1966, 18, 857.
V orland S. К. Дефекты структуры рутила, содержащего малые добавки оки-
си алюминия.— Acta chem. Scand., 1966, 20, 2573.
,F rederikse H. P. K., Candela G. А. Магнитная восприимчивость тита-
ната стронция.— Phys. Rev., 1966, 147, 583.
; F r a n с о i s e C.-M. Изучение условий излучения титанового ильменита MgTiO3.. '_
активированного четырехвалентным марганцем.— Compt. rend., 1967, 265,
В1047.
-G a d а 1 1 а А. М. М., White J. Равновесия в системе СиО — Си2О — TiO2.—
— Trans. Brit. Ceram. Soc., 1966, 65, 157.
G esemann R. Смешанные кристаллы титаната — цирконата свинца.— Wiss.
Z. Humboldt-Univ., Berlin Ges.-sprachwiss. Reihe, 1964, 13, Beil I, 17.
231
Ghorbanian J., Poix P. Получение окислов с общей формулой
[Fe^Fe/J^Ti^] О4.—Compt rend., 1965, 261, 3625.
Guillen M., Bertaut E. F. Структура La2TiO5.— Bull. Sos. franc. cc-
ram., 1966, 72, 57.
Guillen M., Bertaut E. F. Структура La2TiO5.— Compt. rend., 1966
AB262, B962.
Har ar i A., Thery J., Col longues M. R. О получении монокристал-
лов окислов MO2.— Rev. internat. noutes temperat et refract., 1967, 4, 207.
Hagenmuller P., Guillaud Ch., L e с e r f A., Rault M., Vil-
lers G. Синтез и исследование структуры титановых шпинелей.— Bull.
Soc. chim. France, 1966, 8, 2589.
Hanykyr V., Tyrolerova P., Vich J. Кинетика спекания чистого
BaTiO3 — Sb.— Vysokeskoly chem.-technol. Praze, 1967, BIO, 79.
Hanykyr V. Кинетика реакций образования SrTiO3.— Silikaty, 1966, 10, 17.
Hauser O., Schenk M. Изменение кристаллической структуры брукита и
BaTiO3 при облучении нейтронами.— Wiss. Z. Humboldt-Univ., Berlin Ges.-
sprachwiss. Reihe, 1964, 13, Beil 1, 59.
Harris N.H., Tennery V. J. Структурные и диэлектрические исследова-
ния в системе PbTiO3 — BaZrO3.— J. Am. Ceram. Soc., 1967, 50, 404.
Holzapfel H., Sieler J. Получение и структура титанатов редкоземель-
ных элементов.— Z. anorg. Chem., 1966, 343, 174.
Kato М., К u b о Т. Реакции в твердой фазе в системе Y2O3 — TiO2.—
J. Chem. Soc. Japan. Industr. Chem. Sec., 1967, 70, 840.
Kato M., К u b о T. Двойной окисел в системе CuO — TiO2.— J. Chem. Soc.
Japan. Industr. Chem. Sec., 1967, 70, 844.
Kasper H. Поглощение света Co2+, Ni2+ и Cu2"^ в псевдобруките и трипсевдо-
брукитных минералах.— Z. anorg. Chem.," Г967, 354T*2oS.
К о u t n i k V. Изучение образования титаната свинца.— Sb. vedec. praci Vy-
sokeskoly chem.-technol. Pardubice, 1964, 2, 45.
Koizumi H. Рентгенографическое изучение системы РЬО — Nb2O5 — TiO2.—
Japan J. Appl. Phys., 1966, 5, 198.
Krause R. H., Reamer H. W-, Martin J. L. Твердый раствор на базе
рутила.— Mater Res. Bull., 1968, 3, 233.
Куниги M., Кон иси А., Фукутани С. Восстановление титаната
алюминия.— J. Soc. Mater. Sci., Japan, 1966, 15, 517.
Кубо T., Като М., Фудзита Т. Реакции в твердой фазе между TiO2 и
ВаСО3.— J. Chem. Soc. Japan. Industr. Chem. Sec., 1967, 70, 847.
Кубо T., Като M., Фудзита T. Мокрый способ синтеза BaTiOs.—
J. ChemTSoc. Japan. Industr. Chem. Sec., 1968, 71, 1147
L e c e r f A., Rault M., Villers G.— Получение, кристаллическая струк-
тура и магнитные свойства шпинелей состава Mn2J^x Ti?+X Ti4+t)4 для зна-
чений х = 0 — 1.— Compt. rend., 1965, 261, 749.
Louis В., Hubert F. Диаграмма состояния А12О3 — ТьО3.— Compt. rend.,
1964, 258, 4282.
Loan Р. R., Chakrabarty M. R. Кристаллохимия и диэлектрические
свойства в системах BaTiO3 — UOo и BaTiO3 — BaUO3.— J. Am. Ceram. Soc.,
1966, 49, 276.
Loye O., Laruelle P., Harari А. Кристаллическая структура низко-
температурной формы двойного окисла ThTi906.— Compt. rend., 1968, С266,
454.
L е j u s M.-A., Goldberg D., Revcolevschi А. Некоторые соедине-
ния рутила с окислами трех- и четырехвалентных металлов.— Compt. rend.,
1966, С263, 1223.
Marks S. W., Atoniak С. E. Диэлектрические свойства поликристалл и-
ческих трититаната и тетратитаната бария.— J. Phys. Chem., 1968, 72, 1172.
Matsuo Y., Sasaki H. Образование твердых растворов цирконат свинца —
титанат свинца.— J. Am. Ceram. Soc., 1965, 48, 289.
232
ДО е i s е 1 A., К б s t 1 е г М., Merkel А. Влияние химической связи на рент-
геновский Ка-дублет титана.— J. prakt. Chem., 1966, 34, 112.
до u m m е W. G., Wadsley A. D. Кристаллическая структура
NaTi2Al5O12.— Acta Crystallogr., 1967, 23, 754.
доег t e n L. Направленная дисперсия оптических колебаний тетрагонального
BaTiO3 при комнатной температуре.— Phys, status solidi, 1968, 25, 125.
до е s n ar d G., Uz an R., Caboud В. Масс-спектроскопическое иссле-
дование продуктов испарения двуокиси титана и титаната бария.— Rev.
Phys. Appl., 1966, 1, 123.
до и д з у н а М., Н о г у т и Т. Система ZrO2 — TiO2-— Rept. Govt Indastr.
Res. Inst. Nagoya, 1967, 16, 179.
Newnham R. E. Кристаллическая структура ZrTiCL.— J. Am. Ceram. Soc.,
1967, 50,216.
Nilsen W. G., Skinner J.G. Спектр комбинационного рассеяния титана-
та стронция,— J. Chem. Phys., 1968, 48, 2240.
Ouchi H., Nagano K-, Hayakawa S. Пьезоэлектрические свойства
твердых растворов Pb (Mg1/3Nb2/3) O3 — PbTiO3 — PbZrO3.— J. Am. Ceram.
Soc., 1965, 48, 630.
(O’Shea D. S., Kolluri R. V., Cummins H. Z. Температурная зави-
симость спектра KP титаната стронция.— Solid State Communs, 1967, 5, 241.
Оц у к а А. Исследование титано-хромовых шпинелей, содержащих ионы СО2~^
и№2+—J . Ceram. Assoc. Japan, 1966, 74, 153.
Perry F. W. Рост монокристаллов (Ba, Pb) TiO3 из расплавов боратов.—
J. Phys. Chem. Solids, 1967, 1, 483.
Payne W. H., Tennery V. J. Диэлектрические и структурные исследо-
вания системы BaTiO3 — ВаНЮ3.— J. Am. Ceram. Soc., 1965^ 48, 413.
Payne D.A., Anderson H.U. Торможение роста зерен титаната бария.—
J. Am. Ceram. Soc., 1967, 50, 491.
Prezedmojski J., Chmielewska J. Рентгеновское исследование ти-
таната бария с примесями металлов и окислов металлов.— Zesz. nauk Poli-
techn. Warsz., 1965, 117, 113.
Queyroux F. О существовании нового соединения Yb6TiO11 и диаграмма рав-
новесия Yb2O3 — TiO2.— Bull. Soc. franc, mineral, et crystallogr., 1965, 88, 519.
Raveau B., Thomazeau J.-C. Новые фазы типа пирохлора.— Compt.
rend., 1968, C266, 540.
Reid A. F., Sienko N. J. Некоторые характеристики натрий-титановой
бронзы.— Inorg. Chem., 1967, 6, 321.
Reid A. F., Sienko M. J. Титанит скандия ScTiO3. —Inorg. Chem., 1967,
6, 520.
Reid A. F., Wadsley A. D., Sienko M. J. Кристаллохимия NaScTiO*
и изоморфных с ним соединений.— Inogr. Chem., 1968, 7, 112.
Remoissenet M., Godefroy L. Термическая диффузия в твердых ра-
створах титаната бария и титаната стронция.— Compt. rend., 1966, АВ262,
В56.
R u d о г f f W.; Luginsland H.-H. Исследование двойных окислов в си-
стемах TiO2 — NbO2 и TiO2 — TiTaO4.— Z. anorg. Chem., 1964, 334, 125.
Rogers M. D. Рентгенографическое исследование облученного титаната строн-
ция.— J. Nucl. Mater., 1967, 22, 223.
Reu d m an IP. W., О r i e 1 1 у W., Banerjee S. К- Объяснение магнит-
ных свойств Fe2TiO3.— Phys. Letters, 1967, A25, 446.
Schaufele R.F., Weber M. J. Спектр KP первого и второго порядка
. кристалла SrTiO3—J. Chem. Phys., 1967, 46, 2859.
Sieler J., Hennig H. Магнетохимия титанатов редкоземельных элемен-
тов.— J. prakt. Chem., 1966, 34, 168.
$ a i k a 1 i Y., Paris J.M. Получение метатитанатов некоторых двухвалент-
ных металлов при низких температурах.— Compt. rend., 1967, с265, 1041.
3 th г е у F. Влияние pH на химическое получение титаната бария — строн-
ция.— J. Am. Ceram. Soc., 1965, 48, 401.
233
Schenk M. Фазовые превращения титаната бария, вызванные облучением_____
Kristall Technik, 1966, 1, 305.
Schenk М. Фазовый переход в титанате бария, вызванный облучением.— bja.
turwis, 1965, 52, 537.
S t е г z е 1 W. Получение и свойства тиотитаната свинца.— Naturwis., 1966
53, 199.
S и с h о w L. Титанат висмута и магния — новый необычный перовскит.-^.
J. Inorg. a. Nucl. Chem., 1968, 30, 87.
Sugai Т., Hasegawa S., Ohara G. Рост монокристаллов титаната
стронция из солевых расплавов.— Japan J. Appl. Phys., 1968, 7, 358.
Такэда Т., Ватанабэ А. Места замещения редкоземельных элементов в
ВаТЮ3.— Nat. techn. Rept., 1966, 12, 155.
Tanaka Y., Ikeda T., Toyoda H. Пьезоэлектрическая керамика в
цирконатно-титанатных системах свинца.— Rev. Electr. Commun. Lab., 1965,
13, 744.
Tien T. Y., Hummel F. А. Твердые растворы в системе SrTiO3 — (La2O •
• 3TiO2).— Trans. Brit. Ceram. Soc., 1967, 66, 233.
Tien T. Y., Moratis C. J. Диэлектрические свойства твердого раствора ти-
таната стронция, содержащего ниобий.— J. Amer. Ceram. Soc., 1967, 50, 392.
Т г о а и А. Переход пароэлектрической фазы в сегнетоэлектрическую и элемен-
тарный ферроэлектрический диполь в титанате бария. — Bull. Soc. Sci.
Bretagne Sci. mathn., phys. et natur, 1967, 41, 125.
Tillmans E. Новое соединение в системе ВаО — TiO2.— Naturwis., 1968,
55, 81.
Waring J.L., Schneider J.J. Фазовые равновесия в системе Cd2O —
TiO2.— J. Res. Nat. Bur. Standards, 1965, A69, 255.
WaringJ.L., Roth R.S. Влияние окисных добавок на полиморфизм пяти-
окиси тантала.— Phys, and Chem., 1968, 72А, 175.
Walters L. C., Grace С. E. Образование точечных дефектов в титанате
стронция.— J. Phys. Chem. Solids, 1967, 28, 239.
Walters L. C., Grace R.E. Диффузия точечных дефектов в титанате
стронция.— J. Phys. Chem. Solids, 1967, 28, 245.
Winkler E. R., S a r v e г J.F., Cutler LB. Твердые растворы двуокиси
титана в окиси алюминия.— J. Am. Ceram. Soc., 1966, 49, 634.
Weber М. J., Schaufele R. F. Излучательные и безизлу нательные пе.
реходы спектра флуоресценции Еи3“^ в SrTiO3 .— J. Chem. Phys., 1965, 43,
1702.
Webster A. H., McDonald R. C., Bowman W. S. Система
PbO — ZrO2 — TiO2 при 1100° C.— J. Canad. Ceram. Soc., 1965, 34, 97.
Vaughan R. W., Drickmer H. G. Исследование эффекта Массбауера в
a — Fe2O3, FeTiO3 и FeO при высоких давлениях.— J. Chem. Phys., 1967,
47, 1530.
Vincent Н., J о u b е г t J.-С., D u r i f А. Структурные исследования упо-
рядоченных форм ортотитанатов цинка и марганца.—• Bull. Soc. chim. France,
1966, 1, 246.
Zitkova J., Zdansky K., Sroubek Z. ЭПР в кристаллах восстанов-
ленного BaTiO3.— Czechosl. Phys. J., 1967, B17, 636.
ГЛАВА VI
ГАЛОГЕНИДЫ ТИТАНА
Титан образует галогениды, в которых проявляет валентность
2, 3 и 4. Галогениды титана низшей валентности, по-видимому,
следует отнести к металлическим соединениям. По своим свойст-
вам они резко отличаются от тетрагалогенидов.
ж Природа связи титана с галогенами ковалентная. По мере уве-
личения ионного радиуса галогена возрастает полярность его свя-
зи с титаном. Свойства галогенидов при этом закономерно изме-
няются от легколетучего фторида к твердому иодиду, плавяще-
муся при температуре 150° С.
Тетрагалогениды титана неустойчивы к действию воды, под-
ергаются гидролизу с отщеплением, обычно, в начале двух ато-
мов галогена, а затем двух остальных. В качестве промежуточных
^продуктов образуются оксигалогениды, содержащие устойчивую
^группу TiO (титанил). В оксигалогенидах, как и во многих дру-
гих соединениях, титанил выполняет роль устойчивого двухва-
лентного катиона, что объясняется большой энергией связи ки-
слорода с титаном.
[ ФТОРИДЫ ТИТАНА
Г Известны фториды титана, имеющие состав TiF, TiF2, TiF3 и
FiF4. Фториды титана низшей валентности исследованы еще не-
Юстаточно и могут существовать только в экстремальных условиях.
Золее изучен тетрафторид, в котором титан проявляет валент-
иость 4, характерную для его положения в периодической системе
Элементов.
ФТОРИДЫ ТИТАНА НИЗШЕЙ ВАЛЕНТНОСТИ
L Монофторид титана TiF обнаружен в спектре поглощения TiF4,
нагретого до 1000—2000° С [373]. Он образуется из TiF4 вслед-
ствие термической диссоциации. В чистом виде не получен и свой-
ства его не исследованы.
I При нагревании фтортитаната калия K2TiF6 в сухом водороде
Наблюдается образование темно-фиолетовых призматических кри-
сталлов, которые, согласно [360], являются дифторидом титана
235
TiF2. В чистом виде кристаллы TiF2 не получены. Присутствие
дифторида титана в виде труднорастворимых двойных солей обна-
ружено в хлоридно-фторидных расплавах [145]. Двойные фториды
титана получаются при электролизе и имеют состав M2TiF6 (М —
щелочной металл).
Вебер [592] при восстановлении водородом нагретого до крас-
ного каления фтортитаната калия K2TiF6 получил спек, окрашен-
ный в интенсивный фиолетовый цвет. Окраска спека, по мнению
автора, вызвана присутствием двойного трифторида титана K2TiF5.
Трифторид титана TiF3 в чистом виде получают нагреванием гид-
рида титана с фтористым водородом при температуре 700° С [269].
От сопутствующих примесей TiF3 очищают сублимацией в высоком
вакууме при температуре 950° С. Кристаллы TiF3 окрашены в си-
ний цвет и имеют ромбоэдрическую структуру с параметрами ре-
шетки (2 == 5,523 А, ОС == 58,88 , Z == 2, Ррентг — 3,00, Ризм
= 2,98 г!см\ На воздухе они не окисляются, устойчивы к
действию воды и разбавленных кислот. При нагревании выше
950° С подвергаются диспропорционированию по реакции
4TiF3 = Ti + 3TiF4.
Трифторид титана способен присоединять хлор, образуя сме-
шанный галогенид TiClF3 в виде желтого порошка [563]. Это со-
единение на воздухе неустойчиво и под действием влаги гидроли-
зуется с образованием TiOF2.
Глятцель [323] считает, что в отличие от соляно- и сернокислых растворов,
плавиковая кислота реагирует с металлическим титаном без образования соеди-
нений низшей валентности. Однако Вуди Кокрель [568] придерживаются противо-
положного мнения. По их наблюдениям, титан растворяется в плавиковой кислоте
с образованием раствора фиолетового цвета, содержащего ионы Ti3+ . В присут-
ствии кремния происходит комплексообразование и раствор титана в плавиковой
кислоте окрашивается в зеленый цвет. Растворы низших фторидов титана получены
электролизом фтортитаната калия [482] и восстановлением металлами в присут-
ствии соляной кислоты — цинком [500], амальгамой натрия [481 ], кадмием [478].
В результате восстановления раствор K2TiF6 в нагретой соляной кислоте окраши-
вается в зеленый цвет.
Трифторид титана с фторидами щелочных металлов образует
двойные соли. Вебер [592] получил фтортитанат калия K2TiF5.
При взаимодействии TiF3 с KF в водном растворе образуется фтор-
титанат калия KsTiF6. [483, 484].
Фтортитанат аммония (NH4)2TiF5 получают в виде темно-фиоле-
товых кристаллов, плохо растворимых в воде [483, 484]. При элек-
тролитическом восстановлении водного раствора (NH4)2TiFe, со-
держащего примесь NH4F, выделяется фтортитанат аммония
(NH4)3TiF6 [484, 485, 491]. Эта соль в виде красно-фиолетовых
легкорастворимых в воде кристаллов образуется в результате взаи-
модействия TiCl3 с раствором NH4F [478].
При электролизе фтортитанатов калия и натрия в расплаве
NaCl + КС1 (эвтектическая смесь) можно получить смешанный кя-
f
13В
лий-натриевый фторид трехвалентного титана K2NaTiF6 и двой-
ные фториды — Na3TiF6 и KsTiF6. Калий-натриевый офтор-титанат
имеет кубическую решетку с параметром а = 8,3669 А, показатель
преломления 1,408 [201]. У двух других титанатов также куби-
ческая, но несколько искаженная решетка.
Во фторидных расплавах трехвалентный титан существует в
виде гексафторкомплекса TiFe"”. Константа образования этого ком-
плекса и зависимость изобарного потенциала от температуры имеют
вид 1142]
‘ log К- 10,154 + 5,946т-1,
AZ — —27190 — 27,513(1,694+ log [F])T [кал/г-ион].
Бедон и соавторы [193] рассчитали в рамках метода молекулярных орбиталей
уровни энергии иона Ti3^- (3d1) в октаэдрическом поле шести ионов F~ ; о- и л-мо-
лекулярные орбитали комплекса строились в виде линейных комбинаций атомных
орбиталей центрального иона и молекулярных орбиталей лигандов. Найдена ор-
битальная заселенность титана: 3d2’754s0,37 4р0,34. Эффективный заряд на титане
+0,54, а на фторе —0,59.
ж; Фенске [293] отмечает, однако, что последовательное исполь-
зование гибридизированных орбиталей лигандов в качестве сг-ор-
биталей не всегда дает верные значения 10£>g и энергий переходов.
К Растворы TiF3 в плавиковой кислоте, в которых титан нахо-
дится в виде комплексов [TiFn (Н2О)б_л13-л зеленого цвета, дают
сигнал ЭПР [31]. Ширина сгн-линии 20 э при частоте 9430 мгц и
температуре 295° К, G-фактор равен 1,946. Появление сигнала
ЭПР объясняется некубичностью симметрии электрического поля
.лигандов смешанных водно-фторидных комплексов Ti3+.
Н Кроме зеленых комплексов при действии плавиковой кислоты
на кристаллический трихлорид титана получен коричневый раст-
I вор [134], который дает сигнал ЭПР (концентрация 0,25 молъ/л)
при частоте 9320 мгц с 6Н = 18 э.
Негой [446] исследовал видимый спектр поглощения (NH4)3 TiFe
и дал интерпретацию его в рамках модели кристаллического поля,
а также рассчитал нижние электронные уровни ионов [TiF6]"~3 и
IlTi (NH3)6]3+ [447].
Изучена зависимость времени спинрешеточной релаксации
Ти и 7\ протонов водных растворов [TiFn (Н2О)б-пГ-3 [49].
ЧЕТЫРЕХФТОРИСТЫЙ ТИТАН
№ Четырехфтористый титан впервые получили нагреванием ме-
г таллического титана с фтором или со смесью фтористого водорода
• и водорода [515, 517]. Фтор реагирует с металлическим титаном с об-
I разованием TiF4 при температуре около 200° С [358].
Четырехфтористый титан образуется при действии трехфтористого брома на
двуокись титана при температуре около 500° С [197], а также при прокаливании
безводного фтортитаната бария BaTiF6 в результате его термического разложе-
ния [264].
237
Четырехфтористый титан — кристаллическое вещество, при
20,5° С удельный вес его 2,798 [517], температура кипения
284° С [517].
Холл с соавторами [349] измерил упругость пара TiF4, устано-
вил зависимость упругости пара от температуры
logP = — 2,567 log Т+ 16,631 [атм].
По упругости пара определена температура кипения TiF4 283,1° С,
что хорошо согласуется с данными [517]. Для T1F4tb они также
вычислили образования АЯ°98ТВ — 22,87 кксифлоль, S°98ra3 = 73,2 и
5298,., = 31,3 кал!моль град.
Установлено, что тетрафторид титана реагирует с сероводоро-
дом при нагревании, образуя дисульфид титана TiS2, с хлорокисью
фосфора при температуре около 30° С вступает в обменное разло-
жение с образованием POF3 и TiCl4 [515]. Тетрафторид титана
хорошо растворяется в 100%-ной серной кислоте. Металлические
Na, Mg, Са, Fe, Al, В при нагревании восстанавливают TiF4 до
металла. Продуктом взаимодействия TiF4 с медью и кремнием
является TiCl3. Силицилхлороформ при температуре около 100° С
вступает в реакцию обменного разложения [516]
3TiF4 4- 2SiHCl3 = 4SiHF3 + 3TiCl4.
Четырехфтористый титан способен образовывать со многими ор-
ганическими и неорганическими соединениями продукты присо-
единения и двойные (комплексные) соли.
Из спиртового раствора TiF4 после упаривания выделяется амор-
фная масса, имеющая состав TiF4 • QHsOH [515]. С пиридином
TiF4 дает продукт присоединения TiF4 • C5H5N. Получены продук-
ты присоединения TiF4 с нитрилами состава TiF4 • CH3CN и
TiF4 • (C6H5CN)2. [262, 498]. На холоду TiF4 поглощает четы-
ре моля аммиака; при температуре 120° С выделено соединение
TiF4 • 3NH3.
Шандлер с соавторами [226] синтезировал гексафториды тита-
на типа (LH)2 TiF6, где L — NH (изо-С3Н7)2, NH (н-С3Н7)2,
NH (С2Н5)2, N (CgHgJg, пиридин и морфин. Пиперидин (Pip) обра-
зует гексафтортитанат [PipH2] TiF6.
Вторичные амины R2NH, например диизопропиламин и диэтил-
амин, при добавлении их к ацетоновому раствору TiF4 реагируют с
образованием твердых продуктов TiF3R2N + (R2NH2)2 TiF6. [227,228].
Третичные амины в этих условиях дают только продукты присо-
единения. Пиридин, например, образует соединение (C6H6N • TiF4)x,
которое является, вероятно, полимерным продуктом.
Раствор TiF4 в плавиковой кислоте извлекает из н-гептана
растворенные в нем м- и n-ксилол, проявляя свойства сильной
кислоты [428]. При этом одновременно повышается растворимость
TiF4 в плавиковой кислоте.
238
Система TiF4 — HF — H2O
в ^к°1ДНвЙ фтсрнстоводородной кислоте TiF4 растворяется
плохо 1515 J. В водных растворах плавиковой кислоты он раство-
ряется хорошо. Руфф и Ипсен [515] получили из водных раство-
ров гидрат TiF4 • 2Н2О, однако существование этого соединения
не нашло подтверждения в работах других авторов.
СИ£Г^а —Н2О в пределах концентрационного треугольника ТЮ,_
HF — HjU изучена методом растворимости [7] (рис. 56, табл. 27). Растворимость
Рис. 56. Изотерма растворимости в системе TiOa — HF — Н2О при 25* С.
* Ией до концентрации 25, 95 вес.% изменяется линейно, отношение концентра-
®1Й F : Ti в растворе 4,01—4,33. На этом основании авторы [7] считают, что в вод-
растворе плавиковой кислоты титан существует в виде комплексного иона
OF2-. Существование комплексного иона подтверждается данными потенцио-
^р и чес ко го и кондуктометрического титрования. Равновесной твердой фазой
1 Разбавленных плавиковокислых растворах является гидроокись титана, состав
которой, однако, точно не определен.
% Изучено равновесие фторидных комплексов в водных раство-
fex. По мнению [224], титан в плавиковокислых растворах
I
239
Таблица 27
Растворимость в системе HF—TiO2—Н2О при температуре 25° С
Состав жидкой фазы, вес. % Остаток, вес. % Твердая фаза
HF TiO2 HF: TiO2 HF TiOa
3,11 4,54 3,01 4,11 1 - - - TiO2
4,43 4,09 1,48 77,46 То же
6,24 6,07 4,10 — — » »
10,04 9,99 4,01 —- > »
12,56 12,31 4,07 3,48 89,54 » »
16,27 15,17 4,28 5,27 80,92 > >
18,27 17,93 4,07 • — 1. — » »
20,56 18,96 4,33 4,25 75,89 » »
20,44 20,35 4,01 И - — » »
22,00 21,82 4,03 — — » »
24,34 22,74 4,27 8,68 80,09 » »
26,52 25,30 4,18 9,14 79,18 » »
25,95 25,84 4,01 25,02 60,41 TiO, + TiOF2-H,O
26,94 26,33 4,08 32,51 65,53 TiOF,-H»O '
31,75 29,35 4,32 32,63 64,23 То же
34,15 31,20 4,37 33,70 65,67 » »
36,57 32,92 4,44 34,46 58,23
37,29 33,16 4,49 — L » »
38,79 34,99 4,43 36,55 57,10 » »
37,86 35,08 4,31 1 "Ч » »
39,60 36,32 4,35 36,06 53,88 » »
существует в виде фторидных комплексов с зарядом от +1 до —2,
находящихся между собой в состоянии равновесия:
TiOF2- TiOF? + F~,
TiOFr i TiOF2 + F~,
TiOF2 TiOF+ + F-.
Из результатов потенциометрического титрования раствора
TiO (С1О4)2 фтористым натрием авторы вычислили константы рав-
новесия: log К, = 12,03, log К2 = 3,31, log Ks = 4,35.
Ранее константа нестойкости комплекса TiOF+ определена Клей-
нер [51] равной р/С3 = 6,44.
Морган [425] считает, что в водных растворах фторид титана
существует в виде ионов Ti (H2O)4F2^, которые вступают в протон-
ный обмен со средой по двум реакциям:
Ti (H2O)4Fi+ + н3о+ = Ti (Н2О)4 (HF)F2+ + Н2О
и
Ti (Н2О)4Н+ = Ti (H2O)3F22+ + Н2О.
Вудс и Коккерел [568], изучив поглощение ионов из водны*
растворов ионообменными смолами, высказали предположение °
240
существовании в растворах ионов TiF5 или еще более высококоор-
динированных.
Основываясь на данных потенциометрического титрования, кондуктометрии
криоскопии и измерения электропроводности, авторы [7] считают, что в плавико-
вокислых растворах существуют фтортитановые кислоты состава Н [TiF4 (ОН)НоО]
H[TiF5. Н2О] и Н2 [TiF6].
При концентрации в растворе HF более 25,95 вес. % в системе
TiO2 — HF — Н2О кристаллизуется гидрат оксифторида титана
TiOF2 • Н2О. Область системы с малой концентрацией воды не изу-
чена и природа образующихся в ней твердых фаз неизвестна.
Воррес и Доногуе [564] получили безводный оксифторид тита-
на TiOF2 и по данным рентгенограммы порошка определили его
структуру. Оксифтсрид титана кристаллизуется в кубической си-
стеме, фед. гр. РтЗт, параметры решетки а = 3,798 А, ризм =
= 2,92, Ррентг ~ 3,09 г/см3, 2=1. Структура TiOF2 состоит из
октаэдров TiXe (X — атомы F и О), в которых каждая вершина
является общей для двух октаэдров.
По данным [154], в инфракрасных спектрах TiOF2 • Н2О имеется полоса при-
мерно 1600 см~х, относящаяся к деформационным колебаниям молекул воды. Связь
между атомами металла и кислорода в TiOF2 цепочечная. Это подтверждается на-
личием в ПК-спектрах широкой полосы с частотами в пределах 750—950 см~~\
Наиболее вероятное строение оксифторида титана выражается схемой
Частоты валентных колебаний цепей высоки (до 950 см *), из чего можно заключить,
что кратность связей Ti —О значительно выше единицы.
ДВОЙНЫЕ ФТОРИДЫ ТИТАНА
Четырехфтористый титан и оксифторид титана с фторидами ам-
мония, щелочных, щелочноземельных и некоторых других метал-
лов образует двойные (комплексные) соли. Для этих солей харак-
терно присутствие комплексного иона [TiX6]2~, где X = F и х/2 О.
Растворы гексафторидных комплексов титана в присутствии фто-
Ридов щелочных металлов весьма стойки. Вследствие этого двой-
ные фториды титана могут быть получены не только из неводных,
Но также и из водных плавиковокислых растворов. Оксифторид-
Ные комплексы титана обычно образуются в результате окисления
йли гидролиза гексафторидов при высокой температуре.
16 9-614 241
Кроме гексамерных известны двойные соли, содержащие гегь
тафторидные комплексы титана TiF?”. Они устойчивы при избыт-
ке в растворе фторидов щелочных металлов.
Фтортитанаты аммония
При действии аммиака на раствор двуокиси титана в плавико-
вой кислоте получается гексафтортитанат аммония (NH4)2 TiF6
[175, 301, 437, 487]. Эта соль образует гексагональные кристаллы,
неустойчивые на воздухе и подвергающиеся гидролитическому раз-
ложению под действием влаги [301]. При нагревании до темпера-
туры 300° С (NH4)2 TiF6 на воздухе разлагается по реакции
(NH4)2 TiF6 + О2 - 2NH4F + TiO2 + 2F2.
Соль хорошо растворима в воде. Растворимость ее при темпера-
туре 20—22° С равна 25 г на 100 мл воды [301, 303].
Пицини [487], нагревая раствор (NH4)2 TiF6 в воде, получил
оксифторид 3 (NH4)2 TiF6 • 2TiO2 • 3NH4F, индивидуальность кото-
рого требует подтверждения.
Мариньяк [437] из раствора фторида аммония выкристаллизо-
вал гептафтортитанат аммония (NH4)3 TiF7. Эту соль получил так-
же Бекер [199, 200] выпариванием водного раствора.
Титан образует двойные фториды с гидразином и гидроксиламином. Добавляя
к раствору TiF4 в плавиковой кислоте гидроксиламин (20%-ный раствор), Эблер и
Шот [265] получили гексатитанат гидроксиламина (NH2OH)2 TiFe. Эти же авторы
[266] синтезировали фтортитанат гидразина в виде дигидрата (NaH4)2 TiF6 •
• 2Н2О.
Фтортитанаты лития
При действии карбоната лития на плавиковокислый раствор
TiF4получается дигидрат фтортитаната лития Li2TiFe • 2Н2О в виде
длинных игольчатых кристаллов, хорошо растворимых вводе [255].
При нагревании до температуры 105° С дигидрат теряет воду
и превращается в безводный фтортитанат [301]. Безводный фтор-
титанат лития Li2TiF6 плавится около 480° С [303], в воде раст-
воряется хорошо. Растворимость его при температуре 20—22( С
около 48 г на 100 мл воды.
Как показали криоскопические исследования [364], при раст-
ворении в эвтектической смеси LiCl + КС1 фтортитанат лития дис-
социирует
Li2TiFe = 2Li+ 4- TiF< + 2F~.
Фтортитанаты натрия
Гексафтортитанат натрия Na2TiF6 впервые получил Берцелиус
[175]. Хунтер [327] эту соль приготовил нейтрализацией раст-
242
£ора двуокиси титана в плавиковой кислоте едким натром. Осадок
сыпавших кристаллов отфильтровывался и промывался разбав-
ленной плавиковой кислотой. Гинсберг и Гольдер [301, 303] вы-
делили фтортитанат натрия высаливанием из водных растворов
спиртом.
Гексафтортитанат натрия, как установлено по кривой охлаж-
дения, плавится при температуре около 700° С [303]. При длитель-
ном нагревании на воздухе он окисляется кислородом воздуха и
разлагается на TiO2, NaF и F2 [301, 303]. В воде Na2TiFe растворя-
ется плохо. Растворимость его в 100 мл воды при температуре
20—25° С равна 6,5 г [303].
Мариньяк [438] в результате кристаллизации из водного раст-
вора Na2TiF6, содержащего избыток NaF, получил ромбические
кристаллы кислой соли, имеющие состав 3NaF • TiF4 • HF. Струк-
тура состоит из октаэдров [TiF6]2~, а также ионово HF^ и Na+,
параметры решетки а = 7,182, b = 13,86, с = 6,538 А, ризм = 2,80,
ррентг = 2,76 г!см\ фед. гр. СтСт, z = 4.
Оксифтортитанат натрия Na2TiOF4 образуется в результате не-
посредственного взаимодействия двуокиси титана с кремнефтори-
стым натрием при температуре около 600° С по реакции обменного
разложения [19]
3Na2TiF6 + 2ТЮ2 = 2Na2TiOF4 + SiO2 + SiF4.
Фтортитанаты калия
Фтортитанаты калия относятся к числу наиболее хорошо изу-
ченных двойных фторидов титана. Впервые фтортитанат калия
K2TiF6 • Н2О выделил Берцелиус [175] нейтрализацией раствора
двуокиси титана в плавиковой кислоте едким кали. Вёлер [585]
эту соль синтезировал нейтрализацией раствора рутила в плави-
ковой кислоте карбонатом калия. Фтортитанат калия получен
сплавлением рутила с бифторидом калия KHF2 [580], а также из
чистой TiO2, плавиковой кислоты и KF [302, 325].
Кристаллы K2TiF6 • Н2О относятся к моноклинной системе; от-
ношение а : b : с = 0,9924 : 1 : 1,0520, Р = 98°42' [437]. Они изо-
морфны с кристаллами оксифторидов K2NbOFe-H2O и K2WO2F4 •
• Н2О [432]. Удельный вес соли 2,992 [551].
Добавляя бифторид калия к концентрированному раствору дву-
окиси титана в плавиковой кислоте авторы [325, 416, 580] получи-
ли безводный фтортитанат калия.
Структура фтортитаната калия окончательно еще не установлена. Хоард и
JJapTHH [328] нашли, что K2TiF6 имеет структуру типа фторгерманата калия K2GeF6.
Оо мнению [325], кристаллы K2TiF6 имеют гексагональную симметрию, а соглас-
110 [435, 436) — ромбоэдрическую. Как сообщается в [212], при комнатной темпе-
ратуре K2TiFe получается в виде тригональных кристаллов, которые при темпера-
туре 300—350° С превращаются в кубические.
1®* 243
Температура плавления K2TiF6, по данным [68], 822° С, по дан-
ным [65], 850° С; удельный вес 3,02 при 15° С; показатель прелом-
Рис. 57. Изотерма растворимости в системе
TiF4 — KF — Н2О при 20° С.
ления 1,475 [599]. На ос-
новании термического ана-
лиза, авторы [66, 68] при-
шли к выводу, что K2TiF6
существует в виде трех по-
лиморфных модификаций
в интервале температур
а — 820—680° С, у — 680^
620° Сир — ниже 620° С.
Безводный K2TiF6 при
нагревании на воздухе
примерно до 500° С пре-
вращается в оксифторид
K2TiOF4, который при бо-
лее высокой температуре
разлагается на TiO2, KF и
F2 [301].
Оксифторид K2TiOF4 об-
разуется при нагревании
с водяным паром до темпе-
ратуры 400° С [6], а также при спекании двуокиси титана с
кремнефтористым калием [18]. Энтальпия и энтропия гидроли-
за K2TiF6 при температуре 300° С равны 36,4 ккал/молъ и
36,0 кал/молъ • град соответственно.
Структура K2TiOF4 изучена методом инфракрасной спектроскопии [155].
Связь TiO в K2TiOF4 кратная. Строение может быть представлено в виде трех
схем:
С
Наиболее вероятно строение K2TiOF4 выражается схемами b и с.
Гинсберг и Гольдер [301,303] получили кислую соль K2TiF6 • НК
Бамбуров с соавторами [20] изучил систему TiF4—KF—Н2^
методом растворимости при 20° С (рис. 57, табл. 28]. По их данным.
244
Таблица 28
Растворимость в системе TiFd — KF — Н 20° С [20] 2О при температуре
Состав жидкой фазы, вес.% Состав твердой фазы, вес.% Твердая фаза
KF TiF4 KF TiF4
11,020 0,041 44,70 47,60 K2TiF6-H2O
6,750 0,052 44,10 46,90 То же
0,753 2,825 32,25 34,50 » »
0,652 4,880 34,40 37,30 » »
2,937 0,087 35,15 36,80 K2TiFe-2H2O
1,551 0,169 34,10 36,45 То же
0,211 0,948 39,30 41,66 2KoTiFe-3H20
0,830 6,360 30,00 34,30 ' K2TiFe
1,050 13,950 26,50 34,06 То же
из водных растворов в этой системе кристаллизуются четыре фтор-
титаната: K2TiF6, K2TiF6 • Н2О, 2K2TiF6 • ЗН2О и K2TiF6 • 2Н2О.
Одноводный фтортитанат калия кристаллизуется при одной изо-
терме (20° С) в двух формах — кубической при содержании в раст-
1 - K2TiFe • 2Н2О; 2 - 2K2TiFe • 3HeO; 3 - K2TiFe • Н2О.
Воре более 3 вес. % KF, и в виде тонких пластинчатых кристаллов
При концентрации около 0,7 вес. % KF.
Дигидрат K2TiF6 • 2Н2О образуется в виде чешуйчатых кристал-
лов моноклинной формы, полуторагидрат (2K2TiFe . ЗН2О) —
Правильных гексагональных призм.
245
Безводный фтортитанат калия кристаллизуется только из раст.
воров с высокой концентрацией солевой массы (с недостатком KF)
По-видимому, одна из модификаций моногидрата при температуре 20° С яв-
ляется метастабильной, так как поле этой фазы, как видно из рис. 59, разобщено
вклинившимися двумя фазами — K2TiF6 • 2Н2О и 2K2TiF6 • ЗН2О. Существова-
ние двух равновесных фаз одинакового состава при одной и той же температур
невозможно также и по термодинамическим соображениям.
Кристаллогидраты с двумя и тремя молекулами воды при на-
гревании до 100—200° С превращаются в моногидрат K2TiF6 • Н2О
(рис. 58). Последний при температуре 420—680° С гидролизуется
с образованием оксифтортитаната калия K2TiOF4.
Николаев и Буслаев [103] методом потенциометрического ти-
трования показали, что в растворе K2TiF6 + KF + HF + Н2О
титан находится в форме иона TiFe-. Нейтрализация раство-
ров K2TiF6 основаниями приводит вначале к образованию осад-
ков основных солей состава TiOF2 • пН2О и TiO (ОН) F • пН20
[69]. Основные фториды титана устойчивы до pH 5,2—7,0. В более
щелочных растворах они превращаются в гидроокись титана.
Система K2TiF6—NaF эвтектического типа изучена методом плавкости [458]
Эвтектическая точка лежит при температуре 660° С и концентрации 85 вес.%
K2TiF6.
Система K2TiF6—NaCl простого эвтектического типа изучена методом плав-
кости [66—68, 457, 458]. Эвтектическая точка лежит при температуре 535° С и
содержании K2TiF6 65 вес.%.
Аотани с соавторами [ 164] обнаружил в этой системе инконгруэнтно плавяще-
еся соединение 3K2TiF6 • NaCl, а Вурм с соавторами [577]—3K2TiFe • NaF.
Система K2TiF6—KF эвтектического типа [458]. Эвтектическая точка лежит
при температуре 710° С и содержании 65 вес.% K2TiF6.
Система K2TiF6—КС1 относится к простому эвтектическому типу [488]. Эв-
тектическая точка лежит при температуре 640° С и содержании 57 вес.% K2TiF6.
Однако в этой системе методом термического анализа найдено соединение K2TiF6 •
• КС1 с температурой плавления 696° С [164].
Система K2TiF6—TiO2 исследована методом термического анализа до содер-
жания 12,5 вес. % TiO2 [65]. В системе обнаружена эвтектическая точка при тем-
пературе 681° С и содержании 7 вес.% TiO2.
Система K2TiF6—NaCl—TiO2 изучена методом термографии, кристаллооп-
тики, рентгеноструктурного анализа [115] и измерения возкости [116]. В ней
установлена область расслаивания двух жидкостей [118]. Растворимость TiO2 в
системе изменяется от 4,1 вес.% (80% K2TiF6 20% NaCl) до 2,2 вес.% (50%
K2TiF6 50% NaCl) [117]. Вязкость при температуре 700—750° С и содержании
50—80 вес.% K2TiF6 равна 1,5—2,5 спз, что значительно ниже вязкости криолот-
глиноземных расплавов в ваннах для электролитического получения алюминия.
Система K2TiF6—NaCl—ВаС12. Аотани и Концума[165] построили диаграмму
состояния этой системы. Они обнаружили существование в ней соединения
2K2TiF6 • ВаС12.
Фтортитанаты меди
Мариньяком [433] получен фтортитанат меди CuTiF6 • 4Н2О;
изучена его рентгеноструктура [583]. Он кристаллизуется в моно-
246
ДЛИННОЙ системе, параметры решетки а — 7,332, b = 9,828, с =
L 5,326 А, ₽ = 104°20, Ррентг = 2,22, ризм = 2,23 г/см3, z = 2,
фед. гр. Р21/а. Октаэдры TiF6 двумя вершинами связаны с иска-
женными октаэдрами Cu (Н2О)4 Fe.
К Фтортитанаты рубидия
I Пеннингтон [465], добавляя RbF к раствору ТЮ2 в плавиковой
кислоте, получила фтортитанат рубидия Rb2TiF6. Эта соль выде-
лена добавлением к плавиковокислому раствору титана уксусно-
кислого рубидия [255] и нейтрализацией раствора карбонатом ру-
бидия [301].
Кристаллы Rb2TiF6 на воздухе неустойчивы, легко гидролизуются под дей-
ствием влаги. Температура плавления их около 750° С [301]. При длительном на-
гревании около 500° С на воздухе они превращаются в оксифторид рубидия
Rb2TiOF4. При более высокой температуре Rb2TiF6 разлагается на TiO2, RbF и F2.
В воде Rb2TiF6 растворяется плохо, при температуре 20—22° С растворимость
0,8 а на 100 мл воды.
Фтортитанаты цезия
I Пеннингтон [466], добавляя CsFк концентрированному раствору
двуокиси титана в плавиковой кислоте, получила фтортитанат це-
зия Cs4TiF8. Однако столь высокое координационное число (равное
В) для фторидных комплексов титана необычно, из чего следует
заключить, что состав соли, по-видимому, определен неточно и
нуждается в проверке. Энгелькирхен [255] этот фтортитанат це-
зия не обнаружил, а получил только гексафтортитанат Cs2TiF6.
Кристаллы Cs2TiF6 гексагональной системы, на воздухе не очень устойчивы,
под действием влаги легко гидролизуются. Температура плавления их около 690° С.
При нагревании до 500° С в течение 8,5 ч они превращаются примерно на 80% в
оксифторид Cs2TiOF4. При более сильном нагревании, каки фтортитанаты дру-
гих щелочных металлов, разлагаются на TiO2, CaF и F2. Растворимость в воде не-
большая — 2,5 г на 100 мл воды при 20—22° С [301, 303].
В Фтортитанаты металлов II группы
В Фтортитанаты металлов II группы периодической системы эле-
ментов описаны только в старых работах. Индивидуальность со-
единений этого типа нуждается в дополнительном изучении с по-
мощью современных методов исследований.
Берцелиус [174, 175] получил гексагидрат гексафтортитаната магния
MgTiF6 • 6Н2О. Хассель и Сальвезен [329] установили, что кристаллы этой соли
имеют гексагональную решетку с параметрами а — 9,77, с = 9,85 А.
Берцелиус [175], а затем Мариньяк [439], действуя на раствор двуокиси ти-
рана в плавиковой кислоте карбонатом кальция, выделили дигидрат фтортитаната
кальция CaTiF6 • 2Н2О.
247
Мариньяк [439] этим же методом получил дигидрат гексафтортитаната строн,
ция SrTiF6 • 2Н2О. У кристаллов данной соли моноклинная симметрия с отноше-
нием осевых параметров а : b : с — 1,2066 : 1 : 1,2295, р = 11 Гб'.
При действии ВаС12 на раствор двуокиси титана в плавиковой кислоте образу,
ется гексафтортитанат бария BaTiF6. Кристаллы BaTiF6 плохо растворяются в
воде, но легко — в соляной и азотной кислотах.
Оксифтортитанат BaTiOF4 получен нагреванием пероксифтортитаната ба-
рия при температуре 150—160° С [489].
При взаимодействии карбоната цинка с фторидными растворами двуокиси
титана образуется гексагидрат гексатитаната цинка [329], который имеет гекса-
гональные кристаллы с параметрами решетки а = 9,55 и с — 9,88 А. Мариньяк
[434], видимо, получил эту соль раньше и установил изоморфность ее кристаллов
с ZnZrF6 • 6Н2О.
Энгелькирхен [255], действуя карбонатом кадмия на раствор двуокиси тита-
на в плавиковой кислоте, выделил гексагидрат фтортитаната кадмия CdTiF6-6H2O.
Пероксофтортитанаты
Гексафтортитанаты при действии на них в водных растворах
перекиси водорода дают перекисные соединения.
Используя этот метод Фишер и Гольдох [277] получили перок-
сифтортитанат аммония (NH4)3 TiO2F4. Эта соль образуется при
окислении кислородом воздуха фтортитаната аммония низшей ва-
лентности в водном растворе [484, 486, 490, 491]. Пероксифтор-
титанат аммония выделили также Шварц и Гизе [538] и Пейро-
нель [468]. Он образуется в виде октаэдрических кристаллов, изо-
морфных с кристаллами (NH4) NbOF6 и (NH4)3 ZrF4. Параметры
кристаллической решетки таковы: « = 9,20 А, г = 4, фед. гр.
Fm3m, Ррентг = 1,95 г!см? [568]. Пероксифтортитанат аммония
очень устойчив и хорошо растворяется в воде.
Пейронель [468] синтезировал пероксифтортитанат натрия
Na3TiO2F4.
Из водного раствора гексафтортитаната калия после добавления
перекиси водорода получается пероксифтортитанат калия [482,
490]. Он кристаллизуется в виде мелких желтых кристаллов.
При действии на раствор пероксифтортитаната аммония
(NH4)3 TiO2F4 раствором ВаС12 кристаллизуются пероксифтортита-
наты бария — BaTiO2F6 и 3BaF2 • 2TiO2F2 [483, 488].
ХЛОРИДЫ ТИТАНА
Известны четыре хлорида титана, имеющие состав: TiCl, TiCU
TiCl3 и TiCl4.
Монохлорид титана при низких температурах неустойчив. Он,
как и соединения многих металлов низшей валентности, например
алюминия, кальция и других, устойчив только при высоких темпе-
ратурах. Присутствие монохлорида титана установлено по спектрам
248
поглощения в парах четыреххлористого титана, нагретых выше
1500° С, где он образовался в результате термической диссоциа-
ции TiCl4 [423].
Четыреххлористый титан устойчив в широком интервале тем-
ператур. Низшие хлориды титана TiCl2 и TiCl3, в отличие от четы-
реххлористого титана, могут существовать только в равновесии
с продуктами диспропорционирования [513] по реакциям
2TiCl3iB^TiCl2TB+TiCl4ra3,
2TiCl3TB TiTE + TiCl4ra3.
Диспропорционирование TiCl3 по вышеприведенным реакциям в термодинами-
ческом отношении изучали многие авторы [278, 288, 356, 531]. Хартман и Ринк
[356] охарактеризовали его давлением упругости пара TiCl4
log Р =— 7761/7’4-10,787 [мм рт. ст.]
(540—700° С).
По данным [531], в интервале температур 593—721° К давление пара TiCI4
при реакции диспропорционирования TiCl3 выражается уравнением
log Р = — 6897/Т + 8,407 [мм рт. ст.].
Изменение энтальпии и свободной энергии характеризуется уравнениями (кал/моль)
АН = 4,0 . 104 — 3,557 — 2,5 . 10~372 — 0,34 . 1057-1,
AF = 4,0 . 104 + 3,557 In Т + 2,5 . 10~372 — 0,17 1057~’ — 67,757.
Выше температуры 450° С равновесие сдвинуто в сторону распада TiCl3, а ниже
450° С — в сторону образования TiCl3 из TiCl2 и TiCl4.
Дихлорид титана начинает диспропорционировать на металлический титан
и четыреххлористый титан при температуре выше 475° С [537].
ДВУХЛОРИСТЫЙ ТИТАН
Способы получения
Двухлористый титан получают восстановлением четыреххло-
ристого титана или диспропорционированием тетрахлорида титана.
, Впервые двухлористый титан, загрязненный TiCl4, выделен вос-
становлением безводного жидкого тетрахлорида титана амальга-
мой натрия [481].
Чистый двухлористый титан получили [279, 513, 537] нагреванием TiCl3
в стеклянной трубке при температуре 475° С, из которой пары образующегося при
Диспропорционировании TiCl4 непрерывно откачивались. Вместо чистого TiCl3
Можно использовать смесь хлоридов TiCl3 + TiCl2, которая получается при на-
гревании TiCl4 с металлическим титаном при температуре 700—900° С [12, 390],
или нагреванием металлического титана с сухим газообразным хлористым во-
дородом 300—400° С [281 ].
Двухлористый титан образуется при пропускании газообразного TiCl4 в сме-
си с 3—5-кратпым количеством водорода через тихий безэлектродный разряд по
Реакции [321]
TiCl4 + H2 = TiCl2 + 2HC1.
249
Установлено, что восстановление водородом по приведенной реакции с обра-
зованием TiCl2 происходит при температуре выше 800° С [288]. Изменение свобод-
ной энергии при этой реакции выражается уравнением
AF = 68,56 • 103 — 43,787 + 0,397 In 7 — 0,0000572 [кал/моль].
Эрлих с соавторами [273] разработал метод получения TiCl2 нагреванием ме-
таллического титана в парах TiCl4 при температуре 1050° С. Оптимальными ус-
ловиями получения TiCl2 по этому методу являются температура 1030—1050° С
и давление TiCl4 0,3—0,5 атм [36].
Двухлористый титан образуется также из TiCl3 в солевых расплавах
2TiCl3 + Ti 3TiCl2,
2TiCl3 TiCl3+ TiCl4.
Каменецкий [61] изучил состояние равновесия в расплаве КС1 NaCl при
температуре 675—900° С и пришел к выводу, что образование TiCl2 в этих усло-
виях возможно только по первой реакции. Равновесие по второму уравнению
сдвинуто влево. Количество TiCl4, находящегося в равновесии с низшими хлори-
дами титана в расплаве NaCl — КС1, при температуре 700—800° С менее 1% об-
щего содержания титана в расплаве [374].
Равновесие между хлоридами титана в расплаве солей представ-
ляет большой интерес для получения металлического титана ме-
тодом электролиза и поэтому оно изучалось многими авторами.
Существенное влияние на состояние равновесия
2TiCl3 + Ti 3TiCl2
в расплаве солей оказывает комплексообразование между низши-
ми хлоридами титана и хлоридами щелочных металлов [9, 15,
93, 140, 141, 417]. Константы равновесия между хлоридами двух-
и трехвалентного титана в расплавах солей приводятся в рабо-
тах [9, 17, 144, 146, 177, 374, 417].
Показано, что величина константы равновесия в интервале тем-
ператур 750—975° С непостоянна [106, 111 ]. Она колеблется в пре-
делах от 0,034 • 10“2 до 0,258 • 10”2 и не проявляет явной зависи-
мости от температуры. Это объясняется неидеальностью расплава.
Авторы [139], исходя из напряжения разложения TiCl2 в гипоте-
тическом идеальном расплаве КС1 — NaCl, вычислили теплоты взаи-
модействия TiCl2 и TiCl3 с расплавом, оказавшиеся равными — 2,3
и 9,32 ккал!молъ соответственно (800—1000° С). Отсюда следует,
что TiCl2 с расплавом КО — NaCl практически не реагирует, а
TiCl3 проявляет заметное взаимодействие.
Равновесие между хлоридами титана в солевых расплавах при
температуре выше 600 — 700° С обычно сдвинуто в сторону обра-
зования TiCl2. В системе NaCl — КО, по данным [94], около 80—
90% титана находится в форме TiCl2. В расплаве MgCl2, согласно
[386], практически весь титан находится в форме TiCl2 и незначи-
тельное количество его содержится в форме ТЮ3 и TiCl4. Марков
и Белякова [94] нашли, однако, что в расплаве MgCl2 равновесие
устанавливается при равных концентрациях TiCl2 и TiCl3.
250
Несмотря на благоприятное состояние равновесия для образо-
вания TiCl2 в солевых расплавах, методы получения TiCl2, основан-
ные на диспропорционировании, не разработаны. Разработка их
осложняется трудностями, возникающими при отделении кристал-
лов TiCl2 от солей.
В водных растворах хлориды четырех- и трехвалентного тита-
на не восстанавливаются до двухвалентного известными методами
из-за малой устойчивости дихлорида. Потенциометрическим ме-
тодом установлено, что продукты растворения TiCl2 в воде не содер-
жат ионов Ti2+, а состоят из окрашенных комплексов Ti3+ [215].
Эти комплексы имеют максимумы на кривых светопоглощения при
длинах волн 415, 500 и 600 нм, характерные для комплексов Ti3+
со смешанными аддендами Н2О и СГ". Хлорид двухвалентного ти-
тана в водных растворах может существовать только при низких
температурах. Образование его наблюдалось при растворении в
соляной кислоте моноокиси титана TiO [471]. Наряду с ионами
Ti3+ получалось от 10 до 18% ионов Ti2+. Раствор, содержащий
TiCl2, неустойчив и при температуре 0° С может сохраняться всего
несколько минут.
Физические свойства
По данным [176], двухлористый титан TiCl2 имеет структуру
типа Cdl2 с параметрами решетки а = 3,561, с = 5,875 A, z = 1,
Ррентг ==z 3,06, Ризм = 3,13 г /см3, что согласуется с [27] (а = 3,56,
с = 5,88 А, ризм = 3,15 г/см3).
Бирюкова и Саксонов [12] обнаружили, что TiCl2 существует
в двух кристаллических модификациях. Одна из них (I) устой-
чива ниже 600° С, а другая (II) — при более высокой температуре.
Авторы нашли, что рентгенограмма TiCl2-I аналогична рентгено-
грамме TiCl3, приведенной в работе [279]; рентгенограмма TiCl2-II
сходна с рентгенограммой TiCl2 этих же авторов. Рентгенограмма
Банцигера и Рунгле [176] и соответственно [27] идентичны с рент-
генограммой a-TiCl3. Параметры решетки TiCl2-I и TiCl2-II Бирю-
кова и Саксонов не рассчитали.
Дихлорид титана — вещество парамагнитное. Удельная маг-
нитная восприимчивость его 4,30 • 10-6 (15° С) [390], эффектив-
ный магнитный момент 1,08|Лв (300° С) [401].
Температура плавления TiCl2 1035° С [275].
( Теплоемкость TiCl2, вычисленная методом усреднения, в интер-
Нале температур 298—1278° К [1383 выражается уравнением
Ср = 16,34 -f- 0,328 • 10 2Т [кал/молъ • град].
Энтальпия образования TiCl2 АТ/гэв, по данным различных
авторов, такова (в ккал/молъ)'. —118,3 [525], —123,3 [216], —123,7
1531],—120,1 [379], —123,0 1138], -121 [356], —120,6 [168];
251
энтропия TiCl2S298 (кал/моль . град)— 22,6 [138], 23,8 [531], 24,7
[168], 26,2 [356].
Определена зависимость изобарного потенциала Т1С12тв от тем-
пературы [95, 99]:
AZ = — 120,1 + 0,0369 Т [ккал/моль]
(600—1200° С).
Двухлористый титан начинает сублимироваться при темпе-
ратуре 700° С [288]. Давление сублимации его выражается урав-
нением
log Р = — 8,500/7" 4- 9,32 [мм pm. cm.].
Химические свойства
В двухлористом титане между атомами металла и хлора дей-
ствует, по-видимому, смешанная ковалентная и металлическая
связь. Ковалентная природа связи проявляется в способности
TiCl2 вступать в реакции присоединения (комплексообразования)
со многими органическими и неорганическими веществами. На при-
сутствие доли металлической связи указывает высокая температу-
ра плавления TiCl2, характерная для интерметаллических соеди-
нений, и малая растворимость в эфире, сероуглероде и хлорофор-
ме [286].
Кроме реакций присоединения для TiCl2 характерны окисли-
тельно-восстановительные реакции и менее характерны реакции
взаимного обмена.
В результате обработки кристаллов TiCl2 жидким аммиаком
при температуре —78° С образуется соединение TiCl2 • 4NH3 в
виде темно-серого порошка [537]. На воздухе это соединение сильно
пахнет аммиаком. При нагревании до температуры 300° С под ва-
куумом выделяется аммиак и хлористый аммоний, в остатке по-
лучается нитрид титана.
Согласно [462], в жидком аммиаке тетрамин подвергается разложению по
реакции
3TiCl2 . 4NH3 2TiCl3 • 6NH3 + Ti + 2NH3,
в результате чего можно получить металлический титан. Электролитическим вос-
становлением хлоридов титана в аммиаке действительно удается выделить метал-
лический титан, но в пирофорном виде.
Действием алюминиевого порошка на раствор TiCl4 и А12С1б Б
бензоле образуется TiCl2 • А12С16 • С6Н6 [424].
Двойные соли. Двухлористый титан образует двойные хло-
риды с хлоридами щелочных металлов, за исключением хлорида
лития, с которым дает только взаимные твердые растворы (рис. 59).
В системе NaCl — TiCl2 обнаружены два соединения: 2NaCl • TiClg
и NaCl • TiCl2, первое из которых разлагается ниже точки плав-
252
Рис. 59. Диаграмма состояния
системы LiCl — TiCh [274].
Рис. 60. Диаграмма состояния
системы NaCl— TiCh [386].
Рис. 61. Диаграмма состояния
системы КО — ТЮ2 [275].
Рис. 62. Диаграмма состояния
системы RbO —TiCk [274].
ления при температуре 548° С, а второе плавится инконгруэнтно
при 628° С [386] (рис. 60).
Установлено, что NaTiCl3 получается при температуре 650° С,
a Na2TiCl4 разлагается при 585° С [70].
В системе КС1 — TiCl2 обнаружено инконгруэнтно плавящееся
^единение 2КС1 • TiCl2 и конгруэнтно плавящееся соединение
КС1 • TiCl2 (рис. 61). Чижиков и соавторы [10, 71] при повторном
253
изучении этой системы установили, что если смеси сплавлять в
железных тиглях, то построенная по данным этих опытов диаграмм
ма состояния согласуется с [527]. Если же плавление проводить
в титановом тигле, в системе КС1 —
Рис. 63. Диаграмма состояния
системы CsCl — TiCh [274].
TiCl2 обнаруживается только одно
соединение ЗКС1 • TiCl2. Однако
авторы не объяснили причину раз-
личного поведения хлоридных
сплавов.
В системе КС1 — TiCl2 обнару-
жены соединения ЭКС1 • TiCl2 и
КС1 • TiCl2, плавящиеся соответ-
ственно при температурах 766 и
804° С [70].
В системах RbCl — TiCl2 и
CsCl—TiCl2 найдено по два соеди-
нения состава 2МС1 • TiCl2
и MCI • TiCl2, где М = Rb, Cs
(рис. 62,63).
Система MgCl2 — TiCl2 исследо-
вана Гальпериным с соавторами
[28], а также Коршуновым и Ио-
новым [57, 58] методами плавкости, электропроводности и измере-
ния плотности сплавов, но в ней установлена только взаимная
растворимость и не обнаружены двойные соли хлорида натрия
с TiCl2.
Взаимодействие двухлористого титана с водой. По
наблюдениям Фриделя и Гюрина [286], двухлористый титан реа-
гирует с водой очень энергично с выделением водорода и об-
разованием окрашенного в желтый цвет раствора. Получен также
коричневый раствор TiCl2 в воде [481]; согласно [547], свежепо-
лученный раствор TiCl2 бесцветный, при стоянии на воздухе
окрашивается в коричневый цвет.
Из приведенных сообщений следует, что взаимодействие дву-
хлористого титана с водой является сложным окислительно-вос-
становительным процессом, нуждающимся в специальном иссле-
довании.
В соляной кислоте TiCl2 растворяется плохо [481].
ТРЕХХЛОРИСТЫЙ ТИТАН
Способы получения
Треххлористый титан получается восстановлением четыреххло-
ристого титана водородом, двухлористым титаном, металлическим
титаном, многими металлами и металлоорганическими соединениями-
254
Состояние равновесия реакции
TiCl3TB+ НС1газ TiCl4ra3 + 4-H2ra3
интервале температур 350—475° С характеризуется следующим
уравнением [384]:
log = 1,82 — 2,27 • 103Т-1
и сдвинуто в сторону образования TiCl3.
Четыреххлористый титан восстанавливается водородом до TiCl3
при пропускании смеси паров через трубку, нагретую до темпера-
туры 600—900° С [153, 221, 537], а также над вольфрамовой про-
волокой при 1000° С [529], через вольтову дугу [362] и в распла-
ве солей [13].
Клемм и Крозе [391] получили TiCl3, пропуская пары Т1С14
над нагретым до 800° С двухлористым титаном.
Четыреххлористый титан восстанавливается до TiCl3 металли-
ческим титаном в газообразной фазе и в расплаве солей [12, 14, 33»
34, 36, 43,126, 206, 440]. Реакция идет при температуре выше 300° С.
Взаимодействие металлического титана с TiCl4 начинается около
400° С с образованием TiCl3. Однако одновременно с треххлорис-
тым получается и двухлористый титан. Продукты реакции при
температуре 500° С состоят из смеси TiCl3 и TiCl2- При 700° С
невозогнанными продуктами является TiCl2, а возгон состоит из
TiCl3 [446].
Оптимальные условия для получения Т1С13 восстановлением
Т1С14 металлическим титаном: температура 900° С, давление паров
TiCl4 0,3—0,5 атм [36].
Четыреххлористый титан также восстанавливается до Т1С13 ме-
таллическим натрием в газообразном виде [3311 и в расплаве хло-
ристого натрия [2]; металлической ртутью [513, 552]; металличес-
ким серебром при 180—200° С [286]; при нагревании паров TiCl4
с мышьяком и сурьмой при 300—400° С [207, 513, 547]; металличес-
ким алюминием в стеклянной эвакуированной трубке при 200° С
и в присутствии А1С13 как катализатора [513, 547]; металлическим
магнием [530].
Треххлористый титан получают при пропускании паров TiCl4
над смесью двуокиси титана и углерода при температуре 800—
1100° С [160, 495].
Четыреххлористый титан восстанавливается электролитически
До TiCl3 в расплаве солей [169].
Многие металлоорганические соединения, например Hg (С2Н5)2
14061, А1 (С2Н5)2С1 [166, 167], А1 (С2Н5)3 [217], при смешении с
TiCl4 восстанавливают его до TiCl3. В растворе ацетонитрила,
Диметилформамида, нитробензола и безводной уксусной кислоты
TiCl4 бурно восстанавливается металлическими магнием и алю-
минием до Ti3+ [276].
255
В водных растворах Ti4+ восстанавливается до Ti3+ электро-
литически на ртутном или свинцовом катодах и при действии ме-
таллов и амальгам цинка, кадмия, висмута, натрия и др. [324,
429, 430, 471, 499, 590, 594].
Треххлористый титан образуется при растворении металличе-
ского титана в соляной кислоте [276].
Физические свойства
Треххлористый титан имеет четыре полиморфных модификации,
области кристаллизации которых окончательно еще не уста-
новлены.
Согласно [537], при восстановлении TiCl4 водородом при тем-
пературе 650° С получаются кристаллы TiCl3 двух модификаций —
таблитчатые и игольчатые. Клемм и Крозе [392] провели рентге-
ноструктурное исследование кристаллов TiCl3 и показали, что они
имеют гексагональную решетку с параметрами а = 6,12, с =
= 17,50 A, z ~ 6 или ромбоэдрическую с параметрами а ==
= 6,82 А, а - 53° 20', z - 2.
Натта и соавторы [454, 455] обнаружили три модификации
Т1С13 — а, р и у.
а-ПС13 образуется при восстановлении TiCl4 водородом при
температуре 800—900° С. Эта модификация, по данным рентгено-
структурных исследований [12, 454, 455], оказалась аналогична
гексагональной модификации Клемма и Крозе [392].
P-TiCl3 получен при обработке TiCl4 органическими соеди-
нениями алюминия (алюминий алкиламин). Кристаллы P-TiCl3
имеют волокнистое строение, параметры решетки а = 6,27, с =
= 5,82 А, фед. гр. Dln [455].
При температуре 230—250° С P-модификация переходит в у.
В другой работе Натта и соавторы [456] открыли четвертую
модификацию 6-TiCl3, которая получается из а- и y-TiCl3 путем
продолжительного размола. 6-Модификация имеет слоистое строе-
ние и содержит структурные элементы а- и у-модификаций.
В результате взаимодействия Al (С2Н5)3 с TiCl4 в растворе
изооктана при температуре 80° С образуется коричневый осадок
P-TiCl3, а при 170° С — пурпурный осадок y-TiCl3 [166].
Крее [217] получил TiCl3 с P-структурой по реакции
(С2Н5)2 Al + TiCl4 -> TiCl3 + (С2Н6) А1С12 + 4 С2Н4 4- 4С2Н6.
Люис и соавторы [401] считают, что при комнатной температуре TiCl3 су-
ществует в виде фиолетовой и коричневой модификаций, причем последняя менее
парамагнитна. Изучив магнитные свойства TiCl3 в интервале температур 80
300° С, они не обнаружили фазового перехода фиолетовой модификации в ко-
ричневую. Выводы этих авторов не согласуются с данными Огава [459], наблю-
давшего фазовый переход при температуре 217° К.
256
г Дюкграаф [248] снял спектры диффузного отражения а- и
y-TiCl3, а также спектр поглощения |3-TiCl3. Полосы поглощения
ее- и y-TiCl3 с максимумами при 1900 и 14300 см~х сходны с поло-
сами поглощения при 18500 и 15 400 см~~х ионов Ti3+ во фториде
лития, которым приписывается модель локализованного электро-
да. Положение электронных уровней (3-TiCl3 отличается от тако-
вых а- и y-TiCl3. Автор считает, что в кристаллах а-, |3- и у-моди-
фикаций у ионов Ti3+ октаэдрическое окружение. Полоса погло-
щения 19 000 слС1 обязана переходу 27\ ->2Е .
О
Гарднер [314] обнаружил, что растворы а- и p-форм TiCl3B 1,3-н. серной кис-
лоте, этаноле и ацетонитриле имеют различные спектры поглощения, аналогичные
твердым образцам. По его наблюдениям, структура P-TiCl3 может сохраняться и в
растворенном состоянии. Если раствор p~TiCl3 окислить, а затем в течение корот-
кого времени подвергнуть катодному восстановлению, то снова образуется корич-
невая модификация. После длительного окисления p~TiCl3 катодное восстановле-
ние приводит к образованию a-формы фиолетового цвета.
Треххлористый титан парамагнитен [580]. Удельная магнитная
восприимчивость его имеет сложную зависимость от темпера-
туры [391 ].
/°, к х-юв t°, К Х.10«
9 0,97 273 7,20 .
156 1,60 292 6,73
195 2,60 373 6,23
212 4,00 423 5,80
232 7,08 560 4,84
253 7,35 685 4,10
При температуре фазового перехода TiCl3 (217° К) Огава [459]
наблюдал резкое понижение удельной магнитной восприимчивости
в параллельном и перпендикулярном направлении оси с. По его
мнению, это отличает TiCl3 от обычных антиферромагнетиков и
вызвано, вероятно, резким изменением параметров решетки а и с
при температуре перехода. Изменения параметров решетки он при-
писывает образованию связи ионов Ti3+ в плоскости, перпендику-
лярной оси с, при температуре ниже точки перехода.
Эффективный магнитный момент TiCl3 при температуре 300° К
Равен 1,31 [401].
Измерена упругость пара TiCl3 в интервале температур 628—
•° К и установлена зависимость ее от температуры
Е log? =-------= 10,40 мм рт. ст.
Вычислена теплоемкость TiCl3 методом усреднения и установ-
лена зависимость теплоемкости от температуры
I Ср= 19,52 4- 1,02 • 10-2Г [кал/моль • град]
(Г = 298— 1273° К).
V 9’6’4 257
Энтальпия образования TiCl3 ДЯгэв» по данным разных авто-
ров, следующая (ккал/моль): —172,2 [531], —170,0 [525, 218]
—169,1 [3791, —170,7 [168].
Шефер с соавторами [536] определил для фиолетовой модифц,
нации TiCl3 ДЯ29я =— 161,35 ккал/моль. Теплота превращения
TiCl3TB в Т1С14ж 20,3 ккал/моль [536].
Сандерсон и Маквуд [531] для TiCl3 нашли 5^8 = 32,3,
Альтман с соавторами [531] — 34,4 кал/моль • град.
Вычислены термодинамические константы TiCl3 [143] и изме-
нение изобарного потенциала TiCl3 с температурой, выражающе-
еся уравнением [95, 99]
AZ = — 170,4+ 0,0541 Т [ккал/моль].
Химические свойства
С увеличением валентности титана доля ковалентной связи в
его соединениях увеличивается и уменьшается доля металличе-
ской связи. В соответствии с этой закономерностью треххлористый
титан больше склонен к реакциям присоединения (комплексообра-
зования), чем двухлористый титан. Однако в треххлористом ти-
тане металлическая связь проявляется еще в значительной мере,
что оказывается определенным образом на физических свойствах
соединения (высокая температура плавления, металлический блеск
кристаллов и др.).
Треххлористый титан способен присоединять многие органиче-
ские и неорганические валентно-насыщенные соединения.
При пропускании над треххлористым титаном паров безводного аммиака он
присоединяет его, образуя белый порошок состава TiCl3 • 6NH3 [537]. На воздухе
этот продукт теряет аммиак и темнеет. При нагревании четыре молекулы аммиака
отщепляются при температуре около 300° С, а получившийся продукт при даль-
нейшем нагревании претерпевает более глубокое превращение, в результате кото-
рого образуется нитрид титана.
Получен комплекс трех хлор исто го титана с мочевиной TiCl3 • 6СО (NH2)s
[550].
При взаимодействии TiCl3 с пиридином выделен аддукт TiCl3 • C5H5N в виде
светло серого порошка, который легко гидролизуется и отщепляет пиридин при
нагревании до 150° С [326].
Продукт присоединения сернистого азота 2TiCl3 • N4S4 оранжевого цвета,
разлагается на воздухе [235, 263].
С водой треххлористый титан образует кристаллогидрат TiCl3 • 6Н2О [473J-
Треххлористый титан присоединяет многие органические производные —
эфиры, амины, кетоны и др.
Получен продукт присоединения изопропилового эфира в виде светло-жел-
того вещества состава TiCl3 • О (цзо-С3Н7)2 [330].
Шлефер и Гетц [549] синтезировали TiCl3 • 4Alk, где Aik — изопропиловая,
вторичный бутиловый спирты, циклогексанон. С тетрагидрофураном (ТГФ) TiCh
дает комплекс состава TiCl3 • ЗТГФ.
Получены продукты присоединения третичного метиламина TiCl3 .2М(СНзЬ
в виде вещества голубого цвета. Оно образуется при восстановлении TiCl4 за счет
избытка амина. С этилендиамином треххлористый титан дает TiCl3 • 4C2H8N-2'
бледно-голубое вещество с дипольным моментом 1,66 [1в [263].
258
фоульс с соавторами [300], нагревая TiCl4 с избытком 2,2'-дипиридила (Dipy)
з запаянной ампуле при температуре 150° С в течение 20 ч, получил комплекс
TiCls- Dipy. Этот комплекс растворяется в метил- и этилнитриле, образуя трой-
ные соединения — TiCls • Dipy • CH3CN и TiCh • Dipy • QjHsCN.
Дукворт с соавторами [234] синтезировал мономерные комплексы с октаэд-
рическим строением состава TiCls • 3RCN, где R = СН3, QHs, Я-С3Н7.
Соединения эти окрашены. Максимумы спектров поглощения лежат в интер-
вале длин волн 580—670 нм. Метиловое производное в УФ-области имеет полосу
поглощения при X = 230 и 285 нм, обусловленную переносом зарядов.
Получены соединения TiCls с диоксаном и его аналогами (TiCls • ЗС4Н8О2,
TiCls • 2С4Н8О2, TiCI3 • С4Н8О2, TiCl3 • C4H8OS, TiCh • C4H9ON), ацетоном
(TiCh • ЗСН3СОСН3) и уксусным альдегидом (TiCl3 • 1,5 (СН3СН2О)2 [297]. Соеди-
нения с диоксаном сготношениём"Т!С1з': диоксан == 1 : 3 й Т: 2 получил Кларк
с соавторами [321 ]. Этими авторами из раствора TiCls в тетрагидрофуране выделе-
ны TiCls • 3CH2CN и TiCl3 • ЗОС (СН3)2.
Чатт и Хейтер [214] синтезировали темно-коричневое вещество состава
TiCls • С2Н4 [Р (С2Н5)2]2 с температурой плавления 312—319° С.
Много продуктов присоединения получили Шлефер и Гетц [550] и изучили их
магнитные свойства.
Двойные соли. С хлоридами щелочных металлов TiCl3 обра-
зует двойные соли, содержащие комплексные ионы [TiCU j~~______и
[TiCl6]3~. Существование комплексных ионов титана в солевых
’расплавах Марков и Чернов [98, 100] установили с помощью
расчетов по уравнению Шредера. По их данным, в расплаве
NaCl — TiCl3 существует ион TiCll~. В сплавах КС1 — ТiCl3, богатых
КС1, титан также находится в форме иона TiClg-*. Сплавы в систе-
ме RbCl — TiCl3 содержат титан в форме комплексов TiClT* и TiCl|~>
в системе CsCl — TiCl3 преобладают комплексы состава T1C1J" .
£ Груен и МакБет [310] в результате исследования ИК-спектров
поглощения установили, что титан в солевых расплавах находится
в форме ионов ТЮб” и ТЮГ-
I Изучена электропроводность треххлористого титана в расплаве
хлоридов щелочных металлов и показано существование в них
соединений, отвечающих диаграммам состояния [37, 159]. Двой-
ные хлориды титана в расплаве подвержены частичной диссо-
циации, которая уменьшается при переходе от хлоридов натрия
к хлоридам цезия. В системе LiCl — TiCl3 двойные соединения не
обнаружены.
к Система NaCl — TiCl3 эвтектического типа [62]. Эвтектическая
точка лежит прйГтемпературе 540° С и 22,4 мол.% TiCl3. По мне-
нию [62]. в ней существует соединение Na3TiCl6. Методами тер-
мического и рентгеноструктурного анализов установлено, что в
^нной системе образуется инконгруэнтно плавящееся соединение
”и3Т1С16, однако на построенной диаграмме состояния (рис. 64)
ут эвтектической точки. Вместо эвтектики обнаружена переход-
ная точка при температуре 553° С [267].
Существование соединения Na3TiCl6 подтверждено при исследовании тройной
[о£Темь1 TiCl3 — TiCl2 — NaCl [57] и при изучении электропроводности сплавов
0,Ч> что противоречит данным [60, 98].
17*
259-
Система КС1 — TiCl3. Каменецкий [62] в этой системе обна-
ружил конгруэнтно плавящееся соединение K3TiCle, что подтверж-
Рис. 64. Диаграмма состояния
системы NaCl — TiCl3 [267].
Рис. 65. Диаграмма состояния'
системы КС1 — TiCl3 [98].
Рис. 66. Диаграмма состояния
системы RbCl — TiCl3 [98].
Рис. 67. Диаграмма состояния
системы CsCl — TiCl3 [98].
дено в работах 159, 63, 64, 267]. Марков и Чернов [98], исследовав-
шие систему КС1 — TiCl3 методом термического анализа, устано-
вили существование в ней двух соединений — K3TiCl6 и KTiCla
(рис. 65).
Система RbCl — TiCI3 изучена методом термического анализа
[98]. В ней обнаружено два конгруэнтно плавящихся соедине-
ния — Rb3TiCl6 и RbTiCl4 (рис. 66).
260
Система CsCl — ИС1з- Марков и Чернов [98] в этой системе
установили два конгруэнтно плавящихся соединения — Cs3TiCle
и CsTiCl4 (рис. 67). Показано, что соединение CsTJCl4 образуется
при смешении растворов CsCl и TiCl3 в абсолютном спирте [161].
Реакция разложения
На влажном воздухе TiCl3 разлагается с образованием TiO2
и НС1 [513]. При нагревании на воздухе окисляется с образовани-
ем TiCl4 и TiO2 [256,257].
Треххлористый титан TiCl3 присоединяет серу и селен, об-
разуя сульфиды и селениды.
г___В воде и кислотах TiCl3 растворяется, окрашивая их в фиоле-
товый цвет [481 ]; в Т1С14, СНС13, СС14, CS2 и С6Н6 не растворяет-
ся [286].
Металлический натрий восстанавливает TiCl3 в расплаве NaCl
сначала до TiCl2, а затем до металла [125]. В числе промежуточных
продуктов авторы наблюдали образование так называемой черной
соли, приближенный состав которой 9NaCl • 2TiCl3 • TiCl2.
Треххлористый титан — сильный восстановитель. Водные раст-
"воры его восстанавливают гараяс-изомеры азобензола до бензиди-
на и анилина, 4-аминобензол — до анилина и п-фенилдиа-
мина [407].
Взаимодействие с водой. Система TiCh — НС1 — Н2О
Глятцель [305] сообщил о получении гидрата TiCl3 • 4Н2О зе-
леного цвета, но в работах других авторов [473, 545] это соедине-
ние не получено.
При растворении безводного TiCl3 в соляной кислоте образуют-
ся фиолетовые кристаллы гексагидрата TiCl3 • 6Н2О[473], из смеси
соляной кислоты и абсолютного эфира — кристаллы гексагидрата
Таблица 29
Растворимость в системе TiCl3 — НС1 — Н.2О при температуре 0° С
Состав жидкой фазы, вес. % Состав остатка, вес. % Состав жидкой фазы, вес. % Состав остатка, вес. %
TiCl3 НС1 TiCl, HCI TiCl3 НС1 TiCl, НС1
32,90 2,98 47,40 1,23 7,47 25,70 38,30 10,90
31,60 4,11 — 3,32 32,20 — —
31,15 4,32 45,00 1,87 2,96 34,40 40,70 11,10
27,90 7,25 39,00 4,73 2,41 36,70 - —
26,65 8,79 * 2,59 37,45 — —-
24,20 10,40 45,80 4,01 2,81 40,60 34,94 17,35
21,45 12,75 - - 3,60 42,10 32,40 19,43
18,40 14,80 41,50 6,80 3,39 42,90 —
15,20 18,30 36,20 9,76 4,07 42,95 .1
14,25 19,70 — — 45,15 —
Примечание. Твердая фаза TiCl3 • 6Н2О.
261
зеленого цвета [546]. Последние неустойчивы и легко переходят
в фиолетовую модификацию.
Морозов и Топтыгина [92] изучили систему TiCl3—НС1— Н2О методом раство-
римости (табл. 29, рис. 68). В качестве твердой фазы во всей области изученной сис-
темы кристаллизуется гексагидрат TiCl3 • 6Н2О фиолетового цвета. Жидкая фаза
60± TlCl3-6Hz0
до концентрации 37% НС1 в растворе окраше-
на в фиолетовый цвет. Растворы с концентра-
цией НС1 37—44% имеют переходную окра-
ску, содержащие более 42% НС1 — окраше-
ны в зеленый цвет. Приводя эти наблюдения,
авторы, однако, не дают объяснения, чем вы'
50-
40-
Рис. 68. Изотерма растворимости
в системе TiCl3 — НС1 — Н2О
[92].
звано изменение окраски растворов трех-
хлористого титана в концентрированной со-
ляной кислоте.
Кристаллы TiCl3 • 6Н2О под ми-
кроскопом прозрачны, имеют форму
многогранников и бледно-фиолетовую
окраску. Оптически двухосны, Ng =
= 1,635, Np = 1,573 [90].
По данным термического анализа, кристал-
лы TiCl3 - 6Н2О плавятся при температуре
80° С в своей кристаллизационной воде [92].
На термограммах нагревания, снятых на воз-
духе, имеются эндотермические эффекты при
температуре 115 и 190° С, а в атмосфере арго-
на и НС1 — при 125° С. Конечный продукт
прокаливания TiO2.
В атмосфере аргона при температуре 125° С TiCl3 • 6Н2О полностью гидро-
лизуется, превращаясь в TiO2. При этом выделяется эквивалентное количество
НС1 и Н2.
При нагревании до температуры 95° С под небольшим вакуумом TiCl3 • 6Н2О
разлагается, превращаясь в гидроокись трехвалентного титана черно-синего цвета.
После полного испарения воды в токе аргона при температуре 95° С в остатке
получается смесь двуокиси и гидроокиси трехвалентного титана.
В кристаллах TiCl3 • 6Н2О ион титана имеет неспаренный элек-
трон, о чем можно судить по данным исследования ЭПР [30,
571, 578].
Предпринята попытка выяснить состояние трехвалентного титана в водных
растворах. Так, Морозов и Топтыгина [92] показали, что над раствором безвод-
ного TiCl3 в соляной кислоте упругость пара выше, чем над соляной кислотой, в ко-
торой растворен этот хлорид. При растворении TiCl3 • 6Н2О изменения упругос-
ти пара НС1 над раствором соляной кислоты не наблюдается. Отсюда авторы [92]
пришли к выводу, что в соляной кислоте титан существует в виде аквокомплекса
[Ti (Н2О) 6]3+.
Стромберг и Кортушинская [132] методом полярографии показали, что в со-
лянокислых растворах треххлористого титана существуют ионы Ti3-^ и TiOHCl3-^
Гартман с соавторами [357] считает по изменению отношения ан4-: cTi3-p,
что в водных растворах существуют ионы [TiCl^ (Н2О)Л (ОН)^]^ .
Гарифьянов и соавторы [32] отмечают, что в фиолетовых спектрах поглощения
растворов TiCl3 присутствует также и зеленая модификация. На основании этого
они высказали предположение, что зеленая модификация образуется в результа-
262
-j«e замещения молекул воды во внутренней сфере комплекса на ион хлора. Как
доказательство авторы приводят следующее соображение. В фиолетовых спирто-
вых растворах TiCl3 • 6Н2О интенсивность линий ЭПР мала. Сигнал ЭПР возни-
кает только за счет небольшого количества ионов [Ti (СН3ОН)4С12]~^, отвечающих
за поглощение света в зеленой области спектра.
Исследование спектров поглощения водных растворов TiCl3
позволило установить, что как в твердом состоянии, так и в раст-
воре титан находится в виде комплексных ионов с координацион-
ным числом 6 типа [Ti (Н2О)6]3+. Показано, что основной ^-элек-
тронный уровень Ti3+, имеющий в незаполненной 3£/-оболочке
один Зй-электрон, в электростатическом поле симметрии Oh, D4h
и C2v расщепляется соответственно на 2; 4 и 5 подуровней [355,
363]. При октаэдрической симметрии Oh в соответствии с этим мо-
жет существовать только одна полоса поглощения, вызванная
электронным переходом Т2ё — Её. В водных растворах TiCl3 дей-
ствительно наблюдается одна широкая полоса поглощения с мак-
симумом около 500 нм [52, 357], что и доказывает существование
комплексных ионов с шестью координированными группами. Од-
нако тонкая структура этой полосы оказалась сложной, что слу-
жит признаком частичной деформации октаэдрической структуры
комплексного иона. Так, например, Каратаева с соавторами [52]
при изучении спектров поглощения TiCl3 в водных растворах раз-
ложила основную полосу на две компоненты, имеющие форму кри-
вой вероятности Гаусса. Йоргенсен [370] на крыльях основной
полосы обнаружил перегибы. Хартман с соавторами [357], изу-
чивший спектры поглощения комплексов типа [TiA6]3-r, где А =
= Н2О, СН3ОН, С2Н5ОН и СО (NH2)2, отметил дублетную структуру
полосы поглощения, за которую ответственен ион Ti[CO(NH2)2]3+.
Часар с соавторами [222] в спектре поглощения [TiCl6]3+ обнару-
жил три полосы типа «в» по классификации Кишша [396] (пере-
ходы между компонентами одного терма в ионах с незаполненными
оболочками).
Сложная структура полосы поглощения водных растворов TiCl3 позволила
многим авторам [52, 222, 357, 370] высказать предположение, что в результате
проявления эффекта Яна-Теллера (симметрия комплексных ионов и молекул
понижается таким образом, чтобы основной терм был невырожденным), комплекс-
ные ионы титана в водных растворах претерпевают тетраэдрическую деформацию
и приобретают симметрию D4h . Тетраэдрическая деформация заключается в том,
что два адденда, находящиеся в транс-положении, удалены от центрального иона
на большее расстояние, чем остальные четыре.
Двойные соли. Из водных растворов получены двойные хло-
риды трехвалентного титана с хлоридами калия, рубидия, цезия
и аммония.
Штелер [545], добавляя к солянокислым растворам TiCl3 хло-
риды рубидия и цезия, выделил двойные соли 2RbCl • TiCl3 • Н2О
и 2CsCl • TiCl3 • Н2О. Морозов и Топтыгина [91] получили
263
двойные хлориды калия и аммония аналогичного состава. Эти
же авторы изучили системы MCI — TiCl3 — НС1 — Н2О, где М =
= NH4, К, Rb, Cs, методом растворимости (табл. 30—33) и построили
Таблица 30
Растворимость в системе NH4C1 — TiCl3 — НС1 — Н2О при
температуре 0°С
Состав жидкой фазы, вес. %
НС1 TiCl3 NH4C1 Твердая фа за
44,53 44,37 41,58 40,59 39,80 39,41 37,37 36,25 35,61 30,88 30,76 25,92 0,674 0,814 0,941 1,090 1,060 1,880 2,030 2,350 3,416 5,690 7,640 4,240 0,46 0,57 0,65 0,75 0,73 1,24 1,41 2,84 1,67 3,76 4,68 2,07 (NH4)2TiCl5 • То же » » » » » » » » (NH4)2TiCl5 • Н2О То же TiCl3 • 6Н2О + То же » » Н2О + NH4C1 nh4ci
Таблица 31
Растворимость » в системе КС1 — TiCl3 — HCI — при температуре 0° С Н2О
Состав жидкой фазы, вес. %
НС1 TiCl3 КС1 Твердая фаза
43,93 1,501 1,46 K2TiCl5 . Н2О
42,87 1,702 1,16 То же
42,69 1,753 1,12 » »
42,28 1,764 1,15 » »
42,06 1,809 1,23 » »
38,83 2,840 1,30 » »
35,91 3,560 1,20 » »
35,52 3,436 1,16 КС1
35,33 3,601 1,20 То же
30,76 2,260 1,17 » »
диаграмму растворимости пентахлораквотитанатов в соляной кис-
лоте (рис. 69). Пентахлораквотитанаты — кристаллические веще-
ства зеленого цвета со следующими показателями преломления 191k
Np
Cs,TiCl5 • H2O 1,678 1,645
Rb2TiCl5 • H2O 1,682 1,638
(NH4)2TiCl5 • H2O 1,694 1,664
264
Таблица 32
Растворимость в системе RbCl — TiCl3 — НС1 — Н2О
при температуре 0°С
Состав жидкой фазы, вес. % Твердая фаза
НС1 TiCl3 RbCl
44,54 0,265 0,42 Rb»TiCL • Н„О
42,64 0,270 0,42 То же
42,38 0,261 0,41 »
41,38 0,227 0,36 » »
38,74 0,470 0,75 » »
37,72 0,439 0,69 » »
37,68 0,429 0,67 » »
34,87 0,960 1,50 » »
33,92 0,865 1,36 » »
32,59 1,176 1,84 »
30,91 1,660 2,59 » »
36,42 1,686 2,60
30,26 1,629 2,19 Rb.TiCL • I- 120 -н RbCl
29,85 1,847 2,24 То же
27,90 3,598 3,69 »
24,60 4,498 6,96 » »
Таблица 33
Растворимость в системе CsCl — TiCl3 — НС1 — Н2О
при температуре 0°С
Состав жидкой фазы, вес. % Твердая фаза
НС1 TiCl3 CsCl
43,93 0,354 0,77 Cs2TiCl6 • Н2О
41,32 0,423 0,92 То же
41,17 0,473- 1,22 » »
41,00 0,473 1,22 » »
36,11 0,876 1,91 » »
35,51 0,868 1,90 » »
35,04 0,988 2,16 » »
34,18 1,053 2,50 » »
33,06 1,166 2,54 » »
31,14 1,749 3,79 » »
29,84 2,222 4,66 » »
28,96 2,040 3,58 Cs2TiCl5 • Н2О + TiCl3 • 6Н2О
27,07 2,760 6,15 То же
25,81 2,880 5,88 » »
25,81 3,040 6,52 » »
20,33 4,960 9,89 » »
19,18 4,740 9,47 TiCis • 6Н2О + CsCl
Рентгенограммы пентахлораквотитанатов сходны с рентгено-
граммой TiCl3 • 6Н2О, являющегося комплексом [Ti (Н2О)в1 С13.
Основываясь на сходстве рентгенограмм, Морозов и Топтыгина
265
{91 ] склонны пентахлораквотитанатам приписывать комплексное
строение. Состав комплексного аниона они выражают формулой
lTiCl5H2O]2~
па3,бес.%
Рис. 69. Изотерма растворимости
пентахлораквотитанатов в соляной
кислоте при 0° С:
1— Cs2TiCl5 • Н2О; 2 — Rb2TiCl6- Н2О;
3—(NH4)TiCl5 • Н2О; 4 — K2TiCl5 - Н2О.
Сплошные кривые — твердая фаза
M2TiCl5-H2O, пунктирные кривые — две
фазы [89].
Устойчивость пентахлораквотгь
танатов возрастает от калия к це-
зию. Они отщепляют воду при сле-
дующих температурах (° С): 270—.
Cs2TiCl5-H2O, 212 —Rb2TiCl6-H2O,
116 _ (NH4)2 TiCl5 • H2O и 112-
K2TiCl5 • Н2О.
Эффективные магнитные момен-
ты Cs2TiCl5 • Н2О и Rb2TiCl5 • Н2О
равны 1,64 и 1,69[1в 1550].
Оксихлориды титана-3
Оксихлориды титана-3 еще ма-
ло изучены. Фридель и Гюрин
[287] при получении двухлористо-
го титана диспропорционировани-
ем TiCl3 наблюдали образование
в качестве побочного продукта ок-
сихлорида TiOCl— вещества жел-
то-коричневого цвета. Он образует-
ся также в небольшом количестве при нагревании двуокиси титана
с TiCl4 и Н2 до красного каления. Оксихлорид получили и Шефер
с соавторами [535].
Коршунов и Ионов [57] наблюдали образование TiOCl в виде золотисто-жел-
тых кристаллов при нагревании сплавов TiCl3 с хлоридами щелочных металлов
в кварцевой посуде. По их мнению, TiOCl получается в результате взаимодействия
треххлористого титана с кварцем по реакции
2TiCl3 + SiO2 = 2TiOCl + SiCl4.
ЧЕТЫРЕХХЛОРИСТЫЙ ТИТАН
Способы получения
Впервые четыреххлористый титан получил Георг [306] хлор*1*
рованием карбонитрида титана газообразным хлором. В последу*0'
щем было разработано много препаративных методов получения
четыреххлористого титана, основанных на хлорировании разлиЧ'
ных титансодержащих материалов газообразным хлором и нек°'
торыми хлорпроизводными.
266
Двуокись титана реагирует с газообразным хлором при температуре около
/1200° С по реакции [119, 393]
| TiO2 + 2С1о = TiCl4 + О2.
1 реакция эта обратима. Чтобы сдвинуть равновесие в сторону образования TiCl4,
хлорирование необходимо проводить в присутствии восстановителей, связыва-
ющих кислород.
Четыреххлористый титан получен хлорированием смеси двуокиси титана или
рутила с углем [413, 474]; смеси двуокиси титана с сахарным углем при темпера-
туре 1000—1200° С [206], древесным углем при температуре около 1050° С [120,
121], а также при хлорировании двуокиси титана хлор производными углеводоро-
дов, например четыреххлористым углеродом, гексахлорэтаном, хлороформом [219,
252—254, 584], хлоридами серы [74, 195, 196, 237] и др. Четыреххлористый угле-
род реагирует с двуокисью титана при температуре около 440° С. Хлориды серы
(S2C12, SOC12) вступают в реакцию с TiO2 при красном калении, но при этом полу-
чается продукт присоединения 2TiCl4 • SC14.
Георгес и Штелер [312] получили TiCl4 хлорированием карбида титана га-
зообразным хлором. Хлорирование карбида титана начинается при температуре
около 200° С [129].
Четыреххлористый титан может быть получен также в результате хлориро-
вания ферротитана [566] и металлического титана [429].
К Четыреххлористый титан, полученный любым из описанных
методов, бывает загрязнен примесями SiCl4, СОС12, С12 и др. Для
очистки от них технический продукт подвергается ректификаци-
онной и дистилляционной перегонке [206]. От хлорокиси ванадия,
содержащейся в виде примеси в техническом тетрахлориде, полу-
ченном из ванадийсодержащего сырья, четыреххлористый титан
очищается обработкой восстановителями — амальгамой натрия, ме-
таллическим натрием, порошкообразной металлической медью.
Следует отметить, что препаративные методы получения четыреххлористого
титана, описанные выше, в настоящее время имеют только исторический интерес.
{Четыреххлористый титан теперь производится во многих странах в крупных про-
мышленных масштабах и поэтому для исследовательских целей нет необходимости
получать его препаративными методами.
Физические свойства
Молекула газообразного TiCl4 имеет тетраэдрическое строение
1178, 382, 408, 572]. Вира [572] определил расстояние между ато-
мами Ti — Cl 2,21, Cl — Cl 3,61 А, Листер и Суттоон [408] между
Ti-—Cl 2,18, Кимура с соавторами [382] — 2,185 А.
Решетка кристаллов TiCl4 моноклинная с параметрами (при
температуре — 32° С) а = 9,70, b - 6,48, с = 9,75 А, ₽ = 102°4Г,
г = 4, фед. гр. Р^/с [181].
й ИК-спектры поглощения жидкого TiCl4 имеют максимумы
при длинах волн 3,13; 6,18; 10,19; 11,41 и 12,15 мкм [420]. В
РК-спектре газообразного TiCl4 к основным частотам отнесены
^38 (vj, 119 (v2), 498,5 (v3) и 139 (v4) см~1 [335]. По ПК-спектрам
Поглощения в техническом четыреххлористом титане можно
267
определять примеси НС1, VOCI3, СОС12, СС1Н2СОС1, СС12НСОС1
СС13СОСС1 и SiCl4 [367].
Переход Т1С14 из жидкого состояния в твердое сопровождается
небольшим снижением частоты vx (Л), повышением частоты v2 (£)
и расщеплением линии v3 (F2) на две компоненты [418].
Тетрахлорид титана при комнатной температуре бесцветная
жидкость, дымящая на влажном воздухе. Температура кипения
его в старых, работах приводится в пределах от 135 до 136° с.
Температура кипения TiCl4, определенная из уравнения упруго-
сти пара при 760 мм рт. ст., по данным различных авторов, сле-
дующая (в °C): 135,9 [170], 136,5 [534], 135,2 [123], критическая
температура 631° К [315]. Температура плавления четыреххлори-
стого титана — 24,8, по [453], и —25° С, по [411].
Зависимость удельного веса жидкого TiCl4 от температуры та-
кова [170]:
t 0 с Удельный вес t, ° с Удельный вес
—18,10 1,79167 49,85 1,67701
—9,81 1,77800 59,86 1,65974
0 1,76139 69,77 1,64260
9,60 1,74514 79,77 1,62517
19,84 1,72796 90,18 1,60688
24,90 1,71935 99,97 1,58919
29,82 1,71112 109,60 1,57150
39,97 1,69371
Таблица 34
Теплоемкость TiCl4
t ° с с, кал/г - град СР • кал/моль • град Литература
0,112
0,163
0,164
21,3
30,9
31,0
13—99 j
21—136 |
411
411
411
Жидкий
0,191 | 36,3
Газообразный
0,129 | 24,5
Теплота плавления TiCl4, по Латимиру [411], равна 11,77 кал!г
или 2233 кал/моль, по Назу [453], 12,90 кал/г или 2440 кал!моль,
энтропия плавления 9,01 кал/моль - град [411].
Упругость пара Т1С14 измеряли несколько авторов. Зависи-
мость упругости пара от температуры (°К) они выразили уравне-
ниями:
Удельный вес твердого TiCl4 при разных температурах сле-
дующий: 2,06 (—79° С), 217 (—195° С), 2,20 (экстраполяцией,
-273° С) [208]. V F
Поверхностное натяжение (у) TiCl4 равно [3131:
Л °C 13 27,5 39,5 52 75
у,дин[см 34,03 32,16 30,78 29,08 26,15
log Р = — 1764,65/7+ 1,75 log Г —0,0006657 +
+ 2,90055 [мм рт. ст.] [170]
(293 —409° К),
] og Р = — 2919,4/7 — 5,7872 log 7 + 25,1289 [мм рт. ст.] [534]
(40—84° С),
logP = — 2017,8/7 + 7,82116 [мм рт. ст.] [123].
Зависимость вязкости (ц) TiCl4 от температуры такова:
t, °C vj. диН‘Сек/см* tс т], дин-сек/см3
—15 0,0126 20 0,00826
—10 0,0117 24 0,00794
— 4 0,0106 28 0,00771
0 0,01012 32 0,00743
4 0,00965 35 0,00727
8 0,00927 40 0,00702
12 0,00892 50 0,00645
16 0,00862
Теплоемкость TiCl4 приведена в табл. 34.
268
Келли [388] нашел упругость пара TiCl4 равной:
t,° К 225,5 290,0 337,0 361,5 383,5 409,0
Р, атм 0,001 0,01 0,1 0,25 0,5 1,0
Теплота испарения TiCl4 следующая:
t, ° С 25,0 25,0 135,2 135,8 136,5
Теплота ис-
парения,
ккал/моль 8,960 9,932 9,230 8,620 8,650
Литература [170J [365] [123] [170] [365]
269
Зависимость теплоты испарения TiCl4 от температуры выражае-
тся уравнением [388]
L = 23050 — 11,57" [кал/моль],
константа Трутона = 21,07 [170] и 22,4 [510].
* S
Эбуллиоскопическая константа определена из растворимости
антрацена в TiCl4 и равна 66 [511].
Энтальпия образования TiCl4 (ккал!моль), приведенная в рабо-
тах разных авторов, такова: ДЯ2эзж — —186,0 [493]; ДЯг91ж =
= —183,6 [554]; ДЯ^ -—190,3 [311], —190,0 [525]; —191,2
[168], —192,1 [531], —192,3 [365], —192,5 [280]; ДН^газ =
= —182,4 [531 ].
Энтропия Т1С14 (ккал/моль • град) = 47,90 [411], 60,3
[531], 84,4; S2°98ra3 = 60,4 [388], 59,51 [411].
По литературным данным вычислен изобарный потенциал
реакции
TiCl4ra3 = TiTB + 2С12газ
в интервале температур 300—1800° К:
AZ = — 175 600 + 29,137 [кал/моль] [419],
AZ — — 180,94 + 0,02917 [ккал/моль] [95,99].
Из спектроскопических данных рассчитаны термодинамические
функции Т1С14 в интервале температур 50—1600° К.
Энергия диссоциации TiCl4 по реакции
TiCl4 + hv TiCl3 + Cl *,
вычисленная по границе спектра поглощения в ультрафиолетовой
части спектра, составляет 87 [541] и 81,7 ккал/моль [359].
Показатель преломления света в TiCl4 с увеличением длины
волны уменьшается.
X, нм N X, нм ЛГ
358 1,7345 3061 1,563
394 1,6867 3628 1,562
441 1,6528 4045 1,561
480 1,6353 5084 1,558
508 1,6257 6084 1,554
533 1,6189 7010 1,550
589 1,6076 8071 1,545
670 1,5967 9003 1,538
768 1,5886 10620 1,523
1012 1,577 13710 1,482
1524 1,569 14100 1,471
2064 1,566 14500 1,457
2500 1,565
270
Показатели преломления до X = 768 нм определены при темпе-
в ратуре 18° С [526], остальные — при 20,3° С [420]. ч
J Удельная магнитная восприимчивость TiCl4 при температуре
35° С равна — 0,287 • 10~6 [567], диэлектрическая проницаемость
жидкого TiCl4 при 24° С—2,73 [421].
I Дьякграаф [247], Олдердик [173] и Хаякава [334] исследовали
спектры поглощения TiCl4.
Химические свойства
Связь титана с атомами хлора в молекуле TiCl4 осуществляется
за счет спаренных электронов и имеет ковалентную природу. В ва-
лентном отношении молекула TiCl4 насыщена. Однако она может
проявлять дополнительное сродство и вступает в реакции соеди-
нения со многими молекулами неорганических и органических
производных.
ft. Межмолекулярная связь TiCl4c лигандами может носить донор-
но-акцепторный характер, как например, в комплексных соеди-
нениях с сероорганическими производными [29], о чем можно су-
дить по величине дипольных моментов. Возможно также образо-
вание связи за счет переноса зарядов с л-орбиталей лигандов на
^-уровень металла [284]. По типу переноса зарядов образованы
комплексы тетрахлорида титана с хлоропроизводными метана и
.этана. Спектры поглощения их имеют полосы в области частоты
1(45 000 см~\ относящиеся к переходам С1(л)v5. Пики в спек-
« трах поглощения алкилцианидных производных тетрахлорида ти-
I тана с более низкими частотами относятся к переносу л-электронов
/лигандов на несвязующую тб-орбиталь.
I; Тетрахлорид титана вступает также в реакции взаимного об-
J мена и реакции замещения атомов хлора на различные атомы или
Ьрадикалы. В результате замещения атомов хлора на органические
«радикалы образуются многочисленные титанорганические со-
г единения.
К Соединения с аммиаком. Взаимодействие TiCl4 с аммиаком
впервые наблюдал Розе [502]. Пропуская газообразный аммиак через
|жидкий TiCl4, он выделил продукт красно-коричневого цвета, со-
гстав которого не установил. Штелер [548], пропуская газообразный
'аммиак через эфир, в котором суспендирован TiCl4, получил жел-
тый порошкообразный продукт. Состав его, по данным [501, 548],
^выражается формулой TiCl4 . 6NH3. Продукт такого же состава
^получили и другие авторы [285, 596]. На воздухе он неустойчив,
। Под действием влаги легко разлагается с отщеплением аммиака и
^образованием ТЮ2 и NH4C1.
При смешении TiCl4 с жидким аммиаком при температуре —30° С образуются
Тетрагональные кристаллы желтого или желто-красного цвета. При нагревании
До 200° С они переходят в продукт серо-зеленого цвета, а при 350—400° С превра-
|1Цаются в темно-фиолетовый титаннитрохлорид TiNCl.
271
Титаннитрохлорид имеет решетку типа FeOCl, параметры ре-
шетки а = 3,93, b = 3,25, с = 7,80 А, фед. гр. D^h 1369].
Желтый или желто-красный продукт имеет состав TiCl4 • 4NH3
и образует кристаллы с тетрагональной решеткой типа высоко-
температурной модификации NH4C1. Продукт серо-зеленого цвета
образует кубические кристаллы со структурой типа низкотем-
пературной модификации NH4C1 с параметром решетки а =
= 3,86 А [1291.
Четыреххлористый титан присоединяет гидразин, образуя
соединение TiCl4 - 2NH2NH2 [445], которое неустойчиво и ча-
стично разлагается, восстанавливая титан до трехвалентного
состояния.
Получены также продукты присоединения замещенных гидрази-
на — TiCl4 • 2L' (L' — фенил гидр азин) и TiCl4 • 4L? (L" — 1,1' —
диметил гидразин).
Соединения с хлоридами сурьмы, фосфора и фосфином. Четырех-
хлористый титан с треххлористым фосфором образует желтые кристаллы с тем-
пературой плавления 85,5° С состава TiCl4 • РС13 [182]. Существование этого
соединения наблюдал Роедер [510] при криоскопическом и эбуллиоскопическом
изучении системы TiCl4 — РС13. Кристаллы TiCl4 • РС13 ромбической системы,
параметры решетки таковы: а ~ 12,61, b = 13,59, с — 12,70 A, z = 8,
фед. гр. V,,5 [532].
Вебер [595], пропуская смесь паров TiCl4 и РС15 через нагретую стеклянную
трубку, получил соединение TiCl4 • РС15. Верелин и Гирауд [573] выделили это
соединение нагреванием эквимольной смеси TiCl4 и РС15 , Тютчев [559] — нагре-
ванием 1 моля TiO2 с 3 молями РС15 , Гевекке [304] — нагреванием фосфида ти-
тана в токе газообразного хлора. Методом кондуктометрического титрования
установлено существование в системе TiCl4 — РС15 еще одного соединения —
TiCl4 • 2РС15 [308, 309]. Однако оно не было выделено в чистом виде. В более
поздней работе [307] при исследовании системы TiCl4 — РС15 методом термиче-
ского анализа в ней вообще не обнаружено образования соединений,что противо-
речит работам предыдущих авторов.
Соединение TiCl4 • РС15 — желтое гигроскопическое вещество, сублимирую-
щееся при нагревании без плавления, водой и спиртом разлагается с образованием
резиноподобной массы состава TiCl(OH)3 • Н3РО4.
Вебер [595], добавляя по каплям РОС13 к TiCl4, получил кристаллический,
очень гигроскопический осадок состава TiCl4 • РОС13. Гутманн [320] выделил
соединение TiCl4 • 2РОС13.
Система TiCl4— РОС13 изучена методом термического анализа [307]. На кри-
вой плавкости были обнаружены два максимума, соответствующие соединениям
TiCl4 • РОС13 и TiCl4 • 2РОС13 (т. пл. 104 и 105° С соответственно).
Соединение TiCl4 • РОС13 кристаллизуется в ромбической системе в виде
желтых бипирамидальных кристаллов, параметры решетки которых а — 12,68,
Ь = 13,65, с = 12,60 А, ризм. = 1,92 г!см3. Продукт TiCl4 • 2РОС13 также крис-
таллизуется в ромбической системе с параметрами решетки а = 13,4, b — 13,5,
с = 7,7 А, фед. гр. Рппт [188].
Существование TiCl4 • РОС13 и Т1С14 • 2РОС13 подтверждено при исследо-
вании системы TiCl4 — РОС13 методом эбуллиоскопии [104] и с помощью ИК-спект-
ров [26, 156]. Связь в этих соединениях осуществляется за счет группы фосфо-
рила и атома металла, энергия связи 6,4 ккал!моль.
Соединение TiCl4 • РОС13 обнаружено в тройных системах ТiCl4—РОС13 —А1С1з,
TiCl4— SiCl4 — РОС13 и TiCl4 — VOC13 — РОС13 [5, 25].
272
в системе TiCl4—SbCl6 — Р0С13 найдено тройное соединение TiCl4 • SbCl6 •
. ЗРОС13 с температурой плавления 194° С [21], а в системах TiCl4 — (Nb, Та)
cu-pocls.— тройные соединения аналогичного состава: TiCl4 • NbCl5 • ЗРОС1Э,
Titl* • ТаС15 • ЗРОС13 с температурами плавления 96,0 и 113,5° С соответственно
(24, 96].
Образование соединений тетрахлорида титана с фосфином наблюдал впервые
розе [503]. Хельтье [336], пропуская фосфин через жидкий четыреххлористый
Рис. 70. Диаграмма состояния системы
SO2 — TiCl4 [187].
!Лы TiCl4 • BrCN, Обергаузер и Шормюллер [464] смешением растворов TiCl4 и
BrCN в сероуглероде — кристаллы TiCl4 • 2BrCN.
Соединения с окислами серы, селена и хрома и их галогенопроиз-
вйетыреххлористым титаном и построил диаграмму состояния SO2 — TiCl4 (рис.* 70).
*В этой системе существуют в равновесии две несмешивающиеся жидкости и твердая
даза. Твердая фаза при избытке SO2 состоит из сольвата 2TiCl4 • SO2.
Трехокись серы при смешении растворов SO3 и TiCl4 в хлор«v ж мт
Два соединения TiCl4 • 2SO3 и TiCl4 • 3SO3 [74, 403]. При нагревании растворов
тони разлагаются с образованием сульфата титана Ti (SO4)2 [345].
о Соединение TiCl4 • 2SO3_— кристаллическое вещество с удельным весом 2,012
рением на TiCl4 и SO3 [74, 403].
ЛучинСКИЙ [76] iiuuipun^i диаграмму ииихмлпии системы ли
(рис.^71). В ней обнаружено конгруэнтно плавящееся соединение TiCl4
JLJ vrikzitIVIClA OV7V>12 1 I'-«*4 rl OV24j1c
Довали равновесие жидкость — пар.
$гитан, выделил твердые продукты желтого цвета, имеющие состав TiCL • РН3 и
Т1С14 • 2РН3.
Соединения с цианистым водородом. При смешении TiCl4 с HCN полу-
чаются желтые кристаллы TiCl4 • 2HCN [381], с хлорцианом —TiCl4 • 2CNC1 в
виде твердого гигроскопического вещества [577].
Шнайдер [523], нагревая смесь TiCl4 с бромцианом, получил желтые кристал-
Ьы TiCl4 • BrCN, Обергаузер и Шормюллер [464] смешением растворов TiCl4 и
BrCN в сероуглероде — кристаллы TiCl4 • 2BrCN.
1 ' л___________
модными. Бонд с соавторами [185, 187] изучил взаимодействие двуокиси серы с
ретыреххлористым титаном и построил диаграмму состояния SO2 — TiCl4 (рис. 70).
В этой системе существуют в равновесии две несмешивающиеся жидкости и твердая
~ : д " 4 - SO2.
др Трехокись серы при смешении растворов SO3 и TiCl4 в хлористом тиониле дает
Два соединения TiCl4 • 2SO3 и TiCl4 • 3SO3 [74, 403]. При нагревании растворов
|№ разлагаются с образованием сульфата титана Ti (SO4)2 [345].
Соединение TiCl4 • 2SO3 — кристаллическое вещество с удельным весом 2,012
С), нерастворимо в СНС13 и SOC13. При нагревании сублимируется с разло-
Лучинский [76] построил диаграмму состояния системы TiCl4 — SO2CL
“ Л • 2SO2C12:
В системах SOC12 — TiCl4 и SO2C12—TiCl4 Деларова и Заварицкая [38] иссле-
Розе [502, 504] и Вебер [595], вливая хлористую серу с содержанием 68,9%
дора в раствор четыреххлористого титана в хлористом сульфуриле, получили
£лтые кристаллы TiCl4 • SC14 с температурой плавления 62—64° С. Это соедине-
ние неустойчиво на воздухе, хорошо растворяется в SO2C12, СНС13, CS2 и петро-
**ейном эфире.
V
189—614
273
При смешении хлористого селенила с тетрахлоридом титана образуются бель
кристаллы TiCl4 • 2SeOCl2 [196, 225, 589]. Методами кондуктометрии и криоскс_.
пии установлено существование в системе TiCi4— SeOCl2 комплексных ионов ти-
тана TiCl^", TiCl|~, TiCl^~ и TiCl|~ [593].
Хлористый хромил СгО2С12 с TiCl4 в растворе четыреххлористого углерод(
дает шоколадно-коричневый осадок, который, по мнению авторов [261], состоит
из двух соединений —TiCL.
• 2СгО2С12 и TiCi4 • СгО2С1
Согласно [319], Т1С14при
растворении в хлористом
сульфуриле реагирует с ним
по реакциям:
TiCl4 + 2SO2CI2 =
2
TiCll- + СГ = TiCl?~,
образуя хлоркомплексы тита-
на с координационными чи-
слами 6,7 и 8.
Соединения с двуо-
кисью и хлорокисью азо-
та. Жидкая четырехокись
азота с TiCl4 при температуре
ниже—20° С образует соеди-
нение TiCl4 • 3N2O4[505] . Это
вещество неустойчиво и пре-
вращается в нитрат титана
Ti (NO3)4 с выделением хло-
ристого нитрозила.
При пропускании паров
NO3F через раствор TiCl4 в
четыреххлористом углероде
образуется нестойкое соеди-
нение, которое не было выде-
лено в чистом виде [514].
о
Рис. 71.
и NOC1
20 40 60 60 100
Вес. °/Q 50? а2
Диаграмма состояния системы TiCl4—
SO2CI2 [76].
в четырех хлор истом углероде — TiCl4 •
Вебер [593] и Хампе
[337], пропуская пары TiCl4
через царскую водку, полу-
чили TiCl4 • NOCI, а Рейн-
болт и Вассефур [512]
смешением растворов TiCU
2NOC1. Рейнгельм и Хаке [505]
синтезировали последнее по реакции
2TiCl4 + С (NO2)4 = TiCl2CO3 + TiCl4.2NOC1 + N2O3
в виде белых октаэдрических кристаллов, нерастворимых в хлористом нитрози-
лэ [189].
Соединения с сероводородом и азотистой серой. Четь?
реххлористый титан при комнатной температуре очень энергично рея*
гирует с жидким сероводородом. Вначале образуется коричнево6
274
►твердое вещество, которое растворяется в жидком сероводороде,
Докрашивая его в красный цвет. Затем сероводород восстанавливает
КпС14 до низших хлоридов с выделением серы. Соединения TiCli с
HgS устойчивы только при низкой температуре.
По изменению упругости пара в системе TiCl4 — H2S установлено существо-
вание соединений TiCl4 • 2H2S и TiCl4 • H2S [211]. Первое вещество лимонно-
желтого цвета получено растворением TiCl4 в жидком сероводороде при темпе-
ратуре 78,5° С, второе — при —50 30° С, но под давлением может существо-
I вать и при комнатной температуре.
Ролстон и Вилкинсон [509] при температуре —77° С выделили желтые крис-
таллы 2TiCl4 • H2S.
Вельблинг [588] смешением растворов TiCl4 и N4S4 в четыреххлористом угле-
роде получил красно-коричневые кристаллы TiCl4 • N4S4, которые на влажном
воздухе становятся желтыми и разлагаются.
\ Взаимодействие с галогенами. С фтором TiCl4 продуктов
Присоединения не образует, а вступает в реакции замещения.
К Хлор с TiCl4 не взаимодействует. Система TiCl4 — С12 (рис. 72)
относится к простому эвтектическому типу.
Хлористый водород с TiCl4 дает два конгруэнтно плавящихся
^соединения: TiCl4 • 6НС1 и TiCl4 . 2НС1 (рис. 73).
Четыреххлористый титан присоединяет бром.
> В системе TiCl4 — Вг найдено два соединения: TiCl4 • Вг и
TiCl4 • Вг4 (оранжево-коричневого и гранатово-красного цвета соот-
ветственно) [75]. На существование
TiCl4 • Вг в системе TiCl4 — Вг
"Рис. 72. Диаграмма состояния си-
стемы TiCl4 — Cl [210].
Рис. 73. Диаграмма состояния си-
стемы TiCl4— НС1 [524].
•1' -
Указывают Абарбарчук и Костицына [3], обнаружившие максимум
да кривой вязкости, отвечающий составу этого соединения.
К Двойные соли. Четыреххлористый титан с хлоридами щелоч-
ных металлов дает двойные соли. Двойные хлориды титана отно-
сятся к гексакомплексам, содержащим ион [TiCl6]~2 [475]. Полу-
чены двойные хлориды титана с калием, рубидием, цезием, аммо-
нием и одновалентным таллием. Натрий и литий двойных солей
с четыреххлористым титаном не образует.
Двойные хлориды титана получают из концентрированных со-
лянокислых растворов TiCU или из неводных растворителей при
добавлении к ним хлоридов металлов [283].
Двойные соли титана со щелочными металлами образуются так-
же при нагревании хлоридов этих металлов с TiCU, однако вы-
ход их из-за невысокой термической устойчивости обычно мал
[88].
Мерц [413], смешивая растворы TiCl4 и NH4C1 в концентрированной соляной
кислоте, наблюдал под микроскопом образование октаэдрических кристаллов
двойного хлорида. Двойной хлорид титана и аммония также получается при на-
гревании гексааммиаката TiCl4 • 6NH3. Розенгейм и Шютте [532] состав двойного
хлорида после сушки кристаллов над концентрированной серной кислотой вы-
разили формулой (NH4)2 TiCl6 • 2Н2О. Скрейнемакерс [521] с помощью метода
остатков уточнил состав гексахлорида, показав, что из концентрированных со-
лянокислых растворов кристаллизуется безводная соль.
Фиреман [289] гексахлорид аммония получил нагреванием смеси TiCl4 Д-
Д- NH4C1 в автоклаве под давлением при температуре 280° С. Гексахлортитанат
аммония кристаллизуется из солянокислых растворов [258, 522].
На термограмме нагревания (NH4)2 TiCl6 имеется два тепловых эффекта при
температуре 337 и 370° С [89]. Первый тепловой эффект отвечает началу термиче-
ской диссоциации соли, второй — образованию нитрида титана.
Кристаллы K2TiCle имеют кубическую решетку с параметром а = 9,779 А,
по [258], и а — 9,972 A, z — 4, фед. гр. Fm3m, по [190].
Гексахлорид K2TiCl6 синтезирован взаимодействием TiCl4 с КС1 в расплавлен-
ной треххлористой сурьме [322], пропусканием паров TiCl4 через эвтектический
расплав КС1 + LiCl [422].
Упругость пара диссоциации K2TiCl6 по реакции
К2Т1С14 = 2КС1 + TiCl4
измеряли в работах [88, 268, 282, 422]. По данным [282], давление диссоциации
K2TiCl6 выражается уравнением
logР = — 5930/Т Д- 9,363 [мм рт ст.]
(Т = 370 — 580° С).
Энтальпия образования K2TiClG по реакции
КС1ТВ + TiCl4ra3 = К2Т(С16тв
ДН2д8 = —27,1 ккал!моль, что согласуется с более ранними данными [268].
Температура разложения К2ПС1б, по [268] — 532, по [88]—580° С. На тер'
мограмме нагревания K2TiCle отмечены тепловые эффекты при температуре 530
(начало термической диссоциации) и 770° С (плавление K2TiCl6) [89].
Энгель [258] получил кристаллы Rb2TiCle с кубической структурой тип3
K2TiCl6 и параметром решетки а = 9,922 А. Согласно [410], гексахлортитанят
рубидия — вещество светло-зеленого цвета с удельным весом 2,92; параметр ре'
шеткиа= 10,011 А, фед. гр. РтЗт.
276
По данным измерения упругости пара определена температура разложения
р Rb2TiCl6, равная 615, по [268], и 745° С, по [88]. Судя по термограмме нагревания,
I Rb2TiCl6 плавится с разложением при 658° С. Энтальпия образования Rb2TiCle
ДН^98 = “35 ккал/моль [268].
I Гексахлортитанат цезия Cs2TiCl6, полученный [89, 147, 258, 410], вещество
желто-зеленого цвета с удельным весом 3,24. Он кристаллизуется в кубической
системе, параметр решетки а = 10,229 А, фед. гр. FmZm, по [410], и 10,219 А,
цо [258].
Кристаллы Cs2TiCl6 разлагаются при температуре 795° С [88]. На термограм-
ме нагревания Cs2TiCl6 отмечено два тепловых эффекта при температуре 650 (плав-
ление соли) и 685° С (термическая диссоциация соли). Гексахлортитанат цезия
пока единственный известный двойной хлорид титана, плавящийся без разложения.
Листер и Фленгас [409] в системе CsCl — TiCl4 обнаружили второе соединение,
умеющее состав 4CsCl • TiCl4. Авторы определили упругость пара над двойными
хлоридами цезия в интервале температур 870—1040° С.
Энгель [258] и Гюентхер [322] синтезировали гексахлортитанат таллия Т12Т1С1б~
Его кристаллы имеют кубическую решетку с параметром а = 9,849 [258] и
9,804 А, фед. гр. Fm3m [322].
Утенейбл и Атой [570] получили гексахлортитанат гексааммиаката кобальта
[Co(NH3)6] [TiCl6].
Реакции присоединения с органическими
К производными
Четыреххлористый титан присоединяет многие органические
производные, содержащие группы с донорными свойствами, на-
пример > С = 0, = N, > С = S и др.
| Соединения с ароматическими углеводородами. Четы-
реххлористый титан с л-донорными растворителями образует
окрашенные комплексы в видимой области света [400]. Методом
спектрофотометрии показано, что в растворах TiCh с бензолом,
Толуолом, ксилолом и мезитиленом и в смесях TiCl4 с гекса-
метиленбензолом, нафталином фенантреном и антраценом образу-
ются соединения с отношением компонентов 1:1. Гексаметилен-
бензол в избытке Т1С14 также дает комплекс с отношением
|TiCh : СН3)6С6 = 2:1.
В спектрах поглощения комплексов TiCl4 и TiBr4 с бензолом, нафталином,
антраценом, фенантреном, хризеном, пиреном и периленом, обнаружена длинно-
волновая полоса, вызванная переносом заряда между молекулами галогенидов ти-
тана и ароматических углеводородов [249]. Некоторые из перечисленных комплек-
сов имеют по несколько полос поглощения, расстояния между которыми не зави-
сят от природы акцептора. Автор полагает, что появление этих полос соответству-
, ст переходу из основного состояния комплекса на уровень переноса заряда, когда
Молекула донора возбуждена.
Соединения с эфирами. Бедсон [191] получил соединение Т1С14 с димети-
ловым эфиром TiCl4 • (СНз)2О.
С этиловым эфиром известен продукт присоединения TiCl4 • 2 (С2Н5)2О [191,
238, 540, 569]. Это желтое гигроскопическое вещество, плавится при температуре,
42—45° С и разлагается около 60° С [191 ].
Четыреххлористый титан с диоксаном дает светло-желтые кристаллы TiCl4 •
>О(СН2СН2)2О [330, 540, 569].
Отт с соавторами [461] установили методом спектрофотометрии, чтоТЮ4 обра -
Зует соединения с анизолом и дифениловым эфиром с отношением компонентов
277
1 : 1; 1 : 2 (анизол) и 1 : 1 (дифениловый эфир). Вертипорох и Альтман [569] вы.
делили препаративным методом светло-желтые кристаллы с температурой плавле-
ния 44—45° С, имеющие состав TiCl4 • СН3СОС6Н5.
Тетр а гидрофур ан с TiCl4 дает два соединения: TiCl4 • С4Н8 О, разлагающийся
при 138—139°С, и TiCl4 • 2С4Н8О с температурой плавления 122—124° С [330]^
Тетрагидропиран с TiCl4 образует светло-желтые кристаллы TiCl4 • CJI1dq
с температурой плавления 124 — 125° С и кристаллы TiCl4-2 С5Н10 О [330].
Демарсей [250] сообщил о получении продуктов присоединения типа 2TiCl4 . э
где Э — метиловый и этиловый эфиры бензойной кислоты. Соединения этого ти-
па неустойчивы, о существовании их свидетельствуют спектрофотометрические
исследования [86].
Лысенко и Осипов [84] некоторые из них выделили в индивидуальном состоя-
нии, например: 2TiCl4CH3COOC2H5,2Т1С14 СвН5СООС2Н5, 2TiCl4.«-C6H5COOC4H9,
2TiCl4 i/ao-CeHgCOOCgHn. Существование соединений подобного типа
установлено также при изучении методом плавкости двойных систем из
четыреххлористого титана и сложных эфиров амилового и гексилового спиртов
с валериановой, капроновой, энантовой, пеларгоновой, капроновой и стеарино-
вой кислотами [87].
Более стойки эфираты типа TiCl4 • Э, где Э — сложные эфиры органических
кислот. В настоящее время известно уже большое количество соединений, которые
получены препаративным методом или обнаружены при физико-химических ис-
следованиях растворов.
Лысенко с соавторами [82] изучили методом плавкости 15 двойных систем
TiCl4 со сложными эфирами муравьиной, уксусной и хлоруксусной кислот. Во
всех системах образуются соединения типа TiCl4 • Э. С увеличением спиртовых
радикалов в молекулах эфиров температура плавления их понижается. Препара-
ты, содержащие спиртовые радикалы с разветвленной цепью, плавятся при более
высокой температуре, чем эфираты со спиртовыми радикалами нормального
строения.
Измерением вязкости, плотности и электропроводности установлено существо-
вание соединений в растворах TiCl4 с муравьино-н-бутиловым и муравьиноизо-
амиловыми эфирами с отношением компонентов 1:1 и 1 : 2 [78]. Синтезирован
эфират TiCI4 • п -НСООС3Н7 [84].
В системе четыреххлористый титан — этилацетат отмечено существование со-
единения TiCl4 • CHgCOOCgHg [80, 223]. Взаимодействие TiCl4 • CHgCOOC^Hg
со второй молекулой этилацетата приводит к резкому повышению полярности,
что указывает на образование комплекса TiCl4 • 2СН3СООС2Н5, имеющего цис-
строение [48]. Более двух молекул сложного эфира, судя по изменению диэлектри-
ческой проницаемости, к молекуле TiCl4 не присоединяется.
В системах TiCl4 — я-СН3СОО3Н7 и TiCl4 — н-СН3СООС4Н9 методами фи-
зико-химического анализа обнаружены соединения с отношением компонентов
1:1 и 1:2 [79, ПО], в системе TiCl4 — изо-СЩСООСзНц— соединение с
отношением компонентов 1:1 [111].
Лысенко и Осипов [85] изучив изменения вязкости и электропроводности
в системе TiCl4 — СН3СООСН2С1 установили существование соединений с отноше-
нием компонентов 1 : 1 и 1 : 2. Шанд и Ровест [223] получили кристаллические
продукты присоединения TiCl4 • СН3СООСН2СН2С1, TiCl4 • 2СН3СООСН2СН2С1,
TiCl4 • СН2С1СООС2Н5 и TiCl4 • CHC^COOC^Hg, плавящиеся при температуре
78—80, 74—76, 109—111 и 82—84° С соответственно.
Синтезированы неустойчивые эфираты сложных эфиров трихлоруксусной
кислоты TiCl4 • СС13СООС2Н5, TiCl4 • изо-СС13СООС3Н7 иТ1С14 • цзо-СС13СООС4Н9
[84]. Эфират Т1С14 • СС13СООС2Н5 легкоплавкое вещество с точкой плавления
23—24° С [78]. Существование этих эфиратов в растворах обнаружено при иссле-
довании их методами измерения вязкости, плотности и электропроводности [81 ]•
Эфираты TiCl4 • н-СС13СООС4Н9 и TiCl4 • цзо-СС13СООС5Нц в чистом виде не по-
лучены.
В результате изучения вязкости, электропроводности, плотности и плавкости
в системах TiCl4 — С3Н7СООС4Н9 и TiCl4 — С3Н7СООС5НП установлено образование
соединений с отношением компонентов 1 : 1 [112]. В концентрированных раство-
278
pax молекулы эфиратов ассоциированы (по данным криоскопических измерений).
Ассоциированные молекулы, по мнению авторов, диссоциируют по схеме
(TiCl4 • Э)2 ±3 [TiClJ2- 4- [TiCl2- 2Э]2+.
Ривест с соавторами [506] получил соединения типа TiCl4 • L и TiCl4 • 2L,
•где L — этиловые эфиры масляной и капроновой кислот.
Смешением эквимольных количеств веществ, растворенных в бензоле, выделен
продукт TiCl4 • С6Н5СООСН3 желтого цвета с температурой плавления 150° С
(426, 427].
При изучении вязкости и электропроводности в системе TiCl4 — СН3(СН2)16«
ICOOQjHg установлено существование соединения с отношением компонентов 1:1.
Ряд эфиратов четыреххлористого титана с хлорзамещенными эфирами одно-
основных кислот описаны Джейном и Ривестом [371].
Ривест и соавторы [506] получили продукт состава TiCl4 • Э и TiCl4 • 2Э,
где Э — диэтиловые эфиры двухосновных карбоновых кислот: щавелевой, мале-
иновой, янтарной, адипиновой, пимелиновой, пробковой, азелаиновой и себа-
циновой.
Хертель и Деммер [343] синтезировали продукты присоединения TiCl4 с ди-
этиловым эфиром фумаровой кислоты (Fum) — 2TiCl4 • Finn, TiCl4 • Fum, TiCl4 •
•Fum • СбН6 и TiCl4 • 2Fum.
Ацетоуксусный эфир присоединяется к TiCl4 с образованием TiCl4 •
• СН3СОСН2СООС2Нб [23]. Кристаллы оранжевого цвета с температурой плавле-
ния 115—117° С, хорошо растворяются в дихлорэтане, ацетоне и четыреххлористом
углероде.
Лысенко и Осипов [84] смешением растворов TiCl4 и эфиров бензойной кис-
лоты в бензоле выделили эфираты TiCl4 • С6Н5СООС2Н5, TiCl4 • пзо-CeHsCOOQHc)
TiCl4 • U30-C6H5COOC5H1]L. Аналогично получены эфираты TiCl4 •
.СН3С6Н4СООСН3, TiCl4 • 2,6-(СН3)2С6Н3СООСН3, TiCl4 • 2,6-(ОН3)3СвН2СОО'СН3
|533].
Скаглиарини и Тартарини [543] синтезировали желтые кристаллы TiCl4 с
диметилфталатом TiCl4 • СвН4 (СООСН3)2.
Эфираты четыреххлористого титана способны присоединять хлористый цинк
(127]. Образующиеся при этом соединения имеют состав TiCl4 • Э • ZnCl2,
где Э — эфир.
Эфираты четыреххлористого титана в расплавленном состоянии частично
диссоциированы. Осипов и Лысенко [113] провели электролиз расплавленных
эфиратов TiCI4 • н-НСООС2Н5, TiCl4 • CH3COOC5Hn, TiCl4 • НСООС4Н9 и TiCl4 •
• СН3СООС2Н5, при этом в катодном пространстве наблюдалось накопление
TiCl3, а на аноде выделялся хлор. Результаты опытов подтверждают приведен-
ную выше схему диссоциации эфиратов четыреххлористого титана.
При нагревании эфираты и формиаты четырех хлор истого титана подвергают-
ся термическому разложению [84]
TiCl4 • RCOORi -> RC1 + RCOOTiCi3,
HCOOTiCL -> HC1 4- CO + TiOCl2.
Эфираты четыреххлористого титана с бензилформиатом и бензилтрихлорацета-
Том разлагаются с образованием желтой смолы состава С7Нб.
Строение эфиратов исследовано еще недостаточно. Осипов с соавторами [105,
108] обнаружил, что эфираты TiCl4 со сложными органическими эфирами имеют
большие дипольные моменты. Увеличение полярности при образовании этих
соединений, по мнению авторов, может служить признаком комплексного их
Строения.
Взаимодействие TiCl4 со сложными эфирами неорганических кислот почти
Не изучено.
Славинская [133] при действии этилнитрата на TiCl4 при комнатной темпера-
туре наблюдала выделение NO и NO2 и образование бурой массы, состав которой
еще не установлен. Реакция идет с саморазогреванием и взрывом.
279
Методом криоскопического титрования четыреххлористого титана этилнит-
ратом в хлорбензоле установлено образование соединения C2H5ONO2 • TiCI
Из раствора хлорбензола через 30—35 мин после смещения TiCl4 и C2H2ONo
выпадает желтый мелкокристаллический осадок, который со временем
темнеет.
Соединения с альдегидами и кетонами. Взаимодействие TiCl4 с альде-
гидами и кетонами обнаруживается с помощью исследования ИК-спектров [106].
Бензальдегид с TiCl4 образует желтый осадок неустановленного еще состава
[569], ацетон в избытке — солеобразные сольваты, а при недостатке — продукты
конденсации [569]. Сольват состава TiCl4 • СН3СОСН3 плавится при температуре
93—97° С, слабоокрашенные гигроскопичные кристаллы TiCl4 • СН3СОС2ЦГ>
и TiCl4 • СН3СОС6Нб имеют температуры плавления 105—ПО и 120—127° С
соответственно [393, 539].
Эдвард [259 , 260], Хертель и Деммер [343] получили светло-желтые кристал-
лы TiCl4 • ОС (СбН5)2 с температурой плавления 150° С и продукт
TiCl4 • 2ОС<
ХСН=СНС6Н5.
Известно соединение TiCI4 с’гелиотропином в виде красных кристаллов сос-
тава TiCi4 • 2СсН3(ОСН)
{у СН2 (543].
Соединения с карбоновыми кислотами. Реакция TiCl4 с
карбоновыми кислотами относится к числу мало характерных. Из-
вестно только несколько соединений этого типа.
Кемпа и Ли [380] получили продукт присоединения к Т1С14этиленкарбоната —
TiCl4 • 2 (СН2 = СН — О — СООН), а Лысенко [78, 83] — комплексы следующего
состава: TiCl4 . НСООС4Н9, TiCl4 • HCOOC5HU, TiCI4 • 2НСООС4Н9 и TiCl4 •
• 2НСООСН2С4Н-изо. При исследовании системы TiCl4 —С2НбСООН методами
вязкости, электропроводности и плотности обнаружены соединения TiCi4 •
.2HC(X)ii2H5 [109].
Геринг и Зусц [324] изучили комплексы фенил-, о-, м- и n-толилацетатов с
TiCl4.
Соединения с органическими основаниями. Кроме продук-
тов соединения, органические основания с TiCl4 дают гексахлороком-
плексы (RN)2TiCl6 типа двойных солей, где RN — органическое
основание. Эти продукты, подобно гексахлортитанатам щелочных
металлов, устойчивы в концентрированных растворах соляной
кислоты.
Из раствора TiCl4 и (CH3)3N в бензоле выделен желтый, диамагнитный про-
дукт TiCl4 • (CH3)3N [298].
Гутманн [317] по изменению электропроводности растворов тетраметил хлор-
амина и TiCl4 в треххлористом мышьяке установил существование комплексов
(CH3)4NTiCl5 и [(CH3)4N]2TiCl6. Последнее соединение, а также [(CH3)2NH]2 TiCl6
получили авторы [339, 283].
Трост [557, 558] синтезировал 2TiCl4 • (C2H5)3N и TiCl4 • C2H5NH2. Хеги
[339] из хлороформного раствора выкристаллизовал солеподобный продукт
(C2H5N)2TiCl6. Этот гексахлортитанат и (CH3NH3)2TiCle получено из солянокислого
раствора [283].
< Методом криоскопического титрования раствора TiCl4 в бензоле этилендиа-
мином установлено существование комплексов TiCl4 • 2En, TiCl4 • ЗЕп, TiCl4 *
280
L4En, гДе Еп — NH2CH2 — CH2NH2[136]. Соединения аналогичного состава об-
разуют тетраметилендиамин NH2(CH2)4NH3 и гексаметилендиамин NH2 (CH2)6NH<>
[137]- Наряду с реакцией комплексообразования, при взаимодействии TiCl4 с
f4H2(CH2)eNH2 протекает также аминолиз, в результате которого один атом хло-
ра замещается на остаток гексаметилендиамина с образованием комплексов
fiCl3HN (CH2)6NH2 . NH2 (CH2)6NH2 и TiCl4NH (CH2)6NH2 . 2NH2 . (CH2)6NHg.
Пиридин c TiCl4 дает желтый продукт TiCl4 • 2C5H5N [245, 262, 498, 501]. Из
солянокислых растворов получен гексахлортитанат пиридина (C5H5N)2TiC’6 [283].
Дермер и Фернелиус [239] выделили продукт соединения TiCl4 с хинолином
коричневого цвета, имеющий состав TiCl4 • 2CgH7N, Хеги [339], Фоульс и Ни-
хольс [283] — гексахлортитанат (C9H7N)2TiCl6.
Хеги [339] из хлороформных растворов получил серию комплексов желтого
цвета:
(C6H5H4N)2TiCl6, 4C6H5NH3C1 . 4TiCl4, (C6H5NH3)2TiCl6, [(CH3CH2)4NCl]2TiCl6,
3(CH8)2NH2C1CHC13 • TiCl4. Все они хорошо растворяются в спирте и ацето-
не, но разлагаются под действием воды.
Синтезированы комплексы типа TiCl4 • L с 2,2'-дипир и ди лом и о-фенантрали-
ном. Реакция проводится в бензоле или четыреххлористом углероде [214, 232].
Йолучен комплекс этого же типа с N,N-диметилформамидом [172].
Лидс [412] выделил соединение TiCl4 • (C6H5)NH2 в виде светло-зеленого
осадка.
Известны соединения четыреххлористого титана с гексаметилентетрамином
(уротропином) и дихлорэтиленом в виде белых кристаллов, имеющих состав
2TiCl4 • 7C6H12N4 • CoHgC^ [542]. В другой работе [544] описано TiCl4 • 12СвН12О4 •
,ЗСНС1Я.
Дермер и Фернелиус [239] получили серию комплексов TiCl4 с органическими
основаниями следующего состава: TiCl4 • 2(C6H5)2NCOCH3 (светло-желтый),
TiCl4 • 2C4H4O2NH (темно-коричневый), TiCl4 • CH3C6H4SO2NH2 (темно-корич-
невый), TiCl4 • C6H5CH : NC6H5 (светло-желтый), TiCl4 • (СбН5)2С: NC8H5 (жел-
тый), 3TiCl4 • 2C6H5N2C6H5 (оранжево-желтый), TiCl42C6H5: NC6H4OCOC(5Hf>
(оранжево-красный), TiCl4 • C6H5N2NHC6H5 (светло-желтый), 3TiCl4 • 2CcH5N2 •
f N (CeH5)2 (темно-зеленый), TiCl4 • 4C6H7N (у-пиколин, белый), TiCl4 • Cr,HnN
(пиперазин, желтый).
Леопард [404] из раствора в гептане осадил соединение TiCl4 с пиперидином
зеленого цвета.
Шмитц-Думонт и Мотцкус [527] описали светло-желтые кристаллы состава
TiCl4 • 2CgH7N (с индолом) и желто-фиолетовые TiCl4 • 2(C5H7N)3 (с тринизолом).
При длительном нагревании растворов TiCl4 и диэтиламмонийбромида в хло-
роформе образуются смешанные хлорбромтитанаты TiCl6_x ВгхМ2, где М — ам-
мин; х изменяется в пределах от 0 до 6 в зависимости от состава раствора, из ко-
торого кристаллизуется продукт [342]. Смешанные титанаты получены также из
*iBr4 и диэтиламмонийхлорида. По данным рентгеноструктурных исследований,
смешанные гексахлорбромтитанаты являются изоморфными соединениями хло-
ридов и бромидов.
Пиразин и 2,6-диметилпиразин с TiCl4 дает соединения TiCl4 • 2C4H4N2 и
*iCl4 . 2 (CH3)2C4H2N2 [299].
Комплексы TiCl4 с пиразином и 2,6-диметилпиразином образуют слабо про-
водящие растворы в метилцианиде. Электропроводность растворов объясняется
Диссоциацией комплексов по схеме
№ TiCl4 • 2L + CH3CN --- [TiCl3 2LCH3CN]+ -]- СГ.
Исмаилов с соавторами [45] синтезировал соединения, имеющие состав
4 С14 • L, где L — 2 -диметиламинометил-4-метилфенол; 2-диэтил амино метил-
^pem-аминофенол; 2,4,6-(тридиметиламинометил)-фенол; 1,Г -диокси-2-ди-
л аминометил; 4,4'-ди-(трет-бутил)-дифенил-6,6'-сульфид. Эти соединения,,
fe мнению авторов, образуются за счет пары л:-электронов кислородных атомов
иН-групп.
281
Получены аддукты TiCl4 с шиффовыми основаниями состава TiCl4 • 2А, где
А — салицилал ванилин, салицилаль-п-толуидин, салицилаль-п-нитроанилиц
салицила л ь-л£-нитроанилин, салицилаль-л«-нитроанилин, бензал ванилин, 2-ме-
токсибензалванилин. Все эти соединения диамагнитны [55].
Соединения с амидами кислот. Описанные [507] комплексные соединения
TiCl4 с мочевиной и тиомочевиной и некоторыми их производивши имеют состав
TiCl4 • 2CO(NH2)2, TiCl4 • CO(NCH3)2, TiCl4 • 2CO(NHCH3)2, 2TiCl4 . 2NH2CONy
X(C6H5)2, 2TiCl4 • 2CO(NHC6H5)2, TiCl4 • 2CS(NH2)2, TiCl4 • CS(NHC2H5)2
2TiCl4 • CS(NHC2H5)2, 2TiCl4 • NH2CSN(C6H5)2.
По данным исследований ИК-спектров установлено, что координация моче-
винных комплексов с титаном осуществляется через кислород, связв титана с тио-
мочевиной — через атом азота. Фенильные производные дают двуядерные ком-
плексы с атомами титана через мостики атомов хлора и азота.
Ионова и Головня [44] синтезировали три- и тетрамочевинные комплексы
составов TiCl4 • ЗСО (NH2)2 и TiCl4 • 4СО (NH2)2. По их мнению, строение этих
соединений выражается формулами СО (NH2)2H[TiCl4CONH2NH • СО (NH^J
и 2СО (NH2)2H [TiCl4 • СО (NH2)2 . СО (NH2)2].
Взаимодействие с нитросоединениями. Четыреххлористый титан с
мононитросоединениями образует продукты присоединения состава TiCl4 • X,
где X — нитросоединения [476].
Известны комплексы этого состава с нитрометаном (желтые кристаллы, т. пл.
51° С) и 'нитроэтаном (т. пл. 51° С) [505], с нитробензолом (т. пл. 77,5° С) [476,
505], л£-хлор — нитробензолом (т. пл. 61° С), л-хлорнитробензолом (т. пл. 54,5° С),
jw-бромнитробензолом (т. пл. 72° С), о-нитротолуолом (т. пл. 62,5° С), ^-нитрото-
луолом (т. пл. 75° С), n-нитротолуолом (т. пл. 72,3° С), 1,3,4-нитроксилолом
(т. пл. 64° С) [476]. уи-Динитробензол и ;и-динитротолуол дают продукты присоеди-
нения типа TiCl4 • X и TiCl4 • 2Х. Так, Гертель [344] описал продукта присое-
динения с л1-динитробензолом типа TiCl4 • 4Х (т. пл. 45° С), TiCl4 • 2Х (т. пл.
65° С) и 3TiCl4 • 2Х.
В системе TiCl4 — CH3CH2CH2NO2 методами измерения диэлектрической про-
ницаемости и криоскопии установлено существование соединений TiCl4 •
• CH3CH2CH2NO2 и TiCl4 • 2CH3CH2CH2NO2 (561].
Соединения с органическими производными серы и селена. Демарсей
[250, 251] выделил темно-фиолетовые кристаллы TiCl4 • C2H5SH, TiCl4 • 2CaH5SH,
TiCl4 • S (QH^, TiCl4 • 2S (C2H5), которые, в свою очередь, могут давать про-
дукты присоединения TiCl4-CQH5SH-C6H5CO2QH5 и TiCl4 • S (С>Н5)2 • СеН5СО2 —
“ ОгН5.
Установлено существование соединений TiCl4 • 2 S(C4H9)2, TiCl4 • 2C4H8S,
TiCl4 • C4H8S [29], TiCl4 • 2 (CH3)2S, TiCl4 • TiCl4 • 2C4H8S, TiCI4 X
X C4H8S2CH3SCH2SCH3, TiCl4 • C6H5SCH2—CH2SC6H5, TiCl4 - (C2H5)2PSPS(C2H5)2
[198].
Связь титана с лигандами в этих соединениях, по предположению авторов,
реализуется за счет пары электронов серы.
Смешением растворов TiCl4 в бензоле и соответствующих лигандов синтези-
рованы продукты присоединения серо- и селенопроизводных TiCl4 • (СН3)2$»
TiCl4 • 2 KQH^Sl, TiCl4 TiCl4 • (QH^Se и TiCl4 • (C6H5)2Se.2 • СЛ
[579]. Это твердые вещества с температурой плавления 55—57, 69,5—70, 122—132,
55 (разлагается) и 83° С (разлагается) соответственно.
Тиоксан с TiCl4 образует соединение TiCl4 • 2 (CH2CH2S)2 [299], которое, судя
по ИК-спектрам, имеет tfuc-строение. Связь тиоксана с титаном осуществляется
через атом серы. Джайн и Ривест [372] взаимодействием раствора TiCl4 и соответ-
ствующих лигандов в гексане получили продукты TiCl4 • 2C2H5SCN, TiCl4 X
X QjHgSCN, TiCl4 • NCS (CH2)2SCN, 2TiCl4 • NCS (CH2)2SCN, TiCl4 - C,H5NCS
и TiCl4 . 2C2H5NCO.
Соединения с нитрилами. Выделены бесцветные возгоняющиеся кристаЛ-
лы TiCl4 • 2CH3CN [262, 341]. При добавлении воды к кипящему раствору этого
соединения в токе аргона образуется продукт гидролиза в виде желтого порой1'
кообразно го вещества, имеющего состав Ti2Cl6O • 4CH3CN [294]. При нагревания
его с метил нитрилом он дает продукт состава Ti2Cl6O • 6,5CH3CN.
282
Синтезировано соединение TiCl4 • C2H2CN желтого цвета [561]. Путин с
г соавторами (477] показал, что оно плавится инконгруэнтно при 100° С.
В системе TiCl, ~
соединения с отношением TiCl4: пропилнитрил‘= 1 : 1; 5 : 6; 2 : 3: 17 2: 2 : 5 и
1:3 [343].
Продукт TiCl4 • 2C2H5CN в чистом виде представляет белые кристаллы с тем-
J ператУР°й плавления 112° С, разлагающиеся при температуре возгонки [341],
В системе TiCl4 — фенилцианид на-
блюдается образование соединений с от-
ношением компонентов 1:1, 1:2, 1:3,
2:3 и 5 : 6 [343]. Эмелеус и Рао [262]
комплекс TiCl4 . 2C6H5CN выделили в чи-
стом виде.
Четыреххлористый титан с п-толил-
цианидом дает комплекс TiCl4 • 2(п-
CeH4CH3CN) с температурой плавления
153° С [477], а с нитрилом фталевой ки-
слоты аддукты TiCl4 . С6Н4 (CN) (СООН)
К 2TiCl4 • С6Н4 (CN) (СООН) [299].
Соединения с органическими
Производными кремния и олова.
.Четырех хлор истый титан с силанами об-
разует нестойкие соединения типа
4 — C3H7CN, по данным термического анализа, существуют
Рис. 74. Диаграмма плавкости си-
стем:
1 — TiCl< — CCI3COCI; 2 — T1CI4 —
CHjCICOCI; 3 — TiCl4 —СН3СОС1.
Cl3Ti( >SiR3
где R = С2Н5, С6Н5, С2Н5О, CH3SiCl2.
Они неустойчивы и распадаются на R3SiCl
и HTiCl3.
Существование «титанхлороформа»
подтверждено исследованием ИК-спектров.
Он является соединением нестойким и через несколько дней разлагается в растворе
на водород и TiCl3 [ 520].
По изменению диэлектрической проницаемости и поляризации установлено
образование комплекса TiCl4 с тетраэтоксисиланом в неполярных растворителях
^отношением компонентов 1:1. Анализ УФ-спектров и определение молекуляр-
ных весов указывают на существование сильно диссоциированного комплекса со-
става 2TiCl4 . 3Si (ОС2Н5)4 [47].
Изучено взаимодействие TiCl4 с Sn (С2Н5)4, Sn (С2Н5)3С1, Sn (С2Н5)2С12 и
$п(СаН5)С13 в растворе бензола. На основании криоскопических исследований по-
казано, что в бензольных растворах образуются практически недиссоциированные
I неассоциированные комплексы с отношением компонентов 2 : 1 [53].
Соединения с органическими производными фосфина. Чатт и Хейтер
1214] из бензольного раствора выделили следующие соединения в кристалличе-
ском виде: TiCl4 • 2Р(С2Н5)3 (темно-красное, т. пл. 144,5—148° С), TiCl4 • 2Р(СвН5)3
ремно-красное, т. пл. 151,5—153° С), TiCl4 • С2Н4 [Р(СН3)2]2 (оранжевое, т. пл.
$81—285° С), TiCl4 . о-С6Н4 [Р(С2Н5)2]2 (красное, т. пл. 256,5—259,0° С).
I Фоульс и Уолтон получили соединения TiCl4 • 2(С6Н5)3Р [299] и TiCl4 х
Х2(С6Н5)2РС2Н5 [296], Уестлэнд [579] смешением бензольных растворов TiCl4 и
соответствующих лигандов — в виде оранжевых или темно-красных кристаллов
ледующие: TiCl4 • (СбН5)3Р (т. пл. 147—149° С), TiCl4 . 2(С6Н5)3Р (т. пл. 149—
61° С), 3TiCl4.2(СбН5)4Р2С2Н4 (т. пл. 139-143° С), TiCl4.2(С6Н5)4Р2С2Н4 (178° С
Излагается), 2TiCl4 • 3(С6Н5)4РС2Н4 (т. пл. 138—140° С).
В ПК-спектре 2TiCl4 • (СбН5)3Р имеются две полосы с максимумами при
(винах волн 325 и 380 см~~1 [299]. Появление этих полос указывает на цис-
!Троение комплекса.
283
В системе TiCl4— ТБФ (трибутилфосфат) обнаружено соединение TiCl4 . ТБф
[107], а в системе TiCl4—НС1—ТБФ-соединения H2TiCl6 • ЗТБФ и SiCl4 • 2ТБф
[130].
Соединения с галогенопроизводными. Бетранд [192] в результате не-
посредственного соединения компонентов выделил кристаллы TiCl4 • CH3COCI
температурой плавления 25—30° С. Растворяются в ацетилхлориде и сероуглероде.
Войтович и Звагольская [22] изучили системы TiCl4 — СС13СОС1, TiCl4
СН2С1СОС1 и TiCl4 — СН3СОС1 методом плавкости и построили диаграммы состо-
яния этих систем (рис. 74). Первая система оказалась простого эвтектического ти-
па, а в двух других образуются комплексы с отношением компонентов 1:1.
Известны продукты присоединения TiCl4 с бензохлоридом TiCl4 • о-(СН3).э .
• СбН3СОС1 [528].
Соединения с арсинопроизводными. Получены соединения TiCI4 с
трифениларсином TiCl4 • (C6H5)3As и 2TiCl4 • (C6H5)3As [299, 579] и тетрафенил-
этилендиарсином 2TiCl4 • (CeHs^A^CgH* (т. пл. 145—149° С) [579].
о-Фениленбисдиметиларсин (R) с TiCl4 образует желто-оранжевое соединение
TiCl4 • R [230] и оранжевое TiCl4 • 2R [229, 230].
Битл и Уебстер [194] пытались с помощью инфракрасной спектроскопии ис-
следовать строение продуктов присоединения органических производных к те-
трахлориду титана.
С этой целью были изучены ИК-спектры семнадцати соединений. Однако по
данным только инфракрасной спектроскопии решить вопрос с геометрической кон-
фигурации титановых комплексов оказалось невозможным.
Системы, в которых не образуются химические соединения с
TiCl4
Четыреххлористый титан не дает химических соединений с че-
тырехбромистым титаном. В системе TiCl4 — TiBr4 образуется не-
прерывный ряд твердых растворов [511].
Рис. 75. Диаграмма температур кипения системы
СС14 — TiCl4 [451].
Установлено, что система TiCl4 — I относится к простому эвтек-
тическому типу [402, 442]. Растворы четыреххлористого титана с
иодом окрашены в фиолетовый цвет. Максимум светопоглощения
284
лежит при 520 нм [353]. Растворы четырехиодистого титана с
’TiCl4 окрашены в коричневый цвет.
Четыреххлористый титан растворяется в IC1. Раствор обладает
электропроводностью, что может быть результатом существования
ионов TiCl|“ [318].
Рис. 76.
кипения
Система TiCl4— СС14. В системе наблюдается полная взаимная растворимость.
Диаграмма состояния ее относится к эвтектическому типу, эвтектическая точка
отвечает температуре — 66° Си 59 мол. %
СС1Д450, 453]. Ридер с соавторами [497]
измерил давление пара над смесями
TiCl4 — СС14 при температуре 30, 40 и
50° С. Диаграмма кипения смесей TiCl4—
СС14 не имеет экстремальных точек
(рис. 75).
Система TiCI4—SiCl4. Диаграмма
плавкости относится к эвтектическому ти-
пу [448, 450, 452, 453]. На кривой тем-
пературы кипения экстремальные точки
отсутствуют (рис. 76). Измерено давление
пара над растворами SiCl4 в TiCl4 при тем-
пературе 30, 40 и 50° С и вычислены ко-
эффициенты активности SiCl4 и TiCl4 в
ф растворах [213].
Система TiCl4—SiHCl3. Система близ-
ка к идеальной, хотя имеется небольшое
отклонение от идеальности [72]. Упру-
гость пара в интервале температур 342—
436° К выражается уравнением
= 1487,3/7 + 6,5350 [мм рт. ст.].
Диаграмма температуры
системы TiCl4—SiCl4 [449,
451].
Растворимость в четыреххлористом титане хлоридов некоторых металлов.
Изучена растворимость TiCl4 хлоридов железа и алюминия FeCl3 и А1С13 [122,
124]. Зависимости логарифмов концентрации А1С13 и FeCl3 в TiCl4 от температуры
выражаются уравнениями:
I 1о^ ЛгА1С1з- —3214,1/7+ 6,29113
i^g^FeCI = “ 3465,7/7 + 5,82427,
*3
тде /V — мольная доля.
При совместном присутствии растворимость FeCl3 и А1С13 в четырех хлор истом
титане выше, чем отдельных компонентов.
Растворимость сероокиси углерода COS в TiCL при температуре 0° С равна
9,5, а при 100° С - 1,1 вес.% [41].
Марков с соавторами [97] по изменению электропроводности растворов
TiCl4 и VOC13 в нитробензоле сделал вывод, что TiCl4 с VOC13 не вступает в химиче-
ское взаимодействие.
Изучена методом термического анализа система TiCI4 фенантрен [114]. Обра-
зование химических соединений между компонентами не обнаружено.
Реакции восстановления
Четыреххлористый титан восстанавливается многими металла-
ми и водородом до низших хлоридов или металла.
Металлический натрий при нагревании с газообразным TiCl4 восстанавливает
его до металла [202, 241, 340, 387, 405, 467, 469, 479, 492]. Изобарный потенциа,
реакции
TiCl4ras + 4Naras = TiTB + 4NaClJK
выражается уравнением [419]
AZ = 226 200 + 65S2T [кал]
(T = 300— 1800° К).
Исследование кинетики процесса показало, что восстановление TiCl4 натрием
в интервале температур 550—800° С носит сорбционно-автокаталитический харак-
тер [128]. Энергия активации по мере использования восстановителя понижается.
Металлический натрий реагирует с TiCl4 при низкой температуре. Он взаимо-
действует с жидким TiCl4 под слоем углеводородов при 105° С [597]. В растворе
жидкого аммиака в присутствии калия реакция идет при температуре — 50
-4--60° С. Продукт реакции после высушивания на воздухе и спекания под ва-
куумом при температуре 800° С представляет собой металлический титан [463].
Металлический магний при нагревании с TiCl4 восстанавливает его также до
металла [394, 397, 398]. Изобарный потенциал реакции
“Ь — TiTB + 2MgCl2^.
выражается уравнением
AZ = — 129 200 + 45,47 [кал]
(Т = 300 — 1800° К).
Амальгама цинка восстанавливает TiCl4 до TiCl3 [316], металлические титан
и алюминий при нагревании с Т1С14 до температуры кипения (136° С) —также
до TiCl8.
Водород реагирует с TiCl4 с образованием низших хлоридов при нагревании
до высокой температуры. Реакция взаимодействия TiCl4 с водородом начинается
при температуре около 500° С [46] и протекает энергично выше 1000° С [46, 288].
Гутманн и Новотный [316], действуя на TiCl4 водородом в электрическом раз-
ряде, получили на катоде из жидкого натрия металлический титановый порошок.
Четыреххлористый титан восстанавливается до низших хлоридов сплавами
титана с алюминием, молибденом, ванадием, оловом, хромом, марганцем, железом,
азотом (нитрид титана), а также загрязненным титановым порошком [35]. Карбид
титана на TiCl4 не действует. Кремний и его сплавы восстанавливают TiCl4 до ме-
талла [395]. Брикеты из смеси TiO2 + С реагируют с TiCl4 при температуре
900—1100° С с образованием низших хлоридов титана.
На солнечном свету раствор TiCl4 в эфире восстанавливается фотохимиче-
ски [472]. »
Реакции обменного разложения
Атомы хлора в TiCl4 относительно подвижны и могут замещать-
ся многими неорганическими и органическими радикалами. Про-
дукты замещения хлора в TiCl4 на органические радикалы мы от-
носим к титанорганическим соединениям. В этом разделе описаны
реакции TiCl4 только с неорганическими заместителями.
Четыреххлористый титан неустойчив к действию воды и легко
ею разлагается. При растворении TiCl4 в воде вначале образуется
молекулярно-дисперсный раствор, однако он скоро стареет и пре-
вращается в коллоидно-дисперсный [203, 204]. Реакция TiCl4 о
286
!родой сопровождается выделением большого количества тепла, теп-
ловой эффект ее 57,8 ккал!моль [556]. По наблюдениям [493], раст-
ворение TiCl4 в воде сопровождается одновременным образованием
молекулярно-дисперсного и коллоидно-дисперсного раствора.
I В результате гидролиза TiCl4 парами воды в парагазовой фазе
при температуре 25—100° С вначале образуется оксихлорид
Ti (ОН)2 С12, а затем — смесь жидкости с продуктами гидролиза
[332]. В интервале температур 150—300° С гидролиз TiCl4 в па-
рогазовой фазе идет до образования TiO2.
При растворении TiCl4 в соляной кислоте получаются оксихло-
риды титана [171, 377].
В кислороде при температуре выше 1000° С TiCl4 сгорает с об-
разованием TiO2 и С12; при красном калении продуктом окисления
?TiCl4 является оксихлорид TiO • TiOCl2 [555].
I Путин с соавторами [476] показал, что TiCl4 энергично реа-
гирует с такими окислителями, как а-нитронафталин, о-нитроани-
лин, динитроанилин, о-нитроанизол и .м-нитробензойная кислота.
Реакция между TiCl4 и о-нитрофенолом, тринитрофенолом и п-ни-
трозодиметиламином протекает со взрывом.
Метан при температуре выше 1000° С взаимодействует с TiCl4
по реакции
К TiCl4 + СН4 = TiC + 4НС1.
Равновесие этой реакции сдвинуто вправо только в присутствии
углерода, связывающего свободный титан в карбид [39].
Безводная азотная кислота с TiCl4 образует смешанный окси-
хлорид-нитрат титана TiOClNO3 [366].
д Атомы хлора в TiCl4 замещаются на группу NO3 при дейст-
вии N2O4 [470, 505] и на сульфогруппу при действии SO3 в раст-
воре хлористого сульфурила [345].
В Хлорсульфоновая кислота реагирует с TiCl4 [220] по реакции
В TiCl 4 + HOSO2C1 = TiCl3OSO2Cl + НС1.
| С фторсульфоновой кислотой получен продукт замещения двух
{атомов хлора на кислотный остаток — TiCl2 (OSO2F)2 [346].
Четыреххлористый титан вступает в реакции обменного разло-
жения с окислами магния, кальция, алюминия, железа, кремния,
редкоземельных элементов и др. [56, 149]. В результате взаимо-
действия образуются хлориды металлов и двуокись титана.
В присутствии угля продуктами реакции являются хлориды метал-
лов и TiCl3. Температура начала взаимодействия TiCl4 с окислами
металлов следующая: 380 — СаО, 320 — СеО2, 284 — La2O3,
347 — Nb2O5, 291° С — лопарит. Двуокись титана и двуокись ♦
Кремния с TiCl4 в интервале температур 400—900° С не реаги-
руют [56].
Алюмолитийгидрид Li (А1Н4)4 с TiCl4 в растворе эфира при тем-
пературе —110° С образуют нерастворимый алюмогидрид титана
Ti(AlH4)4. Продукт имеет вид белых или желтоватых кристалликов,
287
нерастворимых в эфире и устойчивых до температуры —85° С. Вы-
ше этой температуры он медленно разлагается на титан, алюминий
и водород [575, 576].
Оксихлориды и гидроксихлориды титана
Оксихлориды четырехвалентного титана получаются по реак-
циям обменного разложения между TiCl4 и окислами металлов ц
неметаллов, гидроксихлориды — при действии на TiCl4 воды.
Труст и Отефейт [555], нагревая TiCl4 с кислородом при слабо красном ка-
лении, получили оксихлорид, которому приписали состав TiO2 • TiOCL. Однако
продукты реакции, по-видимому, состояли из смеси оксихлорида TiOCl2 с дву-
окисью титана.
Лучинский [77], действуя на двуокись титана сжиженным и сухим газообраз-
ным хлористым водородом при температуре 200° С, выделил желтый порошко-
образный продукт, состав которого выражается формулой TiO2 • НС1. Водой он
разлагается до оксихлорида титана TiOCl2 • ЗН2О5. Состав этого продукта также
нуждается в уточнении.
Эрлих и Энгель [270, 271, 272] приготовили оксихлорид титана ТЮС12, дей-
ствуя на TiCl4 окислами As2O3, Sb2O3, Bi2O3, HgO, 12Об. Реакция между TiCl4
и As.2O3, Bi2O3 и Sb2O3 протекает непосредственно при растворении окислов в
жидком TiCl4 по схеме
М2О3 + 3TiCl4 -> 3TiOCl2 + 2МС13.
Осадок ТЮС12 отфильтровывался и промывался сухим пентаном.
Оксихлорид титана получен также по реакции [42, 240]
TiCl4 + Cl2O -> TiOCl2 + 2С12
при действии тихого электродного разряда на пары TiCl4 в азоте, загрязненном
кислородом [375] и в результате частичного гидролиза TiCl4 под действием воды
[206, 351].
Кристаллы TiOCl2 имеют кубическую решетку с параметрами
а = 4,51 А, ррентг ~ 2,44, Ризм — 2,45 г/см3, 2=1 [240]. Они очень
разла-
Таблица 35
Растворимость Ti ОС12
в четыреххлористом ти-
тане [272]
t, ° с Вес. % Мол. %
25 2,5 3,5
50 3,2 4,4
75 4,0 5,6
100 5,1 7,0
110 5,1 7,0
120 5,3 7,3
130 5,6 7,6
в зна-
35).
180° С
гигроскопичны, на влажном воздухе
гаются.
Оксихлорид титана растворяется
чительных количествах в TiCl4 (табл.
При нагревании до температуры
TiOCL частично диссоциирует на Ti02 (ана-
таз) и TiCl4 [240].
Оксихлорид титана вступает в реакции
присоединения, образуя, например: TiOCLX
Х2РОС13, TiOCl2 • 2C5H2N [240], TiOCLX
X2N (C2H5)4C1 [295] и др.
Коениг и Афордтен [377] и Юньис соавто-
рами [562] сообщили, что при гидролизе TiCL
под действием концентрированной соляной
кислоты получаются гидроксихлориды титана
TiOCl2 • 2C5H2N [240], TiOCLX
Ti (ОН)2С12 и Ti (ОН) С13.
Ti (ОН)3С1,
При частичном гидролизе TiCl4 образуется гидрат гидроксихло-
рида титана Ti (ОН)3С1 • 2Н2О [171 ].
288
Топтыгина и Морозов [148] изучили систему TiCl4 — НС1 —
' н2О методом растворимости и построили изотерму растворимо"
сти (табл. 36). В качестве твердых фаз в изученной части системы
авторы установили существование гидроокиси титана и двух гид-
роксихлоридов.
Таблица 36
Растворимость в системе TiCl4 — НС1 — Н2О при температуре О ° С
Состав жидкой фазы, вес. % Твердая фаза Состав жидкой фазы, вес. % Твердая фаза
тю2 НС1 TiO, НС1
0,018 \ 0,050 0,055 0,125 0,200 2,74 4,69 5,61 6,72 10,32 TiO2 • пН2О То же » » » » » ,я» 30,28 28,64 27,62 27,10 23,73 36,74 37,07 37,49 38,17 40,12 Ti (ОН)2 С12-2Н2О То же » » » » » »
0,250 0,601 0,930 12,04 14,12 16,20 » » » » » » 23,64 24,73 24,85 41,74 41,59 45,01 » » » » » »
к V Гидроокись титана находится в равновесии с разбавленными
растворами соляной кислоты. Состав ее после высушивания
над концентрированной серной кислотой выражается формулой
3TiO2.2Н2О.
\ Из растворов с концентрацией около 36% НС1 кристаллизуется
гидроксихлорид Ti (ОН)2 С12 • 2Н2О в виде прямоугольных бесцвет-
ных кристаллов с сильным двойным лучепреломлением; из более
концентрированных растворов — гидроксихлорид Ti (ОН)3 С1 х
X 2Н2О в виде одноосных шестиугольных призм. Кристаллы этой
соли, хотя и имеют одинаковый состав с гидроксихлоридом ти-
тана, полученным Аугером [171], однако отличаются от него по
внешней форме.
R I Гидроксихлориды титана при нагревании до температуры
1125° С полностью гидролизуются.
В В средней области концентраций НС1 (16—36%) при растворе-
нии TiCl4 получаются вязкие, высококонцентрированные по со-
держанию титана растворы, из которых Топтыгиной и Морозову
1148] не удалось выкристаллизовать твердые фазы.
В солянокислых растворах TiOCl2 с хлоридами рубидия, це-
зия и аммония образует двойные соли, которые устойчивы в соля-
нокислых растворах средней концентрации. В разбавленных раст-
норах они гидролизуются, а в концентрированных превращаются
в гексахлортитанаты.
Система НС! — K2TiCl6 — Н2О. Изучена методом растворимости [89 J. При со-
держании более 19% НС1 и температуры 0° С в ней кристаллизуется гексахлорти-
танат калия, двойные оксихлориды не образуются (табл. 37).
• I9 9-614 289
Таблица 37
Растворимость в системе НС! — K2TiCl6 — Н2О при
температуре 0°С
Состав жидкой фазы, вес. %
НС1 TiCl4 КС1
19,63 26,53 1,36
25,00 19,16 1,83
26,08 16,02 1,43
35,51 3,78 1,05
41,80 0,46 0,37
42,61 0,22 0,172
44,06 0,152 0,119
Твердая фаза
K2TiCl6 + КС1
То же
» »
» »
» »
» »
» »
Система НС! — RbaTiOCIe — Н2О. Кроме гексахлортитаната в ней кристалла
зуется двойной оксихлорид рубидия Rb2TiOCl4H2O (табл. 38) [89].
Оксихлортитанат рубидия — белое кристаллическое вещество.
При нагревании до 155° С отщепляет молекулу воды, превращаясь в безводную
соль Rb2TiOCl4. Последняя плавится без разложения при температуре 590° С.
Таблица 38
Растворимость в системе НС! — Rb2TiCl6 — Н2О при
температуре О°С
Состав жидкой фазы, вес. % X
Твердая фаза
НС1 TiCl4 RbCl •
Rb2TiOCl4- Н2О +
14,79 12,65 12,30 + RbCl
16,19 9,26 12,43 Rb2TiOCl4-H2O
21,39 5,57 9,50 То же
21,67 5,59 10,34 » »
24,84 3,40 6,00 » »
28,19 2,14 3,87 » »
29,90 1,61 3,00 » »
30,35 0,67 1,40 Rb2TiCl,
30,92 1,20 1,51 То же
35,52 0,031 0,04 » »
36,25 0,024 0,030 » »
42,29 0,007 0,009 » »
44,50 0,005 0,006 » »
Система НС! — Cs2TiCl6 — Н2О. Кристаллизуется оксихлортитанат состава
Cs2TiOCl4 • Н2О (табл. 39) в виде белых кристаллов с показателями преломления
Ng = 1,730, Nm = 1,722 и Np = 1,710 [89]. При температуре 200° С двойной
оксихлорид цезия теряет кристаллизационную воду, превращаясь в безводную
соль Cs2TiCl6, которая плавится без разложения при 535° С.
290
Таблица 39
Растворимость в системе НС1 — C^TiCle — Н2О при
температуре 0° С
Состав жидкой фазы, вес. % Твердая фаза
НС1 TiCl4 CsCl
10,59 7,00 17,49 CS, ТЮС14.Н2О
13,13 5,57 13,92 То же
16,77 4,23 10,58 » »
25,20 1,26 3,14 » »
25,45 1,14 2,85 » »
25,54 1,24 3,00 » »
26,16 0,99 2,47 » »
26,72 0,92 2,31 » »
27,72 0,81 2,02 » »
27,92 0,76 1,89 » »
28,33 0,74 1,86 » »
29,30 0,72 1,27 Cs9TiCle
30,05 0,54 0,95 То же
31,70 0,38 0,67 » »
32,20 0,26 0,46 » »
33,85 0,105 0,18 »
34,78 0,100 0,18 » »
35,20 0,083 0,15 %
39,97 0,024 0,04 » »
44,20 0,024 0,04 » »
44,53 0,021 0,04 »
Таблица 40
Растворимость в системе НС! — (NH4)., TiCle — Н2О при температуре
0°С [89] .
Состав жидкой фазы, вес. % Твердая фаза
НС1 TiCl4 NH4C1
17,53 9,84 3,79 NH4C1
17,82 13,23 3,80 3NH4C1 • 2TiOCl2 • 4Н2О + NH4C1
21,78 8,80 5,29 То же
22,80 6,43 5,16 » »
27,44 2,53 3,53 3NH4Cl-2TiOCl2-4H2O
28,23 2,26 2,75 То же
30,80 2,29 2,02 » »
31,15 1,74 1,80 » »
32,99 0,94 2,40 » в
35,08 0,45 1,82 » »
35,23 2,98 0,86 (NH4)2TiCle
36,32 1,25 0,70 То же
37,47 1,03 0,58 в в
38,43 0,84 0,47 в »
39,13 0,61 0,27 в »
40,27 0,47 0,23 » »
44,70 0,20 0,118 в »
291
Система НС1 — (NH4)2TiCl6 — H20. Кроме гексахлортитаната в ней кристалла-
зуется оксихлортитанат состава 3NH4C1 • 2TiOCl2 • 4Н2О (табл. 40) [89]. Криста л.
лы оксихлортитаната аммония белого цвета, Ng = 1,737, Nm = 1,704 и Np = 1,674.
При нагревании до 155° С оксихлортитанат аммония полностью гидролизуется
с образованием TiO2.
Тиохлорид титана
Эрлих и Энгель [271 ], нагревая на водяной бане смесь TiCl4 ц
As2S3, получили тиохлорид титана в виде гигроскопического ве-
щества светло-коричневого цвета. Состав его точно не установлен.
Состояние титана в солянокислых растворах
Растворение TiCl4 в солянокислых растворах может сопровож-
даться реакциями разложения и комплексообразования.
В разбавленных солянокислых растворах Т1С14 гидролизуется
до окси- или гидроксихлоридов и катионных комплексов. В кон-
центрированных солянокислых растворах образуются анионные
комплексы вследствие присоединения к TiCl4 и ТЮС12 молекул
НО с последующей диссоциацией комплекса.
Природа частиц, в виде которых титан находится в солянокис-
лых растворах, и состояние равновесия изучены еще недостаточно,
но полученные экспериментальные данные уже позволяют делать
качественные, а в отдельных случаях — и количественные выводы.
л
Соловьев и соавторы [135] полагают, что в солянокислых растворах титан
находится в форме катионов TiO2"^, адсорбирующихся на ионообменных смолах.
Алимарин с соавторами [4,8] показал, что в 0,1—0,02-н. НС1 титан образует
катионные формы, поглощающиеся катионитом, в концентрированных солянокис-
лых растворах — анионные комплексы, поглощающиеся анионитами [399].
Набиванец [101 ] методом электромиграции установил, что в растворах с кон-
центрацией менее 2-н. НС1 титан находится главным образом в форме катиона.
Анионные комплексы существуют в растворах с концентрацией более 10-н. НС1
и более 0,01 моля!л TiCl4.
Цитович [158], исследовавшая распределение титана между раствором и ионо-
обменными смолами, считает, что катионные комплексы титана присутствуют в
растворах с концентрацией менее 0,1—0,5-н.НС1, а анионные — при концентрации
НС1 более 8-н. Из растворов с концентрацией НС1 более 1-н. титан на смолах КУ-1»
КУ-2 и СБС не сорбируется. Эго указывает на то, что титан в солянокислых раст-
ворах средней концентрации находится в виде нейтральных частиц, например,
недиссоциированных молекул TiOCl2, или в форме анионных комплексов [157].
Набиванец [102] по количеству ионов водорода, выделяющихся в результате
поглощения титана катионитом в водородной форме, а также по влиянию кониен-
трации Нойонов на сорбцию титана катионитами сделал вывод, что при pH
в растворе хлоридов доминируют катионы ТЮ2+ а при pH 1,4—1,6 — однова-
лентные катионы TiO (ОН)^~.
Стромберг и Картушинская [132, 133] методом полярографии установил
существование в солянокислых растворах ионов Ti (ОН)^. В электродном пр°'
цессе Ti4+
Ti3-^ в растворе НС! принимают участие комплексы Ti (OH)Ci
292
Бано и Кавасита [16] на смоле Дауэкс-50 наблюдали адсорбцию из водно-____
* спиртовых растворов ионов титана с двойным положительным зарядом. Жуков и
<П1азаров 140J нашли, что при pH 0,65 из 0,025-М. хлоридного раствора титан сор-
бируется на смоле КУ-1 в виде двухзарядных ионов TiO2+, а при pH < 0,32 —
Б виде ионов Ti4+.
Ковалевский [376] исследовал растворы TiCl4 в концентрированной соляной
кислоте методом электропроводности и установил существование в них ионов
Румпф [494] тщательно изучал спектры комбинационного рассеивания
растворов TiCl4 и на основании аналогии их со спектрами комбинационного рас-
сеивания H2SnCl6 заключил, что в концентрированных растворах соляной
кислоты титан находится в виде гексахлортитановой кислоты H2TiCl6.
Морозов и Топтыгина [92] изучили упругость пара НС1 над солянокислыми
растворами TiCl4 при температуре 0 и 25° С. Присутствие TiCl4 в растворе повышает
упругость пара НС1, что может быть следствием повышения концентрации НС1 в
растворе из-за гидролиза четыреххлористого титана и гидратации ионов
Добавление к раствору TiCl4 в концентрированной соляной кислоте хлорис-
того аммония не вызывает заметного повышения упругости пара НС1 из-за обра-
зования (NH4)2TiCl6. Из этого авторы заключают, что в концентрированных со-
лянокислых растворах титан находится в виде иона TiCl|—. Морозов и Топтыгина
[89] предполагают, что в разбавленных растворах титан находится в форме иона
TiJOH)2f , а в концентрированных солянокислых — образуются комплексные
гйоны [Ti (ОН)2С14]2~ и [TiClel2-.
Старцев с соавторами [130] установил методом изомолярных серий, что три-
бутилфосфат экстрагирует из солянокислых растворов хлорид титана и соляную
кислоту в отношении TiCl4 : НС1 = 1:2.
Пероксохлортитанаты
I Пероксохлортитанаты образуются при добавлении перекиси во-
дорода к растворам хлоридов титана.
Никольсон и Рейтер [444], приливая к раствору TiCl4 в этилацетате без-
водную перекись водорода, выделили оранжевые кристаллы с отношением
|Ti :Ci: Н2О2 =1:1:1. Массужели и Пантанелли [414, 415], добавляя к раствору
|:TiCi4B соляной кислоте перекись водорода и хлористого натрия, получили перок-
;сититанат натрия Na2 [TiO2Cl4] • 9Н2О.
БРОМИДЫ ТИТАНА
Известны четыре бромида титана — TiBr, TiBr2, TiBr3 и TiBr4.
Монобромид титана изучен еще недостаточно.
V Бромиды титана разной валентности в системе Ti — TiBr4 на-
ходятся между собой в состоянии термодинамического равновесия.
Юнг и Шумб [598] это равновесие выражают уравнениями
И 2TiBr3 = 2TiBr2 + TiBr4,
2TiBr2 = Ti + TiBr4.
। Трибромид титана разлагается по вышеприведенному уравнению
при нагревании под вакуумом до температуры 400° С. В атмосфере
^Водорода TiBr4 возгоняется над твердым TiBr2 при температуре
1280- -380° С.
• 293
Дибромид титана начинает разлагаться на металлический тц,
тан и TiBr4 при 500° С. Выше 600° С разложение TiBr2 протекает
с большой скоростью.
Измерения упругости пара показывают, что металлический титан
начинает реагировать с жидким бромом при температуре 20° с,
конечным продуктом окисления является TiBr4 [152]. Эта реакция
может, однако, протекать ступенчато. ^Вначале получается TiBr?,
который в присутствии избытка TiBr4 при температуре ниже 350° С
дает TiBr3; при избытке металлического титана TiBr2 реагирует
с образованием монобромида TiBr. 'i
Диспропорционирование низших бромидов титана Фунаки с со-
авторами [151 Г представляет в виде следующей схемы:
1 - 2TiBr3TB = TiBr2TB + TiBr4ra3,
log P = — 3000/7 + 6,3 [мм pm. cm.],
&F = 137 100— 15,63Г [кал на 2 моля TiBr3];
2. 3TiBr2TB = 2TiBrTB 4- TiBr4ra3,
log 7 = — 5140/7 + 8,38 [мм pm. cm.],
AF = 23490— 25,147 [кал на 3 моля TiBr2];
3. 4TiBrTB ~ 3TiTB + TiBr4 ,
log P — — 8350/7 + 9,13 [mm pm. cm.],
&F — 38 160 — 28,967 [кал на 4 моля TiBrJ.
Щукарев с соавторами [163] диспропорционирование низших
бромидов титана характеризует"следующими данными:
1. Т1Вг2ти = 1/2TiBr4ra3 4- VaCC-TixB,
log F — — 3200/7 + 4,65 [мм рт. ст.]
(7 = 750 — 900° С);
2. TiBr^ = 7aTiBr4ra3 + ViTiBr^,
log P = — 6400/7 + 9,97 [мм pm. cm.]
(7 = 700 — 850° C).
Холл и Блохер [490] для реакции диспропорционирования
2TiBr3TB = TiBr4ra3 + TiBr2™
нашли log Р = — 8930/7 4* 11,6 [мм рт. ст.]
(7 = 720 — 870° К).
Авторы по зависимости упругости пара от состава TiBra — TiBfs
установили, что дибромид и трибромид титана образуют между со-
шбой твердые растворы.
294
I
Приведенные термодинамические данные различных авторов,
котя и различаются между собой, позволяют определить направ-
ление процесса диспропорционирования низших бромидов титана
L выбрать условия для получения желаемого продукта.
I ДВУБРОМИСТЫЙ ТИТАН
Двубромистый титан получен по реакции диспропорционирования (598]
К 7 2TiBr3 = TiBr2 + TiBr4.
Он образуется также из металлического титана и брома в эвакуированной
трубке при комнатной температуре [390].
Шукарев с соавторами [163] дибромид титана получил восстановлением TiBr4
металлическим титаном. Для этого смесь брома с порошкообразным металлическим
титаном, взятыми в стехиометрических количествах, нагревали в течение двух
г.
недель при температуре 500° С и вакууме
5 - 10~5лм£ рт. ст. Примесь TiBr3 отго-
няли при 470° С.
Рис.|77. Диаграмма состояния
системы RbBr — TiBr2 [163].
Рис. 78. Диаграмма состояния
системы CsBr — TiBr2 [163].
1'
Л
Двубромистый титан — черный порошок с удельным весом 4,31,
кудельная магнитная восприимчивость его 3,1 • 10~ь при 15° С
390]. На воздухе воспламеняется. В воде растворяется с выделе-
f нием водорода. В атмосфере водорода при температуре 500° С с
( Парами брома получается TiBr3. С безводным бромистым водоро-
дом при температуре 160° С также дает TiBr3.
f Энтальпия образования TiBr2 из Вгж и а-Птв Д#298 =
= —95,3 ккал/моль [347].
295
Двубромистый титан проявляет способность вступать в реак-
ции присоединения с образованием двойных солей и комплексных
соединений, но изучены они еще недостаточно.
Щукарев с соавторами [163] исследовал системы RbBr — TiBr2 и CsBr — TiBr,
методом термического анализа и установил существование в них конгруэнтно пла-
вящихся соединений RbTiBr3 и CsTiBr3 (рис. 77, 78). Эти соединения были полу-
чены также и препаративным методом при нагревании эквимолекулярной смеси
титана с бромистым рубидием при температуре 830° С и бромистым цезием при
температуре 910° С в течение 2 ч. Двойной бромид титана и рубидия — вещество
серо-черного цвета, цезиевая соль — коричнево-черного цвета. Энтальпии об-
разования двойных бромидов равны —211,8 (RbTiBr3) и —222,6 ккал!моль
(CsTiBr3).
Как и TiBr2, двойные бромиды титана с рубидием и цезием раст-
воряются в воде и кислотах с выделением водорода.
ТРЕХБРОМИСТЫЙ ТИТАН
Методы препаративного синтеза трехбромистого титана разра-
ботаны Юнгом и Шумбом [598] и Щукаревым с соавторами [163].
Трехбромистый титан образуется при восстановлении TiBr4 металлическим
титаном или бромистым водородом [598]. Металлический титане парами TiBr4 при
температуре 500° С реагирует медленно. В атмосфере водорода реакция идет
^достаточно энергично уже при 280—380° С. Сухой бромистый водород восстанавли-
вает TiBr4 доТ1Вг3 при 160° С. Водород реагирует с TiBr4 при более высокой
температуре. Обычно восстановление TiBr4 водородом проводится при пропускании
смеси паров TiBr4 + Н2 через трубку, нагретую до 750° С. На холодном конце
трубки при температуре 250° С образуются темно-коричневые кристаллы TiBr3.
Трехбромистый титан получается также при нагревании в течение двухГнедель
до температуры 500° С порошкообразного металлического титана с TiBr4 в запа-
янной ампуле под вакуумом 5 • 10”“5 мм рт. ст. [163]. Тетрабромид титана берут
с небольшим избытком по сравнению со стехиометрическим количеством. Избы-
ток его отгоняется после окончания опыта при нагревании ампулы до температуры
200° С под вакуумом. В результате получались листочкообразные сверкающие
кристаллы черно-синего цвета с зеленоватым оттенком.
Трибромид титана существует в виде двух модификаций: гек-
сагональных листочкообразных и игловидных кристаллов [598].
Структура их методами рентгеноструктурного анализа еще не изу-
чена и физические свойства измерялись безотносительно к опре-
деленной полиморфной модификации.
Удельный вес TiBr3 4,18 [267]. По данным [348], энталь-
пия образования Т1Вг3тв ДН298 = —130,6 ккал/моль, S298 =
= 43,4 кал!моль • град. Щукарев с соавторами [162] определили
энтальпию образования TiBr3, равную — 104,5 ккал!молъ. Удель-
ная магнитная восприимчивость TiBr3 при температуре 90° К
0,4 • 10“6 возрастает до 2,01 • 10“6 при 213° К и затем убывает
до 1,43 • 10~6 при 44° К [391].
296
В результате электролитического восстановления TiBr4 в раст-
воре бромистоводородной кислоты образуется гексагидрат трибро-
мида титана TiBr3 • 6Н2О [460,
545]. Кристаллы этой соли
окрашены в светло-фиолето-
вый цвет, плавятся в кристал-
лизационной воде при темпе-
ратуре 165° С. На воздухе
дымят и разлагаются, приоб-
ретая коричневую окраску.
Растворы TiBr3 • 6Н2О в кон-
центр ированной бр омистово-
дородной кислоте имеют фио-
летовую окраску. Разбавлен-
ные бромистоводородные рас-
творы окрашены трибромидом
титана в светло-коричневый
цвет. Кристаллы TiBr3 • 6Н2СН
растворяются в метаноле, эта- V
Рис. 79. Диаграмма состояния системы ноле и ацетоне, нерастворимы
КВг —TiBr3 [267]. ----
Трехбромистый титан вступает
плексообразования) и обменного
реакции еще недостаточно.
~в эфире, четыреххлористом
углероде и бензоле.
в реакции присоединения (ком-
разложения, но изучены эти
Рис. 80. Диаграмма состояния си-
стемы RbBr — TiBr3.
Рис. 81. Диаграмма состояния си-
' стемы CsBr — TiBr3.
Получены комплексы трехбромистого титана типа TiBr3 • 3RCN, где R =
СН3, СаНб, н-СэН7 [234] и TiBr3 • 4А1с, где А1с — изопропиловый и вторбу-*
^иловый спирты и циклогексанол [549]. ~ ~~ ~~
297
Известны соединения трехбромистого титана с тетрагидрофураном TiBr3 у
X 3 (СН2)4О [549] и этилендиамином TiBr3 • 4C2H8N2 [263].
Изучена система КВг — TiBr3 методом термического анализа и построена диа,
грамма состояния (рис. 79). В ней обнаружено соединение K3TiBr6.
Щукарев с соавторами [163] построил диаграммы состояния систем RbBr^
TiBr3 и CsBr — TiBr3 (рис. 80, 81). В этих системах образуются двойные бромиды
титана с бромидами рубидия и цезия, имеющие состав Rb3TiBr6, Cs3TiBrr
Rb3Ti2Br9 и Cs3Ti2Br9.
Двойные бромиды рубидия и цезия Rb3TiBr6 и Cs3TiBr6 синтезированы также
и препаративным методом [162] нагреванием стехиометрических количеств бро-
мидов в эвакуированной трубке при температуре 700° С в течение двух дней. Крис-
таллы их окрашены в красно-оранжевый цвет.
Соли Rb3Ti2Br9 и Cs3Ti2Br9 приготовлены нагреванием стехиометрических
количеств бромидов в эвакуированной трубке при температурах 680 и 750° С со-
ответственно. Кристаллы этих солей шоколадно-коричневого цвета. Авторы опре-
делили для двойных бромидов титана ДЯ298— —455,1 (Rb3TiBr6),—463,1 (Cs3TiBr6.t
—606*4 (Rb3Ti2Br9) и —623,0 ккал!моль (Cs3Ti2Br9).
В табл. 41 приведены параметры кристаллических решеток
двойных бромидов титана.
Таблица 41
Параметры кристаллической решетки двойных бромидов
титана с рубидием и цезием [54]
Формула о a, A о c, A z Фед. гр.
RbTiBr3 7,46 6,08 2 РЪ31ттс
CsTiBr3 7,64 6,16 2 Ptytnmc
Rb3TiBr6 7,26 11,64 2 Ptymmc
Cs3TiBr6 7,60 12,04 2 РЪ31ттс
Rb2TiBr6 10,39 1 4 ———
Cs2TiBr6 10,57 — 4 —-
Rb3Ti2Br9 7,50 18,36 2 РЪ31ттс
Cs3Ti2Br9 7,58 18,51 2 P^z/mtnc
> ЧЕТЫРЕХБРОМИСТЫЙ ТИТАН
Способы получения
Способы получения тетрабромида титана обычно сводятся к
бромированию различных титановых соединений и металлическо-
го: титана элементарным бромом.
‘ Дуппа [242] получал TiBr4 нагреванием смеси двуокиси титана с углем до
красного каления в токе паров брома и двуокиси углерода.
Бердоносов с соавторами [11] бромировал элементарным бромом смесь дву-
окиси титана с сахарным углем и очищал TiBr4 перегонкой при температуре 230°С-
Листер и Суттон [408] и Бердоносов с соавторами [11] бромировали смесь
окиси титана TiO с сахарным углем в токе газообразных Br2 -R СО2.
Штелер [545], Руфф и Айзнер [519], Олсон и Риян [460] и Торпе [353] получа-
ли TiBr4 бромированием карбида титана.
Бильтц и Кеунекке [211] синтезировали TiBr4 по реакции обменного разло*
жения между четыреххлористым титаном и бромистым водородом в результат6
смешения их при температуре —50° С. Реакция протекает очень энергично с вы-
298
^Е
[делением газообразного хлористого водорода! Эту реакцию некоторые авторы [186,
г4б0, 553] проводили пропусканием паров бромистого водорода через жидкий че-
Еырех^лористый титан. Образующийся TiBr4 выкристаллизовывался из раствора.
Муассан [429] получал TiBr4 бромированием металлического титана парооб-
разным бромом при температуре 360° С, Бекстер и Батлер [205] —нагреванием ,
i титана втоке паров брома с гелием, а Масажер и Бустело [431 ] — в растворе эфира. <
-При этом кристаллизовался гексабромтитанат водорода H2TiBr^ ———~~
Щукарев с соавторами [163] бромировал металлический титан в запаянной
’ «трубке. По их наблюдениям бром с металлическим_титаном реагирует очень бурно. /
Поэтому реакция проводилась в аппарате специальной 1к6н^укцйи.“ДЗн состоял
уз реакционной трубки, в которую помещался металлический титан, и отростка с
замороженным бромом, припаянного к трубке. Трубка откачивалась и запаивалась.
Затем та часть ее, в которой находился титан, нагревалась до температуры 300° С,
а бром в отростке постепенно размораживался. Пары брома, реагируя с титаном,
давали TiBr4, кристаллизовавшийся на холодных внутренних стенках трубки.
Н Четырехбромистый титан от примесей очищают многократной
перегонкой [186, 205, 460]. Хассель и Крингштад [333] выделили
большие кристаллы TiBr4 из раствора в сероуглероде.
Физические свойства
Молекула TiBr4 в газообразном состоянии имеет тетраэдриче- }
ское строение [408]. Длина связи Ti — Вг равна 2,31 А.
ж При комнатной температуре TiBr4 — кристаллическое вещество
лимонно-желтого цвета. Температура плавления Т1Вг4, по данным
различных авторов, следующая: 38,2 [186], 38,5 [209], 37,0 [408],
39,5 [2901 и 38,2° С [350], температура кипения 230 [186, 205, 209]
и 228q С [460].
Т Кристаллы TiBr4 имеют форму октаэдров, параметр решетки
а = 14,250 A, z = 8, ррентг = 3,406 г/см3 [333], d4° = 3,254 [460]
и d? = 3,370 [389]. '
В ультрафиолетовой части спектра поглощения TiBr4 обнару-
|жены три полосы, границы окоторых со стороны длинных волн
[ежат при 3760, 3060 и 4290 А [541 ].
Теплота диссоциации TiBr4 по реакции
TiBr4 + hv - TiBr3 + Вг,
ычисленная из границы полосы поглощения в ультрафиолетовой
бласти, равна 76 ккал/моль [541].
Давление пара TiBr4 [378] в интервале температур 131—206° С
писывается уравнением
log Р = — 2501/7" + 7,8083 [мм рт. ст.}.
Вычисленная из этого уравнения температура кипения TiBr4
Тфи давлении 760 мм рт. ст. равна 234° С.
к Фунаки с соавторами [152] определил давление пара TiBr4 в
^Интервале температур 112—220° С и выразил зависимость его от
Температуры уравнением
В; logP = — 2258/7" + 2,38 [мм рт. ст.}.
299
Из этой зависимости вычислена температура кипения TiBr4 230°с
и определена энтальпия испарения 10,81 ккал/моль. В интервале
температур 336—486° К зависимость упругости пара TiBr4 от тем-
пературы выражается уравнением [350]
logP- —3707,29/7 —6,2424 log 7+24,119 [атм\.
Температура кипения TiBr4, вычисленная из этого уравнения,
составляет 233,4° С.
Энтальпия образования Т1Вг4тв из TiTB и Вг2ж Д/У^в = —147,40
[443] и —162,7 ккал/моль [162].
Теплоемкость TiBr4 при температуре 299° К Ср = 24,0 кал/моль.
Энтропия TiBr4 S298 определена равной 95,02 [243] и
57,6 кал/моль • град [350]. Теки [560] вычислил стандартную эн-
тропию TiBr4 из спектральных данных 94,61 кал/моль • град.
Дьякграаф [247] изучил электронные спектры поглощения TiBr4.
Химические свойства
По химическим свойствам четырехбромистый титан проявляет
большое сходство с TiCl4. Он вступает в реакции комплексообра-
зования со многими неоргани-
ческими и органическими ве-
ществами с донорными свой-
ствами и способен замещать
часть или все атомы брома на
различные радикалы.
Реакции присоединения
Руфф и Айзнер [519], смешивая
твердый TiBr4 с жидким аммиаком,
получили соединение Т1Вг4 • 8NH3
красно-коричневого цвета. Этот ам-
миакат при температуре около
270° С в вакууме разлагается с об-
разованием тяжелого черноватого
порошка бромнитрида титана TiNBr-
Бромнитрид титана имеет струк-
туру типа FeQCl с ромбической
решеткой, параметры которой а =
=3,92, 6 = 3,34, с = 8,33 А [369].
При длительном нагревании
TiNBr разлагается, превращаясь в
нитрид
8TiNBr =
- 6TiN + 2TiBr4 + N2.
Рис. 82. диаграмма состояния системы В жидком состоянии TiNBr ре-
SO2 — TiBr4 [186]. агирует с амидом калия, образуя
Ti (NH)NK [291, 518].
С гидразином и 1,1'-диметилгидразином четырехбромистый титан дает про-
дукты присоединения TiBr4 • 2NH2NH2 и TiBr4 • 4NH2N (СН3)2 [445].
300
Прасанд и Сривастава [480] получили продукт присоединения мочевины к
.четырехбромистому титану TiBr4 • 4СО (NH2)2. Построена диаграмма состояния
Системы SO2 — TiBr4 (рис. 82). В ней образуются две несмешивающиеся жидкости,
твердая фаза состоит из сольвата 2TiBr4 . SO2 [185].
Известны соединения тетрабромида титана с фосфином — TiBr4 • 2РН3,
TiBr4 • РН3 [338] и трехбромистым фосфором — TiBr4 • 2РВг3 [510]. Последний
образует кристаллы розового цвета ромбической системы с параметрами решетки
13,84, Ь = 8,61 ис = 7,24 А [533].
Фоульс и Нихольс [283] смешением растворов Т1Вг4 и NH4Br в жидком бро-
ме получили гексабромтитанат аммония (NH4)2TiBr6. Он также образуется при
Рис. 83.
добавлении бромистого аммония к
раствору тетрабромида титана в кон-
центрированной бромистоводород-
ной кислоте [501] и в безводном
бромистом водороде [283].
Уатанейбл и Атой [570] синтези-
ровали соединение [Со (NH3)6] TiBr6.
у/ Скаглиарини и Монти [542]
смешением раствора TiBr4 в СН2С12
с гексаметилентетрамином (уротро-
пином) на холоду получили соеди-
нение TiBr4 - 7C6H12N4.
Четырехбромистый титан при- *
соединяет триметиламин, образуя
TiBr4 • N (СН3)3 [298]. Вторичный
этиловый амин в растворе бромисто-
водородной кислоты дает гексабро-
мид титана [NH (CH3CH2)2]2TiBr6
1339]. Фоульс и Нихольс[283] сме-
шением раствора TiBr4 в абсолют-
ном спирте с растворами пиридина
и хинолина в этом же растворителе
после насыщения газообразным бро-
мистым водородом выкристаллизо-
вали гексабромиды титана (C5H5N)2TiBr6, (C9H7N)2TiBr6 и смешанный
логенид титана (C5H5NH)2TiCl4Br2. Кристаллы (C5H5N)2TiBr6 выделил Рао [498].
Синтезированы соединения TiBr4 • 2HCN и TiBr4 • 2CNBr, а также эфират
TiBr4 • (СН3СН2)2О [464].
Методом изомолярных серий установлено образование соединения TiBr4 X
X СНС13 [11]. >
Получены комплексы TiBr4 с N,N'-диметил формами дом и М,Ы'-дифенилфор-
мамидом состава TiBr4 • 2L [405].’
о-Фениленбисдиметиларсин [R] с четырехбромистым титаном дает кристал-
лы оранжевого цвета состава TiBr4 • R и коричневого цвета состава TiBr4 • 2R
1229].
системы
Диаграмма состояния
TiBr4 — Вг2 [209].
гексага-
Известны также соединения TiBr4 с пиразином TiBr4 . 2N (CH)4N [296], ди-
оксаном TiBr4 • (СН2СН2)2О и TiBr4 • (СН2СН2)2О [508], уксусноизопропиловым
эфиром TiBr4 • СН3СООС3Н7-нзо-[565], нитрилами TiBr4 • 2CH3CN, TiBr4 •
- 2C2H5CN, TiBr4 • 2C6H5CN и с пиридином TiBr4 • 2C5H5N [262].
Бекер и Фоульс [198] синтезировали продукты присоединения к TiBr4
производных серы: TiBr4 • 2 (CH3)2S, TiBr4 • 2 (C2H5)2S, TiBr4 • 2C4H8S,
TiBr4 • (w-C3H7)2S, TiBr4 • C4H8S2CHoSCH3, TiBr4 • C6H5SCH2CH2SC6H5, TiBr4 •
4QH5)2—PSPS(C2H5)2, Джайн и Ривест [372] — TiBr4 • 2C2H5SCN и TiBr4 •
• NCS(CH2)2SCN.
Щукарев с соавторами [162] получил двойные бромиды титана с бромидами
рубидия и цезия Rb2TiBr6 и Cs2TiBr6— при нагревании смеси TiBr4 с эквивален-
тными количествами бромидов в эвакуированной трубке при температуре
•520—530°С в течение двух недель. Энтальпия образования двойных бромидов
ДЯО298 = —386,3 (Rb2TiBr6) и —392,9 ккал!моль (Cs2TiBr6).
301
Система тетрабромида титана с бромом эвтектического типа
(рис. 83).
Реакции разложения
Под действием влаги TiBr4 легко гидролизуется до TiO2 и НВг
[242]. В воде растворяется с выделением тепла, растворы про.
зрачные [553].
При нагревании паров TiBr4 на воздухе до температуры 800° с
он сгорает, давая TiO2 и бром [150].
Азотная кислота разлагает TiBr4 с выделением NO2 и Вг2 и
переводит титан в раствор в виде нитрата [460].
Розенгейм и Шютте [501 ], растворяя гидроокись титана в бро-
мистоводородной кислоте, выделили белый осадок гидроксиброми-
да титана Ti (ОН)3Вг. Из раствора в абсолютном спирте полу-
чен гидрат Ti (ОН)3Вг • 1,5 Н2О, который образуется также и при
упаривании раствора четырёххлористого титана в НВг над металли-
ческим натрием [581 ].
Оксибромиды титана не изучены. Розенгейм и Шютте [501 ] дей-
ствием на спиртовый раствор TiBr4 бромидом пиридина синтезирова-
ли продукт соединения титана с оксибромидом TiOBr2-3C6H5NHBr.
Стромберг и Кдртушинская [133] показали методом полярогра-
фии, что в растворе бромистоводородной кислоты преобладают
комплексы Ti (ОН) Вг+ и Ti3^ а в электродной реакции прини-
мают участие комплексы TiBr3+ и TiBr2+.
ИОДИДЫ ТИТАНА
Иод с титаном, как и другие галогены, образует соединения,,
в которых титан проявляет валентность 2, 3 и 4. Йодиды титана
в системе Ti — Til4 находятся в состоянии термодинамического
равновесия, характеризующегося следующими уравнениями:
2TiI3 = Til2 + Til4,
2TiI2 = Ti + Til4.
Равновесие между иодидами титана изучали Фаст [292] и Фунаки
с соавторами [151]. По данным [292], в результате взаимодействия
металлического титана с иодом при комнатной температуре полу-
чается смесь трех иодидов. Иодид Til3 разлагается по вышеприве-
денной реакции при 350° С, Til4 восстанавливается металличе-
ским титаном до Til2 при температуре около 550° С.
Фунаки с соавторами [151] предложил иную схему равновесия
между иодидами, включающую образование Til. По его данным,
металлический титан при температуре выше 400° С реагирует с те-
траиодидом по реакции
Ti + Til4 = 2TiI2.
302
В случае недостатка Til4 при температуре выше 300° С образу-
ется трехиодистый титан
| TiTB+ ТП4газ -> TiI3TB .
или одноиодистый титан
i TiTB + ТП2тв ТПтв.
. ^Наряду с этим протекают реакции диспропорционирования по
следующей схеме:
1.2TiL =TiI2 + Tib ,
отв ^тв * 4газ’
К log 7 == 227QIT + 6,16 [мм рт. ст.},
AF = 12 700— 15,07 [кал на 2 моля Til3];
К 2. ЗТП2тв =. 3TiITB + Т114тв,
log Р = — + 6,24 [мм рт. ст.],
К AF = 14800— 15,47 [кал на 3 моля Til2];
I 3. 4TiITB — 3TiTB + Ti4ra3,
log 7 =»— 10690/7 + 11,02 [мм pm. cm. 1,
В AF = 48 900 — 37,37 [кал на 4 моля Til],
Однако в основу этой схемы положено образование при дис-
пропорционировании одноиодистого титана, индивидуальность ко-
7торого нуждается в экспериментальной проверке.
I ДВУИОДИСТЫЙ ТИТАН
К Фаст [292] для получения двуиодистого титана рекомендует
|вначале готовить смесь трех иодидов, а затем нагревать ее при
[температуре 440° С. Содержащийся в этой смеси Til3 разлагается
на Til2 и Til4. Последний можно отогнать и получить в' остатке
чистый Til2.
Клемм и Гримм [390] получили Til2 из металлического титана
|И элементарного иода нагреванием в бомбе.
1 Двуиодистый титан образуется при нагревании смеси паров TiBr4
V ртути в атмосфере водорода [244], нагревании TiCl4 с иодистым
жали ем [352].
Двуиодистый титан получается в виде черных блестящих крис-
таллов в форме лепестков [244]. Большие кристаллы имеют ме-
таллический блеск и темно-коричневую окраску [292].
I Известны две кристаллические модификации Til2. У одной
гз них структура СсП2 с параметрами решетки а = 4,4, с ~ 6,82 А,
грентг = 4,99 г!см3 [390]. Эта модификация парамагнитна, удель-
ная магнитная восприимчивость ее 5,5 • 10~6 (15° С) [390]. Свойст-
ва другой модификации еще не изучены.
I 303
Двуиодистый титан очень гигроскопичное вещество, на воздуХе
легко разлагается, энергично реагирует с водой с выделением во,
дорода и образованием пурпурного раствора Ti3+ [292].
Водород при температуре белого каления восстанавливает Til
до металла. В атмосфере кислорода Til2 при температуре красного
каления сгорает до TiO2. Хлор при слабом нагревании хлорирует
Til2 до TiCl4.
Азотная кислота окисляет Til2 на холоду с выделением иода.
. Солянокислый раствор Til2 окрашен в голубой цвет, аммиак из
4 него осаждает гидроокись титана Ti (ОН)3.
Нерастворим в безводных спиртах, эфире, хлороформе, четьь
реххлористом углероде и бензоле [244].
ТРЕХИОДИСТЫЙ ТИТАН
Фаст [292] рекомендует получать трехиодистый титан нагре-
ванием эквимолярной смеси Til2 + Til 4 при температуре около
700° С, Клемм и Крозе [391 ] — смеси металлических титана и иода,
взятых в стехиометрическом отношении.
Кристаллы Til3 темно-фиолетового цвета с металлическим блес-
ком, растворяются в воде с выделением водорода вследствие час-
тичного окисления титана. Из раствора в иодистоводородной кис-
лоте выделены фиолетовые кристаллы гексагидрата Til3 • 6Н2О
[1, 545]. На воздухе они дымят и разлагаются.
Раствор Til3 • 6Н2О в иодистоводородной кислоте, как и соот-
ветствующего хлорида, окрашен в фиолетовый или зеленый цвет.
Зеленый раствор, в котором титан находится в форме ионов
[ТП„ (Н2О)б_п13-л, дает сигнал ЭПР. Ионы фиолетовой модифи-
j кации [Ti (Н2О)6]3+ сигнала ЭПР не обнаруживают.
Получен гексакомплекс с мочевиной Ti [CO(NH2)21613 [3571.
ЧЕТЫРЕХИОДИСТЫЙ ТИТАН
Четырехиодистый титан впервые получил Вебер [592] нагре-
ванием металлического титана с иодом, а затем Муассан и Кемпбел
[183], иодирование титана проводили при температуре 525° С.
Синтез Til4 проводится также действием на TiCl4 сухого
[354, 361] или жидкого [211] йодистого водорода, охлажденного
до —50° С.
Четырехиодистый титан очищается от примесей перегонкой под
вакуумом [389] или в токе водорода [354].
Блюменталь и Смит [180] описали метод получения Til4, осно-
ванный на обработке сплава А1 — Ti в сероуглероде металлическим
иодом при нагревании. Образующиеся иодиды титана и алюминия
растворяются в сероуглероде, который затем отгоняется. К остатку
иодидов для связывания в труднолетучие соединения добавляется
KI, после чего отгоняется Til4.
304
4
Из расплава Til 4 вначале выделяются октаэдрические кристаллы
611. Через несколько дней они переходят в другую модификацию,
фуктура которой еще не изучена [354].
При медленной сублимации ТП4 образуются крупные кристаллы V
^мнбчфасновато-коричневого цвета;
Октаэдрические кристаллы ТП4 ~имеют кубическую структуру
типа Snl4 с параметром решетки а = 12,002 А, Ррентг = 4,27 г!см\
Б = 8 [354], удельный вес 4,40 [389].
F Измерена упругость пара Til4 в интервале температур 160—
370° С [183]. Зависимость упругости пара от температуры выра-
жается уравнением
К, logP = — 3054/Т + 7,57 [мм рт. ст.].
Из этого уравнения вычислена температура кипения Til4 при дав-
лении 760 мм рт. ст., равная 377,2° С. Теплота испарения Til4
113975 кал!молъ [183]. Температура плавления Til4 150° С [540], v
'энтропия Til4, вычисленная из спектральных данных, S°98 —
== 102,38 кал!моль • град.
К При нагревании Til4 выше 1100° С он разлагается на метал-
лический титан и элементарный иод [368]. Кинетика разложения
Til4 на нагретой вольфрамовой проволоке в интервале температур
11100—1500° С описывается уравнением реакции первого порядка
[179].
На воздухе Til4 сильно дымит, а при нагревании сгорает с об-
разованием двуокиси титана и элементарного иода; в воде раст- \
Воряется с выделением тепла и гидролизуется [361].
С аммиаком Til4 дает гексааммиакат^ТП4 • 6NH3, который при нагревании до^
(180° С превращается в нитронодид титана T1NI. Кристаллы его имеют решетку
[типа FeOCl, фед. гр. параметры решетки а = 3,94, b = 3,51, с — 8,95 А.
С гидразином Til4 в жидком аммиаке образует взрывчатый аддукт, с 1,Г-ди-
иетилгидразином — бледно-коричневые кристаллы состава Til4 • 4H2N (СН3)2
И445].
Жидкий сероводород и фосфин с ТП4 в отличие от TiCl4 и TiBr2 соединений
е дает [211, 338]. Треххлористый фосфор с Til4 вступает в реакцию обменного раз-
ожения [385].
I N, N'-Дифенил формамид (L) с Til4 образует продукт присоединения Til4 •
ГлГ (172], в растворе бромоформа с гексаметилентетрамином (уротропином)—v
единение ТП4 • 4С6Н12 • 5СНВг3 [544].
Эмелеус и Рао [262, 498] получили продукт присоединения пиридина Til4,
Имеющий состав Til4 • 4C5H5N. Соединение это неустойчиво и при температуре
№—130° С диссоциирует.
Карантасис [385] из раствора Til4 в чистом иодистом водороде выделил крис-
таллы гидрата гидрокси ио дида титана_Т1 (ОН)31 . 2Н2О^
L Пропусканием кислорода и азота через раствор Til4 в циклогексане получено
— коричневое рентгеноаморфное вещество, очень гигроскопичное [246]. у
4рй нагревании до температуры 125° С в вакууме TiOI2 разлагается на иод и*
Fioi.
Соединение TiOI — парамагнитное, гигроскопичное вещество черного цве-
R, с пиридином образует аддукт TiOI • 3C5H5N коричневого цвета.
9-614
305
Литература
1. А ввакумов В. И., Гарифьянов Н. С., СеменоваЕ. Ц
Физ. металлов и металловедение, 1961, 12, 624.
2. Австралийский пат. № 220435 от 20.03 1958 г.
3. А ба р ба р чу к И. Л., Костицына К. П.— Укр. хим. ж., 193^
19, 618.
4. Алимарин И. П., Белявская Т. А.— Труды Всесоюзного Со^
вещания по хроматографии. Изд-во АН СССР, М., 1960, 373.
5. Барабанова А. С., Войтович Б. А.— Укр. хим. ж., 1965, 31, 352.
6. Буслаев Ю. А., Давидович Р. Я., Бочкарева В. Д
Изв. АН СССР. Неорг. материалы, 1965, 1, 483.
7. Буслаев Ю. А., Бочкарева В. А., Николаев Н. С_________
Изв. АН СССР. Отд. хим. наук, 1962, 3, 388.
8. Белявская Т. А., Алимарин И. П., Колосова И. ф._
Ж. анал. хим., 1958, 13, 668.
9. Белов С. Ф., Скляренко С. И.— Цветные металлы, 1958,11,37.
10. Будневский А. М., Ли Си-чан, Чижиков Д. М., Зви-
ад а д з е Г. Н. Физическая химия расплавленных солей и шлаков. ГНТИ
М., 1962, 344.
11. Бердоносов С. С., Лапицкий А. В., Власов Л. Г. Вести.
/МГУ. Сер. II, хим., 1963, 1, 38.
12. Бирюкова Л. В.,Саксонов Ю. Г.— Ж- неорг. хим., 1960, 5, 993.
13. Бирюкова Л. В., Иванов А. И.— Труды ВАМИ, 1958, Л., 41,84.
14. Б и р ю к о в а Л. В.— ЖВХО, 1962, 1, 119.
15. Бай маков Ю. В.— Труды Всесоюзного совещания по физической хи-
мии расплавленных солей и шлаков. ГОНТИ, М., 1962, 22.
16. Боно Ю., Кавасима Т. — Repts Govt Industr. Res. Inst., Nagoya,
1964, 13, 193.
17. Баймаков Ю. В., Каменецкий M, В., Черны Ф.— Изв.
высш. уч. зав. Цветная металлургия, 1960, 2, 102.
18. Бамбуров В. Г., Фотиев А. А.— Изв. Сиб. отд. АН СССР, 11. Сер.
хим. наук, 1963, 3, 42.
19. Бамбуров В. Г., Фотиев А. А.— Труды Ин-та химии, 7. Уральский
филиал АН СССР, Свердловск, 1963, 195.
20. Бамбуров В. Г., Деменев Н. В., Полякова В. М.— Изв. Сиб.
отд. АН СССР, 1962, 4, 73.
21. Войтович Б. А., Лозовская Н. Ф.— Укр. хим. ж., 1965, 31, 1136.
22. Войтович Б. А., Звагольская Е. В.— Изв. АН СССР. Метал-
лы, 1965, 2, 72.
23. В о л ь н о в Ю. Н., Глезер П. М., Райкина И. Я.— Сборник
статей по общей химии, 2. Изд. ЖОХ, М.— Л., 1963, 976.
24. Войтович Б. А., Барабанова О. С.— ДАН УРСР, 1963, 8, 1068-
25. Войтович Б. А., Барабанова А. С.— Укр. хим. ж.,1963,29, 1264.
26. В о й т о в и ч Б. А., Барабанова А. С., Клочков В. П.,
Ш а р к и н а Э. В.— Укр. хим. ж., 1966, 32, 167.
27. Гальперин Е. Л., Сандлер Р. А. — Кристаллография, 1962-
7, 217.
28. Г а л ь п е р и н Е. Л., Ключникова Е.Ф., Пашкевич Л. А.,
Сандлер Р. А.— Труды ВАМИ, Л., 1962, 48, 164.
29. Г у р ь я н о в а Е. Н., Гольдштейн И. П., Харламо-
ва Е. Н.— В кн.: Химия неорганических соединений, содержащихся в нефтях
и нефтепродуктах, 6. «Химия», М., 1964, 261.
30. Гарифьянов Н. С., Федотов В. Н., Тимеров Р. X.— Ф113’
тв. тела, 1962, 4, 96.
31. Гарифьянов Н. С., Семенова Е. И.— ДАН СССР, 1961, И »
157.
32. Гарифьянов Н. С., Данилова А. В., Ш а г и д у л '
л и н Р. Р.— Оптика и спектроскопия, 1962, 13, 212.
306
33. Гопиенко
34. Гопиенко
35. Гопиенко
16. Гопиенко
1'37. Делимарс
795.
£
В. Г.—Труды ВАМИ, Л., 1960, 46, 159.
В. Г.— Изв. высш. уч. зав., 1963, 1, ПО.
В. Г.— Ж- прикл. хим., 1963, 36, 963.
В. Г.— Ж. прикл. хим., 1960, 33, 2600.
кий Ю. К., Чернов Р. В.—ДАН УССР, 1960, 6,
прикл. хим.,
1963, 36, 1621.
39. Е л ю т и н В. .
зав. Черная металлургия, 1963, 11,5.
/40. Жуков А. И., Назарова А. С.— Ж- неорг. хим., 1964, 9, 1465.
* 41. 3 а в а р и ц к а я Т. А., Зевакин V * XTZ -- ----
П., Пенекин Г. И., Лысов Б. С.— Изв. высш. уч.
41. Заварицка
34, 2783.
42. Заварицка
1958, 31, 503.
43. Звиададзе
АН СССР, М., 1960.
я
Зевакин И. А.— Ж- прикл. хим., 1961,
Пустовалова С. С.— Цветные металлы,
Др.— В кн.: Титан и его сплавы, 4. Изд-во
V44.
45.
46.
• 47.
48.
49.
50.
51.
/52.
53.
55^
ib •
(57.
1.
►7.
Ионова Е. А-., Головня В. А.— Ж. неорг. хим., 1966, 11, 138.
Исмайлов Х.М. и др.— ДАН Азерб. ССР, 1965, 21, 341.
Исино Т., Тамура X., Накагава О.— Zasshi J. Chem. Soc.
Japan, Industr. Chem. Sec., 1961, 64, 1344.
Колодяжный Ю. В., Осипов О. А., Каширени-
н о в О. Е.— Ж- физ. хим., 1965, 39, 1771.
Клетеник Ю, Б., Осипов О. А.—Ж-общ. хим., 1961,31,710.
Кучерявенко Н. С.— В кн.: Материалы научных конференций.
Изд-во Казанского гос. педагог, ин-та, Казань, 1963, 385.
К а з а й н А. А.— В кн.: Титан и его сплавы, 8. Изд-во АН СССР, М.,
1962, 207.
Клейнер К. Е. — Ж- общ. хим., 1952, 22, 17.
Каратаева И. И., Ванштейн Э. Е., Куценко Ю. И.—
Ж. неорг. хим., 1961, 6, 2329.
Каши ренинов О. Е.— В кн.: Материалы 3-й научной конферен-
ции аспирантов. Изд-во Ростовского ун-та, Ростов-на -Дону, 1961, 158.
Кожина И. И., Ко рол ьковД. В.— Ж- структ. хим., 1966, 6, 97.
Коган В. А., Осипов О. А., Минкин В. И., Соко-
лов В. П.— Ж- неорг. хим., 1965, 10, 83.
К е т о в А. Н., И е ч к о в с к и й В. В., Колесов И. М.— Сб.
научн. трудов Пермского политехи, ин-та, 1963, 14, 7.
Коршунов Б. Г., Ионов В. И.— Изв. высш. уч. зав. Цветная
металлургия, 1961, 1.
Коршунов Б. Г., Ионов В. И.— Изв. высш. уч. зав. Цветная
металлургия, 1961, 2, 102.
Каменецкий М. В., Костюков А. А., Попов А. Н.—
Изв. высш. уч. зав. Цветная металлургия, 1960, 1, 119.
Каменецкий М. В., Костюков А. А., Корга нов В. А.—В кн.:
Физ. химия расплавленных солей и шлаков. Металлургиздат, М., 1962, 60.
Каменецкий М. В.— Научн. докл. высшей школы. Л1еталлургия,
1958, 2, 104.
Каменецкий М. В.— Цветные металлы, 1958, 2, 39.
Каменецкий М. В., Шевляков Л. И.— Изв. высш. уч. зав.
Цветная металлургия, 1962, 3, 89.
Каменецкий М. В., Костюков А. А., Сяо Дэ-чуа н.—В кн.:
Физ. химия расплавленных солей и шлаков. Металлургиздат, М., 1962, 54.
Коломицкий Ф. М., Милов А. И., Пономарев В. Д.—
Изв. АН Каз. ССР. Сер. металлургии, обогащения и огнеупоров, 1961, 1, 26.
Коломицкий Ф. М., Пономарев В. Д.— В кн.: Научные
труды Казахского горнометаллургического ин-та, 15. Алма-Ата, 1957, 259.
Коломицкий Ф. М., Пономарев В. Д.— Бюлл. техн, инфор-
мации, МЦМ Каз. ССР, Алма-Ата, 1956, 4, 40.
307
' 68. Коломицкий Ф. М.— Изв. АН Каз. ССР. Сер. металлургии, обо
гашения и огнеупоров, 1958, 2, 7.
69. Л а с т о ч к и н а А. А., Шека И. А.— Укр. хим. ж., 1964, 30, 898
70. Л и С и - ц я н.— Acta metalurg. cinica, 1965, 8, 187.
71. Ли Си-чан, Чижиков Д. М.— Изв. АН СССР. ОТН.
гия и топливо, 1961, 5, 22.
Металл ур.
72. Л а п и д з е И. И., Ни сель сон Л. А., К а р а т а е в а А. А
Ж- неогр. хим., 1965, 10, 2372. ""
73. Л у ч и н с к и й Г. П.— Ж. физ. хим., 1935, 6, 607.
74. Л у ч и н с к и й Г. П.— Ж. общ. хим., 1936, 6, 1108.
75. Л у ч и н с к и й Г. П.— Ж. общ. хим., 1936, 6, 439.
76. Л у ч и н с к и й Г. П.— Ж- общ. хим., 1937, 7, 207.
77. Л у ч и н с к и й Г. П.— Ж. общ. хим., 1940, 10, 769.
78. Л ы с е н к о Ю. А.— Ж. общ. хим., 1956 , 26, 2963.
79. Л ы с е н к о Ю. А., О с и п о в О. А., Федосеев И. Н.— Ж. бич
хим., 1954, 28, 700.
80. Л ы с е н к о Ю. А., Осипов О. А.— Ж- общ. хим., 1954, 24, 53.
81. Л ы с е н к о Ю. А.— Ж- общ. хим., 1956 , 26, 3273.
82. Лысенко Ю. А., Осипов О. А., Акопов Е. К.— Ж. неорг.
хим., 1956, 1, 536.
83. Л ы с е н к о Ю. А.— Ж- общ. хим., 1956, 26, 3297.
-84. Лысенко Ю. А., Осипов О. А.— Ж. общ. хим., 1958, 28, 1724.
85. Л ы с е н к о Ю. А., Осипов О. А.— Ж- неорг. хим., 1961, 6, 1656.
86. Лысенко Ю. А.— Научн. зап. Луганского с.-х. ин-та, 1963, 9, 271.
87. Л ы с е н к о Ю. А., О с и п о в О. А., Кравцов Е. Е.— Ж. неорг.
хим., 1963, 8, 663.
88. М о р о з о в И. С., Топтыгин Д. Я.— Ж- неорг. хим., 1960, 5, 88.
* 89. М о р о з о в И. С., Топтыгина Г. М.— Ж. неорг. хим., 1960, 5,
2518.
90. Мо р о з о в И. С., Топтыгина Г. М.—Ж- неорг. хим., 1960,5,
1637.
91. Морозов И. С., Топтыгина Г. М., Липатова Н. П.—
Ж. неорг. хим., 1961, 6, 2528.
n 92. М о р о з о в И. С., Топтыгина Г. М.— Ж. неорг. хим., 1957, 2,
1929.
93. М а р к о в Б. Ф., Гитман Е. Б., Тишура Т. А.— Укр. хим.
ж., 1961, 27, 718.
94. М а р к о в Б. Ф., Белякова Е. П.— Укр. хим. ж., 1961, 27, 146.
95. М а р к о в Б. Ф., Тишура Т. А.— Укр. хим. ж., 1963, 29, 150.
96. Марков Б. Ф., Барабанова А. С., Войтович Б. А.—
Укр. хим. ж., 1963, 29, 1035.
97. М а р к о в Б. Ф., Войтович Б. А., Барабанова А. С.—
Укр. хим., ж., 1961, 27, 151.
98. М а р к о в Б. Ф., Чернов Р. В.— Укр. хим. ж., 1959, 25, 279.
99. Марков Б. Ф., Тишура Т. А.— В кн.: Титан и его сплавы, 9.
Изд-во АН СССР, М., 1963, 199.
100. Марков Б. Ф., Чернов Р. В.— Укр. хим. ж., 1961, 27, 34.
101. Набиванец Б. И.— Ж. неорг. хим., 1962, 7, 412.
л» 102. Набиванец Б. И.— Ж- неорг. хим., 1962, 7, 417.
103. Николаев Н. С., Буслаев Ю. А.— Ж- неорг. хим., 1959,
4, 543.
104. Нисельсон Л. А., Соколова Т. Д.— Ж- неорг. хим., 1961, 6,
1645.
105. Осипов О. А.— Ж. общ. хим., 1956, 26, 322.
106. Осипов О. А., Гайворонский В. И.— Ж- общ. хим., 1963,
33, 1346.
107. Осипов О. А., Гайворонский В. И., Швец А. А-^
Ж- неорг. хим., 1963, 8, 2190.
108. Осипов О. А. и др.— Ж- общ. хим., 1962, 32, 1368.
308
Г109. бс и п о в О. А., Кравцов Е. Е.— Сборник статей по общей химии,
t 1. Изд-во АН СССР, М.— Л., 1953, 216.
|110. Осипов О. А., Лысенко Ю. А., Акопов Е. К.— Ж- общ.
хим., 1955, 25, 249.
fill- Осипов О. А., С и ч к о в В.— Ж. общ. хим., 1952, 22, 1132.
ГЦ2. Осипов О. А., Семенов А. Д.— Ж- общ. хим., 1956, 25, 2059.
ВщЗ* О с и п о в О. А., Лысенко Ю. А.— Ж- неорг. хим., 1958, 3, 1605.
• Ц4. П у ш и н Н. А.— Гласник хемиского друштва, 1953, 18, 469.
Г115. Путилин Ю. М., Мелихов В. Д., Пономарев В. Д.—
Изв. АН Каз. ССР. Сер. металлургии, обогащения и огнеупоров, 1961, 2, 18.
116. Путилин Ю. М., Пономарев В. Д., Милов А. И.— Изв.
АН Каз. ССР. Сер. металлургии, обогащения и огнеупоров, 1961, 3, 66.
1117. Путилин Ю. М., Галузо В. Н., Пономарев В. Д.—
Труды Ин-та металлургии и обогащения АН Каз. ССР, 1962, 5, 95.
118. Путилин Ю. М. и др.— Труды Ин-та металлургии и обогащения
АН Каз. ССР, 1962, 5, 82.
.119. Памфилов А. В., Ш и х е р М. Г.— Ж- общ. хим., 1937, 7, 2760.
! 120. Памфилов А. В., Штандель Е. Г.— Ж- общ. хим., 1937,
7, 258.
121. Памфилов А. В., Худяков А. С., Штандель Е. Г.—
Ж- общ. хим., 1935, 5, 605.
'122. Рубан Н. Н., Пономарев В. Д., Виноградова К. А.—
Изв. АН Каз. ССР. Сер. металлургии, обогащения и огнеупоров, 1961, 1, 33.
123. Рубан Н. Н., Пономарев В. Д.— В кн.: Цветная металлургия.
Алма-Ата, 1962, 19.
424. Рубан Н. Н., Пономарев В. Д., Виноградова К. А.—
Труды Ин-та металлургии и обогащения Каз. ССР, 1963, 6, 22.
125. Савин В. Д., Резниченко В. А.— В кн.: Исследование ме-
таллов в жидком и твердом состоянии. «Наука», М., 1964, 41.
126. Сандлер Р. А., Подзоров Б. Н.— Ж- прикл. хим., 1966, 39, 7.
127. Склярова Р. А.— Научн. зап. Луганского с.-х. ин-та, 1963, 9, 274.
128. Савин В. А., Резниченко В. А.— В кн.: Титан и его сплавы, 9-
Изд-во АН СССР, М., 1963, 172.
1129. Сырокомский В. С. Титан и его соединения. Изд. Комиссии при
АН СССР, Л., 1926.
1130. Старцев В. Н., КрыловЕ. И., Козьмин Ю. А.— Ж. не-
орг. хим., 1965, 10, 2367.
j 131. Славинская Р. А.— Изв. АН Каз. ССР. Сер. хим., 1962, 2 (22), 26.
<432. Стромберг А. Г., Картушинская А. И.— Изв. Сиб. отд.
АН СССР, 1961, 11, 88.
133. Стромберг А. Г., Картушинская А. И.— В кн.: Физико-
химический анализ. Изд-во Сиб. отд. АН СССР, Новосибирск, 1963, 315.
134. Семенова Е. И.— ДАН СССР, 1962, 143, 1368.
1135. Соловьёв С. И., Крылов Е. И., Кононова Л. П.—
Ж. неорг. хим., 1956, 1, 660.
136. Сумарокова Т. Н., Сакенова Д. С.— Ж- общ. хим., - 1962,
32, 3.
I 137. Сумарокова Т. Н., Сакенова Д. С.— Ж- общ. хим., 1964,
34, 2696.
; 138. Скляренко С. И., Белов С. Ф.— Сборник научных трудов. Ги-
редмет, 1. Металлургиздат, М., 1959, 329.
I 139. Скляренко С. И., Белов С. Ф.— Изв. высш. уч. зав. Химия
и хим. технология, 1962, 5, 383.
' 140. Скляренко С. И., Белов С. Ф.— Изв. высш. уч. зав. Химия
и хим. технология, 1962, 3, 383.
I 141. Скляренко С. И., Белов С. Ф.— Ж. неорг. хим., 1962, 7, 1636.
/ 142. Смирнов М. В., Краснов Ю. Н.— Труды Ин-та электрохимии.
Уральский филиал АН СССР, Свердловск, 1960, 1, 23.
309
143. Смирнов М. В., Логинов Н. А.— Труды Ин-та электрохимии
Уральский филиал АН СССР, Свердловск, 1965, 6, 3.
144. Смирнов М. В., Циовк и на Л. А., Логинов Н. А^_
ДАН СССР, 1961, 136, 1388.
145. СмирновМ. В., Логинов Н. А., Циовкина Л. А.— Труды
Ин-та электрохимии. Уральский филиал АН СССР, Свердловск, 1961, 2, 2g
146. Смирнов М. В., Логинов Н. А.— Изв. Сиб. отд. АН СССР
1962, 4, 64.
147. Топтыгина Г. М., Морозов И. С.— Ж- неорг. хим., 1961
6, 1479.
148. Топтыгина Г. М., Морозов И. С.— Ж- неорг. хим., 1961
6, 1685.
149. Чижиков М. Д., Корсунская В. Н.— Изв. АН СССР. ОТН,
Металлургия и топливо, 1962, 5, 74.
150. Французский пат. № 1049140 от 28. 12. 1953 г.
151. Ф унаки К-, Ути мура К., Мацу на га X.— J. Chem. Soc.
Japan. Insutr. Chem. Sec., 1961, 64, 129.
152. Фунаки K-, Ути мура К., Ку н и я Я-— J. Chem. Soc. Japan.
Industr. Chem. Sec., 1961, 64, 11, 1914.
153. Хромов А. Д., К а й з а н A. A.— В кн.: Титан и его сплавы, 4.
Изд-во АН СССР, М., 1960.
154. Харитонов Ю. Я., Буслаев Ю. А.— Изв. АН СССР. Отд.
хим. наук, 1962, 3, 393.
155. Харитонов Ю. Я-, Буслаев Ю. А.— Изв. АН СССР. Сер.
хим., 1964, 5, 808.
156. Цеховольская Д. И.— Ж. неорг. хим., 1964, 9, 1387.
157. Цитович И. К.—ЖВХО, 1961, 6, 233.
158. Цитович И. К.— ДАН СССР, 1959, 128, 970.
159. Чернов Р. В., Делимарский Ю. К-— Ж- неорг. хим., 1961,
6, 2749.
160. Чижиков Д. М. и др.— Цветные металлы, 1960, 12, 42.
161. Шевченко Ф.Д., Ахрамеева T.I.— Bichhk Китського ун-ту 4.
Сер. астрон., ф!з., х!м., 1961, 1, 92.
162. Щукарев С. А., Василькова И. В., Корольков Д. В.—
Ж. неорг. хим., 1964, 9, 1810.
163. Щукарев С. А., Василькова И. В., Корольков Д. В.—
Ж. неорг. хим., 1963, 8, 1933.
164. Aotani К., Hasegawa К., Migaza wa Y.— J. Electrochem.
Soc. (Japan), 1958, 27, E34.
165. Aotani К., Koizumi T.— J. Electrochem. Soc. (Japan), 1958,26, El 10.
166. A r 1 m a n E. J., Jong J. R., Beintema J., Reijen L. L.—
Rec. trav. chim., 1961, 80, 1129.
167. A r 1 m a n E. J., Jong J. R.— Rec. trav. chim., 1960, 79, 910.
168. Altman D., Farber M., Masson D. M.— J. Chem. Phys., 1956,
25, 531.
169. Alpert M. et al:— J. Electrochem. Soc., 1957, 104, 555.
170. A r i i K.— Sci. Rep. Tohoku, 1933, 22, 182.
171. Auger V.— Bull. Soc. chim., 1924, 35, 18.
172. Archambault J., Rivest R.— Can. J. Chem., 1960, 38, 1331.
173. Alderdice D. S.— J. Molec. Spectrosc., 1965, 15, 509.
174. Berzelius J. J.— Svenska Akad. Handl., 1824, 2, 344.
175. Berzelius J. J.— Pogg. Ann., 1825, 4, 1.
176. Baenziger N. C., Rungle R. E.— Acta Crystallogr., 1948, 1, 274.
177. В о о z e n у A.— Trans. Metallurg. Soc. AIME, 1962, 224, 950.
178. Bergmann E., Engel L.— Z. phys. Chem., 1931, B13, 232.
179. Bowling K. McG.—Austr. J. Chem., 1963, 16, 66.
180. Blumenthal W. B., Smith H.— Ind. Eng. Chem., 1950, 42, 249.
181. Brand P., Sackmann H.— Z. anorg. Chem., 1963, 321, 262.
182. Bertrand A.— Bull. Soc. chim., 1880, 33, 566.
310
183- Blocher J. M., Campbell J. E.— J. Am. Chem. Soc., 1947, 69, 2100.
^84. Bra ger A.— Acta physicochim., 1939, 10, 887.
vj85. Bond P. A., В e t n о n W. E.— J. Am. Chem., Soc., 1945, 67, 1691.
:'|8б. Bond P. A., Crone E. B.— J. Am. Chem. Soc., 1934, 2028.
087. Bond P. A., Stephens W. R.— J. Am. Chem. Soc., 1929, 51, 2910.
j88. В r a n d e n C.-I.— Acta chem. Scand., 1962, 66, 1318.
[189. Burg A. B., Campbell G. W.— J. Am. Chem. Soc., 1948, 70, 1964.
1190- Bland J. A., Flengas S. fJ.— Can. J. Phys., 1961,39, 941.
[191. Bedson P. P.— Lieb. Ann., 1876, 180, 235.
L 492. В e t r a n d A.— Bull. Soc. chim., 1880, 33, 403.
1193. В e d о n D. H., Horner S. M., T у r e 1 S. Y.— Inorg. Chem., 1964,
U 3,647.
[194. Beattie I. R., Webster M.— J. Chem. Soc., 1964, 3507.
f 195. Bourion F. — Compt. rend., 1907, 145, 621.
E196. Bourion F. — Ann. chim. et phys., 1910, 20, 547.
j 197. Baugarter P., Bruns W.— Ber., 1941, 74, 1232.
198. Baker K., F о w 1 e s G. W. A.— J. Less-Common Metals, 1965, 8, 47.
[199. Baker R. H.— J. Chem. Soc., 1879, 35, 763.
ft200. Baker R. H.— Lieb. Ann., 1880, 202, 233.
h201. Bright N. F. H., Wurm J. G.— Can. J. Chem., 1958, 36, 615.
K02. Botts E. D., Krauskopf F. C.— J. Phys. Chem., 1927, 31, 1404.
к'2ОЗ. В r i n t z i n g e r H., Brintzinger W.— L. anorg. Chem., 1931,
196, 44.
204. Brintzinger H., Troemer B.— Z. anorg. Chem., 1929, 184, 97.
205. Baxter G. P., Butler A. Q.— J. Am. Chem. Soc., 1928, 50, 408.
|206.*B illy M.— Ann. Chim., 1921, 16, 5.
>207. Billy M., В r a s e u r P.— Compt. rend., 1935, 200, 1765.
1208. В i 1 t z W., Sapper A.— Z. anorg. Chem., 1932, 203, 277.
L209. В i 1 t z W., Jeep K-— Z. anorg. Chem., 1927, 162, 32.
210. В i 1 t z W., M e i n e с к e E.— Z. anorg. Chem., 1923, 131, 1.
E211. В i 1 t z W., Keunecke E.— Z. anorg. Chem., 1925, 147, 171.
212. Cox B., Sharpe A. G.— J. Chem. Soc., 1953, 1783.
[213. Crookston 1^. B., Canjar L. N.— J. Chem. Eng. Data, 1963, 8, 544.
214. Chatt J., Hayter R. G.— J. Chem. Soc., 1963, 1343.
[315. Cervone E., Furlani G.— Ann. chim., 1961, 51, 838.
216. Clifton D. G., M c W о о d G. E.— J. Phys. Chem., 1956, 60, 309.
217. Cras J. A.—Nature, 1962, 194, 678.
218. Cameron D. J., Shelton R. A. J.— J. Inorg. a. Nucl. Chem., 1966,
28 77
219. Camboulives P.— Bull. Soc. chim., 1910, 7, 616.
220. Clausnitzer F.— Ber., 1878, 11, 2011.
221. Craiu V. e t. a 1.— Dincomun. ICECHIM, 1957—1962,2; РЖХим., 1965,
15, Б667.
Csaszar J., Balog J., Lehotai L.— Acta phys. et chim. Szeged.,
1956 2 56.
Chand S., R i v e s t R.— Can. J. Chem., 1962, 40, 2243.
Caglioti V., Ciavatta L., Liberti A.— J. Inorg. a. Nucl.
Chem., 1960, 15, 115.
Clausnitzer F.— Ber., 1878, 11, 2011.
Chandler J. A., Drago R. S., Latham R.— J. Inorg. a. Nucl.
Chem., 1961, 21, 283.
Chandler J. A., Wuller J. E., Drago R. S.— Inorg. Chem.,
1962, 1, 65.
Chandler J. A., Drago R. S.— Inorg. Chem., 1962, 1, 356.
Clark R. J., Lewis J., N у h о 1 m R. S.— J. Chem. Soc., 1962, 2460.
Clark J. H. et al.— Nature, 1961, 192, 222.
Clark J. H., Lewis J., Machin D. J., N у holm R. S.—
J. Chem. Soc., 1963, 379.
Clark R. J.— J. Chem. Soc., 1963, 1377.
311
233. Demoly A.— Compt. rend. Trav. chim., 1849, 5, 325.
• 234. Duckworth M. W., Fowles G. W., Hoodless R.
J, Chem. Soc., 1963, 5665,
235. Davis О. С. M.— J. Chem. Soc., 1906, 1575.
236. Davis О. С. M.— Proc. Chem. Soc., 1907, 22, 261.
237. Darzens G., Bourion F.— Compt. rend., 1911, 153, 270.
238. Dmitrios A., Ladikos E.— Practica, 1930r 5, 449.
239. Dermer О. C., Ferneli us W. C.— Z. anorg. Chem., 1934, 221, 83
240. D e h n i с к e K-— Z. anorg. Chem., 1961, 309, 266.
241. Deville H. S. C.— Compt. rend., 1855, 40, 1034.
242. D u p p a F. B.— Proc. Roy. Soc., 1856, 8,42.
243. Delwaulle M., Francois F.— Compt. rend., 1945, 220, 173.
244. Defacaz E., Copaux H.— Compt. rend., 1908, 147, 65.
245. Dunn P.— Austr. J. Chem., 1960, 13, 225.
246. D e h n i с к e K-— Z. anorg. Chem., 1965, 338, 279.
247. D i j к g r a a f C.— Spectrochem. Acta, 1963, 21, 769.
248. D i j к gr a a f C.— Nature, 1964, 201, 1121.
249. D i j к g г a a f C.— J. Phys. Chem., 1965, 69, 660.
250. Dema r cay E.— Bull. Soc. Chim., 1873, 20, 127.
251. Demarcay E.— Compt. rend., 1873, 76, 1414.
252. Demarcay E.— Compt. rend., 1885, 100, 740.
253. Demarcay E.— Ber., 1887, 20, 96.
254. Demarcay E.— Compt. rend., 1887, 104, 111.
255. Engelskirchen P. Diss., Berlin, 1903, см. H m e 1 i n s, Handbuch,
Titan, 1951, 41.
256. E b e 1 m e n J. J.— Lieb. Ann., 1947, 63, 269.
257. E b e 1 m e n J. J.— Ann. chim. et phys., 1847, 20, 385.
258. Engel G.— Z. Kristallogr., 1935, 90, 341.
259. Edvard F.— Compt. rend., 1933, 196, 2007.
260. Edvard F.— Bull. Soc. chim., 1933, 53, 1206.
261. Elenius E. P. J., Mereto j a A., Kosohen P.— Suomen kem.,
1964, 37, B114.
262. E m e 1 e u s H. J., Rao G. S.— J. Chem. Soc., 1958, 4245.
263. Edwards D. A., Fowles G. W., Walton R. A.— J. Inorg. a.
Nucl. Chem., 1965, 27, 1999.
264. Emich F.— Monatsh., 1904, 25 , 907.
265. Ebler E., Schott E.— J. prakt. Chem., 1908, 78, 289.
266. Ebler E., Schott E.— J. prakt. Chem., 1910, 81, 552.
267. Ehrlich P., Каира G., Blankenstein K-— Z. anorg. Chem.,
1959 229 213.
268. Ehrlich P., F r a m E.— Z. Naturforsch., 1954 , 9B, 326.
269. Ehrlich P., Pietzka G.— Z. anorg. Chem., 1954 , 275, 121.
270. Ehrlich P., Engel W.— Naturwis., 1961, 48, 716.
271. Ehrlich P., Engel W.— Z. anorg. Chem., 1963, 322, 217.
272. Ehrlich P., Engel W.— Z. anorg. Chem., 1962, 317, 21.
273. Ehrlich P., Hein H. J., К u h n 1 H. Z. anorg. Chem., 1957, 292, 139.
274. Ehrlich P., Schmitt R.— Z. anorg. Chem., 1961, 308, 91.
275. Ehrlich P., Kuhnl H.— Z. anorg. Chem., 1957, 292, 146.
276. Fraklin T. C., Seklemian H. V.— J. Inorg. a. Nucl. Chem., 1959,
12, 181.
277. Fischer F., Goldoch A.— Helv. chim. acta, 1930, 13, 1200.
278. Farber M., Darnell A. J.— J. Phys. Chem., 1955, 59, 156.
279. Farber M., Darnell A. J., Brown F.— J. Chem. Phys., 1955,
23, 1556.
280. Farber M., Darnell A. J.— J. Chem. Phys., 1955, 23, 1460.
281. Forbes G. S., Hall L. P.— J. Am. Chem. Soc., 1924, 385.
282. F 1 e n g a s S. N., Ingraham T. R.— Can. J.. Chem., 1960, 38, 813-
283. Fowles G. W., Nicholls D.— J. Inorg. a. Nucl. Chem., 1961,18, 130-
284. Fowles G. W. A., Walton R. A.— J. Chem. Soc., 1964, 2840.
312
fe85. Fowles G. W.A., Pollard F. H.— J. Chem. Soc., 1953, 2588.
1286. Friedel C., Guerin J.— Compt. rend., 1875, 81, 889.
[\287. Friedel C., Guerin J.— Ann. Chim. Phys., 1876, 8, 24.
£288. F u n a к i K.,’ Uchimura K. Bull. Tokyo inst. Technol., 1961, 42, 27.
[289. Fireman P.— J. Amer. Chem. Soc., 1904, 26, 741.
1^290. Fritsch P.— Compt. rend., 1943, 217, 447.
E291. F г а и к 1 i n *E. C., Hine T. B— J. Amer. Chem. Soc., 1912, 34, 1497.
I 292. Fast J. D.— Rec. trav. chim., 1939, 58, 174.
> 293. Fenske R. F.— Inorg. Chem., 1965, 4, 33.
ь'294. F e 1 t z A.— Z. anorg. chem., 1963, 323, 35.
295. Fe 1 t z A.— Z. anorg. Chem., 1965, 334, 242.
296. Fowles G. B. A., Walton R. A.— J. Less-Common Metals, 1963,5, 510.
297. Fowles G. W. A., Hoodless R. A., Walton R. A.— J. Chem.
Soc., 1963, 5873.
298. Fowles G. W., Hoodless R. A.— J. Chem. Soc., 1963, 33.
299. Fowles G. W. A., Walton R. A.— J. Chem. Soc., 1964, 4330.
300. Fowles G. W., Hoddless R. A., Walton R. A.— J. inorg. a. Nucl.
Chem., 1965, 27, 391.
1301. Ginsberg H., Holder G.— Z. anorg. Chem., 1931, 201, 193.
302. Ginsberg H., Holder G— Z. anorg. Chem., 1931, 196, 188.
303. Ginsberg H.— Z. anorg. Chem., 1932 , 204, 225.
304. G e w e с к e J.— Lieb. Ann., 1908, 361, 79.
305. G 1 a t z e 1 E.— Ber., 1876, 9, 1829.
306. George E. S.— Pogg. Ann., 1825, 3, 171.
307. Groeneveld W. L., Spronsen J. W., Ko u wen ho ven H. W.
Rec. traw. chim., 1953, 72, 950.
1'308. Groeneveld W. L., Zuur A. P.— Rec. traw. chim., 1953, 72, 617.
[309. Groeneveld W. L.— Rec. traw. chim., 1952, 71, 1152.
310. G r u e n D. M., McBeth R. L.— Nature, 1962, 194, 468.
11. Gross P., Hayman C., Levi D. L.— Trans. Farad. Soc., 1955,51,626.
t312. Georges H., Stabler A.—Ber., 1909, 42, 3200.
[•313. Gardner F. B., Sugden S.— J. Chem. Soc., 1929, 1298.
*314. Gardner H. J.— Nature, 1964, 204, 282.
>315.
316.
317.
318.
319.
20.
321.
22.
30.
24.
5.
6.
27.
8.
Gut m a n n
Gut m a n n
Gut m a n n
Gut m a n n
Gut m a n n
Gut m a n n
С. M.— Forh. Videnskabs. Selsk. Christiania, 1882, 20.
V., Nowotny H.— Monatsh.
V— Z. anorg. Chem., 1951, 266, 331.
V.— Z. anorg. Chem., 1561, 264, 151.
V .— Monatsh., 1954, 85, 393.
V .— Z. anorg. Chem., 1952, 269, 279.
V ., Nowotny H., Ofner G.— Z. anorg. Chem., 1955
1958, 89, 331.
278, 78.
Guenther K. F.— Inorg. Chem., 1964, 3, 923.
G I a t z e 1 E.— Ber., 1876, 9, 1829.
Goring J., S u s z В. P.— Helv. chim. acta, 1966, 49, 486.
Ginsberg H., Holder G.— Z. anorg. Chem., 1930, 190, 407.
Garrasi G., Lanielli E.— Chim. e indu., 1965, 47, 307.
Hunter M. A.— J. Am. Chem. Soc., 1910, 32, 330.
Hoard J. L., Martin W. J.— J. Am. Chem. Soc., 1941,63, 11.
Hassel O., Salvesen J. R.— Z. Phys. Chem., 1927, 128, 345.
Hamilton P. M. et al.— J. Am. Chem. Soc., 1953, 75, 2881.
Heller W., Polanyi M.— Trans. Farad. Soc., 1936, 32, 633.
Hudson R. F.— Proc. Intern. Congr. Pure and Appl. Chem., 1947, 11, 297.
Hassel O., Kringstad H.— Z. phys. Chem., 1932, B15, 274.
Hayakawa S.— Bull. Chem. Soc. Japan., 1955, 28, 447.
Hawkin N. J., Carpenter D. R.— J. Chem. Phys., 1955, 23, 1700.
. H б 1 t j e R.— Z. anorg. Chem., 1930, 190, 241.
7. H a m p 1 e W.— Lieb. Ann., 1863, 126, 43.
8. H б 1 t j e R.— Z. anorg. Chem., 1930, 190, 241.
39. H a e g i W.— Bull. Soc. chim., France, 1955, 4, 490.
3(3
340. Hunter M. A.— J. Am. Chem. Soc., 1910, 32, 330.
341. Henke W— Lieb. Ann., 1858, 106, 280.
342. H a e g i W.— Bull. Soc. chim. France, 1955, 4, 494.
343. Hertel E., Demmer A.— Lieb. Ann., 1932, 499, 134.
344. Hertel E.— Lieb. Ann., 1942, 553, 285.
345. Hayek E., Engelbrecht A.— Monatsh., 1949, 80, 640.
346. Hayek E., Puschmann J., Czaloun A.— Monatsh., 1954,85
359. ’
347. Hall E. H., Blocher J. M— J. Phys. Chem., 1959, 63, 1525.
348. Hall E., Blocher J.— J. Electrochem. Soc., 1958, 105, 40.
349. Hall E. H., Blocher J. M., Campbell I. E.— J. Electrochem.
Soc., 1958, 105, 275.
350. H a 1 1 E., Blocher J., Campbell I.— J. Electrochem. Soc., 1958
105, 271.
.351. Hock L., Knauf f W.— Z. anorg. Chem., 1936, 228, 200.
352. Hock L., К n a u f f W.— Z. anorg. Chem., 1936, 228, 204.
353. Hock L., Knauff W.— Z. anorg. Chem., 1936, 228, 209.
354. Hassel O., Krings tad H.— Z. phys. Chem., 1931, B15, 274.
355. Hartmann H., Schafer H. L.— Z. Naturforsch., 1956, 6a, 760.
356. Hartmann H., R i nek G.— Z. Phys. Chem., (BRD), 1957, 11, 213.
*357. Hartmann H., Schlater H. L., Hansen К- H.— Z. anorg.
Chem. 1957 289 40.
358. Haendl er H.’m. et al.— J. Am. Chem. Soc., 1954 , 76, 2177.
359. Hukumoto Y.— Sci. Rep. Tohoku, 1933, 22, 868.
360. Hautefeuille P.— Compt. rend., 1863, 57, 148.
361. Hautefeuille P.— Bull. Soc. chim., 1867, 7, 201.
362. Ingraham T. R., Downes K- W., M a r r i e r P.— Cane. J. Chem.,
1957, 35, 850.
363. Ilse F. E., Hartmann H.— Z. phys. Chem., 1956, 197, 239.
364. J a n z G. J. et al.— J. Phys. Chem., 1958, 62, 823.
365. Johnson W. H., Nelson R. A., Prosen E. J.— J. Res. Nat.
Bur. Stand., 1959, 62, 49.
366. Jander G., Wendt H.— Z. anorg. Chem., 1949, 258, 1.
367. Johannesen R. B., Gordon G. L., Stewart J. E., Gilch-
rist R.— J. Res. Nat. Bur. Stand., 1954, 53, 197.
368. Ingracham T. R., P i d g e о n L. M.— Can. J. Chem., 1952, 30, 694.
369. J u z a R., H e n e r s J.— Z. anorg. Chem., 1964, 332, 159.
370. Jorgensen С. K-— Acta chem. Scand., 1957, 11, 73.
371. J a i n S. C., R i v e s t R.— Can. J. Chem., 1962, 40, 2243.
372. Jain S. C., R i v e s t R.— Can. J. Chem., 1965, 43, 787.
373. Jenkins F. A., Rochester G. D.— Phys. Rev., 1938, 53, 213.
374. К г e у e W. C., Kellogg H. H.— J. Electrochem. Soc., 1957, 104, 504.
375. Kungle E., Arnell F.— J. Soc. Chem. Ind. Trans., 1928, 47, 376.
376. Kowalevsky W.— Z. anorg. Chem., 1900, 25, 189.
/377. Koenig T., Afordten O.—Ber., 1888,21, 1708.
378. Като H., Абэ M.— J. Chem. Soc. Japan. Pure Chem., Sec., 1955, 76,1182.
379. Krieve W. F., Van go S. P., Mason D. M.— J. Chem., Phys.,
1956 25 519.
380. К e m p a R. F., Lee W. H.— Z. anorg. Chem., 1961, 311, 140.
381. Karantassis T.— Compt. rend., 1932, 194, 461.
382. Kimura M. et al.— Bull. Chem. Soc. Japan, 1956, 29, 95.
383. Kangro W., Jahn R.— Z. anorg. Chem., 1933, 210, 325.
384. Krieve W. F., Mason D. M.— J. Chem., Phys., 1956, 25, 524.
385. Karantassis T.— Ann. chim., 1927, 8, 71.
386. Ko mare к K-, Herasymenko P.— J. Electrochem. Soc., 1958,
105, 210.
387. Kern S.— Chem. News, 1876, 33, 57.
388. Kelly К- K — Bull. Bur. Mines., 1935, 383, 106.
314
g89. Klemm W., T i 1 к W., Mill 1 ennei m S.— Z. anorg. Chem., 1928,
f 176, 1.
1091. Klemm W., К г о s e E.— Z. anorg. Chem., 1947, 253, 209.
392- Klemm W., К г о s e E.— Z. anorg. Chem., 1947, 253, 218.
393. KI i ко г к a J., Pavlik J. Sb. vedec. praci Vysoke skoly chem.- technol.
Pardubice, Praha, 1959, 79.
394. Kroll W.— Trans. Am. Electrochem. Soc., 1940, 78, 35.
395. Kroll W. —Chem. Eng., 1960, 43, 1314.
^396. Kiss A.— Z. anorg. Chem., 1955, 282, 141.
397. Kroll W., Schlechten A. W.— Met. Ind. London, 1946, 69, 319.
398. Kroll W. et al.— Trans. Am. Electrochem. Soc., 1947, 92, 187.
399. Kraus K., Nelson J.— J. Phys. Chem., 1954 , 58, 11.
400. Krauss H.-L., Hiittmann H— Z. Naturforsch., 1963, B36, 199.
401. Lewis J. et al.— J. Chem. Soc., 1962, 2036.
402. Lutschinsky G. P.— Z. anorg. Chem., 1935, 224, 420.
403. Lutschinsky G. P.— Z. anorg. Chem., 1936, 226, 333.
- 404. Leonard C. S.— J. Am. Chem. Soc., 1921, 43, 2618.
405. Levy D., Hamburger L— Z. anorg. Chem., 1914, 87, 209.
406. Levy L.— Ann. Chim. et phys., 1892, 25, 433.
407. Large N. R. Stubbs F. J., Hinschelwood C.— J. Chem. Soc.,
1954, 2736.
iftO8. Lister M. W., Sutton L. E.— Tran. Farad. Soc., 1941, 37, 393.
409. Lister R. L., F 1 e n g a s S. N.— J. Electrochem. Soc., 1964, 111, 343.
410. Lister R. L., F 1 e n g a s S. N— Can. J. Chem., 1963, 41, 1548.
>411- Latimer W. M.— J. Am. Chem. Soc., 1922, 44, 90.
412. Leeds A. R.— J. Am. Chem. Soc., 1881, 3, 145.
'413. Merz V.— J. prakt. Chem., 1866, 99, 157, 159, 174.
\414. Mazza cc he 11 i A., Pantanelli E.— Atti Line., 1909, 18, 608.
415. Mazzacchelli A., Pantanelli E.— Gazz., 1910, 40, 666.
16. Marchetti G.— Z. anorg. Chem., 1895, 10, 66.
i417. Mellgren S., Opie W.— J. Metals., 1957, 9, 266.
418. Moszynska B.— Compt. rend., 1963, 256, 1261.
419. Munster A., Ruppert W.— Z. Elektrochem., 1953, 57, 558.
420. Marvin H. H.— Phys. Rev., 1912, 34, 161.
(421. Mathews J. H.— J. Phys. Chem., 1905, 9, 641.
422. Mui J. H., F 1 e n g a s S. N.— Can. J. Chem., 1962, 40, 997.
K23. More K. R., Pokker A. H.— Phys. Rev., 1937, 52, 115.
'424. M a r t i n H., Vohwinkel F.— Chem. Ber., 1961, 94, 2416.
125. Morgan L. Advances Chem. Coordinat. Compounds. N. Y., Macmillan Co,
1961, 471; РЖХим., 1965,11, Б673.
426. Mori B. G., Cassimatis D., Susz B.— Arch. Sci., 1960, 13, 518.
427. Mori B. et al.—Helv. chim. acta, 1962, 45, 77.
428. M с C a u 1 a у D. A., Higley W. S., Lien A. P.— J. Am. Chem. Soc.,
1956, 78, 3009.
429. M о i s s a n H.— Compt. rend., 1890, 120, 290.
430. M о i s s a n H.— Bull. Soc. chim., 1895, 13, 963.
431. Masaquer F. J. R., В u s t e 1 о D. A.— Ann. Real Soc. esp. fis. у guim
1959, B55, 823.
432. M a r i g n a с C.— Compt. rend., 1865, 60, 234.
433. M a r i g n a с C.— Ann. Chim. Phys., 1866, 4, 6.
434. M a r i g n a с C.— Compt. rend., 1860 , 50 , 954.
;35. M a r i g n a с C.— Ann. Chim., 1860, 60, 257.
436. M a r i g n a с C.— J. prakt. Chem., 1861, 83, 201.
;37. Marignac C.— Ann. Mines, 1859, 15, 221.
438. M a r i g n a s C.— Ann. Mines, 1859, 15, 239.
P9. Marignac C.— Ann. Mines, 1859, 15, 249.
440. Nettle J., Baker D. H., Wartman F. S.— U. S. Bur Mines
Rep. Invest., 1957, 5315, 1.
441. Nagarajan G.— Bull. Soc. chim. belg., 1963, 72, 346.
442. N i g i s h i G. R., Donna 11 у L. H., Hildebrand J. H.— J.
Chem. Soc., 1933, 55, 4793.
443. Nelson R. A., Johnson W. H., P г о s e n E. J.— J. Res. Nat
Bur. Stand., 1959 , 62, 67.
444. Nicholson D. Y., Reiter M. A.— J. Am. Chem. Soc., 1937, 59, 15}
445. Nicholls D., Swindells R.— J. Chem. Soc., 1964, 4204.
446. N ego i u D.— St. cer. chim. Acad. RPP, 1963, 11, 161.
447. N e g о i u D.— St. cer. chim. Acad. RPP, 1963, 11, 71.
448. N a s u N.— Bull. Chem. Soc. Japan, 1933, 8, 195.
449. N a s u N.— Bull. Chem. Soc. Japan, 1933, 8, 392.
450. N a s u N.— Sci. Rep. Tohoku, 1933, 22, 972.
451. N a s u N.— Sci. Rep. Tohoku, 1933, 22, 987.
452. N a s u N.— Bull. Chem. Soc. Japan, 1934, 9, 198.
453. N a s u N.— Sci. Rep. Tohoku, 1934, 23, 374.
454. N a t t a G. et al.— J. Polymer. 1958, 31, 181.
455. N a t t a G. et al.— Atti Line., Cl. sci. fis., mat. e natur., 1958, 24, 121.
456. Natta G., Corrodini P., A 1 legr a G.— J. Polymer Sci., 1961
51, 399.
457. Ono К., H a t a K-, К u r i у a m a T.— Bull. Res. Inst. Mineral. Dres-
sing. Met. (Japan), 1954, 10, 39.
458. Ono K. et al.— Sci. Rep. Res. Inst. Tohoku Univ., 1957, A9, 227.
459. Ogawa S.— J. Phys. Soc. Japan, 1960, 15, 1901.
460. Olson J. C., Ryan E. P.— J. Am. Chem. Soc., 1932, 54, 2215.
461. О t to J. B. et al.— J. Inorg. a Nucl. Chem., 1965, 27, 2005.
462. О s h i b a T.— J. Chem. Soc. Japan Ind. Chem. Sec., 1959, 62, 985.
463. О s h i b a T.— J. Chem. Soc. Japan Ind. Chem. Sec., 1963 , 66, 670.
464. Oberhauser F., Schor mill 1 er J.— Ber., 1929, 62, 1436.
465. Pennington M. E.— J. Am. Chem. Soc., 1896, 18, 58.
466. Pennington M. E.— J. Am. Chem. Soc., 1896, 18, 60.
467. P о d s z u s E.— Z. anorg. Chem., 1917, 99, 123.
468. Peyronel G.— Gazz., 1941, 71, 620.
469. Potvin R., Farnham G. S.— Trans. Can. Inst. Min. Metallurgy,
1946 49 516.
470. Partington J. R., Why nes A. L.— J. Chem. Soc., 1949,3135.
471. Patscheke G., Schaller W.— Z. anorg. Chem., 1938, 235, 257.
4 472. P u t ed du E.— Gazz., 1929,59, 161.
473. P о 1 i d о r i A.— Z. anorg. Chem., 1899, 19, 306.
474. Pierre J.— Ann. Chim. Phys., 1847, 20, 5.
475. Pistorius C. W. F. T.— J. Chem. Phys., 1958, 29, 1328.
476. P u s c h i n N. A. et al.— Lieb. Ann., 1942, 551, 259.
477. P u s c h i n N. A., R i s t i с M., Parcomenko J., Ubovic L—
Lieb. Ann., 1942, 553, 278.
478. Petersen E.— J. prakt. Chem., 1889, 40, 44.
479. Petterson R. A.— Phys. Rev., 1925, 10, 56.
480. Prasand S., Sr i v as t a v a R. C.— J. Indian Chem. Soc., 1962, 39, 11-
481. Pfordten O.— Lieb. Ann., 1887, 237, 201.
482. P i с c i n i A.— Atti Lins., 1885, 1, 86.
483. P i с c i n i A.— Gazz., 1886, 16, 105.
484. P i с c i n i A.— Atti Line., 1885, 1» 47.
485. P i с c i n i A.— Gazz., 1885, 14, 38.
486. P i с c i n i A— Ber., 1883, 16, 3060.
487. P i с c i n i A.— Atti Line., 1890, 6, 568.
488. P i с c i n i A.— Atti Line., 1885, 1, 682.
489. P i с c i n i A.— Gazz., 1887, 17, 479.
490. P i с c i n i A.— Z. anorg. Chem., 1895, 10, 438.
491. Piccini A.— Compt. rend., 1883, 97, 1064.
492. Robinson F. C., Cutchins С. C.— Am. Chem. J., 1884, 6, 74.
493. Roth W. A., Becker G.— Z. phys. Chem., 1932, A159, 1.
316
94. R ump I M.-E.— Ann. chim., 1937, 8, 456.
95. Ruehrwein R. A., Skinner G. В.— Пат. США № 2720445, 1955.
,96. Regnauet V.— Ann. chim. et phys., 1841, 1, 129.
97. Ryder G. A., Kamal M. R., C a n j a r L. N.— I. Chem. Eng. Data,
1961, 6, 594.
98. R a о G. S.— Z. anorg. Chem., 1960, 304, 176, 351.
499. Rammelsberg C.— Ber. Bed. Akad., 1874, 480.
500. R a m m e 1 s b e r g C.— Ber. Ber 1. Akad., 1874, 490.
501. Rosenheim A., Schutte O.— Z. anorg. Chem., 1901, 26, 239.
502. Rose H.— Pogg. Ann., 1829, 16, 57.
503. Rose H.— Pogg. Ann., 1832, 24, 141, 159, 259.
504. Rose H.— Pogg. Ann., 1837, 42, 517.
505. Reinheim H., Hake A.— Lieb. Ann., 1927, 452, 47.
506. Rivest E., Aubin R., R i ves t R.— Can. J. Chem., 1961, 39, 2343.
507. Rivest R.— Can. J. Chem., 1962, 40, 2234.
508. Ralston R. F., S i s 1 e r H. H.— J. Am. Chem. Soc., 1957, 79, 1068.
509. Ralston A. W., Wilkinson J. A.— J. Am. Chem. Soc., 1928,50,258.
510. Roeder M. G.— Norske Vidensk. Selsk. Skr., 1929, 3.
511. Roeder M. G.— Z. anorg. Chem., 1933, 210, 145.
512. Rheinboldt H., Wassefuhr R.— Ber., 1927, 60, 732.
513. Ruff O., Neumann F.— Z. anorg. Chem., 1923, 128, 81.
514. Ruff O., Kwasnik W.— Angew. Chem., 1935, 48, 238.
^515. Ruff O., Ipsen R.— Ber., 1903, 36, 1777.
516. Ruff O., Allert C.— Ber., 1905, 38, 53.
517. Ruff O., Plato W.— Ber., 1904, 37, 673.
518.
519.
520.
521.
522.
523.
524.
525.
526.
527.
528.
529.
530.
531.
532.
533.
534.
535.
Ruff O., Friedel O-— Ber., 1912, 45, 1364.
Ruff O., Eisner F.— Ber., 1908, 41, 2250.
Shiihara I., lyoda J., Sugita K-—Bull. Chem. Soc. Japan,
1962, 35, 364.
Schreinemakers. — Z. phys. Chem., 1906, 55, 73, 77.
Seidel W., Fischer W.— Z. anorg. Chem., 1941, 247, 367.
Schneider E. A.— Z. anorg. Chem., 1895, 8, 81.
Shretien A., Varga G.—Compt. rend., 1935, 558.
Skinner G. B., Ruehrwein R. A.— J. Phys. Chem., 1955, 59, 113.
Stilfelhagen A. Diss., Rostok, 1905.
Schmitz-DuMont O., Motzkus E.— Ber., 1929, 62, 466.
S u s z В.- P., Cassimatis D.— Helv. chim. acta, 1961, 44, 395.
Sherfey J. M.— J. Res. Nat. Bur. Stand., 1951, 46, 299.
Seubert K-, Schmidt A.— Lieb. Ann., 1892, 267, 218.
Sanderson B. S., McWood G. E.— J. Phys. Chem., 1956, 60, 314.
S о r u m H.— Norske Vidensk. Selsk. Forh., 1944, 17, 17.
S о r u m H.— Norske Vidensk. Selsk. Forh., 1944, 17, 21.
Schafer H., Zeppenrick F. — Z. anorg. Chem., 1953 , 272, 274.
Schafer H., Wertenpfuhl W., Weise L.— Z. anorg. Chem.,
1958, 295, 258.
536. Schafer H.,Breil G., Pfeffer G.— Z. anorg. Chem., 1954,276, 325.
537. SchumbW. C., Sunderstrom R. F.— J. Am. Chem. Soc., 1933,
55, 596.
538. Schwarz R., Giese H.— Z. anorg. Chem., 1928, 176, 227.
539. Schwartz D., Larson B.— J. Less-Common Metals, 1963, 5, 365.
540. -Schwartz D., Reski P.— J. Inorg. a. Nucl. Chem., 1965, 27, 747.
541. S h a r m a R. S.— Bull. Acad. Sci. Allahabad, 1933/34, 3, 87.
‘542. Scagliarini G., Monti E.— Atti Line., 1926, 4, 210.
543. Scalgiarini G., Tar t ar i ni G.— Atti Line., 1926, 4, 318.
544. Scagliarini G., Bras si E.— Atti Line., 1925, 2, 269.
545. Stabler A.— Ber., 1904, 37, 4405.
546. Stabler A., W i r t h w e i n H.— Ber., 1905, 38, 2619.
547. Stabler A., Bachram F.— Ber., 1911, 44, 2906.
548. Stabler A.— Ber., 1905, 38, 2626. j
317
549. Schlafer H. L., Go tz R.— Z. anorg. Chem., 1964, 328, 1.
550. Schlafer H. L., Go t z R.— Z. phys. Chem., 1964, 41, 95.
551. T о p s о e H.— Arch. Phys. Nat., 1872, 45, 223.
552. Thorpe T. E.— Chem. News, 1885, 51, 260.
553. Thorpe T. E.— J. Chem. Soc., 1885, 47, 108.
554. Thomsen J.— Pogg. Ann., 1870, 139, 193.
555. Troost L., Hauterfeuille R.— Compt. rend., 1871, 73, 573.
556. Thomson J.— Pogg. Ann., 1870, 139, 193.
557. Trost W. R.— Can. J. Chem., 1952, 30, 835.
558. Trost W. R.— Can. J. Chem., 1952, 30, 842.
559. Tuttschew J.— Lieb. Ann., 1867, 141, 111.
560. T e с к P.— Verhandel Koninkl. Vlaam. Acad. Wetenschap. Belg., 1949, 11, 5.
561. U 1 i c h H., Hertel E., Nespital W.— Z. phys. Chem., 1932
B17, 21.
562. Junji Kishi, Shinsaku Kato. — Rept. Osana Pref. Ind. Rese-
arch Inst 1950 2 42
563. V a r r e s ’k. S.,’ Dutton F. B.— J. Am. Chem. Soc., 1955, 77, 2019.
564. Varres K-, Donohue J.— Acta Crystallogr., 1955, 8, 25.
565. Verma I.D., Mehr о t r a R. C.— J. Less-Common Metals, 1959, 1, 263.
566. Vigouroux E., Arrivant G.— Bull. Soc. Chim., 1907, 1, 19.
567. Vaidninathan V. I.— Nature., 1931, 128, 189.
568. Woods P. H., Cockerell L. D.— J. Am. Chem. Soc., 1958, 80, 1534.
569. Wertyporoch E., Altmann B.— Z. phys. Chem., 1934, 168. 1.
570. W a t e n a b 1 T., A t о j i M.— Sci. (Japan), 1950, 20, 422.
571. Waters E. L., Maki A. H.— Phys. Rev., 1962, 125, 233.
572. W i e r R.— Ann. Phys., 1931, 8, 521.
573. Wehrelin E., Giraud E.— Compt. rend., 1877, 85, 288.
574. Woolf A. A.— J. Chem. Soc., 1954 , 252.
575. Wiberg E., Lacal R. U.— Rec. acad. ciens. exact, fis. guim у nat.
Zaragoza, 1951, 6, 15.
576. Wiberg E., Uson R.— Z. Naturforsch., 1951, 6b, 392.
577. Wurm J. Y., Grovel L., Potvin R.— J. Electrochem. Soc., 1957,
104, 301.
578. Wong Y. E.— J. Chem. Phys., 1959, 32, 598.
579. W e s t 1 a n d A. D., Wes 11 a n d L.— Can. J. Chem., 1965, 43, 426.
580. Weiss L., Kaiser H.— Z. anorg. Chem., 1910, 65, 345.
581. Walter — Levy L., Ferey G. S., Syed H. I.— Compt. rend.,
1967, C264, 700.
582. Weiss R., Fischer J., Chevrier B.— Compt. rend., 1965,
260, 3401. Acta crystallogr., 1966, 20, 534.
583. W e i s s R., F i s c h e r J., Keid G. —Compt. rend., 1964, 259,112.
584. W a t t s G. W., В e 1 1 G. A. — J. Chem., Soc., 1878, 442.
585. Wohler F.— Lieb. Ann., 1850, 74, 212.
586. Wedekind E., Hausknecht P.— Ber., 1913, 46, 3763.
587. Whitehouse N. — J. Soc. Chem. Ind., 1907, 26, 738.
588. W б 1 b 1 i n g H. — Z. anorg. Chem., 1908, 57, 281.
589. Wise C. R— J. Am. Chem. Soc., 1923, 45, 1233.
590. Wohler F.— Lieb. Ann., 1849, 73, 34.
591. Wohler F.— Lieb. Ann., 1850, 73, 226.
592. Weber R — Pogg. Ann., 1863, 120, 291.
593. Weber R.—Pogg. Ann., 1863, 118, 471.
594. Weber R.—Pogg. Ann., 1863, 120, 287.
595. Weber R.— Pogg. Ann., 1867, 132, 452.
596. Y о s h i n о T., Kijima L, Masuda Y., Sugiyama I.—
J. Chem. Soc. Japan. Ind. Chem. Sec., 1959, 62, 77.
597. Y a m g u c h i S.— Angew. Chem., 1955, 67, 748.
598. Yong R.C., Schumb W. C.— J. Am. Chem. Soc., 1930, 52, 4233.
599. Zambonini M. F.— Bull. Soc. Min., 1930, 53, 443.
318
Дополнительная литература
Онтипин Л.Н., Верховцев В. Т., Нерубашенко В. В., Ко-
ломицкий ф. М.— Диаграмма плавкости системы КС1—K2TiF6.—В кн.:
Сборник трудов. Всесоюзный научно-исследовательский ин-т титана . Ме-
таллургия, М., 1967, 1, 220.
[Александровский С. В., Гуськов Б. И. Изменение свободной энер-
гии при взаимодействии TiCfe и TiCl3 с расплавом NaCl.— Изв. высш. уч.
зав. Цветная металлургия, 1966, 2, 55. I
Антипин Л. Н., Нерубащенко В. В., Кулешова А. В., Ры-
сьева Ю. И., Вовк В. Н. Диаграммы плавкости фторидно-хлоридных
систем калия, натрия и титана.— В кн.: Сборник трудов. Всесоюзный на-
учно-исследовательский ин-т титана. Металлургия, М., 1968, 2, 204.
Антипов И. В., Коршунов В. Г., Гофман Л. М. Кинетика взаимо-
действия четыреххлористого титана с кислородом.— Ж- прикл. хим., 1967,
40, 11. | j
|VJ5_y латов Н. Е., М а к ру ш и н С. Г. Кинетика и механизм гидролиза со- v
I лянокислых растворов Ti-4‘. —"Ж. неорг. хим., 1966, 11, 468.
Буслаев Ю. А., Щербаков В. А. Изучение гидролиза тетрафторида
титана методом ЯМР19 F.— ДАН СССР , 1966, 170, 845.
у Б и р ю к о в а Л. В., Саксонов Ю. Г. Исследование продуктов взаимо-
действия металлического титана с четыреххлористым титаном.— Ж- неорг.
хим., 1960, 5, 993.
(Верховец В. Т., Антипин Л. Н. Плотность и потери веса при улету-
чивании расплавов систем КС1 — K2TiF6 и NaCl — K2TiF6.— В кн.: Сборник
трудов. Всесоюзный научно-исследовательский ин-т титана. Металлургия,
М., 1967, 1, 226.
' Владимирова А. М. Растворимость Ог, Иг, СО и СОг в четыреххлористом
титане.— Труды ВАМИ, 1965, 54—55, 397.
[Войтович Б. А., Лозовская Н. Ф. Взаимодействие хлористого суль-
фурила с четыреххлористым титаном.— Изв. АН СССР. Металлы, 1967, 6, 81.
Войтович Б. А., Звагольская Е. В. Термический анализ тройных
систем TiCl4—РОС13—СС13СОС1 и TiCl4—РОС13—CH2CICOCI.— Укр. хим. ж.,
1967, 33, 1258.
г Галицкий Н. В., Цеховольская Д. И. О растворимости хлоридов
хрома в четыреххлористом титане.— В кн.: Сборник трудов. Всесоюзный на-
учно-исследовательский ин-т титана. Металлургия, М., 1967, 1, 98.
рарновский А. Д., Минкин В. И., Осипов О. А., Панюш-
кин В. Т., Исаева Л. К-, Княжанский М. И. Комплексные со-
единения титана-4, олова-4 и тория с ароматическими и гетероциклическими
азометинами.— Ж- неорг. хим., 1967, 12, 2443.
| Д у л о в а В. И., Муригина Н. Г. О силе апротонных кислот некоторых
галогенидов IV и V групп в трибутилфосфате.— Изв. АН Каз. ССР. Серия
хим., 1968, 1, 34.
|Е л ю т и н В. П., Лысов Б. С., П е п е к и н Г. И. Исследование процес-
сов восстановления TiCl4 смесью сероводорода и метана.— Изв. высш. уч.
зав. Цветная металлургия, 1967, 5, 86.
[Ж иганова Е. С., Б а т а ш о в К- П. Травление титана в соляной кислоте.—
। ‘ “'ЖТприкл. хим?, Т968,'417206. : * F 4 I F
13 аварии кая Т. А., Подгойская М. Н. Исследование термического , .
разложения оксихлорида титана.— Ж- прикл. хим., 1968, 41, 948.
|И онова Е. А., Буслаев Ю. А., Харитонов Ю. Я- Исследование *
процесса гидролиза тетрахлорида титана в неводных растворах. — Изв.
АН СССР. Неорг. материалы, 1968, 4, 71.
|К о з л о в И. А., Осипов О. А. Взаимодействие тетрахлорида титана с эта-
нолдиамидами жирных кислот.— Ж- общ. хим., 1966, 36, 1484.
|К оршунов Б. Г., Безуевская Б. Н. О взаимодействии гексахлорида v
вольфрама с хлоридами титана, кремния, олова-2,4, сурьмы-4 и оксихлори-
дом фосфора.— Ж. неорг. хим., 1967, 12, 3280.
319
Коршунов Б. Г., Безуевская Б.Н.О взаимодействии хлоридов воль-
фрама с хлоридами сопутствующих примесей.— Изв. высш. уч. зав. Цветная
металлургия, 1967, 5, 105.
Кричевский И. Р., Ивановский Г. Ф.» Сафронов Е. К. Раст-
воримость тетраиодида титана в бензоле.— Ж- физ. хим., 1965, 39, 2684.
4 Кольцо в С. И., Алее к о в с кий В. Б. Изучение взаимодействия четы-
реххлористого титана с силикагелем.— Ж- прикл. хим., 1967, 49, 907.
Кольцов С. И., Алесковский В. Б. Влияние степени дегидратации
силикагеля на механизм гидролиза адсорбированного четыреххлористого
титана.— Ж- физ. хим., 1968, 42, 1210.
Корольков Д. В., Захаржевская В. О. Энтальпия образования
хлортитанатов-2, рубидия и цезия.— Ж- неорг. хим., 1967, 12, 2951.
Корольков Д. В., Кудряшова Г. Н. Системы RbCl — TiCls и CsCl—.
TiCl3.— Ж. неорг. хим., 1968, 13, 1626.
Ласточкина А. А., Шека И. А., Мал и н ко Л. А. Об основных ком-
плексных фторидах элементов подгруппы титана и лития, калия, рубидия,
цезия.— Изв. Сиб. отд. АН СССР. Серия хим. 1968, 1, 37.
Лещенко А. В., Панюшкин В. Т., Горновский А. Д., Оси-
пов О. А. Комплексы TiCl4 с анилино- и хинаминопроизводными.—
Ж- неорг. хим., 1966, 11, 2156.
Лысенко Ю. А. О диссоциации эфиратов четыреххлористого титана. —
Ж- общ. хим., 1967, 37, 11.
vМаксимов В. С., Смирнов М. В. Растворимость тетрахлорида титана
в расплавах хлорида натрия и эквимольной смеси хлоридов натрия и калия.—
Труды Ин-та электрохимии. Уральский филиал АН СССР, Свердловск, 1966,
9, 41.
Мар дыкин В. П., Бадаев В. К-, Гапончик П. Н. Исследование
реакций четыреххлористого титана с эфирами алюминийорганических сое-
динений.— Изв. высш. уч. зав. Химия и хим. технология, 1967, 10, 1258.
ч'Милько В. И., Петрик А. Г., Родя кин В. В., Трублев М. Г.,
Горовой М. А. К вопросу о восстановлении титана и его соединений ато-
марным водородом.— В кн.: Сборник трудов. Всесоюзный научно-исследова-
тельский ин-т титана, 1967, 1, 205.
Милов А. И., Байтенев Н. А., Пономарев В. Д. Некоторые фи-
зико-химические свойства системы Na2TiF6—NaCl—Т1О2.— Труды Ин-та
металлургии и обогащения. АН Каз. ССР, 1966, 18, 23.
Милов А. И., Кучанская О. Ф., Байтенев Н. А., Понома-
ре в В. Д. Некоторые физико-химические свойства расплавов системы
Na2TiF6—NaCl — TiO2.— Труды Ин-та металлургии и обогащения. АН Каз.
ССР, 1966, 18, 14.
4 Н а б и в а н е ц Б._РГ,_ К уд р и ц к а я Л. Н. Изучение полимеризации Ti-4
в солянокислых растворах.— Ж- неорг. хим., 1967, 12, 1165.
1/ Набиванец Б. И., Кудрицкая Л. Н. Определение состава и констант
диссоциации хлоридных комплексов титана.— Ж- неорг. хим., 1967, 12, 1500.
Нисельсон Л. А., Третьякова К- В. Плотность и критическая тем-
пература TiCl4.— Ж. неорг. хим., 1967, 12, 857.
Нерубащенко В. В., Антипин Л. Н., Горовой М. А. Исследо-
вание удельной электропроводности системы КС1 — КгТ1Рб.— В кн.: Сборник
трудов. Всесоюзный научно-исследовательский ин-т титана. Металлургия,
М., 1967, 1, 234.
Осипов О. А., Гайворонский В. Н., Швец А. А. Инфракрасные
спектры и взаимодействие эфиров фосфорных кислот с тетрахлоридами ме-
таллов.— В кн.: Применение молекулярной спектроскопии в химии. «Наука»,
М., 1966, 117.
Подорван I.M., Пенкало Й. Й. Электрохимическое исследование раст-
воров четырехвалентного титана и хлорида лития в нитрометане.— Bichiik
Кишського ушверситету. Сер. ф13. та xiM., 1967, 7, 97.
мПугачевич П. П., Т р етьякова К- В., Нисельсон Л. А. По-
верхностное натяжение тетрахлорида титана.— Ж* физ. хим., 1966, 40, 2902-
320
<
бан Н. Н., Исаева С. М. Вязкость и плотность тетрахлорида титана
в смеси с хлорокисью ванадия.— Труды Ин-та металлургии и обогащения.
АН Каз. ССР, 1966, 18/38.
ние низших хлоридов титана.— В кн.: Сборник трудов. Всесоюзный науч-
но-исследовательский ин-т титана. Металлургия, М., 1968, 2, 171.
«С м и р н о в М. В., Максимов В. С. Растворимость тетрахлорида титана
I в расплавленном хлористом магнии.— Труды Ин-та электрохимии, 10. Ураль-
ский филиал АН СССР, Свердловск, 1967, 49.
Смирнов М. В., Максимов В. С. Взаимодействие тетрахлорида титана
с расплавленным хлоридом цезия.— Труды Ин-та электрохимии, 8. Ураль-
ский филиал АН СССР, Свердловск, 1966, 35.
и Р н.° 5 М. В., Максимов В. С., Хайме нов А. П. Растворимость
вТ1С14 в расплавленны?; хлоридах щелочных металлов.— Ж. неорг. хим.,
1966, 11, 1765.
Старцев В. Н., Крылов Е. И. Экстракция титана-4 и комплексообразо-
вание в системе TiCl4 — НС1 — ТБф.— Труды Уральского политехнического
ин-та, 1966, 148, 147.
Старцев В.Н., Крылов Е.И., Кузьмин Ю. А. Экстракция титана-4
трибутилфосфатом из солянокислых растворов.—Ж- неорг. хим., 1966, 11, 2312.
Харитонов Ю. Я •, Буслаев Ю. А., Ионова Е. А. Инфракрасные . V
спектры поглощения соединении четырех хлор истого титана с мочевиной.—
Ж- неорг. хим., 1966, 11, 2396.
Харламова Е. М., Гурьянова Е. Н. Строение и свойства комплексов
четыреххлористого титана с сероорганическими соединениями.— Ж. структ.
К хим., 1965, 6, 859.
Ц еховольская Д. И., Меренкова Б. М. Исследование инфракрас-
ных спектров оксихлорида титана в растворе четыреххлористого титана.—
Ж- физ. хим., 1966, 40, 495.
Чжан Гу-чэн, Чэнь Нянь-и. Растворимость TiCl4 в некоторых рас-
ж плавленных хлоридах.— Acta metallurg. sinica, 1965, 8, 530.
(Чернов Р. В., Нога П. В. Взаимодействие фтортитанатных растворов с
В аммиаком.— Укр. хим. ж., 1968, 34, 351.
Шарова А. К-, Д е м е н е в Н. В., Полякова В. М., Милюти-
на М. И. Растворимость фтортитаната калия в минеральных кислотах.—
В кн.: Разделение близких по свойствам редких металлов. Металлупгиздат,
й М., 1962, 118.
Ш а р о в а А. К., Полякова В. М., Бамбуров В. Г., Черняв-
ская Е. И. Высаливание фтортитаната калия хлористым калием из суль-
фатно-фторидных растворов.— Труды Ин-та химии. Уральский филиал
К АН СССР, Свердловск, 1963, 7, 7.
|Ш в е ц А. А., Осипов О. А., Кузнецова Л. И., Швец Э. А.—
Комплексные соединения галогенидов металлов IV группы с эфирами а-ке-
тонофосфиновых кислот.— Ж- неорг. хим., 1966, 11, 342.
fce г р о в Л. Н. О некоторых свойствах продуктов частичного гидролиза тет- </
Г рахлорида титана.— Труды Всесоюзного научно-исследовательского ин-та
хим. реактивов и особо чистых хим. веществ, 26, 1964, 160.
Ш1 е в ч е н к о Ф. Д., К у з i н а Л. О. Сольволиз тетрахлорида титана в эта- к
F ноле.— В1сник Кшвського ушверситету. Сер. ф!з. та х!м. 1967, 7, 127.
till е л о м о в И. К-, Осипов О. А., Потатуев А. А. Взаимодействие
В, тетрахлорида титана с этаноламинами жирных кислот.— Ж. общ. хим., 1966,
36, 744.
ГА 1 у е a Е. С., Terrible Е. G. Взаимодействие галогенидов титана с цик-
лическими органическими лигандами, содержащими кислород и серу.—
Canad. J. Chem., 1965, 43, 3468.
(Baker К-, Fowles G. W. Серусодержащие комплексы титана.— Proc.
Chem. Soc., 1964, 362.
|B a r d a у G. A., Gregor I. K-, Lambert M. J., Wild S. В. Мышья- < *
ковые комплексы галогенидов титана.— Austr. J. Chem., 1967, 20, 1571.
9-614 321
В a Kd i”n i G., P о 1 1 i n i I., S p i n о 1 о G. Оптические свойства Гц.
p-TiCl3.— Phys, status solidi, 1968, 27, 95. kW
Beatjie I. R., F a w c e 11 V. Некоторые реакции оксихлоридов титанa. _
J. Chem. Soc., 1967, A, 10, 1583. т i
Becke-Geohring M., Slawisch А. Аддукты тетрахлорида титана
с фосфориловыми соединениями.— Z. anorg. Chem., 1966, 346, 295. к/i
‘Bernd Р., Schwartz D. Реакция хлорида четырехвалентного титана с
некоторыми альдегидами.— Proc. N. Dakota Academy Sci., 1965, 18.
Boustany К. S., Bernauer K-, Jacot Gllarmod А. Изучение
некоторых комплексов TiCl4 с пиридином и его производными.— Helv Chim
acta, 1967, 50, 1120.
Boni А. А. Давление пара тетрабромида титана.— J. Electrochem. Soc., 1966
113, 1089.
jBrouckere Guy. Рассмотрение гидратированного иона титана методом
МО.— Bull. Soc. chim. belg., 1967, 76, 407.
Brun L. Кристаллическая структура (TiCl4 • CH3COOC2H5)2.— Acta crystal-
logr., 1966, 20, 739.
Brandt P., Schmidt J. Кристаллическая структура модификации TiBr4>
устойчивой при комнатной температуре.— Z. anorg. Chem., 1966, 348, 257.
В u s 1 a u v G. A., Dyer D. S., R a gsda11 R.O. Гидролиз тетрафторида
титана.— Inorg. Chem., 1967, 6, 2208.
Clark R.J., Entington W., Lewis J., Nу ho Im R. S. Реакции
бидентатных арилов с четырехвалентными галогенидами IV группы___________.
J. Chem. Soc., 1966, А, 989.
Dahl J. Р., Johansen Н. Электронное строение тетраэдрических СгО|~
VO4~ и TiCl4.— Theoret. chim. acta, 1968, 11, 26.
Dehnicke K-, Weidlein J. О существовании катионов VOJ, TiO2^
и ZrO2\— Angew. Chem., 1966 , 78, 1065.
Dehnicke К. Получение и свойства азидхлоридов SnCl3N3, TiCl3N3 и VOC2N3.—
J. Inorg. a. Nucl. Chem., 1965, 27, 809.
Dehnicke К- Оксибромид титана ТЮВгг.— Chem. Ber., 1965, 98, 290.
Dehnicke К- О дифтор дихлориде и оксифториде титана.— Naturwis.,
1965, 52, 660.
Dickson F. Е., G о w 1 i n g E. W., Bentley F. F. Влияние заместите-
лей на валентные колебания азот — кислород и титан — кислород.— Inorg.
Chem., 1967, 6, 1099.
Dijkgraaf С., Rowsseau J. Р. G. Спектры кристаллов o;-TiCl3 и
a-TiBr3.— Spectrochim. acta, 1967, 23A, 1267.
Dijkgraaf C., Heel J. P. C., Rousseau J. P. G. Электронные пе-
реходы в гексахлоркоординированных титане и хроме.— Nature, 1966, 21 ,
5045, 185.
Dijkgraaf С. Общие черты электронных спектров TiCl4 и VOC13.— Spectro-
chim. acta, 1965, 21, 1419.
Dyer D. S., Ragsball R.O. Дш>трянс-изомерия для аддуктов тетрафто-
рида титана с замещенными 1-окисями пиридина.— Chem. Communs, 1966,
17, 601.
Dyer D. S., Ragsdale R.O. Изучение ядерного магнитного резонанса г
в смешанных аддуктах тетрахлорида титана.— Inorg. Chem., 1967, 6, 8.
Dyer D., Ragsdale R.O. Механизм обмена 19F в комплексах TiF4 • 2 (Д°'
нор).— J. Am. Chem. Soc., 1967, 89, 1528.
Ehrlich P-, Kiihnl H., Muller G. Попытка синтеза под давлением
гексахлортитанатов.— Z. anorg. Chem., 1968, 357, 172.
:F а г ber М., Darnell Аж. J. Энтропии и энтальпии хлоридов титана.-"'
J. Chem. Phys., 1965, 25, 526.
Feltz A., Grohmann Н. Стабилизированный сольватацией оксибром^Д
четырехвалентного титана ТЮВг6 • 4CH3CN.— Z. Chem., 1966, 6, 388.
Feltz А. Тетрахлороксотитанеат этил аммония.— Z. Chem., 1967, 7, 158.
322
r f F е 1 t z А. Тетрахлороксотитанаты [N (C2H5)4]2 TiCl4O и Rb2TiCl4O.— Z. anorg.
♦ Chem., 1965, 334, 242.
* Fisch er H. Исследование методом ЭПР реакции треххлористого титана с пе- v
рекисью'водорода.— Ber. Bumsenges phys. Chem., 1967, 71, 685.
i F о w 1 е s G. W. A., Walton R. А. Комплексы трехвалеитного титана. —
J. Chem. Soc., 1964, 4953.
BFowles G. W. A., Russ B. J. Гексахлоргалогениды-3-титана.— J. Chem.
Soc., 1967, A, 517.
|F о wles G. W., Lester T. E., W a 1 1 t о n R. А. Координационные ком-
плексы двухвалентного титана.— J. Chem. Soc., 1968, A, 1081.
. F о w 1 e s G. W., G r e e h e P. T., Lester T. E. Эфирные комплексы трех-
валентного титана.— J. Inorg. a. Nucl. Chem., 1967, 29, 2365.
\ F u n a k i K-, Saeki Y. Свойства титана, полученного восстановлением нат-
рием.— Bull. Tokyo Inst. Technol., 1965, 64, 41.
[ Q a r d n e r H. J. Хлораквокомплексы трехвалентного титана. —Austral. J. *
Chem., 1967, 20, 2357.
fi а У e r K.H., P a v k о v i c S. F., Tennenhouse G. J. Реакция те- J
Г трахлорида титана с уксусной кислотой и 1,4-диоксаном.-— Z. anorg. Chem.,
1967, 354, 74.
Hathaway В. J., HolahD. G. Галогениднометилцианидные комплексы
титана.— J. Chem. Soc., 1965, 537.
I J a i n S. C., R i v e s t R. Координационные комплексы галогенидов элемен-
тов IV группы.— J. Inorg. a. Nucl. Chem., 1967, 29, 2787.
I Jonson J.W., Shen K-J-, James W. J. Изучение кинетики окисления
Ti-3 до Ti-4 в растворе фтористоводородной кислоты. — J. Less-Common Me-
tals, 1968, 15, 177.
KhanMohd Mahfooz, Ahmad Nasser. 1, 10-Фенантролиновые
комплексы трехвалентного титана.— Bull. Chem. Soc. Japan, 1967, 40, 1254.
! К о 1 1 о r L., Schwarz G., Kollo D. Исследование каталитических сис-
тем Циглера-Натта.— Magyar kern, folyoicat, 1963, 72, 140.
hKrauss H. L., Hiittman H., Deffner U. О магнитном поведении
комплексов с переносом заряда TiCl4 с органическими л-донорами.— Z. anorg.
Chem., 1965, 341, 164.
|'К г a u s s H. L., Nickl J. О комплексах с переносом заряда четыреххлорис-
того титана с замещенными этилена.— Z. Naturforsch., 1965, 20b, 630.
(Lorenzo- S., Gianni M., Eugenio T., Luigi О. Электронные
спектры TiCl4, TiBr4, Til4.— Gazz. chim. ital., 1966, 96, 1785.
Lange G., Dehnicke К- Трифторид-бромид титана и гексафтортитанат
титанила.— Naturwis., 1966, 53, 38.
Machin D. J., M u r r a u K- S., Walton R. А. Магнитные свойства неко-
торых комплексов трехвалеитного Ti.— J. Chem. Soc., 1968, A, 195.
,M e h г о t г a R. С., Misra R. А. Взаимодействие тетра (mpem-бутилата) —
титана с ацетил хлоридом и тетрахлорида титана с трет-бути л ацетатом.—
Indian J. Chem., 1965, 3, 500.
Nagarajan S. Амплитуда колебания молекул тетрафторида и тетраиодида
титана.— Indian J. Pure and Appl. Phys., 1964, 2, 205.
Nitsche R. Синтез соединений титана при реакциях переноса.—J. Phys.
Chem. Solids. Suppl., 1967, 1, 215.
IN о r b u г у A. N., Sinha A. I. P. Координационные соединения тетрахло-
рида титана с нитрометаном.— J. Chem. Soc., 1966, А, 1814.
। R etitpierre В., Susz В. Р. Комплексы присоединения ароматических
кислоте TiCl4.— Helv. chim. acta, 1967, 50, 392.
[Pearce M. L., McCabe N. R. Давление пара тетрахлорида титана.— J.
Inorg. a. Nucl. Chem., 1965, 27, 1876.
P a v 1 i k L, К 1 i k о r k a J., H an dl ir, K-, Sedlmayer P. Донор-
но-акцепторные комплексы ацетил и 1,1'-диацетилферроцена с бромидом
четырехвалентного титана.— Z. Chem., 1967, 6, 427.
iPrasad S., Devi S. Изучение комплексообразования безводного хлорида
трехвалентного титана с моно- и диаминами.— J. Indian Chem., 1966, 4, 495.
/21* 323
Prasad S., Devi S. Изучение комплексообразования безводного хлорида
трехвалентного титана с вторичными и третичными аминами.— J. and Ргос
Inst. Chemists, 1966, 38, 178.
Rossetti G. Р., Susz В. P. Продукты присоединения паразамещенных
производных ацетофенона к ZnCL и TiCl4.— Helv. chim. acta, 1964, 47, 289.
Rossetti G. P. Продукты присоединения метазамещенных ацетофенонов с
TiCl4.— Helv. chim. acta, 1964, 47, 2053.
Russ В. T., Fowles G. S. Гексагалогекотитанаты.— Chem. Communs
1966, 1, 19.
Schlafer H. L., Schroeder W. Взаимодействие хлоридов и бромидов
трехвалентного титана с жидким аммиаком.— Z. anorg. Chem., 1966, 347, 45.
Schlafer Н. L., Schroeder W. Термическое разложение TiCl3 • 6NH3________
Z. anorg. Chem., 1966, 347, 59.
Schlafer H. L., Fr i t z H. P. О структуре соединений TiCl3 • 6H2O и
TiBr3 • 6H2O — Spectrochim'. acta, 1967, 23A, 1409.
* Schwartz D., Bernd P. Реакции тетрахлорида титана с некоторыми аль-
дегидами.— J. Less-Common Metals, 1964, 7, 108.
Slawisch A., Berke-Goehring М. Аддукты TiCl4 с РС15.— Z. Na-
turforsch., 1966, 21b, 589.
Takashi Y. Взаимодействие трихлорида титана с аминами. — Bull. Chem.
Soc. Japan, 1967, 40, 999.
Tornquist E. S. M., Richacdson J. T., Wilchinsky Z. W.,
Looney R. W. Образование твердых растворов в системе TiCl3 — А1С13_____
J. Catalisis, 1967, 8, 189.
* Takakura К., Ranby В. Исследование образцов свободных радикалов,
возникающих при взаимодействии ионов трехвалентного титана с перекисью
водорода.— J. Phys. Chem., 1968, 72, 164.
Walton R. А. Некоторые донорные свойства 1,4-тиоксана.— Inorg. Chem.,
1966, 5, 643.
Walter-Levy L., Ferey G. S., Syed H. I. Об основных бромидах
титана.— Compt. rend., 1967, C264, 700.
* Walter - Levy L., Ferey G. Исследование основных галогенидов четы-
рехвалентного титана.— Compt. rend., 1968, С266, 99.
Wendling E. Исследование титанатов методом ИК-спектроскопии.— Bull.
Soc. Chim. France, 1967, 1, 16.
Wendling E. Расчет d-d спектра гексафтортитаниатиона трехвалентного ти-
тана.— Bull. Soc. Chim. France, 1967, 8, 2738.
Wieker W., Crimmer A.-R. Исследование твердых продуктов присоеди-
нения TiCl4 и SnCl4 PCI5 методом ЯМР 31 Р.— Z. Naturforsch., 1967, 22b, 1220.
Wilchinsky Z. W., R о 1 p h R. W. Исследование процесса образования
твердого раствора при размоле смесей порошков TiCl3 — А13С13.— Advances
X-ray Analysis, 1964, 7, Denver, Colo., 203.
Zikmund M., Wai ent A., Stepnikova L. Взаимодействие хлорида
трехвалентного титана с алкилпроизводными пиридина.— Chem. zvesti 1967,
21, 901.
Zmbov К. F., Margrave J.L. Упругость паров TiF3 и устойчивость TiF2
и TiF.— J. Phys. Chem., 1967, 71, 2893.
ГЛАВА VII
СУЛЬФАТЫ ТИТАНА
Титан образует сульфаты, в которых проявляет валентность
3 и 4. Штелер и Виртвейн [135] сообщили о получении ими суль-
фата двухвалентного титана при растворении металлического ти-
тана в серной кислоте. Однако результаты их работы не подтвер-
дились в более поздних исследованиях [87, 126]. При растворении
металлического титана в серной кислоте образуется только сульфат
трехвалентного титана [87]. Неустойчивость сульфата двухвалент-
ного титана в сернокислых растворах объясняется высоким зна-
чением нормального окислительно-восстановительного потенциала
Ti2+/Ti3+ — —0,376 в. Ионы двухвалентного титана в сернокислых
растворах легко окисляются ионами водорода и не могут накапли-
ваться в значительной концентрации.
Сульфат четырехвалентного титана (средняя соль или нормаль-
ный сульфат) под действием воды, подобно сульфатам других мно-
говалентных металлов (V, Sb, U), гидролизуется с образованием
основных солей. Как и галогениды, сульфаты титана образуют
двойные соли с сульфатами щелочных, щелочноземельных и не-
которых других металлов. Нормальный сульфат титана в безвод-
ных средах, по-видимому, может присоединять органические и не-
органические адденды, давая комплексные соединения.
Вследствие этих особенностей, сульфаты титана представляют
обширный класс соединений, исследованный, однако, только в об-
щих чертах.
СУЛЬФАТЫ ТРЕХВАЯЕНТНОГО ТИТАНА
Существование сульфатов трехвалентного титана в сернокислых
растворах при восстановлении железом, цинком и оловом известно
было еще первооткрывателю титана Грегору. В твердом виде их
впервые получили, правда еще в недостаточно чистом виде, Эбель-
мен [85] и Глятцель [92].
ВОССТАНОВЛЕНИЕ ТИТАНА В СЕРНОКИСЛЫХ РАСТВОРАХ
В сернокислых растворах титан восстанавливается только до
трехвалентного состояния. Получить растворы сульфата титана
с валентностью менее 3 не удалось даже в результате применения
325
самых сильных восстановителей, например амальгамы цинка [14].
Нормальный окислительно-восстановительный потенциал Ti3+/Ti4^
при температуре 18° С равен — 0,05 в [84], поэтому до трехвалент-
ного состояния титан может восстанавливаться многими металлами.
Наиболее часто титан восстанавливают железом, цинком [120],
кадмием и алюминием. В химико-аналитической практике восста-
новление титана в сернокислых растворах проводят амальгамиро-
ванным цинком и жидкими амальгамами цинка [119], висмута
[133] и кадмия. Комплексообразователи (сульфат аммония, уксус-
нокислый аммоний, фтор-ион и др.) сдвигают окислительно-вос-
становительный потенциал Ti3+/Ti4+ в отрицательную сторону
[68]. В их присутствии восстановление титана до трехвалентного
состояния возможно только с помощью жидких амальгам цинка
и кадмия.
Для препаративного получения сульфатов трехвалентного титана удобно поль-
зоваться электролитическим восстановлением. Этот метод нашел применение во
многих работах [31, 84, 94, 112, 121, 122, 127, 134, 135]. Деменев с соавторами
[31], например, проводил электролитическое восстановление титана в растворах,
содержащих 105 г/л ТЮг и 250 г!л H2SO4. Катодом служили свинцовые пластины,
плотность тока на катоде была 0,015 а/см2. Катодное пространство отделялось от
анодного, заполненного разбавленной серной кислотой, пористой диафрагмой.
СПЕКТРЫ ПОГЛОЩЕНИЯ СЕРНОКИСЛЫХ РАСТВОРОВ
ТРЕХВАЛЕНТНОГО ТИТАНА
Растворы сульфата трехвалентного титана окрашены в фиоле-
товый или голубой цвет. Окраска растворов зависит от концент-
рации серной кислоты. Растворы фиолетового цвета имеют полосу
поглощения в пределах 460—580 нм [91, 134]. Она простирается
от 0 до 60—80% H2SO4, максимум поглощения лежит при 'к —
= 527 нм (рис. 84) [15]. Оптическая плотность растворов с одина-
ковой концентрацией Ti3+ повышается с увеличением концентра-
ции серной кислоты.
Растворы сульфата титана-3 в серной кислоте с концентрацией
более 60—80 вес. % окрашены в голубой цвет. Максимум свето-
поглощения приходится на длину волны 608 нм. Голубая окраска
простирается до концентрации серной кислоты 97% и более. В оле-
умной области сульфат титана-3 растворяется в незначительном
количестве, и из-за малой интенсивности окраски зарегистриро-
вать в этих растворах спектры поглощения в видимой части спектра
оказалось невозможным. Анализ спектров поглощения сернокис-
лых растворов сульфата трехвалентного титана еще не проведен,
и причины появления двух полос поглощения не выяснены. Мож-
но предполагать по аналогии с хлоридами, что фиолетовая окрас-
ка растворов обусловлена присутствием в них гексагидратов
[Ti (Н2О)6]3+- В концентрированных растворах H2SO4 происходит
замещение части молекул воды на сульфогруппы, что вызывает
переход окраски из фиолетовой в голубую.
326
Сульфаты четырехвалентного титана в сернокислых растворах
р концентрации серной кислоты 90% не обладают избирательным
Рис. 84. Спектры поглощения растворов сульфатов титана в H2SO4
I [15]:
сплошные кривые — растворы трехвалентного титана, прерывистые — четы-
рехвалентного. Концентрация титана в пересчете на ТЮ2 — 0,8 г/л, длина
кюветы — 50,17 мм. Цифрами на кривых обозначена концентрация серной
кислоты (вес. %).
{поглощением света в видимой области спектра (см. рис. 84). Поло-
са поглощения сульфата титана-4 лежит в ультрафиолетовой об-
Рис. 85. Смещение минимума
^пропускания света при добавле-
нии Ti4+ к сернокислому раство-
| ру Ti3^~ в 30% -ной H2SO4. Сум-
марная концентрация титана в
растворе 8 г/л ТЮг [15].
Рис. 86. Изомолярные серии оптической
плотности растворов сульфатов титана в
20% -ной H2SO4 (оптическая плотность из-
мерена на ФЭК-М с синим светофильтром
№ 3 и отнесена к кювете длиной 1 см) [15].
327
4.
ласти спектра. Однако при добавлении к фиолетовому раствору
сульфата титана-3 раствора сульфата титана-4 появляется темное
окрашивание. Максимум светопоглощения при этом сдвигается
с 527 до 472 нм, когда концентрация Ti44" составляет 50% валового
содержания ионов титана (рис. 85). Дальнейшее добавление ионов
Ti4+ к раствору сульфата трехвалентного титана не приводит к сме-
щению максимума светопоглощения. Окраска голубого раствора
сульфата титана-3 от прибавления ионов Ti4+ не изменяется и мак-
симум полосы светопоглощения не смещается.
Увеличение оптической плотности раствора и смещение макси-
мума светопоглощения при добавлении раствора сульфата титана-4
к раствору сульфата титана-3 свидетельствуют о взаимодействии
между ионами трех- и четырехвалентного титана. Изменение фиоле-
товой окраски растворов сульфата титана-3 при добавлении к ним
растворов сульфата титана-4 Шенк [134] приписал образованию
комплексного соединения с отношением Ti3+ : Ti4+ = 4:3. Го-
рощенко и Годнева [15] по сдвигу максимума светопоглощения
(см. рис. 85) и по изменению оптической плотности в изомолярных
смесях (рис. 86) заключили, что отношение Т*3+ : Ti4+ в этом
комплексе равно 1:1. Попытки выделить из растворов и полу-
чить в чистом виде этот комплекс оказались тщетными.
ПОЛУЧЕНИЕ СУЛЬФАТОВ ТРЕХВАЛЕНТНОГО ТИТАНА
Эбелемен [85], упаривая раствор треххлористого титана с
серной кислотой, получил кислый сульфат титана. В чистом виде
продукт не выделен. Содержание SO3 в нем большее, чем соответст-
вует формуле Ti2 (SO4)3.
Глятцель [92] из раствора металлического титана в серной кис-
лоте получил продукт, которому приписал состав Ti2 (SO4)3 • 8Н2О.
Электролитическим восстановлением раствора сульфата ти-
тана-4 в 50%-ной серной кислоте на амальгамированном свинцо-
вом катоде выделены кристаллы фиолетового цвета, которые были
отфильтрованы, промыты ледяной уксусной кислотой, а затем эфи-
ром. В сухом виде состав кристаллов выражается формулой
2Ti2 (SO4)3 • H2SO4 • 25Н2О [84, 93, 102, 135, 141]. Фиолетовую
соль можно получить при длительном кипячении металлического
титана и низшего окисла (ТЮ) в 40%-ной серной кислоте [112,
126].
Нейманн [118] полагает, что Ti2(SO4)3 образуется как проме-
жуточный продукт на катализаторе из двуокиси титана при окис-
лении сернистого газа по реакции
4TiOSO4 + 2SO2 = 2Ti 2 (SO4)3.
При упаривании сернокислого раствора фиолетовой соли по-
лучен осадок голубых и зеленых кристаллов, которым приписы-
вают состав Ti2(SO4)3 [141].
328
Зеленые кристаллы нерастворимы в воде, концентрированной
серной кислоте, спирте и эфире; в разбавленных серной и соляной
кислотах растворяются с образованием фиолетовой окраски.
Деменев с соавторами [31] по данным химических анализов
фиолетовым кристаллам сульфата трехвалентного титана припи-
сывает состав Ti2 (SO4)3 • H2SO4 • 8Н2О. Исследуемые образцы ав-
торы готовили электролитическим восстановлением сульфата ти-
тана в сернокислом растворе, кристаллы отмывались от маточной
жидкости спиртом и эфиром и высушивались в эксикаторе над
серной кислотой.
Блондель и соавторы [75], судя по потерям веса при прокали-
вании, приписывают фиолетовой соли состав Ti2 (SO4)3 • H2SO4 •
• 5Н2О, т. е. считают ее пентагидратом. В интервале темпера-
тур 75—140° С, по их наблюдениям, пентагидрат превращается
в безводную соль Ti2 (SO4)3 • H2SO4, а при 230—300° С — в средний
сульфат Ti2 (SO4)3. На кривой потери веса при температуре 200° С
отмечено образование сольвата 3Ti2 (SO4)3 • 2H2SO4.
Средний сульфат Ti2 (SO4)3 образует тетрагональные кристаллы,
изоморфные с Сг2 (SO4)3, с параметрами решетки а = 8,44, с —
= 21,95 А, Ризмер = 2,78, рреНтг = 2,80 г! см3. Соль устойчива до
300° С, в интервале температур 300—480° С полностью разлагается
до ТЮ2.
Регидратация Ti2 (SO4)3 при температуре 10° С приводит к об-
разованию неустойчивого кристаллогидрата Ti2 (SO4)3 • 8Н2О, ко-
торый превращается со временем в более прочный пентагидрат
Ti2(SO4)3 • 5Н2О. Последний при температуре 80—180° С превра-
щается в безводную соль Ti2(SO4)3.
Соль Ti2 (SO4)3 • H2SO4 • 8Н2О из-за малой растворимости в
серной кислоте средней концентрации предложена для отделения ти-
тана от примеси ниобия [52, 73]. По методу, описанному в работах
[52, 73], титан рекомендуется восстанавливать электролитически
в растворе с концентрацией 200 г/л H2SO4, а затем укреплять кон-
центрированной серной кислотой до содержания 600—900 г/л. Об-
разовавшиеся при этом кристаллы фиолетовой соли отфильтровы-
ваются. Ниобий остается в растворе и при желании может быть
получен в виде соли [(NH4)8Nb6O3 (SO4)12] • 21Н2О после дополни-
тельного восстановления и добавления сульфата аммония.
' СИСТЕМА Ti2O3 — SO3 — Н2О
Милютина с соавторами [51] изучила эту систему методом растворимости и
построила изотерму (рис. 87, табл. 42).
Из сернокислых растворов в системе Т1гО3 — SO3 — Н2О кристаллизуются два
кислых сульфата титана — Т1г (SO4)3 • H2SO4 • 8Н2О фиолетового и Tia (SO4)3 •
• 2H2SO4 • 5НгО голубого цвета. Состав твердых фаз авторы определили мето-
дом Скрейнемакерса. Кристаллы фиолетовой соли были получены также и препа-
ративным методом [31 ]. Голубой сульфат титана образует очень мелкие кристаллы,
Которые выделить в чистом виде не удалось.
329
<
Двойную точку на изотерме растворимости, в которой происходит кристал'
лизация двух сульфатов титана (фиолетового и голубого) одновременно, автору
{51 ] точно не определили. Она находится в интервале концентраций SO3 55_64%
Области кристаллизации сульфатов титана — фиолетового и голубого — на диа-
грамме растворимости соответствуют данным измерения спектров поглощения раст-
воров [15].
330
а о л и ц a
Растворимость в системе Ti2O3 — S03 —Н20 при температуре 25° С
ВКидкая фаза, вес. % Смесь жидкой и твердой фаз, вес. % Твердая фаза
Ti2O3 SO3 Ti2O3 so3
| 2,52 18,20 ——— TiJSOX • H,SO4 • 8Н,0
2,45 21,96 — — То же
2,39 27,17 — 1 » »
2,34 31,04 — — » »
1,10 31,32 19,10 47,74 » »
0,43 33,32 18,14 47,20 » »
0,14 36,66 — — » »
0,07 39,81 16,26 47,77 » »
0,05 42,25 — — » »
0,03 45,22 16,94 49,95 » »
0,02 47,97 — » »
0,01 49,41 15,37 50,52 » »
0,02 53,01 — — » »
0,02 55,21 16,02 52,77 » »
0,07 60,99 15,26 61,16 Ti,(SO4), • 2H,SO4 • 5Н..О
0,17 62,83 — — То же
0,43 64,46 12,41 63,56 » »
0,53 66,08 11,28 64,75 » »
0,58 67,84 11,55 65,64 » »
0,63 70,79 10,94 67,30 » »
0,72 72,99 10,50 69,24 »
0,68 74,89 10,11 70,14 »
0,46 76,79 12,59 70,75 » »
Сульфат титана Т12 (SO4)3 • H2SO4 • 8Н2О дает пластинчатые кристаллы ром-
бической формы . В работе [51 ] приведена рентгенограмма с набором межплоскост-
[ых расстояний, снятая с кристаллов этой соли. На воздухе сульфат Ti2 (SO4)3 •
Нг5О4 • 8Н2О окисляется до сульфата четырехвалентного титана. При нагре-
вании отщепляет воду, а затем SO2 и SO3. В остатке после прокаливания получае-
ся двуокись титана (рис. 88).
I При растворении безводного сульфата трехвалентного титана
I жидком аммиаке в течение 24 ч образуется белый осадок аммиа-
ката Ti2(SO4)3 • 6NH3 [141].
ДВОЙНЫЕ СУЛЬФАТЫ ТРЕХВАЛЕНТНОГО ТИТАНА
Сульфат трехвалентного титана с сульфатами аммония и натрия
ает двойные соли, а с сульфатами рубидия и цезия — квасцы,
(войные сульфаты титана-3 с литием и калием еще не получены.
(NH4)2SO4 • 3Ti2(SO4)3 • 18Н2О. Соль получена смешением вод-
ных растворов фиолетового сульфата титана и сульфата аммония
> эквимольных количествах [135]. Кристаллы осаждали из ки-
пящих
растворов,
отфильтровывали,
промывали водой
и спиртом.
остав их определен с помощью химического анализа.
331
Двойные сульфат титана-3 и аммония более стойки к окислению
кислородом воздуха, чем кристаллы простого сульфата титана-3,
В воде и в серной кислоте они растворяются плохо, лучше — в со-
ляной кислоте. При кипячении с концентрированной серной кисло-
той превращаются в сульфат титана зеленого цвета, которому
авторы [114] приписывают состав Ti2(SO4)3.
Na2So4 • Ti2 (SO4)3 • 5H2O. Двойной сульфат этого состава описал Кнехт [109]
Rb2SO4 • Ti2 (SO4)3 • 24Н2О. Титано-рубидиевые квасцы получены кристаллиза-
цией из раствора сульфата трехвалентного титана после добавления сульфата ру-
бидия [84, 121, 122]. Они образуют кубические кристаллы с показателем свето-
преломления 1,4649 [121, 122]. На воздухе выветриваются и трехвалентный титан
окисляется до четырехвалентного. Упругость водяного пара над титанорубидц-
евыми квасцами такова [86]:
/°,С 40 50 60 71 80 90 100 105
р, мм рт. ст. 7 18 43 89 153 268 525 700
Cs2SO4 • Ti2 (SO4)3 • 24Н2О. Титано-цезиевые квасцы приготовлены по ана-
логичному методу [84, 112, 121, 122], промыты холодной водой и высушены между
листами фильтровальной бумаги. Они также имеют кубическую симметрию, по-
казатель преломления 1, 472 [112]. На воздухе неустойчивы, быстро выветривают-
ся и титан окисляется до четырехвалентного. Упругость пара над кристаллами
следующая [86]:
/°,С 25 30 38 45 52 61 80 90 95
р, мм рт. ст. 7 14 31 55 83 146 244 516 613
Измерен спектр поглощения кристаллов титаноцезиевых квас-
цов [98]. Он оказался идентичным спектру поглощения водных
растворов, что указывает на одинаковое окружение ионов Ti3+
в кристалле и растворе.
СУЛЬФАТЫ ЧЕТЫРЕХВАЛЕНТНОГО ТИТАНА
Растворы сульфатов титана впервые получил Грегор, обраба-
тывая серной кислотой остаток после выщелачивания из ильменита
железа и марганца. В твердом виде сульфат четырехвалентного
титана выделили Берцелиус и Розе [82], нагревая двуокись ти-
тана с серной кислотой. Однако состав полученной «сернокис-
лой титановой соли», растворимой при нагревании в воде, авторы
точно не определили.
В литературе описано более 20 простых сульфатов титана,
полученных препаративными методами, но подтверждена индиви-
дуальность методами физико-химического анализа только шести
соединений, кристаллизующихся из сернокислых растворов в ин-
тервале температур 75—330° С и имеющих состав: TiOSOr
TiOSO4 • Н2О, TiSO4 • 2Н2О, TiOSO4 . H2SO4 • ЩО, TiOSO4 •
• H2SO4 • 2H2O и Ti (SO4)2 [3—5, 16].
Индивидуальность остальных описанных соединений либо не
проверена методом физико-химического анализа, либо отвергнута
как ошибочная.
332
Кроме простых сульфатов известны двойные сульфаты титана
ро щелочными и щелочноземельными металлами. Исследованы они
недостаточно и можно ожидать, что будет открыто еще много не-
известных в настоящее время двойных солей.
Горощенко и Белофкосков [3, 16] провели классификацию суль-
фатов титана на следующие три вида: нормальный сульфат титана,
гитанилсульфаты, кислые титанилсульфата.
Нормальный сульфат титана Ti (SO4)2 образуется в результате
замещения двух атомов кислорода в двуокиси титана на сульфо-
группы; титанилсульфат TiOSO4— при замещении на сульфогруп-
пу одного атома кислорода. Титанилсульфат в концентрированных
сернокислых растворах присоединяет одну молекулу H2SO4, давая
кислые титанилсульфата в виде кристаллогидратов с одной и двумя
молекулами воды.
Кислые титанилсульфата рассматривают и как комплексные
кислоты, образующие двойные (комплексные) соли типа
(NH4)2 TiO (SO4)2. Нормальный сульфат титана кислых солей не
образует, хотя двойные соли известны, например (NH4)2 Ti (SO4)3.
Лучинский [50] предложил рассматривать сульфаты титана как
продукты замещения кислорода в анионе ТЮз~ на сульфо-группы.
В соответствии с его схемой, в результате взаимодействия сульфо-
групп должны получаться следующие ионы: [TiO2SO4]2-,
[TiO(SO4)2]2~, [Ti (SO)4J3~. Но эта схема мало правдоподобна. Су-
ществование иона [TiO2SO4]
этот ион, никем не подтверждено. Замещение же атомов кислоро-
да, связанных с титаном, на сульфо-группы возможно только
в кислой среде, где ионы ТЮз~ полностью отсутствуют.
I
2—
или солеи, в состав которых входит
МЕТОДЫ ПОЛУЧЕНИЯ РАСТВОРОВ СУЛЬФАТОВ ТИТАНА
Все известные методы получения сульфатов титана сводятся
к кристаллизации их из сернокислых растворов. В свою очередь
растворы сульфатов титана могут быть приготовлены взаимодейст-
вием серной кислоты с различными титансодержащими материа-
лами.
Технические растворы сульфатов титана в крупных промышлен-
ных масштабах получаются нагреванием концентратов природных
минералов (ильменита, рутила, аризонита, сфена и др.) или ти-
тановых шлаков с концентрированной серной кислотой.
, Растворы чистых сульфатов титана для препаративных целей
получают из гидроокиси и двуокиси титана.
По данным [61], гидроокись титана (метатитановая кислота) растворяется в
60—70%-ной серной кислоте (отношение H2SO4 : ПОг равно 3—4) при нагревании
до температуры 100—120° С.
При растворении метатитановой кислоты образуются весьма концентрирован-
ные растворы сульфатов титана без нерастворимого остатка. Недостатком этого
метода служит то, что метатитановая кислота, получаемая в промышленных
333
масштабах только на лакокрасочных заводах, содержит примеси органических Вс
ществ, которые вместе с гидроокисью титана переходят в раствор и загрязняют ег0~
Двуокись титана с серной кислотой реагирует менее энергично, чем метатита1
новая кислота. Полностью перевести двуокись титана в раствор или в растворимый
сульфаты нагреванием с серной кислотой обычно не удается. Продукты реакции
всегда содержат остаток непрореагировавшей двуокиси титана, отделить которую
от раствора довольно трудно.
В литературе описано много различных методов получения растворов судь,
фата титана из его двуокиси. Приводим описание одного из них [27]. 100 г двуокиси
титана смешивают с 220 мл концентрированной серной кислоты (удельный вес 1,8з\
и нагревают, периодически помешивая, до температуры 200° С примерно в течение
1 ч, пока содержимое не примет сметанообразную консистенцию.
По окончании реакции сплав охлаждают до 50—60°С, выливают тонкой струей
в дистиллированную воду при непрерывном перемешивании. На растворение спла-
ва расходуется 700—800 мл воды, температура не должна превышать 60—70° С.
Для полного растворения сплава раствор нагревают 1—2 ч до температуры 60—-
70° С. После этого остаток непрореагировавшей двуокиси титана отфильтровывают
на воронке Бюхнера через бумажный фильтр или отделяют отстаиванием в течение
1—2 суток. Метод позволяет получать раствор сульфата титана, содержащий
80—100 г/л ТЮг и около 500 г/л H2SO4.
Растворы сульфатов титана для препаративных целей можно
приготовить из TiCl4 и губчатого металлического титана. Четырех-
хлористый титан обрабатывают концентрированной серной кис-
лотой в платиновой посуде до полного удаления хлористого водо-
рода. Полученную сыроподобную массу затем растворяют в раз-
бавленной серной кислоте [135, 141].
Металлический титан можно перевести в раствор нагреванием
с 20—40%-ной серной кислотой, однако получить концентрирован-
ные растворы не удается, так как по мере накопления ионов Ti3+
растворение титана замедляется. Поэтому удобно вначале рас-
творять титан в минимальном количестве концентрированной со-
ляной кислоты, которая реагирует с титаном при нагревании
очень бурно. В остатке после растворения металла находится
небольшое количество нерастворимых примесей, которые необхо-
димо отфильтровать. Затем к фильтрату добавляют концентриро-
ванную серную кислоту и кипятят до полного удаления хлористого
водорода. Из раствора при этом кристаллизуется сульфат трех-
валентного титана Ti2 (SO4)2 • H2SO4 • 8Н2О. Фиолетовые кристал-
лы его отфильтровывают на воронке Бюхнера .через стеклянный
фильтр, промывают 50%-ной серной кислотой, растворяют в воде
и трехвалентный титан окисляют до четырехвалентного добавле-
нием рассчитанного количества перекиси водорода.
В сернокислых растворах титан находится в форме различных
сульфатов в зависимости от концентрации раствора и содержания
в нем свободной серной кислоты.
ПРОСТЫЕ СУЛЬФАТЫ ЧЕТЫРЕХВАЛЕНТНОГО ТИТАНА
Ti (SO^. Нормальный сульфат титана очень часто упоминают
в литературе и нередко, без особых на то оснований, считают,
334
что в сернокислых растворах титан находится в форме этого суль-
фата. В чистом виде он получен по реакции [72, 95]
1— а 1 ^^4/2 “I ^2^Ь^12у
которая протекает в две стадии. Вначале из TiCl4 и серного ангид-
[ рида в хлористом сульфуриле образуются продукты присоединения
(TiCl4 • 2SO3 и TiCl4 • 3SO3) в виде темно-желтых игловидных
> кристаллов. При нагревании смеси в колбе с обратным холодиль-
ником в течение 12 ч продукты присоединения разрушаются и суль-
I фат титана выпадает в осадок. Кристаллы Ti(SO4)2 отфильтровы-
вают (в отсутствие паров воды), промывают хлористым сульфурилом
и сушат в течение суток в токе азота, предварительно очищенно-
го от паров воды пропусканием над Р2О5.
Белоскосков и Горощенко [4, 16] нормальный сульфат титана
приготовили кристаллизацией из сернокислого раствора, укреплен-
ного олеумом до содержания SO3 более 80%, в виде мелких крис-
таллов сферической формы с шелковистым блеском.
Кристаллы Ti (SO4)2 очень гигроскопичны, хорошо растворимы
в воде и разбавленных минеральных кислотах, разлагаются спир-
том, эфиром и ацетоном. В чистом виде авторы получить их не
смогли. Состав кристаллов определен без отделения их от маточ-
ной жидкости по методу Скрейнемакерса.
Образцы препаратов, полученные Хайком и Энгельбрехтом [95],
при нагревании до 150° С не изменялись, при более высокой тем-
пературе разлагались с потерей SO3. Отщепление SO3 протекает
в две стадии и заканчивается при температуре красного каления.
Конечным продуктом разложения нормального сульфата титана
является чистая двуокись титана.
' В работах некоторых авторов сообщается о получении гидратов
нормального сульфата титана, например Ti (SO4)2 • ЗН2О [92],
Ti (SO4)2 • 4Н2О [6], Ti (SO4)2 • 9Н2О [142]. Существование соеди-
нений этого типа нам кажется сомнительным. Нормальный сульфат
титана, как отмечают все, изучавшие его авторы [4, 16, 72, 951,
под действием воды легко гидролизуется и по этой причине не
может существовать в форме кристаллогидратов. Вероятно, упо-
мянутые соединения имеют иное строение, не являясь кристал-
логидратами.
Е • TiOSO4. Впервые оксисульфат титана или титанилсульфат по-
лучил Мерц [111] упариванием раствора двуокиси титана в серной
кислоте. Образование титанилсульфата при нагревании сернокис-
лых растворов двуокиси титана выше 200° С наблюдали Блондель
[761 и Вёлер с соавторами [152]. Титанилсульфат образуется в ре-
зультате прокаливания Ti2(SO4)3 при температуре 500—550° С [88,
; 112]. По мнению [118], титанилсульфат образуется на каталити-
ческой массе по реакции
TiO2 + SO3 = TiOSO4.
335
По внешнему виду TiOSO 4—белый кристаллический порошок
с желтым оттенком. Блондель [76] относит кристаллы TiOSO
к ромбической системе. В работе [16] приведена дебаеграмма крис-
таллов титанилсульфата; показатели преломления Ng — 1,712
Np = 1,628 [31.
Баррадух [77] в инфракрасных спектрах титанилсульфата об-
наружил широкие полосы с частотой 810 и 920 см~\ относящиеся
Рис. 89. Потери веса при прокаливании сульфатов титана:
1 — TiOSO4; 2 — TiOSO4 • H2O; 3л— TiOSO4 • 2Н2О [5].
к связи титан — кислород. Высокая частота валентных колебаний,
по мнению автора, свидетельствует о близости этой связи к двойной.
При нагревании ТЮ5О4выше 550° С он разлагается по реакции
(рис. 89)
TiOSO4 - TiO2 + SO3.
Упругость пара над TiOSO 4, по данным [152], следующая:
t°, С 468 490 530 550 560 570 580
р, мм рт. ст. 13 14 28 85 238 448 >760
Титанилсульфат устойчив на воздухе, плохо растворяется в во-
де и в разбавленных кислотах, разлагается растворами едких
щелочей и аммиака с образованием гидроокиси титана.
T1OSO4 • Н2О. Моногидрат титанилсульфата получили, по-ви-
димому, еще Берцелиус и Розе [82].
Сагава [136, 137] и Белокосков [4] выделили его кристал-
лизацией из сернокислых растворов при исследовании системы
TiO2 — SO3 — Н2О методом растворимости. Приводим описание
разработанного нами препаративного метода получения TiOSO4 •
• Н2О кристаллизацией из сернокислого раствора.
336
Рис. 90. Внешний вид кристаллов [4] (X 100):
а — TiOSO, • Н2О; б — TiOSO4 • Н2О.
Рис. 103. Внешний вид користаллов
(NH4)2Ti (SO4)3 [19] (Х55).
Рис. 106. Внешний вид кристаллов
(NH4)2SO4 . 3TiOSO4 - Ti (SO4)2 [19]
(X90).
Рис. 108. Внешний вид кристаллов
a-(NH4)2TiO (SO4)2 [19] [Х48).
Рис. ПО. Внешний вид кристаллов
(NHJaTiO (SO4)2 • Н2О [19].
Рис. 122. Внешний вид кристаллов
CaTi (SO4)3 [16].
Рис. 127. Внешний вид кристаллов
FeTi (SO4)3 [27].
Раствор сульфата титана упаривают до температуры кипения около 140° С.
В интервале температур 140—145 ° С из раствора начинает кристаллизоваться од-
новодный титанилсульфат. Кристаллы TiOSO4 • НгО белого цвета выпадают в
течение 2—3 ч в интервале температур 140—150° С. При более высокой температу-
ре возможна кристаллизация безводного титанилсульфата, поэтому нагревание
выше 150° С недопустимо. После окончания кристаллизации раствор охлаждают
на воздухе до 15—20° С и декантируют. Осадок смешивают с 3—4 кратным ко-
личеством ледяной воды, дают кристаллам в течение 0,5 ч осесть на дно. Жидкость
с осадка декантируют, а осадок переносят на воронку Бюхнера с бумажным филь-
тром, промывают водой и спиртом или ацетоном. Кристаллы сушат в сушиль-
ном шкафу при 70—100° G.
Иногда при получении TiOSO4 • Н2О описанным методом из сернокислого
раствора при температуре около 120° С кристаллизуется двухводный титанилсуль-
фат в виде тонких игольчатых кристаллов. Раствор в этом случае надо разбавить
водой до полного растворения кристаллов и повторить опыт.
Одноводный титанилсульфат — белый кристаллический поро-
шок, устойчивый на воздухе. В холодной воде и в разбавленных
Рис. 91. Идеализированная структура TiOSO4 • Н2О [ПО].
кислотах при комнатной температуре он растворяется хорошо,
но растворение протекает медленно и насыщение растворов на-
ступает через 1—5 суток. Нагревание до температуры 50—60° С
ускоряет растворение, сокращая время насыщения до 2—3 ч. Кон-
центрированные растворы TiOSO4 • Н2О при комнатной темпера-
туре устойчивы и не подвергаются гидролизу.
Под микроскопом кристаллы TiOSO4 • Н2О имеют вид утол-
щенных пластинок (рис. 90, см. вклейку) с сильным двойным лу-
чепреломлением, Ng — 1,830, Np= 1,705 [3].
22 9-614
337
Лундгрен [ПО] исследовал структуру TiOSO4 • Н2О методОм
рентгеноструктурного анализа. Соль кристаллизуется в ромбиче-
ской системе с параметрами решетки а = 9,788, Ь = 5,120, с
= 8,598 А, 2 = 4, (£ед. гр. Р2^^г.
Расстояния между атомами в идеализированной структуре сле-
дующие (A): Ti — Ti = 3,60; Ti—Ox = 1,80; Ti — O3 = 2,00-
Ti —O4=l,93; Ti — O5 = 1,93; Ti —Oe-2,01.
На рис. 91 приведена идеализированная структура TiOSO4 .
* Н2О, имеющая симметрию фед. гр. Рпта, которую надо слегка
Рис. 92. Термограммы нагревания [16]:
'а — TiOSO4 • Н2О; б — TiOSO4 . 2Н,О.
деформировать, чтобы получить реальную структуру. Она состоит
из цепочек (ТЮ)^+, параллельных оси b и соединенных друг
с другом группами SO1“. Координация Ti — О дополняется до
искаженных октаэдров за счет соседних молекул воды.
Судя по инфракрасным спектрам поглощения, связь Ti—О
в TiOSO4 • Н2О, как и в безводном TiOSO4, близка к двойной.
Присутствие ионов гидроокиси в кристаллах TiOSO4 • Н2О не об-
наружено.
Молекула воды в TiOSO4 • Н2О связана очень прочно. При
нагревании (рис. 89, 92) отщепление ее происходит выше 300° С.
Моногидрат при этом превращается в безводный титанилсульфат.
Выше 420° С безводный титанилсульфат разлагается с отщепле-
нием SO3 до двуокиси титана со структурой анатаза. На термограм-
ме нагревания при температуре 975° С зафиксирован эндотерми-
ческий тепловой эффект превращения анатаза в рутил [96].
TiOSO4 • 2Н2О. Дигидрат титанилсульфата впервые получил
Блондель [76] кристаллизацией из раствора двуокиси титана
в 50%-ной серной кислоте при температуре 100° С, а затем и другие
авторы [7, 8, 61, 99, 100, 101] кристаллизацией из сернокислых
растворов при температуре выше 100° С. Кристаллизация TiOSO4 •
• 2Н2О из водных растворов при комнатной температуре проис-
338
ходит только после длительного выстаивания растворов (более
месяца). Как потом оказалось при исследовании системы TiO2 —
SO3 — Н2О методом растворимости [4, 16, 136, 137], кристаллиза-
ция TiOSO4 • 2Н2О из сернокислых растворов выше 100° С являет-
ся метастабильным процессом. По этой причине при температуре
выше 100° С TiOSO4 • 2Н2О иногда не кристаллизуется.
Двухводный титанилсульфат как технический продукт можно
кристаллизовать из сернокислых растворов, получаемых из мало-
железистых титановых минералов, например из перовскита [17].
Для препаративного получения TiOSO4 • 2Н2О мы рекомендуем следующий
метод. К раствору сульфата титана, содержащему около 100 г!л TiO2 и 600 г!л
H2SO4 (общей), добавляют «затравку» кристаллов TiOSO4 • 2Н2О в количестве
1—2% и нагревают 3—4 ч при температуре 120° С. За это время TiOSO4 • 2Н2О
обычно выкристаллизовывается на 80—90%. Кристаллы его отфильтровывают
на воронке Бюхнера через фильтр из стеклоткани и промывают 40%-ной серной
кислотой, а затем 96%-ным этиловым спиртом. Кристаллы сушат при температуре
100° С в сушильном шкафу. Полученный продукт имеет состав, близкий к теоре-
тическому.
Маточную жидкость от кристаллов TiOSO4 • 2Н2О можно от-
мывать и холодной водой, но выход соли при этом уменьшается
из-за частичного растворения.
Двухводный титанилсульфат образует длинные игловидные крис-
таллы (см. рис. 90), симметрия и параметры решетки которых
еще не определены. В работе [3] приведен набор межплоскостных
расстояний, показатели преломления кристаллов TiOSO4 • 2Н2О
Ng = 1,878, Np = 1,75. Богуславская [7] для кристаллов титанил-
сульфата, которые являлись, по-видимому, дигидратом, нашла
Ng= 1,873, Np = 1,79. В другой работе [8] приводятся очень
малые величины показателей преломления.
В воде TiOSO4 • 2Н2О растворяется хорошо, но медленно. Вод-
ные растворы его метастабильны, но сохраняются при комнатной
температуре длительное время. На воздухе капли этих растворов
высыхают, превращаясь в стекловидную массу. В серной кислоте
с концентрацией более 40% двухводный титанилсульфат мало раст-
ворим. Концентрированная серная кислота и олеум дегидрати-
руют TiOSO4 • 2Н2О.
Термическое разложение TiOSO4 • 2Н2О начинается при тем-
пературе около 300° С (рис. 89, 92). Вначале отщепляются одно-
временно две молекулы воды и образуется безводный титанилсуль-
фат, который при температуре около 450° С начинает разлагаться
на ТЮ2 и SO3. Разложение заканчивается несколько выше 500° С.
На термограмме нагревания (см. рис. 92) зарегистрированы эндо-
термический эффект при 200° С (отщепление воды); экзотерми-
ческие эффекты при 540 и 590° С, вызванные, по-видимому, обра-
зованием из аморфного безводного титанилсульфата кристалличе-
ского продукта, и эндотермический эффект при 660° С, отвечающий
разложению титанилсульфата с отщеплением SO3. Образование
22*
339
аморфного титанилсульфата в результате обезвоживания дигид-
рата и последующую кристаллизацию его наблюдал Ханрахан [96]
в интервале температур 225—500° С. Разложение TiOSO4, по дан-
ным термографических исследований этого автора, происходит
при 625° С, а при температуре 975° С на термограмме нагревания
зафиксирован тепловой эффект, вызванный превращением анатаза
в рутил.
Кристаллическая структура двухводного титанилсульфата еще
не исследована.
TiOSO4 • H2SO4 • Н2О. Это соединение впервые получили Бе-
локосков и Горощенко [3—5, 16] при исследовании системы TiO2 —
SO3 — Н2О методом растворимости. Кристаллы его выделены из
сернокислых растворов с концентрацией около 70% SO3 при тем-
пературе 100 и 150° С. Они имеют вид мелких иголок, хорошо
растворимых в воде; спиртом, эфиром и другими полярными орга-
ническими растворителями разлагаются. В чистом виде кристаллы
этой соли не выделены. Состав их определен графически по методу
Скрейнемакерса без отделения от маточной жидкости.
T1OSO4 • H2SO4 • 2Н2О. Продукт этого состава описал Глят-
цель [92]. Он был получен действием на раствор сульфата трех-
валентного титана азотной кислоты с последующим упариванием
I до превращения в желтую желеобразную массу приведенного выше
состава. Данные химического анализа автором не приведены.
Сульфат титана этого же состава получили Белокосков и Го-
рощенко [3—5, 161 при исследовании системы TiO2 — SO3 — Н2О
методом растворимости. Он кристаллизуется из сернокислых раст-
воров с концентрацией около 65% SO3 при температуре 100° С
в виде бесцветных мелких игольчатых кристаллов, хорошо рас-
I творимых в воде и в разбавленной серной кислоте. По внешнему
виду продукт Белокоскова и Горощенко отличается от описанного
Глятцелем. В чистом виде получить его не удалось. Состав его
определен с помощью индифферентной добавки, в виде которой
использован сульфат меди.
Кроме сульфатов титана, индивидуальность которых подтверждена работами
многих авторов, описано большое число соединений, индивидуальность которых
нуждается в подтверждении.
Блондель [76] описал сульфаты титана, имеющие состав 7TiC>2 • 2SO3 • хН2О,
Е 5ТЮ2 • SO3 • хН2О, 2TiO? • SO3 • хН2О и 2TiO2 • 3SO3 • ЗН2О. Первые три сое-
динения получены из разбавленных сернокислых растворов, а последний — при
взаимодействии концентрированной серной кислоты с двуокисью титана при 120—
225° С. По-видимому, все продукты являются смесями нескольких фаз, так как
область концентраций в системе TiO2 — SO3 — Н2О в интервале температур, при
которых они получены, изучена методом физико-химического анализа и существо-
вание этих соединений не было подтверждено.
Сагава [136, 137] при исследовании системы TiO2—SO3—Н2О описал сульфаты
титана 6TiO2 • 5SO3 • 4Н2О, 2TiO2 • 3SO3 • 5Н2О, TiO2 • 4SO3, TiO2 • 4SO3 •
r 4,5H2O и 2TiO2 • 5SO3 • 5H2O. Состав этих сульфатов определен по методу
| Скрейнемакерса. Как показали Белокосков и Горощенко [4, 5, 16], описанные
Сагавой фазы йвляются индивидуальными соединениями, но состав их определех!
340
недостаточно точно из-за больших погрешностей в графических построениях, вы-
званных большой «влажностью» остатков.
Гейсов [89, 91] выделил из сернокислых растворов при температуре 340° С
сульфат титана 4TiO2 • 3SO3, а при 420° С — 2TiO2 • SO3.
Богуславская [6] описала тетрагидрат сульфата титана Ti (SO4)2 • 4Н2О.
Она получила его нагреванием фтортитаната калия K2TiFe с концентрированной
серной кислотой до выделения белых паров SO3 и образования стекловидной массы.
Последняя растворялась в холодной воде и из раствора кристаллизовались белые
ромбические кристаллы, нерастворимые в воде, спирте, эфире, ацетоне, но хорошо
растворимые в разбавленных кислотах.
В монографии Торнтона [142] упоминается девятиводный сульфат титана
Ti (SO4)2 • 9Н2О — мелкокристаллический порошкообразный продукт.
Описан тетрагидрат титанилсульфата TiOSO4 • 4Н2О в виде гранул с глянце-
вым блеском [138].
Розенгейм и Шютте [131 ] нагреванием смеси раствора сульфата титана и спир-
та в колбе с обратным холодильником получили соединение TiOSO4 • 5Н2О.
В патенте США [66] приводится метод получения сульфата TiO2 • 3SO3 " 5Н2О
нагреванием нитрида титана с 50%-ной серной кислотой. Продукт в виде стекло-
видной массы получается упариванием раствора при температуре 130° С.
f , СИСТЕМА ТЮ2 — SO3 — Н2О
Система TiO2 — SO3— Н2О исследована методом растворимос-
ти несколькими авторами [3—5, 16, 128, 136, 137J.
Рейндерс и Киз [128] изучили растворимость в серной кислоте
различных концентраций двух фаз — гидроокиси титана и TiOSO4 •
• 2Н2О при температуре 25° С. Двухводный титанилсульфат, по
данным авторов, в разбавленной серной кислоте растворяется ин-
конгруэнтно, образуя метастабильные растворы (рис. 93). Равно-
весной твердой фазой в этих растворах является гидроокись ти-
тана. Точка двунасыщения растворов двухводным титанилсульфа-
том и гидроокисью титана лежит около 35% SO3 и 4% TiO2. В сла-
бокислых растворах растворимость TiOSO4 • 2Н2О очень велика.
С увеличением концентрации серной кислоты растворимость в ней
TiOSO4 • 2Н2О уменьшается до 0,1% ТЮ2 при содержании 45%
SO3. В серной кислоте большей концентрации растворимость
TiOSO 4 • 2Н2О снова возрастает.
Сагава [136, 1371 построил изотермы растворимости в системе
TiO2 — SO3 — Н2О при температурах 100 и 150° С. Данные Сага-
вы по растворимости сульфатов титана в серной кислоте подтверж-
дены [4, 5 и 16], однако состав твердых фаз, определенных им,
оказался неточным.
Белокосков и Горощенко [3—5, 16] изучили растворимость
в системе TiO2 — SO3 — Н2О в широком интервале концентраций
при 25—300° С (табл. 43) и построили изотермы и политерму раст-
воримости (рис. 94, 95). Насыщенные растворы при температуре
выше 100° С получали кристаллизацией твердых фаз из пересы-
щенных растворов. Установление равновесия проверялось насы-
щением растворов твердыми фазами. Ниже 100° С скорость крис-
таллизации сульфатов титана из пересыщенных растворов мала.
341
Рис. 93. Диаграмма растворимости системы TiO2 — SO3 — Н2О
при 25° С:
а — TiO2 . хН2О; б — TiOSO4 • 2Н2О.
Рис. 94. Изотерма растворимости системы TiO2 — SO3 — Н2О
при температуре 100° С:
а — ТЮ2 - хН2О; в — TiOSO4 • Н2О; с — TiOSO4 • H.SO4 2Н.О;
d - TiOSO4 . H2SO4 . H2O; e - Ti (SO4)2; f - TiOSO4- 2H2O
(метастабильная).
Таблица 43
Растворимость в системе TiO2 — SO3 — Н2О (вес. %)
1 Состав жидкой фазы Остаток Твердая фаза
| Ti°2 SO3 тю2 so3
Температура 20° С
0,362 5,75 - ТЮ2-хН2О
0,405 13,12 —— — То же
0,814 22,07 — •—- » »
2,610 29,35 — » »
V 26,78 26,70 — TiOSO4-2H2O метает.
18,56 26,62 1 « —- То же
10,11 27,03 —• » »
5,15 29,52 1 ' — * » »
1,61 34,98 1 , — — TiOSO4-2H2O
0,108 40,27 —* — То же
0,030 47,52 — ‘ -—. TiOSO4.2H2O метает.
0,203 52,44 —- То же
s 27,55 27,61 — TiOSO4-H2O метает.
15,25 27,61 — —- То же
8,28 29,25 г—— — » »
3,15 34,92 — » » —
1,53 38,44 ** » »
0,248 ,41,15 - — — » »
0,063 43,70 •—- — » »
0,020 48,46 —• —11 — TiOSO4-H2O
0,012 54,91 — — То же
0,045 58,90 — » »
0,227 62,15 — - « TiOSO4-H2O метает.
0,264 58,11 — " — — TiOSO4 • H2SO4 • 2Н2О метает.
0,093 62,35 — —— TiOSO4-H2SO4-2H2O
0,064 65,10 — —- То же
0,142 69,50 — ——— TiOSO4 • H2SO4 • 2Н2О метает.
0,263 63,92 —— ТiOSO4 • H2SO4. Н2О метает.
0,066 69,05 — —- TiOSO4-H2SO4.H2O
0,035 74,32 —. — То же
0,130 80,46 — —- » »
0,662 81,40 1 1 Ti(SO4)2 метает.
0,390 83,97 —— - 1 То же
0,427 87,71 — — '1 » Ti(SO4)2
0,575 88,80 •— То же
Температура 50° С
0,256 11,87 — ТЮ2-хН2О
0,56 21,35 То же
1,07 30,75 — —...» » »
2,25 36,86 — TiO2«xH2O метает.
27,02 27,10 - —- TiOSO4-2H2O метает.
25,37 27,06 “ 1 — —-- То же
24,18 27,13 —- — ~* » »
12,66 27,48 *— т * 1 1 » »
4,10 29,36 —— — » »
2,05 30,48 — •— TiOSO4-2H2O
0,477 34,48 —- То же
343
Продолжение табл. 43
Состав жидкой фазы Остаток Твердая фаза
TiO2 so8 TiO2 so8
0,182 37,36 —* TiOSO«-2H,O
0,084 41,36 — - — Тоже
0,056 45,51 — В В
0,051 50,17 — TiOSO4-2H2O метает.
0,167 54,96 —- I •—• То же
1,32 59,30 — - —— в »
27,94 27,96 — TiOSO4-H2O метает.
10,90 29,48 — То же
5,94 31,26 — в в
1,16 37,22 —-• в »
0,050 44,94 —— 1 ! * в в
0,035 49,03 — TiOSO4.H2O
0,015 54,94 —* То же
0,024 57,20 —- в в
0,045 59,95 —1 ~ — в в
0,439 66,52 ——— — Т 0S04-Ho0 метает.
1,04 72,65 — 11 То же
0,413 54,59 - * '» TiOSO4 • HoSO4- 2НоО метает.
0,145 59,31 — 1 То же
0,104 63,30 ~ — TiOSO4-H2SO4-2H2O
0,096 65,90 — — То же
0,195 67,71 - — ТiOSO4 • H2SO4 • 2Н2О метает.
0,180 69,97 • . —— То же
0,142 65,54 — — Тi0S04 H2SO4 • Н2О метает.
0,078 69,09 — — TiOSO4.H2SO4H2O
0,049 71,68 - 11 То же
0,049 73,75 —- » в
0,055 77,20 в в
0,145 81,42 — — в в
0,461 80,15 Ti (SO4)2 метает.
0,287 83,53 — Ti (SO4)2
0,333 85,71 — | То же
Температура 75°С
0,120 11,71 * ~ TiO2-xH2O
0,301 21,27 — * “ То же
0,622 26,48 - в в
2,13 38,14 — — TiO2-xH2O метает.
0,640 33,52 TiOSO4-2H2O
0,182 38,04 — —- То же
0,088 45,13 —— * — TiOSO4-2H2O метает.
0,127 53,01 — То же
0,440 56,21 —- В В
2,13 58,80 ——1 Ч в в
0,364 37,38 —- TiOSO4‘H2O метает.
0,043 45,66 " - То же
0,033 49,38 —— в в
0,036 51,57 — ' 1- в В
0,051 58,62 — в В
344
Продолжение табл. 43
' Состав жидкой фазы Остаток Твердая фаза
TiO2 SO3 TiO, Л so3
0,818 69,13 — TiOSO4-H2O метает.
0,352 57,38 Т" Т iOSO, • H2SO4 • 2Н2О метает.
0,180 61,20 1 То же
0,142 64,91 TiOS04-H2S04-2H2O
0,201 68,25 ' TiOSO4.H2SO4-2H2O метает.
0,310 70,53 —— ——1 То же
0,311 63,67 м ——1 - в в
0,142 68,64 — TiOSO4-H2SO4.2H2Q
0,133 72,05 — • То же
0,144 75,14 — - TiOSO4-H2SO4-H2O
0,203 78,26 —— То же
2,14 79,90 -— Ti (SO4)2 метает.
0,228 82,91 — Ti (SO4)2
0,274 84,42 То же
Температура 100°С
0,0009 4,73 68,31 1,52 TiO2-xH2O
0,0008 6,64 60,23 2,45 То же
0,0013 8,30 56,40 3,73 » »
0,006 9,75 62,71 3,97 в в
0,011 11,52 63,25 4,80 в в
0,016 13,05 64,43 4,67 в в
0,027 16,21 47,26 8,84 в в
0,032 18,03 52,61 8,91 в в
0,055 21,51 48,60 11,40 в в
0,061 23,11 50,72 11,61 в в
0,079 25,80 53,75 12,40 в в
0,111 27,35 52,20 13,92 в в
0,168 29,35 53,00 14,30 в в
0,260 32,01 52,75 15,87 в в
0,314 34,72 23,04 39,77 TiOSO4.H2O
0,298 35,15 28,83 41,55 То же
0,256 36,06 24,01 40,85 В в
0,182 38,04 23,05 41,68 В В
0,150 39,50 25,95 42,53 в в
0,113 41,10 20,78 42,75 в в
0,088 42,05 * — в в
0,073 43,75 22,97 43,58 TiOSO4-H2O
0,054 45,34 26,10 45,10 То же
0,042 46,92 • * в в
0,041 47,71 26,62 46,34 В В
0,039 47,74 " 1 в в
0,040 48,88 23,65 47,02 В в
0,037 49,64 21,25 47,51 в в
0,033 50,82 21,29 47,95 в в
0,030 52,01 23,50 48,34 в в
0,027 53,33 24,31 48,49 в в
0,030 55,02 24,08 49,62 в в
0,024 56,58 22,19 50,89 в в
0,023 57,69 22,87 51,31 в в
345
Продолжение табл. 43
Состав жидкой фазы Остаток Твердая фаза
тю2 so3 тю2 SO3
0,024 59,52 23,60 51,94 TiOSO4H2O
0,039 60,78 21,74 53,23 То же
0,075 61,85 — » в
0,132 62,31 21,55 54,30 » в
0,162 63,52 3,62 62,45 TiOSO4-H2SO4-2H2O
0,176 64,20 2,30 63,71 То же
0,198 65,67 4,38 64,03 в в
0,209 66,52 3,04 65,51 » в
0,231 67,18 2,11 66,75 в в
0,212 68,38 9,10 65,17 TiOSO4-H2SO4-H2O
0,206 69,51 9,56 65,84 То же
0,211 72,15 8,00 68,40 » в
0,268 74,49 7,54 70,31 » в
0,373 76,53 6,87 72,43 » »
0,441 77,40 8,36 72,16 » в
0,429 78,16 11,91 74,12 TiO(SO4)2
0,322 78,61 13,70 73,86 То же
0,288 79,10 11,15 74,90 в в
0,238 80,39 10,51 76,34 » в
0,238 81,38 7,02 78,38 » в
0,285 82,83 10,10 77,68 » »
0,375 83,35 5,68 80,78 » »
0,351 84,04 8,65 79,50 » в
0,374 84,20 7,77 80,38 » »
0,560 87,03 9,86 81,26 » в
Температура 150 °C
0,060 51,80 30,31 47,18 TiOSO4.H2O
0,052 53,72 28,73 48,02 То же
0,054 56,45 27,50 49,64 » в
0,052 57,64 29,65 49,43 в в
0,063 59,79 25,34 51,34 в в
0,75 61,90 27,10 51,80 в в
0,090 63,51 — ** — TiOSO4- H2O-f-TiOSO4
0,081 64,73 32,08 54,23 TiOSO4
0,076 65,91 30,61 55,95 То же
0,082 67,05 32,80 56,10 в в
0,101 67,68 29,32 54,20 в в
0,120 68,02 30,80 56,88 в в
0,131 68,60 28,03 57^2 в в
0,179 68,73 28,28 58,60 в в
0,301 70,44 19,83 66,04 TiOSO4.H2SO4.H2O
0,310 71,70 9,31 68,17 То же
0,308 72,92 9,57 68,02 В В t
0,327 74,98 10,55 68,53 в в
0,361 76,65 8,90 71,25 В в
0,347 77,78 7,81 75,70 Ti(SO4)2
0,295 77,85 9,16 77,05 То же
0,238 78,68 6,18 76,42 в в
0,206 80,22 8,82 76,50 в в
0,185 81,43 5,05 79,21 в в
346
Продолжение табл. 43
Состав жидкой фазы Остаток Твердая фаза
f TiOa so3 тю2 SO3
Температура 175°С
0,098 58,37 21,98 51,72 TiOSO 4*H2O
0,094 59,38 23,61 52,03 То же
0,090 60,53 21,37 52,97 »
0,094 61,02 20,65 53,70 »
0,101 61,45 24,10 52,61 » »
0,096 62,40 27,50 55,47 TiOSO*-Н2О + TiOSO*
0,066 63,32 — TiOSO.
0,060 65,97 25,20 57,84 То же
0,050 66,22 — — » »
0,054 67,60 26,28 58,17 » »
0,057 68,88 28,03 58,52 »
0,060 69,96 25,13 59,92 » »
0,088 72,03 24,40 61,32 » »
0,109 72,84 27,35 60,04 » »
0,152 73,94 24,00 62,65 » »
0,201 74,74 25,93 62,10 » »
0,262 75,65 25,78 63,50 » »
0,256 76,72 11,21 77,12 Ti (SO*).
0,213 78,05 12,30 73,84 To же
0,193 79,72 11,85 75,33 » »
0,178 80,90 12,43 75,52 » »
0,166 81,65 10,12 77,08 »
Температура 200° С
0,050 65,08 33,26 55,10 TiOSO4
0,051 67,32 36,46 54,80 To же
0,048 68,83 32,02 56,91 » »
0,050 69,40 34,30 56,31 » »
0,054 70,28 30,11 55,02 »
0,070 71,76 31,07 55,62 » »
0,092 73,38 29,01 56,65 » »
0,113 75,05 29,70 57,42 » »
0,155 .75,91 28,34 61,37 » »
0,201 77,52 31,98 57,92 » »
0,233 78,60 33,52 57,60 »
0,212 79,45 12,21 74,53 Ti (S0.)2
0,204 79,92 9,87 76,10 To же
0,182 81,25 9,03 77,65 » »
Температура 250° С
0,042 72,10 21,18 62,45 TiOSO4
0,044 73,79 27,72 60,49 То же
0,046 74,97 24,43 62,76 » »
0,048 76,08 28,79 61,11 » »
0,053 77,50 25,31 63,50 » »
0,061 78,91 30,76 61,02 » »
0,099 80,38 30,09 62,15 » »
0,250 81,02 17,32 73,53 Ti (SO4)2
0,247 81,32 11,50 76,27 То же
347
Продолжение табл- 43
Состав жидкой фазы Остаток Твердая фаза
тю2 SO3 TiO2 SO3
Температура 300рС
0,038 77,98 22,78 65,36 TiOSOg
0,040 80,14 26,60 64,20 То же
0,045 80,41 25,40 65,01 » в
0,046 80,88 26,45 64,84 » в
0,056 81,63 22,04 67,12 в в
Температура 100°С
0,713 34,35 19,59 37,05 TiOSO4-2H2O метает-
0,476 34,89 25,25 38,50 То же
0,354 35,91 27,51 38,95 в в
0,238 37,73 19,44 39,63 В В
0,180 40,03 18,58 41,04 в в
0,122 42,83 19,90 42,02 в в
0,104 44,50 — в в
0,101 47,00 16,57 44,47 в в
0,141 49,48 16,32 46,22 в в
0,190 51,57 - в в
0,292 53,94 18,32 48,50 в в
| 0,402 55,56 19,82 48,84 « В
Температура 125° С
0,470 64,01 12,45 61,83 ТiOSO4 • H2SO4 • Н2О метает.
0,314 65,61 11,48 62,90 То же
1 0,265 67,50 11,35 64,06 в в
Температура 150°С
0,330 67,65 10,11 64,80 в в
0,313 68,50 , 6,35 65,72 в в
Температура 100°С
0,429 63,10 9,36 61,65 в в
Г 0,331 64,25 10,18 61,98 в в
0,275 65,62 8,31 63,72 в в
0,601 78,90 6,40 75,23 в в
0,270 67,69 2,70 66,82 Т iOSO4 • H2SO4 • 2НаО метает-
к 0,320 68,83 3,97 67,14 То же
348
I
о
300-
250-
200л
150
100
50-
95.
о
Н20
Рис.
мы TiO2 — S03 — Н20. Проекция на грань
призмы Н2О — SO8 — Т [3].
20 40 о60 дО ' 100
Вес. Ж 303
Политерма растворимости систе-
Шоэтому насыщенные растворы готовились растворением в серной
к «кислоте различных концентраций твердых фаз, заранее полученных
кристаллизацией из растворов выше 100° С. Так как установление
равновесия при температуре ниже 100° С кристаллизацией твердых
фаз из растворов проверить невозможно» политерму растворимости
I -ниже 100° С, приведенную на
рис. 95, следует рассматри-
вать как продолжение фазо-
| вых полей, существующих
I выше 100° С.
I Состав твердых фаз Бело-
I косков и Горощенко опреде-
I ляли по методу Скрейнема-
гкерса с помощью индифферен-
Гтной добавки и химическим
В анализом образцов, выделен-
I ных в чистом виде. Вслед-
I ствие усовершенствования ме-
I. тодики отбора проб для ана-
|,лиза, в отличие от работ Са-
I гавы, остатки получались с
малым содержанием жидкой
фазы, что позволило опреде-
! лить состав твердых фаз бо-
лее надежно. Примеры гра
|фических построений, по дан-
I ным которых определен со-
четав твердых фаз, приведены
на рис. 96.
I В изученной области си-
стемы обнаружено семь твердых фаз: ТЮ2 • хН2О (гидроокись ти-
рана), TiOSO4, TiOSO4 • Н2О, TiOSO2 - 2Н2О, TiOSO4 - H2SO4 •
I H2O, TiOSO4 • H2SO4 • 2H2O и Ti (SO4)2.
Гидроокись титана на политерме растворимости занимает об-
ширное поле при содержании SO3 от 0 до 33,8% (табл. 44). В облас-
|ти этих концентраций сульфаты титана склонны к гидролитиче-
TiOSO4 • Н2О, TiOSO2 • 2Н2О, TiOSO4 • H2SO4 •
мному разложению, чем пользуются в производстве двуокиси
титана сернокислотным методом.
L Безводный титанилсульфат TiOSO4— высокотемпературная фа-
за, кристаллизующаяся выше 100° С из растворов с концентрацией
более 63% SO3. Моногидрат титанилсульфата является равновес-
ной твердой фазой с сернокислыми растворами до температуры
р200° С, а дигидрат — ниже 100° С. Однако эти две фазы нередко
ймогут кристаллизоваться в виде неравновесных.»Особенно харак-
терна кристаллизация в качестве метастабильной фазы для
TiOSO4 • 2Н2О. Зарегистрированы случаи его образования из раст-
воров при температурах 150 и 175° С.
349
Рис. 96. Определение состава твердых фаз в системе ТЮг —
SO3 — Н2О методом Скрейнемакерса [5, 16]:
а — изотерма 100° С, б — изотерма 175° С.
Таблица 44
Точки двунасыщения в системе Ti02 — SO3 — Н2О
Твердая фаза, находящаяся в равновесии
Состав насыщен- ного раствора, вес. %
TiO, SO,
Температура 25° С
TiO2 + TiOSO4.2H2O
TiOSO4 • 2Н2О + TiOSO4. Н2О
Ti0S04.H90 + TiOSO4-H2SO4.2H2O
TiOSO4. H2SO4.2H2O + TiOSO4 . H2SO4. H2O
TiOSO4 • H2SO4. H2O + Ti(SO4)2
Температура 50°C
TiO2 + TiOSO4.2H20
TiOSO4.2H2O + TiOSO4.H3O
TiOSO4. H2O + TiOSO4 • H3SO4.2H2O
TiOSO4. H2SO4.2H2O + TiOSO4 • H2SO4. H2O
TiOSO4 • H.SO4 • H2O + Ti(SO4)2
Температура 75°C
TiO2 + TiOSO4.2H3O
TiOSO4 -2H2O + TiOSO4.H2O
TiOSO4 • H2O + TiOSO4. H2SO4 . 2H2O
TiOSO4 • H2SO4.2HoO + TiOSO4 • H2SO4. H2O
TiOSO4. H2so4. H2O + Ti(SO4)2
Температура 100°C
TiO2-xH2O + TiOSO4-H2O
TiOSO4. H2O + TiOSO4. H2SO4-2H2O
TiOSO4- H2SOr 2H2O + TiOSO4 - H2SO4. H2O
TiOSO4- H,SO4. H2O + Ti(SO4)2
Температура 125°C
TiOSO4- H2O + TiOSO4- H2SO4. H2O
TiOSO4. H2SO4. H2O + Ti(SO4)2
Температура 150°C
TiOSO4.H2O + TiOSO4
TiOSO4 + TiOSO4. H2SO4. HpO
TiOSO4- H2SO4. H2O + Ti(SO4)2
Температура 175°C
3,70 31,3
0,03 47,0
0,12 61,3
0,09 67,8
0,38 85,0
1,16 31,6
0,05 45,7
0,11 62,0
0,11 67,0
0,29 83,0
1,00
0,10
0,15
0,16
0,31
0,36
0,16
0,23
0,60
I 0,26
I 0,58
0,090
0,31
0,40
32,2
41,5
62,0
67,3
81,0
32,8
62,8
67,2
78,0
68,5
78,0
63,51
70,0
76,0
TiOSOrH3O + TiOSO4
Tioso4 + Ti(so4)2
TiOSO4 + Ti(SO4)2
Температура 200°C
0,113
0,30
I 0,23
61,98
76,0
| 79,0
351
Продолжение табл. 44
Твердая фаза» находящаяся в равновесии
Состав насыщен-
ного раствора, '
вес. %
TiO3
SO.
«5
Температура 225°С
TiOSO4 4- Ti(SO4)2 | 0,24 | 79,2
Температура 250° С
TiOSO4 + Ti(SO4)2 | 10,27 | 80,0
Кислые титанилсульфаты относятся к числу низкотемператур-
ных фаз. Они кристаллизуются из растворов с концентрацией
SO3 от 62 до 85% при температуре ниже 150° С. Однако соль
TiOSO 4 • H2SO4 • Н2О, подобно TiOSO4 • 2Н2О, в качестве мета-
стабильной фазы кристаллизуется и при более высоких темпера-
турах (175 и 200° С).
Нормальный сульфат титана кристаллизуется из растворов
в олеумной области концентраций и устойчив при температуре
ниже 250° С.
Растворимость сульфатов титана в системе TiO2 — SO3 — Н2О
в равновесных состояниях, как правило, мала и редко превышает
1% TiO2. В неравновесных условиях в области существования
гидроокиси титана сульфаты титана могут образовывать высоко-
концентрированные растворы. Высокой растворимостью в этой об-
ласти системы обладают все простые сульфаты титана, за исклю-
чением безводного титанилсульфата. При температуре ниже 50—
60° С растворы сульфатов титана сохраняются длительное время
без видимых изменений и гидролиза с выпадением осадка гидро-
окиси титана.
Безводный титанилсульфат в воде и в разбавленной серной кислоте растворя-
ется плохо. Из него нельзя получить прямым растворением концентрированные
растворы, что следует учитывать при разработке методов сернокислотного вскры-
тия титанового сырья.'Разложение титановых концентратов серной кислотой нуж-
но вести в условиях, обеспечивающих образование в качестве продуктов реакции
водорастворимых сульфатов титана. Наиболее подходящим из них является
TiOSO4 • Н2О. Он кристаллизуется из растворов при сравнительно высокой тем-
пературе (до 175—200°С), при которой скорость взаимодействия серной кислоты
с титановыми минералами велика. Одноводный титан и л сульфат хорошо растворя-
ется в воде и на его образование расходуется минимальное количество серной кис-
лоты (1 моль H2SO4 на 1 моль TiO2).
СОСТОЯНИЕ ТИТАНА В СЕРНОКИСЛЫХ РАСТВОРАХ
Состояние титана в сернокислых растворах отличается большой
сложностью равновесий, вследствие протекания процессов ступен-
чатой диссоциации, гидролиза и комплексообразования.
Гидролиз нормального сульфата титана в сернокислых раство-
рах можно, например, представить как процесс, протекающий
352
с разрывом одной и двух связей групп SO4 с титаном по следующим
схемам:
,TiOH(SO4) (OSO2OH)
Ti (SO4). + H2O(
^TiO (SO4) + H2SO4,
,TiO (OH) (OSO2OH) + H2so4
Ti (SO4)2 + 2H2O(
^Ti(OH)2SO4+h2so4,
Ti (SO4)2 + H2O -> Ti (OH)3 (OSO2OH) + H2SO4.
В свою очередь продукты первой ступени гидролиза нормаль-
ного сульфата титана могут претерпевать дальнейшее разложение
Ti (ОН)2 (OSO2OH)2
TiOH (SO4) (OSO2OH) + H2O-*Ti (OH)2 SO4 + H2SO4
i 4TiO(OH)(OSO2OH) + H2SO4, .
Ti (OH)2 (OSO2OH)2 + H2O = Ti (OH)3 (OSO2OH) + H2SO4,
Ti (QH)3 (OSO2OH) + H2O = Ti (OH)4 + H2SO4 и т. д.
Как нормальный сульфат титана, так и продукты его гидролиза,
в сернокислых растворах могут подвергаться электролитической
диссоциации. Не исключена возможность, что диссоциация соеди-
нений, содержащих группу OSO2OH, протекает по двум механизмам
. 7 TiO (ОН) OSO2O“ + Н+
TiO(OH) (OSO2OH)/
^TiO (ОН)+ + OSO2OH~ (HSOT).
Судя по способности сульфатов титана к образованию двойных
солей, в сернокислых растворах возможно также и протекание
реакций комплексообр азования
Ti (SO4)2 + H2SO4 = [Ti (SO4)3]2- + 2H+,
TiOSO4 + H2SO4 = [TiO (SO4)2]2~ + 2H+.
Одновременное протекание гидролиза, диссоциации и комплек-
сообразования приводит к появлению в сернокислых растворах
титана большого количества нейтральных и заряженных частиц.
На эти процессы накладываются также явления гидратации и по-
лимеризации частиц.
Хотя обоснованной теории ионных равновесий в сернокислых
растворах титана еще не существует, по данным ряда опубликован-
ных статей о природе и поведении частиц титана в растворах можно
составить некоторое представление.
Парравано и Каглиоти [124], Зборовский и Гермогенова [41] считают, что в
сернокислых растворах титан находится в коллоидно-дисперсном состоянии.
23 9-614
353
Делафос [90] полагает, что в растворах с pH < 0,3 титан существует преиму-
щественно форме ионов Ti (ОН)^“, а с pH >0,4 — в форме TiO2+.
Беукенкамп и Херрингтон [78] изучили комплексообразование титана в раз_
бавле шых сернокислых растворах методом ионного обмена. Основываясь на дан.
ных элюирования титана с анионита растворами серной кислоты различных кон-
центраций, авторы высказали предположение, что равновесие в сернокислых раст-
ворах титана характеризуются следующими уравнениями:
Ti (OH)t + Н+ = Ti (ОН)2+ + Н2О,
Ti (ОН)^ + HSOf == Ti (OH)S HSO4,
Ti (OH)2+ + HSOT = Ti (OH)2 HSOt.
Константы равновесий, по расчетам авторов, = 2,0, Кг = 11,3, К3 = 0,64.
Основное количество титана в разбавленных растворах находится в форме частиц
Ti (OH)3HSO4, имеющих брутто-формулу двухводного титанилсульфата.
Рис. 97. Электромиграция ионов тита-
на в сернокислых растворах [59].
Содержание титана в растворе: 1—катио-
ны, 2 — анионы.
Рис. 98. Распределение титана ме-
жду различными формами в зави-
симости от концентрации сульфат-
иона в растворе [58].
Стромберг и Картушинская [69], исследуя растворы сульфата титана методом
полярографии, пришли к выводу, что в электродном процессе принимают непо-
средственное участие ионы Ti (HSO4)3-^, а в растворе преобладают комплексы
Ti (OH)HSO4+.
Методом ионного обмена установлено, что сульфатные ацидо-комплексы тита-
на существуют в сернокислых растворах с концентрацией менее 0,5-н. [70].
Методом электромиграции показано, что в сернокислых растворах с концентра-
цией менее 0,5 моль!л титан находится в катионной форме [59]. Увеличение кон-
центрации серной кислоты приводит к образованию нейтральных и анионных
комплексов по схеме
TiO2+ + SO4-
[TiOSOJ0 + soi~ -» [TiO (SO4)2[2-.
На рис. 97 приведена диаграмма распределения катионных и анионных ком-
плексов титана в зависимости от концентрации серной кислоты [59].
354
= 4,02 • 10~3 ((1 = 0,45-4 1,1, 18° С),
ЕГПо данным электромиграции и поглощения ионов титана ионитами, рассчита-
ны константы нестойкости сульфатных комплексов титана [58]:
[TiO2+] [SO2-]
- [TiO (SO4)]°
_ [TiOSOd [SO2-]
2“ [TiO (SO4)2-]
= 6,35 • КГ2 (18° С).
Из уравнений констант нестойкости вычислено распределение различных форм
титана в растворе (рис. 98).
Бело косков [3] за состоянием титана в сернокислых растворах пытался про-
следить по спектрам поглощения в ультрафиолетовой области света. Оказалось,
что спектры поглощения зависят от метода приготовления растворов сульфата
титана. Из этого можно заключить, что равновесие в водных растворах сульфата
титана устанавливается не мгновенно, а за некоторое время. На скорость установле-
ния равновесия оказывают влияние температура и концентрация серной кислоты.
В равновесных растворах спектры поглощения зависят от концентрации серной
кислоты. Растворы сульфата титана, содержащие 5—20% SO3, имеют идентичные
полосы поглощения. С увеличением концентрации серной кислоты полосы погло-
щения сдвигаются в длинноволновую часть спектра. Растворы с концентрацией
55—60% SO3 составляют вторую область с идентичными полосами поглощения.
При болыцей концентрации SO3 происходит повторный батохромный сдвиг спек-
тров поглощения.
Растворы сульфата титана в разбавленной серной кислоте, полученные раство-
рением TiOSO4-2H2O при комнатной температуре, судя по изменению спектров по-
глощения, являются неравновесными. Они приходят в состояние равновесия толь-
ко спустя пять-шесть месяцев. Полосы поглощения за это время сдвигаются в ко-
ротковолновую область и спектры поглощения их становятся идентичными спек-
трам поглощения растворов, приготовленных при температуре 60—70° С [3].
Брагина и соавторы [9] смещение полосы поглощения в область длинных волн
связывает с гидролизом, комплексообразованием и полимеризацией титана в раст-
ворах.
Ривс и Блоун [129] наблюдали различие во взаимодействии с винной кисло-
той свежеприготовленных и постаревших растворов сульфата титана. По их мне-
нию, во время стояния растворов образуются димеры с кислородными мостиками
между атомами титана. Хиксон и Фредериксон [101] наблюдали медленное вза-
имодействие половины титана, содержащегося в растворах титанилсульфата,
с перекисью водорода, что, по их мнению, служит признаком полимеризации.
В хлоридных растворах полимеризация титана не обнаружена.
Описанные закономерности характеризуют состояние сернокис-
лых растворов с малой концентрацией в них титана. Менее изучены
растворы с высокой концентрацией титана. При комнатной тем-
пературе эти растворы могут существовать длительное время в ме-
тастабильном состоянии. Так, например, Памфилов с соавторами
[62] не мог обнаружить в течение шести месяцев заметных измене-
ний в состоянии растворов сульфата титана, полученных из мета-
титановой кислоты и ильменита. Концентрированный раствор двух-
водного титанилсульфата, содержащий 14,10% TiO2 с отношением
TiO2 : SO3 » 1,0, хранился более четырех лет и за это время не
подвергся видимым изменениям [16].
Водные растворы одноводного и двухводного титанилсульфа-
тов, в которых отношение TiO2 : SO3 = 1, имеют низкое pH. В них
23* 355
может быть оттитровано щелочью без образования осадка гидро-
окиси около 50% серной кислоты, связанной сульфатом титана.
Точка замерзания растворов титанилсульфата в воде понижается
до такой степени, как если бы они подверглись полному гидролизу
с выделением всей связанной сульфатом титана серной кислоты
[46].
Основываясь на этих наблюдениях, некоторые авторы считают, что сульфат
титана в водных растворах подвергается далеко идущему гидролизу [41]. В по-
следнее время этот взгляд на состояние сульфата титана в водных растворах раз-
вили Галинкер и Леках [45—49]. Согласно их представлениям, концентрированные
водные растворы титанилсульфата, выдержанные более трех месяцев, гидролизо-
ваны на 60—65% и представляют коллоидные растворы основного сульфата при-
мерного состава ТЮз • (0,4—0,35) SO3 • пН2О. К этомув ыводу авторы пришли в
результате изучения растворов титанилсульфата методами криоскопии, тензи-
метр ии и др.
В последние годы получила распространение идея полимери-
зации титана вводных растворах, высказанная в работах [101, 129].
Жерман [103, 105, 106, 107, 108], основываясь на представлениях о
полимеризации, пытается объяснить особенности некоторых свойств
растворов сульфата титана: изменение объема, вязкость, реакцион-
ную способность.
Долматов [37] полагает, что концентрированные растворы суль-
фата титана содержат полиионы в виде гидроксокомплексов с оло-
вой связью. На ассоциацию моноионов титана в сернокислых
растворах в полиионы, по его мнению, указывают непропорцио-
нальное увеличение полярографической волны от концентрации
и характер изменения кривых удельной электропроводности и
вязкости.
Величину заряда и число полимеризации гидроксокомплексов
титана в сернокислых растворах титана Долматов пытался опре-
делить экспериментально. Судя по угловому коэффициенту зави-
симости 1g ,-1—:---£ на полярограммах в разбавленных растворах
1а 1
сульфата титана (5 г/л ТЮ2) существуют ионы с зарядами +г/2
и +х/з 138]. Заряды ионов титана, рассчитанные по содержанию
свободной и связанной серной кислоты в растворе, изменяются
в широких пределах концентрации (0,7—6,6 мол/л H2SO4cbo6, 0,15—
3,3 мол/л Ti) от +2 до +1. Повышение концентрации титана сопро-
вождается уменьшением заряда его ионов до +1.
Методом сравнительного диализа определено число полимери-
зации титана в сернокислых растворах (226—240 г/л TiO2), кото-
рое равно 2 [34]. Расчеты, основанные на измерении вязкости
сернокислых и солянокислых растворов титана, показывают, что
при температуре 25° С числа полимеризации титана в сернокислых
растворах изменяются от 1,55 до 8,6. Исходя из содержания сво-
бодной и связанной серной кислоты в сернокислых растворах
с концентрацией 0,4—1,0 г-ат/л Ti, последний существует преиму-
щественно в виде димеров, находящихся в равновесии с полииона-
356
1 ми [37]. При концентрации 2,5 г-ат!л Ti и более число полимери-
| зации повышается до 8—9. Нагревание растворов приводит к депо-
[ лимеризации ионов. Содержание свободной и связанной серной
I кислоты в растворах сульфата титана определялось методом тит-
рования в присутствии маскирующей добавки комплексообразу-
ющих агентов KCNS + KNaC4H4O6 [36].
Нд основании этих представлений Долматов [37] считает, что
гидролиз сульфата титана при нагревании сводится к образованию
из полиионов гидроксокомплексов, содержащих по 8—9 атомов
титана, монокристаллов гидроокиси титана размером по 5—12 нм,
которые затем коагулируют в агрегаты размером 0,2—1,0 мкм.
Зарождение монокристаллов лимитируется центрами кристаллиза-
ции. Введение титановых зародышей, состоящих из агрегатов с мо-
нокристаллами размером по 2—2,5 нм, ускоряет процесс гидролиза.
Жерман [104] считает, однако, что титановые зародыши действуют
не как центры „кристаллизации, а служат источником структурной
информации на молекулярном уровне, не поясняя, к чему сво-
дится сущность этого понятия.
В химическом отношении гидролиз сульфата титана, по пред-
положению [37], сопровождается частичной деполимеризацией оло-
вых соединений, превращением мостиковых оловых связей в ок-
сосвязи и частичным замещением оловых групп анионами.
Изучение системы ТЮ2 — SO3 — Н2О методами растворимости,
кинетическим и светорассеяния позволяет развить иную точку
зрения на состояние титана в растворах.
Равновесные титановые растворы в системе TiO2 — SO3 — Н2О
не отличаются какими-то особенностями от водных растворов дру-
гих твердых фаз. Судя по скорости взаимодействия содержащегося
в них титана с комплексообразующими агентами, светорассеянию
и другим свойствам, они ведут себя как типичные ионные растворы
солей в минеральных кислотах. Своеобразные свойства присущи
только неравновесным (пересыщенным) растворам гидроокиси ти-
тана, простирающимся в системе TiO2 — SO3 — Н2О при темпе-
ратуре 25—100° С от 0 до 31,3—33,8% SO3 (табл. 43). Растворение
сульфатов титана в этой части системы происходит инконгруэнтно,
т. е. с разложением. Если температура не превышает 60—70° С,
то при инконгруэнтном растворении сульфата титана в осадке
не образуется равновесная твердая фаза — гидроокись титана. По-
лучающиеся при этом растворы, имея высокую концентрацию TiO2,
могут сохраняться в метастабильном состоянии неограниченно дол- «
гое время. Исследования метастабильных растворов в системе
TiO2 — SO3 — Н2О кинетическим методом и методом светорассея-
ния [12, 43, 44] показывают, что титан в них находится в двух
формах — молекулярно- и коллоидно-дисперсной. Содержание мо-
лекулярно-дисперсного титана в растворе можно определить по
скорости взаимодействия его с перекисью водорода по следующей
методике.
357
В мерную колбу на 200—250 мл наливают 1 мл 30%-ной Н2О2, 5 мл 0,1%.Го
раствора полиакриламида (коагулянт) и объем доводят до метки 5%-ной серц0^
кислотой. После перемешивания из колбы отбирают пипеткой объем жидкости
равный объему анализируемого раствора. Затем в колбу добавляют анализируй
мый раствор, содержимое перемешивают встряхиванием в течение 30 сек, и произ-
водят отсчет времени. Если раствор мутный, его быстро фильтруют через склад-
чатый фильтр и осветленную жидкость наливают в кювету фотоэлектроколоримет-
ра, на котором производят 5—7 замеров оптической плотности (светофильтр № 3)
с интервалом 30—60 сек. По результатам замеров строят график зависимости оп-
тической плотности раствора от времени и кривую оптической плотности экстра-
полируют на нулевое время. Пересечение ее с ординатой укажет оптическую плот-
ность раствора, вызванную содержащимся в нем титаном в молекулярно-дисперс-
ном состоянии. Концентрацию титана находят по калибровочному графику.
По этой методике определяется концентрация титана в растворе в ионной
форме. Гидроокись титана (продукт гидролитического разложения титана) с пе-
рекисью водорода реагирует медленно. Растворимость ее в разбавленных серно-
кислых растворах мала и поэтому она может в них находиться преимущественно-
в коллоидно-дисперсной форме. Концентрацию коллоидно-дисперсного титана (гид-
роокиси) находят по разности между общим его содержанием в растворе и содер-
жанием в молекулярно-дисперсной форме.
Типичные графики изменения оптической плотности пероксоти-
такового комплекса со временем приведены на рис. 99. В случае,
если весь титан в растворе находится в молекулярно-дисперсном
состоянии, график зависимости D от t имеет вид горизонтальной
прямой, продолжение которой отсекает на ординате отрезок On,
соответствующий общей концентрации титана в растворе. При со-
вместном присутствии в растворе молекулярной и коллоидно-дис-
персной форм титана графическая зависимость D от t со временем
ассимптотически приближается к горизонтальной прямой (показа-
на пунктиром), отвечающей оптической плотности раствора, содер-
жащего весь титан в молекулярно-дисперсном состоянии. Экстра-
полируя ее на нулевое время, отсечем на ординате отрезок От,
соответствующий содержанию в растворе молекулярно-дисперсно-
го титана.
Коэффициенты рассеяния /?90 при больших величинах (более
5 • 102 см-1) рассчитывали по результатам замеров оптической
плотности, вызванной светорассеянием, на спектрофотометре
СФ-4А, а при малой интенсивности светорассеяния — по интен-
сивности рассеянного света под углом 90° к направлению падаю-
щего луча на специальной установке. Установка состояла из спект-
рофотометра СФ-4А, использовавшегося в качестве монохромато-
ра, и приставки ПЗ-01, в которую помещалась стеклянная кювета
с боковым окошком для выхода рассеянного света. Световой поток
регистрировали гальванометром типа М95, включенным в цепь
фотоумножителя ФЭУ-19М.
Исследования состояния титана в сернокислых растворах ки-
нетическим методом и с помощью измерения светорассеяния
показывают, что концентрированные растворы титанилсульфата
(200—300 г/л TiO2) даже при стехиометрическом отношении в них
SO3 : TiO2 = 1 содержат титан преимущественно в молекулярно-
358
дисперсном состоянии. Разбавление их водой вызывает снижение
концентрации молекулярно-дисперсного титана до 20% и менее
по отношению к общему его содержанию. Растворы титанилсуль-
фата в серной кислоте с концентрацией TiO2 выше 100 г/л и отно-
шением SO3 : TiO2 более 1,5 при комнатной температуре имеют
не менее 90% титана в молекулярно-дисперсном состоянии.
В технологических растворах сульфата титана с концентрацией
130—140 г!л TiO2 и 260—300 г!л H2SO4 активной (титрующейся
Рис. 99. Изменение оптической плотности рас-
творов сульфата титана с перекисью водорода:
1 — раствор, в котором титан находится в молекулярно-
дисперсном состоянии; 2 — раствор, содержащий титан
в молекулярно- и коллоидно-дисперсном состоянии.
щелочью по метиловому оранжевому) на разных стадиях процесса
концентрация коллоидно-дисперсной гидроокиси титана состав-
ляет в среднем от 8,5 до 21,1 % по отношению к общему его содер-
жанию.
Сернокислые растворы, концентрация которых приходится на
область кристаллизации из жидкой фазы сульфатов титана, со-
держат весь титан только в молекулярно-дисперсном состоянии,
что и обнаруживается пероксидным методом.
Молекулярно-дисперсная природа растворов сульфата титана
подтверждается также измерением светорассеяния. По данным
автора и Лыкова, раствор сульфата титана с концентрацией
100 г!л TiO2 общей и молярным отношением SO3 : TiO2 = 1,5
при температуре 14° С содержал 91 г/л TiO2 в молекулярно-дис-
персной и 9 г/л TiO2 в коллоидной форме. Коэффициент рассеяния
его Т?9о = 46,3 • 10~6 саГ"1 (X = 500 нм), то есть соизмерим с коэф-
фициентами молекулярного рассеяния типичных молекулярно-дис-
персных веществ, например бензола, для которого Т?90 =
10,7 • Ю“6 см~1 (15° С, А = 546,1 нм).
359
Высокую кислотность растворов сульфата титана, в которых
он находится в молекулярно-дисперсной форме, можно объяснить,
не прибегая к гипотезе глубокого гидролиза, допустив, что титан
в водных растворах существует в форме сульфотитановой кислоты,
диссоциирующей по схеме
HOTiO — OSO2OH -> HOTiO — oso2o~ + н+,
или олигомеров на ее основе состава НО(ТЮ)„—OSO2OH. В под-
тверждение такой гипотезы можно привести тот факт, что автору
и Сидоренко удалось получить соли лития и натрия сульфотита-
новой кислоты в виде стекловидной хорошо растворимой в воде
массы по реакции
HOTiO — OSO2OH + MCI = HOTiO — OSO2OM + HC1 f
при многократном вакуумном выпаривании раствора одноводного
титанилсульфата с эквимольными количествами хлоридов лития
или натрия.
Нагревание метастабильных растворов сульфата титана или
разбавление их водой приводит к гидролитическому разложению
сульфотитановой кислоты
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
HOTiO — OSO2O“ + Н2О = (НО) TiO (ОН) + HSO? + Н+
с образованием гидроокиси титана, что подтверждается кинети-
ческим методом и измерением светорассеяния. Так, нагревание
упомянутого выше раствора титанилсульфата от комнатной тем-
пературы до 80° С за 5 мин привело к повышению содержания
в нем коллоидно-дисперсной гидроокиси титана с 9 до 47,5 г/л,
а коэффициента рассеяния — с 46,3 • КГ”6 до 65,0 • 10~6 см~х.
Данные измерений светорассеяния и содержания коллоидно-
дисперсной гидроокиси титана поддаются математической обра-
ботке по уравнению Релея
1Р — 10
24л3
X4
где ip — интенсивность рассеянного света, i0 — интенсивность па-
дающего света, % — длина волны, — коэффициент светопрелом-
ления дисперсной фазы, п0 — коэффициент светопреломления дис-
персионной среды, N — концентрация частиц, V — объем частицы.
Подставив в уравнение Релея К = ~ (коэффициенту погло-
щения), NV = Ср, где С — концентрация коллоидно-дисперсной
фазы, р — плотность ее, и приняв А = 500 нм, = 2,317, п0 =
= 1,333, р = 3 г/см?, получим следующее выражение для радиу-
са коллоидной частицы:
3 / к
R- 0,335 у~ • 10~7 см.
360 \
1
Обработка экспериментальных данных по изучению гидролиза
сульфата титана при нагревании, проведенная автором и Лыковым
(табл. 45, рис. 100), позволила установить ряд закономерностей,
проливающих свет на механизм этого процесса. Гидролиз сульфата
1 — концентрация молекулярно-дисперсного титана; 2 —
cpeji&nti. радиус частиц; 3 — 1g Roo ; 4 — lg R.
титана в водных растворах является сложным физико-химическим
процессом, в котором следует различать собственно гидролиз как
химическую реакцию, протекание которой характеризуется изме-
нением концентрации молекулярно-дисперсного титана, а также
образование и рост коллоидных частиц, коагуляцию их и выпаде-
ние в осадок. Из табл. 45 видно, что примерно половина содержа-
щегося в растворе при комнатной температуре сульфата титана
гидролизуется при нагревании до температуры 80° С. Дальнейшее
нагревание раствора при постоянной температуре сопровождает-
ся небольшим изменением концентрации молекулярно-дисперсного
361
Таблица 45
Кинетика гидролиза сульфата титана (температура 80° С, X ~ 500 нм)
Т, мин Концентр, молекулярно- дисперсного титана ТЮ2, г/л К-103 СМ 1 Я-103, СМ 1 К90Л06, СМ 1 R, средний нм исправлен ный
Исходный раствор при комнатной температуре
91,0 1,32 0,770 1 46,3 1 1,47 1 —
0 52,5 Раствор 2,17 в период г 1,09 идролиза 65,0 0,99 1,78
5 46,0 2,75 1,37 81,6 1,03 1,85
10 41,0 3,57 1,80 107,2 1,10 1,93
20 35,0 5,87 2,98 174,8 1,25 2,09
30 зо,о 9,00 4,61 275,0 1,41 2,24
40 30,0 12,5 6,44 384,0 1,57 2,35
60 30,0 29,7 15.50 922,0 2,05 2,74
80 30,0 94,5 51,20 3060,0 3,21 3,33
100 30,0 332,4 214,00 12750 4,46 —
120 29,0 521,0 407,00 24250 5,46 —
140 29,0 760,0 640,00 38100 6,17 —
титана за первые 30 MUH, П( эсле чег( э гидрол [из прио станавливае-
тся. Изменение концентрации молекулярно-дисперсного титана
при гидролизе за этот период описывается уравнением реакции
первого порядка [44].
Коэффициент рассеяния и средний радиус коллоидных частиц
по мере нагревания раствора непрерывно возрастают даже после
окончания гидролиза как химического процесса. Логарифмиче-
ские зависимости R90 и R от времени нагревания в период между
40 и 100 мин от начала опыта изображаются прямыми линиями
(рис. 100). Прямолинейный характер логарифмических кривых R90
и R объясняется, по-видимому, тем, что на этой стадии гидролиза
изменяется только размер коллоидных частиц, а концентрация
их остается постоянной.
В первые 30—40 мин от начала опыта логарифмические зна-
чения R90 и R не укладываются на прямую, что объясняется не-
постоянством на этой фазе гидролиза концентрации коллоидно-
дисперсной фазы и относительно большой величиной молекулярно-
го рассеяния дисперсионной среды по сравнению с рассеянием
света на частицах. Экстраполируя прямолинейный участок лога-
рифмической зависимости R90 на нулевое время, можно уточнить
численное значение R90. По уточненным данным коэффициент рас-
сеяния света частицами R90 = 28,8 • 10~6 см~х\ по разности нахо-
дим коэффициент молекулярного рассеяния дисперсионной среды*,
который равен 36,2 • КП6 см~1.
* Включая и немицеллярную гидроокись титана.
362
| Через 100 мин после начала опыта наблюдается повторное
отклонение логарифмической зависимости R90 и R от прямоли-
нейной. Оно вызвано изменением кинетики роста частиц. Стадия
роста коллоидных частиц при постоянной их концентрации сме-
няется коагуляцией за счет взаимного слипания. В согласии с тео-
рией коагуляции Смолуховского, кинетика такого процесса опи-
сывается уравнением реакции второго порядка.
В период, когда логарифмические зависимости R90 и R от вре-
мени изменяются прямолинейно, как уже отмечалось, изменение
среднего радиуса частиц происходит при постоянной концентрации
коллоидно-дисперсной фазы. Аналогия кинетики на этой фазе про-
цесса с реакцией первого порядка указывает на существование
в растворе гидроокиси титана в двух различных формах: мицел-
лярной и немицеллярной (молекулярно-дисперсной?), вызывающей
небольшое светорассеяние, но так же медленно, как и мицеллярная,
реагирующей с перекисью водорода. Пероксидный кинетический
метод поэтому дает нам возможность определять только суммарное
содержание в растворе гидроокиси титана, образовавшейся при
гидролизе сульфата титана. Рассчитанный по данным таких ана-
лизов размер коллоидных частиц является средним и на начальной
стадии гидролиза имеет заниженные значения. Численное значе-
ние радиуса коллоидных частиц на начальной стадии гидролиза
можно уточнить, исходя из следующих соображений. Рост мицелл
за счет немицеллярной гидроокиси титана продолжается до сред-
него радиуса частиц 4—5 нм. После этого наступает их взаимная
коагуляция и выпадение в осадок. Если принять, что к началу
коагуляции немицеллярный материал оказывается израсходован-
ным практически полностью, то можно вычислить концентрацию
коллоидных частиц, образовавшихся при гидролизе. Она будет
в нашем опыте
д/ __ 100с _ 100 • 0,070 _
™ " а4/3л/?3р ~ 58 • % . 3,14 (4,5 . 10-7)3 . 3 -
— 1,05 • 1017 частиц в 1 см3,
В этом уравнении принято R = 4,5 нм, р = 3 г/см3, а = 58%
(среднее содержание ТЮ2 в гидроокиси титана), С = 0,070 г/см3
(см. табл. 45). Полагая, что за все предшествующее время опыта
вычисленная концентрация коллоидных частиц сохранялась по-
стоянной, и подставив ее в уравнение Релея, можем вычислить
исправленный радиус частиц в этот период времени. Результаты
проведенного расчета помещены в табл. 45. Из нее видно, что в
нашем опыте к моменту нагревания раствора до 80° С коллоидные
частицы выросли до R = 1,78 нм.
Расчет N на основе недавно развитой автором и Лыковым
теории мицеллообразования гидроокиси титана дал величину на
порядок меньше.
| Гидролиз сульфата титана можно представить в виде схемы
363
OSsH + г(но)и(огн) oil OSH + posu(ozh) Oil
о: HO—TiOH: O< /О :• HO—Ti—OSO2O~
По этой схеме при гидролизе вначале получается гидратирован-
ная гидроокись титана. Затем она отщепляет гидратную воду,
связанную с титаном, и отдельные молекулы гидроокиси объеди-
няются в агрегате мицеллы посредством водородных связей с учас-
тием воды. Мицеллы могут включать, наряду с гидроокисью титана,
некоторое количество ионов сульфотитановой кислоты, сообщающих
им отрицательный заряд. На один атом титана в мицелле прихо-
дится до 2,5 молекул воды и двух гидроксильных групп, что со-
гласуется с данными 113].
В одной мицелле, образовавшейся в нашем опыте, перед на-
чалом коагуляции содержится
MTiOa
0,070 • 6,02 • 1023
79,9 1,05 • 101?
= 5070 атомов титана.
’ Так как число частиц до начала коагуляции в растворе остае-
тся примерно постоянным, а каждая частица начинает расти
из одного зародыша, то при гидролизе сульфата титана один за-
родыш мицеллообразования возникает примерно на 5000 атомов
титана, находящихся в растворе. Введение в раствор перед гид-
ролизом искусственно приготовленных зародышей ускоряет ми-
целлообразование. Это приводит к увеличению угла наклона ло-
гарифмической зависимости R и сокращению периода гидролиза,
предшествующего коагуляции, что и наблюдается на опыте. Графи-
ки зависимости lg/?9o и lg R могут быть использованы для оценки
эффективности титановых зародышей для растворов данного состава.
Наши опыты по изучению механизма гидролиза сульфата ти-
тана позволяют охарактеризовать этот процесс следующей схемой:
Сульфотитановая кислота
Y
Гидратированная гидроокись титана
Зарождение и рост мицелл
Y
Коагуляция мицелл
Осадок гидроокиси титана
На первый взгляд может показаться, что ойа аналогична [37],
но по существу они принципиально различны. Отличие их со-
365
стоит, во-первых, в том, что по нашей схеме исходным материалом
для формирования коллоидных частиц служит гидратированная
гидроокись титана, которая благодаря наличию оболочки из мо-
лекул воды имеет повышенную растворимость, тогда как по схеме
[37] мицеллы формируются из полиионов. Не отрицая принци-
пиальной возможности существования полиионов в растворе суль-
фата титана, необходимо отметить ненадежность метода, исполь-
зовавшегося в работе [37], для определения чисел полимеризации
и величин зарядов ионов. Концентрации последних, по методике
[35, 36], определяют без учета скрытого гидролиза растворов суль-
фата титана и потому достоверность результатов анализа в ряде
случаев может оказаться сомнительной.
Во-вторых, коллоидные частицы рассматриваются в работе
[37] как монокристаллы. Кристаллическое строение их в началь-
ный период гидролиза достоверно не доказано. Гидроокись титана
обнаруживает кристаллическое строение только после определен-
ного периода старения. Нельзя поэтому процесс мицеллообразо-
вания рассматривать как кристаллохимический.
В заключение следует отметить, что гидролиз сульфата титана
в водных растворах представляет интересный коллоиднохимический
процесс. На его протяжении представляется возможным наблю-
дать зарождение, рост и коагуляцию коллоидных частиц. Изу-
чение этой системы показывает, что свойства золей существенно
зависят от размеров коллоидных частиц. При малых размерах
(менее 4—6 нм) они отличаются повышенной стабильностью к дей-
ствию коагулирующих веществ. По-видимому, кроме электриче-
ского заряда на поверхности раздела в качестве фактора стабили-
зации золей с малым размером частиц выступает и броуновское
движение.
ЭКСТРАКЦИЯ ТИТАНА ИЗ СЕРНОКИСЛЫХ РАСТВОРОВ
ОРГАНИЧЕСКИМИ ЖИДКОСТЯМИ
Органические жидкости экстрагируют титан из сернокислых
растворов обычно в незначительной степени [39, 40], в частности,
титан плохо экстрагируют спирты, кетоны и эфиры уксусной кис-
лоты. Он хорошо экстрагируется только из концентрированных
растворов серной кислоты смесью трибутилфосфата с четырех-
хлористым углеродом.
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ТИТАНА С ПЕРЕХОДНЫМИ МЕТАЛЛАМИ,
ОБРАЗУЮЩИЕСЯ В СЕРНОКИСЛЫХ РАСТВОРАХ
j В сернокислых растворах титан дает комплексы с переходными
металлами и даже с собственными ионами с окислительным числом,
равным трем. Образование комплексов титана с переходными ме-
таллами в сернокислых растворах обнаруживается по изменению
366
растворимости солей, замедлению кристаллизации твердых фаз
и смещению полос в спектрах поглощения. В настоящее время
известны комплексы этого типа, существующие только в растворах.
Из растворов в виде равновесных твердых фаз они кристаллизую-
тся не всегда. Судя по константам нестойкости, комплексы
титана с металлами — соединения непрочные. Изменение свобод-
ной энергии при образовании этих комплексов составляет 1—
2 ккал/моль. Следовательно, энергия связи между титаном и ме-
таллами в этих комплексах невелика, меньше, чем энергия водо-ч
родной связи. Механизм взаимодействия титана с металлами в
сернокислых растворах и природа химической связи еще не
выяснены.
Образование нестойких комплексов титана с переходными ме-
таллами в сернокислых растворах представляет большой интерес
для химии комплексных соединений, так как, по-видимому, по-
добные соединения дают и другие металлы. Наличие взаимодей-
ствия между титаном, железом, хромом и другими металлами
следует также учитывать при разработке технологии получения
различных титановых продуктов из природного сырья сернокис-
лотными методами.
Впервые образование комплексов титана в сернокислых раст-
ворах типа металл — металл наблюдал Шенк [134] при исследо-
вании спектров поглощения сульфатов трех- и четырехвалентного
титана. Наблюдения Шенка нашли подтверждение в работе [15].
Горощенко и Андреева [23] установили образование комплек-
сов титана с ниобием и танталом по изменению растворимости
сульфатов ниобия и тантала в сернокислых растворах, содержа-
щих примесь сульфата аммония. Соотношение титана с ниобием
и танталом в этих комплексах, по-видимому, изменяется со вре-
менем. Из растворов после длительного стояния (около 1,5 г.)
при комнатной температуре спиртом высаливаются продукты, имею-
щие состав xNH4NbO2SO4 • (NH4)2TiO (SO4)2 и #NH4TaO2SO4 •
• (NH4)2TiO(SO4)2, где x 8, г/ ~ 6. Таким образом, комплек-
сные соединения титана с ниобием и танталом являются продукта-
ми взаимодействия двойных солей, или точнее — комплексных
и^црв [TiO (SO4)2 ]2“ и [MO2SO4P, на которые двойные соли дис-
социируют в растворе.
Беукенкамп и Херрингтон [78] наблюдали образование в
сернокислых растворах комплексов желтого цвета между Ti-4 и
Fe-2. Методом непрерывных изменений авторы установили, что в
1-мол. серной кислоты железо и титан входят в состав комплексов
желтого цвета в отношении 2:1.
Независимо от них образование комплексов желтого цвета меж-
ду Ti-4 и Fe-2 в сернокислых растворах установили Горощенко
и Сикорская [28]. Они изучили спектры поглощения Ti-4 и Fe-2
в сернокислых растворах с концентрацией от 0 до 98%. Растворы
смесей сульфатов Ti-4 и Fe-2 в серной кислоте с концентрацией
367
от 0 до 75—80% окрашены в желтый цвет и имеют полосу погло-
щения в области длин волн менее 620 нм (рис. 101). Растворы с кон-
центрацией более 75—80% H2SO4 окрашены в сиреневый цвет.
Сиреневому комплексу отвечает полоса поглощения с максимумом
при длине волны 450 нм. которая по форме идентична с полосой
♦
1 — 0,15 моль/л FeSO4 в 30%-ной H2SO4, кювета 10,10 мм; 2 — 0,010 моаь}л TiOSO4 в
30%-ной H2SO4, кювета 9,98 мм; 3 — 0,10 молъ/л TiOSO4 + 0,15 моль/л FeSO4 в 30%-ной
H2SO4, кювета 10 мм; 4 — 0,0367 моль/л TiOSO4 в 80%-ной H2SO4, кювета 9,99 мм; 5 —
0,0367 моль] л TiOSO4 + 0,000319 молъ]л FeSO4 в 78%-ной H3SO4, кювета 9,99 мм.
поглощения растворов Fe-2 в серной кислоте, но сдвинута отно-
сительно ее в более длинноволновую область спектра.
1 Желтый комплекс существует только в растворе. Состав его
в 30 %-ной серной кислоте определен по методу изомолярных се-
рий (рис. 102), отношение Fe-2 : Ti-4 = 3:2. Константа нестой-
кости /этого комплекса
тг_ [F*e (Н)]3 [Ti (IV)]2 _ 3 19 10~“5 И А°
[3Fe (П) • 2Ti (IV)] — д>19'10 (16 С).
Он относится к малопрочным соединениям, определение состава
которых методом изомолярных серий не может быть выполнено
с высокой точностью. Этим, по-видимому, объясняется несоответ-
ствие наших данных о составе комплекса с данными [78] и с резуль-
368
татами последующих работ, в которых отношение Fe-2 : Ti-4 со-
ставляет 3 : 2; 1 : 1 [18] и 2 : 1; 1 : 1 [28].
Рейнольдс [132] считает, что в сернокислых растворах
(1,85 моль/л H2SO4) Fe-2 и Ti-4 образуют два комплекса с отно-
шением 2:1 и 1:1.
Комплекс сиреневого цвета удалось выделить из раствора и оп-
ределить его состав, который выражается формулой FeTi (SO4)3.
Константа нестойкости его в серной кислоте с концентрацией 80%
H2SO4
[FeSO4] [Ti (SO4)2]
[FeTi (SO4)3]
(16° C).
Горощенко с соавторами [26] по замедлению высаливания двой-
ного сульфата титанила и аммония (NH4)2TiO(SO4)2 • Н2О из
сернокислых растворов в присут-
ствии сульфата трехвалентного
железа установил образование
в этих растворах комплексов
Ti-4 с Fe-ЗЛ^ отсутствие суль-
фата трехвалентного железа вы-
саливание двойного сульфата
титанила и аммония из серно-
кислых растворов после добав-
ления сульфата аммония при
комнатной температуре продол-
жается не более 24 ч. При со-
держании же в растворе приме-
си Fe2 (SO4)3 более 20 г/л выса-
ливание двойного сульфата ти-
танила и аммония длится более
месяца. Изменение оптической
Рис. 102. Изменение оптической
плотности в изомолярных растворах
TiOSO4 + FeSO4 в 30%-ной H2SO4[28]
при различных концентрациях изомо-
лярных серий (л/оль/л):
/ — 0,07295; 2 — 0,1459; 3 — 0,2918.
плотности в сернокислых рас-
творах Ti-4 и Fe-З, как показа-
ли^Корощенко и Сикорская [29],
отклоняется от аддитивности.
По отклонению оптической плот-
ности от аддитивности в изомо-
лярных растворах установлено, что Ti-4 и Fe-З образуют комплексы с
отношением компонентов 1 : 1 (раствор с концентрацией 30% SO3)
и 1 : 2 (53% SO3). Константы нестойкости этих комплексов равны
соответственно 1,9 • 10“2 (18° С) и 5,9 10~4 (19° С). Комплексы
Ti-4 с Fe-З из сернокислых растворов не были выделены.
В растворах сульфатов титана-4 и хрома-3, судя по отклонению
оптической плотности в изомолярных сериях от аддитивности,
образуется комплекс с отношением Сг-3 : Ti-4 = 2:1. Константа
нестойкости его равна 2,3 • 10“4 (75%-ная H2SO4, 20,5° С).
24 9-614
369
Горощенкр и Сидоренко [301 методом спектрофотометрии уста
новили существование комплексов Ti-4 с Fe-З и Сг-3 в сернокислых
растворах, содержащих примесь сульфата аммония. Отношение
компонентов в этих комплексах равно 1 : 1 (растворы с концент-
рацией 200 г/л H2SO4 и 100 г/л (NH4)2SO4).
Характерно, что при образовании комплексов Ti-4 с Сг-3 зе-
леная и фиолетовая модификации хрома ведут себя неразличимо.
Отсюда вытекает, что различия в строении комплексных ионов
хрома не оказывают влияния на состав их комплексов с титаном
ДВОЙНЫЕ СУЛЬФАТЫ ТИТАНА
Двойные сульфаты образуют нормальный сульфат титана и ти-
танилсульфат, в состав которых входят ионы Ti4+ и TiO2+.
Нормальный сульфат титана вступает в соединение с сульфата-
ми щелочных и щелочноземельных металлов, лантанидов, некото-
рых переходных металлов в двухвалентном состоянии (Fe, Ni, Со,
Мп, Zn, Си и др.) и аммония. Состав двойных сульфатов нормаль-
ного сульфата титана большей частью выражается общей формулой
(Мл+)1/л Ti(SO4)3, но имеются и соединения с иным отношением
компонентов. Двойные сульфаты титана неустойчивы к действию
воды, подвергаются гидролизу. Из водных растворов они обычно
не кристаллизуются, поэтому получают их нагреванием растворов
сульфата титана и сульфата соответствующего металла в концент-
рированной серной кислоте.
Титанилсульфат образует двойные сульфаты с сульфатами ще-
лочных металлов и аммония. Они кристаллизуются из сернокислых
растворов средней концентрации. В разбавленных сернокислых
растворах гидролизуются подобно титанилсульфату, а в концент-
рированной серной кислоте превращаются в двойные сульфаты
нормального сульфата титана. Состав двойных солей титанилсуль-
фата выражается большей частью формулой M2TiO (SO4)2 • nH2O,
где М — щелочной металл и группа аммония.
Изучены двойные сульфаты титана еще недостаточно. Сведения
о некоторых соединениях имеются только в патентных публика-
циях, которые, как известно, отличаются малой достоверностью
Двойные сульфаты титана и натрия
Розенгейм и Шютте [131] пытались синтезировать двойные
сульфаты титана и натрия сплавлением двуокиси титана с бисуль-
фатом натрия. Однако из бисульфатных сплавов соединения опре-
деленного состава не были выделены.
При нагревании четыреххлористого титана с бисульфатом нат-
рия образуется двойная соль Na2SO4 • Ti(SO4)2 • ЗН2О [123].
Методы синтеза двойного сульфата NaHSO4 • Ti(SO4)2 • хН2О
приведены в патентах [1, 42]. В авторском свидетельстве [1 ] описан
370
способ получения этой соли взаимодействием рассчитанных коли-
честв водного раствора бисульфата натрия и четыреххлористого
титана. Четыреххлористый титан добавляется к раствору бисуль-
фата натрия постепенно с одновременным охлаждением раствора
до температуры 20° С.
В немецком патенте [60] говорится о получении двойной соли
Na2SO4 • TiOSO4 сплавлением титансодержащих материалов с би-
сульфатом натрия. Соль выделяется в виде белых игольчатых
кристаллов, плохо растворимых в холодной воде и разлагающихся
горячей водой.
В американском патенте [65] описан двойной сульфат 4Na2SO4 •
• 5TiOSO4 • 2TiO2, образующийся при смешении концентриро-
ванных растворов сульфата натрия и сульфата титана.
Недавно синтезирована двойная соль с молярным отношением
Na2SO4 : TiOSO4 = 2:3 [10]. Она кристаллизуется из водных
растворов титанилсульфата с концентрацией более 100 г/л TiO2
после добавления к ним хлорида или сульфата натрия и нагрева-
ния при температуре 60—70° С в течение 1—2 суток или стояния
при комнатной температуре более недели. Из растворов, не содер-
жащих свободной серной кислоты (SO3 : TiO2 = 1:1), двойная
соль кристаллизуется в виде гексагидрата 2Na2SO4 • 3TiOSO4 •
6Н2О. В присутствии свободной серной кислоты из раствора
выпадают кристаллы состава 2Na2SO4 • 3TiOSO4 • xH2SO4 • #Н2О
с переменным содержанием H2SO4 и Н2О, но сходные по кристал-
лооптическим свойствам (Ng = 1,670, Np = 1,549).
Двойные сульфаты титана и калия
*
Сплавлением двуокиси титана с бисульфатом калия получен
двойной сульфат K2SO4 • Ti(SO4)2 [131, 149, 150].
Глятцель [92], упаривая эквимольную смесь сульфатов калия
и титана в сернокислых растворах, выделил плохо растворимые
в £вде кристаллы, которым приписывает состав K2SO4 • Ti(SO4)2 •
• ЗН2О. В американском патенте [65] описан двойной сульфат
4K2SO4 • 5TiOSO4 • 2TiO2, полученный смешением холодного на-
сыщенного раствора сульфата калия с сернокислым раствором
сульфата титана.
Деменев с соавторами [33] синтезировал соль 2K2SO4 •
• 2TiOSO4 • 5Н2О, которая кристаллизуется из сернокислых раст-
воров титанилсульфата после добавления к ним сульфата калия.
К раствору титанилсульфата, содержащему 40—50 г/л TiO2 и 200—
250 г/л H2SO4, добавляется в сухом виде сульфат калия из расчета
K2SO4 : TiO2 = 4:1 и смесь нагревается до растворения соли.
Затем раствор быстро охлаждается до комнатной температуры.
По мере его охлаждения выпадает белый осадок. Кристаллизация
заканчивается за 2 ч. Осадок соли отфильтровывается, промывается
24* 371
• абсолютным спиртом и сушится в эксикаторе над серной кислотой
до постоянного веса.
Препарат двойного сульфата титанила и калия состоит из мелких
коллоидно-дисперсных частиц [32]. Размер отдельных частиц, со-
бранных в агрегаты, по данным электронно-микроскопических ис-
следований, равен 10—30 А. На электронограммах дифракцион-
ные линии не были обнаружены.
Двойная соль 2K2SO4 • 2TiOSO4 • 5Н2О кристаллизуется из
растворов с концентрацией серной кислоты 19—25% при расходе
сульфата калия K2SO4 : TiO2 = 4:1. В иных условиях из раст-
воров образуются другие продукты, являющиеся, по предполо-
жению [33], солями переменного состава.
Полякова и Милютина [63] изучили систему ТЮ2 — H2SO4 —
K2SO4 — Н2О при температуре 20° С методом растворимости.
В интервале концентраций 12,6 — 22,7% H2SO4 и 5—18% K2SO4
в ней обнаружили только одну двойную соль с отношением
K2SO4 : TiOSO4 =1:1. Состав соли, в отличие от [33], они выра-
жают формулой K2SO4 • TiOSO4 • Н2О, рассматривая ее как мо-
ногидрат.
В воде двойная соль K2SO4 • TiOSO4 • Н2О растворяется ин-
конгруэнтно и без добавления к раствору серной кислоты не может
быть перекристаллизована. При нагревании водных растворов гид-
ролизуется с образованием осадка гидроокиси титана.
Осаждение двойного сульфата титанила и калия предложено
использовать для выделения титана из сернокислых растворов [33]
и для очистки титана от примеси хрома [64]. Ниобий соосаждается
с титаном в виде изоморфной примеси и после восстановления
титана до трехвалентного может быть выкристаллизован в виде
двойной соли KNbO (SO4)2 • 2Н2О [74].
Двойные сульфаты титана и аммония
Двойные сульфаты титана и аммония наиболее хорошо изуче-
ны по сравнению с двойными сульфатами титана и других ме-
таллов вследствие большого интереса, который они представля-
ют для технологии переработки некоторых видов минерального
сырья.
(NH4)2Ti(SO4)3. Впервые эта соль получена нагреванием дву-
окиси титана со смесью серной кислоты и сульфата аммония [19].
50 г двуокиси титана смешивают с 250 г сульфата аммония и 500 г
концентрированной серной кислоты. Смесь нагревают около 2 ч
до температуры 300—320° С. Образовавшиеся кристаллы двойного
сульфата после охлаждения промывают большим количеством хо-
лодной воды, спиртом и сушат при 100—110° С. Выход 80—90%.
Двойной сульфат (NH4)2Ti(SO4)3 образуется также при нагре-
вании со смесью серной кислоты и сульфата аммония различных
372
титановых минералов — рутила, ильменита, перовскита, лопарита
и сфена — если весовое отношение H2SO4 : (NH4)2SO4 в сплаве
близко к двум [16].
Соль (NH4)2Ti (SO4)3 образует крупные кристаллы моноклинной
системы (рис. 103, см. вклейку) белого цвета с жирным блеском.
Угол погасания прямой, Ng — 1,754, Np = 1,684. В работе [16]
с кристаллов соли. В холодной
приведена дебаеграмма, снятая
Рис. 104. Потери веса при прокалива-
нии (NH4)2 Ti (SO4)3 [16].
воде кристаллы (NH4)2Ti (SO4)3
растворяются медленно, на чем
основан метод очистки их от из-
бытка плавней. При длительном соприкосновении с водой или
парами воды соль подвергается гидролизу по реакции
(NH^ Ti (SO4)3 + Н2О = (NH4)2 TiO (SO4)2 + H2SO4.
В герметически закрытой посуде она сохраняется без измене-
ний неограниченное время.
Судя по потерям веса и по составу промежуточных продуктов
реакции, двойной сульфат (NH4)2Ti (SO4)3 разлагается в интерва-
ле температур 350—600° С непосредственно до двуокиси титана
(рис. 104). На термограмме нагревания (рис. 105), однако, отме-
нно два экзотермических эффекта при температурах 525 и 610° С,
происхождение которых еще не выяснено.
5(NH4)2SO4 - 2Ti(SO4)2. Соль получена при исследовании си-
стемы TiO2 — SO3 — (NH4)2SO4 — Н2О методом растворимости в
виде гексагональных кристаллов в сплавах двуокиси титана с суль-
фатом аммония и серной кислотой [18]. Она образуется в сплавах
с большим содержанием сульфата аммония и малой концентрацией
воды. Кристаллы ее в воде и в водно-спиртовых растворах хорошо
растворяются. Состав их определен без выделения из сплавов
в чистом виде методом Скрейнемакерса.
2(NH4)2SO4 • 3TiOSO4 • Ti(SO4)2. Получена также нагреванием
двуокиси титана со смесью серной кислоты и сульфата аммония
[19]. Она кристаллизуется из сплавов с температурой кипения
около 220° С. При более высокой температуре кипения сплавов,
373
т. е. при меньшем содержании в них воды, кристаллы этой соли
растворяются и из расплава кристаллизуется соль (NH4)2Ti (SO4)3.
Кристаллы двойной соли 2(NH4)2SO4 • 3TiOSO4 • Ti (SO4)2 бе-
лого цвета, анизотропные, по-видимому, ромбической симметрии,
Ng — 1,755, Np = 1,670 (рис. 106, см. вклейку). В холодной воде
растворяются очень медленно, разлагаясь по реакции
2 (NH4)2 SO4 • 3TiOSO4 • Ti (SO4)2 + H2O =
= 2 (NH4)2 TiO (SO4)2 + 2TiOSO4 + H2SO4.
В интервале температур 400—550° С, судя по потерям веса при
прокаливании, соль разлагается до двуокиси титана [16]. Разло-
Рис. 107. Термограмма нагревания
2 (NH4)2SO4 • 3TiOSO4 • Ti (SO4)2 [16].
жение ее происходит ступен-
чато; на термограмме нагре-
вания (рис. 107) отмечено два
эндотермических эффекта при
температурах 508 и 685° С.
Первый эффект вызван раз-
ложением двойной соли до
безводного титанилсульфата,
а второй — термической дис-
социацией титанилсульфата.
Соль 2(NH4)2SO4 •
• 3TiOSO4 • Ti(SO4)2 интерес-
на тем, что в состав ее входят
одновременно нормальный
сульфат титана и титанил-
сульфат. Она является пере-
ходным соединением между
двойными сульфатами титана двух типов — на основе нормально-
го сульфата и титанилсульфата.
(NH4)2TiO (SO4)2. Безводный сульфат титанила и аммония впер-
вые, по-видимому, получили Розенгейм и Шютте [131] в виде мел-
ких игловидных кристаллов из смеси концентрированных раство-
ров сульфата аммония и сульфата титана. Однако на основе данных
химического анализа авторы приписали полученной соли состав
(NH4)2TiO (SO4)2 • Н2О. Такой же состав этой соли приписывает
Таки [145, 146], рассматривая ее как моногидрат, хотя при ис-
следовании методами протонного магнитного резонанса и инфра-
красной спектроскопии присутствие связанной воды в препарате
не обнаружено.
Индивидуальность безводного сульфата титанила и аммония
установлена нами [19]. Эта соль существует в двух кристалличе-
ских модификациях. a-(NH4)2Ti(SO4)2 кристаллизуется в системе
TiO2 — SOs — (NH4)2SO4 — Н2О при температуре выше 40° С.
Физрастворов с высокой концентрацией серной кислоты она крис-
таллизуется и при более низких температурах. Кристаллы
cc-(NH4)2TiO(SO4)2 образуются в результате разложения природных
374
титансодержащих материалов нагреванием с серной кислотой и
Ьсульфатом аммония при температуре около 180° С [16]. а-Моди-
Iфикация — белый кристаллический порошок, не гигроскопичный,
I устойчивый на воздухе. Кристаллизуется в виде мелких удлинен-
г ных пластинок или тонких иголок (рис. 108, см. вклейку), с прямым
I погасанием, Ng = 1,707, Np = 1,600. В работе [16] приведена де-
баеграмма кристаллов a-(NH4)2TiO (SO4)2. р-Модификация впер-
вые получена обезвоживанием в сушильном шкафу моногидрата
у (NH4)2TiO(SO4)2 • Н2О при температуре 200° С. Кристаллы р-моди-
| фикаиии сохраняют форму тетраэдров моногидрата, но обладают
слабым двойным лучепреломлением с показателями преломления
около 1,629. На воздухе кристаллы £-(NH4)2TiO (SO4)2 неустой-
[ чивы, поглощают пары воды и превращаются в моногидрат
(NH4)2TiO (SO4)2 • Н2О.
Мотов [541 наблюдал образование p~(NH4)2TiO (SO4)2 при ис-
следовании системы ТЮ2 — SO3 — (NH4)2SO4 — Н2О методом раст-
воримости. Кристаллы этой соли получались в результате дегид-
[* ратации моногидрата в маточной жидкости при 0° С.
Двойной сульфат титанила и аммония (а) хорошо растворим
I в воде, но скорость растворения его при комнатной температуре
мала. Концентрированный водный раствор соли можно пригото-
вить только за время растворения около 24 ч. В смесях серной
f кислоты и сульфата аммония с концентрацией более 200—300 г/л
растворимость соли мала. Она также малорастворима в абсолют-
' ном спирте и в водно-спиртовых растворах, содержащих более
40% спирта. На этом основан метод получения. Для выделения
кристаллов a-(NH4)2TiO (SO4)2 в чистом виде их промывают на
воронке Бюхнера смесью серной кислоты и сульфата аммония
(350г/л H2SO4, 200 г/л (NH4)2SO4), затем 40%-ным раствором спирта
и безводным или 96%-ным спиртом. Промытые кристаллы сушат
на воздухе или в сушильном шкафу до полного испарения спирта.
Термическое разложение a-(NH4)2TiO (SO4)2 происходит в ин-
тервале температур 300—600° С. На кривой потери веса при нагре-
в#Йтш [57] около 400е С наблюдается перегиб, вызванный, вероятно,
образованием в качестве промежуточного продукта реакции
безводного титанилсульфата. На термограммах нагревания
a-(NH4)2TiO (SO4)2 в интервале температур 265—703° С имеется
несколько эндотермических и экзотермических эффектов (рис. 109),
свидетельствующих о сложности процессов, протекающих при тер-
мическом разложении этой соли.
(NH4)2TiO(SO4)2 • Н2О. Образование этой соли в виде тетраэд-
рических кристаллов впервые наблюдал Блондель [79]. Он полу-
чил их при упаривании разбавленного раствора сульфата титана
под вакуумом после частичной нейтрализации серной кислоты.
Препарат, по-видимому, был загрязнен гидроокисью титана и по-
этому состав его выражен формулой (NH4)2SO4 • TiOSO4 • TiO2 •
- ЗН2О.
375
Таки и Кунитома [144] получили эту соль смешением водных
растворов четыреххлористого титана, серной кислоты и сульфата
аммония. По результатам химического анализа состав соли выра.
жен формулой (NH4)2TiO (SO4)2 • 2Н2О. В последующих работах
Таки [147, 148] продолжает рассматривать эту соль как дигидрат.
Индивидуальность моногидрата двойного сульфата титанила
и аммония впервые установлена автором [21 ]. В этой же работе
Рис. 109. Термограмма нагревания
a-(NH4)2TiO (SO4)2 [57].
предложены методы синтеза моногидрата из двуокиси титана,
серной кислоты и сульфата аммония. Условия кристаллизации
(NH4)2TiO (SO4)2 • Н2О в системе TiO2 — SO3 — (NH4)2SO4 — Н2О
изучены Мотовым [53, 54, 56].
Метод получения моногидрата двойного сульфата титанила и аммония из
раствора сульфата титанила следующий [16].
К раствору сульфата титанила, содержащему 80—100 г!л ПОг и 400—500 г!л
HaSO4, добавляют сульфат аммония в твердом виде из расчета полного связыва-
ния сульфата титанила в двойной сульфат с сульфатом аммония плюс избыток.
Избыток сульфата аммония необходим для понижения растворимости (высали-
вания) моногидрата. Последний обладает малой растворимостью в смесях серной
кислоты и сульфата аммония с суммарной концентрацией (NH4)2SO4 + H2SO4 =
= 500—600 г!л.
Содержимое перемешивают и оставляют на 12—24 ч др окончания кристалли-
зации. Чтобы из раствора вместо моногидрата не выпала безводная соль, высали-
вание следует проводить при температуре не выше 30—35° G, а отношение H2SO4 :
: (NH4)2SO4 должно быть не более 2. Кристаллизацию моногидрата можно уско-
рить, охладив раствор до —5 10° С. При этой температуре кристаллизация
длится всего 1—2 ч.
Осадок двойной соли отфильтровывают на воронке Бюхнера и промывают
40%-ным спиртом, затем 90%-ным спиртом и сушат при температуре 100—110° G
до постоянного Beca.j
Полученная соль по химическому составу соответствует моно-
гидрату (NH4)2TiO(SO4)2 • Н2О.
376
Технический продукт может быть получен из ильменита, пе-
ровскита, сфена, лопарита и других видов титанового сырья
[20,22, 24, 25].
Одноводный сульфат титанила и аммония образует кристаллы
в форме тетрагонтритетраэдров (рис. 110,см. вклейку). При мед-
ленной кристаллизации выпа-
дают крупные белые кристал-
лы размером до 10 мм. Они име-
ют кубическую решетку с пара-
метром а = 17,57 A, d\5= 2,005
[144], N = 1,580 [19]. Дебае-
грамма приведена в работе [16].
На воздухе при комнатной тем-
пературе кристаллы моногидрата
сохраняются неизменными дол-
гое время, удерживая 3—4 %
гигроскопической воды (рис. 111).
Высушенная при температуре
110° С двойная соль содержит
Рис. 111. Потери веса при высушива-
нии (NH4)2TiO (SO4)2 • Н2О на воздухе
и при температуре 110° С [16].
только одну молекулу кристал-
лизационной воды. Кристалли-
ческая структура соли при суш-
ке не изменяется. Кристаллиза-
ционная вода отщепляется при нагревании соли до температуры
200° С (рис. 112). На кривой потери веса, снятой динамическим
методом, составу моногидрата соответствует площадка.
а — динамический метод [57], б — изотермическое взвешивание [16].
Мотов [57] изучил обезвоживание двойного сульфата титанила и
аммония при температурах 20 и 80° С тензиметрическим методом Ван-
Баммелена. Он проводил опыты с образцами соли, содержавшими
377
9,53 и 6,73% fH20 (теоретическое содержание воды в моногид-
рате 5,81%). При температуре 20° С препараты двойной соли
(9,53% Н2О) над растворами серной кислоты с концентрацией
менее 58% H2SO4 оводня-
РисЛНЗ. Изменение веса
(NH4)2TiO (SO4)2 • Н2О над растворами
серной кислоты при температуре 20° С
[57]. Концентрация серной кислоты (%):
1— 21,04; 2—31,27; 3 — 41,65; 4 — 51,26;
5 — 60,87; 6 — 69,74; 7 — 80,40; 8 — 90,06;
9 — 96,06.
лись, а при большей кон-
центрации — обезвоживались
(рис. 113). Оводнение двойно-
го сульфата титанила и аммо-
ния над растворами разбав-
ленной серной кислоты
(>21,04% H2SO4) сопровож-
дается гидр ол и зом.
Двойной сульфат титани-
ла и аммония очень прочно
удерживает гигроскопическую
влагу и для установления
равновесия образцы соли при-
ходилось выдерживать над
растворами серной кислоты
длительное время. Однако
выдерживание препаратов да-
же над концентрированной
серной кислотой не привело
к полному обезвоживанию
их до состава моногидрата
(NH4)2TiO (SO4)2 • Н2О.
При температуре 80° С равновесие устанавливалось быстрее.
Оводнение препаратов наблюдалось до концентрации серной кис-
Рис. 114. Диаграмма давления пара над
(NH4)2TiO (SO4)2 • Н2О при 20 (/) и 80° С (2) [57].
лоты 82%. Над растворами серной кислоты большей концентрации
препараты двойной соли обезвоживались до моногидрата.
37S
На диаграмме упругость пара — содержание воды (рис. 114)
имеется изгиб, который, по мнению Мотова, указывает на сущест-
вование полуторагидрата (NH4)2TiO (SO4)2 • 1,5Н2О. Однако эта диа-
грамма имеет относительный характер, так как адсорбция воды
на поверхности кристаллов зависит от их поверхности, и потому
изгиб на кривой может быть обусловлен различием в величине
поверхности кристаллов испытуемых образцов.
Термическое разложение моногидрата, судя по
веса при нагревании (см. рис.
И2),
ператур 100—600° С. На термо-
грамме нагревания (рис. 115) фик-
сируется большое число тепловых
эффектов. Отщеплению кристал-
лизационной воды отвечает эндо-
термический эффект в интервале
кривым потери
интервале тем-
происходит
в
Рис. 116. Политерма растворимости
(NH4)2TiO (SO4)2 • Н2О—Н2О [57].
182—210° С. В интервале температур 400—500° С, судя по излс-
м^на кривой потери веса и термограмме нагревания, в качестве
промежуточного продукта реакции образуется титанилсульфат.
Разложению последнего отвечает эндотермический эффект при
496° С. Конечным продуктом прокаливания моногидрата является
двуокись титана со структурой анатаза. Выше 1000° С анатаз
превращается в рутил.
Одноводный сульфат титанила и аммония хорошо растворим
в воде. Растворимость его в воде при 20° С равна 150,2 г!л ТЮ2
[21 ], с понижением температуры растворимость уменьшается
(рис. 116). Диаграмма растворимости в системе(NH4)2TiO (SO4)2 •
• Н2О — Н2О относится к эвтоническому типу [57]. Эвтониче-
ская точка лежит при температуре —10,6° С и содержании
29,0 вес.% (NH4)4TiO (SO4)2. Концентрированные водные растворы
моногидрата при комнатной температуре устойчивы неограничен-
но долгое время. В разбавленных растворах одноводный сульфат
379
титанила и аммония гидролизуется. Нагретые до кипения растворы
двойного сульфата титанила и аммония, подобно растворам ти-
танилсульфата, гидролизуются практически нацело. Из одновод-
ного сульфата титанила и аммония гидролизом получают титановые
пигменты высокого качества.
Кристаллизуясь из сернокислых растворов, одноводный суль-
фат титанила и аммония увлекает в осадок небольшое количество
Рис. 117. Система TiO2 — SO3 — (NH4)2SO4 — Н2О
с малой концентрацией воды [18]:
а — (NH4)2TiO (SO4)2; х — 3 (NH4)2SO4 . 7TiOSO4; t —
TiOSO4 - H2SO4 • H2O; c—2 (NH4)2SO4 . 3TiOSO4 • Ti (SO4)2;
6 — (NH4)2Ti (SO4)3; у - 5 (NH4)2SO4 • 2Ti (SO4)2.
примесей, поэтому эту соль предложено использовать для отделе-
ния титана от ниобия, тантала, хрома и других примесей при пере-
работке минерального сырья [16].
Кинетика кристаллизации (NH4)2TiO (SO4)2 • Н2О зависит от со-
держания в растворе примесей. По данным [71 ], ниобий замедляет,
а тантал ускоряет кристаллизацию двойной соли. Мы наблюдали
380
значительное ускорение кристаллизации (NH4)2TiO(SO4)2 • Н2О из
растворов в присутствии сульфата Fe-2 по сравнению с растворами,
не содержащими посторонних примесей. Длительность кристал-
лизации двойной соли из технологических растворов с примесью
железа при комнатной температуре составляет 15—30 мин.
Одноводный сульфат титанила и аммония, вследствие простоты
его получения, устойчивости на воздухе, хорошей растворимости
Рис. 118. Диаграмма растворимости системы TiO2— SO3 (NH4)2SO4— Н2О при
температуре 20° С [53]:
а]— кривая моновариантного равновесия a-(NH4), TiO (SO4)2 и (NH4)2TiO (SO4)2 • H2O;
б — кривая моновариантного равновесия гидроокиси титана и (NH4)2TiO (SO4)2 • Н2О.
Замкнутые кривые — изогидры, выражающие отношение числа молей воды на 100 молей
солевой массы.
в воде и другим свойствам, со временем становится одним из на-
иболее распространенных титановых препаратов. Технический про-
дукт, кроме производства титановых пигментов, может найти при-
менение для дубления кож и других целей.
3(NH4)2SO4 • 7TiOSO4. Двойной сульфат этого состава получен
Горощенко и Андреевой [18] при исследовании системы TiO2 —
SO3 — (NH4)2SO4 — Н2О методом растворимости. Он имеет вид
мелких пластинчатых кристаллов, видимо, ромбической системы.
381
00
ьэ
Таблица 46
Растворимость в системе Т10а — S08 - (NH4)2 S04 - На0 при температуре 20° С [63]
Состав раствора, вес. % Состав раствора, мол. % Число молей воды на 100 молей со- левой массы
тюя SO, (NH4)a SO4 тю2 so8 (NH4)2 SO4
0,19 9,87 34,25 0,66 27,75 71,59 854
0,22 7,67 38,82 0,75 20,87 78,38 789
0,38 7,33 31,72 1,50 23,38 75,12 1052
0,40 4,54 37,65 1,49 13,77 84,74 948
0,43 3,49 42,22 1,50 9,88 88,62 829
2,46 5,49 35,52 8,66 15,75 75,59 883
2,58 5,94 27,60 10,70 20,08 69,22 1175
0,12 13,22 34,18 0,38 34,13 65,49 738
0,13 15,78 28,59 0,42 42,47 57,11 813
0,73 7,68 23,70 3,42 29,32 67,26 1412
4,62 9,02 20,58 18,93 30,09 50,98 1196
0,07 19,07 29,25 0,22 46,65 53,13 687
0,22 12,99 27,10 0,82 38,91 60,27 974
0,52 12,73 19,66 2,28 45,51 52,21 1308
2,25 9,08 17,35 11,19 36,71 52,10 1570
0,09 21,35 24,08 0,27 54,29 45,44 754
0,15 18,68 22,68 0,52 52,34 47,14 892
1,87 12,76 12,94 9,31 51,73 38,96 1600
7,29 12,73 15,88 26,74 38,02 35,02 1025
0,05 26,72 19,31 0,14 65,00 34,85 714
0,06 25,06 24,69 0,16 57,68 42,16 629
0,13 23,50 18,29 0,42 63,11 36,47 850
0,45 17,55 15,78 1,84 58,87 39,29 1209
6,32 15,81 11,24 24,31 49,54 26,15 1137
10,58 16,86 13,52 32,56 42,28 25,16 806
0,03 31,98 14,28 0,09 75,04 24,87 686
Твердая фаза
(NH4)2TiO(SO4)2 • Н20
То же
в
(NH4)2TiO(SO4)2 • Н20 + гель TiO
(NH4)2TiO(SO4)2 • Н20
То же
в
в
в
в
в
в
в
в
в
в
в
в
a-(NH4)2TiO(SO4)2
(NH4)2TiO (S04)a . Н20 +
+ a- (NH4)2 TiO (SO4)2
a- (NH.k TiO (SO. к
То
»
»
в
»
в
в
в
»
»
»
»
в
»
»
2
То же
a- (NH^ TiO (SOA,
СО
оо
< 0,05 30,89 19,94 0,13^ 67,52 32,35 585
0,13 29,03 13,98 0,40 73,37 26,23 783
0 46 22,28 11,95 1,79 70,25 27,96 1121
2,24 17,38 10,22 9,92 62,71 27,37 1379
7,46 21,05 10,53 24,10 55,34 20,56 873
0,012 42,03 10,83 0,03 83,90 16,07 512
0,022 37,63 10,40 0,06 82,93 17,01 623
0,05 36,57 15,45 0,12 76,04 23,84 543
0,13 34,45 10,10 0,37 81,83 17,80 715
0,34 26,28 7,87 1,30 80,74 17,96 1095
3,08 21,36 8,25 12,11 68,31 19,58 1172
4,20 25,96 6,98 14,22 71,51 14,27 943
7,42 22,07 8,52 24,30 58,83 16,87 900
7,95 25,38 7,51 23,98 62,33 13,69 791
8,87 27,88 7,26 24,66 63,14 12,20 691
0,017 42,70 6,24 0,044 90,18 9,78 587
0,028 47,92 6,14 0,065 91,26 8,68 476
0,086 39,05 6,01 0,25 89,52 10,23 685
1,21 31,46 5,78 3,98 84,50 11,52 900
5,30 29,53 6,04 16,05 72,88 11,07 795
6,72 32,81 5,71 18,22 72,42 9,36 659
7,73 36,03 5,02 19,28 73,13 7,59 566
9,25 30,58 5,86 24,54 66,07 9,39 639
0,07 48,66 1,90 0,18 97,00 2,82 536
0,15 0,81 43,70 53,19 2,08 2,49 0,41 1,77 96,20 94,93 3,39 3,30 648 424
2,27 35,60 3,93 6,75 86,20 7,05 768
3,25 3,43 48,14 40,16 4 2,16 2,23 7,43 9,14 89,59 87,26 2,98 3,60 471 641
же
»
»
To
в
в
(NH4)2 TiO (SO4)2 . H20
To же
a-(NH4)2 TiO
To
в
в
в
(SO4)2
же
в
в
в
(NH4)2 TiO (SO4)2 . Н20 +
+ а- (NH4)3 TiO (SO4)2
a- (NH4)2 TiO (SO4)2
(NH4)2 TiO (SO4)2 .
a- (NH4)2 TiO (S04)
же
в
в
в
в
в
в
в
в
в
в
в
в
в
в
То
в
в
»
в
в
в
в
в
в
в
в
в
в
в
Х12
2
В воде растворяется хорошо, но медленно. Под действием воды
частично разлагается, что затрудняет получение в чистом виде.
Система ТЮ2 — S03 — (NH4)2SO4 — Н2О изучена методом раст-
воримости в области с малой концентрацией воды [16, 18] и в об-
ласти водных растворов [53, 56, 57]. На рис. 117 представлена
диаграмма растворимости в области малой концентрации воды,
построенная по методу Иенеке. На равностороннем треугольнике
отложен солевой состав в вес.%, а перпендикулярно к нему —
отношение воды на 100 г солевой массы.
В изученной области концентраций обнаружено пять двойных
сульфатов титанила и аммония. Вблизи угла SO3 кристаллизуется
двухводный титанилсульфат и кислый титанилсульфат TiOSO4 •
•H2SO4 • Н2О. Эта область системы детально не исследовалась,
так как в ней не образуются двойные сульфаты титана.
Наиболее обширное поле на диаграмме растворимости зани-
мает двойная содь a-(NH4)2TiO (SO4)3 (а). Фаза 3 (NH4)2SO4 •
• 7TiOSO4 (х) кристаллизуется в замкнутой области. С увели-
чением концентрации воды эта фаза превращается в кислый ти-
танилсульфат (т).
Соль 2(NH4)2 SO4 • 3TiOSO4 • Ti (SO4)2 также кристаллизуется
в замкнутой области (с) и с уменьшением содержания воды пре-
вращается в двойной сульфат (NH4)2Ti (SO4)2 (b). Второй двойной
сульфат, кристаллизующийся в области с малым содержанием воды,
имеющий состав 5(NH4)2SO4 • 2Ti(SO4)2 (у), устойчив в сплавах
с содержанием сульфата аммония более 40 вес.%.
Мотов [53, 56, 57] изучил растворимость в системе ТЮ2 —
SO3 — (NH4)2SO4 — Н2О в области водных растворов и по-
строил изотермы растворимости при 0; 20; 40 и 80° С (табл. 46,
рис. 118, 119). В изученной области системы существуют три равно-
весные твердые фазы — (NH4)2TiO (SO4)2 • Н2О, a-(NH4)2TiO (SO4)2
и гидроокись титана.
Область кристаллизации (NH4)2TiO(SO4)2 тяготеет к углу
H2SO4, так как серная кислота способствует дегидратации. Поле
одноводного сульфата титанила и аммония тяготеет к углу
(NH4)2SO4. Область его существования расширяется с пониже-
нием температуры и около 40° С выклинивается. Выше 40° С мо-
ногидрат сульфата титанила и аммония не кристаллизуется. Эта
фаза является низкотемпературной, безводная соль, наоборот,—
высокотемпературная фаза. Область ее кристаллизации прости-
рается до температуры около 220° С.
Поверхность насыщения одноводного сульфата титанила и ам-
мония переходит в поверхность кристаллизации безводной соли
плавно. Изогидры пересекаются на моновариантной кривой под
большим углом, практически равным 180° С.
На диаграмме растворимости системы TiO2 — SO3 —
(NH4)2SO4 — Н2О, построенной по методу Иенеке, поверхность
насыщения двойных сульфатов имеет форму холма с крутым ска-
384
том со стороны грани H2SO4 — (NH4)2SO4 — Н2О и более пологим
у сечения TiOSO4 — (NH4)2SO4 — Н2О.
Со стороны грани TiO2 — (NH4)2SO4 — Н2О поверхность на-
сыщения моногидрата граничит с поверхностью насыщения гид-
роокиси титана (Щ), расширяющейся при понижении темпера-
РисЛПЭ. Диаграмма растворимости системы TiO2 — SO3—(NH4)2SO4 — Н2О при
20° С в координатах (NH4)2SO4 — H2SO4cbo6. Изоконцентраты (NH4)2TiO (SO4)2 да-
• ны в г/л:
I.— ппле a-(NH4)2TiO(SO4)2; II — поле (NH4)2TiO(SO4)2 • Н2О; а — кривая моновариант-
ного равновесия (NH4)2TiO (SO4)2 • Н2О и гидроокиси титана [55’
4’
туры и уменьшении концентрации воды в сторону угла H2SO4.
По мере разбавления растворов водой поверхность насыщения
гидроокиси титана ассимптотически приближается к грани призмы
H2SO4 — (NH4)2SO4 — Н2О.
Сечение поверхности насыщения в системе TiO2 — SO3 —
(NH4)2SO4 — Н2О при постоянном содержании сульфата аммония
25 9-614
385
схематически показано на рис. 120. Разрывы на поверхности
насыщения на разрезах, отвечающих содержанию сульфата ам-
мония 15, 20 и 30% при температуре 20° С, появились вследствие
того, что поля кристаллизации двойных солей простираются в
область системы TiOSO4 — TiO2 — (NH4)2SO4 — Н2О, в которой
растворимость не изучалась. Любопытно, что в пределах этой
системы двойные сульфаты должны находиться в равновесии с
растворами, в которых отношение SO3 : TiO2 (за вычетом сульфата
503 TiOz S03 П0г S03 ПОг 503 ТсОг S03 Ti02
^H4)2S0^7o^ 20 30 55 65
t, С 20 20 20 200 200
Рис. 120. Схематическое сечение поверхности насыщения системы TiO2 — SO3 —
(NH^SO, — Н2О, изображенной по методу Иенеке в виде треугольной приз-
[181:
а — (NH4)2TiO2(SO4)2; at — (NH4)2TiO (SO4)2 • H2O; x — 3 (NH4)2SO4 • 7TiOSO4; t —
TiOSO4 • H2SO4 - H2O; b — (NH4)2Ti (SO4)3; c — 2 (NH4)2SO4 • 3TiOSO4 • Ti2(SO4)2;
у — 5 (NH4)2SO4 • 2 Ti (SO4)2; d — гидроокись титана.
аммония) менее единицы. Это может служить подтверждением
нашей гипотезы о существовании в водных растворах сульфоти-
тановой кислоты и ее солей. Фигуративная точка состава аммоний-
ной соли сульфотитановой кислоты лежит в системе TiOSO4 —
TiO2 — (NH4)2SO4 — Н2О. На изотермах 40 и 80° С поля двой-
ных сульфатов титанила и аммония за пределы системы TiOSO4 —
H2SO4 — (NH4)2SO4 — Н2О не распространяются. Разрывы на се-
чениях поверхности насыщения отсутствуют из-за неустойчивости
аммонийных солей сульфотитановой кислоты.
На разрезах, отвечающих содержанию сульфата аммония 55
и 65%, пунктиром проведены сечения, которые при температуре
200° С могут быть реализованы только под давлением.
Моногидрат (NH4TiO(SO4)2 • Н2О при температуре 0 и 20° С
образуется в качестве метастабильной фазы почти во всей области
кристаллизации безводной соли a-(NH4)2TiO(SO2)2, за исключени-
ем небольшого участка вблизи угла SO3. Со временем тетраэдри-
ческие кристаллы (NH4)2TiO(SO4)2 • Н2О превращаются в моно-
зяь
клинные a-(NH4)2TiO(SO4)2.
в a-фазу зависит от состава
ления от пограничной кри-
вой. Граница распростра-
нения области сравнитель-
ной устойчивости моноги-
драта при температуре 20°C
на рис. 119 отмечена пунк-
тирной кривой. Слева от
пунктирной кривой пре-
вращение моногидрата в сс-
фазу происходит в течение
нескольких суток.
Система TiOSO4 —
(NH4)2SO4 — Н2О. Мотов
[56] построил изотерму
растворимости этой системы
при температуре 20° С
(рис. 121). Из растворов в
этой системе кристаллизую-
тся сульфаты TiOSO4 •
• 2Н2О, a-(NH4)2TiO (SO4)2
и (NH4)2TiO (SO4)2 • Н2О.
Безводная соль растворяе-
тся инконгруэнтно. Моно-
гидрат двойного сульфата
Скорость превращения моногидрата
раствора и повышается по мере уда-
(NH^TL0(S04)2
Tl0S04 мал, % (NH4)2 so4
Рис. 121. Диаграмма растворимости системы
TiOSO4 — (NH4)2 SO4 — Н2О при температу-
ре 20° С (по методу Иенеке).
титанила и аммония раст-
воряется конгруэнтно. Точка d на кривой растворимости отвечает
началу гидролиза двойного сульфата титанила и аммония. Учас-
ток диаграммы, правее d, относится к четверной системе.
Двойные сульфаты титана с сульфатами металлов II группы
Сульфаты металлов II группы образуют двойные соли, по-ви-
димому, только с нормальным сульфатом титана. Соединения эти
неустойчивы к действию воды, легко подвергаются гидролизу,
разлагаясь при этом на титанилсульфат и сульфаты щелочнозе-
мельных металлов.
Мента и Патель [116] получили двойной сульфат титана и маг-
ния MgTi (SO4)3 в виде кубических кристаллов белого цвета. При
нагревании раствора двуокиси титана и сульфата кальция в серной
кислоте образуется двойная соль CaTi(SO4)3 в виде белых кристал-
лов кубической формы [151] (рис. 122, см. вклейку). Кристаллы
двойного сульфата титана и кальция получаются при разложении
серной кислотой перовскита [17, 22] и сфена [25]. ' 1
Двойной сульфат титана и кальция кристаллизуется в системе
ТЮ2 — СаО — SO3 — Н2О из растворов с высокой концентрацией
23*
387
S03 (рис. 123). Растворимость его в концентрированных раство-
рах серной кислоты мала и растворение носит конгруэнтный ха-
рактер. При содержании в растворе менее 67% SO3 двойная соль
разлагается; одновременно происходит частичный гидролиз по
реакции
CaTi (SO4)3 + Н2О - CaSO4 + TiOSO4 + H2SO4.
Кристаллы CaTi (SO4)3 в воде растворяются инконгруэнтно
(с разложением), но при температуре ниже 30° С вода с двойным
СаО
Рис. 123. Диаграмма растворимости системы TiO2 —
СаО—SO3 — Н2О в области устойчивости CaTi (SOJ3
при температуре 150° С [16].
сульфатом титана и кальция реагирует медленно. Соль нераствори-
ма в спирте и не подвергается заметному разложению.
Приводим описание препаративного метода получения двой-
ного сульфата титана и кальция.
К ЮО жл концентрированной серной кислоты добавляют 10 г
раствора титанилсульфата в воде (14% ТЮ2) и 3 г ангидрита и смесь
нагревают. Осадок ангидрита и частично высолившегося титанил-
388
сульфата растворяется и при температуре 130—150° С из прозрач-
ного раствора начинает кристаллизоваться двойная соль. Кристал-
лизация заканчивается после нагревания до температуры 190—
210° С. Содержимое охлаждают, раствор с осевшего осадка двой-
ного сульфата декантируют и отбрасывают. Остаток смешивают
с 20—30-кратным количеством холодной воды (4—5° С). Во избе-
жание разложения двойной соли температура во время смешения
не должна превышать 20° С. Кристаллы двойной соли отфильтро-
вывают на воронке Бюхнера через бумажный фильтр, промывают
водой, 96%-ным спиртом и сушат при температуре 100—110° С.
Высушенная соль должна храниться
в закрытой посуде без доступа па-
ров воды. /
Кристаллы CaTi (SO4)3 устойчивы /
до температуры около 600° С (рис. 124). у
В интервале температур 600—800° С о У
они разлагаются по реакции <
CaTi (SO4)3 = TiO2 + ° /
/ 675 о
+ CaSO4 + SO3. / 740
Выше 1100° С двуокись титана реа- у
гирует с сульфатом кальция, обра- у
зуя титанат кальция. ~ m
Вейнланд и Кюль [151 ] сообщили Рис’
также о получении двойного сульфа-
' та титана и стронция SrTi(SO4)3 и двойного сульфата титана и
бария 2BaSO4 • 3Ti(SO4)2. Однако Мента и Понча [117], повто-
рившие их опыты, не смогли получить эти соли.
Горощенко и Солиев [11 ] показали, что сульфаты титана и бария
образуют двойную соль MTi (SO4)3 в виде белых нитевидных крис-
таллов. Она кристаллизуется из сернокислых растворов с кон-
центрацией SO3 более 78%, насыщенных сульфатом бария, при
нагревании с двуокисью титана (2—3%) до температуры 300—
305° С. В воде и серной кислоте кристаллыMTi (SO4)3 растворяются
инконгруэнтно, однако при температуре ниже 20° С с водой реаги-
руют медленно и поэтому легко выделяются в чистом виде после-
довательным промыванием осадка охлажденной водой, ацетоном
или безводным спиртом. Сульфаты стронция, ртути-2 и бериллия
с сульфатом титана двойных солей не образуют [11].
Сопоставляя литературные данные с собственными исследова-
ниями, авторы [11] установили зависимость образования двойных
солей титана с двухвалентными металлами от величины их ион-
ных радиусов. Согласно этой зависимости, металлы с валентностью
2+ и ионными радиусами менее 1,06, за исключением бериллия
(ионный радиус 0,34), образуют двойные сульфаты состава
MTi (SO4)3 с кубической структурой. Двухвалентные металлы с
389
большими ионными радиусами либо вовсе не дают двойных солей
с титаном [Sr, Hg-2], либо образуют двойные соли аналогичного
состава, но с решеткой более низкой симметрии (барий).
Двойные сульфаты титана с сульфатами
редкоземельных элементов
Сульфаты редкоземельных элементов с сульфатом титана обра-
зуют двойные соли состава (TR)2 (SO4)3 • 4Ti(SO4)2, где TR — ред-
коземельные элементы. Кристаллы двойных сульфатов этого типа
Cez03
Рис. 125. Диаграмма растворимости системы TiO2 —
Се2О3 — SO3 — Н2О в области устойчивости
Се2 (SO4)3 • 4Ti (SO4)2 при температуре 100° С [16].
гексагональной формы получаются при разложении лопаритового
концентрата серной кислотой [16].
По своим свойствам двойные сульфаты титана и редкоземельных
элементов сходны с двойным сульфатом титана и кальция. Подобно
последнему, они мало растворимы в концентрированной серной
кислоте и неустойчивы в присутствии воды. Под действием воды
390
разлагаются на простые сульфаты с одновременным гидролизом
сульфата титана.
Система TiO2 — Се2О3 — SO3 — Н2О изучена в области крис-
таллизации двойной соли методом растворимости [16]. Двойная
соль титана и церия Се<> (SO4)3 • 4Ti (SO4)2 кристаллизуется в этой
системе из концентрированных сернокислых растворов (рис. 125).
Растворимость ее в концентри-
рованной серной кислоте мала.
При содержании в растворе SO3
менее 51% двойной сульфат ти-
тана и церия разлагается по
реакции
Се2 (SO4)3 - 4Ti (SO4)2 + 4Н2О -
= Се2 (SO4)3 • 3H2SO4 +
+ 4TiOSO4 + H2SO4.
По внешнему виду двойной
сульфат титана и церия — мел-
кокристаллический продукт све-
тло-желтого цвета. На воздухе
поглощает пары воды и разлага-
ется на простые сульфаты. В гер-
метически закрытой посуде сохраняется без изменения. Судя по
термограмме нагревания (рис. 126), устойчив до температуры 600° С.
Разлагается ступенчато на двуокись титана и сульфат церия, ко-
торые затем реагируют выше 1100° С с образованием титаната
церия.
Двойные сульфаты титана с сульфатами
переходных металлов
Титан образует двойные сульфаты с сульфатами некоторых
переходных металлов в двухвалентном состоянии. По свойствам
двойные сульфаты титана с переходными металлами аналогичны
двойному сульфату титана и кальция.
Мента и Патель [116], нагревая титанилсульфат с сульфатами
переходных металлов в серной кислоте выше 200° С получили
двойные сульфаты титана с цинком — ZnTi (SO4)3, марганцем —
MnTi(SO4)3, кобальтом — CoTi(SO4)3, медью — CuTi(SO4)3. Соеди-
нения MnTi (SO4)3 и CoTi (SO4)3 окрашены соответственно в желтый
и розовый цвета, а остальные белого цвета.
Мента и Понча [117] аналогичным методом получили двойные
сульфаты титана с никелем NiTi (SO4)3 и кадмием CdTi(SO4)3.
Двойные сульфаты титана и переходных металлов при нагре-
вании разлагаются с отщеплением SO3 на двуокись титана и окислы
или сульфаты переходных металлов. Температура начала разло-
жения равна: CuTi (SO4)3 — 375° С, ZnTi(SO4)3 и CoTi'(SO4)3 —
391
400° C, MnTi (SO4)3 — 450° С. Температура начала разложения
солей находится в пределах 375—450° С.
С целью изучения взаимодействия между сульфатами титана
и железа-2 Горощенко и Сикорская [27] исследовали систему
TiO2 — FeO — SO3 — Н2О при температуре 125° С методом раст-
воримости. В ней обнаружено образование одной двойной роли
Рис. 128. Изотерма растворимости системы FeO — ТЮ2 — SO3 — Н2О при 125° С
[27]. Линии однонасыщения в тройных системах:
&А — TiOSO4 • H2O; Ас — TiOSO4 • H2SO4 . H2O; Сс — Ti(SO4)2; hH — FeSO4 . H2O;
НК — тв. раствор FeSO4 • H2SO4; KI — тв. раствор Fe2S2O7 • 2H2SO4; LI — FeS2O7.
Линии двунасыщения в четверной системе:
еЕ — FeSO4 - H2O-|-TiOSO4 • Н2О; ED — FeTi (SO4)3+TiOSO4 • H2O; DB — FeTi(SO4)3-f-
+ TiOSO4 • H2SO4 . H2O; Bb — FeTi (SO4)3 + Ti (SO4)2; EF — FeTi (SO4)2 + FeSO4 .
• H2O; FG — FeTi (SO4)s + тв. раствор FeSO4 H2O; Cg — FeTi2(SO4)2 4- тв. раствор
Fe,So07 • 2H2SO4, KG — тв. раствор FeSO4 • H2SO4 + тв. раствор FeS2O7 • 2H2SO4;
AE — TiOSO4 • H2O + TiOSO4 . H2SO4 • H2O; BC — TiOSO4 • H2SO4 • H2O + Ti2(SO4)2;
HF — FeSO4 • H2O+ тв. раствор FeSO4 • H2SO4.
состава FeTi (SO4)3 в виде псевдокубическиx кристаллов сирене-
вого цвета (рис. 127, см. вклейку). Под микроскопом у этих кри-
сталлов обнаруживается слабое двойное лучепреломление накрест
между диагоналями. Они кристаллизуются из растворов упомя-
нутой системы в области высокой концентрации SO3 (рис. 128).
В растворах с содержанием менее 68% SO3 двойная соль титана
и железа разлагается на простые сульфаты.
Эти же авторы описали препаративный метод получения крис-
таллов FeTi(SO4)3 [28]. В колбу, заполненную СО2, вливают 50 мл
1,5-мол. раствора FeSO4, 20 мл 3-мол. раствора TiOSO4 и 300 мл
концентрированной серной кислоты. При смешении выпадают в оса-
док кристаллы FeSO4 • Н2О, а раствор окрашивается в желтый
цвет. Затем смесь нагревают. При температуре около 160° С раст-
вор темнеет, осадок сульфата железа начинает растворяться и
из раствора выпадают кубические кристаллы сиреневого цвета
FeTi (SO4)3. По мере испарения воды и повышения температуры
392
кипения раствора (смесь закипает при 210—215° С), количество
осадка двойной соли увеличивается. Кристаллизация двойного
сульфата титана и железа заканчивается при температуре около
240° С. Титан и железо из раствора практически полностью пе-
реходят в осадок из-за малой растворимости сульфатов в концент-
рированной серной кислоте. В осадке наряду с кубическими крис-
таллами FeTi(SO4)3 имеются гексагональные кристаллы FeSO4 •
•H2SO4. Для получения кристаллов FeTi (SO4)3 в чистом виде,
осадку дают отстояться, жидкость декантируют. Кристаллы про-
мывают большим количеством холодной воды, чтобы отмыть от
маточной жидкости и растворить кислый сульфат железа, не до-
опуская повышения температуры выше 20° С. Наконец кристаллы
FeTi(SO4)3 промываются 96%-ным спиртом и сушат при темпера-
туре 100—110° С.
Двойной сульфат титана и железа, полученный по описанному методу, содер-
жит небольшую примесь окисного железа (2—4% РегО3). Избежать частичного
окисления двухвалентного железа в трехвалентное во время синтеза двойной со-
ли не удается даже в атмосфере СО2 или инертных газов.
На воздухе кристаллы FeTi(SO4)3 неустойчивы и под действием
паров воды разлагаются по реакции
FeTi (SO4)3 + Н2О = FeSO4 + TiOSO4 + H2SO4.
Одновременно закисное железо окисляется кислородом воздуха
до окисного. В закрытой посуде FeTi (SO4)3 сохраняется без изме-
нения.
Судя по потерям веса при нагревании, в интервале температур
550—700° С двойной сульфат титана и железа разлагается до TiOo
и Ре,О3 [27].
Ранее кристаллы двойного сульфата титана и железа были
получены Хайдером и Бегумом [97], но эти авторы состав их вы-
разили формулой 2FeSO4 • Ti (SO4)2. Еще раньше [16, стр. 86]
кристаллы этой соли обнаружены в сплавах лопарита и перовскита
с сульфатом аммония и серной кислотой. Однако состав их уста-
новлен также неточно. Фактически из сплавов кристаллизовалась
двойная соль FeTi (SO4)3, загрязненная окислами редкоземельных
элементов и аммония. Двухвалентное железо попадало в сплав
в результате растворения стальных стенок котла.
Демодарен и Неургаонкар [83] описали двойной сульфат титана и железа
состава 3FeSO4 • TiOSO4 • ПН2О, полученный ими в виде желтого продукта после
упаривания сернокислых растворов титанилсульфата и сульфата железа-2 до тем-
пературы около 150° С. Существование этого сульфата при исследовании системы
TiOs — FeO — SO3 — Н2О методом растворимости не подтвердилось. Вероятно, ав-
торы приняли за индивидуальное соединение смесь двух сульфатов — FeSO4 •
• Н2О и TiOSO4 • Н2О, кристаллизующихся из сернокислых растворов, с кото-
рыми они проводили опыты.
393
Горощенко и Сикорская [29] изучили методом растворимости
также четверную систему TiO2 — Fe2O3 — SO3 — Н2О (рис. 129),
однако не обнаружили двойных сульфатов титана и железа-3,
кристаллизующихся из растворов. В качестве твердых фаз в чет-
верной системе кристаллизуются те же соединения, которые су-
ществуют в частных тройных системах. В интервале температур
Рис. 129. Изотерма растворимости системы TiO2 — Fe2O3 — SO3 — Н2О прп тем-
пературе 150° С. Линии насыщения растворов твердыми фазами:
аА — TiOSO4 • H2O; АВ — TiOSO4, ВС — TiOSO4 • H2SO4 • Н2О; Сс — Ti (SO4)2; Kk —
HFe (SO4)2 • H2O; Kf — Fe2(SO4)3; Dd — TiOSO4 • H2O + HFe (SO4). • H2O; DE —
HFe (SO4)2 • H2O + TiOSO4; EL — HeTiO(SO4)2 • H2O -f- HFe (SO4)2 • H2O; LM —
H2TiO (SO4)2 • H2O + Fe2(SO4)s. Линии KL и CM проведены ориентировочно [27].
150—190° С железо в четверной системе кристаллизуется в виде
кислой соли HFe (SO4)2 • Н2О, а при более высокой температуре —
в виде нормального сульфата Fe2 (SO4)3. Сульфаты титана и железа
образуют между собой взаимные твердые растворы ограниченного
состава.
ПЕРОКСОСУЛЬФАТЫ ТИТАНА
При добавлении к сернокислым растворам титана перекиси во-
дорода образуются перекисные комплексы, содержащие группу
Ti\ | Ричардсон [1301 установил методом ионного обмена су-
ществование в сернокислых растворах частиц, имеющих состав
394
находящихся между собой
Патель и Мохан [125], упаривая раствор свежеосажденной
гидроокиси титана с рассчитанным количеством серной кисло-
ты и избытком перекиси водорода, получили красное вещество
TiO2SO4 • ЗН2О. Этот пероксисульфат титана хорошо растворяется
в воде и абсолютном спирте. Строение соли авторы изображают
структурной формулой
Н2О
Присутствие двух молекул воды во внутренней сфере доказывает-
ся более трудным удалением их из препарата, тогда как третья мо-
лекула воды удаляется легче. Наличие трехчленного цикла перо-
ксогруппы подтверждается образованием озона при хранении соли.
Максимум светопоглощения в разбавленном сернокислом раст-
воре перекисного титанового комплекса лежит при длине волны
410 нм, а в абсолютном спирте — при 390 нм. Строение спиртового
комплекса авторы изображают формулой
С М AU -q
С2Н5ОН
0,5Н2О.
Маццужелли и Пантанелли [114, 115] в результате добавления
к раствору титанилсульфата перекиси водорода и сульфата лития
с последующим высаливанием спиртом получили перекисную соль
Li2[TiO2(SO4)2] • 7Н2О в виде аморфного порошка желтого цвета.
Аналогичным методом авторы получили перекисную соль ти-
тана и натрия Na2[TiO2 (SO4)2] • Н2О (желто-красные кристаллы),
титана и калия K2[TiO2 (SO4)2] • ЗН2О (гигроскопический
аморфный порошок красного цвета) и титана и аммония
(NH4)2 [TiO2 (SO4)2] • %Н2О (маслянистая жидкость красно-желтого
цвета, затвердевающая в гигроскопичную аморфную массу).
Синтезированы перекисные комплексы, которым приписывают
состав K2SO4 [Ti(OH)2(H2O) (Н2О2) ]SO4 и 4K2SO4 [Ti(OH)3(H2O2) ]2SO4
[113]. Перекисные соединения титана и калия получили Билли
[81] и Шварц с соавторами [139, 140].
395
Перекисные комплексы титана образуются в сернокислых раст-
ворах при облучении их у-лучами 60Со [80]. Японские исследова-
тели [143] предложили использовать раствор титанилсульфата
(0,02-м. TiOSO4 и 0,8-н. H2SO4) для дозиметрии больших доз
у-лучей (105—108 р). Действие дозатора основано на окрашивании
раствора в желтый цвет за счет взаимодействия титана с перекисью
водорода, образующейся в результате радиолиза.
О ХИМИЗМЕ СУЛЬФАТИЗАЦИИ ТИТАНСОДЕРЖАЩИХ МАТЕРИАЛОВ
В производстве двуокиси титана сернокислотным методом возникает необхо-
димость в разложении (сульфатизации) различных видов титанового сырья. Ис-
торически сложилось так, что из-за недостаточной изученности химии титана,
методы разложения ильменита при создании производства титановых пигментов
разрабатывались чисто эмпирически. Химизм взаимодействия ильменита с серной
кислотой оставался при этом неизвестным, либо о нем имелись ошибочные представ-
ления. Чтобы понять химические процессы, протекающие при разложении тита-
нового сырья серной кислотой, необходимо было изучить физико-химические си-
стемы, состоящие из серной кислоты, воды и основных компонентов, входящих в
состав этого сырья.
Титановая промышленность в качестве сырья для производства двуокиси ти-
тана может использовать ильменит (FeTiO3), аризонит (Fe2O3 • STiCX), перовскит
(CaTiO3), рутил (ТЮг) и титановые шлаки (минералы на основе Ti3O5). Основными
компонентами, входящими в эти виды сырья, являются TiO2, Ti2O3, FeO, Fe2O3
и СаО. Как следует из вышеприведенных материалов, к настоящему времени уже
изучены физико-химические системы, в состав которых входят серная кислота, во-
да и перечисленные компоненты титанового сырья. Основываясь на данных иссле-
дования этих систем, можно объяснить химизм разложения титановых минера-
лов серной кислотой и указать оптимальные условия" проведения процесса.
Разложение рутила и титановых шлаков. Основным компонентом, входящим
в состав рутила и титановых шлаков, является двуокись титана. В титановых шла-
ках, кроме двуокиси титана, содержится небольшое количество полутораокиси
титана и примесь окислов железа, магния, кальция и кремния. Разложение рутила
и титанового шлака серной кислотой можно проводить по реакциям, протекающим
между двуокисью титана и серной кислотой в системе ТЮг — SO3 — Н2О. В этой
системе при температуре выше 100° С имеется два сульфата титана, хорошо раство-
римых в воде — TiOSO4 • Н2О и TiOSO4 • H2SO4 • Н2О,— которые практически
можно получить из рутила и титанового шлака, обрабатывая их серной кислотой.
Два других водорастворимых сульфата титана — TiOSO4 • H2SO4 • 2НгО и
Ti(SO4)2 — получить подобным путем из рутила и титанового шлака практически
невозможно, так как первый из них устойчив при очень низкой температуре, а
второй образуется только в олеумной области системы. Следовательно, рутил и
титановый шлак можно сульфатизировать серной кислотой по реакциям:
TiO2 + H2SO4 = TiOSO4 • Н2О,
TiO2 + 2H2SO4 - TiOSO4 . H2SO4 • H2O.
Условия сульфатизации должны соответствовать условиям существования (тем-
пература и концентрация) моногидрата и кислого сульфата титанила в системе
TiO2 — SO3 — Н2О.
Желательно сульфатизацию рутила и титанового шлака вести по первой ре-
акции, требующей минимальных затрат серной кислоты.
Разложение ильменита. Основные компоненты ильменита — TiO2 и FeO.
Процессы, протекающие при разложении ильменита серной кислотой, реализую-
тся в системе TiO2— FeO — SO3 — Н2О. Из диаграммы равновесия этой четвер-
396
ной системы следует, что сульфатизацию ильменита серной кислотой можно осуще-
ствить по следующим реакциям:
FeTiO3 + 2H2SO4 = TiOSO4 • H2O + FeSO4 • Н2О,
FeTiO3 + 4H2SO4 = TiOSO4 H2SO4 • H2O + FeSO4 • H2SO4>
FeTiO3 + 3H2SO4 = FeTi (SO4)3 + 3H2O.
На современных титановых заводах сульфатизации ильменита ведется при
температуре 180—200° С и протекает по первой реакции. Вторая и третья реакции
протекают при большом расходе и высокой концентрации серной кислоты.
Разложение аризонита. Основные компоненты аризонита.— ПО2 и F2O3.
Процессы, протекающие при разложении аризонита серной кислотой реализуются
в четверной системе ПО2 — Fe2O3 — SO3 — Н2О. В этой системе двойные сульфаты
титана в виде твердых фаз не кристаллизуются. Продуктами реакции аризонита
с серной кислотой могут быть сульфаты, образующиеся в частных тройных сис-
темах.
Равновесными процессами, по которым может быть проведено разложение
аризонита серной кислотой, являются ,
Fe2O3 • 3TiO2 + 7H2SO4 =
= 3TiOSO4 • H2O 4- 2HFe (SO4)2 • H2O + H2O,
Fe2O3 • 3TiO2 + 10H2SO4 =
= 3TiOSO4 • H2SO4 • H2O + 2HFe (SO4)2 . H2O + H2O.
Однако аризонит реагирует с серной кислотой со значительной скоростью при
температуре выше 180—190° С, при которой моногидрат титанилсульфата и кислый
титанилсульфат кристаллизуются только в качестве метастабильных фаз. Поэтому
реально аризонит можно разлагать только по реакции
’ Fe2O3.3TiO2 + 6H2SO3 = 3TiOSO4 • H2O + Fe2 (SO4)3 + 3H2O,
^протекающей выше 190° С, по которой одноводный титанилсульфат получается
в виде метастабильной фазы. Процесс этот осложняется образованием безводного
титанилсульфата TiOSO4, являющегося равновесной фазой в системе Т1О2 — SO3 —
Н2О выше 175—180° С.
Разложение перовскита. Основные компоненты перовскита — ПО2 и СаО.
Процессы разложения перовскита серной кислотой реализуются в системе ПОг —
СаО — SO3 — Н2О. Они могут протекать по следующим реакциям:
CaTiO3 + 2H2SO4 - TiOSO4 • Н2О + CaSO2 + Н2О,
CaTiO3 4- 3H2SO4 = TiOSO4 • H2SO4 • H2O + CaSO4 + H2O,
CaTiO3 + 3H2SO4 - CaTi (SO4)3 + 3H2O,
Наибольший интерес представляют первая и третья реакции. По первой реакции
процесс протекает с минимальным расходом серной кислоты, но он возможен толь-
ко при температуре ниже 200° С, когда скорость процесса мала. Разложение по
третьей реакции протекает с большой скоростью и после выщелачивания сплава
водой получаются крупнокристаллические осадки. Аналогично перовскиту раз-
лагается сфен.
Сплавление титансодержащих материалов с сульфатом аммония и серной кис-
лотой. Сульфатизации рутила, аризонита и перовскита облегчается, если серная
кислота содержит примесь сульфата аммония. Небольшая примесь сульфата ам-
мония не изменяет химизма сульфатизации титановых минералов, но повышает
извлечение титана из продуктов разложения в раствор, так как в четверной сис-
теме Т1О2 — SO3 — (NH4)2SO4 — Н2О область существования безводного титанил-
сульфата по сравнению с тройной системой уменьшается.
397
Литература
1. Авторск. свид. СССР, № 122743 от 10.10. 1959 г.
2. Авторск. свид. № 15806 от 8.6. 1956 г.
3. Белокосков В. И.— Автореф. канд. дисс., ИОНХ АН УССР, К-, 1965.
4. Белокосков В. И.— Ж- неорг. хим., 1961,6, 1443.
5. Белокосков В. И.— Ж- неорг. хим., 1962, 7, 379.
6. Богуславская Б. Е.— Ж. общ. хим., 1939, 9, 1084.
7. Богуславская Б. Е.— Ж. общ. хим., 1939, 9, 1077.
8. Богуславская Б. Е., Оттомановская О. М.— Ж. общ. хим.,
1940, 10, 673.
9. Брагина М. И., Бобыренко Ю. Я-, Долматов Ю. Д.—
Ж- физ. хим., 1966, 40, 2964.
10. Горощенко Я-Г., Сидоренко И. А.— В кн.: Материалы 6-й Ук-
раинской республиканской конференции по неорганической химии. Тезисы
докладов. Донецк, 1968, 152.
11. Горощенко Я. Г., .Солиев Л.— Ж- неорг. хим., 1968, 13, 3205.
12. Г о р о щ е н к о Я- Г-— В кн.: Научно-техническая конференция по обсуж-
дению исследовательских работ в области производства пигментной двуокиси
титана сернокислотным методом. Тезисы докладов. Сумское обл. управл. пе-
чати, Сумы, 1968, 10.
13. Горощенко Я-Г., Бирюк Л. И.— В кн.: Научно-техническая кон-
ференция по обсуждению исследовательских работ в области производства
пигментной двуокиси титана сернокислотным методом. Тезисы докладов. Сум-
ское обл. управл. печати, Сумы, 1968, 7.
14. Горощенко Я-Г., Андреева М. И., Бабкин А. Г.— Ж- прикл.
хим., 1959, 32, 1664.
15. Горощенко Я-Г., Годнева М. И.— Ж- неорг. хим., 1961, 6, 1453.
16. Г о р о щ е н к о Я. Г. Физико-химические исследования переработки ред-
коземельных титанониобатов сернокислотным методом. Изд-во АН СССР,
М.—Л., 1960.
17. Горощенко Я-Г., Белокосков В. И., Фомин Ю. А., Анд-
реева М. И.— В кн.: Сборник трудов по химической технологии мине-
рального сырья Кольского полуострова, 1. Изд-во АН СССР, М.— Л., 1959, 5.
18. Горощенко Я-Г-» Андреева М. И.— Ж- неорг. хим., 1956,2, 1413.
19. Г о р о щ е н к о Я* Г-— ДАН СССР, 1956, 109, 532.
20. Горощенко Я-Г-, Белокосков В. И., Фомин Ю. А.— В кн.:
Труды по химической технологии минерального сырья Кольского полуостро-
ва, 1. Изд-во АН СССР, М.— Л., 1959, 40.
21. Горощенко Я- Г- Автореф. канд. дисс., Кольский филиал АН СССР,
Кировск, 1951.
22. Горощенко Я-Г-, Белокосков В. И., Фомин Ю. А., Анд-
рее в а М. И.— В кн.: Сборник трудов по химической технологии минераль-
ного сырья Кольского полуострова, 1. Изд-во АН СССР, М.— Л., 1959, 25.
23. Горощенко Я-Г-, Андреева М. И.— Изв. Карельского и Коль-
ского филиалов АН СССР, 1959, 3, 115.
24. Горощенко Я-Г-» Белокосков В. И., Фомин Ю. А., Мо-
тов Д. Л.,— В кн.: Сборник трудов по химической технологии минераль-
ного сырья Кольского полуострова, 1. Изд-во АН СССР, М.— Л., 1959, 148.
25. Горощенко Я- Г., Мотов Д. Л., Трофимов Г. В.— В кн.:
Сборник трудов по химической технологии минерального сырья Кольского
полуострова, 1. Изд-во АН СССР, М.— Л., 1959, 67, 79.
26. Горощенко Я- Г., Майоров В. Г., Федюшкина С. А.—
В кн.: Титан и его сплавы, 9. Изд-во АН СССР, М., 1963, 158.
27. Горощенко Я-Г., Сикорская Э. К-— Ж. прикл. хим., 1967, 49,
1941.
28. Горощенко Я-Г-, Сикорская Э. К«— Ж. неорг. хим., 1966,
11, 1951.
29. Горощенко Я -Г., Сикорская Э. К.— Укр. хим. ж., 1967,33, 36L
398
I
30. Горощенко Я- Г., Сидоренко И. А.— Ж- неорг. хим., 1966,
11, 1302.
31. Демен ев Н. В., Милютина М. И., Шарова А. К.,
Штин А. П.— Ж- неорг. хим., 1957, 2, 465.
32. Д е м е н е в Н. В., Буйнов Н. И., Полякова В. М.— ДАН СССР,
1952, 87, 965.
33. Д е м е н е в Н. В., Шарова А. К-, Полякова В. М.—ДАН СССР,
1952, 87, 751.
34. Д о л м а т о в Ю. Д-, Бобыренко Ю. Я-— Лакокрасочные материалы
и их применение, 1965, 1, 27.
35. Д о л м а т о в Ю. Д.— Лакокрасочные материалы и их применение, 1966,
2, 57.
36. Д о л м а т о в Ю. Д.— Лакокрасочные материалы и их применение, 1966, •
5, 29.
37. Д о л м а т о в Ю. Д.— Автореф. канд. дисс., Уральский политехнический
институт, Свердловск, 1967.
38. Долматов Ю. Д., Бобыренко Ю. Я-— ЖВХО, 1965, 10, 352.
39. Жаровский Ф. Г., Вязовская Л. М.— Укр. хим. ж., 1967,33, 737.
40. Жаровский Ф. Г., Вязовская Л. М.— Укр. хим. ж., 1965, 31,
839.
41. Зборовский М. Е., Гермогенова Е. В.— Труды ВИМС. Тита-
номагнетиты, 1935, 68, 29.
42. Канадский пат. № 30614, 1930.
43. К о з а ч е к Н. Н., Горощенко Я-Г-, Парахневич Л. А.—
В кн.: Научно-техническая конференция по обсуждению исследовательских
работ в области производства пигментной двуокиси титана сернокислотным
методом. Тезисы докладов. Сумское обл. управл. печати, Сумы, 1968, 9.
44. Лыков Е. П., Горощенко Я- Г.— В кн.: Научно-техническая кон-
ференция по обсуждению исследовательских работ в области производства
пигментной двуокиси титана сернокислотным методом, тезисы докладов. Сум-
ское обл. управление печати, Сумы, 1968, 12.
45. Л е к а х Н. Б. Автореф. канд. дисс., Изд-во ХПИ, Харьков, 1964.
46. ’Леках Н. Б., Галинкер И. С.— Лакокрасочные материалы и их
применение, 1963, 5.
47. Лек а х Н. Б., Галинкер И. С.— Доклады научн. конф. Харьковского
с.-х. ин-та, 3, 1963.
48. Леках Н. Б., Галинкер И. С. — Лакокрасочные материалы и их
применение, 1964, 3.
49. Л е к а х Н. Б., Галинкер И. С.— Доклады науч. конф. Харьковского
с.-х. ин-та, 3, 1964.
50. Л у ч и н с к и й Г. П.— Химия титана. Госхимиздат, М.— Л., 1941.
51. М и л ю т и н а М. И., Штин А. П., Шарова А. К.— В кн.: Титан
и его сплавы, 5. Изд-во АН СССР, М., 1961, 301.
52. М и л ю т и н а М. И., Шарова А. К. Химия и технология редких ме-
таллов, 7. 1963, 85.
53. М о т о в Д. Л.— Ж- неорг. хим., 1957, 2, 2797.
54. Мотов Д. Л.— Изв. Карельского и Кольского филиалов АН СССР, 1958,
3, 139.
55. Мотов Д. Л.— В кн.: Сборник трудов по химической технологии минераль-
ного сырья Кольского полуострова, 1. Изд-во АН СССР, М.— Л., 1959, 101.
56. М о т о в Д. Л.— Ж- неорг. хим., 1957, 2, 2661.
57. М о т о в Д. Л.— Ж- неорг. хим., 1959, 4, 2026.
58. Набиванец Б. И.— Ж- неорг. хим., 1962, 7, 690.
59. Набиванец Б. И.— Ж- неорг. хим., 1962, 7, 412.
60. Немецкий пат. № 123860, 1901.
61. П а м ф и л о в А. В., Худякова Г. А.— Ж. общ. хим., 1949, 19, 1443.
62. Памфилов А. Б., Пельтихин С. В., Соболева И. М.—
Ж. прикл. хим., 1947, 20, 1—2.
399
63. Полякова В. М-, Милютина М. И.— Труды ин-та химии. Ураль-
ский филиал АН СССР, Свердловск, 1966, 14, 73.
64. П о л я к о в а В. М., Шейкманф. П., Чернявская Е. И.,
Щ епликова Н. Г., Бакулова Е. Б.— Труды ин-та химии. Ураль-
ский филиал АН СССР, Свердловск, 1966, 10, 31.
65. Пат. США № 1783689, 1930.
66. Пат. США № 1783684, 1930.
67. С о л и е в Л., Горощенко Я- Г.— В кн.: Украинская республикан-
ская конференция по неорганической химии. Тезисы докладов. Донецк,
1968, 153.
68. С ы р о к о м с к и й В. С., Силаева Е. В., Авилов В. Б.—
Зав. лабор., 1949, 15, 896.
69. Стромберг А. Г., Картушинск а я А. И.— Ж. физ. хим., 1963,
37, 1793.
70. Цитович И. К., Лапина Т. А.— Изв. высш. уч. зав. Химия и хи-
мическая технология, 1963, 3, 370.
71. Хамский Е. В., Подозерская Е. А.— Ж- прикл. хим., 1968,
41, 252.
72. Ч у х л а н ц е в В. Г.— Ж- неорг. хим., 1957, 2, 2014.
73. Ш а р о в а А. К-, Милютина М. И.— Труды ин-та химии, 7. Ураль-
ский филиал АН СССР, Свердловск, 1963, 79.
74. Ш а р о в а А. К-— В кн.: Ученые Урала в борьбе за технический прогресс.
Свердловск, 1959, 137.
75. В 1 о n d е 1 В., Tido J., Tridot G.— Bull. Soc. chim. France, 1968,
1, 101.
76. В 1 о n d e 1 B.— Bull. Soc. chim., 1899, 21, 262.
77. В a r r a d о u g h C. G., Lewis J., N у ho Im R. S.— J. Chem. Soc.,
1959, 3552.
78. Beukenkamp J., Herrington K. D.— J. Am. Chem. Soc., 1960,
82, 3022.
79. В 1 о n d e 1 B.— Bull. Soc. chim., 1899, 21, 264.
80. Bagno O., Pucheault J.— J. Chem. Phys, et phys.-chim. biol., 1961,
58, 465.
81. Billy M.— Ann. chim., 1921, 16, 5.
82. В e r z e 1 i u s J. J., Rose H.— Pogg., Ann., 1825, 4, 3.
83. Damodaran V., Neurgaonkar V. G.— J. Sci. Ind. Res., 1960,
BC19, B364.
84. Diethelm B., Foerster F.— Z. phys. Chem., 1908, 62, 129.
85. E b e 1 m e n J. J.— Ann. chim. et phys., 1847, 20, 385.
86. Ephraim F., Wagner P.— Ber., 1917, 50, 1088.
87. F r a n k 1 i n T. C., Sek1e mi a n H. V.— J. Inorg. a. Nucl. Chem.,
1959, 12, 181.
88. Farber P.— Chem. Ztg., 1907, 31, 263.
89. G e i s о w G.— Diss., Munchen, 1902, 12.
90. D e 1 a f о s e H.— J. Am. Chem. Soc., 1950, 72, 525.
91. G m e 1 i n s Handbuch. 1951, 41, 345.
92. G 1 a t z e 1 E.— Ber., 1876, 9, 1829.
93. H a a s W. J., Schultz В. H.— Phys., 1939, 6, 481.
94. H 6 1 e m a n n H.— Z. anorg. Chem., 1934, 220, 33.
95. Hayek E., Engelbrecht A.— Monatsh., 1949, 80, 640.
96. Handrahan E. S.— J. Inorg. a. Nucl. Chem., 1964, 26, 1757.
97. H e i d e r S. Z., В e g u m N. A.— J. Inorg. a. Nucl. Chem., 1961, 16, 365.
98. Holmes O. G., M с C 1 a r e D. S.— J. Chem. Phys., 1957, 26, 1686.
99. H i x о n A. W.> P 1 e c h n e r W. W.— Ind. Eng. Chem., 1933, 25, 262.
100. Hixon A. W., Stetkewicz J. D.— Ind. Eng. Chem., 1940, 32, 1009.
101. Hixon A. W., Frederickson R. E. C.— Ind. Eng. Chem., 1945,
37, 678.
102. J о n n a r d R.— Chim. ind., 1947, 57, 551.
103. J e r m a n Z.— Collect. Czechosl. Chem. Communs, 1966, 31, 2481.
400
104. J er man Z.— Collect. Czechosl. Chem. Commons, 1966, 31, 3280.
105. J er m a n Z— Collect. Czechosl. Chem. Commons, 1967, 32, 260.
106. Jerman Z., Figar J.— Collect. Czechosl. Chem Commons, 1966,31,
1222.
107. J e г m a n Z., Figar J.— Collect. Czechosl. Chem. Commons, 1966,31,
1214.
108. Jerman Z., Blechta V., Blechta Z.— Chim. priim., 1966, 16
(41), 410.
109. Knecht E.—Ber., 1903, 36, 166.
110. Londgren G.— Ark. kemi, 1957, 10, 397.
111. M e r z D. V.— J. prakt. Chem., 1866, 99, 157.
112. Meyer J., Meissner H.— J. prakt. Chem., 1935, 143, 70.
113. Mori M., Shi bat a M., Kyono E., Ito S.— Boll. Chem. Soc.
Japan, 1956, 29 , 904.
114. Mazzocchelli A., P a n t a n e 1 1 i E.— Atti Line., 18, 608 (1909).
115. Mazzocchelli A., Pantanelli E.— Gazz., 1910, 40, 666.
116. M e n t a S. M., Patel S. R.— J. Am. Chem. Soc., 1951, 73, 224.
117. M e n t a S. M., P о n c h a R. P.— J. Am. Chem. Soc., 1951, 73, 227.
118. N e о m a n n B.— Z. Elektrochem., 1929, 35, 42; 1932, 38, 306.
119. N a к a z о n o T.— Sci. Rep. Tohoku, 1925, 14, 109.
120. Newton H. D.— Z. anorg. Chem., 1908, 58, 378.
121. P i с c i n i A.— Gazz., 1895, 25, 542.
122. P i с c i n i A.— Z. anorg. Chem., 1898, 17, 355.
123. P i c h о n M.— J. prakt. Chem., 1928, 9, 338.
124. Parravano N., Caglioti V.— Gazz., 1934, 64, 429.
125. Patel С. C., Mohan M. S.— Natore, 1960, 186 , 803.
126. Patscheke G., Schaller W.— Z. anorg. Chem., 1938, 235, 257.
127. Raschig F., Prahl W.— Z. anorg. Chem., 1929, 42, 253.
128. R e i n d e r s W., Kies H. L.— Rec. trav. chim., 1940, 59, 785.
129. Reeves R. E., В 1 о о i n F. A.— J. Am. Chem. Soc., 1954, 76, 5233.
130. Richardson E.— J. Less-Common Metals, 1960, 2, 458.
131. Rosenheim A., Schutte O.— Z. anogr. Chem., 1901, 26, 239.
, 132. Reynolds M. L— J. Chem. Soc., 1965, 2993.
133. S о m e у a K-— Z. anorg. Chem. 1924, 138, 291.
134. Schenk M.—Helv. chim. acta, 1936, 19, 625.
135. Stabler A., Wi r t h wei n H.— Ber., 1905, 38, 2619.
136. S a g a w a T.— J. Soc. Chem. Ing. Japan Suppl., 1938, 41, 27B.
137. S a g a w a T.— Sci. Rep. Tohoku K. Honda annivers. vol., 1936, 480.
138. S h i n r i к i К., К u bo T., Kato M.— J. Chem. Soc. Japan. Ind. Chem.
Sec., 1953, 56, 832; Ch. A., 1954, 48, 14137 e.
139. Schwarz R., Giese H.— Z. anorg. Chem., 1928, 176, 209.
140. S c h w a г z R., Sexaeur W.— Ber., 1927, 60, 500.
141. Schitz-DuMont O., Simons P., В г о j a G.— Z. anorg. Chem.,
1949, 258, 307.
142. Tornton W. M.— Titanium. Amer. Chem. Soc., Monograph series. N. Y.
1927.
143. T а к a n о b u S., Kuni hi ко H.— Nature, 1962, 195, 592.
144 Taki S., Kunitomi M.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1954,
57, 534; РЖХим., 1957, 1406.
145. T a к i i S.— Naturwis., 1958, 45, 10.
146. Taki S.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1956, 59, 1288.
147. Taki S.— J. Chem. Soc. Japan. Ind. Chem. Sec., 1958, 79, 1210.
14Я Taki S.— Rept. Fac. Engng. Yamanashi Univ., 1958, 9, 43; РЖХим.,
J959, 48965
149. Warren С. M.— Pogg. Ann., 1957, 102, 449.
150. Wohler F.— Pogg. Ann., 1826, 7, 417.
151. W e i n 1 a n d R. F., Kuhl H.— Z. anorg. Chem., 1907, 54, 253.
152. Wohler L., Pliiddemann W., Wohler P.— Ber., 1908, 41 , 70
26. 9-61
401
ГЛАВА VIII
ДРУГИЕ КЛАССЫ НЕОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ ТИТАНА
В настоящей главе рассматриваются мало изученные и менее
характерные для титана классы химических соединений. К ним
относятся фосфорнокислые соединения, арсенаты, нитраты, кар-
бонаты, хлораты и др.
ФОСФОРНОКИСЛЫЕ СОЕДИНЕНИЯ
Фосфорнокислые соединения титана изучены еще не настолько
хорошо, чтобы можно было провести четкую классификацию.
В литературе описаны преимущественно соединения титана с фос-
форными кислотами и гипофосфиты титана. Строение некоторых
фосфорных соединений еще не выяснено.
При взаимодействии растворимых солей титана (сульфаты и
хлориды) с ортофосфорной кислотой и ее солями получаются про-
дукты, в которых отношение между ТЮ2 и Р2О5 зависит от условий
опыта [48]. Переменный состав фосфатов титана — следствие не-
стойкости их к гидролитическому разложению.
Розе [53] получил фосфат титана нейтрализацией водного раствора треххло-
ристого фосфора и хлорида титана аммиаком в виде белого осадка, состав которого
точно не был определен.
При действии раствора сульфата титана-3 на раствор фосфата натрия выпа-
дает фиолетовый осадок. После промывания водой он становится белым и содер-
жит только небольшое количество фосфорной кислоты. Пфорден [47] выделил фос-
фат титана-3 в виде светло-голубого осадка.
В растворах солей трехвалентного титана и фосфорной кислоты наблюдается
полоса светопоглощения с максимумом при % = 545 нм [35].
Фосфаты четырехвалентного титана впервые получены в виде объемистых
белых осадков [54].
В английском патенте [1] описан фосфат титана состава 3TiO2-P2O5 • 6Н2О.
Из растворов хлорида титана и фосфата натрия выпадает в осадок ортофосфат
титана 2TiOa • Р2О5 • ЗН2О [15, 26, 28, 45], который при промывании водой не-
прерывно теряет фосфорную кислоту. Прокаливанием его Гхош [28] приготовил
безводный фосфат 2TiO2 • Р2О5.
Состав фосфата титана изменяется при промывании водой вследствие гидро-
лиза от соотношения Ti : Р = 1 : 1 до 1,5 : 1 [10]. Из фосфорнокислых растворов
осаждается фосфат титана переменного состава, близкого к TiO (Н2РО4)о •
. TiOHPO4 • ЗН2О при pH 3—3,7 [6].
Отефейт и Марготтет [33] растворением гидроокиси титана в фосфорной кис-
лоте и последующим прокаливанием образовавшегося из раствора осадка получи-
ли безводный фосфат титана TiO2 • Р2О5. Он имеет кубическую решетку с пара-
402
метрами а = 7,80 А, Ррентг — 3,106 г!см\ z — 4, фед. гр. РаЗ [43]; по данным
[66], — а = 23,592 А.
Курбатов и Павлова [5], нагревая раствор фторида титана с фосфорной кис-
лотой, наблюдали по мере удаления HF образование кристаллического осадка
фосфата титана Ti (НРО4)2. Из кипящих растворов фосфата титана в 5—10 мол.
фосфорной кислоте получены кристаллы Ti (НРО4)г • НгО [13]. Моногидрат
Ti (НРО4)2 • Н2О образуется также при нагревании с концентрированной фосфор-
ной кислотой четыреххлористого титана [7]. При нагревании до 800° С он превра-
щается в безводный пирофосфат TiP2O7.
Присутствие пирофосфата титана отмечено в силикатно-фосфатно-титановых
стеклах [12].
Харрисон и Хуммель [34] сообщили о фосфате Ti5P4O20, плавящемся конгру-
энтно при температуре 1260° С; показатель преломления его 1,97. Верелин и Ги-
рауд [70], действуя на TiCl4 • РС1б и TiCl4 • РОС13 метиловым к этиловым спирта-
ми, получили белые осадки, нерастворимые в эфире, но растворяющиеся в спирте.
Состав их, по данным химического анализа, выражается формулами:
TiO (СН3О)2Н3РО4, TiO (С^О^ • Н3РО4, TiCl (С2Н4О)3 • Н3РО4 и TiO2 • Н3РО4.
При нагревании металлического титана с расплавленной пятью-
окисного фосфора и однометаллической аммонийной солью
NH4H2PO4 образуются триметафосфат титана Ti (РО3)3 и поли-
фосфат Ti4 (Р4О12)3 [44]. Полифосфат титана имеет кубическую ре-
шетку с параметрами а = 13,82 А, рРснТг=г 2,84 г /см3, z = 4, по-
казатель преломления 1,584. Метафосфат титана кристаллизуется
в моноклинной системе, параметры решетки а — 10,42, b = 19,35,
с = 9,54 А, Ррентг “ 2,93 г/см3, z = 12, показатели преломления
яр « 1,635, Пу— 1,651, 2v = 10°.
. Пирофосфат трехвалентного титана, по-видимому, получается
при полярографическом восстановлении в водных растворах с
pH 0—6,15 по реакции
TiO (Н2Р2О7)2- + е + 2Н+ = Ti (Н2Р2О,)7 + Н2О.
Этому процессу отвечает четкая обратимая волна с =
= —0,475 b относительно насыщенного каломельного электрода
[14].
Из расплава двуокиси титана в метафосфате натрия выделены
бесцветные кристаллы состава Na5(TiO)5P7O25 с показателем пре-
ломления 1,78 [4].
При нагревании двуокиси титана с метафосфатом натрия в
окислительном пламени образуются кристаллы квадратной формы,
имеющие состав Na2Ti(PO4)3 [69].
Добавлением раствора сульфата титана к 10—20%-ному раст-
вору гипофосфита натрия получен гипофосфит титана [23]. Осадок
этой соли нерастворим в органических растворителях, но незна-
чительно растворяется в разбавленных и хорошо — в концентри»
рованных минеральных кислотах, особенно при нагревании. Водная
суспензия гипофосфита титана имеет кислую реакцию, состав гид-
ратированной соли выражается формулой Н2 [Ti (ОН)4 • (Н2РО2)2].
При нагревании до 100° С и высушивании над концентрированной
серной кислотой он дегидратируется, превращаясь в продукт
Н2[ ТЮ2 (Н2РО2)2].
26*
403
Фосфаты титана проявляют ионнообменные свойства [49, 59].
По обменной емкости они превосходят хроматографическую окись
алюминия [9].
АРСЕНАТЫ И АРСЕНИТЫ
Розе [54] наблюдал образование арсенатов при взаимодейст-
вии гидроокиси титана с мышьяковой кислотой. Состав их, однако
не был установлен.
Райхард [55] получил титаниларсенит по реакции
5TiOSO4 + К2О • 2As2O3 + 4Н2О =
= 5ТЮ2 • 2As2O3 + K2SO4 + H2SO4.
Титаниларсенит — белый кристаллический порошок, при нагре-
вании окрашивается в интенсивно темный цвет, а затем разлагается
с выделением мышьяковой кислоты. Бокатов и Штин [3] выделили
продукт 4TiO2 • As2O6 • 13Н2О.
Арсенаты титана, подобно фосфатам, обладают ионнообменными
свойствами [9].
СЕЛЕНАТЫ
Впервые образование селенатов титана наблюдал Дитт [25],
но соединений определенного состава не получил.
При смешении сернокислого раствора титанилсульфата с экви-
мольным количеством селеновой кислоты образуются саленаты
в виде хлопьевидных осадков состава TiO2 • SeO3 • Н2О ьЛ2ТЮ2 •
• SeO3 • Н2О [16]. Осадок селенатов титана плохо растворим
в воде, но хорошо—в минеральных кислотах. Селенат TiO2 •
• SeO3 • Н2О прочно удерживает кристаллизационную воду и от-
щепляет ее лишь после нагревания до температуры 130° С [17].
НИТРАТЫ
Нитрат титана Ti(NO3)4 получили Райхельм и Хаке, [58] сме-
шением жидкой четырехокиси азота с четыреххлористым титаном
при температуре —60 4-----20° С. Первоначально четыреххлорис-
тый титан вступает с четырехокисью азота в реакцию соединения.
Образующийся продукт TiCl4 • 2N2O4 разлагается при нагревании
до Ti (NO3)4 и NOC1.
»
При взаимодействии четыреххлористого титана с четырехокисью азота об-
разуется смесь нитрата титана с продуктом присоединения к четыреххлористому
титану — хлористого нитрозила [46].
Шмайсер [60] синтезировал нитрат титана смешением растворов четыреххло-
ристого титана и азотного ангидрида в четыреххлористом углероде, Фильд и Гарди
[27] —нагреванием гидроокиси титана с азотным ангидридом. Избыток азотного
ангидрида отгонялся перегонкой под вакуумом 0,1 мм рт. ст. при температуре
21° С.
404
Нитрат титана — бесцветное кристаллическое вещество с тем-
пературой плавления 58° С [27], плавится с частичным разложе-
нием, полностью разлагается до нитрата титанила TiO'(NO8)2 при
температуре 100° С [60].
Молекула Ti'(NO3)4 имеет симметрию £>2</ [711 и параметры мо-
ноклинной решетки а = 7,80, b — 13,57, с = 10,34 А, Р = 125°s
Ризм = ЬЭ, ррентг = 2,17 г!см3, z = 4, фед. гр. Р2г!с.
С углеводородами нитрат титана образует алкилнитраты [27],
жидким аммиаком — небольшое количество оранжево-желтого осад-
ка состава Ti(NH2)2>83 (NH3)X (NO3)bl7, где х = 1,08—1,15. По ре-
акции обменного разложения между нитратом титана и амидом
калия получен коричневый осадок татрамида титана Ti(NH2)4 [63].
Гидроокись титана в значительных количествах растворяется
в азотной кислоте. Соединения определенного состава из раство-
ров не получены. Методом электродиализа показано, что в азотно-
кислых растворах до 11-н. концентрации титан мигрирует только
к катоду, т. е. существует в катионной форме [8]. В растворах
с большей концентрацией азотной кислоты, по-видимому, сущест-
вуют и нейтральные частицы, анионные комплексы не обнаружены.
КАРБОНАТЫ
Известен только карбонат трехвалентного титана. Он образует-
ся при действии кристаллического карбоната натрия на насыщен-
ный раствор треххлористого титана [11]. По внешнему виду кар-
бонат титана — крупообразная масса черного цвета, состав ко-
торой точно не определен. Судя по данным химического анализа,
карбонат титана является основной солью состава Ti (ОН)СО3 или
Ti2O (СО3)2. Несмотря на присутствие неспаренного электрона в
ионе трехвалентного титана, осадок карбоната титана-3 не дает
сигнала ЭПР.
Растворы карбоната титана в плавиковой и иодистоводородной
кислотах окрашены в травянисто-зеленый цвет. С хлористо- и бро-
мистоводородной кислотами получены растворы фиолетового цвета.
ПЕРХЛОРАТЫ
Образование перхлоратов титана в водных растворах отмечено
в работах [18, 22, 24].
Уаррир с соавторами [68] получил перхлорат титанила в виде гексагидрат
TiO (С1О4)2 • 6НаО. В инфракрасных спектрах поглощения его наблюдается сме-
щение полос поглощения воды, указывающее на наличие очень сильной водород-
ной связи.
Кришнан и Патель [37] синтезировали эту соль действием хлорной кислоты на
четыреххлористый титан.
Гексагидрат перхлората титанила представляет собой гигро-
скопическую массу, плохо растворимую в воде, бензоле, четырех-
405
хлористом углероде и диоксане, но хорошо растворимую в ацетоне
и этаноле. Он кристаллизуется в ромбической системе, параметры
решетки а = 7,66, b - 12,27, с - 14,69 А, ризм = 1,722, рРентг =
= 1,751 г/сж3, z — 4 [38].
Методами криоскопии, кондуктометрии, ионного обмена и спект-
рофотометрии в растворе перхлората титана установлено сущест-
вование комплексных ионов [Ti0C104 • од]+.По данным термогра-
фических исследований, при нагревании гексагидрата перхлората
титана в интервале температур 130—140° С образуется пентагид-
рат, а при 160—180° С — тригидрат перхлората [37].
С диоксаном перхлорат титана дает сольват состава TiO (С1О4)2 •
• 6Н2О • 4С4Н8О2 в виде бесцветных кристаллов [67]. При нагре-
вании до 110° С кристаллы разлагаются со взрывом.
Льюис с соавторами [42] получил карбамидный комплекс пер-
хлората титанила Ti [CO(NH2)2]6(C1O4)3, эффективный магнитный
момент которого 1,72 рв •
При добавлении TiO (С1О4)2 • 6Н2О к избытку диметилсульфида
(L) с последующим высаливанием бензолом образуется соединение
TiO (С1О4)2 • 5L в виде белых гигроскопических кристаллов, раст-
воримых в полярных растворителях [36]. При нагревании до тем-
пературы 190° С кристаллы взрываются.
Методом ионного обмена установлено, что в хлорной кислоте
с концентрацией до 1,5 моля НС1О4 имеет место равновесие
Ti (ОН)^ + Н+ = Ti (OH)f+ + Н2О, [ 18 ]
константа которого равна 2,0 [18]. При более высокой концентрации
хлорной кислоты идет комплексообразование титана с ионами C1CV•
Согласно [24], экстракция титана метилизобутилкетоном из пер-
хлоратного раствора протекает по уравнению
TiOH3+ + 400^ + н+ = Ti (C104)4s + H2OS.
Изучив распределение ионов титана между катионитом
Дауэкс-50Х1 и растворами перхлоратов, Либерти и Виседомини
[41] пришли к заключению, что титан в хлоридных растворах
существует в форме ионов TiOHClO2^, TiOH (С1О4)^ и нейтраль-
ных частиц TiOH(ClO4)3.
По данным распределения титана между водной и органической
фазами, определена константа равновесия этого процесса
[тюн+3]о,[ао4-]^[н+]д?
А [Ti0(C104)4]s ’ *
Спектры поглощения растворов гидроокиси титана в хлорной
кислоте, в отличие от сернокислых растворов, не зависят от спо-
соба их приготовления [2]. Следовательно равновесие между ион-
ными формами титана в хлорной кислоте устанавливается сра-
внительно быстро уже при комнатной температуре.
406
ЦИАНИДЫ
I J
При действии цианистого калия на раствор TiBr3 в жидком
аммиаке [61] образуется комплексная соль К3 [Ti (CN)6] • 2KCN
зелено-серого цвета [61]. На влажном воздухе она неустойчива
и быстро гидролизуется.
РОДАНИДЫ
Растворяя гидроокись титана в свежеполученной 10%-ной ро-
данистоводородной кислоте, Розенгейм и Кон [57] получили крас-
но-коричневый порошок состава TiO(CNS)2 • 2Н2О.
При смешении Ti2 (SO4)3 • 6NH3 и KCNS в жидком аммиаке
в мольном отношении 1 : 6 выделяется осадок, состоящий из ро-
данидов титана Ti (CNS)3h K3Ti (CNS)3 • 6H2O [64]. В чистом виде
роданид титана не получен. Двойной роданид титана в чистом виде
приготовлен из раствора TiCl3 в эфире с роданидом калия KCNS [62 ].
Из смеси Ti2(SO4)3 • 6NH3 с KCNS в мольном соотношении 1 : 12
в жидком аммиаке получены зеленые кристаллы аммиаката двой-
ного роданида титана и калия K3Ti(CNS)6 • 6NH3, который хо-
рошо растворим в жидком аммиаке [64]. Концентрированные его
растворы окрашены в фиолетовый цвет, разбавленные — в зеленый.
Из раствора KCNS и TiCl4 в CH3CN после упаривания под
вакуумом получены красно-черные кристаллы К2 [Ti (CNS)6] •
• CH3CN [65]. При нагревании до 95° С молекула метил нит-
рила отщепляется, давая безводную соль K2[Ti(CNS)]6, медленно
гидролизующуюся на воздухе. В растворе жидкого аммиака
К4 ITi (CNS)6] реагирует с KNH2, образуя объемистый желто-зе-
леный осадок титандиамина Ti (NH)2 и комплекса K[TiNH2 (NH)21.
Двойной роданид титанила и калия К2 [TiO (CNS)4] • Н2О имеет
вид ромбических кристаллов светло-коричневого цвета [57]. В воде
они легко растворяются и гидролизуются с образованием осадка
гидроокиси титана.
Штелер и Уиртвейн [62] получили аммонийную соль
(NH4)2 TiO (CNS)4 коричневого цвета.
При растворении гидроокиси титана и роданистого аммония
в роданистоводородной кислоте образуются желтые кристаллы
двойного роданида титанила и аммония (NH4)2 TiO (CNS)4 [57].
Аналогично получена бариевая соль BaTiO(CNS)4 • Н2О.
Розенгейм и Шютте [56] из смеси TiCl4, НС1 и Pb(CNS)2 в эфир-
ном растворе выделили игловидные желто-красные кристаллы ти-
тановой соли полиродановой кислоты Ti(C3N3S3H2)4.
ИОДАТЫ И ПЕРЙОДАТЫ
Из смеси растворов двуокиси титана в плавиковой кислоте
и йодноватой кислоты получены йодаты титана, которым припи-
сывают состав Ti (Ю3)4 • 2Н2О и Н2 [Ti (Ю3)61 . 2Н2О [511.
407
Нагреванием дигидрата синтезирован безводный перйодат ти-
тана Ti (Ю3)4 в виде бесцветного порошкообразного вещества с
dl2 === 4,0798. Дигидрат подвергается при высушивании гидро-
лизу, в результате которого образуется иодат титанила TiO(IO3)2.
Смешивая растворы йодноватой кислоты и гидроокиси титана
в азотной кислоте, Рай [38] приготовил иодат титана состава
Ti (ОН)2 (Ю3)2 . 2Н2О.
Гексаиодат титана Н2 [Ti (Ю3)61 • 2Н2О — белый кристалли-
ческий порошок, d^ = 3,991. В интервале температур 95—98° С
он теряет две молекулы воды, превращаясь в безводную соль [511.
Эта соль является комплексной кислотой. Рай и Сага [50, 51] по-
лучили соли этой кислоты со щелочными металлами и аммонием
следующего состава:
Формула ^32
Li2 [Ti (Ю8).] 4,155
Na2 [Ti (IO3)6 3,955
K2 [Ti (IO3)e[ 4,392
Rb2 [Ti <IO3)e] 4,432
Cs2 [Ti (IO3)6] 4,623
(NH4)2 [Ti (IO3)el 4,131
Приготовлена также калиевая соль состава К3 [Ti(IO3)7] [21].
При смешивании раствора гидроокиси титана в азотной кислоте
с раствором перйодата натрия в азотной кислоте выпадает белый
осадок перйодата состава 7TiO2 • 12О7 [52]. ^Перйодаты титана
изучены еще мало. *
ФЕРРО- И ФЕРРИЦИАНИДЫ
Раствор сульфата титана Ti2 (SO4)3 с феррицианидом калия*
по данным [291, образует желто-коричневый осадок, который при
промывании разлагается с выделением HCN. .
Ферроцианид калия с сульфатом титана-3 дает осадки от темно-
коричневого до желто-коричневого цвета [30, 47].
Получены смешанная соль KTi[Fe(CN)6] [31] по реакции TiCl5
с K4[Fe(CN)6] и нитропрусидный комплекс Ti [Fe(CN)5NOl •
• 2Н2О [39].
Четырехвалентный титан с феррицианидом калия дает красно-
коричневый, а с ферроцианидом — желтый осадки [30, 47].
Гексацианоферроат-2-титана получен в виде продукта пример-
ного состава (TiO)2(OH) • HFe(CN)6 • 4Н2О и обладает ионообмен-
ными свойствами [19, 40]. .с
ХРОМАТЫ, МОЛИБДАТЫ, ВОЛЬФРАМАТЫ И ПЕРМАНГАНАТЫ
Растворимые хроматы, молибдаты, вольфраматы и перманга-
наты с солями титана образуют неустойчивые соединения в виде
хлопьевидных окрашенных осадков. Под действием воды и кислот
эти соединения разлагаются и из них вымываются анионы. 191.
403
Концентрированная хромовая кислота растворяет значительные
количества гидроокиси титана [20]. Разбавляя растворы гидро-
окиси титана в хромовой кислоте водой при температуре 50—
100° С, Блондель [20] получил осадки основных хроматов следу-
ющих составов: 3TiO2 • 2CrOs • Н2О, TiO2 (TiO) СгО4 • 2Н2О,
2TiO2 • (TiO)CrO4 • ЗН2О.
Мы наблюдали образование основных хроматов титана желто-
зеленого цвета при нейтрализации аммиаком раствора, содержа-
щего соль сульфата титана и бихромата калия. По внешнему виду
основные хроматы титана похожи на осадки основных хроматов
свинца и цинка, получивших в лакокрасочной промышленности
название крона. Титановый крон, по сравнению с цинковым и
свинцовым, менее стоек к действию воды и нагреванию. При дли-
тельном промывании водой из осадка титанового крона вымывае-
тся СгО4~"-ион, а при высушивании он превращается в плотную
стекловидную массу грязно-зеленого цвета.
Растворением TiOCl2 в избытке SbF5 и нагреванием в течение 10 ч при
температуре 200—210° С в высоком вакууме образуется TiO(SbF6)2 [72 ] .
Литератур а
1. Английский патент № 261051, 1926.
2. Белокосков В. И. Автореф. дисс., ИОНХ АН УССР, 1965.
3. Бокатов Г. М., Штин А. П. Труды Ин-та химии, 10. Уральский фи-
лиал АН СССР, Свердловск, 1966, 45.
4. Делимарский Ю. К., Андреева Б. Н., Капцова Т.Н.—
Изв. АН СССР. Неорг. материалы, 1965, 1, 150.
5. Курбатов Д. И., Павлова С. А. Труды Ин-та химии, 10. Ураль-
ский филиал АН СССР, Свердловск, 1966, 73.
6. Курбатов Д. И., Павлова С. А. Труды ин-та химии, 10. Ураль-
ский филиал АН СССР, Свердловск, 1966, 65.
7. Невская Ю. А., Нурмакова А. К-, Сумарокова Т. Н.—
Изв. АН Каз. ССР. Сер. хим., 1968, 2, 20.
8. Набиванец Б. И.— Ж. неорг. хим., 1962, 7, 413.
9. Рябчиков Д. И., Цитович И. К-, Торпуджиян М. К-—
ДАН СССР, 1964, 156, ПО.
10. С п и ц ы н В. И., Ипполитова Е. А.— Ж. анал. хим., 1951, 6, 5.
11. Семенова Е. И.— ДАН СССР, 1962, 1368, 143.
12. С ы р и ц к а я 3. М., Фейкнер С. Я-— Изв. АН СССР. Сер. хим.,
1966, 5, 515.
13. Alberti G., Cardini-Galli Р., Gonstantino U.. Ter-
ra с с а Е.— J. Inorg. a. Nucl. Chem., 1967, 29, 571.
14. A t m a R., Kumar S., Sinha B.— Ind. J. Chem., 1964, 2, 314.
15. В i 1 1 у M.— Compt. rend., 1921, 172, 1411.
16. В r enek H.— Z. anorg. Chem., 1913, 80, 448.
17. В e r g R., Teitelbaum M.— Z. anorg. Chem., 1930, 189, 101.
18. Beukenkamp J., Herrington K. D.—- J. Am. Chem. Soc., 1960,
82, 3025.
19. Bastian J., Lieser К. H.— J. Inorg. a. Nucl. Chem., 1967, 29, 827.
20. В 1 о n d e 1 M.— Bull. Soc. chim., 1898, 19, 218.
21. Beane H. T., M a s s m a n D. R.— J. Am. Chem. Soc., 1932, 54, 1905.
22. C a g 1 i о t i V., Ciavatta L., Liberti A.— J. Inorg. a. Nucl.
Chem., 1960, 15, 115.
23. Cast a gno u R., Guilhem-Ducleo n.— Bill. traw. Soc. pharm.
Bordeaux, 1950, 88, 18.
409
24. Delafosse D.— Compt. rend., 1955, 240, 1991.
25. D i t t A.— Compt. rend., 1887, 104, 175.
26. Farber P.— Z. anorg. Chem., 1907, 46, 277.
27. F i e 1 d В. O., Hardy C. J.— J. Chem. Soc., 1963, 5278.
28. Ghosh J. C.— J. Indian Chem. Soc., 1931, 8, 695.
29. G 1 a t z e 1 E.— Ber., 1876, 9, 1829.
30. Grossmann H.— Chem. Ztg., 1906, 30, 907.
31. Garner C. D., Wa 1 1 wo г к S. C.— J. Chem. Soc., 1966, A, 1496.
32. G a u r J. N.— Z. anorg. Chem., 1963, 324, 304.
33. Hautefeuille P., Margottet J.— Compt. rend., 1886, 102, 1017.
34. Harr ison D. E., Hummel F. A.— J. Am. Ceram. Soc., 1959, 42, 487.
35. Kolbe E.— N. Jb. Min. A Beilagebd., 1935, 69, 183.
36. Krishnan V., Patel С. C.— J. Inorg. a Nucl. Chem., 1964, 26, 220.
37. Krishnan V., Patel C.— Ind. J. Chem., 1964, 2, 425.
38. Krishnan V., Patel С. C.— J. Inorg. a. Nucl. Chem., 1964, 26, 1614.
39. Khan M., Ahmad N.— Z. anorg. Chem., 1967, 354, 301.
40. L i e s t e г К. H., Bastian J., Hecker А. В., H i 1 d W.—
J. Inorg. a. Nucl. Chem., 1967, 29, 815.
41. Liberti A., Vicedomini M.— Ricerca Sci., 1966, 36, 851.
42. Lewi s J., Machin D. J., N e wnha m I. E., N у h о 1 m R. S.—
J. Chem. Soc., 1962, 2036.
43. Levi G. R., Pyeronel G.— Z. Kristallogr., 1935, 92, 190.
44. L i e b a u F., Williams H. P.— Angew. Chem., 1964, 76, 303.
45. Merz V.— J. prakt. Chem., 1866, 99, 157.
46. P a r t i n g t о n J. R., W h у n e s A. L.— J. Chem. Soc., 1949, 3135.
47. Pfordten O.— Lier. Ann., 1887, 237, 201.
48. P u g h A. J.— Soil Sci., 1938, 38, 315.
49. P i r e t J., Henry J., Bacon G., В eau det C.— Bull. Soc. ''him.
France, 1965, 12, 3950. 1
50. R а у P. R.— Z. anorg. Chem., 1933, 210, 304.
51. R а у P. R., Saha H.— Z. anorg. Chem., 1932, 208, 100.
52. Raychoudhury P. C.— J. Indian Chem. Soc., 1941, 18, 335.
53. Rose H.— Pogg. Ann., 1827, 9, 23.
54. Rose H.— Gilb. Ann., 1823, 73, 67.
55. Reichard C.— Ber., 1894, 27, 1019.
56. Rosenheim A., Schutte O.— Z. anorg. Chem., 1901, 26, 239.
57. Rosenheim A., Cohn R.— Z. anorg. Chem. 1901, 28, 167.
58. R e i n h e 1 m H., Hake A.— Lieb. Ann., 1927, 452, 47.
59. J. v a n S m i t, R о b b W., Jacobs J. J.— Nucleon., 1959, 17, 116.
60. Schmeisser M.— Angew. Chem., 1955, 67, 493.
61. S c h a f e r H. L., G 6 t z R.— Z. anorg. Chem., 1961, 309, 104.
62. Stabler A., Wirthwein H.— Ber., 1905, 38, 2619.
63. Schmitz-DuMont O., Mietens G., Ross B.— Angew. Chem.,
1963, 75, 792.
64. Schmitz-DuMont O., Simons P.,Broja G.— Z. anorg. Chem.,
1949, 258.
65. Schmitz-DuMont O., Ross B.— Z. anorg. Chem., 1966, 342, 82.
66. Vo lien к le H., Wittmann A., N о w о t n у H.— Mongtsh., 1963,
94, 956.
67. V i c e n t i n i G., Perrier M., C a m a r g о W. G. R., Couti-
nho J. M.—Chem. Ber., 1961, 94, 1063.
68. W a r r i e г A. V. R., Ramamurthy P., Patel С. C., Naraya-
nan P. S.— Indian J. Chem., 1964, 2, 131.
69. Wunder G.— J. prakt. Chem., 1871, 4, 339.
70. W e h r e 1 i n E., G i r a u d E.— Compt. rend., 1877, 85, 288.
71. Wied lei n J., Muller U., Dehnicke K-—Spectrochim. acta,
1968, A34, 253.
72. Wieldein J., Dehnicke K-— Z. anorg. Chem., 1966, 348, 278.
ОГЛАВЛЕНИЕ
Предисловие ............................................ 3
Глава I. Общие сведения о титане........................ 5
История открытия титана ............................. 5
Изотопный состав и атомный вес ...................... 6
Физические свойства ................................. 8
Нахождение в природе ............................... 11
Коррозионные свойства технического титана и его спла-
вов ................................................ 12
Физиологическое действие титана и его соединений . . 17
Геохимия и геология ................................ 18
Титановые минералы ................................. 20
Особенности химических свойств титана и классифика- 27
ция его химических соединений..........................
Литература......................................... 28
Глава II. Металлические соединения титана .... 35
Металлические соединения титана бинарного состава 36
Титан — водород .................................. 36
Титан — бериллий ................................. 40
Титан — бор ...................................... 41
Титан — углерод .................................. 43
Титан — азот ..................................... 46
Титан — кислород ................................. 48
Титан — алюминий ................................. 59
Титан — кремний................................... 61
Титан — фосфор ................................... 62
Титан — сера...................................... 63
Титан — хром ..................................... 66
Титан — марганец ................................. 67
Титан — железо ................................... 68
Титан — кобальт .................................. 69
Титан — никель ................................... 70
Титан — медь...................................... 71
Титан — цинк ..................................... 72
Титан — галлий ................................... 72
Титан — германий ................................. 73
Титан — мышьяк.................................... 73
Титан — селен .................................... 73
Титан — ниобий ................................... 74
Титан — рутений, титан — палладий ................ 75
Титан — серебро .................................. 75
Титан — индий..................................... 76
Титан — олово .................................... 76
Титан — сурьма.................................... 77
Титан — теллур ................................... 78
Титан — рений..................................... 78
Титан — осмий, титан — иридий, титан — платина 78
411
Титан — золото.................................. 79
Титан — ртуть .................................. 80
Титан — свинец ................................. 80
Титан — висмут ................................. 81
Титан — уран ................................... 81
Растворимость элементов в металлическом титане в 82
твердом состоянии .............................. 82
Многокомпонентные металлические соединения титана 84
Литература........................................ 87
Глава III. Гидроокиси титана .........................106
Гидроокиси титана низшей валентности .............106
Гидроокись титана-4...............................107
Получение гидроокиси титана.....................108
О составе гидроокиси титана......................ПО
О строении гидроокиси титана .................. 111
О состоянии гидроокиси титана в водных растворах 115
Адсорбционные свойства гидроокиси титана .... 116
Коллоидно-химические свойства гидроокиси титана 117
К вопросу о промышленных методах получения гидро-
окиси титана гидролизом сульфатов титана в водных
растворах ..................................... 119
Гидроперекись титана ...........................120
Литература .......................................121
Глава IV. Двуокись титана ............................126
Кристаллическое строение..........................126
Методы получения двуокиси титана .................131
О превращении анатаза и брукита в р^уил .......132
Превращение анатаза в рутил . .<.........132
Превращение брукита в рутил.....................136
Физические свойства ..............................136
Влияние примесей на цвет двуокиси титана .... 136
Люминесценция ..................................138
Поглощение и отражение света ..........138
Светопреломление ...............................139
Магнитные свойства .............................140
Диэлектрические свойства .......................140
Электропроводность .............................141
Полупроводниковые свойства .....................141
Работа выхода электрона ........................141
Поляризация титана в рутиле ....................142
Точка плавления ................................142
Линейное расширение и теплопроводность..........143
Упругость пара................................ 143
Теплоемкость....................................143
Стандартная энтропия и энтальпия образования 144
Свободная энергия ..............................145
Прочие свойства ................................145
Химические свойства................................145 *
Термическая устойчивость .......................146
Действие кислот и оснований ....................146
Растворимость в солевых расплавах ..............147
Действие сульфатов и сульфитов..................148
Действие восстановителей........................148
Восстановление водородом......................148
Восстановление окисью углерода................149
Действие углерода ............................149
412
Действие аммиака.............................150
Восстановление гидридом титана и металлическим
титаном .....................................150
Восстановление натрием ......................151
Восстановление магнием ......................151
Восстановление кальцием и другими металлами . 151
Действие галогенов и их соединений ............151
Фотохимическая активность ......................154 •
Хемосорбция....................................156
Литература.......................................157
Глава V. Титанаты ...................................168
Титанаты металлов I группы.......................169
Титанаты лития.................................169
Титанаты натрия................................170
Титанаты калия.................................173
Титанаты рубидия...............................175
Титанаты цезия.................................175
Соединения с низшими окислами титана ..........176
Пероксотитанаты щелочных металлов .............177
Титанаты металлов II группы .....................178
Титанаты бериллия..............................178
Титанаты магния................................179
'! Титанаты кальция ..............................182
Титанаты цинка ................................184
; Титанаты стронция..............................186
Титанаты кадмия................................188
Титанаты бария ................................189
Соединения с низшими окислами титана ..........196
Пероксотитанаты металлов II группы ............197
‘ Титанаты металлов III группы.......................197
Титанаты алюминия..............................197
Титанаты иттрия............................... 198
Титанаты редкоземельных элементов .............198
Титанаты актинидов ........................... 199
Соединения металлов III группы с низшими окислами
титана ........................................200
Титанаты металлов IV группы .....................200
Титанаты циркония .............................200
Титанаты гафния ...............................201
Титанаты свинца .........................202
Титанаты металлов V группы ....................202
Титанаты ниобия ...............................203
Титанаты висмута ..............................204
Соединения металлов V группы с низшим.; окислами
титана .......................................204
Титанаты металлов VI группы .....................205
, Титанаты металлов VII группы ......................205
Титанаты металлов VIII группы ...................206
Титанаты железа ...............................206
Титанаты кобальта и никеля ....................210
Смешанные титанаты.............................211
Литература ......................................217
Глава VI. Галогениды титана........................ 235
Фториды титана ..................................235
Фториды титана низшей валентности .............235
Четырехфтористый титан.........................237
413
Система TiF4 —HF —Н2О.......................239
Двойные фториды титана ...........................241
Фторититанаты аммония.......................242
Фтортитанаты лития..............................242
Фтортитанаты натрия.............................242
Фтортитанаты калия..............................243
Фтортитанаты меди...............................246
Фтортитанаты рубидия ...........................247
Фтортитанаты цезия..............................247
Фтортитанаты металлов II группы ................247
Пероксофтортитанаты.........................248
Хлориды титана..................................248
Двух лор истый титан..........................249
Способы получения ..............................249
Физические свойства.............................251
Химические свойства.............................252
Треххлористый титан ..............................254
Способы получения ..............................254
Физические свойства ............................256
Химические свойства ............................258
Оксихлориды титана-3............................266
Четыреххлористый титан ...........................266
Способы получения ..............................266
Физические свойства ............................267
Химические свойства ............................271
Оксихлориды и гидроксихлориды титана .... 288
Тиохлорид титана................................292
Состояние титана в солянокислых растворах . . . 292
Пероксохлортитанаты . ... л.....................293
Бромиды титана.................<....................293
Двубромистый титан............................295
Трехбромистый титан . ............................296
Четырехбромистый титан ...........................298
Способы получения...............................298
Физические свойства ............................299
Химические свойства ............................300
Йодиды титана...................................302
Двуиодистый титан ................................303
Трехиодистый титан................................304
Четырехиодистый титан.........................304
Литература .........................................306
Глава VII. Сульфаты титана . . . ....................325
Сульфаты трехвалентного титана......................325
Восстановление титана в сернокислых растворах 325
Спектры поглощения сернокислых растворов трех-
валентного титана ................................326
Получение сульфатов трехвалентного титана .... 328
Система Ti2O3 — SO3 — Н2О.........................329
Двойные сульфаты трехвалентного титана .... 331
Сульфаты четырехвалентного титана ..................332
Методы получения растворов сульфатов титана . . . 333
Простые сульфаты четырехвалентного титана . . . 334
Система TiO2 — SO3 — Н2О .........................341
Состояние титана в сернокислых растворах .... 352
Экстракция титана из сернокислых растворов органи-
ческими жидкостями . . /..........................366
Комплексные соединения титана с переходными ме-
таллами, образующиеся в сернокислых растворах. . . 366
411
Двойные сульфаты титана.......................370
Двойные сульфаты титана и натрия.............370
Двойные сульфаты титана и калия .............371
Двойные сульфаты титана и аммония ...........372
Двойные сульфаты титана с сульфатами металлов
II группы ...................................387
Двойные сульфаты титана с сульфатами редкозе-
мельных элементов ....................... . 390
Двойные сульфаты титана с сульфатами переходных
металлов.....................................391
Пероксосульфаты титана .......................394
О химизме сульфатизации титансодержащих матери-
алов .........................................396
Литература ......................................398
Глава VIII. Другие классы неорганических соединений
т итана ........................................... 402
Фосфорнокислые соединения........................402
Арсенаты и арсениты .............................404
Селенаты........................................ 404
Нитраты .........................................404
Карбонаты .......................................405
Перхлораты...................................... 405
Цианиды..........................................407
Роданиды.........................................407
Ио даты и перйодаты .............................407
Ферро- и феррицианиды............................408
Хроматы, молибдаты, вольфраматы и перманганаты . . 408
Литература ......................................409
V.
Яков Гаврилович Горощенко
ХИМИЯ ТИТАНА
Печатается по постановлению ученого,- совета
Института общей и неорганической химии АН УССР
Редактор Л. П. Кругляк
Художественный редактор И. В. Козий
Оформление художника В. И. Мохнатова
Технический редактор Н. П. Рахлина
Корректоры Р. С. Б о р ис о в а, Р. С. К о г а и.
БФ 36328. Зак. № 9.614. Вид. 85. Тираж 1800.
Формат бумаги № 1 60Х90*/1в. Печ. физ. листов
26+1 вкл. Усл. печ. листов 26,125. Обл. изд. ли-
стов 30,87. Подписано к печати 25/XI I 1969 г.
Цена 3 руб. 30 коп.
Издательство «Паукова думка>, Киев, Репина, 3.
Киевский полиграфический комбинат Комитета по
печати при
1ете Министров УССР, Киев,
л. Довженко, 3.