/



















































































































































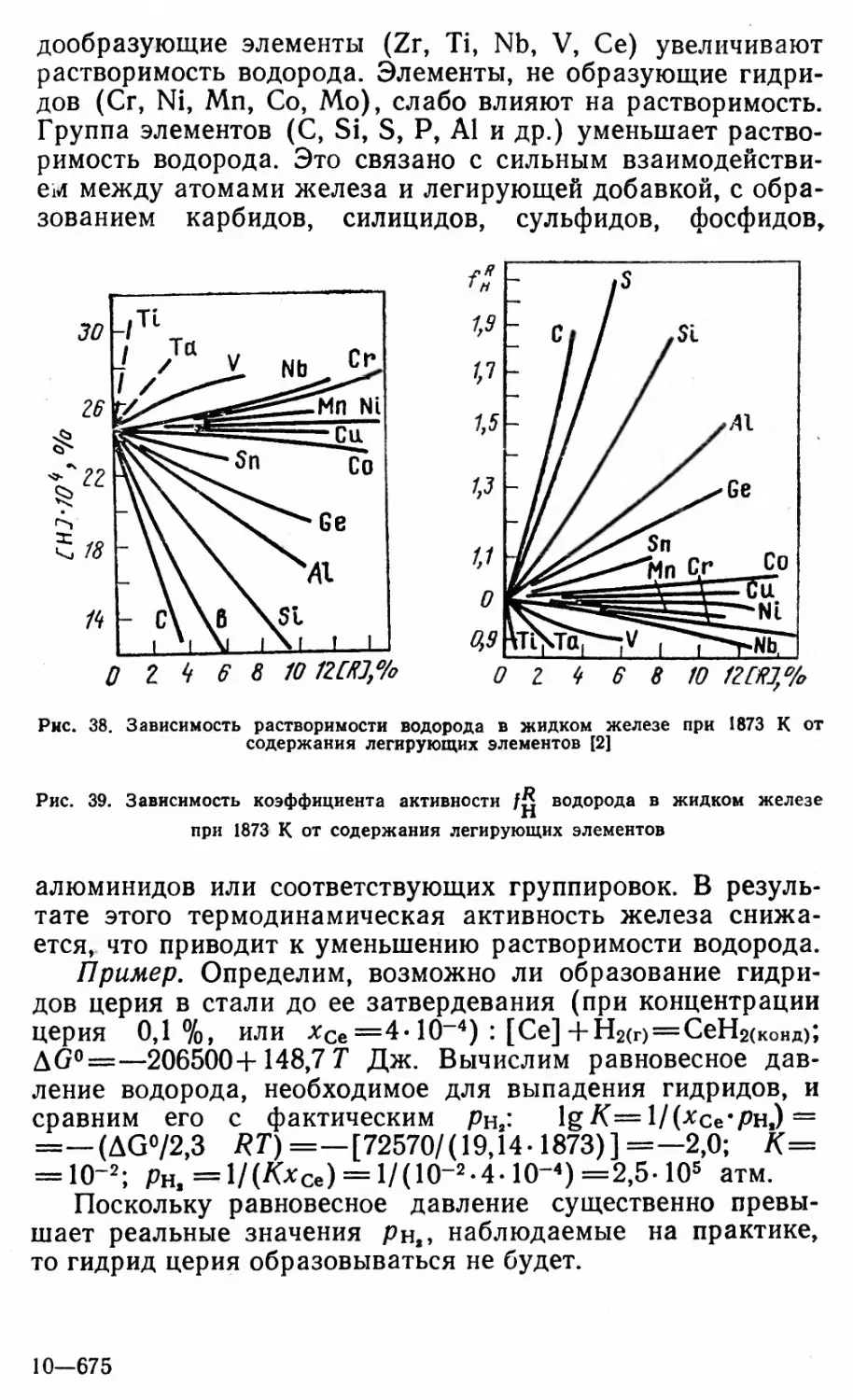
































































































































Author: Григорян В.А Белянчиков Л.Н. Стомахин А.Я.
Tags: металлургия электросталеплавильное производство
Year: 1987




Text
Периодическая система
ПЕРИОДЫ РЯДЫ РУППЫ
- 1 R20 П - R0 Ш R2O3 rh4 ro2 V RH R205
1 ] к Н 1s’ ВОДОРОД 1,0079 1
2 л L К . . 3 Li 2s’ ЛИТИЙ 1 6.941 2 4 Be 2sz БЕРИЛЛИЙ 2 9,01218 2 5 2s22p1 В з БОР 2 10,81 б л 2s22p2 C 4 УГЛЕРОД 2 12,011 7 2s22p3 N 5 АЗОТ 2 14,0057
3 ш М L К 11 Na .зз’ НАТРИИ g 22,9897" 2 12 Mg 3s% МАГНИИ в 24,305 2 13 A , 3s23Pi Al 8 АЛЮМИНИЙ 2 26,98154 14 3s23p2 S1 8 КРЕМНИЙ 2 28,086 15 _ 5 3S23p3 P 8 ФОСФОР 2 30,97376
4 N М L К к 19 , |\ 4S1 1 КАЛИЙ | 39.С96 2 „ 20 Са 4sz г КАЛЬЦИЙ I 40,08 2 „ 21 SC 3d'4s22 СКАНДИЙ в 44,9559 2 . 22 Tl 3d24S22 ТИТАН 47,90 2 23 V 3d34s2 2 ВАНАДИЙ 1J 50,9414 2
V N М L К 29 1 3d’°4s’ С U МЕДЬ 2 63,546 зо - 2 3d,04S2 ZD ’5 цинк 2 65,38 31 ~ 3 4S24p1 G И 15 ГАЛЛИЙ 2 69,72 32 4 4S24p2 G6 ГЕРМАНИЙ 2 72,59 33 5 4S24p3 AS МЫШЬЯК 2 74,9216
5 0 Ь м L К HL 37 1 Rb 5s1 6 РУБИДИЙ 85,4678 2 _ 38 2 Sr 5S2 8 СТРОНЦИЙ 1в 87,62 2 V, 39 2 Y 4d'5s2 9 ИТТРИЙ 88,9059 2 _40 2 Zr 4dz5s210 ЦИРКОНИЙ '» 91,22 2 Nb 4d45S112 НИОБИЙ 92,9064 2
vn 0 N М L К 1 <7 . to 4d,05s’ Ад ’! СЕРЕБРО 2 107,858 2 48 _ . 18 4d'°5s2 Cd КАДМИЙ 2 112,49 з 49 . 16 5S25p1 J П '2 ИНДИЙ 2 114,82 4 50 18 5S25p2 SO ’5 олово 2 118,69 5 51 18 5s25p3 SO '5 СУРЬМА 2 121,75
6 Р 0 N М L К < 55 1 CS ’ 6S1 18 ЦЕЗИЙ 13 132,9054 2 56 2 Ba 6S2nfi • БАРИЙ ’2 137,34 2 57 2 IS 5d’6s4 ЛАНТАН ’? 138,9055 2 72 2 Hf 5d26s4i ГАФНИЙ ’S 178,49 2 73 2 Ta 5d36s212 ТАНТАЛ 4 180,9479 2
VI III
J Р 0 N М L К 1 79 32 5d*36s1 Дц золото 2 196,9665 18 80 . I 32 5d,06s2 Hq РТУТЬ 3 2 200,59 3 81 _ 32 6S26p' T1 ТАЛЛИЙ 2 204,37 4 62 32 6S26p2 РЬ СВИНЕЦ 2 207,19 18 83 r, . 32 6S26p3 □ | ВИСМУТ г 208,9804
7 X ”Т“ р 0 N М L К г- 87 а Ff 7sj ’! ФРАНЦИИ ’§ (223) г _ S8 j Ra 7szi| РАДИЙ 18 226,0254 ? . 88 9 AC 6d’7s2’« АКТИНИЙ 1} (227) 1 .. 704 io Ku 6d27S2 32 КУРЧАТОВИИ ’5 (261) 1 105 ’’ Ns 6d37s2« НИЛЬСБОРИЙ’» (261) f
ЛАНТАНОИДЫ p 0 N M L К 58 2 Ce 4f’3d’6S2iB ЦЕРИЙ 'S 140,12 2 59 2 Pr 4f36S2 2? ПРАЗЕОДИМ ’S 140,9077 2 60 2 Nd 4f46S222 НЕОДИМ 'S 144,24 2 Г 61 2 Pm 4f36s22j ПРОМЕТИЙ 'S (145) 2 62 2 Sm 4f«6s224 САМАРИЙ 150,4 I
P 0 N M L К 55 2 T b 4f 96s2 27 ТЕРБИЙ ’S 158,9254 2 r, 66 8 DV 4f’°6S228 Диспрозий '? 162,50 2 . 67 2 Ho 4fl16S229 ГОЛЬМИЙ 164,9304 2 88 I ЕГ 4f’26S230 ЭРБИЙ 167,26 2 69 ? Tm «f'Wa’ ТУЛИИ 4 168,9342 2 I
’АКТИНОИДЫ p 0 N M L К 80 1Q Th 6d27s2’6 ТОРИИ 18 232,0381 ? г, 91 I Ра 5f26d’7S2i§ ПРОТАКТИНИЙ ’J 231,0359 2 92 g U 5f36d’7s2S УРАН 15 238,029 2 93 g ND 5f46d’7s2|| Нептуний ij 237,0482 ? „ 94 1 PU 5f67S2|; ПЛУТОНИИ ij (244) ! “1
p 0 N M L К 97 I В К sf’eaWg БЕРКЛИЙ 18 (247) J 98 Т Cf 5f’°7s2|| КАЛИФОРНИЙ 14 (251) г ~S9 Г ES 5f”7s2^ ЭЙНШТЕЙНИЙ 18 (254) ? 100 Fm st’2?23 ФЕРМИИ ’ (257) г г 101 i Md 5f”7s23 МЕНДЕЛЕВИИ T (258) [
элементов Д.И. Менделеева
ЭЛЕМЕНТОВ
RH2 R03 RH R207 Г »|ТГ ro4 0
(Н) He 2 .is2 ГЕЛИИ 4,00260 2
2s:2p4 О б КИСЛОРОД 2 15,9994 9 г- 2s?2ps F 7 ФТОР 2 16,99840 Ne W2sv НЕОН 6 20,179 2
16 л 3s23p4 S В СЕРА 2 32,06 - 3s23p5 С] i ХЛОР 2 35,453 16 Ar 3s23p6 АРГОН e 39,946 2
„ 24 С Г 3d’4s’ 1 ХРОМ 18 51,996 2 .... 25 МП 3d54s22 МАРГАНЕЦ1» 54,9380 2 _ 25 F6 3d64s2 2 ЖЕЛЕЗО 1й 55,847 2 л 27 С 0 3d74s2 2 КОБАЛЬТ 58,9332 2 KI- 28 N1 3d84S2 2 НИКЕЛЬ 56,71 2
34 л б 4s24p4 S Б \э СЕЛЕН 2 78,96 35 7 4s24p5 В Г * * БРОМ ? 79,904 КГ 4s:4p6 S КРИПТОН 18 83,80 2
кл 42 1 МО 4d55s1i3 МОЛИБДЕН ’S 95,94 2 _ 43 ' 2 Т С 4d55S213 ТЕХНЕЦИЙ ’S 98,9062 ? п 44 1 RU 4d75sj 15 РУТЕНИЙ 101,07 2 45 1 Rh 4d®5s1 w РОДИИ ’1 102,9055 • 2 _ . 46 0 Ptl 4dW5S°18 ПАЛЛАДИИ g 106,4 2
6 52 _ ;«5s4p* Те з ТЕЛЛУР 2 127,60 7 53 _ 18 5s25p5 J J ЙОД 2 126,9045 54 » Xe w» КСЕНОН '» 131,30 2
74 2 W 5d46S2 32 ВОЛЬФРАМ ’5 183,85 2 75 2 Re 5d’6s4i РЕНИЙ .186,207 2 76 2 OS 5a66s232 ОСМИЙ 190,2 2 77 2 | Г 5d’6s2 32 ИРИДИЙ 192,22 2 78 1 Pt 5d’6s’ll ПЛАТИНА '? 195,09 2
и 6s26p*4 РО 1 ПОЛОНИЙ 2 (209) 1 85 ? 6s26p5 Af J АСТАТ 2 (210) „ 86 » RD РАДОН ’f (222) 2
•
— 63 2 Eu 4f’6s22! ЕВРОПИЙ 1S 151,95 2 64 2 Gd 4f75d16S225 ГАДОЛИНИЙ 157,25 2
70 2 Yb if’W? ИТГгРбИЙ 173,04 z 71 2 LU4f”5d16s232 ЛЮТЕЦИЙ 174,97 ?
Атомный номер Распределение электронов
—.......... по застраивающимся и по-
следующим застроенным
подуровням
Число электронов в слоях
. 95 I АГП 5F7S2?’ АМЕРИЦИЙ is (243) « 98 I Cm 5f76d’7s2‘5 КЮРИЙ 1g (247) §
NO 15fl47S232 НОБЕЛИЙ (255) I . 103 i Lr51,46d 7s2 |2 ЛОУРЕНСИЙ 18 (256) I
Q
к
Р
О
N
М
Fe 3d64Sz 2
ЖЕЛЕЗО 12-
у55,647 2
Атомная масса
I электронный слой К
H L
HI 99 M
1У 99 N
V 99 0
и 99 P
,4 *9 Q
Целое число в скобках- массовое
число наиболее устойчивого изотопа
искусственного радиоактивного элемента
Целое число без скобок-массовое число
наиболее распространенного изотопа
природного радиоактивного элемента.
В. А. ГРИГОРЯН, Л. Н. БЕЛЯНЧИКОВ,
А. Я. СТОМАХИН
ТЕОРЕТИЧЕСКИЕ
ОСНОВЫ
ЭЛЕКТРОСТАЛЕПЛАВИЛЬНЫХ
ПРОЦЕССОВ
Издание 2-е, переработанное и дополненное
&
МОСКВА «МЕТАЛЛУРГИЯ» 198 7
УДК 669 : 541 (075.8)
Рецензент докт. техн, наук, проф. Б. В. Линчевский
УДК 669:541(075.8)
Григорян В. А., Белянчиков Л. Н., Стомахин А. Я.
Теоретические основы электросталеплавильных процессов. М.: Металлур-
гия, 1987, 272 с.
Во втором издании (первое — в 1979 г.) изложены физико-химичес-
кие основы процессов, протекающих в электросталеплавильных печах.
Приведены основные понятия химической термодинамики, кинетики ме-
таллургических процессов, теории шлаков. Описаны методы расчета рав-
новесий в системе металл — шлак — газовая среда. Значительное внима-
ние уделено поверхностным явлениям в сталеплавильных процессах. Рас-
смотрены особенности рафинирования металла при вакуумно-дуговом,
плазменно-дуговом и электрошлаковом переплавах, а также теория кри-
сталлизации слитков разового формирования и получаемых в водоохлаж-
даемых кристаллизаторах. Приведены справочные данные по термодина-
мическим, кинетическим, поверхностным и другим свойствам.
Для научных и инженерно-технических работников. Может быть по-
лезна студентам металлургических вузов. Ил. 69. Табл. 23. Библиогр.
список: 234 назв.
2602000000—159
040(01)—87
29—87
© Издательство «Металлургия», 1987
ОГЛАВЛЕНИЕ
Предисловие ...............................
Глава I. Основы химической термодинамики
5
6
1.1. Энергия Гиббса химической реакции ...................... 6
1.2. Направление и полнота химической реакции.................Ю
1.3. Растворы. Состав. Термодинамические функции .... 12
1.4. Уравнение Гиббса — Дюгема...............................16
1.5. Законы Рауля и Генри....................................17
1.6. Совершенные растворы....................................1®
1.7. Идеальные разбавленные растворы.........................20
1.8. Активность..............................................22
1.9. Экспериментальное определение активности ..... 26
1.10. Реальные растворы.......................................29
1.11. Переход от одного стандартного состояния к другому . . 33
1.12. Регулярные растворы.....................................34
1.13. Субрегулярные и квазирегулярные растворы , . , , 40
1.14. Квазихимическая теория................................ 47
1.15. Параметры взаимодействия ...............................49
1.16. Расчет равновесия реакций с участием растворов ... 63
Глава II. Сталеплавильные шлаки ............................ 68
II. 1. Технологические функции и характеристики шлаков . . 69
II.2. Молекулярная теория шлака ......... 71
II.3. Теория совершенных ионных растворов.....................76
II.4. Теория регулярных ионных растворов ...... 77
II.5. Термодинамические функции шлака как фазы, имеющей кол-
лективную электронную систему.................................80
II.6. Распределение элементов между металлом и шлаком . . 85
Глава III. Поверхностные явления в сталеплавильных процессах 90
II 1.1. Основные понятия.................................... 90
II 1.2. Поверхностное натяжение жидких металлов ..... 92
II 1.3. Уравнения зависимости поверхностного натяжения расплавов
от состава ............................................ 93
III.4. Поверхностное натяжение расплавов на основе железа . 97
II 1.5. Вычисление концентрации в поверхностном слое . 98
II 1.6. Поверхностное натяжение шлаков....................100
II 1.7. Межфазное натяжение между металлом и шлаком . 101
III .8. Смачивание и растекание...........................* 103
Глава IV. Термодинамические характеристики некоторых рас-
плавов на основе железа и никеля..............................109
IV . 1. Кислород в расплавах................... 109
IV.2. Углерод в расплавах......................... 113
IV.3. Взаимодействие углерода и кислорода в расплавах . . 115
IV.4. Обезуглероживание хромсодержащих расплавов . . . 120
IV.5. Сера в расплавах.................. 123
IV.6. Фосфор в расплавах................................133
IV.7. Азот в железе, никеле и их сплавах ....... 135
IV.8. Водород в расплавах..................................* 143
Глава V. Раскисление стали и сплавов..........................146
V.I. Влияние элементов-раскислителей на активность кислорода в
жидком железе................................................146
1*
3
V .2. Анализ кривой раскисления
V. 3, Раскисление железа и его сплавов
V.4 . Комплексные раскислители
V.5. Раскислительная способность шлака
Глава VI. Неметаллические включения
147
149
154
155
156
VI . 1. Образование неметаллических включений ..... 1а/
V I.2. Укрупнение неметаллических включений . «... г 159
VI .3. Удаление включений в гравитационном поле . . . . 151
VI. 4. Влияние конвекции на удаление неметаллических включений 163
VI.5 . Связь между свойствами поверхностей раздела фаз и скоро-
стью удаления включений........................................164
VI. 6. Переход границы раздела металл — шлак...................166
VI .7. Механизм удаления неметаллических включений . 169
VI. 8. Поведение неметаллических включений в условиях вакуума 171
Глава VII. Кинетика металлургических реакций 173
VII. 1,. Молекулярная диффузия...............................173
VI 1.2. Конвективная диффузия ......................... ... 176
VII .3. Критериальные уравнения в задачах по кинетике ... 179
VI 1.4. Признаки, по которым определяют лимитирующую стадию
гетерогенного процесса..................................... 180
VII .5. Кинетика растворения, плавления ....... 182
VI 1.6. Влияние ПАВ на кинетику гетерогенных процессов . . 184
VI 1.7. Кинетика обезуглероживания . ....................186
VII. 8. Образование и удаление газовых пузырей из жидкой стали 189
VII .9. Массоперенос компонентов в шлаке ....... 191
Глава VIII. Особенности рафинирования металла при пере-
плавных процессах ......................................... 194
VIII .1. Общие закономерности рафинирования металла при пере-
плаве .......................................................195
VII 1.2. Рафинирование металла при ВДП ....... 202
VIII.3. Рафинирование металла при ЭШП........................218
VIII.4. Рафинирование металла при ЭЛП........................231
Глава IX. Кристаллизация и формирование электросталепла-
вильного слитка 236
IX. 1. Затвердевание слитка разового формирования .... 237
IX.2. Затвердевание непрерывного и полунепрерывного слитков . 241
IX.3. Двухфазная область ....................................247
IX.4. Концентрационное переохлаждение....................... 250
IX.5. Дисперсность дендритной структуры и дендритная ликвация 252
IX.6. Физическая неоднородность стального слитка .... 255
Библиографический список................................... 259
Предметный указатель.........................................267
ПРЕДИСЛОВИЕ
В решениях XXVII съезда КПСС отмечается необходи-
мость существенного расширения объема проводимых на-
учных исследований и разработок, а также резкого повы-
шения эффективности их практического применения.
В СССР и за рубежом опубликовано значительное число
монографий, посвященных теоретическим основам стале-
плавильного производства в целом или его отдельным раз-
делам (газы в стали, раскисление, неметаллические вклю-
чения и др.). В настоящей книге коротко изложены теоре-
тические закономерности металлургических процессов,
причем значительное внимание уделено основным физико-
химическим понятиям. Тем не менее книга по содержанию
существенно отличается от учебников и пособий по физиче-
ской химии, поскольку отобран лишь материал, необходи-
мый для изложения теоретических основ сталеплавильного
производства.
В разделе химической термодинамики описаны методы
расчета равновесий при помощи различных моделей метал-
лических растворов. В гл. II излагаются основы теории
шлаков и рассматриваются термодинамические функции
шлака как фазы, имеющей коллективную электронную
систему. Значительное внимание уделено также физико-хи-
мическим основам поверхностных явлений в сталеплавиль-
ном производстве, термодинамическим характеристикам не-
которых расплавов на основе железа и никеля, раскисле-
нию, образованию и удалению неметаллических включений,
рафинированию металла при переплавных процессах.
Применение законов физической химии к металлургиче-
ским процессам встречает трудности при проведении коли-
чественных расчетов и оценок. С учетом этого в книге при-
водится необходимое число примеров по каждому разделу.
Их цель — помочь читателям в освоении расчетных мето-
дов. Для этого приведено большое число справочных и таб-
личных данных. В решении ряда задач принимал участие
Ю. И. Уточкин.
Во втором издании переработаны некоторые разделы,
добавлены материалы, делающие изложение более строгим
и последовательным. Приведены новые примеры, которые
позволяют читателю лучше понять излагаемую теорию.
В. А. Григоряном написаны главы III, V, VI, VII и так-
же разделы IV.7, IV.8, Л. Н. Белянчиковым — главы VIII
и IX, А. Я. Стомахиным — гл. I и разделы IV. 1—IV.6; гл. II
написана А. Г. Пономаренко.
5
Глава I
ОСНОВЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
При изучении термодинамических характеристик химиче-
ских и в том числе металлургических процессов использу-
ют обычно два подхода: феноменологический (классическая
термодинамика) и молекулярно-статистический (статисти-
ческая термодинамика). Основное внимание в настоящей
главе уделено классической термодинамике, которая со-
ставляет основу большинства расчетных методов, приме-
няемых на практике. Предполагается, что основы термоди-
намики уже известны читателю из курса физической хи-
мии [1]. Основная задача настоящей главы состоит в том,
чтобы помочь читателю в освоении практического примене-
ния термодинамики для решения задач сталеплавильного
производства.’
1.1. ЭНЕРГИЯ ГИББСА ХИМИЧЕСКОЙ РЕАКЦИИ
Основной термодинамической характеристикой химической
реакции является вызываемое ею изменение энергии Гиб-
бса (ДО)* *1 системы. Для определенности исходные веще-
ства и продукты реакции принимают обычно находящими-
ся в их стандартных состояниях. Соответствующую величи-
ну ДО называют стандартной энергией Гиббса химической
реакции и обозначают Дб° (или ДГО°).
Вещество в стандартном состоянии — это обычно чистое
вещество при давлении 1 атм*2 в том агрегатном виде, ко-
торый устойчив при данных условиях (если нет специаль-
ной оговорки). Для меди, например, при 1000 К стандарт-
ным состоянием обычно является Cu(T>, а при 1400 К —
Си(ж). Однако, если это удобно, при 1000 К можно принять
за стандартное состояние и жидкую (переохлажденную)
медь. Возможны и другие варианты. Например, в справоч-
нике [206], при вычислении констант реакции за стандарт-
ное принято состояние одноатомного газа (при всех темпе-
ратурах). В теории сталеплавильных процессов за стан-
*1 В литературе встречаются и другие названия: свободная энер-
гия, изобарно-изотермический потенциал. Кроме обозначения G, реко-
мендованного АН СССР [3], используют символы Z, F.
*2 Выбор внесистемной единицы измерения 1 атм= 101325 Па яв-
ляется в этом случае общепринятым; переход к другому стандартному
давлению (например, 1 Па) потребовал бы переработки многих спра-
вочников.
6
дартное часто принимают состояние вещества в растворе.
Подробно этот вариант рассмотрен ниже. Стандартные со-
стояния веществ, участвующих в реакции, выбирают про-
извольно, для удобства расчетов. Однако, этот выбор дол-
жен быть четко определен (например, в уравнении реак-
ции), иначе приводимая величина Дб° не имеет смысла.
Часто фактические (рассматриваемые) состояния реаген-
тов не совпадают со стандартными. Во избежание неточно-
стей в уравнениях следует указывать стандартные состоя-
ния. Например, если рассматривается реакция с участием
кремнезема в шлаке, а за стандартное состояние в расчете
принимается чистый жидкий кремнезем, то в уравнении
реакции должен быть записан SiOs(x), а не (ЗЮг).
Значения Дб° можно найти при помощи таблиц, в кото-
рых приведены данные о стандартных энергиях Гиббса об-
разования веществ из элементов при различных темпера-
турах [2, 49, 205, 206, 230 и др.]. В последних справочных
изданиях [206] приводят иногда не энергии Гиббса веществ
GT, а приведенные энергии Гиббса Фт, которые связаны с
GT соотношением:
Фт =• — {Gt — Но)/Т = St — (Нт — Но)/Т. (1)
Для определения Дб° необходимо определить суммар-
ную энергию Гиббса образования всех продуктов реакции
(с учетом стехиометрических коэффициентов) и вычесть
из полученного значения суммарную энергию Гиббса об-
разования всех исходных веществ. В некоторых справочни-
ках приведены стандартные значения энтальпий (Д//°) и
энтропий (Д5°) образования веществ из элементов. Для вы-
числения Дб° в этом случае используют уравнение:
Д6° = Д/У° — ТД5°. (2)
Во многих практических случаях значения ДЯ° и Д5° ре-
акций в определенном температурном интервале можно
считать не зависящими от температуры. При этом урав-
нение (2) характеризует зависимость Дб° от температуры.
Стандартные энергии Гиббса образования некоторых со-
единений, имеющих значение для сталеплавильного произ-
водства, приведены в табл. 1 в виде коэффициентов урав-
нений типа уравнения (2). Коэффициенты в указанном
температурном интервале приняты постоянными. Для бо-
лее строгих расчетов необходимо учитывать зависимость
ДЯ° и AS0 от температуры, однако необходимость в этом
возникает довольно редко. В качестве примера определим
при помощи табл. 1 значение Дб° реакции восстановления
Таблица 1. Стандартные энергии Гиббса образования некоторых
соединений из элементов Д/О°=Л+ВГ, Дж/моль при температурах
сталеплавильных процессов (1800—2000 К)*1
Соединение >4 В Соединение A В
Ka [рбиды
A14C3 —258000 97 Mn7Co2 —66100 —58
В4С —69500 9
—213000 MO2C*2 66100 —110
СаС?4 61
M0C*2 40600 -59
СГ9оС< —380860 —37 SiC —123000
zoD 38
сг7с;3 —168000 —30 Ta2C*2 —142300 5,5
Сг3С23 —84400 —15 TaC*2 —161000 5,5
СгН2(г) 220000 —50 TiC —179000 15
CHi(r) —88400 108,5
♦О ZrC*3 —200000 12
MgC2‘ —49800 102
Нитриды
A1N —330000 117 M63N2(V) —855000 504
BN —248000 86 Mo2N*2 —71550 58
BC3N2 —604000 196 S13N4 —922000 457
—918400 490
CeN*? —336800 114 TaN*2 —234700 79
Cr2N*3 —115000 64 TiN*2 —334500 93
CrN*3 —113600 73 VN*3 —207500 78
NHs(r) —55800 117,6 ZrN*2 —360200 90
NbN*?(600K) —235600 91
Оксиды
А1гО(г) —195000 —44 Ce90o2 —1826300 337
АЮ(Г) 41000 —58,10 2 0
A102(r) —108000 4 CeO92 —1029000 214
Al2O3<a) —1681000 324
B2O3 —1220000 204 СоО(ж) —249700 80
BaO —555000 101 СГ2ОЗ —1131000 250
ВеО(а) —619000 104 СиО(ж) —1132000 58,3
CO(r) —118000 —83,77 Cu2O(>K) —136600 48,5
CO3(r) —397000 0,2 FeOf») Fe2O3 —245000 53
CaO —790000 194 —747000 210
CaO(r) —134000 38,4 Fe3O4 —992000 247
*’ В основном по данным (206] и автоматизированной системы ИВТАНТЕРМО
[230]. За стандартные фазовые состояния во всех случаях, кроме специально
обозначенных, приняты для карбидов, нитридов, оксидов, сульфидов и для эле-
ментов С, В, Сг, Mo, Nb, Та, Ti, W, V, Zr — твердая фаза; для хлоридов, фто-
ридов и для элементов N2, О2, S2, Cl2, F2, Н2, Р2, Са, Mg, Na — газ; для осталь-
ных элементов — жидкость. О вычислении Дуб0 реакций для веществ в другом
фазовом состоянии и с участием элементов, растворенных в металле, см. с. 66, 67.
8
Продолж. табл. 1
Соединение A В Соединение A В
Н2О(г) —251070 57,65 ЗпО2(ж) —560000 197
La2O3 —1784000 278 Та2О5 —1984000 390
MgO' —729000 204 ТЮ(Р) —527000 84
MgO(r) —99000 23,9 ТЮ(Г) 28500 —77
MnO —408400 90 тю2 —936000 176
Мп2О3 —982000 269 TiO2(r) —344000 3
Mn3O4(p) —1790000 652 TigOsfP) —1481000 246
MoO2 —557000 156 TiaOs(P) —2410000 410
МоО2(г) —30000 —24 VO —406000 73
МоО3(г) —370000 65 VO(r) 131000 —80
NO(r) 91500 —12,72 V2O3 —1186000 233
NO2(r) 35600 61,5 ^^О4(ж) —1263000 234
Na2O —531000 250 VO2(r) —251000 —8,3
NbO(r) 196000 —84 V2O5 —1441000 323
МЬ2О5(Ж) —1744000 340 wo(r) 387000 —93
NiO —249800 92 wo2(r) 20000 —22
PO(r) —99600 —11,51 ^О3(ж) —731000 180
P —1235000 331 wo3(r) —323000 61
P 4^10(r) —3112000 983 Y2O3 —1920000 292
SiO(r) —162000 —46,3 ZrO(r> 72000 —77,3
SiO2(P) —947000 198 ZrO2(r) —339000 2
SnO —241000 70 ZrO2 —1081000 176
Сульфиды
AI2S3 —752000 212 MnS —276000 63
BaS —527000 ПО —386060
CS(r) 208500 —86,05 MoS> A* 173
CS2(r) COS(r) —12300 —204400 —5,6 8,9 Na2S*3 —555400 240
CaS —691900 190 —264300 77
CuS(900K) —113100 82 SiS(r) —20000 -47,1
FeS(>K) HS(r) MgS —135000 75000 —90500 —535000 43,2 —15 49,2 187 SiS2(r) SnS(r) SO(r) SO2(r) sQtfr) —177000 24000 —60000 —361000 —455100 27 —45,68 -4,8 72,9 163
Фториды
AIF3 —1230000 80 PF6 —1646000 300
b2f4 —1450000 103 PbF2 —447000 -0,1
СаРгСж) —1312000 210 SF * —48200 —12
CaF2 —970000 84 SiF4 —1665000 170
FeF —120000 —78 SnF2 —500000 2
FeF, —473200 —13 SnF4 —1088600 149
MnF —140000 —63 TiF3 —1164000 45
MnF, —592000 0,5 TiF4 —1566600 134
MnF, —800000 54 ZrF 49600 —87
NaF —405000 40 ZrF43 —1677500 128
9
Продолж. табл. 1
Соединение А В Соединение A В
Хлориды
А1С13 —603000 68 РЬС1 —11000 —59
ВС13 —407000 52 РЬС12 —262000 285
СаС12(Ж) —893000 190 РЬС14 —705000 155
РеС1з(ж) —306600 83 SiCl4 —351000 130
МпС1 —39000 —66 SnCl2 —220000 —3
МпС12 —305000 —6 TiCl4 —768500 125
NaCl(>K) —453000 130 WC1*3 748400 —213
NbCl5 —688000 17 ZrCl4 —870700 116
РС1В —418100 266
*2 По данным [2].
♦3 По данным [49].
По данным [234]: A/GcaCf =-60250-26,28 Т.
оксида титана графитом до карбида титана: TiO2(T> +
+ 3C(rp)==TiC(r)+2CO(r). Просуммировав взятые из табл. 1
выражения для AG0 реакций образования соответствую-
щих соединений, получим: AG°=2AG£0 +AG^iC —AG^.iO =
= [2(— 119700—83,057) + (-1858004-14,277’)] —(—935500+
+176,1 ОТ) = 510800—328,27’ Дж.
1.2. НАПРАВЛЕНИЕ И ПОЛНОТА
ХИМИЧЕСКОЙ РЕАКЦИИ
Согласно второму закону термодинамики, энергия Гиббса
при необратимых процессах может только убывать. Отсю-
да следует, что AG0 является критерием направления и
полноты протекания реакции между веществами, взятыми
в стандартном состоянии. Реакция может протекать толь-
ко в том направлении, которое характеризуется отрица-
тельным значением AG0, т. е. реакция всегда идет в на-
правлении уменьшения энергии Гиббса системы. Если
AG0—0, то это указывает на равновесие между реагента-
ми в их стандартных состояниях (реакция не идет). Полно-
та протекания реакции характеризуется константой равно-
весия К, связанной с AG0 уравнением изотермы:
AG0 = —In/С. (3)
Пользуясь уравнением (3), можно по известному зна-
чению AG0 определить константу равновесия реакции, и
найти необходимые для практики равновесные концентра-
10
ции и другие характеристики. Например, для реакции по-
лучения TiC, исходя из уравнения (3) и температурной за-
висимости AG£, можно получить следующее выражение для
константы равновесия:
дс2.
1g К — 1g (рсо) — — 2 3037?г —
510800 328,27 26700 j
“ 19,147 19,147 “ 7 "Г ’ *
Отсюда следует, что при 1873 К равновесное давление СО
в рассматриваемой системе составляет ~25 атм.
Величина AG0 является однозначной характеристикой
направления и полноты реакции только при данной темпе-
ратуре. Для оценки температурной зависимости полноты
реакции удобнее пользоваться теплотой реакции. Непра-
вильный подход может привести к серьезным ошибкам. На-
пример, исходя из температурной зависимости AG0 реак-
ции окисления углерода [С]+[О]=СОГ, AG°=—22400—
—39,7 Т Дж [2], делают иногда заключение, что реакция
должна более полно протекать при высоких температурах,
при которых величина ДС° более отрицательна. В действи-
тельности реакция идет более полно при пониженной тем-
пературе. Правильный вывод можно сделать при помощи
уравнения изобары Вант-Гоффа:
d In R/dT = MR/RT2. (4)
Из уравнения (4) видно, что знак температурной зави-
симости константы совпадает со знаком Д/7. В приведен-
ном примере ДЯ=—22400 Дж, т. е. меньше нуля, и, следо-
вательно, при повышении температуры константа умень-
шается, реакция идет менее полно. Такой вывод можно
сделать и используя принцип Ле-Шателье. Если температу-
ра повысится, то в системе будет протекать процесс, сопро-
вождающийся поглощением тепла. В приведенном приме-
ре это соответствует протеканию процесса в обратном на-
правлении, т. е. уменьшению полноты реакции. Согласно
второму закону термодинамики, в изолированных системах
все необратимые процессы сопровождаются увеличением
энтропии. Таким образом, по значению Д5° также можно
судить о направлении реакции, но только в изолированной,
системе. Реальные объекты обычно нельзя считать изоли-
рованными системами, поэтому для оценки направления и
полноты реакции пользуются значениями AG°.
if
1.3. РАСТВОРЫ. СОСТАВ.
ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ
Для описания состава растворов в металлургии использу-
ют обычно массовые доли, выраженные в процентах. В тео-
рии широко пользуются также мольными (атомными) до-
лями и процентами. Связь между различными единицами
выражается формулой:
_______Ч_______
ni + «у + пк + • • •
___________c_jlM±_________
CilMi+ Cj/Mj + Ck/Mk+ ...
сат-у
100 ’
(5)
где Xj — мольная доля компонента /; п,, п/, п* — числа мо-
лей компонентов; Ci, с,, Ck — массовые доли компонен-
тов, %; Л1,-, Л4/, Mk — молекулярные (атомные) массы;
Сат,/ — мольная атомная доля компонента /, % • Для разбав-
ленных растворов, в которых и/, пь... очень малы по срав-
нению с количеством молей растворителя п, можно исполь-
зовать упрощенную формулу:
=‘(CJ = • (6)
Из формулы (6) видно, что в разбавленном растворе
мольная доля пропорциональна массовой концентрации.
Это относится и к другим способам выражения концентра-
ции в разбавленных растворах.
Для термодинамического описания состояния компонен-
та в растворе пользуются парциальными мольными харак-
теристиками gi, под которыми понимают частные производ-
ные от соответствующих экстенсивных*1 свойств раствора
по числу молей данного компонента при постоянных темпе-
ратуре, давлении и числах молей других компонентов:
dg
dni
&
Таким образом, gi представляет собой приращение свой-
ства бесконечно большого количества раствора при добав-
ке 1 моля i-того компонента. Например, парциальный моль-
ный объем компонента в растворе заданного состава пока-
зывает, на какую величину изменится объем этого раствора
(7)
4,1 Экстенсивными называют свойства, которые зависят от количе-
ства вещества: V, Н, S, G и т .д. Такие свойства, как температура и
давление, не зависящие от количества вещества, называют интен-
сивными.
12
(взятого в бесконечно большом количестве) при добавлении
1 моля данного компонента. Как видно из определения, пар-
циальные мольные величины являются интенсивными ха-
рактеристиками; они зависят не от количества раствора,
а от его состава. Для определения экстенсивного свойства
g раствора необходимо просуммировать парциальные моль-
ные характеристики компонентов gi, умноженные на соот-
ветствующие числа молей п/. Например для энтальпии
раствора имеем Н=Т.(Н1П1), или для 1 моля раствора** 1
!
В некоторых случаях парциальная мольная характерис-
тика может быть равна соответствующей мольной харак-
теристике чистого компонента. Так, если суммарная энер-
гия взаимодействия частиц при образовании раствора из
чистых компонентов не изменяется (такие растворы назы-
вают совершенными), парциальные объемы компонентов и
их парциальные энтальпии будут равны соответствующим
мольным характеристикам: Vt = V?; В этом слу-
чае объем и энтальпия раствора аддитивно складываются
из мольных величин: V=S(V?nJ); Однако
I I
такие случаи крайне редки. В большинстве растворов пар-
циальные мольные характеристики существенно отличают-
ся от соответствующих мольных характеристик компо-
нентов.
Некоторые свойства (в частности, энтропия и энергия
Гиббса) не являются аддитивными даже в совершенном
растворе. Иногда в результате сильного взаимодействия
компонентов в растворе парциальные мольные характери-
стики принимают отрицательные значения. Это означает,
что добавление одного из компонентов к раствору приво-
дит не к увеличению, а к уменьшению определенного экс-
тенсивного свойства. Так, если добавить немного сульфата
магния к водному раствору этой соли, объем раствора
уменьшится, поэтому Vwgso, <0- Еще одним примером мо-
гут служить разбавленные растворы кремния в расплавах
железа и никеля, в которых парциальные мольные энталь-
пии кремния равны соответственно —41 и —111 кДж. Эти
величины найдены путем суммирования мольной энтальпии
*1 Под молем раствора понимают такое количество раствора, в
котором числа молей компонентов равны их мольным долям (rn=xt),
к следовательно, суммарное число молей компонентов равно 1. Масса
I моля раствора Mm=Ex/M/=100/S(c;7M/).
/ /
13
кремния (#i873 — Я^), равной 91 кДж [2], и теплот рас-
творения кремния при 1873 К в железе (—132 кДж) и в
никеле (—202 кДж). Выделение тепла при взаимодействии
кремния с железом и никелем превышает тот вклад в эн-
тальпию раствора, который вносит добавка 1 моля крем-
ния, нагретого до 1873 К, поэтому парциальная мольная
энтальпия кремния в обоих случаях отрицательна. Приве-
денный пример показывает, что парциальная мольная
величина характеризует не только данное растворенное ве-
щество, но и растворитель, точнее состояние данного веще-
ства в определенном растворе. Особое место среди парци-
альных мольных величин занимает парциальная мольная
энергия Гиббса, называемая химическим потенциалом ком-
понента:
Hi — ^i —
P-T,nj(j-i-i)'
(8)
Величина ц, характеризует стремление компонента по-
кинуть раствор или перейти в него из другой фазы (с боль-
шим значением ц,). Если химический потенциал компонен-
та ц/ в растворе выше, чем в паровой фазе (при данном
давлении pi), то компонент переходит в пар. При повыше-
нии давления химический потенциал компонента в паре
может стать выше, чем в растворе. Это вызовет переход
компонента обратно в раствор.
Наряду с рассмотренными абсолютными значениями
интегральных и парциальных характеристик gm и gi в тео-
рии растворов часто пользуются относительными величи-
нами, которые более четко выражают особенности раство-
ров по сравнению с механическими смесями. Относительная
интегральная характеристика представляет собой раз-
ность между gm и соответствующей величиной, полученной
путем аддитивного сложения характеристик чистых компо-
нентов g°{: &gm=gm—gm=gm—SgiXi- Так, ОТНОСИТеЛЬНЗЯ
i
интегральная мольная энтальпия представляет собой раз-
ность между энтальпией моля раствора и суммарной эн-
тальпией чистых компонентов, взятых в тех же количест-
вах: —'ZHiXi, т. е. характеризует интегральную
i
теплоту образования 1 моля раствора из чистых компонен-
тов (интегральную теплоту смешения). Относительная
парциальная величина Lgt представляет собой разность
между парциальной и соответствующей мольной характери-
стиками данного компонента: kgi=gi—gi. В качестве при-
14
мера можно привести относительную парциальную энталь-
пию t±Hi=Ht—Н(, которая представляет собой теплоту
растворения 1 моля i-того компонента в бесконечно боль-
шом количестве раствора.
Наблюдаемые в реальных растворах отклонения от ад-
дитивности, которые характеризуются относительными зна-
чениями термодинамических функций, иногда могут быть
полностью или частично объяснены теорией идеальных (на-
пример, совершенных) растворов, поэтому относительные
величины принято делить на две составляющих — идеаль-
ную и избыточную: &gm=kg"m + Agm6; ^gi=^gT‘+^gr6‘
Очевидно, что в идеальных растворах избыточные функции
Рис. 1. Связь между интегральными и парциальными характеристиками в двух-
компонентном растворе: _
а — абсолютные величины: интегральные (gTO) и парциальные (g.); б —относи-
тельные величины: интегральные Mg =g —^аЛД==Д5иД_|_Д£гизб\ и парциаль-
\ tn tn tn т tn }
ные ^A«z=gz-e®=Ag^ +Ag"3<5)
равны нулю. Связь между различными характеристиками
раствора иллюстрируется графиками, приведенными на
рис. 1. На рис. 1,а кривая с точкой М характеризует из-
менение интегрального свойства gm при изменении состава
раствора во всем интервале конструкций. На ординатах
*2=0 и х2=1 кривая отсекает мольные характеристики
компонентов g^ и g°. Если в точке М провести касатель-
ную к кривой, то точки пересечения этой касательной с
ординатами будут соответствовать парциальным характе-
ристикам раствора gi и g2 при концентрациях компонен-
тов, соответствующих точке М. Такой графический метод
определения парциальных характеристик очень удобен для
практических расчетов. Соответствующие аналитические
15
выражения имеют следующий вид:
= + (9)
дхг
= + (10)
ёт = ii + (1 — *1) (ёг —ii) = *1£1 + Ш (11)
На рис. 1,6 кривая с точкой М характеризует относи-
тельные значения того же интегрального свойства в
той же бинарной системе 1—2. Относительные парциаль-
ные величины Agi и Д§2 (в точке М) могут быть определе-
ны по этому графику аналогичным способом — проведени-
ем касательной. Аналитические выражения для Д§ имеют
тот же вид, что и для g. Пунктирные кривые на рис. 1, а, б
характеризуют значения gm и Д£т, следующие из теории
совершенных растворов. Такой вид имеют, например, эн-
тропия раствора и взятая с обратным знаком энергия Гиб-
бса. Точки пересечения этих кривых с ординатой точки М
делят величину \gm на две составляющие: идеальную и
избыточную. Касательная к пунктирной кривой, проведен-
ная на рис. 1,6, делит на соответствующие части и парци-
альные характеристики Д£1 и Д^2.
1.4. УРАВНЕНИЕ ГИББСА — ДЮГЕМА
Связь между парциальными характеристиками компонен-
тов в бинарном растворе может быть выражена уравне-
нием:
«1 dg! + п2 dg2 = 0, (12)
которое является одной из форм уравнения Гиббса—Дю-
гема. Разделив это уравнение на п\ + п2, получим аналогич-
ное соотношение с мольными долями:
Xid£i + x2dg2 = 0. (13)
Разделив уравнение (13) на dxx с учетом dxi=—dx% по-
лучим другую форму уравнения Гиббса—Дюгема (для по-
стоянных р и Т):
х = х(_^2_) . (14)
\ dXi )р,т z\ дх2 )р,т
Уравнение Гиббса—Дюгема имеет большое значение в
термодинамике растворов, так как позволяет рассчитать
парциальные характеристики одного компонента раствора
16
по известным характеристикам другого1. Уравнения (12)г
(13) могут быть распространены на многокомпонентные
растворы путем добавления членов, соответствующих i=
=3, 4 и т. д.
1.5. ЗАКОНЫ РАУЛЯ И ГЕНРИ
Основой для описания свойств реальных растворов служат
законы Рауля и Генри, определяющие характер зависимо-
сти давления пара компонента над
става. Первоначально эти зависи-
мости были установлены эмпириче-
ски, как закономерности, выполня-
ющиеся с довольно высокой точно-
стью в ограниченных (для боль-
шинства растворов) интервалах
концентраций. В дальнейшем были
разработаны модели (так называе-
мые идеальные растворы), в кото-
рых указанные законы выполняют-
ся с абсолютной точностью. Эти мо-
дели играют большую роль в совре-
менной теории растворов, посколь-
ку являются основой для обобще-
ния экспериментального материала
и дальнейшего развития теории.
Рассмотрим зависимость давления
пара одного из компонентов бинар-
раствором от его со-
Рис. 2. Давление пара ме-
ди над расплавом Fe—Си
при 1823 К [5]
ного раствора от его концентрации на примере системы
Fe—Си (рис. 2). Из рис. 2 видно, что давление пара меди
над раствором всегда меньше, чем давление пара чистой
меди, и повышается при увеличении концентрации. Наряду
с кривой А, характеризующей изменение реи в реальном
растворе Fe—Си, пунктиром проведены две линии /, 2,
соответствующие двум типам идеальных растворов. Для
идеального раствора первого типа справедлив закон Рау-
ля (см. рис. 2, кривая /), согласно которому давление па-
ра каждого из компонентов прямо пропорционально его
мольной доле:
(15)
причем коэффициент р°с в уравнении (15) представляет со-
1 Пример такого расчета приведен на с. 30, 31.
2—675.
17
•бой давление пара чистого компонента (pt=pi при х,= 1).
Идеальные растворы первого типа называют совершенны-
ми. Некоторые из реальных растворов очень близки к со-
вершенным. Как правило, такие растворы образуются из
компонентов, мало различающихся своими физико-химиче-
скими свойствами, например расплавы Fe—Мп, Fe—Со,
FeO—МпО и т. д. Очевидно, что возможность описания ре-
ального расплава законом Рауля зависит от требуемой
точности: чем она ниже, тем больше растворов можно от-
нести к совершенным. Для идеального раствора второго
типа (разбавленного раствора) справедлив закон Генри,
согласно которому давление пара растворенного вещества
прямо пропорционально его концентрации (рис. 2, кри-
вая 2):
p, = r,Ci(.Vl)«. (16)
Поскольку в разбавленных растворах между концен-
трациями, выраженными в различных единицах, существу-
ет приблизительная прямая пропорциональная зависи-
мость (см. с. 12), то формула (16) практически справедли-
ва при выражении концентрации в любых единицах. Чаще
всего используют массовые доли. Коэффициент Г, (кон-
станта Генри) зависит от химической природы компонентов
раствора, а также от выбранных единиц измерения концен-
трации. В общем случае константа Генри не равна
величине р^, которая служит коэффициентом пропорцио-
нальности в законе Рауля. Исключение составляют только
совершенные растворы (при условии выражения концентра-
ции в мольных долях). Концентрационный интервал, в ко-
тором реальные растворы подчиняются закону Генри, ко-
леблется от долей процента до нескольких процентов. Как
видно из рис. 2, давление пара меди над расплавом под-
чиняется закону Рауля только в небольшой области кон-
центраций вблизи чистой меди. В остальном концентриро-
ванном диапазоне наблюдаются положительные отклоне-
ния от закона Рауля: реальное давление значительно боль-
ше теоретического. Отклонения от закона Генри обычно
имеют противоположный характер. В данном случае они
отрицательны.
Характер отклонения от идеального поведения связан с
химической природой компонентов раствора. Если компо-
ненты проявляют тенденцию к химическому взаимодейст-
* Индексом 1=1 обозначен растворитель
18
вию (например, если при понижении температуры в систе-
ме могут образовываться соединения), то раствор харак-
теризуется отрицательными отклонениями от закона
Рауля. К этому типу относятся сплавы железа с кремнием,,
алюминием, титаном, ванадием и другими элементами.
Как видно из фазовых диаграмм [6—8], в этих сплавах при
охлаждении образуются химические соединения. Если в
растворе проявляется тенденция к отталкиванию молекул,
то отклонения от закона Рауля будут положительными
(как в системе Fe—Си). При сильных положительных от-
клонениях от закона Рауля наблюдается расслоение на две
несмешивающиеся жидкости. Примерами могут служить
системы Fe—Ag и Fe—Pb [6—8].
1.6. СОВЕРШЕННЫЕ РАСТВОРЫ
Как отмечалось, идеальный совершенный раствор пред-
ставляет собой такую модель, в которой закон Рауля выпол-
няется с абсолютной точностью во всем интервале соста-
вов. Суммарная энергия взаимодействия частиц при обра-
зовании такого раствора из чистых компонентов не
изменяется. Тепловой эффект и изменение объема при об-
разовании совершенного раствора равны нулю: ДЯт=0;
Д/7(=0; ДУт=0; ДУ(=0. Фактором, который вызывает
самопроизвольное образование совершенного раствора из
чистых компонентов, является изменение энтропии. Оно
обусловлено тем, что термодинамическая вероятность рас-
твора (в котором каждая из молекул может находиться в
любой части занимаемого им объема) выше, чем вероят-
ность системы, где каждый из компонентов занимает толь-
ко свой объем, составляющий лишь часть общего объема.
Никакого дополнительного изменения энтропии, связанно-
го с упорядочением хаотичного расположения молекул,
при образовании совершенного раствора не происходит. Оп-
ределение энтропии образования совершенного раствора с
использованием формул молекулярной статистики [1] дает
следующие выражения:
Д\п=— In х;);
Д5; =—7?lnx;.
(17)
(18)
Отсюда следует
Дбт = ДЯМ - ТД5т - RT V Xi In xf; (19)
i
2*
19
AGi = Д/7г — T&St = RT In xh (20)
Д6. = = H® + RT In x., (21)
где — химический потенциал компонента в стандартном
состоянии (при Xi—1). Из приведенных выражений (с уче-
том х/<1) видно, что образование совершенного раствора
всегда сопровождается увеличением энтропии. При этом
энергия Гиббса системы всегда уменьшается, так как ее
изменение полностью определяется энтропийной состав-
ляющей. Приведенные уравнения показывают также, что
для расчета относительных термодинамических функций
совершенного раствора требуется лишь задание состава и
температуры. Для вычисления изменения энтропии доста-
точно знать только состав. Из уравнения (17) следует,
в частности, что в бинарном совершенном растворе Д5т ха-
рактеризуется симметричной кривой с максимумом в точ-
ке xi=x2=0,5, для которой Д5т=5,77 Дж/(моль-К).
1.7. ИДЕАЛЬНЫЕ РАЗБАВЛЕННЫЕ РАСТВОРЫ
Экспериментальной основой модели идеального разбавлен-
ного раствора является закон Генри. Особенность модели
заключается в том, что она (в отличие от модели совершен-
ного раствора) не определяет величину энергии взаимо-
действия растворенного вещества с растворителем. Для
упрощения (необходимого в любой модели) не учитывает-
ся взаимодействие молекул растворенного вещества друг
с другом (в связи с малой их концентрацией). Таким об-
разом, добавление растворенного вещества в разбавленный
раствор вызывает такое же изменение внутренней энергии
и энтальпии системы, как добавление его в чистый раство-
ритель (при условии, что раствор и после добавления оста-
ется разбавленным), т. е. парциальная мольная энтальпия
растворенного вещества в разбавленном растворе Н2,
а следовательно, и теплота растворения &Н2 не зависит от
концентрации: Н2=Н2 =const, \Н2—\Н™ =const. Знак
оо означает, что величина относится к начальной области
концентраций и имеет такое же значение, как в бесконечно
разбавленном растворе*.
Изменение энтропии Д32 при образовании разбавлен-
ного раствора представляет собой сумму двух величин.
* В литературе иногда обозначают такие величины тем же индек-
сом, что и значения, относящиеся к стандартным состояниям (напри-
мер Н{').
120
Первая величина связана с изменением состояния молекул
и, следовательно, в области разбавленных растворов не за-
висит от состава. Вторая велична, как и в случае совер-
шенных растворов, обусловлена увеличением объема, пре-
доставленного молекулам растворенного вещества при пе-
реходе их из индивидуального компонента в раствор. Эта
величина пропорциональна логарифму объема раствора,
приходящегося на единицу количества компонента 2, т. е.
обратно пропорциональна логарифму концентрации с2, вы-
раженной в любых единицах. Таким образом, Д52=Л—
—In с2 (Л — постоянная величина, не зависящая от со-
става раствора, но зависящая от выбора единиц измере-
ния с2). Отсюда
AG, - Д/7~ — АТ + RT In с2; (22)
р, - рО + ДЯ.Г — АТ + RT In с2 = + RT In с2. (23)
Из уравнения (23) видно, что ф2 = Р2 при с2 = 1. Отсю-
да следует, что велична ф^ = рО + ДЯ“—АТ представляет
собой химческий потенциал компонента 2 в стандартном
состоянии второго типа, т. е. в идеальном разбавленном
растворе с концентрацией с2=1. Из приведенных формул
видно, что для расчета относительных парциальных харак-
теристик растворенного вещества на основе модели иде-
ального разбавленного раствора требуется знание пара-
метров, характеризующих взаимодействие. Так, из уравне-
ния (23) следует, что для расчета ДС2 при произвольно за-
данных температуре и составе необходимо не менее двух
экспериментальных определений этой величин при разных
температурах, по которым можно найти значения постоян-
ных &Н™ и А (если принять, что они не зависят от Т).
В случае, когда требуется только концентрационная зави-
симость энергии Гиббса (при данной температуре) можно
ограничиться одним экспериментальным значением Дб2,
необходимым для расчета постоянной (при 7'=const) ве-
личины (ДЯ~—АТ).
В отличие от растворенного вещества термодинамиче-
ские функции растворителя в идеальном разбавленном рас-
творе могут быть вычислены по формулам теории совер-
шенных растворов. Докажем это при помощи уравнения
Гиббса—Дюгема. Для упрощения примем, что концентра-
ция растворенного вещества выражена в мольных долях
(с2=х2). Дифференцируя уравнение (23) получим: (дц2/
ldx2)PiT=RTIx2. Подставляя это выражение в уравнение
21
(13), записанное для химических потенциалов, найдем:
I Фх \ __ / 5ц.2 \ ___ RT
I I — х2[ “7 I — х2 —
\ 0X1 /Р.Т \ дх2 /Р'Т х2
(24>
Разделив переменные, получим уравнение dp,i=RT(dxi/
/xi), из которого после интегрирования получим gi =
=RT In Xi + const. Значение константы в этом выражении
можно определить из условия gi = pi при х» = 1. Получен-
ное в результате выражение pi = gi+^r In Xi подчиняется
теории совершенных растворов. Иллюстрацией к сказанно-
му может служить рис. 2, из которого видно, что в области
малых концентраций поведение меди соответствует модели
идеального разбавленного раствора (закону Генри); в об-
ласти высоких концентраций, когда медь является раство-
рителем, ее поведение соответствует модели совершенного
раствора (закону Рауля), поведение железа в первом слу-
чае описывается законом Рауля, во втором — законом
Генри.
Интегральные характеристики для разбавленных рас-
творов могут быть найдены по известным парциальным ве-
личинам при помощи соотношений (9) — (11), или графи-
чески (см. рис. 1).
1.8. АКТИВНОСТЬ
В реальных растворах способность компонента переходить
в пар или вступать в химическую реакцию, как правило, не
определяется только концентрацией. Это проявляется в от-
клонениях от закона Рауля и Генри, а также в том, что
константы реакции с участием растворов, выраженные че-
рез отношения концентраций, не остаются постоянными при
переходе от одних концентраций к другим. Причиной таких
отклонений является взаимодействие компонента с осталь-
ными веществами, находящимися в растворе. Количествен-
ной характеристикой, учитывающей как концентрацию, так
и взаимодействие компонента с другими веществами в рас-
творе, является его активность. В общем виде под актив-
ностью понимают отношение парциального давления пара
компонента над раствором р, к аналогичной величине для
стандартного состояния компонента pJT. Очевидно, что в
стандартном состоянии активность равна 1. Если рассмат-
ривают отклонения от закона Рауля, то за стандартное со-
стояние принимают чистый компонент. В этом случае актив-
ность (обозначим ее а?) определяют выражением:
tr
= PjPl (25)
22
Связь между определенной таким образом активностью
и мольной долей выражают соотношением:
а* = х,, (26)
тде у/ — коэффициент активности. Если соблюдается закон
Рауля, то как видно из уравнений (15) и (25), a? =xt.
В этом случае у/=1. При отрицательных отклонениях от
идеальности y/Cl, при положительных отклонениях у(>1.
Таким образом, коэффициент активности количественно
характеризует отличие реального раствора от совершенно-
го при той же концентрации. Из рис. 2 видно, что это отли-
чие для системы Fe—Си наиболее сильно проявляется в
разбавленном растворе и постепенно уменьшается по мере
увеличения концентрации. Такая картина наблюдается и
во многих других растворах, поэтому коэффициент актив-
ности в бесконечно разбавленном растворе по отношению
к чистому компоненту, обозначаемый у“, имеет, как прави-
ло, минимальное или максимальное значение (в зависимо-
сти от знака отклонений от идеальности). С увеличением
концентрации i величина у,- приближается к 1. Величина
характеризующая отклонение закона Генри от закона
Рауля, имеет большое значение в практических расчетах и
может быть использована для определения активности af
во всем концентрационном интервале соблюдения закона
Генри. Эта величина служит также основой для расчета
активностей компонентов в концентрированных и много-
компонентных растворах.
Наряду с рассмотренным способом определения актив-
ности существует другой способ, в котором объектом опи-
сания являются отклонения не от закона Рауля, а от за-
кона Генри. Стандартным состоянием компонента в этом
случае является идеальный разбавленный раствор единич-
ной концентрации (при этом обеспечивается равенство 1
активности в стандартном состоянии).
Как отмечалось выше, концентрацию в законе Генри
можно выражать в любых единицах. При использовании
мольных долей стандартным состоянием оказывается ги-
потетический разбавленный раствор с концентрацией х, = 1
(чистый компонент с такими же свойствами, как в идеаль-
ном разбавленном растворе). Активность при таком выбо-
ре стандартного состояния обозначим а[, коэффициент ак-
тивности — ip,. В разбавленных растворах (подчиняющих-
ся закону Генри) af =х,, в остальных растворах а[ =
=ф1х1-. Если раствор подчиняется закону Генри, то ф/=1.
23
При отрицательных отклонениях от закона Генри ф(<1,
при положительных — (р>1. Заметим, что в этом последнем
случае а[ может быть больше единицы (в отличие от af).
Найдем отношение а*7а£, не зависящее от концентрации
[220]:
Vi
ф/
Vi \
ф/ <₽i-*1
= (VA..-0 = ?Г. (27)
Соотношение (27) можно использовать для перехода от
одних активностей и коэффицентов активности к другим.
В большинстве расчетов удобнее выражать массовые
концентрации в процентах. Если рассматривать отклонения
от закона Генри с таким выражением концентраций, тс
стандартным состоянием будет 1 %-ный раствор с такими
же свойствами, как при бесконечном разбавлении. В неко-
торых случаях, когда отклонения от закона Генри наблюда-
ются только при концентрациях >1 %, стандартный рас-
твор соответствует реальному 1 %-ному раствору. В других
случаях стандартное состояние является гипотетическим.
Обозначим активность при таком выборе стандартного со-
стояния а*, коэффициент активности ft. По аналогии с пре-
дыдущим вариантом для идеального разбавленного раство-
ра справедливо соотношение ael=ci(f'i> =1); при положи-
тельных отклонениях от закона Генри f/>l, при отрица-
тельных //-<1. Величина a\ (подобно aj) может быть как
больше, так и меньше 1.
Определим соотношение между аг{ и а?, а также связь
fi с yi. По аналогии с предыдущим, используя выражение
(6) в виде (Ci/xi) = где Мт — масса моля ос-
новного сплава (без компонента i), получим:
Vt ЛЬ 100 xt
Mm Cf
Отсюда
(29)
Подставляя вместо Xi/ct выражение, найденное по формуле
(5), которая справедлива для всего диапазона концентра-
ций, получим:
24
f _ Ti 100M, ________1/Afj______=
Ь Y~ ’ Mm ‘ a/Mi + (100-ci) Mm
_ Tf___________lOOAlt_____
~ Y~ CiMm + (100 — Ci)Mt
Аналогичное соотношение в форме:
А; Н W+1 + ММ#!,)] (30)
было получено (для бинарного раствора) К. Люписом
[220]. Оба приведенных выражения отличаются от приме-
няемой обычно в расчетах [221] формулы:
А, = W- (31)
Однако, уточнение в большинстве случаев ничтожно ма-
ло — гораздо меньше погрешности имеющихся значений
коэффициентов активности.
Рассмотрим графики, приведенные на рис. 3. В рас-
плаве Fe—Si наблюдаются сильные отрицательные откло-
а S
Рис. 3. Активности кремния при 1873 К (а) и меди при 1823 К (6) в жидких спла-
вах с железом, выраженные двумя способами: по отношению к чистому компо-
ненту (на основных графиках) и по отношению к 1 %-ному разбавленному рас-
твору (на вставках) [4]
нения от закона Рауля (от лини /), которые можно харак-
теризовать величиной =0,0011. Расплавы Fe—Си
характеризуются положительными отклонениями. В этой си-
стеме для всех составов уСи >1 (у“и =10,1). Стандарт-
ным состояниям на этих графиках соответствуют точки <?;
активности аГ на графиках не показаны. Стандартные со-
стояния для этого варианта определения активности нахо-
25
дятся в точках пересечения кривых закона Генри с ордина-
тами х, = 1. Вставки на рис. 3 показывают в увеличенном
виде части кривых для малых долей кремния и меди. Коор-
динатными осями на этих вставках служат массовая доля
компонента (абсцисса) и активность аг. (ордината). Пря-
мые линии 2 (см. рис. 3) соответствуют закону Генри. От-
клонения от закона Генри противоположны по знаку откло-
нениям от закона Рауля. В системе Fe—Si при 4 % Si
asi =4,6 и, следовательно, Л=1,15 (>1); в расплаве
Fe—Си при 4 % Си агСц =3,6, что соответствует fCa =0,9
(<1). В системе Fe—Si, так же как и во многих других
случаях, небольшие отклонения от закона Генри обнару-
живаются даже при концентрациях <1 %. При этом в ка-
честве стандартного состояния используется гипотетиче-
ский 1 %-ный раствор, обладающий свойствами идеального
разбавленного раствора. Это состояние характеризуется
на графиках точками 3' пересечения ординат 1 % с ли-
ниями закона Генри. Очевидно, что в таких растворах ко-
эффициент активности компонента fi в реальном растворе
при 1 %-ной концентрации не равен 1.
В литературе активности о?, и а. часто обозначают
одним и тем же символом а,. Принятые стандартные со-
стояния указывают при этом либо в тексте, либо в выра-
жениях констант равновесия (вводя yt, <р* или fi). В урав-
нениях реакций стандартные состояния реагентов также
обозначают определенным способом. Для кремния, напри-
мер, чистому компоненту соответствует запись Si>K (или
SiT), 1 %-ному раствору — [Si] или [Si]i% . Вопрос о выбо-
ре стандартного состояния решают в каждом конкретном
случае*. Для основных компонентов металла и шлака чаще
применяют стандартное состояние «чистый компонент».
При рассмотрении систем с ограниченной растворимостью
(например, растворов газов в железе) в качестве стандарт-
ного состояния выбирают обычно 1 %-ный идеальный раз-
бавленный раствор. Такой выбор удобен и для металлов,
когда рассматриваются расплавы с относительно неболь-
шой концентрацией растворенных компонентов.
1.9. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
Различные экспериментальные методы и аппаратура для определения
активностей компонентов в металлургических расплавах подробно опи-
саны в работах [9, 10], поэтому ограничимся рассмотрением лишь не-
* Для удобства расчетов.
26
скольких методов. Наиболее прямым методом определения активности
компонента является измерение давления его пара. Чтобы определить
активность, необходимо измерить давление пара компонента не менее
двух раз: над исследуемым расплавом и над расплавом, принятым за
стандартное состояние. Если таковым служит чистый компонент, то тре-
буемое значение р^ можно принять по справочным данным. Вычисление
активности производится в этом случае непосредственно по формуле
(25). Если стандартным состоянием служит 1 %-ный идеальный разбав-
ленный раствор, то для определения р£т требуется по меньшей мере од-
но значение давления пара компонента над раствором в области дейст-
вия закона Генри. Обозначив эту величину pf при [1% i]*, получим
= —. „ст _ р*
Р,
ai — fi I — ст
Pi
*
Pi
На практике в этом случае выполняют серию измерений при различ-
ных концентрациях. Это повышает точность определения р£т, а также
позволяет найти концентрационный интервал, в котором соблюдается за-
кон Генри. Если растворенное вещество в чистом виде при температуре
опыта является газом, то его активность в расплаве определяют по раст-
воримости. Некоторое отличие этого метода от предыдущего связано с
тем, что молекулы газов, представляющих наибольший интерес (Н2, N2,
О2) являются двухатомными и при растворении диссоциируют. Связь
между давлением газа и его концентрацией выражается при этом квад-
ратичной зависимостью, которая следует из уравнения реакции растворе-
ния. Для азота, например, имеем V2N2=[N]; K=fN[% WI^Pn2- В обла-
сти, где раствор остается разбавленным, fN = l и> следовательно:
[%N] = K]/pNf .
(32)
Зависимость (32), называемая законом Сивертса, является формой
закона Генри для случая, когда растворение сопровождается диссоциа-
цией молекул на два атома. В растворах азота и водорода в железе за-
кон Сивертса соблюдается практически для всех концентраций (откло-
нения можно ожидать только при сверхвысоких давлениях). Если же
экспериментальные значения растворимости азота [% N] не связаны ли-
нейной зависимостью с J/"pNa (как, например, для растворов азота в хро-
ме), то это указывает на отклонение от идеальности. Характеристикой
этого отклонения может служить коэффициент активности азота fa
(стандартное состояние: 1 %-ный идеальный разбавленный раствор
азота в хроме). Для определения необходимо построить по экспери-
ментальным данным график зависимости К' = [ pNj от [% N] и
проэкстраполировать полученную кривую на нулевое значение [% NJ,
W /N = l , и, следовательно, /(' = /<. Пример такого графика в полулога-
рифмических координатах показан на рис. 4. Экстраполяция опытных
данных к [% N]=0 дает 1g К= 1,24. При давлении 1 атм имеем lg=
==lgK—lgK'=l,24—0,71=0,53, /n=3,4, что соответствует положитель-
ным отклонениям от закона Генри (Сивертса). Как отмечалось выше, та-
кой характер отклонений является обычным для систем с относительно
сильным взаимодействием компонентов.
Исследованиями установлено, что при растворении азота и тем бо-
27
лее водорода в расплавленной стали, включая легированную, заметных
отклонений от закона Сивертса не наблюдается. В связи с этим иногда
вызывает удивление тот факт, что при расчете растворимости газа ис-
пользуют значения коэффициентов активности fN и существенно от-
личающиеся от единицы. Это объясняется тем, что при вычислении ко-
Рис. 4. К определению коэффициентов
активности азота в хроме по данным
о растворимости при 2018 К [11]
должны быть равны: fN [% N]Fe
ГС
вая, что = 1, получим:
эффициентов активности газов и
других элементов во многокомпо-
нентных расплавах за стандартное
состояние часто принимают раз-
давленный раствор элемента не в
данном расплаве, а в железе. Рас-
четную формулу для определения
коэффициента активности fN со-
ответствующего такому выбору
стандартного состояния, легко по-
лучить из следующих рассужде-
ний. Пусть в равновесии с азотом
при даннОхМ давлении находятся
два расплава: железо и сплав.
Очевидно, что активности азота в
обоих расплавах при одинаковом
выборе стандартного состояния
[% N] сплав. Отсюда, уЧИТЫ-
cl о
В рассмотренном примере, если выбрать за стандартное состояние
1 %-ный раствор азота в железе, коэффициент активности азота в хроме
будет равен: /N=0,045/5,2—0,009. Полученное значение намного меньше
единицы. Это указывает на то, что азот взаимодействует с хромом зна-
чительно сильнее, чем с железом.
Еще один способ определения активности по данным о равновесии
с газовой фазой — это изучение равновесия химической реакции одного
из компонентов со специально выбранной смесью газов. Примером может
служить реакция Ст+СО2(г)=2СО(г); ^=Рсо/(Рсо/ас)- Для определе-
ния активности углерода в расплаве ас по отношению к графиту нужно
подставить в приведенное уравнение найденное из опыта значение Рсо/
/Рсо, и вычисленную при помощи таблиц константу реакции Ст+СО2(г)=
— 2СО(Г). Если требуется определить коэффициент активности углерода по
отношению к идеальному разбавленному раствору, то н\жно (как и в
предыдущем примере с азотом) проэкстраполировать значение К'=р со/
/(Рсо, [% С]) к нулевой концентрации углерода, а затем найти К и вы-
числить /с из соотношения fc =К7К. Несмотря на значительное число
известных методик, определение активностей в расплавах остается до-
вольно сложным. Существенно облегчает задачу возможность расчета
активности одного из компонентов раствора по данным об активности
другого (или других) при помощи уравнении Гиббса—Дюгема. Пример
такого расчета приведен ниже.
28
1.10. РЕАЛЬНЫЕ РАСТВОРЫ
Уравнения, описывающие поведение компонентов в реаль-
ных растворах, можно получить из уравнений для идеаль-
ных растворов путем замены концентраций соответствую-
щими значениями активностей. Если за основу выбирают
уравнения, выведенные для совершенного раствора, то ис-
пользуют активность по отношению к чистому компоненту.
Выражение для химического потенциала принимает в этом
случае следующий вид:
р£ = RT In at = р? + RT In у£ 4- RT In xt. (34)
Изменение энергии Гиббса компонента при изотермиче-
ском переходе из стандартного состояния в раствор дан-
ного состава по уравнению 1<Ж)=[»](хр составляет:
ДО/ = RT In at = RT In yt + RT In (35)
Первое слагаемое в этой сумме представляет собой из-
быточную энергию Гиббса растворения:
ДСГ6 = RT In уь (36)
которая может быть обусловлена либо теплотой растворе-
ния (Д/Л=/=0), либо отклонением в энтропийном члене
(TASJ136 #= 0), либо и тем и другим. Выражение для интег-
ральной энергии Гиббса легко получить из уравнения (35)
с учетом выражения (11):
д<7т = Дбх + х2 Д<?2 — RT (*i In «1 + хг In а2). (37)
Можно легко показать, что активность, введенная нами
в уравнения для совершенных растворов формально, сов-
падает с ранее введенной активностью, определяемой как
отношение давления пара компонента над раствором к дав-
лению пара над чистым компонентом р£/р°. Рассмотрим
раствор, находящийся в равновесии с паром. Очевидно, что-
химический потенциал компонента в растворе равен при.
этом химическому потенциалу компонента в газовой фазе:
р,1(ж) = |ХЦг) = |ЛЦг) +RT In pi.
Аналогичное уравнение можно написать и для стандарт-
ного состояния: Ц£(ж)=Ццг) + ^7’ In р°г Вычитая второе
уравнение из первого, получим выражение: р£(ж)— р£(ж) =
=RT\n(Pi/p°i)=RT\n сц, эквивалентное уравнению [34].
Если за основу для описания реальных растворов выби-
рают уравнение, выведенное для бесконечно разбавленно-
го раствора, то используют активность по отношению к
29>
1 %-ному раствору, сохраняющему свойства разбавленного.
Выражение для химического потенциала при этом имеет
следующий вид:
= ф° + RT In а* = фо + RT Inf, + RT In c{. (38)
Изменение энергии Гиббса при переходе 1 моля компо-
нента из стандартного 1 %-ного раствора в раствор с дан-
ной концентрацией по уравнению [i](i%) =[Л(с.) составляет:
&Gi=RT In аМПп fi+RT In сь
Отметим, что введение активностей — это удобная по-
становка, но еще не решение задачи об описании термоди-
намических свойств реальных растворов. Активности или
коэффициенты активности, как правило, приходится нахо-
дить из опыта. Однако, определив их по данным о каком-
либо одном свойстве раствора, можно далее рассчитать
ряд других его свойств. Так, измерив парциальное давле-
ние пара компонента над раствором, можно рассчитать его
равновесные концентрации в различных химических реак-
циях. Определив концентрационную зависимость активно-
Рис. Ъ. Графическое интегрирование
уравнения Гиббса — Дюгема для оп-
ределения (при 1873 К) по данным
о концентрационной зависимости у_
he
можно использовать данные
расплаве для получения доп>
сти одного компонента рас-
твора, можно при помощи
уравнения Гиббса — Дюге-
ма определить то же для
другого компонента. Если
определить два значения ак-
тивности при различных тем-
пературах, то по ним можно
оценить теплоту растворе-
ния вещества. Измерив теп-
лоту растворения (при по-
мощи калориметра), можно
по одному значению актив-
ности рассчитать ее величи-
ну при другой температуре.
Это уменьшает число опы-
тов, необходимое для полу-
чения достаточно полной
информации об изучаемой
системе. Приведем два при-
мера, показывающие, как
об активности компонента в
лнительной информации.
Пример 1. В работе [12] были экспериментально определены коэф-
фициенты активности железа в расплавах с различным содержанием уг-
лерода. Эти данные (в сочетании с известной растворимостью углерода
30
в железе) были использованы авторами для расчета коэффициентов ак-
тивности второго компонента — углерода. Удобная для таких расчетов
форма уравнения Гиббса — Дюгема может быть получена из выражения-
[13], если в качестве парциальной характеристики gi взять AG*?36 =
==/?71пу<. В результате такой подстановки получим выражение'
xFedlnTFe+xcdlnyc =0, которое после интегрирования от хс (мольной:
доли насыщения) до хсдает следующую формулу:
Jg Vc = l^Vc —
VFe
f л,
J
(39)
Необходимое для расчетов значение коэффициента активности угле-
рода в насыщенном растворе ус можно найти по известной величине-
растворимости углерода в железе, которая при 1873 К составляет х*с =
=0,21 (5,4 %). Учитывая, что в состоянии насыщения а^=у*сх*с =1 (как.
у чистого графита), получим Yc = l/Xc =4,76. Вычисление входящего в
формулу интеграла удобно производить графическим методом, который,
иллюстрируется рис. 5. На рис. 5 показана в соответствующих координа-
тах часть экспериментальных данных для высоких концентраций угле-
рода. Две заштрихованные площади соответствуют двум значениям ин-
VFe
Г xFe
теграла I ------j 1g« (для растворов, которые характеризуются
J хс
*Fe
точками 1 и 2), Пунктиром показано отношение x^Jxq (в насыщенном*
растворе)*1. Построить на одном графике всю кривую без ущерба для
точности расчетов трудно (требуется график очень большого размера).
В области малых концентраций углерода использование такого графика*
затрудняется еще и тем, что при Xq—>0 кривая уходит в бесконечность.
Для ее построения требуется очень высокая точность исходных экспери-
ментальных данных, которая не всегда может быть достигнута, поэтому
в диапазоне малых Xq для определения коэффициентов активности про-
ще воспользоваться экстраполяцией полученной концентрационной за-
висимости активности углерода к нулю. Исходные данные и результаты
расчета коэффициентов активности углерода описанным методом пред-
ставлены в табл. 2.
Положение точки 0, соответствующей насыщенному раствору,,
определено здесь (как по-видимому, и в работе [12]) путем графической
экстраполяции кривой до пересечения с пунктирной горизонталью. Для
более точных расчетов рекомендуется [222] экстраполировать концент-
рационную зависимость не 1g yFe, а величины lg yFe/(1—xFe)2, кото-
рая значительно меньше зависит от концентрации. В рассмотренном
примере такая экстраполяция дает для точки 01gyFe = —0,128 (вместо^
—0,113).
31/
Таблица 2. Коэффициенты активности углерода в расплавах Fe—С
при 1873 К, определенные при помощи уравнения Гиббса—Дюгема [12]
*с *Fe *Fe/*C 9 IgVFe 1g Vc Vc
0,005 0,995 199 0 - ♦
0,0138 0,986 71 —0,003 —0,172 0,673 0,0093
0,0270 0,973 36 —0,004 —0,112 0,772 0,0208
0,0500 0,950 19 —0,006 —0,012 0,974 0,0486
0,0708 0,929 13,1 —0,012 0,088 1,225 0,0867
0,0920 0,908 9,9 —0,019 0,178 1,506 0,1385
0,1340 0,866 6,47 —0,044 0,358 2,28 0,306
0,1565 0,843 5,4 —0,069 0,458 2,87 0,452
0,1590 0,841 5,3 —0,067 0,478 3,00 0,477
0,1830 0,817 4,47 —0,086 0,568 3,76 0,677
0,210 0,79 3,76 0,678 4,76 1,0
Пример 2. Определим теплоту растворения кремния в железе по двум
.известным значениям .коэффициента активности Т§ц17ооос) =0,00162;
Tsi(i420oC) =0,00049 [2]. Выражение, необходимое для такого расчета,
можно получить из уравнения AGi=RT\nai = AHi—TASi9 если принять,
что в заданном интервале температур ДЯ< и ASi можно считать не за-
висящими от Т. Решая уравнение, получим:
In = [AHi/(RT)] - (ASf /R), (40)
'ИЛИ
In — In — In — In —
= (ДЯ£//?)[(1/Г2)-(1/Л)]. (41)
Наряду с выражением (41) для определения AHi можно воспользо-
ваться и дифференциальным уравнением:
d In d In уi AHj
~dT = dF~ RT* ’
(42)
которое является результатом дифференцирования выражения (40).
Чтобы найти среднее (для заданного температурного интервала) значе-
ние AHi можно заменить производную отношением конечных прираще-
гний:
/ dlgT~ \ Д1б7~ -2,79-(-3,31)
-------- I = --------= -------------------= U.UUloO.
\ dT /сп ДТ 1973—1693
(43)
Подставив найденную величину в уравнение (42), получим
dT
ДЯ^ =— 2,303/?^р
=— 120000 Дж.
=— 19,14-18332-0,00186 =
ср
32
Знак оо означает, что величины у“ и ДЯ~ относятся к области раз-
бавленных растворов, где они не зависят от концентрации. Анализ при-
веденных уравнений показывает, что в растворах, которые образуются с
выделением тепла (ДЯг<0) и в которых коэффициент активности у» как
правило, меньше единицы, с повышением температуры cti и у$ увеличива-
ются. Наблюдаемые в таких растворах отрицательные отклонения от за-
кона Рауля с повышением температуры уменьшаются. В растворах, где
ДЯ<>0 и у,>1, активность и коэффициент активности с повышением
температуры уменьшаются, что также приводит к снижению степени от-
клонения от идеальности. Таким образом, при повышении температуры
Рис. 6. Зависимость активностей и коэффициентов активностей в расплавах
Fe—Si от температуры [2]
все растворы приближаются к совершенным. Это обусловлено тем, что
упорядочивающая роль энергии межчастичного взаимодействия в раство-
ре по отношению к разупорядочивающему действию температурного фак-
тора с повышением температуры уменьшается. Сказанное иллюстрирует
рис. 6. .
1.11. ПЕРЕХОД ОТ ОДНОГО
СТАНДАРТНОГО СОСТОЯНИЯ К ДРУГОМУ
В термодинамических расчетах часто возникает необходи-
мость перехода от одного стандартного состояния к друго-
му. Например, для определения энергии Гиббса реакции
окисления кремния, растворенного в железе [Si](i%)+O2(r)=
= SiO2(r), требуется из табличного значения стандартной
энергии Гиббса реакции чистого кремния с кислородом
Si(«)+O2(r)=SiO2(r) вычесть свободную энергию перехода
Si(jK)=[Si]<p^.
Для вывода расчетной формулы рассмотрим идеальный
3—675
33
разбавленный раствор компонента А в растворителе В. Хи-
мический потенциал компонента А в этом растворе можно
выразить двумя способами. Если за стандартное состояние
принять чистый компонент А, то по уравнению (34),
учитывая, что в разбавленном растворе уа=уа, получим:
р,л=!Лх + RT 1п(улХд). Если за стандартное состояние при-
нять 1 %-ный разбавленный раствор, то согласно выраже-
нию (38) с учетом того, что в рассматриваемом растворе
[а = 1, можно записать: рд=фл + RT In сА. При выборе в
обоих случаях одного и того же начального уровня отсчета
энергии величина р.д не зависит от способа ее выражения.
Приравнивая правые части полученных уравнений и учи-
тывая, что согласно выражению (6) в разбавленном рас-
творе Ха— (са-Мв)/(Ма- 100), найдем искомое изменение
энергии Гиббса при переходе компонента А из одного стан-
дартного состояния в другое -<4ж=|/!l](i%):
AG = ^-^ = RTln-i^- = Wln(^^-). (44)
Некоторые другие случаи перехода из одного стандартного
состояния в другое (от жидкого компонента к твердому или
газу) рассмотрены в разделе 1.16.
1.12. РЕГУЛЯРНЫЕ РАСТВОРЫ
Понятие регулярного раствора было впервые введено в
1929 г. Гильдебрандом. В предложенной им модели откло-
нение раствора от совершенного, т. е. появление избыточной
энергии Гиббса обусловлено только энтальпийным членом:
AG"36 = ДЯт; ДО”’6 = ДЯ; (45)
Из выражения (45) с учетом уравнения (36) можно вы-
вести важное для практических расчетов соотношение:
ДЯ^ЯПпу,-, (46)
которое позволяет оценить температурную зависимость ко-
эффициента активности, если известно хотя бы одно его
значение. Принимая, что Д//( не зависит от температуры, из
выражения (46) можно получить, например:
1ПТ((П = (1873/7) In
Vf(1873) • (47)
Предполагается, что энтропийная составляющая энер-
гии Гиббса в регулярном растворе такая же, как в совер-
шенном, и может быть рассчитана, исходя из допущения о
34
полной хаотичности в расположении атомов. Результаты
расчета выражаются уравнениями (17) и (18). Таким обра-
зом, энергия Гиббса в регулярном растворе определяется
следующими выражениями:
Д6т = ДЯт + RTZxt In (48)
= AHi + RT In xh (49)
Термин «регулярные растворы» не связан с существом
рассмотренной модели и был введен Гильдебрандом для
обозначения семейства растворов, в которых, как показал
эксперимент, с определенной регулярностью изменялось
одно из свойств (растворимость йода или серы). Оказалось,
что предложенная модель удовлетворительно описывает
свойства таких растворов. В результате модель тоже полу-
чила название регулярного раствора. Допущение о хаотич-
ности расположения атомов при определенном взаимодей-
ствии между ними является главным противоречием моде-
ли регулярного раствора, ограничивающим область ее
применения. Очевидно, что наилучшие результаты модель
регулярных растворов может дать для систем с относитель-
но слабым взаимодействием. В таких системах величина
АН, характеризующая тенденцию к упорядочению (к опре-
деленной группировке атомов), должна быть не больше
(или не намного больше), чем энергия теплового движения
атомов RT, определяющая тенденцию к разупорядочению.
С повышением температуры область возможного примене-
ния теории регулярных растворов (ТРР) увеличивается.
Ряд полезных соотношений теории регулярных раство-
ров может быть получен из статистического анализа моде-
ли, предполагающей хаотическое расположение атомов по
узлам некоторой решетки, существование которой допус-
кается как в твердом теле, так и в жидкости. Одним из ре-
зультатов такого анализа [1] является следующее выраже-
ние для избыточной энергии Гиббса образования одного
моля регулярного раствора из чистых компонентов 1 и 2:
AG"86 = АНт = (Д/2) = [(гУд Д)/2]х.х2, (50)
где z — число ближайших соседей каждого атома; Na —
число Авогадро; А — энергия образования двух связей меж-
ду разноименными атомами: Д=2е/_2—8/_/—82—2; Р/~г—
число связей 1—2-, при хаотическом расположении атомов
Р/-2= ZN АХ1Х2.
Из уравнения (50) видно, что отклонение раствора от
идеальности (знак AG™6) определяется знаком величи-
з*
35
ны Д. При Д = 0Д6« и ДЯт обращаются в нуль, что со-
ответствует совершенному раствору. Для этого не обяза-
тельно, чтобы энергии связи между всеми атомами в рас-
творе были одинаковыми. Достаточно, если е/-г будет
средним арифметическим энергией е/_/ и &2—г. Если энер-
гия связи разнородных атомов больше (по абсолютной ве-
личине), чем полусумма энергий связи атомов одного сорта,
то Д<0 и, следовательно, ДЯт<0, т. е. раствор образуется
с выделением тепла. Парциальные теплоты растворения при
этом также отрицательны, а отсюда следует, согласно урав-
нению (46), что в таком растворе yi и у2 меньше 1. В про-
тивоположном случае, когда притяжение разноименных
атомов меньше, чем одноименных (Д>0), раствор харак-
теризуется положительными отклонениями от идеальности.
Рассмотрим концентрационную зависимость величины
ДЯт, следующую из уравнения (50). Величну zNa&I2, не
зависящую от состава, обозначим Й (энергия смешения).
Подставляя, получим:
ДЯт = Йххх2, (51)
что соответствует параболе с экстремумом в точке Х\ =
=х2=0,5. Экстремальное значение Д/7т в этой точке рав-
но У4Й. Для определения парциальных энтальпий компо-
нентов Д/Д и Д//2 необходимо продифференцировать урав-
нение (51), переписав его для раствора, содержащего ni + n2
молей компонентов, следующим образом:
ДЯ = ДЯт (пх + Лг) = Й I(ni«2)/(ni + "г)]-
Отсюда получим
Д//х = = Q--------------= йх2 = Й (1 — *i)2,
1 дП1 (П14-п2)г
ДЯ2 = = йх? = Q (1 - х/.
дп->
(52)
(53)
(54)
Как видно из уравнений (53) и (54), энергия смешения
Й равна предельному значению Д//“ (при х,—>0). Учитывая
связь между теплотой растворения и коэффициентом актив-
ности по уравнению (46), можно получить следующее вы-
ражение для зависимости коэффициентов активности от
концентрации:
1пух = (Й/7?Т)(1—хх)2;
1пу2 ОДТ)(1-х2)2.
(55)
(56)
Из уравнений (55) и (56) следует, что при х2—>0 коэф-
фициент уг стремится к у“, a уг-И. Таким образом,
в теории регулярных растворов возможно только прибли-
женное выполнение законов Генри (уг=const) и Рауля
(у1 = 1) в области малых значений хг, отличных от нуля.
Для иллюстрации расчетных возможностей теории ре-
гулярных растворов перепишем уравнение (48), (49) для
энергии Гиббса с учетом выражений (51) — (54) для Д//.
В результате получаем (для бинарного раствора):
AGm = QXi х2 4- RT (Xi In хг + х2 In х2); (57)
AGi-Q(l — хх)2 + RTInXj; AG2 - Q(1 —x2)2+
4- RT In x2. (58)
Как видно из уравнений (57), (58) все основные термо-
динамические характеристики регулярного раствора, вклю-
чая температурную и концентрационную зависимость ак-
тивности, могут быть рассчитаны, если известен один пара-
метр — энергия смешения й. Величину й, принимаемую в
простейшем случае не зависящей от температуры и концен-
трации, можно рассчитать по данным одного эксперимента.
Такой расчет может быть выполнен при помощи уравнений
(53), (54) по известной величине Д/Л или по уравнениям
(55), (56), если известно одно значение коэффициента ак-
тивности у(- (при заданных концентрации и температуре).
Отметим для сравнения, что в теории идеальных разбав-
ленных растворов для вычисления ДС как функции темпе-
ратуры требуется не менее двух значений у; при разных Т
(см. с. 21).
Таким образом, теория регулярных растворов предо-
ставляет довольно широкие возможности для расчета тер-
модинамических характеристик. Однако довольно ограни-
чен круг расплавов, которые могут быть описаны этой тео-
рией с удовлетворительной степенью точности и в широком
интервале температур и концентраций. На практике эти
уравнения часто используют в качестве интерполяционных
формул в ограниченном интервале температур или концен-
траций. При этом величину й подбирают таким образом
(например при помощи метода наименьших квадратов),
чтобы уравнения соответствовали всей данной совокупно-
сти экспериментальных значений. Далекая экстраполяция
найденных таким способом зависимостей в область неис-
следованных температур и концентраций, как правило, не-
допустима. Тем не менее во многих случаях теория регу-
лярных растворов может быть использована для обработки
37
экспериментальных данных и практических расчетов. Наи-
лучшие результаты эта теория дает при высоких темпера-
турах и в применении к системам с относительно слабым
взаимодействием (с малым различием свойств компонентов).
Приведем в качестве примера результаты расчетов, вы-
полненных при помощи формул теории регулярных раство-
ров для системы Fe—Си. В качестве исходной величины в
этом расчете использован коэффициент активности
?"и(182зк) == полученный в работе [5], что соответству-
ет й=RT In у00=35060 Дж. Большие значения коэффи-
циента активности и энергии смешения (более чем 2RT)
указывают на то, что система Fe—Си, строго говоря, не
может быть отнесена к системам со слабым взаимодействи-
ем. Исходя из этого, можно априори сказать, что в данном
случае при использовании модели регулярного раствора
нельзя ожидать хорошего согласия с экспериментом. По-
смотрим тем не менее, что эта модель может дать. Резуль-
таты расчета приведены в табл. 3 и на рис. 7. Для удобства
Таблица 3. Пример расчета термодинамических характеристик
раствора Fe—Си при 1823 К при помощи теории регулярных растворов*
*Си *Fe T&Sm— RT (*Cu I*1 xCu “b 4-xFe In xFe), Дж/моль AHm=Q xCu Xpe, Дж/моль Дж/моль AHCu=Q (l-xcu)2, Дж/моль AHFe=Q (l~*Fe)2, Дж/моль 1 о и Д csl^ о я О
0 1,0 0 0 0 35060 0 10,1 1,0
0,1 0,9 4940 3160 — 1780 28370 335 6,5 1,03
0,2 0,8 7615 5610 —2010 22425 1425 4,4 1,1
0,3 0,7 9290 7360 —1925 17195 3138 3,1 1,2
0,4 0,6 10210 8410 —1800 12635 5605 2,3 1,45
0,5 0,5 10500 8790 — 1715 8785 8785 1,8 1,8
0,6 0,4 10210 8410 —1800 5605 12635 1,45 2,3
0,7 0,3 9290 7360 — 1925 3138 17195 1,2 3,1
0,8 0,2 7615 5610 —2010 1425 22425 1,1 4,4
• 0,9 0,1 4940 3160 —1780 335 28370 1,03 6,5
-1,0 0 0 0 0 0 35060 1,0 10,1
* Исходные данные:
VCu (1823 К) -10’1 151> следовательно fl-AHCu =
=7? 7'1 п у 2? =35060 Дж/моль.
-38
сопоставления с экспериментальными данными, относящи-
мися к 1823 К, расчет выполнен для той же температуры.
Как видно из рис. 7, расхождения между эксперименталь-
ными и расчетными значениями некоторых характеристик
Рис. 7. Термодинамические
характеристики расплавов
Fe—Си при 1823 К (сплош-
ные линии — эксперимен-
тальные данные [5], пунк-
тирные — результаты расче-
та по теории регулярных
растворов, точечные — ре-
зультаты расчета по квази-
химической теории)
не очень велики. Это относится к энтропии, а для концен-
траций <0,2 и >0,8—и к остальным характеристикам.
Для многих практических целей полученные расчетные дан-
ные можно считать вполне приемлемыми, несмотря на то,
что модель регулярного раствора была выбрана в данном
случае с явным нарушением установленных границ ее при-
менимости. Вместе с тем в области концентрированных рас-
творов (0,2<х,<0,8) заметные отклонения расчетных зна-
чений ДЯШ от экспериментальных привели к тому, что на
расчетной кривой Дбт появились два дополнительных экс-
тремума в точках k и I. Такая форма кривой Дбт и соот-
ветствующая ей форма расчетной кривой активности аСи
39
отвечают расслоению сплава на две несмешивающиеся
жидкости. Следующее из расчета расслоение раствора (не
наблюдаемое в эксперименте) обусловлено не тем, что рас-
чет дает большие отклонения от опытных данных, а свойст-
вами самого расплава Fe—Си, который в действительности
находится как бы на грани расслоения. Подтверждением
этого являются наблюдаемые в системе Fe—Си большие
положительные отклонения от идеальности.
1.13. СУБРЕГУЛЯРНЫЕ
И КВАЗИРЕГУЛЯРНЫЕ РАСТВОРЫ
Отмечая определенную полезность теории регулярных рас-
творов, следует признать, что- большинство реальных сис-
тем не может быть описано этой теорией с достаточной точ-
ностью и полнотой. Проведенный в работе [13] ана-
лиз имеющихся экспериментальных данных о 175
бинарных металлических системах показал, что только во-
семь из них можно удовлетворительно описать теорией ре-
гулярных растворов в ее простейшем варианте.
Одно из наиболее часто встречающихся противоречий
заключается в том, что реальные системы характеризуются
обычно несимметричными зависимостями термодинамиче-
ских характеристик от состава, а теория регулярных рас-
творов предсказывает симметричные кривые (см. рис. 7).
Чтобы устранить это расхождение теории с экспериментом,
в работе [14] был предложен один из усложненных вариан-
тов модели, который получил название субрегулярного рас-
твора. Отличие этого раствора от регулярного заключается
в том, что его энергия смешения Q не является константой,
а изменяется в зависимости от состава и принимает значе-
ния от Й1=Д/У“ в чистом компоненте 1 до Й2=Д#1° в
компоненте 2. Это можно записать в виде уравнения:
Q = «!%! + Q2x2 = Д//Г X! + Д#? Х2.
(59)
Подставляя выражение (59) в уравнение (51), получим:
Mim = Mi? xi х? + ДЯГ xi xl
(60)
Для определения парциальных теплот растворения вос-
.пользуемся формулой (9):
ДЯг = ДЯт + (1-х{)
а (АНТО)
dXi
40
В результате получим
ДЯХ = Д/7Г + (ДЯГ — 2ДЯГ) + xi (5ДЯГ + 4Д//-Г) +
4-2х?(ДЯГ-ДЯГ); (61)
ДЯ2 - ДЯ“ + 2х2 (ДЯГ — 2ДЯГ) + xl (5Д77” — 4Д#Г) +
+ 2х2(ДЯГ —Д//Г). (62)
Остальные формулы теории субрегулярных бинарных
растворов следует непосредственно из выражений (46) —
(49):
In Ji = kH^RT; (63)
Д6т = ДЯт + RT (хг in 4- х2 In х2); (64)
^Gi = &Ht + RT\nXi. (65)
Значения ДЯт и Д/Д в этих формулах определяются
выражениями (60)-(62).
Как видно из полученных уравнений, усложнение моде-
ли привело к тому, что в расчетные формулы входит
теперь не один параметр (как в теории регулярных раство-
ров), а два: й1=Д/7“ и Й2=Д//Г. Таким образом, повы-
шение точности расчетов достигается в этом случае в ре-
зультате увеличения количества исходной информации об
объекте. Во многих случаях это не только приемлемо, но и
необходимо для лучшего согласования расчетных данных
с практическими. Чтобы найти необходимые для расчетов
два параметра й] и й2 в случае, когда неизвестны значе-
ния ДЯ" и ДЯ~, можно воспользоваться любыми двумя
известными характеристиками: yi, А.Нт, АНAGm или Дб,.
Усложнение расчета сводится при этом к необходимости
решения системы двух соответствующих линейных уравне-
ний типа (60)—(65).
В некоторых случаях улучшения согласования теории
с экспериментом достигают введением избыточного энтро-
пийного члена в формулы для регулярного раствора. Од-
ним из вариантов модели такого типа является раствор, в
котором предполагается линейная зависимость й от тем-
пературы. Уравнения, вытекающие из этой модели, и ре-
зультаты расчетов приведены в работе [13], в которой по-
казано, что одновременный учет концентрационной и тем-
пературной зависимости энергии смешения позволяет уве-
личить число систем, удовлетворительно описываемых тео-
рией, с 8 до 70.
Эффективной является модель квазирегулярного рас-
41
твора [16]. В этом случае поправка вводится как избыточ-
ная энтропия, связанная с теплотой образования раствора
простым соотношением
ДН„ = TAS™8, (66)
или
ДЯ, = tAS"36 . (67)
С учетом уравнений (2), (36), (51) и (53) выражение
(66) приводит к важным расчетным формулам:
до£б = ДЯт (1 - —) = ЙХ! х2 f 1 - —(68)
\ т / \ т /
АС1/3® = ЯТЧп = дя71 ——) == Q (1 — Xi)2 (1 — —У
\ т / \ т /
(69)
Из выражения (69), как и из уравнения (46) для регу-
лярных растворов, можно получить формулу для расчета
температурной зависимости коэффициента активности по
его значению при одной температуре. При обычном допу-
щении Д//(т=Alleys) имеем:
, 1873 (т — Т) ,
7 (т—1873) п^(187з>- (70)
Из сопоставления выражений (47) и 70) видно, что при
очень больших значениях т (по сравнению с Г и 1873 К)
формулы теории квазирегулярного раствора (ТКР) и ТРР
эквивалентны.
Как и для субрегулярных растворов, в формулы (68),
(69) входят два параметра, в данном случае Q и т, для
определения которых необходимо не менее двух экспери-
ментально полученных значений AG^36, AG^36 или у«- Что-
бы определить параметры, как и в предыдущем случае, не-
обходимо решить систему двух линейных уравнений типа
(68) или (69). Указанное минимально необходимое коли-
чество исходных данных достаточно для выполнения рас-
чета, но, как правило, не обеспечивает высокой точности
получаемых результатов. В связи с этим для определения
параметров используют обычно максимально возможное
количество экспериментальных данных. Вычисления при
этом выполняют методом наименьших квадратов.
Важным моментом теории квазирегулярных растворов
является то обстоятельство, что параметр т в первом при-
ближении можно считать независящим от индивидуальных
свойств компонентов раствора. Его можно заранее опреде-
42
лить для широкого круга систем по имеющимся данным о
теплотах и энтропиях образования некоторых из них. Фор-
мально т представляет собой температуру, при нагреве до
которой раствор приобретает свойства идеального. Это сле-
дует из того, что при Т=т формулы теории квазирегуляр-
ных растворов (68), (69) переходят в выражения для со-
вершенного раствора. Расчеты [16] показали, например,
что корреляция между и Д3~<изб) в 90 растворах
(жидких и твердых) на основе целого ряда растворителей
(Au, Cd, Zn, Ag, Bi, Cu, Hg и др.) при температуре от 293
до 1426 К удовлетворительно характеризуется одним зна-
чением параметра т, равным 3000 К. Далекая экстраполя-
ция зависимостей, полученных на основе модельных пред-
ставлений, обычно не дает хороших результатов, поэтому
трудно предположить, что найденное в работе [16] значе-
ние параметра т может быть с успехом использовано при
температурах сталеварения (1773—1973 К). Большое раз-
нообразие сплавов, по которым найдено значение т, также
должно сказываться на его применимости. Расчеты [15,
208] по данным о 19 расплавах на основе железа и никеля
показали, что значение параметра т для данной более уз-
кой группы растворов составляет 7000 К. Это значение
можно использовать для практических расчетов.
В табл. 4 представлены имеющиеся экспериментальные
данные о разбавленных растворах на основе железа и ни-
келя И результаты расчетов, выполненных по уравнениям
для регулярного и квазирегулярного раствора. При отборе
для табл. 4 экспериментальных значений Д/7“ предпочте-
ние отдавали данным, которые получены калориметриче-
ским методом. Значения коэффициентов активности эле-
ментов, растворенных в железе, принимали в основном,
согласно данным [17]. Значения в никеле принимали
главным образом по данным В. В. Аверина (см. табл. 4)
и работы [24]. Значения Д7/“, приведенные в табл. 4 (гра-
фа 4), рассчитывали по формулам (46) и (69) с учетом
т=7000 К.
Как видно из табл. 4, экспериментальные значения Д//“
удовлетворительно согласуются с рассчитанными по тео-
рии квазирегулярных растворов. Результаты, полученные
по теории регулярных растворов, значительно хуже согла-
суются с экспериментальными данными. В связи с этим для
вычисления неизвестных значений ДЯ“ или у~, а также при
составлении уравнений для вычисления Д6“ в системах,
43
Таблица
4 Экспериментальные и расчетные термодинамические данные о жидких разбавленных
растворах на основе железа и никеля*1 [15]
Растворенный элемент i Коэффициент актив- “ОСТИ*2 Теплота растворения, ДИ?0, Дж/моль X I
экспериментальные данные результаты расчета по извест- ному значению tf
Рекомендуемое уравнение* •
для вычисления
Дб?° (Дж/моль) перехода
'чист***1 (1 %)
Растворитель — железо
Ag
А1*5
В*5
Стр
Са
Се
Со
Сг
Си*5
Ge
1/2Н2(г)
Hf
Mg
Мп
Мо
1/2N?r)
Ni
1/2 О2(г)
1/2 Р2(г)
РЬ
Pd
200 [17]*4
0,049 {18]
0,040 [21
0,57(17
2270 [23] *4
0,03 [212]
1,07 [17]
1,0 [17]
8,6 [24
0,034*6
0,0043*®
43 [213]*4
1,3 [17]
1,0 [17]*7
—62800(19,20]
—73200 [22]
22600 [15]
47200 [20]
—72000 [25]
36500 [17
—115000 [19]
0,66
850 [28] *4
2,8 [24]
10700 (7<1973К)
21000 (7> 1973К)
—10000 [215]
—117000 [17]
—140000 [15]
212500 [28]
22000 [24]
82400/111700
—46900/—63600
—50200/—67800
-/-
120500/163200
—54600/—74000
1050/1420
0/0
33500/48800
-/-
-/-
-/-
58600/80000
4100/5500
0/0
• -/-
-/-
—6500/—8800
105000/142000
16000/21700
111700—59,4 Т
—62800—23, ST
—73200—12,3 Г
22600—42,3 Г [17]
163200—58,6 Г
—74000—35,57
1420—38,97
—37,66Т
47200—46,65 7
—72000—30,2 7*®
36500+30,46 7 [17]
— 115000—31,4 7*®
80000—42,9 7
5500—39,1 7
—42,87*7
10700+20,3 7 (7< 1973К)
21000+15,1 7 (7>1973 К)
— 10000—36,8 7
— 117000—2,897 [17]
—140000—9,67(17, 15]
212500—106,3 7
22000—46,4 7
А/г Sa(r)
Si
Sn
Ti
U
V
W
Zr
Al*5
В
Crp
Ca
Cr*5
Cu*5
Fe*°
Ge
Hf
Mn
г/г Мг(г)
Nb
1 /2 Ог(г)
г/г Рг(г)
Pb
Pd*5
1 /2 ^2(г)
Si*5
Sn
0,0013(17]
2,15 [28]
0,037 [17]
0,027 [17
0,17 29]
1,0 [17]*7
0,28*«
0,014 [212]
0,00025*’
0,0083*®
0,32 [30]
0,61 [24]
0,6*®
2,21 [24]
0,355 [24]
0,0024*®
0,000036*®
1,0*»
0,0017*®
2 6*®
1,68 [24]
0,00014*®
0,055*®
—72000 [19]
— 131800 [15]
18800 [28]
—69500(15, 19]
—42300 (17]
—26800 [214]
—80300 [19]
-/-
—103800/—140600
11900/16000
—51000/—69500
—58200/—78700
—27600/—37200
0/0
—66500/—90000
Растворитель — никель
— 153100(15]
— 100400(22]
41000(15]
— 13800(19]
271
15700
—32200 [215]
—128400 [32
20100(31]
—216300 [19]
69000 [33]
—134300 [19]
—71500 [34]
—252300(15]
20100(216]
11700(24]
— 123500(19]
—201700(15
—61500 [217
— 128900/—174100
-/-
-/-
—7800/—10600
—7900/—10900
12000/16200
—16150/—21750
8000/10900
—138100/—187000
-/-
—72000—9.92T [17, 19]
— 131800—17,32 T
18800—48,5 7
—69500—27,28 7
—78700—39,4 7
—42300—29,2 7
—48,17*7
—26800—19,2 7*®
—80300—34,97
—153100—18,87
— 100400—10,5 7*®
41000—47,97(9, 15]
— 10600—33,7 7
—13800—34,3 7
15700—4,1 7
—32200—29,3 7
—128400—22,8 7*®
201004-35,1 7(31]
—216300—17,45 7*«
—38,2 7
690004-18,62 7(33]
— 134300—23,26 7*®
-715004-1,84 7 [34]
20100—51,57*»
11700—45,197
-1235004-17,72 7(19, 35]
-2017004-1,8 7
—61500—63,81 7*®
Продолж. табл. 4
Теплота растворения АН.00, Дж/моль
растворенный
элемент i
Коэффициент актив* *
Н0СТИ*’ Т(1873 К)
экспериментальные
данные
результаты расчета по из-
вестному значению у®*10
рекомендуемое уравнение*’ для
вычисления AG” (Дж/моль)
перехода Гчист=[<] (1 %)
Та
Ti«
У*Б
0,00082*»
0,00019*»
0,011*®
0,094*0
0,00003 [36]
— 149400 [19]
—183700 [15
—103800(19
—50200 [218
—202100 [19
-/-
—133500/—179900
—70300/—95000
-/-
—161900/—219700
—149400—26,827* «
—183700—9,83 7
— 103800—19,257
—50200—34,56 7*«
—202100—20,57
Стандартным состоянием для всех элементов, кроме углерода и газов, служит чистая жидкость (Т—1873 К, р=1 атм). Для
приведения данных к этому стандартному состоянию использовали сведения о теплотах плавления элементов, опубликованные в
работе [37].
w Значения у?0при других температурах могут быть определены из выражения AG® =*RT In у?0 +RT In х
ГДе Xi (1 %)
=Afi/100M., с использованием данных о температурной зависимости AG?0, приведенных в графе 5.
w При составлении уравнений использовали значения AG® или у®, приведенные в соответствующих работах. Величину АЯ®
в основном принимали по экспериментальным данным (главным образом, калориметрическим). Если такие данные отсутствовали или
были недостаточно надежны, то АЯ~ рассчитывали по известным значениям у® с использованием теории квазирегулярных рас-
творов — по формуле (69).
♦4 Рассчитано по данным о растворимости.
•5 Экспериментальные значения у ~ и АЯ^° в данной системе были использованы для определения параметра теории квазирегу-
лярных растворов т.
Рассчитано по экспериментальному значению АЯ® с использованием теории квазирегулярных растворов — по формуле (69).
*7 Предполагается идеальный раствор.
♦8 См. п. IV.7.
♦9 Аверин В. В. Растворимость кислорода, азота и активность элементов-раскислителей в расплавах на основе железа, кобальта,
никеля и хрома. Автореф. докт. дис. М., 1968.
*10 В числителе — данные, полученные по уравнению (46) с использованием теории регулярных растворов, в знаменателе — дан-
ные, полученные по уравнению (69) с использованием теории квазирегулярных растворов.
по которым имеются неполные данные, было использовано
уравнение теории квазирегулярных растворов (69). Из это-
го можно заключить также, что уравнение (70) лучше про-
гнозирует температурную зависимость коэффициентов ак-
тивности, чем выражение (47).
Дальнейшее повышение точности расчетов (применимо-
сти теории для описания реальных систем) может быть
достигнуто при объединении допущений, принятых в мо-
делях субрегулярного и квазирегулярного растворов.
1.14. КВАЗИХИМИЧЕСКАЯ ТЕОРИЯ
Как уже отмечалось, основное допущение теории регулярных растворов,
которое заключается в хаотичности расположения частиц, находится в
противоречии с тем, что теплота смешения принимается в этой теории
отличной от нуля. В самом деле, если при замене одних связей между
атомами (1—1 и 2—2) на другие связи (1—2) энергия системы изменя-
ется, естественно предположить, что в растворе будет больше тех свя-
зей, которые энергетически более выгодны. В квазихимической теории,
разработанной Гуггенхеймом, предполагается, что число имеющихся в
растворе связей каждого типа обусловлено равновесием реакции
(1-1) + (2-2) = 2(1 -2). (71)
Константу равновесия этой реакции: К=P2(i—2)/[Pg-i)P(2-2)] можно
представить как произведение двух констант /Схаот и Куп. соответствую-
щих хаотическому и упорядоченному расположению атомов в растворе.
Можно показать, что /Схаот=4 [1, с. 245], и следовательно, К=4 Куп. Со-
гласно квазихимической теории, константа Куп по аналогии с константой
химической реакции определяется энергией упорядочения:
in Куп =- (А/КТ) =- (Na b/RT), (72)
или, учитывая, что Й = 1/г zNa& ,
1пКуп=— (2Q/zRT). (73)
Формальная аналогия между уравнением (72) и уравнением изотермы4
химической реакции (3) нашла отражение в термине «квазихимцческая
теория растворов». Очевидно, что при Й=0 Куп=1. Все расчетные фор-
мулы должны при этом соответствовать уравнениям для совершенных
растворов. При й>0 Куп < 1 и, следовательно, К<4. Это свидетельству-
ет о сдвиге равновесия реакции (71) влево. При й<0, наоборот связи
1—2 более энергетически выгодны — равновесие реакции (71) сдвинуто
вправо. Таким образом, квазихимическая теория, учитывающая опреде-
ленное упорядочение атомов в растворе, является некоторым уточнением
теории регулярных растворов. Гуггенхейм называл эти теории соответст-
венно первым и нулевым приближением. Квазихимическую теорию он
именовал также теорией строго регулярных растворов. Вывод основных
уравнений квазихимической теории довольно сложен [38, 39]. Приведем
для краткости только конечные результаты, необходимые для практичес-
ких расчетов. Интегральная теплота образования 1 моля бинарного
раствора описывается в квазихимической теории уравнением:
ЛЯт Р + 1
Qjq х2.
(74)
47
Коэффициент активности компонентов по отношению к чистым
жидким веществам имеют следующий вид:
Р — 1 + 2xj \*П
(75)
где Xi — мольные доли компонентов: Q — энергия смешения; z— коор-
динационное число; ₽= {1+4XiXg(r)2—l)}lj<2, = Величина т]
является параметром приведенных уравнений, который должен быть оп-
ределен из экспериментальных данных. Этот параметр непосредственно
связан с энергией смешения и, как будет показано ниже, с характери-
стиками разбавленных растворов. Соответствующие формулы имеют вид:
ДЯ^°=Й=г/?Г1пт]; = г)2. Из приведенных формул следует, что в со-
вершенном растворе (при Q=0) т] = 1 и Р=1. При бесконечном разбавле-
нии (Хг->0)Р также стремится к 1. Координационное число z для жид-
ких растворов, строго говоря, также является параметром теории, по-
скольку кристаллической решетки (в обычном смысле этого слова) в
жидкости нет, поэтому величину z иногда варьируют, добиваясь наилуч-
шего совпадения теоретических и экспериментальных значений (при этом
z может принимать как целые, так и дробные числовые значения). При
выполнении упрощенных расчетов значение z обычно выбирают на осно-
вании данных рентгеноструктурного определения числа ближайших сосе-
дей атома в растворе. Для расплава на основе железа, никеля, и кобаль-
та можно в первом приближении принимать z=10.
При использовании формулы (75) для вычисления предельного значе-
ния коэффициента активности у™(в разбавленном растворе) необходимо
иметь в виду, что прямой расчет в этом случае невозможен. Подстанов-
ка Xi=0 и соответственно Р=1 в выражение (75) дает неопределенность
(0/0). Чтобы раскрыть ее, воспользуемся правилом Лопиталя — заменим
предел отношения функций пределом отношения их производных. При
этом учтем, что
dp
dX;
V
Р
В результате получим
А(1]2_1)(1_2х.) + 2
Р__________________
2^-(T12_i)(1_2xi)4-p+l
Р
(76)
=Т]2
и, следовательно, Пту»=у t =т)х. Как уже отмечалось, это выражение
очень удобно для определения параметра т), если известно значение од-
ного из предельных коэффициентов активности (?£°). Заметим что ис-
пользование формулы (75) для расчета у, при малых концентрациях
(Xi<0,05) также имеет свои особенности. Вычисления в этом случае
следует проводить с повышенной точностью.
Определив при помощи уравнений (74), (75) значения ДЯ™ и у»
можно вычислить по известным формулам и остальные характеристики
раствора: ai=yiXi\ AGm = /?7(xilnai+A'2lna2); T&Sm = &Hm—&Gm. Для
48
определения парциальных теплот растворения воспользуемся формулой:
= &Нт + (1 - Xi) d (;Н-— • (77)
иЖ j
Дифференцируя уравнение (74), с учетом полученного выражения
для dfydxi найдем:
d(Wn) гп( 1-2х2 , 2X1x2W-\)(2x2-l) )
dx2 I р+1 P(P+1)2 J
Из этих выражений следует, что в разбавленном растворе при (и
соответственно 0-И) теплота растворения ДЯг стремится к Д//^=£2. Это
отношение можно использовать для определения параметров теории Q
и т), если из эксперимента известно значение одной из предельных теп-
лот растворения ДЯ^°. В табл. 5 приведен пример использования формул
квазихимической теории для расчета термодинамических характеристик
расплавов Fe—Си. В качестве исходной величины, как и в приведенном
выше расчете при помощи теории регулярных растворов, принято значе-
ние Ycu(1823K) =10Д [5]. Координационное число г принято равным 10.
Результаты расчета приведены на рис. 7, из которого видно, что полу-
ченные данные в целом значительно ближе к экспериментальным, чем
результаты расчета по теории регулярных растворов (исключение со-
ставляет лишь энтропия). Однако достигнутое уточнение оказалось не-
достаточным, чтобы полностью ликвидировать перегибы на кривых ДОт
и аСи, из которых следует ошибочный вывод о расслоении расплава на
две несмешивающиеся жидкости.
Таким образом, квазихимическая теория является уточнением теории
регулярных растворов, позволяющим иногда значительно улучшить со-
гласование между расчетными и экспериментальными данными. Это уточ-
нение достигается практически без введения дополнительных параметров,
так как координационное число г может быть принято для широкого
круга расплавов равным 10. Вместе с тем квазихимическая теория не
всегда обеспечивает удовлетворительное соответствие расчетных значе-
ний экспериментальным. Так же как и теория регулярных растворов, она
нс может описать системы с несимметричными зависимостями термоди-
намических характеристик от состава. Для улучшения соответствия тео-
ретических и экспериментальных значений в квазихимическую теорию
тоже можно ввести допущения о концентрационной и температурной за-
висимости параметра (т) или Q). Для этого требуются дополнительные
исходные данные (например, второе значение у£° или ДЯ^°). Ряд полез-
ных формул усложненной квазихимической теории приведен в работах
[13, 40].
1.15. ПАРАМЕТРЫ ВЗАИМОДЕЙСТВИЯ
Практическое использование экспериментальных данных
о коэффициентах активности и других термодинамических
характеристиках бинарных расплавов затрудняется тем,
что реальные сплавы одновременно содержат несколько
примесных или легирующих элементов. Чтобы учесть вза-
имное влияние компонентов раствора на их термодинами-
4—-675
49
Таблица 5. Пример расчета термодинамических характеристик
помощи квазихими
*Cu *Fe 3 Дж/моль ?Fe ?Cu aFe *3 °Cu ДСт- Дж/моль
0 1,0 1,0 0 1 10,1*2 1 0 0
0,1 0,9 1,1 1,173 ЗОЮ 1,023 5,96 0,921 0,596 — 1916
0,2 0,8 5160 1,1 3,98 0,88 0,797 —2305
0,3 0,7 1,223 6620 1,23 1,43 2,88 0,861 0,864 —2272
0,4 0,6 1,25 7470 2,14 0,858 0,857 —2335
0,5 0,5 1,26 7760 1,72 1,72 0,859 0,859 —2305
0,6 0,4 1,25 7470 2,14 1,43 0,857 0,858 —2335
0,7 0,3 1,223 6620 2,88 1,23 0,864 0,861 —2272
0,8 0,2 1,173 5160 3,98 1,1 0,797 0,88 —2305
0,9 0,1 1,1 ЗОЮ 5,96 1,023 0,596 0,921 —1916
1,0 п 0 p и м e ч 1,0 а н и e. 0 Для расчет 10,1*2 а использов 1 али следую 0 щие фо! 1 эмулы: 0
!₽=/ 1 4~ (r АНт - , , й*Си *Fe* VFe= р-Н d (ДЯ~) = ( P —*+2*Fe p2. I *Fe (P+1» J ( 1—2xr„
+xCulnaCu); “ASm d <ДНт> =ДНт+ 21- (1 <*Cu ““xCtP* = 22 J — m <*Cu I 04-1
2 10; VCu(1823K) 10,1
[5]. Следовательно, tj=
♦’ Исходные данные:
Дж/моль.
♦2 О вычислении предельных значений v см. с. 48.
*3 Приведенные значения определены из условия симметрии соответствующих
ческие характеристики, пользуются аппаратом параметров
взаимодействия, который был предложен Вагнером [41],
а затем развит в работах [16, 42, 43]. Основу метода со-
ставляет разложение соответствующей избыточной термо-
динамической функции (ДО,-, ДЯ,- или ASi) в ряд Тейлора
около точки, соответствующей чистому растворителю. Если
за стандартное состояние принять чистый компонент I, то
для избыточной энергии Гиббса, точнее для In у,=Дб“зб/ЯЛ
получим:
In у. = In у™
I I
д In Vj
dxj
50
раствора Fe—Си при 1823К при
ческой теории*1
™sm’ Дж/моль d(AWOT) 4*Cu A«Ctr Дж Д^ре’ Дж
моль Си моль Fe
0 35060 35 060 0
4929 25480 25940 464
7464 17320 19020 1695
8891 11460 14 650 3180
9807 5607 10840 5230
10067 0 7760 7760
9807 —5607 5230 10840
8891 —11460 3180 14 650
7464 —17320 1695 19020
4929 —25480 464 25 940
0 —35060 0 35060
flFe=VFe xFe <*Fe InflFe+
д* In у,-
dxj дх^
т *•!
(79)
2 Хре Xqu (l)1—1) (2 Xqu 1) 'J
PCP+D1
(v“ 1/г -1.261 и Q-гЯПпп-35060
кривых для обоих компонентов (рис. 7).
В большинстве случаев
требуемая точность позво-
ляет ограничиться членами
ряда нулевой и первой сте-
пени. При этом уравнение
(79) можно представить в
виде: In72(^2; х3; х4;...) =
= 1пу2(ж1-и) "Ип/г +1п/2 +
+ In /2 +—, который совпа-
дает с предложенным Чип-
маном [42] выражением:
V2(x,;x,;x4....) — ?2(х,;х,=
Для расчетов повышенной точности (при наличии до-
статочно точных исходных данных) в формулы вводят чле-
ны второй степени. Остальными членами ряда пренебрега-
ют. Входящие в уравнение (79) производные (с коэффи-
циентами), взятые при бесконечно малых концентрациях
растворенных элементов (хг->1), называют параметрами
взаимодействия и обозначают соответственно е{, р{ и р[,Л.
С учетом этих обозначений выражение (79) преобразуется
следующим образом:
п п пп
In = In ъ 4- 2 el Xj + 2 pl+ 2 2 p* Xk '
/=2 /=2 /=2 fe=2
/<*
4*
51
Во многих случаях удобнее принять за стандартное со-
стояние однопроцентный идеальный разбавленный раствор
компонента i в растворителе 1. При этом разложение lg fi =
=Дб"зб/2,3 RT будет иметь вид:
п п п п
ig h = 2 41 % /1 + 2 ИI0/0 i]2 + 22 И’*[%/П%Я (81)
/=2 /=2 /=2 /г=2
i<k
тде
[%1] 400 ’
4 = -^-
<П%/1
[%1J-1CO ’
J.Л
' i
d2 lg fi
d[%;] д[%Л]
[%1J—loo’
Свободный член в выражении (81) отсутствует, так как
в бесконечно разбавленном растворе f*=l, и, следователь-
но, lgf“=O.
Основные соотношения между введенными параметрами
взаимодействия имеют вид [17]:
Е< = Е;; е;--2.зо^е;; ;
Мг ML
1 M} 230Mj ’
(82)
pi = -^- 1100MJr! + Mj(M,-M,)e{] + 4"^'
Mj 2 \ Ml ]
(83)
Для определения табличных значений параметров взаи-
модействия используют экспериментальные данные об из-
менении коэффициентов активности элементов в зависимо-
сти от концентрации соответствующих компонентов раство-
ра. Покажем для примера, как определить параметры
и г* по данным о влиянии хрома на растворимость азота
в жидком никеле. Исходные экспериментальные данные
приведены на рис. 8. Поскольку все значения растворимо-
сти относятся к одному и тому же давлению, активность
азота во всех расплавах можно считать одинаковой (не за-
висящей от концентрации хрома): fN[N]=const. Отсюда
52
следует
0,5/5 — 2,824 ___q 112
20 ’
д Ig [Nj
Параметр второго порядка,
равный
д21g fN
<5[%Cr]2
[%Cr]=0
d2 1g f N[
d [%Cr]2
[%Cr]=o
dig a
a [% Cr]
[%Cr]=0*
характеризует изменение tg а в
зависимости от [% Сг].
Найдем tga' (в точке, со-
ответствующей 20 % Сг):
tga'=(0,475 — 1,209)/(30 —
—20) =—0,073. В результате
д [ % Cr] [%Сг]=о
Рис. 8. Определение параметров
взаимодействия по данным о рас-
творимости азота в сплавах Ni—
Сг при температуре 1873 К и дав-
лении 1 атм
получим:
tga' — tga
[%Сг]
— 0,073 —(—0,112)
2-20
= 0,00097.
0N =
Данные работы [17] о параметрах взаимодействия
и rl в расплавах на основе железа при 1873 К представле-
ны в табл. 6—8. Для удобства табл. 6—8 воспроизведены
с некоторыми сокращениями, в них не включены некоторые
приведенные в работе [17] значения параметров для эле-
ментов: Ag, As, Au, Ge, Pd, Pt, Rh, Sb, Se. Данные о пере-
крестных параметрах r]-ft в работе [17] отсутствуют, по-
скольку сведения о них ограничены и не всегда достаточно
надежны. Расчет ft- при помощи табл. 6—8 (при наличии
в них необходимых параметров) очень прост. Например, в .
расплаве, содержащем 1,0 % С и 1,5% Cr, lgfc =е£ [С] +
+ес [СгН+Ус [С]2+ г<?[Сг]2 = 0,14-1 + (—0,024)-1,5 +
+ 0,0074-12 + 0-1,52 = 0,1114; fc =1,29. Для определения
Ус можно воспользоваться формулой (31) и данными
табл. 4. В рассмотренном примере: ус — fc =0,57-1,29=
=0,74. Однако для многих пар элементов табличные зна-
53
Таблица 6. Параметры взаимодействия первого порядка е{*100 в железе при 1873 К [17]
Элемент i Элемент /
Al в с Са Со Сг Си н Мп Мо N Nb
Al 4,5*х 9,1 -4,7 — - м_ 24 — —5.8*1 1 11
В — 3,8 22 - мм 49 7,4*1 м—
С 4,3 24 14*1 -9,7 0,76 -2,4 1,6 67 -1,2 —0,83 11 —6
Са -7,2 —34 (-0,2) — М— м.. — мм — мм мм
Со ММ 2,1 — 0,22 —2,2 "" — 14 » мм 3,2 1 III
Сг —- — 12 — -1,9 —0,03 1,6 —33 - 0,18 —19 мм
Си —м — 6,6 — 1,8 2,3 —24 — 2,6 —
Н 1,3 5 6 —— 0,18 —0,22 0,05 0 —0,14 0,22 —• —0,23
Мп — — —7 — — мм —- -31 0 мм -9,1 —
Мо — -9,7 - 0,03 — —20 — — 10 1
N —2,8*1 9,4*1 13 — 1,1 —4,7*2 0,9 — —2 -1,1 0 —6*1
Nb — —• —49 — мм» — —61 — — —42*1 (0)
Ni —— —• 4,2 -6,7 —— —0,03 —25 мм — 2,8 ——
О —390*1 —260 —45 0,8 —4 -1,3 —310 -2,1 0,35 5,7 —14
Р — — 13 —— *3 —з*3 2,4*3 21 О*3 *3 9,4 *3
РЬ 2,1 6,6 —— 0 2 -2,8 — —2,3 0 —
S 3,5 13 11 — 0,26 —1,1*1 —0,84 12 —2,6 0,27 1 -1,3
Si 5,8 20 18*1 -6,7 — —0,03 1,4 64 0,2 — 9 —
Sn 37 — — 1,5 * 12 — 2,7 мм
Та
Ti
V
W
Zr
—37
—34
—15
—440
— 110
—59
8,8
—47* *1
—180*1
—35*1
-7,2
—410
•' Имеются данные о температурной зависимости е'(см. с. 58, 59).
*2 По данным [44]: =0,022; =0,009; е^г = 0,046; =q>0q(;3 jcm также дополнение табл. 8).
*3 По данным [219]:
/................ Ti
е/(1673К)Х10° * ’ ’• ~5’6
“4,2
Мп Со Ni Си Nb Мо
—3,2 —0,46 —0,59 —3,5 —4,0 -1,7
W
—2,3
Таблица 7. Параметры взаимодействия первого порядка е^-100 в железе при 1873 К [17]*4
Элемент i Элемент
Ni о р РЬ S Si Sn Та Ti V W Zr
Al —— —660*1 —— 0,65 3 0,56 — —— — 1
В —180 —— — 4,8 7,8 — — — — —
С . 1,2 —34 5,1 0,79 4,6 8*1 4,1 -2,1 — —— -7,7 -0,56
Са —4,4 — — — -9,7 — — —— ——
Со 1,8 0,3 0,11 —- —— — — ' —
Сг 0,02 —14 —5,3 0,83 —2*1 —0,43 0,9 5,9 —• ——
Си 1 —6,5 4,4 —0,56 -2,1 2,7 — t — ——
сл
Элемент i
Продолжение табл. 7
Элемент
Ni О P Pb s Si Sn Ta Ti У w Zr
н
Мп
Мо
N
Nb
Ni
О
Р
РЬ
S
Si
Sn
Та
Ti
V
W
Zr
0 — 19 1,1 — 0,8 2,7
— —8,3 -0,35 —0,29 -4,8 0
—0,07 —• 0,23 —0,05 —
1*2 5 4,5 0,7 4,7
—83 —- — —4,7
0,09 1 —0,35 —0,23 -0,37 0,57
0,6 —20*i 7 « —13,3 — 13,1
0,02*3 13 6,2 1,1 2,8 12
-1,9 4,8 —32 4,8
0 —27 29 -4,6 —2,8*1 6,3
0,5 —23 11 1 5,6 11*1
1 1 3,6 3,5 -2,8 5,7
—— « —2,1 ——
*2 — 180 . —11 -
— —97 —• — -2,8 4,2
— 5,2 —— 0,05 3,5 "
— 16
0,53 —2 -1,9 —0,74 0,48 —
—* 1 ' 1 —— — —•
—- — — * —— —
0,7 —3,2*1 —53*1 —9,3*1 —0,15 —63
1 —
-1,11 —60 —30 0,85 (—300)
1,3 *3 *3 *3 --
5,7 — —— — 0
—0,44 —0,02 -7,2 -1,6 0,97 —5,2
1,7 — — 2,5
0,16 —— —— - 1111 ——
— — — —— —
- — 1,3 —
— 1 1,5 —• —
’
Имеются данные о температурной зависимости е|(см. с. 58, 59).
*2 См. примечание *2 к табл. 6.
" См. примечание*3 к табл. 6.
** Дополнения [17, 212, 213]:
Щ............... Се/Н kLa/H j Nd/H U/O Zr/O Ce/O La/O AC/U
e/.100 ......... 0,0 —2,7 —3,8 '—4,4 —4,4 —57 —57 5,9
i
U/U Zr/Zr Cr/Mg Ni/Mg Al/Mg Si/Mg C/Mg
1,3 2,2 0,81 —2,6 —1,3 —0,04 —2,8
чения ej отсутствуют. В этом случае не следует исключать
неизвестные параметры (принимать их равными нулю),
лучше попытаться оценить их сравнительным методом,
пользуясь значениями параметров для элементов-аналогов
(ближайших соседей i или / в Периодической системе эле-
ментов Д. И. Менделеева). При значительном различии
атомных масс элементов лучше принимать равенство пара-
метров е, а не е; пересчет можно выполнить при помощи
формул (82). Расплавы на основе никеля и других метал-
лов исследованы значительно хуже, чем на основе железа.
Параметры взаимодействия для них менее надежны, а ча-
сто вообще неизвестны. Некоторые имеющиеся сведения по
расплавам на основе никеля приведены в гл. IV. Иногда
для расчета коэффициентов активности в сплавах на осно-
ве никеля можно воспользоваться параметрами взаимо-
действия для сплавов на основе железа (см. с. 63).
Наряду с параметрами взаимодействия, которые пред-
ставляют собой коэффициенты в разложении ДОузб//?Т,
применяют также энтальпийные и энтропийные параметры,
являющиеся коэффициентами в аналогичных разложениях
А/Л и AS?36. По аналогии с параметрами е, е, р, г энталь-
пийные параметры обозначают: т), h, Л, I, а энтропийные —
о, s, л, р. Ряд соотношений между этими параметрами при-
веден в работе [43], например
= П/; rii = 100 (М/Mi) hi. (84)
Практическое значение энтальпийных параметров обу-
словлено тем, что они определяют температурную зависи-
мость параметров и Соответствующее выражение
легко получить из уравнения Гиббса — Гельмгольца:
а(дс‘1зб/т)______
дТ Т2
Учитывая уравнение
(д In у JdT) =-(^Hi/RT2),
вания по X, дает
(36), получим выражение:
которое после дифференциро-
д dlnyf =___________1 dbHj .
дТ ' dxj ~ RT2 ' dxj ’
д^!дТ=— П</(№). (86)
Подставляя в уравнение (86) соотношения (82) и (84),
можно получить удобное для практических расчетов выра-
жение
деЦдТ^—hi/(2,3RT2).
(87)
СТ -7
Таблица 8. Параметры взаимодействия
Элемент i
Al в с Co Сг Си | Мп Mo Nb
Al -0,1* — -0,4 — — —
В — 0 0 — -
С —0,07 —— 0,74* 0 0 0 0 0
Са 0,07 — 1,2 —— —• — —— —
Со - — — 0 — -
Сг — — — 0 0 0 - 0
Си — —— — —• — 0,01 — — — !
Н 0 0 0 0 0 0 0 0 0
Мп - — — — 0 O=— •
N 0 0 0 0 0,04** 0 0 0 0
Ni — - — —
О 170 0 0 0 0 0 0 0 0
Р — — — 0,08 0 0 — ——
РЬ 0 0 0 0 0 0 0 —
S 0,09 0,74 0,58 0 0 0 0 0 —0,01
Si — — — — —
Sn — - — - —
Ti — —— 1 —— — 0** — — —
V — — - — "
* rfJ=0,17/T—0,0011;
AI
=8,94/7 + 0,0026;
г S' = 1,94/7- 0,0003;
г Si =6,5/7-0,0055.
♦» По данным [44]: rgr=0,00032; г Сг, Ni =-0,00008; г gi = 0,00007; г^г =
Как уже отмечалось, теплота процесса значительно сла-
бее зависит от температуры, чем изменение энергии Гиббса,
поэтому энтальпийные параметры hl по сравнению с пара-
метрами взаимодействия е>{ в относительно небольшом
температурном интервале также можно считать постоянны-
ми. Это позволяет выражать температурную зависимость-
параметров взаимодействия уравнением el = (А/Т) +В, ко-
торое получается в результате интегрирования выражения
(87).
Зависимость параметров в железе от температуры
[17] характеризуется следующими выражениями: е^} =
= (63/7)+0,011; е" = (1650/7)—0,94; eg = (714/7)—0,307;
eg =(158/7’) +0,0581; е£г = (—153/7) +0,062; = (859/
/Т)—0,487; egb =—(260/7)+0,0796; е™ =—(152/7)+0,049;
eV =-(350/7)+ 0,094; е"ь =-(1720/7)+0,503; eg =
58
второго порядка -100 в железе при 1873 К [17]
Элемент /
Ni р S Si Sn Та Ti V W Zr
— —0,06 — III М—
> — - — 0 II III м
0 0,41 — 0,07* 0,02 —0,02 — 0,01 0
0 — 0,09 — — — ——
— — м
0 0,25 — 0 0 •— — —
— — — —0,03 —— —
0 0 0 0 0 0 0 0 0
— — — 0 — — —
о** 0 0 0 0 0 0 0 0 0
0 — — —
0 0 0 0 0 3,1 0 0 (0)
0 -0,1 — -0,1 0,01 ——
0 0 0 0 0 — 0
0 0,06 —0,09 0,17 0 0 0,01 0 0,01 —0,02
0 — — —0,21* — — —*1
— 0 0 — —• • —
** — — —- — — -0,1 —
- - — - " —0,06 — —0,01 — * -
-0.0001; i-Cr, Ni =—0.0006; rNi=0 0005
= — (1750/7’)+0,734; <?» =—(94,2/Т) +0,0396; £§,=(380/
/Г)—0,023; = (34,5/Т)+0,089; £?« =— (13900/Т)+5,61;
£$ =—(1270/7')+0,33; е® =-(34740/7’) +11,95; е$ =
= — (20600/7')+7,15; е»1 = (162/Т)+0,008; е|=(233/7’) —
-0,153; £?в= (975/7’)-0,4; £?" =—(1960/7’) + 0,581; е£ =
= — (4070/7’) +1,643. Эти данные так Же, как и сведения©
параметрах взаимодействия второго порядка (см. табл. 8),
немногочисленны, поэтому при проведении расчетов часто
возникает потребность вычисления параметров более высо-
кого порядка по параметрам нулевого или первого поряд-
ка, а также определения энтальпийных параметров по из-
вестным значениям е или е. Соотношения, необходимые для
таких расчетов, можно получить с использованием различ-
ных теоретических представлений или эмпирически (стати-
стической обработкой большого числа экспериментальных
данных). Примером эмпирического подхода может служить
59
соотношение Чипмана и Корригана [45]:
Ли = 4,184" 15000 б^(187зю = 62760 6n(1873K) ,
(88>
полученное по экспериментальным данным для растворов
азота в железе, содержащем различные легирующие эле-
менты. При помощи этого соотношения можно легко полу-
чить уравнение для пересчета параметров 6'N на произ-
вольную температуру. Воспользуемся для этого соотноше-
нием между параметрами:
2,3/?7’6((Т) = h'nг) — Т's'i(T), (89)
которое следует из уравнения: Дб“з6 =Mii—TAS"36. Под-
ставляя выражение (88) и Т=1873 К в уравнение (89), по-
лучим. 2,3 RT 1873 £n(1873iq =62760 £^(187зю 1873 s^1873Kj,
откуда
sn = 14,376^(1873X1. (90)
Подставив уравнение (90) в выражение (89) и приняв,
что параметры h>N и s^ в рассматриваемом интервале тем-
ператур постоянны, получим
2,37?Тбы(Г) = 62760би(187зк) — 14,377’6n(1873x),
или
вк(Т) = 1(3280/7’)— 0,75]бы(187зк).
(91)
При помощи выражения (91) можно вычислить Ig/bicn в
сплаве заданного состава и далее с учетом уравнений (32,
33) получить выражение для расчета растворимости азота
в нем при заданной температуре:
-----™ - '.25-
/ 3280 n 7К\ v / /
— I—-----------0,75 j S ( 6n(1873)
(92);
Это выражение называют уравнением Чипмана — Корри-
гана *.
Ряд полезных соотношений между различными парамет-
рами получен в работах [16, 43, 46] на основе модельных
представлений. Дифференцируя по Xi уравнения теории ре-
* Принятая в выражении (92) температурная зависимость раство-
римости азота в железе (lg[N]=—188/7'—1,25) несколько отличается,
от результатов последних исследований, однако коэффициеннты в выра-
жении (91) были рассчитаны в работе [45] с использованием именно
этого выражения. При его уточнении надо уточнять и уравнение (91)..
60
гулярных растворов (53) — (56), можно получить следую-
щие выражения: е‘ =—21пу“; р‘ =1пу°°; т]*=—2A//J°;
Х‘=АЯ°°.
Продифференцировав выражение (47) по [% /] с учетом
формулы (31), получим
4(т> =--(1873/Т)е<( 1873) . (93)’
Согласно теории квазирегулярных растворов из уравнения
(70) с учетом т=7000К получим
т) = [(2557/Т) — 0,365] 4<1873). (94):
Из изложенного следует, что выражение (94) ближе к дей-
ствительности, чем формула (73). Квазихимическая теория
дает следующие формулы [16, 43]:
inTr-=-i-,nG-—У
2 \ г /
4 = г [ 1 - (тГ)2/М 2 in Т“- -Mln vZT; (95).
Из формулы (96) путем подстановки выражения (95) и
соотношения /?Т1пу~=й, полученного из уравнения (55),
можно получить выражение:
ej = г 1 — ехр
Qjj -f- ^ij
zRT
[46],
(97)
которое позволяет иногда оценить параметр е! приняв
энергии смешения равными соответствующим предельным
геплотам растворения (Qh=A//«bi) и т. д.). Выражение
(97) можно использовать также для вычисления парамет-
ра е/ при произвольной температуре, если известно его
значение при 1873 К. Уравнение для таких расчетов имеет
вид:
i
7(1873)
1873/7''
(98)
Приведем некоторые другие формулы, следующие из
61
квазихимической теории [16, 43, 46]:
P/’fc = ~~ (в/ е? + е? 8/) — е;л;
=— 2Д/7Г exp (2Д/7Г/г/?Т);
T)t =— z/?T [ 1 — (е|7г)] In [ 1 — (ej/z)].
(99)
(100)
(101)
(Ю2)
Как видно из формул (99) — (Ю2), применение квази-
химической теории ограничено системами, в которых е[ <
<2. В работах [46, 47] получены соотношения между па-
раметрами для растворов внедрения, в которых атомы i-
того компонента располагаются не в узлах решетки (как
у модели замещения — в теории регулярных растворов
и т. п.), а в междоузлиях (порах). Рассмотрены поры окта-
и тетраэдрического типа, для которых атомы, расположен-
ные в ближайших узлах, образуют соответственно октаэдр
и тетраэдр [47]. Полученные выражения имеют следующий
вид:
pi- = V2 (е|)2 [ 1 — р exp (2Q1}/zRT) — ф];
11! =— 6RT [1 - (size) ] In f 1 — (в<7«)],
(103)
(104)
где о1/=д/7“; ц, ф и 6 — геометрические параметры, зна-
чения которых для некоторых типов структуры ближнего
порядка приведены в табл. 9.
Таблица 9. Значение параметров для некоторых типов структур
Структура Тип пор г ц Ф б
О. ц. к. Октаэдрические 8 0 1/2 2
Тетраэдрические 8 1/2 1/4 2
Г. ц. к. Октаэдрические 12 2/3 1/6 6
Тетраэдрические 12 3/4 0 4
Как показано в работе [48], применение к растворам
азота в расплавах на основе железа формулы (104) (в ва-
рианте с 6 = 6) дает выражение, близкое к полуэмпириче-
скому уравнению Чипмана — Корригана. Аналогичный ре-
зультат получается также при использовании выражения
(104) в варианте для растворов замещения (с заменой 6-
«2
числа узлов, окружающих пору, на г — координационное
число, равное 8 для о. ц. к.-решетки). Дальнейшее развитие
этих работ позволило получить важную формулу для рас-
чета растворимости азота в многокомпонентных сплавах
[209]:
[ %N] = [ %N]Me [1 - V 2 (105)
/Иуд L V
где Afe — металл-растворитель; Afm=Sx;Af/=100/S(C//
/М/) — мольная масса сплава.
Как показано в работах [210, 211], формула (105), не-
смотря на использование только параметров первого по-
рядка (е0, обеспечивает довольно высокую точность рас-
чета растворимости азота в расплавах на основе железа и
никеля. Особым достоинством выражения (105) является-
то, что за условный растворитель в расчете можно прини-
мать любой компонент сплава и даже элемент, в нем не
присутствующий вообще. Согласно теории важно только,,
чтобы в этом растворителе при Xi->1 реализовалась модель-
раствора с теми же значениями потенциалов межатомного
взаимодействия, которые имеют место в сплаве. Пользуясь,
этой особенностью выражения (105), можно, например,,
преодолеть существенные трудности, связанные с недостат-
ком известных значений параметров взаимодействия для.
сплавов на основе никеля и других, металлов. Как показа-
ли расчеты [2П], для расплавов на основе никеля можно-
выбрать в качестве условного растворителя железо и вос-
пользоваться при расчете параметрами для сплавов на его-
основе. Точность оценки растворимости азота при этомг
оказывается удовлетворительной. Для определения при по-
мощи выражения (105) [% N] при температурах, отличаю-
щихся от 1873 К, в работе [209] рекомендовано для расчета >
e/j т) соотношение типа (98) с заменой z на 6=6.
1.16. РАСЧЕТ РАВНОВЕСИЯ РЕАКЦИЙ
С УЧАСТИЕМ РАСТВОРОВ
Целью расчета, как правило, является определение равно-
весной концентрации или равновесного давления одного из-
реагентов по уравнению константы равновесия реакции.
Для остальных реагентов концентрации или давления
Должны быть заданы или приняты с учетом условий проте-
кания реакции, иначе результатом расчета будет не число,
а уравнение, связывающее неизвестные равновесные харак-
63
теристики. Например, рассчитывая равновесную концентра-
цию кислорода в металле при раскислении, необходимо
задаться определенной концентрацией раскислителя, в про-
тивном случае можно рассчитать только величину произве-
дения концентраций [R][O], стоящую в знаменателе кон-
станты равновесия реакции:
IRJ + (О) = ДО(Т); X = aro/|Zs[Rl-^ol0)|. (106)
Иногда целью расчета является определение возможно-
сти (направления) протекания реакции в заданных усло-
виях. Такая задача легко сводится к первой, так как ответ
можно дать, исходя из сопоставления заданной (фактиче-
ской) концентрации реагента с равновесной (реакция
всегда идет в направлении их сближения).
Для расчета равновесия реакции, в которой один или
несколько реагентов находятся в растворе, недостаточно
знать стандартную энергию Гиббса взаимодействия чистых
веществ. Кроме нее необходимы сведения о коэффициентах
активности реагентов в растворе. Если такие данные име-
ются или могут быть определены расчетом, то определение
параметров равновесия не представляет больших трудно-
стей. Рассчитаем для примера равновесную концентрацию
алюминия в железе в условиях вакуумной плавки в тигле
из оксида алюминия, приняв парциальное давление кисло-
рода в газовой фазе Ро,= 10~6 атм и 7'=1873 К. Прежде
всего полезно убедиться в правильности постановки задачи
с точки зрения правила фаз. В данном случае система со-
держит три компонента: железо, алюминий и кислород
(К=3) и три фазы: жидкую, твердую и газообразную
(ф=3). При постоянной температуре число независимых
переменных в такой системе, согласно правилу фаз, состав-
ляет С=К—Ф+1 = 1. Это означает, что при данной тем-
пературе в системе можно выбрать произвольно только
один параметр, остальные характеристики будут при этом
определены. В рассматриваемой задаче такой произвольно
выбираемой величиной является давление кислорода. Со-
гласно табл. 1, изменение энергии Гиббса для реакции
окисления чистого жидкого алюминия
2А1|Ж1 + %р2(г, = А12О3(т) (107)
можно вычислить при температурах сталеварения по урав-
нению
AQo ==_ 1681000 + 324 Т Дж.
(Ю8)
64
В соответствии с уравнением изотермы реакции (3) по-
лучим
ig к=ig ,
1681000 — 324Т
19,147
(109)
Для вычисления искомой концентрации алюминия xAi
остается определить коэффициент активности удь Прене-
бречь этой величиной в данном случае нельзя (даже при
грубом расчете), так как раствор алюминия в железе об-
наруживает значительные отклонения от теории совершен-
ных растворов. Полагая раствор идеальным разбавленным,
воспользуемся значением у А1 =0,049 (при Т=1873 К),
приведенным в табл. 4. Из уравнения (109) получим %ai —
=6,2-10~10 (3-10~8%). Такой результат (малое значение
л'дО не противоречит принятому допущению о разбавлен-
ности раствора и поэтому может считаться окончательным.
Если бы расчетная концентрация оказалась за пределами
диапазона, в котором раствор можно считать разбавлен-
ным, то расчет следовало бы повторить, воспользовавшись
другим способом оценки удь Так, при pot = 10-17 атм мы
получили бы хд1=0,11 (5,3%)- Оценим величину ум при
такой концентрации при помощи известного значения пара-
метра е*} =0,045 (табл. 6, 7). Получим lgfA1 =0,045-5,3=
=0,24; fA1 = ?А1/у~= 1,74; у А1 =0,049-1,74=0,085. Оче-
видно, что такое отличие уд1 от величины, принятой в рас-
чете, является существенным и не может быть допущено.
Для уточнения расчета необходимо подставить в уравнение
(109) вместо коэффициента yAj его концентрационную за-
висимость. Такую зависимость можно получить, например,
при помощи параметров взаимодействия, приведенных в
табл. 6—8:
lg Taj = 1g Taj + ед} 1 % АП + r% [ % А1Р.
(ИО)
В первом приближении можно для упрощения расчета
пренебречь квадратичным членом. Однако даже в этом
случае вычисления оказываются громоздкими, поэтому
проще воспользоваться методом последовательных прибли-
жений. Для этого нужно повторить два — три раза расчет
по уравнению (109), подставляя в него каждый раз то зна-
чение yAj, которое соответствует концентрации алюминия,
найденной в предыдущем расчете. Вместо уравнения (НО)
можно было бы воспользоваться концентрационной зави-
симостью коэффициента удь которая следует из каких-ли-
В—675
65
бо модельных представлений: теории регулярных раство-
ров с ее разновидностями или квазихимической теории.
В приведенном расчете заданная температура (1873 К)
соответствовала той, для которой известны табличные зна-
чения коэффициентов активности и параметров взаимодей-
ствия. При выполнении аналогичных расчетов для других
температур можно воспользоваться данными о темпера-
турной зависимости у” и е[ приведенными в табл. 4 на
с. 44,45. При отсутствии необходимых данных следует поль-
зоваться соотношениями (47), (70), (93), (94), которые
следуют из теоретических представлений. Зависимость, ана-
логичную формуле (47), можно использовать и для коэффи-
циентов активности ft. В этом случае величина ДЯ;, входя-
щая в исходное уравнение (46), будет иметь смысл теплоты
перехода в раствор 1 моля компонента i не из чистого ве-
щества t, а из стандартного 1 %-ного разбавленного рас-
твора.
•При расчете равновесия между растворенным алюмини-
ем и газообразным кислородом в качестве стандартного
состояния для алюминия был выбран чистый компонент.
Если бы нас интересовало взаимодействие растворенного
кислорода с алюминием, то для кислорода такой выбор
стандартного состояния был бы неудобен. Для веществ,
которые при температурах сталеварения являются газами,
в качестве стандартного состояния выбирают обычно иде-
альный разбавленный раствор (1 %-ной концентрации).
Такой выбор стандартного состояния часто применяют и
для остальных реагентов. При этом в выражение для кон-
станты равновесия входят процентные концентрации ве-
ществ и коэффициенты активности ft, учитывающие откло-
нения растворов от идеальных разбавленных. В ряде
случаев (при небольших концентрациях) этими коэффици-
ентами можно пренебречь. Рассчитаем для примера равно-
весие реакции:
2 [АП + 3 [OJ = А12О3(т) . (111)
Очевидно, что ДО0 для этой реакции будет отличаться
от ранее приведенного Д6° реакции (107) на величину из-
менения энергии Гиббса при переходе от одного стандарт-
ного состояния к другому (для алюминия и кислорода).
Чтобы найти выражение для Дб° реакции (111), нужно
суммировать уравнение (108) и соответствующие уравне-
ния, взятые из табл. 4:
2А1(Ж) + 3/8О2(г) = А12О3(т) ; ДС° =— 1681000 + 324Г Дж,
66
2 [Al] = 2AL • AG° = — 2 (— 62800 — 23,8T) Дж,
{Ж/ ' г
3[О] = 8/2О2(г); Лб° =— 3 (— 117000 — 2,89Т) Дж,
2 [А1] + 3 [О] = А12О3(Т); AG® =— 1203700 + 376,2т Дж
Выражение для расчета равновесия имеет вид:
lgK = lg__________?________=__=l1203700 ± 37612T 2
° /д! [%А1]2 Zq [%А1]3 19,147
Определим концентрацию алюминия, которую необходи-
мо обеспечить для того, чтобы в расплаве содержалось
не более 0,003 % кислорода (при 1873 К). Примем Ддл =
=fo=l. Расчет по уравнению (112) дает [А1]=0,0006 %.
Обе концентрации (алюминия и кислорода) оказались, та-
ким образом, достаточно малыми, чтобы раствор можно
было считать разбавленным. В противном случае (а так-
же при Т=/= 1873 К) уравнение (112) пришлось бы услож-
нить путем ввёдения, как и в предыдущем расчете, концен-
трационных (или температурных) зависимостей коэффици-
ентов /д1 и /о. При наличии в расплаве других элементов,
кроме железа, алюминия и кислорода, необходимо при по-
мощи параметров взаимодействия учесть и их влияние на
коэффициенты и /о-
Если бы в аналогичном примере требовалось рассчитать
равновесную концентрацию не алюминия, а титана, который
при 1873 К находится в твердом состоянии, то для опре-
деления Дб° реакции Т1ж+О2(г)=ТЮ2(т) потребовалось бы
найти Дб° перехода Ti«->TiT. Это легко сделать, если из-
вестны температура и теплота кристаллизации титана.
Д5кр определяют, исходя из того, что в точке плавления пе-
реход твердого титана в жидкий и наоборот происходит без
изменения энергии Гиббса (фазы находятся в равновесии):
ДСкр=Д77кр—ТкрД5Кр=0. Отсюда
ASKp — Д/7 Кр/Ткр; Д(?кр — А//кр Т кр/Гкр)- (113)
Приведем некоторые справочные данные [2] для таких рас-
четов:
Элемент , в Сг Fe Мо Nb
Гкр, К . . 2300 2171 1809 2880 2741
АЯКр, кДж/моль . . 22,2 20,9 15,2 27,6 (26,4)
Продолжение
Элемент а • • • в • Ni Ti V W Zr
Гкр, К 1726 1940 2185 3650 2123
-ДЯкр, кДж/моль . . 17,6 15,5 (17,6) 35,2 16,7
5*
67
Для оксидов имеем:
Оксид.................
Ткр, К ...............
ДЯкр, кДж/моль . . .
А120з СаО SiOp (кристобалит) ТЮ2
2303 2873 1986 2113
108,8 79,5 13,0 64,9
Аналогичный вид имеет уравнение для расчета допол-
нительной энергии Гиббса, которую приобретает жидкость
при перегреве выше точки кипения: ДбИсп=Д#исп—
—Т (ДЯисп/ТКип). Некоторые данные для таких расчетов
приведены ниже [2]:
Элемент .............
Гкип, К (1 атм) • . . >
ДНисп, кДж/моль . . .
Са Mg S Р (красн.)
1765 1390 716 704
150,0 128,7 9,62 30,1
Глава II
СТАЛЕПЛАВИЛЬНЫЕ ШЛАКИ
Шлак представляет собой многокомпонентный оксидный
расплав, непосредственно контактирующий с металлом в
сталеплавильном агрегате и выполняющий важные техно-
логические функции. Развитие теории шлаков связано с ра-
ботами В. А. Ванюкова, П. Герасименко, Г. Шенка,
М. И. Темкина, Л. А. Шварцмана, О. А. Есина, В. А. Ко-
жеурова и многих других отечественных и зарубежных уче-
ных. Первые попытки термодинамического описания шла-
ков были основаны на предположении о том, что структур-
ными единицами в них являются молекулы оксидов (СаО,
SiO2, А120з и др.), а также силикатов, фосфатов и других
соединений. На этой основе была создана так называемая
молекулярная или химическая теория шлака, не потеряв-
шая полностью своего значения для упрощенных количест-
венных оценок и в настоящее время. Молекулярная модель
не объясняет многих физических свойств шлаков, в частно-
сти электрической проводимости и некоторых других, хоро-
шо интерпретируемых на основании представления об
электролитической диссоциации, поэтому дальнейшее разви-
тие количественной теории было направлено главным обра-
зом по пути перехода к ионной модели как более совершен-
ной. Обладая большой гибкостью, ионная модель позволила
дать качественное объяснение многим экспериментальным
фактам и способствовала развитию электрохимии шлаков.
Значительный прогресс в развитии количественной тео-
•8
рии шлаков был связан с применением статистической тер-
модинамики. Разработанные на этой основе методы расче-
та равновесий позволяют делать наиболее обоснованные
численные оценки. Одним из перспективных направлений
дальнейшего развития теории шлаков в настоящее время
представляется последовательный учет характеристик их
электронного строения. На протяжении последних десяти-
летий именно этот путь был связан с наибольшими успеха-
ми физики и химии конденсированного состояния, теории
катализа, теории полупроводников. Все химические связи
осуществляются посредством электронов, поэтому централь-
ный вопрос теории шлаков, касающийся особенностей меж-
атомного взаимодействия, является в конечном счете во-
просом об их электронном строении. Учет характеристик
электронного строения делает количественную теорию бо-
лее последовательной и в ряде случаев приводит к качест-
венно новым результатам.
11.1. ТЕХНОЛОГИЧЕСКИЕ ФУНКЦИИ
И ХАРАКТЕРИСТИКИ ШЛАКОВ
Важнейшей функцией шлака является сорбция (погло-
щение) вредных примесей — серы, фосфора и некоторых
других элементов. Сорбционная способность шлака по от-
ношению к примесям зависит от его состава, температуры
и степени раскисленности системы металл — шлак. Удале-
ние примесей требует различных условий. Так, удаление се-
ры полнее протекает к раскисленной системе, удаление фос-
фора — в окислительных условиях. Для процессов, проте-
кающих в дуговых печах и других открытых агрегатах,
важную роль играют защитные (покровные) свойства шла-
ка. Проницаемость шлака по отношению к азоту, водороду
и кислороду, содержащимся в атмосфере, в значительной
мере определяет газонасыщенность металла и развитие
процессов окисления. Процессы массоперепоса в шлаке свя-
заны с диффузионной подвижностью примесей, вязкостью
шлака, его составом.
В электросталеплавильных процессах существенную
роль могут играть явления, связанные с тепловым и элек-
трохимическим действием электрического тока, проходящего
через слой шлака. Например, в процессах ЭШП шлак вы-
полняет функции нагревательного элемента, а при исполь-
зовании постоянного тока значительное развитие получают
электрохимические эффекты. Шлаки могут выполнять ряд
Других функций в зависимости от специфики конкретных
69
процессов: поддержание заданного теплового режима, обес-
печение хорошей поверхности слитка, стабилизация электри-
ческих дуг и др. Например, в процессе ВДП используется
добавка очень небольшого количества шлака для ассими-
ляции и удаления неметаллических включений.
Важной технологической характеристикой шлака явля-
ется его основность. Мерой основности служит отношение
суммы концентраций основных оксидов к сумме концентра-
ций кислотных, в простейшем случае (CaO)/(SiOs). В ли-
тературе можно встретить большое число других способов
выражения основности, которые могут быть более удобны-
ми в тех или иных случаях. Однако не существует универ-
сальной шкалы основности и в общем случае следует поль-
зоваться по возможности более простыми критериями. Ре-
альные шлаки имеют сложный состав и часто более
основной шлак в ряду, построенном, например, по десуль-
фурирующей способности, может оказаться менее основ-
ным, если критерием основности шлака является активность
ионов кислорода [50]. Более точно зависимость свойств
шлака от состава характеризуют формулы термодинамиче-
ской теории растворов, которые будут рассмотрены ниже.
Основность широко используется в практической метал-
лургии в качестве простой обобщающей характеристики.
Высокоосповные шлаки обладают хорошей сорбционной спо-
собностью по отношению к примесям, жидкоподвижны, га-
зопроницаемы, способствуют стабилизации электрических
дуг. Эти шлаки «короткие», т. е. имеют короткий темпера-
турный интервал перехода от жидкоподвижного состояния
в твердое. Кислые шлаки в тех же условиях более вязки,
обладают хорошими изолирующими свойствами, большим
интервалом перехода от жидкого к твердому состоянию
(«длинные») (рис. 9).
Важнейшими физическими свойствами сталеплавильных
шлаков являются поверхностное натяжение, вязкость, плот-
ность, электрическая проводимость.
Вязкость шлака существенно влияет на скорость массо-
обмена в сталеплавильной ванне, поэтому связана с пока-
зателями всего процесса. Обычные значения вязкости элек-
тросталеплавильных шлаков при 1600 °C находятся в пре-
делах 0,01—0,06 Па-с (вязкость воды при 25°C 0,9 мПа-с,
стали при 1600 °C 25 мПа-с). Вязкость в значительной сте-
пени зависит от температуры (см. рис. 9).
Плотность сталеплавильных шлаков при 1600 °C колеб-
лется в пределах 3,5—2,0 г/см3. Повышенную плотность
имеют железистые и марганцовистые шлаки. При прочих
70
равных условиях основные шлаки имеют несколько более
высокую плотность, чем кислые. С повышением температу-
ры плотность расплавленных шлаков уменьшается пример-
но на 0,2 г/см3 на каждые 100 °C.
Электрическая проводимость шлаков зависит от их со-
става. Обычно электрическая проводимость шлаков при
1600°C находится в пределах
0,0001—1Ом-1-см-1. Более электро-
проводны основные шлаки. Осо-
бенно сильно увеличивают электри-
ческую проводимость шлака добав-
ки оксидов щелочных металлов и
галогенидов, в частности плавико-
вого шпата. Электрическая прово-
димость, как и жидкоподвижность,
экспоненциально увеличивается при
повышении температуры: х=ЛХ
^е-Е/ят, где д — постоянная для
данного шлака; Е — энергия акти-
вации электрической проводимости.
Приведенные данные следует
рассматривать лишь как ориенти-
ровочные, поскольку лабораторные
условия, в которых проводятся-из-
мерения, не всегда полностью вое-
Рис. 9. Зависимость вязко-
сти кислых (/) и основных
(2) шлаков от температуры
производят условия промышленных
агрегатов. Особенно сильно на некоторые свойства шлака
может влиять различие в окислительно-восстановительных
условиях.
11.2. МОЛЕКУЛЯРНАЯ ТЕОРИЯ ШЛАКА
Молекулярная или химическая теория шлака явилась пер-
вой попыткой научного обобщения обширного опыта, на-
копленного практической металлургией. Благодаря просто-
те и наглядности исходных положений этой теории основ-
ные ее принципы широко используются для простых
ориентировочных оценок и в настоящее время. Многие по-
ложения молекулярной теории, касающиеся строения шла-
ка, сугубо условны, поэтому рассмотрим лишь конечные за-
висимости, применение которых в рамках формальной ко-
личественной теории вполне обоснованно. Как обычно,
первым шагом термодинамического описания равновесия
является составление уравнения реакции. Например,
[Мп] + (FeO) = (МпО) + [Fe]. (114)
71
На основании уравнения реакции при помощи идеального
закона действующих масс (ЗДМ) записывается выражение
для константы:
KMn = (MnO)/{(FeO)[Mn]}.
(115)
Специфика молекулярной теории заключается в способе
подсчета «активных» или «свободных» концентраций окси-
дов, входящих в выражение (115). Рассмотрим первона-
чальный, наиболее полный вариант молекулярной теории,
разработанный Г. Шенком с сотрудниками. Структурными
единицами шлака считаются молекулы оксидов (CaO, FeO,
БЮг и др.) и их соединений. Из всего многообразия возмож-
ных соединений рассматриваются лишь пять: (FeO)2-SiO2;
(СаО)з-РезО4; (MnOJa-SiOj; СаО-БЮг; (СаО)4.Р2О5. Этот
набор структурных единиц является минимумом, достаточ-
ным для передачи основных характерных особенностей шла-
ка, определяющих распределение элементов в системе ме-
талл—шлак.
Используя выражения для констант, например формулу
(115) [(FeO) и (МпО)—«свободные» концентрации, опре-
деляемые степенью диссоциации перечисленных выше пяти
«соединений»], на основании обширных опытных данных о
распределении элементов -между металлом и шлаком в ре-
зультате многочисленных пробных расчетов Г. Шенк нашел
численные значения констант распределения и диссоциации,
наилучшим образом удовлетворяющие экспериментальным
данным. Ниже приводятся значения констант, вычислен-
ные Г. Шенком для 1873 К:
Ig^(FeO)t-SiO2
(FeO)?-(SiO2) = j 77.
((FeO)2-SiO2) ’ ’
(MnO)2-(SiO2) 0 70.
's^(MnO),-sio. '& ((MnO)2-SiO2) ’ ’
1g Dc о sio -= (Ca0)' (Si"~ = 0,91;
8 CaOSiO, & (CaO-SiO2)
£FeO = [FeO]: (FeO) = 0,0149;
IgK = 1g (MnF (FeO) =_0 299;
8 xMn 8 (Mn0)
[Si]-((FeO)2 „1>43;
(H6)
(И7)
(H8)
(П9)
(120)
(121)
(122)
Ig^ = lg (Sio2)
lg/< = 1g ISPb(FeO)s(CaO)«
(SP2Os)
4- 0,060 (SP2O8) = 7,42.
72
Обычно принимают, что феррит кальция диссоциирует
полностью, а фосфат кальция не диссоциирует совсем. Оче-
видно, что, располагая этими значениями констант, по за-
данному составу шлака можно с определенной степенью
достоверности предсказать распределение элементов в ус-
ловиях, близких к тем, для которых были определены эти
значения. Таким образом, рассмотренная теория суммиру-
ет в терминах условной молекулярной модели определен-
ный практический опыт и поэтому позволяет делать обосно-
ванные предсказания. Однако следует отметить исключи-
тельную громоздкость практических расчетов по этой схеме.
Для определения «свободной» концентрации какого-ли-
бо оксида требуется совместное решение большого числа
уравнений, поскольку концентрации всех оксидов и их со-
единений оказываются взаимосвязанными. Представление
об образовании соединений между оксидами оказалось
очень полезным и широко используется при различных ори-
ентировочных оценках. Добавление в шлак основных окси-
дов должно приводить к связыванию кислотных оксидов
(SiOj, Р2О5 и др.) и способствовать переходу соответствую-
щих элементов из металла в шлак. Элементы, образующие
основные оксиды (МпО, СгО и др.), при этом будут полнее
переходить в металл.
Было предложено большое число вариантов этого ме-
тода, различающихся допущениями относительно вероятно-
го молекулярного состава шлака (состав соединений, харак-
тер их диссоциации). В теории, выдвинутой Н. М. Чуйко,
наряду с диссоциацией на оксиды допускается частичная
диссоциация соединений на ионы. П. Герасименко исходил
из полной диссоциации всех соединений на простые и слож-
ные ионы. Подбором соединений и параметров их диссоциа-
ции исследователям, как правило, удается удовлетворитель-
но описывать имеющиеся экспериментальные данные при
самых различных моделях строения шлака.
Отсутствие независимых методов определения фактиче-
ского молекулярного состава шлака не позволяет предпо-
честь какой-либо из указанных вариантов, поэтому ни од-
на из теорий, основанных на подборе молекулярного соста-
ва с последующим его использованием в идеальном ЗДМ,
преимущественного распространения не получила. Вместе
с тем общая схема описания равновесий, развитая на ос-
нове представления о существовании в шлаке молекул ок-
сидов FeO, CaO, AI2O3 и др., введением активностей кано-
низирована и служит основой термодинамики шлаков.
Введение активностей заключается в простой замене «сво-
73
бодных концентраций в выражении (115) активностями со-
ответствующих оксидов: Кмп==«(МпО)/(Я[Мп]й(О)).
В настоящее время накоплен значительный объем экс-
периментальных и оцененных теоретически значений актив-
(Зс0г)+(Рг05)
(Сао)+(мпо)+(мдо) (Feo)
(СйО)+(ПпО)+(МдО), °/о(пол.)
Рис. 10. Активность оксида железа
FeO в шлаке (цифры у кривых —
значения активности FeO в моль-
ных долях)
ностей компонентов главней-
ших шлаковых систем. Наибо-
лее полная сводка таких дан-
ных приводится в справочнике
[2]. На рис. 10 и 11 приведе-
ны диаграммы активностей
МпО и FeO в шлаке по дан-
ным Тэркдогана [52, 53].
Пример. Вычислить содер-
жание кислорода и марганца
в металле (железе), находя-
дящемся в равновесии со шла-
ком следующего состава
(плавка Е-39 из работы [54],
1600°C): 39,94 % СаО; 15,38 %
SiO2; 10,59% FeO; 1,77%
Fe2O3; 5,23 % MgO; 10,60 %
МпО; 16,09 % P2O5; 0,06 % S.
Воспользуемся следующими
выражениями для констант
[52, 53]:
Ко = ; 1g ко = (6320/7) - 2,734;
^0(1873) ~ 4,367;
„ _ [%Мп][%О]. 1 к __ 12760 5
«(МпО) ’ 8 Мп 7
^мп(1873) = 0,074;
(123)
(124)
__ ^(МпО) • \0 К = 7406
Mn/Fe~ «(FeO)l%Mnl ’ МП/1?е Т
KW1B73) = 3,296. (125)
Для определения арео и ампо п0 диаграмме рис. 10 и И
необходимо выразить мольный состав шлака в процентах
(% М^. Находим числа молей (т,) каждого из компонен-
тов в 100 г шлака, например тСа0 =% СаО/Мсао =39,94/
/56=0,713. Суммируем найденные /и,- в нашем примере
Sm(= 1,531. Находим мольные проценты %Л41=100(т1/
I'Lmi). Например: % МСаО = 100-(0,713/1,531) =46,5 %•
74
Мольный состав шлака следующий*, %: 46,5 СаО; 16,7 SiO2;
11,0 РеОобЩ; 8,5 MgO; 7,4 P20g; 9,7 МпО. Суммируем кон-
центрации основных и кислых оксидов, согласно размерно-
сти координат диаграмм: % МСао + % MMgo + % Ммпо=
= 64,7 %; % Мсао + % AfMgo = 55,0 %; % ^siq, +
+ % -Мрао, =24,1 %. По диаграммам находим йрео =0,50,
смпо =0,260. Содержание кислорода в металле по уравне-
нию (123) составляет [О](12з)=0,50/4,367=0,114 %; по
(Si02)+(P205)
Рис. 11. Активность оксида
марганца МпО в шлаке (циф-
ры у кривых — значения актив-
ности МпО, %):
л — Мред —0,05±0,01; б —
MFeO “0.10±0,0»6; =
-0,20±0,02
15
W
15 70 65 60 55
(СаОЙ(мдо) (rW
' (СаО)ЧМдО),°ЛИг/7.)
(SiO2)+(P2O5)
чО
10
10
15
го
25
50
55
40
*45
-50
55 -
70 65 60 55 50
(СаО)+(мдо) (МпО)
10 65 60 55
(Сао)+(мдО) (МпО)
(СаО)+(МдО);%^77.}
уравнению (124) [О](124)= (0,074-0,260)/0,212=0,090 %. Со-
держание марганца по уравнению (124) составляет
[Мп](124>= (0,074-0,260)/0,082 = 0,234 %, по уравнению
(125) [Mn](i25)=0,26/(0,5-3,296) =0,157 % • Отклонения вы-
* РеОобщ в исходном шлаке вычисляется по формуле: % РеООбщ=
= % FeO+ (72/80) % Fe2O3.
75
численных значений от экспериментальных [0]ЭКсп=0,082 %,
[Мп] эксп — О,212 % не превышают 25%. Такое согласие в
расчетах равновесий металл — шлак считается хорошим.
11.3. ТЕОРИЯ СОВЕРШЕННЫХ ИОННЫХ РАСТВОРОВ
Естественным путем развития количественной теории был
переход от молекулярной модели к ионной как более со-
вершенной. Однако уже первые работы [55, 56] показали,
что простая замена концентраций оксидов концентрациями
ионов в ЗДМ не приводит к заметному улучшению теории
[57, 58]. Причной этого является ограниченная примени-
мость идеального ЗДМ к таким системам, как шлак. Наи-
более обоснованный путь использования данных о строении
взаимодействующих фаз при расчете равновесий указыва-
ет статистическая физика. ЗДМ является частным решени-
ем статистической задачи об энергии системы химически
инертных частиц, поэтому он хорошо описывает поведение
газов. Для конденсированных фаз ЗДМ строго выполняет-
ся лишь в некоторых предельных случаях. Частицы, обра-
зующие такие фазы, сильно взаимодействуют, что приво-
дит к новым закономерностям. Первая удачная попытка
применения ионной модели для статистического вычисле-
ния термодинамических функций шлака принадлежит
М. И. Темкину [59]. При статистическом подсчете энтропии
ионного раствора М. И. Темкин предположил, что раствор
полностью диссоциирован на ионы, ионы одного знака
энергетически эквивалентны, в процессе теплового движе-
ния возможны перестановки только между ионами одного
знака. Подсчет конфигурационной (перестановочной) эн-
тропии ведется по формуле Д5=7? In W. Вероятность W
(полное число вариантов пространственного размещения
частиц раствора, возникающих при их взаимных переста-
новках) оказывается равной произведению соответствую-
щих величин для'катионов и анионов: W=WK-Wa. При
этом в выражении для химических потенциалов компонен-
тов шлака, например для CaS, растворенного в шлаке,
•UCaS ~ ^Cas ^CaS ^^CaS’
~™SCaS = RT ln(xCa-xs), (126)
где x$=ns/Zna-, xCa — nCa/ZtiK; nCa; ns—числа конов каль-
ция и серы в расплаве соответственно; Sna и SnK — об-
щее число анионов и катионов. Величины х, получили на-
звание ионных долей по Темкину. Принимая AHi = 0, полу-
76
чаем pCaS = |x£aS + /?7’ln(xCa-xs), откуда находим оконча-
тельное выражение для активности: acas=*ca,*s-
Как показали Л. А. Шварцман и А. М. Самарин [60, 61],
теория совершенных ионных растворов вполне применима
для расчета распределения серы между металлом и силь-
но основным шлаком. Сравнительно небольшие содержания
оксидов кремния, алюминия и фосфора вызывают сущест-
венные расхождения теоретических и экспериментальных
данных. В практических расчетах равновесий теорию со-
вершенных ионных растворов применяют сравнительно ред-
ко, однако ее значение для теории шлаков исключительно
велико. Метод подсчета энтропии, предложенный М. И. Тем-
киным, характеризует важную особенность подобных рас-
плавов и используется в более полных статистических тео-
риях, в частности в теории регулярных ионных растворов
В. А. Кожеурова.
11.4. ТЕОРИЯ РЕГУЛЯРНЫХ ИОННЫХ РАСТВОРОВ
В отличие от обычной теории регулярных растворов
(см. гл. I) энтропия в регулярном ионном растворе не счи-
тается идеальной, а вычисляется по методу Темкина, как
было описано выше. Вторым важным моментом теории Ко-
жеурова является то, что шлак рассматривается как сис-
тема простейших атомных ионов. Это приводит к важному
результату, заключающемуся в том, что в выражение кон-
станты равновесия концентрации (или активности) распре-
деляющегося элемента, относящиеся к металлу и шлаку,
входят в одинаковой степени, что более правильно отражает
действительность. Шенк постулировал такое написание кон-
станты для фосфора, ссылаясь на опыт. Авторы работы
[62] пытались обосновать одинаковую степень активности
различными предположениями о формах существования
фосфора в контактирующих фазах.
К наиболее простым зависимостям теория регулярных
ионных растворов приводит в случае растворов с общим
ионом, какими являются, например, чисто оксидные шла-
ки. Единственным анионом в последних оказывается ион
кислорода О2-, поэтому конфигурационная энтропия ани-
онной подсистемы равна нулю. При этом отличие получае-
мых конечных формул от обычных формул теории регуляр-
ных растворов состоит лишь в замене концентраций, выра-
женных в атомных долях, концентрациями катионов по
Темкину (х(). Так, химический потенциал FeO в бинарном
77
расплаве FeO—МпО определяется выражением:
f*FeO — ^FeO 4" ’n ^Fe 4" ^Mn ^Mn— Fe’
(127)
Где XFe=nFe/(nFe + ПМп); *Mn = «Mn/(«Fe + «Мп) — КЭТИОННЫе
доли по Темкину.
Сравнивая выражение (127) с термодинамическим опре-
делением активности:
= Н? + PT In at =
(128)
можно найти выражение для коэффициента активности FeO
в шкале:
' '(Fe) Мп ^Fe—Мп*
(129)
Кожеуров обобщил теорию для шлака произвольного со-
става:
PT In T(Fe) = xi 4" 2 xi Qh xi xi Qi'
i=i i=z+i f=i /=i+i
(130)
Численные значения энергий смешения Qi/, необходи-
мые для вычисления активностей, находятся обратным рас-
четом по имеющимся опытным данным. Так, исходя из экс-
периментальных данных [54], для шлака, состоящего из
FeO, МпО, CaO, MgO, SiO2, Р2О5, В. А. Кожеуров рекомен-
дует следующие значения энергий смешения (индексы со-
ответствуют номерам оксидов в приведенном порядке запи-
си): Q25=—42 кДж, Q35==Q45=—ИЗ кДж, Q36=
=—201 кДж, остальные Q,;=0. Из данных [54] получены
значения констант:
м»=пГГ--—; ^№=-Т—3-12;
Мп [Мп] xFe ?(Fe) Т
ъ 2
-----р--=2,29.10-2.
(131)
[PJ2 *Fe T(Fe)
(Кр мало изменяется с температурой);
= ->т(ре); lgKn= —--------2,724.
0 [О] [О] & ° Т
Подставляя приведенные выше значения Q/у в уравне-
ние (130), получим выражения для коэффицентов актив-
78
IgT(Fe) ~ (1000/T) [2,18xMnxsi + 5,90 (xCa + xsl +
+ 10,50xCaxpl; - (132)
IgY(Mn) = IgT(Fe) -(2180/T)xSI; (133)
lg ?(₽) = fe ?(Fe) - (10500/7) xCa. (134)
Пример. Определим содержание кислорода, марганца и
фосфора в металле по исходным данным предыдущего при-
мера. Выражаем состав шлака в числах молей на 100 г
шлака, но в отличие от предыдущего примера пренебрегаем
содержанием РегОз в шлаке:
Оксид . .........FeO МпО CaO MgO Р2О5 S1O2
Число молей на 100 г
шлака ........... 0,147 0,149 0,713 0,131 0,113 0,256
Затем подсчитываем общее число молей катионов в
6
100 г шлака*1:^ г+=0,147+0,149+0,713+0,131+2-0,113+
z=i
+ 0,256=1,735 и определяем ионные доли катионов, напри-
мер хре=0,147/1,735=0,085.
Катион ..........
Fe Мп Са Mg Si Р
0,085 0,086 0,410 0,076 0,148 0,130
Вычисляем коэффициенты активности FeO, МпО, Р2О5.
Коэффициент активности FeO при 1600 °C вычисляем по
уравнению (132)'; lg y<Fe)= 1,17*0,086-0,148+3,15*0,410+
+ 0,076*0,148+ 5,60*0,410*0,130 = 0,540; у(ре)=3,467. Коэф-
фициент активности МпО вычисляем по формуле (133):
lgТ(Мп)= 0,540—1,17*0,148=0,367; у (Мп)= 2,328. Коэффици-
ент активности Р2О5 вычисляем по уравнению (134):
1 g у (Р)=0,540—5,60• 0,410=—1,756; у (Р) = 0,018.
Решая уравнения (131) относительно [Мп], [Р], [О] и,
подставляя значения х» и yi получам:
[Мп] 0>086‘2>328--------—0,201 %(0,212%);
3,37-0,085*3,467 v ’
0,130-0,018
[Р]= 7---------7wr~U,V1^--------«Г = 0,324% (0,111 %);
(2,29-10~2)1/2-0,0855/2-3,4675/2 '
[О] = °>08^3,467 = 0>065о/о (0,082%).
♦! V,- — число атомов металла i в «молекуле» оксида.
79
В скобках приведены опытные значения*1.
Как и метод Шенка, теория регулярных ионных раство-
ров является по существу интерполяционным методом. Рас-
чет ведется на основании коэффициентов (энергий смеше-
ния), найденных из имеющихся экспериментальных данных
по равновесию в рассматриваемой системе. Однако функ-
циональная связь между составом и термодинамическими
характеристиками в теории регулярных ионных растворов
устанавливается несколько более обоснованным способом,
поэтому точность и надежность предсказаний оказывается
выше.
11.5. ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ ШЛАКА
как фазы, имеющей коллективную
ЭЛЕКТРОННУЮ СИСТЕМУ
Традиционный подход к описанию равновесий основывает-
ся на представлении о шлаке, как взаимном растворе раз-
личных химических соединений — FeO, Ре20з, CaS, CaSiO<
ит. п. Непременным свойством химического соединения,
как индивидуального, так и растворенного в шлаке, считает-
ся определенный состав, строго соответствующий стехио-
метрической формуле и не зависящий от внешних условий.
Записывая выражение закона действующих масс через кон-
центрации (активности) таких соединений, мы, кроме того,
постулируем их существование в виде соответствующих мо-
лекул, совершающих самостоятельное тепловое движение
(это следует из статистического вывода ЗДМ). В действи-
тельности конденсированные оксиды не только не содержат
обособленных молекул, но и стехиометрические пропорции
в них соблюдаются лишь приближенно. Стехиометрические
фазы строго фиксированного состава являются абстракци-
ей и возможны лишь при абсолютном нуле. Нагревание
приводит к частичной или полной дезорганизации струк-
туры (структурный беспорядок) и способствует нарушению
стехиометрических пропорций (нестехиометрия). Изучение
этих явлений связано с именами крупнейших химиков — Бер-
толле, Менделеева, Курнакова, Френкеля, Вагнера, Шот-
тки и др. Однако лишь исследования электронного строения
таких фаз в последние десятилетия позволили более полно
описать закономерности их равновесного существования и
*’ Экспериментальные данные по фосфору характеризуются повы-
шенным разбросом, поэтому полученное расхождение не следует счи-
тать слишком большим.
80
открыли возможность создания последовательной термоди-
намической теории конденсированного состояния, об успе-
хах которой уже упоминалось.
Оксид МеОх, находящийся в равновесии с окружающей
средой, в общем случае хотя бы в малой степени нестехио-
метричен. Независимо от механизма этого явления, изме-
нения состава окружающей среды сопровождаются соот-
ветствующими изменениями состава оксида. Из этого сле-
дует, что термодинамически он может рассматриваться как
раствор металла и кислорода. Если кристалл MgO находит-
ся в равновесии с собственным паром, то химические потен-
циалы кислорода и магния в обеих фазах равны:
hwg = ^(Mg) > (155)
Но ~ Н(О) • (136)
Химический потенциал кислорода в газовой фазе можно
выразить через парциальные давления различных молеку-
лярных составляющих газа:
Но = Й + RT In РО. (137)
ИЛИ
Ро = М>, + |п/>£• (138)
Как и большинство других оксидов, MgO устойчив в
широком диапазоне давлений кислорода, причем изменения
состава во всей области гомогенности настолько малы, что
обнаруживаются лишь специальными методами и совер-
шенно недоступны для обычных методов аналитической хи-
мии. Однако, как бы малы ни были изменения состава, они
оказываются достаточными для того, чтобы поддерживать
равновесие при любых изменениях давления кислорода в
пределах устойчивого существования оксида. Если выра-
зить химический потенциал кислорода в оксидной фазе че-
рез активность
Н'(О) = ^(О) “l* ВТ Ina^Qy (139)
то, приравнивая правые части уравнения (139) и выраже-
ний (137), (138) на основании уравнения (136), можно по-
казать, что активность кислорода в оксиде следует за из-
менениями ро, и может изменяться в широких пределах:
G(O) *оРо- *оЛС Здесь для постоянных введены обо-
значения
’‘О “ Ц(О) ~
Ло = е rt ; К =е-------------«г----. (140)
6—675
81
Изменения активности кислорода в конденсированном
оксиде сопровождаются интенсивными изменениями актив-
ности металлического компонента, в данном случае маг-
ния. Из уравнения Гиббса—Дюгема следует, что
^Mg= (^o^Mg) ^(О) > xo!xt\g 1* (141)
Выразив химические потенциалы через соответствующие
парциальные давления можно получить рм т. е.
активности и равновесные парциальные давления компонен-
тов оксидной фазы могут изменяться в самых широких пре-
делах при практически неизменном составе фазы. Приве-
денные рассуждения можно повторить для фазы сложного
состава и шлакового расплава. Рассмотренный пример ил-
люстрирует важную особенность оксидных фаз, заключаю-
щуюся в том, что активности их компонентов, как и многие
физические свойства (характер и величина электронной про-
водимости, каталитическая активность, транспортные и ад-
гезионные характеристики и др.), могут изменяться в самых
широких пределах при практически неизменном составе
оксида (шлака). Состояние оксидной фазы, а значит и весь
комплекс ее физико-химических свойств не могут быть пол-
ностью охарактеризованы указанием лишь температуры,
давления и обычного химического состава. Необходимы до-
полнительные данные, фиксирующие уровень окислительно-
восстановительного потенциала.
Удобный критерий окислительно-восстановительного по-
тенциала указывает теория электронного строения конден-
сированных веществ. Согласно современным представлени-
ям [63, 64], электронные уровни (электронные состояния)
всех атомов, образующих фазу, составляют единую кванто-
механическую систему. Термодинамически эта система ха-
рактеризуется парциальной энергией Гиббса электронов р,
(уровнем Ферми)*1, являющейся наиболее общей количе-
ственно мерой окислительно-восстановительного потенциа-
ла. Уровень Ферми р, имеет простой физический смысл —
это работа, которую нужно затратить, чтобы ввести один
электрон в коллективную систему фазы (или удалить — с
обратным знаком). В количественных расчетах будем от-
носить р к молю (6,03-1023) электронов. Уровень Ферми в
общем случае может быть вычислен методами квантовой
статистики на основании данных об энергетическом (элек-
тронном) спектре конкретного вещества. Ограничимся вве- **
** Обозначение р (без индекса) для уровня Ферми («уровня хи-
мического потенциала») было введено А. Ф. Иоффе [65].
82
дением р в термодинамические формулы лишь на чисто
формальном уровне. Это позволит проследить возможности
метода.
В работе [66] показано, что полное выражение для хи-
мического потенциала компонента*1 i оксидной фазы
должно содержать член, учитывающий характеристики
электронной системы фазы: р,— р°+RT In a'tчлен
RT In а'{ определяется лишь номинальным*2 составом фазы
и не зависит от окислительно-восстановительного потенциа-
ла системы. Штрих в обозначении а/ введен для того, что-
бы подчеркнуть, что а'{ представляет лишь часть полной
активности элемента i. Член р,(р) в простейшем случае
может быть представлен [66] произведением валентности
элемента i в оксидной фазе vf и химического потенциала
электронов:
= + + (142)
Ниже будут рассмотрены конкретные приложения этой
формулы. Для этого сначала выразим р через измеримые в
опыте величины.
Уровень Ферми в оксидных фазах. Очевидно, что газо-
вая фаза, находящаяся в равновесии с оксидной, всегда со-
держит кислород, поэтому уровень Ферми удобно выразить
через равновесное парциальное давление кислорода как
величину, доступную для прямых измерений. В условиях
равновесия химический потенциал кислорода в шлаке ра-
вен химическому потенциалу кислорода в газе: Р(О)=Ро-
Химический потенциал атомарного кислорода в газовой фа-
зе p0=|Aq+/?T In ро и в шлаке
P(O) ~ Il(O) “1" В? 1° a(O) “I" V(O) !*•
(143)
Приравнивая правые части и преобразуя, получим вы-
ражения для зависимости р от давления кислорода:
р = ро _ RT in a4/v(0) + RT In pJJv(o>; f p° = •
(144)
Для фазы, мало изменяющей состав в широком диапа-
*’ Компонентами оксидной (как и металлической) фазы здесь и
далее считаются элементы Периодической системы элементов Д. И. Мен-
делеева.
*2 Т. е. обычным составом, например, по данным химического ана-
лиза.
6*
83
зоне давлений кислорода, a'Q = const и выполняется соот-
ношение
р, = А — RT In ptf. (v(O)=—2; А = const), (145)
или, используя известную связь между равновесными дав-
лениями ро и ро, в газовой фазе, можно р выразить через
давление двухатомного кислорода:
р = Л -RTlnp'tf.
(146)
Сорбционная способность шлака по отношению к при-
месям. Если газовая фаза, равновесная со шлаком, содер-
жит малорастворимый в шлаке газ i, то, записав условие
равновесия р(1)=ц,- и выражения для химических потенциа-
лов элемента i в газе и в шлаке, после преобразований мож-
но получить выражение растворимости (равновесной кон-
центрации) газа в шлаке:
хт = 1<,Р1Р^‘-
(147)
Если исходить из парциального давления двухатомного
газа, то получим аналог закона Сивертса для шлаков:
,п = К, р№ pvi'\
(») »г к1г J ГО,
(148)
Специфика оксидных фаз проявляется здесь в наличии
V4 , V./2
дополнительного множителя ро, (или Я[о])-
Этот результат наглядно иллюстрируется, например,
опытом насыщения шлака азотом. Шлак системы СаО—
Рис. 13. Сорбционная способность шлака по отношению к примесям
SiOg—AI2O3 выдерживается
при постоянной температуре в
атмосфере с Pn,=const и из-
0,001 0,01 0,1 Ю]?ъ{раств,)}°1о
Рис. 12. Зависимость растворимости азота в шлаке от окислительно-восстанови-
тельного потенциала
И
ней
(%0/[%П в различных окислительно-восстановительных условиях (в верх-
части рисунка дана ориентировочная шкала окисленности железа)
£4
меняющимся рог. В соответствии с обычным законом Си-
вертса в этих условиях следовало бы ожидать постоянства
содержания азота в шлаке. В действительности х<ы) изме-
няется в широких пределах вместе с равновесным давлени-
ем кислорода в полном соответствии с формулой (148)
(рис. 12) {67]. Наклон прямых в таких координатах опре-
деляется валентностью примеси, поэтому подобные опыты
могут использоваться как экспериментальный метод опре-
деления валентности примесей в шлаках.
Сорбционная способность шлака по отношению к при-
месям зависит и от номинального (аналитического) соста-
ва шлака. Однако один и тот же шлак может изменять
свою сорбционную способность на несколько порядков ве-
личины в зависимости от окислительно-восстановительного
потенциала среды. Очевидно, что зависимость, аналогичная
выражению (148), получится, если поддерживать постоян-
ной концентрацию (активность) примесного элемента в
контактирующем со шлаком металле:
*.-= Мо = Мп (149)
На рис. 13 приведены кривые зависимости сорбционной
способности шлака 50 % СаО, 50 % А120з от рог при 1550 °C
по отношению к различным элементам. Ki вычислены по
данным различных авторов, ащ отнесены к активности эле-
мента i, соответствующей концентрации его в железе, рав-
ной 1 % (стандартное состояние— 1 %-ный раствор i в же-
лезе) .
11.6. РАСПРЕДЕЛЕНИЕ ЭЛЕМЕНТОВ
МЕЖДУ МЕТАЛЛОМ И ШЛАКОМ
Если две или несколько фаз находятся в равновесии, то все
элементы, образующие данную систему, содержатся в каж-
дой из фаз. При этом для любого элемента должно выпол-
няться условие равновесия: Ц(о=|1[»]- Химический потен-
циал элемента i в шлаке, согласно выражению (142), равен:
а(о + т< И- Для металла будем исходить из
выражения р[(] = ц^]+/?Г In a[tj. Выполнив преобразования,
получаем выражение для константы распределения эле-
мента i между металлом и шлаком:
<150>
Чтобы исключить множитель, содержащий неконтроли-
руемую обычным химическим составом величину ц, возве-
дем выражение (150) в степень 1/v/ и поделим на аналогич-
85
ное выражение для любого другого компонента (/):
^i/i ~ (аи^аИ)У^‘ (151)
Полученное выражение характеризует константу рас-
пределения элементов i и / в системе металл — шлак. Обыч-
но выражения констант получаются из рассмотрения тех
или иных реакций и ЗДМ. Написание реакции требует
знания конкретных соединений, находящихся в шлаке.
Однако отсутствие такой информации во многих случаях
существенно усложняет задачу и вносит элемент неопреде-
ленности. Рассматриваемый путь позволяет избежать этой
трудности.
При описании распределения фосфора в уравнении (151)
вторым элементом (/) может быть любой из присутствую-
щих в системе, например кислород:
^р/о = (a<P)^a[P])1/5 (a(o/a[O] )1/2
vo=—2; vp=4-5).
Это соответствует Кр, полученной из балансового уравне-
ния окисления фосфора, записанного в следующем виде:
’/б[Р] + VsfO] == Р1/5О1/2. Если j = Fe, то KP/Fe = («<Р)/
/ц[Р])1/5 (a[Fej/a(Fe))1/2 (vpe =+2), что соответствует реак-
ции обмена 7s [Р]4~72 (Fe) =75 (Р) +7г [Fe], или Vs [Р] +
+ 72 (FeO) =7б (РО2,5)4-72 [Fe] и т. д.
Оценка активностей. Активности элементарных компо-
нентов шлака а'{ входящие в приведенные выше формулы,
эквивалентны активностям оксидов по Кожеурову а' =
=Xi-yi или по Темкину ащ =xi. Оба эти метода могут быть
использованы для оценки активностей катионных
>0) компонентов шлака. В качестве а'{ могут быть также
использованы экспериментальные значения активностей
соответствующих оксидов.
Метод теоретической оценки активностей компонентов
шлака, полностью обеспеченный численными параметрами,
приводится в работе [66]. Согласно этому методу, актив-
ность компонента шлака выражается формулой а'} ~
—хщф,- где хи) — полная аналитическая концентрация эле-
мента i в шлаке, выраженная в атомных долях; ф(- — атом-
ный коэффициент активности, равный
(152)
/=1
86
k— полное число компонентов шлака (число сортов ато-
мов); ex, — энергия обмена, равная ец—'к (хИ2— х//2)»
%; — атомные параметры элементов i и /. Значения х
[66] приведены в табл. 10. Формула (152) учитывает лишь
конфигурационную энтропию смешения, однако, как пока-
зали многочисленные расчеты, она дает правильные оценки
равновесий в системе металл — шлак.
Т а б л и ц а 10. Энергетические параметры элементов
Элемент X Элемент X Элемент X Элемент X
н 448 Мп 251 Cd 280 Ac 117
Li 88 Fe 335 Sn 280 U 88
Be 218 Co 280 Sb 310 Pu 163
В 197 Ni 464 Те 427 Am 163
С 29 Cu 418 I 653 Cm 163
N 732 Zn 243 Cs 54 Bk 163
О 1255 Ga 247 Ba 79 Cf 163
F 1544 Ge 385 La 138 Ce 138
Na 79 As 385 Hf 188 Pr 138
Mg 146 Se 556 Ta 113 Nd 163
Al 126 Br 812 W 238 Pm 163
Si 172 Rb 59 Os 427 Sm 163
P 205 Sr 96 Ir 427 Eu 163
S 791 138 Pt 469 Gd 163
Cl 992 Zr 226 Au 510 Tb 163
К 63 Nb 280 Hg 314 Dy 163
Ca 105 Mo 276 Bi 314 Ho 163
Sc 163 Ru 385 Po 385 Er 163
Ti 134 Rb 427 At 469 Tu 163
V 184 Pd 427 Fr 46 Yb 163
Cr 251 Ag 360 Ra 79 Lu I 163
Пример. Рассчитаем содержание примесей в металле по
исходным данным примера, рассмотренного ранее (с. 79).
Для этого сначала выразим состав шлака в атомных долях.
Определяем числа молей оксидов в 100 г шлака (mi). На-
пример, /псао = % СаО/Мсао =39,94/56,1=0,713. Находим
Далее число молей элементов в 100 г шлака (ni), например
Пса= /ПсаО = 0,713; «Fe = wFeO 4" 2 ^Fe,O3 = 0,147 -р 2 X
X 0,011 = 0,169; «о = тСао -\-2т5Ю, + mFe0 + 3mFflo3 +
+ wMgo+5mPjOt+mMnO =2,250. Находим сумму всех пг- —
общее число молей в 100 г шлака. Sn=3,894. Вычисляем
атомные доли элементов: Х(о=п»72п; x<Fe) =0,169/3,894=
=0,043. Для вычисления атомных коэффициентов активно-
сти -ф* сначала оцениваем величины ег/. Например, epe-si =
87
= ss._Fe = 72(х^-Я^)г=,/2(335’/2-172’/2)2 = 13,509 кДж;
еСа_Р = еР_са =1/2(1051/2—205’/2)2 = 8,368 кДж; Затем на-
ходим множители e~(eij,RT’t
£~"eFe—Si/ЯГ _ ^—13.509/8,31.10“3.1873 __ g“0’ 8716 = 0,4179,
g“eCa—Р/КГ _ £-8,368/8,31.10“3.1873 _ 0,5821
И, наконец, находим ф/. Например,
= 2х</> = *(С<) +
/=1
+ X(si) e-eFe-Si/7?7+...+ Х(О) e“£Fe-O/KT =
= 0,183 -0,1218 + 0,066.0,4179 + 0,043 - 1,000 +
+ 0,034-0,2858 + 0,058-0,6012 + 0,038-0,821 +
+ 0,578 -0,000 = 0,1699. фРв = 1/0,1699 ••= 5,886.
Таблица 11. Состав шлака и валентность элементов
Элемент xi vi a(Z) Элемент xi V. 1 “(/)
Са 0,183 2 3,154 0,577 p 0,058| 5 3,139 "0,182
Si 0,066 4 2,919 0,193 Mn 0,0381 i 2 3,736 '0,142
Fe 0,043 2 5,886 0,253 0 0,578 -2 1,7301 "1,000
Mg 0,034 2 2,873 0,098
Примечания, х. — выражено в атомных яслях.
В табл. 11 приведены вычисленные таким образом зна-
чения ф* и «(о=х(>)'Ф‘, а также вычисленный элементный
состав шлака и типичные для условий сталеварения валент-
ности элементов. Значения констант Kt/j могут быть взяты
из термодинамических справочников или оценены по имею-
щимся данным о распределении элементов. Распределение
кислорода в соответствии с уравнением (151) описывается
выражением:
^о// —
'-1/2 1/V, (1/2 1/V i
С(О) а[/]2 _ (а[О] а[/з °)
a-ip.'1/Vj '1/2 '1/v,
а[О] а(/) } а(О) а(П 3
(153)
В качестве второго элемента j в рассматриваемом при-
мере могут быть использованы железо, марганец и фосфор,
88
поскольку известны концентрации этих элементов в обеих
фазах. Учитывая, что й(0) = 1 (см. табл. 11), п[О]«[0]и
O[Fe]=l, получим
^O/Fe ~
„1/2 .„1/2
Q[O] “(Fe) _
'1/2 '1/2
°(O) “(Fe)
[01 \
а№)/
(154)
Константу Ko/Fe можно определить, используя, напри-
мер, известные данные о равновесии железа с собственными
оксидами. Согласно данным работы [2], [О]187з=0,23 %.
Подставив эти данные в выражение (154), находим K'o/Fe =
=0,23/1=0,23. В рассматриваемом примере [O]=Ko/Fe-
•a(Fe) =0,23-0,253=0,058 %. Вычислим концентрацию кис-
лорода по данным о распределении марганца:
К = “[О]‘“[Мп] . к _ [01-[Мп]
^О/Мп *1/2 *1/2 ’ 'О/Мп *
fl(O) "“(Мп) “(Мп)
Учитывая, что аМпО=«('мп)^о^мп). Для
Ко/мп воспользуемся данными работы [2]:
„2 _ г _ [%О][%Мп]
^О/Мп '
1g ^О/Мп
(155)
определения
(156)
Mn “(МпО)
= (— 12760/7") + 5,68;
мп(1873) » ^о/мп = 0.0737; [%О] = -^^> =
0,0737-0,142
0,212
= 0,049%.
При помощи этого уравнения, очевидно, можно вычис-
лить концентрацию марганца по заданному содержанию
кислорода в металле:
[Мп] — ^О/Мпа(Мп) _ 0,0737-0,142 __ q 128%
[%О]
0,082
Рассмотренные примеры иллюстрируют основные прин-
ципы всех разработанных в настоящее время практических
методов расчета равновесий. Опыт показывает, что для
большинства массовых технологий и методов производства
существующая теория позволяет делать обоснованные
предсказания с достаточной для практики степенью точ-
ности.
89
Глава III
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
В СТАЛЕПЛАВИЛЬНЫХ ПРОЦЕССАХ
Поверхностные явления играют огромную, а во многих слу-
чаях решающую роль в процессах производства и обработ-
ки стали и сплавов [69—74]. Непосредственно с поверхно-
стными характеристиками границы раздела фаз связаны
процессы зародышеобразования при кипении и дегазации
металла, зарождения, укрупнения и удаления неметалличе-
ских включений. Смачивание и растекание в системе ме-
талл— шлак определяют такие важные технологические-
характеристики, как прилипаемость, отделение металла от
шлака, их проникновение в капиллярно-пористые материа-
лы, пропитку огнеупоров и др. Способность к ценообразо-
ванию и устойчивость шлаковой пены связаны с образова-
нием при поверхностном кипении на пузырях СО прочных
адсорбционных плевок.
111.1. ОСНОВНЫЕ ПОНЯТИЯ
Поверхностное натяжение. Особенности границы раздела
фаз проявляются в том, что свойства фаз вблизи поверхно-
сти существенно отличаются от свойств в объеме. Это отли-
чие связано прежде всего с тем, что энергия молекул на
границе отличается от энергии молекул в объеме. Межча-
стичные силы, действующие на молекулы находящиеся в
объеме, уравновешены, в то время как силовые поля моле-
кул на поверхности несимметричны. Оба типа молекул име-
ют различные координационные числа, различный характер
теплового движения. Для описания термодинамических
свойств системы с учетом эффектов, связанных с наличием
поверхности раздела фаз, Гиббс ввел понятие поверхност-
ного сгущения. Поверхностное сгущение свободной энергии
характеризует рост свободной энергии системы при увели-
чении межфазной поверхности на единицу. Известно, что
для того, чтобы создать или увеличить поверхность разде-
ла фаз, нужно затратить работу. Величина, численно
равная работе обратимого изотермического образования
единицы поверхности, называется коэффициентом поверх-
ностного натяжения |о|. В этом случае поверхностное на-
тяжение имеет размерность работы (джоуль) на единицу
поверхности: |о| =Дж/м2 = 103 мДж/м2. Часто коэффици-
ент поверхностного натяжения определяют как силу, дей-
90
Рис. 14. К вычислению работы ад-
гезии
ствующую на единицу длины. В этом случае [о]=Н/м=
=Дж/м2.
Работа адгезии. Для того чтобы разделить две несме-
шивающиеся жидкости (металл—шлак) по поверхности
их соприкосновения, равной, например, 1 см2 (рис. 14),
нужно затратить работу на со-
здание двух новых поверхно-
стей жидкость — газ по 1 см2,
равную 014-02 (где oi и <т2 —
поверхностные натяжения обе-
их жидкостей на границе с
газом). Однако в результате
такого разобщения фаз ликви-
дируется поверхность их со-
прикосновения, в результате
чего выигрывается работа, рав-
ная межфазному натяжению
01,2- Работа адгезии (U7a) равна №a=oi + o2—01,2- Чем боль-
ше работа адгезии между металлом и шлаком, тем сцепле-
ние двух фаз больше, т. е. тем труднее отделить металл от
шлака. Условием полной смешиваемости двух жидкостей
является №а=0. В этом случае межчастичные силы каждой
из жидкостей не препятствуют смешению, так как каждая
из жидкостей притягивает молекулы другой с той же силой,
что и собственные молекулы.
Работа когезии. Работой когезии (№к) называют рабо-
ту разрыва столба жидкости с поперечным сечением в
1 см2. В этом случае работа, равная 2ож_г, затрачивается
на создание двух новых поверхностей: 1^к=2ож_г. Сопо-
ставление работы адгезии и когезии позволяет судить о со-
отношении сил притяжения между частицами различ-
ных фаз.
Адсорбция. Изменение поверхностного натяжения ме-
таллического или шлакового расплава под действием до-
бавки одного из компонентов связано с изменением состава
поверхностного слоя, который обогащается i-тым компо-
нентом, характеризующимся более слабым силовым полем.
Рассмотренное явление принято называть адсорбцией Г.
Адсорбцию на поверхности раствора обычно вычисляют по
формуле Гиббса, моль/см2:
Г,=——. (157)
RT det
Для растворов, которые нельзя отнести к классу разбав-
ленных, рекомендуется уравнение Гугенгейма — Адама, в
91
котором в отличие от уравнения (157), где Г1=0 для двух-
компонентной системы Г1 = Г2
Г- = . — (158)
* RT ’ dat ‘ '
Пример. Вычислить величину адсорбции серы на поверх-
ности жидкого железа при 1873 К по экспериментальным
данным зависимости поверхностного натяжения а расплав-
ленного железа от концентрации серы [S]:
[S], % • 0 0,03 0,09 0,14 0,17 0,22 0,33
о, мДж/м2 . . . 1790 1410 1210 1115 1090 1045 980
По этим данным строим график зависимости поверхно-
стного натяжения от состава. Затем проводим касательные
к кривой и находим тангенсы угла наклона в заданных точ-
ках 1 [S] =0,05 %; 2 [S] =0,10 %; 3 [3] =0,15 % и 4 [3] =
=0,25%. Они соответственно равны tg«i=3700; tg«2=
=2100; tga3=1400; tg «4=620. Величина адсорбции в
каждой точке равна Г1= (0,05 -3700) /(8,31 ♦ 103-1873) =
= 1,19-10-5 моль/м2; Г2=1,28-10-5; Г3= 1,35-10~5; Г4=
=0,99-10~5 моль/м2. Адсорбция Г$ с увеличением концен-
трации серы растет и при некотором значении [S] дости-
гает максимума (Fsmax). С последующим увеличением
[S] численное значение Ts снижается. Это связано с тем,
что уменьшение d^fdc опережает рост концентрации [S].
Из уравнения (157) следует, что если добавка компонента
к расплаву приводит к снижению поверхностного натяже-
ния, т. е (da/dG)<0, то адсорбция будет положительной
(ПОО). Это означает, что концентрация i-того компонента
на поверхности будет больше, чем в объеме. Конечные объ-
емные и поверхностные концентрации определяются равно-
весием между движущей силой диффузии, стремящейся
выравнять концентрацию, и молекулярными силами, стре-
мящимися создать поверхностный слой, соответствующий
повышенной свободной энергии.
111.2. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
ЖИДКИХ МЕТАЛЛОВ
Поверхностное натяжение жидкого железа, измеренное
различными методами при 1550 °C, составляет 1800—
1880 мДж/м2. На сгре существенное влияние оказывают
примеси, особенно поверхностно активные, даже при нич-
тожном их содержании. Значения о для некоторых жидких
металлов приведены в табл. 12. Температурная зависимость
92
поверхностного натяжения хорошо описывается уравнением
а = о0 + do/dT (Т — Тпл), (159)
где о и со — поверхностные натяжения при искомой темпе-
ратуре Т и температуре плавления Т™; defdT— темпера-
турный коэффициент поверхностного натяжения, daldT<iQ.
Таблица 12. Поверхностное натяжение чистых металлов
Металл -do/dT t, °C а, мДж/м1 Металл -do/dT t, °C 0. мДж M1
Fe 0,35 1550 1850 Ti 0,26 1720 1330
Al 0,14 700 850 Si 0,09 1550 750
Мп 0,31 1550 1000 Со 0,40 1550 1830
Ni 0,39 1500 1750 Zr 0,20 1880 1330
Сг 0,32 2020 1590 Mo 0,18 2607 2220
V 0,31 1910 1950 W 0,29 3377 2500
111.3. УРАВНЕНИЯ ЗАВИСИМОСТИ
ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ РАСПЛАВОВ
ОТ СОСТАВА
Для расчета поверхностного натяжения растворов предло-
жено большое число уравнений [75]. В большинстве случа-
ев эти уравнения применимы лишь для двойных систем и в
более ограниченном масштабе — для тройных.
Формула Шишковского удовлетворительно описывает
концентрационную зависимость поверхностного натяжения
растворов, для которых выполняется уравнение Лангмюра:
а = а1 —z/?Tln(l 4-&с), (160)
где а и oi — поверхностные натяжения раствора и чистого
растворителя; с — концентрация растворенного вещества;
z и Ь— постоянные коэффициенты в уравнении Лангмюра.
Для металлургических расплавов уравнение (160) мож-
но применять вплоть до нескольких процентов растворен-
ного вещества.
Уравнение Шишковского часто записывают следующим
образом:
<т = СТ1 — (£77©) In [1 + х (F — 1)], (161)
где «о — поверхность, занимаемая одним молем растворите-
ля (мольная поверхность); тогда: 1/о— число молей на
1 см2 поверхности; F — параметр, характеризующий ка-
пиллярную активность растворенного вещества; х — моль-
ная доля растворенного вещества.
S3
Пример. На основе формулы (160) выведем уравнение,
описывающее поверхностное натяжение расплавов системы
Fe—О при 1600 °C, по следующим данным: оте =
= 1800 мДж/м2, площадь, занимаемая одним атомом кис-
лорода, (о=8,1 • 10-20 м2. Величину z в уравнении (160)
можно определить по формуле: z= 1/со Wa; z= 1/(8,1 • 10~20Х
Х6,02 • 1023) =2,05-10-5 моль/м2.
Исходя из экспериментальных данных o=f(c) коэффи-
циент b можно вычислить двумя способами. При малых с,
раскладывая 1п(1+&с) в ряд, имеем 1п(1 + &с)=&с. Тогда
o=oi—zRTbc. Отсюда b=(oi—o)lzRTc. При С[О]=0,005 %;
tri—о=200 мДж/м2; д = (200-10~3)/(2,05-10~5-8,3• 1873><
Х0,005) = 126.
Расчетная формула для о имеет вид о=1800—732 1g
(1+126 с). Коэффициент b можно найти также по уравне-
нию Дангмюра:
Г = z&c/(l + be),
(162)
где z — число адсорбционных мест; уравнение (162) можно
представить в следующем виде: С[о]/Г[о]=£[о]/2+ 1/bz. От-
кладывая значение С[о]/Г[о] по оси ординат против С[о] по-
лучим прямую линию. Начальная ордината этой прямой
равна \/bz, а угловой коэффициент 1/z. Из этих данных
можно вычислить Ь. А. А. Жуховицкий [76] предложил ряд
уравнений, при помощи которых можно вычислить поверх-
ностное натяжение растворов в более широкой области кон-
центраций, чем на основе формулы Шишковского.
Уравнение Жуховицкого. Поверхностное натяжение би-
нарного совершенного раствора (о) от состава зависит сле-
дующим образом:
а = О! — П!/?Т1п(1—х2 + х2С), (163)
где Oi — поверхностное натяжение чистого первого компо-
нента; Х2—мольная доля второго компонента; C=eOl~a,fniRT
ni — число молей первого компонента на 1 см2 поверхности.
Из уравнения (163) следует, что при x2=0 o=oi, а при
Л'2= 1 0=02.
В ряде случаев хорошее совпадение экспериментальных
и расчетных данных наблюдается при использовании урав-
нения Жуховицкого для реальных растворов, в котором
учитывается различие между активностями компонентов в
объеме и на поверхности [77].
Пример. Найти уравнение зависимости поверхностного
натяжения от концентрации марганца в системе Fe—Мп,
образующих совершенный раствор, на основе следующих
94
данных: при /=1550 °C, оре =1850 мДж/м2; омп —
= 1060 мДж/м2; rFe =0,126 нм; гМп=0,130 нм. Поскольку
ре—Мп образует совершенный раствор, то воспользуемся
уравнением (163). Вычислим Пре, приняв, что Гре =
=0,126 нм:
1 1 _
Fe “Fe Na juje Na
—--------------5-------------- 3,3 • 10~5 моль/м2;
3,14-1,262-10-20 •6,02- 1023
Q _ g(°Fe- aMn)/"Fe^T = e( 1850-1060) 10“8/3,ЗЛО-5.8.31 . 1823 _ 4,85;
a = 1850.10-3 —3,3-IO-5-8,31-1823-2,31g (l—xMn +
+ 4,85xMn);
a = 1850 • 10~3 — 11501g (1 — xMn + 4,85xMn) Дж/м2. (164)
На рис. 15 приведены рас-
четные значения а, вычислен-
ные по уравнению (164).
Уравнение Попеля и Пав-
лова. В отличие от уравнений
(160) и (163), которые для
расчета а раствора требуют
знаний лишь характеристик
чистых компонентов Oi и о
уравнение С. И. Попеля и
В. В. Павлова [75] требует
знания большого числа пара-
метров. Уравнение имеет вид:
k
— V = 1
V
1=1
<5,МДж/м*
1800
1600
1400
1200
1000
О 0,2 ОД ОД ОД ХМп
Рис. 15. Изотермы поверхностного
натяжения системы Fe—Мп при
1823 К, вычисленные по уравнению
Жуховицкого (/) и Попеля — Пав-
лова (2)
(165)
где о — поверхностное натяжение раствора; Vю, V—моль-
ные объемы раствора в поверхностном слое и в объеме;
©,— парциальная мольная площадь i-того компонента; зна-
чение ©, вычисляют по формуле • В первом
приближении fi=l; Ai— работа выхода моля вещества в
поверхностный слои; Ai=A/—cuRTinyi, где Д/=07©/, сц,
©9 — поверхностное натяжение и мольная площадь чистых
компонентов соответственно. Величина а характеризует до-
95
лю недостающих связей для элементов Fe, Ni, Мп, Сг, V,
Tia=!/4; для Si, С, Р, На = ‘/б-
Пример. Вычислим поверхностные натяжения расплавов
по уравнениям Попеля и Павлова при 1550 °C: oFe =
= 1850 мДж/м2; оМп=1060 мДж/м2. Расчетная формула
для двухкомпонентной системы имеет вид:
ХРев F F + XMnе Mn М =
_ XFe FFe -Ь xMn ^Mn
xFe FFe + 4 VMn ’
(166)
Вычислим последовательно VFe, Умп, ©Fe, “мп, Аре, Амп.
1. Поскольку железо и марганец образуют раствор,
близкий по свойствам к совершенному, то AV=0 и Vj =
= УГ, V2=V<2 (парциальные мольные объемы компонен-
тов равны мольным объемам чистых компонентов). Тогда
VFe=AlFe/pFe =55,85/7,15=7,81 см3/моль; VMn =54,94/
/6,75=8,29 см3/моль.
2. Парциальная мольная площадь ©z=f»WxVi/3 прини-
маем fi= 1, тогда <oFe =8,4-107-8,252/3=3,4-108 см2; сомп =
=8,4-107-8,932/3=3,7-108 см2.
3. А«=Д/—a^Tlnyz, так как раствор близок к совер-
шенному, то Д<=Д/= о?<o?AFe = 1850-3,32-104 = 6,14\
X10~7 мДж, ДМп = 1060-3,45-104=3,68-107 мДж.
Ae-AFeW_r 3,32-10’оМ>, 14.1071823-8,31.10»
Fe XFe е “ AFeC
(167)
-Y Хмп-Дмп/W _ Y з-^-ю^-з-бв-ю’Двгз.в.зыо’
AMn — AMn e — *Mn e
~ B.
Mn
И, наконец, вычислим л"е и x$n:
(168)
X-, = x«7(x«; + <„); = </« + <). (169)
Подставив полученные выражения (168) в уравнение
(166), получим:
Axf, 4- ВлМп = (8,25Хр + 8,93хм ) /( +
г® Mn \ ’ Fe ‘ ’ Мп/ / | л». । 1
/ \^Fe-j-DXMn
8,93хМв
^Fe + ВхМп
46
или окончательно расчетная формула для вычисления име-
ет вид, мДж/м2:
7,81xFe + 8,29xMn = 0(136xFce3':wi5i5 + 0,731xMn
(171)
Вычисленные по этой формуле значения о расплавов
Fe—Мп при различных концентрациях хмп приведены на
рис. 15.
111.4. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ РАСПЛАВОВ
НА ОСНОВЕ ЖЕЛЕЗА
Все легирующие и примесные элементы (кроме вольфрама)
являются поверхностно-активными элементами. Введение
10% (ат.) вольфрама увеличивает поверхностное натяже-
ние железа примерно на 150 мДж/м2. Сера и кислород об-
ладают наибольшей поверхностной активностью. Поверх-
ностное натяжение чистого сульфида (FeS) при 1550 °C
равно 550 мДж/м2. Поверхностное натяжение FeO при
1450 °C равно 600 мДж/м2. Для вычисления поверхностного
натяжения сплавов на основе железа (исключая растворы
серы и кислорода) С. И. Попель и В. В. Павлов [75] реко-
мендуют пользоваться обобщенным уравнением Шишков-
ского (161):
ст = стре —2000 lg [1 +S(Fz-l)xz]. (172)
При о= 17,4-103 м2 множитель, стоящий перед опера-
тором 1g, равен (2,3RT)/® = (2,3-8,31 -103-1873)/(17,4X
ХЮ3) =2000 мДж/м2. Для расчетов ст при 1600°C по урав-
нению (172) нужно пользоваться значениями Fi [75], ко-
торые приведены ниже:
Элемент .... Fe С Si Мп Ni Сг Ti
£<(расч^ .... 1 2,3 2,0 2,7 3,6 1,4 2,1 0,12
Р • 1 t( эксперт .... 1 2,2 5,0 1,4 2,5 0,12 Продолжение
Элемент .... V Мо Р S О Н N
^1(расч^ .... 0,19 — 1,8 150 800 — ——
эксперт .... 0,60 0,45 1,5 500 1000 1,0 150,0
Значения F, при температурах, отличных от 1873 К, можно
приближенно вычислить по формуле: FT=F^/T. Экспери-
ментальные значения ст и расчетные, полученные по фор-
муле (172), различаются не более чем на 5%. Для оценки
7—675
97
поверхностного натяжения жидких сталей и чугуна реко-
мендуется следующее уравнение:
о = aFe - 20001g 2 F. х., (173)
где i — элементы: Fe, С, Мп, Si, S, Р и др.
Уравнение (173) удовлетворительно описывает экспери-
ментальные данные, если концентрация кислорода и серы
не превышает 0,05 и 0,1 % соответственно. Авторы работы
[75] рекомендуют уравнение, в котором для кислорода и
серы используется со=34,8- 10~4 м2: a=<TFe —20001g 2 Лх,—
- 1000[lg(l + Foxo)+lg(l+FsXs)].
Пример. Вычислить о для стали следующего состава:
0,15-% С; 1,16 % Мп; 0,54 % Si; 17,68 % Сг; 8,87 % Ni;
0,014 % S; 0,0075 % О при 1873 К. ост = oFe —2000 lg(FFe•
•XFe + FcXc + + ^Si*Si + FcrXcr + ^Ni^Ni 4" F$XS -f-
4-FqXo); oCT =1800-2000 lg(l-0,724+2-0,007+5-0,012+
+ 2,2-0,0126 + 2,5-0,192 + 1,4-0,084 + 1000-0,0003 4- 500 X
X0,0003) = 1240 мДж/м2.
Оценка размеров адсорбирующихся частиц. Зная мак-
симальную адсорбцию Г™ах, можно вычислить площадь Q(-
частицы, адсорбированной на поверхности раствора: й,=
= 1/(Г™ахдгл) Например, размер площади, приходящейся
на один атом серы в поверхностном слое ее расплавов с
железом при Г™х = 13,1 • 10'6 моль/м2, равен Qs= 1/(13,IX
X10-e-6,02-1023) = 12,6-10-20 м2.
III.5. ВЫЧИСЛЕНИЕ КОНЦЕНТРАЦИИ
В ПОВЕРХНОСТНОМ СЛОЕ
Как известно, адсорбция по Гиббсу Гг (моль/см2) характе-
ризует избыток второго компонента в поверхностном слое
по сравнению с его содержанием в одинаковой порции объ-
емного раствора, т. е. адсорбция по Гиббсу характеризует
не абсолютную поверхностную концентрацию, а лишь из-
быточную. Возникает вопрос — какую выбрать толщину
поверхностного слоя, чтобы можно было бы сопоставить по-
верхностные и объемные концентрации в одинаковых пор-
циях растворов. Гиббс предложил толщину поверхностного
слоя отсчитывать от некоторой разделящей поверхности,
для которой Гг=0. Таким образом, адсорбцию по Гиббсу
экспериментально можно определить, измеряя изменение
концентрации объемного раствора в результате адсорбции.
Для практических расчетов такой способ определения
поверхностного слоя связан с рядом неудобств. Существует
98
несколько способов определения поверхностной концентра-
ции. Сравнивают содержание второго компонента в таких
порциях поверхностного и объемного раствора, которые
равны по массе, по объему или содержат одинаковое число
молей всех компонентов. Последний способ сравнения по-
лучил наибольшее распространение. В этом случае в раз-
бавленном растворе поверхностную концентрацию i-того
компонента можно вычислить по уравнению
xf — х( -1- <оГр
(174)
где Г/ — адсорбция по Гиббсу; х® и х,-— мольные доли
i-того компонента в поверхностном слое и объеме раствора;
со — поверхность, занимаемая одним молем раствора, рав-
ная со=Л^зу2/3 для того чтобы по формуле (174) вычис-
лить поверхностную концентрацию, необходимо по экспе-
риментальным данным определить адсорбцию Г/.
Пример. Вычислить поверхностные концентрации мар-
ганца в сплаве с железом при 3 и 10 % [Мп]. По методике,
описанной выше, вычисляем Гмп для расплавов указан-
ных концентраций: ГМп(з%) =3,5-10“6 моль/м2, Гмп(ю%) =
=9,3-10~6 моль/м2.
Из уравнения (174) имеем:
= гм„ + <> = N'P V‘£ = N'f .
(175)
Поскольку атомные массы железа (55,85) и марганца
(54,94) близки, то мольная доля будет приблизительно рав-
на массовой:
ХМп(3%)
= 0,03 + (6,02- 1023)V3
54,94-108
6,75-10—3
З.б.Ю-6
= 0,16 или 16%;
xMn(io%) — +
+(6,02. Ю23)1/3 /-L4-’-94'103-^ 9,3 • IO-6 = 0 45 или = 45о/
' \ 6,75-10-3 )
Таким образом, различают два понятия: адсорбцию —
избыток адсорбированного компонента на поверхности по
сравнению с объемом и поверхностную концентрацию, со-
ответствующую абсолютной величине концентрации на по-
верхности. Если поверхностное натяжение раствора описы-
вается уравнением Шишковского, то поверхностную кон-
центрацию i-того компонента (xf) можно вычислить по
формуле:
х“ = х. FJ'Zx. F..
i • I I
(176)
7*
99
Пример. Вычислим поверхностную концентрацию серы,
если объемная концентрация серы равна 0,05%. Для это-
го определим мольную долю растворенной серы:
х =---------°-05/32-----= 0,88-IO-3
s 0,05/32 + 99,95/55,85
Из приведенных данных для железа Л=1, для серы
Fi=500. Тогда по формуле (176)
= 0,88-10-3-500 = 0 308 или 20 03%
s 0,88-10—9 500+1
111.6. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ ШЛАКОВ
Поверхностные свойства расплавов оксидов в эксперимен-
тальном и теоретическом отношениях изучены хуже, чем
поверхностные характеристики чистых металлов и сплавов.
Это связано с тем, что шлак является более сложным объ-
ектом. Строение и структура шлаковых расплавов изучены
в меньшей степени, чем металлических. Ниже приведены
значения о для некоторых жидких соединений, важных для
сталеплавильного производства.
Соединение
t, °C . .
а, мДж/м2
AI0O3 FeO CaF2 FeS
2050 1400—1450 1400 1200
690 580—670 280 350
SiO2
1750—1800
400
Рассматривая поверхностное натяжение расплавов ок-
сидов, следует отметить одну общую закономерность — за-
мена одного основного оксида другим, как правило, мало
влияет на о. Значительные изменения наблюдаются лишь
при введении компонентов, являющихся поверхностно ак-
тивными (FeO, FeS, Р2О5 и др.). Значения поверхностного
натяжения о различных шлаков приведены ниже, мДж/м2.
Окислительный электросталеплавильный шлак (35—45 %
СаО, 10—20 % SiO2, 3—7 % А12О3, 8—30 % FeO, 2—8 %
Р2О6, 4—10 % МпО, 7—15 % MgO............................. 250—350
Восстановительный электросталеплавильный шлак (55—
60 % СаО, 20 % SiO2, 2—5 % А12О3, 8—10 % MgO, 4—8 %
CaF2) ................................................... 400—450
Синтетические известково-глиноземистые шлаки для обра-
ботки металла в ковше (55 % СаО, 20—40 % А12О3, 2—15 %
SiO2, 2—10% MgO).......................................... 450—60
Синтетические шлаки для разливки стали (10—20 % СаО,
20—25 % SiO2, 1—2 % FeO, 5—20 % МпО, 30—35 % CaF2) 250—30
Шлаки ЭШП................................................. 300—40
Шлаки индукционных печей (60 % СаО, 10 % MgO, 30 %
Са F2)................................................ 350—450
100
111.7. МЕЖФАЗНОЕ НАТЯЖЕНИЕ МЕЖДУ МЕТАЛЛОМ
И ШЛАКОМ
Межфазное натяжение между металлом и шлаком (ом-ш)
тесно связано с природой и характером взаимодействия
двух фаз. Значение ам-ш всегда меньше наибольшего зна-
чения поверхностного натяжения металла или шлака. Это
связано с тем, что частицы каждой из жидкостей притяги-
вают молекулы другой и тем самым снижают взаимодейст-
вие со стороны второй жидкости ее собственных молекул.
Межфазное натяжение зависит от химического состава фаз,
их строения, ориентации молекул и ионов. Чем ближе кон-
тактирующие фазы по свойствам, тем межфазное натяже-
ние меньше. Межфазное натяжение на границе раздела
жидкостей, характеризующихся значительной взаимной
растворимостью, может достигать ничтожных значений.
Говоря об изменении ом_ш в зависимости от состава шлака,
следует отметить следующее. Изменение концентрации, а
точнее замена оксидов SiC>2, СаО, А120з друг на друга, не
приводит к сколько-нибудь заметным изменениям ом-ш. Та-
ким образом, если компонент шлака малорастворим или
практически нерастворим в жидкой стали, то межфазное
натяжение велико: как правило, >1000 мДж/м2. При вве-
дении FeO, МпО, FeS и других компонентов, которые распре-
деляются между металлом и шлаком, наблюдается сущест-
венное снижение ом-ш до 200—600 мДж/м2. Добавление
3 % СаС2 к восстановительному шлаку электроплавки сни-
жает Ом—ш с 1300 до 750 мДж/м2, а изменение (FeO) от 0,1
до 95 % снижает ом-ш с 1250 до 180 мДж/м2. Шлаки, содер-
жащие оксиды железа (FeO, Ре20з), характеризуются
большей работой адгезии к жидкому металлу, чем безже-
лезистые шлаки. Элементы металлического расплава, вли-
яющие на Ом—ш можно разделить на три группы [75]:
1. Мало влияющие на ом-ш и практически не переходя-
щие в шлаковую фазу (С, W, Mo, Ni);
2. Характеризующиеся сравнительно низкой капилляр-
ной активностью (Si, Мп, Cr, Р и др.), но которые могут
быть переведены в шлак в значительных количествах с об-
разованием соответствующих оксидов. Из формулы Wa =
=oi + o2—Oi.2 следует, что если под влиянием добавки ка-
кого-либо компонента в металлическую фазу ом и Ом-ш
снижаются па одну и ту же величину, то работа адгезии
по сравнению с №я исходной системы не изменится. Однако
в практических условиях так не бывает. Указанные компо-
ненты (Si, Мп, Cr, Р) характеризуются той особенностью,
101
что они сильнее влияют иа сгм-ш, чем на ом и это приводит
к увеличению Wa;
3. Обладающие очень высокой поверхностной и меж-
фазной активностью (S, О). Так же, как и в предыдущем
случае, сера и кислород сильнее влияют на ом_ш, чем
на (Тм«
Например, в результате насыщения жидкого металла
кислородом при 1550°С ([% О] =0,19) поверхностное на-
тяжение в соответствии с формулой (172) снижается в два
раза, а межфазное натяжение снижается более чем в семь
раз. Таким образом, сера и кислород способствуют сущест-
• венному увеличению адгезии между металлом и шлаком.
Как показано выше, Wa определяется величинами ом-г, Ош-г
и Ом-ш. Однако поскольку поверхностное натяжение метал-
ла больше, чем шлака, то и вклад в межфазную энергию,
вносимый металлом, больше. Это означает, что поверхност-
ная активность компонента в металле влияет на ом_ш, а
следовательно, и на Wa в большей степени, чем поверхно-
стная активность этого же компонента в шлаке.
Говоря о поверхностных свойствах промышленных ме-
таллических и шлаковых расплавов, следует иметь в виду,
что реальные стали и шлаки содержат примесные компонен-
ты, концентрации которых изменяются в широких пределах.
Даже ничтожные изменения концентрации поверхностно-
активных компонентов могут привести к изменению ом-ш,
которое в ряде случаев может быть значительно больше,
чем влияние, вызванное изменением основного компонента.
В табл. 13 приведены значения ом-ш для реальных метал-
лургических систем. В обычных условиях измеряют так на-
зываемое статическое межфазное натяжение оМф на грани-
це раздела двух фаз, находящихся в термодинамическом
равновесии. При протекании межфазной химической реак-
ции или переходе компонента из одной фазы в другую име-
ют дело с межфазным натяжением, которое меньше соот-
ветствующего статического значения оМф. Межфазное на-
Таблица 13. Значение (Гм-ш для реальных металлургических
систем [69—73]
Металл Шлак О'д! МДж/М*
ШХ15 10X18Н9Т Известково-глиноземистый Белый электропечной Электропечной АНФ—6 1100—1300 1000—1200 900—1000 1100
102
тяжение, измеренное при градиенте химического потенциала
Др, принято называть динамическим или неравновесным.
Снижение межфазного натяжения До определяется сте-
пенью удаления системы от состояния равновесия. Количе-
ственная связь между До и Др описывается уравнением
[78]: До=МДр. В результате рассматриваемого эффекта
межфазное натяжение при переходе серы из металла в
шлак может быть снижено до 1—5 мДж/м2. В водных рас-
творах о удается снизить до такой степени, что происходит
автоэмульгирование. Изменение межфазного натяжения
при химическом взаимодействии или переходе компонентов
из одной фазы в другую в определенных условиях приводит
к увеличению поверхности раздела двух фаз. Наиболее на-
глядно это наблюдается в системах, где одна из взаимодей-
ствующих фаз находится в эмульгированном состоя-
нии [79].
111.8. СМАЧИВАНИЕ И РАСТЕКАНИЕ
Если каплю жидкого расплава, например металла или шла-
ка, поместить на твердую подложку (графит, огнеупоры
и др.), то в случае ограниченного смачивания наблюдается
картина, изображенная на рис. 16. По периметру капли со-
Рис. 16. Условия равновесия сил
поверхностного натяжения в различных систе-
мах:
а— твердое тело—жидкость — газ; б — две несмешивающиеся жидкости — газ;
1 — газ; 2 — жидкость; 3 — твердое тело
прикасаются три фазы: жидкость, газ и твердое тело. На
поверхности раздела каждых двух фаз действуют силы
поверхностного натяжения, которые стремятся сократить
эти поверхности. К единице длины окружности капли при-
ложены три силы, которые численно равны коэффициентам
поверхностного натяжения от-г, от-ж, ож-г. В состоянии
равновесия эти три силы связаны уравнением:
СТт-г ~ СТт-ж = аж-г C0S 0> (177)
103
где 0 — краевой угол смачивания. Если жидкость смачива-
ет твердую подложку, то она будет натекать на твердое
тело до тех пор, пока не установится равновесный краевой
угол. Процесс натекания жидкости связан с затратой рабо-
ты на создание новой межфазной поверхности между твер-
дым телом и жидкостью. При создании единицы поверхно-
сти (1 см2) затрачиваемая работа равна от_ж. Однако при
этом ликвидируется поверхность раздела твердое тело —
газ, и это приводит к выигрышу энергии, равной от-г. Отсю-
да следует, что количественные характеристики растекания
определяются соотношением величин от-г, от-ж и Ож-r. Та-
ким образом, в момент соприкосновения жидкости с твер-
дым телом создается некоторая неустойчивая система. По
границам раздела фаз действуют три силы сгт-г, От-ж, Ож-г
и конечное равновесное положение капли определяется од-
ним из двух условий.
1. Если
(178)
то смачиванию соответствует затрата работы (увеличение
свободной энергии капли), т. е. энергетические затраты,
связанные с созданием новой поверхности, превалируют
над выигрышем от ликвидации поверхности твердое те-
ло—газ и жидкость—газ, поэтому условию (178) соответ-
ствует стягивание капли до некоторого равновесного со-
стояния, описываемого уравнением (177).
2. Если
о > СГ 4-0
т—г т—-ж 1 ж—г »
(179)
то жидкость растекается, пока не покроет всей поверхности
твердого тела. Этот случай соответствует полному смачи-
ванию (0=0). Из уравнения (177) имеем
cos0 = (о_ _ — о_ ж )/о (180)
Подставляя в уравнение (180) разность от-г—от-ж из
уравнения ТГа=От-г4-сгж-г—от-ж, получим
cos 0 = Га ~ ; cos 0 = ----1. (181)
СГ>к—г °ж-г
Поскольку величина ож-г, стоящая в знаменателе урав-
нения (181), характеризует когезию, то краевой угол сма-
чивания (cos 0) определяется работой адгезии и когезии.
В первом случае !Fa характеризует силы сцепления между
жидкостью и твердой подложкой, во втором — силы сцеп-
ления между частицами жидкости. Если Н7а>ож_г, то
104
cos0>O и 0<9O°; если №а<(Тж-г, то cos0<O и 0>9О°.
Если имеем систему из двух несмешивающихся жидкостей
Жд и Жв, то условием равновесия сил поверхностного на-
тяжения (см. рис. 16 6) является аЖ£_г=оЖл_ЖБ-со8 (3 +
<тжл-г• cos а. Вместе с тем аЖд-г-sin а=оЖА_Ж£-sin (3. Из
двух последних уравнений имеем расчетную формулу для
вычисления межфазного натяжения сгЖл_Ж£для случая, по-
казанного на рис. 166:
ажл-же = Кстжл + аж Б~ а. (182)
Различные характеристики смачивания в системе твер-
дое тело—жидкость приведены на рис. 17. В большинстве
о=о еооГ е>90°
7/77/^77, 77^777/77/
а Б в
Рис. 17. Характеристики смачивания в системе твердое тело — жидкость:
« — полное смачивание; б — хорошее смачивание; в — плохое смачивание; г —
полное несмачивание
случаев с повышением температуры угол смачивания
уменьшается и жидкость растекается более полно.
Смачивание оксидов жидкими металлами. Приведенные
соотношения характеризуют процесс смачивания лишь с
точки зрения сил поверхностного натяжения. В. Н. Еремен-
ко и Ю. В. Найдич [80] показали, что процесс смачивания
в значительной мере определяется химическим взаимодей-
ствием между смачивающей жидкостью и материалом под-
ложки. Если жидкий металл (Me) контактирует с оксидом
(МеО), то полнота химического взаимодействия между ни-
ми Ме+Ме'О=Ме'+МеО определяется степенью химиче-
ского сродства соответствующих металлов к кислороду.
Если химическое сродство смачивающего металла к кисло-
роду меньше, чем металла оксида подложки, то химическое
взаимодействие протекает слабо и этому соответствует ту-
пой краевой угол (0>9О°). Так, например, железо имеет
меньшее химическое сродство к кислороду, чем магний,
алюминий, кальций, поэтому жидкое железо на оксидах
из MgO, А120з, СаО образует краевой угол 140—145°.
С уменьшением различия химического сродства к кислоро-
ду краевой угол уменьшается. В частности, при смачива-
нии жидким железом кремнезема 0=126°. Рассмотрим, как
105
влияют различные компоненты (С, Si, Мп и др.), содержа-
щиеся в жидком железе, на краевой угол при смачивании
оксидов. Характеристики смачивания определяются хими-
ческим взаимодействием не только основного металла (Fe),
но и легирующих компонентов с оксидом. Очевидно, что
компоненты, характеризующиеся высоким химическим
сродством к кислороду (Si, Ti, Al), в результате взаимо-
действия обеспечат существенное снижение 6.
На рис. 18 [75] приведены данные, показывающие влия-
ние компонентов, растворенных в жидком железе, на сма
Рис. 18. Влияние компонентов, раство-
ренных в жидком железе, на угол сма-
чивания (0) к AI2O3 [7]
чиваемость 0 к AI2O3. Фор-
мально характер влияния
того или иного компонента
описывается уравнением
(181), из которого следует,
что краевой угол определя-
ется показателями адгезии
и когезии. Наименьшее вли-
яние на 0 оказывают слабо
поверхностно-активные ком-
поненты никель и молибден.
Они практически не изме-
няют ни когезию, ни адге-
зию и в рамках уравнения
(181) мало влияют на 0.
Остальные элементы (Si,
Mn, С и др.) умеренно уменьшают когезию (сравнительно
слабо снижают поверхностное натяжение железа) и силь-
нее влияют на адгезию. Это приводит к уменьшению 0 с
ростом концентрации элементов. Увеличение концентра-
ции кислорода в жидком железе улучшает смачиваемость
(0 уменьшается, адгезия между металлом и оксидом уве-
личивается). Нераскисленный металл лучше смачивает фу-
теровку, чем раскисленный.
Смачивание графита жидким железом и шлаком. Гра-
фит хорошо смачивается жидким железоуглеродистым рас-
плавом. Образующиеся углы смачивания имеют значе-
ния: a=32-j-35°, 0=404-45° (см. рис. 16). Значение <тж_г
для расплава, насыщенного углеродом, составляет 1470
мДж/м2; ат-ж=1200—1400 мДж/м2; №а=3000 мДж/м2
[81]. Краевой угол смачивания графита жидким никелем
составляет 60°. Характеристики смачивания сильно зави-
сят от кристаллографических плоскостей, которые облада-
ют различными значениями от-г. Приведенные выше дан-
ные относятся к поликристаллическому графиту, от-г кото-
106
рого оценивается в 2500 мДж/м2. Шлаковые расплавы
плохо смачивают графит. Оксидный расплав системы
СаО—SiO2—AI2O3 на графите дает угол 125°. При длитель-
ной выдержке вследствие образования СаСг в шлаке угол
смачивания снижается до 100°.
Растекание металла и шлака. Если каплю жидкого шла-
ка положить на поверхность металлического расплава, то
она может либо растечься, либо остаться в виде нерасте-
кающейся капли. Это однозначно определяется поверхност-
ными натяжениями обеих жидкостей и межфазным натя-
жением. Сказанное справедливо и для случая, если ниж-
ней фазой является твердое тело (графит, огнеупор и др.).
Характеристики растекания обычно находят по разности
работ адгезии и когезии. В. Д. Харкинс эту разность на-
звал коэффициентом растекания:
S — — Wк = 0>м—г Ощ—г — Ом—щ — 2ош-г;
S = о — о — о
м—г ш—г м—ш
(183)
Если коэффициент растекания положителен (S>0, т. е.
№’а>1Гк), то шлак растекается по металлу. Если S<0 и
Wa соответственно меньше WK, то шлак на поверхности
образует линзу.
Пример. Вычислим коэффициент растекания для систе-
мы шлак—металл со следующими поверхностными харак-
теристиками: поверхностное натяжение стали ом-г=
= 1400 мДж/м2 и шлака ош-г=400 мДж/м2. Межфазное
натяжение ом-ш=900 мДж/м2. На основании этих данных
получим S = Ом—г—Ош—г—Ом-ш = 1400—400—900 = 100
мДж/м2. Поскольку $>0, то шлак будет растекаться по
металлу. Таким образом, растекание происходит при усло-
вии Ом-г^Ош-г+^м-ш. Это возможно в том случае, когда
шлак содержит поверхностно-активные компоненты (FeO,
FeS и др.). К таким системам относятся, в частности, окис-
лительные сталеплавильные шлаки с высоким содержани-
ем оксидов железа. Отметим, что в уравнение (183) мы
подставили значение работы когезии, равное 2ош-г и полу-
чили коэффициент растекания S шлака по металлу. Оче-
видно, что в уравнение (183) можно подставить значение
когезии, равное 2ом-г. В этом случае получим коэффици-
ент растекания металла по шлаку: 5'=ом-г+ош-г—Ом-ш—
—2ом-г=ош-г—Ом-г—Ом-ш. В случае растекания жидкости
по поверхности твердого тела имеем следующее соотно-
шение:
W ~о
а т—г
(184)
107
Подставляя значение разности от-г—от-ж из уравнения
(177) в уравнение (184) имеем: S = V^a—И7к=дж_г-|-
+ ож-г cos 0—2стж-г; 5=ож-r(cos 0—1). Видно, что если
0=0, то cos 0=1 и 5=0.
Проникновение металла и шлака в капиллярно-порис-
тые материалы. Огнеупорные материалы в процессе служ-
бы на значительную глубину пропитываются шлаком, ме-
таллом и его оксидами. Так, например, в зоне шлакового
пояса в основной электродуговой печи шлак проникает в
огнеупорную кладку на 50—100 мм. Подина печи пропи-
тывается оксидами железа на глубину до 150 мм. Футеров-
ка тигля индукционной печи на */з толщины пропитывает-
ся оксидами железа и легко окисляющихся элементов, со-
держащихся в стали. Многочисленные исследования по
изучению скорости проникновения металла и шлака в ка-
пиллярно-пористые огнеупорные материалы показывают,
что глубина проникновения I в зависимости от времени т
удовлетворительно описывается параболической кривой:
Z2 = (ocos0/?t)/2t],
(185)
где о— поверхностное натяжение проникающего расплава;
0 — угол смачивания на поверхности расплав—твердое те-
ло; R— радиус капилляра; т] — вязкость расплава.
Во многих случаях капиллярная пропитка футеровки
протекает при наличии градиента температуры в направ-
лении пропитки. В этом случае для количественных расче-
тов скорости или глубины пропитки следует учесть измене-
ние вязкости расплава с изменением температуры. Помимо
проникновения шлака в огнеупоры, для практики представ-
ляет интерес определение радиуса R пор в огнеупорах, в ко-
торые жидкая сталь проникнуть не может вследствие по-
верхностного натяжения. Формула имеет следующий виц:
/?= (2ocos 0)/р. Поверхностное натяжение стали в зависи-
мости от состава изменяется в пределах 1200—1700 мДж/м2
(нижний-предел для нераскисленной стали).
Пример. Вычислим радиус пор в огнеупорной кладке
ванны сталеплавильной печи, в которые жидкая сталь не
может проникнуть (сг= 1450 мДж/м2). Глубина сталепла-
вильной ванны 0,6 м. Угол смачивания металлом огнеупор-
ной кладки составляет 0=120°. Ферростатическое давле-
ние на глубине 0,6 м составляет 0,6/1,42=0,42 атм=4,12Х
ХЮ4 Па. Если о=1,45 Дж/м2, то R — (2-1,45cos 120°)/
/(4,12-104) =0,35-10-4 м=0,035 мм.
Глава IV
ТЕРМОДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ
НЕКОТОРЫХ РАСПЛАВОВ
НА ОСНОВЕ ЖЕЛЕЗА И НИКЕЛЯ
В обобщенном виде основные термодинамические свойства
бинарных расплавов на основе железа и никеля приведены в
гл. I (см. табл. 4). Настоящая глава посвящена более
подробному рассмотрению характеристик отдельных рас-
плавов, имеющих большое значение для практики электро-
сталеплавильного производства.
IV.1. КИСЛОРОД В РАСПЛАВАХ
Прямое экспериментальное исследование взаимодействия
кислорода с расплавленным железом затруднено тем, что
равновесное давление газа в условиях опыта имеет очень
малую трудноизмеримую величину (10~8 атм), поэтому в
опытах вместо чистого кислорода используют обычно газо-
вую смесь, например Н2 + Н2О, для которой равновесное
значение рог, легко рассчитать, воспользовавшись таблич-
ной величиной AG° реакции
На(г) + V,O2(r) = Н2О(Г); ДО» =- 252000 +
+ 58,367 Дж 12|. (186)
Искомое значение рог определяют из выражения для
константы равновесия реакции (186):
IgK = 18(РнЖ) ('/ Кп) =
(187)
зная состав равновесной смеси (рн,о/рн2)- Как показывают
результаты экспериментов, представленные на рис. 19, рав-
новесная концентрация кислорода в железе при относитель-
но небольших его содержаниях линейно зависит от квад-
ратного корня из давления кислорода в газовой фазе. Эта
зависимость, которую принято называть законом Сивертса,
согласуется с выражением для константы равновесия ре-
акции: УгОгс-)=[ОД К==/о[0]/Крог , откуда при fo = l
следует [О]=ККрог. Это согласование свидетельствует о
том, что процесс растворения протекает в соответствии с
написанной реакцией, т. е. сопровождается диссоциацией
молекул на атомы (или другие одноатомные частицы: ио-
ны, FeO и др.) В области повышенных содержаний кисло-
109
рода наблюдаются отклонения экспериментальных точек
от пунктирных прямых, иллюстрирующих закон Сивертса.
Эти отклонения можно количественно выразить концен-
трационной зависимостью коэффициента активности
lg fo =—0,2[% О], найденной в работе [82] при помощи
метода, приведенного на с. 27. Для многокомпонентных
расплавов значения fo можно определить при помощи па-
раметров взаимодействия (см. 1.15).
Увеличение равновесной концентрации кислорода в рас-
плаве при возрастании ро, ограничено образованием плен-
ки оксида. При помощи правила фаз легко показать, что
при постоянной температуре
появление третьей фазы в
двухкомпонентной системе де-
лает ее нонвариантной. Число
степеней свободы при этом об-
ращается в нуль: С=К—Ф+
+ 1 = 2—3+1=0. На рис. 19
такое состояние системы ха-
рактеризуется точками 3, по-
ложение которых определяет-
ся только температурой. Если в
равновесной трехфазной систе-
ме повысить давление кислоро-
да, то его концентрация в ра-
сплаве не изменится (это по-
казано горизонталями). Состо-
яние, которое возникает при
этом, будет неравновесным —
система вернется к прежнему
(равновесному) состоянию
вследствие связывания избыт-
VpT '105,атм1/г
°г
Рис. 19. Зависимость растворимости
кислорода в железе [68, 82] и ни-
келе [341 от 1/ р газовой фазы:
г ия
1,2 — экспериментальные значения
растворимости при 1823 К по дан-
ным [68] и [82]; 3 — пределы рас-
творимости
ка кислорода в оксид.
Чтобы отличить раствори-
мость кислорода (в двухфаз-
ной области) от концентрации
его в равновесии с оксидом, последнюю называют преде-
лом растворимости (или растворимостью оксида). Таким
образом, растворимость и предел растворимости представ-
ляют собой совершенно различные понятия, которые ха-
рактеризуют разные состояния равновесия. Недооценка
этого различия и неточность формулировок приводят иног-
да к серьезным ошибкам.
Обобщенной количественной характеристикой взаимо-
действия расплавленного железа с кислородом газовой фа-
110
зы служит следующая температурная зависимость Дб°
реакции:
V2O2(r) = I°hl%BFe>; ДС° = “ 117200 = 2’89Г ДЖ 121-
(188)
Отсюда
1g К = 1g (/о101/1/Рог) = (6120/7) + °, 151, (189)
где [О] — растворимость кислорода в железе при данном
Ро,, %•
Как видно из приведенных уравнений и рис. 19, раство-
рение газообразного кислорода в железе сопровождается
сильным тепловыделением (Д7/°°=—117 200 Дж/моль) и,
следовательно, — отрицательной зависимостью раствори-
мости от температуры.
Комбинируя уравнение (188) с выражением для Д6°
реакции образования FeO из элементов [2], можно полу-
чить характеристики равновесия:
Fe О . = Хре(ж) + 101(1 о/)'.
л (ж; (1 /о)
ДС°- 121000 — 52,387 Дж.
Отсюда следует, если принять xFe=l,
1g К = lg (/о 1 % 01) — (~ 6372/7) + 2,73;
lg [ %О] = (— 6320/7) + 2,735 [4],
(19°)
(191)
где [О] — предел растворимости, %.
Как показывает уравнение (190) процесс растворения
кислорода в железе из оксидной фазы сопровождается
поглощением тепла, поэтому предел растворимости кисло-
рода (в отличие от растворимости) с повышением темпе-
ратуры увеличивается. Иллюстрацией этого может слу-
жить рис. 19. Значения пределов растворимости при 1923
и 1973 К на рис. 19 не показаны; согласно уравнению (191),
эти значения равны соответственно 0,28 и 0,34 %.
Отметим, что полученные расчетом данные о пределах
растворимости кислорода в железе подтверждаются как
прямыми наблюдениями за образованием оксидной плен-
ки при определенном значении Рнго/рнг [83], так и специ-
альными опытами [84] по изучению равновесия расплава
со шлаком из FeO (в индукционной печи с вращающимся
тиглем, исключающим контакт шлака с огнеупорами).
Никель обладает значительно меньшим химическим сродством к кис-
лороду, чем железо, поэтому как видно из рис. 19, растворимость кис-
лорода в никелевом расплаве (при данном Pq2) намного ниже, чем в же-
111
лезном. Равновесное давление кислорода (при данной его концентрации)
у никелевого расплава, наоборот, значительно выше. При определенных
условиях оно может достигать значений, превышающих остаточное дав-
ление газов в промышленных вакуумных индукционных печах
133 мПа). Так, в работе [85] показано, что при вакуумной плавке ни-
келя, содержащего более 0,2 % кислорода, последний может выделяться
из расплава в виде молекул О2 (без участия углерода). При вакуумной
плавке железа, даже если оно насыщено кислородом (0,23 % при
1600°C), такое выделение невозможно, поскольку равновесное давление
в этом случае составляет 665 мкПа.
Термодинамической характеристикой взаимодействия жидкого нике-
ля с газообразным кислородом может служить уравнение
1^2°2(г) —
Отсюда
bG°° =— 71600 + 1,847 Дж [34]. (192)
1gк =lg(fo I%О]/рО1)=(3740/Т) - 0,0961. (193)
Коэффициент fo в никеле, по данным [34], может быть принят равным
1 в довольно широком диапазоне концентраций. Для многокомпонент-
ных расплавов на никелевой основе значения можно найти при по-
мощи параметров взаимодействия [86], приведенных в табл. 14.
Таблица 14. Параметры взаимодействия в сплавах на основе
никеля Ni—О—/
Эле- мент / Концент- рация % 4 'о Эле- мент / Концент- рация % е'о i
Сг 0—11 —0,231 0,009 V 0—5 —0,394 0,037
Мо 0—19 —0,024 0 Si 0—0,35 —5,38 9,32
W 0—20 —0,004 0 Ti 0—5 -0,51 0,036
Мп 0—14 —0,072 0 Al 0—2 —1,06 0,27
Fe 0—25 —0,025 0 0—0,05 —21,6 111,8
Со 0—55 —0,004 0 р 0—2 0 0
Си 0—58 —0,0099 0,0001 S 0—5 —0,089 0
Комбинируя уравнение (192) с выражением для AG0 реакции обра-
зования NiOT из элементов [2], получим
NiO(T) = NiM + [О](1% в Ni); AG°= 181100 — 92,82 7 Дж. (194)
Отсюда следует, что предел растворимости кислорода в никеле оп-
ределяется уравнением
lg([%O]-xNi) = (- 9460/7) + 4,847. (195)
Допущение xNJ = l по аналогии с уравнением (191) здесь нежелательно,
поскольку значения пределов растворимости кислорода в никеле, опре-
деленные по формуле (195), существенно выше, чем в железе. Так, при
1600°C [О] =0,64 %. Прямые измерения растворимости NiO(T), обзор ко-
торых приведен в работе [87], дают еще более высокие значения [О] —-
около 1 %
112
IV.2. УГЛЕРОД В РАСПЛАВАХ
При растворении углерода (графита) в расплавах железа
и никеля по достижении равновесия образуются насыщен-
ные растворы, состав которых соответствует фазовым диа-
граммам и характеризуется уравнениями:
1g хс = (— 560/7’) — 0,375 [88],
для никеля
1g хс = (— 1820/7’) + 0,097 [88].
(196)
(197)
В области температур сталеплавильных процессов мож-
но применять более простые соотношения:
[ %C]Fe = 1,34 + 2,54 • 10-3 /°C; [ % C]Ni = - 3,04 +
+ 3,8-IO-3/°C.
(198)
В работе [94] приведены несколько иные данные о рас-
творимое гн углерода в никеле. Сведения о влиянии «треть-
их» элементов на растворимость углерода в железе и
никеле приведены работах [2, 94]. Очевидно, что актив-
ность углерода по отношению к чистому графиту в насы-
щенном растворе равна 1. Коэффициенты активности -ус
могут быть найдены при этом как величины, обратные рас-
творимостям графита хс (см. с. 31). Для определения ак-
тивностей углерода в ненасыщенных растворах пользуют-
ся обычно методом, основанным на изучении равновесия
расплавов с газовыми смесяси СО4-СО2 или Н2 + СН4. При
этом активности cic=fc [% С] (по отношению к 1 %-ному
идеальному разбавленному раствору) находят из выраже-
ний для констант равновесия реакций:
IC1 + СО,(Г) = 2СО(Г); К = р’с0/(рс0, ас) • С")
ICI + 2HJ(r) = СН1(Г); К = рсн,/(й,ас). (200)
Значения К определяют из экспериментальных данных
по аналогии с примером, приведенным на с. 28. Темпера-
турная и концентрационная зависимость найденных таким
образом коэффициентов fc в сплавах на основе железа
характеризуется значениями параметров, приведенных в
табл. 6—8 и на с. 54—56, 58, 59.
Если по значениям констант реакций (199), (200) най-
ти значения соответствующих AG0 и сопоставить их с таб-
личными значениями AG0 аналогичных реакций с участи-
ем графита, то можно определить изменение энергии Гиб-
8—675
113
<5са при переходе
Id(n>) = 1О(1Я , и); ДС“ = 22600 - 42,37 Дж 12]. (201)
Первый член в полученном выражении представляет
собой теплоту растворения графита в железе с образова-
нием бесконечно разбавленного раствора (Д//“). Значение
ДЯ~ =22 600 Дж подтверждено прямыми калориметриче-
скими измерениями (см. табл. 4). По данным ряда авторов
[89], с ростом концентрации углерода величина АЯс уве-
личивается. Одно из предложенных уравнений имеет
вид: Д/7с=22 600+24 330(1—Хре) Дж [2]. Используя вы-
ражение (155) и соотношение
Дб“ = RT In ас = 2,3/?7' 1g -^c( i%> =
= 2,3RT f 1g у” + 1g ------------'I, (202)
s iC -r s 1/12 _j_99/55>85 Д ’
можно найти, что коэффициент активности углерода в бес-
конечно разбавленном растворе по отношению к чистому
графиту при 1873 К равен Тс<вре)=^>^- Умножив эту ве-
личину на соответствующее значение /с можно определить
Ус при любой концентрации. Значения коэффициентов ак-
тивности углерода в жидком железе были определены еще
одним способом — измерением скорости испарения железа
из углеродсодержащих расплавов в вакууме [12]*. Полу-
ченные результаты удовлетворительно согласуются с дан-
ными других исследований.
Этот же метод был использован для определения активностей угле-
рода в расплавах на основе никеля. Авторы работы [30] приводят сле-
дующее уравнение для расчета коэффициентов активности углерода ус
в жидком никеле при температуре ~1823 К:
lgYc=-°>5+ 11хс. (203)
Из уравнения (203) следует, что Yc(bN1) = ^,32. Используя уравне-
ние (203) в сочетании с данными о теплоте растворения графита в жид-
ком никеле, полученными калориметрическим методом (см. табл. 4),
можно получить выражение для AG перехода графита в раствор:
С(гр) = [С](1О/о в Ni) ; = 41000 - 47,94 Т Дж. (204)
Результаты сравнения активностей углерода в расплавах железа и
никеля представлены на рис. 20. Как видно из рис. 20 при концентраци-
ях >1,5 % активность углерода в никеле существенно выше, чем в желе-
зе. При малых содержаниях углерода наблюдается обратное соотноше-
ние: в никеле у“ =0,32, а в железе у с =0,57. В работе [90] показано, что
знакопеременные отклонения кривой активности ас (по отношению к
* Пример расчета ас по этим данным приведен на с. 30.
J14
графиту) от кривой 1 для совершенного раствора не связаны с каким-
либо принципиальным изменением свойств раствора. Если выбрать за
стандартное состояние вместо твердого графита его гипотетический пере-
охлажденный расплав*, который, очевидно, ближе по свойствам к раст-
воренному углероду, то кривая 2 для совершенного раствора оказывается
выше линии активности ас . Это соответствует отрицательным отклоне-
ниям от идеальности (при всех возможных концентрациях раствора
вплоть до насыщения графитом).
Рис. 20. Активность углерода в рас-
плавах железа и никеля при 1873 К по
данным (2, 95]:
— по
отношению к графиту;
отношению к гипотетичсс-
~ж
кому жидкому углероду; /, 2 — а
сгр
и а г в соответствующих совер-
шейных растворах; 3, 4 — в разбавлен-
ных растворах (в железе и никеле)
о 1 г з «fC7/A
IV.3. ВЗАИМОДЕЙСТВИЕ УГЛЕРОДА
И КИСЛОРОДА В РАСПЛАВАХ
Термодинамические характеристики реакции
= ®^(г) (205)
в железе и никеле можно получить из вышеприведенных
данных о растворении кислорода и углерода в соответст-
вующих расплавах, комбинируя уравнения (188), (192),
(201), (204) с выражением для ДО0 реакции
Сг„ + = С0<гр ДС” =~ 118000 - 84’4 Т Дж 12)-
__________ (206)
* При построении шкалы принято, что температура плавле-
ния графита составляет 4020 К, а теплота 125 600 Дж/моль [91, 92].
AG плавления графита при 1873 К найдена по уравнению (113). В ре-
зультате для раствора на основе железа получено =0,008. Цена
деления для шкалы аСж принята большей, чем для шкалы ^сгр»
в у г /у~ =0,57/0,008 = 71 раз.
ьгр Сж
8*
115
Полученные таким способом уравнения температурной
зависимости Дб° и 1g К реакции (205) в железе хорошо
согласуются с экспериментальными данными, которые име-
ют следующий вид:
Д6°=-22400 - 39,7 7 Дж; 1g К = 1g (рсо//с1С1/о[О])^
--=(1168/7) 4-2,07 [2]. (207)
Для никеля расчет дает
ДС° =— 87500 — 38,37 Дж; 1gК = (4570/7) + 2,0. (208)
Результаты расчета по уравнению (208) (рис. 21, кри-
вая /) сопоставлены с экспериментальными данными [95]
(рис. 21, кривая 2). Для сравнения приведены данные о
равновесии кислорода с кремни-
ем в никеле (рис. 21, кривая 3)
и кислорода с углередом в желе-
зе (рис. 21, кривая 4). Как видно
из рис. 21, расчетные концентра-
Г07, %
о,Off
о,ог
0,01
0,005
0,002
0,001
0,0005
О 0,05 0,10 0,15 0,20
£0]. °!о
Рис. 21. Сопоставление расчетных данных о взаимодействии [С]+[О]=СОГ в ни-
келе (при 1798 К и р - =1 атм) с экспериментальными
Рис. 22. Зависимость концентрации кислорода в металле от содержания углерода:
/ — равновесная кривая, соответствующая уравнению (207); 2 — фактические кон-
центрации для дуговых печей вместимостью 40 и 100 т [96]; 3 — то же, для мар-
теновских печей [62]; 4 — равновесные концентрации с фактическими мартенов-
скими шлаками [62]
ции кислорода в никеле несколько ниже опытных. Это мо-
жет быть связано как с погрешностями расчета, так и с экс-
периментальными ошибками при определении очень малых
концентраций кислорода (см. с. 120). Как видно из рис. 21,
в никеле равновесные с углеродом концентрации кислоро-
да значительно меньше, чем в железе. Углерод по своей
116
раскислительной способности в никеле близок к кремнию
или даже его превосходит.
Из уравнений (207), (208) видно, что окисление угле-
рода растворенным кислородом сопровождается выделени-
ем тепла и поэтому полнее протекает при пониженной тем-
пературе. В расплавах на основе железа температурная
зависимость константы выражена довольно слабо, так как
значение Д//° здесь невелико. Однако экзотермический ха-
рактер реакции (205) в железе установлен достаточно точ-
но. Его часто используют на практике, добиваясь, напри-
мер, более полного раскисления металла углеродом при
вакуумной индукционной плавке в результате «подстужи-
вания».
В связи с этим возникает вопрос: почему в некоторых
случаях влияние температуры на процесс окисления угле-
рода оказывается противоположным. Так, в дуговых пе-
чах после расплавления металла кипение практически не
происходит (даже при введении окислителя — руды) до
тех пор, пока ванна не нагрета. Казалось бы, что противо-
речит экзотермическому характеру реакции (205), однако
в действительности никакого противоречия нет. При кипе-
нии металла в печи окисление углерода определяется не
только протеканием реакции (205), но и поступлением
кислорода из шлака в соответствии с эндотермической ре-
акцией (190). Суммарный процесс (FezO)+[C]=x[Fe] +
Н-СОГ; Дб°=98600—92,08 т Дж также является эндотер-
мическим и при повышении температуры протекает более
полно. Следует добавить, что наряду с термодинамическим
фактором существенное влияние на интенсивность кипения
ванны оказывают кинетические факторы. С повышением
температуры скорость окисления углерода увеличивается.
Из выражения (207) следует, что при постоянных Т и
Рсо произведение ос^о в расплаве является константой.
В области сравнительно небольших содержаний углерода
и кислорода, где произведение коэффициентов /с/о близко
к 1, произведение равновесных концентраций [С][О] =т
также можно считать постоянным, равным при 1873 К
0,002 (%)2 [2J. Графически такая связь между равновес-
ными концентрациями углерода и кислорода выражается
гиперболой (рис. 22, кривая 1). Для точных расчетов и
вне указанного на графике диапазона концентраций вели-
чину m нельзя считать постоянной. Расчеты следует вы-
полнять в этом случае по уравнению (207), определяя
коэффициенты активности при помощи параметров взаимо-
действия. В реальных сталеплавильных процессах значе-
117
ние т в конце окислительного рафинирования металла
также примерно одинаково. Это обусловлено, с одной сто-
роны, слабой температурной зависимостью константы ре-
акции (205) (в связи с малым ее тепловым эффектом), а с
другой — характером выделения СО (в виде пузырей). Ус-
ловием выделения СО в пузырь является неравенство
Рсо > Par + Рм Shu + Рш ghw + (2о/г). (209)
В выражении (209) рсо означает равновесное давление
СО, определяемое выражением (207), а правая часть пред-
ставляет собой сумму действующих на пузырь внешних
сил: атмосферного давления, гидростатического давления
металла и шлака и капиллярного давления. При кипении
ванны без введения окислителей («чистый кип») в резуль-
тате снижения концентраций углерода и кислорода значе-
ние рсо постепенно уменьшается. В конце периода чис-
того кипа Рсо приближается к некоторому пределу, ко-
торый определяется правой частью неравенства (209).
Предельное значение рсо в различных сталеплавильных
агрегатах (за исключением вакуумных) приблизительно
одинаково — немного более 1 атм, поэтому конечные зна-
чения т в различных агрегатах и па разных плавках (пе-
ред раскислением ванны) довольно близки. Они несколь-
ко превышают равновесное значение т при рсо =1 атм
и не зависят от содержания СО в газовой фазе.
Концентрации углерода и кислорода в металле после
кипа сопоставлены с равновесными на рис. 22. Как видно
из рис. 22, реальные содержания кислорода в металле за-
нимают промежуточное положение между двумя равновес-
ными концентрациями, которые определяются, с одной сто-
роны, взаимодействием с углеродом, а с другой — перехо-
дом кислорода из шлака. Высокий окислительный
потенциал шлака (или при продувке — струи газа) обес-
печивает при кипении ванны постоянное поступление в ме-
талл кислорода, где он расходуется на образование СО.
Степень переокисления ванны (по отношению к кривой /)
определяется соотношением скоростей поступления и рас-
ходования кислорода. В конце периода, когда введение
окислителей прекращают, ванна самораскисляется — со-
держание кислорода понижается, приближаясь к равновес-
ному. Как видно из рис. 22, в дуговых печах такое прибли-
жение лучше, чем в мартеновских. Это связано с тем, что
технология электроплавки предусматривает более высо-
кую температуру металла в конце окислительного перио-
да, необходимую для обеспечения скачивания шлака и про-
118
ведения восстановительного периода. При повышенной
температуре реакция (205) идет быстрее и ближе подхо-
дит к равновесию.
Анализ уравнения (207) показывает, что уменьшение
рсо должно приводить к пропорциональному снижению
равновесного значения произведения [С]-[О]. На практике
это используется в вакуумных процессах и при обработке
металла инертными газами. В результате снижения равно-
весного значения рсо всего до 0,1 атм резко повышается
раскислительная способность углерода, а также обеспечи-
вается глубокое обезуглероживание расплава без пере-
окисления.
Как следует из законов термодинамики, при снижении
Рсо Щ = [С][О] должно уменьшаться неограниченно, до-
стигая очень малых значений. Например, для рсо =
= 10-5 атм, которое можно легко обеспечить при вакуум-
ной индукционной плавке, равновесное значение т со-
ставляет ~10-8 (%)2. Реально наблюдаемые концентра-
ции углерода и кислорода в железе вакуумной плавки зна-
чительно выше: т не уменьшается обычно ниже 10-4 (%)2,
соответствующего Рсо =0,1—0,01 атм. Как показали
А. М. Самарин и Р. А. Карасев [223], одной из основных
причин такого несоответствия являются кинетические труд-
ности протекания реакции (205), связанные с условием
(209). При малых значениях рсо силы гидростатического
и капиллярного давления делают невозможным кипение
металла, а беспузырьковое выделение СО (через поверх-
ность ванны) является очень медленным процессом, кото-
рый в крупных промышленных агрегатах обычно не дает
ощутимых результатов. Не отрицая правильности этого
объяснения, следует однако заметить, что оно не подходит
для опытов с малыми массами металла (10—50 г) и в осо-
бенности для экспериментов с висящей каплей (1,5 г)*.
При уменьшении массы металла его удельная поверхность
значительно возрастает и, как показывают кинетические
расчеты, оказывается в большинстве опытов достаточной
для протекания реакции (205) почти до равновесия даже
в беспузырьковом режиме. Несмотря на это, конечная ве-
личина т не снижалась в таких опытах ниже 10~4. Для
объяснения причин указанного несоответствия теории с
результатами даже лабораторных опытов было выдвинуто
несколько гипотез. Согласно одной из них все объяснялось
поступлением кислорода из тигля. Для проверки были про-
* В обобщенном виде эти данные приведены в работе [93].
119
ведены опыты с применением бестигельной плавки (во взве-
шенном состоянии и в медном водоохлаждаемом кристал-
лизаторе с электронно-лучевым нагревом). Результаты
оказались практически такими же.
Правильной оказалась другая гипотеза. Как показала
проверка, выполненная в работе [93], неполное протекание
реакции (205) в лабораторных опытах часто является ка-
жущимся, и связано оно с недостаточной чувствительностью
анализа металла на содержание углерода и кислорода.
Определение малых содержаний [О] и [С] сильно осложне-
но поверхностным загрязнением образцов. Применение спе-
циальных методов анализа позволило зафиксировать [93]
в железе после 60-мин выдержки в вакууме (при массе ме-
талла 20—50 г) произведение [С]-[О]= 1,3-10~10 (%)2. По-
лученное значение близко к теоретической величине т, со-
ответствующей давлению в печи при проведении плавок
(6,6-10—8 атм). Характерно, что наблюдаемая величина т
систематически понижалась по мере перехода к более чув-
ствительным методам анализа. Поступление кислорода из
тигля влияло на конечное значение [О], но произведение
[С]-[О] при этом оставалось близким к теоретическому,
вследствие соответствующего уменьшения [С] согласно
уравнению (207). Обнаруженная принципиальная возмож-
ность практически полного протекания реакции (205) в
глубоком вакууме (которая в последние годы иногда ста-
вилась под сомнение) имеет большое практическое значе-
ние и определяет перспективность попыток активизации
массообменных процессов в условиях промышленных агре-
гатов (например, при электроннолучевой плавке) с целью
получения сталей и сплавов со сверхнизким содержанием
углерода и кислорода.
IV.4. ОБЕЗУГЛЕРОЖИВАНИЕ
ХРОМСОДЕРЖАЩИХ РАСПЛАВОВ
Окисление углерода в расплавах с высоким содержанием
хрома является важной проблемой при выплавке корро-
зионностойких, жаропрочных и других сталей и сплавов.
При обычных (для других сталей) условиях плавки хром
в таких расплавах обладает более высоким химическим
сродством к кислороду, чем углерод, поэтому обезуглеро-
живание в обычных условиях сопровождается большими
потерями хрома. Для решения проблемы нужно создать та-
кие условия плавки, которые способствовали бы преиму-
щественному окислению углерода. Качественный выбор не-
120
обходимых условий можно сделать, исходя из различия
свойств оксидов хрома и углерода. Как видно из табл. 1,
образование СггОз сопровождается значительно большим
выделением тепла, чем образование СО (приблизительно в
три раза, если отнести значения Д//° к 1 молю кислорода).
В соответствии с изобарой Вант-Гоффа (см. с. 11) отсюда
следует, что термодинамическая устойчивость СггОз с по-
вышением температуры понижается значительно быстрее,
чем устойчивость СО, поэтому одним из условий эффек-
тивного обезуглероживания металла в присутствии хрома
является повышение температуры. Вторым существенным
различием между оксидами углерода и хрома является то
обстоятельство, что СО в условиях плавки представляет
собой газ, а оксиды хрома — твердые вещества. Согласно
принципу Ле-Шателье, образованию газообразного оксида
должно способствовать понижение давления.
При количественной оценке необходимых условий обез-
углероживания хромсодержащих расплавов следует учи-
тывать, что хром, во-первых, существенно понижает коэф-
фициенты активности углерода и кислорода (см. табл. 6)
и, во-вторых, ограничивает концентрацию кислорода в ме-
талле вследствие образования твердых оксидов: РеО-СггОз
или СГ3О4 [2]. В результате уменьшения коэффициентов
/с и /о произведение [С]-[О], достигаемое при обезуглеро-
живании хромсодержащих расплавов, всегда выше, чем в
железе. Равновесные значения т можно рассчитать по
уравнению (207) при помощи параметров взаимодействия.
Например, для расплава, содержащего 18 % Сг и 10 % Ni
при 1873 К и р—\ атм получим lgw=lgPco —lgfc—
—lgfo— (1168/T) —2,07 = —eg .18 —.Ю— eg -18—
—• 10—2,693. Подставляя в это выражение значения е[
из табл. 6—8, находим, что /п=0,019. Коэффициенты
К /8 и /с в этом расчете не учтены, поскольку при малых
[С] и [О] они близки к 1.
Предельные концентрации углерода, которые могут быть
достигнуты при обезуглероживании хромсодержащих рас-
плавов, определяются равновесием с соответствующими
оксидами. Экспериментальные данные для расплавов на
основе железа выражаются следующими уравнениями [2].
При ^9 % Сг:
FeO•Сг2О3(т) 4- 4 [Cl = [Fe] + 2[Сг] 4- 4СО(Г); (210)
AG0 = 830 000 — 541Т Дж; (211)
lg №/4 = IgdCrl' -Pco^c IC]) = (- 10830/Т) 4- 7,06. (212)
121
При >9 % Cr:
Сг3О4(Т) ~|" 4 [С] = 3 [Cr] + 4C0(r);
ДС° = 882500 — 585Т Дж.
1g /<>/4 = 1g ([Сг]3/4 -рсо//с [С]) = (- 11520/7" + 7,64).
(213)
(214)
(215)
Активности оксидов хрома в этих уравнениях приняты
равными 1 в связи с тем, что как в лабораторных, так и в
производственных условиях обезуглероживание высокохро-
pCQ-0,01amn p^OJamri
1873 2073 2273 Т,К
I—I----1____I_। i i
1600 1800 2000 t,°C
Рис. 23. Равновесные значения
[Сг]/[С] для расплавов Fe—
18 % Сг (/), Fe—18 % Cr—
10 % Ni (2); Fe—10 % Сг—
10 % Ni (3)
мистых расплавов сопровожда-
ется образованием вязких (ге-
терогенных) шлаков, насыщен-
ных оксидами хрома. Резуль-
таты расчетов по приведенным
уравнениям, как правило, удо-
влетворительно согласуются с
данными опытов и производст-
венных плавок. Используя!
уравнения (210) — (215), мо-
жно определить, какие усло-
вия требуются для достиже-
ния необходимой степени обез-
углероживания расплава при
заданном содержании хрома.
При таких расчетах удобно
пользоваться параметром*
[Сг]/[С]. Зависимость этого
параметра от температуры,
давления СО и состава сплава.
рассчитанная по уравнению
(215) с использованием данных табл. 6, 7 и уравнения
(47)*, представлена на рис. 23.
Из рис. 23 видно, что в обычных условиях (при рсо —
= 1 атм) для достижения необходимого значения [Сг]/[С]=
= 10/0,05=200 (которое примерно соответствует концу обез-
углероживания в случае выплавки стали типа 12Х18Н10Т)
требуется температура ~ 1850 °C. Для нагрева расплава до>
такой температуры применяют газообразный кислород.
При обезуглероживании в вакууме или в условиях про-
дувки инертным газом эффективность процесса значитель-
но повышается. Достигаемые при этом результаты соответ-
ствуют обычно значениям рсо =0,1—0,01 атм. В таких ус-
ловиях даже при температурах <Л700 °C можно обеспечить.
* Об использовании (47) для расчета /<(Т)—см. с. 66.
122
очень высокие значения [Сг]/[С] 1000), необходимые,
в частности, для выплавки низкоуглеродистых коррозион-
ное гойких сталей, содержащих <0,03 % С.
Как видно из рис. 23, условия обезуглероживания за-
метно улучшаются в присутствии никеля. Это связано с
тем, что никель увеличивает коэффициент активности угле-
рода (см. табл. 7) и, следовательно, облегчает его взаимо-
действие с кислородом.
IV.5. СЕРА В РАСПЛАВАХ
Равновесное давление паров серы над расплавами желе-
за и никеля очень мало, поэтому при исследовании таких
расплавов, так же как и при изучении растворов кислоро-
да, используют соответствующие газовые смеси, обычно
H2 + H2S. Например, в работах [35, 224, 225] теплоты рас-
творения серы в железе и никеле (ДТ/s) определяли по
температурным зависимостям константы реакции
- IS] (216)
при помощи уравнения изобары Вант-Гоффа (4). Было про-
ведено также определение теплот растворения серы в же-
лезе и никеле калориметрическим методом [97]. При этом
серу вводили в расплавы в виде соответствующих сульфи-
дов: FeS и NisSz. Для никеля значение &Н$ =—123,5 кДж,
определенное калориметрическим методом, практически
совпало с результатами равновесных опытов: —117,9 кДж
[227] и —118,6 кДж [35]. Для железа различие оказалось
значительным: калориметрическим методом было получе-
но Д/Ts =—72 кДж, а в равновесных опытах Д/Zs =
=—131,9 кДж [2]. Калориметрический метод является наи-
более надежным для определения тепловых эффектов, по-
этому полученное с его помощью значение теплоты раство-
рения серы в железе можно считать, по-видимому, более
правильным, оно лучше согласуется с данными [226] о бо-
лее высокой растворимости серы в никеле по сравнению с
железом (при том же значении рн^/рн,. Кроме этого, из
табл. 4 следует, что все элементы (за исключением угле-
рода, азота и кислорода — элементов второго периода Пе-
риодической системы элементов Д. И. Менделеева) раство-
ряются в никеле более экзотермично, чем в железе. По-ви-
димому сера не отличается в этом от остальных элементов
третьего периода: алюминия, кремния и фосфора.
Приняв за основу полученные калориметрическим ме-
тодом значения теплоты реакции (216) и определенные в
123
равновесных опытах значения AG00 этой реакции при
1873 К [2, 35], получим следующие выражения темпера-
турной зависимости ДО00 реакции (211) в железе и ни-
келе:
3760
72000 — 9>92 т Дж;
lg' ——
Vps,
4-0,518;
(217)
=— 123500 + 17,727' Дж;
. fs 1 %S]
г Ps2
6450
— 0,926.
(218)
Влияние различных элементов на коэффициент актив-
ности серы в железе характеризуется параметрами взаимо-
действия, приведенными в табл. 6—8. Следует отметить
значительное увеличение коэффициента активности серы
под влиянием добавок углерода, кремния и фосфора. Это
является причиной того, что в чугуне коэффициент актив-
ности в несколько раз выше, чем в железе. Удаление серы
из чугуна, а также из высокоуглеродистой или высококрем-
нистой стали протекает значительно лучше, чем из обыч-
ного низкоуглеродистого металла. В никеле влияние доба-
вок на коэффициент активности серы характеризуется сле-
дующими значениями параметров (1848 К) [35]:
Элемент j
Параметр е$
S Fe Gr .Mo Ti Al
—0,035 0,005 0,030 0,053 0,160 0,133
Для лучшего понимания поведения серы в сталепла-
вильных процессах представляет интерес рассмотрение тер-
модинамических характеристик взаимодействия растворен-
ной в металле серы со шлаком, с газовой фазой и с суль-
фидообразующими элементами.
Взаимодействие серы со шлаком. Как показали опыты
[99, 100], сорбционная (поглотительная) способность шла-
ков по отношению к сере в значительной степени зависит
от окислительного потенциала. Из рис. 24, А видно, что за-
метное растворение серы в шлаке наблюдается в двух об-
ластях значений равновесного давления кислорода: <10-6и
>10“4атм. Это соответствует двум различным валентнос-
тям, которые может проявлять сера в шлаке: —2 (сульфид-
ная) и +6 (сульфатная). В соответствии с ионной моделью
124
шлака можно написать две реакции
сульфидной и сульфатной форме:
% S2(r) + (О2-) = i/aO2(r) + (S^-);
растворения серы в
(219)
+ \О2(Г> + (°2-) = (SO2-);
1/2. „3/2
S, Ро,
(220)
Если учесть, что для данного шлака при небольших кон-
центрациях серы ее коэффициент активности и значение
«о?---практически не зависят от (%S),to выражения
(219), (220) можно преобразовать следующим образом:
K;=(%S)(₽0/p )«;
(221)
(222)
Полученные константы называют соответственно суль-
фидной и сульфатной емкостью шлака. Из рис. 24, Б видно,
что эти характеристики действительно не зависят от р$, и
ро , но каждая в своей области: — при малых значениях
Ро, когда реальный процесс описывается реакцией (219), а
К'п—при больших ро, когда процесс описывается реакци-
ей (220).
При помощи выражений (221), (222) легко объяснить,
почему в различных диапазонах изменения ро, его значение
по-разному влияет на растворимость серы. Влияние темпе-
ратуры также имеет противоположный характер (см. рис.
24, Л). Это согласуется с различными знаками теплот реак-
ций [99]: CaO(T)4-1/2S2(r)=CaS(T)+1/2O2(r); Af/noo—гооок =
= 100,5 кДж; CaO(T)+1/2S2(r)+3/2O2(D=CaSO4(T);
^^1700—2000К= 816,5 кДж.
Для условий взаимодействия шлака с расплавом стали
при po,<10-8aTM первостепенное значение имеет суль-
фидная емкость шлака (обозначаемая обычно Cs). Не-
которые из имеющихся экспериментальных значений Cs,
для различных шлаковых расплавов представлены на
рис. 25. Из рис. 25 видно, что сорбционная способность
шлаков по отношению к сере резко возрастает с повыше-
нием температуры и основности. Последнее согласуется с
вытекающим из уравнения (219) увеличением сульфидной
125
«мкости шлака при повышении активности анионов кис-
лорода (напомним, что главными донорами ионов О2- в
шлаке являются основные оксиды). Замена СаО на МпО
и SiC>2 на AI2O3 приводит к увеличению Cs. Однако более
высокая сульфидная емкость шлаков системы МпО—SiO2
Рис. 24. Растворимость серы в шлаках, содержащих 37 % СаО4-27 % AI2O3+
+3ь % SiO2 (шлак 7) и 41 % СаО+52 % AI2O3+7 % SiO2 (шлак 2), в условиях
равновесия с газовой смесью SO2+H2+CO2+50 % N2 при различных pq* и
[99]:
А — опыты со шлаком 7 при 1 % SO2 в газе (изгиб линий связан с непостоянст-
вом р ); 5 —опыты при 1773 К; а — область . при которых кислород
и,
находится в растворе (не образует оксидов); б — область малой точности анали-
за; 7 — шлак 7, 1 % SO2; 2 —шлак 7, 2 % SO2; 3 — шлак 2, 1 % SO2; 4 — шлак
2, 2 % SO2
(а также основных мартеновских шлаков) не обеспечивает
им более высокой десульфурирующей способности по срав-
нению со шлаками системы СаО—SiOg. Это объясняется
тем, что величина Cs включает равновесное давление кис-
лорода ро,> а значение ро,для области, в которой могут су-
ществовать шлаки с высоким содержанием МпО или FeO
довольно велико.
126
Для расчета сульфидной емкости шлаков, отличающих-
ся по составу от изученных, можно воспользоваться реко-
мендациями гл. II. Задача сводится к вычислению актив-
ности кислорода [a(02-}, Д(СаО)Или а<о)] и коэффициента
активности серы в двух шлаках: в заданном и в одном из
изученных (с наиболее близким составом к заданному).
О 0,2 Oft 0,6 0,8 хосн
Рис. 25.
Дальнейший расчет выпол-
няется при помощи выра-
жения (219). Подставив в
него вычисленные характе-
ристики изученного шлака и
известное значение Cs на-
(sh%
fsC% SJ
от мольной
Сульфидная емкость различных шлаков в зависимости
доли основного оксида *осни температуры [100]:
Номер кривой Шлак т, к Номер кривой Шлак Г. К
1
Г
2
2*
3'
4
4'
СаО + SiO,
СаО + SiO.
СаО 4- AljOa
СаО 4- ALO;
MgO + SiO,
МпО + SiO,
1773
1923
1773
1923
1923
1773
1923
МпО 4- Al,О,
СаО 4- AlrO3 + CaF,
л _ =1,3:П
I, А1,О3 CaF, )
СаО 4- CaF,
СаОт
FeO
1923
1773
1773
1923
1823
8
А — промышленные шлаки: о.м. — основной мартеновский (1923 К); к.м. — кис-
лый мартеновский (1923 К); о.д. — основной доменный (1773 К)
Рис. 26. Зависимость коэффициента распределения серы при 1823—1973 К от со-
держания FeO для шлаков с долей свободных оснований R, равной 0,22 (/);
0,15 (2) и 0,10 (3) [2]:
А — доменные шлаки; Б — восстановительные шлаки электросталеплавильного
процесса; В — окислительные шлаки
127
ходят константу Къ а затем обратным расчетом вычисля-
ют Cs для заданного шлака (с другими значениями актив-
ности кислорода и коэффициента активности серы).
Зная сульфидную емкость шлака, можно оценить тер-
модинамический коэффициент распределения серы между
шлаком и металлом Ls = (%S)/[%S]. Для такой оценки
величину Cs нужно сначала преобразовать при помощи
выражений (189) и (217) в коэффициент Ls-oв
= (%S) [%O]/[%SJ, который представляет собой видоиз-
менную константу равновесия реакции
[S] + (О2-) = (S2-) + [О]; к = as2- [ % О]/1 % S]. (223)
(при заданных значениях а^г-и коэффициента активности
серы). Для нахождения Ls требуется определить содер-
жание в металле растворенного кислорода. Очевидно, что
лрн данном значении С5 десульфурация металла будет
проходить тем лучше, чем полнее он раскислен. Реакция
(223) идет с поглощением тепла (100—200 кДж). Это сле-
дует из температурной зависимости Cs (см. рис. 25) и
уравнений (188) и (217). Исследования влияния темпера-
туры на Ls_o дают аналогичные результаты.. Рассчитаем
коэффициент распределения серы между железом и извест-
ково-глиноземистым шлаком для условий, исследованных в
работе [101]. Из сопоставления выражений для Cs и Ls-o
следует __ ____________
lg = lg Cs + 1g (Ю1//ро_) - Ig (IS)//^). (224)
Согласно рис. 25, значение lgCs для шлака, содержа-
щего 53.% СаО и 47 % AI2O3 (хсао =0,673), при 1873 К
составляет примерно —2, 4. С учетом выражений (189) и
(217), считая fo=fs=l получим lgLs_o =—2,4 + 3,42—
—2,53=—1,51; Ls_o=0,03. Для перехода к Ls оценим
концентрацию кислорода в металле по содержанию алю-
миния, использовав график раскислителыюй способности
элементов [2]. Результаты расчетов и экспериментов при-
ведены в табл. 15. Сопоставление показывает, что расчет-
Таблица 15. Расчетные и экспериментальные значения Ls
[А1], % [О], % LS [А1]. % [О]. %
0,02 0,00034 90/35 0,05 0,00019 160/150
0,03 0,00026 115/70 0,1 0,00012 250/500
* В числителе — расчетные значения, в знаменателе—экспериментальные [101,
102[.
128
ные значения могут служить лишь для ориентировочной
оценки Ls.
Экспериментальные значения коэффициента распределе-
ния серы для различных шлаков приведены на рис. 26 [2].
Мерой основности шлаков, представленных на рис. 26, слу-
жит R — доля «свободных» оснований, которая вычисля-
ется по следующим формулам: для восстановительных ус-
ловий электроплавки и доменной плавки /?=«сао +
+2/3«MgO—rtSiO»=^Al1O, — число молей оксидов в
100 г шлака), для условий окислительного рафинирования
металл a R=Нсао+Лмво+Пмпо—2nsio,—4пР1о4.
Взаимодействие серы с газовой фазой. Переход серы из
металла в газовую фазу наблюдается главным образом при
окислительном рафинировании расплавов. Он происходит
также в некоторых процессах вакуумной металлургии, од-
нако, широкого практического применения десульфурация
в вакууме не имеет. Термодинамические характеристики ре-
акций; сопровождающихся переходом серы из металла в
газовую фазу, можно найти при помощи табл. 14. Приве-
дем выражения для констант равновесия некоторых реак-
ций такого типа (табл. 16).
Расчетные давления различных серосодержащих газов
над расплавами железа с заданными значениями активнос-
ти серы приведены на рис. 27, из которого видно, что зна-
чительное давление чистой газообразной серы, превышаю-
Та блица 16*. Коэффициенты в уравнении lgK=A/T+B
Реакция
Г /г п2(г) = п;
+ [Н] = HS(r)
НгСг) —
4- 2[Й] = H2S(r)
+ [С] = CS(r)
IS] + [О] = SO(r)
IS] + 2(0] = SO2(r)
IS] + 3(0] = SO3(r)
IS
(г)
В
—3670 —0,518
—7718 0,283
—5812 1,874
973 —3,12
4786 0,066
—13672 1,845
—5714 —3,002
2003 —2,397
—4731 —4,510
—6803 —0,426
2662 —4,563
1528 —9,450
—12908 2,735
—8113 —1,873
* Расчеты выполнены А. В. Богдановым по данным [4, 206].
9—675
129
щее остаточное давление при вакуумной индукционной
плавке (~0,1 Па), наблюдается лишь при высокой актив-
ности серы в расплаве. Такое значение активности можно
ожидать только в чугуне (при высоком содержании серы,
а также углерода, кремния, фосфора, повышающих величи-
ну fs). В этих условиях возможно также удаление серы в
виде COS, H2S, HS, SO2, CS, SiS.
Приведем для примера расчет удаления серы из рас-
плава, содержащего 0,02 % S; 0,1 % С; 0,002 % О, в случае
0,001 0,01 0,1
Рис. 27. Равновесное давление серосодержащих газов (рр (см. табл. 16) для
сплавов на основе железа при 1873 К при различных условиях расчета:
2, *-Рн -1 атм; 3, 5-[Н]—0,001 %; 6, 7 —[С]—0,1 %: 8-[С]=0,1%; [О]—
-0,23 % (условно— на поверхности металла); 8' — [С]-0,1 %, [О]=0,022 %; 9 —
12 — [О]—0,23 %; 9'—121 — [0]=0,022 %; 13, 14— [Si]=0,l %
[8], %
0,50,151,0 1,5 2,0 2,53,0 5,0
Рис. 28. Зависимость растворимости сульфида марганца в чугуне от содержания
марганца и температуры [4]
пропускания над ним водорода со скоростью 1 м3/мин (при
1873 К и рн, =101,325 кПа=1 атм). Результаты определе-
ния (при помощи приведенных на с. 129 уравнений) содер-
жания серы в удаляемом газе приведены в табл. 17.
Максимально возможная скорость удаления серы из
расплава равна 5,3288* 10-5 (32* 103/22,4) =0,076 г серы на
1 м3 водорода, или 0,076 г/мин.
Взаимодействие серы с сульфидообразующими элемен-
тами. Иногда для десульфурации расплавов используют
высокое химическое сродство некоторых металлов к сере,
130
Таблица 17. Содержание серы в удаляемом газе
Газ
H2s
HS
cos
Равновесное
давление газа
атм
5,018
0,2905
0,0188
{SJ -10\
моль S
моль Н,
5,018
0,2905
0,0188
Газ
Равновесное
давление газа
р.«10\ атм
0,0004
0,0007
{S}-10%
моль S
моль Н,
0,0008
0,0007
Примечание. {S}_ —5,3288-10“*'
сопоставимое с химическим сродством к кислороду или да-
же более высокое (Na, К). При введении таких элементов-
десульфураторов в металл удаление серы происходит ана-
логично процессу глубинного раскисления. Имеющиеся све-
дения об устойчивости некоторых наиболее прочных
сульфидов представлены ниже в виде значений AG0 реакций
их образования [2], кДж/моль:
Сульфид . . . .
—ДС?298 , , . . .
—ДО1873 . . . .
A12S3 BaS CaS
275 464 513
Нет св. 340
401
Нет св.
Сульфид , . , .
—AGms • . . .
—AG1873 • . . .
MgS
455 389
Нет св. 184
Продолжение
MnS Na2S TiS
248 400 254
140 Нет св.
При использовании относительно слабых десульфурато-
ров, например марганца, заметное удаление серы может
быть достигнуто только в условиях пониженной температу-
ры. Результаты одного из исследований десульфурации чу-
гуна (согласующиеся с данными термодинамического рас-
чета при /s = 5-?-6) представлены на рис. 28, из которого
видно, что несмотря на большое (по сравнению со сталью)
значение коэффициента активности серы в чугуне и более
высокие концентрации марганца (~1 %) и серы (<С0,1 %),
заметная десульфурация расплава может иметь место толь-
ко при <1600 К. Такие условия реализуются в миксерах и
ковшах, в которых и наблюдается некоторое удаление се-
ры в виде сульфида марганца, всплывающего в шлак. При
выплавке стали марганец не может служить десульфура-
тором. Ослабление вредного действия серы, которое дости-
гается в присутствии марганца, связано с образованием
MnS при охлаждении слитка. Замена, даже частичная,
9*
131
вредных легкоплавких сернистых соединений (сульфидов
и оксисульфидов железа) на значительно более тугоплав-
кий сульфид марганца обеспечивает снижение краснолом-
кости стали и другие положительные последствия. Анало-
гичным образом взаимодействует с серой алюминий и не-
которые другие элементы. Для удаления серы из стали за
счет образования сульфидов применяют более сильные де-
сульфураторы — кальций, РЗМ (Се, La) и др. Эффектив-
ными десульфураторами являются и соединения сульфид-
образующих элементов: карбид и цианамид кальция (СаС2,
CaCN2), сода (Na2CO3) и др. Термодинамические характе-
ристики реакций взаимодействия десульфураторов с рас-
плавами могут быть определены при помощи справочных
таблиц. Рассчитаем для примера минимальную концентра-
цию серы, которую можно получить при обработке стали
карбидом кальция. При помощи табл. 1, 4 найдем:
CaCj(T) = Са(г) 4- 2С(гр); Д6° = 213000 - 61,057 Дж.
[S] = 4zS2(r}-, AG° = 72000 + 9,927 Дж
Са.г. + = CaS,т); Д6° =- 702500 + 193,37 Дж
2Q.P = 2[С]; ДС° = 45200 — 84,67 Дж
СаС2(т) 4- IS] = CaS. . + 2 [С];
х\*/ I1/
4- 57,617 Дж
Д6° =— 373 700 4-
(225)
Отсюда при 1873 К:
lgK=lg
[С]2 * 4 * /с Qcag
IS] /saCaC,
264 000 2
19,14-1873
7,352.
(226)
Если принять, что СаС2 и CaS находятся в самостоятельных
фазах, то их активности можно считать равными 1. Коэф-
фициенты активности /с и fs при небольших [С] и [SJ
также можно приравнять 1. В результате получим:
[C]2/|jS]=2,25-107, откуда следует, что равновесная кон-
центрация серы в расплаве для рассмотренных условий
очень низка (при 1 %С — 4-10~8 %). Таким образом, рас-
чет показывает, что термодинамические ограничения для
процесса десульфурации стали в результате обработки кар-
бидом кальция (например, вдувания порошка) практически
отсутствуют.
IV.6. ФОСФОР В РАСПЛАВАХ
Фосфор имеет высокое химическое сродство к железу и ни-
келю, которое проявляется, например, в большом тепловом
эффекте образования соответствующих растворов. Калори-
метрическими измерениями [204] энтальпий растворения
фосфида никеля в железном и никелевом расплавах полу-
чены следующие значения ДЯ реакции */2Р2(г)= [Р]:
ДЯ“Ре) = ~ 140 ± 35 КДЖ> A^P(Ni) =—252 ± 30 кДж.
(227)
Полученное значение ДДр в железе хорошо согласуется
с результатами равновесных измерений (—123 кДж) [2].
Если рассчитать температурную зависимость ДО00 реакции
(227), приняв за основу калориметрическое значение Д/7~
и величину Дб~73, по данным [2], то уравнение будет
иметь следующий вид: AG°°=—140 000—9,6 Т Дж. Для
никеля в связи с отсутствием равновесных данных значение
Дб°° реакции (227) определить нельзя.
Растворы фосфора в железе характеризуются значитель-
ными положительными отклонениями от закона Генри, что
согласуется с данными о сильном химическом взаимодейст-
вии компонентов. Коэффициент активности фосфора в би-
нарном растворе может быть определен из выражения
lgfp=0,062[% Р]. Влияние других присутствующих в рас-
плаве элементов на fp можно учесть при помощи парамет-
ров взаимодействия, приведенных в табл. 6—8.
Удаление из стали фосфора производят путем его окис-
ления и перевода в шлак. Для понижения активности про-
дуктов реакции в шлак добавляют известь. Согласно пред-
ставлениям молекулярной теории шлака, процесс дефосфо-
рации металла можно описать уравнением
2 [Р] + 5 [FeO] + 4 (СаО) = (4СаО -РД) + 5Fe. (228)
Экспериментальные данные о равновесии реакции
(228), полученные Уинклером и Чипманом, имеют следу-
ющий вид [54]:
1g К = 1g х<4СаО'р»°»)— = — 15,06. (229)
[Р]2 FeO) ‘ rfcaO')
Величина Х(сао') представляет собой мольную долю
«свободной» СаО (не связанной в соединения с кислотны-
ми оксидами): х(Сао') = *(CaO) — 2x(Sio2) — 4x(PiOs)—
133
—2х(а1,о,). Графическая интерпретация результатов рабо-
ты [54] представлена на рис. 29. При этом Ар=Х(4сао Р,о,)/
/[%PJ2; В= [ (СаО) 4- (MgO) + (МпО) ] /[ (SiO2) +2 (Р2О5) +
+ V2 (Fe2O3) + '/2 (А12О3) ] % (мол).
Как видно из приведенных данных, для дефосфорации
металла требуются пониженная температура и шлак с до-
статочно высоким содержанием FeO и СаО. Практическую
Рис. 29. Зависимость коэффициента распределения фосфора Lp от основности
шлака В, содержания (FeO) и температуры [54]:
а — 1873 К; б —Lp=100
оценку необходимых условий дефосфорации можно сделать
на основе упрощенного графика, приведенного на рис. 30
или при помощи эмпирического уравнения: lg(%P)/
/[ % PJ = (22350/Т) —16,0+2,51g (% Feo6l4) +0,08 (% СаО) ±
±0,4, предложенного в работе [105] для шлаков (СаО) +
+ (FeO) + (SiO2) с нормальным содержанием (MgO),
(МпО) и (А12О3).
В последние годы рядом исследований показано, что фосфор подоб-
но сере может переходить в шлак не только в окисленной (фосфатной)
’форме, но и в восстановленной (фосфидной). Это имеет большое значе-
ние в связи с необходимостью разработки процесса дефосфорации высо-
колегированных расплавов без окисления содержащегося в них хрома
и других элементов. Экспериментальные данные о влиянии окислитель-
ного потенциала на коэффициент распределения фосфора между шлаком
я металлом приведены на рис. 31. Три левые линии на рис. 31 относятся
к восстановительным условиям (расплав Fe—Al—Р, шлак СаО : А120з =
= 1:1), правая линия — к окислительным условиям [54J (расплав Fe—Р,
шлак основностью CaO/SiO2^3). При построении графика равновесные
значения определяли при помощи термодинамических характеристик
[2] следующих реакций:
в восстановительных условиях
2 [А1] +3/2О2(г)
а(А12О,)
а[А1]
82000
+ 13,9 ;
134
в окислительных условиях
[Fe] + V2O2(r) =(FeO);
SPO, ~2 ^(FeO) “
12500
+ 2,6
Активность AI2O3 в шлаке определяли по экспериментальным данным:
[2], активность FeO — по формуле В. А. Кожеурова (см. гл. II). Как вид-
но из рис. 31, дефосфорация в восстановительных условиях требует высо-
кой температуры и очень сильной раскисленности металла и шлака. По>
сравнению с кривыми для серы (см. рис. 24) кривые для фосфора су-
щественно сдвинуты в область более низких значений Pq*. Для обеспе-
Рис. 30. Зависимость коэффициента распределения фосфора
основности и степени окисленности шлака при 1873 К [1051
основность (CaO)/[(SiOa) 4- (Р2О5)]
Lp-(%PiOs)/[p]2 от
[цифры у кривых —
Рис. 31. Зависимость коэффициента распределения фосфора между шлаком и
металлом от окислительного потенциала [1061
чения необходимой раскисленности системы требуется присадка значи-
тельного количества элементов, обладающих высоким химическим срод-
ством к кислороду. При использовании алюминия (как в опытах, представ-
ленных на рис. 31) его концентрация достигает 10—20 %. Это затрудня-
ет применение процесса в сталеплавильной технологии. Значительно
лучшие результаты достигаются при использовании расплава Са—CaF2>
который обеспечивает хорошее раскисление системы без существенного
перехода кальция в металл [202]. Необходимо отметить, что пока можно
говорить лишь о принципиальной возможности восстановительной дефос-
формации металла в условиях сталеплавильного производства. Практи-
ческая реализация процесса связана с рядом трудностей, которые пока
не преодолены.
IV.7. АЗОТ В ЖЕЛЕЗЕ, НИКЕЛЕ И ИХ СПЛАВАХ
Растворимость азота в железе подчиняется закону Сиверт-
са (закону квадратного корня):
V. N2=INJ; IN1 = Xn1/7^, (230)
135
где [N] — растворимость
циальном давлении ры/,
пение которой зависит от
Рис. 32. Зависимость раствори-
мости азота в железе от тем-
пературы
азота в железе при данном пар-
— константа, численное зна-
температуры и способов выраже-
ний концентраций и давления.
Выполнимость закона Сивертса
указывает на идеальность обра-
зующегося раствора. Результаты
измерений растворимости азота
в жидком железе, выполненные
различными методами разными
авторами, хорошо согласуются
[107—111]. Авторы работы [112]
рекомендуют следующее уравне-
ние температурной зависимости
растворимости азота в жидком
железе до 2550 °C:
lgl%N] =(-850/73-0,905 4-V2 lg pNj. (231)
Из уравнения (231) следует, что при 1873 К и Pn, =
= 1 атм растворимость азота в жидком железе составляет
0,044 %. Температурный коэффициент растворимости азота
равен 1,0 • 10-5 %/К. Зависимость растворимости азота в
железе от температуры приведена на рис. 32. Энтальпия
растворения азота в жидком железе ДТ/n представляет
разность двух противоположных по знаку величин: энталь-
пии диссоциации молекулярного азота на атомы (Д//ДНс) и
энтальпии растворения атомарного азота в жидком желе-
зе (ДЯР). Первый процесс является эндотермическим, вто-
рой — экзотермическим.
Поскольку ДЯдис>ДЯр, то и суммарный процесс, опи-
сываемый уравнением (230), протекает с поглощением теп-
ла (Д/7н = 16280 Дж/моль (см. табл. 4).
Анализ 47 исследований, выполненных различными ав-
торами методами Сивертса и отбора проб показывает, до
статочно хорошее совпадение растворимости азота в желе-
зе при 1873 К [N] =0,044+0,002 %. В то же время энталь-
пия растворения обнаруживает значительный разброс.
Результаты статистической обработки массива из 513 экс-
периментальных точек можно описать двумя уравнениями
[98]: при < 1973Klg[%N] =—560/7 — 1,06; при >1973К
lg[%N]=—1100/7— 0,79. Таким образом энтальпия рас-
творения азота в жидком металле до 1973 К Д#№ 10700
Дж/моль, а выше 1973К Д// =21000Дж/моль. А. М. Са-
марин [161], первый обративший внимание на это, связы-
1973 К
136
вал изменение Д/Zn при изменении температуры со струк-
турными превращениями в жидком состоянии [233]. Извест-
но, что при растворении кислорода в жидком железе оксид-
ная фаза появляется при очень низком парциальном давле-
нии кислорода в газовой фазе (Ро, = 10-8атм). В отличие
от этого при растворении азота в жидком железе даже при
давлениях £n,>1 атм самостоятельная нитридная фаза не
образуется. Это означает, что в условиях открытой плавки
азот с жидким железом образует истинный раствор.
Образование нитридов типа Fe2N и Fe4N имеет место
лишь в твердом металле в температурном интервале суще-
Рис. 34. Зависимость коэффициента активности азота в жидком железе при
1873 К и =1 атм от содержания легирующих элементов [/?] [2]
Рис. 33. Зависимость растворимости азота [N1 в жидком железе при 1873 К и
а™ ® от содержания легирующих элементов [/?] [2]
ствования аустенита. Реальные стали и промышленные
сплавы содержат легирующие элементы, которые оказыва-
ют существенное влияние на термодинамическое поведение
азота. Присутствие нитридообразующих элементов значи-
тельно упрочняет силы связи с азотом и может привести
к образованию нитридов в расплаве в виде самостоятельной
фазы. Влияние легирующих элементов на растворимость
азота в сплавах на основе железа показано на рис. 33. По
характеру влияния на растворимость азота элементы мож-
но разделить на следующие группы:
1. Титан, цирконий, ванадий, ниобий и другие элемен-
137
ты, образующие с азотом прочные нитриды. С увеличением
концентрации этих элементов растворимость азота в жид-
ком железе возрастает.
2. Хром, марганец и молибден, характеризующиеся
большим химическим сродством к азоту, чем железо. При
pNj =1 атм эти элементы в жидком железе при концентра-
циях, встречающихся в стали, нитридов не образуют, одна-
ко заметно увеличивают растворимость азота в жидком
расплаве.
3. Углерод, фосфор, никель, медь, снижающие раствори-
мость азота в сплавах с железом.
4. Кремний и алюминий, являющиеся нитридообразую-
щими элементами. Первый снижает, а второй не изменяет
растворимости азота в железе.
5. Кислород и сера, мало влияющие на растворимость
азота в железе. Являясь сильно поверхностно-активными,
они оказывают существенное влияние на кинетику погло-
щения азота.
Коэффициент активности, описывающий влияние леги-
рующего элемента [/?] на термодинамическое поведение
азота, выражается формулой (33). На рис. 34 показано
влияние легирующих элементов на коэффициент активнос-
ти азота в жидком железе при 1873 К- При помощи урав-
нения (33) можно вычислить растворимость азота в сплаве
любого состава. Для этого прологарифмируем уравнение
(33), а коэффициент активности /n, выразим через пара-
метры взаимодействия. При наличии нескольких легирую-
щих элементов уравнение имеет вид:
lg I%N]cna = lg[°/oN]Fe (232)
Пример. Вычислить растворимость азота [%N]Crw в кор-
розионностойкой стали 12Х18Н8 при 1600 °C и Pn, =1 атм.
Ввиду малых содержаний углерода, кремния и марганца
их влиянием можно пренебречь. В соответствии с уравне-
нием (232) имеем: lg[%N]Cra=lg[%N]Fe — eNr[o/oCr]—
-ей [%Ni]; lg[%N]cnJI = IgO,0444-0,047-18-0,010-8=
= -0,59; [%N]c™=0,25 %.
Применение параметров взаимодействия первого поряд-
ка в большинстве случаев обеспечивает необходимую точ-
ность расчета. Однако в ряде случаев нужно учесть также
параметры взаимодействия второго порядка. Это улучша-
ет совпадение расчетных данных с экспериментальными.
Для рассматриваемой задачи г£г=4-10-4; г^1 =0. В этом
случае lg[ % N] спл = lg[ % N]Fe-^r[ % CrJ-ей1 [ % Ni] -
138
-rN [%Cr]2; lg[%N]cna = lg0,044 + 0,047-18-0,01-8 -
—4-10"4-182=—0,683; [%NCIHi] =0,207. В работе [210] на
основе анализа экспериментальных данных по 43 сплавам
на основе железа показано, что при расчете растворимости
азота с использованием парамет-
ров взаимодействия только пер-
вого порядка погрешность со-
ставила 28 %. При учете пара-
метров второго порядка средняя
погрешность снижается до 8%.
Расчет по формуле (105) дает
погрешность 5 %. Растворимость
азота при температурах, отлич-
ных от 1873 К, можно вычислить,
пользуясь данными по темпера-
турной зависимости параметров
взаимодействия или при помощи
уравнения Чипмана — Коррига-
на (см. с. 60).
Условия нитридообразования.
Для описания фазового равно-
весия в системе Fe—R—N при
Рис. 35. Зависимость количест-
ва поглощенного азота ЫпогЛ
°т/^:
ОА — растворимость азота в
жидком железе; ОВ и ОЕ—рас-
творимость азота в присутст-
вии нитридообразующего эле-
мента [Я'] и [Я"Ъ [Я"]>[Я'];
BCD — суммарное количество
поглощенного азота в растворе
и нитриде: Е и В — точки нит-*
ридообразовання
некоторой постоянной,
температуре Т рассмотрим зависимость количества азота,
поглощенного расплавом (Ь1ПОгл) от УрмДрис. 35). Прямая
ОА характеризует изменение растворимости азота в жид-
ком железе в зависимости от|/ры,> в соответствии с зако-
ном Сивертса. Ранее указывалось, что нитриды железа при
этом не образуются, поэтому имеет место линейное увеличе-
ние концентрации растворимого азота с ростом его парци-
ального давления. Введение в жидкое железо элементов
[/?], обладающих большим химическим сродством к азоту,
чем железо, изменяет ход изотермы на рис. 35. В этом слу-
чае увеличение растворимости азота в расплаве Fe—R с
КPn,наблюдают лишь до определенного предела. В точках
В и Е из расплава выделяются нитриды и равновесие в
трехфазной системе определяется реакцией
[/?] + IN] = tfNK0W.
(233)
Таким образом, линии ОВ и ОЕ соответствуют равнове-
сию растворенного азота с газовой формой в расплавах
Fe—R. С увеличением концентрации легирующего элемен-
та образование нитридов происходит при меньшем парци-
альном давлении азота. Следовательно, линия ОЕ соответ-
139
ствует расплаву с большей концентрацией легирующего
элемента, чем ОВ. Точки В и Е соответствуют трехфазно-
му равновесию в системе (газ, расплав, нитрид). При по-
стоянном значении Pn, (линия 1—1) с увеличением кон-
центрации легирующего элемента растворимость азота
возрастает вследствие упрочнения связи R—N, так как ко-
эффициент активности азота уменьшается. Отсюда следует,
что с увеличением концентрации легирующего элемента уг-
ловой коэффициент прямых ОА и ОБ будет возрастать.
Рассмотрим ход кривой на рис. 35 за точкой В. Если
жидкое железо содержит небольшое количество [7?], то
зависимость Nnor.n otVPn, изменяется по кривой ВС. На
этом участке нитридообразующий элемент в результате
взаимодействия с азотом по реакции (233) полностью пе-
рейдет в нитрид. Таким образом, на участке CD расплав
не содержит нитридообразующего элемента и концентрации
растворенного азота будут возрастать по тому же закону,
что и по линии ОА, т. е. с тем же угловым коэффициентом.
Выпадение нитридной фазы при ри, =1 атм и 1873 К по
расчету наблюдается при следующих концентрациях нитри-
дообразующих элементов: титана 0,29 %, алюминия 0,82 %;
циркония 0,093 %.
С учетом фазового равновесия условия нитридообразо-
вания можно описать при помощи трех независимых пере-
менных: температуры, ри, и концентрации легирующего
элемента [/?]. Вместо давления Pn, можно задать связан-
ную с ним равновесную концентрацию азота в расплаве
[%N]. Если в рассматриваемой системе образуется нитрид,
то число фаз возрастает до 3, число степеней свободы
уменьшается до 2. Это означает, что из трех указанных
независимых переменных достаточно задать две для того,
чтобы определить состояние системы. Третья переменная в
этом случае зависит от двух других. Следовательно, для
того, чтобы определить условия существования нитридной
фазы, достаточно записать уравнение, связывающее меж-
ду собой три указанные переменные. Такая задача, в част-
ности, была решена в работе [113].
Пример. Определить минимальную концентрацию тита-
на, при которой образуются нитриды титана (TiN) в рас-
плаве Fe—Ti—N при 1873 К и Pn, = 1 атм. Коэффициент
активности титана в разбавленном растворе железа при
1600 °C равен =0,037 (стандартное состояние — чистый
жидкий титан). Запишем уравнение реакции образования
нитрида титана:
140
Tix + = TiN(T); AG?873 =- 135140 Дж;
lg % = 1g 1 _ AG° = 135140 =
Vti’N-ti-Pn.2 19.15.1873 19,15-1873 ’ ’
!g*Ti =— ]g —ig Tn—tepfe
lg*Ti =~ 3,76 — lg 0,037; x = 3-10-3; [Ti] = 0,29%.
Пример, Определить концентрацию циркония, при кото-
рой образуются нитриды циркония при 1873 К и рн, = 1 атм.
Zr(T) + V2N2(r> = ZrN(T); AG° = — 351800 + 91,297;
Zr(}H) = Zr(T,; AG° =-16736 + 7,8837;
___ [Zr](1%) =Zr(xt); ДО0 = 80330 + 34,97’.
[Zr](1./e) + »/2N2(r) = ZrN(T); ДС°=*- 288206 + 134,0737
IgK = lg----------------= = - 7,004.
l°/oZr].fZr./PNt T
При 1873K lgX=l,03fZr =1, pN, = l, тогда lg[%Zr] =
=—lgK=—1,03; [Zr] =0,093 %.
Растворимость азота в никеле. Никель является основой
многих жаропрочных сплавов и входит в состав многих
коррозионностойких и конструкционных сталей. Темпера-
турная зависимость растворимости азота в жидком никеле
описывается следующим уравнением [33]:
VsNjj = [N]Ni; AG° = 69000 + 18,627;
(234)
lg f «N1n, = (- 3610/T) - 0,973 + 0,51g pN[. (235)
«
Из уравнения (235) следует, что при р^г = 1 атм и
1873 К растворимость азота составляет 0,0015 %. Большая
теплота растворения азота в никеле (69000 Дж/моль), чем
в железе (16280 Дж/моль) свидетельствует о более силь-
ной температурной зависимости растворимости. Вместе с
тем это указывает на то, что растворение азота в никеле
энергетически затруднено в большей мере, чем в железе.
Химическое сродство никеля к азоту меньше, чем железа.
В связи с этим влияние легирующих элементов на раство-
римость азота в сплавах на никелевой основе проявляется
несколько сильнее, чем в сплавах на железной основе. Ни-
же приведены значения параметров взаимодействия c;N в
141
сплавах на основе никеля при 1873 К, определенные в раз-
ных работах:
/......... А1 Се Ti Zr V Nb
(1873) • • 0 —0.53 —°»21 —°’23 —°.13 —°.O74
Продолжение
.......... Ta Cr Mo W Fe Co
«Lmi” 0,065 —0,10 —0,038 —0,022 —0,018 0,0054
Для перехода к другим температурам рекомендуется
уравнение
4((Т) = [(2440/7) - 0,305] 41873). (236)
Параметры взаимодействия второго порядка для нике-
левых сплавов, как правило, отсутствуют, исключая г£г »
=0,00103 [207]. Сопоставление экспериментальных дан-
Рис. 36. Активность азота в жид-
ком хроме при 2018 К [4]
ных по растворимости азота в
20 никелевых сплавах с ре-
зультатами расчета по пара-
метрам взаимодействия пока-
зывают, что средняя погреш-
ность составляет 48 % [211].
Более высокая погрешность,
чем для расплавов на основе
железа, связана не только с
недостатком данных по пара-
метрам взаимодействия и их
меньшей надежностью, но еще
и большими абсолютными зна-
чениями коэффициентов ак-
тивности.
Азот в хроме. Поведение раствора азота в хроме
отклоняется от закона Сивертса (рис. 36). На правой оси
ординат отложено значение р|^2. Линейная зависимость
между [%N] и р]^ наблюдается лишь до 1 % N. Таким об-
разом, закон Сивертса соблюдается лишь при концентра-
циях, не превышающих 1 %. При более высоких концентра-
циях наблюдаются положительные отклонения от закона
Генри, связанные с уменьшением межчастичной связи меж-
ду азотом и хромом. При Pn, = 1 атм, 2018 К в жидком
хроме растворяется 5,2 % N. Это позволяет получить в про-
мышленных условиях азотированный феррохром с содер-
жанием азота до 6 %. Термодинамическое поведение азота
в жидком хроме характеризуется той особенностью, что
142
процесс растворения сопровождается сильным химическим
взаимодействием и протекает с выделением значительного
количества тепла, поэтому с повышением температуры ра-
створимость азота уменьшается. Тем не менее нитриды хро-
ма в виде самостоятельной фазы даже при столь больших
концентрациях азота не образуются. Расчеты показывают,
что при 1873 К CrN выпадает лишь при ры, =20,5 атм.
Растворимость азота в жидких сплавах Fe—Сг с повыше-
нием концентрации хрома увеличивается. Температурный
коэффициент растворимости азота в жидком железе при
увеличении концентрации хрома изменяет знак. В сплавах
Fe—Сг при концентрации хрома <2,3 % с повышением
температуры растворимость азота растет. В сплавах, со-
держащих >2,3 % Сг, с повышением температуры раство-
римость уменьшается. Очевидно, что в сплаве, содержащем
— 2,3 % Сг, растворимость азота в жидком железе от тем-
пературы не зависит (Д#ы=0). Растворимость азота в
сплавах Fe—Ni с увеличением концентрации никеля сни-
жается. Таким образом, термодинамическое поведение азо-
та в расплавах Fe—Сг—Ni определяется равнодействующей
двух взаимопротивоположных влияний, оказываемых нике-
лем (снижает растворимость) и хромом (повышает раство-
римость).
IV.8. ВОДОРОД В РАСПЛАВАХ
Растворимость водорода в жидком железе подчиняется
закону Сивертса:
V2H2(r) = (И; Кн = [%Н]//^;. (237)
Согласно работе [115],
lg I %H]Fe - (— 1900/7") — 1,577. (238)
Величина Кн, как следует из уравнения (237), числен-
но равна растворимости водорода при рн, = 1 атм и задан-
ной температуре. При 1873 К и рн, = 1 атм [Н] =
=0,0027 % *. Энтальпия растворения водорода ДЯн =
=32100 Дж/моль. Растворимость водорода в жидком ни-
келе описывается уравнением [116]:
lg I %H]N1 = (- 924/7) - 1,88. (239)
Зависимость растворимости водорода в железе от тем-
пературы приведена на рис. 37, из которого видно, что при
* 29 см3/100 г, или 27 ppm, 1 ppm= 10-4%.
143
температуре кристаллизации наблюдается скачкообразное
изменение растворимости водорода от 28 до 8 см^/100 г.
Это имеет большое значение для металлургической прак-
тики. В процессе кристаллизации водород выделяется из
раствора и это позволяет получать литой металл (слитки
или непрерывную заготовку)
Ш.смЧЮОг
Рис. 37. Зависимость растворимо-
сти водорода в железе от темпе-
ратуры
с меньшей концентрацией рас-
творенного водорода. Раство-
римость водорода в аустените
выше, чем в б-Fe. Это означа-
ет, что стали, кристаллизую-
щиеся в форме y-Fe и имею-
щие аустенитную структуру,
могут содержать больше водо-
рода в твердом растворе, по-
этому такие стали менее под-
вержены пористости. Уравне-
ние (237) пригодно для расче-
та растворимости в жидком
железе, находящемся в равновесии с газовой фазой, содер-
жащей молекулярный водород. Однако в реальных услови-
ях парциональное давление молекулярного водорода в печ-
ной атмосфере ничтожно мало, а содержание водорода в
металле определяется парциальным давлением паров во-
ды и влажностью шихтовых и шлакообразующих материа-
лов. Влияние легирующего элемента на растворимость ко-
личественно описывается коэффициентом активности /g
(рис. 38, 39). Зная параметры взаимодействия (е£ ) и рас-
творимость водорода в железе ([% H]Fe), можно вычис-
лить растворимость водорода в сплаве данного состава
при заданной температуре:
lg I %Н]СПЛ = Igl %H]Fe— Sag [ % fl].
(240)
Коэффициент активности водорода при любой темпе-
ратуре Т для сплава заданного состава можно вычислить
на основе приближения Чипмана и Корригана по следую-
щей формуле:
IgfS(T) = 1(6945/7) — 2,481 SeS(I(ra) [%Я.
(241)
Количественные изменения растворимости водорода в
жидком железе в присутствии третьего элемента опреде-
ляются соотношением сил химического взаимодействия
между частицами растворителя (железа), растворенного
вещества (водорода) и легирующей добавки (/?). Гидри-
144
дообразующие элементы (Zr, Ti, Nb, V, Се) увеличивают
растворимость водорода. Элементы, не образующие гидри-
дов (Cr, Ni, Мп, Со, Мо), слабо влияют на растворимость.
Группа элементов (С, Si, S, Р, А1 и др.) уменьшает раство-
римость водорода. Это связано с сильным взаимодействи-
ем между атомами железа и легирующей добавкой, с обра-
зованием карбидов, силицидов, сульфидов, фосфидов.
Рис. 39. Зависимость коэффициента активности водорода в жидком железе
н
при 1873 К от содержания легирующих элементов
Рис. 38. Зависимость растворимости водорода в жидком железе при 1873 К от
содержания легирующих элементов [2]
алюминидов или соответствующих группировок. В резуль-
тате этого термодинамическая активность железа снижа-
ется, что приводит к уменьшению растворимости водорода.
Пример. Определим, возможно ли образование гидри-
дов церия в стали до ее затвердевания (при концентрации
церия 0,1 %, или хсе=4« 10-4) : [Се]+Н2(г)=СеН2(конд);
AG°=—206500+148,7 7 Дж. Вычислим равновесное дав-
ление водорода, необходимое для выпадения гидридов, и
сравним его с фактическим Рн,: 1g Я= 1/(хсе’Рн,) =
=— (AG°/2,3 RT)=—[72570/(19,14-1873)]=— 2,0; К=
= 10-2; рн, =1/(/СхСе) = 1/(10“2-4-10-4)=2,5-105 атм.
Поскольку равновесное давление существенно превы-
шает реальные значения рн,, наблюдаемые на практике,
то гидрид церия образовываться не будет.
10—675
Глава V
РАСКИСЛЕНИЕ СТАЛИ И СПЛАВОВ
Раскисление является заключительной операцией перед
разливкой, которая в значительной мере определяет свой-
ства готового металла. Задачами раскисления являются:
1) снижение растворимости кислорода присадками эле-
ментов-раскислителей, характеризующихся большим хими-
ческим сродством к кислороду, чем железо, до уровня,
обеспечивающего получение плотного металла; 2) созда-
ние условий для возможно полного удаления образующих-
ся продуктов раскисления из жидкой стали. Термодинами-
ческие характеристики раскисления хорошо изучены и до-
статочно полно изложены в работах [118—121].
V.I. ВЛИЯНИЕ ЭЛЕМЕНТОВ-РАСКИСЛИТЕЛЕЙ
НА АКТИВНОСТЬ КИСЛОРОДА В ЖИДКОМ ЖЕЛЕЗЕ
На рис. 21 представлена зависимость концентрации кисло-
рода от уpot. Наклонные линии соответствуют равнове-
сию растворенного в металле кислорода с газовой фазой.
Точки перегиба отвечают давлениям ро, , при которых
образуются оксиды данных металлов, и нонвариантному
равновесию трех фаз: газовой, металлической и оксидной.
Углы наклона прямых характеризуют константы равнове-
сия реакции растворения газообразного кислорода в соот-
ветствующем металле */2 О2(г)=[О]. При заданном значе-
нии ро, для металла с большим химическим сродством к
кислороду характерна более высокая концентрация раство-
ренного кислорода. Вместе с тем, чем больше химическое
сродство данного металла к кислороду, тем при более низ-
ких парциальных давлениях происходит образование ок-
сидной фазы. Если к исходному металлу Me добавить не-
которое количество раскислителя /?, то линии [О] =f[yrрог)
будет соответствовать больший угловой коэффициент.
В этом случае оксидная фаза появится при более низком
давлении кислорода. Равенству парциальных давлений
кислорода соответствует равенство активности кислорода
в исходном металле Me и в сплаве, содержащем рас-
кислитель (Me — R): ClQ(Me) — аО(Ме—Ц}\ [% О]ме/(О](Ме) =
= [%О] (Aie-₽)f[o](Me-R). Поскольку для Me выполняется
закон Сивертса, то /[О](Ме) =1. Коэффициент ffo] = ffO](Me)
характеризует влияние раскислителя на коэффициент ак-
146
тивности кислорода, растворенного в сплаве Me—R. Сле-
довательно, /[о]= [% О]Ме/[ % О](Л1е-Я)-
Экспериментальные данные по изменению коэффициен-
тов активности кислорода в жидких сплавах на основе же-
леза обычно представляют в координатах lg— [% Я].
Угловые коэффициенты прямых характеризуют параметры
взаимодействия первого порядка (е£). Чем сильнее рас-
кислитель, тем в большей мере он уменьшает коэффициент
активности кислорода.
V.2. АНАЛИЗ КРИВОЙ РАСКИСЛЕНИЯ
Результаты экспериментов по определению раскислитель-
ной способности элементов во многих случаях представля-
ют графически в виде зависимости активности растворен-
ного кислорода О[о] от активности элемента-раскислителя
Я[к]. При этом монотонному снижению О(о] соответствует
увеличение Ц[Л]. Если по оси ординат откладывать не ак-
Рис. 40. Зависимость содержа-
ния кислорода и его активно-
сти в расплавах Fe—Сг при
1873 К от количества вводимо-
го хрома [4]
тивность, а концентрацию раство-
ренного кислорода, а по оси абс-
цисс — остаточную концентра-
цию элемента-раскислителя, то
на кривой растворимости кисло-
рода для большинства раскисли-
телей наблюдают минимум. На
рис. 40 такие данные приведены
для раскисления жидкого железа
хромом. Видно, что с увеличени-
ем содержания хрома до 8 % кон-
центрация растворенного кисло-
рода уменьшается до минималь-
ного значения (0,03 %), а с даль-
нейшим ростом [% Сг] значение
[% О] возрастает, в то время как
Ц[О] непрерывно снижается. Физические причины такого
изменения [% О] связаны с уменьшением коэффициента
активности кислорода с увеличением [ % Я]. Увеличение
концентрации кислорода после точки минимума некоторые
исследователи связывают с растворением продуктов реак-
ции раскисления в жидком железе. Проанализируем зако-
номерности, приводящие к минимуму на кривой раствори-
мости кислорода. Рассмотрим реакцию раскисления
₽ОконД=[7?] + [О]. Примем, что отклонения раствора от
идеальности связаны с поведением кислорода Я[О] #= [ % О].
В этом случае Л=[% Я]«1О] = [% Я] [% O]ffo]. По опре-
10*
147
делению параметра взаимодействия 7[о]=е ° тогда
К=[°/о7?Н%0] [%R1; [%О1 = (242)
Из формулы (242) следует, что зависимость [% О] от
[%/?] носит сложный характер. В реальных условиях при
небольших концентрациях [Я] роль знаменателя проявля-
ется сильнее, и концентрация кислорода с увеличением
[% Я] убывает. С дальнейшим увеличением [/?] (после
точки минимума) роль экспоненты возрастает и это при-
водит к увеличению концентрации растворенного кислоро-
да. Из данных, приведенных на рис. 40, следует, что увели-
чение концентрации железа (правая ветвь кривой) приво-
дит к снижению концентрации кислорода. Однако отсюда
нельзя сделать вывод, что железо может быть раскислите-
лем для жидкого хрома. На самом деле с увеличением
концентрации железа активность растворенного кислорода
увеличивается, что свидетельствует о повышении окисли-
тельного потенциала расплава.
В работе [120] установлена связь между параметром
взаимодействия ео и концентрацией раскислителя xr, ко-
торый соответствует минимальная концентрация кисло-
рода:
=—(т/п)(1/хЛ), (243)
где xr — мольная доля раскислителя; т и п — число ато-
мов R и О в молекуле RO.
При помощи уравнения (243) на основе эксперимен-
тальной кривой [% О] =<р(хн) можно вычислить ео. Если
продукты раскисления имеют сложный состав (FeO-RxOy),
то равновесная концентрация кислорода будет описывать-
ся уравнением [ЮЗ]:
1%О] = (е«жя)/[(* — + xR) К,], (244)
где b=Ki/K2 (/Ci и Кг — константы равновесия реакции
окисления железа и раскислителя).
В этом случае в точке минимума кривой растворимости
кислорода имеем
<245>
Анализ кривой раскисления показывает, что она может
иметь не только минимум, но и быть замкнутой [232]. Ми-
хайлов Г. Г. сделал полезное обобщение и систематизацию
литературных и собственных данных в виде диаграмм со-
148
стояния, устанавливающих связь между составами жидко-
го металла и равновесных неметаллических фаз. Состав
равновесного металла определен на поверхности раство-
римости компонентов в жидком металле.
V.3. РАСКИСЛЕНИЕ ЖЕЛЕЗА И ЕГО СПЛАВОВ
По увеличению раскислительной способности элементы
можно расположить в следующей последовательности: Ni,
Fe, Мп, Si, Al. В этом ряду каждый последующий элемент
является раскислителем по отношению к предыдущему.
Существуют различные способы оценки раскислительной
способности элементов, которые выражают одну и ту же
термодинамическую сущность. В частности с увеличением
раскислительной способности равновесная остаточная ак-
тивность кислорода, соответствующая одинаковым оста-
точным активностям элементов-раскислителей, уменьша-
ется. Если продуктом раскисления является чистый оксид
/?тОп, то его активность вя_о#=1. В общем случае следу-
ет учитывать сложность образующегося оксида FeOX
X^mOn. Чем выше раскислительная способность, тем при
меньших концентрациях элемента-раскислителя образуют-
ся сложные продукты раскисления типа FeO-RmOn. Место
углерода в ряду раскислителей определяется давлением
Рсо. При рсо = 1 атм углерод располагается между эле-
ментами Мп и Si. При низких значениях рсо углерод мо-
жет быть более сильным раскислителем, чем например,
алюминий.
Раскисление марганцем
Марганец является сравнительно слабым раскислителем.
При 1600 °C и концентрациях марганца 0,2 и 0,8 % жидкое
железо содержит 0,15 и 0,1 % кислорода. При этой же тем-
пературе под шлаками из чистых оксидов железа [О]нас=
=0,23 %. При введении марганца в сталь образуются про-
дукты раскисления, состоящие из FeO и МпО. Концентра-
ция МпО зависит от содержания марганца в металле и
температуры. В соответствии с диаграммой состояния
FeO—МпО продукты раскисления в зависимости от содер-
жания марганца образуют жидкие или твердые растворы
[122]:
[Мп] + (FeO) = (МпО) + Fe,K; AG° =— 123260 +
+ 56,467. (246)
149
Оксиды FeO и МпО образуют практически совершен-
ный раствор. Тогда
Км„ = WW I %Мп]; '8 *м„ = (6440/7-) - 2,95.
(247)
Справедливость соотношения (247) подтверждается
ЛИНеЙНОЙ ЗаВИСИМОСТЬЮ X(MnO)/*(FeO) °т [% Мп] или по-
стоянством Лмп при заданной температуре. Процесс рас-
кисления жидкого железа марганцем изучен сравнительно
полно. Результаты различных авторов достаточно хорошо
согласуются. Во многих случаях реакцию раскисления мар-
ганцем записывают в следующем виде:
(Мп] + [О] = (МпО); /СМп = Д(Мп0) /Д[О] ’^мпр (248)
Выражение (248) можно получить сложением уравне-
ний (246) и (190). В этом случае
1g = (12770/Т) — 5,69. (249)
Пример. Определить состав продуктов раскисления ста-
ли при 1600 °C и 0,5 % [Мп] в жидком металле. Из урав-
нения (247) следует, что мольная доля (хмпо) при посто-
янной температуре определяется концентрацией марганца.
Принимаем, что образующиеся продукты раскисления об-
разуют совершенный раствор x(FeO) + *(МпО) = 1- Констан-
та равновесия реакции (247) при 1600°С равна lg/Смп =
= (6440/7')—2,95=0,48; 3,02=х(мпО)/*щеО)• [% Мп]. От-
сюда при [Мп] =0,5 % образующиеся продукты раскисле-
ния имеют следующий состав: (МпО) =60 %, (FeO) =
=40%.
Раскисление кремнием
Кремний — более сильный раскислитель, чем марганец.
Термодинамические характеристики реакции раскисления
стали кремнием выражаются следующим уравнением
[126]:
SiO2(T) = ISi] + 2 [OI; Д<3" = 596785 —231,217; (250)
lgKsl = (- 31180/7) + 12,08; KS1 = =
= [%Sil 1%0Г7,...£о. = 1% Sill«/oOP. (251)
Многочисленные исследования показывают, что произ-
ведение растворимости [% Si] [% О]2 остается постоянным
при изменении концентрации кремния вплоть до 2%. Это
связано с тем, что с увеличением концентрации кремния
в расплаве /[sij увеличивается, a f[oj уменьшается. Ука-
150
занные изменения происходят так, что их произведение
f(Si]f[O] = 1- При малых содержаниях кремния в стали и
высоком содержании кислорода продуктом раскисления
является не чистый кремнезем, а силикаты (FeO)x-
• (SiOz) различного состава. Количественные соотношения
между X(FeO) и X(sio,) можно вычислить на основе кон-
станты равновесия уравнения: 2 (FeO) + [Si] = (SiOs) +
+ 2Ре<ж); Дб°=—354470+126,32 7' полученного комбина-
цией уравнений (249) и (250):
1g К = x(SiO1)M?FeO) • I %Sil = (18520/Т) - 6,6. (252)
Расчеты по уравнению (252) показывают, что при 0,1
и 0,3 % Si содержание FeO в продуктах раскисления со-
ставляет 10 и 4 % соответственно. Переход от одних про-
дуктов раскисления (железистых силикатов) к другим
(чистый кремнезем) по мере увеличения концентрации
кремния в жидком железе происходит непрерывно.
Пример. Вычислим концентрацию кремния, соответст-
вующую минимальной концентрации кислорода при
1600 °C.
В соответствии с уравнением (251) имеем:
In [ %О] = V, In К । - V. In [ % Si] - V2 In fg _ in /gj.
(253)
Чтобы найти экстремум функции, продиффундируем
уравнение (253) по концентрации кремния и приравняем
нулю:
dln[%0] 1____1____1_ dln/gj] dlnfgj
d[%Si] ‘ 2 ’ [%Si] 2 ‘ d[%Si] d[%Si] ’ ' ’
Используя данные по параметрам взаимодействия
(egi =0,11; е^ =—0,131) и решая уравнение (254) отно-
сительно [% Si] при минимальной концентрации кислоро-
да, имеем [% Si] =2,86 %. По формуле (251) вычисляем
[O]mln = 4,19-10-3 %.
Раскисление алюминием
Алюминий является очень сильным раскислителем и в свя-
зи с этим широко применяется для раскисления стали.
Раскислительная способность алюминия экспериментально
определялась многими исследователями. Трудности экспе-
риментального изучения раскислительной способности алю-
миния связаны со сравнительно большой погрешностью
определений малых концентраций кислорода и раскисли-
151
теля в расплаве, а также низкого окислительного потенци-
ала (ро,) в газовой фазе. Например, при остаточной кон-
центрации алюминия 0,001 % содержание растворенного
кислорода составляет 0,001 %, что приближается к преде-
лу определения кислорода. При малых добавках алюминия
образуются сложные включения, состоящие из шпинели
РеО-А12Оз (герценита) или раствора оксидов А120з и FeO.
При избытке алюминия [% А1]/[% О]^25 образуется чи-
стый А120з. Для большинства сталеплавильных процессов
это условие выполняется:
А12О3 = 2 [АН 4- 3 [О]; AG° = 124220 — 394,85Т. (255)
На основе обобщения результатов различных исследо-
ваний авторы работы [119] рекомендуют следующее вы-
ражение для температурной зависимости константы рав-
новесия реакции (255):
lg КА1 - lg j• = (- 64900/7) + 20,63. (256)
Для большинства практических расчетов можно при-
нять, что a[Ai] = [% А1] и а [о] = [% О].
Другие раскислители
Цирконий. Относится к одному из наиболее сильных рас-
кислителей. Продуктом раскисления является ZrO2:
ZrO2(T) = [Zr] + 2 [О]; AG° = 790700 — 230,697. (257)
lgKZr = lg[%Zr][%0]2; lgtfzr = (-41370/7) +
+ 12,07 [119]. (258)
Бор. Температурная зависимость константы равновесия
реакции раскисления бором
В2О3 = 2 [В] + 3 [О]; Д6° = 855460 — 295,337 (259)
выражается уравнением
lg Кв = 1g [ % В]2 [ % О]3 = (— 44695/7) + 15,431119]. (260)
Ванадий — сравнительно слабый раскислитель. При
1600°С и <0,2 % V образуется шпинель РеО-У2Оз. При
>0,2 % образуется оксид У20з:
V2O3 = 2 [V] + 3 [О]; AG° = 877340 — 336,027 (261)
lg Kv = I% VI21 % °13; /[О] = (— 44793/7) +
+ 17,5561121]. (262)
Малые добавки ванадия вводят в металл не для рас-
152
кисления, а для регулирования размера зерна и связыва-
ния азота с целью предотвращения старения.
Титан. При изучении раскислительной способности ти-
тана наибольшие трудности связаны с идентификацией
продуктов раскисления. При <0,2 % [Ti] образуется
Ti3O5 [124, 125]:
Ti8O6 = 3 [Ti] + 5 [О]; AG° = 793735 — 65,07Т; (263)
lg ^Ti = lg I % Ti]31 °/o°l6 =* (- 41470/T) + 3,4. (264)
Кальций и магний являются очень сильными раскисли-
телями. Трудности использования этих раскислителей для
обработки жидкой стали связаны с тем, что при темпера-
турах сталеварения оба элемента находятся в газообраз-
ном состоянии и характеризуются очень низкой раствори-
мостью в жидком железе. Растворимость кальция при
1600 °C и ро =1 атм равна 0,02—0,03%, а магния — ты-
сячные доли процента. Раскислительная способность маг-
ния, по данным [119], определяется уравнением:
MgOb = Mg(r) 4- [О]; AG° = 622050 — 201,35 7; (265)
lg р* [ % О] = (— 32500/7) 4-10,52. (266)
Раскислительная способность кальция, соответствую-
щая СаО(Т)= [Са] + [О], определена при 1600°С в магне-
10~4________I-----------------1--------1--------
1(Г< 10"J 10~z IO'1 1 10
СЮ,%
Рис. 41. Раскислительная способность элементов в жидком
железе при 1873 К [121]
153
зитовом тигле. Получено следующее значение произведе-
ния: [% Са] • [% О] = 1,2-10~:. В реальных условиях маг-
ний используют для раскисления металла в виде лигатур,
а кальций в виде сплавов с кремнием и алюминием. Очень
сильными раскислителями являются лантаноиды (лантан,
церий и др.), которые в последнее время широко применя-
ются в сталеплавильном производстве [119, 121]. Раскис-
лительная способность элементов в жидком железе при
1873 К приведена на рис. 41.
V.4. КОМПЛЕКСНЫЕ РАСКИСЛИТЕЛИ
В практике сталеварения широко применяют комплексные
раскислители, представляющие собой сплавы двух или не-
скольких компонентов (силикомарганец, силикокальций,
сплав кремния, марганца и алюминия и др.). Преимуще-
ства, связанные с применением комплексных раскислите-
лей, обусловлены двумя обстоятельствами: существенным
улучшением термодинамических условий раскисления и бо-
лее благоприятными кинетическими условиями зарожде-
ния, укрупнения и удаления неметаллических включений.
Например при 1600 °C в жидком железе с 0,2 % Si нахо-
дится в равновесии 0,012 % растворенного кислорода. Вме-
сте с тем при добавке 0,5 % Мп содержанию 0,2 % Si соот-
ветствует более низкая равновесная концентрация кисло-
рода: 0,008%. Таким образом, добавка марганца приводит
к повышению раскислительной способности кремния. Мар-
ганец и кремний раздельно и совместно повышают раскис-
лительную способность алюминия. Рассматриваемый эф-
фект увеличения раскислительной способности под влия-
нием второго компонента объясняется уменьшением
термодинамической активности образующегося оксида в
сложных продуктах раскисления, которые существенно
отличаются от продуктов при раздельном раскислении.
Если сталь одновременно раскислить кремнием, марган-
цем, то продуктами раскисления являются железомарган-
цовистые силикаты. Состав силикатного раствора и его
агрегатное состояние определяется температурой, а также
концентрацией кремния и марганца в железе. В этом слу-
чае реакцию раскисления кремнием следует записать сле-
дующим образом:
[Si] + 2 [О] = (SiO^. (267)
В рассматриваемых условиях (SiOa) образует раствор
с (МпО). Константа равновесия реакции (267) в отличие
154
от (251) имеет вид:
К ~ fl(sios) /(«[si] °[О]) • (268)
В уравнении (268) активность кремнезема aSiot мень-
ше единицы. Поскольку константа равновесия при данной
температуре постоянна, то уменьшение asio, приводит к
снижению произведения растворимости «^‘«гор а сле-
довательно, к уменьшению концентрации остаточного кис-
лорода в жидкой стали. Таким образом, в присутствии бо-
лее слабого раскислителя (марганца) раскислительная
способность кремния повышается. Активности компонентов
в бинарных и тройных оксидных системах приведены в ра-
ботах [2, 123]. Если продуктом раскисления является
шлак, насыщенный кремнеземом (asio, =1) или чистый
SiO2 (flsio, = 1), то марганец не влияет на раскислитель-
ную способность кремния. Марганец увеличивает раскис-
лительную способность алюминия лишь в том случае, ког-
да жидкий металл находится в равновесии со шлаком и
шпинельными растворами. В области, где металл находит-
ся в равновесии с твердым корундом, содержание [ % О]
определяется только концентрацией алюминия.
V .5. РАСКИСЛИТЕЛЬНАЯ СПОСОБНОСТЬ ШЛАКА
Для оценки раскислительной способности восстановитель-
ных шлаков необходимо сопоставить фактическую актив-
ность кислорода «[о]* в металле под шлаком данного со-
става с равновесным значением fl(ojp. Если «(ou>«[ojp»
то шлак оказывает раскисляющее действие. Исследования,
проведенные Ф. П. Еднералом, А. М. Левиным, Д. Я. По-
волоцким, А. И. Холодовым и др., показали, что диффузи-
онное раскисление стали играет сравнительно малую роль
в снижении концентрации кислорода в средне- и высоко-
углеродистых сталях. В первой половине восстановитель-
ного периода активность кислорода в этих сталях опреде-
ляется углеродом и в большинстве случаев ниже активно-
сти кислорода, равновесной со шлаком. Так, например,
если в металле имеется 0,7 % [С], то равновесная с угле-
родом 0(0] = [% О] =0,003. Этой концентрации кислорода
соответствует равновесная активность FeO в шлаке:
«(FeO) =[% О]/[% О]нас=0,003/0,23=0,013, или 1,5 %
(FeO). Вместе с тем в первую половину восстановительно-
го периода содержание (FeO) >1,5 %. Такой шлак по от-
ношению к металлу не проявляет раскисляющего действия.
155
Активность кислорода в металле для низкоуглеродистых
сталей в большинстве случаев превосходит равновесную
активность со шлаком. Однако и в этом случае при диф.
фузионном раскислении углеродом содержание кислорода
в металле не уменьшится ниже равновесного с углеродом.
Для обеих групп сталей существенное уменьшение содер-
жания кислорода в металле при раскислении шлака по-
рошком ферросилиция наблюдается лишь при одновремен-
ном повышении концентрации кремния в металле. В ре-
альных условиях равновесие между расплавом и газовой
фазой, как правило, не достигается, а глубинное раскисле-
ние имеет то преимущество, что заданная степень раскис-
ления достигается за более короткое время. Восстанови-
тельный период протекает в условиях постоянного попол-
нения ванны кислородом. По данным А. М. Левина, в этот
период из воздуха в ванну поступает до 0,3 % кислорода
от массы металла. Это в 5—10 раз больше концентрации
кислорода в металле в начале восстановительного периода.
В этом смысле восстановительный шлак обладает защит-
ными свойствами.
Таким образом, диффузионное раскисление металла яв-
ляется малоэффективным процессом. Концентрация кисло-
рода в металле в восстановительный период плавки опре-
деляется не законом распределения кислорода и не
степенью развития реакции раскисления в шлаке и на гра-
нице раздела металл — шлак, а в основном процессами глу-
бинного раскисления.
Глава VI
НЕМЕТАЛЛИЧЕСКИЕ ВКЛЮЧЕНИЯ
Получение стали, чистой от неметаллических включений, является важ-
ной и актуальной задачей современной металлургии. Повышение требова-
ний к качеству металла определяет поиск способов снижения количества
неметаллических включений и ослабления их вредного влияния. На осно-
вании большого числа исследований [129—135] можно заключить, что
экзогенные включения достаточно полно удаляются из жидкой стали, а
основная масса неметаллических включений, встречающихся в твердой
стали, принадлежит к классу эндогенных, образующихся в результате
сложных физико-химических процессов в жидком, затвердевающем и
твердом металле. Это прежде всего изменение растворимости кислорода,
более полное протекание реакций раскисления, нитридообразования при
понижении температуры. Чтобы различить эндогенные включения, обра-
зующиеся в различные периоды плавки и при затвердевании металла,
В. И. Явойский с сотрудниками [136] предложил следующую классифи-
156
нацию неметаллических включений: 1) предкристаллизационные: первич-
ные, образующиеся при раскислении стали, и вторичные, образующиеся
при’охлаждении жидкого металла до температуры, соответствующей ли-
нии ликвидуса; 2) кристаллизационные: третичные, образующиеся в зат-
вердевающей стали в температурном интервале между линиями ликви-
дуса и солидуса; 3) послекристаллизационные: четвертичные, образую-
щиеся в затвердевающей стали при охлаждении металла до комнатной
температуры.
VI .1. ОБРАЗОВАНИЕ
НЕМЕТАЛЛИЧЕСКИХ ВКЛЮЧЕНИЙ
Закономерности зарождения неметаллических включений в жидком ме-
талле изучены в недостаточной степени. Объясняется это тем, что даже
для более простых объектов количественные расчеты интенсивности за-
родышеобразования связаны со многими трудностями. Сложности эти
существенно возрастают при переходе к техническим объектам — сталям
и чугунам. Основной моделью, используемой большинством исследова-
телей, является образование новой фазы в рамках классической теории
Гиббса — Фольмера. Согласно этой теории система может находиться в
метаста би льном состоянии '(переохлажденном, перенасыщенном и др.)
сколь-угодно долго, если внешние условия остаются неизменными. Гцббс
первым указал на то, что возникновение зародыша и его рост до крити-
ческого размера связаны с увеличением свободной энергии. Для случая
кристаллизации рост свободной энергии обусловлен затратой работы на
образование новой поверхности между зародышем и матрицей. Исходя из
теории флуктуации, допускающей такой процесс, Фольмер показал, что
скорость образования J числа зародышей в единице объема за 1 с опи-
сывается уравнением:
J = A exp (— AG*/JfeT), (269):
где ДО* — изменение свободной энергии при образовании зародыша кри-
тического размера: k — постоянная Больцмана;
ДО* = (16л<£_вУ2)/[3 (kT)2 (In а)2], (270)
Ом—в — межфазное ‘натяжение на ^границе металл — включение; V •—
мольный объем образующейся фазы; а — степень пересыщения.
При образовании продуктов раскисления под степенью пересыщения
понимают отношение произведений растворимости фактических концент-
раций элемента раскислителя и кислорода [% /?]£•[% O]£k равновесным.
Предэкспоненциальный множитель характеризует частотный фактор.
Уравнения (269) и (270) устанавливают связь между степенью пересы-
щения, значением ам-в и интенсивностью зародышеобразования. При по-
мощи уравнений (269) и (270) можно установить относительную вели-
чину вклада, вносимого ом-в и а для спонтанного образования зароды-
ша критического радиуса. Приведенные формулы применимы лишь для
расчета скорости зарождения новой фазы в однокомпонентных системах,
в частности, для процессов кристаллизации, конденсации и др. Тем не
менее эти закономерности используют и для оценки скорости зарожде-
ния неметаллических включений. В связи с этим расчеты являются при-
ближенными и в ряде случаев носят полуколичественный характер.
Из уравнения (270) следует, что чем больше степень пересыщения,
тем легче происходит процесс зародышеобразования. По данным'
157
С. И. Попеля, FeO и МпО (ом-в=180 мДж/м2) из окисленного металла
могут выделяться самопроизвольно при сравнительно небольшой степе-
ни пересыщения. При этом диаметр зародыша составляет около четырех
параметров решетки. С увеличением Ом-в требуется существенно боль-
шая степень пересыщения. С повышением межфазного натяжения до
500 (расплавы FeO—МпО—SiO2) и 700 мДж/м2 интенсивность зароды-
шеобразования близка к нулю даже при трех- и десятикратном пересы-
щениях соответственно. Необходимость пересыщенья для зародышеоб-
разования подтверждается не только расчетом, но и экспериментами.
В работе [137] пересыщения расплава, содержащего алюминий и кисло-
род, достигали быстрым охлаждением жидкого железа на 350 °C. После
этого расплав выдерживали 20 мин и затем быстро охлаждали до кри-
сталлизации. Было показано, что при выдержке образуются включения
А120з. В тех случаях, когда пересыщение отсутствовало, включения ко-
рунда не образовывались вплоть до температуры затвердевания.
Ряд исследователей, исходя из предложения о существенном раз-
личии между От-г и Ож-г. заключают, что образование твердых включе-
ний оксидов при введении сильных раскислителей в соответствии с фор-
мулой (270) может происходить лишь при значительном пересыщении.
Однако это не так. Основанием для такого утверждения является боль-
шее значение межфазного натяжения в системе твердый оксцд — жид-
кий металл по сравнению с межфазным натяжением жидких продуктов
раскисления на границе с жидкой сталью, При прочих равных условиях
этот факт имеет значение, но переоценивать его не следует. Известно, что
поверхностные натяжения веществ в твердом и жидком состояниях срав-
нительно близки и различаются в большинстве случаев не более чем на
10—20 %. В этом смысле агрегатное состояние вещества не играет ре-
шающей роли. Картина изменяется, если в продуктах раскисления име-
ются компоненты (например, FeO), которые существенно снижают меж-
фазное натяжение. Это приводит к уменьшению необходимой степени пе-
ресыщения для образования зародышей критического радиуса г*. Расче-
ты показывают, что при одинаковой скорости зарождения включения
А12О3 требуют гораздо большей степени пересыщения, чем включения
SiO2 или силикаты (FeO)x(SiO2)v. На этом основании, казалось бы,
можно сделать заключение, что гомогенное зарождение включений ко-
рунда затруднено в большей степени, чем зарождение включений сили-
катов. Однако в рассматриваемом случае важна не только степень, но и
концентрация пересыщения [138]. Например, при содержании элементов-
раскислителей 0,03 и 0,1 % концентрация кислорода в пересыщенном
растворе должна составлять соответственно 0,037 и 0,013 % при образо-
вании А12О3 и 79 % (т .е. больше предела растворимости кислорода)
при образовании SiO2. Исходя из этого, можно заключить, что глинозем
образуется гомогенно, а кремнезем — гетерогенно на посторонних приме-
сях.
Известно, что наличие готовых поверхностей существенно облегчает
процесс зародышеобразования. В соответствии с этим степень пересыще-
ния, необходимая для гетерогенного образования зародышей, всегда
меньше, чем для гомогенного. Это обстоятельство играет очень важную
роль в практике раскисления. Промышленные раскислители содержат не-
металлические включения, устойчивые при температурах сталеварения,
которые являются подложками для гетерогенного зародышеобразования
продуктов раскисления. Вместе с тем эти раскислители содержат при-
месные концентрации 'более сильных элементов-раскислителей, которые
обеспечивают образование соответствующих подложек, облегчающих
дальнейшее образование неметаллических включений. Например при рас-
кислении стали алюминием и кремнием вначале образуются включения
158
глинозема, на поверхности которых формируются образования силикатов.
Гомогенное образование зародышей SiO2 может иметь место в процессе
кристаллизации металла, при которой достигаются требуемые концент-
рации критических пересыщений. При содержании в стали азота нитри-
ды титана, как правило, образуются на подложках включений оксидов
титана. После образования зародыша критического размера его рост но-
сит молекулярный характер и связан с диффузией растворенных приме-
сей (кислорода и раскислителя) в жидкой стали с последующим выделе-
нием новой фазы на имеющуюся подложку. Наличие зародыша снижает
степень пересыщения, однако вследствие обеднения ванны кислородом
этот процесс сравнительно быстро заканчивается. Дальнейший рост за-
родыша связан с коагуляцией частиц, движущихся под действием пото-
ка или силового поля. В ряде случаев первичные продукты раскисления
в стали образуются в результате расслоения исходного расплава при
введении раскислителя*.
Многочисленные данные показывают, что основные трудности полу-
чения чистой стали связаны с укрупнением и удалением включений. По
данным ряда исследователей, продолжительность процесса образования
и удаления легко коалесцирующих включений составляет 0,5—2 мин.
Процесс удаления мелких включений (~5 мкм) более медленный. Уда.
ление последней сотой доли кислорода занимает основное время рафини-
рования и не зависит от начальной концентрации кислорода.
VI.2. УКРУПНЕНИЕ
НЕМЕТАЛЛИЧЕСКИХ ВКЛЮЧЕНИИ
Укрупнение частиц возможно при их столкновении. Это может произой-
ти в результате естественной миграции частиц или под воздействием на-
правленных сил. Различают два типа коагуляции: перикинетическую и
ортокинетическую (термины введены Вигнером). Под перикинетической
коагуляцией понимают укрупнение частиц размером ~ 1 мкм, движу-
щихся по законам случайных блуждений (например, броуновского дви-
жения). Под ортокинетической коагуляцией понимают укрупнение частиц
в жидком металле, когда частицы находятся под воздействием силового
поля: гравитационного (всплывание), электрического (электрофорез),
концентрационного (движение под действием адсорбционных сил). К ор-
токинетической коагуляции относится также укрупнение неметаллических
включений в результате столкновений движущихся с потоком жидкости
частиц в условиях естественной и принудительной конвекции.
Расчеты показывают, что для частиц диаметром >0,1 мкм ор-
токинетическая коагуляция имеет преобладающее значение [139]. Роль
двух типов коагуляции одинакова для очень мелких частиц ( — 1 мкм).
Скорость слияния двух капель одинакового радиуса v прямо пропор-
циональна межфазному натяжению ом—в и обратно пропорциональна
вязкости 1): о=/С(ом—в/т|). По данным работы [147] скорость слия-
ния составляет 15—17 м/с. В реальных условиях для различных вклю-
чений Ом—в изменяется в 10—15 раз, а вязкость — в 103—105 раз,,
поэтому вязкость может оказать ббльшее влияние на скорость слия-
ния, чем межфазное натяжение на границе металл—включение. Распро-
странено мнение, что более эффективно укрупняются включения, ха-
* Менделев В. А. Термодинамический анализ и эксперименталь-
ное изучение условий выделения неметаллической фазы при раскисле-
нии и легировании сплавов на основе железа. Автореф. канд. дис. М.,
1981.
159
рактеризующиеся высоким межфазным натяжением, поскольку этому
случаю соответствует значительное снижение свободной энергии. Это
подтверждается результатами сравнения жидких включений, которые
различаются значениями Ом-в при прочих равных условиях. Дейст-
вительно, предпочтительнее будут сливаться включения, имеющие более
высокие значения ом-в- Слиянию двух жидких неметаллических ча-
стиц предшествует образование металлического перешейка. Зависимость
радиуса перешейка а от времени т при слиянии двух соприкасающихся
вязких капель радиусом г описывается формулой Френкеля:
а2 = 3/2 [(^т)/(лт))], (271)
где а—радиус кривизны; а — поверхностное натяжение; т) — вязкость.
При различных радиусах кривизны капель скорость их слияния
определяется скоростью слияния капель с меньшим радиусом, а при
различных вязкостях — скоростью слияния капель с меньшей вязко-
стью [147]. Уравнение (271) хорошо передает соответствие опытных
и расчетных данных при т]>0,7 Па-с. При меньших значениях вязко-
сти, когда сопротивление носит инерционный характер, следует пользо-
ваться формулой:
a2 = xl^(m)/Ti . (272)
Опытные данные, а также расчеты по формулам (271) и (272)
показывают, что капли из неметаллических фаз различного состава
(СаО, AI2O3, CaF2, SiO2, MgO) с вязкостью от 0,005 до 0,26 Па-с сли-
ваются со скоростями увеличения площади перешейка d(na2)[d% от
единицы до десятков квадратных сантиметров в секунду. Особенно
важны закономерности укрупнения твердых включений. Во многих
случаях наблюдаемые в стали крупные включения глинозема (<3 мм)
состоят из множества мелких частиц размером в несколько микромет-
ров. Моделированием и математическими расчетами показано, что ча-
стицы в этих скоплениях удерживаются силами поверхностного натя-
жения, возникающими вследствие уменьшения межфазной поверхности
металл—включение при выдавливании манжеты жидкой стали между
частицами. Расчеты показывают, что две соединяющиеся частицы раз-
мером 5 мкм могут быть разорваны потоком движущегося металла,
если скорость потока относительно частиц >1,4 м/с.
Образование скоплений возможно при условии самопроизвольного
установления контакта между включениями. Этому предшествует ста-
дия, связанная с появлением манжеты из жидкого металла. Дальней-
шее поведение этих включений в существенной степени определяется
капиллярными силами. Удержание неметаллических включений при
помощи капиллярных сил связано с адгезией жидкого металла к соеди-
няемым поверхностям. Этот вопрос достаточно полно рассмотрен в
работах Ю. В. Найдича, С. И. Попеля, А. А. Дерябина. Жидкая ман-
жета может быть удалена лишь при отрицательном значении измене-
ния свободной энергии этого процесса: AG=2(oB-r—Пм-в^О, или
ДО=2ом-г cos0<O (0 — краевой угол смачивания металлом поверх-
ности включения).
Таким образом, смачиваемость является важнейшей характеристи-
кой прочности образования сложных конгломератов. Если 0>9ОЭ, то
образование скоплений возможно; если 0<9О°, то процесс термоди-
намически невозможен. Расчеты, выполненные в работе [140], показа-
ли, что при 0>1ОО° силы, действующие в турбулентном потоке, не мо-
гут разорвать связи и разобщить скопления включений.
При турбулентных пульсациях в струе металла (и>10 м/с) вклю-
160
чения крупных размеров (г=10 мкм) будут отрываться друг от друга,
если их вязкость превышает 0,04 Па-с, а маленькие частицы (г=
^=1 мкм) при т]>5-109 Па-с, о=1000 мДж/м2 [141].
VI.3. УДАЛЕНИЕ ВКЛЮЧЕНИИ
В ГРАВИТАЦИОННОМ ПОЛЕ
Перенос неметаллических включений в жидкой стали к межфазной
поверхности раздела металл—шлак и металл—футеровка осуществля-
ется либо движением частицы относительно самой среды (всплывание,
броуновская диффузия, инерционное смещение и др.), либо конвектив-
ными потоками металла. Всплыванию частиц в вязкой жидкости под
действием гравитационных сил посвящено большое число теоретических
и экспериментальных исследований. Для количественной оценки ско-
рости всплывания v сферического твердого включения радиусом г ши-
роко используют формулу Стокса, выведенную при условии равенства
сил сопротивления движению шара в вязкой среде (Г=6лгт]и) и Ар-
химеда /?,=4/зЛг3^(рм—Рв):
v [(рм - Рв)/Л]- (273)
Формула (273) строго выполняется, когда критерий Рейнольдса
<0,6. Если при Re<0,6 вычислить предельные размеры включений, то
получится, что формула Стокса пригодна лишь для расчета скоростей
всплывания твердых включений диаметром <100 мкм. Однако практика
показывает, что экспериментальные данные хорошо согласуются с рас-
четными для включений размером <10 мкм. Д. Я. Поволоцкий [118]
показал, что при раскислении стали кремнием экспериментальные зна-
чения скорости удаления включений размером от 2 до 10 мкм хорошо
согласуются с рассчитанными по формуле Стокса (от 0,5 мм/мин при
выдержке 2 ч до 6 мм/мин при выдержке 13 мин). Для включений
большого размера формула (273) дает завышенные результаты. Для
такого типа включений можно пользоваться формулой, которая легко
выводится, если силу F' приравнять силе сопротивления по закону
Ньютона (F2=Kr2\\v2). Тогда
02 = gr (4л/ЗК)[(рм - рв)/Л 1, (274)
где /C=24/Re— коэффициент, зависящий от критерия Рейнольдса, Re.
Скорости всплывания жидких включений вследствие движения жид-
кости в капле и различных граничных условий отличаются от скорости
движения твердых частиц такого же размера, поэтому для количествен-
ной оценки скорости всплывания жидких включений нужно пользо-
ваться не уравнениями (273) и (274), а формулой Рыбчинского —
Адамара:
V» = 2/з^2 [(Рм — Рв)/Пм][(Пм + Лв)/(2Чм + Зт]в)1. (275)
где 1]в — динамическая вязкость жидкого включения, кг/(м-с).
Сравнивая уравнения (273) и (275), имеем:
^ж/^т = 3 (Лм + Лв)/(2Лм 4“ ЗЛв) > 1 • (276)
Таким образом, при одинаковом размере частиц скорость всплыва-
ния жидкого включения больше скорости всплывания твердого вклю-
чения. При Лв^*Лм формула (275) переходит в формулу (273), поэто-
му для очень вязких жидких включений (л>0,2 Па-с) можно пользо-
ваться формулой Стокса. Различия расчетных и опытных значений v
могут быть вызваны не только размером, но и формой всплывающих
11—675
161
включений. Для включений корунда действительно скорость всплывания
даже для малых частиц меньше, чем по формуле Стокса [118]. Это
учитывается фактором формы х=гэЛс (fa— эквивалентный радиус
шара, объем которого равен объему включения; гс — радиус шара по
Стоксу с той же плотностью и скоростью движения). Фактор формы х
для различных моделей варьируется в пределах 2—5. С учетом х ско-
рость всплывания можно вычислить по формуле:
V = я/8 grz (Рм — Рв)/(Пм X) • (277)
Включения А120з всплывают в четыре — восемь раз медленнее,
чем равные по размерам включения силикатов. Это связано с непра-
вильной формой А12О3, а также изменением ориентации включений гли-
нозема во время всплывания, приводящим к изменению х [118]. Анализ
работ по удалению неметаллических включений позволяет заключить,
что закон Стокса удовлетворительно описывает удаление включений в
спокойной ванне. В условиях реальной плавки достаточно крупные
включения (>15 мкм) сравнительно быстро всплывают из жидкой
стали в течение первых минут после введения раскислителей. Вслед-
ствие развитых конвективных потоков в жидкой стали для мелких
включений непременимы законы Стокса и Рыбчинского—Адамара.
Пример. Рассчитаем скорости всплывания неметаллических частиц
в жидкой стали в гравитационном поле при 1600 °C. Покажем, в каком
интервале скоростей движения для включений радиусом 50 мкм в спо-
койном металле выполняется формула (273):
Re = (vdpM)/T]M< Ь (278)
где d — характеристический размер, в данном случае равный диаметру
частицы: t/<(Re-T]M)/(pM-d) = (b6.10-3)/(2.5.10-67,15-103) = 0,8Х
Х10“2 м/с==0,8 см/с. По формуле (273) вычислим скорость подъема
частицы глинозема радиусом 10 мкм: и=2/д-9,81[(7,15—3,97) • 103/6-10“3]
(10-5/2)=30 мкм/с.
Таблица 18. Скорости всплывания различных включений
Тип включения Скорость всплывания 105 (м/с) включений диаметром, мкм Тип включения Скорость всплывания 105 (м/с) включений диаметром, мкм
10 20 50 10 20 50
А12О3 SiO2 3 5 12 20 75 125 A1N TIN 4 2 16 8 100 50
Полученное значение скорости удовлетворяет условию Re<l. Сле-
довательно, для частиц такого размера можно применять формулу
Стокса. Скорости всплывания частиц размером 20 и 50 мкм соответст-
венно равны 120 и 750 мкм/с. Для вычисления скорости всплывания
жидкого силикатного включения воспользуемся формулой (275):
2 (7,15 — 2,5)
□ _. — .-------------
3 6-Ю-3
= 40 мкм/с
6-10—8 + 5-10—1
2-6- 10—3 + 3- 1О-*
9,81-10—5
2
Для частиц радиусом 20
всплывания 160 и 1000 мкм/с.
и 50 мкм соответственно имеем скорость
Для вязкой частицы силикатов (т)в Эи]*)
162
поправочный множитель в уравнении Рыбчинского—Адамара примерно
равен !/з. Таким образом жидкие силикатные включения в спокойном
металле всплывают несколько быстрее твердых частиц глинозема тако-
го же размера. Значения скоростей всплывания включений приведены
р табл. 18.
VI .4. ВЛИЯНИЕ КОНВЕКЦИИ НА УДАЛЕНИЕ
НЕМЕТАЛЛИЧЕСКИХ ВКЛЮЧЕНИЙ
Лабораторными и промышленными экспериментами показано, что кон-
вективные потоки в жидком металле улучшают условия для удаления
неметаллических включений. С ростом интенсивности конвективного
перемешивания увеличивается частота столкновений между включения-
ми, приводящая к укрупнению частиц, растет вероятность выхода вклю-
чений к поверхности раздела металл — шлак, увеличивается частота
прилипания включений к футеровке и т. д. Этот процесс протекает осо-
бенно интенсивно в период кипения жидкой ванны. Скорость конвек-
тивных потоков (м/с) в различных металлургических агрегатах приве-
дена ниже:
Мартеновская печь
Кислородный кон-
вертер .........
Дуговая электро-
печь ...........
0,1—1
1—2
0,5—1
Индукционная печь
ЭШП.............
ВДП, ПДП, ЭЛП
Ковш...........
Изложница . . .
3__g
0,05—0,10
0,05—0,20
0,05—0,50
10—3—Ю
Количественная сторона вопроса о влиянии перемешивания на уда-
ление включений изучена недостаточно. Это в первую очередь связано
с тем, что сталеплавильная ванна относится к числу гидродинамически
неопределенных задач. Закономерности распределения скоростей пото-
ков жидкого металла и шлака в сталеплавильной ванне в большинстве
случаев неизвестны. Они сложны, и их трудно количественно описать.
Имеющиеся в литературе уравнения носят эмпирический или полуэмпи-
рический характер. Например, учет конвективного переноса для верти-
кальной составляющей скорости приводит к появлению поправочного
слагаемого vM cosy в уравнении (273) (им — скорость движения ме-
талла; у — угол между вертикалью и направлением вектора скорости
движения металла). По расчетам авторов работы [142] для включений
размерами 10—70 мкм конвективное слагаемое превалирует над сток-
совским, если им cos у >0,046—2,27 мм/с. Принимая во внимание ско-
рости движения, наблюдаемые на практике, следует отметить, что про-
цессы удаления включении будут в основном определяться конвектив-
ным массопереносом. Однако положительное влияние турбулизации име-
ет предел, превышение которого вызывает повторное вовлечение вклю-
чений в жидкий металл.
Практика показывает, что при кипении ванны и обильном газовы-
делении происходит интенсивное удаление неметаллических включений
[143]. Это связано с улучшением условий укрупнения включений и их
доставки на границу раздела металл — шлак вследствие развитой кон-
векции, а главное — удаления флотацией. Включения прилипают к
всплывающим пузырям и выносятся на поверхность. При расчете ра-
боты прилипания включений к пузырям газа W нужно учитывать раз-
мер включений. Неметаллические включения малых размеров легче
прилипают, чем крупные. Существенное влияние на величину W оказы-
вает 6. При небольших краевых углах смачивания необходимо затра-
11*
163
тить меньшую работу [144]. Авторы работы [145] рассматривают три
возможных механизма прилипания неметаллических включений к по-
верхности всплывающих пузырей СО: инерция, зацепление и миграция
включений. Расчеты показывают, что для мелких включений (rd мкм)
преобладающим механизмом осаждения является диффузия из турбу-
лентного потока, для частиц радиусом г<10 мкм — зацепление и для
более крупных — инерционное отклонение от линии тока.
VI.5. СВЯЗЬ МЕЖДУ СВОЙСТВАМИ
ПОВЕРХНОСТЕЙ РАЗДЕЛА ФАЗ
И СКОРОСТЬЮ УДАЛЕНИЯ ВКЛЮЧЕНИЙ
Связь между скоростью движения включений и межфазным натяжением
на границе металл — включение. Сопротивление, которое испытывает
недеформируемая частица при движении в вязкой жидкости, не зави-
сит от природы тела и характера его поверхности. Высказывается пред-
положение, что с увеличеним работы адгезии (или уменьшением ам-в)
скорость движения должна снижаться. Некоторые исследователи пы-
таются установить количественную связь между скоростью всплывания
и поверхностными характеристиками, вводя в формулу Стокса попра-
вочный множитель, учитывающий влияние ам-в. Однако точка зрения
о существовании связи между скоростью всплывания включений и
межфазным натяжением, между включением и металлом является
ошибочной. Наблюдаемые на практике различия скоростей движения
при изменении межфазного натяжения следует объяснить другими при-
чинами. Например этот эффект может быть связан с неконтролируемым
газовыделением на поверхности движущегося объекта и др. Вопрос о
возникновении скольжения между жидкостью и движущимся объектом
в случае отсутствия смачивания рассмотрен Б. В. Дерягиным [146],
который показал, что эффект скольжения в жидкостях, не смачивающих
твердое тело, имеет место лишь в очень узких зазорах (~1 мкм). Та-
ким образом, различия в поверхностных характеристиках в системе
включение — расплав могут проявиться лишь в пограничных слоях тол-
щиной ~1 мм, что существенно меньше обычно наблюдаемых неметал-
лических включений [147]. Для определения скорости движения твер-
дого шара в жидкости с учетом скольжения на границе раздела фаз мо-
жет быть использовано уравнение, предложенное Ламбом:
V = vCt <3т1 + /(2т> + М ’ (279)
где k — коэффициент трения скольжения; уСт —скорость движения
по формуле Стокса. Расчеты по уравнению (279), выполненные для
двух типов включений А12О3 и SiO2 показывают, что поправочный мно-
житель к стоксовой скорости для включений радиусом 0,01; 0,1; 1 и
10 мкм составляет примерно 0,76 — 0,84; 0,93 — 0,97; 0,99; 1,0 [148].
Следовательно, скольжение на границе металл — включение может
влиять на сопротивление движению лишь для очень мелких неметалли-
ческих включений (0,01—0,001 мкм). Для более крупных включений
(>1 мкм) поправочный множитель близок к 1. Таким образом, лучшее
удаление включений, характеризующихся меньшей смачиваемостью
(А120з и др.), чем силикаты, связано не с эффектом скольжения, а с
особенностями перехода межфазной границы.
Адсорбционное торможение. Присутствие в жидком металле по-
верхностно-активных компонентов (серы, кислорода и др.) приводит к
адсорбции этих компонентов на поверхности неметаллических частиц
и существенному снижению межфазного натяжения на границе ме-
164
талл — включение. А. Н. Фрумкин показал, что присутствие поверхно-
стно-активных веществ (ПАВ) может вызывать торможение движущей-
ся частицы в результате перемещения молекул адсорбционного вещества
с лобовой части включений в кормовую. Это приводит к снижению
поверхностного натяжения в кормовой части. При градиенте межфазно-
го натяжения возникает сила, тормозящая движение капли и тем самым
предотвращающая накопление ПАВ в кормовой части. Расчеты пока-
зывают, что эффект адсорбционного торможения заметно влияет на
скорость перемещения для включений размером 1 мкм.
Движение неметаллических включений в полях диффузии ПАВ.
В жидком металле существуют локальные области, характеризующиеся
градиентом концентрации ПАВ. Такие области появляются в резуль-
тате введения в жидкий металл раскислителей, легирующих добавок
и др. Присутствие неоднородных концентрационных полей (полей диф-
фузии) приводит к неравномерной адсорбции вдоль поверхности части-
цы. Возникающий градиент поверхностного натяжения обусловливает
возникновение силы, вызывающей движение неметаллического включе-
ния [149]. Расчеты показывают, что для оксидных частиц сложного со-
става (А120з—СаО—SiO2—FeO) даже при незначительном градиенте
концентрации поверхностно-активного кислорода (1О“2О/о/м) в расплав-
ленном железе реализация адсорбционного механизма движения вместо
гравитационного приводит к увеличению скорости частиц размером
3—8 мкм в пять — десять раз, а частиц размером 10—20 мкм — в пол-
тора — три раза. Лабораторными опытами показано, что полнота уда-
ления оксидных включений зависит от градиента концентрации. Скоро-
сти всплывания продуктов раскисления в полях диффузии кремния и
марганца превышают скорости всплывания под действием гравитацион-
ных сил в 30—50 и 10—15 раз соответственно. Больший эффект для
кремния связан с тем, что он является более капиллярно активным по
отношению к железу, чем марганец. По рассмотренному механизму
происходит укрупнение включений в основном в локальных объемах
металла. Удаление включений из объема металла происходит под дей-
ствием гравитационных сил и конвективных потоков.
Пример. Рассчитаем скорость движения жидкой неметаллической
частицы размером 20 мкм, содержащей до 40 % А12О3 в жидком ме-
талле в поле диффузии ПАВ. Действием выталкивающей силы можно
пренебречь. Концентрация кислорода в металле равна 0,015 %, для
такого содержания кислорода Г^Г—ЗОО мДж/м2 (Г—адсорбция кис-
лорода на поверхности неметаллического включения; R — универсаль-
ная газовая постоянная; Т — абсолютная температура). Значение Г7?Г
получено расчетом из зависимости межфазного натяжения на границе
включение — металл от содержания кислорода в последнем. Среднее
значение градиента концентрации кислорода в окрестности неметалли-
ческого включения dc/dx=0,5* 1О”1О/о/м. Вязкость металла т)=0,5 Па-с.
Выражение для силы, вызывающей перемещение частицы в поле диф-
фузии, описывается формулой:
F' =— S grad с. (280)
В соответствии с уравнением Гиббса:
grad а =— Г grad р. (281)
В свою очередь
др дс ПТ дс
grad р = —— • —— =-------- • —— , (282)
дс дх с дх
где с — концентрация вещества.
165
Подставляя уравнения (281) п (282) в выражение (280), получим
„ TRT de
F = S----------.
с dx
(283)
Выражение для скорости движения включения получим, если силу F
приравняем силе сопротивления среды:
F 4лг2Г/?Т (dc/dx) 2 rTRT de
v = ------=---------—--------— = — ----------------— • (284)
блгт] бл/ri] 3 Л] dx
Подставляя значения величин в уравнение (284), получим: и=
= 2-10“5*300* 10~3-0,5-Ю^/З-1,5* 10~2«5-10~3» 1,4-10“3 м/с = 1,4 мм/с.
Для сравнения \ кажем, что скорость всплывания неметаллического
включения такого же размера в гравитационном поле равна 0,4 мм/с.
VI.6. ПЕРЕХОД ГРАНИЦЫ РАЗДЕЛА МЕТАЛЛ—ШЛАК
Термодинамическое условие устойчивости неметаллических включений.
Для того, чтобы ответить на вопрос, останется неметаллическое включе-
ние в жидкой стали или перейдет в шлаковую фазу (без учета кине-
тики процесса), необходимо сформулировать общее термодинамическое
условие устойчивости включений в жидком металле. Условия самопроиз-
вольного перехода неметаллических включений через границу раздела
металл — шлак обычно записывают в виде соотношения:
(°гм-в+ °м—ш) > в’ (285)
или в виде эквивалентного выражения До=ош-в—Ом-В—ом-ш. Переход
границы раздела может осуществляться при условии Да<0. Для боль-
шинства оксидных систем До<0 и, следовательно, частицы могут пе-
реходить границу раздела. Соотношение (285) является приближенным,
тем не менее им часто пользуются для практических расчетов.
Краевой угол смачивания и работа адгезии в системе жидкий ме-
талл— неметаллические включения для некоторых сталей и сплавов
приведены в табл. 19 [69—74]. Значения межфазного натяжения и
Таблица 19. Значения краевого угла смачивания и работы
адгезии W& в системе жидкий металл — неметаллические включения
Включе- ние Жидкий металл 6, град Включе- ние 1 Жидкий металл 0. град wa’ г мДж/м2
А12О3 Железо 130 A1N ШХ15 ПО 960
А12О3 Fe—4 % С 120 В\ ШХ15 114 875
SiO2 Железо ПО TiN ШХ15 ПО 1007
Сг2О3 » 88 A1N 12X18Н9 125 1
СаО » 132 А1оО3 12Х18Н9Т 570
ТЮ2 » 84 А12О3 Никель 130 —
краевого угла на границе раздела жидкое железо — А12О3 приведены
в табл. 20. Ю. А. Минаев, А. И. Русанов [150] на основе термодинами-
ческого рассмотрения условий устойчивости включений в заданной фазе
предложили следующую формулу, при помощи которой можно пред-
166
Таблица 20. Межфазное натяжение (ом-в) и краевой угол (0)
на границе раздела жидкое железо — А12О3
Металл 0, град ав—Г’ мДж/м2 ам—г. мДж/м2 GM—В’ мДж/м2
Железо 140 900 1850 2280
Fe- И 4 % С . . . 133 900 1730 2080
Fed н 0,002 % S . 140 900 1390 1965
сказать переход неметаллического включения из металла на поверхность
металл — шлак или металл — газ:
К = ам_в- aM_r = V2 (1 + cos 0M_B) - 0,3ов_г. (286)
Расчеты по формуле (286) показывают, что твердые включения
AI2O3 образуют наименее устойчивую взвесь и лучше удаляются из ме-
талла, чем другие твердые включения (SiC>2» MgO и др.). Стабильность
эмульсий, образованных жидкими продуктами раскисления, также
уменьшается с увеличением содержания А12О3. Например при 1560 °C
плавильным и рафинировочным шлаком с высоким содержанием А12О3
К=—1380, для 75 % А120з и 25 % SiO2 /(=—740; для SiO2 /<=—450
(расчетные значения). Все металлургические взвеси (твердые включе-
ния) и эмульсии (жидкие включения) характеризуются отрицательными
значениями /С Это означает, что при отсутствии химических процессов
растворения и изменения условий адсорбции в процессе выхода на
границу раздела фаз включения являются неустойчивыми дисперсными
системами. Следовательно, при К<0 возможен самопроизвольный пере-
ход частиц из металла в шлак. Если рассматривать устойчивость вклю-
чений в условиях, когда жидкий металл покрыт шлаком (все продукты
раскисления растворяются в шлаках, а на границе раздела включение —
шлак ов-ш->0), то приближенно коэффициент К можно определить по
формуле:
К«-ав_г-ам_г. (287)
Расчеты по формулам (286) и (287.) показывают, что по мере сни-
жения содержания А12О3 в жидких продуктах раскисления стабиль-
ность образованных им эмульсий возрастает. Под белым электростале-
плавильным и рафинировочным шлаком с высоким содержанием А12О3
и СаО неметаллические включения менее устойчивы, чем под карбидным.
Под карбидными шлаками сталь получается более загрязненной по двум
причинам: включения в металле более устойчивы; карбидный шлак
имеет большую склонность к запутыванию в металле вследствие повы-
шенной работы адгезии. Таким образом, металлургические системы ха-
рактеризуются той особенностью, что неметаллические включения в жид-
кой стали образуют термодинамически неустойчивую систему. Переходу
включений из металла в шлак соответствует снижение свободной энер-
гии. К числу факторов, снижающих термодинамическую устойчивость не-
металлических включений в жидком металле и способствующих пере-
ходу границы раздела металл — шлак, металл — газ, относятся: 1) вы-
сокое межфазное натяжение на границе металл — включение <гм_в;
2) высокое поверхностное натяжение жидкого металла ом-г; 3) низкое
поверхностное натяжение неметаллического включения Ов-г.
Образование устойчивых пленок металла на границе раздела фаз.
167
В предыдущем разделе при оценке устойчивости неметаллических
включений не учитывалась возможность образования смачивающих
пленок металла на поверхности частицы. Вместе с тем закономерности
перехода частиц из металла в шлак следует оценивать с учетом этих
пленок. Образование устойчивой пленки может привести к появлению
энергетического барьера при переходе границы раздела фаз. Исследо-
ваниями показано, что тугоплавкие неметаллические включения (напри-
мер, А120з) удаляются из металла более эффективно, чем жидкие вклю-
чения силикатов. Это связано с различными условиями преодоления
тонкой пленки металла на границе раздела металл — шлак и металл —
газ включениями различных типов. Например выходу силикатных вклю-
чений на границу металл—газ предшествует деформация тонкой пленки
металла. В. В. Хлынов с сотрудниками экспериментально установи-
ли, что в месте выхода включения SiC>2 на поверхность металла появля-
лась выпуклость, которая сопровождалась оттеканием металла с по-
верхности оксида под действием поверхностных сил. В случае удаления
включений типа А12О3 разделительная пленка металла «прокалывалась»
без предварительной деформации, а поверхность расплава при этом не
искажалась (рис. 42). Было показано, что наблюдаемые явления не свя-
Рис. 42. Переход частицами корунда (/) и силикатов (2) границы раздела шла-
ковой (3) и металлической (4) фаз
Рис. 43. Схема возникновения расклинивающего давления:
1 — шлак; 2 — металл; 3 — неметаллические включения; 4 — пленка
заны с адсорбционно-кинетическим эффектом (Марангони), так как при
медленном растяжении поверхности металла диффузия успевает обес-
печить равновесную концентрацию ПАВ на межфазной поверхности.
Устойчивость пленок связана с расклинивающим давлением, впер-
вые обнаруженным Б. В. Дерягиным. Первоначальное понятие раскли-
нивающего давления было применено к случаю тонкого слоя жидкости,
разделяющего поверхности двух твердых тел. Расклинивающее давление
тонкого плоскопараллельного слоя жидкости, разделяющего два твердых
тела, равно давлению р, с которым действует в состоянии равновесия
слой жидкости на ограничивающие его тела, стремясь их раздвинуть
или сжать (если рп<0). Расклинивающее давление появляется по сле-
дующей причине. При утончении жидкости до пленки (рис. 43) поверх-
ностные слои жидкости сближаются до такой степени, что начинает
проявляться потенциальная энергия их взаимодействия, которую нель-
зя отнести ни к одной из граничных поверхностей. Это вызывает появле-
ние расклинивающего давления рп, которое для данной системы зависит
только от толщины пленки Хп. Формулы для расчета рп сложны. Они
включают ряд характеристик, которые для металлических систем (жид-
кий металл — неметаллические включения) неизвестны. Для низкотем-
пературных жидкостей рп начинает проявляться при Хп = 0,1-т-1 мм и
168
достигает 10 кПа. Таким образом, переход неметаллическим включе-
нием границы раздела фаз (металл — газ, металл — шлак) связан с
преодолением тонкой пленки металла. Расклинивающее давление игра-
ет также существенную роль при укрупнении включений в результате
их столкновения.
Рассмотрим термодинамическую устойчивость пленки при смачива-
нии включений жидкими расплавами. Для металлургических систем
представляет интерес случаи, когда ом>огв. Такая ситуация реализуется
при контакте жидкого металла с большинством оксидов, нитридов, суль-
фидов и др. При оценке устойчивости неметаллических включений не-
обходимо учесть работу образования пленки. В этом случае уравнение
(286) принимает вид:
К °М-гР/2 0 cos ®в—м) “Ь Рц *
(288)
где Хп — толщина пленки; рп — расклинивающее давление, равное
д ,
Рп — (ав_г~г ам_г)»
(289)
Поскольку Ом—г всегда положительно, то при К=0 можно получить
приближенную оценку отрицательного значения ра для равновесной
пленки. Пленка устойчива, если любое изменение хц приведет к уве-
личению натяжения. В работе [151] разработан экспериментальный
метод оценки устойчивости пленок, использование которого позволяет
иллюстрировать влияние жидких пленок на процесс перехода неметал-
лическими включениями границы раздела фаз.
При оценке влияния расклинивающего давления на переход вклю-
чениями границы раздела фаз отмечается, что наибольшую устойчивость
имеют пленки на жидких включениях FeO [148]. В процессе раскисле-
ния металла кремнием устойчивость пленок снижается (образуются
силикаты) и совершенно исчезает при 1 % Si. При выходе на поверх-
ность продуктов раскисления силикомарганцем также образуются
устойчивые пленки металла, хорошо смачивающие неметаллические
включения. При подходе к поверхности раздела продуктов раскисления
Т1О2, MgO, А120з пленки не образуются и частицы за ничтожно малое
время (10-4—10“6 с) самопроизвольно выходят на границу раздела
фаз, не испытывая барьерного действия.
VI.7. МЕХАНИЗМ УДАЛЕНИЯ
НЕМЕТАЛЛИЧЕСКИХ ВКЛЮЧЕНИИ
Длительное время считали, что порядок раскисления жидкой стали дол-
жен быть таков, чтобы первоначально обеспечить получение жидких
неметаллических включений, применяя предварительное раскисление
стали марганцем и кремнием с добавками очень малых количеств алю-
миния. При этом исходили из того, что, поскольку преобладающая
часть включений удаляется по закону Стокса, согласно которому ско-
рость удаления пропорциональна квадрату радиуса, нужно добиваться
формирования включений, способных к быстрому укрупнению и удале-
нию. Этим требованиям соответствуют жидкие продукты раскисления:
они легко коалесцируют, в результате чего увеличиваются размеры ча-
стиц, и быстро всплывают в гравитационном поле; хорошо ассимилиру-
ют твердые включения (типа А120з), которые образуются на второй
стадии раскисления при введении сильных раскислителей. Такая точка
зрения на процесс раскисления и механизм удаления неметаллических
169
включений широко распространена и в настоящее время. Последующие
исследования поверхностных свойств на межфазных границах металл—
неметаллическое включение — шлак — газовая фаза [152, 153] показали,
что особенности удаления продуктов раскисления связаны главным об-
разом не с размером, а с типом включений. Было показано, что продук-
ты раскисления сильными раскислителями (А12О3) удаляются из жидкой
стали быстрее, чем силикаты. Быстрое удаление включений корунда
объясняли высоким межфазным натяжением А12О3 (ав-м=2000 мДж/м2)
и явлением скольжения. Наряду с этим было показано, что силикаты,
характеризующиеся хорошей смачиваемостью и меньшим значением
Ов-м=600-н800 мДж/м2, при тех же самых размерах удаляются зна-
чительно медленнее.
Истинная причина более быстрого удаления из стали включений
глинозема связана не с тем, что металл выталкивает неметаллические
включения глинозема на поверхность, а с особенностями перехода гра-
ницы раздела фаз, которые существенно различаются для разных про-
дуктов раскисления. Эксперименты, проведенные лишь в условиях гра-
витационного поля, показали, что скорость всплывания включений
глинозема в спокойной ванне в пять — десять раз меньше скорости
всплывания силикатов того же размера.
На основе исследований, проведенных в последнс время в лабо-
раторных и промышленных условиях по изучению поверхностных не-
металлических включений, механизм удаления включения можно пред-
ставить следующим образом. После введения раскислителей в жидкой
стали образуются неметаллические включения, размеры которых прибли-
женно можно описать кривой нормального распределения. Процесс
удаления из стали неметаллических включений включает две стадии:
1) перенос включений на межфазную поверхность металл — шлак,
металл — газ, металл — футеровка; 2) переход границы раздела фаз.
Крупные включения (10—20 мкм и более) достаточно полно удаляются
из металла под действием выталкивающей силы и конвективного пере-
носа в течение первых минут после введения раскислителей. Даже с
нижних горизонтов сталеплавильных печей включения размером 100 мкм
всплывают за 1 мин, а включения размером <30 мкм — за 5—10 мин.
Для частиц размером <10 мкм действие выталкивающей силы подав-
ляется конвекцией. Перенос мелких включений на границу раздела
металл — шлак осуществляется в основном движущейся жидкостью,
причем с увеличением интенсивности перемешивания (до некоторого пре-
дела) частота столкновений и «прилипания» к границе возрастает. Кон-
векция способствует также укрупнению включений. После того как
включение подведено к межфазной поверхности, начинается вторая ста-
дия — переход границы.
Существуют различные мнения о том, какая из двух указанных
стадий является лимитирующей. Под переходом границы раздела фаз
понимают преодоление пленки жидкого металла между включением и
шлаковым расплавом. При этом если пленка преодолена и неметалли-
ческое включение коснулось шлака, то в большинстве случаев созда-
ются условия, обеспечивающие дальнейшее извлечение включения из
стали. Неметаллические включения (глинозем, силикаты и др.) из числа
тех, которые остались после всплывания крупных частиц, к границе раз-
дела фаз доставляются конвективными потоками металла со скоростью
движущегося расплава. При этом плохо смачиваемые включения (А12О3,
ZrO2 и др.) разделительную пленку металла проходят беспрепятственно.
Следовательно, для этих типов включений процесс удаления лимитирует-
ся доставкой их на границу раздела фаз. Включения (FeO, МпО, SiO2).
хорошо смачивающиеся металлом, разделительную пленку преодолева-
170
ют во времени. Включения такого типа могут накапливаться на меж-
фазной границе, и вероятность их уноса вглубь металла существенно
больше, чем в первом случае. Для этих типов включений в большинстве
случаев реализуются условия, при которых процесс удаления лимити-
руется переходом границы раздела фаз.
Таким образом, на первой стадии размер включений в основном
определяет способ доставки к межфазной границе. Крупные включения
всплывают сами, мелкие переносятся конвективными потоками жидко-
сти. На этой стадии роль поверхностных свойств в основном проявля-
ется в таких процессах, как укрупнение (слияние, слипание, агрегация),
адсорбционное торможение, движение включений в концентрационном
поле и др. На второй стадии удаления включений, связанном с перехо-
дом границы, поверхностные свойства системы (ом-в, Пм-ш, Ош-в) и
характеристики смачиваемости, определяют термодинамические и ки-
нетические закономерности перехода. Размеры включений в этом случае
играют меньшую роль. Рассмотренная схема носит общий характер и
не может отразить многообразия частных случаев, которые имеют место
при выплавке отдельных групп сталей и сплавов, характеризующихся
образованием включений различных типов.
VI .8. ПОВЕДЕНИЕ НЕМЕТАЛЛИЧЕСКИХ ВКЛЮЧЕНИИ
В УСЛОВИЯХ ВАКУУМА
При использовании вакуума создаются дополнительные термодинамиче-
ские и кинетические возможности для удаления из жидкой стали не-
металлических включений. К ним относятся: 1) термическая диссо-
циация неметаллических включений (оксидов, нитридов, сульфидов)
при высокой температуре и малом остаточном давлении; 2) восстановле-
ние оксидных неметаллических включений растворенным в металле уг-
леродом и последующее удаление СО; 3) удаление неметаллических
включений в результате интенсивного перемешивания жидкого ме-
талла и прилипания включений к пузырям при обезуглероживании и
удалении растворенных газов; 4) удаление кислорода в результате
испарения субоксидов.
При внепечной вакуумной обработке из жидкой стали удаляются
неметаллические включения главным образом вследствие их переноса
на границу фаз движущимся потоком расплава и всплывающими пузы-’
рями газа. При этом в зависимости от состава металла, степени его
раскисления и газонасыщенности удаляется до 30 % включений. При
плавке металла в глубоком вакууме (0,133—0,013 Па) степень удаления
возрастает до 50—60 %. В тех случаях, когда металл затвердевает в
водоохлаждаемых кристаллизатороах, наряду с удалением существен-
но изменяются форма, размер и характер расположения оставшихся
включений. Они, как правило, дробятся и распределяются более рав-
номерно. Рассмотрим возможность удаления кислорода из жидкой стали
в результате термической диссоциации оксидов:
МехОу = х [Me] + у [О]. (290)
Получающийся по реакции (290) растворенный кислород диффун-
дирует к поверхности раздела металл — газ и десорбируется в газовую
фазу в виде О2. Таким образом, суммарный процесс связан с осуществ-
лением реакции:
Мех Оу = х [Me] + у/2О2. (291)
С уменьшением остаточного давления в системе реакция (291) про-
171
текает более полно. Однако этот процесс может получить развитие лишь
в том случае, когда давление диссоциации pOj, МехОу оксида больше
парциального давления кислорода р££т в вакуумной камере с задан-
ным значением остаточного давления рост. Значения давлений диссо-
циаций оксидов (СаО, MgO, А12О3, FeO и др.) настолько малы
(< 1,3 мкПа), что в условиях вакуумной плавки (рост=1,3-?1,3-10-2Па)
следует скорее ожидать поступления кислорода в расплав из атмосферы,
чем его удаления в газовую фазу. Вместе с тем расчеты показывают,
что из расплава жидкого никеля уже при 24 Па и 1600 °C возможна
прямая десорбция кислорода в газовую фазу [85]. Присутствие углеро-
да существенно изменяет картину:
Мех Оу + у [С] = х [Me] + у СО. (292)
С уменьшением остаточного давления реакция (292) протекает бо-
лее полно. В некоторых работах показано соответствие между коли-
чеством удаляемых включений и снижением концентрации углерода.
Однако в ряде случаев такого соответствия не наблюдают, что можно
объяснить очень малыми изменениями концентраций углерода
(—0,005 %), которые могут остаться незамеченными. О возможности
восстановления включений свидетельствуют присутствие монооксида
углерода СО в отходящих газах, а также углеродный конденсат на
водоохлаждаемых стенках кристаллизатора, покрытых возгонами мар-
ганца и железа, которые являются хорошими катализаторами реакции
разложения СО (реакции Белла).
Взаимодействие оксида с углеродом может происходить по двум
схемам. В первом случае реакции (292) предшествуют диссоциация
оксида, диффузия углерода и кислорода к поверхности раздела фаз
и последующая реакция между ними. Во втором случае восстановление
оксида связывается с образованием зародыша СО на поверхности вклю-
чений, а последующий рост пузыря с диффузией углерода. В этом
случае характеристики восстановления оксидов зависят от их смачивае-
мости (на А12О3 зародышу легче образоваться, так как он хуже сма-
чивается), соответствия пузыря СО размеру неметаллического вклю-
чения и др. В рассматриваемых условиях могут получить развитие
также процессы всплывания неметаллических включений, увлекаемые
газовыми позырями на поверхность жидкого металла. Возможность
восстановления взвешенных частиц растворенным углеродом зависит
от температуры, остаточного давления и состава металла, глубины зале-
гания включения. Расчеты показывают, что при 1600 °C частично могут
восстановиться включениям SiO2 и достаточно полно МпО, Сг2О3. Воз-
можность восстановления неметаллических включений с образованием
газообразных субоксидов связано с протеканием, например, следующих
реакций:
А12О3(т) + 2 [CJ = А 12О(Г) Ч- 2СО; AG0 = 287 800 — 110,317;
А12О3(т)+ [С] - 2А1Огг) Ч~ СО; AG0 = 375150 — 113,707;
SiO2(T) + [С] = SiO(r) + СО; AG0 = 156 040 — 68,997.
Расчеты показывают, что если содержание углерода составляет
0,1 %, указанные реакции при 1600 °C получают развитие при рост,
равном 21,32 и 87 Па. Несмотря на термодинамическую возможность
восстановления включений углеродом, тем не менее указанные реакции
вследствие различных кинетических трудностей получают ограниченное
развитие. Доля удаленных включений в результате восстановления уг-
леродом не превышает 20 %. В частности, наличие оксидной пленки
172
на поверхности жидкого металла при ВИП, ВЛП и ЭЛП также указы-
вает на то, что процессы диссоциации включений и взаимодействия с
углеродом доминирующей роли не играют.
Глава VII.
КИНЕТИКА МЕТАЛЛУРГИЧЕСКИХ РЕАКЦИИ
Металлургические реакции в большинстве случаев явля-
ются сложными многостадийными процессами, протекаю-
щими на границе металл — шлак — газовая фаза. Изуче-
ние кинетики гетерогенных реакций сводится, как правило,
к последовательному решению двух задач: установлению
лимитирующего звена и определению количественных ха-
рактеристик этого звена (константы скорости процесса, ко-
эффициента массопередачи, влияния температуры и др.).
Реакции сталеплавильного производства характеризуются
тем, что их скорость в большинстве случаев лимитируется
процессами переноса веществ. Различают внешнюю и вну-
треннюю массопередачу. В реакциях между текучими фа-
зами с твердым телом или между жидкостью и газом при
сравнительно малых скоростях потока газа лимитирующей
стадией является внешняя диффузия, удельной количест-
венной характеристикой которой является коэффициент
массопередачи. Данные термодинамики не ограничены
масштабными факторами, в то время как кинетические
данные существенно зависят от геометрических характе-
ристик системы, поэтому скорости металлургических ре-
акций, легко измеряемые в лабораторных условиях, дают
информацию, которую в большинстве случаев нельзя пе-
ренести на промышленные условия. Во многих случаях ре-
шение подобных задач связано с использованием теории
подобия и критериальных уравнений.
VII .!. МОЛЕКУЛЯРНАЯ ДИФФУЗИЯ
Количественные характеристики диффузии описываются
двумя уравнениями Фика. Если количество вещества dm
продиффундировало в единицу времени dx через площадь
S при градиенте концентрации dc/dx, то первый закон Фи-
ка выражается следующим уравнением:
dmtdx =—DS (dc/dx), (293)
где D — коэффициент диффузии, м2/с. Если S=1 м2, а
173
dcldx=\ моль/м4, то D =—dm/dx. Таким образом, коэф-
фициент диффузии равен скорости диффузии через единич-
ную поверхность при градиенте концентрации, равном 1.
Минус указывает, что диффузия происходит в направле-
нии уменьшения концентрации. Под диффузионном пото-
ком П понимают количество вещества (в граммах, молях
и др.), которое диффундирует через единицу площади
(1 м2) за единицу времени (с). Уравнение (293) мало при-
годно для определения D, так как диффузионный поток П
моль/(м2-с) нужно измерять в условиях постоянного гра-
диента, что экспериментально трудно осуществить. Значи-
тельно проще проследить за распределением концентрации
диффундирующего элемента вдоль направления диффузии
за определенный промежуток времени или измерить изме-
нение концентрации во времени в определенной точке. Для
вычисления коэффициента диффузии по эксперименталь-
ным данным используют второй закон Фика, который для
одномерной задачи выражается уравнением:
дс/дх = D(d2 с/дх2). (294)
Интегрируя выражение (294) при заданных начальных
и граничных условиях, которые определяются конкретной
постановкой задачи, можно получить выражение для ско-
рости диффузии [154; 155]. Для приближенных расчетов
представляет интерес уравнение x2=2Dx. При помощи это-
го уравнения зная значение D можно вычислить путь диф-
фузии х за заданное время, или время, необходимое для
того, чтобы фрбнт диффузии переместился на величину х.
Приведем еще одно полезное соотношение: q=k]' х. Про-
порциональность количества продиффундировавшего веще-
ства q квадратному корню из времени является убедитель-
ным доказательством диффузионного характера исследуе-
мого процесса. Значения коэффициентов диффузии в
твердых телах составляют 10-12—10~16 жидкостях 10-8—
10-10 и газах 10~s—10-4 м2/с.
В табл. 21. приведены значения коэффициентов диффу-
зии некоторых элементов в жидком железе. Зависимость
коэффициента диффузии от температуры выражается урав-
нением D=D0 e~E/RT, где Е — энергия активации диффу-
зии, Дж/моль. Величину Е экспериментально определяют
из углового коэффициента прямой в координатах lg D—
— 1/Т. Энергия активации диффузии в газовой фазе, как
это следует на кинетической теории газов, равна приблизи-
тельно RTj2. Таким образом, при температурах металлур-
гического производства энергия активации диффузии в га-
174
Таблица 21. Значения коэффициента диффузии D компонентов и
энергия активации (£) в жидком железе
Диффунди- рующий элемент Т. К D • 109, м*/с Е, кДж/моль Диффунди- рующий элемент т, к D • 10’, М*/С Е, кДж/моль
N 1873 5,0 38,6 1863 7,2 50,4
Н 1873 300,0 16,8 Si 1833 3,8 37,8
О 1873 12 21,0 Мп 1953 0,61 67,1
S 1873 4,5 42,0 Сг 1843 0,4 29,4
р 1823 4,7 33,6 W 1873 1,2 100,7
зах составляет ~8,0 кДж/моль. Для диффузии компонен-
тов в жидкой стали энергия активации диффузии
варьируется в пределах 20—80 кДж/моль. Увеличение ко-
эффициента диффузии с температурой связано с уменьше-
нием вязкости т), температурная зависимость которой опре-
деляется соотношением: i] —x\^E^ RT, где Еп —энергия ак-
тивации вязкого течения жидкости.
В табл. 22 приведены значения динамической вязкости
для некоторых жидкостей. Зависимость коэффициента диф-
фузии D от вязкости т) для частицы радиусом г определя-
ется формулой Стокса—Эйнштейна:
D = RT/6nn]NA, (295)
где Na — число Авогадро.
Значения коэффициентов диффузии, вычисленные по фор-
муле (295), удовлетворительно согласуются с эксперимен-
тальными данными. По формуле (295) решают и обратную
задачу — по заданному значению D определяют радиус
диффундирующей частицы. Однако, погрешности, связан-
ные с применением формулы (295), существенны и могут
достигать одного порядка.
Таблица 22. Значения динамической вязкости т] для некоторых
жидкостей
Жидкость т, К кПа-с Жидкость Т, К п. кПа с
Вода Глицерин . . . Касторовое масло Жидкое железо . Жидкая сталь . . 298 298 298 1873 1873 0,89 500 800 4,5 2,5 Жидкий чугун . . Жидкий шлак . . Средний шлак . . Густой шлак . . 1673 1873 1873 1873 1,5 2,0 20,0 >200
175
VII.2. КОНВЕКТИВНАЯ ДИФФУЗИЯ
В металлургических процессах с участием жидких и га-
зообразных фаз молекулярная диффузия в чистом виде
встречается сравнительно редко. На самом деле имеет
место конвективная диффузия, когда на молекулярную
диффузию накладывается перенос вещества потоком. В тео-
рии конвективной диффузии различают два пограничных
слоя: гидродинамический бг и дифузионный бд. Под гид-
родинамическим пограничным слоем понимают толщину
жидкости или газа на границе с конденсированной фазой,
в которой скорость потока жидкости изменяется от а=0
Рис. 44. Схема гидродинамического Сг и диффузионного 6Д пограничных слоев
при набегании жидкости со скоростью а на плоскую пластину
Рис. 45. Изменение концентрации в пограничном слое (с*—концентрации насы-
щения, вблизи твердой фазы; с — концентрация вещества в объеме жидкости):
1 — твердая фаза; 2 — жидкость
(на поверхности) до значения а в ядре потока. Таким об-
разом, вследствие трения скорость жидкости на поверхно-
сти равна 0, а на некотором расстоянии от поверхности она
равна скорости течения в объеме. Если на плоскую плас-
тину набегает поток жидкости со скоростью а, то распре-
деление скоростей вблизи твердого тела на расстоянии у
от точки набегания струи имеет вид, показанный на рис. 44.
В этом случае толщина гидродинамического слоя опреде-
ляется уравнением:
бг = 5,2 К (vy)/a (296)
где v — кинематическая вязкость. В кинетических задачах
интересуются в основном не распределением скоростей,
а распределением концентрации вблизи межфазной по-
верхности. Толщину слоя жидкости, в которой наблюдают
градиент концентрации компонента, называют диффузион-
ным пограничным слоем (6Д). Если изучается растворение
твердого тела в жидкости и процесс лимитируется массо-
176
передачей атомов растворенного вещества, то к поверхности
твердого тела прилегает слой жидкости, в которой кон-
центрация соответствует насыщению (рис. 45). На некото-
ром расстоянии от поверхности концентрация вещества со-
ответствует концентрации с в объеме. Толщину слоя, в пре-
делах которой наблюдают изменение концентрации от с*
до с называют диффузионным пограничным слоем. При
данных значениях у и а толщина гидродинамического слоя
в соответствии с формулой (296) определяется вязкостью v,
а толщина диффузионного слоя — коэффициентом диффу-
зии D. Во многих случаях количественная связь между тол-
щинами гидродинамического и диффузионного погранично-
го слоев удовлетворительно описывается уравнением:
6д/бг = (D/v)1/3. (297)
Коэффициент диффузии компонентов в жидкой стали £)«
«10-9 м2/с, а кинематическая вязкость жидкой стали v~
« 10-6м2/с. Тогда £)/v=10-3 и в соответствии с уравнением
(297) бд«0,1 бг. Таким образом, в рассматриваемых ус-
ловиях толщина диффузионного слоя составляет всего
10 % от толщины гидродинамического слоя. В общем ви-
де бд=бг/Рг1/3. Для газов критерий Прандтля Рг=1, по-
этому бд = бг.
Коэффициент массопередачи
При изучении гетерогенных металлургических реакций в
большинстве случаев истинная толщина пограничного диф-
фузионного слоя неизвестна. Для практических расчетов
пользуются коэффициентом массопередачи р. В этом слу-
чае формулу для определения диффузионного потока 77 за-
писывают в следующем виде:
П =— D (дс/дх) =—D (Дс/б) =— рДс,
(298)
где p=D/6.
Распределение концентрации вещества вблизи поверх-
ности твердого тела показано на рис. 45. Поток вещества
через плоскость х=0 определяется уравнением:
П =— D (дс/дх)X=Q. (299)
Если сложное распределение концентрации в пределах
истинной толщины диффузионного слоя бд заменить линей-
ным распределением, то
(дс/ дх)х=0 = — (с* — с)/б, (300)
12—675
177
где 6 — эффективная толщина пограничного диффузион-
ного слоя.
Величну б можно определить по отрезку, который отсе-
кает касательная к кривой в точке с(х=0) на оси абсцисс.
Подставив значение производной из уравнения (300) в
уравнение (299), получим:
П = (Р/б)(с* — с) = § (с* — с). (301)
Под эффективной толщиной пограничного диффузион-
ного слоя понимают значение б, которое обеспечивает та-
кую же величину потока, что и действительное значение
бд при переносе вещества в результате чисто молекуляр-
ной диффузии.
Поскольку диффузионный поток выражает количество
вещества на единицу поверхности за единицу времени, то
Р имеет размерность скорости (м/с), а концентрация выра-
жается в кг/см3. Если массовая концентрация выражена в
процентах, то р имеет размерность кг/(м2«с«%). От мас-
сового содержания, выраженного в процентах, к размерно-
сти кг/м3 можно перейти при помощи выражения с=
= (%) (р/ЮО), где р плотность среды, кг/м3. Однако, в ря-
де случаев р рассчитывают не по величине потока на 1 м2
поверхности, а на 1 м3 объема пористого или зернистого
материала. В этом случае поток имеет размерность
кг/(м3-с), а р—1/с. Уравнение (301) широко применяют
при изучении кинетики гетерогенных металлургических ре-
акций. Если измеряют изменение концентрации реагирую-
щего компонента, то уравнение (301) записывают следую-
щим образом:
dc/dt = (— DS/6V)(c — с*), (302)
где V — объем; DS/(6V) =р. Рассмотрим температурную
зависимость р. Поскольку р=£)/б, а б в соответствии с
формулой (296) пропорциональна вязкости б~т)1/2, то
Efr=ED+ Таким образом, энергия активации массо-
передачи Ер имеет сложный физический смысл и отражает
температурную зависимость коэффициента диффузии и вяз-
кости.
При внешней диффузии количественные закономерно-
сти переноса вещества в направлении координаты х опи-
сываются уравнением конвективной диффузии:
П ~— D (дс/дх) + ах с. (303)
Первое слагаемое правой части уравнения (303) характе-
ризует перенос вещества в результате молекулярной диф-
178
фузии, второй — перенос вещества потоком. Анализ урав-
нения (303) показывает, что в жидкостях перенос вещест-
ва осуществляется потоком. Даже в условиях естественной
конвекции не имеет места чисто молекулярный перенос.
Слагаемым конвективного переноса можно пренебречь лишь
при малых значениях критерия Пекле (Pe=PrRe). Такие
условия реализуются, в частности, при массопередаче в га-
зовой фазе [157]. Уравнение конвективной диффузии мож-
но решить только для гидродинамически определенных за-
дач, т. е. для задач, в которых известны распределение ско-
ростей и концентрации. Вместе с тем большинство
металлургических задач являются гидродинамически неоп-
ределенными. Закономерности движения жидкого распла-
ва в сталеплавильной печи и в ковше сложны и в большин-
стве случаев неизвестны. С этим обстоятельством связаны
трудности решения уравнения конвективной диффузии для
многих задач металлургического производства. Точные ре-
шения конвективной диффузии получены лишь для ограни-
ченного числа задач, в частности, для набегания потока
на плоскую пластину и на поверхность вращающегося
диска.
V1I.3. КРИТЕРИАЛЬНЫЕ УРАВНЕНИЯ
В ЗАДАЧАХ ПО КИНЕТИКЕ
Коэффициент массопередачи, как правило, определяют экс-
периментально и устанавливают связь между значением £
и различными параметрами. Например, если переход серы
из металла в шлак лимитируется массопереносом в шлако-
вой фазе, то нужно знать зависимость £ от интенсивности
перемешивания шлака, вязкости, характеристических раз-
меров системы, температуры и других величин. Количествен-
ную связь между £ и соответствующими параметрами срав-
нительно просто можно устанавливать' на основе теории по-
добия [156—160]. Значение коэффициента массопередачи
определяется критерием Нуссельта Nu = £d/£> (d — харак-
теристический размер, м; D — коэффициент диффузии,
м2/с). Иногда диффузионный критерий Нуссельта называ-
ют критерием Шервуда и обозначают Sh. Критерий Нус-
сельта характеризует перенос вещества в неподвижной сре-
де или ламинарном потоке. Для случая, когда поток жид-
кости или газа обтекает тело (например, растворение куска
ферросплава в сталеплавильной ванне), критерий Нуссель-
та имеет постоянное значение и зависит лишь от геометри-
ческой формы тела. В частности, при обтекании шара по-
12*
179
током Nu=2. При ламинарном течении жидкости или га-
за критерий Nu также имеет постоянное значение.
Например, для труб Nu=3,66. Количественные характе-
ристики между р и соответствующими параметрами вязко-
стью v, скоростью течения а, характеристическими разме-
рами (du др.) сравнительно просто можно установить на
основе критериальных уравнений Nu=&Re"Prm. Количест-
венную связь между критериями Nu, Re и Рг обычно на-
ходят по экспериментальным данным. Пользуясь изложен-
ной методикой, можно написать критериальные уравнения,
которые описывают зависимость коэффициента массопере-
дачи р от парметров опыта:
pd/D = b (ad/v)m (v/D)n-
р Аат vn~m.
(304)
Если показатель степени tn изменяется в пределах 0,4—
0,6, то это указывает на диффузионную природу процесса.
При помощи критериальных уравнений типа (304) в лите-
ратуре описана кинетика перехода серы из металла в шлак,
обезуглероживания, растворения твердых тел, восстанов-
ления оксидов и др. [156—159].
VII.4. ПРИЗНАКИ,
ПО КОТОРЫМ ОПРЕДЕЛЯЮТ
ЛИМИТИРУЮЩУЮ СТАДИЮ
ГЕТЕРОГЕННОГО ПРОЦЕССА
Наиболее распространенные признаки, по которым можно
выявить лимитирующее звено, рассмотрены ниже.
1. Влияние перемешивания или изменение скорости по-
тока газа или жидкости на кинетику изучаемого процесса.
Если изменение скорости потока а или интенсивности пере-
мешивания не приводит к изменению скорости процесса в
целом, это значит, что внешняя диффузия не является ли-
митирующей стадией. В этом случае процесс контролиру-
ется либо внутренней диффузией, либо химико-адсорбци-
онным звеном. Зависимость скорости реакции от интенсив-
ности перемешивания в большинстве случаев наиболее убе-
дительно свидетельствует в пользу внешней диффузии.
2. Отсутствие градиентов концентрации взаимодейст-
вующих компонентов в фазах, прилегающих к межфазной
границе, указывает на то, что реакция на поверхности раз-
дела фаз протекает в кинетическом режиме. Градиент кон-
центрации в одной из фаз указывает на диффузионный ха-
180
рактер процесса. В этом случае имеет место термодинами-
ческое равновесие реакции, протекающей на границе
раздела фаз.
3. Вид кинетической кривой. Во многих случаях харак-
тер режима определяют по виду кинетической кривой или
соответствию экспериментальных данных определенным ре-
шением кинетических уравнений. Рассмотрим кинетику по-
глощения азота жидким железом, который включает три
стадии: внешнюю диффузию — массоперенос азота в газо-
вой фазе к поверхности жидкого металла; химико-адсорб-
ционное звено, включающее процессы диссоциации моле-
кулярного азота, адсорбции, десорбции и др.; внутреннюю
диффузию—массоперенос азота в жидком расплаве.
Эксперименты показывают, что первая стадия, как пра-
вило, осуществляется довольно быстро и не является лими-
тирующей. Если лимитирующей является вторая стадия, то
прежде всего нужно установить, какой частный процесс
контролирует адсорбцию в целом. Допустим, что такой ста-
дией является диссоциация молекул азота на атомы и ад-
сорбция последних на поврехности расплавов. В этом слу-
чае
d [Nl/dT = К. pNt — К2 [N]2,
(305)
где KiPn, — характеризует скорость прямого процесса;
/(2[N]2— скорость обратного процесса.
Поскольку ры, =[N]|//<n и в состоянии равновесия
KiPn, =/G[N]2, откуда Ki/K2=Ku, то
d[N]/dT = (^/2<n)([N]2 — INF).
(306)
Интегрируя уравнение (306) в пределах О и [NJo и
t[N], получим
In Wp + IN]---[N]P-[N]_o = 2 i [N]р т. (307)
[N]p —[N] [N]p + [N]0 P
Если лимитирующей стадией является внутренняя диффу-
зия, то
d [N]/dt = рж (IN]P — [N])
(308)
и после интегрирования получим
(309)
Для определения лимитирующей стадии эксперимен-
тальные результаты обрабатывают при помощи уравнений
(307) и (309) и полученные данные представляют гра-
181
фически. График, на котором получена линейная зависи-
мость, указывает на то, что данная стадия является лими-
тирующей. Из приведенных уравнений следует, что при
т-»-0 скорость адсорбции пропорциональна р^г (процесс
лимитируется второй стадией) и Урйа (процесс лимитиру-
ется третьей стадией). Однако рассмотренные признаки в
некоторых случаях не дают однозначного ответа. В частно-
сти, решение для первой стадии будет таким же, как и для
второй.
4. Температурная зависимость скорости реакции. По-
скольку энергия аткивации химической реакции Ек'боль-
ше энергии активации диффузии Ед, то обычно при низких
температурах процесс лимитируется актом химического
взаимодействия, при высоких температурах — массопе-
реносом. Таким образом, с повышением температуры кине-
тические ограничения устраняются и процесс, как правило,
реализуется в диффузионном режиме.
5. Пропорциональность количества прореагировавшего
вещества квадратному корню из времени (<7=^Кт) яв-
ляется доказательством того, что процесс лимитируется
диффузией.
6. Если добавки ПАВ влияют на скорость процесса, то
в большинстве случаев это указывает, что реакция проте-
кает в кинетическом режиме.
7. Порядок реакции, определяемый экспериментально.
Если на основе опытных данных установлено, что реакция
имеет первый порядок, то на основе этого нельзя сделать
однозначного вывода о лимитирующей стадии, поскольку
скорость диффузии также описывается реакцией первого
порядка. Дробный или второй порядок реакции в боль-
шинстве случаев указывает, что диффузионное сопротив-
ление либо устранено, либо существенно уменьшено.
Если реакция по реагирующему компоненту имеет ну-
левой порядок, то это указывает, что скорость реакции не
зависит от концентрации, которая во времени изменяется
линейно.
VII.S. КИНЕТИКА РАСТВОРЕНИЯ, ПЛАВЛЕНИЯ
Растворение твердого тела в жидкости включает две ста-
дии: 1) разрушение кристаллической решетки и переход
атомов твердого тела в жидкий раствор (кинетическая ста-
дия); 2) массопередачу растворенных атомов из погранич-
ного слоя в объем раствора (диффузионная стадия). Рас-
182
смотрим кинетический режим растворения. В этом случае
dddt = (Л\ Sn0/V)(1 — с/снас), (310)
где «о — число атомов на единичной поверхности.
Интегрируя уравнение (310) при условии, что при т=0,
с=0, и приняв, что (KiHoS)/V=K получим
нас
Лт^снас)
л
(311)
Если процесс реализуется в кинетической области, то
зависимость концентрации от времени выражается экспо-
ненциальной зависимостью. В этом случае константу рас-
творения можно определить графически по угловому коэф-
фициенту прямой в координатах 1g с—т [162]. Если ско-
рость растворения лимитируется массопередачей, то к
поверхности твердого тела прилегает слой жидкости толщи-
ной бд (см. рис. 45), в котором концентрация растворенно-
го вещества изменяется от сНас на границе с твердым те-
лом до с, равного концентрации растворенного вещества
вдали от поверхности сОб. Градиент концентрации (сНаС—
—с)/б определяет размер диффузионного потока
dc/dx = DS 1(енас — с)/б!. (312)
Интегрирование уравнения (312) при условии, что при т=0
с=0, дает
( — \
е = енаси — е v6 J. (313)
Из уравнения (313) следует, что при диффузионном ре-
жиме растворения также наблюдается экспоненциальная
зависимость концентрации от времени, поэтому только по
виду кинетической кривой нельзя сделать вывод о лимити-
рующей стадии, поскольку для обоих режимов характерна
одинаковая зависимость концентрации от времени. В про-
межуточном режиме растворения с=сНас(1—еКх), где К =
=^Р/(^+Р)- При р^>/г получаем уравнение, соответствую-
щее кинетическому режиму, при р<£& — диффузионному ре-
жиму. Если в жидкости растворяется монокристалл, то
скорость растворения существенно зависит от кристалло-
графической плоскости. Большие скорости растворения со-
ответствуют граням, которые характеризуются большим
значением поверхностного натяжения. Обычно рассматри-
вают два случая растворения твердых тел в жидкостях.
В первом случае твердое тело имеет более низкую темпе-
ратуру плавления, чем жидкость (например, растворение
алюминия в жидкой стали). При этом твердое вещество пе-
183
реходит в жидкое состояние, а последующее распределение
по объему связано с диффузией или конвективным массо-
переносом. Во втором случае температура плавления твер-
дого тела выше температуры расплава и растворение свя-
зано с химическим взаимодействием, диффузией компонен-
тов, проникновением жидкого расплава в поры твердого
тела и др.
В последние годы широкое распространение получила
диффузионная теория плавления твердых тел. Эта теория,
в частности, описывает плавление лома в жидком чугуне.
Растворенный в жидком расплаве углерод диффундирует к
поверхности скрапа и насыщает до некоторой концентра-
ции, что приводит к снижению температуры плавления в со-
ответствии с диаграммой системы Fe—С и переходу твер-
дой фазы в жидкую. Таким образом, поверхностная кон-
центрация углерода определяется соотношением двух
потоков углерода: внешнедиффузионного, к границе разде-
ла фаз и внутридиффузионного, связанного с переходом
углерода внутрь твердой фазы. Наряду с указанным меха-
низмом существенную роль играет межкристаллитное про-
никновение жидкого расплава, который под действием ка-
пиллярных сил располагается по границам зерен или в раз-
витых порах, что приводит к распаду твердого тела на
отдельные куски. По такому механизму происходит плавле-
ние шлакообразующих, растворение извести и др. Если на
границе раздела фаз между жидкостью и твердым телом
имеет место взаимодействие с образованием химических со-
единений (растворение железа в алюминии), то подвиж-
ность компонентов описывается законами реактивной диф-
фузии [164]. В большинстве случаев растворение добавок
в жидких металлах и шлаках проходит по диффузионному
механизму, поэтому для ускорения растворения необходи-
мо перемешивание расплава.
VII.6. ВЛИЯНИЕ ПАВ НА КИНЕТИКУ
ГЕТЕРОГЕННЫХ ПРОЦЕССОВ
Влияние ПАВ на скорость гетерогенного процесса в боль-
шинстве случаев наблюдают, если суммарная скорость ли-
митируется химико-адсорбционной стадией. Если процесс
реализуется в области внешней и внутренней диффузии, то
на границе раздела фаз имеется термодинамическое рав-
новесие и любые изменения состояния границы (например,
в результате добавок ПАВ) не могут быть замечены и вли-
ять на скорость реакции [165]. Однако, если добавки ПАВ
184
тем или иным способом влияют на гидродинамические ус-
ловия в пограничном слое, даже в диффузионном режиме
можно наблюдать изменение скорости под действием ПАВ.
Влияние ПАВ на скорость гетерогенных реакций количест-
венно учитывается введением в кинетическое уравнение ве-
личины 0, которая характеризует число занятых мест. Так,
например, скорость реакции обезуглероживания vc газо-
образным кислородом в присутствии ПАВ можно описать
уравнением: uc=^i(l—0)[Q|Po,» где множитель (1—0)
характеризует число мест, незанятых ПАВ. В большинстве
случаев удовлетворительное
соответствие эксперимен-
тальных и расчетных дан-
ных наблюдают в рамках
лангмюровский кинетики.
Говоря о влиянии ПАВ на
кинетику гетерогенной реак-
ции, обычно имеют в виду
блокировку границы разде-
ла фаз, т. е. уменьшение ак-
тивной поверхности. Однако
если процесс протекает в
диффузионом режиме, то до-
бавки ПАВ вследствие
Рис. 46. Зависимость константы ско-
рости обезуглероживания от концент-
рации поверхностно-активных веществ
уменьшения поверхности
взаимодействия могут смес-
тить реакцию в кинетичес-
кую область. На рис. 46 по-
казано влияние серы и фос-
и скорости перемешивания:
/ — добавки серы, ип=2,5 м/с: 2—5 до
бавки фосфора при соответственно рав
ной 2,5; 1,6; 1 и 0,75 м/с
фора на кинетику обезуглероживания жидкого железа в
условиях электрокапиллярного движения металла в шла-
ке. Из рис. 46 следует, что обезуглероживание развивается
в диффузионном режиме, так как ускорение движения ме-
таллической капли приводит к увеличению значения К\.
В рамках диффузионного режима небольшие добавки ПАВ
не изменяют характера процесса. Однако чем в большей
мере устранено диффузионное сопротивление, тем меньшие
концентрации ПАВ нужны для перевода процесса в кине-
тическую область, которая соответствует линии АВ (см.
рис. 46). При достаточной скорости движения капли про-
цесс развивается в кинетическом режиме. Известно, что
скорость абсорбции азота, протекающая обычно в диффу-
зионном режиме, в присутствии растворенного в металле
кислорода по причинам, указанным выше, лимитируется
кинетическим звеном. Во многих случаях ПАВ играют роль
185
не примесных добавок, а сами являются участниками ре-
акции.
Очевидно, что если реакция протекает на границе раз-
дела фаз, то поверхностные концентрации этих компонен-
тов существенно отличаются от объемных и кинетика рас-
сматриваемых реакций имеет ряд особенностей. Рассмот-
рим реакцию окисления серы, растворенной в металле или
шлаке газообразным кислородом. В этом случае в кинети-
ческое уравнение следует представлять не объемную, а по-
верхностную концентрацию серы. В рамках модели Ланг-
мюра имеем:
— dcs/dx = KpOt [(zbcs)/(l + 6cs)]. (314)
После интегрирования получим:
In + b (с — й ) =— Кро zbx,
где cs—исходная концентрация серы в расплаве; ро, —
парциальное давление кислорода. При малых значениях с|
In (сс/с2 ) + b = In сс +
Линейная зависимость между Incs + ^Cs от т указы-
вает на выполнимость уравнения (314). Если окисление се-
ры происходит в режиме максимальной адсорбции и при
этом диффузионные ограничения устранены, т. е. достав-
ка частиц серы из объема на границу раздела фаз и кис-
лорода не лимитируют процесса в целом, то реакция бу-
дет протекать с постоянной скоростью. При заданном зна-
чении ро, этому соответствует нулевой порядок реакции по
сере. Это выполняется, если bcs >1 и правая часть фор-
мулы (314) является постоянной величиной. С уменьшени-
ем объемной концентрации до с$, при котором Ьс$ <1, на-
блюдается изменение концентрации серы по экспоненци-
альному закону: cs = с^е~Крог. Таким образом, большие
скорости взаимодействия на границе раздела фаз между
компонентами, один из которых является поверхностно-ак-
тивным, наблюдаются лишь в том случае, когда диффузи-
онное торможение либо устранено, либо существенно умень-
шено. В присутствии ПАВ распределение кислорода меж-
ду различными компонентами определяется химическим
сродством и поверхностной концентрацией [11].
VII.7. КИНЕТИКА ОБЕЗУГЛЕРОЖИВАНИЯ
В практике электросталеплавильного производства для
окисления примесей существует два способа подвода окис-
лителя. В первом случае кислород поступает из шлака,
186
в котором высокая активность кислорода O(FeO) поддержи-
вается добавками железной руды, агломерата и других
окислителей, при этом скорость окисления углерода состав-
ляет 0,5—1,0 % С/ч; во втором — расплав продувают газо-
образным кислородом, что обеспечивает vc=2-j-3 % С/ч.
Кинетические закономерности обезуглероживания связаны
с особенностями этой реакции, являющейся многостадий-
ной. Лабораторные и промышленные плавки свидетельст-
вуют о существовании температурного порога (1450—
1500°C), ниже которого скорость окисления углерода рез-
ко снижается и наблюдается интенсивное окисление железа
(расплав переокисляется). По данным М. Я. Меджибож-
ского, соотношение кислорода, расходуемого на окисление
углерода и железа при 1250—1280 °C, составляет 1 : 1, при
1400°C 4:1 и при более высокой температуре — более
10: 1. Снижение скорости окисления с понижением темпе-
ратуры связано с уменьшением диффузионного потока кис-
лорода (Ло), вследствие уменьшения коэффициента мас-
сопередачи (0о) и уменьшения градиента концентрации
([% 0]пов-[% О]об) ••
по = ₽О [%О1ов = ₽o(Lo,FeO) — I%OJo6). (315)
Переход кислорода из шлака в металл является эндо-
термическим процессом и с понижением температуры пол-
нота перехода FeO уменьшается, а следовательно, снижа-
ется [% OJnoB, соответствующая равновесию £=[% О]Пов/
/fl(FeO)-
Температурный порог обезуглероживания наблюдают и
при продувке металла газообразным кислородом. В отли-
чие от рассмотренного случая здесь растворение кислорода
1/2О2=[О] является процессом экзотермическим, поэтому
снижение температуры будет приводить к увеличению
[% О]пов, а следовательно, росту градиента концентраций.
Одинаковый характер кинетических закономерностей окис-
ления углерода в обоих случаях позволяет заключить, что
в рассматриваемых условиях коэффициент массопередачи
оказывает более сильное снижающее действие на поток
По. Можно допустить также, что окисление углерода в от-
личие от кремния, марганца, фосфора и других шлакооб-
разующих элементов осуществляется в результате взаимо-
действия с кислородом не шлаковой фазы, а растворенным
в металле [95]. Это также объясняет одинаковые законо-
мерности окисления при двух способах подвода окисли-
теля.
Большая скорость обезуглероживания газообразным
187
кислородом связана с существенным увеличением поверх-
ностного раздела металл—шлак, так как на поверхности
пузырей образуется пленка (FeO), что объясняет кипение
не только на границе раздела металл—футеровка, но и во
всем объеме.
Важным вопросом кинетики обезуглероживания явля-
ется установление лимитирующей стадии. Ряд исследовате-
лей формулируют крайние точки зрения на природу лими-
тирующей стадии (или диффузия, или химическое звено!).
Вместе с тем закономерности окисления следует рассмат-
ривать в заданных конкретных условиях. Достаточно хоро-
шо объясняет экспериментальные данные теория критиче-
ских концентраций, разработанная С. И. Филипповым [167].
При высоких концентрациях углерода ([% С]>[% С]Кр)
процесс обезуглероживания лимитируется диффузией окис-
лителя. При этом наблюдается поверхностное (беспузырь-
ковое) обезуглероживание. Скорость окисления углерода
не зависит от концентрации углерода и состава металличе-
ского расплава. Обезуглероживание протекает при избыт-
ке реагирующего углерода и недостатке кислорода. Опыт-
ные данные хорошо аппроксимируются уравнением прямой
линии:
— d [C]/dx = (11 V)(Wo,) > (316)
где V — объем металлической ванны; т] — коэффициент ис-
пользования окислителя; w — скорость потока окислителя;
ро, — парциальное давление кислорода.
С уменьшением концентрации растворенного углерода
до значений, меньших, чем [С]Кр, закономерности окисле-
ния существенно изменяется. В этих условиях макроскопи-
ческая скорость окисления определяется концентрацией уг-
лерода:
— d[C]/dT = К [%С]. (317)
Этот период характеризуется меньшими скоростями
окисления углерода, превышением концентрации кислоро-
да в металле, существенным развитием процесса окисления
железа. Ниже критической концентрации углерода полу-
чает развитие процесс объемного обезуглероживания. Тео-
рия критических концентраций вполне удовлетворительно
описывает и промышленные плавки. Вместе с тем изложен-
ная теория полностью исключает стадию химического вза-
имодействия, в то время как в определенных условиях эта
стадия является лимитирующей [163, 168]. Показано, что
роль стадии химического взаимодействия возрастает с
188
уменьшением концентрации углерода, увеличением скоро-
сти подвода окислителя, снижением температуры и добав-
кой ПАВ.
VII .8. ОБРАЗОВАНИЕ И УДАЛЕНИЕ
ГАЗОВЫХ ПУЗЫРЕЙ ИЗ ЖИДКОЙ СТАЛИ
В сталеплавильном производстве важную роль играют про-
цессы, связанные с образованием, ростом и удалением из
жидкого металла газовых пузырей во время газовыделения,
обезуглероживания, продувки жидкого металла в печи и в
ковше и др. Эти процессы получают особое развитие при
вакуумной плавке и обработке жидкого металла. Для об-
разования пузыря необходимо соблюдение условия, описы-
ваемого формулой (209), из которой следует, что равновес-
ный радиус пузыря
т 2<у/(ратgpM ftM-J-gpm/1Ш). (318у
Таким образом, чем больше поверхностное натяжение,
тем больше радиус пузыря при одном и том же равновес-
ном давлении. Известно, что спонтанное гомогенное за-
рождение газовых пузырей связано с существенным пере-
сыщением. Развитые соображения лежат в основе общепри-
нятого положения, что пузыри газа могут образовываться
по гетерогенному механизму лишь на границе разде-
ла фаз. Для пузыря данного объема краевой угол смачи-
вания 0 определяет радиус равновесного зародыша, а сле-
довательно, и равновесное давление внутри пузыря
(рис. 47). Рассмотрим два случая. Если расплав плохо сма-
чивает контактную поверхность (0-»-0), то центрами зарож-
дения пузырей газа могут быть трещины, углубления, поры,
заполненные газом. Эти полости являются центрами обра-
зования пузырей газа при обезуглероживании и дегазации.
Очевидно, что равновесное давление рсо зависит не только
от 0, но и от формы углубления. Для радиуса основания
конусообразной трещины 7? возможные варианты приведе-
ны на рис. 47 [128], из которого следует, что если 0<9О°,
то пузырь может расти, так как г в процессе увеличения-
объема остается большим r>/?/sin 0. В этом случае (при
заданном значении 0) наименьший радиус пузыря опреде-
ляется соотношением r—R/sinG. На с. 108 приведен пример
определения значений R и 0, при которых металл не запол-
няет газовую полость в футеровке. Если R больше расчет-
ного, значения, то для того, чтобы такие полости могли
быть постоянными источниками пузырей, необходимо, что-
189!
бы угол в вершине конуса при 0<9О° не был острый, а за-
кругленный. Рассмотрим условия роста пузырей. Мини-
мальное давление внутри пузыря, необходимое для его су-
ществования, можно найти из условия p=pgr+ (2о/г):
dpi dr — pg — (2o/r2) — 0; rmin — V 2d pg. (319)
Минимальное давление Pco(min) в пузыре, образующем-
ся в вакуумной камере непосредственно под зеркалом ме-
талла, равно рсо = 2 V2opg. Для стали о=1400 мДж/м2,
при этом pco(min) =0,01 атм, что обеспечивается концентра-
цией кислорода, равной 0,01 % при 0,02 % [С]. Для пузы-
е<9о°
в >90°
Рис. 47. Схема образования пузырей на границе раздела жидкость — твердое
тело
рей азота и водорода соответствующие равновесные кон-
центрации равны 0,04 % [N] и 0,0025 % [Н]. При равновес-
ных давлениях 0,01 атм радиус пузыря равен 6 мм. Для
того чтобы обеспечить интенсивное кипение или газовыде-
ление, Pco(min) должно быть больше. Скорость подъема га-
зового пузыря зависит от гидродинамических условий жид-
кой ванны, формы пузыря, массообмена между пузырем и
жидкой ванной и др. Многие исследователи упрощают кар-
тину, принимая, что форма пузырей остается шарообраз-
ной, или вводят различные поправки с учетом грибовидной
формы. Для шарообразных пузырей в жидкой стали, ради-
ус которых превышает 1 мм для скорости подъема v мож-
но пользоваться выражением: v^V^l^rg.
Переход пузырями границы раздела фаз, например ме-
талл—шлак, происходит по двум механизмам [169]. В пер-
190
вом случае переход осуществляется в среду с меньшим зна-
чением оМф. Такой тип перехода правилен для любых раз-
меров частиц (газовых пузырей). По второму механизму
существует некоторый критический размер частиц, меньше
которого переход не осуществляется, так как увеличение
поверхностной энергии не компенсируется уменьшением
потенциальной энергии частиц. В сталеплавильной прак-
тике различают поверхностное и донное кипение. В первом
случае пузыри зарождаются на границе футеровка—металл
и при своем движении растут как в результате уменьшения
ферростатического давления, так и вследствие реакции,
протекающей на границе пузырь—металл. При достиже-
нии ими поверхности шлак—газ в атмосферу выбрасыва-
ется значительное число металлических корольков. В слу-
чае поверхностного кипения газовые пузыри, образующиеся
на межфазной границе металла со шлаком, имеют зна-
чительно меньшие размеры. Такой тип кипения не приво-
дит к выносу корольков. Наряду с этим рассмотрим зако-
номерности, исходя из двух механизмов перехода границы
раздела фаз. Пузыри газа, образующиеся на подине и под-
ходящие к границе металл—шлак с объемом менее крити-
ческого, не могут ее перейти из-за металлической пленки
на поверхности. Пузыри с пленкой могут перейти только по
второму механизму. Они останавливаются на границе до-
тех пор, пока их объем не достигнет критического значения.
Это может произойти в результате протекания реакции на
поверхности пузырей, укрупнения их при слиянии, а также
вследствие слипания с образованием комплекса ячеистой
структуры.
В случае поверхностного кипения вследствие возникно-
вения газовой полости непосредственно на границе ме-
талл—шлак возможно образование пузырей без пленки.
Переход таких пузырей соответствует уменьшению свобод-
ной энергии системы для частиц любого размера.
VII.». МАССОПЕРЕНОС КОМПОНЕНТОВ В ШЛАКЕ
Согласно закону Фика, диффузионный поток примеси в.
шлаке определяется его градиентом концентрации. Одна-
ко в реальных условиях (см. гл. II) поведение примеси в
шлаке определяется не только градиентом концентрации,
но и градиентом окисленности расплава оксидов. Суммар-
ный поток i-того компонента определяется соотошением
_ _ ( d In ci . v,- . d In pn \ ,onn,
П,- KB.RTc, +-J-+—j’St. . (320)
191
где Bi — подвижность диффундирующих частиц; V,- — ва-
лентность.
Из уравнения (320) можно легко получить два предель-
ных случая. Первый соответствует равномерной окислен-
ности (d\npojdx=0) и приводит к закону Фика. Второй
случай рассмотрим на примере переноса азота через слой
расплавленного шлака. Пусть два газовых объема разде-
лены пленкой шлака толщиной Д<?=1. В газовых объемах
поддерживаются постоянный перепад парциальных давле-
ний азота Дры, =const (например при разбавлении инерт-
ным газом) и одинаковое давление кислорода. Перепишем
уравнение Фика, заменив производную конечной разнос-
тью:
П — D Ar = D (с’ —с"
(N) N“°N N (CN ‘'N
(321)
Но согласно уравнению (148), c<n) зависит от ро,: С(Ы) =
=WРо!'4• Следовательно, /7(N) =Р(1Ч)р^/4Др^2. Та-
ким образом, если при ApN,=const изменять ро, (сохра-
няя его одинаковым в обоих объемах), будет также изме-
няться проницаемость шлакового слоя по отношению к азоту.
Так, в условиях сталеплавильных процессов проницае-
мость слоев шлака, омываемых печной атмосферой (ро, «
л: 10-1 атм) в 106 раз ниже, чем слоев, контактирующих с
металлом (р^=10-9 атм), поэтому диффузионное сопротив-
ление оказывается сосредоточенным в тонком поверхност-
ном слое шлака. Аналогичные выводы можно сделать и по
отношению к другим элементам. При этом характер влия-
ния ро, на проницаемость шлака определяется значением
и знаком валентности рассматриваемого элемента vt- поэто-
му, например, для водорода (vh=,+ 1) наблюдается обрат-
ная картина: перенос лимитируют слои, прилегающие к ме-
таллу.
Для сталеплавильной практики важное значение име-
ет взаимодействие потоков компонентов в шлаке, обус-
ловленное градиентами концентрации и окисленности. На-
пример, в условиях открытой плавки существует непре-
рывный поток кислорода из атмосферы к границе раздела
металл — шлак, который существенно влияет на поведе-
ние примесей в шлаке. Используя методы термодинамики
необратимых процессов, запишем выражения для потоков
кислорода По и азота в расплаве оксидов: По —
= PJN—L2,idiio^-L2,2d[iN, где go и Hn —
градиенты химических потенциалов кислорода и азота.
Пусть шлак вначале равномерно насыщен азотом, т. е.
192
duo =0. Тогда при наличии потока кислорода возникает по-
ток азота /7ы = (Ь2,1/Ь1,1)По. Этот поток приведет к воз-
никновению градиента химического потенциала азота, ко-
торый противодействует породившему его потоку П^. При
некотором предельном значении поток азота прекра-
тится (77n = 0) и будет достигнуто стационарное состоя-
ние. Таким образом, если в шлаке существуют только два
потока — азота и кислорода, выполняется соотношение
Пл/По=— 3/2; = 3/2. Из этих соотношений следу-
ет, что поток азота направлен навстречу инициирующему
его потоку кислорода, а создаваемый при этом градиент
химического потенциала азота совпадает по знаку с гра-
диентом химического потенциала кислорода. Следователь-
но, система работает как насос, создавая перепад хими-
ческого потенциала азота. Обобщая эти результаты, мож-
но записать:
Пс/По =— VVO’ d^0/d^i = vi/vo-
(322)
Из соотношений (322) следует, что направление пото-
ка примеси и знак возникающих при этом градиентов р,<
определяются законом валентности. Например, поток во-
дорода (vh=H~1) в отличие от потока азота совпадает по
направлению с потоком кислорода. Рассмотрим действие
азотного «насоса» на конкретном примере. Известно, что
шлак, контактирующий с печной атмосферой (р=
= 10132,5 Па) практически не растворяет азота. Находя-
щиеся в контакте с металлом слои шлака раскислены
([0]«0,1 %; ро,» 10,13-10 Па) и растворяют заметное ко-
личество азота.
При конвективном движении отдельные порции шлака
попеременно оказываются с областях с пониженной и по-
вышенной окисленностью, поглощая азот вблизи металла
и выбрасывая его в атмосферу. Перемешивание шлака иг-
рает активную роль в работе азотного «насоса», резко по-
вышая его производительность. Однако и в отсутствие пе-
ремешивания эффект «накачки» должен существовать и
подчиняться тем же закономерностям. Из сказанного сле-
дует, что фактическая стационарная концентрация азота в
металле всегда ниже, а водорода выше равновесной с печ-
ной атмосферой. В пределе это отклонение может дости-
гать значительных величин:
INU = INIP (Ро, /Р'о,'! = INI, • 10-6; (323)
|Н|,„„ = [Н1р (10 "9/1(Г,),/4 = [Н1р-10!. (324)
13—675
193
Рассмотренные закономерности подтверждаются прак-
тикой — содержание водорода в металле открытых печей
либо близко к равновесию, либо превышает его. Содержа-
ние азота более чем в 10 раз ниже равновесного даже в ду-
говых электропечах, где происходит интенсивное насыще-
ние металла азотом в области дуг. В процессе выдержки
под шлаком содержание водорода в металле увеличивает-
ся, а содержание азота снижается. Перемешивание резко
ускоряет поступление водорода и удаление азота.
Глава VIII
ОСОБЕННОСТИ РАФИНИРОВАНИЯ МЕТАЛЛА
ПРИ ПЕРЕПЛАВНЫХ ПРОЦЕССАХ
Металл ответственного назначения после выплавки и раз-
ливки подвергается еще одному металлургическому пере-
делу — переплаву в специальных плавильных агрегатах.
В настоящее время в промышленных масштабах применя-
ются следующие методы переплава: вакуумный дуговой
(ВДП), электрошлаковый (ЭШП), электроннолучевой
(ЭЛП) и плазменно-дуговой (ПДП). Разрабатывается и ос-
ваивается вакуумно-плазменно-дуговой переплав (БПДП).
Основной задачей каждого из этих процессов является по-
вышение качества металла, улучшение его служебных ха-
рактеристик. Для этого необходимо выполнение следующих
требований: 1) удаление из металла вредных или нежела-
тельных примесей в результате создания развитой контакт-
ной поверхности металла с рафинирующей средой (разре-
женный газ, инертный газ, шлак); в некоторых случаях,на-
пример при ПДП, наряду с удалением примесей возможно
легирование металла компонентами рафинирующей среды
(например, азотом); 2) создание благоприятных условий
для кристаллизации металла при его наплавлении в мед-
ном водоохлаждаемом кристаллизаторе. Процессы рафини-
рования наряду с особенностями, характерными для каж-
дого метода переплава, имеют ряд общих закономерностей,
связанных, в частности, с кинетикой рафинирования.
VIII .1. ОБЩИЕ ЗАКОНОМЕРНОСТИ
РАФИНИРОВАНИЯ МЕТАЛЛА ПРИ ПЕРЕПЛАВЕ
Кинетическим закономерностям процесса рафинирования
металла при переплавных процессах посвящен ряд работ
[171—173, 229]. Характерной чертой всех переплавных про-
цессов является многостадийность процессов рафинирова-
ния, которые происходят на торце оплавляемой заготовки,
в каплях металла, проходящих рафинирующую фазу, и в
ванне металла. При анализе процессов рафинирования
удобно использовать в качестве характеристик режима мас-
совую скорость плавки g линейные скорости оплавления
электрода vs и наплавления слитка оо- Между этими ве-
личинами имеют место следующие соотношения:
°о = S/(PC„ Р) = £/(«/%. Р); »э = ё/(Рэ Р) =
= ё/(лг2э р); и0 = (d2 • V9)/D2, (325)
где Fcn, F3 — площади поперечных сечений слитка и элек-
трода; /?сл, Гэ—радиусы слитка и электрода; D, d — диа-
метры слитка и электрода; р — плотность металла.
Площадь поверхности рафинирования на стадии элек-
трода (S) может отличаться от площади F3 в связи с тем,
что поверхность торца электрода может иметь форму ко-
нуса, полусферы ит. п. В этом случае S>F3, и можно при-
нять S=tnF3, где гп^\. Кроме многостадийности, пере-
плавные процессы характеризуются существенной неизо-
термичностью. Во-первых, могут значительно различаться
по температуре различные стадии процесса. Во-вторых, в
пределах одной стадии обычно имеет место неравномерный
перегрев поверхности рафинирования. Имеются участки
локального высокого перегрева (фокальные и электродные
пятна). В связи с этим все расчеты для определенной ста-
дии процесса проводят обычно для какой-то средней ха-
рактерной температуры. В-третьих, как правило, наблюда-
ется существенное различие температур поверхности вза-
имодействия металла с рафинирующей фазой и самой
рафинирующей фазы. Все это необходимо учитывать при
расчетах. Анализ процесса удаления примесей полезно про-
водить на основании балансовых уравнений на каждой ста-
дии переплавного процесса. При этом необходимо учиты-
вать порядок реакции лимитирующего звена. Удаление ис-
паряющихся цветных примесей лимитируется в любом
случае реакцией первого порядка (стадиями внутреннего
массопереноса, испарения или внешнего массопереноса).
13*
195
Балан-
общем
Вместе с тем в общем случае процесс удаления газооб-
разных примесей (азота и водорода) может лимитировать-
ся реакциями как первого так и второго порядков,
совое уравнение на i-той стадии процесса в этом
случае имеет вид:
rlr- (а. К, ТС pS.) С?
М, -%- = gc,, - gc, - ( +
аТ Ai + Ao С j
(326)
металла; Со/ — исходное
содер-
— текущее (конечное)
Ml "Г М2 vin
где М, — масса жидкого
жание примеси на данной стадии; Ci
содержание примеси на данной стадии; с1П — содержание
примеси на поверхности металла, равновесное с рафини-
рующей фазой; а/ — доля площади поверхности металла,
незаблокировапная ПАВ; /G, К2— константы скорости
процесса реакции первого и второго порядка. При
процесс лимитируется реакцией первого порядка,
при TGc/C/G— реакцией второго порядка, то есть при
прочих равных условиях вопрос о лимитирующем зве-
не определяется содержанием примеси в металле на дан-
ной стадии. При этом, если условия в среднем соответству-
ют некоторому переходному состоянию, когда то
не исключено, что при переходе от одной стадии к другой
порядок реакции изменится с первого на второй. Измене-
ние произойдет при Ci=K\IK2, и это содержание является
критической массовой долей примеси. В. В. Авериным
[107] для азота установлена при 1873 К зависимость [N]Kp
от ди, и показано, что в вакууме при pn, ->-0 в железе
N]KP->0,05 %. Хотя определенное в работе [230] значение
;N]KP и представляется слишком высоким из-за принятого
в расчетах очень высокого значения коэффициента массо-
переноса 0=Ki=3,5-10-4 м/с, однако роль химического
звена при удалении азота в вакууме может быть сущест-
венной. Особенно это относится к металлу с повышенной
чистотой по азоту (выплавленному в ВИП и подвергаемо-
му ВДП и ЭЛП и т. п.). Решение уравнения (326) в об-
щем виде достаточно сложно для наглядного анализа, и
к нему следует прибегать только для условий переходного
режима. Если известен преобладающий характер реакции,
то для установившегося режима переплава при dCi/dx=Q
можно пользоваться упрощенными выражениями. Обозна-
чая линейную скорость оплавления или наплавления на
данной стадии через и,-, получим: для реакции первого по-
196
рядка, когда /С1</С2С/
- („ । mi Ki ai cin\ //1 । mi ai\
ci — Coi "1 I / И “r I •
\ V/ // \ Vi /
для реакции второго порядка, когда
__i , п; Cpj |_2 ______Ei_
Ci ~ V 4m2 K2 af + mi *2 ai + C‘n 2mi K2 «i '
I Zr I
(327)
(328)
Наиболее часто используют трехстадийные переплав-
ные процессы, включающие стадии: 1) жидкая пленка на
торце оплавляемой заготовки (электрода); 2) падающие
капли; 3) ванна жидкого металла. Возможны процессы с
большим числом стадий и с использованием промежуточ-
ных емкостей, холодного пода и т. п. При рассмотрении
всех этих процессов для упрощения анализа необходимо
исключить из рассмотрения вторую стадию (падающих ка-
пель) вследствие незначительности степени рафинирова-
ния металла на этой стадии.
Для примера рассмотрим простейший случай, когда
процесс лимитирует реакция первого порядка, а величиной
с(П можно пренебречь. Тогда на стадии капли имеем для
установившегося режима:
с,о = g+ Ki F« pct или = ci0/ [1 + (/Ci Fk P)/g], (329)
где Ff — суммарная поверхность всех одновременно суще-
ствующих и падающих капель, число которых будет п.
При радиусе капли RK имеем Fk=4jthFk. При про-
должительности падения тПад с высоты L всегда одновре-
менно существует п капель:
п = (^пад)/(4/3"Як р) = [(3g)/(4 р) ] /2Z7i“, (330)
где go — ускорение свободного падения.
Отсюда с{ = с.о/[1 + (З/СуЯи) У 2L/gJ, а степень рафи-
нирования (%):
f ____ (clo Ю0
(О = --------------------
= 100
(331)
Оценим радиус капли. Примем, что в момент ее отрыва
радиус шейки равен среднему радиусу капли RK. Отрыв
происходит, когда сила тяжести капли превышает силы
межфазного натяжения в шейке и выталкивания капли из
окружающей среды:
4/qJIRk go (р Рср) 2л7?к О*мф»
(332)
197
где рСр — плотность окружающей среды; оМф — межфазное
натяжение на границе капли с окружающей средой (для
вакуума оМф совпадает с поверхностным натяжением ме-
талла). Отсюда
Як > /Зомф/[2^0(р-рср)Ь (333)
В условиях вакуума для железа при оМф=1,7 Дж/м2
и р=6900 кг/м3 получаем Як^б мм. При этом значении
Як и L=2 см, a Ki=0,01—0,1 мм/с получим о=0,03—
—0,3 %, т. е. стадией капель при расчетах можно пренебречь.
Тогда для лимитирующей реакции первого порядка с уче-
том стадии пленки и ванны, считая для ванны т=Л, име-
ем:
со + 1т^пл апл ^пл.п)/уэл
апл
^эл
(/Св £в.п)/Ц)
. (334)
Индексы «пл» и «в» относятся соответственно к пленке
на торце электрода и к ванне, а индекс «п» относится к
процессам на поверхности металла. В практических расче-
тах часто пренебрегают величинами сПл.п и св.п. Кроме то-
го, как правило, (тЯпл аПл)/оэл<1 и KBaB/vo<l- В этом
случае можно воспользоваться подстановкой при
х<1, и получить выражение:
In со/св = (1/и0)[Кв ав + Япл апл т (<Р/ЕР)].
(335)
Выражением (335) часто пользуются в литературе, объ-
единяя для простоты выражение в скобках в одну констан-
ту. Наряду с этим используется и упрощенный вариант
формулы (334):
тКпп апл
^эл
(336)
В этом случае степень рафинирования металла от при-
меси не зависит от ее исходной массовой доли Со, %:
Независимость со от с0 является в данном случае приз-
наком первого порядка реакции лимитирующего звена. Аб-
солютное снижение массовой доли примеси в этом случае
198
имеет вид:
Дс = с0 — св = Сд 1 — 1 +
апл
иэл
(337)
т. е. при построении зависимости Дс от Со вида Дс=Лсо
для реакции первого порядка всегда К<1. Если процесс
лимитируется реакцией второго порядка, также существу-
ют упрощенные выражения для расчетов. Допустим, что
для каждой стадии различие между величинами с0 и с не-
велико, а величиной с1П можно пренебречь. Тогда для не-
установившегося процесса балансовое уравнение примет
вид:
Mi (dci/dx)«— а{- Ki St pel
(338)
Интегрируя ОТ Т=0 ДО Т=Т И ОТ Со до СПл, получим, счи-
тая Мпл/5плрт=уэл//п:
1 1 __ т^ПП ®ПЛ . „ __ Z,
» ^дл ^0
Сдл со уэл
тКпл апл go\
^эл /
(339)
Аналогично для стадии ванны
_______L_ — . г =с
> °в °пл
СВ СПЛ ^0
Отсюда в конечном результате
1_________1 ^Кдл °&дл
СВ ^0 ^эл
Кв С&В ^ПЛ
в = С0
___*В ав _
^0
d2 \
ПЛ Д2 у ’
d2
(341)
(342)
»0
В этом случае
(343)
гдеа=(1/у0) [ЛвССв+Аплт апл (d2/D2)].
При ас0<С1 имеем Дс«асо, а при ас0>1 имеем Дс«
«Со—(1/а), т- е- если зависимость Дс от с0 в определен-
ном интервале значений с0 описывается выражением Дс=
=А(со—с'), где то это свидетельствует о том, что
процесс лимитируется реакцией второго порядка. Наличие
в этом выражении члена c'=\fa не следует рассматривать
как некое блокирование реакционной поверхности. По при-
199
роде своей, как следует из приведенных выше расчетов,
этот член не связан с содержанием примеси в поверхност-
ных слоях металла. Степень рафинирования при лимити-
рующей реакции второго порядка существенно зависит от
исходной массовой доли примеси %:
со — (&с/с0) • 100 [(ас0)/( 1 + ас0)] • 100.
(344)
Таким образом, характер зависимости Дс и со от с0 оп-
ределенный экспериментально, позволяет судить о поряд-
ке лимитирующей реакции. Во всех упрощенных расчетах
величиной с1П пренебрегали. Однако в процессах, в кото-
рых имеет место дуговой разряд (ВДП, ПДП, ВПДП), не-
обходимо учитывать наличие на поверхности металла так
называемых особых зон возможного контакта металла с
плазмой дугового разряда. Если локальное поверхностное
содержание примеси в особой зоне составляет с'п, а вне
зоны с'п, то расчетное значение сп=пс„J- (1—п)сп (п— до-
ля площади особой зоны). Обычно Cn*Ccn и
Особенностью процессов, идущих с использованием ду-
гового разряда, является высокая степень их неизотермич-
ности. Степень диссоциации двухатомных газов в этих про-
цессах, а следовательно, и состав газовой атмосферы оп-
ределяется температурой столба дуги. Процесс поглоще-
ния (или выделения) газа из металла идет при температу-
ре металла, которая существенное ниже температуры стол-
ба дуги. При ПДП температура плазмы в столбе сущест-
венно выше даже температур катодного и анодного пятен
на поверхности металла. При ВДП дуга горит в парах ме-
талла, и температура ионов и нейтральных частиц в усло-
виях вакуума определяется температурой испаряющих по-
верхностей, т. е. температурой катодных и анодных пятен
(Тк и Та). Для железа и никеля ориентировочно можно при-
нимать при ВДП Тк = Тл+175 К и Та = Тл+400 К (Тл — тем-
пература ликвидуса). Соответствующие температуры име-
ют место в прикатодных и прианодных частях столба дуго-
вого разряда. Состав газовой фазы, контактирующей непос-
редственно с металлом, определяется на основании оценки
критерия Дамкелера: De = /С/(р2б2) =тх/тд, где р— дав-
ление; 6 — расстояние, которое должны пройти частицы
газа; тх — продолжительность химического акта рекомби-
нации атомов в молекулу; тд — продолжительность про-
хождения расстояния б; К — коэффициент, равный 108Х
ХПа3-м2(10-3 атм3-см2).
При De^>l атомарные газы достигают поверхности ме-
200
талла, не успевая рекомбинировать. В случае ПДП при
р=1 атм (0,1 МПа) и 6=2 см, De=2,5- 10-4<С1, в случае
ВДП при р = 1 Па и 6=2 см, De=2,5- 10и^>1.
Исходя из оценки значения De при ПДП В. И. Лаком-
ский [173] считает наиболее вероятным контакт металла
при ПДП не с атомами азота, а с его возбужденными мо-
лекулами N2. Дополнительная энергия возбуждения этих
молекул ДО в по оценке В. И. Лакомского составляет
898 кДж/моль: {N2 }={N2}, Д6°=—898 кДж/моль. Отсю-
да для реакции V2 {N2}=[N]n в железе имеем
lg Kn = lg I/n I % Nln/(A)1/2} = (2264/7’) + 1,16. (345)
Для ВДП De^>l и металл контактирует с газовой фа-
зой, имеющей состав плазмы столба. В зоне катодных и
анодного пятен процесс взаимодействия металла и газа
при ВДП протекает при равенстве их температур. Неизо-
термичность возможна при различии температур особых
зон и остальной реагирующей поверхности.
В связи с этим в зонах катодных и анодного пятен
[%Г]=(адг) где Кг и fr определяются при темпе-
ратуре соответствующего пятна. Однако площадь сечения
столба дуги в вакууме значительно больше площади анод-
ного пятна и общей площади многочисленных катодных
пятен. Отсюда при De+>1 имеет место вероятность подхо-
да газа плазмы к холодному металлу вне площади элек-
тродных пятен. В этом случае процесс поглощения (или
удаления) газа неизотермичен. Водород и азот в газовой
фазе диссоциирует по реакциям: {Н2}=2{Н}, AG°=434080—
—98,0 Т Дж/моль; {N2} = 2{N), AG° = 946000—115,0 Т Дж'
/моль. Константа диссоциации =р^/рг , для водорода
1g Ка = (—22700/7’) + 5,11, для азота 1g Ка = (—49400/7) --
+ 6,00. При общем давлении атомарного и молекулярною
газов рхг, = Рг+рг, и степени диссоциации а имеем:
а=1/|/ l + (4pIrs/K„) ; (346)
Рг, = М -«)]/(!+«); рг = (2р1Г1 а)/(1 + а). (347)
Парциальное давление атомарного газа составляет:
Рг“2р1г_/[1+1/ l+(4pIri/KJ .
При Т’=2000 К для водорода Ка = 5,76-10“7 атм, для
азота /Са =2,0-10-19 атм. При р2н,= 1 Па (1 • 10“5 атм) и
/72^=0,14-1,0Па (Ы0-6—1-10-5атм) имеем 4 р &•»//<« S1 •
201
В ЭТИХ условияхРг ~ У У КаРг* . ТОГДЗ ПО реЭК-
ции {Г)=[Г] имеем 1%Г1 = (Kr/fr) рг « (^Сг//г) V ^СаРг,»
где Кг и fr — определяются при температуре металла, а
Ка — при температуре соответствующей зоны столба дуги,
т. е. при Тк для катода и Та для анода. Для железа lg/С н =
= lg(M%H]/pH) = (9750/Т)—4,28; lg Kn =lg(fN[%N]/
/ры) = (24155/Т)—4,065. Для никеля lgKH= (10380/7)—
— 4,45; lg KN = (21000/7)—3,960. При 7мет = 1773 К и
Тплаз=2000 К, рн,=2,5 Па (2,5-10 5 атм) и рл,= 1 Па (IX
Х10-5атм) в чистом железе имеем [%Н]П=6,2-10-5 %;
I % N]n=0,017 % • Соответственно в никеле [ % Н]п = 1,0 X
Х10-4%; [%N]n=l,3-10-4 %. Такие значения [Г]п могут
оказывать определенное влияние на кинетику процесса
при существенных значениях доли площади контакта и.
Величина п при одной и той же линейной плотности тока
j=I/D уменьшается с ростом D так как где
i — плотность тока в столбе дуги; F — площадь поверхности
металла на соответствующей стадии процесса. На промыш-
ленных установках влиянием Cia в кинетических расчетах
можно пренебречь.
Однако местные, локальные перенасыщения металла
азотом при содержании в металле нитридообразующих
элементов могут приводить к образованию нитридных пле-
нок на поверхности металла в особой зоне. Если эта плен-
ка при перемещении особой зоны по поверхности металла
не успевает диссоциировать, она может вместе с каплями
металла перейти в ванну и, достигнув фронта кристалли-
зации, стать причиной образования нитридных скоплений
в металле.
VIII.2. РАФИНИРОВАНИЕ МЕТАЛЛА ПРИ ВДП
Температурный режим процесса ВДП
Для оценки среднего перегрева металла пленки на торце
электрода над температурой ликсидуса (А7Э) можно ис-
пользовать следующее выражение:
где 7\„K, Геол — температуры ликсидуса и солидуса; б —
средняя толщина жидкой пленки; а — коэффициент темпе-
202
ратуропроводности металла; С — удельная теплоемкость
металла при ТСол; р—плотность металла при Геол; епр—
приведенная степень черноты в системе электрод — стенка
кристаллизатора (корона слитка); Тст — температура стен-
ки кристаллизатора (короны слитка); сто — постоянная Сте-
фана — Больцмана. Величину 6 можно оценить по фор-
муле:
б = V ам/(6р£0), (348)
где р — плотность жидкого металла; ом — поверхностное
натяжение металла при Глик", go—ускорение свободного
падения. Среднее значение б составляет 1,0—1,5 мм, а зна-
чения ДТэ находятся в пределах 20—50 К в зависимости
от режима процесса.
В зонах каплеобразования толщина пленки примерно в
два раза больше б, а температура перегрева также в два
раза больше ДТэ. Средняя скорость течения металла в
пленке «=36иэ. Число центров каплеобразования и=
^л[гэу' (pgo)/(24стм)—1]. Коэффициент массопереноса при-
меси в пленке определяется по формуле:
(349)
Коэффициент массопереноса примеси в зоне каплеобразо-
вания составляет:
Рк — гъ
(350)
Для поверхности зеркала жидкой ванны среднюю сте-
пень перегрева над температурой ликвидуса можно оце-
нить по формуле:
ДТПОВ = Д7\пах [1 - (а - bD + cD2)/!], (351)
где ДТтах — теоретически максимально возможная сте-
пень перегрева металла: I — ток переплава.
Величины ДТтах, «, Ь, с являются функциями свойств
переплавляемого металла, величин d, D и длины межэлек-
тродного промежутка L. Для сталей при £«20 мм, D =
= 100^-1000 мм и d/D=0,74-0,85 можно принимать
ДТтах=485 К, а=1000 А, 6=2500 А/м, с= 15100 А/м2.
Степень перегрева металла существенно зависит от отно-
шения d/D. При малых отношениях d/D велики потери теп-
203
ла на излучение и испарение с поверхности ванны, при
больших значениях d/D эти потери снижаются, но появ-
ляется утечка тока на стенку кристаллизатора, поэтому су-
ществует определенное значение d/D, при котором темпе-
ратура ванны при данном токе максимальна. Температу-
ра снижается при удалении от поверхности в глубь ванны
[174] (рис. 48, с. 216). При отсутствии электромагнитных
полей средняя скорость движения металла в ванне, вызван-
ного падением в ванну капель металла, составляет и =
=8а/КСл (а. — эффективная температуропроводность ме-
талла). Коэффициент массопереноса примеси в ванне при
центральном падении капель составляет:
0, = (4W«.) /(2aD,)/n. (352>
При падении капель на расстоянии г от центра ванны
0, = (4/Ям) 11 + (2гЖс„)1 /(2aD,)M. (353)
Удаление водорода
При оптимальных режимах процесса ВДП наблюдается
значительное удаление водорода из металла. При перепла-
ве сплавов на никелевой основе удаляется 80—90 %, а при
переплаве коррозионностойких сталей 70—80 % водорода.
При реакции второго порядка константа скорости процес-
са удаления водорода определяется по формуле, м/ (с - %Н):
К = [1013/н,/(рКн,)] У Мн,/(2лЯП (354)
где Кн, — константа в формуле [ %Н1 = (Кн//н)Кр^пРи
рн,» атм; р — плотность жидкого металла.
Оценим критическую концентрацию водорода в чистом
железе и в стали типа 12Х18Н10Т для условий пленки и
зоны каплеобразования на электроде при 7=1773 К При
р=7 г/см3 имеем для железа К=217 м/(с-°/о), а для
стали 12Х18Н10Т К=176 м/(с-%). В железе рпл =
*-=81,4 мкм/с в каплях при d=20 см и и=9 рк=0,33 мм/с.
Соответственно в стали 12Х18Н10Т рпл = 0,325 мм/с и
Эк =1,31 мм/с. Отсюда в железе [Н]кр=3,8-10-7—1,5Х
ХЮ"6 %, или 4,2-10-3—1,7-10-2 см3/100 г. В стали
12Х1810Т соответственно [Н]Кр= 1,85-10-6—7,5-10 6 %,
или 2,1-10-2—8,4-10-2 см3/100 г. Из этого следует, что ли-
митирующим звеном при реально существующих концент-
рациях водорода должен быть внутренний массоперенос.
Для небольших лабораторных и полупромышленных
установок необходимо учитывать влияние на кинетику
процесса поверхностных концентраций водорода в особых
204
зонах. В работе [175] установлена экстремальная зависи-
мость степени удаления водорода от отношения d/D для
коррозионностойких сталей при £>=1254-150 мм. При ма-
лых значениях d/D<S№ велики потери тепла ванной в
результате излучения и испарения, малы степень перегре-
ва зеркала ванны и константа скорости процесса. При
d/D>Gfi мала пропускная способность зазора между элек-
тродом и кристаллизатором, водород плохо откачивается
из зоны плавления, в ней растут рн, и поверхностные мас-
совые доли [Н]пл.п, [Н]в.п. Это должно ухудшать условия
рафинирования. Однако на крупных промышленных печах
проведение процесса при d/D=0,804-0,83 обеспечивает
хорошее удаление водорода. Это объясняется тем, что роль
процесса откачки на малых и крупных печах различна.
Поток удаляемой примеси (газа) при лимитирующей ре-
акции второго порядка для многостадийного процесса со-
ставляет:
77= —аР2^Лс?пПг-
i=i i=i
(355)
Величина с)П определяется по формуле:— 7^]^Kaiprt,
где рГ1== (77Vpp0T)/(7Wrt-STf-298); — молярный объ-
ем; ро — атмосферное давление; 7ИГ, — молекулярная мас-
са; Srj —скорость откачки газа Г2 из зоны плавки. Таким
образом величина с1П является функцией 77 как и 77 явля-
ется функцией С1Л. Отсюда
i=k
i=k
«Ум Ро рт VI
Мгг-298
1=1
к, Г, Ki, ка
(356)
Характер зависимости П от Sr, зависит от соотношения
между Srs и величиной
А = («V„P0pT)/(Mri.298)2\,F,K?1^I't?-
1=1
При плавке у устья кристаллизатора (ЭЛП, конечная ста-
i=k
дия ВДП) и 77 « a V7</Fic^, т. е. не зависит от Sr,.
4=1
При плавке в глубине кристаллизатора (ВДП) соотно-
205
шение между Sr и А зависит от п,. На малых печах при
i=k
STt<A в глубине кристаллизатора/? «(aSr>M) V
z=i
Поскольку ni уменьшается с увеличением D, то зависи-
мость П от ni на печах с диаметром кристаллизатора
£>>200 мм практически отсутствует. В связи с этим пред-
ставляет интерес определение значения рг> в различных
участках зоны плавления. При этом определении необхо-
димо учитывать газопоглотительное действие парового
конденсата нй стенках кристаллизатора. В общем случае
£?кр п ^0
$кон 4*
^КР ^0 + i/n ^0~Ь ^кр t/n
(357)
1?кр 50
t/пр (t^KP 4“ Unp $0 — Un ^o)
где So — быстрота действия вакуумного насоса; SKOh —
быстрота поглощения газа Г2 конденсатом; Un, UBp— про-
пускная способность вакуумной системы печи, зазора меж-
ду кристаллизатором и электродом и подэлектродного про-
межутка.
Вопрос о влиянии Sr, на давление в зоне плавления из
всех переплавных процессов актуален только для ВДП,
так как при ЭЛП отсутствует дуговой разряд, а при
ВПДП, как при ЭЛП, процесс идет у устья кристалли-
затора. По аналогии с испарительно-геттерными насосами
S«>H — лер)/ (RT)/(2nMr2) ВИкои,
ГДС Лкон высота конденсата; <р — коэффициент прилипа-
ния. В геттерных насосах коэффициент прилипания харак-
теризует вероятность захвата молекулы газа пленкой гет-
тера при соударении. По смыслу <р^1. В условиях плавки
в медном водоохлаждаемом кристаллизаторе при темпе-
ратуре конденсата ~500 К для азота <ры, «0,38, для СО
Фсо«0,59, для водорода <рн,~0,02, т. е. поглощением во-
дорода можно пренебречь.
Обычно величины So и Un велики по сравнению с ос-
тальными и не лимитируют процесса. Если торец электро-
да имеет вид конуса (полусферы), то Unp также не лими-
тирует процесса. Тогда для водорода Sh, «U„v, а для
азота SN>« (/kp-J-Skoh. Для плоского торца Sh,« (uKPU„p)/
/(f?Kp-|-t/np); SN1«[£/пр(£?кр-1-5кон)]/(^/кр+5Кон-}-^пр)•
Для молекулярного режима течения газа
206
*4р = V, К(лЯТ)/(2Мг.) (D/L) [ 1 - (d/D)] [ 1 - (d?/D*)]; (358)
t/np = 32/з К(n/?T)/(2Mr>) I(Z2 x)/(d - 2х)], (359)
где х — расстояние от оси электрода до точки замера; I—
расстояние между торцом электрода и зеркалом ванны.
Распределение давления газа в подэлектродном про-
межутке при условии его равномерного выделения по всей
поверхности торца электрода и зеркала находится из усло-
вия dp=—dn/Unv. При интенсивности газовыделения.
^(Па-м/с, л-атм/с-см2) имеем
Р = Рзаз + (3^/16Z2) /(2лМг,)/(Я7’) [(d/2) - х]2,
где рзаз — давление в зазоре между электродом и кристал-
лизатором на уровне зоны плавки.
Удаление азота
Степень удаления азота при ВДП меньше степени удале-
ния водорода и существенно зависит от исходной массовой,
доли азота. Ориентировочно из металла удаляется 20—
40 % азота. А. Б. Сергеевым для ряда сталей и сплавов
установлена зависимость A[N]=K[N]0—[N]'), где К»1.
Это свидетельствует о том, что процесс лимитируется в
основном реакцией второго порядка. По-видимому, диффу-
зионно-десорбционное звено (реакция первого порядка)
лимитирует процесс удаления азота только при ВДП ста-
лей и сплавов с очень высоким (0,04—0,06 %) содержанием
азота. При массовой доле азота 0,02—0,04 %) имеет место-
смешанный режим. При более низкой массовой доле азота
лимитирующая стадия имеет второй порядок реакции. Су-
щественную роль играют также условия гидродинамики на
данной стадии процесса. Например, расчеты показывают,,
что для стали 08Х18Н10Т при с?=0,4 м и 0=0,5 м при
температуре на торце электрода 1773 К и на поверхности
ванны 1873 К в пленке [N]Kp=0,06 %, в каплях [N]Kp=
=0,25 %, в ванне ,{N]KP=0,0077 %.
Константа скорости процесса в данном случае сущест-
венно зависит от активности азота в металле. Как и в слу-
чае водорода для реакции второго порядка константа ско-
рости составляет м/ (с • %)
К = [(Ю13$)/(рКУ] /Л4ы,/(2лЯТ), (360)
где К n,— константа равновесия процеса V2{N2}=[N].
Чем ниже значение /и, тем выше роль химико-десорбцион-
ного звена. Особенно это относится к сплавам на никеле-
20Т
вой основе, в которых массовая доля титана достигает не-
скольких процентов. При расчетах необходимо учитывать
также блокировку поверхности раздела металл — газ по-
верхностно-активными веществами и вводить коэффициент
а, показывающий долю площади адсорбционных центров
на поверхности металла, на которых идет химическая реак-
ция, от общей площади поверхности раздела металл — газ.
Величина а должна зависеть от содержания в метал-
ле поверхностно-активных веществ (кислорода и серы).
При оценке значения а необходимо учитывать содер-
жание в металле не только ПАВ, но и элементов, влияю-
щих на активность ПАВ. Зависимость К. от /и приводит к
тому, что при введении в металл элементов, понижающих
активность азота (в частности, титана) резко возрастает
кажущаяся энергия активации процесса. Это заметно
влияет на характер зависимости степени удаления азота
от электрического режима переплава, определяющейся
критериями Стэнтона 31э=/Спл/1’э и StB=KB/i>o- В пределах
используемых значений j—1/D (11—25 кА/м) в сталях без
титана величины Кпл и Ав растут с увеличением тока мед-
леннее, чем v9 и Vo. Критерии St9 и StB при этом уменьша-
ются, и удаление азота ухудшается. Если же в результате
введения нитрообразующих элементов значение /Св растет
быстрее, чем ц0, то при уменьшении St3 критерий StB может
расти. При этом отмечается либо улучшение удаления
азота при повышении тока, либо независимость степени
рафинирования от режима переплава.
В литературе отмечается возможность улучшения уда-
ления азота при введении в металл титана, что связывают
[229] с всплыванием нитридов титана в ванне. Вместе с
тем данные работы [231] показывают, что даже при высо-
кой массовой доле титана (до 3 %) в никелевых сплавах
практически все нитриды титана диссоциированы уже на
стадии пленки на торце электрода. Появление пленки нит-
ридов титана на поверхности жидкой ванны связано не с
всплыванием нитридов, а с образованием их в месте кон-
такта металла с плазмой дуги вне анодного пятна.
Одной из причин улучшения удаления азота при введе-
нии в металл титана может быть понижение активности
ПАВ, главным образом, серы.
Нитридные пленки могут возникать и на торце электро-
да по реакции
[Ti] + {N} = TiNT;
_ gTiN
T,N ’ ZTI[%TiJpN
208
________Г™______ . (361)
hi I%Til /KapNt
Для сплавов на основе железа lg JCtin = (39497/7’)—
—9,704), для никелевых lgKtin= (33497/7’)—8,802. Отсюда
при Ojin =1 образование нитридной пленки возможно при
условии Pn,>Pn„ где
(362)
где Ка определяется при температуре катодного пятна
(для торца электрода) или анодного пятна (для зеркала
ванны), а К tin и f п для температуры поверхности метал-
ла на соответствующей стадии.
Дуга непрерывно перемещается в подэлектродном про-
межутке, и образующаяся пленка по выходе из особой
зоны растворяется в металле. Если пленки возникают в
районе каплеобразования, они вместе с каплями перено-
сятся в ванну к фронту кристаллизации. Капля, проходя
жидкий металл, превращается во вращающийся тор с
уменьшающимся по мере опускания радиусом кругового
сечения. В результате этого в металле образуются спира-
левидные скопления нитридов титана (дефект «улитка»).
Такие же капли, отброшенные электродинамическими си-
лами на стенку кирсталлизатора в районе короны слитка
приносят пленки нитридов в корону. Опадение таких уча-
стков короны в ванну также может привести к фиксации
в металле нитридных скоплений, но уже не спиралевидных
(дефект «корона»). Оба дефекта имеют общую первопри-
чину, и для их предотвращения необходимы следующие
условия: 1) уменьшение вероятности перекрытия зон кап-
леобразования и горения дуги; 2) перемещение зоны кап-
леобразования в район наилучших условий откачки (на
периферию торца электрода); 3) уменьшение активности
титана в пределах возможных колебаний марочного соста-
ва; 4) понижение [N]o при выплавке исходного металла.
Периферийное каплеобразование при d^240 мм (£)>
>320 мм) обеспечивается поддержанием короткого под-
электродного промежутка и плотной короны слитка, обес-
печивающей хороший лучистый теплообмен с боковой
поверхностью торца электрода. Это обеспечивает форми-
рование «куполообразного» торца с периферийным капле-
образованием и преимущественно подэлектродным горе-
нием дуги.
При нарушении этих условий и формирования плоского
14-675
209
торца капли образуются в районе центра тяжести участ-
ков металлосбора, т. е. на расстоянии примерно 4/з от
оси электрода. В этом же районе располагается в данном
случае и зона наиболее вероятного горения дуги, что резко
увеличивает опасность брака по «улитке» («короне»). При
£)^180 мм (с^150 мм) электроды плавятся, имея кони-
ческий торец с центральным каплеобразованием и дугой,
горящей преимущественно на середине образующей кону-
са. Наряду с хорошей откачкой азота при такой форме
торца это приводит к тому, что соответствующие дефекты
в этих случаях, как правило, не возникают. При D =
=2004-280 мм (d= 1604-200 мм) имеет место переход от
одноцентренного каплеобразования к многоцентренному и
от конического торца к плоскому. В этих переходных усло-
виях наиболее трудно развести в пространстве зоны наи-
более вероятного каплеобразования и горения дуги, поэто-
му в слитках такого размера возможно появление дефекта
«улитка» («корона»).
Для каждого сплава можно оценить максимально до-
пустимое значение [N]o, превышение которого при данном
расположении зон каплеобразования вызывает опасность
появления дефектов «улитка» и «корона». Дефекты отсут-
ствуют при Pn,kt <CPnP, • Для периферийного каплеобразо-
вания на промышленных печах :
ппер = = gAl%N]^p0T =
*4 Ч-298 *4 (% + 5кор) 29800
s° [ %N]? р„ т
"n.(%+S„,)(>+“I«NJ)298<X)-
При подэлектродном каплеобразовании на расстоянии
х от центра торца p^,KT=PN3,= PnT+APn,,
ApN> = W[(dM/2)-x]2, (363)
где
где Л= [(УцТрор)/16].1О-‘(а/<пл[%Кплр +
+ аКв I % NJ2) /2n/(/?TA4N1) «
« 1(^Ро£а)/(4Г)2)] /277(Л flMN1)l%N]3 • 10“4.
При подэлектродном каплеобразовании значение [N]Hp
меньше, чем при периферийном.
Пример. При ВДП хромоникелевого сплава в кристал-
лизаторе диаметром £)=400 мм содержание азота PV]q по-
210
низилось с 0,01 до 0,0056 %. Определить конечное содержа-
ние азота в этом же сплаве при переплаве его в кристалли-
заторе диаметром £)=500мм. Массовую скорость перепла-
ва считаем прямо пропорциональной D. Определим коэф-
фициент a=([%N]o-[7oN]KOH)/([%N]o[%N]KoH) = (0,01 -
—0,0056)/(0,01-0,0056) = 78,5’/%.
При переходе к другому типоразмеру, считая I/D =
=const и температурные условия на всех стадиях близки-
ми, примем ajazttv'v/vo—(g2D\)/(g\/D2)=D\/D2. От-
сюда a2=ai (Pz/Di) =78,5 (500/400) =98,0.
В новом кристаллизаторе [N]Koh=0,01/(14-98,0-0,01) =
=0,0051 %.
Пример. Определим парциальное давление азота в зо-
не плавки, при котором на поверхности катода образуется
нитрид титана при получении никелевого сплава методом
ВДП.
Исходные данные: температура плазмы 1830 К, темпе-
ратура катода 1675 К, титана в сплаве 2,6 %, lg fn =
= (4488/Т)—0,518. Определяем константу диссоциации
азота в плазме: 1g Ка (—49400/1830)4-6,00 = —21;
jSfa=10-21. Константа нитридообразования на поверхности
металла составляет: lg Ktin = (33497/1675)—8,802=11,22;
/(пи =1,66-10п. Коэффициент активности титана на по-
верхности металла определяем из выражения: lg fa =
= (4488/1675)—0,518=2,16; fri = 145. Критическое давле-
ние азота составляет: р^=(Ка Ктт /т1[%Т1])-1= (10~21Х
X 1.662-1022-1452-2,62)—1 = 2,6-10-6 атм=0,26 Па.
Пример. Определить максимально допустимую массо-
вую долю азота в хромоникелевом сплаве, гарантирующую
отсутствие дефекта «улитка» («корона») при периферий-
ном и подэлектродном каплеобразовании. Критическое
парциальное давление азота составляет 0,26 Па. Исходные
данные: Р=38 см; rf=30 см; g=5,5 г/с; суммарная быст-
рота откачки насосами и конденсатом у периферии элект-
рода 5,5 м3/с, коэффициент а=62’/%. При подэлектрод-
ном каплеобразовании /=2см. Температура плазмы 1830 К.
Определим давление азота на периферии подэлектродного
промежутка:
5,5-10~?-62-22,4-105-1830 [%N]g
Pn’ ~ 298-28-5,5-100(1 4-62 (%N]0)
3,05-103 t°/oN^
1 + 62 [%N]0
14*
211
При подэлектродном каплеобразовании примем х=
=4/3=0,1 м. Тогда перепад давления Дрк, равен:
= 22,4-10ь-5,5-10—3-62 2-1830 %Ng=
rN, 4.104-0,38?-22-10-4 V л-81830-28
= l,2-104[%N]2.
Давление азота в зоне лодэлектродного каплеобразова-
ния составляет: р Nj=p^eP+ApNj=3,05-10~3 {[% N0]2/(l
+62 [% N]o)} + 1,2-104 [% N]o . В случае периферийного
каплеобразования при условии р™? =Pnp, имеем: (3,05X
ХЮ3 [% N]o)nep (1 + 62 [% N]о) = 0,26; [% N]B -5,2х
X io-3 [% N]o—8,4-10-5=0. Отсюда [% N]o=O,O121 %.
При подэлектродном каплеобразовании уравнение отно-
сительно [% N]o является кубическим. Поскольку Дрма>
>Pn,p, то пренебрегаем величиной рйаР . Тогда 1,2- 104Х
Х[% N].2=0,26 Па, отсюда [% N]o~4,6-10“3 %- Подставим
это значение в знаменатель формулы для pnp : {(3,О5Х
XЮ3 [% N]o/(l+62-4,6-10-2)}+1,2-104 [% N]o=O,26 Па,
отсюда [% N]o=4,2-lO-3 %.
Удаление оксидных включений при ВДП
Снижение в металле кислорода и, соответственно, оксид-
ных включений при ВДП может достигать 50—70 %- Ко-
нечное содержание оксидов при этом является результатом
ряда процессов, каждый из которых по-своему зависит от
режима переплава, диаметра электрода, кристаллизатора и
состава металла. От этих же факторов зависит и относи-
тельная роль каждого процесса. К таким процессам отно-
сятся всплывание включений в ванне и взаимодействие ра-
створенного кислорода с углеродом на стадиях торца и
ванны, диссоциация и ассоциация включений. Взаимодейст-
вие углерода с недиссоциированными оксидами при ВДП
заметного развития не получает.
Г. Кнюппель [119], исследуя процесс обезуглероживания
в вакууме при отсутствии кипения (что характерно для
ВДП), установил в области массовых долей кислорода
0,012—0,53 % зависимость константы скорости процесса
от содержания растворенного кислорода в металле: К=
= р(1+с [% О]), где 0 — константа скорости диффузион-
ной ступени. При этом зависимость К от [% О] довольно су-
щественна. При 1873 К (1,44-10~3)/(1 + 174 [% О]) м/с.
С учетом этой зависимости К от массовой доли растворен-
ного кислорода получено приближенное выражение для
212
конечного содержания оксидов в металле, %:
где Кэ, Кв — соответственно константы скорости углерод-
ного расселения на стадии торца и ванны; хэ, хв—степень
диссоциации оксидов на стадии образования пленки на тор-
це электрода и на стадии ванны; w—скорость всплывания
оксидов в ванне.
Формула верна для рэ/цэ>»1 и Рв/г>о^>1, что обычно
имеет место. Для устойчивых, недиссоциирующих включе-
ний, при хэ=Хв=0 [%HB]koh=[%HB]o/[1 + (w/o0)]. Для
другого предельного случая, когда включения полностью
диссоциированы уже в пленке, при хэ=хв=1 имеем:
[ % НВ]кон = [ % НВ] [ 1+ (КА)1“111 + (ЯвЧ)Ь1 • (364)
В первом случае удаление включений определяется про-
цессами их всплывания, во втором — раскислением метал-
ла углеродом. Для шаровидного включения х=1—(г/
/го)3 (го — исходный радиус включения; г — текущий ра-
диус). Величины г и х могут быть оценены на основании
решения дифференциального уравнения процесса раство-
рения шаровидного включения. Без учета конвективной
диффузии примем, что за время dr растворяется слой окси-
да толщиной dr. Отсюда:
4лг2 у dr - (— 4л£>/100) {] %О]П — [ %О]об} pr dr, (365)
где у— плотность кислорода в оксиде, кг/м2; р— плотность
жидкого металла; D — коэффициент диффузии кислорода
в металле, м2/с; [%О]П—массовая доля кислорода в ме-
талле на границе с оксидом, равновесная с оксидом при
данной температуре; %; [% О] об —массовая доля кислоро-
да в объеме металла, %.
В свою очередь [% О]0б=х[°/о O]z , где [% O]z —об-
щее содержание в металле кислорода, связанного в оксиды.
В пределе равновесная при данной температуре степень
диссоциации составляет:
Хравн = I %О]п/1 %О]2 = 1 - (гкон/г0)3. (366)
Следовательно, конечный радиус включения гКон сущест-
венно зависит от общего содержания кислорода в метал-
ле, связанного в оксиды. Чем больше количество оксидов
в металле, тем они крупнее.
21$
Таким образом содержание оксидов в исходном метал-
ле обусловливает интенсивность протекания процессов уг-
леродного раскисления и всплывания. Чем грязнее исход-
ный металл, тем большую роль играют процессы всплыва-
ния, и наоборот. Принимая, что величина [% О]п является
равновесной, определяемой по реакции диссоциации окси-
да: VnAlemOn^^AitAle] + [О], получим [О]п==Я[аметоп/
/ (fo а^еп ]. Следовательно, на характер механизма удаления
оксидов влияет и массовая доля элемента Me в металле и
активность оксида МетОп. Если не учитывать влияние ве-
личины [ % Me] на состав оксидов, то повышение величины
[% Ale] уменьшает х и повышает роль процесса всплыва-
ния до появления минимума [% О] на кривой раскисле-
ния. Роль всплывания повышается при содержании оксида
МетОп в сложном комплексном оксиде, что понижает
аметОп. Например, для силлиманита А12Оз+5Ю2 = А12ОзХ
XSiO2; AG° = —188550—1,25Г Дж/моль; 1g К1873 = 5,24;
<*sio2 =яа1,оэ =К—1/* =0,0024, для андалузита AI2O3+SiO2 =
=Al2O3«SiO2; AG°=—158000— 1,95 Дж/моль; lgK1873 =
= 4,82; flsios= aAitoa = K~1/2 = 0,0039. При 1873 К для сили-
ката кальция CaO-SiO2 «sio, — 0,056, для силиката 2СаОх
XSiO2 flsio2 =0,072 для силиката MgO-SiO2 asio, =0,29,
для силиката 2MgO-SiO2 flsio, =0,224.
Таким образом термодинамическая устойчивость SiC>2
существенно повышена в силикатах кальция и магния, но
особенно резко повышена в алюмосиликатах. Для алюмо-
силикатов характерно и резкое снижение активности гли-
нозема. Для шпинели типа СаО-А12О3 имеем: CaO-f-
Ч-А12Оз = СаО-А12Оз; AG°=—15420+23,4 Т Дж/моль, т. е.
при 7^660 К эта шпинель неустойчива и разлагается.
Также неустойчивы и другие алюминаты кальция.
Укрупнение слитка, увеличивая пребывание металла в
жидком состоянии и повышая х, способствует развитию
процессов углеродного раскисления и снижению роли про-
цессов всплывания. Повышение степени диссоциации х с
ростом диаметра кристаллизатора (или с повышением то-
ка при том же D) приводит к росту доли включений, вто-
рично образовавшихся по реакции [7Ие]+п[О] —МетОч.
Размеры таких «вторичных» включений не зависят от
размеров исходных включений и определяются только ус-
ловиями кристаллизации слитка ВДП. Всплывание частиц
оксидов в ванне возможно в результате действия трех ос-
новных механизмов: конвективных потоков, закона Сток-
са, движения в поле ПАВ. Хотя скорость конвективных
214
потоков металла превышает скорость частиц по двум дру-
гим механизмам, конвективные потоки являются циркуля-
ционными; они могут увлекать включение на поверхность
или в глубь металла. Два других механизма являются не-
циркуляционными, а следовательно, определяющими про-
цесс всплывания включений. В диффузионном поле ПАВ
включения движутся со скоростью: w= (2r/3q) (Г/?7/с) X
X(dc/dx). Поскольку TRT/c——daM-s/dc (оы-в — межфаз-
ное натяжение на границе включения с металлом), то
w =— (2г/3т|) (doM_B/dx) =— (2г/3г|) (dcM-JdT) (dT/dx).
(367)
С учетом выражения ом-в = Св—ом cos0 получим
= ^в _ cos0 о in 0 d0 j^m cos0
dT dT dT dT dT
(368)
Значения do^/dT по данным различных авторов суще-
ственно различаются для одного и того же металла. Так,
для железа do^dTравно—3,5-10~4 Дж/(м2-К) (см. табл. 12)
и—5-10~4 Дж/(м2-К) [177]. Для никеля duM/dT равно
—3,9-10~4 Дж/(м2-К) (см. табл. 12) и—2-10~4 Дж/(м2- К)
[177]. Поскольку dctn/dT<iQ, то всплывание включений в
направлении повышения температуры (dTldx^>0) возмож-
но только, если cos0<O, т.е. если 0>л/2 (90°). Для SiO2
в жидком железе 0=126° для MgO, СаО, А120з 0= 140-ь
-ь145°. Компоненты сплава, имеющие высокое химическое
средство к кислороду (Al, Ti) резко понижают 0 и cos0.
Так, в сплавах на основе железа с 2,5—3 % Ti для А120з
0 уменьшается до 90°, a cos 0 до 0.
Всплывание частиц в направлении повышения темпе-
ратуры связано с увеличением в этом направлении степе-
ни диссоциации оксидов и ростом массовой доли раство-
ренного кислорода. Средний температурный градиент по
объему жидкой ванны можно приближено оценить из вы-
ражения: dT/dxw (tn/R) (ц?/12)[1—(и?/4)][1 + 3/8(/?2/^)].
где /ц — температура в центре зеркала ванны; R —
радиус слитка; Нк— глубина конической части ванны; ц? —
коэффициент, определяемый условиями теплообмена и теп-
лофизическими свойствами металла. Соотношение скоростей
всплывания частиц по закону Стокса и в диффузионном
поле кислорода в условиях ВДП составляет:
215
(369)
При меньшем значении гв
Рис. 48. Распределение температур
по глубине жидкой ванны I при
ВДП стали IHX15 в кристаллизато-
ре диаметром 280 мм при токе
6,0 (/), 5,6 (2), 5,0 (.?), 4,2 (4) и
3,3 кА (5)
где gQ — ускорение свободного падения; R—радиус слитка.
Принимая go — 9,8 м2/с; dGuldT =—4-10~4 Дж/(м2-К) cos 0 =
= 0,7; р? = 1=2; R/HK=\-, рм = 7 г/см3; рв = 3 г/см3, получа-
ем Wct/^пав = (2,5 = 3,3) • 105ГвЯ, где гв и R — выражено
в метрах.
Если выразить R в метрах, а гв в микрометрах, то ра-
венство скоростей имеет место при условии гв = (3 = 4)//? мкм.
включения всплывают под дей-
ствием поверхностных сил,
при большем — по закону
Стокса. Для /? = 20 см части-
цы с гв^ 154-20 мкм всплыва-
ют главным образом в поле
кислорода. Температурный
градиент в ванне резко колеб-
лется по ее высоте. Анализ
кривых рис. 48 показывает, что
dT/dx у поверхности зеркала
ванны в 10—90 раз превыша-
ет значение dT/dx у фронта
кристаллизации. Отсюда и
роль механизмов всплывания
должны быть различной по вы-
соте ванны. В глубине ванны,
у фронта кристаллизации мо-
жет преобладать всплывание
по закону Стокса, в то время
ванны преобладает всплывание
в диффузионном поле кислорода.
Сравним теперь скорости процесов всплывания и угле-
родного раскисления. Учитывая, что обычно в металле
ВДП общее содержание кислорода <0,01 %, а содержание
растворенного кислорода еще меньше, примем по Г. Кнюп-
пелю при 1873 К для молекул СО в железе 0=14,4 У
ХЮ-3 м/с. Для среднего по объему ванны значения темпе-
ратурного градиента, принятых выше значениях парамет-
ров процесса и физических величин и считая т] = 4 г/(м-с),
имеем Топав ~ (6,5 = 8,5)Гв//? м/с. Отсюда рв/а’пдв =(1,7-:-
= 2,2)-RlrB-10~4. Считая, что в среднем в ванну с каплями
приходят оксиды с гв«2 мкм, получим $B/w^R-102. Это
как в поверхностных слоях
216
означает, что практически для всех кристаллизаторов вос-
становительные процесы быстрее процессов всплывания.
Скорости обоих процессов могут быть сравнимы только
для случая всплывания крупных (гв>10 мкм) оксидов в
малых (7?<5 см) лабораторных кристаллизаторах.
Изменение химического состава при ВДП
Как правило, в процессе ВДП не наблюдается значитель-
ного изменения химического состава металла. Только ле-
тучие компоненты, такие как марганец, могут удаляться
из металла в заметных количествах. Поведение марганца
имеет при ВДП особое значение, так как его концентра-
ция в некоторых конструкционных и коррозионностойких
сталях достигает нескольких процентов, а снижение кон-
центрации марганца после ВДП составляет 25—30 %.
Удаление марганца лимитируется главным образом внут-
ренней диффузией. В работе [174] показано, что связь
между исходной и конечной концентрацией марганца ха-
рактеризуется следующей формулой:
In [Mn]o/[Mn]K = (рм n/v0) [ 1 - (rf2/r>2)] (370)
Формула (370) показывает, что марганец испаряется
преимущественно с незаэкранированной электродом части
поверхности жидкой ванны. Кажущееся неучастие в испа-
рении остальной поверхности ванны и торца электрода свя-
зано, по-видимому, с тем, что под электродом испарение
марганца лимитируется внешним массопереносом. Кроме
того, некоторую роль может играть частичная конденса-
ция паров марганца на отдельных участках торца электро-
да вследствие значительного различия температур близко
расположенных поверхностей электрода и ванны. Констан-
та скорости испарения марганца рМи по данным работы
[174], составляет 0,6 мкм/с для спокойной ванны и
~1,2 мкм/с для ванны, вращаемой электромагнитным по-
лем соленоида.
Особый интерес в смысле поведения марганца пред-
ставляют участки слитков, наплавленные в нестационар-
ном режиме (нижняя и верхняя зоны). Нижняя часть
слитка наплавляется, как правило, при повышенном токе,
что способствует сохранению марганца. В верхней части
слитка, когда требуется понижать скорость плавления для
выведения усадочной раковины, потери марганца резко
возрастают. В тех случаях, когда в стали оговорен нижний
предел по марганцу, приходится либо совсем отказывать-
ся от выведения усадочной раковины, либо проводить этот
217
процесс в атмосфере инертного газа для понижения скоро-
сти испарения марганца.
Пример. Определим конечное содержание марганца в
конструкционной стали при ВДП в кристаллизаторе диа-
метром 500 мм при токе 9 кА. Исходные данные: g =
= 90 г/с; d=0,4 м; [Мп]0= 1,5 %; р = 7 г/см3; рмп = 6Х
XIО"3 см/с. 1). v0= (4g)/(nD2p) = (4-90)/(3,14-502-7) =
= 6,7-Ю"3 см/с; 2). 1п[Мп]о/[Мп]к =(Рмп/о0) [1— (d2/D2)] =
= [(6-10~3)/(6,7-10~3)] (1—0,82) =0,32; 3) [Мп]0/[Мп]к =
==ео,з2=1>38. 4) [Мп]к = [Мп]о/1,38 =1,5/1,38 =1,09 %.
Из цветных металлов, присутствующих в виде примеси
в конструкционных сталях, подвергаемых ВДП, в неболь-
ших концентрациях присутствует медь, содержание кото-
рой составляет 0,1—0,3 %. В результате ВДП содержание
меди понижается на 10—30 %. По данным работы [174],
испарение меди подчиняется зависимости, аналогичной той,
что получена для марганца [формула (342)]. Для крис-
таллизатора диаметром 320 мм при ВДП стали 50Х2НМ
при токе 5,5 кА величина Реи составляла 4,0-10-3 см/с, а
при 6,5 кА Реи =8,8-10-3 см/с. Более сильная, чем у мар-
ганца, зависимость р от температуры вызвана, по-видимо-
му, тем, что испарение меди в большей степени, чем испа-
рение марганца, лимитируется стадией десорбции атомов
с поверхности расплава. Это предположение подтвержда-
ется повышенной активностью меди в расплавах железа.
VIII.3. РАФИНИРОВАНИЕ МЕТАЛЛА ПРИ ЭШП
Температурный режим процесса ЭШП
Измерение температуры шлака и металла при ЭШП сви-
детельствует о неравномерном распределении температу-
ры по объему шлаковой ванны и по поверхности ванны ме-
талла. Максимальные температуры (1750—1900 °C) соот-
ветствуют подэлектродной зоне. Температура шлака в
кольцевом зазоре между электродом и стенкой кристалли-
затора достигает 1675—1725°С. Температура открытой по-
верхности шлаковой ванны и слоя шлака, прилегающего к
металлу, на 100—150 °C ниже температуры шлака в коль-
цевом зазоре. Температура шлака и металла при ЭШП за-
висит от двух факторов: электрического режима перепла-
ва и электрической проводимости шлака, определяющей
размер межэлектродного промежутка.
Для определения эффективной температуры, характер-
ной для процессов рафинирования при ЭШП, может быть
использована зависимость, аналогичная ВДП:
218
5Р“’’.„ + ДТт„[1-(Р;д/Руд)], (371)
где РуД — удельная мощность, выделяемая в шлаковой
О °
ванне; руд — удельная мощность, при которой перегрев
металла практически отсутствует.
Величина Руд определяется из выражения Руд=
—Р/(НШО) (Р — полная мощность, выделяемая в шлаке;
D — диаметр кристаллизатора; Нш— расстояние между
торцом электрода и поверхностью металлической ванны).
Пример. Определим эффективную температуру при
ЭШП стали ШХ15 в кристаллизаторе диаметром 400 мм
при токе 10 кА, напряжении на шлаковой ванне 60 В и
глубине шлаковой ванны /7Ш= 150 мм. Исходные данные:
/лик=1460°С; Тлик=1733 К; Р°уд=400 Вт/см; Д7'тах=
= 500К. 1) Руд = = (60-10000)/(15.40) =
= 1000 Вт/см; 2) Т= 1733+500[1— (400/1000)] =2033 К
(1760 °C).
Удаление неметаллических включений при ЭШП
Вопросам рафинирования металла при ЭШП посвящен
ряд работ. Из них наиболее интересными являются работы
[179—185]. Значительное внимание во всех работах уделе-
но вопросу удаления включений. При ЭШП удаление не-
металлических включений может достигать 65—75 % • При
этом главную роль в процессе удаления включений при
ЭШП играет рафинирование металла шлаком. Удаление
включений из металла может сопровождаться вторичным
загрязнением металла включениями вследствие окисления
электрода кислородом воздуха, перехода раскислителей из
шлака в металл и т. п. При переплаве под шлаками, со-
держащими глинозем, возможно загрязнение металла
AI2O3 по следующим схемам: 1) 2(А13+) + 2(О2-) + [О]4-
+ [Fe] = А120з(Т) + (Fe2+); 2) 2(А13+) + 2(О2~) + [О] +
+ [Мп]=А120з(Т)+(Мп2+); 3) 2(AF+)+9/2(O2-)+3[O]-|-
+ 3/2[Si] =А12Оз(т)4-3/2(5Юз ). Растворенный кислороде
металле появляется в результате разложения оксидов
кремния и марганца, а малая концентрация в шлаке катио-
нов железа и марганца и кремнекислородных комплексов
способствует сдвигу этих реакций в сторону раскисления
металла алюминием шлака. Появление в шлаке катионов
железа и марганца, т. е. металлов с переменной валентно-
стью, в свою очередь способствует своеобразной «перекач-
ке» кислорода из газовой атмосферы в металл. На поверх-
ности шлаковой ванны происходит адсорбция кислорода
(О) аде- Адсорбция сопровождается ионизацией
219
атомов кислорода и окислением присутствующих в шлаке
катионов низшей до катионов высшей валентности:
(О)да + 2 (М^+) + 3 2 (Л-К^). (372)
Металлом Me с переменной валентностью могут быть
например, железо, марганец, титан. Анионы FeO7, МпО-Г,
тюг диффундируют через шлак к металлу. Конвективные
потоки в металле значительно ускоряют подход анионов
к металлу. На границе шлак — металл анионы трехвалент-
ных металлов восстанавливаются до двухвалентных:
2 (Me О-) + [Fe] 2 (Ме2+) + (Fe2+) + 4 (О2-). (373)
С учетом перехода кислорода нз шлака в металл по
реакции (Fe2+) + (O2-) = [Fe] + [O] суммарный процесс
«перекачки» кислорода имеет вид: |/2{02} = [0].
Возможна также транспортировка кислорода из газо-
вой фазы в металл в результате растворения в шлаке во-
дяных паров:
{Н2О} + (О2-) = 2 (ОН-);
2 (ОН") + (Fe2+) = [Fe] + 2 [О] + 2 [Н];
(Н2О) 4- (Fe2+) + (О2-) = [Fe] +2 [О] + 2 [Н]. (374)
При этом в металл поступает не только кислород, но и
водород. Таким образом, всегда имеются источники появ-
ления в металле (даже предварительно раскисленном)
растворенного кислорода, который может стать причиной
образования вторичных глиноземистых включений. Кроме
железа, марганца и кремния, вторичному загрязнению ме-
талла глиноземом при ЭШП заметно способствует углерод
в металле. Поскольку глинозем в шлаке диссоциирован по
реакции (А12О3)ч=±(А1О7) + (А13+) + (О2-), то «перекачка»
кислорода из атмосферы в металл анионами (А1ОГ) в при-
сутствии углерода происходит по схеме: 3/2{Ог}+ 2(А1О7) +
.+ 2 ГС] =А12Оз(Т)+2(О2-) +2 {СО}.
Обработка экспериментальных данных по ЭШП угле-
родистых сталей дает следующее выражение для мини-
мального содержания глинозема в неметаллических вклю-
чениях при ЭШП под шлаком АНФ-6: [% Al2O3]min=
=0,0029+9- 10-4а£ + 2,6- 10-3о3{2 где 0,0029 % — количест-
во глинозема, образующегося в результате раскисления
металла алюминием, восстановленным из шлака железом
и марганцем. С учетом того факта, что в металле всегда
после ЭШП присутствует определенное минимальное ко-
220
?
личество глинозема, характерное для данного шлака и
данного металла, формула для оценки степени удаления
оксидов при ЭШП принимает вид:
г о/ pjgi __ [%НВ] К^И^пл) Ab] [%^2^з]ш1п I
° кон [ 1 + (тшпл) /v9] [ 1 + (wB/v0)]
। (wB/t>o) 1%А120з]т1п
[1 + (и>в/«о)
где адПл, Wb — линейные скорости перехода включений из
металла в шлак на стадии пленки и ванны. Для включе-
ний, самопроизвольно переходящих границу раздела фаз
(AI2O3, MgO, ЛОг), лимитирующей является скорость
подвода включений к границе раздела. Для силикатов
(при <1 % Si), оксидов марганца и железа, преодолева-
ющих при межфазном переходе расклинивающее давле-
ние, процесс лимитируется стадией перехода. Принимая в
первом случае, что включения перемещаются в диффузи-
онном поле ПАВ, имеем при самопроизвольном переходе
критерий Стэнтона:
St = wt/vi = — [(2rB)/(3i]a)l (d<rM/dT) (Т{ — To) cos 6, (375)
где Ti — температура поверхности металла на соответст-
вующей стадии.
Как следует из выражения (375) при таком переходе
лучше удаляются более крупные частицы. Повышение
подводимой мощности, увеличивая Т, и тем самым умень-
шая гв сложным образом влияет на конечное содержание
включений [% НВ]кон. Для включений системы
FeO—МпО—SiO2 решающую роль при переходе границы
играют поверхностные свойства системы. Размеры вклю-
чений играют здесь меньшую роль. Однако, поскольку
малому включению легче преодолеть расклинивающее дав-
ление, в данном случае лучше должны удаляться включе-
ния с меньшим Гв, так как значения о>Пл и wB в этом слу-
чае. слабо связаны со скоростью переплава, критерии
Wilvi с увеличением Оо уменьшаются, и рафинирование от
этих включений должно при этом ухудшаться. Для хоро-
шей экстракции включений шлаком при ЭШП кроме
указанных кинетических условий необходимо: 1) хорошее
смачивание неметаллических включений шлаком; фторид-
ные расплавы предпочтительнее оксидных в качестве осно-
вы для шлаков ЭШП, так как обладают лучшей смачивае-
мостью; 2) плохое смачивание неметаллических включений
металлом; 3) оптимальное значение поверхностного натя-
жения металла; величина <тм в условиях ЭШП зависит
221
металлических включений
50
<t0
o'
20
LVf/o
Рис. 49. Зависимость степени уда-
ления оксидных включений ф при
ЭШП от содержания углерода в
стали
О 1 2
главным образом от химического состава металла и в сис-
теме Fe—С в присутствии растворенного кислорода имеет
максимум при ~0,2 % [С].
На рис. 49 приведена зависимость степени удаления не-
от массовой доли углерода в
слитках диаметром 100 мм при
7=1,5 кА под шлаком АНФ-6.
Как видно из рис. 49, наибо-
лее эффективный процесс уда-
ления включений должен иметь
место при ЭШП среднеуглеро-
дистых сталей (0,3—0,5 % С).
Пример. Определить конеч-
ное содержание включений
глинозема после ЭШП стали
под шлаком АНФ-6 при их ис-
ходном содержании 0,009 %;
Z)=40 см; <7=20 см: £=90 г/
/с. В стали содержится 1,0 % С
и 0,2 % Si. Средний радиус
включений гв=5 мкм. Тэ=1783 К; Тв=2033 К; 70=300 К;
«=1,5-10 5 м2/с, т]=0,025 Па-с, коэффициент т=2.
1. Определим [% Al2O3]mln: lg fc =eg[% С] g1 [% Si] =
=0,14-1,0+ 0,08-0,2= 0,156; lg fSi=escl [% C] +ef| [Si] =
=0,18-1,0+0,11-0,2=0,202; fc = 1,43; fsi = 1,6; ac = l,43;
asl =0,32; [ % A12O3] mm=0,0029+9 -10 4 -1,432 + 2,6-10~3 X
X 0,32 3/2 =0,0052.
2. Примем для железа doM/dT=—3,5-10~4 Дж/(м2-К)
и cos 0=cos 130°=—0,765.
3. т St8 = — ^,10~е'3,^10~4 (1783 — 300) -0,765 = 7,0.
3-2,5-10—М ,5-10—* ' ' ’ ’
4. StB =
2-5-10—е-3,5-10—*
3-2,5-10-2.1,5-10-5
(2033 —300). 0,765 = 4,1.
5. [%А12О3]Н0Н —
0,009 + 7-0,0052
8-5,1
4,1-0,0052
= 0,0053%.
Из других процессов, происходящих в ванне и имею-
щих отношение к неметаллическим включениям, необходи-
мо отметить процессы укрупнения включений. Те включе-
ния, которые не были экстрагированы в шлак и не успели
222
диссоциировать в области высоких температур, попадая в
низкотемпературную зону ванны, становятся центрами
формирования новых включений по реакции: х[Ме] +
Ч-£/[О]->Л4вЛО1/. Процесс осаждения продукта МехОу на
поверхность включения — подложки лимитируется диффу-
зией растворенного кислорода. Поток П(г/с) вещества
МехОу на частицу равен: П=4ягвОо С[о] (7И/16), где Do-
коэффициент диффузии кислорода; qoj — концентрация
кислорода в расплаве, г/см3; М — молекулярная масса
МехОу-, 16 — атомная масса кислорода.
Скорость увеличения размера включения drB/dx при
этом составит: drBldx—Do сЛ1/(16рвгв), где рв—плотность
включения. Таким образом, сохранившиеся в металле мел-
кие включения укрупняются быстрее, чем более крупные.
Этот процесс наряду с явлением более интенсивной экстра-
поляции мелких включений в шлак приводит к тому, что
одним из результатов ЭШП является относительно более
резкое снижение содержания мелких неметаллических
включений по сравнению с крупными.
Поведение серы
Обычно целью ЭШП является понижение содержания се-
ры в металле. Степень удаления серы при ЭШП, как пра-
вило, составляет 50—75 % и зависит от содержания серы
в исходном металле, состава шлака и атмосферы над
шлаком, режима переплава, рода и полярности тока.
При использовании высокоосновных шлаков с большим
содержанием СаО, имеющих в своем составе карбид каль-
ция СаСг, содержание серы в металле может быть сниже-
но до 0,0025 % • Обычное содержание серы в металле после
ЭШП составляет 0,006 — 0,010%. Наиболее существенное
влияние на степень десульфурации при ЭШП оказывает
состав шлака.
При ЭШП на фторидных шлаках содержание серы в
отработанном шлаке, как правило, не только не повыша-
ется, а даже заметно снижается, если процесс идет в атмо-
сфере, содержащей кислород.
Данное явление связано с тем, что существует два типа
реакций, управляющих содержанием серы в процессе
ЭШП — между шлаком и металлом (I) и между газом и
шлаком (2): 1) [S] + (О2-) = (S2~) + [O]; 2) (S2~) +
-НМСМ = {SO2} + (О2-). Суммарная реакция процесса де-
сульфурации в данном случае имеет вид:
IS] + % {О2} = {SO2} + [О]. (376)
223
Таким образом, процесс десульфурации при ЭШП со-
стоит из следующих стадий: 1) диффузии серы из объема
металла к поверхности раздела металл — шлак; 2) пере-
хода серы из металла в шлак; 3) переноса серы от по-
верхности раздела металл — шлак к поверхности раздела
шлак — атмосфера; 4) окисления серы на границе шлака
с атмосферой; 5) отвода продуктов окисления серы от гра-
ницы раздела шлак — атмосфера.
Равновесное с данной атмосферой содержание серы в
металле равно: [S]p=([O]fo/Kfs)(p so, /Р о-2) ’ где Я—кон-
станта равновесия реакции (376); р so,— парциальное
давление SO2 в атмосфере над шлаком; р о, — парциаль-
ное давление кислорода в атмосфере над шлаком; /о» /s~“
коэффициенты активности кислорода и серы (по Генри).
Таким образом, равновесное
1С,ММ
Рис. 50. Зависимость степени и
десульфурации металла при ЭШП
от введения аргона (пунктирная
линия — исходное содержание се-
ры, I с — расстояние от низа слит-
ка):
содержание серы в металле
должно зависеть только от
степени раскисленности метал-
ла и состава газовой фазы.
Установлено [179], что пре-
кращение доступа воздуха к
шлаковой ванне, достигаемое
подачей в кристаллизатор ар-
гона, тормозит процесс десуль-
фурации (рис. 50) и ведет к
накоплению серы во фторид-
ном шлаке. Содержание серы
в исходном шлаке АНФ-1П
составляет 0,018, в шлаке пос-
1 — с аргоном; 2 — без аргона
ле ЭШП на воздухе 0,015, пос-
ле ЭШП в атмосфере аргона
0,100 %. Зависимость степени десульфурации от состава
шлака связана с тем, что состав шлака влияет на степень
достижения равновесия на границе раздела металл —
шлак.
Чем больше в шлаке анионов О2-, тем легче и полнее
осуществляется анионный обмен между серой и кислоро-
дом на границе металл — шлак и тем ближе концентрация
серы в поверхностном слое металла на границе этого раз-
дела приближается к равновесной с данной атмосферой.
Наибольшее приближение к равновесию достигается при
использовании шлаков с высоким содержанием СаО, наи-
меньшее — при использовании силикатных шлаков. Следо-
вательно, в реальных процессах содержание серы в метал-
ле на его границе со шлаком по отношению к атмосфере
224
является не равновесным, а квазиравновесным ,[SJK.p-
Квазиравновесной эту величину можно считать потому,
что для каждого состава шлака эти величины, очевидно,
должны быть постоянной характеристикой и влиять на
скорость перехода серы из металла в шлак. При данном
составе газовой атмосферы скорость этого перехода может
лимитироваться диффузией серы либо в шлаке, либо в ме-
талле. В условиях ЭШП с использованием жидкоподвиж-
ных фторидных и известково-глиноземистых шлаков наи-
более вероятным лимитирующим звеном является диффу-
зия серы в металле.
Считая, что условия массопереноса серы в металле на
торце электрода и на поверхности ванны примерно одина-
ковы, поведение серы при ЭШП можно описать уравне-
нием:
In ^исх [$1к.р _ Ps (1 । т
[$1кон [S]k.p vo \ D2 J
или
A [S] _ [S]hCX — [$]koh /1 lS]B.p
[S]
ИСХ [S]ИСХ \ [S]koh.
(377)
e“₽S /»о<1+т </1
где Ps—средний коэффициент массопереноса серы на тор-
це электрода и на поверхности ванны; [S]K.p — концентра-
ция серы в диффузионном слое, квазиравновесном с дан-
ным составом шлака и атмосферы.
Нами установлено, что при ЭШП под шлаком АНФ-6
металла, содержащего 0,03—2,0 % С, 0,22—1,47 % Мп,
0,2—0,95 % Si и 0,51 % А1 величина [S]K.p не зависит от
состава металла и режима переплава и равна 0,002±0,001.
Коэффициент массопереноса серы р8зависит главным
образом от концентрации углерода в металле. Какой-либо
заметной зависимости Ps от содержания в металле мар-
ганца, кремния и алюминия (в исследованных пределах)
не обнаружено. Зависимость Ps от содержания углерода
приведена на рис. 51. Величина Ps имеет максимум при
0,2 % С, что соответствует максимуму поверхностного натя-
жения в системе Fe—С. Для сталей с>0,5 % С величина
Ps от содержания углерода практически не зависит. Оче-
видно, что понижение содержания серы в металле при
ЭШП возможно только до величины ,[S]K.p и только, если
исходное содержание серы [S]Hcx превышает [S]K.p. Если
[S]k.p> [S]hcx, то удаления серы из металла не происхо-
дит, и даже может иметь место загрязнение металла серой
шлака.
15—675
225
На рис. 52 приведены данные, характеризующие влия-
ние исходного содержания серы в стали ШХ15 на содер-
жание серы после ЭШП под шлаком АНФ-6 [179]. Из
рис. 52 следует, что для стали ШХ15 при ЭШП под шлаком
АНФ-6 [S]K.p=0,004-4-0,005 %.
Рис. 51. Зависимость коэффициента
массопереноса серы в металле при
ЭШП от содержания углерода
Рис. 52. Связь между исходным
ISJ л и конечным содер-
жанием серы в металле при ЭШП
Электрический режим, определяя массовую скорость
плавки и линейную скорость наплавления слитка о0, дол-
жен заметно влиять на десульфурацию при ЭШП. Повы-
шение скорости плавки и линейной скорости наплавления
слитка должно приводить к ухудшению удаления серы из
металла.
Поскольку в реакции десульфурации принимают уча-
стие электрически заряженные частицы (ионы серы и кис-
лорода), род тока и его полярность должны влиять на
процесс удаления серы из металла. В первую очередь не-
обходимо учитывать возможность протекания процесса
электролиза. На аноде при этом происходит реакция
(S2-) = [S] + 2е, одновременно на катоде возможна обрат-
ная реакция: [S]-f-2e= (S2-). При использовании пере-
менного тока эти реакции как на оплавляемом торце
электрода, так и на поверхности ванны взаимно компенси-
руют друг друга, и электролиз серы из шлака не получает
развития. При использовании постоянного тока может
иметь место электрохимический перенос серы от катода к
аноду через шлаковый расплав. Кроме того, необходимо
иметь в виду, что шлаки ЭШП содержат серу, которая
может выделяться на аноде. Шлак АНФ-6 до переплава
содержит 0,03—0,04 S % [179]. При таком содержании
серы в шлаке ее электролитическое выделение на аноде
226
может существенно влиять на процесс удаления серы из
металла.
Кроме электролизных явлений на электродах, исполь-
зование постоянного тока приводит к возникновению в
шлаковой фазе направленного потока анионов серы (от
катода к аноду). Поскольку в процессе десульфурации при
ЭШП большую роль играет окисление серы на границе
шлака с газовой фазой, условия транспортировки серы к
этой границе должны существенно влиять на ход процесса.
При переменном токе, когда направленный электрохи-
мический перенос серы в шлаке отсутствует, сера перено-
сится к границе с газовой фазой главным образом благо-
даря конвективным потокам в шлаке. При наличии интен-
сивных потоков в шлаке этот процесс не лимитирует общего
процесса десульфурации, который в данном случае
лимитируется диффузией серы в металле. При постоянных
токе и полярности электрод — анод электрохимической
перенос серы от ванны к электроду способствует транспор-
тировке анионов серы к границе с газовой фазой. В случае
противоположной полярности электрод — катод электрохи-
мический перенос препятствует поступлению серы в газо-
вую фазу, что должно привести к ухудшению условий де-
сульфурации в целом,
В табл. 23 приведены данные работы [179] о влиянии
рода тока и полярности на десульфурацию среднеуглеро-
дистой стали (0,4 % С, 0,3 % Si, 0,69 % Мп, 0,032 % S,
0,017 % Р) при ЭШП под различными шлаками. Наилуч-
шие результаты по десульфурации достигаются при ис-
пользовании переменного тока. При постоянном токе и по-
лярности электрод — анод степень десульфурации несколь-
ко снижается. Ухудшение десульфурации связано с
электролитическим выделением на аноде серы, содержа-
Таблица 23, Влияние рода тока на десульфурацию при ЭШП [179]
Род тока Полярность Содержание серы после ЭШП под шлаками, %
АНФ-1 АНФ-6 АНФ-9 АНФ-7
Переменный Постоянный Электрод — катод Электрод — анод 0,017 0,032 0,018 0,013 0,036 0,015 0,009 0,039 0,011 0,006 0,034 0,015
Примечание. Исходное содержание серы в металле 0.032 %. Состав шла-
ков: АНФ-1 95 % CaFs; АНФ-6 70 % CaFs+30 % А12О3; АНФ-9 80 % CaF3+
+20 % MgO; АНФ-7 80 % CaF3+20 % СаО.
15*
227
щейся в шлаке. Однако этот процесс в значительной степе-
ни скомпенсирован интенсификацией транспортировки се-
ры с границы с газовой фазой. При полярности элект-
род—катод условия такой транспортировки резко ухудшены
и электролиз серы на аноде—ванне не только приоста-
навливает процесс десульфурации, но и пирводит к неко-
торому насыщению металла серой из шлака.
Поведение азота
Поведение азота при ЭШП зависит от многих факторов,
в частности от исходного его содержания в металле и
формы, в которой он в нем присутствует, от состава шлака,
режима переплава и т. д. При ЭШП сталей, не содержа-
щих нитридообразующих элементов, степень удаления азо-
та тем выше, чем больше его исходное содержание. При
ЭШП сталей, содержащих такие нитридообразующие
элементы, как титан, ниобий, азот из металла почти не
удаляются. Имеют место даже случай некоторого повыше-
ния азота в металле после ЭШП таких сталей. Определя-
ющим моментом в данном случае является способность
шлака адсорбировать нитридные включения. Фторидные
шлаки типа АНФ-Ш и АНФ-6 обладают слабой адсорби-
рующей способностью по отношению к нитридам ниобия и
титана.
На поведение азота при ЭШП влияет состав шлака.
Шлаки ЭШП являются безжелезистыми, а такие шлаки
обладают повышенной способностью растворять азот. При
переплаве углеродистых сталей под шлаками систем
AI2O3—СаО; CaF2—AI2O3; AI2O3—СаО возможны образо-
вание в шлаке ионов CN- и перенос ими азота из газовой
фазы в металл. Введение в шлак SiO2 снижает раствори-
мость азота в шлаке и улучшает удаление азота из металла
при ЭШП. Поведение растворенного азота при ЭШП мо-
жет быть описано уравнением:
In [NJhcx [N]paBH __ Pn
[NJkoh [NJpaBH fo
т. e. лимитирующим звеном удаления азота является вну-
тренняя диффузия азота в металле к границам раздела
металл — шлак. Таким образом, предельное нижнее со-
держание азота после ЭШП определяется его равновесной
концентрацией на границе металла и шлака, т. е. величи-
ной [N] равн.
Для случая ЭШП среднеуглеродистых сталей под шла-
ком АНФ-6 равновесная между металлом и шлаком кон-
d2
D2
(378)
228
центрация азота [N] равн — 0,006+0,03[% N]m, где [% N]u —
концентрация азота в шлаке. Коэффициент массопереноса
азота в металле в условиях ЭШП 0 n= (5,7±1) • 10~5 м/с.
При содержании углерода в металле ~0,2 % величина 0n
понижается до (4±1)-10-5 м/с. Это явление может быть
связано как с влиянием самого углерода на 0n, так и
с тем, что при содержании в стали 0,2 % С ухудшаются
условия удаления из металла серы и неметаллических
включений, а по данным работы [187], повышение серы и
кислорода в металле понижает 0n-
Такое значение 0n соответствует эффективной темпера-
туре процесса на поверхности раздела металл — шлак
7Эф==1750°С. Влияние электрического режима на значения
[К]равн и 0ы обнаружено не было. Режим переплава ока-
зывает сложное влияние на поведение азота при ЭШП.
Поскольку 0ы практически не зависит от режима, т. е.
от скорости переплава, повышение линейной скорости на-
плавления слитка и0 должно приводить к ухудшению ра-
финирования металла от азота. Вместе с тем, режим
переплава влияет на величину заглубления оплавляемого
конца электрода в шлак и на прогрев электрода. При плав-
лении электрода вблизи поверхности шлаковой ванны мо-
жет происходить не только усиленное окисление металла,
но и поглощение им азота из атмосферы воздуха. Таким
образом, режим переплава может влиять на величину
[N],1CX.
Если вести ЭШП при неизменных значения силы тока
и напряжения на источнике питания, то к концу плавки в
связи с нарастанием напряжения на шлаковой ванне за-
глубление электрода в шлак уменьшается и возможно по-
вышение содержания азота в верхней части слитка [188].
Для предотвращения этого явления переплав необходимо
вести на дифференцированном режиме, постепенно пони-
жая силу тока и напряжение на источнике питания по ходу
плавки.
На удаление азота при ЭШП может влиять процесс
восстановления алюминия из шлака. В работе [179] в ре-
зультате ЭШП стали ЗОХГСНА содержание азота повыси-
лось при увеличении остаточного алюминия до 0,3 %• При
дальнейшем повышении содержания алюминия содержа-
ние азота в металле после ЭШП понижалось. Влияние
титана аналогично влиянию алюминия. Удовлетворитель-
ное объяснение этого явления пока не найдено.
Таким образом, содержание азота в металле ЭШП
можно регулировать подбором шлака и режима перепла-
229
ва. Степень удаления азота при ЭШП сталей, в которых
азот находится в растворенном состоянии, находится в
пределах 15—30 %, т. е. несколько хуже, чем при способе
ВДП.
Поведение водорода
Поведение водорода при ЭШП подчиняется тем же кинети-
ческим закономерностям, что и поведение азота:
d*
D*
(379)
металле.
массопе-
1п 1 %Н]исх [%Н]
равн __ Рн
[%Н]
КОН [%Н]равн
где Рн—коэффициент массопереноса водорода в
При 1750°C рн=(3,3±0,2).10-2 см/с.
При таком высоком значении коэффициента
реноса поведение водорода определяется главным обра-
зом величиной [% Н]Равн — концентрацией водорода, рав-
новесной со шлаком.
Удаление водорода из металла при ЭШП возможно
только, если [% Н]равн<[% Н]Исх» иначе будет иметь ме-
сто насыщение металла водородом. Поскольку содержа-
ние водорода в шлаках может достигать 20 мл/100 г,
очень важна специальная подготовка шлаков ЭШП (про-
дувка аргоном во время выплавки шлака, прокаливание
шлака перед ЭШП).
Концентрация водорода в шлаке в процессе ЭШП опре-
деляется не только исходным содержанием водорода, но и
водородом, попадающим в шлак из атмосферы, по реак-
ции: {Н2О} + (О2-) =2(ОН-). С повышением содержания
водяных паров над шлаковой ваной в металле увеличива-
ется содержание водорода. Это увеличение значительно
при использовании известковистых шлаков и чистого фто-
рида кальция. Наилучшими защитными свойствами обла-
дают фторидно-глиноземистые шлаки типа АНФ-6 и сили-
катные типа АН-8. Защита поверхности шлака аргоном
позволяет понизить содержание водорода в металле. Для
шлаков системы СаЁ2—СаО (АНФ-7) и CaF2—А12О3—
—СаО—MgO (АН-291) обычной выплавки при ЭШП без
защитной атмосферы связь между [% Н]равн и содержани-
ем водорода в шлаке (Н) имеет вид: [% Н]раВн=3,бХ
ХЮ-5 (Н)* %; для шлака АНФ-6:
. равн
% Н равн — 2,0Х
XЮ-5 (Н)* %; для чистого фторида кальция: [% Н]равн =
= 2,5-10-5 (Н)* %. При ЭШП в атмосфере аргона
* (Н) в мл/100 г.
230
величина [% Н]раВн понижается в 1,7—1,8 раза. Содержа-
ние водорода в металле изменяется по мере наплавления
слитка. Очень часто металл самой нижней части слитка
имеет наибольшую концентрацию водорода, так как при
наплавлении этой части из шлака недостаточно удален
водород. По мере наплавления происходит некоторое ра-
финирование шлака от водорода и в системе атмосфера —
шлак — металл устанавливается равновесие. По степени
удаления водорода процесс ЭШП уступает ВДП. В зави-
симости от качества подготовки шлаков и режима перепла-
ва степень удаления водорода колеблется в пределах
О—30 %. При использовании плохо подготовленных, сырых
шлаков может иметь место насыщение металла водородом.
VIII.4. РАФИНИРОВАНИЕ МЕТАЛЛА ПРИ ЭЛП
ЭЛП по сравнению с ВДП по условиям рафинирования
металла имеет следующие особенности:
1. Меньшую толщину жидкой пленки на торце заготов-
ки, составляющую при ЭЛП ~0,1 мм, что связано с осо-
бенностями стекания металла с конического или скошен-
ного торца. Отсюда большая скорость движения металла
в пленке и в 3—4 раза больший коэффициент массопере-
носа. Для конического торца средний коэффициент мас-
сопереноса равен:
4 (1 + К2 ) sin ф/2 ।3/
п^2 У
g0cosф/2 Зг—
—-------1 иэ>
3r8v
(380)
где ф — угол при вершине конуса; v — кинематическая
вязкость. В зоне каплеобразования величина р определя-
ется по формуле (350) при п=1.
2. Более высокую эффективную температуру процесса
на поверхности ванны.
3. Возможность более длительной выдержки металла в
вакууме, так как процессы плавления заготовки и подогре-
ва ванны при ЭЛП могут осуществляться раздельными ис-
точниками тепла независимо друг от друга. Это позволяет
наплавлять слиток и проводить рафинирование металла
в ванне при сколь угодно малой скорости плавки.
4. Большую удельную поверхность металла на кониче-
ском или скошенном торце по сравнению с плоским торцом
при ВДП.
231
Температурный режим процесса ЭЛП
Так же, как при ВДП и ЭШП, температура металла на
поверхности ванны при ЭЛП распределяется неравномер-
но. Она максимальна в центре и минимальна на перифе-
рии. Перепад температур между центром и периферией
достигает 100—200 К. Управление разверткой электронно-
го луча, дающее возможность дополнительно прогревать
периферийные зоны, позволяет снижать этот перепад до
50 К. Подробно вопрос о температуре металла при ЭЛП
рассмотрен в работе [189]. Средняя эффективная темпера-
тура поверхности ванны при ЭЛП является функцией
удельной мощности, поступающей на поверхность ванны:
Рул.ъ=Р^Г (Рв — общая мощность электронных лучей, обо-
гревающих ванну; F—площадь поверхности ванны). Для
оценки эффективной температуры ТОф при Руд.в^ 1,2 кВт/см2
может быть использована формула:
Т = Т 4- АТ ГР i(P I ро 11
эф 1 лик ’ ш max [гуд.вД уд.в т гуд.в)р
(381)
где Тлин — температура ликвидуса, К; АРшах — максималь-
но возможный перегрев К. Для железа ДТтах«580 К;
РуД.в — постоянная величина. Для сталей Р уД.в ~
«4600 кВт/м2. Обычно плавки проводят при Руд.в= 10004-
4-6000 кВт/м2, что соответствует перегреву ванны на
100—330 К.
Удаление растворенных газов и летучих примесей
При ЭЛП происходит значительное удаление из металла
азота, водорода, неметаллических включений и цветных
примесей [189—191]. Из работ, посвященных механизму
удаления примесей из металла при ЭЛП, необходимо от-
метить [187, 192, 193]. Если удаление примеси при ЭЛП
лимитируется внутренней диффузией, то кинетика процес-
са описывается выражением:
In Со/скон = (1 /о0) [₽в + трпЛ (d2/D2) J. (382)
Равновесной поверхностной концентрацией примеси
здесь можно пренебречь, так как в условиях низких оста-
точных давлений (Рх<0,1 Па)с;п<Сс«, поэтому, если удале-
ние примеси лимитируется реакцией первого порядка, что
характерно в частности для процесса испарения, то для
расчетов следует пользоваться выражениями (334), (335)
и (337). Если же процесс лимитируется реакцией второго
порядка (удаление азота), то рекомендуется пользовать-
ся формулами (342) и (343). При этом величины Апл опре-
232
деляются при температуре пленки Тпл = ГлИк+ (1—2) К,
а /Св при Тэф. Отличительной особенностью ЭЛП является
управляемое распределение общей мощности Р между за-
готовкой (Р3) и ванной (Рв). Поскольку Р=Р3 + РВ, то
соответственно относя удельные мощности к площади зер- .
кала ванны, имеем Руд=Руд.з=Руд.в. Если обозначить
Рв/Р=Руд.в/Руд через п, то
т^ = тт + [ („руд)/ (пр„ + j ]. (383)
Видно, что при малых значениях Руд увеличение п силь-
нее сказывается на росте температуры, чем при большой
Руд. Если принять, что массовая скорость плавки при ЭЛП
g=(P3/Q), где Q—количество тепла, необходимое для
нагрева, расплавления и некоторого (на 1—2 К) перегрева
металла, то
л/?2р
Р 3 __ Р уД.з
Qpn R2 Qp
Qp
(384)
Отсюда для процесса испарения примесей, считая К=
=Koe~E/RT получим:
In (е-£^гпл + е-Е*тэф) (385)
С Руд(1—«)
Для процессов удаления растворенных газов справедли-
во выражение:
Руд (1 w)
QpK0
(g £/Я^пл g Р/РТ’еф)
(386)
Из приведенных формул видно, что степень рафиниро-
вания металла при ЭЛП определяется такими факторами
электрического режима как полная удельная мощность
Руд и доля мощности, поступающей в ванну п.
На рис. 53 показана зависимость степени удаления во-
дорода от п для различных значений Руд. Из рис. 53 вид-
но, что степень рафинирования непрерывно увеличивается
с ростом п. Наиболее резкое увеличение степени рафини-
рования наблюдается при п> 0,74-0,75. Это положение
справедливо для всех случаев рафинирования металла при
ЭЛП от растворенных газов и летучих примесей. Если пе-
реплавляется металл, не содержащий летучих составляю-
щих (например, марганца), потери которых нежелательны,
величину п следует выбирать в пределах 0,75—0,85, обес-
печивая хорошее удаление водорода, азота и вредных лету-
чих примесей. В противном случае п выбирается в преде-
233
лах 0,4—0,6. При одном и том же значении п полная
удельная мощность Руд оказывает сложное влияние на
механизм рафинирования. С одной стороны, увеличение
РуД ускоряет процесс наплавления и снижает длительность
рафинирования, а с другой — с уве-
личением Руд повышается темпера-
тура поверхностного слоя металла
в ванне и ускоряется массоперенос
примесей. Это приводит к тому, что
сначала с ростом Руд степень рафи-
нирования металла снижается а за-
тем, пройдя через минимум, начина-
ет возрастать. Минимальная сте-
пень рафинирования металла от
примеси будет иметь место при ус-
ловии РуД.В==Ркр.В И РуД = РуД.кр:
АТ’щах х
>0 гр
УД.В 1 Лик
1 = пР
кр.в О1уд.кр
Рис. 53. Зависимость степе-
ни удаления водорода от п
при ЭЛП
/ — =6000 кВт/м2; 2 —
*руД=зооо кВт/м2 Таким образом, наихудшие ус-
ловия рафинирования определяют-
ся не полной удельной мощностью,,
а удельной мощностью, подаваемой на поверхность ванны.
Рабочие значения Руд, Руд.в и Рул.3, кроме условия хо-
рошего рафинирования, выбирают также из условия полу-
чения определенной желаемой структуры слитка. Характер
структуры во многом определяется формой профиля кони-
ческой части ванны, т. е. коэффициентом формы ванны
K$=Hk/R. В частности, отсутствие осевой пористости!
обеспечивается при условии Кф^1. При заданной величи-
не Хф удельную мощность при ЭЛП определяют по фор-
мулам:
(387):
^д.з = (4р(?аКф)/Ясл; (388>
Рум = 1 - п)] Руд.3 = [п/( 1 - «)] «4р Qa Кф)//?сл1; (389)
Руд = *W(1 - п) = (4р Qa /Сф)/( 1 - П) Ясл. (390>
Значения эффективного коэффициента температуропро-
водности находятся в пределах (3—4) • 10-6 м2/с, что соот-
ветствует значениям произведения 4pQa, равным 1200—
1600 кВт/м. Следовательно, при п«0,7 и /Сф=0,5-т-2,0
рабочие значения Руд.в для установок ЭЛП с радиусом
234
слитка >5 см, как правило, ниже значений Ркр.в. В этих
случаях понижение Руд.в, а следовательно, при данном п
понижение Руд.в и Руд должно приводить к улучшению ра-
финирования. Вместе с тем в ряде случаев бывает необхо-
димо вести процесс так, чтобы сохранять ценные легиру-
ющие, например, хром. В этом случае необходимо, чтобы
Руд.в== Ркр.в, а между радиусом слитка и величиной п име-
ло место соотношение:
&Тmax Ра QK<t>
Т Р
УД.в 1 пл хсл
(391)
Таким образом, при увеличении радиуса слитка и при
неизменном коэффициенте формы ванны для обеспечения
минимальных потерь летучих легирующих, необходимо по-
вышать долю мощности, подаваемой на ванну. Если же
эта доля остается постоянной, то для сохранения мини-
мальных потерь необходимо увеличение коэффициента
формы ванны. В целом ЭЛП позволяет удалять из метал-
ла до 85—95 % водорода, 70—90 % азота, 80 % меди, 90—
95 % сурьмы, 98—99 % свинца. Потери марганца в сталях
могут достигать 60—80 %, а в никелевых сплавах состав-
ляют 30—50 %. Вместе с тем потери при оптимальных с
точки зрения этих потерь режимах переплава могут не пре-
вышать 20 %. Содержание кремния, серы и фосфора после
ЭЛП практически не изменяется. Общие потери металла
на испарение при ЭЛП составляют 4—5 %.
Удаление неметаллических включений
Закономерности удаления неметаллических включений при
ЭЛП аналогичны тем, которые имеют место при ВДП при
аналогичных температурных режимах. Вместе с тем при
ЭЛП в области фокального пятна, где температура превы-
шает среднюю эффективную, может иметь место активное
испарение некоторых субоксидов, имеющих высокое дав-
ление насыщенного пара (NbO, ВО, ZrO, SiO, А1О), обра-
зующихся по реакциям:
МеД(т,Ж) = х{МеО} + (у — х) 101; (392)
МехОу{т,х} + (у - х) [С] = х {МеО} + (у-х) {СО}. (393)
Общее снижение содержания неметаллических включе-
ний при ЭЛП составляет от 50 до 90 % в зависимости от
характера включений и режима переплава. Более низкие
остаточные давления в зоне плавки, чем при ВДП, более
быстрый массоперенос в пленке на торце заготовки, более
235
длительная выдержка металла в жидком состоянии и воз-
можность более высокого перегрева создают при ЭЛП
благоприятные условия для восстановления неметалличе-
ских включений углеродом. При этом возникает возмож-
ность глубокого вакуумного обезуглероживания особониз-
коуглеродистых сталей [194].
Глава IX
КРИСТАЛЛИЗАЦИЯ И ФОРМИРОВАНИЕ
ЭЛЕКТРОСТАЛЕПЛАВИЛЬНОГО СЛИТКА
В настоящее время распространены различные методы по-
лучения слитков. Основную часть слитков все еще получа-
ют при разливке металла в изложницы. Однако с каждым
годом все большее распространение получают машины не-
прерывного литья заготовок (МНЛЗ) и машины полуне-
прерывного литья заготовок (МПНЛЗ), а также методы
специальной электрометаллургии. Во всех этих случаях
металл затвердевает в медных водоохлаждаемых кристал-
лизаторах. Закономерности формирования слитков во всех
перечисленных случаях имеют, с одной стороны, много
общего, а с другой — характеризуются рядом особеннос-
тей. Например, при разливке металла в изложницы фор-
мирование слитка практически начинается после того, как
вся изложница заполнена металлом. Поступления новых
порций металла в изложницу в процессе затвердевания
слитка не происходит. По аналогии с терминологией ли-
тейного производства, где отливка, получаемая в подобных
условиях, называется отливкой разового формирования,
такой слиток можно характеризовать как слиток разового
формирования (СРФ). В слитках же, получаемых на
МНЛЗ, МПНЛЗ и в переплавных процессах, затвердевание
металла сопровождается непрерывным поступлением в
кристаллизатор новых порций жидкого металла. При этом
в зависимости от конкретных условий массовая скорость
затвердевания может быть равна массовой скорости по-
ступления жидкого металла, может быть меньше ее и, на-
конец, может ее превышать.
В случае равенства этих скоростей имеет место так на-
зываемый квазистационарный процесс формирования слит-
ка. В случае их неравенства процесс формирования слитка
нестационарен. Квазистационарный процесс имеет место
при получении слитков на МНЛЗ и при разных видах про-
236
цессов переплава при малой скорости наплавления слит-
ков. Слитки, полученные в этих условиях, можно характе-
ризовать как слитки непрерывного формирования. Слитки,
полученные МПНЛЗ, а также при переплаве с повышенной
скоростью наплавления, формируются в нестационарных
условиях затвердевания. Такие слитки можно отнести к
слиткам полунепрерывного формирования. Одной из важ-
нейших задач, стоящих перед металлургами, является по-
лучение слитков с максимальной химической и физической
однородностью металла.
IX.1. ЗАТВЕРДЕВАНИЕ СЛИТКА
РАЗОВОГО ФОРМИРОВАНИЯ
Слиток разового формирования, отлитый в изложницу, ха-
рактеризуется тем, что основная часть его тела кристалли-
зуется при радиальном отводе тепла к стенкам изложницы.
Вертикальная составляющая скорости кристаллизации при
этом практически отсутствует. Точка на фронте кристалли-
зации движется горизонтально, и вектор ее скорости по
величине и направлению совпадает с вектором скорости
утолщения твердой корочки металла в данном горизон-
тальном сечении слитка. Обычно для определения толщи-
ны корочки затвердевшего металла £, образующейся при
кристаллизации стального слитка, пользуются так назы-
ваемой формулой квадратного корня; £=/<рт (т — вре-
мя; К — постоянный коэффициент, подобранный эмпири-
чески). К, по данным различных авторов, колеблется в
зависимости от марки стали в пределах 1,7—2,7 см/мин0'5
(0,220—0,362 см/с0-5). Однако практика показывает, что
закон квадратного корня выполняется не всегда. Достаточ-
но хорошо описывая начальную стадию затвердевания пе-
риферийной зоны слитка, этот закон дает отклонения от
эксперимента при рассмотрении затвердевания централь-
ной зоны.
Кроме того, значения коэффициента К, подобранные
эмпирически, не могут полностью отразить влияние таких
факторов, как теплофизические свойства затвердевающей
стали и условий разливки, в частности температуру раз-
ливаемого металла.
Для вывода зависимости между величинами £ и т в
работе* были сделаны следующие допущения: 1) теплоем-
1 Белянчиков Л. Н. Формирование слитка спокойной стали (раз-
дел курса «Производство стали и ферросплавов». Курс лекций). М.,
МИСнС, 1975. 143 с. с ил.
237
кость и теплопроводность металла не зависят от темпера-
туры и фазового состояния металла; 2) температура в
затвердевшей корке изменяется по линейному закону.
С учетом принятых допущений решение уравнения тепло-
вого баланса имеет следующий вид:
/ & У_______2_ S + 2С (Д/кр + Д/) / I У +
\ *сл / 3 2 + С (Д/кр + Д/) л \
*СЛ /
С (Д/вр + А/) / I у ss
2 -f- С (Д/цр Д/) \ Ren. )
* X Д/с—и т*
(394)
где 2 — скрытая удельная теплота кристаллизации; С —
удельная теплоемкость жидкого металла; Д/кр— темпера-
турный интервал кристаллизации; Д/— перегрев металла
над температурой ликвидуса; X — коэффициент теплопро-
водности твердого металла; Д/с-и— перепад температур
между слитком и изложницей, в среднем Д/с-и=275К;
р — плотность металла; т— время; Bi — критерий Био.
За исключением последнего момента, когда в центре
слитка затвердевает двухфазная зона, затвердевание слит-
ка (отливки) протекает при постоянном значении крите-
рия Био [195]. Обработка экспериментальных данных пока-
зывает, что для стальных слитков, отлитых в изложницу,
можно принять Bi=0,74. Длительность полного затверде-
вания слитка обозначим через те и определим из условия
т=т2при £=ЯСЛ:
«, = рШ2 + 2С (Д(кр + Д/)]/(6ШмЫ).
(395)
Известна эмпирическая формула В. М. Тагеева, обоб-
щившего многочисленные экспериментальные данные по
затвердеванию спокойной стали в изложнице: те —
=0,112 /?сл, где те —продолжительность затвердевания
•слитка, мин; Rcn— радиус слитка, см. Проведем сравне-
ние результатов, получаемых по формуле (395) и по фор-
муле В. М. Тагеева, на примере стали ШХ15. Примем для
этой стали следующие значения теплофизических констант:
2 = 270 кДж/кг; Х = 34Вт/(м-К); С = 645 Дж/(кг-К),
Д^кр=120К; р=6950 кг/м3. Перегрев № принимаем рав-
ным 0—50 К. Получим те = (0,1074-0,123) R2 мин. При
Д/=20 К результат совпадает с данными В. М. Тагеева.
В безразмерных координатах £//?—т/те уравнение затвер-
«38
девания слитка принимает вид:
( I у______2_ 2 4- 2С (Д/ир + AQ / Е V ,
^/?сл/ 3 24-С(Д/кр + Д/) ’\/?сл/
С (Д/кр + ДО ( В У =
S + С (Д^кр 4" Др \ Ren /
_ 2 4- 2С (Д/вр + Др , т (396)
3 is + С (Д/кр + Д/)] ’ т2
На рис. 54 показана зависимость £/7?сл от т/т2 для
стали ШХ15 при различном перегреве металла над тем-
Рис. 54. Расчетное изменение толщины твердой коронки в процессе затвердева-
ния слитка разового формирования стали ШХ15 при перегреве 0 (/), 20 (2), 70
(3), 120 К (4) и теоретическая зависимость, соответствующая закону квадратного
корня (5)
Рис. 55. Сравнение экспериментальных (1—3) и расчетных (4, 5) данных об из-
менении толщины твердой корочки в процессе затвердевания слитка разового
формирования диаметром 125 (/), 200 (2) и 400 (3) мм:
4 — закон квадратного корня; 5 — расчет по формуле (256)
пературой ликвидуса при разливке. Наряду с результата-
ми, полученными по формуле (396), на рис. 54 приведен
график, соответствующий закону квадратного корня. За-
кон квадратного корня тем лучше описывает процесс за-
твердевания слитка, чем выше перегрев металла, шире ин-
тервал кристаллизации, больше удельная теплоемкость
металла и меньше скрытая удельная теплота кристаллиза-
ции. На рис. 55 приведены экспериментальные данные, по-
лученные Н. И. Хвориновым [196] в сравнении с расчетными
кривыми. Расчеты выполнены по формуле (396). Сов-
падение расчетных данных с экспериментом удовлетвори-
239
тельное. Полученная зависимость толщины затвердевшей
корочки от времени позволяет определить скорость продви-
жения фронта кристаллизации в слитке разового формиро-
вания. Если слиток имеет форму цилиндра, то вектор ско-
рости продвижения фронта кристаллизации по величине и
направлению совпадает с вектором скорости затвердева-
ния. В этом случае
dt
S + 2С (Л/кр + AQ #сл
__________________6 [S -г С (А/кр + AQ] ' ______________________
Е Г Z + 2С (А/КР + AQ I 2С (А/кр + AQ
^?сл . 2 -f- С (AfKp А/) /?сл S -f- С (А/цр -р А/) \ /?сл / .
На рис. 56 показано изменение безразмерной скорости -
кристаллизации по сечению слитка стали ШХ15 при раз-
личных степенях перегрева металла. Под безразмерной
скоростью кристаллизации подразумевается величина
ft v //?сл- Как видно из рис. 56, скорость кристаллизации
в начальный момент затвердевания слитка бесконечно ве-
Рис. 56. Изменение скорости кри-
сталлизации по сечению слитка ра-
зового формирования при перегре-
ве 0 (/). 20 (2), 70 (3) и 120 К (4)
лика. Затем по мере нараста-
ния твердой корочки скорость
кристаллизации снижается.
При малых значениях перегре-
ва скорость кристаллизации
проходит через минимум при
определенном значении £//?Сл,
а затем начинает возрастать.
При больших степенях пере-
грева скорость кристаллиза-
ции от периферии к центру
слитка убывает монотонно.
Условие появления минимума
на кривой скорости кристал-
лизации определяется выраже-
нием Д/4-Д/Кр<2/ЗС, т. е. по-
явление минимума значения v
тем вероятнее, чем меньше степень перегрева металла, уже
его интервал кристаллизации, меньше удельная теплоем-
кость и больше скрытая удельная теплота кристаллизации.
Интерес для современной специальной металлургии пред-
ставляет процесс получения полых слитков (трубных за-
готовок) в медных водоохлаждаемых кристаллизаторах.
В этом процессе затвердевание слитка проходит как по на-
.240
правлению от наружных слоев к внутренним, так и от внут-
ренних слоев к внешним. При этом, если в первом случае
площадь поверхности затвердевания постоянно уменьша-
ется, то во втором случае она возрастает. Изменение тол-
щины затвердевающей корочки при затвердевании, идущем
от периферии в глубь заготовки, описывается уже извест-
ной формулой (396). Для корочки, образующейся при за-
твердевании внутренней полости заготовки, эта зависимость
несколько видоизменяется:
/_Ц? _i_ 2 + 2С (&кр + AQ /_£\з _
\г ) 3 ’ S + С (Д/кр + ДО. \ г )
______С (Д/Ир + ДО г /_g_\< _ 2 Bi A.Afc_H т ,
2 [2 + С (Д/кр + Др] Яо-Д'/ р [S + С (Д/кр + Др] г? ’
где г — радиус внутренней цилиндрической полости; Ro—
радиус окружности, на которой встречаются фронт затвер-
девания, идущий от периферии к центру, с фронтом, иду-
щим от центра к периферии.
IX.2. ЗАТВЕРДЕВАНИЕ НЕПРЕРЫВНОГО
И ПОЛУНЕПРЕРЫВНОГО СЛИТКОВ
Затвердевание слитков, получаемых на МНЛЗ и МПНЛЗ,
характеризуется тем, что передачей тепла в осевом направ-
лении можно пренебречь, поскольку длина слитка в десят-
ки (для МПНЛЗ) и даже сотни (для МНЛЗ) раз превы-
шает его радиус. Для слитков, полученных переплавом, при
длине их, в 2,5—3 раза превышающей радиус, осевым от-
водом тепла также можно пренебречь. Ванна жидкого ме-
талла в непрерывных и полунепрерывных слитках в общем
случае состоит из двух частей. Верхняя часть, в которой
жидкий металл непосредственно прилегает к стенке кри-
сталлизатора или гарнисажу из паров и' брызг, образую-
щемуся на стенке, называется цилиндрической частью.
В этой части ванны металл, отдавая тепло перегрева, еще
не кристаллизуется. Глубину этой части ванны обозначим
Нц (рис. 57). Нижняя часть ванны, условно называемая
конической, соответствует участку затвердевания металла.
Здесь жидкий металл контактирует с корочкой затвердева-
ющего металла. Глубину этой части ванны обозначим Нк
(см. рис. 57).
Для квазистационарных условий в непрерывном слитке
HK=g/(4npa), или HK=Rlnvo/4a, где g — массовая ско-
рость подачи металла в кристаллизатор, кг/с, кг/мин, г/с;
16—675
241
vq — линейная скорость наплавления слитка; а — эффек-
тивный коэффициент температуропроводности. Массу кони-
ческой части ванны при квазистационарном режиме мож-
но определить из выражения: GCt.K=gRcn /8а; (?ст.к=
=л/?слрао/8а. Зависимость
скорости плавки на приме-
ре слитков ВДП показана
на рис. 58. При нестационар-
ном процессе в полунепре-
величины Ост.к от массовой
1 Z 3 4 3 6 7 В
д, кг/мин
Рис. 58. Зависимость массы конической части слитка ВДП от массовой скорости
плавки при квазистационарном режиме для разных сталей и сплавов
—380 мм):
1 — ХН77ТЮР; 2—12Х21Н5Т; 3 —08Х18Н10Т
Рис. 57. Фронт кристаллизации в непрерывном слитке
рывном слитке
Ск= —
8а
лп2 и2
Я^сл Mi
2 ,
+ 2L? /
(и2
।__1*1
3
(397)
где gvp — массовая скорость кристаллизации; L — длина
накристаллизовавшегося слитка; /Сол— температура соли-
дуса; pi — коэффициент, зависящий от теплофизических
свойств металла и условий теплообмена между слитком и
кристаллизатором: |л5ф=Ц14-(л2/?сл/4£2). Коэффициент
Hi зависит от критерия Био и является первым корнем
уравнения: Jo(p)//i(pi) =p./Bi, где /о(ц) и Л(л)—цилин-
дрические функции первого рода нулевого и первого по-
рядка соответственно. Изменение общей массы жидкой
242
ванны G подчиняется следующему закону:
где Мах—максимальная температура поверхности ванны
при данном режиме переплава; /п.с— средняя температура
поверхности ванны.
На рис. 59—61 приведены примеры изменения величин
G, GK и Оц (масса цилиндрической части ванны) при ВДП
Рис. 60. Изменение массы конической части ванны по мере наплавления слитка
ВДП сплава ХН77ЮР (цифры у точек — ток переплава)
Рис. 59. Изменение общей массы жидкой ванны по мере наплавления слитка
ВДП сплава ХН77ТЮР (цифры у точек — ток переплава)
коррозионностойких сталей. Аналогичная закономерность
имеет место и при других видах переплава (ЭШП, ЭЛП,
ПДП) с той только разницей, что в этих процессах получа-
ет большое развитие цилиндрическая часть ванны. Это свя-
зано с тем, что при ЭШП, ЭЛП и ПДП доля тепла, посту-
пающего в ванну путем нагрева, больше, чем при ВДП.
16*
243
При ВДП основная доля тепла поступает в ванну с кап-
лями жидкого металла.
Такой вид зависимости Gх от L связан с изменением
тепловых потерь ванны по мере наплавления слитка. Поте-
ри тепла через поддон при этом уменьшаются. Через боко-
вую поверхность слитка потери тепла сначала возрастают
Рис. 62. Изменение массовой скорости кристаллизации по мере наплавления
слитка ВДП сплава ХН56ВМКЮ (£>кр "“105 мм):
/ — 1,2 кА; 6,3 г/с; 2— 1,6 кА, 10,5 г/с; 3 — 2 кА, 15,2 г/с; 4 — 2,4 кА, 17,8 г/с
Рис. 61. Изменение массы цилиндрической части ванны по мере расплавления
слитка ВДП сплава ХН77ТЮР (цифры у точек — ток переплава)
пропорционально L, а затем их рост замедляется, так как
нижние (более холодные) части слитка имеют меньшую
теплоотдачу, чем верхние. Общие потери в единицу време-
ни при этом минимальны при определенном значении L, а
затем возрастают, ассимтотически приближаясь к некото-
рому стабильному значению. При малой общей глубине
ванны (плавка на относительно малых токах) затухание
роста потерь через боковую поверхность начинается рано,
и общие потери монотонно (без минимума) уменьшаются
по мере наплавления слитка. Соответственно общая масса
ванны либо имеет максимум (при повышенных токах),
либо монотонно возрастает, стремясь к некоторому стабиль-
ному значению. Коэффициент а зависит от условий охлаж-
дения слитка, а следовательно, и от его диаметра (радиу-
са). Ориентировочно можно принимать, что lga=lgo<j—
— 1,8ЯСЛ, где ао — коэффициент температуропроводности ме-
талла при /сол; /?сл — радиус слитка, м.
При наплавлении слитка в вакууме критерий Био мож-
244
но оценить из второго выражения, связывающего щ и Bi:
gj _ 2/?сл (z °о епр^сол)
лА. {^сол [ 1 4* Jo (Hi)] 300}
где епр — приведенная степень черноты епр~0,56; о0 — по-
стоянная Стефана — Больцмана; х=1260 (1570—^сол),
Вт/м2.
Наличие системы двух уравнений, связывающих ц! и
Bi, позволяет определить значения этих величин в каждом
конкретном случае.
На рис. 62 приведен пример изменения массовой скоро-
сти кристаллизации по мере наплавления слитка в неста-
ционарном режиме. Под профилем жидкой ванны метал-
ла обычно понимают профиль конической части ванны. От-
ношение HK/R называется коэффициентом формы ванны.
Найти уравнение профиля конической части можно из урав-
нения (396). Пусть фронт кристаллизации продвигается
вверх вдоль оси слитка со скоростью и0. На оси слитка век-
тор продвижения фронта кристаллизации по величине и по
направлению совпадает с вектором скорости продвижения
твердой фазы в осевом направлении.
Коническая часть жидкой ванны полностью закристал-
лизуется при этом за время —Hk/vq, которое соответ-
ствует длительности продвижения твердой фазы от края
слитка до его оси. Если на фронте кристаллизации выбрать
точку D (см. рис. 57) с координатами г и у, то твердая ко-
рочка толщиной £ нарастает за время, пока точка D пере-
мещается по вертикали на величину Нк—у, т. е. за время
т=Нк—y/v0. Отсюда т/тх = 1— (у/Нк).
Профиль конической части жидкой ванны удобнее вы-
ражать не в координатах £//?—у/Нк, а в координатах
r/R—у/Нк. В этих координатах уравнение профиля жид-
кой ванны имеет вид:
У = ! 3 [S + С (Д/кр + AQ] Л/\2
Нк S + 2С (Д/ир + ДО \ Дел/
, 2 А_____{_\3 _ ЗС (Д/Кр -I- ДО Л_____
\ ^?СЛ ') S 4-2С (Д/кр + др \
/?СЛ /
Если учесть, что С(Д/Кр+А0<2, то эту формулу мо-
жно существенно упростить:
у/Нк «(1 4- п) (гЧИи ~ п ’ <399)
где п колеблется от 0 до 2. Профиль конической части ван-
ны, рассчитанный по уравнению (399) для п—\, показан
на рис. 63.
245
Непрерывные и полунепрерывные слитки непрерывно
наращиваются в кристаллизаторах. Твердая фаза в этом
случае продвигается не только от периферии к центру, но
и вдоль оси слитка. Осевая составляющая скорости про-
движения фронта кристаллизации влияет на процесс за-
твердевания непрерывного и полунепрерывного слитков.
Рис. 63. Профиль конической
части ванны в непрерывном слитке
сечению непрерывного слитка
Рис. 64. Изменение скорости кристаллизации по
(Л'ф = Я 7?=1)
Под скоростью в данном случае следует понимать ли-
нейную скорость продвижения фиксированной точки на
фронте кристаллизации в направлении, перпендикулярном
поверхности фронта. Таким образом, эта точка движется
одновременно и по вертикали, и по горизонтали.
При этом радиальная и вертикальная составляющие
вектора скорости кристаллизации не совпадают по вели-
чине с радиальной и вертикальной скоростями продвиже-
ния твердой фазы в данном горизонтальном или вертикаль-
ном сечении.
Рассмотрим схему на рис. 57. На нем представлены в
вертикальном разрезе два положения поверхности фронта
кристаллизации. Пусть их разделяет во времени 1 с. Тогда
путь, пройденный любой точкой от первой поверхности до
второй, равен скорости этой точки в данном направлении.
Возьмем на первой поверхности точку Л. За 1с она пе-
реместится на вторую поверхность в точку Л'. Скорость
перемещения точки Л, нормальная к поверхности фронта
кристаллизации (vn) численно равна отрезку ЛЛ'. В ра-
диальном направлении изменение координаты точки Л опи-
шется отрезком АС, а в вертикальном — отрезком АВ. Вме-
246
сте с тем твердая фаза за это время переместилась в ра-
диальном направлении на расстояние АС'. Этот отрезок
численно равен скорости перемещения твердой фазы в ра-
диальном направлении vr. В вертикальном направлении
твердая фаза переместилась па расстояние АВ', численно
равное скорости продвижения твердой фазы вдоль оси слит-
ка Vo.
Поскольку при квазистационарном процессе фронт кри-
сталлизации переместится параллельно самому себе, то ве-
личина у0 постоянна по всему сечению слитка. Нетрудно
заметить, что АВ<АВ' и АС<АС'. Таким образом, ско-
рость перемещения точки на фронте кристаллизации в ра-
диальном и горизонтальном направлениях меньше скорости
продвижения твердой фазы в этих направлениях.
В этом существенное отличие условий затвердевания не-
прерывного и полунепрерывного слитков от условий за-
твердевания слитков разового формирования.
Вместе с тем скорость оттеснения примесей фронтом
кристаллизации определяется величиной и направлением
вектора vn (т. е. отрезком АА'). Следовательно этот век-
тор и его составляющие АВ и АС являются определяющи-
ми скоростями при кристаллизации в непрерывном и
полунепрерывном слитках. Проведем касательную к поверх-
ности фронта кристаллизации в точке А. Пусть она обра-
зует с горизонталью угол а. Тогда очевидно, что ДЛ'=='
=АВ' cos а, т. е. yn = y0cosa. Если профиль фронта кри-
сталлизации описывается уравнением у=у(Нк, r/Rc:i), то
tg a=i/', отсюда vn — vjV 1 + (t/')2.
На рис. 64 показано изменение величины уп/у0 по се-
чению слитка, когда профиль фронта кристаллизации (про-
филь конической части ванны) описывается уравнением
(399) для различных значений п. Если и>0, то скорость
продвижения фронта кристаллизации имеет минимум в се-
чении слитка, а на периферии и на оси слитка она совпа-
дает по направлению и величине с вектором у0-
Если п«0, то величина vn максимальна на оси слитка,
где она совпадает с у0, а от оси к периферии монотонно
уменьшается.
IX.3. ДВУХФАЗНАЯ ОБЛАСТЬ
При затвердевании чистых металлов или эвтектических
сплавов в слитке образуется перемещающийся фронт кри-
247
сталлизации, отделяющий твердый металл от жидкого.
В сплавах, кристаллизующихся в интервале температур,
затвердевание происходит в переходной, так называемой
двухфазной области. Ширина двухфазной области ф зави-
сит от интервала кристаллизации Д6ф=6шк—/сол и тем-
пературных градиентов в районе изотерм ликвидуса и со-
лидуса (57лик и Vco.n).
Доля твердого металла в двухфазной области изменя-
ется от 0 при (лик до 100 % при /Сол. При этом достаточно
прочный скелет из сросшихся осей развивающихся ден-
дритов формируется при образовании в жидкости пример-
но 20 % твердой фазы. Этому состоянию соответствует тем-
пература, которую обозначим /0>2.
Двухфазную область в интервале температур /Сол—to.?.
назовем твердо-жидкой зоной (областью), а область в ин-
dr
R
Рис. 65. Схема распределения фаз в затвердевшем слитке
1,0
r/fi
Рис. 66. Изменение протяженности двухфазной области в непрерывном
(Кф=Я;Я=1)
слитке
тервале температур /0,2—Лдик назовем жидко-твердой зо-
ной (областью). Наибольшее число различных дефектов
возникает в слитке при его охлаждении в двухфазной об-
ласти. Рассмотрим процесс кристаллизации металла в ча-
сти слитка высотой dl (рис. 65).
Пусть на расстоянии г от оси слитка градиент темпе-
ратуры равен V, а на расстоянии r-\-dr градиент равен
V-J-dV. Поскольку жидкий металл в двухфазной области
пронизан растущими кристаллами и не принимает участия
в циркуляции, то конвективной передачей тепла здесь мо-
жно пренебречь и считать, что теплопередача в этой области
осуществляется только теплопроводностью. Решение для
248
этого случая дано М. И,- Мусатовым Количество тепла,
проходящего за время dr через цилиндрические поверхно-
сти радиусом г и r-\-dr, обозначим соответственно dQi и
rfQ2. Тогда dQi=—2nXrVdldr\ dQ2=—2n%(r+dr) (V+
. За время dr во всей двухфазной зоне закри-
сталлизуется металл массой dM. В рассматриваемом эле-
ментарном кольцевом объеме толщиной dr выделится ко-
личество тепла, пропорциональное изменению доли закри-
сталлизовавшегося в данном кольце металла. Обозначим
это количество тепла через dQ3. Тогда dQ3='£dMdN (S —
скрытая удельная теплота кристаллизации; dN — измене-
ние доли закристаллизовавшегося металла в элементарном
кольце за время dr). Это количество тепла пройдет через
цилиндрическую поверхность, ограничивающую область со
стороны полностью затвердевшего металла и имеющую ра-
диус (7?сл—£)• Обозначим количество прошедшего через
эту поверхность тепла через dQt: dQ* = —2лА, (/?Сл —
—D^condNdldr, где Vco.n— градиент температур на гра-
нице двухфазной области с полность затвердевшим метал-
лом, т. е. градиент температур при температуре солидуса.
Очевидно, что dQs=—dQ^. Тепловой баланс в рассматри-
ваемом элементарном объеме двухфазной области можно
представить в виде любого из двух следующих уравнений:
dQ2—d(b = dQs, (400)
dQ2 — dQr=— dQ3. (401)
Применительно к слитку разового формирования с не-
стационарным температурным полем удобнее воспользо-
ваться уравнением (400), так как в величину dQt входит
градиент VСол, величина которого для слитка разового
формирования легко определяется. Для наплавляемого
слитка, температурное поле которого квазистационарно,
удобнее пользоваться уравнением (401). Как уже говори-
лось выше, для слитка разового формирования можно
принять линейное распределение температуры в корочке пол-
ностью затвердевающего металла. Тогда \7Сол=(^сол—
—^пов)/£, где /пов — температура боковой поверхности слит-
ка. Но, как показано в работе [197], /Пов=^сол/(1+ЕИ)..
Следовательно,
V», = ® 0.425 Ьь. (402)
I w чЭ
1 Мусатов М. И. Некоторые металлургические основы дуговой
плавки титана с расходуемым электродом. Автореф. канд. дис. М.»
1961.
249
Решение уравнения (400) для слитка разового форми-
рования дает следующие результаты:
(403)
где т — отношение перепадов концентраций между изотер-
мами ликвидуса и солидуса при температурах ликвидуса
и солидуса для данного состава сплава.
Поскольку для большинства бинарных сплавов железа
/п=0,2ч-0,5, то велиична фт_ж составляет в этих сплавах
90—95 % от ф. Решение уравнения (401) для непрерывных
слитков проведено в работе1. Протяженность двухфазной
области в данном случае равна:
ф ж
{4/7K/[p?J/ 1 +ОТ]}.
(404)
Из выражения (404) следует, что величина ф в непре-
рывных и полунепрерывных слитках изменяется по более
сложному закону, чем в слитке разового формирования.
На рис. 66 показано изменение безразмерной протяженно-
сти двухфазной области <р/фо (фо—протяженность двух-
фазной области на оси слитка) по сечению непрерывного
слитка. Принято, что профиль жидкой ванны описывается
уравнением (399). Из рис. 66 следует, что в сечении слит-
ка имеет место минимум ширины двухфазной области в на-
правлении, нормальном к фронту кристаллизации. Положе-
ние этого минимума соответствует условию у"=0. При
этом неравномерность протяженности двухфазной области
увеличивается с ростом относительной глубины жидкой
ванны. Только при n—Q протяженность двухфазной области
максимальная в центре слитка и монотонно убывает от
центра к периферии.
1Х.4. КОНЦЕНТРАЦИОННОЕ ПЕРЕОХЛАЖДЕНИЕ
В сплавах перед фронтом растущих кристаллов имеет мес-
то концентрационное уплотнение, т. е. слой с повышенным
содержанием примеси. Допустим, что температура ликви-
дуса представляет собой прямую линию с тангенсом угла
наклона т. Тогда на расстоянии х от фронта кристалли-
зации температура ликвидуса /Л1Ш должна быть равна:
^ппк(х)=^о—/псж(х), где /0 — температура плавления чис-
1 Белянчиков Л. Н. Закономерности кристаллизации наплавляемого
слитка. М., МИСнС, 1981. 108 с. с ил.
250
того растворителя; сж(х)—концентрация примесив точ-
ке х.
Концентрация примеси изменяется в концентрационном
уплотнении вдоль оси х по следующему закону:
(1 + ЦД e~vx'D* j, (405)
где v— скорость продвижения фронта кристаллизации;
К — эффективный коэффициент распределения примеси;
DM— коэффициент диффузии примеси в жидкой фазе;
сук — концентрация примеси при х большем, чем толщина
дифузионного слоя б, т. е. при х^Ь. Тогда
U. (х) = t0 - сж {1 + К1 - K)/KI e~vx,D*). (406)
Поскольку сж(х) уменьшается с увеличением х, равно-
весная температура 6шк(х) должна при этом увеличивать-
ся. Однако реальное распределение температуры в жидкой
фазе /ж(х) не обязательно должно совпадать с равновес-
ным. В простейшем случае распределение температуры мо-
жно записать /ж(х) = /ж(0)4-^Я(х, где /ж(0)—температу-
ра на фронте кристаллизации; V>k — температурный гра-
диент в жидкой фазе. Величину можно определить
из формулы (406) при условии х=0: Лк(0) =/Лик(0) =
— to—тсж(}/К). Отсюда
^ж (х) — Iq /лсж/К 4" Vh< х.
(407)
Очевидно, что если в каком-то объеме жидкости будет
соблюдаться соотношение /ж(х) </лпк(х), то жидкость в
этом объеме оказывается переохлажденной по отношению
к равновесной температуре ликвидуса. Это явление полу-
чило название концентрационного переохлаждения в от-
личие от переохлаждения термического. В районе, приле-
гающем к фронту кристаллизации, при х=0 условие соз-
дания концентрационного переохлаждения принимает вид
#ж (х)
dx
х=0
*^лик (*)
dx
(408)
Продифференцировав выражения (406) и (407) по х
получим на основании уравнения (408) условие существо-
вания концентрационного переохлаждения в районе изо-
термы ликвидуса:
?лшЛ < (mcn/Dn) [(1 - К)/К1. (409)
И, наборот, концентрационное переохлаждение должно
отсутствовать при условии
?лИК/У > (tncJDH<) [(1 - к)/К].
(410
251
IX.5. ДИСПЕРСНОСТЬ ДЕНДРИТНОЙ СТРУКТУРЫ
И ДЕНДРИТНАЯ ЛИКВАЦИЯ
В черной металлургии приходится иметь дело со сталями
и сплавами, у которых концентрации примесей легирую-
щих элементов так велики, что в реальных слитках плос-
кий и ячеистый фронт кристаллизации существуют малое
время. Структура таких слитков формируется как дендрит-
ная структура. Важным вопросом при рассмотрении про-
цесса дендритной кристаллизации является вопрос о раз-
мерах дендритов, расстоянии между их главными осями
л1 и осями второго порядка х11. Концентрация примеси на
оси дендрита соответствует ее концентрации в вершине
дрендрита, кристаллизующейся в первую очередь и долж-
жна быть минимальной. Обозначим эту концентрацию
cm)n. Баланс примеси в концентрационном уплотнении, при-
веденной в работе [198], дает следующее выражение для
Cmlib
^mln = К KVniiK D)K/tnv) -|- Сж]. (411)
Отношение Cmrn/Сж назовем эффективным коэффициен-
том перераспределения и обозначим КЭф, т. е. КЭф=ст1п/сж.
Тогда
Кэф — К [1 4“ (Vлик 01-
(412)
Эффективный коэффициент распределения КЭф, как впер-
вые было показано в работе [199], связан с равновесным
коэффициентом распределения Ко следующим образом:
КэФ = КДо + (1 - Ко) ехр (- пбад. (413)
После того как дендрит выбросил ось второго порядка,
вершина главной оси продолжает расти. Концентрация
примеси в жидком металле вдоль главной оси по мере уда-
ления от оси второго порядка меняется в соответствии с
формулой (405). Когда концентрация сж(х) достигнет зна-
чения cmin/K, дендрит выбросит следующую вторичную ось.
Приравнивая выражение для cm(x) и стш, получим
х11 = (Рж/цп) In [(1 - К)/(Кэф - К)],
(414)
где о11 — скорость утолщения осей второго порядка.
Обозначим скорость роста осей первого порядка че-
рез пт.
А. Е. Волков показал, что соотношение между и11 и п1
подчиняется закономерности: ип/о1=(Кэф—К)/(1—К).
Отсюда, подставляя это выражение в формулу (413) и рас-
252
крывая значение КЭф, получаем:
-<мож
~ /ПСЖ у(1 —К
Улик Кд
е
д.п__ /1 — Ко
Улик \ Ко
Конкретный вид функции хп(и) во многом определяет-
ся видом функции V.4hk(v) и Vaiffi/n. В слитке разового
формирования при линейном распределении температуры
в твердой корочке имеем VnHK«VCOn= (W£)[Bi/(l +
4-Bi)] и с учетом выражения для v получим:
Улик _ Р^сол [2 С (А^кр 4~ AQ1 । 2 -j- 2С (Д/Кр Д/)
v ХД/q—и (1 -}- Bi) _ 2 + С (Д/кр 4“ АО
х I 2С(Д/КР + Д/) £ Я
Ксл 2 + С (Д/кр + ДО ’ •
V*1
• (415)
На рис. 67 показано изменение Vnm(/v по сечению слит-
ка стали ШХ15 при различной степени перегрева. От пери-
Рис. 67. Изменение отношения V лик/^ по сечению слитка разового формирова-
ния стали UIX15 при перегреве 0 (/), 70 (2), 120 (3) и 150 (4) К
Рис. 68. Зависимость степени дисперсности дендритной структуры при кристал-
лизации сплава ХН77ТЮР (/) и стали типа 12Х18Н10Т (//) в кристаллизаторе
диаметром 380 (/), 320 (2) и 500 (3) мм
ферии к центру величина VnaK/n практически всегда моно-
тонно убывает, хотя при условии А/^ (S/2C) — А/Кр она
может проходить через минимум.
Для непрерывного слитка имеем
v™ = (н?rr+OT; vU<-„ = (4HKJZ
/(«.)•
где a — эффективный коэффициент температуропроводно-
сти. Совместный анализ выражений для х11, \7Лик и о в слу-
чае слитка разового формирования показывает, что при
253
прочих равных условиях величина х11 прямо пропорцио-
нальна радиусу слитка, поэтому удобно рассматривать
связь безразмерных величин xTI/Rcn и £//?Сл. От перифе-
рии к центру слитка величина хп//?Сл изменяется в общем
случае сложным образом, проходя через максимум. В ра-
боте* показано, что этот максимум имеет место при
£ _ 2 4-С (Д/КР + А/)
Ясл ~ S + 2C(AfKP + A0
В непрерывных слитках
(14-/2.10-3^).
°п *сл С1 ~ X
4нХиЛо
X In
*о) g—воп/Ож
_ 4Н ^ж а^лт ^о)
Максимум значения xn/R при этом имеет место при ско-
рости продвижения фронта кристаллизации vn, равной ко-
эффициенту массопереноса легирующих примесей р=£)ж/б,
т. е. при оп=р=Рж/6. При малых скоростях кристаллиза-
ции, когда Оп-<р, условие сж(х) =cmin/K обеспечивается
при малых значениях х11, благодаря малому значению ко-
эффициента распределения R. При больших скоростях кри-
сталлизации, когда оп>р, благодаря чему в концентраци-
онном уплотнении резко возрастает градиент концентра-
ции примеси, условие сж (х) = Cmin/К при малых значениях
х11 обеспечивается вследствие роста сж. Выше [см. форму-
лу (352)] было показано, что в наплавляемом слитке ко-
эффициент массопереноса примеси обратно пропорциона-
лен радиусу слитка, т. е. (6оп)/2)ж^= (и/?сл )/Л, где А —
для данной примеси в данном металле есть величина по-
стоянная при данной температуре, т. е. применительно к ус-
ловиям фронта кристаллизации при температуре ликвиду-
са. Отсюда следует, что для данного сплава безразмерная
величина хп//?Сл является функцией произведения vaR. На
рис. 68 показана зависимость между параметрами xn/Rca
и RcnVn для никелевого сплава и коррозионностойкой
стали.
Рассматривая случай диффузии примеси с конечной
скоростью в твердой фазе при неограниченной скорости
диффузии в жидкой фазе, И. Н. Голиков [201] получил
* Белянчиков Л. Н. Формирование слитка спокойной стали (раздел
курса «Производство стали и ферросплавов». Курс лекций). М.: МИСиС,
1975. 143 с.
254
следующее выражение для степени дендритной ликва-
ции гр
(^max ^min)/^o — ^0
и11 х11
Ло
4-1
(416)
где с0 — концентрация примеси в ковшовой пробе; Стах —
концентрация примеси в межосном объеме; стщ — концен-
трация примеси в оси дендрита; Ко— равновесный коэф-
фициент распределения. В реальных условиях коэффици-
ент Ко должен быть заменен эффективным коэффициен-
том К, определяемым для условий затвердевания между
осями второго порядка при и = ип. В этих условиях К~Ко
и формула (416) применима и при ограниченной диффузии
в жидкой фазе.
Отсюда следует, что степень дендритной ликвации опре-
деляется главным образом' фактором ппхп. В непрерыв-
ном слитке
о11 Xй = D
Ж
Ко /
(417)
Это выражение имеет максимум при ип=2р, т. е. оди-
наковая степень дендритной ликвации может быть получе-
на как при малых, так и при больших скоростях кристал-
лизации.
IX.6. ФИЗИЧЕСКАЯ НЕОДНОРОДНОСТЬ
СТАЛЬНОГО СЛИТКА
Усадочная раковина
Понижение температуры жидкой стали, а также ее затвер-
девание сопровождаются уменьшением объема металла.
Различие между объемом, занимаемым ранее жидким ме-
таллом, и объемом твердого металла приводит к возник-
новению в слитке усадочной раковины. Располагается уса-
дочная раковина в районе так называемого теплового цен-
тра, т. е. в самой горячей зоне слитка, затвердевающей в
последнюю очередь. Особенностью наплавляемых слитков
является возможность путем снижения по определенному
закону мощности, подаваемой в зону плавления, практи-
чески полностью вывести усадочную раковину из тела
слитка. При этом в головной части слитка создаются ус-
ловия кристаллизации, близкие к нормальным. Если при
этом не происходит подпитки головной части слитка оплав-
ляющимся электродом, а только греется поверхность ван-
255
ны, что возможно при ЭЛП и ПДП, то длительность вы-
ведения раковины определяется по формуле: тр=/?сл /4а.
В случае ВДП и ЭШП при снижении мощности элек-
трод продолжает оплавляться, и металл продолжает посту-
пать в кристаллизатор, что увеличивает длительность вы-
ведения раковины. В работе (см. сноску на с. 250) показа-
но, что в этом случае
(418)
где go — массовая скорость плавки до начала выведения
раковины; Lo— высота слитка до начала выведения рако-
вины.
В течение Т1=/?сл/4а« (0,554-0,60)тр производят сни-
жение мощности, а при Т2=тР—Ti«(0,40—0,45)тр произ-
водят выдержку при постоянной мощности. При этом пе-
риоды ti и т2 могут быть разделены на несколько стадий.
Пример. Определим длительность выведения усадочной
раковины в слитке стали 12Х18Н10Т при ВДП в кристалли-
заторе диаметром 0,5 м. Исходные данные: а=4,2 10-6 м2/
/с; 2=2,78-10s Дж/кг; %=30 Вт/(м-К); р=7400 кг/м3;
^сол= 1390°C; p,i = l,36; go=O,O9 кг/с; Lo=2,5 м. По табли-
цам цилиндрических функций находим /0(Ц1) =/о(1,36) =
=0,588. Т1=7?^/4а=0,252/(4-4,2-ICb6) =3720с=62мин=
= 1 ч 02 мин.
тр = 3720 1 +
4-4,2-10~«-2,78-106-7400
1,86-30-1390-0,588
М1 -
0,09-2,78-105
6,28-1,86-30-1390-0,588-2,5
= 6708 С = 111,8 МИН «
« 1 ч 42 мин; т2 = тр — = 40 мин.
Осевая пористость
В реальных условиях затвердевающего стального слитка
имеет место двухфазная область, в которой температура
изменяется от ликвидуса до солидуса. Вблизи температу-
ры солидуса кристаллы срастаются, и это приводит к ра-
зобщению ячеек, заключающих в себе остатки жидкой фа-
зы. Поскольку затвердевание таких ячеек происходит без
подпитки жидким металлом, в каждой из них образуется
небольшая усадочная раковина. Множество таких раковин
в совокупности образуют пористость.
256
В слитке особенность этого процесса состоит в том, что
до момента полного срастания кристаллов часть жидкой
фазы успевает профильтроваться через сетку кристаллов из
осевой зоны в периферийную, где металл имеет более низ-
кую температуру. В результате периферийные ячейки по-
лучают некоторое питание от осевых. Осевые ячейки такого
питания не имеют, поэтому в осевой части слитка металл
получается более рыхлым. Дефект этот получил название
осевой пористости. Пористость, развивающаяся в осевой
части слитка непосредственно под зоной усадочной ракови-
ны, называют усадочной рыхлостью. Ширина пористой зо-
ны фпор определяется шириной двухфазной области в мо-
мент достижения фронтом ликвидуса центра слитка. В этом
момент имеет место условие ф=фпор=(7?—фпор) [(1-j-Bi)/
/Bi] (Д/Кр//кр). Отсюда можно найти
^сол
/(0,425 +
^сол // \ ^сол
(419)
т. е. относительная ширина пористой зоны в слитке разо-
вого формирования не зависит от радиуса слитка, а зави-
сит только от интервала кристал-
лизации и температуры солиду-
са. Поскольку повышение содер-
жания углерода в стали расши-
ряет интервал кристаллизации,
то сталь с повышенным углеро-
дом проявляет большую склон-
ность к осевой рыхлости. В не-
прерывных и полунепрерывных
слитках условия подпитки за-
твердевающих ячеек жидким ме-
таллом значительно лучше, чем в
слитках разового формирования.
Здесь над фронтом двухфаз-
ной области всегда имеется ван-
на жидкого металла, который по
капиллярам, образуемым зазора-
Рис. 69. Ширина пористой зоны
в непрерывном слитке:
1 — линия изоликвидуса; 2-ли-
ния изосолидуса
ми между растущими дендрита-
ми, проникает в глубь двухфаз-
ной области. Однако в центральной части слитка процесс
фильтрации жидкого металла затрудняется. На рис. 69
представлен разрез ванны жидкого металла в непрерывном
слитке. Видно, что дендриты, сходящиеся своими вершина-
17—675
257
ми в центре фронта ликвидуса, как бы отсекают некоторую
область в центре слитка, условия подпитки которой хуже,
чем у остальных участков двухфазной области. Очевидно,
что эта зона будет в наибольшей степени поражена усадоч-
ной пористостью. Ширина пористой зоны фПОр в данном
случае может быть определена следующим образом. С од-
ной стороны, из рис. 69 видно, что
“Фпор = ф0 cos a -sin а = фо {//I 1 + (/)2]} • (420)
С другой стороны, значение производной у' = dy/dr в
точке А можно найти из формулы (399) при г=фПОр и п=
=0. Отсюда в условиях затрудненной подпитки имеем:
Фпор __ 1 1 2 ______(421)
Rex | |Xj ^сол 4ЯК
При этом осевая пористость в непрерывном слитке бу-
дет отсутствовать при условии:
1 / /СОЛ
——у — т;
Асл ° а‘КР
Пример. Определим максимально допустимое с точки
зрения осевой рыхлости значение коэффициента формы
ванны • при ВДП стали 12Х18Н10Т. Исходные данные:
Д(кр=56°С; (сол=1390°С; gi=l,86. Отсюда
HK/Rcn < К( 1,86/8) (1390/50) = 2,54.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Жуховицкий А. Л., Шварцман Л. Л. Физическая химия. М.: Ме-
таллургия, 1976. 544 с.
2. Эллиот Д. Ф., Глейзер М., Рамакришна В. Термохимия сталепла-
вильных процессов. М.: Металлургия, 1969. 252 с.
3. Термодинамика. Вып. 85, 97, 103 (Комитет научно-технической
терминологии АН СССР. М.: Наука, 1973; 1980; 1984.
4. Производство стали в электропечах. Пер. с англ. (Под ред. Гри-
горяна В. А. М.: Металлургия, 1965. 424 с.
5. Morris L Р., Zellars G. R.— J. Metals, 1956, v. 8, № 8, р. 1086—
1090.
6. Хансен М., Андерко К. Структуры двойных сплавов: Справочник.
Т. I, II. М.: Металлургиздат, 1962. 1488 с.
7. Эллиот Р. П. Структуры двойных сплавов: Справочник. Т. 1, 2. М.:
Металлургия, 1970. 454; 472 с.
8. Шанк Ф. Л. Структуры двойных сплавов: Справочник. М.: Метал-
лургия, 1973. 760 с.
9. Линчевский Б. В. Техника металлургического эксперимента. М.:
Металлургия, 1967. 344 с.
10. Физико-химические методы исследования металлургических процес-
соъ!Филиппов С, И., Арсентьев П. П., Яковлев В. В., Крашенинни-
ков М. Г. М.: Металлургия, 1968. 552 с.
11. Умбер Д., Эллиот Д. Ф. — Проблемы современной металлургии,
1961, №1 (55), с. 3—23.
12. Сюй Цзен-цзи, Поляков Л. Ю., Самарин Л. М. — Изв. вузов. Чер-
ная металлургия, 1959, № 11, с. 3—12.
13. Срывалин И, T.t Есин О. А., Ватолин Н. Л. и др. — ЖФХ, 1968,
т. 42 № 3 с. 717____722.
14. Наг di Н. К. —Acta Met., 1953, v. 1, р. 202—209.
15. Островский О. И., Стомахин Л. Я., Григорян В. А. — Изв. АН
СССР. Металлы, 1977, № 1, с. 81—85.
16. Lupis С. Н. Р., Elliott J, Г. —Acta Met., 1967, v. 15, № 2, р. 265—
276.
17. Sigworth G. К., Elliott J. F.— Metal Science, 1974, v. 8, p. 298—
310.
18. Ямаучи T., Ичизе Э., Мори 7\ — В кн.: Процессы раскисления и
образования неметаллических включений в стали. М.: Наука, 1977,
с. 147—160.
19. Дюбанов В. Г., Стомахин Л. Я., Филиппов А. Ф.—Изв. вузов.
Черная металлургия, 1975, № 3, с. 5—7.
20. Wolley F., Elliott J. F. —Trans. Met. Soc. AIME, 1967, v. 239,
p. 1872—1883.
21. Fruehan R.— Met Trans., 1970, v. 1, p. 2083—2088.
22. Есин Ю. О., Баев В. M.t Петрушевский M, С., Гельд П, В —
Изв. АН СССР. Металлы, 1975, № 4, с. 82—86; 1974, № 4, с. 73—76.
23. Sponseller D. L.t Flinn R. Л., —Trans, Met. Soc. AIME, 1964,
v. 230, p. 876—887.
24. Selected Values of the Thermodinamic Properties of Binnary Allo-
ysJHultgren R., Desai P. D.t Hawkins D. T.t a. o. ASM, Metals
Park, Ohio, 1973. 1435 p.
25. Есин Ю. О., Завьялов В. К., Петрушевский М. С. и др. —Изв.
. вузов. Черная металлургия, 1973, № 12, с. 23—27.
26. Явойский В. И., Свяжин Л. Г., Вишкарев А. Ф., Чурсин Г. М
В кн.: Взаимодействие газов с металлами. М.: Наука, 1973, с. 98
108.
17*
259
27. Tozaki У., Iguchi У., Ban—ja S., Fuwa T. — Int. Sympos. Met. Chem.
Appl. Ferrous Met, Sheffield, 1971, p. 2—7.
28. Wagner S.f Pierre G. R.— Met. Trans., 1972, v. 3, p. 2873—2878.
29. Furukawa T., Kato E.— Tetsu to Hagane, 1975, v. 61, № 15, p. 3050—
3059.
30. Пупынин Г. П., Сюй Цзен-цзи, Поляков А. Ю., Самарин А. М.—
Металлургия, металловедение, физико-химические исследования:
Науч. тр. ИМет. им. А. А. Байкова. М.: Наука, 1962, № 10, с. 155—
162.
31. Weinstein М., Elliott J. F. — Trans. Met. Soc. AIME, 1963, v. 227,
p. 285—286.
32. Есин Ю. O.t Баев В. M., Гельд П. В., Петрушевский М. С. — Изв.
АН СССР. Металлы, 1974, № 4, с. 73—77.
33. Стомахин A. Я-, Байер П., Поляков А. Ю. — Изв. АН СССР. Ме-
таллы, 1965, № 4, с. 37—45.
34. Аверин В. В., Поляков А. Ю., Самарин А. М.— Изв. АН СССР.
ОТН, 1957, № 8, с. 120—122.
35. Venal Ж V., Geiger G. Я. —Met. Trans., 1973, v. 4, р. 2567—
2573.
36. Триполитов А. И., Крючков О. Н., Стомахин А. Я-, Филип-
пов А. Ф. — Изв. вузов. Черная металлургия, 1969, № 5, с. 57—61.
37. Margrave Л L. — Collog. Int. CNRS, 1972, № 205, р. 71—77.
38. Guggenheim Е. А.— Mixtures. Oxford, 1952, 270 с.
39. Шахпаронов М. И. Введение в молекулярную теорию растворов.
М.: Гостехтеориздат, 1956, 507 с.
40. Бурылев Б. П.— ЖФХ, 1967, т. 41, с. 676—680.
41. Вагнер К. Термодинамика сплавов.: Пер. с англ. М.: Металлург-
издат, 1957. 179 с.
42. Chipman J. — J. Iron and Steel Inst, 1955, № 180, p. 97.
43. Lupis С. H. P.t Elliott J. F. — Acta Met., 1966, v. 14, p. 529, 1019.
44. Wada H., Pehlke R. D.— Met Trans., Ser. 1977, v. 8 B, p. 443—
450.
45. Chipman J., Corrigan D. A. — Trans. Met. Soc. AIME, 1965, v. 233,
№ 7, p. 1249—1252.
46. Большов JI. А., Григорян В. А., Стомахин A. Я. — Изв. вузов.
Черная металлургия, 1973, № 1, с. 53—55.
47. Большов JI. А., Григорян В. А., Стомахин А. Я. — ФММ, 1972, т. 34,
вып. 6, с. 1139—1144.
48. Большов J1. А., Григорян В. А., Стомахин А. Я. — Изв. вузов. Чер-
ная металлургия, 1972, № 11, с. 60—62.
49. Stull D. R., Prophet Н.— JANAF Thermochemical Tables., 2—d ed.,
US NBS, Wash., 1971, 1141 p.; Suppl.: J. Phys, and Chem. Rev.
Data, 1974, v. 3, p. 311—480; 1975, v. 4, p. 1—175; 1978, v. 7, p. 793—
940; 1982, v. 11, p. 695—940.
50. Шварцман Л. А., Томилин И. A.—Успехи химии, 1957, № 26,
с. 554.
51. Пономаренко А. Г. — ЖФХ, 1972, т. 46, № 8, с. 1950—1958.
52. Turkdogan Е. Т., Pearson J. — J. Iron and Steel Inst, 1953, v. 173,
№ 3, p. 217—223.
53. Turkdogan E. T„ Pearson J. — J. Iron and Steel Inst, 1953, v. 173,
№ 12, p. 393—398.
54. Winkler T. B., Chipman J. —Trans AIME, 1946, v. 167, p. 111.
55. Herasymenko P. — Transactions of the Faraday Soc., 1938, № 34,
p. 1245.
56. Herasymenko P., Speit G. E. — J. Iron and Steel Inst., 1950, v. 166,
p. 169.
260
57. Есин О. А, Электролитическая природа жидких шлаков. Сверд-
ловск: Дом техники Уральского индустриального ин-та им. С. М.
Кирова, 1946, 41 с.
58. Кожеуров В. А. Термодинамика металлургических шлаков. Сверд-
ловск: Металлургиздат, 1955, 163 с.
59. Темкин М. Я. —ЖФХ, 1946, т. 20, с. 105.
60. Самарин А. М., Шварцман Л. А. — Изв. АН СССР, ОТН., 1948,
№ 9, с. 1457.
61. Самарин А. AL, Шварцман Л. А. — Успехи химии, 1954, № 1,
с. 24.
62. Явойский В. И. Теория процессов производства стали. М.: Метал-
лургия, 1967. 518 с.
63. брмонт Б, Ф. Введение в физическую химию и кристаллохимию
полупроводников. М.: Высшая школа, 1973. 655 с.
64. Киреев П. С. Физика полупроводников. М.: Высшая школа, 1975,
584 с.
65. Иоффе А. Ф. Полупроводники в современной физике. М.—Л.: АН
СССР 1954. 356 с.
66. Пономаренко А. Г. — ЖФХ, 1974, т. 48, № 7, с. 1668—1671.
67. Пономаренко А. Г., Козлов Ю. Е., Морозов А. Н. — Изв. АН
СССР. Металлы, 1974, № 3, с. 64—69.
68. Gokcen N.— J. of Metals, 1956, v. 8, № 11, p. 1558—1567.
69. Поверхностные явления в металлургических процессах: Труды
межвузовской конференции. М.: Металлургиздат, 1963, 267 с.
70. Поверхностные явления в расплавах и возникающих из них твер-
дых фазах. Нальчик: Кабардино-Балкарское книжное изд-во, 1965,
652 с.
71. Поверхностные явления в расплавах. Киев: Наукова думка, 1968,
488 с.
72. Смачиваемость и поверхностные свойства расплавов и твердых
тел. Киев: Наукова думка, 1972, 347 с.
73. Адгезия расплавов. Киев: Наукова думка, 1974, 222 с.
74. Физическая химия конденсированных фаз сверхтвердых материа-
лов и их границ раздела. Киев: Наукова думка, 1975, 231 с.
75. Попель С. И. Теория металлургических процессов. М.: ВИНИТИ,
1971, 131 с.
76. Жуховицкий А. А. —ЖФХ, 1944, т. 18, № 5—6, с. 113—120.
77. Хабаиш Ф. Основы прикладной металлургии. Т. I. М.: Метал-
лургия, 1975, 231 с.
78. Жуховицкий А. А., Григорян В. А., Михалик Е. — ДАН СССР, 1964,
т. 155, № 2, с. 392—394.
79. Григорян В. А., Минаев Ю. А. — В кн.: Физико-химические основы
производства стали. М.: Наука, 1971, с. 582.
80. Еременко В. М., Найдич Ю. В.—ЖФХ, 1959, т. 33, № 6, с. 1238—
1245.
81. Ниженко В. И., Флока Л. И. — В кн.: Поверхностные явления в
расплавах. Киев: Наукова думка, 1968, с. 130—138.
82. Floridis Т. Р., Chipman J. — Trans. Met. Soc. AIME, 1958, v. 212,
p. 549.
83. Аверин В. В., Поляков А. Ю., Самарин А. М. — Изв. АН СССР.
ОТН, 1955, № 3, с. 90—107.
84. Taylor С. R., Chipman J. — Trans. AIME, 1943, v. 154, p. 228.
85. Линчевский Б. В, — Изв. АН СССР. Металлы, 1971, № 4, с. 79—83.
86. Janke D„ Fischer W. Л, —Arch. Eisenhuttenwesen, 1975, Bd. 46,
№ 5, S. 297—302.
261
87. Tankjns E. C., Gokcen N. A,, Belton G. R.— Trans. Met. Soc. AIME,
1964, v. 230, p. 820—827.
88. Turkdogan E. T., Hancock R. A., Herlitz S. J. — J. Iron and Steel
Inst., 1956, v. 182, pt. 3, p. 274.
89. Schurniann E., Rinikus H. J. — Giessereiforschung, 1974, Bd. 26,
№ 4, S. 175—179.
90. Григорян В. А., Жуховицкий A, A. — Изв. АН СССР. Металлы,
1969, № 5, с. 61—66.
91. Bundy F. D. — J. Chem. Phys., 1963, № 38, p. 618—630.
92. Leider R., Krikorian О. H., Young D. A. — Carbon, 1973, № 11,
p. 555—563.
93. Кочетов А. И., Стомахин А. Я., Григорян В, А., Натансон К. Ю.—
Изв. вузов. Черная металлургия, 1976, № 1, с. 65—71.
94. Schenck Н., Frohberg М. G., Steinmetz Е. — Arch. Eisenhuttenursen,
1963, Bd. 34, № 1, S. 37—42.
95. Поляков А. Ю. Теоретические основы рафинирования сталепла-
вильной ванны. М.: Наука, 1975, 207 с.
96. Электрометаллургия стали и ферросплавов/Поволоцкий Д. Я-, Ро-
щин В. Е., Рысс М. А. и др. М.: Металлургия, 1984. 568 с.
97. Вайсбурд С. Е., Дюбанов В. Г., Зедина И. Н. и др. — Изв. вузов.
Черная металлургия, 1972, № 1, с. 53—55.
98. Юрин В. В., Котельников Г. И., Стомахин А. Я., Григорян В. А. —
Изв. вузов. Черная металлургия, 1986, № 11, с. 40—45
99. Финчем С., Ричардсон Ф. — Проблемы современной металлургии,
1955, № 1, с. 5—24.
100. Richardson F. D., Physical Chemistry of Melts in Metallurgy, v. 1,
2, L—N. Y, 1974 Acad. Press, 537 p.
101. Сазонов M. Л., Шалимов А. Г., Меркулов Ю, M. и др. Теория ме-
таллургических процессов: Науч. тр./ЦНИИЧМ. М.: Металлургия,
1967, № 50, с. 14—22.
102. Рафинирование стали синтетическими шлаками /Воинов С. Г., Ша-
лимов А. Г., Косой Л. Ф., Калинников Е. С. М.: Металлургия,
1970. 464 с.
103. Жуховицкий А. А., Григорян В. А. — Изв. АН СССР. Металлы,
1970, № 4, с. 27—33.
104. Сталеплавильное производство: Справочник. Т. 1, 2/Под ред.
А. М. Самарина. М.: Металлургия, 1964, 527; 1039 с.
105. Healy G. IT. — J. Iron and Steel Inst., 1970, № 7, p. 664—668.
106. Петрухин С. H., Григорян В. А., Стомахин А. Я- и др.— Изв. ву-
зов. Черная металлургия, 1975, № 3, с. 76—80.
107. Азот в металлах/Аверин В. В., Ревякин А. В., Федорченко В. И.,
Козина Л. Н. М.: Металлургия, 1976, 221 с.
108. Морозов А. Н, Водород и азот в стали. М.: Металлургия, 1968,
281 с.
109. Взаимодействие газов с металлами — В кн.: Труды III советско-
японского симпозиума по физико-химическим основам металлурги-
ческих процессов. М.: Наука, 1973, 215 с.
110. Взаимодействие металлов и газов в сталеплавильных процессах:
Науч. тр./МИСиС. М.: Металлургия, 1973, № 79, 292 с.
111. Pehlke R. D., Elliott J. F. —Trans. Met. Soc. AIME, 1963, v. 227,
№ 5, p. 849—855.
112. Свяжин А. Г., Чурсин Г. M., Вишкарев А. Ф., Явойский В. И.—
Металлы, 1974, № 5, с. 24—35.
113. Стомахин А. Я-, Поляков А, Ю, — Изв. вузов. Черная металлур-
гия, 1967, № 3, с. 116—121.
262
114. Проблемы дегазации металлов/Кунин Л. Л., Головин А. М., Суро-
вой Ю. Н., Хохрин В. М. М.: Наука, 1972, 324 с.
115. Бан-я Ш„ Фува Т. — В кн.: Физико-химические основы металлур-
гических процессов. М.: Наука, 1973, с. 225—235.
116. Lange К. W.> Schenk H. — Z. Metallkunde, 1969, Bd. 60, № 8,
S. 638.
117. Явойский В. И., Лузгин В. П., Вишкарев А. Ф. Окисленность ста-
ли и методы ее контроля. М.: Металлургия, 1970, 285 с.
118. Поволоцкий Д. Я- Раскисление стали. М.: Металлургия, 1972,
207 с.
119. Кнюппель Г, Раскисление и вакуумная обработка стали: Пер. с
нем. М.: Металлургия, 1973, 311 с.
120. Аверин В. В., Самарин А. М. — Изв. АН СССР. Металлургия и
топливо, 1961, № 5, с. 3—10.
121. Куликов И. С. Раскисление металлов. М.: Металлургия, 1975,
504 с.
122. Fischer A., Fleischer FL J. — Archiv Eisenhiittenwesen, 1961,
Bd. 1, S. 1—10.
123. Мияшита И., Нишикава K-, Немото X.— В кн.: Взаимодействие
газов с металлами. М.: Наука, 1973, с. 50—59.
124. Chino Н. — Tetsu to Hagane, 1966, v. 52, p. 559—966.
125. Segawa K.t Tsunetomi — Trans. Iron and Steel Inst. Japan, 1969,
№ 9, p. 89—95.
126. Suzuki Kenichiro, Sanbongi Koji — J. Iron and Steel Inst. Japan,
1972, v. 58, № 12, p. 1594.
127. Metallurgical Chemistry of Iron and Steel. Symposium. July, 1971,
Sheffield; Iron and Steel Inst., 1973, 425 p.
128. Фаст Дж. Д. Взаимодействие металлов с газами. Т. 2. М.: Метал-
лургия, 1958, 351 с.
129. Сталь, 1973, № 7, с. 590—616.
130. Шульте Ю. А. Неметаллические включения в электростали. М.:
Металлургия, 1964, 207 с.
131. Виноград М. И., Громова Г. П. Включения в легированных ста-
лях и сплавах. М.: Металлургия, 1972, 214 с.
132. Кислинг Р., Ланге Н. Неметаллические включения в стали: Пер.
с англ. М.: Металлургия, 1968, 121 с.
133. Нарита Н. Кристаллическая структура неметаллических включе-
ний в стали: Пер. с яп. М.: Металлургия, 1969, 191 с.
134. Литвинова Т. И., Пирожкова В. FL, Петров А. К- Петрография не-
металлических включений. М.: Металлургия, 1972, 184 с.
135. Шпис X. И. Поведение неметаллических включений в стали при
кристаллизации и деформации: Пер. с нем. М.: Металлургия, 1971,
126 с.
136. Явойский В. И., Близнюков С. А., Горохов Л. С., Вишкарев А. Ф.—
В кн.: Рафинирование и кристаллизация стали. Ч. I. М.: Наука,
1974, с. 1—15.
137. Turpin М. L., Elliot J. F. — Iron and Steel Inst., 1966, v. 204, № 3,
p. 217.
138. Мчедлишвили В. А., Гонгадзе Г. А., Самарин A. M. — Изв. АН
СССР. Металлы, 1972, № 5, с. 10—19.
139. Поппель С. И. — В кн. Физико-химические основы производства
стали. М.: Наука, 1964, с. 15—18.
140. Баптизманский В. И„ Бахман Н. К., Дмитриев Ю. В. — Изв. ву-
зов. Черная металлургия, 1969, № 3, с. 42—45.
141. Хлынов В. В., Сорокин Ю. В., Горновой В. А. — В кн.: Физиче-
263
ская химия поверхностных явлений в расплавах. Киев: Наукова
думка, 1971, с. 234—237.
142. Строганов А. И., Дробышевский А. С., Поволоцкий Д. Я. — Изв.
вузов. Черная металлургия, 1972, № 2, с. 64—66.
143. Обработка металла инертными газами/Опкс Г. Н., Степанов А. В.г
Мелехов 77. И. и др. М.: Металлургия, 1969, 112 с.
144. Хуснояров К. Б., Власов Н. И. Производство стали: Науч, тр./
/НИИМ. М.: Металлургия, 1972, № 16, с. 11—18.
145. Еланский Г. И., Кудрин В. А., Агеев Е. Е., Бабич В. К- — Изв.
вузов. Черная металлургия, 1974, № 5, с. 44—48.
146. Дерягин Б. В. Что такое трение? М.: АН СССР, 1963, 227 с.
147. Попель С. И., Хлынов В. В., Дерябин А. А. — В кн.: Физико-хими-
ческие основы металлургических процессов. М.: Наука, 1973, 308 с.
148. Хлынов В. В., Сорокин Ю. В., Стратонович В. И. — В кн.: Физико-
химические исследования металлургических процессов. Сверд-
ловск: УПИ, 1973, с. 114—126.
149. Минаев Ю. А. — ЖФХ, 1969, т. 23, № 9, с. 2379—2383.
150. Минаев Ю. А., Русанов А. И.—Изв. АН СССР. Металлы, 1971,
№ 5, с. 59—66.
151. Минаев Ю. А., Уточкин Ю. И., Григорян В. А. — Изв. АН СССР.
Металлы, 1971, № 6, с. 15.
152. 77лекингер Е., Вальстер М. — Проблемы современной металлур-
гии, 1960, № 6 (54), с. 82—107.
153. Plockinger Е. — J. Iron and Steel Inst, 1963, v. 201, № 7, p. 576.
154. Ленинских Б. M., Кайбычев А. В., Савельев Ю. А. Диффузия эле-
ментов в жидких металлах группы железа. М.: Наука, 1974,
с. 191.
155. Бокштейн Б. С., Бокштейн С. 3., Жуховицкий А. А. Кинетика диф-
фузии в твердых телах. М.: Металлургия, 1974, 280 с.
156. Астарита Дж. Массопередача с химической реакцией. Л.: Химия,
1971, 329 с.
157. Левич В. Г. Физико-химическая гидродинамика. М.: Физматгиз,
1959, 659 с.
158. Эккерт Э. Р., Дрейк Р. М. Теория тепло- и массообмена и перено-
са. М.—Л.: Госэнергоиздат, 1961, 680 с.
159. Франк-Каменецкий Д. А. Диффузия и теплопередача в химической
кинетике. М.: Наука, 1967, 491 с.
160. Физико-химические основы металлургических процессов/Жуховиц-
кий А. А., Белащенко Д. К, Бокштейн Б. С., Григорян В. А.,
Григорьев Г. А., Гугля В. Г.: Металлургия, 1973, 391 с.
161. Самарин А. М. — В кн.: Физико-химические основы металлургиче-
ских процессов/Труды 1-го советско-японского симпозиума. М.:
Наука, 1969, с. 128—145.
162. Вигдорчик Е. М., Шейнин А. Б. Математическое моделирование
непрерывных процессов растворения. Л.: Химия, 1971, 246 с.
163. Попель С. И., Павлов В. В. — Журнал Всесоюзного химического
общества им. Д. И. Менделеева, 1971, т. XVI, № 5, с. 523—528.
164. Процессы взаимной диффузии в сплавах/Под ред. Гурова К. П.
М.: Наука, 1973. 359 с.
165. Григорян В. А. — Журнал Всесоюзного химического общества
им. Д. И. Менделеева, 1971, т. XVI; № 5, с. 535—541.
166. Григорян В. А., Алеев Р. А. — В кн.: Смачиваемость и поверхност-
ные свойства расплавов и твердых тел. Киев: Наукова думка,
1972, с. 189—192.
167. Филиппов С. И. Теория металлургических процессов. М.: Метал-
лургия, 1967, 279 с.
264
168. Травин О. В., Мокрова В. П., Паршин С. И. — Теория металлурги-
ческих процессов: Науч. тр./ЦНИИЧМ. М.: Металлургия, 1971,
с. 33—37.
169. Григорян В, А., Швиндлерман Л. С,, Алеев Р. А. — Изв. вузов.
Черная металлургия, 1968, № 5, с. И —17.
170. Wahlster М.— Proceeding of the IV International conference on
vacuum metallurgy, Tokyo, June 4—8, 1973, The Iron and Steel
Inst, of Japan, Tokyo, 1973, p. 56—61.
171. Патон Б. E., Тихоновский А. Л. — В кн.: Рафинирующие пере-
плавы. Киев: Наукова думка, 1974, с. 150—162.
172. Тихоновский А. Л. — В кн.: Рафинирующие переплавы. Киев: Нау-
кова думка, 1974, с. 167—179.
173. Лакомский В. И. Плазменно-дуговой переплав. Киев: Техника,
1974, 335 с.
174. Сергеев А, Б., Швед Ф. И., Тулин Н. А. Вакуумный дуговой пере-
плав конструкционной стали. М.: Металлургия, 1974, 191 с.
175. Серебрийский 3. И,, Полин И. В, — Металлургия. Л.: Судпромгиз,
1959, № 2, с. 22—32.
176. Есин О. А., Гельд П. В. Физическая химия пирометаллургических
процессов. Ч. I. М.: Металлургиздат, 1962, 703 с.
177. Еременко В. Н.—Украинский химический журнал, 1962, т. XXVIII,
вып. 4, с. 427—449.
178. Белянчиков Л, Н. — Изв. вузов. Черная металлургия, 1965, № 7,
с. 74—76.
179. Латаш Ю. В., Медовар Б. И. Электрошлаковый переплав. М.: Ме-
таллургия, 1970, 239 с.
180. Клюев М. М., Каблуковский А. Ф. Металлургия электрошлаково-
го переплава. М.: Металлургия, 1969, 256 с.
181. Волков С. Е. — В кн.: Специальная электрометаллургия. Киев:
Наукова думка, 1972. Ч. I, с. 9—17.
182. Кей А — В кн.: Специальная электрометаллургия. Киев: Наукова
думка, 1972. Ч. 2, с. 47—63.
183. Медовар Б. И., Емельяненко Ю. Г., Тихонов В. А.—В кн. Рафи-
нирующие переплавы. Киев: Наукова думка, 1975, с. 73—81.
184. Kojima У., Kato М., Toyoda Т., Inouye М. — Tnans. Iron, and Steel
Inst, of Japan, 1975, v. 15, № 8, p. 397—406.
185. Вычугов Г. А., Хлынов В, В., Хасин Г. А.—Сталь, 1967, № 6,
с. 514—516.
186. Абрамов О. В. Кристаллизация металлов в ультразвуковом поле.
М.: Металлургия, 1972, 320 с.
187. Лемпицкий В. В., Окороков Г. Н., Бояршинов В, А. — Сталь,
1972, № 3, с. 224—229.
188. Каменский Ю. М., Сухотин Б. Н., Явойский В. И. — Автоматиче-
ская сварка, 1965, № 10, с. 69—71.
189. Смелянский М. Я-, Малышев С. А., Ткачев Л. Г., Гуттерман К- Д-—
Электротермия, 1964, № 39, с. 18—20.
190. Заборонок Г. Ф., Зеленое Т. И., Ронжин А. С., Соколов Б, Г. —
Электронная плавка. М.: Металлургия, 1966, 291 с.
191. Антропов О. Ф., Устин К. П.— Электротермия, 1969, № 87, с. 21 —
22.
192. Патон Б. Е., Мовчан Б. А., Тихоновский А. Л. — В кн.: Рафини-
рующие переплавы. Киев: Наукова думка, 1974, с. 179—195.
193. Тихоновский А. Л. — В кн.: Рафинирующие переплавы. Киев: Нау-
кова думка, 1975, с. 125—131.
194. Дюбанов В. Г., Ломберг Б. С., Герасимова Т. Д. и др. — Изв. ву-
зов. Черная металлургия, 1975, с. 63—66.
265
195. Литейное производство/Под ред. Куманина И. Б. М.: Машино-
строение, 1971, 319 с.
196. Хворинов Н. И. Кристаллизация и неоднородность стали. М.: Маш-
гиз, 1958, 392 с.
197. Вейник А. И. Теория особых видов литья. М.: Машгнз, 1958,
300 с.
198. Flemings М. S.— Modern Castings, 1964, v. 46, № 1, р. 31—36.
199. Бартон, Прим и Слихтер.— В кн.: Германий, М.: ИЛ, 1955, с. 74—
81.
200. Белянчиков Л. Н., Савченко Г. Г., Григорян В. А. — Изв. вузов.
Черная металлургия, 1975, № 7, с. 71—74.
201. Голиков И, Н. Дендритная ликвация в стали. М.: Металлургиздат,
1958, 206 с.
202. Петрухин С. Н., Григорян В. А., Стомахин А. Я. и др. — Изв. ву-
зов. Черная металлургия, 1977, № 5, с. 65—67.
203. Пономаренко А. Г., Козлов Ю, С. — Изв. вузов. Черная металлур-
гия, 1975, № 5, с. 20—24.
204. Островский О, И., Вайсбурд С. Е., Григорян В. А. и др. — Изв.
вузов. Черная металлургия, 1976, № 3, с. 55—58.
205. Термические константы веществ/Atedeedee В. А., Бергман Г. Л.,
Гурвич Л. В. и др.: Справочник/Отв. ред. В. П. Глушко. М.:
ВИНИТИ, 1965—1981. Вып. 1 — 10.
206. Термодинамические свойства индивидуальных веществ/Г ур-
вич Л. В., Вейц И, В„ Медведев В. А и др.: Справочное издание.
Т. 1—4/Отв. ред. В. П. Глушко. М.: Наука, 1978—1982.
207. Суровой Ю. Н., Окороков Г. H.t Нефедерова С. Л., Кузнецов А. Л.—
В кн.: Теория металлургических процессов. М.: Металлургия, 1972,
с. 29—38.
208. Stomakhin Л. Ya., Grigoryan V. A. The Eighth Japan — USSR Joint
Symposium on Physical Chemistry of Metallurgical Processes. June-
16—18, 1981. Tokyo, Japan, p. 129—143.
209. Большов Л. A. — Изв. вузов. Черная металлургия, 1982, № 1,
с. 8—10.
210. Соколов В. М., Большов Л. А. — Изв. вузов. Черная металлургия,
1982, № 9, с. 5—8.
211. Большов Л. А., Стомахин А. Я-, Соколов В. М., Тетерин В. Г.—
Изв. АН СССР. Металлы, 1984, № 5, с. 60—62.
212. Elliott J. F.— Ellectric Furnace Steel Conf. Proc. v. 32, AIME, 1975,
p. 62—74.
213. Агеев Ю. Л., Арчугов С. A. — Изв. АН СССР. Металлы, 1984, № 3,
с. 78—80.
214. Рысс Г. М., Строганов А. И., Есин Ю. О., Гельд П. В.—ЖФХ,
1976, т. 50, № 3, с. 771—772.
215. Kubaschewskl О., Geiger К.—Н., Hack К. — Z. Metallkunde, 1977,
v. 68, № 5, р. 337—341.
216. Бертман А. А., Самарин А. М. Свойства расплавов железа. М.:
Наука, 1969, 280 с.
217. Есин Ю. О., Баев В. М., Морозов С. Н. — Физические свойства ме-
таллов и сплавов: Науч. тр./УПИ. Свердловск: УПИ, 1976, № 1,
с. 66—72.
218. Баталин Г. И., Стукало В. А., Нещименко Н. Я- и др. — Изв. АН
СССР. Металлы, 1977, № 6, с. 44—45.
219. Ban—ya S., Maruyama N., Kawase Y.— Tetsu to Hagane, 1984,
v. 70, p. 65—72.
220. Lupis С. H. P.— Chemical Thermodynamics of Materials. N.—Y.:
Elsevier Sci. Publ. Co., Jnc., 1983, 581 p.
266
221. Григорян В. А., Белянчиков Л. Н., Стомахин А. Я- Теоретические
основы электросталеплавильных процессов. 1-е изд-е. М.: Метал-
лургия, 1979. 256 с.
222. Даркен Л. С., Гурри Р. В. Физическая химия металлов. Пер. с
англ./Под ред. акад. Н. Н. Сироты. М.: Металлургиздат, 1960,
582 с.
223. Самарин A. М., Карасев Р. Л. —ДАН СССР, 1958, т. 119, № 5,
с. 990______992.
224. Sherman С. W., Elwander Н. Chipman Л —Trans AIME, 1950,
v. 188, р. 334.
225. Ban—ya S., Chipman J. — Trans. Met. Soc. AIME, 1968, v. 242,
p. 942.
226. Alcock С. B., Cheng L. L.— J. Iron and Steel Inst., 1960, v. 195,
pt. 2, № 6, p. 169—173.
227. Cordier J. A., Chipman J. — J. Metals, 1955, v. 7, № 8, p. 905.
228. Уточкин Ю. И„ Менделев В. А., Григорян В. Л., Вестфаль С. В. —
Изв. вузов. Черная металлургия, 1983, № 7, с. 5—9.
229. Рафинирующие переплавы стали и сплавов в вакууме/Боярши-
нов В, Л., Шалимов Ал. Г., Щербаков Л. И. и др. Под ред.
В. А. Бояршинова. М.: Металлургия, 1979. 304 с.
230. Гурвич Л. В. — Вестник АН СССР, 1983, № 3, с. 54.
231. Доронин Г. И., Гузенков В. Л., Белянчиков Л. Н. — Изв. вузов.
Черная металлургия, 1982, № 7, с. 152.
232. Менделев В. Л., Уточкин Ю. И., Григорян В. Л.— Изв. вузов.
Черная металлургия, 1980, № 7, с. 40—44.
233. Изв. вузов. Черная металлургия, 1986, № 9, с. 132—141.
234. Туркдоган Е. Т. Физическая химия высокотемпературных процес
сов. Пер. с англ./Под ред. В. А. Григоряна. М.: Металлургия, 1985,
344 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
А
Аверина зависимость 196
Авогадро число 36
Автоэмульгирование 103
Адсорбция 91
Азот в расплавах 135—143
Азотопроницаемость шлака 69
Активность кислорода в жидком
металле 146, 156
— межфазная 102
— методы определения 26—28
— поверхностная 102
— растворов 22—26
— чистого оксида 149
— элементов-раскислителей 149—
155
Архимеда формула 161
Б
Белла реакция 172
Био критерий 238
Быстрота действия вакуумного
насоса 206
— поглощения газа 206
В
Вант-Гоффа уравнение 11
Водород в расплавах 143—145
Водородопроницаемость шлаков 69
Вязкость динамическая 175
— кинематическая 175
— шлаков 70
Взаимодействие серы с газовой
фазой 129, 130
----с сульфидообразующими эле-
ментами 130—132
Г
Газопоглотительное действие паро-
вого конденсата 206
Газопроницаемость шлака 69
Генри законы 17—19, 20, 22
— константа 18
Герценит 152
Гиббса формула 91
Гиббса энергия 6
----избыточная 50
----образования соединений 8—
10
---- приведенная 7
267
____ свободная 6
_— стандартная 6, 7
Гиббса—Гельмгольца уравнение
57
Гиббса—Дюгема уравнение 16, 17,
21, 28, 30, 31
Гиббса—Фольмера теория 157
Гильдебранда понятие регулярно-
го раствора 34, 35
Гуггенхейма теория 47
Гугенгейма—Адама уравнение 91,
92
Графический метод определения
парциальных характеристик 15, 16
Д
Давление расклинивающее 168
Дамкелера критерий 200
Десульфурация при ЭШП 223—
229
Дефекты слитка ВДП 209, 210
Дефосфорация металла 133—135
Диффузия конвективная 176—179
— молекулярная 173—175
Доли компонента атомные 12
----массовые 12
----молекулярные 12
----мольные 12
Е
Емкость шлака сульфатная 125
----сульфидная 125—129
Ж
Жидкоподвижность шлаков 70
Жуховицкого уравнение 94
3
Закон действующих масс идеаль-
ный 72, 73, 76
— квадратного корня 135
Зародышеобразование гетероген-
ное 158
— гомогенное 159
И
Избыточная энергия Гиббса 34,35
Изобарно-изотермический потен-
циал 6
Интенсивность зародышеобразова-
ния 157
К
Кислород в расплавах 109—112
----взаимодействие с углеродом
115—120
Кислородопроницаемость шлака
69
Кнюппеля зависимость 212
Коагуляция ортокинетическая 159
— перикинетическая 159
Кожеурова теория 77, 78
— формула 78
Константа равновесия реакции 64
— скорости процесса удаления
азота 207
--------- водорода 204
Концентрация равновесная 64
Коэффициент активности 23. 28
--- водорода 144
--- серы 124
— диффузии 173, 175, 177
— массопередачи 173, 177—179
— массопереноса серы 225
— перераспределения эффектив-
ный 252
— поверхностного натяжения 90,
91
— распределения серы 127—129
---фосфора 134, 135
— растворимости азота 136
— растекания 107
— температуропроводности 203,
253
— - формы ванны 245
Краевой угол смачивания 104,106,
166
Критическая концентрация водо-
рода 204
Л
Дамба уравнение 164
Лангмюра уравнение 93, 94. 186
Ле-Шателье принцип 11
Ликвация дендритная 252—255
Лимитирующая стадия гетероген-
ного процесса 180—182
Лопиталя правило 48
Люписа соотношение 25
М
Массоперенос компонентов в шла-
ке 191—194
Машина непрерывного литья заго-
товок 236
— полунепрерывного литья заго-
товок 236
Межфазное натяжение между ме-
таллом и шлаком 101—103
---------динамическое 102. 103
--------- неравновесное 103
--------- статическое 102
Метод калориметрический 123
— определения термодинамичес-
ких функций шлака 76—78
— теоретической оценки активно-
стей компонентов 86
Механизм образования пузырей
189—191
— удаления включений 169—171
268
-------в условиях вакуума 171 —
173
Моль 13
— масса 13
Мольная доля насыщения 31
Н
Направление химической реакции
11
Насосы испарительно-гетерогенные
206
Неметаллические включения 156 —
173
----классификация 157
Нитридообразование 139, 140
— условия 139
Нуссельта критерий 179
О
Обезуглероживание 120, 185—189
— температурный порог 187
Основность шлаков 70
Отклонение от законов Рауля и
Генри 22
---------отрицательное 23, 24
---------положительное 24, 27
Отливка разового формирования
236
П
Параметры взаимодействия эле-
ментов 49—63
-------второго порядка 58, 59
первого порядка 54—56
— энтальпийные 57
— энтропийные 57
Пекле----------------------критерий 179
Перемешивание шлака 193
Переокисление расплава 187
Переохлаждение концентрацион-
ное 250, 251
— термическое 251
Плотность шлака 70, 71
Поведение элементов при ЭШП:
азота 228, 229
водорода 229—231
серы 223—228
Поверхностно-активные вещества
165, 184—186
Поверхностное натяжение жидких
металлов 90—92
----коэффициент 90, 91
----чистых металлов 93
----шлаков 100
Поверхностное сгущение 90
Подвижность примесей диффузи-
онная 69
Полнота протекания реакции 10,
11
Попеля—Павлова уравнение 95
96
Пористость осевая 256, 257
Поток азота 192, 193
— диффузионный 174, 177
— кислорода 192, 193
Правило фаз 64
Предел растворимости ПО
Продолжительность полного за-
твердевания слитка 238
Процесс гетерогенный 180— 182,
184—186
— квазистационарный 236
— переплава трехстаднйный 197
Р
Работа адгезии 91
— когезии 91
— разрыва столба жидкости 91
Равновесие химических реакций
63—68
Радиус капли 197, 198
— пузыря равновесный 189
Раковина усадочная 255, 256
Раскисление металла 146—156
---- глубинное 156
----диффузионное 155, 156
Раскислители комплексные 151,
155
Раскислительная способность эле-
ментов 149—155
----шлака 155, 156
Распределение элементов между
металлом и шлаком 85—89
----константа 85, 86
Растворимость азота в железе 136,
137
----в никеле 141, 142
----в хроме 142, 143
— водорода в железе 143—145
— кислорода в железе ПО, 111
---- в никеле 111, 112
— оксида ПО
— серы в шлаках 126
Растворы 12—16
— активность 22—26
— бесконечно разбавленные 29;.
30
— идеальные 17—22
— квазирегулярные 41, 42
— насыщенные 31
— разбавленные 18, 20—22
— реальные 22—26, 29—33
— регулярные 34—40
— свойства интенсивные 13
----экстенсивные 12
— совершенные 13, 18—20
269
— состав 12
— субрегулярные 40, 41
Растекание 107, 108
— коэффициент 107
— металла и шлака 108
Рауля законы 17—19, 22
Рафинирование металла при пере-
плавных процессах 194—236
----при ВДП 202—218
----при ЭЛП 231—236
----при ЭШП 218—231
Реакция раскисления 147
— химическая, направление 10,
11
---- полнота протекания 11
Режим температурный процесса
ЭЛП 232
----ЭШП 218, 219
— электрический процесса ЭШП
226, 227
Рыбчинского—Адамара формула
161, 162
Рыхлость усадочная 257
С
Свободная энергия 6
Свойства раствора интенсивные 13
---- экстенсивные 12
— шлаков защитные 69
Сергеева зависимость 207
Сера в расплавах 123—132
— взаимодействие с газовой сре-
дой 129
----с сульфидообразующими эле-
ментами 130—132
----со шлаком 124, 125
Сивертса закон 27, 109, 135, 136
Скорость всплывания включений
161—163
— диффузии 174
— конвективных потоков 163
Слитки непрерывного формирова-
ния 237
— полунепрерывного формирова-
ния 236
— полые 240
— разового формирования 236,
237
Слой пограничный гидродинамиче-
ский 176
----диффузионный 177
Смачивание 103—108
— графита железом и шлаком
106, 107
— оксидов жидкими металлами
105, 106
Сорбция 69
Способ подвода окислителей 186,
187
Способность шлака десульфуриру-
ющая 126
----сорбционная 69, 84, 85, 121
Стадия диффузионная 182, 183
— кинетическая 182, 183
Стандартная энергия Гиббса об-
разования соединений 8—10:
карбидов 8
нитридов 8
оксидов 8, 9
сульфидов 9
фторидов 9
хлоридов 10
Стандартное состояние веществ
6,7
----компонентов 22
Степень дендритной ликвации 255
— перегрева над температурой
ликвидуса 202, 203
— пересыщения 157
— рафинирования 197, 198, 200
— удаления азота 207
----водорода 205
Стефана—Больцмана постоянная
203
Стокса формула 161
Стокса—Эйнштейна формула 175
Стэнтона критерий 208
Т
Тагеева формула 238
Темкина метод 76—78
Температура ликвидуса 202
— солидуса 202
Температурный коэффициент по-
верхностного натяжения 93
Теория квазирегулярных раство-
ров 42
— квазихимическая 47
— плавления твердых тел диффу-
зионная 184
— регулярных ионных растворов
77—80
— регулярных растворов 35—40
— совершенных ионных растворов
76,77
— строго регулярных растворов
47
— субрегулярных растворов 41
— шлаков ионная 68
----молекулярная 68, 69, 71—76
— электронного строения конден-
сированных веществ 82
Термодинамическое условие устой-
чивости включений 166
270
Толщина пограничного диффузи-
онного слоя эффективная 178
Торможение адсорбционное 164,
165
У
Углерод в расплавах 113—115
----взаимодействие с кислоро-
дом 115—120
Удаление при ЭЛП включений
235, 236
----газов и летучих примесей
232—235
Уинклера—Чипмана формула 133
Уравнение конвективной диффу-
зии 178
Уровень Ферми 82
----в оксидных фазах 83
— химического потенциала 82
Устойчивость пленок металла на
границе раздела фаз 167—169
Ф
Фаза рафинирующая 195
Ферми уровень 82
Фика второй закон 174
— первый закон 173
Филиппова теория 188
Формула квадратного корня 237
Фосфор в расплавах 133—135
Френкеля формула 160
Фронт кристаллизации в непре-
рывном слитке 242
Функции шлаков термодинамиче-
ские 80—85
----технологические 69
X
Характеристики мольные интег-
ральные 14, 15
----относительные 14, 15
---- парциальные 13, 15
— шлака технологические 70
Химический потенциал компонен-
та 14, 30
----кислорода 81, 83
----магния 81
---- электронов 83
Ч
Чипмана—Корригана соотношение
59, 60
— уравнение 60, 61
Чуйко теория 73
Ш
Шенка значения констант распре-
деления и диссоциации 72
— теория 72
Шервуда критерий 179
Шишковского формула 93, 97
Шлаки сталеплавильные 68—69
----высокоосновные 70
----кислые 70
— фторидные 223, 228
Э
Электрическая проводимость шла-
ка 70, 71
Элементы гидридообразующие 144,
145
— диффундирующие 174, 175
— нитридообразующие 137, 138,
140
— поверхностно-активные 97
Элемепты-десульфураторы 131
Элементы-раскислители 146—156:
алюминий 151, 152
бор 152
ванадий 152
кальций 153
кремний 150, 151
магний 153
марганец 149, 150,
титан 153
цирконий 152
Энергетические параметры эле-
ментов 87
Энергия активации массопередачи
178
— обмена 87
— смешения 36
Энтальпия мольная интегральная
14
---- парциальная 14
Энтропия образования разбавлен-
ных растворов 20, 21
----совершенных растворов 19
— смешения 87
Эффективная толщина погранич-
ного диффузионного слоя 178
Я
Явления электролизные на элект-
родах 227
Явойского классификация неме-
таллических включений 156, 157
271
НАУЧНОЕ ИЗДАНИЕ
Вули Аршакович ГРИГОРЯН
Лев Николаевич БЕЛЯНЧИКОВ
Александр Яковлевич СТОМАХИН
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
ЭЛЕКТРОСТАЛЕПЛАВИЛЬНЫХ ПРОЦЕССОВ
Редактор издательства Е, К. Полторацкая
Художественный редактор /О. И. Смурыгин
Технический редактор М. И. Воскобойникова
Корректор В. М. Гриднева
ИБ № 2802
Сдано в набор 03.10.86. Подписано в печать 13.04.87. Т-11321. Формат бумаги
B4X108V32. Бумага типографская № 1. Гарнитура литературная. Печать высокая.
Усл. печ. л. 14,28. Усл. кр.-отт. 14,49. Уч.-изд. л. 16,48. Тираж 4600 экз.
Заказ 675. Цена 2 р. 90 к. Изд. № 1113
Ордена Трудового Красного Знамени издательство «Металлургия»
119857, ГСП, Москва, Г-34, 2-й Обыденский пер., д. 14
Владимирская типография Союзполиграфпрома при Государственном
комитете СССР по делам издательств, полиграфии и книжной торговли
600000, г. Владимир, Октябрьский проспект, д. 7
Периодическая система
ПЕРИОДЫ РЯДЫ ГРУППЫ
I - R20 п - R0 ш R2O3 IV rh4 ro2 V RH R205
1 I к Н !S’ ВОДОРОД 1,0079 1
2 л L К . . 3 Li 2s’ ЛИТИЙ 1 6.941 2 4 Be 2sz БЕРИЛЛИЙ г 9,01218 2 5 25г2р1 В з БОР 2 10,81 б л 2522рг С 4 УГЛЕРОД 2 12,011 7 2s22p3 N 5 АЗОТ 2 14,0057
3 ш М L К 11 Na .зз’ НАТРИИ J 22,9897" 2 12 Мд 35% МАГНИИ в 24,305 2 13 А , 3s23Pi Д] б АЛЮМИНИЙ 2 26,98154 14 л. зз2зр2 Si 8 КРЕМНИЙ 2 28,086 15 _ 5 3S23p5 Р 8 ФОСФОР 2 30,97376
4 IV N М L К к 19 , КАЛИЙ | 39.С96 2 „ 20 Са 4s2 г КАЛЬЦИЙ б 40,08 2 „ 21 SC 3d'4s22 СКАНДИЙ в 44,9559 2 . 22 Ti 3d24s22 ТИТАН 47,90 2 lf 23 V 3d34s2 2 ВАНАДИЙ 1J 50,9414 2
V N М L К 29 1 3d’°4s’ Cl! МЕДЬ 2 63,546 зо - 2 3d,04s2 ZD ’5 цинк 2 65,38 31 ~ 3 4S24p1 G 3 15 ГАЛЛИЙ 2 69,72 32 4 4S24p2 (Зб ГЕРМАНИЙ 2 72,59 33 5 4S24p3 AS МЫШЬЯК 2 74,9216
5 0 ь м L К 37 1 Rb 5S1 6 РУБИДИЙ 85,4678 2 п 38 2 Sr 5S2 8 СТРОНЦИЙ 1в 87,62 2 V, 39 2 Y 4d'5s2 9 ИТТРИЙ 'S 88,9059 2 _ 40 2 2Г 4d25S210 ЦИРКОНИЙ '? 91,22 2 Nb 4d45S112 НИОБИЙ 92,9064 2
vn 0 N м L К 1 47 . to 4d,05s’ Ад ’! СЕРЕБРО 2 107,858 2 48 _ . 18 4d’°5s2 Cd '? КАДМИЙ 2 112,49 з 49 . 16 5S25p1 1 П '2 индий 2 114,82 4 - 53 18 5S25p2 SO ’5 олово 2 118,69 5 51 „L 18 5s25p3 SO '5 СУРЬМА 2 121,75
6 7Ш Р 0 N М L К < 55 1 CS ’ 6S1 18 ЦЕЗИЙ 132,9054 2 56 2 Ba 6s2,? ’ БАРИЙ ’2 137,34 2 57 2 IS 5d’6s4 ЛАНТАН ’? 138,9055 2 72 2 Hf 5d26S2^ ГАФНИЙ 'S 176,49 2 73 2 Tfil 5d36s2»2 ТАНТАЛ 4 180,9479 2
|у Р 0 N М L К 1 79 32 5d*36s1 Дц золото г 196,9665 18 80 . . г? 5d,o6s2 Нд '2 РТУТЬ 2 200,59 з 81 _ 32 6S26p’ TI ТАЛЛИЙ 2 204,37 4 82 32 6S26p2 РЬ СВИНЕЦ 2 207,19 18 83 r, . 32 6S26p3 □ | ВИСМУТ 2 208,9804
7 X ”7Г р 0 N М L К _ 07 1 Fr 7SJ ’! ФРАНЦИИ ’§ (223) 1 _ 88 , 5 Ra 7s2i| РАДИЙ 18 226,0254 2 on 2 Ac 6d’7s2j АКТИНИЙ if (227) 5 , . 104 Ku 6d27S2 j2 КУРЧАТОВИИ 18 (261) ? .. 105 ,5 Ns 6d37s’« НИЛЬСБОРИЙ’» (261) f
-D Ct 0 p 0 N M L К 58 2 Ce 4f’5d16S2ig ЦЕРИЙ 140,12 2 59 2 Pr 4f36s2 21 ПРАЗЕОДИМ 140,9077 2 60 2 Nd 4^3 НЕОДИМ '5 144,24 2 Г 61 2 Pm 4f56s22§ ПРОМЕТИЙ ’S (145) 2 62 2 Sm 4f«6s224 САМАРИЙ 150,4 ‘г
P 0 N M L К 55 2 T b 4f 96s2 27 ТЕРБИЙ 158,9254 2 r, 66 8 DV 4f’°6S228 Диспрозий 162,50 2 . 67 2 Ho 4fl16S229 ГОЛЬМИЙ 164,9304 2 88 I ЕГ 4f’26s23® ЭРБИЙ 167,26 2 69 2 ТГП 4F136s23i ТУЛИЙ 168,9342 2 I
’АКТИНОИДЫ p 0 N M L К Th 9M ТОРИИ 14 232,0381 J _ 91 I Pa 5f26d’7s22» ПРОТАКТИНИЙ ’в 231,0359 2 92 g U 5f36d’7s232 УРАН « 238,029 2 93 | ND 5f46d’7s2|| Т1ЕПТУНИЙ 18 237,0482 ? 94 5 Pu 5f67s2|4 ПЛУТОНИЙ ij (244) I
p 0 N M L К 97 § В К sf’edWg БЕРКЛИЙ 18 (247) J 98 Г Cf 5f107s?2f КАЛИФОРНИЙ 14 (251) 2 ~99 Г ES 5f”7s2$ ЭЙНШТЕЙНИЙ 18 (254) ? 100 j Fm 5f’27S23 ФЕРМИИ i§ (257) § 101 3 Md 5f»7s2t’ МЕНДЕЛЕВИИ i| (258) ?
элементов Д.И. Менделеева
ЭЛЕМЕНТОВ
RH2 R03 УК RH R207 ro4 0
(Н) He 2 .is2 ГЕЛИИ 4,00260 2
2s:2p4 О б КИСЛОРОД 2 15,9994 9 г- 2s?2ps F 7 ФТОР 2 16,99840 Ne °2S22ps НЕОН 6 20,179 2
16 л 3s23p4 S В СЕРА 2 32,06 17 - 3s23p5 С] i ХЛОР 2 35,453 16 АГ 3s23p6 АРГОН e 39,946 2
„ 24 С Г 3d’4s’1 ХРОМ 18 51,996 2 .... 25 МП 3d’4s2? МАРГАНЕЦ1» 54,9380 2 _ 25 Fe 3d64s2 2 ЖЕЛЕЗО 1в 55,847 2 л 27 C 0 3d74s2 2 КОБАЛЬТ 58,9332 2 M. 28 N1 3d84S2 2 НИКЕЛЬ 56,71 2
34 л б 4s24p4 S е \э СЕЛЕН 2 78,96 35 7 4s24p5 В Г * * БРОМ ? 79,904 КГ 4s:4p6 S КРИПТОН 18 83,80 2
кл 42 1 Мо 4d55S113 МОЛИБДЕН ’S 95,94 2 _ 43 ' 2 Тс 4d55S213 ТЕХНЕЦИЙ ’J 98,9062 ? n 44 1 RU 4d75sj 15 РУТЕНИЙ ’f 101,07 2 45 1 Rh 4d®5s1 16 РОДИИ 1g 102,9055 • 2 _ . 46 0 P(1 4dW5S°18 ПАЛЛАДИЙ 106,4 2
6 52 ;$ 5s25P4 Те 3 ТЕЛЛУР 2 127,60 7 53 _ ifl 5s25p5 j 'e Йод 2 126,9045 54 в Xe 5sV16 КСЕНОН 1g 131,30 2
74 2 W 5d46s2 32 ВОЛЬФРАМ ’? 183,85 2 75 2 Re 5d’6s4i РЕНИЙ .185,207 2 76 2 Os 5d66s232 осмий igo,2 2 77 2 | Г 5d76s2 32 ИРИДИЙ 192,22 2 78 1 Pt 5d’6S’I? ПЛАТИНА ’f 195,09 2
32 6S26p*4 РО 1 ПОЛОНИЙ 2 (209) 18 85 .1 32 6S26p5 At 'S АСТАТ 2 (210) „ 86 » RD РАДОН ’J (222) 2
•
- 63 Z Eu 4f’6S22I ЕВРОПИЙ 151,95 2 64 2 Gd 4f75d16S225 ГАДОЛИНИЙ 157,25 2
70 2 Yb >f’W2 ИТГгРбИЙ 173,04 z 71 2 Ltl4fw5d16s23z ЛЮТЕЦИЙ 174,97 ?
Атомный номер Распределение электронов
—4-------------' по застраивающимся и по-
следующим застроенным
подуровням
Число электронов в слоях
Am 5^7$гц АМЕРИЦИЙ (243) « 96 | Cm 5f76d’7s2‘S КЮРИЙ 1$ (247) §
NO 15fl47s232 НОБЕЛИЙ 18 (255) I . 103 i Lr5f,a8d17s2 32 ЛОУРЕНСИЙ 18 (256) I
Q
Р
О
N
М
Fe 3d64Sz 2
ЖЕЛЕЗО
у55,647 2
Атомная масса
I электронный слой К
H L
HI 99 M
E 99 N
V 99 0
и 99 P
,4 *1 Q
Целое число в скобках- массовое
число наиболее устойчивого изотопа
искусственного радиоактивного элемента.
Целое число без скобок-массовое число
наиболее распространенного изотопа
природного радиоактивного элемента.